/












































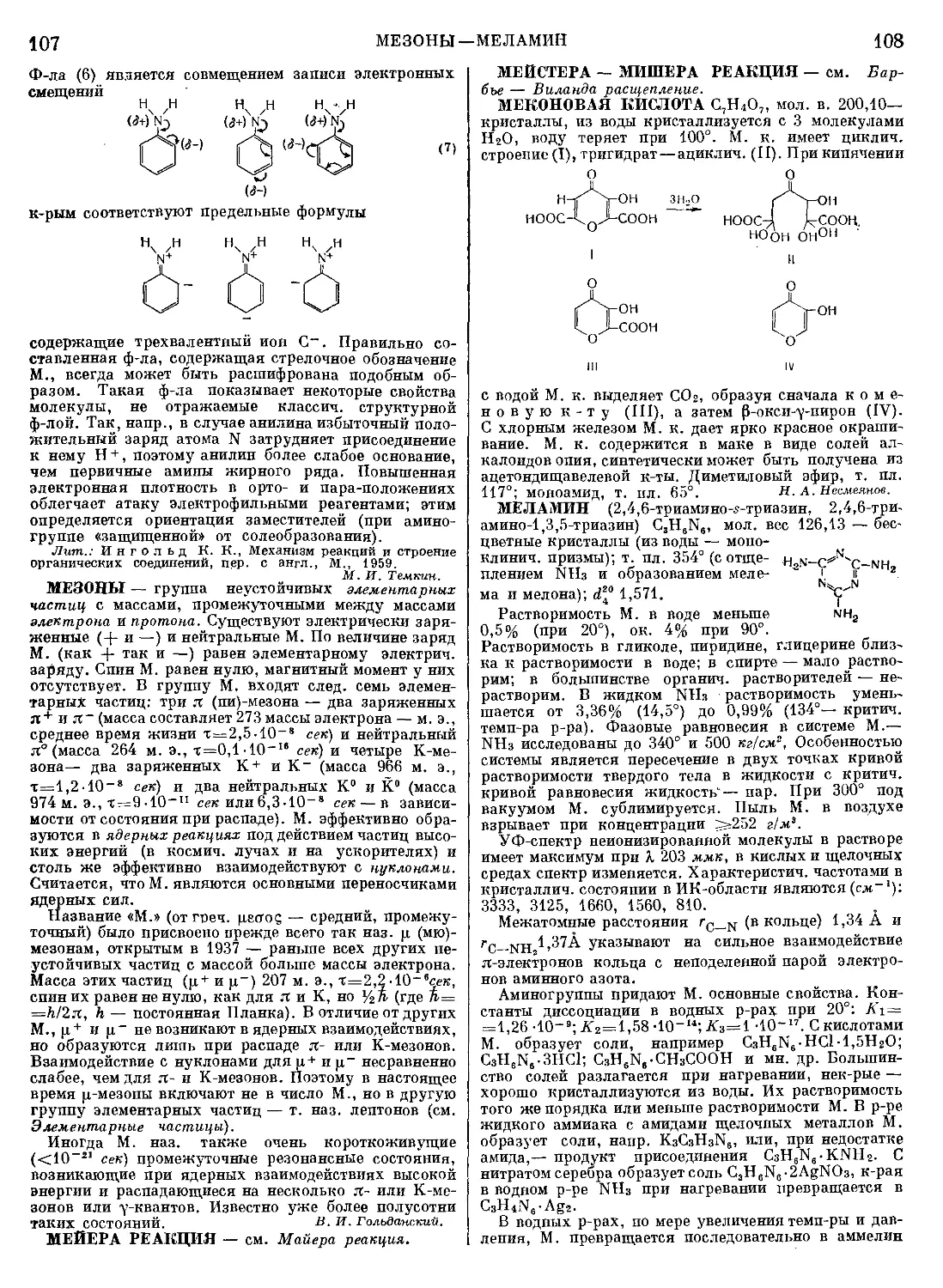





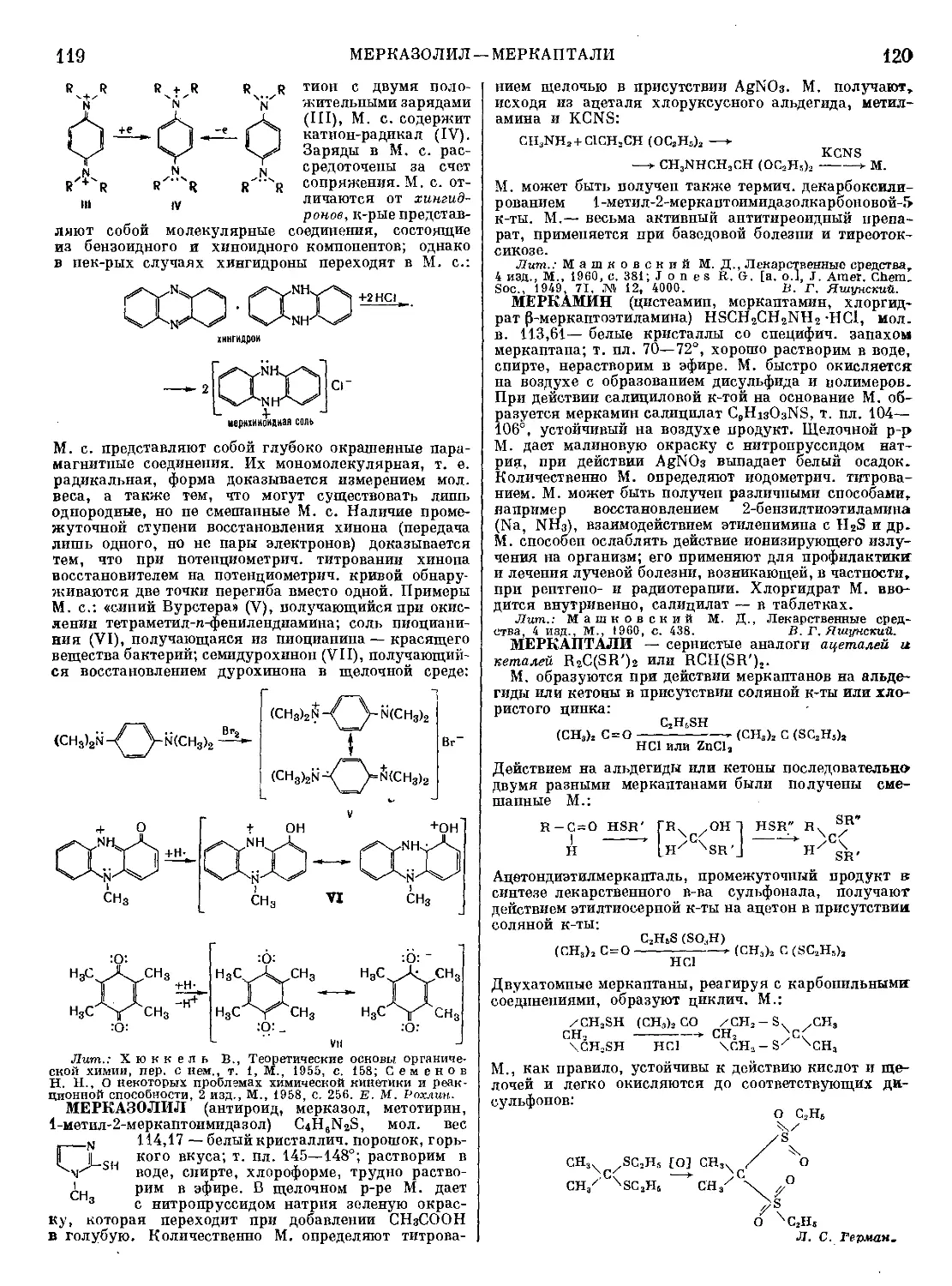







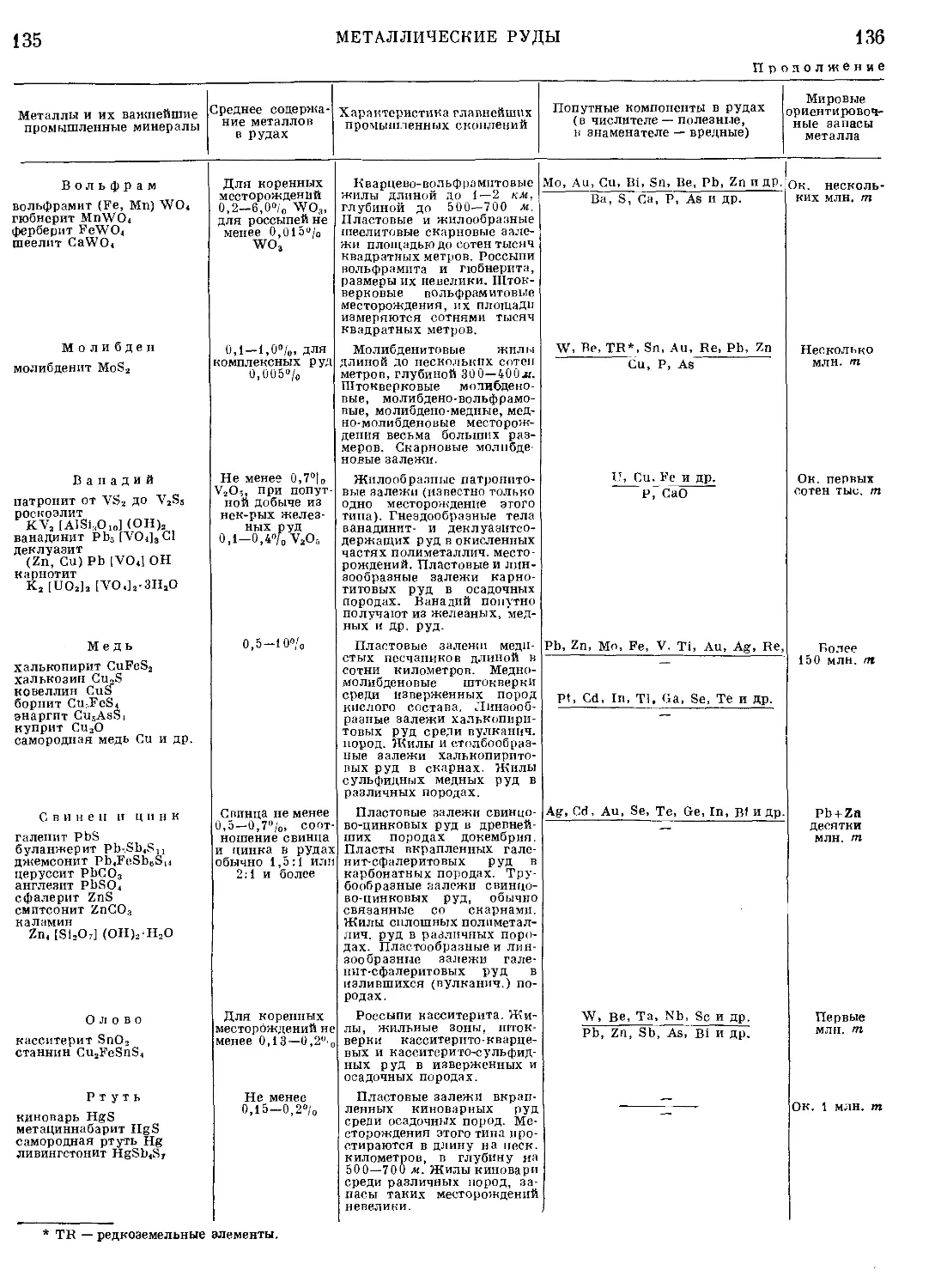
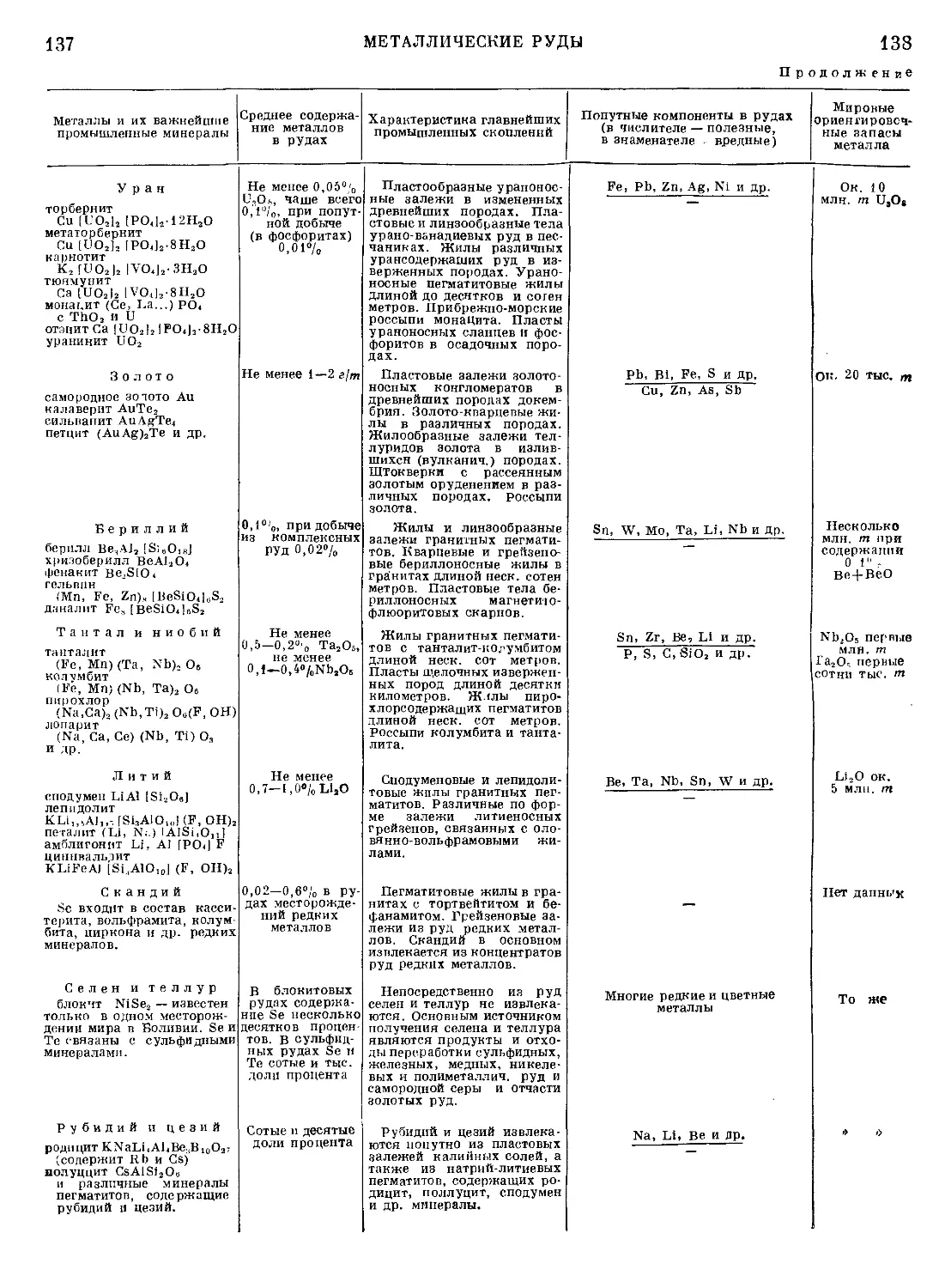























































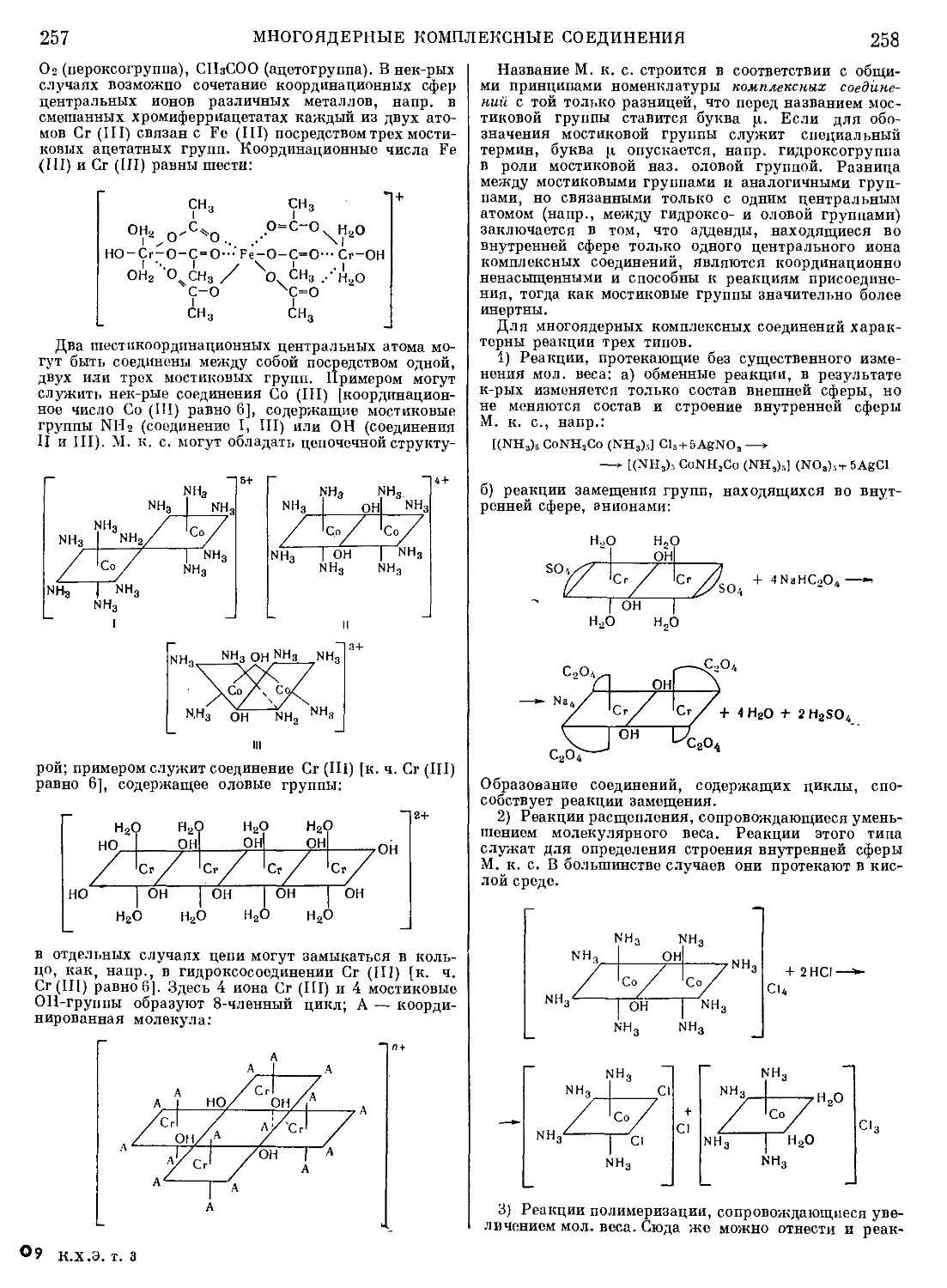


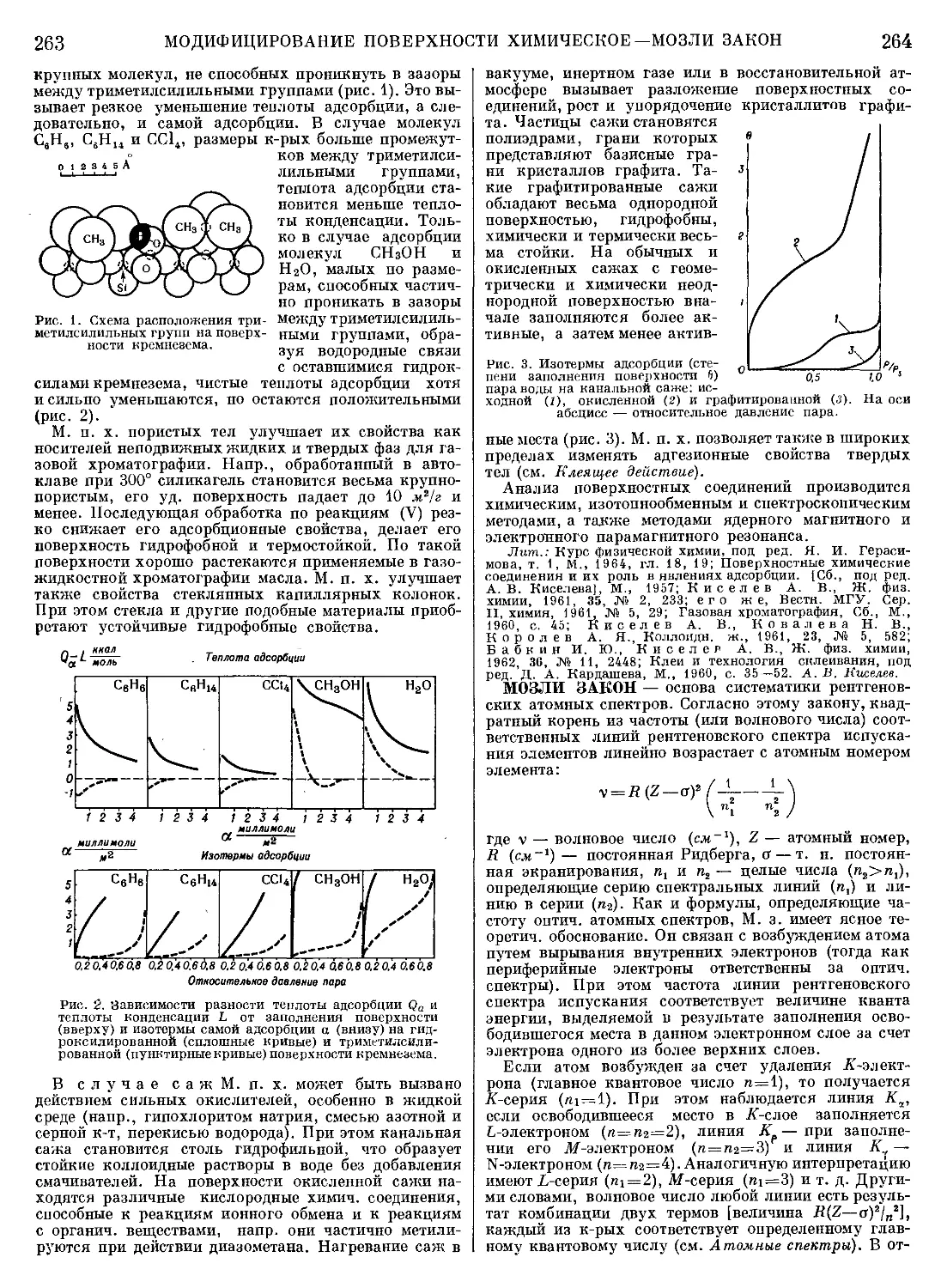



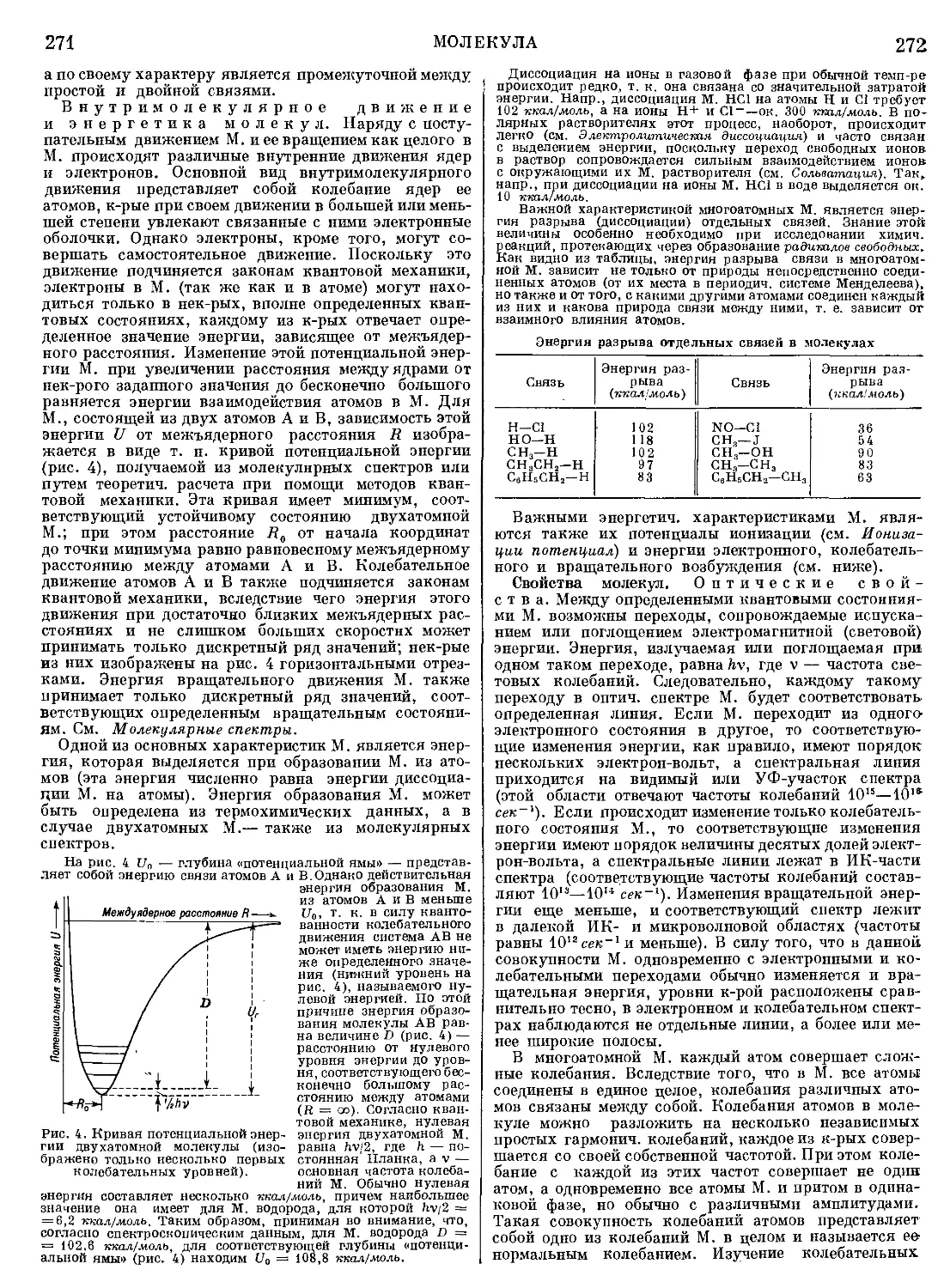





















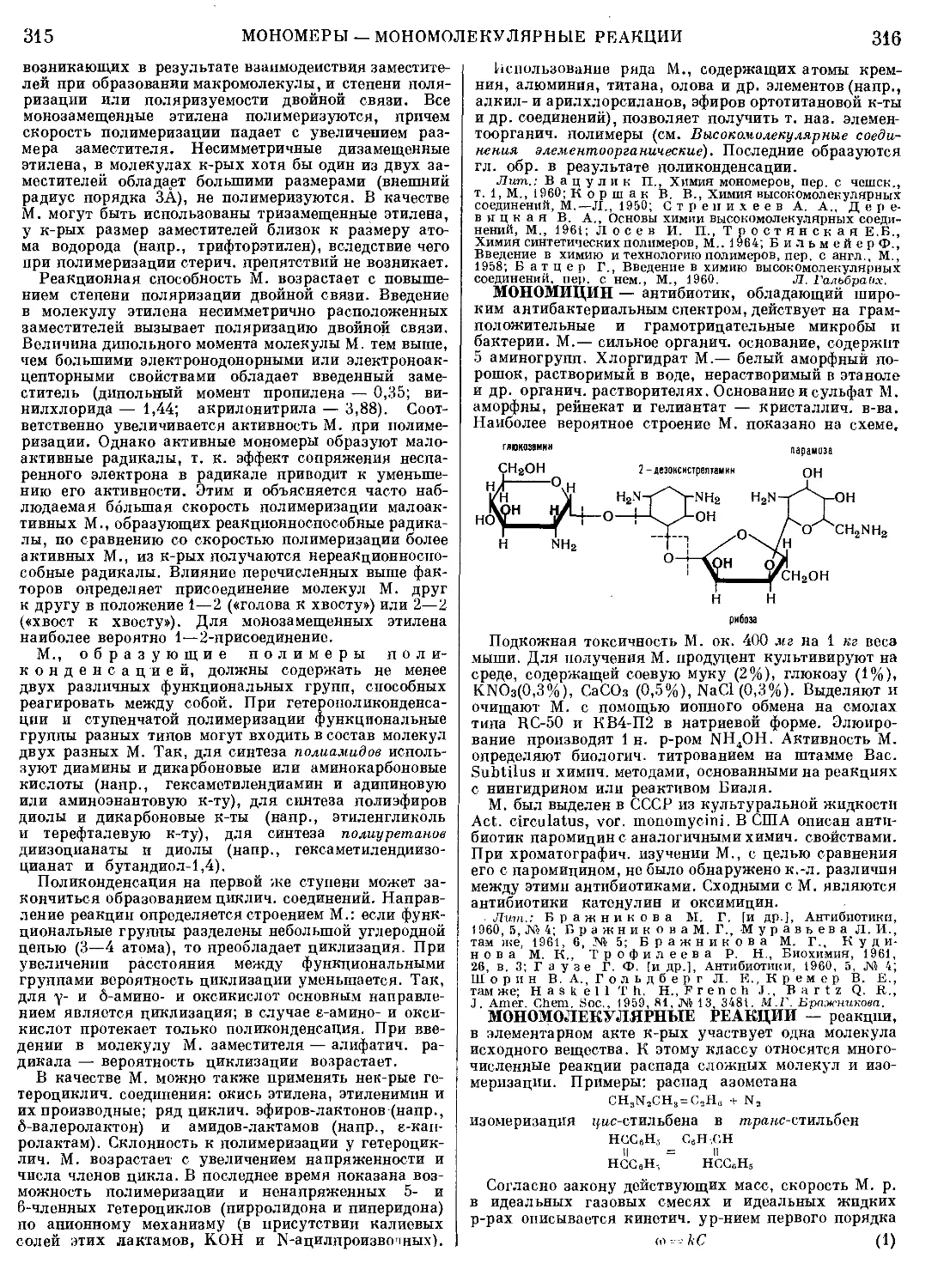















































































































































































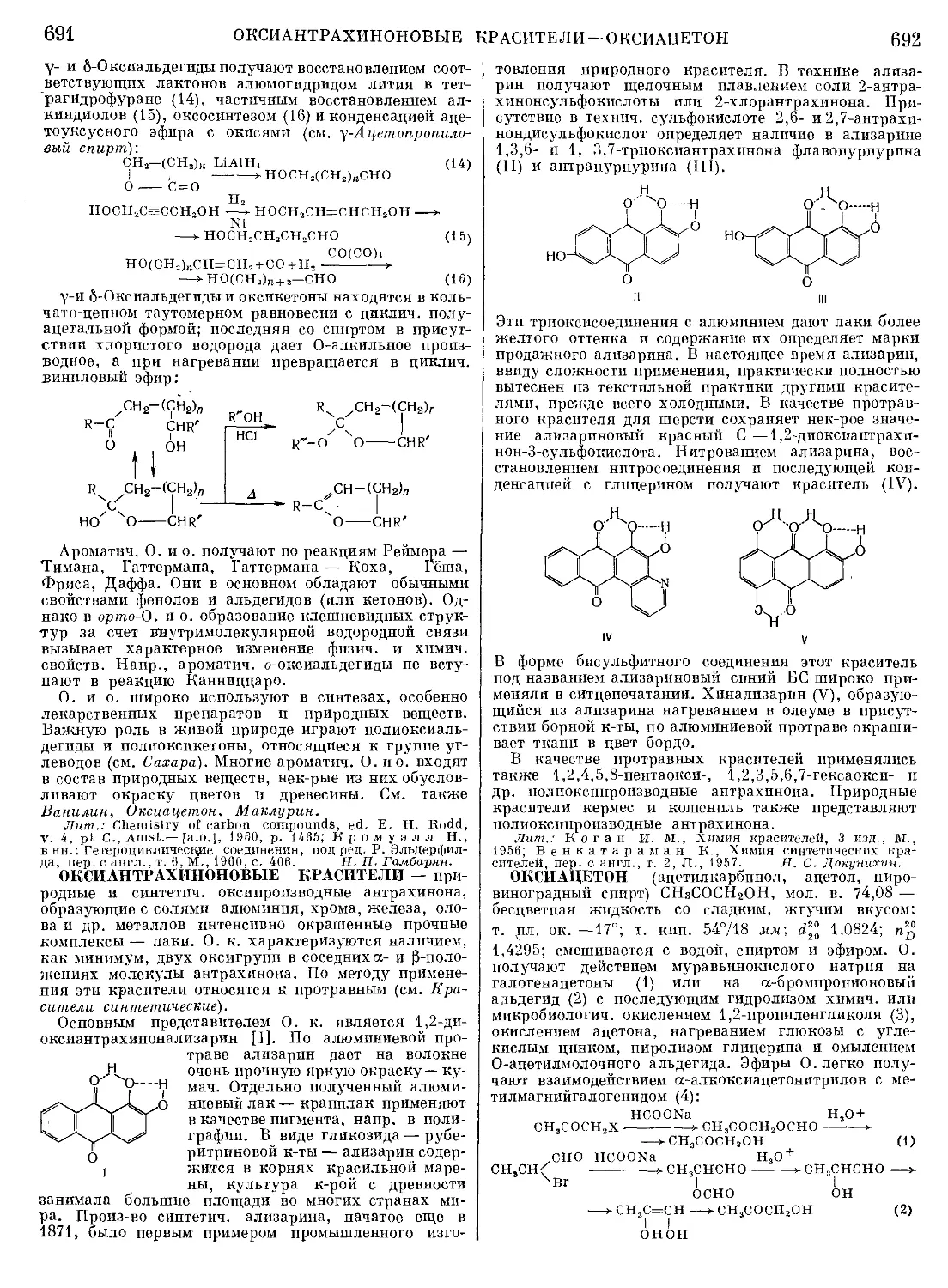






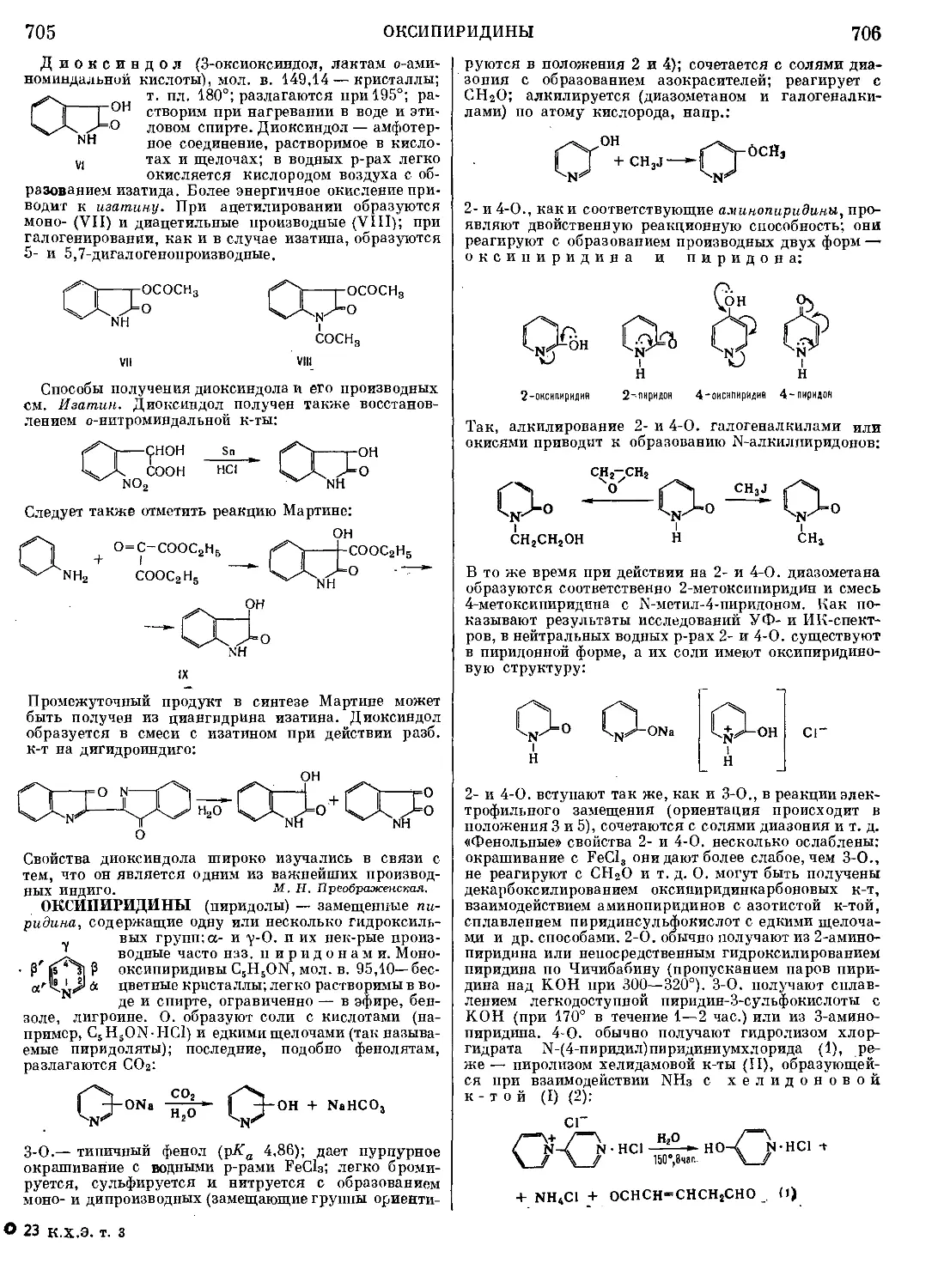






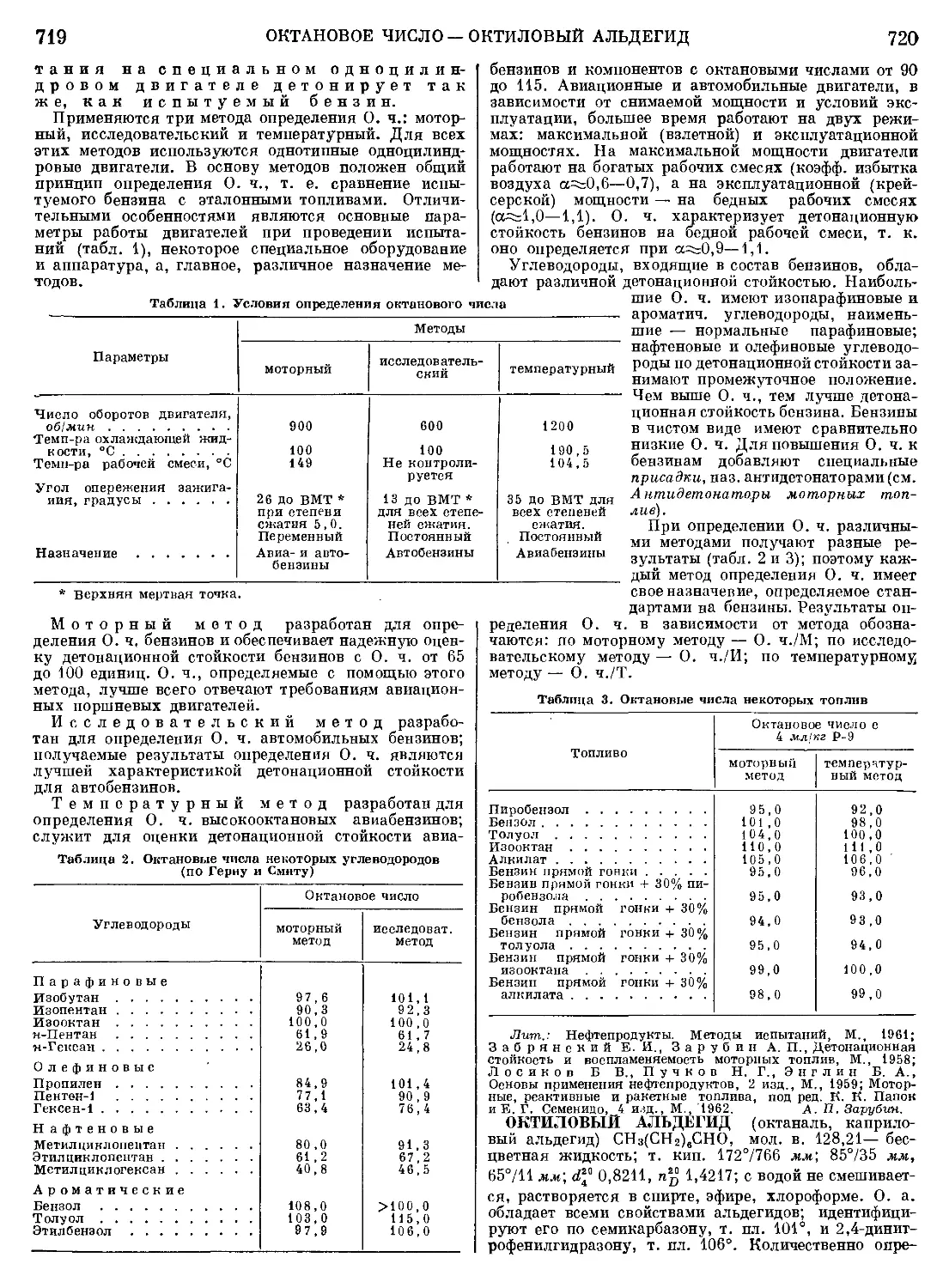
























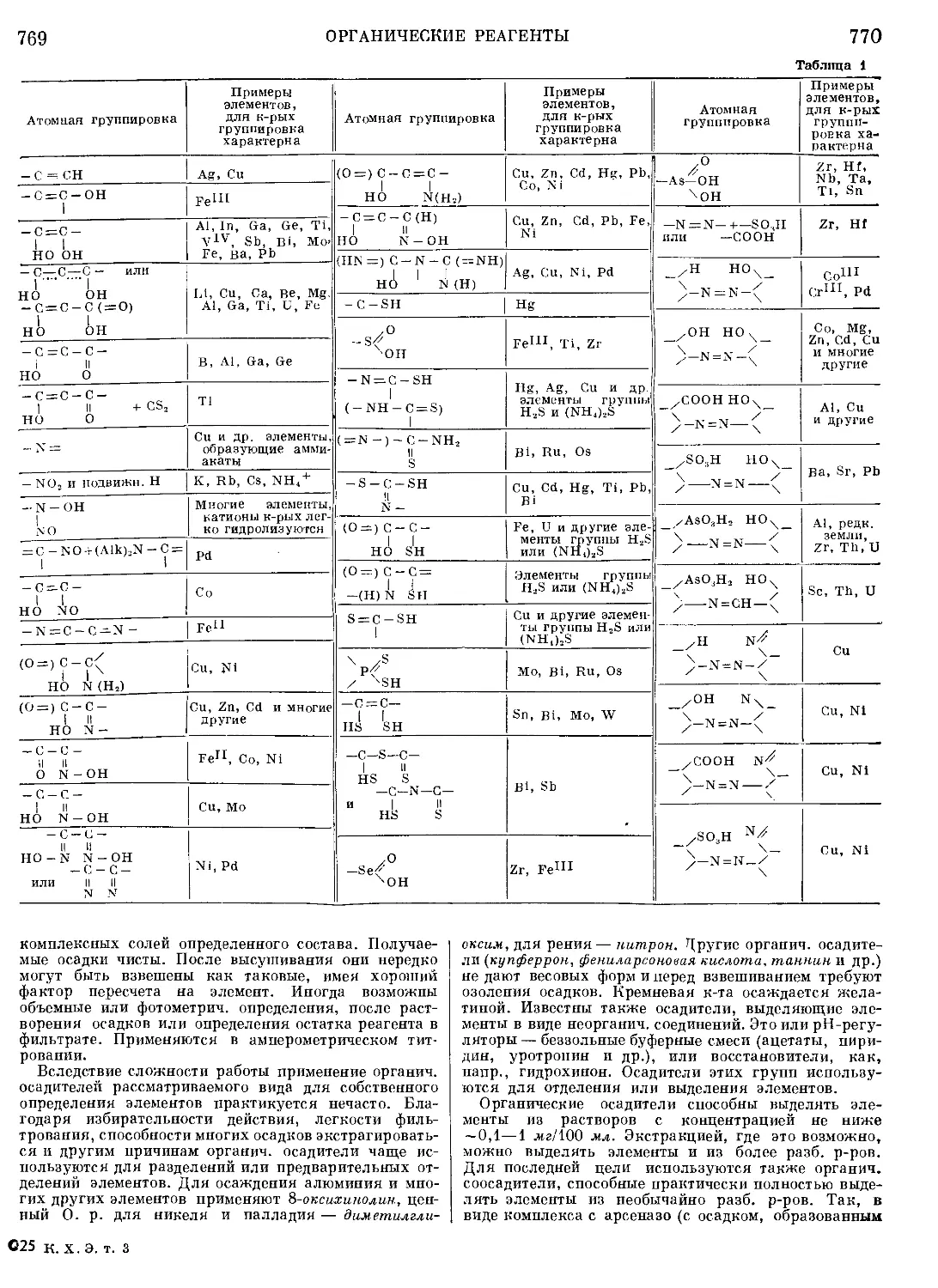






























































































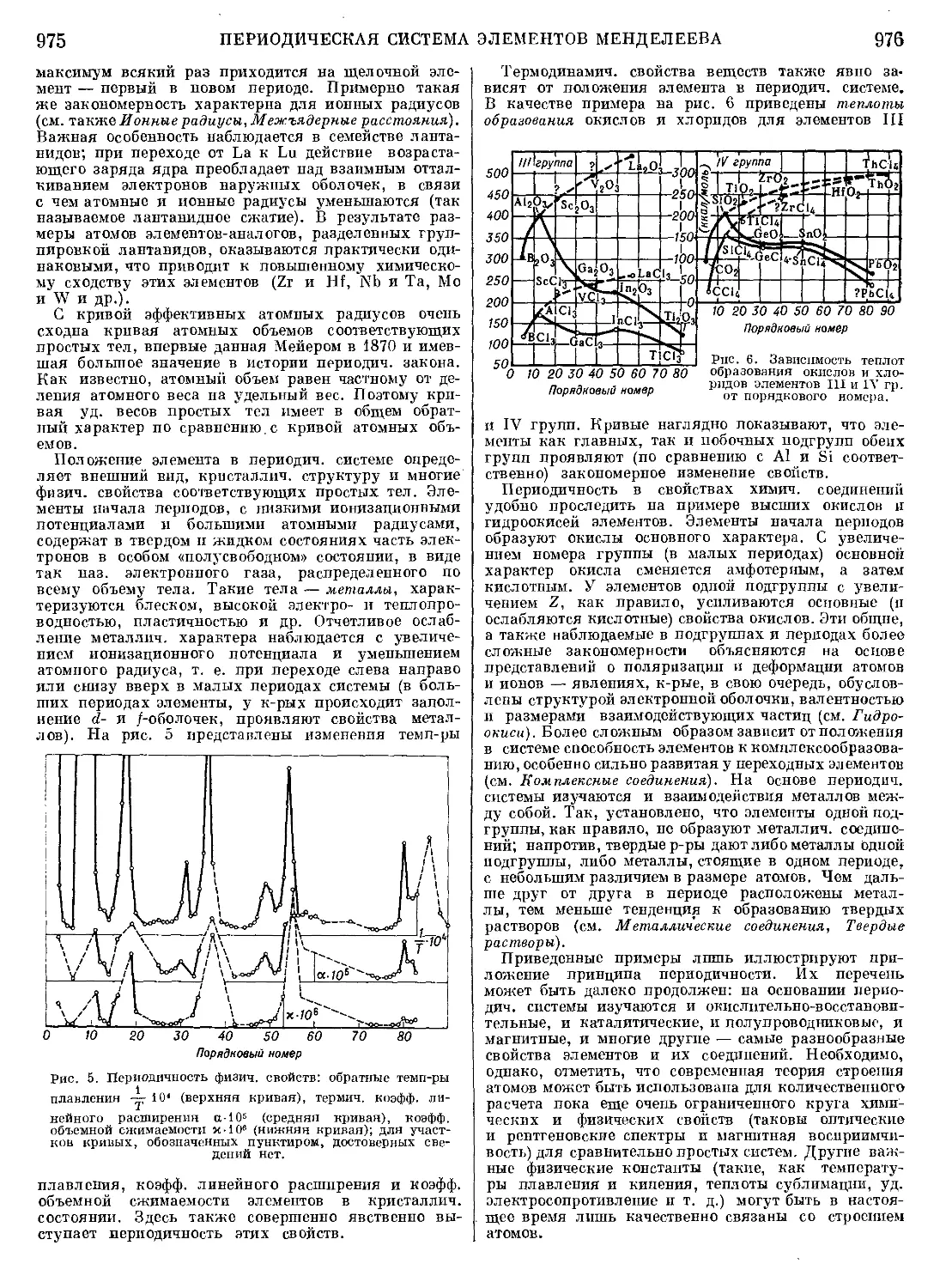






































































Текст
КРАТКАЯ
ХИМИЧЕСКАЯ
ЭНЦИКЛОПЕДИЯ
РЕДАКЦИОННАЯ КОЛЛЕГИЯ
II. Л. КНУНЯНЦ (главный редактор), Г- Я. БАХАРОВСКИЙ (зам. главно-
го редактора), А. И. БУСЕВ, Я. М. ВАРШАВСКИЙ, Н. И. ГЕЛЬПЕРИН,
П. II. ДОЛИН, В. А. КИРЕЕВ, Ю. А. КЛЯЧКО, Г. А. МЕЕРСОН, А. Н.
МУРИН, С. А. ПОГОДИН, П. А. РЕБИНДЕР, Г. Л. СЛОНИМСКИЙ,
М. II. ТЕМКИН, Д. А. ЭПШТЕЙН
Мальтаза—Пиролиз
ИЗДАТЕЛЬСТВО «СОВЕТСКАЯ ЭНЦИКЛОПЕДИЯ»
ИЗДАТЕЛЬСТВО «СОВЕТСКАЯ ЭНЦИКЛОПЕДИЯ»
ЭНЦИКЛОПЕДИИ
СЛОВАРИ
СПРАВОЧНИКИ
НАУЧНЫЙ СОВЕТ ИЗДАТЕЛЬСТВА
А. П. АЛЕКСАНДРОВ, А. А. АРЗУМАНЯН, А. В. АРЦПХОВСКИИ,
Н. В. БАРАНОВ, А. А. БЛАГОНРАВОВ, Н. Н. БОГОЛЮБОВ,
Б. А. ВВЕДЕНСКПП (председатель Научного Совета), Б. М. ВУЛ,
Г. Н. ГОЛИКОВ, И. Л. КНУНЯНЦ, Ф. В. КОНСТАНТИНОВ,
Б. В. КУКАРКИН, Ф. Н. ПЕТРОВ, В. М. ПОЛЕВОЙ, А. И. РЕВИН
(заместитель председателя Научного Совета), Н. М. СИСАКЯН,
А. А. СУРКОВ, Л. С. ШАУМЯН (заместитель председателя Научного
Совета)
МОСКВА . 1964
54(03)
К 78
РЕДАКТОРЫ-КОНСУЛЬТАНТЫ
А. К. БАБКО В. Н. БЕЛОВ , II. П. БУДНИКОВ, В.Н. БУКИН, А. И. ГОЛЬ-
БИНДЕР, Л. В. ГОРДОН, Б. А. ДОГАДКИН, М. А. ЕЛЬЯШЕВИЧ, Д. Д. ЗЫ-
КОВ, В. А. КАБАНОВ, А. В. КИСЕЛЕВ, А. И. КОРОЛЕВ, А. Д. КУЗОВКОВ,
О. Ю. МАГИДСОН, Н. Н. МЕЛЬНИКОВ, И. И. НОВИКОВ, А. Б. ПАКШВЕР,
Н. Г. ПУЧКОВ, А. С. ХОХЛОВ, А. И. ШЕРЕШЕВСКИЙ, В. А. ЯКОВЛЕВ
РЕДАКЦИЯ КРАТКОЙ ХИМИЧЕСКОЙ ЭНЦИКЛОПЕДИИ
Научные редакторы - Д. Н. ВАСКЕВИЧ, Е. В. ВОНСКИЙ, 3. И. ГОДИН,
Н. П. МОСТОВЕНКО; редактор —М. Е. ТРУХАНОВА; литературные редакторы—
Л. Д. КИРИЛЛОВА, Ю. А. ГОРЬКОВ, В. Л. КИСЛОВ; младшие редакторы —
В. А. ЛЕБЕДЕВА, В. А. СОЛОМЕННИКОВА
Редактор-библиограф — Е. И. ЖАРОВА
Художественный редактор — И. Н. САХАРОВА
Технический редактор — И. Д. КУЛИДЖАНОВА
Корректор — А. В. МАСЛОВА
Указатель составил — А. Б. ДМИТРИЕВ
м
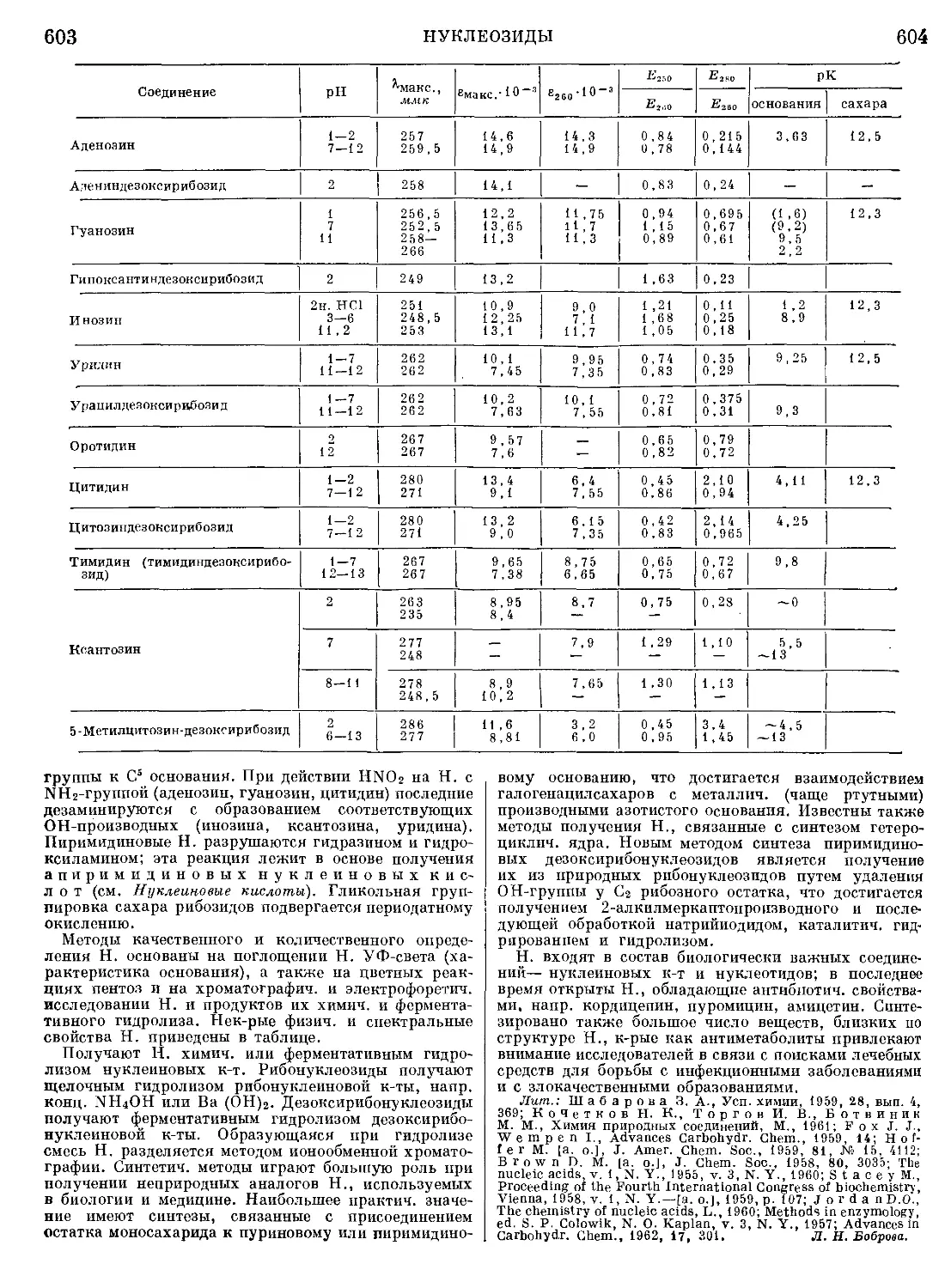
МАЛЬТАЗА — фермент, катализирующий гидро-
литпч. расщепление мальтозы на две молекулы D-глю-
козы; относится к группе а-гликозидаз. М. расщепляет
нек-рые производные мальтозы, а также различные
алкил- и арил-а-П-глюкопиранозиды. Кроме реакций
гидролитич. расщепления, М., подобно другим гли-
козидазам, катализирует также реакции трансглпко-
зилированпя, т. е. переноса глюкозного остатка от
одного субстрата на другой, с образованием высоко-
молекулярных олигосахаридов и изомальтозы. При
высокой концентрации D-глюкозы М. катализирует
синтез мальтозы из глюкозы.
М. — широко распространенный фермент, входит
в состав слюны, поджелудочного и кишечного сока,
присутствует в крови, печени и скелетных мышцах.
М. встречается также в растительных тканях и у бак-
терий; очень богаты М. дрожжи. М. не получена
в кристаллич. состоянии. Препараты М., полученные
из различных источников, отличаются активностью,
субстратной специфичностью и pH-оптимумом. Наи-
более очищенная М. выделена из дрожжей. Оптимум
активности М. соответствует pH 6,75—7,25; фермент
утрачивает активность при нагревании до 55°, инак-
тивируется спиртом, ацетоном и толуолом. Константа
Михаэлиса для мальтозы 1,35—2,0 -10-1 М, для глю-
козы 1,6—2,0 •10-2М. Активность М. определяют
поляриметрически, по изменению вращения плоскости
поляризованного света р-ром в результате превраще-
ния мальтозы в D-глюкозу. М. играет важную роль
в процессах переваривания, расщепления и обмена
углеводов.
Лит.: Диксон М., Уэбб Э., Ферменты, пер. с англ.,
М., 1961; The enzymes. Chemistry and mechanism of action, ed.
by J. B. Sumner and K. Myrback, v. 1, pt 1, N Y., 1950:Methods
in carbohydrate chemistry, cd. R. L. Whistler..., v. 1, N. Y.,
1962. В. Б. Спиричев.
МАЛЬТОЗА [4-(а-О-глюкозидо)-О-глюкоза, солодо-
вый сахар] С12Н22О11, мол. в. 360,19 — восстанавлива-
ющий дисахарид, построенный из двух остатков
D-глюкозы, связанных а-1,4-глюкозидной связью.
М. существует в а- и p-формах, отличающихся про-
странственным расположением полуацетального гид-
роксила. В кристаллич. виде М. получена в виде гид-
рата P-формы, т. пл. 102—103°, [а]р=+И1,7°, и
в виде безводной а-формы, т. пл. 108°, [а]д=-|-173°.
В водных р-рах вследствие мутаротации образуется
равновесная смесь различных форм М. с [а]д=
=+130,4°. М. очень хорошо растворима в воде, раст-
ворима в безводном пиридине и смеси пиридина с во-
дой, нерастворима в спирте и эфире.
М. восстанавливает раствор Фелинга, аммиачный
р-р AgNOs, образует фенилозазон, р-нафтиозазон
и др. производные, характерные для восстанавливаю-
щих сахаров. При окислении концевой альдегидной
группы из М. получается мальтобионовая
к-та, при гидролизе к-рой образуются D-глюкоза и
D-глюконовая к-та. М. гидролизуется кислотами и
а-глюкозидазами — мальтазами; эти ферменты
широко распространены в растениях, животных и
микроорганизмах. М. сбраживаются дрожжами. В сво-
бодном состоянии М. содержится в нек-рых растениях.
Производят М. ферментативным гидролизом — «оса-
хариванием» — крахмала:
Р-амплаза
(С[2П[оО-,) £---* хС,,2Н22О;t
крахмал мальтоза
Синтетически М. получена из D-глюкозы. М. обра-
зуется как промежуточный продукт в произ-ве спирта
из картофеля и других крахмалосодержащих продук-
тов. Применяют М. в винокурении и пивоварении,
в пищевой пром-сти в виде мальтозной патоки. Очи-
щенная М. применяется в микробиологии для изго-
товления сред, используемых для идентификации
микроорганизмов.
Лит.: Р и х е А., Основы технологии органических ве-
ществ, пер. с нем., М., 1959; Methods in carbohydrate chemist-
ry, ed. R. L. Whistler..., v. 1, N. Y., 1962; The carbohydrates.
Chemistry, biochemistry, physiology, ed. W. Pigman, N. Y.,
1957. Л. И.Линевич.
МАНГАНАТЫ — соли марганцовистой кислоты
H2MnOi (см. Марганцовистая кислота и манганаты).
МАНГАНИТЫ — соли марганцоватистой кислоты
ЩМпОз (или Н2МпОз) (см. Марганец).
МАННИТ СбНцО8 — шестиатомныи спирт, изве-
стен в двух стереоизомерных формах: D-маннпт (I)
и L-маннит (II).
н н он он он он и н
1 2| >| 41 S| в 1111
НОН,С-С-С-С-С-СН2ОН НОН2С-С—с—С—С-СН2ОН
I I I I 1111
(I) ОН ОН НН (П) НН он он
а также в виде рацемич. соединения П,Ь-маннита.
L-и DL-спирты в природе не встречаются; под наиме-
нованием М. обычно подразумевают D-M.— бесцвет-
ные кристаллы сладкого вкуса, мол. в. 182,17, т. пл.
166°; т. кип. 276—280°/1 -и.» рт. ст.-, [а]р =—0,244°
(вода); <Z^°1,487; уд. теплоемкость 0,327 кал/г.град
(28—100°); теплота сгорания 4,0 кал!г; М. нетоксичен,
растворим в воде, анилине, малорастворим в пири-
дине, диоксане и абс. этиловом спирте; нерастворим в
эфирах и углеводородах; кристаллизуется в виде игл
или призм. М. обладает всеми свойствами многоатом-
ных спиртов. Неполные эфиры М. с высшими жир-
ными к-тами обладают поверхностно-активными свой-
ствами. Продукты реакции с борной к-той являются
стекловидными смолами. Гексанитрат М. — взрывча-
тое вещество; его используют также как лекарствен-
11
МАННИХА РЕАКЦИЯ—МАННОЗА
12
ное средство. В условиях каталитич. гидрирования
М. при темп-pax выше 150° и повышенном давлении
водорода происходит его гидрогенолиз с разрывом
связей С—С преим. между 3 и 4 атомами углерода
с образованием соответственно низших полиолов,
а также небольшого количества первичных спиртов.
М. хорошо утилизируется микроорганизмами с обра-
зованием различных продуктов биохимия. синтеза.
D-М. очень распространен в природе. Он является
основной частью «манны» ясеня и др. деревьев, содер-
жится в значительных количествах в водорослях и
высушенных грибах, обнаружен во многих растениях.
В пром-сти D-М. получают из морских коричне-
вых водорослей или каталитич. гидрированием саха-
розы, в процессе к-рого происходит ее инверсия с об-
разованием глюкозы и D-фруктозы, восстанавливаю-
щихся соответственно в сорбит и М. Для выделения
М. из смеси с другими полиолами используют его спо-
собность легко кри-
сталлизоваться из рас-
творов. М. можно полу-
чить также из гидро-
лизатов хвойной дре-
весины,содержащей до
10% маннана. М. и
его ангидриды могут
служить исходным ма-
териалом для получе-
ния поверх ностно-
RNH2 + СН2О
н+_ г V' г ±н2о
[-^RNHCH2OH=^:[RNHCH2OH^=tRNHCH2OH2J < , I
1 + + н If
-*-RN=CH2 [rn=CH2—— RNH-CHa]*
II + н н _______11 IV
-*RNHCH2N.HRS=E:[RNHCH2NHR3=-RNHCHs>.NHRf—±RNy^
III ‘ 2
-CH + R№CH2—«--^-CH2.NHR,+ h+
активных веществ, олиф, смол, лаков и т. д.; приме-
няется также в пищевой пром-сти, фармации и пар-
фюмерии.
Лит.: The carbohydrates. Chemistry, biochemistry, physio-
logy, ed. W. Pigman, N. Y., 1957; Stanek J. [и др.I, Mono-
sacharidy, Praha, 1960; Ч e п и г о С. В., Получение из расти-
тельного сырья многоатомных спиртов и их применение, М.,
1964 (в печати). С. В. Чепиго.
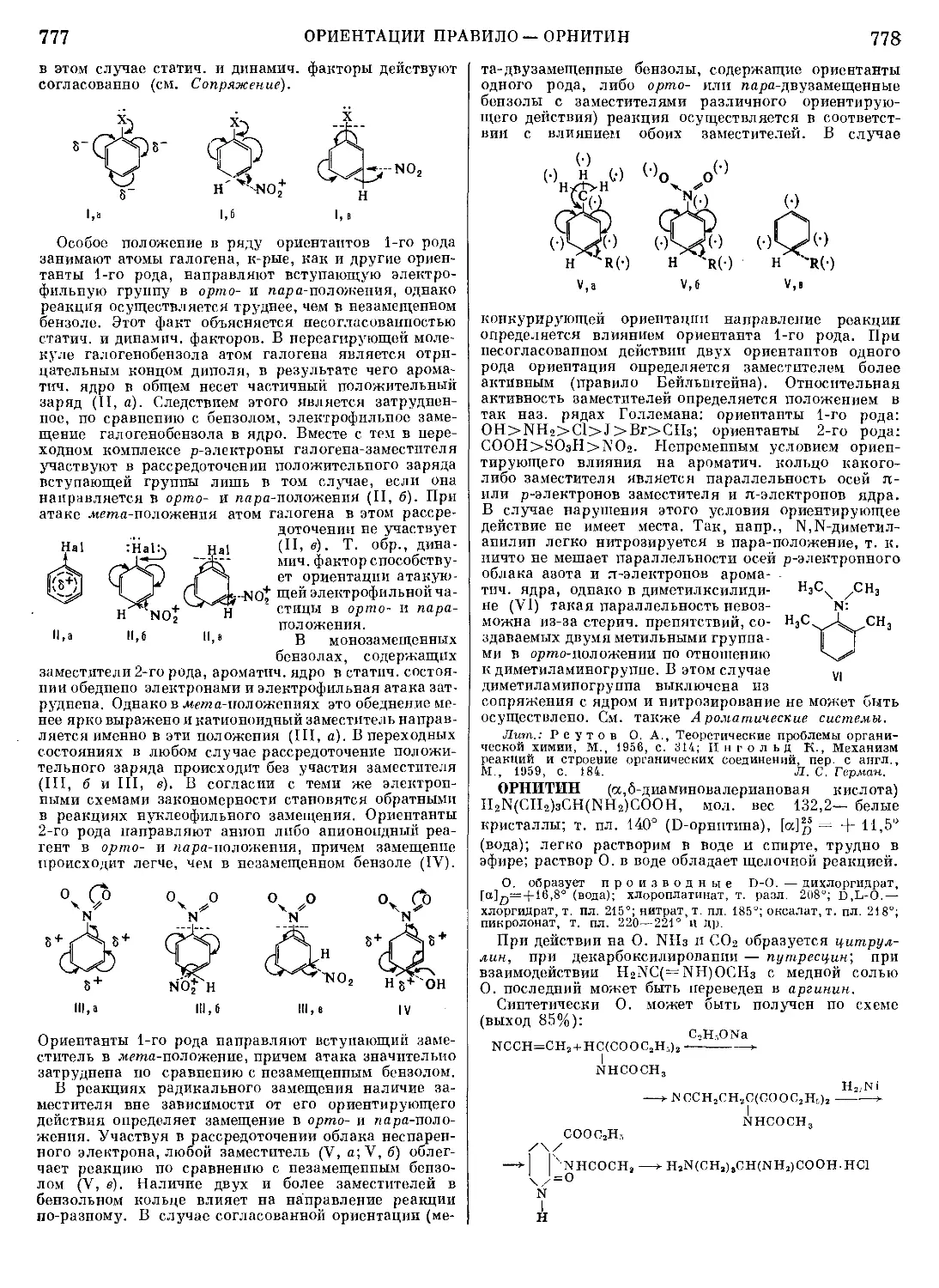
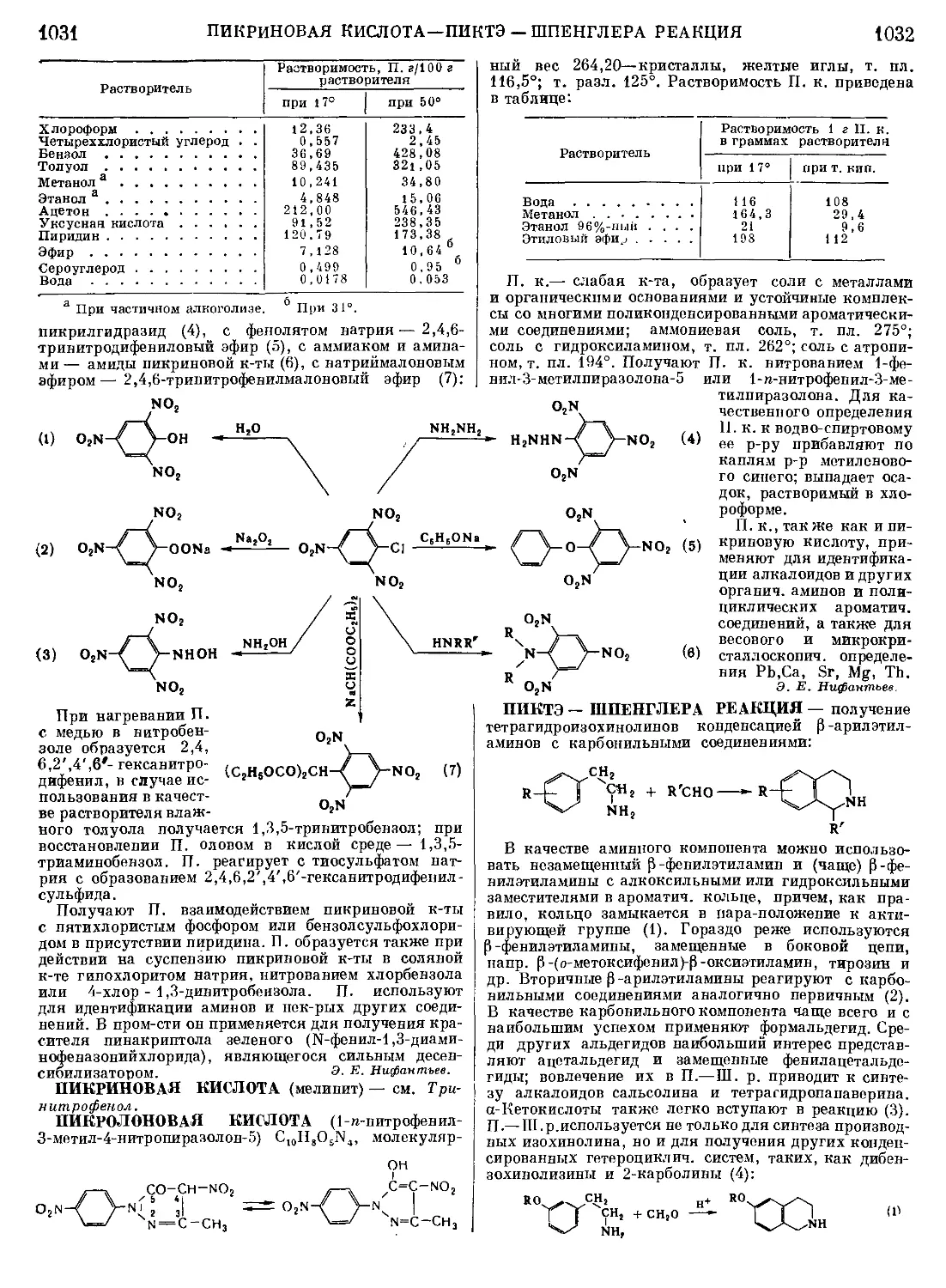
МАННИХА РЕАКЦИЯ — замена подвижного ато-
ма водорода в соединениях с С—Н-связью на амино-
метильную группу взаимодействием этих соединений
с формальдегидом и аммиаком (или аминами);
1 / I /
-CH+CH.O+HN <-»-c-ch2-n<
I 4 I 4
С первичными аминами и особенно аммиаком часто
образуются смеси аминов;
I I
—CH+CH2O + HNH3-> —с—ch2nh2+
• I I
, ( 1 \ Г 1 \
+ — С-СН2 NH+( -С-СН2 ) N
\ I /2 \ I
К еще большим осложнениям реакции приводит нали-
чие нескольких подвижных атомов водорода в конден-
сируемом соединении; при этом образуются смеси
продуктов моно-, ди- и полиаминометилирования.
Однако при строгом соблюдении условий реакции п
соотношения компонентов обычно удается добиться
образования индивидуальных веществ. Область при-
менения М. р. чрезвычайно велика. Соединения самых
разнообразных классов, как, например, алифатпч.
а, ^-ненасыщенные альдегиды и кетоны, жирноарома-
тич. алициклич. и гетероциклич. кетоны, кислоты
типа циануксусной и малоновой и их эфиры, ароматич.
и гетероциклич. соединения с достаточно подвижными
атомами водорода кольца, а-николины и хинальдины,
монозамещенные ацетилены и др., аминометилируются
с большей или меньшей легкостью:
RCOCH + СН20 + hn(-RCOCCH2N^
ROCOs, z z
С + CH2O + HN. -----
Xх ЧН v
ROC<Xcz
X' 4ch2n(
_RCHCH + СН2О + Hi/ •- RC=C-CH2Nf
Формальдегид в ряде случаев может быть заменен
другими алифатпч., ароматич. и жирноароматич.
альдегидами, что еще более увеличивает разнообразие
соединений, к-рые могут быть синтезированы по М. р.
Наиоолее вероятный механизм М. р., объединяющий
схемы с промежуточным образованием оксиметилами-
нов (I), оснований Шиффа (II) или метиленбисаминов
(III), включает промежуточное образование катиона
(IV). Основания Манниха находят очень большое при-
менение в органич. синтезе (см., напр., Михаэля ре-
акцию), особенно в синтезе фармакологии, препаратов
и природных веществ. Реакция открыта Маннихом
в 1917.
Лит.: М erz К. W., Pharmazie, 1956, 11, Н. 8, 505; Блик
Ф. Ф., в кн.: Органические реакции. Сб. I, пер. с англ., М.,
1948, 399; Wagner Е. С., J. Organ. Chem., 1954, 19, Ks 12,
1861; R е 1 с h е г t В., Die Maimich — Reaction, В. — [u. a.J,
1959. H. П. Гамбарян.
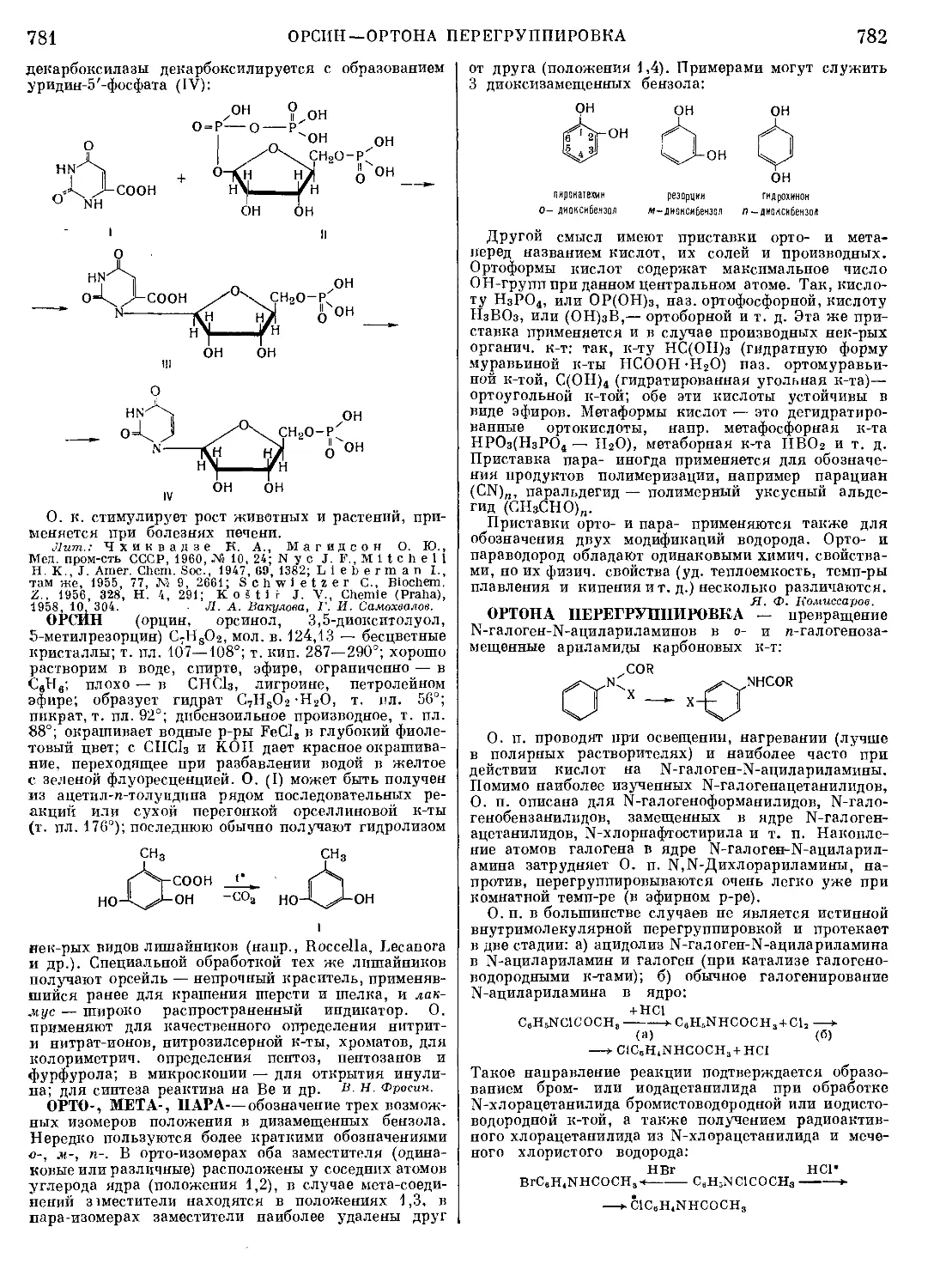
МАННОЗА CeH12Oa, мол. в. 180,16 — одна из вось-
ми изомерных альдогексоз, от глюкозы отличается
только пространственным расположением атома водо-
рода и гидроксильной группы у второго атома угле-
рода (эпимер глюкозы):
ОНО
НО-С-Н
НО-С-Н
н-с-он
н-с-он
СН2ОН
открытая альдегидная
форма D-маннозы
циклическая пнранвзная
Форма D- маннозы
М. существует в D- и L-формах, каждая из к-рых
образует открытую альдегидную и несколько цик-
лич. таутомерных форм. В кристаллич. виде получены
a-D-M„ т. пл. 133°; [a]D=+29,3°; 0-D-M., т. пл. 132°,
[a]D=17,0°; P-L-M., т. пл. 132°, [a]D = +14,0°. Для
D-M. [a|D= +14,2° (равновесная система в воде).
В природе встречается D-М.; она входит в состав по-
лисахаридов растений и микроорганизмов — манна-
нов, галактоманнанов, глюкоманнанов; является ком-
понентом некоторых сложных белков (овомукопд
кровп и др.).
М. растворима в воде, нерастворима в эфире п
большинстве органич. растворителей. М. дает реакции
альдоз: восстанавливает р-р Фелинга, аммиачный р-р
AgNOs, образует гликозиды — маннозиды; гид-
разоны, озазоны. Фенилгидразон М. очень плохо
растворим. Озазоны М. идентичны озазону глюкозы
и фруктозы. При окислении альдегидной группы М.
13
МАННОЗИДОСТРЕПТОМИЦИН—МАРГАНЕЦ
14
образуется одноосновная манноновая кис-
лота, при окислении обеих концевых групп —
двухосновная маннаровая кислота, окис-
ление только первичной спиртовой группы приводит
к образованию маннуроновой кислоты;
при восстановлении М. образуется шестиатомный
спирт маннит (м а н н и т о л). М. образует лег-
кокристаллизующееся соединение с хлористым кальци-
ем C6Hi2Os-CaCl2-4H2O.D-M.сбраживается дрожжами.
Получают D-М. гидролизом природных веществ, напр.
маннанов скорлупы каменного ореха (отходы пугович-
ного произ-ва), глюкоманнана эремурана. D- и L-M.
и их рацемат получают синтетически из арабинозы
циангидринным методом, эпимеризацией глюкозы и
др. способами.
Лит. см. при ст. Идоза. Л. И. Линевич.
МАННОЗИДОСТРЕПТОМИЦИН (стрептомицин В,
а-В-маннозидо-ТЧ-метил-а-Ь-глюкозаминидо-Р-Ь-стреп-
тозидострептидин) — антибиотик, образуемый наряду
со стрептомицином штаммами Streptomyces griseus.
М. является трехкислотным основанием, образует,
напр., кристаллич. трирейнекат (т. разл. 178—179°)
и трихлоргидрат, кристаллизующийся с 2 мол. воды
в виде гексагональных призм. Последняя соль при
нагревании выше 100° теряет воду, переходя в безвод-
ный трихлоргидрат [а]р= — 54,1° (вода).
В химич. отношении М. очень близок к стрептоми-
цину, отличаясь от последнего наличием в молекуле
остатка D-маннозы. М. дает положительную Сакагучи
реакцию, взаимодействует с реагентами на карбониль-
ную группу, при нагревании с р-ром щелочи образует
мальтол. При каталитич. гидрировании М. в присут-
ствии платины происходит восстановление альдегид-
ной группы с образованием дигидроманнозидо-
стрептомицина. От стрептомицина М. может быть
отделен хроматографией на А120з или противоточ-
ным распределением между водным буферным р-ром
с pH 7,15 и смесью амиловых спиртов в присутствии
«переносчиков» — высших жирных к-т. Для отделения
от стрептомицина в пром-сти используют неспособ-
ность М. давать комплекс с СаС1».
По биологич. действию М. аналогичен стрепто-
мицину. Он подавляет микобактерии, грамположитель-
ные и грамотрицательные микроорганизмы, но слабее,
чем стрептомицин. В опытах in vivo, напр. на живот-
ных, зараженных Mycobacterium tuberculosis, стреп-
томицин также примерно в 3 раза активнее М. Токсич-
ность этих двух веществ приблизительно одинакова.
Лит.: Шемякин М. М. [и др.], Химия антибиотиков,
т. 1, 3 изд., М., 1961, с. 715—20. Г, С. Розенфельд,
МАННУРОНОВАЯ КИСЛОТА — см. Гексуроновые
кислоты.
МАРГАНЕЦ (Manganum) Мп — химич. элемент
VII гр. периодич. системы Менделеева; п. н. 25, ат. в.
54,9381. Состоит из одного устойчивого изотопа
Мп55. Сечение захвата тепловых нейтронов 12,6 ±
±0,6 барн (на атом). Из искусственно радиоактивных
изотопов важнейшие Мп5*(Т>/2=5,72 дня) и Мп58 (Г12=
=2,567 часа). Конфигурация внешних электронов ато-
ма 3d±2. Энергии ионизации (в ав): Мп°-^Мп + -*
-*Мп2 + -«-Мп’+ соответственно равны 7,432; 15,636 и
33,69.
Марганцовые руды были известны с древних времен и
применялись при получения стекла и керамич. изделий. В 1774
Шееле выделил М. из руд в виде сплава с железом. В относи-
тельно чистом виде М: получен в 1894. Название «М.» принято
во всех странах с начала 19 в.
Содержание М. в земной коре составляет 0,09 вес.%.
В свободном виде в природе М. не встречается. Входит
в состав большого числа минералов, преимущественно
окисного типа; наибольшее распространение имеют
пиролюзит и псиломелан — окисные минералы, в
к-рых преобладает 4-валентный М., а также браунит,
манганит и черная охра. Пиролюзит МпО2—
серовато-черный минерал, содержащий в чистом виде
ок. 63% М.; сингония тетрагональная; плотность
4,8—5,6, твердость 2,0—2,5. Псиломелан —
буровато-черный минерал, содержит 45—60% М.,
представляет собой коллоидную форму МпО2, в к-рой
адсорбированы примеси: вода и окислы калия, нат-
рия и бария. Плотность минерала 3,7—4,7, твердость
2,5. Браунит ЗМп20з-Мп8Юз—буровато-черный
минерал, содержит 62% Мп и 8—10% SiO2; синго-
ния тетрагональная, плотность 4,7—4,9, твердость
6—6,5. Манганит Мп2Оз-Н2О —темно-серый (до
черного) минерал, содержит 62,4% Мп; сингония ром-
бическая, плотность 4,2—4,4, твердость 4. Черная
охра — земляннстая аморфная смесь, мягкая и лег-
кая, состоящая из окисловМ., железа, воды и др. ве-
ществ. Плотность 3,0—4,2. Большинство марганцо-
вых руд обнаружено в виде вторичных отложений,
реже встречаются первичные месторождения. Круп-
нейшие месторождения марганцовых руд находятся
в СССР (Чиатурское, Никопольское и др.), а также
в Индии, Гане, Южно-Африканском Союзе и других
странах.
Физические и химические свойства. Компактный
М. — серебристо-белый хрупкий металл; на воздухе
покрывается тонкой пленкой окислов. Имеет 4 кри-
сталлич. модификации: ниже 700° устойчива а-форма
с кубич. объемноцентрированной решеткой (58 атомов
на элементарную ячейку), а=8,9118 А; в интервале
700—1079° устойчива P-форма с объемноцентриро-
ванной кубич. решеткой (20 атомов на элементарную
ячейку), а=6,3016 А; в интервале 1079—1143° стабиль-
на у-форма с гранецентрированной кубич. решеткой,
а=3,8627—3,9372 А (при комнатной темп-ре у-модифи-
кация образует гранецентрированную тетрагональ-
ную решетку, а = 3,7815 А); выше 1143° существует
6-форма с объемноцентрированной кубич. решеткой.
а = 3,0812—3,0932 А. Атомный радиус М. 1,30 А. Ион-
ные радиусы (в А): Мп2+0,91, Мп’+ 0,70, Мп4 + 0,52,
Мп,+ 0,46. Плотность модификаций М. при комнатной
темп-ре: а 7,44, р 7,29, у 7,21. Т. пл. 1244°; т. кип.
2095°. Теплота плавления 3,5 ккал/г-атом. Теплота
испарения 53,7 ккал/г-атом (при т. кип.). Жидкий М.
обладает заметным давлением пара (.и.ц рт. ст.)'.
7,1 (1427°), 173 (1827°), 541 (2027°).
Уд. теплоемкость модификаций М. при 25° (кал/г-
•град)', а. 0,114, |3 0,115, у 0,120; термич. коэфф, линей-
ного расширения при 20°: а 22,3-10_6, Р24,9-10_в, у
14,8- 10_в;теплопроводность (25°) 0,159 кал/см - сек град.
Уд. электросопротивление мком-см: а 150—260, р 90,
у 40. Термич. коэфф, электросопротивления соответ-
ственно равны: (2—3) -10—4; 12-10-4; 60-10-4. М. пара-
магнитен; атомная магнитная восприимчивость (20°)
9,6-10-в. Вследствие сложности структур а- и Р-мо-
дификацийМ. при комнатной темп-ре тверд и хрупок,
в области устойчивости у-фазы пластичен, но дефор-
мированный М. при охлаждении разрушается из-за
объемных изменений; твердость М. по шкале Роквелла
(Rc): а—70, у—20.
В химич. соединениях М. проявляет положитель-
ную валентность от 2 до 7, причем, как правило,
15
МАРГАНЕЦ
16
производные 2- и 7-валентного М. наиболее прочны.
По мере увеличения валентности М. его основные
свойства ослабевают, а кислотные усиливаются, что
иллюстрирует приводимая схема:
усиление основных свойств
Мп(ОН)2 Мп (ОН), Мп (ОН). Н2МпО< НМпО.
усиление кислотных свойств
На воздухе металл покрывается тонкой окисной
пленкой, к-рая предохраняет его от дальнейшего
окисления даже при нагревании. Напротив, в мелко-
раздробленном состоянии М. окисляется довольно
легко. С кислородом М. образует ряд окислов — основ-
ных, амфотерных и кислотных (см. Марганца окисли).
Водород адсорбируется М. — до 250 см3 Н2 на 100 г
М., с образованием твердых р-ров внедрения; химии,
соединений в этой системе не обнаружено. Взаимо-
действие М. с галогенами протекает весьма энергично
и ведет к образованию солей —галогенидов
МпХ2, розовых кристаллов, хорошо растворимых
в воде (за исключением MnF2). Важнейшим из них
является марганца хлорид МпС12. При действии F2
на MnF2 при 250° образуется винно-красный MnF3,
к-рый при более высокой темп-ре распадается на ис-
ходные вещества. При низких темп-pax получен весь-
ма неустойчивый черно-зеленый МпС1з. Галогениды
4-валентного М. в чистом виде не выделены, но полу-
чены соединения типа MnFi 2KF, МпСЦ • 2КС1.
Образование продуктов совместной кристаллизации
с соответствующими солями других металлов вообще
характерно для галогенидов М.: MnF2-KF, МпС12-
• 2КС1, MnF3 • 2KF и т. д. Галогенопроизводные 6-
и 7-валентного М. не получены. В ряду С1—Br—J ус-
тойчивость галогенидов М. уменьшается. При нагре-
вании М. непосредственно соединяется также с дру-
гими типичными неметаллами — серой, азотом, фос-
фором, углеродом, кремнием и бором. Известны
сульфиды MnS, MnS2 и Mn3Si, из к-рых наиболее
устойчив MnS (см. Марганца сульфиды). С азотом М.
образует нитриды Mn3N2, Mn2N и МщА’; раство-
римость N2 в а- и P-Мп незначительная. В системе
М. — фосфор имеются два фосфида Мп2Р и МпР.
С углеродом М. образует карбиды МпзС, Мп23Св
и Мп;Сз, максимальная растворимость углерода в
а-Мп 1,3%, в Р-Мп 0,4%, в у-Мп 2,0%. М. и кремний
образуют силициды состава Mn2Si, MnSi и
Mn2Si3, играющие важную роль в процессах получе-
ния технически чистого металла (см. ниже).
В ряду напряжений М. стоит между Mg и Zn.
Вода в обычных условиях действует на металл очень
медленно, при нагревании — быстрее. М. растворяет-
ся в разбавленной соляной и азотной к-тах, а также
в горячей конц. серной к-те (в холодной H2SO4 он
нерастворим), образуя катионы Мп2+. Соли 2-валент-
ного М. являются наиболее устойчивыми производ-
ными этого элемента в кислой среде (см. Марганца
карбонат, Марганца нитрат, Марганца сульфаты).
Кристаллогидраты солей 2-валентного М. и их водные
р-ры имеют бледно-розовый цвет, обусловленный ио-
ном [Мп(Н2О)4]2 + . Из р-ров солей Мп2+при pH 8,7
осаждается Мп(0Н)2 — слабое основание, малораст-
воримое в воде. Гидроокись 3-валентного М. Мп(0Н)3
почти нерастворима в воде и является очень слабым
основанием; соли ее весьма неустойчивый встречаются
редко. Также практически нерастворима и Мп(0Н)4;
она обладает амфотерными свойствами, причем и
основная и кислотная функции выражены очень слабо.
Соли, отвечающие кислотной функции Мп(0Н)4,
наз. манганитами, а сама кислота Н4МПО4—
ортомарганцоватистой (а Н2МпОз —
метамарганцоватистой). Выделить ман-
ганиты в чистом виде весьма трудно, т. к. при их син-
тезе сухим путем (вакаливанием окислов металлов
с МпО2) и при осаждении из р-ров получаются смеси
различных продуктов. От кислоты Н4МПО4 и основа-
ния Мп(0Н)2 производится встречающийся в природе
темно-красный минерал гаусманит: 2Мп(ОН)2 -ф
+ ЩМпО*— Мп3О4 + 4Н2О. Соли, отвечающие ос-
новной функции Мп(0Н)4, неустойчивы. Так, МпСЦ,
образующийся при взаимодействии МпО2 с крепкой
НС1, сразу распадается: МпО2 -ф 4НС1=МпС14 -ф
-ф2Н2О; МпС14=МпС12 -ф С12. Производные 6-ва-
лентного М. — марганцовистая кислота
Н2МпО4 и ее соли — манганаты, являются силь-
ными окислителями и легко восстанавливаются до
МпО2 (в щелочной среде) или солей Мп2+ (в кислой).
С другой стороны, они могут быть окислены до произ-
водных 7-валентного М.— марганцовой кис-
лоты НМпО4 и ее солей — перманганатов,
окрашенных, как правило, в характерный фиолетово-
красный цвет иона Мп04 (см. М арганцовистая кислота
и манганаты, Марганцовая кислота и перманганаты).
Аналитическое определение. М.
относится к аналитич. группе сульфида аммония,
т. е. его сульфид MnS нерастворим в воде, но раство-
рим в разб. к-тах. При осаждении Мп2+ сульфидом
аммония сначала образуется светло-розовый осадок
MnS • пН20, к-рый при нагревании с избытком
(NH4)2S переходит в зеленую модификацию MnS.
Чаще всего качественно М. открывают по фиолетово-
красной окраске р-ров, содержащих ион МпО4 ;
предварительно Мп (II) переводят в Мп (VII) дейст-
вием (NH4)2S2Os (в присутствии AgNO3), действием
NaBiO3, РЬО2, KSO4 и других окислителей. М. откры-
вают также по зеленому цвету массы, полученной
после окислительного плавления: MnO + Na2CO3 -ф
-ф 2KNO3=Na2MnO4 + 2KNO2 -ф СО2. Для коли-
чественного определения М. применяют гл. обр. ме-
тоды объемного анализа, также основанные на окис-
лении Мп2+ (висмутатный, перманганатометрический,
персульфатный и др.). Широко используются фотомет-
рии. методы, основанные на измерении светопоглоще-
ния раствора МпО4 , а также спектральные,
потенциометрические и др. инструментальные методы
анализа, к-рые позволяют определять М. в присут-
ствии др. элементов.
Получение. В пром-сти М. получают электролизом
водных р-ров сернокислого М. или восстановлением
окислов М. кремнием в электрич. печах; алюмино-
термич. способ применяется ограниченно. Исходным
сырьем для электролитич. получения М. могут служить
как богатые, так и бедные руды. В случае окисленных
руд их предварительно восстанавливают для перевода
М. в 2-валентную форму, а затем обрабатывают ис-
пользованным анолитом с добавками сульфата аммо-
ния и серной к-ты. Электролиз ведут в электролитных
ваннах со свинцовыми анодами и катодами из нержа-
веющей стали, разделенными диафрагмами (водоне-
проницаемое полотно); для устранения осаждения
МпО2 на аноде последний делают из свинцовых спла-
вов, содержащих, напр., Sn и Со или 1% Ag; с той
же целью уменьшают поверхность анодов и повышают
анодную плотность тока. Исходный электролит содер-
жит обычно 35—40 г М. и ок. 150 г (NH4)2SO4 на 1 л
р-ра; осаждение ведут при pH 8,0—8,5 (католита).
Снятые с катодов осадки переплавляют в индукционной
печи в слитки. По этому методу может быть получен
М. чистотой до 99,9% . Электролитич. метод применя-
ют для нанесения защитных покрытий М. на металлы.
В Советском Союзе М. получают также в электро-
печах силикотермич. методом, к-рый более экономи-
чен по сравнению с электролизом, но уступает ему по
чистоте продукта. Технология состоит из 3 стадий.
17 МАРГАНЦА КАРБОНАТ—МАРГАНЦА МЕТИЛЦИКЛОПЕНТАДИЕНИЛ ТРИКАРБОНИЛ 18
Вначале путем плавки руды и кварцита с недостатком
углеродистого восстановителя получают марганцовый
шлак с пониженным содержанием фосфора ( —0,012%Р)
и железа ( — 0,6% FeO), к-рый используют как сырье
при производстве М. и для выплавки кремнистого
восстановителя — силикомарганца (>36% Si); послед-
ний получают в дуговой электропечи восстановлением
окислов бесфосфористого шлака и кварцита углеро-
дом (коксом) в присутствии флюса (СаГг)- Выплавку
М. из шлака и силикомарганца с добавкой извести ве-
дут в дуговой электрич. печи с магнезитовой футеров-
кой; для уменьшения газонасытценности жидкий М.
в ковше вакуумируют. Полученный по этой техноло-
гии металл содержит ок. 97% Мп, 0,1% Си 0,5% Si.
Техника безопасности. При электротермия, получении М.
вторичное напряжение печи достигает 250 и 350 в, что представ-
ляет большую опасность для обслуживающего персонала. Для
предупреждения несчастных случаев ограждают подводящую
сеть и принимают другие меры. Для защиты работающих
от тепловых излучений печей применяют передвижные водо-
охладительные экраны и другие устройства. Наибольшую опас-
ность представляют соединения М., в том числе и кислородные,
весьма ядовитые, действующие на центральную нервную си-
стему и другие органы. Характерны хронич. отравления,
проявляющйеся через 2—3 года систематич. работы с М. Ис-
точниками отравления являются пыль (при добыче и переработ-
ке марганцовой руды), аэрозоль, образующийся при плавке
стали. Пути проникновения в организм — дыхательные ор-
ганы, пищевод, кожа, в соответствии с этим строится и техни-
ка безопасности, прежде всего защита дыхательных путей,
предупреждение попадания соединений М. с пищей и водой,
а также защита рук, частая смена белья и платья.
Применение. М. является оДним из основных ме-
таллов, применяемых для раскисления и легирования
стали (о ферромарганце см. Железа сплавы). Чистый
М. используют в небольших количествах при получе-
нии алюминиевых и др. сплавов (см. Алюминия спла-
вы), до 20% М. содержится в специальных сплавах
типа манганин (см. Магния сплавы, Меди сплавы).
М. применяют также для создания антикоррозионных
защитных покрытий на металлах.
Лит.: С а л л п А. X., Марганец, пер. с англ., М., 1959;
Производство ферросплавов, 2 изд., М., 1957; Гиллебр анд
В. Ф. [и д р.], Практическое руководство по неорганическому
анализу, пер. с англ М.. 1957; Mellor, v. 12, L.—N.Y.—
Toronto, 1932; то же, v. 12, 1953; Pascal, t. 16, P., I960;
Ullmann, 3 Aufl., Bd 12, Mtinch.—B., 1960, S. 201—235;
Ki rk,v. 8, N. Y., 1952, p. 718—64. JO. А. Павлов.
МАРГАНЦА КАРБОНАТ МпСОз—белый или
светло-розовый порошок; плоти. 3,125. Встречается
в природе (минерал родохрозит или марганцовый
шпат). Почти нерастворим в воде. При кипячении
разлагается водой. На воздухе окисляется с образо-
ванием МпО2 и отщеплением СОг. В от-
сутствие воздуха разлагается при 300°
с образованием СО и высших окислов
марганца. Получают осаждением р-ров
сернокислого марганца содой или кар-
бонизацией аммиачной суспензии окиси
марганца. При осаждении бикарбонатом
натрия из раствора MnSO4, насыщенного
СОз, осаждается гидрат МпСО3’Н2О. М. к.
применяют для изготовления марганцо-
вых пигментов, сиккативов и т. п.
Д. С. Стасиневич.
МАРГАНЦА КАРБОНИЛ Мп2(СО)10 —
золотисто-желтые прозрачные кристал-
лы; плотн. 1,75, т. пл. 154°. Разложение
М. к. с отщеплением СО начинается при
110°, но в атмосфере СО он стоек до 250°.
Растворим в большинстве органич. растворителей, не-
растворим в воде. М. к. получают, обрабатывая гало-
генид марганца окисью углерода под давлением в
присутствии металлоорганич. соединений [напр.,
C5H5MgBr, А1(С2Н5)з и др.]. Реагируя с галогенами
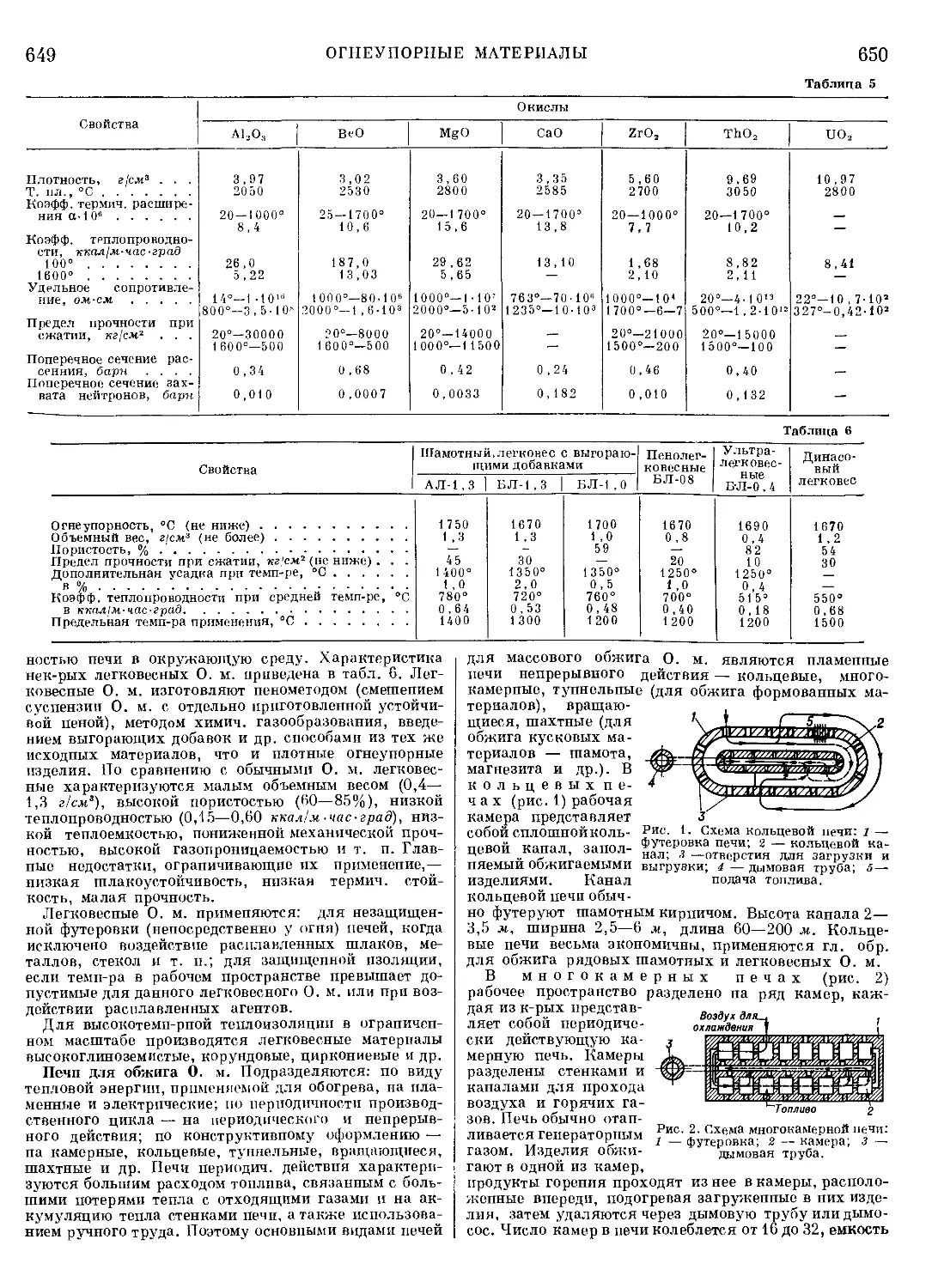
(иод, бром), М. к. образует карб они лгалоген fl-
fl ы Мп(СО)5Х. При действии Na на М. к. или его гало-
гениды получается NaMn(CO)s. Последний при разло-
жении кислотами дает НМп(СО)5. При действии гало-
геналкилов на NaMn(CO)s образуются алкильные
производные М. к., напр. CH3Mn(CO)s, бесцветные
кристаллы, т. пл. 95°, стойкие на воздухе. При взаимо-
действии циклопентадиена с М. к. образуется цикло-
пентадиенилтрикарбонил марганца CsHsMn(CO)3. М. к.
и его нек-рые производные (см. Марганца мепгилцик-
лопентадиенилтрикарбонил) являются сильными ан-
тидетонаторами, сравнимыми по эффективности с ТЭС,
НО мало ТОКСИЧНЫМИ. Д- С. Стасиневич.
МАРГАНЦА МЕТИЛЦИКЛОПЕНТАДИЕНИЛТРИ-
КАРБОНИЛ (МД-СМТ, АК-ЗЗХ) CH3CsH4Mn(CO)3 —
антидетонатор моторных топлив, маловязкая жид-
кость светло-янтарного цвета; плотн. 1,39; темп-ра
замерзания 1,5°; темп-ра кип. 233°; нерастворим в
воде, хорошо смешивается с органич. растворителя-
ми. Антидетонационные свойства М. м., установленные
по исследовательскому методу в расчете на металл
(Мп), значительно лучше, чем у тетраэтилсвинца (ТЭС),
а по моторному методу — примерно равны. Антпдето-
национные свойства по исследовательскому методу
лучших автомобильных неэтилированных бензинов
при добавлении М. м. в количестве 0,52 г на 1 л, счи-
тая на Мп, возрастают в зависимости от их химич.
состава на 4,5—15 октановых единиц (см. Октановое
число). Антидетонационные свойства топлив, содержа-
щих М. м., улучшаются мало, если в топливе присут-
ствуют ароматич. углеводороды; заметно улучшаются
при содержании в топливе алкилатов. Олефины и сер-
нистые соединения не ухудшают восприимчивости
топлив к М. м. В авиабензинах М. м. более эффекти-
вен, чем тетраэтилсвинец (ТЭС), при оценке детона-
ционной стойкости (сортности) богатых смесей и ме-
нее эффективен на бедных смесях.
Хорошие результаты получаются при использова-
нии М. м. в этилированных топливах. В таблице при-
ведены результаты определения антидетонационных
свойств этилированных топлив по исследователь-
скому методу, содержащих различные количества М.м.
При испытаниях выявлено, что при добавлении М. м.
в малых концентрациях к бензину, содержащему
ТЭС, увеличиваются отложения на изоляторах свечей
и снижаются сроки службы выхлопных клапанов дви-
гателей. Для устранения этого отрицательного влия-
ния к топливу добавляют в небольших количествах
фосфорную добавку, которая благоприятно дейст-
вует на срок службы свечей и уменьшение нагара.
Стоимость М. м. значительно превышает стоимость
Топливо Октановое число Увеличение октанового чис- ла топлив, содержащих ТЭС 0,792 м,л;л, при добавлении М. м., г Мп в 1 л
без ТЭС с 0,792 мл ТЭС в 1 л
0,066 0,1 32 0,264
Алкилат 93,8 104,7 7,5 9,2 11,3
Газовый бензин 71,0 88,0 4,2 5,0 5,8
Изомеризационный компонент Тяжелый бензин каталитич. 85,0 97,0 5,0 5,8 7,4
крекинга Бензины каталитич. рифор- минга, содержащие арома- тику: 9'3,4 98,6 0,5 0,8 1,1
35,0% 88,1 98,9 1,2 1,3 1,8
62,5% 97,8 102 ,1 — о,1 -0,1 —0,1
Бензин обычного сорта . . 83,7 93,6 2,0 2,2 2,9
Бензин высшего сорта . . . 91,6 98,5 0,7 1 ,1 1,8
ТЭС, что препятствует его самостоятельному приме-
нению. Предполагается добавлять М. м. в небольших
концентрациях в топлива, содержащие оптимальное
количество ТЭС. При этом повышается детонационная
стойкость топлива при сравнительно небольшом его
удорожании.
Лит.: Гибсон X. Д., Лиджетт У.Б., Уоррен
Т. У., в кн.: Труды пятого международного нефтяного кон-
гресса, Нью-Йорк, 1959, т. 4, М., 1961, с. 166; Научно-техни-
ческий бюллетень н.-и. ин-та ГСМ, М., 1959. В. В. Панов.
19
МАРГАНЦА НИТРАТ—МАРГАНЦА СУЛЬФАТЫ
20
МАРГАНЦА НИТРАТ Mn(NO3)2 — кристаллизует-
ся ниже 24° с 6, выше 24°—с 3 молекулами воды.
Mn(NO3)2-6H2O — розово-красные моноклинные кри-
сталлы, плотность 1,82; т. пл. 25,8°. В воде М. н.
хорошо растворим. В насыщенном р-ре содержится
Mn(NO3)2 50,5% при 0°, 76,8% при 35,5°. Тригидрат
можно обезводить умеренным нагреванием с конц.
HNO3. Безводный М. н. выше 180° разлагается:
Mn(NO3)2 = МпО2 + 2NO2. М. н. можно получить
обработкой марганцовой руды, предварительно обож-
женной в восстановительной печи азотной к-той.
Разложением р-ра Mn(NO3)2 или безводной соли полу-
чают очень чистую двуокись марганца, используемую
как деполяризатор. Д- С. Стасиневич.
МАРГАНЦА ОКИСЛЫ — соединения марганца с
кислородом; в свободном состоянии получено 5 М. о.,
причем увеличение валентности Мп от 2 до 7 сопро-
вождается усилением кислотных и ослаблением основ-
ных свойств окислов. Обычным исходным продуктом
для получения прочих М. о. служит природный пиро-
люзит МпОг-
Закись марганца МпО — серо-зеленые
кристаллы с кубич. решеткой, а = 4,436 А; плоти.
5,18; т. пл. 1650°; возгоняется с диссоциацией при
3400°. При темп-pax ниже 200° МпО образует с кисло-
родом твердые р-ры с областью гомогенности от
МпО до МпО, 1S. Окисление при 200—350° приводит
к образованию’ МпгОз. Свежеполученный тонкодис-
персный порошок МпО окисляется на воздухе уже
при комнатной темп-ре. МпО — основной окисел;
в воде практически нерастворим; легко растворяется
в кислотах с образованием солей 2-валентного Мп.
Гидроокись Мп(0Н)2 — слабое основание, малорас-
творимое в воде; выпадает в виде белого осадка, бурею-
щего на воздухе вследствие окисления. В природе
МпО встречается в виде минерала манганозита.
Искусственно его можно получить восстановлением
высших окислов Мп водородом при 300— 600°, разло-
жением карбоната или оксалата Мп в вакууме при
350—800° и др. способами. МпО обладает полупровод-
никовыми свойствами с дырочным механизмом про-
водимости. Применяется в производстве ферритов с
высокой диэлектрич. проницаемостью стали, а так-
же красок.
Закись-окись марганца Мп3О4 — наи-
более устойчивый при высоких температурах окисел
марганца; черно-коричневые кристаллы, существую-
щие в трех модификациях: при обычных условиях
устойчива а-Мп304 (природный гаусманит)
с тетрагональной решеткой, а=8,148 А, с=9,410А;
плоти. 4,718; при 890—960° а-Мп304 необратимо
переходит в f}-Mn3O4, что сопровождается поглоще-
нием тепла. 0-Мп3О4 образуется также при прока-
ливании различных соединений Мп. Решетка Р-
Мп3О4 аналогична а-Мп3О4. При нагревании до 1170°
Р-Мп3О4 обратимо переходит в у-Мп3О4 с кубич. ре-
шеткой, a=8,7 А. Т. пл. Мп3О4 ок. 1590°; теплота плав-
ления ок. 38 ккал/молъ. Взаимодействие с кислородом
приводит к образованию твердого р-ра с областью
гомогенности от МпО4,33 до MnOi,4 с последующим
окислением до Мп203. Восстановление Мп304 до
МпО в атмосфере Н2 происходит при 500°. В воде
Мп3О4 нерастворим. В холодной конц. H2SO4 раство-
ряется с образованием сульфатов 2- и 3-валентного
Мп. В природе Мп3О4 встречается в виде минерала
гаусманита. Искусственно его можно получить вос-
становлением МпО2 водородом при 220°.
Окись марганца, т. наз. курнакит,
Мп203 — бурые кристаллы, обладающие полимор-
физмом. a-Курнакит, получаемый термич. разложением
МпО-ОН или окислением МпО кислородом воздуха
при 350—450°, имеет тетрагональную центрированную
решетку, а=8,85 А, с=9,95 А, плоти. 4,94. р-Кур-
накит, образующийся при термич. разложении пиро-
люзита (f}-MnO2) выше 600°, имеет сложную кубич.
решетку, а=9,41 А. а-Мп203 переходит в f}-Mn2O3
при 420°. При нагревании выше 750° Мп203 разла-
гается на Мп3О4 и О2. С кислородом Мп203 образует
твердые р-ры с областью гомогенности от МпО, 5
до МпО, 6. При последующем окислении образуется
МпО2. Окись марганца — основной окисел, практи-
чески нерастворимый в воде. В конц. кислотах рас-
творяется с образованием неустойчивых солей 3-валент-
ного Мп. Черно-коричневая гидроокись Мп(0Н)3—
очень слабое основание, нерастворимое в воде; теряет
воду выше 100°. По составу к Мп203 приближается
минерал браунит Mn(Mn,Si)O3.
Двуокись марганца МпО2 — наиболее
устойчивый при обычных условиях окисел марганца;
черные кристаллы, обладающие полиморфизмом. Наи-
более изучены f}-MnO2 (пиролюзит) — тетрагональные
кристаллы типа рутила, а=4,398 А, с=2,867 А,
плоти. 5,026. При 540—600° f}-MnO2 разлагается
с образованием f}-Mn2O3; при 940—1025° переходит
в f}-Mn3O4. Модификация у-МпОг имеет ромбич. ре-
шетку, а = 4,533 А, 5=9,27 А, с = 2,866 А. При
нагревании выше 325° у-МпО2 необратимо переходит
в р~МпО2.
Двуокись марганца является сильным окислителем.
Переводит S в SO2, НС1— в Cl2, NH3 и соли аммония —.
в N2, углерод и органич. соединения — в СО2 и т. д.
Двуокись марганца обладает слабовыраженными
кислотными и основными свойствами. В воде и щело-
чах нерастворима. В кислотах легко растворяется
с образованием неустойчивых солей 4-валентного Мп,
к-рые быстро превращаются в соли 2-валентного Мп.
При прокаливании МпО2 с окислами металлов обра-
зуются м а н г а н и т ы, наир. СаМпО3 — соли, от-
вечающие кислотной функции гидроокиси Мп(0Н)4
(производные метамарганцоватистой к-ты Н2МпО3).
Темно-бурая гидроокись Мп(0Н)4 практически нера-
створима в воде.
В природе р-МпО2 встречается в виде минерала
пиролюзита; природная у-МпО2 — рамсдел-
лит, встречается редко. Искусственно МпО2 получают
термич. разложением Mn(N03)2, электролизом солей
закиси марганца и др. способами. МпО2 служит основ-
ным сырьем для получения марганца и его соединений,
применяется в произ-ве сухих гальванич. элементов,
для приготовления катализаторов типа гопкалита,
как окислитель в химич. пром-сти и т. д. Ангидрид
марганцовистой к-ты (МпО3) в свободном виде не вы-
делен. О марганцовом ангидриде Мп20, см. Мар-
ганцовая кислота и перманганаты.
Лит.: Роде Е. Я., Кислородные соединения марганца,
М., 1952; Г е л ь д П. В., Есин О. А., Процессы высоко-
температурного восстановления, Свердловск, 1957.
МАРГАНЦА СУЛЬФАТЫ — сернокислые соли
2-, 3- и 4-валентного марганца. Сульфат 2 - в а-
лентного марганца MnSO4 — бесцветные
ромбич. кристаллы, периоды решетки: а = 4,86 А,
Ъ = 6,84 А, с = 8,58 А; плотн. 3,25; т. пл. 700°; раз-
лагается при 850°. Растворимость в воде (г MnSO4
на 100 г воды): 52,9 (0°), 64,8 (25°), 35,5 (100°). В при-
сутствии серной к-ты растворимость М. с. в воде сильно
снижается. Образует кристаллогидраты с 1, 4, 5 и
7 молекулами Н2О. Продажный препарат представляет
собой MnSO4-4H2O — моноклинные кристаллы розо-
вого цвета, выпадающие из водных р-ров при 30—40°.
MnSO4-5H2O устойчив в интервале 9—25°, a MnSO4-
7Н2О ниже 9°-MnSO4 • Н2О получается при нагре-
вании других гидратов до 200°. Все гидраты, кроме
MnSO4 • 4Н2О, при стоянии на воздухе постепенно
выветриваются. MnSO4 получают растворением МпО
или МпС03 в серной к-те. Применяют в текстильной
пром-сти, в фарфоровом произ-ве, а также в качестве
21
МАРГАНЦА СУЛЬФИДЫ—МАРГАНЦОВИСТАЯ КИСЛОТА И МАНГАНАТЫ
22
электролита при получении МпО2 и чистого металлич.
Мп. М. с. является эффективным микроудобрением.
Сульфат 3-валентного марганца
Мп2(804)з — зеленый аморфный порошок. Разла-
гается при 160°. Сильный окислитель, его смеси с ор-
ганич. веществами взрывчаты. Получается раство-
рением МпО2 в серной к-те при повышенных темп-рах.
Сульфат 4-валентного марганца
Mn(SO4)2 известен только в виде растворов (коричне-
вого цвета), образующихся при окислении MnSO4
при помощи КМпО4 в сернокислой среде. Растворим
в серной к-те, выше 80° разлагается.
Лит,: П о з и н М. Е., Технология минеральных солей,
2 изд., Л., 1961; Руководство по препаративной неорганической
химии, под ред. Г. Брауера, пер. с нем., М., 1956.
Д. С. Стасиневич.
МАРГАНЦА СУЛЬФИДЫ — соединения марганца
с серой; получены MnS, MnS2 и Mn3S4. В природе MnS
встречается в виде минерала алабандина, MnS2—
в виде минерала гауерита.
Марганец сернистый MnS известен в
трех модификациях: красный MnS — кубич. решетка
типа сфалерита, а = 5,611 А; розовый MnS — гекса-
гональные кристаллы, а=3,976 А, с=6,432 А; зеленый
MnS — кубич. решетка типа NaCl, а —5,21 А. Красный
и розовый MnS переходят в зеленый при нагревании
в токе СО 2 до 300—320°. Теплота образования зеле-
ной модификации АЯ298 = —48,8 ккал/моль. Плот-
ность MnS 3,99—4,08; т. пл. 1600° ± 10°. При
нагревании на воздухе окисляется доМп3О4 и MnSO4.
В воде труднорастворим: 0,00623 г/л. Растворяется
в кислотах (с разложением и выделением H2S) и в
р-рах солей аммония. М. с. может быть получен дей-
ствием (NH4)2 S на водные р-ры солей 2-валентного Мп.
Вначале выпадает светло-розовый аморфный осадок,
к-рый при стоянии превращается в зеленый. Эта
реакция используется в качественном анализе для
обнаружения марганца.
Марганец двусернистый MnS2 — чер-
ные кубические кристаллы типа пирита, а=6,097 А,
плотн. 3,463. При нагревании на воздухе разлагает-
ся. В воде нерастворим, растворяется в кислотах
с образованием H2S, S и соответствующей соли мар-
ганца.
Смешанный сульфид 2- и 4-валентного
марганца Mn3S4 весьма неустойчив. Получен при
прокаливании MnSiOs в токе CS2 при темп-ре белого
Каления Н. Ханда-иирова.
МАРГАНЦА ХЛОРИДЫ — соединения марганца
с хлором. Хлорид 2-валентного мар-
ганца МпС12 — розовые гексагональные кри-
сталлы, а=6,20 А, а=34°32'; плотн. 2,977; т. пл.
690°, т. кип. 1190°. Теплота образования ДН°298 =
= — 115,3 ккал/моль. Растворимость в воде (в %):
38,8 (0°); 42,5 (20°); 53,5 (100°). Образует гидраты
с 2, 4 и 6 молекулами воды. МпС12 • 6Н2О стоек ниже
—2°. Технический препарат представляет собой устой-
чивый при обычной темп-ре МпС12 • 4Н2О — светло-
розовые моноклинные кристаллы, плавящиеся в крис-
таллизационной воде при 58,089°; плотн. 2,01.
МпС12 • 2Н2О стоек выше 58,089°; теряет кристаллиза-
ционную воду при нагревании до 200°. М. х. раство-
рим в этиловом спирте и образует алкоголят МпС12 •
• ЗС2Н5ОН. На воздухе М. х. в присутствии влаги
•окисляется с образованием МпО2 и выделением НС1.
М. х. получают растворением МпО2 в соляной к-те.
Для выделения безводного продукта МпС12 • 4Н2О
прокаливают в токе N2 или НС1. Хлорид 3-в а-
леитного марганца МпС13 содержится в
темно-красных или коричневых р-рах, получаемых
при растворении МпО2 в соляной к-те. Легко разла-
гается на МпС12 и С12 уже при низких темп-рах. Выде-
лить МпС1з из р-ров в чистом виде не удалось. Хло-
рид 4-валентного марганца МпСЦ
в свободном состоянии не получен ввиду крайней
неустойчивости, но известны его комплексные соли,
напр. К2МпС18 (см. также Марганец).
МАРГАНЦОВАЯ КИСЛОТА И ПЕРМАНГАНА-
ТЫ— кислота HMnOi, отвечающая 7-валентному
марганцу, и ее соли. Анион МпО4 имеет фиолетово-
малиновый цвет. НМпО4 известна лишь в водных
р-рах. В свободном состоянии получен ее ангидрид
Мп20,— зеленовато-черная маслянистая жидкость
с плоти. 2,396 и темп-рой затвердевания +5,9°.
Мп20, начинает разлагаться прибл. при 55°, вызывая
саморазогрев системы, что может привести (при
— 90°) к взрывному распаду. Благодаря исключитель-
но сильным окислительным свойствам Мп20, эфир,
спирт и др. органич. вещества при соприкосновении
с ним воспламеняются. Раствор НМпО4, образующий-
ся при взаимодействии Мп20, с водой, может быть упа-
рен лишь до 20%-ного содержания НМпО4; при даль-
нейшем концентрировании НМпО4 разлагается с обра-
зованием МпО2 и кислорода. НМпО4 — очень сильная
к-та, она диссоциирована примерно в такой же сте-
пени, как НС1 и HN03. Перманганаты Na и 2-валент-
ных металлов легкорастворимы в воде, перманганаты
Rb, Cs, К сравнительно труднорастворимы. Марган-
цовая к-та и перманганаты — очень сильные окисли-
тели, причем степень восстановления Мп’+ зависит от
среды; в кислой:
2Мпо7 + 6Н-(-> 2Мп2 + +ЗН;О + 2'/2О2
в нейтральной или слабощелочной:
2МпО~+Н,О —> 2МпО2+2ОН- + 1‘/=О2
в сильно щелочной:
2МпО~ + 2ОН-—(-2МпО4-+Н2О+1/2О2
Т. обр., окислительные свойства производных
7-валентного Мп наиболее сильно выражены в кислой
среде. Перманганаты широко применяются как окис-
лители, в медицине — для дезинфекции, при ожогах
и проч. Наибольшее практич. значение имеет калия
перманганат.
МАРГАНЦОВИСТАЯ КИСЛОТА И МАНГАНА-
ТЫ — кислота Н2МпО4, отвечающая 6-валентному
марганцу, и ее соли. Анион Мп04 окрашен в зеленый
цвет. Н2МпО4 и ее ангидрид МпОз в свободном виде
не выделены. Образующаяся при подкислении раст-
воров манганатов марганцовистая к-та неустойчива
и тотчас распадается на МпО2 и марганцовую к-ту
НМпО4. Аналогичный самопроизвольный распад ха-
рактерен и для манганатов, но протекает значительно
медленнее (особенно в сильнощелочной среде). Все
производные 6-валентного Мп являются сильными
окислителями и восстанавливаются до МпО2 (в щелоч-
ной среде) или солей Мп2+ (в кислой). С другой сто-
роны, действием очень сильных окислителей (напр.,
свободного хлора) манганаты могут быть окислены до
солей марганцовой к-ты — перманганатов: 2К2МпО4+
+ С12=2КС1 + 2КМпО4. Манганаты Na и К легко-
растворимы в воде, манганат Ва труднорастворим.
Наибольшее практич. значение из манганатов имеет
К2МпО4, служащий для получения КМпО4 по приве-
денной выше реакции.
Манганат калия может быть получен сплавлением МпО2
с КОН и KN03. Продукт реакции дает с водой раствор зеленого
цвета. При окислении кислородом воздуха раствор становится
синим, фиолетовым и, наконец, малиновым (окраска аниона
МпО4 ). Вследствие этих перемен цвета К2МпО( был назван
впервые получившим его К. Шееле (1774) минеральным ха-
мелеоном (по цвету ящерицы, легко меняющей свою окраску).
Со второй половины 19 в. минеральным хамелеоном стали на-
зывать калия перманганат.
МАРГАРИН—МАРКОВНИКОВА ПРАВИЛО
23
МАРГАРИН — пищевой продукт, приготовленный
из смеси растительных масел, животных жиров, молока
и нек-рых др. компонентов (соль, сахар, красители,
ароматизаторы и др.). В состав М. входит: 82% жиров,
ок. 0,5% белков, ок. 0,5% сахара, 0,2—0,7% соли и
16,5% воды (не больше).
Различают след, виды М.: 1) собственно
маргарин (столовый, сливочный, кондитерский и
др.), представляющий собой, как и масло сливочное,
охлажденную до кристаллич. состояния жира и под-
вергнутую механич. обработке водно-жировую эмуль-
сию; 2) кухонные жиры (комбижир, марту-
селин, растительное сало), представляющие собой
закристаллизованную смесь твердых и жидких живот-
ных и растительных жиров, предназначенную для
приготовления пищи, а также для применения в
хлебопекарной, кондитерской и пищевой пром-сти.
Основным сырьем для произ-ва М. служат натуральные
и гидрированные растительные масла (подсолнечное,
хлопковое, соевое, арахисовое, кокосовое и др.), гид-
рированный китовый жир, говяжье и свиное топленое
сало (последние только в кухонных жирах). В состав
сливочного М. входит 25% сливочного масла. Помимо
жиров, при произ-ве М. используют молоко, эмуль-
гаторы, витамины, ароматизаторы и др. Молоко,
являющееся основным компонентом нежировой фазы
М., предварительно сквашивают чистыми культурами
молочнокислых заквасок; образующиеся в результате
брожения продукты (молочная к-та, эфиры и др.)
придают М. характерный молочнокислый аромат.
В качестве эмульгаторов, необходимых для получения
стойких и дисперсных жиро-водных эмульсий, приме-
няют: эмульгатор Т-2 — продукт этерифика-
ции твердых жирных к-т с полиглицерином; эмуль-
гатор Т-Ф — смесь моно- и диглицеридов с фосфа-
тидами; жиросахара — моно- или дистеарат
сахарозы.
М. получают по след, технология, схеме: подготов-
ленную жировую основу, состоящую из рафиниро-
ванных растительных масел, саломасов и пищевых
жиров, смешивают с молоком и др. составными ча-
стями. Полученную смесь эмульгируют при ,32—35° и
быстро охлаждают до 3—5° на вращающихся бараба-
нах. Закристаллизовавшаяся масса поступает в аппа-
рат для уплотнения, после чего М. расфасовывают
автоматами или упаковывают в бочки. При произ-ве
кухонных жиров подготовленные рафинированные
растительные масла и пищевые животные жиры тща-
тельно смешивают, охлаждают и сливают в ящики или
бочки. Все шире начинают применять метод произ-ва
М., по к-рому прошедшую через гомогенизатор тонко-
дисперсную эмульсию переохлаждают в закрытом ци-
лпндрич. аппарате, после чего сразу подают на рас-
фасовку.
По методу Н. И. Козина и В. И. Варибруса из жи-
ровой основы и молока в присутствии белка — казеи-
на (эмульгатора) готовят 60—70%-ную эмульсию типа
сливок. В полученную эмульсию вводят молочно-
кислую закваску и недостающее до 82% количество
жира. После дополнительного эмульгирования эмуль-
сия поступает в вытеснительный охладитель, в к-ром
происходит перемешивание, охлаждение и частичная
дестабилизация поступившего продукта; в результате
он приобретает структуру, сходную со сливочным ма-
слом. Благодаря наличию непрерывной водно-мол оч-
ной фазы этот продукт имеет более приятный вкус и
аромат по сравнению с др. видами М. Полученный
по этой схеме М. должен храниться в охлаждаемых
складах при ±2°. Н. И. Козин, В. И. Варибрус.
МАРГАРИНОВАЯ КИСЛОТА (гептадекановая кис-
лота) СНз(СН2)15СООН, мол. в. 270,46 — кристаллы
(пластинки из петролейного эфира); т. пл. 61,3°;
т. кип. 363,87760 мм, 230,7716 мм’, d’e 0,8532; п™
ch3-ch=Qh2
г
i Г
сн3-сн=сн2
и
24
1,4342; в 100 г воды при 20° растворяется 0,00042 г;
растворяется в эфире, частично — в спирте. В при-
роде М. к. не найдена. М. к. получают из гексадецил-
бромида или хлорида превращением их в нитрил
с последующим гидролизом; реакцией гексадецилмаг-
нийбромида с СО2 и др. Глицерид М. к. используют
в медицине для изучения метаболизма при диабете.
„ . _ Л. С. Поваров.
МАРКОВНИКОВА ПРАВИЛО — правило, опре-
деляющее порядок присоединения элементов галоге-
новодородов к несимметричным олефинам. Согласно
М. и., атом водорода присоединяется к более гидроге-
низированному, а атом галогена —к менее гидроге-
низированному атому углерода, напр.:
СН3-СН = СН24-НХ —►СНз-СНХ-СН,
Теоретич. истолкование М. п. заключается в сле-
дующем. Двойная связь несимметрично построенного-
олефина поляризована, что подтверждается наличием
дипольного момента у подобных соединений, напр.
дипольный момент пропилена равен 0,35 D. Направле-
ние поляризации определяется электронно-донорным
влиянием алкильных групп (I). Указанная поляри-
зация и обусловливает в общем случае порядок
присоединения элементов галогеноводородов (II). Та-
ким образом, катион, при-
соединяющийся к молеку-
ле олефина, атакует бо-
лее гидрогенизованный(за-
ряженный отрицательно)
атом углерода, а анион —
менее гидрогенизованный
(заряженный положительно). В соответствии с М. п.
в его обобщенной, только что приведенной форме идут
ионные реакции присоединения к олефинам галоге-
новодородов, H2SO<, воды, хлорноватистой к-ты, со-
лей ртути и нек-рых других соединений.
Для объяснения М. п. в случае реакций присоеди-
нения к молекулам олефиновых углеводородов были
рассмотрены только статич. факторы (строение исход-
ной молекулы олефина и строение реагента). К объяс-
нению М. п. можно прийти и другим, независимым
путем, рассматривая динамич. факторы (строение
активированного комплекса). Известно, что присоеди-
нение галогеноводородов по двойной углерод-
углеродной связи представляет собой двухстадийную
электрофильную реакцию, начинающуюся с присоеди-
нения протона, напр.:
Н+ + X-
СН3—СН = СН2-----> СН3—СН—СН3------> CH —CTI -сн»
медленно быстро |
X
Из двух катионов, к-рые могут образовываться н
результате присоединения протона к пропилену —
изопропил-катиона и н-пропил-катиона,—более устой-
чив первый; он и образуется в действительности.
Т. обр., из рассмотрения динамич. факторов выте-
кает второе, независимое объяснение М. п.: реакции
присоединения по двойной связи идут в направлении
образования наиболее устойчивого промежуточного
катиона. В рассмотренных случаях присоединения
к олефинам статич. и динамич. факторы действуют
согласованно, благоприятствуя одному и тому же
направлению реакции; трудно судить, какой из фак-
торов решающий. При переходе к хлорзамещенным
этиленам становится ясным, что решающее влия-
ние оказывает динамический фактор. Присоединение
НВг к хлористому винилу проходит в соответствии
с М. п.:
Н2С = СН-С1+НВг —> СН3-СНВг-С1
в то время как в нереагирующей молекуле хлористого
винила (III) двойная связь поляризована так, что
25
МАРКОВНИКОВА ПРАВИЛО—МАСКИРОВКА
26
наиболее гидрогенизованный атом углерода несет не-
который положительный заряд. В реакции хлористо-
_______ то винила с НВг статический фактор
СН?^СН^-"*~ С1 (характер поляризации двойной связи
под влиянием атома хлора) благопри-
щ ятствует присоединению НВг против
М. п.
Для того чтобы составить суждение о динамич.
факторах данной реакции, необходимо рассмотреть
два катиона:
+ ,, +
Н2С—CH—С1: и Н2С—СН2—С1:
IV V
Катион IV возникает при атаке молекулы хлористого
винила протоном в соответствии с М. п., а катион
V — при атаке против М. п. И в обоих случаях на
атоме углерода возникает положительный заряд, что
связано с повышением энергии системы. Однако в
катионе IV положительный заряд частично компен-
сирован смещением внешнего электронного облака
атома хлора:
CH2-CH-£i:
н
В результате этого положительный заряд оказывается
нелокализованным на атоме углерода (в нек-рой сте-
пени положительно заряженным становится и атом
хлора), что и является причиной снижения энергии
системы и, следовательно, причиной повышения устой-
чивости катиона IV.
В катионе V вследствие его структурных особен-
ностей положительный заряд на атоме углерода не
может быть хотя бы отчасти компенсирован смещением
электронного облака к атому хлора, т. к. последний
расположен далеко. Наличие же локализованного
положительного заряда на атоме углерода обуслов-
ливает значительную энергию системы и, следователь-
но, неустойчивость катиона V. Т. обр., катион IV
более устойчив, поэтому рассматриваемая реакция
протекает по М. п., несмотря на то, что поляризация
исходной молекулы (статпч. фактор) благоприятствует
обратной реакции. Однако статпч. фактор, заключаю-
щийся в неблагоприятной поляризации молекулы
для присоединения НВг по М. п., сказывается на
скорости реакции. Т. к. приближение протона к атому
углерода, несущему нек-рый положительный заряд,
затруднено, то скорость присоединения НВг к хло-
ристому винилу во много раз меньше скорости при-
соединения НВг к незамещенному этилену:
Г <Г
СН2=СН—О
н
Аналогично можно объяснить направление присо-
единения бромистого водорода к трихлорэтилену:
СС12 = СНС1+НВг —> СС12Вг—СН2С1
Из этих примеров ясно, что решающую роль играют
не статич., а динамич. факторы.
Поэтому наиболее общая современная формули-
ровка М. п. должна быть дана с учетом этих факторов,
а именно: присоединение элементов
галогеноводородов (и нек-рых
других веществ) к непредельным
соединениям происходит таким
образом, что промежуточно обра-
зуются катионы, наиболее устой-
чивые нз всех возможных. Такая об-
щая формулировка охватывает также и случаи при-
соединения (представлявшиеся ранее аномальными)
галогеноводородов к непредельным соединениям, в
молекулах которых ненасыщенные атомы углерода
связаны с сильными электронно-акцепторными груп-
пами, напр.:
(CH3)3N - CH = CH2 + HJ —» (CH3),N - СН2 — СН2 — J
CF3 - CH=CH2 + HBr —► CF3 ~ CH2 - CH2 - Br
В случае присоединения НВг к олефинам в присут-
ствии перекисей М. п. неприменимо, т. к. реакция
в этом случае имеет не гетеролитич., а свободно-ра-
дикальный характер.
Лит.: Марковников В. В., Материалы по вопросу
о взаимном влиянии атомов в химических соединениях, в сб.:
Избранные труды, М., 1955; Реутов О. А., Теоретические
проблемы органической химии, М., 1956, гл. IV. О. А. Реутов.
МАСКИРОВКА (в химическом анализе) — связы-
вание мешающих ионов в малодиссоциирующий ком-
плекс при открытии, определении и отделении к.-л.
компонента. При этом часто исключаются трудоемкие
процессы отделения мешающих ионов осаждением
или экстракцией. Иногда относят к М. также переве-
дение мешающего иона в другое валентное состояние,
в к-ром этот ион не мешает определению. Во всех
случаях М. позволяет сократить затраты времени и
труда на отделение мешающих ионов. Кроме того, во
многих случаях при использовании М. получаемые
результаты более точны. М. широко применяют при
осаждении определяемого компонента, при титри-
метрич., фотометрич. и электрохимич. методах опре-
деления.
До сих пор не существует классификации методов
и общепринятых критериев оценки различных веществ
для М. одного и того же иона в присутствии других.
В принципе, М. мешающего (препятствующего) иона
в присутствии данного определяемого иона может
быть решена расчетным путем. В случае реакций осаж-
дения определяемого иона для расчета необходимо
знать произведения растворимости осадков мешаю-
щего и определяемого ионов с данным осадителем,
константы кислотной диссоциации осадителя и маски-
рующего вещества и, наконец, константы диссоциации
комплексов препятствующего и определяемого ионов
с маскирующим веществом. Однако эти характеристи-
ки обычно недостаточно изучены, особенно в сложных
равновесных системах. Поэтому поиски маскирую-
щего вещества и оптимальных условий его применения
обычно ведут опытным путем, на основании общих
данных об отношении ионов к данному осадителю
и различным маскирующим веществам; иногда при-
меняют частичные расчеты отдельных равновесий,
графич. интерполяцию и т. п. Выбор маскирующего
вещества зависит от конкретных условий анализа.
Так, железо (Fe11^) мешает определению никеля осаждением
диметилглиоксимом, т. к. при pH осаждения диметилглиокси-
мата никеля осаждается и гидроокись железа. В этом случае
для М. железа применим широкий круг маскирующих веществ,
образующих с ним устойчивый комплекс, напр. пирофосфат,
винная к-та п др. Однако, напр., комплексон III неприменим,
т. к. в этом случае задерживается осаждение никеля. Не
применяют также фторид, т. к. возможно осаждение гексафто-
роферриатов щелочных металлов. Если же необходимо замас-
кировать железо при иодометрич. определении меди, то послед-
нее обстоятельство не имеет значения, и фторид вполне приме-
ним. Наконец, если необходимо замаскировать железо при
фотометрич. определении кобальта, то не применяют тартратов
из-за окраски их комплексов с железом; наоборот, фторид,
пирофосфат или др. вполне допустимы. В последнем случае
удобно применять восстановители 3-валентного железа, что,
разумеется, недопустимо при иодометрич. определении меди.
Одно и то же маскирующее вещество может приме-
няться для различных, а иногда и для противополож-
ных целей, в зависимости от выбранной реакции. Так,
при фотометрич. определении вольфрама роданидным
методом ниобий можно маскировать щавелевой к-той.
Однако при осаждении ниобия купфероном щавелевая
к-та хорошо маскирует вольфрам.
Применение М. значительно расширилось в связи
с введением в практику комплексонов. Так, этилен-
диаминтетрауксусная кислота (комплексон III) об-
разует малодиссоциирующие комплексы с ионами
27
МАСЛА ВЫСЫХАЮЩИЕ
28
многих металлов, а с ионами уранила и бериллия —
весьма слабые комплексы. Поэтому большие коли-
чества мешающих ионов можно связать комплексоном
III и определять бериллий или уран. При комплек-
сометрия. титровании металлов нек-рые мешающие
ионы можно связать цианидом калия, триэтанолами-
ном и др.
В использовании М. появилось два новых направ-
ления: а) Буферная М.— свободное маски-
рующее вещество, иногда действует слишком сильно,
вследствие чего неполностью проходит основная реак-
ция. В таких случаях прибавляют комплекс маски-
рующего вещества с к.-л. другим ионом, не мешаю-
щим основным процессам, но несколько уменьшающим
концентрацию свободного маскирующего вещества.
Так, для отделения бария в виде хромата от больших
количеств стронция рекомендуется вводить комплек-
сон III для маскировки стронция; в результате умень-
шается его соосаждение с хроматом бария. Однако ком-
плексон III значительно связывает также барий, что
приводит к его неполному осаждению. Поэтому од-
новременно вводят также ионы магния, к-рые обра-
зуют с этим реактивом соединение невысокой проч-
ности. Комплексон III в достаточной степени связы-
вает стронций, но не мешает количественному осаж-
дению хромата бария. Аналогично, фторид в качестве
маскирующего вещества иногда слишком сильно
связывает определяемый компонент; в этом случае
используют ионы тетрафторобората, б) Демаски-
ровка — применяется, в частности, при титровании
смеси ионов металлов растворами комплексонов. Так,
при титровании смеси свинца и цинка сначала ионы
цинка маскируют цианидом и титруют ионы свинца.
Затем прибавляют формальдегид, к-рый реагирует
с цианидом, разрушая, т. обр., цианидный комплекс
цинка, т. е. демаскирует цинк. Освободившийся цинк
титруют комплексоном III; такой прием позволяет
определить оба компонента в одной порции раствора.
Лит.: Ф а й г л ь Ф., Капельный анализ, пер. с нем., М.,
1937; Cheng К. L., Analyt. Chem., 1961, 33, 783.
А. К. Бабко.
МАСЛА ВЫСЫХАЮЩИЕ — растительные масла,
способные отверждаться (высыхать) с образованием
на поверхности, на к-рую они нанесены тонким слоем,
твердых и эластичных пленок. Благодаря этому свой-
ству М. в. применяют в качестве пленкообразующих
веществ для приготовления лакокрасочных материа-
лов (олиф, масляных красок, лаков, эмалей, грунтовок
и шпатлевок).
Растительные масла в зависимости от способности
к высыханию делят на высыхающие, полувысыхающие
и невысыхающие. Кроме того, в зависимости от их
состава, скорости высыхания и свойств пленок, об-
разующихся при высыхании, их делят на пять групп
(название группы определяется по наиболее характер-
ному для нее виду масел): 1) группа тунгового масла
(тунговое, ойтисиковое); 2) льняного (льняное, ко-
нопляное, перилловое, лаллеманциевое и др.); 3) ма-
кового (маковое, подсолнечное, кукурузное, соевое,
рыжиковое, сафлоровое и др.); 4) оливкового (олив-
ковое, хлопковое, рапсовое, арахисовое); 5) касторо-
вого масла. О способности масел к высыханию судят
по различным химич. показателям: иодному числу,
родановому числу, характеризующему не-
насыщенность масла по количеству иода (в г), экви-
валентному количеству родана, присоединенному к
100 г масла; гексабромному числу, ха-
рактеризующему содержание в маслах жирных к-т
с тремя и более двойными связями; диеновому
числу, характеризующему содержание соединений
с сопряженными двойными связями в масле по коли-
честву малеинового ангидрида (выраженному в г иода),
прореагировавшего со 100 г масла (об основных физич.
и химич. свойствах, составе М. в. и их показателях
см. Жиры растительные, Жиры).
Тунговое масло высыхает быстрее других и дает
наиболее прочную пленку с рисунком, напоминающим
ледяные узоры. Образования узоров избегают пред-
варительной совместной полимеризацией с льняным
маслом. Льняное масло наиболее распространено из
М. в. Оно быстро высыхает в присутствии сиккативов
и дает прочные пленки. Маковое масло характеризует-
ся замедленным высыханием и дает пленку меньшей
прочности, оливковое и касторовое масла пленок
не образуют. Пленки масел группы льняного после
высыхания становятся нерастворимыми и неплавкими,
что указывает на образование полимера сетчатого
строения. С повышением темп-ры пленка обугливается,
не переходя в жидкую фазу. Пленки масел группы
макового при нагревании плавятся, иногда они размяг-
чаются и без нагревания; эти пленки растворимы.
Масла типа макового используются в смеси с актив-
ными маслами — тунговым, льняным и др. Пленки
из масел группы оливкового могут быть получены
только после глубокой химич. переработки масел.
Эти масла используются только в смеси с М. в. в ка-
честве небольших добавок. Вследствие того что ка-
сторовое масло без термич, обработки не высыхает,
оно с успехом применяется как пластификатор в
произ-ве целлюлозных и нек-рых смоляных лаков.
В обработанном виде, после дегидратации, касторовое
масло может быть отнесено по ряду свойств к группе
льняного и к группе тунгового масел.
По химич. природе М. в. являются смесью полных
сложных эфиров глицерина (триглицеридов). Способ-
ность высыхать обусловлена у М. в. тем, что они со-
держат в составе молекул триглицеридов значитель-
ное количество остатков высоконепредельных жирных
К-т. К ним относятся: линолевая кислота, линоленовая
кислота, олеостеариновая, ликановая и др. (см.
Высшие жирные кислоты). Они присутствуют в М. в.
в сочетании с предельными к-тами (пальмитиновой,
стеариновой и др.) и непредельными к-тами, имею-
щими одну двойную связь (в частности, с олеиновой
к-той). В процессе высыхания (продолжающемся
несколько дней) М. в. поглощают кислород (10—17%),
в результате чего образуются перекисные и гидропере-
кисные соединения и одновременно выделяются лету-
чие продукты окислительного распада молекул. Коли-
чество присоединившегося кислорода, к-рый содер-
жится в высыхающем слое, наз. кажущимся кисло-
родным числом, в отличие от истинного кис-
лородного. числа, характеризующего общее коли-
чество присоединившегося кислорода с учетом той
его доли, к-рая удаляется с летучими продуктами.
Образующиеся органич. перекиси и гидроперекиси
инициируют окислительную полимеризацию М. в.
по местам двойных связей. Слой масла становится все
более вязким и превращается в мягкую пленку. Эта
пленка постепенно отвердевает, становясь у нек-рых
М. в. неплавкой и нерастворимой вследствие образо-
вания сетчатого полимера. Пленка льняного масла
наз. линоксином. Скорость высыхания масел
увеличивается с повышением содержания ненасыщен-
ных к-т, количества двойных связей и их расположе-
ния. Сопряженные двойные связи обеспечивают более
быстрое высыхание, чем изолированные. Продолжи-
тельность высыхания может быть значительно сокра-
щена введением сиккативов или нагреванием до
60—100°.
Высыхание масла ускоряется под действием сол-
нечного света (УФ-лучи), при уменьшении влажности
воздуха, а также от добавления солей свинца, мар-
ганца, кобальта, к-рые могут присутствовать в пленке
в качестве сиккативов или пигментов. К веществам,
действующим в качестве антиоксидантов, замедляю-
29
МАСЛА ВЫСЫХАЮЩИЕ —МАСЛА МИНЕРАЛЬНЫЕ
30
щих высыхание, относятся фенолы, амины и нек-рые
пигменты (сажа, крапплаки и др.). При отверждении
пленка желтеет вследствие образования хромофорных
групп (что нежелательно в практич. отношении).
В процессе высыхания наблюдаются нек-рые явления,
характерные для старения пленки (напр., окисли-
тельная деструкция масел под влиянием кислорода
воздуха). Со временем эти процессы приводят к потере
пластичности, эластичности и к разрушению пленок.
Нек-рые лаки стареют в течение 2—3 недель, другие
лаки и масляные краски служат 5—7 лет.
Ввиду того, что большинство растительных масел
служит пищевым сырьем, они постепенно утрачивают
роль самостоятельных пленкообразователей. Их ис-
пользуют в качестве модифицирующих и пластифици-
рующих добавок к синтетич. лаковым смолам. Для
этого наряду с М. в. применяют также полувысыхаю-
щие и невысыхающие масла. Помимо М. в., большое
количество высоконепредельных жирных к-т содержат
Пэйн Г. Ф., Технология органических покрытий, пер. с
англ., т. 1— Масла, смолы, лаки и полимеры, Л., 1959; Шам-
пе т ь е Г., Рабата Г., Химия лаков, красок и пигментов,
пер. с франп., т. 1, М., 1960; Лакокрасочные материалы. Сы-
рье и полупродукты. Справочник, под ред. И Н. Сапгира, М.,
1961. И. И. Головастиков.
МАСЛА ДРЕВЕСНО-СМОЛЬНЫЕ ФЛОТАЦИОН-
НЫЕ — см. Смола древесная.
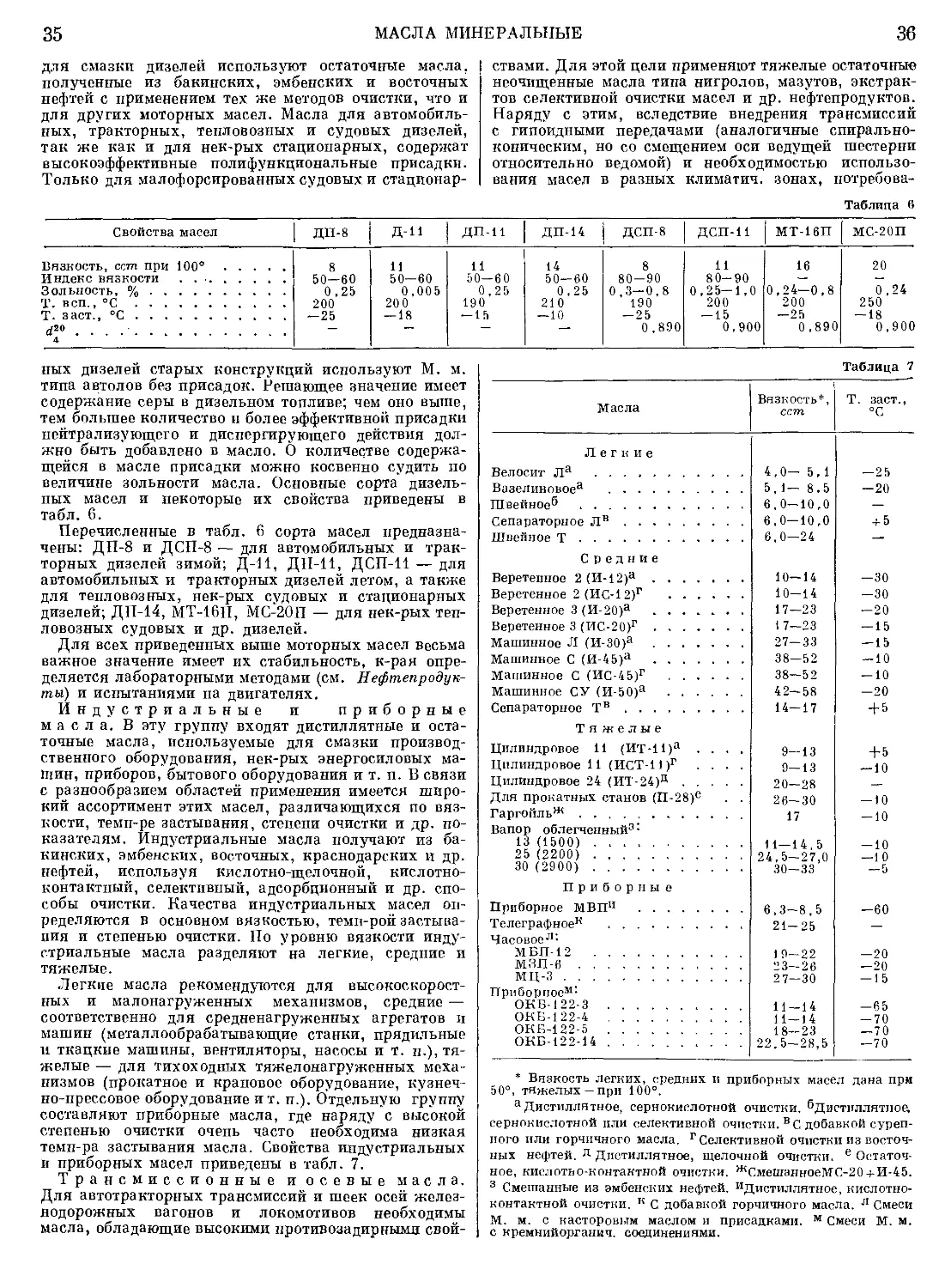
МАСЛА КАМЕННОУГОЛЬНЫЕ ТЕХНИЧЕСКИЕ—
смесь ароматич. углеводородов, гл. обр. двух- и
трехкольчатых и гетероциклич. соединений с приме-
сью фенолов. М. к. т. — маслянистые жидкости от
светло-желтого до темно-коричневого цвета с харак-
терным фенольным запахом; выделяются из фракций
каменноугольной смолы, или из масел, полученных из
фракций этой смолы путем отделения из них компонен-
тов, кристаллизующихся при охлаждении, и извле-
чения из них фенолов и оснований. Наименования,
источники получения, свойства и назначение важней-
ших М. к. т. приведены в таблице.
Наименование. Источники получения Назначение Плот- ность Пределы кипения, “С Темп-ра начала кристалли- зации, °C
Поглотительное масло. Из поглотительной фрак- ции путем ее обесфеноливания Извлечение бензола из коксового газа, приготовление других технич. масел. 1,05 230—300 Ниже + 15
Нафталиновое поглотительное масло. Из антра- ценовой фракции после кристаллизации Извлечение нафталина из коксового газа 1,06-1,10 270-370 То же
Шпалопропиточное масло. Из антраценовой и поглотительной фракции при совместной крис- таллизации Пропитка шпал и строительных изде- лий из дерева для предохранения от гниения 1,06—1,10 210—400 Ниже + 35
Отопительное масло. Из пековых дистиллятов и различных каменноугольных масел и фракций Для энергетических целей 1,10-1,20 230-400 Ниже + 40
Масло для подсвечивания пламени. На раз- личных фракций и пека Карбюрирование газа, идущего на обогрев мартеновских печей 1,05-1,15 200—400 + 20
Масла для производства сажи. Из пек-дистил- лятов, антраценовых масел и фракций Получение активной сажи 1,10-1,15 210-400 + 40
Газгольдерное масло. Из узких фракций, полу- ченных при редистилляции антраценовой фракции, после их кристаллизации Для заполнения затворов сухих газ- гольдеров 1,10—1,20 300-380 Ниже -30
Тяжелый сольвент. Из фенольной фракции после извлечения фенолов и оснований и кристал- лизации Для изготовления битумных лаков 1,02—1,05 170—240 Ниже 0
Тяжелые растворители. Из поглотительной и антраценовой фракций Для изготовления дегтей и клебе- масс 1,06-1,10 210-400 + 35
Масла для ядохимикатов. Из поглотительной и антраценовой фракций Изготовление карболинеума ц ядо- химикатов * 1,06-1,10 210-400 + 35
Масла для флотореагентов. Из поглотительной фракции Для обогащения угля и руд путем флотаций 1,05 230-300 От +5 до —5
Дизельные масла. Из поглотительного масла Дизельное топливо 1,05 230-300 От +5 ДО —5
И. М. Носалевич.
жиры рыб и морских животных (ворвань), однако из-за
высокого содержания предельных и низкопредель-
пых кислот они образуют мягкие неводостойкие
пленки и используются только после предваритель-
ной селективной обработки.
Лит.: Зиновьев А. А., Химия жиров, М.—Л., 1939;
Киселев В. С., Олифа и лаки, 3 изд., М.—Л., 1940;
Дринберг А.Я. иВар л амов В.С., Жиры и масла как
плеекообразователи, М.—Л., 1940; Дринберг А. Я.,
Технология пленкообразующих веществ, 2 изд., М., 1955;
МАСЛА МИНЕРАЛЬНЫЕ (нефтяные) — смеси вы-
сокомолекулярных углеводородов различных клас-
сов, применяемые для смазки двигателей, промыш-
ленного оборудования и приборов, для электроизо-
ляционных целей, в качестве рабочих жидкостей
в гидросистемах, при обработке металлов, в медицине,
парфюмерии и др.
Сложный химич. состав М. м. исключает возмож-
ность разделения их на индивидуальные углеводороды,
31
МАСЛА МИНЕРАЛЬНЫЕ
32
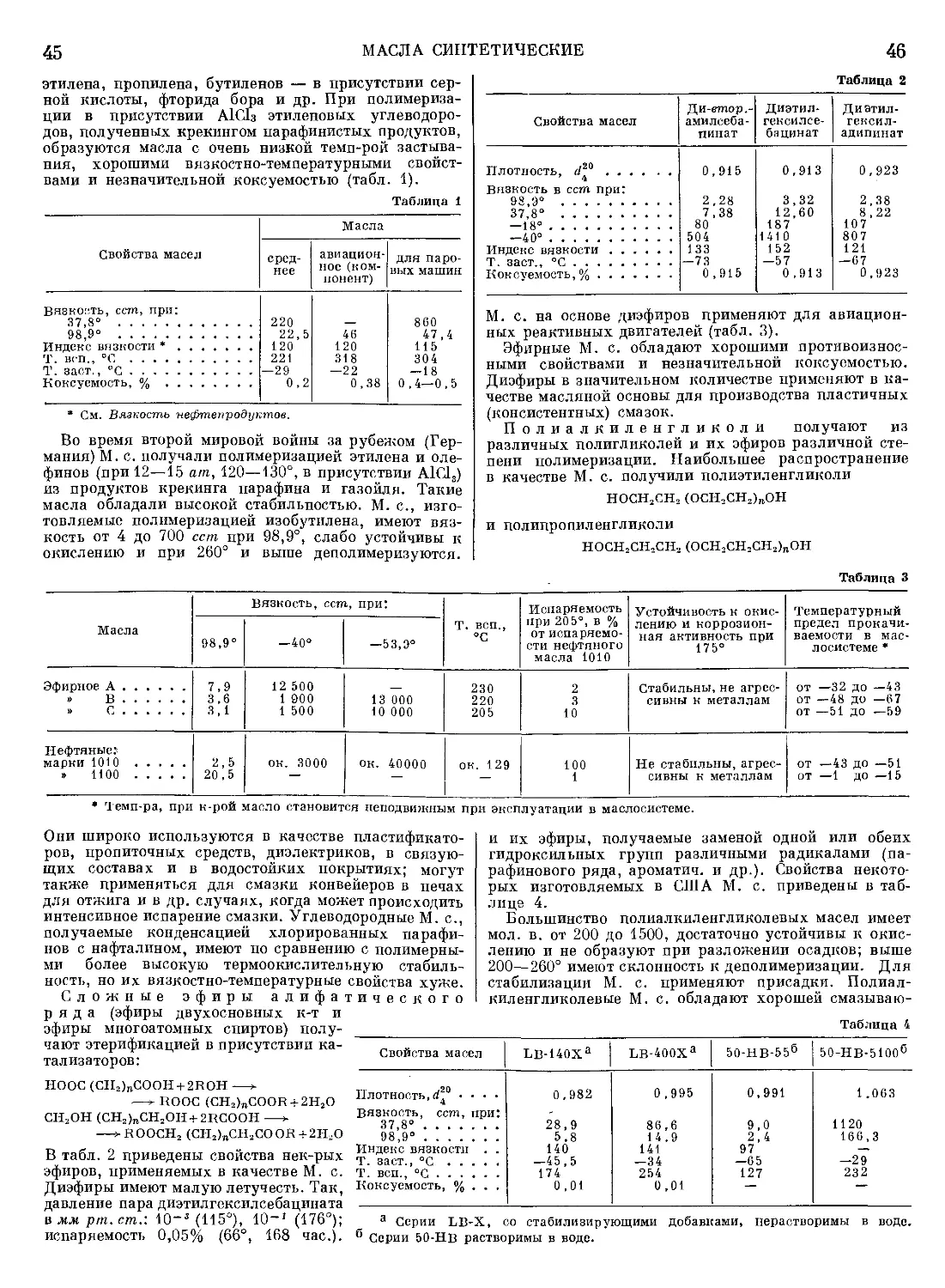
Таблица 1
поэтому о химич. составе масел судят по содержанию
в них отдельных групп углеводородов: парафиновых,
нафтеновых, ароматических, а также асфальто-смоли-
стых веществ, выделяемых хроматография, способом.
Групповой химич. состав одного из типичных образ-
цов М. м. (об.%):
Масла т. Вязкость в сст при темп-ре °C
заст., °C 100° 0° | —5° —10° —15» —20“
Моноциклич. нафтены...................... 18,4
Бициклич. нафтены......................... 9.9
Три-и более циклич. нафтены.............. 16.5
Моноциклич. ароматич. с нафтеновыми кольцами 10,5
Дициклич. ароматич. с нафтеновыми кольцами 8,1
Трициклич. ароматич. с нафтеновыми кольцами 6,6
Полициклич. ароматич. с большим числом колец 8,0
н-Парафиновые цепи .......................13,7
Изопарафины............................... 8,3
АС-6 (бакинск.) . -32 7 , 1 1434 2843 5205 22665
АС-6 (вост.) . . -32 6,6 670 1069 1716 2996 5771
СУ (бакинск.) . . —25 7,6 2162 3842 7385 16800 44200
ДС-8 (восточн.) . -32 8,3 1189 1700 2892 5920 15830
ДП-11 (бакинск.) — 18 11,0 4124 —— 15287 34123 —
ДСП-11 (вост.) . — 18 10,6 1800 6400 13700 —-
содержится больше нафтеновых углеводородов с более
короткими боковыми цепями (табл. 2).
Таблица 2
Чем выше плотность и вязкость масла, тем обычно
сложнее его химич, состав, т. к. с увеличением мол.
веса углеводородов резко возрастает количество их
изомеров. В общем случае содержание отдельных
групп углеводородов и др. соединений в М. м. опре-
деляется природой нефтяного сырья, глубиной и спо-
собом очистки. Высокомолекулярные парафины, а
также нек-рые ароматич. и нафтеновые углеводороды
при достаточно высокой темп-ре растворимы в более
легких углеводородах. Однако при понижении темп-ры
они выделяются из р-ра в виде твердой фазы, об-
разуя кристаллич. решетку и снижая текучесть масла
(см. Вязкость структурная). Поэтому процессы депа-
рафинизации, а также очистки масел от смолистых
продуктов являются основными при их производстве.
По способу получения М. м. делят на дистиллятные
и остаточные. В первом случае для получения М. м.
используют дистилляты вакуумной перегонки ма-
зутов, во втором — т. наз. масляные гудроны — тя-
желые остатки от перегонки нефти. Многие товарные
М. м. получают смешением дистиллятных и остаточ-
ных масел. Такие масла называют смешанными,
или компаундированными. По способу
очистки М. м. условно делят след, образом: сернокис-
лотной очистки, селективной очистки, адсорбционной
очистки, гидрогенизационной очистки. Условность
такой классификации определяется тем, что в произ-
водстве масел возможно сочетание селективной очист-
ки с гидрогенизационной, селективной с кислотной
или адсорбционной доочисткой и т. д. Выбор способа
очистки масел определяется химич. составом сырья,
назначением масла и экономии, целесообразностью.
Основную массу М. м. очищают селективным или
сернокислотным способами, иногда с применением
адсорбционной доочистки. На основании изучения
эксплуатационных свойств масел установлено, что
масла адсорбционной и гидрогенизационной очистки
обладают более высокой стабильностью и дают мень-
шее количество нагаров, лака и низкотемпературных
отложений в двигателях.
Современные методы переработки нефти и, в част-
ности, деструктивная переработка и гидрирование
позволяют получать М. м. практически из любой
нефти. Однако, исходя из высоких требований к экс-
плуатационным качествам М. м. и соображений стои-
мости, для их производства используют специально
отобранные т. н. масляные нефти с высоким содержа-
нием масляных дистиллятов и остатков, с минималь-
ным содержанием смол, парафина, серы и др. неже-
лательных компонентов. В Советском Союзе для
производства М. м. используют нефти бакинских,
эмбинских и гл. обр. восточных (Урал и Поволжье),
месторождений. М. м. из восточных нефтей имеют
преимущества перед бакинскими по вязкостно-тем-
пературным свойствам (табл. 1), стабильности и
нек-рым другим эксплуатационным качествам, особен-
но в случае использования тех и других с присадками.
По химич. природе масла из восточных нефтей су-
щественно отличаются от бакинских. В последних
Масла Индекс вязко- сти Содер- жание серы, % Кольцевой состав по методу п — d - m*, %
арома- тич. , Сар. на фте- новых, ^наф. боковых парафино- вых цепей, спар.
Автол 10 (бакинск.) 41 0,21 И 33 56
Автол 10 (восточн.) 93 0,88 14 19 67
* Состав определен по показателю преломления (п), плот-
ности (d) и мол. весу (т) (метод в — d — т).
Поэтому бакинские масла характеризуются низким
индексом вязкости (см. Вязкость нефтепродуктов).
Исходя из различия в химич. составе, понятно, почему
окисление бакинских и восточных масел идет по-раз-
ному. Возможно также, что различное направление
окисления этих масел зависит от содержания в них
серы. В условиях работы масла в двигателях в бакин-
ских маслах образуются преимущественно карбены
и карбоиды, а в маслах из восточных нефтей —
гл. обр. оксикислоты и асфальтены. В масла для
современных двигателей вводят присадки, улучшаю-
щие свойства масла.
Товарные М. м. характеризуются рядом физико-
химич. показателей, определяемых условиями приме-
нения, химич. природой сырья и способом очистки;
важнейшие из них: вязкость, зольность, коксуемость,
темп-ра вспышки, стабильность, темп-ра застывания
(см. Нефтепродукты). По назначению М. м. разде-
ляют на смазочные и несмазочные. Каждый из указан-
ных классов включает ряд групп и подгрупп масел,
предназначенных для использования в различных об-
ластях техники. К смазочным относятся след,
масла: а) для двигателей внутреннего сгорания (мо-
торные): авиационные, автотракторные (автолы), ди-
зельные; б) для оборудования. промышленных пред-
приятий и приборов (индустриальные, приборные);
в) для высоконагруженных механизмов (трансмис-
сионные, осевые); г) для поршневых паровых машин
(цилиндровые); д) для паровых и водяных турбин
(турбинные); е) для воздушных компрессоров и холо-
дильных машин (компрессорные). К несмазочным
относятся след, масла: а) электроизоляционные (транс-
форматорные, конденсаторные, кабельные); б) для
гидравлических систем; в) для технологии, целей
(абсорбенты, для закалки металлов, мягчители и т. п.);
г) для фармакопеи и парфюмерии (белые масла).
В связи с тем, что высоконапряженные карбюраторные
двигатели иногда требуют масел столь же высоких качеств,
как и дизели, возникла необходимость в классификации мо-
торных масел по эксплуатационным качествам, условиям при-
менения, невависимо от типа двигателя, подобно тому, как это
принято в большинстве зарубежных стран. В соответствии с
этим разработан проект новой классификации, в основу к-рой
положено деление масел на группы: а) по моторным качествам,
оцениваемым стандартными методами на специальных двига-
телях; б) по вязкостной характеристике.
В табл. 3 приведен проект новой классификации моторных
масел в сопоставлении с группами масел, принятыми за рубе-
жом. В каждой колонке приведены марки масел, рекомендуе-
мые по новой классификации, и указаны сорта, к-рым они со-
ответствуют по классификации SAE и APJ.
33
МАСЛА МИНЕРАЛЬНЫЕ
34
Таблица 3
Группы масел
Вязкость при 100°, ест А Б В Г Д Е по класси фикации SAE
6 + 0,5 М-6А М-6Б М-6В SAE-20
8±0,5 М-8А М-8Б М-8В М-8Г М-8Д — SAE-20
10±0,5 М-10А М-10Б М-10В М-10Г М-10Д — SAE-30
12 + 0,5 М-12Б М-12В М-12Г М-12Д — SAE-30
14 + 0,5 — М-14Б М-14В М-14Г М-14Д — SAE-40
16±0,5 — М-16Б М-16В М-16Г М-16Д М-16Е SAE-40
20 + 0,5 М-20А М-20Б М-20В М-20Г М-20Д М-20Е SAE-50
По классифи- кации APJ ММ MS DG DM DS - -
Назначение масел: А — для карбюраторных двигателей и дизелей, работаю-
щих на малосернистых топливах; Б — для форсированных карбюраторных дви-
гателей и малофорсированных дизелей; В — для дизелей средней напряженности;
Г — для форсированных дизелей; Д — для высокофорсированных дизелей; Е —
для двигателей с лубрикаторной цилиндровой смазкой, работающих на высоко-
сернистых тяжелых топливах. Масла групп Б—Е содержат специальные присад-
ки в возрастающих количествах (от 3 до 25%).
За рубежом эти группы масел имеют след, наименования: А — Premium;
Б — Hevi Dutl; В — Supplement I; Г — Series II; Д — Series III; E — Marine
Diesel.
Товарный ассортимент включает более 130 наиме-
нований различных масел. На современных нефте-
перерабатывающих заводах получают обычно 4—6 по-
токов очищенных масел, от самых легких дистиллят-
ных до самых тяжелых остаточных. Смешивая эти
масла в различных сочетаниях и количествах, добав-
ляя в них необходимые присадки, получают товарные
масла разного назначения. Физико-химич. свойства
и важнейшие технические характеристики М. м.
строго регламентированы Государственными стан-
дартами (ГОСТ).
Масла смазочные. Авиационные масла.
В поршневых и реактивных авиационных двигателях
смазочные масла используются при высоких темп-рах
и уд. нагрузках. Темп-pa масла на выходе из дви-
гателя достигает 120°, а уд. нагрузки в нек-рых уз-
лах трения поршневых двигателей 900 кг/смг. В ра-
боте масло соприкасается с различными металлами,
в том числе цветными, что способствует интенсив-
ному окислению масла, засорению его продуктами,
карбонизации масла и износа двигателя. Авиацион-
ные масла получают из дистиллятов и остатков от
перегонки отборных масляных нефтей (бакинских,
эмбенских, восточных), подвергая их тщательной
очистке. Основные методы очистки: сернокислотный
(бакинское М. м.) и селективный (грозненское и вос-
точное М. м.). Масла, предназначенные для эксплуа-
тации в летних и зимних условиях, отличаются по
уровню вязкости, темп-ре застывания и др. свойствам.
Сорта авиационных масел и нек-рые их свойства при-
ведены в табл. 4.
Масло МС-20С вырабатывается из восточных сер-
нистых нефтей. Для реактивных двигателей наряду
с маслом МК-8 предназначается также масло МС-8
из восточных сернистых нефтей. Авиационные масла
МС-20, МС-20С, МК-22 используются также для не-
которых быстроходных форсированных дизелей.
В этом случае к маслам добавляют
присадки, позволяющие применять их
при использовании высокосернистых
дизельных топлив и в особо тяжелых
условиях работы двигателей.
Автотракторные масла
(автолы). По способу получения ав-
тотракторные масла разделяют на ди-
стиллятные и компаундированные, по
способу очистки — на сернокислотные
и селективные. В зависимости от ти-
па двигателя, условий эксплуатации
и времени года применяют тот или
иной сорт масла. Зимой используют
масло малой вязкости с низкой тем-
пературой застывания. Летом масло
должно быть более вязким. Для дви-
гателей с высокой степенью сжатия
необходимы масла с присадками.
Автотракторные масла получают из
бакинских и восточных нефтей. Масла
селективной очистки отличаются вы-
соким индексом вязкости, малой кок-
суемостью и хорошей приемистостью
к присадкам. Сорта автолов разделяют по вязкости,
способу очистки (АК — кислотной очистки; АС — се-
лективной очистки) и наличию присадки [3—загу-
щающей (вязкостной), П — полифункциональной].
Свойства нек-рых автотракторных масел приве-
дены в табл. 5.
Перечисленные в табл. 5 масла применяют: АКЗп-6
и АКЗп-10 — для автомобильных двигателей в се-
верных районах зимой; АСп-6 — для автомобильных
двигателей в средней полосе зимой; АКп-10, АС-10,
АСп-10 — для автомобильных двигателей летом и
тракторных зимой; АК-15— для тракторных карбю-
раторных двигателей летом.
Дизельные масла. Эти масла предназна-
чены: для быстроходных транспортных высокофор-
сированных дизелей, имеющих вкладыши подшип-
ников из корродирующих сплавов; быстроходных
транспортных и стационарных дизелей средней форси-
ровки с достаточно устойчивыми против коррозии под-
шипниками; стационарных дизелей различной мощно-
сти со средним и малым числом оборотов. Дизельные
масла получают компаундированием. В ряде случаев
Таблица 4
Свойства •Д' -20С •Д' МК-22 <эо
масел мс МС МС мс
Вязкость при 100°, ест 14 20 20 24 22 8,3
Температура вспышки, °C 200 225 250 240 230 135
Температура застывания, °C -30 -18 — 18 -17 — 14 -55
Плотность, 4 0,890 0,895 0,895 0,900 0,905 0,885
Индекс С означает, что масло селективной очистки, К —
кислотной очистки.
Таблица 5
Свойства масел Бакинские Восточные
АКЗп-6 АКЗп-10 АСп-6 АКп-10 АК-15 АСп-10 АС-10 АСп-6 АСп-10
Вязкость, сст: при 100° 6 10 6 10 15 10 10 6 10
при 0° 600 1000 1500 — —— — 700
Индекс вязкости . . . >100 >100 50 35 — 60 80-90 80-90 80-90
Зольность, % 0,26 0,26 0,26 0,26 — 0,26 0.005 0,4 0,4
Т. заст., °C -40 -40 -35 -25 -5 -25 -15 —30 -15
Т. всп.. °C 170 170 185 200 220 200 200 180 200
°2 К.Х.Э. т. 3
35
МАСЛА МИНЕРАЛЬНЫЕ
36
для смазки дизелей используют остаточные масла,
полученные из бакинских, эмбенских и восточных
нефтей с применением тех же методов очистки, что и
для других моторных масел. Масла для автомобиль-
ных, тракторных, тепловозных и судовых дизелей,
так же как и для нек-рых стационарных, содержат
высокоэффективные полифункциональные присадки.
Только для малофорсированных судовых и стационар-
ствами. Для этой цели применяют тяжелые остаточные
неочищенные масла типа нигролов, мазутов, экстрак-
тов селективной очистки масел и др. нефтепродуктов.
Наряду с этим, вследствие внедрения трансмиссий
с гипоидными передачами (аналогичные спирально-
коническим, но со смещением оси ведущей шестерни
относительно ведомой) и необходимостью использо-
вания масел в разных климатич. зонах, потребова-
Таблипа В
Свойства масел ДП-8 д-и ДП-11 ДП-14 ДСП-8 ДСП-11 МТ-16П МС-20П
ВЯЗКОСТЬ, СС771 при 100° 8 И и 14 8 11 16 20
Индекс вязкости ........ 50-60 50-60 50-60 50—60 80-90 80-90 — —
Зольность, % 0,25 0,005 0,25 0,25 0,3—0,8 0,25-1,0 0,24—0,8 0,24
Т. всп., °C 200 20 0 190 210 190 200 200 250
Т. заст., °C -25 — 18 -15 — 10 -25 -15 —25 -18
</2О ... 4 — — — 0,890 0.900 0.890 0,900
ных дизелей старых конструкций используют М. м.
типа автолов без присадок. Решающее значение имеет
содержание серы в дизельном топливе; чем оно выше,
тем большее количество и более эффективной присадки
нейтрализующего и диспергирующего действия дол-
жно быть добавлено в масло. О количестве содержа-
щейся в масле присадки можно косвенно судить по
величине зольности масла. Основные сорта дизель-
ных масел и некоторые их свойства приведены в
табл. 6.
Перечисленные в табл. 6 сорта масел предназна-
чены: ДП-8 и ДСП-8 — для автомобильных и трак-
торных дизелей зимой; Д-11, ДП-11, ДСП-11 — для
автомобильных и тракторных дизелей летом, а также
для тепловозных, нек-рых судовых и стационарных
дизелей; ДП-14, МТ-16П, МС-20П — для нек-рых теп-
ловозных судовых и др. дизелей.
Для всех приведенных выше моторных масел весьма
важное значение имеет их стабильность, к-рая опре-
деляется лабораторными методами (см. Нефтепродук-
ты) и испытаниями на двигателях.
Индустриальные и приборные
масла. В эту группу входят дистиллятные и оста-
точные масла, используемые для смазки производ-
ственного оборудования, нек-рых энергосиловых ма-
шин, приборов, бытового оборудования и т. п. В связи
с разнообразием областей применения имеется широ-
кий ассортимент этих масел, различающихся по вяз-
кости, темп-ре застывания, степени очистки и др. по-
казателям. Индустриальные масла получают из ба-
кинских, эмбенских, восточных, краснодарских и др.
нефтей, используя кислотно-щелочной, кислотно-
контактный, селективный, адсорбционный и др. спо-
собы очистки. Качества индустриальных масел оп-
ределяются в основном вязкостью, темп-рой застыва-
ния и степенью очистки. По уровню вязкости инду-
стриальные масла разделяют на легкие, средние и
тяжелые.
Легкие масла рекомендуются для высокоскорост-
ных и малонагруженных механизмов, средние —
соответственно для средненагруженных агрегатов и
машин (металлообрабатывающие станки, прядильные
и ткацкие машины, вентиляторы, насосы и т. п.), тя-
желые — для тихоходных тяжелонагруженных меха-
низмов (прокатное и крановое оборудование, кузнеч-
но-прессовое оборудование ит. п.). Отдельную группу
составляют приборные масла, где наряду с высокой
степенью очистки очень часто необходима низкая
темп-pa застывания масла. Свойства индустриальных
и приборных масел приведены в табл. 7.
Трансмиссионные и осевые масла.
Для автотракторных трансмиссий и шеек осей желез-
нодорожных вагонов и локомотивов необходимы
масла, обладающие высокими противозадирными свой-
Таблица 7
Вязкость*, Т. заст.,
Масла сст °C
Легкие
Велосит Ла 4,0- 5,1 -25
Вазелиновое3 5,1- 8,5 -20
Швейное® 6,0-10,0 —
Сепараторное Лв 6,0-10,0 + 5
Швейное Т 6,0—24 —
Средние
Веретенное 2(И-12)а 10-14 -30
Веретенное 2 (ИС-12)г 10-14 -30
Веретенное 3 (И-20)а ....... 17—23 -20
Веретенное 3 (ИС-20)г 17—23 -15
Машинное Л (И-30)а 27—33 -15
Машинное С (И-45)а 38—52 -10
Машинное С (ИС-45)Г 38-52 -10
Машинное СУ (И-50)а 42-58 -20
Сепараторное Тв 14-17 + 5
Тяжелые
Цилиндровое И (ИТ-11)а .... 9—13 + 5
Цилиндровое И (ИСТ-11)Г .... 9—13 -10
Цилиндровое 24 (ИТ-24)Д 20—28 —
Для прокатных станов (П-28)е . . 26-30 -10
Гаргонльж 17 -10
Вапор облегченный3*
13 (1500) 11—14,5 — 10
25 (2200) 24,5-27,0 —10
30 (2900) 30—33 — 5
Приборные
Приборное МВП11 6,3—8,5 -60
Телеграфное1* 21-25
ЧасовоеЛ;
МБП-12 19-22 — 20
МЗП-6 23—26 -20
МЦ-3 2 7—30 -15
Приборное311
ОКБ-122-3 11 — 14 -65
ОКБ-122-4 11 — 14 -70
ОКБ-122-5 18—23 -70
ОКБ-122-14 22.5-28,5 -70
* Вязкость легких, средних и приборных масел дана при
50°, тяжелых-при 100°.
а Дистиллятное, сернокислотной очистки, ®Дистпллятпое,
сернокислотной пли селективной очистки. вС добавкой суреп-
ного или горчичного масла. г Селективной очистки из восточ-
ных нефтей. д Дистиллятное, щелочной очистки. е Остаточ-
ное, кислотно-контактной очистки. жСмешанноеМС-20+ И-45.
3 Смешанные из эмбенских нефтей. ИДистиллятное, кислотно-
контактной очистки. к С добавкой горчичного масла. л Смеси
М. м. с касторовым маслом и присадками. м Смеси М. м.
с кремнийорганич. соединениями.
37
МАСЛА МИНЕРАЛЬНЫЕ
38
лись масла на базе очищенных дистиллятных и оста-
точных продуктов с хорошими вязкостными качест-
вами и высокими противозадирными свойствами. Для
этой цели к маслам добавляют серу-, хлор- и фосфор-
содержащие присадки.
В товарном ассортименте трансмиссионных масел
сохранились прежние масла и появились новые, с
присадками, к-рые постепенно, по мере обновления
техники, полностью заменят старые. Основные сорта
летних и зимних масел этой группы приведены в табл.8.
Таблица 8
Масла Вязкость, сст т. заст., °C Содер- жание серы, %
Трансмиссионные3
Автотракторное, зимнее .... 18—22 -20 —
» летнее .... 28—32 -5
Для коробки передач и руля . . 20,5—32,4 — 20 1,2
Гипоидное 20,5-32,4 -20 1 ,5
Для редукторов троллейбусов:
зимнее с присадкой 20—28 -10 0,9
летнее • ... 28—36 — 5 0,9
Автотракторное (ТАП-1 5) . . . 15 -30 0,9 0,9
» (ТАП-10) . . . 10 -40
Осевые®
Осевое Л (летнее) 36—52 — 15 —
>> 3 (зимнее) 20—25 —40 —
» С (северное) 12—14 — 55 —
а Вязкость при 100°. б Вязкость при 50°.
Масла для паровых машин (цилин-
дровые). Цилиндровые масла разделяют на две группы:
а) для машин, работающих насы-
щенным паром; б) для машин, ра-
ботающих перегретым паром. В
первом случае масло находится в
относительно легких условиях: да-
вление пара ок. 16 кг/см2, темп-ра
ок. 200°. Во втором случае масло
работает в контакте с паром, пере-
гретым до 400° и выше. В этих усло-
виях предъявляются чрезвычайно
высокие требования к испаряемо-
сти, коксуемости, зольности и дру-
гим качествам масла, определяю-
щим прочность и антифрикционные
качества смазочного слоя. Для
приготовления цилиндровых масел * Натровая проба характеризует степень чистоты (промывки) масла, выражают
используют тяжелые дистилляты и ее в условных единицах (баллах).
остаточные продукты (гудроны)
бакинских и эмбенских нефтей. Способ очистки ки- I давления (до 225 ат) используют высоковязкие авиа-
слотно-контактный или селективный (табл. 9). | ционные масла МК-22 или МС-20. Для компрессо-
ров холодильных машин вырабаты-
Таблица 9 вают три сорта: масло ХА (фригус),
для смазки охлаждаемых водой подшипников паровых
машин морских и речных судов.
Турбинные масла. Масла для паровых и
водяных турбин работают в условиях циркуляцион-
ной системы смазки при повышенной темп-ре и кон-
такте с влагой. Большая емкость масляных систем
(от 150 до 400 г на 1 кв) и значительные затраты вре-
мени и средств на смену масла в них требуют приме-
нения масел, стойких против окисления. Применяемые
масла должны также обладать высокой деэмульги-
рующей способностью, чтобы быстро и полностью от-
делять попадающую в масляную систему воду. Полу-
чают турбинные масла из высококачественных масля-
ных нефтей, бакинского и эмбенского месторождений.
Для повышения стабильности одной из марок турбин-
ного масла (22) к нему добавляют 0,009—0,015%
антиокислительной присадки ВТИ-1 (параоксидифе-
ниламин), позволяющей в 2—3 раза увеличить срок
работы масла при большей надежности эксплуатации
машин. Основные сорта и свойства турбинных масел
приведены в табл. 10.
Компрессорные масла предназначены
для поршневых и ротационных компрессоров и возду-
ходувок, а также для компрессоров холодильных
машин. Эти масла должны быть стойкими против окис-
ления, иметь достаточно высокую темп-ру вспышки
и в необходимых случаях хорошую подвижность при
низких темп-pax. В ассортименте имеются три сорта
компрессорных масел: сорт 12 (М) для компрессоров
низкого давления (до 40 ат), сорт 19 (Т) для компрес-
соров высокого давления (выше 40 ат) и сорт КС-19
из восточных нефтей для этих же целей. В отдельных
случаях для многоступенчатых компрессоров высокого
Таблица 10
Свойства масел Тур- бинное 22П (Л) Тур- бинное 22 (Л) Тур- бинное 30 (УТ) Тур- бинное 46 (Т) Турбинное 5 7 (турбо- редуктор- ное)
Вязкость, сст, при 50° 20—23 20—23 28—32 44-48 55—59
Кислотное число 0,02 0,02 0,02 0,02 0,05
Стабильность: осадок после окисления, % 0,05 0.10 0,10 0,35 0,15 0,45
кислотное число . 0,20 0,35 8 —
Скорость деэмульгирования, мин .... 8 8 8 8
Натровая проба, баллов (не более)* . . . 9 9 9 9 2
Т. заст., °C — 15 — 1 5 — 10 -10 —
Свойства масел Для насыщенногопара применяемое для аммиачных и угле- Для перегретого пара кислотных комппессопов. и мае-
цилиндро- вое- 1 1 цилиндро- вое-24 цилиндро- вое-38 цилиндро- вое-50 цилиндро- вое- 5 2 (вапор) цилипдро- ла ХФ-12 и ХФ-22, используемые вое-65 для фреоновых компрессоров. Масло (вапор) ХФ-12 содержит до 0,02% присад-
Вязкость, сст, при 100° . . . Коксуемость, % . Т. всп., °C . . . Т. заст., °C . . . 9—13 0.8 215 + 5 20—28 2,5 240 32-44 3 300 + 17 44—56 3 300 + 17 44-59 3 310 -5 ки ВТИ-1 (параоксидифениламин), йп_70 а масло ХФ-22 — загущающую Ь° 2 5 присадку винипол (полимер ви- 325' нил-н-бутилового эфира). В табли- +3° це 11 приведены основные свой- ства масел, применяемых для этой
К группе цилиндровых масел условно можно
отнести два сорта судовых масел с присадками:
одно — с 20% сурепного и горчичного масла, другое—
с добавкой синтетич. эфирного масла и канифоли.
Эти масла имеют вязкость 80— 90 сст при 100°,
т. всп. 210° и т. заст. 0°. Применяются судовые масла
цели.
Масла несмазочные. Электроизоляцион-
ные масла. В электротехнике М. м. выполняют
роль диэлектрика и теплоотводящей среды. Для транс-
форматоров, помимо высоких диэлектрич. свойств, тре-
буется химически стойкое масло с хорошей подвиж-
ностью при низких темп-pax. Масло для заливки и
2*
39
МАСЛА МИНЕРАЛЬНЫЕ
40
Таблица 11
Свойства масел Для компрессоров Для холодильных машин
12 (М) 19 (Т) КС-19 ХА ХФ-12 ХФ-22
Вязкость, ест* 11-14 17-21 17-21 И ,5-14,5 18 24,5-28,4
Стабильность: осадок после окисления, % 0,3 0,02 0.005 0,005
кислотное число . . . . —. — 0,5 — 0,05 —
Т, всп., °C 216 242 270 160 160 125
Т. заст., °C — — -15 -40 -40 —58
* Для компрессорных масел при 100°, для холодильных машин при 50°.
пропитки конденсаторов (конденсаторное) должно быть
стойким к газовыделению при ионизации, иметь ста-
бильные диэлектрич. и хорошие антикоррозийные ка-
чества. Важнейшие параметры для оценки качества
кабельных масел — минимальное значение тангенса
угла диэлектрич. потерь и малая зависимость его от
темп-ры. Масла, используемые для заполнения и про-
питки силовых кабелей высокого напряжения, дол-
жны быть стойки к выделению газов, особенно к во-
дороду, иметь достаточно низкую темп-ру застывания
и высокую стабильность при контакте с медью, свин-
цом и другими металлами, являющимися катализато-
рами окисления. В электроизоляционных маслах
не должно быть асфальтово-смолистых веществ.
Кабельные масла должны представлять собой прак-
тически чистые нафтено-парафиновые углеводороды.
Для повышения стабильности трансформаторных ма-
сел применяют присадку ВТИ-1 (параоксидифенил-
амин) и ионол (топанол, 2,6 дитретичный бутил-4-ме-
тилфенол). Сорта и основные свойства электроизоля-
ционных масел приведены в табл. 12.
зано с улучшением смазочной спо-
собности масла. Перечисленные ка-
чества масла должны сочетаться
с высокими антикоррозионными
свойствами, малой изменяемостью
вязкости с темп-рой, взрывобезо-
пасностью и другими свойствами.
По-видимому, такие масла возмож-
ны только на базе узких нефтяных
фракций из низкозастывающих от-
борных нефтей при широком исполь-
зовании различных присадок. Ко-
личество и характер последних
зависят от назначения масла и условий его приме-
нения. К группе гидравлических относят также
масла для форвакуумных и высоковакуумных па-
роструйных насосов. Эти масла применяют в не-
сколько иных условиях, чем чисто гидравлические.
В частности, в форвакуумных насосах масло зали-
вают в коробку, где расположен корпус насоса.
Помимо смазки трущихся частей, масло создает уплот-
нение и выполняет роль теплоотводящей среды.
В пароструйных высоковакуумных насосах приме-
няемые в качестве рабочей жидкости масла должны
иметь очень низкое давление паров. Нек-рые свойства
гидравлич. масел приведены в табл. 13.
Масла для технологических де-
ле й. К группе технологических отнесены М. м., не
имеющие общего характерного признака, определяю-
щего условия их применения. К их числу относятся:
а) соляровое масло, применяемое для пропитки кож,
закалки металлов, охлаждения режущих инструмен-
тов и др. целей; б) поглотительное, используемое в
качестве абсорбента для поглощения сырого бензола
Таблица 12
Свойства масел Трансформаторные Конденса- торное Кабельные
без присадки с ВТИ-1 восточное с ионолом МН-2 С-220 С-110
Вязкость, сстп: при 50° 9,6 9,6 8-9 9-12 9,6 50 80—120
при 20° 30 30 23-28 37-45 37,3 800 680-800
при 0° —— — 2200 (—30°) 3000—5000 5000
Кислотное число 0,03 0,05 0,02 0,02 0,04 0,02 0 ,01
Стабильность: осадок после окисления 0,05 0,10 0.35 0,10 0,08
кислотное число после окисления 0,20 0,10 —. 0,35 — —-
Электрич. прочность, кв! см при ча- стоте 50 гц 1 ...... 200 180 200 180
Тангенс угла диэлектрич. потерь при частоте 50 ей и °C 0,003 (20°) 0,003 (20°) 0,003(20°) 0,005(100°) 0,003 (100°) 0,003 (100°) 0,003 (100°)
Т. заст., °C -45 -45 -45 -45 -45 -30 —15
Примечания: 1. Для пропитки кабелей напряжением до 35 ив используют также высоковязкое индустриальное
масло П-28 в смеси с 20% канифоли.
2. Кабельные масла средней вязкости (С-220 и С-110) идут для заполнения и пропитки кабелей на 110 кв и выше
с изоляцией, работающей при высоком давлении (до 14 ат). Маловязкое масло (МН-2) используют для заполнения кабелей
с изоляцией, рассчитанной на низкое н среднее давление (до 3 ат).
Гидравлические масла. Масла для
гидравлич. систем составляют довольно значительную
группу в ассортименте нефтяных масел. Высокая
стабильность и возможность получения М. м. с раз-
личными вязкостными и низкотемпературными свой-
ствами способствовали широкому их применению
в качестве рабочих жидкостей в различных гидравлич.
системах. С внедрением комплексной механизации
и развитием поточных линий возрастает широта ис-
пользования гидравлич. масел. Напр., выявилась
потребность в гидравлич. маслах, способных дли-
тельно работать в точнейших механизмах при тя-
желых условиях без заметного изменения своих ка-
честв. В большинстве случаев гидравлич. масло яв-
ляется также антифрикционной жидкостью, что свя-
из коксового газа; в) висциновое, применяемое для
очистки воздуха в вентиляционных фильтрах; г) масло
для обработки хлопка; д) «мягчитель», применя-
емый в резиновой пром-сти. Масла для технология,
целей получают из обычных масляных нефтей извест-
ными методами очистки. Исключение составляет
«мягчитель» для резиновой пром-сти (т. заст. —60°),
к-рый получают из специальных низкозастывающих
нефтей или глубокой депарафинизацией дистиллятов
парафинистых нефтей. Масло, используемое при об-
работке хлопка, содержит эмульгатор — натриевые
мыла окисленного петролатума.
Белые масла (для фармакопеи и парфюме-
рии). Эти масла получают глубокой очисткой дымящей
серной к-той или серным ангидридом соответствую-
41 МАСЛА МИНЕРАЛЬНЫЕ —МАСЛА ПРЕПАРИРОВАННЫЕ 42
Таблица 13
Масла Вязкость, сст при темп-ре, °C Т. всп,, °C (не ниже) Т. заст., °C (не выше) Примечания
АМГ-10 10 (50°) 1250 (—50°) 92 -70 Масло с вязкостной присадкой
Гидротормозное (ГТН) 10 (50°) 1500 (-50°) 92 —60 То же
Веретенное АУ 12-14 (50°) 163 -45 Дистиллятное
Для механизма опрокидывания ваго- нов-самосвалов 18,5—20,5 (50°) 135 -40 Масло с вязкостной присадкой
Для шагающих экскаваторов (ЭШ-406) 20 (50°) 150 -50 То же
Для прессов 10 (100°) 200 —15 Смешанное масло
Для форвакуумных насосов (ВМ-4) . . 47—57 (50°) 8—11 (100°) 213 -15 Дистиллятное масло с давлением пара при 20° 4-10 —5 мм рт. ст.
Для высоковакуумных пароструйных насосов ВМ-1 65-69 (50°) 248 — И Давление пара при 20° 4-10“8 мм рт. ст.
То же ВМ-2 65—69 (50°) 248 —11 То же 2-10“® мм рт. ст.
Для гидропередач автомобилей'(ГТМ-З) 4,8(100°),540(0°), 5700 (—20°) 170 -40 Масло с присадками
щих дистиллятов с последующей нейтрализацией и
доочисткой отбеливающими землями. Белые масла
представляют собой бесцветную маслянистую, про-
зрачную нефлуоресцирующую жидкость без запаха
и вкуса; по составу — это чистые нафтено-парафино-
вые углеводороды без примесей ароматики и смол.
Вырабатываются два вида белых нефтяных масел:
медицинское вазелиновое и парфюмерное. Первое
применяется в лечебных целях в чистом виде и в ка-
честве компонента при изготовлении лекарств; второе
используют в парфюмерной пром-сти. В табл. 14
приведены основные свойства белых масел.
Таблица 14
Масла ВЯЗКОСТЬ при 50°, сст Т. всп., °C
Медицинское вазелиновое Парфюмерное 28—36 16-24 185 160
Нормируются также кислотность, зольность, при-
меси, запах, вкус и ряд других свойств.
Регенерация масел — восстановление эксплуата-
ционных свойств масел с целью повторного
их использования. При работе в двигателях и меха-
низмах свойства М. м. изменяются под воздействием
высоких темп-p, давлений, электрич. поля, света, ка-
талитич. действия металлов и др. факторов. Степень
и характер изменений, к-рые происходят в маслах, за-
висят от условий их применения: для моторных масел
это термин, разложение, полимеризация и разбавление
топливом; для индустриальных — засорение механич.
примесями; для электроизоляционных — накопле-
ние продуктов окисления и т. д. Совокупность процес-
сов, к-рые происходят в М. м. при их эксплуатации,
наз. старением масла.
Существующие методы регенерации масел можно
разделить на физич., физико-химич. и химические.
К физич. методам относятся сепарация, фильтрование
и отстой. В необходимых случах может быть произ-
веден отгон легких фракций топлива. К физико-хи-
мич. методам регенерации относятся: адсорбция (с по-
мощью глин, силикагеля, окиси алюминия и др. сор-
бентов); коагуляция растворимых в масле асфальтово-
смолистых веществ и коллоидов, не отделяемых
фильтрованием и сепарацией (в качестве коагуляторов
используют жидкое стекло, водный р-р крахмала,
хлористый цинк и др.); очистка с помощью селектив-
ных растворителей. В качестве химич. методов исполь-
зуют сернокислотную и щелочную очистку. Во многих
случаях пользуются сочетанием различных методов.
Для регенерации больших количеств масел может
быть использована обычная нефтезаводская аппара-
тура; для выполнения этой операции в местах потреб-
ления нефтепродуктов (заводы, автохозяйства и т. п.)
созданы специальные установки различной конструк-
ции и назначения. К числу наиболее рациональных и
эффективных методов следует отнести регенерацию
М. м. в процессе их применения. Достигается это вклю-
чением в смазочные системы двигателей и машин спе-
циальных фильтров, непрерывно очищающих масло
в процессе его использования. Для разных условий
применения масел существуют фильтры самых раз-
личных конструкций и назначений: щелевые, сеточ-
ные, матерчатые, центробежные, термосифонные, ад-
сорбционные, электромагнитные и др.
Нормы качества регенерированных масел, как пра-
вило, несколько ниже, чем у исходных.
Лит.: Черножуков Н. И., Очистка нефтепродуктов
и производство специальных продуктов, Зизд.,М,—Л., 1952
(Технология нефти, ч. 3); Ч ер ножук о в Н. И., К р е й в
С. Э., Л о с и к о в В. В., Химия минеральных масел, 2 изд.,
М., 1959; Моторные топлива, масла и жидкости, под ред. К. К.
Папок и Е. Г. Семенидо, т. 2, 3 изд., М., 1957; Л о с и к о в
В. В.,Пучков Н. Г., Энглин В. А., Основы применения
нефтепродуктов, 2 изд., М., 1959; Технические нормы на нефте-
продукты, под ред. Н. Г. Пучкова, 16 изд., М., 1957; Крейн
С. Э., К у л а к о в а Р. В., Нефтяные изоляционные масла,
М.—Л., 1959; Шашкин П. И., Регенерация отработанных
нефтяных масел, М., 1960. Н. Г. Пучков.
МАСЛА ПОГЛОТИТЕЛЬНЫЕ — см. Масла Ка-
менноугольные технические.
МАСЛА ПРЕПАРИРОВАННЫЕ — растительные
масла, подвергнутые специальной обработке с целью
улучшения свойств пленок, образующихся после вы-
сыхания, а также ускорения высыхания или превра-
щения полувысыхающих и невысыхающих масел в
масла высыхающие. Давно известными способами пре-
парирования являются: рафинация масел, их поли-
меризация (уплотнение) путем длительного нагревания
при 280—300°, окисление продуванием через них
воздуха при 130—150°, вулканизация хлористой или
элементарной серой. Различают масла: лаковые, по-
лимеризованные, оксидированные и фактизированные
(вулканизованные). В результате уплотнения повыша-
ются вязкость, плотность и кислотное число масел,
понижается иодное число. Скорость уплотнения воз-
растает с повышением содержания двойных связей в
жирных к-тах масел и при сопряженном расположе-
нии этих связей; невысыхающие масла не уплотня-
ются. Применение уплотненных масел в лаках и эма-
лях улучшает блеск, твердость, водостойкость покры-
тий и их стойкость к атмосферным воздействиям.
43
МАСЛА ПРЕПАРИРОВАННЫЕ —МАСЛА СИНТЕТИЧЕСКИЕ
44
Разработаны физич. и химич. способы препариро-
вания масел: рафинация, разделение ненасыщенных
компонентов масел и насыщенных, переэтерификация,
изомеризация, дегидратация, сополимеризация, вве-
дение в них различных катализаторов и др.
Для удаления из масел присутствующих в них раз-
личных нежирных примесей, слизистых и красящих
веществ и др. их очищают, или рафинируют, следую-
щими методами: отстаиванием, нагреванием до темп-ры
270—300°, обработкой кислотами (серной и соляной),
щелочами, восстановителями или окислителями, гид-
ратацией и адсорбционной отбелкой природными или
искусственными землями. Наиболее распространен-
ными в лакокрасочной промышленности являются
методы адсорбции и гидратации. Комбинированный
метод очистки для получения высококачественного
т. наз. лакового масла заключается в гидратации, ще-
лочном рафинировании, адсорбционной отбелке и
фильтрации.
Компоненты масел разделяют, используя разли-
чие их растворимости в ацетоне, метилэтилкетоне,
спирте, фурфуроле и др. растворителях при разных
темп-pax. Применяется также разделение глицеридов
льняного масла кристаллизацией их при низкой тем-
пературе из смеси эфира с ацетоном.
Переэтерификация масел, имеющая целью ускоре-
ние их высыхания, заключается в расщеплении масел
с последующей этерификацией полученных (разде-
ленных и очищенных) жирных к-т глицерином, пен-
таэритритом, сорбитом, триметилолпропаном и др.
многоатомными спиртами. Для расщепления масел
используют омыление, действие контакта Петрова
или реактива Твитчеля, а также пропускание через
колонку противотоком масла и воды (непрерывный
способ) и др. Жирные кислоты разделяют кристалли-
зацией их из 90%-ного метанола, очищают дистилля-
цией с последующей кристаллизацией из раствора.
Изомеризацию масел производят с целью переориен-
тации двойных связей из изолированного в сопряжен-
ное положение. Для изомеризации масел применяют
последовательную обработку их едким натром и сер-
ной к-той или каталитич. обработку (катализатор —
никель на угле). Изомеризованное льняное масло
полимеризуется в 3—5 раз быстрее, чем исходное.
Изомеризованные масла лучше высыхают, а их пленки
имеют повышенную щелоче- и водостойкость.
Дегидратацией можно превратить невысыхающее
касторовое масло в высыхающее масло, сходное с
тунговым по способности образовывать пленку с
«ледяными узорами». Дегидратацию проводят в при-
сутствии катализаторов кислого, основного или ней-
трального характера. Наряду с отщеплением гидро-
ксильной группы при дегидратации происходит раз-
ложение рицинолевой к-ты на энантовый альдегид
и ундециленовую к-ту, что сопровождается изомери-
зацией и полимеризацией, вызывающими изменение
физико-химич. свойств масла. После дегидратации
образуются изомеры линолевой к-ты с сопряжен-
ными и несопряженными двойными связями, а также
цис- и транс-изомеры. Транс-изомер окисляется
труднее и высыхает медленнее, чем цис-изомер. Это
задерживает высыхание дегидратированного масла.
При обработке малеиновым ангидридом образуются
т. наз. малеинизированные масла — высыхающие
продукты, получаемые в результате присоединения
малеинового ангидрида к ненасыщенной части мо-
лекулы масла. В результате повышается способность
молекулы к полимеризации при нагревании. Дли-
тельность воздушной сушки малеинизированных ма-
сел не сокращается. Однако по сравнению с исходным
маслом они быстрее полимеризуются, меньше темнеют
при полимеризации, а пленки, получающиеся при
высыхании, обладают лучшей водостойкостью. Обычно
малеиновым ангидридом обрабатывают льняное и
соевое масла. При нейтрализации малеинизирован-
ных масел щелочами, аммиаком или аминами получают
растворимые в воде масла, к-рые можно применять
в произ-ве водо-эмульсионных красок. Пленки малеи-
низированных масел после высыхания обладают хо-
рошей водостойкостью. Обычно 2—10% (по отноше-
нию к весу масла) малеинового ангидрида достаточно
для повышения у масел с несопряженными связями
способности к полимеризации при нагревании. Если
же 10% малеинового ангидрида ввести в масло, в
к-ром есть сопряженные связи, наир, в дегидратиро-
ванное касторовое масло, то аддукт очень быстро обра-
зует гель. В зависимости от характера обрабатывае-
мого масла процесс проводят при темп-ре от 200 до 280°.
Сополимеризованные масла образуются также в
результате присоединения молекул мономеров по
месту непредельных связей молекулы масла. В качест-
ве мономеров, способных полимеризоваться, приме-
няют стирол, а-метилстирол и ццклопентадиен. Масла,
имеющие сопряженные связи, соединяются с этими
мономерами легко, в отличие от масел с несопряжен-
ными двойными связями. Сополимеризация стирола
с тунговым маслом идет очень быстро; дегидратиро-
ванное касторовое масло реагирует медленнее, т. к.
оно имеет меньше сопряженных связей; с льняным
и соевым маслами, не имеющими совсем сопряжен-
ных связей, реакция идет так медленно, что стирол
успевает заполимеризоваться, в результате чего он
отделяется от масла, т. к. полистирол с маслом несов-
местим. Однако смеси льняного и соевого масел
с тунговым и с дегидратированным касторовым мас-
лами дают удовлетворительные сополимеры. Про-
дукты сополимеризации могут быть жидкими или
твердыми в зависимости от соотношения масла и сти-
рола и степени полимеризации. Продукты сополиме-
ризации дегидратированного касторового масла со
стиролом остаются жидкими, если содержание сти-
рола не превышает 70%, и твердыми, когда стирола
больше. Для сополимеризации целесообразно приме-
нять смесь, состоящую из 50—55% стирола и 45—
50% масла. Сополимеризацию стирола с маслом про-
водят в р-ре ксилола или же без растворителя в при-
сутствии перекиси бензоила или гидроперекиси
третичного бутила в качестве инициаторов полимери-
зации.
«Катализированные» масла представляют собой
высыхающие масла, в к-рые для сокращения продол-
жительности полимеризации вводится катализатор.
В качестве катализаторов, не замедляющих высыха-
ния масла и не влияющих на его цвет и химич. стой-
кость, применяют fJ-метилантрахинон, дифенилкарб-
оксилантрацен, тиоф-пафтол и продукты окисления
углеводородов кам.-уг. смолы. Катализированные
масла полимеризуются почти в два раза быстрее не-
катализированных масел и дают более светлые про-
дукты с низким кислотным числом. В пром-сти взамен
М. и. из пищевого сырья все шире используют тал-
ловое масло и продукты его переработки.
Лит. см. в ст. Масла высыхающие. И. И. Головистиков.
МАСЛА СИНТЕТИЧЕСКИЕ — органич. или эл'е-
ментоорганич. соединения, применяемые в качестве
смазочных масел и рабочих жидкостей в различных
машинах и приборах. М. с. по сравнению с высоко-
качественными нефтяными маслами более стабильны
при термич. окислении, обладают лучшей смазочной
способностью и подвижностью при низких темп-рах
и др. ценными свойствами. В пром-сти применяют
следующие М.с.: 1) углеводородные; 2) сложные эфи-
ры алифатич. ряда; 3) полиалкиленгликоли; 4) поли-
силоксаны; 5) фторуглероды.
Углеводородные М. с. получают поли-
меризацией низших олефиновых углеводородов —
45
МАСЛА СИНТЕТИЧЕСКИЕ
46
этилена, пропилена, бутиленов — в присутствии сер-
ной кислоты, фторида бора и др. При полимериза-
ции в присутствии А1С1з этиленовых углеводоро-
дов, полученных крекингом парафинистых продуктов,
образуются масла с очень низкой темп-рой застыва-
ния, хорошими вязкостно-температурными свойст-
вами и незначительной коксуемостью (табл. 1).
Таблица 1
Свойства масел Масла
сред- нее авиацион- ное (ком- понент) для паро- вых машин
Вязкость, сст, при: 37,8° 220 860
98,9° 22,5 46 47,4
Индекс вязкости * 120 120 115
Т. всп., °C 221 318 304
Т. заст., °C Коксуемость, % -29 -22 -18
0,2 0,38 0,4—0,5
* См. Вязкость нефтепродуктов.
Во время второй мировой войны за рубежом (Гер-
мания) М. с. получали полимеризацией этилена и оле-
финов (при 12—15 ат, 120—130°, в присутствии А1С13)
из продуктов крекинга парафина и газойля. Такие
масла обладали высокой стабильностью. М. с., изго-
товляемые полимеризацией изобутилена, имеют вяз-
кость от 4 до 700 сст при 98,9°, слабо устойчивы к
окислению и при 260° и выше деполимеризуются.
Таблица 2
Свойства масел Ди-етор,- амилсеба- пинат Диэтил- гексилсе- бацинат Диэтил- гексил- адипинат
Плотность, 0,915 0,913 0,923
Вязкость в сст при: 98,0° 2,28 3,32 2,38
37,8° 7,38 12,60 8,22
—18° 80 187 107
-40° 504 1410 807
Индекс вязкости 133 152 121
Т. заст., °C -73 —57 —67
Коксуемость, % 0,915 0.913 0,923
М. с. на основе диэфиров применяют для авиацион-
ных реактивных двигателей (табл. 3).
Эфирные М. с. обладают хорошими противоизнос-
ными свойствами и незначительной коксуемостью.
Диэфиры в значительном количестве применяют в ка-
честве масляной основы для производства пластичных
(консистентных) смазок.
Полиалкиленгликоли получают из
различных полигликолей и их эфиров различной сте-
пени полимеризации. Наибольшее распространение
в качестве М. с. получили полиэтиленгликоли
НОСН2СН2 (ОСН2СН2)ПОН
и полипропиленгликоли
НОСН2СН,СН2 (ОСН2СН2СН2)ПОН
Таблица 3
Масла 98,9° Вязкость, ССГГ —40° 1, при: —53,9° Т. всп., °C Испаряемость при 205°, в % от испаряемо- сти нефтяного масла 1010 Устойчивость к окис- лению и коррозион- ная активность при 175° Температурный предел прокачи- ваемости в мас- лосистеме *
Эфирное А » В » с 7,9 3,6 3,1 12 500 1 900 1 500 13 000 10 000 230 220 205 2 3 10 Стабильны, не агрес- сивны к металлам от —32 до —43 от —48 до —67 от —51 до —59
Нефтяные: марки 1010 » 1100 2,5 20,5 ок. 3000 ок. 40000 ок. 129 100 1 Не стабильны, агрес- сивны к металлам от —43 до —51 от —1 до —15
• Темп-pa, при к-рой масло становится неподвижным при эксплуатации в маслосистеме.
Они широко используются в качестве пластификато-
ров, пропиточных средств, диэлектриков, в связую-
щих составах и в водостойких покрытиях; могут
также применяться для смазки конвейеров в печах
для отжига и в др. случаях, когда может происходить
интенсивное испарение смазки. Углеводородные М. с.,
получаемые конденсацией хлорированных парафи-
нов с нафталином, имеют по сравнению с полимерны-
ми более высокую термоокислительную стабиль-
ность, но их вязкостно-температурные свойства хуже.
Сложные эфиры алифатического
ряда (эфиры двухосновных к-т и
эфиры многоатомных спиртов) полу-
чают этерификацией в присутствии ка-
тализаторов:
НООС (CH2)nCOOH + 2ROH —>
—> ROOC (CH2)nCOOR + 2H2O
СН,ОН (CH2)nCH2OH + 2RCOOH —>
—>ROOCH2 (CH2)nCH2COOR +2H,0
В табл. 2 приведены свойства нек-рых
эфиров, применяемых в качестве М. с.
Диэфиры имеют малую летучесть. Так,
давление пара диэтилгексилсебацината
влип рт.ст.: 10~s (115°), 10-1 (176°);
испаряемость 0,05% (66°, 168 час.).
и их эфиры, получаемые заменой одной или обеих
гидроксильных групп различными радикалами (па-
рафинового ряда, ароматич. и др.). Свойства некото-
рых изготовляемых в США М. с. приведены в таб-
лице 4.
Большинство полиалкиленгликолевых масел имеет
мол. в. от 200 до 1500, достаточно устойчивы к окис-
лению и не образуют при разложении осадков; выше
200—260° имеют склонность к деполимеризации. Для
стабилизации М. с. применяют присадки. Полиал-
киленгликолевые М. с. обладают хорошей смазываю-
Таблпца 4
Свойства масел LB-140Xa LB-400Xa 50-НВ-556 50-НВ-51006
Плотность,^0 « • « • 4 Вязкость, сст, при: 0,982 0,995 0,991 1.063
37,8° 28,9 86,6 9,0 1120
98,9° 5,8 14,9 2,4 166,3
Индекс вязкости . . 140 141 97
Т. заст., °C —45,5 -34 —65 -29
Т. всп., °C 174 254 127 232
Коксуемость, % . . . 0,01 0,01 — —
а Серии LB-Х, со стабилизирующими добавками, нерастворимы в воде.
® Серии 50-НВ растворимы в воде.
47
МАСЛА СИНТЕТИЧЕСКИЕ —МАСЛО КОРОВЬЕ
48
щей способностью, инертны к металлам. Эти масла
широко применяют для смазки двигателей и других
машин, зубчатых передач, компрессоров, приборов
и др., а также в качестве антифризов, гидравлич.
жидкостей и теплоносителей. М. с., получаемые кон-
денсацией окисей этилена и пропилена со спиртами,
отличаются низкой стоимостью.
П олисилоксановые М. с. (силиконовые жид-
R R R
। R —S1 —О — 1 । — Si—О — । — Si— R
1 R 1 R n R
кости) содержат силоксановую группу—Si—О—Si—.
I I
В качестве смазочных масел применяют метил-, этил-
и метилфенилсилоксаны. Введение в силоксаны фе-
нильных групп значительно повышает их устойчи-
вость к окислению, но ухудшает вязкостно-темпера-
турную характеристик/^ В табл. 5 приведены свой-
ства типичных силоксановых масел разной вязкости.
По вязкостно-температурным свойствам силоксаны
превосходят все известные классы органич. соедине-
ний и значительно лучше неф-
тяных масел, что видно из сра-
внения вязкости двух масел,
обладающих одинаковой вязко-
стью при 37,8° (табл. 6).
Силоксаны отличаются очень
низкой температурой застыва-
ния, малой испаряемостью, хо-
рошей термоокислительной ста-
бильностью. Нек-рые из них
термостабильны до 275—300° и
выше. Применение этих масел
ограничено низкими смазочны-
ми свойствами в условиях больших скоростей и на-
грузок, а также при нек-рых сочетаниях металлов
в узлах трения. Для повышения смазочных свойств
в молекулу силоксанов вводят группы, содержащие
хлор, олово и др.; использование присадок затруд-
няется низкой растворяющей способностью силиконов.
Таблица 5
Масла
Свойства I 11 III
Плотность, 0,950 0,968 0,971
Вязкость, сст, при: 37,8° 15,9 70,1 147,8
98,9° 6,3 32,0 58,0
Индекс вязкости .... 212 156 142
Т. заст., °C -57 —54 -51
Т. всп., °C 271 315 324
Таблица 6
Показатели
Температура, °C
98,8 ....
37,8 ....
-18 ....
Вязкость, сст
силоксан
нефтяное
масло
40,2
95,6
333
562
150
11,0
95,6
10 600
137 000
100
-34
Индекс вязкости . .
Силоксановые жидкости широко применяют в ка-
честве антипенных присадок и добавок, повышающих
термич. стабильность и моющие свойства нефтяных
масел; расходы присадки при этом ничтожно малы
(0,0005-0,005%).
Фторуглеродные М. с. состоят из угле-
водородов, в которых атомы водорода замещены фто-
ром. Используются также продукты, содержащие
в молекуле, кроме фтора, также и хлор. Фторугле-
родные М. с. чрезвычайно устойчивы к воздействию
высоких темп-p, окислителей (азотная, хромовая к-ты,
перекись водорода и др.), хлора, щелочей и др.
химич. активных веществ. Они не воспламеняются
и не взаимодействуют с Кислородом, имеют очень
малое давление пара, достаточно высокую смазоч-
ную способность. Легкие фторуглероды не изме-
няются при нагреве до 400—500°. Недостатками фтор-
углеродов являются очень резкое повышение их вяз-
кости с понижением темп-ры и высокая темп-ра за-
стывания. Свойства некоторых фторуглеродных ма-
сел, выпускаемых в США, приведены в таблице 7.
Некоторые хлорфторуглероды являются более цен-
ными смазочными веществами, чем фторуглероды.
У них в меньшей степени возрастает вязкость с тем-
пературой и весьма высоки смазочные свойства даже
в условиях граничной смазки. Фторуглеродные мас-
ла получают фторированием нефтепродуктов в присут-
ствии C0F3 или других фторидов металлов (AgF2,
Таблица 7
Масла Приблизи- тельный состав Мол. вес. Плот- ность (25°) Вязкость в сст при Индекс вязкости т. заст.,°C
20° | 60°
Легкое (FCX-330) . . Среднее (FCX-512) . Среднее (S-30) .... C14F30 Cao-F 42 (CF2-CFC1)7 738 1038 775 1 ,994 2,059 1,960* 19 1600 350 3,5 1 34 / 36 от —500 до —1600 —400 -29 + 8 -15
* При 20°.
HgF2, MnFa и др.) при 200—350°. Каталитич. поли-
меризацией хлортрифторэтилена с последующим фто-
рированием при 200° в присутствии CoF3 получа-
ют хлорфторуглероды: (CF2—CFC1)S, (CF2—CFC1)7,
(CF2—CFC1)8 и др. Фтор- и хлорфторуглеродные М. с.
применяют для смазки компрессоров, кранов, кла-
панов и различных устройств, работающих при очень
высоких темп-pax и в условиях соприкосновения
с активными химич. веществами.
Лит.: Основы технологии нефтехимического синтеза.
Сб. статей, под ред. А. И. Динцеса и Л. А. Потоловского, М.,
1960; Джорджи К. В., Моторные масла и смазка двигате-
лей, пер. с англ., М., 1959; В а ж а н т В., Хвал о в с к и
В., Ратоуски И., Силиконы, [пер. с чешек.], М., 1960;
Браудо Е. Е., Д инц ее А. И., Химия и технология топ-
лив и масел, 1960, № 2; Новейшие достижения нефтехимии и
нефтепереработки, под ред. К. А. Побей Д. Д. Мак-Кета, пер.
с англ., т. 2, М., 1961; Моторные топлива, масла и жидкости,
под ред. К. К. Папок и Е. Г. Семенидо, т. 2, 3 изд., М., 1957.
В. В. Панов.
МАСЛА ШПАЛОПРОПИТОЧНЫЕ — см. Масла
каменноугольные технические.
МАСЛО АНТРАЦЕНОВОЕ — см. Каменноуголь-
ная смола.
МАСЛО КОРОВЬЕ — пищевой продукт, в к-ром
главной составной частью является молочный жир.
В сливочном масле его содержится 81—82,5%,
в топленом масле— 98%. М. к. обладает хорошей
усвояемостью (97%) и высокой калорийностью (ок.
7800 ккал/кг)', в сливочном масле содержится ок. 1%
белка, в соленом — дополнительно 1,5% поваренной
соли. Специфич, вкус и аромат масла принадлежат
гл. обр. глицеридам, входящим в состав молочного
жира, летучим жирным к-там, лецитину, белку.
Молочный жир имеет т. пл. 28—36°, т. заст. 18—23°.
В молочном жире содержится ок. 0,26—0,42% сво-
бодных жирных к-т. Основными жирными к-тами,
глицериды к-рых входят в состав жира, являются
(в скобках приведены средние данные): олеиновая
(32%), пальмитиновая (24%), миристиновая (11%),
49
МАСЛО СОСНОВОЕ ФЛОТАЦИОННОЕ — МАСЛЯНАЯ КИСЛОТА
50
стеариновая (9,5%); остальных к-т содержится от
0,2 до 3,6%. Всего в молочном жире находятся 20
различных жирных к-т (в твердых животных жирах
этих к-т 5—7, в растительных 5—8). В состав
молочного жира входят также фосфатиды (лецитин,
кефалин и сфигномиэлин) и стериды (холестерин).
В масле содержатся все жирорастворимые витамины,
количество к-рых колеблется в зависимости от сезона,
характера кормления животных и породы. Масло
является важнейшим источником витамина А и каро-
тина. В среднем в 100 г масла содержится (в интернац.
единицах): ок. 3000 витамина А и каротина, ок. 80
витамина D и ок. 2000 мкг% витамина Е. В СССР для
консервирования М. к. добавляют поваренную соль.
М. к.вырабатывают след, видов: 1) сладкосливочное соленое,
2) сладкосливочное несоленое, 3) вологодское сливочное несо-
леное, 4) кислосливочное соленое, 5) кислосливочное несоленое,
6) шоколадное, 7) любительское, 8) топленое. Каждый вид мас-
ла должен иметь характерный для него вкус, без посторонних
примесей и запахов. Для произ-ва масла применяют 2 способа:
непрерывно-поточный и сбивания в маслоизготовителях перио-
дического действия. В обоих случаях молоко сепарируют. При
непрерывно-поточном методе горячие сливки сепарируют пов-
торно при 83—92° для получения высокожирных сливок, со-
держащих 83—85% жира. Полученные сливки в случае необ-
ходимости нормализуют до установленной жирности и направ-
ляют в маслообразователь для охлаждения и механич. обра-
ботки. Готовое масло выходит из охладителя при темп-ре 12—
15°. При получении масла методом сбивания сливки пастери-
зуют, охлаждают, выдерживают в ваннах для «созревания»,
затем сбивают, образовавшееся масляное зерно промывают, по-
лученное масло обрабатывают. При выработке кислосливочно-
го масла сливки после заполнения согревательных ванн за-
квашиваются закваской, приготовленной на чистых культурах
молочнокислых бактерий. Аромат и вкус этого вида масла обу-
словлены наличием диацетила и др. летучих веществ, образую-
щихся при сквашивании сливок. При выработке соленого
масла после промывания зерна производят посолку. Для повы-
шения стойкости масла, т. е. способности сохранять вкусовые
качества свежего масла без к.-л. заметных изменений в течение
длительного срока, имеет важное значение распределение влаги
в нем. Качество масла определяется по 100-балльной системе
на основе органолептич. показателей и химич. состава (содер-
жания жира, воды и соли). В зависимости от оценки масло
относят к высшему или первому сортам. В. И. Сирик
МАСЛО СОСНОВОЕ ФЛОТАЦИОННОЕ — жид-
кий продукт лесохимии, производств, получаемый
при ректификации скипидаров, извлекаемых из про-
смоленных сосновых пней. Основные физико-химич.
свойства М. с. ф.: плотность 0,91—0,95 (20°); кислот-
ное число 0,5—2,0; содержание терпеновых спиртов
50—75%; содержание влаги 0,5—1,5%; Пр1,477 —
1,495 (20°); пределы кипения основных погонов
170—225°. М. с. ф. состоит из высокомолекулярных
терпеновых спиртов (до 75%), гл. обр. а-терпинео-
ла и небольшого количества цис- и трянс-дигидротер-
пинеолов, борнеола и др. Кроме того, в М. с. ф. со-
держится до 10% других кислородсодержащих терпе-
нов (камфары, эфиров: анетола, эстрагола и др.).
Вследствие значительного содержания в М. с. ф. тер-
пеновых спиртов оно обладает свойствами поверх-
ностно-активных веществ и оказывает эффективное
вспенивающее действие при флотации.
Различают экстракционное М. с. ф.,
получаемое путем экстракции сосновых пней, и с у-
хоперегонное, получаемое при сухой пере-
гонке последних. Стандартом (ГОСТ 6792—53) пре-
дусматривается выпуск нескольких марок М. с. ф.
(в зависимости от содержания терпеновых спиртов).
Экстракционные М. с. ф. (<<пайп-ойль>>) — один из наи-
более высококачественных широко распространенных
флотореагентов (из лесохимии, продуктов). Помимо
М. с. ф., для флотации применяют древесно-
смоляное флотационное масло,
имеющее совершенно другой химич. состав (фенолы,
кетоны, спирты). Наряду с натуральными флотацион-
ными сосновыми маслами применяют и синтетические,
получаемые гидратацией содержащегося в скипидаре
пинена и его последовательного превращения в терпи-
неол. Е. Г. Быховский.
МАСЛЯНАЯ КИСЛОТА С,Н,СООН, мол. в. 88,10—
известны 2 изомера: н-масляная к-та и изомасляная
к-та. н-М асляная кислота (бутановая, этил-
уксусная к-та) СНзСН2СН2СООН — бесцветная жид-
кость; т. пл. — 5,26°; т. кип. 163,25°; d™ 0,9577; Пд
1,3980; т)15 1,814 спуаз', о 26,74 дин!см (20°); т. Крит.
354,7°; константа диссоциации А=1,508 • 10-s(25°);
т. воспл. в закрытой чашке 76,70°; при —4,1° сме-
шивается с водой и органич. растворителями во
всех отношениях; перегоняется с водяным паром; об-
разует азеотропную смесь с водой (18,5% н-М. к., т.
кип. 99,4°). Из водных р-ров н-М. к. высаливают NaCl,
СаС12 или лучше Al2(SO4)s. н-М. к. обладает общими
свойствами предельных карбоновых к-т. Бромирование
в присутствии красного фосфора приводит к а-бром-
пропионовой к-те; пропусканием паров н-М. к. над
СаСОз, МпО (350—450°) получают дипропилкетон
С3Н,—СО—С3Н7; этот кетон образуется также при
прокаливании ее кальциевой соли. и-М. к. гладко
этерифицируется; с галогенными соединениями фос-
фора дает галогенангидриды; может быть получена
всеми общими способами получения карбоновых к-т.
В пром-сти н-М. к. получают окислением бутилового
спирта, а также сбраживанием (35—40°) отходов,
содержащих крахмал (маслянокислое брожение).
н-М. к. — низшая из к-т, входящих в состав жиров
(в коровьем масле ее содержится 3—4%), находится
также в мясном экстракте, нек-рых эфирных маслах
и т. д. н-М. к. используют для получения ее эфиров;
нек-рые из них применяют в качестве веществ, при-
дающих пищевым продуктам определенный фруктовый
запах. Так, метиловый эфир пахнет яблоками, изо-
амиловый — грушами. Полный глицериновый эфир
(трибутирин) служит пластификатором в лаках на
основе эфиров целлюлозы. Бутирил- и бутирилацетил-
целлюлозу применяют в качестве основы для атмос-
фероустойчивых покрытий. Метиловый, . этиловый,
изоамиловый, гептиловый, гераниловый и цитронел-
лиловый эфиры используют в качестве душистых
веществ с фруктовым и цветочным запахом (табл. 1).
Таблица 1
Эфир Мол. в. Т. кип., °C da "д
Метиловый . . . 102,13 102,6 0,8984 Q°) 1,3780в
Этиловый .... 116,16 121,6 0,8788 0°) 1,4000
н-Пропиловый . . 130,18 143 0,879 (15) 1,4005
«-Бутиловый . . 144,21 166,6 °,872 (”) 1,4049
Изобутиловый 144,21 156,8 0,8606 (25) 1,4035
н-Амиловый . . . 158,23 185,0 0,8713(1 5) 1,4110
Изоамиловый . . 158,23 178,6 0,882 (0) —
н-Гексцловый . . 172,26 207,9 0,8567(30) 1,41886
«-Гептиловый . . 186,29 225.9 0,8555 (30) 1,4288°
Фениловый . . . 164,20 227—228 1,0363(9 —
Бензиловый . . . 178,22 238—240 1,0135 (15) 1,4920
Гераниловый . . 224.33 142- 143/13 мм 0,9014(15) 1,4593
Цитронеллиловый 226,35 134— 135/12 мм 0,8874(15) 1,4449
а В скобках приведена темп-pa. б При 15°.в 25°.
Изомасляная кислота (диметилуксус-
ная к-та) СНзСН(СНз)СООН, т. пл. —46,1°; т. кип.
154,70°; 0,9530; т)15 1,126 спуаз', о 25,55 дин/см
(15°); константа диссоциации 7Г=1,44 • 10-5 (18°);
т. воспл. в закрытой чашке 74°. При 20° в 100 г воды
растворяется 20 г изо-М. к.; с органич. растворителями
смешивается во всех отношениях. Химич, свойства
изомасляной к-ты аналогичны свойствам к-М. к-ты;
51
МАСЛЯНЫЙ АЛЬДЕГИД —МАССООБМЕН
52
в пром-сти ее получают окислением изобутилового
спирта. Эфиры uso-М. к. с гликолями и глицерином
предложено применять для коагуляции латекса, ре-
гулирования вулканизации, в качестве пластификато-
ра лаков и др.; эфиры изо-М. к. так же, как и эфиры
н-М. к., обладают фруктовым и цветочным запахом
(табл. 2).
Таблица 2
Эфир Мол. в. Т. кип., °C da nD
Метиловый . . . 102,13 92,3 0,8890 Q°) 1,3840
Этиловый .... 116, 16 101,1 0,8662 (30) 1,3903
н-Пропиловый . . 130,18 135,4 0.8843 (°) —
Изопропиловый 130,18 120,8 0.8687 (°) —
Изобутиловый . . 144,21 148,7 0,8749 (°) 1,3999
н-Амиловый . . . 156,23 155 0,8592 (13) 1,4076
Изоамиловый . . 158,23 168,8 0.8759 (°) —
Бензиловый . . , 178,22 229 1,0058 (23) —
а Б скобках приведена темп-ра.
Лит.: Ullman п, 3 Aufl., Bd 4, Munchen—В., 1953,
S. 769; Вайсбергер А. [и д р.1, Органические раство-
рители, пер. с англ., М., 1958, с. 136. Н. А. Несмеянов.
МАСЛЯНЫЙ АЛЬДЕГИД (бутиральдегид, бута-
наль) СНзСН2СН2СНО, мол. в. 72,10—бесцветная
жидкость с острым запахом альдегида; т. пл. —97,1°;
т. кип. 74,78°; d30 0,8016; Пр 1,3791; давление паров
91,5 мм рт. ст. (20°); вязкость 0,433 спуаз (20°);
теплота испарения 104,4 кал/г (т. кип.); теплота сго-
рания 586,1 ккал!моль', т. воспл. в закрытой чашке
—6,7°; минимальная взрывоопасная концентрация
в воздухе 1,5%; М. а. образует азеотропные смеси
с водой (94 вес. % М. а., т. кип. 68°), с этиловым
и метиловым спиртами; растворимость М. а. в воде
(%): 8,7 (0°), 7,1 (20°), 5,4 (40°); растворимость воды в
М.а. (%): 3,01 (0°), 3,17 (20°), 3,39 (40°); с большинст-
вом органич. растворителей (спирт, эфир, толуол и др.)
М. а. смешивается во всех отношениях.
М. а. обладает всеми свойствами, характерными
для насыщенных алифатич. альдегидов; при восста-
новлении образует бутиловый спирт, при окисле-
нии —• масляную кислоту. Окисление в присутствии
солей кобальта приводит к ангидриду масляной к-ты,
применяемому в произ-ве ацетобутирата целлюлозы.
М. а. легко образует бутилбутират. В присутствии
кислот происходит полимеризация М. а. в пара- или
мета-масляный альдегид. Под действием щелочей М. а.
в результате альдольной конденсации образует бутир-
альдоль; с аммиаком — бутиральдегидаммиак; с
анилином или бутиламином — продукты конденса-
ции, применяющиеся как ускорители вулканизации
каучука. С альдегидами и кетонами в присутствии
разб. щелочей М. а. дает продукты конденсации:
с ацетальдегидом — ацетобутиральдоль, а с ацето-
ном — 4-ОКСП-2-гептан. С поливиниловым спиртом
М. а. образует поливинилбутираль. Эфиры М. а. с
фталевой к-той являются пластификаторами многих
виниловых смол, эфиров целлюлозы и синтетич. кау-
чуков.
Промышленными способами получения М. а. яв-
ляются частичное восстановление кротонового альде-
гида, реже — дегидрирование бутилового спирта.
Известны другие методы получения М. а., напр. ча-
стичное окисление бутана; лабораторные способы —
пиролиз смешанных солей масляной и муравьиной
к-т; действие солей натрия илн кальция на 1-нитро-
бут ан (Нефа реакция), гидрокарбонилирование про-
пилена, перегруппировка этилвинилового эфира и др.
М. а. применяют в произ-ве поливинилбутираля
(бутвара), используемого в произ-ве безосколочного
стекла. При конденсации в кислой среде образуются
масло-, спирторастворимые смолы (при конденсации
с мочевиной). Практич. значение имеет также образо-
вание бутиральдоля, продуктов конденсации с ани-
лином и др. (см. выше).
Лит.: К 1 г k, V. 2, N. Y., 1948, р. 684; Н о u b е n-W е у 1,
4 Aufl., Bd 7, Т1 1, Stuttgart, 1954. Н. И. Гамбарян.
МАССОВОЕ ЧИСЛО — целое число, ближайшее
к атомному весу изотопа данного химич. элемента.
Обозначается М или А. М. ч. равно общему числу
нуклонов в атомном ядре. При обозначении атомных
ядер М. ч. ставят сверху справа (или слева) у знака
соответствующего элемента; напр., обозначение атом-
ного ядра изотопа урана с М. ч. 238 записывается так:
U238 (или 238U). См. также Изотопы, Дефект массы.
МАССООБМЕН - явление переноса вещества в пре-
делах одной или нескольких фаз, лежащее в основе
процессов разделения разнообразных смесей (сорбция
и десорбция, дистилляция и ректификация, экстрак-
ция, сушка, растворение и кристаллизация). Типич-
ным примером М. может служить ректификация,
когда компоненты разделяемых смесей, находясь
в жидкой и паровой фазах, перераспределяются в них
т. обр., что компоненты с низкой темп-рой кипения
концентрируются преим. в паровой фазе, а компонен-
ты с высокой темп-рой кипения — в жидкой фазе.
Интенсивность М., или диффузионный поток qD (коли-
чество вещества, передаваемое с единицы поверхности
в единицу времени), прямо пропорциональна движущей
силе процесса Ас, т. е.
q^—KAc (1)
где К — коэфф, массопередачи. За время т через
поверхность фазового контакта F перейдет количество
вещества:
Ga~KAcFx кг-моль (2)
Движущая сила процесса Ас может быть выражена
через разность концентраций (рабочей и равновесной
или, наоборот, в зависимости от направления потока),
разность парциальных давлений, разность темп-р,
разность активностей и т. п. Термодинамич. выражение
движущей силы представляет собой разность хими-
ческих потенциалов. В зависимости от выражения
движущей силы процесса коэфф, массопередачи имеет
различные размерности, а количественное его зна-
чение зависит от гидродинамич. обстановки процесса,
свойств смесей, условий фазового равновесия и кон-
структивной характеристики аппарата.
Так как обычно поверхность фазового контакта не-
известна, коэфф, массопередачи связывают в виде
единого комплекса с фиктивной поверхностью фазо-
вого контакта (а), развиваемой в единице объема ап-
парата, выражая его как Ко а или Кох а, в зависимо-
сти от того, по газовой или жидкой фазе рассчитывает-
ся движущая сила. Тогда при рабочем объеме аппара-
та V=Hf, где Н — высота рабочей зоны аппарата,
/ — площадь его поперечного сечения:
Ga = KOyaHf!Ayl..p, кг-молъ/час (3)
Средняя движущая сила процесса М. может быть отнесена
к любой из двух фаз рассматриваемой системы. Напр., в про-
цессах ректификации и абсорбции она может быть отнесена
либо к паровой (газовой) (Аус ), либо к жидкой фазе (А\.р )
При этом
ду (4): Ах =- -- /и- (5)
ср. С dy ' ср. о dx
Ьр"» J x~xv
где у и х — рабочие, а 1/ и х — равновесные концентрации
компонента, переходящего из одной фазы в другую; у у хн
и хк—соответственно начальные и конечные концентрации пере-
ходящего компонента в газовой и жидкой фазах.
53
МАССООБМЕН
54
В случаях, когда линии фазового равновесия и рабочие
линии процесса являются прямыми
Дун-
Ду =—51—г-5
СР- Ду„
2,31g —»
ДУК
AyH = yPH-yH; Дук = урК-уи
(6)
(7)
(8)
О)
где?/ и у —начальные и конечные концентрации, отвечаю-
щие состоянию равновесия. Аналогично
Дх — Дх
Дх п
ср. Дх„
2,3 Iff —?
s Дх
к
Дх =х —х ; Дх —х —х
нн рн’ к к рк
Из ур-ний (3), (5) и (6), учитывая, что Ga=G
ходим рабочую высоту аппарата:
л — &а - & С
J Ур~~У
(1/к—Уя). на'
(10)
али
Ga
Н Kuxaf\xQV_ К
dx
Р
где G и L — потоки жидкости и газа, поступающие на обработ-
ку, a Ktlya и Киха — объемные коэфф, массопередачи в газовой
и жидкой фазах, выраженные в кг-моль/час-м3-и, где и — еди-
ница движущей силы.
Интегралы правой части ур-ний (10) и (И) представляют со-
бой нек-рое безразмерное число N, определяемое величиной
движущей силы и пределами изменений концентраций, а вели-
чины G/K<ty(if и L/Kitxaf имеют размерность высоты, на к-рой
переносится вещество; они характеризуют кинетику процесса,
носят название высоты единипы переноса, обо-
значаются соответственно (ВЕП)0& и (ВЕП)оа; и определяются
на основании опытных данных.
Таким образом,
Н = (ВЕП)^^ = (ВЕП)0а.2У0Ф
Jdij Р dx
и Nox=\ ——выра-
у J р
жают перепады концентраций переходящего компонента, при-
ходящиеся на единицу локальной движущей силы, и наз. чис-
лами единиц переноса. Число единиц переноса
при диффузии в одном направлении (абсорбция, экстракция)
определяется ур-нием:
У1
Noy =
Уг
(l-p)f dy
(1 -v)(y-yp)
(12)
где
(1 -У) (1 -уо)
(-У)Г—
1ПТ^7
Если (1—y)f может быть заменено средним арифметическим,
то: *
Уг
N0J,= C-^+l,151g
.5 V~Vp
У1
(1 ~ Уа)
(1 ~У1)
(13)
Вторым слагаемым часто можно пренебречь.
Если диффузия в обоих направлениях проходит с одинако-
вой скоростью (дистилляция), то ур-ние (13) упрощается путем
1 —у2
исключения отношения т—— и приводится к виду;
1—У\
У1
Noy -
V.
dy
(14)
Соотношение между Nny и N()X может быть найдено из соот-
ношений между тангенсами угла наклона линии равновесия
(т) и рабочей линии процесса L/G:
/V о у L mG
(15); (ВЕП)оу = (ВЕП)оа, — (16)
Эффективность масеообменного аппарата может быть вы-
ражена также через число теоретич. ступеней контакта или
число теоретич. тарелок, а кинетич. характеристика — через
кпд или через высоту, эквивалентную теоретич. ступени кон-
такта.
Число теоретич. ступеней контакта или число теоретич.
тарелок может быть найдено аналитически или графически
путем совместного решения ур-ний равновесия и рабочей линии
процесса. В последнем случае строятся ступени в пределах
заданных концентраций. Следует иметь в виду, что одна теоре-
тич. тарелка выражает одно изменение движущей силы по га-
зовой Ду и одно по жидкой Ах фазам, причем число теоретич.
тарелок и движущая сила процесса находятся в обратном соот-
ношении, т. е чем больше движущая сила, тем меньше потре-
буется теоретич. тарелок для данного разделения. Действи-
тельное число тарелок, к-рое необходимо установить в аппара-
те, определяется с помощью кпд. Различают: кпд колонны т|,
кпд тарелки т|т и точечный кпд т]0.
Кпд колонны — отношение числа теоретич.
тарелок и к числу тарелок nD, практически установ-
ленных в колонне, т. е.
H = n/nD (17)
Средняя эквивалентная высота теоретич. ступени
контакта (теоретич. тарелки) h3 применяется чаще
всего для анализа кинетики насадочных, пленочных
и др. колонн и предстанляется в виде отношения:
h3=H/n (17а)
где Н — полная высота слоя насадки в м, п — число
теоретич. тарелок.
Кпд тарелки (кпд Мерфри) — отношение
разности средних составов пара, поднимающегося
с тарелки уп (см. рисунок) и поступающего на та-
релку (уп+1), к разности у,— где (/р— состав
пара, отвечающий равновесию с жидкостью, поки-
дающей эту тарелку
цт„ = _ к Т ) (18)
У Vp~Vn+i
Кпд тарелки, выраженный в концентрациях жидкой
фазы, принимает вид:
где хр — состав жидкости данной тарелки (р„).
Т очечный кпд Т|о — отношение разности
концентрации пара, поднимающегося в данной точке
тарелки, и среднего состава пара, поступающего на
эту тарелку, к разности равновесной концентрации
пара, отвечающей жидкости, покидающей эту та-
релку, и среднего состава пара, поступающего на эту
тарелку.
Согласно рисунку
у' - 1/n+i , " _ y"-Vn+i ,
Ч0 Ур-Уп+1 ’ 'о Ур-Уп+1
Соотношение между тр, и т]г определяется степенью
перемешивания жидкости на тарелке и выражается
ур-нием:
Ви”1 -у- (1 — J)
Т)г = е__________=1 (20)
G ,
т — (I — J)
где J — фактор перемешивания. При J = 0,r|r дости-
гает максимального значения:
nr = ^(^mG/L-D (21)
При J — 1, т|г достигает минимального значения Т)г = Т)о-
55
МАССООБМЕ Н—МАСС-СП ЕКТР АЛЬНЫЕ ПРИБОРЫ
56
На практике 7=0,25—0,75.
Связь между различными способами выражения
движущей силы и кинетич. характеристики устанав-
ливается в виде соотношений
l_T)o=e'^a/-H''G=e-;Vo!' (22)
Птх=Л’0Х/(1+ДГ0Ж) (23)
1 — I L
h - G . -(ВЕП1 (24>
Пэ — к^а i-mGjL ( >°У 1-mGjL
Если линия равновесия и рабочая линия парал-
лельны, то
mG/L—i и 7гэ = (ВЕП)(у, = (ВЕП)0Х (25)
При анализе явлений М. в двухфазных системах
необходимо учитывать механизм переноса вещества
через границу раздела фаз. Различают два механизма
переноса вещества: молекулярную диф-
фузию, определяемую законами взаимодействия
молекул (микрокинетика) и вихревую, или
турбулентную диффузию, определяе-
мую законами макрокинетики. В неизотермич. усло-
виях в однофазных системах может иметь место
термодиффузия. В однофазных системах М. описы-
вается обычными ур-ниями диффузии.
Процесс М. в однофазных системах обычно описы-
вается критериальным ур-нием вида:
Nu = ARem-Prn (26)
где Nu=KdJD — диффузионный критерий Нуссельта;
К — коэфф, массопередачи, — эквивалентный диа-
метр или характерный линейный размер, D — коэфф,
молекулярной диффузии; 7?е= ivdjv—критерий Рей-
нольдса для движущегося потока, Pr=vjD — диф-
фузионный критерий Прандтля; v — кинематич. вяз-
кость потока.
Для систем с фиксированной границей раздела (присут-
ствие твердой фазы) существенным является наличие погра-
ничного с твердой фазой слоя насыщенного р-ра, в к-ром
преобладает молекулярная диффузия. При неподвижном со-
стоянии фаз (отсутствие вынужденной конвекции) механизм
молекулярного переноса становится преобладающим.
По т. наз. двупленочной теории Люииса и Уитмана прини-
мается преобладающее влияние молекулярного переноса и
для движущихся потоков фаз. Согласно этой теории, со стороны
каждой фазы на границе раздела фаз, где устанавливается рав-
новесие, образуются 2 пленки. Так, в процессах абсорбции со
стороны газа образуется газовая пленка, со стороны жидко-
сти — жидкостная пленка. Перенос вещества в этих пленках
лимитируется молекулярной диффузией. Общее сопротивление
переносу вещества 1/Xogm в стационарном процессе соответ-
ственно складывается из сопротивления газовой пленки 1/Аг
и сопротивления жидкостной пленки 1/К :
1 1 , <р
тГ-Г~ = тГ+к (27)
Аобщ. Кг кж
где — общий коэфф, массопередачи, К?, — пленочные
коэфф, массопередачи, <р — коэфф, пропорциональности, выра-
жающий соотношения равновесных концентраций в газовой и
жидкой фазах. Благодаря простоте и наглядности схемы дву-
пленочная теория имела широкое распространение. Однако
исследования последнего времени не подтверждают основных
положений двупленочной теории для движущихся потоков фаз.
Для объяснения явлений переноса вещества через границу
раздела фаз выдвинуты другие теории. Так, Хигби исходит из
нестационарности процесса М. через поверхность раздела фаз
и их кратковременных контактов и пропорциональности коэфф,
массопередачи коэфф, молекулярной диффузии в степени %.
Данквертц и независимо от него Кишеневский выдвинули
гипотезу непрерывного обновления поверхности массообмена
между 2 потоками.
По Данквертцу, в течение периода обновления поверх-
ностного слоя, т. е. за время, в течение которого рассматривае-
мый элемент поверхности находится в контакте с газовой фа-
зой, молекулы поглощаемого газа продвигаются с поверхности
в объем жидкости путем мол. диффузии. При этом, как и в тео-
рии Хигби, коэфф, массопередачи пропорционален коэфф,
молекулярной диффузии в степени ’/2.
Кишеневский исходит из наличия не только молекулярной,
нои турбулентной диффузии, и качестве кинетич. характе-
ристики вводит коэфф, эффективной диффузии, равный сумме
коэфф, молекулярной и вихревой диффузии. Из этого следует,
что коэфф, массопередачи пропорционален коэфф^ молекуляр-
ной диффузии в степени, меньшей %, что соответствует процес-
сам, осуществляемым при значительной турбулизации.
Представления о межфазной турбулентности при-
водят к кинетич. уравнениям М. в след, виде:
Nu ~ A Rem • Prn (1 + /) (28)
иЙЧЖП-Ш" (29>
где /—фактор гидродинамич. состояния двухфазной
системы, зависящий от величины р, определяемой
соотношением данной и предельной скорости газа,
от соотношения потоков фаз L/C, их плотностей Yp'Yk>
вязкости рж/рг и поверхностного натяжения (01,02)
каждой фазы на границе с воздухом и между фазами.
При / = 0 уравнение (29) согласуется с пленочной
теорией, т. е. выражает эффект М. в пределах одной
фазы за счет молекулярной и вихревой диффузии.
Коэфф. / учитывает количество переносимого вещества
за счет взаимодействия потоков. Допуская аналогию
между трением и М. в однофазном газовом потоке,
можно определить порядок величин показателей сте-
пени т и п в ур-нии (29).
Для ламинарного режима при т = 0 и п = 1 / 3
получим:
“Э=А (б-ум+л,) (30)
откуда
A~L>2/, (31)
Так как в условиях очень малых скоростей потока
фактор /0 —весьма малая величина (значительно мень-
ше единицы), то ур-ние (31) приводится к виду:
Nu — const (32)
На практике этот случай соответствует массопере-
даче в колоннах со смоченными стенками и со смо-
ченной насадкой.
Для турбулентного режима при т = 0,8 и п = 2/3
получим
KcL . ZwcL\ 0,8 / v \2/,
— =АТ(__ J (1+/i) (33)
откуда
(34)
Для режима развитой свободной турбулентности
т - 1, п = 1, получим:
^- = А2(1+/2);А~Л°
где —критерий Маргулиса.
Лит.'. Treybal R. Е., Mass-transfer operations, N. Y.—
Toronto — L., 1955; К а фаров В. В., Основы массопередачи,
М., 1962; Lewis W. К., Whitman W. G., Industr. and
Ergng. Chem., 1924,16, 1215; H 1 g b 1 e R., Trans. Amer. Inst.
Chem. Engrs, 1935, 31, 365; Кишиневский M. X.,
Панфило в А. В., Ж. прикл. химии, 19 49, 22, МИ, 1173;
Кишиневский М. X., там же, 1951, 24, № 5, 542;
Danckwerts Р. V., Trans. Faraday Soc., 1950, 46, pt 4,
300, 701; К а ф a p о в В. В , Ж. прикл. химии, 1957, 30, №10,
1449; 1958, 31, № 5, 706; 1960,33,№ 7, 1495; 1961, 34,вып. 1
(совм. с В. С. Муравьевым); Фран к-К аменецкий ДА.,
Диффузия и теплопередача в химической кинетике, М — Л.,
1947. В. В. Кафароо.
МАСС-СПЕКТРАЛЬНЫЕ ПРИБОРЫ — приборы,
предназначенные для анализа химич. и изотопного
состава нейтральных веществ и ионизованных газов,
для исследования структуры молекул, определения
масс ядер и т. д. (см. Масс-спектрометрия). Действие
М.-с. п. основано на разделении ионов исследуемогс
вещества по величинам тУе (отношение массы иона w
к его заряду е) и измерении этих величин и tokoi
разделенных ионов. Как правило, М.-с. п. состоит
из следующих частей: 1) системы подготовки веществ!
57
МАСС-СПЕКТРАЛЬНЫЕ ПРИБОРЫ
58
к введению в масс-спектрометр; 2) ионного источника;
3) разделительного устройства (масс-анализатора);
4) коллекторной системы, на к-рую попадают разде-
ленные ионные пучки; 5) системы вакуумных насосов
с ловушками, обеспечивающей достаточно глубокий
вакуум во всей вакуумной системе масс-спектро-
метра, охватывающей ионный источник, вакуумную
часть разделительного устройства и коллекторную
систему; 6) набора питающих и измерительных уст-
ройств, предназначенных для стабилизированного
питания ионного источника, электрического или (и)
магнитного разделительного устройства, высокочувст-
вительного усилителя (усилителей) для измерения
ионных токов и т. д.
Разделение ионов в М.-с. п. может быть основано
на двух основных принципах: статическом и динами-
ческом. Статич. принцип основан на разделенном или
совмещенном в пространстве воздействии на пучок
ионов статич. полей — электрического и магнитного.
Отклонение заряженной частицы в статич. электрич.
поле является функцией величины тг?2/е, где v —
скорость иона; отклонение иона, движущегося в маг-
нитном поле, является функцией величины mv/e\
поэтому комбинированием этих двух типов полей
можно разделить ионы по величине m/е. Комбинация
осуществляется обычно т. обр., чтобы наряду с разде-
лением ионов происходила фокусировка пучка. Ди-
намич. принцип разделения по величине т/е основан
на том, что ионы, обладающие одинаковой кинетич.
энергией тсг/2, но разной массой, имеют разные ско-
рости и, следовательно, проходят равные расстояния
за разное время. Применение динамич. принципа
позволяет создавать М.-с. п. без магнитов. Вариантом
динамич. принципа является также применяемый
в М.-с. п. циклотронный метод разделения ионов
по массам, в к-ром для такого разделения исполь-
зуется зависимость от массы иона периода его обра-
щения (Т) в постоянном магнитном поле (Я), т. е.
Т = 2лт/еН.
Основной характеристикой разделительного уст-
ройства является его разрешающая сила (способ-
ность). Под разрешающей силой понимается относи-
тельная разница в величине т/е ионов, к-рая может
быть достаточно четко обнаружена прибором. Напр.,
разрешающая сила ’/200 (иногда под тем же назва-
нием употребляется обратная ей величина 200)
означает, что линии однозарядных ионов с массами
199 и 200 могут быть различимы с достаточной чет-
костью. В мировой литературе, а также в системе
стандартов СССР не существует единого определения
того, что следует в данном случае понимать под до-
статочной четкостью. Часто ее определяют, так же
как в оптич. спектроскопии, по ширине максимума
ионного тока, измеренной на его полувысоте. Поэтому
в технич. условиях, относящихся к отечественным
М.-с. и., принято оговаривать способ измерения разре-
шающей силы (напр., разрешающая сила измеряется
на высоте масс-спектроскопич. максимума, равной
5% от шкалы регистрирующего масс-спектр прибора).
Разрешающая сила зависит не только от параметров
разделительного устройства, но и от ряда других
факторов (разброс ионов по энергиям в нек-рых
видах приборов и т. д.). Сообщаемая в описаниях
приборов разрешающая сила обычно является ре-
зультирующей всех этих факторов.
Ионный источник предназначен для перевода
нек-рой части исследуемого вещества в ионизованное
состояние. Он необходим во всех случаях, кроме тех,
когда исследуемая смесь уже находится в ионизован-
ном состоянии (последнее имеет место при изучении
ионного состава электроразрядных плазм, при иссле-
довании ионов, образующихся в пламенах, а также
при изучении состава ионизованной составляющей
в ионосфере с помощью М.-с. п., непосредственно
помещенного на спутнике или ракете). В зависимости
от назначения М.-с. и. в ионных источниках исполь-
зуются след, способы ионизации: ионизация электрон-
ным ударом, фотоионизация, ионизация ионным уда-
ром, ионизация в вакуумном искровом разряде,
термин, ионизация на поверхности нек-рых металлов
или полупроводников, ионизация в очень сильном
поле, ионизация в плазме низкого давления и др.
Основным методом ионизации при газовом анализе
является электронный удар, при изотопном анализе
твердых тел с низкими потенциалами ионизации —
поверхностная термин, ионизация, при анализе труд-
ноиспаряемых веществ со сравнительно высокими по-
тенциалами ионизации — вакуумный искровой раз-
ряд или плазма низкого давления.
Регистрация ионных токов проводится большей
частью электрич. методами; приборы с такой регист-
рацией наз. масс-спектрометрами. Менее
распространен метод регистрации с помощью фото-
эмульсии; приборы этого типа наз. масс-спект-
рографами, они сохранили свое значение только
как один из вариантов М.-с. п. для прецизионного
измерения масс ядер, а также для элементарного ана-
лиза твердых веществ. При фотография, регистрации
масс-спектр образует на пластинке нек-рый набор
черточек-линий, изображающих в «свете» ионов с дан-
ным отношением т/е выходную щель ионного источ-
ника. При электрич. регистрации линиям соответ-
ствуют максимумы ионного тока или, как их часто
называют, пики. Однако и при электрич. регистрации
часто говорят о «линиях» масс-спектра и об их интен-
сивности, т. е. об относительной величине токов, со-
ответствующих высотам пиков, или, когда формы
максимумов в спектре не подобны друг другу, пло-
щадям пиков.
Основной электрич. метод регистрации ионного тока
состоит в приеме разделенных ионов на коллектор,
соединенный с входом электрометрии, усилителя по-
стоянного тока. Во многих случаях с помощью того
или другого метода автоматически осуществляют
последовательный вывод на коллектор ионов разных
масс, и автоматич. потенциометр, расположенный
на выходе усилителя, регистрирует, в результате,
масс-спектр. В случае, когда надо измерять отношение
ионных токов двух масс с очень большой точностью
(напр., при измерении малых вариаций изотопного
состава), применяют двухколлекторные (двухлуче-
вые) масс-спектрометры и компенсационный метод
измерения (лучше всего — при быстром попеременном
напуске анализируемой смеси и эталона). Когда
необходимо иметь особо высокую чувствительность
прибора, т. е. измерять очень малые ионные токи,
для их усиления применяют вторично-электронные
умножители, к-рые позволяют регистрировать ионные
токи, соответствующие десяткам ионов в минуту
(Ц)-1»—10-2° а). Вторично-электронные умножители
применяют также в том случае, когда надо зарегист-
рировать масс-спектр в течение очень короткого про-
межутка времени (вплоть до десятых долей милли-
секунды).
В качестве примера на рисунках приведены уст-
ройства двух распространенных вариантов масс-
спектрометров статич. и динамич. типов. Масс-
спектрометр статич. типа (рис. 1) предназначен для
целей газового анализа; анализируемый газ из на-
пускного баллона 1 через тонкое отверстие в фольге 2
поступает в ионный источник 3, где при давлении
10~5—10”’ мм рт. ст. происходит ионизация элект-
ронами с энергией в несколько десятков электрон-
вольт, летящими с раскаленного катода 4. Получен-
ные ионы вытягиваются из области ионизации сла-
бым электрич. полем, ускоряются и формируются
59
МАСС-СПЕКТРАЛЬНЫЕ ПРИБОРЫ—МАСС-СПЕКТРОМЕТРИЯ
60
системой диафрагм 5 в слабо расходящийся пучок
и движутся в вакуумном пространстве разделитель-
ного устройства 6 при давлении 10_8 мм рт.. ст.
или ниже. Магнит разделительного устройства 7
делит ионы по массам, одновременно фокусируя их
2
3
Рис. 1. Масс-
спектрометр ста -
тического типа:
1 — напускной
баллон; 2 — на-
пускная диаф-
рагма; 3 — ИОН-
НЫЙ источник; 4 —катод; 5 — ди-
афрагмы; 6 — вакуумная камера
масс-анализатора; 7 — магнит масс-
анализатора; 8—сфокусированный
пучок ионов; 9 — щель коллектор-
ской системы; 10 — коллектор ио-
нов; 11 — усилитель ионного тока;
12 — вакуумная ловушка; 13 — ва-
куумные насосы.
в виде острого пучка
8 на щель коллектор-
ной системы 9. Когда
разность потенциалов
ускоряющего ионы
электрич. поля равна
U в, а напряженность
магнитного поля рав-
на И эрстед, на кол-
лектор масс-спектро-
метра попадают ионы
с массой т=4,82-
• \0~szr2H2/U атомных
единиц массы, где
г — радиус централь-
ной траектории при-
бора в см, a z—чис-
ло зарядов иона.
В динамич. масс-
спектрометре (рис. 2)
ионы выталкиваются
из ионного источника в виде узкого пакета, к-рый
по мере дрейфа его в эквипотенциальном про-
странстве делится на пакеты ионов с разными вели-
чинами т/е. Приход этих ионов на коллектор регист-
рируется с помощью вторично-электронного умно-
жителя и катодного осциллографа, на экране к-рого
в результате сразу возникает масс-спектр.
Рис. 2. Масс-спектрометр динамического типа:
1 — напускной баллон; 2 — напускная диафраг-
ма; 3 — ионный источник; I — сетки; 5 — катод,
через который ионы вытягиваются, и области
ионизации; 6 — эквипотенциальное вакуумное
пространство, в к-ром более легкие ионы «дрей-
фуют» быстрее тяжелых; 7 — система сеток; 8 —
вторично электронный умножитель; S — осцилло-
граф; ПИ — неразделенный пакет ионов, только
что выброшенный из ионного источника; Р11И —
разделенные пакеты ионов разной массы.
Основными характеристиками М.-с. п., определяю-
щими возможность и выбор прибора химиком-экспе-
риментатором, являются: 1) тип веществ, для кото-
рых предназначен прибор (он определяется в основ-
ном конструкцией системы напуска и типом ионного
источника); 2) способ разделения ионов по массам:
3) область массовых чисел, на которую рассчитан
М.-с. п.; 4) разрешающая сила прибора; 5) чувстви-
тельность прибора; 6) способ регистрации масс-спек-
тра; 7) наличие или отсутствие автоматической ре-
гистрации спектра в широком диапазоне массовых
чисел.
Приведем в качестве примера характеристики оте-
чественного масс-спектрометра МХ-1304. Этогазоана-
литич. однолучевой масс-спектрометр статич. типа
с однородным магнитным полем, ионизацией электрон-
ным ударом, на область массовых чисел 1—150,
с разрешающей силой 150—300, с автоматич. разверт-
кой спектра в диапазонах 1—12, 12—44, 44—75 или
2—24, 24—88, 88—150, с чувствительностью 5 •10-5%
по аргону. Последнее означает, что в любой газовой
смеси, не содержащей веществ, ионы к-рых совпадали
бы по массам с ионами аргона, прибор может почувст-
вовать 5 -10-5% аргона. В случае же, если в смеси
имеются такие вещества, то чувствительность по ар-
гону может быть ниже. Чувствительность к любому
другому веществу может быть вычислена, исходя
из того, что она, грубо говоря, пропорциональна
числу электронов в молекуле и доле, к-рую представ-
ляет в молекулярном масс-спектре данного вещества
его наиболее интенсивная линия.
В Советском Союзе пром-сть выпускает масс-спект-
рометры для анализа химич. и изотопного состава
вещества. Назначение прибора индексируется с по-
мощью двух букв «МИ» или «МХ» и четырех цифр.
Индекс «МИ» относится к приборам для анализа
изотопного состава и индекс «МХ»— к приборам для
анализа химич. состава. Первая из цифр обозначает
принцип разделения ионов: 1 — прибор с разделением
ионов в однородном магнитном поле; 2 — в неодно-
родном магнитном поле; 3 — резервный; 4 — маг-
питно-динамический; 5 — по времени полета; 6 —
радиочастотный. Вторая цифра обозначает область
применения: 1 — индикаторы; 2 — приборы для про-
изводственного контроля; 3 — приборы для лабора-
торных исследований; 4 — приборы для специальных
условий; последние две цифры обозначают номер
модели этого .типа.
Начало развитию масс-спектроскопии было поло-
жено в 1910 Дж. Дж. Томсоном, в приборе к-рого
разделение по массам производилось с помощью
электростатич. и магнитного полей. Спектр регист-
рировался на фотопластинке. Опыты Томсона при-
вели к открытию изотопов неона, что явилось одним
из основополагающих доказательств существования
изотопов вообще. В 1919 Ф. Астон впервые применил
в масс-спектрографе элементы фокусировки и иссле-
довал изотопич. состав большого числа элементов.
А. Демпстер (1918) построил первый масс-спектрометр.
В. Н. Кондратьев (1922—24) впервые применил
масс-спектрометр для решения химич. задач. В после-
военные годы в Советском Союзе был разработан ряд
оригинальных конструкций масс-спектрометров (Н.И.
Ионов и сотр.— новый тип прибора, основанный
на различии во временах полета ионов; Н. Е. Алек-
сеевский и сотр.— применение неоднородного маг-
нитного поля для создания масс-спектрометра с вы-
сокой разрешающей силой и др.).
Лит.: Р и к Г. Р., Масс-спектроскопия, М., 1953 (готовит-
ся новое издание); Барнард Д ж., Современная масс-
спектрометрия, пер. е англ., М., 1957; Успехи в масс-спектро-
метрии, сб. докладов, М., 1964 (готовится к печати); Пав-
ленко В. А. [ид р. ], Приборы и техн, эксп., 1960, № 6,
89; В е у п о n J. Н., Mass spectrometry and its application to
organic chemistry, Amst. — [a. o.], 1960 (готовится рус.
перевод). В. Л. Тальрозе.
МАСС-СПЕКТРОМЕТРИЯ (масс-спектроскопия,
масс-спектрография) — способ исследования веще-
ства путем определения массы (чаще, отношения
массы к заряду т/е) и относительного количества
ионов, получаемых из исследуемого вещества или уже
присутствующих в изучаемой смеси. Исследования
проводятся с помощью масс-спектралъных приборов.
М.-с. применяется для прецизионного определения
масс ядер, анализа изотопич. и химич. состава ве-
ществ, исследования элементарных химич. реакций,
структуры и энергетики молекул, анализа состава
ионов и нейтральных частиц в электроразрядиых
плазмах, пламенах и ионосфере [см. также Ионы, (в
газах), Изотопы и др.].
Прецизионное определение масс
ядер. Прецизионное определение масс ядер произ-
водится обычно с помощью масс-спектральных при-
боров высокой разрешающей силы. Основным мето-
61
МАСС-СПЕКТРОМЕТРИЯ
62
дом при этом является метод дублетов.
Дублетом в М.-с. наз. пара линий, образуемых ио-
нами, в к-рых отношение числа нуклонов к заряду
одинаково, но состав ядер различен. Величины mie
для компонентов дублета различаются весьма мало —
на доли атомной единицы массы (сокращенно а.е.м.).
Напр., для дублета Н2+—D+ разница в массах состав-
ляет Am = (1,5503:р0,0015) ТО-3 а.е.м.; для дублета
,2С3Н4+— 40Аг+ Ат = (67,93 ±0,07)-10-3 а. е. м.
В методе дублетов измеряется малая величина Am
и, следовательно, очень точно может быть определена
масса одного из ионов, если масса другого известна.
Поочередно подбирая дающие дублет пары ионов,
у одного из к-рых масса известна, и начиная с пары,
включающей атом, масса к-рого принята за стандарт
(1SO~, а с недавнего времени 12С), можно определить
массу практически любого атома. Особенно удобны
для этой цели углеводородные ионы, встречающиеся
в весьма широком диапазоне масс. Наряду с дубле-
тами в ряде случаев используются триплеты, т. е.
три близко расположенные линии масс-спектра.
По точности определения масс ядер масс-спектральный
метод уступает в большинстве случаев в области лег-
ких и средних масс методу ядерных реакций, а так-
же р- и у-спектроскопии, однако в области тяжелых
масс масс-спектральный метод оказывается наиболее
применимым.
Анализ изотопного состава. Ана-
лиз изотопного состава вещества производится обычно
путем измерения отношения токов ионов с одинаковым
зарядом и химич. составом, но с различным изотопным
составом. Эта задача оказывается наиболее простой
в случае одноатомных газов. Напр., для определения
изотопного состава Ne надо ионизовать Ne в ионном
источнике методом электронного удара и, разделив
ионы по массам, измерить отношения токов ионов
2tNe + , 21Ne+ и 22Ne+. При анализе изотопного состава
ряда элементов используются их газообразные сое-
динения, состоящие из двух-, трех- и вообще много-
атомных молекул (Н—в виде Н2, N — в виде N2,
С — в виде СО2 и т. д.). В этом случае приходится
учитывать небольшое влияние изотопного эффекта
на вероятность диссоциации при ионизации, а в случае
молекул, содержащих химически разнородные ато-
мы,— учитывать вклад в интенсивность соответст-
вующих масс-спектральных линий ионов, обуслов-
ленных изотопами других элементов. При ионизации
методом электронного удара веществ, к-рые не могут
быть испарены полностью, а также при использова-
нии метода поверхностной или искровой ионизации
необходимо учитывать поправки на изотопный эф-
фект, связанный с разницей в скоростях испарения
в первом случае и с разницей в вероятностях эмпссии
изотопных ионов в двух других случаях. Изотопные
эффекты наиболее существенны в случае легких
элементов. Для получения точных данных стремятся
применять метод сравнения с пробой строго извест-
ного изотопного состава. Такой метод сравнения при-
меняется также при необходимости определять малые
изменения в изотопном составе. Напр., в случае
определения изотопного состава газообразных ве-
ществ устраивают быстрый попеременный впуск
в масс-спектрометр смеси неизвестного и близкого
к ней известного изотопного состава, непрерывно
регистрируя отношения интенсивностей соответствую-
щих масс-спектральных линий.
В случае анализа водородсодержащих веществ или
их присутствия в смеси наряду с анализируемым
веществом очень вероятно образование вторичных
ионов, представляющих собой по составу первичные
ионы с присоединенным к ним атомом водорода.
Такие ионы возникают в ионном источнике в резуль-
тате ионно-молекулярных реакций типа: Н2.+ Н2 =
= Н + +Н. Эти вторичные ионы могут имитировать
соответствующие изотопные ионы (в приведенном
примере Н* имитирует HD+). Чтобы выявить истин-
ные первичные изотопные ионы, приходится либо
снимать зависимость распределения интенсивностей
в масс-спектре от давления, либо, что дает более
надежные результаты, применять для анализа масс-
спектральный прибор с высокой разрешающей силой,
достаточной для разрешенпя образующегося мульти-
плета (в приведенном примере дублет Н2—HD +
требует разрешающей силы 1/2000).
Масс-спектральный изотопный анализ находит ши-
рокое применение не только в химич. исследованиях,
но и в геологии (определение абс. возраста), биологии
и особенно в атомной технике, где он имеет серьезное
значение как метод производственного контроля.
Исследование элементарных про-
цессов. Элементарные процессы, происходящие
при образовании ионов и возбужденных частиц
и их реакциях с молекулами в ионном источнике
масс-спектрометра, являются вместе с тем важными
элементарными реакциями радиационной химии, хи-
мии высоких темп-p, химии электроразрядной плазмы,
ионосферы и космохимии. Так, напр., исследуя обра-
зование ионов из молекул при электронном ударе,
изучают тем самым одну из основных первичных
элементарных реакций радиационной химии или
электроразряда. Под действием удара электронов
с энергией несколько десятков электрон-вольт, наи-
более часто применяемой в ионных источниках,
происходит как ионизация первичных молекул, так
и диссоциативная ионизация. Напр., при ударе элект-
ронов с энергией, лежащей в диапазоне 4-101—102 эв,
о молекулы метана образуются след, ионы: СН
(47% случаев), СН+ (39%), СН 2 (7%), СН+ (4%),
С + (1,3%). В результате получается характерный
масс-спектр метана, дающий качественную и, веро-
ятно, количественную характеристику набора пер-
вичных ионов, образующихся при радиолизе газо-
образного метана (см. Радиационная химия). Сово-
купность масс-спектральных линий ионов, обра-
зующихся при ионизации молекул индивидуального
вещества с характерным распределением интенсив-
ности, наз. молекулярным масс-спектром данного
вещества.
Исследование структуры и энер-
гетики молекул. При исследовании струк-
туры и энергетики молекул методами М.-с. исполь-
зуются, во-первых, влияние структуры молекул
на распределение интенсивностей в молекулярном
масс-спектре, полученном бомбардировкой электро-
нами с энергией более 40—50 эв, когда это распре-
деление уже мало зависит от энергии электронов.
Во-вторых, с помощью М.-с. определяют критич.
энергию электронов (потенциал появления), при
к-рой в спектре появляется линия соответствующих
ионов. Потенциал появления линии однозарядного
молекулярного недиссоциированного иона в боль-
шинстве случаев равен потенциалу ионизации моле-
кулы. Потенциал появления осколочного иона R *
в результате элементарного процесса R,R2 + е =
= R*+R2+2e равен в общем случае Pr + = Tr,-}-
+ jD(Ri -R2) + £Ri+£r2+^, где D — энергия диссо-
циации молекулы по связи R, — R2, /Rj — потен-
циал ионизации радикала R„ Er * и 7?r2— энергии
возбуждения иона R * и соответственно радикала R2,
Ek— суммарная поступательная энергия R | и R2,
63
МАСС-СПЕКТРОМЕТРИЯ
64
получаемая ими в акте диссоциативной ионизации.
В ряде случаев удается в независимых опытах изме-
рить часть входящих в эту сумму слагаемых и ис-
пользовать измерение потенциала появления для из-
мерения других слагаемых, гл. обр. энергии разрыва
связи Щ—В2.
Для определения критич. потенциалов находят
все большее применение методы фотоионизации и
ионного удара. Для теоретич. описания процесса
диссоциативной ионизации двухатомных молекул
удается использовать метод потенциальных кривых
в соединении с принципом Франка — Кондона [см.
Фотохимия, Ионы, (в газах)]. Количественной теории
диссоциативной ионизации (и. следовательно, теории
масс-спектров) для многоатомных молекул не сущест-
вует. Один из теоретич. подходов предполагает, что
процесс диссоциации происходит значительно позднее
процесса ионизации, после того как энергия возбуж-
дения успела квазиравновесно распространиться по
степеням свободы молекулярного иона. С помощью
этого подхода удалось полуэмпирич. путем рассчи-
тать молекулярные масс-спектры нек-рых веществ
и получить удовлетворительное согласие с экспери-
ментальными данными. Однако для многих случаев,
особенно для длинных молекул, как показывают
опыты, идея квазиравновесного распределения энер-
гии возбуждения не находит подтверждения.
Анализ химического состава
смесей. М.-с. дает возможность проводить анализ
молекулярного состава смесей и в нек-рых случаях
элементарный анализ. При молекулярном масс-
спектральном анализе анализируемую газообразную
смесь пропускают (обычно в виде непрерывного по-
тока) через ионный источник масс-спектрометра таким
образом, чтобы наименьшая доля продуктов разло-
жения анализируемых веществ на раскаленном катоде
могла попасть в зону ионизации и исказить истинный
состав анализируемой смеси. Качественный молеку-
лярный масс-спектральный анализ основан либо на
измерении массы недиссоциированного молекуляр-
ного иона, либо на характеристичности распреде-
ления интенсивности между линиями в спектре каж-
дого индивидуального вещества. Степень характерис-
тичности такова, что она позволяет в случае индиви-
дуальных веществ различать между собой практи-
чески любые химич. соединения и во многих случаях
изомеры. На рисунке в качестве примера приведены
масс-спектры н-бутана и изобутана. Наглядно видны
различия в соотношениях интенсивностей линий этих
масс-спектров.
Основным способом ионизации при молекулярном
анализе является ионизация электронами с энергией
в несколько десятков электрон-вольт. Для получения
больших различий между масс-спектрами уменьшают
степень диссоциации при ионизации и тем самым
снижают число осколочных ионов или полностью
исключают их путем применения ионизации электро-
нами низкой энергии (7—15 эв), фотоионизации,
ионизации ионным ударом, ионизации в сильном
электрич. поле. Из этих усовершенствованных методов
ионизации достаточное практич. распространение
получил пока только удар электронами малой энер-
гии; применение других методов ведет к значительным
технич. усложнениям аппаратуры. Количественный
молекулярный масс-спектральный анализ основан
на пропорциональности интенсивности всех линий
масс-спектра каждого из веществ его парциальному
давлению в области ионизации. Суммарный масс-
спектр смеси является поэтому аддитивным наложе-
нием масс-спектров каждого из компонентов смеси.
Для того чтобы состав смеси в области ионизации
не отличался от состава исходной смеси, стремятся
обеспечить молекулярное (кнудсеновское) натекание
из объема, в к-ром содержится газообразная анали-
зируемая смесь (напускной объем), в ионный источник.
Во избежание ошибок, связанных с эффектами разде-
ления при испарении смеси, стремятся везде, где это
возможно, чтобы проба, введенная в напускной объем,
была бы до начала анализа полностью испарена.
Изо-Бутан
58
57
I 42 40 38 29 27 15 ”.
43 41 39 37 28 28 14 <
Для веществ с низким давлением пара напускной
объем делают поэтому подогреваемым. Градуировки
при масс-спектральном молекулярном анализе в об-
щем случае состоят из снятия масс-спектров инди-
видуальных компонентов смеси и относительных или
абсолютных коэфф, чувствительности масс-спектро-
метра к данному веществу. Под абсолютным коэфф,
чувствительности понимают обычно отношение ин-
тенсивности либо всего масс-спектра индивидуального
вещества, либо линии, принятой за эталонную,
к количеству этого вещества в напускном объеме при
стандартном режиме работы масс-спектрометра. За
относительную чувствительность принимают отноше-
ние коэфф, абсолютной чувствительности для двух
данных веществ.
Распределения интенсивностей в масс-спектрах
индивидуальных веществ, снятые с помощью разных
масс-спектрометров при соответствующих стандарт-
ных условиях (темп-ра ионного источника, энергия
электронов, условия развертки спектра) и приводи-
мые в научных статьях или каталогах, различаются
в большинстве случаев на несколько относительных
процентов и в редких случаях на 10% и более. Отно-
сительные чувствительности для одного прибора
меняются с течением времени при условии регулярной
чистки прибора в пределах нескольких процентов.
Абс. чувствительности колеблются часто значитель-
но больше. Разработаны методы, позволяющие ис-
ключать большую или меньшую часть градуировок
(метод полной ионизации и др.). Молекулярный масс-
спектральный анализ получил значительное рас-
пространение в органич. химии. Он охватывает смеси
практически всех классов органич. веществ. Значи-
тельное распространение он получил в нефтехимии,
65
МАСС-СПЕКТРОМЕТРИЯ —МЕДИ ГАЛОГЕНИДЫ
66
где с его помощью анализируются смеси углеводородов
до Cso. Основные методич. затруднения доставляют
при масс-спектральном анализе сильно адсорбирую-
щиеся полярные вещества (явление «памяти» прибора),
а также вещества, обладающие особенно сильным
агрессивным действием к нек-рым деталям вакуумной
части прибора (напр., нек-рые фторсодержащие).
Чувствительность масс-спектрометра зависит от сте-
пени различия масс-спектров компонентов смеси
и совершенства прибора. В лучших случаях она
достигает 10~’—10_9%, в большинстве случаев лежит
в интервале 10-4—1%. При условии прямого соеди-
нения масс-спектрометра с химич. реактором моле-
кулярный масс-спектральный анализ позволяет опре-
делить короткоживущие активные частицы, свободные
радикалы и атомы, в реагирующих смесях при давле-
ниях до нескольких десятков мм рт. ст. При этом
должны быть предусмотрены меры предотвращения
гибели активных частиц до их попадания в область
ионизации (стеклянные напускные диафрагмы, тех-
ника молекулярных пучков). Эффективность М.-с.
как метода молекулярного анализа резко возрастает
при его комбинации с другими методами, особенно
хроматографией. Особенно эффективным является
непосредственное присоединение масс-спектрометра
в качестве датчика к выходу газового хроматографа.
Молекулярный масс-спектральный анализ находит
нек-рое применение как метод производственного
контроля в химич. и нефтяной пром-сти. Присоеди-
нение масс-спектрометра к коммуникациям и аппара-
там позволяет осуществлять непрерывный контроль
и в нек-рых случаях автоматич. управление произ-
водственным процессом. Элементарный масс-спект-
ральный химич. анализ применяется для исследования
многих типов твердых веществ, в первую очередь,
металлич. сплавов и полупроводников. В связи с ма-
лой летучестью большинства таких веществ их одно-
временное испарение и ионизация осуществляются
в вакуумном искровом разряде, что приводит к не-
которому усложнению прибора в целом. Измерение
состава пара над твердым телом с помощью М.-с.
находит важное применение как метод измерения
теплот испарения и нек-рых других характеристик
твердых тел.
Исследование верхних слоев ат-
мосферы, электрического газового
разряда и ионизации в пламенах.
М.-с. является важнейшим методом для прямого
анализа состава нейтральных и заряженных частиц
в верхних слоях атмосферы. Для этого на спутниках
и ракетах устанавливался один из вариантов («радио-
частотный») динамич. масс-спектрометров, к-рый бла-
годаря отсутствию магнита, ненужности в верхних
слоях атмосферы откачной системы и умеренным тре-
бованиям к разрешающей силе и чувствительности
прибора удается сделать очень компактным.
Масс-спектральное исследование газового разряда
и ионов, образующихся в пламенах, позволило выя-
вить целый ряд важных черт механизма происходя-
щих процессов и, в частности, установить исключи-
тельно важную роль в них «утяжеленных» и «соль-
ватированных» ионов.
Лит.: Р и к Г. Р., Масс-спектроскопия, М., 1953 (гото-
вится новое издание); Барнард Д ж., Современная масс-
спектрометрия, пер. с англ., М., 1957; Успехи в масс-спектро-
метрии, сб. докладов, М., 1964 (готовится к печати); Таль-
розе В. Л., в сб.: Сессия Академии наук СССР по на-
учным проблемам автоматизации производства. 15—20 октя-
бря 1956 г. Научные основы построения технических средств
автоматики, М., 1957 (Труды, т. 3); Т а л ь р о з е В. Л. [и
др.], веб.: Органический анализ. М., 1963 (Труды комис-
сии по аналитической химии, т. 13), с. 456; Истомин
В. Г., веб.: Искусственные спутники Земли, вып. 3, М., 1959;
В е у и on J. Н., Mass spectrometry and its application to
organic chemistry, Amst.—[a. o.], 1960 (готовится рус. пер.),
В. Л. Тальрозе.
сн2он
Сн2 но сн3
с—он ____
СН^з ~~
срон
I л
МЕВАЛОНОВАЯ КИСЛОТА (3-метил-3,5-диокси-
валериановая кислота) CeHI204 (I), мол. в. 148,16;
существует в равновесии с ее
лактоном CeHI0Os(II)—кри-
сталлы, т. пл. 27—28°. М. к.
хорошо растворима в воде и
органич. растворителях; об-
разует производные: бензги-
дриламид (III)— кристаллы,
т. пл. 92—93°, [а]д= — 2°;
N, Nдибензилэтилендиаммо-
ниевая соль М. к. (IV) — кристаллы, т. пл. 125—
126°, хорошо растворима в воде. Количественно М. к.
определяют микробиологич. методом по ее действию
как ростового фактора для мутанта Lactobacillus
aciolofilus, а также по цветной реакции ее гидро-
ксамата с Fe’+.
СН2ОНСН2С (ОН) CH2CONHCH(CaH5);
сн3
III
[CH2OH(CH2)2C(OH)CH2COO-]2-CeH5CH2NH2(CH2)2NHaCH2CeH,
сн3
IV
Физиологически активная форма М. к. является
(+)-изомером М. к.— промежуточный продукт при
биосинтезе соединений изопреноидной структуры
(стеринов, каротиноидов, терпенов, каучуков и др.).
Синтетически рацемич. М. к. может быть получена по
Реформатского реакции'.
Zn
СН,СООСН2СН2СОСН3 + ВгСН2СООС2Н, —>
--> СН3СООСН2СН2С (СН3) CH2COOC2HS —> (I) (II)
он
М. к. открыта в 1956; выделена из сухой барды, куль-
туральной жидкости Aspergillus oryzoe и корнеплодов
(морковь).
Лит.: Труды V Международного биохимического конгрес-
са. Москва, 10—16 авг. 1961, т. 7 — Биосинтез липидов, М.,
1962; Pop j a k G-., CornforthJ. W., Advances ih enzymo-
logy and related subjects of biochemistry, 1960, 22; Wagner
A. F., там же. 1961, 23. Г. И. Самохвалов.
МЕДИ ГАЛОГЕНИДЫ — соединения одно- и двух-
валентной меди с галогенами, СиХ и СиХ, (где X—F,
Cl, Br, J).
Все галогениды одновалентной
меди кристаллизуются в кубич. системе по типу
цинковой обманки; периоды решетки для Си F, CuCl,
CuBr, CuJ соответственно равны: 4,264 А; 5,418 А;
5,961 А; 6,059 А. Плотности их составляют: 7,07;
4,13; 5,12; 5,67. Хлорид, бромид и иодид одновалент-
ной меди бесцветны, плавятся соответственно при
430°, 500°, 605°, заметно летучи и кипят без разло-
жения. Их теплоты образования — ДЯ°98 (ккал/моль
соответственно равны: 32,2; 25,1; 16,2. CuCl, CuBr
и CuJ трудно растворимы в воде (соответственно
1,1-10-3, 2-10-4, 2-10-6 моль/л). Несколько особ-
няком стоит CuF, отличающийся красной окраской
и более высокой т. пл. (908°). Из галогенидов закисной
меди фторид наименее устойчив. Так, он мгновенно
разлагается водой, легко восстанавливается водоро-
дом (следует отметить, что в последнее время ста-
вится под сомнение само существование этого соеди-
нения). Способность раствора CuCl в конц. НС1 аб-
сорбировать на холоду СО с образованием коорди-
национного соединения CuCICO используется в газо-
вом анализе (при нагревании СО вновь выделяется).
Из галогенидов 2-в алентной меди
известны CuF2, CuC12, CuBr2. Их теплоты образования
—АЯ208 (в ккал/молъ) соответственно равны: 126,9;
49,2; 33,2. Наиболее устойчивым из этих соединений
О3 к.х.э. т. з
67
МЕДИ КАРБОНАТЫ—МЕДИ ОКИСЛЫ
68
является CuF2 (плавится при 950° с разложением:
CuF2-* CuF-P/jFj). Хлорид CuCL, распадается на
CuCl и '/filz ок. 500°. Бромид СиВг2 диссоциирует при
более низкой темп-ре, CuJ2 разлагается уже при об-
разовании (ДЯ°08= — 1,7 ккал/моль). CuF2— бес-
цветные кубич. кристаллы, а = 5,417 А, плотность
4,23. СнС12— темно-коричневые моноклинные кри-
сталлы, а=6,70 А, 6=3,30 А, с=6,67 А, р=118°23';
плотность 3,44.
CuBr2— черные моноклинные кристаллы, а=
=7,14 А, 6=3,46 А, с=7,02 А, Р=119°О5'. CuF2
незначительно растворим в холодной воде (4,7 г
в 100 г Н2О). Горячей водой разлагается с образова-
нием CuOHF. Напротив, СиС12 и СпВг2 хорошо рас-
творимы в воде и расплываются на воздухе. Раствори-
мость СнВг2 составляет 122 г в 100 г Н2О (15°). Рас-
творимость СиС12 в г на 100 г Н2О: 39,8 (—40°); 40,9
(0°); 42,7 (20°); 53,7 (100°). Из растворов галогениды
2-валентной меди выделяются в виде кристалло-
гидратов СнХ2-2Н2О и др. В отличие от дигидратов
фторида (голубого) и бромида (коричнево-зеленого
цвета), СиС12-2Н2О при прибавлении незначительных
количеств воды меняет свою окраску от темно-корич-
невой через зеленую до голубой. При прибавлении
конц. НС1 даже сильно разбавленные р-ры вновь
становятся зелеными. Конц, р-ры СнВг2 и CuCL, спо-
собны присоединять значительные количества окиси
азота с образованием [Cu(NO)X3]_.
Из галогенов двухвалентной меди небольшое при-
менение находит СиС12, используемая как протрава
при крашении, катализатор, в медицине. В промыш-
ленности ее получают растворением СнО или СиСО3
в НС1 или обменной реакцией ВаС12 с медным ку-
поросом. А- В. Ванюкое.
МЕДИ КАРБОНАТЫ — медные соли угольной
кислоты. Простой нейтральный карбонат медп до сих
пор не удалось получить. Хорошо известны основные
карбонаты меди. Они часто встречаются в природе.
Широко распространены изумрудный малахит состава
СнСО, -Си(ОН)2 и голубой азурит 2СнСО3 -Си(ОН)2,
кристаллизующиеся в моноклинной системе. В виде
особенно красивых кристаллов азурит найден в Чесси
(близ Лиона) и поэтому называется чессилитом. Дей-
ствием Na2CO3 на CuS04 в водном р-ре получается
основной карбонат состава СнСОз -Си(ОН)2 -0,5Н20.
При нагревании он переходит в малахит. Все кар-
бонаты меди очень неустойчивы и при температуре
выше 200° разлагаются на СнО, СО2 и Н2О. Так же
легко они восстанавливаются до металлич. меди.
Карбонат меди способен образовывать двойные соли;
в частности, К2СОз -СнСОз может кристаллизоваться
как в безводном состоянии, так и с 1 или 4 молекулами
воды. Благодаря образованию этой двойной соли
осадок основных карбонатов можно перевести в р-р
большим избытком щелочного карбоната. В аммиаке
карбонаты меди растворяются только в присутствии
солей аммония. Азурит применяется под названием
медной лазури в качестве краски, а также в пиро-
технике; малахит — для изготовления поделок и юве-
лирных изделий. А. В. Ванюкое.
МЕДИ НИТРАТ (азотнокислая медь)
Си(ХОз)з— белые (чуть зеленоватые) кристаллы.
Теплота образования АЯ298 = —73,4 ккал/моль. Без-
водная Си(ИОз)2 может быть получена из конц.
азотнокислых р-ров. Чаще всего из р-ров выделяется
Си(И0з)2-6Н2О — синие кристаллы с плотн. 2,07, при
обычной темп-ре расплывающиеся на воздухе, а при
26,4° плавящиеся с потерей 3 молекул Н2О. Более
устойчива при обычной темп-ре Сн(ИОз)2-ЗН2О —
темно-голубые кристаллы плотн. 2,32; т. пл. 114,5°.
Cu(NOa)2 легко растворяется в воде и спирте. Рас-
творимость безводной соли в 100 г Н2О: 52,2 (—20°),
81,8(0°), 125,2(20°). При добавлении щелочи к р-ру М.н.
кристаллизуется основной нитрат Сн(ХОз)2-ЗСн(ОН)а
в виде зеленовато-голубых кристаллов с моноклин-
ной решеткой, нерастворимых в воде, но легко рас-
творимых в кислотах. Основной нитрат встречается
в природе в виде минерала герхардтита. При нагре-
вании Cu(NOa)2 разлагается на CuO, NO2 и О2, что
служит одним из способов получения СнО. Нитрат-
меди используется для приготовления красок.
Нитрат одновалентной меди существует только
в р-рах и в свободном состоянии не выделен. Однако
известно его устойчивое комплексное соединение с
тиомочевиной состава Cu[CS(NH2)2]2NO3.
А. В. Ванюков.
МЕДИ ОКИСЛЫ — соединения меди с кислородом:
закись Сн2О, окись СнО, перекись СнО2 и Сн2Оа
(см. Медь). Перекись меди—неустойчивое соедине-
ние и полностью разлагается даже в атмосфере кисло-
рода при 100—150°. Наибольшее значение имеют
первые два окисла. Окись меди СнО встре-
чается в природе в виде минерала тенорита или ме-
лаконита (черного цвета), являющегося продуктом
выветривания сульфидных минералов. СнО имеет
моноклинную решетку (а = 5,108 А; b = 3,410 А;
с = 4,653 А; Р = 99°29'); плотность 6,45. Теплота,
образования А76298 = —37,1 ккал/моль. СнО — не-
устойчивое соединение, заметно разлагается начиная
с 800°, в атмосфере чистого кислорода полностью
разлагается при 1100°. Уже при 150° СнО начинает
восстанавливаться водородом, при 450° восстановле-
ние идет быстро и до конца. Примерно так же легко
восстанавливается СнО окисью углерода. При 600 —
700° CuO взаимодействует с кремневой кислотой,
образуя моносиликат меди. При прокаливании СнО
с Fe20a (800—900°) образуется феррит, устойчивый
ниже 1000—1100°. Феррит меди относится к числу
легко восстановимых соединений. CuO хорошо рас-
творяется в цианидах, в конц. и разб. к-тах. Последнее
используется в гидрометаллургии меди. В чистом
аммиаке CuO практически нерастворима. Однако рас-
творимость ее в аммиачных растворах резко воз-
растает по мере увеличения в растворе концентрации
связанного аммиака.
CuO легко вступает во взаимодействие с р-рами солей
3-валентного железа с образованием соответствующих
солей 2-валентной меди и Ее(ОН)з. Окись меди рас-
творяется также в горячих конц. р-рах едких щелочей,
но слабые растворы щелочей действуют на нее незна-
чительно. При взаимодействии солей 2-валентной
меди со щелочами выпадает Си(ОН)2 в виде объе-
мистого голубого осадка. Обычно он получается
в виде геля, к-рый высыхает в порошок, а при нагре-
вании переходит в CuO. Гидроокись Си(ОН)2 можно
получить и в кристаллич. форме. Свежевыпавший
Си(ОН)2 заметно растворим в щелочах, однако его
кислотный характер выражен очень слабо. В водном
аммиаке образует раствор [Си(МНз)4](ОН)2 синего
цвета, способный растворять целлюлозу. При раз-
бавлении или подкислении раствора целлюлоза вновь
выпадает в осадок, это свойство находит применение
при производстве искусственного шелка. Окись меди
может быть получена нагреванием меди на воздухе
до красного каления. Она применяется в стекольной
и эмалевой пром-сти в качестве зеленого и голубого
красителей, а также для получения рубинового стекла;
используется как деполяризатор в элементах, как
окислитель в органич. элементарном анализе.
Закись меди встречается в природе в виде-
минерала куприта (печеночно-красного цвета). Си2О
имеет кубич. решетку (а = 4,261 А); плотность 5,8—
6,11. Теплота образования АЯ298 = —39,84 ккал/моль.
Плавится при 1230°. Давление диссоциации при
69
МЕДИ СПЛАВЫ
70
1260° составляет 12 мм рт. ст. Ниже 375° Си2О
неустойчива и разлагается на СиО и Си. При нагре-
вании на воздухе устойчива лишь выше 1070°. Ниже
этой темп-ры начинается ее окисление в окись. При
темп-ре 800° и продолжительном прокаливании пере-
ходит в СиО почти полностью. С РегОз закись меди
образует феррит, устойчивый выше 1100°. Известны
также силикаты закиси меди, образующиеся из соот-
ветствующих окислов при 600° и плавящиеся при
1055—1200°. Так же как и окись меди, Си2О легко
восстанавливается водородом и окисью углерода.
В разб. H2SO4 СигО растворяется полностью только
в присутствии кислорода. Без доступа воздуха в рас-
твор переходит лишь половина меди. В конц. H2SO4
закись меди растворяется с выделением SO2.
Закись меди может быть получена прокаливанием
меди при недостатке воздуха, а также действием
щелочи и восстановителя (гидразин, гидроксиламин,
виноградный сахар) на р-р сульфата меди. При полу-
чении чистой Си2О применяют электролиз в растворе
хлоридов щелочных металлов с использованием мед-
ного анода. СигО применяется для окрашивания
стекла, эмалей и для борьбы с вредителями с. х-ва.
Интересным является использование ее в паре с ме-
таллич. медью для изготовления «купроксных» вы-
прямителей Переменного тока. А. В. Ванюков.
МЕДИ СПЛАВЫ — сплавы на основе Си, содер-
жащие Zn, Sn, Al, Ni, Fe, Mn, Si, Be, Cr, Pb, P и др.
легирующие элементы (в сумме до 50%). В двухком-
понентных М. с. легирующий элемент входит в твер-
дый р-р замещения на основе Си (обычно обозначаемый
а), а также может образовывать электронные соеди-
нения, характеризующиеся определенной электрон-
ной концентрацией (отношением суммарного числа
валентных электронов к числу атомов, к-рое может
быть равно 3/2, 2I/1S или 7/4); этим соединениям условно
приписывают формулы CuZn, CusSn, Cu3iSn8, CU3AI,
Си^АЦ, CuBe и др. (см. Металлические соединения).
В многокомпонентных М. с. часто присутствуют слож-
ные металлич. соединения точно неустановленного
состава. Легирующие элементы вводят в Си для повы-
шения прочности и твердости, улучшения антифрик-
ционных свойств и стойкости против коррозии и для
получения сплавов с заданными физич. свойствами
(уд. электросопротивлением, термоэдс и пр.). М. с.
делят на латуни, бронзы и медно-никелевые сплавы.
Латуни — М. с., в к-рых главным легирующим
элементом является Zn. Структура двойных латуней,
не содержащих, кроме Zn, других добавок, состоит из
а-раствора на основе Си или из двух фаз (а+Р),
где [5-фаза (условно CuZn) — электронное соеди-
нение, характеризующееся электронной концентра-
цией ’/2. Из двойных а-латуней наиболее распрост-
ранены сплавы с 96,90 и 80% Си (томпаки) и патронная
латунь с 68—70% Си, а из (а+Р)-латуней — сплавы
с 62 и 59% Си. Многокомпонентные (специальные)
латуни, кроме Zn, содержат Al, Fe, Mn, Ni, Si, Sn,
Pb и др. элементы (табл. 1).
Таблица 1. Составы, свойства и назначение латуней
Марка сплава Содержание легирующего элемента, % Типичные механич. свойства3 Примерное назначение
Си Zn А1 Fe мп прочие аь, кГ/мм2 б, % НВ, кГ/мм2
Л96 Л90 95,0-97,0 88,0-91,0 — Д в о й п ы е лат) н и 24 26 50 45 111 53 Радиаторные трубки Листы и ленты для плакировки
Л80 Л70 79,0—81,0 69,0-72.0 0) о д д 03 — — 32 32 52 55 54 Листы, ленты и проволока Полосы и ленты специального назначения
Л62 60,5—63,5 О * 33 49 56 Полосы, листы, ленты, трубы, прутки и прово- лока
Специальные латуни
ЛА 85-0,5 . . . ЛА 77-2 .... 84-86 76,0—79,0 0,4—0,7 1,75—2,50 — — — 30 40 60 55 54 60 Заменитель золо- та при изготов- лении знаков отличия Трубы конденса- торные
ЛАЖ 60-1-1 . . 58,0-61,0 0,75—1,50 0,75—1,50 0,1—0,6 45 45 95 Трубы и прутки
ЛАЖМц 66-6-3-2 64—68 0) Q 6-7 2,0—4,0 1,5-2, 5 65 7 160 Литые массивные червячные вин- ты, гайки на- жимных винтов
ЛАН 59-3-2 . . 57,0—60,0 К 2,50—3,50 — 2,0—3,0 N1 38 50 75 Трубы и прутки
ЛЖМц 59-1-1 . 57,0—60,0 Л я о 0,1-0,2 0,6—1,2 0,5-0,8 0,3—0,7 Sn 45 50 88 Полосы, проволо- ка, прутки и трубы
ЛН 65-5 .... 64,0-67,0 О — — — 5 ,0-6,5 N1 40 65 70 Трубы маномет-
ЛО 70-1 .... 69,0-71,0 — — 1,0—1,5 Sn 35 60 59 Трубы
ЛС 74-3 .... 72,0-75,0 — — — 2,4-3,0 Pb 35 50 57 Полосы, ленты, прутки для ча- сового произ-ва
ЛК 80-3 .... 79,0-81,0 — — — 2,5-4,0 Si 30 58 60 Поковки п штам- повки
ЛКС 80-3-3 . . . 79—81 — — 2—4 Pb 2,5-4,5 SI 35 20 95 Литые подшипни- ки и втулки
а Свойства латуней, обрабатываемых давлением, указаны для рекристаллизованного состояния (отжиг при 600°).
ав — предел прочности при растяжении, 6 —относительное удлинение, НВ—твердость по Бринеллю,
71
МЕДИ СПЛАВЫ
72
Все М. с., исключая латуни и сплавы с большим
содержанием никеля, называют бронзами. По
основному легирующему элементу бронзы подразде-
ляют на оловянные, алюминиевые, кремнистые, мар-
ганцовые, бериллиевые, хромовые, свинцовые и др.
(табл. 2).
бронзы. Алюминиевые бронзы широко применяют
как в литом, так и в деформированном состояниях.
Структура однофазных алюминиевых бронз состоит
из кристаллитов а-раствора А1 в Си, а двухфазных —
из a-раствора и эвтектоидной смеси («+у), где у —
электронное соединение с электронной концентра-
Таблица 2. Составы, свойства л назначение некоторых бронз и медноникелевых сплавов
Название и марк а сплава Содержание легирующих элементов Типичные механич. свойства3 Примерное назначение
ab, кГ/лш2 6, % нв, к Г/мм2
Оловянные бронзы
Бр.010 9 - 11 % Sn 25 10 80 Сложное фасонное литье, армату-
Бр.ОФ 10-1 9-11% Sn; 0,8-1,2% Р 25 3 90 Р Литые подшипники, шестерни, венцы
Бр.ОФ 4-0,25 3,5-4,0% Sn; 0,25% Р 34 52 60 Трубки для манометрия, пружин
Бр.ОЦС 6-6-3 5,0—7,0% Sn; 5,0—7,0% Zn; 2,0-4,0% Pb 17 10 60 Литые антифрикционные детали
Бр.ОЦСН 3-7-5-1 Алюминиевые бронзы 2,5—4,5% Sn; 6,0-9,5% Zn; 3,0—6,0% Pb; 0,5-1,5% Ni 18 8 60 Литая арматура, работающая в морской и пресной воде и атмо- сфере пара до 25 ат
Бр.А7 6-8% Al 42 70 70 Ленты, полосы для пружинящих деталей
Бр.АЖ 9-4б 8—10% Al; 2—4% Fe 60 40 НО Прутки, поковки, ответственное литье
Бр.АЖМц 10-3-1,5 9—11 % Al; 2—4%Fe; 1—2%Mn 61 32 130 Прутки и толстостенные трубы для изготовления втулок, шестерен, подшипников, ответственное литье
Бр.АЖН 10-4-4 Свинцовая бронза 9,5-11,0% Al; 3,5-5,5% Fe; 3,5—5,5% NI 60 35 150 Седла клапанов, втулки, шестер- ни, ответственное литье
Бр.С 30 Кремненикелевая б р о н- 27-33% Pb 7,6 5 25 Литые вкладыши подшипников
за Бр.КН 1-3 Бериллиевая бронза 0,6-1,1% Si; 2,4-3,4% Ni; 0,1—0,4% Mn 60 12 180 Прутки, направляющие втулки
Бр.Б2в 1.9—2,2% Be; 0.2—0,5% Ni 135 1,5 350 Пружины и пружинящие детали ответственного назначения
Мельхиор МНЮ 18,0-20,0% (Nl+Co) 35 35 70 Медицинский инструмент, детали точной механики, изделия широ- кого потребления
Мельхиор МНЖМц 30-0,8-1 . . . 29,0—33,0% (Ni + Co); 0,6—1,0% Fe; 0,8—1,3% Mn 38 45 70 Трубы для конденсаторов
Нейзильбер МНЦ 15-20 13,5—16,5% (Ni + Co); 18,0—22,0% Zn 40 45 70 детали приборов точной механи- ки, технич. посуда, художест- венные изделия и изделия широ- кого потребления
°Ь — предел прочности при растяжении, 6 — относительное удлинение, НВ — твердость по Бринеллю.
’ а Свойства деформируемых сплавов указаны для мягкого (рекристаллизованного) состояния. ® Свойства указаны для
прутков. Свойства указаны после старения.
Оловянная бронза — самый древний
сплав (бронзовый век). Структура оловянных бронз
состоит из «-раствора Sn в Си и эвтектоидной смеси
(a-|-Cu3jSng). Фосфор, вводимый в бронзы для рас-
кисления и улучшения антифрикционных снойстн,
образует включения фосфида СизР. Благодаря хоро-
шим литейным, механич. и антифрикционным свой-
ствам, стойкости против коррозии на воздухе и в мор-
ской воде и красивому внешнему виду сплавы Си с Sn
(часто с добавками Zn, Pb, Ni и Р) на протяжении
столетий занимали ведущее место в разнообразных
отраслях произ-ва (машинная или пушечная бронза
с 10% Sn, художественная бронза с 5% Sn, колоколь-
ная бронза с 20% Sn). В настоящее время применение
оловянных бронз в машиностроении все более сокра-
щается из-за дефицитности олова и в связи с тем, что
они уступают по нек-рым свойствам безоловянным
бронзам.
Алюминиевые бронзы с 5—10% А1
и добавками Fe, Мп, Ni и др.— наиболее распрост-
раненные безоловянные М. с. По прочности и стой-
кости против коррозии они превосходят оловянные
цией 21/'i3. Добавка Fe образует включения интер-
металлида (возможно, FeAls), измельчает зерно, по-
вышает прочность и улучшает антифрикционные
свойства.
Подавляющее большинство М. с. не упрочняется
закалкой и старением. Сравнительно малочисленная,
но очень важная группа М. с. подвергается закалке
и упрочняющему отпуску (старению). Характерным
представителем этой группы являются берил-
лиевые бронзы. Сплав Си с 2% Be закали-
вают в воде с 780°; при этом образуется пересыщенный
твердый раствор Be в Си. Закаленный сплав стареет
при 320° в течение двух часов. При старении проте-
кает частичный распад пересыщенного раствора и вы-
деляются в дисперсном виде частицы электронного
соединения СиВе; в результате происходит сильное
дисперсионное твердение, повышающее твердость в 3
раза. Подвергнутая старению бериллиевая бронза
по прочности не уступает лучшим сортам легирован-
ной стали. Сочетание высокой прочности, твердости,
сопротивляемости усталости, стойкости против кор-
розии, теплопроводности, электропроводности и оса-
73
МЕДИ СПЛАВЫ—МЕДИ СУЛЬФИДЫ
74
бенно очень высокого предела упругости делает бе-
риллиевые бронзы незаменимыми в произ-ве разного
рода пружин, мембран, электрич. контактов и других
ответственных изделий. Инструмент из бериллиевой
бронзы не искрит при ударах. К М.с., упрочняемым
термин, обработкой, относятся также кремпеникеле-
вые, хромовые и нек-рые другие бронзы.
В группу медноникелевых сплавов
входят такие М. с., в к-рых Ni является главным
легирующим элементом, оказывающим решающее
влияние на свойства. Медноникелевые сплавы под-
разделяют на конструкционные и электротехнические.
К конструкционным сплавам относятся мельхиоры
и нейзильберы (табл. 2). Мельхиоры содержат
20—30% Ni и часто дополнительно легируются Fe
и Мп. Они отличаются очень высокой стойкостью
против коррозии в морской воде и парах воды, что
сочетается с хорошими механич. свойствами; поэтому
их широко применяют в кораблестроении, в част-
ности для конденсаторных труб, работающих при
повышенных темп-рах и давлениях. Нейзиль-
беры относятся к тройной системе Си—Ni—Zn
и содержат 5—35% Ni и 13—45% Zn. Они имеют
красивый серебристый цвет и высокую стойкость
против коррозии. К электротехнич. медноникелевым
сплавам относятся константан (40% Ni, 1,5%
Мп) и манганин (3% Ni, 12% Мп), обладающие
низкими температурными коэфф, электросопротив-
ления, и термопарный сплав копель (43% Ni,
0,5% Мп), дающий большую термоэдс в паре с хро-
мелем, а также нек-рые другие сплавы.
Механич. свойства М. с. изменяются в широких
пределах при холодной обработке давлением и при
отжиге. Холодной деформацией можно увеличить
твердость и предел прочности М. с. в 1,5—3 раза
при одновременном снижении пластичности, а после-
дующий отжиг — рекристаллизация — позволяет ча-
стично или полностью восстановить исходные (до
деформации) свойства (см. Термическая обработка
металлов). Смягчающий отжиг латуней и бронз после
холодной обработки давлением проводят при 600—
700°.
Все М. с. отличаются хорошей стойкостью против
атмосферной коррозии. Кислород при комнатной
темп-ре не действует на М.с. Окись углерода не реа-
гирует с М. с. Сухой аммиак при комнатной темп-ре
не реагирует с М.с., но в присутствии паров воды вызы-
вает сильную коррозию. Незагрязненный пар, сухой
или влажный, действует на бронзы очень слабо. Серо-
водород уже при незначительной влажности и особен-
но при повышенных темп-рах сильно реагирует с М.с.
Сернистый ангидрид во влажной атмосфере и в элект-
ролитах вызывает сильную коррозию бронз и латуней.
Галогены и их водородные соединения в сухом виде
при невысоких темп-рах не действуют на латуни
и оловянные бронзы; в присутствии влаги они сильно
взаимодействуют с М. с. Азотная н соляная к-ты дей-
ствуют на латуни и оловянные бронзы очень сильно,
а серная—значительно слабее. В щелочах и жирных
к-тах латуни и оловянные бронзы корродируют весьма
сильно. Оловянные бронзы не корродируют в боль-
шинстве органич. растворителей. Широко распрост-
раненными специфич. видами коррозии латуней,
содержащих более 30% Zn, являются обесцинкова-
ние и коррозионное («сезонное») растрескивание.
Последнее проявляется в образовании трещин по
границам зерен; трещины возникают при хранении
или работе изделий во влажной атмосфере со следами
аммиака и сернистого газа, при наличии в металле
растягивающих напряжений. Отжиг латуни при 260°
снимает остаточные напряжения и предотвращает
коррозионное растрескивание. Алюминиевые бронзы
значительно более стойки против коррозии, чем
латуни и оловянные бронзы. Медноникелевые сплавы
сильно корродируют в минеральных к-тах (особенно
в азотной) и очень слабо — в органич. кислотах и
щелочах.
Хорошие механич. свойства, высокая стойкость
против коррозии во многих средах, ценные физич.
свойства в сочетании с простотой плавки, литья
и обработки давлением обусловили широкое приме-
нение М. с. в многочисленных отраслях техники:
в авиа-, авто- и судостроении, химич. пром-сти, стан-
костроении, электротехнике, приборостроении, в
ироиз-ве разнообразной водяной и паровой арматуры,
посуды, художественных и других изделий.
Лит.: Б о ч в ар А. А., Металловедение, 5 изд., М., 1956;
Справочник по машиностроительным материалам, т. 2, М.,
1959. И. И. Новиков.
МЕДИ СУЛЬФАТ (сернокислая медь)
CuSC>4— бесцветные кристаллы, плотность 3,64. Теп-
лота образования Aff°9g= — 184,0 ккал/моль. При
нагревании диссоциирует: CuSOi—CuO-^SOi-^-'^Oi
с образованием в качестве промежуточного продук-
та основного сульфата CuO-CuSO4. При 7604 дав-
ление диссоциации CuSO« достигает 287 мм рт. ст.,
а СиО -CuSO4—84 мм рт.ст. Растворимость CuSCH
в г на 100 э Н2О составляет: 14(0°); 23,05 (25°); 73,6
(100°). В присутствии свободной H2SO4 растворимость
понижается. При pH 5,4—6,9 CuSCh гидролизуется
с образованием основных солей. CuSCh очень гигро-
скопична, поэтому применяется как осушающее ве-
щество; присоединяя воду, синеет, что иногда ис-
пользуется для обнаружения воды в спирте, эфире
и др.
Из водных р-ров сульфата медн кристаллизуется
CuSO4-5H2O — медный купорос, ярко-синие
кристаллы триклинной системы с параметрами решет-
ки: а=7,15 А, 6=10,70 А, с=5,97 А; а=97°44',
(5 = 125°20', у = 94°19'; плотность 2,29. При нагре-
вании выше 105° плавится с потерей части кристалли-
зационной воды и переходит в CuSCh -ЗН2О (голу-
бого цвета) и CuSO4-H2O (белого цвета). Полностью
обезвоживается выше 258°. При действии сухого
NH3 на CuSCH образуется CuSCH-SNHs, обмени-
вающий во влажном воздухе NH3 на И2О. С суль-
фатами щелочных металлов CuSCH образует двойные
соли типа Me2SC>4 -CnSCh -6Н2О, окрашенные в зе-
леноватый цвет.
В природе медный купорос встречается в виде ми-
нерала хальконтита основные сульфаты меди — в виде
минералов брошантита CuSO4-3Cu(OH)2, вернадскита
3CuSC>4 Cu(OH)2.4H2O, лангита CuSO4-3Cu(OH)2-H2O
и др., двойной сульфат CuSO4-Na2SO4-2H2O —
в виде минерала кронкита. В промышленности мед-
ный купорос получается растворением металлической
меди в нагретой разб. H2SC>4 при продувании
воздуха: Cu-|-H2SO4+ Т/2О2 = CuSO4 + Н2О. Он
является также побочным продуктом электролити-
ческого рафинирования меди. Медный купорос — са-
мая важная технич. соль меди. Он находит приме-
нение при получении минеральных красок, пропитке
древесины, для борьбы с вредителями и болезнями
растений в сельском хозяйстве, для протравливания
зерна, при выделке кож, в медицине, в гальванич.
элементах; служит исходным продуктом для полу-
чения других соединений меди.
Лит.: По зин М. Е., Технология минеральных солей,
2 изд., Л., 1961. А. В. Ванюков.
МЕДИ СУЛЬФИДЫ — соединения меди с серой
Cu2S и CuS. На большом сродстве медик сере и обра-
зовании легкоплавкого штейна (Cu2S-FeS) основаны
пирометаллургич. методы получения меди. Свойство
меди легко соединяться с серой используется так-
же при очистке свинца от меди и в ряде других
процессов.
75
МЕДИНАЛ — МЕДЬ
76
Сульфид одновалентной меди (т.
наз. полусернистая медь) Cu2S встречается в природе
в виде минерала халькозина (медного блес-
ка) — кристаллов темно-серого цвета. Известны 3 мо-
дификации Cu2S — одна низкотемпературная, устой-
чивая ниже 91°, ромбич. сингонии (р-халькозин)
и две высокотемпературные: гексагональная и куби-
ческая (а-халькозин). Природный минерал нередко
представляет собой смесь а- и p-форм. Плотность
халькозина 5,5—5,8; т. пл. 1127°; теплота образования
a-формы ДЯ°98= —19,0 ккал/моль. При нагревании
Cu2S на воздухе образуются CuO, CuSO4 и SO2. В воде
и разб. к-тах Cu2S практически нерастворима. Только
горячая HNO3 растворяет ее с выделением элементар-
ной серы. Аммиачные р-ры также не действуют на
Cu2S. Хорошими растворителями для нее являются
подкисленные р-ры солей 3-валентного железа, рас-
твор хлорной меди и растворы цианидов.
Сульфид 2-валентной меди (сернистая
медь) CuS встречается в природе в виде минерала
ковелина — кристаллов сине-черного цвета ,с
гексагональной решеткой (а=3,802 А, с=16,43 А)
и плотностью 4,68. Теплота образования ДН298 =
= — 11,6 ккал/моль. Довольно хорошо проводит
электрич. ток. Начиная с 450° в нейтральной или вос-
становительной атмосфере CuS энергично диссоции-
рует с выделением серы и образованием Cu2S (давле-
ние диссоциации составляет 80,0 мм рт. ст. при
449,9° и 595 мм рт. ст. при 502,2°). При нагревании
на воздухе CuS легко окисляется до СиО; во влажном
состоянии на воздухе частично окисляется до CuSO4.
При пропускании H2S через нейтральные или слабо-
кислые р-ры солей меди CuS выпадает в виде черного
осадка. В отсутствии кислот очень легко образует
коллоидные р-ры. В воде CuS практически нерас-
творима, легко растворима в горячей умеренно разб.
азотной к-те, плохо — в кипящей разбавленной.
Растворяется в солях 3-валентного железа, в растворе
хлорной меди и в щелочных цианидах. Практически
нерастворима в р-рах сульфидов, но хорошо рас-
творима в полисульфидах щелочных металлов благо-
даря образованию полисульфидов меди.
Лит. см. при ст. Медь. А. В. Ванюков.
МЕДИНАЛ (натриевая соль 5,5-диэтилбарбитуро-
вой кислоты —веронала) С8НцОзМ2Ха, мол. в. 206,19—
бесцветные кристаллы; раство-
рим в холодной, лучше — в го-
рячей воде; трудно растворим
в большинстве органич. раство-
рителей. Водный раствор М. об-
ладает щелочной реакцией на
фенолфталеин. М. получают нейтрализацией верона-
ла концентрированным р-ром NaOH в этиловом
спирте. М.— снотворное и успокаивающее средство.
ME ДНО-АММИАЧНОЕ ВОЛОКНО — гидратцеллю
лозное волокно, получаемое из медно-аммиачного р-ра
целлюлозы. М.-а. в. появилось в конце 19 в. и было
первым практически ценным искусственным волок-
ном; в настоящее время произ-во М.-а. в. составляет
лишь ок. 1% от общего мирового произ-ва химич.
волокон. М.-а. в. получают растворением целлюлозы
в р-ре гидроокиси или основной соли меди в конц.
водном р-ре аммиака.
При растворении гидроокиси или основной соли меди
образуется активное медно-аммиачное основание, способное
растворять целлюлозу:
Си (OH)2 + mNH3^z±[Cu (NH3)m]2 + + 2 (ОН)“
или
5Cu (OH)2-2CuSO4 + wNH3 7—>
7 [Си (NH3)mp + + 2SO“- + 10 (ОН)-
Б присутствии значительного количества ОН-ионов происхо-
дит присоединение этих ионов гидроксильными группами
ZCO-N
с—ONa
C2H5Z CO-NZ
I
Н
целлюлозы через водородные связи. При этом ослабляются
межмолекулярные связи и повышается подвижность макромо-
лекул, что ведет к переходу целлюлозы в раствор. При приме-
нении гидроокиси меди растворение происходит в одну стадию,
если же применяется основная соль меди, то растворение целлю-
лозы протекает в две стадии: после набухания целлюлозы в мед-
но-аммиачном реактиве необходимо добавлять NaOH для связы-
вания SO2 -ионов и для увеличения концентрации ОН-ионов.
Для растворения обычно применяют хлопковую,
реже древесную целлюлозу. При этом образуется очень
вязкий прядильный р-р (1000—1500 пуаз), который
содержит ок. 10% целлюлозы, 4% меди и 7% аммиака.
После удаления воздуха и избытка аммиака раствор
фильтруют через никелевые сетки и направляют
на формование в прядильные воронки, заполнен-
ные водой. Формование производится со скоростью
50 м/мин с большой вытяжкой (в 200—300 раз) при
40—50° в мягкой воде, содержащей не более 1 г/л
аммиака п 0,5 г/л меди. В качестве прядильной
ванны применяют также разб. р-ры едкого натра
или карбонатов. Благодаря высокой вытяжке полу-
чаются очень тонкие волокна с элементарным номе-
ром от 3000 до 18000. Из-за мягких условий формо-
вания волокно имеет круглый срез, не обладает
ориентированным внешним слоем, отличается мяг-
костью и глубоко окрашивается всеми классами кра-
сителей, применяемых для крашения хлопка. Из-за
отсутствия в процессе формования М.-а. в. вторичной
(пластификационной) вытяжки прочность при разрыве
обычно не превышает 12—18 ркм. После формования
свежеспряденные волокна обрабатывают кислотой
для разложения медно-целлюлозного комплекса и для
удаления следов меди, а затем обрабатывают водой
для полной отмывки солей и кислоты. После этого
волокна подвергают авиважной обработке и сушке.
В СССР выпускается гл. обр. штапельное М.-а. в.,
применяемое в смеси с шерстью; за границей вы-
пускается медно-аммиачный шелк высоких номервв,
используемый для изготовления очень тонких три-
котажных изделий, чулок, белья и тканей. Расхо-
дуемые в произ-ве медь и аммиак регенерируются
почти полностью (медь на 95—98%, аммиак на 70%).
Несмотря на это, произ-во М.-а. в. является сравни-
тельно дорогим, гл. обр. из-за применения хлопковой
целлюлозы и малых скоростей формования. Неболь-
шая разрывная прочность М.-а. в. не позволяет при-
менять его для технич. целей, хотя в опытных усло-
виях получен медно-аммиачный шелк с прочностью
при разрыве до 55 ркм, пригодный для изготовления
кордных нитей. Простота и безвредность производст-
венного процесса, тонина и высокое качество М.-а. в.
позволяют надеяться, что при повышении прочности,
полном возврате расходуемых химикалий и улучше-
нии технологии, процесса произ-во М.-а. в. в будущем
сможет развиваться более быстро.
Лит.: Пакшвер А. Б., Технология мсдно-аммиачного
волокна, М., 1947; Роговин 3. А., Основы химии и тех-
нологии производства химических волокон, 2 изд., М., 1957.
_ А. Б. Пакшвер.
МЕДНЫЙ КУПОРОС CuSO4-5H2O — см. Меди
сульфат.
МЕДЬ (Cuprum) Си — химич. элемент I гр. пе-
риодич. системы Менделеева, п. н. 29, ат. в. 63,54.
Природная М. состоит из смеси двух стабильных
изотопов с м. ч. 63 (69,1%) и 65 (30,9%). Сечение за-
хвата тепловых нейтронов атомов М. 3,59 барн. Из
искусственных радиоактивных изотопов в качестве
меченых атомов используются Си81 (Г>/2=3,3 час)
иСии(Т>/3= 12,8 час). Конфигурация внешних эле-
ктронов атома М. Зй1"^1. Энергии ионизации (в эв):
Cu°-)-Cu + -)-Cu2+-)-Cu3+ соответственно равны: 7,724;
20,29; 36,83.
М. относится к числу металлов, известных с глубокой древ-
ности. Латинское название ее происходит от о. Кипр, где древ-
ние греки добывали медную руду. М. и ее сплавы сыграли
большую роль в развитии материальной культуры человека.
77
МЕДЬ
78
М. сравнительно мало распространена в природе.
Содержание ее в земной коре составляет 0,01%.
Встречается в свободном состоянии в виде самородков,
достигающих иногда значительных размеров (до
нескольких тонн). Однако руды самородной М. срав-
нительно мало распространены, и в настоящее время
из них добывается не более 5% М. от общей ее мировой
добычи. М. относится к числу элементов, образующих
халькосферу, расположенную между земным ядром
и литосферой. Присутствие халькофилов в литосфере
современная геохимия объясняет выдавливанием их
из халькосферы вследствие магматич. и гидротермаль-
ных процессов. В связи с этим подавляющая часть
М. (~80%) присутствует в земной коре в виде со-
единений с серой. Около 15% М. находится в виде
кислородных соединений (карбонатов, окислов, си-
ликатов и т. и.), являющихся продуктами выветри-
вания первичных сульфидных медных руд. М. обра-
зует до 240 минералов, однако лишь немногие из них
(—40) имеют промышленное значение.
Важнейшими из промышленных минералов яв-
ляются халькопирит — медный колче-
дан CuFeS2, х а л ь к о з и н—медный блеск
Cu2S, ковеллин CuS, борнит Cu5FeS4, мала-
хит СиСОз-Си(ОН)2, азурит СиСОз-2Си(ОН)2,
хризаколла CuSiOs -2Н2О, брошантит
CuS04-Cu(OH)2. Довольно обычны также арсениды,
антимониды и сульфоарсениды М.
Медные руды по минералогии, составу могут
быть разделены на три категории: самородные, окис-
ленные, сульфидные. Последние, в свою очередь,
делятся на сплошные сульфидные и вкрапленные.
Основным сопутствующим минералом сплошных суль-
фидных руд является пирит FeS2. Вмещающей поро-
дой вкрапленных руд являются силикаты и алюмо-
силикаты. В настоящее время перерабатываются ру-
ды, содержащие 0,7—3% М. Медные руды являются
комплексным сырьем и в зависимости от основного
спутника подразделяются на медноцинковые, медно-
никелевые, медномолибденовые, меднокобальтовые.
Наряду с перечисленными выше ценными спутниками
медные руды обычно содержат S, Se, Те и благородные
металлы (Au, Ag). Медноникелевые руды служат
основным источником получения платиновых метал-
лов. Часто медные руды содержат Ge, In, Tl, Cd, Bi,
Re, Pb, As, Sb и др. ценные спутники.
Наиболее крупные запасы медных руд, кроме СССР
(Казахстан, Урал, Закавказье и др.), сосредоточены
в Африке (Катанга, Северная Родезия), Америке
(Чили, США, Канада).
Физические и химические свойства. М.— металл
красного, в изломе розового цвета, при просвечи-
вании в тонких слоях зеленовато-голубой. Имеет
гранецентрированную кубич. решетку, а = 3,6074 А;
плотность 8,96 (20°^). Атомный радиус 1,28 А; ионные
радиусы Си+ 0,98 А; Си2 + 0,80 А.
Т. пл. 1083°; т. кип. 2600°; теплота плавления
3,11 ккал/г-атом; теплота испарения (при т. кип.)
72,8 ккал/г-атом; уд. теплоемкость 0,092 кал/г -град
(20°). Наиболее важными и широко используемыми
свойствами М. являются ее высокая теплопроводность
(0,941 кал/см -град -сек при 20°) и малое электрич.
сопротивление (1,68 -Ю-8 ом-см при 20°); темпера-
турный коэфф, электрич. сопротивления 4,3-10 ~3
(0—100°). Термич. коэфф, линейного расширения
17,0-10-8. Упругость паров над медью ничтожна,
давление 1 мм рт. ст. достигается лишь при 1628°.
М. диамагнитна; атомная магнитная восприимчи-
вость — 5,27 -10-8. М.— мягкий, ковкий металл;
твердость по Бринеллю 35 кГ/мм2, предел прочности
при растяжении 22 кГ/мм2, относительное удлинение
60%, модуль упругости 13 200 кГ/мм2. Путем наклепа
предел прочности может быть повышен до 40—
45 кГ/мм2, при этом удлинение уменьшается до 2%,
а электропроводность уменьшается на 1—3%. Отжиг
наклепанной М. следует проводить при 600—700°.
Небольшие примеси висмута (тысячные доли %)
и свинца (сотые доли %) делают М. красноломкой,
а примесь серы вызывает хрупкость на холоду.
В химич. отношении М. занимает промежуточное
положение между элементами первой плеяды VIII гр.
и щелочными элементами I гр. системы Менделеева.
Как и Fe, Со, Ni, медь склонна к комплексообразо-
ванию, образует окрашенные соединения, нераство-
римые сульфиды и т. д. Сходство с элементами глав-
ной подгруппы I группы незначительно. Так, М.,
подобно щелочным металлам, обладает высокой элект-
ропроводностью и образует ряд одновалентных со-
единений, но, в отличие от щелочных металлов, для
М. более характерно 2-валентное состояние. Соли од-
новалентной М. в воде практически нерастворимы и
легко окисляются или распадаются до соединений
2-валентной М.; соли 2-валентной М., напротив, хоро-
шо растворимы в воде и в разб. р-рах полностью дис-
социированы. Гидратированные ионы Си2+ окрашены
в голубой цвет.
Известны также соединения, в к-рых М. 3-валентна.
Так, действием перекиси натрия на р-р куприта нат-
рия Na2CuO2 получен окисел Си20з — красный поро-
шок, начинающий отдавать кислород уже при 100°.
Си20з является сильным окислителем — напр., вы-
деляет хлор из соляной к-ты. Этот окиеел имеет кис-
лый характер и образует со щелочами красные, очень
легко разлагающиеся соли типа Ме[Си(ОН)4].
Были получены и другие производные 3-валентной
М.: KCuO2, Ba(CuO2)2, K3[CuFes], K,[Cu(JO6)2]-7Н2О,
Na5H4[Cu(TeOe)2]-18H2O.
Химич, активность М. невелика. Компактный ме-
талл при темп-рах ниже 185° с сухим воздухом и
кислородом не взаимодействует. В присутствии влаги
и СО2 на поверхности М. образуется зеленая пленка
основного карбоната (ядовит). При нагревании М. на
воздухе идет поверхностное окисление; ниже 375°
образуется СиО, а в интервале 375—1100° при непол-
ном окислении М.— двухслойная окалина, в поверх-
ностном слое к-рой находится СиО, а во внутреннем—
Си2О (см. Меди окислы). Влажный хлор взаимодей-
ствует с медью уже при обычной темп-ре, образуя
СцС12, хорошо растворимую в воде. М. легко соеди-
няется ц с другими галогенами (см. Меди галогениды).
Особое сродство проявляет М. к сере и селену; так,
она горит в парах серы (см. Меди сульфиды). С в о-
дородом, азотом и углеродом М.
не реагирует даже при высоких темп-рах. Раствори-
мость водорода в твердой М. незначительна и при
400° составляет 0,06 мг в 100 г М. Присутствие водоро-
да в М. резко ухудшает ее механич. свойства («водо-
родная болезнь»). При пропускании аммиака над
раскаленной М. образуется CusN. Уже при темп-ре
каления М. подвергается воздействию окислов азота,
а именно N2O и NO (с образованием Сц2О) и NO2
(с образованием СиО). Карбиды Сц2С2 и СиС2 могут
быть получены действием ацетилена на аммиачные
р-ры солей М. М. —электроположительный металл. Ее
нормальный потенциал для реакции Cu2+-|-2e-^-Cul)
равен 4- 0,337 в, а для реакции Cu~-|-e->Cu0 равен'
+ 0,52 в. Поэтому М. вытесняется из своих солей
более электроотрицательными элементами (в пром-сти
используется железо) и не растворяется в кислотах —
неокислителях. В азотной к-те М. растворяется
с образованием Cu(NOs)2 и окислов азота, в горячей
конц. H3SO4—с образованием CuSC>4 и SO2. В нагре-
той разб. H2SC>4 М. растворяется при продувании
через раствор воздуха. Все соли М. ядовиты.
М. в двух- и одновалентном состоянии образует
многочисленные комплексные соединения.
79
МЕДЬ
80
обладающие значит, устойчивостью. В большинстве
случаев комплексы Cu(I) прочнее, чем аналогичные
производные Ag(I) и Au(I). Примеры комплексных сое-
динений одновалентной М.: (NH4)2CuBr3, K3Cu(CNk—
комплексы типа двойных солей, [Cu{SC(NH2)2l2Cl
и др. Примеры комплексных соединений 2-валентной
М.: CsCuCI3, K2CuCl4—тип двойных солей [СнЕп3]С12
и др. Кроме того, известно несколько комплексов
Cu(III), напр. KaCuF,.
Для М. характерны переменные координационные
числа (к. ч.): Cu(I) — 2, 3, 4; Cu(II) — 3, 4, 6.Боль-
шинство комплексных соединений Си относятся к
комплексам равновесного типа. Так [Си(1ЧНз)4] Хг,
где,Х=С1, Вг, при нагревании обратимым образом
разрушается по схеме:
135—150° 270о
[Си (NH3)J Cl,7 ’•fCu(NH.,MCl, + 2NH, 7^C.UC1. + 4NH.
При пропускании же тока сухого аммиака над про-
дуктами термич. распада образуются исходные ком-
плексы. В растворе в зависимости от концентрации
аммиака образуются различные аммины, вплоть до
типа [Си(ЫНз)5]2+. Для М. особенно характерно обра-
зование комплексов с азот-, серу-, кислородсодер-
жащими аддендами и с галогенами. В ряду перечис-
ленных ниже соединений Cu(I) устойчивость изме-
няется в следующем порядке:
наибо- найме-
лее ус- нее ус-
TBbi7‘ Cu(CN)r> Cu(NH3)2+ >CuJ7> CuBi7>CuC17 т°^и‘
комп- комп-
лексы лексы
Путем образования комплексных соединений можно
перевести в раствор многие нерастворимые соли М.
Аналитическое определение. Со-
единения М. при нагревании с содой и углем в пламени
паяльной горелки образуют красную металлич. ми-
шуру или королек, растворимые в азотной к-те.
Раствор при прибавлении аммиака становится темно-
голубым. Под действием сероводорода выпадает
черный осадок CuS, а под действием K.4[Fe(CN)6] —
красно-коричневый осадок состава Cu2[Fe(CN)9], Ре-
акция очень чувствительна. В ходе систематич. ана-
лиза М. находят в части, не растворяющейся в по-
лисульфиде аммония. Количественно М. определяют
при высоком ее содержании иодометрич. титрованием
или электролитически. Последний метод наиболее
надежен. Для определения малых количеств М.
применяется калориметрия, метод с использованием
натрийдиэтилдитиокарбамината и микроаналитич. ме-
тоды, основанные на образовании либо KjPbCufNOa),
(чувствительность 0,03у Си), либо внутрикомплексной
соли с рубеановодородной к-той (чувствительность
0,006 у Си).
Получение. В современной металлургии применяют
различные пирометаллургия, и гидрометаллургия,
методы извлечения М. из руд. Наибольшее распро-
странение получили пирометаллургия, методы (более
80% от общей мировой добычи М.), основанные на
большом сродстве М. к сере, а компонентов пустой
породы — к кислороду. При расплавлении серусо-
держащей шихты образуются две жидкие фазы: сплав
сульфидов (штейн), в к-ром концентрируются М. и дру-
гие ценные компоненты (Au, Ag, Ni),H сплав окислов
и силикатов (шлак), в к-рый переходят компоненты
пустой породы. Шлак используется для производства
каменных изделий и шлаковаты или выбрасывается
в отвал. Штейн направляется на окислительную
плавку. Полученную черновую М. рафинируют.
Производство М. из руды складывается из следую-
щих последовательных операций: 1) обогащения,
2) обжига, 3) плавки, 4) конвертирования, 5) огневого
рафинирования, 6) электролитич. рафинирования.
Поскольку содержание М. в руде, как правило, невы-
сокое, а руда наряду с М. содержит другие ценные
компоненты, то обычно целесообразно по возмож-
ности отделить пустую породу и разделить ценные
минералы методами механич. обогащения, т. к. эти
Методы наиболее дешевы. Обычно медные руды обо-
гащают флотацией; в результате получают ряд товар-
ных концентратов (напр., медный, цинковый, пирит-
ный) и отвальные хвосты. Медный концентрат направ-
ляют на пирометаллургич. переработку, первой ста-
дией к-рой является обжиг, к-рый проводят для
окисления части сульфидов железа. Вследствие этого
при последующей плавке большее количество железа
переходит в шлак и содержание М. в штейне повы-
шается. Обжиг проводят в механич. многоподовых
печах или в кипящем слое. Газы обжига содержат
5—7% SOa и используются для производства серной
кислоты. Горячий огарок из печи непосредственно
направляется на плавку. Назначение плавки — рас-
плавление шихты, состоящей из концентратов (огарка)
и флюсов, и разделение продуктов плавки (штейна,
шлака) по плотности. Плавку концентратов на боль-
шинстве заводов проводят в отражательных или
электрич. печах.
Богатые кусковые сульфидные руды (2—3% Си)
с высоким содержанием серы (35—42% S) и малым
содержанием цинка в ряде случаев непосредственно
направляют на плавку в шахтных печах. При этом
плавка в значительной мере происходит за счет тепла,
выделяющегося при окислении сульфидов (пиритная
плавка) и лишь частично за счет тепла от сжигания
кокса. В одной из разновидностей шахтной плавки
(медносерная плавка) в шихту добавляют мелкий
кокс, восстанавливающий в верхних горизонтах пе-
чи SO2 до элементарной серы. В последние годы начи-
нают применяться методы плавки медных концентра-
тов путем их частичного сжигания во взвешенном
состоянии в атмосфере кислорода или подогретого
воздуха. Достоинство этих процессов — малый рас-
ход топлива и получение газов с высоким содержанием
SO2 (до 80% при сжигании в кислороде). Близко при-
мыкает к этим процессам плавка медных концентратов
в циклонных печах; сульфиды окисляются в тонкой
пленке на стенке циклона.
Получающийся при плавке жидкий штейн (в ос-
новном Cu2S • FeS) заливают в конвертер и через
расплав продувают сжатый воздух. Конвертирование
штейнов протекает в две стадии. Сначала окисляется
сульфид железа, для связывания окислов железа
в конвертер добавляют кварц; образующийся кон-
вертерный шлак, вследствие повышенного содержа-
ния в нем меди (1,5—3% Си) возвращается в отра-
жательные печи. Затем окисляется сульфид меди
с образованием металлич. М. и ЭОг. Получающаяся
при конвертировании черновая М. с целью извлече-
ния ценных спутников (Au, Ag, Se, Те, Bi и др.)
и удаления вредных примесей направляется на рафи-
нирование, сначала на огневое, а затем на электро-
литическое. Огневое рафинирование основано на
большем, чем у меди, сродстве металлов-примесей
к кислороду; жидкая М. насыщается кислородом,
при этом Fe, Zn, Со и частично Ni и др. переходят
в шлак, а сера удаляется с газами. После удаления
шлака М. снова восстанавливают погружением в рас-
плавленный металл сырой древесины и разливают
в формы. Полученные отливки являются анодами.
Электролитич. рафинирование М. проводят в серно-
кислых р-рах. Катоды наращиваются на тонких
медных листах, также получаемых электролизом
в специальных матричных ваннах. Для получения
плотных гладких осадков в электролит вводят по-
верхностно-активные добавки (столярный клей, тио-
81
МЕДЬОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ — МЕЕРВЕЙНА РЕАКЦИЯ
82
мочевину и др.). Получающаяся катодная М. после
промывки направляется на переплавку. Благородные
металлы, селен, теллур и другие ценные спутники М.
концентрируются в анодном шламе, из к-рого их
извлекают специальной переработкой. Никель кон-
центрируется в электролите и может быть получен
в виде никелевого купороса путем выведения части
растворов на упаривание и кристаллизацию.
Наряду с описанными пирометаллургическими при-
меняют также гидрометаллургия, методы получения
М. (преимущественно из бедных окисленных и само-
родных руд). Эти методы основаны на селективном
растворении медных минералов обычно в слабых
р-рах H2SO4 и аммиачных р-рах. Из раствора М.
осаждают железом, либо выделяют электролизом
с нерастворимыми анодами. Весьма перспективны
применительно к смешанным рудам комбинированные
гидрофлотационные методы, при к-рых кислородные
соединения М. растворяются в сернокислых р-рах,
а сульфиды выделяются флотацией.
Техника безопасности. В горячих цехах возможны
попадания брызг расплавленных шлака или штейна
на одежду. При соприкосновении расплавленного
штейна с водой происходят сильные взрывы. Недопу-
стимо попадание влаги на рабочие площадки, работа
с влажным инструментом и охлаждение расплавов, со-
держащих штейн, водой. С целью предохранения от
ожогов рабочих снабжают шерстяной одеждой, ва-
ленками, защитными очками. Места большого тепло-
излучения экранируют водяными или воздушными
завесами. В целях устранения загазованности уста-
навливают общие и местные отсосы загрязненного
воздуха, к рабочим местам подают кондиционирован-
ный воздух. Работа в загазованных и пыльных местах
разрешается только в противогазах.
Применение. Широкое применение М. в пром-сти
обусловлено рядом ее ценных свойств и прежде
всего высокой электропроводностью, пластичностью,
теплопроводностью. В частности, хорошая электро-
проводность М. в сочетании с большой пластичностью
делает ее основным материалом для проводов. В связи
с этим свыше 50% добываемой М. применяется в
электротехнич. пром-сти. Все примеси понижают
электропроводность М., а потому в электротехнике
применяют металл высших сортов, содержащий не
менее 99,9% Си. Благодаря высокой теплоцровод-
ности и сопротивлению коррозии М. употребляется
для изготовления наиболее ответственных изделий
(в теплообменниках, холодильниках, вакуумных ап-
паратах и т. п.). Около 30—40% М. используется
прем-стью в виде различных сплавов, находящих
широкое и разнообразное применение. Среди этих
сплавов наибольшее значение имеют латуни (от 0
до 50% Zn) и различные виды бронз: оловянистые,
алюминиевые, свинцовистые, бериллиевые и т. д.
(подробнее см. Меди сплавы). Кроме нужд тяжелой
промышленности, связи, транспорта, нек-рое коли-
чество М., гл. обр. в виде солей, потребляется для
приготовления минеральных пигментов, борьбы с вре-
дителями и болезнями растений, в качестве микро-
удобрений, катализаторов окислительных процессов,
а также в кожевенной и меховой пром-сти и при
производстве искусственного шелка.
Лит.: Смирнов В. И., Металлургия меди и никеля,
Свердловск — М., 1950; Аветисян X. К., Металлургия
черновой меди, М., 1954; Газарян Л. М., Пирометаллур-
гия меди, М., 1960; Справочник металлурга по цветным метал-
лам, под ред. Н. Н. Мурача, т. 1, 2 изд., М., 1953, т. 2, М., 1947;
Gmelin, 8 Aufl., Syst.-Nummer 60, В., 1955, 1958, 1961,
1962; Mellor, v. 3, L.—N. Y.—Toronto, 1956; Pascal,
t. 3, P., 1957; U llmann, 3 Aufl., Bd 11, Munchen—B., 1960,
S. 1 19—260; Kirk, V. 4, N. Y., 1949, p. 391—479.
А. В. Ванюков.
МЕДЬОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ — термиче-
ски нестойкие бесцветные или желтоватые кристаллы,
окисляющиеся на воздухе, общей формулы RCu.
М. с. образуются при взаимодействии магний- или
литийорганич. соединений с галогенными солями меди:
Cu2J2 + 2CH,Li —> 2CH.,Cu + 2LiJ
CuBr2 + C„H5MgBr —> MgBr2 + C0H5C„H5 + CeH5Cu
Разложение борфторидов арилдиазониев порошком
меди приводит к арильным соединениям меди:
NO2CeH4N2BF4 + 2Cu —» NO2C»H4Cu + N2 + CuF + BF3
Устойчивый на воздухе ацетиленид меди CuC^CCu
получают пропусканием ацетилена в аммиачные р-ры
солей меди.
Метилмедь разлагается при 20° со взрывом:
2СНзСи^>-2Си-|-СНзСНз. Более устойчивы арильные со-
единения меди. М. с. реагируют с водой, спиртами, аль-
дегидами, подобно магнийорганич. соединениям, но не
взаимодействуют, в отличие от последних, со сложными
эфирами; с амидами к-т дают устойчивые комплексы
типа RCu-R'CONHa. М. с. реагируют с хлорангид-
ридами карбоновых к-т с образованием кетонов (1),
а с изоцианатами — замещенных амидов кислот (2).
RCOCl + R'Cu—>RCOR'+CuCl (1>
СДЬСи + С.Щ- N = C = O —>- CeH5CONHCeH5 (2)
Известно, что медь или ее соли являются катализато-
рами многих важных реакций. Возможно, что нек-рые
из последних протекают через стадию образования
М. с. Сами по себе М. с. применения не находят.
Лит.: Gilman Н., Woods L. A., J. Amer. Chem.
Soc., 1943, 65, № 3, 435; Во 1th F. А. [а. о.], там же,
1943, 65, № 8, 1456; G 1 1 m a n H., Jones R.G.,Woods
L. A., J. Organ. Chem., 1952, 17, № 12, 1630; Cotton F. A.,
Chem. Rev., 1955, 55, 551; В a w n C.E. H.,Whitby F.J.,
J. Chem. Soc., 1960, October, 3926. H. А. Несмеянов.
МЕЕРВЕЙНА РЕАКЦИЯ—введение арильной груп-
пы ва,Р-непредельные карбонильные соединения кон-
денсацией их с солями арилдиазониев:
ArN2X + RCH=CHCOX —s-RCH = CCOX или RC = CHCOX
I I
Аг Ar
Часто при этом образуются продукты присоединения
арила и галогена к непредельным соединениям:
NO2C6H,N2C1 + CH2 = CHCN —> NO2C„H,CH2 - CHC1CN + n2
M. p. проводят обычно в водном ацетоне, а иногда
в ацетонитриле, пиридине или воде при 5—40°. Соли
двух- и одновалентной меди катализируют реакцию.
Вступление арильной группы в а- или ^-положение
к карбонильной, карбоксильной или нитрильной груп-
пе зависит от наличия или отсутствия у двойной связи
конъюгированного арильного остатка. Так, производ-
ные кротоновой к-ты и акрилонитрил дают [3-арил-
производные:
СН3СН = CHCOOR + ArN3Cl —> CH3CHCHC1COOR
Аг
Напротив, в производных коричной к-ты арильная
группа вступает в a-положение, напр.
С„Н5СН=СНСООСН3 + ArN2Cl —»- С»Н3СНС1СНСООСН3 + N2
Аг
С6Н5СН = СНСООН + ArN2Cl —> С„Н5СН = CHAr + СО2 + N3 + НС1
C„H5CH = CHCHO + ArN2Cl —> C„H5CH = CCHO + N2 + HC1
Ar
Двойная связь влияет аналогично фенильному
кольцу:
сн.сн=снсн=снсоон+c»h5n2ci—>-
—>СН3СН = СН - СН = СНС6Н5 + СО2 + НС1 + Na
Такое направление присоединения согласуется только с ради-
кальным механизмом М. р.:
ArN2Cl —> Аг- + Na+Cl-
RCH = CHCOX + Ar (-RCHCHCOX или RCHCHCOX
A Ar Аг Б
83 МЕЕРВЕЙНА—ПОННДОРФА—БЕРЛЕЯ РЕАКЦИЯ—МЕЖДУНАРОДНАЯ СИСТЕМА ЕДИНИЦ 84
Направление присоединения зависит только от большей устой-
чивости радикала А или Б. В дальнейшем радикалы А и Б
стабилизируются либо выбросом протона, либо присоедине-
нием хлора:
RCHCHCOX RCH=CCOX RCH-CHCOC1
• I —» I ИЛИ I I
Аг Аг С1 Аг
Арилированию солями диазония подвергаются не
только а.Р'непредельные карбонильные соединения,
но и олефины, ацетилен, сопряженные диены, стиролы,
винилацетиленовые углеводороды, виниловые эфиры,
n-хиноны и пр., напр.:
n-O2NC6H4N2Cl + СН2=СН2—— n-O2NC6H4CH2CH2CI + N2
O2NC6H4N2C1 + CHS CH —— O2NC6H4CH=CHC1+N2
CiC6H4N2CI + C6H5CH=CH2 —- ClC6H4CH2CHClC6H6 + n2
ArN2Cl + CH2=CH-CH=CH2—- ArCH2CH=CHCH2Cl + N2
O2NC6H4N2OH + ch2=chor—O2NC6H4N2CH2CH
OH
—*- o2nc6h4nh-n=chcoh
M. p. открыта и подробно изучена в 1939 Г. Меер-
вейном, независимо от нашедшего ее в 1935 Шустера,
и часто называется реакцией Меервейна—Шустера.
Лит.: Franzen V., К ranch Н., Chemlker Ztg.,
1955, 79, № 4, 101; Kochi J. К., J. Amer. Chem. Soc., 1955,
77, № 19, 5090; Geppert А., Справочник по органическим
реакциям, пер. с англ., М., 1962, с. 184; Терентьев А.П.,
3 агоревски й В. А., Ж. общ. химии, 1956, 26, № 1,
2°МЕЕРВЕЙНА — ПОННДОРФА — ВЕРЛЕЯ'" PEAK-
ЦИЯ (восстановление) — селективное восста-
новление карбонильных соединений в спирты в при-
сутствии алкоголятов алюминия:
он
A1(OCHR"R"')3 I
RCOR' 4-R"CHR'''---------> RCHR' 4-R"COR'''
1
ОН
Реакция обратима (см. Оппенауэра реакция); равно-
весие сдвигается в зависимости от природы алкоголя-
та и условий реакции. Для восстановления карбониль-
ных соединений чаще всего используют раствор
изопропилата алюминия в изопропиловом спирте; из
реакционной смеси отгоняют ацетон по мере его образо-
вания. О завершении реакции судят по прекращению
выделения ацетона.
Изопропилат алюминия — очень специфичный вос-
становитель; он восстанавливает лишь альдегиды и ке-
тоны (алифатич., алициклич. и ароматич.). Альдеги-
ды, как правило, восстанавливаются значительно
легче, чем кетоны. Другие группировки, присутствую-
щие в молекуле, напр. нитро-, карбалкокси-, ацеталь-
ные группы, галогены, углерод — углеродные двойные
связи, в том числе и расположенные в а, р-положении
к карбонильной группе, совершенно не затрагивают-
ся. Простота и высокие выходы спиртов обусловлива-
ют широкое применение реакции в лабораторной
практике.
При восстановлении оптически активных кетонов обычно об-
разуются примерно в равных количествах оба диастереоизо-
мера; однако при восстановлении бензила или бензоина полу-
чается 90% мезогидробензоина. При восстановлении неактив-
ного кетона оптически активным спиртом образующийся
спирт также обладает оптич. активностью.
М.—П.—В. р., как и нек-рые другие реакции восстановле-
ния карбонильных соединений (Канниццаро реакция, Тищенко
реакция, восстановление нек-рых карбонильных соединений
реактивами Гриньяра), включает перемещение гидрид-пона.
Такой механизм реакции подтверждается кинетич. данными,
а также тем, что при использовании а-дейтерированного спирта
получается спирт, содержащий дейтерий:
D СНа
R\ \/
)С + С,
R' II | ЧСН3
О ОА1(ОС3Н,)2
R4
)CD
R'z |
ОА1(ОС3Н7)2
СН3
. С(
4 11ЧСН3
о
С другой стороны, в среде О-дейтерированного спирта карбо-
нильное соединение восстанавливается в недеитерированный
продукт.
Реакцию открыли в 1925 Г. Меервейн и А. Верлей
и в 1926 независимо от них Понндорф.
Лит.: Уайлдс А. Л., в кн.: Органические реакции,
сб. 2, пер. с англ., М., 1960, с. 194; Moulton W. N. [а. о.],
J. Organ. Chem., 1961, 26, № 2, 290. Я. Я. Гамбарян.
МЕЖДУНАРОДНАЯ СИСТЕМА ЕДИНИЦ — уни
версальная система физич. величин. XI Международ-
ная конференция по мерам и весам приняла (Париж,
октябрь 1960) Международную систему единиц, одо-
бренную затем другими международными организа-
циями и введенную в ряде государств законодатель-
ными актами и стандартами. Принятие этой системы
(SI — «система интернациональная») диктуется не-
обходимостью устранения множественности единиц
измерения, затрудняющих научно-технич. общение.
В Советском Союзе Комитетом стандартов мер и изме-
рительных приборов Международная система единиц
(СИ) принята (ГОСТ 9867—61) для предпочтительного
применения ее с 1 января 1963 во всех областях
науки, техники и народного хозяйства, а также при
преподавании.
М.с.е. состоит из 6 основных единиц, 2 дополнитель-
ных и 27 важнейших производных единиц, при помо-
щи к-рых и образуется вся система единиц измерения
(см. табл. 1 и 2).
Таблица 1. Основные и дополнительные единицы
Величина Единица из- мерения Сокращенные обозначения
русскими буквами латинскими буквами
Длина метр м m
Масса килограмм кг kg
Время секунда сек s
Сила тока . . . ампёр а А
Термодинамич. градус Цель- °к °к
температура вина
Сила света . . . свеча св cd
Плоский угол радиан рад rad
Телесный угол стерадиан стер sr
Последние две единицы являются дополнительными;
используются для образования единиц угловой ско-
рости и углового ускорения.
В ГОСТ 9867—61 даны следующие определения основных и
дополнительных единиц:
Метр — единица длины, равная 1650763,73 длин волн в
вакууме излучения, соответствующего перехода между уров-
нями 2р10 и 5d-' атома криптона 86;
Килограмм — единица массы, соответствующая массе
международного прототипа килограмма;
Секунда — единила времени, 1/31556925,9747 часть
тропического года для 1900 января 0 в 12 ч эфемеридного вре-
мени ;
Ампер — единица силы тока, сила неизменяющегося
тока, к-рый, проходя по двум параллельным прямолинейным
проводникам бесконечной длины и ничтожно малого кругового
сечения, расположенным на расстоянии 1 м один от другого в
вакууме, вызвал бы между этими проводниками силу, равную
2-10-7 единиц силы Международной системы на каждый метр
длины;
Градус Кельвина — единица измерения темп-ры
по термодинамич. температурной шкале, в к-рой для темп-ры
тройной точки воды установлено значение 273, 16°К, лежащей
выше точки таяния льда на 0,01° К.
Свеча — единица силы света, значение к-рой прини-
мается таким, чтобы яркость полного излучателя при темп-ре
затвердевания платины была равной 60 се на 1 см2;
Радиан — угол между двумя радиусами круга, выре-
зающий на его окружности дугу, длина к-рой равна радиусу;
Стерадиан — телесный угол, вершина к-рого рас-
положена в центре сферы и к-рый вырезает на поверхности
85
86
МЕЖДУНАРОДНАЯ СИСТЕМА ЕДИНИЦ
Таблица 2. Производные единицы СИ
Величина Единица измерения Сокращенные обозначения Размер единицы
русскими бук- вами латинскими буквами
Площадь ....... Единица пространст квадратный метр в а и в р е ь At2 е н и т2 (1 Л{)-(1 м)
Объем, вместимость кубический метр .ч3 т3 (1 Л1).(1 «)(! м)
Частота герц гц Hz 1 :(1 сек)
Линейная скорость метр в секунду м/сек m/s (1 At):(l сек)
Линейное ускорение метр на секунду в квадрате м сек2 m/s2 (1 м/сек):(1 сек)
Угловая скорость радиан в секунду рад/сек rad/s (1 рад):(1 сек)
Угловое ускорение ....... радиан на секунду в квадрате рад/сек2 rad/s2 (1 рад/сек):(1 сек)
Плотность (объемная масса) . . . Механические е килограмм на кубический метр д и н и ц ы кг/м3 kg/m3 (1 кг):(1 .и3)
Количество движения килограмм-метр в секунду кг -м/сек kg-m/s (1 кг)-(1 м/сек)
Момент количества движения . . килограмм-квадратный метр на кг-м2/сек kg*m2,s (1 m)-(i м/сек)-(1 м)
Момент инерции секунду килограмм-квадратный метр кг-м2 kg-m2 (1 кг)-(1 м2)
Момент инерции плоской фигуры (осевой или полярный) . ... . метр в четвертой степени м* m* (1 зс2)*(1 м2)
Сила ньютон н N (1 кг)-(1 м/сек2)
Момент силы, пары сил, статиче- ский момент . . . ньютон-метр н-м N-m (1 И).(1 м)
Давление, напряжение ньютон на квадратный метр н/м2 N/m2 (1 н):(1 At2)
Модуль упругости, модуль сдвига, модуль объемного сжатия . . . ньютон на квадратный метр н/м2 N/'m* (1 н):(1 м2)
Поверхностное натяжение .... ньютон на метр н/м N/m (1 н):(1 м)
Удельный вес ......... ньютон на кубический метр н/м3 N/m3 (1 н):(1 л.3)
Динамическая вязкость ньютон-секунда на квадрат- н-сек/м2 N-s;m2 [(1 н):(1 .м2)]-[(1 .м):(1 .м/сек)]
Кинематическая вязкость .... ный Метр квадратный метр на секунду м2/сек m2 s (1 н-сек/м2у.(1 кг/м3)
Работа, энергия . . ‘ ...... джоуль дж J (1 н)-(1 м)
Мощность . ватт вт w (1 дж):(1 сек)
Э лектрическис и м а г н итные единицы
Количество электричества, элект- рический заряд кулон к c (1 а)-(1 сек)
Плотность электрического тока ампер на квадратный метр а/м2 A/m2 (1 а):(1 At2)
Линейная плотность электрическо- го тока ампер на метр а/м A/m (1 а):(1 л.)
Объемная плотность электрическо- го заряда кулон на кубический метр к/м3 C/m3 (1 к):(1 At3)
Поверхностная плотность электри- ческого заряда кулон на квадратный метр к/м2 C/m2 (1 к):(1 л.2)
-Электрическое смещение ..... кулон на квадратный метр к/м2 C/m2 (1 к):(1 -м2)
Поток электрического смещения кулон к c (1 к/л12)-(1 At2)
Разность электрических потенциа- лов, электродвижущая сила, электрическое напряжение . . . вольт в V (1 вт):(1 а)
Напряженность электрического по- ля вольт на метр в/м V/m (1 »):(! м)
Электрическая емкость фарада ф F (I к):(1 в)
Абсолютная диэлектрическая про- ницаемость, электрическая посто- янная (диэлектрическая прони- цаемость свободного пространст- ва); диэлектрическая восприим- чивость фарада на метр ф/м F/ni (1 w/At2):(l в;м)
Диэлектрическая проницаемость (относительная) отвлеченная единица — — (1 об/At) :(1 (р/м)
Момент электрического диполя . . кулон-метр к-м C-m (1 к):(1 At)
Электрическое сопротивление . . ом ом q (1 в):(1 а)
Удельное электрическое сопротив- ление ом-метр ОМ -31 Q-m (1 OAt)-(l At2):(l At)
Электрическая проводимость . . . сименс сим S (1 а):(1 в)
Удельная электрическая проводи- мость сименс на метр сим/м S/m (1 CUAt)-(l 3t):(l At2)
Магнитный поток вебер вб Wb (1 в)«(1 сек)
Магнитная индукция тесла тл T (1 вб):(1 At2)
Магнитодвижущая сила, разность магнитных потенциалов .... ампер а A (1 а)
Напряженность магнитного поля ампер на метр а/м A/m (1 а) :(1 At)
Векторный потенциал вебер на метр вб/м Wb/m (1 ен/.и)-(1 а)
Индуктивность, взаимная индук- тивность генри гн H (1 вб):(1 а)
Абсолютная магнитная проницае- мость; магнитная постоянная (ма- гнитная проницаемость свободно- го пространства) генри на метр гн/м H/m (1 тл):(1 а/м)
Магнитная проницаемость (отно- сительная) отвлеченная единица — — (1 гн/м):([ гн/м)
Магнитная восприимчивость . . . отвлеченная единица — — (1 а/м):(1 а/.и)
Магнитный момент диполя, элект- рического тока ампер-квадратный метр а-м2 A-m2 (1 а)-(1 jh2)
Намагниченность (интенсивность намагничивания) ампер на метр а/м A/m (1 а):(1 At)
Магнитное сопротивление .... ампер на вебер а/вб A/Wb (1 а):(1 вб)
Магнитная проводимость вебер на ампер вб/а wb A (1 вб):(1 а)
Электрическая энергия джоуль дж J (1 н)-(1 •«)
87
МЕЖДУНАРОДНАЯ СИСТЕМА ЕДИНИЦ
88
Продолжение
Величина Единица измерения Сокращенные обозначения Размер единицы
русскими бук- вами латинскими буквами
Объемная плотность электромаг- нитной энергии джоуль на кубический метр дж/м3 J/ms (1 Зж):(1 At3)
Активная мощность электрической цепи ватт вт W (1 дж):(1 сек)
Реактивная мощность электриче- ской цепи вар вар var (1 а)-(1 e)-(sin ф)
Полная мощность электрической цепи вольт-ампер в>а V-A (1 <А-(1
Вектор Пойнтинга ватт на квадратный метр вт/м2 W/m2 (1 втп):(1 At2)
Угловая частота электрического то- ка радиан в секунду рад/сек rad/s 1:(1 сек)
Частота электрических колебаний герц гц Hz 1 :(1 сек)
Количество теплоты; термодинами- ческий потенциал (внутренняя энергия, изохорно-изотермный потенциал, энтальпия, изобарно- изотермный потенциал) .... Тепловые ед джоуль и н и ц ы дж J (1 н)-(1 Л1)
Удельная теплота (фазового пре- вращения, химической реакции); удельный термодинамический по- тенциал джоуль на килограмм дж/кг J/kg (1 дж):([ кг)
Теплоемкость системы; энтропия системы джоуль на градус дж/град J/deg (1 дж):(1 град)
Удельная теплоемкость; удельная энтропия системы джоуль на килограмм-градус дж/(кг-град) j/(kg-deg) (1 дж)-(1 иг)-(1 град)
Тепловой поток ватт вт W (1 дж);(1 сек)
Поверхностная плотность теплово- го потока ватт на квадратный метр вт/м2 W/m2 (1 em):(l At3)
Коэффициент теплообмена (коэф- фициент теплоотдачи); коэффи- циент теплопередачи ватт на квадратный метр-гра- дус ватт на метр-градус вт!(м2-град) W/(m2deg) (1 вти):(1 At2)‘(l град)
Коэффициент теплопроводности . . вт'(м-град) W/(m-deg) (1 вти):(1 At2)-(1 град/м)
Коэффициент температуропровод- ности квадратный метр на секунду м2/сек m2/s [1 втЦм-град)]-. {[1 дж/(кг- -граЗ)]-(1 кг/м3)| (1 град)'.(1 м)
Температурный градиент .... градус на метр град /м deg/m
Относительный температурный ко- эффициент (линейного или объ- емного расширения, электриче- ского сопротивления, электриче- ской емкости ит. д.) градус в минус первой степе- 1/град tdeg 1: (1 град)
Звуковое давление ни Акустические е ньютон на квадратный метр д и н и ц ы н/м2 N/m2 ms /s (1 н):(1 Л12)
Объемная скорость кубический метр в секунду At3/сек (1 At3): (1 сек)
Акустическое сопротивление . . . ньютон-секунда на метр в пя- н-сек/м5 N-s'm5 (1 н/м2):(1 м3/сек)
Удельное акустическое сопротив- ление той степени ньютон-секунда на кубиче- н-сек/м3 N-s/m3 (1 н/л12):(1 м3/сек)-(1 ма>
Механическое сопротивление . . ский метр ньютон-секунда на метр н-сек/м N -s/m (1 н):(1 м/сек)
Интенсивность звука ватт на квадратный метр вт/м2 W,m2 (1 вти):(1 At2)
Плотность звуковой энергии . . джоуль на кубический метр дж /м3 J/nr1 (1 дж):(1 At3)
Световые единицы и единицы энергетической фотометрии
Световой поток люмен лм Im (1 ce)-(l стер)
Световая энергия л юме н-се кун да лм-сек Ims (1 4At)-(l сек)
Светность ...... люмен на квадратный метр лм/м2 Im/m2 (1 л.ч):(1 At2)
Освечивание свеча-секунда св-сек cds (1 ce)-(l сек)
Яркость нит нт nt (1 ce):(l At2)
Освещенность люкс лк lx (1 ллс):(1 Al2)
Количество освещения люкс-секунда лк-сек Ix-s (1 лк)-(1 сек)
Волновое число метр в минус первой степени 1 /Л1 1/m 1 :(1 м)
Энергия излучения Объемная плотность энергии излу- джоуль дж J (1 н)-(1 м)
чения Поверхностная плотность потока джоуль на кубический метр дж/м3 J/m3 (1 дж):(1 At3)
излучения ватт на квадратный метр етп/л»2 W/m2 (1 ети):(1 At2)
Поток излучения Энергетическая светность, энерге- ватт вт w (1 дж):(1 сек)
тическая освещенность Энергетическое количество освеще- ватт на квадратный метр вт/м2 W/m2 (1 sm):(l -и2)
НИЯ джоуль на квадратный метр дж!м2 J/m2 (1 вти/л12)-(1 сек)
Энергетическая сила света .... ватт на стерадиан вт/стер W/sr (1 em):(l стер)
Энергетическая яркость Спектральная плотность энергии ватт на стерадиан-квадрат- ный метр вт/(стер-м2) W/(sr-m2) (1 emlcmep):(i м2}
излучения (по длине волны) . . Спектральная плотность энергии джоуль на метр дж/м J|m (1 Зж):(1 At)
излучения (по частоте) джоуль на герц дж/гц J /Hz (1 Яле): (1 гц)
Оптическая сила линзы диоптрия (метр в минус пер- вой степени) дптр (1 /м) 1/m 1 :(1 м)
89
МЕЖДУНАРОДНАЯ СИСТЕМА ЕДИНИЦ
90
Продолжение
Величина Единица измерения Сокращенные обозначения Размер единицы
русскими бук- вами латинскими буквами
Единицы ио Поглощенная доза излучения. Энергия любого ионизирующего излучения, поглощенная единицей массы облученного вещества визирующих и 3 л джоуль на килограмм учений дж/кг J/kg (1 Зэ#с):(1 кг)
Мощность поглощенной дозы излучения. Погло- щенная доза излучения, отнесенная к единице времени ватт на килограмм вт/кг W/kg (1 em):(l кг)
Экспозиционная доза рентгеновского и гамма-из- лучений. Количественная характеристика рентге- новского и гамма-излучений, основанная на их ио- низирующем действии в сухом атмосферном воздухе и выраженная отношением суммарного электричес- кого заряда ионов одного знака, образованного из- лучением, поглощенным в некоторой массе воздуха, к этой массе кулон на килограмм Kjm C/kg (1 к):(1 кг)
Мощность экспозиционной дозы рентгеновского и гамма-излучений. Экспозиционная доза рентгенов- ского и гамма-излучений, отнесенная к единице вре- мени ампер на килограмм а/кг A/kg (1 а):(1 кг)
Активность. Число актов распада данного нуклида, происходящих в единицу времени в радиоактивном излучателе 1:(1 сек)
Интенсивность излучения. Энергия ионизирующе- го излучения, падающего в единицу времени на по- верхность элементарной сферы, отнесенная к еди- нице площади поперечного сечения этой сферы . . ватт на квадратный метр втп/м2 W/m2 (1 епг):(1 м2)
Плотность потока ионизирующих частиц или кван- тов. Число частиц или квантов, падающих в еди- ницу времени на единицу поверхности, располо- женной нормально к направлению распространения частиц или квантов — - — — 1:(1 сек)-( 1 л<2)
сферы площадь, равную площади квадрата со стороной, рав-
ной радиусу сферы.
Для удобства пользования можно образовать кратные и
дольные единицы умножением или делением единиц СИ на
степень числа 10, с соответствующими приставками, реко-
мендованными ГОСТ 7663—55 (см. табл. 3).
Таблица 3. Приставки для образования кратных и дольных
единиц
Кратность и дольность Приставки Сокращенное обозначение
русскими буквами латинскими или греч. буквами
10« тера Т т
10й гига Г G-
10° мега м М
103 кило к к
102 гекто 3 h
10 дека да da
10~1 деци д d
10-2 санти с С
ю-3 милли м m
10-6 микро мп Ц
10 -» нано н n
10—12 ПИКО п р
1 0 - 1 • фемто ф f
10-18 атто а а
Последние две приставки рекомендованы Международным
союзом чистой и прикладной физики (1960).
Приставки можно присоединять только к простым наимено-
ваниям, не содержащим приставок; недопустимо удвоение при-
ставок. Нельзя присоединять приставки к кратным и дольным
единицам, имеющим особые наименования, напр. микрон,
тонна, ар и др. Поэтому неправильны наименования: милли-
микрон, мегатонна, гектар, микробар и др. Кратные и доль-
ные единицы не входят в М.с.е. (кроме килограмма, являюще-
гося основной единицей) и пользоваться ими в сложных рас-
четах не рекомендуется.
В М.с.е. важное место занимают переводные множители.
В табл. 4 приведены: переводные множители для кратных и
дольных метрич. единиц, а также, имеющих собственное наи-
менование (микрон, тонна, ангстрем, бар и другие); кратные и
дольные единицы, образованные с нарушением ГОСТ 7663—55
и нек-рые другие. Перевод неметрических единиц (англий-
ских) приводится в табл. 5.
В соответствии с принятой системой единиц реко-
мендуется выражать количество вещества в единицах
массы, а не в единицах веса. По аналогии с этим сле-
дует говорить: атомная масса, молекулярная масса^
закон сохранения массы вещества и т. д., а не атомный
вес, молекулярный вес и т. п. Следует строго разли-
чать такие понятия, как масса, сила, вес, плотность,
удельный вес. Международный комитет мер и весов
принял постановление об отказе от применения литра
в качестве единицы объема при точных измерениях
(1 л = 1,000028 дм). При переходе на единицы СИ
следует отказаться от т. наз. нормальных кубич. метров
Таблица 4. Переводные множители метрических и некоторых внесистемных единиц
Сокращенные обозначения
Единица измерения русскими буквами латинскими или греч. буквами Переводный множитель
Метр Миллиметр Микрометр (микрон) Нанометр Ангстрем Икс-единица Ар Гектар Барн • для едини м мм мкм нм для единиц а га ц длины m mm цт опт А X площади ha 1 — основная единица (см. табл. 2) 1 мм=10 ~3 м 1 AtKAt = 10 ~6 м 1онм=10-9 м 1А = 1О“10 м 1X = 1,00206-10 ~18 м 1 а=100 м2 1 за=104 м2 1 барн=10~28 м3
91
МЕЖДУНАРОДНАЯ СИСТЕМА ЕДИНИЦ
92
Продолжение
Сокращенные обозначения
Единица измерения русскими латинскими или Переводный множитель
буквами греч. буквами
для едини ц объема
Литр л I 1 1 1 л = 1,000028 10 -3 -и3
Миллилитр МЛ ml 1 Л1Л = 1,0000 28 см3
Килолитр КЛ 1 kl 1 1 кл=1,000028 -и3
для единиц пл ос к or о угла
Оборот (полный угол) об — 1 об= 2л ра5 = 6,283185 рад
Прямой угол L L 1^=Л'2 рад=1,570796 рад
Градус о о 1° = (-ду) = 0,01 745329 рад
Минута f г 1'=(^)° = 2,908882-10-' рад
Секунда н ff 1" = (^) = 4,848137 10-» рад
Град (гон) — g 1 град=0,01 L=0,01570796 рад
для единиц времени
Минута мин in in 1 лшн = 60 сек
Час ч h 1 ч = 3600 сек
Сутки d 1 сутки=86400 сек
для единиц лине иной скорое т и
Метр в секунду м/сек m/s (см. табл. 2)
Километр в час км/ч km/h 1 кл«/ч=~^ л</сек=0.2777... м/сек
Сантиметр в секунду СЛ1 !се к cm/s 1 сл1/сек=0,01 м/сек
ДЛЯ единиц угловой скорости
Радиан в секунду рад ‘сек rad s (см. табл. 2)
Оборот в секунду об /сек — 1 об/сек-2п рад/сек
Оборот в минуту об/мин — 1 o6/AtUH=^- рад/сек
Градус в секунду °/сек °/s 1 °/сек = 0,01 74533 рад/сек
для едини и масс ы
Тонна т t 1 m = 1000 кг
Грамм г g 1 г = 10“3 кг
Карат — — 1 карат метрический = 200 л4г=2»10~<кз
Килограмм-сила-секунда в квадрате на
метр — техническая единица массы . . кгс сек2 м kgf-s2 m 1 кгС‘сек2/м = 9,80665 кг
для единиц плотности
Тонна на метр кубический т м3 t,'m3
Килограмм на дециметр кубический . . . кг.Олг3 kgzdma тп'л13 = 1 кг,'б,и3 = 1 г;сл13 = 1000 кг/м*
Грамм на сантиметр кубический .... г;см3 g cm'
Грамм на миллилитр г:мл g ml 1 г/мл = 999,972 кгл13
Килограмм-сила-секунда в квадрате на
метр в четвертой степени кгс-сек^м* kgfs’/m’ 1 кгс-сек2 м* = 9,80665 кг1м3
ДЛЯ единиц массового расхода
Килограмм в час кг/ч kg h [ кг >1 = 278-10 -6 кг/сек
Килограмм в минуту кг лшн ks min 1 кг мин — 1 6,67» 10 кг/сек
Тонна в час т ч t/ll 1 тч = 0,278 кг,сек
Грамм в секунду г сек g S 1 г сек=10“3 кг,сек
Для единиц о б ъ е много расхода
Метр кубический в час м3 ч m3 h 1 .и3,’.'ас= 278 10~6 л<3 сек
Литр в час лч 1'11 1 л'ч = 278-Ю”9 Л43;сек
Литр в минуту л мин 1/min 1 л/мин — 16,67-10 ~в мэ[Сек
для един иц силы (в частности, единиц силы тяжести)
Ньютон н N (см. табл. 2)
Дина дин dyn 1 дин — 10 н
Стен — sn 1 стен = 103 н
Килограмм-сила кгс, кГ kgf 1 кгс = 9,80665 н
Тонна-сила тс — 1 тс=9,80665-Ю3 н
для единиц давления
Ньютон на квадратный метр Н>М2 N m2 (см. табл. 2)
Бар бар bar 1 бар- 10-’ н м2
Миллибар на квадратный метр мбар/м2 mbar.m2 1 Л1бар=100 н.м2
Килограмм-сила на квадратный метр . . кгс'м2 kgf ni2 1 1 кгс м2~1 мм вод. ст ~9 80665 н/м2
Миллиметр водяного столба .ил» вод. ст. mmH2O J
Килограмм-сила на квадратный сантиметр кгс;см3 kgfem2 1 1 кгс'см9 — 1 ат- 98066,5 н/м3
Атмосфера техническая ат at I
Атмосфера физическая атм atm 1 атм= 1 ,01 325 105 н м2
Миллиметр ртутного столба мм рт. ст. mmHg 1 мм рт. ст. = 133,322 n/At2
93
МЕЖДУНАРОДНАЯ СИСТЕМА ЕДИНИЦ
94
Продолжение
Единица измерения Сокращенные обозначения Переводный множитель
русскими буквами латинскими или греч. буквами
Д л Ньютон на метр кубический я единиц удельного веса 1 н/.н8 1 N/m8 (см. табл. 2)
Килограмм-сила на метр кубический . . 1 кгс/м3 kgf/m3 1 кзсДи8=9,80665 н/м3
для единиц динамической вязкости
Ньютон-секунда на квадратный метр . . н-сек/м2 N-s.m2 (см. табл. 2)
Пуаз пз р 1 пз=0,1 н-сек/м2
Сантипуаз спз cP 1 спз = 10“3 н-сек/м2
Килограмм-сила-секунда на квадратный метр кгс-сек/м2 kgfs;m2 1 кгс-сек/м2=0,80665 н-сек/м2
для е д и ниц кинематической вязкости
Квадратный метр на секунду At2, сек m2/s (см. табл. 2)
Стокс ст s* 1 ст= 10 “4 м2/сек=1 см2/сек
Сантистокс сст cS< 1 сст= 10 "° м2/сек
Градус Энглера — °E (см. Энглера градусы)
для единиц работы, энергии и количес т в а теплоты
Джоуль дж J (см. табл. 2)
Эрг эрг erg 1 э рг = 10 “7 дж
Килограмм-сила-метр кгс-м kgf-m 1 кзс-л1=9,80665 дж
Киловатт-час квт-ч kW-h 1 квт-ч=3^- 10е дж
Литр-атмосфера л-атм 1 • atm 1 л • атм =101,328 дж
Электрон-вольт эв eV 1 эе = 1,60 2-10 ~ 19 дж
Калория международная кал са!!^ 1 кал = 4,1868 дж
Калория 20-градусная — cal20 1 кал->о = 4,182 дж
Калория термохимическая — cal (thermochem.) 1 кал (термохим.) = 4,1840 дж
Килокалория ккал kcal 1 ккал = 4186,8 Ож
Мегакалория Мкал Meal 1 Мкал = 4,1868-10“ дж
Термин — th 1 термия = 4,1855-Ю6 дж
для единиц мощи ости и тепло вог о потока
ватт вт w (см. табл. 2)
Эрг в секунду эрг.сек erg,'s 1 эрз/сек=10-7 вт
Килограмм-сила-метр в секунду .... кгс-м [сек kgf-m/s 1 кгс'М;сек=-0,80665 вт
Килокалория международная в час . . . ккал/ч kcal/h 1 ккал/4 = 1,163 вт
Калория в секунду кал,сек cal/s 1 кал/сек = 4,1868 вт
для единиц удельной теплоты
Килокалория на килограмм ..........
Эрг на грамм.......................
ккал/кг I
эрг/г ]
kcal'kg I 1
erg/g | 1
ккал/кг=4186,8 дж/кг
эрг/з=10 —4 дж/г
для единиц теплоемкости и энтропии
Эрг на градус.........
Килокалория на градус
эрг'град 1 erg/deg | 1 эрг[град = 10~7 дж/град
ккал/град | kcal,deg I 1 ккал/град = Ь, 1868-Ю3 дж/град
для единиц удельной теплоемкости и удельной
энтропии
Эрг на грамм-градус....................I эрг'(г-град)
Килокалория на килограмм-градус . . . ккал (кг-град)
erg/(g-deg) 1 1 эрг/(г-град) = 10~* дж(кг-град)
kcal/(kg-deg) 1 ккал/(кг-град) = 1 кал/(г-град) =
I =4,1868-Ю3 дж[(кг-град)
для единиц поверхностной плотности теплового потока
Ватт на квадратный сантиметр..........
Эрг на квадратный сантиметр в секунду
Килокалория на квадратный метр-час . .
Калория на квадратный сантиметр-секунду
вт см?
эрг [(см2-сек)
ккал[(мг-ч)
кал [(см2-сек)
W/cm2
erg/(cm2-s)
kcal/(m2-h)
cal/(cm2-s)
1 em/cAt2 = 104 вт/м2
1 эрг [(см2-сек) = 10-3 вт/м2
1 ккал (м2-ч) = 1,1 63 вт/м2
1 кал,(слг2-сек) = 4,1868-104 вт/м2
для единиц коэффициентов теплообмена (теплоотдачи) и теплопередачи
Ватт на квадратный сантиметр-градус . .
Эрг на квадратный сантиметр-секунду-гра-
дус.....................................
Килокалория на квадратный метр-час-гра-
дус.....................................
Калория на квадратный сантиметр-се-
кунду-градус ...........................
вт,(см2- г рад)
эр г [(см2-сек-град)
ккал1(м2-ч-град)
кал /(см2 сек • град)
W/(cm2-deg)
erg/(cm2-s-deg)
kcal/(m3 h-deg)
cal/(cm2-s-deg)
1 вт (см2-град) = 10* вт/(м2-град)
1 эрг[(см2-сек-град) = 10 ~a вт/(м2-град)
1 ккал,(м2-ч-град) = 1,103 вт/(м2-град)
1 кал/(см2-сек-град) =
= 4,1868-Ю4 вт/(м2-граду
для единиц коэффициента теплопроводности
ватт на сантиметр-градус ..............
Эрг на сантиметр-секунду-градус . . . .
Килокалория на метр-час-градус . . . .
Калория на сантиметр-секунду-градус
вт[(см-град)
эрг [(см-сек-град)
ккал [(м-ч-град)
кал/(см-сек- град)
W/(cm-deg) .
erg/(cm-s-deg)
kcal(m-h-deg)
cal/(cm-s-deg)
1 вт/(см-град) = Ю2 вт/(м-град)
1 эрг/(см-сек-град) = 10 — 5 вт/(м-град)
1 ккал/(м-ч -град) = 1,163 вт!(м-град)
1 кал[(см' сек-град) =
= 4,1868-Ю2 вт/(м -град)
95 МЕЖДУНАРОДНАЯ СИСТЕМА ЕДИНИЦ —МЕЖМОЛЕКУЛЯРНОЕ ВЗАИМОДЕЙСТВИЕ 96
Продол жение
Единица измерения Сокращенные обозначения Переводный множитель
русскими буквами латинскими или греч. буквами
для единиц тем Термодинамич. температурная шкала в градусах Цельсия Температурная шкала Ренкина (термоди- намическая) Температурная шкала Фаренгейта . . . Градус на сантиметр пературы и т °C град/см емпературн °C °R ор deg/ст эго градиента 0°С = 273,15°К; 1°С=1°К 0°R = 0°K; l°R = 5/9eK=5/9°C 0°F=— 17,77°С; 1ЬР=5/9°К=5/9ОС (под- робнее см. Температурные шкалы) 1 зрад/см=100 град/м
для единиц ионизирующего излучения
Поглощенная доза излучения Мощность поглощенной дозы Экспозиционная доза рентгеновского и гамма-излучения (рентген) Активность (кюри) Интенсивность излучения рад рад/сек V эреЦсек-см9) rad rad (s г ci erg/(s-cm2) 1 рад = 0,01 дж{кг 1 рад в секунду=0,01 етп/кз 1 р= 2,5 79 7 6 • 10 _ 1 к/кз (к —кулон) 1 кюри=3,700-101 °единиц CU (см. табл. 2) 1 эрз;(еек-сл12) = 10“8 вт!м2
Таблица 5. Переводные множители для некоторых
английских единиц
Единицы измерения Сокра- щенное обозна- чение Переводные множители
Ярд yd 1 yd = 0,9144 м
Ярд квадратный . . . yd2 1 yd2 = 0,836127 m2
ФуТ ft 1 ft = 0,3048 м
Дюйм in 1 ln = 0,0254 jh = 2,54 c.m
Дюйм квадратный . . . in2 1 ln2 = 0,001046 ai2 =
Род, пфль, перч .... = 6,4516 см2 1 род = 5,0292 м
Миля mile 1 mile= 1609,344 м
Миля морская .... n-mile 1 n-mile = 1852 м
Акр — 1 акр = 4046,86 м2 =
Галлон (Брит.) .... gal(UK) = 0,404686 га 1 gal = 4,54609-10 -3 «' =
Галлон (США) .... gal(US) = 4,55609 дм3 1 gal = 3,78543-10 "3 м3 =
Барелл — (США) (для ке- росина) = 3,78543 Ом3 1 барелл=
Фут в секунду .... ft/s = 158,988-Ю-3 лг3 = = 158,988 дм3 1 ft/s = 0,30 48 At/сек
Миля в час mile/h 1 mile h=0,44704 м сек
Фунт lb 1 lb = 0,45359237 кг
Гран gr 1 gr = 64,79891-Ю-’ кг =
Унция oz = 64,79891-Ю-3 г 1 oz = 28,3495 10~3 кг =
Фунт на кубический фут lb/ft3 = 28,3495 г 1 lb/ft = 16,0185 кг/At3
Фунт-сила Ibf 1 lbf = 4,44822 п '
Фунт-сила на квадрат- ный дюйм lbf/in2 1 lbf/in2 = 6894,76 н/.и2 =
Фут водяного столба ft H2O = 0,07031 хгс/CAt2 1 ftH.O = 2989,07 нй2 =
Дюйм водяного столба in HaO = 304,8 мм вод. cm. 1 in H2O = 249,089 h/m2 =
Дюйм ртутного столба in Hg = 25,4 мм вод. cm. 1 in Hg=3386,39 hm2 =
Тепловая единица бри- танская Btu = 25,4 мм pm. cm. 1 Btu = l,05506-103 дж
(н.ч3), т. к. такую запись можно читать как куби-
ческий нонаметр (1 нм3= 10-2’ м3). В этом случае
следует пользоваться выражением Ин= хм3, что зна-
чит х — метров кубических при нормальных усло-
виях (20°С, 101325 н/м3-- 760 .и.и рт. ст., где
н — ньютон).
Лит.: Международная система единиц. ГОСТ 9867—61; Об-
разование кратных и дольных единиц. ГОСТ 7663—55; Меха-
нические единицы. ГОСТ 7664—61; Электрические и магнит-
ные единицы. ГОСТ 8033—56; Тепловые единицы. ГОСТ 8550—
61; Оветовые единицы. ГОСТ 7932—56; Акустические единицы.
ГОСТ 8849—58; Единицы рентгеновского и гамма-излучений и
радиоактивности. ГОСТ 8848—58; Б у р д у н Г. Д., Едини-
цы физических величин, 2 изд., М., 1962; Единицы измерения и
обозначения физико-технических величин. Справочник, М.,
1961; Таблицы перевода единиц измерений, под ред. Н.П Ши-
рокова, М., 1963; Сто цкий Л. Р., Хим. пром-сть, 1962,
М 7,1 (467).
МЕЖМОЛЕКУЛЯРНОЕ ВЗАИМОДЕЙСТВИЕ —
действие одной электрически нейтральной молекулы
на другую, вызываемое силами притяжения или
отталкивания. М. в. обусловливает отступления от за-
конов идеальных газов, переходы от газообразного со-
стояния к жидкому, существование молекулярных
кристаллов и ряд их свойств, явления переноса (диф-
фузия, вязкость, теплопроводность), термич. релак-
сацию в газах, тушение люминесценции, уширение
спектральных линий, адсорбцию и др. Межмолеку-
лярные силы притяжения, называемые иногда силами
В а н-д е р-В а а л ь с а, много слабее валентных
сил. В то время как энергия, необходимая для раз-
рыва химич. связи, составляет 50—100 ккал/моль,
энергия М. в. составляет от десятых долей до несколь-
ких ккал/моль. Об относительной величине этой энер-
гии для разных веществ можно судить, напр., по теп-
лоте испарения жидкостей. Так, теплота испарения
воды составляет ок. 10 ккал/моль, бензола — ок.
7 ккал/моль, а жидкого азота — 0,67 ккал/моль.
При больших расстояниях между молекулами, при
к-рых их электронные оболочки не перекрываются,
превалируют силы притяжения (силы дальнодейст-
вующие); при малых расстояниях — силы отталкива-
ния (силы короткодействующие). При нек-ром рас-
стоянии между молекулами эти силы взаимно уравно-
вешиваются, причем потенциальная энергия системы
становится минимальной. Это положение равновесия
определяет размеры молекул. Половина равновесного
расстояния между одинаковыми сферически симмет-
ричными молекулами определяется как ван-дер-вааль-
сов радиус молекулы.
Короткодействующие силы имеют ту же природу,
что и силы химич. (валентной) связи и возникают при
условии, когда электронные оболочки молекул сильно
перекрываются. В конечном счете они обусловлены
тем, что при малых межмолекулярных расстояниях
электростатич. отталкивание ядер и отталкивание
электронов начинает превалировать над взаимным
притяжением ядер и электронов. Расчеты при помощи
квантовой механики показывают, что потенциал этих
сил 77ОТТ_ (г) имеет сложную зависимость от меж-
молекулярного расстояния г. Поэтому при решении
конкретных задач (7отт.(г) обычно выражают в при-
ближенной форме, к-рая имеет простой вид и часто
оказывается удовлетворительной:
^огт. (г)= Be~ar (1)
97
МЕЖМОЛЕКУЛЯРНОЕ ВЗАИМОДЕЙСТВИЕ
98
где а и В — постоянные. Как показывает сравнение
расчетов с экспериментальными исследованиями, в
частности с данными о передаче энергии при столкно-
вениях и о термич. релаксации, постоянная а для
большинства молекул варьирует в пределах от 2,5
до 3,5 А-1.
Дальнодействующие силы при достаточно больших
г можно описать строго и выразить через физич.
характеристики отдельных молекул. Как показывает
теория, при r^> d, где d — размер молекулы, различ-
ные составляющие дальнодействующих сил обратно
пропорциональны различным степеням г. Различают
три вида этих сил: электростатические, индукционные
и дисперсионные.
К электростатическим относятся силы,
действующие между различными мультиполями: ди-
полями, квадруполями и т. д. Потенциальная энергия
£/эЛ.(г), отвечающая этим силам, легко получается с
помощью закона Кулона. Если молекула имеет ди-
поль или квадруполь, то зависит также от их
ориентации в пространстве. Так как электростатич.
силы притяжения стремятся ориентировать молекулы
так, чтобы их потенциальная энергия имела минималь-
ное значение, в то время как тепловое движение нару-
шает эту ориентацию, то в среднем энергия взаимодей-
ствия молекул оказывается зависящей от темп-ры. Так,
если расстояние между молекулами много больше их
собственных размеров, то средняя энергия взаимодей-
ствия, напр., молекул, обладающих дипольными мо-
ментами ца и ць, равна:
2 2
7/ — _ i d D
u ДИП.ДИП. ~ ro W
Для энергии взаимодействия молекул, обладающих
дипольным моментом ца, с молекулами, обладающими
квадрупольным моментом Qb, теория дает:
1 »4Qb
^дип. квадр. = ~(3)
Знак минус означает, что при сближении молекул
энергия понижается и, следовательно, молекулы вза-
имно притягиваются. Если взаимодействуют между
собой на большом расстоянии два радикала или два
атома, ни один из к-рых не находится в «-состоянии,
то большое значение приобретает их квадруполь —
квадрупольное взаимодействие.
Индукционные силы имеют также
электростатич. природу, но, в отличие от сил первого
вида, возникают вследствие того, что одна из молекул,
обладающая мультиполем, поляризует другую моле-
кулу и индуцирует в ней дипольный момент, притяже-
ние к-рого к мультипольному моменту и обусловли-
вает взаимодействие молекул. Если одна из электри-
чески нейтральных молекул обладает дипольным мо-
ментом |д.а, а другая неполярна, то средняя энергия
этого взаимодействия равна:
^инд.=-----(4)
где аь— поляризуемость второй молекулы. Эта форму-
ла получается в предположении, что поляризуемость
молекулы изотропна, т. е. не зависит от направления.
Согласно (4), индукционная энергия не зависит от
темп-ры, что связано с предположением об изотроп-
ности молекулы Ь.
Дисперсионные силы имеют более
сложную природу. При простейшей трактовке (в
рамках представлений классич. физики) можно нари-
совать след, картину происхождения этих сил. Рас-
смотрим в молекуле к.-н. один электрон. Вследствие
того, что он движется в среднем на нек-ром конечном
расстоянии от ядер, в молекуле в каждый данный мо-
мент времени возникает диполь, к-рый в случае элект-
рически симметричной молекулы в среднем равен
нулю. Однако этот мгновенный диполь индуцирует
диполь в другой молекуле. Такое же действие оказы-
вают все другие электроны. Взаимодействие между
мгновенными диполями молекул а и b и приводит к
их притяжению. Дисперсионные силы — наиболее
общие силы М. в., действующие между любыми моле-
кулами или атомами при больших расстояниях.
Их название связано с тем, что в соответствующую
формулу для энергии входит выражение, к-рое фигу-
рирует в теории дисперсии света.
Для сферически симметричных молекул теория при-
водит к след, выражению для энергии дисперсионных
сил:
^дисп. — (5)
где „ 3 ЕЪ
С~2£а+Вьаааъ 5а
Е3 и Еь приблизительно равны энергиям ионизации
молекул а и Ь, аа иаь— их поляризуемости. При более
полной трактовке в выражение для {7ДИСП- входят также
члены; содержащие 1/г в более высоких степенях. Ес-
ли молекулы не обладают сферич. симметрией, то вы-
ражение для дисперсионной энергии будет другим.
Так, в случае цепочечных молекул с сопряженными
связями закон г~в начинает действовать только при
условии, что расстояние г между центрами тяжести
молекул больше 2L, где L — длина сопряженной це-
почки. При этом, однако, величина С в формуле (5)
приобретает иной вид и оказывается зависящей от
длины молекул и от их взаимной ориентации. Для
достаточно длинных цепочек при параллельном
расположении при условии, что оба центра тяжести
лежат на одной и той же нормали к осям молекул,
дисперсионная энергия равна:
гг ___
и ДИСП. я» Гф1 W
где I — длина одной связи, е — заряд электрона,
Р — т. н. обменный интеграл (для углеводородных
цепочек он приблизительно равен 40 ккал/моль). На
рис. 1 показано как при L=6Z (гексатриен) при раз-
личных г изменяется ^дисп. при смещении одной из мо-
лекул параллельно другой (варьирование координа-
ты х). Замечательно, что при нек-ром х Uдисп- имеет
Рис. 1. Зависимость дис-
персионной энергии от х
при L=6i (гексатриен)
при различных расстоя-
ниях т между центрами
тяжести молекул.
Рис. 2. Зависимость дисперсии
онной энергии от х при г=8 А
для сопряженных цепочек
различной длины L.
минимум. На рис. 2 изображена зависимость £7ДИСП от х
для молекул различной длины (при г=8 А). Диспер-
сионная энергия зависит также от угла поворота одной
молекулы относительно другой; при этом оказывается,
что при угле, лежащем где-то между 60 и 90°, энер-
гия имеет максимум, к-рый, очевидно, при низких
04 К.Х.Э. т. 3
99
МЕЖМОЛЕКУЛЯРНОЕ ВЗАИМОДЕЙСТВИЕ
100
темп-pax будет препятствовать вращению молекул.
Заметим, что формула (5) перестает быть верной
не только для молекул, лишенных сферич. симметрии,
но и в том случае, если расстояние между их центрами
тяжести становится очень большим, именно, если оно
значительно превосходит длину световой волны Л,,
отвечающую наиболее интенсивному электронному
переходу в молекуле. Истинное значение энергии дис-
персионных сил по абс. величине при этом оказывает-
ся меньше того, к-рое следовало бы ожидать согласно
(5) при таких межмолекулярных расстояниях. Этот
эффект объясняется тем, что скорость распростране-
ния электромагнитного поля имеет конечную величи-
ну. Мгновенный диполь первой молекулы излучает
электромагнитную волну, к-рая распространяется со
скоростью с и через время ч. = г1с достигает второй мо-
лекулы, индуцируя в ней мгновенный диполь. Излу-
чаемая последним вторичная электромагнитная волна,
достигнув через такое же время т первой молекулы, в
свою очередь взаимодействует с ее диполем, к-рый за
время 2т уменьшился и изменил направление, вслед-
ствие чего это взаимодействие будет ослабленным.
Этим эффектом «запаздывания» можно пренебречь в
случае небольших молекул, но он может оказаться
существенным в случае высокомолекулярных соеди-
нений и коллоидных систем.
При г d полная энергия М. в. будет равна сумме
членов, выражающих различные виды М. в. По фор-
мулам (2)\ (4) и (5) легко оценить относительный
вклад дипольного, индукционного и дисперсионного
М. в. Для этого достаточно сравнить величины
гв^дип.дип.> гв^индл 'г6^дисп.- Как показывает расчет,
для слабо полярных молекул (СО, HJ и др.) первые две
величины на несколько порядков меньше, чем г6С/дИСП
Для молекул с большими дипольными моментами
(Н2О, ГШз, НС1) гвС/дИП.днп. начинает превалировать
над дисперсионным членом; индукционный член почти
всегда оказывается весьма малым и в первом при-
ближении им можно пренебречь.
При межмолекулярных расстояниях, сравнимых с
размерами молекул d, приведенные формулы (2—5)
становятся неверными. При этом истинная энергия
взаимодействия уже не складывается аддитивно из
указанных величин и оказывается заметно меньше
той, к-рую следовало бы ожидать, если бы закон г~е
сохранялся вплоть до этих расстояний. Вследствие
этого и само разделение межмолекулярных сил на
электростатические, индукционные и дисперсионные
при средних расстояниях становится чисто условным.
Нахождение U при таких расстояниях связано с очень
сложными расчетами, поскольку здесь существенную
роль начинает играть перекрывание электронных обо-
лочек молекул. Соответствующие вычисления были
проведены только для нек-рых простейших систем
(атом Не, молекулы Н2, N2 и нек-рые др.).
Однако с практич. точки зрения промежуточные
значения г представляют наибольший интерес. В са-
мом деле, в большинстве физич. явлений молекулы
находятся вблизи положений равновесия, когда силы
притяжения и силы отталкивания по абс. величине
становятся сравнимыми между собой. Чтобы воспол-
нить указанный пробел, прибегают к различного рода
полуэмпирич. приближенным ф-лам, к-рые по сути
дела представляют собой более или менее удачную
интерполяцию U(r) в область промежуточных меж-
молекулярных расстояний на основе выражений для
очень больших и очень малых г. Одной из таких ин-
терполяционных ф-л, применимых для сферически
симметричных молекул, является потенциал Букин-
гема: U(r) = Be~ar — сг-*—с'г~а (7)
содержащий четыре параметра, подбираемых эмпири-
чески. Однако практически более удобен потенциал
Леннарда — Джонса, имеющий вид:
7/(г) = 48[(Н-),2-0)в] (8)
(т. наз. «потенциал 6—12»), где 8 имеет смысл глубины
потенциальной ямы, минимуму к-рой отвечает рассто-
яние г0=2 /"а. Вместо экспоненциального закона
здесь выбрана 12-я степень 1/г, что сделано исключи-
тельно с целью облегчения математич. расчетов. В
случае дипольных молекул к ур-нию (8) иногда при-
бавляется член, выражающий диполь — дипольное
взаимодействие — и зависящий от углов между молеку-
лами (Штокмейер). Имеются также и другие интерпо-
ляционные формулы.
Значения параметров, входящих в полуэмпирич.
потенциал U(r), можно найти изданных о таких свой-
ствах веществ, в к-рых играют роль межмолекуляр-
ные силы. С этой целью можно воспользоваться экс-
периментальными данными о втором вириальном ко-
эфф., данными о процессах переноса, значениями ко-
эфф. Джоуля—Томсона, данными о критич. точке,
точке плавления или кипения, о теплоте сублимации
молекулярных кристаллов, их сжимаемости и др.
[Вторым вириальным коэфф. (Р) наз. величина, фигу-
рирующая в теории неидеальных газов и связанная с
постоянными а и b ур-ния Ван-дер-Ваальса след, соот-
ношением: $—2а/кТ— 26]. Критерием качества выбран-
ного для расчета потенциала может служить, напр.,
сравнение вычисленной и измеренной зависимости
рассматриваемой величины от темп-ры или сравнение
значений параметров, получаемых из различных
свойств. Темп-рная зависимость второго вириального.
коэффициента, коэффициентов переноса и других ве-
личин, найденная при помощи разных потенциалов,
обычно получается вполне удовлетворительной. Зна-
чения параметров для данной формы U(r), найденные-
из различных свойств вещества, наоборот, далеко не
всегда совпадают. В качестве примера в таблице при-
ведены значения параметров а и 8 Леннарда—Джонса
для нек-рых газов, вычисленных по значениям второго»
вириального коэфф, и коэфф, вязкости.
Газ Значения параметров, найденных из
данных о втором вириальном коэфф. данных о вязкости
е (кал/моль) П(А) е (кал/моль) О (А)
Ne 55 2,86 69 2,78
Аг 247 3,42 238 3,41
н2 66 2,97 58 2,87
n2 159 3,75 191 3,71
n2o 438 3,88 3 77 4.5 9
Этан 458 4,42 484 3,95
Этпл ен 408 4,23 397 4,52
Столь заметные расхождения связаны, разумеется,
с приближенным характером потенциала. По-видимо-
му, значениями параметров, полученными из данных
о втором вириальном коэфф., следует пользоваться
при расчетах равновесных свойств вещества, а полу-
ченными из данных о вязкости — при расчетах нерав-
новесных свойств (процессы переноса, релаксация и
др.). Однако, как показывает анализ, получаемые из
данных о вязкости потенциальные кривые приводят
к неудовлетворительным результатам, если ими поль-
зоваться для описания передачи энергии и релакса-
ции при сильных столкновениях (ударные волны, вы-
сокие темп-ры, химич. реакции и др.). Для этих про-
цессов предложены специальные способы интерполя-
ции Щг), однако вопрос в целом нельзя считать ре-
шенным. Расчеты различных коэфф, для смесей газов
более затруднительны и результаты сильнее отлича-
101
МЕЖЪЯДЕРНЫЕ РАССТОЯНИЯ
102
ются от экспериментальных данных, чем для чистых
газов.
Частными случаями М. в. являются водородная
связь и т. наз. резонансное взаимодействие. Последнее
возникает между одинаковыми молекулами, если одна
из них находится в электронно возбужденном состоя-
нии. Взаимодействие обусловлено тем, что при сбли-
жении молекул возбуждение, так сказать, переходит
от одной молекулы к другой и обратно. В результате
в возбужденном состоянии оказывается не одна моле-
кула, а. обе, причем уровень энергии этого состояния
уже иной, чем для отдельной молекулы. Энергия
притяжения, обусловленная резонансным взаимодей-
ствием, может достигать 1—3 ккал/молъ и быть замет-
ной при межмолекулярных расстояниях от 5 до 20 А
(в зависимости от природы молекул).
Особый вид М. в. представляет собой т. наз. донор-
но-акцепторное взаимодействие молекул, осуществ-
ляющее одну из форм координационной связи и по
своей природе близкое к ковалентной связи. Типич-
ным примером может служить взаимодействие между
молекулой аммиака и молекулой фтористого бора,
приводящее к образованию устойчивого комплекса
H3N ... BF3. М. в. здесь в основном обусловлено парой
электронов атома N, не участвующих во внутримоле-
кулярной связи в NHs (т. наз. неподеленная пара элек-
тронов). Эта электронная пара становится общей для
атомов N и В, причем первый т. обр. выступает в
роли донора, а второй — в роли акцептора электро-
нов. Энергия донорно-акцепторного взаимодейст-
вия варьирует в широком интервале и иногда дости-
гает энергии ковалентной связи. Различие между по-
следней и донорно-акцепторным взаимодействием за-
ключается в основном в происхождении связывающей
электронной пары: в обычной ковалентной связи не
один атом, а каждый из атомов в молекуле дает в со-
вместное пользований по одному электрону.
М. в. всегда представляет собой первую стадию
элементарного акта химич. бимолекулярной реакции.
Однако зависимость скорости реакции от характера
М. в. изучена очень мало. Приближенно можно при-
нять, что для данного типа реакции: А + В-^ С,
при варьировании одного из компонентов более силь-
ному притяжению А....В при больших расстояниях
будет отвечать более низкая энергия активации реак-
ции. Исходя из этой гипотезы, теоретически были вы-
числены нек-рые характеристики ряда углеводород-
ных молекул с сопряженными связями, влияющие на
энергию М. в. (напр., эффективные заряды атомов
и т. наз. индексы свободной валентности); на основа-
нии полученных результатов оказалось возможным
судить об относительной реакционной способности
различных молекул или различных центров в одной и
той же молекуле.
Лит.: Гиршфельдер Д ж., Кертисс Ч., Берд
Р., Молекулярная теория газов и жидкостей, пер. с англ., М.,
1961. Н. Д. Соколов.
МЕЖЪЯДЕРНЫЕ РАССТОЯНИЯ — расстояния
между ядрами атомов или ионов в молекуле или кри-
сталлич. решетке. Выражаются обычно в ангстремах.
М. р. являются важными структурными характерис-
тиками вещества. Вследствие колебания ядер М. р.
не остаются строго постоянными. В качестве констант
используют равновесные М. р. (ге), т. е. расстояния
между ядрами в положении равновесия, что соответ-
ствует гипотетич. состоянию в отсутствии колебаний (в
минимуме кривой потенциальной энергии атомной сис-
темы). Часто используют также средние М. р. (г0)
для низшего (основного) колебательного уровня. От-
клонение колеблющихся ядер от положения равнове-
сия в сторону их сближения всегда меньше, чем в сто-
рону удаления друг от друга, из-за очень быстрого ро-
ста сил отталкивания при сближении ядер. сПоэтому
г0 несколько больше ге (на тысячные доли А).
М. р. обнаруживают ряд закономерностей. Так, в
двухатомных молекулах элементов в ряду Li—F
М. р., максимальное для Li2, убывает с увеличением
атомного номера, проходит через минимум в азоте и
далее возрастает (см. табл. 1). Аналогичное изменение
Таблица 1. Межъядерные расстояния в двухатомных
молекулах элементов
Молекула re, А Молекула ге, А
Н2 0,7414 рГ 1 ,894
ч 2,6723 с32 Ь2 1,889
ВГ 1.589 С1“ 1,988
с’2 1,3117 К’9 3,923
1 ,094 Сл80 Se2 2,16
°” 1,2074 ВГ’Вг*1 Те3 2,284 2,59
Na” 1,435 3,079 2,667
М. р. наблюдается и в других периодах системы эле-
ментов. Для двухатомных водородных соединений
элементов имеет место четкая периодич. зависимость
М. р. от атомного номера. Для изотопных молекул
М. р. остаются практически неизменными; макси-
мальная разница может составлять лишь десятиты-
сячные доли А. Весьма важное значение имеет прибли-
зительное постоянство (в пределах сотых Долей
А) М. р. для связи двух данных элементов в различных
соединениях, если сохраняются валентное состояние и
вид связи элементов. Такие М. р. характеризуют т. н.
длину связи (г). Нек-рые наиболее характер-
ные длины связей приведены в табл. 2. я','.
Таблица 2. Характерные длины связей 1
Свя зь гЛ Связь
С—Н 1,09 C = N 1,15
N—Н 1 ,03 С-0 1,42
о-н 0,97 с=о 1,21
S—н 1,35 C-F 1,39
с—са 1,54 1 С—С1 1,77
С—Вг 1,92
С-С 1,34 С—J 2,10
с=с 1,20 N—N 1,45
С —С 6 1 ,40 N = N 1.24
C-N 1,46 О—О 1,48
а б
Без сопряжения. Ароматич. соединения.
На характерных значениях М. р. основаны также
такие понятия, как ковалентный радиус и ионный ра-
диус. В случае кристаллов М. р. зависят не только от
вида химич. соединения, но и от типа кристаллич.
решетки (см. Кристаллы). Эти М. р. значительно от-
личаются от М. р. в молекулах тех же соединений.
Напр., М. р. в кристаллах KF, КС1, КВг и KJ .со-
ставляют соответственно 2,664, 3,138, 3,285 и 3,525 А,
тогда как в соответствующих двухатомных молекулах
эти расстояния равны 2,55, 2,79, 2,94 и 3,923 А; в
кристаллич. решётках элементов Li, Na, К, Se, С
(алмаз), С (графит) М. р. составляют соответственно
3,039, 3,716, 4,544, 2,321, 1,5445 и 1,4210 А, вместо
значений, указанных в табл. 1 для двухатомных мо-
лекул.
Наиболее точное экспериментальное определение М. р. ос-
новано на измерении моментов инерции молекул путем анализа
вращательной структуры молекулярных спектров (электрон-
ных, инфракрасных, комбинационных и микроволновых).
4*
103
МЕЗАКОНОВАЯ КИСЛОТА—МЕЗИТИЛЕН
104
Однако молекула обладает не более чем тремя независимыми
моментами инерции, измерение к-рых дает не более трех урав-
нений для вычисления М. р. Непосредственное определение
М. р. указанным путем выполнимо лишь для молекул, в к-рых
количество неодинаковых М. р. не превышает числа незави-
симых моментов инерции (максимум трех). К подобным моле-
кулам относятся все двухатомные молекулы, симметричные
трехатомные молекулы типа линейных (СО2, CS2 и др.), или
треугольных (Н2О, HgS, SO2 и др.), а также особо симмет-
ричные многоатомные молекулы типа СН4, СС14 и др. В более
сложных случаях, когда неодинаковых М. р. больше, чем
независимых моментов инерции, получают дополнительные
данные путем измерения моментов инерции изотопных молекул,
используя неизменность М. р. прн изотопном замещении. Этот
способ применяют в основном для водородных соединений,
т. к. из всех изотопов только изотопы водорода существенно
изменяют моменты инерции молекул. Точность спектроскопия,
измерения М. р. доходит до 10”4 А. На порядок менее точным,
но зато применимым к любым молекулам, а также к кристаллам,
является определение М. р. методами рентгеноструктурного
анализа, электронографии и нейтронографии. Эти методы ос-
нованы на зависимости распределения интенсивностей рент-
геновых лучей, электронных или нейтронных потоков при
дифракции на молекулах или кристаллической решетке от
М. р. Расшифровка подобных рентгено-, электроно- или ней-
тронограмм обычно весьма сложна и сводится к вычислению
расположения ядер методом подбора до совпадения вычис-
ленной дифракционной картины с экспериментально полу-
ченной.
Лит.: Кондратьев В. Н., Структура атомов и моле-
кул, 2 изд., М., 1959; Волькенштейн М. В., Строение
и физические свойства молекул, М.—Л., 1955; Герцберг
Г., Спектры и строение двухатомных молекул, пер. с англ.,
М., 1949; его же, Колебательные и вращательные спектры
многоатомных молекул, пер. с англ., М., 1949; Справочник хи-
мика, т. 1, Л., 1962. С. Э. Вайсберг.
МЕЗАКОНОВАЯ КИСЛОТА — см. Цитраконовая
кислота.
МЕЗАТОН [адрианол, нео-синефрин, хлоргидрат-1-
(3-оксифенил)-2-метиламиноэтанола] CJiuNC^Cl, мол.
в. 203,67 — белый кристал-
(У CHCH2NHCHa на лич- П0???01$,^ез запаха’
. / j 2 3 т. пл. 142—146 ; хорошо
он растворим в воде, спирте и
разб. щелочах; нерастворим
в эфире. При прибавлении FeCls к водному р-ру М.
появляется сине-фиолетовое окрашивание; с AgNOs
М. дает в азотнокислой среде белый осадок AgCl.
Количественно М. определяют титрованием по Фоль-
гарду. М. обычно получают из ацетофенона. М.
близок по характеру действия к адреналину, вызы-
вает повышение кровяного давления, сужение пери-
ферических кровеносных сосудов, расширение брон-
хов и зрачков, торможение перистальтики кишеч-
ника. В одинаковых дозах менее активен, но более
стоек, чем адреналин. М. применяют для повышения
кровяного давления при шоковых состояниях, коллап-
се, гипотония, болезни, кровопотерях и проч., а так-
же для сужения сосудов и уменьшения воспалитель-
ных явлений при вазомоторном и сенном насморке,
конъюнктивитах и др.
Лит.: Машковский М. Д., Лекарственные средства,
4 над., М., 1960, с. 149; Фармакопея СССР, 9 изд., М., 1961.
В. Г. Яшунский.
МЕЗИДИН (аминомезитилен, 2,4,6-триметилани-
лин), CgHisN, мол. в. 135,21 — жидкость, незамерза-
NH ющая при —15°, т. кип. 232—233°;
i2 d’0 0,9633; образует устойчивые соли
СНз В I ~Нз с гал°ген0В0Д0Р°Дными к-тами, ук-
Чу сусиой, щавелевой и тринитробен-
Т.,, зойной к-тами; с SnCh и PtCh дает
“ 3 двойные соли. М. легко ацетилирует-
ся (N-ацетил — призмы, т. пл. 216—217°) и бензо-
илируется (N-бензоил — иглы, т. пл. 204°); при на-
гревании с избытком CH3J дает РГ,М-диметилмезидии.
М. взаимодействует с альдегидами с образованием
азометиновых производных. М. легко диазотируется,
при сочетании с азокомпоиентами получаются азо-
красители. М. вступает в реакции электрофильного
замещения. При нитровании М. при 0° в р-ре H2SO4,
в зависимости от количества HNOs (1 или 2 моля),
образуются 4-нитро- или 4,6-динитро-2-амино-1,3,
5-триметилбензолы. При окислении М., в зависимости
от условий, образуются 2-нитромезителен или 2,6-
диметилбензохинои. Получают М. восстановлением
иитромезитилена; возможно получение каталитич.
алкилированием n-толуидина. Наибольшее значение
М. имеет в произ-ве кислотных антрахиноновых
красителей. В. А. Пучков.
МЕЗИЛ — условное краткое обозначение остатка
метансульфокислоты CH3SO2O—. Эфиры этой к-ты
CH3SO2OR называются мезилатами (ср. Тозил).
МЕЗИТИЛА ОКИСЬ (2-метилпентеи-2-он-4)
(СНз)2С=СН—СО—СНз, мол. в. 98,46— жидкость мят-
ного запаха; т. пл. —59,0°, т. кип.: 130—131°/76О мм,
34—35°/11 мм; 0,8548; Пд 1,4475; в воде ра-
створяется 2,8—3,1 вес. % (20°), растворимость
воды в М. о. 3,1—3,4 вес. %(20°); смешивается
со спиртом и другими органич. растворителями.
Жидкая М. о. представляет собой таутомерную смесь
собственно М. о. со след, ненасыщенным спиртом
(СНз)2С=СН—С(ОН)=СНг, к-рый при обычных усло-
виях содержится в смеси в количестве ок. 6%.
2,4-Динитрофеиилгидразои, т. пл. 200°; оксим суще-
ствует в двух формах: а, т. кип. 84°/11 мм (лабильная
форма), ир, т. пл. 49°, т. кип. 92°/9 мм; a-форма пре-
вращается в p-форму при нагревании гидрохлорида;
P-форма при действии щелочи снова превращается в
а-форму; семикарбазои также существует в двух фор-
мах:», т. пл. 164°, и р, т. пл. 133—134°. М. о. реагирует
как а, р-неиасыщенный кетон: при окислении происхо-
дит расщепление двойной связи; с Н2О2 в щелочной
среде дает соответствующий эпоксид; при гидрирова-
нии дает насыщенный кетон; изопропилатом алюминия
восстанавливается в соответствующий ненасыщенный
спирт; с бензолом в присутствии А1С1з дает метил-
-р-фенилизобутилкетои, с эфиром малоновой к-ты об-
разует 1,1-диметил-3,5-циклогексаидион {димедон);
двойная связь в М. о. отличается высокой реакционной
способностью: легко присоединяются бром, гипогало-
генные к-ты. синильная к-та, аммиак, амины и др.;
при действии бисульфита натрия происходит присо-
единение двух молекул этого реагента — одной по
двойной связи, другой — по карбонильной группе;
при встряхивании с 1%-ной НС1 присоединяется мо-
лекула воды с образованием диацетонового спирта.
При нагревании с разб. к-тами и щелочами М. о. гид-
ролизуется в ацетон.
В пром-сти М. о. производится дегидратацией ди-
ацетонового спирта иодом, щавелевой к-той, минераль-
ными к-тами, NaHSOa, хлоридами и бромидами желе-
за ит. д.; из ацетона, действием таких дегидратирую-
щих агентов, как НС1, H2SO4, ZnC12, AICI3 (при по-
вышенных температурах) и т. д. М. о. является хо-
рошим растворителем для нитроцеллюлозы и по-
ливиниловых смол. При длительном контакте с воз-
духом М. о. осмоляется, стабилизируется диизопро-
пипамином; при длительном хранении могут обра-
зоваться перекиси, что создает опасность взрыва при
перегонке. Токсичность М. о. ниже токсичности
ацетона. Л. С. Поваров.
МЕЗИТИЛЕН (1,3,5-триметилбензол), мол. вес
120,19 — бесцветная жидкость с ароматичным запа-
хом; т. пл. —44,72°; т. кип. 164,72°;
d2° 0,8652; Пд 1,4991; хорошо раство-
рим в спирте, эфире, ацетоне, хло- j |)
реформе, бензоле; плохо — в воде (ме- СНз^Х/^СНз
нее 0,01%); горит и образует взрыво-
опасные паро-воздушные смеси; ядовит (в большей
степени нежели бензол), вызывает острые и хрони-
ческие поражения кроветворных органов (костного
мозга, селезенки). М.— типичное ароматическое
соединение: легко алкилируется, хлорируется, суль-
105
МЕЗО... —МЕЗОМЕРИЯ
106
фируется и нитруется; при окислении превращается
в мезитиловую (I), увитииовую (II) и тримезиновую
(III) к-ты:
М. получают дегидратацией ацетона при действии
серной к-ты; образуется при уплотнении метил-
ацетилеиа; при метилировании л-ксилола по Фриде-
лю — Крафтсу; содержится в жидких продуктах кре-
кинга и пиролиза углеводородов. М.— растворитель
лаков и красок, высокооктановая добавка к бензинам,
исходное сырье для приготовления различных пре-
паратов.
Лит.: Kirk, V. 7, N. Y., 1952, р. 612. Г. А. Сокольский.
МЕЗО... — приставка, применяемая перед названи-
ем соединений, содержащих два асимметрич. центра
и лишенных оптич. активности вследствие внутренней
компенсации. В отличие от рацематов, к-рые могут
быть различным образом разложены на оптически де-
ятельные формы, мезосоединеиия в принципе ие могут
быть расщеплены. Типичными примерами мезо-со-
едииеиий могут служить мезовинная (аитивиииая) к-та
НООССНОНСНОНСООН (см. Винные кислоты), мезо-
форма дибромяитариой к-ты НООССНВгСНВгСООН
и т. д. По физич. свойствам мезосоединеиия отличают-
ся как от оптически деятельных форм, так и от раце-
матов. Приставка ,М. применяется также для обозна-
чения положений в
среднем ядре некото-
рых полициклич. си-
стем. Так, положения
9 и 10 в аитрацеие (I)
обозначаются как ме-
мезосоединениям отно-
зо-положеиия. Аналогично
к
сят вещества, содержащие заместители в положении
9 В акридине (II). я. Ф. Комиссаров.
МЕЗОВИННАЯ КИСЛОТА — см. Винные кислоты.
МЕЗОКСАЛЕВАЯ КИСЛОТА НООС—СО—СООН,
мол. в. 118,05 — двухосновная кетокислота, суще-
ствующая в соединении с Н2О. Благодаря электро-
иоакцепториому влиянию двух соседних карбоксиль-
ных групп, карбонильная группа в М. к. гидратирова-
на (подобно тому, как это имеет место в хлорале). По-
этому М.к. существует в виде диоксималоиовой кислоты
НООС—С(ОН)г—СООН, ст. пл. 121°. При нагревании
вводе М. к. расщепляется иа гликолевую к-ту и СОг,
а при действии щелочи — иа щавелевую и муравьи-
ные к-ты. М. к. вступает в обычные реакции кетоиов,
образует гидразон, оксим, восстанавливается амаль-
гамой натрия (до тартроиовой к-ты) и т. д. М. к. (I)
коидеисируется с мочевиной (II) в аллоксан (III):
СООН NH. CO-NH
I I II
с=о + с=о со со
I I II
СООН NH2 CO-NH
1 II III
М. к. получают окислением малоновой к-ты азотистой
к-той. Производные М. к. по карбоксильным группам
существуют как в виде гидратов (т. е. производных
диоксималоиовой к-ты), так и в виде производных ке-
токислоты. Диметиловый эфир М. к., т. пл. 81°, т.
кип. 106°/40 .ы; диэтиловый эфир, т. пл. 57°, т. кип.
117731 мм: гидразон, т. пл. 80°; феиилгидразон, т.
ПЛ. 160°. Н. А. Несмеянов.
МЕЗОМЕРИЯ, или мезомериый эффект,— постоян-
ное смещение электронных пар в молекуле по отноше-
нию к их расположению, отвечающему классич.
структурной ф-ле, приводящее к тому, что действитель-
ное строение молекулы является промежуточным меж-
ду строениями, описываемыми двумя или более ф-лами
(термин происходит от греч. цёсгод — средний). Эти
ф-лы должны отвечать одинаковому расположению
атомов в пространстве, ио различной локализации
химич. связей. Теория М. разрабатывалась преим.
К. Иигольдом, начиная с 1926, и явилась развитием
более раииих химич. теорий. Физич. природа элект-
ронных смещений, постулировавшихся теорией М.,
была разъяснена Л. Полингом в резонанса теории;
последняя и теория М. в настоящее время ие разли-
чаются.
Наиболее характерные проявления М. наблюдаются
в сопряженных системах, т. е. при чередовании про-
стых и двойных связей (мезомериый эффект называют
также эффектом сопряжения). Напр., в молекуле с
классич. структурной ф-лой:
Н.
)N-с=с-с=О ...
HZ 111 <’>
н н н
согласно теории М., электронные пары смещены в на-
правлениях, указанных изогнутыми стрелками:
1 НуГк Г*
(2)
и Н НН
Доведенное до конца, это смещение дало бы локали-
зацию связей, отвечающую ф-ле:
(3)
Действительно,, если иеподелеииая пара электронов
атома N перейдет в общее обладание атомов N и С,
то иа долю N придется лишь 1 электрон пары, так что
N превратится в N+, и т. д. Предельная ф-ла (3)
согласуется с правилами валентности: иои N+ цро-
электроиен с атомом С и, следовательно, четырехва-
лентен, а ион О_, изоэлектронный с атомом F, одно-
валентен (ср. N+Hi, О-Н). Вместо ф-лы (2) теории М.
для описания строения рассматриваемой молекулы
можно, следуя Полингу, использовать совокупность
ф-л (1) и (3). Стрелочную запись иногда дополняют
знаками (S + ), (6—), показывающими концевые заряды,
созданные электронным сдвигом, при этом буква S
обозначает, что заряды составляют нек-рую долю от
элементарного электрич. заряда:
(6+) р-с=с-с-о(г-)
Н н Н н
В замкнутых цепях сопряжения, содержащихся в
ароматич. соедииеииях, мезомериый эффект может
осуществляться без возникновения концевых зарядов.
Пример — М. в молекуле бензола:
благодаря к-рой все связи в кольце равноценны.
Различные мезомерные смещения электронных пар
могут накладываться друг иа друга. В молекуле
аиилииа наряду с М. бензольного ядра, отвечающей
ф-ле (5), представлен мезомериый эффект, записы-
ваемый след, образом:
(в)
107
МЕЗОНЫ —МЕЛАМИН
108
Ф-ла (6) является совмещением записи электронных
смещений
НЧ /н н\ /н НС/Н
U+) (а+) р (а+) гл
0 (Й->С0
(<Н
к-рым соответствуют
предельные формулы
содержащие трехвалентный ион С-. Правильно со-
ставленная ф-ла, содержащая стрелочное обозначение
М., всегда может быть расшифрована подобным об-
разом. Такая ф-ла показывает некоторые свойства
молекулы, не отражаемые классич. структурной
ф-лой. Так, напр., в случае анилина избыточный поло-
жительный заряд атома N затрудняет присоединение
к нему Н +, поэтому анилин более слабое основание,
чем первичные амины жирного ряда. Повышенная
электронная плотность в орто- и пара-положениях
облегчает атаку электрофильными реагентами; этим
определяется ориентация заместителей (при амино-
группе «защищенной» от солеобразования).
Лит.: Ингольд К. К., Механизм реакций и строение
органических соединений, пер. с англ., М., 1959.
М. И. Темкин.
МЕЗОНЫ — группа неустойчивых элементарных
частиц с массами, промежуточными между массами
электрона и протона. Существуют электрически заря-
женные (+ и —) и нейтральные М. По величине заряд
М. (как так и —) равен элементарному электрич.
заряду. Спин М. равен нулю, магнитный момент у них
отсутствует. В группу М, входят след, семь элемен-
тарных частиц: три л (пи)-мезона — два заряженных
л + и л~ (масса составляет 273 массы электрона — м. э.,
среднее время жизни т=2,5-10_8 сек) и нейтральный
Л° (масса 264 м. э., т=0,1-10-18 сек) и четыре К.-ме-
зона— два заряженных К+ и К- (масса 966 м. э.,
т=1,2-10~8 сек) и два нейтральных К° и К° (масса
974 м. э., т = 9-10-11 се» или 6,3 • 10_8 сек — в зависи-
мости от состояния при распаде). М. эффективно обра-
зуются в ядерных реакциях под действием частиц высо-
ких энергий (в космич. лучах и на ускорителях) и
столь же эффективно взаимодействуют с нуклонами.
Считается, чтоМ. являются основными переносчиками
ядерных сил.
Название «М.» (от гпеч. ftecrog — средний, промежу-
точный) было присвоено прежде всего так наз. ц (мю)-
мезонам, открытым в 1937 — раньше всех других не-
устойчивых частиц с массой больше массы электрона.
Масса этих частиц (ц+иц-) 207 м. э., т=2,2 •10-6се»,
спин их равен не нулю, как для л и К, но У2& (где А=
=/г/2л, h — постоянная Планка). В отличие от других
М., р + и ft- не возникают в ядерных взаимодействиях,
но образуются лишь при распаде л- или К-мезонов.
Взаимодействие с нуклонами для ц+ и несравненно
слабее, чем для л- и К-мезонов. Поэтому в настоящее
время ft-мезоны включают не в число М., но в другую
группу элементарных частиц — т. наз. лептонов (см.
Элементарные частицы).
Иногда М. наз. также очень короткоживущие
(<10-г1 сек) промежуточные резонансные состояния,
возникающие при ядерных взаимодействиях высокой
энергии и распадающиеся на несколько л- или К-ме-
зонов или у-квантов. Известно уже более полусотни
таких СОСТОЯНИЙ. В. И. Гольдамский.
МЕЙЕРА РЕАКЦИЯ — см. Майера реакция.
МЕЙСТЕРА — МИШЕРА РЕАКЦИЯ — см. Бар-
бье — Виланда расщепление.
МЕКОНОВАЯ КИСЛОТА С,Н4О7, мол. в. 200,10-
кристаллы, из воды кристаллизуется с 3 молекулами
Н2О, воду теряет при 100°. М. к. имеет циклич.
строение (I), тригидрат — ациклич. (II). При кипячении
О
Л ОН
соон.
НООН ОнОН
и
с водой М. к. выделяет СО2, образуя сначала к о м е-
новую к-ту (III), а затем p-окси-у-пирон (IV).
С хлорным железом М. к. дает ярко красное окраши-
вание. М. к. содержится в маке в виде солей ал-
калоидов опия, синтетически может быть получена из
ацетондищавелевой к-ты. Диметиловый эфир, т. пл.
117°; моноамид, Т. ПЛ. 65°. Н. А. Несмеянов.
МЕЛАМИН (2,4,6-триамино-х-триазин, 2,4,6-три-
амино-1,3,5-триазин) C3H6Ne, мол. вес 126,13 — бес-
цветные кристаллы (из воды — моно-
клинич. призмы); т. пл. 354° (с отще- HoN-c^'c-nh
плением NH3 и образованием меле- « 2
ма и мелона); d22 1,571. 'у''
Растворимость М. в воде меньше nh2
0,5% (при 20°), ок. 4% при 90°.
Растворимость в гликоле, пиридине, глицерине близ-
ка к растворимости в воде; в спирте — мало раство-
рим; в большинстве органич. растворителей — не-
растворим. В жидком NH3 растворимость умень-
шается от 3,36% (14,5°) до 0,99% (134°— критич.
темп-pa р-ра). Фазовые равновесия в системе М.—
NH3 исследованы до 340° и 500 кг/см2, Особенностью
системы является пересечение в двух точках кривой
растворимости твердого тела в жидкости с критич.
кривой равновесия жидкость'— пар. При 300° под
вакуумом М. сублимируется. Пыль М. в воздухе
взрывает при концентрации 5»252 г/м3.
УФ-спектр неионизированной молекулы в растворе
имеет максимум при X 203 ммк, в кислых и щелочных
средах спектр изменяется. Характеристич. частотами в
кристаллич. состоянии в ИК-области являются (с.м-1):
3333, 3125, 1660, 1560, 810.
Межатомные расстояния rc_N (в кольце) 1,34 А и
ГС—NH 1.37А указывают на сильное взаимодействие
л-электронов кольца с неподеленной парой электро-
нов аминного азота.
Аминогруппы придают М. основные свойства. Кон-
станты диссоциации в водных р-рах при 20°: A’i=
=1,26 -10-9; К2—1,58 -10-14; А3=1 -10-17. С кислотами
М. образует соли, например C3H6N6-HC1-1,5H2O;
C3HeNe-3HCl; C3H6Ne-CH3COOH и мн. др. Большин-
ство солей разлагается при нагревании, нек-рые —
хорошо кристаллизуются из воды. Их растворимость
того же порядка или меньше растворимости М. В р-ре
жидкого аммиака с амидами щелочных металлов М.
образует соли, напр. K3C3H3N6, или, при недостатке
амида,— продукт присоединения CsH6N6-KNH2. С
нитратом серебра образует соль C3H6N6-2AgNO3, к-рая
в водном р-ре NH3 при нагревании превращается в
C3H$N6-Ag2.
В водных р-рах, по мере увеличения темп-ры и дав-
ления, М. превращается последовательно в аммелин
109
МЕЛАНИНЫ — МЕЛАНОТРОПНЫЙ ГОРМОН
110
<1), аммелид (II), циануровую кислоту (III) и, нако-
нец, в NH3 и СО2. Оксиаминопроизводные образуются
NH о О
с .с А
N” XNH HN^nh HN^xNH
h2n-c^nXc=o3 h2n-c^c=o o=<LnJ:=o
1 и in H
также при нагревании M. в водном р-ре в присутствии
щелочи. При кипячении с разб. азотной к-той образует-
ся циануровая к-та. При нагревании паров М. под атмо-
сферным давлением до 600° и выше триазиновое коль-
цо разрушается и образуется цианамид NC—NH2.
М. нитруется смесью азотной и уксусной кислот.
Описаны моно- и дииитромеламины. Галогенирование
.может быть произведено в водных и неводных средах.
Продукты реакции содержат от одного до шести ато-
мов галогена. Ацилирование М. с введением от 1 до
3 ацильных групп производят с помощью ангидридов
кислот. Хлорзамещенные к-ты, используемые обычно
для получения ацилпроизводных, в большинстве слу-
чаев с М. не реагируют. Алкилирование М. протекает
при реакции с аминами или их хлористоводородными
солями, Количество вводимых алкильных групп (от
одной до трех) определяется избытком амина. Под
действием алкилсульфатов, напр. метил- или этил-
сульфата, М. превращается в сульфат моноалкилизо-
меламина. М. легко реагирует с альдегидами,продукты
конденсации с формальдегидом — метилолмеламины
имеют большое промышленное значение. С целлюло-
зой, сахарами, гликолями и др. органич. соединени-
ями, содержащими гидроксильные группы, М. всту-
пает в конденсацию аналогично реакции с альдегида-
ми; получаются смолообразные продукты. Исключени-
ем является глюкоза, одна молекула к-рой с двумя
молекулами М. образует кристаллич. продукт.
В промышленности М. получают из дициандиамида
3(Н2СН)2 —^2C3H6N6 при темп-ре 180—500°, давлении
40—200 кг!см2, без или с применением растворителей:
жидкого аммиака или р-ров аммиака в алифатических
спиртах. Экономически целесообразно получение М. из
мочевины:
6CO(NH2)2—> C3H6N, + 6NH3 + 3CO2
<350—500°, 100—400 кг/см2).
Известны периодич. и непрерывная схемы техноло-
гии. процесса.
Количественно М. определяют весовым способом
в виде солей циануровой или пикриновой к-т. Возмож-
но определение спектрофотометрически в слабокислой
среде по длине волны 236 ммк.
Основная область применения М.: пластич. массы,
лаки и клеи, отличающиеся высокой механич. проч-
ностью, малой электропроводностью, водо- и термо-
стойкостью. Электрич. изоляторы стойки против
дуговых разрядов. В текстильной пром-сти метилол-
меламины используются для изготовления безусадоч-
ных и иемиущихся тканей; в бумажной — для полу-
чения водонепроницаемой бумаги; в деревообрабаты-
вающей — для склеивания древесины и получения
высококачественных лаковых покрытий. Кроме того,
метилолмеламины применяют для приготовления дуби-
телей, ионообменных смол и др. продуктов (см. Смолы
меламино-форм.алъдегидные).
Hum.: Smolin Е. М.. Rapoport L., S-Triazlnes and
derivatives, N Y.—L., 1959; В a n n B., Miller S. A.,
Chem. Revs, 1958, 58, № 1, 131; К аза рновский С. Н.,
Швецова 3. Н., Хим. пром-сть, 1958, № 6, 1; Гольд-
берг Н. А., Заграничный В. И.. Хим. пром-сть,
i960, № 8, 6. В, 0. Заграничный.
МЕЛАНИНЫ — смесь веществ неустановленного
строения темно-коричиевого или черного цвета, встре-
чающихся в волосах, коже, перьях, сетчатке глаз и др.
тканях животных, а также в растениях. Предполагает-
ся следующий механизм образования М. в организмах:
L-тирозин (I) под влиянием фермента тирозиназы окис-
ляется до 3,4-диокси-Ь-фенилаланина (II) и далее до
фенилаланин-3,4-хинона (III); затем происходит за-
мыкание кольца с образованием дигидро-5,6-диоксиин-
долкарбоновой к-ты (IV), к-рая превращается в крас-
ный пигмент хиноидного строения (V) и далее при
pH 5,6—6,8 в 5,6-диоксиин до л (VI); последний через
иидол-5,6-хинои (VII) переходит в М.
М., выделенный из мочи человека или волос, пред-
ставляет собой темио-коричневый или черный порошок,
к-рый разлагается при 185°; плохо растворим в воде
и органич. растворителях, хорошо растворим в ще-
лочах; гидросульфитом натрия восстанавливается в
слегка окрашенное в коричневый цвет лейкосоедиие-
ние, к-рое окисляется вновь при добавлении феррициа-
нида или кислородом воздуха иа свету.
Образование М. в организмах животных регулирует-
ся гормональной системой, гл. обр. с помощью гор-
монов гипофиза (а- и р-мелаиоцитстимулирующие гор-
моны). При иек-рых заболеваниях, связанных с рас-
стройством гормональной системы (напр., аддисонова
болезнь), а также при беременности наблюдается по-
вышенное образование М., сопровождающееся усиле-
нием пигментации кожи. Образование М. в коже уси-
ливается под влиянием солнечных лучей (загар), а
также при злокачественных новообразованиях кожи
(мелаиобластомы).
Лит.: Кретович В, Л., Основы биохимии растении,
3 изд., М., 1961; Schmidli, Helv. chim. acta, 1955, 38, № 4,
1078; F r u t о n J. S., S i m m о n d s S., General biochemist-
ry, 2 ed., N. Y., 1958. E. В. Будницкая-Павлова.
МЕЛАНОТРОПНЫЙ ГОРМОН (меланотропин, ме-
лаиоцитостимулирующий гормон, МСГ, мелаиофорный
гормон, интермедин) — гормон средней доли гипо-
физа, при введении к-рого холоднокровным животным
происходит потемнение кожи вследствие увеличения
количества окрашенных клеток (меланоцитов), содер-
жащих пигмент мелании. В химич. отношении М. г.
различных животных представляют собой сравнитель-
но сходные по строению полипептиды, состоящие из
18—22 аминокислотных остатков, последовательность
расположения к-рых изучена. Напр., последователь-
ность расположения аминокислотных остатков М. г.
из гипофиза свиньи (р-МСГ, глутамил-р-МСГ) следую-
щая: Н-асп.-глут.-глиц.-прол.-тир.-лиз.-мет.-глут.-
гис.- фал.- арг,- трип.- глиц,- сер.- прол.-прол.-лиз.-
асп.-ОН (условные обозначения аминокислот см. в
ст. Аминокислоты). М. г. из гипофиза быка отличается
от М. г. из гипофиза свиньи тем, что место остатка глу-
таминовой к-ты (глут.) в нем занимает остаток серина
(сер.). Гормональную активность М. г. связывают
111
МЕЛАТОНИН—МЕМБРАННОЕ РАВНОВЕСИЕ
112
с наличием в их молекуле характерного участка из 7
аминокислотных остатков:...-мет.-глут.-гис.-фал.-арг.-
трип.-глиц.-...
Препараты М. г. хорошо растворимы в воде и вод-
ном спирте, растворимы в метиловом и этиловом спир-
тах, мало растворимы в бутиловом и изоамиловом спир-
тах, нерастворимы в эфире, ацетоне, этилацетате,
хлороформе, бензоле и ксилоле. М. г. термостабилен
и относительно устойчив к действию разб. к-т и щело-
чей; сильные к-ты и щелочи, а также окислители
(Н2О2, КМпОа) разрушают гормон.
Препараты М. г. получают путем экстракции из-
мельченной ткани гипофиза свиньи или крупного рога-
того скота подкисленной водой с последующим фрак-
ционированием экстракта. Синтетически получен ряд
пептидов со структурой, приближающейся к струк-
туре отдельных участков М. г. и обладающих нек-рой
биологич. активностью, напр. пентапептид гис.-фал.-
арг.-трип.-глиц. и нонапептид карбобензокси-сер.-
тир.-сер.-мет.-глут.-гис.-фал.-арг.-глиц.
Активность препаратов М. г. определяют по дей-
ствию, к-рое они оказывают на окраску кожи опреде-
ленного вида земноводных (напр., лягушки). Физиоло-
гия. роль М. г. у холоднокровных состоит в регуляции
окраски кожи в целях лучшего приспособления к
окружающей среде. Роль М. г. у теплокровных живот-
ных и, в частности, у человека менее ясна. По-видимо-
му, он принимает участие в процессах адаптации глаз
к темноте, стимулируя образование и регенерацию
зрительного пурпура.
Лит.: Современные проблемы биофизики, пер. с англ., т. 1,
М., 1961, с. 51; НапС О., Hormone, 2 Aufl., Jena, 1959, S.
508; В e r s 1 n Th., Btochemle der Hormone, 2 Aufl., Lpz.,
1960, S. 169; Fermente, Hormone, Vitamine..., Bd 2, Stuttg.,
1959; Современные проблемы биохимии, M., 1961, с. 100.
В. Б. Спиричев.
МЕЛАТОНИН [р-(5-метокси-3-индолил)-М-ацетил-
этиламин] CisHleN202, мол. вес 232,38 — гормон
, шишковидной желе-
снз° CH2CH2NHCOCH3 зы; кристаллит, про-
, дукт, т. пл. 116—
1Х 118° (из бензола),
1 растворим в орга-
* нич. растворителях,
плохо — в воде. М. выделен из шишковидной же-
лезы крупного рогатого скота (в 100 г лиофилизиро-
ванной железы содержится 40 мкг М.), в меньших ко-
личествах содержится в периферия, нервах человека,
обезьяны и крупного рогатого скота. Синтетически М.
получен методами, обычными для синтеза производ-
ных триптамина. В организме человека установлено
образование М. из серотонина', биологич. роль М. изу-
чена слабо.
Лит.: Axelrod J., Weissbach Н., Science, 1960,
131, № 3409, 1312. М. Н. Преображенская.
МЕЛИБИОЗА С12Н 220ц, мол. в. 342,31 — диса-
харид, состоящий из остатков глюкозы (I) и галактозы
(II); входит в состав
т рисахарида—рафи-
нозы; встречается и
в свободном виде у
растений. М.— кри-
сталлический про-
дукт, кристаллизу-
ется с двумя молеку-
лами воды, т. пл.
82—85°. Р-рыМ. об-
(в воде) меняется от
до 4-129,5°, в хлороформе [а]д=4-102,5°.
СН2ОН
нО/1 ° °
/и . /|
Кон 1 ° СНа
Г"----- нА —°Х
н он 7/н \,
Кон " н/1н-0"'
НО\1 I/
Н' он.
ладают мутаротацией; [а] д
4-ш,7о д : .... Г ,. . _ .
М. обладает сладким вкусом (в 4 раза слабее саха-
розы); как и другие сахара М. обладает восстана-
вливающей способностью. С помощью метилирова-
ния и перйодатного окисления было показано, что в
молекулах М. первый углеродный атом галактозы
связан с шестым углеродным атомом глюкозы. Сни-
жение [a]D М. и ее производных при гидролизе
свидетельствует о ее a-конфигурации. Т. обр., М.
является a-D-галактопиранозил (1—6)-П-глюкозой. По-
лучают М. неполным кислотным гидролизом рафи-
нозы. В дрожжах низового брожения содержится фер-
мент а-мелибиаза, расщепляющий М. на глюкозу и
галактозу. Распад М. происходит также под влиянием
минеральных к-т. Под действием ферментов нек-рых
бактерий М. образует мелибионовую к-ту.
Лит.: Каррер П., Курс органической химии, пер.
с нем., Л., 1960, с. 451; French D., в кн.: Advances In
carbohydrate chemistry, v. 9, N. Y., 1954, p. 149; The carbohyd-
rates, ed. by W. Pigman, N. Y., 1957, p. 500.
E. Л. Розенфельд.
МЕЛИНИТ — см. Тринитрофенол.
МЕЛЛИТОВАЯ КИСЛОТА (бензолтрикарбоновая
к-та)— см. Бензолполикарбоноеые кислоты..
МЕМБРАННОЕ РАВНОВЕСИЕ — равновесие, ус-
танавливающееся в системе растворов, разделенных
мембраной, непроницаемой хотя бы для одного вида
присутствующих в системе ионов. Это явление было
детально рассмотрено и теоретически обосновано
Ф. Д. Доннаном. Если раствор к.-н. соли, напр.
Na+A-, где А-— анион, неспособный к диффузии
через мембрану (т. наз. недиализуемый ион), отделен
при помощи последней от р-ра NaCl, то начальное со-
стояние этой системы можно изобразить след, схемой:
c^INa + L I INa + ]2=c2
с, = [А-], (1)1(2) (С1~]2=с2
где вертикальная линия представляет собой мембрану,
а индексами (1) и (2) обозначены левая и правая ячей-
ки системы; ci— концентрация ионов в ячейке с ин-
дексом (1), с2— в ячейке с индексом (2). Т. к. мембра-
на проницаема для ионов Na+ и С1_, то они свободно
передвигаются из одной ячейки в другую. Но полное
выравнивание концентраций этих ионов по обе сторо-
ны мембраны не может быть достигнуто, поскольку
имеются ионы (А~), неспособные проходить через пе-
репонку. Постепенно в системе устанавливается рав-
новесие, при к-ром распределение ионов имеет след,
вид:
c,+x=[Na + ]1 1X3+12=02 — х
с, = [А-], [С1-]2=с2-х
* = !«-], (1) (2)
При этом по обе стороны мембраны сохраняется элект-
ронейтральность, т. е. концентрации анионов и катио-
нов равны. Отсюда следует:
[Na + ]2=[Cl-]2 (I) и INa + I^IA-h + ICl-j! (И)
В соответствии с термодинамич. условием равновес-
ного состояния, произведения концентраций катио-
нов и анионов NaCl, находящихся по обе стороны мем-
браны, должны быть равны между собой:
[Na+],-ICl-]1 = [Na+]2-[Cl-]2 (III)
Из соотношения (III) с учетом соотношения (I) сле-
дует:
[Na+],-[Cl-], = [Na+]’ = [Cl-]| (IV)
Из соотношения (II) видно, что [Na+]i> [Cl_1]i, а
из уравнения IV следует;
IC1-], < [С1-]2 и INa+L > (Na+]2
Т. обр., в состоянии равновесия концентрации NaCl
по обе стороны мембраны не равны. Концентрация
NaCl меньше там, где присутствуют ионы, не диффун-
дирующие через мембрану. Эти же соображения при-
водят к заключению, что неравномерность распреде-
ления NaCl по обе стороны мембраны меняется в за-
висимости от концентрации недиффундирующего иона.
113
МЕМБРАННОЕ РАВНОВЕСИЕ —МЕНТЕНЫ
114
Соотношение (IV) можно представить в виде:
(с,+х) х=(с2-х)2 (V)
или
(VI)
X с2 - X
Отсюда
к=с*/(с, + 2с3)
Подставив значение х в правую часть выражения VI,
получим:
В случае, когда ci |>> сз, величиной сз в сумме
щ-)-с2 можно пренебречь:
С2—X _ С1
X ~ с2
Отсюда следует, что чем больше концентрации недиф-
фундирующего иона, тем меньшее количество диффун-
дирующего электролита проникает из второй ячейки
в первую. В другом крайнем случае, когда с2,
можно пренебречь величиной щ по сравнению с с2
Это означает, что диффундирующий
электролит распределяется по обе стороны мембраны
почти равномерно.
Приведенный пример дает распределение электро-
литов по обе стороны неактивной мембраны в простей-
шей системе, когда имеется только один электролит,
состоящий из одновалентных ионов, причем по обе
стороны мембраны присутствует общий ион. Одна-
ко, как было показано Доннаном, полученные соот-
ношения ионных концентраций сохраняются и в
более сложных системах, когда, напр., присутствует
несколько диффундирующих электролитов или диф-
фундирующие ионы многовалентны.
М. р. учитывают при исследовании осмотич, дав-
ления коллоидных растворов, т. к. в последних всегда
присутствуют ионы, способные проникать через мемб-
рану осмотич. ячейки. Если концентрация коллоид-
ных частиц значительно больше концентрации диф-
фундирующих ионов (cij>c2), то в пределе лн/лк=1,
где лн— экспериментально наблюдаемое осмотич.
давление, лк— осмотич. давление без учета диализуе-
мых ионов. В случае, когда cj^c2, jtH/jtK=J4. В си-
стемах с промежуточными значениями соотношения
концентраций наблюдаемое осмотич. давление может
принимать значения в области от лк до лк/2.
Условия доннановского равновесия сохраняют свою
силу и в случае применения активных мембран, пред-
ставляющих собой ионообменные смолы (см. Иониты).
Такая мембрана, погруженная в раствор электролита,
играет роль второго, более конц. р-ра. Обмен ионов
между раствором и мембраной происходит до тех пор,
пока не устанавливается равновесие, полностью отве-
чающее условиям М. р.
Как активные, так и неактивные мембраны широко
используют в практике для решения самых разнооб-
разных вопросов. Сюда относятся процессы диализа
и электродиализа различных препаратов, умягчение
воды для котельных установок, очистка питьевой воды
от вредных примесей, разделение смесей неорганич.
катионов, редкоземельных металлов. Широко поль-
зуются методом ионного обмена для очистки антибио-
тиков, концентрирования сырья, содержащего лекар-
ственные вещества, обработки крови ионообменной
смолой для предохранения ее от свертывания, разде-
ления смесей аминокислот и др. процессов.
Лит.: Don nan F. G., Z. Electrochem., 1911, Bd 17,
М 14, S. 572; Ионный обмен. Сб. статей, пер. с англ., под ред.
К. В. Чмутова, М., 1951; Ионообменные смолы в медицине и
биологии, пер. с англ., под ред. С. Я. Капланского, М., 1956;
В о 1 am Т. R., The Donnan equilibria and their application
to chemical physiological and technical processes, L., 1932.
T. В. Гатове кая.
МЕНДЕЛЕВИЙ (Mendeleevium) — химич. элемент
с п. н.101, относится к числу актинидов.
МЕНТАН С1()Н2(), мол. вес 140,26 — бесцветная
жидкость; известны 3 изомера: n-ментан (1-метил-4-
изопропилциклогексан,
I а и б); о-ментан (1-ме-
тил- 2-изопропилцикло-
гексан, II), т-ментан
(1 -метил-3-изопропил-
циклогексан, III). Каж-
дый изомер существует
СН(СН3)г
в виде цис- и транс-
форм; наиболее изучен
n-ментан. ।
Все три изомера пред-
ставляют собой про- !
зрачную жидкость со 111
слабым запахом, напоминающим керосин. Хорошо рас-
творим в органических растворителях, практически
нерастворим в воде.
Изоме ры T. кип., °C/мм рт. ст. 20 Пй . ,23 [“]о
,, а орто-М 171/760 0,81356 1,447б
метпа-М.а, d . . . 167—168/760 0.8116® 1,446Г + 1,6
1 167— 168/749 0,7938д 1,4358 —0,3
dl 166—167/760 0,7965е 1 ,44ж —
napa-М., ipic- . . 168,5/760, 63/22 0,816 1,4515 —
транс- 161/760, 61 — 62/19,5 0,792 1,4393 —
а Смесь цис- и транс-изомеров. ® При 21°. в d™. г п”
ДЙ 2°. е <7.24. Ж «24
о о О
М. устойчив по отношению к химическим воздей-
ствиям; галогены действуют с трудом, нитрование
проходит труднее нитрования ароматических углево-
дородов. Дегидрирование над Pt при высокой темпе-
ратуре приводит соответственно к п-, о- и лг-цимо-
лу. При окислении n-М. получается гексагидрокуми-
новая (n-изопропилбензойная) и гексагидротолуи-
новая (n-метилбензойная) кислоты. При окислении
М. молекулярным кислородом образуются гидропе-
рекиси.
М. мало распространены в природе. n-М. присут-
ствует в масле из древесины и коры араукарии (Arau-
caria Cunninghamii Ait.). М. можно синтезировать:
цнс-п-ментан — гидрированйем n-цимола в ледяной
СНзСООН в присутствии коллоидной Pt; транс-п-
ментан — электролитич. восстановлением 1-ментона
на свинцовом катоде в слабокислом р-ре.
Лит.: Никитин В. М., Химия терпенов и смоляных
кислот, М.—Л., 1952; Горяев М. И., Характеристика хи-
мических соединений, входящих в состав эфирных масел, Ал-
ма-Ата, 1953. И. И. Бердышев.
МЕНТЕНЫ С!()Н18, мол. в. 138,24 — прозрачная,
подвижная жидкость; т. кип. выше 160°; практически
нерастворимы в воде, хорошо растворимы в органич.,
особенно неполярных растворителях. М.— дегидро-
производные ментанов. Известны о-, м- и n-М. Каж-
дая из указанных форм объединяет углеводороды,
отличающиеся положением двойной связи в моле-
куле. Наличие 6-членного кольца служит причиной
так называемой эндо- и экзоизомерии, а наличие
двойной связи — цис- и транс-изомерии. Наличие
асимметрии, атомов является причиной оптич. изоме-
рии М.
Поэтому число всех возможных изомерных М.
исчисляется несколькими десятками, многие из них
еще не получены в чистом виде. Ниже приведены.
115
МЕНТОЛ —МЕНТОН
116
наир., структурные ф-лы трех М,
Д’-.м-меитеи (II), Д'-п-ментен (III):
СН3 VHs
ZCH3 с
HaC^'tC-CH Н2С -СН2 ZCH3
НоС^СНз СН3 Н2С.ЛН СН
сн2 сн3
Д’-о-ментен
СН
С
н2с" чсн2
Н2С^*/СН2
СН
I
/Сх
СН3ХСН3
III
М. легко гидрируются в соответствующие ментаны
и дегидрируются в о-, м- и n-цимолы. При диспропор-
ционировании водорода из М. получаются соответ-
ствующие ментан и цимол. При окислении КМпОа
образуются соответствующие кислоты. М. реагируют
как непредельные углеводороды, содержащие двойную
связь: они легко присоединяют галогены, галогено-
водороды, озон, хлористый нитрозил, азотистый ан-
гидрид и т. д.
М. сравнительно мало распространены в природе.
Д3-п-ментен входит в состав масла перечной мяты,
тимьянового масла, в хвойное эфирное масло моретон-
ской ели и др.
Оптически активный Д’-п-ментен (с примесью Д2-
и-ментена) лучше всего синтезировать из я-ментола,
дегидратируя его ксантогеновым методом (методом
Чугаева). Различные М. (чаще всего смеси с преобла-
данием одного из изомеров) получаются при частичной
гидрогенизации моноциклич. терпеновых углеводоро-
дов или при диспропорционировании водорода в этих
терпенах.
Лит.: Ч у г а е в Л. А., Иабр.труды, т. 2, М., 1955; Wal-
lach О., Terpene und Kamplier, Lpz., 1914. И. If. Бардышев.
МЕНТОЛ (3 .гексанол, п-м в. 156,26 — б( держит 3 асиу а поэтому им< пых и 4 неде -метил-6-изопропилцикло- ентанол-3) С,0Н20О, мол. ;сцветные кристаллы; со- 1метрич. атома углерода, зет 8 оптически деятель- ятельные формы. 3 СНз X) zCH H3CZ 'сн3 3
1 1
н ме СН, 6 С3Н7 н нтол неор 3 Н СНз 1 6 С3н, аентол 3 Н 1
С,И, из оме 6 н Сзн7- ятол неоизо 6 н ментол
В М. и неоментоле группы СП, ц С3Н7 находятся
в т/)аис-положении, а в изоментоле и неоизоментоле—
Стереоизомеры Т. пл., °C Т. КПП., °C [а]д
d- или 1-ментол 43 216 + 49.9°а 1,4609
(1,1-ментол 38 216 + 0° 1,4615
<1- или 1-неоментол .... __ 9 9 212 + 19,7° 1,4603
<1,1-неоментол 53 212 + 0° 1,4604
d- или 1-изоментол .... 82.5 219 + 27°а —
d ,1-изоментол . 53.5 219 + 0° —
d- или 1 неоизоментол . . . -8 215 ±2.2°а 1,4649
d, 1-неоизоменгол 14 215 ±0° 1,4649
В спирте. 0 d18.
в цис-положении. В М. и неоизоментоле группы СН3
и ОН находятся в цис-положении, а в неоментоле и
изоментоле — в игуане-положении.
М. имеет характерный мятный запах (запах d-M.
слабее запаха l-М.), охлаждающий вкус, вызывает на
коже ощущение холода. Растворим в спирте, эфире,
хлороформе, бензоле, скипидаре, эфирных и жирных
маслах, в уксусной к-те, жидких SO2 и NH3, плохо—
в воде. М. является весьма реакционноспособным ве-
ществом. При нагревании с дегидратирующими аген-
тами М. образует в основном п-ментен-3. М. дегидра-
тируется труднее, чем неоментол. Окисление хромовой
смесью приводит: для М. и неоментола к ментону,
а для изо- и неоизоментола к изоментону (см. Ментон).
При нагревании М. с CuSOa до 270° образуется ци-
мол. При восстановлении с HJ все изомеры М. дают
ментан.
М. и неоментол находятся в эфирных маслах, напр.
в масле перечной мяты (до 80% от веса масла), гера-
ниевом масле. Другие изомеры М. получены синтети-
чески. Для промышленного получения основным ист
точником является масло перечной мяты. Масло
перечной мяты сильно охлаждают, центрифугируют и
получают ок. 40—50% 1-М. По другому способу —
дистилляцией масла — отделяют фракцию М. и нео-
ментола. Обработкой гидроксиламином удаляют из
эфирного р-ра этой фракции примесь ментона. При
выпаривании эфира из раствора кристаллизуется М.
Выделение неоментола из маточного р-ра производят
через кислый сукцинат или кислый фталат. Предло-
жено несколько способов синтетич. получения М.:
восстановлением ментона натрием и спиртом в индиф-
ферентных растворителях, восстановлением пулегона
натрием в эфире, гидрированием тимола, изопулггола.
Экономически выгодным является синтез М. из n-цимо-
ла: и-цимол нитро-п-цимол -> аминоцимол амино-
цимолсульфоновая к-та -> диазосоединение -> п-ци-
молсульфокислота ->• тимол ментол. Наряду с кри-
сталлин. М. образуются жидкие изомеры, от к-рых
М. необходимо хорошо освободить, т. к. они обладают
нек-рой ядовитостью.
Применяют М. в пищевой пром-сти при приготов-
лении ликеров, пряников, конфет; в парфюмерии
при приготовлении зубных порошков и паст, в фар-
мацевтич. пром-сти при получении валидола, бормен-
тола и др.
Лит.: Simonsen J. L and Ross W. C. J., The
terpenes, v. 1, 2 ed., Camb., 1953. Пигулевский Г. В.,
Химия терпенов, Л., 1949; Машковский М. Д., Лекар-
ственные средства, 4 изд., М., I960. И. И. Бардышев.
МЕНТОН (1-метил-4-изопропилциклогексанон-3),
мол. в. 154,24 — существует в виде транс- п цис-
изомеров. Каждый
изомер имеет d-, 1 Н3С Н Ч3С_.Н
и d, 1-формы. Г)
Оба изомера пред- I I Q I J=o
ставляют собой бес- ><;
цветное масло с запа- Н НС(СН3)2 (СН3)2СН Н
ХОМ мяты и горьким, транс-изомер (ментил) цис-изомер (изомвшои)
слегка охлаждающим
Т пл. вкусом; растворяются в большин- стве органич. растворителей, ча-
J15 15 нитробен- стично — в воде.
зоатов, °C Под действием кислот и щелочей,
особенно при нагревании, М. легко
0,904 153 изомеризуется в изоментон, при
0,904 121 этом наступает динамич. равновесие
0,903 130 между двумя изомерами. М. вое-
147 станавливается и окисляется в со-
— 130 ответствующие спирт и кислоту;
0.91Зб 101 образует производные, характерные
— 73 для кетонов (оксим, семикарбазон
и др.), за исключением бисуль-
фитных.
117
МЕПАЗИН—МЕРИХИНОИДНЫЕ СОЛИ
118
Название Т. пл., °C Т. кип., °С1мм t пй
d-Ментон . . . 210,5/760 0,8960а 1,4525 (1 7°) + 20°
1-Ментон .... —7 207/750 0,8954 1 ,4505 (20°) —24,8°
81/10 0,8073° — 28,2°
<1,1-Ментон . . — 204/750 (210°) 0,897 1,4521 (20°) 0.0°
d-Изоментон . . ок.—35 212/760,86—87/12 0,9057е 1,45302 (20°) + 95° (100°)
dJ-Изоментон — 210/760, 90-93/18 — 0,0°
a d20 6 dls0 в d15
4 IS
Для количественного определения М. восстанавли-
вают в ментол и определяют количество последнего.
М. распространен в природе: в масле перечной мяты
(до 30%), в масле полевой мяты (6—18%), бетелевом,
гераниевом, буковом и др. маслах.
Изоментон входит в состав африканского полейного
масла (Hedeoma pulegioides Pers.), содержится в пе-
речном масле из листьев Mentha species и др. Для
получения М. или изоментона из масел выделяют
фракцию с т. кип. 204—212°, переводят в оксим или
семикарбазон кетонов, а разложением их выделяют
чистые М. или изоментон.
М. представляет интерес как сырье для получения
ментола.
Лит.: Пигул евский Г. В., Химия терпенов, Л.,
1949; Бурно К., Терпены, пер. снем., [Л.], 1933; Горя-
е в М. И., Характеристика химических соединений, входящих
в состав эфирных масел, Алма-Ата, 1953; Zeitschel О.,
Schmidt Н., Вег., 1926, 59, № 9, 2307. И. И. Бардышев.
МЕПАЗИН (лакумин, пакатал, пеказин), мол. вес
370,52, C,9H22N2S.CH3COOH — белый кристаллич. по-
рошок со слабым запахом
уксусной к-ты, т. пл. 69—
Г £ 1 ] 70°; хорошо растворим в
воде, спирте, бензоле и то-
q|_| —луоле. При добавлении к
2 1 I • СНзСООН разб. водному раствору М.
(1 : 100) смеси конц. Н3РОа
сн3 и Ю%-ной HJO3 появляет-
ся темно-красное окраши-
вание; конц. HNO3 или H2SO4 вызывает оранжево-
красное окрашивание, переходящее при стоянии в
вишнево-красное. Количественно М. определяют тит-
рованием в водно-эфирной среде NaOH по фенолфта-
леину. М. получают конденсацией фенотиазина в при-
сутствии щелочи с З-хлорметил-1-метилпиперидином.
М.— нейроплегич. препарат, по свойствам близок
к аминазину, применяют гл. обр. при неврозах, пси-
хозах, а также для усиления действия снотворных,
при обезболивании родов и др.
Лит. см. при ст. Адалин. В. Г. Яшунский.
МЕПАНИТ (мерпанит, метилсульфометилат ди-
этиламиноэтилового эфира 1-фенилциклопентан-1-
карбоновой кислоты)
D9eH5 + C20H33NO6S, мол. вес
-COOCH2CH2n(C2h5)2 • SO7 415,56 — бесцветные
СН £Н кристаллы, т. пл. 71—
3 3 72°, растворим в воде
(1 : 5). Мепанит получают из Вг(СН2)4Вги CeH5CH2CN;
образующийся при этом нитрил 1-фенилциклопен-
тан-1-карбоновой кислоты гидролизуют; действием
(С2Н5)2КСН2СН2С1 получают диэтиламиноэтиловый
эфир 1-фенилциклопентан-1-карбоновой к-ты; дейст-
вием на этот продукт (хлоргидрат, наз. пентафен)
диметилсульфатом получают мепанит. М. обладает
сильным периферич. холинолитич. действием, приме-
няется при спазмах гладкой мускулатуры (почечные,
печеночные, кишечные колики), при язвенной болезни
желудка и двенадцатиперстной кишки, при бронхи-
альной астме; противопоказан при глаукоме.
Лит.: Хромо в-Б о р и с о в Н. В., Яновицкая
А. М., Михельсон М. Я., Мед. пром-сть СССР, 1958,
№ 6, 43; Машковский М. Д., Ле-
карственные средства, 4 изд., М., 1960,
с. 136; Tilford С. Н. [а. о.], J. Amer.
Chem. Soc., 1947, 69, № И, 2902.
Л. Ш. Городецкий,
МЕПРОТАН (тадексил, мепав-
лон, мепробамат, мепробан, мепро-
сан, мильтаун, транквил, 2-ме-
снзх .ch2oconh2
CH3CH2CH2Z xCH2OCONH2
тил - 2-н-пропил-1,3- пропандиолди-
карбамат), мол. в. 218,26—белый
кристаллич. порошок без запаха, горького вкуса;
т. пл. 104—106°; легко растворим в этиловом спирте,
ацетоне, бензоле, толуоле; трудно растворим в
холодной и хорошо в горячей воде. Устойчив при
хранении. При нагревании М. с водной щелочью выде-
ляется NH3. Количественно М. определяют по содер-
жанию азота методом Кьельдаля. М. получают, исхо-
дя из пропионового альдегида:
NaOH Н2
СН3СН2СНО------> СН3СН2СН = С- СНО —
| СН2О
—> СН3СН2СН2СНСНО---->
СН,Ч УСН2ОН CH3OCONH2
)CZ--------------> м.
CHaCH2CH2Z ХСН2ОН А1С13
М. может быть получен также при действии на
2-метил-2-н-пропил-1,3-пропандиол мочевины в при-
сутствии Pb (СН3СОО)2. М. оказывает успокаивающее
влияние на центральную нервную систему, обладает
выраженной противосудорожной активностью, уси-
ливает действие снотворных. М. применяют гл. обр.
при лечении нервно-психич. заболеваний.
Лит.: Машковский М. Д., Лекарственные средства,
4 изд. М., 1960, с. 46- Ferrari G.. Chimica е Industrie,
1958, 40, № 1, 13; Y al е H. L. [а. о.], J. Amer. Chem. Soc.,
1950, 72, № 8, 3716. В. Г. Яшунский.
МЕРИДИЛ (центендрин, хлоргидрат метилфени-
дата, риталин) C14H2oNC1O2, мол. в. 269,78 — хлор-
гидрат метилового эфира а-фе-
нил-а-(пиперидил-2)-уксусной к-ты, С6Н.СНСООСН3
т. пл. 193—199,5°, плохо растворим -
в ацетоне, бензоле, почти нераство- | *jIH ’ НС!
рим в эфире и этилацетате. М. по-
лучают путем конденсации а-хлор-
пиридина с натриевым производным CeH5CH2CN в
а-фенил-а-(пиридил-2)-ацетонитрил. Полученный про-
дукт гидратируют, а затем действием СН3ОН и НС1
получают метиловый эфир а-фенил-а-(пиридил 2)-ук-
сусной к-ты. М. является стимулятором централь-
ной нервной системы, применяют при депрессивных
состояниях.
Лит.: PanizzonL., Helv. chim. acta, 1944, 27, fasc. 7,
1748; Машковский M. Д., Лекарственные средства,
4 изд., М., 1960, с. 99. Л. Ш. Городецкий.
МЕРИХИНОИДНЫЕ СОЛИ (семихиноны) — соли,
содержащие ионы-радикалы. М. с. образуются про-
межуточно при восстановлении хинонов в гидрохино-
ны и при обратной реакции —• окислении гидрохи-
нонов в хиноны, однако выделить их в свободном виде
удается лишь при наличии структурных особенностей,
способствующих стабилизации М. с. Если хиноидное
соединение электриче-
ски нейтрально (I), то
соответствующая ему
М. с. содержит анион-
радикал (II); если же
хиноидное соединение
представляет собой ка-
119
МЕРКАЗОЛИЛ —МЕРКАПТАЛИ
120
тион с двумя поло-
жительными зарядами
(III), М. с. содержит
катион-радикал (IV).
Заряды в М. с. рас-
средоточены за счет
сопряжения. М. с. от-
личаются от хингид-
ронов, к-рые представ-
ляют собой молекулярные соединения, состоящие
из бензоидного и хиноидного компонентов; однако
в нек-рых случаях хингидроны переходят в М. с.:
М. с. представляют собой глубоко окрашенные пара-
магнитные соединения. Их мономолекулярная, т. е.
радикальная, форма доказывается измерением мол.
веса, а также тем, что могут существовать лишь
однородные, но не смешанные М. с. Наличие проме-
жуточной ступени восстановления хинона (передача
лишь одного, но не пары электронов) доказывается
тем, что при потенциометрич. титровании хинона
восстановителем на потенциометрич. кривой обнару-
живаются две точки перегиба вместо одной. Примеры
М. с.: «синий Вурстера» (V), получающийся при окис-
лении тетраметил-ге-фенилендиамина; соль пиоциани-
ния (VI), получающаяся из пиоцианина—красящего
вещества бактерий; семидурохинон (VII), получающий-
ся восстановлением дурохинона в щелочной среде:
Лит.: Хю кк ел ь В., Теоретические основы органиче-
ской химии, пер. с нем., т. 1, М., 1955, с. 158; Семенов
Н. Н., О некоторых проблемах химической кинетики и реак-
ционной способности, 2 изд., М., 1958, с. 256. Е. М. Рохлин.
МЕРКАЗОЛИЛ (антироид, мерказол, метотирин,
1-метил-2-меркаптоимидазол) CiHgNaS, мол. вес
____N 114,17 — белый кристаллич. порошок, горь-
f Lc кого вкуса; т. пл. 145—148°; растворим в
воде, спирте, хлороформе, трудно раство-
рим в эфире. В щелочном р-ре М. дает
3 с нитропруссидом натрия зеленую окрас-
ку, которая переходит при добавлении СНзСООН
в голубую. Количественно М. определяют титрова-
нием щелочью в присутствии AgNOs. М. получают,
исходя из ацеталя хлоруксусного альдегида, метил-
амина и KCNS:
ch3nh2+cich,ch (OC2Hs)3 —>
KCNS
—>- CH3NHCH2CH (OC2Hs)2--> M.
M. может быть получен также термич. декарбоксили-
рованием 1-метил-2-меркаптоимидазолкарбоновой-5
к-ты. М.— весьма активный антитиреоидный препа-
рат, применяется при базедовой болезни и тиреоток-
сикозе.
Лит.: Машковский М. Д., Лекарственные средства,
4 изд., М., 1960, с. 381; Jones R. G. [а. о.], J. Amer. Chem.
Soc., 1949, 71, № 12, 4000. В. Г. Яшунский.
МЕРКАМИН (цистеамин, меркаптамин, хлоргид-
рат p-меркаптоэтиламина) HSCH2CH2NH2 -НС1, мол.
в. 113,61— белые кристаллы со специфич. запахом
меркаптана; т. пл. 70—72°, хорошо растворим в воде,
спирте, нерастворим в эфире. М. быстро окисляется
на воздухе с образованием дисульфида и полимеров.
При действии салициловой к-той на основание М. об-
разуется меркамин салицилат C9HisO3NS, т. пл. 104—
106°, устойчивый на воздухе продукт. Щелочной р-р
М. дает малиновую окраску с нитропруссидом нат-
рия, при действии AgNO3 выпадает белый осадок.
Количественно М. определяют иодометрич. титрова-
нием. М. может быть получен различными способами,
например восстановлением 2-бензилтиоэтиламина
(Na, NHs), взаимодействием этиленимина с H2S и др.
М. способен ослаблять действие ионизирующего излу-
чения на организм; его применяют для профилактики
и лечения лучевой болезни, возникающей, в частности,
при рентгено- и радиотерапии. Хлоргидрат М. вво-
дится внутривенно, салицилат — в таблетках.
Лит.: Машковский М. Д., Лекарственные сред-
ства, 4 изд., М., I960, с. 438. В. Г. Яшунский.
МЕРКАПТАЛИ — сернистые аналоги ацеталей it
кеталей R2C(SR')2 или RCH(SR')2-
М. образуются при действии меркаптанов на альде-
гиды или кетоны в присутствии соляной к-ты или хло-
ристого цинка:
(СН3)2 с = о
C2H3SH
НС1 или ZnCl2
(СН3)3 С (SC2H5)j
R. .ОН"] HSR" R, S.R
----------------А-
Hz 'SR'J Hz <Х,
Действием на альдегиды или кетоны последовательно
двумя разными меркаптанами были получены сме-
шанные М.:
R — С = О HSR'
I------------
н
Ацетондиэтилмеркапталь, промежуточный продукт в
синтезе лекарственного в-ва сульфонала, получают
действием этилтиосерной к-ты на ацетон в присутствии
соляной к-ты;
C2HsS (SO3H)
(СН3)2 С=О----------* (СН3)2 С (SC2H5)2
НС1
Двухатомные меркаптаны, реагируя с карбонильными
соединениями, образуют циклич. М.:
/CH2SH (СН3)2СО /CH2-S, .сна
сн2 ------------>- сн2
\CH,SH НС1 \CH2-SZ хсна
M., как правило, устойчивы к действию кислот и ще-
лочей и легко окисляются до соответствующих ди-
сульфонов:
о с2Н6
✓ S
СН3. SC2H5 [О] СН3, / 4 о
► .С Q
ОН/ 4SC2H5 он/ X
о С2Н3
Л. С. Герман.
121
МЕРКАПТАНЫ
122
МЕРКАПТАНЫ (тиоспирты, тиолы) — органич.
производные сероводорода, содержащие углеводород-
ный радикал, связанный с сульфгидрильной группой—
SH. М., особенно низшие члены гомологии, ряда, об-
ладают ярко выраженным специфич. запахом, благо-
даря чему могут быть обнаружены в воздухе в концент-
рации до 2• 10-9 мг/л. Физич. свойства нек-рых М. об-
щей формулы RSH приведены в таблице.
R Т. пл., °C Т. кип., °C п25 nD
СН, —123,0 6.0 0,8599
С2Н5 —147,3 35,0 0,8314 1,4278
н-СаН7 —111,5 67,8 0,8357 1,4351
М-С4Н9 —115,9 98,2 0,8365 1,4401
Я-С12Н23 18—20 153,8(20.5 мм 0,8411 —
Химич, свойства М. определяются наличием подвиж-
ного атома водорода, связанного с атомом серы, а так-
же иеподеленной пары электронов на атоме серы. Яв-
ляясь слабыми кислотами, М. легко растворяются
в щелочах с образованием металлич. производных —
меркаптидов:
NaOH
RSH------> RSNa+H3O
С окислами тяжелых металлов М. также образуют со-
ответствующие меркаптиды. Ртутные производные
служат для целей идентификации М. При действии
алкилирующих агентов щелочные меркаптиды обра-
зуют сульфиды:
R'Hal
RSNa------к RSH' + NaHal
Ацилирование М. в присутствии оснований либо ще-
лочных меркаптидов приводит к соответствующим
S-ацильным производным:
R'C(O)C1
RSH--------->- R — S — С — R'
пиридин 11
О
Мягкие окислители реагируют с М. с образованием
дисульфидов:
HjSO.; NH2OH
C2H5SH------------>-C2HiSSC2H6
Более энергичное окисление (HNOg или С12+Н2О)
приводит к сульфокислотам RSO3H или их производ-
ным, иапр. RSO2C1.
Являясь ярко выраженными нуклеофильными ре-
агентами, М. в присутствии оснований легко присо-
единяются по активированной кратной связи:
RSH
ch2=chcn
--------> RSCH,CH2CsN
CF.— CF,
-------'-> RSCF, CF..H
R'CsN NH
* RC<^
SR
При действии M. на альдегиды или кетоны в при-
сутствии кислых катализаторов образуются соответ-
ствующие тиоацетали или тиокетали:
R. R"SH r SR"
) С = О--> +Н2О
R. НС1 R'x "SR"
При обработке М. нитрозирующими реагентами
(NOCI, HNO2 in statu nascendi) образуются неустой-
чивые тиоиитриты RSNO, обладающие характерной
окраской (цветная проба на М.). Связанная с арома-
тич. ядром SH-группа, подобно оксигруппе, является
ориеитантом 1-го рода.
Основные методы синтеза М.:
1) Действие алкилирующих агентов [RJ, ROSO3K,
(К0)г§02 и др.] иа кислые сульфиды щелочных ме-
таллов (KSH, NaSH).
2) Конденсация галогеиалкилов с тиомочевииой
с последующим щелочным гидролизом образовавшей*
ся соли S-алкилизотиурония:
S = C(NH2)2 z/nh2
R - X----------» R — S — С<
'NH,
H20
x------> R-SH
NaOH
3) Конденсация галогеиалкилов с тиосульфатом
натрия с последующим кислотным гидролизом образо-
вавшейся соли алкилитиосерной к-ты (т. н. соли
Бунтэ):
Na2S,O2 H..SO,
С.Н,СН2С1------» C„H»-CH2-S-S0,Na----> C,H,CH2SH
4) Присоединение сероводорода по углерод-уг-
леродной кратной связи, либо по нуклеофильному
(в случае олефина с активированной кратной связью),
либо по свободно-радикальному механизму. В послед-
нем случае, как правило, образуется смесь теломер-
ных М.:
H2S (избыток)
СН2 = СН - С = N---------->- СН, - СН2 — С = N
п СН2 =СН2-------► H(CH2CH2)n SH
перекиси
5) Пропускание смеси паров спирта и H2S иад
ThO2:
H,S
r-он ——-> r-sh+h2o
Th02
6) Восстановление органич. производных серы,
содержащих связь С—S (сульфокислоты, их хлоран-
гидриды, роданиды, дисульфиды и др.):
[Н] [Н]
R-SO2C1--------> RSH; R-S-S-R---------> 2R — SH
Zn + H2SO. LIA1H,
7) Действие элементарной серы на металлорга-
нич. соединения с последующим гидролизом:
s н,о
R - Mgx - - R - s - MgX--> r - SH
8) Синтез разнообразных р-замещенных алкил-
меркаптанов с раскрытием цикла тиоокисей:
сн2-сн2
НС1
-------> CH2C1-CH.SH
RSH
------> RS- CH2CH2SH
B.NH
-------► R2N-CH2CH2SH
ROH
------->- RO-CH2CH,SH
9) Расщепление кольца а-окиси, либо этилеиими-
на сероводородом; получаются соответственно р-окси-
и р-аминоалкилмеркаптаны:
сн2 - сн2 „ о сн2 - CH2SH СН2 - СН2 „ о СН2 - CH2SH
/ Г13О । n s XI2 О I
О ^0Н JJH
10) Действие щелочных ксантогеиатов на соли
арилдиазония с последующим гидролизом промежу-
точно образующегося арилксантогената:
KS(CS)OC,H5 н2о
CeH5N2Cl---------C,,HsSC (S) ОС2Н3------>- C.H3SH
он-
11) Действие серы на ароматич. углеводород в при-
сутствии хлористого алюминия
s
С„Н«------>C,H5SH + HC1
А1С13
Последние 2 способа являются специфич. для син-
теза ароматич. М.
Лит.: Н о u b е п—W е у 1, 4 Autl., Bd 9, Stuttgart, 1955;
Вейганд К., Методы эксперимента в органической химии,
пер. с нем., ч. 2, М., 1952, с. 322, 642; Кнунянц И. Л. и
Фокин А. В., Усп. химии, 1950, 19, вып. 5, с. 545.
Л. С. Герман.
123
МЕРКАПТИДЫ—МЁССБАУЭРА ЭФФЕКТ
124
МЕРКАПТИДЫ — см. Меркаптаны.
6-МЕРКАПТОПУРИН C5H4N4S, мол. в. 152,13—
желтоватые кристаллы; т. пл. 312—314° (с разл.);
SH
।
н
трудно растворим в воде и в большин-
стве органич. растворителей, легко — в
щелочах. Получают М. взаимодействием
гипоксантина с пятисернистым фосфором
в сухом пиридине, а также при нагре-
вании 6-хлорпурина с тиомочевиной в
абс. спирте:
ОН
SH
H2NC(S)NH2
Cl
I
н
Алкилирование М. в щелочном р-ре в первую очередь
протекает по SH-группе, а не по циклич. азотам. При
нагревании М. с Ni-катализатором образуется пурин.
М. применяют при лечении острого лейкоза. Анти-
лейкемич. активность М. связана с его биологич.
ролью в качестве антиметаболита природных пуринов.
Являясь структурным аналогом 6-оксипурина (см.
Гипоксантин) и 6-аминопурина (см. Аденин), М. ак-
тивно вмешивается в пуриновый обмен и вызывает
нарушение синтеза нуклеиновых к-т.
Лит .: Е 11 о п G.B., Burgl Е., Hitchlngs G. Н.,
J. Amer. Chem. Soc., 1952, 74, № 2, 411; Ben di ch A.,
Russell P. J„ Fox I. J., там же, 1954, 76, № 23, 6073.
P. Г. Глушков.
МЕРКАПТОФЕНИЛТИОТИО ДИАЗОЛ ОН (висму-
тол II) CsH6N2S3, мол. в. 226,31 — белые или жел-
товатые тонкие игольчатые кристал-
свн5~М N лы; растворяется в воде и горячем
с«»С СхсН спирте. Получают кипячением фе-
V SH нилгидразина и CS2 в присутствии
спиртового р-ра щелочи. М. приме-
няют для обнаружения Sb, Bi, Си, Pb, Hg, Ni и Ag;
для фотометрия, определения Bi. я. я Ефимов.
МЕРКАПТОФОС — см. Фосфорорганические инсек-
тициды.
МЕРКУЗАЛ — водный 10%-ный р-р мононатрие-
вой соли меркузаловой к-ты (I) (карбоксиметилового
эфира 2-метокси-З-ртутьоксипропиламида салицило-
вой к-ты) в соединении с вероналом (II) в эквимолеку-
лярных соотношениях; бесцветная прозрачная жид-
кость, обладает щелочной реакцией. М е р к у з а-
ловая кислота С13Н13О3 NHg, мол. в. 465,76—
_ ОСН2СООМа
О
CON HCH2CHCH2Hg
I ОСН3
NH-CO С2Н5
г \ г
о=с хсч
Ч'Н-СО С2Н5
II
кристаллич. порошок, нерастворима в воде, кислотах
и органич. растворителях, растворима в щелочах;
при добавлении к AgNOs выпадает белый осадок, под
действием разб. H2SO4 образуется белый липкий оса-
док. Меркузаловую к-ту получают, исходя из аллил-
амина и салола, салициловой кислоты и аллилгорчич-
ного масла или из амида салициловой к-ты.
М.— эффективное ртутьорганич. мочегонное средст-
во; применяют при хронич. сердечной недостаточно-
сти, циррозах печени, нек-рых нефрозах и т. и. М.
относительно высоко токсичен.
Лит.: Машковский М. Д., Лекарственные средства,
4 изд., М., 1960, с. 227; Гр и н б е Р г Ф. Л., Фармация, 1940,
№6, 12. В. Г. Яшунский.
МЕРКУРИМЕТРИЯ — титриметрич. метод коли-
чественного анализа, основанный на использовании
титрованных р-ров солей двухвалентной ртути (мер-
кури-ионов), к-рые с хлоридами, бромидами, циани-
дами, роданидами образуют малодиссоциированные
соединения. Окончание титрования устанавливают
с помощью индикаторов. В качестве рабочих р-ров
обычно используют нитрат или перхлорат ртути. При
определении хлорид-ионов в качестве индикатора
употребляют натрия нитропруссид, к-рый дает с ио-
нами ртути (II) белый осадок. Заканчивают титрова-
ние при появлении неисчезающей мути. В качестве
индикаторов при меркуриметрич. определении гало-
ген-ионов используют также дифенилкарбазид или ди-
фенилкарбазон, образующие с ионами Hg (II) окрашен-
ные в фиолетовый цвет соединения. При титровании
иодидов образуется устойчивый комплекс [HgJi]2-.
Конечную точку титрования устанавливают по образо-
ванию неисчезающей мути красного цвета HgJ2.
Указанные методы| определения галоген-ионов могут
быть использованы и для определения ртути; для этой
же цели часто используют титрованный р-р роданида,
к-рый дает с ионами Hg (II) малодиссоциированноэ
соединение, индикатором служат ионы Fe (III), об-
разующие с избыточными ионами роданида красное
окрашивание. М. находит широкое практич. приме-
нение благодаря нек-рым преимуществам перед ар-
гентометрией. М. позволяет вести прямое определе-
ние анионов в кислой среде; применяется не только
для определения галогенидов, цианидов, роданидов,
но и ионов Hg (II); дает возможность титрования в при-
сутствии многих посторонних ионов.
Лит. см. при ст. Аналитическая химия и Объемный анализ,.
И. П. Ефимов.
МЕРКУРОМЕТРИЯ — титриметрич. метод коли-
чественного анализа, основанный на применении тит-
рованных растворов солей одновалентной ртути (мер-
куро-ионов). При взаимодействии ионов [Hg2]2+
с хлоридами, бромидами, иодидами и т. д. образуют^
ся малорастворимые осадки Hg2Cl2, Hg2Br2, Hg2J2.
В качестве индикатора в М. используют раствор
Fe(SCN)s. При титровании хлоридов красный р-р
роданида железа обесцвечивается, когда появляется
избыток ионов [Hg2]2 + . При меркурометрич. определе-
нии галоген-ионов в качестве индикаторов исполь-
зуют также дифенилкарбазон, дифенилкарбазид, об-
разующие с ионами ртути осадок синего цвета, а также
ализаринсульфонат натрия, бромфенолсиний и др.
Применение М. для определения ряда анионов (хло-
ридов, бромидов и др.), несмотря на возможность тит-
рования в кислых р-рах и незначительную раствори-
мость соединений, пока ограничено.
Лит.: Т а р а я н В. М., Меркуроредуктометрия, Ереван,
1958; см. также при ст. Объемный анализ. И. П. Ефимов.
МЕРОМИОЗИН — см. Миозин.
МЕССБАУЭРА ЭФФЕКТ — ядерная резонансная
флуоресценция без отдачи, т. е. явление испускания,
поглощения и рассеяния гамма-квантов атомными яд-
рами без расходования энергии на отдачу этих ядер;
открыто в 1958 Р. Л. Мёссбауэром. Важное значение
М. э. заключается в том, что с его помощью можно
обнаруживать исключительно слабые изменения
энергии ядерных переходов, напр., обусловленные из-
менениями состояния окружающих ядро электрон-
ных, в том числе валентных, оболочек. Наиболее бла-
гоприятные условия для наблюдения М. э. создаются,
когда ядра—излучатели и поглотители гамма-квантов,
входят в состав кристаллич. решеток, находящихся
при достаточно низких темп-рах. Поскольку энер-
гия отдачи свободного ядра далеко не достаточна для
разрыва химич. связей в решетке, она становится до-
стоянием всей решетки как целого и может высве-
титься лишь в виде квантов коллективного возбуж-
дения — фононон. При низких темп-рах весьма веро-
ятны случаи, когда возбуждение фононов не происхо-
дит, а потому импульс отдачи при поглощении или
испускании гамма-кванта воспринимается всей решет-
кой. Как при выстреле из бесконечно тяжелого ору-
дия, энергия отдачи оказывается при этом ничтожно
125
МЁССБАУЭРА ЭФФЕКТ —МЕТАЗИД
126
мала, несравненно меньше собственной (естественной)
ширины резонанса Г, а потому отдача не мешает на-
блюдению резонанса.
Естественная ширина тех возбужденных уровней
атомных ядер, для к-рых характерен М. э., состав-
ляет всего лишь 10-1°— 10-15 от величин энергии
возбуждения. Поэтому резонанс нарушается и ядерное
поглощение падает уже при таких чрезвычайно малых
изменениях энергии. Этого можно достичь, приведя
поглотитель в движение относительно источника:
благодаря эффекту Допплера изменяется частота, а
следовательно, и энергия гамма-квантов. Наличие
зависимости скорости счета гамма-квантов в счет-
чике от скорости движения резонансного поглотителя
у-источник у - поглотитель
Рис. 1.
этих квантов, расположенного между источником и
счетчиком, является характерной особенностью М. э.
(см. рис. 1). При этом достаточно двигать поглотитель
г
с весьма малой скоростью V—g" с (где — энергия
кванта, с — скорость света), от нескольких см/сек
до нескольких микрон/сек. Всякие внешние факторы,
изменяющие энергию гамма-квантов даже в весьма ма-
лой степени, нарушают резонанс. Действие этих фак-
торов можно определить количественно, компенси-
руя его допплеровским сдвигом энергии гамма-кван-
тов при движении поглотителя с определенной
скоростью.
Возможность применения М. э. в химии опреде-
ляется влиянием на энергию гамма-квантов строения
окружающих ядро электронных оболочек. Зависи-
мость энергии электростатич. кулоновского притяже-
ния ядра и электронов от плотности электронного об-
лака в области расположения ядра приводит к тому,
что резонансная энергия гамма-квантов несколько ме-
няется как при непосредственном изменении числа
s-электронов в атоме, так и при всякой перестройке
других электронных оболочек, меняющей их влияние
на конфигурацию s-оболочек (волновым функциям
«-состояний отвечает максимум электронной плот-
ности в центре атома, волновым функциям р- и др. со-
стояний — нулевая электронная плотность в центре
атома). Поэтому в случае химически различных излу-
чателя и поглотителя энергии их резонансных пере-
ходов различаются на нек-рую величину ±А£, и со-
ответственно максимум резонансного поглощения
(минимум скорости счета гамма-квантов) наблюдает-
ся при движении поглотителя относительно излуча-
теля со скоростью ±6= с. Величина 6 наз. хими-
ческим (изомерным) сдвигом мёссоауэровскои линии
поглощения. Пример такого химич. сдвига изображен
на рис. 2, где показан спектр поглощения гамма-
квантов ядрами Sn119 в SnCh, когда излучателем слу-
жат возбужденные ядра Sn"9* в SnO2. В данном слу-
чае наличие сдвига отвечает большей степени ионно-
сти Sn—Cl, по сравнению со Sn—О, связью. Валентные
5«р3 электроны оттягиваются от олова сильнее в SnCh,
нежели в SnO2, и плотность электронного облака в
области ядра олова в SnCU меньше, чем в SnO2.
Нарушение сферич. симметрии распределения элект-
рич. заряда в молекуле, приводящее к возникновению
неоднородного электрич. поля в области расположе-
ния ядер—излучателей и поглотителей, обусловливает
квадрупольное расщепление мёссбауэровских спект-
ральных линий типа изображенного на рис. 2 для три-
фенилоловохлорида, в молекулах к-рого одна из свя-
зей (Sn— Cl) является ионной в гораздо большей сте-
пени, чем остальные (Sn—С) связи. Данные о квад-
рупольном расщеплении служат характеристикой
валентных связей и позволяют определить градиенты
внутримолекулярных электрич. полей. Наконец, дейст-
вие внешних или создаваемых собственными электрон-
ными оболочками атома магнитных полей на ядра —
излучатели и поглотители, приводит к магнитному
(зеемановскому) расщеплению мёссбауэровских спект-
ральных линий, по к-рому можно судить о величине
внутримолекулярных магнитных полей.
Совокупность указанных данных о сдвигах и рас-
щеплениях линий открывает широкие возможностй
химич. применений М. э.— позволяет определить
как валентное состояние исследуемых атомов, так
даже и вклад различных электронных оболочек в
валентность, что особенно существенно для установ-
ления природы химич. связи. Основным недостатком
М. э. с точки зрения перспектив его применений в хи-
мии является отсутствие ядерного резонансного по-
глощения без отдачи для ядер легче железа. Поэтому
такие элементы, как водород, углерод, азот, кислород,
нельзя непосредственно исследовать с помощью М. э.
До настоящего времени М. э. особенно подробно изу-
чался для железа и олова. Однако круг элементов,
для к-рых М. э. может наблюдаться, достаточно ши-
рок (напр., железо, никель, цинк, германий, мышьяк,
рутений, олово, сурьма, теллур, иод, ксенон, цезий,
гафний, тантал, вольфрам, рений, осмий, иридий,
платина, золото, ртуть, таллий, почти все лантаниды
и актиниды) и потому М. э. несомненно найдет плодо-
творное применение в химии — особенно в химии
комплексных и элементоорганич. соединений.
Лит.: Гольданский В. И., Эффект Мёссбауэра и
его применения в химии, 1963; Фрауэнфельдер Г., Эф-
фект Мёссбауэра, пер. с англ., М., 1964.
В. И. Гольданский.
МЕТА-----приставка, применяемая для обозначения
положения двух заместителей в бензольном ядре (см.
рис.). Приставка М. указывает также на
состояние полимеризации, напр. в металь- । 3
дегиде. В неорганич. химии приставку М.
применяют в названии кислот и их солей, L JLq_|
к-рые отвечают наименьшему содержанию 3
гидроксильных групп, напр. метафосфорная кислота
РО2(ОН). Подробнее см. Орто-, мета-, пара-.
МЕТАЗИД (1,Г-метилен-бис-изоникотиноилгидра-
зон) Ci3Hi4O2Ne, мол. в. 286,30 — кристаллич. поро-
шок белого или кремового цвета со слабогорьковатым
127
МЕТАКРИЛОВАЯ КИСЛОТА—МЕТАЛЛИРОВАНИЕ
128
^-NHNHCO
СН2
XNHNHCO
вкусом, т,. пл. 175—181° (с разл.), нерастворим
в холодной и горячей воде и в органич. растворите-
лях, растворим в разб. мине-
ральных к-тах. М. обладает хи-
мич. свойствами, характерными
для ряда изоникотиноидгидрази-
да. После кипячения с водой М.
дает с нитропруссидом натрия
в уксуснокислом р-ре оранжевое окрашивание, пе-
реходящее при добавлении 10%-ной соляной к-ты в
желтое или зеленовато-желтое. Охлажденный после
кипячения водный р-р М. дает с фенилгидразином и
нитропруссидом натрия интенсивное синее или сине-
зеленое окрашивание, переходящее при стоянии в
красно-оранжевое. Количественное определение М.
основано на окислении его избытком титрованного
р-ра иода в щелочной среде и титровании выделяю-
щегося при подкислении избытка иода тиосульфатом
натрия. М. получают взаимодействием гидразида изо-
никотиновой к-ты с формалином. М.— эффективный
противотуберкулезный препарат.
Лит.: Сычева Т. П., в кп.: Материалы по обмену пе-
редовым опытом и научными достижениями в химико-фарма-
цевтической промышленности, № 1/12, М., 1958, 55; Grun-
b erg Е., Schnitzel R. J., Proc. Soc. Exptl. Biol, and
Med., 1953, 84, № 1, 220; L arson F.C., Dickie H. A.,
там же, 1953, 84, № 2, 301. Л. И. Яхонтов.
МЕТАКРИЛОВАЯ КИСЛОТА (а-метилакриловая
кислота) сн2=с-соон, мол. в. 86,09 — жидкость;
сн,
т. кип. 160,5°/760 мл; 60°/12 мм-, d“l,0153;n”l,4314;
растворима в эфире, воде, спирте. М. к. полимери-
зуется постепенно уже при комнатной температуре,
особенно быстро при нагревании в присутствии ката-
лизаторов. М. к. восстанавливается амальгамой нат-
рия до изомасляной к-ты; с иодистым водородом дает
р-иодизомасляную к-ту; присоединяет бром, образуя
а, 0-дибромизомасляную к-ту. Производные М. к.
также легко полимеризуются, напр.:
CHj = C(CH3)CN
,CHS zch,
.. .сн2-с-сн2-с
4 GN 4 GN
M. к. и ее эфиры в пром-сти получают из ацетона:
СН3
>C=O+HCN
СН3Х
сн3. ОН—Н2О
СН/ 'GN
СНа = С- CN НОН
|----------► СН2 = С (СН3) COOR
СН3 H2SO,
М. к. может быть получена также нагреванием со ще-
лочью продукта конденсации хлороформа с ацетоном:
кон кон
СН,СОСН3 + НСС13 —► (СН3)3С(СС1..)2ОН —>
---------->- СН. = С(СН3) соон
М. к. и ее производные широко применяют при по-
лучении технически важных полимеров и сополиме-
ров. Особенно большое значение имеет метиловый эфир
М. к., являющийся важнейшим продуктом в производ-
стве органич. стекла (см. Метилметакрилат). Поли-
меры М. к. используют также в текстильной пром-сти,
при изготовлении пигментированных паст, для полу-
чения эластомеров. Полиметакриламид применяют
в качестве защитного коллоида в фотографии; сопо-
лимеры используют при получении фотоэмульсий, а
сополимеры с акриловой и метакриловой к-тами — для
отделки текстильных изделий. Сополимеры нитрила
М. к. с рядом непредельных соединений находят неко-
торое применение для получения волокон и пленок.
Производные М. к.: этиловый эфир, т. кип-
1180,'760 мм, 30°/18 мм', амид, т. пл. 102—106°; нитрил,
т. кип. 90—92°. И. А. Несмеянов.
МЕТАЛЕПСИЯ — последовательное замещение
атомов водорода в насыщенном углеводородном ради-
кале атомами галогена. Замещение происходит по
свободно-радикальному механизму:
ci2 +С12 +С12 +С1
сн4----->• сн3с1 ——> сн2с1а----> сна. ——► ecu
на -на -на
Регулировать металептич. фторирование органич.
соединений, как правило, не удается ввиду крайне
высокого теплового эффекта реакции. При действии
элементарного фтора на органич. соединения проис-
ходит либо исчерпывающее замещение атомов водорода
фтором (в случае СН4 или С2Н6), либо разрушение ор-
ганич. молекулы (в случае более сложных соеди-
нений).
Наиболее разработано металептич. хлорирование.
Этот процесс осуществляется в пром-сти: хлорирова-
ние метана до хлористого метила, метиленхлорида,
хлороформа или четыреххлористого углерода. В жир-
но-ароматич. ряду практич. применение нашло мета-
лептич. хлорирование толуола до хлористого бен-
зила, хлористого бензилидена или бензотрихлорида:
+ С12 +С12
С„Н5 - СН, —> С6Н, - СН2С1--►
-на -на
+а2
—*с6н5 - сна2 ——» с„н3 - са3
-на
Металептич. бромирование насыщенных углеводоро-
дов и их производных проводят в несколько более
жестких условиях нежели хлорирование. М. значи-
тельно ускоряется освещением и повышением темп-ры.
Прямое иодирование насыщенных углеводородов и их
производных является обратимой реакцией:
RH + J2^rtRJ + HJ
Образующийся иодистый водород в условиях реакции
легко восстанавливает иодистый алкил до исходного
алкана. Для успешного металептич. иодирования
в качестве галогенирующего агента применяют смесь
иода с окислителем (HJOs, HgO, HNOs, Н2Ог и др.).
Иодистый водород, выделяющийся в процессе иоди-
рования, окисляется до свободного иода и т. обр. рав-
новесие смещается в сторону образования иодорганич.
соединения. Реакция М. на примере хлорирования
уксусной к-ты последовательно до моно-, ди- и три-
хлоруксусной к-ты была осуществлена Ж. Дюма и
О. Лораном в 1834 и явилась важным фактором в опро-
вержении существовавшей тогда теории радикалов и
послужила толчком к принятию теории типов. В на-
стоящее время термин «М.» является устаревшим и
выходит из употребления.
Лит.: Бутлеров А. М., Избранные работы по органи-
ческой химии, [М.], 1951, с. 11. Л. С. Герман.
МЕТАЛЛИРОВАНИЕ — замещение атома водо-
рода в органич. соединении на металл или металлосо-
держащий остаток с образованием связи углерод—ме-
талл. В зависимости от условий, характера металли-
руемого соединения и рода металлирующего агента
реакция М. имеет различный механизм. В качестве
металлирующих средств применяют металлы, амиды
металлов, металлоорганич. соединения:
НН + Ме —> НМе+ ’;2Н2
RH + NH2Me —н RMe + NH3
RH + R'Me —>- RMe+R'H
Парафиновые углеводороды в реакцию не вступают.
Ароматич. углеводороды не реагируют с Na, К и спла-
вом К — Na, однако металлируются при действии ме-
таллоорганич. соединений жирного ряда:
CbH,+C2H5Na —» CeH5Na+C2H,
129
МЕТАЛЛИРОВАНИЕ
130
Реакцию проводят в избытке ароматич. углеводорода
или в среде парафинового углеводорода (при 0—25°).
Металл вступает в о-положение к таким заместителям,
как СН,О, С6Н5О, CsH5S, R2N. При повышении темп-ры
происходит биметаллирование, причем атомы ме-
талла оказываются в .мета-положении друг к другу:
таллируются в боковую цепь амидами металлов в жид-
ком аммиаке, напр.:
№+С2Н6
Na
СНэ\'а
В случае жирноароматич. углеводородов легче всего
замещается металлом водород в a-положении боковой
цепи. Поэтому металлирующими агентами могут слу-
жить как алкильные, так и арильные соединения ще-
лочных металлов:
C8H3CH3 + C2H5Na —> C8HsCH2Na + C2H„
C8H5CH3 + C8H3Na —C8H3CH2Na + C8He
По этой же причине полученные тем или иным спосо-
бом жирноароматич. соединения с металлом при аро-
матич. ядре быстро переходят в соответствующие со-
единения с металлом в боковой цепи:
К
(n-CH3C8H,)2Hg —> n-CHjCeH.K .—► С„Н3СН2К
Метиленовая группа диарилметанов легко металли-
руется металлоорганич. соединениями (при 20—70°).
М. амидами металлов происходит в более жестких
условиях (120—150°). Водород активной метилено-
вой группы в таких углеводородах, как флуорен, ин-
ден и циклопентадиен, замещается на металл также
действием К, Na и КОН. Замещение третичного водо-
рода в триарилметанах протекает гладко при дейст-
вии амидов щелочных металлов в жидком аммиаке, а
также металлоорганич. соединений, в том числе и
производных диарилметана.
При взаимодействии полиарилметанов и -этанов
с амидами щелочных металлов в жидком аммиаке
образуются металлоорганич. соединения только в тех
случаях, когда углеводороды содержат в качестве
структурной единицы бензгидрильную группировку
(«бензгидрильное правило»):
(С„Н8)2 СН2 —► (С8Н3). СН Ме
(С„Н5)а СН —> (С8Н8)3СМе
(С8Н3)2 СНСН,—> (С,Н5)2 СМеСН3
(С8Н3)2 СНСН (С8Н3)2 (С8Н8)2 СМеСМе (СаН3)2
Такие соединения, как СвН5СН2СН2СвН5 и (С6Н5)3ССН31
с амидами металлов в жидком аммиаке не реагируют.
Замещение атома водорода при углерод — углерод-
ной тройной связи в ацетиленах происходит очень
легко при действии р-ров металлов или их амидов
в жидком аммиаке:
ЗНС = CH + 2Na—> 2НС = CNa + H2C=CH2
НС= CH + NH2Na—> HC = CNa+NH3
М. может протекать также при действии свободных
металлов:
1 00 - 200°
HC = CH + Na ---—HC = CNa+;.,H,
220°
НС = CNa —> NaC = CNa + HC = CH
Ацетиленовые производные щелочноземельных метал-
лов могут быть легко получены с помощью соответст-
вующих металлоорганич. соединений:
HC = CH + 2RMgX—> XMgC = CMgX+2RH
Металлируются также и гетероциклич. соединения
(см. Иоцича реакция), хотя они исследованы в значи-
тельно меньшей степени, чем углеводороды; как пра-
вило, процесс идет с гораздо худшими выходами. 2- и
4-Метильные производные пиридина и хинолина ме-
Известны случаи М. в ядро, напр. в случае тиофена
и дибензофурана:
Рассмотренные реакции имеют определенное значе-
ние для синтетич. органич. химии (получение через ме-
таллоорганич. соединения карбоновых к-т, спиртов
и т. д.). Область применения реакции ограничена, с од-
ной стороны, способностью к образованию металло-
органич. соединений, с другой — наличием в исходном
соединении таких реакционноспособных группиро-
вок, как СО, СООН, NO2 и т. п.
М. с помощью металлоорганич. соединений по
схеме: _
RH + R'Me —> RMe + R'H
аналогично реакции вытеснения кислоты из ее соли
более сильной кислотой, напр.:
H2SO. + CH3COOK •—> KHSOt + CH3COOH
Основываясь на этой аналогии, можно построить след,
ряд относительной кислотности углеводородов:
RC = СН; Аг3СН; Аг2СН2; ArCH3; ArH; AlkH
Металлич. производное каждого члена ряда металли-
рует все соединения, находящиеся справа; следова-
тельно, кислотность убывает слева направо. Таким же
образом можно получить ряд основности (т. е. актив-
ности в реакциях М.) металлоорганич. соединений
(основность убывает слева направо):
НК; RNa; RLi; RCaX; RMgX: R2Zn; R3A1; H2Cd
Другую большую группу составляют реакции М.
производными тяжелых металлов. Сюда можно отне-
сти след, процессы.
1) Образование ацетиленидов серебра и однова-
лентной меди (при пропускании ацетилена в аммиач-
ные р-ры солей серебра и закисной меди) и ацетиле-
нида двухвалентной ртути (при пропускании ацети-
лена в щелочной р-р иодида ртути):
HC^CH + 2Ag+ —> Ag2C2 + 2H +
НС = СН + 2Cu + —> Cu2C2 + 2Н +
НС = CH + HgJ2 + 2KOH —- HgC2 + 2KJ + 2H2O
2) Меркурирование ионизирующимися солями рту-
ти в р-рах сильных к-т происходит по электрофиль-
ному, а слабо диссоциирующей уксуснокислой ртутью
в неполярных растворителях — по радикальному ме-
ханизму:
/—к Н^С1О4 \
°2n-( ) + —► o2n-( ) + НС1О4
, С1О7 '—<
, Н HgCio4
OCOCH3 —.
н + Hgz —- o2n-Z VHgCio4 + сн3соон
ГоСОСНз
0 5 К.х.э. т. 3
131
МЕТАЛЛИЧЕСКАЯ СВЯЗЬ—МЕТАЛЛИЧЕСКИЕ РУДЫ
132
3) Ряд процессов по общей схеме Фриделя—
Крафтса реакции, напр. арсенирование ароматич. сое-
динений галогенными солями мышьяка в присутствии
хлористого алюминия:
А1С13
C»H. + ASC1S-—« CsH5ASC13
А1С13
С»Н34-CeligAsCl2 -► (С3Н3)з AsCl
CftHe4-(CeHs)2 AsCl —> (CeH6)3 AS
Имеются аналогичные примеры аурирования, герма-
нирования, стибирования, таллирования. Таким же
образом получают органич. производные нек-рых эле-
ментов-неметаллов, напр. фосфора:
А1С13
С3Н»4-РС13-----> C„HSPC13
4) М. окислами и гидроокисями тяжелых металлов,
наир.:
NaOH .СН2.
СН2 (COOH)34-HgO------► Hg( )с = о
' о z
Лит.: Синтетические методы в области металлоорганиче-
ских соединений, под ред. А. Н. Несмеянова и К. А. Кочешко-
ва, М.—Л., вып. 1, 1949, вып. 3, 7, 1945, вып. 5, 8, 1947.
Б. Л. Дяткин.
МЕТАЛЛИЧЕСКАЯ СВЯЗЬ — см. Металлы.
МЕТАЛЛИЧЕСКИЕ РУДЫ — горные породы, со-
держащие один или несколько полезных металлов в та-
ких соединениях, концентрациях и количествах, при
к-рых возможно и целесообразно их извлечение при
современном уровне техники и экономики. М. р. яв-
ляются основным сырьем для получения металлов.
М. р. образуются из многочисленных ассоциаций эле-
ментов, находящихся в земной коре либо в состоянии
рассеяния, либо в состоянии концентрации. Подав-
ляющее большинство металлов (железо, медь, свинец,
цинк, марганец, хром, ванадий, вольфрам, олово,
уран и др.) образует в силу своих химич. свойств са-
мостоятельные минералы, имеющие промышленное
значение. Другие металлы, такие, напр., как рубидий,
кадмий, галлий, таллий, индий, германий, гафний,
скандий, рений, хотя иногда и образуют собственные
минеральные формы, но промышленное значение по-
следних ничтожно, т. к. подавляющая часть этих ме-
таллов находится в земной коре в рассеянном состоя-
нии, в малых концентрациях. Нек-рые из них при-
сутствуют в других минералах в виде изоморфной
примеси. Так, рений связан с молибденитом, гафний—
с цирконом, индий — со сфалеритом и касситеритом,
галлий — со сфалеритом, бокситом, рубидий —
с лепидолитом.
Природные соединения металлов в основном срав-
нительно просты. Нек-рые металлы (железо, марга-
нец, хром, вольфрам, олово, ниобий, тантал, уран,
торий и др.) преим. распространены в виде кислород-
ных соединений. Это, напр., основные минералы
железа — магнетит РезО4, гематит Ре20з’, минералы
марганца — пиролюзит МпО2, браунит Мп20з, гаусма-
нит МпзО4; минерал хрома — хромит FeCr2O4; мине-
ралы вольфрама — вольфрамит (Fe, Mn)WO4, шеелит
CaWO4. Другие металлы (никель, медь, цинк, свинец,
кобальт, ртуть, молибден, висмут и др.) встречаются
в виде сернистых, мышьяковистых и сурьмянистых
соединений. Таковы: основные минералы меди — халь-
копирит CuFeS2, халькозин Cu2S, ковеллин CuS; ми-
нералы свинца — галенит PbS, буланжерит Pb5Sb4Sn;
минералы никеля — миллерит NiS, никелин NiAs,
герсдорфит NiAsS. Нек-рые элементы (золото, серебро,
иногда висмут, медь и др.) образуют самородные ме-
таллы, встречающиеся почти в химически чистом
виде.
М. р. добываются из естественных обособленных
промышленных скоплений рудных минералов к.-л.
формы (жила, система жил, пласт, система пластов,
штокверк — совокупность пересекающихся между со-
бой мелких рудных жилок, и др.). Эти промышленные
скопления минералов наз. месторождения-
м и. Размеры месторождений самые разнообразные.
Наибольшие размеры (десятки и сотни километров
длиной или площади в сотни тысяч квадратных мет-
ров) имеют пластообразные залежи железных, мар-
ганцовых, лопаритовых и др. руд, прибрежно-морские
россыпи циркона, монацита, касситерита, уранонос-
ные пегматитовые жилы, штокверки молибдена, меди,
вольфрама. Размеры месторождений определяются их
запасами, т. е. определенным количеством металла,
заключенного в недрах месторождения. Размеры в де-
сятки, сотни метров — до нескольких километров
в длину характерны для жильных месторождений
цветных и редких металлов.
М. р. подразделяются на промышленные и
непромышленные. Руды относятся к про-
мышленным только в том случае, если они по своему
качеству и количеству отвечают требованиям пром-сти.
Главнейшими из этих требований являются: оп-
ределенное среднее процентное содержание металла
в руде, подходящий химич. и минералогич. состав
руды (для сульфидных руд особенно важна степень
их окисленности, т. к. окисленные, полуокисленные и
первичные сульфидные руды требуют различной тех-
нологии в переработке, причем, напр., окисленные мо-
либденовые руды, вследствие значительных трудно-
стей при их обогащении, пока, по состоянию техноло-
гии, почти не имеют промышленной ценности). Не ме-
нее важными для оценки качества руд являются и их
физич. и горно-технич. свойства; темп-ра плавления,
магнитность, кусковатость, радиоактивность и др.
Понятие промышленной руды меняется со временем.
По мере совершенствования техники руды, считав-
шиеся ранее непромышленными, переходят в разряд
промышленных.
Руды состоят из рудных и нерудных (пустой поро-
ды) минералов. Для руд данного металла характер-
ны определенные главные или основные минералы.
Так, для железных руд характерны магнетит, гема-
тит, гетит; для медных руд — халькопирит, халько-
зин, ковеллин; для свинцово-цинковых — галенит,
сфалерит, церуссит, англезит и др. К нерудным ми-
нералам обычно относятся кварц, кальцит, доломит,
барит, гипс, хлорит, полевой шпат, серицит и др.
В зависимости от химич. состава М. р. подразде-
ляются на две разновидности. Простые руды, из
к-рых извлекается один металл, характерны для же-
леза, алюминия, хрома, марганца и др. Сложные,
или комплексные, руды, из к-рых извлекается
сразу несколько металлов, характерны для свинца и
цинка, меди и молибдена, никеля и кобальта, железа
и титана, железа и ванадия и др. При переработке
комплексных руд из них извлекают многие рассеян-
ные металлы, в частности рений, кадмий, германий,
селен, теллур и др. Из никелево-медных руд крупней-
шего в мире месторождения Сёдбери (Канада) извле-
кают, помимо меди и никеля, кобальт, золото, серебро,
платину, палладий, селен, теллур. Многие М. р., по-
мимо полезных компонентов, содержат вредные
компоненты, к-рые в процессе переработки руд долж-
ны быть удалены. Для марганцовых руд вредным ком-
понентом считается фосфор, для железных и хромовых
руд — сера, фосфор и др., для никелевых — свинец,
цинк, висмут, мышьяк и др., для вольфрамовых —
фосфор, сера, мышьяк, олово, медь и др.
В зависимости от процентного содержания основ-
ного металла М. р. подразделяются на сорта: бога-
тые, средние и бедные. Медные руды,
напр., считаются богатыми при содержании меди бо-
лее 2%; средними — если меди больше 1 %; бедными —
133
МЕТАЛЛИЧЕСКИЕ РУДЫ
134
при содержании меди от 0,7 до 1%. В последнее время
бедные и комплексные руды все более широко исполь-
зуются пром-стью.
Руды отличаются текстурами и струк-
турами. Под текстурой руды понимают ее строе-
ние, обусловленное пространственным расположением
слагающих ее рудных и нерудных минералов. При бо-
лее или менее равномерном сочетании минеральных
агрегатов с явным преобладанием рудных минералов
(до 80%) среди пустой породы текстура руды считает-
ся массивной. Такие текстуры характерны для руд же-
леза, хрома, меди, свинца, марганца и др. При преоб-
ладании нерудных минералов текстура руды является
вкрапленной. Вкрапленные текстуры характерны для
большинства руд цветных и редких металлов. В зави-
симости от рисунка, образованного вкрапленностью
рудных минералов на фоне пустой породы, различа-
ются текстуры: полосчатые (вкрапленность рудных
агрегатов образует вытянутые полоски), пятнистые
(вкрапленность рудных минералов слагает массивные
скопления), петельчатые, или сетчатые (тончайшие
полоски рудных минералов пересекаются между
собой в различных направлениях) и мн. др. Много-
образие текстур руд определяется различными спо-
собами происхождения руд. Под структурой руды
подразумевают строение каждого минерального агре-
гата, определяемое формой, размерами и способом
сочетания зерен отдельных минералов, входящих
в состав данного агрегата. По структурам М. р. под-
разделяются на равномернозернистые, неравномерно-
зернистые, порфировые (с отдельными крупными зер-
нами минералов среди равномернозернистой массы),
оолитовые (с концентрически округлыми скопления-
ми рудных минералов), решетчатые (образовавшиеся
в процессе закономерного пересечения минеральных
полос), радиально-лучистые п др. Текстурно-струк-
турные особенности М. р. имеют большое практич.
значение при установлении качества руд с точки зре-
ния их технологич. переработки.
М. р. по условиям образования делятся на м а г-
матогенные, метаморфогенные и
экзогенные. Магматогенное происхождение ха-
рактерно почти для всех руд черных, цветных и ред-
ких металлов. Метаморфогенное происхождение имеют
гл. обр. нек-рые железные, марганцовые и золотые
руды. Экзогенным путем возникли россыпи олова,
золота, хромита, месторождения никеля, алюминия,
марганца, урана, ванадия и др. О происхождении
руд см. Геохимические процессы.
Почти все М. р., за исключением богатых, перед
металлургич. переделом проходят процесс обогащения,
в результате к-рого процентное содержание металла
в сырье увеличивается в десятки и сотни раз за счет
удаления значительной части пустой породы. Конечным
продуктом обогатительного процесса, представля-
ющим наибольшую промышленную ценность, яв-
ляется рудный концентрат данного металла.
О процессах обогащения см. Обогащение полезных ис-
копаемых. Металлургич. передел богатых руд и кон-
центратов заключается в их химич. обработке либо
при высоких темп-pax (пирометаллургиче-
ские процессы), либо в водных р-рах (гидро-
металлургические процессы), а также в со-
четании пиро- и гидрометаллургич. процессов. О ме-
таллургич. переделе руд см. ст. Гидрометаллургия,
Пирометаллургия и Электрометаллургия. Сведения
о важнейших рудах, их промышленных минералах,
средних содержаниях металла в руде, формах про-
мышленных скоплений, сопутствующих полезных и
вредных компонентах, а также ориентировочных ми-
ровых запасах главнейших металлов приведены в
таблице.
Металлы и их важнейшие промышленные минералы Среднее содержа- ние металлов в рудах Характеристика главнейших промышленных скоплений Попутные компоненты в рудах (в числителе — полезные, в знаменателе — вредные) Мировые ориентировоч- ные запасы металла
Железо магнетит FeFeaO( гематит Fe2O3 сидерит FeCO3 лимонит HFeO3aq гетит HFeO2 Не менее 20%, чаще 35—50% Пластообразные залежи гематито-магнетитовых руд в древнейших породах до- кембрия, Пластовые залежи гематито-сидеритовых руд прибрежно-морского проис- хождения. Столбообразные и жилообразные тела магне- титовых руд в породах ос новного и ультраосновного состава. Пластообразные и жилообразные залежи магне- тито-гематитовых руд, свя- занных со скарнами. Mn, Nl, Со, Mo, Т1. Ge, V S, Р, As, Zn, Sn Ок. 75 млрд. т
Марганец пиролюзит МпО2 браунит Мп2Оа псиломелан mMnOMnO3nH3O гаусманит Мп3О< манганит Мпб2Мп(ОН)2 родохрозит МпСО8 родонит MnSIO3 Не менее 15%, чаще 40—50°/о Пластообразные залежи марганцовых руд среди оса- дочных пород прибрежно- морского типа. Длина зале- жей достигает десятков и сотен километров. Плашеоб- разные пирол.юзитовые и псиломелановые залежи, об- разованные процессами вы- ветривания. Размеры место- рождений этого типа весьма значительны. Fe, Nl, Co и др. p Более 1 млрд. т
Никель пентландит (Fe, N1)9S8 миллерит N1S никелин NiAs герсдорфит NIAsS гарниерит Ni, [Si,O10] (ОН),-4Н2О ревденскит 3 (Nl, Mg) O-2S1O2*2H3O Не менее 0,3% Пластовые залежи сплош- ных сульфидных никелевых руд в основных и ультраос- новных породах. Размеры залежей измеряются сотня- ми метров. Пластовые, жи- лообразные и гнездообраз- ные залежи силикатных никелевых руд, образован- ных процессами выветрива- ния. Cu, Co, Pt, Pd, Rh, Au, Ag, Se, Те Pb, Zn, Bi, As и др. Ок. 50 млн. гп
5*
136
Продолжение
135
МЕТАЛЛИЧЕСКИЕ РУДЫ
Металлы и их важнейшие промышленные минералы Среднее содержа- ние металлов в рудах Характеристика главнейших промышленных скоплений Попутные компоненты в рудах (в числителе — полезные, в знаменателе — вредные) Мировые ориентировоч- ные запасы металла
Вольфрам Для коренных Кварцево-вольфрамитовые Mo, Au, Си, Bi, Sn, Be, Pb, Zn и др. Ок. несколь-
вольфрамит (Fe, Мп) WO4 гюбнерит MnWO4 ферберит FeWO4 шеелит CaWO* месторождений 0,2—6,0°/о WO3, для россыпей не менее 0,015% WO3 жилы длиной до 1—2 км, глубиной до 500—700 м. Пластовые и жилообразные шеелитовые скарновые зале- жи площадью до сотен тысяч квадратных метров. Россыпи вольфрамита и гюбнерита, размеры их невелики. Шток- верковые вольфрамитовые месторождения, их площади измеряются сотнями тысяч квадратных метров. Ba, S, Ca, P, As и др. ких млн. т
Молибден молибденит MoS2 0,1—1,0%, для комплексных руд 0,005% Молибденитовые жилы длиной до нескольких сотен метров, глубиной 300—400л. Штокверковые молибдено- вые, молибдено-вольфрамо- вые, молибдено-медные, мед- но-молибденовые месторож- дения весьма больших раз- меров. Скарновые молибде- новые залежи. W, Be, TR*, Sn, Au, Re, Pb, Zn Си, P, As Несколько млн. т
Ванадий патронит от VS2 до V2Ss роскоэлит KV2 [AISi,OI0] (ОН)2 ванадинит PbjfVO.JjCl деклуазит (Zn, Си) Pb [VO.] ОН карнотит К2 [UO2]2 [VO.]2-3H2O Не менее 0,7°|о V2O5, при попут- ной добыче из нек-рых желез- ных руд 0,1—0,4°/о V2O5 Жилообразные патронито- вые залежи (известно только одно месторождение этого типа). Гнездообразные тела ванадинит- и деклуазитсо- держащих руд в окисленных частях полиметаллич. место- рождений. Пластовые и лин- зообразные залежи карно- титовых руд в осадочных породах. Ванадий попутно получают из желеаных, мед- ных и др. руд. и, Cu, Fe и др, P, CaO Ок. первых сотен тыс. т
Медь 0,5-10% Пластовые залежи меди- стых песчаников длиной в сотни километров. Медно- молибденовые штокверки среди изверженных пород кислого состава. Линзооб- разные залежи халькопири- товых руд среди вулканич. пород. Жилы и столбообраз- ные залежи халькопирито- вых руд в скарнах. Жилы сульфидных медных руд в различных породах. Pb, Zn, Mo, Fe, V. Ti, Au, Ag, Re, Более
халькопирит CuFeS2 халькозин Cu2S ковеллин CuS борнит Cu.-,FeS4 энаргит Cu5AsS4 куприт Си2О самородная медь Си и др. Pt, Cd, In, Tl, Ga, Se, Те и др. 150 млн. т.
Свинец и цинк Свинца не менее Пластовые залежи свинцо- Ag, Cd, Au, Se, Те, Ge, In, Bl и др. Pb + Zn
галенит PbS буланжерит Pb-,Sb4Sn джемсонит Pb4FeSb6Si4 церуссит РЬСО3 англезит PbSO4 сфалерит ZnS смитсонит ZnCO.3 каламин Zn4 [Si2O7] (ОН)2 Н2О 0,5—0,7%, соот- ношение свинца и цинка в рудах обычно 1,5:1 или 2:1 и более во-цинковых руд в древней- ших породах докембрия. Пласты вкрапленных гале- нит-сфалеритовых руд в карбонатных породах. Тру- бообразные залежи свинцо- во-цинковых руд, обычно связанные со скарнами. Жилы сплошных полиметал- лич. руд в различных поро- дах. Пластообразные и лин- зообразные залежи гале- нит-сфалеритовых руд в излившихся (вулканич.) по- родах. десятки млн. т
Олово Для коренных месторождений не менее 0,13—0,2% Россыпи касситерита. Жи- W, Be, Ta, Nb, Sc и др. Первые
касситерит SnO2 станнин Cu2FeSnS4 лы, жильные зоны, шток- верки касситерито-кварце- вых и касситерито-сульфид- ных руд в изверженных и осадочных породах. Pb, Zn, Sb, As, Bi и др. млн. т
Ртуть Не менее 0,15—0,2°/„ Пластовые залежи вкрап- — Ок. 1 млн. т
киноварь HgS метациннабарит HgS самородная ртуть Hg ливингстонит HgSb.S, ленных киноварных руд среди осадочных пород. Ме- сторождения этого типа про- стираются в длину на иеск. километров, в глубину на 500—700 м. Жилы киновари среди различных пород, за- пасы таких месторождений невелики.
* TR — редкоземельные элементы.
137
МЕТАЛЛИЧЕСКИЕ РУДЫ
138
Продолжение
Металлы и их важнейшие промышленные минералы Среднее содержа- ние металлов в рудах Характеристика главнейших промышленных скоплений Попутные компоненты в рудах (в числителе — полезные, в знаменателе - вредные) Мировые ориентир овся- ные запасы металла
Уран Не менее 0,05°/0 U,О6, чаще всего 0,'1°/„, при попут- ной добыче (в фосфоритах) 0,01% Не менее 1—2 г/т Пластообразные уранонос- Fe, Pb, Zn, Ag, NI и др. Ок. 10
торбернит Си [UO2]2 [РО4].г12Н2О метаторбернит Си [UO2]2 [РО4]2-8Н2О карнотит К2 [UO2]2 |VO4]2-3H2O тюямунит Са l.UO2|2 IVO<]2-8H2O монацит (Се, La...) РО< с ТЬО2 и U отэнит Са [UО2}21РО4]2-8Н2О уранинит UO2 Золото ные залежи в измененных древнейших породах. Пла- стовые и линзообразные тела урано-ванадиевых руд в пес- чаниках. Жилы различных урансодержащих руд в из- верженных породах. Урано- носные пегматитовые жилы длиной до десятков и соген метров. Прибрежно-морские россыпи монацита. Пласты ураноносных сланцев и фос- форитов в осадочных поро- дах. Пластовые залежи золото- Pb, Bl, Fe, S и др. млн. т U3Og Ок. 20 тыс. т
самородное зотото Au калаверит АиТе2 сильванит AuAgTe4 петцит (AuAg)2Te и др. Бериллий 0,1%, при добыче из комплексных РУд 0,02% Не менее 0,5—0,2% Та2О5, не менее 0,1—0,4%Nb2O6 Не менее 0,7—1,0% Ы8О 0,02-0,6% в ру- дах месторожде- ний редких металлов В блокитовых рудах содержа- ние Se несколько десятков процен- тов. в сульфид- ных рудах Se и Те сотые и тыс. доли процента Сотые и десятые доли процента носных конгломератов в древнейших породах докем- брия. Золото-кварцевые жи- лы в различных породах. Жилообразные залежи тел- луридов золота в излив- шихся (вулканич.) породах. Штокверки с рассеянным золотым оруденением в раз- личных породах, россыпи золота. Жилы и линзообразные залежи гранитных пегмати- тов. Кварцевые и грейзено- вые бериллоносные жилы в гранитах длиной неск. сотен метров. Пластовые тела бе- риллоносных магнетито- флюоритовых скарнов. Жилы гранитных пегмати- тов с танталит-кодумбитом длиной неск. сот метров. Пласты щелочных извержен- ных пород длиной десятки километров. Жилы пиро* хлорсодержащих пегматитов длиной неск. сот метров. Россыпи колумбита и танта- лита. Сподуменовые и лепидоли- Cu, Zn, As, Sb Sn, W, Mo, Ta, Li, Nb и др. Несколько млн. т при содержании 0 1"- Ве4-ВеО Nb2O5 первые
берилл Ве,А12 |S:»О18] хризоберилл ВеА12О( фенакит Be.SiO. гельвин (Mn, Fe, Zn)« [BeSiO.lA даналит Fes [BeSiO4]0S2 Тантал и ниобий Sn, Zr, Be, LI и др.
та нт алит (Fe, Мп) (Та, Nb)2 Об колумбит (Fe, Мп) (Nb, Та)2 Ое пирохлор (Na,Ca)2 (Nb,Ti)2 O0(F, ОН) лопарит (Na, Са, Се) (Nb, Ti) О3 и др. Литий P, S, C, SiO2 и др. Be, Ta, Nb, Sn, W и др. млн. т Га2О-ч первые сотни тыс. т Li2O ок. 5 млн. т Нет данный То же »> >>
сподумен LiAl [Si2Oe] лепидолит KLilcAIi,-. [Si3A10H)! (F, OH)2 петалит (Li, N;.) IAISuOh] амблигонит Li, Al [PO4] F циннвальдит KLiFeAI [Si.tAlO10] (F, OH)2 Скандий Sc входит в состав касси- терита, вольфрамита, колум- бита, циркона и др. редких минералов. Селен и теллур блокчт NiSe2 — известен только в одном месторож- дении мира в Боливии. Se и Те связаны с сульфидными минералами. Рубидий и цезий родицит KNaLi4Ai4Be;iBi0O27 (содержит Rb и Csj полуццит CsAiSi2O0 и различные минералы пегматитов, содержащие рубидий и цезий. товые жилы гранитных пег- матитов. Различные по фор- ме залежи литиеносных грейзенов, связанных с оло- вянно-вольфрамовыми жи- лами. Пегматитовые жилы в гра- нитах с тортвейтитом и бе- фанамитом. Грейзеновые за- лежи из руд редких метал- лов. Скандий в основном извлекается из концентратов РУД редких металлов. Непосредственно из руд селен и теллур не извлека- ются. Основным источником получения селена и теллура являются продукты и отхо- ды переработки сульфидных, железных, медных, никеле- вых и полиметаллич. руд и самородной серы и отчасти золотых руд. Рубидий и цезий извлека- ются попутно из пластовых залежей калийных солей, а также из натрий-литиевых пегматитов, содержащих ро- дицит, поллуцит, сподумен и др. минералы. Многие редкие и цветные металлы Na, LI, Be и др.
139
МЕТАЛЛИЧЕСКИЕ СОЕДИНЕНИЯ
140
Продолжение
Металлы и их важнейшие промышленные минералы Среднее содер- жание металлов в рудах Характеристика главнейших промышленных скоплений Попутные компоненты в рудах (в числителе — полезные, в знаменателе — вредные) Мировые ориентировоч- ные запасы металла
Стронций Не менее Пластовые линзообразные Нет данных
целестин SrSO< стронцианит SrCO3 3-15»,„ SrSO. залежи целестина в осадоч- ных породах. Размеры ме- сторождений измеряются несколькими километрами. Са, Ва
Цирконий и гафний циркон ZrSiO< бадделеит ZrO2 ввдиалит (Na,Ca)e Zr[(SiiiOis)(OH,Cl)], Hf входит в состав этих минералов. Не менее 0,4°1о циркона в россыпях Прибрежно-морские рос- сыпи циркона. Плащеобраз- ные залежи бадделеита в месторождениях коры вы- ветривания. Источником гафния являются цирконо- вые концентраты. — ZrO2 в россы- пях ок. 20 млн. т
Германий германит CujGeS^ аргиродит Ag-GeSe канфильдит AgsSnSe с Ge. Ge входит в состав магне- тита, сфалерита, халько- зина и других минера- лов, находится также в нек-рых углях. Высшее содер- жание Ge 0,1-0,3° 0, в комплексных рудах тысячные доли процента В мире известно только два месторождения германи- товых руд: Тзумеб (Африка) и Принц Леопольд (Респуб- лика Конго). Обычно герма- ний извлекается из отходов произ-ва цинка, свинца и меди а также из ископаемых углей или продуктов и от- ходов их переработки. Многие цветные, редкие и рассеянные элементы Запасы не учтены
Рений Самостоятельных минера- лов не образует, сопутствует многим сульфидным минера- лам, чаще всего молибде- ниту. Содержание рения в молибде- новых концен- тратах 0,05—100 г!т Рений извлекается в ос- новном из отходов перера- ботки молибденовых концен- тратов. - Запасы не учтены
Таллий Содержится в халькопири- те, сфалерите, галените, марказите и др. сульфидных минералах. Сотые доли процента Основным источником по- лучения таллия являются отходы‘переработки свинцо- во-цинковых и колчеданных РУД. — Запасы не учтены
Галлий Самостоятельных минера- лов не образует, встречается в составе германита, сфале- рита и др. минералов. Высшее содер- жание галлия в германите 1,85°|О Галлий в основном извле- кается из отходов алюмини- евого и цинкового произ- водств, при переработке медных и некоторых свин- цово-цинковых руд. In, Т1 и др. То же
Кадмий Входит в состав сфалерита и др. сульфидных минера- лов. Содержание кадмия в сфале- рите 0,1—0,5°/о Кадмий извлекается при переработке цинковых и свинцовых концентратов. — » »
Сведения о редкоземельных элементах см. в статье Лантаниды.
Лит.: Крейтер В. М., Поиски и разведка месторож-
дений полезных ископаемых, ч. 1, М., 1960; Первушин
С. А. [и д р.], Экономика цветной металлургии СССР, М., I960;
Б етехтин А. Г. [и д р.], Текстуры и структуры руд, М.,
1958; Требования промышленности к качеству минерального
сырья. Справочник для геологов, вып. 24—25, 27, 36, 39, 41 —
43, 45—46, 49, 51, 63, 67, 68, 70, 71, М., 1958—61. В. Г. Ростов.
МЕТАЛЛИЧЕСКИЕ СОЕДИНЕНИЯ. Содер-
жание:
Общие особенности...............................140
Классификация...................................142
Характеристика важнейших типов..................142
Практическое значение...........................146
Металлические соединения (металлиды, металло-
подобные соединения, промежуточные фазы в спла-
вах) — химич. соединения, обладающие металлич.
свойствами. М. с. большей частью образуются при
взаимодействии нескольких металлов (интерме-
таллические соединения), но могут так-
же содержать С, N, В, Si, Н и др. неметаллы, в соот-
ветствии с чем к М. с. относят металлоподобные кар-
биды, нитриды, бориды, силициды, гидриды и др.
М. с. рассматриваются в химии металлич. сплавов
(металлохимии), изучающей вопросы химич. взаимо-
действия элементов при образовании металлич. фаз.
Общие особенности. Составы М. с. большей частью
не подчиняются правилам нормальной валентности.
Многие М. с. имеют переменный состав и на диаграм-
мах состояния характеризуются ограниченными об-
ластями гомогенности. М. с., как правило, характери-
зуются упорядоченным расположением атомов и имеют
индивидуальные кристаллич. структуры, отличные от
структур образующих их элементов. В отличие от ион-
ных и ковалентных соединений, в М. с. преобладает
металлическая связь. Для исследования
химич. связей в М. с. проводят определения электрон-
ной плотности, изучение пространственного распреде-
ления (корреляции) электронов, а также определение
физико-химич. свойств. Типы кристаллич. структур
М. с. позволяют косвенно судить о характере химич.
связи в них. М. с. проявляют металлич. свойства
(в меньшей степени, чем чистые металлы) — высокую
электропроводность и теплопроводность, металлич.
блеск; обычно они обладают высокой твердостью и яв-
ляются хрупкими, но при нагревании становятся плас-
тичными. Многие М. с. имеют высокие темп-ры плав-
ления, а нек-рые из них обладают ферромагнетизмом
и свойствами полупроводников.
141
МЕТАЛЛИЧЕСКИЕ СОЕДИНЕНИЯ
142
В таблице приведены значения микротвердо-
сти а М. с., встречающихся в алюминиевых сплавах.
Соединение Микротвер- дость & кг/мм2 j Соединение Микротвердость кг/мм2
при 20° при 300° при 20° при 300°
MgsAl, 281 269 Cu2Al7Fe 595 571
CuMgtAl„ 414 400 Al,Ni 595 585
Mff.zn.,A12 422 350 Al„Mn 686 643
Mg, Si 433 380 1 AhMn 835 794
С11A12 531 481 | AlinSiMn2 880 780
Cu Mg A 1, 564 476 Cu3Al6 Ni 987 857
Mg3Al8SiBFe 585 571 Al3Fe 1147 1074
а Микротвердость определяется путем вдавливания в испы-
туемый образец алмазного пирамидального наконечника
(индентора) под действием малой нагрузки и последующего
измерения диагонали отпечатка с помощью микроскопа.
Число микротвердости есть отношение нагрузки к площади
поверхности полученного отпечатка.
& Нагрузка 10 г, время действия нагрузки 30 сек.
Для многих М. с. характерна способность давать
твердые растворы со своими компонентами, а также
друг с другом; такие фазы наз. металлидными
растворами. Свойства М. с. закономерно изме-
няются с их составом; напр., влияние химич. природы
1600
р 1400
1l200
"I
8 юоо
§
%
2
g 800 •
I 6ОО-
£
с
I 400-
200-
/ ff Ш /V V w
Элементы по группам (подгруппы Ь)
Рис. 1. Зависимость темп-p плавления нек-рых
соединений магния от расположения элементов
по рядам и группам периодич. системы (пунк-
тирные линии связывают соединения с пред-
положительными темп-рами плавления).
элементов, соединяющихся с магнием, на периодич.
изменение темп-p плавления образующихся М. с.
видно из рис. 1. Влияние этого фактора проявляется
NlMnSb
NlgMnSb
®N1 *Mn • Sb
Рис. 2. Кристаллические структу-
ры NlMnSb (тип CaF2) и Ni2MnSb
(тип Cu2AlMn).
также при сравнении
М. с. одинакового
качественного соста-
ва. Напр., при пе-
реходе от NiMnSb к
Ni2MnSb происходит
усиление металлич.
связи, что подтвер-
ждается соответствую-
щим изменением маг-
нитных свойств, воз-
растанием пластично-
сти и переходом кри-
сталлин. структуры
от типа CaF2, характерного для ионных соединений,
к плотноупакованному типу Си2А1Мн, свойственному
металлич. фазам (рис. 2). Устойчивость М. с.
также определяется химической природой состав-
ляющих элементов и возрастает с различием у них
величины сродства к электрону. Признаками устой-
чивых М. с. являются: высокая температура плавле-
ния, повышенная электронная плотность в определен-
ных участках решетки, большая теплота образования,
узкая область гомогенности на диаграмме состоя-
ния и др.
Классификация. Среди М. с. встречаются как даль-
тониды, так и бертоллиды (см. Двойные системы,
Далътониды и бертоллиды). Состав дальтонида харак-
теризуется сингулярной точкой на изотермах свойств
и обычно отвечает простому стехиометрия, отношению
компонентов, напр. Mg2Sn, NiSb, CuMgAl2. Бертол-
лиды не имеют сингулярных точек на изотермах
свойств. Обычно бертоллиды рассматривают как
твердые р-ры на основе «мнимого» (предполагаемого)
М. с., состав к-рого лежит за пределами гомогенной об-
ласти. Атомы компонентов в бертоллидах распреде-
лены частично или полностью неупорядоченно. Приме-
рами бертоллидов являются М. с. на основе FeSb,
NaPbs, MgsZnsAL и т. п. Ионная связь в бертолли-
дах обычно менее выражена, чем в дальтонидах. При
нагревании дальтонидов сингулярная точка может
смещаться, становиться менее отчетливой и даже ис-
чезнуть, что объясняется изменением валентности
элементов и диссоциацией М. с.; в этом проявляется
единство природы дальтонидов и бертоллидов. Помимо
деления на дальтониды и бертоллиды, М. с. классифи-
цируют: 1) по числу элементов, образующих М. с.,—
на двойные, тройные и т. д.; 2) по характеру преобла-
дающего типа химич. связи — на ионные, ковалент-
ные (типичные для полупроводниковых соединений)
и соединения с преимущественной металлич. связью;
3) по признаку зависимости состава фаз от валентно-
сти — на валентные (в к-рых проявляется нормальная
валентность обоих элементов), никель-арсенидные
фазы (в к-рых состав зависит от валентности менее
металлич. элемента), электронные (в к-рых определяю-
щим фактором является электронная концентрация,
т. е. отношение числа валентных электронов к числу
атомов) и на М. с., состав к-рых не связан с валентно-
стью; 4) по типу кристаллич. структур — наиболее
распространенная классификация, согласно к-рой
различают: а) гомодесмические М. с.,
в к-рых нельзя выделить отдельных атомных комплек-
сов, т. к. величина связи между двумя любыми сосед-
ними атомами в кристаллич. решетке является одина-
ковой (типы CusAu, NdSb, TiNi); б) гетероде с-
м ическиеМ. с., в к-рых атомы определенных групп
связаны между собой более сильно; в зависимости от
характера таких групп различают комплексы остров-
ные, линейные, слоистые, пространственные. В гете-
родесмич. структурах 1-го рода в такие комплексы
связаны атомы одного элемента (напр., цепочки
...—В—В—В—В—... в нек-рых боридах), а в гетеро-
десмич. структурах 2-го рода — атомы различных эле-
ментов (напр., комплексы ZrGe2). В зависимости от
компактности кристаллич. структуры М. с.
разделяют на соединения с плотнейшей упаковкой ато-
мов (СизАи и др., производные от типа Си или Mg),
плотной упаковкой (Cu5Zn8 и др., производные от ти-
пов a-W, CsCl, NaTl), структуры внедрения (типы
NaCl, NiAs) и структуры, не связанные с плотными
упаковками; 5) по условиям образования — на М. с.,
кристаллизующиеся из жидких расплавов, возникаю-
щие при перитектич. реакциях, выделяющиеся при
распаде пересыщенных твердых р-ров и образующиеся
при превращениях твердых р-ров (соединения Курна-
кова).
Характеристика важнейших типов (расположены
в порядке уменьшения доли металлич. связи).
143
МЕТАЛЛИЧЕСКИЕ СОЕДИНЕНИЯ
144
а) Соединения Курнакова (упорядо-
ченные твердые р-ры, сверхструктуры). Образуются
при охлаждении некоторых неограниченных (рису-
Си Ю 20 30 40 50 60 70 80 90 Аи
ат^о Аи
нок 3) или ограничен-
ных твердых раство-
ров определенного со-
става. Возникающие
обычно дальтониды
имеют полностью или
частично упорядочен-
ную структуру, при-
чем б. ч. образуются
новые кристаллич.
типы. Мера упорядо-
ченности (ст), харак-
теризующая степень
дальнегопорядка,м.б.
рассчитана по фор-
муле:
<Т = 1-ТУА/СА= '
Pt 9080 7060504030 20 10 CuFe
am. % Pt
Рис. 3. Образование соединений
Курнакова в системах Аи—Си (а)
и Си—Fe—Pt (6).
=1-жв/сВ;
где Wa hWb — веро-
ятности нахождения
атомов А и В в «чу-
жих» узлах решетки;
Саи Св — концентра-
ции компонентов А и
В. При полной упо-
рядоченности И’д =
= И’в=0; о=1. В го-
могенной области, рас-
положенной ниже тем-
пературы превраще-
ния, на изотермах
свойств обнаружива-
ются сингулярные точки, отвечающие составу соеди-
нения Курнакова. Этому составу соответствуют макси-
мальная скорость упорядочения, наибольшая степень
упорядоченности и экстремальные значения свойств,
напр. наибольшая электропроводность или наимень-
шая твердость.
Для соединений Курнакова типичны структуры
плотнейших упаковок (рис. 4). К числу соединений
Курнакова принадлежат нек-рые т. наз. <Т-фазы
(FeCr, FeV и др.), образованные переходными метал-
лами V—VIII групп. По природе химич. связи сг-фазы
близки к электронным соединениям. Они имеют слож-
ную тетрагональную структуру (5-U и отличаются оди-
наковым отношением числа s- и d-электронов к числу
атомов, равным 6,5—7,4. Свойства соединений Курна-
кова существенно отличаются от свойств твердых
р-ров, из к-рых они образовались. Напр., в случае
СпзАп увеличиваются твердость и химич. стойкость; в
соединениях PtCr3, 1гСг и 1гСг3 появляются ферромаг-
нитные свойства; образование cr-фаз сопровождается
Mg3Cd
• Mg
О Cd
Рис. 4. Кристаллические структуры соединений Курнакова.
резким возрастанием хрупкости. Для изучения в
твердых р-рах междуатомного химич. взаимодействия
наряду с термич. анализом применяются также методы
измерения гальвано-магнитных свойств, эффекта Хол-
ла (возникновения разности потенциалов при взаимо-
действии электрич. и магнитных полей) и остаточного
электросопротивления. Этими методами, напр. в си-
стеме Fe—Со—Ni, были обнаружены тройные метал-
лич. соединения FeeCo2Ni, FeCoNi4 и FeCoNi10.
б) Фазы Левее а. Большая группа М. с.,
относящихся к плотно упакованным структурам
с координационным числом 12 и более, была исследо-
вана Лавесом. Главное значение при образовании фаз
Лавеса имеет объемный фактор. Большей
частью фазы Лавеса имеют состав АВг, реже АВ,
причем соотношение атомных радиусов гА /гв =1,10—
1,37, что близко к теоретически рассчитанному.
Н. В. Белов установил, что к фазам Лавеса относятся
также нек-рые М. с. другого состава, напр. Fe,W6.
В структуре фаз Лавеса образуются трехмерные,
плоские, линейные или островные комплексы, что ха-
рактерно для гетеродесмич. М. с. Для них типичны
три разновидности плотноупакованных структур:
MgCu2, MgNi3 и MgZn2, производных от типов цинко-
вой обманки и вюртцита и имеющих родственное
строение (рис. 5, а, б, в).
Подобное строение имеют нек-рые тройные фазы
Лавеса как частично упорядоченные (CuCdZn,
CuMgAl, TiTaCo), так и полностью упорядоченные
(Cu4MgSn, см. рис. 5, г). Для фаз Лавеса ряда тройных
систем характерна определенная последовательность
MgNi2
• Mg oNl
Рис. 5. Кристаллические структуры плотноупакованных
металлических соединений (фаз Лавеса).
чередования структур в направлении MgCu2=MgNi2
-^MgZn2, чему соответствует возрастание элект-
ронной концентрации от 1,33 до 2,2. В фазах Лавеса
преобладает металлич. связь; однако, несмотря на
сходство кристаллич. структур, характер связи мо-
жет существенно изменяться в зависимости от состава,
что дает основание нек-рым авторам не выделять эти
фазы в самостоятельную группу.
в) Электронные соединения. Это.
фазы «латунного» типа, образованные, с одной сто-
роны, Си, Ag, Аи или переходными металлами, а с
другой — элементами Пб—Уб-подгрупп периодич.
системы. Главным фактором, определяющим их со-
став и строение, является электронная концентрация.
Т. обр., валентность имеет большое значение при фор-
мировании этих М. с. На диаграммах состояния элек-
тронные соединения обычно характеризуются широ-
кими областями гомогенности. Юм-Розери установил
для двойных М. с. 4 типа электронных соединений,
к-рым отвечают соответственно 4 кристаллич. типа:
1) тип (5-латуни (кристаллич. структура типа CsCl)
с электронной концентрацией 3/2 (CuZn и др.); 2) тип
(5-Мп с электронной концентрацией 3/2 (y-Cu5Si и др.);
3) тип у-латуни (сложная кубич. кристаллич. струк-
тура) с электронной концентрацией 2I/]3 (Cu5Zn8 и
др.); 4) тип е-латуни (гексагональная кристаллич.
структура) с электронной концентрацией ’/4 (CuZn, и
др.). Образование электронных М. с. объясняется
зонной теорией, по к-рой каждая фаза в дан-
ной системе устойчива до определенной электронной
концентрации, после чего дальнейшее заполнение
145
МЕТАЛЛИЧЕСКИЕ СОЕДИНЕНИЯ
146
энергетич. уровней становится невозможным и необ-
ходимо возникает другой кристаллич. тип.
Правило Юм-Розери имеет много исключений, про-
являющихся особенно, если взаимодействующие эле-
менты различаются по своей химич. природе ив М. с.
возрастает доля ионной связи. Среди тройных М. с.
фазы Юм-Розери встречаются редко (напр., MgSnNi2
и CuAuZn2 с электронной концентрацией 3/2 и струк-
турой (5-латуни). Однако известна группа тройных
электронных М. с. с более высоким значением элект-
ронной концентрации (ок. 2,4). Сходство тройных
М. с., имеющих одинаковую электронную концентра-
цию, нек-рые авторы объясняют исходя из теории ва-
кантных электронов Полинга, по к-рой переходные
металлы способны быть акцепторами электронов, за-
полняющих недостроенные d-уровни; донорами слу-
жат элементы, легко отдающие свои валентные элект-
роны, напр. Al. Т. обр. объясняется строение М. с.
Al80MnnNi4, CusAleNi, Al9FeNi и др.
г) Никель — арсенидные фазы. Эти
фазы образуются при взаимодействии переходных
металлов с анионообразователями IV6—У1б-под-
групп (Sn, Sb, Те и др.), напр. NiAs, FeSb, CoSn,
TiSe2 и др.
По химич. свойствам они стоят ближе к солеобраз-
ным соединениям, чем рассмотренные выше. Для них
характерна значи-
тельная доля на-
правленного, ко-
валентного вида
связи. Многие из
них кристаллизу-
ются в гексаго-
нальной решетке
типа NiAs с коор-
динационным чис-
лом 6 и на диаг-
раммах состояния
имеют широкие об-
ласти гомогенно-
ONi «Те °Ni »Sb ONi «In
Рис. 6. Кристаллические структуры
гомологических металлических соеди-
нений с переменным числом атомов в
элементарной ячейке.
сти. Е. С. Макаров установил, что они представляют
гомологии, систему фаз с переменным числом атомов
в элементарной ячейке. Примером служит ряд соеди-
нений (рис. 6): NiTe»-^NiSb—Ni2In.
В этом гомологии, ряду, с переходом от NiTe2 к
Ni2ln и увеличением относптельного содержания ни-
келя, последовательно уменьшается доля ионно-ко-
валентной связи и усиливается металлич. связь. Со-
ответственно изменяются строение и свойства; кри-
сталлич. решетка переходит из слоистой в NiTe2
(тип CdJ2, характерный для солеобразных веществ)
к типу у-латуни в Ni2In. Свойства меняются от полу-
проводниковых в NiTe2 к металлическим в Nbln.
Аналогичное изменение свойств происходит и в рядах
соединений одинакового качественного состава; напр.,
при переходе от NiS и Ni3S2 появляются признаки ме-
таллич. свойств.
Примерами тройных никель-арсенидных фаз могут
служить соединения BeSiZr, GeMnFe, GeMnCo,
GeFeCo и т. п.
д) Валентные соединения. Эти соедине-
ния образуются элементами различной химич. при-
роды. Составы их постоянны (т. е. они относятся к
дальтонидам) и подчиняются правилам валентности.
Основные виды связи в них — ионная и ковалент-
ная; с уменьшением различия химич. свойств элемен-
тов усиливается доля металлич. связи. Валентные сое-
динения обладают рядом свойств, характерных для
солеобразных соединений, а именно: 1) большой вели-
чиной теплоты образования; 2) высокой, обычно кон-
груэнтной, темп-рой плавления; 3) пониженной элект-
ропроводностью (часто являются полупроводниками);
4) наличием на диаграммах состояния узких областей
гомогенности (практич. отсутствием твердых р-ров со
своими компонентами).
Однако, в отличие от типичных ионных соединений,
эти соединения характеризуются в нек-рой степени
также металлич. свойствами. При изменении сос-
тава валентных М. с. отчетливо проявляется перио-
дичность свойств. Типичные кристаллич. структуры
валентных М.с. показаны на рис. 7.
Рис. 7. Кристаллические структуры некоторых
валентных М. с.
В тройных валентных М.с. ряда: LiMgN-+LiMgP-»-
-^LiMgAs->-LiMgSb-*LiMgBi — свойства последова-
тельно изменяются от солеобразных к металлическим
в связи с усилением металлич. свойств элементов V гр.
и постепенным переходом от ионной связи к металли-
ческой.
М. с. Mg2Sn со структурой CaF2, несмотря на соот-
ветствие правилам валентности, не может быть отне-
сено к типичным валентным соединениям, т. к. в нем
связь не только ионная (валентные электроны маг-
ния не находятся в полном владении аниона Sn4-).
Это соединение, вместе с близкими ему соединениями
Mg2Si, Mg2Ge, Mg2Pb, Li2Se, CuMgSb и т. п., должно
быть отнесено к отдельному типу, куда следует вклю-
чить также нек-рые невалентные соединения (А12Ац)
и ряд других, содержащих Со, Ni, Ir, Pt, с расширен-
ными областями гомогенности на диаграммах состоя-
ния. Свойства их меняются в широких пределах в за-
висимости от химич. природы составляющих эле-
ментов.
Наряду с М. с., относящимися к типичным рассмот-
ренным группам, есть много других, обладающих про-
межуточными строением и свойствами.
Практическое значение. М. с. часто встречаются
в промышленных сплавах и оказывают сильное влия-
ние на их свойства. В ряде случаев они увеличивают
твердость, жаропрочность и жаростойкость сплавов,
сообщают им антикоррозионные, полупроводниковые,
магнитные и другие полезные свойства. В структурно-
свободном состоянии твердость, прочность и жаро-
прочность увеличивают главным образом карбиды,
нитриды и бориды. В частности, твердые сплавы,
изготовляемые металлокерамич. путем, могут состо-
ять из М.с. WC, TiC, связанных пластичной связкой
Со. Силициды повышают жаростойкость и кислото-
упорность.
Выделяясь при старении пересыщенных твердых
р-ров в дисперсном состоянии, многие М.с. значитель-
но увеличивают прочность сплавов. В этом отношении
для медных сплавов имеют значение М. с. СпВе и
CusSi; для магниевых сплавов — Mgn Ali2, Mg2Ce,
MgsTh; для алюминиевых сплавов — CuAl2, CuMgAl2,
MgZn2, MgsAls, MgsZnsAh; для сталей — металлопо-
добные карбиды, нитриды, бориды и др.; для никеле-
вых сплавов — NisAl и Ni3Ti; для кобальтовых
сплавов — AlCo, TiCo2, VCo3, WCo3. Структурно-
свободные М.с. сильно влияют на процессы электро-
химия. коррозии сплавов и в отдельных случаях
увеличивают коррозионную стойкость (напр., А16Мп
и Mg2Al12Cr в алюминиевых сплавах).
М. с. могут служить самостоятельной основой
жаропрочных сплавов (Ti3Al, NisAl, TieAl). Многие
М. с. применяют в качестве полупроводников и ма-
147
МЕТАЛЛКЕТИЛЫ—МЕТАЛЛООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
148
сериалов с особыми термоэлектрическими свойства-
ми. Особенно широко используются для этой цели
селениды, теллуриды, антимониды и др. соединения
элементов II, III и VI групп (напр., валентные InSb,
MgsSb2 или невалентные CoSbs, SbZn).
Нек-рые М. с. сообщают сплавам магнитные свойст-
ва; напр. магнетизм «гейслеровых» сплавов связан с
образованием тройных М. с. Cu2MnAl, Cu2MnGa,
Cu2MnIn, Cu2MnSn; ферромагнитные свойства обна-
ружены также в химически аналогичных соединениях
Ni2MnGe, СогМпОе и Ni2MnSn, имеющих одинако-
вые кристаллич. структуры.
Лит.: К у рнаков Н. С., Избранные труды, т. 1—2,
М., 1960—61; его же, Введение в физико-химический ана-
лиз, 4 изд., М., 1940; Аносов В. Я., Погодин С. А.,
Основные начала физико-химического анализа, М.—Л., 1947;
Корнилов И. И., Металлохимия и некоторые ее задачи,
Изв. АН СССР. Отд. хиЖ. н., 1957, № 4, 397; Агеев Н. В.,
Химия металлических сплавов, М.—Л., 1941; его же,
Природа химической связи в металлических сплавах, М.—Л.,
1947; Теория фаз в сплавах, пер. с англ., М., 1961; Ю м-Р о-
s е р и В., Рейнор Г. В., Структура металлов и сплавов,
пер. с англ., М., 1959; Ю м-Р озери В., Христиан Дж.,
Пирсон В., Диаграммы равновесия металлических сис-
тем, пер. с англ., М., 1956; The physical chemistry of metallic
solutions and intermetallic compounds. Proceedings of a symposi-
um..., v. 1—2, L., 1959; Макаров E. С.. Строение твердых
фазе переменным числом атомов в элементарной ячейке, М.—
Л., 1947; Корнилов И. И., Вульф Б. К., Металличе-
ские соединения, Успехи химии, 1959, 28, вып. 9; Вульф
Б. К., Тройные металлические соединения, там же, 1960, 29,
вып. 6; Хансен М., А н д е р к о К., Структуры двойных
сплавов, пер. с англ., т. 1—2, М., 1962; В о л А. Е., Строение
и свойства двойных металлических систем, т. 1, М., 1959;
Крипякевич П. И., Черкашин Е. Е., Системати-
ка двойных интерметаллических фаз, Изв. СФХА АН СССР,
1954, 24: Бокий Г. Б., Вульф Б. К., Смирнова
Н. Л., Кристаллические структуры тройных металлических
соединений, Ж. структур, химии, 1961, 2, № 1; Механические
свойства металлических соединений, пер. с англ., М., 1962;
Савицкий Е. М., Влияние температуры на механические
свойства металлов и сплавов, М., 1957. Б. К. Вульф.
МЕТАЛЛКЕТИЛЫ — свободные радикалы, содер-
жащие трехвалентный атом углерода типа:
/С — О—Me
Rz
М. получают действием щелочных металлов на карбо-
нильные соединения, не содержащие активного атома
водорода, способного замещаться металлом:
R. R.
)С=О+Ме-—> /С —О—Me
R/ R/
Реакцию проводят в жидком аммиаке либо в абсолют-
но сухом инертном органич. растворителе без досту-
па воздуха. М. имеют глубокую окраску, причем ли-
тиевые производные окрашены в голубой цвет, а нат-
риевые, калиевые, цезиевые и рубидиевые — в темно-
синий. Образование М. наиболее характерно для аро-
матич. кетонов, хотя нек-рые сильно разветвленные
алифатич. кетоны, типа гексаметилацетона или пина-
колина, также дают М., правда с большим трудом.
Ароматич. кетоны способны образовывать и магние-
вые М.
М.— очень активные соединения, напоминающие
по свойствам триарилметильные радикалы; они нахо-
дятся в равновесии со своими димерами — пинаколя-
тами:
ОМе ОМе.
Аг. . Аг. | | /Аг
2 )с-о-ме^± ;с-------------С/
Arz Аг \
хАг
Положение равновесия зависит от растворителя, темп-
ры, природы металла и карбонильного соединения.
М., полученные из алифатич. кетонов, подобно алкиль-
ным радикалам, быстро подвергаются полной необра-
тимой димеризации. М. мгновенно окисляются возду-
хом или галогенами до кетонов, а при гидролизе обра-
зуют смеси, состоящие из кетона, спирта и пинакона:
Аг. . О2 Аг,
ХС — ОМе------♦ ХС = О
Arz или J2 Аг
Аг, . Н2О Аг.
ХС - ОМе-----—» ХС=О +
Аг Аг
Аг. Аг. .Аг
+ JCHOH+ )С-----------С(
Аг' АГ [ Iх Аг
ОН ОН
С металлами и амальгамами М. дают интенсивно
окрашенные диметаллич. производные кетонов:
Na
Na |
(C„H5)2CONa----► (C»H5)2 C-ONa
NH3
к-рые далее реагируют как обычные металлоорганич.
соединения, напр.
.COONa
Na СО2 (CeHs)2C<
I .----> xONa
(C»H5)2 C-ONa-- RC1
I----г yR
(CSH5). C<
' ONa
При взаимодействии M. с галогеналкилом образуется
смесь кетона и третичного спирта; с трифенилхлорме-
таном получается трифенилметильный радикал:
.R
(CeH5)2 CONa+RBr—>(CsH5)2C = O + (C»H5)2Cz
'ONa
(CeHs)2CONa + (C»H5)3 CCl —> (C„H5), CO + (C8HB)3 C-+NaCl
M. парамагнитны, что является бесспорным физич.
доказательством их свободнорадикального характера.
М. открыты и подробно изучены в 1911 В. Шленком.
Лит.: Реутов О. А., Теоретические проблемы органи-
ческой химии, М., 1956, с. 228; Gilman Н., Organic chemi-
stry. An advanced treatise, v. 1, 2 ed., N. Y., 1944, p. 612;
Hamrick P. J., Hauser C. R., J. Amer. Chem. Soc.,
1959, 81, № 2, 493. H. П. Гамбарян.
МЕТАЛЛООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ - co
единения, содержащие одну или более связей металл —
углерод. Выделение М. с. в отдельную группу веществ
в известной мере условно. В последнее время часто
пользуются термином «элементоорганические соедине-
ния», относя сюда, кроме М. с., и органич. производ-
ные неметаллов — фосфора, бора и др. Образование
связи металл — углерод представляет собой общее яв-
ление; все элементы, за исключением инертных газов,
способны образовывать связь с углеводородными груп-
пами. По характеру связи металла с органич. частью
молекулы М. с. делятся на два типа: 1) большая часть
известных М. с., образованных преим. непереходными
металлами, содержит <т-связи металл — углерод; 2) для
переходных металлов более характерны М. с., образо-
ванные за счет заполнения d-оболочек металла л-элек-
тронами различных ненасыщенных систем — аромати-
ческих, циклопентадиенильных, диеновых ит. п. Свой-
ства этих двух групп М. с. различны. Неодновалент-
ные металлы могут образовывать как полные М. с.—
MeR„, содержащие «связей металл — углерод, так и
смешанные R„MeXm, где X—галоген, алкоксил, ки-
слотный остаток, водород и т. п., a n-f-m — валент-
ность металла.
Открытие М. с. обычно связывается с именами Р. Бунзена
(1841) и Э. Франкленда (1849). М. с. сыграли важную роль в
установлении основных представлений химии — валентности,
разграничения понятий «эквивалент» и «атомный вес»; вслед-
ствие значительной летучести многих М. с. с их помощью
впервые были определены истинные атомные веса ряда метал-
лов. Многие М. с. обладают высокой реакционной способно-
стью и приобрели большое значение в синтезе. А. М. Бутлеро-
вым и его учениками синтезы с помощью цинкорганич. соеди-
нений были широко использованы для обоснования выводов
теории химич.строения.
Металлоорганические соединения непереходных ме-
таллов. Наиболее полно изучены производные Li,
149
МЕТАЛЛООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
150
Na, К, Rb, Cs, Be, Mg, Ca, Sr, Ba, Zn, Cd, Hg, B,
Al, Ga, In, Tl, Si, Ge, Sn, Pb, As, Sb, Bi. Эти металлы
образуют наиболее характерные М. с., тогда как М.с.
переходных металлов весьма специфичны как по свой-
ствам, так и по способам получения. Характер связи
металл — углерод в М. с. существенно зависит от при-
роды металла, в частности от его электроотрица-
тельности. Полярность связей М. с. изменяется в весь-
ма широких пределах. Так, алкильные производные
щелочных металлов (MeR), электроотрицательность
к-рых низка, содержат весьма полярные связи Me—С.
Это обстоятельство определяет высокую реакционную
способность М. с. щелочных металлов в гетеролитич.
реакциях. С увеличением электроотрицательности
металла возрастает гомеополярный характер связи
Me—С, и М. с. таких металлов, как Hg, Sn, Pb и др., яв-
ляются, по существу, гомеополярными соединениями.
Соответственно этому, возрастает склонность М. с. к
гомолитич. распаду с образованием свободных радика-
лов. Если термич. распад алифатич. М. с. лития, маг-
ния, алюминия приводит, как правило, к отщеплению
олефина с образованием гидридов соответствующих
металлов, напр. C2H3Li ^C»H4^-LiH, то М. с. свинца,
ртути и других тяжелых металлов при нагревании
легко отщепляют свободные радикалы, выделяя ме-
талл, напр.: (С2Н5)4РЬ-*РЬ+4С2Н5. Помимо приро-
ды металла, устойчивость М. с. зависит также и от
способности органич. радикала к рассредоточению из-
быточной электронной плотности. Ароматич. М. с.
обычно устойчивее алифатич.; это относится и к цик-
лопентадиенильным производным металлов, если пос-
ледние не имеют сэндвичевой структуры типа ферро-
цена.
Способы получения М. с. во многом специфичны для
каждой группы; универсальных методов, пригодных
для синтеза М. с. всех металлов, не существует. Од-
нако нек-рые синтетич. методы являются ключевыми;
они пригодны не только для получения большого чи-
сла М. с., но и позволяют переходить от одних М. с.
к другим. Эти методы могут быть разделены на 2 груп-
пы; к первой относят образование связи металл — уг-
лерод, если в исходных соединениях ее нет; ко второй—
получение одних М. с. с помощью других.
Важнейшими методами первой группы являются: 1) Взаи-
модействие металлов с галогеналкилами (арилами). За исклю-
чением щелочных металлов, все металлы, способные к этой
реакпии (Са, Be, Mg, Zn, Hg, Al и нек-рые др.), образуют при
этом смешанные М. с., напр.
RHal+Mg—>RMgHal; 3RHal+2Al—^RAlHal24-R2AlHal
Этот метод имеет большое значение для синтеза симметрич-
ных (полных) М. с., так как образующиеся смешанные М. с.
различными путями могут быть превращены в полные. Особен-
но широко этот метод применяется для получения литий- и
магнийорганических соединений, с помощью к-рых получают-
ся другие М.с. (см. также Гриньяра реакция, Цинкорганические
соединения и др.). Взаимодействие галогеиалкилов со сплавами
часто применяют для получения симметричных органич. про-
изводных более электроотрицательного металла из числа вхо-
дящих в сплав. Особенно часто для этой цели используют спла-
вы ртути, олова или свинца с натрием или магнием; последняя
реакция лежит в основе промышленных методов получения
алкильных производных Hg, Sn и Pb. 2) Реакция между гало-
генидами металлов и галогеналкилами в присутствии натрия
типа. напр. SbCl.< 4- 3RC1 4-6Na—>6NaCl + SbR3. Метод при-
меняется для синтеза М. с. кремния, германия, олова, мышья-
ка, сурьмы. 3) Диазометод, состоящий в действии порошков
металлов на двойные диазониевые соли типа, напр. ArN2Cl-
•HgCl2; реакция проводится в среде ацетона или этилацетата и
протекает с выделением азота (см. Несмеянова реакция)'.
ArN2Cl HgCl2 + Си —> ArHgCl + N3 + СиС12
Метод широко применяют для синтеза ароматич. М. с. Hg,
Pb. Sn, As, Sb, Bi, в том числе и таких M. с., к-рые не могут
быть получены др. способами. Кроме солей диазония, в реакци-
ях этого типа могут принимать участие и аналогичные соеди-
нения иодония и боргидриды диазония. 4) Присоединение гид-
ридов элементов к кратным связям — препаративный метод
синтеза М. с. бора, алюминия, кремния и др. Напр., присоеди-
нение диборана к олефинам служит удобным способом полу-
чения бортриалкилов: B2H64-6RRC=CH2 -> 2B(CH2CHRR)3.
5) Взаимодействие свободных радикалов с металлами также
приводит к образованию соответствующих М. с. Сюда примы-
кают также реакции солей металлов с диазометаном, дихлор-
карбеном и его аналогами, напр. HgCl2 + CH2N2-*~ClCH2HgCl.
6) Металлирование солями металлов: RH4- МеХ -> RMe+
4-НХ; этот метод играет важную роль в химии ртутьорганиче-
ских соединений. Реакции металлирования такого типа науче-
ны для солей Си, Ag, Аи, Tl, Si, Ge, Pb.Необходимым услови-
ем металлирования солями металлов является наличие в моле-
куле RH достаточно подвижного атома водорода. Важным син-
тетич. методом является присоединение солей ртути и сурьмы
по кратным связям, напр.:
HC=CH4-HgCl2—► ClCH=CH2HgCl
полученные таким путем ртутьорганич. соединения могут быть
широко использованы для получения Д-хлорвинильных про-
изводных других металлов.
Важнейшими методами синтеза одних М. с. с помощью
других являются след.: 1) Взаимодействие М. с. с солями (чаще
всего галогенными), алкоксильными производными и в ряде
случаев с гидридами металлов, приводящее к обмену общего
вида: RMe + М'Х^* RM'4-Me. В качестве исходных М. с.
особенно широко применяют магнийорганич. соединения, а
также М. с. лития, цинка, алюминия; в качестве солей MX —
галогениды металлов. Как правило, при реакциях такого типа
образуются М. с. менее реакционноспособного элемента.
Алкилирующая способность литийорганич. соединений обычно
выше, чем у реактивов Гриньяра; относительно мало реакцион-
носпособные М. с.: Pb, Sn, Hg чаще применяют для получения
смешанных М. с., напр. R2Hg+ AsCl3 -> R2AsC1+ HgCl2.
2) Индивидуальные M. с. щелочных металлов, М. с. магния,
бериллия, алюминия и др., отличающиеся высокой реакционной
способностью, получают нагреванием ртутноорганич. соедине-
ний с соответствующими металлами, напр. R2Hg4- 2Li ->
2RLi 4- Hg. В отличие от первого метода, реакции такого
типа обычно служат для получения более реакционноспо-
собных М. с. 3) Реакция перераспределения радикалов между
М. с. различных металлов: nMeRm 4- uiMe'Rn^± nMeRw+
4- mMe'Rn; равновесие может быть смещено в нужную сторону
удалением одного из компонентов смеси. Метод успешно приме-
няется, напр., для получения виниллития:
(СН2 = СН)1 Sn4-CeH5 Li —(CeH5)t Sn + 4CH = CHLi
Свойства М. с. изменяются в широких пределах,
поскольку в различных М. с. существенно изменяется
характер связи металл — углерод. Многие М. с. крайне
чувствительны к кислороду [М. с. щелочных и щелоч-
ноземельных металлов, Be, Mg, Zn, Cd, В, Al, Ga,
In, P(III), As, Sb, Bi] и к воде (M. с. щелочных и ще-
лочноземельных металлов, Be, Mg, Zn, Cd, Al, Ga,
In). Низшие алкильные производные Li, Na, К, Rb,
Cs, Ca, Sr, Ba, Be, Mg, Zn, B, Al, Ga, Sb,Bi самовос-
пламеняются на воздухе, а нек-рые из них, напр.
соединения Li, Mg и Be,— также и в атмосфере углеки-
слого газа. Напротив, полные (симметричные) М. с.
таких металлов, как Pb, Hg, Sn, устойчивы к действию
кислорода и воды и могут быть перегнаны с водяным
паром. Как правило, низшие алкильные производные
металлов являются перегоняющимися жидкостями (за
исключением твердых соединений щелочных и щелоч-
ноземельных металлов и магния), в то время как аро-
матич. М.с. — обычно твердые вещества. Большинство
полных М. с. (за исключением полимерных) раствори-
мо в индиферентных органич. растворителях и имеет
заметное давление паров; большинство М. с. обла-
дает высокой токсичностью; особенно это относится
к летучим соединениям ртути, свинца, олова и др.
Основными типами химич. превращений М. с., кроме упо-
мянутых реакций их синтеза, являются: 1) Диспропорциони-
рование смешанных М. с., приводящее к образованию более
симметричных М. с. и галогенидов, напр.:
3(CH3)2A1J->2(CH3)3A1 4- A1J3
Реакция обычно обратима: равновесие сдвигается при
удалении или связывании (химически) одного из компонентов
равновесной смеси, напр.:
2RHgX + 4KJ->R2Hg + 2КХ + K2HgJ4
Наиболее характерно диспропорционирование (симметри-
зация) для М. с. элементов II и III групп. К этой группе
реакций примыкает также симметризация под действием раз-
личных восстанавливающих агентов, напр. металлич. натрия,
имеющая промышленное значение в химии М. с. алюминия
RAlX2 + R,AlX + 3Na —> R3Al+Al + 3NaCl
2) Реакции, обратные диспропорционированию, общего вида
MROT 4- MXW -> MRnXp (n 4- p — m) охватывают большое
151
МЕТАЛЛООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ—МЕТАЛЛОПРОТЕИДЫ
152
число М. с. и имеют важное синтетич. значение в химии М. с.
В, Al, Hg, Sn. Эти реакции, как правило, экзотермичны, одна-
ко для их завершения часто требуется последующее нагрева-
ние. 3) Различные реакции М. с. с органич. веществами, связан-
ные с разрывом имеющихся связей металл—углерод. К ним от-
носятся многочисленные синтезы с помощью литий- и маг-
нийорганич. соединений, а также радикальные процессы,
инициированные свободными радикалами, образующимися
при термин, распаде М. с. 4) Окисление М. с. непереходных
металлов I—III групп (кроме ртути и таллия) обычно проте-
кает с внедрением атома кислорода по связи Me—С (по пере-
кисному механизму), напр.: R3B (RO)3B; М. с. элемен-
тов IV группы и ртути к окислению устойчивы, тогда как М. с.
3-валентных элементов V группы присоединяют атом кислорода
с увеличением валентности: В3М -> R3MO. 5) Взаимодействие
с углекислым газом, приводящее к образованию солей карбоно-
вых к-т, характерно для М. с. щелочных и щелочноземельных
металлов, Be, Mg и Al, напр.: RMgX + СО2-> RCOOMgX.
8) Дезалкилирование под действием различных неорганич.
реагентов составляет важную группу реакций М. с.; легкость
этих реакций сильно зависит от прочности соответствующих
связей Me—С. Общим свойством большинства М. с. является
способность к ступенчатому дезалкилированию под действием
галогенов, кислот и, в ряде случаев, воды: RnM +2пХ3->
-> ;R„_mMXwJ -+ МХ„ + nRX, RnM + nHX -> nRH + MX,.
Наиболее устойчивы к действию галогенов и галогеноводородов
связи Me—С в М. с. элементов V группы; соединения типа MR3
проявляют при этом своеобразные металлич. свойства, напр.:
RsSb + X2 —> R3SbX2; R3Sb + 2HCl —* R3SbCl3 + Ha
Гомолитич. дезалкилирование многих М. с. вызывают также
соли многих переходных металлов (FeCl3, СоС1а и др.). Гидри-
рование М. с., часто катализируемое свободными металлами,
также приводит к разрыву связей Me—С; зти реакции особенно
важны в химии М. с. бора и алюминия, напр.-. 2R3B + 6H2-»
-» 6RH + В2Н8. 7) Высокая реакционная способность многих
М. с. проявляется также в легкости реакций комплексооб-
разования. М. с. бериллия, магния и металлов группы алюми-
ния с эфиром образуют весьма устойчивые зфираты и анало-
гичные комплексы с аминами; борорганич. соединения типа
R3B являются в этом отношении переходными; давая комплекс-
ные соединения с аминами, они не образуют эфиратов. Цент-
ральный атом в М. с. таких металлов, как Al, Zn, вообще об-
ладает значительной тенденцией к использованию возможных
гибридизированных орбит; в случае алюминийорганич. соеди-
нений (R3A1) это качество проявляется, в частности, в образо-
вании димерных молекул мостиковой структуры за счет пере-
крывания зр’-орбит алюминия и углерода. По этой же причине
цинк-, бор- и алюминийорганич. соединения склонны к образо-
ванию комплексов типа «обратных ониев» — солеподобных
NaAIR,, NaZnR3, а также NaB(C8Hs)4, K(AlR2Hal2) и др. На-
против, М. с. элементов V группы дают «ониевые» комплексы,
напр. соли замещенных стибониев типа (R4Sb)J, однако и
сурьма способна к образованию комплексных соединений, ана-
логичных обратным ониям, наир. L1 [Sb(C8H8)e]. Известны так-
же соединения, образованные обоими типами органич. ионов.
(С8Н5)5В1 + (СвН5)3В —► [(С8Нв)4В1]+[(СвН5)4В]-
Реакции многих М. с., особенно лития и магния,
с различными органич. соединениями широко исполь-
зуются в органич. синтезе (получение углеводородов,
спиртов, карбонильных соединений и др.).
Металлоорганические соединения переходных ме-
таллов. Эти соединения, содержащие простые о-связи
металл — углерод, изучены относительно мало; для
этих металлов характерно образование М. с., имею-
щих структуру л-комплексов. Сюда относят ацети-
лениды переходных металлов, в том числе и такие,
как K4Fe(CsCH)6, циклопентадиенильные сэндви-
чевые соединения типа ферроцена и родственные пос-
ледним бензольные производные (тип дибензолхрома).
По характеру связи углерод — металл к ним же при-
мыкают и карбонилы металлов. Общей чертой всех
этих М. с. является образование связей за счет л-элек-
тронов органич. молекулы и незаполненных d-оболо-
чек металлов. Нек-рые циклопентадиенильные про-
изводные обладают ароматич. характером. Известны
л-циклопентадиенильные соединения Ti, Zr, Y, Nb,
Ta, Cr, Mo, U, Re, Fe, Ru, Os, Co, Rh, Ir, Ni (нек-рые
из них образованы катионами металлов) и различные
карбонилы Ti, Hf, Th, Сг, Mo, W, Mn, Re, Fe, Ru,
Os, Co, Rh, Ir, Ni. Характерно, что в обоих типах
соединений окись углерода и циклопентадпен способны
вытеснять друг друга либо полностью, либо частично,
образуя смешанные соединения, напр. CsH5Mn (СО)3.
Окись углерода в карбонилах металлов может быть
вытеснена другими молекулами, также способными
представить этому металлу необходимые для образова-
ния связи электроны (окись азота, амины, нитрилы,
фосфины, ацетилены, диены и т. п.). М. с. переход-
ных металлов, Ag, Си и Au, содержащие сг-связи
металл — углерод, в большинстве случаев неустой-
чивы и склонны к гомолитич. распаду и отщепляют
свободные радикалы уже при невысоких темп-рах.
Это относится также к алкильным производным;
арильные и а-алкенильные производные более устой-
чивы, тогда как ацетилениды и ионогеннопостроенные
циклопентадиенильные соединения переходных ме-
таллов (V, Sc редкие земли) обладают значительной
устойчивостью. Необычайно устойчивы также ме-
тильные производные платины (СНз)зРи, (CHaJtPt.
Обычным способом получения алкильных и ариль-
ных М. с. переходных металлов является действие на
их галогениды различных М. с. по реакции обмена
радикал — галоген, наир.:
(СаН,)4РЬ+Т1С14 —> (C2Hs)3PbCl + C2H5TiCl3
М. с. имеют важное практич. значение. Ртутьор-
ганич. соединения применяют в качестве антисеп-
тиков, бактерицидных и лекарственных препаратов;
оловоорганич.— в качестве антиоксидантов и стаби-
лизаторов полихлорвинила и каучуков, инсектофун-
гицидов и гербицидов, противоглистных препаратов.
Весьма актуальным является получение чистых ме-
таллов для полупроводников, сплавов и металлопок-
рытий с помощью М. с. и карбонилов металлов. М. с.,
обладающие высокой реакционной способностью, на-
ходят обширные применения в синтезе различных
органич. соединений, высокоэффективных катализато-
ров полимеризации (напр., для получения полиэти-
лена, полипропилена, полиизопрена и др.).
Лит.: Синтетические методы в области металлоорганиче-
ских соединений, под общей редакцией А. Н. Несмеянова и
К. А. Кочешкова, вып. 1—5, 7—8, М.—Л., 1945—50; Бело-
зерский Н. А., Карбонилы металлов, М., 1958; Несме-
янов А. Н., Перевалова Э. Г., Усп. химии, 1958,
27, вып. 1, 3; Coates G. Е., Organo-metallic compounds,
L.— N.У.,1956; Рохов Ю., Херд Д., Льюи с Р., Хи-
мия металлоорганических соединений, пер. с англ.. М., 1983;
Cotton F. A., Chem. Revs, 1955, 55, № 3. Л.И.Захаркин.
МЕТАЛЛОПРОТЕИДЫ — белковые вещества,
необходимой составной частью к-рых являются ионы
металлов. Связь металла с белковой частью молекул
М., как правило, весьма прочная, может осуще-
ствляться различными способами. Во многих М. ион
металла непосредственно связан с белком. Молекула
карбоксипептидазы А — фермента, отщепляющего
аминокислоты от карбоксильного конца полипептид-
ной цепи, содержит прочно связанный атом цинка.
Отщепление цинка под действием сильных комплек-
сообразователей приводит к инактивации фермента,
к-рую можно обратить добавлением ионов цинка.
При замещении цинка др. ионами, напр. кобальтом,
изменяется активность и специфичность фермента.
По-видимому, к М. относятся и другие пептидазы,
проявляющие активность в присутствии ионов цинка,
марганца, магния, кобальта. Типичным М., содер-
жащим прочно связанный цинк, является карбоан-
гидраза, катализирующая разложение угольной к-ты
на СО2 и воду (выделена из эритроцитов крови).
В плазме крови найден синий М.церулоплаз-
мин (мол. в. 150 000), в молекулу к-рого входят
8 атомов меди, поддающиеся обратимому отщепле-
нию. Церулоплазмин катализирует окисление кисло-
родом аскорбиновой к-ты, ароматич. диаминов и ди-
фенолов; в этом отношении близок многочисленным
медьсодержащим окислительным ферментам, к-рыми
особенно богаты растения (аскорбатоксидаза, дифенол-
оксидаза и др.). В плазме крови содержится М.
трансферрин (мол. в. 90 000), к-рый переносит
железо от желудочно-кишечного тракта к месту обра-
153
МЕТАЛЛОТЕРМИЯ
154
зования эритроцитов. В его молекулу входит два иона
Ге’ + , непосредственно связанные с белком.
Обширный класс составляют М., в к-рых металл
является составной частью комплексной простетич.
группы, связанной с белком. К этому классу относятся
многочисленные гемопротеиды. В состав про-
стетич. группы этих М. входит атом железа, комплекс-
но связанный с порфирином. Чрезвычайно широко
распространенными гемопротеидами являются цито-
хромы, основная биологич. функция к-рых состоит
в переносе электронов и (или) водорода посредством
обратимого изменения валентности иона железа в
простетич. группе — геме. К гемопротеидам относятся
способные переносить молекулярный кислород дыха-
тельные белки — гемоглобины крови, миоглобины
мышц, внеклеточные гемоглобины беспозвоночных —
эритрокруорины, отличающиеся большим молеку-
лярным весом, достигающим нескольких миллионов.
Гемопротеидами являются и нек-рые ферменты, напр.
каталаза и пероксидаза. К М. относится фермент
ксантиноксидаза, являющаяся металлофлавопротеи-
дом; в ее состав входит молибден. М. широко распро-
странены в растительных и животных организмах и
выполняют ряд ответственных функций в процессах
жизнедеятельности.
Лит.: Белки, пер. с англ., под ред. Г. Нейрата и К. Бейли,
т. 3, ч. 1—2, М., 1958—59. В. М. Степанов.
МЕТАЛЛОТЕРМИЯ — восстановление металлов
из их соединений другими металлами, химически бо-
лее активными, при повышенных темп-pax. Впервые
металлотермия, реакции изучил и подробно описал
Н. Н. Бекетов (1865). Основные закономерности М.
определяются соотношениями изобарных по-
тенциалов образования соответствую-
щих соединений (окислов, хлоридов, фторидов и др.)
восстанавливаемого металла и металла-восстанови-
теля.
На рисунке показана температурная зависимость
изобарных потенциалов образования AZ окислов
Температура. ’С
Зависимость изобарных потен-
циалов образования окислов от
темп-ры (AZ в ккал/моль Оа):
1 _ 4Cu + О2=2Си2О;
2 — 2Ni+O2=2NiO;
3 — 2Co-(-Oa=2CoO;
4 — 2Fe+O2=2FeO;
5 — ‘/3Cr-|-O2=2/3Cr2Oa;
« — 2Mn-|-O, = 2MnO;
7 — Si + O2=SiO2;
8 — Tl + O2=TiO2;
» — </3А1-|-02=2>зА120з;
10 — 2Mg+O2=2MgO;
11 — 2Ca-|-O2=2CaO;
12 -- 2C + O2 = 2CO.
нек-рых металлов и оки-
си углерода, рассчитан-
ных на 1 моль О 2 (при
парциальном его давле-
нии 1 атм). Все кривые
приближаются к прямым
линиям в тех пределах,
где нет фазовых превра-
щений металла или окис-
ла. В точках плавления
и кипения эти прямые
претерпевают изломы.
Изобарный потенциал
металлотермич. реакции
Мв! + Ме2О -» МехО +
-)-Ме2 равен разности
изобарных потенциалов
реакций Me, + ’/2 О2
-*МехО и Ме2 + ’/2 О2-*
-*Ме2О, т. е. разности
AZ этих окислов. Чем
больше по абсолютной
величине AZ окисла,
т. е. чем ниже на диа-
грамме проходит прямая
для данного металла,
тем более устойчивым
является данный окисел
и тем сильнее восстано-
вительная способность
восстанавливает из окислов
металла. Напр., кремний
все вышележащие металлы, алюминий восстанавли-
вает кремний из SiO2, а кальций является самым
активным из восстановителей.
Диаграмма делится на две части линией образова-
ния окиси углерода, 2С + О2 -> 2СО. Выше этой
линии находится область карботермии(т. е.
область, в к-рой соответствующие металлы по термо-
динамич. параметрам процесса могут восстанавли-
ваться углеродом), а ниже — область М., в к-рой
углерод не может быть использован как восстанови-
тель и где располагаются Кривые AZ для большин-
ства легких и редких металлов. Чем больше сродство
металла к Кислороду (измеряемое величиной AZ),
тем более смещается вправо точка пересечения прямой
AZ этого металла с кривой AZ реакции 2С + О2
-» 2СО и тем, следовательно, выше темп-ра восстанов-
ления этого металла из окисла углеродом. Кроме
того, металлотермическое восстановление металлов,
склонных к образованию карбидов — Mn, Сг, V,
Nb, Та, Zr, Ti, Mo, W, позволяет получать эти ме-
таллы или их сплавы с железом (ферросплавы), сво-
бодными от углерода. Поэтому М. имеет большое зна-
чение для получения безуглеродистых легирующих
присадок, используемых в производстве прецизион-
ных сплавов с весьма малым содержанием углерода.
В зависимости от теплового баланса восстановле-
ния различают два вида металлотермич. процессов —
внепечные и печные. Последние разде-
ляются на процессы, протекающие при обычном дав-
лении, и процессы вакуумной М. При внепечной М.
уд. теплового эффекта реакции (на единицу веса
шихты) достаточно для ее самопроизвольного проте-
кания. После разогрева одного участка шихты (внеш-
ним нагревом в начальный период или воспламенением
зажигательной смеси, заложенной в верхний слой
шихты в реакторе) реакция быстро распространяется
по всей ее массе, причем расплавление и перегрев
образующихся продуктов не требуют внешнего подо-
грева реактора. При внепечных алюминотермия, про-
цессах развиваются темп-ры до 3000°.
Нек-рые металлотермич. процессы, отличающиеся
высоким уд. тепловым эффектом, протекают настолько
бурно, что интенсивное выделение газов может вызвать
выброс части шихты из реактора или даже его разру-
шение. В этих случаях интенсивность процесса необ-
ходимо уменьшать введением в шихту флюсующих
добавок или применением комплексных восстанови-
телей, обладающих пониженной активностью (напр.,
сплавов Al—Si вместо чистого А1). Наоборот, если
нужно несколько повысить уд. тепловой эффект реак-
ции, в шихту вводят вспомогательные реагенты (по-
догревающие добавки), к-рые реагируют с восстанови-
телем с большим выделением тепла. Напр., при алю-
минотермии. процессах с этой целью в шихту вводят
CaSOi, КСЮз и др. реагенты с большим содержанием
кислорода. К внепечным относится большое число
алюминотермия, процессов, напр. восстановление
алюминием нз окислов железа (при термитной сварке),
марганца, хрома, получение большинства ферроспла-
вов (см. Алюминотермия, Железа, сплавы), а также ряд
процессов восстановления редких металлов (U, Та,
Nb) из их галогенидов при помощи Са и Na.
Печные металлотермич. процессы характеризуются
тем, что уд. теплового эффекта реакции недостаточно
для ее самопроизвольного протекания, что вызывает
необходимость внешнего подогрева массы реагентов.
Большей частью для этого применяют электрич.
печи. Напр., при силикотермич. производстве ферро-
ванадия и нек-рых сортов ферромарганца шихту на-
гревают в дуговых электропечах. Границы между об-
ластями применения внепечной и печной М. не всегда
могут быть четко проведены, т. к. тепловой баланс
металлотермич. процесса изменяется в зависимости
от масштаба плавки (определяющего соотношение
между уд. тепловым эффектом и потерями тепла во
внешнюю среду), от состава восстановителя,
155
МЕТАЛЛОТЕРМИЯ
156
применения упомянутых выше добавок и характера
получаемого продукта (чистого металла или сплава).
В зависимости от рода применяемого восстанови-
теля различают алюминотермию, си л и-
котермию (кремний обычно применяют в виде
ферросилиция), магниетермию, кальцие-
термию и т. п. Выбор восстановителя опреде-
ляется не только термодинамич., но и технологии, и
экономии, условиями. Если нужно получить чистый
металл, то необходимо, чтобы восстановитель не обра-
зовывал с ним сплавов и соединений, а избыток вос-
становителя и побочный продукт (шлак) легко отде-
лялись от восстанавливаемого металла (механич.
путем, ошлаковыванием, отмывкой, отгонкой и т. п.).
Необходимо также, чтобы стоимость полученного
металла оправдывала расходы на восстановитель. При
восстановлении химически активных металлов реак-
торы заполняют инертными газами. Наиболее актив-
ным восстановителем окислов и галогенных соедине-
ний является кальций. Восстановление окислов гид-
ридом кальция также представляет собой кальциетер-
мич. процесс, т. к. выделяющийся водород лишь в
малой степени служит восстановителем. Кальциетер-
мич. восстановление химически прочных окислов —
TiO2, ZrO2, ThO2—наиболее полно протекает при
1000—1100°. При этих темп-pax давление паров Са
высокое; чтобы исключить испарение кальция, вос-
становление проводят в герметически закрытых сталь-
ных реакторах, заполненных аргоном. Кальцием вос-
станавливают La, Се, Pr, Nd из их хлоридов, Th, V,
У, Sc, Gd, Tb, Dy, Ho, Er, Tu, Lu—из фторидов. Маг-
нием восстанавливают из окислов U, V, В, из хлори-
дов — Ti, Zr, Th, Та, из фторида — U. При восста-
новлении тугоплавких металлов (Та, Nb, Zr) соот-
ветственно из фтортанталата, фторниобата и фтор-
цирконата калия в качестве восстановителя приме-
няют натрий, т. к. образующийся при реакции NaF
растворим в воде, что облегчает его отделение от по-
рошков Та, Nb и Zr, в то время как MgF2 и CaF2 в
воде нерастворимы.
Иногда при восстановлении тугоплавких металлов
с целью получения компактного слитка в шихту вво-
дят специальные добавки, образующие с восстанавли-
ваемым металлом сплавы с пониженной темп-рой плав-
ления. Напр., при восстановлении ThFi кальцием в
шихту добавляют ZnCl2 для образования сравнительно
легкоплавкого сплава Th—Zn. Из полученного сплава
Zn отгоняют в вакууме при 1100°. При восстановлении
YFs кальцием в шихту вводят Mg для образования
иттрий-магниевого сплава и затем в вакууме отгоняют
из него магний.
Выбор соединения, из к-рого восстанавливают ме-
талл, имеет большое значение в М. Так, нек-рые
окислы металлов можно восстановить алюминием
только до низших окислов (напр., TiO2 до TiO), обла-
дающих большой химич. прочностью. Значительно
легче получать металлотермич. путем сплавы, по-
скольку их образование сопровождается уменьшением
изобарного потенциала системы. Так получают, напр.,
ферротитан и феррованадий (при присадке железной
руды), сплавы Ni—Ti, Си—Ti, Zr—Сг и др. При вос-
становлении хлоридов (напр., TiCL, ZrCU, ТаС15,
SiCh) образуются сравнительно легкоплавкие шлаки,
основная масса к-рых легко отделяется от корольков
или губки восстановленного металла в самом реакторе.
В нек-рых случаях, как, напр., в производстве титана
и циркония, остатки исходных солей и восстанови-
теля, относительно летучих, удаляют из восстановлен-
ной губки отгонкой в вакууме (см. Титан и Цирко-
ний). Темп-ры металлотермич. плавки при восстанов-
лении хлоридов достигают 1100°. При восстановлении
более тугоплавких фторидов развиваются темп-ры до
1400—1500°. Слиток восстановленного металла от-
деляют от фторидного шлака после охлаждения ме-
ханич. путем и промывкой водой. Фториды как исход-
ные материалы для восстановления более дороги, чем
хлориды, но менее гигроскопичны.
Металлотермич. процессы применяют также при
рафинировании металлов и в производстве сплавов.
Напр., из чернового олова удаляют примесь свинца
по реакции:
Pb + SnCIa —► Sn + PbClj
Алюминиевые сплавы очищают от примеси магния об-
работкой криолитом:
3Mg + 2Na3AlFe —► 6NaF + 3MgF2 + 2А1
Кальциевый баббит, представляющий собой сплав РЬ
с Са и Na, а также свинцово-кальциевые сплавы
получают обработкой сплава РЬ—Na расплавленным
СаС12:
2Na (РЬ) + СаС12 —> Са (Pb) + 2NaCI
Поскольку изобарные потенциалы образования хло-
ридов NaCl и СаС12 близки, эта реакция энергично
протекает только в присутствии свинца как раствори-
теля кальция.
Вакуумная металлотермия приме-
няется для восстановления относительно летучих
металлов из химически прочных соединений. Созда-
ние в системе вакуума сдвигает реакцию в сторону
образования летучего продукта, давление пара к-рого
в данных условиях выше остаточного давления, соз-
даваемого в аппарате. Напр., равновесие реакции:
Si + 2MgO Ц 2Mg + SiO2 при атмосферном давле-
нии и умеренно высоких темп-pax (ниже 1200°) сдви-
нуто влево в соответствии со значениями AZ MgO и
SiO2, показанными на рисунке. Между тем, в вакууме
порядка 10-1—10_2.и.»1 рт. ст. при 1100—1200° рав-
новесие этой реакции сдвинуто вправо, т. е. в сторону
восстановления магния кремнием, т. к. выделяющиеся
пары Mg уходят из сферы реакции. При восстанов-
лении летучих металлов создание в системе вакуума
обычно позволяет снизить рабочую темп-ру металло-
термич. процесса на 300—400°. Кроме того, создание
вакуума в значительной степени уменьшает опасность
взаимодействия паров металлов с кислородом и азо-
том. В вакуумной М. применяют гл. обр. нелетучие
восстановители — Al, Si, сплавы А1—Si, СаС2. Крем-
ний применяют обычно в виде 75%-го ферросилиция.
Известны также комплексные восстановители, напр.
ферросилиций — карбид кальция.
Характер конденсации паров восстановленных ме-
таллов определяется давлением их паров в тройной
точке и остаточным давлением в системе. Пары Mgr
Са, Sr и редкоземельных металлов обычно конденси-
руют в кристаллы, а пары Ва и щелочных металлов —
в жидкость.
В отличие от внепечных и обычных печных метал-
лотермич. процессов, протекающих с образованием'
жидких шлаков, большинство восстановительных про-
цессов в вакуумной М. сопровождается образованием
твердых силикатных или алюминатных шлаков (или
извести), напр., по реакциям:
2CaO + 2MgO + Si —► 2Mg +2СаО-SIO2
6CaO + 2AI —> ЗСа +ЗСаО-А12О3
MgO + CaC2 —> Mg + CaO +2C
4BaO + 2Al —> 3Ba +BaO-AI2O3
2Li3O-AI2O3+SI —> 4LI+Al2O3 SiO2
Шихту для вакуумных металлотермич. процессов-
обв1чно загружают в печи или реторты в виде брикетов
из смеси порошков исходных окислов и восстановите-
лей. Вакуумным металлотермич. восстановлением
при соответствующем подборе исходных соединений
и восстановителей также можно непосредственно»
получать сплавы, напр. Са—Mg, Li—Mg и др.
157
МЕТАЛЛУРГИЯ—МЕТАЛЛЫ
158
Лит.: Векетов Н. Н., Исследования над явлениями
вытеснения одних элементов другими, Харьков, 1885; Елю-
тин В. П. [и др ], Производство ферросплавов, 2 изд., М.,
1957; М е е р с он Г, А , 3 е л и к м а н А Н., Металлургия
редких металлов, М., 1955, Основы металлургии, под ред.
Н. С. Грейвера [и др.), т, 3, М.„ 1962; М у р а ч. Н Н., Ва-
рятин У. Д., Внепечная металлотермия, М., 1956, Зе-
ликман А. Н., Металлургия редкоземельных металлов
тория и урана, М., 1961; П а з у х и н В. А., Фиш ер А Я,
Вакуум в металлургии, М., 1956; Крестовников А Н.,
Владимиров Л. П., Гуляницкий Б. С., Фишер
А. Я., Справочник по расчетам равновесий металлургических
реакций, М., 1963; Е 1 1 1 n g h a m Н. I, Т., J. Soc Chem.
Ind 1944, 63, № 5, 125; D a u t z enb erg W., Z. Erzbergbau
und Metallhiittenwesen, 1950, 3, № 10, 341. А. Я. Фишер.
МЕТАЛЛУРГИЯ — наука о промышленном полу-
чении металлов из природного сырья. Долгое время
М. считалась искусством выплавлять металлы из руд.
Лишь в 19 в. М., под влиянием развития химии, стала
превращаться в науку, что завершилось в начале
20 в., гл. обр. благодаря широкому внедрению в М.
физич. химии и химич. термодинамики.
Современная М. охватывает: 1) все технич, про-
цессы подготовки металлич. руд к металлургич. об-
работке (см. Измельчение, Обогащение полезных иско-
паемых, Флотация)', 2) собственно металлургич. обра-
ботку их, к-рая производится либо сухим путем,
при высоких темп-рах (см. Пирометаллургия), либо
мокрым путем (см. Гидрометаллургия)', 3) очистку
металлов от вредных примесей (см. Рафинирование
металлов)', 4) науку о металлич. сплавах, изучающую
зависимости их свойств от состава и строения (ме-
талловедение); 5) термин, и химико-термич.
обработку металлов и сплавов, имеющую цель —
повышение их механич. свойств, химич. стойкости и
др. (см. Термическая обработка металлов). Особые
виды металлургич. процессов составляют: 1) электро-
металлургия', в ней электрич. энергия используется
либо как источник тепла (электротермия), либо для
восстановления металлов из водных р-ров или рас-
плавленных сред (см. Гальванотехника и Электрохи-
мия)', 2) алюминотермия (восстановление металлич.
окислов алюминием) и металлотермия (восстановле-
ние их магнием, кальцием и др.); 3) порошковая метал-
лургия. О способах получения отдельных металлов см.
соответствующие статьи.
МЕТАЛЛЫ. Содержание:
Введение....................................157
Кристаллическая структура...................158
Тепловые свойства...........................161
Электрические и магнитные свойства..........161
Механические свойства.......................163
Действие излучения..........................164
Окисление...................................165
Металлические фазы сложного состава.........166
Металлы как важнейшие материалы современной тех-
ники .................................. ..167
Металлы — вещества, основной отличительной осо-
бенностью которых в конденсированном состоянии
является наличие свободных, не связанных с опреде-
ленными атомами, электронов, способных переме-
щаться по всему объему тела. Эта особенность ме-
таллического состояния вещества опре-
деляет собою всю совокупность свойств М.
Введение. В атомах элементов, образующих М.,
внешние, валентные, электроны удерживаются значи-
тельно слабее, чем в атомах других — «неметаллич.»
элементов. Это обстоятельство приводит к тому, что
при конденсации из парообразного состояния валент-
ные электроны таких элементов утрачивают связь
с отдельными атомами, обобществляются и удержи-
вают положительные ионы в сближенном состоянии.
Связь атомов в твердом или жидком М., так наз. м е-
таллическая связь, основанная на обобще-
ствлении наружных электронов, является одним из
видов химич. связи. Наличие свободных электронов
определяет собою высокую электро- и теплопровод-
ность М., положительный температурный коэфф.
электросопротивления, хорошую отражательную спо-
собность к световому излучению (блеск и непрозрач-
ность), высокую пластичность, термоэлектронную
эмиссию (т, е. способность испускать электроны при
нагревании), магнитные свойства и т. д.
Между металлич. и ковалентной связью имеется
сходство, т. к. оба типа связи основаны на обобще-
ствлении валентных электронов. Однако ковалентная
связь соединяет только два соседних атома, общие
электроны все время пребывают в непосредственной
близости от соединяемых атомов и прочно с ними свя-
заны. При металлич. связи все атомы, присутствую-
щие в данном теле, участвуют в обобществлении ва-
лентных электронов. В связи с этим ковалентная
связь между атомами в твердом теле практически не
допускает взаимного смещения атомов без разрыва,
связей, т. е. без разрушения тела, тогда как металлич.
связь позволяет осуществлять довольно значительные
взаимные смещения атомов без нарушения связи.
Этим объясняется, в частности, хрупкость кристал-
лов с ковалентной связью между атомами и высокая
пластичность М.
Прочность металлич. связи определяется из значе-
ний теплот сублимации. Теплота сублима-
ции М., т. е. энергия, к-рую нужно затратить для
разделения твердого металлич. тела на изолирован-
ные атомы, обычно очень велика; она изменяется н
пределах от ок. 19 ккал/г-атом для цезия до ок. 210
ккал/г-атом для вольфрама. Особенно прочна связь в
тяжелых М., соответствующих переходным элементам,.
что обусловливает высокие темп-ры плавления и кипе-
ния этих М. и их большую механич. прочность. Вмести
с тем высокие значения теплот сублимации указывают,
что твердые М.— весьма устойчивые тела, к-рым н.
обычных условиях свойственно очень малое давление
пара. Атомные теплоемкости М. (за исключением обла-
сти низких темп-p) очень близки к ЗЛ кал/г-атом-град'
(закон Дюлонга и Пти); при сопоставлении со значе-
нием средней энергии гармонии, колебаний, даваемым,
кинетич. теорией, это показывает, что основными
видами движения атомов в М. являются классич. гар-
монии. колебания в трех измерениях,
В жидком состоянии М. полностью сохраняют свои
электрич., оптич. и тепловые свойства; взаимное
расположение ближайших соседних ионов, т. н.
ближний порядок, в металлич. жидкости сохраняется,
примерно таким же, каким он был в твердом М. (вбли-
зи темп-ры плавления).
Кристаллическая структура. В твердом состоянии
большинство М. имеет высокосимметричную кристал-
лич. структуру (с большим координационным числом)
одного из трех типов: кубич. объемноцентрированную
(все щелочные М., а также р-титап, ванадий, хром,.
a-железо и др.), кубич. гранецентрированную (алю-
миний, медь, серебро, золото, у-железо, р-кобальт,
никель, палладий, платина и др.) и плотную гексаго-
нальную (бериллий, магний, кадмий, цинк, а-титан,
а-кобальт и др.). Кристаллич. решетка М. может рас-
сматриваться как система правильно расположенных
в пространстве положительных ионов и перемещаю-
щихся среди них свободных электронов. Связь элек-
тронов со всей системой кристалла характеризуется
т. н. работой выхода электрона. Она
может быть измерена при термоэлектронной эмиссии
или фотоэффекте. Работа выхода электрона для М.
имеет обычно малые значения (2—5 эв), она значи-
тельно меньше, чем энергия ионизации (см. Иониза-
ции потенциал) изолированного атома М.,что указы-
вает на возрастание кинетич. энергии электронов при
образовании кристаллич. решетки М.
Схематический квантовомеханич. анализ проблемы
образования кристаллич. структуры М. показывает,
что по крайней мере при низких темп-рах метал-
159
МЕТАЛЛЫ
160
лич. связь должна приводить к плотноупакованной
гексагональной или гранецентрированной кубич.
структурам. Однако вопрос о возникновении той или
иной структуры в различных частных случаях остается
открытым, а наличие у нек-рых М. неплотноупакован-
ных структур (кубич. объемноцентрированная,тетраго-
нальная и др.) остается совершенно не объясненным.
Интересный подход к объяснению структуры ме-
таллических кристаллов основывается (Юм-Розери)
на представлении об электронной концен-
трации. В чистом металле или сплаве электрон-
ная концентрация равна среднему числу валентных
электронов, приходящихся на один атом, т. е. пол-
ному числу валентных электронов, присутствующих
в кристалле, деленному на число образующих его
атомов. Т. к. свободные электроны статистически рав-
номерно распределены в объеме металла, то каждый
атом окружен, в среднем, числом электронов, равным
электронной концентрации. Следовательно, можно
ожидать, что в различных металлах и сплавах, имею-
щих одинаковую электронную концентрацию, силы
связи должны быть также одинаковы и обеспечивать
образование одинаковых структур. Действительно,
все металлы 1а подгруппы (главной подгруппы)
периодич. системы — Li, Na, К, Rb и Cs — кристалли-
зуются в объемноцентрированной кубич. системе.
Металлы, находящиеся в 1 b подгруппе (побочной) —
Си, Ag и Au,— кристаллизуются в гранецентрирован-
ной кубич. системе. Но уже во II группе элементов
наблюдаются исключения из этого правила, а в
III группе А1 имеет гранецентрированную кубич.
решетку, Ga — орторомбическую, In — тетрагональ-
ную и Т1 — гексагональную.
Аналогичное положение имеет место и в металлич.
сплавах. Так, напр., р-латунь содержит в среднем
1 атом цинка на 1 атом меди. С каждой такой парой
атомов связаны 2 электрона цинка и 1 электрон меди.
Следовательно, электронная концентрация р-латуни
равна */2; кристаллич. же структура ее — объемноцен-
трированный куб. Известно много сплавов, к-рые
имеют ту же электронную концентрацию и ту же кри-
сталлич. структуру, что и р-латунь. Однако суще-
ствует также много сплавов, в к-рых электронная
концентрация равна 3/2, а структура иная. Такими
сплавами являются, напр., Ag3Al, АизА1, CusSi,
Со/пз. Вместе с тем существует много сплавов с объем-
ноцентрированной кубич. структурой, в к-рых элек-
тронная концентрация не равна 3/2, напр. LiAg и
СоА1.
Плотноупакованная гексагональная структура ши-
роко распространена среди сплавов с электронной кон-
центрацией 7/4. Структура у-латуни (сложная решет-
ка) обычно сопутствует электронной концентрации
21/1з- Однако как в том, так и в другом случае изве-
стно много сплавов, имеющих ту же электронную кон-
центрацию, но иную структуру, или ту же структуру,
но иную электронную концентрацию. Отсюда следует,
что электронная концентрация не является единствен-
ным фактором, определяющим структуру чистого
металла или сплава. Однако представление об элек-
тронной концентрации безусловно полезно и дает
возможность в ряде случаев сделать правильные вы-
воды относительно структуры сложных сплавов.
Электроны в М. не остаются в покое: они беспоря-
дочно движутся, переходя от атома к атому. Скорость
движения электронов практически не связана
темп-рой и сохраняется почти без изменения при аб-
солютном нуле.
В классич. электронной теории М., разработанной Друде
и Лоренцом в начале XX в. на основе статистики Больцмана,
предполагалось, что средняя кинетич. энергия свободных элект-
ронов в М. определяется обычной формулой кинетич. теории
газов, т. е. e=3/2kT, где Т — абс. темп-ра, a k — постоянная
Больцмана. Такое уподобление свободных электронов в М.
идеальному газу привело к противоречию с опытом. Дальней-
шая разработка теории была проведена Зоммерфельдом, к-рый
применил к электронному газу квантовую статистику
Ферми.
В теории Зоммерфельда все электроны в М. распределяются!
по дискретным энергетич. уровням (подобно тому, как распре-
деляются по квантовым уровням электроны отдельного атома)
в соответствии с Паули принципом, причем энергия электрона
изменяется в зависимости от длины волны электрона: е=
=h2/2m№, где h — постоянная Планка, т — масса электрона и
K—h/mv — длина волны электрона (г — скорость движения
электрона). Эти дискретные энергетич. уровни определяются на
основе упрощенного предположения, что потенциал электрич.
поля внутри М., в к-ром движутся электроны, постоянен во
всех точках. Несмотря на свою ограниченность, эта теория
дала правильное описание многих электрич., магнитных и те-
пловых свойств М.
Дальнейшим этапом в развитии электронной теории М.
явилось создание так наз. зонной теории, в основу к-рой
было положено представление о периодически меняющемся по-
тенциале электрич. поля внутри М., в к-ром движутся электро-
ны. По этой теории энергетич. спектр электронов делится на зо-
ны, каждая из к-рых заключает в себе 2N состояний (N— число
атомов в единице объема). В трехмерном К-пространстве (К—
волновой вектор, для свободных электронов определяемый ра-
т, 2nmv . _
венством К== —-—» гдеу — вектор скорости) зоны изобразят-
ся в виде охватывающих друг друга многогранников (зоны
Бриллюэна). Внутри каждой зоны энергия меняется практи-
чески непрерывно.
Вблизи границы зоны энергия е электронов меняется более
сложно, также сложно меняется и функция п(е), показываю-
щая число электронов, приходящихся на единичный интервал
энергии, в зависимости от энергии е. Для вычисления общей
энергии кристаллич. решетки М. наиболее успешно применяет-
ся т. н. ячейковый (целлулярный) метод (метод Вигнера —
Зейтца—Слэтера). Все пространство решетки делится на рав-
ные многогранники (ячейки) около каждого из атомов. Если
рассмотреть с помощью методов квантовой механики атом,
заключенный в многогранник, то энергия электронов окажется
меньше, чем у свободного атома за счет изменения граничных
условий. Это понижение энергии и создает межатомную связь
в кристаллах. Энергия имеет минимум при определенных рас-
стояниях между центрами атомов; отсюда могут быть найдены
нормальные расстояния между атомами и периоды кристаллич.
решетки, к-рые для исследованных кристаллов, гл. обр. щелоч-
ных металлов Li, Na, К, находятся в хорошем согласии с опы-
том. Хуже согласуются с экспериментальными данными вычис-
ленные значения энергии решетки атомов таких М., как медь,
где трудно правильно учесть взаимодействие положительных
ионов друг с другом и обменную энергию.
На основании зонной теории удалось объяснить для спла-
вов устойчивость ряда кристаллич. структур, иногда со слож-
ной решеткой и сложным расположением атомов. В случае
образования твердых р-ров в сплавах элементов разной валент-
ности число валентных электронов, приходящихся на атом,
может быть переменным. Вместе с тем меняется и степень за-
полнения зон Бриллюэна, структура и размеры к-рых опреде-
ляются строением атомной решетки. По мере приближения к
заполнению первой бриллюэновой зоны рост кинетич. энергии
сперва замедляется, а затем с увеличением электронной кон-
центрации начинает увеличиваться. Это ограничивает устой-
чивость кристаллич. решеток твердых р-ров определенными
концентрациями разновалентных компонентов. С помощью этих
же представлений можно объяснить существование дефектных
структур, в к-рых отдельные узлы остаются не занятыми ато-
мами (напр., в сплаве N1A1), устойчивость таких сложных
структур, как решетка типа у-латуни, а-марганца и др.
Многие М., особенно переходного типа, способны
к полиморфным превращениям (см. Полиморфизм).
Известны следующие чистые М., существующие в двух
полиморфных модификациях, устойчивых в различных
температурных областях: железо, никель, кобальт,
вольфрам, титан, уран, цирконий, родий, скандий,
таллий, церий, лантан, празеодим и олово. По край-
вей мере относительно четырех М. можно утверждать,
что они имеют три полиморфных модификации — это
хром, кальций, литий и селен. Марганец имеет четыре
модификации. Полиморфные превращения наблю-
даются также и в сплавах на основе вышеуказанных М.
Во всяком М. и сплаве полиморфное превращение
влечет за собой изменение периода решетки, уд.
объема, а также уд. электросопротивления, уд. теп-
лоемкости, твердости, прочности и пластичности. Эти
изменения свойств имеют очень большое значение
в процессах производства и применения М.
При обычных способах получения М. являются по-
ликристаллич. телами, состоящими из многих мелких
беспорядочно ориентированных и сросшихся друг с
другом кристаллов. Границы между кристаллами
161
МЕТАЛЛЫ
162
в чистом М. составляют переходную зону толщиной
в несколько атомных слоев с сильно искаженной ре-
шеткой. В связи с этим границы зерен обладают из-
бытком свободной энергии. Это обстоятельство приво-
дит к резкому повышению химич. активности границ,
что при наличии коррозионной среды вызывает меж-
кристаллитную коррозию М. Вместе с тем высокая
активность границ используется в металлографии
для травления шлифов и выявления микроструктуры
М., т. е. для определения размеров отдельных кри-
сталлов, составляющих поликристаллический М. Сами
кристаллы имеют мозаичную структуру, т. е. состоят
из отдельных блоков (размеры 10“5 — 10-6 см), неско-
лько отличающихся по ориентировке (на доли углового
градуса). Блоки мозаики хорошо различимы в элект-
ронном микроскопе при увеличении в 30 000 и более
раз. В настоящее время образование блоков мозаики
связывают с дислокационными скоплениями.
Тепловые свойства. Тепловые колебания атомов
в М. не могут происходить независимо друг от друга
из-за наличия сильной связи, и потому колебания
распространяются в виде системы упругих (тепловых)
волн. Спектр частот этих волн зависит от типа решет-
ки, от скорости распространения колебаний (скорости
звука) в разных направлениях и т. д. Спектр тепловых
колебаний можно изучать опытным путем, используя
явление дифракции рентгеновских лучей на тепловых
волнах. Для идеальной кристаллич. решетки отраже-
ние монохроматич. рентгеновских лучей происходит
лишь при строго определенных положениях кристал-
ла. При наличии теплового движения рассеяние про-
исходит также и вблизи положений правильного от-
ражения. Интенсивность этого рассеяния (диффузного
фона) связана с распределением амплитуды колебаний
по направлениям и частотам тепловых волн. При
помощи подобных опытов можно определить скорости
распространения тепловых волн в кристалле и вычис-
лять упругие свойства решетки.
Как правило, коэффициент термиче-
ского расширения больше у легкоплавких
металлов, что соответствует эмпирически установлен-
ному правилу (правило Грюнейзена): относительное
увеличение объема от 0° К до темп-ры плавления для
всех металлов приблизительно постоянно и равно
0,06. Анизотропия теплового расширения для некубич.
М. (Zn, Cd и др.) может быть довольно значительной.
Практически она играет существенную роль, вызывая
у многих М. в поликристаллич. состоянии при изме-
нении темп-ры значительные внутренние напряже-
ния. Сравнение теплоты плавления и теп-
лоты сублимации показывает, что при плавлении
прочность связей между атомами меняется лишь нез-
начительно (приблизительно на 2—5% от общей энер-
гии связи).
Теплопроводность составляется из пере-
дачи тепла: 1) за счет колебаний атомной решетки и
2) электронами М. Главную роль в М. играет элект-
ронная теплопроводность, обусловленная наличием
свободных электронов. При средних темп-pax тепло-
проводность мало зависит от темп-ры, при низких —
она возрастает приблизительно обратно пропорцио-
нально 3-й степени абсолютной темп-ры.
Электрические и магнитные свойства. Хорошо из-
вестным свойством М. является малое значение уд.
электросопротивления. Однако еще более характерно
возрастание электросопротивления металлич. провод-
ников с темп-рой (положительный температурный
коэфф, сопротивления). Это свойство тесно связано
с тем, что электрич. ток в М. переносится свободными
электронами. Для прохождения электронов правиль-
ная кристаллич. решетка не представляет сопротив-
ления. Последнее возникает тогда, когда вследствие
неравномерности теплового движения образуются
местные сгущения, флюктуации плотности, на к-рых
рассеиваются электронные волны. Аналогичное дей-
ствие оказывают всякого рода неоднородности в струк-
туре М.: наличие чужеродных атомов, границы зерен,
дефекты структуры, внутренние напряжения, вызван-
ные деформацией М., нт. п.
Большое значение для выяснения природы М. име-
ют разнообразные гальваномагнитные и термоэлект-
рич. явления. К этим явлениям относятся эффект
Холла — возникновение поперечного электрич. поля
в металлич. пластинке, вдоль к-рой течет электрич.
ток при наличии перпендикулярного к направлению
тока магнитного поля; эффект Зеебека — появление
электродвижущей силы в цепи, состоящей из разно-
родных М., если темп-pa спаев этих М. различна;
эффект Пельтье — поглощение или выделение тепла
на стыке двух М. при прохождении электрич. тока,
и т. д. Все эти явления указывают, что как в случае
электрического, так и в случае теплового тока носи-
телями тока являются электроны. Это подтверждается
также существующей для всех М. закономерной
связью между коэфф, электропроводности а и тепло-
проводности х (закон Видемана — Франца):
сг/хГ — const 2,2-10“8 эрг -см~’ -град~г
Электросопротивление чистых М.
при приближении к 0° К стремится к нулю, как
Т\ Для нек-рых М., напр. Hg, Pb и др., оно, однако,
при нескольких градусах абсолютной шкалы внезапно
падает до исчезающе малых значений (сверхпро-
водимость). Для твердых р-ров или для М.,
содержащих малые загрязнения, электросопротивле-
ние стремится не к нулю, а к нек-рому конечному зна-
чению. Это остаточное сопротивление связано с не-
правильностями кристаллич. решетки. То же самое
наблюдается и в чистых М. при наличии в них оста-
точных напряжений (следы прошедшей пластич. де-
формации, наклепа и пр.). Измерение электросопро-
тивления при низких темп-рах — весьма чувствитель-
ное средство для обнаружения этих искажений. Мно-
гие металлич. соединения (напр., SnSb, AuPb2, РЬТ12
и др.), подобно чистым М., являются сверхпровод-
никами.
Термоэлектронная эмиссия — ха-
рактерная для М. способность испускать электроны
при высокой темп-ре. Плотность тока насыщения,
возникающего при термоэлектронной эмиссии, опре-
деляется формулой Ричардсона: i = ВТ2ехр (—(р/кТ),
где В — постоянная, Т — абс. темп-pa, <р — работа
выхода и к — постоянная Больцмана. Т. к. <р равно
нескольким электронвольтам (2—5 эв), а кТ при ком-
натной темп-ре составляет 0,03 эв, то множитель
ехр (—<р/кТ) в этих условиях ничтожно мал — состав-
ляет ~10-30. При 1500° К величина Л-Т7 равна 0,15 эв
и в этих условиях можно наблюдать заметный элек-
трич. ток. Явление термоэлектронной эмиссии широко
используется в радиотехнике (электронные лампы).
Магнитные свойства М. имеют суще-
ственное значение для теории, характеризуя состоя-
ние, в к-ром находятся атомы в М. Большинство М.,
ионы к-рых имеют замкнутую электронную оболочку,
оказываются диамагнитными (т. е. коэфф, магнитной
проницаемости ц<1), так как диамагнетизм в силу
общих законов электродинамики свойствен всем
вообще атомам, не имеющим собственного магнитного
момента. Напротив, М. переходных групп (Ti, V...
Ni; Nb, Mo ... Pd), атомы к-рых имеют недостаток
электронов в с?-оболочке и тем самым обладают атом-
ным магнитным моментом, оказываются парамагнит-
ными (ц>1). Щелочные и щелочноземельные М.
слабо парамагнитны за счет электронного газа, к-рый0
как показывает электронная теория, должен обла-
дать парамагнетизмом. Магнитные свойства М. в целом
Э 6 к.х.Э. т. 3
163
МЕТАЛЛЫ
164
согласуются с представлением о том, что внеш-
ние, валентные, электроны в металлич. кристалле от-
делены от атомов и движутся в периодич. поле кри-
сталлич. решетки, состоящей из ионов. Среди чистых
М. четыре — железо, кобальт, никель и гадолиний —
обладают значительной способностью сгущать маг-
нитные силовые линии (обладают высоким значением
коэфф, магнитной проницаемости: |л^>1) и могут по-
стоянно сохранять состояние намагничения. Эти М.
наз. ферромагнитными. Предельная намагниченность
этих металлов (магнитное насыщение) уменьшается с
повышением темп-ры. Темп-pa, при к-рой предельная
намагниченность обращается в нуль, наз. точкой
Кюри.
Ферромагнетизм связан со спином элек-
тронов и в М. группы железа обусловлен (/-электро-
нами. Подтверждение этому можно видеть в том фак-
те, что введение в никель такого количества меди (или
другого М.), к-рое обеспечивает заполнение электро-
нами d-уровня, приводит к исчезновению ферромагне-
тизма никеля.
Механические свойства. К числу главных механич.
свойств М. относятся: упругость, пластичность,
ползучесть и прочность. Эти свойства М. являются
основной их характеристикой как конструкционных
материалов.
Упругость — свойство восстанавливать свою
форму после снятия деформирующих сил; характери-
зуется пределом упругости, т. е. напряже-
нием в кГ/мм2, выше к-рого в М. появляются остаточ-
ные деформации, не исчезающие после снятия нагруз-
ки. В упругой области деформаций имеет место за-
кон Гука: Р — ЕЛ1/1, где Р — напряжение,
А/// — относительное удлинение (в простейшем случае
растяжения стержня) и £ — коэфф, пропорциональ-
ности, называемый модулем Юнга (модулем
нормальной упругости). Упругие свойства М. сильно
разнятся для монокристаллов и поликристаллич.
агрегатов. Упругая деформация у предела упругости
для монокристаллов составляет 10-4—10_5, т. е. сотые
и тысячные доли процента, тогда как для поликри-
сталлов она повышается до 1%. Модуль Юнга метал-
лич. монокристаллов имеет явно выраженную анизо-
тропию. В поликристаллич. М. анизотропия отсут-
ствует в результате усреднения по множеству различно
ориентированных кристаллов. По абсолютной величи-
не модуль Юнга поликристалла практически не отли-
чается от усредненного модуля монокристалла и ме-
няется для наиболее употребимых в технике М. от
~7000 кПмм2 (для алюминия) до —40 ООО кГ/мм2 (для
вольфрама). У большинства М. обнаруживаются бо-
лее или менее значительные отступления от идеально
упругого поведения под нагрузкой даже при напря-
жениях, малых по сравнению с пределом упругости.
Эти отступления, наз. упругими несовершенствами
или неупругостью, вызываются неоднородностью
строения реальных М. и дефектами их структуры.
Пластичность — состояние М., в к-ром он
способен сохранять изменение формы, вызванное воз-
действием деформирующих сил после того как силы
сняты. Это остаточное изменение формы наз. пласти-
ческой, или остаточной, деформацией.
В отличие от пластичности, хрупкость представляет
собой такое состояние М., когда остаточные дефор-
мации вовсе не возникают вплоть до разрушения
либо они весьма малы. Пластичность и хрупкость
нельзя рассматривать как свойства М., т. к. один и
тот же М. может, в зависимости от условии деформа-
ции (темп-ры, внешней среды и т. п.), переходить от
состояния пластичности к состоянию хрупкости и
наоборот. То наименьшее напряжение в кГ/мм2, после
снятия к-рого обнаруживается остаточная деформа-
ция, наз. пределом текучести данного М.
Т. к. возможность обнаружения остаточной дефор-
мации зависит от точности выбранного метода, то по-
нятие предела текучести — условное понятие. Обычно-
за предел текучести принимают такое напряжение^
к-рое при кратковременном действии вызывает пла-
стин. деформацию в 0,2% расчетной длины образ-
на (Ро,2).
Для большинства чистых М. Ро 2 составляет 10—
25 кГ/мм2. Пластин, деформация ’М. всегда сопро-
вождается упрочением, т. е. ростом напряжения
по мере роста пластин, деформаций. М,— наиболее
пластичные твердые тела. Особенно высокой пластич-
ностью обладают металлич. монокристаллы с гексаго-
нальной решеткой; они пластически удлиняются
перед разрывом в 6—8 раз. В настоящее время теоре-
тически и экспериментально показано, что способ-
ность М. к пластин, деформации определяется нали-
чием в их структуре дислокаций — линейных дефек-
тов структуры, способных перемещаться под действием
малых напряжений и генерироваться внутри тела а
процессе деформации.
Ползучесть М. проявляется в медленном
пластин, течении при напряжениях, значительно-
меньших предела текучести. Механизм ползучести в.
большинстве случаев принципиально не отличается
от механизма пластин, течения, т. е. заключается
в движении дислокаций. Лишь при очень высоких
темп-рах, близких к плавлению М., ползучесть может
реализоваться движением вакансий, т. е. механизмом
диффузионного переноса вещества или скольжения
по границам зерен. Ползучесть — очень опасное яв-
ление для конструкционных М. при их использовании
при повышенных темп-рах, т. к. приводит в конце
концов к разрушению М. Разработаны различные
способы борьбы с этим явлением, главным из к-рых
является добавка к данному М. пли сплаву малых ко-
личеств других М.— легирование. Так, для сталей
такими легирующими М. являются молибден, воль-
фрам, ванадий, для алюминия — медь и т. д.
Всякий процесс деформации как упругой, так и
пластической при достаточном повышении напряже-
ния (или при достаточно длительном действии посто-
янного напряжения) заканчивается разрушением М.
Сопротивление М. разрушению наз. прочностью
данного М. и измеряется в кГ/мм2 поперечного сече-
ния. Если разрушение М. рассматривать как одно-
временный разрыв связей между всеми атомами, рас-
положенными по обе стороны площадки в 1 мм2, вы-
бранной перпендикулярно растягивающей силе, то
величина прочности М. может быть теоретически оце-
нена и оказывается равной 105—101 кГ/мм2. Однако
на практике прочность М. не превосходит 100—
200 кГ/мм2. Такое расхождение теории с опытом опре-
деляется наличием в М. множества дефектов струк-
туры, возникающих в процессе деформации М. или
присутствовавших в нем ранее. Существенное влия-
ние на прочность М. оказывают размеры образцов
(масштабный эффект). Опыт показывает, что с умень-
шением размеров прочность повышается. Весьма
тонкие металлич. нити (несколько микрон в диамет-
ре), получившие название «усов», обнаруживают очень
высокую прочность, близкую к теоретической.
Действие излучения. Остаточные изменения свойств
М. под действием излучения возникают в результате
образования дефектов решетки — вакансий и сме-
щенных атомов. При этом модуль упругости М. прак-
тически не меняется, тогда как предел текучести,
прочность при растяжении и пластичность могут
испытать значительные изменения. Так, напр., железо,
облученное нейтронами (1—1,5 -Ю20 н/см2) при темп-ре
300°, обнаружило увеличение предела текучести
(Р„2) на 70%, временного сопротивления (Рв) — на
65%, а удлинение (б) перед разрывом снизилось в.
165
МЕТАЛЛЫ
166
три раза. Значительная потеря пластичности является
характерным следствием облучения. Вместе с тем ме-
няется и темп-рный порог перехода М. из пластичного
состояния в хрупкое. Так, у молибдена этот порог
находится около —30°, тогда как после облучения
потоком 1,5 -1020 н/см2 он повышается до +70°.
через слой окисла. Примером может служить окисле-
ние меди при темп-pax > 550° и окисление цинка при
темп-pax до 350°. Логарифмич. закон роста, как пра-
вило, приводит к образованию защитной окисной
пленки и особенно характерен для алюминия при
темп-pax от комнатной до ок. 300° и хрома в широкой
Изменение свойств нек-рых металлов под действием нейтронного облучения
Металл Темп-ра облуче- ния, °C Поток, н,'см2 Рв, кГ!мм2 Рс,2. кГ[мм2 | Удлинение
до облу- чения после облуче- ния 1 измене- ние, % до облу- чения после обл уче- ния измене- ние, % до облу- чения о о С =5 >»- О EJ Я ОС S cos | измене- ние, %
Углеродистая сталь .... Сталь 1Н18Н9Т Никель Цирконий Медь Алюминий Молибден 80 80 80 80 100 65 90 2 -1 О19 1 -IO20 1 -1 О20 1-1020 5 • 10 10 1 , 3 • 1 О22 5 • 1 О20 53 65 39 26,5 19,0 9.6 70 69 100 82 36 24 18,4 107 + 30 + 54 + 110 + 36 + 26 + 91 + 53 28 24 14 16 5,9 4.8 66 65 92 7 3 34 21,5 12,1 107 + 132 + 283 + 420 + 112 + 265 + 150 + 62 25 66 37 34 42 38 42 6 43 5 15 27,5 21 0 -76 -35 -87 -56- -3 5- -45- - 1 0»
Изменение механич. свойств М., вызванное облу-
чением, снимается полностью лишь при темп-ре ре-
кристаллизации. Механизм действия облучения на
механич. свойства М. связан с образованием как ат-
мосфер Коттрелла из вакансий и смещенных атомов
вокруг дислокаций, так и с торможением различных
источников генерирования дислокаций (см. Дисло-
кации).
Окисление. Это — наиболее распространенная хи-
мическая реакция, свойственная всем М. Окисление
приводит к образованию на поверхности М. пленки
окисла. Скорость наращивания окисной пленки опре-
деляется при высокой темп-ре скоростью диффузии
реагирующих веществ через слой окисла и подчиняет-
ся диффузионному закону Фика, по к-рому прираще-
ние толщины Дж пленки за единицу времени опреде-
ляется ур-нием: Интегрирование этого
ур-ния дает Дж2 = 2а’t, или, если перейти от прира-
щения толщины пленки к приращению ее массы на
1 см2: (1m)2 = 2а't. Этот параболич. закон роста
окисной пленки действительно имеет место на многих
М. при высоких темп-pax. Однако, как показал Мотт,
наращивание толщины, окисной пленки может осуще-
ствляться не только в результате тепловой подвиж-
ности ионов. При наличии на поверхности М. тончай-
шей пленки окисла толщиной 8—10 А проникновение
электронов через этот слой в результате туннельного
эффекта и захват их адсорбированным слоем кисло-
рода, всегда покрывающим окисную пленку, приводят
к образованию конденсатора (положительная пласти-
на — металл, отрицательная — ионы адсорбирован-
ного кислорода, захватившие электроны) с разностью
потенциалов, по расчетам Мотта, до 1 в. В связи с
этим внутри такого конденсатора образуется градиент
потенциала порядка 10’ в/см. Столь сильное поле
значительно облегчает перенос ионов М. сквозь окис-
ную пленку даже при низких темп-pax. С возрастанием
толщины окисной пленки градиент потенциала быстро
падает и рост пленки прекращается. Толщина такой
пленки может составить 20—50А при комнатной
темп-ре и скорость ее наращивания подчиняется
логарифмич. закону. В последнее время Мотт и Каб-
рера теоретически указали условия, приводящие к
различным законам роста во времени окисных пленок
на М.: параболическому, логарифмическому, кубиче-
скому и линейному. Параболич. закон роста, как уже
указывалось, встречается наиболее часто и наблю-
дается в том случае, когда процессом, определяющим
скорость реакции, является процесс диффузии ионов
области темп-p. Линейный закон роста наблюдается
в тех случаях, когда образующиеся на М. окисные
пленки весьма пористы и обеспечивают легкий доступ
газообразной фазы к поверхности М. По этому закону
растут пленки окислов на щелочных и щелочноземель-
ных М. И, наконец, кубич. закон роста наблюдается
на нек-рых сплавах, напр. на сплавах цинка с неболь-
шим количеством лития.
В сплавах при низкотемпературном окислении наб-
людается образование смешанных окислов, напр. на
сплавах Fe—Ni—Сг и Fe—Си—Al образуются сме-
шанные окислы типа шпинелей. Однако вследствие
различия в подвижности ионов состав окисного слоя
часто бывает неоднороден. Так, при окислении сплавов
А1—Fe образуются пленки, к-рые вблизи М. состоят
из почти чистого А12О3, а по мере удаления от М.—
из смешанных окислов. При высоких темп-pax тече-
ние процесса окисления определяется доминирующей
диффузией одного из ионов, и при этом возникают
пленки чистого окисла. При окислении сплавов
Fe—Ni—Сг на поверхности образуется пленка чистой
Сг20з. На сплавах Fe—Сг—А1 при темп-pax выше-
900° образуется пленка почти чистой А120з; на сплавах
Си—Be образуются пленки чистого ВеО.
Как правило, окисные пленки на М. обладают ми-
кропористостью (рыхлой структурой), и их образова-
ние не защищает М. от дальнейшего окисления. Только
хром, алюминий и кремний образуют на своей по-
верхности очень плотные защитные окисные пленки.
В связи с этим Сг, А1 и Si широко используются в ка-
честве легирующих добавок к сталям для повышения
их жаростойкости, т. е. для повышения их сопротив-
ляемости окислению.
Металлические фазы сложного состава. В сплавах
М. образуются кристаллы, сложные по химич. составу,
металлич. фазы различного типа. Наиболее распро-
страненными средп них являются металлич. растворы
(жидкие и твердые) и металлические соединения. Ме-
таллич. твердые р-ры образуются путем замещения
части узлов в кристаллич. решетке одного М. атомами
другого (т. н. твердые растворы замеще-
ния). Такие фазы образуются химически близкими
элементами, имеющими малое различие атомных диа-
метров. При однотипности решеток компонентов и
близости их атомных структур и диаметров твердые
р-ры замещения образуются при любой концентрации
(полная взаимная растворимость), напр. в системах
Ag—Au или Мо—W; при меньшем сходстве структур-
атомов — лишь в ограниченной области составов
(ограниченная взаимная растворимость), например в
6'
167
МЕТАЛЛЫ
168
системах Ag—Си или Fe—Ti. Как правило, М., рас-
положенные в разных группах пли рядах периодич.
системы, при различии атомных диаметров более чем
на 8—10% способны давать между собой только
ограниченные твердые р-ры. Чередование атомов
различных элементов в твердом р-ре не является упо-
рядоченным и подчиняется статистич. закономерно-
стям, а средний период решетки изменяется в зависи-
мости от состава р-ра.
В ряде случаев при достаточно медленном охлажде-
нии твердого р-ра замещения из него образуются ме-
таллич. соединения, характеризующиеся упорядочен-
ным чередованием атомов компонентов в решетке
сплава. Впервые это было обнаружено Н. С. Курни-
ковым с сотрудниками в системе Си—Ап (фазы СпзАп
и CuAu). Чем дальше друг от друга в периодич. систе-
ме находятся взаимодействующие элементы, тем
большую склонность проявляют они к образованию
металлич. соединения. Это обнаруживается, в част-
ности, в расширении температурного интервала суще-
ствования этих соединений. Так, напр., темп-pa обра-
зования соединений железа с другими М. VIII группы
последовательно возрастает в системе Fe—Ni, Fe—Pd,
Fe—Pt от 580° до 1320°. Темп-pa образования соеди-
нений железа с другими М. одного и того же периода
точно так же растет от 580° в системе Fe — Ni до 1230°
в системе Fe—V; при еще большем взаимном удалении
элементов в периодич. системе соединение образуется
при еще более высокой темп-ре и выделяется непосред- <
ственно при кристаллизации сплава из жидкости
(напр., в системе Fe—Ti). Металлич. соединения могут
быть постоянного состава — дальтониды и
переменного состава — бертоллиды (см. Далъ-
тониды и бертоллиды). Нек-рые наиболее устойчивые
соединения с металлич. характером связи являются
переходными к соединениям ионного типа. Так, напр.,
соединение MgsSb2 имеет металлич. связь, а соедине-
ние магния с элементами того же ряда — теллуром
MgTe, не проявляет признаков металлич. характера,
является полупроводником, соединение же магния с
иодом MgJ2 является уже типично ионным. Таким
образом, по мере возрастания электрохимия, разли-
чия атомов реагирующих веществ металлич. связи
переходят постепенно в ионные.
Весьма важны для техники соединения переходных
М. с бором, углеродом и азотом, элементами-неме-
таллами, имеющими малый атомный диаметр. При
условии определенного соотношения между атомными
диаметрами М. и неметалла соединения кристалли-
зуются по типу т. н. фаз внедрения. Эти фазы
сохраняют многие металлич. свойства: простую кри-
сталлич. решетку, высокую электропроводность, ме-
таллич. блеск, весьма высокую твердость и тугоплав-
кость (особенно это относится к группе карбидов).
Кроме того, переходные М., в частности железо, спо-
собны образовывать с теми же элементами (бором,
азотом, углеродом) твердые р-ры внедрения в широ-
кой, но все же ограниченной области концентраций
и темп-ры. В таких р-рах внедрения сохраняется тип
кристаллич. решетки основного М., а атомы углеро-
да или азота размещаются между узлами, увеличи-
вая период решетки пропорционально своей концен-
трации.
Металлы как важнейшие материалы современной
техники. Большая способность М. к взаимному раст-
ворению, образованию многочисленных соединений
различного типа, разнообразным фазовым превраще-
ниям создает благоприятные условия для получения
большого числа сплавов, отличающихся различной
структурой и самыми разнообразными сочетаниями
полезных свойств. Все это делает металлич. сплавы
важнейшими материалами, значение к-рых в технике
непрерывно возрастает. Применение того или иного М.
или сплава в значительной мере определяется практич.
ценностью комплекса механич., физич. и химич.
свойств, к-рыми обладает данный М. Однако сущест-
венную роль играют и другие обстоятельства, и в
первую очередь запасы соответствующих М. в земной
коре, доступность и рентабельность добычи. Из наи-
более ценных и важных для современной техники М.
лишь очень немногие имеются в большом количестве
в составе земной коры. К числу их относятся: железо
(составляет 5,1% от веса земной коры) — главный
материал современной техники, основной компонент
всех видов стали и чугуна; алюминий (8,8%) и магний
(2,1%), являющиеся главными компонентами т. н.
легких сплавов, созданных лишь в 20 в. и имеющих
особое значение для авиации; титан (0,6%), начинаю-
щий приобретать в последние годы самостоятельное
значение в качестве основы для новых высококаче-
ственных сплавов.
В земной коре содержатся в весьма больших коли-
чествах натрий, калий и кальций, но они, в силу своей
большой химич. активности и низкой механич. проч-
ности, самостоятельного значения не имеют и иногда
используются в металлургии лишь как вспомогатель-
ные добавки для раскисления сплавов и для других
целей. Нек-рые весьма важные для техники М. содер-
жатся в земной коре в количестве сотых долей про-
цента (напр., Си, Mn, Cr, V, Zr) или даже тысячных
долей (напр., Zn, Sn, Pb, Ni, Со, Ce, Nb). Прочие М.,
обладающие ценными технич. свойствами, присут-
ствуют еще во много раз меньших количествах. Так,
содержание в земной коре урана — важнейшего М.
для получения атомной энергии составляет по ориен-
тировочной оценке ок. 3-10-4 %, а вольфрама — осно-
вы твердых режущих сплавов — всего 1 10_4% и
т. д. Особенно мало содержание в земной коре драго-
ценных М. (напр., платины или золота— 5-10“’%) и
нек-рых редких и естественно-радиоактивных М.
Помимо запасов того или иного М. в земной коре,
возможности его использования в значительной мере
определяются характером геохимич. распределения
элемента, химич. и минералогии, составом соответ-
ствующих руд, мощностью и географии, расположе-
нием рудных месторождений, а также прочностью
химич. связи данного М. в его природном соединении и
другими особенностями металлургии, переработки
руд. Соединения таких важнейших для техники М.,
как железо, алюминий, встречаются в виде весьма
мощных месторождений относительно богатых руд,
содержащих 30—60% М., что значительно облегчает
их добычу в больших масштабах. Однако производ-
ство алюминия сопряжено с большими затратами
электрич. энергии и освоено пока лишь для нек-рых
видов руд.
Относительное значение отдельных М. в технике
может быть охарактеризовано их долей в общем миро-
вом производстве М., если последнее принять за
100%. Так, в 1939 производство чугуна составляло
ок. 94% , меди—ок. 2% , свинца и цинка —примерно по
1,5% каждого, алюминия ок.—0,6%, прочих металлов
гораздо меньше. В последующие годы производство
алюминия и особенно магния значительно увеличи-
лось, а доля этих М. в общем металлургии, производ-
стве намного повысилась. После второй мировой войны
1939—45 возникло производство титана, молибдена,
циркония, ниобия и нек-рых других М., приобретаю-
щих большое значение в современной технике. Весьма
сильно возросло также производство т. н. легирующих
элементов, применяемых при выплавке легированных
и высоколегированных сталей и сплавов типа нержа-
веющих сталей, жаропрочных сталей, а также алю-
миниевых сплавов и сталей с особыми физич. свой-
ствами. В технике в настоящее время применяется не-
сколько тысяч сплавов; в их состав входит более 40
169
МЕТАЛЬДЕГИД —МЕТАНИЛОВАЯ КИСЛОТА
170
химич. элементов в самых различных сочетаниях.
Широко распространены многокомпонентные сплавы,
специфич. свойства к-рых достигаются путем опреде-
ленного сочетания нескольких М. В качестве М., на
основе к-рых создаются многочисленные сплавы для
конструкционных п др. целей, используются гл. обр.
Fe, Al, Си, Mg и в последнее время Ti, Mo, Nb. О свой-
ствах, способах получения и применении М. см. статьи
о соответствующих элементах.
Лит.: Смит М. К., Основы физики металлов, пер. с
англ., М., 1959; Ю м-Р о з е р и В., Рейнор Г. В., Струк-
тура металлов и сплавов, пер. с англ., М., 1959; Гуляев
А. П., Металловедение, 2 изд., М., 1951; М е л в и н-Х ь ю з
Э. А., Физическая химия, пер. с англ., кн. 2, М., 1962; Лихт-
м а н В. И., Щ у к и н Е. Д., Р еб и н д ер П. А., Физико-
химическая механика металлов, М., 1962; Фридман Я. Б.,
Механические свойства металлов, 2 изд., М., 1952; X а у ф ф е
К., Реакции в твердых телах и на их поверхности, пер. с нем.,
ч. 1—2, М., 1962—63; Барретт Ч. С., Структура металлов,
[пер. с англ.], М., 1948; Успехи физики металлов, Сб. статей,
пер. с англ., т. 1, М., 1956; Агеев Н. В., Природа химиче-
ской связи в металлических сплавах. М.—Л., 1947; Н е-
к р а с о в Б. В., Курс общей химии, 13 изд., М.—Л., 1962.
В. И. Лихтман,
МЕТАЛЬДЕГИД — см. А цеталъдегид.
МЕТАМЕРИЯ — частный случай изомерии, связан-
ный с положением гетероатома в цепи алифатич. сое-
динения. Метамерны, напр., приведенные ниже про-
стые эфиры (I и II) или алифатич. амины (III, IV
и V):
СН,-О-СН.СН.СН. (I) (CH3)3N (III)
C.H5NHCH3 (IV)
СН3СН2-О-СН2СН3 (II) C3H,NH2 (V)
Термин «М.» предложен Я. Берцелиусом в 1830.
В настоящее время практически не употребляется.
Лит.: Бутлеров А. М., Избранные работы по органи-
ческой химии, М., 1951, с. 97, 468. Л. С. Герман.
МЕТАМИЗИЛ (хлоргидрат 2-диэтпламинопропило-
вого эфира бензиловой кислоты), мол. вес 377,91,
(C6H5)2C(OH)COOCH2CH(CH3)N(C2HS)2 -НС1 — белый
кристаллич. порошок, т. пл. 159 —161°; растворим в
воде, хлороформе, спирте, мало растворим в бензоле,
нерастворим в эфире. Качественное определение М.
основано на появлении оранжево-красного окрашива-
ния при прибавлении к М. конц. серной кислоты.
Для количественного определения М. гидролизуют
кипячением с 30%-ной НС1, раствор подщелачивают
30%-ноп КОН, отгоняют с паром 2-диэтиламинопропа-
нол в 0,1 н. р-р H2SOj и избыток последней оттитро-
вывают 0,1 н. р-ром КОН по метиловому красному.
М. получают из 1-диэтиламино-2-хлорпропана и
бензиловой к-ты:
CHsCHC1CH2N(C2H3)2 + (C»H5)2C(OH)COOH —> м
При этом одновременно образуется фармакологи-
чески менее активный изомер — хлоргидрат 1-ди-
этиламиноизопропилового эфира бензиловой кислоты
(C,H5)2(COH)COOCH(CH3)CH2N(C2H5)2-HC1, который, в
отличие от М., хорошо растворим в горячем ацетоне,
что используется для разделения смеси изомеров.
М. может быть также получен переэтерификацией
метилового эфира бензиловой к-ты 2-диэтиламинопро-
пиловым спиртом:
(C.H5)2C(OH)COOCH3 + CH.,OHCH(CH3)N(C2H5)2 —> м
М. обладает преимущественно центральным холи-
нолитич. действием и принадлежит к сильнейшим
представителям центральных холинолитиков. М. при-
меняется в качестве успокаивающего средства при
состоянии страха, при психич. возбуждении, при раз-
личных видах психоза, для потенцирования наркоза,
при язвенной болезни и как противоядие при отрав-
лении антихолинэстеразными препаратами.
Лит.: Денисенко П. П., Фармакология и токсико-
логия, 1960, 23, № 3, с. 206; Торф С. Ф., Хромов-
Борисов Н. В., Инденбом М. Л., Мед. пром-сть
СССР, 1961, № 12, с. 19; К у з н е Ц о в С. Г., Ельцов
А. В., Ж. общ. химии, 1962, 32, вып. 2, с. 511.
Н. В. Хромов-Борисов.
МЕТАН СНа, мол. в. 16,03 — первый член гомо-
логия. ряда предельных углеводородов. Бесцветный,
не имеющий запаха газ; т. пл. —182,48°; т. кип.
—161,49°; = 0,0160 (760 л.и); плотность жидкого
0,436 (—170°), плотность газа по отношению к воздуху
0,554 (20°); 1 г М. занимает объем 1520 мл (25°,760 .и.и);
коэфф, темп-рного расширения 0,003681 (0°—100°); те-
плоемкость газа 0,530 кал/г-град\ теплота испарения
121,87 кал/г-, теплота сгорания газа 11954 ккал/кг (25°);
Ркрит. 45-8 ат’ гкрит. —82,5°; ^крит. °’162; давление
пара при —180° 119 мм рт.ст., при—100° 27,2 ат.
Растворимость в воде: 0,05563 (0°), 0,03308 (20°);
0,0170 (100°); в 100 мл спирта растворяется 52 мл (0°);
в 100 мл диэтилового эфира 106,6 Л1л (0°); в 100 мл СС14
60 мл (30°, 760 мм) и 540 мл (30°,8 ат)', в 100 мл
40%-ной H2SOi 1,58 мл М., в 60%-ной 1,3 мл М.; в
96%-ной 3,1 мл М. (20°).
М. является наиболее прочным и устойчивым к
химич. воздействиям углеводородом. Однако он легко
загорается; т. воспл. 695—742°. Смеси М. с воздухом
чрезвычайно взрывоопасны; пределы взрываемости:
нижний 5 об. %; верхний 15 об. %. При неполном
сгорании или каталитич. окислении М. образует ме-
танол, формальдегид, ацетилен (см. Газы природные
горючие). При разложении М. в электрич. дуге при
1000° получается ацетилен. В этих же условиях М.
реагирует с азотом, образуя HCN. Галогены энергич-
но реагируют с М., замещая водород, напр.
СН, + С12 —> СН3С1 + НС1 + 23,9 ккал;г-моль
Взаимодействием М. с серой при 500—700° в присут-
ствии катализаторов получают CS2.
М. является основным компонентом природного
газа (60—99%), рудничного газа, а также различных
газообразных продуктов анаэробного разложения ор-
ганич. веществ, напр. болотный газ, газы полей оро-
шения. Атмосфера Сатурна, Юпитера, спутника по-
следнего — Титана — состоит в основном из М.
В больших количествах М. образуется при коксовании
каменного угля, гидрирования угля, нефти при дей-
ствии воды на карбид алюминия:
А1,С3+6Н2О —> ЗСН, + 2А12О3
М. используют как топливо при газификации, а
также для получения водяного газа: С Hi + Н2О
СО -j- ЗН2, применяемого для промышленного
синтеза (на основе СО) углеводородов большего мол.
веса, спиртов и др. продуктов, для получения водо-
рода. В меньших масштабах М. используют для по-
лучения ацетилена электрокрекингом или термоокис-
лительным крекингом (подробнее см. Ацетилен), а
также в произ-ве сажи, хлористого метила, хлорбром-
метана, нитрометана, а также синильной кислоты.
Лит.: Ullman п, 3 Aufl., Bd 10, Miinclien—В., 1958,
S. 18. М. И. Розенгарт.
МЕТАНИЛОВАЯ КИСЛОТА (анилин-3-сульфокис-
лота) C6H7OsNS, мол. в. 173,20— кристаллизуется
с Р/2 мол. воды в виде бесцветных игл
или призм; кристаллизационная вода 7" 2
постепенно теряется при стоянии на
воздухе. В 100 мл водного р-ра содер- L Lso ц
жится безводной М. к. (в г): 1,276 (7°), 3
1,9571 (9°), 6,5 (85°). Водные р-ры М. к. на воздухе
окрашиваются в красный цвет. Константа диссоциа-
ции в воде К = 1,8-10-4(25°). Щелочные соли М. к.
хорошо растворимы в воде. Соли М. к. с сильными
к-тами, напр. с Н3РО4, легко разлагаются водой.
М. к. вступает в реакции электрофильного замеще-
ния. При бромировании М. к. бром замещает водород
последовательно в 6-, 4- и 2-положениях. Действием
хлориода на М. к. в разб. НС1 получают 4,6-дииод-
анилин-3-сульфокислоту. Сульфирование М. к. с по-
мощью дымящей H2SO4 при 180° приводит к анилин-
171
МЕТАНОВОЕ БРОЖЕНИЕ —МЕТАСТАБИЛЬНОЕ СОСТОЯНИЕ
172
3,5-дисульфокислоте. Из реакций нуклеофильного
замещения в М. к. практич. значение имеет замена
HSOs-группы на оксигруппу при сплавлении с NaOH
при 280° (получение .и-аминофенола). Функциональные
свойства аминогруппы проявляются в реакциях
ацетилирования и диазотирования; при диазотирова-
нии М. к. легко образует диазоаминосоединение — ди-
азоаминобензол-3,3'-дисульфокислоту. Поэтому М. к.
диазотируют в условиях, исключающих образование
диазоаминосоединения, пропусканием через водную
суспензию окислов азота при сильном охлаждении.
В пром-сти М. к. получают восстановлением 3-нит-
робензолсульфокислоты; ее широко применяют в
произ-ве синтетич. красителей. В. А. Пучков.
МЕТАНОВОЕ БРОЖЕНИЕ — см. Брожение.
МЕТАНОЛ СНзОН — название простейшего али-
фатич. спирта — метилового спирта по женевской
номенклатуре органич. соединений.
МЕТАСТАБИЛЬНОЕ СОСТОЯНИЕ — состояние,
не являющееся термодинамически устойчивым, но
способное длительно сохраняться во времени. При
узком применении этого понятия сюда относят состоя-
ния пересыщенного раствора, пере-
сыщенного пара, переохлажденной
или перегретой жидкости и др., подоб-
ные им.
Возможность длительного существования вещества
в таких состояниях связана с тем, что самопроиз-
вольное образование частичек новой фазы требует
значительно большей степени пересыщения, переох-
лаждения или перегрева, чем в присутствии этой фазы
или частиц, облегчающих ее выделение. Так, водя-
ной пар при отсутствии в нем каких-нибудь частиц,
к-рые могли бы послужить центрами конденсации
(напр., пылинок), может быть сжат до концентрации
в несколько раз более высокой, чем концентрация
насыщенного пара. Это объясняется тем, что очень
маленькие капли воды, возникающие первоначально,
обладают более высоким давлением насыщенного пара,
чем давление его над плоской поверхностью жидкости
при данной темп-ре; обычный насыщенный и даже не-
много пересыщенный пар является ненасыщенным
в отношении таких очень маленьких капель, и они ис-
паряются. Присутствие центров конденсации снижает
степень пересыщения, необходимую для образования
частиц новой фазы.
Подобным же образом при кристаллизации веще-
ства из раствора в отсутствии готовых центров кри-
сталлизации образование очень мелких кристалликов
новой фазы требует пересыщения раствора из-за бо-
лее высокой растворимости их по сравнению с раство-
римостью крупных кристаллов. Аналогичные явле-
ния имеют место при переохлаждении чистой жид-
кости до темп-p ниже ее темп-ры кристаллизации
или при перегреве ее до темп-p выше ее темп-ры кипе-
ния при данном давлении. В последнем случае обра-
зование пузырьков пара в объеме жидкости может
облегчаться также выделением растворенных газов.
Кроме того, стенки сосуда, в зависимости от материа-
ла его и состояния поверхности, часто могут облег-
чать выделение вещества в новой фазе как при кипе-
нии, так и при конденсации, кристаллизации и
проч.
Вещества в М. с. всегда обладают более высоким
значением мольного изобарного потенциала (см.
Термодинамические потенциалы), чем в устойчивом
состоянии, и, в соответствии с этим, они обладают
большей способностью к различным переходам и пре-
вращениям — большим давлением насыщенного пара,
более высокой растворимостью, большей активностью
при химич. взаимодействиях и пр. Нередко возможно
бывает осуществить переход вещества из одного М. с.
в другое, являющееся тоже метастабильным, но отно-
сительно более устойчивым, чем первое в данных
условиях. Можно с помощью химич. реакции или фа-
зового перехода получить или выделить данное веще-
ство сразу в М. с., проводя процесс в условиях, доста-
точно далеких от равновесных.
Более устойчивая форма всегда менее растворима,
чем менее устойчивая. Напр., Na2SOi образует два
гидрата: 1) стабильный Na2SO4-10H2O и 2) метаста-
бильный Na2SO4‘7H2O. При 20° растворимость пер-
вого (считая на безводный Na2S04) равна 16,2 вес. %,
а второго—30,9 вес. %; поэтому раствор, насыщенный
по отношению к Na2SO4-7Н2О, будет пересыщенным
по отношению к Na2SO4-ЮН20. Однако в обычных
условиях, т. е. при доступе воздуха, в к-ром носятся
мельчайшие частички Na2SO4-10Н2О, последние, по-
падая в р-р, играют роль центров кристаллизации,
вызывающих выпадение кристаллов Na2SO4-ЮН20.
Но если раствор Na.2SO4, насыщенный при темп-ре
ок. 34° (т. е. выше 32°— верхней температурной гра-
ницы устойчивого существования Na2SO4-10Н2О),
охлаждать, тщательно защищая его от попадания ча-
стичек Na2SO4-ЮН20, то ниже темп-ры 24°, к-рая
является верхней границей существования Na2SO4-
7Н2О, могут выделиться кристаллы последнего.
Два М. с. данного вещества могут быть связаны устой-
чивым равновесием между собой. Так, переохлажден-
ная вода находится в устойчивом равновесии со своим
насыщенным паром. Но пар этот является пересыщен-
ным по сравнению с равновесным паром льда при этой
темп-ре (при —10° давление насыщенного пара пере-
охлажденной воды равно 2,143 мм рт. ст., а льда —
1,946 мм рт. ст.).
На практике М. с. встречаются в самых различных
формах. В облаках вода в виде жидких капелек может
сохраняться до —40° (и ниже), не замерзая при от-
сутствии центров кристаллизации. Способность пере-
сыщенного водяного пара конденсироваться на заря-
женных частицах лежит в основе работы широко из-
вестной камеры Вильсона, используемой в ядерной
физике. Нек-рые группы веществ, напр. силикаты,
допускают переохлаждение до темп-p, при к-рых теп-
ловое движение частиц уже не может легко разрывать
возникающие между ними связи. В результате этого
происходит сильное уменьшение подвижности частиц
в жидкости, и она переходит в стеклообразное состоя-
ние. Стекла являются М. с. вещества по отношению
к соответствующим кристаллам. Однако, в отличие от
рассмотренных выше случаев, здесь переход в устой-
чивое (кристаллическое) состояние тормозится необ-
ходимостью разрыва значительного числа связей,
существующих между частицами, что в кинетич. от-
ношении играет роль нек-рого энергетич. барьера.
Поэтому при темп-pax, достаточно низких по сравне-
нию с нормальной темп-рой кристаллизации данного
вещества, стекла являются практически устойчивыми
в отношении кристаллизации и только при повышении
темп-ры эта способность у них усиливается.
В связи с подобным же торможением перехода из
одной кристаллич. формы в другую во многих слу-
чаях модификации данного вещества, не являющиеся
наиболее устойчивыми в рассматриваемых условиях,
могут сохраняться долгое или практически неограни-
ченнее время, сохраняя, однако, способность само-
произвольно переходить в более устойчивую форму.
Так, белое олово может сохраняться, не переходя в
серое, и при темп-pax ниже 13°, но в этих условиях
оно является метастабильным в отношении такого пе-
рехода. Хотя устойчивой кристаллич. формой SiO2
при комнатных темп-pax является кварц, а тридимит
и кристобалит (устойчивые при высоких темп-рах)
отвечают при комнатных темп-рах М. с. SiO2, однако
практически они могут сохраняться в этих условиях
неограниченное время.
173
МЕТАХРОМАТИЧЕСКОЕ ОКРАШИВАНИЕ —МЕТЕОРИТЫ
174
Применяя понятие М. с. в широком смысле, к нему
можно отнести и такие состояния, из к-рых система
может перейти в более устойчивое состояние в резуль-
тате химич. реакции. Так, система из металлич. же-
леза и свободного кислорода является в обычных
условиях менее устойчивой, чем соответствующие
окислы железа. Поэтому можно рассматривать такую
систему находящейся в М. с. по отношению к переходу
в окислы. Торможение такого перехода, вызываемое
факторами кинетич. характера, дает возможность
пользоваться металлич. железом в обычных условиях.
Но именно способность к самопроизвольному переходу
его из М. с. в более устойчивое лежит в основе про-
цессов ржавления железа, его пирофорных свойств
и пр.
Лит-' Киреев В. А., Курс физической химии, 2 изд.,
М.. 1956. В. А. Киреев.
МЕТАХРОМАТИЧЕСКОЕ ОКРАШИВАНИЕ —
изменение цвета р-ра красителей под влиянием
некоторых химич. веществ. Вещества, вызывающие
М. о., получили название хромотропов. В боль-
шинстве случаев они являются линейными полианио-
нами с высоким мол. вес. или низкомолекулярными
веществами, способными ассоциироваться в крупные
агрегаты. Наиболее распространенными хромотропа-
ми среди веществ животного происхождения являются
кислые мукополисахариды (гепарин, хондроитинсуль-
фаты, гиалуроновая к-та), нуклеиновые к-ты, некото-
рые анионные липиды; из веществ растительного
происхождения к хромотропам относится агар, из
неорганических и синтетических веществ — силикаты,
полифосфаты, карбоксиметилцеллюлоза, полиакрила-
ты и др.
Красители, дающие М. о., являются катионами.
Для гистохимии, целей наиболее широко используют-
ся тиазиновые красители (толуидиновый синий, ажур
А, тионин и др.). Особенность водных р-ров таких кра-
сителей состоит в том, что
Ламберта — Бэра: при
50000
30000
10000
500 580 660 ММН
Поглощение света растворами
толуидинового синего (по Ми-
хаэлису и Гранику):
-------4,63*10“° М в воде
•— ---- 2,31-10“* М в воде
— • — 2,31-10“* М в 1%-ном
агаре
они не подчиняются закону
увеличении концентрации
красителя пик абсорб-
ции в видимой части
спектра смещается в сто-
рону более короткой
длины волны (см. рису-
нок). Отклонение от за-
кона Ламберта — Бэра,
по-видимому, лучше все-
го объясняется тем, что
эти красители в р-ре
могут существовать в
виде моно-, ди- и поли-
мерных форм. Увеличе-
ние концентрации кра-
сителя приводит к сдви-
гу равновесия и образо-
ванию большого количе-
ства полимерных форм,
что обусловливает изме-
нение спектра абсорбции и соответственно — цвета
раствора: от синего к фиолетовому и красному.
Механизм М. о. до сих пор окончательно не выяс-
нен. Согласно гипотезе Михаэлиса и Граника, в при-
сутствии хромотропа происходит образование поли-
мерных форм красителя, пик абсорбции смещается
в сторону коротких длин волн и эти соединения окра-
шиваются в красно-фиолетовый (р- м е т а х р о м а-
з и я) или красный (у-метахромазия) цвет (см. рису-
нок, в качестве хромотропа использован агар). Веще-
ства, с к-рыми хромотроп не взаимодействует, приоб-
ретают синий цвет, характерный для мономерных
•форм. Это позволяет легко идентифицировать на сре-
де структуры, образующие метахроматическое окра-
шивание.
В гистохимии М. о. получило значительное рас-
пространение как один из методов выявления кислых
мукополисахаридов. Наиболее яркое и стойкое М. о.
вызывают сульфатированные кислые мукополисаха-
риды (хондроитинсерные к-ты и гепарин); гиалуроно-
вая к-та дает более слабую окраску. Для усиления
М. о. гиалуроновой к-ты ее превращают в эфир серной
к-ты. При практич. использовании М. о. в гистохи-
мии важно учитывать относительную неспецифичность
М. о. и ставить соответствующие контрольные опыты.
Идентификация веществ, дающих М. о., обычно про-
изводится путем предварительной обработки парал-
лельных срезов ферментами, разрушающими хромо-
тропы. Получаемые с помощью М. о. результаты не
постоянны, поэтому следует пользоваться идентич-
ными препаратами красителей и стандартизировать
все гпстохпмич. процедуры.
Лит,: Виноградов В. В., Фукс Б. Б., Архив па-
тологии, 1961, 23, № 2, 74; Пирс Э., Гистохимия теоретиче-
ская и прикладная, пер. с англ., М., 1956; Kramer Н.,
Win drum G. М., J. Hlstocliem. and Cytochem., 1954, 2,
№ 3, 196; Schubert M., Ha merman 1)., J. Histo-
chem. and Cytochem., 1956, 4, № 2, 158; Michaelis L.,
G r a n i c k S., J. Amer. Chem. Soc., 1945, 67, № 7, 1212.
Ю. В. Наточин.
МЕТАЦИН (иодметилат Д-диметиламиноэтилового
эфира бензиловой к-ты)
(C6H6)2C(OH)COOCH2CH2S(CH3)3J“
мол. в. 441,32 — бесцветные кристаллы, т. пл. 193—
195°; почти нерастворим в эфире, этилацетате, бен-
золе, ацетоне и хлороформе, растворимость в воде
(35—40°) 1 : 100. Количественно М. определяют арген-
тометрически (по Фольгарду). Получают М. этери-
фикацией бензиловой кислоты Д-диметиламиноэтил-
хлоридом в толуоле с последующим взаимодействием
образовавшегося Д-диметиламиноэтилового эфира бен-
зиловой к-ты с иодистым метилом в этилацетате. М.
применяют в качестве холпнолитич. и спазмолитич.
средства при бронхиальной астме и спазмах органов
брюшной полости. Р- Г. Глушков.
МЕТГЕМОГЛОБИН — см. Гемоглобины.
МЕТЕОРИТЫ — железные и каменистые тела,
падающие на земную поверхность и представляющие
собой остатки метеорных тел, вторгающихся из меж-
планетного пространства в атмосферную оболочку
Земли. М. делятся на три класса: железные,
железокаменные и каменные. Железо-
каменные подразделяются на палласиты (в к-рых же-
лезо образует губчатый каркас, пустоты же каркаса
заполнены оливином) и мезосидериты (в к-рых железо
рассеяно в виде несвязанных между собой включе-
ний). Каменные подразделяются на хондриты (содер-
жащие округлые образования, наз. хондрами, диамет-
ром в среднем 1 мм) и ахондриты (не содержащие
хондр). Однако существуют переходные типы и по-
этому можно проследить непрерывный ряд от «чисто
железных» до «чисто каменных» М. В настоящее время
общепринятой является классификация метеоритов
Прайора. В основу ее положено разделение М. по ми-
неральному составу, к-рый, конечно, связан и с хи-
мич. составом; с тем и другим, в свою очередь, свя-
зана и структура М.
Главнейшими признаками М. являются: кора
плавления и регмаглипты. Кора пред-
ставляет собой наружный переплавленный слой,
к-рый покрывает М. со всех сторон и имеет толщину
не свыше миллиметра. Регмаглиптами наз. характер-
ные ямки на поверхностях М., образующиеся в ре-
зультате «сверлящего» действия атмосферы на М. во
время движения его с космич. скоростью. В М. были
найдены практически все известные на Земле хими-
ческие элементы, хотя большинство их содержит-
ся в М. в ничтожных количествах, как это видно из
табл. 1.
175
МЕТЕОРИТЫ
176
Таблица 1. Средний состав метеоритного вещества
(по Месону)
Поряд- ковый номер и символ Число атомов на 10* атомов Si Поряд- ковый номер и символ Число атомов на 104 атомов Si
в метеори- тах в Ь’осмо се в метеори- тах в Кос- мосе
1 н 4.0-1 о8 44 Ru 0,016 0,015
2 Не 3.1-10’ 45 Rb 0,003 0,002
3 Li 0,47 1,0 46 Pd 0,016? 0,007
4 Be 0,18? 0,20 4 7 Ag 0,0015 0,003
5 В 0,30 0,24 48 Cd 0,0 07? 0,009
6 с 35000 49 In 0.00001 0,001
7 N 66000 50 Sn 0,014 0,013
8 О 34200 215000 51 Sb 0,0014 0,002
9 F 2,60? 16 52 Те 0,013 0,047
10 Ne 86000 53 J 0,0005 0 ,008
И Na 490 440 54 Xe — 0,040
12 Mg 9450 9100 55 Cs 0,0012 0,005
13 Al 680 950 56 Ba 0,041 0,037
14 Si 10000 10000 57 La 0,0040 0,020
15 P 69,50 100 58 Ce 0,0062 0,023
16 s 1070 3750 59 Pr 0,0015 0,004
17 Cl 5? 90 60 Nd 0,0074 0,014
18 Ar 1500 61 Pm —
19 К 43 32 6 2 Sin 0,0025 0,007
20 Ca 576 490 63 Eu 0,00097 0,002
21 Sc 0,33 0,28 64 Gd 0,0036 0,007
22 Ti 28 24 6 5 Tb 0,00056 0,001
23 V 2,1 2,2 66 Dy 0,0039 0,006
24 Cr 96 78 67 Ho 0,00078 0,001
25 Mn 60 69 68 Er 0,0021 0,003
26 Fe 8490 6000 6 9 Tu 0,00039 0,0003
27 Co 28 18 70 Yb 0,0019 0,002
28 Ni 474 270 71 Lu 0,00036 0,0005
29 Cu 2,6 2,1 72 Hf 0,014 0,004
3 0 Zn 1 ,25 4,9 73 Ta 0,0019 0 ,0007
31 Ga 0,12 0,11 74 W 0,0013 0,005
32 Ge 0,23 0,51 75 Re 0,0008 0,001
3 3 AS 0,04 0,04 76 OS 0,0100 0,010
34 Se 0,19 0,68 77 It 0,0043 0,008
35 Br 0,21? 0,13 78 Pt 0,043? 0,016
36 Kr — 0,51 79 Au 0,0025 0,001
37 Rb 0,06 0,0 7 80 Hg 0,008? 0,003
38 Sr 0,21 0,19 81 Tl 0,000003? 0 ,001
39 Y .0,07 0,09 82 Pb 0,0012 0,005
40 Zr 0,75 0,55 83 Bi 0,00002? 0,001
41 Nb 0,009 0,01 90 Th 0,00028 0,0004
42 MO 0,028 0,02 92 U 0,00097 0,0001
43 Tc — —
В табл. 1 представлены данные о среднем составе
всего вещества М. (по Месону), соотношения отдель-
ных фаз в котором приняты следующие: никелистое
железо 19,6, каменистая часть 80,4. Для сравнения
в таблице приведены также данные о распростра-
ненности химических элементов в Космосе (по Зюсу
и Юри), опубликованные в 1956 и
основанные на составе М., а также
Таблица 3. Средний химический состав железных метеоритов разных типов (в вес. %>
на составе солнечного и звездно- го вещества. Поэтому, естественно, указанная распространенность зна- чительного числа элементов как в М., так и вообще в Космосе близ- ка. Тем не менее можно видеть то существенное различие, что веще- ство М., равным образом и Земли (см. Геохимия), а также планет зем- ной группы отличается от солнеч- ного и звездного вещества мень- шим содержанием летучих химич. элементов, таких, как Н, Не, С, N, Аг, Хе и нек-рых других, что объ- ясняется условиями образования планетной системы. Наиболее рас- Элементы Типы железных метеоритов
гексаэдри- ты о ктаэдриты атакситы
весьма грубо- струк- турные грубо- струк- турные средне- струк- турные тонко- струк- турные весьма тонко- струк- турные богатые никелем бедные нике- лем
Fe Ni Со Си Р S С Прочие 93.59 5,57 0,66 0,35 0,29 0,06 0,19 0,19 92,33 6,54 0,50 0,01 0,16 0,02 0,23 0,27 91 ,22 7,39 0,54 0,18 0,18 0,08 0,21 0,54 90,67 8,22 0,59 0,03 0,18 0,09 0,08 0,30 90,53 9.00 0,57 0,05 0,17 0,08 0,61 0,18 86,75 11,65 0 ,61 0,11 0,24 0,63 0,01 0,45 79.63 18,85 1 ,01 0,05 0,12 0,08 0,10 0,19 91 . 07 6,88 0,54 0,19 0 , 18 0,06 0,08 0,17
пространенными в М. химич. эле-
ментами являются: Fe, Ni, S, Mg, Si, Al, Ca, Na, О.
В табл. 2 приведен средний химич. состав М. разных
классов и групп по данным М. И. Дьяконовой и
А. А. Явнеля.
Состав железных М. приведен только для никели-
стого железа, без учета различных минеральных вклю-
Таблица 2. Средний химический состав метеоритов разных классов и групп (в вес. %)
Элементы и соедине- ния Железные Железокаменные Каменные
никелис- тое желе- зо палла- ситы мезоспде рпты хонд- риты ахондриты
бедные Са бога- тые Са
Fe 90,5 48,0 43,0 11,2 2,3 0,9
Ni 8,7 5,1 4,5 1.4 0,2 —
Со 0,56 0,3 0,2 0,07 — —
Си 0,02 0,03 — — — —
р 0,20 0,05 0,07 — —
с 0,08 — 0,22 —
FeS — 6,3 1.8 0,60
SiO2 — 18 24 37.0 49 4 9
MgO — 21 13,5 23,6 30,5 1 1
FeO — 6,5 7,3 13,5 12 17
A12O. — 0,12 3,7 2,4 1 ,2 11
CaO — __ 3 , 7 1 , 9 1 ,4 10
Na2O — 0,03 0,2 0,9 0,25 0,5
K2O 0,006 0,11 0,05 0,09
Cr2O3 — — 0,34 0,8 0,4
MnO — 0,1 — 0,23 0,3 0,4
T1O2 —- — . — 0,12 0,1 0.4
P,O5 — 0,23 0,1 0.1
H,0 — — — 1 ,5 —' —
ченип, встречающихся в М. Содержание Н2О в угли-
стых хондритах может достигать 20,1% (метеорит
Orgueil). Насколько может колебаться химич. состав
отдельных М. или даже типов М., можно видеть из
табл. 3, в к-рой приведен химич. состав отдельных
типов железных М.
Никель является характерной и обязательной со-
ставной частью метеоритного железа, наз. никели-
стым, причем его содержание колеблется от 4,0% до
18,9%. Однако известно пять богатых никелем ата-
кситов, в которых содержание никеля значительно
больше и достигает: в метеорите Freda 23,49%, Lime
Creek 29,99%, San Cristobal 25,60%, Santa Catharina
33,97%, Oktibbeha Comity 59,69%.
Согласно прежним определениям среднее содер-
жание в М. драгоценных металлов и редких элементов
на 1 т вещества М. в граммах составляет: RulO,
Rh5, PdlO, Ag5, Os3, Ir5, Pt20, Au5. Установлено,
что содержание таких химич. элементов, как Ga, Ge,
Pd и др., присутствующих в М. в ничтожных коли-
чествах, тесно связано как с содержанием в них дру-
гих элементов, так соответственно и со структурой.
Так, оказалось, что чем выше содержание Ni в М., тем
меньше содержится в нем Ga и т. д.
В табл. 4 приведен средний минеральный состав
основных классов и групп М., вычисленный Дэли по
данным химич. анализов.
Каких-либо следов присутствия в М. органич. ве-
ществ биогенного происхождения не установлено.
Наиболее резкие отличия М. от объектов земного
177
МЕТЕОРИТЫ
178
Таблппа 4. Средний минеральный состав метеоритов разных классов и групп (в вес. %)
Минералы Желез- ные Палласиты | Мезосидериты Хондриты Ахондриты
всего метеорита камени- стой части всего метеорита камени- стой части всего метеорита камени- стой части всего метеорита камени- стой части
Никелистое железо .... 98,34 50,0 45,0 __ 10,58 1 , 57
Оливин — 48,0 96,0 1 ,5 2,7 42.31 47,34 12.82 13,03
Пироксены —— — — 30,6 55.6 28,01 32,40 62,25 63,24
Анортит — — — 16,4 30,0 3,34 3,73 13,23 13,44
Альбит — — 7,37 8,20 5,83 5,92
Ортоклаз — — — — 1,11 1,24 1 .69 1 , 72
Троилит 0,12 0.3 0,6 3,1 5,6 5,01 5 , 58 1 ,53 1 ,55
Шрейберзит ........ 1,1? 0,2 0,4 2,6 4,7 — — 0,40 0,41
Хромит —— — — 0,8 1 .4 0,70 0 , 78 0,68 0.69
Когенит 0,42 1 ,5 3,0 — —- —
Апатит — — — - 0,67 0,73 — —
происхождения проявляются в их внутренней струк-
туре. Так, у большинства железных М. (октаэдритов)
при травлении полированных поверхностей распила
выявляется сложный рисунок, называемый видман-
штеттеновыми фигурами. Он состоит из пересекаю-
щихся между собой полосок-балок камасита, окай-
мленных узкими блестящими лентами тэнита. Же-
лезные М. более редкого типа (гексаэдриты) представ-
ляют собой монокристаллы кубич. системы, у к-рых
при травлении выявляются тонкие параллельные
между собой линии, наз. неймановыми; могут быть
видны линии до 12 систем. Подавляющее большин-
ство каменных М. относится к хондритам (см. выше),
отличающимся своеобразной структурой, чуждой
земным горным породам. Каменные метеориты ред-
кого типа (эвкриты) по составу и структуре более все-
го похожи на изверженные земные горные породы
(габбро).
По современным представлениям, М. рассматри-
ваются в качестве обломков астероидов — малых пла-
нет, движущихся вокруг Солнца, гл. обр. между ор-
битами Марса и Юпитера. Огромное количество мел-
ких астероидов, диаметром много меньше километра,
составляет переход астероидов к М. Вследствие со-
ударений, происходящих между мелкими астероидами
при пх движении, идет непрерывный процесс, их дроб-
ления на все более мелкие части, пополняющие ме-
теорные тела в межпланетном пространстве. Возмож-
ны случаи падения на Землю комет, единственным за-
регистрированным примером чего является, по-видп-
мому, т. н. Тунгусский метеорит, изучение падения
к-рого еще не окончено. Нек-рые исследователи от-
носят к М. и тектиты, своеобразные стеклянные
тела, к-рые находят в разных частях земной поверх-
ности.
М. являются единственными образцами твердого
вещества внеземного происхождения, доступными для
непосредственного изучения. Разнообразные иссле-
дования М. с помощью современных методов и технич.
средств имеют чрезвычайно важное научное и практич.
значение как для изучения условий происхождения и
дальнейшей истории планетной системы, так и для
изучения состава и строения внутренних частей Земли
и происходящих в них процессов. Последнее понятно,
если учесть, что составы каменных М. (хондритов) и
мантии Земли тождественны. Таким образом, каменные
М. могут служить образчиками первичного вещества,
из к-рого образовалась Земля и другие планеты зем-
ной группы. Поэтому в последнее время изучение
М. приобретает все большее развитие. Так, напр.,
определение в М. радиоактивных химич. элементов и
продуктов их распада позволило определить возраст
вещества, слагающего М., оказавшийся в пределах
4—4,5 млрд. лет. Для определения возраста М. при-
меняют свинцовый изотопный, рубидиево-стронцие-
вый, рений-осмиевый, калий-аргоновый и урано-ге-
лиевып методы (см. Возраст геологический абсолют-
ный).
Измерения изотопного состава С, О, Si, Cl, Fe, Ni,
Co, К, Cu, S и др. химич. элементов М. показали
тождество с изотопным составом тех же элементов
земного происхождения. Вместе с тем было установле-
но, что отношение изотопов урана U235 : U238, а также-
калия К39: К40: К41 идентично с изотопными отноше-
ниями урана и калия земного происхождения. Эти
результаты говорят об одновременном образовании
атомных ядер Земли и М. и служат одним из важных
доказательств единства вещества М. и планет солнеч-
ной системы.
Однако, находясь в межпланетном пространстве
в виде самостоятельных космич. тел (с момента отде-
ления — в результате дробления родоначальных
тел — до падения на Землю), М. подвергаются воз-
действию космич. лучей. В результате происходящих
при этом реакций между частицами больших энергий
и элементами, содержащимися в М., образуются ста-
бильные и нестабильные космогенные изотопы, в том
числе Н3, Не3, Не4, Li6, Be10, Ne20, Ne21, Ne22, Al26,
Ar36, Ar38, Ar39, К 40 и другие, вновь открываемые при
более углубленных исследованиях М. По содержанию
космогенных изотопов определены т. н. космич.
возрасты М., т. е. промежутки времени самостоятель-
ного существования М., изменяющиеся в очень широ-
ких пределах, от немногих миллионов до сотен мил-
лионов и миллиардов лет. Измерения космогенных изо-
топов пока в немногих М. позволили также опреде-
лить и земные возрасты М., т. е. промежутки времени
с момента падения М. на Землю, оказавшиеся от сотен
до десятков тысяч лет.
Содержание в М. космогенных изотопов начинают
использовать для изучения вариации интенсивности
космич. лучей в пространстве и во времени, а измере-
ние содержания их в М. с глубиной (определение-
глубинного эффекта) — для определения первичных
(до падения на Землю) масс М. железного класса
(для каменных М. методы еще не разработаны). С изу-
чением М. связано решение и многих других задач,
касающихся Земли и Космоса (см. также Космо-
химия).
Лит.: Кринов Е. Л., Основы метеоритики, М., 1955;
Заварицкий А. Н., Кваша Л. Г., Метеориты СССР,
М., 1952; Кринов Е. Л., Тунгусский метеорит, М., 1949;
Фесенков В. Г., Метеорная материя в междупланетном
пространстве, М.—Л., 1947; Метеоритика. Сб. статей, под ред.
В. Г. Фесенкова, вып. 1—23, М.—Л., 1941—63; Вииогра-
д о в А. П., Происхождение оболочек Земли, «Вести. АН
СССР», 1962, № 9, 16—29; Mason В., Meteorites, N. Y.,
1962; Researches on meteorites, ed. by С. B. Moore, N. Y.,
1962; Heide F., Kleine Meteoritenkunde, B., 1957; Wood
J. A., Meteorites: physics and chemistry. The solar system, ed.
G. P. Kuiper, B. Middlehurst, v. 4, [N. Y.], 1963 ch. 12, P r 1-
o r G. T„ Catalogue of meteorites, ed. M. H. Hey, 2 ed., L., 1953.
E. Л. Кринов.
179
МЕТИЛАКРИЛАТ -N-МЕТИЛ АНИЛИН
180
. МЕТИЛАКРИЛАТ (метиловый эфир акриловой ки-
слоты) СН2=СНСООСН3, мол. в. 86,09 — бесцвет-
ная прозрачная жидкость; т. кип. 80,27760 мм
•(с разл.); 61,87400 мм; 28,07100 мм; —13,5710 .ii.it;
rf” 0,9564; Пд, 1,4040; теплота полимеризации 18,7 —
120,2 ккал/моль.
По химическим свойствам и способам получения
М. подобен метилметакрилату; химич. свойства его
обусловлены наличием сложноэфирной группировки
и кратной связи, находящейся в сопряжении с карбо-
ксильной группой. М. является диенофилом в диено-
вом синтезе. М.— мономер, полимеризующийся под
действием свободных радикалов (подробнее см. По-
лиакрилаты). М. используется гл. обр. в виде сополи-
мера, напр. со стиролом.
Впервые М. получен в 1873 при дебромировании
метилового эфира а, p-дибромпропионовой к-ты.
Для промышленного произ-ва М. наибольшее
значение имеют три способа: 1) Из нитрила акриловой
к-ты (акрилонитрила), который гидролизуют разб.
H2SO4 до акриловой к-ты; последнюю, без выделе-
ния, этерифицируют метанолом; 2) из этиленциангид-
рина, одностадийной обработкой метанолом с дегид-
ратацией его до акронитрила и превращение акронит-
рила в М.:
160“
HOCH2CH2CN + CH3OH-----л СН—СН-СООСН.
H2SOt
3) прямое карбонилирование ацетилена в присутствии
метанола:
СН=СН + СО + СН3ОН —> СН2 = СНСООСН3
Разработаны два варианта процесса: при атмосферном
давлении в водном р-ре соляной или уксусной к-ты
с использованием тетракарбонила никеля в качестве
источника окиси углерода:
4CH=CH + Ni (CO)t + 4CH3OH + 2HCl —
NiCl3 + H3 + 4СН2 = СН - СООСН3
или из ацетилена и СО (1 : 1); ацетилен вводят в р-р
катализатора (напр., NiBrs) в метиловом спирте при
150—180° и 30 ат;
СН^СН + СО + СН,ОН —>. сн, = снсоосн
При получении, хранении и транспортировке М. ста-
билизируют солями меди и др. М. обладает наркотич.
и общеядовитым действием, пары его раздражают сли-
зистые оболочки носа и глаз, а при концентрации
25 мг/л воздуха происходит сильное выделение мок-
роты.
Лит. см. при ст. Метилметакрилат. В. Н. Александров.
МЕТИЛАЛЛЕН (бутадиен-1,2) СН2=С=СН—СН3,
мол. в. 54,09 — первый гомолог ряда алленовых угле-
водородов; бесцветная жидкость, т. кип. 17—19°;
d°i 0,676; nD 1,4205. М. можно получить дехлорирова-
нием тетрахлорбутана:
СН2С1 - СС12 - СНС1 - СН3 —> сн, = с = сн - сн3
или синтезировать из кротилового (кротонилового)
спирта СН3—СН=СН—СН2ОН через бромиды. М.
образуется в качестве побочного продукта прп по-
лучении дивинила (бутадиена) из этилового спирта
по методу С. В. Лебедева; он является последним
звеном в цепи превращений, в результате изомериза-
ции к-рого и образуется дивинил. М. можно изомери-
зовать в присутствии флоридина при 205—330°; при
низких темп-pax образуется ок. 3% этилацетилена
СН = С—С2Н5, а при высоких возникает и дивинил
СН2=СН—СН=СН2 (ок. 20%). Под влиянием
флоридина происходит частичная полимеризация М. с
образованием жидких продуктов. Аналитич. опреде-
ление основано на образовании жидких тетрабромидов
М., в отличие от тетрабромидов этилацетилена и бу-
тадиена-1,3, получающихся в кристаллич. виде.
Лит.: Слободин Я. М., Ж. общ. хим., 1935, 5, вып.
1, 48. А. К. Никитин.
МЕТИЛАМИН — СНзМН2, мол. в. 31,06 — газ с
резким тошнотворным запахом, чувствующимся в кон-
центрациях 0,005%; т. пл.— 93,46°; т. кип. — 6,32°;
Й4~10’8 0,699; 1крит. 156,9°; рКриг. 73,6 ат; теплота об-
разования (при p = const) 25§,3 кал/молъ (газ), 258,1
кал/моль (жидк.); теплота парообразования 6169
ккал/моль; легко горит и образует взрывоопасные газо-
воздушные смеси в интервале 4,95—20,75%; ядовит
(угнетение функции дыхания); смешивается со спир-
том, эфиром, ацетоном, бензолом, хлороформом; рас-
творимость в воде 42,5% (40°, 1 ат); 40%-ный вод-
ный р-р имеет т. пл.— 38°, rfj4 0,905. М.—сильное
основание, А=4,25-10-4 (25°). М.—типичный предста-
витель первичных аминов: при взаимодействии с ан-
гидридами, галогенангидридами и эфирами к-т об-
разует метиламиды к-т (1); алкилируется спиртами,
алкилгалогенидами и сложными эфирами минеральных
к-т с образованием соответствующих вторичных и
третичных аминов (2); окисью этилена превращается
в метилдиэтаноламин (3); при обработке М. азотистой
к-той количественно выделяется элементарный азот
(4); при действии сероуглерода и соли тяжелого ме-
талла образуется метплгорчичное масло (5); при дей-
ствии хлороформа и щелочи — метилизонитрил (6):
2СН3 - ХН2 + Ас - Hal — + Ас-НН- СН3 + [CH3NH3]Hal (1)
RJ RJ RJ
CH3-NH,—► СН3- NH- R—«-CHj-NR.—> [CH3-NR3]J
<2)
CH3-NH2 + 2СН2-СН2—> СН3-N(CH.CH.OH).
\ / (3)
О
CH3-NH3 + HNOt—>СН3ОН + Н,0 + N2 (4)
CH3-NH2 + CS2 + HgCl—*CH3 —N —C = S + HgS + 2HC1 (5)
CH3-NH2 + CHC13+3KOH—>CH3-N = C + 3KC1 + 3H2O (6)
M. легко образует: гидрат CHaNH2.3H2O, т. пл. —35,8°;
хлоргидрат, т. пл. 225—226° (из этанола); бромгидрат, т. пл.
250—251°; пикрат, т. пл. 215° (из этилацетата); 2,4-динитрофе-
нил-М-иетпламмОнпйхлорпд, т. пл. 178°. Соли типа хлоропла-
тината (CH3NH3)3H3PtCl. и хлораурата CHaNH2-HAuCl,
нерастворимы в воде.
Получают М. нагреванием смеси формалина с хло-
ристым аммонием:
2СН3О + NH.C1 —> CH3NH, HC1 + НСООН
а также восстановлением HCN и алкилроданидов, ал-
килированием аммиака и др. М.; выпускается про-
мышленностью в виде хлоргидрата или водных р-ров.
М. содержится в нек-рых растениях и селедочном
рассоле.
М. широко применяют при приготовлении фарма-
цевтич. препаратов (антиспазматических, симпатоми-
метических, диуретиков и др.), алкалоидов группы
тропана, красителей антрахинонового ряда, фото-
реактивов аналогов метола (продукта взаимодействия
М. с гидрохиноном), поверхностно-активных веществ
на основе N-метилтаурина, продукта взаимодействия
М. с изетионовой к-той (2-оксиэтансульфоновая к-та),
N-метиланилина (взаимодействием с хлорбензолом),
метилдиэтаноламина (и исходное сырье для получения
канцеролитич. препаратов и заменителей морфина).
М. предложен также в качестве селективного абсор-
бента кислых газов.
Лит.: Kirk, V, 9, N. Y., 1952, р. 62. Г. А. Сокольский.
N-МЕТИЛАНИЛИН (метилфениламин) CeH5NHCH3,
мол. в. 107,15 — бесцветная жидкость, окрашиваю-
щаяся на воздухе в желтый и далее в коричневый
цвет; т. пл. —57°; т. кип. 195,7°/760 мм, 95°/25 мм,
79,2°/10 мм; d.™ 0,9868; «д 1,5714; хорошо растворим
в спирте, эфире, бензоле, хлороформе, ацетоне; пло-
хо — в воде. М., подобно анилину, токсичен. М.—
181
2-МЕТИЛ АНТРАХИНОН — 2-МЕТИЛ-5-ВИНИЛПИРИДИН
182
органич. основание более сильное, чем анилин, и менее
сильное, нежели алифатич. амины, константа диссоциа-
ции ЛГ=2,5-10 —10 (18°). М.— типичный представитель
вторичных аминов: алкилируется алкилгалогенидамп
и сложными эфирами минеральных к-т (1); с ангид-
ридами и галогенангидрпдами кислот образует соот-
ветствующие амиды к-т (N-метилацетанилпд, т. пл.
104°, N-метилтозиланилид, т. пл. 95°) (2); действием
азотистой к-ты превращается в N-нптрозометпланн-
лин (3):
RJ .СНз RI
C»HS-NH-CH3—>C»H5-N° —► [C»H3N(CH3)R2]J (1)
R
с«н5
2С»Н5 —NH —CH3 + Ac - Hal—>Ac-N^ +
CH3
+ [C«H3N(CH3)H2] Hal (2)
CH3
CSH3-NH-CH3 + HNO2—►C.Hs-n/ + H.O (3)
NO
При нагревании хлоргидрата M. до 250—300° проис-
ходит перегруппировка Гофмана и образуется соль
n-толуидина. М. легко образует: хлоргидрат, т. пл.
122° (из смеси эфир—хлороформ); бромгпдрат, т. пл.
99°; оксалат, т. пл. 113°; пикрат, т. пл. 144,5°; соли
типа хлороплатината (CeH3NHCH3)2 -Н2Р1С1б и хлор-
аурата CeH3NHCH3 -HAuCli, плохо растворимые в
воде (используется в качественном и количественном
анализе).
М. получают нагреванием смеси формалина и соли
анилина, алкилированием анилина метанольным р-ром
НС1 и последующим разделением смеси первичного,
вторичного и третичного аминов обработкой п-толуол-
сульфохлоридом или хлорсульфоновой к-той. М.—
исходное сырье при произ-ве .\,К'-диметилкарба-
нилида (стабилизатора пироксилиновых порохов —
продукта взаимодействия М. с фосгеном), тетрила
(нитрованием), красителей дифенил- и трифенилме-
танового рядов.
Лит.: К 1 г k, V. 1, N. Y., 1952, р. 713. 923. Г. А. Сокольский
2-МЕТИЛАНТРАХИНОН (p-метилантрахинон), мо-
лекулярный нес 222,25, С13Н10О2 — почти бесцветные
0 иглы; т. пл. 177—179°; растворяет-
|| _ ся в спирте, бензоле, ледяной
г<г|Г/х>|Чт-СН3 СНзСООН, в конц. H2SO4; легко
L JL JI J возгоняется.
II При перегонке с цинковой
0 пылью или при нагреваниис IIJ-+-Р
М. восстанавливаете я до 2-метилантрацена; восста-
новление металлами в серной к-те приводит гл. обр.
к З-метилантрону-9. При пропускании хлора через
расплав 2-М. образуется 2-дихлорметилантрахинон;
бромирование при 170° приводит к моно- или дибром-
метильным производным 2-М. В то же время хлориро-
вание 2-М. в присутствии SO2C12 или иода при 95—100°
дает 1-хлор-2-метилантрахинон; аналогично при бро-
мировании 2-М. в олеуме в присутствии SO2C12 полу-
чается 1-бром-2-метилантрахинон. Окисляется 2-М.
в зависимости от условий в антрахинон-2-карбоновую
кислоту или в антрахинон-2-альдегид. Нагревание
2-М. с КОН в спирте при 150—170° дает антрафла-
вон, ранее применявшийся в качестве кубового кра-
сителя.
В пром-сти 2-М, получают конденсацией фталевого
ангидрида с толуолом в присутствии А1С13, с после-
дующей циклизацией образующейся п-толуилбен-
зойной к-ты под действием конц H,SO4 или слабого
олеума. 2-М. и особенно его замещенные широко
применяют н произ-ве кубовых, кислотных и серни-
стых красителей. Следующий гомолог антрахинона —
2-этилантрахинон — применяют в произ-ве водорода
перекиси. В. А. Пучков.
МЕТИЛАЦЕТАТ (метиловый эфир уксусной кис-
лоты) СНзСООСН3, мол. в. 74,08 — бесцветная про-
зрачная горючая жидкость, со своеобразным запа-
хом; т. пл. —98,05°; т. кип. 56,32°; с/2’ 0,9390; п2£
1,3619; давление пара мм рт. ст.: 100 (9,4°); 200
(24,0°); 400 (40,0°); теплота испарения 7,268 ккал/моль;
теплоемкость (p=const) 37,19 кал/молъ-град (18—42°);
1крит. 233,70°; ркрит, 46,3 ат; ц 0,362 спуаз (25°);
поверхностное натяжение о 24,76 дин/см (25,70°),
т. воспл. —10° в закрытой чашке; с водой образует
азеотропную смесь (т. кип. 56,4°, 96,3—96,8 вес. %).
М. получают этерификацией уксусной к-ты
метанолом (катализатор — ионы водорода) или из
уксусного ангидрида и метанола. Применяют М. гл.
обр. как растворитель при приготовлении лаков,
пленок, клеев, ацетил- и нитроцеллюлозы, растворов
различных смол; М. входит в качестве составной ча-
сти во многие растворители, имеющие различные
торговые и фирменные названия. М. является слабым
наркотиком; предельно допустимая концентрация
0,1 .иг/.г воздуха. в. Н. Александров.
п-МЕТИЛАЦЕТОФЕНОН (метил-п-толилкетон) —
п-СН3СеН4СОСН3, мол. в. 134,18 — бесцветная или
бледно-желтая жидкость с цветочно-фруктовым запа-
хом, напоминающим запах ацетофенона, т. пл. 28°,
т. кип. 2277760 мм, 112,5711 мм; d™ 1,0051;
п р 1,5335. М. нерастворим в воде и ограниченно рас-
творим в спирте. По химич. свойствам М. подобен аце-
тофенону. М. синтезируют в пром-сти взаимодействием
толуола с уксусным ангидридом в присутствии А1С13.
Применяют М. для создания парфюмерных компози-
ций с цветочным запахом, предназначенных для мыл
И реже ДЛЯ духов И одеколонов. в. М. Дашунин.
2-МЕТИЛ-5-ВИНИЛПИРИДИН (5 -винпл-а-пико-
лин) CsH9N, мол. вес 119,17 — бесцветная жид-
кость, т. кип. 75°/15 мм; с/2°
0,9579; п” 1,5454; выше 163° ки- 2 Q J_CH
пит с разложением и быстро N 3
полимеризуется. За счет двойной связи в боковой
цепи М. вступает в реакции, характерные для оле-
финов; способен полимеризоваться и сополимеризо-
ваться, в частности с бутадиеном. Для предотвраще-
ния самопроизвольной полимеризации М. стабилизуют.
Для этого можно применять серу, пикриновую к-ту,
а-нитрозо-р-нафтол, хлоранил, метиленовый голу-
бой, метол и др. При добавлении 0,001% метола М.
сохраняется при комнатной темп-ре в течение не-
скольких недель без следов полимеризации, а н
присутствии 0,01% метола он не полимеризуется н
течение многих месяцев. В процессе выделения М.
из продуктов реакции в условиях повышенных темп-р
применяют для стабилизации добавление 0,1 %S.
В производственных условиях М. получают ката-
литич. дегидрированием при 575—625° и разбавле-
нии водяным паром (1 : 6—8) 2-метил-5-этилпири-
дпна (5-этил-а-пиколина):
СН3 - G3H3N - СН2СН3 —► СН3 - G3H3N - СН = СН2
Последний синтезируют в жидкой фазе взаимодей-
ствием NH3 и паральдегида, под давлением.
Кроме М., образуются метилэтилпиридин, а-пико-
лин, 3-этилпиридин, 2,5-лутидин, 3-винилпиридин,
аммиак и др. В качестве катализатора применяют
никель-фосфатный катализатор и др. Удовлетвори-
тельные результаты дают катализаторы, где основ-
ным компонентом являются окись цинка или окись
железа.
Конденсат, состоящий н основном из М. и метил-
этилпиридина ректифицируют на многоколонной си-
стеме под вакуумом (остат. давление 5 мм рт. ст.).
183
МЕТИЛГЛИОКСАЛЬ—я-МЕТИЛИЗОПРОПИЛБЕНЗОЛ
184
Трудность разделения определяется близостью темп-р
кипения основных компонентов (разность их состав-
ляет ок. 9°). Выделенный продукт содержит 98—
99% М. Для аналитич. определения М. применяют
бромид-броматный метод, а также фракционирован-
ное осаждение пикратов. Более точные результаты
дает метод ИК-спектроскоппи. М. применяют для
получения синтетич. каучуков и латексов сополиме-
ризацией его с бутадиеном, для синтеза ионообмен-
ных смол, а также в произ-ве химич. волокон.
Лит.: Фарберов М. И. [и др.], Ж. прикл. хим.,
1961, 34, № 3, 632. А. К. Никитин.
МЕТЙЛГЛИОКСАЛЬ (альдегид пировиноградной
к-ты, а,р~диоксопропан) СН3СОСНО, мол. в. 72,06 —•
простейший представитель а,р-кетоальдегидов али-
фатического ряда. Свежеприготовленный М.— желтая
резко пахнущая, вызывающая головную боль жид-
кость, бимолекулярна по определению мол. веса в
бромоформе и бензоле; очень гигроскопична; поли-
меризуется за неск. часов и затвердевает в хрупкую
смолоподобную массу, т. кип. 72°, при кипении об-
разует желтовато-зеленые пары, однако перегоняет-
ся только ок. 70%, остальное превращается в поли-
мер. В растворителях, не содержащих гидроксиль-
ных групп, М. растворяется с образованием окрашен-
ных в желтый цвет растворов, в гидроксилсодер-
жащих соединениях растворяется с разогреванием,
образуя бесцветные растворы. При встряхивании М.
растворяется в воде с обесцвечиванием и сильным
разогреванием.
Водные р-ры М. имеют слабокислую реакцию, мед-
ленно восстанавливают федингов раствор. Для М.
свойственны все характерные реакции карбонильных
соединений. При окислении Н2О2 в щелочи дает му-
равьиную и уксусную к-ты; разбавл. р-р щелочи ко-
личественно превращает М. в молочную к-ту; анало-
гично действует нагревание с водой до 100°. Полу-
чать М. удобнее всего гидролизом его монооксима —
изонитрозоацетона в присутствии H2SO4. Образо-
вание М. наблюдается в слабых аммиачных и слабых
содовых р-рах различных моносахаридов. Под влия-
нием М. аланин, а-аминоизомасляная к-та, лейцин
и а-аминофенилуксусная к-та переходят соответ-
ственно в ацетальдегид, ацетон, изовалериановый
альдегид и бензальдегид; цистин переходит в ацет-
альдегид; гуанидин, гликокол, креатин, аргинин,
лизин, креатинин, аденин, гуанин и гуанозин также
дезаминируются под влиянием М. В организме М.
является промежуточным продуктом распада угле-
водов; под влиянием фермента метилглиоксалазы
превращается в молочную к-ту.
М. дает цветные реакции с индолом и соляной
к-той, с карбазолом и серной к-топ. Определяют М.
в виде диоксима, лучше в виде никелевой соли, или
титрованием после перевода его в молочную к-ту под
действием щелочи. Бис-фенилгидразон М. имеет т. пл.
153 —154°. В. Н. Александров.
М ЕТИЛДИХЛОРА РСИ Н CH3AsC12, мол. вес
160,86— бесцветная тяжелая жидкость с резким
раздражающим запахом; т. пл.—59°, т. кип. 134,5°,
rf” 1,84, п'д5 1,5677. М. плохо растворяется в воде
и хорошо — в органич. растворителях; медленно гид-
ролизуется холодной и быстро — горячей водой с
образованием токсичного метиларсиноксида; легко
окисляется до нетоксичной метилмышьяковоп к-ты.
М. образует с H2S нерастворимый в воде метилар-
синсульфид CHsAsS; эта реакция используется
для качественного обнаружения М. Основным ме-
тодом получения М. является Майера реакция. По-
добно другим галогенарсинам, М. обладает кожно-
нарывным действием без скрытого периода, раздра-
жающим действием на верхние дыхательные пути и
общетоксич. действием. При попадании капель М.
на кожу (3—5 лг/с.и2) образуются пузыри. Средством
терапии поражений М. является димеркаптопропа-
нол и его производные.
Лит. см. при ст. Люизит. Р. Н. Стерлин.
МЕТИЛ ДИХЛ ОРФ ОСФИН — см. Хлоралкилфос-
фиты.
МЕТИЛЕНОВЫЙ СИНИЙ (метиленовая синь,
метиленовый голубой, тригидрат хлорида тетраме-
Н3С <- сн3-
нзс^СОО^снз-
СГ-ЗН2О
тилентионина) CleH18N3SCl-ЗН2О, мол. в. 373,91 —
темно-зеленые кристаллы с бронзовым блеском или
мелкий порошок темно-зеленого цвета (водные р-ры
имеют темно-синий цвет); легко растворим в горячих
спирте и воде, труднее — в холодных. М. с. раство-
ряется в конц. H2SO4, образуя желтовато-зеленый
р-р, цвет к-рого при разбавлении водой переходит
в голубой. Количественное определение М. с. осно-
вано на его взаимодействии с К2Сг2О7 и титровании
избытка последнего Na2S2O3 в присутствии KJ
в кислой среде.
М. с. получают след, образом:
nh2
(CHg)2N-{ y-NHa —-
*—2-S-SO3H
(CH3)2N
Na2S2O3
K2Cr2O7.ZnCI2
ZnCl2
HCl
M. c.
M. с. может быть получен также окислением диме-
тил-я-фенилендиамина в присутствии H2S.
М. с. применяют для крашения хлопка по тани-
новой протраве; плохо выбирается шерстью, хоро-
шо — шелком; светопрочность умеренная. М. с. ин-
тенсивно окрашивает нек-рые органы живого орга-
низма, в частности периферийную нервную систему;
поэтому его широко используют как витальное кра-
сящее вещество в микроскопии. В аналитич. практике
М. с. применяют при определении хлоратов, пер-
хлоратов, катионов ртути, олова, титана, а также
при анализах мочи, молока, крови; служит индика-
тором при иодометрич. титровании. М. с. широко при-
меняют как антисептик (в виде 1—3%-ного спирто-
вого р-ра) при ожогах, фолликулитах и др.; при ци-
ститах и уретритах применяются внутрь (для промы-
вания) в виде 0,02%-ных водных р-ров. Двойную
соль М. с. и AgNO3 используют как сильнодействую-
щее дезинфекционное средство. М. с., обладая оки-
слительно-восстановительными свойствами, может
в организме превращать гемоглобин в метгемогло-
бин и обратно; на этом основано применение М. с.
в качестве антидота при отравлении цианидами,
окисью углерода, сероводородом, а также нитрита-
ми, анилином и его производными.
Лит.: Коган И. М., Химин красителей, 3 изд., М., 1956;
Каррер П., Курс органической химии, пер. с нем., Л.,
1960; Машковский М. Д., Лекарственные средства,
4 изд., М., 1960, с. 576; Фармакопея СССР. 9 изд., М.,
19 я-МЕТИЛИЗОПРОПИЛБЕНЗОЛ (я-цимол^ с“н'14,
мол. в. 134,21 — бесцветная жидкость с запахом кам-
фары; т. пл.—68,07°; т. кип. 176,9°/760 мм;
ИО/ЮО.мл; с1г° 0,8571; п“ 1,4903; хорошо .1
растворяется в спирте, эфире, ацетоне, | 1
ароматич. и алициклич. углеводородах;
плохо растворим в воде (менее 0,01%); сн(Сн )
горит и образует взрывоопасные паро-воз- 3 2
душные смеси; т. всп. 59°; ядовит (в большей степени,
нежели бензол и толуол) — при ингаляции вызывает
острые и хронич. отравления кроветворных органов
185
МЕТИЛИНДОЛЫ—МЕТИЛИРОВАНИЕ
186
(костного мозга, селезенки). п-М.— типичное арома-
тич. соединение: легко алкилируется, хлорируется,
сульфируется и нитруется. При окислении превращает-
ся в «-метилацетофенон, «-СН3—С6Н4—СО—СН3,
n-толуиловую п-СНз—С6Н4СООН, «-изопропилбен-
зойную п-(СН3)2СН—С6Н4—СООН и терефталевую
н-СбН4(СООН)2 к-ты. При гидрировании превращается
в ментол. «-М. выделяют из сульфитного скипидара
(отходы произ-ва сульфитной целлюлозы из древе-
сины хвойных деревьев); получается при пиролизе
а-иинена (составной части первичного скипидара),
при нагревании камфары с P2OS. Содержится /г-М.
во многих эфирных маслах (напр., в тимиановом, эв-
калиптовом) и является структурным фрагментом
большинства ациклич. и моноциклич. терпенов. п-М.
используют для приготовления фармацевтпч. препара-
тов (как растворитель, наполнитель мазей, душистое
вещество, исходное сырье для синтеза), при произ-ве
терефталевой к-ты (окислением) и «-крезола (окисле-
нием до гидроперекиси «-М. и последующим ее разло-
жением серной к-той):
О2
СН:,—СВН,-СН(СН3), —>
H,SO,
—>СН3-СвН4—С (СН3)3-----► п-СН3--С,,Н,- ОН + (СН3)2СО
I
ООН
Лит.: Kirk, V. 7, N. Y., 1951, р. 612.
Г. А. Сокольский.
МЕТИЛИНДОЛЫ — гомологи индола (I), содержа-
щие метильную группу в пирольном или бензольном
ядрах индольного цикла. Известны все
---------лр) возможные изомеры М.: 1-метилиндол
< Jk 1 Вм») (N-метилиндол), 2-метилиндол (а~мети-
линдол, метилкето л), 3-метилиндол
Н । (р-метилиндол, скатол), а также 4-, 5-,
6- и 7-метилиндолы. М. растворимы в
обычных органич. растворителях. По химич. свой-
ствам М. аналогичны индолу. Наибольший интерес
представляют 1-, 2- и 3- метилиндолы, т. к. их широ-
ко применяют в синтетич. органич. химии. Для И.
характерна флуоресценция в УФ-свете; спектры по-
глощения в УФ-свете имеют след, значения:
7. ммк... шах, £ ........ 1-метилпндол 2-метилпндол 3-метплцндол
219 34500 283 5650 218 32600 272 6700 222 32000 281 5300
2-, 3-, 4- и 5- М. выделены из каменноугольной смолы.
I-Метилиндол, желтое масло, т. кип. 240—241°/720 мм,
113°/5 мм; d2° 1,0476; 1,5993; летуч с водяным паром, не-
растворим в воде. С реактивом Эрлиха (подкисленный
спиртовой р-р n-диметиламинобензальдегида) дает красное
окрашивание. 1-М. получают действием метилирующих аген-
тов на р-р Na-соли индола в жидком аммиаке:
I
Na
<СНэ)25О^
Свежепсрегнанный препарат не обладает неприятным запахом.
2-М етилиндол, бесцветные кристаллы, иглы (из воды)
или пластинки (из водного спирта); т. пл. 61°, т. кип. 271 —
272°; слабо растворим в горячей воде. С реактивом Эрлиха
2-М. дает красное окрашивание; обладает неприятным запахом,
усиливающимся при хранении. 2-М. может быть легко получен
из фенилгидразона ацетона (Фишера реакция) или из ацето-о-
толуидина (Маделунга реакция).
3-М етилиндол (скатол), бесцветные кристаллы —
пластинки (из лигроина); т. пл. 95°; т. кип. 265—266°; раство-
рим в горячей воде; обладает отвратительным запахом. С реак-
тивом Эрлиха дает фиолетово-голубое окрашивание. Скатол,
являясь продуктом распада триптофана под влиянием гни-
лостных бактерий кишечника, содержится в фекальных
массах, а также встречается в нек-рых видах растений. Полу-
чают 3-М. нагреванием индола с метилатом натрия и метанолом
до 100° в автоклаве:
Си ССТ"1
н н
Он может быть получен также действием йодистого метила на
индолнлмагнийгалогениды и реакцией Фишера из фенилгидра-
зона пропионового альдегида. В сильном разведении скатол
приобретает запах жасмина, применяется в парфюмерной
пром-сти как вещество, усиливающее запах душистых компо-
нентов.
Индолы, содержащие метильные группы в бензоль-
ном ядре, получают методами, аналогичными методам
получения самого индола.
Лит.: Дж ул иен П., Мейер Э., Принти Э.,
в кн.: Гетероциклические соединения, под ред. Р. Эльдерфилда,
пер. с англ., т. 3, М., 1954; Morton A. A., The chemistry
ol heterocyclic compounds, N. Y.—L., 1946; В e г t 1 G., D a
S e t t i m о A., S e g n i n i D., Gazz. chlm. Ital., 1961, 91,
6, 571. M. H. Преображенская.
МЕТИЛИРОВАНИЕ — замещение атомов водорода,
металлов или галогенов в органич. соединениях ме-
тильной группой; обычно под М., в более узком смыс-
ле, понимают введение в органич. соединения метиль-
ной группы к электроотрицательным атомам О, N, S
и др. М. является простейшим примером алкилирова-
ния. Известно М. как неорганич. соединений (солей
галогеноводородных к-т, NH3, РЬС12 и т. д.) с обра-
зованием соответственно СН3Х, CH3NH2, (СНз)4РЬ
ит. д., так и разнообразных органич. соединений.
М. осуществляется действием различных метили-
рующих агентов. Простейшим, самым доступным ме-
тилирующим агентом является метанол. Образование
сложных эфиров из кислот и СНзОН можно рассмат-
ривать как М. кислоты с введением СНз-группы к
атому О. Метанол, также как и диметиловый эфир,
применяют на произ-ве для N-метилирования, напр.
для получения моно- и диметиланилина из анилина.
Однако во многих случаях требуются более эффектив-
ные агенты М., в качестве к-рых часто применяют ме-
тиловые эфиры минеральных к-т, в том числе метил-
галогениды. Самым доступным нз таких метилирую-
щих агентов является метилсерная к-та CH3OSO2OH.
Однако применение метилсерной к-ты носит ограни-
ченный характер, т. к. она обладает сильными кислот-
ными свойствами, нерастворима в органич. раствори-
телях и представляет собой сравнительно слабый ме-
тилирующий агент. Гораздо более эффективен полный
эфир серной к-ты, диметилсулъфат (CH3O)2SO2,
к-рый по метилирующему действию не уступает менее
доступному CH3J. Диметилсульфат широко приме-
няется для М. многих органич. соединений не только
в лабораторной практике, но и в пром-сти (М. ксан-
тина в кофеин и теобромин, N-метилирование бен-
зилиденаминоантипирина в произ-ве анальгина и
др.). Нередко вместо диметилсульфата применяют эфи-
ры арилсульфокислот, напр. «-СН3СбН4ЗО2ОСН3,
к-рые нетоксичны и по своей эффективности не усту-
пают диметилсульфату. При О-метилировании мор-
фина в кодеин возникают затруднения, вызываемые
возможностью одновременного N-метилирования тре-
тичного атома N в морфине. Во избежание этого для
М. веществ типа морфина рекомендовано (В. М. Ро-
дионов) применение четвертичных аммониевых соеди-
нений типа C6HSN (СНз)3Х и, в частности, пользуются
[C6H5N(CH3)3] + [SO3C6H4CH3-«]-.
М. нередко достигается особым приемом, заключаю-
щимся в действии формальдегида и восстановитель-
ного агента, обычно муравьиной к-ты. В частности,
этот прием применяется в произ-ве пирамидона М.
аминоантипирина:
нсоон
rnh2 + СН2О —>RNHCH2OH-------^RNHCH, + со2 + н=о
RNHCH3 + СН2О-> RN (СН3)СН2ОН -> RN (СН3)2 + СО2 + Н2О
187
МЕТИЛКАУЧУК — МЕТИЛМЕТАКРИЛАТ
188
Своеобразным метилирующим агентом, в особен-
ности для О-метилирования, является диазометан
CH2N2, применяемый в виде эфирного р-ра. Введение
СНз-групп вместо галогенов в вещества типа РС1з
легко достигается применением гриньярова реактива
CHsMgX. Так, действием CH3MgJ на AsCls можно
получить (СНз)зАв (менее алкилированные производ-
ные получить т. о. трудно); из CHsMgJ и Si(OCH3)4
можно синтезировать (CHs^Si и т. д.
C-Метилирование обычно осуществляется с трудом.
Однако СНз-группу удается легко ввести вместо нат-
рия в натрийорганич. соединения, напр. в натрийма-
лоновый эфир:
NaCH (СООС2Н5)2 + СН3Х —>СН3СН (СООС2Н3)2 +NaX
Ароматич. углеводороды можно легко метилировать
действием СНзХ в присутствии А1С1з (Фриделя —
Крафтса реакция). С-метилирование наблюдается и
при действии CHsMgXна альдегиды или кетоны, напр.:
СН3СОСН3 + CH3MgJ —> (CH3)3COMgJ —> (СН3)3СОН
К особого рода агентам М. принадлежат перекись
ацетила и тетраацетат свинца, позволяющие осуще-
ствить в нек-рых случаях С-метилирование. Так, при
действии этих реагентов на 2-метилнафтохинон обра-
зуется 2,3-диметилнафтохинон-1,4. Реакция основана
на промежуточном образовании свободных метиль-
ных радикалов, напр.:
(СН3СОО), Pb —► СН,- + СО2 + СН3СОО- + Pb(OCOCH3)2
Исчерпывающее N-метилирование представляет
собой распространенный прием изучения природных
и синтетич. аминов. Для этого исследуемый амин пе-
реводят в четвертичное аммониевое соединение, по
продуктам термич. разложения к-рого (гофманский
распад) можно сделать выводы о строении исход-
ного амина. Межмолекулярный перенос СНз-груп-
пы (пе реметилирование) играет важную биохимич.
роль. Основным агентом М. в организме служит
одна из незаменимых а-аминокислот — метионин
CH3SCH2CH2CHNH2COOH. См. также Алкилирова-
ние. Я- -А - Комиссаров.
МЕТИЛКАУЧУК — полимер 2,3-диметилбута-
диена-1,3. Свое название М. получил в связи с нали-
чием в исходном мономере
Г СН3 СН3 1 лишней метильной группы
[—сгт. —с---г._Отт,—J п. по сравнению с изопреном—
структурной единицей кау-
чука натурального. М. производился в Германии
в период первой мировой войны: было выработано
всего 2350 т. М. имел очень низкое качество. Про-
цесс полимеризации продолжался неск. недель или
месяцев.
М. применялся для изготовления резиновых изде-
лий (преим. эбонита) и в произ-ве кабелей.
А. К. Никитин.
МЕТИЛМЕРКАПТАН (метантиол, меркаптометан)
CH3SH, мол. в. 48,10 — бесцветный газ с сильным
отвратительным запахом, т. пл.—123,0°; т. кип.
6,00°; rf4s0,8599; ограниченно растворим в воде: 23,3 г/л
(20°), хорошо растворим в спирте и эфире; теплота
сгорания 298,81 ккал/л4оль(р=соп81);образуеткристал-
логидрат. Важнейшие производные М.: меркаптид
ртути (CH3S)2Hg, т. пл. 175°, 2,4-динитрофенилметил-
сульфид, т. пл. 128°; 2,4-динитрофенилметилсульфон,
т. пл. 189°. М. находится в продуктах термич. кре-
кинга нефти; образуется при гидролизе кератина и
гниении белковых веществ. М. применяется в синтезе
метионина, для одорацип светильного газа (по запаху
можно обнаружить 2,1 -10“8л*г М. в 1 л воздуха).
МЕТИЛМЕРКАПТОФОС — см. Фосфорорганиче-
ские инсектициды.
МЕТИЛМЕТАКРИЛАТ (метиловый эфир метакри-
ловой к-ты, метиловый эфир а-метилакриловой кис-
лоты) СН2=С (СНз)—СООСНз, мол. в. 100,11 — бес-
цветная прозрачная жидкость; т. пл. —48°; т. кип.
—10°/5 мм, 1Г/20 мм, 47,0°/100 мм, 82,0°/400 ллц
101,0°/760 мм', d2^ 0,945; п2£ 1,4162; вязкость ц
0,6322 (20°) спуаз, т. воспл. 10°, теплота испарения
9,1 кка.г/молъ; уд. теплоемкость (кал/г-град): 0,450
± 0,002 (20°); 0,480 0,002 (50°); пределы взрывае-
мости смеси М. с воздухом 4,9—12,5 об.% (25°), рас-
творимость в воде 1,5 вес. % (30°).
Химпч. свойства М. обусловлены наличием сложно-
эфирной группировки и кратной связи, находящейся
в сопряжении с карбоксильной группой. Вода медлен-
но гидролизует М., кислоты и щелочи ускоряют
гидролиз. При нагревании М. с органич. к-тами (му-
равьиной и др.) в присутствии нелетучих минеральных
к-т (катализаторы) происходит переэтерификация с
образованием метакриловой к-ты (один из способов ее-
получения) и метилового эфира оргднич. к-ты. При
нагревании М. со спиртами (катализаторы сильные
к-ты — серная, арилсульфоновая, но лучше алкого-
ляты), происходит переэтерификация с образованием
эфира метакриловой к-ты и взятого в реакцию спирта;
процесс используют для получения нек-рых эфиров,
метакриловой к-ты.
М. присоединяет по двойной связи хлор, бромистый
водород (атом брома всегда становится в р-положе-
ние), хлорноватистую к-ту (гидроксил — обычно в
а-, а хлор в p-положение); при каталитич. гидрирова-
нии водород присоединяется количественно. В при-
сутствии сильных оснований (алкоголяты, феноляты,,
гидроокись трпметилбензиламмония — тритон В) М.
присоединяет по месту двойной связи многие соедине-
ния с подвижным атомом водорода, водород становит-
ся в a-положение к карбоксильной группе. Напр.,
к М. присоединяются меркаптаны и тиофенолы, тио-
уксусная к-та, этандитиол, алифатич. нитросоедине-
ния, аммиак, амины, амиды к-т, синильная к-та, ке-
токислоты и т. п. Эти реакции широко используют
в препаративных целях. М. является диенофильным
компонентом в диеновом синтезе. Из М. и диолефинов
образуются эфиры замещенных тетрагпдробензойных
к-т; из М. и ненасыщенных альдегидов —• 6-членные
кислородсодержащие гетероциклы, из М. и алифатич.
дпазосоединений — 5-членный пиразолиновый цикл;
из М. и тетрафторэтплена — эфир тетрафторбутанкар-
боновоп к-ты. И. вступает в реакцию с солями диа-
зония — Меервейна реакция', с SnC14 образует устой-
чивый окрашенный комплекс, имеющий состав
SnCl4-2CH2= С(СН3)СООСН3. М,—мономер, полиме-
ризующийся под влиянием свободных радикалов. Ос-
новным потребителем М. является пром-сть пластпч.
масс; в основном его применяют для пропз-ва органич.
стекла. Нек-рое количество М. используется в качестве
сополимера, а также для лаков и клеев.
Для синтеза М. используют многие общие методы
полученпя эфиров ненасыщенных к-т: отщепление
галогеноводорода от метиловых эфиров а- или р-гало-
генированных изомасляных к-т, дегидратацию эфи-
ров а- или Р-оксиизомасляных к-т и др. М. можно
получить также деполимеризацией его полимера при
нагревании в вакууме до 300—400°. В пром-сти М.
получают из ацетона и синильной к-ты через стадию
ацетонциангидрина и амида метакриловой к-ты:
ОН
HCN + СН3 - С = О —> СН3 - С - CN —>
I I
сн3 сн3
+ СН3ОН
—>CH2 = C-CONH2-------» СН2 = С- COOCHj.
I I
CH, СН,
189
МЕТИЛНАФТАЛИНЫ—МЕТИЛОВЫЙ СПИРТ
190
При хранении М. в качестве стабилизаторов (0,005—
0,5%) применяют серу, медь, резинат меди, фенолы,
гидрохинон, пирогаллол, дифениламин, га-н-бу-
тиламинофенол. Для анализа М. используют либо
каталитич. гидрирование, либо бромирование (бромид-
бромат, пиридинсульфатдибромид). М. обладает об-
щеядовитым и наркотич. действием на организм; пары
его раздражают слизистые оболочки при концентра-
ции 0,5—1,0 мг/л воздуха. См. также П олиметилмета-
крилат. В. Н. Александров.
МЕТИЛНАФТАЛИНЫ — гомологи нафталина
(1-метилнафталин, 2-метплнафталии, 1,3-диметилнаф-
талин, 1,4-диметплнафталин и др.) — бесцветные
жидкости или твердые продукты, нерастворимые в
воде, растворимые в органич. растворителях. Реак-
ции М. (сульфирование, нитрование, гидрирование,
алкилирование и др.) сходны с аналогии, реакциями
нафталина. Так, сульфирование при низких (до 80°)
темп-рах приводит, как и в случае нафталина,
к а-сульфокислотам, переходящим с повышением
темп-ры (до 160—170°) в 0-изомеры; из 1-М. (а-метил-
нафталина) при комнатной темп-ре образуется: 4-суль-
фокислота, при 110° — смесь 3- и 7-изомеров, при
170° — гл. обр. 7-сульфокислота; из 2-М. (0-метил-
нафталина) при 40° образуется преим. 8-сульфокпс-
лота, при 90—95° — 6-сульфокислота и при 160°—
7-сульфокислота. Окисление 1-М. над V2OS приводит
к тем же продуктам, что и окисление нафталина —
к фталевой кислоте (30%) и малеиновому ангидриду
(10%). М. содержатся в кам.-уг. смоле, причем неко-
торые, напр. 1-М. и 2-М., в значительных количествах
(1% и 1,5 % соответственно); 2-М. выделяют из фрак-
ции с т. кип. 230—250° вымораживанием, а 1-М.— в
виде пикрата, к-рый затем разлагают щелочью. М.
могут быть получены также из продуктов каталитич.
крекинга, нефти и синтетически
С10н, + СН3С1—>с,он,сн3 + НС1
С,0Н7Вг + СН3Вг + 2Na—>Ci0HTCH3 + 2NaBr
1-М. входит в состав смесей с цетаном, применяемых
для определения цетановых чисел дизельных топлив,
может быть использован для синтеза а-нафтллуксусной
к-ты (см. Регуляторы роста растений)', 2-М. применя-
ют для получения р-метилпафтохипопа-1,4. Сульфо-
кислоты высших гомологов нафталина (амилнафталина
и др.) используют в качестве детергентов и эмульга-
торов; полиметилнафталины являются инсектицида-
ми. В табл, приведены основные свойства нек-рых М.
Пд 1,3748. М. плохо растворим в воде (ок. 3,5% при
20°), хорошо — в эфире и спирте, желатинирует
нитроцеллюлозу. При нагревании до ~200° и выше
М. самовоспламеняется. Получают М. этерификацией
метилового спирта азотной к-той или серно-азотной
смесью (см. Нитроэфиры). М.— мощное, высокочувст-
вительное, весьма опасное в обращении взрывчатое
вещество; его практич. применение ограничено. Вдыха-
ние паров М. вызывает головную боль. Б- Н. Кондриков.
МЕТИЛОВЫЙ СПИРТ (метанол, карбинол, древес-
ный спирт) СНзОН, мол. в. 32,04 — простейший пред-
ставитель предельных одноатомных спиртов. Впервые
в чистом виде М. с. был выделен в 1834 из продуктов
сухой перегонки дерева Ж. Дюма и Э. Пелиго, к-рые
установили его химич. формулу. В 1857 М. Бертло.
синтезировал М. с. омылением хлористого метила.
Физические свойства. М. с.— бесцветная, легко-
подвижная жидкость, с запахом, подобным запаху
этилового спирта; т. пл. —97,88°; т. кип. 64,509°;.
<7° 0,81009, 0,78687,+° 0,76761;n2D° 1,3286; n% 1,3188;
вязкость г]20 0,584 спуаз (подробнее см. табл. 1);
Таблица 1. Вязкость метилового спирта
°C п °C п °C П
-20 1,16 10 0,68 40 0,45
— 10 0.97 20 0,584 50 0,396
0 0,817 30 0,514 60 0.351
г°крит. 240,1°; ркрит. 78,67 am; 0,272 г/см3; диполь-
ный момент 1,63-10”18; диэлектрич. проницаемость.
32,63 (25°); теплота образования ДЯ2°8= —48,10
ккал/молъ (газ), —57,0 ккал/молъ (жидкость). Теп-
лоемкость М. с. в жидкой фазе для темп-р 0—50° оп-
ределяют по ф-ле Ср=0,5688 + 0,0014 1°С кал/г-гра&
в паровой фазе для темп-р 0—1200° — по 6-ле:
С = 5,9814+ 12,0199 • 10”’ Т. + 14,1229 10”6 Тг —
-10,8068-10”’ Т3 кал/молъ-град; теплота парообра-
зования 9,19 ккал/молъ (20°); теплота сгорания
5420 ккал/г (25°) или 173,650 ккал/молъ; т. всп.
15,6° (в открытой чашке), границы взрывоопасных
концентраций в воздухе 6,72—36,5 об.% М. с.
М. с. смешивается во всех отношениях с водой .
а также со спиртами, бензолом, ацетоном и другими,
органич. растворителями, не смешивается с алифатич..
Положение заместителей Т. пл., °C Т. кип., °C /мм 4 nD Т. пл. пикрата. °C
1 -30 ,77 244,78(760! 151,3/58,78 1,0163 (25°) 1 ,61494 (25°) 141-142
2 34,44 241,44/760 1 48.07/58.78 0,99045 (40°) 1,60 1 92 (40°) 115 — 116
1,2 -3,5 1 39 — 1 40/15 1 ,015 (20°) 1 ,6135 (20 °) 130 — 131
1,3 107/1 — — 118
1,4 — 262—264/760; 145/40; 108 — 1 09/1 1,01803 (16°) 1,61 56 7 (16°) 144
1,5 80—80,5 — — 138—139
1,6 262—263/760 1 ,00 56 (1 5°) — 114
1,7 1,8 261 — 262,760; 1 47—1 49/1 5 1,0115 (20°) 1 ,60831 (20°) 121
63 140/18 — —— 148
2.3 104 — 104,5 возг. —— — 123-124
2,6 11 0 — 111 261—262 — — 142-143
2,7 . . . 96-97 262 — 136-1 36,5
1,2,7 — 14 8/1 6 1,008 (1 5°) 1,6093 (1 5°) 129,5—130
1-метил-7-изопропил . . — 281/720 — — 95
1,6-диметил- 4-изопропил — 291—292/720 — — 11 5
В. Н. Фросин.
Р-МЕТИЛНАФТОХИНОН — см. Витамины К.
МЕТИЛНИТРАТ CHsONO,, мол. вес 77,04 —
бесцветная, подвижная, легколетучая жидкость с
характерным запахом, т. кип. 64,6°, плотн. 1,21,
скрытая теплота испарения 7,73 ккал/молъ, т. пл.
—83°, скрытая теплота плавления 1,97 ккал/молъ,
углеводородами. М. с. образует азеотропные смесиг
с ацетоном (12%, т. кип. 55,7°); с бензолом (39,1%,.
т. кип. 57,5°); с сероуглеродом (14%, т. кип. 37,65°);
с четыреххлористым углеродом (20,66% , т. кип. 55,7°),
а также со многими другими соединениями. М. с.
горит несветящимся слабо-голубым пламенем.
191
МЕТИЛОВЫЙ СПИРТ
192
Таблица 2. Давление паров метилового спирта
при разных температурах
°C и.н рт. стп.1 °C мм рт. сш.| °C ат
—20 6,3 40 243,5 111,8 5
10 13,5 50 381,7 137,4 10
0 26,8 60 579,9 166,6 20
+ 10 50,1 80 1238,5 200,2 40
20 88,7 100 2405,1 214 50
30 150,0 — — 224,4 60
Таблица 3. Температура замерзания водных растворов
Содержание метило- вого спирта, вес. %. . 10 20 30 40 50 60 64
Темп-ра замерзания, °C -6,5 -15,0 — 26,0 —39,7 —55.4 — 75 , 7 -83,4
Химические свойства. М. с., как и другие одно-
атомные спирты, сочетает в себе свойства очень
слабого основания и еще более слабой кислоты. При
действии щелочных металлов на М. с. образуются
метилаты, нанр.: 2CHsOH+2Na -+ 2CH3ONa +Н2;
водой метилат омыляется до СНзОН и NaOH. При
действии кислот на М. с. образуются сложные эфиры;
реакция ускоряется в присутствии сильных минераль-
ных к-т. Азотистая кислота с М. с. количественно об-
разует метилнитрит: СНзОН- Н.\О2^-СНз.\'О2—Н2О;
реакцию применяют для иодометрич. определения М. с.
Серная к-та, реагируя с М. с. при темп-ре ниже 100°,
образует метплсульфат:СНзОН ;112 80,—СНз8О3ОНл-
+ Н2О; при перегонке последнего в вакууме в
присутствии дымящейся H2SO4 образуется диметил-
.сулъфат. Органич. к-ты с М. с. образуют метиловые
эфиры: СНзОН+НСООН = НСООСН3+Н2О.
При неполном окислении М. с. кислородом воздуха
при 500—600° в паровой фазе на серебряном или
медном катализаторе образуется формальдегид:
СНзОН-(1,5О2-^НСНО—Н2О. Формальдегид полу-
чается также при дегидрировании М. с.: СН3ОН->-
-S-HCHO+ Н2. В пром-сти формальдегид получают со-
четанием обеих реакций. М. с. реагирует с окисью
углерода. Пропуская над катализатором при повы-
шенной темп-ре и под давлением смесь метанола,
окиси углерода и водяного пара, получают метил-
формиат: СН3ОН+СО->-НСООСНз. Из М. с. и окиси
углерода каталитически можно получить также ук-
сусную к-ту; СН3ОН-СО^-СН3—СООН. Процесс
идет либо в паровой фазе при 300—500° и давлении
300 ат, либо в жидкой при 200—240° и 700 ат.
С водяным паром на катализаторе М. с. может
быть разложен до водорода и двуокиси углерода:
СНзОН+Н2О->ЗН2+СО2. Эту эндотермич. реакцию
можно вести под давлением до 20 ат. После очистки от
СО2 водород имеет 98%-ную чистоту. Процесс приме-
няют в передвижных установках для получения водо-
рода и удовлетворения маломощных потребителей.
М. с. с аммиаком образует метиламины в присутствии
дегидратирующих катализаторов в паровой фазе
при 370 — 400° и 60—200 ат'.
СН3ОН — NH3—► CH3NH, + Н-0
СН3ОН + CH3NH2 —>(СН3), NH + Н3О
Ароматич. амины метилируются М. с. в присут-
ствии H2SO4. Так, при действии М. с. на анилин
при 200° и 30 ат получают дпметиланилпн: C6H5NH2+
+ 2СНзОН-»С|;Н..\'(СН3)2—2Н2О. М. с. применяли так-
же при произ-ве толуола из бензола: С6Н6-)-СН3ОН->-
-*С6Н5СНз+ Н2О. Реакция идет на катализаторе при
35 ат и 340—380°. Дегидратацией М. с. при повы-
шенных темп-pax на катализаторе получают димети-
ловый эфир СН3ОСН3. С галогеноводородными к-тами,
хлористым сульфурилом и хлорокисью фосфора М. с.
образует соответственно иодистый метил, хлористый
метил и т. д. Взаимодействием М. с. с иодом и фос-
фором получают иодистый метил: ЮСНзОН-|-5 J 2-+-
+ 2Р^10СНз1 + 2НзРО4.
При обычной темп-ре М. с. устойчив; при 350—
400° и атмосферном давлении на катализаторе раз-
лагается на СО и Н2.
Получение. Начало получения синтетич. М. с.
относится к 1923. До этого М. с. выделяли из под-
смольной воды при сухой перегонке дре-
весины. Содержание М. с. в подсмоль-
ной воде зависит от породы перерабаты-
ваемой древесины и составляет 2—3%.
Древесный спирт-сырец отгоняют и за-
тем ректифицируют; т. обр. получают
технически чистый метанол. Основной
современный способ промышленного по-
лучения М. с.— синтез непосредственно
из водорода и окиси углерода. Исходным сырьем яв-
ляются природный, коксовый и другие углеводородсо-
держащие газы, напр. «синтез-газ», образующийся
при термоокислительном пиролизе метана (ем. Ацети-
лен), а также кокс, бурый уголь. Из них получают смесь
СО и Н2 в молярном соотношении 1 : 2. Синтез М. с.
из СО и Н2—обратимая экзотермич. реакция:
СО + 2Н2^л±СН3ОН ДН°98 = — 21684 кал'моль
Равновесие смещается в сторону образования М. с.
при повышении давления и понижении темп-ры. Но
на применяемых в пром-сти катализаторах реакция
протекает со скоростью, удовлетворяющей требованиям
произ-ва лишь при темп-pax выше 300° и давлениях
300—500 ат.
Константа равновесия реакции синтеза М. с. может быть
найдена по ур-нию Темкина и Чередниченко, полученного тер-
модинамич. расчетом с учетом заметной ассоциации паров
метанола:
lgtf = 3971-T-‘ - 7,492-lgT + 0,001770-T- 0,0731 1 - 7’2 + 9,218.
В табл. 4 приведены равновесные концентрации метанола в
зависимости от давления и темп-ры с учетом летучести компо-
нентов.
Таблица 4. Равновесные концентрации метанола
Давле- ние, ат Концентрация СН3ОН (%) при темп-ре (°C) Давле- ние, ат Концентрация СНаОН (%) при
темп-ре (С)
300° 350° 300° | 350°
100 26,3 8,2 300 75 , 5 40,4
150 40,8 16.0 400 86,0 55,5
200 54,0 24,0 500 89,6 66 , 7
250 66,0 31,9
В промышленных установках М. с. синтезируют
обычно при 300—375° и 300—500 ат на катализаторе;
ранее применяли также давление 900—1000 ат.
Основой катализатора б. ч. является смесь окислов
хрома и окиси цинка. Применяют также медь —
цинк — хромовый катализатор, работающий при
более низких темп-pax (ок. 300°), но весьма чувст-
вительный к ядам, в частности к серусодержащим
соединениям, и не допускающий перегревов.
Натта для катализатора, содержащего 89% ZnO и 11%
Сг2О3, предложил следующее ур-ние скорости реакции:
V .„ .V2 ych3ohpch3oh
YCO ?СО VH3 ?Н, к________________
'PH.'£>VCH3OH рсн3он)3
где г — скорость реакции; К — константа равновесия; угп
VH2’ VCH3OH~ летучести компонентов; Рс0> Рсн0£^
парциальные давления компонентов. Постоянные А. В, С и О
СН3ОН
(А + Byco”co+Cyh3
193
МЕТИЛОВЫЙ СПИРТ—МЕТИЛПИПЕРИДИНЫ
194
определяют экспериментально, пользуясь ур-ниями:
д=----------5------------
в=----------------------г
(‘^S2'cl„-kCO'k2h2)/s
с_
(42K-s*.c к к* )‘/з
\ Ljq СО -Н-2 '
с__ ксн3он______________
(>;2K-s» cL к к* )*/>
\ Liq G и -П-2 /
где К — константа скорости прямой реакции в хемосорбиро-
ванной стадии; 8 — количество активных центров, соседних
с центрами, участвующими в хемосорбции; — молярная
концентрация всех активных центров на единицу массы ката-
лизатора; Kqq, Kqjj qjj— константы равновесия ад-
сорбции соответственно СО, Н2 и СН3ОН.
В промышленных условиях концентрация М. с.
после зоны катализа значительно ниже равновесной
и для рабочего давления в 300 ат обычно составляет
3—4 об. %. Синтез М. с. ведут с циркуляцией непрореа-
гировавшей газовой смеси, подобно процессу синтеза
аммиака; произ-во и аппаратура обоих процессов
сходны. Процесс идет при объемной скорости 30 000—
40000 л13 газа на 1 ма катализатора в час. Наряду с
М. с. образуются побочные продукты — диметиловый
эфир, высшие спирты, метан, органич. к-ты. Для
уменьшения выхода побочных продуктов в циркуля-
ционном газе обычно поддерживают избыток водорода:
Н2 : СО=6 : 1 (мол.). Снижение выхода М. с. при
повышенном содержании водорода и применяемых
объемных скоростях значительно меньше, чем по
термодинамич. расчетам, т. к. водород адсорбируется
катализатором медленнее окиси углерода.
Получаемый М. с.-сырец содержит 89—92% СНзОН,
2% диметилового эфира, высших спиртов и др. при-
месей и 6—9% воды. Ректификацией получают рек-
тификат, содержащий 99,9% М. с. Присутствующие
в сырце трудно отгоняемые, но легко окисляемые
ненасыщенные соединения удаляют обработкой р-ром
перманганата калия, после того как из сырца был
отогнан диметиловый эфир.
М. с. может быть получен из двуокиси углерода и водо-
рода;
СО2 + ЗН2 СН2ОН + Н2О ДН298= — 1 1830 кал/моль
В пром-сти М. с. получают также неполным окислением
высших предельных углеводородов (этана и пропана), содер-
жащихся в попутных нефтяных газах при 340—360° и 50—
100 ат с применением воздуха или кислорода в качестве окис-
лителя. Концентрация кислорода в смеси ок. 3%. Реакция
протекает как на катализаторе, так и без него, длительность
реакции — несколько секунд. Наряду с М. с. образуются
формальдегид и ацетальдегид, в небольших количествах —
этиловый спирт и ацетон, а также окись и двуокись углерода
и вода. Неполное окисление ведут по проточной, циркуляцион-
ной либо ступенчатой схемам. При проточной схеме непро-
реагировавшие газы после выделения органич. продуктов ре-
акции направляются на дальнейшее использование. При
циркуляционной схеме непрореагировавшие газы после вы-
деления органич. продуктов циркуляционным насосом вновь
возвращаются в процесс (часть газа выдувается из цикла
во избежание чрезмерного накопления СО). При ступенчатой
схеме непрореагировавшие газы после отделения органич.
продуктов поступают на след, ступень окисления. Количество
ступеней окисления 5—6. После последней ступени непрореа-
гировавшие газы идут на дальнейшее использование, напр. на
синтез аммиака. При некаталитич. неполном окислении
примерно 65% вступивших в реакцию углеводородов идет на
образование органич. продуктов. Образовавшиеся органич.
продукты разделяют ректификацией. Неполное окисление,
несмотря на сложность выделения индивидуальных продуктов,
находит применение в пром-сти.
Применение. М. с. перерабатывается гл. обр. в
формальдегид, используемый для выработки пластич.
масс, уротропина и мочевино-формальдегидных удоб-
рений. Он применяется также как добавка к жид-
ким топливам для повышения октанового числа и
для приготонления растворителей. М. с. идет на вы-
работку метакрилатов, метиламинов, а также диме-
тилтерефталата (произ-во синтетич. волокна лавсан).
М. с. применяют в качестве антифриза, а также для
произ-ва галогеналкилов.
М. с.— сильный яд. Он действует преим. на нерв-
ную и сосудистую систему, обладает резко выражен-
ным кумулятивным действием. Прием внутрь 5—10 мл
М. с. приводит к тяжелому отравлению, а прием
30 мл и более — смертелен. В парообразном состоя-
нии М. с. сильно раздражает дыхательные пути и
слизистые оболочки глаз, проникает через кожу,
поражает зрительные нервы и сетчатку глаз.
Лит.: Долгов Б. Н., Методы химического использова-.
ния окислов углерода, Л., 1936; его же, Катализ в орга-
нической химии, 2 изд., Л., 1959; N a t t a G., Synthesis о£
methanol, в кн.: Catalysis, ed. Р. Н. Emmet, v. 3, N. Y., 1955;
Казарновская Д. Б., Казарновский Я. С.,
Сидоров И. П., Тр. Гос. ин-та азотной пром-сти, 1960,
вып. 11,16; Чередниченко В. М., Темкин М. И.,
Ж. физ. химии, 1957, 31 вып. 5, 1072; N a t t a G., Chiml.
са е Industrie, 1953, 35, Ki 10, 705; Р а з q и о n I., там же,
1960, 42, № 4, 352; U с h 1 d а Н., О g i п о Y., Bull. Chem.
Soc. Japan, 1958, 31, 1, 45; Ullman n, 3 Aufl., Bd 12,
Munch.—B., 1960, S. 398; Kirk, v. 9, N. Y., 1952, p. 31.
E. Я. Мельников.
МЕТИЛОВЫЙ ФИОЛЕТОВЫЙ (метилвиолет) —
кристаллич. порошок с зеленым металлич. блеском.
М. ф. принадле-
жит к триарил-
метановым кра-
сителям (пента-
метилзамещен -
CH3NH-CeH,
'с
(CH3)2NC6h/
N(CHg)2
Ci-
ный парарозани-
лин C24H28N3C1, мол. в. 393,97 с примесью гекса- и,
возможно, тетразамещенного). Растворяется в воде,
спирте, глицерине; максимум поглощения в воде X
587,0 и 535 Л4Л1К. При прибавлении к нодному р-ру
красителя разб. соляной к-ты, в зависимости от
количества ее, р-р становится синим, зеленым или
желтовато-бурым. Через неск. минут разб. подкис-
ленные р-ры почти обесцвечиваются. Со щелочью
краситель дает буровато-красное окрашивание и оса-
док. М. ф. в пром-сти получают окислением диметил-
анилина.
Как органич. катион М. ф. осаждает многие тя-
желые комплексные анионы; в кислых р-рах это
осаждение сопровождается изменением окраски. При-
меняют М. ф. для обнаружения и для фотометрия,
определения Zn, Cd, Hg, Sb, TI, In, Ga, Au, Sn, Та и
др., образующихc CNS-,J-, Br“, Cl~, F~ комплекс-
ные анионы; для отделения Zn, Cd, Sb, осаждаемых
в виде комплексных анионов; при концентрировании
следов ряда элементов; в качестве кислотно-основного
индикатора в интервале pH от 0,13 до 0,5 и в интер-
вале pH от 1,5 до 3,2 (переход окраски соответствен-
но от желтой к зеленой и от синей к фиолетовой),
окраска в кислой среде неустойчива, чем выше кислот-
ность, тем быстрее она обесцвечивается; в микро-
скопии — для окрасок препаратов; для изготовления
чернил, лент для пишущих машинок, фаналевых
лаков и т. д., ограниченно применяется для крашения
шелка, шерсти, хлопка по таниновой протраве,
окраска не светопрочна. И. п. Ефимов.
МЕТИЛПИЙЕРИДИНЫ (гексагидрометилпипери-
дины) — гомологи пиперидина, содержащие одну
или несколько метильных групп
(монометилпиперидины, т. наз. п и-
пеколины, диметилпипериди-
ны — лупетидины, триметил-
пиперидины — копеллидины,
и т. д.). В таблице приведены свой-
ства некоторых метилпиперидинов.
жидкости; растворимы в воде, спирте, эфире; об-
ладают сильно основными свойствами. М. получают
из соответствующих метилпиридинов гидрированием
г
СН3
а' Н2С^1^СН2 а
2 NH 2
М.— бесцветны!
0 7 К.Х.Э., т. 3
195
МЕТИЛПИРИДИНЫ
196
(натрий в спирте, каталитич. восстановление в
присутствии коллоидной платины в уксусной к-те
и др.). М. встречаются в природе, напр. 2,6-ди-
метилпиперидин найден в нек-рых растениях; от-
дельные М. и их производные нашли примене-
ние в медицине (сальсолин, дикалин и др.). К М.
может быть отнесен и N-метилпиперидин — бесцвет-
ная жидкость, т. кип. 107°; <72° 0,8159; n|J’e 1,4378.
Р-пипеколин; из 4-М.— у-пипеколин и т. д. Окисление
М. перманганатом в щелочной среде, кислородом
воздуха над V2O5, азотной к-той и др. окислителями
приводит к пиридинкарбоновым к-там:
Соединения Т. кип., "С/мм nD Т. пл. производ- ных, °C
соляно- кислый пикрат
Метилпиперидины (пипе- колины) CeH13N,Mon. в. 99.17 2 (а) 3 0) 4 (V) 117-118/747 125—126/763 132—134/756 0,8436(23,6°) 0,8446(24,3°) 0,8674(0°) 1,4464 1,4463 210 171-172 134-135 136—138
Диметилпиперидин (лу- петидин) C7H15N, 2,6 (а, а'), мол. в. 113,20 . 127,5-128,3/768 0,8158(25°) 1,4366 281 162-164
Триметилпиперидин (ко- пеллидин) C6H17N, 2,4,6 (а, а', у), мол. в. 127,23 165—166/760 0,8430(4°) — — —
Так, из 2-М. образуется пиколино-
вая к-та, из 3-М.—никотиновая, из
4-М.— изоникотиновая и т. д. Ка-
талитическое окисление М. кисло-
родом воздуха в отсутствие аммиа-
ка дает соответствующие цианпи-
ридины:
NH3(O2)t
veO5
Амидирование М. по Чичибабину
приводит к а-аминометилпириди-
нам, напр.
N-М. может быть получен из 1,5-дибромпентана и
CH3NH2, из пиперидина и CH3J и другими спосо-
бами. Многие соединения растительного происхож-
дения содержат в своей структуре N-метилпипери-
диновое кольцо, например производные тропана —
атропин и кокаин, алкалоиды — ареколин, лобелии,
N-метпланабазин и др. Среди синтетич. производных
N-М. следует отметить лилол и промедол, нашедшие
применение в качестве заменителей морфина.
В. Н. Фросин.
МЕТИЛПИРИДИНЫ — гомологи пиридина,
содержащие одну или несколько метильных групп:
монометилпирпдпны (пиколин ы), ди-
метилпиридины (л у т и д п н ы), триме-
’ F Д Тилпиридины (коллидин ы) и т. д.
Известны все три возможных монометил-
ппрпдпна, все диметилпиридины, 5 из (5
триметилпиридинов, 2 из 3 тетраметилпиридинов.
Важнейшие из них приведены в таблице. М.— бес-
цветные жидкости с пиридиноподобным запахом;
хорошо растворимы в спирте, эфире и др. органич.
растворителях; растворимость в воле иония ается с
увеличением числа метильных групп (пиколины хо-
рошо растворимы, лутпднны—ху >е, коллидины —
плохо или совсем нерастворимы). Подобно пиридину,
М. с сильными к-тамп образуют устойчивые соли;
с галогеналкилами — галогеналкилаты. Надуксусной
к-той М. легко окисляются в N-окиси; каталитически
возбужденным водородом (напр., коллоидной пла-
тиной в СНдСООН) или натрием в спирте М. восста-
навливаются в гексагидрометплпиридины (метилпи-
перидины); из 2-М. получают а-иппеколин; из 3-М.—
Ряд реакций М. обусловливается подвижностью ато-
мов водорода метильных групп в а- и у-положениях.
Так, 2-М. при действии амида натрия или фенил-
лития образует соли, взаимодействие к-рых с га-
логеналкилами может служить способом получения
гомологов пиридина:
+ RX
zNaX'
0^~CH2R
2-М. конденсируется с альдегидами, кетонами, нит-
розосоединениями и др. соединениями (аналогично,
хотя и с большим трудом, реагирует 4-М.):
Соли пирпдпипя вступают в такого рода реакции
гораздо легче. Благодаря подвитиостп атомов водо-
рода в метильных группах, находящихся в а- и у-по-
лояениях, 2-М. и 4-М. могут давать производные,
обладающие пиридонметиленовой структурой. Гак,
Г. пл., °C Т. КПП., °C d20 4 90 nD К (25°) Т. пл. пик- рата, °C
Пиридин СП N, мол. в. 79,10 -41 .55 115.26 0,98310 1,51020 5.23 163—164
Метилпиридины CHHTN, мол. в. 93,12 2-метилпиридии (п-ппколпв) -66.55 129,44 0,94432 1 ,50101 3,210-* 5.96 169 — 171
З-метилпиридин (0-пиколин) -17,7 144,00 0,95658 1.50582 1 .1 ю-’* 5.66 1 49 — 1 50
4-метилпиридин (у-пиколин) — 4,3 145,3 0.95478 1 .50584 1 ,1 -1 0 — 4 6,05 1 68
Диметилпиридины C-IL.N, мол. в. 107,15 2.6-диметилпиридин — 5,9 144,00 0.92257 1.49767 — 6.62 163—164
Тримстилпиридин C.HHN, мол. в. 121,18 2,4,6-триметилпиридин (симм. коллидин) . . . -46,0 170.0 0,9142 1.4981 2.05-1 0-’ 7.45 1 55-1 56
2-метил-5-этилпиридин (альдсгидколлидпн) . . . -70,3 1 74 — 176 0,9208 1,4970 — — 16 4 — 165
197
МЕТИЛСУЛЬФАТ —МЕТИЛТРАНСФЕРАЗЫ
198
напр., при обработке галогеиалкилатов 2-М. и 4-М.
щелочами образуются т. и. метиленовые основания
(ангидроосиования):
В случае 3-М. четвертичное основание устойчиво и
не отщепляет воду.
В пром-сти М. выделяют из кам.-уг. смолы, где они содер-
жатся вместе с пиридином и др. пиридиновыми основаниями в
количестве 0,1 % (по весу). После обработки легкого и среднего
масел разб. серной к-той и последующей нейтрализации смесь
пиридиновых оснований подвергают разгонке. При этом пря-
мой ректификацией удается выделить только пиридин и
а-пиколин; остальные пиридиновые основания получают в виде
многокомпонентных фракций, разделение к-рых производят
ректификацией азеотропов, образуемых М. с различными рас-
творителями (водой, уксусной к-той, формалином и др.) и раз-
делением М., основанным на избирательном комплексообразо-
вании М. с нек-рыми органич. и неорганич. соединениями.
Так, напр., 3-М. выделяют в виде комплекса с CnSO4. Окон-
чательно свободное основание очищают через комплекс с
Cu2Cl2. 4-М. также выделяют в 2 стадии (комплексообразова-
нием с N1C12 с последующим разложением комплекса и очист-
кой полученного основания через комплекс с СаС12). 2,6-Ди-
метилпиридин выделяют и очищают в виде комплекса с моче-
виной.
М. могут быть получены конденсацией аммиака
с многими соединениями — альдегидами, кетонами,
ацетиленами, ацетоуксусным эфиром (Ганч), термич,
разложением галогенометилатов пиридина (Ладен-
бург):
СН3
и многими другими способами (подробнее см.
Пиридиновые основания). Так, |3-пиколин можно
получить из аммиака и акролеина или генерирующих
его веществ; 2,6-лутидин — из NHg, ацетоуксусного
эфира и СН2О; а-пиколин с хорошим выходом обра-
зуется из пиридина и диазометана.
Способ получения 2-метил-5-этилпиридина осуще-
ствлен в пром-сти:
4СН3СНО + NH3
Побочным продуктом этого процесса является
а-пиколин. Дегидрированием 2-метил-5-этилпиридина
получают 2-метил-5-винилпиридин. Для качествен-
ного и количественного определения М. пригодны все
способы, применяемые при идентификации пиридина.
Многие М. являются важными исходными продук-
тами для приготовления различных фармацевтич.
препаратов. Так, из 3-М. получают витамин РР
(см. Никотиновая кислота), кордиамин, цезол (хлор-
метилат метилового эфира никотиновой к-ты), рони-
кол (3-пиридилкарбинол); из 4-М.— фтивазид и ан-
тисептич. средства (напр., 4-метил-1М-лаурилпириди-
ниумхлорид); из 2,6-диметилпиридина — алкалоид
лобелии и лекарственные препараты — нанофин и
дикалин. Кроме того, в смеси с другими пиридиновыми
основаниями М. используются как растворители
(напр., для очистки антрацена от примесей), а также
для денатурирования спирта. в. н. Фросин.
МЕТИЛСУЛЬФАТ (метилсерная кислота) — см.
А лкилсульфаты.
ME ТИЛ ТЕСТОСТЕРОН (17а-метилтестостерон,
17а-метил-Д4-андростенОл-17р-он-3) C20HS0O2, мол. в.
302,46— синтетич. аналог мужского полового гор-
мона — тестостерона. М.— бесцветные кристаллы,
т. пл. 161-167°, [а]о=+(82-85°) (1%-ный р-р в
спирте), уд. показатель погло-
щения е=520—540 при >.макс. ц,С
240 льмк (0,001%-ный р-р в
спирте, 1 см). М. практически н3С [ | I
нерастворим в воде, раство-
ряется в спирте, ацетоне, эфи- 1з I J
ре. Характерными производны-
ми М. являются: ацетат, т. пл.
173—176°, оксим, т. пл. 210—216°. М. обладает фи-
зиология. свойствами тестостерона, но несколько
менее активен. В отличие от последнего, М. активен
при приеме внутрь; он стимулирует функции муж-
ских половых органов. Применяют М. в случаях на-
рушения функций мужских половых желез.
Лит.: Государственная фармакопея СССР, 9 изд., М., 1961,
с. 295; Назаров И. Н., Бергельсон Л. Д., Химия
стероидных гормонов, М., 1955, с. 167; F 1 е s е г L., F 1 е s е г
М., Steroids, N. Y , 1959, р. 519. И. И. Суворов.
MEТИЛТИОУРАЦИЛ (6-метил-2-тиоурацил)
C5HeON2S, мол. в. 142,18 — желтоватый порошок,
т. пл. ок. 330° (с разл.); трудно рас- HN_C0
творим в воде и в большинстве органич. । ;
растворителей, хорошо — в р-рах ще- S=C сн
лочей. Получают М. конденсацией тио- HN—с—СН
мочевины с ацетоуксусным эфиром в 3
спирте в присутствии этилата натрия. М. обладает
антитиреоидным действием, связанным с уменьше-
нием синтеза тироксина в щитовидной железе. При-
меняют М. при лечении базедовой болезни, тирео-
токсикоза и гипертиреоза.
Лит.: Anderson G. W., [а. о.], J. Amer. Chem. Soe.,
1945, 67, № 12, 2197. Р. Г. Глушков.
МЕТИЛТРАНСФЕРАЗЫ (трансметилазы) — фер-
менты, катализирующие реакции переноса метиль-
ных групп; относятся к классу трансфераз. В боль-
шинстве процессов метилирования, катализируемых
М., донором метильной группы является S-адено-
зилметионин (т. н. активный метионин),
метильная группа к-рого переносится на тот или
иной акцептор:
—► CH3R +
НО . ОН
К числу М. относятся: а) гуанидинацетат-
метилтрансфераза, катализирующая об-
разование креатина путем метилирования гуани-
динуксусной к-ты (гликоциамина):
NH2 - С - NH - СН2СООН + S-аденозилметионин —>
II
NH
—> S-аденозилгомоциетеин + NH2—С—N—СН2СООН
И I
HN СН3
б) никотинамид-метилтрансфера-
з а, катализирующая метилирование никотинамида;
в) ацетилсеротони н-м етилтрансфе-
7*
199
МЕТИЛФОСФИН—МЕТИЛЦИКЛОГЕКСАН
200
раза, катализирующая превращение N-ацетил-
серотонина в 1М-ацетил-5-метокситриптамин. Оптимум
действия этой М., выделенной из шишковидной железы
в высокоочищенном состоянии путем фракциониро-
вания (NH4)2SO4 и очистки на геле А120з, находится
при pH 7,5—8,3. Константа Михаэлиса (А'м) для N-
ацетилсеротонина 5,4-10~3 М., а для S-аденозилме-
тионина 4,6-IO-3 М.
М. играют важную роль в обмене веществ и широко
распространены в различных животных и раститель-
ных тканях.
Лит.: Диксон М., Уэбб Э., Ферменты, пер. с англ.,
М., 1961; Классификация и номенклатура ферментов, М., 1962;
Axelrod J., Weissbach Н., J. Biol. Chem., 1961,
236, № 1, 211. Б. В. Спиричев.
МЕТИЛФОСФИН — см. Фосфины.
МЕТИЛФОСФИНОВАЯ КИСЛОТА (метилфос-
фоновая кислота) — см. Хлоралкилфосфины.
МЕТИЛХОЛАНТРЕН (20-метилхолантрен) С21Н16,
мол. в. 268,34 — кристаллы, светло-желтые иглы;
т. пл. 179—180° (из бензола или эфира); нераство-
рим в воде, растворяется в бензоле, эфире и др.
органич. растворителях, флуоресцирует в УФ-све-
те; т. пл. пикрата 180—181°. Для М. применяют
две системы нумерации: I и II:
Впервые М. был получен при расщеплении дезокси-
холевой кислоты. М. образуется также в качестве од-
ного из продуктов расщепления холевой кислоты и
одного из видов холестерина. Синтетически М. полу-
чают пиролизом кетона III:
ыетилхолантрен
М. обладает мощным канцерогенным действием, уступая по
своей канцерогенной активности только 9,10-диметил-1,2-
бензантрацену. М. широко применяют при изучении рака, гл.
обр. на мышах и крысах. У кроликов, морских свинок, собак
и кошек рак с помощью М. вызвать пока не удается. При сма-
чивании кожи мышей 0,3%-ным р-ром М. в бензоле злокачест-
венные опухоли возникают в 75—85% случаев. Первые папи-
ломы появляются через 1 месяц, а рак — через 3—3,5 месяца.
В нек-рых случаях достаточно одного смазывания кожи, что-
бы вызвать рак. При подкожном введении мышам М. в количе-
стве от 0,125 до 1 мг удалось вызвать рак в 100% случаев. Ла-
тентный период при этом, в зависимости от дозы, составляет
3,3—2,4 месяца.
Лит.: Chemistry of carbon compounds, ed. E. H. Rodd, v. 3,
pt B, Amst.— [a. o.J, 1956; Ф и з e p Л., Ф и з e p M., Химия
природных соединений фенантренового ряда, пер. с англ.,
М.—Л., 1953, с. 155; Ш а б а д Л. М., Очерки эксперименталь-
ной онкологии, М., 1947, с. 135. Л. С. Поваров.
МЕТПЛЦЕЛЛЮЛОЗА — метиловые эфиры цел-
люлозы различной степени замещения общей ф-лы
[СвН7О2(ОН)3_д.(ОСНз)д.]„; предельное содержание ме-
токсильных групп 45,6%. Препараты М. представ-
ляют собой продукты, неоднородные по степени
этерификации и по мол. весу. В холодной воде
растворима М., содержащая от 14 до 33% метоксиль-
ных групп; в горячей воде растворима лишь низко-
молекулярная М. Характерным свойством М. является
изменение вязкости ее р-ров с увеличением темп-ры:
сначала вязкость падает, а затем резко возрастает.
При охлаждении наблюдается значительная петля
гистерезиса и значение вязкости не совпадает с пер-
воначальной величиной. Повышение вязкости тем
интенсивнее, чем выше степень полимеризации и
концентрация М. в р-ре; при достаточно высокой
концентрации растворы застудневают. С повышением
содержания метоксильных групп гигроскопичность М.
увеличивается:
Содержание метоксиль-
ных групп, %........ 0 2,55 9,6 13,6
Гигроскопичность, вес.% 7,9 8,2 10,3 10,5
Метоксильные группы М. весьма устойчивы к омыле-
нию и отщепляются количественно лишь при темп-рах
110—130° под действием конц. р-ров HJ с выделе-
нием CHaJ и одновременным разрушением целлюлозы.
Эту реакцию используют для определения степени
этерификации М.
Промышленные способы получения М. основаны
на действии на целлюлозу в присутствии р-ров щелочи
(или на щелочную целлюлозу) диметилсульфата или
йодистого метила (реакция необратима);
[CeH7O2(OH)3]^ + n(OCH3)2 SO2 + nNaOH—>
—►[С,Н7О2(ОН)2(ОСН3)1п + n(OCH3)(NaO)SO2 + nH,0
Обработку диметилсульфатом проводят при ком-
натной температуре; реакция экзотермична. Метилиро-
вание иодистым метилом ведут под давлением при
80—100° в течение 5—10 час. Цроцесс сопровождается
значительным расходом щелочи на побочные реакции
[омыление CH3J или (ОСНз)2ЗО2].
Для получения М. обычно используют щелочную
целлюлозу, содержащую значительно большие коли-
чества NaOH (35—45%), чем щелочная целлюлоза
вискозного произ-ва (18%). Метилирование проводят
также в гомогенной среде, растворяя целлюлозу в
8—12%-ном растворе щелочи, содержащем 1,0—1,5%
цинката натрия. Реакция в растворе идет с более вы-
сокой скоростью и меньшим расходом реагентов,
а получаемая М. более однородна. Так, М., по-
лученная в гомогенной среде, растворяется в воде
при значительно меньшем содержании метоксильных
групп.
Известны лабораторные методы получения М.: 1) метили-
рование целлюлозы или ее ксантогената диазометаном (эфир-
ный р-р); получаются низкоэтерифицированные препараты М.
(2—19% метоксильных групп); 2) метилирование медно-щелоч-
ного соединения целлюлозы или ее р-ров в четвертичных ам-
мониевых основаниях диметилсульфатом; 3) переэтерификация
ацетилцеллюлозы в ацетоновых р-рах в присутствии щелочи
диметилсульфатом при 55°; за одно метилирование можно по-
лучить триметилцеллюлозу, содержащую 45,6% метоксильных
групп.
Водорастворимую М. применяют гл. обр. для за-
мены крахмала в шлихте и в аппретах в текстильной
пром-сти.
Лит.: Роговин 3. А., Шорыгина Н. Н., Химия
целлюлозы и ее спутников, М.—Л., 1953. Ю. В. Васильев.
МЕТИЛЦИКЛОГЕКСАН (гексагидротолуол)
С7Н14, мол. вес 98,18— алициклич. (нафтеновый)
углеводород; бесцветная жидкость с за- '___
пахом бензина; т. пл.—126,59°; т. кип. / \-сщ
100,93°; 0,76939, d“ 0,76506; п% X-J
1,42312; вязкость (спуаз) 0,734 (20°), t крИт. 299, 13°;
Ркирт. 34,322 ат', Ср0,3287кал/г-град (25°); теплота па-
рообразования (ккал/молъ): 8,451 (25°), 7,58 (т. кип.);
теплота сгорания 11113,3 кал/г (Н2О жидк.); т.
воспл.—3,9° (закр. чашка); анилиновая точка 40,3; ок-
тановое число 74,8. М. горит некоптящим светящимся
пламенем. В присутствии катализаторов (Pt, Pd при
200—300° или МоО2, Сг20з при 400—550°) М. дегид-
рируется в толуол СвНцСНз СвН6СНз4-ЗН2. При
мягком окислении под действием УФ-лучей или в
присутствии перекисей из М. образуется Ггидропе-
рокси-1-метилциклогексан (I), HNOa или Al(NOs)3
нитруют М., при этом образуется смесь 1-нитро-1-ме-
тилциклогексана (II) и циклогексилнитрометана (III).
201
МЕТИЛЦИКЛОПЕНТАДИЕНИЛТРИКАРБОНИЛМАРГАНЕЦ—МЕТИОНИН
202
Под действием брома в присутствии А1Вгз получает-
ся пентабромтолуол.
।
in
М. содержится в нефтях и отогнанных из них
бензинах, особенно в кавказских. В чистом виде М.
получают в лаборатории гидрированием толуола,
тщательно очищенного сульфированием и последую-
щим гидролизом и четкой ректификацией, в авто-
клавах в присутствии скелетного никеля; образуется
также при действии HJ на метилциклогексанол или
циклогептанол с сужением цикла последнего:
М., содержащийся в узких фракциях бензинов,
является основным сырьем для промышленного полу-
чения толуола каталитич. риформингом бензинов
или узких бензиновых фракций (гидроформинг,
платформинг); рекомендуется в качестве сырья для
нек-рых синтезов. М. применяют как компонент
в растворителях. Благодаря доступности высокоочи-
щенного препарата М. служит эталонной жидкостью
для проверки различных лабораторных измеритель-
ных приборов; считается наилучшим горючим для
карманных зажигалок. М. И. Розенгарт.
МЕТИЛЦИКЛОПЕНТАДИЕНИЛТРИКАРБОНИЛ -
МАРГАНЕЦ — см. Марганца метилциклопен та-
диенилтрикарбонил.
МЕТИЛЦИКЛОПЕНТАН С6Н12, мол. в. 84,16—
алициклический (нафтеновый) углеводород; бесцвет-
ная жидкость с запахом бензина, т. пл.
ГД —142,455°; т. кип. 71,812°; o'2’ 0,74864; п‘°
СНз 1,40970; <крит. 259,61°; ркрит. 37,36 ат- теп-
лота парообразования 7,560 ккал/моль
(25°), 6,916 (т. кип.), теплота сгорания (Н2О жидк.)
11183,3 кал/г.
Под действием А1С1з-|-НС1 М. изомеризуется в
циклогексан:
С повышением темп-ры равновесие сдвигается влево.
При 27° равновесная смесь содержит 11,4% М. и
88,6% циклогексана, при 100° — соответственно
33,5% и 66,5%. При действии водорода в присутствии
катализаторов (Pt и др.) протекает гидрогенолиз цик-
лопентанового кольца с образованием смеси изомер-
ных гексанов:
j ^-СНЭ —- СН3(СН2)4СН3 + (СН3)2СН(СН2)2СН3 +
+ (СН3СН2)2СНСН3
При дегидрировании М. на окисных катализаторах
образуется смесь изомерных метилциклопентенов, а
из них — метилциклопентадиенов и даже циклопен-
тадиена.
М. содержится в нефтях и отогнанных из них
легких бензинах. В чистом виде может быть получен
в пром-сти и лаборатории ректификацией дезарома-
тизированного бензина на высокоэффективных ко-
лоннах. В лаборатории М. синтезируют изомеризацией
циклогексана в автоклавах в присутствии А1С13-|-НС1
или из циклогексанола последовательно через ци-
клогексен и метилциклопентен. М. наряду с цикло-
гексаном является основным сырьем промышленных
процессов получения бензола из бензиновых фракций
на платиновых катализаторах под давлением водо-
рода — платформинг (см. Риформинг). В последние
годы служит сырьем для получения инсектофунги-
цидов, синтетич. смазочных масел, устойчивых к
окислению, и в других синтезах. Применяют М. как
компонент в растворителях органич. соединений.
М. И. Розенгарт.
МЕТИЛЭТИЛКЕТОН (бутанон-2, метилацетон)
СН3СОСН2СНз, мол. в. 72,10 — бесцветная жид-
кость, по запаху напоминает ацетон; т. пл. —-85,9°;
т. кип. 79,57°; <72’ 0,8047; п^ 1,3789; давление паров
(20°) 71,2 мм рт. ст.', теплота испарения 106,0 кал/г-,
теплота парообразования (ккал/моль)-. 7,643 (20°),
7,639 (т. кип.); т. воспл. 1,1° (в открытой чашке);
взрывоопасная концентрация паров в воздухе 1,97—
10,2%. Растворимость при 20°: М. в воде 26,8%, во-
ды в М. 12,5%. С обычными органич. растворителями
М. смешивается во всех отношениях; образует азео-
тропную смесь с водой, т. кип. 73,41°, содержание
воды 11,27%. М. обладает всеми свойствами, харак-
терными для алифатич. кетонов, хотя наличие двух
различных активированных групп (метильной и ме-
тиленовой) и придает ему нек-рую специфичность.
Атака различными реагентами направляется на
метиленовую или метильную группу в зависимости
от применяемого катализатора, а также от природы
реагента. Карбонильная группа в М. участвует в
реакциях, обычных для кетонов. В пром-сти М.
получают дегидрированием или окислением втор,
бутилового спирта. Меньшее значение имеют такие
методы получения М., как разложение гидроперекиси
втор, бутилбензола, омыление 2,2-дигалогенбутанов,
гидратация диметилацетилена, синтез через металлоор-
ганич. соединения, пиролиз солей уксусной и пропио-
новой к-т, окисление олефинов, кетонное расщепление
метилацетоуксусного эфира. М. применяют в качестве
растворителя, а также для синтеза метилизопропил-
кетона, втор, бутиламина и нек-рых физиологически
активных веществ. н. П. Гамбарян.
МЕТИОНИН (а-амино-у-метилтиомасляная кис-
лота) CH3SCH2CH2CH(NH2) — СООН,мол. в. 149,2 —
моноаминомонокарбоновая аминокислота, содер-
жащая метилированную сульфгидрильную группу.
М. существует в виде D- и L-форм и рацемической
Б,Ь-формы. L-М. входит в состав большинства
белков животного и растительного происхождения;
особенно богат М. казеин. М.—-бесцветные кристаллы,
т. пл. L-M. 283° (с разл.), D,L-M. 281° (с разл.).
М. умеренно растворим в воде и спирте, плохо рас-
творим в абс. спирте, нерастворим в эфире. М. амфо-
терен, растворяется в к-тах и щелочах. Для L-M.
рАа, 2,28; рКа2 9,21; р! 5,74.
М. дает обычные реакции аминокислот (нингидри-
новую и др.). При восстановлении М. иодистоводород-
ной к-той (в присутствии красного фосфора) от М.
отщепляется метильная группа и образуется гомо-
цистеин. Перекись водорода с хлорной к-той
и др. окислители окисляют М. до метионинсульфона
CHsSO2CH2CH2CH(NH2)—СООН. С нитропруссидом в
сильнощелочной среде М. дает окрашивание. Эта
реакция используется для колориметрия, количествен-
ного определения М.
Получают М. из гидролизатов белков; синтез М.
может быть осуществлен несколькими способами, напр.:
COOK соок
| С1СН2СН2С1 | (CH,)2S
NaCNHCOCH3----------> C1CH2CH2CNHCOCH3------>
COOK COOK
COOK
I H2o
—> CH3SCH2CH2CNHCOCH3—> CH3SCH2CH2CH (NH2)COOH
COOK
203
МЕТОЛ —МЕХАНИЧЕСКИЕ СВОЙСТВА МАТЕРИАЛОВ
204
М.— незаменимая аминокислота; потребность в нем
у человека довольно велика (до 2,5—3 г в день).
В животном организме М. может использоваться для
биосинтеза цистеина и является источником метиль-
ных групп в биосинтезе холина, саркозина, ансерина
и др. биологически активных веществ. Применяется
М. для лечения заболеваний печени и атеросклероза
(способствует удалению из печени излишнего жира
и снижает содержание холестерина в крови больных
атеросклерозом).
Лит.: Машковский М. Д., Лекарственные средства,
4 изд., М., 1960; Капланский С. Я., О 3 е р е ц к о в-
с к а я Н. Е., Ширвиндт Б. Г., Вопросы питания, 1953,
12, № 6, 21; Янг Л., Моу Дж., Метаболизм соединений
серы, пер. с англ., М., 1961. Л. И. Линевич.
МЕТОЛ (сернокислая соль п-метиламинофенола) —
бесцветные кристаллы, иглы; т. пл. ок. 250° (с разл.).
В 100 г воды растворяется: 5 г
CH3-NH • | H2SO4 (25°), 16,5 г (100°). В эфире, хло-
роформе и бензоле М. не раство-
Г j ряется. В водном, особенно в ще-
лочном растворе М. окисляется бы-
ОН стро, медленно — в присутствии
Na2SOs. М.— фотография, прояви-
тель. При взаимодействии с галогенным серебром
М. окисляется до n-хинонмонометилимина (I). Однако
конечным продуктом превращения М. при проявле-
нии является натриевая соль п-метиламинофенол-
сульфокислоты (II). М. прояв-
ляет не только в щелочной, но
и в слабокислой среде (pH 6),
что позволяет составлять бес-
щелочные метоловые проявите-
ли. Особенно часто М. приме-
няют в комбинации с други-
ми проявляющими веществами
(гидрохинон). В промышленности М. получают из
n-оксифенилг лицина нагреванием его до 150° в цик-
логексаноне для отщепления СО2.
• -со2
п-НО - С6Н, - NHCH2COOH-> п-НО - С6Н, - NHCH3
После подкисления рассчитанным количеством H2SO<
из реакционной смеси выделяется чистый М. М. дает
ряд цветных реакций, напр. с меркуриацетатом
водный р-р М. приобретает фиолетовую окраску.
Количественно М. определяют иодометрич. методом,
основанным на окислении М. иодом до п-хинонмоно-
метилимина, или методом диазотирования (образо-
вание N-НПТрОЗОПрОИЗВОДНОГО). В. А. Пучков.
МЕТРИКА ХИМИЧЕСКОЙ ДИАГРАММЫ -
нахождение количественных зависимостей между
элементами физико-химич. диаграммы, а именно:
1) между различными измеримыми свойствами си-
стемы и 2) между величинами свойств и составом
системы. Главный вопрос М. х. д.— отыскание
ур-ния, выражающего зависимость состав — свой-
ство для систем, в к-рых образуется химич. соеди-
нение. К М. х. д. относятся также обобщенный Рауля
закон и Шредера уравнение. М. х. д. начал системати-
чески разрабатывать Н. И. Степанов (1924—38); он
указал следующие общие пути решения этого вопроса:
1) отыскание ур-ния, связывающего выход соеди-
нения (т. е. количества, образовавшегося из 1 моля
исходной смеси) с составом последней; 2) нахождение
т. наз. ур-ния связи, выражающего зависимость
свойства от выхода соединения; 3) получение ур-ния
состав — свойство путем исключения выхода со-
единения из указанных выше ур-ний. Уравнение
выхода связывает количество {у) соединения из од-
ного моля исходной двухкомпонентной смеси
?/[1 — (? + ?—1)у]/’+?-‘—К(х—ру)Р — (1 — х— qy)9 = 0,
в к-рой идет химич. реакция:
рА -|- дБ АрВд
с составом х, выраженным в мольных долях компонен
та А исходной смеси; К — константа равновесия реак
ции. Исследование этого ур
что: 1) если соединение
не образуется (диссоци-
ировано целиком, К = 0),
то кривая выхода совпа-
дает с осью состава ВА;
2) если соединение совсем
не диссоциировано {К— оо),
то кривая выхода распа-
дается на два пучка: один
из них состоит из р, а
другой из q совпадающих
прямых; эти пучки пере-
секаются в сингулярной
точке D, отвечающей хи-
мич. соединению; 3) при
частичной диссоциации
соединения (оо > К >0) кри-
вая выхода—плавная кри-
ния показывает (см. рис.),
Изотермическая диаграмма
выхода соединения АВ и в
системе В—А (по Н. И. Сте-
панову).
вая, проходящая между осью состава и двумя ука-
занными выше пересекающимися прямыми.
Исходя из положения, согласно к-рому отношение
числа молей растворенного твердого вещества к числу
молей растворителя, хотя бы и сложного, постоянно,
Степанов вывел ур-ние связи, выражающее зависи-
мость между выходом соединения и растворимостью
третьего индифферентного вещества в двойной си-
стеме. С помощью этих двух ур-ний он получил за-
висимость между этой растворимостью, выраженной
числом молей, растворенных в 1 моле растворителя,
с составом исходной двойной системы:
(А(р + д-1)х + РП + Ар]Р (А (1 - р - д) х + дт; + k (р - 1) J4
где к — число молей третьего индифферентного ком-
понента, растворенных в 1 моле растворителя, причем
расчет ведется на 1 моль равновесной смеси, а рас-
творимость т) рассчитана на 1 моль исходной двой-
ной смеси. Исследование этого ур-ния показывает,
что по своим общим свойствам кривая растворимости
(пунктирные кривые на рисунке) несколько сходна с
кривой выхода.
Нек-рые выводы М. х. д. были независимо от
Степанова даны в 1935 М. И. Усанновичем. Е. Е. Чер-
кашин разработал М. х. д. систем с ассоциирован-
ными компонентами.
• Лит.: Аносов В. Я. иПогодин С. А., Основные
начала физико-химического анализа, М.—Л., 1947, с. 824—39;
Степанов Н. И., Метрика равновесной химической диа-
граммы, Усп. химии 1936, 5, вып. 7—8; Черкашин Е. Е.,
Метрика равновесной химической диаграммы систем с ассоции-
рованными компонентами, Львов, 1958. В. Я. Аносов.
МЕХАНИЧЕСКИЕ СВОЙСТВА МАТЕРИАЛОВ.
Содержание:
Упругость .......................................205
Пластичность и хрупкость.........................207
Усталость (динамическая).........................209
Усталость (статическая)..........................209
Ползучесть ......................................210
Твердость........................................211
Прочность........................................211
Механические свойства материалов — комплекс
свойств, определяющий поведение твердых тел при
воздействии внешних деформирующих сил. До начала
20 в. М. с. м. изучались преим. в рамках теории
упругости и теории пластичности (механики сплошных
сред), т. е. без учета дискретной структуры твердых
тел. Это направление исследований, оказавшееся
чрезвычайно плодотворным при решении многих
конкретных задач практики, продолжает развиваться
и в наст, время, однако наряду с ним возникло новое
направление в изучении М. с. м., к-рое можно назвать
205
МЕХАНИЧЕСКИЕ СВОЙСТВА МАТЕРИАЛОВ
206
физич. механикой и в основе к-рого лежат представ-
ления об атомном (молекулярном) строении твердых
тел и о разнообразных дефектах их структуры.
К числу основных М. с. м. относятся: упру-
гость, пластичность и хрупкость,
усталость (динамич. и статич.), ползучесть,
твердость и прочность. Каждое из
зтих свойств зависит от темп-ры, внешней среды и
характера напряженного состояния.
Упругость — свойство твердого тела обратимо
восстанавливать свою форму после снятия деформи-
рующих сил. Характеризуется пределом упругости,
т. е. напряжением (в кГ/мм2), выше к-рого в теле по-
являются остаточные деформации, не исчезающие
после снятия нагрузки. В упругой области деформаций
имеет место прямая пропорциональность между прило-
женным напряжением и относительной деформацией —
закон Гука: Р = Е(Д1/1) = Е -е , где Р — напряжение,
Д1/1 =е — относительное удлинение (в простейшем
случае растяжения стержня) и Е — коэфф, пропор-
циональности, наз. модулем Юнга (модулем нормаль-
ной упругости). Закон Гука для упругой дефор-
мации сдвига: Т — Gg, где Т — напряжение сдвига,
§ — относительная деформация сдвига, a G — модуль
сдвига. Аналогично для всестороннего сжатия:
F = /сх, где F — напряжение сжатия, х — относи-
тельное изменение объема тела и к — модуль сжатия.
Между этими тремя модулями упругости имеется два
соотношения (для изотропных тел): Е = 2G(l-J-p) и
Е = ЗА(1 — 2ц), где ц — коэфф. Пуассона, равный
отношению поперечной и продольной деформации при
одноосном упругом растяжении стержня. Для боль-
шинства металлов 0,25 ej 0,35, для стекла р==0,25.
В таблице приведены значения основных модулей
упругости для нек-рых материалов. Переводные
коэфф, на международные единицы см. в ст. Между-
народная система единиц.
Материал Е, кГ1мм2 G, яТ/лш2
Алюминий 7200 2700
Медь 12100 4400
Железо 22000 8400
Вольфрам 40000 —
Бетон 1000-3000 600—1900
Дерево вдоль волокна 1000-1 500 —
» поперек » 50 — 1 20 —
Стекло 6000—10000 —
Опыт показывает, что при растяжении или сжатии (одно-
осное напряженное состояние) происходит трехосная дефор-
мация, связанная с одновременно протекающими поперечными
деформациями. В связи с этим описание процесса упругой де-
формации элементарным законом Гука является неполным.
Необходимо установить зависимость не только между главным
напряжением Рх и удлинением в направлении его действия ех,
но и учитывать влияние на каждое из удлинений в трех глав-
ных направлениях (х, у, z) каждого из трех главных напря-
жений (Рх, Ру, PzY
Для удлинений будем иметь:
Рх -Чрр-рг)1; %=^1РР -Ц(Рт -Рг)1
% 4lPz ~ЧРх ~Рр)1
и для сдвигов
g IG; g = Т IG; g = Т 'G
ху ху syz yz zx zx
Эти шесть ур-ний являются развернутой записью обобщен-
ного закона Гука. Не менее важна другая форма обобщенного
закона Гука, когда ур-ния решены относительно главных
напряжений:
Р =кп + 2Ge ; Р + 2Ge ; Р = Xk + 2Ge2
х х у у z
где G — модуль сдвига, Л=Ец/(1 + ц) (1 —2ц), K=ex+eJ, + e2—
относительное изменение объема.
Для сдвиговых напряжений и деформаций имеем:
Т — Gg , Т — Gg , Т — Gg
ху ху yz yz zx zx
Следовательно, каждое нормальное напряжение вызывает у
изотропного тела как изменение объема (Х.х), так и изменение
формы (2Gex, 2Gey, 2Ge2), тогда как сдвиговые напряжения
вызывают только изменение формы и не вызывают изменения
объема.
Упругая деформация распространяется
в теле со скоростью звука. Ф-ла Ньютона связывает
скорость распространения звука (продольных упру-
гих волн) в данном твердом теле с модулем Юнга;
v = УE/q, где р — плотность материала. На этой
зависимости основаны наиболее точные динамич.
методы определения модуля Юнга. Статич. методы,
основанные на прямом измерении упругой деформации,
вызванной данным напряжением, достаточно точны
лишь для малопластичных материалов.
У большинства реальных материалов обнаружи-
ваются более или менее значительные отступления
от идеально упругого поведения под нагрузкой даже
при напряжениях, весьма малых по сравнению с
пределом упругости. Эти отступления, наз. упругими
несовершенствами или неупругостью, вызываются
неоднородностью строения твердых тел и дефектами
их структуры. К упругим несовершенствам относятся
упругое последействие, упругий гистерезис и внут-
реннее трение.
Упругое последействие выражается
в том, что деформация тела не сразу достигает той
величины, к-рая соответствует приложенному напря-
жению, а сравнительно медленно«дорастает» до нужной
величины (упругое последействие нагрузки), или же
деформация не сразу спадает до нуля при снятии
напряжения (упругое последействие разгрузки). Осо-
бенно отчетливо упругое последействие проявляется
в материалах сетчатой структуры (пластмассы, кол-
лоидные структурированные системы, ткани и т. п.).
У металлов последействие выражено гораздо слабее,
но возрастает с увеличением темп-ры. В хорошо обра-
зованных, т. е. обладающих минимальным количеством
дефектов, кристаллах (кварц, каменная соль, метал-
лич. монокристаллы и т. п.) упругое последействие
проявляется в очень слабой степени лишь при темп-рах,
близких к плавлению.
Упругий гистерезис проявляется в об-
разовании «петли гистерезиса» на диаграмме напряже-
ние — деформация в упругой области при нагруже-
нии и разгрузке. Наличие упругого гистерезиса ука-
зывает на появление небольших остаточных (пласти-
ческих) деформаций в нек-рых участках деформируе-
мого материала, объем к-рых мал по сравнению с
упруго напряженным объемом. Величина петли гис-
терезиса растет с ростом напряжения и при нек-рой
его величине может оказаться незамкнутой, что ука-
зывает на появление более значительных пластич.
деформаций. Так же как упругое последействие,
упругий гистерезис наиболее отчетливо выражен у
неоднородных материалов, нек-рые структурные
элементы к-рых обладают более низким пределом
упругости, чем основная масса.
Внутреннее трение проявляется в эф-
фекте затухания свободных колебаний, вызванных
в материале внешним воздействием, очень малым по
сравнению с пределом упругости. Примером может
служить камертон. Энергия его колебаний постепенно
рассеивается и звучание ослабевает. Лишь малая
доля энергии камертона переходит в энергию звуко-
вых волн; основная часть энергии рассеивается вслед-
ствие внутреннего трения. Причиной возникновения
внутреннего трения в материалах является упругий
гистерезис, т. е. появление весьма малых пластич.
деформаций в нек-рых более слабых элементах струк-
туры. По величине внутреннего трения нек-рые ме-
таллы можно расположить в следующий нисходящий
ряд: чугун, никель, свинец, олово, медь, кадмий,
железо, магний. Повышение темп-ры очень резко
увеличивает внутреннее трение.
207
МЕХАНИЧЕСКИЕ СВОЙСТВА МАТЕРИАЛОВ
208
Пластичность и хрупкость. Пластичность — со-
стояние твердого тела, в к-ром оно способно сохранять
изменение формы, вызванное воздействием внешних
сил, после того как силы сняты. Мерой пластично-
сти является величина остаточной (пластической)
деформации перед разрушением, выраженная обычно
в процентах. Хрупкость представляет собой такое
состояние твердого тела, когда остаточные деформации
либо вовсе не возникают вплоть до разрушения, либо
они весьма малы. Переход от состояния пластичности
к состоянию хрупкости можно осуществить либо
изменением темп-ры, либо соответствующим выбором
внешней среды, в к-рой происходит деформация дан-
ного твердого тела, либо изменением характера на-
пряженного состояния. В связи с этим пластичность
и хрупкость нельзя рассматривать как свойства тел,
одинаково им присущие при всех условиях деформа-
ции. Даже такое «абсолютно» хрупкое в обычных
условиях тело, как кристаллич. кварц, в условиях
весьма значительного всестороннего сжатия способно
пластически деформироваться под действием растяги-
вающих или сдвиговых напряжений.
То наименьшее напряжение, после снятия к-рого
в теле обнаруживаются остаточные деформации, наз.
пределом текучести данного материала.
Т. к. возможность обнаружения остаточной деформа-
ции зависит от точности выбранного метода (а также
от времени действия напряжения на тело), то понятие
предела текучести — условное понятие. Обычно за
предел текучести принимают такое напряжение, к-рое
при кратковременном действии вызывает пластич.
деформацию в 0,2% расчетной длины образца (Р02).
Пластич. деформация твердого тела всегда сопровож-
дается его упрочнением, т. е. ростом напряжения по
мере роста пластич. деформации. Упрочнение в про-
цессе пластич. деформации характеризуется коэфф,
упрочнения K=dP!dz кГ/см2, где Р— напряжение,
е — пластич. деформация. Коэфф, упрочнения наз.
иногда модулем нормальной пластичности. Его ве-
личина на 2—3 порядка меньше модуля нормальной
упругости и непостоянна вдоль кривой напряжение —
деформация: по мере роста пластич. деформации
величина коэфф, упрочнения падает.
Наиболее пластичными материалами являются
металлы. Металлич. монокристаллы с гексагональной
решеткой (цинк, кадмий и др.) пластически удлиня-
ются перед разрушением в 6—8 раз (при оптимальной
темп-ре и ориентации). На металлич. монокристаллах
наиболее отчетливо проявляется зависимость сопро-
тивления пластич. деформации от темп-ры. У моно-
кристаллич. кадмия предел текучести повышается
примерно в 4 раза с изменением темп-ры от -|-300°
до —252°, а у монокристаллич. алюминия при пони-
жении темп-ры с +600° до —185° — повышается в
8 раз. Коэфф, упрочнения меняется с темп-рой еще
в большей мере. У кадмия, напр., в интервале темп-р
от +200° до —200° он изменяется в 400 раз.
Влияние темп-ры как на предел текучести, так и
на коэфф, упрочнения проявляется гл. обр. в области
средних темп-p, тогда как при очень низких темп-рах
или при темп-рах, близких к плавлению, это влия-
ние невелико. В этой же области средних темп-р
имеет место влияние скорости деформации. С ростом
скорости деформации заметно увеличиваются как
предел текучести, так и коэфф, упрочнения. Влияние
темп-ры и скорости деформации на сопротивление
пластич. деформированию обусловливается одновре-
менным протеканием процессов упрочнения и разуп-
рочнения (отдыха). Пластич. деформация вызывает ис-
кажение плоскостей сдвига, появление дефектных
участков на этих плоскостях, приводящее к росту
напряжения при дальнейшем деформировании, что и
проявляется в упрочнении. При низкой темп-ре это
Рис. I. Зависимость предела
текучести Рт и предела проч-
ности а хладноломких мате-
риалов от темп-ры Т (схема
А. Ф. Иоффе).
высоких, так и более низ-
упрочнение развивается в полной мере, т. к. диффузи-
онные процессы, приводящие к «залечиванию» воз-
никающих дефектов структуры (отдых), заторможены.
Поэтому и влияние темп-ры в этих условиях мало.
При очень высоких темп-рах, близких к плавлению,
скорость диффузионных процессов столь велика, что
они успевают уже в процессе самой деформации лик-
видировать возникающие искажения решетки и
упрочнение практически не возникает. Следовательно,
и в этих условиях влияние темп-ры незначительно.
Но в интервале средних темп-р, когда в процессе
пластич. деформации имеет место конкурирующее
развитие и упрочнения и отдыха, предел текучести
и коэфф, упрочнения оказываются наиболее чувстви-
тельными к изменениям темп-ры и скорости дефор-
мации.
Многие материалы с понижением темп-ры перехо-
дят от пластичного состояния к хрупкому. Такие
материалы наз. хладноломкими. По А. Ф. Иоффе,
это обусловлено тем, что предел текучести таких
материалов в гораздо большей мере зависит от темп-ры,
чем предел прочности (см. рис. 1). При нек-ром зна-
чении темп-ры раньше до-
стигается предел прочно-
сти, чем предел текучести,
и материал хрупко разру-
шается без заметных пла-
стич. деформаций. При вы-
соких темп-рах, наоборот,
всегда ранее достигается
предел текучести, и мате-
риал до разрушения во-
влекается в пластич. де-
формацию. Температурная
область, в к-рой соверша-
ется переход от пластич-
ности к хрупкости (п о-
рог хладноломко-
сти) может быть сдви-
нута как в область более
ких температур изменением характера напряжен-
ного состояния. Так, глубокая и острая круговая
выточка на поверхности растягиваемого стержня
делает его хрупким при более высоких темп-рах, чем
для гладкого стержня. С другой стороны, замена рас-
тяжения кручением понижает темп-ру перехода в
хрупкое состояние.
В дислокационной теории пластичности порог хладнолом-
кости определяется след, образом:
Т =(Ц/й)[1Дп (v6)J
кр
где V — энергия активации, необходимая для освобождения
заблокированной дислокации, зависящая от напряжения Р,
действующего на дислокацию; k — константа Больцмана; б—
среднее время, протекающее до освобождения дислокации (или
величина, обратная вероятности освобождения дислокации за
единицу времени); v — постоянная с размерностью частоты.
Для зависимости порога хладноломкости от скорости деформа-
ции теория дислокации дает: 1/’РКр=—(A/U) 1пв Д- с, где v —
скорость деформации, с — постоянная.
Чрезвычайно резкое смещение порога хладно-
ломкости наблюдается на металлах в присутствии
сильных адсорбционноактивных веществ -т- расплавов
легкоплавких металлов. Так, монокристаллы цинка
в обычных условиях переходят в хрупкое состояние
при понижении темп-ры до минус (70—100°). Если же
их покрыть тонким слоем ртути (толщиной ~1 лк),
то этот переход осуществляется уже при +150—
+170°, что связано со значительным снижением по-
верхностной энергии на внутренних поверхностях
зародышевых трещин разрушения (см. Адсорбционное
понижение прочности').
Теоретически и экспериментально показано, что
способность твердых тел к пластич. деформированию
определяется наличием в их структуре дислокаций —
209
МЕХАНИЧЕСКИЕ СВОЙСТВА МАТЕРИАЛОВ
210
Рис. 2. Зависимость разру-
шающего напряжения Р от
числа циклов нагружения 7V;
— предел усталости.
л инейных дефектов структуры, способных переме-
щаться под действием малых напряжений и генери-
ро ваться внутри тела в процессе деформации. Испи-
та ния материалов на пластичность и хрупкость в
условиях растяжения, сдвига или кручения прово-
дятся обычно на специальных испытательных машинах.
Для прозрачных материалов (каменная соль, стекло,
плексиглас и др.) разработаны оптич. методы наблю-
дения возникающих деформаций в поляризованном
свете. Для более точных исследований малых оста-
точных деформаций (особенно в металлах) исполь-
зуется метод рентгенография, съемок.
Усталость (динамическая) — проявляется
в разрушении материала при его многократном нагру-
жении, хотя величина напряжения в каждом отдельном
нагружении может быть меньше предела текучести
(Р02). Чем выше напряжение в каждом отдельном
цикле нагружения, тем при меньшем числе циклов
происходит разрушение материала (см. рис. 2). То
напряжение в кГ/мм*, при
к-ром материал не разру-
шается при бесконечно
большом числе циклов,
наз. пределом уста-
лости. Усталость мате-
риалов мало зависит от
закона изменения напря-
жений в течение одного
цикла и от частоты изме-
нения напряжений (до
100 гц). В основном на
усталость влияет характер
напряженного состояния
(растяжение, кручение, из-
гиб) и температура. Предел усталости наиболее вы-
сок при изгибе н наименее — при кручении. С ро-
стом темп-ры предел усталости обычно понижается.
Механизм усталостного разрушения связан с нали-
чием упругих несовершенств в реальных твердых
телах. Хотя напряжения в каждом цикле нагружения
и очень малы, но в результате действия большого
числа таких циклов (10’—107 циклов) в наиболее
слабых структурных элементах твердого тела про-
исходит локальное накопление пластич. деформаций,
и материал в этих местах предельно упрочняется.
Здесь образуется первая микротрещина разрушения,
к-рая постепенно растет с числом циклов нагружения
и в конце концов приводит к поломке образца.
Динамич. усталость свойственна всем твердым
телам, но наибольшее технич. значение имеет уста-
лость металлов, в связи с неизбежным эксцентриси-
тетом при установке валов и осей в разного рода
машинах и двигателях. Особенно большую опасность
усталость металлов представляет в тех случаях,
когда подвергаемый циклич. нагружениям металл
находится в коррозионноактивной среде. Коррозия,
протекающая одновременно с усталостным нагруже-
нием,-резко снижает предел усталости и быстро при-
водит к поломке в результате энергичного разъедания
металла, гл. обр. по границам зерен. Для повышения
сопротивляемости металла усталостным нагружениям
в технике используется поверхностное упрочнение,
вызываемое обдувкой металла дробью или обкаткой
роликами. Деформация поверхностного слоя, вы-
званная такой обработкой, приводит к возникновению
в этом слое сжимающих напряжений, к-рые в даль-
нейшем, в условиях усталостного нагружения, сни-
жают величину растягивающих напряжений и тем
самым увеличивают усталостную прочность металла.
Усталость (статическая) — проявляется в
разрушении материала при длительном действии по-
стоянного напряжения, величина к-рого может быть
намного ниже предела прочности и даже ниже предела
текучести Р0,2. Изучение статич. усталости материалов
в широкой области темп-р и напряжений показало, что
время до разрушения в зависимости от напряжения
и темп-ры для очень широкого круга материалов под-
чиняется общей закономерности:
т=т0 exp [(Z70—yP)]kT]
где Uo — уР = U (энергия активации процесса раз-
рушения); Р — растягивающее напряжение и Т —
абс. темп-ра; Z70, у и т0 — постоянные коэфф., зави-
сящие от природы вещества. Следовательно, дли-
тельность процесса разрушения при заданном напря-
жении определяется темп-рой и величиной актива-
ционного барьера U = UQ — уР. Величина U за-
висит от нагрузки и уменьшается с увеличением
растягивающего напряжения. Оказалось, что энергия
U0 совпадает для разных материалов с их энергией
сублимации, а величина т0 по порядку величины
равна периоду собственных колебаний атомов в
решетке.
Механизм статич. усталости связан с кинетикой
роста трещины разрушения и активацией этого
процесса за счет тепловой подвижности атомов твер-
дого тела. Возникшая при заданном напряжении в
наиболее слабом месте испытуемого тела зародыше-
вая трещина далее медленно подрастает в результате
флюктуационного ослабления межатомных связей в
своей тупиковой части. Этот процесс подрастания
трещины до критич. величины, когда приложенного
напряжения окажется достаточно для ее лавинного
распространения на все сечение образца, и опреде-
ляет собой в основном время жизни образца под
напряжением. Молекулярный механизм роста заро-
дышевой трещины связывают также с наличием ва-
кансий в кристаллич. решетке. Возникновение
перенапряжений на краях трещины приводит к сни-
жению энергии активации образования в этих местах
вакансий, что значительно ускоряет самодиффузию
и обеспечивает нужную скорость подрастания тре-
щины.
Испытания материалов на динамич. усталость
проводят на специальных машинах, позволяющих
циклически нагружать испытуемые детали с заданной
частотой (обычно 3000 циклов в минуту, т. е. 50 гц).
При этом определяется как ограниченная выносли-
вость материала (при напряжениях выше предела
усталости), так и величина предела усталости. Испы-
тания на статич. усталость проводятся т. о., чтобы на
образец действовало постоянное по величине напря-
жение. Но так как сечение образца может умень-
шаться, если в нем развивается пластич. деформация,
то, следовательно, должна меняться и сила, прило-
женная к образцу. Это достигается использованием
различных механизмов непрерывной разгрузки.
Ползучесть — проявляется в медленном пластич.
течении твердого тела при длительном действии таких
напряжений, к-рые при кратковременном действии
вызывают лишь упругие деформации. Наиболее общая
картина ползучести наблюдается в той области темп-р,
в к-рой процессы отдыха (разупрочнения) протекают
с достаточной скоростью. При этих темп-рах (тем
более высоких, чем выше темп-ра плавления данного
твердого тела) отчетливо обнаруживаются три стадии
ползучести: 1) нестационарное течение с уменьшаю-
щейся скоростью, связанное с развивающимся упроч-
нением; 2) стационарное течение с постоянной мини-
мальной скоростью в результате компенсации нара-
стающего упрочнения отдыхом и, наконец, 3) стадия,
обнаруживаемая при очень длительном наблюдении,—
нестационарное течение с возрастающей скоростью,
приводящее к разрушению испытуемого образца и
связанное с развитием трещины разрушения. При низ-
ких темп-рах, когда отдых практически не развивается,
211
МЕХАНИЧЕСКИЕ СВОЙСТВА МАТЕРИАЛОВ
212
Рис. 3. График ползучести ма-
териала под действием постоян-
ного напряжения: I — началь-
ная стадия ползучести; II —
основная стадия ползучести —
пластическое течение с постоян-
ной скоростью; III — заклю-
чительная стадия ползучести —
течение с возрастающей скоро-
стью, завершающееся разрывом.
имеет место лишь первая стадия ползучести, за-
вершающаяся прекращением ползучести в резуль-
тате развивающегося упрочнения. На рис. 3 приведена
типичная кривая зависимости деформации от времени
при постоянном напряжении в процессе ползучести.
При достаточно высоких
темп-рах ползучесть твер-
дых тел может разви-
ваться по механизму
вязкого течения, связан-
ного с диффузионным пе-
реносом вещества. У ме-
таллов при высоких
темп-рах ползучесть мо-
жет реализоваться в
скольжении по границам
зерен, что сходно с тече-
нием аморфных материа-
лов — стекла или смолы.
Всякого рода примеси
обычно повышают сопро-
тивление материала пол-
зучести. При испытаниях
на ползучесть испыта-
тельные машины кон-
струируются таким образом, чтобы можно было
одновременно проводить испытания многих образцов.
Это вызвано большой продолжительностью таких ис-
пытаний, длящихся обычно сотни и тысячи часов.
Твердость — сопротивление местным деформациям
поверхностных слоев материала. Величина твердости
материалов обычно оценивается сопротивлением вдав-
ливанию в их поверхность шарика из более твердого
материала (твердость по Бринеллю или Роквеллу).
Кроме того, твердость оценивается методом вдавли-
вания пирамиды, царапания, маятниковым методом и
методом Шора. Простейший метод оценки относитель-
ной твердости двух тел заключается в царапании
одного другим. Более твердое тело оставляет царапину
на более мягком. На этом принципе построена шкала
твердости минералов по Моосу: 1. Тальк; 2. Гипс;
3. Кальцит; 4. Флюорит; 5. Апатит; 6. Полевой шпат;
7. Кварц; 8. Топаз; 9. Корунд и 10. Алмаз. В этой
шкале твердость характеризуется номером и не имеет
размерности. За меру твердости по Бринеллю Нв
принимается среднее сжимающее напряжение в
кГ/мм2, вычисляемое на единицу поверхности шаро-
вого отпечатка. При замене шарика одного диаметра
другим нагрузка Р на шарик меняется так, чтобы
отношение P/d2 оставалось постоянным (на основании
законов подобия). С помощью измерений твердости
и микротвердости (вдавливание алмазной пирамиды,
но при очень малой нагрузке) можно оценить степень
упрочнения поверхностных слоев твердых тел в раз-
личных процессах обработки, провести несложный
фазовый анализ металлич. сплавов, контролировать
М. с. м.
Прочность — сопротивление материала разруше-
нию. Всякий процесс деформации, как упругой, так и
пластич., при достаточном повышении напряжения
(или при достаточно длительном действии постоянного
напряжения) заканчивается разрушением материала.
Если разрушение материала рассматривать как одно-
временный разрыв связей между всеми атомами, рас-
положенными по обе стороны площадки в 1 см2,
выбранной перпендикулярно растягивающей силе,
то величина напряжения отрыва одной части тела от
другой может быть теоретически оценена и оказывается
весьма значительной. Это теоретич. значение проч-
ности твердого тела, вычисляемое на основе той или
иной модели межатомных сил для «идеальной», т. е.
не содержащей дефектов, кристаллич. решетки, ока-
зывается равным: Рт—а\^Еа1Ь, где Е— модуль
Юнга, о — поверхностное натяжение, b — постоян-
ная решетка и а—коэфф., равный по порядку
величины 1. Отсюда получаем, напр., для каменной со-
ли Рт = 200 кГ/мм2, а для металлов == 103—10*кГ/мм2.
Но экспериментально определяемая прочность ка-
менной соли на разрыв не превосходит 0,5 кГ1мм2,
а у металлов — 10—100 кГ1мм2, т. е. всегда значи-
тельно ниже, чем теоретич. Такое расхождение
связывается обычно с присутствием в реальном твер-
дом теле различных дефектов структуры и прежде
всего микротрещин.
Наиболее простую оценку разрывного напряжения хруп-
кого тела при наличии в нем трещин можно получить следую-
щим образом. До появления трещины плотность упругой энер-
гии в теле была равна W — Р2/2Е. С появлением трещины дли-
ной I происходит разгрузка на площади = /2; убыль упругой
энергии составляет при этом и Р2Р/2Е. Вместе с тем наличие
трещины связано с дополнительной энергией 21а, где а — сво-
бодная поверхностная энергия стенок трещины. Общее изме-
нение энергии при наличии трещины будет &W=21а—Р212/2Е.
Кривая £РЛ’--Ш) имеет максимум при gj-(MV)=0. Следова-
тельно, пока длина трещины меньше того критпч. значения,
к-рое соответствует максимуму ДЖ, равновесие остается ус-
тойчивым, т. е. трещина не имеет тенденции удлиняться. Но
как только длина трещины становится равной или больше
критпч., энергия начинает убывать с удлинением трещины и,
следовательно, трещина будет удлиняться до полного разрыва
тела.
(Д1У)-= 2а-Р21,Е = 0
Отсюда, при заданном значении длины трещины I, получаем
разрывное напряжение Р—V2Еа/1. Сравнивая это выражение с
теоретич. значением прочности Р —аУЕо/Ь, получаем Р/Рт ~
к Vb/l. Т. к. Ъ — постоянная решетки ~10-8аи, то достаточно
в теле иметь трещинку длиной 1~1 мк, чтобы прочность тела
понизилась в 100 раз по сравнению с теоретич. ее значением.
Однако наличие микротрещин в твердом теле
может снизить его прочность только в условиях
хрупкого разрушения. В тех случаях, когда разру-
шению предшествует пластич. деформация, степень
опасности этих трещин резко снижается в результате
выравнивания напряжений по сечению (снятие пере-
напряжений на концах трещины), вызываемого
пластич. деформацией. Но вместе с тем сама пластич.
деформация на нек-рой стадии своего развития
создает условия для зарождения трещин в деформи-
руемом теле. Решающее значение имеет при этом то
обстоятельство, что пластич. деформация в твердом
теле развивается всегда крайне неоднородно, что
тесно связано с дислокационной природой пластич.
течения. Эти неоднородности деформации прояв-
ляются, в частности, в возникновении незавершенных,
т. е. не распространившихся на все сечение кристалла
или зерна пластич. сдвигов. В дислокационной трак-
товке это означает, что в одной или нескольких
близко расположенных плоскостях скольжения обра-
зовались скопления дислокаций перед прочными
препятствиями — двойниковыми границами, грани-
цами блоков мозаики, «сидячими» дислокациями
и т. д. Такое скопление дислокаций всегда связано с
возникновением высоких локальных концентраций
напряжения, во много раз превосходящего напря-
жение, приложенное извне. Эти высокие местные
напряжения приводят на нек-рой стадии пластич.
деформации к возникновению и постепенному раз-
витию микротрещин — зародышей разрушения.
Экспериментальное изучение закономерностей раз-
рушения кристаллов и теоретич. анализ полученных
закономерностей приводят к выводу, что появление
первых зародышевых трещин разрушения связано
только со сдвигом по плоскостям скольжения и опре-
деляется величиной скалывающего напряжения т. Но
в дальнейшем, когда эти зародышевые трещины до-
стигнут в своем развитии определенной величины, ре-
шающая роль переходит к нормальным напряжениям,
к-рые «дорывают» трещину до полного разрушения
213
МЕХАНИЧЕСКИЕ СВОЙСТВА ПОЛИМЕРОВ
214
образца. Самый начальный момент возникновения
зародышевой трещины, не имеющей еще вполне раз-
витой свободной поверхности, можно представить
как процесс объединения первых (ведущих) дисло-
каций скопления, приводящий к образованию узкого
полого дислокационного ядра. Такие полые ядра
могут образовываться в процессе пластич. дефор-
мации на очень ранних ее стадиях при весьма малом
скалывающем напряжении т (извне приложенном
к кристаллу). С ростом т растет и длина микротре-
щины, причем каждому т соответствует своя вели-
чина равновесной трещины I (равновесной в том
смысле, что ее величина еще недостаточна для пол-
ного разрушения тела при данном нормальном к пло-
скости трещины напряжении). Расчет показывает, что
при данном т в деформируемом кристалле может воз-
никнуть трещина с максимальной длиной /^рт21,2/Е<т,
где L — диаметр кристалла или величина зерна в
поликристаллич. материале, Е — модуль Юнга и
а — уд. свободная поверхностная энергия; р — коэфф.,
величина к-рого близка к 1. С другой стороны, при
наличии в материале трещины длиной I он разру-
шается, когда нормальное к плоскости трещины напря-
жение достигает величины Р=аУ Eall. Сравнивая
оба эти выражения, получаем закон разрушения твер-
дого тела Рт--.уУ Ea/z=const, т. е. произведение
из нормального напряжения на скалывающее в момент
отрыва есть величина постоянная для данного твер-
дого тела. Этот закон разрушения хорошо подтвер-
ждается экспериментально во всех тех случаях, когда
разрушение кристалла происходит по определенной
кристаллография, плоскости после нек-рой пластич.
сдвиговой деформации.
Существенное влияние на прочность материала
оказывают размеры испытуемых образцов (масштаб-
ный фактор). С уменьшением размеров прочность
повышается и для весьма тонких нитей (до 1 мк)
достигает теоретич. значения. Так, прочность стек-
лянных нитей диаметром 22 мк равна 20 кГ/мм2,
диаметром 12,5 мк —140 кГ/мм2 и диаметром 2,5 мк —
~560 кГ/мм2. Зависимость прочности топких нитей
от диаметра характеризуется следующей эмпирич.
ф-лой: Р = А — В/а, где А и В — постоянные. Анало-
гичные явления наблюдаются и у металлов. Весьма
тонкие металлич. нити (несколько микрон в диаметре),
получившие название «усов», обнаруживают чрез-
вычайно высокую прочность, приближающуюся к
теоретич. Так, напр., прочность нитевидного кри-
сталла железа диаметром 1,6 мк равна 1350 кГ/мм2.
При этом модуль упругости таких тонких нитей не
отличается от модуля упругости массивных образ-
цов. То обстоятельство, что прочность различных
материалов как хрупких, так и пластичных зависит
от размеров испытуемых образцов, указывает на
значительную роль дефектных участков структуры
твердых тел, как уже имеющихся, так и возникающих
в процессе деформации. С уменьшением размеров
образцов вероятность возникновения таких дефект-
ных участков уменьшается, в результате чего воз-
растает прочность.
Лит.: Кузнецов В. Д., Физика твердого тела, т. 1,
2, 4, 2 изд., Томск, 1937—47; Беляев Н. М., Сопротивление
материалов, 12 изд., М., 1959; Фридман Я. В., Механиче-
ские свойства металлов, 2 изд., М., 1952; Шмид Е., Боас
В., Пластичность кристаллов, в особенности металлических,
пер. с нем., М.—Л., 1938; Ильюшин А. А., Пластичность,
М.—Л., 1948; Давиденков Н. Н., Некоторые проблемы
механики материалов, Л., 1943; Иоффе А. Ф., Физика кри-
сталлов, М.—Л., 1929; ЛихтманВ. И., Ребиндер
П. А., К а р п е н к о Г. В., Влияние поверхностно-активной
среды иа процессы деформации металлов, М., 1954; Гарбер
Р. И., Гиндин И. А., Усп. физ. наук, 1960, 70, вып. 1;
Лихтман В. И., Щукин Е. Д., Усп. химии, 1960, 29,
вып. 10; Ш р е й н е р Л. А., Твердость хрупких тел, М.—Л.,
1949; Машиностроение, Энциклопедический справочник, т.
1 —3, М., 1947—48; Одинг И. А., Современные методы испы-
тания металлов, 4 изд., М., 1944; Авдеев Б. А., Техника
определения механических свойств металлов, м., 1949; Ш а-
пошников Н. А., Механические испытания металлов,
М.—Л., 1951; Жданов Г. С., Физика твердого тела, М.,
1961; Савицкий Е. М., Влияние температуры на механи-
ческие свойства металлов и сплавов, М., 1957.
МЕХАНИЧЕСКИЕ СВОЙСТВА ПОЛИМЕРОВ.
Содержание:
Введение.......................................214
Деформационные свойства полимеров..............214
Релаксационные свойства полимеров..............219
Вязкое течение полимеров.......................220
Прочность полимеров............................222
Временная зависимость прочности полимеров . . ,22 3
Фрикционные свойства полимеров.................225
Введение. Механические свойства полимеров —
комплекс свойств, определяющих поведение поли-
меров при действии механич. сил. В отличие от других
твердых тел, для полимеров характерны: 1) сильно
выраженные релаксационные свойства, проявляю-
щиеся в запаздывании деформации и релаксации
напряжения; 2) двойственная природа обратимых
деформаций (обычная упругость, характерная для
твердых тел, и высокоэластичность); 3) влияние
молекулярной ориентации на прочность; 4) большое
влияние физико-химич. процессов (действие агрес-
сивных сред, процессы старения); 5) активирующее
влияние механич. сил на химич. процессы в полиме-
рах, приводящее к изменению их структуры и разви-
тию явлений утомления. У полимеров, кроме того,
наблюдаются два вида необратимой деформации:
1) при малых напряжениях молекулярный механизм
течения в общих чертах тот же, что и для обычных
жидкостей (диффузионный); 2) при больших напря-
жениях наблюдается «химическое» течение (см.
Вязкость полимеров).
К основным М. с. п. относятся деформационные,
прочностные и фрикционные свойства. Все полимеры
в зависимости от их химич. природы, строения, мол.
веса и темп-ры находятся в кристаллическом или в
одном из аморфных состояний — стеклообразном,
высокоэластическом (каучукоподобном) или вязко-
текучем.
Кристаллич. и твердые аморфные полимеры при-
меняют обычно в виде волокон, пленочных материалов
и массивных изделий. На их основе изготовляют также
различные материалы, содержащие, помимо полимера,
также и др. вещества (напр., стеклопластики, пено-
пласты, поропласты и др.). Каучукоподобные поли-
меры используют для изготовления резин.
В процессе переработки полимеров в изделия они
переводятся в вязкотекучее или пластич. состояние
(растворением, пластификацией, нагреванием).
Деформационные свойства полимеров. Кри-
сталлические полимеры при малых
деформациях (см. Деформация полимеров) ведут себя
как обычные твердые тела, при больших — претер-
певают фазовое превращение от исходной, обычно
изотропной, фазы к ориенти-
рованной с расположением це-
пей вдоль оси растяжения.
Фазовое превращение сопро-
вождается значительным изме-
нением длины образца при по-
стоянном значении растягиваю-
щего напряжения авэ, полу- Рис t диаграмма ра-
чившего название предела стяжения кристаллине-
вынужденной эл а- ского полимера,
стичности; овэ зависит
от темп-ры, скорости растяжения и природы поли-
мера. На рис. 1 показана зависимость цвэ от уд-
линения. На стадии I происходит равномерное не-
большое растяжение образца, при достижении точки
А в образце внезапно возникает «шейка», к-рая
при дальнейшем растяжении образца на стадии
Удлинение
215
МЕХАНИЧЕСКИЕ СВОЙСТВА ПОЛИМЕРОВ
216
II постепенно распространяется на весь образец.
Начиная с точки Б, образец вновь растягивается как
целое вплоть до разрыва. С уменьшением мол. веса
прочность кристаллич. полимера падает, и точка
разрыва В смещается по кривой деформации влево.
Предел прочности при этом может стать меньше авэ, и
тогда полимер испытывает только хрупкий разрыв
без фазового превращения. Большие деформации
кристаллич. полимеров по своей природе — высоко-
эластические, т. к. связаны с изменением конфор-
маций и ориентацией макромолекул.
Аморфные полимеры при малых напря-
жениях могут находиться в трех состояниях: стек-
лообразном (твердом), высокоэластиче-
ском и вязкотекучем (рис. 2, кривая 1).
Темп-ры перехода
Рис. 2. Термомеханическая кривая (Г)
и кривая механических потерь (2)
аморфного полимера.
Тс (темп-ра стекло-
вания) и Тт (темп-ра
текучести) из одно-
го состояния в дру-
гое зависят от типа
и структуры поли-
мера, молекулярно-
го веса, пластифика-
торов, введенных в
полимерные матери-
алы, и т. д., а также от частоты или времени дей-
ствия силы. От частоты деформации v темп-ра стекло-
вания Тс, выраженная в абс. шкале темп-р, зависит
след, образом: Т~1=А —Blgv, т. е. возрастает с
увеличением частоты. Высокоэластичный линейный
полимер можно перевести в стеклообразное или вязко-
текучее состояние при заданной темп-ре, если изме-
нять частоту деформации. Поэтому эти состояния
полимера рассматриваются в связи с режимом меха-
нич. воздействий. В отсутствие последних аморфные
полимеры ведут себя как жидкости, обладая теми
же термодинамич. свойствами, и обнаруживают при
охлаждении переход из жидкого структурного в
стеклообразное состояние («структурное» стеклование).
Все факторы, уменьшающие роль или величину
межмолекулярного взаимодействия (напр., пластифи-
кация), приводят к снижению темп-р стеклования и
текучести. Образование небольшого числа химич.
связей между макромолекулами (напр., вулкани-
зация каучуков) приводит к резкому увеличению
Тт или полной потере текучести, но практически
не изменяет Тс. Уменьшение мол. веса линейных
полимеров до определенного предела не изменяет
Тс, но снижает Тт. При достаточно низком мол. весе
высокоэластичность полностью теряется, и такое
вещество при нагревании сразу переходит из стекло-
образного в вязкотекучее состояние.
В стеклообразных полимерах при малых напряже-
ниях возникает только упругая деформация с модулем
Юнга 200—600 кТумм2 (для стали модуль Юнга равен
20 000 к Г/мм2). В высокоэластич. полимерах встре-
чаются оба вида обратимой деформации, однако высо-
коэластичная превышает упругую примерно в 103—
104 раз и поэтому практически всю деформацию
полимера относят за счет высокоэластичной. Модуль
высокоэластичности при одноосном растяжении ра-
вен 0,2—0,01 кГ/мм2. (Переводные коэфф, на между-
народные единицы см. в ст. Международная система
единиц).
Аморфные стеклообразные полимеры при больших
напряжениях имеют более сложные деформационные
свойства. Растяжение таких полимеров происходит
аналогично растяжению кристаллических (рис. 3).
В точке А, когда напряжение достигает предела
вынужденной эластичности аЕЭ, в наиболее слабом
месте образца образуется «шейка», в к-рую по мере
растяжения переходит весь образец, затем на участке
БВ образец вновь целиком растягивается до разрыва.
Предел вынужденной эластичности тем больше, чем
ниже темп-ра и выше
скорость деформации
(рис. 4), но при низких
темп-рах (ниже темп-ры
хрупкости ТХр) он ста-
новится больше прочно-
сти полимера и послед-
ний хрупко разрывается,
не достигая точки А
(рис. 3). С уменьшением
мол. веса также дости-
гается состояние, когда
точка разрыва В (рис. 3),
гает точки А, и материал
рыв. Полимерные стекла,
кулярных, обладают
меньшей хрупкостью
и даже гибкостью,
что связано с их
структурой (неплот-
ность упаковки жест-
ких цепей) и наличи-
ем вынужденно-эла-
Рис. 3. Диаграмма растяжения
стеклообразного полимера.
перемещаясь влево, дости-
испытывает хрупкий раз-
в
отличие от
низкомоле-
Область £ о
эксплуатации g с
аэл 5 _
8
Тхр. Тс
Температура
Высоко-
ластические
УпрУгие^е3-9ефОрмации
деформации \
Г»
Тс — температура стеклования;
температурной области высокой
Рис. 4. Диаграмма де-
формапионно - прочност-
ных состояний аморфных
полимеров: ТХр — гра-
ница перехода от темпе-
ратурной области хруп-
кости к температурной
области разрыва в ори-
ентированном состоянии;
Тп — граница перехода от
эластичности к области пластичности; Тт — температура те-
кучести; сгХр — хрупкая прочность; сгвЭ — предел вынужден-
ной эластичности; о9л — прочность высокоэластичного ма-
териала (напряжение рассчитано на поперечное сечение об-
разца при разрыве); ап — предел пластичности.
стич. деформаций. При напряжениях ниже авэ
стеклообразный полимер испытывает в основном уп-
ругую деформацию. Только под действием внешних
сил, превышающих авэ, развивается высокоэластич.
деформация.
Это явление объясняется релаксационной теорией следую-
щим образом. Время релаксации 6, характеризующее скорость
перегруппировки элементов структуры (сегментов полимер-
ной цепи), а следовательно, и скорость высокоэластич. де-
формации, зависит от напряжения <3 и абс. темп-ры Т:
9 = Лехр^-^Т—) (О
где и0— энергия активации, определяемая потенциальными
барьерами, к-рые преодолевают элементы структуры при пере-
ходе из одного равновесного положения в другое; а — объем
элемента структуры; h — постоянная Больцмана. Из (1) сле-
дует, что при малых темп-рах и напряжениях 9 может быть
гораздо больше времени наблюдения; поэтому высокоэластич.
деформации цепей «заморожены». Время релаксации 9, срав-
нимое с временем наблюдения (или скоростью деформации),
при к-ром высокоэластич. деформация «размораживается»,
может быть достигнуто повышением либо темп-ры до Т , либо
напряжения до <?вэ. Отсюда следует, что Тс и свэ зависят от
времени наблюдения или скорости деформации. Из (1) следует
также, что <?вэ является лищь условной границей напряжен-
ного состояния, а высокоэластич. деформация медленно раз-
вивается и при меньших напряжениях.
Ползучесть твердых полимеров
проявляется в медленном накоплении при малых
напряжениях высокоэластич. деформации, что и
отличает это явление от ползучести металлов, когда
происходит медленное накопление истинной остаточ-
ной деформации. Высокоэластич. деформация поли-
меров после разгрузки остается «замороженной»
неограниченно долгое время, но при нагреве выше
217
МЕХАНИЧЕСКИЕ СВОЙСТВА ПОЛИМЕРОВ
218
Тс полностью исчезает, и образец принимает преж-
нюю ферму. Действие излучений высокой энергии
на полимеры аналогично резкому нагреванию —
ползучесть ускоряется во много раз, но после пре-
кращения излучения ее скорость сразу возвращается
к прежнему значению.
В высокоэластическом состоянии
при темп-рах выше Тс время релаксации мало, по-
этому высокоэластич. деформация развивается сразу
при любых малых напряжениях. Она характеризуется
малым модулем высокоэластичности (0,01—0,2 кГ/мм2)
и большими механически обратимыми деформациями,
к-рые могут во много раз превышать начальные разме-
ры образщ. Каучуки и резины являются типичными
высокоэластич. материалами в области темп-р от
—70 до -J-1000 и выше. При малых деформа-
циях (несколько %) напряжение пропорционально
деформации во всей области высокоэластичности.
В случае неравновесных деформаций коэфф, про-
порциональности (модуль высокоэластичности Е)
зависит от временного режима испытания вследствие
релаксационных свойств полимера. При больших
растяжениях, достигающих сотен процентов, проис-
ходит значительная ориентация макромолекул. Про-
цесс растяжения заканчивается разрывом, который
наступает тем раньше, чем выше темп-ра.
При нек-рой темп-ре Т„ и выше ее вплоть до
темп-ры текучести Тт материал при растяжении
переходит через предел пластичности ап (рис. 4).
В этой области линейный полимер характеризуется
диаграммой растяжения, сходной с диаграммой рас-
тяжения пластичных металлов (рис. 5). До точки А
(предела пластичности) наблюдается гл. обр. высоко-
эластич. деформация, а после нее — также и необра-
тимое течение материала. Перед разрывом (точка В)
происходит образование шейки, в к-рой и разрывается
Рис. 5. Рис. 6.
Рис. 5. Диаграмма растяжения линейного полимера в
пластическом состоянии: 1 — напряжение рассчитано на
начальное; 2 — напряжение рассчитано на истинное по-
перечное сечение образца.
Рис. 6. Кривая растяжения высокоэластичного пространст-
венно-структурированного полимера: 1—напряжение рас-
считано на начальное; 2 — на истинное сечение.
Гибкость макромолекул, к-рая особенно отчетливо
проявляется выше 7’с, где тепловое движение доста-
точно интенсивно, является причиной высокоэластич.
свойств полимеров. Упругая деформация полимеров
в стеклообразном и кристаллич. состояниях связана
с изменением средних расстояний между частицами,
а высокоэластическая — с перемещением звеньев
гибких цепей без изменения среднего расстояния
между цепями. Высокоэластич. деформация наиболее
четко проявляется у пространственно-структуриро-
ванных полимеров, получаемых при поперечном
«сшивании» линейных макромолекул (напр., в про-
цессе вулканизации). Не очень большое «сшивание»
препятствует необратимым перемещениям макромоле-
кул и текучести полимера. Поэтому пространственно-
структурированные полимеры в высокоэластич.
состоянии способны восстанавливать свою форму
после разгрузки, также как и упругие твердые тела.
По другим свойствам они близки к жидкостям.
Жидкости и пространственно-структурированные
полимеры являются аморфными веществами, их коэфф,
теплового расширения и сжимаемости близки и
намного меньше, чем у твердых тел. Те и другие под-
чиняются закону Паскаля. В то же время природа
высокоэластич. деформации пространственно-струк-
турированных полимеров отлична от природы дефор-
мации твердых тел, но сходна с природой упругости
газа. Так, напряжение в деформированной резине,
как и давление сжатого газа, пропорционально абс.
темп-ре. Такое сочетание в высокоэластич. полимерах
свойств твердого тела, жидкости и газа связано со
строением макромолекул и их гибкостью (см. Гиб-
кость цепных молекул).
Высокая эластичность, обусловленная
гибкостью цепных макромолекул, наблюдается у
полимеров самой различной химич. природы: у ти-
пичных углеводородов, напр. расплава полиэтилена,
полиизопрена (натурального каучука), полиизобу-
тилена; кремнийорганич. каучуков, напр. полидиме-
тилсилоксана, а также у неорганич. полимеров, напр.
полифосфонитрилхлорида, и даже у неорганич. сте-
кол (слабая высокая эластичность наблюдается выше
темп-ры стеклования).
Если к полимеру приложена внешняя сила, то
молекулярные цепи вследствие их гибкости будут
развертываться и цепи будут принимать вытянутые
конформации. После прекращения действия внешней
силы цепи испытывают под действием теплового
движения беспорядочные толчки, стремящиеся изог-
нуть и свернуть их в клубки, отвечающие наиболее
вероятным конформациям. Термодинамически переход
в более вероятное состояние описывается как процесс
возрастания энтропии. Поэтому возвращение дефор-
мированного образца пространственно-структуриро-
ванного полимера в начальное состояние сопровож-
дается увеличением энтропии. В этом отличие высо-
коэластич. деформаций от деформаций твердых тел,
когда энтропия практически не меняется. В первом
приближении принимается, что при деформации
высокоэластичного полимера его внутренняя энергия
не изменяется. Поэтому из термодинамич. соотноше-
ний следует, что истинное напряжение а зависит
только от изменения энтропии в процессе равновес-
ной изотермич. деформации:
а = -ПЦ (2)
где Т — абс. темп-ра, Л — кратность растяжения,
5 — энтропия единицы объема, убывающая по мере
растяжения.
Простейшая статистич. теория равновесной высокоэластич.
деформации, не учитывающая взаимодействия между цепями
пространственного полимера, приводит к следующему выраже-
нию высокоэластич. потенциала трехмерного полимера:
W=>;2g(^ + Л2 + Ь2) (3)
где W — упругая энергия 1 с.м.3 деформированного полимера,
&=NkT— модуль сдвига, N— число цепей сетки в 1 см3,
k — постоянная Больцмана, , Л.2, Л.3— главные кратности рас-
тяжений по трем осям координат (при растяжении они больше,
а при сжатии меньше единицы). В недеформированном состоя-
нии Л, 2== 1 •
Для однородных напряженных состояний (одноосного,
двухосного или чистого сдвига) соответствующие главные
истинные напряжения 01>сг2>сг3 находятся между собой
в след, соотношениях:
„ „_4 4 „ „-4^4 QW
01 a ‘ 0Л, ’ 2 0Х2 ?'а0/.а
Для одноосного растяжения или сжатия в направлении 1,
обозначая о1=ц(о2=сУз=0); и учитывая, что Х2=Л3=Л
219
МЕХАНИЧЕСКИЕ СВОЙСТВА ПОЛИМЕРОВ
220
так как из условия несжимаемости резины Х.1Х.2Х.3=1), имеем
O=G(V-1/M (4)
Для двухосного симметричного растяжения в направлениях
1 и 2 с1=О2=о; (Оа=0); Х.,=Х.2=>. и Х.3=Х.-2, поэтому:
O=G(M-1/V) (5)
Т. к. эта теория лишь качественно согласуется с опытом,
то предложены различные ур-ния, выведенные из нестрогих
теоретич. соображений, но лучше описывающие деформацию
сетчатых полимеров. Кривая растяжения резины, полученная
в равновесных условиях (рис. 6), почти линейна до 150—200%
растяжения (если напряжение рассчитывать на истинное се-
чение образца), т. е. а=Ех(к—1), где равновесный модуль
Ех и число цепей N пространственной сетки в 1 cat3 находятся
в след, соотношении: N=C(Eo0/T)3/2. Эта ф-ла позволяет оп-
ределять частоту пространственной сетки по механич. данным.
Если Ех выражать в кГ/см2, то для каучуков константа С
имеет'значения 1-1023—З'Ю22.
Релаксационные свойства полимеров. Релакса-
ционный характер деформации наиболее ярко прояв-
ляется в переходной области от стеклообразного
к высокоэластич. состоянию, а для линейных полиме-
ров и при переходе материала в вязкотекучее состоя-
ние. Релаксационные свойства обусловливают многие
особенности механич. свойств полимерных материалов.
Как атомы и молекулы в низкомолекулярных жидко-
стях под действием теплового движения переходят
из одного равновесного положения в другое, так и
участки линейных макромолекул (сегменты) переме-
щаются из одного положения в другое. При этом
частота перехода сегментов из одного равновесного
состояния в соседнее зависит от величины потен-
циальных барьеров и темп-ры, а также от напряже-
ния — чем выше напряжение, тем легче совершается
переход сегментов в направлении силы и труднее в
противоположном. Развитие деформации цепи про-
исходит путем последовательного перемещения сег-
ментов, т. е. во времени. Поэтому высокоэластич.
деформация отстает от напряжения. При периодич.
изменении напряжения при каждом цикле деформа-
ции происходят механич. потери, изображаемые на
диаграмме петлей гистерезиса (см. Гистерезис меха-
нический). Иначе говоря, феноменологической причи-
ной как отставания деформации от напряжения, так
и механических потерь, является внутреннее трение.
Несколько выше температуры стеклования высоко-
эластическая деформация за период колебания или
за время наблюдения не сразу достигает максималь-
ного значения, так как изменение конфигурации цепи
под действием приложенной силы из-за внутреннего
трения происходит постепенно. При повышенных
температурах время «оседлой жизни» каждого сег-
мента для каучуков настолько мало, что цепи после
приложения нагрузки деформируются очень быстро
(при 20°С за время 10-3—10-5 сек в зависимости от
типа каучука).
Релаксационные свойства полимеров с достаточным при-
ближением в общей форме описываются уравнением после-
действия Больцмана (для одноосного напряженного состояния
или сдвига):
е = + \ ф (1 — О') с (О') d’O (6)
е0 J
о
где е — деформация, <3 — напряжение, <р(£—О') — ядро инте-
грального уравнения (в нем учтено строение полимера и влия-
ние температуры), t —время, О' — переменная интегрирования,
Е<,— модуль упругости твердого полимера, характеризующий
быструю деформацию полимера со скоростью звука с в этом
полимере (для пластич. масс с= 1000 м/сек). Он рассчитывается
по формуле E0=qc2, где q — плотность полимера. Для каучу-
коподобных полимеров (резин) упругой частью деформации,
составляющей 0,1%, можно пренебречь и рассматривать толь-
ко высокоэластич. деформацию, к-рая, как показывает опыт,
состоит из двух составляющих: быстрой, развивающейся
со скоростью звука (50 —100 м/сек), и замедленной,
развивающейся во времени.
Для качественного описания процессов деформации поли-
меров иногда пользуются различными упрощенными ур-ниями,
и
а=Ее + $ (8)
известными (соответственно) как ур-ние Максвелла (для упру-
го-вязких тел) и ур-ние Кельвина—Фогта (для вязко-упругих
тел). Здесь 9 — время релаксации, вязкость т]=Е5.
Релаксационные свойства полимеров выражаются
в релаксации напряжения, упругом последействии
(ползучести), механич. потерях и в отличии дина-
мич. свойств от статических. При заданной дефор-
мации напряжение с течением времени падает. Если
к полимеру приложено постоянное или периодич.
(постоянное по знаку) напряжение с неизменной ампли-
тудой, то величина деформации возрастает с течением
времени. В первом случае наблюдается статическая,
а во втором — динамич. ползучесть (упругое после-
действие). Как в процессе релаксации напряжения,
так и в процессе упругого последействия модуль
высокоэластичности уменьшается, стремясь к равно-
весному значению Еа> (для линейных полимеров
Ео>=0).
Механич. потери аморфных полимеров достигают
наибольшего значения (рис. 2, кривая 2) тогда, когда
время релаксации 6 становится сравнимым с периодом
колебаний [оно существенно зависит от темп-ры,
согласно ур-нию (1)]. Максимум механич. потерь на-
блюдается в области темп-ры стеклования Тс (рис. 2,
кривая 2). В высокоэластич. состоянии механич.
потери полимера зависят от частоты и скорости дефор-
мации. Динамич. модуль высокоэластичного полимера
Е так же, как и механич. потери, зависит от темп-ры,
частоты и скорости деформации. Это связано с тем,
что при динамич. режимах работа внешних сил
совершается не только против высокоэластич. сил, но и
против сил внутреннего трения. Соответственно этому,
для статич. и динамич. режимов деформации высоко-
эластич. модуль состоит из двух частей E=Ea>+Elt
где Еж— равновесный модуль и Ех — неравновесная
часть модуля, соответственно дающие вклад высоко-
эластич. сил и сил внутреннего трения в сопротивление
деформированию. Равновесный модуль пространст-
венно-структурированных полимеров зависит гл. обр.
от степени поперечного сшивания макромолекул.
Внутреннее трение почти всегда (за исключением
амортизирующих устройств) играет в эксплуатации
отрицательную роль, т. к. приводит к теплообразо-
ванию. При многократных и ударных деформациях
механич. потери приводят к разогреву, оказываю-
щему вредное влияние на усталостную прочность и
износ высокоэластич. материалов.
Вязкое течение полимеров. Наряду с упругой
и высокоэластич. деформацией при определенных
условиях в полимерах могут развиваться необратимые
деформации, с к-рыми связан определенный комплекс
свойств полимеров (иногда называемых реологич.
свойствами). К последним относится все, что связано
с процессами течения (см., напр., Вязкость полиме-
ров). Изучение текучести самих полимеров практиче-
ски важно для технологии процессов переработки.
Обычно исследования текучести ведутся при дефор-
мации сдвига, реже при растяжении или сжатии.
Кристаллич. полимеры выше темп-ры плавления
становятся аморфными и могут течь при темп-рах,
превышающих их темп-ру текучести. Аморфные ли-
нейные полимеры при малых напряжениях начинают
заметно течь выше темп-ры текучести, а пространст-
венно-структурированные — нетекучи вплоть до
темп-ры химич. распада пространственной сетки. При
больших напряжениях (в условиях сложного напря-
женного состояния) аморфные полимеры могут течь
при любых темп-рах. При малых напряжениях (обычно
не превышающих несколько кГ/см2) механизм тече-
ния заключается в многократном перемещении сег-
ментов макромолекул под действием внешних сиц
221
МЕХАНИЧЕСКИЕ СВОЙСТВА ПОЛИМЕРОВ
222
и теплового движения. На это указывает, во-первых,
небольшая энергия активации процесса течения
(см. Вязкого течения энергия активации), сравнимая
с энергией активации течения низкомолекулярных
органич. соединений, и, во-вторых, независимость
энергии активации от длины цепи. В сущности, это
диффузионный механизм вязкого течения, но, в от-
личие от простых жидкостей, осложненный полимер-
ным строением. Большая вязкость полимеров обус-
ловлена, следовательно, не самим механизмом тече-
ния, а большой длиной макромолекул. Влияние
последней на вязкость ц выражается различными эм-
пирии. формулами, напр. ф-лой Фокса и Флори;
ц=ХМ3’4, где М — средневесовой мол. вес линей-
ного полимера, К — константа. Последняя ф-ла
пригодна для сравнительно больших значений М
(св. 20000—50000), при меньших значениях М вязкость
обычно пропорциональна М. Переход с увеличением
М от одной зависимости к другой связан с появлением
в линейном полимере пространственной сетки с вре-
менными узлами, образованными перепутыванием
цепей и вторичными поперечными связями.
Необратимое перемещение макромолекул проис-
ходит в условиях более или менее сильной их дефор-
мации, т. е. сопровождается высокоэластич. дефор-
мацией. В первый момент после приложения напря-
жения скорость деформации сдвига у максимальна
так как одновременно разви-
вается и высокоэластич. дефор-
мация и течение. Скорость вы-
сокоэластич. деформации затем
быстро убывает до нуля (точ-
ка В), где высокоэластич. на-
пряжение, достигнув макси-
мального значения, уравнове-
шивает внешнее. На участке
ВВ наблюдается только необ-
ратимое течение, скорость ко-
торого увеличивается, вероят-
но, из-за частичного разру-
шения вторичных поперечных
связей, пачек и т. п. и дости-
гает постоянного значения на
установившейся стадии тече-
ния на участке В. При весьма
малых скоростях течения раз-
рушение межмолекулярной структуры линейного
полимера практически не происходит, и зависимость
(кривая 2) имеет монотонный вид, часто наблюдаемый
при исследованиях течения при растяжении и сжатии.
В процессе растяжения скорость необратимой со-
ставляющей деформации даже замедляется, а истин-
ная вязкость возрастает до определенной величины
вследствие увеличения внутреннего трения из-за
выпрямления макромолекул в направлении растя-
жения. Развитие высокоэластич. деформации в про-
цессе течения полимера имеет своим следствием высо-
коэластичность текущей полимерной струи и появле-
ние всего комплекса релаксационных явлений.
Вязкость т) установившегося течения линейных по-
лимеров в определенных пределах изменения темп-ры,
напряжения сдвига Р, мол. веса и содержания ак-
тивного наполнителя х подчиняется логарифмич.
аддитивности:
In т] = С + 1п гц (Т) + In т)2 (Р) 4-]п T)s (М) +1п гц (х)
где С — константа, зависящая от природы полимера,
1li(^) = exp(J7/A7’) выражает температурную зависи-
мость вязкости, U — энергия активации вязкого тече-
ния, г)2(Р)=ехр(—аР)— для ненаполненных линейных
и ц (Р)—Р~т— для наполненных и разветвленных
линепных полимеров, тр(М) = №’4 — для полимеров
с высоким мол. весом и г)3(М) = М — для полимеров
, рисунок
Время t
Рис. 7. Изменение скоро-
сти деформации сдвига
высокоэластичных поли-
меров при заданном на-
пряжении сдвига: i —
при больших; 2 — при
малых скоростях дефор-
с низким мол. весом. Все указанные параметры
влияют на вязкость независимо друг от друга, в
частности энергия активации не зависит от напря-
жения, мол. веса, природы и количества активного
наполнителя.
При больших напряжениях полимер даже при тех
темп-рах, при к-рых в обычных условиях он является
высокоэластическим или твердым материалом, при-
обретает способность к необратимому «химическому»
течению (см. Вязкость полимеров, Механохимия поли-
меров). Т. обр., помимо течения, состоящего в пере-
мещении макромолекул относительно друг друга и
имеющего место в линейных полимерах при неболь-
ших напряжениях, при больших напряжениях пре-
обладает течение, связанное с разрывом цепных макро-
молекул или химич. поперечных связей между ними,
перемещением образовавшихся «обрывков» макро-
молекул (свободных бирадикалов) и комбинацией
их в новую полимерную систему.
В нек-рых процессах переработки полимеров в
изделия изменяется структура материала (напр.,
при отверждении термореактивных смол, вулканиза-
ции каучуков и т. д.), что вызывает значительное
повышение вязкости.
Прочность полимеров. Под прочностью полимеров
в широком смысле принято понимать как сопротивле-
ние возникновению вынужденной высокоэластпч.
(стеклообразные и кристаллические полимеры) или
пластич. (высокоэластич. полимеры) деформации, так
и сопротивление полимеров разрыву или растрески-
ванию. Прочность полимеров тесно связана с их
деформационными свойствами. Разрыв полимеров
в большинстве случаев происходит в ориентиро-
ванном состоянии, полученном либо предварительно
(напр., в волокнистых материалах), либо возникаю-
щем в процессе испытания на разрыв, т. к. всем ви-
дам разрыва, за исключением хрупкого, предшествует
высокоэластич. деформация. Хрупкая прочность
одного и того же полимера может, однако, разли-
чаться по величине в зависимости от степени пред-
варительной вытяжки полимера.
У кристаллических полимеров (рис. 8) при низ-
ких темп-рах в области .
хрупкий разрыв. Начиная с
темп-ры Тщ, в области ВВ,
где при растяжении обра-
зуется шейка, полимер име-
ет два значения прочности
в соответствии с тем, где
происходит разрыв—в шей-
ке или в широкой части. Вы-
ше разрыв происходит
только в предельно ориен-
тированном состоянии пос-
ле перехода всего образца
в шейку и дополнительной
вытяжки. Разрывная дефор-
мация здесь остается при-
мерно постоянной вплоть до
температуры плавления Тпл.
Предварительно ориентированный полимер представ-
ляет собой высокопрочный анизотропный волокнистый
материал во всей температурной области ОН. Проч-
ность волокон высока вследствие значительной моле-
кулярной ориентации цепей и др. структур полимера.
Последняя является основным фактором упрочнения
ориентированных полимеров, независимо от того, на-
ходятся волокна в кристаллич. или аморфном сост< я-
нии. Однако структура кристаллич. полимера харак-
теризуется различными степенями надмолекулярной
упорядоченности (вторичные структуры), в связи с
чем прочность кристаллич. полимеров зависит пэ
только от степени и совершенства ориентации, нс
наблюдается только
Температура
Рис. 8. Температурные зави-
симости прочности кристал-
лического полимера при ра-
стяжении: верхняя кривая
для ориентированного, ниж-
няя — для неориентирован-
ного состояния.
223
МЕХАНИЧЕСКИЕ СВОЙСТВА ПОЛИМЕРОВ
224
и от типа их вторичных кристаллич. структур.
Волокнистые материалы испытывают хрупкий разрыв,
при к-ром излом часто имеет своеобразный «волокни-
стый» вид, определяемый их строением и структурой.
Аналогичный характер разрыва наблюдается у кри-
сталлич. и аморфных полимеров, подвергнутых
холодной вытяжке.
У аморфных полимеров вид разрыва меняется в
зависимости от темп-ры и скорости деформации.
Диаграмма механич. состояний аморфных полимеров
{рис. 4), полученная при испытаниях на разрыв с
постоянной скоростью растяжения, отличается от
схемы А. Ф. Иоффе для твердых тел (см. Механические
свойства материалов) тем, что хрупкая и пластич.
области разделены двумя новыми — вынужденно-
эластической (в интервале Тхр, Тс) и высокоэласти-
ческой (в интервале Тс, Тт). Ниже темп-ры Тхр
материал испытывает хрупкий разрыв; выше — раз-
рыву предшествует высокоэластич. деформация, раз-
вивающаяся, начиная с нек-рого напряжения — т. наз.
предела вынужденной эластичности овэ (рис. 3).
Выше Тс разрыву тоже предшествует высокоэластич.
деформация, но ее развитие начинается с момента
приложения нагрузки.
Особым случаем является хрупкое разрушение
полимеров. Этот вид разрушения наиболее опасен
и в условиях эксплуатации обычно проявляется в
виде растрескивания изделий. Разрушение хрупких
полимеров протекает в две стадии: на медленной
растут одна или несколько наиболее опасных (пер-
вичных) трещин, на быстрой — одновременно растет
большое число вторичных трещин. На поверхности
разрыва ясно различаются зоны двух типов: зеркаль-
ная и шероховатая. Нек-рые органич. стекла при
малых нагрузках разрушаются в результате медлен-
ного и одновременного роста (с самого начала нагру-
жения) большого количества трещин «серебра» (псев-
дотрещин) от поверхности в глубь образца. Эти тре-
щины отличаются от обычных трещин в хрупких
телах тем, что их поверхности скреплены тяжами, воз-
никшими в результате местных вынужденно-эластич.
деформаций и представляющими собой высокоориен-
тированный материал. Тяжи мешадот трещинам рас-
крываться и принимают на себя часть нагрузки. Это
является одной из причин того, что трещины «серебра»
растут почти с постоянной скоростью, в отличие от
настоящей трещины. Этот вид длительного разрушения
является промежуточным между хрупким разрывом
полимеров и длительным разрушением высокоэлас-
тичных пространственно-структурирован, полимеров,
на первой стадии к-рого образуется дефект иного
рода — надрыв, перерезающий образец на две части
путем образования в вершине надрыва тяжей и их
разрыва. В результате первая зона поверхности
длительного разрыва, напр. резины, в противополож-
ность хрупкому разрыву, шероховатая, а вторая —
гладкая, иногда покрытая линиями скола, образо-
ванными встречей трещин, возникающих на второй
стадии разрыва. На медленной стадии в результате
разрыва тяжей и их сокращения на поверхностях
разрушения образуются бугорки и впадины, в сово-
купности образующие шероховатую зону. С умень-
шением скорости растяжения и уменьшением числа
поперечных связей пространственной сетки шерохо-
ватая зона постепенно вытесняет гладкую.
Все три типа разрушения связаны с ростом раз-
ного рода дефектов, причем скорость их роста
зависит от напряжения. Поэтому для всех полимеров
характерна временная зависимость прочности.
Временная зависимость прочности полимеров (пласт-
масс и волокон). Эта зависимость выражается ф-лой:
т = тпехр - Yg (9)
kT
где т — долговечность, а — заданное напряжение растяжения,
Т — абс. темп-pa, т0 и у — постоянные, и0— энергия актива-
ции процесса разрушения, экстраполированная на о=0. Энер-
гия активации при данном напряжении есть U— Uo—ус, сле-
довательно, она уменьшается с увеличением напряжения.
Временные зависимости прочности при различных темп-рах
(Тг>Т2>Т3) образуют в полулогарифмич. координатах семей-
ство прямых, выходящих веерообразно из одного полюса
(рис. 9). Чем нижетемп-ра, тем слабее выражен временной фак-
тор. При относительно низких темп-рах характер разрыва
близок к катастрофич. разрушению. Это означает, что время
нагружения при низких темп-рах практически не влияет на
величину разрывного напряжения, и в этом случае разрушение
материала можно характеризовать одной величиной — преде-
лом прочности оКр (рис. 9). Вообще же нельзя говорить о
прочности без указания времени, в течение которого образец
или деталь находились в
напряженном состоянии до
разрушения. Энергия ак-
тивации и0 характеризует
энергию разрыва химич.
связи и поэтому не зави-
сит ни от предварительной
ориентации, ни от изме-
нения межмолекулярного
взаимодействия при вве-
дении пластификаторов и
растворителей, ни от моле-
кулярного веса, тогда как
у зависит от всех этих фак-
торов и, следовательно, яв-
ляется чувствительной к
структуре константой.
Процессы длительного
разрушения и ползучести
полимеров взаимосвязаны
и идут одновременно. Од-
Рис. 9. Временные зависимости
прочности твердых полимеров при
различных температурах.
нако процесс разрушения
определяется не только
процессом ползучести; он
более сложен и поэтому, несмотря на сходство зависимостей
(1) и (9), константы в них имеют различный физич. смысл и по-
разному зависят от межмолекулярного взаимодействия. Моле-
кулярная ориентация не изменяет и0 в обоих процессах. В то
же время постоянная а в ф-ле (1) не меняется, а у в ф-ле (9)
уменьшается. Пластификация уменьшает энергию активации
процесса деформации, не меняет энергию активации процес-
са разрушения полимера, но приводит к увеличению кон-
стант а и у.
Высокоэластичные пространственно-структуриро-
ванные полимеры не только по механизму длитель-
ного разрушения, но и по специфич. виду временной
зависимости прочности представляют собой особый
класс полимеров. Временная зависимость прочности
выражается приближенной ф-лой:
X = Co-beU,hT (10)
где константы Ъ и С не зависят от темп-ры, о — за-
данное истинное напряжение растяжения. Энергия
активации U процесса разрушения, в противополож-
ность твердым полимерам, не зависит от напряжения,
как и энергия активации процесса течения линейных
полимеров. С увеличением межмолекулярного взаимо-
действия (роста полярности, поперечного сшивания
и введения наполнителя) временная зависимость
прочности каучукоподобных полимеров постепенно
переходит в зависимость (9).
Разрушение при многократных деформациях су-
щественно определяется механохимич. процессами
утомления, активируемыми или инициируемыми пере-
менным напряжением. Воздействие агрессивных сред
(озон, кислоты и др.) на полимер в ряде случаев
(напр., резин) приводит к растрескиванию. Стой-
кость высокоэластич. материалов к растрескиванию
зависит от величины их деформации и при неболь-
ших растяжениях порядка 10—20% достигает наи-
меньшего значения, а затем увеличивается.
Влияние мол. веса на прочность твердых полимеров
существенно до степеней полимеризацпи />=200—
300, затем прочность практически не зависит от мол.
веса. Полимеры с низким мол. весом весьма хрупки
и легко растрескиваются, прочность их в значитель-
ной мере определяется силами межмолекулярных
взаимодействий. Прочность полимеров с большим мол.
весом (высокополимеры) зависит в основном от химич.
225
МЕХАНОХИМИЯ ПОЛИМЕРОВ
226
связей в цепях главной валентности и надмолекуляр-
ной структуры. В случае каучуков их исходный
мол. вес влияет на прочность резин, но еще большее
влияние на их прочность оказывают поперечные
связи (степень вулканизации) и активные наполни-
тели. С увеличением числа поперечных связей проч-
ность резины, рассчитанная на истинное разрывное
сечение, проходит через максимум.
Фрикционные свойства полимеров. Сила трения
F= cS, где с — коэффициент, зависящий от условий
опыта и природы трущихся поверхностей, 5 — пло-
щадь фактич. контакта, зависящая от механических
свойств фрикционной пары, шероховатости поверх-
ностей и величины нормальной нагрузки. Природа
трения твердых полимеров близка к природе трения
твердых тел. Отличие заключается лишь в том, что
у твердых полимеров площадь фактического контак-
та формируется главным образом в результате
вынужденно-эластич. деформации в местах контакта,
а у металлов — в результате пластической дефор-
мации.
Трение высокоэластичных пространственно-струк-
турированных полимеров по твердым поверхностям
имеет свои специфич. особенности по сравнению с
трением других твердых тел. Цепи пространствен-
ной сетки полимера в местах контакта находятся
в сцеплении с поверхностью твердого тела ограни-
ченное время, а затем отрываются и переходят в
новые места контакта. Этот процесс совершается под
действием теплового движения и аналогичен пере-
бросу сегментов макромолекул из одного равновесного
положения в другое в самом полимере. Каждый
элементарный акт перехода связан с затратой энергии
(энергия активации U), необходимой для преодоле-
ния сил прилипания. Вероятность переходов цепей
в новые места контакта неодинакова по всем направ-
лениям. В направлении тангенциальной силы F
вероятность перехода наибольшая. Скорость сколь-
жения резины по твердой поверхности в установив-
шемся процессе равна
где С и & — постоянные, зависящие соответственно
от структуры резины и нормальной нагрузки, а
также шероховатости поверхностей трения, Т —
абс. темп-ра.
Лит.: Каргин В. А., Слонимский Г. Л., Крат-
кие очерки по физико-химии полимеров, М., 1960; К о б е к о
П. П., Аморфные вещества, М.—Л., 1952; Трелоар Л.,
Физика упругости каучука, пер. с англ., М., 1953; А л ф р е й
Т., Механические свойства высокополимеров, пер. с англ., М.,
1952; Tobolsky А. V., Properties and structure of poly-
mers, N. Y., 1960; Flory P. J., Principles of polymer chemist-
ry, N. Y., 1953; E i r i c h F. R., Rheology. Theory and appli-
cations, v. 1—3, N. Y.—L., 1956—60; Реология, пер. с англ.,
под ред. Ю. Н. Работнова и П. А. Ребиндера, М., 1962; Fer-
ry J. D., Viscoelastic properties of polymers, N. Y.,1961;
Бартенев Г. M., ДАН СССР, 1956, 110, № 5, 805—07;
его же, Усп. химии, 1955, 24, вып. 7, 815—41; ДАН СССР,
1960, 133, № 1, 88—91; Журков С. Н., Вести. АН СССР,
1957, вып. И, 78—82; Журков С. Н., Абасов С. А.,
Высокомолек. соединения, 1961, 3, № 3, 441—56; Бессо-
нов М. И., Кувшинский Е. В., Пластические массы,
1961, вып. 5, с. 56—65; Каргин В. А., Вести. АН СССР,
1961, вып. 4; Schallamach A., Proc. Phys. Soc. В.,
1953, 66, pt 5, 386—92; Бартенев Г. М., Стыран
3. Е., Высокомолек. соединения, 1959, 1, № 7, 978—89.
Г. М. Бартенев.
МЕХАНОХИМИЯ ПОЛИМЕРОВ — отрасль хи-
мии, изучающая химич. превращения полимеров под
действием механич. сил. Иногда термином М. п.
охватываются и обратные явления — возникновение
деформаций полимеров под действием химич. реаген-
тов (напр., способность макромолекул полиэлектро-
литов сокращаться или растягиваться при изменении
pH среды); однако целесообразнее выделить эту
область явлений, имеющих большое значение для
биологии и электрохимии полимеров, в х е м о м е-
ханику полимеров.
Элементарным механохимич. актом является разрыв
химич. связей в полимерных цепочках под действием
механич. сил (вальцевание, дробление, перетирание,
перемешивание р-ров и т. д.) — механокре-
к и н г. Макромолекулы полимеров иногда легче
разорвать, чем оторвать друг от друга, т. к. при их
большой длине энергия межмолекулярного взаимо-
действия во много раз превышает энергию химич.
связи. В результате разрыва цепочки образуются
активные частицы — обрывки макромолекул. Они,
в зависимости от химич. природы полимера и окру-
жающей среды, скорости разрыва и т. д., могут быть:
макрорадикалами R", макроионами R+ и R- или
ион-радикалами +R' и _R’. Наиболее полно экс-
периментально исследованы случаи разрыва с обра-
зованием макрорадикалов. Последние в дальнейшем
могут претерпевать все типичные для свободных ра-
дикалов превращения — стабилизироваться (при вза-
имодействии с акцепторами или при диспропорцио-
нировании), инициировать химич. цепные процессы.
Различают следующие разновидности механохимич.
процессов: механо деструкция, механосинтез, механо-
химич. изомеризация.
Механодеструкция включает в себя
процессы разрыва макромолекул под действием меха-
нич. сил и последующей стабилизации образовавших-
ся макрорадикалов. В результате механодеструкции
образуются стабильные обрывки исходных молеку-
лярных цепочек полимера. В качестве акцепторов
обычно используют тиофенолы и их га логе но произ-
водные, дисульфиды, хиноны, амины, кислород и др.
низкомолекулярные вещества. При взаимодействии
с макрорадикалом такой акцептор, в свою очередь,
отщепляет низкомолекулярный радикал (остаток ак-
цептора); последний, однако, столь малоактивен, что
не способен самостоятельно инициировать цепные
процессы в данной системе, а стабилизируется сам
в результате рекомбинации либо с аналогичным ему
радикалом, либо с макрорадикалом. Если же отщеп-
ляемый акцептором радикал достаточно активен для
инициирования цепных процессов в данной системе,
то наряду с линейными продуктами деструкции об-
разуются разветвленные и трехмерные фрагменты
из цепочек данного полимера. Как свободные макро-
радикалы, так и радикалы — остатки акцепторов —
могут в определенных условиях инициировать раз-
витие цепных процессов свободнорадикальной де-
струкции, усугубляя тем самым эффект собственно
механич. деструкции.
Как уже отмечалось выше, макрорадикалы могут
стабилизироваться также при диспропорционирова-
нии. В этом случае возможно образование как ли-
нейных полимеров вследствие преимущественного
разрыва наиболее длинных цепочек и рекомбинации
с более короткими обрывками, так и разветвленных
и трехмерных продуктов при взаимодействии макро-
радикалов с функциональными группами макромо-
лекул (напр., двойными связями, а-метиленовыми
группами и т. д.).
Кинетика механодеструкции полимеров может быть
описана следующим ур-нием:
= — Мх) +
где Mt — мол. вес полимера в момент времени 4,
Ма — мол. вес исходного полимера, — мол. вес
наименьшего отрезка цепочки, для к-рого энергия
межмолекулярного взаимодействия с соседними це-
почками равна энергии химич. связей в главной
цепочке (так наз. предел деструкции), к — константа
скорости деструкции, t — время деструкции.
Q8 к.х.э, т. з
227
МЕХАНОХИМИЯ ПОЛИМЕРОВ
228
Механодеструкция отличается от других видов
деструкции полимеров наличием Мх и отрицатель-
ным темп-рным коэфф, скорости. Значения к и Мх
для нек-рых полимеров приведены в таблице, а ти-
пичные кинетич. кривые на рис. 1.
Полимер k
Полиметилметакрилат. . 0,1200 9000
Полистирол 0,0945 7000
Полпвинилацетат .... 0 ,0468 11000
Ацетилцеллюлоза .... — 10000
Поливиниловый спирт . 0,0237 4000
В результате механо деструкции, вследствие уко-
рачивания
повышается
качественно
полимеров,
и т. д.; у
600
240
120
80
40
20 40 60 80 ((часы)
Рис. 1. Кинетика механодеструк-
ции полимеров при размоле: 1— по-
лиметилметакрилат; 2 — полисти-
рол; 3 — поливинилацетат; 4 — по-
цепочек и возникновения новых концевых
групп, изменяются механические свой-
ства полимеров, в частности
пластичность, возрастает и
изменяется растворимость
падает вязкость растворов
белков меняется аминокислотный состав,
у белков и углеводов снижается устой-
чивость к действию ферментов, меняются
электрохимич. свойства и проч. Меха-
нодеструкция используется в промыш-
ленности для пластикации (повышения
пластичности) каучуков, а также для ре-
гулируемого снижения мол. веса, уско-
рения ферментативных процессов и т. д.
Интенсивные механич. воздействия
вызывают ряд побочных независимых и
сопряженных химич.
и физич. эффектов,
сопутствующих меха-
нической деструкции
полимеров. Одним из
них является меха-
нич. активация поли-
мерных цепочек вслед-
ствие их растяжения,
изменения валентных
углов, а также меж-
атомных и межмолеку-
лярных расстояний,
параметров структуры
ливиниловый спирт; 5 — ацетил-
целлюлоза; t — продолжительность
деструкции, —мол. вес полимера
после деструкции в течение t час.
Такое отклонение различных
от равновесных значений повышает свободную энергию
цепочек и делает их
более реакционно-
способными. Други-
ми эффектами меха-
нодеструкции явля-
ются снижение плот-
ности молекулярной
упаковки, превра-
щение кристаллич.
структур в аморф-
ные и т. д.
Механосин-
тез может быть
проведен в систе-
* ф мах, состоящих из
А/WWW + ДО —*- чААААЛЛЛЛ + М
*• V|.. Рис. 2. Схема струк-
м*+пМ— М„ । турных превращений
' 4 п+1 ПрИ механосинтезе по-
лимеров в системах;
а — полимер — полимер; б — полимер — мономер; М — мо-
лекула мономера. Точками обозначены свободные радикалы.
двух (и более) полимеров или полимера и мономеров.
Взаимодействие макрорадикалов (рис. 2,а, I и II), воз-
никающих при совместной механодеструкции разных
полимеров, приводит к образованию блок-сополимеров
(III) или трехмерных продуктов (V). Если механокре-
кингу в данной системе преим. подвержен один из по-
лимеров (II), а цепочки другого содержат активные
центры, способные реагировать с макрорадикалами»
то образуются блок-сополимеры (IV) или фрагменты
сеток одного полимера (напр., хлоропренового ка-
учука) с привитыми обрывками другого (напр., нату-
рального каучука). При механич. деструкции полимера
в присутствии мономера М (рис. 2, б) свободные
макрорадикалы могут инициировать полимеризацию:
наращивать «блоки» мономеров (VII); передачей цепи:
вызвать гомополимеризацию мономеров (VIII). Могут
образоваться и другие, самые разнообразные по
структуре сополимеры. Вследствие трудности регу-
лирования механосинтеза обычно получается смесь
сополимерных продуктов, во многих случаях с
преимущественным содержанием одной из структур.
На скорость и направленпе механохимич. сополи-
© 50
О 15 30 45
Длительность пластикации (мин.)
Рис. 3. Кинетика механосополи-
меризации натурального каучука
при пластикации с 33% различ-
ных мономеров: 1—метакриловая
к-та; 2—метилметакрилат; 3 —
хлоропрен; 4— стирол.
меризапии в системе полимер — полимер влияют
вязкость системы (чем выше вязкость, тем больше:
скорость), активность образующихся макрорадикалов
(чем они активнее, тем
вероятнее разветвление
и сшивание), прочность
валентных связей в
главных цепях макро-
молекул (деструкти-
руется преим. полимер
с менее прочными свя-
зями), интенсивность
механич. воздействия и
др. Соответственно, в
системе полимер — мо-
номер на скорость и
направление механохи-
мич. синтеза оказывает
заметное влияние пла-
стифицирующее дейст-
вие вводимых мономе-
ров (чем выше их кон-
центрация, тем меньше
скорость), гибкость на-
ращиваемых «блоков»
(наращивание жестких
наоборот), активность макрорадикалов и мономеров
[первыми наращиваются блоки более активного моно-
блоков ускоряет процесс и
мера (рис. 3), напр. блоки полихлоропрена в случае:
натурального каучука, набухшего в смеси хлоропре-
на с метилметакрилатом] и др.
Механосинтез имеет ряд преимуществ перед обыч-
ным синтезом полимеров (простота аппаратуры,
большая скорость, экономичность, отсутствие ряда
ограничений в выборе компонентов, свойственных
химич. методам). Недостатком его является неопре-
деленность структуры получаемых полимеров.
Механохимическая изомеризация
полимеров происходит в присутствии изомеризаторов,
напр., тиоловых к-т RCOSH. Предположительная
схема изомеризации: R^ -f- RCOSH-^ RjH -f- RCOS'.
Свободный радикал — остаток тиоловой к-ты, возни-
кает в результате взаимодействия последней с макро-
радикалом полимера и, обратимо присоединяясь к
полимеру по двойным связям, позволяет реализовать
цис-транс-изомеризацию звеньев. Такое нарушении
пространственной упорядоченности цепочек затрудня-
ет кристаллизацию полимера.
Механохимич. процессы могут быть и причиной
усталостных изменений и разрушения полимерных
материалов при многократных деформациях. Одной
из важных областей М. является т. наз. химиче-
ское течение полимеров, под к-рым понимают
непрерывно , происходящее под действием механич.
229
МЕЧЕНЫЕ АТОМЫ — МИГРАЦИЯ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
230
сил разрушение и восстановление химич. структуры
макромолекул полимера. При сильном и быстром
механич. воздействии происходит глубокое химич.
разрушение макромолекул. Возникшие свободные
радикалы и бирадикалы вследствие большой подвиж-
ности перемещаются, что приводит к возникновению
текучести. Благодаря высокой химич. активности
таких частиц процесс сопровождается их рекомби-
нацией с образованием линейной или пространствен-
ной структуры, в среднем тождественной исходной.
Явление химич. течения позволяет в определенных
условиях осуществлять пластич. деформацию, а
следовательно, и необратимое изменение формы трех-
мерных полимеров (напр., формовать из них изделия).
Возникнув как область полимерной химии, М. п.
начинает распространяться и на низкомолекулярные
тела: соли, окислы, металлы и др., виброразмол
к-рых в присутствии мономеров приводит к вскрытию
новых активных поверхностей, что и возбуждает
полимеризацию.
Лит.: Барамбойм Н. К., Механохимия полимеров,
М., 1961; Каргин В. А., Соголова Т. И., Ж. физ.
химии, 1957, 31, вып. 6; ДАН СССР, 1956, 108, № 4, 662;
Плата Н. А., Прокопенко В.В., Каргин В.А.,
Высокомолекулярные соединения, 1959, 1, № 11, 1713; Бер-
лин А. А Усп. химии, 1958, 27, вып. 1,94; Слоним-
ский Г. Л., Хим. наука и пром-сть, 1959, 4, вып. 1, 73;
Догадкин Б. А., Кулезнев В. Н., Коллоидн. ж.,
1958, 20, вып. 5, 674; Каргин В. А., Коварская Б. М.,
Голубенкова Л. И., Акутин М. С., Сло-
нимский Г. Л., ДАН СССР, 1957, 112, № 3, 485; Хим.
пром-сть, 1957, № 2, 13; Кузьминский А. С., Май-
зелье М.Г.,Лежнев Н. Н., ДАН СССР, 1950, 71, № 2,
319; Angler D. J., Ceresa R. J., Watson W. F.,
Chem. Ind., 1958, № 20, 593; Grohn H., Bischof К.,
Chemische Technik, 1959, 11, № 7, 384; L e В г a s J., С о m-
pagnon P., D ela 1 a n (1 e A., Rev. g6n. caoutchouc,1956,
33, № 2, 148. H. К. Барамбойм.
МЕЧЕНЫЕ АТОМЫ—см. Изотопные индикаторы.
МЕЧЕНЫХ СОЕДИНЕНИЙ СИНТЕЗ — синтез
химич. соединений, в к-рых один или несколько атомов
имеют отличный от природного изотопный состав.
Меченые соединения (содержащие как стабильные,
так и радиоактивные изотопы) приобрели большое
значение в физике, химии, биологии, медицине,
сельском хозяйстве, а также в различных областях
техники (см. Изотопные индикаторы). Номенклатура
меченых соединений основана на общепринятой
номенклатуре соответствующих неизотопных соеди-
нений. Положение изотопа в химич. соединении ука-
зывается обычно после названия соединения. Напри-
мер, (СНз)2С14НСООН — изомасляная-2-С14 кислота;
CH3S8SCH2CH2CH(NH2)COOH — у-метилтио-а-амино-
масляная-S88 кислота; H2NCH2CH2CH(N1SH2)COOH —
а, у-диаминомасляная-а-N18 кислота, и т. д. Иногда
положение изотопа в структурной ф-ле соединения
указывают не массовым числом, а звездочкой.
В нек-рых случаях положение «метки» однозначно мо-
жет быть охарактеризовано только специальным по-
яснением, напр. о-СвН4(СН2СООН)СН2СН2С'4ООН —
о-фениленуксусная-Р-пропионовая кислота (С11 в
карбоксильной группе остатка пропионовой к-ты);
C14H3SCH2CH2CH(NH2)COOH — у-метилтио-а-амино-
масляная кислота (С14 в метилтиогруппе) и т. д.
Каждая конкретная задача исследования предъяв-
ляет свои специфич. требования к строению меченого
соединения как с точки зрения природы метки, т. е.
выбора стабильного или радиоактивного изотопа, так
и к положению метки в молекуле; существенное зна-
чение при этом имеет степень изотопной чистоты са-
мого вещества и концентрация изотопа в нем.
В качестве метки наиболее часто применяют след,
изотопы (в скобках даны тип излучения и период полу-
распада): D (стабилен); Т (Р; 12,26 года); С1’ (ст.); С14
(Р; 5600 лет); N1S (ст.); О18 (ст.); Р82 (Р; 14,22 дня);
S8S (Р; 86,3 дня); Cl” (Р; 3,08-10s лет); Вг” (ст.);
Вг81 (ст.); Вг82 (Р; у; .35,-8 часа); J181 (Р, у; 8,08 дня).
8*
Синтез меченых соединений подчинен требованиям
основного исследования и, как правило, проводится
обычными методами препаративной химии (с учетом
правил техники безопасности в случае работы с ра-
диоактивными изотопами). Относительно высокая
стоимость изотопного сырья приводит к необходимо-
сти экономного расходования реактивов, в связи с
чем при М.с.с. широко применяется микро- и полу-
микросинтез, а также используется вакуумная тех-
ника.
До проведения М. с. с. с изотопным исходным ве-
ществом, в целях экономии, все детали синтеза,
включая переработку побочных продуктов реакции,
должны быть тщательнейшим образом отработаны на
модельном синтезе, с использованием не содержащих
изотопов исходных веществ, но в масштабе опыта,
выбранном для изотопного синтеза. Часто самым удоб-
ным способом получения меченого соединения может
служить метод изотопного обмена, к-рый наиболее
применим для получения соединений, меченных D, Т
и О18. В отдельных случаях, особенно для получения
меченых природных соединений, пользуются мето-
дами биологич. синтеза.
Учитывая незначительную энергию излучения, ма-
лую длину пробега и слабую проницающую способ-
ность [J-частиц, излучаемых Т, С14 и Sss, работа с ними
не требует столь серьезных мер предосторожности,
к-рые приходится применять при манипуляциях с
изотопами, обладающими у-активностью. Достаточной
мерой предосторожности является работа в хорошо
вентилируемых вытяжных шкафах, снабженных спе-
циальными фильтрами, и применение приборов с
поглотителями для газообразных побочных продук-
тов реакции и аэрозолей. fJ-излучение Р82 более жест-
кое и требует экранирования листом меди (1 мм),
цинка (4 лглг) или органич. стекла (8 лг.н). Для работы
с у-излучателями, как правило, требуется применение
дистанционных манипуляторов. В соответствии с
санитарными нормами для лиц, работающих с радио-
активными изотопами, допустимо облучение 0,1 р
в неделю. Посуда после синтеза с применением ра-
диоактивных соединений моется под вытяжным шка-
фом (обязательно в спецодежде). Отходы от синтеза,
обладающие радиоактивностью выше 0,1 мкюри!л,
сохраняются в свинцовых контейнерах и сдаются на
захоронение в соответствии с нормами санинспекции.
Отходы с незначительной активностью или кислоты
после мытья посуды сливаются в канализацию после
разбавления до 0,1—0,7 мкюри/л (см. также Защита
от излучений радиоактивных веществ).
Лит.: Мэррей А., Уильямс Д., Синтез органи-
ческих соединений с изотопами углерода, пер. с англ., ч. 1,
М., 1961; Аронов С., Изотопные методы в биохимии, пер.
с англ., М., 1959; Бродский А. И., Химия изотопов,
2 изд., М., 1957. Н. С. Вульфсон.
МИАРСЕНОЛ — см. Сальварсана препараты.
МИГРАЦИЯ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ (гео-
химическая) — перемещение химич. элементов в зем-
ной коре, приводящее к перераспределению элемен-
тов, концентрации одних и рассеянию других, к их
разделению и новым сочетаниям. М. х. э. лежит в
основе всех геохимич. процессов; с непрерывно про-
текающими явлениями миграции связан круго-
ворот веществ в природе. М. х. э.
происходит в виде свободных атомов (инертные газы,
пары ртути) или ионов, находящихся в растворах и
расплавах (р-ры солей в природных водах) в виде
молекул (газы типа НС1, SiFa и молекулы в силикат-
ных и сульфидных расплавах), а также в коллоидном
состоянии (золи, илистые частицы и т. д.).
Причиной М.х.э. является непрерывное изменение
термодинамич. условий в процессах земной коры.
Согласно А. Е. Ферсману, к-рый ввел данное понятие,
факторы М. х. э. подразделяются на внутренние и
231 МИГРАЦИЯ ЭНЕРГИИ—МИКРОБИОЛОГИЧЕСКИЕ МЕТОДЫ ХИМИЧЕСКОГО АНАЛИЗА 232
внешние. К внутренним относятся радиоактивные
свойства элементов, природа связи в образуемых ими
соединениях и энергия кристаллич. решетки минера-
лов. К внешним — внутренняя теплота Земли и тепло,
получаемое от Солнца земной поверхностью; давление
вышележащих слоев Земли, способствующее либо
удержанию, либо (при его понижении) удалению
летучих компонентов и смещающее равновесие химич.
реакций, присутствие других веществ, обусловли-
вающих щелочность или кислотность среды, окисли-
тельно-восстановительные реакции и т. п.; вязкость
среды и т. д. Сочетание этих факторов и приводит к
М- х. э. с образованием наиболее устойчивых в данных
термодинамич. условиях систем. Несколько особо
стоят биогенные факторы миграции (фотосинтез, кон-
центрирование веществ из весьма ненасыщенных
р-ров под действием живых организмов и т. д.),
играющие в зоне земной поверхности и в гидросфере
очень существенную роль и значительно расширяю-
щие явление М. х. э.
Различные химич. элементы, в зависимости от гео-
химии. условий, обладают разной миграционной спо-
собностью (Б. Б. Полынов, 1944) или разной подвиж-
ностью (Д. С. Коржинский, 1942). Полынов, приняв
для коры выветривания в целом миграционную спо-
собность иона хлора за 100, из данных анализов по-
лучил для S04-60; Са2+3; Na+2,5; Mg2 + 1,3; К + 1,3;
SiO2 (силикатов) 0,20; Fe2 Оз 0,04 и т. д.
Основываясь на работах Полынова, А. И. Перель-
ман вывел «коэффициент водной миграции»
g тПд-100
где тх— содержание элемента X в речной воде (в
г/л); а— сумма минеральных веществ, содержащихся
в воде данной реки (в г/л); пх— среднее содержание
элемента X в горных породах бассейна рассматри-
ваемой реки (в %).
Коржинским подвижность элемента определяется
как произведение коэфф, диффузии на максимальную
концентрацию, достигаемую для данного элемента
в изучаемом процессе. Им введены, на основе правила
фаз, понятия «вполне подвижного компонента» (НгО,
СОа, иногда щелочи) и «инертного компонента» (напр.,
SiOa, А1аОз, TiO2). В. В. Щербина изучал формы
переноса мигрирующих элементов; так, вольфрам мо-
жет переноситься в газовой фазе в виде WO2CI2 или
WOFa, а в водных р-рах — в виде анионов WO*-
или [Si(WsOI0)4]4-; золото— в виде АиЭ~или, реже,
в виде АиС14 ; свинец — в виде РЬС14_, уран—в виде
[UO2(CO3)2]4-или [иР2(СОз)з]4-, бериллий — в виде
BeF4~ и т. д. Миграционная способность высока для
элементов, образующих легколетучие, растворимые
и легкоплавкие соединения, и низка в случае трудно-
растворимых, тугоплавких, химически инертных со-
единений. К числу элементов с очень высокой мигра-
ционной способностью относятся такие, как, напр.,
С1, Вг, J, N, В, Na, Ra,..., с высокой— К, Са, Ge,
U, Fe,..., средней—Al, Si, Mg,... редкоземельные
элементы, низкой — Zr, Nb, Та, Sb,..., очень низ-
кой — платиновые металлы.
Лит.: Ферсман А. Е., Геохимия, т. 2, Л., 1934,
с. 5—99; Коржинский Д. С., Зап. Всероссийского
минералогия, об-ва, 1942, ч. 71, № 3—4, 160—67; Полы-
нов Б. Б., Изв. АН СССР. Сер. геол., 1944, № 2, 3—14:
Перельман А. И., Очерки геохимии ландшафта, М.,
1955, с. 20 — 28. В. В. Щербина.
МИГРАЦИЯ ЭНЕРГИИ — см. Перенос энергии.
МИЕЛОСАН (мильеран, 1,4-диметан-сульфонилок-
сибутан) CH3SO2OCH2CH2CH2CH2OSO2CH3, мол. в.
246,31— белый кристаллич. порошок; т. пл. 116—
1^7°; трудно растворим в ацетоне и спирте, нераство-
рим в воде. При нагревании М. со спиртовой щелочью
образуется соль метансульфокислоты, к-рая выпадает
в виде белого студенистого осадка; на этом основано
количественное определение М. Получают М. взаимо-
действием 1,4-диоксибутана в присутствии пиридина
с метансульфохлоридом CH3SO2CI. М. применяют
в виде таблеток для лечения хронич. миелоидной лей-
кемии.
Лит.: Машковский М. Д., Лекарственные средства,
4 изд , М., 1960, с. 592; Haggis G. A., Owen L. N.,
J. Chem. Soc., 1953, 389. В. Г. Яшунский.
МИКРОБИОЛОГИЧЕСКИЕ МЕТОДЫ ХИМИ-
ЧЕСКОГО АНАЛИЗА — методы анализа, основан-
ные на количественной зависимости между содержа-
нием нек-рых веществ, вносимых в специальную син-
тетич. питательную среду, и интенсивностью роста и
размножения микроорганизмов в этой среде, или
между содержанием определяемого вещества, вноси-
мого в сахаро-фосфатный р-р, и интенсивностью бро-
жения. М.м.х.а. не являются универсальными и
позволяют определять лишь те вещества, к-рые отно-
сятся либо к обязательным факторам роста микроор-
ганизмов, либо к ингибиторам их роста.
Потребляемые микроорганизмами витамины и ви-
таминоподобные в-ва отличаются большим разнооб-
разием. Многие микроорганизмы обладают высокой
способностью к синтезу витаминов из простых соеди-
нений и полностью обеспечивают свои потребности в
них; нек-рые же способны лишь к частичному их
синтезу. В качестве индикаторных культур для коли-
чественного определения витаминов отбирают обычно
штаммы резко гетеротрофные по отношению к опре-
деляемому витамину (или витаминоподобному веще-
ству), т. е. полностью не способные к синтезу его.
Если внесено ничтожно малое количество посевного
материала, то такие культуры совершенно не растут
на средах, в к-рых отсутствует требуемый фактор
роста; на добавление витамина к среде соответствую-
щая культура реагирует резкой ростовой (или бро-
дильной) реакцией. Подобного типа микроорганизмы
обладают наиболее высокой чувствительностью к
определяемому витамину.
С помощью микроорганизмов могут быть количе-
ственно определены вещества витаминной природы,
относящиеся к факторам роста индикаторного микро-
организма, напр. витамины группы В, аминокислоты,
пуриновые и пиримидиновые основания. Также коли-
чественно могут быть определены ингибиторы роста—
антибиотики и различные микробные яды. Пользуясь
определенными культурами дрожжей, путем броже-
ния количественно определяются след, сахара: 1) глю-
коза и др. моносахариды; 2) глюкоза и сахароза;
3) глюкоза, сахароза и мальтоза; 4) глюкоза, галак-
тоза, сахароза, '/, рафинозы и мальтоза; 5) глюкоза,
галактоза, сахароза, */з рафинозы и лактоза; 6) глю-
коза, галактоза, сахароза, рафиноза полностью,
включая и мелибиозу; 7) глюкоза, галактоза, саха-
роза, ‘/з рафинозы, мальтоза, мальтотриозы (простые
декстрины). По разности может быть вычислено со-
держание отдельного сахара в растворе.
Устанавливают зависимость между количествами
размножившихся клеток и внесенного вещества (ви-
тамина, аминокислоты, пуринового или пиримиди-
нового основания) и по полученным данным строят
калибровочную кривую. При использовании бро-
дильной реакции в кратковременных опытах и в от-
сутствие размножения клеток находят зависимость
между количествами добавляемого витамина, напр.
витамина В, и интенсивностью брожения, определяе-
мой по объему выделившегося углекислого газа (газо-
метрический метод).
Чувствительность М.м.х.а. иллюстрируется след,
данными:
233
МИКРОКРИСТАЛЛОСКОПИЯ—МИКРОУДОБРЕНИЯ
234
Витамины Чувствитель- ность3 мкг/мл Витамины Чувствитель- ность3 мкг/мл
Биотин .... Фолиевая к-та Цианкобала- мин Пиридоксин rt-Аминобен- зойная к-та Пантотеновая к-та (вита- мин В3) . . до 0,0001 0.001—0,0001 0.001—0,0001 0.004—0,001 0,02—0,002 0,03—0,02 Тиамин . . . Тиамин^ . . Никотиновая к-та .... Рибофлавин . Холин .... Инозит .... Аминокислоты Сахара .... 0.1—0,005 0,5—0,001 0,15—0,03 0,2—0.02 0,5—0,05 2-1 100—25 не ниже 0,5% не выше 4—6%
а Чувствительность метода определяется степенью физиоло-
гия. активности витамина или витаминоподобного в-ва. Для
определения названных соединений предложены индикаторные
культуры из групп бактерий, дрожжей или грибных микро-
организмов. 6 Определено бродильным методом; результат
зависит от чувствительности микробродильного прибора.
Наибольшее число известных М. м. х. а. разработа-
но для определения витаминов группы В.
С помощью М.м.х.а. можно количественно опреде-
лять оптич. изомеры; способ основан на свойствах
микроорганизмов усваивать только право- или лево-
вращающий изомер.
В М.м.х.а. пользуются рекомендуемым микроорга-
низмом и условиями его культивирования таким об-
разом, чтобы в отсутствие в искусственной питатель-
ной среде определяемого вещества не происходил рост
культуры (или были бы только следы роста), а в при-
сутствии его скорость роста культуры изменялась
в зависимости от количества анализируемого вещест-
ва. Количественные определения рекомендуется про-
водить по стандартным р-рам, концентрации, близкой
к максимальной дозе, т. к. малый стимулирующий
эффект могут вызвать многие неспецифич. вещества.
Для определения ингибиторов роста применяют
индикаторные штаммы микроорганизмов (бактерии),
наиболее чувствительные к данному ингибитору.
Эталоном служит доза ингибитора, с к-рой рост куль-
туры почти полностью задерживается.
Интенсивность размножения микроорганизмов (ко-
личество клеток) определяют: а) весовым путем (оп-
ределение массы клеток или мицелия); б) по степени
мутности суспензии (экспрессный метод); в) в случае
применения молочнокислых бактерий также и по на-
растающей кислотности среды титрованием р-ром ще-
лочи; г) при размножении на твердых питательных
средах измеряют диаметр колоний; д) интенсивность
брожения определяют газометрич. способом (приборы
Варбурга или Одинцовой).
Для расчета сравнивают результаты опыта со стан-
дартной (калибровочной) кривой зависимости интен-
сивности размножения роста культуры (или интен-
сивности брожения) от известных количеств опреде-
ляемого вещества. Кривая должна быть получена
одновременно, точно в тех же условиях.
Лит.: Омелянский В. Л., Микроорганизмы, как
химические реактивы, Л., 1924; Кудрявцев В. И.,
Систематика дрожжей, М., 1954, с. 143—44; М а р д а-
шев С. Р., в кн.: Успехи биологической химии, т. 1, М.,
1950; Микробиологические методы определения витаминов
и аминокислот, пер. с англ., М., 1954; Одинцова Е. Н.,
Микробиологические методы определения витаминов, М.,
1959; Кудрявцев В. И., Агатов П. А., Кло-
пов с к а я К. И., Микробиология, 1947, 16, вып. 2, с. 155;
Г р о в Д. С., Р е н д а л л В. А., Руководство по лабора-
торным методам исследования антибиотиков, пер. с англ., М.,
1958; Егоров Н. С., Выделение микробов-антагонистов
и биологические методы учета их антибиотической активности,
М., 1957. Е. Н. Одинцова.
МИКРОКРИСТАЛЛОСКОПИЯ — метод качест-
венного микрохимич. обнаружения неорганич. и
органич. веществ по образованию характерных крис-
таллич. осадков при действии соответствующих реак-
тивов на каплю анализируемого р-ра. В случае
органич. соединений исследование можно произво-
дить и без введения реактивов путем медленного испа-
рения капли водного, этанолового, бензольного
и т. п. р-ров. По внешнему виду кристаллов осадка
(форма, окраска, размер, взаимное расположение),
а также их нек-рым кристаллография, и кристалло-
оптич. константам (углы между гранями кристаллов,
угол погасания, плеохроизм и др.) можно сделать
заключение о принадлежности кристаллов опреде-
ленному веществу и, следовательно, о составе ана-
лизируемого объекта. Кристаллы выпадающего в оса-
док соединения приобретают характерный для них
внешний вид только при умеренных и малых концен-
трациях исследуемого р-ра, когда осаждение кристал-
лов протекает не моментально, а в течение 0,5—1 мин.
При очень малой концентрации исследуемого объекта
кристаллы появляются медленно, гл. обр. по краям
капли.
Каплю анализируемого р-ра, объемом ок. 0,001 мл и
меньше, смешивают на предметном стекле с такой же по объему
каплей реактива или вводят в каплю анализируемого р-ра
крупинку реактива в твердом виде (диаметр крупинки не
более 0,1 мм). Исследуемый р-р и р-р реактива отбирают
и переносят на предметное стекло при помощи стеклянного
капилляра. В нек-рых случаях обрабатывают каплю газо- или
парообразным реактивом (хлористый водород, бром, аммиак,
пиридин и др.). Реакции с газообразными реактивами или
обнаружение газообразных веществ производят в газовой
камере. Газовую камеру применяют и для микровозгонки
нек-рых неорганич. и особенно органич. соединений; возгоны
многих соединений состоят из характерных кристаллов.
Каплю с кристаллами образовавшегося осадка рассматривают
под микроскопом при увеличении обычно в 80—200 раз. Для
определения кристаллография, и кристаллооптич. констант,
наблюдения и исследования плеохроизма и интерференционных
окрасок пользуются поляризационным микроскопом. Иногда
для наблюдения и исследования кристаллов применяют
ультрафиолетовую или электронную микроскопию. В осталь-
ном техника эксперимента (фильтрование, нагревание, выпари-
вание и др.) такая же, как и при других методах качественного
микрохимического анализа.
Микрокристаллоскопич. реакции в большинстве
случаев отличаются высокой чувствительностью (не-
большим открываемым минимумом). В капле раствора
можно обнаруживать десятые и сотые доли микрограм-
ма, а нередко и значительно меньшее количество того
или иного вещества. Характерные признаки кристал-
лов наряду с условиями их получения делают микро-
кристаллоскопич. реакции достаточно специфиче-
скими.
М. находит применение гл. обр. для качественного
анализа очень небольших по размерам объектов (вклю-
чения в металлы и минералы, мельчайшие металлич.
частицы, пыль, содержимое растительных и животных
клеток ит. п.). Применение специфич. реакций делает
М. очень удобным методом анализа минералов, спла-
вов и др. объектов, а также простым способом иденти-
фикации органич. соединений. Выполнение качест-
венных реакций и других операций в этом методе
осуществляется легко и быстро.
Лит.: Коренман И. М.. Микрокристаллоскопия,
М„ 1955; Маляров К. Л., Качественный микрохимиче-
ский анализ, М., 1951; Аншелес О. М., Бура-
кова Т. Н., Микрохимический анализ на основе кристалло-
оптики, Л., 1948. И. М. Коренман.
МИКРОУДОБРЕНИЯ — удобрения, в составе ко-
торых имеются т. н. микроэлементы (бор, медь, марга-
нец, молибден, цинк, кобальт и др.), необходимые ра-
стениям в малых количествах для повышения их уро-
жайности, улучшения качества растений, а также
предохранения растений и животных от нек-рых за-
болеваний. М. повышает активность ферментов, ка-
тализирующих биохимич. процессы в организме, спо-
собствует синтезу сахара, крахмала, белков, нуклеи-
новых кислот, витаминов и ферментов. Бор, марганец,
медь и молибден оказывают положительное действие
на фотосинтез, ускоряют развитие растений и созре-
вание семян. Наибольшее значение среди М. имеют
борные удобрения.
235
МИКРОХИМИЧЕСКИЙ АНАЛИЗ
236
Медные удобрения оказывают значитель-
ный эффект на развитие и рост растений на многих
торфяных, песчаных и дерново-глеевых почвах, повы-
шают устойчивость растений к неблагоприятным ус-
ловиям внешней среды (засуха, повышение и пони-
жение темп-ры) и к нек-рым бактерицидным и гриб-
ным болезням. В качестве медных удобрений приме-
няют пиритные (колчеданные) огарки, в к-рых, кроме
меди (0,3—0,6%), содержатся также др. микроэле-
менты (Zn, Со, Мо). Пиритные огарки вносят в поч-
ву 1 раз в 4—5 лет по 5—6 ц/га.
В качестве марганцовых удобрений,
потребность в к-рых для растений чаще всего прояв-
ляется на карбонатных и др. нейтральных или слабо-
щелочных почвах, используются: нерастворимые в
воде марганцовые шламы — отходы обогащения мар-
ганцовых руд, содержащие 15—18% марганца;
легкорастворимый сульфат марганца — отход неф-
тяной пром-сти, содержащий 21—22% марганца; т. н.
марганизированный суперфосфат, содержащий 17,5—
18% усвояемой Р2О5 и 1,5—2% марганца. Маргани-
зированный суперфосфат изготовляют добавлением к
суперфосфату перед его грануляцией 10—15% марган-
цового шлама. Марганцовые удобрения вносят в почву
из расчета 5—25 кг/га марганца (в зависимости от
растворимости удобрения).
В качестве молибденовых удобрений
применяют молибденовокислый аммоний (гл. обр.
в виде раствора для предпосевной обработки семян
и опрыскивания растений), а также молибденизирован-
ный суперфосфат, содержащий 18—20% усвояемой
Р2О5 и 0,05—0,2% молибдена. Молибденизированный
суперфосфат готовят путем добавки к суперфосфату
соединений молибдена. Молибден участвует в реак-
циях связывания молекулярного азота микроорганиз-
мами и в азотном обмене растений; его вводят в кис-
лые и железистые почвы. Этот элемент необходим для
жизни большинства растений, особенно нуждаются
в нем бобовые растения.
В качестве цинковых удобрений, не-
обходимость в к-рых наблюдается на песчаных, су-
песчаных, гравийных и карбонатных почвах, приме-
няют различные промышленные отходы, содержащие
соли цинка. Цинковое голодание чаще всего наблю-
дается у плодовых и цитрусовых деревьев, кукурузы,
бобовых и др.
Лит.: Каталымов М. В., Унанянц Т. П.,
Производство и применение микроудобрений в СССР и за
рубежом, М., 1960; Микроэлементы и урожай. Об. статей, под
ред. Я. В. Пейве, Рига, 1961; П е й в е Я. В., Ж. Всес. хим.
о-ва 1962, 7, № 5, 542; Микроэлементы в сельском хозяйстве
и медицине. Труды Всесоюзного совещания по микроэле-
ментам, Рига, 1955, Рига, 1956. А. И. Шерешевский.
МИКРОХИМИЧЕСКИЙ АНАЛИЗ (микроанализ)—
совокупность методов и приемов выполнения качест-
венного и количественного анализа малых количеств
неорганич. и органич. веществ (от миллионных до
сотых долей грамма); является частью микрохимии.
В М. а. обычно используются те же реакции обна-
ружения и химич. методы определения, что и в мак-
роанализе, но работу ведут с малыми объемами рас-
творов обычных концентраций в небольшой по размеру
посуде, часто с применением специальной аппаратуры
и методики эксперимента.
Возникнув в 18 в., М. а. стал систематически разрабаты-
ваться с конца 19 в. Особенно широко он применяется с на-
чала 20 в. при всестороннем исследовании малых образцов
любой природы и состава, когда из-за малого количества
вещества обычные приемы работы становятся недостаточными
и ненадежными.
Для М. а. характерна быстрота выполнения, малый
расход реагентов, портативность посуды и др. Эти
преимущества делают целесообразным применение
микрометода в ряде случаев и при наличии большого
количества исследуемого вещества.
В зависимости от количества анализируемого дан-
ным методом вещества различают: полумикро-, микро-
и ультрамикрометоды (порядок навески соответствен-
но 10“2г, 10—а г и 10“’ г). Каждый из названных мето-
дов отличается аппаратурой и приемами выполнения
операций.
При выполнении количественного микроанализа
неорганич. веществ малую навеску вещества перево-
дят в раствор и затем определяют в нем содержание
различных элементов. Пользуясь методами количест-
венного органич. М. а., можно определять содержание
отдельных элементов в органич. соединении (элемен-
тарный анализ), функциональных групп (функцио-
нальный анализ) и мол. вес соединения.
В количественном М. а. неорганич. и органич.
веществ (как и в макроанализе) в зависимости от
принципов, положенных в основу определений, разли-
чают весовой, объемный (по объему осадка или газа),
титриметрический, фотометрический и др. методы.
Ниже описаны наиболее общие приемы и аппаратура
микрометода химич. анализа.
Реакции качественного микрохимии, обнаружения эле-
ментов выполняются: под микроскопом на предметном стекле,
причем элемент идентифицируют по форме кристаллов образуе-
мого им с реактивом соединения (микрокристаллоскопия); на
фильтровальной бумаге (капельный анализ); на фарфоровых
пластинках — по окраске того или иного соединения, обра-
зуемого идентифицируемым элементом; в капиллярных про-
бирках — по цвету, характеру, растворимости и др. свойствам
образующегося осадка. Существуют и другие приемы, напр.,
обнаружение элементов по способности их соединений светиться
в УФ-свете (люминесцентный анализ). Вообще в М. а. исполь-
зуют преим. наиболее чувствительные химич. реакции, харак-
теризуемые наименьшими открываемым минимумом и предель-
ной (минимальной) концентрацией (см. Чувствительность
реакции). Это позволяет наряду с определением основных
компонентов в малом образце определять в нем и примеси
других веществ.
При выполнении весового анализа используют микровесы,
позволяющие взвешивать с точностью до 10“’ г; особенно
широко весовые методы определения применяют в микроана-
лизе органич. веществ. Взвешивание производят в специаль-
ных микроаппаратах и сосудах.
При микротитровании используют микробюретки с точ-
ностью отмеривания до 10 “’ мл и сосуды для титрования не-
большого объема (микростаканы, микрочашки). В титриметрич.
М. а. предпочитают объективные методы регистрации конечной
точки титрования (потенциометрия и др.).
Большое практич. значение приобрели фотометрич. методы
М. а. При их выполнении используют микрокюветы.
Методика и техника микрохимич. эксперимента
позволяют с достаточной точностью (часто не уступаю-
щей точности макроанализа) выполнять различные
определения с малым количеством вещества. В М. а.
необходимо учитывать увеличение относительного
влияния ошибок, связанное с уменьшением количест-
ва исследуемого вещества. Увеличение поверхности
соприкосновения раствора со стенками микрососуда
ведет к загрязнению пробы материалом стенок и к
повышению количества вещества, сорбирующегося из
раствора на стенках. Поэтому при микрохимич. оп-
ределениях применяют аппаратуру, поверхность и
объем к-рой изменяются примерно в том же отноше-
нии, как и масса образца. При микротитровании отно-
сительно велико влияние индикаторной ошибки, к-рая,
однако, полностью устраняется, если конечную точку
титрования устанавливать физико-химич. методами.
От уровня техники микрохимич. эксперимента
зависит минимально определяемое количество эле-
мента. Оно уменьшается по мере совершенствования
техники эксперимента и способов наблюдения и сос-
тавляет ок. 10-8 г. На практике встречается много
случаев, когда количество исследуемого вещества
настолько мало, что оно может быть проанализирова-
но только при использовании техники М. а. Примера-
ми являются включения в минералах и сплавах,
ценные объекты, небольшие детали разнообразных
устройств, различные объекты судебно-химич., био-
химич. и клинич. исследований, вновь синтезируемые
органич. вещества. В последние 15—20 лет техника
микрохимич. эксперимента стала широко использо-
237
МИЛЛОНА РЕАКЦИЯ—МИНЕРАЛЫ
238
ваться в радиохимии при изучении свойств новых эле-
ментов, а также из соображений безопасной работы
экспериментатора с радиоактивными веществами.
При наличии больших количеств исследуемого ве-
щества методы М. а. целесообразно использовать,
напр., для определения примесей в том или ином
объекте; в этом случае можно существенно уменьшить
исходную навеску вещества. В трудоемком анализе
сложных органич. соединений в последнее время все
большее значение начинает приобретать использова-
ние микротехники химич. эксперимента. См. также
Бесстружковый метод анализа.
Лит.: Ал и марин И. П., Фрид Б. И., Количест-
венный микрохимический анализ минералов и руд, М., 1961;
Бенедетт и-Пихлер А., Техника неорганического
микроанализа, пер. с англ., М., 1951; Коренман И. М.,
.Микрокристаллоскопия, М., 1955; его же, Количественный
микрохимический анализ, М.—Л., 1949; Кульберг Л. М.,
Альтерзон Г. С., Вельтман Р. П., Капель-
ный анализ, М.—Л., 1951; Feigl F., Spot tests in Inor-
ganic analysis, Amst.— [a. o.], 1958; Ф а й г л ь Ф., Капельный
анализ органических веществ, пер. с англ., М., 1962; Кор-
шун М. О., Гельман Н. Э., Новые методы элемен-
тарного микроанализа, М.— Л., 1949; Нидерль Дж.,
Нидерль В., Микрометоды количественного органиче-
ского анализа, пер. с англ., М.— Л., 1949; Тана-
на е в Н. А., Капельный метод, 6 изд., М.—Л., 1954; Сто-
ляров К. И., Методы микрохимического анализа, Л.,
1960; Бобранский Б., Количественный анализ органи-
ческих соединений, пер. с польск., М., 1961; Ч е р о н и с Н.,
Микро- и полумикрометоды органической химии, пер. с англ.,
М., 1960. М. И. Петракова.
МИЛЛОНА РЕАКЦИЯ — качественная реакция
на белки. При добавлении реактива Миллона (р-р
азотнокислых солей окиси и закиси ртути в HNOs,
содержащей примесь азотистой к-ты) к р-ру белка
образуется белый осадок денатурированного белка.
При стоянии, быстрее при слабом нагревании (не
выше 50°), осадок окрашивается сначала в розовый,
а затем в пурпурно-красный цвет; жидкость также
может окраситься в красный цвет. Красное окрашива-
ние при М. р. обусловлено образованием ртутных
солей нитропроизводных, содержащих фенильную
группу. М. р. дают только белки, содержащие фе-
нильную группу тирозина. Триптофан при нагрева-
нии с реактивом Миллона дает буро-красное окраши-
вание. Реакции мешают хлориды, Н2О2, спирт, боль-
шие количества минеральных солей. Щелочные р-ры
белков для проведения М. р. следует нейтрализовать
азотной или уксусной к-той во избежание выпадения
осадка окиси ртути.
Для приготовления реактива Миллона 40 г ртути
растворяют в 57 мл конц. HNO3 сначала при комнатной темп-ре,
а затем при нагревании на водяной бане. Раствор разбавляют
двумя объемами воды и жидкость сливают с отстоявшегося
осадка. С. И. Пехтерева.
МИНДАЛЬНАЯ КИСЛОТА (оксифенилуксусная,
фенилгликолевая кислота) C6HSCH(OH)COOH, мол.
в. 152,14 — простейшая ароматич. оксикислота с
•одним асимметрич. атомом. Левовращающая (L-М.к.)
образуется при гидролизе амигдалина (генциобиозида
нитрила М.к.), содержащегося в горьком миндале.
Правовращающая (D-М.к.) лежит в основе другого
гликозида — самбунингрина (к-рый может быть выде-
лен из бузины); т. пл. L- и D-изомеров 133,3°, [а] д=
= + 156,57° (в воде). Рацемич. М. к. (параминдаль-
ную к-ту) получают из бензальдегида через циангид-
рин:
[N+]
С,Н5СНО +HGN —> C8H5GH(OH)GN------>- G8H5GH (ОН) GOOH
Т. пл. параминдальной к-ты 120,5°; константа диссо-
циации К = 4,17-10-4 (25°). При проведении напи-
санной выше реакции в присутствии эмульсина (энзи-
ма горького миндаля) образуется больше левовращаю-
щей М. к. Асимметрический синтез удается провести и
в присутствии хинина. Параминдальную к-ту можно
расщепить на антиподы кристаллизацией цинхонино-
вых солей. Как оксикислота, М. к. дает производные
по обеим функциональным группам обычным образом.
Хлористый тионил расщепляет М. к. на бензальдегид
и окись углерода. Под действием фосгена и пиридина
М. к. образует циклич. сложный эфир (I). В присут-
ствии серной к-ты конденсируется с фенолом в соеди-
нения II, а с ацетоном в тех же условиях дает III.
/О-х
С6н5-сн с=о
О=с^ хСН-СвН5
О
L
свн5-сн-о
О=СхохС(СН3)г
и
Перманганат калия окисляет М. к. до фенилглиокса-
левой к-ты CeHsCOCOOH.
М. к. обладает антисептич. свойствами; находит
нек-рое применение в медицине. Из М. к. и тропина
получают гоматропин, к-рый менее токсичен, чем
аналогично действующий атропин.
Параминдальная к-та образует производные:
Т. пл., °C Т. кип., °C
Метиловый эфир 58 250
Этиловый эфир 37 253—255
Амид 123—124 —
Нитрил Метиловый эфир (а-Метоксифенпл — уксусная 21,5—22 —
кислота) 71-72 —
Диметиловый эфир — 246
Н. А. Несмеянов,
МИНДАЛЬНОЕ МАСЛО — см. Жиры раститель-
ные.
МИНЕРАЛЫ. Содержание:
Классификация .............................238
Морфология и физические свойства...........239
Физико-химическая природа, состав и кристалличе-
ская структура ...........................242
Генезис и парагенезис......................243
Минералы — простые и сложные химий, инди-
виды, растворы (жидкие и твердые) и коллоидные си-
стемы, образовавшиеся в результате происходящих
в природе физико-химич. процессов. Это определение
охватывает не только М. земной коры, образующиеся
при геохимических процессах, но и М., встречающиеся
в телах внеземного происхождения (напр., в метео-
ритах). По происхождению и составу различают не-
органич. и органич. М. К последним еще ок. 25 лет
назад относили нефть, торф, бурые и каменные угли,
ископаемые смолы (асфальт, янтарь), озокерит. Од-
нако новейшие исследования показали,что перечислен-
ные природные органич. продукты являются сложными
смесями различных органич. М., пока еще недостаточно
изученных. Поэтому здесь будут рассмотрены только
неорганич. М. Число известных в настоящее время не-
органич. М. достигает 2 тыс. По агрегатному состоя-
нию М. делятся на твердые и жидкие; жидких М.
очень немного (вода, ртуть, жидкая СОа в виде вклю-
чений в кристаллах кварца и др.); находящиеся в
природе газы (см. Газы природные) называть М. не
принято. Подавляющее большинство М.— твердые
кристаллич. тела.
Классификация. Первую рациональную классифи-
кацию М., основанную на их химич. составе, предло-
жил И. Я. Берцелиус (1814). Его классификацию
видоизменил Дж. Дэна (1837). Эта классификация
с нек-рыми улучшениями была общепринятой еще
в первых десятилетиях 20 в. и нередко применяется
и в настоящее время, особенно в зарубежной литера-
туре. Дэна распределяет М. по 8 классам: 1) самород-
ные элементы*, 2) сульфиды, селениды, теллуриды,
* Этот термин общепринят, хотя его нельзя признать
правильным. Вместо «самородные элементы» было бы лучше
говорить «самородные простые тела» — ред.
239
МИНЕРАЛЫ
240
арсениды, антимониды, 3) сульфосоли, 4) галогениды,
5) окислы, 6) соли кислородных к-т, 7) соли органич.
к-т, 8) углеводороды.
Современная классификация неорганич. кристал-
лич. М. должна, кроме химич. состава, учитывать
их кристаллич. структуру и типы химич. связи между
структурными единицами. Наиболее последовательно
этот принцип провел Г. П. Барсанов (1959). В его
классификации типы М. выделяются по типам химич.
связей, классы — по качественному составу анионов
(простых или комплексных), связанных в кристаллич.
решетку при помощи того или иного вида связи. Ниже
приводится схема классификации М. по Барсанову.
Тип 1. Атомные (частью ионные) решетки с металлич. и
ковалентной связью. Классы: а) свободные атомы элементов —
самородные Au, Ag, б) интерметаллич. соединения — дискра-
зит Ags Sb, амальгама Hg,Ag2.
Тип 2. Ионные решетки с ковалентной (реже с ионной
связью). Классы: а) сульфиды и сульфосоли — пирит FeS2,
тетраэдрит Cu12Sb4S13, б) арсениды — никелин NIAs, в) антимо-
ниды — брейтгауптит NlSb, г) селениды — науманнит Ag2Se,
S) теллуриды — гессит Ag2Te, е) хлориды, бромиды, иодиды —
галит NaCl, эмболит Ag (Cl, Вг), ж) фториды — флюорит CaF2.
Тип 3. Типичные ионные решетки без комплексных анион-
ных групп в структурах. Классы: а) окислы — корунд А12О3,
б) гидроокислы — гетит HFeO2.
Тип 4. Ионные решетки с'комплексными анионными
группами — соли кислородных к-т. Классы: а) танталонио-
баты — колумбит (Fe, Mn) [Nb2O6], б) силикаты, алюмосили-
каты — оливин (Mg, Fe)2[S104], полевые шпаты К [AlSlaOa],
в) бораты — борацит Mg3B7OI3Cl, гидроборацит MgGaB6OH-
• 6Н2О, г) фосфаты — апатит Ca5[PO4]sF, д) арсенаты — скоро-
дит Fe[AsO4]-2H2O, е) ванадаты — ванадинит Pb5[VOt]aCl,
ж) сульфаты — барит BaSO4, гипс CaSO4-2H2O, з) хрома-
ты, вольфраматы, молибдаты — крокоит РЬСгО4, вольфрамит
(Mn, Fe),WO4, повеллит СаМоО,, и) карбонаты — кальцит
GaGO3, к) нитраты — чилийская селитра NaNO3. Внутри
классов в зависимости от химич. типа соединений, сходства
кристаллич. решеток, структурного типа катионных групп
и др. признаков выделяются подклассы и группы.
По главным типам число минеральных видов рас-
пределяется следующим образом (в %):
Силикаты и алюмосиликаты................ 25,8
Фосфаты, арсенаты, ванадаты............. 18,0
Сульфиды и сульфосолп................... 13,3
Окислы и гидроокиси..................... 12,7
Сульфаты................................. 9,4
Галогениды............................... 5,8
Карбонаты................................ 4,5
Самородные элементы...................... 4,3
Бораты................................... 2,9
Карбиды, нитриды, фосфиды, хроматы и проч. 3,3
Содержание различных М. в исследованной части
земной коры выражается след, числами (в вес. %):
Силикаты..........................ок. 75
Окислы и гидроокиси (в т. ч. 12,6%
S1O2)............................ок. 17
Карбонаты ........................ок. 1,7
Фосфаты, арсенаты, ванадаты.......ок. 0,7
Галогениды.........................ок. 0,5
Сульфиды, сульфаты.................ок. 3,0—4,0
Самородные элементы................ок. 0,1
Морфология и физические свойства. Из морфоло-
гия. свойств М. особенно важен габитус (облик)
кристаллов. Обычно в природе твердые М. распростра-
нены в виде зерен неправильной формы; хорошо обра-
зованные кристаллы, ограниченные естественными гра-
нями, встречаются сравнительно редко. Среди крис-
таллов обычно выделяют следующие морфология,
типы: изометрия, формы, т. е. формы, одинаково раз-
витые во всех трех направлениях в пространстве,
например кристаллы пирита FeS2, пиропа (разновид-
ность граната) MgsAblSiOah, магнетита ЕезОа, шпи-
нели MgA^Oa; формы, вытянутые в одном направле-
нии,— таковы призматич., шестоватые, игольчатые
кристаллы, например, аквамарин ВезА12[316О18], тур-
малин (Na,Ca)(Mg,Al)6[B3A13Si6(O,OH)3!,], антимонит
SbaSs, эпидот Ca2(Al,Fe)3[Si2O,][SiO4jO(OH); фор-
мы, вытянутые в двух направлениях, таблитчатые,
пластинчатые, листоватые, чешуйчатые кристаллы,
напр. слюды КА12[А1й1зО10] (ОН)2, гипс CaSO4-2H2O,
отэнит Ca[UO2]2[PO4]2 -8Н2О. Для нек-рых М. харак-
терны двойники, т. е. закономерно сросшиеся
кристаллы того же минерала. Двойники типичны для
рутила ТЮ2, касситерита SnO2, гипса CaSCU -2Н2О
и др.
Чаще всего М. встречаются в виде множества срос-
шихся кристаллич. зерен — минеральных агрегатов.
Бывают агрегаты мономинеральные, со-
стоящие из зерен одного М., и полиминерал ь-
н ы е, представленные несколькими различными по
составу и свойствам М. Различают несколько типов
минеральных агрегатов в зависимости от их строения
и морфология, признаков. Главнейшие типы следую-
щие. Зернистые агрегаты, образованные
кристаллич. зернами различных М., напр. руды
полезных ископаемых. Друзы — сростки хорошо
образованных кристаллов, наросших на стенках пус-
тот (друзы кварца SiO2, кальцита СаСОз). Конкре-
ции — сферич. стяжения, возникшие в рыхлых оса-
дочных породах; в виде конкреций часто встречаются
пирит FeS2, марказит FeS2, фосфорит и др. Натеч-
ные формы возникают из гелей в пустотах, как,
напр., сталактиты и сталагмиты кальцита СаСОз в
больших пещерах. Морфология М., облик кристаллов,
характер минеральных агрегатов, а также структуры
агрегатов (зернистые, скрытокристаллич., колломорф-
ные, натечные и др.) часто дают возможность устано-
вить историю образования М.
Из оптических свойствМ. обычно выде-
ляют прозрачность, цвет, блеск и показатель прелом-
ления. В зависимости от степени прозрачности все М.,
встречающиеся в крупных кристаллах, принято делить
на прозрачные — горный хрусталь SiO2, ис-
ландский шпат СаСОз, топаз Al2[S1O4] (F,OH)2 и др.;
полупрозрачные — изумруд ВезА12[8цО18],
сфалерит ZnS, киноварь HgS и др.; непрозрач-
ные— пирит FeS2, магнетит РезО4, графит С и др.
Многие минералы, кажущиеся в больших кристал-
лах или обломках непрозрачными, просвечивают в
тонких осколках или в тонких шлифах — биотит
K(Mg,Fe)8[SisAlO10](OH,F)2, рутил TiO2. В тонко дис-
персных агрегатах даже совершенно прозрачные М.
кажутся непрозрачными, благодаря рассеянию света.
Цвет для многих М. очень характерен и постоя-
нен. Различают идиохроматич., аллохроматич. и псев-
дохроматич. окраску М. Идиохроматиче-
ская окраска (т. е. свойственная данному М.) обу-
словлена внутренними свойствами М. Такие М. ни-
когда не встречаются в природе в виде бесцветных
образований. Примеры: черный магнетит ЕезСЦ, ла-
тунно-желтый пирит FeS2, карминово-красная кино-
варь HgS и др. Чаще всего идиохроматич. окраска М.
вызвана присутствием в М. т. н. хромофора,
т. е. химич. элемента, приносящего окраску. К числу
хромофоров относятся Сг, Ti, V, Mn, Fe, Со, Ni, в
меньшей степени W, Mo, U, Си, редкоземельные эле-
менты. Сг — наиболее сильный краситель. Содержа-
ние даже незначительных количеств Сг, в зависимости
от валентности иона и положения его в кристаллич.
решетке, приводит к интенсивной и разнообраз-
ной окраске М., напр. красный цвет имеют пироп
MgsAlHSiCHh, рубин А120з и др., зеленый — изумруд
ВезА12[Si6Ola], фуксит КА12[AlSi3O10] (ОН)2 и др.>
фиолетовый — родохром (Mg,Fe)sAl[AlSi3Oi0](OH)8,
кеммерерит (разновидность родохрома) и др. Реже-
идиохроматич. окраска вызывается нарушением элек-
тростатич. однородности кристаллич. решетки под
влиянием изменения темп-ры облучения радиоактив-
ными веществами и др.; пример — синяя каменная
соль NaCl. Наконец, идиохроматич. окраска может
быть обусловлена внедрением целых групп ионов
внутрь кристаллич. решетки. Так, синяя окраска
лазурита Na8[AlSiOJ6[SO4] предположительно свя-
зана с ионами серы.
241
МИНЕРАЛЫ
242
Аллохроматическая окраска (т. е. чуж-
дая для данного М.) зависит не от химич. природы са-
мого М., а связана в большинстве случаев с тонко
рассеянными механич. примесями органич. и неорга-
нич. происхождения. Напр., красный цвет барита
BaSOa и кварца SiO2 вызывается примесью Ре20з,
черный цвет кальцита СаСОз— присутствием суль-
фидов или органич. веществ, серый и дымчатый цвет
кварца — распыленными газовыми и жидкими вклю-
чениями.
Псевдохроматическая (т. е. ложная)
окраска обусловлена интерференцией света при его
отражении от внутренних поверхностей трещин в М.
или от поверхности к.-л. включений. В нек-рых не-
прозрачных М. псевдохроматизм наблюдается в виде
характерной «игры цветов», напр. в лабрадоре (М.
из группы плагиоклазов, см. ниже) при нек-рых уг-
лах поворота наблюдаются местами синие и зеленые
переливы.
Блеск М., вызванный отражением света от зер-
кально гладких поверхностей (граней кристаллов),
зависит от показателей преломления М. Обычно раз-
личают стеклянный блеск, характерный для
М. с показателями преломления п = 1,3—1,9,— та-
ковы лед, криолит NasAlFj, флюорит CaF2, кварц SiO2,
гранат Mg3Al2[SiOa]3 и др.; алмазный блеск,
типичный для М. с п == 1,9—2,6,— касситерит SnO2,
сфалерит ZnS, алмаз С и др.; полуметалличе-
ский блеск, свойственный М. с пй"3 (куприт Сп2О,
киноварь HgS и др.), металлический блеск,
характерный для М. с га>3 (молибденит MoS2, анти-
монит Sb2S3, галенит PbS и др.). Металлич. блеск
очень характерен для многих важных в промышленном
отношении М. Блеск, обусловленный отражением
света от поверхностей излома М., бывает жирный,
восковой, матовый, перламутровый
и др. Типичным матовым блеском обладают, напри-
мер, мел, каолинит Ala[Si4O10](OH)8 и др. Перламут-
ровый блеск или отлив характерен для мускови-
та КА12[А181зО10](ОН)2, гипса CaSO4-2H2O, талька
Mg3[Si4O10](OH)2 и др. минералов со слоистой плос-
копараллельной структурой.
Из механических свойств важней-
шими для диагностики М. являются спайность, плот-
ность, твердость. Спайность — способность кри-
сталлических зерен или кристаллов раскалываться
по определенным кристаллографическим плоскостям.
Различают спайность: 1) весьма совершенную — та-
ковы слюды; 2) совершенную — кальцит СаСОз, гале-
нит PbS; 3) среднюю — полевые шпаты №[А181зО8];
4) несовершенную—апатит Са.[РО4]зР; касситерит
SnO2; 5) весьма несовершенную (спайность практи-
чески отсутствует) — самородные золото, платина
и др.
Удельные веса (плотности) М.
колеблются в широких пределах. Главная масса
окислов и солей легких металлов обладает уд. весом
от 1,0 до 3,5; таковы, напр., гипс CaSO4-2Н2О (2,3),
кварц SiO2 (2,65), алмаз (3,5). М. тяжелых металлов
характеризуются уд. весом от 3,6 до 9,0; примерами
служат сидерит ЁеСОз (3,9), гематит Fe2Oa (5,25),
галенит PbS (7,5). Наибольшие уд. веса имеют само-
родные тяжелые металлы: Си (8,9), Ag(10), Аи (ок. 19),
1г (22,6—22,8).
Твердость М. определяют посредством ца-
рапания острием, более твердым, чем исследуемый М.
Для точных измерений твердости М. служат специаль-
ные приборы (склерометры), имеющие острие или на-
конечник в виде конуса из алмаза, твердого сплава,
стали. Минералоги обычно определяют относительную
твердость М. по шкале Мооса. Эта шкала представлена
десятью минералами — эталонами, из к-рых каждый
последующий своим острым концом царапает все
предыдущие. Эталонами шкалы Мооса служат след.
М. в порядке возрастания твердости.
1. Тальк Mg3 [Si4O10] (ОН)а 6. Ортоклаз К [AlSi,O8l
2. Гипс Са8О4-2Н2О 7. Кварц SiO2
3. Кальцит СаСО3 8. Топаз Al2 [SiO,] (F, ОН)а
4. Флюорит CaF2 9. Корунд А12О3
5. Апатит Са5[РО(]3 F 10. Алмаз С
Из других свойств для определения М. важны:
магнитные свойства, радиоактивность, растворимость,
вкус, запах, электро- и теплопроводность.
Физико-химическая природа, состав и кристалли-
ческая структура. Лишь немногие М. являются прос-
тыми телами; таковы, напр., алмаз, графит, самород-
ные Fe, Ni, Pt, Ir, Rh, Ru, Os, Cu, Ag, Hg, Pb, S,
As, Sb, Bi, Pd, Au; впрочем, самородные металлы
почти всегда содержат примеси обычно в виде твердых
р-ров. В подавляющем большинстве М. состоят из
химич. соединений, твердых р-ров и коллоидных си-
стем. К простейшим соединениям, состоящим только
из атомов двух различных элементов, принадлежат:
1) окислы—куприт Си2О, периклаз MgO, гематит Ге20з,
кварц SiO2 и др., 2) галогениды— галит NaCi, флюо-
рит CaF2, 3) сульфиды— миллерит NiS, галенит PbS,
антимонит Sb2Ss и др. Соли кислородных к-т более
сложны; кроме кислорода, они могут содержать 2,3
и большее число различных элементов. Сюда относятся
т. наз. «простые соли» — кальцит СаСОз, ангидрит
CaSO4, форстерит Mg2SiO4, а также двойные соли —
глауберит Na2Ca[SO4]2 и комплексные соли. Многие
М. имеют переменный состав вследствие способности
к образованию твердых растворов, являющейся об-
щим свойством кристаллич. веществ. Среди М. встре-
чаются все 3 структурных типа твердых р-ров, а
именно: 1) замещения, 2) внедрения, 3) вычитания.
Наиболее распространены твердые р-ры замещения,
образование к-рых основано на изоморфизме.
Различают изовалентный и гетеровалентный изо-
морфизм. Изовалентный изоморфизм, характеризую-
щийся взаимным замещением в кристаллич. структуре
ионов с одинаковой валентностью, проявляется при
условии, если свойства и размеры этих ионов близки
друг к другу. Примерами совершенного изоморфизма
являются ряды: магнезит — сидерит (MgCOs—FeCOs),
ковеллин—клокманит (CuS—CuSe). Гетеровалентный
изоморфизм отличается тем, что в кристаллич. струк-
турах происходит замещение целых групп ионов груп-
пами ионов другой валентности при условии оди-
наковой суммы всех валентностей. Напр., мона-
цит (Ce,La,Th,Ca)[PO4,SiO4,SC>4] с примесями MgO,
МпО, РЬО, Ре2Оз,А12О3 и Н2О или ряд плагиоклазов,
альбит — анортит Na[AlSisO8]— Ca[Al2Si2O8]. Благо-
даря изоморфизму многих М. в них, кроме элементов,
входящих в их типовую формулу, обычно присут-
ствуют посторонние химич. элементы. Например,
в сфалерите ZnS изоморфная примесь железа может
достигать 20%, кадмия — десятых долей процента,
индия — сотых долей процента; кроме того, в сфа-
лерите присутствуют обычно Са, Mn, Hg.
Физич. и химич. свойства твердых р-ров плавно
изменяются в зависимости от содержания второго
компонента. Так, в ряду плагиоклазов, в к-ром на-
считывается 6 членов, по мере изменения состава от
чистого альбита до чистого анортита (крайние члены
ряда), плотность М. этого изоморфного ряда увеличи-
вается от 2,61 до 2,76. В отличие от описанных выше'
истинных твердых р-ров, в твердых псевдо растворах
содержатся тонкодисперсные механич. примеси посто-
ронних веществ (напр., розовый кварц SiO2, красный
кальцит СаСОз). Твердые р-ры внедрения и вычитания
встречаются среди М. значительно реже. Примером
твердого р-ра внедрения могут служить цеолиты, в
структуре к-рых пустоты располагаются «столбами»,
образуй «трубы», в к-рые и могут попадать молекулы.
243
МИНЕРАЛЫ —МИНЕРАЛЬНЫЕ УДОБРЕНИЯ
244
воды. Пример твердого р-ра вычитания — магнитный
колчедан или пирротин Fe, _XS, где z=0,00—0,16.
В его структуре атомы S располагаются по местам
шаров гексагональной плотнейшей упаковки как при
соотношении Fe:S, равном единице, так и при избытке
атомов S. Поэтому при избытке S против ф-лы FeS
в структуре появляются свободные октаэдрич. пусто-
ты; образуется т. наз. дефектная или дефицитная
структура.
М. могут содержать воду, как связанную, вхо-
дящую в их кристаллич. структуру, так и свободную,
не участвующую в строении кристаллич^ вещества.
В первом случае молекулы воды занимают в кристал-
лич. решетке строго определенные места, причем
число молекул ШО находится в простых отношениях
с другими компонентами соединения, напр. кристал-
логидраты— сода КагСОз-ЮН20, гипс CaSOa'2Н2О,
айНабергит NiafAsOah -8Н2О. Кристаллогидраты при
нагревании теряют воду скачкообразно, причем их
физич. свойства также изменяются скачкообразно.
В цеолитах же содержание воды может изменяться без
нарушения кристаллич. однородности вещества и при
плавном изменении физич. свойств (прозрачности,
показателей преломления и пр.). Это показывает, что
вода в цеолитах находится в состоянии твердого р-ра.
Молекулы воды часто удерживаются в тонких трещи-
нах, порах и порошковатых массах М. силами по-
верхностного натяжения (т. н. гигроскопичес-
кая вода). Часто свободная вода находится в М.
в виде механич. примесей — мельчайших газовожид-
ких включений, захваченных кристаллами во время
.их роста
Кроме явно кристаллич. образований, в природе'
широко распространены гели и золи. Примеры М.—
гелей: опал—гель кремнезема SiO2 'aq, лимонит—гель
гидроокисей железа HFeO2-aq. Примеры М.— крис-
талловолей: красный барит BaSOa, молочно-белый
кварц SiO2.
Генезис и парагенезис. В природе М. распростра-
нены в виде образовавшихся совместно групп или
парагенетич. ассоциаций. Это явление, называемое
парагенезисом, объясняется взаимодействием рудо-
носных р-ров с окружающей средой на каждой стадии
развития процессов минералообразования. Примером
парагенетич. ассоциации может служить ряд рудных
минералов: галенит PbS, сфалерит ZnS, халькопи-
рит CuFeS»; другая типичная ассоциация: пирротин
Fe,_xS, пентландит (Fe, Ni)9S8, магнетит Fe3O4, халь-
копирит. Знание типичных парагенетич. ассоциаций
помогает при поисках полезных ископаемых.
Все особенности М.— химич. состав, структура,
морфология, химич. и физич. свойства, парагенезис,
распространение в природе — обусловлены их г е-
иез и сом. Под генезисом М. понимают совокуп-
ность следующих явлений: 1) собственно образование
М., охватывающее все моменты их возникновения —
нарождение, кристаллизация, изменения, а также
явления разрушения и уничтожения М., 2) геоло-
гия. процесс минералообразования — магматический,
гидротермальный, осадочный и др. (См. Геохимические
процессы).
Отдельные минералы, их свойства и области приме-
нения в народном хозяйстве см. в соответствующих
статьях о химич. элементах.
Лит.: Вернадский В. И., Избранные сочинения,
т. 2, М., 1955; Б е т е х т и н А. Г., Курс минералогии, 3 изд.,
М., 1961; Минералы. Справочник, т. 1, М., 1960; Григорь-
ев Д. П., Что такое минерал?, Зап. Всес. Мин. о-ва, 1961, 90,
Л 4; Б а р с а н о в Г. П., Принципы современной класси-
•фикации минералов, Тр. Минералогического музея, 1959,
.вып. 9; Григорьев Д. П., Онтогения минералов,
-Львов, 1961; Геологический словарь, т. 1—2, М., 1960; Бо-
жий Г. Б., Введение в кристаллохимию, М., 1954;
Дэна Дж. Д. [и др.], Система минералогии т. 1, полутом
1—2, т. 2, полутом 1—2, М., 1951—54; Handbuch der Mi-
aieralchemle, hrsg. v. C. Doelter, Bd 1—4, Dresden — Lpz.,
1912—31; Hintze C., Handbuch der Mineralogle, Bd 1,
Lpz., 1915. В. Г. Ростов.
МИНЕРАЛЬНЫЕ УДОБРЕНИЯ — неорганич. ве-
щества, содержащие необходимые для растений эле-
менты, вносимые для воздействия на режим пита-
ния растений. При правильном использовании М. у.
увеличивается урожай и улучшается качество с.-х.
продуктов: повышаются технология, свойства волок-
на хлопчатника, льна и других лубяных культур,
увеличивается содержание сахара в сахарной свекле
и винограде, крахмала в картофеле, белков в злаках
и кормовых травах, повышается сортность табака и
т. д. По агрономия, назначению различают прямые
и косвенные М. у.
Прямые М. у. предназначены для непосредст-
венного питания растений и содержат необходимые
для этого элементы (гл. обр. азот, фосфор, калий, а
также магний, серу, микроэлементы — бор, медь,
марганец, молибден, цинк и др.). Прямые М. у. раз-
деляют на односторонние и комплексные. К односто-
ронним удобрениям, содержащим один питательный
элемент, относятся: азотные, фосфорные, калийные,
углекислые, магниевые, серные, борные удобрения
и др. Комплексные удобрения содержат два или
большее число питательных элементов. Различают
двойные удобрения, напр. азотно-фосфорные, азот-
но-калийные, фосфорно-калийные, и тройные удоб-
рения, напр. азотно-фосфорно-калийные, к-рые на-
зывают также полным М. у. Большую часть прямых
М. у. изготовляют промышленным путем. Комплекс-
ные удобрения делят также на смешанные и сложные
удобрения. Смешанные удобрения пред-
ставляют собой механич. смеси различных удобрений,
состоящие либо из различных по своему составу ча-
стиц, либо из гранул, в состав которых включены
компоненты смеси. Сложные удобрения
получают в результате химич. взаимодействия, а
также путем совместной кристаллизации, сплавления
основных компонентов.
Косвенные М. у. предназначаются гл. обр.
для улучшения свойств почвы. К косвенным удоб-
рениям относятся средства для нейтрализации кислот-
ности почв (известкование) и мелиорации солонцов
(гипсование), а также удобрения для подкисления
почв с целью повышения растворения фосфорных
удобрений (бисульфат натрия).
По влиянию на реакцию почвы различают физио-
логически кислые, щелочные и нейтральные М. у.
К физиологически кислым относят удобрения, катионы
к-рых лучше поглощаются растением, чем анионы, а
последние подкисляют почвенный р-р. К физиоло-
гически щелочным принадлежат удобрения, анионы
к-рых лучше ассимилируются растением, а катионы,
постепенно накапливаясь, подщелачивают почвенную
среду. Физиологически нейтральные удобрения не
изменяют режима почвы.
Большую часть М. у. выпускают в порошковид-
ном или гранулированном виде. Гранулированные
М. у. менее слеживаются, чем порошковидные, и
лучше рассеиваются. Гранулированные водораствори-
мые фосфорные удобрения полнее используются на
кислых почвах, чем порошковидные. Все большее
значение приобретают жидкие удобрения. М. у. оце-
нивают гл. обр. по содержанию в них питательных
элементов в усвояемой для растений форме. Важней-
шими из этих элементов являются N,P2O5 и К2О.
Содержание питательных элементов в наиболее рас-
пространенных М. у. приведены в табл. 1.
М. у. вносят перед посевом (основное удобрение),
во время посева (припосевное удобрение) и в период
роста растений (подкормка) (подробнее см. Удобрения).
Дозы М. у., вносимых в почву на разных стадиях, при-
ведены в табл. 2.
245
МИНЕРАЛЬНЫЙ ОБМЕН—МИРЦЕН
246
Таблица 1
Наименование М. у. Примерное содержание (в %%)
N Р2О3 К2О
Аммиачная селитра ..... 34-35
Натриевая селитра 15-16 —
Кальциевая селитра .... 15,5-17 — —
Сульфат аммония ...... 20.5—21 —
Мочевина 46-46.3 —- —
Суперфосфат порошковид- ный 14-20
Суперфосфат гранулирован- ный 14-21
Суперфосфат аммонизиро- ванный 1 ,5-7 14-21
Суперфосфат двойной грану- лированный 42—48
Преципитат — 33—40 —
Обесфторенный фосфат . . . — 26-3 6 —
Томасшлак — 1 4 — 18 —
Фосфоритная мука — 16-25 —
Хлористый калий — — 52—60
40%-пая калийная соль . . — — 40
Сульфат калия — —— 45—52
Аммофос 10 — 12 47—52 —
Диаммофос 18—21 48—52 —
Нитрофос 14-20 14—20 —
Нитрофоска 12-16 12-16 12—16
Таблица 2
Вид удобрения Средние дозы удобрений, кг[га
N Р2О3 К2О
Основное удобрение .... 30—90* 30—90* 40-120
Прмпосевное удобрение . . 10-15 10—20 10-15
Подкормка 15—30 20—30 20—40
* Под хлопчатник, чай, виноград и цитрусовые культуры
вносят и большие дозы удобрений.
Лит' Справочник по минеральным удобрениям. Теория
и практика применения, М., i960; ПрянишниковД. Н.,
Избр. сочинения, т. 1, Агрохимия, М., 1952; Кол-
лингс Г. X., Промышленные удобрения, их производство
и применение, пер. с англ., М., 1960; Вольфкович С. И.,
Ж. Всес. хим. о-ва, 1962, 7, № 5, 482; П о з и н М. Е., Техно-
логия минеральных солей, 2 изд., Л., 1961.
А. И. Шсрешевский.
МИНЕРАЛЬНЫЙ ОБМЕН — см. Обмен веществ.
МИОЗИН — фибриллярный белок, основной белко-
вый компонент мышечного волокна, в комплексе с
актином образует актомиозин, являющийся сократи-
тельной системой мышц. Данные об аминокислотном
составе М. приведены ниже. Макромолекулы М. легко
комплексуются между собой, в силу чего литератур-
ные данные о мол. весе М. колеблются в пределах
от 450 000 до 1 000 000; по той же причине для р-ров
Аминокислотный состав актина, миозина и меромиозинов
(в г аминокислот на 100 г белка)
Аминокислота Актин Миозин Н -Меро- миозин L-Mepo- миозин
Аланин 6.31 6,94 6,50 6,75
Глицин 5,02 2,92 3,38 1 ,80
Валин 4,92 4,92 5.27 4,57
Лейцин 8,25 10,35 10,20 11,13
Изолейцин 7,46 5,50 5.50 4,58
Пролин 5,06 2,53 3,34 0,97
Фенилаланин 4.78 4,46 6,60 1 ,58
Тирозин 5,80 3,25 3,80 2,17
Триптофан 2,04 0,80 0,61 1 ,22
Серин 5,88 4,31 4,52 3,89
Треонин 7,02 4,88 5,83 4,52
Цистин 12 1,34 1,03 1 ,31 0,67
Метионин 4,47 3,28 2,83 2.08
Аргинин 6,61 7,13 5,04 8,87
Гистидин 2,94 2,32 1 ,78 2,94
Лизин 7.60 12,40 11,98 12,11
Аспарагиновая к-та . . 10,90 11,40 11,80 10,24
Глутаминовая к-та . . . 14,85 22,80 20,30 25,60
мышечного М. константы седиментации в единицах
Сведберга колеблются в пределах от 6,4 до 7,2. По-
добно глобулину М. не растворяется в воде, хорошо
растворим в 0,1 М р-ре КС1 и полностью выпадает
при снижении его концентрации до 0,02 М. На этом
принципе основано выделение студня М. из солевых
р-ров. М. может быть получен и в виде удлиненных
псевдокристаллов. Характерным для М. является
присутствие свободных SH-групп. М. обладает свой-
ствами фермента аденозинтрифосфатазы. При от-
щеплении от АТФ под действием миозина остатка
фосфорной к-ты освобождается энергия, необходимая
для сокращения мышечных элементов.
При кратковременном действии на р-р М. трипсина
макромолекула М. распадается на два компонента,
названных меромиозинами. Меромиозин с
мол. в. ок. 230 000 назван тяжелым, или Н-меромио-
зином, а меромиозин с мол. в. ок. 96 000 —L-меро-
миозином.
Н-меромиозин сохраняет аденозинтрифосфатазную
активность и способность соединяться с актином, L-
меромиозин отличается отчетливо выраженными оп-
тич. свойствами. Полагают, что в состав макромоле-
кулы М. входит один Н- и два Ь-меромиозина.
Лит.: Белки, под ред. Г. Нейрата и К. Бейли, пер.
с англ., т. 3, ч. 2, М., 1959, с. 460—8; Любимова М. Н.,
Энгельгардт В. А., Биохимия, 1939, 4, вып. 6, 716—35;
Kiel 11 ng W. W., Harrington W. F., Blochein.
et blophys. acta, 1960, 41, Ki 3, p. 401—21; S z e n t-G у б г g у
A. G., Arch. Blochein. and Blophys., 1953, 42, 2, 305.
В. O. IHnuKumep.
МИП0РА — поропласт на основе смолы карбамид-
ной (см. Поропласты).
МИРИСТИНОВАЯ КИСЛОТА СН3(СН2)12СООН,
мол. в. 228,36—т. пл. 54,40; т. кип. 250,57100 мм,
199716 мм; d™ 0,8533; п^ 1,4268; кислотное число
245,7°; плохо растворима в спирте и эфире. М. к. со-
держится в виде глицеридов (в основном в виде три-
миристина) в мускатном и кокосовом маслах, в семенах
Virola venezuelensis, в небольшом количестве в коро-
вьем масле. Под влиянием грибков Penicillium glau-
cum глицериды М. к. и сама к-та «прогоркают»
(подобно другим высшим жирным к-там), при этом
образуется метилундецилкетон. Полный глицерид
М. к. (тримиристин), т. пл. 56,2°; метиловый эфир
т. кип. 155—15777 .и.и; этиловый эфир т. кип. 295°.
МИРИЦИЛОВЫЙ СПИРТ (мелиссиловый спирт)
Сз1НвзОН, мол. в. 452,85— твердое вещество, т. пл.
87°, растворимое в органич. растворителях, кристал-
лизуется из них в виде пластинок. При нагревании в
присутствии щелочи М. с. превращается с выделением
водорода в мелиссиновую к-ту СНз(СН2)29 СООН и др.
продукты. М. с. содержится в виде эфира пальмити-
новой и церотиновой к-т в пчелином и карнаубском
восках.
Ранее считалось, что мирициловый и мелиссиловый спирты
являются различными соединениями. Предположение было
сделано вследствие неоднородности различных образцов М. с.
Считают, что М. с. представляет смесь гомологов Сзо и С32.
А. В. Арбатский.
МИРЦЕН С10Н1в, мол. в. 136,23— прозрачная жид-
кость с приятным запахом, легко изменяющаяся на
воздухе и при нагревании; М. хорошо растворим в
большинстве органич. растворителей, в воде практи-
чески нерастворим. Известны 2 изомера:
1) а-мирцен (2-метил-6-метилен-октадиен-1,7)
2) 0-мирцен (2-метил-6-метилен-октадиен-2,7)
н=с\
насх
с-сн2-сн2-сна-с-сн=сн2
и
сн2
(1)
(2)
н3сх
Hacz
с=сн-сн2-сн2-с-сн=сн2
247
МИХАЭЛИСА КОНСТАНТА
248
а-М. мало изучен. 0-М. имеет т. кип. 1687760 мм,
65,5720 мм; d,s’s 0,7966; n2D° 1,4650; XMaKC 224,5 ммк. Для
количественного определения 0-М. в смеси с другими
углеводородами, не содержащими сопряженной сис-
темы двойных связей, используют реакцию его с ма-
леиновым ангидридом. М. (гл. обр. 0-форма) с при-
месью небольшого количества a-формы встречается
в составе многих эфирных масел, напр. в байевом
масле (Pimenta acris), в масле багульника (Ledum
palustre), в масле плодов амурского и японского бар-
хатного дерева и др. М. содержится в скипидаре из
сосны обыкновенной и в скипидарах из других хвой-
ных. 0-М. получают дегидратацией линалоола; осо-
бенно удобна термич. изомеризация 0-пинена. 0-М.
применяют для синтеза душистых веществ и гидро-
перекисей И др. и. II. Бардышев.
МИХАЭЛИСА КОНСТАНТА характеризует зави-
симость скорости ферментативной реакции от кон-
центрации субстрата в стационарном состоянии про-
цесса; численно равна концентрации субстрата, при
к-рой скорость реакции составляет половину макси-
мально возможной. Использование М. к. в качестве
величины, характеризующей ферментативные реак-
ции, базируется на представлении, впервые выдви-
нутом Брауном в 1902 и получившем окончательное
обоснование в работах Михаэлиса и Ментен в 1913,
согласно к-рому во всяком ферментативном процессе
первым этапом является обратимая реакция между
ферментом (Е) и субстратом (А) с образованием ком-
плекса (ES), к-рый в ходе последующих превращений
дает продукты ферментативной реакции (S') с реге-
нерацией исходного фермента. Реакции эти характе-
ризуются соответствующими константами скорости
k2.
А, й.
E+S ^ES --,S’ +Е
k — 1
I II
Очевидно, что константа равновесия реакции I вы-
разится соотношением
к h,_____[ES]
Р Л_, [В] [S]
Величина Хр может быть найдена из измерений зави-
симости скорости реакции от концентрации субстра-
та. Если начальная концентрация фермента (точнее,
активных центров фермента) равна [Ео], то
[£] = [£„] —[ЕА]; тогда
К №8]
₽“([Eo]-[ES])[S]
Принимается, что [А] == [Ао], т. к. обычно [<У0]^>[Е0].
Из ур-ния (1)
(1)
KD [Bo] [S]
= l+Kp[S]
Скорость (v) реакции, измеряемая по образованию
продуктов реакции S' (реакция II), выразится соот-
ношением ’
v = и, следовательно,
йгкр[В] [S]
v== l+Kp[S]
и Ментен в своем выводе использовали не
Ар, а обратную ей величину — константу
(2)
Михаэлис
величину
диссоциации комплекса ES на £ и А; т. о., константа
Михаэлиса, Ам=1/Ар. Если в ур-нии (2) заменить
К„ на К„, то
[Во] [S]
Km + [S]
Анализ этого ур-ния показывает, что при значитель-
ном избытке субстрата, т. е. при [У]^>АМ, когда рав-
новесие в реакции I сдвинуто вправо и практически
(3)
весь фермент Е находится в виде комплекса ES,
v = *2 l^o] = const = Емакс. (4)
Иначе говоря, при значительном избытке субстрата
наблюдается максимальная скорость ферментативной
реакции, не зависящая от концентрации субстрата.
При малых концентрациях [А], когда [А] 7: Kw,
= М-ЕД [5]
т. е. скорость реакции находится в линейной зависи-
мости от концентрации субстрата. В действительности
очень большое количество ферментативных реакций
подчиняется этим соотношениям.
Из ур-ний (3) и (4) следует:
г /макс,^
KM + [S]
(5)
Если положить v = 0,5 Емакс , то Ам= [А], откуда
следует, что М. к. численно равна концентрации суб-
страта, при к-рой скорость ферментативной реакции
составляет половину максимальной.
Бриггс и Холден показали, что вывод Михаэлиса и
Ментен справедлив при допущении, что константа
скорости реакции II k2 существенно меньше констан-
ты klt что не всегда оправдывается. Бриггс и Холден
вывели ур-ние зависимости скорости ферментативной
реакции от концентрации субстрата для общего слу-
чая любых значений klt k_t и k2. Они показали, что
в условиях стационарного состояния системы
[ЕА] =
k, [E„][S]
+ + [S]
Если обозначить = К, то скорость реакции
выразится:
v_^h2K[E„] [S] 6.
1+K [S] ' 7
Ур-ние (6) аналогично ур-нию (2), но в него, вместо
константы равновесия Ар, входит константа К, учи-
тывающая скорости всех реакций. Если k2<^.k_t, то
т. е. А=Ар=1/Ам. Т. обр., ур-ния (2) и (3)
являются частными случаями ур-ния (6).
Аналогично ур-нию (4), k2 [EJ = EMaKC-; тогда
V К [S]
У__ макс. *• J
~ 1+K[S]
Обозначая K=i/KM, получим:
У ^MaKcJ8-^
KM + [S]
(7)
Ур-ние (7) вполне аналогично ур-нию (5), причем
константа может быть названа М..к., исправленной
по Бриггсу и Холдену.
Она действительна для
стационарного состояния
ферментативного процес-
са, в котором один актив-
ный центр фермента реа-
гирует с одной молекулой
субстрата, давая активиро-
ванный комплекс, распа-
дающийся с образованием
продуктов реакции и ре-
генерацией фермента.
Для определения величины М. к. исследуют зави-
симость скорости энзиматич. реакции от концентрации
субстрата, беря широкий ряд [А], и выражают эту
зависимость графически, откладывая по оси абсцисс
249
МИХАЭЛИСА—БЕККЕРА РЕАКЦИЯ — МИХАЭЛЯ РЕАКЦИЯ
250
1/[А], а по оси ординат i/V (метод двойных обратных
величин Лайнуивера и Бэрка, см. рисунок). Если в
ур-нии (7) взять обратные величины, то оно примет
вид:
1 к 1 1
—м-------1-----
v V [8]TV
' макс. 1 J макс.
В случае применимости к исследуемой реакции теории
Михаэлиса зависимость -j- = / выразится ли-
нейной функцией, причем прямая отсечет на оси орди-
нат отрезок, равный 1/7макс , а на оси абсцисс отрезок,
равный 1/Ам. Т. обр. графически могут быть вычисле-
ны величины Км и 1'макс. Из этих величин, если из-
вестна концентрация активных центров фермента, по
ур-нию (4) может быть вычислена k2.
Как следует из приведенных соотношений, М. к.
имеет размерность концентрации и выражается в
молях/л (М). Значения Кк и k2 для нек-рых фермен-
тативных реакций приведены в таблице.
Фермент Субстрат Тем- пера- тура, °C pH Км, М *2, сек 1
Трипсин Стурин 24,5 7,5 2,5-Ю-3 13100
Трипсин Бензоил-L-ap- гинин 25,0 8,0 8,0-10-5 26,7
Апенозинтри- фосфатаза Аденозинтри- фосфат 25,0 7,0 1 ,3-10-5 104
Уреаза Мочевина 20,8 8,0 4,0-10-3 30800
Лит.: L а 1 d 1 е г К. J., The chemical kinetics of enzyme
action, Oxf., 1958. В. А. Яковлев.
МИХАЭЛИСА — БЕККЕРА РЕАКЦИЯ — широ-
ко используемый метод синтеза фосфорорганич. сое-
динений взаимодействием натриевых или др. солей
кислых фосфитов и фосфинитов с галогеналкилами
(или другими алкилирующими реагентами). Напр.,
при взаимодействии натрийдиэтилфосфита с йодис-
тым этилом образуется диэтиловый эфир этилфос-
финовой к-ты:
(C2H5O)2P-ONa + C2H3J —>С2Н5РО (OC2H5)2 + NaJ
В настоящее время не существует единого взгляда
на механизм М.—Б. р. Можно рассматривать эту реак-
цию как двухстадийный процесс: присоединение гало-
геналкила к трехвалентному фосфорному соедине-
нию и отщепление соли галогена от промежуточного
соединения:
RO ROx'(^-Me ROx 7/.
4P-O-Me + R'Hal —»- + МеНа!
ROZ Rt/j^Hal ROZ V
Возможно, алкилирование солей диалкилфосфитов и
подобных соединений протекает с перенесением реак-
ционного центра:
Ме^О)
ROX\
R ;P:+R'-Hal ----- (RO)2P = O + MeHal
R'
В случае аллилгалогенидов М.—Б. р. часто сопровож-
дается аллильной перегруппировкой:
-,R-CH2-CH = CH-PO(OR')2
R - СН=СН - СН2Х + NaPO(OR ')а\
*R-CH = CH-CH2PO(OR')2
М.—Б. р. имеет общий характер. Кроме солей диалкил-
фосфитов, во взаимодействие с галогеналкилами всту-
пают производные тиофосфористой, моно- и диалкил-
фосфинистых к-т, причем образуются фосфорорганич. ]
соединения с одной, двумя или тремя Р—С-связями:
/S
(RO)3 P-SNa + R'Hal —► (RO)3 Р"
4R'
/zO
(RO) R'P - ONa + R"Hal —> (RO) R'PC
XR"
RR'P-ONa+R"Hal —>RR'P<?
4R"
В M.—Б.р. хлористые алкилы вступают менее энер-
гично, чем бромистые, а последние — труднее, чем йо-
дистые. Фтористые алкилы в эту реакцию не вступа-
ют. Скорости реакции дибутилфосфористого натрия с
хлористым, бромистым и иодистым бутилом в толуоле
относятся как 1:125:475. Скорость реакции алкилбро-
мидов с дибутилфосфористым натрием резко падает
при переходе от бромистого метила к бромистому бу
тилу. При дальнейшем увеличении числа атомов
углерода в углеводородном радикале (до додецила)
она остается примерно на одном уровне; приблизи-
тельно такая же закономерность существует и для
других галогеналкилов.
Введение нуклеофильных групп в молекулу гало-
геналкила ускоряет реакцию, электрофильные груп-
пы замедляют ее. Вторичные галогеналкилы вступают
в М.—Б. р. с меньшей скоростью, чем первичные; в этом
случае наблюдаются побочные процессы. Из третич-
ных галогенопроизводных только триарилхлорметаны
образуют фосфорорганич. соединения. Другие гало-
гениды этого типа реагируют иначе, так, при взаимо-
действии натрийдиэтилфосфита с трифенилброммета-
ном образуются тетраэтилсубфосфат и трифепилметил,
стабилизирующийся в присутствии кислорода в виде
перекиси:
(С0Н5)3СВг + (С2Н,О),Р - ONa —►
/О
—с (СГ,Н5)3 с- +(С2Н5О)2 P<f
ЧО-Р(ОС,Н5)2
2(C,HS),C+O3—*(С,Н5)3С-О-О-С (С.Н3),
При взаимодействии натрийдиалкилфосфитов с гало-
геналкилами, имеющими функциональные группы в
алкоксильном радикале, могут образоваться фосфор-
органич. соединения, содержащие соответствующие
функциональные группы или продукты из последую-
щих превращений.
Во взаимодействие с натрийдиалкилфосфитами мо-
гут вступать и галогенацилы с образованием эфиров
кетофосфиновых кислот, однако в этом случае имеют
место побочные процессы. М.—Б. р. используется для
получения соединений, содержащих связь фосфора не
только с углеродом, но и другими элементами. В этом
случае применяются галогенангидриды элементоорга-
нич. к-т и подобные соединения, напр.
/О
(RO)2 Р —ONa + (R'O)s S1C1 —> (RO)2 Р"
4S1 (OR')s
(RO)3 P-ONa+R’SCl —>- (RO3)"
M.—Б.р. применяется для получения многих фосфор-
органич. пестицидов, лекарственных средств и других
физиологически активных фосфорорганич. соедине-
ний, экстрагентов для ряда металлов и др. целей.
Реакция открыта в 1897.
Лит.: Плец В. М., Органические соединения фосфора,
М„ 1940; Michaelis A., Becker Th., Вег., 1897, 30,
1, 1003; Nylen Р., Studien tiber organische Phosphorverbln-
dungen, Uppsala, 1930; Kosolapoff Organophosphorus
compound, N.Y.—L., 1950. Э. E. Нифантьев.
МИХАЭЛЯ РЕАКЦИЯ — присоединение к акти-
вированной двойной связи соединений с подвижным
атомом водорода при углероде:
। ।
RCH =CHCOR'+ —СН—с - С-CHRCH.COR'
II 1
251
МИХЛЕ РА КЕТОН—МИЦЕЛЛЫ
252
С этилатом натрия в качестве катализатора М. р.
осуществляется уже при комнатной темп-ре, с пипе-
ридином—при незначительном нагревании. М. р. обра-
тима, чем объясняется образование в нек-рых случаях
продуктов замещения, а не присоединения:
СН(СООС2Н5)а СН(СООС2Н5)2
/ /
CNCHaCOOC2H3+CH CH-CH(CN)COOC2H3—с
Хс (COOC2HS)2 \н(СООС2Н,)2
СН (СООС2Н5)2
/
—>СН CN + СН2(СООС2Н5)а
Хс/
\
соосан3
М. р. имеет широкое распространение; производные
малоновой к-ты, эфиры циануксусной и ацетоуксусной
к-т, цианистый бензил, динитрометан и мн. др. при-
соединяются к а,р-непредельным альдегидам, кето-
нам, нитрилам и кислотам, к алкилиденмалоновым
к-там и пр.:
ch3coch2cooc2h5+ch2=chcn —> сн3соснсоос2н5
CH2CH2CN
СН2(СООС,Н5)а + С,Н5СН = СНСООС2Н5 —>
—> СвН5СН-СН2СООС2Н5
I
СН (СООС2Н5)а
CNCH2COOC2H5 + (CH3)2C = CHCOOC2Hj —>
—> [СН3|2 С-СН2СООС2П5
I
СН (CN) СООС2Н5
NO2CH2NO3 + 2CH2 = CHCN —> (NO2)2C[CH2CH2CN]2
Частным случаем М. р. является цианэтилирование.
Внутримолекулярная М. р. приводит к образова-
нию циклич. соединений:
GCH=CHCOOCH3
ZCH2COOCH3
сн-сн.,соосн3
СНСООСНз
Z = O, S. NCH3
Механизм М. р. до сих пор твердо не установлен,
однако предполагается, что под действием основания
образуется анион псевдокислотного агента, к-рый
далее присоединяется к p-углеродному атому моле-
кулы а,Р-ненасыщенного соединения
। ।
-СН+В- —►-С-+ВН
। ।
।
-С- + RCH = CHCOR' —> RCH-CHCOR' —> RCHCH2COR'
Такой механизм реакции согласуется с тем, что
Р-алкильные группы в ненасыщенном соединении
понижают, а Р-карбэтоксильная — значительно уве-
личивает скорость присоединения. Реакция открыта
Михаэлем в 1887.
Лит.: С е р р е й А., Справочник по органическим реак-
циям, пер. с англ., под ред. Н. С. Вульфсона, М., 1962, с.
189; Ингольд К. К, Механизм реакций и строение
органических соединений, пер. с англ., М., 1959, с. 553;
Bergmann Е. D., Ginsburg D., Рарро R.,
в кн.: Organic reactions, ed. R. Adams, v. 10, N. Y., 1959,
p. 179. H. П. Гамбарян.
МИХЛЕРА КЕТОН [ди-п-диметиламинобензофе-
нон, тетраметилдиаминобензофенон, 4,4'-бис-(диме-
_______________________ тиламино) бензофе-
(CH3)aN-^\A-CO-^^X-N(CH3)g нон1 9c’?2°0N2’
' 13/2 \5J_g-7 \g._s/ 3 i мол. в. 268— кри-
сталлы, блестящие
серебристые или зеленоватые листочки; т. пл. 179°;
т. кип. выше 360° (с разл.); растворимость при 20—
25°: в воде 0,04%, в спирте 0,63%, в пиридине 10%,
в хинолине 9,72%; очень мало растворим в эфире,
хорошо растворяется в теплом бензоле; растворяется
в водных р-рах кислот, однако соли имеют склон-
ность гидролизоваться в горячей воде; растворы в
спирте обладают слабой флуоресценцией; т. пл. пи-
крата 156—157°.
При восстановлении М. к. действием Na в спирте
образуется 4,4'-тетраметилдиаминобензгидрол — гид-
рол Михл ера; при действии амальгамы алюминия об-
разуется пинакон; с NH3 дает желтый краситель—
аурамин. С диметиланилином в присутствии хлороки-
си фосфора дает кристаллический фиолетовый', кон-
денсируется со многими ароматич. аминами, образуя
триарилметановые красители', с Na в бензоле полу-
чается Na-производное, к-рое реагирует с соедине-
ниями, содержащими метильную или метиленовую
группы, с образованием карбинолов. Напр., с толу-
олом получается бензил-4,4'-тетраметилдиаминодифе-
нилкарбинол, а с галогенарилами — триарилкарби-
нолы, с хлорбензолом — основание малахитового
зеленого — тетраметил-п-диаминотрифенилкарбинол.
Качественно М. к. определяют по желтой окраске его
раствора в ледяной СНзСООН, окраска после добав-
ления зернышка Zn и нагревания переходит в зеленую
или сине-зеленую.
Получают М. к. действием фосгена на диметилани-
лин (промышленный метод); действием диметиламина
на 4,4-дихлорбензофенон в присутствии соединений
меди и др. М. к. является важным промежуточным
продуктом для получения красителей — производных
трифенилметана.
Лит.: Ворожцов Н. Н., Основы синтеза промежу-
точных продуктов и красителей, 4 изд., М., 1955, с. 360, 693,
705. Л. С. Поваров.
МИЦЕЛЛЫ — частицы дисперсных фаз в коллоид-
ных системах. М. являются сложными комплексами
из многих тысяч атомов, молекул, ионов. Средний раз-
мер М.—от 10-5до 10-7«.M. образуются как в про-
цессах диспергирования (особенно самопроизвольного)
фазы в данной среде, так и в процессах образования
новой фазы — при конденсации из отдельных моле-
кул и ионов. Исследование механизма конденсацион-
ного образования М. при помощи электронного микро-
скопа показало, что первоначально зародыши новой
фазы могут образоваться в виде аморфных агрегатов
(микрокапелек), в дальнейшем же происходит их
кристаллизация, сопровождающаяся диспергирова-
нием на отдельные ультрамикрокристалликп.
М. типичных лиофобных золей состоят из нераст-
воримого в данной среде ядра (представляющего собой
чаще всего ультрамикрокристаллич. образование),
окруженного двойным электрическим слоем ионов.
Один слой ионов, т. н. адсорбционный, находится на
поверхности ядра, сообщая ему электрич. заряд. Этот
слой образуется в результате адсорбции какого-либо
одного вида ионов из раствора или диссоциации ионо-
генных групп в поверхностном слое ядра. Эти ионы,
сообщающие ядру заряд, наз. потенциалоопределяю-
щими. В состав адсорбционного слоя входит также
часть ионов противоположного знака — компенсирую-
щих ионов; основная масса этих ионов образует второй
слой (слой противоионов), к-рый вследствие теплового
движения размыт (диффузный слой). М. обычно окру-
жена ориентированными молекулами растворителя —
сольватной оболочкой.
М. лиофильных коллоидов представляют собой ас-
социированные комплексы молекул (или ионов) ряда
веществ (напр., мыл), имеющих ярко выраженную
полярную асимметрию. Такие молекулы (или ионы)
состоят обычно из двух частей — длинного углеводо-
родного радикала и полярной группы. В водных р-рах
молекулы (или ионы) мыла вследствие межмолеку-
253
МНОГОАТОМНЫЕ СПИРТЫ
254
лярного взаимодействия ориентируются так, что
углеводородные цепи образуют внутреннее жидко-
образное гидрофобное ядро М., а наружная поверх-
ность ее состоит из гидратированных полярных (гид-
рофильных) групп. В разб. р-рах мыл такие М. имеют
сферич. форму и состоят из сотен плотно упакованных
молекул (ионов) мыла. Обратное строение имеют М.
мыл в углеводородных средах — с гидрофильным
олеофобным ядром и углеводородной сольватирован-
ной оболочкой. В более конц. р-рах мыл М. принимают
сложную форму с той же ориентацией молекул или
ионов, превращаясь в зародыши кристалликов мыла.
Ядро мыльных М. может поглощать значительное
количество жидкости, противоположной дисперсион-
ной среде по полярности и потому нерастворимой в
этой среде (углеводороды в воде, вода в углеводородах,
см. Солюбилизация). Т. обр., строение М. лиофильных
коллоидов в принципе такое же, как у лиофобных:
инактивное по отношению к дисперсионной среде яд-
ро, окруженное стабилизующей оболочкой.
Вне электрич. поля М. электрически нейтральна.
Под действием электрич. поля происходит разрыв
М. примерно на границе между адсорбционным и
диффузным слоями. Ядро М. с адсорбционным слоем
(коллоидный ион) перемещается к одному электроду,
противоионы диффузного слоя — к другому. При
изображении формул М. состав ядра заключают в
квадратные скобки, а состав ядра и адсорбционного
слоя — в фигурные. Напр., строение М. золей йо-
дистого серебра, гидроокиси железа и кремневой
к-ты изображается след, формулами:
[AgJ]n Ag+ (n-x) NO3 |’t+xNO3
{m [Fe (OH)3]n FeO+ (n-x) 01-}»+ xCl-
|m [SIO2-l/H2O]n S102- -2 (n-x) H+ |23c-2xH +
По форме M. бывают шарообразными (напр., час-
тицы золей благородных металлов, галогенидов се-
ребра, сульфидов) и анизодиаметричными — пластин-
чатыми, палочкообразными и т. д. (напр., частицы
золей VsO,., А1(0Н)3, Fe(OH)3, SiO2 -пН20). Форма
М. обусловливается кристаллич. строением ядра.
У анизодиаметричных М. окружающая их сольватная
оболочка неравномерна. При столкновении такие
М. притягиваются друг к другу наименее сольвати-
рованными участками, что приводит к образованию
рыхлой сетчатой структуры. В такой пространствен-
но структурированной системе М. теряют способность
свободно перемещаться и золь переходит в гель.
Важнейшим фактором устойчивости М. является лио-
фильность наружной поверхности их оболочки.
Лит.: Песков Н. П., Александров а-П рейс
Е.М., Курс коллоидной химии, 2 изд., М,—Л., 1948; Пасын-
ский А. Г., Коллоидная химия, М., 1959.
МНОГОАТОМНЫЕ СПИРТЫ — спирты°₽жирного
ряда, содержащие в молекуле 4 и более гидроксиль-
ные группы; нек-рые авторы относят к М. с. также и
глицерины. М. с. разделяются на ациклич. (альдиды,
глициты или сахарные спирты) и алицикли< (цик-
литы). К первому классу относятся высшие много-
атомные спирты (полиолы). Общие названия М. с.
соответствуют числу атомов углерода в их молекуле,
оканчиваются на «ит»: тетриты, пентиты, гекситы,
гептиты, октиты, нониты и дециты. Число стереоизо-
меров М. с. резко возрастает с увеличением числа асим-
метрии. атомов углерода. Высшие М. с., генетически
связанные с простейшими сахаристыми веществами,
в частности с соответствующими им по числу атомов
углерода моносахаридами, образуются при их вос-
становлении и сами при осторожном окислении дают
моносахариды. Эти спирты — бесцветные кристаллич.
продукты от сладковатого до очень сладкого вкуса,
со слабым оптич. вращением, встречаются в природе
во многих растениях, а также образуются в результате
воздействия нек-рых микроорганизмов на углеводы.
Для качественного и количественного определения
М. с. пользуются хроматографией, перйодатным ме-
тодом, а также способами, основывающимися на спо-
собности этих спиртов к образованию в определенных
условиях медного и других комплексов.
М. с. обладают всеми свойствами одноатомных
спиртов, и наиболее важным является образование
простых и сложных эфиров. Нитраты М. с. обладают
взрывчатыми свойствами. При каталитич. гидриро-
вании (выше 150° под давлением) происходит гидро-
генолиз высших М. с. с образованием низших; при
жестких условиях могут также образовываться пер-
вичные спирты, дезоксипроизводные и углеводоро-
ды. Восстановление полиолов конц. иодистоводород-
ной к-той приводит к образованию в основном вторич-
ного алкилиодида и нек-рого количества алкена без
изменения углеродной цепи. Полиолы реагируют с
альдегидами и кетонами с образованием преимущест-
венно циклич. ацеталей и кеталей.
Высшие М. с. могут быть синтезированы на основе
моноз, сахарных к-т. Способы промышленного полу-
чения этих спиртов основаны на восстановлении аль-
доз или кетоз. Альдозы дают один продукт, кетозы —
два эпимера. Для восстановления моноз лучше всего
применять каталитич. гидрирование.
Тетриты. Простейшим тетритом является эритрит
СН2ОН (СНОН)2СН2ОН. Известны три стереоизомерных фор-
мы: D- и L-эритриты, или треиты, и мезоэритрит:
СН2ОН
I
но-с-н
I
н-с-он
I
СН2ОН
L-эритрит
СН2ОН
I
Н-С-ОН
I
но-с-н
сн2он
D-эритрит
CH..OII
I
Н-С-ОН
I
н-с-он
I
сн2он
мезоэрптрит
В природе был найден только мезоэритрит, содержащийся в свободном виде в водорослях (Roccella tinctoria). К тетритам относят также пентаэритрит С(СН2ОН)4, являющийся че- тырехатомным спиртом с разветвленной углеродной цепью. Пентиты СН2ОН (СНОН)3СН2ОН существуют в виде двух оптически активных и двух нерасщепляемых, оптически недеятельных пентитов:
СН2ОН СН2ОН СН2ОН СН2ОН
НО-С-Н Н-С-ОН Н-С-ОН Н-С-ОН
но-с-н Н-С-ОН Н-С-ОН НО-С-Н
н-с-он НО-С-Н Н-С-ОН Н-С-ОН
СН2ОН СН2ОН СН2ОН СН2ОН
D-арабит (+лик сит) L-арабит (—ликсит) адонит (рибит) ксилит
Метилпентиты образуются при восстановлении метилпен-
тоз; так из рамнозы получается оптически активный рамнит
ОН OH H H
СН3- С — С — С — С — СН2ОН
1111
И Н ОН ОН
Пентиты содержатся в ряде растений и грибах. Наибольшее
практич. значение имеет ксилит, могут также найти широкое
применение арабит и адонит.
Гекситы СН2ОН (СНОН),СН2ОН известны в виде
десяти стереоизомерных форм, получаемых восстановлением
соответствующих альдоз и кетоз.
СН2ОН СН2ОН СН2ОН
Н-С-ОН НО-С-Н НО-С-Н
НО-С-Н НО-С-Н НО-С-Н
Н-С-ОН Н-С-ОН НО-С-Н
Н-С-ОН Н-С-ОН Н-С-ОН
СН2ОН D-сорбит (D-гл.оцит) СН2ОН D-маннит СН2ОН D-талит (D-аЛЬтрит)
255 МНОГОКОМПОНЕНТНЫЕ СИСТЕМЫ —МНОГОЯДЕРНЫЕ КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ 256
СН2ОН сн2он сн2он
но-с-н но-с-н н-с-он
н-с-он н-с-он н-с-он
но-с-н н-с-он н-с-он
н-с-он но-с-н н-с-он
СН2ОН сн2он сн2он
D-идит (сорбьерит) дульцит (галактит) аллит (аллодульцит)
СН2ОН сн2он сн2он сн2он
НО-С-Н н-с-он н-с-он н-с-он
н-с-он н-с-он он-с-н н-с-он
но-с-н но-с-н н-с-он н-с-он
но-с-н но-с-н но-с-н но-с-н
сн2он сн2он сн2он сн2он
L-сорбит L-маннит L-идит L-талит
Гекситы широко распространены в природе, присутствуют
во многих растениях, плодах и водорослях. Наибольшее зна-
чение и практич. применение имеют D-сорбит, D-маннит и
дульцит.
Г е п т и т ы СН2ОН (СНОН)5СН2ОН теоретически воз-
можны в 16 стереоизомерных формах. Два спирта —персеит
и волемит — встречаются в природе, остальные — синтези-
руют восстановлением альдогептоз. Из последующих высших
многоатомных спиртов известны четыре октита и по одному
нониту, дециту и додециту; в природе зти спирты не встре-
чаются.
М.с. и их ангидриты широко применяют в пром-сти
органич. синтеза (получение полимеров, поверхност-
но-активных веществ, лаков, клеев, пластификаторов
и т. д.), их используют как стабилизаторы влажности
в произ-ве бумаги, целлофана, красок и др. М. с.
нетоксичны, поэтому их применяют в пищевых изде-
лиях, парфюмерии и в фармацевтии.
Лит.: The carbohydrates. Chemistry, biochemistry, physio-
logy, ed. W. Pigman, N. Y., 1957; Ч e п и г о С. В. [и др.],
Хим. наука и пром-сть, 1957, № 4. С.В.Чепиго.
МНОГОКОМПОНЕНТНЫЕ СИСТЕМЫ — физико-
химические системы, содержащие более трех компо-
нентов (т. е. составных частей, количества к-рых могут
изменяться независимо друг от друга). Из М. с. лучше
других, хотя и далеко не полно, изучены системы,
состоящие из 4 компонентов. Изучение же систем с
числом компонентов более 4 лишь начинается. Состав
четверных систем изображается по методу, предло-
женному Дж. Гиббсом в 1878. Этот метод основан на
свойстве правильного тетраэдра; сумма длин перпен-
дикуляров, опущенных на грани тетраэдра из точки,
взятой внутри него, равна высоте тетраэдра. Если
выразить состав системы в процентах и, приняв вы-
соту тетраэдра за 100, восстановить к 3 граням тетра-
эдра перпендикуляры, отложить на них процентное
содержание компонентов и провести через концы этих
перпендикуляров плоскости, соответственно парал-
Рис. 1. Изображение
состава четверной си-
стемы.
лельные граням, то пересечения
этих трех плоскостей дают точку,
выражающую состав данной си-
стемы. Другой способ изобра-
жения состава четверной си-
стемы, предложенный Б. Розе-
бомом (1894), состоит в следую-
щем. Делят ребро правильного
тетраэдра на 100 частей; от од-
ной из его вершин А (рис. 1)
откладывают по трем ребрам
отрезки АВ', AC, AD', изо-
бражающие процентное содер-
жание компонентов, и через концы этих отрезков
проводят три плоскости B'PML, C'PMN и D'LMN,
соответственно параллельные трем его граням; пере-
сечение этих плоскостей дает точку М, изображаю-
Рис. 2. Простейшая
диаграмма состояния
четверной системы.
охлаждении выделяют
щую состав системы. Если в одном и том же тетраэдре
построить обоими способами точки, изображающие
состав системы, то эти точки будут совпадать друг
с другом. Точки диаграммы состава, отвечающие оди-
наковой величине свойства, обычно соединяют поверх-
ностями. Однако часто при построении диаграмм со-
стояния этого и не делают, а ограничиваются проведе-
нием в тетраэдре поверхностей, отделяющих друг от
друга т. н. объемы кристаллизации.
На рис. 2 изображена диаграмма состояния четверной
системы А — В — С — D для простейшего случая, когда
компоненты в жидком состоянии неограниченно растворимы
друг в друге и, кроме того, не обра-
зуют ни твердых р-ров, ни химич.
соединений. Все внутреннее простран-
ство тетраэдра разделено шестью
поверхностями esE9EEx, ехЕхЕЕ4,
esE3EEit е:.ЕзЕЕ2, etExEE2 и е2Е4ЕЕ2
на четыре т.н. объема кристаллиза-
ции (или пространства кристаллиза-
ции отдельных компонентов); это зна-
чит, что если, например, точка, изоб-
ражающая состав смеси, попадает в
пространство между поверхностями
еьЕзЕЕх, еяЕзЕЕ4 и е,ЕхЕЕ4, то при
охлаждении жидкого расплава пер-
вым начинает кристаллизоваться
компонент А. Расплавы, точки соста-
ва к-рых попадают на поверхности,
разделяющие объемы кристаллиза-
ции, при охлаждении выделяют одно-
временно 2 компонента; расплавы,
точки состава к-рых попадают на
линии ЕХЕ, Е2Е, ЕзЕ и EiE, при
одновременно 3 компонента. Наконец, если состав расплава
изображается точкой Е (т. н. четверная эвтектич. точка, или
просто четверная эвтектика), то при отнятии от системы теп-
лоты кристаллизуются одновременно все 4 компонента, причем
темп-pa и состав расплава остаются постоянными. Этот расплав
наз. четверным эвтектич. расплавом, или четверным эвтектич.
раствором (или просто четверной эвтектикой). Расплав произ-
вольного состава застывает так: после кристаллизации 1-го
компонента происходит совместная кристаллизация 1-го и 2-го,
затем f-го, 2-го и 3-го и, наконец, 1-го, 2-го, 3-го и 4-го компо-
нентов. Эта последняя т. н. эвтектич., кристаллизация про-
исходит при постоянной, т. н. эвтектич., темп-ре и из жидкости
одного и того же состава, независимо от состава исходного
сплава. Иногда, в частных случаях, некоторые из указан-
ных стадий могут выпадать. Если в системе образуются
химические соединения, диаграмма состояния ее усложняет-
ся, при образовании же твердых растворов — может и упро-
ститься.
Если в системе возможна реакция обмена (тип АХ + BY =
= AY + ВХ) или вытеснения (тип АХ + В = А + ВХ),
то такая система наз. взаимной. Система, образованная
двумя солями без общих ионов и водой, является четверной
взаимной системой, т. к. в ней имеются пять составных частей
(вода, две исходные соли и две, образующиеся в результате
реакции обмена) и возможна одна химич. реакция. Для изобра-
жения взаимных солевых систем указанный выше способ не-
применим и для этой цели служат другие способы, к-рые
описаны в работах, перечисленных в указателе литературы.
Советскими учеными (В. Н. Лодочниковым, В. П. Радищевым
и др.) предложен ряд способов изображения систем с чис-
лом компонентов больше четырех; с этими методами можно
познакомиться по тем же источникам.
Лит.: Аносов В. Я., Погодин С. А., Основные
начала физико-химического анализа, М.—Л., 1947, с. 719—94;
Радищев В. П., О методах изображений, применяемых
в физико-химическом анализе, в кн.: Курнак ов Н. С.,
Введение в физико-химический анализ, 4 изд., М. —Л., 1940,
с. 194—241; Воловик Б. Е., Захаров М. В., Трой-
ные и четверные системы, М., 1948, с. 117—227; Радищев
В. П., К теоретическому изучению многокомпонентных взаим-
ных систем, ст. 1, Изв. Сектора физ.-хим. анализа, 1953,
22, 33—62; его же, ст. 2, там же, 23, 46—60; Пе-
рельман Ф. М., Методы изображения многокомпонент-
ных систем. Системы пятикомпонентные, М., 1959; П а-
латник Л. С., Л а н д а у А. И., Фазовые равновесия
в многокомпонентных системах, Харьков, 1961.
В. Я. Аносов.
МНОГОЯДЕРНЫЕ КОМПЛЕКСНЫЕ СОЕДИНЕ-
НИЯ — комплексные соединения, в состав молекул
к-рых входит несколько центральных атомов с окру-
жающими их координационными сферами, соединен-
ных отдельными атомами или группами атомов, т. н.
мостиками, или мостиковыми группами. Если в состав
М. к. с. входят две координационные сферы, то соеди-
нение наз. двухъядерным, если три — трехъядерным
и т. д. Мостиковыми группами могут служить ОН
(оловая группа), NH2 (аминогруппа), О (оксогруппа),
257
МНОГОЯДЕРНЫЕ КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ
258
О» (пероксогруппа), СНзСОО (ацетогруппа). В нек-рых
случаях возможно сочетание координационных сфер
центральных ионов различных металлов, напр. в
смешанных хромиферриацетатах каждый из двух ато-
мов Сг (III) связан с Fe (III) посредством трех мости-
ковых ацетатных групп. Координационные числа Fe
(Ш) и Сг (Ш) равны шести:
СН3 СН3
О1^о^о.. ,..^-°хн2о
НО —СгуО—С = О’" Fe—О—С=О"’ Ст—ОН
ОН2 'О сн3 / Чо СН3 / н о
с-о хс=о
сн3 сн3
Два шестикоординационных центральных атома мо-
гут быть соединены между собой посредством одной,
двух или трех мостиковых групп. Примером могут
служить нек-рые соединения Со (III) [координацион-
ное число Со (III) равно 6], содержащие мостиковые
группы NHs (соединение I, III) или ОН (соединения
II и III). М. к. с. могут обладать цепочечной структу-
рой; примером служит соединение Сг (III) [к. ч. Сг (III)
равно 6], содержащее оловые группы:
в отдельных случаях цепи могут замыкаться в коль-
цо, как, напр., в гидроксосоединении Сг (III) [к. ч.
Сг (III) равно 6]. Здесь 4 иона Сг (III) и 4 мостиковые
ОН-группы образуют 8-членный цикл; А — коорди-
нированная молекула:
Названием, к. с. строится в соответствии с общи-
ми принципами номенклатуры комплексных соедине-
ний с той только разницей, что перед названием мос-
тиковой группы ставится буква ц. Если для обо-
значения мостиковой группы служит специальный
термин, буква ц опускается, напр. гидр оксогрупп а
в роли мостиковой наз. оловой группой. Разница
между мостиковыми группами и аналогичными груп-
пами, но связанными только с одним центральным
атомом (напр., между гидроксо- и оловой группами)
заключается в том, что адденды, находящиеся во
внутренней сфере только одного центрального иона
комплексных соединений, являются координационно
ненасыщенными и способны к реакциям присоедине-
ния, тогда как мостиковые группы значительно более
инертны.
Для многоядерных комплексных соединений харак-
терны реакции трех типов.
1) Реакции, протекающие без существенного изме-
нения мол. веса: а) обменные реакции, в результате
к-рых изменяется только состав внешней сферы, но
не меняются состав и строение внутренней сферы
М. к. с., напр.:
[(NH3)s CoNH3Co (NHs)J Cls + 5AgNO3 —>
—- [(NH,), CoNH2Co (NH3)5J (NO3)5t 5AgCl
б) реакции замещения групп, находящихся во внут-
ренней сфере, анионами:
+ 4 NaHC2O4 —х
Образование соединений, содержащих циклы, спо-
собствует реакции замещения.
2) Реакции расщепления, сопровождающиеся умень-
шением молекулярного веса. Реакции этого типа
служат для определения строения внутренней сферы
М. к. с. В большинстве случаев они протекают в кис-
лой среде.
3) Реакции полимеризации, сопровождающиеся уве-
личением мол. веса. Сюда же можно отнести и реак-
°9 к.Х.Э. т. 3
259
МОДЕЛИ МОЛЕКУЛЯРНЫЕ
260
ции образования двухъядерных соединений из одно-
ядерных:
Ясно, что при образовании двухъ- или трехъядер-
ных соединений происходит образование мостиковых
групп из заместителей, находящихся в цис-положе-
нии. Полимеризация акво- и аминосоединений про-
текает в щелочной среде. Процессу образования М.к.с.
способствует повышение темп-ры. М. к. с. мотут да-
вать геометрии, или оптич. изомеры (см. Комплексные
соединения).
Лит. см. при ст. Комплексные соединения.
МОДЕЛИ МОЛЕКУЛЯРНЫЕ *(м о д 'еля°а то м-
ные)— наглядное изображение молекул органич.
соединений, позволяющее судить о взаимном распо-
ложении атомов, входящих в ту или иную молекулу.
М. м. используют в тех случаях, когда по структур-
ной ф-ле трудно или практически невозможно пред-
ставить пространственное расположение атомов в мо-
лекуле. В частности, широко пользуются построением
М. м. при изучении пространственной изомерии,
в конформационном анализе, для оценки стерич. пре-
пятствий и т. д.
Все М. м. могут быть отнесены к одному из двух ос-
новных типов: 1) модели, приближенно отражающие
ориентацию валентностей в пространстве, но не даю-
щие представления об относительных размерах ато-
мов; 2) т. наз. объемные модели, отображающие ва-
лентные углы, ковалентные радиусы атомов и радиу-
сы действия (эффективные радиусы).
К первому типу относятся обычные модели из ша-
риков, соединенных отрезками проволоки, т. наз.
модели Кекуле — Вант-Гоффа. Дрейдингом разрабо-
таны более совершенные модели, состоящие из стерж-
ней и трубок, соединенных в точке, изображающей
ядро атома, под углами, равными валентным. Дли-
ны трубок и стержней пропорциональны длинам свя-
зей между атомами водорода и данного элемента (1 см
соответствует 0,4А). Свободные концы трубок и стерж-
ней изображают ядра атомов водорода, поэтому каж-
дый фрагмент в отдельности является моделью моле-
кулы простейшего водородного соединения данного
элемента (СЩ, NHa, Н20 и т. д.). Для сборки модели
более сложного соединения вставляют стержень од-
ного фрагмента в трубку другого; благодаря ограни-
чительному устройству расстояние между их центра-
ми пропорционально соответствующему межатомному
расстоянию. Модели Дрейдинга верно отражают
межатомные расстояния и валентные углы в моле-
кулах. Они позволяют имитировать внутреннее вра-
щение, оценивать энергетич. выгодность различных
конформаций, а также измерять расстояния между не-
посредственно не связанными атомами. Моделями
Дрейдинга особенно широко пользуются при изуче-
нии стереохимии полициклич. систем типа стероидов.
Объемные модели, правильно передающие раз-
меры и форму молекул, были разработаны в 1934
Стюартом и позднее усовершенствованы Бриглебом.
Каждый фрагмент, изображающий атом определенного
элемента, представляет собой шаровой сегмент. Ра-
диус шара пропорционален радиусу действия атома,
расстояние от центра шара до плоскости среза про-
порционально ковалентному радиусу. В случае мно-
говалентных атомов делают соответственно 2, 3 или 4
среза, причем угол между перпендикулярами из цен-
тра шара на плоскость среза равен валентному (см.
рис.). Для атомов, соединенных .кратными связями,
сегменты изготовляют не из шаров, а из эллипсоидов,
большая полуось к-рых соответствует эффективному
радиусу, обусловленному наличием л-электронного,
а малая — ст-электронного облаков. Модели изготов-
ляют из дерева, пластмассы или резины и окраши-
срезов.
могут вращаться
вают в различные цвета, установленные для каждого
элемента. При сборке моделей молекул сегменты
соединяют между собой по плоскостям
В случае простых связей сегменты
один относительно другого. Моде-
ли Стюарта — Бриглеба верно пе-
редают валентные углы, меж-
атомные расстояния и эффектив-
ные радиусы; они позволяют изме-
рять расстояния между различ-
ными атомами и группами (модели
выпускаются в 2 масштабах, 1 А
соответствует 1,5 или 2 см). Вели-
чины эффективных радиусов или радиусов действия,
принятые в моделях Стюарта — Бриглеба, на 10—15%
меньше значений вандерваальсовых радиусов, полу-
чаемых из кристаллографии, данных. Это связано
с тем, что модели предназначены для рассмотрения
стерич. эффектов в молекуле, находящейся при обыч-
ных условиях, а не при темп-ре абсолютного нуля.
Известны модели, подобные стюартовским, отли-
чающиеся от последних масштабом, ассортиментом,
раскраской и нек-рыми конструктивными особенно-
стями. Из числа таких моделей следует отметить
М. м. Хартли — Робинсона, выпускаемые в Англии
под названием модели типа Куртолд. Эти модели бла-
годаря эластичному соединению сегментов допускают
значительные деформации валентных углов (до 15°)
и позволяют, в отличие от М. м. Стюарта—Бриглеба,
собирать модели напряженных молекул, в к-рых ве-
личина валентного угла существенно отличается от
стандартной. Такие же возможности дают и модели
Стюарта—Бриглеба, сделанные из резины. См. также
Валентные углы.
Лит.: Briegleb в кн.: Houben — Weyl,4Aufl.,
Bd 3, Т1 1, Stuttgart, 1955; Drelding A. S., Helv. chlm.
acta, 1959, 42, fasc. 4, S. 1339; Темникова T. И., Курс
теоретических основ -органической химии, 2 изд., Л., 1962,
с. 190; Хюкк ель В., Теоретические основы органической
химии, пер. с нем., т. 1, М., 1955, с. 54. Л. И. Беленький.
МОДИФИЦИРОВАНИЕ ВЫСОКОМОЛЕКУЛЯР-
НЫХ СОЕДИНЕНИЙ — направленное изменение
физич., механич. или химич. свойств высокомолеку-
лярных соединений под воздействием различных фак-
торов. Наиболее часто для М. в. с. применяют поли-
мераналогичные превращения, а также макромолеку-
лярные реакции, ориентацию и кристаллизацию по-
лимера.
Модифицирование методом по-
лимераналогичных превращений.
Полимераналогичные превращения заключаются в
замене одних функциональных групп полимера
другими без изменения его структуры и длины цепи.
В результате такого превращения значительно изме-
няются растворимость, степень кристаллизации, тем-
пература стеклования, текучесть, прочность и др.
свойства полимера. К числу таких реакций относятся,
напр., получение простых и сложных эфиров целлю-
лозы, синтез поливинилового спирта омылением поли-
винилацетата.
261
МОДИФИЦИРОВАНИЕ ПОВЕРХНОСТИ ХИМИЧЕСКОЕ
262
К этой группе методов относится также внутримо-
лекулярное отщепление функциональных групп, или
циклизация. Таким путем осуществлен синтез поливи-
ниленов и др. полимеров, содержащих систему сопря-
женных двойных связей и обладающих повышенной
теплостойкостью. Внутримолекулярная циклизация,
протекающая в условиях, благоприятствующих обра-
зованию ненапряженных циклов, приводит к увеличе-
нию жесткости и температуры стеклования полимера.
Пользуясь этой реакцией, получают так называе-
мый циклокаучук — твердый и хрупкий продукт,
применяемый в производстве клеев и защитных по-
крытий.
Модифицирование методом макро-
молекулярных реакций. К макромолеку-
лярным реакциям можно отнести блок-сополимериза-
цию, процесс получения привитых сополимеров,
«сшивание» полимеров.
Макромолекула блок-сополимера содержит цепоч-
ки (блоки) одного химического состава, разделенные
полимерными участками другого химического соста-
ва или низкомолекулярными «сшивающими» груп-
пами. Блок-сополимеры построены по общей схеме:
—А—А—А—Б—Б—А—А—А—Б—Б—-Б—Б..., где А
и Б — группы различного химич. состава.
Привитые сополимеры получаются в результате
«прививки» к главной цепи полимера боковых цепей.
Схематически привитой сополимер можно представить
след, образом:
. . .—А —А—А —А —А-А—. . .
I I
Б Б
I I
Б Б
I I
Б Б
I I
Сшивание макромолекул заключается в образова-
нии между ними химич. связей, что обусловливает
создание жесткой структуры. Типичным примером по-
лучения сшитого полимера является вулканизация
каучука, осуществляемая различными агентами: се-
рой, окислами или солями карбоновых к-т полива-
лентных металлов и др. Образование трехмерной
структуры повышает устойчивость материала к тер-
мич., химич. или механич. воздействиям, обусловли-
вает нерастворимость и почти полное отсутствие набу-
хания. Полимеры, содержащие комплексообразующие
группы, при взаимодействии с солями металлов дают
межмолекулярные координационноковалентные (клеш-
невидные) связи (см. Координационные полимеры).
Возникновение наряду с ионными связями координа-
ционных повышает устойчивость соединений к гидро-
лизу и воздействию высоких темп-р.
Ориентация и кристаллизация
полимера. В результате увеличения межмолеку-
лярных связей при кристаллизации возрастают мо-
дуль упругости и твердость полимера, а также повы-
шается прочность при разрыве. Ориентация полимера
(как аморфного, так и кристаллич.) приводит к появ-
лению анизотропии механич. и физико-химич. свойств:
так, прочность ориентированных полимеров в направ-
лении ориентации выше, чем в перпендикулярном
Направлении.
Ориентируют полимер обычно вытягиванием в од-
ном направлении. Растяжение неориентированного
кристаллич. полимера осуществляется при темп-рах,
более низких, чем темп-pa плавления кристаллич.
участков, но более высоких, чем темп-pa размягчения
аморфных. При этом наряду с ориентацией кристаллич.
участков возможно увеличение степени кристаллично-
сти полимера за счет кристаллизации макромолекул
аморфных областей. Аморфные полимеры подвергают-
ся значительной ориентации при темп-рах, близких
к темп-ре размягчения.
Лит.: Стрепихеев А. А., ДеревицкаяВ. А.,
Основы химии высокомолекулярных соединений, М., 1961;
Лосев И. П., Трост янская Е. Б., Химия синтети-
ческих полимеров, М., 1960; Б 6 р л е н т У.,- Хофман А.,
Привитые и блок-сополимеры, пер. с англ., М., 1963.
А. II. Поляков.
МОДИФИЦИРОВАНИЕ ПОВЕРХНОСТИ ХИМИ-
ЧЕСКОЕ — реакция присоединения (прививка) к
поверхности твердых тел различных химич. соеди-
нений с целью изменения в нужную сторону свойств
поверхности. Модифицирование применяется для улуч-
шения химич. и физич. свойств высокодисперсных на-
полнителей, пигментов, загустителей смазок, по-
верхностей адсорбентов и различных материалов, напр.
для придания им гидрофобных или гидрофильных
свойств (см. Гидрофобные покрытия). Химич, модифи-
цирование окисей и гидроокисей, напр. гидратиро-
ванной и дегидратированной поверхности кремнезема
(силикагеля или аэросила — высокодисперсного крем-
незема, состоящего из непористых частиц сферич.
формы, см. Адсорбенты), производят с помощью сле-
дующих реакций: дегидратация (гидратация), напр.
ОН ИО' х /°\ х
4$|Z 'Siz - 'si 'Si^+H2O
' 'o OZ 4 " \ О (I>
'sZ S'
/ x ' '
этерификация, напр.
он o-R
i i
-Si -+ROH -Si-+H2O (II)
I I
метилирование диазометаном, напр.
о-н O-CHS
। ।
-Si-+CH2N2—>-Sf-+N2 (III)
I I
хлорирование тионилхлоридом, напр.
о-н ci
। i
-S1- + SOC12—> -Si-+SO3 + HCl (IV)
I I
алкилсилирование, напр.
Si (СЩ),
I
о-н о
I I
-Si- +(CH3)sSICl—«-S1-+HC1 (V)
I I
Поверхность м. б. модифицирована образованием
слоя химически привитых к кремнезему органич.
соединений, содержащих определенные функциональ-
ные группы (ОН, NH3, SO3 и т. п.) или реакционно-
способные звенья, напр. с двойными связями. В ре-
акциях (I)—(IV) образуются поверхностные группи-
ровки, нестойкие к воздействию воды, реакция (V)
приводит к образованию покрытий очень стойких как
к воздействию воды, так и к нагреванию (до 300—
350°). Поэтому реакции типа (V), в результате к-рых
образуется прочная кремнийорганич. связь Si—С,
служат для придания кремнезему, а также другим
окислам, стеклам, армирующим волокнам и т. п.
стойких гидрофобных свойств. Подобное М. п. х. пре-
дохраняет частицы наполнителей и пигментов от сли-
пания (аггрегирования), облегчает их введение в угле-
водородные и многие полимерные среды. М. п. х.,
напр. по реакциям типа (V), создает на поверхности
кремнезема довольно плотный слой триметилсилиль-
ных групп, зазоры между к-рыми не могут превышать
размеров этих групп. Этот слой, отодвигая адсорби-
руемые молекулы от скелета собственно кремнезема,
резко уменьшает энергию адсорбции, в особенности
9*
263
МОДИФИЦИРОВАНИЕ ПОВЕРХНОСТИ ХИМИЧЕСКОЕ—МОЗЛИ ЗАКОН
264
О 1 2 3 4 5 А
Рис. 1. Схема расположения три-
метилсилильных групп на поверх-
ности кремнезема.
крупных молекул, не способных проникнуть в зазоры
между триметилсилильными группами (рис. 1). Это вы-
зывает резкое уменьшение теплоты адсорбции, а сле-
довательно, и самой адсорбции. В случае молекул
С9Н8, С6Н14 и СС14, размеры к-рых больше промежут-
ков между триметилси-
лильными группами,
теплота адсорбции ста-
новится меньше тепло-
ты конденсации. Толь-
ко в случае адсорбции
молекул СНзОН и
Н2О, малых по разме-
рам, способных частич-
но проникать в зазоры
между триметилсилиль-
ными группами, обра-
зуя водородные связи
с оставшимися гидрок-
силами кремнезема, чистые теплоты адсорбции хотя
и сильно уменьшаются, но остаются положительными
(рис. 2).
М. п. х. пористых тел улучшает их свойства как
носителей неподвижных жидких и твердых фаз для га-
зовой хроматографии. Напр., обработанный в авто-
клаве при 300° силикагель становится весьма крупно-
пористым, его уд. поверхность падает до 10 л12/г и
менее. Последующая обработка по реакциям (V) рез-
ко снижает его адсорбционные свойства, делает его
поверхность гидрофобной и термостойкой. По такой
поверхности хорошо растекаются применяемые в газо-
жидкостной хроматографии масла. М. п. х. улучшает
также свойства стеклянных капиллярных колонок.
При этом стекла и другие подобные материалы приоб-
ретают устойчивые гидрофобные свойства.
0,20.40.60,8 0.20,40.60,8 0.2 0.4 0.60,8 0.20.4 0,60.8 0,2 0.4 0.60.8
Относительное давление пара
Рис. 2. Зависимости разности теплоты адсорбции Qa и
теплоты конденсации L от заполнения поверхности
(вверху) и изотермы самой адсорбции а (внизу) на гид-
роксилированной (сплошные кривые) и триметилсили-
рованной (пунктирные кривые) поверхности кремнезема.
В случае сажМ. п. х. может быть вызвано
действием сильных окислителей, особенно в жидкой
среде (напр., гипохлоритом натрия, смесью азотной и
серной к-т, перекисью водорода). При этом канальная
сажа становится столь гидрофильной, что образует
стойкие коллоидные растворы в воде без добавления
смачивателей. На поверхности окисленной сажи на-
ходятся различные кислородные химич. соединения,
способные к реакциям ионного обмена и к реакциям
с органич. веществами, напр. они частично метили-
руются при действии диазометана. Нагревание саж в
вакууме, инертном газе или в восстановительной ат-
мосфере вызывает разложение поверхностных со-
единений, рост и упорядочение кристаллитов графи-
та. Частицы сажи становятся
полиэдрами, грани которых
представляют базисные гра-
ни кристаллов графита. Та-
кие графитированные сажи
обладают весьма однородной
поверхностью, гидрофобны,
химически и термически весь-
ма стойки. На обычных и
окисленных сажах с геоме-
трически и химически неод-
нородной поверхностью вна-
чале заполняются более ак-
тивные, а затем менее актив-
Рис. 3. Изотермы адсорбции (сте-
пени заполнения поверхности 9)
пара воды на канальной саже: ис-
ходной (2), окисленной (2) и графитированной (':). На оси
абсцисс — относительное давление пара.
ные места (рис. 3). М. п. х. позволяет также в широких
пределах изменять адгезионные свойства твердых
тел (см. Клеящее действие).
Анализ поверхностных соединений производится
химическим, изотопнообменным и спектроскопическим
методами, а также методами ядерного магнитного и
электронного парамагнитного резонанса.
Лит.: Курс физической химии, под ред. Я. И. Гераси-
мова, т. 1, М., 1964, гл. 18, 19; Поверхностные химические
соединения и их роль в явлениях адсорбции. [Сб., под ред.
А. В. Киселева], М., 1957; Киселев А. В., Ж. физ.
химии, 1961, 35, № 2, 233; его же, Вести. МГУ. Сер.
II, химия, 1961, № 5, 29; Газовая хроматография, Сб., М.,
1960, с. 45; Киселев А. В., Ковалева Н. В.,
Королев А. Я., Коллоидн. ж., 1961, 23, М 5, 582;
Бабкин И. Ю., Киселев А. В., Ж. физ. химии,
1962, 36, № И, 2448; Клеи и технология склеивания, под
ред. Д. А. Кардашева, М., 1960, с. 35—52. А. В. Киселев.
МОЗЛИ ЗАКОН — основа систематики рентгенов-
ских атомных спектров. Согласно этому закону, квад-
ратный корень из частоты (или волнового числа) соот-
ветственных линий рентгеновского спектра испуска-
ния элементов линейно возрастает с атомным номером
элемента:
v = 7?(Z-<i)2M------Н
где v — волновое число (см-1), Z — атомный номер,
R (см-1)— постоянная Ридберга, о — т. н. постоян-
ная экранирования, п1 и пг — целые числа (п2>п1),
определяющие серию спектральных линий (п4) и ли-
нию в серии (п2). Как и формулы, определяющие ча-
стоту оптич. атомных спектров, М. з. имеет ясное те-
оретич. обоснование. Он связан с возбуждением атома
путем вырывания внутренних электронов (тогда как
периферийные электроны ответственны за оптич.
спектры). При этом частота линии рентгеновского
спектра испускания соответствует величине кванта
энергии, выделяемой в результате заполнения осво-
бодившегося места в данном электронном слое за счет
электрона одного из более верхних слоев.
Если атом возбужден за счет удаления А-элект-
рона (главное квантовое число н=1), то получается
A-серия (п1=1). При этом наблюдается линия А7,
если освободившееся место в К-слое заполняется
L-электроном (п=п2=2), линия Кр—при заполне-
нии его М-электроном (п=п2=3) и линия К., —
N-электроном (п=п2=4). Аналогичную интерпретацию
имеют £-серия (п\=2), М-серия (щ=3) и т. д. Други-
ми словами, волновое число любой линии есть резуль-
тат комбинации двух термов [величина R(Z—о)г1п2],
каждый из к-рых соответствует определенному глав-
ному квантовому числу (см. Атомные спектры). В от-
265
МОЛЕКУЛА
266
личие от ф-лы для частот оптич. спектра водородопо-
добного атома, М. з. включает вычитаемую поправку
а к заряду ядра (атомному номеру) Z, связанную с эк-
ранирующим действием электронов на заряд ядра;
в простейшем случае А^-линии, когда экранирование
вызвано единственным электроном А'-слоя, близко рас-
положенного к ядру: <т=1. В отличие от спектров ис-
пускания, рентгеновские спектры поглощения непре-
рывны и имеют резкие границы (края поглощения)
только у длинноволновых концов серий. Это обус-
ловлено любой возможной величиной поглощаемого
кванта энергии сверх определенного минимума, тре-
буемого на вырывание электрона из данного слоя;
остаток энергии кванта переходит в кинетич. энер-
гию вырванного электрона. Для краев поглощения
М. з. принимает очень простой вид:
(n, = n, n2=oo): v=--7?(Z—п)2^ .
Закон был установлен Г. Мозли в 1913. Он позволил
непосредственно определять экспериментальным пу-
тем (по частоте линий рентгеновского спектра) атом-
ный номер элемента и поэтому сразу же был исполь-
зован для подтверждения правильности расположения
элементов в периодич. системе. В частности, было
окончательно установлено расположение редкозе-
мельных элементов, а также с полной достоверностью
доказано существование именно 92 элементов от водо-
рода до урана. См. также Атомные спектры.
Лит..: Шпольский Э. В., Атомная физика, 1, 4 изд.,
М.—Л., 1951, 2, 3 изд., М.—Л., 1951; Кондратьев В. Н.,
Структура атомов и молекул, 2 изд., М., 1959; Б о -
ровский И. Б., Физические основы рентгеноспектраль-
ных исследований, М., 1956. С. Э. Вайсберг.
МОЛЕКУЛА. Содержание:
Введение...................................265
История изучения молекул...................266
Строение молекул ...................... 267
Свойства молекул...........................272
Молекула — наименьшая частица данного веще-
ства, обладающая его основными химич. свойствами,
способная к самостоятельному существованию и со-
стоящая из одинаковых или различных атомов, сое-
диненных в одно целое химич. связями. Число атомов,
входящих в состав М., колеблется в очень широком
интервале: от двух (напр., М. водорода, азота, окиси
углерода и др.) до сотен и тысяч (напр., М. полиэти-
лена, белков и других высокомолекулярных соедине-
ний).
Введение. Вследствие малых размеров М. их нель-
зя видеть непосредственно или при помощи оптич.
микроскопа. Имеется, однако, много косвенных дан-
ных, убедительно свидетельствующих о молекуляр-
ном строении вещества. Существование М. (и атомов)
наиболее наглядно проявляется в свойствах газов.
Так, большая сжимаемость газов обусловлена тем,
что газ представляет собой не сплошное вещество, а
совокупность беспорядочно движущихся частиц —
М., среднее расстояние между к-рыми велико по срав-
нению с размерами самих частиц и сравнительно лег-
ко уменьшается при увеличении давления. Явление
диффузии, к-рое наблюдается не только в газах, но и
в жидких и твердых телах, также обусловлено атомно-
молекулярным строением тел. Одним из наиболее
убедительных доказательств существования М., как
было показано Ж. Перреном, является броуновское
движение. Наглядным доказательством существова-
ния М. служит также дифракция рентгеновских лу-
чей и дифракция электронов на молекулярных крис-
таллах и на М. газов. В пользу реального существо-
вания М. свидетельствует совокупность огромного ко-
личества данных, полученных в результате химич.
исследований.
М. состоит из атомов, связанных между собой в оп-
ределенной последовательности и определенным обра-
зом расположенных в пространстве. О составе М. и
последовательности связи ее атомов можно судить на
основании того, к каким химич. превращениям спо-
собна данная М. В процессе химич. превращения (хи-
мич. реакции) происходит исчезновение нек-рых меж-
дуатомных связей в исходных М. и возникновение но-
вых связей, в результате чего образуются новые М.
Сравнительное изучение исходных и конечных веществ
химич. реакций представляет собой главный метод
определения состава и структуры М. Существенную
помощь в определении структуры М. оказывают раз-
личные физич. методы исследования (см. ниже).
Прочность межатомных связей в М. характеризует-
ся той энергией, к-рую надо затратить на разрыв от-
дельных связей. Энергия связей атомов в М. состав-
ляет обычно несколько десятков ккал/моль. Атомы
в М. непрерывно совершают сложные колебательные
движения; при определенных условиях, напр. в газо-
вой фазе, М. в целом, кроме того, совершает враща-
тельное и поступательное движения.
Устойчивое существование М., находящейся в
нек-рой среде, зависит от ее способности к химич. взаи-
модействию с веществом среды, а также от темп-ры,
давления и других внешних факторов. Вещество в
газообразном состоянии при обычной темп-ре, как
правило, состоит из М. Исключение составляют инерт-
ные газы, а также пары ртути и нек-рых других метал-
лов, при обычных условиях состоящие из атомов; при
достаточно высоких темп-рах М. всех газов распадают-
ся на атомы. В конденсированных системах М. сущест-
вуют в том случае, если работа разрыва связи внутри
М. значительно больше, чем работа, необходимая для
удаления одной М. из конденсированной системы.
Так, напр., для воды теплота испарения в расчете на
одну М. составляет ок. 7-10“13 эрг, а работа отрыва
от М. воды одного атома водорода— ок. 8-10-12
эрг, т. е. приблизительно в 10 раз больше; в соответст-
вии с этим вода состоит из М. во всех трех агрегатных
состояниях. К конденсированным системам, состоя-
щим из М., относится большинство жидкостей, а также
молекулярные кристаллы. Для этих веществ, так же
как и для газов, химич. формулы выражают непосред-
ственно количественный и качественный состав М.
В противоположность этому, в таких кристаллич. те-
лах, как металлы, сплавы, ионные и атомные кри-
сталлы, а также в их расплавах, как правило, М.
не существует, поскольку в этих веществах любой
атом (или ион) взаимодействует со всеми соседними
атомами (или ионами) приблизительно одинаково.
В природе довольно широко распространены вещест-
ва, состоящие из М. с очень большим числом атомов
(т. наз. макромолекул), напр. каучук, целлюлоза и
др. За последнее время искусственно получено боль-
шое число макромолекулярных соединений.
Физич. и химич. свойства веществ связаны с соста-
вом и строением их М. Однако химич. и физич. свой-
ства данного вещества, как правило, не представляют
собой суммы свойств составляющих его М. Совокуп-
ность М. обладает особенностями, не присущими от-
дельным М. Так, напр., химич. реакции в газах могут
протекать обычно только при наличии совокупности М.
История изучения молекул. Представление о том,
что все тела состоят из движущихся частиц, возникло
еще в древности. Однако впервые понятие о М., как
о частице, состоящей из нескольких атомов, появи-
лось лишь в начале 17 в. в работах П. Гассенди. В 18 в.
понятие о М. («корпускуле») развивалось М. В. Ломо-
носовым, к-рый на основе молекулярных представле-
ний объяснил ряд физич. и химич. свойств веществ.
Представление о М., как о наименьшем количестве вещества,
вступающего в химич. взаимодействие, и отличие М. от атома
было впервые четко сформулировано А. Авогадро в 1811.
Его идеи, однако, были окончательно признаны только в 1860
благодаря трудам С. Канниццаро. Работы С. Канниццаро,
267
МОЛЕКУЛА
268
Ш.Жерара и Э. Франкленда позволили установить одно из важ-
нейших понятий химии — понятие валентности как свойства
атома соединяться с определенным числом других атомов.
На основе этого понятия в начале 2-й половины 19 в. А. Кекуле,
Й. Лошмидт и А. Купер независимо один от другого предло-
жили способы написания структурных формул М. органич.
соединений. Однако Кекуле не был последователен в своих
воззрениях и иногда утверждал, что эти ф-лы не отражают
действительного строения М., а Лошмидт и Купер не сумели
связать эти структурные ф-лы с данными химич. исследований.
Новый этап в развитии представлений о М. начинается с созда-
ния А. М. Бутлеровым теории химич. строения,
к-рая легла в основу современной теоретич. химии. Согласно
Бутлерову, в М. существует определенная последовательность
химич. связи атомов, причем качественное отличие свойств М.
от свойств составляющих ее атомов обусловлено в основном
этими химич. связями и взаимным влиянием непосредственно
не связанных атомов в М.; строение М. проявляется в химич.
свойствах данного вещества, и поэтому изучение химич.
свойств есть важнейшее средство познания строения М. Теория
химич. строения М. дала объяснение изомерии, показав, что
причина этого явления заключается в различном порядке сое-
динения одних и тех же атомов в М. изомеров, и предсказала
число изомерных форм. В дальнейшем представление об изо-
мерии было расширено и, благодаря учению Я. Вант-Гоффа
и А. Ж. Ле-Беля о различном пространственном расположении
атомов в М., были заложены основы стереохимии М. С помощью
химич. методов удалось установить последовательность связей
атомов в М. и их расположение в пространстве для большого
числа органич. соединений. В области неорганич. химии
стереохимия, представления были разработаны в конце 19 в.
А. Вернером, создавшим координационную теорию строения
комплексных соединений. Важнейшую роль в развитии учения
о М. сыграло открытие Д. И. Менделеевым периодич. закона,
к-рый дал возможность систематизировать существующие
химич. соединения и предвидеть многие свойства М., исходя
из свойств образующих их атомов. Дальнейшие исследования
М. при помощи химич. методов были направлены в основном
на решение задачи о связи строения М. с ее реакционной спо-
собностью и с другими свойствами вещества.
Учение о природе сил, соединяющих в одно целое атомы
внутри М., возникло еще в начале 19 в., когда Я. Берцелиус
предложил электрохимия, теорию, согласно к-рой атомы при-
тягиваются между собой благодаря наличию у каждого из них
двух противоположных и не равных по абс. величине электрич.
зарядов, находящихся один от другого на нек-ром расстоянии.
Для изучения природы химич. связи основное значение имело
открытие электрона и разработка электронной теории строения
атома и М. в конце 19 — начале 20 вв. Первые электронные
теории химич. связи были предложены В. Косселем (1915)
и Г. Льюисом (1916). Теория Косселя относилась в основном
к М. неорганич. соединений и до нек-рой степени была род-
ственна представлениям Берцелиуса, поскольку основывалась
на предположении, что все М. состоят из заряженных ионов,
взаимодействующих по закону Кулона (ионная связь). В основе
теории Льюиса лежало представление о том, что при образо-
вании связи валентные электроны могут не только полностью
переходить от одного атома к другому (ионная связь), но и
принадлежать одновременно обоим атомам, причем в этом слу-
чае в зависимости от валентности атомов связь осуществляется
одной, двумя или тремя парами электронов (гомеополярная,
или ковалентная, связь; подробнее см. Валентность). Однако
представления Льюиса оставляли открытым фундаментальный
вопрос о природе сил, обусловливающих притяжение нейт-
ральных атомов и образование между ними химич. связи.
Природа химич. связи была в основном выяснена только
после создания в 1925 квантовой механики. В 1927 В. Гейтлеру
и Ф. Лондону удалось при помощи квантовомеханич. методов
удовлетворительно рассчитать энергию связи и равновесное
междуатомное расстояние М. водорода (см. ниже). Эта работа
положила начало квантовой химии, развитие к-рой дало более
глубокое понимание основных закономерностей, ранее уста-
новленных в химии экспериментально, и значительно содей-
ствовала объяснению взаимосвязи между различными свойст-
вами М. Квантовомеханич. теория М. смогла в значительной
степени выяснить природу насыщаемости и направленности
валентностей, природу кратных связей и связей в М. ароматич.
соединений, природу межмолекулярных сил, устойчивость
свободных радикалов, закономерности взаимного влияния
атомов в М. и мн. др. Развитие различных физич. методов ис-
следования позволило с высокой точностью определять струк-
туру, оптические, электрические, магнитные и другие свой-
ства М.
Строение молекул. Природа химической
связи. Для образования химич. связи из всех суще-
ствующих в природе сил имеют значение только элект-
рич. силы, т. е. силы взаимодействия электрич. заря-
дов, носителями к-рых являются электроны и ядра
атомов. Специфика химич. связи определяется волно-
выми свойствами электронов и особой корреляцией
в их движении. Согласно квантовой механике, со-
стояние электрона отображается волновой функцией,
квадрат абс. значения к-рой характеризует вероят-
ность нахождения электрона в том или ином элементе
объема. В связи с этим для электрона в атоме или М.
имеет смысл представление об «электронном облаке»,
плотность к-рого в различных точках пространства
пропорциональна вероятности нахождения в них
электрона (см. Атом). Наиболее распространенный
тип химич. связи — гомеополярная (ковалентная)
связь, осуществляемая в простейшем случае двумя
валентными электронами (по одному от каждого из
взаимодействующих атомов). Простейшим примером
гомеополярной связи может служить связь в моле-
куле Н2. При сближении двух атомов водорода может
быть два случая. Если собственные моменты количе-
ства движения (спины) электронов антипараллельны,
то получается притяжение атомов, приводящее к об-
разованию молекулы Н2; если же спины параллельны,
то молекула Н2 не образуется. При сближении двух
атомов с антипараллельными электронными спинами
электронные оболочки этих атомов благодаря волно-
вым свойствам электронов под влиянием обоих ядер
деформируются так, что образуется единое электрон-
ное облако М., причем его плотность в пространстве
между ядрами оказывается большей, чем сумма соответ-
ствующих электронных плотностей двух изолирован-
ных атомов. При этом энергия взаимного электроста-
тич. притяжения между электронами и ядрами оказы-
вается больше, чем сумма энергии взаимного оттал-
кивания электронов и взаимного отталкивания ядер
и приращения кинетич. энергии электронов. При
пек-ром межъядерном расстоянии силы притяжения
уравновешиваются силами отталкивания, что при-
водит к устойчивому равновесию и, т. о., к обра-
зованию молекулы Н2.
Если спины электронов двух сближающихся атомов
параллельны, то при сближении атомов в простран-
стве между ядрами электронная плотность, наоборот,
уменьшается и результирующим эффектом оказывает-
ся отталкивание атомов один от другого, вследствие
чего молекула Н2 не образуется. Химич, связи, как
правило, пе образуется во всех случаях, когда в обеих
или в одной из взаимодействующих частиц (атомов
или М.) спины всех электронов скомпенсированы, т. е.
имеют попарно противоположные направления. Та-
кие частицы взаимно притягиваются только сравни-
тельно слабыми ван-дер-ваальсовскими силами (см.
Межмолекулярное взаимодействие). Это объясняет
способность химич. связей к насыщению. Исключе-
ние составляют те атомы (и М.), у к-рых сравнительно
легко происходит возбуждение электронов, нару-
шающее компенсацию спинов. Так, напр., атом угле-
рода в нормальном состоянии имеет два непарных
электрона, по в результате возбуждения одного из
спаренных электронов он становится четырехвалент-
ным. При взаимодействии атомов (или М.), имеющих
по одному или по нескольку непарных электронов, хи-
мич. связь, как правило, образуется; при этом проис-
ходит компенсация спинов электронов, первоначально
принадлежавших различным частицам. При образо-
вании связи электронная оболочка системы прини-
мает такую конфигурацию, к-рая отвечает наиболь-
шей энергии связи. Если энергия связи имеет тот же
порядок величины, что и средняя тепловая энергия М.,
то образовавшаяся М. не будет устойчивой. Устойчи-
вость М. данного вещества зависит также и от при-
роды среды, в к-рой она находится, поскольку при
столкновении с М. этой среды при определенных ус-
ловиях может произойти химич. реакция.
В случае Н2 и других двухатомных М., состоящих из
одинаковых атомов, «центр тяжести» электронного
облака совпадает с «центром тяжести» положительного
заряда и М. не имеет дипольного момента. Такие М.
называются гомеополярными. Если М. со-
стоит из различных атомов, то «центр тяжести» элект-
ронного облака обычно смещен к одному из них и М.
269
МОЛЕКУЛА
270
(НС1, СО и др.) оказывается полярной, т. е.
имеет дипольный момент. В предельном случае, когда
«центр тяжести» валентных электронов очень близок
к «центру тяжести» одного из атомов, возникающая
М. наз. ионной. Химич, связь в таких М. обус-
ловлена в основном кулоновским притяжением про-
тивоположно заряженных ионов. К этому типу приб-
лижаются, напр., М. галогенных солей щелочных ме-
таллов, существующие в газовой фазе (CsF, NaCl и
др.). Для комплексных соединений характерно, что
в них, помимо обычного взаимодействия между заря-
дами ионов и электрич. диполями М., в образовании
связей в сложной М. важную роль может играть т. н.
донорно-акцепторное взаимодействие, осуществляю-
щее одну из форм координационной связи и по своей
природе близкое к ковалентной связи.
Связь между двумя атомами в М., осуществляемая
одной парой электронов, наз. простой. Между много-
валентными атомами могут возникать кратные связи:
двойные и тройные, обусловленные соответственно
двумя и тремя парами электронов. По мере увеличе-
ния кратности прочность'связи между одними и теми
же атомами возрастает и соответственно этому сокра-
щаются длины связей (т. е. равновесные расстояния
между ядрами непосредственно связанных атомов).
Так, напр., энергия разрыва простой связи С—С в
этане равна 83 ккал/моль, тогда как энергия разрыва
двойной С=С связп в этилене составляет приблизи-
тельно 140 ккал/моль. Длина простой связи С—С (пре-
дельные углеводороды) равна 1,54А, а двойной связи
С=С (этилен)—1,33 А. М., в к-рых имеются две или нес-
колько двойных связей, чередующихся с простыми, как,
напр., в гексатриене Н2С=СН—СН=СН—СН=СН2,
наз. М. с сопряженными связями. Благодаря взаимо-
действию двойных связей распределение электрон-
ной плотности в них уже не отвечает простой структур-
ной ф-ле. Это несоответствие проявляется особенно
резко в случае ароматич. соединений. Напр., обычная
структурная ф-ла бензола не передает истинного рас-
пределения электронов в его М., т. к. в действитель-
ности связи между всеми атомами С равноценны и М.
имеет ось симметрии шестого порядка.
Размеры и форма молекул. Размеры
молекулы приближенно можно определить при по-
мощи числа Авогадро, если известны молекулярный
вес и плотность вещества. Взяв одну грамм-молекулу
к.-л. вещества (напр., 18 г воды), заключающую в себе
6,02-1023 М. (число Авогадро), и поделив объем, за-
нимаемый этим количеством вещества, на число М.,
можно найти объем, приходящийся на одну М. В слу-
чае жидкостей (и молекулярных кристаллов) можно
приближенно принять, что найденный объем равен
объему М. Для воды этот объем равен:
~б"о^= 29,7-10-24сл13, откуда диаметр М. равняет-
ся ^/29,7-Ю-24 ^3-10-8 см или ЗА. Как пока-
зывают измерения, произведенные при помощи раз-
личных методов, линейные размеры большинства
простых М. имеют такой же порядок величины.
Для каждой устойчивой М. существует такое взаим-
ное расположение образующих ее атомов, которое
отвечает минимуму потенциальной энергии М. Это
расположение атомов соответствует равновесной кон-
фигурации М., определяющей ее форму и размеры,
т. е. геометрию М. Основными экспериментальными
методами исследования геометрии, конфигурации М.
служат рентгено- и электронография, а также нейтро-
нография, молекулярная спектроскопия и ядерный
магнитный резонанс. При пользовании этими мето-
дами обычно привлекаются также химич. данные о
последовательности связи атомов в М.
Геометрия М. однозначно определяется, если из-
вестны длины связей М. и углы между связями (ва-
лентные углы). Так, напр., в М. воды Н—О—Н длина
каждой связи в О—Н равна 0,9584 А и валентный угол
между ними равен 104°27'. Форма и размеры М.
определяются в основном строением электронной обо-
лочки образующих ее атомов и взаимным влиянием по-
следних. Форма многоатомных М. весьма разнооб-
разна. Во многих случаях они обладают той или иной
симметрией (см. Симметрия молекул). В М. нек-рых
веществ атомы расположены линейно, напр. в аце-
тилене Н—С=С—Н, в двуокиси углерода О=±С=О и
др. Однако большинство трехатомных М. имеет тре-
др. Однако большинство трехатомных
угольную форму, напр. М. воды
Н2О, сероводорода H2S, озона Оз
и др. Многие М. построены пи-
рамидально. Сюда относятся, на-
пример, М. аммиака NHs, к-рая
представляет собой трехгранную
равнобедренную пирамиду. Пяти-
валентный фосфор РС15, соеди-
няясь с пятью атомами галогена,
дает М. в виде тригональной би-
пирамиды (рис. 1). Четырехвалент-
ный атом углерода в предельных
углеводородах, напр. в метане
СЩ, образует связи, направлен-
Рис.
1. Схема моле-
кулы пятихлористого
фосфора РС15.
ные к вершинам тетраэдра, причем углы между свя-
зями равны 109°28' (рис. 2). Если в метане нек-рые
атомы водорода заместить другими атомами или атом-
ными группами, то на-
блюдается небольшое
искажение тетраэдра.
Если в вершинах те-
траэдра находятся че-
тыре различных атома
(как, напр., в М. ви-
да CHXYZ, имеющей
так наз. асимметрич-
ный атом углерода), то
для такого соединения
Рис. 2. Модель молекулы метана
СН,: а — атомы Н, изображен-
ные в виде шариков, отодвинуты
от центрального атома С вдоль
линии связей, благодаря чему
видно их тетраэдрич. располо-
жение; б — атом С и атомы Н
находятся на равновесных рас-
стояниях друг от друга; вслед-
ствие сильного перекрывания
электронных оболочек их пер-
воначальная сферич. форма силь-
но искажена.
существуют два про-
странственных изоме-
ра (поскольку суще-
ствуют два различных
способа расположения
в пространстве четы-
рех атомов: Н, X, Y, Z,
не переходящих один
в другой ни при каких
поворотах М., причем
один является зеркаль-
ным отображением другого. Такого рода изомерия
приводит к появлению активности оптической, т. е.
обусловливает способность данного
вещества вращать плоскость поляри-
зации света.
В олефиновых углеводородах, на-
пример этилене,
н. .н
/с=с<
нх н
каждый из атомов углерода связан
с тремя соседними атомами, располо-
женными в одной плоскости с ато-
мами С, причем одна из этих связей
(со вторым атомом углерода) двойная;
углы между связями равны 120°.
В М. бензола (рис. 3) все 6 атомов
углерода (и 6 атомов водорода) рас-
положены в одной плоскости и обра-
зуют правильный шестиугольник.
Каждый из атомов С, так же как и
Рис. 3. Схема и
модель молекулы
бензола СеНб.
в этилене, связан с тремя соседними атомами; однако,
в отличие от этилена, каждая из связей СС не двойная,
271
МОЛЕКУЛА
272
а по своему характеру является промежуточной между
простой и двойной связями.
Внутримолекулярное движение
и энергетика молекул. Наряду с посту-
пательным движением М. и ее вращением как целого в
М. происходят различные внутренние движения ядер
и электронов. Основной вид внутримолекулярного
движения представляет собой колебание ядер ее
атомов, к-рые при своем движении в большей или мень-
шей степени увлекают связанные с ними электронные
оболочки. Однако электроны, кроме того, могут со-
вершать самостоятельное движение. Поскольку это
движение подчиняется законам квантовой механики,
электроны в М. (так же как и в атоме) могут нахо-
диться только в нек-рых, вполне определенных кван-
товых состояниях, каждому из к-рых отвечает опре-
деленное значение энергии, зависящее от межъядер-
ного расстояния. Изменение этой потенциальной энер-
гии М. при увеличении расстояния между ядрами от
нек-рого заданного значения до бесконечно большого
равняется энергии взаимодействия атомов в М. Для
М., состоящей из двух атомов А и В, зависимость этой
энергии U от межъядерного расстояния R изобра-
жается в виде т. н. кривой потенциальной энергии
(рис. 4), получаемой из молекулярных спектров или
путем теоретич. расчета при помощи методов кван-
товой механики. Эта кривая имеет минимум, соот-
ветствующий устойчивому состоянию двухатомной
М.; при этом расстояние Ro от начала координат
до точки минимума равно равновесному межъядерному
расстоянию между атомами А и В. Колебательное
движение атомов А и В также подчиняется законам
квантовой механики, вследствие чего энергия этого
движения при достаточно близких межъядерных рас-
стояниях и не слишком больших скоростях может
принимать только дискретный ряд значений; нек-рые
из них изображены на рис. 4 горизонтальными отрез-
ками. Энергия вращательного движения М. также
принимает только дискретный ряд значений, соот-
ветствующих определенным вращательным состояни-
ям. См. Молекулярные спектры.
Одной из основных характеристик М. является энер-
гия, которая выделяется при образовании М. из ато-
мов (эта энергия численно равна энергии диссоциа-
ции М. на атомы). Энергия образования М. может
быть определена из термохимических данных, а в
случае двухатомных М.— также из молекулярных
спектров.
На рис. 4 и0 — глубина «потенциальной ямы» — представ-
ляет собой энергию связи атомов А и В.Однако действительная
энергия образования М.
из атомов А и В меньше
170, т. к. в силу кванто-
ванности колебательного
движения система АВ не
может иметь энергию ни-
же определенного значе-
ния (нижний уровень на
рис. 4), называемого ну-
левой энергией. По этой
причине энергия образо-
вания молекулы АВ рав-
на величине D (рис. 4) —
расстоянию от нулевого
уровня энергии до уров-
ня, соответствующего бес-
конечно большому рас-
стоянию между атомами
(R = оо). Согласно кван-
товой механике, нулевая
энергия двухатомной М.
равна hv/2, где h — по-
стоянная Планка, a v —
основная частота колеба-
ний М. Обычно нулевая
энергия составляет несколько ккал/моль, причем наибольшее
значение она имеет для М. водорода, для которой hv/2 =
= 6,2 ккал/моль. Таким образом, принимая во внимание, что,
согласно спектроскопическим данным, для М. водорода D =
= 102,6 ккал/моль, для соответствующей глубины «потенци-
альной ямы» (рис. 4) находим Uo = 108,8 ккал/моль.
Рис. 4. Кривая потенциальной энер-
гии двухатомной молекулы (изо-
бражено только несколько первых
колебательных уровней).
Диссоциация на ионы в газовой фазе при обычной темп-ре
происходит редко, т. к. она связана со значительной затратой
энергии. Напр., диссоциация М. НС1 на атомы Н и С1 требует
102 ккал/моль, а на ионы Н+ и С1“—ок. 300 ккал/моль. В по-
лярных растворителях этот процесс, наоборот, происходит
легко (см. Электролитическая диссоциация) и часто связан
с выделением энергии, поскольку переход свободных ионов
в раствор сопровождается сильным взаимодействием ионов
с окружающими их М. растворителя (см. Сольватация). Так,
напр., при диссоциации на ионы М. НС1 в воде выделяется ок.
10 ккал/моль.
Важной характеристикой многоатомных М. является энер-
гия разрыва (диссоциации) отдельных связей. Знание этой
величины особенно необходимо при исследовании химич.
реакций, протекающих через образование радикалов свободных.
Как видно из таблицы, энергия разрыва связи в многоатом-
ной М. зависит не только от природы непосредственно соеди-
ненных атомов (от их места в периодич. системе Менделеева),
но также и от того, с какими другими атомами соединен каждый
из них и какова природа связи между ними, т. е. зависит or
взаимного влияния атомов.
Энергия разрыва отдельных связей в молекулах
Связь Энергия раз- рыва (ккал/моль) Связь Энергия раз- рыва (ккал/.моль)
Н—С1 102 NO-C1 36
но—н 1 18 СН3—J 54
сн3—н 102 сн3-он 90
СН3СН2-Н 97 сн3-сн3 83
С„Н5СН2-Н 83 С,Н5СН2-СН3 63
Важными энергетич. характеристиками М. явля-
ются также их потенциалы ионизации (см. Иониза-
ции потенциал) и энергии электронного, колебатель-
ного и вращательного возбуждения (см. ниже).
Свойства молекул. Оптические свой-
ства. Между определенными квантовыми состояния-
ми М. возможны переходы, сопровождаемые испуска-
нием или поглощением электромагнитной (световой)
энергии. Энергия, излучаемая или поглощаемая при
одном таком переходе, равна hv, где v — частота све-
товых колебаний. Следовательно, каждому такому
переходу в оптич. спектре М. будет соответствовать
определенная линия. Если М. переходит из одного-
электронного состояния в другое, то соответствую-
щие изменения энергии, как правило, имеют порядок
нескольких электрон-вольт, а спектральная линия
приходится на видимый или УФ-участок спектра
(этой области отвечают частоты колебаний 1015—10,s
сек~'). Если происходит изменение только колебатель-
ного состояния М., то соответствующие изменения
энергии имеют порядок величины десятых долей элект-
рон-вольта, а спектральные линии лежат в ИК-части
спектра (соответствующие частоты колебаний состав-
ляют 1013—10й сек~'). Изменения вращательной энер-
гии еще меньше, и соответствующий спектр лежит
в далекой ИК- и микроволновой областях (частоты
равны 1012сек-1 и меньше). В силу того, что в данной
совокупности М. одновременно с электронными и ко-
лебательными переходами обычно изменяется и вра-
щательная энергия, уровни к-рой расположены срав-
нительно тесно, в электронном и колебательном спект-
рах наблюдаются не отдельные линии, а более или ме-
нее широкие полосы.
В многоатомной М. каждый атом совершает слож-
ные колебания. Вследствие того, что в М. все атомы
соединены в единое целое, колебания различных ато-
мов связаны между собой. Колебания атомов в моле-
куле можно разложить на несколько независимых
простых гармонии, колебаний, каждое из к-рых совер-
шается со своей собственной частотой. При этом коле-
бание с каждой из этих частот совершает не один
атом, а одновременно все атомы М. и притом в одина-
ковой фазе, но обычно с различными амплитудами.
Такая совокупность колебаний атомов представляет
собой одно из колебаний М. в целом и называется ее
нормальным колебанием. Изучение колебательных
273
МОЛЕКУЛА
спектров М. (см. Молекулярные спектры) дает возмож-
ность судить о наличии в М. тех или иных элементов и
их изотопов, о природе и прочности связей, о харак-
тере и силе взаимодействия связей. Это позволяет,
в частности, применять колебательные спектры для
качественного и количественного анализа веществ.
Подобный анализ облегчается в тех случаях, когда
в М. имеются такие атомные группы, колебания кото-
рых обладают характеристичностью, т. е. почти не
изменяют своей частоты (и интенсивности) при струк-
турных изменениях в остальных частях М. Нек-рые
изменения частот и интенсивности характеристич. ко-
лебаний могут происходить не только под влиянием
структурных изменений в М., но также и под влия-
нием межмолекулярного взаимодействия, в частности,
водородной связи.
Электронные спектры поглощения М. представляют
собой обычно совокупность полос различной интен-
сивности. Наиболее длинноволновые полосы принад-
лежат обычно отдельным атомным группам, т. н. хро-
мофорам (напр., CO,NO2 и др.), в пределах к-рых про-
исходят переходы электрона. Полосы поглощения
предельных углеводородов лежат в УФ-области;
в М. с сопряженными связями спектр смещен к види-
мой области, причем часто появляется окраска. Изу-
чение электронных спектров поглощения и люминес-
ценции имеет большое значение как для решения
вопросов строения М., так и для аналитич. целей.
Так, на основании присутствия или отсутствия в спек-
тре тех или иных характерных полос можно делать
заключения о наличии в данной смеси тех или иных
М. или о наличии в М. определенных атомных групп,
а также идентифицировать малоустойчивые частицы
(напр., радикалы). Исследования молекулярных спект-
ров дают, кроме того, необходимые данные для теоре-
тич. расчета теплоемкостей и других термодинамич.
характеристик вещества.
Из других оптич. свойств М. следует упомянуть
оптич. активность (см. выше). Исследования оптич.
активности позволяют изучить ряд особенностей
в строении М. Следует отметить также, что измерения
показателя преломления газов дают возможность
определить рефракцию молекулярную и тем самым
электронную поляризуемость М. (см. ниже).
Электрические и магнитные свой-
ства молекул. Если М. попадает во внешнее
электрич. поле, то «центры тяжести» зарядов — отри-
цательных (электроны) и положительных (ядра) —
смещаются из своих первоначальных положений и М.
поляризуется, т. е. она приобретает индуцированный
дипольный момент Ринд. Поскольку смещение цен-
тра тяжести электронов значительно больше смещений
тяжелых ядер, указанный дипольный момент в основ-
ном обусловлен деформацией электронной оболочки,
причем Ринд =аЕ, где Е — напряженность электрич.
поля, а — т. н. поляризуемость М., характеризующая
способность электронной оболочки М. к деформации
под действием внешнего электрич. поля. Значения по-
ляризуемостей для разных М. составляют, как пра-
вило, от IO-24 до 10~23 см3.
Если М. имеет постоянный дипольный момент, то
в электрич. поле она будет не только поляризоваться,
но также и определенным образом ориентироваться.
При этом тепловое движение М. будет, однако, стре-
миться нарушить эту ориентацию. Таким образом, по-
ляризация всей совокупности М. будет зависеть от
темп-ры. Знание дипольных моментов М. дает воз-
можность путем их сопоставления делать различные
выводы о строении М. — о ее симметрии в целом и рас-
пределении электронной плотности, о присутствии тех
или иных характерных групп и их расположении.
В качестве простого примера можно указать на М.
двуокиси углерода СОг, к-рая не имеет дипольного
274
момента и потому может иметь только линейную фор-
му О=С=О, что подтверждается также и другими
опытными данными. Наоборот, наличие дипольного-
момента у М. воды и аммиака означает, что эти М.
должны иметь соответственно треугольное и пирами-
дальное строение. Дипольные моменты большинства
М. имеют порядок IO-18 (CGSE).
Ценные сведения о строении электронной оболочки
М. дают также исследования ее магнитных свойств,
(см. Магнетохимия, Магнитные свойства). Большин-
ство М. диамагнитны, т. е. не имеют постоянного маг-
нитного момента. Диамагнитная восприимчивость не-
зависит от темп-ры и характеризует поведение элект-
ронной оболочки М. в магнитном поле. Парамагне-
тизм М. обусловливается наличием у них постоянного-
магнитного момента, благодаря к-рому М. во внеш-
нем магнитном поле стремится ориентироваться в на-
правлении поля; однако тепловое движение М. в той
или иной мере нарушает эту ориентацию, вследствие
чего парамагнитная восприимчивость совокупности
М. зависит от темп-ры. Постоянным магнитным мо-
ментом могут обладать как электронная оболочка,
так и атомные ядра. У ядра магнитный момент по по-
рядку величины в 10s раз меньше магнитного момен-
та электрона. Магнитный момент электронной обо-
лочки М. имеет двоякое происхождение: он может
быть обусловлен либо собственным магнитным момен-
том электрона, связанным со спином, либо орбиталь-
ным магнитным моментом, связанным с движением:
электронов по молекулярной «орбите».
Спиновый магнитный момент М. отличен от нуля
в тех случаях, когда в М. число электронов, имеющих
спин определенного направления, превосходит число
электронов со спином противоположного направле-
ния. Такая ситуация осуществляется всегда в М. с не-
четным числом электронов (напр., NO и др. радикалы),
а также в нек-рых других М. (напр., Ог, органич.
бирадикалы и др.). Измерение парамагнитного момен-
та дает возможность делать заключение о числе не-
спаренных электронов в М. и об ее электронном строе-
нии. Применение метода электронного парамагнитного
резонанса значительно расширило возможности иссле-
дования свойств парамагнитных молекул и радика-
лов (см. Радиоспектроскопия).
Химические свойства молекул.
Если сталкиваются две частицы (М., атомы), одна пз
к-рых или обе не имеют валентных электронов и по-
тому они не могут соединиться между собой химич.
связью, то, как было сказано выше, при сближении они
отталкиваются одна от другой. Это отталкивание про-
исходит, однако, только при условии, если кинетич.
энергия относительного движения частиц мала и пото-
му наименьшее расстояние между ними при столкнове-
нии сравнительно велико. При больших энергиях
столкновения частицы могут сблизиться настолько,
что на короткое время химич. связи делокализуются и
возникает т. н. переходное состояние. Дальнейшее дви-
жение атомов приводит к новой локализации связей
между ними и образуются новые частицы, т. е. проис-
ходит химич. реакция. Если одна из сталкивающихся
частиц представляет собой М., а другая — однова-
лентный атом (или радикал), то для преодоления ука-
занного отталкивания и осуществления химич. реак-
ции необходима сравнительно небольшая энергия
столкновения частиц (небольшая их активация); в
соответствии с этим М. может реагировать с атомом
или радикалом уже при сравнительно невысоких
темп-рах. Наоборот, если сталкиваются две М., у к-рых
спины всех электронов скомпенсированы, то для пре-
одоления отталкивания и обеспечения реакции, как
правило, необходима значительно большая актива-
ция и, следовательно, более высокая темп-pa (см.
Энергия активации, Переходного состояния метод).
275
МОЛЕКУЛЯРНАЯ БИОЛОГИЯ
276
Химич, свойства М. проявляются при химич. реак-
циях и определяются ее строением; они зависят также
ст строения М. второго компонента, от свойств среды
(природа растворителя, агрегатное состояние, ката-
литич. свойства и др.) и внешних условий. Одна из
важнейших проблем химии заключается в выяснении
зависимости химич. свойств М. от их химич. строения,
т. е. от состава М., от последовательности связи и
пространственного расположения атомов в М. и от
характера взаимного влияния атомов.
Как правило, влияние внешних факторов приводит
к изменению механизма реакции, т. е. к изменению
природы ее промежуточных процессов, изменению их
относительных и абс. скоростей и т. д. Так, напр.,
в газовой фазе реакции обычно идут через образова-
ние нейтральных активных частиц (атомов, радика-
лов), а в полярных растворителях многие М. реаги-
руют лишь после того, как распадаются на ионы про-
тивоположного знака. В нек-рых случаях это приво-
дит к изменению направления реакции, т. е. к изме-
нению природы конечных М.
Изучение М. имеет большое научное и практич.
значение, т. к. огромное число явлений и процессов
в природе связано с взаимодействием и превращением
М. Учение о М. лежит в основе химии и ряда фи-
зич. дисциплин. Подробнее о химич. свойствах М. см.
Кинетика химическая, Термодинамика химическая,
Катализ.
Лит.: Ломоносов М. В., Полное собрание сочинений,
т. 1—3, М.—Л., 1950—52; Бутлеров А. М., Сочинения,
т. 1, М., 1953; Меншуткин Б. Н., Химия и пути ее
развития, М.—Л., 1937; Состояние теории химического строе-
ния в органической химии. Доклад комиссии отделения хими-
ческих наук АН СССР, М., 1954; Некрасов Б. В.,
Курс общей химии, 14 изд., М., 1962; Чичибабин А. Е.,
Основные начала органической химии, 7 изд., т. 1, М., 1963;
Волькенштейн М. В., Строение и физические свой-
ства молекул, М.— Л., 1955; Кондратьев В. Н., Строе-
ние и химические свойства молекул, М., 1953; его же,
Структура атомов и молекул, 2 изд., М., 1959; РеутовО. А.,
Теоретические проблемы органической химии, М., 1956;
Г л ест он С., Теоретическая химия, пер. с англ., М., 1950;
Ельяшевич М. А., Атомная и молекулярная спектро-
скопия, М., 1962; Веселов М. Г., Элементарная квантовая
теория атомов и молекул, 2 изд., М., 1962; Шусторо-
вич Е. М., Природа химической связи, М., 1963.
И. Д. Соколов.
МОЛЕКУЛЯРНАЯ БИОЛОГИЯ — область био-
логии. науки, изучающая свойства и структурные
особенности химич. соединений, гл. обр. макромоле-
кулярных, в связи с их биологии, функциями, а также
природу физич. процессов и химич. реакций, лежа-
щих в основе жизнедеятельности. М. б. как наука воз-
никла сравнительно недавно на стыке биологии, хи-
мии и физики. В соответствии с этим она, с одной сто-
роны, базируется на достижениях современной био-
логии, а с другой — широко использует идеи и ме-
тоды органич. химии, физики и кибернетики.
В основе всех биологич. явлений лежат химич.
процессы, в к-рых так или иначе участвуют белки.
Поэтому познание молекулярных основ жизнедея-
тельности немыслимо без изучения молекулярной
структуры этих сложных биополимеров и их свойств,
определяемых структурой. Бурно развивающиеся
структурная химия и физика белковых макромоле-
кул являются фундаментом М. б. Установлено, что син-
тез разнообразных по свойствам белков в живых
организмах связан с другим классом биополимеров—
нуклеиновыми кислотами. В результате возникла не-
обходимость изучения структурной химии и физики
нуклеиновых к-т, а также молекулярных механиз-
мов их участия в сложном, многостадийном процессе
биосинтеза белков. Естественным развитием проблемы
явилось познание механизмов биосинтеза самих нукле-
иновых кислот, строение к-рых, как оказалось, самым
непосредственным образом связано с механизмами пе-
редачи наследственных признаков. При этом сущест-
венно, что, несмотря на огромное многообразие форм
живой материи, реализация наследственных призна-
ков во всех живых организмах, начиная от бактерио-
фагов и вирусов и кончая растениями и животными,
осуществляется по сходному пути.
Как известно, при делении живых клеток обра-
зуются две дочерние клетки, получающие идентичную
наследственную информацию. Кардинальные проб-
лемы, затрагивающие самую сущность жизни, состоят
в том, чтобы выяснить, каким образом происходит
этот процесс точного копирования и каким путем на-
следственная информация реализуется в клетке. За по-
следние годы, базируясь на прочной основе современ-
ной физики, химии и генетики, оказалось возможным
приблизиться к пониманию процессов, происходящих
на молекулярном уровне (передача наследственной
информации, механизм, при помощи к-рого эта ин-
формация реализуется).
На основе этих главнейших направлений современ-
ной М. б. изучаются следующие проблемы: структура
биополимеров и их комплексов, природа биологич.
катализа, механизмы реакций, лежащих в основе
физиологии, функций (работа мышц, зрение, слух,
нервная деятельность и др.), молекулярные механизмы
гормональной регуляции, природа наследственных
заболеваний и др. Изложенными задачами М. б., ее
предметом, определяются используемые объекты и
методы исследования. Часто М. б. исследует механизмы
сложных биологич. явлений в упрощенных и модель-
ных системах (бесклеточные системы, смеси индивиду-
альных веществ и т. п.). В этих системах исключается
большая часть вторичных факторов, непосредственно
не связанных с изучаемым процессом, но мешающих
его исследованию в сложном живом организме.
Вместе с тем, М. б. учитывает неизбежность извест-
ной модификации явления при его искусственном
изолировании. Поэтому представления о молекуляр-
ном механизме того или иного биологич. процесса,
полученные в изолированной системе, проверяются,
как правило, на целостной живой системе.
Наряду с изолированными бесклеточными систе-
мами важнейшими объектами изучения М. б. являются
простейшие живые образования — вирусы и бактерии.
Относительная несложность их строения, быстрый
рост, возможность быстрого получения многочислен-
ных поколений, а также получения и исследования му-
тантов сделали вирусы и бактерии неоценимым объек-
том исследований в области молекулярной генетики,
биосинтеза нуклеиновых к-т и белков. Большая часть
важнейших результатов в этом направлении получена
при изучении вирусов и бактерий. Есть основания счи-
тать, что основные закономерности, установленные
М. б. при исследовании простейших организмов, дей-
ствительны и для высших организмов.
М. б. в своих исследованиях использует современные
методы установления строения сложных полимерных
веществ, а также изучения кинетики и механизма хи-
мич. реакций: рентгеноструктурный анализ, электрон-
ная микроскопия, денситометрич. и седиментационный
анализ в мощных гравитационных полях (реализуе-
мых в ультрацентрифугах), изотопные методы, хро-
матография, электрофорез, оптическая спектрометрия,
спектрополяриметрия и др. Наиболее интересные дан-
ные были получены при сочетании этих методов с био-
логическими. Нек-рые из основных и наиболее харак-
терных для М. б. исследований излагаются ниже.
Структурные особенности и функции белков. Од-
ной из основных биологич. функций белков является
их каталитич. действие. Ферменты, к-рые ускоряют
все реакции в живых клетках, являются белками, при-
чем каждый фермент оказывает специфич. каталитич.
действие избирательно на определенную химич. ре-
акцию. Таким образом, необходимым условием жиз-
недеятельности клетки является ее способность свое-
277
МОЛЕКУЛЯРНАЯ БИОЛОГИЯ
278
временно синтезировать необходимый набор ферментов
в нужных количествах. Помимо того, белки являются
структурным материалом, из к-рого состоят живые
ткани — мышцы, кожа, кровеносные сосуды и т. д.
Они также выполняют и многие другие физиология,
функции. Несмотря на высокий мол. вес, основ-
ная химич. структура белков относительно проста.
Молекулы белков представляют собой длинные
полипептидные цепочки, состоящие из многократно
повторяющихся структурных элементов — аминокис-
лотных остатков. Все встречающиеся в природе белки
включают остатки ок. 20 различных аминокислот;
общая длина белковой цепи составляет от нескольких
десятков до нескольких сотен остатков. Каждый белок
характеризуется строго определенной и специфич.
для него последовательностью расположения амино-
кислот (первичной структурой). Первым белком, для
к-рого была полностью расшифрована первичная
структура, явился инсулин (Р. Сейнджер). Позднее
была раскрыта первичная структура нек-рых других
белков (рибонуклеаза, белок вируса табачной мозаи-
ки и др.).
Отдельные участки полипептидной цепочки могут
свертываться в спираль (нторичная структура). Вся
молекула в целом в свою очередь свертывается весьма
сложным, но вполне определенным образом в простран-
стве. В результате каждый белок обладает специ-
фич., ему одному присущей трехмерной конфигура-
цией (третичная структура).
Информация об аминокислотной последовательности
каждого из белков содержится в зашифрованном виде
в молекулах нуклеиновых к-т. Реализация этой ин-
формации обеспечивает своевременный синтез нужных
количеств белков, необходимых для жизнедеятельно-
сти клетки.
Генетическая роль дезоксирибонуклеиновой кис-
лоты (ДНК). Хранение и передача генетич. информа-
ции во время деления клетки, а также в процессе раз-
вития образующихся клеток осуществляются находя-
щимися в ядрах молекулами ДНК (см. Нуклеиновые
кислоты). Молекулы ДНК представляют собой очень
длинные цепочки, содержащие до 10 тысяч (а воз-
можно и больше) мономерных единиц — нуклеоти-
дов,— и характеризуются мол. весом порядка несколь-
ких миллионов; в отдельных случаях мол. вес ДНК
достигает десятков миллионов. В отличие от бел-
ков, состоящих из 20 различных структурных единиц,
в молекулах ДНК большинства организмов их все-
го 4. Генетич. информация зашифрована (закодиро-
вана) последовательностью расположения четырех
оснований в нуклеотидах ДНК — аденина, гуанина,
тимина и цитозина. Молекулы ДНК в большинстве
случаев состоят из двух цепочек, свернутых в одну
двойную спираль. Принято считать, что благодаря
такой структуре клетка имеет возможность в процессе
своего деления воспроизвести точную копию каж-
дой из молекул ДНК, поскольку порядок чередова-
ния нуклеотидов в одной из цепочек однозначно оп-
ределяет порядок их чередования в другой цепочке:
аденину одной цепочки соответствует тимин, гуанину
соответствует цитозин.
Генетич. роль ДНК экспериментально доказывается
следующими фактами. При введении чистой ДНК од-
ной разновидности бактерий в бактерии другого вида
последние приобретают наследуемые признаки тех бак-
терий, из к-рых была взята ДНК (явление трансфор-
мации бактерий). При заражении бактерий бактери-
альными вирусами-фагами, к-рые состоят, как изве-
стно, только из нуклеиновых к-т и белков, в бактери-
альную клетку попадает ДНК фага (белок остается
вне клетки), к-рая вызывает внутри бактериальной
клетки синтез специфич. белков, свойственных фагу,
но не клетке. Половая рекомбинация у бактерий,
заключающаяся в перетекании ДНК из мужской бак-
терии в женскую, приводит к появлению у жен-
ской бактерии способности синтезировать те белки,
к-рые были свойственны мужской бактерии. Чистая
ДНК (или РНК) вирусов обладает инфекционностью,
т. е. при ее введении в клетку можно наблюдать раз-
множение вирусных частиц так, как будто в клетку
был введен весь вирус; при этом синтезируются бел-
ки вируса.
Отдельные участки ДНК, являющиеся функцио-
нальными единицами, носят названия ц и с т р о -
нов. Различают структурные цистроны и цистроны-
регуляторы. Последовательность нуклеотидов внутри
одного структурного цистрона однозначно определяет
аминокислотную последовательность в молекуле од-
ного белка. Цистрон-регулятор продуцирует вещества-
репрессоры (химич. природа к-рых пока еще неясна),
способные взаимодействовать со структурными цист-
ронами, и снижает их активность. Если репрессор бло-
кирован определенными клеточными веществами, то
информация от структурного цистрона передается в
органоиды клетки, осуществляющие синтез соответст-
вующего белка. Изменения порядка чередования нук-
леотидов вДНК (мутации) приводят к изменениям
структуры цистронов, обусловливающим изменения
первичной структуры соответствующих белков, что,
в свою очередь, приводит к изменениям или полной ут-
рате их биологич. функции. Мутагенное действие таких
соединений, как H2O2,NH2OH, (C2H5)2SO4,HNO2,HCOH
и др., состоит в том, что они химически взаимодейст-
вуют с азотистыми основаниями нуклеотидов и при-
водят к образованию неправильных нуклеотидных пар
(напр., вместо пары аденин — тимин может получиться
пара гуанин — цитозин). Одна точечная мутация (изме-
нение одного нуклеотида) соответствует изменению
одной аминокислоты в белковой цепочке.
Все эти факты свидетельствуют о том, что нуклеотид-
ная последовательность отдельных участков молекулы
ДНК данной клетки определяет аминокислотную по-
следовательность синтезируемых в ней белков. При
этом осуществляется как бы перевод информации, за-
писанной с помощью 4-буквенного нуклеотидного
кода, на 20-буквенную аминокислотную последова-
тельность белков. Сущность проблемы кодирования
сводится к механизму, при помощи к-рого осущест-
вляется такой перевод.
Как указывают работы Крика, Виттманна и др.,
код является триплетным и неперекрывающимся. Это
означает, что одной аминокислоте в молекуле белка
соответствует определенное сочетание трех нуклеоти-
дов в ДНК, причем нуклеотиды одного триплета не
используются, даже частично, для другого соседнего
триплета, т. е. все они независимы и структурно не
перекрываются. Характерной особенностью кода яв-
ляется его вырожденность, проявляющаяся в том, что
многим аминокислотам соответствует не один триплет,
а несколько разных триплетов. Код «считывается»
в определенном направлении от точки отсчета (старто-
вый пункт). Каждый из триплетов ничем не отделяется
от соседнего, т. е. в нем отсутствуют «запятые». Сис-
тема кодирования является, вероятно, универсаль-
ной. т. е. действует одинаково во всем живом мире.
Механизм биосинтеза белка. Синтез белка проис-
ходит в ультрамикроскопич. частицах клетки (диа-
метром ок. 250А), наз. рибосомами, к-рые состоят
почти полностью из высокомолекулярной РНК и
белка. Наследственная информация передается из
ядра в рибосомы при помощи специального вида
РНК,т.н. «информационной», «матричной» РНК (temp-
late, messenger, informational RNA), образующейся
на ДНК с помощью фермента РНК-полимеразы. По-
сле переноса в рибосомы «информационная» РНК уча-
ствует в синтезе белка в качестве матрицы.
219
МОЛЕКУЛЯРНАЯ БИОЛОГИЯ—МОЛЕКУЛЯРНЫЕ СИТА
280
В 1961—62 были раскрыты связи между химич.
строением полинуклеотидной цепи, играющей роль
матрицы, и аминокислотным составом образующегося
белка (Ниренберг, Очоа и др.). Если к бесклеточной
системе, содержащей все компоненты, необходимые для
биосинтеза белка, добавить синтезированные полину-
клеотиды заданного состава, то удается наблюдать
образование полипептидных цепей. При этом, если до-
бавить полиуридиловуюк-ту,то полипептид оказывает-
ся состоящим из остатков только одной аминокисло-
ты — фенилаланина. Таким образом, тринуклеотид, со-
стоящий из трех уридиловых остатков, кодирует фенил-
аланин, к-рый оказывается в тех участках белковых
молекул, где в матрице встречается последователь-
ность оснований урацил — урацил — урацил. Подобным
образом могут быть расшифрованы кодирующие три-
нуклеотиды и для других аминокислот. Напр., ами-
нокислоте лизину соответствует тринуклеотид из аде-
ниловых остатков и другой триплет, содержащий 2
уридиловых и 1 адениловый остаток. Разрабатывают-
ся химич. и генетич. методы, позволяющие установить
последовательность оснований внутри триплетов.
Аминокислоты непосредственно не присоединяются
к «информационной» (матричной) РНК. Прежде чем
оказаться в составе белковой молекулы, аминокисло-
ты испытывают превращения, переводящие их в реак-
ционноспособное состояние и обеспечивающие уста-
новку на полирибонуклеотидную матрицу («информа-
ционную» РНК) в определенном, специфич. участке.
В присутствии содержащихся в клетке специальных
ферментов (активирующие рН5-ферменты, амино-
ацил РНК-синтетазы) и ионов Mg2+ аминокислоты реа-
гируют с аденозинтрифосфорной к-той (АТФ) с обра-
зованием аминоациладенилатов, т. е. смешанных ан-
гидридов аминокислот с адениловой к-той, освобож-
дая при этом пирофосфат. Такой аминоациладенилат
является активированной по карбоксилу формой ами-
нокислоты и под действием того же фермента перено-
сит остаток последней на молекулы «транспортной» или
«растворимой», РНК (soluble, transfer, acceptor, adap-
tor RNA), образуя эфир с третьим углеродным атомом
рибозы конечного (аденилового) нуклеотида (амино-
ацил-РНК).
Для каждой аминокислоты существует свой особый
«pH5-фермент» и своя особая «транспортная» РНК.
Для аминокислот с вырожденным генетич. кодом од-
ной аминокислоте соответствует несколько таких
РНК. Молекулы «транспортной» РНК (мол. в. ок.
25 000) состоят приблизительно из 70 нуклеотидов.
У всех молекул этой РНК один конец, реагирующий
с аминокислотой, имеет последовательность основа-
ний: аденин—цитозин — цитозин. Аминоацил РНК пе-
реносится в рибосомы; в присутствии специальных «пе-
реносящих» ферментов и гуанозинтрифосфорной к-ты
аминокислотный остаток отделяется от «транспортной»
РНК и реагирует с другим аминокислотным остатком,
образуя пептидную связь. Рост полипептидных це-
пей осуществляется постепенно, начиная с МН? кон-
цевой аминокислоты и нарастая с карбоксильного кон-
ца. Согласно гипотезе Крика — Хоглэнда, установка
определенной аминокислоты в особом специфич. для
нее месте полинуклеотидной матрицы обеспечивается
взаимодействием молекулы-адаптора («транспортной»
РНК) с высокомолекулярной матричной РНК, осу-
ществляющимся за счет водородных связей. Амино-
кислота сама по себе не влияет на место, занимаемое
ею на матрице, что доказано экспериментально. Вы-
яснено, что в ряде случаев синтез одной молекулы
белка осуществляется при участии нескольких рибо-
сомальных частиц (полизома, эргосома), связанных
одна е другой полинуклеотидной цепью «информацион-
ной» РНК. Число рибосом, входящих в этот актив-
ный комплекс, зависит от размеров синтезируемого
белка (и тем самым от мол. веса «информационной»
РНК). Последней стадией образования белка следует
считать его отделение от рибосомных частиц и пере-
ход в цитоплазму клетки. Возможно, что этот процесс
так же, как и процесс самого синтеза, совершается при
участии специальных ферментов.
Описанный выше механизм биосинтеза белка, ка-
ким он представляется в настоящее время, может слу-
жить типичным примером, характеризующим пред-
мет исследования и подходы современной М. б. За
последние годы изучен механизм многих других био-
логически важных процессов, к-рые так же, как и
процесс биосинтеза белка, явились объектами иссле-
дования на молекулярном уровне.
М. б., как наука, находится в стадии бурного роста.
Ее дальнейшее развитие позволит интерпретировать
на молекулярном уровне важнейшие процессы, проте-
кающие в живых организмах, что приведет к теоретич.
обобщениям огромного познавательного и приклад-
ного значения. По мере развития М. б. ее общетеоре-
тич. достижения будут все шире использоваться в ме-
дицине, сельском хозяйстве, химии и технологии
полимерных материалов. Разработка эффективных
средств борьбы с раковыми и вирусными заболевания-
ми, новых технологии, схем с использованием фермен-
тов, синтез больших количеств белков для их исполь-
зования в с. х-ве и пищевой пром-сти и многие другие
важные практич. проблемы в скором времени будут
решаться с учетом достижений М. б.
Общенаучное и философское значение М. б. состоит
в том, что она знаменует собой синтез отдельных нап-
равлений современного естествознания, изучающих
живую и неживую материю, и создание единой кар-
тины окружающего нас материального мира. Именно
поэтому теоретич. и практич. достижения М. б. на-
носят удар по идеалистич. и, в первую очередь, по ви-
талистич. представлениям и означают победу материа-
листич. взгляда на мир.
Лит.: Химические основы наследственности, пер. с англ..
М., 1960 (Тр. Международного симпозиума); Современные про-
блемы биофизики, т. 1—2, М., 1961; Анфинсен К., Моле-
кулярные основы эволюции, пер. с англ., М., 1962; Энгель-
гардт В. А., Некоторые проблемы современной биохимии,
М., 1959; Ж. Всес. хим. об-ва, 1961, 6,№ 3; 1963, 8,№ 1; 3 б а р-
ский И. Б., Успехи соврем, биол., 1962, 54, 265; Труды
V Международного биохимического конгресса. Симпозиум 1,
М., 1962; Молекулярная генетика. Сб. статей, пер. с англ,
и нем., М., 1963; Бреслер С. Б., Введение в молекуляр-
ную биологию, М.—Л., 1963; Kosower Е. М., Molecular
biochemistry, N. Y.—[a. o.J, 1962; Horizons in biochemistry,
ed. M. Kasha, B. Pullman, N. Y., 1962; Progress in nucleic acid
research, v. 1, N. Y., 1963. Я. M. Варшавский, Л. Л. Киселев.
МОЛЕКУЛЯРНАЯ ДИСТИЛЛЯЦИЯ — см. Ди-
стилляция молекулярная.
МОЛЕКУЛЯРНЫЕ СИЛЫ (межмолеку-
лярные силы) — см. Межмолекулярное взаимо-
действие.
МОЛЕКУЛЯРНЫЕ СИТА — пористые адсорбен-
ты, у к-рых размеры пор или входов в поры близки
к размерам молекул. Такие адсорбенты способны рез-
ко избирательно адсорбировать более мелкие моле-
кулы. Относительно более крупные молекулы ими
практически не адсорбируются. Т. о., при адсорбции
происходит как бы отсеивание более мелких молекул
от более крупных. Такое молекулярно-ситовое дейст-
вие свойственно различным адсорбентам, напр. мел-
копористым активным углям, пористым стеклам и в
особенности пористым алюмосиликатным кристаллам—
природным и синтетич. цеолитам (диаметр входов в
поры от 3 до 9А). По мере приближения размеров мо-
лекул к размерам пор адсорбента или размерам вхо-
дов в поры резко уменьшается скорость проникания
молекул в поры благодаря возрастанию энергии акти-
вации процесса. Поэтому молекулярно-ситовое дей-
ствие адсорбентов во многих случаях носит кинетич.
характер и усиливается с понижением темп-ры. М. с.
281
МОЛЕКУЛЯРНЫЕ СПЕКТРЫ
282
позволяют наиболее четко производить адсорбцион-
ное разделение смесей веществ в газообразных и жид-
ких фазах.
Лит.: Дубинин М. М. [и др.], Изв. АН СССР.
Отд. хим. н., 1961, № 1, 29: Жданов С. П., Кор о-
мальди Е. В., ДАН СССР, 1961, 138, № 4, 870; В а г-
гег R. М., Brit. Chem. Engng, 1959, 4, № 5; Синтетическге
цеолиты, М., 1962. М. М. Дубинин.
МОЛЕКУЛЯРНЫЕ СПЕКТРЫ — электромагнит-
ные (оптические) спектры испускания и поглощения, а
также комбинационного рассеяния, возникающие при
квантовых переходах между энергетич. состояниями
молекулы.
Согласно квантовой теории, энергия hv испускаемо-
го или поглощаемого фотона (кванта света) частоты v
определяется основной ф-лой (условием частот Бора,
см. Атом):
hv = E'—E" (1)
переходах
। к-рых дает
а л ь н у ю
определяет
и с п у с к а-
молекулы.
г = 10----*
10^- s
-+--IQ
О
:5
‘0-
Рис. 1. Схема уровней энергии
двухатомной молекулы: а иб —
электронные уровни, v' и v" —
квантовые числа колебательных
уровней, J' и J" — квантовые
числа вращательных уровней.
==5
где Е’ и Е" — значения энергии молекулы в двух
состояниях, между которыми происходит квантовый
переход. Если энергия начального состояния больше
энергии конечного, то молекула испускает фотон; со-
вокупность частот свето-
вых колебаний, испус-
каемых молекулой при
различных
(каждая из
спектр
лини ю),
спектр
ния
Если энергия начально-
го состояния меньше
энергии конечного, то
молекула поглощает фо-
тон; совокупность частот
световых колебаний, по-
глощаемых молекулой
при различных перехо-
дах, определяет спектр
поглощения мо-
лекулы. На схеме
уровней энергии
молекулы (рис. 1) пере-
ходы в испускании изо-
бражаются стрелками,
направленными сверху
вниз, переходы в погло-
щении—снизу вверх; со-
ответственно говорят о переходах с верхних уров-
ней на нижние и с нижних на верхние. Верхние
уровни принято в молекулярной спектроскопии обо-
значать одним штрихом ('), нижние — двумя ("); в
ф-ле (1) Е'>Е".
При комбинационном рассеянии света энергия
AVpacc. рассеянного фотона частоты vpacc_ определяет-
ся ф-лой:
^vpacc.“ ^пад. (2)
где TiVnaa.— энергия падающего фотона частоты vnaj.,
a Ei—Е2 — разность энергий состояний молекулы до
и после рассеяния; если E2>Ei, то молекула получа-
ет энергию от падающего фотона и vpa<.c.<vll;ln. (сток-
сово рассеяние, аналогичное поглощению све-
та молекулой); если E2<Elt то молекула получает
энергию и vpacc,>vnan. (антистоксово рас-
сеяние, аналогичное испусканию). Совокупность
частот световых колебаний, рассеиваемых молеку-
лой при различных переходах, определяет спектр
комбинационного рассеяния мо-
лекулы.
М. с. имеют сложную структуру. Типичными яв-
ляются полосатые М. с. испускания и погло-
щения, наблюдаемые в ультрафиолетовой, видимой и
близкой инфракрасной областях в виде совокупности
более или менее широких полос, распадающихся при
достаточной разрешающей силе спектрального при-
бора (когда можно раздельно наблюдать спектральные
линии, близкие по частоте) на совокупность тесно
расположенных линий. Сложность полосатых
М. с. по сравнению с линейчатыми атом-
ными спектрами определяется тем, что внут-
реннее движение в молекулах более сложно, чем в ато-
мах, для к-рых уровни энергии определяются только
движением электронов относительно ядра атома: в мо-
лекуле наряду с движением электронов относительно
ядер атомов, образующих молекулу, происходит ко-
лебательное движение самих ядер относительно их по-
ложений равновесия и вращательное движение моле-
кулы как целого. Соответственно М. с. разделяют j на
электронные, колебательные и вра-
щательные. Конкретные структуры М. с. раз-
личны для различных молекул и, вообще говоря,
усложняются с увеличением числа атомов в молекуле.
Для весьма сложных молекул, однако, в ультрафио-
летовой и видимой областях вместо спектров, состоя-
щих из систем сравнительно узких полос, распадаю-
щихся на отдельные линии, наблюдаются широкие
сплошные полосы испускания н поглощения; спектры
при этом упрощаются и выявляется их сходство для
различных молекул.
Виды движения в молекуле и типы молекулярных
спектров. Трем видам движения в молекуле —
электронному, колебательному и
вращательному — соответствуют три типа
квантовых состояний и уровней энергии. Полная энер-
гия молекулы принимает определенные значения, со-
ответствующие различным электронно-колебательно-
вращательным состояниям, и с хорошей степенью
приближения может быть представлена, как показы-
вается квантовомеханич. методами, в виде суммы кван-
тованных значений энергий электронного, колеба-
тельного и вращательного движений:
Е = •Ё’эл. + ^кол. + ^вр. (3)
По порядку величин отношение этих энергий равно:
£эл.:Якол.:Явр.= 1: (4)
где т — масса электрона, а М имеет порядок массы
ядер атомов, входящих в состав молекулы, поэтому
т/М~10~3—10“5, следовательно:
^эл. -^кол. Э5* ^вр. (°)
Обычно Еэл имеет порядок величины от неск. десят-
ков ккал/моль до ста—двухсот ккал/моль (неск. эв
на молекулу), Екол.— от сотен кал/моль до неск.
ккал/моль (10~2—10-1 эв), Ев„—от долей кал/молъ до
неск. десятков кал/моль (IO’-5—10-3 эв).
В соответствии с (5) для молекулы получается сово-
купность далеко отстоящих электронных уровней
энергии (различные значения £’эл_ при EK0JI.=Eflp =0),
более близких колебательных уровней (различные
значения Екол. при заданном Еэл и при Евр.=0) и
еще более близких вращательных уровней (различные
значения Евр. при заданных Еэл и EK0!i), как схемати-
чески показано на рис. 1 для двухатомной молекулы.
Для многоатомной молекулы схема уровней еще
усложняется.
Значение электронной энергии Еэл. в (3) и на схеме
рис. 1 соответствует равновесной конфигурации моле-
кулы (в случае двухатомной молекулы характеризуе-
мой равновесным значением междуядерного расстоя-
ния Яо, см. рис. 4 в ст. Молекула). Для каждого элект-
ронного состояния получаются определенная равно-
весная конфигурация и определенное значение Еал^
283
МОЛЕКУЛЯРНЫЕ СПЕКТРЫ
284
наименьшее значение Еэл. соответствует о с н о в н о-
м у (нормальному) электронному состоянию — са-
мому нижнему электронному уровню энергии.
Различные типы молекулярных спектров возни-
кают при различных типах переходов между уровнями
энергии молекул. При переходе, связанном с испуска-
нием или поглощением,
А-ЕдЛ. А^КОЛ. А^вр, (6)
(расстояния между уровнями того же порядка, что и
энергии Еэл<, £кол_, ^в₽.)-
При ДЕзл,#0 получаются электронные М.с.,
наблюдаемые в видимой и УФ-областях как в испус-
кании (в частности, в виде спектров люминесценции,
см. Люминесцентный анализ), так и в поглощении.
Обычно одновременно ДЕкол.^0 и А^вр.т=О; различ-
ным ДЕКОЛ при заданном ДЁЭЛ соответствуют различ-
ные колебательные полосы, а различным ДЕвр. при за-
данных Еэл. и Екол, — отдельные вращательные линии,
на к-рые распадается данная полоса; получается ха-
рактерная полосатая структура (рис. 2). Совокупность
Рис. 2. Полосатый спектр испускания молеку-
лы N2 в близкой ультрафиолетовой области;
видны группы полос т. наз. второй положи-
тельной системы азота, соответствующие раз-
личным значениям Ди = v’—v".
полос с заданным ДЕЭЛ. (соответствующая заданному
чисто электронному переходу с частотой
vM,= ДЕЭЛ./А) наз. системой полос. Электронные (точ-
нее’ электронно-колебательно-вращательные) спектры
изучаются экспериментально при помощи спектро-
графов со стеклянной (для видимой области) и квар-
цевой (для УФ-области) оптикой, в к-рых для разло-
жения света в спектр применяются призмы или ди-
фракционные решетки.
При ДЕЭЛ. =0, а ДЕкол т=0 получаются колеба-
тельные М. с., наблюдаемые в близкой (до неск.
.чк) и в средней (до неск. десятков мк) ИК-областп,
обычно в поглощении и при комбинационном рассея-
нии света. Как правило, одновременно ДЕвр> = 0п при
заданном АЕКОЛ. получается колебательная полоса,
распадающаяся на отдельные вращательные линии,
как и в случае полос в электронных спектрах. Наблю-
дение колебательных (точнее колебательно-вращатель-
ных) спектров производится в ИК-области в погло-
щении ИК-спектрометрами с призмами, прозрачными
для ИК-излучения (см. Инфракрасная спектроскопия),
и в видимой области в комбинационном рассеянии
светосильными спектрографами со стеклянной опти-
кой (см. Комбинационное рассеяние света).
При ДЕЭЛ = 0 и Д^Кол.= 0, а ДЕвр 5Д 0 получаются
состоящие из отдельных линий чисто вращательные
М. с., наблюдаемые в поглощении в далекой инфра-
красной и особенно в микроволновой областях, а так-
же в комбинационном рассеянии света. Наблюдение
инфракрасных вращательных спектров производится
спектрометрами со специальными дифракционными
решетками (эшелеттами) для далекой ИК-области, а
наблюдение микроволновых вращательных спектров—
микроволновыми (СВЧ) спектрометрами (см. Радио-
спектроскопия). Для наблюдения вращательных спект-
ров комбинационного рассеяния применяются свето-
сильные спектрографы для видимой, а также для УФ-
области.
Возможные переходы между уровнями энергии дан-
ной молекулы зависят от свойств симмет-
рии ее равновесной конфигурации и от связанных с
ними свойств симметрии соответствующих электрон-
ных, колебательных и вращательных состояний (см.
Симметрия молекул)', соответствующие правила от-
бора, определяющие возможные (разрешенные) пере-
ходы с испусканием или поглощением фотона, а также
переходы, возможные при комбинационном рассеянии,
для нек-рых случаев будут рассмотрены ниже при
разборе различных типов М. с. Интенсивности спект-
ральных линий и полос для возможных переходов про-
порциональны заселенностям начальных
уровней энергии (нижних при поглощении и стоксо-
вом комбинационном рассеянии и верхних при испус-
кании и антистоксовом комбинационном рассеянии) и
вероятностям переходов, различным
для М. с. различных типов и зависящим от конкретных
свойств данной молекулы. Заселенности уровней, т. е.
числа молекул в соответствующих энергетич. состоя-
ниях, определяются законом распределения Больц-
мана:
ni = giAe-E‘hT (7)
где Ej — энергия состояния, g; — его статистик, вес,
а Л — постоянная [см. Квантовая статистика, ф-ла
(3), в к-рой надо положить е;=Е;, а=—in А и $=1/кТ].
Вероятности разрешенных переходов определяются
изменением при переходе дипольного момента моле-
кулы (для спектров поглощения и испускания) или ее
поляризуемости (для спектров комбинационного рас-
сеяния) и могут в простейших случаях быть рассчита-
ны квантовомеханич. методами. В ряде случаев относи-
тельные вероятности различных переходов одного
типа определяются, как и правила отбора, свойст-
вами симметрии комбинирующих состояний моле-
кулы.
Вращательные спектры (чисто вращатель-
ные). Эти спектры получаются при переходах между
различными вращательными уровнями энергии, к-рые
можно найти путем квантования кинетической энер-
гии вращения молекулы, приближенно рассматри-
вая ее как твердое тело с определенными моментами
инерции.
В простейшем случае двухатомной молекулы ее
энергия вращения Евр = ’ гДе М — вращатель-
ный момент количества движения молекулы, а
I — момент инерции вокруг осп, проходящей через
центр тяжести перпендикулярно оси молекулы (т. е.
линии, соединяющей ее ядра), равный ц/<2 [где ц=
=MiM2/(Mi+M2) — т. наз. приведенная масса моле-
кулы, выражающаяся через массы ядер М, и М2, /<0—
равновесное междуядерное расстояние]. Квадрат мо-
мента количества движения может принимать зна-
чения, определяемые квантовомеханич. формулой:
Л72=^ J (/-|-1) (ср. Атом, стр. 313); это дает
h 2
EBp. = ^J(J + l) = hBJ(J + l) (8)
где
7=0, 1, 2, 3, ... (9)
— вращательное квантовое число, а
В = /г;'8лЧ = /г./8л2(1Я2 (10)
— вращательная постоянная, определяющая масштаб
расстояний между уровнями энергии, расположение
к-рых показано на рис. 3.
Чисто вращательные спектры поглощения и испус-
кания возникают, в силу правила отбора Д7=7'—7"=
= 1, при переходах между соседними уровнями, что
дает равноотстоящие вращательные линии с интер-
валами 2В (см. рис. 3, слева). Такие спектры имеют
285
МОЛЕКУЛЯРНЫЕ СПЕКТРЫ
286
лишь молекулы с отличным от нуля постоянным
дипольным моментом D\ для состоящих из двух одина-
ковых атомов молекул типа А2 D=0, и эти молекулы,
напр. Н2 и О2, не способны
испускать или поглощать
свет при изменении состоя-
ния вращения.
Чисто вращательные спек-
тры комбинационного рас-
сеяния имеют все двухатом-
ные молекулы; в этом слу-
чае правило отбора AJ—J'—
—J"=2, что также дает рав-
ноотстоящие вращательные
линии, но с интервалами
4В (см. рис. 3, справа).
Для линейных многоатомных
молекул вращательные уровни
также определяются ф-лой (8)
и получаются аналогичные вра-
щательные спектры; в этом слу-
J=0
J=2
20 8
12 В
зо в
6B
28
О
2 3 4 5
0 12 3
J* 1234
J" 0123
Рис. 3. Вращательные уров- чае момент инерции 1—2 MiR*,
ни энергии, переходы между i
ними и вид спектров для где М;— масса i-того атома, а
двухатомной молекулы. Ri— расстояние его ядра от
центра тяжести молекулы.
Для нелинейных многоатомных молекул возможны три
случая, в зависимости от соотношения между тремя глав-
ными моментами инерции Ix, ly, Iz вокруг трех
взаимно-перпендикулярных осей х, у, z, связанных с моле-
кулой.
1. Если молекула является сферическим волч-
ком, 1Х = что имеет место для молекул тетра-
эдричес.кой симметрии типа АВ< (напр., СН4)
и октаэдрической симметрии типа АВв, то фор-
мула (8) остается справедливой; однако, в этом случае в силу
высокой симметрии молекула не обладает чисто вращатель-
ными спектрами ни поглощения и испускания, ни комбина-
ционного рассеяния.
2. Если молекула является симметричным волч-
ком Ix ~ что имеет место при наличии выделенной
оси симметрии порядка не ниже третьего (см. Симметрия моле-
кул), напр. NH3, то формула (8) заменяется несколько более
сложной, однако чисто вращательный спектр по-прежнему
состоит иэ равноотстоящих линий, интервалы между к-рыми
зависят только от значений момента инерции 1х=1у, но не
от Iz (линейные молекулы также являются симметричными
волчками, но с 12=0; они имеют при этом ось симметрии z
бесконечного порядка).
3. Если молекула является асимметричным волчком, 1Х^
^Iy^Iz, что имеет место для молекул достаточно низкой сим-
метрии, в частности для всех нелинейных трехатомных молекул
(типа АВ2, напр. Н2О, и типа АВС), то как расположение вра-
щательных уровней, так и расположение вращательных линий
в спектре весьма сложны и зависят от значений всех трех
моментов инерции— Ix, Iy, Iz.
Изучение чисто вращательных спектров молекул,
особенно методами радиоспектроскопии в микровол-
новой области, позволяет по найденным из опыта зна-
чениям моментов инерции определять в большой точ-
ностью параметры равновесной конфигурации — дли-
ны связей и углы между связями. Для увеличения
числа определяемых параметров исследуют вращатель-
ные спектры изотопич. молекул, имеющих одинако-
вые параметры равновесных конфигураций, но раз-
личные моменты инерции.
Колебательные спектры (колебательно-
вращательные). Эти спектры получаются при
переходах между различными колебательными уров-
нями энергии, к-рые можно найти квантованием коле-
бательной энергии. Приближенно колебания можно
считать гармоническими: двухатомная молекула (одна
колебательная степень свободы, соответствующая из-
менению междуядёрного расстояния R) рассматри-
вается как гармония, осциллятор (см. Квантовая ме-
ханика), а TV-атомная молекула (r=37V—6 колеба-
тельных степеней свободы в случае нелинейной и
г—ЗК—5 колебательных степеней свободы в случае
линейной равновесной конфигурации) рассматри-
вается как совокупность г связанных между собой
гармония, осцилляторов, совершающих нормаль-
ные колебания (см.* Молекула).
Для двухатомной молекулы, совершающей
колебания около положения равновесия, соответст-
вующего минимуму кривой потенциальной энергии
(рис. 4 в ст. Молекула), вблизи минимума кривая по-
тенциальной энергии имеет вид параболы:
= W (И)
где q—R—Ro — колебательная координата, а к=
fd>U\
= h~d2 — силовая постоянная.
\^R2J R = R0
Формула (11) получается разложением в ряд потенциальной
энергии U(R) около точки R~R0:
С7(Д) = и(Яо)+(^)д = До(Д-До) +
1 /d2t7\ 1 /’№Т1\
+т(^}й = йо(й-й“)2 + т(«з)й = йо<й-й“>3+---<12>
Линейный член разложения равен нулю в силу условия ми-
нимума ( } =0, а кубические и более высокие члены
для колебаний малой амплитуды приближенно отбрасываются:
они определяют ангармоничность колебаний (см. ниже). Для
минимума U(R0) и U(R)—U(R0) = U(R) — Езл = ЕКОл,
Гармония, осциллятор с потенциальной энергией (11 j
обладает равноотстоящими уровнями энергии:
EK01i. = hve (у+!/2) (13)
где ve=T к/и—основная частота гармония, колеба-
ний молекулы (р — приведенная масса молекулы),
v — колебательное квантовое ч и с-
л о, принимающее значения 0, 1, 2, 3, при и=О
получается отличная от нуля и равная hve/2 нулевая
энергия колебаний.
Как для переходов при поглощении и испускании,
так и для переходов при комбинационном рассеянии
имеет место правило отбора Дг=г'— if=l, что дает
hv = EBon— Екоя.=^е (переход У-f-l^y) (14>
т. е. частота перехода между любой парой соседних
колебательных уровней совпадает с основной часто-
той колебаний.
Реальная кривая потенциальной энергии, вблизи
минимума близкая к параболе, затем начинает зна-
чительно от нее отличаться, и колебания с увеличе-
нием их амплитуды перестают быть гармоническими—
проявляется ангармоничность колебаний.
Вследствие ангармоничности колебательные уровни
энергии с увеличением колебательного квантового,
числа и постепенно сближаются (сходятся) к г р а -
нице диссоциации, соответствующей беско-
нечно большому расстоянию R. Ф-ла (11) заменяется
двухчленной ф-лой
EKOJt. = hve (г + '/2) — hvexe (г + ‘/2)2 (15)*
в к-рой ангармоничность характеризуется постоян-
ной
Ф-ла (15) соответствует учету в разложении (12) членов,
с (R—Rq)3 и (R—Rq)4 и так же, как и ф-ла (11), является при-
ближенной. При определенном конечном значении v=v0
расстояние между соседними уровнями уменьшается до нуля,
что соответствует границе диссоциации; подстановка v0 в (15)
дает значение энергии диссоциации De (отсчитываемой от дна
потенциальной ямы и отличающейся от действительной энер-
гии диссоциации D на величину нулевой энергии). Для
получается ф-ла:
De=/ive/4xe (16)
по к-рой можно приближенно определить энергии диссоциации
по известным ve и хв. Для Н2 этот метод определения энергии
диссоциации дает хорошие результаты, однако для боль-
шинства молекул вычисленные значения превышают дейст-
вительные до 20—30%.
При учете ангармоничности становятся возможны-
ми переходы с Ау>1— обертоны, а частоты пе-
реходов lj?0, 2^1, 3^2, ... уже не совпадают друг
с другом. Общая ф-ла для частот переходов имеет вид:
Vv’v” =т(£’кол.—Con.)=[ve-ve*e (2г"+ДН-1)1 Дг(17>
287
МОЛЕКУЛЯРНЫЕ СПЕКТРЫ
288
т. е. наряду с переходом с основной частотой
ve—2vexe будет появляться 1-й обертон 2^0 (Дг=2)
с частотой 2(ve—3vexe), 2-й обертон 3^:0 (Дг=3)
с частотой 3(ve—^vexe) и т, д.; интенсивности обер-
тонов быстро убывают с увеличением Др.
Вращательная структура колебательной полосы
определяется изменением вращательной энергии (8)
при колебательном переходе:
З'-Ю 9 8 7 6 5 4 3 2 ) J-I 2 345678910
Р-ветвъ(т»~/) R-ветво(т~ J')
Рис. 4. Схема переходов и вид
спектра для колебательно-вра-
щательной полосы.
£вр V + (J” +1) (18)
Для заданного значения разности \J=J'—J"
получается совокупность линий, соответствующих раз-
личным значениям /' и
J" — ветвь той или
иной полосы. Разностям
Д7=—2,—1, 0, +1, +2
соответствуют ветви, обо-
значаемые буквами О,
Р, Q, R, S,
В случае полос по-
глощения и испуска-
ния, соответствующих
переходам между коле-
бательными уровнями
основного электронного
состояния, обычно на-
блюдаются две ветви:
Р-ветвь (ДР=—1) и
.R-ветвь (Д7=+1); соот-
ветствующая схема пе-
реходов и вид спектра
показаны на рисунке 4.
Частоты вращательных
линий определяются формулой:
= (£вр.-£вр.)=(-В'+-В") т + (В'—В") т* (19)
где m=J' = l, 2, 3, ... для R-ветви (положительной
ветви) и т=—J" =—1,—2,—3, ... для Р-ветви (от-
рицательной ветви).
Вращательная постоянная В убывает с увеличением
v согласно приближенной ф-ле:
+ (20)
где 0<ае<<;1. Поэтому В'—В"<0 и |В'—В"|<^В'-|-В";
в результате в R-ветви линии постепенно сходятся,
а в Р-ветви постепенно расходятся с увеличением
]zn|. В нек-рых случаях наблюдается, помимо Р- и
К-ветвей, Q-ветвь (нулевая ветвь, ДУ=0), для к-рой
vm = (B'—В") т (m-pl) (m=Z'=Z' = l, 2, 3, ...) (21)
состоящая из близко расположенных вращательных
линий.
В случае комбинационного рассеяния света ДУ=
=0,±2 и получаются Q-, S- и О-ветви, причем обычно
наблюдается лишь Q-ветвь в виде одной достаточно
интенсивной неразрешенной линии, a S- и О-ветви
отсутствуют из-за недостаточной интенсивности от-
дельных вращательных линий.
Для многоатомных молекул в при-
ближении гармония, колебаний
г
•^кол. — У1 ^'vi (у| “1“ '1г) (22)
1=1
где V,-— частота i-того нормального колебания, а
р(=0, 1, 2, 3, ... — соответствующее колебательное
квантовое число; для каждого нормального колеба-
ния получается своя последовательность колебатель-
ных уровней. Для совокупности гармония, осцилля-
торов может одновременно изменяться при переходе
лишь одно из квантовых чисел на единицу (Аг,=
=гл—^=1, Дуу=О, где и возможны лишь пере-
ходы с частотами v,-, что дает колебательные полосы,
соответствующие разным нормальным колебаниям (ос-
новные полосы). При учете ангармоничности в коле-
бательных спектрах могут появляться обертоны
основных частот колебаний (Ду,>1, напр. Ду,=2,
Дг2=4, что обозначают как 2vn 4v2) и т. наз. с о-
ставные частоты, возникающие при одновре-
менном изменении двух или более колебательных
квантовых чисел (напр., Ду^!, Ду2=1 или Ду,=2,
Дг2= — 1, что обозначают как Vj-|-v2, 2vi—v2). Как
для основных частот колебаний, так и для обертонов
и составных частот имеются правила отбора, зави-
сящие от симметрии колебательных состояний (см.
Симметрия молекул) и различные для спектров погло-
щения и испускания и спектров комбинационного рас-
сеяния. В частности, если молекула обладает цент-
ром симметрии, например СОг, СвН6, то имеет
место так называемый альтернативный за-
прет — переходы, возможные в поглощении и ис-
пускании, запрещены в комбинационном рассеянии
и наоборот.
Набор основных частот колебаний молекулы, опре-
деляемый путем изучения ИК-спектров поглощения,
а также спектров комбинационного рассеяния, яв-
ляется существенной характеристикой молекулы.
Особо важное значение имеет наличие у молекул х а-
рактеристических частот колебаний,
соответствующих наличию в молекуле определенных
связей и определенных типов валентных углов и их
комбинаций, различных групп в молекуле и различ-
ных пространственных структур.
Электронные спектры (электронно-коле-
бательн о-вращательные). Эти спектры
получаются при переходах между различными элект-
ронными уровнями энергии, к-рые находятся путем
квантования электронной энергии молекулы, т. е.
энергии молекулы при неподвижных ядрах. Кванто-
вомеханич. методы позволяют в принципе рассчи-
тать электронную структуру молекулы, определить ее
электронные уровни энергии и их характеристики.
Однако данная задача весьма сложна и решается
с достаточной точностью лишь для молекулы водорода
Нг. Для других двухатомных молекул и для много-
атомных молекул приходится применять различные
приближенные квантовомехапич. методы (подробнее
см. Квантовая химия).
Ряд характеристик электронных состояний моле-
кул и переходов между ними может быть найден пу.
тем рассмотрения свойств симметрии молекулы. В наи-
более простом случае двухатомных и линейных мно-
гоатомных молекул электронные состояния характе-
ризуются, в силу наличия симметрии относительно оси
молекулы — аксиальной симметрии, значе-
ниями квантового числа А, определяющего абс. вели-
чину проекции суммарного орбитального момента
электронов на ось молекулы. Состояния с А=0, 1, 2,
3, 4 обозначают прописными греч. буквами2,П,Д,Ф,...
(аналогично обозначениям <S, Р, D, В,... состояний
атома с 1 = 0,1,2,3,..., см. Атомные спектры); индексом
слева сверху указывают мультиплетность х=25Ч-1,
где квантовое число S определяет спин молекулы —
величину суммарного спинового момента электронов
S. Напр., 2П обозначает электронный уровень энергий
с А=1, <9='/г, а 32— с А=0, 5=1. Для подавляю-
щего большинства химически устойчивых молекул
основным является состояние '2 (А=0, 5=0) с пол-
ным электронным моментом, равным нулю. Для пе-
реходов в поглощении и испускании имеет место пра-
вило отбора ДА=0, ±1; соответственно для линей-
ных молекул возможны переходы типов
2—2, 2 —П, П —2, П —П, П —Д, Д —П, ...
289
МОЛЕКУЛЯРНЫЕ СПЕКТРЫ—МОЛЕКУЛЯРНЫЙ ВЕС
290
Принято сперва писать символ верхнего уровня, а
затем нижнего и указывать стрелкой направление
перехода; напр. 2->П означает переход в испускании
с верхнего уровня 2 на нижний уровень П, а 2+-II—
переход в поглощении с нижнего уровня П на верхний
уровень 5.
Наряду с характеристикой электронного состояния
двухатомных и линейных многоатомных молекул
в целом квантовым числом Л существенное значение
имеет характеристика состояний отдельных электро-
нов в этих молекулах при помощи квантового числа Л,
определяющего абс. значение проекции орбитального
момента электрона на ось молекулы; состояния с Л=
=0, 1, 2, 3, ... обозначают строчными греч. буквами
сг, л, б, ф... (аналогично обозначениям s-,p-,d~,/-состоя-
ний электрона с 1=0, 1, 2, 3, .... см. Атом). Такая ха-
рактеристика важна при рассмотрении электронных
оболочек молекул в соответствии с Паули принципом
и теорией химической связи.
Для нелинейных многоатомных молекул можно
классифицировать электронные состояния молекулы
в соответствии со свойствами симметрии равновесной
конфигурации (см. Симметрия молекул).
Колебательная структура электронных спектров
определяется изменением колебательной энергии при
электронном переходе. Для двухатомной молекулы это
изменение определяется ф-лой (17); при этом Ду=
= г'—v" может принимать различные значения и по-
а бе
Рис. 5. Система электронно-колебательных полос: а — об-
щая схема переходов, б — поперечная серия, в — про-
дольная серия.
лучается система полос, состоящая из поперечных
серий (p"=const) и продольных серий (v’= const)
(рис. 5). Для молекулы N2 такая система полос видна
на рис. 2. Распределение интен-
и1т сивностей в системе полос опреде-
ляется свойствами кривых потен-
1 циальной энергии комбинирующих
\ электронных состояний в соответ-
I ствии с принципом Фран-
I___ ка — Кондона. Согласно это-
I- -4***“*^ му принципу, наиболее вероятны-
г--f ми являются электронно-колеба-
V—-Г . тельные переходы, при к-рых ме-
—k-Z_______ждуядерное расстояние R и ско-
рость движения ядер не меняются;
вероятные электрон- такие переходы соответствуют вер-
но - колебательные тикальным линиям, соединяющим
переходы; показаны потенциальные кривые комбини-
дыга7доТлетТоря“’ РУ’ощих электронных состояний
щие принципу (рис. 6). Для многоатомных моле-
Франка — Кондона, кул колебательная структура элек-
тронных спектров усложняется по
сравнению с соответствующей структурой спектров
двухатомных молекул. Для сложных молекул, со-
держащих большое число атомов, вместо систем узких
полос получаются широкие сплошные полосы.
Вращательная структура электронно-колебательных
полое для двухатомных молекул определяется ф-лой
(18) и благодаря правилу отбора ДУ=0,4-1 получаются
три ветви —Q, R и Р, частоты линий для к-рых даются
формулами (21) и (19) (для 2—S-переходов ДУ=0
запрещено и Q-ветвь отсутствует). Однако, в отличие
от колебательно-вращательных спектров, В' я В"
относятся к разным электронным состояниям и могут
сильно отличаться за счет изменения междуядервого
расстояния Ro, различного для таких еостояний
(см. рис. 6). Поэтому |jB'—В"| может быть сравни-
мо с В' и В", наряду со случаем В' <В" возможен
и случай В’>В". В результате в одной из ветвей
(в В-ветви при В' <В" и в Р-ветви при В'>В")
вращательные линии сгущаются, образуя резкую гра-
ницу полосы — к а н т, и полоса оттененав про-
тивоположную сторону; на рис. 2 видно такое отте-
нение полос.
Для линейных многоатомных молекул получается
аналогичная вращательная структура, для нелиней-
ных молекул вращательная структура усложняется
и ее удается наблюдать лишь для сравнительно про-
стых молекул.
Лит.: 1) Кондратьев В. Н., Структура атомов и
молекул, 2 изд., М., 1959; 2) Е л ьяшевич М. А., Атомная
и молекулярная спектроскопия, М., 1962; 3)Г ерцберг Г.,
Спектры и строение двухатомных молекул, пер. с англ., М.,
1949; 4) е г о же, Колебательные и вращательные спектры
многоатомных молекул, пер. ’с англ., М., 1949; 5) Применение
спектроскопии в химии, под ред. В. Веста, пер. с англ., М.,
1959; 6) В а г г о w &. М., Introduction to molecular spectro-
scopy, N. Y., 1962. M. А. Ельяшевич.
МОЛЕКУЛЯРНЫЙ ВЕС — сумма произведений
атомных весов всех атомов, составляющих молекулу
вещества, на число атомов данного вида в молекуле.
М. в. характеризует массу молекулы в единицах атом-
ного веса. Поскольку разные молекулы данного ве-
щества могут включать различные изотопы составляю-
щих их атомов, то М. в. представляет собой среднюю
массу молекулы с учетом изотопного состава всех
элементов, образующих данное химич. соединение.
Для полимеров и различного рода ассоциатов, не яв-
ляющихся строго индивидуальными соединениями,
М. в. есть средняя величина по всем молекулам
соединения (см. Молекулярный вес высокомолекуляр-
ных соединений). Кроме того, понятие «М. в.» в каче-
стве средней величины применяют иногда в отноше-
нии смесей'веществ с определенным составом (напр.,
для воздуха можно принять средний М. в. 29).
М. в. является одной из самых фундаментальных
характеристик вещества. М. в. лежит в основе опре-
деления понятий грамм-молекулы (моля) и мольных
величин (т. е. физико-химич. характеристик при рас-
чете на один моль) — таких, как молярные (мольные)
концентрация, объем, теплоемкость, электропровод-
ность, теплота реакции и т. д. М. в. входит во многие
ур-ния и соотношения, выражающие связь между
различными физико-химич. величинами. Многие уни
версальные законы и величины, не зависящие от ин-
дивидуальных свойств вещества, приведены к одному
молю и, следовательно, также содержат М. в. в скры-
том виде. М. в. используется для установления химич.
ф-лы вещества, применяется во всех стехиометрия,
расчетах по химич. ф-лам и ур-ниям.
Понятием, в. как величины, характеризующей массу моле-
кулы, было четко сформулировано в результате работ С. Нан
ннццзро (1858), к-рый в свою очередь использовал и развил
ранее не признанные взгляды А. Авогадро (1811). На основе
закона Авогадро был дан простой способ определения М. в.
газа по его плотности. Работы Авогадро и Канниццаро полу-
чили всеобщее признание на международном съезде химиков
в 1860. С этого времени стали широко разрабатываться различ-
ные методы измерения М. в., на основе к-рых определялись ат.
веса элементов. Однако после работ Ф. Астона (1920) и после
дующего развития техники масс-спектрометрич. определений
для измерения ат. весов стали применять в основном масс-
спектрометрию, а измерение М. в. для этой цели утратило свое
былое значение. Тогда же понятие М. в., как и ат. веса, было
О 10 К.Х.Э. т. 3
291 МОЛЕКУЛЯРНЫЙ ВЕС—МОЛ. ВЕС ВЫСОКОМОЛЕКУЛЯРНЫХ СОЕДИНЕНИЙ 292
уточнено в связи с явлением изотопии. При этом М. в. состав-
лялись по химич. шкале ат. весов. Переход на новую единицу
ат. веса — */12 массы изотопа С12, принятую международным
съездом химиков (Монреаль, 1961), не внес значительных изме-
нений в величины ат весов оо химич, шкале, а следовательно,
и в величины М. в , поскольку разница в массах между новой
единицей и единицей по химич. шкале весьма мала. Развитие
химии высокомолекулярных соединений привело к разработке
и широкому использованию специальных методов определения
М. в. полимеров.
Методы определения М. в. В основу
экспериментального определения М. в. вещества могут
быть положены различные соотношения между физи-
ко-химич. величинами, включающие М.в. или мольную
величину. Выбор же наилучшего метода определяется
в каждом случае индивидуальными свойствами веще-
ства (агрегатное состояние, растворимость, возмож-
ность химич. воздействия на измерительные приборы
и т. д.). Для повышения точности определения М. в.
проводят не абсолютные, а относительные измерения
тех величин, с к-рыми связан М. в. в исходном соот-
ношении, т. е. определяют М. в. исследуемого вещест-
ва по отношению к другому веществу (стандарту),
М. в. к-рого точно известен. Ниже перечислены наи-
более распространенные методы измерения М. в.
Метод измерения по плотности и давле-
нию газа применим только к газообразным ве-
ществам. Он основан на ур-нии состояния идеаль-
ного газа, к-рое можно записать в виде M=RT(>/p,
где М—М. в., 72 = 0,082054 л-атм/град-моль— уни-
версальная газовая постоянная, Т—абс. темп-ра,
Q—плотность (г/л) газа, р — его давление (атм).
Поскольку к реальному газу это ур-ние применимо
лишь при бесконечно разреженном состоянии, то де-
лают ряд измерений величины q/p при постепенно
уменьшающемся давлении и затем экстраполируют
результат к нулевому давлению, т. е. находят величину
(e/p)0=lim(g/p). Соответствующие измерения (на од-
р-*0
ном и том же приборе и при одной и той же темп-ре)
проводят для исследуемого вещества (qx/px) и стандар-
та (qJpj) и находят М.в. по ф-ле Мх= M1(p1/q1)b(qxIpx)b.
Замеры можно проводить также, уравновешивая на
газовых микровесах исследуемое вещество со стандарт-
ным; в этом случае QX=Q] и Мх=М1(р1/рх). Точность
метода может превышать 0,01%.
Для растворенных веществ, молекулы к-рых не дис-
социированы в растворе, можно применить аналогич-
ный метод, основанный на осмотическом дав-
лении. При этом используется закон Вант-Гоффа,
к-рый можно записать в виде M=(c/n,)RT, где с —
весовая концентрация (г/л) растворенного вещества,
а П — осмотич. давление (атм). Поскольку закон
Вант-Гоффа справедлив для бесконечно разб. р-ров,
то экстраполяцией результатов измерений находят
величину (с/П)0=Иш(с/П). Определяя в одном и том же
с->0
растворителе при одинаковых условиях величину
(с/П)0 для исследуемого и стандартного веществ, нахо-
дят искомый М. в. (см. Осмотическое давление).
Более распространены, однако, в случае р-ров не-
летучих веществ (без диссоциации молекул) крио-
скопический и эбулиоскопический
методы определения М. в., т. е. методы, основанные
на определении понижения точки замерзания и соот-
ветственно повышения точки кипения достаточно разб.
р-ров по сравнению с точкой замерзания или кипения
чистого растворителя. При этом используются зави-
симости М=Х(с/Л7’3) или М=Е(с/Л7’А), где Л7'3 и
Л 7^— понижение точки замерзания и соответственно
повышение точки кипения, а К и Е — криоскопия, и
соответственно эбулиоскопия, постоянные растворите-
ля, определяемые по стандартному веществу с точно
известным М. в. Измерения также проводят по от-
ношению к р-ру стандартного вещества и экстраполи-
руют на бесконечное разбавление, т. е. определяют ве-
личину (c/A7)0=lim(c/A7’). Методы обладают весьма
с-*0
высокой точностью, поскольку при помощи тер-
мометра Бекмана можно очень точно измерять малые
изменения темп-ры (см. Криоскопия и Эбулиоскопия).
Для определения М. в. можно использовать также за-
висимость понижения давления пара над раствором от
мольной доли растворенного вещества (закон Рауля)
(Ро'~Р)/Ро=Лг, гДе Ро— давление пара чистого ра-
створителя, р — давление пара над раствором,
N=(m/ М)/(т/М-\- mJ Мв)— молярная доля растворен-
ного вещества, т — его навеска, т0— навеска раство-
рителя, Мв— М. в. растворителя. При этом также
проводят экстраполяцию к бесконечно разб. р-ру.
Используют и другие физико-химич. соотношения,
напр. зависимость давления пара вещества (р атм) от
теплоты испарения (г кал/г): RT2 р =Мг и т. д.
Следует указать, что при известной химич. формуле
вещества нет надобности экспериментально опреде-
лять его М. в., поскольку он известен как сумма ат.
весов атомов, составляющих молекулу. Поскольку
атомные веса всех элементов известны, то лишь при
необходимости их уточнения прибегают иногда к пре-
цизионным измерениям М. в., однако и в этом случае
чаще используют более точные физич. методы измере-
ния непосредственно атомного веса. Когда же химич.
ф-ла вещества неизвестна, то определение М. в. окон-
чательно устанавливает, является ли найденная по
химич. анализу простейшая ф-ла истинной или имеет
место определенная кратная ф-ла (если измеренный
М. в. в целое число раз больше соответствующего ф-ле).
Измерение М. в. (если он не в целое число раз больше
соответствующего ф-ле) может указывать на присут-
ствие наряду с обычными молекулами вещества также
ассоциированных молекул. Особое значение имеет
определение мол. веса полимеров, поскольку он ха-
рактеризует длину их цепи и размеры молекул (т. е.
степень полимолекулярности).
Лит.: Некрасов Б. В., Курс общей химии, 14 изд.,
М„ 1962; Киреев В. А., Курс физической химии, 2 изд.,
М., 1956; Гуггенгейм Э. А. и Пру Дж., Физико-
химические расчеты, пер. с англ., М., 1958. С. Э. Вайсберг
МОЛЕКУЛЯРНЫЙ ВЕС ВЫСОКОМОЛЕКУЛЯР-
НЫХ СОЕДИНЕНИИ — важнейшая характеристика
высокомолекулярного соединения (полимера), опре-
деляющая его физич. и технология, свойства. Т. к.
при синтезе полимеров получается всегда смесь мак-
ромолекул разных мол. весов (разной степени поли-
меризации), то имеет смысл говорить о средних зна-
чениях мол. веса; полное же описание полимера дает-
ся функцией распределения по мол. весам (МБР).
Под функцией (плотностью) распределения Ф„(Л7)
понимают число п молекул, имеющих мол. вес М.
Если и0 — общее число молекул, то
2 Фп(м)=,го
м=о
Учитывая, что для полимеров М принимает большие
значения и потому может считаться непрерывно из-
меняющейся величиной, это равенство часто заменя-
ют интегралом:
00
и0 = ,f Фа (М) dM, где Фа (М) =
о
Выражение <pn(M)dM — число молекул, мол. вес
к-рых лежит в пределах от М до M+dM.
Функция распределения может быть пронормиро-
вана к единице:
ОС
ф = <р„ (М) '.па = <р„ (М) / f<p,2 (М) dM
О
293
МОЛЕКУЛЯРНЫЙ ВЕС ВЫСОКОМОЛЕКУЛЯРНЫХ СОЕДИНЕНИЙ
294
Наряду с численной функцией распределения мож-
но рассматривать весовую функцию распределения
где с — количество полимера, выраженное в весовых
единицах.
Функции <р„ и ф№ связаны зависимостью q>w(M)=
Зная функцию распределения ф(М),
можно легко найти средние мол. веса:
средпечи елейный:
Мп= J' М<р„ (М) dM '^j ф„ (Л/) dM = J' Л/ф (Л/) dM
ООО
и средневесовой:
<ю .»
Mw = ]' M<9W {М) dMj^' Ф№ (М) dM =
О о
= J М2ф (Л/) (W/J' Л/ф (Л/) dM
о о
а также ряд других средних значений. При экспери-
ментальном определении мол. веса полимера находят
то или другое среднее значение, в зависимости от вы-
бранного метода измерения.
Свойства р-ров полимеров, положенные в основу то-
го или иного метода измерения мол. веса, могут зави-
сеть как от числа растворенных макромолекул поли-
мера данного мол. веса, так и от их весовой концент-
рации. Если полимер монодисперсен (т. е. макромо-
лекулы не отличаются помол, весу), то вес полимера
будет прямо пропорционален числу макромолекул и
средние мол. веса, измеренные разными способами,
будут иметь одно и то же значение. Если же полимер
полидисперсен, то средние мол. веса не совпадают.
Напр., осмотич. давление, понижение темп-ры за-
мерзания, повышение темп-ры кипения определяются
числом растворенных макромолекул полимера. В слу-
чае полидисперсных полимеров, пользуясь этими ме-
тодами, можно получить среднечисленный мол. вес.
Рассеяние света раствором пропорционально ие числу
молекул, а весовой доле растворенного вещества. Сле-
довательно, измерением рассеяния света в р-ре поли-
мера определяется средневесовой мол. вес. В других
методах измерения определяются другие средние
мол. веса полимеров. Т. о., значения мол. веса полиди-
сперсного полимера, измеренные разными методами,
не должны быть одинаковыми во всех случаях.
Зная два различных средних мол. веса полимера,
можно судить о функции распределения ф„(М). Так,
напр., при радикальной полимеризации часто полу-
чается функция распределения типа Флори:
ф„(М) = ае~“м
При этом М„=1/а, a Mw=2/a, т. е. отношение Мш/Мп —
=2. Это отношение характерно для широких молеку-
лярно-весовых распределений при радикальной по-
лимеризации и механизме обрыва без рекомбинации.
При рекомбинационном механизме обрыва ф„(М)=
=а!Мг’Л и Мт/Мп=*1г. При каталитич. полимери-
зации получаются иногда очень узкие молекулярно-
весовые распределения и 1<МЮ/М„<1,2. В нек-рых
случаях двухфазной полимеризации, когда полимер
выпадает из гомогенной среды, распределения мол.
весов особенно широкие и значение величины MwjМп
может достигать 20 (полиэтилен низкого давления).
Методы измерения молекулярного веса. Осмо-
тический метод. С помощью осмометра (рис. 1)
измеряют осмотич. давление р-ра полимера. Очень
важным является подбор хорошей мембраны, непро-
10*
Рис. 1. Осмометр: 1 —капилляр-
ный манометр; 2,3,4 — детали
корпуса осмометра; 5 — перфо-
рированный диск; 6—мембрана;
7 — ртутный затвор; 8, 9, ю —
резервуар с растворителем.
ницаемой для макромолекул и не создающей слишком
большого сопротивления току жидкости. Лучшие
мембраны изготовляют из омыленных пленок нитрата
целлюлозы (или других
эфиров) или из пори-
стого стекла. Мол. вес
определяют по вели-
чине измеренного дав-
ления, пользуясь ура-
внением состояния р-ра:
где р — осмотическое
давление, с — весовая
концентрация полиме-
ра, Т— абс. темп-ра,
М —мол. вес, R — га-
зовая постоянная, В2—
т. наз. второй вириаль-
ный коэфф., учитываю-
щий силы взаимодей-
ствия макромолекул
друг с другом и с ра-
створителем. В сильно
сольватирующих раст-
ворителях («хороших»)
это преимущественно
силы отталкивания,
обусловленные соб-
ственным объемом ма-
кромолекул. Поэтому второй вириальный коэффици-
ент в хороших растворителях всегда положителен.
Если растворитель слабо сольватирует растворен-
ные молекулы, то силы сцепления между ними
начинают играть заметную роль и частично ком-
пенсируют эффект сил отталкивания. Чем хуже
растворитель, тем коэфф. В2 меньше. Зависимость В2
г> ь
от темп-ры выражается соотношением В2=а — — .
Понижая темп-ру, можно в нек-рых плохих’раствори-
телях полностью компенсировать сцепление отталки-
ванием, т. е. достигнуть состояния, при к-ром В2=0.
Соответствующая темп-ра наз. 9-точкой, а среда —
О-растворителем. Если вести измерения не в О-раст-
ворителе, то необходимо определить в осмометре ряд
точек для одного полимера при разных его концентра-
циях. Отсюда независимо определяются Мп и В2.
Химический метод. В случае полимеров,
несущих на конце цепи характерную функциональную
группу, можно определить М„ химически с помощью
анализа па соответствующие группы.
Метод, основанный на рассеянии
света. Для определения средневесового мол. веса
измеряют рассеяние света раствором полимера под уг-
лом 90° к падающему световому лучу. Причина рассея-
ния света р-ром заключается в тепловых флюктуа-
циях концентрации макромолекул, создающих флюк-
туации показателя преломления среды. Согласно тео-
рии Эйнштейна — Смолуховского:
!><>* 2”2vo ^УМс
J„ rUW(l + 2B3Mc)
где /9о°— интенсивность (поток энергии на 1 смг)
рассеянного света под углом 90°, /0 — интенсивность
падающего света, X — длина волны света, г — ра-
диальное расстояние от центра кюветы до точки на-
блюдения, .V — число Авогадро, v0— показатель пре-
ломления растворителя, v — показатель преломления
р-ра. В принципе метод рассеяния света является
абсолютным, он дает средневесовой мол. вес М^.
295
МОЛЕКУЛЯРНЫЙ ВЕС ВЫСОКОМОЛЕКУЛЯРНЫХ СОЕДИНЕНИЙ
296
При пользовании этим методом необходимо устано-
вить зависимость рассеяния света от концентрации
(для исключения второго вириального коэфф.) или же
работать в в-точке. Основная экспериментальная
Рис. 2. Оптическая схема прибора Дебая — 4
Бюхе для измерения светорассеяния: 1— ис- S)
точник света; 2 — линза; 3 — светофильтр; „
4, 5, 6 — диафрагмы; 7 — кювета с раство-
ром полимера; 8 — лабиринт для устранения отраженного
света; 9 — фотометр (фотоэлектрический или визуальный).
трудность заключается в необходимости тщательной
очистки р-ра от пыли и взвешенных частичек. Для
этой цели пользуются фильтрованием через стеклян-
ные фильтры и центрифугированием. Схема прибора
приведена на рис. 2.
Два других метода измерения мол. веса основаны на
использовании ультрацентрифуги (рис. 3). За седимен-
тацией макромолекул в поле центробежной силы сле-
дят с помощью оптич. методов, дающих возможность
Рис. 3. Схема ультрацентрифуги Сведберга (с масляны-
ми турбинами): 1 — центрифуга; 2 — баллон с водородом;
3 — фотокамера; 4 — мотор; 5 — вакуумный насос; в — ре-
зервуар с маслом; 7 — компрессор; 8 — мотор; 9 — масло-
охладитель; 10 — масляный резервуар для подшипников.
измерить производную от показателя преломления
среды (вдоль радиуса) dv/dr или пропорциональную
величину de! dr (с — концентрация полимера). Методы
основаны на измерении седиментационного равнове-
сия или скорости седиментации.
Метод седиментационного равно-
S е с и я. Если макромолекулы осаждаются в не слит-
ном большом центробежном поле (ускорение 20 000—
30 000 g, где g — ускорение силы тяжести), то они
не оседают на дно, а распределяются благодаря тепло-
вому движению в поле центробежной силы. Это рас-
пределение напоминает распределение плотности в ат-
мосфере с высотой и определяется законом Больцмана:
•— = е
Со
где с — концентрация в точке с радиальной коорди-
натой г, с0— концентрация полимера у дна кюветы,
где координата равна r0, г=(г0+г)/2— средний ра-
диус, <о — угловая скорость ротора, V — уд. объем
полимера, измеренный пикнометрически по плотно-
сти р-ра, q — плотность растворителя, R — газовая
постоянная, Т — абс. темп-ра; величина (1—V) учи-
тывает закон Архимеда. Все величины в ф-ле Больц-
мана известны из опыта, кроме мол. веса, к-рый отсю-
да легко вычислить. Обычно откладывают lg(c/c0)
как функцию г—г,.'. получается приблизительно пря-
мая линия, из наклона к-рой находят среднее зна-
чение Для применения этого метода нет нужды
дожидаться установления равновесного распределе-
ния, для чего требуется несколько десятков часов;
применяя несколько более сложные расчеты, мож-
но определить значение молекулярного веса и из дан-
ных по неустановившемуся равновесию (метод Арчи-
бальда).
Метод скорости седиментации.
При осаждении полимерных молекул в очень боль-
ших полях (центробежное ускорение ш-/г — 100 000—
300 000 g, где g — уско-
рение силы тяжести) из-
менения концентрации
(с) и ее производной
del dr в разные моменты
времени от начала опыта
приведены на рис. 4. В
нек-рой точке кюветы
будет находиться грани-
ца между чистым раство-
рителем и седиментирую-
Рис. 4. Экспериментальные кри-
вые, получаемые при измерении
скорости седиментации полиме-
ра (а — кривые распределения
концентрации полимера в кюве-
те; б — непосредственно изме-
ренные величины — производ-
ная от концентрации полимера
по координате).
щим полимером. Грани-
ца будет постепенно раз-
мываться и вместе с тем
продвигаться по напра-
влению к дну кюветы.
Определяемая экспери-
ментально кривая del dr
будет выглядеть как
пик, равномерно движущийся в направлении от цент-
ра к периферии ротора. Измерение скорости движе-
ния этого пика drK3KQ Jdt—v позволяет определить кон-
станту седиментации раствора б1:
S = р/<о2т ==
drMaKc
dt ‘
<o2r
Константа седиментации имеет размерность времени и
величину порядка 10-1’ сек. Эта единица наз. с в е fl-
бе р г о м. Константа седиментации связана с мол.
весом ур-нием:
S = M(1 — Vq)IJN
где / — коэфф, поступательного трения макромолеку-
лы о среду, N — число Авогадро. Так как / зависит
от мол. веса, то метод в таком виде не является абсо-
лютным. Для перехода от S к М необходима калиб-
ровка прибора или независимое измерение величины /.
Последнее можно сделать, измеряя коэфф, диффузии
D макромолекул в растворе. Коэфф, диффузии свя-
зан с коэфф, трения / соотношением Эйнштейна /=
=kT/D, где к—константа Больцмана. Измерив в двух
независимых экспериментах константу седиментации
S и константу диффузии D, находят мол. вес полимера
вполне строго и точно.
Пользуясь ультрацентрифугой, можно получить
в одном эксперименте всю функцию распределения
данного полимера если предварительно прове-
сти калибровку. Для этого полидисперсный образец
полимера фракционируют на ряд фракций с относи-
тельно узким молекулярно-весовым распределением
в каждой, пользуясь фракционным осаждением, к-рое
проводят несколько раз. Наряду с фракционным осаж-
дением применяют и другие методы. Для фракциони-
рования очень малых количеств полимера удобно по-
степенное фракционное растворение в растворителе,
297 МОЛЕКУЛЯРНЫЙ ВЕС ВЫСОКОМОЛЕКУЛЯРНЫХ СОЕДИНЕНИЙ —МОЛИБДАТЫ 298
состоящем из смеси хорошего растворителя и осади-
теля, состав к-рой постепенно изменяется. Еще один
метод основан на экстракции. Как показывает тео-
рия Брепстеда, коэфф, распределения между фазами
экспоненциально зависит от мол. веса. Проводя эк-
стракцию многократно, можно полимер разбить на
достаточно узкие фракции. Для фракционирования
полимеров с успехом применяется хроматография,
пользуясь к-рой можно получить очень узкие фрак-
ции.
Каждая узкая фракция может быть изучена в цент-
рифуге, а константа / определена измерением диффу-
зии. Определив для каждой фракции мол. в. М/,
находят функциональную зависимость Si=Sj(Mi),
строго справедливую для данного полимера в дан-
ном растворителе. Для того чтобы получить правиль-
ные результаты, необходимо: 1) проводить измерения
по возможности в разб. р-рах полимеров и 2) исклю-
чить межмолекулярное взаимодействие, работая в
0-растворителе или, по крайней мере, приближаясь к
нему насколько удастся. После предварительной ка-
либровки для данного полимера можно в одном экс-
перименте определить всю функцию распределения
для полидисперсного образца.
Проделав седиментационный опыт в ультрацентри-
фуге, получают экспериментальную кривую, к-рая
размыта вследствие полидисперсности образца. Эта
кривая есть весовая функция распределения, т. е.
q)w(6’) = rfc/d<S, где с—весовая концентрация полиме-
ра. От с помощью найденной предварительно
связи между константой седиментации и мол. весом
легко перейти к функции распределения по мол. весам:
, Я(Г. de de dS . dS
)= dM~ds'’ dM'==<P“’^dM
Рис. 5. Конструкции
простейших вискози-
метров.
Для получения МБР можно поступать иначе. По
известным количествам и характеристикам фракций
можно реконструировать МБР. Мол. веса отдельных
фракций могут быть измерены не только в центрифуге,
но и осмометрически, по светорассеянию или по вяз-
кости. Метод надежен только при условии, что фрак-
ции узкие и их МБР мало перекрываются. В этом от-
ношении лучший метод фракционирования — хрома-
тографический.
Вискозиметрический метод. Этот
метод прост экспериментально, но не является абсо-
лютным, для каждой системы
полимер-растворитель он нуж-
дается в калибровке. Наиболее
часто применяемый вискозиметр
Оствальда служит для опреде-
ления времени протекания за-
данного объема жидкости через
капилляр (рис. 5). Вискозиметр
калибруют с помощью чистого
растворителя. Очень важно, что-
бы в нем условия течения были
далеки от начала турбулентно-
сти: число Рейнольдса Re
=рф/т|<<;2000, гдер—линейная
скорость жидкости, d — диаметр
капилляра, q — плотность жид-
кости, т| — вязкость. При вис-
козиметрия. определениях уста-
навливают зависимость вязкости
р-ра от концентрации при низ-
вычисляют экстраполяцией т. наз.
ких ее значениях и
характеристич. вязкость:
[ц] = Ит [±(П/По-1)]^О
где т| — вязкость р-ра, т|0— вязкость растворителя.
Величина [т|] непосредственно связана с мол. весом.
Штаудингер первым обратил внимание на эту зави-
симость и нашел, что для производных целлюлозы она
линейна, т.е. [т|]=А -М. Однако результаты измерений
в широком диапазоне мол. весов для разных полимеров
показали, что эта связь более сложная. При рассмот-
рении зависимости вязкости от мол. веса следует учи-
тывать особенности поведения макромолекул в р-ре.
Действительно, макромолекулы линейных полимеров
представляют собой статистич. клубки, содержащие
значительные количества растворителя. Для диамет-
ра (среднего) клубка статистич. физика дает выра-
жение (в случае в-растворителя):
d2_a2Z = a2M'M0
где а — длина отдельного звена цепи, Z — степень
полимеризации, М — мол. вес, Мо — мол. вес звена
цепи. С другой стороны, вязкость взвеси шариков оп-
ределяется законом Эйнштейна т|/т|0 = 1 + 2,5 be/М,
откуда [т|]=2,56/М, где Ъ — объем, занимаемый клуб-
ком. Так как каждый клубок движется как одно це-
лое с заполняющим его растворителем, то
Ь = 4- 63 = -^г М*11
6 бм’/2
откуда
[п] =0,4 М12 = АЛ/0’5
^0
В 0-точке характеристич. вязкость пропорциональна
корню квадратному из мол. веса. Это правило подтвер-
ждается опытом. Если несколько отступить от 0-точ-
ки, то [т|]=АЛР, гдеа=0,6—0,8. Обе константы А и а
определяют эмпирически для данной системы полимер-
растворитель. Зная этот так наз. закон вязко-
сти, можно пользоваться измерением величины [р]
для характеристики мол. веса. Для полидисперсного
образца снова получается среднестатистич. характери-
стика — средневязкостный мол. вес). Закон вяз-
кости зависит от взаимодействия макромолекул с рас-
творителем, а также от жесткости цепи полимера.
У абсолютно жестких палочек, напр. у синтетич. по-
липептидов, теория предсказывает, что [т|]=АМ1’7;
эта зависимость хорошо подтверждается опытом. Су-
ществует ряд полимеров с цепями промежуточной
жесткости, для к-рых 0,5<а<1,7. Напр., у эфиров
целлюлозы а=0,9—1,1.
Лит.: Flory Р. J., Principles of polymer chemistry,
N. Y., 1953; Цянь Ж э н ь-ю а н ь, Определение молеку-
лярных весов полимеров, пер. скит., М., 1962; Методы исследо-
вания полимеров, пер. с англ., под ред. А. Н. Праведникова,
М., 1961; Френкель С. Я., Усп. физ. наук, 1954, 53,
№ 2, 161; Успехи химии и технологии полимеров, сО. 3, М.,
1960; Бильмейер Ф., Введение в химию и технологию
полимеров, пер. с англ., М., 1958. С. Е. Бреслер.
МОЛЕКУЛЯРНЫХ ОРБИТ МЕТОД — см. Кван-
товая химия.
МОЛИБДАТЫ — соли молибденовых кислот. Из-
вестно несколько типов М.: мономолибдаты, или нор-
мальные молибдаты (Н2МОО4, В — одновалентный
металл), — соли Н2МоО4; изополимолибдаты (zB2O-
уМоОз, где х<у) — соли не выделенных в свободном
состоянии изополикислот молибдена; аквополимолиб-
даты (гВгО-уМоОз-зНгО, где х<у) — соли акво-
поликислот молибдена; гетерополимолибдаты — соли
гетерополикислот молибдена (см. Молибдена окислы
и кислоты).
М ономолибдаты щелочных металлов полу-
чают сплавлением стехиометрич. количеств щелочи
и МоО, или растворением МоО, в избытке щелочного
р-ра; мономолибдаты др. металлов — при длительном
нагревании смеси соответствующих окислов стехио-
метрич. состава при 450—700° или осаждением из
р-ров щелочных мономолибдатов путем добавления
солей соответствующих металлов. Мономолибдаты
щелочных металлов, аммония, Mg, Be, Т1 хорошо
299
МОЛИБДАТЫ —МОЛИБДЕН
300
растворимы в воде, другие мономолибдаты очень
мало растворимы. Мономолибдат натрия Na2MoO4—
бесцветные кристаллы, плотн. 3,28(18°), т. пл. 687°.
Растворимость вводе(вес. %): 30,65 (0°), 45,57 (100°).
Из растворов выше 10° кристаллизуется Na2MoO4-
• 2Н2О, ниже 10°—Na2MoO4-10H2O. Мономолибдат
аммония (NH4)2MoO4 устойчив в водных р-рах, со-
держащих избыток NH3; может быть выделен в виде
белых моноклинных призм при создании давления
NH3 над раствором; плотн. 2,27. Легко теряет NH3
при комнатной темп-ре, превращаясь в полимолибдат
(см. ниже).
Среди нерастворимых мономолибдатов практич.
интерес представляют молибдаты Са, Си, Zn, Fe, Pb.
Их свойства приведены в таблице. СаМоО4 и РЬМоО4
встречаются в природе в виде минералов повеллита
и вульфенита.
Молибдат Цвет Плотн., 0/СЛ13 Т.пл., "С Растворимость в воде, г'л Теплота обра- зования -AW°jeg, ккал/моль
20° 100°
СаМоО< СиМоО* белый желто- 4,38-4,53 1520 0,0058 0,02 369,9
зеленый —- 820 0,162 1 ,61 225,7
ZnMoO. розоватый 700 1,57 1,94 272,4
FeMoO< темно-сеоый 850 0,034 0,16 258,1
PbMoO. белый 6,7-7,0 1060— 1070 0,08 — 249 + 0,9
С р-рами аммиака СаМоО4 и РЬМоО4 практически
не взаимодействуют, СиМоО4 и ZnMoO4 образуют
растворимые комплексы типа [Me(NH3)4](OH)2, а
FeMoO4 медленно реагирует с переходом Мо в рас-
твор. В кислотах указанные М. растворяются, раство-
рами соды и щелочей разлагаются с образованием
растворимого щелочного М.
Изополимолибдаты образуются при
сплавлении мономолибдатов с различным количеством
МоО3. Известны ди-, три- и тетрамолибдаты, напр.
Na2Mo2O7, К2Мо3О10, Li2Mo4Oi3 (о строении см.
И зополисоединения).
Аквополимолибдаты получают нейт-
рализацией до определенных значений pH водных
р-ров мономолибдатов (см. Аквополисоединения). Из-
вестны ди-, пара-, три-, тетра (мета)-, окта-, дека- и
16-молибдаты, pH образования к-рых изменяются
соответственно от 6,1 до 0,9. Практич. значение имеют
описанные ниже аквополимолибдаты натрия и аммо-
ния. Парамолибдат натрия 5Na2O •
•12MoO3-zH2O (по др. данным, 3Na2O-7MoO3.zH2O)
выделяется из р-ров при рН=“5 в виде белых моно-
клинных призм. В зависимости от темп-ры соль кри-
сталлизуется с 7, 22, 36 и 38 мол. Н2О. Раствори-
мость в воде 46,9% (на безводную соль) при 24°.
Парамолибдат аммония 3(NH4)2O.7MoOs-
• zH2O выделяется на холоду при pH“5 с 11 молеку-
лами Н2О, а при выпаривании с 4 Н2О в виде бесцвет-
ных моноклинных призм. Растворимость (в г/л) ок.
300 (20°) и ок. 500 (80—90°) (для соли z=4). При 150°
парамолибдат разлагается с выделением NH3, превра-
щаясь в тетрамолибдат (NH4)2O-4MoO3, при 350°
удаляется весь NH3. Метамолибдаты R2Mo4OI3-zH2O
или H6H4[H2(Mo2O7)6]-zH2O (В — одновалентный ме-
талл) образуются при подкислении р-ров щелочных
молибдатов до pH— 3,2—3,5. Особенностью метасолей
является хорошая растворимость в воде (в т. ч. мета-
молибдатов Fe, Са, Си, Pb, Zn).
Г етерополимолибдаты образуются при
подкислении р-ров молибдатов, содержащих фосфат-,
силикат-, арсенат- и ряд других ионов. Практич.
значение имеет фосфоромолибдат аммония (NH4)3PO4-
• 12МоО3-6Н2О или (NH4)3-H4[P(Mo2O7)e]-4H2O (см.
Гетерополисоединения).
Молибдат кальция используется для легирования
сталей; молибдат натрия — в произ-ве лаков и кра-
сок. Парамолибдат аммония применяют для произ-ва
трехокиси молибдена; в аналитич. химии — для оп-
ределения РО’", SiO4_ и др. См. также Аммония
фосфомолибдат.
Лит.: К I 1 1 е f f е г D. Н., Linz A., Molybdenum com-
pounds, N. Y.—L., 1952; Зеликман A. H., Беляев-
ская Л. В.. Ж. прикл. химии, 1954, 27, № .11, 1151; 1956,
29, № 1, 11; Зеликман А.Н., Просенкова Т. Е.,
Ж. неорг. химии, 1961, 6, вып. 1. Л. В. Беляевская.
МОЛИБДЕН (Molybdaenum) Мо—химич. эле-
мент VI гр. периодич. системы Менделеева, п. н. 42,
ат. в. 95,94. В природном М. семь стабильных изото-
пов с массовыми числами: 92 (15,86%), 94 (9,12%),
95 (15,70%),. 96 (16,50%), 97 (9,45%), 98 (23,75%),
100 (9,62%). Сечение захвата тепловых нейтронов ато-
мом М. 2,4 барн. Из искусственных радиоактивных
изотопов М. в качестве радиоак-
тивных индикаторов используют
Мо93 (7’1/г=6,95 час) и Мо" (7%=
=66 час). Конфигурация внешних
электронов атомаМ. 4d55s1. Энер-
гии ионизации (в ав): Мо°-+-Мо+->-
->Мо2+-+-Мо’+-г- Мо4+ -+- Mos + ->
Мо9+-г- соответственно равны:
7,10; 16,15; 27,13; 40,53; 55,6; 71,7.
М. открыт в 1778 Шееле, к-рый
при обработке минерала молибде-
нита азотной к-той выделил молиб-
деновую к-ту и получил нек-рые ее соли. Металлич.
М. впервые получен в 1782 Гьельмом восстановлением
МоО2 углеродом. М.—мало распространенный элемент.
Среднее содержание его в земной коре 3-IO-4 вес. %.
Известно ок. 20 минералов М. Важнейший из них— мо-
либденит MoS2, являющийся единственным первич-
ным минералом. Остальные минералы вторичного
происхождения. Среди них промышленное значение
имеют повеллит СаМоО4, молибдит Fe2(MoO4)3-иН2О и
вульфенит РЬМоО4.
М олибденит MoS2 — основной промышлен-
ный сырьевой источник М. Это мягкий свинцово-се-
рый минерал с металлич. блеском. Кристаллич. ре-
шетка гексагональная, слоистого типа, плотн. 4,7—
4,8, твердость по минералогия, шкале 1. Важнейшие
месторождения молибденита связаны с гидротермаль-
ными образованиями. Особенно широко распростра-
нены месторождения в кварцевых жилах и окварцо-
ванных породах. В рудах молибденит ассоциируется
с минералами: шеелитом, вольфрамитом, касситери-
том, сульфидами меди и железа, иногда бериллом.
Часто молибдениты содержат изоморфную примесь
редкого металла рения (от 0,04 до 0,0001 %). Из молиб-
денитовых руд, содержащих обычно 0,1—1% М.,
флотацией получают концентраты, содержащие 85—
90% MoS2. Кроме собственно молибденовых руд, ис-
точниками М. служат нек-рые медные и медно-свин-
цово-цинковые руды. Крупные молибденовые место-
рождения находятся в западной части США, Мексике,
Чили, Канаде, Норвегии, Австралии. Ряд крупных
месторождений имеется в СССР.
Физические и химические свойства. М.— светло-
серый металл; кристаллизуется в кубич. объемноцент-
рированной решетке с периодом а=3,14 А. Атомный
радиус 1,40 А (для координационного числа 12); ион-
ные радиусы Мо4+ 0,68 А, Мо6 + 0,62 А; плотн. 10,2;
т. пл. 2620±10°; т. кип. ок. 4800°; теплота плавления
50 кал/г. Давление пара над твердым М. при различных
темп-рах (в мм рт. ст.): 2,40-10-8 (1627°), 1,88-10_’
(1727°), 6,35- 10-е(1927°),1,16• 10-* (2127°);теплота суб-
лимации 1620кал/г. Уд. теплоемкость 0,065 кал/г-град
(20—100°), теплопроводность 0,35 кал! см-сек-град
(20°), термич. коэфф, линейного расширения (5,8—
6,2)-10-6 (25—700°), удельное электросопротивление
301
МОЛИБДЕН
302
4,8-10-6 ом-см (25°), термич. коэфф, электросопро-
тивления 0,00479 (20—2600°). Мощность излучаемой
энергии поверхностью М. при высоких температу-
рах (в вт/см2): 0,55 (730°), 6,3 (1330°), 19,2 (1730°),
70 (2330°). Работа выхода электронов 4,37 эв. М.
парамагнитен. Атомная магнитная восприимчивость
~90-10-6(20°).
Механич. свойства М. в большой степени зависят
от предшествующей обработки. Так, твердость по
Бринеллю равна 150—160 кГ/мм2 (для спеченного
штабика), 200—230 кГ/мм2 (для кованого штабика)
и 140—185 кГ/мм2 (для отожженной проволоки);
предел прочности при растяжении 80—120 кГ/мм2
(для отожженной проволоки) и 140—260 кГ/мм2 (для
неотожженной проволоки в зависимости от диаметра).
Модуль упругости М. 28500—30000 кГ/мм2. М. более
пластичен, чем вольфрам. В отличие от вольфрама,
рекристаллизующий отжиг М. не приводит к его хруп-
кости.
На воздухе при обычной темп-ре М. устойчив.
Начало окисления (цвета побежалости) наблюдается
при 400°. Выше 600° металл быстро окисляется с об-
разованием высшего окисла МоО3. Пары воды при
700° интенсивно окисляют М. до двуокиси МоОз.
С водородом М. химически не реагирует вплоть до
плавления, однако при нагревании в водороде наб-
людается его поглощение с образованием твердого
р-ра (при 1000° поглощается 0,5 см3 Н« в 100 г Мо).
При 1500° с азотом М. образует нитрид (вероятно
M02N). Твердый углерод и углеводороды, а также
окись углерода при 1100—1200° взаимодействуют с ме-
таллом с образованием карбида М02С (плавится с раз-
ложением при 2400°, см. Карбиды). Если реакция с СО
проводится при 200° и 200—250 атм, образуется кар-
бонил Мо(СО)6. Карбонил сублимирует при 30—
40° и разлагается в интервале темп-р 150—400°. С наи-
большим выходом Мо(СО)6 получается при взаимодей-
ствии хлорида или оксихлорида молибдена с СО
в присутствии порошка А1 в абсолютном эфире при
70—100° и 145—220 атм. Двуокись углерода окис-
ляет М. при 1200°. Фтор действует па М. при обычной
темп-ре, хлор — выше 250°, причем образуются лету-
чие галогениды MoF6 и МоС15 (см. Молибдена гало-
гениды). Бром взаимодействует с М. при нагревании,
пары иода с М. не реагируют.
При действии паров серы (при 440°) и сероводорода
(выше 800°) образуется дисульфид M0S2 (см. Молиб-
дена сульфиды). Сернистый газ при 700—800° окис-
ляет М. Выше 1200° М. реагирует с кремнием, обра-
зуя силицид M0S12, обладающий высокой устой-
чивостью на воздухе вплоть до темп-р 1500—1600°; его
микротвердость 1410 кГ/мм2. При комнатной темп-ре
М. устойчив в соляной и серной к-тах, но несколько
растворяется в них при 80—100°. Азотная к-та и цар-
ская водка медленно растворяют металл на холоду,
быстро — при нагревании. Хорошим растворителем
М. служит смесь из 5 объемов HNO3, 3 объемов H2SO4
и 2 объемов воды (вольфрам в этой смеси не раство-
ряется). В холодных р-рах щелочей М. устойчив, но
несколько корродирует при нагревании.
Для М. наиболее характерны 6-валентпые соеди-
нения. Известны также соединения, отвечающие ва-
лентности + 5, —(-4, —|—3 и 4-2. Среди кислородных со-
единений высшей валентности важнейшие: молибде-
новый ангидрид МоОз (белый с зеленоватым оттенком),
молибденовая к-та Н2МОО4 и ее соли — молибдаты
(см. Молибдена окислы и кислоты). Все средние молиб-
даты, за исключением молибдатов щелочных металлов
и магния, мало растворимы в воде. Молибденовая к-та
склонна к образованию высокомолекулярных изо-
поли- и аквополикислот и их солей, а также гетеро-
поликислот с кремневой, фосфорной и мышьяковой
к-тами (см. Гетерополисоединения). Среди ряда окис-
лов низших валентностей важнейшие — двуокись
МоОз (темно-коричневый) и промежуточный окисел
МО4О11 (сине-фиолетовый). При действии восстанови-
телей (SO2, H2S, Zn и др.) на молибденовую к-ту или
кислые р-ры молибдатов образуется молибденовая
синь. Ее состав близко отвечает формуле Мо5О14.
Аналитическое определение. Для
качественного открытия М. большей частью приме-
няют след, реакции: 1) образование окрашенного
в красный цвет комплексного соединения Мо5 + с ро-
данидом калия или аммония; для восстановления Мо’+
до Mos + служат SnCl2, тиомочевина и др.; 2) образова-
ние красно-фиолетового комплексного соединения
Мо6 + с этилксантогенатом в слабокислом р-ре. Для
количественного определения М. в рудах и рудных
концентратах навеску материала разлагают сплавле-
нием с NaOH или ХазОз с последующим выщелачи-
ванием сплава водой или применяют обработку HNO3,
отделяя затем железо от М. осаждением аммиаком.
При малых содержаниях М. (до десятых долей про-
цента) обычно применяют колориметрий, роданидный
метод определения. Для устранения влияния примеси
вольфрама в раствор добавляют винную к-ту. Боль-
шие количества М. (напр., в рудных концентратах,
солях, сплавах) определяют весовым методом, осно-
ванным на осаждении молибдата свинца в уксусно-
кислом р-ре. Кроме того, применяют весовое определе-
ние в виде трисульфида М., осаждаемого сероводоро-
дом из слабокислого р-ра. Иногда используют объем-
ный метод определениям., к-рый состоит в восстанов-
лении Мо8+ цинком или амальгамами и последующем
титровании соединения М. низшей валентности пер-
манганатом.
Получение. Основным сырьем для производства М.,
его сплавов и соединений служат стандартные молибде-
нитовые концентраты, содержащие 47—50% Мо, 28—
32% S, 3—5% SiO2 и примеси минералов Fe, Си, Са и
других элементов. Первоначально концентрат подвер-
гают окислительному обжигу при 550—600° в много-
подовых печах или в печах кипящего слоя. Продукт
обжига — огарок — содержит МоО3, загрязненную
примесями. Чистую трехокись М., необходимую для
производства металлич. М., получают из огарка двумя
путями: 1) возгонкой МоОз при 950—1000°; 2) химич.
методом, к-рый состоит в том, что огарок выщелачи-
вают аммиачной водой, извлекая М. в раствор. Из
р-ра молибдата аммония (после очистки его от при-
месей Си, Fe) выделяют полимолибдаты аммония (б. ч.
парамолибдат 3 (NHJ2O ^МоОз-пНзО) методом нейт-
рализации или выпарки с последующей кристаллиза-
цией. Прокаливанием парамолибдата аммония при
450—570° получают чистую МоО3, содержащую не
более 0,05% суммы примесей.
Металлич. М. получают первоначально в форме по-
рошка восстановлением МоО3 в токе сухого водорода.
Процесс проводят в трубчатых печах в две стадии:
первая при 550—600°, вторая — при 950—1000°.
Молибденовый порошок превращают в компактный ме-
талл металлокерамич. методом или методом дуговой
плавки. Металлокерамич. метод служит для получе-
ния сравнительно небольших заготовок (сечением 2—
9 см2 при длине 450—600 мм). Порошок М. прессуют
в стальных прессформах под давлением 3 т/см2. По-
сле предварительного спекания при 1000—1200° в ат-
мосфере водорода заготовки (штабики) подвергают вы-
сокотемпературному спеканию при 2200—2400°. Спе-
ченный штабик затем поступает на механич. обработку
давлением (ковка, протяжка,прокатка). Для получе-
ния крупных заготовок весом до 2000 кг применяют
дуговую плавку в печах с охлаждаемым медным тиг-
лем и расходуемым электродом, в качестве к-рого слу-
жит пакет спеченныхштабиков. Для произ-ва ферро-
молибдена (сплава 50—70% Мо, остальное Fe),
303
МОЛИБДЕНА ГАЛОГЕНИДЫ
304
служащего для введения присадок М. в сталь, приме-
няют метод восстановления обожженного молибдени-
тового концентрата (огарка) ферросилицием в при-
сутствии FSO„.
Применение. Начало широкого применения М. от-
носится к первому десятилетию 20 в., когда было от-
крыто влияние добавок М. на свойства сталей. В на-
стоящее время 75—80% производимого М. исполь-
зуется в произ-ве легированных сталей (см. Железа
сплавы). Остальное количество применяется в форме
чистого металла, сплавов с цветными и редкими метал-
лами (см. Молибдена сплавы), а также в виде химич.
соединений.
Металлич. М.— важнейший конструкционный ма-
териал в произ-ве электроосветительных ламп и элект-
ровакуумных приборов (радиолампы, генераторные
лампы, рентгеновские трубки и др.), где из молибдено-
вой проволоки, ленты и прутков изготовляют различ-
ные детали: аноды, сетки, катоды, вводы тока, держа-
тели нити накала в электролампах. Молибденовая про-
волока и лента используются в качестве нагревателей
для высокотемпературных печей. В последние годы,
в связи с освоением произ-ва крупных заготовок, М.
стали применять (в чистом виде или с легирующими
добавками др. металлов) в тех случаях, где необхо-
димо сохранение прочности при высоких темп-рах,
напр., для изготовления лопаток турбин и других де-
талей реактивных двигателей. Для предохранения М.
от окисления при высоких темп-рах применяют пок-
рытия деталей силицидом М. и другие способы защиты.
М. применяют также как конструкционный материал
в энергетич. ядерных реакторах, т. к. он имеет срав-
нительно малое сечение захвата тепловых нейтронов.
Важную роль М. играет в ряде жаропрочных и кисло-
тоустойчивых сплавов, в к-рых он сочетается гл. обр.
с никелем, кобальтом и хромом (см. Жаропрочность,
Химически стойкие материалы). В технике исполь-
зуются нек-рые химич. соединения М. Так, M0S2 при-
меняют как смазочный материал для трущихся частей
механизмов; молибдат натрия — в произ-ве лаков и
красок; окислы использут как катализаторы в химич.
и нефтяной пром-сти. В последние годы стали приме-
нять растворимые молибдаты в качестве микроудобре-
ний, стимулирующих рост нек-рых с.-х. культур.
Лит.: Зеликмав А. Н., Металлургия вольфрама и
молибдена, М., 1949; М е ер с он Г. А., ЗеликманА. Н.,
Металлургия редких металлов, М., 1955; Молибден, пер.
с англ., М., 1959; Молибден. Сб. статей, пер. с англ., М., 1962;
К 1 1 1 е f I е г D. Н., Linz A., Molybdenum compounds,
N. Y., 1952; G m e 11 n, Syst.-Nummer, 53, Molybdan, B.,
1935; Mellor, v. 11, L —N. Y—Toronto, 1948; то же,
v. 11, 1954;U liman n, 3 Aufl., Bd 12, Munchen — B., 1960,
S. 555—572; Pascal, t. 14, P., 1959; К 1 г к, v. 9, N. Y.,
1952, p. 191—214; Rare metals handbook, ed. C. A. Hampel,
2 ed., L., 1961. A. H. Зеликман.
МОЛИБДЕНА ГАЛОГЕНИДЫ — ряд галогенидов
и оксигалогенидов, в к-рых молибден проявляет ва-
лентности от +2 до +6. Устойчивость галогенидов
уменьшается в ряду F-(-Cl->-Br->-J.
С фтором молибден образует фториды MoF6
и MoF3 (возможно и MoF4) и оксифториды MoOF4,
MOO2F2, MoOF«4H2O. Известны комплексные соеди-
нения, производные от MoF3, такие как NH4[MoF4]-
•Н2О, (NH4)3[Mo2F91, K3[Mo2F9]-2H2O, а также ряд
фторомолибдатов (производные молибдатов). При дей-
ствии фтора на порошкообразный металл образуется
гексафторид MoFe— белоснежные кристаллы,
т. пл. 17,5°, т. кип. 35°; плотность жидкого MoFe
изменяется от 2,55 при 17,5° до 2,47 при 34°. Теплота
образования Д/7298=^ —450 ккал/моль. Трифторид
MoF3 — темно-розовые кристаллы; при нагревании на
воздухе образует МоО3 и HF, в отсутствии воздуха не
плавится и не сублимирует до 800°; незначительно
растворим в воде, не разлагается кислотами. MoF3
получен действием HF на МоВт3 при 600°.Оксифториды
образуются при гидролизе MoFe или при галогенном
обмене между MoF, и соответствующим оксихлоридом.
С хлором молибден образует хлориды МоС15,
M0CI4, МоС13, МоС1г, оксихлориды МоОгСЬ, МоОС12,
M00CI4, гидраты оксихлоридов МоОС1.4НгО, МоО2С1-
• НгО. Известны комплексные соединения, производ-
ные от МоС13 и M0CI2 такие, как KfMoCh], К3[МоС16|
и др. При действии хлора на металлич. порошок Мо,
а также при хлорировании МоО3 избытком СС14 об-
разуется пентахлорид МоС15 — фиолетово-
черные кристаллы, т. пл. 194°, т. кип. 268°, плотн.
2,928. Теплота образования ДЯ°298= —126,5 ±
±2,2 ккал/моль. Низшие хлориды молибдена могут
быть получены восстановлением МоС15 водородом. Те-
трахлорид МоС14 (коричневого цвета) устойчив до
130°, выше разлагается (до плавления): 2МоС142г
д?МоС13+МоС15. Т рихлорид МоС13 (красно-бурого
цвета) устойчив до 500°, выше диспропорционирует
в основном по ур-нию: 2МоС13^МоС12+МоС14. Д и-
хлорид МоС12 (желтого цвета) диспропорционирует
выше 700°: 2МоС12;гМо+МоС14. Имеются данные о том,
что дихлорид молибдена является гексамером Мо6С112;
известны его производные, в частности Мо6С18Вг4.
Хлориды молибдена растворимы в спирте, эфире и ряде
других органич. растворителей; чувствительны к вла-
ге; окисляясь, дают окси- и гидрооксихлориды. Из ок-
сихлоридов молибдена изучены след.: желтовато-бе-
лый MOO2CI2, т. пл. 170°, растворим в воде, ДЯ°298=
= — 173,О±О,7 ккал/моль; зеленый MoOCU, т. пл. 104°,
разлагается водой, 1^Н°г>л=—153,5±0,7 ккал/моль;
зеленый МоОС13, сублимирует ниже 100°, разлагается
водой; МоОгОг-НгО в виде зеленоватых игольчатых
кристаллов, т.пл. 214°, Д/7°298=—247,6 ± \ fiккал/моль.
Из соединений молибдена с бромом известны
МоВг4, МоВг3, МоВг2 и ряд оксибромидов; для МоВг3
известны производные (NH4)2[MoBr5]-H2O, К.г[МоВг5]
и др. При бромировании порошка Мо 100%-ным га-
зообразным бромом образуется МоВг4, разбавление
паров брома СО2 приводит к получению МоВг3; при
диспропорционировании МоВг3 при высокой темп-ре
образуется МоВг2.
Тетрабромид М0ВГ4 — черные игольчатые
кристаллы, легко разлагается на МоВг3 и бром; лег-
коплавок, летуч. Теплота образования ДЯ°298=
=—45 ккал/моль. Т рибромид МоВг3— черно-
зеленые игольчатые кристаллы, малолетуч. Д и б р о-
м и д МоВг2 — аморфный порошок желтого цвета,
тугоплавок. МоВгг фактически является полимером,
ему приписывают ф-лу Мо3Вг6 или Мо8Вг12. Соответ-
ственно его производные имеют ф-лы Mo3Bi4J2 или
Mo6Br8J4. В холодной воде бромиды нерастворимы,
М0ВГ4 разлагается горячей водой.
Существование соединений молибдена с иодом
ставилось под сомнение. Однако в последние годы вы-
делены M0J4, MoJ3 и M0J2- Т етраиодид M0J4
получен взаимодействием паров J2 под давлением 1 атм
с твердым M0J2 при 330°. M0J4 — черные кристаллы,
т. пл. ок. 400°; при комнатной темп-ре устойчив, при
незначительном нагревании разлагается на M0J2
и J2. Теплота образования Д77°298^г—48 ккал/моль.
Трииодид MoJs приготовлен нагреванием поро-
шка Мо с избытком иода при 300°; черные, волокнистые
кристаллы, т. пл. ок. 900°. Дииодид M0J2 об-
разуется при термич. диссоциации MoJ3, а также при
непосредственном взаимодействии Мо с парами иода
при 700—800°; коричневое аморфное вещество, т. пл.
ок. 700°. Устойчив до 550° в вакууме, на воздухе раз-
лагается с образованием МоО3 и J2. Широкого прак-
тич. применения М.г.не имеют. Описано использова-
ние MoFe в процессе разделения изотопов. MoCL
служит промежуточным продуктом при получении
карбонила молибдена, применяемого для нанесения
покрытий из газовой фазы, а также может непосредст-
305
МОЛИБДЕНА ОКИСЛЫ И КИСЛОТЫ
306
венио использоваться для этой цели. Перспективными
для нанесения покрытий Мо могут оказаться оксихло-
риды. Описано получение электролитич. покрытий мо-
либдена из К3МоС16 в расплаве солей.
Лит.: Щукарев С. А., Василькова И. В.,
Шар у пин Б. Н., Вести. ЛГУ. Сер. физ. хим., 1959,
№ 4, вып. 1; 1960, № 10, вып. 2; Ж. общ. химии, 1956, 26, вып.8,
2093; Killeffer D. Н., Linz A., Molybdenum compo-
unds, N. Y.—L., 1952; Pascal, t. 14, P„ 1959; P ы с с И. Г.,
Химия фтора и неорганических соединений, М., 1956; В о 1-
sten R. F., Iodide metals and metal iodides, N. Y.—L.,
1961; Lewis J. [a. o.], Chem. Ind., 1960, № 10, 259.
МОЛИБДЕНА ОКИСЛЫ И КИСЛОТЫ — кисло-
родсодержащие соединения молибдена. Окислы мо-
либдена существуют в виде определенных фаз, состав
к-рых изменяется в нек-рой узкой области. Наиболее
устойчивы МоО3 (т. наз. a-фаза) и МоОг (б-фаза).
Окислы промежуточного состава: М04ОЦ (у-фаза),
Мо8Огз (0-фаза) и Мо9О2в (0'-фаза), Мо17О47 (х-фаза)
и МоО2 8() (v-фаза), термически неустойчивы и выше
700° разлагаются с образованием МоОг и М0О3.
Трехокись молибдена МоО3 — бес-
цветные кристаллы с зеленоватым оттенком, ортором-
бич. решетка слоистого типа с параметрами: а=3,9А,
6=13,8А, с=3,7А; плотн. 4,69, т.пл. 795°, т.кип. 1155°.
Выше 650° МоОз заметно сублимирует. Теплота об-
разования Д//°298=—178,2-|-1,5 ккал/моль. В газо-
вой фазе молекулы МоОз полимеризованы — при
900°—1000° (МоО3)3. Трехокись слабо парамагнитна,
в твердом состоянии является диэлектриком, в рас-
плавленном — хорошо проводит электрич. ток. Рас-
творимость МоОз в воде, по данным разных авторов,
колеблется от 0,4 до 2 г/л при комнатной темп-ре. МоОз
растворяется в водных р-рах щелочей и аммиака с об-
разованием молибдатов, а также в р-рах минеральных
к-т, причем поведение МоОз при растворении ана-
логично молибденовой к-те (см. ниже). МоОз восста-
навливается водородом и углеродом до металла при
700—1000°. Получается прокаливанием молибдено-
вой к-ты или парамолибдата аммония при 450—500°
или нагреванием металлич. Мо на воздухе выше 400°.
Промежуточный окисел Мо40п (область
гомогенности от Мо02,5, что соответствует Мо4Оп,
до Мо02,0) — ромбич. кристаллы сине-фиолетового
цвета: а=24,4 А; 6=5,45 А; с=6,7 А; плотн. 4,15;
обладает полупроводниковыми свойствами. М04О11
практически нерастворим в воде, мало растворим в сер-
ной или соляной к-тах, а также в разб. р-рах щело-
чей. Азотная к-та окисляет МО4О11 до МоОз- Полу-
чают М04О11 осторожным окислением Мо02 или вос-
становлением МоОз водородом, содержащим пары
НгО, а также длительным прокаливанием в инерт-
ной среде при 500—600° 1 моля МоОг с 3 молями
М0О3.
Двуокись молибдена М0О2 (область го-
могенности от Мо02 8() до МоОг) — темно-коричневые
моноклинные кристаллы с .лиловым оттенком; а—
=5,61 А; 6=4, 84 А; с=5,53 А; 0=119, 6°; плотн. 6,47.
Теплота образования ДЯ°298= —140,8 ккал/моль. Вы-
ше 1000° М0О2 частично сублимирует без разложения,
однако большая часть ее диспропорционирует на газо-'
образную МоОз и твердый Мо. Двуокись молибдена —
проводник электрич. тока. Не растворяется в воде,
водных р-рах щелочей, НС1, H2SO4. Азотная к-та окис-
ляет МоОг до МоО3. Выше 700° М0О2 восстанавливает-
ся водородом и углеродом до металла. МоОг может
быть получена восстановлением водородом МоО3
при 450—470° или прокаливанием в инертной атмосфе-
ре, выше 400° стехиометрич. смеси порошкообразного
Мо с М0О3.
Трехокись молибдена образует простые кис-
лоты молибдена — моногидрат Н2М0О4,
дигидрат Н2Мо04-Нг0 и аквополикислоты—гНгО-
•уМоОз-гНгО (гдеу>г). Крометого, известно несколь-
ко надаислот—НгМоОг (х—от 5 до 8) и комплексных
гетерополикислот с Р, As, Si, В и рядом др. элементов
в роли центрального атома.
Моногидрат Н2Мо04 — белые игольчатые
кристаллы, существуют в двух модификациях. Ни-
же 60° кристаллизуется (Х-Н2М0О4, выше 60°—
0-Н2МОО4. Полное обезвоживание (Х-Н2М0О4 про-
исходит только при 220°, 0-Н2МОО4—при 115°.
Растворимость Н2М0О4 в воде при 25° ок. 2,6 г/л
(по МоОз). Моногидрат выделяется при длительном
стоянии сильно подкисленного горячей HNO3 р-ра
молибдата или при разложении горячими к-тами не-
растворимых молибдатов. Дигидрат НгМо04- НгО—
желтые моноклинные кристаллы; плотн. 3,124; рас-
творимость в воде при 18° ок. 1,1г/л (по МоОз). Ди-
гидрат выделяется при длительном стоянии р-ров мо-
либдатов, подкисленных при комнатной темп-ре. При
нагревании водной суспензии дигидрата до 60—70°
последний переходит в моногидрат.
В щелочных р-рах молибденовой к-ты существует
анион Мо04 при любых концентрациях Мо. В сильно
разб. р-рах (конц. Мо<0,0003 моль/л) при всех значе-
ниях pH существует также анион Мо04~. В более конц.
р-рах при pH от 6,0 до 0,9 образуются анионы акво-
поликислотМозО*7> HMosOjj , НзМоцО’, , Н,Мо240,8 ,
Н9Мо24О7~, в еще более кислых р-рах молибден нахо-
дится в виде катиона (возможно Мо09+). При действии
перекиси водорода на р-р молибденовой к-ты образует-
ся ряд надмолибденовых к-т НгМоОг (х—от 5 до 8).
Для молибденовой к-ты характерно образование ге-
терополикислот с кремневой, фосфорной, мышья-
ковой и рядом других кислот, напр. Н7[Р(Мог07)в]
(см. Гетерополисоединения).
Описаны гидроокиси молибдена низших валентно-
стей Мо(ОН)3 и МоО(ОН)3, осаждающиеся из р-ров 3- и
5-валентного Мо при добавлении щелочи. Мо(0Н)3—
черный порошок; практически нерастворим в воде,
трудно растворяется в разб.серной и соляной к-тах.
МоО(ОН)3 — коричнево-черный хлопьевидный оса-
док; нерастворим в щелочах, легко растворяется в кис-
лотах. Растворимость в воде ок. 2 г/л при комнатной
темп-ре.
Молибденовая синь — соединение ярко-
синего цвета, образующееся при действии восстанови-
телей (SO2, H2S, Zn и др.) на р-ры молибденовой к-ты
или кислые р-ры молибдатов (при pH от 2 до 6). По
другим источникам, под названием молибденовая
синь объединяются различные вещества, в к-рых Мо
находится в степени окисления, промежуточной
между 5 и 6. Молибденовой сини приписывают ф-лы:
Мо802з-8Н20, М04ОЦ.Н2О, Мог05-НгО (вода консти-
туционная). Молибденовая синь находится в раство-
ре в коллоидном состоянии и легко адсорбируется
поверхностно-активными веществами, окрашивая
их в ярко-синий цвет. Из раствора может быть
выделена сильным подкислением НС1. Практически
нерастворима в 3,5—4% HCI, р-рах щелочей и аммиака.
Трехокись молибдена является основным полупро-
дуктом для получения металлич. Мо и большинства
его соединений. Кроме того, МоОз и М0О2 служат ката-
лизаторами в ряде производств. Образование солей
иадмолибденовой к-ты используется при колориметрии,
определении молибдена. На реакции образования мо-
либденовой сини основано применение ряда соединений
для окраски тканей. Эта реакция используется и в ана-
литич. практике.
Лит.: Killeffer D. Н., Linz A., Molybdenum
compounds, N. Y.—L., 1952; Glemser O.. Lutz G.,
Z. anorgan. und allgem. Chem., 1950, 263, H. 1—3, 2—14;
1956, 285, № 3—6, 173—180; Kihlborg L., Acta chem.
scand., 1959, 13, Ks 5, 954—62. Л. В. Беляевская.
307
МОЛИБДЕНА СПЛАВЫ—МОЛИБДЕНА СУЛЬФИДЫ
308
МОЛИБДЕНА СПЛАВЫ— сплавы на основе (с пре-
обладанием) молибдена. Наибольшее практич. значе-
ние имеют сплавы Мо с W, Re, Zr, Ti, V, Се и La,
а также углеродом. Пределом, ограничивающим со-
держание легирующих добавок, служит способность
М. с. к пластич. деформации. Темп-ра перехода Мо
из вязкого в хрупкое состояние зависит от содер-
жания в нем примесей внедрения (в первую очередь
кислорода) (см. таблицу).
Марка молибдена Содержание кислорода, % Темп-ра перехода в хрупкое состояние, °C
Технический 0,02 300
Очищенный 0,002 25
Особо чистый 0,0001 ниже —196
Особенно вредно залегание хрупких и легкоплавких
окислов молибдена по границам его кристаллов. Не-
легированный Мо имеет достаточно высокие механич.
свойства при длительной нагрузке выше 1000° (пре-
дел длительной прочности за 100 часов при 1100°
порядка 20 кГ/ммг). Поэтому главное назначение до-
бавок других элементов к Мо — его раскисление,
связывание кислорода и других примесей внедрения
в форму легкоудаляемых газообразных или менее вред-
ных для пластичности тугоплавких неметаллич. сое-
динений. Для этой цели используются более активные
к кислороду, чем Мо, взаимозаменяющие друг дру-
га элементы — Ti, Zr, V, Al, Hf, С (образующаяся
СО удаляется в вакууме) —в количествах менее0,5%
каждого. В последнее время показано эффективное
влияние на пластичность Мо обработки его распла-
ва редкоземельными металлами, особенно La и Се
(0,1-0,2%).
Для измельчения кристаллов литого Мо начинают
применять бор в количествах десятых долей процента.
Таким образом, основное применение находит Мо,
легированный раскислителями, избыток к-рых после
раскисления входит в твердый р-р замещения на основе
Мо. К основным методам изменения механич. свойств
легированного Мо относится наклеп и рекристалли-
зация. Предел прочности молибденового листа после
холодной прокатки составляет 108 кГ/ммг, а По-
сле отжига при 1000° он снижается до 82 кГ/мм2
при соответственном увеличении относительного уд-
линения с 5 до 13%. Легирующие элементы тем силь-
нее повышают темп-ру рекристаллизации твердого
р-ра на основе Мо, чем больше размер атома добавки
по сравнению с атомом Мо; наибольший эффект вызы-
вают В, Si, Ti, Zr.
Тугоплавкие металлы, имеющие аналогичную с Мо
объемноцентрированную кубич. решетку и немного
отличающиеся размерами атомов, образуют с ним не-
прерывные ряды твердых р-ров; сюда относятся: W,
Nb, Та, V, Ст, 0-TL.C большинством остальных метал-
лов Мо образует системы с перитектич. превращением
и образованием промежуточных фаз на основе метал-
лич. соединений (Мо3А1, С02М03, FeMo, NiMo, ZrMo2,
НегМоз). Между твердым р-ром на основе Мо и соеди-
нениями МоВег, Мо2В, М02С и MosSi образуются эв-
тектики. Дисилицид молибдена MoSi2 устойчив на
воздухе до 1700° и применяется для изготовления
нагревателей электропечей сопротивления. Исследо-
вания, проведенные по изучению влияния легирую-
щих добавок других металлов на твердость и ковкость
Мо, показали, что горячая деформируемость Мо со-
храняется при введении не свыше 1% добавок. Исклю-
чение составляют Та, Re и W, содержание к-рых в твер-
дом молибденовом р-ре допускается соответственно
10 и 50 и почти 100%. За последнее время разработаны
прочные и пластичные сплавы на основе системы
Мо—W—Re. Введение значительных количеств Re
в Мо (напр., сплав 1:1) снижает темп-ру перехода
Мо в хрупкое состояние до —196° (т. кип. жидкого
азота) за счет введения дополнительного к скольже-
нию механизма деформации путем двойникования.
Такие сплавы легко прокатываются в фольгу и про-
волоку при комнатной темп-ре и хорошо свариваются.
Заготовки и слитки легированного Мо или М.с. (иду-
щие, после обработки давлением, на различные по-
луфабрикаты) могут быть получены как методами
порошковой металлургии, так и путем вакуумной ду-
говой или электроннолучевой плавок. В самое послед-
нее время большое научное и практич. значение приоб-
рели монокристаллы Мо и нек-рых его сплавов бла-
годаря их исключительной способности к пластич.
деформации и отсутствию хрупкости при низких
темп-рах.
К недостаткам легированного поликристаллич. Мо
(кроме сплавов с Re), помимо хрупкости при обычных
темп-рах, относятся плохая свариваемость и низкая
жаростойкость, требующая нанесения специальных за-
щитных покрытий (MoSi2 и др.).
Легированный Мо и его сплавы благодаря высокой
жаропрочности имеют след, области осваиваемого или
возможного применения: авиационная и ракетная тех-
ника (рабочие и сопловые лопатки газовой турбины,
выхлопные сопла и камеры сгорания прямоточных
реактивных двигателей, передние кромки ракет),
атомная пром-сть, электровакуумная пром-сть (то-
ковводы, подогреватели, катоды электронных ламп),
стекольная, химическая (трубы) и некоторые другие
отрасли пром-сти.
Лит.: Моргунова Н. Н., Жаропрочные сплавы на
основе молибдена, М., 1959; Молибден. Сб. статей, [переводы],
М., 1962; Савицкий Е. М., Изв. АН СССР, Отд. техн. н.
Металлургия и топливо, 1960, № 5, 52; 1961, № 6, 74; 1962,
№ 1, 157; 1962, A's 4, 133; Тылкина М. А., Поваро-
ва К. Б., Савицкий Е. М., Рекристаллизация и меха-
нические свойства сплавов системы вольфрам — молибден —
рений, там же, 1962, № 5; Э с п е В., К н о л ь М., Техноло-
гия электровакуумных материалов, пер. с нем., М,—Л., 1939.
Е. М. Савицкий.
МОЛИБДЕНА СУЛЬФИДЫ — соединения молиб-
дена с серой, из к-рых известны четыре: M02S3, MoS2,
M02S5 и M0S3. Молибден образует также ряд оксисуль-
фидов, в к-рых сера частично замещается кислородом.
M0S2 встречается в природе в виде минерала молибде-
нита, а также может быть синтезирован. Остальные
М. с. получают искусственно.
Полуторный сульфид M02S3 — иголь-
чатые кристаллы серо-стального цвета, плотн. 5,9;
теплота образования ДЯ°298=—102 ±7,0 ккал/молъ.
M02S3 образуется при быстром нагревании MoS2 до
1700—1800° в отсутствии воздуха; может быть получен
из сернокислого р-ра 3-валентного молибдена при
длительном насыщении его H2S. Не растворяется в во-
де, НС1 и H2SO4, конц. HNO3 окисляет M02S3 до
МоОз и H2SO4.
Дисульфид MoS2 — кристаллы серого цвета;
напоминает по виду графит. Природный M0S2 (а-МоБг)
имеет гексагональную структуру слоистого типа; па-
раметры кристаллич. решетки: а=3,15 А, с=12,30А;
плотн. 4,8. Характерной особенностью структуры яв-
ляется расположение шести атомов S вокруг каж-
дого атома Мо не в вершинах октаэдра, а в вершинах
тригональной призмы. Искусственно полученный
M0S2 имеет структуру также слоистого типа, но либо
ромбоэдрическую (P-M0S2) с параметрами а=3,15 А,
с= 18,38 А, либо промежуточную между a-MoSj и
P-MoS2. Приведенные ниже данные о свойствах M0S2
относятся к природному минералу. При быстром
нагревании в защитной атмосфере M0S2 плавится
при 1700—1800° (с разл.); темп-ра начала сублимации
1050°. Твердость M0S2 по Моосу 1. Теплота образова-
ния ДЯ°298=— 56,27 ккал/молъ.
309
МОЛОЧНАЯ КИСЛОТА—МОНОАЗОКРАСИТЕЛИ
310
Термич. диссоциация M0S2 при атмосферном давле-
нии начинается выше 1300°, при этом в качестве про-
межуточного продукта образуется M02S3; при дли-
тельной выдержке при 1700—1800° M0S2 диссоци-
ирует полностью с образованием Мо. В вакууме M0S2
диссоциирует при 1100°. На воздухе M0S2 устойчив
до 300—350°. В интервале темп-р 400—600° MoS2
окисляется на воздухе непосредственно до МоОз,
появление МоОг в продуктах окисления объясняется
вторичным взаимодействием МоОз с M0S2 в отсутст-
вии воздуха. M0S2 практически нерастворим в воде,
НС1, разбавл. H2SO4, растворяется в царской водке,
горячих конц. HNO3 и H2SO4. Активно взаимодейст-
вует с хлором и фтором, образуя МоС15 и MoF6. Во-
дород выше 800° восстанавливает M0S2 до металла.
Описан ряд способов синтеза M0S2: нагревание без
доступа воздуха высших сульфидов молибдена; дей-
ствие паров S или H2S на металл при 800—900°;
сплавление МоО3 или CaMoOi с поташом п серой;
взаимодействие МоС15 с H2S и др.
Пентасульфид Мог85-ЗН20 образуется в ви-
де тригидрата при обработке раствора 5-валентного
молибдена H2S; представляет собой темно-коричне-
вый аморфный порошок, нерастворимый в воде, но
растворимый в сильных к-тах и щелочах. Мог85-ЗНгО
легко теряет часть воды при нагревании в токе СО2,
полная дегидратация достигается трудно.
Трисульфид M0S3— темно-коричневый хло-
пьевидный порошок; неустойчивое соединение, при
нагревании теряет серу, образуя M0S2. Теплота об-
разования ДЯ°298=—61,48 ккал/моль. M0S3 нераство-
рим в воде, разб. НС1, H2SO4, растворяется в щелочах
и щелочных сульфидах и HNO3. Осадки M0S3 часто
содержат нек-рое количество О2, являясь по существу
смесью M0S3 и оксисульфидов молибдена (MoOS2,
МоОгЗ). M0S3 образуется при пропускании H2S или
добавлении сернистых щелочей в подкисленный р-р
молибдатов. В щелочной или нейтральной среде при
добавлении сульфид-иона к раствору молибдатов об-
разуются сульфомолибдаты (напр., Na2MoS4), раство-
римые в воде. При подкислении сульфомолибдаты раз-
лагаются с образованием M0S3.
Наибольшее значение из М. с. имеет природный
M0S2 — главный источник получения молибдена.
Чистый M0S2 применяют как смазку в подшипниках
и других истирающихся деталях. Реакция осаждения
M0S3 используется в аналитич. практике, а также
в производстве молибдена для его извлечения из бед-
ных р-ров и отделения от вольфрама. M02S3 и M02S5
практич. применения не имеют.
Лит.: К i 1 1 е f f е г D. Н., Linz A., Molybdenum com-
pounds, N. Y.—L., 1952; Cannon P., Nature, 1959, 183,
.№_> 4675; Зеликман A. H., Беляевская Л. В.,
Ж. неорг. химии, 1956, 1, № 10; Герасимов Я. И.,
Крестовников А. Н., Шахов А. С., Химическая
термодинамика в цветной металлургии, т. 3, М., 1963.
Л. В. Беляевская.
МОЛОЧНАЯ КИСЛОТА (а-оксипропионовая, про-
панол-2-овая-1 кислота), мол. вес 90,08 — окси-
сн кислота; существует в оптически изомер-
, / 3 ных формах; 1-молочная к-та и левовра-
н с соон щающая, d-молочная (мясомолочная) к-та;
он рацемич. М. к. обычно наз. молочной
к-той брожения. 1- и d-изомеры обладают
одинаковыми физич. свойствами (т. пл. 25—26°) и рав-
ным по абс. величине, но противоположным по знаку
оптическим вращением. В 10 %-ном растворе воды
[а]д= ±3,82, в 2,5%-ном р-ре [а]д = ±2,67°. Оп-
тически-активные М. к. рацемизуются при 130—150°.
1- М. к. может быть получена брожением сахаристых
веществ при посредстве Bacillus acidi laevolactici;
мясомолочная к-та содержится в мускульном соке
животных.
Обыкновенно молочнокислым брожением получают
недеятельную М. к., т. пл. 18°, т. кип. 122714 мм
константа . диссоциации, К=1,38-К)-4 (25°); может
быть разделена на оптич. антиподы с помощью особых
плесневых грибков, а также кристаллизацией молоч-
но-кислых солей оптически деятельных алкалоидов.
В присутствии минеральных к-т происходит само-
этерификация М. к.:
СН3СН (ОН) СООН —> СН„СН (ОН) СООСН (СН,) соон —>
—► СН3СНСООСН (СН3) со
1-----О-----1
Дегидратация М. к. приводит к акриловой кислоте.
М. к. брожения применяют в протравном крашении,
в кожевенном произ-ве, в бродильных пронз-вах (до-
бавляется для предохранения от заражения посто-
ронними бактериями) и в фармакологии.
Производные М. к. брожения: метиловый эфир,
т. кип. 144,8°; этиловый эфир, т. кип. 154,5°; нитрил,
т. кип. 182—1847760 .о; 102730 мм; амид, т. пл.
49—51°. Н. А. Несмеянов.
МОЛЬ — см. Грамм-молекула.
молюскоциды — см. Лимациды.
МОЛЯЛЬНОСТЬ раствора — концентрация
раствора, выраженная числом молей растворенного
вещества, содержащегося в 1000 г растворителя.
См. также Концентрация.
МОЛЯРНОСТЬ раствора — концентрация
раствора, выраженная числом молей растворенного
вещества, содержащегося в 1 л раствора. См. также
Концентрация.
МОЛЯРНЫЙ КОЭФФИЦИЕНТ ПОГЛОЩЕНИЯ —
см. Поглощение света.
МОНОАЗОКРАСИТЕЛИ — азокрасители, содер-
жащие только одну азогруппу—N=N—. Большая
часть азокрасителей принадлежит к М. Получе-
ние М. обычно проще, чем красителей с большим
числом азогрупп, и заключается в диазотировании
амина и сочетании образовавшегося диазосоединения
с к.-л. азосоставляющей. Реже получение М. до-
полняется другими реакциями. Так, в азокрасителях,
содержащих нитрогруппы, последние иногда восста-
навливают до аминогрупп. Амино- и гидроксильные
группы в М. в ряде случаев алкилируют и ацилируют,
чем достигается большая яркость и повышенные проч-
ности окрасок, напр. кислотный хром синий 2К (I).
М., содержащие в орто-положении в разных арильных
остатках две группы ОН или одну ОН и одну из групп
NH2, СООН, ОСНз, СНгСООН и др. превращают в ком-
плексы с Сг, Со, Си или№, отличающиеся повышен-
ными прочностями, напр. кислотный зеленый ЖМ (II).
При получении М. выходы часто близки к количест-
венным (90—95%). М. преим. желтого, оранжевого,
красного цветов, но известны и более глубоко окра-
шенные, в том числе синие, зеленые, коричневые и
черные. М. отличаются от дисазокрасителей и полиа-
зокрасителей яркостью оттенков, как правило, луч-
шей растворимостью, часто большей устойчивостью
к действию света и химич. реагентов. М. составляют
большую часть кислотных красителей и широко при-
меняются для окраски шерсти, а также шелка, кожи,
бумаги, древесины и др. Простейшие из них, типа
кислотного желтого светопрочного, образуют на про-
теиновых волокнах окраски, недостаточно прочные
к мокрым обработкам. Значительно прочнее кислот-
311
МОНОКРИСТАЛЛЫ
312
ные М., имеющие большой мол. вес, напр. карболан
желтый ЗГС (III), металлсодержащие, напр. кислот-
ный зеленый ЖМ (II), «активные» красители, обра-
зующие при крашении ковалентные связи с волокном.
К последним принадлежат, в частности, М., содержа-
щие группу — СОСН2С1, реагирующие с аминогруп-
пами шерсти:
R-NHCOCH2C1 + H2N-R' —> r-nhcoch2nh-r'
краситель шерсть
К М. принадлежит большая часть протравных азо-
красителей, неярких, но очень прочных и поэтому ши-
роко применяемых для крашения шерсти, напр. кис-
лотный хром синий 2К (I). Почти все пигменты и ла-
ковые азокрасители являются М.; их широко исполь-
зуют в окраске пластич. масс, текстильных изделий,
в лакокрасочной пром-сти и т. п.
Для окраски ацетатного, полиамидного, полиэфир-
ного и других химич. волокон применяют «дисперсные»
красители, молекулы к-рых не должны быть больших
размеров. Для окраски этих волокон в желтые, оран-
жевые и красные цвета главную роль играют М., обыч-
но производные азобензола, напр. алый для ацетат-
ного шелка (IV).
.2„4ОН
NaO3S^\
^'SOgNa ОН
V
М., за исключением производных 2-амино-5-нафтол-
7-сульфокислоты, не обладают практически субстан-
тивными свойствами; по этой причине в группе пря-
мых азокрасителей М. крайне редки. Для окраски
целлюлозных волокон выпущены «активные»
красители (см. выше), образующие в процессе краше-
ния эфиры целлюлозы. Окраски очень ярки и прочны
к мокрым обработкам и трению. Большая часть «ак-
тивных» красителей принадлежит к М., напр. актив-
ный оранжевый (V). Прочие свойства М. и литературу
СМ. В ст. Азокрасители. м- А. Чекалин.
МОНОКРИСТАЛЛЫ. Монокристалл — кристал-
лич. индивидуум, зарождающийся из единичного за-
родышевого центра; в монокристалле между любыми
двумя точками всегда можно провести прямую ли-
нию, не пересекающую поверхность, где наблюдается
резкое изменение ориентировки. Характерными осо-
бенностями М., отличающими их от поликристаллич.
твердых тел, являются: 1) способность самоограняться,
т. е. принимать многогранную форму при свободном
росте в подходящих условиях; 2) симметрия строения,
к-рая проявляется не только во внешней форме М. и
его структуре, но и в физич. явлениях, протекающих
в М. при их взаимодействии с внешней средой (см.
Кристаллы); 3) однородность, т. е. наличие одинако-
вых свойств в параллельных направлениях (однород-
ность М., являющаяся основой их все более широкого
применения, зависит от условий их выращивания,
чистоты и природы материала); 4) анизотропия.
т. е. изменение свойств в зависимости от направления;
проявлением анизотропии является, напр., наличие
в М. определенных плоскостей скалывания, вдоль
к-рых они наиболее легко разрушаются под действием
напряжений. Анизотропия физич. свойств М. опре-
деляется их симметрией.
Внешняя форма М. чистых веществ зависит от усло-
вий выращивания М. Различают равновесную и не-
равновесную формы роста М. Равновесная форма ро-
ста соответствует такой огранке, к-рая обеспечивает
минимум свободной поверхностной энергии. Равно-
весные формы могут быть получены при росте и рас-
творенииМ. (любойформы), когда все стадии процесса
близки к равновесию. Рост М. в неравновесных усло-
виях может приводить к резкому изменению их внеш-
ней формы.
Значительное влияние на свойства М. оказывают со-
держащиеся в них примеси и структурные дефекты.
Неоднородное распределение примесей и структур-
ных несовершенств в объеме М. приводит к неоднород-
ности их физич. свойств. Особенно резкая зависимость
свойств от чистоты наблюдается в М. полупроводни-
ков: даже содержание 10-’—10-8 % примеси сильно
изменяет электрич. свойства М. На резкой зависимости
электрич. свойств М. полупроводников от природы и
концентрации примесей основаны методы их легиро-
вания. Основными структурными несовершенствами
в М. являются дислокации и точечные дефекты (см.
Дислокации в кристаллах и Дефекты структуры).
Структурные дефекты в значительной степени опреде-
ляют механич., электрич. и оптич. свойства М., осо-
бенно полупроводников, и влияют на диффузию при-
месей в них. Многие свойства М. определяются слож-
ным взаимодействием структурных дефектов с при-
месями.
Получение М. Можно выращивать М. самых раз-
личных веществ. Большую часть М., широко исполь-
зуемых в науке и технике, получают искусственным
путем. В зависимости от свойств вещества (характер
химич. связи, темп-ра плавления, диссоциация при
нагреве, растворимость, агрессивность и др.) приме-
няют различные методы изготовления М., а разрабо-
танная для каждого конкретного случая технология
определяет размеры, форму и совершенство М. Почти
все методы изготовления М. включают к.-л. фазовое
превращение: твердая фаза — жидкость, твердая фа-
за — пар; твердая фаза — твердая фаза. К превра-
щениям типа твердая фаза — жидкость относятся ме-
тоды выращивания М. из расплавов и растворов.
Выращивание М. из расплавов является основным
методом получения М. полупроводников и металлов.
Применяют 2 основных способа выращивания М. из
расплава: направленную кристаллизацию и зонную
плавку. В первом случае исходный материал сначала
полностью расплавляется, а затем за счет направлен-
ного снижения темп-ры расплав постепенно затвер-
девает. Во втором случае расплавляется лишь часть
исходного поликристаллич. материала — создается
сравнительно узкая расплавленная зона, к-рая затем
перемещается по всей длине образца. Различают ти-
гельные и бестигельные методы получения М. из рас-
плава. Чтобы сделать процесс более воспроизводи-
мым п получить М. заданной кристаллографии, ори-
ентировки, кристаллизацию проводят с примене-
нием монокристаллич. затравки, к-рую размещают
в зоне начала затвердевания. В качестве затравки
обычно используют кусочек монокристалла кристал-
лизуемого вещества, вырезанный в определенном кри-
сталлографии. направлении. Широкое распростра-
нение при получении М. нашел метод вытягивания:
исходная поликристаллич. шихта расплавляется в тиг-
ле; затем в расплав, поддерживаемый при температу-
ре, близкой к точке затвердевания, погружается мо-
313
МОНОКРИСТАЛЛЫ — МОНОМЕРЫ
314
нокристаллич. затравка, являющаяся единственным
центром кристаллизации; после того как на затравке
начинает кристаллизоваться расплав, ее постепенно
поднимают вверх, при этом на нее наращивается мо-
нокристалл. Регулируя темп-ру расплава и скорость
вытягивания, можно получать М. заданного диаметра.
Для получения М. исключительно высокой чистоты,
применяемых в полупроводниковой технике, исполь-
зуют бестигельные методы выращивания, наиболее
распространенным из к-рых является метод «плаваю-
щей зоны»: на границе между закрепленными верти-
кально в специальных держателях монокристаллич.
затравкой и по лик ристал лич. заготовкой создается
расплавленная зона, удерживаемая силами поверх-
ностного натяжения; зона перемещается по длине за-
готовки, переводя ее в М.
При выращивании М. из р-ров в качестве раствори-
телей могут быть использованы вода, органич. рас-
творители, расплавы неорганич. соединений, металлов
и сплавов. Выращивание М. из р-ров можно осущест-
вить удалением части растворителя испарением при
постоянной темп-ре либо охлаждением насыщенного
р-ра. В обоих случаях при этом образуется пересы-
щенный р-р, из к-рого растворенное вещество выде-
ляется в виде кристаллов. Выращивание, как пра-
вило, производится с применением монокристаллич.
затравок. Скорость выращивания М. из растворов
меньше, чем из расплавов, т. к. она лимитируется ско-
ростью диффузии растворенного вещества через рас-
твор к зоне кристаллизации. Проводя достаточно мед-
ленную кристаллизацию в условиях хорошего пере-
мешивания, малого пересыщения р-ра и строгого
температурного контроля, можно получить однород-
ные М., имеющие форму правильных многогранни-
ков. Выращиванием из растворов получают М. квар-
ца, сегнетовой соли, квасцов и др. солей, а также М.
полупроводниковых материалов.
Широкое распространение в последние годы полу-
чило выращивание М. из парообразной фазы. Т. к.
в парах молекулярная диффузия протекает значи-
тельно быстрее, чем в жидком р-ре, рост М. из паров
происходит быстрее, чем из растворов, но медленнее,
чем из расплава. Наиболее существенными парамет-
рами кристаллизации из паровой фазы являются:
давление паров кристаллизуемого вещества в рабочей
камере, темп-ра и состояние поверхности вещества
(подложки), на к-ром растет М. При непосредственном
выращивании М. из паров данного вещества источник
паров помещают в область с высокой темп-рой, а
кристаллизация происходит в области с более низкой
темп-рой. Пары переносятся к зоне кристаллизации
либо за счет создания определенного темп-рного гра-
диента в рабочей камере, либо потоком инертного га-
за. Если росту кристалла предшествует реакция в па-
рообразной фазе, то в рабочей камере, как правило,
создается газовый поток. Кристаллизацией из паро-
образной фазы выращивают М. многих металлов и по-
лупроводников, особенно в виде тонких (десятки или
сотни микрон) пленок. Этим путем получают также
нитевидные М., или усы. Как правило, усы имеют диа-
метр несколько десятков микрон, длину несколько
миллиметров и обладают очень высокой прочностью.
Выращивание М. в твердой фазе нашло ограничен-
ное распространение: собирательная рекристаллиза-
ция (рост М. из поликристаллич. заготовки при ее
длительном нагреве до высоких темп-р) и рекристал-
лизация обработки (рост М. при длительном отжиге
предварительно деформированного поликристалла).
Наряду с М., полученными искусственным путем,
существуют природные М., образовавшиеся в земной
коре при процессах породообразования. Широкое
распространение в природе находят М. кварца, ис-
ландского шпата, каменной соли, флюорита, алмаза
и ряда других элементов и соединений. Природные М.
часто достигают гигантских размеров и весят несколь-
ко сот килограммов.
М. применяют в различных областях науки и тех-
ники: М. полупроводников (германия, кремния, ар-
сенида галлия и др.) служат для изготовления диодов,
силовых выпрямителей, триодов, солнечных батарей,
фотосопротивлений, термогенераторов и др.; из М. ру-
бина, фтористого кальция и нек-рых полупроводнико-
вых соединений изготавливают квантовые генераторы
(лазеры и мазеры). Широко используются М.
в качестве линз, работающих в УФ-и ИК-областях
спектра (кварц, каменная соль, фтористый литий,
кремний, германий и т. д.), а также в качестве поля-
ризаторов (исландский шпат). Пьезоэлектрич. М.
кварца применяются для стабилизации радиочастот,
генерации ультразвука, а сегнетовой соли для кон-
денсаторов, громкоговорителей, микрофонов. М. ал-
маза широко используют при обработке сверхтвер-
дых материалов, а рубина — в часовой пром-сти.
Области применения М. с каждым годом все более
расширяются.
Лит.: Шубников А. В., Образование кристаллов,
М.—Л., 1947; Кузнецов В. Д., Кристаллы и кристалли-
зация, М., 1953; Бакли Г., Рост кристаллов, пер. с англ.,
М., 1954; Варма А., Рост кристаллов и дислокации, пер.
с англ., М., 1958; Процессы роста и выращивание монокристал-
лов, со. статей, пер. с англ., под ред. Н. Н. Шефталя, М.,
1963; Най Дж., Физические свойства кристаллов.. , пер. с
англ., М., 1960. М. Г. Мильвидекий.
МОНОМЕРЫ — низкомолекулярные соединения,
применяемые для синтеза полимеров. В качестве М.
могут быть использованы соединения, молекулы к-рых
вступают в химич. реакцию не менее, чем с двумя дру-
гими молекулами (би- или полифункциональные со-
единения). Функциональность М. определяется числом
функциональных групп, а также количеством подвиж-
ных атомов водорода или кратных связей и зависит
от характера реакции и ее условий. Из бифункциональ-
ных М. получают линейные полимеры; синтез полиме-
ров на основе полифункциональных М. приводит к
образованию разветвленных или пространственных
структур.
М., участвующие в цепной полиме-
ризации, содержат в молекуле кратные связи,
способные при определенных условиях разрываться
с образованием активных частиц (радикалов или ио-
нов). При полимеризации М., содержащих между ато-
мами углерода одну двойную связь, получаются карбо-
цепные высокомолекулярные соединения, не содержа-
щие двойных связей в основной цепи макромолекулы
(к таким М. относятся этилен, монозамещенные произ-
водные этилена типа RHC=CH2, пропилен, акрило-
нитрил, стирол, акриловая к-та и др.; нек-рые несим-
метричные дизамещенные этилена типа H2C=CRR',
винилиденхлорид, винилиденцианид и др.). Полимери-
зация М., имеющих сопряженные углерод-углеродные
двойные связи (бутадиен-1,3 и его гомологи и произ-
водные — 2-метилбутадиен-1,3; 2-хлорбутадиен-1,3
и др.), ведет к образованию карбоцепных полимеров,
у к-рых в основной цепи имеются двойные связи. При
наличии в молекуле М. изолированных двойных свя-
зей при полимеризации получаются либо линейные
полимеры с циклами в основной цепи, либо развет-
вленные или сетчатые полимеры. Полимеризоваться по
механизму цепных реакций могут также М. с кратной
связью между атомом углерода и каким-либо другим
атомом — кислорода, азота и др. (напр., формальде-
гид и формальоксим); в этом случае получают гетеро-
цепные полимеры. Скорость цепной полимеризации и
структура образующегося полимера зависят от строе-
ния и свойств исходного М. Решающее влияние на
рост цепи оказывает количество, взаимное расположе-
ние и строение заместителей в М. Активность М. при
полимеризации зависит от стерических напряжений,
315
МОНОМЕРЫ — МОНОМОЛЕКУЛЯРНЫЕ РЕАКЦИИ
316
возникающих в результате взаимодействия заместите-
лей при образовании макромолекулы, и степени поля-
ризации или поляризуемости двойной связи. Все
монозамещенные этилена полимеризуются, причем
скорость полимеризации падает с увеличением раз-
мера заместителя. Несимметричные дизамещенные
этилена, в молекулах к-рых хотя бы один из двух за-
местителей обладает большими размерами (внешний
радиус порядка ЗА), не полимеризуются. В качестве
М. могут быть использованы тризамещенные этилена,
у к-рых размер заместителей близок к размеру ато-
ма водорода (напр., трифторэтилен), вследствие чего
при полимеризации стерич. препятствий не возникает.
Реакционная способность М. возрастает с повыше-
нием степени поляризации двойной связи. Введение
в молекулу этилена несимметрично расположенных
заместителей вызывает поляризацию двойной связи.
Величина дипольного момента молекулы М. тем выше,
чем большими электронодонорными или электроноак-
цепторными свойствами обладает введенный заме-
ститель (дипольный момент пропилена — 0,35; ви-
нилхлорида — 1,44; акрилонитрила — 3,88). Соот-
ветственно увеличивается активность М. при полиме-
ризации. Однако активные мономеры образуют мало-
активные радикалы, т. к. эффект сопряжения неспа-
ренного электрона в радикале приводит к уменьше-
нию его активности. Этим и объясняется часто наб-
людаемая большая скорость полимеризации малоак-
тивных М., образующих реакционноспособные радика-
лы, по сравнению со скоростью полимеризации более
активных М., из к-рых получаются нереакционноспо-
собные радикалы. Влияние перечисленных выше фак-
торов определяет присоединение молекул М. друг
к другу в положение 1—2 («голова к хвосту») или 2—2
(«хвост к хвосту»). Для монозамещенных этилена
наиболее вероятно 1-—2-присоединение.
М., образующие полимеры поли-
конденсацией, должны содержать не менее
двух различных функциональных групп, способных
реагировать между собой. При гетерополиконденса-
ции и ступенчатой полимеризации функциональные
группы разных типов могут входить в состав молекул
двух разных М. Так, для синтеза полиамидов исполь-
зуют диамины и дикарбоновые или аминокарбоновые
кислоты (напр., гексаметилендиамин и адипиновую
или аминоэнантовую к-ту), для синтеза полиэфиров
диолы и дикарбоновые к-ты (напр., этиленгликоль
и терефталевую к-ту), для синтеза полиуретанов
диизоцианаты и диолы (напр., гексаметилендиизо-
цианат и бутандиол-1,4).
Поликонденсация на первой же ступени может за-
кончиться образованием циклич. соединений. Направ-
ление реакции определяется строением М.: если функ-
циональные группы разделены небольшой углеродной
цепью (3—4 атома), то преобладает циклизация. При
увеличении расстояния между функциональными
группами вероятность циклизации уменьшается. Так,
для у- и 6-амино- и оксикислот основным направле-
нием является циклизация; в случае е-амино- и окси-
кислот протекает только поликонденсация. При вве-
дении в молекулу М. заместителя — алифатич. ра-
дикала — вероятность циклизации возрастает.
В качестве М. можно также применять нек-рые ге-
тероциклич. соединения: окись этилена, этиленимин и
их производные; ряд циклич. эфиров-лактонов (напр.,
6-валеролактон) и амидов-лактамов (напр., е-кап-
ролактам). Склонность к полимеризации у гетероцик-
лич. М. возрастает с увеличением напряженности и
числа членов цикла. В последнее время показана воз-
можность полимеризации и ненапряженных 5- и
6-членных гетероциклов (пирролидона и пиперидона)
по анионному механизму (в присутствии калиевых
солей этих лактамов, КОН и N-ацилпроизвочных).
Использование ряда М., содержащих атомы крем-
ния, алюминия, титана, олова и др. элементов (напр.,
алкил- и арилхлорсиланов, эфиров ортотитановой к-ты
и др. соединений), позволяет получить т. наз. элемен-
тоорганич. полимеры (см. Высокомолекулярные соеди-
нения элементоорганические). Последние образуются
гл. обр. в результате поликонденсации.
Лит.: В а ц у л и к П., Химия мономеров, пер. с чешек.,
т. 1, М., 1960; Коршак В. В., Химия высокомолекулярных
соединений, М.—Л., 1950; Стрепихеев А. А., Д е ре-
ви ц к а я В. А., Основы химии высокомолекулярных соеди-
нений, М., 1961; Лосев И. П., ТростянскаяЕ.Б.,
Химия синтетических полимеров, М.. 1964; Бильмейер Ф.,
Введение в химию и технологию полимеров, пер. с англ., М.,
1958; Б ат ц ер Г., Введение в химию высокомолекулярных
соединений, пер. с нем., М., 1960. Л. ГальбраИх.
мономицин — антибиотик, обладающий широ-
ким антибактериальным спектром, действует на грам-
положительные и грамотрицательные микробы и
бактерии. М.— сильное органич. основание, содержит
5 аминогрупп. Хлоргидрат М.— белый аморфный по-
рошок, растворимый в воде, нерастворимый в этаноле
и др. органич. растворителях. Основание и сульфат М.
аморфны, рейнекат и гелиантат — кристаллич. в-ва.
Наиболее вероятное строение М. показано на схеме.
Подкожная токсичность М. ок. 400 мг на 1 кг веса
мыши. Для получения М. продуцент культивируют на
среде, содержащей соевую муку (2%), глюкозу (1%),
KN03(0,3%), СаСОз (0,5%), NaCl (0,3%). Выделяют и
очищают М. с помощью ионного обмена на смолах
типа RC-50 и КВ4-П2 в натриевой форме. Элюиро-
вание производят 1 н. р-ром NH40H. Активность М.
определяют биологич. титрованием на штамме Вас.
Subtilus и химич. методами, основанными на реакциях
с нингидрином или реактивом Биаля.
М. был выделен в СССР из культуральной жидкости
Act. circulatus, vor. inonomycini. В США описан анти-
биотик паромицин с аналогичными химич. свойствами.
При хроматографич. изучении М., с целью сравнения
его с паромицином, не было обнаружено к.-л. различия
между этими антибиотиками. Сходными с М. являются
антибиотики катенулин и оксимицин.
Лит.: Бражникова М. Г. [и др.], Антибиотики,
1960, 5, № 4; Б р а ж н и к о в а М. Г., Муравьева Л. И.,
там же, 1961, 6, № 5; Бражникова М. Г., Куди-
нова М. К., Трофилеева Р. Н., Биохимия, 1961,
26, в. 3; Г а узе Г. Ф. [и др.], Антибиотики, 1960, 5, № 4;
Шорин В. А., Г о л ь д б е р г Л. Е., Кремер В. Е.,
там же; Haskell Th. Н.,French J., Bartz Q. R.,
J. Amer. Chem. Soc., 19 59,81,№13,3481. М.Г. Бражникова.
МОНОМОЛЕКУЛЯРНЫЕ РЕАКЦИИ — реакции,
в элементарном акте к-рых участвует одна молекула
исходного вещества. К этому классу относятся много-
численные реакции распада сложных молекул и изо-
меризации. Примеры: распад азометана
снн~ с. 11,, -I- n2
изомеризация гщс-стильбена в тракс-стильбен
НСС6Н3 С6Н,СН
и = и
НССеН-, нсс„н,
Согласно закону действующих масс, скорость М. р.
в идеальных газовых смесях и идеальных жидких
р-рах описывается кинетич. ур-нием первого порядка
<о — АС (1)
317 МОНОМОЛЕКУЛЯРНЫЕ РЕАКЦИИ — МОНОМОЛЕКУЛЯРНЫЙ СЛОЙ 318
где со — скорость реакции, С — концентрация исход-
ного вещества, к — константа скорости (зависящая
от темп-ры по ур-нию Аррениуса).
Если реакция происходит при постоянном объеме,
dC
то (0=—^-, где t — время, и интегрирование дает
С = Сое-« (2)
(Со— значение С при 7=0). Отсюда время половинного
превращения 7,^=(1п 2)/к, т. е. 7,,^ не зависит от С.
Не только М. р. подчиняются ур-нию (1), напр.
существуют цепные реакции первого порядка. С другой
стороны, не всегда скорость газовых М. р. следует
ур-нию (1), причины этого рассмотрены ниже. Пред-
экспоненциальный множитель в ур-нии Аррениуса
для константы первого порядка к у большинства М. р.
имеет значение 1012—10’4 сек-1, т. е. по порядку
величины близок к частотам молекулярных колеба-
ний. Резко уменьшенные значения этого множителя,
наблюдающиеся у нек-рых М. р. (напр., порядка
105 сек-1), указывают на неадиабатический (в кванто-
во-механич. смысле) характер элементарного акта ре-
акции, т. е. на то, что соответствующий электронный
переход имеет весьма малую вероятность.
Возможность протекания газовых М. р. по ур-нию
первого порядка не очевидна. На первый взгляд, по-
скольку молекула исходного вещества получает энер-
гию, необходимую для преодоления активационного
барьера, в результате столкновения с другой молеку-
лой, то скорость реакции должна быть пропорциональ-
на числу столкновений, а следовательно С2, а не С.
Эта трудность была устранена Линдеманом (1922),
принявшим, что момент химич. превращения отделен
нек-рым промежутком времени от момента активирую-
щего столкновения. Если среднее значение этого
промежутка времени много больше среднего времени
между двумя последовательными столкновениями
данной молекулы, то энергетизированная (или
активная) молекула, т. е. молекула, обладающая
достаточной для реакции энергией, в подавляющем
большинстве случаев теряет энергию при первом же
столкновении, не претерпевая химич. превращения.
В результате протекания взаимнообратных процессов
передачи энергии — активации и дезактивации, с
почти равными скоростями, осуществляется статистич.
равновесие между энергетизированными молекулами
и всеми молекулами, так что концентрация энергети-
зированных молекул, а следовательно и скорость ре-
акции, пропорциональна общей концентрации моле-
кул исходного вещества. Из представлений Линдемана
следует, что при понижении концентрации, приводя-
щем к увеличению среднего времени между столкнове-
ниями, должны возникать отклонения от ур-ния
первого порядка, и при достаточно низких концент-
рациях, когда практически все энергетизированные
молекулы успевают прореагировать за время между
столкновениями, реакция должна протекать по ур-нию
второго порядка. Этот вывод полностью подтверждает-
ся опытными данными. Дальнейшим подтверждени-
ем служит эффект инертных газов. В области низких
давлений, когда скорость реакции лимитируется ско-
ростью активации при столкновениях, добавление
инертного газа ускоряет реакцию, потому что молеку-
лы этого газа, сталкиваясь с молекулами исходного
вещества, передают им энергию. В области высоких
давлений, когда концентрация энергетизированных
молекул является равновесной и реакция протекает
по ур-нию первого порядка, добавление инертного
газа не изменяет скорость реакции.
При М. р. в жидких р-рах молекулы исходного ве-
щества получают энергию от молекул растворителя,
поэтому равновесная концентрация энергетизирован-
ных молекул достигается при всех концентрациях ис-
ходного вещества и отклонения от ур-ния первого
порядка, не происходят.
Существование промежутка времени между момен-
том получения молекулой энергии активации и момен-
том реакции обусловлено тем, что мономолекулярным
превращениям подвергаются многоатомные молеку-
лы, обладающие значительным числом колебательных
степеней свободы. Согласно теории Касселя (1928)
и другим сходным теориям, в энергетизированной мо-
лекуле энергия непрерывно перераспределяется между
колебательными степенями свободы до тех пор, пока
случайно не сосредоточится в том колебании, к-рое,
при достаточно большой амплитуде, приводит к реак-
ции (напр., в случае изомеризации цис-стильбена та-
ким колебанием будет крутильное колебание вокруг
двойной связи). В теории Слейтера (1953) принимает-
ся, что энергия каждой колебательной степени сво-
боды молекулы изменяется только при столкнове-
ниях, реакция происходит в результате определенного
сочетания фаз и амплитуд колебаний энергетизирован-
ной молекулы. Теории Касселя и Слейтера позволяют
количественно описать наблюдающийся у газовых
М. р. переход от второго к первому порядку при уве-
личении давления. Переход происходит при тем более
низких давлениях, чем больше число колебательных
степеней свободы молекулы. Новейшие эксперимен-
тальные данные указывают на возможность незатруд-
ненного обмена энергией между различными колеба-
тельными степенями свободы, т. е., по-видимому, го-
ворят в пользу исходных представлений теории Кас-
селя.
Лит.: Кондратьев В. Н., Кинетика химических
газовых реакций, М., 1958; Касс ел ь Л. С., Кинетика го-
могенных газовых реакций, пер. [с англ.], Л., 1937.
МОНОМОЛЕКУЛЯРНЫЙ СЛОЙ (монослой)М —
слой поверхностно-активного вещества толщиной в
одну молекулу, образующийся на границе раздела
фаз в результате адсорбции или нанесения вещества
из летучего растворителя и поверхностной диффузии.
Так, органич. поверхностно-активные вещества, прак-
тически нерастворимые в воде (высшие жирные к-ты
и спирты и др.), образуют на поверхности воды слои
толщиной только в одну молекулу, а слои заданной
по расчету большей толщины самопроизвольно рас-
падаются на М. с. и микрокристаллики или капельки
того же вещества в виде обычной объемной фазы. Та-
кие М. с. образуют т. наз. двухмерную фазу, в к-рой,
благодаря гидратации и наличию только одного слоя
молекул, ослаблено взаимодействие их между собой
по сравнению с объемным кристаллом. Молекулы али-
фатич. соединений с прямой цепью в М. с., близких
к предельной плотности покрытия, при достаточно
сильном боковом сжатии ориентируются так, что их
полярная группа погружена в воду, а углеводородная
цепь расположена вертикально в газовой фазе; при
разной длине цепи и разных полярных группах они
занимают одинаковую наименьшую площадь на мо-
лекулу (ао=20,5 А2), близкую к сечению углеводо-
родной цепи для объемных кристаллов тех же веществ
(18,5 А2).
Измерениями двухмерного давления F=aQ— а
(дин-см~' = эрг-см~г), т. е. понижения поверхностного
натяжения чистой воды <т0, вызванного М. с., уста-
новлено, что при малых F М. с. представляют собой
разреженные двухмерные газы, подчиняющиеся
ур-нию состояния идеального газа
Fa--kT
где а — площадь на молекулу в М. с., a=A/FN (Г —
адсорбционное покрытие в моль/см\ N — число Аво-
гадро), Т — абс. темп-pa, к — постоянная Больцмана.
В таких слоях а велико и составляет тысячи А2 и угле-
водородные цепи расположены горизонтально — они
319
МОНОМОЛЕКУЛЯРНЫЙ СЛОЙ—МОНОСАХАРИДЫ
320
касаются поверхности воды. При постепенном сжатии
М. с. переходят из двухмерного газообразного состоя-
ния в двухмерную жидкость (жидко-расширенный мо-
нослой) и, наконец,— в двухмерную конденсирован-
ную фазу (при пониженных темп-рах минуя жидко-
расширенное состояние). Конденсированных двух-
мерных фаз может быть несколько, они могут быть
жидкими и твердыми и отличаться различным накло-
ном цепи и гидратацией полярной группы. Фазовые
переходы между двухмерными фазами могут быть как
1-го, так и 2-го рода. Учет взаимного притяжения угле-
водородных цепей в М. с., т. е. отклонения от идеаль-
ного газового состояния с переходом к конденсирован-
ным фазам, приводит к ур-нию двухмерного состояния
типа Ван-Дер-Ваальса. Для жидко-расширенного
состояния это ур-ние имеет вид: (F—Р0)(а—а0)=кТ,
где Ро и а0— константы. Ро обусловлено притяжением
углеводородных цепей, аналогичным существующим
в жидком углеводороде равного мол. веса; а0— пре-
дельная площадь, близкая, но не обязательно равная
площади плотно упакованных цепей в конденсирован-
ном состоянии. Для больших площадей в состоянии
реального газа при условии Ро<; F (если пренеб-
речь силами молекулярного притяжения) получаем
F(a—aQ)=kT— ур-ние типа Фольмера — Ленгмюра, в
к-ром учитываются лишь собственные площади мо-
лекул а0, которые благодаря стремлению к горизон-
тальной ориентации могут быть большими, чем наи-
меньшие аа, соответствующие конденсированному со-
стоянию.
Исследование М. с. важно для характеристики
свойств, строения и размеров органич. молекул, а
также строения адсорбционных слоев, являющихся
в основном М. с. Особенности таких слоев определяют
свойства многих дисперсных систем и биологич. струк-
тур, явления смачивания, трения и т. д. Способность
М. с. нек-рых веществ, особенно высших жирных спир-
тов, понижать скорость испарения воды дает возмож-
ность, путем нанесения таких слоев, уменьшать испа-
рение воды в водоемах.
Методы перенесения М. с. с поверхности воды на
твердую подкладку позволяют установить, что уже
первый М. с. очень резко снижает коэфф, трения ме-
таллов и изменяет их смачиваемость. Последователь-
ное нанесение большого числа М. с. жирных кислот на
пластинку с периодически меняющейся ориентацией
иозволяет довести такой «мультимолекулярный слой»
до определенной значительной толщины.
Большой интерес представляют М. с. высоко-
полимерных веществ. Так, макромоле-
кулы растворимых белков, имеющие правильную про-
странственную или глобулярную структуру, попадая
на поверхность раздела фаз, развертываются, в них
разрываются многие связи, гидрофильные (полярные)
группы остаются в воде, а гидрофобные группы выхо-
дят наружу, что приводит к своеобразной (поверх-
ностной) денатурации — белок становится нераство-
римым (однако эта денатурация не эквивалентна теп-
ловой денатурации). На концентрированной солевой
подкладке М. с. многих белков переходят в состояние,
близкое к газообразному. Это позволяет в ряде слу-
чаев определить мол. веса по ур-нию состояния иде-
ального двухмерного газа. Нек-рые линейные поли-
меры, напр. полиметилсилоксаны, хорошо распро-
страняются по воде до полного развертывания клуб-
кообразно свернутых в растворе (в органич. раствори-
теле) молекул, другие, как натуральный каучук, рас-
текаются на воде, только до толстых (неоднородных)
слоев (260—360 А), а на ртути под действием более
сильных молекулярных полей — до полного развер-
тывания цепей и толщины пленки в ~3 А. Метод мо-
нослоев позволяет исследовать строение нуклеиновых
к-т, моделировать процессы проникновения токсичных
веществ в белково-липоидные М. с., напоминающие
стенки клетки.
Лит.: Мономолекулярные слои, пер. с англ., М., 1956;
Адам Н. К., Физика и химия поверхностей, пер. с англ.,
М.—Л., 1947; Р а й д и л Э. К., Химия поверхностных явле-
ний, пер. с англ., Л., 1936; Марселей А., Поверхностные
растворы, пер. с франц., М.—Л., 1936; Davies J. Т.
Rideal Е. К., Interfacial phenomena, N. Y.—L., 1961.
МОНОНУКЛЕОТИДЫ —
МОНОСАХАРИДЫ (гликозы, монозы, простые
сахара) — группа углеводов, не способных к гидро-
лизу. М. представляют собой полиоксиальдегиды
(альдозы) или полиоксикетоны (кетозы), к-рые реа-
гируют в открытой альдегидной или кетонной форме
и в виде циклич. полуацеталей. Типовая формула
М. в открытой форме СН2ОН(СНОН)„—СНО для аль-
доз и СН2ОН(СНОН)„—СО—СН2ОН для кетоз, где
п>1. Полуацетальные формы М. схематически можно
представить ф-лами I для альдоз и II для кетоз (т=
=2, 3 или 4).
| —снон [СН2ОН
I । ।
О (СНОН)т |-2СНОН
1-СН (1) (СН0Н)„
R I—с'н
I
I R II
Большинство М. содержит не разветвленную углерод-
ную цепь и одну карбонильную группу, причем в
кетозах карбонил в углеродной цепи занимает обычно
положение 2. В зависимости от числа атомов углерода
в цепи М. делятся на тетрозы (Ct), пентозы
(С5), гексозы (Св), гептозы (С,), о к т о з ы
(С8), н о н о з ы (С9), д е к о з ы (С1с) и т. д. Соеди-
нения с 3 углеродными атомами в цепи (триозы),
аналогичные М., глицериновый альдегид и диокси-
ацетон относят в группу М. условно, поскольку
эти вещества генетически связаны с углеводами и
схожи с ними по нек-рым свойствам. В названии в
случае необходимости отражают принадлежность М.
к альдозам или кетозам добавлением соответствую-
щей приставки «альдо» или «кето» или вводя в на-
звание кетозы суффикс «ул». Так, М. с альдегид-
ной группой называют альдопентозами, альдогексо-
зами, альдогептозами и т. д., а М. с кетонной
группой—кетопентозами, или пентулозами, кетогексо-
зами, или гексулозами, кетононозами, или нонулоза-
ми и т. д.
Группировка СНОН в М. содержит асимметрия,
атом углерода; поэтому М. образуют ряд оптически
активных стереоизомерных форм, число к-рых равно
2" (где п — число асимметрии, атомов в молеку-
ле). Так, существуют 2 альдотриозы, 4 альдотетрозы,
8 альдопентоз, 16 альдогексоз и т. д. Для графич.
изображения стереоизомерных форм М. пользуются
ф-лами Фишера, представляющими проекции тетра-
эдрич. моделей ф-л на плоскость:
СНО 1 СНО
неон 2 н— -ОН
носн 3 но- —н
неон 4 н— -он
неон 5 н- -он
сн2он 6 с Н20Н’
По предложению Розанова, те М., к-рые получаются
из D-глицеринового альдегида путем последова-
тельного применения циангидринного синтеза, т. е.
те М., в к-рых конфигурация у углеродного атома, наи-
более удаленного от карбонила, совпадает с конфи-
гурацией у асимметрич. углерода D-глицериновогс
321
МОНОСАХАРИДЫ
322
— ______________Н ОСН2— СН(ОН)-СНО
1 сно 1 НСОН 1 НСОН СН2ОН D-эритроза D-глицериновый альдегид (D-глицероза) 1 сно 1 носн 1 НСОН СН2ОН D-треоза
А
сно НСОН 1 ' НСОН НСОН 1 НСОН 1 СН.ОН 1 1 1 сно сно сно 1 1 1 НСОН НОСН НСОН 1 1 1 НСОН НСОН носн 1 1 ‘ НСОН НСОН НСОН 1 1 1 сн2он сн2он сн2он D-рибоза D-арабиноза D-ксилоза / \ Z \ Z \ сно сно сно сно СНО II II 1 НОСН НСОН НОСН НСОН носн НСОН носн НОСН НСОН НСОН II II 1 НСОН НСОН НСОН носн носн It II 1 НСОН НСОН НСОН НОСН НСОН СН2ОН СН2ОН СП2ОН СН2ОН СН2ОН 1 сно 1 носн 1 носн . 1 НСОН 1 сн2он D-ликсоза сно сно 1 1 НСОН носн 1 1 НОСН НОСН 1 1 НОСН НОСН 1 1 НСОН Н 0 I 1 1 СН2ОН СН2ОН
D-аллоза D-альтроза D-глюкоза D-манноза D-гулоза D-идоза D-галактоза D-талоза
Схема 1. D-ряд альдоз.
альдегида, относят к D-ряду (см. Глицериновый аль-
дегид); их антиподы относят к L-ряду, т. е. к ряду
L-глицеринового альдегида. D-ряд альдоз приведен на
схеме 1, D-ряд кетоз — на схеме 2 (см. стр. 323).
Группа D-альдоз, представленных на схеме 1, и их
антиподы L-ряда рассматривают как ключевые вещест-
ва для всех М. Напр., М., содержащие дополнительные
функциональные группы — аминогруппу, карбоксиль-
ную группу и др., также рассматривают как произ-
водные ключевых альдоз (напр., глюкозамин, галак-
тозамин и т. д.).
Для обозначения остатков М., входящих в состав олиго-
сахаридов и полисахаридов, пользуются условными сокраще-
ниями, аналогично условным обозначениям аминокислот в
белках. Для триоз и гексоз используют 3—4 первые буквы
названия и только для глюкозы используют одну букву «Г»,
напр.:
Ф-лы Хеуорса дают более правильное представление
о конфигурации. Допускается, что атомы углерода и
кислородный атом, образующий цикл, лежат в одной
плоскости, а остальные группы оказываются либо под
плоскостью кольца, либо над нею.
СН2ОН
сн.он
Алл — аллоза
Альт — альтроза
Араб—арабиноза
Г — глюкоза
Гал — галактоза
Гул — гулоза
Идо — идоза
Ксил — ксилоза
Лик — ликсоза
Риб — рибоза
Маи — манноза
Тал — талоза
Трео — треоза
Фрук — фруктоза
Эрит — эритроза
Циклич. полуацетальные формы М. представляют
ф-лами Фишера—Толленса или Хеуорса. Ф-лы Фише-
ра—Толленса указывают на характер цикла М. и на
конфигурацию гидроксила (см. Аномеры), но дают
неправильное представление о взаимном расположе-
нии атомов в пространстве. Так, по Фишеру—Тол-
ленсу, различные циклич. формы D-глюкозы можно
представить ф-лами:
неон—
неон
носн о
неон
।
нс----
I
сн,он
a-D-глюкопира-
ноза
НОСН----:
неон
НОСН о
I
неон
I
НС-----
I
СН2ОН
P-D-глюкопи-
раноза
НСОН—
НСОН |
I О
НОСН
нс-----1
I
НСОН
сн,он
a-D-глюкофу-
раноза
НОСН-----
НСОН |
I О
НОСН I
I
НС-----1
НСОН
I
СН2ОН
P-D-глюкофу-
раноза
В формах М. с 5-членными циклами (пиранозных)
атомы углерода действительно лежат в одной плоско-
сти, в случае же 6-членных циклов пираноз возмож-
но существование креслообразных и ваннообразных
конформаций. Креслообразные конформации М. бо-
лее устойчивы. На соотношения конформационных
структур в М. влияют заместители у атомов углеро-
да кольца.
М.— кристаллич. продукты, хорошо растворимые
в воде, плохо — в спирте и нерастворимые в эфире.
В твердом состоянии М. существуют в виде циклич.
форм. В водных р-рах устанавливается равновесие
между таутомерами (см. Мутаротация). М. дают ре-
акцию на карбонильную группу, обладают восстанав-
ливающими свойствами, например восстанавливают
AgNOs в аммиачном р-ре, щелочной р-р окиси меди.
При окислении альдегидной группы альдоз образу-
ются альдоновые кислоты (гликоновые) (I); окисление
первичной спиртовой группы приводит к образова-
нию сахарных (гликаровых) кислот (II). При окисле-
нии только первичной спиртовой группы (при защи-
соон
(СНОН)„
।
СН2ОН
I
соон
(СНОН)„
соон
II
Oil К.Х.Э. т. 3
223
МОНОСАХАРИДЫ—МОРИН
324
щенной альдегидной группе) образуются уроновые
кислоты (гликуроновые) (III). Восстановление альдоз
приводит к образованию соответствующих сахарных
спиртов—альдитолов (IV); при восстановлении ке-
.тозы образуется смесь двух стереоизомерных аль-
дитолов.
сно СН..ОН
(СНОН)„ (СНОН)„
। ।
СООН СН2ОН
III IV
За счет полуацетального гидроксила М. образуют
гликозиды: альдозиды (V) и кетозиды (VI):
।—сноп
I (СНОН)„
о |
।
X
СН3ОН
I
j—С-OR
I (СНОН)„
О I
а также олигосахариды и полисахариды. М. образует
озазоны и др. производные (за счет карбонильной
группы).
Качественное и количественное определение М.
производят обычно, используя их восстанавливающие
свойства (метод Хагедорна и Йенсена и др.) и при-
меняя некоторые специфические цветные реакции
сахаров. Широко используется для разделения М.
метод хроматографии на бумаге и на колонках с
адсорбентами.
сн3он
।
со
।
СН2ОН
Диоксиацетон
СН2ОН
I
с=о
неон
СН2ОН
D-глицеро-тетрулоза
(D-эритрулоза)
(D-треулоза)
У \
СН2ОН СН2ОН
с=о с=о
неон носн
неон неон
снаон сн2он
D-эршпро-пентулоза D-трео-пентулоза
(D-рибулоза) (D-ксилулоза)
(адоноза) (D-ликсулоза)
/ \ /
СН20Н сн2он снаон снаон
с=о с=о с=о с=о
неон носн неон носн
неон неон I IOCH носн
неон неон неон неон
сн2он сн2он сн2он сн2он
D-рибо-гексу- D-арабинс-гексу- D-ксило-гексу- D-ликсо-
лоза лоза лоза гексулоза
(D-аллулоза) (D-фруктоза) (D-сорбоза) (D-тагатоза)
(D-псикоза) (левулоза)
Схема 2. D-ряд кетоз.
Получают М. из природных источников: многие М.
встречаются в растениях и в животных организмах
(D-глюкоза и др.) в свободном виде и в виде раз-
личных производных: гликозидов, фосфорных эфиров,
олигосахаридов, полисахаридов. В природе более ши-
рокое распространение имеют пентозы и гексозы.
М. с разветвленной цепью встречаются в нек-рых
растениях, напр. апиоза (VII) и гамамелоза (VIII),
и в нек-рых микроорганизмах, в частности они явля-
ются компонентами антибиотиков [стрептоза (IX),
микароза (X), кладиноза (XI) и кордицепоза (XII)]
сно сно
I I /СН2ОН
неон с(
I । 'он
сон неон
Z\ I
нон2с сн2он неон
VII 1
сн2он
VIII
сно
I
неон
оно-сон
I
носн
CHS
IX
сно
сн2
I /СН3
с.
I 'ОСН;
неон
неон
сн3
XI
сно
I
СН,
। ;сн3
с\
I 'он
неон
неон
I
СН,
X
СНО
I
СНОН
I
СН
НОН2С/ЧСН2ОН
XII
Встречаются в природе модифицированные М.—
дезоксисахара, гликозамины и др. Синтетически М.
получают путем взаимных превращений, напр. циан-
гидринным синтезом, Лобри-де Бруина и Ван-Эккен-
штейна реакциями и др. способами. См. также Сахара,
Гексозы, Пентозы, Гликозиды, Углеводы и статьи об
отдельных представителях М.
Лит.: Кочетков Н. К., Торгов И. В., Бот-
ви ник М. М., Химия природных соединений, М., 1961;
Степаненко Б. Н., Усп. химии, 1959, 28, вып. 5, 521;
The carbohydrates, ed. W. Plgman, N. Y., 1957; Michel F.,
Chemie der Zucker und Polysaccharide, 2 Aufl., Lpz., 1956;
Percival E.G.V., P e г с 1 v a 1 E., Structural carbohyd-
rate chemistry, L., 1962; Shafizadeh F., Wolfrom
M. L., в kh.: Handbuch der Pflanzenphyslologle, Bd 6, B.—
[u. a.], 1958, S. 10; Comprehensive biochemistry, v. 5, Amst.—
N. Y., 1963; Methods in carbohydrate chemistry, v. 1, N. Y.,
1 962. Л. II. Линевич.
МОНОТЕКТИКА — см. Двойные системы.
МОНТАННЫЙ ВОСК (монтанвоск) — см.
Воски.
МОРИН (3, 5, 7, 2', 4'-пентаоксифлавон) C,sH10O.,
мол. в. 302,24 — желтый краситель класса оксифлаво-
нов (ср. кверцетин), содер-
жится в экстракте желтого
дерева Morns tinctoria (кра- но
сильное тутовое дерево) на-
ряду с 2,4,6,3',4'-пентаок-
сибензофеноном (маклури-
ном). М. — бледно - желтые
иглы или порошок, т. пл. 290°; может кристаллизо-
ваться с 1—2 молекулами Н2О; мало растворим в
воде, эфире и уксусной к-те; растворим в спирте и
щелочах с темно-желтой окраской. В конц. H2SO4
растворяется с желтой окраской, давая яркую сине-
вато-зеленую флуоресценцию. При добавлении к спир-
-ОН
325
МОРФИН—МОТОРНОЕ топливо
326
товому р-ру М. р-ра соли алюминия появляется ин-
тенсивная зеленая флуоресценция. М. был использо-
ван для качественного и количественного обнаруже-
ния и определения элементов III группы, с к-рыми
он образует прочные комплексы ярко-желтого цвета,
флуоресциирующие даже при днецном свете (за ис-
ключением редкоземельных элементов). М. исполь-
зуют также для фотометрия, и флуоресцентного опре-
деления элементов IV группы (Zr, Th, Ge, Sn, Ti);
для флуоресцентного определения Be, Мо и т. д.
Лит.: The chemistry of flavonoid compounds, ed. T.A. Geiss-
man. L., 1962. И. П. Ефимов.
МОРФИН (1) C17H19O3N— главный алкалоид опия,
млечного сока несозревших плодов снотворного мака
Papaver somniferum, культиви-
руемого в Индии, Китае, на юге
.К СССР н др. странах. М. (моно-
0' j; hoi н гидрат)— бесцветные кристаллы,
ПРИ теРяет кристаллизаци-
н р I is, 3 онную воду, т. пл. 254° (с разл.,
испр.); [а]” = — 130,9° (мета-
1 нол), —70,23° (водный NaOH).
Растворимость М. в г/100 г растворителя: вода 0,019
(20°), 0,04 (40°), 0,217 (100°); эфир—0,016 (кипящий),
90%-ный спирт — 0,37 (10 6°), 2,99 (78°); метанол—
1,67 (10,8°), 8,466 (50°); хлороформ — 0,04 (9,4°),
1,235 (56°); бензол 0,02 (9,4°), 0,011 (кипящий); 0,2 н.
раствор аммиака — 0,123 (25°). М. растворим в щело-
чах; сильное основание с кислотами дает соли: хлор-
гидрат (ЗН2О) и безводный, т. пл. 200°, [а]д =
=—111,5° (вода), растворим в воде (1 : 17,2 при 25°
и 1 : 0,5 при 80°); сульфат (H2SO4 -5Н2О), [а]д =
=—100,47° (вода), растворим в воде (1 : 15,5 при 25°
и 1 : 0,7 при 80°); пикрат, т. пл. 163—165°. Метиловый
эфир М.— алкалоид кодеин — при однократном гоф-
мановском распаде образует метилморфиметин (II),
к-рый при повторном гофмановском распаде дает ме-
тилморфенол (3-метокси-4,5-оксидофенантрен), а при
обработке уксусным ангидридом переходит в метил-
морфол (З-метокси-4-оксифенантрен) (м о р ф о л ь-
ный распад). При действии на М. НС1 или
Н3РО4 (140° в автоклаве) образуется апоморфин (III).
Вещество, имеющее циклич. скелет М., но без функцио-
нальных групп и двойной связи, называется N-метил-
морфинан (IV); получено синтетически. При действии
на М. окислителей происходит связывание двух
молекул М. и образуется ф-морфин, имеющий строение
2,2'-биморфина. ф-М. также выделен из опия. Из
многочисленных производных М. следует отметить
также героин (О,О-диацетат), дионин (ароматич.—
О-этилморфин), анторфин (Кт-десметил-К-аллилмор-
фин), дилаудид (дигидроморфинон). М. обладает
5 асимметрия, атомами углерода, а также аллильной
системой и имеет ряд изомеров (изоморфины). а-Изо-
морфин имеет обратную конфигурацию у С(8), р- и
у-изоморфины — ОН-группу у С(8) и двойную связь
С(в) и отличаются между собой конфигурацией у С(8).
Описано несколько полных синтезов М. Выделение
алкалоидов из опия производят многократной экст-
ракцией горячей водой. Упаренный водный экстракт
обрабатывают спиртом и аммиаком и из выпадающего
осадка (после удаления водным спиртом меконово-
кислого аммония) при подкислении уксусной к-той
в фильтрате получают соль М., к-рую действием
аммиака переводят в основание.
М. является сильным аналгетиком, обладает не-
большим снотворным действием и вызывает эйфорию,
чем обусловлено привыкание к М. (морфинизм).
Тормозит условные рефлексы и усиливает действие
наркотических снотворных н местноанестезирующих
средств. Возбуждает рвотный центр и понижает возбу-
димость дыхательного н кашлевого центров, тормозит
двигательную и секреторную активность желудочно-
кишечного тракта. Понижает основной обмен. Прини-
мается в виде хлоргидрата внутрь нли подкожно как
болеутоляющее средство. М. входит также в состав
омнопона (смесь хлоргидратов алкалоидов опия, 48—
50% М.). Синтезированы многочисленные заменители
М., напр. лидол, промедол, фенадон и др.
Лит.: Bentley К. W., The chemistry of the morphine
alkaloids, Oxt., 1954; S m a 1 1 L. F., Chemistry of the opium
alkaloids, Washington, 1932. См. также при ст. Алкалоиды.
А. Е. Васильев.
МОТОРНОЕ ТОПЛИВО — топливо, используе-
мое в двигателях внутреннего сгорания: бензины.,
дизельное топливо, лигроин, керосин и др. (по преж-
ней терминологии и ГОСТ М. т. наз. топливо для ти-
хоходных дизелей: 4- и 2-тактных, бескомпрессорных,
компрессорных судовых и др.). В качестве М. т.
используют гл. обр. смеси дистиллятных и остаточных
продуктов прямой перегонки и крекинга нефти;
применяют также продукты полукоксования, коксова-
ния и гидрогенизации углей, сланцевые и торфяные
смолы, нек-рые растительные масла и от. Плотность
нефтяных и буроугольных М. т. 0,85—0,95, сланцевых
1,0. Содержание углерода 82—86%, водорода 9—13%.
Теплотворная способность М. т.: нефтяных ок.
10000 ккал/кг; полукоксования бурых углей ок.
9700 ккал/кг; сланцевых ок. 9500 ккал/кг; кам. уг. ок.
8700 ккал/кг. Чем выше отношение Н : С и больше их
суммарное содержание, тем выше теплотворная спо-
собность и другие качества М. т. Поверхностное натя-
жение М. т. в среднем 30 дин/см при 15°, при нагре-
вании уменьшается приблизительно на 1 дину на
каждые 10°. Теплоемкость 0,47—0,50 ккал/кг, теплота
испарения ок. 40 ккал/кг. В табл. 1 приведены другие
технич. константы разных марок М. т.
Таблица 1
Константы ДТ-1 (М-3) ДТ-2 (М-4) ДТ-3 (М-5) ДТ-1 (из восточных нефтей)
Вязкость при 50°, сст . . . 36,0 55,3 66,6 20,5
Коксуемость, %, не более 3,0 3,5 4,0 5,2
Зольность, % » » 0,04 0,08 0,08 0,05
Содержание серы,%,не более 0,5 0,5 0,5 1 ,5
Т. всп., °C, не ниже .... 65 65 65 65
Т. заст., °C, не выше . . . —5 -5 + 5 -5
Необходимый сорт М. т. выбирают в зависимости
от быстроходности двигателя и условий его использо-
вания (темп-ра окружающего воздуха, наличие подо-
грева топлива, его фильтрации, центрифугирования
и т. д.). При использовании М. т. с высоким содержа-
нием серы (1,5—3%), особенно в свободно-поршневых
генераторах газа (СПГГ), необходимо применять спе-
циальные дизельные масла с присадками.
М. т. применяют также в газовых турбинах. В этом
случае исключительно большое значение имеют вели-
чина и состав зольного остатка. По существующим
требованиям зольность топлива для газовых турбин
11*
327
МОЧЕВАЯ КИСЛОТА — МОЧЕВИНА
328
не должна превышать 0,03%, а содержание в золе ва-
надия 0,0005—0,001%. Чем легче фракционный состав
газотурбинного топлива, тем меньше зольность и луч-
ше качество, при этом колебания в содержании серы
решающего значения не имеют. Основные свойства и
состав золы низкокачественного (А) и высококачест-
венного (Б) М. т. для газовых турбин приведены в
табл. 2.
Таблица 2
Константы Топливо А Топливо Б
Плотность при 20° 0,939 0,857
Коксуемость, % Теплотворная способность, 4,4 0,6
ккал/кг .. ........ . 9980 10194
Содержание серы, % 1,07 1 ,28
Содержание золы, % 0,02 0 ,0036
Наиболее перспективными М. т. для газовых турбин
могут быть дистиллятные продукты контактного и за-
медленного коксования нефтяных остатков, к-рые
практически не содержат ванадия и других ---------------
металлов. °C ... .
Лит.: Пучков Н. Г., Дизельные топлива,
М., 1953; Моторные, реактивные и ракетные тон- г/100 г воды
лива. Сб. под ред. К. К. Папок и Е. Г Семе- ------------
нидо, М., 1962. Н. Г. Пучков.
МОЧЕВАЯ КИСЛОТА (2,6,8-триоксипурин)
CjHaOsNa, мол. в. 168,12 — продукт азотистого обмена
в организме; бесцветные кристаллы; разлагается ниже
темп-ры плавления; плохо растворима в воде
(6,45 л1г/100 мл при 37°), спирте и эфире, растворима
в глицерине и горячей серной к-те. М. к. существует
в двух таутомерных формах — енольной (I) и кетонной
(II), причем равновесие сильно смещено в сторону по-
следней. Как кислота М. к. образует два ряда солей
(уратов) — MeCjHsOaNi и Ме2С5Н20зМ4. Двуметаллич.
ураты щелочных металлов легко растворимы в воде,
однометаллич. (за исключением литиевой соли) —
трудно. М. к. легко алкилируется по атомам азота,
с РОС1з образует 2,6,8-трихлорпурин. При окисле-
нии азотной к-той М. к. частично превращается в ал-
локсантин (III), к-рый при обработке аммиаком дает
N
мурексид (IV) пурпурно-красного цвета (м у р е к-
сидная реакция). Эта реакция, применяемая
для идентификации М. к , характерна для многих
пуриновых соединений. При окислении М. к. в ней-
тральном или щелочном р-рах разрушается пирими-
диновое кольцо и образуется аллантоин (V). М. к.
выделяют из гуано; синтетически получается из бар-
битуровой кислоты или из мочевины и циануксусного
эфира.
В нормальной моче человека и млекопитающих
М. к. содержится в незначительных количествах, но
при нек-рых нарушениях обмена веществ (подагра)
выделение М. к. резко возрастает. Камни мочевого пу-
зыря и почек состоят из М. к. или уратов. Для их
разрушения применяют соединения лития. У человека
М. к.— конечный продукт обмена пуринов, но у жи-
вотных она расщепляется в аллантоин под влиянием
фермента уриказы. У птиц и пресмыкающихся М. к.—
основной продукт азотистого обмена и поэтому ее со-
держание в гуано доходит до 25%. Применяют М. к.
для синтетич. получения кофеина.
Лит.: Каррер П., Курс органической химии, пер.
с нем., Л., 1960. А. Е. Васильев.
МОЧЕВИНА (карбамид) CO(NH2)2, мол. в. 60,06—
кристаллы (тонкие иглы или плоские призмы) без
цвета и запаха, холодящие на вкус; т. пл. 132,7°;
d23 1,335; 1,484; сублимируется в вакууме при 120—
130° (без разл.). Аэровзвесь с концентрацией до
500 г/м3 воздуха не взрывается. М. растворима в во-
де, спиртах, жидких аммиаке и сернистом ангидриде;
мало растворима в эфире, нерастворима в хлороформе.
Для водных р-ров характерна способность к пересы-
щению; растворимость в воде:
0 | 10 | 20 | 30 | 40 | 50 | 60 | 80 |100
67 | 84 | 104,7 | 135,3| 165,3 | 205 | 246 | 400 | 733
Растворимость (20°): 20 г в 100 г этанола, 50 г в 100 г
глицерина. Теплоемкость 22,4 кал/молъ -град (24,8°),
теплота образования (из элементов) 79 ккал/молъ',
теплота сгорания 151,6 ккал/молъ', теплота плавле-
ния 57,8 кал/г, теплота растворения в воде и спир-
те: 57,8 и 50,2 кал/г соответственно. Кристаллы М.
обладают пьезоэлектрич. свойствами.
Н. .Н
Структурная ф-ла /N—с—и плоское строение
о
подтверждены методами рентгенография., нейтроно-
графия. протонного магнитного резонанса и по спект-
рам поглощения в ИК-области. Таутомерная форма
Н—N=C—О—Н
(изомочевина) | проявляется в реакциях
нек-рых производных, напр. О-алкиловые эфиры
изомочевины могут быть получены реакцией соля-
нокислого цианамида со спиртами:
ст сн,о.
)C = NH+CH3OH—> >C=NH-HC1
H2NZ h,nz
Подобно иминоэфирам производные изомочевины яв-
ляются сильными основаниями. Ионы М. в водных
р-рах имеют строение H2NCONH3 и H2NC(NH)O~.
Константа диссоциации А=1,5-10“14 (25°). М.—
полный амид угольной к-ты, амид карбаминовой
к-ты, весьма реакционноспособна, образует комплекс-
ные соединения с многими веществами, напр.
NaCI CO (NH2)2.H .O; CaSO4-CO (NH2)2-H2O
H202 CO (NH2)2; CH.OH-CO (NH1),
Нек-рые из этих соединений устойчивы, напр. с
Н2О2 до 85°.
При нагревании до 150° и выше М. последователь-
но превращается в NH4NCO, NH3, СО2, биурет, циану-
ровую к-ту; в замкнутом сосуде при развивающемся
давлении, особенно с добавкой NH3, образуются также
продукты амминирования циануровой к-ты: аммелин,
аммелид, меламин. Расплавленная М. реагирует с
щелочными металлами и их амидами с образованием
солей цианамида:
H2NCONH2 + Na —> NaHCN2 + 0,5H2 + NaOH
H2NCONH2 + NaNH2 —> NaHCN24 H,O+NH,
При сплавлении с содой М. разлагается с образовани-
ем NaCNO, СО2, NH3 и Н2О. При сплавлении с
329
МОЧЕВИНА
330
NH4NO3 в присутствии силикагеля образуется гуани-
дин, NHs и СО2.
В водных растворах М. при нагревании протекают
реакции изомеризации, гидролиза и деаминирования;
образуются NH4NCO, NHs, СО2, биурет, циануровая
к-та. Гидролиз ускоряется в присутствии щелочей и
особенно кислот. Под действием фермента уреазы,
находящегося во многих организмах и семенах нек-рых
растений (соевые бобы и др.), а также конц. H2SO4
гидролиз протекает количественно. С HNOs, НС1,
Н3РО4, Н2С2О4 и др. кислотами М. образует соли.
В солеобразовании участвует только одна аминогруп-
па. Нек-рые из этих солей мало растворимы в воде,
напр. 1 часть H2NCONH2 ЩСгСи в 23 частях Н2О
при 16°. Нитрат М. СО (NH2)2-HNOs при нагрева-
нии разлагается со взрывом; при обработке на холоду
серной кислотой превращается в нитромочевину
H2NCONHNO2.
При взаимодействии М. с хлорсульфокислотами об-
разуются амидосульфокислоты, с конц. олеумом —
сульфаминовая к-та, с уксусным ангидридом при
140°— ацетамид и диацетамид (но в присутствии конц.
H2SO4 при 60°—ацетилмочевина с количественным вы-
ходом), с хлором на холоду — хлор- и дихлормочеви-
на [H2NCONHCI и CO(NHC1)2], с бромом — циануро-
вая к-та, NH4Br и N2, с гипохлоритом Na в щелочном
р-ре— гидразин (выход 50% и более), с CS2 при 110°—
NHiCNS и COS. К действию Н2О2 и КМпСН М. устой-
чива .
При действии на М. алкилирующих средств образу-
ет алкилзамещенные:
RJ+H2NCONH, —RNHCONH3 + HJ
при действии спиртов — уретаны:
ROH + H2NCONH2—> h2ncoor 4- nh2
с кислотами, их ангидридами, хлорангидридами и
сложными эфирами — уреиды:
RCOC1 + H2NCONH2 —> rconhconh2 + hci
Если эти к-ты двухосновные, то уреиды могут быть
кислыми и циклич. (напр., с натриймалоновым эфиром
в алкогольном р-ре образуется циклич. уреид — бар-
битуровая кислота). При нагревании М. с нек-рыми
арилапгидридами двухосновных к-т образуются соот-
ветствующие имиды. Напр., из фталевого ангидрида
при 150°— фталимид с выходом 90%. О реакциях М.
с альдегидами и кетонами см. Смолы карбамидные.
С аминами М. образует высокомолекулярные продукты
поликонденсации типа H2N(CH2)„[NHCONH(CH2)„]m,
с анилином — фенил- и дифенилмочевину; с гидра-
зином — семикарбазид (H2NCONHNH2) и гидразо-
формамид (H2NCONH)2.
Для открытия М. используют образование осад-
ков солей CO(NH2)2-HNO3, CO(N Н 2) 2 • Н 2С2О4 и
2CO(NH2)2-Hg(NO3)2-3HgO (последняя образуется
при действии р-ром азотнокислой окиси Hg па под-
кисленный р-р М.). Со свежеосажденной окисью Ag
образуется осадок CH2N2OAg, растворимый в NH3.
Солянокислые р-ры М. окрашиваются в фиолетово-
пурпурный цвет при добавлении фурфурола. Возмож-
но также использование биуретовой реакции, посколь-
ку биурет образуется при нагревании М. и ее р-ров.
Для количественных определений М. гидролизуют в
присутствии H2SO4 или уреазы и определяют NH3 или
окисляют титрованным слабощелочным р-ром гипо-
бромита калия с определением избытка гипобромита
или объема выделяющегося азота (А. П. Бородин).
Весовое определение (чувствительность 0,4 мкг) осно-
вано на выделении из подкисленного уксусной к-той
раствора М. в метаноле осадка диксантилмочевины.
По интенсивности желто-зеленого окрашивания,
появляющегося при взаимодействии подкисленного
соляной к-той раствора М. с п-диметиламинобензаль-
дегидом, можно определять М. в концентрациях от
50 до 240 частей на 1 млн., с ошибкой, не превышаю-
щей 1 %.
Промышленный способ получения М. основан на
реакции, открытой А. И. Базаровым (1870), и состоит
во взаимодействии аммиака и двуокиси углерода;
промежуточным продуктом в этом синтезе является
карбамат аммония:
2NHS+CO3 = H3NCOON1I4; ДН298=-38,0 ккал/г-моль (1)
H2NCOONH4 = H3NCONH2 + H3O; 4Н°9!= + 6,7 ккал/г-моль (2)
Реакции образования карбамата аммония и его пре-
вращения в М. обратимы. Степени превращения СО2
в карбамат аммония и последнего в М. зависят от
темп-ры, давления, весового соотношения между ис-
ходными веществами, примесей инертных газов и во-
ды, а также времени пребывания реагирующих веществ
в реакционном объеме. Обычно обе реакции проводят-
ся непрерывно и одновременно в проточном реакцион-
ном аппарате — колонне синтеза. При этом теплота,
выделяющаяся по реакции (1), используется для нагре-
вания реагирующих веществ до темп-ры процесса и
компенсации теплового эффекта реакции (2). В раз-
личных промышленных способах синтез М. осуществ-
ляется при 160—200° и давлении 100—400 кг/см2.
После снижения давления из продуктов реакции —
четверного р-ра карбамата аммония и М. в Н2О и
NH3 отгоняется NH, (свободный и связанный в кар-
бамате аммония) и СО2. Из дстатка — водного рас-
твора М.— продукт выделяется в виде кристаллов
или гранул.
М. широко применяется в пром-сти и с. х-ве. Она
является исходным материалом для получения карб-
амидных смол, цианатов Na и К, гидразина, гидразо-
формамида (при получении порофора азодикарбонами-
да), циануровой к-ты и ее эфиров (напр., аллилового),
фармацевтич. снотворных препаратов: веронала, люми-
нала, бромурала и мн. др., нек-рых красителей.
В нефтяной пром-сти применяют для депарафиниза-
ции масел. М. входит в состав гигиенич. средств (зуб-
ные пасты) и косметич. препаратов (кремы). Нек-рые
производные (напр., сим.-диэтилдифенилмочевина под
названием централит) — стабилизаторы бездымных
порохов. Продукты конденсации М. с нек-рыми диа-
минами [напр., H2N(CH2)sNH2] стали применяться для
получения синтетич. волокна «урилон» (Япо-
ния). Экономически целесообразно получение мелами-
на из М.
В растениеводстве М. применяется в качестве
высококонцентрированного (46,55% N2), легко усваи-
ваемого азотного удобрения; пригодна на всех почвах
и под все культуры (особенно для внекорневой под-
кормки растений).
В животноводстве М. применяется в качестве до-
бавки к кормам для жвачных животных. Такая добав-
ка позволяет заменить в кормовых рационах 25—30%
белковых веществ, содержащихся в фуражном зерне,
бобовых культурах, масличных жмыхах, люцерновой
муке и т. д. Попадая в рубец (часть преджелудка
жвачных животных), мочевина гидролизуется с об-
разованием аммиака, из к-рого бактерии микрофлоры
преджелудка синтезируют белок собственного тела.
Суточная доза мочевины 20—25 г на 100 кг живого
веса. Скармливание производится в смеси с кормами
(но не в виде пойла).
Мочевина — конечный продукт белкового обмена у
многих беспозвоночных животных и у большинства
позвоночных — рыб, земноводных, млекопитающих,
человека. У позвоночных выделяется почками и,
частично, потовыми железами. В животных организ-
мах содержится в небольших количествах в мышцах,
331
МОЩНОСТЬ ДОЗЫ—МОЮЩЕЕ ДЕЙСТВИЕ
332
крови, лимфе, слюне, молоке, слезах. Обнаружена в
тканях растительных организмов (грибы, нек-рые
высшие растения).
М. открыта И. Руэллем (1773) в моче, идентифициро-
вана У. Праутом (1818), впервые синтезирована Ф.
Велером (1828) из хлористого аммония и циановокис-
лого серебра. Этот синтез оказал существенное влия-
ние на становление современной органической химии.
Н. А. Гольдберг.
Производные М.— активные гербициды,
применяют для борьбы с сорными растениями.
Практически для борьбы с сорными растениями ис-
пользуют след, производные М.:
Т. пл., °C Растворимость в воде, %
Ы-фенил-Ы', N'-диметилмочеви- на (феиидим, фенурон) . . . Ы-4-хлорфенил-Ы', N'-диметил- мочевина (хлорфенидим, мо- нурон) 136 0,29
171,5 0,023
М-3,4-дихлорфенил-Ы', N-диме- тилмочевина (диурон, ди- хлорфенидим) . 158—159 0,0042
N-З ,4-дихлорфенил-М'-метил- N'-бутилмочевина (небурон) . 101,5-103 0 ,00048
М-3,4-дихлорфенил-Ы'-метил • N'-метоксимочевина (герби- цид 326) 93 0,0075
N -циклооктил-N' ,N' • диметил- мочевина 138 0,11
М-хлорборнил-М',Ы'-диметил- мочевина (геркулес 7175) . . 203—204 —
N-тетрагидродициклопентадие- нил-Н',Н'-диметилмочевина (геркулес 7 531) 168
Почти все перечисленные соединения применяют в
виде смачивающихся порошков для обработки почвы
перед всходами; содержание действующего начала
50—80%, нормы расхода 0,5—5 кг!га (при больших
нормах расхода они проявляют себя как сплошные
гербициды).
Получают производные М. взаимодействием соот-
ветствующих изоцианатов с аминами:
ArNCO + HN (СН3)2 —> ArNHCON (СНа)2
Все производные М. умеренно токсичны.
Н. Н. Мельников,
Лит.: Кретов А. Е. Кальций-цианамид и продукты
его переработки, М.—Л., 1934, с. 166—89; Гольдберг
Н. А., Ж. Всес. хим. о-ва им. Д. И. Менделеева, 1961, 6,
№ 1, с. 49—58; Гольдберг Н. А. [и др.], Хим. наука и
пром-сть, 1956, 1, № 6, 669; Ш м а н е н к о в Н. А., Исполь-
зование мочевины в животноводстве, М., 1960; Мельни-
ков Н. Н., Баскаков Ю. А., Химия гербици-
дов и регуляторов роста растений, М., 1962; ЗотовА. Т.,
Мочевина, М., 1963; Z b i rovsky М., Му ska J., Z е-
m A n е k J., Herblcidy, Praha, 1960.
МОЩНОСТЬ ДОЗЫ (в радиационной химии) —
мера интенсивности поля рентгеновского или гамма-
излучения в данном месте, определяемая по скорости
ионизации, к-рая может быть вызвана этим излуче-
нием. Произведением, д. на время действия излучения
дает дозу, или дозу облучения, измеряе-
мую в рентгенах. Поэтому единицей М. д. является
р/сек. Наряду с дозой облучения рассматривают
поглощенную дозу, определяя эту величи-
ну по энергии излучения, поглощенной единицей
массы вещества. Это понятие может применяться ко
всем видам излучения радиоактивных веществ. Соот-
ветственно, количество энергии излучения, поглощае-
мое единицей массы вещества в единицу времени, наз.
мощностью поглощенной дозы. Еди-
ницы измерения; радДек и зв/г-сек. В настоящее
время измерение дозы облучения в рентгенах выходит
из употребления и вытесняется измерением поглощен-
ной дозы в радах. Поэтому мощность поглощенной
дозы часто для краткости называют простом, д. При
энергиях излучения 0,3—3 Мэе для воды и биологич.
тканей 1 р/сек=0,93 ра5/сек=5,87 • 10” зв/г-сек. М. д.
пропорциональна активности источника излучения и
зависит от энергии излучения, а также геометрии пре-
парата. Точечный гамма-источник активностью в
1 кюри создает на расстоянии 1 м М. д. порядка 1 р/час.
М. д. от источника гамма-лучей в ряде случаев можно
рассчитать. Однако вычисления весьма громоздки и
обычно дают лишь приближенные результаты. Вслед-
ствие этого для получения точных данных всегда при-
ходится прибегать к экспериментальным измерениям.
Для определения М. д. применяют те же методы, что
и для определения интегральной дозы (см. Доза
ионизирующего излучения. Дозиметрия}. Для воды и
водных р-ров широко применяется дозиметрия хи-
мическая.
М. д. определяет скорость генерирования активных
частиц в системе и, следовательно, скорость химич.
процесса. Когда все образующиеся активные частицы
захватываются акцептором, скорость процесса прямо
пропорциональна М. д. С ростом М. д. при постоянной
концентрации акцептора могут возникнуть условия
для рекомбинации активных частиц; в этих условиях
зависимость скорости процесса от М. д. носит более
сложный характер. Изучение подобных зависимо-
стей в ряде случаев позволяет сделать выводы о соот-
ношении констант скорости реакций активных частиц
с акцептором и процесса рекомбинации.
Лит.: Аглинцев К. К., Дозиметрия ионизирующих
излучений, 2 изд,, М., 1957; Б р е г е р А. X. [и д р.), Проб-
лемы физической химии, 1959, вып. 2, 132; С 1 1 ngm а п
W. Н., J. Chem. Phys,, 1957, 27, № 1, 322; Б у р д у н Г. Д„
Единицы физических величин, 2 изд., М., 1962; В е р е-
щинский И. В П и к а е в А. К., Введение в радиа-
ционную химию, М., 1963. В. Н. Шубин.
МОЮЩЕЕ ДЕЙСТВИЕ — способность моющих
веществ и их растворов удалять прилипшие к отмывае-
мым поверхностям частицы загрязнений и переводить
их во взвешенное состояние в виде эмульсии или сус-
пензии. Моющие вещества относятся к группе поверх-
ностно-активных веществ. Состав и свойства загряз-
нений крайне разнообразны: одни из них жидки, дру-
гие — твердообразны; органич. соединения (нефтяные
и растительные масла, жиры, смолы, сажа) гидрофоб-
ны, т. е. не смачиваются водой, минеральные же час-
тицы обычно гидрофильны, но могут быть гидрофоби-
зированы жировой частью загрязнения. Резко отличны
друг от друга также и природа, и свойства отмывае-
мых поверхностей — тканей (хлопок, шерсть, синте-
тич. полимерные волокна), металлов, стекла, кера-
мики, кожи человека и животных и т. д. Поэтому,
хотя поверхностно-активные моющие вещества дейст-
вуют по одному универсальному механизму, адсор-
бируясь на всех поверхностях раздела, участвующих
в процессе отмывания, эффективность их действия
весьма различна.
Для проявления достаточно эффективного М. д.
поверхностно-активные вещества должны обладать
определенным молекулярным строением и свойства-
ми, к-рые и позволяют отнести их к особому классу
моющих веществ. Эти свойства и структура
характерны для одного из лучших моющих средств—
обычного жирового мыла, и поэтому все многочислен-
ные синтетич. моющие вещества рассматриваются как
типичные мыла. Помимо мыл, сложные моющие средст-
ва включают еще и разного рода активирующие до-
бавки — электролиты, гидрофильные защитные кол-
лоиды-стабилизаторы, препятствующие обратному
прилипанию отмытых частиц загрязнения к очищен-
ной поверхности.
Наряду с сильной поверхностной активностью и
смачивающим действием моющим веществам свойст-
венна высокая стабилизующая способность по отно-
шению к частицам загрязнений, что и отличает их от
333
МОЮЩЕЕ ДЕЙСТВИЕ—МОЮЩИЕ СРЕДСТВА
334
поверхностно-активных веществ вообще. Все поверх-
ностно-активные вещества содержат молекулы с гид-
рофильной функциональной (полярной) группой и
длинной неполярной углеводородной цепью; у мою-
щих веществ последняя содержит не менее 12 и не
более 18 атомов углерода. Именно при такой структу-
ре у ионогенных моющих веществ, молекулы к-рых
диссоциируют в водных р-рах с образованием длинно-
цепочечных органич. ионов (напр., Ci2H2SSO3Na;t
С12Н2580з_+ Na+), одной полярной группы оказы-
вается достаточно для того, чтобы создать необходимое
сочетание гидрофобных и гидрофильных свойств
(т. наз. «молекулярный баланс»), В молекулярных же
р-рах неионогенных веществ, напр. C12H2S—О—
—(С2Н4О)ПОН, для этого необходимо несколько неио-
низированных полярных групп на каждый углево-
дородный радикал.
При правильно подобранном балансе свойств мыла
приобретают необходимую растворимость, позволяю-
щую получать их растворы достаточно больших кон-
центраций с оптимальными коллоидо-химич. свойст-
вами. Эта коллоидизация проявляется в том, что
в объеме растворов происходит мицеллообразование,
а адсорбционные слои на поверхностях раздела при-
обретают особые прочностные свойства: в соответствии
с общим термодинамич. принципом минимума свобод-
ной поверхностной энергии, в этих растворах, начи-
ная с нек-рой определенной «критической» концент-
рации, углеводородные цепи изолированных ранее
молекул (ионов) ассоциируют друг с другом, образуя
мицеллы с внутренним углеводородным жидкообраз-
ным ядром и наружной поверхностью, состоящей из
гидратированных полярных групп. В относительно
разб. р-рах, обычно применяемых на практике,
такие мицеллы сферичны, состоят из 70—100 плотно
упакованных молекул (ионов) и имеют диаметр,
равный их удвоенной длине (порядка 30—50 А).
Мицеллообразование вызывает очень резкие изме-
нения как объемных, так и поверхностных свойств
растворов и, в частности, приводит к возникновению
у них нового свойства — солюбилизирующей способ-
ности (коллоидной растворимости, см. Солюбилиза-
ция). Указанные особенности моющих веществ и их
коллоидо-мицеллярных р-ров и определяют их повы-
шенное М. д. Оно слагается из двух последовательных
стадий: 1) смачивания отмываемой поверхности р-ров
моющего вещества, вследствие чего частицы загряз-
нения переходят в объем раствора; 2) стабилизации
частиц, что предохраняет их от взаимного слипания
(коагуляции) и повторного осаждения на поверх-
ность (см. Стабилизация дисперсных систем).
Хорошее смачивание обеспечивает контакт раствора
с загрязненной, обычно гидрофобной, поверхностью,
что необходимо для дальнейшего воздействия на нее
моющего вещества. Вследствие высокой поверхностной
активности мыл в их растворах уже при концентрации
— 0,1—0,2% можно получить предельно сниженное
поверхностное натяжение (28—30 эрг/см2 на границе
раздела раствор — воздух и ниже 5 эрг/см2 на грани-
це раствора и углеводородной фазы). Адсорбция из
таких растворов, в соответствии с ур-нием Гиббса (см.
Гиббса уравнение адсорбции), приводит к покрытию
поверхности частиц загрязнения и твердой подкладки
насыщенными плотно упакованными адсорбционными
слоями, что нарушает прилипание (адгезию) между
ними. Механич. воздействие (стирка ткани, трение по
твердой поверхности) и солюбилизация частиц облег-
чают этот процесс. Эта стадия процесса отмывания —
необходимое, но недостаточное условие для эффектив-
ного М. д., что следует хотя бы из того, что она может
быть проведена с помощью многих поверхностно-
активных веществ, не относящихся к моющим ве-
ществам.
В отличие от смачивающей способности, другая важ-
ная технология, функция мыл — стабилизирующее
действие — уже непосредственно связана с коллоидо-
химич. свойствами их растворов. Для образования
устойчивых эмульсий и суспензий частиц загрязне-
ний, отделенных от подкладки, необходимо, чтобы ад-
сорбционные слои обладали определенными струк-
турно-механич. (защитными) свойствами — повышен-
ной вязкостью, упругостью или прочностью. Эти свой-
ства, характерные для типичных мыл жирных к-т,
образующих гелеобразные адсорбционные слои, в
значительной мере определяются поверхностным ми-
целлообразованием, к-рому благоприятствует сильно
повышенная концентрация мыла на поверхностях раз-
дела и, как следствие этого, сильное взаимодействие
адсорбированных молекул (ионов) на близких рас-
стояниях. Особенно резко проявляется такое взаимо-
действие у высоких гомологов (С12—С18) с неразветв-
ленными цепями и не содержащих ненасыщенных свя-
зей, способных образовывать компактно упакованные
мицеллы. Отмывание загрязнений обычно сопровож-
дается вспениванием растворов, возникающим вследст-
вие адсорбции моющих веществ на поверхности раздела
раствор—воздух. Однако пенообразование не является
необходимым для осуществления М. д., хотя и спо-
собствует удалению отмытых частиц загрязнения.
Синтетич. ионогенные моющие вещества, в отличие
от жирового мыла, образуют адсорбционные слои с
слабо выраженными защитными свойствами, и поэто-
му, чтобы проявить стабилизирующее действие, они
должны применяться, особенно в процессах мытья
тканей, в виде композиций в смеси с электролитами
и т. н. защитными коллоидами (напр., с карбоксиме-
тилцеллюлозой). Эти добавки сильно повышают аг-
регативную устойчивость частиц загрязнений в ре-
зультате повышения поверхностной активности мою-
щих веществ, облегчения мицеллообразования в их
растворах, повышения прочности и вязкости их ад-
сорбционных слоев. Особое значение в качестве акти-
вирующих электролитов наряду с сульфатом, фосфатом
и силикатом натрия и др. имеют полифосфаты общей
ф-лы Na„+2P„OJn+l. Их высоковалентные ионы уси-
ливают М. д., связывая путем комплексообразования
катионы солей, сообщающих жесткость воде, повышая
пептизацию частиц неорганич. загрязнений и тем
самым снижая возможность их прилипания к отмывае-
мой ткани. М. д. обладают не только растворимые по-
верхностно-активные вещества, но и нерастворимые
твердые эмульгаторы (напр., бентонитовые глины),
высокодисперсные частицы к-рых (размерами в доли
микрона) в водных суспензиях покрывают капельки
жидких загрязнений и устойчиво эмульгируют их
(см. Эмульсии).
Указанные закономерности явлений, лежащих в
основе М. д., имеют лишь качественный характер и не
позволяют пока установить однозначной связи между
эффективностью М. д. и многочисленными факторами,
влияющими на него. Механизм М. д. очень сложен, что
связано с различиями в составе, молекулярном строе-
нии и коллоидо-химич. свойствах моющих веществ, с
многообразием свойств отмываемых поверхностей и
загрязнений, а также с характером молекулярных
взаимодействий между компонентами системы, в
к-рой осуществляется М. д. См. также Адсорбция,
Дисперсные системы, Пены, Суспензии, Флотация.
Лит.: Шварц А., Перри Дж.. Берч Дж.,
Поверхностно-активные вещества и моющие средства, пер.
с англ., [2 изд.], М., 1960; Штюпель Г., Синтетические
моющие и очищающие средства, пер. с нем., М., 1960; Клей-
тон В., Эмульсии, пер. с англ., М., 1950; Хим. наука
и пром-сть, 1959, 4, №5 (статьи П. А. Ребиндера, А. Б. Тауб-
мана и др.); Ребиндер П. А., Поверхностно-активные
вещества, М., 1961. А. Б. Таубман.
МОЮЩИЕ СРЕДСТВА — технические препараты,
применяемые в водных р-рах для отмывания загряз-
335
МОЮЩИЕ СРЕДСТВА
336
нений с поверхности различных материалов, кожи и
волос человека и животных. К М. с. принадлежат мыла
и разнообразные, сравнительно сложные по составу
синтетич. препараты. В состав синтетич. М. с. в ка-
честве активной основы входят поверхностно-актив-
ные вещества и различные добавки: щелочные и нейт-
ральные электролиты, перекисные соединения, т. н.
оптич. отбеливатели, вещества, препятствующие ре-
сорбции загрязнений, и др. Применяемые органич.
поверхностно-активные вещества делят на 4 основных
класса: анионоактивные, катионоактивные, неионо-
генные и амфотерные.
К анионоактивным веществам относят, кроме обыч-
ных мыл, алкилсульфаты (первичные и вторичные),
алкиларилсульфонаты, алкилсульфонаты.
Алкилсульфаты получают обычно из жир-
ных спиртов, олефинов и парафиновых углеводо-
родов.
Для получения алкилсульфатов из жирных спиртов послед-
ние сульфатируют. В качестве сульфирующих агентов исполь-
зуют серную к-ту (моногидрат, олеум), хлорсульфоновую к-ту
или серный ангидрид. Сульфатирование проводят при возмож-
но более низкой темп-ре и при интенсивном охлаждении реак-
щгонной массы. Получаемые сульфоэфиры нейтрализуют
щелочами (NaOH, КОН, NHtOH) или органич. основаниями
(моно-, ди- и триэтаноламины). После нейтрализации сульфо-
эфиров получают пастообразный продукт, состоящий из ал-
килсульфатов (ок. 40%), сульфата натрия, хлористого натрия
(при сульфатировании хлорсульфоновой к-той) и воды.
У алкилсульфатов с прямой углеводородной цепью
моющая способность, возрастающая с длиной цепи,
проявляется, начиная с децилсульфата, и достигает
оптимума (при темп-ре до 40°) у алкил сульфатов с 12—
14 атомами углерода. Алкилсульфаты с 16 атомами
углерода в цепи и выше очень мало растворимы в воде
при комнатной темп-ре; при взаимодействии с жесткой
водой образуются малорастворимые кальциевые соли.
С повышением темп-ры растворимость натриевых и
кальциевых солей повышается. Вторичные алкилсуль-
фаты обладают более низкой моющей способностью по
сравнению с первичными (чем полярная группа ближе
к центру молекулы, тем хуже моющая способность).
При расположении полярной группы у второго или
третьего атома углерода вторичные алкилсульфаты
при достаточной длине цепи по своим свойствам мало
отличаются от первичных. Вторичные алкилсульфаты
значительно уступают первичным в отношении термо-
устойчивости, и они тем более гигроскопичны, по
сравнению с первичными, чем ближе к центру молеку-
лы расположена полярная группа. Алкилсульфаты
гидролизуются тем легче, чем больше длина цепи.
Вторичные алкилсульфаты применяют гл. обр. для
произ-ва жидких М. с.; их получают сульфатирова-
нием олефинов, образующихся при крекинге парафи-
нов. В продажу вторичные алкилсульфаты выпуска-
ются в виде водного 20—40%-ного р-ра.
Алкилсульфаты применяют для стирки шерстяных
и шелковых тканей и для приготовления шампуней.
Наибольшее распространение среди синтетич. М. с.
получили алкиларилсульфонаты, преим.
моноалкилбензолсульфонаты общей ф-лы RC6H4SOsNa,
известные в СССР под названием сульфонолов.
Моноалкилбензолсульфонаты получают след, способами:
1) Непрерывным фотохимич. хлорированием парафиновых
углеводородов (очищенной деароматизированной керосиновой
фракции парафинистой нефти, жидких парафинов или синте-
тич. углеводородов) до присоединения ок. 12% хлора. Полу-
ченными алканхлоридами алкилируют бензол; алкилбензолы
фракционируют, а затем сульфируют олеумом или серным ан-
гидридом. 2) Алкилированием бензола полимерами пропилена
(преим. тетрапропиленом) в присутствии А1С13. Алкилбензолы
фракционируют (обычно отбирают фракцию, выкипающую
впределах 280—315°, средний мол. в. 235—240). Сульфирова-
ние алкилбензола и последующие операции проводят так же,
как и в первом случае. Количество несульфированных в ал-
килбензолсульфонате не должно превышать 2,5%.
У алкилбензолсульфонатов с алкильной цепью нор-
мального строения моющая способность начинает
проявляться с нонилбензолсульфоната и повышается
с увеличением длины цепи. Однако начиная с доде-
цилбензолсульфоната, снижается растворимость в воде
натриевых и тем более кальциевых солей; поэтому
применение алкилбензолсульфонатов с длинной пря-
мой цепью требует относительно высоких темп-р и
наличия в рецептуре водосмягчающих средств. В слу-
чае разветвленной алкильной цепи закономерности
усложняются, т. к. в этом случае имеет значение как
степень разветвленности, так и длина наиболее длин-
ной части цепи. Моющая способность диалкилбензол-
сульфопатов зависит от длины каждой из алкильных
цепей и степени их разветвленности. Алкилбензолсуль-
фонаты с разветвленной цепью гигроскопичны, а с
прямой — не гигроскопичны.
Алкилсульфонаты получают сульфо-
хлорированием или сульфоокислением парафиновых
углеводородов:
RHJ-SCb+Ch —s-RSO2C1 + HC1
RSO2Cl + 2NaOH —> RSO3Na+NaCl +H2O
Присоединение группы SO2CI может происходить к
любому атому углерода по всей длине цепи, поэтому
в результате реакции сульфохлорирования и сульфо-
окисления образуется смесь изомеров. Для получения
алкилсульфонатов используют синтетич. углеводоро-
ды или очищенную фракцию нефти, выкипающую в
пределах 180—310°. Поверхностно-активные свойства
и моющая способность алкилсульфонатов, как и у
вторичных алкилсульфатов, определяются общей дли-
ной алкильной цепи и положением полярной группы.
Катионактивные вещества имеют сравнительно
меньшее значение в качестве М. с. Они обладают бак-
терицидным действием и применяются совместно с
неионогенными соединениями в качестве дезинфици-
рующих М. с., а также используются в ряде отраслей
пром-сти (текстильная, флотация, дорожное строитель-
ство и др.). Наиболее распространенными продуктами
данного класса являются соли четвертичных аммоние-
вых оснований (I) и пиридиновые соединения (II).
Одним из исходных
продуктов для получе-
ния четвертичных ai -
мониевых соединений
служат жирные к-ты.
При взаимодействии их
с аммиаком получают
R', R", R'", R""— углеводород-
ные радикалы; X _ — анион(С1~,
Вг-, J-).
аммонийные мыла, ко-
торые при нагревании
переходят в амиды, а
затем в нитрилы. Гид-
рогенизацией нитрилов получают амины, взаимодей-
ствием амина с хлористым метилом — водораствори-
мые четвертичные аммониевые соединения.
Для получения неионогенных веществ используют
органич. соединения с активным водородом (жирные
спирты, жирные к-ты, жирные амины, алкилфенолы,
меркаптаны, а также талловое масло). Эти соединения
конденсируют с различным количеством молекул оки-
си этилена:
R
ОН + пСН2-СН2
R
-О-(СН2-СН2О)„Н
В зависимости от выбранной гидрофобной части моле-
кулы (R) и числа присоединенных молекул окиси
этилена можно получить продукты с различными фи-
зич. свойствами — от текучих жидкостей до твердых
тел. Свойства неионогенных продуктов зависят от
соотношения гидрофобной и гидрофильной частей мо-
лекулы и от природы гидрофобной части.
Амфотерные соединения могут быть приготовлены
из высокомолекулярных аминов путем конденсации
их с окисью этилена с последующим сульфатирована-
337
МОЮЩИЕ СРЕДСТВА—МУ КОВЕЩЕСТВА
338
ем полученного соединения, а также при взаимодей-
ствии аминов с хлоруксусной к-той:
rnh2 +С1СН2СООН —> RNHCII2COOII + IIC1
Амфотерные соединения в изоэлектрич. области ве-
дут себя как неионогенные вещества, в щелочной сре-
де — как анионактивные, а в кислой — как катион-
активные. Эти соединения обладают хорошей раст-
воримостью и смачивающей способностью и высо-
кой поверхностной активностью. При большой вели-
чине pH амфотерные соединения обладают хорошим
моющим действием; они являются ингибиторами кор-
розии.
Синтетич. поверхностно-активные вещества в чис-
том виде недостаточно эффективны для стирки белья и
других хозяйственных нужд. Для придания товарно-
му продукту надлежащего моющего эффекта в рецеп-
туру вместе с синтетич. поверхностно-активными ве-
ществами вводят такие вещества, как триполифосфат
натрия, углекислую соду, двууглекислую соду, ме-
тасиликат натрия, сульфат натрия, пирофосфаты нат-
рия (ди-, тетра-), тринатрийфосфат, гексаметафосфат,
перборат натрия, сульфат натрия. Чтобы предотвра-
тить осаждение на ткань загрязнений (ресорбция), к
М. с. добавляют в качестве стабилизатора карбокси-
метилцеллюлозу (подробнее см. Защитные коллоиды
и Моющее действие).
В качестве добавок к М. с. применяют а л к и л од-
ами д ы — неионогенные соединения, получаемые
конденсацией жирных к-т с моно- или диэтанолами-
Таблица 1
Компоненты Содержание компонентов в рецептуре, %
I 11 III IV V VI VII
Алкилбензолсульфонат .... Сульфат полиоксиэтиленового — 15 8 22—35 20 15 —
эфира жирного спирта. . . . — 5 8 — — — —
Алкилсульфаты Смесь алкилбензолсульфонатов — — 8 — — 10 —
и алкилсульфатов 22 — — — — —- 20
Алкилоламиды 5 — 3 — 2,5 — —
Триполифосфат натрия .... 30 40 40 37—48 50 30 25
Тетрапирофосфат натрия . . . — 10 — — —— 5 ——
Углекислая сода 1 5 — —— — — — 10
Силикат натрия 3 5 5 5-9 3 6 2
Силикат магния — — —— —— —— 2 3
Перборат натрия Конденсат жирных к-т с белко- — — — — — 16 15
выми веществами — — —— — — 2 —
Сульфат натрия 20 15 25 12-19 24 13 15
Карбоксиметилцеллюлоза . . . 2 0,5 0,03 1 0,5 0,5 1 —
Оптический отбеливатель . . . 0,02—0,2 0,03 — — —
Отдушка Вода 0,03—0,2 Ос таток до 100 % — —
ном. Моноэтаноламиды применяют преим. в порошко-
образных М. с., а диэтанолампды — в жидких. В ка-
честве жирных к-т используют кислоты кокосового
масла или синтетич. жирные к-ты из окисленного пара-
фина фракции С1о—С14. Моноэтаноламиды нераство-
римы в воде, но растворяются в растворах М. с. Ди-
этанолампды обладают заметной растворимостью в
воде. Алкилоламиды повышают пеноустойчивость и
моющее действие синтетич. М. с.; нек-рые из них после
сульфирования обладают самостоятельным моющим
действием. В М. с. вводят от 0,1% до 0,05% оптич.
отбеливателей, поглощающих коротковолновый УФ-
свет и излучающих голубой свет, что и повышает бе-
лизну ткани.
Синтетич. М. с. изготовляют преим. в порошкооб-
разном виде, реже — в виде жидких препаратов.
В табл. 1 приведены нек-рые рецептуры синтетич.
порошков для стирки хлопчатобумажных и льня-
ных тканей, в табл. 2 — для шерстяных и шелковых
тканей.
Таблица 2
Компоненты Содержание компонентов в рецептуре, %
1 1 11
Смесь алкилсульфатов и алкил* бензолсульфонатов 40
Конденсированные фосфаты . . 10 —
Сульфет натрия 47 60
Алкилбензолсульфонат .... — 30
Метафосфат натрия —- 10
Вода 3 —
Рецептуры нек-рых жидких синтетич. М. с., приме-
няемых для стирки шерстяных и шелковых тканей и
мытья предметов домашнего обихода, приведены ниже.
Рецептура I
Вторичные алкплсульфаты.............15%
Неионогенное вещество ОП-7..........10%
Триполифосфат натрия................ 4%
Сульфат натрия.....................2,7%
Вода.............................. остаток
до 100%
Рецептура II
Калийная соль алкиларилсульфокислоты 12%
Неноногенное соединение............. 2%
Диэтаноламид........................ 5%
Ксилолсульфонат (натриевая соль) . . . 15—18%
Пирофосфат калия....................12%
Спирт, карбоксиметилцеллюлоза, оптич.
отбеливатель .................... остаток
до 100%
Лит.: Т о в б и н И. М., Производство
синтетических моющих средств, М., 1949;
Неволин Ф. В., Синтетические мою-
щие средства, М., 1957;Товбин И. М.,
Пути развития производства синтетических
жирозаменителей и моющих средств, М.,
1959; Науменко П. В., Синтетические
жирозаменители, поверхностно-активные ве-
щества и моющие средства, М., 1960; Хим.
наука и пром-сть, 1959, 4, № 5; S t u р е 1
Н., Syuthetische Wasch- und Reinigungsmit-
tel, Stuttgart, 1954; Lindner K., Textil-
hilfsmittel und Waschrohstoffe, Stuttgart,
1954; Schwartz A. M., Perry J. W.,
В er ch J., Surface active agents, v. 1—2,
N. Y.—L., 1949—58. Ф. В. Неволин.
МУКОВЕЩЕСТВА(мукосоединения,
мукокомпле^сы, мукоидные веще-
ства) — природные высокомолекуляр-
ные соединения, построенные из эле-
ментов различных групп биополиме-
ров — белков, полисахаридов и ли-
пидов. Единой общепринятой класси-
фикации М. не существует, чаще
всего М. делят на группы, представ-
ленные в таблице:
Муковещество Компоненты Примеры
Мукополисаха- рид Мукопротеин Липопротеин Муколипоид Липополисахарид Полисахарид2 + бе- лок Белока+углевод Бе лок а+липои д Липои да + углевод (возможен и пептид или белок) Полисах арида+ли- поид Вещества групп крови Глобулины криви Липовителлин Антигены микро- организмов Воски микобактерий
а Преобладающий компонент.
Связи между компонентами в М. могут быть кова-
лентными или ионными. М. широко распространены
в природе, многие из них являются биологически
важными соединениями. См. также Белки и Полиса-
хариды.
Лит. см. при ст. Мукополисахариды. Л. И. Линевич.
339
МУКОНОВАЯ КИСЛОТА—МУРАВЬИНАЯ КИСЛОТА
МУКОНОВАЯ КИСЛОТА (гексадиен-2,4-дикарбо-
в. 142,11 — существует в
нс------------сн
и и
с с
н/Чсоон ноос н
цис, цис
новая-1,6 кислота), мол.
виде трех изомеров:
нс —сн
и и
ноос-сн нс-соон
транс, транс
НС------- СН
II II
нс-соон н-с-соон
цис, транс
Наиболее устойчивой и изученной является транс,
транс-М. к., т. пл. 305° (с разл.). Она может быть
получена осторожным окислением фенола надуксус-
ной к-той или нагреванием fl, р'-дибромадипиновой
к-ты с пиридином или едким кали наряду с другими
продуктами. Транс, транс-М. к. количественно осаж-
дается ацетатом меди из нейтральных водных р-ров, в
виде медной соли. М. к. может быть восстановлена до
гидромуконовой к-ты НООССН2СН=СНСН2—СООН
и до адипиновой кислоты; полимеризуется при 200°
под давлением. Цис, цис-М. к., т. пл. 187—188°, по-
лучается окислением е-хинона надуксусной кислотой.
Ее эфиры уже при комнатной темп-ре постепен-
но изомеризуются в эфиры транс, транс-М. к. Ди-
метиловый эфир транс, транс-М. к., т. пл. 159°;
цис, цис-М. к., т. пл. 75°; диэтиловый эфир соответ-
ственно 64° и 13°. н. А. Несмеянов.
МУКОПОЛИСАХАРИДЫ (кислые аминополиса-
хариды) — высокополимерные природные вещества,
построенные из остатков уроновых кислот и гликоз-
аминов (аминосахаров), аминогруппа к-рых ацетили-
рована или сульфатирована; ацилированы в М.
могут быть также и гидроксилы в различных положе-
ниях. М. широко распространены в животных тка-
нях — в соединительной ткани, в печени, легких и в
слизистых секретах (муцинах), а также в микроор-
ганизмах. Различные М. отличаются моносахаридны-
ми компонентами, характером связей между ними, дли-
ной и степенью ветвления полисахаридной цепи,
ацильными группировками и их положением. Наибо-
лее хорошо изученными М. являются гепарин, гиал-
уроновые кислоты и хондроитинсерные кислоты, ком-
поненты к-рых представлены в таблице:
Мукополисахарид Компоненты (молярные соотношения)
Гиалуроновая к-таа N-ацетил-П-глюкозамин (1), D-глюкуроновая к-та (1)
Хондроитин3 М-ацетил-Б-галактозамин (1), D-глюкуроновая к-та (1)
Хондроитинсернац кисло- та Аа (хондроитин-4-суль- фат) N-ацетил-D-галактозамин (1), D-глюкуроновая к-та (1), серная к-та (1)
Хондроитинсерная кисло- та Са Ы-ацетил-О-галактозамин (1), D-глюкуроновая к-та (1), серная к-та (1)
Хондроитинсерная кисло- та Ва (дерматансульфат) М-ацетил-О-галактозамин (1), L-идуроновая к-та (1), сер- ная к-та (1)
Гепарин^ D-глюкозамин (1), D-глюк - уроновая к-та и L-идуроно- вая к-та (1), серная к-та (2-3)
Кератосуль<5атв N-ацетпл-D-глюкозамин (1), галактоза (1), серная кис- лота (1)
а Гликозидные связи £-1,3 и Р-1,4, б Гликозидные связи а-1,3 и а-1,4. в Гликозидные связи не установлены.
340
М.— бесцветные вещества, они хорошо растворя-
ются в воде, образуя вязкие р-ры, плохо растворя-
ются в большинстве органич. растворителей. С орга-
нич. основаниями и белками М. образуют соли. Ос-
новными красителями (толуидиновый синий и др.)
М. окрашиваются метахроматически. Нек-рые пред-
ставители М. окисляются иодной к-той и тетраацета-
том свинца с образованием полиальдегидных соеди-
нений, которые легко выявляются восстановлеиием
AgNO3 или реактивом Шиффа. Эти реакции и мета-
хроматич. окрашивание используют для гистохимия,
выявления М. Мукополисахариды чувствительны к
щелочам и к-там; последними гидролизуются обычно
до альдобиуроновых к-т. Ферментативный гидролиз
М. идет под влиянием ряда специфич. ферментов.
М. являются биологически активными веществами:
гепарин и хондроитинсерная к-та В препятствуют
свертыванию крови, хондроитинсерные к-ты А и С
и гиалуроновая к-та принимают участие в водносоле-
вом обмене, гиалуроновая к-та во многих случаях
определяет проницаемость тканей для микробов и раз-
личных веществ. Нек-рые М. применяют в качестве
лечебных препаратов. В литературе М. называют так-
же группу природных веществ — высокополимеры,
построенные из связанных ковалентными связями
аминокислот и моносахаридов (см. Муковещества).
Лит.: Степаненко Б. Н., в кн.: Углеводы и угле-
водный обмен в животном и растительном организмах, М.,
1959, с. 5; Ciba foundation symposium on the chemistry and
biology of mucopolysaccharides, L., 1958; Ken t P. W.,
Whitehouse M. W., Biochemistry of the aminosugars, L.,
1955; Advances in protein chemistry, v. 13, N. Y., 1958, p. 39;
The biochemistry of mucopolysaccharides of connective tissue,
L., 1961; International Symposium onbiologically active mucoids,
Warsaw, 1960; Biochimica et biophysica acta, 1963, 69, № 3;
Comprehensive biochemistry, v. 5, Amst.—N. Y., 1963; Ливе-
вич Л. И., Усп. биол. химии, 5, 298. Л. И. Линевич.
МУКОПРОТЕИДЫ (мукопротеины) — высоко-
молекулярные природные вещества, построенные иа
белков и углеводов; в М. преобладают белковые ком-
поненты, обусловливающие белковый характер всей
молекулы в целом. Так, М. осаждаются реактивами,
осаждающими белки, денатурируются в условиях,
в к-рых денатурируются белки, и т. д. М. широко рас-
пространены в природе; к ним относят сложные белки
крови, простетич. группа к-рых представлена углево-
дом (гликопротеиды), белки молока, гонадотропные
гормоны, ряд белков яйца, М. подчелюстных желез,
мочи. Многие М. обладают высокой биологич. актив-
ностью, напр. М. подчелюстных желез и мечи, к-рые
подавляют гемаглютинацию крови вирусом гриппа,
гонадотропные гормоны и т. д. См. также Муковещест-
ва, Белки.
Лит. см. при ст. Мукополисахариды. Л. И. Линевич.
МУЛЬТИПЛЕТНАЯ ТЕОРИЯ КАТАЛИЗА — см.
Катализ гетерогенный.
МУРАВЬИНАЯ КИСЛОТА НСООН, мол. в. 46,03—
бесцветная жидкость с резким запахом; т. пл. 8,25°;
т. кип. 100,77760 м; 507120 мм; d™ 1,21961;
Ид 1,37140; ц25 1,966 спуаз; константа диссоциации
7^=1,765-10-4 (20°); скрытая теплота плавления
66 кал/г; теплота испарения (в кал/г): 115,5 (100,5°),
103,3 (25°); плотн. водных р-ров (<7^s): 1,0256 (10%);
1,1225(50%); 1,1902 (80%); 1,2102 (90%); 1,2191 (95%);
смеси М. к. с воздухом (1 : 6 и 1:2) взрывоопасны.
М. к. во всех отношениях смешивается с водой, эфи-
ром, спиртом; ограниченно растворима в бензоле;
с водой образует азеотропную смесь (77,4 вес. %)
М. к., т. кип. 107,2°. Остаток М. к.—формил; соли
ее — формиаты (натрия, калия и т. д.).
М. к.— простейшая карбоновая к-та и отличается
от других кислот тем, что карбоксильная группа
соединена не с углеводородным радикалом, а с атомом
водорода. В связи с этим она, с одной стороны, замет-
341
МУРАВЬИНАЯ КИСЛОТА—МУСКАРИН
342
но сильнее других алифатич. к-т, с другой стороны,
проявляет восстановит, свойства, характерные для аль-
дегидов, напр. образует «серебряное зеркало» (с амми-
ачным раствором окиси серебра). М. к. иногда исполь-
зуется в синтезах в качестве восстановителя, напр.:
(СвН5)3 сон + нсоон —» (С„Н5)3 сн + со2 + н2о
При нагревании ( — 160°) М. к. разлагается на СО2
и Н2, серной к-той расщепляется на СО и Н2О;
окисляется Н2О2 до НСОООН. Нагревание формиата
натрия приводит к оксалату натрия NaOOC— COONa.
При прокаливании смешанных кальциевых солей му-
равьиной и других к-т образуются альдегиды:
НСООСаООССНз—> СН3СНО + СаСО3
Хлорангидрид М. неизвестен, неустойчивый фторан-
гидрид получается в результате обменной реакции:
HCOOH + CeHsCOF —> HCOF + CeHsCOOH
Со спиртами в присутствии серной к-ты М. к. дает
сложные эфиры. Наибольшее значение имеют метил-
формиат, этилформиат. Из других производных нахо-
дят применение формамид и диметилформамид.
М. к. распространена в природе, содержится в хвое
ели, в крапиве, в едких выделениях муравьев и пчел.
В пром-сти формиат натрия получают нагреванием
твердой щелочи с окисью углерода (или генераторным
газом, свободным от СО2) при 100—105° и 5—10 ат
(СО + NaOH—>-HCOONa). Безводную М. к. получа-
ют разложением формиата натрия серной к-той при
охлаждении р-ра. Известны способы получения М. к.
каталитическим окислением метана, а также прямым
синтезом из окиси углерода и воды.
М. к. применяют в текстильной пром-сти при изго-
товлении протрав и крашении шерстяной и хлопчато-
бумажной пряжи из кислой ванны. Она употребляет-
ся как коагулирующее средство в произ-ве природного
каучука, в медицине, для формилирования аминов и
др. Значительное количество М. к. расходуется на
приготовление водно-аммиачных р-ров формиата меди,
применяемых для очистки под давлением газовых сме-
сей от окиси углерода. М. к. используют также в про-
из-ве катализаторов, содержащих никель и кобальт.
М. к. обладает антисептич. свойствами, что исполь-
зуется для консервирования фруктов и пр. М. к. и ее
метиловый, этиловый, пропиловый, бутиловый и изо-
бутиловый эфиры применяют как растворители; ал-
лиловый, бензиловый, n-метоксибензиловый эфиры
используют в качестве душистых веществ с запахом
фруктового или цветочного характера.
Эфир Мол. вес Т. кип., “С d (в гк< темп-р nD збках а, °C)
Метиловый 60,05 31,5 0,9867 (15) 0,9168 И) —
Этиловый 74,08 54,3 1,3598 (20)
н-Пропиловый . . . 88,10 81,3 0,90 58 (24) 1,377 (20)
изо-Пропиловый . . 88,10 71 ,3 0.883 —
н-Бутиловый .... 102,13 106,8 0 ,'9108 (22) 1 .389 (20)
н-Амиловый .... 116,16 130,4 0,8926 (15) 1,3951 (И.5)
н-Гексиловый . . . 130,18 153,6 0,898 (0)
н-Октиловый .... 158,23 198 0,872 (12,5) 1,414 (20)
Аллиловый Бензиловый .... 86,09 136,14 83 20 2—203 (747 мм) 0,932 (17) 1,081 (23)
Лит.: Ullman п, 3 Aufl., Bd 3, Mfinch.—В., 1953,
S. 436. Н. А. Несмеянов, В. М. Дашунин.
NH-CO
ос
NH-C
I
ONH,
CO-NH
С0-Н,0
\ г
CO-NH
МУРЕКСИД (пурпурат аммония) CgHgOaNg-H2O,
мол. в. 302,21—темно-красный порошок, плохо раство-
римый в воде, водный
0,05%-ный р-р окрашен в
фиолетово-красный цвет.
Окраска М. изменяется
от розовой в кислой
среде до фиолетовой в
щелочной. Окраска раз-
C-N=C
/
рушается в кислых р-рах при концентрации к-ты
более 1 н. Изменение окраски объясняется отщеплени-
ем протонов иминогрупп. М. можно получить окис-
лением мочевой к-ты (I) азотной к-той до аллоксантина
(II) с последующей обработкой аммиаком (аллоксан-
тин может быть получен также восстановлением
СО
HN С----Nil
I II I
ОС Сч СО
''Nff NH
I
/NH-CO CO-NH
ОС Cy---------C co
NH-CO OHHO 'со-NH
II
аллоксана}. M. реагирует с катионами многих метал-
лов. Лишь в случае Са, Ni, Со и Си, а также редкозе-
мельных элементов устойчивость комплексов достаточ-
на для их использования в анализе. М. образует ком-
плексы с молярным отношением 1:1. Они менее ус-
тойчивы, чем комплексы этих же металлов с комплек-
соном. Поэтому при титровании комплексоном в ко-
нечной точке титрования происходит отчетливое
изменение красной окраски в сине-фиолетовую. М.
применяют для фотометрич. определения кальция;
для обнаружения Sc, Zr, Th и редкоземельных эле-
ментов; в качестве индикатора при комплексометрич.
титрованиях при определении Са, Ni, Со, Си; для оп-
ределения жесткости воды и т. д.
См. также Мурексидная реакция.
Лит.: S с h w а г z е n b а с h G., Gy s 1 I n g H., Helv.
chim. acta, 1949, 32, 1314; Пршибил P., Комплексоны
в химическом анализе, пер. счет., М., 1960; Комплексометрия.
Об. переводов, М., 1958. И. П. Ефимов.
МУРЕКСИДНАЯ РЕАКЦИЯ — реакция, приме-
няемая для идентификации мочевой кислоты. Раствор
мочевой к-ты выпаривают с разбавленной HNOa и
смачивают р-ром аммиака; появляется пурпурно-
красная окраска вследствие образования мурексида—
аммониевой соли пурпурной кислоты. Окраска стано-
вится сине-фиолетовой после добавления р-ра едкого
натра. М. р. дают также и другие пурины.
Д. Н. Васкевич.
МУСКАРИН (I) [C9H20O2^]X_ — алкалоид, основ-
ное ядовитое вещество красного мухомора (Amani-
ta muscaria); четвертичное основание, образует кри-
20
сталлич. соли: хлорид (Х=С1), т. пл. 182°, [а]о=+7°
(вода); хлораурат (Х=АиС14), т. пл. 122°, и др. Сте-
реохимия М. установлена на основе данных рентгено-
структурного анализа, абс. конфигурация М. и его
н Н
Н3<^ кСНг",сн^ чо-1------------рон
ч н нон^с^Са cooir
биогенетич. связь с аминогексозами установлена син-
тезом иодида М. нз L-хитаровой к-ты (II). М. изби-
рательно стимулирует холинреактивные структуры
эффекторных органов, иннервируемых постганглио-
нарными холинэргич. нервами. Такое действие, про-
являемое другими веществами, называют «мускарино-
подобным». М. замедляет, а в больших дозах прекра-
щает сердцебиение, усиливает секрецию слюны, слез,
пота, сужает зрачок и т. д. Чувствительность человека
343
МУСКУС—МУТАРОТАЦИЯ
344
к М. велика тяжелое отравление при приеме 5—
8 мг М.), противоядие против М.— атропин.
А. Е. Васильев.
МУСКУС — сильно пахучий продукт сложного
строения животного или растительного происхожде-
ния. Обладает исключительно ценными парфюмерны-
ми свойствами (тонкий запах, облагораживающее и
фиксирующее действие на композицию и т. д.). М.
животный (продукт выделения мускусных желез
нек-рых парнокопытных) — зернистая блестящая масса
бурого цвета, горькая на вкус и с сильным своеобраз-
ным запахом. В состав животного М., помимо души-
стых веществ, относящихся к группе макроциклич.
кетонов: мускона из желез кабарги (см. ниже), ци-
бетона из циветты (см. ниже) и дигидроцибетона (из
ондатры), входят также белки, жиры, холестерин
и нек-рые др. продукты. Душистым началом расти-
тельного М. являются макроциклич. лактоны: напр.
тибеттолид (см. ниже), амбреттолид и др.
Установлено, что мускусным запахом обладают и
многие другие макроциклич. соединения, содержащие
в цикле 14—18 звеньев (диэфиры а, со-гликолей и
нек-рых двуосновных к-т, имины и ангидриды двух-
основных к-т и т. п.).
Синтезированы также оксалактоны общей ф-лы
<сн2)т—о—(СН,)„-С=о, имеЮщИе (при определенном
значении пит) тот же характер запаха, что и макро-
циклич. лактоны, а по стойкости превосходящие их.
Мускусный запах присущ многим органич. соеди-
нениям, не относящимся к группе макроцикличе-
ских. Так, еще в прошлом столетии получены нек-рые
нитросоединения ароматич. ряда, обладающие му-
скусным запахом (т. наз. «нитромускусы»), напр.:
мускус амбровый, мускус-кетон, мускус-ксилол (см.
ниже). Сравнительно недавно найдено, что соедине-
ния с мускусным запахом встречаются и среди пред-
ставителей других типов органич. соединений. К та-
ким «искусственным» М. относятся, напр., мустерон
(см ниже), а также нек-рые производные полиалкил-
инданов и тетрагидронафталипа. Среди них особен-
но интересны: 7-ацетил-1,1,4,4-тетраметил-6-этилтет-
рагидронафталин («версалид», I) и 6-ацетил-1,1,2,3,
3,5-гексаметилиндан («фантолид», II).
и
Благодаря развитию синтетич. методов как ис-
кусственные М., так и макроциклич. лактоны и осо-
бенно оксалактоны являются в настоящее время
доступными душистыми веществами («нитромускусы»
производятся в промышленных масштабах уже в те-
чение многих десятилетий).
Важнейшие представители мускусных препаратов:
М у с к о н (3-метилциклопентадеканон, 111), мол. в.
238,40 — бесцветная густая жидкость; т. кип. 130°(0,5 «и.м;
d '4’0,9221; 1,4802; [а] д = — 13,01°; выделяют из желез
самца мускусной кабарги отгонкой с водяным паром; семи-
карбазон, т. пл. 134°; фенилсемикарбазон, т. пл. 158—160°.
(СН2)12-С = О СН-(СН2),Ч
I I II )С = О
Н-С------- СН2 ОН —(СН2),/
I
СН3 III IV
Ц и б е т о н (циклогептадецен-9-он, IV) С17Н30О, мол. в.
250,41 — бесцветные кристаллы; т. пл. 32,5°; т. кип.
342°/742 мм, 158—160°/2 леи; семикарбазон, т.пл. 187°, п-нптро-
фенилгидразон, т. пл. 125°; оксим, т. пл. 92°; при гидрировании
образует дигидроцибетон.
Тибетолид (экзальтолид, пентадеканолид, лактон
пентадеканОЛ-15-овой к-ты, V) С15Н2ьОа> мол. вес 240,37 —
кристаллы; т. пл. 31 —32°; т. кип. 176°/15 .и; содержится в кор-
не аптечного дягиля Angelica Archangelica L.; может быть по-
лучен окислением экзальтона; в СССР производят двумя про-
мышленными способами: из дикарбоновых к-т и на основе тет-
рахлоралканов, получаемых теломеризацпей этилена с СС1,.
11 ОН'
С1 (СН,СН2), СС13—► С1 (СН2СН,), СООН —>
Н2О Н2О
—> НО (СН2СН2), СООН —> — (СН2СН2),-С = О
Лактонизация в обоих случаях происходит через промежу-
точную стадию — образование полиэфира, к-рый затем под-
вергается термич. деполимеризации в присут-
ствии щелочных катализаторов. |-------1
Экзальтон (циклопентадеканон, VI) (СН2)ц С=О
C15H2SO, мол. в. 224,37 — бесцветные крпстал- |________।
лы; т. пл. 63°, т. кип. 120°/0,Зл1м; обладает наи- VI
более интенсивным запахом среди всех извест-
ных М.; семикарбазон, т. пл. 187—188°; 2,4-динитрофенилги-
дразон, т. пл. 105°; оксим, т. пл. 75—76°.
Мускус амбровый (мускус-амбретт, 2,6-динитро-З-
метокси-4-трет-бутилтолуол, VII) C12HlBO5N2, молекулярный
вес 268,36 — зеленовато-желтые крис-
таллы; т. пл. 84—86°; нерастворимые в СН3
воде, ограниченно растворимые в спир-
те (2,5%); получают из At-крезола, кото-
рый сначала превращают в его метиловый
эфир и затем действием изобутилового
спирта в присутствии H2SO4 в 3-меток-
си-4-трет-бутилтолуол; последний ни-
уксусного анги-
С(@Н3)3
труют смесью HNOS и
дрида.
М у с к у с-к е т о н
(3,5-динитро-2,6-диметил-4-трет-бу-
СОСНз
Н3С
o2n
СНа
тилацетофенон, VIH) ChH^OjNo, мол. вес 294,30 — зелено-
вато-желтые кристаллы; т. пл. 134,5—
136,5°, нерастворим в воде, ограничен-
но растворим в спирте (1,8%); получа-
ют нитрованием 2,6-диметил-4-тпрет-бу-
тилацетофенона; последний готовят из
м-ксилола, к-рый предварительно пере-
водят способом, указанным выше, в 1,3-
диметил 5-тр ст -бути л бен зол и зат ем
С(СНз>з
ацетилируют уксусным ангидридом в v...
присутствии А1С13. v
М у с к у с-к с и л о л (2,4,6-тринитро-1,3-диметил-5-трет-
бутилбензол, IX) C12H,SOBN3, мол. в. 297,26 — кристаллы;
т.пл. 112—114,5°; получают нитрованием 1,3-диметил-5-тре?п-
бутилбензола.
NO2
IX
Мустерон (пзоборнил-2-метилциклогексанон, X), мол.
вес 250,41 С17Н30О — бесцветная жидкость; т. кип. 134—
137°/2 AtAt; d4°0,9779, 1,4955; получают в промышленности
окислением изоборнил-2-метилциклогексанола хромовой к-той;
последний готовят гидрированием продукта конденсации
о-крезола с камфеном; оксим, т. пл. 137,5—138,5°; 2,4-динитро-
фенилгидразон, т. пл. 182—183°.
Лит,: Белов В. Н. [и др.], Химия и технология души-
стых веществ, М., 1953; Ж. Всес. хим. о-ва им. Д. И. Менде-
леева, 1960, 5, № 4, 367—68, 371 — 76. В. Н.Ф-росин.
МУТАЗЫ — см. Фосфомутазы.
МУТАРОТАЦИЯ (мультиротация) — постепенное
изменение оптич. активности нек-рых веществ после
их растворения до установления постоянного вра-
щения. М. характерна для моносахаридов, восстанав-
ливающих олигосахаридов, нек-рых производных са-
харов, напр. N-гликозидов, а также оксикислот и лак-
тонов. Причиной М. является превращение таутомер-
ных форм в р-рах до установления динамич. равнове-
сия между ними. Напр., тотчас после растворения
в водеа-П-глюкозы (I), к-рая в кристаллах существует
в виде циклич. a-D-глюкопиранозы, в растворе [а]д=
=+112,2°, оптич. вращение р-ра постепенно умень-
шается и в течение дня при комнатной темп-ре дости-
гает постоянной величины +52,5°. Изменение враще-
ния a-D-глюкозы в р-ре происходит вследствие превра-
щения этой формы сахара в аномер — P-D-глюкопира-
345
МЫЛА
346
нозу (II), у к-рой [а]о=+18,5°, и др. таутомерные фор-
мы D-глюкозы—открытую альдегидную (III), а-
и Р-фуранозную формы (IV, V) и др.
Н-С-ОН
Н-С-ОН
СН2ОН
IV
В равновесной системе в воде содержится ок. 36%
tz-D-глюкопиранозы и ок. 64% P-D-глюкопиранозы, а
остальные таутомерные формы присутствуют в не-
значительных количествах. При растворении в воде
fi-D-глюкозы первоначальное [а]д=-1-18,5° изменяет-
ся до той же постоянной величины +52,7°. Другие
моносахариды мутаротируют в р-рах аналогично
D-глюкозе.
Как видно из схемы, этот процесс относится к двух-
ступенчатым прототропным превращениям одной тау-
томерной (глюкопиранозной) формы в другую через
стадию образования открытой оксоформы сахара.
С точки зрения этого механизма понятной является
зависимость скорости М. и соотношения таутомеров
в равновесной системе от природы сахара, влияние
на М. природы растворителя (в полярных раствори-
телях М. идет быстрее, чем в неполярных), а также
причины каталитич. действия кислот, оснований и
особенно бифункциональных катализаторов типа ок-
сипиридина.
М. некоторых моносахаридов, напр. D-глюкозы и
L-галактозы, ускоряется под влиянием фермента
мутаротазы, который получен пз некоторых микроор-
ганизмов.
Лит.: Степаненко Б. Н., Усп. химии, 1959, 28,
вып. 5, 521; Кочетков Н. К., Торгов И. В. и
Ботвиник М. М., Химия природных соединений, М.,
1961; The Carbohydrates. Chemistry, biochemistry, physiology,
ed. W. Pigman, N. Y., 1957. Л. Й. Линевич.
МЫЛА — соли высших жирных к-т, а также наф-
теновых и смоляных к-т. В качестве моющих средств
применяют М., растворимые в воде: калиевые, натрие-
вые, аммонийные или триэтаноламиновые, соли жир-
ных к-т, содержащих от 10 до 20 атомов углерода. Со-
ли с меньшим мол. весом к-т моющим действием
не обладают, с большим мол. весом — трудно раст-
воримы в воде. Растворимость натриевых М. нена-
сыщенных жирных к-т выше растворимости М.
насыщенных жирных к-т с таким же числом атомов
углерода. Высокомолекулярные М. растворяются
лучше в присутствии низкомолекулярных. В зависи-
мости от катиона растворимость в воде соответствую-
щих М. располагается в ряд: NH4>K>Na. Соли ще-
лочноземельных и тяжелых металлов, условно назы-
ваемые металлич. М., нерастворимы в воде, но раст-
воримы в углеводородах; такие соли применяют,
напр., при изготовлении пластичных (консистентных)
смазок, лаков, в качестве пластификаторов и др.
Безводные натриевые М.— твердые продукты, т.
пл. их колеблется в пределах 225—260°. При добав-
лении к М. воды темп-pa его плавления снижается.
Товарные М. плавятся примерно при 100°. Плотность
твердого М. ок. 1,05; безводные М. гигроскопичны.
В водных р-рах М., являющиеся солями слабых к-т
и сильного основания, гидролизуются; гидролиз уси-
ливается с повышением темп-ры и понижением кон-
центрации; р-ры М. имеют щелочную реакцию.
Под влиянием минеральных к-т М. разлагаются с вы-
делением свободных жирных к-т. В очень разб.
р-рах М. полностью диссоциируют на ионы. При более
высоких концентрациях, характерных для каждого
М., происходит ассоциация ионов жирных кислот в
мицеллы. Концентрация, при к-рой происходит обра-
зование мицелл, наз. критической концен-
тр ацией мицеллообразования; в
области критич. концентрации свойства р-ров М.
(поверхностное натяжение, электропроводность, осмо-
тич. давление и др.) резко изменяются.
М. получают действием едкой щелочи на нейтраль-
ные жиры или нейтрализацией жирных к-т едкими или
углекислыми щелочами. Металлич. М. чаще всего по-
лучают обменным разложением М. щелочных метал-
лов. По консистенции М. делят на твердые, мазеобраз-
ные, жидкие и порошкообразные; по целевому на-
значению — на хозяйственные, туалетные и техниче-
ские, а по способу получения — на клеевые и ядровые.
Клеевые М. получают охлаждением их конц.
водных р-ров (без нарушения однородности раствора);
они содержат от 30 до 50% жирных к-т в виде солей.
Ядровые М. получают охлаждением «ядра», вы-
деляемого из мыльного р-ра при обработке последнего
электролитами; ядровые М. содержат выше 60% жир-
ных кислот.
Сырьем для приготовления М. являются: жиры
растительные в натуральном и гидрогенизированном
виде (см. Гидрогенизация жиров), жиры животные,
жирные кислоты синтетические (см. Высшие жирные
кислоты), утильные жиры, отходы от рафинирования
жиров, канифоль и мылонафт. Для приготовления
М. используют нейтральные и расщепленные жиры
(см. Расщепление жиров).
Произ-во М. слагается из варки М. и придания ему товар-
ного вида (охлаждение, формование, штамповка). При варке
М. из нейтральных жиров их омыляют при кипячении водным
р-ром каустич. соды; расщепленные жиры сначала нейтрали-
зуют кальцинированной содой при кипячении и интенсивном
перемешивании (нейтрализуетсяпримерно 70% свободных жир-
ных к-т), а затем оставшиеся жирные к-ты и нейтральный жир
доомыляют р-ром каустич. соды. В образующемся при варке
М. концентрированном р-ре (т. наз. мыльном клее) поддержи-
вается небольшой избыток свободной щелочи (порядка 0,2%).
При выпуске клеевых М. массу доводят в варочном котле до
установленного для данного сорта М. содержания жирных к-т,
после чего ее охлаждают и режут на куски. При варке ядро-
вого М. в полученный мыльный клей добавляют крепкий р-р
электролита (каустич. соды или поваренной соли), после чего
масса разделяется на два слоя. Верхний слой, так наз.
ядро,— очищенное М. с высоким содержанием жирных к-т
(после отстаивания—не ниже 60%), и нижний слой, так наз.
подмыльный щело к,— р-р электролита, содержащий
небольшие количества М. и загрязнения, находившиеся в ис-
ходном сырье (а также глицерин, если М. варится из нейтраль-
ных жиров). Подмыльный щелок освобождают от увлеченного
М. и направляют для утилизации глицерина или в канализа-
цию. Отделившееся ядро для его очистки и осветления обраба-
тывают два — три раза р-ром электролита.
Хозяйственное М. охлаждают при помощи холодильных
прессов. Образовавшиеся при этом плоские твердые плиты М.
разрезают на механизированных резальных машинах на
куски, к-рые маркируют на автоматич. штампрессах, и затем
укладывают в ящики. Более прогрессивными являются непре-
рывно действующие механизированные вакуумные установки
для охлаждения и последующего формования М. Горячее М.
под давлением ок. 3 ат через трубчатый подогреватель посту-
пает в вакуумную башню. Распыливаемое через сопла горячее
М., попадая в камеру, теряет часть воды, за счет испарения
к-рой охлаждается и частично подсушивается; содержание
жирных к-т повышается при этом до 66—68%. Охлажденное
347
МЫЛО СУЛЬФАТНОЕ—МЫШЬЯК
348
М. падает на дно башни, откуда поступает в шнекпрессы, из
к-рых выходит в виде непрерывного бруска, разрезаемого
автоматич. резальной машиной на куски; последние марки-
руют и упаковывают в ящики.
Для произ-ва твердого туалетного М. используют «ядро»,
сваренное из лучшей по составу жировой основы (большее
количество — 72—80% —животных жиров, кокосовое масло
или соответствующие фракции синтетич. жирных к-т). Охлаж-
дение и формование сваренного туалетного М. производят по
более сложной технология, схеме, чем ядровое М.
Жидкие туалетныеМ. содержат 18—20% калийных или
смешанных калийно-натриевых М. в водноспиртовом р-ре (со-
держание этилового спирта 10—15%). К М., изготовляемым
для специальных целей, относятся, напр., олеиновое мыло для
текстильной пром-сти и для флотации, зеленое М. для медицин-
ских и ветеринарных целей, М. с ДДТ (смесь 95% твердого
хозяйственного клеевого М. с 5% ДДТ) и др.
Порошкообразные М. выпускаются в чистом виде
или в смеси со щелочными электролитами (кальцинированной
содой, конденсированными фосфорными солями, силикатом
натрия и др.), к-рые вводят для умягчения воды; при этом
снижается расход М. и повышается эффект стирки (см. Моющее
действие). Для получения порошкообразных М. их водный р-р
в смеси с электролитами в горячем состоянии подают через
распылительные форсунки или при помощи вращающегося
диска в сушильно-распылительную башню, куда одновременно
вдувают воздух. Твердые хозяйственные М. (в зависи-
мости от вида) должны содержать (в % к весу): 40—72%
жирных к-т в виде солей, 0,1—0,2% свободной едкой щелочи,
1—2% свободных карбонатов натрия или калия, 0,5—1,5%
нерастворимого в воде остатка, 2 —4% неомыляемых веществ
и неомыленного жира; температура застывания жирных к-т
28—43°.
Лит.: Тютюнников Б. Н., Юхновский Г. Л.,
Маркман А. Л., Технология переработки жиров, М.,
1950; Технология переработки жиров, под ред, Б. Н. Тютюн-
никова, 2 изд., М., 1956; Зиновьев А. А., Химия
жиров, М., 1952; Thomssen Е. G., Me Gu t с h е on
J. W., Soaps and detergents, N. Y., 1949; Thomssen
E. G., Kemp C. R., Modern soap making, N. Y., 1937;
Martin G-., Themodem soap and detergent industry, v. 1—2,
3 ed., L., 1950—51; Marti nenghi G. B., Chimica e
technologia degli oli, grass! e derivati, 2 ed., Milano, 1948.
Ф. В. Неволин.
МЫЛО СУЛЬФАТНОЕ — побочный продукт суль-
фатной варки целлюлозы из хвойной древесины; всплы-
вает на поверхности упаренного черного щелока
(80—100 кг на 1 т целлюлозы из сосны, 40—50 кг —
из кедра; из ели и пихты получается незначительное
количество М. с.). М. с. содержит 50—55% смоляных
и жирных к-т и перерабатывается на талловое масло.
Л. В. Гордон.
МЫЛОНАФТ (асидол-мылонафт) — технич. про-
дукт, состоящий в основном из смеси натриевых солей
нафтеновых к-т. Асидол-М., кроме натриевых солей
нафтеновых к-т, содержит и свободные нафтеновые
к-ты. М. имеет мазеобразную консистенцию, цвет от
светло-коричневого до темно-коричневого, легко раст-
воряется в воде; водные р-ры М. (натриевых солей на-
фтеновых к-т) обладают поверхностно-активными
свойствами, моющим действием (поверхностное натя-
жение 0,5%-ных р-ров 36,9 эрг/смг, смачивающая спо-
собность по краевому углу смачивания cos0=
=0,7623), хорошей эмульгирующей способностью и
устойчивостью к элементам жесткости воды. По сорт-
ности М. должен удовлетворять след, требованиям:
1-й сорт 2-й сорт 3-й сорт
Содержание нафтеновых к-т, % (не менее) 70 70 67
Содержание неомыляемых веществ (минерального масла) в расчете на органич. часть, % (не более) Кислотное число (мг КОН на 1 г нафтеновых к-т, не менее) . . . 9 13 15
220 210 190
Содержание минеральных солей, % (не более) 1 1 1,5
В том числе хлоридов, % (не более) 0,3 0,3 0,5
Важным свойством водных р-ров М. является их
более слабая диссоциация по сравнению с мылами
жирных к-т. М. и асидол-М. получают из продуктов
переработки нефти: 1) из щелочных отходов после
щелочной промывки дистиллятов, очищенных серной
к-той; 2) обработкой соответствующих дистиллятов
нефти (керосинового, солярового) р-ром едкого натра.
Последний метод обусловливает больший выход про-
дукта. Полученный р-р натриевых солей нафтеновых
к-т концентрируют упариванием, после чего М. вы-
саливают р-ром NaCl; при получении асидол-М.
обработкой серной к-той частично выделяются сво-
бодные нафтеновые к-ты. М. и асидол-М. применяют
в мыловарении в качестве заменителя жиров; М. так-
же используют в качестве эмульгатора в др. отраслях
пром-сти. Основными недостатками М. являются при-
сущие ему неприятный запах нефти, темный цвет и
значительное содержание минерального масла.
Лит.: II архоменко В. Е., Переработка нефти, 2 изд.,
М.—Л., 1948. А. Ю. Рабинович.
МЫШЬЯК (Arsenicum) As — химич. элемент V гр.
периодич. системы Менделеева; п. н. 33, ат. в. 74,9216.
Природный М. состоит из одного стабильного изото-
па As7S. Из искусственно радиоактивных изотопов
наиболее важны: As75 дней), As74 (Г>/2=
= 17,5 дней) и As76 (Т’>/2= 26,8 часа). Поперечное
сечение поглощения тепловых нейтронов атомом М.
4,1 барна. Конфигурация внешних электронов 4№4р3.
Энергии ионизации (в эв): As°->-As+ 9,81; As+->-As2+
18,63: As2+-»-Ass+ 28,34. .Атомный радиус 1,48А;
ионные радиусы: As’+ 0,69 A, Ass+ 0.47А, As8- 1,91А.
Природные соединения М.— золотисто-желтый аурипиг-
мент As2S3 и темно-красный реальгар As4S4 — были известны
древним народам, к-рые пользовались этими минералами для
приготовления красок и лекарств. Соединения М. вследствие
своих сильно ядовитых свойств назывались dQcrevixdv (от греч.
iQff-HV— сильный, мужественный); отсюда название arsenicum.
Русское название «мышьяк» произошло от «мышь» и «яд», т. к.
препараты М. применялись для истребления мышей. Получе-
ние свободного «металлического» М. обычно приписывается
А. Больштедту (ок. 1250), хотя несомненно, что до него гре-
ческие и арабские алхимики получали М. нагреванием As2O3
с органич. веществами. В 1789 франц, химик А. Лавуазье приз-
нал М. химич. элементом.
Земная кора содержит 0,0005 вес. % М. Свободный
(самородный) М. встречается редко. В природе М.
находится преим. в виде сульфидов и сульфоарсенидов,
реже — в виде арсенатов и арсенидов. Известно св.
120 минералов, содержащих М.; наиболее распрост-
раненные из них: мышьяковый колчедан
(миспикель, арсенопирит) FeAsS (46,0% As); мышь-
яковистый колчедан (леллингит) FeAs2
(72,8% As); реальгар AS4S4 (70,1% As); аури-
пигмент AsaSa (61,0% As). Наибольшее про-
мышленное значение имеет FeAsS. Обычно он со-
держит примеси как изоморфного характера (напр.,
Со, Ni), так и связанные с субмикроскопич. прорас-
таниями сульфидов висмута и серебра, с самородным
золотом и т. д. Богатые кобальтом разновидности
мышьякового колчедана носят название д а н а и т а
(до 3—9% Со) и глаукодота (до 9—18% Со).
Кристаллизуется мышьяковый колчедан в моноклин-
ной системе, блеск металлический, цвет оловянно-
белый, плоти. 5,9—6,2; твердость 5,5—6; при ударе
издает резкий чесночный запах. Встречается в рудных
жилах совместно с сульфидами, а также с вольфра-
митом, касситеритом, самородным золотом и др. Ти-
пичный гидротермальный минерал золоторудных
кварцевых жил. При выветривании и окислении на
земной поверхности переходит в скородит
FeAsO4-2H2O и другие водные арсенаты железа.
Мышьяковый колчедан служит основным источником
для получения М. и его соединений.
За мышьяковым колчеданом по важности следуют
сульфиды М. и мышьяковистый колчедан. Мышьяко-
вые цветы (природная АзгО3), встречающиеся обычно
в зоне окисления мышьяковых месторождений, и
самородный М. самостоятельного промышленного зна-
чения не имеют. Очень важны полпметаллич. руды,
в к-рых М. связан с золотом или др. металлами. Глав-
349
мышьяк
350
пая масса М. (более 90% всей мировой добычи) полу-
чается попутно при переработке теннантита 3Cu2S-
•As2S3, энаргита 3Cu2S-As2S5, прустита 3Ag2S-As2S3,
шмальтина CoAs2 и др. руд.
В подавляющем большинстве образование месторож-
дений руд М. генетически связано с кислыми магмами.
Соединения М. чаще всего встречаются в комплексе
с благородными и цветными металлами (Au, Ag, Pb,
Си, Sb и др.). По промышленной классификации
месторождения М. группируются в следующие типы:
чисто мышьяковые (реальгаро-аурипигментные и
арсенопиритовые), золото-мышьяковые, полиметал-
лическо-мышьяковые, медно-мышьяковые, мышьяко-
во-кобальтовые и мышьяково-оловянные. Месторож-
дения руд М. имеются в СССР, США (Бьютт, Голд-
хилл и др.), Швеции (Булиден), Мексике (Метеуала
и Чиуауа), Японии (Кашиока и Сасачатани), Боливии
(Потоси), Конго и др. странах.
Физические и химические свойства. М. существует
в нескольких аллотропных модификациях. Наиболее
устойчив при обычных условиях т. н. металли-
ческий, или серый, М. (a-форма); он кристал-
лизуется в ромбоэдрич. решетке (гексагональная сис-
тема) с параметрами а=4,123 А и Да = 54°6'; плотн.
5,72 (20°). Серый М. образует серо-стальную хрупкую
кристаллич. массу с металлич. блеском в свежем из-
ломе (на воздухе быстро тускнеет вследствие окисле-
ния). Пары М. бесцветны, до 800° состоят из моле-
кул As4, от 800° до 1700° — из смеси As4 и As2, выше
1700° — только из As2. При быстрой конденсации
паров на поверхности, охлаждаемой жидким возду-
хом, образуется т. н. желтый М.— прозрачные,
мягкие, как воск, кристаллы кубич. системы с плотн.
1,97. По свойствам желтый М. напоминает белый фос-
фор, однако он гораздо менее устойчив и под дейст-
вием света или при нагревании легко переходит в
серый М. Известны также аморфные формы М.—
Р, у и 6 с плотностью соответственно 4,73, 4,97 и 5,1.
Выше 270° все эти формы переходят в металлич. М.
При 615° М. возгоняется не плавясь; т. пл. 817°
(в запаянной трубке под давлением 36 атм). Теплота
плавления 88,5 кал/г\ теплота сублимации 102 кал/г.
Уд. теплоемкость (в кал/г-град): 0,078 (18°); 0,0822
(0—100°). Термич. коэфф, линейного расширения
5,6 -IO-’ (40°). Характеристич. темп-ра 224. Серый
М. (в отличие от других его аллотропных модифика-
ций) обладает металлич. электропроводностью, состав-
ляющей ок. 4% от электропроводности серебра. Уд.
электрич. сопротивление металлич. М. 33,3 мком-см
(20°). М. диамагнитен, его атомная магнитная вос-
приимчивость при комнатной темп-ре —5,5-10-в; она
почти одинакова для различных аллотропич. моди-
фикаций М. Твердость металлич. М. по Моосу 3,5;
по Бринеллю 147 кГ!ммг.
М. более электроположителен, чем водород, а также
чем сурьма и висмут. Его нормальный электродный
потенциал +0,3 в. В соединениях обычно проявляет
валентности +5, +3 и —3. Серый М. обладает сред-
ней химич. активностью. При накаливании па воз-
духе легко окисляется в мышьяковистый ангидрид
As2O3. Измельченный М. быстро сгорает ярким голу-
боватым пламенем, выделяя белый дым As2O3. Азот-
ная к-та и царская водка окисляют М. в мышьяковую
к-ту H3AsO4, к-рой соответствуют мышьяковый ан-
гидрид As2Os и соли — арсенаты. При сплавлении
со щелочами М. образует AsH3 и соли мышьяковистой
к-ты (не выделенной в свободном состоянии) — арсе-
ниты, напр. KAsO2 (см. Мышьяка окислы и кислоты,
Арсенаты и арсениты). М. непосредственно соеди-
няется с галогенами (см. Мышьяка галогениды) и с
серой (см. Мышьяка сульфиды).
При сплавлении с металлами М. нередко образует
химич. соединения — арсениды. Наиболее типичные
из них, напр. Cu3As, Ca3As2, Mg3As2, Zn3As2, Cd3As2,
отвечают по составу мышьяковистому водороду AsH3.
Кроме того, известны арсениды типа гидрида AsH
(ZnAs2, CdAs2), а также другие арсениды, напр.
MnAs, FeAs, CoAs, NiAs, Fe3As2, Co3As2, NiAs2,
Co5As2, NisAs2 (см. Арсениды металлов). Способность
M. к образованию твердых р-ров невелика. Непре-
рывные твердые р-ры он дает только с сурьмой (III
типа Розебома, с минимумом при 87 вес. % Sb и 605°);
в большинстве же случаев наблюдается небольшая
растворимость М. в твердых металлах (напр., в Cu,Fe)
или ее практич. отсутствие.
Аналитическое определение. Наи-
более общий способ открытия М. основан на том, что
все его соединения восстановляются цинком и разб.
НС1 или H2SO4 до AsH3. При пропускании AsH3 через
накаленную, наполненную водородом стеклянную
трубку на ее стенках осаждается М. в виде черно-
бурого зеркала, к-рое легко растворяется в свеже-
приготовленном щелочном р-ре NaClO (отличие от Sb).
При систематич. ходе качественного анализа М. в
виде смеси As2S3 и As2Ss осаждают из кислого р-ра
сероводородом вместе с металлами IV и V аналитич.
групп. Осадок сульфидов обрабатывают многосер-
нистым аммонием. Сульфиды As, Sb и Sn переходят
в виде тиосолей в р-р; при действии на него конц.
НС1 выпадает As2Ss. Смесью НС1 с КС1О3 (или NH4OH+
+ Н2О2) его переводят в H3AsO4; к р-ру прибав-
ляют небольшой избыток NH4OH и затем магне-
зиальную смесь (MgCl2+NH4Cl); образование белого
осадка MgNH4AsO4-6H2O доказывает присутствие
AsO’-. Можно также, прибавив к аммиачному р-ру,
избыток HNO3, осадить ион AsO’- т. наз. молибде-
новой жидкостью (р-р, содержащий парамолибдат
аммония, NH4NO3 и HNO3); получается желтый
мелкокристаллич. осадок арсеномолибдата аммония
(NH4)sAsO4 • 12МоО3 6Н2О.
Весовым путем М. определяют в виде пироарсена-
та магния Mg2As2O7, получаемого прокаливанием
MgNH4AsO4-6H2O, или же в виде As2Ss. Объемное
определение 3-валентного М. может быть произведено
титрованием иодом (в нейтральном р-ре) или броматом
калия (в солянокислом р-ре в присутствии метило-
вого оранжевого до его обесцвечивания). Очень не-
большие количества М. (до 10_в г As2O3) определяют
колориметрически.
Получение. Исходным материалом для получения
М. и его соединений служит As2O3, к-рая образуется
при окислительном обжиге руд М., а также полиме-
таллич. сульфидных руд, содержащих соединения
М. как примесь. Улавливаемая при этом сырая As2O3
в основном идет на произ-во соединений М. и лишь
в незначительном количестве служит для получения
серого («металлического») М. Пыль, содержащую ок.
30% As2O3, являющуюся побочным продуктом об-
жига полиметаллич. руд, смешивают с небольшим
количеством пирита FeS2 или свинцового блеска PbS
и подвергают обжигу в пламенных печах. Отходящие
газы пропускают через систему камер, в к-рых кон-
денсируется As2O3 чистотой 90—95%; для очистки
ее подвергают возгонке и получают продукт чистотой
не менее 99%. Металлический М. получают восста-
новлением As2O3 древесным углем или коксом, а
также термическим разложением мышьякового кол-
чедана.
Техника безопасности. Чистый М. неядовит, но все его
соединения, растворимые в воде или могущие перейти в р-р
под действием желудочного сока, чрезвычайно ядовиты; осо-
бенно опасен мышьяковистый водород. При работе с М. и его
соединениями необходима безукоризненная герметизация ап-
паратуры, удаление пыли и газов вентиляцией, соблюдение
личной гигиены (противопылевая одежда, очки, перчатки,
частые души, ванны), частый медицинский контроль, недопу-
щение к работе с М. женщин н подростков.
351
МЫШЬЯКА ГАЛОГЕНИДЫ—МЫШЬЯКА СУЛЬФИДЫ
352
При остром отравлении М. наблюдаются
боли в животе, рвота, понос, угнетение центральной
нервной системы, падение кровяного давления. По-
мощь при отравлении заключается в промывании же-
лудка и приеме внутрь противоядия (водного раство-
ра сульфидов натрия и магния); весьма эффектив-
но также внутримышечное введение дитиопропанола
(CH2(SH)CH(SH)-CH2OH].
Применение. Металлич. М. применяют только как
добавку в нек-рые сплавы. Напр., в медь, используе-
мую для паровозных топок, вводят 0,25—0,50% М.
В свинец, идущий для произ-ва дроби, прибавляют
0,5—1,6% М., к-рый повышает поверхностное натя-
жение жидкого свинца, что способствует получению
дробинок сферич. формы; кроме того, добавка М.
увеличивает твердость свинца. М. входит в состав
нек-рых антифрикционных и типографских сплавов,
не содержащих олова.
Соединения М. применяют значительно более ши-
роко. As2O3 служит для обесцвечивания стекла,
консервирования кож, мехов и чучел, а также служит
исходным материалом для получения мышьяковых
препаратов. Натрия арсенат, кальция арсенат и ар-
сенит, швейнфуртскую зелень Cu(C2H3O2)2-3Cu (AsO2)2
применяют как инсектициды. Нек-рые соединения
М. используют как лекарственные вещества. Неорга-
нич. соединения М. (As2O3, KAsO2 и Na2HAsO4)
применяют при малокровии, истощении, неврасте-
нии, в стоматология, практике и др. Органич. пре-
параты М. (новарсенол, миарсенол, осарсол и др.)
применяют в основном для лечения сифилиса, реже—
возвратного тифа, малярии и др.
Лит.: ГиллебрандВ. Ф. [и др.], Практическое руко-
водство по неорганическому анализу, пер. с англ.,М., 1957;
Gmelin, 8 Aufl., Syst.-Nummer 17, Arsen, Weinheim, 1952;
Ullmann, 3 Aufl., Bd 3, Munch.— B., 1953; Химически
вредные вещества в промышленности, под общ. ред. Н. В. Ла-
зарева, ч. 1, Л.— М., 1951. С. А. Погодин.
МЫШЬЯКА ГАЛОГЕНИДЫ — могут быть полу-
чены взаимодействием As с F, Cl, Вг, J. Физич. свой-
ства М. г. приведены в таблице.
Соеди- нение Цвет Плотн., г/см3 Т. пл., °C Т. кип., °C Теплота обра- зования, Atf°2ob, ккал/моль
AsF3 бесцветный 2,64 — 13° 63° — 226,8 (жидк.)
AsF, » 7,71 (г/л) — 80° — 53° —
AsCl, AsCls >> 2,163 — 18° 130° —80,2 (жидк.)
» — ок. —40° ок. — 28° Разлагается —
AsBr3 » 3,54 33° 221° — 46,61 (тв.)
AsJ3 красный 4,39 141° 403° — 13,7 (тв.)
As2 j, темно-крас- ный 136° (с разл.) — —
Для всех М. г. характерна способность к присоеди-
нению самых разнообразных веществ; напр., извест-
ны соединения AsF3-3NH3, AsCl3-4NH3, AsF3 SCl4.
При действии воды М. г. гидролизуются с образо-
ванием галогеноводорода и кислот — мышьякови-
стой или мышьяковой. Практическое значение имеет
только AsCl3. Его получают по реакции As2O3+
+6NaCl+3H2SO4-*2AsCl3+3Na2SO4+3H2O. Хлорид
AsCl3 (как и все М. г.) очень ядовит. Воздух при
содержании 0,1 мг/л и выше AsCl3 сильно раздражает
слизистые оболочки дыхательных органов, жидкий
AsCl3 вызывает на коже язвы. AsCl3 применяется в
произ-ве фармацевтич. препаратов и в др. синтезах.
Лит. см. при ст. Мышъяк. С. А. Погодин.
МЫШЬЯКА ОКИСЛЫ И КИСЛОТЫ — соеди-
нения 3- и 5-валентного мышьяка с кислородом и
отвечающие им кислоты.
Мышьяковистый ангидрид As2O3
встречается в природе в виде минералов арсенолита
и клодетита. Арсенолит — кристаллы кубич. сии-
гопии, плотн. 3,865, устойчив при темп-рах ниже
—13°; между этой темп-рой и т. пл. (315°) устойчив
клодетит — кристаллы моноклинной сингонии, плотн.
4,15; теплота образования АЯ°298= —156,4 ккал/моль.
Превращение арсенолита в клодетит протекает очень
медленно; присутствие водяного пара ускоряет
его. При нагревании As2O3 легко возгоняется; ес-
ли темп-ра поверхности, на к-рой конденсируются
его пары, выше 310°, то получается т. наз. «мышьяко-
вое стекло», или стекловидная As2O3, с плотн. 3,738;
если же конденсация происходила при более низкой
темп-ре, получается мелкокристаллич. мука As2O3.
Плотность пара мышьяковистого ангидрида отвечает
ф-ле As40e, при 1800° — ф-ле As2O3. Растворимость
кристаллич. As2O3 в воде (в г на 100 г Н2О): 1,2 (0°),
2,1 (25°), 6,0 (75°); раствор имеет сладковатый вкус
с металлич. привкусом. As2O3— соединение амфотер-
ное с преобладанием кислотных свойств. Со щело-
чами As2O3 образует соли (арсениты), отвечаю-
щие неизвестным в свободном состоянии к-там: орто-
мышьяковистой H3AsO3 и (что чаще) метамышьяко-
вистой HAsO2. При действии НС1 или НВг па As2O3
получаются AsCl3 и AsBr3 с катионом As3 + (см. Мышь-
яка галогениды). О получении As2O3 см. Мышьяк.
Мышьяковый ангидрид As2O5— бес-
цветная аморфная масса, плотн. 4,086; теплота обра-
зования АЯ°298= —218,6 ккал/моль; при 315° раз-
лагается на As2O3 и О2; легко растворима в воде
(150 г в 100 г Н2О при 16°). С водой As2O5 образует
ортомышьяковую к-ту H3AsO4, дающую кристалло-
гидрат 2H3AsO4-H2O, плотн. ок. 2,5, т. пл. 35,5°;
при нагревании ок. 120° переходит в As2O5. Мышья-
ковая (ортомышьяковая) к-та H3AsO4
относится к трехосновным кислотам средней силы.
Константы ее электролитич. диссоциации при 25° соот-
ветственно равны: 6-Ю-3, 2-10~7 и 3-10-12. В кислом
р-ре используется как окислитель. Мышьяковая к-та
может быть получена окислением мышьяка или As2O3
по реакциям;Аз2О3+2Н]ХО3+2Н2О = 2H3AsO4 + N2O3
или 3As+5HNO3+2H2O = 3H3AsO4 + 5NO. Мышья-
ковая к-та H3AsO4 образует три ря-
да солей (арсенатов): Na3AsO4;
Na2HAsO4 и NaH2AsO4. Известны так-
же соли к-т пиромышьяковой H4AsO7
и метамышьяковой HAsO3, не полу-
ченных в свободном состоянии.
As2O3 применяют для осветления
стекла, для консервирования мехов,
в произ-ве мышьякорганических соеди-
нений (лекарственные средства), в пи-
ротехнике. As2O3, а также арсениты
и арсенаты применяют как лекарства,
а также для борьбы с вредителями
c. х-ва. As2O3 и арсениты очень ядовиты; арсенаты го-
раздо менее ядовиты. См. также Арсенаты и арсени-
ты, Кальция арсенат и арсенит, Натрия арсенаты.
Лит. см. при ст. Мышъяк. С. А. Погодин.
МЫШЬЯКА СУЛЬФИДЫ — соединения мышьяка
с серой: AsS, As2S3 и As2S5. Односернистый
мышьяк AsS встречается в природе в виде мине-
рала реальгара, оранжево-красного цвета;
плотн. 3,5—3,6; моноклинная решетка, а = 9,27,
b = 13,50, с = 6,56 A; fJ = 106°37'; структура построе-
на из отдельных молекул As4S4. Т. пл. 320°, т. кип.
565°. Трехсерн петый мышьяк As2S3
встречается в природе в виде минерала аурипиг-
мента, от бледно-желтого до золотисто-желтого
цвета; плотн. 3,43; моноклинная решетка; а = 11,46,
Ь = 9,56, с = 4,21 А; р = 90°; структура сходна со
структурой AsS. Т. пл. 310°, т. кип. 707°. Теплота
образования АЯ°298= —35 ккал/моль. П я т и с е р-
н истый мышьяк As2S5— вещество желтого
цвета, ок. 500° разлагается на As и S.
353 МЫШЬЯКОВИСТЫЙ ВОДОРОД—МЫШЬЯКОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ 354
Сульфиды AsaSa и As3S3 могут быть получены сплав-
леппем As с S. Из кислых р-ров соединений 3-валент-
ного As сероводород осаждает As2S3; в тех же условиях
из соединений 5-валентного As выпадает смесь AS2S3
и As2S5. Сульфид AS2S5 количественно осаждается
сероводородом из р-ров мышьяковой к-ты и ее солей
в дымящей НС1 при охлаждении льдом. В воде и
разб. к-тах М. с. практически нерастворимы. Сильные
окислители (конц. HNO3, Н2О2, НС1+КС1О3) пере-
водят их в смесь H3AsOa и НгЗОа- Сульфид AS2S3
растворяется в сернистом аммонии, в аммиаке и кар-
бонате аммония по схемам: As2S3+3S2_ 2AsS’~;
As2S3+60Н - -^3Н 20 + 2 AsO+A s As2S,+ЗСО2--
—>3CO2+AsS3- +AsO2“. При этом образуются соли
мышьяковистой к-ты H3ASO3 и тиомышьяковистой
к-ты H3AsS3, легко растворимые в воде. При действии
на AS2S3 желтого (NH4)2SX получается легко раство-
римая соль тиомышьяковой к-ты H3AsSa и сера. Смесь
AS4S4 с Са(ОН)г применяется для удаления волос со
шкур при произ-ве высших сортов кожи.
Лит. см. при ст. Мышьяк. С. А. Погодин.
МЫШЬЯКОВИСТЫЙ ВОДОРОД (арсин)
AsH3 — бесцветный газ, в чистом состоянии запаха
не имеет (с сопутствующими примесями пахнет чес-
ноком); т. кип.—62,4° (760 лш рт. ст.), т. пл.—113,5°;
плотность по воздуху 2,7. Теплота образования
АЯ°298= + 41,0 ккал/моль. Непрочен, при нагревании
св. 500° нацело разлагается на водород и мышьяк,
что используется для обнаружения последнего (Мар-
ша способ). Совершенно чистый М. в. может быть
получен действием воды на арсениды, Na3As или
СазАвг; нечистый М. в. (с примесью водорода) обра-
зуется при растворении сплавов As с Zn (или Fe) в
разб. НС1 или H2SO4, а также при действии водорода
в момент выделения на растворимые в кислотах соеди-
нения мышьяка. Известны также гидриды мышьяка
другого состава: AS2H4 — газообразное соедине-
ние, чрезвычайно легко разлагающееся на AsH3;
твердый гидрид (Ав2Н)х— аморфный красный поро-
шок; кроме того, получается твердый гидрид AsH—
аморфный коричневый порошок.
М. в. очень ядовит. В организм М. в. проникает
только через дыхательные пути, а оттуда — в ток
крови. В произ-ве М. в. образуется как вредный по-
бочный продукт при взаимодействии загрязненных
мышьяком кислот и металлов (травление металлов,
получение Н2 действием разб. кислот на Zn, содержа-
щий As, и т. п.), при воздействии воды, влажного
воздуха на металлы, содержащие As, и др. Предельно
допустимая концентрация М. в. в воздухе производ-
ственных помещений по действующим нормам
0,0003 мг/л. Отравления М. в. всегда носят острый
характер и являются неожиданными. Основное дей-
ствие яда — гемолиз, поражение нервной системы
и выделительных органов. Отравления М. в. обычно
тяжелые, нередко со смертельным исходом.
Лит. см. при ст. Мышьяк. С. А. Погодин.
МЫШЬЯКОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ —
класс металлоорганических соединений, характери-
зующихся содержанием атомов мышьяка, непосред-
ственно связанных с атомами углерода. Иногда к
М. с. относят все органические соединения, содер-
жащие As, в том числе и эфиры мышьяковистой
и мышьяковой кислот [As(OR)3 и OAs(OR)3], без
С—As-связи.
Первые М. с. были синтезированы еще в 1760 Л. Ка-
дэ, к-рый нагреванием смеси мышьяковистого ангид-
рида с ацетатом калия получил жидкость с отвра-
тительным запахом («жидкость Кадэ»); впоследствии
было установлено, что эта жидкость содержит гл.
обр. какодил (CH3)2As — As(CH3)2 п окись какодила
(C1I3)2AsOAs(CH3)2. Исследования какодиловых со-
единений сыграли большую роль в развитии теории
радикалов.
В начале 20 в. в результате обширных исследова-
ний П. Эрлиха в области М. с. в медицинскую прак-
тику был введен сальварсан и затем неосальварсан
(препарат 606 и 914) для эффективного лечения сифи-
лиса и др. болезней, вызываемых спирохетами (воз-
вратный тиф, сонная болезнь), что явилось крупным
успехом химиотерапии. К числу других М. с., при-
менявшихся в медицине, относятся соль метиларси-
новой к-ты CH3AsO(ONa)2-6H2O (арреналь), 3-амино-
4-оксифениларсиноксид (мафарсен), 3-ацетиламино-
4-оксифениларсиновая к-та (стоварсол, спироцид),
n-N-фенилглицинампдарсиновая к-та (трипарсамид)
и др. В первую мировую войну 1914—18 нек-рые лету-
чие М. с., содержащие трехвалентный As, были ис-
пользованы в качестве боевых отравляющих веществ
общетоксич., кожного и раздражающего действия
(метилдихлорарсин, фенилдихлорарсин, дифенилциан-
арсин и др.; см. также люизит, адамсит).
Разработан ряд общих методов создания мышьяк-
углеродной связи. Так, действием алкилирующих
агентов на соли мышьяковистой к-ты можно получить
алкиларсиновые (алкиларсоновые, алкилмышьяко-
вые) кислоты RAsO(OH)2 (см. Майера реакция). При
взаимодействии солей мышьяковистой кислоты с
арилдиазониевыми солями образуются соли арилар-
синовых кислот ArAsO(ONa)2 (см. Барта реакция).
Аналогичным образом можно получить и вторичные
диариларсиновые к-ты АггАзО(ОН) действием диа-
зониевых солей на ArAs(ONa)2. Третичные арсины
удобнее всего получать действием реагентов Гринья-
ра (или других металлоорганич. соединений, напр.
RaHg) на AsX3 или RASX2. Помимо этих общих ме-
тодов, известны и частные способы образования связи
С—As. Так, при действии ацетилена на AsCl3 обра-
зуется fJ-хлорвинилдихлорарсин (люизит); реакция
мышьяковой к-ты с анилином ведет к получению
арсаниловой к-ты (см. Вешана реакция); при действии
AsCl3 на бензол в присутствии А1С13 образуется фенил-
дихлорарсин; при действии А1С13 на дифениламин —
адамсит.
В зависимости от числа радикалов, связанных с
атомом As, и от степени окисленности атома As в
М. с. они могут быть разбиты на следующие основ-
ные группы:
1) Первичные RAsH2, вторичные R2ASH, третич-
ные R3As арсины и соли четырехзамещенного арсо-
ния R4AsX. Первичные и вторичные арсины обычно
получают действием сильных восстановителей на
кислородные М. с. Четвертичные соли образуются
при присоединении алкилгалогенидов к третичным
арсинам. Известен и представитель соединений типа
R5As — пентафенилмышьяк (т. разл. 150°), получен-
ный (Виттиг, 1952) действием фениллития на тетра-
фениларсоний — хлорид [(CeH5)4AsCl]. Первичные и
вторичные арсины — сильные восстановители, по-
глощают кислород воздуха без самовоспламенения.
Арсины обладают неприятным запахом и значительной
токсичностью. Третичные арсины присоединяют кисло-
род, серу и галогены с образованием соответственно
R3AsO, R3AsS и R3AsX2. Арсины, в отличие от аминов,
почти лишены основных свойств и способности к соле-
образованию с к-тами. Четвертичные арсониевые ос-
нования R4ASOH представляют собой сильные осно-
вания .
2) Арсеносоединения ArAs=AsAr (Аг — ароматич.
радикал), аналоги азосоединений, образуются при
восстановлении ArAsO или ArAsO(OH)2. Арсепосо-
единения получаются и при взаимодействии АгАэНг
с ArAsO или ArAsX2. К этому типу М. с. относятся,
в частности, сальварсан (в полимерной форме) и нео-
сальварсан.
О^2 К.Х.Э. т. 3
355
МЯГЧИТЕЛИ—МЯТНОЕ МАСЛО ПЕРЕЧНОЕ
356
3) Соединения типа R2As—AsR2 (тетраалкилдиар-
сины) образуются при восстановлении вторичных
М. с.— (R2As)2O или R2As(O)OH — в сравнительно
мягких условиях.
4) Арсипоксиды RAsO представляют собой анги-
дриды алкиларилмышьяковистых (алкиларсинистых)
к-т RAs(OH)2. Арсиноксиды как и галогенангидриды
указанных к-т [алкил (арил) дихлорарсины RAsC12]
обычно получают восстановлением RAsO(OH)2.
К рассматриваемому типу соединений примыкают и
эфиры алкил (арил) мышьяковистых к-т RAs(OR)2,
алкил(арил)арсинсульфиды RAs=S, алкил(арил)ар-
синимины RAs=NH и др. Известны и многочис-
ленные вторичные М. с. рассматриваемого типа, на-
пример хлористый какодил (CH3)2AsC1, ди-|3-хлорви-
ниларсинхлорид (C1CH=CH2)2AsC1, какодисульфид
(CH3)2As—S—As(CH3)2, а также соединений типа
R2AsX, где X — псевдогалоген, напр. роданистый ка-
кодил (CH3)2AsSCN, дифенилцианарсин (CeH5)2AsCN.
5) Алкил(арил)арсиновые кислоты RAsO(OH)2
как наиболее доступные М. с. имеют большое значе-
ние, т. к. от них можно легко перейти к другим раз-
личного рода М. с. Известны и производные этих кис-
лот, напр. эфиры RAsO(OR)2, амиды RAs(O)(NH2)2
и т. д. Галогенангидриды этих кислот RAsOX2 или
R2As(O)X неустойчивы и легко отщепляют галоген-
алкилы. Эфиры алкиларсиновых к-т RAsO(OR)2 при
нагревании изомеризуются в эфиры мышьяковистой
к-ты (RO)3As с понижением валентности атома As
и уничтожением связи С—As. Этот процесс противо-
положен перегруппировке эфиров фосфористой к-ты
в эфиры алкилфосфиновой к-ты [P(OR)3-*RPO(OR)2]
(см. Арбузова реакция).
6) Алкил(арил)арсинтетрагалогениды RAsX4 или
R2AsX3 и R3AsX2, образующиеся при присоединении
галогенов к хлорарсинам или к R3As, неустойчивы
п легко, в особенности в жирном ряду, отщепляют
RX с образованием соединений трехвалентного As,
напр. (CH3)2AsC13—*CH3AsC12+CH3C1. При гидро-
лизе RAsX4 и R2AsX3 образуются соответствующие
первичные и вторичные к-ты RAsO(OH)2 и R2As(O)OH.
Известно много соединений, обладающих оптич.
активностью, обусловленной атомами As, вызываю-
щими асимметричность молекулы, напр. 10-метилфе-
ноксиарсинкарбоновая-2 кислота и др.
Лит.: ФрейдлинаР. X., Синтетические методы в об-
ласти металлоорганических соединений мышьяка, М,—Л.,
1945; Raiziss G. W., Gavron J. L., Organic arsenical
compounds, N.Y.. 1923; Органические реакции, пер. с англ., сб.
№ 2, М., 1950, с. 448—90; Комиссаров Я. Ф., Соро-
к о у м о в А. С., Малеева А. Я., ДАН СССР, 1947, 56,
№ 1, 51. Я. Ф. Комиссаров.
МЯГЧИТЕЛИ (пластификаторы) — компоненты ре-
зиновых смесей, понижающие внутреннее трение в сис-
теме. Введение М. облегчает обработку каучуков,
улучшает диспергирование наполнителей, снижает
продолжительность и темп-ру смешения. М. влияют
на свойства вулканизатов: последние становятся мяг-
че, эластичнее, уменьшаются гистерезисные потери,
но прочность их обычно снижается. При правильном
выборе М. достигается повышение морозостойкости,
сопротивления утомлению, улучшение других свойств
резин.
М.— один из первых классов ингредиентов, ис-
пользованных для приготовления резиновых смесей.
К ним относились различные воски, масла, смолы и
дегти, к-рые вводили в натуральный каучук (НК).
С появлением разнообразных синтетич. каучуков (СК)
значительно возрос ассортимент М. (ок. 700 наименова-
ний). Большинство М.— продукты переработки неф-
ти, каменного угля, торфа, сланцев, древесины; при-
меняют также растительные масла и синтетич. соеди-
нения (гл. обр. сложные эфиры, напр. дибутилфта-
лат). В большинстве случаев М. являются смесью угле-
водородов алифатич., ароматич. и нафтенового рядов
или их производных, лишь нек-рые М. представляют
собой индивидуальные соединения.
Эффективность действия М. зависит от способности
полимеров набухать в них. Пластификация заключает-
ся в проникновении М. в каучук, увеличении меж-
молекулярных расстояний, ослаблении межмолеку-
лярных сил, облегчении подвижности звеньев кау-
чуковой цепи, что приводит к изменению темп-ры стек-
лования полимера.
Поведение полярных и неполярных СК по отношению к М.
различно. В случае полярных каучуков и полярных М. по-
нижение темп-ры стеклования пропорционально молярной
доле М. В случае неполярного каучука темп-ра стеклова-
ния понижается пропорционально объемной доле М., т. е.
при одинаковом мол. весе линейные молекулы М. действуют
эффективнее сферических. Для сополимерных СК наблюдается
отклонение от указанных правил.
Насыщенные и ароматич. М. сравнительно инертны по от-
ношению к вулканизации, но они могут улучшать распреде-
ление серы и ускорителей в резиновой смеси. Такие М., как
канифольные к-ты, ненасыщенные алифатич. растительные
масла, алкилариловые эфиры, сосновая смола, кумароно-
инденовые смолы, могут замедлять вулканизацию. В случае
СК с высокой степенью насыщенности (напр., бутилкаучук)
применение непредельных М. недопустимо, так как они могут
исключить эффект вулканизации. При введении в смесь М.,
ограниченно совмещающихся с каучуками, уменьшается при-
липание резиновых смесей к поверхности оборудования, облег-
чается каландровапие и шприцевание, улучшается поверх-
ность каландрованной резиновой смеси.
Применение М. связано также с экономия, сообра-
жениями. В резиновые смеси вводят до 30—35 вес.
ч. М. на 100 вес. ч. каучука. В случае высокомолеку-
лярного бутадиен-стирольного каучука применяют
М. нефтяного происхождения в указанном количестве
без потери прочностных свойств вулканизатов. Для
получения маслонаполненных каучуков применяют
СК, не содержащие или содержащие меньше, чем обыч-
но, низкомолекулярных фракций. Место низкомоле-
кулярных фракций каучука в резиновой компози-
ции занимают М. (см. Каучуки масло- и саженапол-
ненные).
Отдельные М. специфически влияют на свойства ре-
зин и резиновых смесей. Напр., рубракс, получаемый
окислением асфальтов или гудронов, сообщает рези-
нам водостойкость, парафин — стойкость к свету
и озону, трикрезилфосфат понижает горючесть и т. д.
Лит.: Кошелев Ф. Ф., К л н м о в Н. С., Общая техно-
логия резины, 2 изд., М., 1958; Ж у р к о в С. Н. и Л е р м а н
Р. И., ДАН СССР, 1945, 47, № 2, 109; Ж у р к о в С. Н., там
же 1945, 47, .№ 7, 493; Каргин В. А., Малинский
Ю.’ М., там же, 1950, 73, J6 5, 967; Storey Е. В., Rubber
Chem. and Technol., 1961, 34, № 5, 1402. В. С. Юровский.
МЯТНОЕ МАСЛО ПЕРЕЧНОЕ — см. Эфирные
масла.
НАБУХАНИЕ полимеров — изменение веса
и объема полимера при контакте его с низкомолеку-
лярными жидкими или газообразными соединениями,
обусловленное поглощением последних полимером.
Возможность Н. полимера в том или ином веществе
зависит от сродства между ними. Неполярные полиме-
ры сильно набухают в неполярных жидкостях и их
парах, полярные — в близких нм по полярности ве-
ществах. Н. полимеров можно рассматривать как од-
ностороннее смешение, т. е. проникновение низкомо-
лекулярного компонента в полимер практически без
проникновения полимера в низкомолекулярный ком-
понент. Это обусловлено большим различием по-
движностей больших и малых молекул. Н. всегда со-
провождается раздвижением макромолекул или их
пачек (внутри- и межпачечное Н.). Объем набухшего
полимера может быть равен или меньше (в большин-
стве случаев) суммы объемов компонентов. Н.— про-
цесс самопроизвольный, и набухший полимер пред-
ставляет собой истинный термодинамически устой-
чивый р-р низкомолекулярного компонента в поли-
мере.
Различают неограниченное и ограниченное Н. Н е-
ограниченное Н., характерное для линейных
и разветвленных полимеров, при избытке низкомоле-
кулярного растворяющего вещества приводит к обра-
зованию истинного р-ра полимера. Ограничен-
ное Н. свойственно линейным и разветвленным по-
лимерам при контакте с плохими растворителями, а
также полимерам, имеющим пространственную струк-
туру. При изменении темп-ры ограниченное Н. ли-
нейных и разветвленных полимеров может перейти
в неограниченное. Пространственные полимеры набу-
хают только ограниченно.
Ограниченное Н. оценивается степенью Н., т. е.
отношением объема или веса набухшего образца к
объему или весу исходного образца. Степень Н. изме-
няется во времени. При ограниченном Н. достигается
максимальная или равновесная ст -
пень Н., не меняющаяся во времени. Сравнивать
полимеры по их способности к Н. следует только по
значениям равновесной степени Н., к-рая у простран-
ственных полимеров уменьшается с возрастанием ча-
стоты трехмерной сетки (в случае предельно частой
сетки Н. практически не происходит). У остальных
полимеров равновесная степень Н. зависит от сродства
к жидкости или ее пару. Еслп образец набухающего
полимера со всех сторон ограничить жесткими стен-
ками, оставив небольшое отверстие для проникнове-
ния растворителя, то набухающий образец не может
увеличиваться в объеме. Он оказывает на ограничи-
вающие его стенки очень большое давление, наз. дав-
лением Н.
При эксплуатации изделий из полимерных материа-
лов следует учитывать возможность их Н. и подбирать
для различных жидких и парообразных сред полимеры,
минимально набухающие в этих средах. При перера-
ботке полимеров в изделия очень важно, чтобы до-
бавляемые к полимеру компоненты хорошо с ним
совмещались, т. е. чтобы полимер в этих компонентах
хорошо набухал.
Лит.: Тагер А. А., Растворы высокомолекулярных
соединений, М.— Л., 1951; В о ю Ц к и й С., Растворы высо-
комолекулярных соединений, 2 изд., М., 1960; Т а г е р А. А.,
Физико-химия полимеров, М., 1963. А. А. Тагер.
НАДИЗАН [алентин, оранил, М-(4-аминобензол-
сульфонил)-М'-и-бутилкарбамид] СпН170зКз8, мол. в.
271,35 — белый мелко- __________
кристаллич порошок; h2N-/>-so2NHCONHC4H9
т. пл. 138—141°; хорошо * \ / 9
растворим в спирте и
ацетоне, хуже — в дихлорэтане; плохо растворим в
эфире, хлороформе, воде и разб. кислотах. Н. хорошо
растворяется в щелочах, давая натриевую соль; при
гидролизе Н. серной к-той и подщелачивании появ-
ляется характерный запах бутиламина. Количест-
венно Н. определяют титрованием щелочью по тимол-
фталеину в спирте. Н. может быть получен конденса-
цией натриевой соли N-ацетилсульфаниламида с
н-бутилизоцианатом и последующим омылением аце-
тильной группы:
CHgCONH
+ и—C4h9nco ——
CH3CONH4 y-SO2NHCONHC4H3
Н.
Н. применяют для лечения сахарного диабета; кроме
того, Н. обладает антибактериальным действием.
По химич. строению и лечебному действию Н. бли-
зок к бутамиду [М-(4-метплбепзолсульфонил)-
N' - н - бутилкарбамид]
CizHjgOsNsS, мол. вес н с
270,34—белый кристал- 3
лич. порошок, т. пл.
126—129°; нерастворим в воде, растворим в спирте,
ацетоне, диоксане и щелочах. Бутамид не обладает
антибактериальным действием, не нарушает микро-
флоры кишечника; поэтому ему часто отдают предпо-
чтение по сравнению с Н.
Лит.: Машковский М. Д., Лекарственные средства,
4 изд., М., 1960, с. 352. В. Г. Яшунский.
НАДКИСЛОТЫ (перкислоты) — см. Перекиси, и гид-
роперекиси.
НАДСМОЛЬНАЯ ВОДА — водная часть конден-
сата, образующегося при охлаждении сырого коксо-
вого газа, получаемого в процессе производства
кокса из каменного угля. При отстое конденсата
(rf=l,0) Н. в. располагается над смолой (<7=1,15—1,21),
образуя верхний водный слой конденсата. Общее коли-
чество Н. в., выделяющейся в процессе коксования,
слагается из пирогенетич. воды, образующейся при
пиролизе кислородсодержащих соединений перего-
няемого угля (выход на сухой уголь 2—4%), и влаги,
12*
359
НАЙЛОН—НАПАЛМ
360
содержащейся в угле, пошедшем на коксование (обыч-
но влажность шихты 7—9%). Основная масса Н. в.
конденсируется вместе со смолой в газовых холодиль-
никах (см. Коксохимическое производство), откуда
конденсат через гидрозатворы отводится в отстой-
ники. При конденсации в Н. в. растворяется часть
водорастворимых составляющих коксового газа (ам-
миак, углекислый газ, сероводород и др.).
Н. в.— прозрачная легкоподвижная жидкость буро-
вато-зеленоватого цвета, с характерным запахом;
выход 9—12%, считая на сухую шихту. Общее содер-
жание аммиака в Н. в. 4—6 г/л (и более). В надсмоль-
ной воде различают: л е т у ч и й аммиак, к к-рому
относят свободный аммиак и легко разлагающиеся
при кипячении соли аммония [(NH4)2S, NH4CN,
(NH4)2CO3 и др.]; связанный аммиак, соли
к-рого при кипячении не разлагаются [NH4C1,
(NH4)2SO4, NH4CNS и др.]. В Н. в. содержатся также
фенола, в основном одноатомные — фенол, крезолы
(0,8—2 г/л), двухатомные — пирокатехин, резорцин
(0,01—0,2 г/л), легкие пиридиновые основания (0,2—
0,5 г/л), а также мельчайшие капельки смолы (в ка-
честве механич. примеси).
Из Н. в. путем ее переработки получают аммиак.
До разработки способа получения аммиака синтетич.
путем Н. в. была одним из основных источников полу-
чения этого продукта. Ввиду наличия в Н. в. фено-
лов, цианидов и др. токсических веществ сброс ее в
близлежащие к коксохимии, заводу водоемы без пред-
варительной очистки (с целью обезвреживания) не
допускается.
Переработка и очистка Н. в. производятся в амми-
ачной колонне. При наличии в Н. в. от 1 г/л (и более)
связанного аммиака для разложения его солей колон-
на имеет известковый смеситель, куда подается из-
вестковое молоко, и вспомогательную колонну. В ре-
зультате взаимодействия связанных солей аммиака
с известью образуются соли кальция — СаС12, CaSO4,
Ca(CNS)2 и др. и свободный аммиак. Отгоняемый ам-
миак получают в виде конц. аммиачной воды (17—-
18%-ной) или передают в газопровод коксового газа
для извлечения в виде солей, преимущественно суль-
фата аммония. Для отгонки аммиака в колонну пода-
ют острый пар. Если в Н. в. содержится менее 1 г/л
связанного аммиака, то известь обычно не применяет-
ся и необходимость в установке смесителя и вспомога-
тельной колонны отпадает.
Для удаления фенолов из Н. в. ее подвергают обес-
феноливанию в специальном скруббере, где Н. в.
обрабатывают острым паром и р-ром NaOH, или про-
мывают Н. в. до подачи ее на аммиачную колонну
органич. растворителем (обычно бензолом) с последую-
щей отмывкой фенолов из экстракта конц. р-ром
NaOH. При повышенных требованиях к очистке Н. в.
от фенолов ее после обесфеноливания по указанным
выше способам очищают биохимич. способом; при этом
способе фенолы полностью разрушаются микробами
и безвозвратно теряются. После отгонки аммиака и
удаления фенолов смесь Н. в. и введенных в нее вод,
отстоявшаяся от нерастворимых солей кальция и раз-
бавленная дополнительно другими, безвредными во-
дами коксохимии, производства, используется обыч-
но для тушения кокса либо сбрасывается в водоемы.
Лит.: Колян др Л. Я., Улавливание и переработка хи-
мических продуктов коксования, Харьков — М., 1953; Мед-
ведев К. П. [и др.], Кокс и химия, 1962. №10; Соколов
Б. М., там же, 1958, К- 6; ЛевинЕ. Б., там же, 1957, № 1;
Ненпч В. Н. [и др.], там же, 1960, № 1; Handbuch des
Kokereiwesens, hrsg. v. О. Grosskinsky, Bd 1—2, Dusseldorf,
[1955—58]. А. А. Ревякин.
НАЙЛОН — CM. Полиамиднае волокна.
НАЛОРФИН (анторфин, летидрон, наллин, хлор-
гидрат N-аллилнорморфина) C,9H2]O3N -НС1, мол. в.
347,85 — бесцветные кристаллы, т. пл. 260—263°,
р-рах едких щелочей, труд-
[а]д =—122—125° (2%-ный водный р-р), хорошо рас-
творим в воде и в вод!
но растворим в спир-
те, практически не-
растворим в эфире и
хлороформе. Являясь
производным морфи-
на, Н. дает харак-
терные реакции с ал-
калоидными реакти-
вами, с реактивом Фреде дает фиолетовое окрашивание,
постепенно переходящее в сине-зеленое. В отличие от
морфина Н. растворяется в избытке водного аммиака.
Количественное определение Н. основано на титрова-
нии хлорной к-той в безводной уксусной к-те в при-
сутствии ацетата ртути по кристаллическому фиоле-
товому. Н. получают из морфина через цианнорди-
ацетилморфин и норморфин. Н. является антагони-
стом морфина и других анальгетиков (промедол, фе-
надон, пальфиум и др.), снимает вызванное последни-
ми угнетение дыхания, снижение артериального дав-
ления, аритмию сердца и т. д. Н. применяют в каче-
стве антидота при передозировке морфина и других
анальгетиков, а также для предупреждения и лечения
асфиксии новорожденных при обезболивании родов
анальгетиками. Н. не влияет на нарушения дыхания
и кровообращения, вызванные барбитуратами, эти-
ловым эфиром или циклопропаном.
Лит.: U n п а К., J. Pharmacol, and Exptl. Therap., 1943,
79, 27; M с C a w 1 e у E. L. [a. o.], J. Amer. Chem. Soc., 1941,
63, 314; G r e e n A. F. [a. o.], J. Pharmacy and Pharmacol.,
1954, 6, 390; Rentsch G., Pharmazie, 1956, 11, 580; M a ni-
ковский M. Д., A p у т ю н я н Г. С., Мед. пром-сть,
СССР, 1958, №s 6, 37. Л. H. Яхонтов.
НАМАГНИЧЕННОСТЬ — см. Магнитнае свой-
ства.
НАМЕТКИНА ПЕРЕГРУППИРОВКА — см. Кам-
феновае перегруппировки.
НАПАЛМ — загущенное жидкое горючее (бензин,
керосин, газолин и др.), иногда со специальными до-
бавками, применяемое в качестве зажигательных и
огнеметных смесей. Часто термином Н. обозначают
также сам загуститель, при растворении к-рого в жид-
ком горючем последнее загустевает и превращается
в гель. В качестве загустителя применяют алюминие-
вые соли (мыла) нафтеновой, пальмитиновой, лаури-
новой, стеариновой и др. органич. к-т. Количество
загустителя, вводимого в бензин (газолин), составляет
от 4 до 11%; консистенция получаемого при этом Н.
может быть от вязкой жидкости до практически нете-
кучего студня. Скорость горения Н. можно регули-
ровать добавлением асфальта, древесной муки и раз-
личных смол. Н.— вязкий, липкий продукт; горит
медленнее бензина, что позволяет использовать его
при поджоге деревянных строений. Теплота горения
Н. ок. 10 000 ккал/кг, темп-pa горения 800—1000°.
В рецептуру Н. могут быть добавлены также белый
фосфор, асфальт и смесь перхлората калия с алюми-
нием или магнием; при горении такой смеси темп-ра
достигает ок. 1980°.
Н. может быть применен в зажигательных авиабом-
бах, ракетах, минах, ручных гранатах и огнеметах.
При применении Н. из огнеметов дальность огнемета-
ния и поражающее действие значительно выше, чем
при использовании для этих целей жидкого горючего.
Если к Н. добавить сплавы легких металлов (напр.,
натрия), смесь самовоспламеняется на цели (особенно
на воде или снегу). Такие смеси наз. с у п ер на-
ла л м; при ночном огнеметании они не демаскируют
бойца. Возможно также добавление к Н. небольшого
количества 82%-ной Н2О2; при этом образуется эму-
льсия. Н. применялся в годы второй мировой войны
авиацией США в зажигательных бомбах; в 1950—5.3
войска США применяли Н. в агрессивной войне про-
361
НАПОЛНИТЕЛИ —НАПРЯЖЕНИЯ ТЕОРИЯ
362
тив корейского народа в зажигательных бомбах, ог-
неметах и др.
Лит.: Шидловский А. А., Основы пиротехники,
2 изд., М., 1954; Woods D. Е., Taylor A. J., J. Appl.
Chem., 1958, 8, 237; Н a j е k Н. V., Explosivstoffe, 1957, № 6,
121. А. А. Шидловский.
НАПОЛНИТЕЛИ (для резин) — твердые тонкоди-
сперсные вещества, вводимые в резиновую смесь или в
латекс для улучшения (усиления) технических свойств
резин, а также для их удешевления. Н. различают:
по происхождению — природные и синтетич.; по сос-
таву — органич. и неорганич.; по действию на механич.
свойства резин — активные и неактивные. К актив-
ным Н. относят: сажи (углеродные), смолы, лигнин,
аморфную двуокись кремния и нек-рые сорта сили-
катов кальция, алюминия, циркония, окислы железа,
алюминия, титана и др. Такие Н. характеризуются
высокой дисперсностью, способностью хорошо дис-
пергироваться в полимерах, высокой адсорбционной
активностью ио отношению к полимерам, в отдельных
случаях — способностью катализировать вулкани-
зацию, а иногда и окисление резины. К неактив-
ным Н. относят: природный мел, каолин, тальк,
слюду, асбест и др.
Органические Н. Основными активными
наполнителями являются углеродные сажи, получае-
мые при неполном сгорании углеводородов. Разли-
чают сажи: газовые — из природного газа, и жидкост-
ные — из ароматизированных масел переработки
нефти и каменного угля. Как те, так и другие могут
быть получены или при ламинарном потоке газа (па-
ров) в пламени щелевых горелок, или в турбулентном
пламени в специальных реакторах. Особенно ценны
«турбулентные» сажи, придающие резинам высокую
износостойкость. Различают подгруппу полуусили-
вающих саж, повышающих прочностные характери-
стики резин без существенной потери их эластичности.
В последнее время в качестве Н. используют
нек-рые синтетич. смолы — продукты конденсации фор-
мальдегида с резорцином, фенолом, анилином и т. д.,
а также эпоксидные и кумарон-инденовые смолы.
Неорганические Н.Из этих Н. наибольшее
распространение имеет двуокись кремния. Известны
продукты жидкофазного гидролиза р-ра силиката
натрия и продукты газофазного гидролиза четырех-
хлористого кремния; первые — под названием б е-
л а я сажа (СССР), хайсил (США), ультрасил
(ФРГ) и т. д., вторые — аэросил (ФРГ), карбосил
(США), франсил (Франция) и др. Существенное
значение имеют продукты, полученные «гидрофобиза-
цией» поверхности двуокиси кремния и некоторых дру-
гих Н. органич. поверхностно-активными веществами.
Углеродные сажи до сих пор незаменимы для соз-
дания износостойких резин. Смолы усиливают ре-
зины без снижения эластичности. Неорганические Н.
особенно ценны для специальных резин на основе по-
лярных каучуков. Нек-рые Н. этого класса резко по-
вышают теплостойкость резин.
Лит.: К е л ь ц е в В. В., ТеснерП. А., Сажа. Свойства,
производство и применение, М.—Л.,1952; Кошелев Ф. Ф.,
К л и м о в Н. С., Общая технология резины, М., 1958; Par-
kinson D., Reinforcement of rubbers, L., 1957.
П. H. Орловский, H. H. Лежнев.
НАПРЯЖЕНИЕ РАЗЛОЖЕНИЯ — напряжение
между электродами, погруженными в р-р электро-
лита, при к-ром становится возможным процесс элек-
тролиза, сопровождающийся выделением продуктов
разложения на катоде и на аноде. Н. р. равно разно-
сти потенциалов выделения соответствующих веществ
на обоих электродах (<ранояа—Фкатода)! напр., при элек-
тролизе р-ра хлорида цинка Н. р. должно быть рав-
ным (+1,36) — (—0,76) = 2,12 в; при электролизе
хлорида меди (+ 1,36) — (+0,35) = 1,01 в. В раство-
рах большинства кислородсодержащих к-т и щело-
чей Н. р. определяется потенциалами выделения водо-
рода на катоде и кислорода на аноде, так как анионы
кислородсодержащих к-т и катионы щелочных метал-
лов обладают более отрицательными (катионы) и бо-
лее положительными (анионы) потенциалами, чем по-
тенциалы разряда Н+ и ОН-. Поэтому Н. р. кисло-
родсодержащих к-т и щелочей примерно равно 1,7 в;
хотя разность потенциалов выделения кислорода и
водорода составляет примерно 1,2 в {кислородный
электрод), в действительности Н. р. достигает 1,7 в
вследствие перенапряжения на катоде и на аноде.
В р-рах сульфатов, нитратов и других кислород-
содержащих солей и щелочных и щелочноземельных
металлов Н. р. достигает 2,2 в, так как в этих случаях
в процессе электролиза
изменяется состав электро- '°
лита у катода и у анода: у /
катода электролит подщела- /
чивается за счет разряда /
ионов Н + , а у анода — под- /
кисляется за счет разряда /
ОН-. Вследствие этого по- /
тенциал катода становится /_____
более отрицательным, а по- ________________|
тенциал анода — более по- ---—
ложительным, и Н. р. воз- р' t e
растает. Н. р. определяют экспериментально пу-
тем снятия поляризационной кривой (см. рис.), вы-
ражающей зависимость силы тока I от приложенного
напряжения Е. При этом предполагается, что оба
электрода имеют одинаковый размер и не взаимодей-
ствуют с электролитом. Чаще всего применяют пла-
тиновые электроды.
Термин «Н. р.» был введен Лебланом в конце прош-
лого века. По современным воззрениям, Н. р. являет-
ся лишь приближенной величиной, характеризую-
щей то напряжение, при к-ром начинается интенсив-
ный разряд ионов данного вида; в действительности
же разряд нек-рой части этих ионов возможен и при
меньших напряжениях, т. к. в соответствии с Больц-
мана законом распределения не все ионы данного вида
обладают одинаковой энергией, вследствие чего и
разряд их происходит при различных значениях по-
тенциала. Н. р. соответствует тому напряжению, при
к-ром разряжается основная масса ионов, причем си-
ла тока резко возрастает (см. рис.).
Лит.: Скорчеллетти В. В., Теоретическая электро-
химия, 2 изд., Л., 1963; Делимарский Ю. К., Г о-
родыский А. В., Электродные процессы и методы иссле-
дования в полярографии, Киев, 1960; Левин А. И., Тео-
ретические основы электрохимии, Свердловск, 1960; Г л е с-
стон С., Введение в электрохимию, пер. с англ., М.,
1951. О. А. Сангина.
НАПРЯЖЕНИИ РЯД — СМ. Ряд напряжений.
НАПРЯЖЕНИЯ ТЕОРИЯ — теория, объясняю-
щая различия в устойчивости циклич. систем отклоне-
нием их валентных углов от нормальных. Н. т. пред-
ложена А. Байером (1885), к-рый исходил из того, что
алициклич. молекулы должны быть плоскими и что
отклонение валентных углов от тетраэдрического
(109°28') является мерой напряженности цикла. В
дальнейшем Заксе (1890) и Мором (1918) было пока-
зано, что 6-членные и высшие циклы могут быть не-
плоскими и, следовательно, почти не иметь углового
напряжения. Отклонения валентностей (половина
разности между 109°28' и величиной внутреннего угла
многоугольника) составляют: 24°44' для циклопро-
пана, 9°44'— циклобутана, 0°44'— циклопентана,
0° — циклогексана и высших алициклов (неплоские
циклы). Соответственно возрастает устойчивость при
переходе от 3-членных к 6-членному циклу. Теплоты
сгорания при постоянном объеме (в ккал/моль):
166,6 циклопропан; 163,9 циклобутан; 158,7 цикло-
пентан; 157,4 циклогексан; 158,3 циклогептан; 158,6
циклооктан и т. д. Соответственно легче всего раз-
рывается 3-членный цикл циклопропана (напр., при
363
НАРКОЛАН — НАСАДКИ
364
действии Вг, Ш, каталитич. гидрированием при
100°) и труднее — 4-членный в циклобутане (дейст-
вие HJ, каталитич. гидрирование при 180°). Пятичлен-
ный цикл в циклопентане не раскрывается при дей-
ствии Вг и HJ, а гидрогенолиз 5-членного цикла про-
исходит лишь при значительно более высокой темп-ре
(выше 310°). В общем виде Н. т. с успехом может
быть применена к самым разнообразным циклич. сис-
темам, а также при рассмотрении стереохимии нек-рых
соединений с открытой цепью. Следует, однако,
учесть, что классич. Н. т. объясняет далеко не все осо-
бенности циклич. соединений, т. к. не учитывает вза-
имодействия валентно не связанных атомов.
Наряду с классическим, байеровским, напряжением,
связанным с деформацией валентных углов, большую
роль в циклах играет конформационное напряжение,
связанное с наличием энергетически невыгодных
конформаций (см. Конформационный анализ). Так, во-
преки Н. т., 5-членное кольцо в циклопентане яв-
ляется неплоским из-за отталкивания атомов водорода
(торсионное напряжение). Конформационное напряже-
ние оказывает заметное влияние на реакционную
способность циклич. систем. Нарушение валентных
углов и конформаций при образовании переходного
состояния и при образовании в ходе реакции карбонил
ионов может весьма значительно сказаться на скоро-
сти реакции. Так, напр., такого рода изменением внут-
реннего напряжения объясняется отличие в реакцион-
ной способности циклопентанона и циклогексанона
(первый присоединяет реагенты по карбонильной груп-
пе намного медленнее, чем второй).
Лит.: Домнин Н. А., Строение циклических соедине-
ний в свете теории напряжения, Л., 1936; X ю к к е л ь В.,
Теоретические основы органической химии, пер. с нем., т. 1,
М., 1955, с. 71; Г о л ь д ф а р б Я. Л., Беленький Л. И.,
Успехи химии, 1960, 29, вып. 4, 470. М. Е. Вольпин.
НАРКОЛАН (авертин, ректанол, трибромэтанол)
СВГ3СН2ОН, мол. в. 282,77 — мелкокристаллич. по-
рошок белого цвета; т. пл. 80—82,5°; т. кип. 92—
94°/10 мм; трудно растворим в воде (1:35), легко —
в спирте, эфире и бензоле; неустойчив на воздухе и
в воде. Водные растворы Н. при нагревании выше 40°
разлагаются с образованием НВт и СНВг2СНО. При
нагревании Н. с реактивом Несслера выпадает крас-
ный осадок, окраска к-рого переходит в серую. Коли-
чественно Н. определяют по Фольгарду после кипя-
чения со щелочью. Н. может быть получен из СВгзСНО
восстановлением А1(изо-СзН7О)з в изо-СзН7ОН. Н.
применяют в качестве наркотич. средства (в виде
водно-спиртового р-ра) в комбинации с другими пре-
паратами.
Лит.: Bud ё sinsky Z., Protiva М., Synthetlsche
Arzneimittel, В., 1961, S. 234. В. Г. Яшунский.
НАРКОТИН (8-метоксигидрастин) С22Н2зО7Х, мол.
в. 413,43 — алкалоид опия, где его содержание дохо-
дит до 9%; кристаллы; т. пл. 176°, почти нерастворим
в воде, растворим при 15° в бензоле (1:22), этилаце-
тате (1:31) и эфире (1:170); [а]д = — 198° (хлоро-
форм). Растворим в горячих щелочах, расщепляю-
щих лактонный цикл с образованием солей —н а р-
к о т а т о в. Соли Н.— оксалат, т. пл. 174°, [а]д=
= + 39,5°; фталат, т. пл. 160°, [а]д = + 115° (хло-
роформ) и др. нестойкие; в водных р-рах гидролизу-
ются, так что Н. можно извлекать растворителями
из слабокислой среды. Природный левовращающий
Н. (1-а-наркотин) при действии метанольного р-ра
КОН переходит в изомер с более слабым вращением
(рацемизация у одного из асимметрия, атомов С — в
l-fj-наркотин); полный рацемат Н. носит название гно-
скопин (т. пл. 232—233°).
От других алкалоидов опия Н. отделяется благо-
даря слабой основности. При нагревании с Ва(ОН)2
наркотин (I) распадается на опиановую к-ту (II) и
гидрокотарнин (III), а при действии восстановителей
вместо (II) образуется меконин (IV), который также
содержится в небольшом количестве в опии. Окисле-
ние Н. разб. азотной к-той дает котарнин. Строение
IV
Н. подтверждено синтезом гноскопина и разделением
последнего на антиподы. При конденсации метилового
эфира 2,3-диметоксибензойной к-ты с хлоралгид-
ратом получается трихлорметиллактон, СС1з-группа
к-рого омыляется щелочью; образовавшаяся СООН-
группа удаляется декарбоксилированием. Продукт
реакции —меконин (IV) — при конденсации с котар-
нином дает гноскопин. Возможен обратный синтез Н.
из меконина и опиановой к-ты.
Физиология, активность Н. подобна активности
других алкалоидов опия (напр., кодеина), но слабее.
Н. усиливает действие морфина. В медицине Н. не
применяется и служит сырьем для получения котар-
нина и гидрастинина.
Лит. см. при ст. Алкалоиды. А. Е. Васильев.
НАСАДКИ — тела различной формы и размеров,
помещаемые в аппараты для тепло- и массообмена с
целью развития поверхности контакта между двумя
фазами: жидкостью и газом, жидкостью и паром,
жидкостью и жидкостью, а также для выравнивания
потоков, отделения брызг, изменения характера пере-
мешивания. Н. широко применяются в аппаратах для
абсорбции, ректификации, экстрагирования, в гра-
дирнях и др.; изготовляют Н. чаще всего из керамики,
в ряде случаев из фарфора, в малых установках —
из стекла; применяют также Н. пз металла, дерева,
реже из пластмасс, графита и др. материалов.
Характеристики Н. (их типы, размеры, уд. поверх-
ность и др.) приведены в таблице. Н. загружают в
аппарат навалом или укладывают в определенном
порядке. Н., загружаемые навалом (рис. 1), в пром,
практике применяют размером 10—80 мм. В некото-
рых случаях в качест-
ве Н. используют куски
различных материалов
(кокс, кварц и др.); чаще
всего применяют кольца
с высотой, равной диа-
метру (кольца Рашига).
Иногда применяют коль-
ца с перегородками,
с отверстиями в стенках
(кольца Паля), седлооб-
разные Н., спирали из
металлич. ленты и др. В
лабораторных условиях
применяют более широ-
кий ассортимент Н. (раз-
мером 2—15 мм), осо-
бенно в ректификацион-
ных колонках (кольца,
шарики, бусы, прово-
лочные кольца, седла,
спирали и др.). Н. из
Промышленные типы
Лабораторные типы
Рис. 1. Насадки, загружаемые
навалом: 1 — кольца Рашига;
2 — кольца Паля; 3 — седла
Берля; 4 — кольца Диксона из
сетки; 5 — спирали Фенске;
б — шарики.
365
НАСАДКИ
366
Тип насадки Размеры, мм са б пв
Н., загружаемые навалом
Кольца Рашита (ке- рамич.) 50х50х5г 90 0,785 5500
То же 25X25X3 20 0 0,74 45000
То же 15X15X2 330 0,7 210000
То же 10X10X1,5 440 0,7 700000
Кольца Рашига (стеклянные) . . 4 х 6 ;< о, 6 1110 0,7
То же 2X2X0,5 2140 0,5
Кольца Рашига (стальные) . . . 50X50X1 1 10 0,95 5600
То же 25X25X0,8 220 0,92 46000
То же 15X15X0,5 350 0,92 210000
То же 8X8X0,3 630 0,9 900000
Кольца Паля (кера- мич.) 50x50X5 110 0,78 5500
То же 25x25x3 250 0,74 45000
Кольца Паля (ме- тал лич.) .... 50X50X1 110 0,95 5600
То же 25X25x0,8 220 0,92 46000
Седла Берля (кера- мич.) 50 120 0,7 8000
То же 25 260 0,7 75000
Спирали из метал- лич. проволоки . 4X4X0,4 1500 I 0,9—0,95
То же 2X2X0,2 4500 /
Спирали из метал- лич. ленты . . . 0,2x19x380^ 130 0,99
Насадки, загружаемые в определенном
порядке (регулярные Н.)
Кольца Рашига (ке- рамич.) юохюохю 60 0,72 940
Тб же 80x80X8 80 0,72 1900
То же Хордовая (деревян- 50x50x5 120 0,72 7800
ная) 100х10Х20е 65 0,68
То же Плоскопараллель- 100x10x10 толщина 5, 100 0,55
ная (из тенстоли- 0,8
товых пластин) зазор 21
То же из стальных толщина 2, 133 0,87
пластин .... зазор 13
Насадка мультифил
(из металлич. сет- ки) 2000 0,95
Насадка Стедмена
из стальной про- волоки диаметром 0,23 мм .... — 715 0,645
а Уд. поверхность (поверхность насадки, з^/лг3). ® Доля
свободного объема (доля пустот в слое насадки). в Число
элементов насадки в 1 л3. г Диаметрхвысотахтолщина.
Д Толщина лентыхдиаметр спиралиХдлина отрезка спира-
ли. е Высотах толщинах зазор.
проволоки или стеклянных нитей обладают самым
большим свободным объемом и весьма развитой по-
верхностью.
Н., укладываемые в определенном порядке (регу-
лярные насадки) (рис. 2), отличаются, как
правило, низким гидравлич. сопротивлением и более
высокой пропускной способностью, чем Н., загружен-
ные навалом. Такие Н. применяют для снижения со-
противления, уменьшения энергетич. затрат, либо
при проведении процесса в вакууме. Для этих целей
применяют укладываемые упорядоченно кольца Ра-
шига, Н. в виде керамич. блоков, хордовую Н. из дере-
вянных досок, поставленных на ребро. Наименьшее
гидравлич. сопротивление оказывает Н. в виде па-
раллельных листов большой высоты (плоско-парал-
лельная Н.). Некоторые типы регулярных Н. изго-
товляют из металлич. сеток или перфорированных
листов.
Вес 1 ма Н. (gH) можно определить по формуле:
й’н=й'м(1 — уо)
где gM — плотность материала, из которого сделана
насадка.
В абсорбционных и ректификационных насадоч-
ных колоннах по Н. стекает пленка жидкости, омы-
ваемая потоком газа
или пара. Так как ско-
рость массообмена про-
порциональна поверх-
ности контакта фаз, то
выгоднее пользоваться
мелкими Н., имеющи-
ми большую удельную
поверхность. Коэфф,
массопередачи также,
как правило, больше в
случае более мелкой Н.
Однако с уменьшением
размеров насадочных
тел ухудшается их сма-
чивание и уменьшается
доля активной поверх-
ности Н., участвующая
в массообмене, что ос-
лабляет эффект исполь-
Рис. 2. Насадки, укладываемые
в определенном порядке (регу-
лярные насадки): I — кольца Ра-
шига; 2 — керамические блоки;
3 — деревянная хордовая; -I —
спрейпак (из перфорированных
металлических листов).
зования мелких Н. В
мелких Н. (размерами
менее 20—30 мм) эф-
фективно смоченной бы-
вает менее 0,5, а в
нек-рых случаях даже
менее 0,1 всей поверх-
ности. В крупных, особенно регулярных, Н. степень
смоченности может доходить почти до 100%.
Гидравлич. сопротивление Н. может быть вычислено
по формуле:
ДР = С
н_
2gt>J
где Ар — гидравлич. сопротивление, Н — высота
слоя Н., d3— ivjo — эквивалентный диаметр, g —
ускорение силы тяжести, у — плотность газа, —
скорость газа (отнесенная к сечению аппарата без Н.).
Коэфф, сопротивления £ в обычных условиях слабо
зависит от скорости газа (для регулярных Н. £ зна-
чительно меньше, чем для Н., загруженных навалом).
При орошении Н. ее сопротивление возрастает вслед-
ствие уменьшения свободного объема за счет находя-
щейся в Н. жидкости. Этот рост может быть описан
приближенной ф-лой:
Сорош. ?сух. (1 4* AL)
где L — плотность орошения ма'мг -час; для большого
числа Н. коэффициент А колеблется в пределах 0,03—
0,06. Кроме увеличения гидравлич. сопротивления,
количество жидкости, удерживаемое Н., оказывает
влияние и на массопередачу. Это количество жид-
кости растет с увеличением плотности орошения и уд.
поверхности Н.; оно меньше в регулярных Н., чем в
загруженных навалом. При малых скоростях встреч-
ный поток газа мало влияет на количество жидко-
сти, удерживаемое Н., но с увеличением скорости
выше нек-рого предела начинается накопление
жидкости в Н. (подвисание). При дальнейшем уве-
личении скорости газа наступает момент, когда жид-
кость перестает стекать вниз и выбрасывается из
Н., этот момент обозначается как «захлебывание»
Н.; вблизи точки захлебывания ускоряется массо-
передача.
Лшп.: Жаворонков Н. М., Гидравлические основы
скрубберного процесса и теплопередача в скрубберах, М.,
1944; К р е л ь Э., Руководство по лабораторной ректифи-
кации, пер. с нем., М., 1960, с. 441; Leva М., Tower packings
and packed tower design, 2 ed., Akron, 1953; E 1 11 s S. R. M.,
VarjavandlJ., Chem. and Process Engng, 1958, 39, № 7,
239; R a n d a 1 1 D. G., там же, 1959. 40, № 9, 306; Жаво-
367
НАСЫЩЕННЫЙ РАСТВОР—НАТРИЙ
368
р о н к о в Н. М., Мал ю сов В. А., Научн. докл. высшей
школы. Химия и хим. технология, 1958, К» 1. 185. См. также
лит. к ст. Абсорбция и Ректификация. А. Ю. Закгейм.
НАСЫЩЕННЫЙ РАСТВОР — см. Раствори-
мость.
НАТР ЕДКИЙ — см. Натрия гидроокись.
НАТРИЙ (Natrium) Na — химич. элемент I гр.
периодич. системы Менделеева, принадлежит к под-
группе щелочных металлов; п. н. 11, ат. в. 22,9898.
В природе встречается лишь стабильный изотоп Na23.
Сечение захвата тепловых нейтронов атомом Н.цэф =
= 0,49±0,02 барн. При бомбардировке Н. тепловыми
нейтронами образуется ^-активный изотоп Na24
(fi/2 = 15,06 час.), применяемый в биологич. и меди-
цинских исследованиях. Конфигурация внешних
электронов атома Н. 3s1; энергия ионизации (в эв):
Na°=Na + =Na2+=Na3 + --Na4 + соответственно состав-
ляет 5,09; 46,65; 71,3; 99,0. Н.с имеет характерные
спектральные линии 5890 и 5896 А (интенсивный жел-
тый цвет).
Природный карбонат Н., образующийся в египетских со-
ляных озерах по реакции GaGO,+2NaGl = 2Na3GO34-GaGl2,
применялся в глубокой древности как моющее средство под
названием neter (древнееврейское), virpov (греч.), nitrum (лат.),
от к-рых произошли названия натр (окись натрия) и натрий
(металл). Различие между содой и поташом установил только
в 1736 А. Л. Дюамель дю Монсо (опубл. 1747). Металлический
Н. получил в 1807 Дэви электролизом едкого натра. В 1808
Гей-Люссак и Тенар получили Н. восстановлением едкого
натра железом при темп-ре красного каления.
Весовое содержание Н. в земной коре составляет
2,64%; по распространенности Н. занимает 6 место
среди других элементов. Наличие Н. установлено в
атмосфере Солнца и в межзвездном пространстве. В
гидросфере Н. содержится в виде растворимых солей
в количестве ок. 2,9% (при общей концентрации солей
в морской воде 3,5—3,7%). Абс. содержание Н. в мор-
ской воде составляет ок. 1,5 -1016 т. Вследствие своей
большой реакционной способности Н. в земных усло-
виях находится только в виде солей с различными
анионами. Важнейшие из них: хлористый натрий NaCl
(каменная соль, галит), сульфат натрия
Na2SO4 -ЮН20 (мирабилит, глауберова
соль), нитрат натрия NaNOa (чилийская се-
литр а), гексафторид натрия-алюминия 3NaF -AlFa
(криолит), тетраборат натрия Na2B4O7 -ЮН20
(бура, т и н к а л) и др.
Кроме того, Н. в сочетании с Са, Sr, Ba, Mg, Al и
редкоземельными металлами входит в состав многих
природных силикатов. Небольшие количества Н. на-
ходятся в растениях; напр., в свежей траве тысяче-
листника (Achillea millefolium) обнаружено всего
0,0006% Na. Морские растения значительно богаче
Н.; так, свежая морская трава (Zostera morina) содер-
жит 0,547%, а ее зола 16,78% Na. Соединения Н. вхо-
дят в состав животных организмов, главным обра-
зом в виде NaCl; так, напр., в плазме крови че-
ловека ионы Н. составляют 0,320%, в костях 0,6%,
в мышечной ткани 0,6— 1,5%. Восполняя естест-
венную убыль Н. из организма, человек ежедневно
должен потреблять с пищей 4—5 г Н. в виде поварен-
ной соли.
Месторождения Н. широко распространены на Зем-
ле. Богатые залежи каменной соли имеются в СССР
(район Брянск—Бахмач, мощность пласта св. 100 л»;
Илецкая защита близ Оренбурга—чистый NaCl;
Усолье близ Перми, мощность пласта 38,5 ж; Арме-
ния; Сибирь; солевые озера — Эльтон с концентра-
цией 26,5% NaCl, Баскунчак и др.); США (штаты
Техас, мощность пласта до 200 м, Нью-Мексико,
Оклахома, Канзас и др.); Австрия и др. страны. Ми-
рабилит, тенардит встречаются в СССР (Кара-Богаз-
Гол, Сибирь), США (зап. штаты), на о-ве Сицилия,
в Испании, в Сев. Африке. Залежи селитры имеются
в Юж. Америке (Перу, Чили). Богатейшие залежи
троны (минерал состава Na2CO3 -NaHCOa -2Н2О) име-
ются в Сев. Африке (сев.-зап. Каира, Алжир, Судан,
Ливан), а также в СССР (Армения, Сибирь), США (шт.
Невада, Калифорния) и др.
Физические и химические свойства. Н.— серебрис-
то-белый металл (в тонких слоях — с фиолетовым от-
тенком), кристаллизующийся в объемноцентрирован-
ной кубич. решетке с периодом а = 4,28А; плотн.
0,96842 (19,7°). Температурная зависимость плотности
твердого Н. выражается ур-нием: =0,9725—
—0,0002011г—0,00000015г2; для жидкого Н.: =
= 0,9490— 0,000223г—0,0000000175г2. Ат. радиус
1,86А; ионный радиус Na+ 0,92А. Т. пл. 97,83°; т.
кип. 882,9°. Теплота плавления 622,2 кал/г-атом;
теплота испарения 20642 кал/г-атом. Вязкость
жидкого Н. вычисляется из ур-ния lgr|=—1,09127+
+ 382/(г+313); напр., для 147° ц= 0,5475 спуаз. Атом-
ная теплоемкость Н. (в кал/г-атом-град) в твердом
состоянии рассчитывается по ур-нию: Сртв.=9,93555—
—0,028053 Т + 0,000057883 Тг, и в жидком — по
ур-нию: Срж =8,95811—0,0045788 Т+0,0000025409 7’2
(Т в °К). Поверхностное натяжение (дин/см); у = 202—
—0,10г. Теплопроводность (кал/см -сек -град) для твер-
дого Н. &тв.=0,324—0,00040 г (0—97,83°), для жид-
кого Н. кЖ'= 0,2166—0,000116 г (до 550°). Электро-
сопротивление (.нко.и •<?.«): для твердого Н. гтв =
= 4,477+0,01932+0,00004г2 (0—97,83°), для жидкого
Н. гук =6,225+0,0345 г (до 400°); например, 4,477
(0°); 4’,879 (20°); 5,543 (50°); 6,750 (97,83° — тв.Н.);
9,600 (97,83° — ж. Н.); 13,125 (200°); 16,575 (300°);
20,025 (400°).
Пары Н. состоят гл. обр. из одноатомных молекул,
однако имеются и димеры Na2, причем содержание их
увеличивается с темп-рой (600°К — 0,008 часть Na->;
650°К — 0,013; 700°К — 0,019; 750°К — 0,025).
Давление паров Н. весьма низко (в лип рт. ст.):
1,199-10-’ (100°); 1,848-10-3 (250°); 3,958 (500°);
1988 (1000°); гкрит.2000°; ркрит.343 атм. Чистый Н. па-
рамагнитен; уд. магнитная восприимчивость 9,2 -10-6;
диэлектрич. проницаемость 60.
Химич, активность Н. весьма высока. На воздухе
металл интенсивно окисляется с поверхности; с водой
энергично реагирует, образуя NaOH и Н2. При боль-
шой поверхности контакта Н. с водой реакция идет
со взрывом. Нормальный электродный потенциал
Н.—2,74 в. При 200° Н. начинает поглощать водород,
образуя весьма гигроскопич. порошок гидрида Н.
NaH (см. Натрия гидрид). При взаимодействии с
кислородом в зависимости от условий образуется
окись Na2O или перекись Na2O2 (см. Натрия
перекись). Na2O— бесцветные кристаллы с плотн.
2,314; т. пл. 920°, т. кип. 1350°. Натрийамин (амид
натрия) NaNH2 образуется при взаимодействии Н.
с NHa при 300—400° или же в присутствии металлич.
катализаторов при охлаждении до —30°. Действием
на амид Н. азотной к-ты при 175° или закиси азота
при 190° образуется азид NaNa, применяющийся
как азотирующее средство. Нитрид NaaN может
быть получен взаимодействием паров Н. с азотом в
поле тихого электрич. разряда. Реакционная способ-
ность Н. с галогенами падает в ряду F--Cl--L>r=.J;
в атмосфере фтора Н. воспламеняется, в атмосфере
хлора Н. горит при обычной темп-ре; реакция с бро-
мом идет лишь при нагревании; с иодом Н. не реаги-
рует в обычных условиях (см. Натрия фторид, Нат-
рия хлорид и др.). С большинством галогенидов Н.,
особенно в диспергированном состоянии, энергично
реагирует, восстанавливая катион до металла, что на-
ходит применение в произ-ве ряда металлов (напр.,
титана, калия и др.). Реакция Н. с серой идет бурно
в твердой фазе при растирании их в ступне (см. Нат-
рия сульфид). При 800— 900° пары Н., соединяясь с
369
НАТРИЙ
370
углеродом, образуют карбид Na2C2; известны
также и другие карбиды — NaC8 и NaC16.
И. легко растворяется в жидком аммиаке, образуя
аммиачные комплексы; растворимость (в г Н. на 100 г
NH3): 34,6(0°), 31,9 (—50°), 29,4(—105°). Изучена
растворимость Н. в расплавленном едком натре
(в г И. на 100 г NaOH): 25,3 (480°); 10,1(600°); 7,9
(760°); 6,9 (800°) и в расплавленном хлористом натрии
(в вес. % Н.): 3,4—4,2 (800°); 15-20 (850°).
Со многими металлами Н. образует сплавы. Широкое
применение имеют сплавы, находящиеся при ком-
натной темп-ре в жидком состоянии в пределах со-
держания от 10 до 60 вес. % Н. Амальгамы легко
получаются путем прибавления небольшими порция-
ми Н. к ртути, находящейся под слоем керосина или
минерального масла. На воздухе поверхность амаль-
гам покрывается пленкой, состоящей из окиси или
карбоната Н. Амальгамы Н. легко разлагаются во-
дой, разбавленными кислотами, щелочами. Добавле-
ние незначительных количеств амальгам меди, се-
ребра, свинца или олова пассивирует очень чистый
Н., резко повышая его эффективность в органиче-
ских реакциях. О сплавах Н. с калием см. в статье
Калий.
Большое и разнообразное применение находит II.
в органич. синтезе. Н. хорошо реагирует со спиртами
(особенно низшими) с образованием алкоголятов; с
помощью Н. производят восстановление органич.
соединений, реакции конденсации, полимеризации
(см. Натрийорганические соединения).
Аналитическое определение Н.
Качественно Н. определяют по характерному желтому
окрашиванию пламени. Концентрация Н. менее
3 мг/л в р-ре объемом менее 1 мл может быть опреде-
лена спектроскопически с применением угольных
электродов, а также осаждением Н. триэтаноламин-
динитроциклогексилфенолятом в виде золотистых че-
шуек или пикролоновой кислотой в виде характер-
ных кристаллов. Количественное определение Н.
производят осаждением натрий-цинк-уранилацетата
NaZn(UO2)3(CH3COO)9-6Н2О или натрий-магний-
уранилацетата NaMg(UO2)3(CH3COO)9-6Н2О, которые
либо непосредственно взвешивают, либо в них коли-
чественно определяют уран и полученные данные пе-
ресчитывают на Н. В смеси хлоридов щелочных ме-
таллов Н. определяют след, образом: навеску перево-
дят в перхлораты, Н. и литий извлекают смесью
н-бутилового спирта и этилацетата, после чего Н. оса-
ждают в виде NaCl добавлением н-бутилового спирта,
насыщенного газообразным НС1. Существует также ряд
специальных методов определения Н.—хроматогра-
фический, нефелометрический, колориметрический,
радиометрический и т. п.
Получение. Н. может быть получен действием на
его соли углерода или других восстановителей при
темп-рах выше темп-ры их плавления. Однако эти
способы не имеют промышленного значения. Основ-
ным современным методом получения Н. является
электролиз расплава поваренной соли. Трудность
получения по этому способу заключается в незначи-
тельной разнице между темп-рой плавления NaCl
(800°) и темп-рой кипения Na (882,9°). При темп-ре
плавления NaCl давление паров Н. настолько велико,
что металлич. Н. почти полностью испаряется. Кро-
ме того, Н. при этих темп-рах легко растворяется в
расплаве, активно взаимодействует с футеровкой ванн,
кислородом воздуха. При 800° и выше практически
получить Н. из NaCl невозможно. Эта трудность пре-
одолевается добавлением к NaCl таких солей, как
КС1, NaF, СаС12 и др., чем удается значительно сни-
зить темп-ру плавления электролита и электролиз
вести при темп-ре порядка 575—585°. Электролиз
проводят в электролизерах (принципиальная схема
одного из них приведена на рис.) с одновременным
получением Na и С1.
Вторым по значимости способом получения натрия
является электролиз
соб и значительно
уступает электроли-
зу NaCl, но он по-
явился раньше и до
сих пор еще оконча-
тельно не вытеснен.
Основной его недо-
статок — дороговиз-
на исходного сырья;
его достоинство —•
низкая температура
процесса разложе-
ния NaOH (порядка
320—330°).
едкого натра; хотя этот спо-
Электролизер Даунса:
1 — стальной цилин-
дрич. кожух; 2 — огне-
упорная кирпичная футеровка; 3 — графитовый анод с то-
копроводом; / — кольцеобразный железный или медный ка-
тод с токоподводом; .5 — сетка-диафрагма; 6 — катодный
колпак; 7 — стояк с трубой для слива Н.; е — сборник для
Н.; 9 — хлоросборная камера с отводной трубой; 10 — люк
для загрузки соли; 11 — крышка.
Техника безопасности. Вследствие высокой реак-
ционной способности Н., особенно по отношению к
воде, хранение и работа с ним требуют мер предосто-
рожности. Хранить Н. следует под слоем инертной
жидкости (керосина и т. п.). Перевозить Н. можно
только в запаянных сосудах или специально обору-
дованных цистернах. При работе с Н. сотрудники
должны быть защищены спецодеждой, резиновыми
перчатками, очками или масками. Обработку больших
количеств Н. следует производить в герметич. бок-
сах с инертной атмосферой. Все рабочие места должны
быть оборудованы противопожарным инвентарем,
песком, кальцинированной содой, огнетушителями,
снаряженными совершенно сухими хлористым на-
трием, углекислым натрием, графитом и пр. В присут-
ствии натрия пользоваться водой для тушения огня
строжайше воспрещается, т. к. это может привести
к взрыву.
Применение. Металлический Н. (чистый или в виде
сплава с другим металлом) находит разнообразное
применение в качестве теплоносителя в клапанах
авиационных двигателей, в машинах для литья под
давлением (для охлаждения плунжера), а также в
ряде химич. процессов, где возникает необходимость
равномерного обогрева в пределах 450—650°. Особое
место занимает применение Н. и его сплава с калием
в качестве жидкометаллич. теплоносителя в ядерных
энергетич. установках благодаря малым эффектив-
ным сечениям поглощения нейтронов, высокой темп-ре
кипения, высокому коэфф, теплопередачи, очень хо-
рошей теплостойкости, а также отсутствию взаимо-
действия с обычными конструкционными материала-
ми при высоких темп-рах, развиваемых в современ-
ных энергетич. ядерных реакторах.
Характерное свечение паров Н. используют в спе-
циальных светильниках. При запуске советской
космич. ракеты в сторону Луны 3 января 1959 было
выпущено облако из Н., нагретого до необходи-
мой температуры; образовавшаяся «искусственная ко-
мета» была использована для уточнения траектории
ракеты.
В металлургии Н. применяют как восстановитель,
а также для упрочнения сплавов; так, напр., сплав
на основе свинца, идущий на изготовление железно-
дорожных осевых подшипников, содержащий 0 58%
Na, 0,04% Li, 0,73% Са, имеет твердость по
371
НАТРИЙАМИН — НАТРИЙ-БУТАДИЕНОВЫЙ КАУЧУК
372
Бринеллю 34 кГ/мм*, прочность на сжатие 1800—2200
кГ/см* и может сжиматься на 28% без растрескива-
ния. Сплав 10% Н. и 90% Pb (соединение NaPb) при-
меняется в произ-ве тетраэтилсвинца — наиболее
эффективного из антидетонаторов моторных топлив.
Соединения Н. входят в состав многих лекарствен-
ных препаратов (растворимый норсульфазол, сали-
цилат натрия и др.), а также физиология, р-ра, при-
меняемого при больших кровопотерях с целью под-
нятия кровяного давления. Радиоактивный изотоп Н.
Na24 применяют для лечения нек-рых форм лейке-
мии, а также для диагностпч. целей. В виде селитры
Н. широко применяют для удобрения почвы. О при-
менении соединений Н. см. также Натрия карбонаты,
Натрия сульфаты и др.
Лит.: Ситтиг М., Натрий, его производство, свой-
ства и применение, [пер. с англ.], М., 1961; Алабышев
А. Ф., Производство натрия, М., 1943; Натрий и калий, Л.,
1959; Жидкометаллические теплоносители (Натрий и натриево-
калиевый сплав), пер. с англ., М., 1958; Андреев П. А.,
К а н а е в А. А., Федорович Е. Д., Жидкометаллические
теплоносители ядерных реакторов, Л., 1959; Жидкие метал-
лы. Сб. ст., М., 1963; G m е 1 I п, 8 Aufl., Syst.-Num. 21,
Natrium, В., 1936—1938; то же, Natrium, Anhangband, 1952,
Nachdruck, 1953; Mellor, v. 2, L.—N. Y.—Toronto, 1946;
то же, v. 2, 1952; U 1 1 m a n n, 3 Aufl., Bd 12, Milnchen —
B., 1960; Kirk, v. 1, N. Y., 1947; v. 12, N. Y., 1954.
И. А. Реформатский.
НАТРИЙАМИН NaNH2—производное аммиака, в
к-ром атом Na соединен с аминогруппой (—NH2);
обычно неправильно иаз. амидом натрия.
Бесцветные кристаллы; т. пл. 210°, начинает возго-
няться при 400°, энергично разлагается при 500—600°.
Теплота образования ДЯ298 — — 28,4 ккал/моль. Прак-
тически нерастворим в жидком NH3 и обычных органич.
растворителях. По своей структуре является солью с
анионом NH“; в расплавленном состоянии проводит
электрич. ток. Бурно разлагается водой по реакции:
NaNH2+H2O-rNaOH+NH3. На воздухе постепенно
окисляется. Окисленные препараты окрашены в жел-
тый или коричневый цвет и могут самопроизвольно
взрываться. При хранении в инертной атмосфере бе-
зопасен. Н. получают взаимодействием Na с жидким
NH3 при —30° в присутствии катализаторов, напр.
мелкодисперсного железа, или с газообразным NH3
при 300—400°. Применяют при получении цианида
натрия, синтетич. индиго, аминопиридинов, витамина
А и др.
Лит.: Ситтиг М., Натрий, его производство, свойства
и применение, [пер. с англ.], М., 1961. Д. С. Стасиневич.
НАТРИЙ-БУТАДИЕНОВЫЙ КАУЧУК (СКВ) -
продукт полимеризации бутадиена-1,3, получаемый
в гомогенной среде в присутствии металлич. натрия.
Среднечисловой (осмометрический) мол. вес колеб-
лется в пределах 85 000—200 000. При полимериза-
ции молекулы бутадиена могут соединяться по любой
двойной связи, в результате чего получаются поли-
меры с различными положениями звеньев в
макромолекуле:
12 3 4
п сн2 = сн -сн = сн2 —>
1 2 3 4 1 2 3 4
--> ...-СН2-СН = СН-СН2-СН2-СН = СН-СН2“...
(конфигурация 1,4)
ИЛИ
12 12
—► ...-сн2-сн-сн2-сн-...
I I
з GH з СН
II II
4 СН2 4 СН2
(конфигурация 1,2)
Чаще всего образуется смешанная структура,
в к-рой половина двойных связей находится
в боковых цепях. Повышенпе темп-ры спо-
собствует увеличению содержания конфигура-
ции 1,4.
Н.-б. к.— эластичный продукт желтоватого цвета,
плотность 0,90—0,92; удельное объемное электри-
ческое сопротивление каучука средней пластичности
1014—1015 олсси; т. стекл.—48°. Н.-б. к. хорошо
растворяется в бензине, бензоле, хлороформе, серо-
углероде и др. органич. соединениях. Вследствие
наличия большого числа двойных связей в боко-
вых цепях Н.-б. к. при нагревании и при действии
света (особенно УФ-лучей) структурируется; нату-
ральный каучук (НК) в этих условиях деструкти-
руется.
В зависимости от способа полимеризации Н.-б.к.
делится на бесстержневой и стержневой. По стержне-
вому методу бутадиен полимеризуют под давлением
10 атм в автоклаве из обычной углеродистой стали.
Металлич. Na наносят тонким слоем па стальные стер-
жни, к-рые равномерно распределяют по всему объему
реактора. Незаполимеризовавшпеся продукты уда-
ляют из каучука под вакуумом.
Товарный Н.-б. к. содержит ряд примесей, введен-
ных в него в процессе получения. Щелочность его (в
пересчете на ЙагСО3) 0,6—1,2% для стержневого
каучука и 0,1—0,6% для бесстержневого, зольность
2,5—4,0%, содержание неозона Д (антиоксидант)
0,5—1,0%, стеарина 0,5—1,0%, газообразных угле-
водородов 0,05—0,10%, влаги <1%.
Помимо Н.-б. к. общего назначения, вырабаты-
ваются также специальные марки: полпдпеновый (П),
диэлектрический (Д), балонный (б) и пищевой (щ).
Полидиеновый каучук содержит низкомолекулярные
жидкие полимеры гомологов бутадиена (пиперилена,
гексадиена и др.): стержневой 6,5%, бесстержневой
17%. Диэлектрич. Н.-б. к. отличается пониженным
содержанием щелочи (не более 0,2%), что обеспечи-
вает получение резин с хорошим электросопротивле-
нием. В пищевой Н.-б. к., предназначенный для
изготовления резин, соприкасающихся с пищевыми
продуктами, вместо неозона Д вводят вазелиновое
масло. Н.-б. к. обычно вулканизуют с помощью серы;
иногда применяют хлористую серу, диазоаминосое-
динения, галогенохиноны, полинптросоединения и др.
Можно проводить также термовулканизацию Н.-б. к.
Для получения резин с хорошей прочностью в резино-
вые смеси вводят активные наполнители — сажу, ак-
тивную окись кремния (белая сажа), активную окись
алюминия, окись магния, каолин и др. При много-
кратных деформациях вулканизаты Н.-б. к. харак-
теризуются повышенным теплообразованием, что объ-
ясняется разветвленностью и недостаточной регуляр-
ностью их структуры.
По той же технологии, что и для СКВ, но под дей-
ствием других инициаторов, вырабатывают и другие
бутадиеновые каучуки — СКВ и СКБМ. Основные
свойства вулканизатов СКВ, СКВ, СКБМ и НК пред-
ставлены в таблице.
Показатели Сажевые вулканизаты (60 вес. ч. сажи на 100 вес. ч. каучука) а
СКВ СКВ СКБМ н. к.
Предел прочности при раз- рыве, -кг/см* при 20° при 100° Относит, удлинение, % . Остаточное удлинение, % Истирание, см^кв-т-ч . . Эластичность по упругому отскоку, % Коэфф, морозостойкости (100%-ное растяжение, температура —3 5°) . . 125-155 (18-22) 84—104 500—650 50—75 210—260 26—28 0,2—0,3 140—180 (20-25) 52-67 500—650 35-55 210—260 28—32 0,35—0,40 140 — 170 (30) 59—72 500 — 650 28—40 210 — 260 35—40 0,50—0,61 240—310 (230—300) 170—220 600 — 700 25-40 240—280 50-55 0,85
а В скобках приводятся данные для ненадолненных вулканизатов
373
НАТРИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ —НАТРИЯ АРСЕНИТ
374
Н.-б. к. относится к числу каучуков общего назна-
чения. На его основе можно изготовлять широкий ас-
сортимент резиновых изделий (автомобильные шины,
транспортные ленты, рукава, обувь и др.), к к-рым
не предъявляется таких, напр., требований, как масло-
п бензостойкость, газонепроницаемость и др.
Лит.: Смирнов Н. И., Синтетические каучуки, 2 изд.,
Л., 1954. А. К. Никитин.
НАТРИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ —
органич. соединения, содержащие связь углерод —
натрий. Название Н. с. строится из названия органич.
радикала и слова «натрий» — этилнатрий, бензилнат-
рий и 'Т. д. Н. с.— твердые ассоциированные веще-
ства, содержащие сильно поляризованные связи
Na+ — С-; нерастворимы в инертных растворителях,
при нагревании разлагаются не плавясь, на воздухе
самовоспламеняются.
Своеобразным типом Н. с. являются ионнопостроен-
ные продукты присоединения натрия к ароматич.
углеводородам, образующиеся в среде нек-рых прос-
тых эфиров (лучшая среда — дпметоксиэтан). Сильно
окрашенные р-ры этих соединений электропроводны
и парамагнитны; в них присутствуют сольватирован-
ные растворителем катионы металла и анион-радпка-
лы углеводорода, напр. С10Н~.
Олефины, по меньшей мере дважды арилированные
при двойной связи, присоединяют натрий по одному
из двух типов:
1) (С,Н3)2С = С (G»H5)2 + 2Na —> (C.IEhCCfaHs)’-Na* +
(«нормальное» присоединение)
2) (C„HS)2C = CH2 +Na —► [(C«II5)2GGH2]Na + ,
2[(C.eH5)2GGH2] Na + —► 1(G6H3)2GGH2GH2C (C»H5)2] Na’ +
(«димеризующее» присоединение, если один из ато-
мов двойной связи неарилирован).
По химич. свойствам Н. с. весьма сходны с литий-
органическими соединениями, однако значительно
превосходят их по активности. Все работы с Н. с.
следует проводить в атмосфере инертного газа (азот,
аргон). Соединениями, содержащими подвижный атом
водорода, Н. с. немедленно разлагаются; двуокисью
углерода — карбоксилируются до солей соответст-
вующих карбоновых к-т. Вследствие исключительной
реакционной способности Н. с. карбоксилирование
часто осложняется побочными процессами, напр.:
СО2 GBH3GH2Na
GeH5CH2Na —>- G»Il3GH2COONa —-----»
СО..
—> G»HsGH (Na) GOONa —> C6H5CH (GOONa)2
Важнейшими способами получения Н. с. являются:
а) Нагревание натрия с ртутьорганич. соединениями
(обычно в среде петролейного эфира); это практически
единственный способ синтеза индивидуальных Н. с.:
R2Hg+2Na—>Hg+2RNa. б) В ряде случаев удается
получать Н. с. из галогенарилов (алкилов) п Na:
RX+2Na—>NaX + RNa (X, как правило, Cl); синтез,
особенно в жирном ряду, осложняется реакцией Вюр-
ца. в) Исключительное значение для синтеза Н. с.
имеет металлирование, приводящее к замене радикала
R (по Шорыгину): RH + R'Na—> R'H + RNa. Исходя
из относительно доступных Н. с., напр. C2H5Na,
C4H,Na, этим путем могут быть получены другие Н. с.,
если водород в соединении RH более подвижен, чем
в R'H. Характерен в этом отношении синтез фенил-
натрия из дибутилртути и натрия в бензоле:
(CtH9)2Hg + 2Na—*Hg + 2C1IlaNa;
C,H,Na + С„Н„—> CsHsNa + G(HI0
и перегруппировка толилнатрия в бензилнатрий:
CHjCoHiNa —> CJi5CH3Na
Последний образуется также и при нагревании фенил-
натрия в толуоле. Одним из способов получения Н. с.
является расщепление простых эфиров (гл. обр. аро-
матич. и жирноароматич.) натрием (Шорыгин), напр.:
С„Н5ОС2Н3 + Na —> C2H5ONa + C»H>ONa + G«H3Na + C2H3Na
Расщепление проводят нагреванием эфиров с натрием
или действием Na в жидком NH3. д) В ряде случаев,
когда в радикале R имеются особо благоприятные
условия для рассредоточения избыточной электрон-
ной плотности, кроме методов «б» и «в», пригодно так-
же непосредственное замещение водорода натрием
(обычно в жидком аммиаке), напр.: (СвН5)3СН +
+ Na(NaNH2) —'(CeH5)3CNa+I/2H2(NH3). Получаемые
этим методом Н. с., в отличие от обычных нат-
рийалкилов (-арилов), имеют глубокую окраску,
растворимы в эфире, более стабильны и менее реакци-
онноспособны. Типичным соединением этого типа
является бензилнатрий. К этой же группе реакций
примыкают также образование циклопеитадиенилнат-
рия, в котором устойчивый анион циклопеитадиени-
лия обладает ароматическими свойствами: CeH5+Na—►
—C5H5Na+I/2H2, и образование ацетиленидов.
Реакции Н. с. с карбонильными соединениями,
эфирами, неорганич. соединениями и т. п. аналогичны
соответствующим реакциям литийорганич. соедине-
ний, но проходят более энергично. Н. с. применяют
в качестве алкилирующих агентов (обычно в тех слу-
чаях, когда реакционная способность реактивов Гринь-
яра и литийалкилов оказывается недостаточной).
Лит.: Кочешков К. А. и Талалаева Т. В., Син-
тетические методы в области металлоорганических соединений
лития, натрия, калия, рубидия и цезия, М.—Л., 1949 (Синте-
тич. методы в области металлоорган. соединений, вып. 1.
ч. 1—5); С о a t е s G. Е., Organo-metallic compounds, 2 ed.,
L.—N. Y., 1960; Рохов Ю., Херд Д., Льюис Р., Хи-
мия металлоорганических соединений, пер. с англ., М., 1963.
О. Ю. Охлобыстин.
НАТРИЯ АМИД — см. Натрийамин.
НАТРИЯ АРСЕНАТЫ — натриевые соли мышья-
ковой к-ты H3AsO4- Средний Н.а. — три натри й-
арсенат, кристаллизуется из водных р-ров, со-
держащих 10,4—20% As2Os и 7,6—12,5% Na2O в
виде Na3AsO4-12H2O — бесцветных кристаллов,
плотн. 1,76; в 100 г воды растворяется 26 г водной
соли (20°). При 85,5° плавится в кристаллизационной
воде. При более высокой концентрации щелочи кри-
сталлизуются гидраты с 8, 4 и 3 молекулами Н2О.
Водные р-ры тринатрийарсената пмеют щелочную
реакцию и поглощают СО2 из воздуха.
Двухзамещенный кислый Н. а.— д и н а т р и й-
арсенат, кристаллизуется из растворов, содер-
жащих 20—41% As2Os и 12—15% Na2O в виде
Na2HAsO4-7H2O. В воде растворяется 24,8 вес. %
безводной соли (20°).
Однозамещенный кислый Н. а.— мононатрий-
ар с е н а т, кристаллизуется из растворов, содер-
жащих 41—61% As2Os и 8—14,5%ХагО в виде
NaH2AsO4-7H2O. В 100 г воды растворяется 200 г
водной соли (20°). При нагревании обезвоживается,
превращаясь при 230° в метаарсенат натрия (NaAsO3)x
с т. пл. 615°. Все арсенаты натрия сильно ядовиты.
Их получают растворением As2Os в щелочи или оки-
слением NaAsO2 воздухом. Применяют для борьбы
с с.-х. вредителями, а также для антпсептпрования
древесины.
Лит.: По зин М. Г., Технология минеральных солей,
2 изд., Л., 1961. Д. С. Стасиневич.
НАТРИЯ АРСЕНИТ NaAsO2 — натриевая соль
метамышьяковистой к-ты; белый порошок; хорошо
растворим в воде (156 г Н. а. в 100 г Н2О при 25°);
в кристаллич. виде не выделен. Получают в виде
влажной пасты при упаривании р-ров As2O3 в водных
р-рах соды. Применяют для тех же целей, что и
натрия арсенаты.
375
НАТРИЯ БОРГИДРИД —НАТРИЯ ГИДРООКИСЬ
376
НАТРИЯ БОРГИДРИД NaBH4 — бесцветные кри-
сталлы кубич. системы, а =6,14 А; плотн. 1,074.
Теплота образования ЛЯ298= — 43,82 ккал/моль.
При нагревании в вакууме стоек до500°, выше раз-
лагается без плавления с образованием В, Na и Н2.
Гигроскопичен; растворимость в воде 55 г на 100 г
Н2О (20°). Образует гидрат NaBH4-2H2O, устойчи-
вый ниже 36,5°. Водные р-ры Н. б. устойчивы до 50°.
Выше начинается разложение с выделением Н2.
Разложение сильно ускоряется в присутствии кислот
и катализаторов (Ni, Со, Pt). Щелочные р-ры Н. б.
более устойчивы. Н. б. растворим в жидком аммиаке
(104 г на 100 г NH3 при 25°), в диметиловых эфирах
этиленгликоля, ди-, три- и тетраэтиленгликолей, в
метиловом и этиловом спиртах (с частичным разло-
жением), в алифатич. аминах, диметилформамиде и др.
На сухом воздухе Н. б. стоек до 300°, выше окис-
ляется; при поджигании спокойно сгорает. Н. б.—
сильный восстановитель, с солями Ni, Со, Fe, Mn, Pt
образует бориды этих металлов, с галогенидами
В, Ge, Sn, As, Sb — гидриды этих элементов. Служит
селективным восстановителем для ряда функциональ-
ных групп в органич. соединениях. При попадании
на кожу вызывает раздражение.
Получают Н. б. взаимодействием NaH с борпо-
метиловым эфиром при 250—270° Или же при действии
Н2 и металлич. Na на сплав буры с SiO2. В обоих
случаях Н. б. извлекают из реакционной массы жид-
ким NH3. Применяют в качестве восстановителя в
химико-фармацевтич. пром-сти, как газообразователь
в произ-ве пенопластов, для получения водорода в
полевых условиях.
Лит..- Бор, его соединения и сплавы, под общ. ред. Г. В.
Самсонова, Киев, 1960; Гейлорд Н., Восстановление ком-
плексными металлическими гидридами металлов, пер. с англ.,
М., 1959. Д. С. Стасиневич.
НАТРИЯ БРОМИД (бромистый натрий) NaBr —бес-
цветные кристаллы с кубич. решеткой, а =5,97299А;
плотн. 3,20; т. пл. 760°, т. кип. 1390°. Теплота обра-
зования АН298=—86,030 ккал/моль. Р астворимость
в воде (%):44,5 (0°); 48,6 (25°); 54,8 (100°). Криоги-
дратная точка— 28° (40,3% NaBr). Насыщенный р-р
(55,1%) кипит под давлением 760 мм рт. ст. при
121°. Плотность водных р-ров:
% NaBr ... 10 20 30 40
dia......... 1,0803 1,1745 1,2841 1,4138
Образует гидраты: NaBr-5H2O, кристаллизующий-
ся из водных растворов от —28° до —23,5° и
NaBr-2H2O, устойчивый от —23,5° до 51°. Н. б.
растворим в жидком аммиаке, метиловом спирте,
этиловом спирте и ацетоне. Бром в NaBr и NaBr-2H2O
изоморфно замещается хлором. Н. б. получают дей-
ствием брома на р-ры NaOH в присутствии восстано-
вителей (муравьиная к-та, аммиак) либо обменной
реакцией между FeBr2 и Na2CO3 или СаВг2 и Na2SO4.
Применяют в медицине и фотографии.
Лит.: Ксензенко В. И., Стасиневич Д. С.,
Технология брома и иода, М., 1960; ПозинМ. Е., Техноло-
гия минеральных солей, 2 изд., Л., 1961. Д. С. Стасиневич.
НАТРИЯ ГИДРИД NaH — бесцветные кристаллы;
структура аналогична NaCl (кубич. гранецентриро-
вапная), постоянная решетки а=4,88 А, плотн. 1,364.
Теплота образования АН298=—13,60 ккал/моль.
Водород в Н. г. содержится в виде аниона Н-. При
нагревании Н. г. разлагается па металлич. Na и
Н2 без плавления; давление диссоциации достигает
1 атм при 430°. Н. г. нерастворим в обычных инерт-
ных растворителях, растворяется в расплавленном
NaOH и в сплаве К — Na. Водой Н. г. энергично раз-
лагается с выделением водорода. Жадно притя-
гивает влагу и во влажном воздухе может воспламе-
ниться. На сухом воздухе стоек до 230°; при поджига-
нии сгорает. Сильный восстановитель, восстанавли-
вает металлы из окислов, хлоридов и др. Н. г. полу-
чают в виде порошка или суспензии в минеральном
масле взаимодействием металлич. Na с Н2 при 250—
400° и атмосферном пли повышенном давлении. Хра-
нят его в герметичной таре, а при работе с ним приме-
няют защитные маски и плотные перчатки. С больши-
ми количествами Н. г. рекомендуется работать в
азотных камерах. Для тушения горящего Н. г.
применяют сухие вещества: песок, соду, хлористый
натрий. Масляные суспензии Н. г. безопасны в обра-
щении. Н. г. применяют для получения натрия
боргидрида, для восстановления титана из его хлори-
да, для снятия окалины с металлов, а также как
конденсирующее средство в органич. синтезах (см.
также Гидриды).
Лит.: Ситтиг М., Натрий, его производство, свойства
и применение, [пер. с англ.], М., 1961; Херд Д., Введение
в химию гидридов, пер. с англ., М., 1956. Д. С. Стасиневич.
НАТРИЯ ГИДРООКИСЬ (едкий натр) NaOH —
бесцветные кристаллы; при обычной темп-ре устой-
чива ромбич. решетка (а-NaOH), а = 3,397 А,
6=11,32 А, с =3,397 А; выше 299,6° превращается
в fJ-NaOH. Плотн. 2,13; т. пл. 320°; т. кип. 1378°.
Теплота образования А//298= —101,99 ккал/моль.
Давление пара (мм рт. ст-): 0,1 (618°); 1 (738°);
10 (896°); 100 (1111°). Теплоемкость 19,2 кал/моль-
град. Технич. продукт— белая твердая непрозрачная
масса с лучистым изломом. Н. г. гигроскопична;
при ее соприкосновении с водой выделяется большое
количество тепла. Водные р-ры имеют сильно щелоч-
ную реакцию. Растворимость в воде (%): 29, 6(0°);
52,2 (20°); 59,2 (50°); 75,8 (80°); 83,9 (192°). Крио-
гидратная точка —28° (19% NaOH). Плотность вод-
ных р-ров:
% NaOH .. 1 0 20 30 40 50
dm..........1,100 1,219 1,328 1,430 1,525
Давление пара над водными р-рами NaOH (в .ч.ч
рт. ст. при 20°); 2 (50%-ный р-р), 8,8 (30%-ный).
Темп-ра кипения водных р-ров (°C): 107,7 (20%-ный
р-р); 128,0 (40%); 144,0 (50%). Известны гидраты
Н. г., кристаллизующиеся из водных растворов в
след, температурных интервалах: NaOH-7H2O (от
—28° до —24°); NaOH-5H2O (от—24° до —17,7°);
a-NaOH-4H2O (от -17,7° до +5,4°); Na(OH)-3,5Н2О
(5,4°—12,3°); NaOH-H2O (12,3°—61,8°). Выше 61,8°
кристаллизуется безводный NaOH. Известен мета-
стабильный [J-NaOH-4H2O. Гидрат NaOH-3,5H2O
конгруэнтно плавится при 15,5°, a NaOH-H2O при
64,3°. Н. г. поглощает СО2 из воздуха, превращаясь
в натрия карбонат-
В присутствии больших количеств NaOH раство-
римость солей резко снижается: так, растворимость
NaCl (см. Натрия хлорид) в 50%-ном р-ре NaOH
составляет 0,91%. Н. г. вытесняет также многие ор-
ганич. соединения из их водных р-ров,что используют
для высушивания органич. жидкостей. Н. г. раство-
рима в метиловом (23,6% NaOH) и этиловом (14,7%
NaOH) спиртах. Практически нерастворима в жидком
аммиаке и большинстве органич. растворителей. Рас-
плавленная Н. г. растворяет металлич. натрий и гид-
рид натрия. Н. г. разрушает кожу, бумагу и другие
материалы органич. происхождения. На кожу и сли-
зистые оболочки действует прижигающе. Особенно
опасно попадание даже малейших ее количеств в
। глаза. Все работы с Н. г. должны производиться в
защитных очках и резиновых перчатках.
Получают Н. г. электрохпмич. или химич. спосо-
бами. Электрохимии, получение основано на элек-
тролизе р-ров NaCl; одновременно получается хлор.
В пром-сти для получения щелочи и хлора применяют
два типа ванн: с твердыми электродами и проточным
377
НАТРИЯ ГИДРОСУЛЬФИТ—НАТРИЯ КАРБОНАТЫ
378'
электролитом, движущимся от анода к катоду, и
со ртутным катодом (см. Хлор). Ванны с проточным
электролитом конструировались различной формы с
горизонтальным и вертикальным расположением
электродов, с фильтрующей диафрагмой и без нее.
Электролиз ведут при 60—90°. Ванны питают почти
насыщенным р-ром NaCI (310—320 г/л) и получают
щелока, содержащие 100—140 г/л NaOH и 150—180
г/л NaCI. Выход по току порядка 90%. Щелока
упаривают до концентрации 40—50% NaOH, при
этом NaCI кристаллизуется. Его отделяют и полу-
ченный крепкий щелок упаривают в плавильных
котлах при темп-рах до 440° и остаточном давлении
350 мм рт. ст- и получают твердый продукт, содер-
жащий до 1—2% NaCI и 96—98% NaOH. Ванны
со ртутным катодом обычно имеют горизонтальное
расположение электродов. Ртуть и рассол протекают
через электролизер в одном направлении. Электролиз
ведут при темп-рах до 70°. Образующуюся на катоде
амальгаму натрия, содержащую 0,2—0,3% Na, обра-
батывают водой при 70—110° и получают щелока,
содержащие 50—70% NaOH, к-рые в случае надоб-
ности упаривают до твердого продукта. Щелочь,
получаемая по этому способу, содержит минимальное
количество NaCI.
Химич, способ получения Н. г. основан на взаимо-
действии горячего р-ра карбоната натрия с известью,
в результате к-рого образуются 10—12%-ные р-ры
NaOH. Ранее применялся также способ, основанный
на спекании Na2CO3 с Fe2O3 п гидролизе получен-
ного феррита натрия Na2Fe20a.
Н. г. является одним из основных продуктов химич.
пром-сти и широко применяется как в самой химич.
пром-сти, так и во мн. др. производствах.
Лит.: Генин Л. С., Ковалев Н. М., Современное
производство хлора и каустической соды,М., I960; Генин
Л. С., Электролиз растворов поваренной соли, М., 1960; Бил-
литер Ж., Промышленный электролиз водных растворов,
пер. с нем., М., 1959. Д. С. Стасиневич.
НАТРИЯ ГИДРОСУЛЬФИТ (гидросернистокис-
лый натрий) Na2S2Oa-2H2O — блестящие бесцветные
призматич. кристаллы; в безводном состоянии белый
кристаллич. порошок, устойчивый на воздухе в те-
чение нескольких дней, во влажном состоянии быстро
окисляется. Н. г. легко растворим в воде (25,4 г
в 100 г Н2О при 20°), нерастворим в спирте. Водный
р-р сильно поглощает кислород. Н. г. может быть
получен по следующим реакциям:
Zn + 2SO2 = ZnS,O,
ZnS2O( + Na2CO3=Na2S2O< + ZnCO3
При получении Н. г. принимают меры, предупреждаю-
щие окисление препарата. Используют Н. г. в ана-
лизе как восстановитель; в газовом анализе для пог-
лощения кислорода; при фотометрия, определении Se
и др.; применяют также в текстильной, пищевой
пром-сти.
Лит. см. при ст. Натрия гипофосфит. И. П. Ефимов.
НАТРИЯ ГИПОФОСФИТ (фосфорноватистокис-
лый натрий) NaH2PO2-H2O — бесцветные, очень ги-
гроскопичные кристаллы; при нагревании выше 200°
разлагается; хорошо растворим в воде (100 г в 100 г
прп 25°), растворим в спирте, в сухом состоянии устой-
чив при хранении. Н. г. является сильным вос-
становителем; восстанавливает соли Au, Ag, Pt, Hg, As;
с сильными окислителями бурно реагирует. Н. г. мо-
жет быть получен по реакциям:
ЗСа (ОН)3 + 8Р + 6Н2О = ЗСа (Н2РО,)2 + 2РН3
Са (Н2РО2)2 + Na2CO3=2NaH2PO2 + СаСО3
Н. г. в химич. анализе используют в качестве вос-
становителя; применяют для определения мышьяка;
прп фотометрия, определении сурьмы и др.
Лит.: Карякин Ю. В., Ангелов И. И., Чистые
химические реактивы, [3 изд.], М., 1955, с. 410.
И. П. Ефимов.
НАТРИЯ ИОДИД (иодистый натрий) NaJ — бес-
цветные кристаллы с кубич. решеткой, а =6,475 А;
плотн. 3,665; т. пл. 662°; т. кип. 1304°. Теплота обра-
зования АН298 = —68,84 ккал/моль. При хранении
на свету желтеет вследствие окисления кислородом
воздуха с выделением свободного иода. Гигроскопи-
чен; растворимость в воде (%): 61,4 (0°); 64,1 (20°);
75,1 (100°). Насыщенный водный р-р кипит при
141° (ок. 76,3%). Плотность водных р-ров:
% NaJ . . 10 20 28 40 50 60
d20 .... 1,0808 1,1769 1,2663 1,4271 1,5942 1,8038
Из водных р-ров кристаллизуются гидраты:
NaJ-5Н2О, устойчивый от—31,5° (криогидратная точ-
ка, ок. 39,5 вес. % NaJ) до —13,5°, и NaJ-2Н2О,
устойчивый от —13,5° до 65,5°. Растворим в метило-
вом п этиловом спиртах и ацетоне. Н. и. получают
по обменной реакции между Fe3J8 и Na2CO3. Приме-
няют в медицине.
Лит.: Денисович Б. П„ Иод и его производство, М.,
1938; см. также при ст. Натрия бромид. Д. С. Стасиневич.
НАТРИЯ КАРБОНАТЫ — натриевые соли уголь-
ной кислоты. Натрия карбонат нормаль-
ный (углекислый натрий, кальцинирован-
ная сода) Na2CO3 — бесцветные кристаллы,
плотность 2,5326; т. пл. 853°. Теплота образова-
ния Д#298 = —2 70,3 ккал/моль. Теплоемкость 26,41
кал/моль-град. Давление диссоциации (в мм рт. ст.)'.
1 (700°), 41 (1200°). Na2CO3 гигроскопичен; растворяет-
ся в воде с сильным разогреванием. Теплота раство-
рения 1 моля Na2CO3 в 1000 молях Н2О равна 5,5 ккал.
Растворимость в воде (%): 6,54 (0°); 17,69 (26°);
32,8 (34,8°); 31,29 (100°). Плотность водных р-ров:
% Na2CO3 . . 5 10 13
d20 ........ 1 , 0502 1 . 1 029 1,1354
Криогидратная точка — 2,05° (5,7% Na2CO3). На-
сыщенный р-р кипит при 105,0° (31,13% Na2CO3].
Из водных р-ров ниже 32° кристаллизуется Na2CO3-
ЮН2О в виде больших прозрачных моноклинных
кристаллов, плотн. 1,45; между 32 и 34,8° кристалли-
зуется ромбич. Na2CO3-7H2O, а выше 34,8° — ромбич.
Na2CO3-H2O. Безводный Na2CO3 кристаллизуется из
водных р-ров выше 112,5°. Растворы Н. к. в воде име-
ют сильнощелочную реакцию, окрашивают фенол-
фталеин в ярко-красный цвет. В большинстве органич.
растворителей Н. к. нерастворим. При хранении на
воздухе Н. к. поглощает СО2 и слеживается.
Произ-во и применение карбоната натрпя см. в ст.
Сода.
Натрия бикарбонат (натрий двуугле-
кислый, питьевая сода) NaHCO3 — моно-
клинные бесцветные кристаллы; плотн. 2,16—2,22;
при нагревании до 50° начинает отщеплять СО2,
а при 100—150° полностью разлагается, превращаясь
в карбонат. Теплота образования бикарбоната
Д#°98 = —226,5 ккал/моль. Растворимость в воде
(%) 6,5 (0°); 9,4 (25°); 13,8 (60°). Водные р-ры имеют
слабощелочную реакцию и окрашивают фенолфта-
леин в бледно-розовый цвет. Выше 20° из р-ров бикар-
боната постепенно выделяется СО2 ; парциальное
давление СО2 над насыщенными водными р-рами
NaHCO3 (мм рт. ст.); 120 (15°); 563 (50°).
Бикарбонат натрия получают насыщением СО2
р-ров карбоната натрия. «Сырой» бикарбонат является
промежуточным продуктом при получении соды; его
растворяют в горячей воде, отгоняют примесь NH3
и осаждают бикарбонат при повторной карбониза-
ции. Бикарбонат применяют в хлебопечении, в пище-
вой пром-сти, в медицине, а также при изготовлении
зарядов для огнетушителей.
379
НАТРИЯ КОБАЛЬТИНИТРИТ—НАТРИЯ ПЕРЕКИСЬ
380
Натрия сесквикарбонат (трона)
КаНСОз-НагСОз^НгО — бесцветные моноклинные
кристаллы; встречается в донных отложениях со-
довых озер. Д- С. Стасиневич.
НАТРИЯ КОБАЛЬТИНИТРИТ Ка3[Со(МО2)в]
•0,5Н2О, мол. в. 412,99 — желтый кристаллич. по-
рошок, устойчивый на воздухе; кристаллизационную
воду нагреванием удалить нельзя, т. к. препарат при
этом разлагается; легко растворим в воде, мало рас-
творим в спирте и эфире; водные р-ры неустойчивы.
Получают по реакции:
СоС12 + 7NaNO,+ 2GH3GOOH =
=Na3 [Go (NO2)«] + 2NaCl + 2GH3GOONa + NO + H2O
Применяют Н. к. для осаждения К, Rb, Cs; для весо-
вого, объемного и фотометрия, определения калия;
для идентификации алкалоидов. И- п. Ефимов.
НАТРИЯ НИТРАТ (азотнокислый натрий, н а-
триевая селитра) NaNO. — бесцветные кри-
сталлы гексагональной системы; а =5,082 А, с =
= 17,602 А; плотн. 2,257; т. пл. 308°. Выше темп-ры
плавления разлагается на NaNO, и Ог. При более
высоких темп-рах продуктами разложения явля-
ются Na2O2 и Na2O. Теплота образования ДН298=
=—111,54 ккал/моль', п™ =1,5874; гигроскопичен.
Растворимость в воде (%): 42,2(0°); 47,6(25°);
64,3 (100°). Криогидратная точка — 18,1°. Насыщен-
ный р-р кипит при 120° (222,0 г NaNO3 на 100 г Н2О).
Плотность водных р-ров:
% NaNO3 . . 10 20 30 40
d2°........ 1,0674 1,1426 1,2256 1,31 75
Н. н. хорошо растворим в жидком аммиаке. Обра-
зует легкоплавкие эвтектич. смеси со многими соля-
ми. Является сильным окислителем.
Н. н. встречается в природе (чилийская селитра).
В пром-сти его получают путем абсорбции окислов
азота р-рамп щелочи с последующим окислением
образовавшегося одновременно нитрита натрия азот-
ной к-той. Другой способ получения Н. н. основан
на обменной реакции между нитратами кальция и ам-
мония и натриевыми солями (напр., NaCl, Na2SO«).
Применяют Н. н. в качестве удобрения, а также в
пищевой пром-сти для консервирования, в металло-
обрабатывающей как компонент солевых закалоч-
ных ванн и в стекольной пром-сти как окислитель.
Лит.: П о з и н М. Е., Технология минеральных солей,
2 изд., Л., 1961; МиниовичМ. А., Соли азотной кислсты.
(Нитраты). М.—Л., 1946. Д. С. Стасиневич.
НАТРИЯ НИТРИТ (азотистокислый натрий)
NaNO, —бесцветные или слегка желтоватые кристал-
лы ромбич. системы, а=5,38 А, Ъ =5,56 А, с =3,55 А,
плотн. 2,17; т. пл. 271°. При нагревании выше 320°
разлагается. Теплота образования ДН298=—85,9
ккал/моль. Растворимость в воде (%): 41,63 (0°);
45,07 (22°); 61,5 (99,9°); 68,7 (128?). Насыщенный р-р
кппит прп 128°. Криогидратная точка лежит при
—26° и 38,0 вес. % NaNCb. Плотность водных р-ров:
% NaNOa . . 10 20 28 40
d2"........ 1,065 1,137 1.198 1.299
Водные р-ры имеют щелочную реакцию (рН~9).
Н. н.— окислитель умеренной силы. Кислотами раз-
лагается с выделением окислов азота. Ядовит. Н. н.
получают при поглощении окислов азота щелочными
р-рами с последующим упариванием р-ра. При охла-
ждении Н. н. кристаллизуется, а одновременно
образующийся NaNO3 остается в маточнике. При-
меняют в пропз-ве красителей, иода, а также в пище-
вой пром-сти и в медицине.
Лит. см. при ст. Натрия нитрат. Д. С> Стасиневич.
НАТРИЯ НИТРОПРУССИД (нитрозопентацино-
ферриат натрия) Na2[Fe(CN)5NO]-ЗНгО, мол. вес
297,97 — рубиново-красные, прозрачные, ромбич. фор-
мы кристаллы, плотн. 1,71; не теряет кристалли-
зационной воды даже при 100°; при 400° разлагается
с выделением NO и (CN)2; растворим в воде: 40 г
в 100 г воды (16°); нерастворим в абс. спирте; водный
р-р довольно быстро разлагается даже на рассеян-
ном свете. В щелочном р-ре Н. н. сильный восстано-
витель; ядовит. Н. н. можно получить по след, реак-
циям: 2К) [Fe (CN)e] + 4NaNO3 + 4H2so4 =
=2KJ[Fe(CN)5NO] + 2NO + (CN)2 + 2Na3SO(+2KISO4+4H2O
К2 [Fe (GN)-,NO] + GuSO, = Gu [Fe (GN)-, NO] + K2SO4
Gu [Fe(GN)3NO] + 2NaHGO3=Na2 [Fe(GN)5NO] + GuGO.+
+ CO2 + H2O
Получение препарата необходимо
проводить под сильной тягой
вследствие чрезвычайной ядови-
тости выделяющихся при реакции
газообразных продуктов (циан и
окислы азота). Прп взаимодействии II. и.
с сульфид-ионами в щелочном р-ре образуется красно-
фиолетовое окрашивание. Применяют Н. н. для
обнаружения и фотометрия, определения H2S и ще-
лочных сульфидов, SO2 и сульфитов, карбонильных
соединений — альдегидов, кетонов, кетокислот и др.,
в качестве индикатора при меркуриметрич. титро-
вании галогенов и цианидов. И- П. Ефимов.
НАТРИЯ ПЕРЕКИСЬ Na20z — одно из перекис-
ных соединений натрия, характеризующееся наличием
молекулярного иона О2~; содержание активного кисло-
рода составляет 20,5 вес. %. Чистая Н. п.— белый
порошок; технич. продукт имеет слабожелтую ок-
раску, обусловленную примесью надперекиси нат-
рия NaOa. Решетка NaaQs гексагональная (искажен-
ная); а =6,207 ±0,004 А, с =4,471 ±0,003 А; плотн.
2,60. Существует в трех модификациях: Q-Na2O2,
устойчива при темп-ре жидкого воздуха, Na3O2 (I),
устойчива до 512 ±1° и NasCb (II), устойчива выше
этой темп-ры. Стандартная теплота образования
ДН298 =—122,1 ±1,2 ккал/моль. Молярная теплоем-
кость С° =21,35 кал/моль-град. Н. п. диамагнитна,
молярная магнитная восприимчивость Хи =—24-10-6.
При нагревании Н. п. при 311—400° наблюдается
нек-рая потеря активного кислорода, бурное разло-
жение начинается при 540°. Плавится Н. п. выше
596° и полностью отдает свой активный кислород
при 675°. Растворяется в воде с выделением
34+0,3 ккал/моль. При этом образуются NaOH, Н2О2
и выделяется нек-рое количество кислорода, т. к. ще-
лочная среда и повышенная темп-ра способствуют
разложению Н2О2 (см. Водорода перекись). С разб.
к-тамп Н. п. реагирует с образованием соответствую-
щих солей и перекиси водорода. Определение актив-
ного кислорода ИагОг проводится обычно перманга-
натометрически в кислой среде и на холоду. Известны
молекулярные соединения Н. п. с водой (октагидрат
Na2O2'8H2O), с перекисью водорода (дипероксигид-
рат Na2O2-2H2O2) и е водой и перекисью водорода
(тетрагидрат дипероксигидрата Na2O2-2H2O2-4H2O).
С влагой и углекислым газом воздуха Н. п. реагирует
с образованием NaOH, На2СОз и с выделением кисло-
рода. На этом основано ее применение для регенера-
ции воздуха в закрытых помещениях.
Н. п. получают окислением расплавленного на
противнях металлич. натрия в противотоке очищенного
от СОг и высушенного воздуха или в форсуночных
аппаратах. Для получения высококачественной Н. п.
рекомендуется восстанавливать Н. п., полученную
окислением металла, до окиси путем нагревания при
381
НАТРИЯ ПИРОСУЛЬФАТ—НАТРИЯ СУЛЬФАТЫ
382
130—200° с небольшими порциями металлич. натрия
в инертной атмосфере, увлажненной парами воды,
а полученную таким образом окись окислять до пере-
киси во вращающихся печах при 250—400°. Получен-
ный продукт содержит 96—98% Na2O2. Поскольку
Н. п. весьма агрессивна по отношению к металлам,
при ее получении пользуются обычно реакторами из
никелевых сплавов, покрытых графитом, и мешалка-
ми из циркония. За рубежом Na2O2 получают также
окислением смеси гидразобензола и алкоголята на-
трия кислородом. Н. п. отделяют фильтрованием, а
регенерирующиеся азобензол и спирт возвращают
в цикл.
Н. п. производят в значительных количествах.
Применяют в основном для отбеливания хлопчатобу-
мажных, льняных и шерстяных тканей, джутовых
материалов. Широко используют для отбелки древес-
ной массы — механич. пульпы (молотой древесины),
сульфатной и сульфитной пульпы, пульпы из старой
бумаги и полухимич. пульпы, а также вискозной мас-
сы, соломы и прочих материалов. В герметически зак-
рытой таре Н. п. не подвержена разложению даже
при продолжительном хранении. Сосуды с Н. п.
следует хранить в прохладном месте, вдалеке от вос-
пламеняющихся материалов. Сама по себе Н.п. ие
воспламеняется, но огнеопасна при соприкосновении
с органич. веществами, напр. деревом, маслом, бу-
магой или с восстановителями в присутствии влаги.
Лит. см. при ст. Перекиси и перекисные соединения.
И. И. Вольнов.
НАТРИЯ ПИРОСУЛЬФАТ Na2S2O7 — см. Натрия
сульфаты.
НАТРИЯ РОДПЗОНАТ (натрий родизоновокислый)
CeOeNa«, мол. в. 214,05 — фиолетовые кристаллы;
q растворим в воде, нерастворим в
и спирте. Водный р-р неустойчив. При
ONa взаимодействии свежеприготовленного
O=k JL-ONa раствора Н. р. с раствором соли ба-
|| рия выпадает красный осадок. Н. р.
О может быть получен взаимодействием
дииатриевой соли диоксивиниой к-ты с Na2SOs и
NaOH при нагревании. Применяют Н. р. для обна-
ружения и определения Ва2+, Pb2 + , Sr2 + , SO®-; как
индикатор при титриметрич. определении Ва2+ и
SOj , для идентификации нек-рых органич. соеди-
нений. И. П. Ефимов.
НАТРИЯ СИЛИКАТЫ — натриевые соли крем-
невых к-т. Принимается существование след, безвод-
ных И. с.: Na2O -SiO2 (или Na2SiOa) — метасиликат
натрия, 2Na2O -SiO2 (или Na4SiOa) — ортосиликат
натрия, 3Na2O -2SiO2 (или NaeSi2O7) — пиросиликат
натрия, Na2O -2SiO2 (или Na2Si2O5) — бисиликат на-
трия и Na2O -3SiO2 (или Na2SiaO7) — трисиликат
натрия. Известны гидраты метасиликата натрия с
1, 5, 6, 8 и 9 молекулами Н2О (есть указания иа ряд
гидратов с иным количеством воды). Получены гид-
раты пиросиликата натрия с 3, 5 и И молекулами
Н2О. Предполагается, что три последние соединения
представляют собой кислые соли, напр. NaeSi2O7 -ЗН2О
является кислой солью состава NaaHSiCH -Н2О,
и т. д. Выделено также соединение NaeSilsO29-11Н2О.
Ортосиликат натрия существует в двух
модификациях с темп-рой превращения 960°; т. пл.
1118° (с разл.). Ппросиликат натрия кри-
сталлизуется в остроугольных пластинках с показа-
телями преломления ng 1,529, пр 1,524; плотн. 2,96,
т. пл. 1122°. Мет а силикат натрия об-
разует игольчатые кристаллы ромбич. системы, дву-
осные, оптически отрицательные, ng 1,528, пр 1,513;
плотн. 2,614; т. пл. 1089°. Бисиликат натрия
образует кристаллы ромбич. системы, двуосные, оп-
тически отрицательные, ng 1,508, пр 1,497; плоти.
2,496; т. пл. 874°. Трисиликат натрия
образует кристаллы моноклинной системы, двуос-
ные, оптически отрицательные, ng 1,502, п0 1,496;
плоти. 2,944; т. пл. 750°. Все эти соединения получа-
ются кристаллизацией стекол соответствующего со-
става.
Водные р-ры Н. с. подвергаются гидролизу вслед-
ствие слабости кремневой к-ты и показывают силь-
нощелочную реакцию. В отличие от обычных солей,
Н. с., растворяясь в воде, не обнаруживают точки
насыщения, причем может быть получен очень кон-
центрированный, густой коллоидный р-р. На практике
для получения р-ров Н. с. пользуются стеклообраз-
ными продуктами с различным соотношением Na2O и
SiO2 (см. Стекло растворимое). Растворы Н. с. при
pH 10,9 становятся неустойчивыми и выделяют
кремнекислоту в виде геля. Принимается, что в обла-
сти pH 10,9—13,6 в растворе существует димерный
ион Si2(OH)2”. Выше pH 13,6 образуется ион Si(OH)2-.
Наличие шестерной координации кремния в этих
ионах обосновывается малым размером группы ОН-
Лит.: Айлер Р. К., Коллоидная химия кремнезема и
силикатов, пер. с англ., М., 1959; Евстропьев К. С.,
Торопов Н. А., Химия кремния и физическая химия
силикатов, 2 изд., М., 1956; Григорьев П. Н., Матве-
ев М. А., Растворимое стекло. (Получение, свойства и приме-
нение), М., 1956; V а 1 1 J. G., Soluble silicates, v. 1—2, N. Y.,
1952. В. П. Барзаковский.
НАТРИЯ СУЛЬФАТЫ — натриевые соли серной
к-ты. Натрия сульфат нормальный
(сернокислый натрий) Na2SO4 — бесцветные кристал-
лы, образующие несколько модификаций. Ромбич.
форма встречается в природе в виде минерала тенар-
дита; плотн. 2,698, т. пл. 884°. Теплота образова-
ния тенардита Д7?298 =—330,9° ккал/моль. Раство-
римость Na2SC>4 в воде (%): 4,76 (0°);16,3 (20°); 33,6
(32,384°); 29,8 (100°). Криогидратная точка лежит
при 3,85 вес. % Н. с. и —1,2°. Из водных р-ров выше
32,384° кристаллизуется ромбический Na2SO4, ниже
этой темп-ры—Na2SC>4-10Н2О, прозрачные моноклин-
ные кристаллы, плотн. 1,464—1,481. В природе
Na2SC>4'10H2O встречается в виде минерала мира-
билита (глауберова соль); при 32,384°
плавится, разлагаясь на тенардит и насыщенный р-р,
содержащий 33,6% Na2SC>4. Теплота образования
мирабилита Дй298 =—1033,48 ккал/моль. Известен ме-
тастабильный гидрат Na2SC>4 ^HsO, выпадающий из
конц. р-ров Н. с. в воде при охлаждении до 12° и
превращающийся в тенардит при 23,5°. Плотность
водных р-ров Na2SO4
% Na2SO, . . 5 10 16
d20 ....... 1,0 4 41 1,09 1 5 1,15 0 6
Na2SC>4 образует твердые р-ры со многими солями
(Li2SC>4, K2SO4,Na2COs), а также двойные соли с
другими сульфатами; нек-рые из них встречаются
в природе: Na2SC>4-MgSCH-4Н2О (астраханит),
Na2SO4 -CaSO4 (глауберит), Na2SC>4 -3K2SO4 (глазе-
рит), 2Na2SC>4-А1а2СОз (беркеит). Значительные ко-
личества Na2SO4 содержатся в рапе и донных отложе-
ниях соляных озер. В холодный период года из рапы
многих озер кристаллизуется мирабилит; его собирают
и используют. Более рационально получение мира-
билита естественным охлаждением рапы в специаль-
ных бассейнах, что дает возможность получать более
чистый продукт и механизировать его сбор. Применяет-
ся также получение мирабилита охлаждением рапы
в заводских условиях. Обезвоживание мирабилита
проводят в естественных или заводских условиях
(наир., путем плавления, выпарки растворов или суш-
ки горячим воздухом и дымовыми газами). Другой
способ получения Na2SO4 заключается во взаимо-
действии NaCl с H2SO4, к-рое проводят в специальных
383
НАТРИЯ СУЛЬФИД —НАТРИЯ СУЛЬФИТ
384
«сульфатных» печах при 500—550°. Одновременно
получается соляная к-та.
Сульфат натрия применяют в стекольном произ-ве,
при получении сульфатной целлюлозы, в текстильной,
мыловаренной и кожевенной пром-сти, в цветной
металлургии. Н. с. служит сырьем для получения
силиката и сульфида натрия. Разработаны способы
получения соды, серной к-ты и сульфата аммония из
сульфата натрия. Он применяется также в медицине
и ветеринарии.
Натрия бисульфат (кислый сернокислый
натрий) NaHSOi — бесцветные кристаллы, плотн.
2,742, т. пл. 186°, при более высокой темп-ре отщеп-
ляет воду с образованием Na2S2O7. Растворимость в
воде (г на 100 г Н2О): 40 (0°), 100(100°). Образует
гидрат NaHSOi -Н2О, плавящийся при 58,5°. При-
меняется как реактив и в качестве флюса в цветной
металлургии. Известны и другие кислые сульфаты нат-
рия, образующиеся, в частности, при взаимодействии
NaCI с H2SOi, напр. Na3H(SO.j)2, NaHa(SO4)2 и др.
Натрия пиросульфат Na2S2O7 — бес-
цветные кристаллы, плотн. 2,658; т. пл. 400,9°. При
темп-ре красного каления разлагается на Na2SO4
и ЭОз. Водные р-ры имеют сильно кислую реакцию.
Получают при нагревании бисульфата натрия до
320°. Применение то же, что бисульфата.
Лит.: П о з и н М. Е., Технология минеральных солей,
2 изд., Л., 1961; Сульфат натрия в СССР. [Сб. статей], М.—Л.,
1946; Бергман А. Г., Лужная Н. П., Физико-хими-
ческие основы изучения и использования соляных место-
рождений хлорид-сульфатного типа, М., 1951; Горбанев
А. И., Николина В. Я., Сульфат натрия, М., 1954; М а-
каров С. 3., Иткина Л. С., Промышленные методы
обезвоживания мирабилита, М.—Л., 1946. Д. С. Стасиневич.
НАТРИЯ СУЛЬФИД (сернистый натрий) Na2S —
бесцветные кристаллы кубич. системы, а =6,539 А;
плотн. 1,856; т. пл. 1180°. Теплота образования ДЯ’д8 =
= —89,2 ккал/моль. Сильно гигроскопичен. Раство-
римость в воде (%): 13,6 (20°); 45,0 (97,5°). Плотность
подных р-ров при 18°: 1,115 (10%), 1,214 (18%).
Из водных р-ров ниже 48° кристаллизуется Na2S -9Н2О,
между 48° и 91,5° Na2S-6H2O, а выше 91,5° Na2S-
•5%Н2О. При охлаждении р-ра, упаренного до т.
кип. 175—185° (63—65% Na2S), кристаллизуется
Na2S-2H2O. Водные р-ры Na2S вследствие гидролиза
имеют щелочную реакцию; степень гидролиза в 0,1 н.
р-ре при 25° равна 86,5%. При действии воздуха и
света Н. с. постепенно окисляется и при этом желтеет.
Получают Н. с. восстановлением Na2SC>4 углем
при 800—1000° в шахтных или вращающихся печах.
Реакционную массу выщелачивают водой и р-р упари-
вают. Восстановление Na2SC>4 производят также при
помощи газов: Н2, СН4, СО или же технич. газов.
Восстановление водородом идет уже при 550—600°;
его проводят во вращающихся печах или во взвешен-
ном слое. При этом сразу получается высокопроцент-
ный продукт. Н. с. может быть получен также при
улавливании сероводорода р-рами щелочи. Н. с.
применяют при получении сернистых красителей,
в текстильной и кожевенной пром-сти и при флота-
ции руд.
Натрия гидросульф ид NaHS — бес-
цветные, очень гигроскопичные кристаллы, известные в
2 модификациях — ромбической (а) и кубической (Р).
Хорошо растворяется в воде: 42% при 20°. Из водных
р-ров кристаллизуется NaHS-3H2O, плавящийся при
22°, или NaHS-2H2O. Водные р-ры постепенно, осо-
бенно прп кипячении, теряют сероводород, при этом
образуется Na2S. Легко окисляется на воздухе. Обра-
зуется при поглощении H2S р-рами щелочи или сер-
нистого натрия; в продажу поступает в виде конц.
р-ров (не менее 22% NaHS). Применяют в пропз-ве
искусственного шелка, при получении этплмеркап-
тана (одоранта) и в кожевенной пром-сти.
Натрия пол и сульфиды Na2Sn (где п=
=2—5) или Na4Sm (где т=3—9) — сильно гигро-
скопичные желто-бурые кристаллы. Темп-ры плавле-
ния: Na2S2 445°, Na2S4 270°, Na2S6 251,8°. Хорошо
растворимы в воде. На воздухе легко окисляются с
выделением элементарной серы. Прочность снижается
с увеличением содержания серы. Токсичны. Полу-
чают при растворении серы в щелочи или в р-рах
Na2S. Применяют в кожевенной пром-сти, при флота-
ции руд, для борьбы с с.-х. вредителями и при полу-
чении полисульфидных каучуков.
Лит.: П о з и н М. Е., Технология минеральных солей,
2 изд., Л., 1961. Д. С. Стасиневич.
НАТРИЯ СУЛЬФИТ (серпистокислый натрий)
Na2SC>3 — бесцветные кристаллы .гексагональной си-
стемы, а =5,441 А, с =6,133 А; плотность 2,63.
Теплота образования ДЯ°д8=—260,6 ккал/моль. Рас-
творимость в воде (%): 12,59 (0°); 20,82 (19,9°);
25,75 (50°); 21,70 (99°). Плотность водных р-ров
d20: 1,0948 (10%), 1,1755 (18%). Криогидратная
точка лежит при —3,45° и 10,48 вес. % Na2SO2.
Между —3,45° и 33,4° из водных р-ров кристалли-
зуется моноклинный Na2SO3-7H2O, плотн. 1,56. Дав-
ление диссоциации 18,59 мм рт. ст. (25°). Выше
33,4° кристаллизуется безводный Н. с. Растворы Н. с.
в воде имеют щелочную реакцию, при их подкисле-
нии выделяется SO2. Н. с.— сильный восстановитель,
в водных р-рах легко окисляется кислородом воздуха;
окисление может быть предотвращено при добавке
ингибиторов, напр. парафенилендиамина или гидро-
хинона, порядка 0,001%. Кристаллич. Na2SO3-7H2O
во влажном воздухе также легко окисляется. В сухом
воздухе кристаллогидрат обезвоживается. Безвод-
ный Н. с. стоек к окислению. Растворы Н. с. погло-
щают SO2 с образованием бисульфита натрия. В вод-
ных р-рах Н. с. присоединяет серу с образованием
натрия тиосульфата.
Н. с. получают взаимодействием р-ров Na2SO3
с SO2. Насыщение ведут до получения р-ра, содер-
жащего 45—47% NaHSOs- Раствор нейтрализуют
содой при 40° и охлаждают для кристаллизации
Na2SC>3-7H2O. Безводный Н. с. получают, выпаривая
конц. р-р при 95—100° до кристаллизации или обез-
воживанием гептагидрата. Н. с. образуется также в
качестве отхода при получении фенола из бензолсуль-
фокислоты методом щелочной плавки. Применяют
Н. с. в фотографии, химико-фармацевтич. пром-сти,
в медицине, при произ-ве искусственного волокна.
Натрия бисульфит NaHSOs — бесцвет-
ные моноклинные кристаллы, плотн. 1,48. На воздухе
легко отщепляет SO2 и окисляется до Na2SO4. Водные
р-ры более стойки. При упаривании водных р-ров
получается Na2S2O5. Насыщенный р-р бисульфита
содержит 39,2% (15°) и 49,1% (97,5°)Na2S2O5.
Получают NaHSOs прп взаимодействии SO2 с Na2CO3
в виде р-ров, содержащих не менее 36,5% NaHSOs.
Применяют в пищевой пром-сти (как консервирую-
щее средство), в кожевенной и текстильной пром-сти
(при белении и крашении тканей).
Натрия пиросульфит (натрия мета-
бисульфит) Na2S2O5 — бесцветные кристаллы, плотн.
1,48. Разлагается выше 150° с отщеплением SO2.
Растворимость в воде (%): 39,77 (22,8°); 49,06 (97,2°).
Образует нестабильный гидрат Na2S2O5-6H2O и стаби-
льный Na2S2O5'7H2O, устойчивый ниже 5,5°. В водных
р-рах превращается в NaHSO3. Пиросульфит полу-
чают, насыщая пульпу сульфита натрия в маточном
р-ре сернистым газом, а также сухим способом —
обрабатывая соду сернистым газом в присутствии
небольшого количества воды. Применяют для тех
же целей, что и NaHSO3.
Лит. см. при ст. Натрия сульфид. Д. С. Стасиневич.
385
НАТРИЯ ТИОСУЛЬФАТ—НАТРИЯ ФТОРИД
386
НАТРИЯ ТИОСУЛЬФАТ (серноватистокислый на-
трий, неправильное название гипосульфит) NaaSaOs —
бесцветные кристаллы, плотн. 2,119. Теплота образо-
вания Д//298=—267,0 ккал/моль. При нагревании
до 300° разлагается на NaaSOs + S, при 600° — на
NaaSO4 + Na2S5. На воздухе стоек до 120°, при более
высокой темп-ре окисляется. Растворимость в воде
(%): 33,40 (0°); 41,20 (20°); 69,86 (80°). Плотность
водных р-ров:
% Na3S3O3 . . 10 20 30 40
d'-°....... 1,0827 1,1740 1,2739 1,3827
От—11° до 47,9° устойчив гидрат NaaSaO3-5HaO,
выделяющийся из водных р-ров в виде прозрачных
моноклинных кристаллов; плотн. 1,715; плавится в
кристаллизационной воде при 47,9°. Теплота образо-
вания пентагидрата Д//298=—621,89 ккал/моль. При
47,9—65° кристаллизуется NaaSaO3-2HaO, при 65—
75° — NaaSaOa • Уа НаО, выше — безводный Н. т.
Известен ряд метастабильных гидратов. Н. т.—
сильный восстановитель. Сильные окислители окис-
ляют его до сульфата, умеренные — до сульфата и
свободной серы, а слабые (напр., иод в нейтральных
р-рах) — до тетратионата NaaSiOj. На этом Основано
применение Н, т. в объемном анализе (см. Иодометрия).
Водные р-ры Н. т. имеют нейтральную реакцию;
при их подкислении выделяется сера.
II. т. получают растворением измельченной серы в
горячем р-ре сульфита натрия: Na2SO.3+S =Ка23зОз;
взаимодействием гидросульфида с бисульфитом на-
трия: 2NaHS+4NaHSO3=3Na2S2O3+3H2O, и др. спо-
собами. Кроме того, Н. т. образуется в качестве по-
бочного продукта в произ-ве гидросульфита, при
очистке промышленных газов от серы и при полу-
чении сернистых красителей и тиокарбапилида.
Обычно получают кристаллический Na2S2O3-5H2O.
Для получения безводного Н. т. гидрат плавят, рас-
плав упаривают и высушивают. Н. т. применяют в
фотографии как закрепитель, в текстильной пром-сти
как антихлор (для уничтожения остатков хлора после
отбелки тканей), в кожевенной промышленности, меди-
цине и ветеринарии, а также как аналитический
реактив.
Лит. см. при ст. Натрия сульфид. Д. С. Стасиневич.
НАТРИЯ ФОСФАТЫ — натриевые соли мета-,
пиро- или ортофосфорной к-ты, соответственно мета-,
пиро- илп ортофосфаты натрия. Ортофосфорная к-та
Н3РО4 образует три соли: однозамещенный натрия
фосфат NaH2PO4, двузамещенный натрия фосфат
NaaHPCH И трехзамещенный натрия фосфат NasPO4.
Эти соли сокращенно называют моно-, ди- и трина-
трийфосфатами. При нагревании NaH2PO4 выделяет
воду и переходит в кислый пирофосфат цатрия
Na2H2P2O7, а при более высокой темп-ре в метафосфат
натрия — полимерное соединение состава (NaPOs)x,
где х=2—6; практнч. значение имеет гл. обр. гекса-
метафосфат (NaPOs)a. Двузамещенный фосфат Na2HPO4
при нагревании и обезвоживании переходит в пиро-
фосфат натрия Na4P2O7. Наибольшее практич. зна-
чение имеет промежуточное соединение между мета-
фосфатом и пирофосфатом натрия — три полифосфат
натрия, или пентанатрийтрифосфат, Na5P3Ot,. Мета-,
пиро- п полифосфаты называют дегидратированными
или конденсированными фосфатами натрия, т. к. они
содержат меньше воды по отношению к фосфорному
ангидриду, чем ортофосфаты. Большинство фосфатов
натрия растворимо в воде.
Промышленные методы произ-ва ортофосфатов нат-
рия основаны на нейтрализации фосфорной к-ты
кальцинированной содой или гидроокисью натрия,
на последующей кристаллизации солей и отделении
их на центрифуге. Мононатрийфосфат вырабатывают
в форме одноводной NaH2PO4-H2O и безводной соли.
Динатрий- и тринатрийфосфаты вырабатывают в форме
двенадцативодных кристаллогидратов Na2HPO4-12Н2О
и Na3PO4-12H2O, а также безводных солей. При
произ-ве дегидратированных фосфатов натрия рас-
творы ортофосфатов быстро выпаривают и сушат,
напр. в распылительной сушилке, и прокаливают.
При производстве гексаметафосфата натрия раствор
мононатрийфосфата выпаривают досуха и затем на-
гревают до плавления. Н. ф. применяют гл. обр.
в качестве моющих и водоумягчающих средств. Осо-
бенно возросло применение для этой цели дегидра-
тированных фосфатов натрия, очень хорошо устра-
няющих жесткость воды благодаря способности к
образованию растворимых комплексов с кальцием,
магнием, барием и др. металлами. В частности, три-
полифосфат натрия признан паилучшей неорганич.
основой синтетич. моющих средств, произ-во к-рых
значительно развивается. В большинство синтетич.
моющих средств триполифосфат натрия входит в ко-
личестве 10—50%. Н. ф. применяют также при обо-
гащении руд, в текстильной и кожевенной пром-сти,
в различных отраслях пищевой пром-сти (произ-во
колбас, сыра, кондитерских изделий и др.), фотогра-
фии, в электролитических процессах и для других
целей.
Лит..: Гофман И. Д., Соли фосфорных кислот и их ис-
пользование, Хим. наука и пром-сть, 1957, 2, № 6, 706—13;
его же, Современные тенденции развития производства
фосфорных солей, Ж. Всес. хим. о-ва им. Д. И. Менделеева,
1962, 7, № 1, 72; Ваггаман В., Фосфорная кислота, фос-
фаты и фосфорные удобрения, пер. с англ., М., 1957; Везер
В а н Д. Р., Фосфор и его соединения, пер. с англ., М., 1962.
А Т/Г / Г / PTIZM/I / 71
НАТРИЯ ФОСФОВОЛЬФРАМАТ (натрий фосфор-
новольфрамовокислый) Na2H5[P(W2O7)s]-гН2О — мел-
кие белые кристаллы, выветривающиеся на возду-
хе; растворим в воде (47,4% при 20°). Н. ф. может
быть получен по реакции:
12Na3WO1 + Na,HPO, + 24HCl = Na3H-,[P(WaO7),;] + 24NaCl +
+ 10Н.О
Применяют Н. ф. для осаждения белков и полипеп-
тидов крови; для нефелометрического определения ко-
феина и др.
Лит. см. при ст. Натрия гипофосфит. И. П. Ефимов.
НАТРИЯ ФТОРИД (фтористый натрий) NaF —
бесцветные кристаллы с кубич. решеткой, а =4,628 А;
плотн. 2,79; т. пл. 992°; т. кип. 1695°. Теплота обра-
зования ДН298 =—136,0 ккал/моль. Растворимость
в воде (%): 3,90 (20°); 4,83 (100°). Водный р-р вслед-
ствие гидролиза имеет щелочную реакцию. В спирте
Н. ф. нерастворим. Сухой Н. ф. поглощает газообраз-
ный HF с образованием бифторида NaHF2. В природе
Н. ф. встречается в виде минерала вильомита; вхо-
дит в состав криолита и др. минералов. В пром-сти
Н. ф. получают спеканием плавикового шпата с
содой и кремнеземом (по реакции CaF2 + Na2COs +
+SiO2=2NaF+Ca3iOs+CO2), разложением кремне-
фторида натрия содой илп растворением соды или
едкого натра в плавиковой к-те. Применяют для кон-
сервирования древесины, для борьбы с вредителями
с.-х. культур, при изготовлении эмалей и флюсов,
для фторирования воды.
Натрия бпфторид NaHF2 — бесцветные
кристаллы, плотн. 2,08; при нагревании до 200°
расщепляется на NaF и HF. Растворимость в воде
3,75% (20°). Водные растворы имеют кислую реак-
цию. Получают взаимодействием плавиковой кислоты
с NaF или NaOH. Применяют в качестве флюса при
пайке.
Лит.: Рысс И. Г.» Химия фтора и его неорганических
соединений, М., 1956; БогачовГ. Н., Г у з ь С. Ю., Про-
изводство криолита, фтористого алюминия и фтористого нат-
рия, М.—Л., 1940. Д. С. Стасиневич.
О 13 К.Х.Э, т. 3
387
НАТРИЯ ХЛОРИД—НАФТАЗАРИН
388
НАТРИЯ ХЛОРИД (хлористый натрий, пова-
ренная соль) NaCl — бесцветные кристаллы
с кубической гранецентрированной решеткой, а=
=5,63874 А; плотн. 2,161; т. пл. 801°, т. кип. 1413°;
диэлектрич. проницаемость 6,12; уд. теплоемкость
0,20 кал/г-град, твердость по Моосу 2. Теплота
образования ДН298 =—98,232 ккал/моль. Раствори-
мость в воде (в %): 26,28 (0°); 26,43 (25°); 28,12 (100°)
(мало зависит от темп-ры). В присутствии NaOH,
НС1, MgCla, СаСЬ и др. солей растворимость Н. х.
в воде сильно снижается. Криогидратная точка
—21,1° (23,0% NaCl). Насыщенный водный р-р кипит
при 108,8° и содержит 40,7г NaCl на 100г НгО.
Плотность водных р-ров:
% NaCl ... 5 10 15 20 25
d30 ........ 1.03460 1,07142 1,10939 1,14876 1,18975
Чистый Н. х. мало гигроскопичен, но в присут-
ствии примесей (напр., солей магния) гигроскопич-
ность его сильно повышается. Из водных р-ров при
темп-рах от —21,1° до +0,1° кристаллизуется дигид-
рат NaCl-2^0 — тонкие иглы или хорошо огранен-
ные моноклинные призматич. кристаллы; плотн. 1,6.
Н. х. растворим в жидком аммиаке; нерастворим в
большинстве органич. растворителей.
В природе Н. х. хорошо распространен в виде
минерала галита (каменная соль). Обра-
зует мощные месторождения, толщина пластов к-рых
достигает сотен и тысяч метров (см. Натрий). Часто
Н. х. залегает совместно с солями К, Mg и т. д. При-
родный галит иногда бывает окрашен в голубой,
синий или фиолетовый цвет, обусловленный присут-
ствием следов металлич. натрия. Огромные количе-
ства Н. х. содержатся в морской воде, рапе соляных
озер и подземных рассолах. В летнее время при
испарении рапы во многих озерах происходит кри-
сталлизация Н. х. (самосадочная соль,
новосадка). Иногда в соляных озерах под
слоем рапы залегает слой соли садки прошлых лет,
не растворяющейся в осенне-зимний период (старо-
садка). В зимнее время из рапы соляных Озер
иногда кристаллизуется дигидрат ПаС1-2НзО (г ид-
рога л и т).
Разработка месторождений каменной соли произ-
водится обычно закрытым (шахтным) способом и
реже — открытым способом. Добыча каменной соли
полностью механизирована. Для извлечения соли
широко применяется метод подземного выщелачива-
ния при помощи скважин, в к-рые нагнетают воду.
Озерную соль добывают механич. способом при по-
мощи скрепперов, бульдозеров, экскаваторов и со-
лесосов. Обычно озерная соль бывает загрязнена
илом, для удаления к-рого ее промывают рапой.
Промытую соль центрифугируют и сушат. Для полу-
чения бассейной соли применяется регулируемое
солнечное испарение морских или озерных рассолов
в системе специально устроенных бассейнов. В ме-
стностях с холодным климатом поваренную соль
получают из рассолов вымораживанием. Каменная и
самосадочная соль обычно содержит нек-рое количе-
ство примесей (CaSCh, MgSCU, MgCL, СаСЬ, не-
растворимые в воде вещества и т. п.). Наиболее
чистая соль получается при упаривании естествен-
ных или искусственно полученных и очищенных рас-
солов в вакуум-выпарных аппаратах (выварочная
или вакуумная соль).
Н. х.— важный пищевой продукт, служит также
для консервирования мяса, рыбы и др., применяется
при кормлении скота. Н. х. — один из главных ви-
дов химического сырья и используется для получе-
ния едкого натра, хлора, соды, сульфата натрия. Ши-
роко применяется Н.х. ив различных других произ-
водствах.
Лит.: П о з и н М. Е., Технология минеральных солей,
2 изд., Л., 1961; Зиновьев А. И., Технология выварочной
и йодированной соли, М., 1957; Kaufmann D. W., Sodium
chloride, N. Y.—L., 1959. Д. С. Стасиневич.
НАТРОВАЯ ИЗВЕСТЬ (неправильно натронная
известь) — смесь гашеной извести с едким натром;
белая пористая масса. Жадно притягивает влагу и
двуокись углерода. Поглощает СО а в количестве
свыше 20% от своего веса. Н. и. получают действием
конц. р-ра NaOH на свежепрокаленную СаО (на
2 вес. ч. СаО 1 вес. ч. NaOH); массу выпаривают
досуха в железной чашке, слабо прокаливают и по
охлаждении измельчают и просеивают сквозь сита
с разным диаметром отверстий. Применяют в хими-
ческих лабораториях для поглощения СОг, напри-
мер при определении углерода в чугуне и стали.
Д. С. Стасиневич.
НАТРОННАЯ ИЗВЕСТЬ — см. Натровая известь.
НАФТАЗАРИН (5,8-диоксинафтохинон-1,4) С10НвО4,
мол. в. 190 — кристаллы желто-зеленого или крас-
ного цвета, возгоняются при 2—10 мм. При возгонке
Н. образуются две модификации — темно-зеленые
иглы и темно-красные призмы; при кристаллизации
из бензола получается третья модификация — светло-
красные пластинки. Модификации кристаллов мож-
но разделить механически. Нафтазарин мало раство-
рим в воде, эфире и спирте; лучше — в ледяной
СНзСООН; растворы окрашены в красный цвет. Н.
растворяется в щелочах с синей окраской. С метал-
лами (Ва2 + , Са2 + , Fe’+ и др.) образует глубокоокра-
шенные лаки.
Рентгенографич. исследованиями установлено, что
две модификации Н. являются центросимметричными
и отвечают структуре А, а одна — нецентросимметрич-
ная, отвечает структуре Б.
При кристаллизации происходит обратимый переход
одной структуры в другую. На рисунке приведены
длины связей центросимметричной модификации Н.
Строение нафталинового ядра обусловлено харак-
тером связей между атомами углерода и кислорода,
из к-рых одна является двойной (1,18 А), вторая —
одинарной (1,35 А) связью. При исследовании спек-
тра паров Н. были обнаружены две полосы, соответ-
ствующие валентным колебаниям группы ОН, что
также подтверждает существование равновесия ме-
жду изомерами А и Б.
Н.— окислитель; его нормальный окислительно-
восстановительный потенциал при 25° равен 0,361 в
(измерено в смеси 0,1 н. соляной к-ты и 50%-ного
спирта в присутствии LiCl). Восстановление Н.
Zn-пылью в разб. H2SO4 или Na2S2O4 в разб.
СНзСООН дает 1,4,5,8-тетраоксинафталин. При обра-
ботке Н. тетраацетатом свинца в СН3СООН на холо-
ду получается нафтодихинон-1,4,5,8. При нагревании
Н. с HNO3 (плотн. 1,2) превращается в щавелевую
к-ту. С уксусным ангидридом и конц. H2SC>4 дает
дпацетильное производное — 5,8-диацетоксинафтохп-
нон-1,4, т. пл. 192—193°. При бромировании Н.
в кипящей СН3СООН 2 молями брома получается
2,3-дибромнафтазарин, а с 4 молями брома — 2,3,
6,7-тетрабромнафтазарин. С фенилгидразином Н. взаи-
модействует с образованием 8- бензолазо-2, 4, 5-три-
оксинафталина. С фенил семикарбазидом образует мо-
389
НАФТАЛЕВАЯ КИСЛОТА—НАФТАЛИН
390
ио- и бисфенилсемикарбазоны (т. пл. 218° и 285—
287° соответственно).
Н. получают из 1,5-динитронафталина при нагре-
вании (40—45°) его с олеумом и серой; образующийся
при этом 1,4-амино-окси-а-нафтохинонимин гидро-
лизуется до Н. разбавленными к-тами. Н.— проме-
жуточный продукт в синтезе ряда ценных красителей.
Лит.: Гольдер Г. А., Жданов Г. С., ДАН СССР,
1958, 118, № 6, 1131. См. также лит. при ст. Метилнафталин.
В. А. Пучков.
НАФТАЛЕВАЯ КИСЛОТА (нафталин-1,8-дикар-
боновая кислота) CnHgOa, мол. в. 216— белые иглы;
при 140—150° теряет воду и плавится в виде ангидри-
да при 274°; диметиловый эфир, т. пл. 102—103°;
днэтиловый эфир, т. пл. 59—60°, дихлорангидрид,
т. пл. 84—86°. Н. к. (I) почти нерастворима в воде
и эфире; растворяется в горячем спирте и кипящей
15%-ной НС1. Ангидрид Н. к. на холоду нерастворим
в воде, эфире и бензоле; при нагревании растворяется
в СНзСООН, хлороформе и бензоле. Ангидрид полу-
чается из Н. к. при нагревании с HNO3 (плоти. 1,4)
или с СНзСООН или H2SO4 в спирте.
При кипячении ангидрида Н. к. с конц. водным NH3
образуется нафталимид, т. пл. 307—308°. Сплавле-
нием последнего с КОН при 290—300° получают
диимид 3,4,9, 10-перилен-тетракарбоновой к-ты (II),
Х,М'-диметильное производное к-рой — прочный кубо-
вый краситель (индантрен красный ЖЖ). Ангидрид
Н. к. сульфируется сначала в положение 3, а затем
в положение 6. Нитрование HNO3 в конц. H2SO4
приводит к образованию 3-нитронафталевой к-ты, а
конечным продуктом является 3,6-динитронафтале-
вый ангидрид. Хлорирование в олеуме при 180—
200° дает тетрахлорпроизводное ангидрида Н. к.
4-Замещенные Н. к. получают окислением соответ-
ствующих замещенных аценафтена, а также из аце-
нафтена при окислении НзСгСН конц. HNO3 или
KMnOd в щелочной среде. Качественное определение
Н. к. основано на взаимодействии ее ангидрида с
резорцином и конц. H2SO4 при 130°; при растворе-
нии в щелочи продукта реакции обнаруживается
интенсивная желто-коричневая (темно-зеленая в УФ-
свете) флуоресценция.
Н. к___промежуточный продукт в синтезе краси-
телей.
Лит. см. при ст. Метилнафталины. В. А. Пучков.
НАФТАЛИН С10Н8, мол. в. 128,16 — кристаллы, бес-
цветные пластинки; т. пл. 80,3°; т. заст. 80,287 ±0,002°;
т. кип. 218°; сублимируется при
507760 мм; d'J 1,1517, d'M 0,9625;
Т г]
nD 1 >58232; скрытая теплота плавле-
~ а а ния 4,54 ккал/моль; теплота испарения
11,31 ккал/моль. Н. мало растворим в
воде (0,0344 г/л при 25°); хорошо растворяется в боль-
шинстве органич. растворителей; перегоняется с вод-
ным паром.
Н. представляет собой конденсированную ароматич.
систему. По данным рентгеноструктуриого анализа, в
молекуле Н., в отличие от бензола, длины связей С—С
неравноценны (1). Расчет порядка кратности всех
связей в молекуле Н. (II) показал, что С—С-связь
1—2 (соответственно 3—4, 5—6, 7—8) ближе по ха-
рактеру к двойной, а связь 2—3 (6—7 и т. д.) ближе
к простой связи, вследствие чего электронная плот-
ность у а-атомов углерода значительно выше, чем у
Р-атомов. В целом же 1, 2, 3, 4-система Н. в известной
мере подобна бутадиену-1,3:
в процентах ло отношению
к двойной связи
Монозамещенные Н.
формах III и IV:
существуют в двух изомерных
III IV S
Нахождение 2 заместителей в полсжении 2,6 наз»
также амфи-положением, в 1,8 — пери-положением.
Наиболее характерными для Н., так же как и для бен-
зола, являются реакции электрофильного замещения.
При нитровании и сульфировании Н. образуются
смеси изомеров, соответственно нитро- и сульфоза-
мещенных (см. Нафталина сульфокислоты, Нитро-
нафталины); заместитель при этом легче вступает в
a-положение. При хлорировании Н. в присутствии
FeCl3 на холоду получаются 1-хлор-, 1,4- и 1,5-ди-
хлорнафталины; при повышенной темп-ре (до 150°)
образуются полихлориды Н. (галоваксы), применяю-
щиеся в качестве электроизоляционных материалов.
Бромирование II. приводит к образованию анало-
гичных соединений. 1-Иоднафталин можно получить
при действии на Н. смеси 1г+ННО3. Н. легко всту-
пает в Фриделя—Крафтса реакцию. С ацетилхлори-
дом в среде дихлорэтана Н. дает 1-ацетилнафталин.
В отсутствие растворителя получается смесь 1- и
2-ацетилна фталинов. При ацилировании Н. бензоил-
хлоридом заместитель вступает только в а-поло-
жения (1,5- и 1,8-изомеры). С фталевым ангидридом
в дихлорэтане образуется 2-а-нафтоилбензойная к-та.
Н. взаимодействует с янтарным ангидридом с обра-
зованием смеси р-1-нафтоил- и Р-2-нафтоилпропи-
оновых к-т (36% и47% соответственно). При алкилиро-
вании Н. спиртами (40—100° в присутствии А1С13
н BF3) получается смесь примерно равных количеств
моно- и диалкилнафталинов. Место вступления алкиль-
ного радикала зависит от условий реакции (темп-ра,
катализатор и др.). Алкилнафталины образуют гомо-
логия. ряд общей ф-лы С1О+ПН8+2П. Гомологи Н. легко
сульфируются, нитруются и вступают в др. реакции
электрофильного замещения; их широко применяют
в синтезе красителей и вспомогательных веществ в
текстильной пром-сти. Хлорметилирование Н. осу-
ществляется нагреванием его с параформом, ледяной
уксусной к-той, фосфорной и концентрированной
соляной к-тами; образуется исключительно а-хлорме-
тилнафталин. Н. легко взаимодействует со свобод-
ными радикалами. С арильным радикалом Н. реаги-
рует с образованием смесей продуктов замещения и
димеризации. Н. обладает большей по сравнению с
бензолом склонностью к присоединению различных
реагентов. Н. образует продукты присоединения с
Na и Li (1,4-), с хлором (1,4-), с озоном (1,2, 3,4-)
(V), с диазоуксусным эфиром (1,2-) (VI); с водоро-
13*
301
НАФТАЛИНА СУЛЬФОКИСЛОТЫ -
392
дом, соответственно, получаются 1,4-дигидронафталин
(VII) (Na+C2H5OH, 80°), 1,2,3,4-тетрагидронафталин,
тетралин (Ha, Ni; 200°) и декагидронафталин, дека-
лин (H2,Pt). Тетралин и декалин применяют в каче-
стве растворителей и компонентов моторного топли-
ва. При окислении Н. смесью СгО3+СН3СООН полу-
чают 1,4-нафтохинон, при действии щелочного пер-
манганата образуется сначала фталоповая к-та, а
затем фталевая кислота. Большое технич. значение
имеют окисление Н. во фталевый ангидрид и декар-
боксилирование последнего в бензойную к-ту. При
биологич. окислении Н. образуется 1,2-диокси-1,2-
дигидронафталин. Атомы водорода в Н. не обмени-
ваются на дейтерий ни в D2O, ни в ND3. Изотопный
обмен протекает лишь в присутствии NaND2 в среде
жидкого ND3. Обменное равновесие при комнатной
темп-ре достигается За десятки минут. Октадейтеро-
нафталин (C10D8, т. пл. 80—81°, пикрат, т. пл. 147°)
синтезирован из дейтероацетилена (C2D2).
Н. является исходным соединением при синтезе
большого числа красителей и промежуточных про-
дуктов анилино-красочной пром-сти; применяется
также в качестве инсектицида, напр. против моли.
Основное количество Н. идет на произ-во фталевого
ангидрида. Основной источник Н.— каменноугольная
смола — содержит 8 —11% Н. В пром-сти Н. полу-
чают кристаллизацией из нафталиновой фракции этой
смолы. В последние годы вследствие огромного спроса
на Н. разрабатываются методы его получения из не-
фтепродуктов. Нек-рые нефти содержат значительные
количества Н. и его гомологов, напр. в нефти
О. Борнео до 6—7% Н. Известен способ получения Н.
(совместно с другими ароматическими соединениями)
пропусканием лигроиновых и нафтеновых фракций
(температура кип. 65—290°) над катализаторами при
630—680°. Однако этот метод пока не может кон-
курировать с получением Н. из каменноугольной
смолы.
Количественное определение Н. основано на обра-
зовании пикрата С10Н8-С6Н2 (NO2)3OH, т. пл. 151°;
содержание Н. в моторном топливе определяют по
поглощению в УФ-свете.
Лит.: Каррер П., Курс органической химии, пер. с
нем., Л., 1960; Ворожцов И. И., Основы синтеза проме-
жуточных продуктов и красителей, 4 изд., М., 1955; До-
налдсон И., Химия и технология соединений нафтали-
нового ряда, пер. с англ., М., 1963; Clar Е., Aromatlsche
Kohlenwasserstoffe, В., 1952; UHmann, Bd 12, В,—[u. а.],
1960, S. 584. В. А. Пучков.
НАФТАЛИНА СУЛЬФОКИСЛОТЫ — производ-
ные нафталина общей формулы — C10Hs_n(SO3H)n, где
п=1, 2, 3 и 4 (более четырех сульфогрупп в молекулу
нафталина не вступает). Известны все возможные
изомеры Н. с., однако технич. значение имеют лишь
сульфокислоты, к-рые получаются при сульфирова-
нии нафталина. Н. с.— кристаллич. продукты, хо-
рошо растворимые в воде и минеральных к-тах.
Растворимость различных солей а- и р-моносульфо-
кислот нафталина в воде и спирте приводится ниже;
1 вес. часть соли растворяется в след, количестве
вес. частей растворителя:
Соль a р
в воде в 8 5 %-ном этаноле в воде в 85%-ном этаноле
к 13 108 15 115
Са 16,5 19,5 76 437
Ва 87 350 290 1950
РЬ 27 11 115 305
Ниже приведена схема образования Н. с. из нафта-
лина. Скорость сульфирования Н. в а~положении
значительно больше, чем в p-положении. Образую-
щаяся при сульфировании при низких темп-рах
смесь с преобладанием а-изомера быстро изомери-
зуется при повышении темп-ры и количество [5-изомера
возрастает. При нагревании выше 150° происходит
обратное превращение р-изомепа в а -изомер.
Цифрами обозначено положение сульфогрупп в
молекуле образующейся Н. с.; стрелками показаны
возможные переходы Н. с. к содержащим большее
число сульфогрупп, а также переходы между Н. с.
с одинаковым числом сульфогрупп.
При сульфировании нафталина до моносульфокис-
лоты в зависимости от концентрации серной к-ты
и темп-ры образуется смесь, содержащая преим.
или а- (не менее 82%) или p-изомер (>85%). При
темп-ре сульфирования выше 163° становится замет-
ным образование дисульфокислот. Обычно ди- ц
трисульфокислоты нафталина получают сульфирова-
нием а- или р- Н. с. олеумом при различных темп-рах.
Прп прочих равных условиях сульфогруппа вводится
в a-положение с применением более концентрирован-
ного сульфирующего агента, при более низкой темп-ре
и меньшей продолжительности, чем при введении
Р-сульфогруппы. При сульфировании нафталина стро-
ение образующихся ди- и полисульфокислот удовлет-
воряет правилу Армстронга и Винна: не получаются
такие сульфокислоты, в к-рых сульфогруппы распо-
ложены одна по отношению другой в орто-, пара-
или пери-(1,8)-положениях. Сульфирование нафта-
лина сопровождается изомеризацией образующихся
Н. с. Межмолекулярный механизм изомеризации
был установлен на примере нафталин-1 (С14)-сульфо-
кполоты:
nh2
aCONH2
сн=снсоон
+ со»
сн=снсоон
Изомеризация протекает межмолекулярно, т. к.
отношение молярных радиоактивностей СО2 и транс-
о-карбамидокоричной- (С'4)-кислоты равняется 0,25:1.
Н. с. образуют различные функциональные произ-
водные (эфиры, хлорангидриды, амиды); эти производ-
ные используют для идентификации. Сульфогруппа
в Н. с. может быть замещена оксигруппой методом
щелочного плавления. Эта реакция имеет важное
технич. значение, в частности для получения [5-
нафтола. При нитровании нафталин-1,3,6-трисуль-
393
НАФТАЦЕН —НАФТЕНОВЫЕ КИСЛОТЫ
394’.
фокислоты получают 8-нитронафталин-1,3,6-трисуль-
фокислоту, необходимую для синтеза Ain-кислоты;
Н. с.— промежуточные продукты для синтеза кра-
сителей.
Лит. см. при ст. Нафталин. В. А. Пучков.
НАФТАЦЕН (2,3-бензантрацен) С18Н12, мол. в.
228 — кристаллы, оранжевые пластинки с бронзовым
оттенком, т. пл. 337° (испр.), сублимируется; пары —
зеленовато-желтые. Н. трудно растворим в большин-
стве органич. растворителей; растворы имеют зеленую
флуоресценцию. В конц. H2SO4 растворяется с интен-
сивным зеленым окрашиванием. Н. представляет со-
бой линейную поликонденсированную ароматич. си-
стему. По сравнению с антраценом (Хмакс, 370 ммк) име-
ет более длинноволновое поглощение (Х,макс_ 460 ммк)
вследствие увеличения л-сопряжения. По химич.
свойствам Н. близок антрацену. Н. был выделен
из сырого антрацена хроматографией на окиси алю-
миния (растворитель бензин). Синтетически Н. на-
ряду с 9,10-дигидронафтаценом может быть полу-
чен перегонкой над Zn-пылью нафтаценхинона-9,10
или 9,10-диоксинафтаценхинона-11,12 (1).
Лит.: С 1 а г Е., Aromatische Kolilenwasserstoffe, 2 Aufl.,
В.— [u. а.], 1952. В. А. Пучков.
НАФТЕНОВЫЕ КИСЛОТЫ — алициклические
карбоновые к-ты (б. ч. одноосновные), содержащие
пяти- и шестичленные углеродные циклы, общей
формулы В(СН2)„(СООН)Л!, где R — нафтеновый ра-
дикал. Природные Н. к. относятся в основном к кис-
лотам циклопентанового ряда, начиная от простей-
ших: циклопентилкарбоновой к-ты (I) и циклопен-
тилуксусной к-ты (II)
Н2С-СН2 Н2С-СН2
| ^снсоон | 'снсн2соон
н2с—сн2 н2с-сн2
I 11
Низшие Н. к. являются моноциклическими, высшие
Н. к. (от Cis)—ди-и полициклическими. Н. к. цикло-
гексанового ряда встречаются реже (бакинская, ка-
лифорнийская, сахалинская нефти). Карбоксильная
группа в Н. к. присоединена к кольцу чаще всего че-
рез метилен или боковую углеродную цепь.
Н. к.— вязкие жидкости мол. веса от 114 до 1000.
Свежеперегнанные в вакууме бесцветны и не имеют
запаха; при стоянии желтеют и приобретают стойкий
неприятный запах. Н. к. кипят от 214° до 300°;
т. заст. ниже —80°. С увеличением мол. веса показа-
тель преломления Н. к. возрастает от 1,45 до 1,52;
плотность при этом обычно уменьшается от 1,05 (для
низших фракций) до 0,96. Однако плотность II. к.,
выделенных из румынской нефти, растет с увеличе-
нием их мол. веса; для нек-рых нефтей эта зависимость
имеет более сложный характер. Коэфф, расширения
Н. к. в пределах 15—20° равен 0,00066. Вязкость
Н. к. растет с увеличением мол. веса. Так, вязкость
по Энглеру при 50° для Н. к., выделенных из кероси-
новой фракции майкопской нефти, равна 2,77, а из
цилиндрового масла той же нефти 49,13. Природные
Н. к. обладают слабой онтич.. активностью; Они
растворимы в нефтепродуктах и других органич.
растворителях, нерастворимы в воде; в свою очередь,
сами хорошо растворяют каучук, смолы, лаки.
Н. к. обладают всеми типическими химич. свой-
ствами карбоновых к-т. Известны разнообразные
производные Н. к., как ангидриды, хлорангидриды,
амиды (последние хорошо кристаллизуются и могут
служить для идентификации), нитрилы и др. Восста-
новлением эфиров Н. к. получены нафтеновые спирты.
Перегонкой Са-солей получают соответствующие ке-
тоны. Эта реакция идет менее гладко, чем для жирных
к-т, сопровождается образованием непредельных угле-
водородов и других продуктов. Имеются производные
Н. к., полученные действием реагента на нафтеновый
радикал, напр. бромпроизводные. Известны разнооб-,
разные соли Н. к.— нафтенаты, из к-рых соли щелоч-
ных металлов хорошо растворимы в воде и спирте,
трудно — в легких нефтяных фракциях. Нафтенаты
щелочноземельных металлов чаще всего плохо раство-
римы в воде, спирте, бензине. Соли А1, Мп образуют
пластич. каучукоподобные массы. Нек-рые соли
(в основном циклопентанового ряда) растворяются
в бензинах, давая окрашенные р-ры, напр. фиолетовые
(Со) или голубовато-зеленые (Си).
Н. к. составляют основную часть кислородсодержа-
щих компонентов нефтей. Из нефтепродуктов Н. к.
извлекают слабым щелочным р-ром, при подкислении
к-рого (серной к-той) Н. к. выделяются в свободном
виде. Содержание Н. к. в нефтях невелико (0,5—3%).
Наиболее богаты Н. к. соляровый и веретенный ди-
стилляты. Индивидуальные Н. к. синтезируют дей-
ствием СО2 на алициклич. магнийорганич. соедине-
ния (см. Гриньяра реакция) или взаимодействием нат-
рий-малонового эфира с 1,4- илп 1,5-дибромидами,
напр.:
Н,С-CH2Br COOR Н2С—СН2 COOR
? I z \ z *2Н2О
’ I * --2N^ I xCv
H2C-CH2Br COOR H2C-CH2 COOR
H2C-CH2
___ j 4CHCOOH * CO2 * 2 ROH
H2c-CH2
H. к. циклогексанового ряда получают также гидриро-
ванием ароматич. к-т. В технике смеси Н. к. получают
каталитич. окислением соответствующих нефтяных
углеводородов.
Сами Н. к. применяются мало, напр. для деэмуль-
гирования нефтей, пропитки шпал; они хорошо ра-
створяют каучук и смолы, поэтому применяются как
растворители при девулканизации каучука, а также
в составах для чистки и в качестве пластификаторов.
Наибольшее применение находят нафтенаты. Соли
щелочных металлов применяют в качестве технич.
мыл — мылонафт, напр. при мытье шерсти, валянии
войлока, а также в качестве эмульгаторов или дезин-
фицирующих средств, т. к. нафтенаты сами обладают
бактерицидным действием. Нафтенаты свинца, мар-
ганца, кобальта, цинка — превосходные сиккативы для
масляных красок. Соли меди применяют для предох-
ранения дерева, канатов, тканей от- гниения и терми-
тов; нафтенат свинца улучшает качества смазочных
масел при работе под высоким и сверхвысоким давле-
нием; соли алюминия служат загустителями мине-
ральных масел, входят в состав разных смазочных
масел.
Лит.: Рыбак Б. М., Нафтеновые кислоты, М.—Л.,
1948; НаметкинС. С., Химия нефти, [3 изд.], М., 1955,
с. 213; ИгонинП. Г. [и др.], Тр. Грозненок, нефт. ин-та,
1960, вып. 7, с. 198; Нафтали М., Х^мия, технология
и анализ нафтенойых кислот, пер. с нем., М., 1934.
М. И. Розенгарт.
395
НАФТЕНЫ—НАФТИЛАМИНА СУЛЬФОКИСЛОТЫ
396
НАФТЕНЫ — алициклические насыщенные угле-
водороды, широко представленные в нефтях, молеку-
лы к-рых содержат пяти- или шестичленные углерод-
ные кольца (нек-рые авторы считают Н. все алициклич.
насыщенные углеводороды или циклопарафины, цик-
ланы). Подобно всем циклич. соединениям, Н. могут
быть однокольчатыми, с двумя или многими изолиро-
ванными кольцами (напр., дициклогексил) или с кон-
денсированными кольцами (напр., декалин).
Как правило, темп-pa плавления Н. ниже, чем у
парафинов близкого мол. веса. Темп-pa кипения,
плотность, показатель преломления — выше соответ-
ствующих констант парафиновых углеводородов, со-
держащих то же число атомов углерода, но ниже кон-
стант ароматич. углеводородов. Последние две кон-
станты у Н. выше, чем у изомерных им олефинов. По
мере увеличения мол. веса эти физич. константы Н.
или возрастают, или проходят через минимум для
метилциклопентана в ряду циклопентановых угле-
водородов и для метилциклогексана в ряду циклогек-
сановых углеводородов.
Химич, свойства Н. близки к свойствам парафино-
вых углеводородов. При действии хлора происходит
замещение атомов водорода с образованием хлорпро-
изводных (металепсия), нитрование приводит к нит-
ропроизводным. При высоких темп-рах происходит
разрушение кольца (термокрекинг), напр. циклогек-
сан распадается с образованием бутадиена: CeHi2^
-^C4He+C2He. При окислении Н. могут образовы-
ваться нафтеновые кислоты за счет алкильных заме-
стителей или же происходит разрушение кольца с об-
разованием двухосновных кислот с тем же числом
атомов углерода, напр. окислением циклогексана
получают адипиновую к-ту. Характерной реакцией
пятичленных Н. является их гидрогенолиз (при дей-
ствии водорода в присутствии металлич. катализато-
ров, напр. Pt на угле) с разрывом кольца и образова-
нием парафиновых углеводородов, напр.:
f----------*СН3СНСН2СН2СН3
! СН3
Н2СтСН2_ _ _---_ СН3СН2 СН2СН2СН2СНа
I--1 уСН-СН3
{ Н2С-СН2
1---------------*СН3СН2СНСН2СН3
сн3
Для шестичленных Н. характерно дегидрирование
в соответствующие ароматич. углеводороды
'CH2-CH2'
н2с' 'сн2
'сн2-сн/
СН = СН
нс' 'сн + зн2
СН-СН
протекающее при 200—400° на металлич. катализато-
рах (Pt, Ni) и при 450—550° на окисных катализато-
рах (МоОг, СггО3). В присутствии металлич. катали-
заторов протекает также обмен водорода между Н.
и ароматич. углеводородами:
В присутствии А1С13 и нек-рых других катализаторов
происходит изомеризация Н., затрагивающая строе-
ние кольца с образованием равновесной смеси угле-
водородов, напр.:
[>“ ~ О
Н. в относительно больших количествах содержатся
в нефтях, от к-рых и получили свое название (В. В.
Марковников, В. Н. Оглоблин). В небольших количе-
ствах Н. содержатся в низкотемпературной части
сланцевой смолы, живицы, смол коксования. Пяти-
членные Н. получают в лаборатории обычно с помощью
магнийорганич. синтеза, исходя из циклопентанона
или алкилциклопентанонов:
R R
+ R’Mgx ------
или циклизацией замещенных дивинилкетонов и по-
следующих реакций:
Циклогексановые углеводороды получают в основном
гидрированием соответствующих ароматич. углево-
дородов, напр. метилциклогексан гладко получается
гидрированием толуола.
Н. являются желательными компонентами мотор-
ных топлив и смазочных масел. Содержание Н. в
бензинах улучшает их антидетонационные характе-
ристики, в керосинах — снижает темп-ру их засты-
вания, в дизельных топливах, кроме того, повышает
теплоту сгорания единицы объема. Н. являются ос-
новными компонентами смазочных масел, снижаю-
щими темп-ру замерзания и обеспечивающими их
высокое качество. В нефтехимич. пром-сти Н. явля-
ются источником получения ароматич. углеводоро-
дов (см. Риформинг) и рекомендуются как сырье
для производства инсектофунгицидов, репелентов,
промоторов горения дизельного топлива. Наиболь-
шее промышленное значение из индивидуальных Н.
приобрел циклогексан как источник производства
капролактама, адипиновой кислоты и других соеди-
нений, идущих для производства синтетического
волокна.
Лит.: И а м е т к и н С. С., Химия нефти, [3 изд.], М., 1955;
Химия углеводородов нефти, под ред. Б. Т. Брукса и др., пер.
с англ., т. 1, М., 1958, с. 451; В го о к з В. Т., The chemislry
of the nonbenzenoid hydrocarbons, 2 ed., N. Y., 1950.
M. И. Розенгарт.
НАФТИЛАМИНА СУЛЬФОКИСЛОТЫ — произ-
водные а- и Р-нафтиламина, содержащие в молекуле
одну, две и три сульфогруппы. Моносульфокислоты
нафтиламинов и их соли умеренно растворимы, ди-
и трисульфокислоты хорошо растворимы в воде. Н. с.
образуют функциональные производные по суль-
фогруппе и аминогруппе; эти производные исполь-
зуют для идентификации Н. с. В таблице приве-
дены темп-ры плавления характерных производ-
ных Н. с.
Наиболее важными реакциями Н. с. являются диазо-
тирование и азосочетание. Сочетание Н. с. с диазо-
соединениями происходит в кислой среде. Благодаря
возможности последовательного сочетания и диазо-
тирования Н. с. используют для получения дисазо-
красителей. В Н. с. сульфогруппа может быть заме-
щена оксигруппой методом щелочного плавления.
Эта реакция имеет технич. значение для получения
аминонафтолсульфокислот. Сульфогруппа может быть
элиминирована при действии цинковой пыли на ще-
лочной р-р Н. с., напр.:
397
ПАФТИ ЛАМИНЫ—НАФТОЙНЫЕ КИСЛОТЫ
398
Положе- ние SO3H- группы Т. пл. производных нафтиламиномоносульфо- кислот, °C
а 6 в г
П р о и з водные а-н а ф т и ламина
Г) 20 5
о 189 —
4 175 232—233 176 221—222
5 197 255 169 264—265
6 157—158 232 — __
7 196 —
8 — 207 138 —
П р о и з в о д н ы е р-н а ф т н ламина
1 178 142 180
4 185 — ——
5 — 119 — 173—174
6 171 243 218 190,5—192
7 265 — 203
8 183 — — 209 — 210
а Пиридиновая соль ацетиламиионафталипсульфокислоты.
б Соль ацетиламинонафталинсульфокпслоты с п-толуиди-
пом.
в n-Нитробензилпиридиниевая соль нафтиламиносульфо-
кие лоты.
г Диэтиламиновая соль нафтиламиносульфокислоты.
Аминогруппа в Н. с. легко замещается оксигруппой
при кислотном гидролизе. Таким способом получают
1-нафтол-3,8-дисульфокислоту из 1-нафтиламин-3,8-
дисульфокислоты, хромотроповую к-ту (1,8-диокси-
нафталин-3,6-дисульфокислоту) из Аш-кислоты и др.
Большое значение имеет реакция N-арилирования
Н. с., в частности для получения арилперикислот:
150-180’
HO3S
NHCeH6
фениплерикислога
Н. с. получают или сульфированием а- и fl-нафтила-
минов, или нагреванием нафтолсульфокислот с аммиа-
ком, или восстановлением нитронафталинсульфокис-
лот. Количественное определение Н. с. основано на
диазотировании. И. с. широко применяются в синтезе
красителей.
Лит. см. при ст. Нафталин. В. А. Пучков.
НАФТИЛАМИНЫ (аминонафталины) Ci0H7NH2,
мол. в. 143,18 — кристаллы, темнеющие при хране-
нии вследствие окисления; мало растворимы в воде;
хорошо растворяются в кислотах; перегоняются с во-
дяным паром. Практич. значение имеют также наф-
тилендиамины Cl0He(NH2)2, мол. в. 158,20. Констан-
ты 1(а)-и 2(Р)-нафтиламинов, 1,5- и 1,8-нафтилендиа-
минов и их характерных производных приведены
в таблице.
Изомер Т. пл., °C Т.кип., °C d nD Характерное про- изводное; т.пл.,°С
1(a) 50 300,8 1,1229 Zd25\ I. 25/ 1,1144 / 50\ K.d507 1,67034 (51°) пикрат, 181—182 ацетил, 1 60 бензоил, 161—162
2(₽) ИЗ 29 4 1 ,0614 (98°) 1,64927 (98°) пикрат, 195 ацетил, 134 бензоил, 162—163
1,5 189,5 — — — диацетил, 360 дибензоил, 3 50
1,8 66,5 — — — дибензоил, 311—312
Н.— типичные ароматич. амины. С минеральными
к-тами Н. образуют устойчивые соли; легко ацили-
руются, образуют N-алкильные замещенные, диазо-
тируются, вступают в реакции азосочетания. Амино-
группа в Н. может быть обменена на оксигруппу
(Бухерера реакция). Из реакций электрофильного
замещения практическое значение имеет только суль-
фирование. При сульфировании Р-Н. сульфогруппа
вступает в положения 1,5,6,7 и 8; при дальнейшем
сульфировании образуются важные 6,8- и 5,7-дисуль-
фокислоты Р-нафтиламина. Из а-Н. получают наф-
тионовую кислоту и 1-нафтиламин-2-сульфокислоту.
При нитровании аминогруппу защищают, напр.,
ацилированием. а-Н. получают восстановлением (в
том числе и каталитич. восстановлением водородом)
а-нитронафталина. Аналогично получают 1,5- и
1,8-нафтиламины из 1,5- и 1,8-динитронафталинов.
Р-Н. получают из Р-нафтола, обрабатывая его аммиа-
ком и сульфитом аммония. Р-Н. обладает канцеро-
генной активностью и вызывает рак мочевого пу-
зыря. Вследствие этого в Советском Союзе снят с
производства. Количественно Н. определяют диазо-
тированием. Н. широко применяют в синтезе краси-
телей.
Лит. см. при ст. Нафталин. В. А. Пучков.
НАФТИОНОВАЯ КИСЛОТА (1-нафтиламин-4-суль-
фокислота) С10НдОзМ8, мол. в. 223 — кристаллы,
иглы (с 0,5 Н2О), хо-
рошо растворяется в nh2 nh2h2so,
воде; растворы Н. к. (I)
и ее солей имеют сине- Г [ J Г I J
ватую флуоресценцию;
K=2,l -10-3 (25°). , so3h п
Н. к. вступает в ре-
акции электрофильного замещения, однако иногда
при этом сульфогруппа вытесняется другим замести-
телем. При бромировании Н. к. образуется сначала
1-амино-2-бромнафталин-4-сульфокислота, а при по-
вышении темп-ры реакции — 2,4-дибром-1-нафтил-
амин. При нитровании Н. к. азотной кислотой по-
лучается 2,4-динитро-1-нафтол. Нагревание Н. к. с
50% NaOH при 250° дает 1-нафтол-4-сульфокислоту;
последняя образуется из Н. к. также по Бухерера
реакции.
Н. к. получают нагреванием кислого сульфата
а-нафтиламина (II) до 160—180° (метод «запекания»),
Н. к. применяют как диазокомпонент и как азоком-
поненту; исключительное значение имеет в произ-ве
красителя конго-красного (индикатор).
Для идентификации Н. к. используют различные
функциональные производные, например пиридино-
вая соль N-ацетильного производного, т. пл. 175°;
n-толуидиновая соль N-ацетильного производного,
т. пл. 232—233°; диэтиламмониевая соль Н. к., т. пл.
221—222°; бензилтиурониевая соль Н. к., т. пл.
195,1°.
Лит. см. при ст. Нафталин. В. А. Пучков.
НАФТОЙНЫЕ КИСЛОТЫ (а-и Р-нафта линка рбо-
новые кислоты) СПН8О2, мол. в. 172, 1 — бесцветные
кристаллы, нераство-
римы в холодной и
умеренно растворимы
в горячей воде; хоро-
шо растворяются в
СООН
спирте и эфире. По а fi
своей силе Р-Н. к.
(А=6,78-10-5 при 25°) соответствует бензойной
к-те, а-Н. к. (7f=2,025-10-4) несколько сильнее по-
следней. Н. к. образует все возможные функциональ-
ные производные; константы Н. к. и нек-рых произ-
водных приведены в таблице (стр. 399).
При электрофильном замещении заместитель всту-
пает в свободное ядро Н. к. а-Нафтойная кислота хло-
399
НАФТОЛ АС —НАФТОЛЫ
400
Кислоты и их производные a Р
т. пл.,° С т. кип., °C /мм т. пл., °C т. кип., 9 С/мм
Нафтойные к-тыа 162(161) 231/50 184(185,5) ок. 300
Метиловый эфир 59,5 165/17 77 —
Этиловый эфир . . 310 171/13 32 178/12
Хлорангидрид . . 26 189/20 51 — 52 304—306
Амид 205 —— 200—202 —
Нитрил 38 229 68 304—305
а И. к. перегоняются без разложения.
рируется и бронируется в среде СН3СООН в положе-
ние 5; Р-Н. к. при бромировании в тех же условиях
дает смесь 5-бром- и 5,8-дибромпроизводных. Нитро-
группа при нитровании в среде СНзСООН в обоих
случаях (а- и Р~Н. к.) вступает в положения 5 и 8.
При сульфировании а-Н. к. конц. H2SO4 при 60—70°
образуется смесь 5-, 6- и 7-сульфо-1-нафтойных к-т.
Сульфирование Р-Н. к. 20%-ным олеумом при 10—
20° приводит к смеси 5- и 8-сульфопропзводных в соот-
ношении 80 : 20; нагревание Р-Н. к. с концентриро-
ванной H2SO4 при 160° дает 7-сульфо-2-нафтойную
кислоту.
а-Н. к. устойчива по отношению к щелочному ра-
створу перманганата, р-р хромовой кислоты в
СНзСООН окисляет ее во фталевый ангидрид. При
перегонке Са-соли а-Н. к. в смеси с формиатом каль-
ция (10%) образуется а-нафтальдегид.
Получают а-Н. к. окислением а-метил- пли а-хлор-
метилнафталинов, либоа-нафтплметплкетона; Р-Н.к.—
окислением Р-алкилнафталинов. Н.к. применяют в
синтезе красителей.
Лит. см. при ст. Нафталин. В. А. Пучков.
НАФТОЛ АС — то же, что азотол А.
НАФТОЛА СУЛЬФОКИСЛОТЫ — производные
нафталина, содержащие в молекуле одну илп не-
сколько оксигрупп и соответственно одну или не-
сколько сульфогрупп. Свойства Н. с. определяются на-
личием двух функциональных групп: ОН и SO3H.
Большинство моносульфокпслот а- и Р-нафтола (и их
соли) хорошо растворимо в воде; они более растворимы
в воде, чем соответствующие сульфокислоты нафта-
лина. Аналогично другим сульфокислотам ароматич.
ряда, Н. с. не имеют четких темп-р плавления; поэтому
их обычно идентифицируют в виде сульфохлорпдов,
Положение rnvnn Растворимость кис- лоты (НА) пли ее соли (МеА) в воде Т. пл. О-карб- этокси- сульфо- хлорида,°C Т. пл.других производных, °C
ОН SO,H
1 2 НА'0,5 Н2О — мало растворим, 2,7 е/100 г (18°) — О-ацетилсуль- фохлорид, 87,5
1 3 — 140 —
1 4 НА — хорошо раст- ворима Амид, 223 (217)
1 5 НА — то же 80 Амид, 263
1 6 — 112 —
1 7 НА и МеА — хорошо растворимы 105 —
2 1 НА и МеА — то же — Сульфохло- рид, 124
6 NaA-2H2O — мало растворима, 1,73 г/100 г — Соль с ди- этиламином 195—196
2 7 НА и МеА — хоро- шо растворимы — 2-Этоксисуль- фохлорид, 1 03
2 8 МеА и МеА2 — то же — 2-Этоксисуль- фохлорид, 93
амидов, солей с аминами и тиомочевиной, в виде карб-
этоксинафтолсульфохлоридов и других производ-
ных. В таблице приведены температуры плавле-
ния наиболее характерных производных моносульфо-
кислот а- п Р-нафтола и данные об их растворимости
в воде.
Для идентификации Н. с. может быть использована
также реакция азосочетания с различными диазосо-
единенпями; азосочетанпе легко протекает в щелочной
среде. Эта реакция имеет технич. значение для получе-
ния многих ценных красителей. И. с. можно разделить
хроматографически в системе н-С^НдОН : СНзСООН:
: НзО=4 : 1 : 5. В пром-сти отдельные изомеры выде-
ляют «высаливанием» из раствора, используя различ-
ную растворимость их солей.
Сульфогруппа в Н.с. может быть обменена на окси-
группу методом щелочного плавления; эта реакция
используется для получения полиоксинафталинов.
Исключительное технич. значение имеет реакция ами-
нирования Н. с. (см. Бухерера реакция). При нагрева-
нии, напр., 2-пафтол-1-сульфокислоты с р-ром аммиака
в присутствии сульфита аммония при 150° оксигруппа
замещается аминогруппой и получается 2-нафтил-
амин-1-сульфокислота. Аналогично получают 2-наф-
тиламин-6,8-дисульфокислоту из 2-нафтол-6,8-дисуль-
фокислоты:
NH3,(NHa)2SO3
185’
Сульфогруппа, находящаяся в мета-положении к
любому гидроксилу, защищает его от аминирования
также, как сульфогруппа в орто-положенип по отно-
шению только к а-гидроксилу. Это обстоятельство
позволяет в диокспнафталипсульфокислотах обмени-
вать на аминогруппу только один гидроксил:
Оксигруппа в Н. с. может быть замещена также арн-
ламипогруппой. При нагревании (190—200°) Н. с. с
первичными ароматич. аминами сначала получаются
сульфокислоты арилированных нафтиламинов, к-рые
при дальнейшем нагревании теряют сульфогруппу и
переходят в арилнафтиламины:
CsHsNH,- KV
-нго
h2O
•HaSO,
NHCeH5
H. с. получают: сульфированием а-или (3-нафтолов;
замещением сульфогруппы оксигруппой в ди- и поли-
сульфокислотах нафталина; из нафтиламинсульфокис-
лот бисульфитной реакцией илп другим методом; гид-
ролизом хлорнафталинсульфокислот; элиминирова-
нием одной a-сульфогруппы из нафтолди- и нафтол-
полисульфокислот.
Н. с. широко применяют в синтезе промежуточных
продуктов и красителей.
Лит. см. при ст. Нафталин. В. А. Пучков.
НАФТОЛКАРБОНОВЫЕ КИСЛОТЫ — см. Ок-
синафтойные кислоты.
НАФТОЛЫ (оксинафталины), соединения общей
ф-лы С10Н8_„(ОН)„, гдеп=1, 2, 3 и более. Н.— бес-
цветные кристаллы с характерным запахом; нераство-
римы в разб. к-тах, умеренно растворимы в воде,
401
НАФТОЛЫ—НАФТОХИНОНЫ
402
хорошо — в щелочах и органич. растворителях;
перегоняются с водяным паром. При нагревании Н.
возгоняются. Константы а- и р-нафтолов и 1,5-,
1,8-, 2,3- и 2,7-диоксинафталинов, имеющих технич.
значение, приведены в таблице.
Изомер Т.пл., °C Т. кип., °C Характерное произ- водное; т.пл., °C
а (1) 96,1 278—280 ацетил, 48—49 бензоил, 56 пикрат, 189
₽ (2) 123 285—286 ацетил, 7 0 бензоил, 107 пикрат, 157
1,5 265 — диацетил, 159 — 160 дибензоил, 235
1.8 140 диапетил, 147—148
2.3 159 — ——
2.7 190 — —
По свойствам Н. очень близки к фенолам бензоль-
ного ряда. Однако гидроксильная группа Н. вступает
в реакции легче, чем фенольный гидроксил. Н. легко
ацилируются, этерифицируются, вступают в реакцию
азосочетания, при этом образуются азокрасители.
При действии HNOa получаются нитрозозамещенные,
напр. р-Н. дает 1-нитрозо-2-нафтол. Н. и особенно их
нитрозо- и азопроизводные способны реагировать
в таутомерной кетоиной форме:
енольная форма
кетонная форма
Напр., нитрозо- и азопроизводные а- и Р-иафтолов
образуют бисульфитные соединения, реагируя при
этом в хиноноксимной и хинонгидр азонной формах:
NO NOH NOH
Н SO3Na
Присоединение бисульфита, в отличие от обычных
кетонов, происходит по С—С-связи. Многие азокра-
сители, производные Н., представляют собой смесь
азосоединения (I), соответствующего енольной фор-
ме, и гидразона (II), соответствующего кетоиной
форме Н.:
Положение таутомерного равновесия зависит от заме-
стителя в дпазосоставляющей и от растворителя и
может быть определено спектральными методами. Н.
легко обменивают оксигруппу на аминогруппу в при-
сутствии бисульфита (см. Бухерера реакция). В пром-сти
таким способом из Р~Н. получают Р-нафтиламин.
Н., так же как и фенолы, легко карбоксилируютсн
(см. Кольбе—Шмидта реакция), что имеет большое зна-
чение длн получения окспнафтойных к-т. Легче, чем
незамещенный нафталин, II. вступают в реакцию
электрофильного замещения. При сульфировании
Р~Н. в зависимости от условий образуются 1-, 6-
и 8-моносульфокислоты, 3,6- и 6,8-дисульфокислоты
2-нафтола. Сульфирование а-Н. дает 4-моносульфо-
кислоту и 2,4-дисульфокислоту. Аналогично при
нитровании а-Н. образуется 2,4-динитро-1-нафтол.
а-Н. и Р-Н. при действии FeCl3 или окисей Fe и Си
переходят в соответствующие диоксидинафтилы:
а-Н. и р-Н. получают в пром-сти щелочным плавле-
нием соответствующих сульфокислот нафталина при
280—320°.
Н. и их производные имеют исключительно важное
значение для синтеза красителей. Метиловый эфир
Р-H., неролин и этиловый эфир Р-Н.— новый неролип—
используют в парфюмерной пром-сти. Нитрозозаме-
щенные Н. применяют в аналитич. химии для откры-
тия железа и др. катиопов.
Лит. см. при ст. Нафталин.
В. А. Пучков.
НАФТОСТИРИЛ (лактам 8-амино-1-нафтойной кис?
лоты) CuH7ON, мол. в. 169 — кристаллы, желтые
иглы (после сублимации); т. пл. 180—
181°; трудно растворим в кипящей воде н _____£
и эфире. Разб. р-ры в органич. раство- i । у
рптелях имеют зеленую флуоресценцию.
Растворяется при нагревании в соляной ц.. II
и разб. серной к-тах без изменения. При
кипячении со щелочами Н. переходит в соль 8-амино-
нафталин-1-карбоновой к-ты.
При нитровании Н. азотной к-той в уксусной к-те
образуется 6-нитронафтостирил; при нагревании с
хлорной водой образуется дихлорзамещенное, веро-
ятно, 5,7-дихлорнафтостирил. Н. с хлористым бен-
зоилом и А1С13 дает 6-бензоилнафтостирил; при за-
мыкании последнего в расплавленной смеси А1С13-|-
+ NaCl образуется производное бензантрона — лак-
там 4-аминобензантрон-З-карбо-
новой к-ты (I). При метилиро- О
вании Н. диметилсульфатом или
CeH5SO3CH3 образуется N-метил- |
нафтостирил. При действии пя-
тисернистого фосфора на Н. и [ _ | |Г |
N-метилнафтостирил получены
тиопафтостирил и N-метилтио- q
нафтостирил. Тионафтостирил,
аналогично тионафтену, вступает в конденсацию
с соединениями, имеющими активную метиленовую
группу, с образованием окрашенных веществ от
желтого до синего цветов.
Н. получают по Лейка рта реакции из 1-нафтилизо-
цианата нагреванием в смеси AlCl3-|-NaCl при 155—
160°. Н.— промежуточный продукт в синтезе поли-
циклич. красителей.
Лит.: Докунихин Н. С., Новые реакции арилизо-
цианатов, арилизотиоцианатов и некоторых продуктов их
превращения, М,, 1958 (Докт. дисс.). См. также лит. при
ст. Нафталин. В. А. Пучков.
РНАФТОХИНОЛИН Ci3H9N, мол. в. 179,22— бе-
лые мелкие кристаллы; т. пл. 93—94°; мало растворим
в воде, хорошо — в спирте, эфире, бен-
[I4] золе, растворяется в разб. минеральных
L Jx. к-тах. Получают р-Н. взаимодействием
f ]| 1 р-нафтиламина с глицерином в присут-
ствии H2SO4 и нитробензола. Приме-
няют как реагент для обнаружения и
осаждения Zn, Bi, Cd, Си, Hg, W и др. и. П. Ефимов.
НАФТОХИНОНЫ С1оН6О2, мол. вес 158,14—
хиноны нафталинового ряда.
403
НАФТОХИНОНЫ—НЕДООКИСЬ УГЛЕРОДА
404
а-Н афтохинон, 1,4-нафтохинон (I) — желтые
иглы, т. пл. 128,5°; легко летуч с водяным паром,
сублимируется при нагревании;' трудно растворим.
в воде (0,35%), легко растворяется в органич. раство-
рителях. Оксим (таутомерен с 4-нитрозо-1-нафтолом),
т. пл. 198°; диоксим, т. пл. 207° (с разл.). а-Н. более
энергично, чем n-бензохинон, вступает в большинство
реакций. В щелочных р-рах под действием воздуха
а-Н. окисляется в 2-окси-1,4-нафтохинон; при действии
Н2О2 образуется 2,3-эпоксид 1,4-нафтохинона (IV).
Более сильные окислители (КМпО«,
HNO3) превращают а-Н. во фталевую
кислоту. а-Н. присоединяет галогены
(С1г и Вгг в кипящей СНзСООН) с обра-
зованием 2,3-дигалоген-2,3-дигидро-1,4-
нафтохинонов; в присутствии иода по-
лучаются 2,3-дигалоген-1,4-нафтохино-
ны. При нагревании а-Н. с анилином образует-
ся 2-анилин-1,4-нафтохинон, а при действии азида
натрия получается 2-амино-1,4-нафтохинон. а-Н. легко
вступает в Дильса— Альдера реакцию. С замещенными
1,3-бут адиенами а-Н. образует производные антра-
хинона. При восстановлении а-Н. (напр., Na2S2O«)
образуется 1,4-диоксинафталин. а-Н. является окис-
лителем. Характер заместителя в ^-положении
1,4-наф тохинона оказывает значительное влияние на
величину окислительно-восстановительного потен-
циала (см. табл.).
Заместитель в ^-положении Ео, мв Заместитель в 0-положении Ео, Мв
NHCH3 —252 NHCOCH3 -67
NH2 —210 с,.и« -32
N (СН»)2 — 181 ОСОСНз —9
ОН — 128 С1 + 24
оснэ -131 SOsNa + 69
СН3 . —76 SO2CeH,CH3(n) . . . + 121
а-Н. получаются окислением нафталина хромовой
к-той или, еще легче, окислением 1,4-диокси- и 1-окси-
4-аминонафталинов; является побочным продуктом
при получении фталевого ангидрида. В природе
встречаются отдельные производные а-Н. К ннм от-
носится юг л он (5-окси-1,4-нафтохинон), выделяе-
мый из незрелых плодов грецкого ореха; л а у с о н
(2-окси-1,4-нафтохинон) — красящее вещество листьев
алканны и др. Биологич. активностью обладают ал-
кильные и алкенильные замещенные а-Н. (вещества
группы витамина К). а-Н.— промежуточный продукт
в синтезе красителей.
р-Н афтохинон, 1,2-нафтохинон — светло-жел-
тые или оранжево-красные иглы; неустойчив в водных
р-рах; с водяным паром не перегоняется. 2-Оксим
(таутомерен с 2-нитрозо-1-нафтолом), т. пл. 162—164°
(с разл.); диоксим, т. пл. 169°. При действии Н2О2,
КМпО« пли НгСгО« р-Н. окисляется в 2-окси-1,4-
нафтохинон и далее в о-карбоксикоричную, фталоно-
вую и фталевую к-ты; в водном р-ре ЕеС1з окисляет
в 3-окси-2,2'-динафтил-1,4,3',4'-дихинон. При восста-
новлении (Na2S2O4) р-Н. превращается в 1,2-диокси-
нафталин. р-Н. нитруется HNO3 в 3-нитро-1,2-нафто-
хинон. При хлорировании в СНзСООН сначала обра-
зуется 3,4-дихлор-3,4-дигидро-1,2-нафтохинон, к-рый
затем отщепляет НС1 и превращается в З-хлор-1,2-
нафтохинон. р-Н. присоединяет бисульфит с образо-
ванием 1,2-диоксинафталин-4-сульфокислоты, имею-
щей технич. значение. Р-Н. получают из 1-амино-2-
нафтола при окислении бихроматом; применяют как
полупродукт в синтезе красителей.
Амфи-нафтохинон, 2,6-нафтохинон —
очень неустойчивое соединение; более сильный окис-
литель, чем а-Н. и р-Н. Получают окисление 2,6-м
диоксинафталина перекисью свинца в кипящем бен-
золе.
Лит. см. при ст. Нафталин. В. А. Пучков.
НЕБЕНЗОЛЬНЫЕ АРОМАТИЧЕСКИЕ СИСТЕ-
МЫ — см. Ароматические системы.
НЕГАТИВ — фотография, изображение, в к-ром
соотношение яркостей различных участков обратно
соотношению яркостей соответствующих участков
объекта. Н. получают при проявлении скрытого фото-
графия. изображения, образовавшегося в светочув-
ствительном слое в процессе съемки. Так как ступени
яркости объекта передаются в фотография, изображе-
нии ступенями потемнения, то в результате первичного
фотохимия, и последующего химич. процесса на месте
действия света и проявителя происходит образование
фотография, потемнения. Поэтому получаемое вместо
скрытого изображения видимое фотографическое яв-
ляется обратным по распределению света и тени, т. е.
негативным. Кроме серебра, веществами, образую-
щими Н. на различных фотография, материалах, могут
служить: азокрасители — в диазотипии; гексациано-
ферриат окисного железа — в цианотипии; азометино-
вые и индоанилиновые красители — в цветной фо-
тографии.
Н. применяют гл. обр. для печати позитивов.
Непосредственно негативные изображения используют
в астрономии, рентгенографии, диазотипии, циано-
типии. В кинематографии для печатания позитивных
копий кинофильмов применяют дубль-негативы (кон-
тратипы), получаемые печатью с промежуточного
позитива. В цветной фотографии зональные цвето-
деленные Н. в трех слоях пленки получают из азоме-
тиновых и индоанилиновых красителей, образую-
щихся из первичных продуктов окисления проявля-
ющего вещества и компонентов цветного проявления.
В фотографии, отношении Н. характеризуется
плотностью, т. е. поверхностной концентрацией ве-
щества, образующего потемнение, а также контраст-
ностью. Эти характеристики Н. определяются фото-
графии. свойствами светочувствительного слоя, ве-
личиной экспозиции и условиями проявления.
Лит.: Н е б л и т К. Б,, Фотография, ее материалы и про-
цессы, пер. с англ., М., 1958. В. С.Челъцов.
НЕГАТИВНЫЙ ПРОЦЕСС — см. Фотография.
НЕДООКИСЬ УГЛЕРОДА (ангидридмалоновойк-ты,
диоксоаллен) О=С=С=С=О, мол. в. 68,03—бесцвет-
ный газ с резким неприятным запахом; т. пл. минус
108—107°; т. кип. 7°. Выше 15° Н. у. образует твердый
темно-красный полимер. Разложение Н. у. начинается
уже при 37°, а при 100° происходит бурно; продукты
термич. диссоциации окрашены в интенсивно красный
цвет. На воздухе Н. у. горит синим пламенем с выде-
лением копоти. Н. у.— один из низших окислов угле-
рода; молекула ее построена линейно. По своей струк-
туре Н. у. принадлежит к классу кетенов, что и опре-
деляет ее химич. свойства. Так, при взаимодействии
Н. у. с соединениями, содержащими активный атом
водорода, образуются производные малоновой к-ты.
ох ,о
О=С=С=С=О+2НХ—> 7С—сн2-с<
Xх хх
Х=ОН, OR, NH2, NHR, NRR', Cl
С водой H. у. образует малоновую к-ту.
Получают Н. у. пиролизом ангидрида диацетилвин-
ной к-ты или этилового эфира щавелевоуксусвой
к-ты: о
СНзСОО-СН-С^
/° П
О = С-СООС3Н5
I
СН3-СООСаН5
405
НЕЗАМЕНИМЫЕ АМИНОКИСЛОТЫ —НЕЙРАМИНИДАЗЫ
406
Н. у. можно также получить перегонкой малоновой
к-ты или ее эфиров в вакууме над пятиокисью фос-
фора:
.соон р3о5
сн3< -------►
ХСООН 150°
О=С=С=С=О
и действием окислов металлов (Al, Zn, РЬ) на хлоран-
гидрид малоновой к-ты или цинка на бромангидрид
диброммалоновой к-ты:
СЩ (СОС1).-—|
—У О=С=С=С=О
СВг2(СОВг)2-^-1
Н. у. образуется из окиси углерода в тихом разряде:
4СО—>С3О3 + СО2
Н. у. сильно раздражает слизистые оболочки, ядо-
вита.
Лит.: Chemistry of carbon compounds, ed. E. H. Rodd,
v. 1, pt B, Amst.—L.—N. Y., 1952; Mellor, v. 5, L., 1946,
там же, 1952. Б. Л. Дяткин.
НЕЗАМЕНИМЫЕ АМИНОКИСЛОТЫ — амино-
кислоты, содержание к-рых в пище необходимо для
нормального роста, развития и для поддержания
нормального физиологич. состояния животных и неко-
торых микроорганизмов. Аминокислоты, к-рые могут
к.-л. путями синтезироваться в организме, наз. за-
менимыми аминокислотами. Основным источником
аминокислот в пище являются белки, к-рые расщеп-
ляются в желудочно-кишечном тракте до аминокислот.
Белки, в состав к-рых входит набор всех Н. а., наз.
полноценными; белки, к-рые не содержат
хотя бы одну из незаменимых аминокислот, являются
неполноценными. Богаты Н. а. животные
белки (молоко, мясо). Резкой границы между Н. а.
и заменимыми аминокислотами провести нельзя.
Потребность в определенных аминокислотах зависит
от вида животного или микроорганизма, от присутст-
вия в пище других факторов, напр. витаминов, ами-
нокислот-антагонистов, от физиологич. состояния
организма, от возраста животного и т. д. Ту или иную
аминокислоту можно отнести и к заменимым и к не-
заменимым в зависимости от того, рассматривать ли
ее с точки зрения необходимости для роста или для
поддержания баланса азота в организме и т. д. Пот-
ребности в Н. а- у человека и различных видов жи-
вотных различны. Восемь аминокислот: лизин, трео-
нин, триптофан, метионин, фенилаланин, лейцин,
валин и изолейцин — являются незаменимыми для
человека и всех видов животных. Для роста молодых
крыс, кроме того, необходим еще аргинин; для
роста цыплят необходимо 15 аминокислот. Суточная
потребность вН. а. для человека в среднем составляет
(в г): 0,15—0,25 триптофана; 0,8—1,1 фенилаланина;
0,4—0,8 лизина; 0,3—0,5 треонина; 0,4—0,8 валина;
0,8—1,1 метионина; 0,5—1,1 лейцина; 0,65—0,7 изо-
лейцина. Потребность в аминокислотах возрастает
при беременности, в период лактации, при нек-рых
заболеваниях и травмах, когда дополнительное коли-
чество аминокислот нужно для синтеза белков тканей,
крови, антител и т. д. Отсутствие или недостаток
Н. а. в пище может привести к остановке роста, потере
веса, нарушению баланса азота; при отсутствии в пище
отдельных Н. а. могут развиться нек-рые заболева-
ния, напр. при отсутствии триптофана развивается
катаракта.
Потребность в аминокислотах у микроорганизмов
различна: нек-рые виды, напр. кишечная палочка,
могут синтезировать все широко распространенные
аминокислоты, для других микроорганизмов необхо-
дим ряд аминокислот, напр. для нормального развития
культуры Leuconostoc необходимо наличие в культу-
ральной среде 18 аминокислот.
Лит.: Майстер А., Биохимия аминокислот, пер. с
англ., М., 1961; Болдуин Э., Основы динамической био-
химии, пер. с англ., М., 1949; G г е е n s t е 1 n J. Р., W 1-
nltzM., Chemistry of the amino acids, v. 1, N. Y.—L., 1961,
p. 245. См. также лит. при ст. Аминокислоты и Белки.
Л. И. Линееич.
НЕЗАМЕНИМЫЕ ЖИРНЫЕ КИСЛОТЫ (вита-
мин F) — группа ненасыщенных органич. жирных
к-т, к к-рым относятся: арахидоновая кис-
лота (5-цис, 8-цис, 11-цис, 14-цис-эйкозатетраеновая
кислота) СНз(СН2)3(СН2СН=СН)4(СН2)зСООН, мол. в.
304,48, линолевая кислота п линоленовая кислота.
Физич. свойства этих кислот приведены в таблице.
Кислота Т.пл., °C Т. кип., °C/мм d2» 4 п20 " D Иодное число
Арахидоновая Линолевая Линоленовая -49 5 -5.2 -И 160—165/1 149,5/1; 182,4/4 184/4; 230/17 0,903 0,914 1 ,4699 1,4780 333,5 181,0 273,5
Для Н. ж. к? характерна способность к легкому
окислению, к-рая возрастает с увеличением количе-
ства двойных связей; под влиянием щелочей происхо-
дит изомеризация с превращением цис-формы в
транс-форму. Н. ж. к. легко гидрируются и присо-
единяют галогены.
Линолевую и линоленовую к-ты выделяют из жир-
ных к-т, полученных из подсолнечного, льняного,
макового, хлопкового, табачного или других масел
кристаллизацией из ацетона или петролейного эфира
при низких темп-рах. Арахидоновую к-ту выделяют
из лецитина печени свиньи.
Н. ж. к. не синтезируются в животном организме
и поступают в организм с пищей; они не могут быть
заменены другими жирными к-тами. Линолевая и ли-
ноленовая к-ты способны в организме переходить в
арахидоновую. Н. ж. к. находятся в фосфолипидах
тканей животных и человека в составе лецитина пе-
чени, кефалина мозга и др. Жир человека содержит
8,2—11% линолевой к-ты и 0,3—1% арахидоновой
к-ты (до отношению ко всем жирным к-там). Биока-
талитич. функции Н. ж. к. изучены мало. Н. ж. к. пре-
дупреждают и излечивают дерматиты. Наиболее ак-
тивна арахидоновая к-та; она в 10 раз активнее ли-
нолевой и линоленовой к-т.
Лит.: Березовский В. М., Химия витаминов, М.,
1959. В. М. Березовский.
НЕЙРАМИНИДАЗЫ (сиалидазы) — ферменты, рас-
щепляющие а-кетозидную связь, образуемую нейра-
миновой кислотой в олигосахаридах, полисахаридах
и углеводных компонентах сложных белков. Нейра-
минидазная активность обнаружена у нек-рых вирусов
(гриппа, ныокастльской болезни и др.), в холерном
вибрионе, пневмококках и др. патогенных микроор-
ганизмах, а также в животных организмах. Н. вирус-
ного происхождения не выделена, но из вирусных ча-
стиц получены субъединицы, обладающие нейтрами
нидазной активностью. Из холерного вибриона Н,
выделена в виде кристаллич. белка с мол. в. 10000—
20000. Нейраминидазная активность проявляется при
действии патогенных микроорганизмов на клетки
животного организма (эритроциты).
Н. используют для установления строения белковых
веществ и сложных липидов, в состав к-рых входит
нейраминовая к-та, для получения сиаловых к-т из
природных веществ, в гистохимия, исследованиях.
Нейраминидазная активность используется как пока-
затель синтеза специфич. белка вирусов в живой клет-
ке. Для измерения нейрамидазной активности опре-
деляют скорость отщепления сиаловой к-ты от три-
сахарида молока — нейраминиллактозы. Н. играет,
по-видимому, большую роль в процессах обмена
407
НЕЙРАМИНОВЫЕ КИСЛОТЫ —НЕЙТРИНО
408
мукопротеинов и муколипидов, содержащих сиаловую
кислоту.
Лит.: ТоварницкийВ. И., в кн.: Актуальные воп-
росы вирусологии, М., 1960 (Итоги науки. Биологические
науки, 4). с. t29; Gottschalk A., Advances in enzymo-
logy, 1958, 20,135; Nol 1 Н., Aoyagi Т„ Orlando J„
Virology, 1961, 14, № 1, 141; см. также лит. при ст. Нейрами-
новые кислоты. Л. И. Линевич.
НЕЙРАМИНОВЫЕ КИСЛОТЫ (нонулозаминовые
кислоты, а-кето-б-аминопентаоксикарбоновые кисло-
ты, аминокарбоксиоктулозы)
HOOC-CO-CH2-CHOH-CH(NH2)-(CHOH)3-CH3OH
мол. в. 267,234—полифункциональные соединения, со-
держащие 9 углеродных атомов в цепи и функциональ-
ные группы углеводов (кетоз) и аминокислот. Теорети-
чески, вследствие наличия 5 асимметрия, атомов в мо-
лекуле, возможно существование 32 стереоизомерных
Н. к., но до настоящего времени доказана конфигура-
ция атомов лишь одной Н. к., к-рую рассматривают
как продукт альдольной конденсации пировиноградной
к-ты и D-маннозамина, образующий открытую и
циклич. таутомерные формы:
СООН СООН
со со он
сн3 1 сн2 н
+ но/ 1\Н
1 - ун нМ
сно носн нИ
h2nch h2nch Н0000_ у NHa
носн носн НСОН
НСОН НСОН НСОН 1
НСОН неон СН2ОН
СН2ОН снгон формула Хеуорса
Эта Н. к. в виде ацилированных производных входит
в состав многих биологически активных мукополиса-
харидов, мукопротеинов, муколипидов (ганглиозидов),
нуклеотидов и встречается во всех органах и тканях
животных (мышцы, почки, печень, мозг, соединитель-
ные ткани, кровь и др.) и в нек-рых микроорганизмах
(кишечная палочка и др.). При отщеплении Н. к. от
нек-рых биологически активных веществ (гонадотроп-
ных гормонов, эритропоетина — белка, стимулирую-
щего кроветворение, — ингибиторов агглютинации
крови при действии вирусов гриппа и др.) биология,
активность этих веществ утрачивается. При нек-рых
патология, состояниях (рак, туберкулез, психич.
заболевания) содержание Н. к. в жидкостях и тканях
организма резко возрастает. Н. к. принимает какое-то
участие во взаимодействии организма животных с ви-
русами (напр., гриппа, ньюкастльской болезни).
Н. к.— нестойкие соединения, в свободном виде не
выделены; они получены в виде кристаллич. производ-
ных: метилгликозида (метоксинейрамоновой к-ты) и
различных ацилпроизводных, называемых сиаловыми
кислотами.
Н. к. дают реакции восстанавливающих сахаров,
дезоксисахаров, на амино- и карбоксильную группы,
реакции а-кетокарбоновых к-т и др. Связь Н. к. с
другими компонентами в сложных природных соеди-
нениях осуществляется обычно за счет а-кетозидной
или карбоксильной группы (сложноэфирная или амид-
ная связи). Для обнаружения и количественного опре-
деления Н. к. используют прямую реакцию Эрлиха
(с л-диметиламинобензальдегидом, без предваритель-
ной обработки щелочью), реакцию с орсином, цветные
реакции с резорцином, дифениламином, триптофаном
и хлорной к-той, с тиобарбитуровой к-той. Этими же
реакциями пользуются для выявления Н. к. на хро-
матограммах.
Получают Н. к. в виде метилгликозида и сиаловых
кислот мягким кислотным или ферментативным гидро-
лизом сложных природных соединений (оросомукоид
крови, овомуцин яйца, трисахарид молока — нейра-
миниллактоза, ганглиозиды и др.). Ферменты, отщеп-
ляющие Н. к., наз. нейраминидазами. Н. к. (ацили-
рованная) получена также синтетически из N-ацетил-
D-глюкозамина и щавелевоуксусной к-ты (при кон-
денсации в щелочной среде идет изомеризация D-глю-
козамина в D-маннозамин) и путем биосинтеза из
пировиноградной кислоты и ацилированного D-ман-
нозамина под влиянием фермента альдолазы
нейраминовой кислоты, или из фосфо-
пировиноградной кислоты и Ы-ацетил-В-маннозами-
на под влиянием специфической ферментативной
системы.
Лит.: Бычков С. М., Успехи соврем, биол., 1960, 49,
вып. 1, 3; Цветкова И. Б., Вопросы мед. химии, 1961,
7, вып. 1, 3; Л и н е в и ч Л. И., Успехи биол. химии, 1962,
4,193; Gottschalk A.,The chemistry and biology of sialic
acids and related substances, Camb., I960; Zil liken F.,
Whitehouse M. W., Advances in carbohydr. Chem., 1958,
13, 237: Whitehouse M. W., 2 i 11 1 k e n F., в кн.: Me-
thods of biochemical analysis, v. 8, L., [1960], p. 199.
Л. И. Линевич.
НЕЙРИН (гидроокись винилтриметиламмония)
(СНз)зМ(ОН)СН=СН2, мол. в. 103,16 — бесцветная
сиропообразная жидкость; прп упаривании в высоком
вакууме образует гигроскопичные кристаллы, к-рые
разлагаются на триметиламин и ацетальдегид. Н. ра-
створим в воде, спирте и эфире; сильное основание, с
кислотами образует соли: хлорид, т. разл. 213°; бро-
мид, т. пл. 193°, и др.
Н. устойчив в разб. водных р-рах, при перегонке
разлагается с образованием диметилвиниламина, а
также триметиламина и ацетальдегида; присоединяет
Вг2 с образованием бромистого триметил(а, Р-дибром-
этил)аммония. С H2SO4 Н. образует тауробетаин
(CH3),nch,ch2so2o, осаждается из водных р-ров фос-
форномолибденовой, фосфорновольфрамовой и пик-
риновой к-тами, хлористой платиной, HgJ2, J2-KJ
и др. реактивами на алкалоиды, что может быть ис-
пользовано для определения Н.
Н. очень ядовит, по своему действию на животный
организм близок к кураре и мускарину. Смертельная
доза Н. для кроликов или собак 0,04—0,05 г/кг (под-
кожно); у человека уже небольшие дозы Н. вызывают
тошноту, головокружение, усиленное отделение пота
и замедление сердцебиения.
Н. может быть получен из бромистого триметил-
(Р-бромэтил)аммония или йодистого триметил-
ф-иодэтил) аммония обработкой их Ag2O или кипяче-
нием со щелочью.
В природе Н. образуется из холина, гл. обр. в резуль-
тате деятельности гнилостных, патогенных или кишеч-
ных бактерий при различных процессах гниения азо-
тистых веществ, в частности в процессе трупного
разложения. В небольших количествах Н. обнаружен
также в надпочечниках и моче здоровых животных
и людей.
Лит.: Guggenheim М., Die biogenen Amine, 4 Aufl.,
Basel —N V., 1951. В. Б. Спиричев.
НЕЙТРИНО — электрически нейтральная эле-
ментарная частица с массой, равной нулю,
и спином У2& (fi=h/2x, где h — постоянная Планка);
обозначается у. Античастицей Н. является антиней-
трино (v). Название дано от итал. neutrino, уменьшит,
от neutrone — нейтрон. При различных процессах
распада элементарных частиц (в том числе при |3-рас-
паде связанных в ядра нуклонов) Н. испускается в
паре с позитроном или ц+-мезоном, антинейтрино —
в паре с электроном или ц“-мезоном (напр, п-г-р+е--}-
4-ve; л ++ Экспериментально доказано, что
Н., сопровождающие процессы с участием электронов
и позитронов (равно, как и соответствующие анти-
нейтрино), нетождественны Н., фигурирующим в про-
409
НЕЙТРОН т-НЕЙТР.ОНОГРАФИЯ
410
цессах с участием р.+- и ц_-мезонов. Т. обр,, существует
два вида Н. и антинейтрино — электронные (уе и v4)
и ц-мезонные (v и v ).
Все распады с испусканием v или v принадлежат
к числу так. наз. слабых взаимодействий. В соответ-
ствии с этим Н.— полностью продольно поляризован-
ные частицы — спин v всегда направлен против, а V —
по направлению его движения. Теоретически суще-
ствование Н. (и антинейтрино) было предсказано еще
в 1931, но прямое наблюдение обусловленного дей-
ствием v процесса: v4-p-*n-|-e + удалось осуществить
лишь в 1955. Нейтрино и антинейтрино чрезвычайно
слабо взаимодействуют с веществом и потому обладают
необычайно высокой проникающей способностью —
длина свободного пробега Н. с энергией в несколько
млн. ае сравнима с радиусом известной части Все-
ленной. На этом свойстве Н. основана возможность
будущего развития «нейтринной астрономии», т. е.
получения ценной космогонич. информации путем
регистрации и изучения свойств приходящих к нам
из Космоса Н. и антинейтрино. См. также Элементар-
ные частицы. В- В. Голъданский.
НЕЙТРОН — электрически нейтральная элемен-
тарная частица с массой 1838,6 масс электрона; обо-
значается и. Открыт в 1932 Дж. Чедвиком. Назван
от лат. neuter — ни тот, ни другой. Свободный Н.
нестабилен и испытывает Р--распад: n-*p-|-e_-]-v;
период полураспада Т\2 = 11,7 мин. Наряду с про-
то нами Н. входят в состав всех атомных ядер. Про-
тоны и Н. носят общее название нуклоны. Спин Н.
равен i/Д (где А.=Л/2л; h — постоянная Планка).
Магнитный момент Н.=—1,9131 ядерных магнетонов.
Благодаря отсутствию электрич. заряда Н. свободно
проникают в любые ядра, вызывая всевозможные ядер-
пые реакции. Особенно эффективны в этом отношении
медленные — в частности, так наз. тепловые Н. с
энергиями порядка k Т (где к — постоянная Больц-
мана, Т — абс. темп-ра; при комнатной темп-ре /;Т=^
^1/40 эв). Очень важным в практич. отношении про-
цессом является деление тяжелых ядер под действием
Н. Поскольку при таком делении в ряде случаев
испускается в среднем от двух до трех Н., возникает
возможность осуществления цепной ядерной реакции
деления — источника ядерной энергии. Наряду с
рентгеновскими лучами Н. используются для струк-
турного анализа молекул (см. Нейтронография),
при помощи Н. изучают также магнитные свойства
вещества, энергетич. уровни твердого тела. Основным
источником медленных Н. служат ядерные реакторы.
Быстрые Н. получают посредством ядерных реакций
под действием у-лучей или заряженных частиц,
испускаемых радиоактивными препаратами или раз-
личными ускорителями. Античастицей Н. является
антинейтрон. См. также Элементарные частицы.
В. И. Голъданский.
НЕЙТРОНОГРАФИЯ — метод изучения структуры
молекул, кристаллов и жидкостей при помощи диф-
ракции нейтронов; имеет много общего с рентгено-
графией. Дифракция нейтронов — ти-
пичное оптич. явление, аналогичное дифракции рент-
геновских лучей, в к-ром ярко проявляются волновые
свойства нейтрона (согласно известному соотношению
де-Бройля, движение частицы с массой т и энергией Е
можно рассматривать как движение волны с длиной
k=h/V2^E~, где h—постоянная Планка). Явление
дифракции характерно для очень медленных нейтро-
нов — с энергиями 0,1—0,01 эе (тепловые нейтроны),
поскольку длина волны этих нейтронов имеет порядок
межатомных расстояний в веществе (А). Дифракция
нейтронов была впервые продемонстрирована в 1936 в
опыте Митчелла — Пауэрса с помощью прибора, схе-
матически показанного на рис. 1.
Радиево-бериллиевый источник нейтронов 1 окружен па-
рафином 2 для получения тепловых нейтронов [в результате
замедления быстрых нейтронов, возникающих в реакции
Ве» (а, п)С12]. Прибор имеет вид цилиндра 3, по внутренней
поверхности к-рого расположено 60 монокристаллов MgO,
Рис. 1. Схема прибора Митчелла — Пауэрса для
демонстрации дифракции нейтронов: 1 — радие-
во-бериллиевый источник нейтронов; 2—пара-
фионовый замедлитель нейтронов; 3 — цилин-
дрическая часть прибора с расположенными
по периферии монокристаллами MgO; 4 — счет-
чик нейтронов.
ориентированных специальным образом. Отражение нейтрон-
ного пучка от монокристаллов при угле скольжения 22° соот-
ветствует выполнению условия дифракции для основной части
нейтронов пучка (с Х=1,6А) и поэтому должно быть макси-
мальным (нейтроны, не участвующие в отражении, детектором
не регистрируются, т. к. они захватываются поглотителем).
Действительно, при небольшом повороте монокристаллов число
регистрируемых детектором нейтронов сильно уменьшается.
Для нейтронографич. исследований требуются пучки
тепловых нейтронов высокой интенсивности. Поэтому
применение дифракции нейтронов к изучению струк-
туры вещества началось лишь после строительства
ядерных реакторов (первые работы по Н. относятся
примерно к 1945). Пучки тепловых нейтронов полу-
чают из ядерных реакторов в результате замедления
нейтронов, возникающих при делении ядер горючего
(U, Ри). Прогресс в реакторостроении определил
быстрое развитие работ по Н. Большинство работ
выполнено на кристаллах.
Дифракция нейтронов заключается в интерференции
между нейтронными волнами, когерентно рассеян-
ными отдельными ядрами кристалла, приводящей к
усилению волн в направлениях, выделяемых условием
Брэгга — Вульфа, и к взаимному погашению в дру-
гих направлениях (обычно говорят об отражении ней-
тронных волн от определенных кристаллич. плоско-
стей). Условие Брэгга—Вульфа связывает углы, при
к-рых происходит отражение, с расстояниями между
отражающими плоскостями (d) и длиной волны излу-
чения (к):
2d sin (8/2) = пк
где п — порядок отражения, 8 — угол рассеяния
(подробнее см. Дифракция рентгеновских лучей).
Интенсивность n-го отражения пропорциональна п~2,
поэтому основным является отражение при п = 1.
Чтобы определить структуру кристалла, надо изме-
рить углы, под к-рыми наблюдаются отражения пер-
вого порядка и интенсивности этих отражений. По-
следние выражаются через т. н. структурный фактор
единичной ячейки кристалла:
Fhki = У вуехр [2ni (hXj-\- kyj^lzj)]
i
где h, k, I — Миллеровские индексы отражающей пло-
скости; сумма берется по всем атомам ячейки кри-
сталла, имеющим координаты xj, уj, zj (в постоянных
решетки); а у — амплитуды ядерного рассеяния нейтро-
нов (дифференциальное сечение рассеяния нейтронов
на единичном свободном ядре: ^=а2). Отражение
нейтронных волн от кристаллич. плоскостей — ре-
зультат взаимодействия нейтронов с ядрами кристал-
ла, константы а являются характеристикой этого
411
НЕЙТРОНОГРАФИЯ
412
взаимодействия. Структурный фактор учитывает ха-
рактер и плотность размещения атомов каждого сорта
в кристаллич. плоскости.
В обычно применяемом методе Н. снимают нейтро-
нограмму образца — зависимость интенсивности ней-
тронов, упруго рассеянных из падающего моноэнерге-
тич. пучка, от угла рассеяния 0. Моноэнергетич. пучки
обычно получают с помощью брэгговского отражения
нейтронов от специально выбранных монокристаллов.
Каждому максимуму на нейтронограмме соответствует
определенная плоскость (hkl). На рис. 2 дана схема
обычно применяемой установки для нейтронография,
исследований.
Узкий направленный пучок тепловых нейтронов из реактора
падает на монокристалл А (напр., РЬ, LiF, NaCl и др.). При
заданной ориентации кристал-
ла и заданном угле рассеяния
6, будут отражаться нейтроны
только одной энергии (опреде-
ляемой соотношением Брэг-
га — Вульфа). Моноэнергетич.
пучок падает затем на обра-
зец В п частично рассеивает-
ся на нем. Детектор нейтро-
нов С (счетчик BF, или сцин-
тилляционный детектор) может
поворачиваться вокруг образ-
ца и т. о. измерять интенсив-
ность нейтронов, рассеянных
на разные углы @. Возникаю-
щие при попадании в детек-
тор нейтронов импульсы напряжения регистрируются после
усиления пересчетной схемой или поступают в интегрирую-
щую схему, соединенную с записывающим прибором.
При исследованиях на монокристаллич. образцах
нейтронограммы снимают для нескольких ориентаций
образца по отношению к пучку; в случае же поликри-
сталлич. образца требуемая информация может быть
получена в одном положении образца. Достоинство по-
ликристаллич. образцов (на них выполнено большин-
ство исследований) — сокращение времени измерений,
а также устранение трудностей, связанных со вторич-
ной экстинкцией (затуханием нейтронной волны в
веществе вследствие отражений), что позволяет уве-
личивать размеры образца; недостаток — ухудшение
разрешения, связанное с двумя причинами: а) полным
и частичным перекрыванием максимумов, поскольку
в отражении одновременно участвует много плоско-
стей, и б) относительным увеличением фона, поскольку
дифракционный пик обусловлен малым объемом об-
разца, включающим лишь определенно ориентиро-
ванные кристаллиты, а в создании фона участвует
весь облучаемый объем образца. На рис. 3 сравнива-
ются нейтронограммы, полученные на моно- и поли-
кристалле
Число отсчетов в минуту
КВг.
Рис. 3. Нейтронограммы, полученные на поли- (а) и моно-
кристаллических (б) образцах.
Н. имеет ряд преимуществ по сравнению с рентгено-
графией. Так, Н. применяют для исследования следу-
ющих структур: водородсодержащих (в частности,
органич. соединений), соединений из элементов с силь-
но различающимися атомными номерами (напр.,
PbS, ТЬОг, WO2), соединений из элементов с очепь
близкими атомными номерами (наир., сплавы Fe—Со,
Ni—Мп), соединений из определенных изотопов одного
и того же элемента. Неудобство (или даже невозмож-
ность) применения рентгеновских лучей для струк-
турных исследований в указанных случаях связано
с тем, что рентгеновские амплитуды рассеяния про-
порциональны атомному номеру рассеивателя Z.
Поэтому в случае соединений из элементов с сильно
различающимися Z (в особенности в случае водород-
содержащих соединений) рентгеновские лучи «не
замечают» легких атомов на фоне более тяжелых и
фиксируют структуру, образованную только тяжелы-
ми атомами. В случае же соединений из элементов с
близкими 1 рентгеновские лучи «не различают» вкла-
дов отдельных компонент соединения. Т. к. ампли-
туды ядерного рассеяния нейтронов обнаруживают
нерегулярные изменения от элемента к элементу и от
изотопа к изотопу, оставаясь в то же время величи-
нами одного порядка, то поэтому тепловыми нейтро-
нами можно пользоваться во всех указанных случаях.
Более того, т. к. рассеяние нейтронов на протонах
аномально велико, то при исследовании водородсо-
держащих соединений нейтроны в первую очередь
«замечают» именно водородную структуру. Как пра-
вило, положение неводородных атомов исследуется
сначала рентгеновскими лучами, а по нейтронограм-
мам затем уточняется положение протонов. Преиму-
щества Н. перед рентгенографией связаны также с изо-
тропностью (отсутствием зависимости от угла рассея-
ния) ядерной амплитуды рассеяния нейтронов. Изо-
тропность объясняется тем, что длина волны тепловых
нейтронов во много раз больше размеров ядра-рас-
сеивателя, вследствие чего можно полагать, что рас-
сеяние происходит на точечном объекте. Из-за изо-
тропности амплитуды рассеяния на нейтронограммах
отсутствует уменьшение интенсивности отражений
при больших углах, характерное для рентгенограмм.
Поэтому нейтронограммы могут оказаться богаче
соответствующих рентгенограмм.
Примером нейтронографич. исследования водород-
содержащих соединений могут служить исследования
структуры льда. Рентгено-
графически лед исследовал- О
ся в течение длительного 0/юд
времени; было установлено,
что атомы кислорода зани-
мают центры и вершины тет-
раэдров, но положение про-
тонов оставалось неопреде-
ленным. Были предложены
4 вероятные модели (рис. 4):
а — протон находится по-
средине каждой ближайшей
пары атомов О; б — протон
смещен к одному из атомов О,
каждый атом О имеет два
приблизившихся и два уда-
лившихся протона; е — пре- Рис. 4. Модели структуры
дыдущая модель с заменой льда'
статич.
нов на
ходится
мени, а
мам О);
топов. Данные нейтронографич. исследования оказа-
лись в хорошем согласии с моделью вив резком про-
тиворечии с остальными моделями.
Методами Н. изучались фазовые переходы второго
рода, наблюдаемые в таких соединениях, как, напр.,
NH4CI, NH4Br, ND4CI, ND4Br. Так, структура ре-
положения прото-
нестатическое (каждая пара протонов на-
вблизи данного атома О половину вре-
затем протоны расходятся к соседним ато-
г — вокруг атома О вращается пара про-
413
НЕЙТРОНОГРАФИЯ
414
щетки ND4Br изменяется при темп-рах —104° и —58°.
С помощью Н. удалось уточнить структуру таких сое-
динений в каждой фазе и понять, какие изменения
происходят при фазовых переходах. Так как при
указанных
60-
» 20
§ so-
1 40-
6
20-
fl-
расположение лег-
ких атомов, то поэто-
му применение рентге-
новских лучей в дан-
ном случае оказывает-
ся неудобным.
Большой интерес
представляют нейтро-
нография. исследова-
ния явления упоря-
дочения в сплавах из
элементов с близкими
Z, таких как Fe—Со,
Ni—Мп, Ni—Fe. В
неупорядоченном со-
стоянии атомы от-
дельных компонентов
сплава хаотически пе-
ю 20 зо ремешаны по узлам
Угол 9. град. кристаллич. решетки.
Рис. 5. Нейтронограммы упорядо-
ченного и неупорядоченного сплава
Fe—Со.
Подходящей термич.
обработкой (медлен-
ным охлаждением от
некоторой темп-ры)
можно добиться перехода сплава в упорядоченное
состояние, в к-ром атомы отдельных компонентов спла-
ва образуют собственную подрешетку. В качестве при-
мера на рис. 5 сравнива-
ются нейтронограммы упо-
рядоченного и неупоря-
доченного сплавов Fe—Со.
На нейтронограмме упо-
рядоченного сплава видны
дополнительные, сверх-
структурные максимумы.
Нейтронография, иссле-
дования оказались плодо-
творными также при изу-
чении текстур (анизотроп-
ного распределения кри-
сталлитов в поликристал-
лич. образце, вызванного
специальной обработкой
образца — прокаткой, во-
лочением, прессовкой).
Рентгенография, исследо-
вание текстур часто за-
труднено из-за сильного
поглощения рентгеновских
лучей, не позволяющего
обследовать внутренние области крупных образцов.
Особый интерес представляют нейтронография, ис-
следования магнитных материалов (магнитная
нейтронография), использующие магнитное
рассеяние тепловых нейтронов, к-рое обусловлено
взаимодействием между магнитными моментами ней-
трона и электронной оболочки атома (или иона).
Поскольку квант рентгеновского излучения не имеет
магнитного момента, то поэтому рентгенография,
исследование магнетиков оказывается невозможным.
Магнитное когерентное рассеяние тепловых нейтронов
чувствительно к степени и характеру упорядочен-
ности магнитных моментов атомов кристалла и может
быть использовано для ее прямого исследования. Дан-
ные по дифракции нейтронов дали прямое подтвержде-
ние существования антиферромагнитных структур
(МпО, NiO, a-FeaOs и др.) и позволили в каждом
случае однозначно решить вопрос о переходе веществ
в антиферромагнитное состояние. Подтвердилось
также существование ферримагнитных структур
(Fe3O4, MgFesO4, NiFeaCH и др.). Только с помощью
дифракции нейтронов можно определить ориентацию
магнитных моментов атомов по отношению к кристал-
лография. осям и плоскостям [напр., было установ-
лено, что в МпО магнитные моменты ориентированы
вдоль осей (100), а в FeO — вдоль (111)]. В случае
бинарных сплавов дифракция нейтронов позволяет
получить сведения о магнитных моментах атомов
каждой из компонент сплава, тогда как иные магнит-
ные измерения могут дать сведения только о среднем
магнитном моменте на атом. Если магнитная струк-
тура имеет элементарную ячейку, совпадающую с
химия, ячейкой, то магнитное рассеяние нейтронов не
приводит к появлению дополнительных по сравнению
с ядерным рассеянием дифракционных максимумов.
В антиферромагнетиках размеры магнитной ячейки
могут в несколько раз превышать размеры химич.
ячейки; в этом случае на нейтронограмме появляются
дополнительные, «магнитные» максимумы.
Н. магнитных структур использует несколько мето-
дов. Один из них заключается в предварительном изу-
чении кристаллохимия, структуры с помощью рентге-
новских лучей. Т. к. последние не испытывают маг-
нитного рассеяния, то поэтому последующее сопостав-
ление с нейтронография, данными позволяет выявить
магнитные максимумы. На рис. 6 приведены рентгено-и
нейтронограмма гематита a-FesOa- Из сравнения
видно, что первые два максимума на нейтронограмме
(не проявившиеся на рентгенограмме) являются маг-
нитными и отвечают антиферромагнитной структуре
Рис. 6. Рентгенограмма и нейтронограмма
антиферромагнитного состояния гематита.
Рис. 7. Нейтронограммы на МпО при 80° К
и 293° К.
гематита. Другой метод заключается в изучении диф-
ракции нейтронов при двух темп-рах образца — ниже
и выше точки Кюри (при повышении темп-ры за точку
Кюри магнетик переходит в парамагнитное состояние).
В последнем случае когерентное магнитное рассеяние
отсутствует. На рис. 7 приводятся нейтронограммы
для МпО, снятые при 80° К и 293° К (точка Кюри
равна 120° К). В первом случае наблюдаются допол-
нительные дифракционные максимумы, к-рых нет на
второй нейтронограмме. Наличие таких максимумов
и положение их по шкале углов указывает на то, что
при 80° К имеет место антиферромагнитная структура
МпО с ячейкой, линейные размеры к-рой в 2 раза
больше размеров химич. ячейки.
Лит.: Б э к о н Д ж., Диффракция нейтронов, пер. с англ.,
М., 1957; Ю з Д., Нейтронная оптика, пер. с англ., М., 1955;
Ringo G. R., Neutron diffraction and interference, в кн.:
Handbuch der Physik, hrsg. von S. Fliigge, Bd 32, B.[u. a.].
1957, S. 552; Shull C. G., W о 1 1 a n E. O., Applications of
neutron diffraction to solid state problems, Solid State Physics,
415
НЕКАЛЬ БХ—НЕОГЕКСАН
416
1956, 2, 138; Озеров Р. П., Структурная нейтронография,
Усп. физ. наук, 1951, 45, К» 4, 481; Wilkinson М. К.,
W о 1 1 a n Е. О., К о е h 1 е г W. С., Neutron diffraction, Ann.
Rev. Nucl. Scl., 1961, 11, 303. Л. В. Тарасов.
НЕКАЛЬ БХ (смачиватель СВ-101, натриевая
соль дибутилнафталинсульфокислоты) C18H2,O,SNa,
мол. в. 342,4 — желтовато-серый порошок, хорошо
растворимый в воде и спирте. Н. (I) представляет
собой смесь изомеров дибутилнафталинсульфокислот;
сульфогруппа преим. находится в a-положении, а
С«Н9-радикалы в различных ядрах. Получают Н.
из нафталина алкилированием его н-бутиловым спир-
том в H2SO4 или олеуме:
Выпускается Н. в виде пасты с содержанием воды
не выше 20%. В качестве примесей присутствуют
NaaSOi (до 36%) и NaCl (до 3—4%).
Применяют Н. как эмульгатор при полимеризации
многих виниловых соединений, а также как смачи-
ватель в текстильной пром-сти. Н. обладает слабым
стимулирующим действием на рост корней пшеницы.
Лит.: Шва рЦ А. М., Перри Дж., Берч Дж., Поверх-
ностноактивные вещества и моющие средства, пер. с англ.,
М., 1959. В. А. Пучков.
НЕМАТОЦИДЫ — химич. средства борьбы с нема-
тодами (круглыми червями). В качестве средств борьбы
с растительными нематодами могут быть применены:
смесь ДД (60—66% смеси изомеров 1,3-дихлор-
пропена, 30—35% 1,2-дихлорпропана и 5% кубовых
остатков); 1,2-дибромэтан; нем а г он (1,2-дибром-
3-хлорпропан); 1,4-дибромбутен-2 (транс); метил-
изотиоцианат; N-метилдитиокарбамат натрия; дихлор-
тетрагидротиофендиоксид; метиловый и этиловый
эфиры диметилдитиокарбаминовой к-ты и др.
Для борьбы с нематодами почву обрабатывают
водной эмульсией или р-ром одного из перечисленных
препаратов (норма расхода 60—300 кг/га). Посев
производят после разложения или испарения Н.
(обычно через 2—3 недели). Нек-рые препараты (напр.,
эфиры диметилдитиокарбаминовой к-ты) применяют
в виде дустов (см. Пестицидные препараты). Для
лечения от нематод человека и животных (кроме про-
филактич. мер) в качестве Н. применяют различные
глистогонные средства (напр., гептилрезорцин, фено-
тиазин, гексахлорэтан, пиперазин и др.).
Лит.: С к р я б и н К. И. [и др.], Нематодология, М.—Л.,
1931; Павловский Е.Н., Руководство по паразитологии
человека..., 5 изд., т. 1, М.—Л., 1946; Advances In pest control
research, ed. R. L. Metcalf, v. 3, N. Y., 1960; N e w h a 1 1 A. G.,
Botan. Rev., 1955, 21, 189. H. H. Мельников.
НЕМБУТАЛ [ этаминал-натрий, пентобарбитал
натрия, 5-этил-5-(1-метилбутил)-барбитурат натрия]
CnH^OsNaNa, мол. в.
NH-СО хС2Н5 248,26 — бесцветныйпо-
ОС^ 'Сх рошок без запаха, горь-
NNa-CO СН-(СНо)г-СН3 кого вкуса; растворим
СИ3 в воде и спирте, прак-
тически нерастворим в
эфире. При добавлении к Н. растворов СаС12 и Co(NO3)3
в спирте появляется сине-фиолетовое окрашивание.
Количественно Н. определяют титрованием к-той в сре-
де вода — эфир с индикатором метилоранжевым или в
смеси метанол — ацетон с индикатором тимоловым си-
ним. Н. получают взаимодействием диэтилового эфира
атил-(1-метилбутил)-малоновой к-ты с (NHa)aCO в при-
сутствии NaOCaHj, а также другими методами, напр.
обессериванием тиопентала. Н.— изомер барбамила и
близок к нему по снотворному действию, однако Н.
разрушается несколько быстрее, действует несколько
менее продолжительно и в ряде случаев переносится
лучше, чем барбамил.
Лит.: Машковский М. Д., Лекарственные средства,
4 изд., М., 1960, с. 16. В. Г. Яшунский.
НЕНАСЫЩЕННЫЙ РАСТВОР — см. Раствори-
мость.
НЕОАБИЕТИНОВАЯ КИСЛОТА СгоН3,Оа, мол.
в. 302,44 — бесцветные кристаллы; т. пл. 167—169°,
йо>=^15/9°’ г ^макс- . (СПИРТ) н3С СООН
250 ммк (по Гаррису); т. пл. ><
179-180; [а]о= + 175°, Хмакс. | | |
(спирт) 250 ммк (по Бардыше-
ву); хорошо растворима в боль- , н3С |
шинстве органич. растворителей,
нерастворима в воде. Соли ще-
лочных металлов и аммония растворимы в поде,
лучше в этаноле.
Н. к. изомеризуется при действии минеральных илп
органич. к-т и при нагревании; конечный продукт
изомеризации — абиетиновая кислота. При длитель-
ном нагревании (180° и выше) происходит диспропор-
ционирование водорода. При нагревании с серой,
селеном и палладированным углем образует ретен.
Аддукт Н. к. с малеиновым ангидридом имеет т. пл.
226—228°, [а]о=—27°. Из всех известных кислот
смоляных только Н. к. имеет характерный максимум
при /. 250 ммк.
Для количественного определения Н. к. исполь-
зуют сочетание хроматография, и спектроскопия,
методов анализа. Н. к. присутствует почти во всех
бальзамах хвойных в количествах: 18% в Pinus
silvestris, 15—20% в Pinus palustris, 10—15% в
Picea excelsa, 14% и 3.1 % в Pinus pithiusa, P. insignis;
присутствует и в промышленных образцах канифоли.
Н. к. можно получить изомеризацией абиетиновой
к-ты, левопимаровой кислоты и полметровой кислоты.
Н. к. выделяют из смесей к-т при помощи труднорас-
творимых в ацетоне (или метилэтилкетоне) солей с
бутаноламином, с 2-амино-2-метил-1,3-пропандполом,
диэтиламином и бруцином. Лучшие результаты дает
хроматография в сочетании с кристаллизацией солей
с органич. основаниями. Н. к. наряду с другими
смоляными к-тами содержатся в канифоли, к-рая
находит широкое применение в пром-сти.
НЕОБРАТИМЫЕ РЕАКЦИИ — см. Обратимые ре-
акции.
НЕОГЕКСАН (2,2-диметилбутан) СН3С(СН3)2СгН5,
мол. в. 86,17 — бесцветная жидкость, т. пл.—99,870°;
т. кип. 49,7417760 мм; dt/dp=O,04117; 0,64916;
Лд 1,36876; теплота сгорания 11506,3 кал/г; теп-
лота испарения 72,96 кал/г (т. кип.); теплоемкость
Ср 33,91 кал/град-молъ (25°); октановое число 91,8
(исследовательский метод). Поверхностное натяжение
16,30 дин/см (20°); критич. константы: ркряг. 30,67 ат;
<крит. 216,2°; йкрит.0,240. Из всех изомерных гексанов
Н. наиболее устойчив к окислению. Н. содержится
в нефтях и бензинах. Синтезируют Н. реакцией тре-
тичного хлористого бутила и цинкдиэтила:
2 (СН3) СС1 + Zn (С3Н5). —> 2 (CH 3)3СС2Н5 + ZnCl2
В пром-сти Н. получают термич. алкилированием
этилена изобутаном при 500—511° и 150—300 ат.
Н3С=СН3+СН(СН3)3—Ь С3Н5С(СН3)3
Н. применяют как высокооктановый компонент для
получения высококачественных (высокооктановый
бензинов. м- И. Розензарт.
417
НЕОДИКУМАРИН—НЕОПЕНТАН
418
ОН
НЕОДИКУМАРИН [этиловый эфир бис- (4-оксику-
маринил-3) уксусной кислоты, пелентан, тромексан]
С2гН1вО8, мол. в.
СОгСгН5 ?н 408,35—бесцветный
СИ г4/4] порошок, т.пл. 175—
о Г I I 178°; труднораство-
рим в воде и боль-
шинстве органич.
растворителей. Получают Н. из о-ацетилсалицило-
вой к-ты (см. Аспирин). Н. уменьшает содержание в
крови протромбина; применяется для профилактики и
лечения тромбозов, тромбофлебитов, эмболий и ин-
фаркта миокарда.
Лит.: F и с 1 к К., ProChazka Z., Cechova V.,
Bull. Soc. chlm. France, 1949, № 1—2, 99. P. Г. Лутков.
НЕОДИМ (Neodymium) Nd — химич. элемент с п.п.
60, относится к лантанидам.
НЕОЗОНЫ — продукты, препятствующие «старе-
нию» резин под действием кислорода воздуха (озона)
и тепла. Н. применяют в качестве добавок к сырым
каучуковым смесям. Обычно Н. являются производ-
ными нафтилампнов или их смесями с различными
веществами. Известны следующие марки Н.: неозон
А—N-фенил-а-нафтиламин; неозон Ц •— смесь 92,5%
N-фенил-а-нафтплампна и 7,5% ле-толу ил ендпа-
мппа, неозон Д — N-фенил-р-нафтиламин и неозон
стандартный — смесь 50% фенил-а-нафтилампна,
25% ле-толуилеидпамина и 25% стеариновой к-ты.
Наиболее важны неозоны Д и А. Их выпускают за
границей под названиями соответственно PBN
и PAN или нонокс Д и нонокс А. Неозон Д получают
взаимодействием р-нафтола с избытком анилина. Для
получения неозона А исходят из а-нафтиламина и
анилина. В обоих случаях процесс ведут при 250—
280° и повышенном давлении; рекомендуется приме-
нение катализаторов: СаС1г, ZnCh, НС1, H2SO4,
KHSO4 или ароматич. сульфокислот. Н. основных
марок могут применяться также как ингибиторы
смоло- и кислотообразования в моторном топливе и
конденсаторных маслах, как ингибиторы полимери-
зации и окисления в процессах очистки олефинов
и их производных, напр. изобутена, бутадиена, изо-
прена и т. п., как антиоксиданты для стабилизации
полиэтилена и высокотемпературных смазочных
материалов, как добавки к антикоррозионным ком-
позициям И Т. Д. °- м- Чернцов.
НЕОМИЦИН — антибиотик, выделенный из куль-
туральной жидкости Str. fradiae; смесь двух изомер-
ных неомицинов В и С, обладающих различным дей-
ствием на микробы. Оба Н. — сильные органич. осно-
вания, содержат по 6 первичных аминогрупп. Мол.
вес свободных оснований 612; [а]д неомицина В = +54°
(1%, вода), неомицина С=+80° (1%, вода). Основа-
ния и сульфаты неомицинов В и С — белые аморфные
порошки, растворимые в воде и нерастворимые в
этаноле и др. органич. растворителях; рейнекат,
гелиантит и пикролонат — кристаллич. продукты.
При метанолизе в абс. метаноле с НС1 Н. распа-
дается на неамин и необиозамины В и С. При гидро-
лизе неамииа образуется 1,3-диамино-4,5-6-триок-
спциклогексан (2-дезоксистрептамин), связанный с
дпаминогексозон, вероятно, эфирной связью. Необиоз-
амин является диаминогексозидопентозой, легко
гидролизующейся до диаминогексозы и D-рибозы. Раз-
личие в биологич. и химич. свойствах необиозаминов
В и С вызвано изомерией диаминогексозы. Строение
неомицинов В и С выражается формулами I и II.
Н. подавляет грамположительные и грамотрицатель-
ные микробы и туберкулезную палочку. Подкожная
токсичность Н. составляет ок. 250 мг на 1 кг веса
(мыши). Н. обладает ототоксич. действием и пора-
жает слуховой нерв. В связи с этим Н. применяется
только как наружное и полостное средство.
Н. выделяют из культуральной жидкости с помощью
карбоксильных катионообменных смол типа IRC-50,
вофатита Ср-300 и КБ4-П2. Активность Н. опреде-
ляется биологически — титрованием на тест-микро-
бах, или химич. методами, основанными на реакциях
Н. с нингидрином, антроном или на образовании
фуРфУРола под действием 36 %-ной H2SO4 при нагре-
вании. Н. был выделен в 1949 С. Ваксманом и Ле-
Шевалье.
В 1951 был получен антибиотик колимициниз особой
разновидности лучистого грибка Str. fradiae var. spiralis, вы-
деленного из почвы. Изучением биологич. и химич. свойств
колимицина показано, что он принадлежит к антибиотикам
«неомицинового комплекса». Применяют колимицин в качестве
местного и полостного средства для лечения нагноительных
процессов, а также для приема внутрь в качестве антисептиков
желудочно-кишечного тракта при хирургич. вмешательствах.
К этой же группе антибиотиков относится выделенный несколь-
ко позже м и ц е р и н.
Лит.: Шемякин М. М. [и др.], Химия антибиотиков,
т. 1, 3 изд., М., 1961; Г а у з е Г. Ф., Лекции по антибиотикам,
3 изд., М., 1958; ж. «Антибиотики», ряд статей за 1959—1961;
Neomycin. Its nature and practical application, ed. S. A. Wak-
sman, Baltimore, 1958; Rinehart K. L. [a. 0.], J. Amer.
Chem. Soc., 1962, 84, № 16, 3216, 3218. M. Г. Бражникова.
НЕОН (Neon) Ne — химич. элемент нулевой груп-
пы периодич. системы элементов Менделеева, инерт-
ный газ; п. н. 10, ат. в. 20,183. Состоит из 3 ста-
бильных изотопов: Ne2’(90,92%), Ne21 (0,257%),
Ne22 (8,82%о); приведенные в скобках числа относятся
к образцу Н. атмосферного происхождения. Попе-
речное сечение поглощения тепловых нейтронов ато-
мом Н. <2,8 барн. Искусственно получены радиоак-
тивные изотопы Н., все они короткоживущие. Кон-
фигурация внешних электронов атома 2s22ps. Энергия
ионизации (в se): Ne°-*Ne + ->-Ne2 + ->Ке’ + соответствен-
но равна 21,56; 41,07; 64,0. Н. состоит из одноатомных
молекул: ат. радиус 1,60 А. Плотн. газа 0,90035г/л
(при 0° и 760 мм рт. ст.).Т. пл.— 248,6°; т. кип.
—245,9° (760 мм рт. ст.). Теплота плавл. 81 кал/моль;
теплота испарения (в точке кипения) 440 кал/моль.
Плотность жидкого Н. (в точке кипения) 1,205 г/см3,
4°крит.—228,6°; ркрит. 27,75 кг/см2 и с7крит. 0,4835 кг/л.
Тройная точка: 24,57° К, 323,5 мм рт. ст.-, плотность
в тройной точке dTB. 1,439 г/см3, dm. 1,247 г/см3.
Как и др. инертные газы, Н. не вступает в обычные
химич. реакции. Получены гидрат Ne-бНгО и нек-рые
др. соединения, в к-рых связь осуществляется моле-
кулярными силами. В пром-сти Н. получают из воз-
духа (см. Воздуха разделение). В электротехнике Н.
используется для заполнения экономичных ламп
накаливания, газосветных и сигнальных ламп (для Н.
характерно красное свечение). Н. применяется также
в различных электронных приборах, в вакуумной
технике и т. д.
Лит. см. при ст. Инертные газы.
НЕОПЕНТАН (2,2-диметилпропан) (СН3)2С(СНз)2,
мол. в. 72,14 — простейший углеводород, молекула
к-рого содержит четвертичный атом углерода; бес-
цветная жидкость; т. пл. —16,55°; т. кип. 9,57760 лм;
о14 к.х.Э. т. з
419
НЕОПРЕН —НЕОРГАНИЧЕСКАЯ ХИМИЯ
420
^’0,591 (при давлении насыщенного пара); 1,342;
октановое число 83 (по моторному методу). Н. тер-
мически более устойчив, чем н-пентан, но менее, чем
изобутан. Из всех изомеров пентана наиболее устойчив
к окислению. Может быть синтезирован из 2,2-дихлор-
пропана и цинкдиметила:
(СН3)2 CCl. + Zn (СН3)2 —+ (СН3)2 С (CH3)2 + ZnCl2
Н. содержится в весьма небольших количествах в
нек-рых нефтях. м- и. Розепгарт.
НЕОПРЕН — см. Хлоропреновый каучук.
НЕОРГАНИЧЕСКАЯ ХИМИЯ — наука о химич.
элементах и образуемых ими простых и сложных хи-
мич. индивидах; соединения углерода (кроме нек-рых
наиболее простых) изучаются в органич. химии. До
конца 18 в. термины «Н. х.» и «органическая химия»
указывали лишь на то, из какого «царства» природы
(минерального или растительного и животного) полу-
чены те или иные соединения. Начиная с 19 в., они
употреблялись как указание на то, присутствует
ли в данном веществе углерод. В настоящее время
эти термины приобрели новое значение в соответствии
с местом, к-рое занимает химия в ряду естественных
наук: Н. х. граничит прежде всего с геохимией и да-
лее с минералогией, петрографией и реологией, т. е.
науками о неорганич. природе; органическая химия
граничит с биохимией и далее с биологией, т. е. с
науками об органич. природе.
Одна из главных задач Н. х.— установление стро-
ения атомов элементов и всестороннее изучение их
свойств в связи со строением, а также исследование
состава и свойств соединений и определение строения
их молекул. Для решения этих задач в современной
Н. х. широко применяются теоретич. представления
и методы физики, кристаллографии и кристаллохимии,
равно как и других областей химии — аналитической,
физической и коллоидной. Другая основная задача
Н. х. состоит в разработке и научном обосновании
способов создания новых материалов, обладающих
нужными для современной техники свойствами —
высокой механич. прочностью, жаропрочностью, стой-
костью к действию сильно агрессивных сред и радио-
активных излучений.
Теоретич. основой Н. х., как и всех прочих отрас-
лей химии, служат законы химич. атомистики и пе-
риодич. закон Д. И. Менделеева. Законы химич.
атомистики определяют весовые соотношения веществ
при химич. превращениях — это законы сохранения
массы, постоянства состава, кратных отношений и
эквивалентов, а также законы простых объемных
отношений при взаимодействии газов. Периодич.
закон и основанная на нем периодическая система
элементов Менделеева составляют незыблемый фун-
дамент всего развития Н. х. и ее изучения в средней
и высшей школах.
Историческая справка. История Н. х,, в
особенности до 2-й половины 19 в., неразрывно переплетается
с общей историей химич. знаний (см. Химия). Достаточно упо-
мянуть, что главнейшие достижения химии конца 18— на-
чала 19 вв., а именно создание кислородной теории горения,
разработка и утверждение химич. атомистики и открытие ос-
новных стехиометрич. законов, были сделаны на основе изу-
чения неорганич. веществ.
Первые сведения о неорганич. веществах, в частности о
тяжелых металлах — золоте, серебре, меди, железе, свинце,
олове, ртути, либо встречающихся в природе в самородном
состоянии, либо сравнительно легко выплавляемых из руд,
относятся к глубокой древности. В этом смысле одним из
древнейших приложений Н. х. является металлургия. Из
неметаллов наиболее давно были известны углерод и сера.
Стремление превратить неблагородные, «несовершенные»,
металлы (медь, свинец и др.) в благородные, «совершенные»
(золото и серебро), было причиной возникновения в 3—4 вв.
алхимии. Алхимики открыли нек-рые важные неорганич.
соединения, напр. минеральные кислоты — соляную, серную,
азотную, получили в свободном состоянии мышьяк, сурьму,
фосфор; однако деятельность алхимиков вследствие ее мистич.
направленности мало способствовала развитию химич. знаний.
Начиная с эпохи Возрождения, все большее значение приоб-
ретают технич. применения Н. х.— металлургия, стеклоделие,
гончарное произ-во, приготовление красок, чернил, окраска
тканей, дубление кож и др., а также приготовление лекарств
(ятрохимия). В этот период были накоплены разнообраз-
ные экспериментальные навыки и наблюдения, а также раз-
работаны и усовершенствованы конструкции печей и других
лабораторных приборов, способы очистки веществ (кристалли-
зация, перегонка, возгонка и др.), получены многие новые хи-
мич. препараты и т. д. В конце 17 в. намечается обособление
сведений о неорганич. веществах в отдельную отрасль химии;
начало этого периода обычно связывают с именем франц,
химика Н. Лемери, к-рый в своем «Курсе химии» (1675) раз-
делил изучаемые химией вещества по их происхождению на
минеральные, растительные и животные. Отсюда произошло
название «минеральная химия», сохранившееся до наших дней
во франц, литературе как синоним Н. х.
В начале 18 в, возникла и прочно укрепилась гипотеза
флогистона — некоей горючей субстанции, свойственной в той
или иной мере всем веществам в природе и выделяющейся из
них при горении. Эта гипотеза, хотя и ошибочная, позволила
тем не менее истолковать большое число химич. явлений с
единой точки зрения. В 1756 М. В. Ломоносов на опыте пока-
зал, что при обжигании металлов происходит поглощение ча-
стичек воздуха, и открыл закон сохранения массы при химич.
реакциях, утвержденный в химии благодаря последующим
трудам А. Лавуазье (см. Сохранения массы закон). В конце
18 в. Лавуазье ниспроверг гипотезу флогистона, доказав, что
обжигание и горение — это реакции соединения веществ с
кислородом. В результате произошел коренной пересмотр
всех теоретич. представлений прошлого, были окончательно
отброшены мистические «элементы — качества» алхимиков
и утвердилось предложенное еще Р. Бойлем (1661) научное
определение химич. элемента как предела разложения веществ
(см. Элементы химические). В 1789 Лавуазье опубликовал
первый список химич. элементов. В 1786—87 Лавуазье (со-
вместно с другими франц, химиками) разработал первую рацио-
нальную систему химич. названий, принципы к-рой в основном
сохранились в современной номенклатуре неорганических
соединений.
Ломоносов и Лавуазье положили начало периоду количест-
венных исследований, характеризующемуся гл. обр. разра-
боткой точной методики химич. анализа и приложением ее к
установлению состава природных и искусственно полученных
веществ. Вторая половина и особенно последняя четверть 18 в.
весьма богаты экспериментальными открытиями в области
Н. х. К началу 18 в. было известно только 13 химич. элемен-
тов, а к концу 18 в.— 32 элемента, т. е. за одно столетие от-
крыто 19 элементов, в т. ч. кислород, водород, азот, хлор.
Кроме того, в 18 в. был установлен состав воздуха и воды,
открыты окись и двуокись углерода, аммиак, окись и закись
азота, сернистый ангидрид, хлороводород, мышьяковистый
водород и др. газообразные соединения; исследование газов
(пневматическая химия) приобрело особенно
широкий размах. Однако на рубеже 18—19 вв. теоретич. пред-
ставления в Н. х., как и в химии вообще, были еще слабо
развиты. Она оставалась по преимуществу наукой аналитиче-
ской.
Следующий этап истории Н. х. (и химии в целом), занимаю-
щий почти весь 19 в., характеризуется разработкой теоретич.
основ, к-рые группируются вокруг центральной проблемы —
атомно-молекулярного учения. Введение в химию атомной
гипотезы (Дж. Дальтон) и ее дальнейшее утверждение (Я.
Берцелиус и др.), работы по определению и уточнению атом-
ных весов, открытие стехиометрич. законов, по к-рым совер-
шаются химич. превращения (см. Постоянства состава закон,
Кратных отношений закон, Эквивалентов закон, Гей-Люссака
законы, Авогадро закон), и, наконец, первый международный
съезд химиков в г. Карлсруэ (1860), где были приняты четкие
определения понятий атома, эквивалента, молекулы и валент-
ности,— таковы основные вехи развития химич. атомистики.
Огромные успехи были достигнуты и в области эксперимента.
Наряду с химико-аналитич. методами все большее значение
в Н. х. приобретают методы физические. Так, благодаря при-
менению электрич. тока для разложения сложных веществ в
первой половине 19 в. были получены в свободном состоянии
калий, натрий, кальций, стронций, барий, магний. Многие
вещества, считавшиеся прежде сложными, оказались просты-
ми, и наоборот. Эти новые методы и открытия явились базой
и для новых теоретич. представлений. Разлагая током соли,
кислоты и щелочи, Берцелиус сделал вывод, что в каждом
атоме, равно как и в их соединениях, имеются два электрич.
полюса, взаимодействие к-рых служит причиной химич. про-
цессов (дуалистическая система, 1812—19).
Берцелиус расположил все элементы в ряд (от наиболее элект-
роотрицательного — кислорода, до наиболее электрополо-
жительного — калия) и назвал электроотрицательные метал-
лоидами, а электроположительные — металлами. По тому же
принципу он классифицировал химич. соединения и минера-
лы. Это была первая единая классификация всех элементов
и их соединений.
Электрохимия, теория Берцелиуса, разработанная им для
неорганич. веществ, послужила толчком к развитию теоретич.
представлений и в органич. химии. Дальнейшие важные тео-
ретич. обобщения в области химии были сделаны в 30—80-х гг.
на основе исследования органич. соединений. С развитием
421
НЕОРГАНИЧЕСКАЯ ХИМИЯ
422
химич. атомистики появились первые попытки систематизации
элементов по их атомным весам, завершившиеся открытием
Менделеевым (1869) периодич. закона химич. элементов.
Периодич. закон внес простоту, ясность и порядок в огромный
запас сведений Н. х., он показал взаимную связь всех элемен-
тов, благодаря чему каждое исследование, посвященное тому
или иному частному вопросу Н. х., приобрело общее значение.
С открытием и утверждением периодич. закона начинается
современный этап развития Н. х.
Мощным толчком для развития Н. х. явились потребности
пром-сти. Еще в конце 18 и начале 19 вв. были изобретены и
реализованы в большом масштабе отдельные выдающиеся по
значению химич. произ-ва, гл. обр. получение солей, кислот
и щелочей (серной к-ты — камерным способом, соды — по
способу Леблана, хлора — окислением соляной к-ты, и т. д.).
Эти произ-ва возникли тогда для удовлетворения нужд других
развивающихся произ-в (текстильной и металлообрабатываю-
щей пром-сти, стеклоделия и др.). Во второй половине 19 в.
на смену им пришли новые, более экономичные и совершенные
технологии, процессы (получение соды по способу Сольвэ, кон-
тактный метод произ-ва серной к-ты, электрохимия, получе-
ние хлора и щелочей ит. д.). В середине 19 в. появились за-
воды искусственных минеральных удобрений, сначала фосфор-
ных, а затем калийных. Во втором десятилетии 20 в. был осу-
ществлен в промышленном масштабе синтез аммиака. В конце
19 —начале 20 вв. повышаются и требования к металлургии.
Если в 19 в. производились лишь чугун, сталь и небольшое
число цветных и благородных металлов, то в начале 20 в.,
в связи с развитием машиностроения, электротехнич. и химич.
пром-сти, число применяемых цветных металлов расширяется;
организуются произ-ва легких (алюминий, магний, натрий
и др.), редких (молибден, ванадий, никель, кобальт, вольфрам
и др.) и благородных (родий, иридий, палладий и др.) металлов.
В состав чугунов, сталей и сплавов цветных металлов начинают
широко вводить легирующие добавки, повышающие химич.,ме-
ханич. и термич. стойкость этих материалов. Наряду с различ-
ными термич. процессами в металлургию внедряются электро-
лиз и «мокрые» процессы (растворение, кристаллизация и т.п.),
сближающие металлургия, процессы с чисто химическими.
Применение теории и методов физической химии,
сформировавшейся к концу 19—началу 20 вв., к не-
органич. объектам подняло Н. х. на более высокую
ступень развития. Выдающиеся открытия физики
конца 19 в., установившие сложную структуру ато-
ма, поставили перед Н. х. новые задачи — изучение
химич. элементов и их соединений с точки зрения
теории строения атома. Благодаря развитию пред-
ставлений об электронно-ядерной структуре атомов
стало возможным решение вопроса о природе хими-
ческой связи. В 1915 В. Коссель, а в 1916 Г. Льюис
предложили первые электронные теории валентности,
описательная сторона к-рых сохраняет свое значе-
ние и теперь. Более глубокое понимание строения
молекул и химич. связи достигнуто в квантовой хи-
мии. Новый, более глубокий смысл и дальнейшее
развитие получил периодич. закон химич. элементов
Менделеева, сохранивший все свое значение как ру-
ководящий принцип Н. х. Именно на основе перио-
дпч. системы возникли новые, современные представ-
ления о строении атомов и молекул.
Если до установления периодич. закона открытие
химич. элементов носило по большей части случай-
ный характер, то после 1869 стало возможным пред-
сказывать, какие элементы еще ие известны и како-
вы их вероятные свойства. Большое значение для
систематич. поисков новых элементов имела кванто-
вая теория строения атома Н. Бора (1913), точно
определившая в периодич. системе число свободных
мест от водорода до урана (к 1913 их оказалось 6).
Изучение естественной радиоактивности (с 1898)
привело к открытию природных радиоактивных эле-
ментов и созданию новой отрасли химии, тесно свя-
занной с Н. х.,—радиохимии. Новейший же этап в
этой области начался с 1934, когда с открытием ис-
кусственной радиоактивности были получены химич.
элементы и изотопы, не найденные в природе вслед-
ствие их крайней неустойчивости. Благодаря этому
удалось не только заполнить последние пробелы в
системе Менделеева (получить прометий, технеций и
астатин), но и достроить систему до 103-го элемента
включительно (лауренсий, 1961).См. Актиниды, Транс-
урановые элементы, Элементы химические.
Одним нз новых направлений Н. х. в 20 в. явилось
изучение химии комплексных соединений. Общая тео-
рия их строения и свойств была создана в 1893
А. Вернером, который наряду с понятием валентно-
сти ввел понятие координации. Дальнейшее изуче-
ние этой области Н. х. (Л. А. Чугаев, И. И. Чер-
няев и др.) внесло много нового в представления о
валентности, химич. связи, пространственном рас-
положении атомов в соединениях и т. д. Оно оказа-
лось очень важным и для практики — пользуясь
способностью к комплексообразованию, можно разде-
лять и анализировать весьма близкие по свойствам
химич. элементы, в частности платиновые металлы.
Большое место в современной Н. х. заняло изучение
соединений, в к-рых атомы проявляют валентность,
не подчиняющуюся классич. представлениям, — гид-
ридов, карбидов, нитридов, боридов и т. д. Современ-
ная Н. х. тесно соприкасается с кристаллохимией.
Систематич. исследования структур кристаллов на-
чались с 1912, после открытия интерференции рент-
геновских лучей в кристаллах. Кристаллохимия,
исследования позволяют связать структуру неорга-
нич. веществ (пространственное расположение и связь
атомов в кристаллах) с их химич. и физич. свойствами
(см. Твердое тело).
В Н. х. широко применяются два основных метода
химич. исследования — анализ и синтез. В конце
18 в. главной задачей химии считалось определение
состава веществ, т. е. их анализ, но уже в начале 19 в.
наряду с анализом химики начали уделять все боль-
шее внимание синтезу новых веществ. Развитию не-
органич. синтеза за последние десятилетия чрезвычай-
но способствовали успехи экспериментальной техники,
в частности возможность пользоваться весьма высо-
кими темп-рами в электрич. печах, осуществлять хи-
мич. реакции при высоких давлениях, в глубоком ва-
кууме и т. п. Неорганич. вещество стремятся получить
в возможно более чистом виде, т. е. свободным от
посторонних примесей; для этой цели, в зависимости
от природы и свойств вещества и примесей, пользуют-
ся самыми разнообразными приемами, напр. осажде-
нием, фильтрованием, кристаллизацией, возгонкой,
перегонкой. Чистоту полученного вещества прове-
ряют химич. или спектральным анализом, после чего
приступают к определению физич. и химич. свойств
вещества и его строения. В современной Н. х., кроме
обычных основных физич. констант веществ (плот-
ности, темп-ры плавления, темп-ры кипения), весьма
часто измеряются и многие другие физич. их свойства:
тепловые, оптические, электрические, магнитные.
Для кристаллич. веществ сложного состава, кроме
того, с помощью рентгеноструктурного анализа оп-
ределяют пространственное расположение атомов в
кристаллах. Химич, свойства вещества исследуются по
его разнообразным взаимодействиям с другими веще-
ствами, а также по отношению к действию физич.
агентов, в особенности теплоты, света и электричества.
Совокупность полученных данных в большинстве слу-
чаев дает полную характеристику состава и свойств
вещества.
Однако такого рода исследования не всегда приво-
дят к цели, т. к. нередко, особенно при изучении раст-
воров и сплавов, образующиеся в них сложные веще-
ства выделить в чистом виде либо весьма трудно, либо
практически невозможно. В таких случаях вместо
выделения и изучения отдельных веществ изучают
физич. свойства систем последовательно изменяюще-
гося состава, в к-рых эти вещества образуются. Пост-
роив диаграмму, изображающую зависимость свойств
системы от ее состава, делают заключения о характере
взаимодействия компонентов системы и о составе и
границах существования всех образующихся в ней
I фаз, без их выделения и химич. анализа. В этом со-
14*
423
НЕОРГАНИЧЕСКАЯ ХИМИЯ
424
стоит сущность метода физико-химического анализа,
разрабатываемого с начала 20 в. Н. С. Курнаковым и
его школой.
Число неорганич. соединений, описанных в лите-
ратуре к середине 20 в., составляет 50—60 тыс.; точно
установить его не представляется возможным, т. к.
многие вещества, которые считались определенными
соединениями, при проверке оказались растворами
или механич. смесями. Несмотря на малочисленность
неорганич. соединений по сравнению с органическими
(число к-рых свыше 1 000 000), классификация их
пока еще весьма несовершенна, т. к. общего принципа,
позволяющего охватить все разнообразие форм и
химич. функций соединений, в Н. х. еще нет.
К числу важнейших неорганич. соединений отно-
сятся соединения элементов с кислородом, водородом,
галогенами — фтором, хлором, бромом, иодом, а также
с серой, селеном и теллуром, азотом, фосфором и мышь-
яком, углеродом, кремнием, бором, соединения метал-
лов между собой, а также кислоты, основания и соли
(см., напр., Окислы, Перекиси и перекисные соединения,
Гидриды, Нитриды, Карбиды, Металлические сое-
динения и т. д., а также Кислоты и основания,
Соли).
Новый этап в развитии Н. х. наметился в после-
военный период в связи с развитием ядерной энер-
гетики, реактивной техники, использованием полу-
проводниковых материалов и новой строительной
техники. Основные задачи, решаемые Н. х. на этом
этапе, связаны прежде всего с изысканием новых
синтетич. материалов, обладающих таким комплексом
физико-химич. и механич. свойств, к-рый не встре-
чается в обычных природных материалах и в их ранее
известных сочетаниях. Кроме того, важнейшей зада-
чей современной Н. х. является разработка новых
технологии, процессов с использованием последних
достижений техники в области очень высоких (а так-
же весьма низких) темп-р, высоких давлений, глубо-
кого вакуума, низко- и высокочастотных механич.
воздействий для получения материалов и изделий,
необходимых для нужд народного хозяйства вообще
и новой техники в особенности.
Требования, предъявляемые новой техникой к
материалам, весьма высоки как в отношении чистоты
препаратов (не более 10-s% примесей для полупровод-
ников и материалов ядерной энергетики), так и в
отношении их прочности, жаропрочности и стойкости
к агрессивным средам (ядерная энергетика, ракетная
техника, химич. пром-сть, промышленность строи-
тельных материалов). В настоящее время намечены
пути получения весьма прочных и жаропрочных мате-
риалов на основе совмещения металлов с тугоплав-
кими окислами (А12О3, Сг2О3, ТйО2 и т. п.). Еще недав-
но присутствие окислов в металле рассматривалось
как дефект в структуре металла, резко снижающий его
свойства. Однако это справедливо лишь в случае, когда
включения окисных пленок разрознены и не состав-
ляют сплошных сеток — каркасов. Если же создать
такой каркас внутри металла (ячеистую структуру) из
весьма тонких (сотни ангстрем) окисных пленок, но
без сплошного капсюлирования металла, то такой
материал оказывается весьма прочным и, что особенно
важно, исключительно жаропрочным. Такого рода
материалы получили общее название сапов, происхо-
дящее от названия «спеченный алюминиевый поро-
шок», где впервые удалось осуществить каркасную
структуру окислов. Для этих целей могут быть ис-
пользованы не только окислы, но и другие соедине-
ния, обладающие высокой темп-рой плавления и вы-
сокой прочностью (карбиды, силициды, бориды).
К материалам этого типа тесно примыкают керме-
ты, отличающиеся от них повышенным содержанием
окислов.
Проблема увеличения прочности строительных
материалов и прежде всего бетонов, их коррозионной
стойкости и влагонепроницаемости относится к числу
важнейших в Н. х. В настоящее время цемент как
связующее используется в бетоне едва на 20—30%
из-за сравнительно высокой крупности помола. Од-
нако тонкий помол цемента лимитируется его высокой
химич. активностью. Исследования в области новой
вибрационной технологии помола и вибрационных
воздействий в процессе изготовления бетона показы-
вают, что прп этом получается высокопрочный и влаго-
стойкий бетон при значительно меньшем расходова-
нии цемента.
В последние годы значительные успехи достигнуты
в произ-ве стекла — одной из старейших отраслей
пром-сти, связанной с Н. х. Здесь получены новые
материалы, обладающие исключительно ценными
свойствами, кристаллич. стекла, или ситаллы, (пи-
рокерам). Обычно появление кристаллич. фазы в
стекле (расстекловывание) рассматривалось как брак
стеклянных изделий. Но в том случае, когда ос-
новная масса стекла оказывается закристаллизо-
вавшейся, возникает новый материал, в к-ром отсут-
ствует характерная для стекла хрупкость, появ-
ляется высокая прочность, способность к пластич.
деформированию (ковке, обработке давлением). Осо-
бый интерес представляют шлакоситдллы, получае-
мые по специальной технологии из отходов металлур-
гич. пром-сти — шлаков. Шлакоспталлы могут быть
широко использованы в самых различных отраслях
народного хозяйства — в строительстве жилых до-
мов и производственных зданий, в химич. пром-сти,
в электро-радиотехнике и т. д.
Использование высоких темп-р и давлений позво-
лило в последнее время организовать промышленное
произ-во искусственных алмазов, обладающих высо-
кими эксплуатационными качествами. Современный
этап развития Н. х. характерен также широким изу-
чением высокомолекулярных неорганич. соединений
как гомоцепных, построенных из одинаковых атомов
(замещенные и незамещенные полисиланы, полигер-
маны, полпсульфаны и т. д.), так и гетероцепных,
макромолекулы к-рых образованы чередующимися
группами из двух и более атомов различных элемен-
тов (полисилоксаны, полититаноксаны, полифос-
фазены, полиарсенаты и т. п.) — среди них прежде
всего следует назвать «неорганический каучук» —
полифосфонитрилхлорид (см. Высокомолекулярные сое-
динения неорганические). Вместе с тем большое зна-
чение приобретает использование высокомолекуляр-
ных соединений в сочетании с неорганич. наполните-
лями. Металлополимеры, графитопласты и др. ма-
териалы подобного рода обладают рядом очень цен-
ных свойств и широко используются промышлен-
ностью.
Исключительно большое значение для современной
техники приобрели в последние годы редкие металлы—
ниобий, титан, молибден, цирконий, вольфрам, тан-
тал, а также нек-рые редкоземельные элементы, не-
обходимые для ядерной энергетики. Получение этих
веществ, изучение их химич. свойств и свойств сплавов
на их основе является весьма важной задачей Н. х.
Вместе с тем продолжается широкая разработка мето-
дов получения ряда элементов в особо чистом виде,
необходимых для полупроводниковой техники: крем-
ния, германия, иттрия, сурьмы, индия и др. Нек-рые
щелочные металлы в последнее время приобрели но-
вую весьма важную область применения в ядерной
энергетике, в связи с чем возникла необходимость в
изучении их свойств в этих новых условиях исполь-
зования.
Огромное значение уделяется Н. х. и как научной
базе основной химич. пром-сти (произ-во солей, кис-
425
НЕПРЕДЕЛЬНЫЕ СОЕДИНЕНИЯ — НЕРНСТА УРАВНЕНИЕ
426
лот и щелочей), необходимой как для развития тя-
желой индустрии, так и для интенсификации с. х-ва
(см. Минеральные удобрения).
Лит.: Менделеев Д. И., Основы химии, т. 1—2,
13 изд., М.—Л., 1947; Некрасов Б. В., Курс общей хи-
мии, 14 изд., М.—Л., 1962; Реми Г., Курс неорганической
химии, пер. с нем., т. 1, [М.], 1963; Remy Н., Lehrbuch
der anorganischen Chemie, Bd 1—2, 10 Aufl., Lpz., 1960; Prog-
ress in inorganic chemistry, ed. A. Cotton, v. 1—2, N. Y.—L.,
1959- 60; E m ё 1 u s H. J., Anderson J. S., Modern
aspects of inorganic chemistry, 3 ed., L., 1960; Филянд M.A.,
Семенова E. И., Свойства редких элементов, 2 изд.,
М., 1964; Sanderson R. Т., Chemical periodicity, N.
Y.—L., 1960; Westermann K., Naser K.—H.,
Gru hl К,—H., Anorganische Chemie, Bd 1—2, Lpz., 1958;
Hackspill L. [e. a.], Chlmie minerale, t. 1, P., 1958;
Day M. C., S e 1 b 1 n J., Theoretical inorganic chemistry,
N. Y.—L., 1962; G m e 1 1 n, 8 Aufl., Syst.—Num. 1—70,
B., 1926—62 (издание продолжается); Mellor J. W., v.
1—16, L., 1946—47, то же, 1952—54; Pascal, t. 1 —19, P.,
1956—63. С. А. Погодин. H. П. Мостовенко.
НЕПРЕДЕЛЬНЫЕ СОЕДИНЕНИЯ (ненасыщен-
ные соединения)— органич. соединения, содержащие
кратные (двойные или тройные) связи. К Н. с. при-
надлежат непредельные углеводороды, содержащие крат-
ные связи между атомами углерода, альдегиды и
кетоны, содержащие двойные связи между атомами
углерода и кислорода, нитрилы и оксимы — между
атомами углерода и азота, азосоединения — между
двумя атомами азота и др. К Н. с. не относят циклич.
соединения, содержащие кратные связи в цикле,
напр. бензол.
НЕПРЕДЕЛЬНЫЕ УГЛЕВОДОРОДЫ (ненасы-
щенные углеводороды)— органич.соединения, содер-
жащие атомы углерода, связанные между собой крат-
ными связями (см. Двойная связь, Тройная связь). Для
Н. у. характерна способность присоединять молекулы
водорода, галогенов и др. с образованием насыщенных
соединений. Полимеризация многих Н. у. (этилена,
пропилена, бутадиена, изопрена, стирола, эфиров
акриловой к-ты и др.) основана на способности моле-
кул таких Н. у. присоединять!я друг к другу.
В отличие от насыщенных углеводородов общей
ф-лы С„Нгге+г, Н. у. содержат меньше атомов водо-
рода. Так, к углеводородам ряда С„Н2ге относятся оле-
фины, содержащие одну двойную связь, напр. этилен
СН2=СН2, изобутилен (СНз)2С=СН2. Таким же со-
ставом обладают циклопарафины с кольцом связанных
между собой атомов углерода, папр. циклопропан (I),
циклогексан (И), не
/СНа СН2-СН2 относящиеся к числу
Н2С—СН2 Н2Сч ХСН2 Н. у. (см. Алицикли-
СН2~СН2 ческие соединения).
। и К углеводородам об-
щей ф-лы С„Н2„_2
принадлежат Н. у. двух типов: 1) ацетилены с трой-
ной связью, например аллилен С113С СН; 2) диены,
содержащие две двойные связи. В зависимости от вза-
имного расположения кратных связей различают ди-
ены типа аллена СН2=С=СН2 с кумулированными
двойными связями (см. Кумулены), соединения типа
бутадиена (дивинила) СН2_ СН—СН=СН2 с сопря-
женными двойными связями и соединения с изолиро-
ванными двойными связями, напр. пентадиен-1,4
СН2=СНСН2СН=СН2. Известны также еще более
Н.у., напр. енины СгеН2ге_1, содержащие одну тройную
и одну двойную связь, полиены и т. д.
Формально по составу к Н. у. относятся также
соединения ароматич. ряда общей ф-лы С„Н2П_в (см.
Ароматические углеводороды). Однако такие соедине-
ния вследствие полной выровненности всех межугле-
родных связей по свойствам резко отличаются от
обычных Н. у. и выделяются в особый класс органич.
соединений. Все рассмотренные незамещенные угле-
водороды содержат четное число атомов водорода.
Углеводороды с нечетным числом атомов водорода,
напр. метил СН3-, трифенилметил (СвН5)3С-, принадле-
жат к числу свободных радикалов,
крайне реакционноспособных и легко превращающих-
ся В насыщенные соединения. я- ф- Комиссаров.
НЕПРЕРЫВНОСТИ ПРИНЦИП — см. Физико-
химический анализ.
НЕПТУНИЙ (Neptunium) Np — химич. элемент
с п. н. 93 из семейства актинидов.
НЕРВОН C18H91NO8, мол. в. 810,26 — белые кри-
сталлы, т. пл. 180° (из СН,ОН); [а]д = —4,33° (в пи-
ридине); иодное число 62,7; хорошо растворим в пи-
ридине и хлороформе; растворим при нагревании
в спирте, бензоле, уксусной к-те; плохо растворим
в этилацетате и ацетоне; нерастворим в эфире, петро-
лейном эфире и воде. Молекула Н. построена из остат-
ков нервоновой (гщс-А15-тетракозеновой) кислоты,
сфингозина и D-галактозы в соотношении 1:1 : 1.
сн2
СН3(СН2)|2СН-СНСНСН
НО NH
СН3(СН2)7СН-СН(СН2),3СО
D-галактоза
сфингозин
иерЬоноеая кислота
Н. осаждается из горячего спиртового р-ра при добав-
лении ацетата РЬ и аммиака; при кипячении с 10%-ным
р-ром H2SOa в спирте гидролизуется с образованием
эквимолярных количеств составляющих его компо-
нентов. При взаимодействии с конц. H2SO4 Н. дает
желтую окраску, переходящую затем в пурпурную,
что связано с присутствием в его молекуле сфингозина
и галактозы. При гидрировании Н. образуется тетра-
гидронервон, т. пл. 185°, [а]д= + 5,58° (в пиридине).
Для количественного определения Н. после его выде-
ления и очистки служат методы, основанные на опре-
делении входящего в его состав сфингозина, в част-
ности с помощью антронового реактива. Н., наряду
с другими цереброзидами, в большом количестве
присутствует в белом веществе головного мозга и
нервных тканей многих животных; в меньших коли-
чествах Н. встречается в сером веществе мозга и др.
тканях (печень, селезенка). Имеются данные о нали-
чии в мозге животных оксинервона, отличающегося от
Н. присутствием а-оксинервоновой к-ты.
Для выделения Н. из смеси с другими липидами
ранее пользовались методами фракционирования рас-
творителями и многократной кристаллизации. В по-
следнее время для этих целей с успехом применяется
колоночная хроматография на силикагеле. Физиоло-
гия. роль Н. окончательно не выяснена. Вместе с дру-
гими цереброзидами он входит в состав миелиновых
оболочек нервных волокон. См. также Цереброзиды.
Лит.: DeuelH. J., The lipids, v. 1—2, N. Y.—L., 1951 —
1955; Hanahan D. J., Lipide chemistry, N. Y.—L., 1960.
В. Б. Спиричев.
НЕРЖАВЕЮЩАЯ СТАЛЬ — см. Железа сплавы..
НЕРНСТА ПРИНЦИП — см. Третий закон термо-
динамики.
НЕРНСТА УРАВНЕНИЕ — соотношение, связы-
вающее равновесный электродный потенциал
с концентрациями реагирующих веществ с. Н. у.
является следствием принципов термодинамики в при-
ложении к электродным процессам и было первона-
чально выведено В. Нернстом (1888) для частного
случая — металлич. электрода, опущенного в раствор,
содержащий одноименные с ним ионы, когда процесс
на электроде заключается в обратимом переходе ионов
427
НЕРОЛ —НЕСМЕЯНОВА РЕАКЦИЯ
428
металла в раствор. Н. у. имеет вид:
. RT ,
<Р = <Ро+^?1пс
где <р,> — нормальный потенциал, характеризующий
данный электрод, R — газовая постоянная, Т — абс.
темп-ра, F — число Фарадея (RT/F = 0,025 в при 18°),
п — заряд (валентность) иона. Часто Н. у. наз. более
общее соотношение:
, RT (А)°(А')°'...
ф — <Ро+ nF (в)ь (В')ь'...
выражающее потенциал электрода, на к-ром происхо-
дит окислительно-восстановительная реакция вида
а А -|- а'А' -(- ...»е_ = ЬВ + Ь'В'...
где А, А'..., В, В'... — компоненты, участвующие в
процессе, е — электрон, п — число электронов, уча-
ствующих в окислительно-восстановительной реакции,
а, а'... Ь, Ъ' ...— стехиометрии, коэфф., (А) (А')... (В)
(В') — активности реагирующих веществ. Для разб.
р-ров, когда коэфф, активности близки к 1, активности
можно заменить концентрациями.
Лит.: Киреев В. А., Курс физической химии, 2 изд.,
М., 1956; Д е л а х е й П., Новые приборы и методы в электро-
химии, пер. с англ., М., 1957. А. И. Молодое.
НЕРОЛ С10Н18О, мол. в. 154,25 — терпеновый
спирт жирного ряда (цис-изомер, транс-изомер —
гераниол). Н. является смесью лимоненной (I) и тер-
пин оленной (II) форм спирта (соответственно а- и
Р-форм).
сн3. СН3.
7с-СН3 (CH,),-C-CHS )С=СН-(СН2)2-С-СН2
СН/ II сн/ II
НО-СН..С-Н • но-сн2-с-н
I (а> 11(3)
Н. — бесцветная жидкость с приятным запахом роз,
входпт в состав эфирных масел: нероли, розового,
бергамотового, иланг-илангового; т. кип. 226—
2277760 мм, 125725 мм; d™ 0,8862; w’° 1,4746; дифе-
нилуретан, т. пл. 52—53°; аллофонат, т. пл. 101,5°.
Н. нерастворим в воде, хорошо растворим в спирте,
эфире. Н. легко присоединяет бром, превращаясь
в тетрабромид, при гидрировании над окисью платины
он образует 2,6-диметплоктан, при окислении пер-
манганатом калия — ацетон и левулиновую к-ту,
окисление хромовой к-той ведет к нералю. Действием
серной к-ты Н. превращается в 1,8-терпингидрат:
Н. получают изомеризацией линалоола в кислой
среде, изомеризацией гераниола при действии бензи-
лата алюминия, при восстановлении цитраля по
Меереейна — Понндорфа — Берлея реакции. Синтетич.
Н. почти всегда загрязнен гераниолом, последний уда-
ляют в виде твердого соединения с СаСЦ; стереоизо-
мерные спирты можно разделять также через их ди-
фенплуретаны. Качественно определяют Н. электро-
форезом его кислого фталата; количественно — по
методу Чугаева — Церевитинова (см. Активный во-
дород). Применяется Н. как компонент парфюмерных
КОМПОЗИЦИЙ. Э- Е. Нифантъев.
НЕРОЛИН (метиловый эфир р-нафтола, яра-яра)
СпН10О, мол. в. 158,19 — бесцветные кристаллы;
т. пл. 72°, т. кип. 274°; комплекс с 1,3,5-тринитробен-
золом, т. пл. 93,5°. Н. летуч с водяным паром, он
хорошо растворяется в эфире, хлороформе, бензоле,
хуже — в этаноле и метаноле, нерастворим в воде.
Н. легко нитруется к первому атому углерода; при
последующем нитровании образует-
ся 1,6-динитро-2-метоксинафталин
с нек-рой примесью 1,8-динитро- П Т аГоснз
2-метоксинафталина. При гидриро-
вании Н. над платиной образуется
гл. обр. 6-метокситетралин, а над нпкелем — нафта-
лин и метанол. Н. получают метилированием Р-наф-
толата натрия или калия галогенметилами или диме-
тилсульфатом; при пропускании паров (5-нафтола и
метанола над окисью тория при 400°. Н. легко по-
лучается при действии на fJ-нафтол метилового спир-
та в присутствии серной к-ты; реакция протекает в
очень мягких условиях, что объясняется, видимо,
способностью Р-нафтола реагировать в таутомерной
кетоформе. Н. обладает приятным запахом, напо-
минающим запах черемухи; используют в качестве
дешевой отдушки туалетных мыл и одеколонов.
НЕСМЕЯНОВА РЕАКЦИЯ — синтез металлоор-
ганич. соединений разложением двойных солей арил-
диазонийгалогенидов с галогенидами металлов в при-
сутствии металлич. порошка:
(ArN2X)OT-MeX„+Me' —> ArmMeXn_m + Me'Xp + N2
где Me — арплпруемый металл, а Me'— металл-вос-
становитель с валентностью р. Реакция впервые
открыта на примере ртутьорганич. соединений:
АгХ2С1 -HgCl2 + Си —► ArHgCl + N„ + СиС12
В дальнейшем эта реакция была распространена на
синтез металлоорганич. соединений олова, свинца,
таллия, висмута, мышьяка, сурьмы, селена и др.
Лучшими растворителями для разложения двойных
диазонпевых солей служат кетоны и сложные эфиры,
хотя иногда применяют и другие органич. раствори-
тели и даже воду; разложение проводят при незначи-
тельном нагревании или охлаждении. В зависимости
от состава исходной двойной соли в основном полу-
чаются продукты различной степени арплироваппя.
напр.:
ArN2Cl-SbCl3+Zn—> ArSBClj (в основном)+Аг,8ЬС1 +
+ Ar3Sb+Ar3SbCl2
[ArN2Cl]2-SbCl3 + Zn —> Ar2SbCl (в основном)+Аг8ЬС12+ArsSb
Вариантом H. p. является разложение металлами
(Hg, Pb, Tl, Sn) двойных солей арилдиазонийфторидов
с трехфтористым бором:
4C»H5N2F •BF3+ ЗРЬ —> (CsHs). Pb + 4N2 + 2Pb (BF,)a
В этом случае арилируется не соль металла, а свобод-
ный металл. Таким же образом разлагаются и нек-рые
другие двойные соли или просто соли диазонин:
3 [ArN2Cl]2-ZnCl2 + Sb —> Ar3SbCl3
ArN2Cl + Me —>- ArMeCl + Na
Другим вариантом диазометода является разложение
сулемой, треххлористой сурьмой и треххлористым
мышьяком солей арилазокарбоновых к-т:
ArN = NCOOK + HgCl2 —> C„H3HgCl + CO2 + KCl
Для синтеза металлоорганич. соединений можно ис-
пользовать и алифатические дпазосоединения, на-
пример:
CHN2COOC2H3 + HgCl2 —! Hg [СС1 (ngCl)COOC2H,]2
RCHN2 + MeX3—>RCHXMeX2 + (RCHX)2MeX + (RCHX)3Me+N>
где Me — мышьяк, сурьма, висмут, фосфор.
Механизм Н. р. еще полностью не выяснен. Увели-
чение скорости реакции с возрастанием электронодо-
429
НЕССЛЕРА РЕАКТИВ —НЕФЕЛОМЕТРИЯ И ТУРБИДИМЕТРИЯ
430
норности заместителя X говорит о гетеролитич. ха-
рактере реакпии:
SbCU + C6H5N2C1 + Fe
X SbCl3 + N2 + FeCl2
C6H5
^NO2<C1<H<CH3<C2H5J
Однако многие факты, как, напр., ориентация заме-
стителей (образование смеси о-, м-, w-изомеров при
разложении борфторида фенилдиазония в пиридине,
нитробензоле и этилбевзоате), свидетельствуют о
гомолитич. механизме реакции:
Ar-:-;N:;.x -- Ar- + N2 + X-
Аг- + МеС1„---АгМеС|„.., +С1-
Н. р. открыта в 1929 А. Н. Несмеяновым.
Лит.: Макарова Л. Г., в сб.: Реакции и методы иссле-
дования органических соединений, кн. 3, М., 1954, с. 75;
Реутов О. А., Теоретические проблемы органической хи-
мии, М., 1956, с. 273; Макарова Л. Г., Матвеева
М. К., Изв. АН СССР, Отд. хим. н., 1960, № И, 1974.
Н. П. Гамбарян.
НЕССЛЕРА РЕАКТИВ — щелочный р-р меркури-
иодида калия К2 [HgJJ. При взаимодействии с ам-
миаком, соединениями аммония или нек-рыми восста-
новителями (первичные и вторичные спирты, альде-
гиды) Н. р. образует окрашенный в красно-коричне-
вый цвет осадок состава HOHgNHHgJ:
NHa+2K3 [HgJJ + ЗКОН —> HOHgNHHgJ + 7К J + 2Н2О
В присутствии очень малых количеств NH3 раствор
окрашивается в желтый цвет, что используют для
колориметрия, определения содержания NH3 мето-
дом сравнения со стандартной серией р-ров, содержа-
щих известные количества NH3. При взаимодействии
Н. р. с восстановителями образуется сначала бурый,
становящийся затем серым, осадок металлич. ртути.
Для приготовления Н. р. предложены разные способы,
напр. 10 г KJ растворяют в 10 мл воды, к раствору
приливают при постоянном перемешивании насыщен-
ный водный р-р HgCl2 до появления нерастворяюще-
гося осадка. Затем прибавляют 30 г КОН, переме-
шивают до его растворения, приливают 1 мл насыщен-
ного р-ра HgCl2, раствор разбавляют водой до объема
200 мл, дают ему отстояться и сливают с осадка. Н. р.
хранят в хорошо закупоренной посуде из темного стекла.
Лит.: Ф а й г л ь Ф., Капельный анализ органических ве-
ществ, пер. с англ.,М., 1962; Гос. фармакопея СССР, 9 изд.,
М., 1961; Пала у зов В, А., Химические реактивы, Харь-
ков — Киев, 1935. А. А. Черкасский.
НЕФА РЕАКЦИЯ — образование альдегидов или
кетонов при гидролизе солей первичных или вторич-
ных нитросоедппеппй:
2 r"ch-n-° —.2RV=y°!'1'-2l’_
Rx
2 .ZC=O t N2O + H2o + 2 MeX
R
Водный р-р соли ацнформы нитросоедпнения прибав-
ляют к холодному водному р-ру минеральной к-ты
(серной, иногда соляной). Альдегид или кетон выде-
ляют обычными способами, иногда в виде производ-
ного (оксим, гидразон и т. п.). В Н. р. используют
натриевые соли нитроалканов, иногда калиевые;
возможно использование других солей. Выходы в ре-
акции обычно хорошие.
С помощью Н. р. получают алифатич. и циклоали-
фатич. альдегиды и кетоны, включая соединения,
содержащие другие функциональные группы, помимо
карбонильной;
CH3CH2NO2 сн3СНО
„ выход 84 % _„
сн3сн сн3----------•- СН3СОСН3
no2
HOCH2(CHOH)4CH2NO2----*- НОСН2(СНОН)4СНО
СН3СН2СН(СН2)2СООСН3 ,-',°д6—»- СН3СН2С(СН2)2СООН
NOS О
выход 84%
CH3CH(CH2)2SO2'CH2)3CH3----------
no2
---- CH3^(CH2)2SO2(CH2)3CH&
При Н. р., кроме альдегида или кетона и закиси азо-
та, побочно образуются также карбоновые к-ты,
гидроксиламин, азотистая к-та, продукты нитрозп-
рования. Для уменьшения количества последних ре-
комендуется добавка мочевины.
Механизм Н. р. в нек-рой степени аналогичен меха-
низму гидролиза оксимов: молекула воды атакует
молекулу ациформы нитросоедпнения и при после-
дующей дегидратации получается оксинитрозосоеди-
нение I, к-рое распадается с образованием альдегида
или кетона:
R''c=£^x0H +нз°,
R’x ЧО' н+
ps 1
---*- C=O + HNO(l/3N.O + >/гНгО)
R'z
Н. р. широко применяют в лабораторной практике для
получения угл вводов, кетокислот, циклич. кетонов и т. д.
Реакция открыта почти одновременно М. И. Коно-
валовым (1893) и независимо от него Дж. У. Нефом
(1894). В литературе известна как Н. р.
Лит.: Noland W. Е., Chem. Revs., 1955, 55, № 1,
137; Hawthorne М. F., J. Amer. Chem. Soc., 1957, 79,
№ 10, 2510. E. M. Рохлин.
НЕФЕЛОМЕТРИЯ И ТУРБИДИМЕТРИЯ — методы
колич. химич. анализа, основанные на измерении
интенсивности света, рассеянного или поглощенного
взвешенными частицами мутной среды; нефело-
метрией называют также метод определения дисперс-
ности частиц на основании измерения интенсивности
рассеянного света. Рассеивание света дисперсными
системами наз. явлением Тиндаля. Теория рассеива-
ния разработана Дж. Редеем, Ми и Гансом. Если
световой поток с интенсивностью J проходит через
кювету со взвешенными частицами, то часть света
рассеивается поверхностью частиц. Интенсивность
рассеянного света в простейшем случае определяется
законом Релея
где к — коэфф, пропорциональности, зависящий от
ряда условий, п — число частиц, рассеивающих свет
431
НЕФЕЛОМЕТРИЯ И ТУРБИДИМЕТРИЯ — НЕФТЕПРОДУКТЫ
432
в единице объема, V — объем каждой из частиц,
Л — длина волны падающего света. Закон выведен
для частиц, малых по сравнению с длиной световой
волны, а также для весьма разб. р-ров, где не наблю-
дается вторичного рассеяния света. Для частиц ре-
альных аналитических осадков выражение услож-
няется.
Если измеряют непосредственно интенсивность рас-
сеянного света, т. е. наблюдение ведут сбоку, перпен-
дикулярно направлению основного светового потока,
тогда метод наз. нефелометрией. Если измеряют
ослабление интенсивности основного светового по-
тока, после прохождения через дисперсную систему,
метод наз. турбидиметрией. Различают несколько
вариантов Н. и т. В одной из них, наз. обычно просто
Н. и т., измеряют непосредственно мутность объекта
исследования, без проведения химич. реакций. Так
измеряют мутность нек-рых производственных р-ров,
прозрачность воды, нефтяных фракций, наличие пыли
в газах и т. п. Вторая, более распространенная
группа методов Н. и т. основана на том, что опреде-
ляемое вещество с помощью подходящих химич.
реакций переводят в малорастворимое соединение,
к-рое остается в виде взвеси в анализируемом р-ре.
После этого измеряют интенсивность рассеянного
света и, пользуясь предварительно построенными
калибровочными графиками, рассчитывают содержа-
ние определяемого вещества. Эта группа методов
близка к колориметрия, фотометрия, методам анализа;
определение нек-рых компонентов основано на одних
и тех же реакциях. Так, определение аммиака Нес-
слера реактивом основано на образовании малорас-
творимого и интенсивно окрашенного соединения;
в этом случае конечное измерение производят как
методом нефелометрии или турбодиметрпи, так и фото-
метрически.
Методы Н. ит. менее точны, чем фотометрические,
т. к. рассеяние и поглощение света частицами дис-
персной фазы зависят не только от ее количества, но
и от размера и формы частиц и от характера их поверх-
ности. Поэтому методы нефелометрия, и турбометрич.
анализа применяют обычно только для определения
таких компонентов, для к-рых нет удовлетворитель-
ных фотометрия, методов, как, напр., для определения
хлоридов, сульфатов и т. п. Для удержания твердых
частиц во взвешенном состоянии, т. е. для сохранения
высокой первичной дисперсности, большое значение
имеют стабилизаторы (напр., желатина и др.).
В химич. анализе применяют, кроме того, нефело-
метрия. и турбидиметрия, титрование. Наиболее рас-
пространенным вариантом этого метода является
титрование определяемого вещества р-ром осадителя.
В процессе титрования интенсивность рассеянного
света растет приблизительно пропорционально коли-
честву образующегося осадка. Интенсивность рассеян-
ного света измеряют с помощью фотоэлемента. В точке
эквивалентности (или вблизи нее) рост помутнения
прекращается. По излому кривой находят объем зат-
раченного на реакцию осадителя. При титровании,
кроме указанных выше факторов, влияет время между
прибавлением реактива и измерением интенсивности
рассеянного света, т. к. многие малорастворпмые
вещества дают пересыщенные р-ры. Далее, вблизи
точки эквивалентности наблюдается обычно измене-
ние знака заряда частиц, что влияет на их размер
и форму. Тем не менее метод нефелометрия, и турби-
диметрия. титрования в ряде случаев с успехом
применяют в химия, анализе, а также для определения
состава веществ, образующихся при реакциях осаж-
дения.
Лит.: Й о у Д ж. Г., Фотометрический химический анализ,
т. 2, Нефелометрия, пер. с англ., М., 1936; Л яликов
К). С., Физико-химические методы анализа, 3 изд., М., 1960.
А. К. Бабко.
НЕФТЕПРОДУКТЫ — смеси газообразных, жид-
ких и твердых углеводородов различных классов,
получаемых из нефти и нефтяных газов. Н. разде-
ляются на след, основные группы: топлива, масла,
твердые углеводороды (парафины, церезины, озоке-
риты), битумы и разные Н. Отдельную группу состав-
ляют пластичные смазки (консистентные смазки), для
приготовления к-рых используют в основном нефтя-
ные масла малой и средней вязкости (реже синтетич.
масла) и загустители (твердые углеводороды, мыла
высших жирных к-т, органич. пигменты, бентонитовые
ГЛИНЫ И др.).
Топлива. В эту группу Н. входят: газы углеводо-
родные (см. Газы нефтепереработки, Газы нефтяные
попутные, Газы природные и Газы природные горючие),
бензины, топливо для воздушно-реактивных двига-
телей (см. Реактивное и газотурбинное топливо),
лигроины, керосины, дизельное топливо, котельное
топливо (мазут) и др.
Масла (см. Масла минеральные) — тяжелые дистил-
лятные и остаточные фракции нефти, подвергнутые
специальной очистке (см. Очистка нефтепродуктов)
и применяемые для смазки двигателей и машин, в ги-
дравлич. системах, в качестве электроизоляционных
сред и др. целей. Масла делят на смазочные и несма-
зочные. Смазочные масла по назначению делятся на
моторные (для двигателей внутреннего сгорания) —
авиационные, автотракторные (автолы) и дизельные;
индустриальные — для смазки станков, электродви-
гателей и др. производственных и энергосиловых
машин; приборные; трансмиссионные и осевые; цилин-
дровые для поршневых паровых машин; турбинные;
компрессорные. Несмазочные масла используются
для технология, целей и при эксплуатации меха-
низмов. К несмазочным маслам относятся: электро-
изоляционные — трансформаторные, конденсаторные,
кабельные; для гидравлич. систем; для технологии,
целей — закалочные жидкости, поглотительные, мяг-
чители и проч., для фармакопеи и парфюмерии (белые
масла — медицинское вазелиновое, парфюмерное).
Твердые углеводороды — парафины, церезины и
озокериты и их смеси с маслами.
Битумы — продукты полутвердой и жидкой конси-
стенции, получаемые после глубокого отгона из гуд-
рона масляных фракций или окислением гудрона
воздухом.
Разные нефтепродукты. К ним относятся: кокс
нефтяной (см. Кокс), сажа, различные продукты пи-
ролиза нефти (бензол, толуол, ксилол и др.), мыло-
нафты, асидолы, деэмульгаторы, хлорпарафин и др.,
сырьем для получения к-рых являются различные
нефтяные газы (попутные, нефтепереработки), а также
различные фракции перегонки нефти.
Методы оценки качества Н. Оценку качества Н.
производят для контроля технологии их произ-ва па
предприятиях нефтяной пром-сти и определения со-
ответствия качества готового продукта технич. усло-
виям и степени пригодности готового продукта усло-
виям его применения. По характеру определяемых
качеств методы оценки удобно разделить на две
группы: 1) методы оценки физич. свойств; 2) мето-
ды оценки химич. свойств.
Оценка физических свойств. Вязкость — важ-
нейший показатель качества Н., имеющий решающее
значение для оценки эксплуатационных качеств топ-
лив и масел. Определение понятия и методы оценки
см. в ст. Вязкость.
Плотность, заменяемая в ряде случаев пока-
зателем удельного веса, является одним из основных
и простейших показателей качества Н. Плотность и
уд. вес Н. определяют пикнометром, ареометром
п весами Мора — Вестфаля (ГОСТ 3900—47). Плот-
ность Н. зависит от вида сырья, способа и глубины
433
НЕФТЕПРОДУКТЫ
434
очистки Н. п возрастает с утяжелением фракционного
состава. В практике показателем плотности поль-
зуются для вычисления веса Н., находящихся в ре-
зервуарах и других емкостях.
Фракционный состав определяют пе-
регонкой Н., производимой со строго определенной
скоростью, из колбы стандартной формы п размеров
(ГОСТ 2177—59). Фракционный состав характеризуется
зависимостью между темп-рой паров Н. в колбе и ко-
личеством Н., сконденсировавшегося в
холодильнике и собранного в приемнике
для конденсата (рис. 1). Фракционный
состав бензинов оценивается обычно по
5 точкам: темп-ре на-
чал а кипения, темп-р е
выкипания 10% ,50%,
90% и 97,5% топ-
лива. Для нек-рых
других Н.,напр. для
дизельных топлив,
часто нормируется
количество топлива,
выкипающего до оп-
ределенно заданной
темп-ры, напр. до300
и 360°. Фракцион-
рис. 1. Аппарат для определения
фракционного состава топлива.
ный состав масел обычно определяют при понижен-
ном давлении (в вакууме), чтобы избежать разло-
жения высококипящих фракций при темп-рах, соот-
ветствующих темп-рам их кипения. Определение
фракционного состава топлив производят для техно-
Рис. 2. Бомба для
определения уп-
ругости паров
бензина: 1 — ма-
нометр; 2 — вода;
3—пары бензина;
4 — испытуемый
бензин.
зывая темп-ру,
ния из топлива
логия. контроля в процессе их произ-ва, а также для
оценки эксплуатационных свойств топлив. От фрак-
ционного состава Н. зависят возможность запуска
двигателя, устойчивая работа его на эксплуатацион-
ных режимах ц экономичность.
Д а в л е н и е (упругость) паров Н. (.и.и рт. ст.)
определяют гл. обр. для топлив, применяемых
в двигателях с искровым зажиганием
(бензинов). Определение производят
в стальной бомбе (рпс.2) при соотно-
шении объемов жидкой и паровой
фаз 1 : 4, при 38° (ГОСТ 1756—52).
Технич. условия на Н. ограничивают,
обычно, верхнее значение давления
паров, как меру предотвращения об-
разования «паровых пробок» в топ-
ливной системе двигателя.
Температуру помутне-
ния определяют визуально, наб-
людая за состоянием охлаждаемого
при строго регламентированных усло-
виях образца Н., находящегося в про-
бирке стандартных размеров (ГОСТ
5066—56). Темп-рой помутнения счи-
тается темп-pa, при к-рой из топлива
начинают выделяться кристаллы вы-
сокоплавких углеводородов или воды.
Этот показатель имеет большое зна-
чение в условиях эксплуатации, ука-
при которой можно ожидать выделе-
твердой фазы; строго нормируется для
бензинов, реактивных и дизельных топлив.
Температура застывания — темп-ра,
при к-рой Н. в условиях опыта загустевает настолько,
что при наклоне пробирки с Н. под углом 45° уровень
продукта остается неподвижным в течение 1 минуты;
определяется визуально путем наблюдения за состоя-
нием образца Н., находящегося в снабженной термо-
метром пробирке стандартных размеров и охлаждае-
мого при строго регламентированных условиях
(ГОСТ 1533—42). Этот показ ательимеет иек-рое зна-
чение для определения условий обращения с продук-
том при хранении, транспортировке и сливе из желез-
нодорожных и автоцистерн, а также как один из
показателей технология, контроля. Его определяют
для большинства смазочных и специальных масел,
тяжелых остаточных топлив (мазутов), а также для
дизельных и реактивных топлив и авиационных бен-
зинов.
Температура плавления твердых Н.
(парафина, озокерита, церезина, дифенила и др.) оце-
нивается путем замера темп-ры в момент полного
затвердевания предварительно расплавленного про-
дукта по методу и в приборе Жукова (ГОСТ 4255—48).
Для битумов взамен темп-ры плавления определяют
т. н. темп-ру размягчения но методу «кольцо и шар»
(ОСТ 7872—39) — темп-ру, при к-рой деформация
битума достигает определенной величины под дейст-
вием заданной нагрузки (вес шара 3,45—3,55 г).
Температура вспышки — минималь-
ная темп-ра, при к-рой смесь паров Н. с воздухом
вспыхивает при поднесении открытого пламени (см.
Вспышки температура). Определение производят
в закрытом сосуде специальной конструкции и формы
(ГОСТ 6356—52) или в открытом тигле (ГОСТ 4333—
48). Разница между значениями температуры вспышки
в открытом и закрытом сосудах составляет 10—25°
и может служить характеристикой однородности
фракционного состава Н. Определение температуры
вспышки производят для оценки пожарной опас-
ности Н.
Температура воспламенения — ми-
нимальная температура, при к-рой вспыхнувший от
постороннего источника пламени (искры) Н. горит
устойчивым, незатухающим пламенем; определяется
в тех же приборах, что и темп-ра вспышки. Темп-ра
воспламенения, как правило, выше темп-ры вспышки
на 15—25°.
Цвет характеризует качество очистки Н. с точки
зрения полноты удаления смолистых и др. окрашен-
ных веществ; определяют путем сравнения цвета Н.
со специальными окрашенными стеклами (ГОСТ
2667—52).
Дуктильность, или растяжимость,
характеризует способность битумов растягиваться,
не обрываясь, в тонкие нити под влиянием приложен-
ной силы; определяется в специальном приборе
(дуктилометре) путем растягивания образца битума
стандартной формы с определенной скоростью при
25° (метод ОСТ 17872, м. и. 6 в—40). В совокупности
с показателем темп-ры размягчения дуктильность
характеризует эксплуатационные свойства битума
как материала для дорожных и иных покрытий.
Оценка химических свойств. Содержание
серы определяют несколькими способами. Наиболее
распространенным для светлых Н. является т. н.
ламповый метод: навеска Н. сжигается в та-
рированной лампочке; продукты сгорания поглощают-
ся титрованным содовым р-ром (ГОСТ 8657—57) с по-
следующим титрованием содового р-ра соляной к-той.
Метод может быть использован и для определения
содержания серы в темных Н., к-рые для этого раз-
бавляют каким-либо легким Н. с известным содержа-
нием серы. Вариантом этого метода, приспособленным
специально для темных Н., является метод, в к-ром
навеска Н. сжигается в трубчатой печи, а продукты
сгорания, как и в первом методе, поглощаются содо-
вым р-ром (ГОСТ 1437—56).
Определение серы в темных Н. производят также
путем сжигания навески Н. в калориметрия, бомбе
в атмосфере кислорода с последующим определением
сульфат-ионов хлористым барием (ГОСТ 3877—49).
Для определения серы в тяжелых топливах и смазочных
маслах навеску исследуемого Н. сжигают в тигле
в смеси с перекисью марганца и содой (метод ВТ И)
435
НЕФТЕПРОДУКТЫ
436
с последующим растворением образовавшихся суль-
фатов и осаждением сульфат-иона в виде сернокислого
бария (ГОСТ 1431—49). Широкое применение в послед-
ние годы получил метод определения серы в Н. сож-
жением навески Н. в трубчатой печи в токе кислоро-
да с последующим окислением образующегося SO 2
в SO3 перекисью водорода и определением сульфат-
иопа прямым титрованием р-ром NaOH.
Качественную оценку присутствия в Н. агрессив-
ных сернистых соединений, напр. элементарной серы
и меркаптанов, производят методом медной
пластинки (ГОСТ 6321—52), в к-ром оценивается
изменение цвета медной пластинки после контакта
с испытуемым Н. в условиях опыта. Для этих целей
пользуются также т. наз. докторской про-
б о й, в к-рой фиксируется изменение цвета элемен-
тарной серы под влиянием продуктов взаимодействия
меркаптанов и сероводорода с плюмбитом натрия
(ГОСТ 6957—57).
Для определения содержания H2S к 10 мл испытуе-
мого Н. добавляют 10мл 2%-ного р-ра NaOH, взбал-
тывают, отделяют водную вытяжку, подкисляют ее
0,4—0,6 мл конц. НС1, нагревают до 70—80° и подно-
сят к краю пробирки бумажку, смоченную уксусно-
кислым свинцом. Окрашивание бумажки в коричне-
вый или черный цвета свидетельствует о присутствии
H2S. Количественное определение меркаптанов, серо-
водорода и элементарной серы в Н. (бензины, реактив-
ные и дизельные топлива) возможно также потенцио-
метрии. способом (ГОСТ 9558—60).
Содержание твердого парафина
определяют растворением навески Н. в подходящем
растворителе, напр. в бензине, с последующим охлаж-
дением р-ра до минус 20—минус 40° и осаждением твер-
дых углеводородов из р-ра этиловым или пропиловым
спиртом. Выделившиеся твердые углеводороды отде-
ляют на фильтре, охлаждаемом до заданной темп-ры,
промывают спирто-бензиновой смесью для удаления
масла, растворяют в петролейном эфире, растворитель
отгоняют и определяют вес остатка. В тяжелых
остаточных Н., содержащих большое количество смо-
листых веществ, препятствующих кристаллизации
твердых углеводородов, применяют предварительную
деструкцию испытуемого образца; при этом смолистые
вещества концентрируются в коксообразном остат-
ке, а твердые углеводороды определяют в получен-
ном при коксовании дистилляте. Практически этими
методами определяют не только твердые парафины,
но сумму высокоплавких углеводородов различного
строения.
Содержание смол в Н. можно определить
их адсорбцией иа к.-л. твердом адсорбенте, чаще всего
на силикагеле, с последующей десорбцией смол под-
ходящим экстрагентом, например спирто-бензольной
смесью. В нек-рых маслах и тяжелых остаточных
топливах определяют т. наз. акцизные смолы,
характеризующие количество веществ, способных
реагировать с концентрированной серной к-той
(ГОСТ 2550—44) в строго регламентированных усло-
виях опыта. В бензинах, реактивных и дизельных
топливах определяют количество т. наз. факти-
ческих смол; для этого навеску топлива
испаряют в струе воздуха (ГОСТ 1567—56) пли водя-
ного пара (ГОСТ 8489—58) и определяют вес остатка.
Испарение в струе водяного пара дает более точное
представление о фактическом содержании смол в Н.,
т. к. при этом исключается окисляющее воздействие
кислорода воздуха на нестабильные компоненты
топлива.
Содержание органических кис-
лот определяют по величине кислотного числа или
кислотности, выражаемых количеством мг КОН,
необходимых для нейтрализации к-т, содержащихся
в 1 г Н. (масла) или в 100 мл Н. (топлива). Навеску
Н. в смеси с предварительно нейтрализованным
спиртом титруют (ГОСТ 5985—59) спиртовым р-ром
КОН в присутствии подходящего индикатора (нит-
розиловый желтый, алкали-блау, фенолфталеин). Ки-
слотное число и кислотность определяют также по-
тенциометрическим титрованием раствора Н. в спир-
то-бензольпой (1 : 4) смеси раствором КОН (ГОСТ
1784—47).
Стабильность против окисления
бензинов и нек-рых других продуктов крекинга ха-
рактеризуется величиной т. паз. индукцион-
ного периода — длительностью времени, в те-
чение к-рого испытуемый Н., находящийся в среде
кислорода иод давлением 7 кг/с.и2 при 100°, практи-
чески не поддается окислению
(ГОСТ 4039—48). Индукционный
период определяют в стальной
бомбе, куда устанавливают ста-
кан с испытуемым продуктом
(рнс. 3). Для нек-рых реактив-
ных топлив о стабильности топ-
лива судят по количеству осадка,
образующегося при жидкофазном
окислении его в специальномпри-
боре в течение 4 часов при 150°
(ГОСТ 9144-59).
Стабильность турбинных и
трансформаторных масел опреде-
ляют по методу ВТИ (ГОСТ 981—
55): образец масла в сосуде спе-
циальной формы окисляют про-
дуванием через масло воздуха
при 140° в течение 14 часов,
Рис. 3. Бомба для оп-
ределения индукцион-
ного периода бензина;
1 — кислород; 2 — бен-
зин; а — вода; 4—элек-
тропечь .
после чего в масле определяют кислотное число и
количество образовавшегося осадка. Для определе-
ния склонности масла к образованию низкомолеку-
лярных кислот окисление масла ведут в том же при-
боре при 120° в течение 6 часов, после чего опреде-
ляют кислотность водной вытяжки из окисленного
масла.
Стабильность к окислению моторных (авиационных,
автомобильных, дизельных) масел оценивают по ве-
личине т. иаз. термоокислительной стабильности
(ГОСТ 4953—49), характеризующей изменение меха-
нических свойств тонкой пленки масла, находящей-
ся на металлической поверхности в контакте с возду-
хом при 260°.
Коррозионные свойства масел оце-
нивают по воздействию на металлич. пластинки
(свинец, медносвинцовый сплав) нагретого до 140°
масла (ГОСТ 5162—49); тонкий слой масла на пла-
стинке периодически соприкасается с кислородом
воздуха. Коррозионные свойства масел оценивают по
изменению веса (г.'.и2) пластинки после ее 50-часового
испытания в указанных условиях. Коррозионные свой-
ства масел оценивают также по потере в весе свинцо-
вой пластинки (ГОСТ 8245—56), к-рая подвергается в
течение 10 часов периодич. воздействию испытуемого
масла и воздуха, нагретых до 140° (метод ДК-2
НАМИ). Оба эти метода позволяют приближенно
оценивать коррозионные свойства масел и защитные
свойства антикоррозионных присадок.
Так как сернистые соединения, присутствующие
в маслах, добываемых из восточных нефтей, в приня-
тых условиях окисления почти полностью устраняют
коррозию свинца, методика ДК-2 НАМИ была уже-
сточена за счет введения катализатора (0,02% нафтена-
та меди) п увеличения длительности окисления масла.
Для оценки коррозионных свойств топлив нет стан-
дартных методов. Качественное обнаружение в топ-
ливе активных сернистых соединений производится
упомянутым выше методом медной пластинки.
437
НЕФТЕХИМИЧЕСКИЙ СИНТЕЗ
438
Моющие свойства масел оцениваются
по ГОСТ 5726—53 на установке ИЗВ, представляющей
собой одноцилиндровый двигатель, вал к-рого при-
водится в движение электродвигателем, а головка
и стенка цилиндра обогреваются специальными
электроподогревателями (до 300° и 225° соответст-
венно). По окончании испытания производится осмотр
поршня и оценка количества и цвета лаковых отложе-
ний на его поверхности с помощью эталонной цветной
шкалы лакообразованпя. Метод ПЗВ дает хорошее
соответствие с поведением масла в реальных двига-
телях.
Коксуемость характеризует способность И.
образовывать углистый остаток (кокс) прп испарении
Н. в стандартном приборе и в строго определенных
условиях нагрева (ГОСТ 5061—49); определяется гл.
обр. в моторных п цилиндровых маслах, в тяжелых
остаточных топливах, а также в 10%-ном остатке от
перегонки дизельных топлив. Коксуемость может
служить технология, показателем качества и глубины
очистки Н.
Высота не коптящего пламени, ха-
рактеризующая осветительную и нагревательную
способности светлых Н. прп сжигании их в лампах и
нагревательных приборах, определяется путем сжи-
гания испытуемого образца в лампе специальной
конструкции (ГОСТ 4338—48) и измерением макси-
мальной высоты некоптящего пламени. Численное
значение этого показателя зависит от группового
химич. состава Н. и, прежде всего, от содержания
в Н. ароматич. углеводородов. Применяется для
оценки качества осветительных керосинов, дизельных
и реактивных топлив.
Наряду с рассмотренными методами оценки свойств
Н., к-рые с достаточным основанием могут быть отне-
сены к группе физич. пли химич., имеется ряд показа-
телей, значение к-рых определяется совокупным дей-
ствием физич. или химич. факторов. К числу пх
должны быть отнесены показатели аитпдетонацион-
ной стойкости топлив, характеризуемой величинами
октанового числа (см. Октановое число) и сортности
на богатой смеси (см. Сортность топлив), показатели
воспламеняемости дизельных топлив, оцениваемые
цетановым илп цетеновым числом или дизельным
индексом (см. Цетановое число), п нек-рые др. Методы
оценки этих показателей рассмотрены в соответст-
вующих статьях.
Лит.: Технические нормы на нефтепродукты, под ред.
Н. Г. Пучкова, 16 изд., М., 1957; Нефтепродукты и продукты
переработки твердых топлив. Технические требования, М.,
1961; Технические условия на нефтепродукты, М., 1960; Мо-
торные, реактивные и ракетные топлива, под ред. К. К. Папок
и Е. Г. Семенидо, 4 изд., М., 1962; Нефтепродукты. Методы
испытаний. [Сборник стандартов], М., 1961; Рыбак Б. М.,
Анализ нефти и нефтепродуктов, 5 изд., М., 1962; К у с а к о в
М. М., Методы определения физико-химических характери-
стик нефтяных продуктов, М.—Л., 1936. Б. В. Лосиное.
НЕФТЕХИМИЧЕСКИЙ СИНТЕЗ — получение
химич. продуктов из нефти и природных газов.
Жидкие нефтепродукты н углеводороды, содержа-
щиеся в газах нефтяных попутных, газах нефтепере-
работки и газах природных горючих, получили боль-
шое значение как сырье для химич. пром-сти. На ос-
нове химич. переработки нефти и газов за последние
десятилетия создана продолжающая быстро разви-
ваться крупная пром-сть, выпускающая около 25%
всей мировой химич. продукции. Для переработки
нефтехимии, сырья приобрели важное значение такие
процессы, как дегидрирование и гидрирование, ал-
килирование, циклизация, изомеризация, полимери-
зация и конденсация, а также галогенирование, нитро-
вание, сульфирование, окисление и др. В пром-сти И .с.
используют в больших масштабах парафиновые, не-
предельные (олефины, ацетилен и др-), ароматические
и значительно меньше нафтеновые углеводороды.
Химическая переработка парафиновых углеводо-
родов. В качестве исходного сырья для Н. с. важней-
шими являются: метан, этан, пропан, бутан и пен-
тан. Парафиновые углеводороды с 6—10 атомами С
применяют в качестве растворителей. Углеводороды
с 10—20 атомами С используют в производстве мою-
щих средств, смазочных масел, эмульгаторов для
синтетич. каучуков и др. Чрезвычайно ценным сырьем
для Н. с. являются парафины, состоящие из углево-
дородов с 18—44 атомами С. В табл. 1 приведены
основные направления промышленной переработки
парафиновых углеводородов. Из них важнейшим
является производство олефинового сырья методами
пиролиза и дегидрогенизации. Основными методами
получения из парафиновых углеводородов различных
продуктов Н. с. являются окисление, хлорирование
и фторирование, нитрование, сульфохлорирование,
реакции с серой и аммиаком.
Окисление. Окисление низкомолекулярных
углеводородов является одним из перспективных
путей получения спиртов, альдегидов, кетонов, ки-
слот. Некаталитическим окислением пропана и бутана
воздухом или кислородом в паровой фазе при 425э
и давлении 7 атм получают метиловый, пропиловый и
бутиловый спирты, формальдегид, ацетальдегид, аце-
тон, метилэтилкетон, смесь кетонов С4—С,, окиси
пропилена и бутилена и др. продукты. Большое разви-
тие получило производство жирных к-т с 6—20 атома-
ми С путем окисления парафинов, из к-рых можно изго-
товлять спирты для производства мягчителей, моющих
средств, смачивающих и эмульгирующих веществ,
синтетич. жиров и др. Окислением парафинов при
95—170° и 10—20 ат получают продукты, исполь-
зуемые для изготовления веществ для пропитки
тканей, антикоррозионной защиты, мягчителей и проч.
Неполное сгорание парафиновых углеводородов, со-
держащихся в природном газе, или их термич. расщеп-
ление используются для получения сажи.
Хлорирование и фторирование.
Парафиновые углеводороды можно хлорировать фото-
химии., каталитич. и термич. способами. В пром-сти
хлорпронзводные метана получают в две стадии; газо-
фазным хлорированием метана получают преимущест-
венно хлористый метил и метпленхлорид; затем
жидкофазным фотохимии, хлорированием метилен-
хлорида получают хлороформ и четыреххлористый
углерод. Хлорирование проводят при 400—450°;
выход хлорпроизводных по метану 85—90%. Термич.
хлорирование пентана в трубчатой печи при темп-ре
ок. 260° дает хлористый амил. Высокомолекулярные
парафиновые углеводороды в большинстве случаев
хлорируются при 100°. Прямое фторирование пара-
финовых углеводородов протекает очень бурно и
сопровождается воспламенением. Для введения фтора
в молекулу углеводорода пары парафинового углево-
дорода, разбавленные азотом, пропускают над трех-
фтористым кобальтом при 200—300°. Для получения
перфторированных соединений фторированный угле-
водород пропускают через фторирующий аппарат
много раз. Фторировать можно, пропуская сильно
разбавленную азотом смесь углеводорода и фтора над
обработанной фтористым серебром медной стружкой
при 150—300°. Таким путем из «-октана получают
перфтороктан. Перфторпарафины получают также при
действии на высокохлорированные углеводороды фто-
ристым водородом в присутствии фтористой сурьмы.
Полученный высокофторированный углеводород об-
рабатывается затем трехфторидом кобальта до обра-
зования перфторида. Фторированные углеводороды
исключительно устойчивы против действия азот-
ной и серной к-т, олеума, нитрующей смеси и т. п.
активных химич. агентов. Они совершенно негорючи
и вполне стабильны (по крайней мере до 500°). Пиро-
439
440
НЕФТЕХИМИЧЕСКИЙ СИНТЕЗ
Таблица 1. Основные химические продукты, получаемые из парафиновых углеводородов
Углеводороды Основные методы переработки Важнейшие продукты и нх производные
Метан СН, Электрокрекинг. Термоокис- лительный крекинг, окисление, хлорирование и фторирование Ацетилен; водород; синтез-газ; аммиак; ацетальдегид; мета- нол —► формальдегид, этанол и др. спирты; хлорвинил; хло- ропрен; хлористый метилен; хлористый метил; метилмеркап- тан; хлороформ—► тетрафторэтилеи—> тефлон; четыреххло- ристый углерод—-> фреоны; сероуглерод; диметилдихлорсилан; дихлордифторметан; диметилсульфид; метилцеллюлоза; сажа
Этан С2Нв Пиролиз, нитрование, хлори- рование Этилен; ацетилен; хлористый этил; нитрометан; нитроэтан; дихлорэтан; синтез-газ
Пропан СаНа Пиролиз, окисление, нитрова- ние, дегидрирование Этилен; пропилен; ацетилен; ацетальдегид; формальдегид; нитрометан; нитроэтан; нитропропан; 1,3-дихлорпропан; нитро- спирты; ацетаты; уксусная кислота
н-Бутан С4Н10 Пиролиз, окисление, дегидро- генизация, изомеризация, взаи- модействие с серой или H2S Этилен; пропилен; бутилен; изобутан; изобутилен; бутадиен; ацетальдегид; бутиловый спирт; уксусная к-тз; ацетон; метил- этилкегон; тиофен
Пентан С5Н1а Хлорирование, нитрование, изомеризация, пиролиз Хлористый амил, а из него: амиловые спирты, амилфенолы, амилнафталин, амилмеркаптан, амиламин; нитрометан; нитро- этан; нитропропан; нитробутан; нитропеитаны; изопентан—► —>изопрен; этилен; пропилен; бутилен
Изопентан С5Н13 Дегидрирование, пиролиз, хлорирование Изоамилен—> изопрен—► каучук; этилен; пропилен; хло- ристый изоамил
Высшие парафины (С1Ь-С..) Крекинг, окисление, хлори- рование, сульфирование, нитро- вание Синтетич. масла; олефины; спирты; высшие жирные к-ты; депрессаторы; пластификаторы; сульфонаты; питропарафины; хлорпарафины, а из них: пластификатор для поливинилхло- рида, парафлоу, хлоровакс для огнестойких материалов, син- тетич, смазочные масла
лизом хлордифторметана получают тетрафторэтилеи,
при полимеризации к-рого образуется исключительно
стойкое вещество — тефлон.
Нитрование. В промышленности для нитро-
вания используют главным образом пропан. Нитро-
вание проводят в газовой фазе при давлении 7 атм и
430—450° в потоке тонко распыленной 75 %-ной
азотной кислоты. При этом получают: нитрометан,
нитроэтан, нитропропан. Из нитроиарафинов мож-
но вырабатывать нитроолефины, алифатические ами-
ны и др.
Сульфохлорирование. При действии
сернистого ангидрида и хлора на парафиновые угле-
водороды образуются алифатич. сульфохлориды:
RH + SO2 + C1, —> RSO3C1 + HC1
Лучше всего реагируют нормальные парафины. Ще-
лочным омылением сульфохлориды переходят в суль-
фонаты RSOjOH. Действием на сульфохлориды ам-
миака получают алифатич. сульфамиды RSOjNHa.
Продукты превращения С12—С1в с фенолятом натрия
используются как пластификаторы поливинилхлорида.
При взаимодействии высокомолекулярных алифатич.
сульфамидов с хлорированными жирными к-тами
в присутствии щелочей образуются алкилсульфамидо-
карбоновые к-ты RSO2NHR'—СООН (эмульгаторы
минеральных масел и др.).
Реакция с серой и аммиаком. При
взаимодействии бутана и бутенов с двуокисью серы
над катализатором (окись молибдена — окись алюми-
ния илн окись хрома — окись алюминия) при
500—595° получают тиофен. При нагревании природ-
ного газа с серой и сероводородом в присутствии
окиси хрома, окиси марганца или пятиокиси ванадия
при 700° образуется сероуглерод. При сжигании при-
родного газа (низкомолекулярных углеводородов)
с воздухом в присутствии аммиака при 1000° над
катализаторами, применяемыми для окисления ам-
миака в окислы азота, образуется синильная к-та.
Большое значение приобрели синтезы на основе
смеси окиси углерода и водорода, так наз. синтез-газа,
являющегося источником получения большого числа
продуктов (см. Оксосинтез). Природный газ, содер-
жащий в основном метан, может быть переработан
в синтез-газ: 1) конверсией с водяным паром, угле-
кислым газом и их смесью: СЩ+ЩО-^СО+ЗЩ;
2) окислительной конверсией при помощи кислорода
или воздуха: СН< + УгОг^-СО + 2Н2; 3) смешанной
парокислородной конверсией. Образующийся водород,
а также водород из газов каталитич. риформинга
нефтепродуктов может быть использован для синтеза
аммиака, а окись углерода после тонкой очистки
от СОг и низкотемпературного фракционирования —
для различных органич. синтезов (подробнее см.
Газы природные горючие).
Химическая переработка непредельных углеводо-
родов. Б пром-сти для этих целей широко используют
олефины, ацетилен н др. непредельные углеводороды.
Наиболее важным источником низших олефинов (эти-
лена, пропилена и др.) являются продукты пиролиза.
Бутилены и амилены извлекают из газов термич.
или термокаталитич. переработки нефтепродуктов
или получают каталитич. дегидрированием бутанов
и пентанов. Все возрастающая потребность в высоко-
молекулярных олефинах с 12—18 (и выше) атомами G
(для получения моющих н эмульгирующих средств
11 ДР-) удовлетворяется за счет крекинга парафинов
при 550—560° или каталитич. ди-, три- и тетрамериза-
цией пропилена или бутилена, а также выделением
из продуктов синтеза по Фишеру — Тропшу (см.
Фишера — Тропша синтез) в присутствии железного
катализатора. Из диенов наиболее ценным являются
бутадиен и изопрен. Важнейшим способом получения
бутадиена является каталитич. дегидрирование н-бу-
441
442
НЕФТЕХИМИЧЕСКИЙ СИНТЕЗ
Таблица 2. Основные химические продукты, получаемые из непредельных углеводородов
Углеводороды Основные методы переработки Важнейшие продукты и их производные
Этилен С2Н< Полимеризация, алкилирова- ние, хлорирование, гидратация, окисление Полиэтилен, этилбензол—> стирол—> каучук; дихлор- этан —> 1,1,2-трихлорэтан —> винилиденхлорид —> саран; хлористый этил; хлористый винил;этиловый спирт —► ацет- альдегид и бутадиен—> каучук; этиленхлоргидрин—> гли- коль—>полиэфирная смола (терилен); окись этилена, а из нее: этаноламины, этиленгликоль, полигликоли; уксусный аль- дегид —> уксусная кислота, этиленхлоргидрин, диоксан, эти- ленциангидрин—> акрилонитрил—> акриловая кислота, поли- акрилонитрил
Пропилен С3Н6 Полимеризация, гидрохлори- рование, оксосинтез, гидрата- ция, алкилирование, окисление Полипропилен; хлористый аллил —* аллиловый спирт —> —► глицерин; альдегиды и спирты; изопропанол —► ацетон; изопропилбензол —► а-метилстирол —каучук; додецилен —> —> додецилбензол —► додецилбензолсульфокислота; окись пропилена —> пропиленглнколь; фенол
-н-Бутилен С,Н8 Гидратация, дегидрогениза- ция, полимеризация, хлориро- вание, гидроформилирование Втор-бутиловый спирт; метилэтилкетон; дивинил—>найлон и каучук; полибутилеп; малеиновый ангидрид; дпхлорбутан; амиловый альдегид —> амиловый спирт
Изобутилен С,Н8 Гидратация, полимеризация, сополимеризация, хлорирова- ние, гидроформилирование Триметилкарбииол; полиизобутилен; диизобутилен —> нони- ловый спирт; бутилкаучук; хлористый металлил; триизобутп- лен —► додецилмеркаптан; трет-бутилфенол —> синтетич. смолы; изоамиловый альдегид —► изоамиловый спирт
Ацетилен С2Н2 Полимеризация, хлорирова- ние, гидратация, реакция с NH3 Бензол; купрен; циклооктатетраен; уксусный альдегид; аль- доль; монохлоруксусная к-та; поливинилхлорид; акриловая к-та; хлоропрен; ацетон; поливиниловые эфиры; эфиры акрило- вой к-ты; акрилонитрил; ацетонитрил; ацеталь; дивинилацети- лен; тетрахлорэтан
тана и и-бутилена, получаемых из газов природных
п нефтеперерабатывающих заводов. Как побочный
продукт бутадиен образуется также (3—5%) при
пиролизе тяжелых бензиновых, керосиновых и газой-
левых фракций с целью получения этилена и пропи-
лена. Изопрен получают высокотемпературным пиро-
лизом нефтяных фракций состава С5 (выход 15—25%).
Чистый изопрен можно выделить экстрактивной пере-
гонкой с ацетоном, содержащим 5% воды. Ацети-
лен получают электрокрекингом и высокотемпера-
турным пиролизом метана и др. предельных угле-
водородов.
Промышленная переработка непредельных углево-
дородов основана гл. обр. на их хлорировании,
гидратации, алкилировании, окислении и полимери-
зации (табл. 2).
Хлорирование. При хлорировании этилена,
пропилена и изобутилена образуются (соответствен-
но) дихлорэтан, являющийся растворителем и исход-
ным продуктом для синтеза многочисленных про-
дуктов, хлористый аллил, хлористый металлил
СН2=С(СН3)СН3С1, обладающий высокой реакцион-
ной способностью, и др. хлорпроизводные.
Гидратация. При гидратации этилена, про-
пилена, бутилена образуются соответствующие
спирты. При непрямой гидратации олефины обрабаты-
вают серной к-той; образующиеся алкилсульфаты
гидролизуют:
h2so, н2о
R-CH = CII2-->- R—СН—СН3 —> R-CH-ClI, + H3SOt
I I
OSO3H on
Второй способ заключается в прямом каталитич.
действии воды и присоединении по двойной связи
Н+ и ОН-:
Н2О
R—СН=СН2 —> R—СН2-СН2ОН
Алкилирование. Этим путем получают
этилбензол, кумол, изододецил- и изопентадецилбен-
зол, алкилфенолы с длинными боковыми цепя-
ми, например изододецилфенол и др. Алкилирование
проводят в присутствии различных катализаторов
(хлористый алюминий, безводная фтористоводород-
ная к-та, фтористый бор, серная кислота и др.). Ви-
нилированием бензола олигомерами пропилена полу-
чают алкилбензол с высокомолекулярными алкиль-
ными цепями.
Окисление. Важнейшим продуктом окисления
олефинов является окись этилена, получаемая оки-
слением этилена:
2СН2=СН2 + О. —>- 2СН2 — С.[I, + 56 к«ал/.иоль
\ /
\/
О
Для проведения реакции применяют обычно серебря-
ные катализаторы.
Полимеризация. Полимеризацию непре-
дельных углеводородов проводят в присутствии ката-
лизаторов и инициаторов при различных давлениях
(от атмосферного до 300 атм). В качестве катализа-
торов применяют окислы хрома, никеля, кобальта
и др., а также металлоорганич. катализаторы (три-
алкилалюминий с TiCh, литийалюминийизобутпл
с TiCh и др.). Инициаторами полимеризации являются
кислород, перекись бензоила, перекись трет.-бутила
и др. Продуктами полимеризации олефинов являются:
полиэтилен, полипропилен, полиизобутилен, каучук
синтетический и др. Известны сополимеры изобути-
лена с изопреном и бутадиеном. Путем теломеризации
из олефинов можно получить ряд ценных материалов.
При действии на ацетилен хлористого водорода в при-
сутствии солянокислого р-ра хлористой меди при
40—50° образуется 2-хлорбутадиен (хлоропрен), к-рый
легко полимеризуется в маслостойкий каучук (см.
также А цетилен).
Химическая переработка ароматических углево-
дородов. Ароматические углеводороды — бензол, то-
луол, этил-, пропил- и бутилбензолы, ксилолы, стирол
и др., широко используются для И. с.; из них наи-
НЕФТЕХИМИЧЕСКИЙ синтез
443
большее значение для Н. с. имеет бензол, к-рый по
объему потребления занимает второе место после
этиленов. Из бициклич. ароматич. углеводородов все
большее значение приобретает нафталин. Важней-
шим источником получения ароматич. углеводородов
является каталитич. риформинг бензиновых и лигрои-
новых фракций прямой перегонки нефти на платино-
вых или окисных (MoOs, Сг20з) катализаторах при
450—550° и давлении от 1 до 50 атм. Наибольшее
распространение получили процессы на платиновых
катализаторах (платформинг и др.), нанесенных на
активной окиси алюминия или на алюмосиликатном
катализаторе, при давлении 35—52 атм. Выходы це-
левых ароматич. продуктов зависят от содержания
в исходном сырье ароматич. и нафтеновых углеводо-
родов. При содержании 20—25% нафтенов выход
бензола составляет 16—18% (см. Ароматизация
нефтепродуктов). Выделение ароматич. углеводоро-
дов и разделение изомеров производятся четкой
ректификацией, азеотропной перегонкой, экстрак-
цией, низкотемпературной кристаллизацией, адсорб-
ционными и др. методами, при этом можно получать
индивидуальные углеводороды с концентрацией
99,5—100%.
Источником ароматич. углеводородов является
также пиролиз бензиновых керосино-газойлевых фрак-
ций и нефтяных остатков при 650—800°. Выход бен-
зола может достигать 8—10% на перерабатываемое
сырье. Ароматич. углеводороды могут извлекаться
из бензинов прямой перегонки нефти и бензинов ка-
талитич. крекинга при помощи селективных раство-
рителей, экстрактивной перегонки и др. Основные
промышленные процессы переработки ароматич. угле-
водородов основаны на алкилировании, дегидрирова-
нии, сульфировании, окислении, хлорировании
(табл. 3).
Алкилирование. При действии хлористых
алкилов пли олефинов на ароматич. углеводороды
пли фенолы в присутствии катализаторов получают
этил- и изопропилбензол, гексил-, гептил- и октилфе-
нолы, mpem-бутилфенол, mpem-амилфенол и др. сое-
динения, широко используемые в пром-сти. Алкили-
рованием бензола полимерами пропена, изододеценом
и пзопентадеценом получают промежуточные продук-
ты для производства синтетич. моющих средств. Ката-
лизаторами являются хлористый алюминий, плавико-
вая к-та, серная к-та и др. Алкилаты для получения
из них моющих средств подвергаются сульфированию
(в большинстве случаев 20%-ным олеумом при темп-ре
ниже 25°), напр. сульфированием додецилбензола
444
получают додецилбензолсульфоновую к-ту, которую
нейтрализуют NaOH:
H.,SO< NaOH
С12Н25С»Н3 ——*C12H2!,CeH<- SO,H-> C12H3r,-C.H1-SO3Na
Дегидрирование. Каталитич. дегидриро-
ванием этилбензола получают стирол. Пары этил-
бензола в смеси с примерно равным количеством
водяных паров пропускают при 600° через заполнен-
ную катализатором трубчатую печь. Катализатор
состоит гл. обр. из окиси цинка с добавкой окиси
алюминия (кальция, магния).
Окисление. При окислении ароматич. углево-
дородов получают ангидриды и кислоты (из бен-
зола — ангидрид малеиновой к-ты, пз о-ксилола —
ангидрид фталевой к-ты, из л1-ксплола — изофталевую
к-ту и из п-ксилола — терефталевую к-ту). Окисление
в промышленных условиях производится обычно
воздухом или азотной к-той. В присутствии вана-
диевого катализатора о-ксилол окисляется воздухом
в ангидрид фталевой к-ты. Другие ксилолы окис-
ляются в этих условиях в бензойную к-ту и ангидрид
малеиновой к-ты. Превращение n-ксилола в терефта-
левую к-ту и л-ксилола в изофталевую к-ту прово-
дится в большинстве случаев в две стадии: вначале
ксилол окисляется в толуиловую к-ту, к-рая в при-
сутствии 1% нафтената кобальта, при теми-ре
100—200° и повышенном давлении окисляется до тере-
фталевой к-ты.
Химическая переработка нафтеновых углеводоро-
дов. Из нафтеновых углеводородов промышленное
значение получил только циклогексан, применяемый
в качестве растворителя и перерабатываемый в кап-
ролактам. Его выделяют четкой ректификацией про-
дуктов прямой перегонки нефти пли гидрированием
бензола. Важнейшей областью использования нафте-
нов является превращение их в ароматич. углеводо-
роды путем каталитич. риформинга.
Основные продукты нефтехимического синтеза. Па
основе нефти и углеводородных газов вырабатывается
более 5000 химич. продуктов. Больших масштабов
достигло производство каучука синтетического, син-
тетических волокон, пластических масс, синтетич.
моющих средств, аммиака и многих др. продуктов.
В 1961—62 мировое потребление синтетич. каучука
в капиталистич. странах превысило уровень потреб-
ления натурального каучука (в 1962—2,3 млн. т из
общего количества 4,4 млн. т).
Мировое производство синтетич. волокон за 10 лет
(1950—60) возросло в 10 раз и достигло в 1960
Таблица 3. Основные химические продукты, получаемые из ароматических углеводородов
Углеводороды Основные методы переработки Важнейшие соединения, их производные и продукты
Бензол С„Н« Алкилирование, дегидрирова- ние, хлорирование, сульфиро- вание, окисление, нитрование Этилбензол—> стирол —*• полистирол; кумол, пз к-рого по- лучают ацетон и эпоксидные смолы или фенол и бакелит; додецилфенол—> иеионогенные моющие средства; додецилбен- зол—> арилсульфонат; фенол, а из него: дезинфекционные вещества, найлон, капролактам, фармацевтич. препараты, сред- ства для борьбы с вредителями, моющие средства; гексахлор- гексан; нитробензол; хлорбензол —> ДДТ; ангидрид ма- леиновой к-ты —> малеиновая к-та
Толуол CelljCUj Окисление, нитрование Бензальдегид (красители, душистые вещества); бензойная к-та (консервирующие вещества, лечебные средства н др.); трини- тротолуолфтороглюцин; сахарин
Ксилолы Св1Г4(С11.,)2 Окисление Фталевая, пзофталевая и терефталевам к-ты
Этилбензол СГ,Н-,С.11.- Дегидрирование, окисление, хлорирование Стирол —> полистирол и др. полимеры; ацетофенон —злако- вые смолы, стирол, о-хлорацетофенон
445
НЕФТЕХИМИЧЕСКИЙ СИНТЕЗ — НЕФТЬ
446
704 тыс. т, а в 1961 830 тыс. т (ок. 5,2% от общего
объема производства текстиля). Намечается еще более
быстрый рост пропз-ва синтетич. волокон. Основными
видами синтетич. волокна, в производстве к-рых
используются нефтехимия, мономеры, являются: по-
лиамидные, типа найлона, полиэфирные, акриловые,
винилхлоридные и винилиденхлоридные. Производст-
во этих волокон связано с выпуском ряда исходных
мономеров — циклогексана, фенола, бутадиена, бен-
зойной кислоты, параксилола, ацетона, адипиновой
к-ты, гексаметилен диамина, капролактама, диметил-
терефталата, этиленгликоля и др. На протяжении
последних лет происходит развитие производства
таких новых видов синтетических волокон, как поли-
пропиленовое, полиуретановое и др.
Производство пластмасс базируется на 50% на
использовании углеводородного нефтяного сырья;
предполагается, что доля этого сырья в произ-ве пла-
стич. масс в ближайшие десятилетия возрастет до
65—85%. В 1960 производство пластич. масс в капи-
талистпч. странах составляло 6,9 млн. т, пз них на
долю США приходилось 37%, на долю стран Западной
Европы 35%, Японии 8%. Чрезвычайно быстро
развивается производство полипропилена, полиэти-
лена и полистирола. Широко развито произ-во по-
ливинилхлорида, значительно расширяется произ-во
винилацетата, поливинилацетата, поливинилового
спирта и поливинилацеталей.
Интенсивное развитие получили синтетич. моющие
средства, к-рые широко применяются в текстильной
пром-сти, горной, металлургической, в полиграфии и
др. В 1962 производство синтетич. моющих средств
в каипталистич. странах составляло ок. 2 млн. т, из
них на долю США приходилось примерно 1600 тыс. т.
За последние годы получило большое развитие
производство водорода, получаемого из углеводород-
ных газов (природных, промышленных) и используе-
мого для синтеза аммиака, к-рый потребляется в боль-
ших количествах для произ-ва различных продуктов
органич. синтеза (мочевины, этпламина, синильной
к-ты, метилвинилппрпдина и др.). В огромных коли-
чествах аммиак потребляется для произ-ва азотной
к-ты и минеральных удобрений. Этот источник полу-
чения водорода в наше время является основным.
* *
Большой интерес представляет доклад А.Шампанья
и др. на 6-м Всемирном нефтяном конгрессе (Франк-
фурт-на-Майне, июнь 1963) о микробиология, депара-
финизации нефти с получением белково-витаминных
концентратов. Биосинтез осуществляется аэробными
формами микроорганизмов, избирательно использую-
щих парафиновые углеводороды нормального строе-
ния. Помимо парафиновых углеводородов, для разви-
тия микроорганизмов необходимы воздух и питатель-
ная водная среда, содержащая неорганич. или орга-
нич. азот, соли фосфора, калия, магния и микроэле-
менты: железо, цинк, медь, марганец и др., к-рые
обычно находятся в пресной и морской воде. Темп-ра
поддерживается в пределах 25—40°. Из парафини-
стых газойлей получается ок. 10% белково-витамин-
ного концентрата, представляющего собой клеточное
вещество микроорганизмов и содержащее до 45—50%
белка, близкого по составу к животным белкам.
Наряду с белками в получаемом продукте присутст-
вуют водорастворимые витамины, гл. обр. группы В.
Проведенные опыты свидетельствуют, что белково-
витампннып концентрат является важным продуктом
питания животных. Во Франции (Лавере) построена
полупромышленная установка для получения белково-
витаминных продуктов из нефти в количествах, доста-
точных для широкой проверки их питательных
свойств на животных.
Одновременно с образованием белковых веществ
в процессе биосинтеза происходит депарафинизация
исходного парафинистого газойля; в результате по-
лучается продукт с низкой темп-рой застывания.
Напр., после микробиология, депарафинизации га-
зойля из иракской нефти (фракция 270—367°) темп-ра
его застывания снижается с +5° до —34°.
Лит.: Основы технологии нефтехимического синтеза, под
ред. А. И. Динцеса и Л. А. Потоловского, М., 1960; Новейшие
достижения нефтехимии и нефтепереработки, под ред. К. А.
Кобе и Д. Д. Мак-Кета, пер. с англ., т. 2—4, М., 1961—63;
Лукьянов П. И., Б а с и с т о в А. Г., Пиролиз нефтяного
сырья. (Ресурсы нефтехимии), М., 1962; Мехтиев С. Д.,
Кам баров Ю. Г., Олефиновые углеводороды и их приме-
нение в нефтехимической промышленности, Баку, 1962;
Г у д к о в С. Ф., Переработка углеводородов природных и по-
путных газов, М., 1960; Аз ингер Ф., Введение в нефте-
химию, пер. с нем., М., 1961; Химическая переработка нефтя-
ных углеводородов, М., 1956 (Труды Всес. совещ. по комплекс-
ной хим. переработке нефтяных газов); Гольдштейн Р.,
Химическая переработка нефти, пер. с англ., 2 изд., М., 1961;
Нефтехимия, 1963, 3, № 5; Д е б и Н. К., Нефтехимиче-
ская технология (Процессы нефтехим. синтеза), пер. с рум.,
М., 1963; Голдинг Б., Химия и технология полимерных
материалов, пер. с англ., М., 1963; Вацулик П., Химия
мономеров, пер. с чеш., т. 1, М., 1960. В. В. Панов.
НЕФТЬ. С о д е р ж а н и е:
Происхождение нефти ............................. 446
Физические свойства............................. 447
Химический состав .............................. 449
Классификация нефтей ........................... 450
Подготовка нефти к переработке ................. 453
Переработка нефти .............................. 454
Влияние группового углеводородного состава неф-
тепродуктов на их свойства.................... 454
Нефть как источник химического сырья............ 455
Нефть — горючее ископаемое, смесь углеводородов
с другими органич. соединениями (сернистыми, азо-
тистыми, кислородными). Н. — важнейший источник
жидкого топлива, смазочных масел и др. нефтепро-
дуктов, а также сырья для химич. пром-сти.
Мировая добыча Н. в 1962 составила св. 1,2 млрд. т.
Ниже приведены основные нефтедобывающие страны
и доля их участия в добыче Н. па 1962.
Страны добыча, млн. m Страны Добыча, млп. т
США 359,0 Ирак 49,0
СССР 186,4 Канада 34
Венесуэла .... 166,5 Индонезия .... 22,5
Кувейт 93,0 Алжир (Сахара) . . 20,4
Саудовская Аравия 75,0 Мексика 16,2
Иран 65,0 Румыния 12,3
В СССР нефтяные месторождения находятся в Коми
АССР, Пермской обл., Удмуртской, Татарской, Баш-
кирской АССР, Куйбышевской, Оренбургской, Волго-
градской, Астраханской областях, Калмыцкой АССР,
Украинской ССР, Чечено-Ингушской АССР, Азер-
байджанской, Казахской, Туркменской, Узбекской
и Таджикской ССР, в Западной и Восточной Сибири
и на о. Сахалине. Наибольшая добыча нефти (порядка
85%) в настоящее время приходится на месторожде-
ния восточных районов СССР (т. наз. Волжско-Ураль-
ской области), гл. обр. на месторождения Татарской
АССР (Ромашкине, Бавлы), Башкирской АССР
(Туймазы, Арлан), Куйбышевской обл. (Муханово).
Происхождение нефти. Было выдвинуто много тео-
рий, объясняющих происхождение Н. Из них основ-
ными являются: неорганическая, космическая и орга-
ническая.
Согласно неорганич. теории, автором к-рой был
Д. И. Менделеев, Н. образовалась в результате взаимо-
действия карбидов металлов, находящихся в ядре
Земли, с водой, проникшей по трещинам к раскален-
ным карбидам. По космич. теории Н. образовалась из
углерода и водорода при формировании Земли. Эта
теория находит подтверждение в наличии метана в
атмосфере нек-рых планет. Согласно органич. теории,
447
448
НЕФТЬ
Таблица 1. Физико-химическая характеристика основных нефтей СССР
Месторождение 4 ВЯЗКОСТЬ при 50°, ссгп Парафины, %/т. пл. °C Сера, % Смолы (ад- сорбирован- ные на сили- кагеле), % Ас- фаль тены, % Коксу- емость, % Выход фракций, вес. %
28- 200° 28- 300° 28 — 350’
Коми АССР
Ярегскоеа 0,945 406,0 1,45/50 1,1 29,4 3,68 8,44 0,4 10,0 18,8
Пермская область
Яринское6 0,817 2,73 5,5/50 0,54—0,70 6,2 отс. 1,28 30,8 49,0 59,2
Павловское8 0,896 13,8 2,04/58 2,95 18,1 6,04 5,03 18,9 32,0 40,4
Башкирская АССР
Туймазинскоеа .... 0,852 4,46 5,90/50 1 .47 10,9 3,90 5,10 25,0 41,0 49,0
Арланскоег 0,893 10,9 4,70/49 2,84 20,3 5,20 7,70 18,0 31,6 39,7
Татарская АССР
Ромашкинское3 .... 0,867 6,54 4,87/50 1,62 11,6 4,16 5,85 22,4 38,9 46,0
Куйбышевская обл.
Мухановское2 .... 0,840 3,46 4,53/52 0,57 7,0 0,12 2,84 26,0 44,8 54,4
Волгоградская обл.
ЖирновскоеД 0,859 8,51 4,60/50 0,27 5,3 0,63 1,85 15,4 36,2 48,0
Украинская ССР
долинскоее 0,849 5,00 16,5/51. 0,35 4.6 0,64 1,00 25,3 44,2 53,5
Краснодарский край
Анастасиевскоеж . . . 0,908 — следы 0,26 6,3 3,20 7.30 30,8 47,0
Ставропольский край
бзексуатское3 .... 0,818 3,73 20,6/52 0,05 2,12 0,49 0 ,80 24,5 43,1 55,9
Чечено-Ингушская АССР
Карабулакское1' . . . 0,819 6,7/52 0 ,15 2,2 0,70 0,71 35,4 55,2 73,9
Азербайджанская
Сураханскоек .... 0,896 11,0 0,96 0,23 — — — 13,2 35,1 46,8
Балахано-Сабунчино-Ра-
манинскоек .... 0,920 149,7 0,15 0,30 — — — 8,0 28,6 42,7
(при 20°)
Нефтяные камни" . . 0,887 9,7 1 ,0/52 0,20 9,9 0,10 2,23 16,0 3 7,2 48,4
КкЧ>овдагскоел .... 0,928 61,0 0,63/51,6 0,35 25,0 7,88 7,12 8,9 25,3 33,7
Казахская ССР
Жетыбайскоем .... 0,855 28,20 10,3/53 0,45 15,8 4,22 5,21 12,9 23,2 30,7
Узеньскоем 0,843 15,78 13,69/49 0,22 12,7 0,71 3,84 16,3 29,9 40,0
Кенькиякскоем .... 0,908 37,98 1,13/58 0,63 11,45 0,27 3,30 3,3 17,ч 29,2
Прорвенскоем .... 0,866 3,49 3,86/54 1 , 30 3,95 1,96 2,92 25,0 47,8 62,2
Узбекская ССР
Южно-Алымашикское11 . 0,824 3,06 4,93/55 0,21 3,72 0,55 1 ,26 27,3 48.8 58,4
Северо-Сохское° . . . 0,838 3,62 7,83/51 0,16 7,54 1,04 2,90 24,4 42,4 51,5
Киргизская ССР
Избаскентское0 .... 0,868 7,11 5,55/54 0,35 9,93 4,13 4,31 21 ,4 37,6 45,6
Туркменская ССР
Котур-Тепе11 0,869 8,90 6,30/47 0,18 12,70 1,08 2,60 15,7 34,7 48,0
Западная Сибирь
Усть-БалыкскоеР . . . 0,871 9,43 3,79,54 1,40 17,93 2,43 3,24 17,8 34,7 43,8
Восточная Сибирь
Марковское12 0,806 2,44 1 ,12/55 0,89 4,68 следы 0,35 34,0 52,0 60,9
о. Сахалин
Эхабинскоет 0,854 2,68 1 ,62/50 0,45 8,00 . о.ао 1,82 31,9 54,7 63,8
Примечание: Здесь и в последующих таблицах эксплуатационными объектами являлись: а девон; б свита Б; в турне;
г карбон; л угленосная сайта; е менилитовая свита; ж меотический горизонт; 3 нижний мел; 11 майкопский горизонт; к продук-
тивная толща; л подкирмакинская свита; м юра; н неоген; ° палеоген; п красноцветная свита; Р готерпв-баррем; с кембрий;
токобыкайская свита.
получившей наибольшее распространение, Н. образо-
валась из остатков морских животных, низших орга-
низмов или растительных остатков, к-рые скаплива-
лись в течение миллионов лет и под давлением нахо-
дившихся над ними пород и под действием тепла
превращались в углеводороды.
Физические свойства. Н.— маслянистая жидкость,
как правило, темно-бурого цвета; плотность Н.
колеблется в пределах 0,73—1,04 г/см3
(обычно 0,82 — 0,95; при плотности ниже
0,9 Н. наз. легкими, выше 0,9— т fl-
же л ы м и). Начало кипения Н. обыч-
но ок. 20°; встречаются и более тяжелые
Н. с началом кипения 100° и выше; т. заст.
+ 23—минус 60°, теплоемкость 0,4—0,5
ккал/кг -град; теплота сгорания 10 400—
11 000 ккал/кг; диэлектрич. проницае-
мость ?—2,5; электропроводность 2 -IO-10—
0,3 -10-1811ом -см. Вязкость Н. при 50° ко-
леблется в пределах 1,2— 55 сст и зави-
сит от химич. состава Н., ее фракционного
состава, содержания асфальто-смолистых
веществ и темп-ры; чем легче фракционный
состав Н. и чем выше ее темп-ра, тем ниже вязкость;
чем больше асфальто-смолистых веществ, тем она
выше. Темп-ра застывания Н. зависит в основном от
содержания парафина (чем его больше, тем выше
темп-ра застывания) и содержания легких фракций
(чем их больше, тем ниже темп-ра застывания). Н.
растворима в органич. растворителях; в воде Н.
практически нерастворима, но может образовать с
Таблица 2. Характеристика сибирских нефтей
Наименование нефти .20 d4 Сера, % Смолы, о/ /о Парафин, %,т. пл. °C Выход фрак- ЦИЙ, %
до 200’ до 300’
Шаимская 0,827 0,46 10,2 2,89/55 29 46
Мартыньинская Мегионская: 0,841 0,55 7,32 3,46,54 27 47
валанжинская .... 0,857 1 ,10 11,65 2,28/56 27 47
юрская 0,856 0,56 6,57 2,17/59 28 50,5
Усть-балыкская 0,871 1,40 17,93 3,79/54 18 35
Западно-сургутская . . . 0,892 2,11 19,06 3,38/5 4 16,5 28
Соснинская 0,839 0.93 8,56 1,27/55 30 52
449
НЕФТЬ
450
Таблица 3. Физико-химическая характеристика нефтей основных месторождений Европы, Азии и Африки
Стр ана ,20 d4 ВЯЗКОСТЬ при 50°, сст Парафин, %/т. пл. °C Сера, % Смолы, о/ /о Асфаль- тены, % \= 1 о4- О £ Ё X О о о £ S Выход фракций, вес. %
до 200° до 300°
Румыния (Марень) 0.884 5,32 0,31/55 0,32 7,23 2,75 29,8 48,0
Кувейт (Бурген) 0,862 6,80 5,94 1.7 — 1 ,97 — 21,8 34,5 (до 2 75°’
Саудовская Аравия (смесь нефтей) . . . 0,852 3,86 2,35,48 1 .7 1,95 4,1 27,0 45,0
Иран (Ачха-Джари) . 0,841 37.8 — 1 ,36 — — 3,9 28,5 67,2
Ирак (Киркук) 0,843 3,98 3,9/47 1,89 9,44 2,86 4.06 27,0 45,4
Индонезия (Лирик) 0,840 30 , 1 — 0,08 — — — —
Китай (Лаодзюньмяо) . . 0,869 15,9 8,4/47,5 0,11 12,3 1 , 4 5 ,1 16,0 31 ,0
Индия (Нахоркатия) 0,851 2,89 15,65/55 5,0/55" 0,22 7,64 0,3 9 1,53 26,6 46,6
Египет (Белапм) Алжир (Хасси-Мессауд) 0,917 0,815 52,6 3 7,8—2.4 4 3,47 0 ,16 19,7 12,9 14,01 12,0 21,4
ней стойкие эмульсии. В табл. 1 приведена харак-
теристика физпко-химич. свойств Н. основных место-
рождений СССР. В последние годы открыт ряд мес-
торождений Н. в Сибири; в табл. 2 дана их крат-
кая характеристика. Характеристика Н. основных
месторождений Европы, Азии, Африки и Америки
приведена в табл. 3 и 4.
Таблица 4. Физико-химическая характеристика основных
нефтей Америки
Месторождение Сера, % Выход фрак- ций, вес. %
до 200° до 300°
США Вилмингтон (Калифорния) Вентура (Калифорния) . , Лулинг (Тексас) ..... 0,913 0,875 0,889 1 ,38 1 ,00 0,92 17,9 30,2 13,1 32,5 44.8 35,0
Канада (смесь нефтей) ...... 0,842 0,42 34 5 1
Мексика Поса-Рика 0,873 1 .81 1 1 (до 150°) 21 ,5
Венесуэла Лагупнльяс (тяжелая) . . Нппа (основная) 0,950 0,840 2,20 0,45 8,0 33,0 18,5 52,0
Химический состав. Основными элементами, вхо-
дящими в состав И., являются углерод и водород.
Содержание углерода в Н. колеблется в пределах
82—87%, водорода 11—14%, серы 0,1 — 5%. Содержа-
ние азота и кислорода у большинства II., как пра-
вило, не превышает десятых долей процента и лишь
у нек-рых Н. (напр., калифорнийской —США) дости-
гает 1,7%N и 1,2%О.
Н. состоит из смеси метановых (алкановых), нафте-
новых (циклановых) и ароматич. углеводородов,
кислородных, сернистых и азотистых соединений.
К кислородным соединениям Н. относятся нафтеновые
к-ты, фенолы, асфальто-смолистые вещества. Серни-
стые соединения содержатся в Н. гл. обр. в виде серо-
водорода, меркаптанов, сульфидов, тиофенов и тио-
фанов; азотистые соединения — в основном в виде
гомологов пиридина, гидропиридпна и гидрохпно-
лина. Компонентами Н. являются растворенные в ней
газы (см. Газы нефтяные попутные), вода и минераль-
ные соли.
Содержание газов в Н., состоящих из углеводородов
от Ci до С4, колеблется от десятых долей до
4% (табл. 5), воды (в Н., отобранных непосредст-
венно из скважин) 0,5—10% п выше, минеральных
015 к.х.э. т. з
Таблица 5. Состав газов, растворенных в нефтях
Месторождение Выход па нефть, % Содержание углеводородов в газе, %
CH.jc.Hb С3н„ и-С,П,, н-С,Н,„
Яринское , . . . . 3,7 0,4 6 , 5 .33,2 16,0 4.3,7
Туймазинское . . . 1,8 1 .5 3,3 26,6 18,6 50,0
Арланское 1 ,2 3,8 8,2 3 9,1 17,3 31,6
Рбмашкпнское . . . 0,9 2,2 2,4 13,1 22,9 59,4
Мухановское . . . 1.6 — 2,3 28,2 12,1 57,4
Жирновское .... 0,53 — 5,5 15,2 30,3 49,0
Жетыбайское . . . 0,78 4,7 27,2 21 ,0 47,1
Прорвенское .... 1,76 7.0 25,4 19,2 48,4
Южно-Аламашпкокое 3,54 — 4,4 21 ,9 16,2 57,5
Избаскентское . . . 1 ,58 — 4.6 26,8 52,4 16,3
Котур-Тепе .... 0,30 — 2,8 17,7 29,6 49,9
Усть-Балыкскос . . 0,87 — 1,8 19,9 15,3 63,0
Марковское .... 3,42 — 5,3 32,3 16,6 45,8
солей 0,1—4000 мг/л и выше. Кроме того, минеральные
вещества содержатся в Н. в виде р-ров солей органич.
к-т, в комплексных соединениях и др. Состав мине-
ральных компонентов в Н. определяется в золе,
получаемой при сжигании Н.; содержание золы обыч-
но не превышает десятых долей процента, считая на
Н. В золе обнаружено до 20 различных элементов
(Са, Fe, Si, Zn, Си, Al, Mg, Ni, V, Na, Sn, Ti, Mn,
Sr, Pb, Co, Ag, Ba, Be, Сг и др.), содержание к-рых,
считая на Н., лежит в пределах 5-10-6—1-10-3 %.
Групповой углеводородный состав бензиновых и
керосиновых фракций основных нефтей СССР приве-
ден в табл. 6; в табл. 7 приведены данные по группо-
вому углеводородному составу масляных фракций,
выкипающих от 300 до 460—500°.
Классификация нефтей. Н. могут быть классифици-
рованы: по содержанию в них углеводородов различ-
ных классов (химическая к л а с с и ф и к а-
ц и я), по содержанию в Н. серы и по качеству полу-
чаемых нефтепродуктов (технологическая
классификация).
В основу химич. классификации Н. положен
групповой углеводородный состав фракции, выки-
пающей при 250—300°. В зависимости от преоблада-
ния в этой фракции углеводородов к.-н. одного класса
(выше 50%) Н. делятся на 3 основных типа: метано-
вые, нафтеновые и ароматические. Прп содержании
в этой фракции 25% (и более) углеводородов других
классов Н. делятся на смешанные типы: метано-наф-
теновые, нафтено-метановые, ароматпческо-нафте-
новые, нафтено-ароматические, ароматическо-мета-
новые, метано-ароматические и метано-ароматическо-
нафтеновые.
По технология, классификации Н. в зависимости от
содержания в них серы делятся па 3 класса: I класс —
малосернпстые II. с содержанием серы от 0 до 0,5%;
451
НЕФТЬ
452
Таблица 6. Групповой углеводородный состав бензиновых
и керосиновых фракций основных нефтей СССР
Месторождение Темп-ра отбора фракций, °C Содержание углеводородов, °|0
арома- тич. нафте- новых мета- новых
Яринское 28-200 13 26 61
2 0 0—30 0 19 44 37
Павловское 28—200 1 2 1 9 69
200—300 30 28 42
Туймазинское 28—200 12 21 67
200—300 25 32 43
Арланское 28—200 7 1 9 74
200-300 22 78
ромашкинское 28—200 11 24 6 5
200—300 27 21 52
Мухановское . . 28—200 20 26 54
200—300 20 27 53
Жнрновское 28—200 3 51 46
200-300 14 34 52
Долинское 28—200 19 30 51
200-300 24 19 57
Анастасиевское 28—200 4 69 27
200 — 300 25 51 24
Озексуатское до 200 И 30 59
Карабулакское 28—200 15 30 55
200—300 18 27 55
Сураханское 28—200 14 40 46
200—300 29 48 23
Ва лахано-Сабунчино- Раманинское ...... 28—200 10 71 19
200-300 32 58 10
Нефтяные камни до 350 15 53 32
Жетыбайское 28—200 6 31 63
200—300 12 32 56
Прорвинсксе 28 — 200 30 30 40
200—300 26 43 31
Северо-Сохское ...... 28-200 22 28 50
200-300 16 23 61
Избаскентское 28-200 17 24 59
200—300 19 21 60
Котур-Тепе 28—200 И 48 41
200—300 12 30 58
Усть-Балыкское 28-200 12 22 66
200—300 16 21 6 3
Марковское 28-200 14 15 71.
200—300 17 27 56
ЭхаСпнское 28—200 14 71 15
200—300 26 61 13
II класс — сернистые Н. с содержанием серы от
0,51 до 1,9%; III класс— высокосернистые Н. с со-
держанием серы более 1,9%. Далее Н. в зависимости
от выходов и качеств получаемых нефтепродуктов,
подразделяются на подклассы. Первый подкласс ха-
рактеризует Н. в отношении выходов и качеств топ-
лива; сюда относятся H.I и II классов, светлые дистил-
Таблица 7. Групповой углеводородный состав масляных
фракций основных нефтей СССР
Месторождение Темп-ра отбора, °C Выход, % Содержание углеводородов
ароматических метано- нафтеновых
всего 1 в том числе твердых
о .0 т.пл.* °C
Яринское 300—350 10,2 28 72
350—450 14,4 34 6 6 8 50
450—490 5,6 43 57 14 53
Павловское 300-350 6,4 45 55
350—450 14,0 53 А 7 8 53
450—480 7,9 57 43 8 57
Туймазинское 300—350 8,3 33 67
350—450 15,0 42 58 7 47
450—500 9,0 50 50 11 57
Арланское 300—350 8,1 52 48
350—450 15,1 57 43 __
450—500 -6 65 35 8 56
Ромашкинское 300—350 7,0 31 69
350-450 14,0 45 55 9 47
450—500 7.0 54 46 12 56
Мухановское 300-350 9.6 29 71 ..
350—450 15,8 39 61 12 56
450—500 5,1 43 57 14 60
Жирновское 300—350 11,3 19,5 21 7 9
350—450 25 75 3,0 49
450—500 4,8 31 69 7.6 61
Долинское 300—350 9,3 19 81 —
Анастасиевское 300-350 16,6 34 66 —
Карабулакское 30 0—35.0 5,1 16 84 — —
Сурахлнсксе 300-350 11,7 32 68 — —
Балахано-Слбун- чино-Рамаипн- ское 300—350 14,1 44 56 — —
Нефтяные камни до 350 — 15 85 —
Озексуатское 300 — 350 12,8 10 90 — -
Жетыбайское 300—350 7,5 13 87 __
350—450 19,5 13 87 27 40
450-500 7.6 18 82 3 4 62
Прорвенское 300—350 12,6 33 67
350—450 19,8 3 7 63 5 55
450—5и0 5,5 4 3 57 7 60
Северо-Сохское 300-350 9,1 7 6
350-450 15,4 28 72 19 46
450—470 7,7 3 4 66 18 59
Избаскентское 300—350 8,3 26 74
350—450 16,5 33 67 15 51
450—480 6,3 36 64 15 62
Котур-Тепе 300—350 10,5 16 84
350—450 20 26 74 17 50
450—490 .8 35 65 16 62
Усть-Балыкское 300—350 8,1 27 73
350—450 19,0 33 67 8 45
450-460 3,6 40 60 9 50
Эхабинское 300—350 9,2 31 69
350—450 14,9 45 55 следы —.
450—496 6,1 54 46 6 52
НЕФТЬ
453
ляты (бензин, реактивное топливо, керосин, ди-
зельное топливо) к-рых отвечают требованиям тех-
нич. норм СССР по содержанию серы. Второй под-
класс характеризует Н. в отношении выходов и ка-
чества базовых дистиллятных и остаточных масел;
к нему относятся Н., в к-рых суммарное содержание
указанных масел не ниже 15 вес. %, считая на Н.,
и не ниже 30 вес. %, считая на мазут (после отбора
фракции до 350°). К третьему подклассу относятся
Н. III класса, все светлые дистилляты к-рых не отве-
чают требованиям ГОСТ по содержанию серы и сум-
марное содержание базовых дистиллятных и остаточ-
ных масел в которых ниже 15%, считая на Н., и ниже
30%, считая на мазут. Подклассы в свою очереди под-
разделяются на сорта Н. в зависимости от выходов
нефтепродуктов и их качеств.
Подготовка нефти к переработке. Н. в нефтяных
месторождениях всегда сопутствует пластовая вода
с высоким содержанием растворенных в ней солей.
При существующих способах добычи Н. пластовая
вода смешивается с ней, образуя эмульсию типа «вода
в нефти». С легкими малосмолистыми Н., за исключе-
нием высокопарафинистых, образуются неустойчивые
эмульсии, легко разрушающиеся при обычном отстое
в резервуарах. Тяжелые смолистые Н. образуют
весьма устойчивые, трудноразрушаемые эмульсии,
способствующие увеличению обводненности Н. и
загрязнению их неорганич. солями. Следует отметить,
что свежеполученные нефтяные эмульсии на промыс-
лах значительно легче разрушаются, чем застаревшие,
после длительного хранения и многих перекачек.
Поэтому сырая Н. перед поступлением на нефтепере-
рабатывающий завод должна быть обезвожена и обес-
солена; содержание в ней воды не должно превышать
0,1%, хлористых солей 40 мг/л.
Полная подготовка Н. к переработке состоит из
след, основных процессов: обезвоживания, обессоли-
вания и стабилизации.
Для обезвоживания Н. с высоким содер-
жанием влаги применяют т. наз. термохимия, уста-
новки, работающие при 50—80° и атмосферном дав-
лении или под давлением 5—10 атм и 120—160°. При
правильном подборе деэмульгатора, являющегося
наиболее важным фактором обезвоживания, можно
легко разрушить самую устойчивую эмульсию и обез-
водить Н. В качестве эффективных деэмульгаторов
нефтяных эмульсий типа «вода в нефти» применяют
различные поверхностно-активные вещества (неионо-
генные, анионные и др.). Однако только одним обез-
воживанием Н. не удается полностью удалить хло-
ристые соли (натрия, магния, кальция), содержащие-
ся в сырой Н.
Пластовая вода восточных нефтяных месторожде-
ний СССР содержит от 15 до 25% хлористых солей,
поэтому даже невысокое содержание в Н. пластовой
воды (0,1—0,3%) связано с высоким содержанием
солей в Н. Наличие хлористых солей пагубно влияет
яа аппаратуру перегонных и крекинговых установок,
вызывая сильную коррозию оборудования нефтепере-
рабатывающих заводов. В присутствии сероводорода,
выделяющегося при переработке сернистых нефтей,
процесс хлористоводородной коррозии значительно
увеличивается.
Для полного удаления хлористых солей из Н.
(обессоливание) применяют электрообессоливающие
установки (ЭЛОУ), на к-рых Н. при тщательном пе-
ремешивании промывают 5—10% пресной воды с до-
бавкой деэмульгатора, образующуюся эмульсию по-
догревают до 80—120° и подают в электродегидраторы.
Под воздействием электрич. поля высокого напря-
жения (2,5—3 кв/см), деэмульгатора и темп-ры эмуль-
сия разрушается, вода отстаивается от Н. и уда-
ляется. После такой обработки Н. содержание влаги
15*
454
в ней достигает 0,1—0,3% и хлористых солей
10—40 мг/литр.
Многие легкие Н., содержащие большое количество
легких углеводородов, после обезвоживания и обес-
соливания подвергают стабилизации, т.е. отгон-
ке пропан-бутановой, а иногда частично и пентановой
фракции углеводородов. Стабилизация Н. произво-
дится на специальных установках, оборудованных
печами для ее подогрева и колонкой для отбора
пропан-бутановой фракции.
Удаление из Н. пропал-бутановой фракции углево-
дородов необходимо для того, чтобы снизить потери
ценных углеводородов при ее транспортировке и хра-
нении, а также обеспечить постоянное давление паров
Н., поступающей на нефтеперегонные установки.
Получаемая при стабилизации Н.пропал-бутановая
фракция углеводородов является ценным сырьем
для химич. пром-сти.
Переработка нефти. Основным (первичным) процес-
сом переработки Н. (после стабилизации, обезвожи-
вания и обессоливания) является ее перегонка (см.
Перегонка нефти), в результате к-рой, в зависимости
от поставленной цели, отбирают след, нефтепродук-
ты: бензин авиационный или автомобильный или
их компоненты; лигроин', реактивное и газотурбин-
ное топливо', керосин' дизельное топливо', мазут.
К вторичным процессам переработки Н., связанным с
изменением структуры углеводородов, входящих в ее
состав, относятся: крекинг, риформинг, гидроформинг,
платформинг, алкилирование, ароматизация нефте-
продуктов, изомеризация, полимеризация, гидрогениза-
ция деструктивная, пиролиз, коксование.' Нефтепро-
дукты, полученные при первичных или вторичных
процессах переработки Н., подвергают очистке (см.
Очистка нефтепродуктов). Для дальнейшего повы-
шения качества полученных нефтепродуктов к ним
добавляют различные присадки.
Влияние группового углеводородного состава неф-
тепродуктов на их свойства. Преобладание углеводо-
родов отдельных классов в различных фракциях Н.
(топливе и маслах) различно сказывается на их
свойствах. Так, напр.: а) бензиновые фракции,
содержащие значительное количество метановых угле-
водородов изомерного строения, нафтеновых и аро-
матич. углеводородов, отличаются высокими октано-
выми числами, в то время как метановые углеводороды
нормального строения отличаются низкими октано-
выми числами, причем, чем длиннее цепь, тем ниже
октановое число; октановое число метановых углево-
дородов изомерного строения повышается с увеличе-
нием разветвленности цепи при одном и том же числе
углеродных атомов;
б) дизельные топлива, в к-рых преобладают мета-
новые углеводороды нормального строения, отли-
чаются высокими цетановыми числами; чем более
разветвлена цепь у метановых углеводородов, тем
ниже их цетановое число. При одинаковом количестве
боковых цепей нафтеновые моноциклич. углеводо-
роды, как правило, обладают более высокими цетано-
выми числами, чем ароматические; с увеличением
количества циклов в молекуле цетановое число
уменьшается. Наличие ароматич. углеводородов, осо-
бенно полициклических, резко снижает цетановое
число дизельного топлива;
в) масла, в состав к-рых входят преим. нафтеновые
углеводороды с невысоким содержанием циклов в мо-
лекуле и длинными боковыми цепями, обладают
высоким индексом вязкости и низким значением вяз-
костно-весовой константы (зависи->
мость между вязкостью и плотностью). Нафтеновые
и ароматич. углеводороды с относительно высоким
содержанием циклов в молекуле обладают более вы-
сокой вязкостью и плотностью по сравнению с цик-
455
НИГРОЗИНЫ —НИКЕЛЬ
456
лич. углеводородами, кипящими в тех же темпера-
турных пределах, с относительно малым количеством
циклов.
Нефть как источник химического сырья. В послед-
ние годы наряду с увеличением выработки топлива и
масел Н. является все более широким источником
сырья для получения различных синтетич. продуктов
(искусственные и синтетич. волокна, пластич. массы,
синтетич. каучук, спирты, кислоты и др.). Необхо-
димое для нефтехимия, пром-сти сырье может быть
получено из Н. след, путями: а) различными физич
методами (перегонка, экстракция, кристаллизация,
адсорбция и др.), при помощи к-рых из Н. выделяют
необходимые индивидуальные углеводороды или их
классы; б) вторичной переработкой (пиролиз, йрекинг
и др.), связанной с изменением структуры углеводо-
родов, входящих в состав Н. Из метановых углеводо-
родов наибольшее применение для нефтехимия,
пром-сти нашли газообразные (при нормальных
условиях) или жидкие низкокппящие углеводороды
[метан, этан, пропан, бутан и пентан), а также
высокомолекулярные углеводороды с 10—20 атомами
углерода в молекуле. Из нафтеновых углеводородов
важнейшим исходным материалом для нефтехимия,
пром-сти является циклогексан, из ароматических —
бензол, толуол, ксилол, этилбензол. Из ненасыщен-
ных углеводородов (полученных при вторичной пере-
работке Н.) основным продуктом в качестве сырья
для нефтехимия, пром-сти являются этилен и ацети-
лен. Подробнее см. Нефтехимический синтез, Гази
нефтяные попутные, Газы природные горючие.
Лит.: НаметкинС. С., Химия нефти, М., 1955; Д о б-
рянскийА. Ф., Химия нефти, Л., 1961; Нефти восточных
районов СССР, под ред. С. Н. Павловой и 3. В. Дриацкой, Л.,
1958и 1962; Ивченко Е.Г., Севастьянова Г. В.,
Сернистые и высокосернистые нефти Башкирской АССР, М.,
1963; Скляр В. Т., Лебедев Е. В., Нефти Украины,
Киев, 1962; Павлова С. Н., Д р и а ц к а я 3. В„ М х чи-
я н М. А., Химия и технология топлив и масел, 1963, № 6;
АшумовГ. Г., Азербайджанские нефти, Баку, 1961; П е т-
р о в А. А., Обессоливание и обезвоживание нефтей, Куйбы-
шев, 1959. С. Н. Павлова, 3. В. Дриацкая, Д. Н. Левченко.
НИГРОЗИНЫ — органические красители черно-
го цвета, получаемые длительным нагреванием
(170—180°) смеси нитробензола и анилина (иногда
других нитро- и аминосоединений) с хлористым или
металлич. железом (стружкой). Образующийся плав
обрабатывают разб. соляной к-той, получая основа-
ние Н. пли Н. спирторастворимый, применяемый для
окраски пластич. масс и приготовления лаков. Кипя-
чением основания Н. с р-ром едкого натра получают
Н. жирорастворимый, к-рый широко используется
для приготовления типографских красок, гуталина,
окраски лент для пишущих машин и др. Сульфирова-
нием основание Н. превращают в Н. водорастворимый,
к-рым окрашивают прсим. кожу, а также древе-
сину, бумагу и др. Н. являются смесью азиновых
красителей. Нигрозины дешевы и вырабатываются
в значительных количествах во всех индустриальных
странах.
Лит.: Коган И. М., Химин красителей, 3 изд., М.,
1956, с. 357; Венкатараман К., Химия синтетических
красителей, пер. с англ., т. 2, Л., 1957, с. 887; Чекалин
М. А., Химия и технология органических красителей, М.,
<956. М. А. Чекалин.
НИКЕЛЬ (Niccolum) Ni — химич. элемент VIII гр.
периодич. системы Менделеева, п. н. 28, ат. в. 58,71.
Природный Н. состоит из смеси пяти стабильных изо-
топов: Ni58 (67,76%), Ni60 (26,16%), Ni61 (1,25%),
Ni63 (3,66%), Ni64 (1,16%). Поперечное сечение погло-
щения тепловых нейтронов атомом Н. 4,8 барн. Из
искусственно радиоактивных изотопов наиболее дол-
гоживущий Ni59 (Ti/^IO5 лет), образующийся при
облучении Н. нейтронами: Ni58 (п, у) Ni59. Конфигура-
ция внешних электронов атома 3 <384№. Энергии иони-
зации (в эв): Ni-^Ni + —Ni2+ -^Ni3+ соответственно
равны 7,633; 18,15; 35,16.
Мышьяковистый Н. NiAs (руда красного цвета) был изве-
стен еще в конце 17 в. После неудачных попыток получить
медь из этой руды ей было дано пренебрежительное название
Kupfernickel (от нем- Kupfer — медь и Nickel — назв. «злого
духа^>). В загрязненном виде Н. впервые получен в 1751
А. Кропштедтом, предложившим и название элемента. Значи-
тельно более чистый металл получил в 1804 И. Рихтер. До
80-х гг. 19 в. Н. использовался лишь как ювелирный металл.
Широкое развитие никелевой пром-сти в последующие деся-
тилетия связано с нахождением мощных месторождений нике-
левых руд в Невой Каледонии и в Канаде и открытием «обла-
гораживающего» его влияния на свойства сталей.
Содержание Н. в земной коре составляет 8.10“5
вес. %. В природе Н. встречается в виде сульфидных
медпоникелевых, окисленных (силикатных) и мышья-
ковистых руд (игследпие имеют второстепенное зна-
чение).
Наибольшие запасы Н. сосредоточены в суль-
фидных медно никелевых рудах,
имеющих магматич. происхождение и связанных с ос-
новными и ультраосновными породами. Руда хорошо
раскристаллизована и потому относительно легко
поддается механич. обогащению. Общее содержание Н.
в сульфидных рудах составляет 0,3—2,0%.
Основной никелевый минерал сульфидных руд — пен-
тландит (Fe, Ni)9S (железо-никелевый колчедан)— кубич.
кристаллы бронзово-желтого цвета с металлич. блеском;
плотн. 4,5—5; твердость по Моосу 3—4. Часть Н. представлена
другими сульфидами, преим. простейшими; к ним, в первую
очередь,относится миллерит NiS (желтый никелевый колче-
дан) — игольчатые кристаллы тригональной структуры; плотн.
5,2—5,6; твердость по Моосу 3—3,5. В рудах всех сульфидных
месторождений заметное количество Н. присутствует в виде
твердого р-ра в пирротине Fej-^S. Часть Н. в сульфидных
рудах (5—10%) находится в силикатной форме, изоморфно
замещая Mg в оливине, серпентине и др. минералах. В рудах
отдельных месторождений имеются железо-никелевые иитер-
металлич. соединения (аварцит Ni2Fe, джозефинит Ni3Fe).
Пустая порода сульфидных руд обычно отличается высоким
содержанием MgO и потому тугоплавка.
Сульфидные медноникелевые руды — ценное поли-
металлич. сырье. Наряду с Н. они содержат Си
(преим. в виде халькопирита СнЕебг. см. Медь), Со,
Au, Ag, платиновые металлы, редкие и рассеянные
элементы. Из этих руд добывается св. 90% Pt от
общей ее добычи. Наиболее крупные месторождения
сульфидных медноиикелевых руд находятся в Канаде
(район Седбери — Верхние озера); известны также
месторождения в Норвегии, Финляндии и др. странах;
в Советском Союзе на Кольском и Таймырском полу-
островах.
Окисленные никелевые руды — про-
дукт выветривания ультраосновных серпентинитовых
пород, представляют собой мелкую землистую массу.
Содержание в них Н. составляет 0,7—2%. В окислен-
ных рудах Н. лишь частично находится в виде само-
стоятельных минералов — водных силикатов или
алюмосиликатов, обычно изоморфно с магнием
(NiSiOs- nMgSiO3-mH2O). Ок. 50% И. изоморфно
замещает другие катионы в различных силикатах
пли просто адсорбировано последними. Вследствие
очень тонкого распределения Н. в окисленных рудах
их эффективное обогащение невозможно, п эти руды,
несмотря па относительно невысокое содержание Н.
(0,7—2%), непосредственно направляются на метал-
лургич. переработку. Ценным спутником Н. в окис-
ленных рудах является кобальт. Известны руды с по-
вышенным содержанием железа, что делает целесо-
образным его попутное извлечение. На долю окислен-
ных руд приходится ок. 30% от общих запасов Н.
в земной коре. Наиболее крупные их месторождения
находятся на острове Куба и в Новой Каледонии;
известны также месторождения в Бразилии, Китае,
Индонезии и др. В СССР основные месторождения
расположены на Среднем и Южном Урале и в Казах-
стане.
Физические и химические свойства. Компактный
Н.— металл серебристо-белого цвета с характерным
блеском, очень ковок и пластичен. Н., выделенный
из соединений пли сплавов,— серо-черный порошок.
457
НИКЕЛЬ
458
Н. образует две модификации: а-Н. с гексагональной
решеткой (а= 2,65 А, с = 4,32 А, ниже 250°) и Р~Н.
с гранецентрированной кубич. решеткой (а=3,5236 А,
при 0°). Переход а->р происходит при нагревании до
250—300°. Н. в виде компактного обработанного
металла (лист, проволока и т. п.) существует обычно
в fl-форме между 0°.и 1200°. Плотн. Н. 8,9 (20°). Атом-
ный радиус 1,24 А, ионные радиусы: Ni2 + 0,79 А,
Ni3 + 0,72 А. Т. пл. 1453°; т. кип. ок. 3000°. Теплота
плавления 73 кал/г’, теплота испарения 1487 кал/г.
Уд. теплоемкость 0,105 кал/г-град (20°); термич.
коэфф, линейного расширения 13,3-10-6 (0—100°);
теплопроводность (кал/см-град-сек) 0,215 (25°);
0,148 (500°). Уд. электросопротивление 6,84 мком-см
(20°); термич. коэфф, электросопротивления 6,8-10“3
(0-100°).
Н., как и его аналоги кобальт и железо, ферро-
магнитен; теряет ферромагнитные свойства при 358°
(точка Кюри). Магнитное насыщение 6084 гаусс (15°).
Коэрцетивпая сила для мягкого Н. 1,6 а. Все сплавы
Н. с другими ферромагнетиками также ферромаг-
нитны. В сплавах с парамагнетиками ферромагнитные
свойства, как правило, исчезают лишь при малых
концентрациях Н. Ферромагнетиками являются и
нек-рые соединения Н. ^NiO, NiS2, Ni3N).
Н,— ковкий и тягучий металл, из пего можно при-
готовить тончайшие листы и трубки. Предел проч-
ности при растяжении 40—50 кГ/мм2, предел упру-
гости 8 кГ/мм2, предел текучести 12 кГ/мм2. Отно-
сительное удлинение 40%; относительное сужение
70% . Модуль нормальной упругости 20 500 кГ/мм2.
Твердость по Бринеллю 60—80 кГ/мм2.
В химич. отношении Н. сходен, с одной стороны,
с железом и кобальтом, с другой — с медью и благо-
родными металлами. Как и др. элементы VIII гр., Н.
проявляет переменную валентность. Разнообразие
валентных состояний Н. особенно характерно для
комплексных соединений (см. Никеля комплексные
соединения). В простых соединениях Н. обычно
2-валентен. Н.— металл средней активности. В обыч-
ных условиях устойчив к действию атмосферных
газов, воды, галогенов, серы и др. При нагревании
(в особенности в измельченном состоянии) легко
реагирует с галогенами, S, Se, Р, As, Sb и др.
Н. (особенно в мелкораздробленном состоянии)
поглощает большие количества газов (Н2, СО и др.);
1 объем Ni поглощает: 3,87 (1600°); 3,50 (1465°);
1,54 (1400°); 0,542 (400°); 0,16 (212°) объемов Н2.
Насыщение Н. газами существенно ухудшает его ме-
ханич. свойства. Растворимость водорода в Ni значи-
тельно выше, чем в Fe, Со и Си, и резко увеличивается
при расплавлении. Определенных химич. соединений
при взаимодействии Н. и водорода не образуется.
Гидрид NiH2 можно получить действием сухого
безводного NiCl2 на NiMgBr в атмосфере На- Описаны
также гидриды NiH, NiH4. Гидриды Н. относятся
к группе металлич. гидридов — они представляют
собой твердый р-р водорода в металле; большая часть
водорода окклюдирована в межкристаллитных тре-
щинах. Теплоты образования NiH и NiH2 AZ7°298
соответственно равны (ккал/моль)’.—2,7 и —6,2. Спо-
собность поглощать водород и при этом его активи-
зировать дает возможность использовать Н. как ката-
лизатор гидрирования и дегидрирования (см. ниже).
Взаимодействие Н. с кислородом начинается при
500°. Однако металл, выделенный в очень мелкодис-
персном состоянии, пирофореп—на воздухе самовос-
пламеняется. Растворимость кислорода в твердом Н.
составляет (вес. %): 0,012 (1200°); 0,014 (1000°); 0,019
(800°) и 0,020 (600°). Из окислов Н. наибольший прак-
тич. интерес представляет NiO, образующаяся при
илавке Н. в присутствии кислорода. Поскольку NiO
в воде нерастворима, гидроокись Ni(OH)2 — основание
средней силы, может быть получена только косвен-
ным путем (см. Никеля окислы и гидроокиси).
В парах галогенов Н. горит, образуя соединения
NiX2 (см. Никеля галогениды). Н. горит также в парах
серы, образуя сплав, близкий по составу к Ni3S2. Из-
вестны и другие никеля сульфиды. С Se Н. образует
селениды NiSe и NiSe2, с Те — теллуриды
NiTe и NiTe2; для последних вероятен непрерывный
переход через твердые р-ры. С азотом Н. не реагирует
даже при высоких темп-рах (до 1400°). Растворимость
азота в твердом Н. приблизительно 0,07 вес. %
(445°). Нитрид Ni3N может быть получен пропу-
сканием NH3 над NiF2, NiBr2 или порошком метал-
лич. Н. при 445°. Ni3N — кристаллы серого цвета,
гексагональная решетка, а = 2,660 А, с = 4,294 А;
плотн. 7,66; на воздухе разлагается при 450°, в ва-
кууме — при 190°.
При действии на Н. при высокой темп-ре паров фос-
фора образуется фосфид Ni3P2 в впде серой массы,
напоминающей графит. В системе Ni—Р установлены
и др. фосфиды. Из них Ni3P (разл. при 970°), Ni5P2
(т. пл. 1175°), Ni2P (т. пл. 1110°) приготовлены пря-
мым сплавлением Н. с фосфором. Неустойчивые выс-
шие фосфиды Ni6Ps, NiP2 и NiP3 образуются при
сплавлении с фосфором низших фосфидов. Примесь
фосфора увеличивает хрупкость Н. и его сплавов и
понижает темп-ру плавления. Поэтому фосфор в ка-
честве раскислителя Н. и его сплавов не применяется.
В системе Ni—As установлено существование трех
арсенидов: Ni5As2, Ni3As2 и NiAs, из них Ni3As2
встречается в природе в виде минерала маухерита.
Известен также минерал раммельсбергит NiAs2 (синте-
зировать искусственно это соединение не удалось).
Растворимость As в твердом Н. составляет приблизи-
тельно 5,5 вес. % (900°).
Неустойчивый карбид Ni3C может быть полу-
чен медленным (сотни часов) науглероживанием (це-
ментацией) порошка Н. в атмосфере СО при 300°.
В жидком состоянии Н. растворяет заметное коли-
чество С, выпадающего при охлаждении в виде гра-
фита. При выделении графита Н. теряет ковкость
и способность обрабатываться давлением. В связи
с этим, несмотря на нек-рое сходство систем Fe—С
и Ni—С, последняя не имеет практич. ценности
с точки зрения улучшения свойств Н. Растворимость
углерода в твердом Н. составляет (вес. %): 0,55
(1318°), 0,5 (1265°) и 0,3 (1055°). При повышенных
темп-рах Н. взаимодействует также с окислами азота,
SO2 и NH3. При действии СО на тонкоизмельченный
порошок Н. при нагревании образуется никеля кар-
бонил Ni(CO)4. Термич. диссоциацией карбонила
получают наиболее чистый Н. (так наз. «карбониль-
ный» Н.). С многими металлами (Bi, Sn, Ti, U, V, W,
Zn, Zr и др.) H. образует иптерметаллич. соединения.
С элементами VIII гр., а также с Си и нек-рыми др.
Н. дает непрерывный ряд твердых р-ров (см. Никеля
сплавы).
В ряду напряжений Н. стоит правее железа (нор-
мальные потенциалы Fe и Ni соответственно равны
—0,44 v и —0,24 v) и поэтому медленнее, чем Fe, рас-
творяется в разб. к-тах. Этому способствует также его
анодная пассивация. По отношению к воде (а также
к совместному действию воды и воздуха) Н. устойчив.
Органич. к-ты — уксусная, щавелевая и др., дейст-
вуют на Н. лишь после длительного соприкосновения
с ним. Серная и соляная к-ты медленно растворяют
Н.; в разб. азотной к-те Н. растворяется очень легко.
Конц. HNO3 пассивирует Н., однако в меньшей сте-
пени, чем железо. Сильные щелочи на Н. не действуют,
но он растворяется в аммиачных р-рах в присутствии
(NH4)2CO3 с образованием растворимых аммиакатов.
При взаимодействии с кислотами образуются соли
2-валентного Н. Почти все соли Ni2+ и сильных к-т
459
НИКЕЛЬ
460
хорошо растворимы в воде, р-ры их вследствие гидро-
лиза имеют кислую реакцию. Труднорастворимы
соли таких сравнительно слабых к-т, как угольная
и фосфорная. Большинство солей Н. разлагается при
прокаливании (600—800°). При действии на р-ры солей
Н. сероводорода в присутствии CH3COONa количест-
венно осаждается сульфид Ni3S. Щелочи осаждают
нерастворимый в избытке реактива Ni(OH)3 яблоч-
ного цвета, не окисляющийся на воздухе. KCN дает
с никелевыми солями светло-зеленый Ni(CN)3, к-рый
при избытке реактива превращается в комплексное
соединение K2[Ni(CN)4J-
Аммиак осаждает из р-ров никелевых солей зеленый
осадок основных солей, к-рый растворяется в избытке
реактива с образованием аммиакатов. Аммиакаты
можно получить при пропускании NH3 через безвод-
ную соль Н. или при приливании водного р-ра NH3
к р-рам солей. Для большинства аммиакатов характер-
но наличие комплексных ионов [Ni(NH3)J2 + и
[Ni(OH)2(NH3)4]z + . Все аммиакаты Н. окрашены
в интенсивно синий цвет. На избирательном образова-
нии аммиакатов Н. основываются гидрометаллургия,
методы его извлечения из руд. Разложение аммиакатов
обычно проводят пропариванием р-ров.
Соли Ni2+ устойчивы по отношению к кислороду
воздуха, медленно окисляются при действии озона.
NaOCl и NaOBr осаждают из р-ров солей Ni2 + черный
Ni(OH)s. О важнейших солях Н. см. Никеля нитрат,
Никеля карбонат. Никеля сульфат.
Аналитическое определение. В си-
стематич. ходе анализа при определении Н. прежде
всего осаждают из р-ра сероводородом металлы серо-
водородной группы и действием брома и аммиака —
гидроокиси Fe, Al и Mg. После этого из элементов,
мешающих определению Н., в р-ре остается один Со.
При действии на р-р (NH4)2S выпадают сульфиды Ni
и Со. От кобальта Н. можно отделить, осаждая его
щелочью и бромом или хлором из р-ра комплексных
цианидов (Со остается в р-ре), или осаждением Н.
в виде диметилглиоксимата. Реакция Н. с диметил-
глиоксимом — реактивом Чугаева — идет только
в аммиачной, нейтральной или слабокислой среде.
Кобальт хотя и не дает с диметилглиоксимом осадка,
но образует с ним растворимое комплексное соеди-
нение; при больших количествах Со это снижает
чувствительность реакции. Кроме того, в присутствии
комплексных соединений повышается растворимость
диметилглиоксимата Н. Если содержание Со превы-
шает содержание Н. более чем в 200 раз, то последний
при осаждении диметилглиоксимом не обнаружи-
вается. Для уменьшения вредного влияния Со (II)
его окисляют в аммиачных р-рах до Со (III). Реактив
Чугаева можно применять в весовом, в объемном
(титрование 1%-ным щелочным или водным р-ром
диметилглиокслма) и в колориметрия, методах, а
также для микроаналитич. определения Н. Для опре-
деления Н. используют также дициандиамидин суль-
фат (C2HsN4O)2-H2SO4— реактив Гроссмана. Он об-
разует с солями Н. в щелочном аммиачном р-ре жел-
тый осадок; Н. определяют также электролитически
на Pt-катоде после тщательного отделения элементов
сероводородной группы и группы сульфида аммония,
а также удаления соляной и азотной к-т.
Получение. Ок. 80% Н. от общего его произ-ва (без
СССР) получают из сульфидных медноникелевых руд.
Извлечение Н. из руд—сложный, многостадийный
процесс. Прежде всего руда подвергается селектив-
ному обогащению методом флотации с выделением
медного и никелевого концентратов (4—5 % Ni).
Никелевый концентрат в смеси с флюсами плавится
в электрич., шахтных или отражательных печах
с целью отделения основной массы пустой породы
и извлечения Н. в сульфидный расплав (штейн), содер-
жащий 10—15% Ni; обычно электроплавке (основной
метод плавки в СССР) предшествует частичный оки-
слительный обжиг и окускование концентрата мето-
дом агломерации или окатывания. Наряду с Н.
в штейн переходит часть Fe, Со и практически пол-
ностью Си и благородные металлы. Для отделения Fe
его окисляют продувкой жидкого штейна в конверте-
рах; в результате получают сплав сульфидов Си и
Ni — файнштейн, к-рый после медленного охлаждения
тонко измельчают и направляют на флотацию для
разделения Си и Ni. Полученный никелевый концен-
трат обжигают в кипящем слое до практически пол-
ного удаления серы и получения NiO. Металлич. Н.
получают восстановлением NiO в электрич. дуговых
печах. Черновой Н. разливают в аноды и подвергают
электролитич. рафинированию. Содержание примесей
в электролитном Н. (марка Н0)~0,01%.
Кроме описанной технологии, для разделения Си
и Ni используют т. н. карбонильный процесс, ос-
нованный на обратимости реакции: Ni + 4СО =
= Ni(CO)4. Получение карбонила Н. проводят обычно
при 100—200 атм и при 200—250°, а его разложение—
при атмосферном давлении и ок. 200°. «Карбониль-
ный» никель получается в виде порошка высокой
чистоты. Разложение Ni(CO)4 используют также для
получения никелевых покрытий и изготовления раз-
личных изделий (разложение на нагретой матрице).
Б последние годы получают распространение авто-
клавные методы растворения сульфидных никелевых
концентратов в аммиачных р-рах в присутствии О3
с последующей обработкой р-ров водородом под дав-
лением для осаждения металлич. порошка Н.
Из окисленных руд Н. также может быть сконцент-
рирован в штейне при введении в шихту плавки серусо-
держащих флюсов (гипса или пирита). Восстанови-
тельно-сульфидирующую плавку проводят в шахтных
или электрич. печах, образующийся штейн содержит
16—20% Ni, 16—18% S, остальное Fe. Технология
извлечения Н. из штейна аналогична описанной
выше, за исключением операции отделения Си, к-рая
часто просто выпадает. При малом содержании в окис-
ленных рудах Со их целесообразно подвергать вос-
становительной плавке с получением ферроникеля,
направляемого в качестве легирующей добавки на
произ-во стали. Выделение Н. из его сплава с желе-
зом — довольно сложная и трудоемкая операция.
Для извлечения Н. из окисленных руд применяются
также различные гидрометаллургии, методы —ам-
миачное выщелачивание предварительно восстанов-
ленной руды; сернокислотное автоклавное выщела-
чивание и др.
Применение. Подавляющая часть Н. используется
для получения сплавов с другими металлами (Fe,
Сг, Си и пр.), отличающихся высокими механич.,
антикоррозионными, магнитными или электрич. и
термоэлектрич. свойствами. Особенно важны в связи
с развитием реактивной техники и созданием газотур-
бинных установок жаропрочные и жаростойкие хро-
моникелевые сплавы. В последние годы сплавы Н.
используются в конструкциях атомных реакторов (под-
робнее см. Никеля сплавы).
Значительное количество Н. расходуется для
произ-ва щелочных аккумуляторов и антикоррозион-
ных покрытий. Ковкий Н. в чистом виде применяют
для изготовления листов, труб и т. д. Он исполь-
зуется также в химич. пром-сти для изготовления
специальной химич. аппаратуры и как катализатор
многих химич. процессов. Н. — весьма дефицитный
металл и по возможности должен заменяться другими,
более дешевыми и распространенными материалами.
А. В. Ванюков.
Никель как катализатор. Металли-
ческий Н. в дисперсной форме широко применяют
461
НИКЕЛЯ ГАЛОГЕНИДЫ —НИКЕЛЯ ДИЭТИЛ ДИТИОФОСФАТ
462
как катализатор. Методы получения дисперсного Н.
различны. Чаще всего используют скелетный Н.,
получаемый сплавлением Н. с алюминием (также
с кремнием) с последующим удалением А1 и Si из
сплава при помощи щелочи. Такой Н. часто называют
никелем Ренея (по имени исследователя,
взявшего на него патент в 1927). В СССР скелетный
Ni-катализатор с неполностью выщелоченным А1
называют катализатором Бага-Егупова. Активные
катализаторы получаются также при нанесении Н.
на носитель: А12О3, MgO, кизельгур и др. Для их полу-
чения либо пропитывают носитель р-ром солп И.
(хлорид, нитрат), либо наносят на носитель NiO или
NiSOs с последующим восстановлением водородом
при низкой темп-ре (150—200°). Реже (напр., при
жидкофазном гидрировании растительных масел)
получают высокодисперсный Н. без носителя путем
восстановления окиси или солей Н. водородом не-
посредственно в реагирующей среде. Металл, выделен-
ный в мелкодисперсном состоянии пирофореп, на воз-
духе самовоспламеняется. Н. — основной катализа-
тор гидрогенизации. На скелетном Н. уже при комнат-
ной темп-ре водород легко присоединяется по связи
С=С. На Ni-катализаторах осуществляют также в ла-
боратории и в пром-сти гидрогенизацию ацетиленовых
и ароматич. углеводородов, связей С=О, C -N, NO2,
деструктивное гидрирование с разрывом связей С—С,
С—S и др. В зависимости от условий опыта (темп-ра,
давление) и формы Ni-катализатора можно осущест-
вить избирательную гидрогенизацию. Напр., ацети-
лен при низких темп-рах на скелетном Н. гидрируется
до этана и на Н., нанесенном на кизельгур,— до эти-
лена. Н. является также активным катализатором
дегидрогенизации, однако на практике используется
для этой цели редко из-за его спекаемости при высокой
темп-ре (в условиях дегидрогенизации) и способности
вызывать побочные реакции (напр., крекинг). На
Ni-катализаторах протекают также реакции окисле-
ния, изомеризации, конденсации и др. Промышленное
значение имеет конверсия метана водяным паром
(см. Конверсия газов) на Н. с добавкой А12О3, синтез
углеводородов из СО + Н2О на Ni 4- А12О3 и др.
О. В. Крылов.
Лит.: Корнилов И. И., Никель и его сплавы, М.,
1958; Туркин В. Д., Румянцев М. В., Структура и
свойства цветных металлов, М., 1947; Цейдлер А. А.,
Металлургия меди и никеля, М., 1958; Орм о нт Б. Ф.,
Структуры неорганических веществ, М.—Л., 1950; К у ба-
in е в с к и й О., Эванс Э,, Термохимия в металлургии, пер.
с англ., М., 1954; Богословский Б. М., Казакова
•3. С., Скелетные катализаторы. Их свойства и применение
в органической химии, М., 1957; Сокольский Д. В.,
Гидрирование в растворах, Алма-Ата, 1962; Mellor, v. 15,
L.—N. Y.— Toronto, 1947; то же, v. 15, 1953; Pascal,
t. 18, P., 1959; Ullmann, Bd 12, Munch. —B., 1960, S.
690—731; К i г k, v. 9, N. Y., 1952, p. 271, 304; Sidgwick
N. V., Th? chemical elements and their compounds, v. 2, Oxf.,
1951; Глазковский А. А., Никель, M., 1963.
НИКЕЛЯ ГАЛОГЕНИДЫ — соединения никеля
с F, Cl, Вг и J, в к-рых Ni обычно 2-валентен. Из
р-ров кристаллизуются гидраты Н. г. Безводные Н. г.
можно получить либо непосредственно из элементов,
либо прокаливанием соответствующих гидратов или
аммиакатов. При обработке р-ров Н. г. щелочью
могут быть получены оксигалогениды Ni.
Никеля фторид NiF2 — прпзматпч. кри-
сталлы от светло-коричневого до зеленого цвета;
тетрагональная решетка типа рутила, а = 4,719 А,
с = 3,124 А; плотн. 4,641 (20°). Теплота образования
ДЯ 298 = —159,5 ккал/моль. Безводный NiF2 очень
плохо растворим в воде; растворимость (в вес.%):
2,49 (10°) и 2,52 (90°); нерастворим в бензоле, спирте
и эфире. Известны кристаллогидраты NiF2c3,4n6 мо-
лекулами Н2О. Светло-зеленые кристаллы NiF2-
• ЗН2О (плотн. 2,014 при 19°) получены при упарива-
нии р-ра плавиковой к-ты, нейтрализованной карбо-
натом никеля; светло-зеленые кристаллы NiF2-4H2O
(плотн. 2,219 при 25°) — при действии 40%-ной HF
на р-р Ni(OH)2. Оксифторид состава 2NiO-2NiF2.
• Н2О образуется при выпаривании с HF р-ра NiCO3
в плавиковой к-те прп избытке карбоната.
Безводный фторид никеля устойчив к действию хо-
лодных соляной и азотной к-т и серной к-ты (даже при
кипении). При нагревании в атмосфере NH3 или Н2
NiF2 восстанавливается до металлич. Ni. Фторид Ni
образует двойные соли с фторидами Na, К, NH4,
Be, Zr, Се, Ti. Трифторид никеля NiF3 образуется при
электролизе р-ра NiF3 в конц. плавиковой к-те.
Никеля хлорид NiCl2 — золотистые гекса-
гональные кристаллы, а=3,527 А, с = 17,31 А,
плотн. 3,508 (25°). Теплота образования ДЯ^ =
= —75,5 ккал/моль. NiCl-з возгоняется при 987°. При
нагревании на воздухе легко превращается в NiO.
При нагревании в токе Н2 или NH3 легко восстанав-
ливается до металла. В воде и в спирте NiCl2 легко
растворим, нерастворим в жидком NH3; в органич.
растворителях NiCl2 гораздо менее растворим, чем
СоС12. Растворимость в воде (вес. %): 34,8 (0°),
39,6 (25°) и 46,7 (100,2°). Ниже перечислены гидраты
NiCl2 и их пределы устойчивости:
NiCl2-7H2O (до -33,3°),
NiCl2-6H2O (от —33,3° до +28,8°),
NiCl2-4H2O (от 28,8° до 64,3°),
NiCl2-2H2O (выше 64,3°),
Первые трп соединения зеленого цвета, дигидрат •—
желтого. С хлоридами щелочных металлов NiCl2
образует двойные солп. Безводный NiCl2 при комнат-
ной темп-ре реагирует с NH3 с образованием NiCl2-
• 6NH3. При нагревании NiCl2-6NH3 до 120° образует-
ся NiCl2-2NH3, при дальнейшем нагревании — NiCl2
NH3. Безводный NiCl2 получают прокаливанием по-
рошка Ni с С12, действием СС14 на NiO и др. способами.
NiCl2 применяют как катализатор — переносчик хлора
в реакциях с участием НС1 или С12 (соответственно
NiBr2 — в реакциях с участием НВг и Br2; NiJ 2 —
в реакциях с участием HJ и J2). При восстановлении
NiCl2 водородом получают высокодисперсный Н.,
используемый в качестве катализатора.
Никеля бромид NiBr2 — желто-коричневые
гексагональные кристаллы, а = 3,715 А, с = 18,30 А;
плотность 5,25. Теплота образования ДЯ298 =
= —54,2 ккал/моль. Хорошо растворим в воде, спирте,
ацетоне. Растворимость в воде (вес. %):: 53, (0°),
57,3 (25°), 60,8 (100°). Образует кристаллогидраты
с 9,6 и 3 молекулами Н2О.Из них NiBr2 6H2O устой-
чив при обычной темп-ре.
Никеля иодид NiJ 2 — черно-зеленые гекса-
гональные кристаллы, а = 3,895 А, с = 19,63 А,
плоти. 6,36. Теплота образования ДЯ°298 =
= —20,5 ккал/моль. Хорошо растворим в воде и орга-
нич. растворителях; растворимость в воде (вес. %):
55,4 (0°),60,7 (25°), 65,3 (90°). Из водных р-ров выпа-
дает в виде NiJ2-6H2O. Бромид и иодид никеля при-
меняют как катализаторы. А. В. Ванюков.
НИКЕЛЯ ДИЭТЛЛДИТИОФОСФАТ,мол. в.429,15,
[(C2HSO)2 PSS]2Ni — кристаллы синего цвета, т. пл.
104—105°; растворимость в воде 6-10-2 моль/л',
растворим в бензоле, эфире, четыреххлористом угле-
роде, дихлорэтане, хлороформе; при нагревании рас-
творяется в спирте и конц. уксусной к-те; негигроско-
пичен, устойчив как в сухом виде, так и в водных
р-рах. Н. д. может быть получен при взаимодействии
р-ра диэтплдитиофосфорной к-ты (из P2Ss и С2Н5ОН)
с насыщенным р-ром хлорида никеля. Н. д. может быть
использован для определения Cu2 + , Cdz+, Pb2 + ,
Bi’ + , Pd2+.
Лит.: Б у с e в А. И., Иваяютин М. И.,«Тр. Комис,
по аналит. химии АН СССР», 1960, 11, 172. И. П. Ефимов.
463
НИКЕЛЯ КАРБОНАТ —НИКЕЛЯ КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ
464
НИКЕЛЯ КАРБОНАТ NiCO3 — в безводном со-
стоянии неустойчив; на воздухе легко присоединяет
воду, образуя кристаллогидрат. Безводный Н. к.
может быть получен нагреванием кристаллогидрата в
запаянной трубке. Безводный Н. к.— голубовато-
зеленые кристаллы, очень плохо растворимые в воде
и холодной НС1, но растворимые в сильных к-тах.
Теплота образования АЯ2В8= —163,2 ккал/моль.
При действии на р-ры солей Ni(II) бикарбонатов
(NaHCOs) выпадает NiCO3-6Н2О — бледно-зеленые
ромбоэдрич. кристаллы. При действии средних карбо-
натов (Иа2СОз)на р-ры солей Ni(II) образуются основ-
ные гидратированные соли типа NiCO3 -2Ni (ОН)2 •
•геН2О. Одна из таких солей NiGO3 •2Ni(OH)2-4H2O
встречается в природе в виде минерала заратита
изумрудно-зеленого цвета. Основные Н. к. практиче-
ски нерастворимы в воде и угольной кислоте, однако
легко растворяются в сильных кислотах, в растворе
K.CN и нагретом р-ре NH4C1. Основной Н. к.— про-
межуточный продукт при аммиачном выщелачивании
никелевых руд и концентратов. Выщелачивание про-
изводят водным р-ром NH3, содержащим (NH4)2GO3,
в присутствии кислорода; после отгонки из р-ра NH3
осаждается основной Н. к., к-рый подвергают об-
жигу. В гидрометаллургии никеля и кобальта основ-
ной Н. к.— промежуточный продукт, используется
для очистки сульфатных никелевых р-ров от Си и Fe
путем нейтрализации избыточной кислотности рас-
ТВОров. А. -В. Ванюков.
НИКЕЛЯ КАРБОНИЛ Ni(CO)4 — бесцветная лег-
коподвижная жидкость с характерным резким и не-
приятным запахом; т. пл. —19,3°; т. кип. 43°; плотн.
1,36153 (0°); показатель преломления п'^ 1,4542;
вязкость 0,27 спуаз (0°). Критич. константы: /крит.
191 —195°, рКТ)иг 30—40amai, с/крит —0,46. Давление па-
ра (в мм рт. ст.): 133,1 (2,0э°);'238,2 (15,27°); 444,2
(29,52°); 647,2 (39,97°). Пары Н. к. в присутствии
воздуха взрывоопасны. В твердом состоянии Н. к,—
игольчатые кристаллы кубич. структуры, а = 10,84А
(—55°); плотн. твердого 1,78 (—55°). Теплота обра-
зования Н. к. по реакции Ni (тв.) + 4СО (газ) =
= Ni(CO)4 (газ), ДЯ,°8 = — 35 ккал/моль. Струк-
тура молекулы Ni(CO)4 отвечает правильному тет-
раэдру с атомом Ni в центре (см. Никеля комплекс-
ные соединения).
В воде II. к. почти нерастворим, однако при со-
прикосновении с водой несколько гидролизуется,
выделяя зеленоватый осадок неопределенного со-
става. Хороню растворяется в органич. растворите-
лях — спирте, бензоле, ацетоне и др. С разб. щело-
чами и кислотами не реагирует. Конц. IINO3 и цар-
ской водкой энергично разлагается. При хранении
II. к. в запаянной трубке он вполне устойчив. С
кислородом воздуха заметно реагирует уже на холо-
де, давая смесь различных продуктов окисления.
При нагревании на воздухе Н. к. разлагается с вос-
пламенением и взрывом. С хлором Н. к. реагирует
также энергично, образуя NiCl2 и фосген. Не менее
энергично идет реакция с бромом и иодом. Аммиак и
азот с Н. к., по-видимому, не взаимодействуют.
Н. к. токсичен. Ядовитость его в пять раз выше,
чем у СО. При содержании в атмосфере 0,5 мол. %
Н. к. воздух становится опасным для дыхания. Для
защиты нужен изолирующий противогаз или маска
с принудительной подачей воздуха.
Н. к. можно получить действием СО на тонкоиз-
мельченный активный порошок Ni при 50—80°. При
180—200° Н. к. разлагается. На обратимости реак-
ции Ni+4CO Ni(CO)4 основан промышленный ме-
тод получения наиболее чистого никеля в виде тонко-
дисперсного порошка. В пром-сти Н. к. получают
обработкой никелевого файнштейна или меднопике-
левых штейнов (см. Никель) окисью углерода под
давлением в 200 атм и темп-ре 200—250°. Разложе-
ние Н. к. ведут при 180—250° при небольшом (200 мм
вод. ст.) избыточном давлении в герметичной аппа-
ратуре. Термич. разложением Н. к. производят
плакированные изделия из металла и керамики. Как
сам Н. к., так и «карбонильный никель» служат ката-
лизаторами в ряде химич. процессов. См. также Кар-
бонилы металлов.
Лит.: Белозерский Н. А., Карбонилы металлов,
М., 1958; Поз ин М. Е., Технология минеральных солей,
2 изд., Л., 1961. См. также лит. при ст. Никель. А. В. Ванюков.
НИКЕЛЯ КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ. Ни-
кель как 2-валентный, так и в менее обычных ва-
лентных состояниях образует многочисленные ком-
плексные соединения. Наиболее распространены
комплексы Ni(II). Комплексы Ni(0), Ni(I), Ni(III)
и Ni(IV) немногочисленны и мало изучены. Для
Ni(0) и Ni(I) характерно координационное число 4;
для Ni(II) — 4 и 6; наконец, Ni(III) и Ni(IV) исклю-
чительно гексакоординационны. Структурно ком-
плексы никеля охарактеризованы неполно.
Как показало изучение дифракции электронов,
карбонил [Ni(CO)4] и нек-рые др. производные Ni(0)
имеют тетраэдрич. строение. Комплексы Ni(I) в ряде
случаев имеют полимерное строение; напр., рентге-
ноструктурные исследования показали, что ион
[Ni(CN)3]2- построен следующим образом:
NC. ZNC. ,CN~I 4 —
xNi( XN1(
NCZ NC' XCNJ
Нек-рые производные Ni(II) с координационным чис-
лом 4 имеют плоское строение. Таковы, например,
[Ni(GN)4]2- и многие внутрикомплексные соединения.
Существуют указания (изучение магнитных свойств,
цветности, спектров поглощения), что аналогичные
по составу соединения [Ni(NH3)4X2], где X—С1, Вг,
SCN, содержат тетраэдрически построенный ион
[Ni(NH3)4]2-. Однако недавно было показано (ре-
зультаты рентгеноструктурного исследования), что
в твердом состоянии такие соеди- х
нения содержат Ni(II) с коорди- I
национным числом 6 (а не 4) и по- ———у
строены октаэдрически: / Ni /
Гексакоординационные Ni(III) и ---------1—NH
Ni(IV) характеризуются окта- 3 3
эдрич. строением.
Для Ni характерно образование следующих типов
комплексных соединений:
1. Комплексы, к-рые формально можно представить
как продукты сочетания простых солей никеля (напр.,
NiGl2) с соединениями, молекулы к-рых содержат
N, S, О и др. атомы, обладающие свободной парой
электронов {напр., с этилендиамином NH2—СН2—
—СН2—NH2 (обозначают еп), пиридином G3H5N (обо-
значают ру), тиомочевиной (NH2)2CS (обозначают
thio)}. Таковы производные: а) гексаминового типа,
напр. [Ni(NH3)6]X2, [Nien3]X2, где X—Cl, Вг, J,
NO3, ‘ASCU и т. п. Известны смешанные гексамины,
содержащие во внутренней сфере молекулы различ-
ных аддендов, напр. [Nipy2thio4](NO3)2; б) пента-
минового типа, среди к-рых описан только [Nipya
thio2Br]Br, строение к-рого не изучено. Ni(0) и
Ni(I) соединений гекса- и пентаминового типов не
образуют. Пента- и гексамины Ni(III) и Ni(IV) пока
неизвестны, хотя их существование принципиально
возможно, в) тетраминового типа, наир. [Nipy4J2],
[Nithio4Cl2], [Ni(CO)4]; г) диаминового типа, напр.
[Nipy2X2], где X—CI, Вг, NO2 и др. Существование
диаминов, содержащих Ni в других валентных со-
стояниях, принципиально возможно. Комплексные
три- и моноамины Ni не получены.
465
НИКЕЛЯ КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ —НИКЕЛЯ ОКИСЛЫ И ГИДРООКИСИ
466
2. Комплексные производные типа двойных солей,
наир.:
K1[Ni(CN)1], К3 [Ni(CN)3], Na, [Nl (СВД
K2[N1FB], K[N1C13], Cs[N’iBr3].
Два последних, по-видимому, существуют только в
твердом состоянии.
3. Внутрикомплексные соединения, довольно мно-
гочисленные у Ni(II), напр.:
.оно ч
CH3-C=N< )N=C—СН3
I > Nl / |
СН3—C=N( >N=C—CH3
хонох
Образование внутрикомилексных соединений Ni(0),
Ni(I), Ni(HI) и Ni(IV) не характерно.
4. Многоядерные соединения, напр.:
PNC, ,NC, .CNT
К, >N1< )Ni(
Lncz NCZ xcnJ
,С1 1
en,Ni; 3,Nien, Cl2
XC1Z J
Существование многоядерных соединений, содержа-
щих Ni в других валентных состояниях, принци-
пиально возможно.
5. Соли поликислот с Ni(II) в качестве комплексо-
образоватсля, напр. (NHiJsH, [Ni(MoO4)6]-3H2O.
Изомеры соединений Ni(0), Ni(I), Ni(III) и Ni(IV)
неизвестны. Связи Ni(II) — адденд носят в значитель-
ной степени ионный характер, поэтому для комплек-
сов Ni(II) также не характерно проявление изомерии.
Комплексы Ni(II), геометрия, изомерия к-рых принци-
пиально возможна (напр., [Nipy2Cl2], [Nipy2thio2Cl2],
как правило, обладают шрояс-строением; соответст-
вующие цис-изомеры неизвестны. Есть сведения отно-
сительно существования оптич. изомеров нек-рых
комплексов Ni(II), напр.:
Природа отдельных описанных типов
плексвых соединений Ni(II) остается
Напр., глноксимины типа
R"-C=N-OH\ 1
1 N1
r'-c=n-o /2 J
изомерии ком-
невыясненной.
существуют в виде двух форм. Предполагается, что
отличие между ними сводится к различному положе-
нию групп R' и R"
.оно, ОНО,
R'-C = N( )N=C-R' r'-c. = n< )n = c-r"
I ; Nl / i и | у Ni ; i
R"-C=N( )N=C-R" R"-C=N( 7N=C-R'
XOHO7 ЩНО7
Наиболее характерно для Ni образование связей
Ni—N, Ni—S, Ni—As и Ni—P. Галогеносоединения
Ni(II) нестойки и мало изучены. Образование свя-
зей Ni—О более характерно, чем Pt—О или Pd—О.
R ряде Pt—Pd—Ni усиливается ионный характер
связи центральный ион—адденд. Поэтому скорость
взаимодействия с водой аналогичных комплексов
Pt(II), Pd(Il), Ni(II), напр. соединений типа [МеА2Х2],
где Me—Pt, Pd, Ni; A—py, NH3; X—Cl, Er, NO2,
увеличивается. В водном р-ре комплексы Ni(II) легко
распадаются по схеме:
[Nipy3Cl2]-*-6H2O [Ni (H2O)„r-+ +2ру + 2С1-
B неводных растворителях (напр., метиловом или
этиловом спиртах) диссоциация комплексов на цен-
тральный ион и аоденды протекает медленнее. Воп-
рос о проявлении трансвлияния в комплексах Ni
пока остается открытым, хотя нек-рые результаты
изучения реакций внутрисферного замещения ком-
плексов Ni(H) подтверждают справедливость зако-
номерности трансвлияния для этих комплексов.
Однако, вследствие уменьшения устойчивости ком-
плексов в ряду Pt—Pd—Ni, направление реакций
внутрисферного замещения в химии Ni(Il) часто опре-
деляется не закономерностью трансвлияния, а зави-
сит от образования малорастворимых веществ, т. е.
происходит в соответствии с правилом Бертолле.
Химия соединений Ni(0), Ni(I), Ni(III) и Ni(IV)
изучена мало. Комплексные производные Ni(0) и
Ni(I), напр. K.4[Ni(CN)4] или K2[Ni(CN)3], получаю-
щиеся при восстановлении комплексов Ni(II) в ам-
миачном р-ре при действии металлич. К или Na, об-
ладают сильно выраженными восстановительными
свойствами. Для нек-рых комплексов Ni(I) характер-
ны реакции присоединения. Напр., комплексный ион
[Ni(CN)s]2— легко присоединяет молекулы СО или
NO с образованием карбонильных или нитрозильных
соединений [Ni(CN)3COj2- или [Ni(CN)3NO]2-, имею-
щих полимерное строение.
Производные Ni(III) и Ni(IV) образуются при окис-
лении соответствующих соединений Ni(ll). Ком-
плексы Ni(III) HNi(lV) характеризуются окислитель-
ными свойствами. н. Н. Желиговская.
НИКЕЛЯ НИТРАТ Ni(NOs)2 — зеленовато-жел-
тые очень гигроскопичные кристаллы. Раствори-
мость в воде (вес. %): 44,2(0°), 50(25°) и 69,2(99,4°).
Из водных р-ров (зеленого цвета) кристаллизуются
гидраты Н. н.: до —3° Ni(NO3)2 -ЭЩО, от —3° до
54° Ni(NO3)2 -6Н2О, от 54° до 85,4° Ni(NO3)2 -4Н2О,
выше 85,4° Ni(NO3)2 -2Н2О. Устойчивый при обычных
условиях гексагидрат имеет плотн. 2,037 (22°); пла-
вится в кристаллизационной воде при 56,7°. При 105°
Н. н. разлагается с образованием чистого тонко-
дисперсного NiO, что используется для получения
последнего. При восстановлении Н. н. водородом
образуется металлич. Ni с высокоразвитой поверх-
ностью, используемый как катализатор Ni(NOs)2, слу-
жит промежуточным продуктом при получении и др.
соединений Ni, напр. Ni(OH)3, применяющегося в.
щелочных аккумуляторах. А. В. Ванюков.
НИКЕЛЯ ОКИСЛЫ И ГИДРООКИСИ — из окис-
лов никеля достоверные сведения имеются только
о моноокиси NiO. Существование соединений NiO2,
NiOs, Ni2Os и NisO4 доказано недостаточно убеди-
тельно; согласно Б. Ф. Ормонту и Н. Сиджвику, выс-
шие окислы никеля при обычной темп-ре и давлении
термодинамически неустойчивы.
Закись NiO при комнатной темп-ре — зеленова-
тые кристаллы с ромбоэдрич. решеткой, а = 2,9518 А,
а = 60°4,2'(18°); плотн. 7,45(20°). Выше 200° пере-
ходит в кубическую модификацию (тип NaCl), а =
— 4,1946 А (275°). Теплота образования ЛЯ°98 =
= — 58,4 ккал/моль. Т. пл. 1950°. На основании рент-
геновского анализа и измерения электропроводности
предполагается, что в решетке NiO может растворять-
ся избыток кислорода с образованием не занятых
никелем узлов, при этом цвет ее становится черным.
В воде NiO практически нерастворима. Если она
приготовлена при не очень высокой темп-ре, то легка
растворима в кислотах. Прокаливание при высокой
темп-ре (—1000°) резко снижает ее растворимость
в разб. к-тах. Водородом и окисью углерода при
нагревании NiO легко восстанавливается до металла.
В природе NiO встречается в виде минерала бун-
зенита. Искусственно ее получают прокаливанием
Ni(OH)2 или NiCOs при 600° или Ni(NOs)2 при 1000°
в отсутствии воздуха. Применяют в керамич. пром-сти
для приготовления красок и эмалей: используют для
467
НИКЕЛЯ СПЛАВЫ
468
получения катализаторов и как полупроводниковый
материал.
Гидроокись 2-валентного никеля Ni(OH)2
выпадает из р-ров никелевых солей при прибавлении
шелочей (рН7) в виде объемного осадка яблочного
цвета, к-рый при долгом стоянии в соприкосновении
с раствором переходит в зеленые .гексагональные
кристаллы с параметрами а = 3,123А, с = 4,604А;
плотн. 4,09. Теплота образования ДЯ2°8=— 128,7
ккал/молъ. При прокаливании при 230° переходит в
NiO. Последние следы воды удаляются при 360°. В кис-
лотах, а также в р-рах аммиака
и солей аммония Ni(OH)2 легко
растворима. Если на р-ры солей
Ni(II) действовать щелочью в при-
сутствии сильных окислителей
(NaOCl), то выпадает черный оса-
док, близкий по составу к Ni(OH)a.
Гидроокись 3-валентного никеля—
сильный окислитель, используется
при изготовлении щелочных ак-
кумуляторов.
Лит. см. при ст. Никель.
A R Pnuviv/ib
НИКЕЛЯ СПЛАВЫ-сплавы на
основе никеля, а также сплавы, в
к-рых присутствие никеля опреде-
ляет их физико-химич. и механич. свойства. Известны
8 металлов, образующие с никелем непрерывные твер-
дые р-ры: Мп, Fe, Со, Си, Rh, Pd, Pt, 1г, и 23 металла,
обладающие ограниченной растворимостью в никеле:
Be, Mg, Al, Ti, V, Cr, Zn, Ga, Ge, Zr, Nb, Mo, Sb,
Ru, In, Sn, La, Ta, W, Re, Os, Bi, U. Наличие интер-
металлич. соединений в системах с непрерывной рас-
творимостью твердо установлено лишь для систем:
Ni—Мп, Ni—Fe, Ni—Pd и Ni—Pt. В системах Ni—Co
и Ni—Си интерметаллиды не обнаружены, а системы
Ni—Rh и Ni—Ir не изучены. Интерметаллич. соеди-
нения обнаруживаются также в системах с ограни-
ченной растворимостью: Ni—Cr, Ni—Мо, Ni—W,
Ni—Ti, Ni—Al и др. В табл. 1 приведены составы
нек-рых интерметаллич. соединений, образующихся в
Н. с.
Таблица 1. Интерметаллические соединения никеля
Система Состав Темп-ра обра- зования, °C Тип решетки •
Ni.,Mn 620 гц.кубич.
Ni — Мп NiMn 900 тетр.
Ni—Fe Ni3Fe 480 гц. кубич.
Ni — Pt Ni.,Pt — гц. кубич.
Ni.Ti гекс.
Ni — Ti NiTi — оц. кубич.
Ni - Cr Ni3Cr 800 гц. кубич.
Ni,Al гц. кубич.
Ni — Al NiAl оц. кубич.
Ni2Al3 — гекс.
Ni — Mo Ni,Mo 980 гекс.
* гц,— гранецентрировапная, оц.— объемноцентрирован-
ная, тетр.—тетрагональная, гекс.— гексагональная.
Исследование и произ-во различных Н. с. особенно
бурно развивались последние 25—30 лет в связи с
потребностями новой техники в высокопрочных и
жаропрочных материалах. В настоящее время (1963)
известно несколько сот марок Н. с. (не считая спла-
вов, в к-рых Ni используется как легирующий эле-
мент), к-рые можно разделить на три группы: жаро-
прочные сплавы, магнитные сплавы и сплавы с осо-
быми физико-химич. свойствами.
Жаропрочные Н. с. В современных тур-
бинах и реактивных двигателях лопатки турбоком-
прессора разогреваются до 850— 900° и выше. Сплавы
на железной основе этих темп-р не выдерживают,
деформируются и быстро выходят из строя. В таких
тяжелых эксплуатационных условиях используются
жаропрочные Н. с., к числу к-рых относятся прежде
всего сплавы типа нимоник, инконель, хасталлой и
Таблица 2. Состав никелевых жаропрочных сплавов (в вес. %)
Название или марка сплава Ni Мп Si Cr Со Мо W Nb Ti Al Fe
Инконель . . . 73 0,6 0,3 15,0 — 0,9 2,4 0,9 7,0
Нимоник .... 59 0,5 0,3 20,0 16,0 2,3 1,4 0,5
Хасталлой . . . 65 0,2 0,6 15,0 2,5 5,0 — 2,5 2,3 7,0
X асталлой Д *) 85 1 ,0 10,0 — — — — 1 ,0
ЭИ 437 .... 75 0,4 0,7 20,0 __ __ 2,2—2,7 до 1°/о 1 ,0
ЭИ 602 .... 73 0,4 0,8 20,0 1,6 0,6 0,6 3,0
ЭИ 617 .... 72 0,5 0,5 15,0 — 2,0 6,0 — 2,0 2,0 —
* Содержит также до 4% Си.
нек-рые марки ЭИ. В табл. 2 приведены составы наи-
более распространенных жаропрочных Н. с.
Как и аустенитные стали (см. Железа сплавы), эти
сплавы изменяют свою структуру в результате термо-
обработки. В равновесном состоянии структура жа-
ропрочных Н. с. представляет собой, в основном,
твердый р-р различных элементов в никеле, содержа-
щий интерметаллич. соединения типа NiaTi. Жаро-
прочные Н. с. применяются обычно в горячедеформи-
рованном виде (прокатка, штамповка и т. п.), т. к.
ТРУДНО поддаются механич. обработке. Сплавы типа
инконель и ЭИ 437 применяют также в реакторо-
строении.
В последнее время находят применение в технике
металлокерамич. жаропрочные сплавы, содержащие
Ni(Ni—Cr и т. п.) в качестве матрицы (связки). Со-
держание Ni колеблется от 30 до 70%, а остальное
составляют высокодисперсные карбиды тугоплавких
металлов — TiC, WC, СгзСг и т. п. В качестве на-
полнителя никелевой матрицы используются также
и бориды нек-рых металлов (напр., борид хрома).
Жаропрочные металлокерамич. Н. с. могут длительно
использоваться при темп-рах до 1100°. Широкое при-
менение в электротехнич. пром-сти в качестве на-
гревательных элементов находят жаропрочные и жа-
ростойкие Н. с. типа нихром, подразделяющиеся
на два класса: 1) двойные нихромы, содержащие толь-
ко Ni (ок. 80%) и Ст (ок. 20%); эти сплавы стойки
к газовой коррозии при темп-рах до 1000—1100°;
2) железистые нихромы, содержащие при том же со-
отношении Ni и Ст еще до 15% Fe; эти сплавы могут
использоваться при темп-рах до —900°.
Магнитные Н. с. Широкое распространение
в технике получили магнитотвердые сплавы типа
алнико, в к-рых коэрцитивная сила достигает 500 э
при остаточной индукции 12 000 гаусс. Лучший из
них — алнико 24, содержит 14% Ni, 24% Со, 9% А1,
3% Си и 50% Fe. Большое значение для этих сплавов
имеет термич. обработка — наиболее благоприятные
магнитные свойства возникают при охлаждении от
1250° с определенной скоростью снижения темп-ры
(10 град-сек~'). В пром-сти для изготовления посто-
янных магнитов используют также более дешевые
тройные сплавы Fe—Ni—Al (не содержащие доро-
гостоящего Со). Эти сплавы, получившие общее
469
НИКЕЛЯ СУЛЬФАТ—НИКЕЛЯ СУЛЬФИДЫ
470
название алии, содержат 22—34% Ni, 11 — 14% Al, ос-
тальное Fe.
Особое значение в слаботочной технике (телефон,
телеграф, радио) приобрел сплав пермаллой, содер-
жащий 78,5% Ni и 21,5% Fe. Он обладает исключи-
тельно высоким значением начальной магнитной про-
ницаемости, достигающей 10 000 гаусс, что обеспе-
чивает его интенсивную намагничиваемость даже в
весьма слабых магнитных полях. Пермаллой отно-
сится к числу т. наз. прецизионных сплавов, т. к.
высокое значение магнитной проницаемости дости-
гается лишь при точном соблюдении состава и техно-
логии термич. обработки.
Н. с. с особыми свойствами. К их
числу следует отнести монель-металл, никелин и
константан, инвар, платинит, элинвар. Монель-ме-
талл — сплав Ni с 30% Си (содержит также неболь-
шие количества Fe и Мп), по своим механич. свой-
ствам превосходит чистый никель, а по стойкости
против коррозии почти не уступает ему. Широко ис-
пользуется в химич. аппаратостроении, для изготов-
ления турбин и насосов, а также в домашнем обиходе
(белый металл). В последнее время в состав монель-
металла вводят до 3% А1, что повышает его прочность
почти в 1,5 раза. Никелин и константан также яв-
ляются сплавами Ni с Си. Они обладают высоким
электросопротивлением, практически не меняющимся
с темп-рой, что очень важно при их использовании в
электроизмерительной аппаратуре. Максимальная ра-
бочая темп-ра для этих сплавов не превышает 500°.
Н. с., содержащий 36% Ni и 64% Fe, практически
не расширяющийся при нагревании (до 100°) и по-
этому названный инвар, используется в электрорадио-
технике и химич. машиностроении. Если железони-
келевый сплав содержит до 48% Ni, то его коэфф,
теплового расширения таков же, как у стекла; этот
сплав — платинит — используется для пайки со
стеклом (электровводы в стеклянные приборы) в ка-
честве заменителя Pt. Тройной сплав Ni—Сг—Fe,
содержащий 36% Ni и 12% Ст, обладает свойством
не изменять своих модулей упругости (модуль Юнга
и модуль сдвига) в широкой области темп-р. Этот
сплав, наз. элинваром, применяют для изготовления
различных точных приборов.
Сплавы Ni с Al (NiAh, NiAl2, NiAl, NioAls) явля-
ются основным сырьем для приготовления скелетных
никелевых катализаторов (см. Никель). Путем выще-
лачивания из них А1 получают активный катализатор
гидрогенизации (т. н. Ni Ренея). Реже для этой цели
используют сплав Ni с Si. Из сплава Ni с Mg для по-
лучения Ni-катализатора Mg удаляют растворением
в уксусной к-те. В Ni-катализаторе Бага-Егупова
содержится значительное количество невыщелочен-
ного Ni—А1-сплава.
Лит.: Корнилов И. И., Никель и его сплавы, М.,
1458; Гуляев А. П., Металловедение, 2 изд., М., 1951;
Кащенко Г. А., Основы металловедения, М,— Л., 1950;
Бочвар А. А., Металловедение, 5 изд., М., 1956.
В. И. Лихтман.
НИКЕЛЯ СУЛЬФАТ NiSCH — лимонно-желтые
ромбич. кписталлы, а = 11,884 А, Ь = 12,104 А,
с = 6,824 А; плотн. 3,652 (25°). Теплота образования
ЛЯ298= — 2 1 3,0 ккал/моль. Во влажном воздухе мед-
ленно окрашивается в зеленый цвет и связи с
поглощением воды. Растворимость в воде (вес. %):
21,40 (0°), 29,99(30°), 33,39 (50°), 43,42 (99°). Раство-
римость в СНзОН и С2Н5ОН (вес. %) соответствен-
но 0,001 и 0,017% (15°). Из водных р-ров Н. с. при
темп-рах от —3,15° до 31,5° кристаллизуется нике-
левый купорос NiSOa -7Н2О—изумрудно-зеле-
ные ромбич. кристаллы, изоморфные с гептагидратами
Mg, Mn, Fe, Со и Zn;. параметры решетки а = 11,8А,
b = 12,0А, с = 6,80А; плотн. 1,948 (25°). При вывет-
ривании NiSO4-7H2O на воздухе, а также при кри-
сталлизации из водных р-ров при 31,5—53,6° образуем-
ся голубовато-зеленый тетрагональный a-NiSO4-6H2O;
при 53,6—99°— зеленый моноклинный P-NiSC>4-6H2O.
При более высоких темп-рах кристаллизуются ме-
тастабильные гидраты с меньшим числом моле-
кул воды (от 5 до 2) и стабильный NiSO4-H2O,
к-рый обезвоживается выше 280°. Обычно при ох-
лаждении насыщенного р-ра вначале выделяется
NiS04 -6Н2О, а затем нек-рое количество NiSO4 -7Н2О.
При взаимодействии р-ров NiSO4 и едкой щелочи
выделяются осадки основных Н. с., состав к-рых
зависит от концентрации р-ров. С сульфатами щелоч-
ных металлов Н. с. легко образует двойные соли
типа сиенитов Me2(NiSO4)2 -6Н2О, где Me—К, Rb,
Cs, NH<, Tl. Наибольший практич. интерес представ-
ляет никель-аммонийсульфат.
В пром-сти никелевый купорос получают растворе-
нием металлич. никеля в серной к-те, к к-рой добав-
ляют незначительное количество азотной к-ты с по-
следующей выпаркой раствора, кристаллизацией и
сушкой кристаллов. Его выделяют также на электро-
литных заводах из р-ра электролита, применявшегося
при рафинировании меди, т. к. в таком р-ре постепенно
концентрируется NiSO4. Наибольшее же количество
Н. с. выделяют из сульфатных р-ров, являющихся
отходами кобальтового произ-ва, где они получаются
при электролитич. растворении никелькобальтовых
анодов. Н. с.— одна из наиболее употребительных
солей никеля; используется для получения чистого
электролитич. никеля; в гальванотехнике для ни-
келирования металлов; в последнем случае исполь-
зуется также никельаммонийсульфат (NB^MNiSChh •
•6Н2О. Н. с. применяют также для изготовления
аккумуляторов, в жировой и парфюмерной пром-сти,
для приготовления катализаторов и пр.
Лит.: П о з и н М. Е., Технология минеральных солей,
2 изд., Л., 1961. А. В. Ванюков.
НИКЕЛЯ СУЛЬФИДЫ — предполагается, что ни-
кель образует с серой соединения составов NiS,
Ni2S, NiS2, NisS2, NisSi и Ni„S5. В природе встре-
чаются NiS (минерал миллерит), NbSi (полидимит),
NiS2 (ваэсит). NisS2 и NieS5— основные компоненты
файнштейна, промежуточного продукта в производ-
стве никеля.
Никеля моносульфид NiS может быть
получен нагреванием NiO с серой и действием на рас-
творы никелевых солей сероводорода или сульфида
аммония в нейтральной или слабокислой среде и др.
методами. В зависимости от условий получения об-
разуются след. 3 модификации: аморфный a-NiS;
р-NiS (высокотемпературная модификация).— гекса-
гональные кристаллы, а — 3,42 А, с = 5,30 А, плотн.
•5,6; у-NiS (низкотемпературная модификация) — гек-
сагональные кристаллы, а = 9,60 А, с = 3,15 А,
плотн. 5,4. Природный у-NiS (миллерит) образует
кристаллы игольчатой формы латунно-желтого цвета
с сильным металлич. блеском. Т. пл. NiS 797°. Теплота
образования АЯ298= — 17,5 ккал/моль (для у-моди-
фикации). В воде NiS практически нерастворим.
а-NiS легко растворим в соляной к-те, [З-NiS раство-
ряется в ней лишь при кипячении, тогда как y-NiS
вообще нерастворим в НС1. На воздухе NiS медленно
окисляется. Водородом не восстанавливается.
Относительно Ni2S существует мнение, что он яв-
ляется твердым р-ром Ni в NiS или простой смесью
веществ. Он может быть получен нагреванием смеси
NiSO4 с S в токе Н2 или восстановлением NiSO4 угле-
родом в электрич. печи. Плотн. Ni2S 5,52—6,05;
т. пл. 645°; нерастворим в воде; с соляной к-той реа-
гирует очень плохо, азотной к-той разлагается с
отщеплением серы. Полидимит №384 — серебристо-
белые кристаллы с сильным металлич. блеском, ре-
шетка кубическая, а = 9,405 А, плотн. 4,5—4,8;
471
НИКОДИН—НИКОТИНОВАЯ КИСЛОТА
472
можея быть получен нагреванием NiCl2 с полисуль-
фидом калия в запаянной трубке при 160°. Ваэсит
NiS2 — серого цвета с металлич. блеском, кубич.
решетка, а = 5,74 А, плотн. 4,45. Получается сжига-
нием смеси NiO, S и K2SO3 и др. способами. Суль-
фид Ni3S2 образуется в системе Ni—S при диссоциа-
ции твердого р-ра; его т. пл. 790°; при медленном ох-
лаждении Ni3S2 распадается с образованием Ni6S5.
Сульфиды никеля являются важными промышлен-
ными катализаторами гидрогенизации и дегидрогени-
зации. Чаще всего первоначально в каталитич. реак-
тор вводится металлический Ni или NiO, к-рые затем
под влиянием сернистых примесей (H2S, CS2 и т. д.)
в реагирующем сырье превращаются в сульфиды.
В качестве стабильных фаз в каталитич. системах
идентифицированы сульфиды NiS и Ni3S2. Сульфиды
никеля применяют для гидрогенолиза связей С—С и
С = С (гидрогенолиз угля, кам.-уг. смолы и тяжелых
масел), дегидрогенизации алифатич. и гидроароматич.
соединений (сульфиды Ni в смеси с сульфидами Мо, Сг,
А7 или W), гидрогенизации сернистых соединений и
обессеривания, восстановления нитрилов и нитросое-
динений В амины И В др. реакциях. А. В. Ванюков.
НИКОДИН (N-оксиметилампд никотиновой кисло-
ты) C,II8O2N2, мол. в. 152,15— бесцветные кристаллы;
т. пл. 141 —142°; растворим (при
CrCONHCH2OH нагревании) в воде и спирте, He-
ll растворим в эфире. При нагрева-
" нии Н. разлагается с выделением
формальдегида. При нагревании в щелочном р-ре Н.
расщепляется с образованием никотиновой к-ты. Ко-
личественно Н. определяют иодометрически. Получа-
ют конденсацией амида никотиновой к-ты с формаль-
дегидом. Применяют Н. для лечения заболеваний желч-
ных путей.
Лит.: G г a f R„ J. Prakt. Chem., 1933, 138, 292.
Р. Г. Глушков.
НИКОТИН [р-(Н-метил-а-пирролидил)-пиридип1
C1cH14N2. мол. вес 162,24 — главный алкалоид та-
__________ бака (Nicotiana tabacum) сем. паслено-
J вых (Solanaceae), содержание Н. 2—10 %;
f встречается в других растениях. Выделен
' в чистом виде впервые в 1828 Поссель-
3 том и Рейманом. Н.—летучая бесцвет-
ная маслянистая жидкость, темнеющая на воздухе,
с характерным запахом табака; т. кип. 246°/730 мм,
124—125°/18 мм; d™ 1,0092; п™ 1,5282; [а]£ =
— — 168,66°. Н. смешивается с водой во всех отноше-
ниях ниже 60° п выше 210° (в пределах 60—210°
растворимость ограничена); хорошо растворим в обыч-
ных органич. растворителях; перегоняется с водяным
ларом. Благодаря наличию двух третичных атомов
азота Н. с минеральными к-тами образует одно- и
двукислотные соли. Соли Н. вращают вправо: для
сульфата [а]п = + 84,8°; для хлоргидрата [а]д =
= + 102,2°. Диппкрат никотина, т. пл. 224°. При
действии сильных окислителей (HNO3, KMnO4 и др.)
в Н. (I) расщепляется пирролидиновое кольцо, угле-
родный атом к-рого, связанный с пиридиновым цик-
лом, окисляется до карбоксильной группы с обра-
зованием никотиновой к-ты (II):
Н. образует с n-диметиламинобензальдегидом розо-
вое окрашивание, переходящее в фиолетовое.
В пром-сти Н. получают из отходов табачного
произ-ва обработкой их известью и экстракцией орга-
нич. растворителем; пз растворителя Н. извлекают
водным р-ром H2SO4 и выделяют в виде сернокислой
соли. Синтетически Н. был получен Шпетом.
II.— сильный яд, действующий на центральную и
периферия, нервную систему. При малых дозах насту-
пает возбуждение центральной нервной системы, уси-
ление дыхания, повышение кровяного давления; при
больших дозах — угнетение и паралич нервной сис-
темы, остановка дыхания и прекращение сердечной
деятельности. Н.— инсектицид, применяется в впде
сульфата для борьбы с вредителями с.-х. растений,
в ветеринарии при накожных паразитарных заболе-
ваниях, а также для получения никотиновой к-ты
п ее амида.
Лит. см. при ст. Алкалоиды. В. М. Березовский.
НИКОТПНАМИДИУКЛЕОТИДНЫЕ КОФЕР-
МЕНТЫ — группа коферментов, включающая нико-
тинамид-аденпн-дппуклеотид (НАД) и пикотинамид-
аденин-динуклеотид-фосфат (НАДФ). В соответст-
вии с современной номенклатурой ферментов (см.
Номенклатура и классификация ферментов) термин
никотинамнднуклеотидные коферменты используется
взамен устаревших терминов кодегидрогеназы или
пирпдиннуклеотидные кофермен-
т ы (пиридпнпуклеотиды). В частности, термин НАД
применяется вместо названий кодегидрогепаза I
(Koi), козпмаза, кофермент I, дифосфопирпдинну-
клеотид (ДПН), а термин НАДФ — вместо названий
кодегидрогепаза II (KoII), фосфокозимаза, кофер-
мент II, трифосфопиридиннуклеотид (ТПН).
Восстановленные формы НАД и НАДФ обознача-
ются соответственно НАД-Н2 и НАДФ-Н2 (см. Коде-
гидрогеназы).
Лит. см. прп ст. Номенклатура и классификация фер-
ментов. В. Б. Спиричев.
НИКОТИНОВАЯ КИСЛОТА (I) (р-пирпдппкарбо-
новая кислота, ниацин, провитамин РР), мол. вес.
123,11 — бесцветные иголь-
чатые кристаллы; т. нл.
235,5—236,5°; константа
кислотной диссоциации Ка
1,4-10-5(25°):изоэлектрич.
точка при pH 4,23—4,25;
спектр поглощения УФ-
света имеет максимум при 261,5 и 385 ммк. Рас-
творимость Н. к. (г/100 мл растворителя): вводе —
1,3 (15°)- 2,47 (38°); 4,06 (61°) и 9,76 (100°); в спирте—
0,92 (15°); 2,10 (38°); 4,20 (61°) и 7,06 (78°). В органич.
растворителях Н. к. растворяется плохо; в водных
р-рах дает кислую реакцию.
Н. к.— амфотерное соединение, образует 2 ряда
солей — с кислотами и основаниями. Соли Ag+, Сн2+ и
Са2 + плохо растворимый воде. Подобно аминокисло-
там, II. к. может образовывать бетаины, напр. при
действии CH3J в щелочной среде образуется N-метпл-
производное пиридина — тригонеллин (II). Н. к.,
подобно карбоновым к-там, образует ангидрид, гало-
генангидриды, эфиры, амиды и т. п., декарбоксили-
руется при нагревании до 260°.
Для определения Н. к. применяют метод осаждения
нерастворимой медной соли или колориметрия, мето-
ды, основанные на образовании окрашенных р-ров
с KCN и хлорамином или KSCN, бромом и анилином.
Для определения Н. к. в биологич. средах часто ис-
пользуют микробиология, методы.
Н. к. может быть получена:
1) Омылением нитрила Н. к., к-рый получают из
пиридина:
сн2
I <1
473
НИНГИДРИН—НИОБАТЫ
474
2) Окислением хинолина с последующим частичным
декарбоксилированием хинолиновой к-ты:
Х-соон
соон
N:
3) Окислением р-замещенных пиридина КМпО4 в
щелочной среде, Н20з, H2SO4, HNO3 или H2SO4 в
присутствии Se в качестве катализатора, а также
окислением никотина и анабазина.
Амид никотиновой кислоты (III)
(истинный витамин РР или антипелларгич. вита-
мин) — бесцветные кристаллы, т. пл. 131 —132°, рас-
творим в воде, спирте и органич. растворителях. Амид
Н. к. получают пропусканием NH3 в Н. к. при 230°
или действием р-ров аммиака па водные или спирто-
вые р-ры эфиров Н. к. Н. к. и ее амид обладают свой-
ствами антипелларгич. витамина; они широко рас-
пространены в растительном и животном мире, гл.
обр. в виде сложных соединений — нуклеотидов. Су-
точная потребность человека 20—30 мг Н. к.; она
ределении аминокислот, аминосахаров, аминов п др.
соединений. См. также Нингидриновая реакция.
Лит.: McCaldinD. J., Chem. Rev., 1960, 60, № 1, 39.
НИНГИДРИНОВАЯ РЕАКЦИЯ (нингидринная ре-
акция) — реакция аминокислот, иминокпелот п
аминов с нингидрином, сопровождающаяся образо-
ванием окраски в щелочной среде:
+ Н^СНКСООН^^~Г‘
QCONH2
111
v
в основном удовлетворяется за счет Н. к., содержа-
щейся в пище (в молоке, мясе, рыбе, дрожжах и др-).
Однако хлебо-булочные изделия из высокосортной
пшеничной муки необходимо витаминизировать Н. к.
Н. к. может образовываться и в организме животных
в результате биосинтеза из триптофана. Нек-рые
соединения, близкие ио строению Н. к., напр. пирп-
дин-р-сульфокислота (IV) и р-ацетплипридин (V),
являются антивитаминами Н. к.
Лит.: Березовский В. М., Химия витаминов, М.,
1959; Одинцова Е. Н., Микробиологические методы
определения витаминов. М.. 1959; Труфанов А. В., Био-
химия и физиология витаминов и антивитаминов, М.. 1959.
В. Л1. Березовский.
НИНГИДРИН (трикетогидрпнденгидрат, 2,2-ди-
окси-1,3-пндандпон) СэНбО4, мол. в. 178,14—светло-
желтые кристаллы; т. пл. 241° (прп
О нагревании до 125° краснеют). Хорошо
С»------с он растворим в воде, щелочах, плохо —
1 q' в спирте. Н. дает реакции, харак-
С' Qu терные для кетонов (восстанавливает
О р-р Фелинга и аммиачный р-р азотно-
кислого серебра), а также ряд цветных
реакций: при действии водного р-ра КОН на холоду
появляется желтое окрашивание, к-рое исчезает прп
стоянии раствора; прп нагревании желтое окрашива-
ние переходит в синее; прп действии NazCOa появ-
ляется красное окрашивание. С различными аминокис-
лотами и с иминокислотамп Н. дает окрашивание
(см. Нингидриновая реакция). Цветные реакции с Н.
дают многие первичные п вторичные амины и солп
аммония; третичные амины с II. не реагируют. С пир-
енрашеннсе соединение
Наиболее вероятным является механизм, в основе
к-рого лежит разложение по Штреккеру. Иминокпс-
лоты и амины реагируют, по-впдпмому, таким же обра-
зом. Большинство аминокислот дает голубое или ли-
ловое окрашивание, аспарагин — оранжево-корпчпе-
вое, пролин и океппролин—желтое. Н. р. широко при-
меняют для колориметрия, определения аминокислот.
Лит.: GreensteinJ. Р., Winitz М;, Chemistry of
the amino acids, v. 2, N. Y.—L., 1961, p. 1301, 1394.
JI. И. Линевич.
НИОБАТЫ — солп ниобиевых к-т, отвечающие
5-валентному Nb. К важнейшим И. относятся мета-
ниобаты MeNbO3, ортониобаты Me3NbO4 и солп типа
Me5NbO5 (где Me — щелочной металл). Ортониобаты
и соли Me5NbO5 растворяются в воде с одновременным
гидролитич. разложением, приводящим к образова-
нию сложных аквополиниобатов различного состава:
4Me2O.3Nb2O5.aq, 7Ме2О-6Nb2O5-aq.
Метаниобаты щелочных и щелочноземельных метал-
лов термически прочны и прп высоких темп-рах
(1200°) нелетучи. Однако в вакууме KNbO3 и NaNbO3
разлагаются прп 400—800° с выделением кислорода
и образованием твердой фазы, обладающей свойст-
вами ниобиевых «бронз» (аналогичных вольфрамовым
«бронзам»); при 800—1200° в вакууме из этих мета-
ниобатов выделяется свободный щелочной металл, а
остаток после наиболее полного разложения состоит
гл. образом из NbO2. Темп-ры плавления пек-рых
И.: LiNbO3 1164°; NaNbO3 ~ 1250°; KNbO3 1184°;
RbNbO3~1200°; GsNbO3-1200°; Na5NbO5 980°. M e-
тан иобат калия KNbO3 имеет объемноцен-
трированную кубич. структуру; а = 4,018 А (при
комнатной темп-ре); темп-ры фазовых переходов
435°, 225° и -10°. Метан иобат натрия
NaNbO3 имеет при комнатной темп-ре ромбич. струк-
туру; темп-ра фазовых переходов 640°, 520°, 470°, 350°
и —203°; при 640* структура переходит в кубическую.
Все безводные метаниобаты щелочных металлов и
нек-рые Н. других элементов плохо растворимы в воде,
причем соли К значительно более растворимы, чем
соли Na.
ролом и его производными нингид-
рин реагирует с образованием пиг-
ментов.
Получают Н. окислением 1-гид-
риндопа, 2-гидрипдопа, 1,3-дикето-
гидриндепа и др. способами. Н. при-
меняют в качестве реактива прп оп-
Растворимость некоторых нпобатов
Метаниобаты LlNbO3 NaNbO3 KNbO3 RbNbO3 GsNbO,,
Растворимость, J при 25° моль/л 1 при 7 5° 2,8-10-* 6,0-10-* 5,9-10-1 3,7’Ю-3 8,7-10-* 9,5-10-* 3,4.10 “< 8,7.10-5 2,3-IO-4
Сложные нпобаты Ве7^'Ьг.Оз7- • 22Н..О Mg7Nb]2O37- •44Н2О Ca7Nb12O37* • 36Н2О Sr,Nb„O37- •ЗЗН3О Ba-Nb,-.O,-7- •26Н=О Pb7Nb,,O37- •26Н;о
Растворимость, моль!л (20°) .... 1 -10-- 4,7-10-» 2,4-10-» 1,1-10-» 5,0-10-» 7,6-10-»
мъ
НИОБИЙ
476
Растворимость гексаниобата натрия 7NaaO-6Nb2Os-
• З2Н2О в воде составляет ок. 1,6 г/л раствора при
20°, причем она резко падает с увеличением щелоч-
ности раствора и при содержании NaOH 1,5 г/100 мл
соль становится практически нерастворимой.
Н. многих металлов обладают сегнетоэлектриче-
скими, а в нек-рых условиях также пьезоэлектрич.
свойствами. Н. получают при сплавлении НЬгО5 с
окислами других металлов или при обработке гидро-
окиси ниобия щелочными р-рами. В технологии нио-
бия и тантала Н. образуются при разложении рудных
концентратов. При pH 5= 13 из растворов кристаллизу-
ются Н. с отношением Ме2О:НЬгО5= 4:3; в интер-
вале pH 13—9 — с отношением 7:6; в интервале pH
9—5 выделяется метаниобат ЗМеО -SNbaOj- aq, к-рый
при прокаливании переходит в MeNbOa. Из Н. некото-
рых металлов изготовляют керамич. сегнетоконден-
саторы и пьезоэлектрич. преобразователи; блокиро-
вочные и емкостные конденсаторы, работающие при
звуковых частотах; малые блокировочные конденса-
торы, работающие до темп-ры 200° (неконтактные
взрыватели, самолетная аппаратура и т. д.); усилите-
ли сигналов изображения (в телевизорах).
Лит.: Лапицкий А. В., Вестник МГУ, 1958, К» 6,
121—26; J a n d е г G., Е г t е 1 D., J. Inorg. and Nucl. Chem.,
1956, 3, № 2, 139—52, 1960, 14, № 1/2; 3 e л и к м а н A. Н.,
О р е х о в М. А., Изв. высш, учебн. заведений. Цветная метал-
лургия, 1963, № 5, 99—107; Колчин О. П., Ниобий и тан-
тал. Области освоенного и возможного применения, М., 1959.
О. П. Колчин.
НИОБИЙ (Niobium) Nb — химич. элемент V гр.
периодич. системы Менделеева; п. н. 41, ат. в. 92,906.
Имеет один природный изотоп Nb9’ (100%). Сечение
захвата тепловых нейтронов атомом Н. 1,15 барн. Из
искусственно получаемых изотопов в качестве радио-
активного индикатора в исследованиях обычно ис-
пользуется Nb95 (p-распад; Г1/2=35 дней). Внешняя
электронная оболочка атома Н. имеет строение 4d45sJ.
Энергии ионизации (в эв): Nb0—Nb + ^Nb2 + ^Nb’ + -^
— Nb4+— Nb5+ соответственно равны 6,88; 13,48; 24,7;
37,7;51,9.
Н. был открыт в 1801 Гетчетом в минерале, найденном в
Колумбии, и получил название «колумбий». В 1844 Розе об-
наружил «новый элемент» и назвал его Н. в честь древнегреч.
богини Ниобеи — дочери Тантала, чем подчеркнул сходство
между Н. и танталом. Позднее было доказано, что Н. и ко-
лумбий — один и тот же элемент. В США элемент и сейчас
иногда называют Columbium (знак СЬ). Металлич. Н. впервые
получен в 1907.
Содержание Н. в земной коре принималось до пос
леднего времени равным 2-10“’ вес. %; фактически
оно выше, если учесть открытие в последние годы но-
вых крупных месторождений Н. в Африке (Нигерия,
Уганда, Кения, Танганьика, Конго), Сев. Америке
(Канада). Свободный металл в природе не встречает-
ся. В минералах Н. почти всегда изоморфно ассоции-
рован с танталом. К чисто ниобиево-танталовым мине-
ралам относятся ок. 80. Минералы Н. делятся на
2 группы: тантало-ниобаты — соли ниобиевой и тан-
таловой к-т, и титано(тантало)-ниобаты — сложные
соли титановой, ниобиевой (танталовой) к-т. Из пер-
вой группы наиболее важны колумбит и танталит, из
второй — лопарит и пирохлор.
Колумбит-танталит (Fe, Мп) (Та, Nb)2Oe
содержит 82—86% суммы (Nb2O5 Та2О5). Если
Nb>Ta, то минерал наз. колумбитом, при Ta>Nb —
танталитом. Из примесей минерал содержит Ti, Sn,
W, Zr, Ca и др. Сингония ромбическая; цвет черный, с
сероватым или коричневым (у танталита) оттенком;
блеск металлический или смолистый; плотность от
5,3 для колумбита до 8,0 для танталита; твердость
ок. 6. Минерал парамагнитен, слабо радиоактивен
(за счет примеси Th).
Лопарит (Na, Се, Са, Sr) (Nb, Ti)Os содержит
8—10% (Nb, Та)2О5 (при соотношении Та : Nb от
1 : 12 до 1 : 20), 39,2—40% ТЮ2, 32—34% редких
земель, а также Fe, Al, Si, Th, Ca, К, Na и др. Синго-
ния кубическая; цвет черный, реже буровато-красный
и зеленовато-бурый; плотн. 4,73 — 4,99; твердость
5,5—6,0. Слабо парамагнитен и слабо радиоактивен
(примесь Th).
Пирохлор (Na, Ca)2(Nb, Ti)2Oe(F, ОН) со-
держит 37,5—65,6% Nb2O5, не более 10% Та2О5, а
также Si, Ti, Sn, Zr, Th, Fe, редкоземельные элемен-
ты, U, Ca, F и др. Сингония кубическая; цвет бу-
рый, черный, желтый разных оттенков; плотн. 4,2—
4,36, твердость 5,0—5,5; слаборадиоактивен (примеси
U, Th).
Месторождения Н. связаны гл. обр. с гранитными
пегматитами (колумбит-танталит), карбонатитом (пи-
рохлор), щелочными нефелин-сиенитовымн пегмати-
тами (лопарит, пирохлор). Кроме коренных (жиль-
ных) месторождений, имеются россыпи (колумбит-
танталит). Крупные ниобиевые месторождения изве-
стны в СССР (лопариты Кольского п-ова и др.), в ука-
занных выше странах Африки, Канаде, Норвегии и др.
Физические и химические свойства. Компакт-
ный Н. — металл светло-серого цвета. Кристаллич.
решетка объемноцентрированная кубич. с, периодом
а=3,294 А. Плотн. 8,57. Ат. радиус 1,47 А. Ионные
радиусы: Nb2+ 0,85 A, Nb’+ 0,78А, Nb4 + 0,67А, Nb3 +
0,66 А. Т. пл. 2500°; т. кип. 4927° (5127°). Давление
пара (мм рт. ст.): 1 • 10-5 (2194°), 1-10-4 (2355°),
6-10-4 (т. пл.), 1-10“’ (2539°). Теплота плавления
6,4 ккал/г-атом', теплота испарения 166,5 ккал/г-атом
(т. кип.). Уд. теплоемкость (кал!г-град)-. 0,0645 (0°),
0,077 (1000°), 0,0904' (2000°); термич. коэфф, линей-
ного расширения 7,1 -10-9 (20°); теплопроводность
(кал/см-сек-град): 0,125 (0°) и 0,156 (600°). Уд.
электрич. сопротивление 15,22-10-6 ом-см (0°); тер-
мич. коэфф, электросопротивления 3,95-10-’(20°);
темп-ра перехода Н. в сверхпроводящее состояние
9,25°К. Металл парамагнитен, уд. магнитная вос-
приимчивость 4-2,28-10-9 (18°). Работа выхода элект-
ронов 4,01 эв — меньше, чем у любого другого туго-
плавкого металла (исключая гафний); положитель-
ная эмиссия 5,52 эв. Коэфф, излучения 0,37 (20°;
Х=6500 А).
Н. легко обрабатывается давлением на холоду;
имеет сравнительно низкий модуль упругости (11 000
кГ/мм2), что позволяет при его легировании вводить
большее количество легирующих металлов без суще-
ственного снижения пластичности. Важным свойст-
вом Н. является сохранение удовлетворительных ме-
ханич. свойств при высоких темп-рах. Его предел
прочности и относительное удлинение при 20° соот-
ветственно равны 34,2 кГ/мм2 и 19,2%, а при 800° —
31,2 кГ/мм2 и 20,7%. Отношение напряжения, вызы-
вающего за сутки остаточную деформацию в 1% при
1000°, к плотности Н. равно 0,5, т. е. больше, чем для
Ti, Zr, V, Та, Сг и W. Сопротивление Н. ползучести
при 100-час. испытании в вакууме при 980° и 1090°
~в 1,5 раза выше, чем у Мо. Нек-рые примеси (осо-
бенно Н, N, С и О) сильно ухудшают пластичность
Н. и повышают его твердость. Чистый Н. имеет твер-
дость по Бринеллю 45 кГ/мм2, технич. 75—180 кГ/мм2.
Темп-ра перехода Н. в хрупкое состояние от —100°
до—200°.
По химич. свойствам Н. близок к танталу. Оба
металла чрезвычайно устойчивы на холоду к действию
многих агрессивных сред, хотя в этом отношении Н.
уступает танталу. Компактный Н. окисляется на
воздухе только выше 200°, порошкообразный — при
150°. При нагревании на воздухе до относительно
высоких темп-р (900—1200°) толщина окисной плевки
на Н. постепенно увеличивается, но скорость раство-
рения кислорода в Н. невелика. В связи с этим оста-
ющийся еще не окисленным Н.сохраняет пластичность,
что важно для тех случаев применения Н., когда он
477
НИОБИЙ
478
должен подвергаться кратковременному нагреванию
до высоких темп-р на воздухе. Фтор действует на Н.
при комнатной темп-ре, хлор — выше 200°, бром и
иод — выше 250°, водород — при 250° (интенсивно—
прп 360°), азот — при 400°, углерод и углеродсодер-
жащие газы — при 1200—1400°. На Н. практически
не действуют хорошо обескислороженные жидкие нат-
рий, калий и их сплавы,литий, висмут, свинец, ртуть,
олово, применяемые в качестве жидкометаллич. теп-
лоносителей в ядерных реакторах. Н. образует спла-
вы со многими металлами. Легирование Н. других
металлов (Ni, Сг, Со, Ti, Zr, W, Мо, Al, Си и др.)
повышает их предел текучести, жаропрочность, жа-
ростойкость, сопротивление ползучести (см. Ниобия
сплавы).
Н. характеризуется хорошей коррозионной стой-
костью против действия многих кислот и р-ров солей.
На Н. не действуют царская водка, соляная и серная
к-ты при 20°, азотная, фосфорная, хлорная к-ты, вод-
ные р-ры аммиака и многие другие неорганич. и ор-
ганич. вещества. Плавиковая к-та, ее смесь с азотной
к-той, а также щелочи растворяют Н. В кислых элект-
ролитах на Н. образуется анодная окисная пленка,
имеющая высокие диэлектрич. характеристики, что
позволяет использовать Н., как и тантал, в радио-
электронике для изготовления электролитич. конден-
саторов.
В своих наиболее устойчивых соединениях Н. име-
ет валентность -)-5; известны также соединения с
валентностью -)-4, -)-3, -|-2 и -f-1, к образованию
к-рых Н. склонен более, чем тантал. Высший окисел
Н.— пятиокись Nb2O5 имеет кислотный характер.
Слабая ниобиевая кислота, или гидро-
окись Н. Nb2O5-xH2O, образует соли— ниобаты.
Гидроокись ниобия полностью обезвоживает-
ся выше 400°; растворима в конц. соляной и серной
к-тах, но вновь выпадает в осадок при разбавлении
р-ров; хорошо растворима в плавиковой к-те и р-рах
КОН; образует комплексы со щавелевой и винной
к-тами, что используется в аналитич. химии Н. В тех-
нологии гидроокись Н. получают как промежуточный
продукт в результате нейтрализации аммиаком пла-
виково-, соляно- и сернокислых р-ров Н., гидролиза
пентахлорида и окситрихлорида Й., при обработке
кислотами р-ров ниобатов и др. способами. Смесь тех-
нич. гидроокисей Н. и тантала получается при
солянокислой обработке продукта разложения тан-
талита щелочью.
Из органич. комплексных соединений Н.важны ком-
плексы с танином, трибутилфосфатом и метилизобу-
тилкетоном. Комплекс с танином оранжевого цвета
выпадает из нейтрального или очень слабокислого
р-ра при избытке танина, в отличие от танталового
комплекса, выпадающего после кипячения из слабо-
кислего р-ра при рН=3,4, что используют в аналитич.
химии для отделения Н. от тантала. Образование
ниобиевых комплексов с трибутилфосфатом и метил-
изобутилкетоном и различие в их растворимости от
соответствующих танталовых комплексов исполь-
зуют в пром-сти для разделения Н. и тантала и очист-
ки их от примесей экстракционным методом.
В системе ниобий — кислород
установлены, помимо Nb2O5, следующие фазы: твер-
дый р-р кислорода в Н.,окись NbO и двуокись NbO2.
Растворимость кислорода в Н. увеличивается с 0,24
вес. % (1,4 ат.%) при 775° до 1,00 вес.% (5,5 ат.%)
при 1100°.
Окись Н.—серый порошок; сплавленная в слиток,
обладает металлич. блеском и электропроводностью
металлич. типа; область гомогенности от NbOf94
до NbOI10l;o структура гранецентрированная кубич.,
а=4,2097А; плотн. 7,26; т. пл. 1935°; начинает испа-
ряться в вакууме с заметной скоростью при 1700°,
скорость испарения резко возрастает с повышением
темп-ры до 2300—2350° и выше, что используют для
вакуумного рафинирования Н. от кислорода электрон-
но-лучевой плавкой или спеканием. Наряду с испаре-
нием диспропорционирует. Теплота образования NbO
АЯ298=—108,8 ккал/моль.
Двуокись Н.— черного цвета, полупровод-
ник. Область гомогенности от NbOli94 до NbO2 09.
Структура тетрагональная, а=4,82 А и с=2,99 А,
т. пл. 2080°; давление насыщенного пара (в.нлс рт.ст.)
6,02.10-“ (1665°); 3,19-10-’(1758°); 7,55-10-’(1808°);
1,12-10-2(1849°). Теплота образования NbO2 ДЯ°98=
= — 193,5ккал/люль; теплота сублимации 138 ккал/моль.
При нагревании на воздухе NbO2 и NbO образуют
Nb2O5. В системе Nb—NbO2 имеются две эвтектики:
твердый р-р Ог в Nb-j-NbO (10,5 вес. % О) ст. пл.
1915° и NbO-|-NbO2 (21 вес. %О) с т. пл. 1810°.
ПятиокисьН. гомогенна в пределах Nb2O4 78—
Nb2O5 (по нек-рым данным, в указанных пределах
существует не одна, а три фазы с весьма близкими
структурными характеристиками: Nb2O5, NbO2>48 и
йЬО2140).По Брауеру, она существует в трех модифи-
кациях: низшей (L) — до 900°, средней (М) — при
900—1100° и высшей (высокотемпературной — Н) —
выше 1100°; по Гольдшмидту — только в двух формах:
аир, причем низкотемпературная a-форма существует
до 900°, метастабильна и необратимо переходит в
p-форму, стабильную при всех темп-рах твердой фазы.
По Шеферу и Рою, имеется еще одна форма — при
темп-ре выше 1280°, не поддающаяся переохлаждению.
Легирующие добавки могут стабилизировать а- или
p-форму, напр. добавка Та2О5 стабилизирует а-форму
пятиокиси Н., а добавки окислов многих переходных
элементов (TiO2, Сг2О8 и др.) стабилизируют р-форму.
Структура низкотемпературной модификации орто-
ромбическая; а=6,16 А, 6=3,65 А, с=3,94А; плот-
ность 4,95. Т. пл. 1510°. При обычной темп-ре пяти-
окись Н.белого цвета,при нагревании приобретает жел-
тую окраску, исчезающую при охлаждении. Вблизи и
при т. пл. имеет черный цвет, слегка диссоциирует
на NbO2 и О2. Умеренные количества примесей TiO2,
SiO2,Fe2O8, Р и S практически пе влияют на т. пл. пяти-
окиси Й., примесь К приводит к существенному сни-
жению т. пл. — на 110° при 1,0 вес. % К. Теплота
образования пятиокиси Н. (p-модификация) ДН298=
= —453,5 ккал/моль. Молярная теплоемкость Ср=
= 38,76-|—3,54-10—а Т—7,318-105Г-2 кал/град-молъ.
Давление паров при 1380° не вып.j8,6 • 10~6 мм рт. ст.,
при 1450°—не выше 1,8-Ю-3 мм рт. ст.; скорость
испарения равна соответственно 2-10-’ и 4,1-10-7
г/см2-сек. Пятиокись Н. склонна к рекристаллизации
при нагревании. В воде она нерастворима, в минераль-
ных к-тах, кроме плавиковой, почти нерастворима.
При 800—1200° Nb2O5 восстанавливается водородом до
NbO2, при взаимодействии с NH3 при 500—800°
образуется нитрид ниобия NbN, с С12 при 1000—1050°
или СС14 при 200—225° — летучие пентахлорид и ок-
ситрихлорид ниобия (см. Ниобия галогениды); с Та2О5
образует непрерывный ряд твердых р-ров. Обычно
пятиокись Н. получается как промежуточный продукт
при переработке рудных концентратов в результате
прокаливания на воздухе гидроокиси Н., а также
окислением металла, карбидов Н. и т. п.
С водородом компактный Н. начинает вза-
имодействовать с заметной скоростью при 250°, быст-
ро — при 360° с образованием вначале а-твердого
р-ра внедрения. Область гомогенности a-фазы дости-
гает 10 ат. % Н. При дальнейшем поглощении водо-
рода существует двухфазная область а-|-р, а от NbH0 7
(41 ат. % Н.) до NbHOj89, а по нек-рым данным— до
NbHc„ или моногидрида NbH, существует p-фаза с
479
НИОБИЙ
480
ромбической (по др., менее надежным данным,— ку-
бической) структурой, а=4,85 А, 6=4,91 А, с=3,46 А-
Растворимость водорода в 98,5 %-ном Н. составляет:
Темп-ра, °C ... . 20 |200 300 400 500 | 700 900
Растворимость Н2, 'смл}г 104 93,3 88,0 76,8 74,4 9,7 4,0
Поглощение водорода носит обратимый характер: при
нагревании, особенно в вакууме при 800—900°, во-
дород вновь выделяется. Выше 1000° можно нагревать
Н. в водороде без опасения заметного его поглоще-
ния. Теплота образования гидрида состава NbH0 7
Д//,98=—23,3 ккал/ моль.
Плотность гидрида, близкого к NbH, равна
6,0—6,6; уд. теплоемкость: 0,0987 кал/г-град (0—100°),
0,0834 кал/г-град (0—400°); темп-ра перехода в состоя-
ние сверхпроводимости 15°К. При комнатной темп-ре
гидрид Н. устойчив на воздухе, нри нагревании
окисляется до Nb2O3; нерастворим в соляной, азот-
ной, разб. серной к-тах и в царской водке. Пользуясь
тем, что Н. при поглощении водорода становится
хрупким, а поглощенный им водород может быть уда-
лен при нагревании в вакууме, гидрирование с после-
дующим измельчением и дегидрированием применяют
в металлургии Н. для получения порошка Н. из ком-
пактного металла (напр., с целью регенерации отхо-
дов компактного Н.).
В системе ниобий — азот установлены
при темп-рах до 1500° 6 фаз: а, 0, у, б, б' и £. а-Фаза—
твердый р-р внедрения азота в Н. (<4,8 ат. % Н.);
растворимость азота в Н. составляет (вес. %) : 0,005,
0,04 и 0,07 соответственно при 300°, 1000° и 1500°.
Азот может быть удален из твердого р-ра в Н. при
нагревании в высоком вакууме выше 1900°. Гексаго-
нальная p-фаза (Nb2N) гомогенна в пределах NbNe 4(|—
NbN0,sl, а = 3,05бА, с=4,955—4,968 А. Тетрагональ-
ная у-фаза (NbiNs) гомогенна в пределах NbNc ,3—
NbN0 79, периоды решетки от а=4,385 А, с=4,31’0 А
до а = 4,386 А, с=4,355 А. Выше 1230° у-фаза нахо-
дится в равновесии с кубич. б-фазой, ниже 1230°—
в равновесии с §-фазой. б-Фаза гомогенна в пределах
NbN0,98—NbN0>88, а=4,393 —4,373 А; при темп-ре
1230°’опа переходит в гексагональную б'-фазу, гомо-
генную в области NbN0 9,— NbN0 ,8, с периодами решет-
ки а=2,968 А, с=5’,548 А. ’б'-Фаза ниже 1000—
1230° превращается в у-фазу (NbN0,79) и псевдогекса-
гональную с §-фазу (NbN, с1) с периодами решетки
а = 2,9591 А, с= 11,2714 А’, б-, б'- и §-Фазы очень
близки по химич. составу и, видимо, представляют
собой результат полиморфных превращений н и т-
р и д a NbN. Этот нитрид имеет светло-серый цвет с
желтоватым оттенком; плотн. 8,40; т. пл. 2300° (с
разл.); давление диссоциации (мм рт. ст.): 1 (1946°),
1-10-3 (1491°), 7,5-10“5 (1230°). Теплота образова-
ния NbN ДЯ298= — 59 ккал/моль: теплоемкость
10,41 кал/моль-град (25°); электросопротивление
(jtKoai-c.w)-. 200 (25°), 450 (2050°); темп-ра перехода в
сверхпроводящее состояние 15,6°К. Образует твердые
р-ры с NbC и с нитридами и карбидами других туго-
плавких металлов; устойчив к действию соляной, азот-
ной, серной к-т и царской водки даже при кипячении;
при нагревании со щелочами разлагается с выделени-
ем аммиака. Нитриды Н. окисляются иа воздухе при
500—800° до Nb2O3. Нитриды Н. получают нагрева-
нием Н. или гидрида Н. в азоте или аммиаке, либо пя-
тиокиси Н. в смеси с углем в токе азота.
В системе ниобий — углерод уста-
новлены при 1800—2000° 3 фазы (а, (5 и б). а-Фаза—
твердый р-р внедрения углерода в Н., содержащий
при наибольшем насыщении при 2335° 2 ат. % С; пе-
риод объемноцентрированной кубич. решетки у гра-
ницы a-фазы, а=3,012 А. Гексагональная |3-фаза
(Nb2C) гомогенна в пределах NbC0 3S—NbCn 50;период.ы
ее решетки, а=3,120—3,128 А, с=4,957 —4,974 А.
Плотность Nb2C 7,85; теплота образования ДН298=
=—46,6 ккал/моль: модуль упругости 30 000 кГ/мм2.
Гранецентрпрованная кубич. б-фаза (NbC) гомогенна
от NbC0,70 до NbC0>91; период ее решетки, а=4,432А—
4,469 А.’ Карбид NbC] 00 может быть получен, по-ви-
димому, при пониженных темп-рах. NbC имеет серый
цвет; плотн. 7,74 (NbC0,7(>), 7,82 (NbCM0); темп-ра
плавления карбида, близкого по составу к стехио-
метрия., 3500±125°; т. кип. 4300°. При 1990—2670°
NbC диссоциирует до NbC0>75, выделяя углерод; дав-
ление пара углерода над NbC: lg Pa[i, = 5,296—
3,276• 104 Т-1 при 2670°, после достижения состава
NbC0,75 карбид испаряется конгруэнтно. Теплота обра-
зования NbC ДЯ298= — 34,8 ккал/молъ: теплопровод-
ность 0,034 кал/см-сек-град: темп-ра перехода в сверх-
проводящее состояние 10,1 — 10,5°К; уд. электросо-
противление (мком-см): 74 (20°), 254 (3500°); микро-
твердость 2400 кГ/мм2: модуль упругости 34 500 кГ/мм2.
Образует твердые р-ры с NbN, карбидами и нитрида-
ми др. тугоплавких металлов; окисляется на воздухе
при 800° до Nb2O3. Карбид получают при взаимо-
действии углерода с Н. (1200°) или с Nb2O3 (1800°).
В системе Nb—С имеются две эвтектики: твердый р-р С
в Nb-f-NboC (1,5 вес. %С) с т. пл. 2335° и NbC-|-C с
т. пл. ~3000ь. Nb2C при 3265° распадается по пери-
тектич. реакции: Nb2C—* жидк.+NbC.
Н. с галогенами образует ряд галогенидов, окси-
галогенидов и комплексных солей (см. Ниобия гало-
гениды). Известны 5 боридов Н.: NbsB, Nb2B, NbB,
NbsBa и NbB2; 3 силицида: NbiSi, Nb3Si3 и NbSi2;
2 фосфида: NbP и NbP2; 2 сульфида: NbS и NbS2 c
узкими областями гомогенности, границы к-рых не
определены.
Аналитическое определение. Ана-
лиз материалов, содержащих Н., — один из наибо-
лее трудных. Разложение рудных материалов произ-
водят сплавлением с пиросульфатом калия, со щело-
чами или растворением в смеси HF и 1I2SO4; сплавы
и металлич. Н. растворяют в смеси HF и HN03. Ка-
чественно Н. открывают по оранжевой окраске соеди-
нения с танином, а также с применением пирогаллола,
пирокатехина и др. органич. реактивов или спект-
ральным методом. Основными способами отделения Н.
являются таниновый, пирогалловый, фениларсоно-
вый, ионообменные и экстракционные.
Для количественного определения Н. описаны ве-
совые, объемные и колориметрия, методы. Последние
приобрели большое значение для определения Н. в ши-
роком диапазоне концентраций. Из них одним из луч-
ших является роданидный метод, основанный на об-
разовании желтого комплекса Н. [NbO (SCNJi],
экстрагируемого органич. растворителями (эфир, этил-
ацетат). Широко применяют спектральный и рентге-
но-спектральный методы. Н.втехнич. металле может
быть определен по привесу пробы при прокаливании
на воздухе с учетом нелетучих примесей, в чистых
сплавах с Та — по плотности сплава.
Получение. Главнейшими видами сырья для произ-ва
II. и его соединений являются лопарит, пирохлор,
колумбит-танталит. Руды комплексные бедны 11.;
содержание Nb2O3 в них составляет 0,05—0,5, реже
2,0%. Их обогащают гравитационными методами с
последующей флотацией, электромагнитной, электро-
статич. или радиометрии, сепарацией. Ппрохлоровые
концентраты содержат не менее 37%, лопаритовые—
8%, колумбитовые — 30—60% Nb2O5', вредными при-
481
НИОБИЙ —НИОБИЯ ГАЛОГЕНИДЫ
482
месями при переработке на металл являются Ti, Si,
Sn. Большую часть концентратов перерабатывают
алюмино-или силикотермич. восстановлением на фер-
рониобий (40—60% Н.) и ферротанталониобий (см.
Металлотермия). Концентраты перерабатывают на
металл в три стадии: 1) вскрытие; 2) разделение Н. и
тантала и получение их чистых соединений; 3) полу-
чение и рафинирование металлов. Основными мето-
дами вскрытия являются: хлорирование; обработка
плавиковой к-той или фтористыми солями; сплавление
со щелочными плавнями — NaOH, КОН, Na2COa,
К2СОз, с кислыми плавнями — бисульфатами К или
Na; обработка серной к-той. Иногда для тех же целей
хлорируют феррониобий. Соединения Н. и тантала
переводят затем в р-р. При хлорировании, напр.,
лопаритовый концентрат смешивают с углем и связу-
ющим, брикетируют, подвергают коксованию при
800° и действию хлора при 700—730°, получая три
группы хлоридов: плав хлоридов редкоземельных
элементов (т. кип. выше 1000°), жидкие при обычной
темп-ре хлориды титана и кремния, твердые хлориды
Н. и тантала. До недавнего времени последние путем
гидролиза превращали в технич. гидроокиси, к-рые
растворяли в плавиковой к-те. При добавлении к по-
лученному р-ру КС1 или KF при концентрации HF
в р-ре<7% (обычно 1—2%) образуются комплексные
фториды K2NbOF5 и K2TaF7; последний выпадает в
осадок. Этот т. наз. фторидный метод разделения Н.
и тантала, или метод Мариньяка, был предложен в
1866. Его недостатки: сложность процесса, низкое из-
влечение и недостаточно полное разделение Н. и тан-
тала, трудности получения чистых соединений Н.
при высоком содержании Ti в сырье. Более эффекти-
вен метод разделения с одновременной очисткой
Nb и Та избирательной экстракцией органич. раство-
рителями. В качестве экстрагентов применяют обыч-
но трибутилфосфат и метилизобутилкетон. В пром-сти
экстракцию ведут, как правило, из плавиковокислых
р-ров. Из чистых р-ров Н. и Та выделяют их комплекс-
ные фториды с калием или гидроокиси. Предложены
также методы ионного обмена, избирательного вос-
становления соединений Н., ректификации галогени-
дов (очень перспективный метод) и др.
Одним из основных методов промышленного произ-ва
Н. является карботермический; применяют так-
же натриетермич. метод; интенсивно изучают элект-
ролиз расплавленных сред, восстановление пентахло-
рида водородом и др. методы. При восстановлении
K2NbF, натрием при 1000° получают порошок Н.,
как и при электролизе. Карботермич. метод обычно
осуществляют в две стадии: вначале из смесн Nb2O5
с сажей в атмосфере Н2 при 1800° получают карбид,
затем из смеси карбида и пятиокиси при 1800—1900°
в вакууме — металл. Спрессованные из порошка
штабики металла спекают в вакууме до 2300°, проко-
вывают на холоду и после кратковременного вто-
рого спекания получают плотные заготовки. Ковкой,
прокаткой, волочением на холоду получают прутки,
пластины, ленту, фольгу, проволоку и т. п. Качество
металла зависит от способа получения. Карботермич.
восстановлением и спеканием получают Н., практи-
чески не загрязненный примесями др. металлов и со-
держащий сотые — тысячные доли % О, С и N.С целью
получения крупных заготовок для выпрессовывания
труб спеченные штабики Н. переплавляют в дуговой
печи в вакууме. Спекание Н. при 2300° в пром-сти
заменяют электроннолучевой вакуумной плавкой,
дающей Н. высокой чистоты (менее чемпо0,01 вес.%
O,N и С) с твердостью по Бринеллю 45—55 кГ/мм2.
Монокристаллы Н. высокой чистоты получают бести-
гельной электроннолучевой зонной плавкой. Ниобие-
вые покрытия на др. металлы и материалы наносят
при восстановлении водородом парообразного NbC^
016 к.х.Э. т. 3
на нагретых покрываемых поверхностях либо тер-
мич. диссоциацией иодидов Н.
Применение. Металлич. Н. стал применяться в
пром-сти лишь последние 10—15 лет, его произ-во и
применение быстро возрастают. Полагают, что в ка-
питалистич. странах произ-во металлич. Н. увели-
чится через 5—7 лет до 5—10 тыс. m/год. Кроме того,
выпускается большое количество феррониобия и фер-
ротанталониобия.Напр., в США 60%всего Н. в 1955—
1956 использовали (в виде ферросплавов) в произ-ве
сталей, до 30% — для спецсплавов и до 10% — в виде
чистого металла, карбида и т. п. Наиболее перспек-
тивными областями применения Н., использующими
такие его свойства, как тугоплавкость, жаропрочность,
коррозионная стойкость (на холоду в кислотах и в
обезкислороженных расплавленных металлах), а так-
же способность поглощать газы, являются области,
связанные с высокими темп-рами, химич. аппарато-
строение, радиоэлектроника, атомная энергетика.
Н. — один из основных компонентов многих ме-
таллич. жаропрочных и коррозионностойких сплавов
(см. Ниобия сплавы); нек-рые сплавы содержат карби-
ды, нитриды, бориды, силициды Н. Введение Н. (в
виде ферросплавов) в нержавеющие хромоникелевые
стали предохраняет их от межкристаллитной корро-
зии и разрушения, улучшает свойства стали др. ма-
рок (см. Железа сплавы). В химич. пром-сти изН. из-
готовляют коррозионноустойчивую аппаратуру, напр.
в произ-ве соляной к-ты. Н. используют в электронике
для изготовления «горячей» арматуры, напр. катодов
мощных генераторных ламп, электролитич. конден-
саторов, в качестве геттера. Н. и сплавы Н. с танта-
лом могут заменить дефицитный тантал в большинст-
ве областей его применения, что дает большой эконо-
мия. эффект (Н. дешевле и почти вдвое легче Та).
Нек-рое применение находят соединения Н. Пяти-
окись — в качестве исходного сырья для получения
металлич. Н., ниобатов, карбидов и др. соед. нио-
бия; как катализатор в химич. пром-сти; в производст-
ве огнеупоров, керметов, а также стекол, не пропу-
скающих инфракрасные лучи и с высоким коэфф,
преломления. Ниобаты — для изготовления керамич.
сегнетоконденсаторов и пьезоэлектрич. преобразо-
вателей. Нитрид — в качестве детектора для радио-
волн, в конструкции трубок для передачи изображе-
ний, в сверхпроводящих болометрах. Карбид может
применяться при произ-ве твердых сплавов, для из-
готовления нагревателей и деталей печей, работаю-
щих при высоких темп-рах в вакууме или инертной
атмосфере, анодов и др.
Лит.: МеерсонГ. А., ЗеликманА. Н., Металлур-
гия редких металлов, М., 1955; Самсонов Г. В., Кон-
стантинов В. И., Тантал и ниобий, М., 1959; Заха-
рова Г. В. (и др.], Ниобий и его сплавы, Л., 1961; К о л-
ч и н О. П., Производство и применение ниобия и тантала за
рубежом, в сб.: Ниобий и тантал, М., 19б0;его же, Нио-
бий и тантал. Области освоенного и возможного применения,
М., 1959; Требования промышленности к качеству минераль-
ного сырья, вып. 49; Зив Е. Ф., ВайсенбергА. И.,
Ниобий и тантал, 2 изд., М., 1959 (Справочник для геологов);
Ч е р н и х о в Ю. А., Горошина В. Г., Завод, лабор.,
1945, 11, № 10, 875; М 1 1 1 е г G. L., Tantalum and Niobium,
L., 1959; Columbium metallurgy, ed. D. L. Douglass, F. W.
Kunz, N. Y.—L., 1961; Rare metals handbook, ed. Ch. A.
Hampel, 2 ed., L., 1961; Mellor, v. 9, L.—N. Y.—Toronto,
1947; U 1 1 m a n n, 3 Aufl., Bd 12, Munch.— B., 1960, S.
736—45; Kirk, v. 4, N. Y., 1949, p. 314—24; Pascal,
v. 12, P., 1958. О. П. Колчин.
НИОБИЯ ГАЛОГЕНИДЫ — простые и комплекс-
ные соединения ниобия с галогенами. Из соединений
Н. с фтором наиболее изучены производные 5-валент-
ного Nb; о низших фторидах в литературе очень мало
данных.
Пентафторид NbFs— расплывающиеся и ды-
мящие на воздухе бесцветные кристаллы; т. пл. 76°;
т. кип. 225°; теплота образования ДЯ°98=—370
483
НИОБИЯ ГАЛОГЕНИДЫ
484
ккал/моль. Водородом выше темп-ры кипения восста-
навливается до голубого низшего фторида, кислород
ниже темп-ры кипения на пентафторид не действует.
Водой интенсивно разлагается с образованием о к-
ситрифторида NbOFs и плавиковой к-ты. Пен-
тафторид получают действием фтора на Nb, а в рас-
творе — при растворении Nb2O5 в плавиковой к-те,
причем с избытком к-ты NbFs образует комплексную
фторониобиевую кислоту H2[NbF7].
При концентрации HF ниже 7% NbFs гидролизуется
с образованием окситрифторида и оксифторо-
ниобиевой кислоты: NbFs-|-H2O NbOF3+
7t2HF-|-H2[NbOFs]. При добавлении в плавиковокис-
лый р-р ниобия солей калия образуются, в зависи-
мости от концентрации к-ты, комплексные фтористые
соли: K2NbF, или K2NbOFs. Фторониобат
калия K2NbF, — моноклинные кристаллы; а =
=5,58 А, 6=12,67.4, с= 8,50 А, 0=90°; плотн. 3,21;
прп нагревании во влажном воздухе переходит в
K2NbOF5-H2O; в воде и р-рах, содержащих менее 7%
HF, гидролизуется, образуя K2NbOF5. Растворимость
зависит от концентрации HF и KF и темп-ры раствора.
Получают фторониобат при добавлении солей калия
в плавиковокислый р-р ниобия, содержащий более
7% HF. Применяют как исходное сырье для восста-
новления Nb натриетермич. методом.
Оксифторониобат «пластинчатый» («нор-
мальный») K2NbOFs-H2O, или NbOF3-2KF-H2O —
кристаллы с моноклинной структурой; а : Ь : с=
=0,992 : 1 : 0,980, 0=1ОЗ°46'. Устойчив на воздухе
при комнатной темп-ре и нагревании до красного ка-
ления, полностью обезвоживается при 180— 200°.
Растворяется в 12,5—13-кратном количестве воды; в
разб. р-рах HF при 20 40, 60 и 75° растворяется лучше
фторотанталата КгТаР, соответственно в 11,9, 11,6,
10,1 и 11 раз. При совместном присутствии K.2NbOF5
и K2TaF7 различие в растворимости этих солей уве-
личивается во много раз, происходит «высаливание»
танталовой соли солью ниобия, на чем основан один
из промышленных методов разделения Nb и Та. Устой-
чив в холодной и кипящей воде и плавиковокислых
р-рах, содержащих менее 7% HF, из к-рых получается
кристаллизацией. В зависимости от условий кристал-
лизации образуются и другиеформы: кубич. оксифторо-
ниобат NbOFs-3KF; моноклинный (иглы или приз-
мы) NbOFs-3KF-HF; гексагональный 3NbOFs-5KF-
Н2О; триклинный 3NbOF3-4KF-2H2O. При перекри-
сталлизации из водных растворов все эти малоизу-
ченные формы переходят в пластинчатый оксифтор-
ониобат.
Из соединений Nb с хлором важны и наиболее изу-
чены пентахлорид NbCl5 и окситрихлорид NbOCls;
низшие хлориды менее изучены.
Пентахлорид NbCl3— кристаллы желтого
цвета, в расплавленном состоянии с красноватым от-
тенком; имеет структуру тригональной пирамиды с
атомом Nb в центре; плотн. 2,75 (20°); т. пл. 204,5°;
т. кип. 247,4°; теплота образования = —190,5
ккал/моль. При высокой темп-ре пентахлорид разла-
1300°
гается: NbClsX Nb-|-5/2C12; в вакууме разложение идет
300°
на нагретых св. 600° поверхностях. NbCls восстанав-
ливается водородом при 400—500° до NbCl3, в отличие
от TaCls, к-рый в этих условиях устойчив, что исполь-
зуется для разделения Та и Nb. При 1000° NbCls
восстанавливается водородом до металлич. Nb (через
промежуточные низшие хлориды), что может быть
использовано для получения металлич. Nb и для на-
несения ниобиевых покрытий на др. металлы и ма-
териалы. NbCls образует непрерывный ряд твердых
р-ров с ТаС15. Пентахлорид ниобия гигроскопичен и
склонен к гидролизу, в результате к-рого образует-
ся гидроокись ниобия. Растворяется в плавиковой
к-те, в конц. соляной и серной к-тах. Получают дейст-
вием хлора на Nb или на смесь Nb2O5+C при нагре-
вании, либо действием НС1 на Nb (при 300—350°),
либо «дохлорированием» NbOCl, и ректификацией
продуктов хлорирования рудных концентратов.
Тетрахлорид NbCl4— игольчатые кристал-
лы черно-бурого цвета; теплота образования ДЯ°98 =
— —166,0 ккал/моль; сублимирует при темп-рах —350—
400°. Диспропорционирует при температурах 200—
1000° по реакции: 2,15 NbCl4 (газ)=1,15 NbCl3,13(TB.)-|-
-f-NbCls (газ). Легко окисляется на воздухе. NbCl4—
промежуточный продукт восстановления NbCls до
металла; может быть получен восстановлением NbCls
ниобием или танталом. Трихлорид NbCls — ве-
щество черного цвета; плотн. 3,7; теплота образо-
вания ДЯ298 = —139 ккал/моль. Диспропорционирует
при 800° по реакциям: 3NbCl2j87=2NbC12-|-NbCl*
и 4,5 NbCl2!97 = 3,5NbCl2 + NbCl3;’ восстанавливает-
ся водородом при 600—1000° до металлич. Nb.
Трихлорид нерастворим в воде, спирте, эфире,
разб. минеральных к-тах, водном растворе аммиака;
азотной к-той окисляется до Nb2Os; взаимодействует
при нагревании с СО2: NbCls+CO2=NbOC13-|-CO;
на воздухе начинает окисляться выше 100°. Получа-
ют восстановлением пентахлорида водородом ниже
600°. Ди хлорид NbCh — вещество коричнево-
го цвета; теплота образования ДЯ298 =—98 ккал/моль;
диспропорционирует при 800° по реакциям: 2NbCl2=
= Nb+NbCh (газ) и 2,5NbCl2=l,5Nb-)-NbCl5 (газ).
Окситрихлорид NbOCls — игольчатые бе-
лые кристаллы; теплота образования ДЯ298=—210,2
ккал/моль. Возгоняется при темп-рах до 400°, выше
распадается на Nb2Os и NbCls. Окситрихлорид незна-
чительно растворим в расплаве пентахлорида; с хлори-
дами нек-рых одновалентных металлов образует двой-
ные соединения типов MeCL NbOCls и 2МеС1-NbOCls;
склонен к гидролизу с образованием гидроокиси нио-
бия. Получается по реакции: 2NbO2-|-3C12=2NbOC13-|-
-|-О2, а также при нагревании моноокиси ниобия в
атмосфере хлора или при взаимодействии НС1 либо
NbCls с Nb2O5. Промежуточный продукт в техноло-
гии ниобия. Оксихлорид NbO2Cl почти не изу-
чен, теплота образования (рассчит.) ДЯ298 =—235
ккал/моль.
При взаимодействии Nb с бромом образуется пен-
табромид NbBrs — кристаллы красного цвета;
т. пл. 265,5°, т. кип. 361,6°, теплота образования
ДЯ298=—135,2 ккал/моль; растворим в воде, спир-
те, бромистом этиле. Известны, но менее изучены
трибромид NbBis и окситрибромид
NbOBis. С иодом Nb образует 2-, 3-, 4- и 5-ва-
лентные соединения. Пентаиодид NbJ6 — кри-
сталлы черного цвета с бронзовым оттенком; струк-
тура орторомбическая; плотн. 5,24; т. пл. 496°, т. кип.
543°, теплота образования ДЯ298= —102 ккал/моль;
очень неустойчив — выделяет иод при нагревании; в
вакууме уже при 200° переходит в низшие иодиды;
легко гидролизуется на воздухе, бурно реагирует с
водой. Тетраиодид NbJ4— кристаллы серого
цвета; структура орторомбическая; легко возгоняется
при 300°; парамагнитен; бурно реагирует с водой,
легко окисляется кислородом. Т р и и о д и д NbJ3—
гексагональные кристаллы черного цвета, подобно
NbCls и NbBrs, довольно устойчивы на воздухе. Д и-
иодид NbJ 2 — кристаллы темно-серого цвета;
плотн. 5,18; нерастворим в ацетоне, спирте и многих
др. органич. растворителях, а также в воде. Хорошо
растворим в азотной к-те. Получают восстановлением
485
НИОБИЯ ПЯТИОКИСЬ—НИРЕНШТЕЙНА РЕАКЦИЯ
486
водородом NbJ3 при 300—400°; при 400—1000° диио-
дид восстанавливается водородом до гидрида ниобия.
Существует окситрииодид NbOJ3— гексаго-
нальные кристаллы рубиново-красного цвета. NbOJa
разлагается при нагревании: 2NbOJ3=2NbOJ2 (тв.)-|-
-f-J2 (газ). Оксидииодид NbOJ2 разлагается вы-
ше 500°, конечная твердая фаза содержит NbO и NbO2.
Оба оксииодида диамагнитны. Йодиды Н. исполь-
зуют при рафинировании металлич. Н. способом их
термич. диссоциации, а также (как и пентахлорид Н.)
при нанесении покрытий Н. на др. металлы и мате-
риалы.
Лит.: Самсонов Г. В., Константинов В. И.,
Тантал и ниобий, М., 1959; Амосов В. М., Изв. высш,
учебн. заведений. Цветная металлургия, 1963, № 2; С а в-
ч е н к о Г. С., Т а н а н а е в И. В., Ж. прикл. химии, 1946,
19, № 10—И, 1947, 20, №5; М е е р с о н Г. А., Зелик-
ман А. Н., Металлургия редких металлов, М., 1955; М а-
r 1g па с С., Ann. chlmie et de physique, 1866, ser. 4, 8, 5—75.
О. П. Колчин.
НИОБИЯ ПЯТИОКИСЬ — см. Ниобий.
НИОБИЯ СПЛАВЫ — сплавы на основе ниобия.
Наиболее важны Н. с. с элементами, образующими
непрерывные ряды твердых р-ров, а также ограничен-
ные твердые р-ры и соединения с металлич. связью.
Строение электронной оболочки, размеры атома, сред-
нее положение в ряду электроотрицательности (по
отношению к Nb ок. 36 металлов являются электропо-
ложительными и ок. 40 — электроотрицательными)
определяют склонность Nb давать со многими ме-
таллами твердые р-ры, а с элементами, отличными
по электроотрицательности,— металлич. соединения.
Непрерывные ряды твердых р-ров Nb образуют с ме-
таллами, имеющими изоморфную кристаллич. струк-
туру, близко расположенными в ряду электроотри-
цательности, отличающимися по размерам атомных ра-
диусов не более чем на 8—10%; с [З-Ti, fi-Zr, V, Та,
Ра, Мо, W. При различии в атомных радиусах от
8—10% до 15—16% и большей удаленности элементов
от Nb в ряду электроотрицательности образуются ог-
раниченные твердые р-ры, а за пределами раствори-
мости — соединения с металлич. связью. Таких эле-
ментов 68; к ним относятся, в частности, La и ланта-
ниды, Hf, Th, Сг, Mn, Re, Fe, Ru, Os, Co, Rh, Ir, Pt,
Cu, Ag, Au, Zn, Cd, Hg, Al.
H. с. с цветными и редкими металлами, а также с
неметаллами обладают рядом ценных свойств — вы-
сокой механич. прочностью при обычной темп-ре,
твёрдостью, жаропрочностью, жаростойкостью, кис-
лотоупорностью, хорошими термоэмиссионными и
магнитными свойствами. Очень большое значение
приобрели жаропрочные Н. с. Ниобий, входящий в
«большую четверку» тугоплавких металлов — Nb, Та,
W, Мо,— один из важнейших компонентов многих
жаропрочных сплавов для газовых турбин, реактив-
ных двигателей, ракет и т. п. Высокотемпературное
применение чистого Nb, как и других металлов «боль-
шой четверки», без специального легирования и за-
щитных покрытий невозможно из-за его склонности
при повышенных темп-рах взаимодействовать с кис-
лородом, азотом и многими др. газами, причем металл
в результате поглощения газов становится хрупким
(см. Ниобий). Исследования по разработке жаропроч-
ных сплавов на основе металлов «большой четвер-
ки» ведутся в двух направлениях — путем легиро-
вания и путем создания защитных покрытий, а сами
сплавы выплавляют, как правило, в условиях вы-
сокого вакуума. Наиболее важны жаропрочные Н. с.
с V, Та, Мо, W, Ti, Zr, Hf, Al, Re и др., содержащие,
напр. (в вес. %): 10Zr; 5Ti и 5Zr; 5Мо и 5Hf; по
1 Ti, Zr, Hf; 5V, 5Mo и IZr; 15 Мо и IZr; 10 Мо и 10Ti;
20W, 10 Ti и 6 Мо; ЗА! и 3V; 15W, 5Мо и IZr. Послед-
ний сплав имеет при 1205° предел текучести 21 кГ/мм2,
предел прочности 34 кГ/мм2, длительную прочность
за 100 час. 11,9 кГ/мм2, удлинение 21%.
Важны Н. с. с танталом, к-рыми во многих об-
ластях техники можно заменять дефицитный Та. Раз-
работаны для ядерных реакторов Н. с. с относитель-
но небольшим поперечным сечением захвата тепловых
нейтронов, содержащие, напр. (в вес.%), ЗА1 и 3V;
7Ti и др. Н. с. с 20 вес.% урана показали высокие
предел прочности и твердость при 870°, что позволяет
повысить температуру эксплуатации ТВЭЛ. Высоки-
ми качествами обладают магнитные и коррозионно-
устойчивые Н. с.
В произ-ве жаропрочных и других высокотемпера-
турных твердых металлокерамич. материалов важ-
ную роль могут сыграть бориды, силициды, нитриды
и карбиды ниобия. Высший карбид NbC находит при-
менение в произ-ве твердых сплавов. Из интерметал-
лич. соединений Nb представляет интерес соединение
с оловом NbsSn, имеющее наивысшую темп-ру перехода
в состояние сверхпроводимости из всех известных
веществ, равную 18,05° К. Н. с. с железом — ферро-
ниобий (см. Железа сплавы.) — применяют для присад-
ки в хромоникелевые нержавеющие, жаропрочные
и другие стали для предотвращения межкристаллит-
ной коррозии, повышения жаропрочности, вязкости,
свариваемости и улучшения других свойств.
Лит.: Корнилов И. И., Полякова Р. С., Изв.
АН СССР. Отд. хим. н., 1962, № 4, 565; Захарова Г. В.
[и др.], Ниобий и его сплавы, М., 1961; Колчин О. П.,
Ниобий и тантал. Области освоенного и возможного примене-
ния, М., 1959; Самсонов Г. В., Константинов В. И.,
Тантал и ниобий, М., 1959; Ниобий и тантал, под ред. О. П. Кол-
чина, М., 1960; Miller G. L.. Tantalum and niobium, L.,
1959; J. Less-common Metals, 1960, 2, № 2—4; Columbium me-
tallurgy, ed. D. L. Douglass, F. W. Kunz, N. Y.—L., 1961.
О. П. Колчин.
НИРЕНШТЕЙНА РЕАКЦИЯ — получение га-
логенометилкетонов взаимодействием галогенангид-
ридов кислот с диазометаном:
RCOX + CH2N2—>-RCOCHaX + Na
Обычно к раствору 1 моля хлорангидрида при ком-
натной темп-ре или при незначительном нагревании
прибавляют диазометан. При обратном порядке при-
бавления получаются диазокетоны (см. А рндта-Эйсте-
рта реакция), к-рые также можно превратить в галоге-
нометилкетоны при действии галогеноводородов; по-
видимому, и при прибавлении дцазометана к р-ру
хлорангидрида промежуточно образуются диазокето-
ны, однако они тотчас же реагируют с галогеноводо-
родом, давая галогенометилкетоны, тогда как при об-
ратном порядке прибавления галогеноводород связы-
вается избытком диазометана:
RCOX + CH2N2—>-RCOCHNa + HX
RCOCHNa + HX —>-RCOCH2X+N2
CHaN3 + HX —>-CH3X + N2
Первая стадия H. p. (образование диазокетона)
включает присоединение диазометана по карбониль-
ной группе с дальнейшим отщеплением галогеноводо-
рода от биполярного иона (I):
— + R. ,О-
RCOX + CH2-N=N—S- )с< + —>-RCOCHN2 + HX
Xх xCH2N=N
I
Вторая стадия аналогична алкилированию кислоты
с помощью алифатич. диазосоединения:
RCOCH —N=N + HX—>-[RCOCH2Ne=N]X- —>-RCOCH2X + N2
Не исключена возможность, что в нек-рых случаях бипо-
лярный ион (I) непосредственно перегруппировывается в
галогенометилкетон подобно тому, как это имеет место при
взаимодействии кетонов с диазометаном (при этом образуются
гомологичные кетоны):
R. ,О“
/С\ +
Xх ^CHaN=N
R , R . ,О~
)C=O + CHaNa—>- NCZ
R'z R'z CHaKsN
rcoch2x
RCOCHaR'+N;
16*
487
НИСТАТИН—НИТРАТЫ
488
Н. р. применяется гл. обр. для получения алифа-
тич. и жирноароматич. галогенокетонов. Побочно
при Н. р. образуются дигалогенодиоксаны; в случае
инертных галогенангидридов эта реакция может стать
основной:
z°- R4 zO4
RCOX + CH2N2 ----- R-CZ ----- CH2
ГСН2 X CH2zC-X
V R
H. p. открыта в 1915 M. Ниренштейном.
Лит.: Бахман В., Струве В., в сб.: Органические
реакции. Сб. 1, пер. с англ., М., 1948, с. d4; Кнунянц И.Л.
[и Др.], Изв. АН СССР. Отд. хим. н., 1956, № 3, 377; Lewis
Н. Н., Nierenstein М., К ich Е. М., J. Amer. Chem.,
Soc., 1925, 47, № 6, 1728. Н. П. Гамбарян.
НИСТАТИН (фунгицидин, микостатин) C46H77NO19,
мол. в. 949,02 — антибиотик из группы антибиоти-
ков-полиенов, продуцируемый штаммом актиномицета
Streptomyces noursei. Аморфные препараты Н. окра-
шены в желтый цвет, кристаллич. высокоочищенные —
бесцветны или слабо окрашены; [а]р= —10° (в ле-
дяной уксусной к-те), +21° (в пиридине), -)- 12°
(в диметилформамиде). Н. хорошо растворим в диме-
тилформамиде, пиридине, ледяной уксусной к-те и
0,05 н. метанольных р-рах NaOH и НС1; в последних
трех растворителях Н. быстро инактивируется; плохо
растворим в воде, хлороформе, гексане; слабораство-
рим в метаноле, этаноле, н-бутаноле и диоксане; рас-
творимость его в этаноле, н-бутаноле и изопропаноле
возрастает в присутствии воды.
Н. частично разлагается при 160°, полностью —
при 250°, медленно окисляется кислородом воздуха
и разрушается на свету. В молекуле Н. содержатся
4 двойные, сопряженные, углерод-углеродные связи;
в УФ-спектре имеет характерные для тетраенов мак-
симумы при 292, 305 и 320 ммк. Н. обесцвечивает р-ры
перманганата калия в воде и брома вСС14. В Н. содер-
жатся функциональные группы кислого и основного
характера, он нестабилен при низких и высоких зна-
чениях pH.
При ацетолизе Н. и продукта его гидрирования в
присутствии H2SC>4 получается аминосахар, мико-
замин (I) в виде его тетраацетата:
н । -------о-----|
I. 1С-СН-СН-СН-СН
Roz 1111
ОН NH, ОН СН3
Микозамив выделен также и в тех же условиях из
антибиотиков амфотерицина В и пимарицина. Отно-
сительно строения безазотистой части молекулы Н.
достоверных давних не опубликовано.
При глубинном культивировании Н. накапливает-
ся в мицелии, откуда может быть извлечен экстрак-
цией низшими жирными спиртами и ацетоном. Из
экстрактов, полученных при обработке мицелиальной
массы спиртами, Н. выделяют фракционным осажде-
нием или концентрированием экстрактов в вакууме с
последующим промыванием выпавшего Н. водой и
хлороформом и сушкой в вакуумсушилке.
Н. обладает противогрибковым действием, особенно
активен по отношению к грибкам рода Candida. Н.
применяют внутрь при кандидомикозах кишечника,
общих микозах и как профилактич. антимикозное
средство при терапии антибиотиками, а также местно
при поверхностных кандидомикозах. Н. используют
также для предохранения культур тканей от загряз-
нения дрожжевой и грибковой флорой. Активность
Н. определяется микробиология, и спектрофотомет-
рия. методами.
Лит.: Шемякин М. М. [и др.], Химия антибиотиков,
3 изд., М., 1961; Трахтенберг Д. М. [и др.], Мед.
пром-сть СССР, 1960, К» 8, 18; ДоскоЧ и лов а Д.,
Гесс И., Антибиотики, 1959, 4, № 1, 50; Трахтенберг
Д. М. [и др.], там же, 1960, 5, № 5, 9; S а 1 t г а М. Н., [а. о.],
J. Amer. Chem. Soc., 1961, 83, Хв 12, 2785. Д. М. Трахтенберг.
НИТРАНОЛ (метамин, нитралетте, пренитрон, ди-
фосфат тринитрата триэтаноламина)СвН12О^4 -2НзРО4,
мол. в. 480,28—белый кристал-
лич. порошок, т. пл. 106—110° ZCH2CH2ONO2
(с разл.), растворим в спиртах n—ch2ch2ono2-2h3po4
и кислотах, плохо растворим в ch2ch2ono2
эфире, ацетоне и хлороформе.
При действии воды Н. гидролизуется с образова-
нием маслообразного тринитрата триэтаноламина,
нерастворимого в воде. Являясь тринитроэфиром,
Н. взрывоопасен. При нагревании со сплавом Де-
варда в 30%-ном р-ре NaOH Н. выделяет аммиак, с
молибдатом аммония Н. образует желтый осадок
(Р04 ). Количественно Н. определяют алкалиметрия,
титрованием фосфат-иона. Получают Н. этерификаци-
ей триэтаноламина азотной к-той с последующим
переводом образующейся азотнокислой соли через
основание в дифосфат. Н. — эффективное спазмоли-
тич. средство, вызывает расширение коронарных
сосудов; применяют гл. обр. для предупреждения
приступов стенокардии, реже — при спазме пери-
ферия. сосудов (перемежающаяся хромота и др.).
Лит.: Bovet D., В о v е t-N 11 t 1 F., Arch. Internal,
pharmacodyn., 1947, 73, 367; Barbiere J., Bull. Soc. chim.
France, 1944, 11, 470; Машковский M. Д., M e д в e-
д e в Б. А., Мед. пром-сть СССР, 1958, № 4, 56.
Л. Н. Яхонтов.
НИТРАТЫ металлов — соли азотной к-ты
HNOa. Н. вполне устойчивы при обычных условиях
и хорошо кристаллизуются. При нагревании Н. ще-
лочных и щелочноземельных металлов, а также Н.
Ag и Т1 разлагаются выше их темп-р плавления. Мно-
гие безводные Н. распадаются ниже темп-ры плавле-
ния. Характер распада Н. зависит от природы катио-
на. Соли щелочных металлов с отщеплением кислоро-
да переходят в соответствующие нитриты, напр.
2KNO3=2KNO2+O2; Н. металлов, стоящих в ряду
напряжений правее Си,— с образованием свободных
металлов, напр. 2AgNO3=2Ag+2NO2-|-O2; Н. осталь-
ных металлов разлагаются до окислов (обычно с
промежуточным образованием основных солей), напр.
2Pb(NO3)2=2PbO-|- 4NO2-|-O2. Н. легко отдают кис-
лород при высоких темп-рах и являются в этих усло-
виях сильными окислителями; смеси их с горючими
веществами сгорают чрезвычайно быстро.
Все Н. элементарных катионов хорошо растворимы
в воде, причем повышение темп-ры обычно сильно
увеличивает растворимость (напротив, нек-рые ком-
плексные Н. и Н. органич. оснований труднораство-
римы, напр. Н. нитрона C20H14N4-HNO3, применяе-
мый для количественного определения других Н.).
Лишь сравнительно немногие Н. (гл. обр. Na, К, Rb,
Cs, Ag, Tl, Ba и Pb) кристаллизуются из водных р-ров
в безводном состоянии. Н. большинства 2-валентных
катионов кристаллизуются с 4, 5, 6, а 3-валентных —
с 9 молекулами воды. На воздухе почти все эти крис-
таллогидраты легко расплываются. Напротив, Н.,
кристаллизующиеся без воды, негигроскопичны (кро-
ме NaNOs). Обезвоживание кристаллогидратов Н.,
образованных слабоосновными катионами, сопро-
вождается гидролизом, удаление HNO3 начинается
при 90—100° (Fe, Al, Сг), 140—150° (Mn, Со, Ni) и
наиболее интенсивно идет на 50—100° выше.
В нейтральных и слабокислых р-рах Н. не прояв-
ляют окислительных свойств. Только в сильнокислой
среде соли Fe2+ восстанавливают Н. до NO, напр.:
2KNO3 + 8FeSO4 + 4H2SO4 = 3Fe2(SO4)3 + K2SO4 +
4-4H2O4-2[Fe(NO)]SO4. Активные металлы (Zn, сплав
Деварда) восстанавливают Н. в щелочной среде до
NH3, что используется для количественного опре-
деления Н.
489
НИТРАТЫ—НИТРИДЫ
490
Вхождение во внутреннюю сферу комплексных со-
единений для иона NO~ мало характерно. Поэтому
нитратные ацидокомплексы известны лишь для срав-
нительно немногих элементов (в особенности они
характерны для Th, La и его аналогов и некото-
рых др.).
Н. получают действием HNOs на металлы, окислы,
гидроокиси и нек-рые соли, по реакциям обмена, дей-
ствием NO2 на основания (наряду с нитритами) или
солИо Н. нек-рых элементов (Na, К, Са) встречаются
в природе. Из них практич. значение имеют только
месторождения NaNOa (чилийская селитра). Н. ши-
роко применяют в различных отраслях пром-сти и
с.-х.: как удобрения (H.NH4, К, Na, Са), в произ-ве
взрывчатых веществ (Н. NH4, Ва) и черного пороха
(KNO3), как протраву при крашении (И. Cr, Fe, Al,
Си) и т. д. Н. аммония, щелочных и щелочноземель-
ных металлов наз. также селитрами.
О наиболее важных Н. см. Аммония нитрат, Ка-
лия нитрат, Натрия нитрат, Алюминия нитрат,
Железа нитраты и т. д.
Лит.: МиниовичМ. А., Соли азотной кислоты. (Ни-
траты), М., 1946; Некрасов В. В., Курс общей химии,
14 изд., М., 1962. Л. М. Ковба.
НИТРАТЫ о р г а нические — производные
азотной к-ты общей ф-лы R—ONO2 (эфиры, сметан-
ные ангидриды и дрв) Эфиры азотной к-ты — алкил-
нитраты — бесцветные или бледно-желтые приятно
пахнущие жидкости, кипящие немного выше соответ-
ствующих спиртов; нерастворимы в воде, хорошо рас-
Алкилнитраты
R Т. кип., °C 4
сн3 65 1,2075 (20°)
С2Нб 87,6 1,1084 (20°)
н-С3На 110,5 1.0548 (20°)
изо-СэНу 101 1,0360 (19°)
H-ChHs 136 1,0153
emop-C4Hg 124 1,0382 (0°)
изо-СШо 123 1,0124 (25°)
изо-С5Нп 147 0,9961 (22°)
— сн2-сн2 - взрывает 1,486 (25°)
СН--СН-СН2 1 1 1,335 (5°)
сн2—сн—сн2 1 1 1 > 1,591 (25°)
а пд 1,3979. 6 п£ 1,4122
творимы в спирте и эфире. Максимум абсорбции Н. ок.
6,1, 7,8 и 11,8 мк-, низшие Н. (до н-бутил) имеют ди-
польный момент~2,9Р;—ONCh-rpynna в Н. копланар-
на, и атом углерода находится в плоскости, перпен-
дикулярной к ONC>2-rpynne. При гидролизе Н. во-
дой и щелочами вследствие параллельно идущего
окисления образуется сложная смесь продуктов:
!RCH2CH2OH + NO3
RCH = CH2 + NC>7 +Н2о
RCbRCHO + NO-+Н2О
2
Действие конц. к-т приводит к образованию соответ-
ствующего спирта и нитроний-катиона:
Г н 1 +
rono2+h2so,—’Ir-onoJ +hso4
1 +
r-oh+no2
H. термически неустойчивы и нек-рые из них при
перегревании паров взрывают. Склонность к взрыву
повышается с увеличением числа ONC>2-rpynn:
2C3Hs (ONO2)3 —> 6СО2 + 5 Н2О + 3N2 + ‘/2О2
Н. — энергичные окислители. С реактивами Гринья-
ра Н. дают 1Х,1Х-диалкилгидроксиламипы:
2RMgJ+CH3CH2ONO2 —*-R2NOH
Н. обычно получают обработкой соответствующих
спиртов на холоду 100%-ной HNO8 или нитрующей
смесью в присутствии небольших количеств моче-
вины (последняя добавляется для удаления HNO2):
ROH + HNO3—>-RONO2
H2NCONH2 + 2HNO2—>3H2O + 2N2 +CO2
H. могут быть также получены взаимодействием AgNO,
с галогеналкилами (диалкилсульфатами), электроли-
зом солей дикарбоновых к-т в присутствии KNO,
(NaNOs) и др. методами. Нек-рые Н. (нитроглицерин,
динитрат этиленгликоля, пентрит, нитроцеллюлоза
и др.) широко применяют для приготовления на их ос-
нове взрывчатых веществ и порохов. Смешанные ан-
гидриды карбоновой и азотной к-т, т. наз. ацплнитра-
ты (напр , ацетил- и бензоилнитраты) иногда приме-
няют для нитрования ароматич. соединений. Ацетил-
нитрат получают прибавлением НгО5 к (СН3СО)2О
с последующей разгонкой в вакууме (22770 мм). Пре-
паративные возможности ацетилнитрата ограниче-
ны его сильными взрывчатыми свойствами. Физио-
логии. действие Н. основано на их сильных окисляю-
щих свойствах. Н. окисляют гемоглобин до метгемо-
глобина, вызывают головные боли, в больших дозах—
рвоту. Нек-рые Н. благодаря их способности расши-
рять кровеносные сосуды и понижать давление на-
шли применение в медицине (нитроглицерин и др.).
Лит.: Chem. Rew., 1955, 55, № 3, 485. В. Н. Фросин.
НИТРИДЫ — соединения азота с электроположи-
тельными элементами, гл. обр. с металлами. По
типу химич. связи между атомами элемента и азота
Н. могут быть подразделены на три основные группы:
ионные (или солеобразные), ковалентные и металло-
подобные. К первой группе относятся Н. сильно
электроположительных металлов I и II групп перио-
дич. системы элементов, атомы к-рых имеют внешние
s-электроны (щелочных, щелочноземельных, металлов
подгрупп Си и Zn). Ковалентные Н. образуются при
соединении с азотом металлов и неметаллов, атомы
к-рых имеют внешние р-электроны (с В, Al, Si, Ga,
Ge и т. п.). Металлоподобные Н. (являющиеся фа-
зами со структурой внедрения) образуются металла-
ми переходных групп, атомы к-рых имеют недостроен-
ные внутренние d- или /-электронные оболочки.
В соответствии с этими типами связи ионные и кова-
лентные Н. имеют ф-лы, отвечающие обычным валент-
ностям элементов (Li3N, K3N, Ca3N2, GaN, InN, BN,
AIN, Si3N4), и могут рассматриваться как производ-
ные аммиака, в к-ром атомы водорода замещены ато-
мами металла; металлоподобные же Н. имеют более
сложные составы, не отвечающие обычным валентно-
стям (TiN, ZrN, Cr2N, MnaN2), причем переходные
металлы образуют в ряде случаев по нескольку Н.
(Nb2N и NbN, Ta2N и TaN, Cr2N и CrN).
Ионные Н. могут быть получены непосредст-
венным взаимодействием металлов с азотом или аммиа-
ком (напр., Li3N — при 450— 460°, Zn3N2 — при
500—600°, BesN2 — при 500°), либо взаимодействием
гидридов с азотом, либо легко восстанавливаемых
окислов металлов с аммиаком (напр., ЗСигО-f- 2NH3=
= 2Cu3N-]-3H2O — при 250—280°) или разложением
высших азотистых соединений металлов — азидов.
Все ионные Н. имеют высокое уд. электросопротивле-
ние и являются полупроводниками с различными
значениями ширины запрещенной зоны— для Cu3N,
напр., она составляет 0,23 эв, для Zn3N2 — 0,09 эв.
491
НИТРИДЫ
492
Таблица 1. Физические свойства некоторых ионных
нитридов
Нит- рид Кристал- лич. струк- тура Плот- ность, г/см3 Т. пл., °C Т. разл., °C Уд. электро- сопротивле- ние, ом-см
Li3N гекс. 1,28 845а 300-350 8,2-10* (446°)
Na3N ромб. 1,85 разл. 350 —
Cu,N куб. ГЦ. 5,84 разл. 450 6-Ю2 (20°)
Zn,N, куб. гц. 6,22 разл. 700—750 4,5-Ю3 (20°)
куб. гц. 2,72 2200 2240 2-10е (20°)
MgaN3 куб. гц. 2,74 разл. 1500 —
Примечание. В этой и других таблицах приняты
след, условные обозначения: гекс,—гексагональная, ромб,—
ромбоэдрическая, куб. — кубическая, ну,— плотноупакован-
ная, гц,— гранедентрированная. а Под давлением азота.
Н. щелочных металлов — малоустойчивые соеди-
нения; при обычных темп-рах устойчивы лишь в су-
хом воздухе и водороде; при темп-рах порядка 300—
350° разлагаются на элементы; под действием воды
легко разлагаются с образованием аммиака (как вооб-
ще все ионные Н.), напр.: Na3N+3H2O=3NaOH-(-
4-NH3; легко реагируют с хлором, фосфором, серой,
с разб. к-тами; при нагревании в водороде переходят
в гидриды. Н. щелочноземельных металлов более
устойчивы; при нормальной темп-ре на воздухе не
изменяются; разлагаются на элементы при темп-рах
плавления или еще более высоких; нек-рые из
них очень медленно разлагаются кипящей водой
(например, BesNz), другие легко разлагаются при
обычной температуре во влажном воздухе (например,
Mg3N2).
Ковалентные Н. получают действием азо-
та или аммиака на порошкообразные металлы или не-
металлы, напр. 2A1+N2=2A1N. В случае трудноиз-
мельчаемых в порошок легкоплавких р-элементов,
Таблица 2. Физические свойства некоторых ковалентных нитридов
Нит- рид Кристал- лич. структу- ра Плот- ность, г/см3 Т. пл., °C Т. разл., °C Уд. элект- росопро- тивление, ом-см
A1N гекс. пу. 3,05 > 2200а 2200 108-10>° (20°)
BN гекс. 2,34 30 00а 2500 1013 (20°)
S13N, гекс. 3,18—3,21 1780— 1820 1780— 1820 105 (350°)
а Под давлением азота-
дающих ковалентные Н. (напр., галлия и индия.),
для ускорения процесса к мелконарезанным кусоч-
кам металла добавляют т. наз. разрыхли-
тели — азотсодержащие вещества, легко
разлагающиеся при нагревании, напри-
мер (NH4)2CO3; продукты их разложе-
ния барботируют через расплавленный
металл, облегчая доступ к нему азота.
Другим способом получения ковалентных
Н. служит восстановление окислов соот-
ветствующих элементов при одновремен-
ном действии азота: B2O3-|-3C-|-N2=
= 2BN-4-3CO.
Н. этой группы являются либо диэлек-
триками, либо полупроводниками; осо-
бенно высокими изоляционными свойст-
вами при высоких темп-рах обладают Н.
алюминия, бора и кремния, а также
нек-рые их сплавы. Н. бора BN изве-
стен в двух кристаллич. модификациях;
при обычных условиях образуется гекса-
гональный BN со структурой типа графита в виде
белого порошка; при 1360° и давлении 60—70 т/см*
он превращается в известную под названием боразо-
на кубич. алмазоподобную модификацию с периодом
решетки а=3,615 А и плотностью 3,45. Боразон —
кристаллы, окрашенные в цвета от красного до чер-
ного, он обладает твердостью, близкой к твердости
алмаза, стоек при нагревании до 2000° (в то время как
алмаз сгорает уже при 900°). Для Н. кремния, алю-
миния и бора характерна высокая термостойкость
при быстрых и частых изменениях темп-ры. Все
ковалентные Н. довольно устойчивы; особенно устой-
чивы AIN, BN, Si3N4, к-рые начинают слабо разла-
гаться на элементы только при темп-рах выше 1000—
1200°, обладают высокой стойкостью против окис-
ления, против действия расплавленных металлов,
горячих кислот, различных агрессивных газов.
Металлоподобные Н. получают гл. обр.
непосредственным действием азота или аммиака на
порошки металлов при 800—1200°, либо восстанов-
лением окислов углем или иными восстановителями
в среде азота, напр.: TiO24-2C-|-l/2N2=TiN+2CO.
Для металлоподобных Н. переходных металлов ха-
рактерно наложение нескольких типов химич. связи,
гл. обр. ионной и металлической, причем доли раз-
личных типов связи меняются в соответствии с изме-
нением степени электроположительности металлич.
компонента, степени достроенности d- или /-элект-
ронной оболочки и потенциала ионизации металла.
Благодаря участию в связях между атомами металлов
и азота не только внешних s-электронов, но и более
глубоко расположенных d-электронов недостроенных
оболочек (образование гибридных spa!-функций); хи-
мич. связь в таких Н. очень прочна. Вследствие это-
го Н. переходных металлов представляют собой, как
правило, весьма тугоплавкие вещества, обладающие
высокой твердостью, металлическими электро- и тепло-
проводностью, химич. стойкостью, жаростойкостью
и жаропрочностью.
В структурной отношении Н. переходных металлов
представляют продукты внедрения атомов азота в кри-
сталлич. решетки металлов с образованием простых в
структурном отношении фаз (с гранецентрированной
кубической или плотноупакованной гексагональной
решетками). Это обусловлено тем, что большая их часть
удовлетворяет правилу Хэгга, согласно к-рому фазы
внедрения образуются при условии An : Яме^0,59,
где 7?n и 7?ме — атомные радиусы азота и ме-
талла. Природа металлоподобных Н., как фаз внедре-
ния, обусловливает их высокую твердость и износо-
стойкость, практическое отсутствие пластичности при
обычных температурах, высокую хрупкость и относи-
тельно невысокие прочие механические (прочностные)
свойства.
Таблица 3. Физические свойства некоторых металлоподобиых нитридов
Нит- рид Кристал- лич. структура Область гомо- генности, ат. % азота Плот- ность, г /см3 Т. пл., °C Микро- твердость, кГ/мм2 Уд. элект- росопро- тивление, мком-см (при 20°)
TIN
ZrN
HfN
V3N
VN
Nb2N
NbN
Ta2N
TaN
Cr2N
CrN
MoaN
куб. гц. 37,5-50 5,43 3200 2000 25
куб. гц. 46-50 7,09 2980 1520 21
куб. гц. не опред. 13,84 3380 1800 33
гёкс. пу. 25—33 5,97 не опред. 1900 123
куб. гц. 41-50 6,04 2360 1520 85
гекс. пу. 28.5—33,5 8,33 2420 1720 142
гекс. пу. 50—50,6 8,40 2300 1400 78
гекс. пу. 28,5—31 15,81 2050 1220 263
гекс. пу. 44,5—47,3 15,46 3087 1060 128
гекс. пу. 32-33,3 6.51 1650 1500 79
куб. гц. не опред. 6,18 1500 1093 640
(разл.)
куб. гц. 32-33 8,04 600 » 630 20
493
НИТРИДЫ — НИТРИЛЫ
494
Характерным свойством Н. этой группы является
существование широких областей гомогенности (см.
табл. 3). В пределах области гомогенности свойства
металлоподобных Н. изменяются, что связано с из-
менением характера распределения электронной плот-
ности в их кристаллич. решетках и соответственным
изменением характера химич. связи. Характер химич.
связи в Н. переходных металлов зависит от степени
недостроенности d-электронной оболочки изолирован-
ных атомов металлов и энергетич. состояния d-элект-
ронов, т. е. от т. наз. акцептирующей или акцептор-
ной способности d-электронных оболочек переходных
металлов. Многие металлоподобные Н. являются
хорошими сверхпроводниками, напр. темп-ры пере-
хода в сверхпроводящее состояние составляют: для
NbN 15°К, для MoN 12°К. Магнитная восприимчи-
вость Н. переходных металлов больше, чем у карбидов
и боридов тех же металлов. Азот в Н. способен до
определенных концентраций замещаться кислородом
с соответственным усилением ионных признаков, с
образованием оксинитридных фаз, обладающих ти-
пичными полупроводниковыми свойствами. Металло-
подобные Н. обладают высокой химич. стойкостью,
особенно против действия холодных и кипящих к-т,
многих расплавленных металлов, а также против
окисления на воздухе; менее устойчивы в растворах
щелочей, быстро разлагаются сплавлением со щелоча-
ми и солями щелочных металлов.
Применение. Многообразие нитридных фаз
с широкими пределами изменения физических свойств
создает возможности их использования в разнооб-
разных отраслях техники. Огне-
упорные свойства ковалентных
и металлоподобных Н. исполь-
зуются для создания футеровок
ванн в процессах электролиза
металлов из расплавленных сред,
защитных чехлов термопар для
контроля темп-р разнообразных
металлургических и высокотем-
пературных химич. процессов и
их автоматизации, изготовления
лодочек для испарения металлов
в процессах металлизации, сопел
для распыления расплавленных
металлов, тиглей для плавки
редких металлов, сверхчистых и
прецизионных сплавов. Высокая жаропрочность и
жаростойкость ковалентных Н. (нитриды А1, В, Si),
а также некоторых металлоподобных Н. (нитриды Ti,
Zr, Hf) в сочетании с умеренными коэффициентами
термического расширения, высокой термостойкостью
позволяют использовать их для создания сплавов
с высокой жаропрочностью для техники высоких
температур, газотурбостроения, энергетики. Полу-
проводниковые свойства ионных, ковалентных и
некоторых металлоподобных Н. позволяют создавать
новые полупроводниковые устройства, способные эксп-
луатироваться при высоких темп-рах. Сверхпро-
водимость нек-рых металлоподобных и ковалентных
Н. может быть использована в технике низких темп-р,
для создания сверхпроводниковых болометров (нитрид
ниобия) и т. п. Высокая электропроводность нек-рых
Н. и малая зависимость их электросопротивления от
темп-ры позволяют использовать их в качестве токо-
проводящих компонентов малогабаритных или мини-
атюрных сопротивлений, проводящих элементов, ок-
сидных катодов. Большие перспективы использова-
ния в технике имеют ковалентные Н. (AIN, BN)
в связи с их высокими диэлектрическими свойст-
вами.
Азотированием металлов (т. е. действием азота или
аммиака на компактные металлы при нагревании) по-
лучаются нитридные покрытия, обла-
дающие высокой твердостью, износостойкостью, кор-
розионной устойчивостью, что широко используется
в машиностроении для поверхностного упрочнения
деталей машин и механизмов. Высокая твердость Н.
позволяет применять их для абразивной обработки
(шлифования) особо твердых материалов — твердых
сплавов, искусственных и природных драгоценных и
полудрагоценных камней. Нек-рые Н. обладают вы-
сокими каталитич. свойствами, к-рые используются в
процессах органич. и металлоорганич. синтеза, в част-
ности при переработке твердого топлива в жидкое.
Лит.: С а м с о н о в Г. В., У м а и с к и й Я. С., Твердые
соединения тугоплавких металлов, М., 1957; Самсонов
Г. В., Портной К. И., Сплавы на основе тугоплавких
соединений, М., 1961; С а м с о н о в Г. В., Ж. структ. хим.,
1960, 1, К» 4, 447; Сам сонов Г. В., Верхоглядова
Т. С., ДАН СССР, 1962, 142, № 3, 608; С а м с о н о в Г. В.,
Верхоглядова Т. С., Львов С. Н., Немченко
В. Ф., там же, 1962, 142, № 4, 862; В е р х о г л я д о в а Т. С.,
Дубовик Т. В., С а м с о н о в Г. В., Порошковая метал-
лургия, 1961, № 4, 9; Б е л и к о в А. М., У м а н с к и й Я.С.,
Научн. докл. высш. шк.Металлургия,1958, № 1,192; Саме о-
н о в Г. В., Верхоглядова Т. С., Ж. структ. химии,
1961, 2, вып. 5, 617; Самсонов Г., Лютая М. Д., Ж.
прикл. химии, 1962, 35, вып. 8, 1680; Киффер Р., Швар-
цкопф П., Твердые сплавы, [пер. с нем.], М., 1957; Руко-
водство по препаративной неорганической химии, под ред.
Г. Брауера, пер. с нем., М., 1956; ЮргенсонА. А., Азоти-
рование в энергомашиностроении, Свердловск — М., 1962;
Уманский Я. С., Карбиды твердых сплавов, М., 1947.
Г. В. Самсонов.
НИТРИЛЫ карбоновых кислот R—C=N,
производные карбоновых к-т, являющиеся органич.
производными синильной к-ты. Физич. свойства важ-
нейших Н. приведены в таблице:
Нитрил Формула Мол. в. T. пл., ’С Т. кип., ’С t nD
ацето- CH.C=N 41,052 —44,9 81,6 1,3442в 0,7828в
пропио- C2HeCsN 55,08 —91,85 97,1 1,3683а 0,78673а
бутиро- h-CsH,C=N 69,10 —111,9 117,6 1,3860а 0,79542а
стеаро- h-C17Hs5C=N 265,47 41 214/13 1,4405д 0.8149Д
акрило- CH2 = CH-CsN 53,03 —83 77,3 1,3911в 0,8060
пропиоло- H-C=C-teN 51 , 05 5 42,5 1,3870s 0,8159s
глутаро- NsC-CH2CH2CH2CsN 94,11 -29 142/10 — 0,9894г
бензо- C»H,CsN 103,12 -13,8 191,1 1,53056а 1,0095а
цианоформ- H-C (CN)3 91,07 55-56 — — —
а При 15°. 6 При 17°. в при 20’. г При 25°. Д При 45’.
Химич. свойства Н. определяются наличием непо-
деленной пары электронов на атоме азота, а также
ярко выраженной полярностью связи углерод — азот.
За счет неподеленной пары электронов на атоме азо-
та Н. легко образуют комплексы с солями Al,Sb и
др. Обработка Н. алкилирующими агентами электро-
фильного характера приводит к нитриллие-
вым солям.
+ — + -----------------------------------
(C2HS)3OBF, + CH3- С =N —> [СН3 - С= N - C2HS] BF.
С„Н5-С s N + SbCl5 + RCl —> [С«Н5-С = N-R'J SbCla
H. взаимодействуют с диеновыми углеводородами
с образованием алкилпиридинов, причем промежуточ-
но образуются соответствующие дигидропиридины:
Конденсация Н. с ацетиленом в присутствии щелочных
металлов в зависимости от условий проведения реак-
ции приводит к алкилпиридинам (I) либо алкилпири-
495
НИТРИЛЫ
496
мидинам (II). Под действием щелочных металлов или
их амидов Н. образуют димеры и тримеры.
R—C=N
н-с=с-н
2С2Н5-CHN
С2Н5—С—NH
СН3—СН—C=N
3C2H5-C=N
nh2
Ароматич. H., а также Н., содержащие электроот-
рицательные группировки под действием кислотных
реагентов, образуют симметрия, триазины:
|_| +
3CeH5-C2N—-
СвН5
Гидратация Н. как в кислой, так в щелочной среде
приводит к амидам, к-рые при дальнейшем гидролизе
переходят в соответствующие карбоновые к-ты:
н2о н2о
R — С = N----------- R - С - NH3--> R - СООН
Н+ или ОН- II
О
Переход от Н. к амидам легко осуществим также при
действии перекиси водорода в щелочной среде.
С безводными галогеноводородами Н. образуют
комплексные имидогалогениды:
НВгГ + 1 Г +1
СН3—C=N —*[СН3—C=NHJBr- илиЬсНз—C=NHJBr--HBr
Спиртовые р-ры галогеноводородов переводят Н. в
галогеноводородные соли иминоэфиров, гидролизом
к-рых могут быть получены сложные эфиры соответ-
ствующих карбоновых к-т:
+
R'OH + IICl zNII2 НоО
R-C=N-----------> R - С/ Cl" —-R — COOR'
XOR' J
H. легко присоединяют по кратной связи различные
нуклеофильные реагенты. Так, действие аммиака
или аминов на Н. приводит к соответствующим ами-
динам:
R,R2NH
R-C = N----->
R-C
/NH
xnr,r2
Обработкой Н. сероводородом получают тиоамиды:
H2S
R — С = N —> R — С — NH3
II
S
Азотистоводородная к-та реагирует с Н. с образова-
нием алкплтетразолов:
R-C=N R-C=NH— R-C—N
Магнийорганич. соединения легко присоединяются
по кратной связи Н., содержащих группировку —
CH2C=N, с образованием магниевых производных
иминов кетонов. Гидролиз последних приводит к са-
мим кетонам:
R'MgX RCh2x h2orch24
r-ch2-csn------> )c=N-Mgx —> ;c=o
R'Z R'Z
H., содержащие циангруппу, связанную co вторич-
ным или третичным атомом углерода, с магнийорга-
нич. соединениями вступают в реакцию двойного об-
мена:
R'MgX
(СН3)3С-С = N----->(СН3)3 CMg X+R' — С =N
Под действием каталитически возбужденного водорода
либо Na в спирте Н. легко восстанавливаются до
первичных аминов:
1) H2/Pt, Ni; 2) Na + R'OH
K-CsN--------------------------->- R — CH2 — NH,
При каталитич. гидрировании в качестве побочных
продуктов могут образовываться также и вторичные
амины:
Нд НгО /уО
R —С= N—> R-CH=NH-------►R-C^
1н2 Н
r-ch2-nh2 rch2nh2
rch2nh-ch2r
H2
-----rch2n=ch-r
Двухлористое олово в кислой среде восстанавлива-
ет Н. до альдегидов с промежуточным образованием
альдиминов (см. Стефена реакция):
SnCl2 + HCl
RC == N-HC1------->RCH = NH-HC1
Ь°
/О
R - С/
'II
Реакции Н., связанные с изменением углеводород-
ного радикала, аналогичны соответствующим реакциям
сложных эфиров карбоновых кислот.
Основные методы синтеза Н.: дегидратация амидов
или аммонийных солей карбоновых к-т, а также окси-
мов альдегидов:
R—С - о—NH, \
о
R—с — nh2 I------------->r-Csn
£ р2о3; so2ci2; p2s3
R—CH = N—OH I
Действие алкилирующих агентов на щелочные соли
синильной к-ты:
R - Hal 1
(RO)2SO2 I KCN; NaCN
RO-SO2-C.,H<-CII3 )
Присоединение синильной к-ты по углерод — углерод-
ной кратной связи:
сн3
СН3-С = СН2 Н —C = N |
|--------------«- СН3 —С —C = N;
СН3 А1,О3; ~ 400° |
СН3
СН.=СН-Сз\
ОН-IHCN
NsC-CH2CH2-CsN
Действие синильной к-ты на альдегиды или кетоны с
образованием Н. а-оксикислот (см. Циангидрины):
СН,. H-C = NCH,. ,ОН
;с=о----------->•
сн/ к2со3 сн/ хс = N
аналогично из а-окисей получают Н. р-оксикислот:
СН2-СН2 Н —C = N
KCN; К2СО3
НО —СН2СН2 —С ==а N
Окислительное аминирование (одновременное амини-
рование — дегидрирование) углеводородов и нек-рых
их производных:
NHS
С»Н5-СН3 -----► С»Н5С == n + h2
Си (Ag)
NHa
С3Н3ОН -------—>СН3-С - N + H2o + H,
МоО3 — А12О3
497
НИТРИЛЬНЫИ КАУЧУК —НИТРИТЫ
498
дегидрирование аминов в присутствии металлич. ка-
тализатора:
— 2Н
CeH5CH2NH2----------->- C6HS - С = N
Nl; 300-350°
Действие металлоорганич. соединений на галогеноциа-
ны или дициан:
Cl —C=N (N==C —C=N)
R - MgX----------------► R - Cs N + CIMgX (N=C-MgX)
теломеризация олефинов галогеноцианами с образо-
ванием Н. со-галогенокарбоновых к-т:
Cl —C = N
п СН2 = СН2----------->-С1 (CH2CH2)nC = N
50-80°; перекись
Специфич. методы синтеза ароматич. Н.: действие
цианистых солей на соли арилдиазониев (см. Занд-
мейера реакция):
CuC s N + KCN
CeH5N2Cl-----------»С6Н5С = N
Обработка ароматич. углеводородов хлорцианом ли-
бо трихлорацетонитрилом в присутствии А1С1з:
C1-C=N
Cells------>СаН5-С = N + HC1
AICI3
CQj-CaN -CHC13
CsHs-------->- CC13-C-CBH5------>CBH,-CsN
A1C13+HC1 II
NH
сплавление солей сульфокислот с щелочными циани-
дами:
к-с=х
CBH5SO3K-------> с»н5- с = N + K2SO3
См. также Ацетонитрил, Акрилонитрил.
Лит.: Houben-Weyl, 4 Aufl., Bd 8, Stuttgart, 1952,
S. 265. Л. С. Герман.
НИТРИЛЪНЫЙ КАУЧУК — см. Бутадиен-нит-
рилъный каучук,
НИТРИТЫ металлов — соли азотистой к-ты
HNO2. Термически Н. обычно менее устойчивы, чем
нитраты. Темц-ры разложения Н. щелочных элемен-
тов несколько выше их темп-р плавления, остальные
Н. разлагаются до плавления, напр. Ba(NO2)2 —
ок. 235°, AgNOa — ок. 120°. Н. малоактивных метал-
лов (напр., Ag) при нагревании распадаются на ме-
талл, нитрат и окислы азота; Н. остальных металлов —
на их окислы, нитраты, окислы азота и азот, напр.;
2 Ba(NO2)2 = BaO + Ba (NO3)2 + NO + '/2N2
2 AgNO2 = AgNO3 + Ag + NO
Термич. распад Н. осложняется побочными реакция-
ми образующихся окислов азота с Н. и окислами ме-
таллов:
2 NaNO2 = Na3O + NO+NO2; NaNO2 + NO2=NaNO,+NO;
Na2O + 2 NO2=NaNO3+NaNO2; 2 NaNOa + 2 NO = 2 NaNO3 + Na
Почти все H. хорошо растворимы в воде; с повыше-
нием темп-ры растворимость увеличивается. Сравни-
тельно мало растворим AgNO3 (в 100 г воды раство-
ряется 0,16 г при 0°,1 г при50°). Из растворов Н. Na,
К и Ag кристаллизуются в виде безводных солей;
Н. Са, Sr, Ва обычно кристаллизуются в виде моно-
гидратов и лишь выше 100° — в виде безводных со-
лей. Нек-рые Н. растворимы в растворе NH3, почти
все Н. (кроме LiNO3-2H3O) плохо растворимы в абсо-
лютном C2HSOH.
Н. проявляют как окислительные, так и восстано-
вительные свойства. Твердые Н., нейтральные и сла-
бощелочные р-ры Н. щелочных и щелочноземельных
металлов не окисляются кислородом воздуха. Менее
устойчивы разбавленные р-ры Н., особенно в случае
менее активных металлов. Сильные окислители
(КМпО4, Н2О2, Оз и т. д.) в слабокислых р-рах окис-
ляют Н. до нитратов. В щелочной среде Н. восста-
навливаются до NHa (восстановители: H2S, сплав Де-
варда, Zn и т. д.):
NaNO2 + 3 Zn + 2 NaOH + 2H2O=NH3+3 NaHZnOj
В слабокислой или нейтральной среде Н. восста-
навливаются до NO (восстановители: Fe2+, Ti3 + ,J~,
H2S), до N3 (NH4+, CO(NH3)3, HSO3NH2), до N2O (вос-
становитель Sn2+), до гипонитритов' (амальгама Na)
и т. д.
Образование коричневого комплексного соедине-
ния [Fe(NO)]SO4 идет на холоду в слабокислой среде
(отличие от нитратов, к-рые дают эту реакцию в при-
сутствии конц. H2SO4). С хлорангидридами и некото-
рыми безводными хлоридами Н. реагируют, образуя
NOC1:
PCl5+NaNO3=POCl3+NaCl + NOCl
Из комплексных Н. наиболее известны соединения
типа Me*[Zn(NO3)4j и Ме|[МеП1(НО3)в] (где Mei —
Na, К, NH4, Rb, Cs, a Mein— Со, Rh, Ir). Кобальти-
нитриты К, NH4, Rb и Cs мало растворимы, что ис-
пользуется в аналитич. химии этих элементов. Раство-
ры комплексных Н. устойчивее растворов простых Н.
Получают Н. действием смеси равных количеств NO
и NO2 на окислы и гидроокиси, восстановлением
нитратов (восстановители Pb, CaSO3, PbS-|-CaO и др.):
NaNO3+Pb=PbO + NaNO2
и по реакциям обмена:
BaCl3 + 2NaNO3 Ba (NO2)2 + 2 NaCl
H. образуются также при действии N2O и NO на пере-
киси:
Na2O2 + 2 N2O = 2 NaNO2+N2
H. применяют гл. обр. в произ-ве азокрасителей.
О строении Н. и их аналитич. определении см. Азо-
тистая кислота. Важнейшие Н. описаны в статьях
Натрия нитрит И др. Л. М. Коеба.
НИТРИТЫ орг анически е—производные азо-
тистой к-ты общей ф-лы R—О—N=O (эфиры и т. д.).
Эфиры азотистой к-ты — алкилнитриты — бесцвет-
ные или бледно-желтые приятно пахнущие жидкос-
ти, плохо растворимы в воде, хорошо — в спирте и
эфире; нек-рые константы этих эфиров приведены в
таблице.
т. кип.,
°с
сн„
с3н.
и-С3Н
изо-С.Н,
н-С4Нэ.
изо-С,Н9
н-С5Н
изо-С5Нц
н-С„Н,а
-СН2-СН2—
- (СН2)3-
-СН2-СН-СН2-
17
57
45
75
67
104
99
130
96
109
взрывает
0,991(15°)
0,991(15°)
0,8861(20°)
0,844(20°)
0,9114(0°)
0,894(0°)
0,8528(20°)
0,880(1 5°)
0,8851(20°)
1,216 (0°)
1,144 (0°)
1 ,291 (10°)
Н. обычно получают нитрозированием спиртов азо-
тистым ангидридом, азотистой к-той, хлористым нит-
розилом и др. Наряду с нитросоединениями Н. об-
разуются также при взаимодействии AgNO2 с га-
логеналкилами, диалкилсульфатами и т. д.:
R — ОН + НО- N = O —>-R-O- N = O
— Н2О
RX + AgNO2 —>-R — О- N = O
-AgX
H. легко гидролизуются; при хранении медленно раз-
лагаются с выделением окислов азота. Связь RO—NO
в Н. легко гомолптически разрывается. Ее прочность
ок. 37 ккал/моль, в то время как прочность связей
499
N-НИТРОАМИНОСОЕДИНЕНИЯ—НИТРОАНИЛИНЫ
500
R—ONO и R—NO2 (в нитросоединениях) ок. 57
ккал/моль. Нек-рые Н. (напр., амилнитрит) широко
используются для введения нитрозогруппы в раз-
личные органич. соединения (см. Н итрозирование). Н.
обладают физиология, действием, сходным с дейст-
вием на организм нитратов", они учащают пульс, рас-
ширяют артерии, вызывают быстрое падение кровя-
ного давления, и поэтому нек-рые из них (этил-,
амил-, циклогексил- и пинаколилнитриты) нашли
применение в медицине (напр., при лечении астмы
и грудной жабы). При более длительном воздействии
Н. возбуждают нервную систему, вызывают образо-
вание метгемоглобина. Наркотич. действие Н. почти
незаметно. В. Я. Фросин.
N-НИТРО АМИНОСОЕДИНЕНИЯ (нитрами-
ны) — продукты замещения атома водорода в ами-
нах нитрогруппой. Н. могут быть первичными
RNHNO2 и вторичными RR'NNOj. Первичные Н. су-
ществуют в двух формах: RNHNO2^RN=N(O)OH,
и в соответствии с этим способны давать N- и О-произ-
Название Формула T. пл., °C T. кип., °С/лш
Метилнитрамин .... CH3NHNO2 38
Этилнитрамин .... c2h5nhno2 3
Пропилнитрамин . . . О-Метилэтилизонитра- c3h7nhno2 — 128/40
мин C2H5N = N(O)OCH, 37/20
Диметилнитрамин . . (CHs)2P-NO2 58 187
Фенилнитрамин . . . C,HSNHNO, 46,5 —
N-Метилфенилнитрамин CBHSN (CH3) no2 39 —
О-Метилфенилнитрамин 2, 4, 6-Тринитрофенил- C,H5NN(O)OCH3 — 112
нитрамин (NO2)aCeH2N INO, 125-130 —
2-Нитраминопиридин C^XnHNOj ^N\NO2 184 —
Циклотриметилентрини-
трамин (гексоген) . . O3N-N^ ^N-NO3 CH2 204,1 —
водные. Первичные Н. алифатич. ряда — слабые од-
ноосновные кислоты, образующие анион RN—NO2.
Первичные Н. ароматич. ряда относительно сильные
кислоты, напр. фенилнитрамин по силе приближается
к уксусной к-те. Соли щелочных металлов этих к-т
реагируют с галогеналкилами, с образованием вторич-
ных нитраминов [диалкил- или алкиларилнитрамины
RN(Alk)NO2], а серебряные соли — с образованием
О-алкилпроизводных (RN=NOAlk). Восстановление I
первичных и вторичных нитроаминов, как
алифатич., так и гетероциклич., цинковой
пылью в уксусной к-те приводит соответ-
ственно к моно- или несимметричным
дизамещенным гидразинам. Арилнитра-
мины в этих условиях дают соли диазо-
ния. При восстановлении в щелочной сре-
де они образуют изодиазотаты (реакция
обратима):
н2
ArN = NO“-^ArN = NO-
I
О
Вторичные нитрамины алифатич. ряда
разлагаются кипящими щелочами с обра-
зованием первичных аминов и альдеги-
дов:
кон н2о
C2H6N (СН3) NOa —>-C2H6N = CH2 —»C2HjNH2 + HCHO
При действии конц. серной к-ты на Н. нитрогруп-
па, связанная с азотом, отщепляется; особенно легко
поддаются разложению первичные Н., вторичные Н.
более устойчивы и разложение их часто наступает
только при высокой темп-ре. При действии разбав-
ленной серной к-ты на первичные Н. образуется спирт
и N2O: RNHNO2-^ROH-|-N2O. Расщепление Н. жир-
ного и гетероциклического ряда конц. серной к-той
сопровождается бурным выделением закиси азота.
Первичные ароматич. Н., за немногим исключением,
являются твердыми продуктами. N-Нитрогруппа в ани-
линах дает гипсохромный эффект. Как и большинство
алифатич. Н., ароматич. Н. при нагревании взры-
вают, причем стабильность их уменьшается с увели-
чением числа нитрогрупп в ароматич. кольце. В кипя-
щей воде они медленно разлагаются, причем .м-нитро-
группы в полинитроарилнитраминах заменяются
группами ОН; очень стойки по отношению к окисли-
телям. Арилнитрамины под влиянием водных р-ров
минеральных к-т способны к перегруппировке. Так,
фенилнитрамин переходит в о-нитроанилин (наряду
с небольшим количеством п-нитроанилина):
СДЬ, - NH - NO3—» NH2 - С.Н. - NO2
Под действием азотистой к-ты первичные арилнитра-
мины превращаются в соответствующие соли диазония.
Моноалкилнитрамины получают: действием конц.
HNOs на ацилпроизводные первичных аминов, напр.
на моноалкилуретаны или диалкилоксамиды, с после-
дующим гидролизом промежуточных продуктов ам-
миаком; нитрованием алкилдихлораминов:
CH3NHCOOCH3 —>- CH3N (NO2) СООСН3 —>- CH3NHNO2
CtH,NCl2—> (C.HgNCINO,) —»C4HSNHNO2
Арилнитрамины получают: прямым нитрованием
соответствующих аминов; окислением в щелочной сре-
де диазосоединений различными окислителями (NaOCl,
KMnOi, Н2О2, KsFefCN), и др.); действием щелочей
на арилдиазонийпербромиды. Нитраминопиридины
получают прямым нитрованием аминопиридинов.
Большинство Н.— взрывчатые вещества, в связи
с чем многие из них находят применение для воен-
ных целей. Так, напр., сильным взрывчатым вещест-
вом является циклотриметилентринитрамин (гексо-
ген), а также 2,4,6-тринитрофенилметилнитрамин
(тетрил).
Лит.: Chemistry of carbon compounds, ed. E. H. Rodd,
v. 3 A, Amst.—[a. o.] ,1954, p. 253 (и сл.);В орожцовН. H..
Основы синтеза промежуточных продуктов и красителей,
4 изд., М., 1955, с. 143 (и ел.). Л. С. Поваров.
НИТРО АНИЛИНЫ — нитропроизводные анилина,
содержащие от одной до пяти нитрогрупп у С-атомов
ядра; кристаллы желтого цвета. Ниже приведены
важнейшие Н.
Нитроанилин Плотность 4 Темпе- ратура пл., °C Давле- ние па- ров, мм рт. ст. 100° Растворимость
е/100 г при 25°
вода этанол бензол
Мононитроанилин C6H4 (NO3) Ж, мол. в. 138,12 орто мета . . . • . . . пара Динитроанилин (2,4) C8H3(NO2). NH2,MOn. в. 183,12 Тринитроанилин (2,4,6) С»Н. (NO2)3 NH2,moh.b. 228,"12 1,442 (1 5°) 1,430 (15°) 1 ,424 (4°) 1,615 (14°) 1, 762 (14°) 71,0 114,0 146,7 178— 179 190- 191 0,732 0,159 0,032 0,1212 0.09 0,0568 не раств. не раств. 27,87 7,8 6,048 0,58 плохая 20.8 2.7 0,5794 хоро- шая
Н., содержащие более одной нитрогруппы, взрыв-
чаты: 2,4-динитроанилин и тринитроанилин взрыва-
ют при ударе. Химич, свойства Н. зависят от числа
и положения нитрогрупп. Введение нитрогрупп в
501
НИТРОАНТРАХИНОНЫ—НИТРОБЕНЗОЙНЫЕ КИСЛОТЫ
502
п- и особенно в о-положения к аминогруппе приводит
к уменьшению основности Н. и снижению их раство-
римости в кислотах. Диазотирование мононитроани-
линов проходит легко с помощью NaNO2 в разб.
к-тах; 2,4-динитроанилин и 2,4, 6-тринитроанилин
диазотируется только в конц. серной к-те. При нагре-
вании с водными щелочами Н., содержащие во- и п-
положениях нитрогруппы, обменивают аминогруппу
на гидроксил тем легче, чем больше нитрогрупп в
о- и «-положениях. Полное или частичное восстанов-
ление Н. приводит соответственно к ароматич. диа-
минам, нитродиаминам, триаминам и др. В пром-сти
с- и n-нитроанилин получают обычно из нитрохлор-
бензолов, обменом галогена на аминогруппу при нагре-
вании с водным р-ром аммиака.Так, из о- и ге-нитро-
хлорбензолов получают о- и ге-H., из 2, 4-динитрохлор-
бензола — 2, 4-динитроанилин и т. п. Пара-Н. полу-
чают также нитрованием форманилида CeH5NHCHO
с последующим отщеплением формильной группы при
нагревании в кислой среде. Произ-во .м-Н. основано
на восстановлении одной из нитрогрупп .и-динит-
робензола посредством NaHS. Применяются Н. преим.
в синтезе азокрасителей, а также для получения
фенилендиаминов.
Н. сильно ядовиты, превращают гемоглобин крови
в метгемоглобин; особенно опасно одновременное
воздействие Н. и спирта. Предельно допустимая кон-
центрация Н. в воздухе 0,005 мг/л. Количественное оп-
ределение моно-Н. основано на реакции диазотиро-
вания; для качественного обнаружения Н. получен-
ные диазосоединения вводят в реакцию азосочетания
с Р-нафтолом и другими азосоставляющими. Концент-
рацию 2, 4-динитроанилина определяют по количеству
аммиака, образующегося при кипячении вещества с
р-ром едкого натра.
Лит.: Фирц-ДавидГ. Э., В ланже Л., Основные
процессы синтеза красителей, пер. с нем., М., 1957; Ворон-
цов И. И., Полупродукты анилинокрасочной промышлен-
ности, М., 1955; Ullmann, 3 Aufl., Bd 12, Munchen — В.,
1960. M. А. Чекалин.
НИТРОАНТРАХИНОНЫ — продукты замещения
атомов водорода в антрахиноне нитрогруппой. Боль-
шинство Н.— кристаллич. продукты; темп-ра плав-
ления нек-рых Н. приведена ниже:
реакционноспособны, однако галоген в положе-
нии 1 более реакционноспособен, чем нитрогруппа.
Нагревание 1-нитроантрахинона с водным р-ром
нейтрального сульфита приводит к замещению нитро-
группы сульфогруппой, образуется антрахинон-1-
сульфокислота. Аналогично из 1,5- и 1,8-динитро-
антрахинонов получают 1,5- и 1,8-антрахинондисуль-
фокислоты.
Получают Н. как прямым нитрованием антрахинона
в серной к-те или олеуме, так и окислением надсерной
к-той аминоантрахинонов. Для прямого нитрования
можно брать' и антрацен, к-рый при этом окисляется
в антрахинон. При нитровании антрахинона при 50°
образуется лишь 1-мононитроантрахинон, к-рый не
имеет практич. значения. При 70—100° образуется
смесь, содержащая гл.обр. 1,5-, 1,8-динитроантрахи-
ноны и в небольших количествах 1,6-, 1,7-, 2,6-и
2,7-динитроантрахиноны. Многие Н. применяют в
произ-ве красителей. Отделенную от примесей смесь
1,5- и 1,8-динитроантрахинонов применяют для син-
теза антраценового синего и кубового желтого. Прак-
тически важен 1-нитро-2-метилантрахинон, используе-
мый в произ-ве кубовых красителей. Дигалогенодини-
троантрахиноны служат промежуточными продуктами
для приготовления широкого ряда полиамино-, окси-
и аминооксиантрахинонов, используемых в произ-ве
красителей.
Лит.: Ворожцов Н. Н., Основы синтеза промежуточ-
ных продуктов и красителей, 4 изд., М., 1955; Chemistry of
carbon compounds, ed. E. H. Rodd, v. 3, pt B, Amst.-L.-N. Y.,
1956, p. 1391. Л. С. Поваров.
НИТРОБЕНЗОЙНЫЕ КИСЛОТЫ — производные
бензойной к-ты. Физич. свойства наиболее известных
Н. к. приведены в таблице.
Н. к. более сильные к-ты, чем бензойная. Восста-
новление Н. к. приводит к аминобензойным к-там.
При повышенной темп-ре происходит декарбоксилиро-
вание Н. к. Технич. значение имеют о-,м- и ге-нитробен-
зойные к-ты. Л1-Н. к. получают нитрованием бензойной
к-ты нитратом калия в конц. H2SO« при нагрева-
нии, причем образуются 3 изомера (22,3% о-;
76,5% л-; 1,2% re-изомера) или, лучше, нитрованием
бензальдегида (темп-ра не выше 100°, процесс взрыво-
опасен) и последующим окислением гипохлоритом
Положение нитрогрупп 1а 2 1.3 1,5 1,6 1,7 1,8 2,6 2,7 10
Т. пл. °C 230 184,5—185 240 384—385 255-257 29 5 311—312 >330 280 (262) 269-270
а т. кип. 270°/7 мм. 6 В положении 2—метильная группа.
Нитрогруппа в Н. может быть
восстановлена в аминогруппу; она
легко замещается другими группа-
ми. В этом отношении поведение
нитрогруппы в Н. аналогично пове-
дению атомов галогенов в галоге-
нантрахинонах. Обычно нитрогруп-
па в положении 1 замещается легче,
чем в положении 2, при нагревании
с ариламинами или р-ром сульфита
натрия; при действии метилата нат-
рия наблюдается обратная картина.
Заместители при атоме углерода,
соседним с нитрогруппой, напр. в
1-нитро-2-метилантрахиноне, на-
столько сильно снижают реакцион-
ную способность нитрогруппы, что
реакция с р-ром сульфита натрия
протекает только под давлением.
В галогенонитроантрахинонах как
галоген, так и нитрогруппа очень
Кислота Т.пл., °C Плот- ность, d20 4 Констан- та диссо- циации (25°) Растворимость, г в 100 г Темп-ра декарбок- силирова- ния, °C
воды (15°) этано- ла (25°) хлоро- форма (20°)
Мононитробензойная CeH4(NO2)COOH, мол. в. 167,12 орто 146,8 1,575 6,2 -10-» 0,625 46,96 0,45 180
мета 141,1 1,494 3,55-10-* 0,238 68,02 (30,5°) 4,07 238
пара , 242,4 1,610 3,61-10-* 0,0213 2,306 0,101 242
3, 5-Динитробензой- ная CSH3 (NO2)2 СООН, мол. в. 212,12 ...... 206,5— 207,2 1,63-ю-3
2,4,6-Тринитробен- зойная СаН2(1\О3)3СООН,мол. вес 257,12 . . . . 228,7 2,05 26,6 100
(23°)
503
НИТРОБЕНЗОЛ
504
л-нитробензальдегида в л-Н. к.; о- и га- изомеры
получают окислением, соответственно, о- или п- нитро-
толуолов бихроматом или двуокисью марганца в сер-
ной к-те. Лабораторный метод получения .м-Н. к.
заключается в нитровании метилового эфира бензой-
ной к-ты и последующем гидролизе метилового эфира
л-нитробензойной к-ты.
Восстановлением .и-Н. к. получают л-аминобензой-
ную к-ту, применяемую при произ-ве азокрасителей.
о-Н.к. применяют для произ-ва бензидин-3,3-ди-
карбоновой к-ты, га-Н. к. служит для произ-ва п-ами-
нобензойной кислоты, га-нитробензоилхлорида (полу-
продукт для азо- и др. красителей, т. пл. 75—75,5°)
и др., а также эфиров и др. производных. л-Н.к.
применяют для осаждения и разделения Th, Се, Zr,
Hg, Hf; для микрокристаллоскопич. обнаружения
алкалоидов, га- и л-Н. к. — бактерициды, обладают
также бактериостатич. действием (задерживают рост
бактерий); нек-рые обладают свойствами инсекти-
цидов. Для количественного определения моно-Н. к.
проще всего восстановить их в кислой среде цинковой
пылью до аминобензойных к-т и титровать последние
нитритом натрия (диазотирование). Метод может
служить также для обнаружения Н. к.; в этом случае
полученные диазосоединения сочетают с различными
азосоставляющими, напр. 0-нафтолом. Н. к., содержа-
щие более одной нитрогруппы, взрывчаты.
Темп-ры плавления (°C) производных мононитро-
бензойных к-т:
Орто Мета Пара
Хлорангидрид . . . 20 35 75
Амид 175 143 200
ДнилиД 155 153 211
Нитрил 110 118 146
Метиловый эфир . . — 13 78,3 96
Этиловый эфир . . . 30 47 57
Лит.: Чичибабин А. Е., Основные начала органиче-
ской химии, т. 2, М., 195"; Ullmann, 3 Aufl., Bd 12,
Munch.— В., 1960. M. А. Чекалин.
НИТРОБЕНЗОЛ (мононитробензол, мирбановое
масло) CgHjNOa, мол. в. 123,11 — простейшее арома-
тич. нитросоединение, содержащее нитрогруппу у
С-атома ядра. Впервые получен Митчерлихом в 1834
нитрованием бензола. Н. — маслянистая жидкость,
по запаху напоминающая горькоминдальное масло;
раствор в воде обладает интенсивно сладким вкусом.
Чистый Н. бесцветен, технический светло-желтого
цвета: т. пл. 5,76°; т. кип. 210,80°/760 ы; 3° 1,22305;
1,20824; </’’ 1,19341; ralD5 1,55457; ra2D° 1,55257;
вязкость т|15 2,165 спуаз\ т|ю 1,634 сгауаз; теплота паро-
образования при темп-ре кипения 9,744 ккал/моль',
теплота плавления 2,78 ккал/моль', теплоемкость
С„ 42,37 (30°) кал/г-град', давление пара: 1 мм (44,4°),
10 мм (84,9°), 100 » (139,9°); 200 мм (161,2°),
400 мм (185,8°); 1крит. 459°; криоскопия, постоянная
6,852°; теплота сгорания при постоянном объеме
734,8 ккал/моль', поверхностное натяжение (дин/см):
43,35 (20°);42,17(30°); диэлектрич.проницаемость 34,82
(25°); т. всп. 83° (в закрытом сосуде); т. самовоспламе-
нения 482°; нижний предел взрываемости смесей
с воздухом 1,8%. Н. смешивается во всех отношениях
с бензолом, этанолом, диэтиловым эфиром и другими
органич. растворителями; в воде при 30° растворяется
0,206 вес. % Н.; в Н. растворяется 0,24 вес. % воды.
Н. хорошо растворяет многие неорганич. соли,
напр. А1С13, смолы и т. п.; перегоняется с водяным
паром; образует азеотропную смесь с водой (12 вес. %
Н., т. кип. 98,6°); сильно притягивает влагу.
Восстановление Н. металлами в присутствии элект-
ролитов, сульфидами, водородом в присутствии ка-
тализаторов и другими методами приводит к анилину,
а металлами в щелочной среде через ряд промежуточ-
ных стадий — к гидразобензолу, к-рый в кислой среде
перегруппировывается в бензидин. При электрохимия,
восстановлении образуется га-аминофенол. Реакции
электрофильного замещения протекают для Н. труд-
нее, чем для бензола, причем второй заместитель
(Cl, NO2, SO3H) вступает преим. в мета-положение
к нитрогруппе. Нуклеофильное замещение проходит
легче, чем в случае бензола, второй заместитель зани-
мает орто- или пара-положение, напр. сплавление
с безводным едким кали при 100° приводит к о-нитро-
фенолу.
В пром-сти Н. получают нитрованием бензола
смесью азотной и серной к-т при темп-ре 40—50’
непрерывным методом; периодич. способы сохрани-
лись только на небольших заводах. На 1 моль бензола
обычно берут 1,01 —1,02 моля азотной к-ты, а серпую
к-ту с таким расчетом, чтобы концентрация отрабо-
танной к-ты была 69—72%. Нитрование бензола про-
текает в гетерогенной среде с большой скоростью
и выделением тепла (28 ккал/моль). При недостаточном
отводе тепла и плохом перемешивании возможны
резкий подъем темп-ры и бурное протекание реакции,
к-рое может привести к взрыву. Поэтому аппараты
для нитрования (нитраторы) оборудуют быстроход-
ными мешалками; охлаждение производят через ру-
башку нитратора или при помощи змеевиков, или
других теплообменных устройств, устанавливаемых
внутри аппарата. Нитратор для непрерывного полу-
чения бензола — котел, внутри к-рого установлен
змеевик в виде вертикального направляющего ци-
линдра, без зазоров между витками змеевика: другой
змеевик размещают между цилиндром и стенками
котла. Эмульсия бензола в отработанной серной к-те
и нитрующая смесь непрерывно поступают в направ-
ляющий цилиндр сверху вниз, затем реакционная
масса перемещается в пространстве между цилиндром
и стенками нитратора снизу вверх; часть реакционной
массы перетекает из верхней части нитратора во 2-й
котел с мешалкой, где нитрование завершается. В по-
следующих аппаратах масса отстаивается, нитробен-
зол промывают водой и разб. щелочью; отработанную
к-ту частично возвращают в нитратор, другую часть
экстрагируют бензолом для извлечения небольших
количеств Н., освобождают от азотной к-ты, концен-
трируют и опять направляют на нитрование. Все
процессы проходят непрерывно. Выход Н. достигает
98%; он содержит до 1% бензола и до 0,1% динитро-
бензола и пригоден для ряда производств. При необ-
ходимости бензол отгоняют с водяным паром и дистил-
лируют Н. при пониженном давлении. Известны и
другие схемы непрерывного нитрования бензола:
устанавливают последовательно 3 нитратора или при-
меняют нитраторы, разделенные на секции, в к-рые
последовательно поступает реакционная масса. При
периодич. методе к бензолу при энергичном переме-
шивании и интенсивном охлаждении постепенно
приливают нитрующую смесь. Отделение и очистку Н.
производят так же, как и при непрерывном процессе.
В реакциях нитрования бензола и его замещенных
смесью азотной и серной к-т электрофильным реаген-
том является нитроний-катион NO*, образующийся
при взаимодействии азотной и серной к-т (подробнее
см. Нитроний-ион)'.
НХО.-2 H2SO, NO* +H3O* + 2 HSO,
Первой, медленной стадией реакции является присо-
единение нитроний-иона к бензолу, второй, быстрой
стадией — отщепление протона:
,н6 +
C.H. + NO. + HSO, —>СвН< 6'+—>C„H5NO2 + H2SO1
1 '.\02
505
НИТРОВАНИЕ ОРГАНИЧЕСКИХ СОЕДИНЕНИИ
506
Н. применяют в больших количествах для получе-
вия анилина, а также бензидина, .и-динитробен-
зола, л-нитрохлорбензола, л-нитробензолсульфокис-
лоты; в произ-ве красителей — индулинов, нигрози-
нов; в качестве растворителя, напр. при очистке неф-
тяных смазочных масел; как мягкий окислитель, напр.
при получении фуксина и хинолина; для отдушки
мыла и др.
Качественно Н. открывают восстановлением до
анилина и окислением последнего разб. р-ром извести
или гипохлорита натрия. Вследствие окисления обра-
зовавшегося анилина возникает интенсивная фиоле-
товая окраска. Для количественного определения Н.
восстанавливают в анилин и затем титруют стандарт-
ным р-ром бромата калия и бромида калия. Н.—силь-
ный яд, окисляет гемоглобин в метгемоглобин, пора-
жает центральную нервную систему, нарушает обмен
веществ, вызывая заболевания печени. Предельно
допустимая концентрация Н. в воздухе 0,003 мг/л.
Лит.: Вайсбергер А. [и др.], Органические раство-
рители, пер. с англ., М., 1958; Ворожцов Н. Н., Основы
синтеза промежуточных продуктов и красителей, 4 изд., М.,
1955; Общая химическая технология, под ред. С. М. Вольф-
ковича, т. 2, М., 1959; Фирц-Давид Г. Э., Б л а н ж е Л.,
Основные процессы синтеза красителей, пер. с нем., М., 1957;
Ullmann, 3 Aufl., Bd 12, Munch.— В., 1960, S. 771.
M. А. Чекалин.
НИТРОВАНИЕ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ.
Содержание:
Нитрование парафиновых соединений .........505
Нитрование ненасыщенных соединений ........50 7
Нитрование ароматических соединений .......508
Нитрование гетероциклических соединений . . . .513
О- и N-нитрование..........................513
О механизме нитрования и реакционноспособности . .513
Нитрование органических соединений — введение
нитрогруппы — NO- в молекулы органич. соединений
при действии азотной к-ты, двуокиси азота и других
нитрующих средств; в более узком смысле — замеще-
ние водорода СН-связи нитрогруппой или присоеди-
нение — NOa по кратным углеродным связям. Н. о. с.
может быть сопряжено с введением других групп:
—ОН (окислительное нитрование), F (нитрофториро-
вание) и т. п. Нитрование — одна из старейших и из-
любленных реакций химика-органика. Систематич.
применение реакции началось с открытия нитрования
бензола Митчерлихом в 1834; оно приобрело большое
научное и практич. значение (см. Нитросоединения
алифатические, Нитросоединения ароматические).
Нитрование может протекать по ионному и ра-
дикальному механизмам в зависимости от условий и
природы реагентов.
Нитрование парафиновых соединений. Нитрование
парафинов R—Н начинается с атаки на них радикало-
подобных агентов -A (-NOa, NOs’, -ОН, -Cl, О2)
и приводит к возникновению свободных алкилов:
R-H+A—^R- + H- А (1)
Образование нитросоединений R—NO2 происходит
по ур-нию (2) и сопровождается вследствие сопряже-
ния в NO2 и атаки по кислороду образованием алкил-
нитритов R—ONO по ур-нию (3):
Qj ✓RN°a (2)
R’ + К \
><RO-N=O (3)
Алкилнитриты RONO при Н. о. с. в жвдкой фазе HNO3
или N2O4 образуют равновесную смесь с ROH, RONO2,
R2O и затем более глубокие продукты окисления —
альдегиды, кетоны, карбоновые к-ты, их сложные
эфиры и др., а также продукты деструкции вплоть
до СО2. Введение кислорода усиливает процессы окис-
ления, в частности образование нитратов RONO2:
О2 -NO- no2
R--->R—0-0---э-R-О-О-NO2—^RONO2 + NO3- (4)
По мере насыщения окисью азота увеличивается
роль соединения R- с - NO, возникновения нитрозосое-
динений R—NO и их превращений, напр. в гем .-ди-
нитропроизводные при нитровании алкилбензолов:
NO
с„н,сн2-->-c6h5ch2-no
no2
C„H3CH = NOH—► С6Н5 —CH(NO2)2 (5)
В соответствии с механизмом реакции, в частности
с влиянием отношения концентраций NO к NO2, повы-
шение степени разбавления N2O4 углеводородом и
понижение темп-ры способствует увеличению выхода
динитропроизводного, нацр. толуол при 100° дает гл.
обр. фенилнитрометан C6H5CH2NO2, а при 20° —
фенилдинитрометан C6H5CH(NO2)2. При насыщении
кислородом, быстро реагирующим с NO, динитросоеди-
нение не образуется.
При нитровании в газовой фазе прп темп-ре ок. 400°
алкилнитриты в основном переходят в карбонильные
соединения и низшие нитропарафины, например при
Н. о. с. этана возникающий по ур-нию типа (3) этил-
нитрит превращается в формальдегид и нитрометан:
-NO
ch3-ch2-ono-----*chs-ch2-o—►
t°
-сн2о -no2
------> СН3-->-СН3 —NOa (6)
Обычно Н. о. с. парафиновой цепи производят HNO3
или N2O4, иногда в присутствии Ог- Активность N2O4
возрастает по мере разбавления ее углеводородом и
повышения темп-ры вследствие сдвига равновесия
N2Oi^2NO2 в сторону NO2; насыщение реакционной
массы кислородом может привести к возникновению
NO3 по схемам (4) и (7):
NO-
NO + O- ON —О-О-----7ON-O-O-NO2 -> NO2+ONO2 (7)
Азотная к-та не обладает нитрующим эффектом в от-
ношении парафиновой цепи и служит лишь источни-
ком возникновения необходимых для реакции окислов
азота -NO2, -ONO2 и средством прогрессивной регене-
рации NO2 из окиси азота:
t
2 HNO. + NO .4 N0. + H..0 (8)
При нитровании по М. И. Коновалову герметизация
реакторов обеспечивает сохранение окислов азота
в сфере реакции, а повышение темп-ры — диссоциа-
цию N2O4 и сдвиг равновесия (8) вправо. Естественно,
что нитровать можно с успехом самой N2O4. При
Н. о. с. безводной или очень крепкой HNOs, а также
при высокой темп-ре 70 %-ной к-той возникает очень
энергично инициирующий агент — трехокись азота
•ONO2:
2 HNO3^=± N2O3 + H2O; N-OsTZS: NO3-О- +-NO2 (9)
Поэтому сам азотный ангидрид очень энергично
нитрует парафины, даже в среде ССП при пониженной
темп-ре.
Активность инициирующих агентов -А пропорцио-
нальна их ненасыщенности и электрофильности:
•ОН> -NO3> -NO2. Реакционная способность атомов
водорода парафиновой цепи при Н. о. с. возрастает с
увеличением стабильности свободного радикала R • и
его электродонорного характера. В соответствии с этим
у парафинов активность третичного водорода R3C—Н,
как правило, выше, чем вторичного R2—СН2, еще более
инертны первичные R—СН3, реакционно-способность
метана — минимальна. Иногда более тонкие влияния
нарушают эти общие закономерности, а при повыше-
нии темп-ры активности всех атомов водорода вырав-
ниваются. Вступление арилов очень резко повышает
активность а-атомов водорода в, напр., дифенилметан
(С6Н5)2СН2 быстро реагирует с NO2 уже при обычной
507
НИТРОВАНИЕ ОРГАНИЧЕСКИХ СОЕДИНЕНИИ
508
темп-ре. При малой концентрации NOs происходит
частичная изомеризация радикалов типа неофила
Аг—С(СН3)2—СЩ-.Нитрозамещенные вследствие вы-
сокой электрофильности NOa-rpynnu, за редкими
исключениями, не вступают в реакцию нитрования;
ациформы, в том числе оксимы по ур-нию (5), их соли
и анионы реагируют с NO2 с большой скоростью благо-
даря их электродонорному характеру и координа-
ционной ненасыщенности а-атомов углерода:
С,Н3С (NOa)-K+ + 2 NO2 —»-CeH5C(NO2), + KNO2
Нитрование активной парафиновой цепи, напр.
в a-положение алкилбензолов или к третичному
атому углерода высших изопарафинов, с успехом про-
текает в открытых аппаратах при темп-рах ок. 100°
посредством N2O4 или HNO3 с выходом R—NO2 до 70—
75%. Нитрование «-парафинов, от додекана и выше,
можно производить в открытых аппаратах при 170—
200°; средние парафины и циклопарафины (С6—С10)
удобнее нитровать в запаянных трубках или автокла-
вах из специальной стали. Нитрование низших пара-
финов (Ci—С5) обычно производят в газовой фазе при
нагревании в проточных системах до 200—500°.
Промышленное Н. о. с. низших парафинов с выхо-
дом до 40% производится двуокисью азота или азот-
ной к-той в газовой фазе при 250—450°, нередко под
давлением; время пребывания в реакционной трубке
проточной системы порядка от нескольких секунд до
долей секунды. Для понижения побочных процессов
применяются покрытия стальных стенок реактора
стеклом «пирекс» или селитрой. Отмечено ускорение
реакций от добавления кислорода, брома, хлора и
покрытия стенок окислами мышьяка и сурьмы. Опи-
сана аппаратура и техника проточного Н. о. с. дву-
окисью азота парафинов (С,—С12) в жидкой фазе под
давлением до 50 ат при 200—250° и временем пребы-
вания в реакционной трубке из стали V 2А 15—70 сек.
Нитрование в газовой фазе при 250—500° дает продукт
с содержанием до 45 мол. % низших нитропарафинов,
напр. при нитровании этана при 460° получают наряду
с нитроэтаном (73%) ок. 27% нитрометана, образую-
щегося по ур-нию (6).
Во всех случаях должны неук-
лонно соблюдаться общие и спе-
циальные правила безопасности
работы. При высоком содержании N2O4, HNO3,
RONO2, RONO, R—СН=О реакция может принять
цепной и взрывоподобный характер с преобладанием
процессов глубокого окисления.
Нитрование ненасыщенных соединений. Олефины
и алкины нитруют по радикальному механизму обычно
двуокисью азота, часто в среде эфира, СС14, CH2CI2
и, напр., для пропилена, реакция протекает в основ-
ном по схеме:
•no2
сн8-сн=сн2—>CH3-CH-CH2NO2 —>
No'2/TCH3-CH(NO2)-CH2NO2 (10)
^СН3-СН(ОМО)-СН2КО2 (И)
Реакция начинается с разрыва л-связи при действии
+6
•NO2 и возникновения свободного 0-нитроалкила I,
наиболее стабильного из двух возможных; затем
присоединяется вторая молекула NO2 с образовани-
ем динитросоединения (10) или нитронитрита (11).
Нитронитрит при избытке NO2, лучше при насы-
щении О2, легко превращается в 0-нитронитрат
CH3CH(ONO2)CH2NO2. В присутствии NO I образует
нитронитрозосоединение СНз—CH(NO) — CH2NO2,
к-рое быстро переходит в димер — т. н. псевдонитро-
зит. Эта и другие побочные реакции в специальных
условиях могут стать даже главными (образование
нитроолефинов, теломеров, реакции свободных 0-нит-
роалкилов с О2, J2, NO2CI, СНВгз, C,HSNO2 и т. и.).
Присоединение NO2 к нитроолефинам осуществить
значительно труднее. Нитрование олефи-
нов и алкинов N2O4 или N2O5 и обра-
ботка продуктов реакции требует
максимальных мер предосторож-
ности.
Нитрование ненасыщенных соединений безводной
HNO3, особенно в присутствии подходящих сильных
к-т, протекает по ионно-комплексному механизму,
по правилу Марковникова, напр. для хлористого
винилидена в р-ре фтористого водорода:
HONO2 + 2HF NO2++H3O + + 2F- (12)
+ +6 F“
O2N + + CH2 = CC12—>O2N«-—СН2 = СС12—>O2N-CII2-CC12F(13)
Азотная к-та ведет себя в HF как основание, превра-
щаясь в нитроний-катион O=N=O. Последний за
счет внедрения л-электронов галогенолефина в сферу
атома N дает комплекс, присоединяющий фторид-
анион с образованием 0-нитрофторида. Нитрофтори-
рование в нек-рых случаях идет очень гладко. Но
обычно присоединение по типу схемы (13) сопровож-
дается реакциями замещения водородов и образова-
нием а- и 0-нитроолефинов, напр. при атаке аниона А~
(F , NOS и т. п.) на комплекс NO* с пропиленом:
. д. O3N-CH=CH-CH3 а (14)
О2и»--сн=сн-сн2-н
н 'д'* o2n-ch2-ch=ch2 S <15>
Кроме того, могут побочно получаться теломеры и по-
лимеры.
Азотный ангидрид в умеренно полярной среде
(CH2CI2 и СНС1з), по-видимому, присоединяется как
в форме относительно свободных ионов NO* и NO~
по типу ур-ния (13), так и в виде ионной пары
+
O2N-<---NO3 . В последнем случае реакция проис-
ходит в ^ис-положении через образование циклич.
комплекса; до нек-рой степени аналогично реагирует
ацетилнитрат O2N—ОСО—СНз в форме сольвата
O2N^ О=С(ОН)—СН3. Образование Р-нитронит-
5ата NO2—CHR—CR2—ONO2 в первом случае и
-нитроацетата во втором сопровождается возникно-
вением а- и Р-нитроолефинов с преобладанием послед-
них. Нитрование по ионному типу нек-рых производ-
ных олефинов с сопряженной С=С-связью, напр.
(СНз)2С=СН—СООН, дает иногда исключительно
а-нитрозамещенные, как и для АгН (см. далее).
Нитрование ароматических соединений. Ароматич.
соединения АгН отличаются от олефинов большей
устойчивостью их системы л-связей. Поэтому у аддук-
тов нитрующих агентов (NO2+, -NO2 и т. п.) к л-связи
молекулы АгН наблюдается во всех стадиях реакции
тенденция к восстановлению ароматич. ядра, осо-
бенно резко выраженная при нитровании по ионному
механизму. Как правило, лишь в случаях нитрования
по радикальному механизму можно было химически
доказать возникновение аддуктов по двойной связи,
быстро переходящих в продукты замещения водо-
рода.
Обычно нитрование АгН происходит по ионному
механизму. Можно различать два важнейших типа
этих реакций АгН, как, впрочем, и для других
ненасыщенных соединений. Нормальное нитрование
509
НИТРОВАНИЕ ОРГАНИЧЕСКИХ СОЕДИНЕНИИ
510
осуществляется энергично нитрующими смесями или
азотной к-той без участия двуокиси азота; нитрующим
агентом служит NO*, возникающий, напр., в смеси
серной и азотной к-т:
HONOa + 2 H2SO4 NO2+ +2 SO4H + H3O+ (16)
Роль аналогичную серной к-те играют другие актива-
торы нитрования (НСЮ4, HF, BF3, SO3, А1С1з, TiCh
и т. и.), a HNO3 можно заменить ее эфирами, соля-
ми, NaOj, а также N2O4. Каталитич. нитрование про-
изводят водной HNO3 через промежуточное взаимодей-
ствие с различными формами двуокиси азота:
t HNO3 ,
O2N'NO2;z± 2NO2JZ±ON-ONO, ^z_rNO + +NO3 (17)
К нему примыкает нитрование самими окислами азота
без активаторов и при низкой темп-ре. Нитрование,
напр. NO*, начинается с образования комплекса II
за счет внедрения л-электронов ядра АгН в сферу вы-
сокоэлектрофильного координационно ненасыщенного
атома азота:
+<5, ,-N°2+
I АгН + NO2+ —- Аг''
^Н+*2
---- Ar-NOZ + Н* (18)
(30-<J,+ <!г>
II
+$
Этот процесс приводит к поляризации связи Аг-н
и он заканчивается отщеплением протона и возникно-
вением Аг—NO2. Аналогично протекает реакция с нит-
розил-катионом N=O с образованием нитрозосоедине-
ния Аг—NO, к-рое под действием NO2 может перейти
в ArNO2
Аг-NO + NO2—»- Ar-NO2 + NO (19)
или, в зависимости от условий, претерпеть ряд иных
превращений (см. далее). Реакция АгН с другими фор-
мами двуокиси азота в общем случае дает смесь
ArNO2 и ArNO, напр. согласно упрощенной схеме
взаимодействия с NO—CNO2, в к-рой пунктиром пока-
заны связи в процессе их возникновения или расщеп-
ления, а стрелками — обязательные смещения элект-
ронных дуплетов:
ZNO-O^ Аг-NOa + HNO2 (20)
Аг-Н + NO-ONO2—- Аг 'No
Н-»-О ^Ar-NO + HNO3 (21)
Образующиеся по ур-нию (19) и (20) NO и HNO2
по реакциям с HNOs превращаются в двуокись азота
и т. обр. она регенерируется, что и оправдывает
отнесение этих процессов к каталитич. Н. о. с.
К ионному нитрованию приложимы общие теории
электрофильного замещения в АгН и правила ориен-
тации. Как это следует из схемы (18), реакционно-
способность и ориентация при нитровании АгН
в первую очередь зависят от способности АгН к пере-
даче электронов через тот или иной атом С ядра.
Активность нитрующих и нитрозирующих агентов
обусловлена их электрофильностью и наличием коор-
динационно ненасыщенного атома азота, напр.:
+ +6
NO2 >NO+>NO-ONO3 (22)
Так как максимальное координационное число атома
азота в его кислородных соединениях равно 3, то мо-
лекулы HONO2 пли ее эфиров, имея координационно
насыщенные атомы азота, как правило, не нитруют
АгН. Вследствие невысокой электрофильности всех
форм NO2 и N2O4 в каталитич. нитрование легко всту-
пают лишь такие электродонорные АгН, как наф-
талин, фенолы, амины с относительным потенциалом
электрона >0,1, если потенциал бензола принять
равным 0 (см. далее).
Нормальное нитрование является более универсаль-
ной реакцией, приложимой даже к нитро- и динитро-
бензолу, к-рые совсем не реагируют с двуокисью
азота; более трех NC>2-rpynn в одно ядро ввести нельзя.
Скорость нормального нитрования азотной к-той резко
уменьшается с ее разбавлением, вследствие сильного
понижения содержания NO*, а затем и быстроты его
образования из молекул HNOs. После понижения
концентрации кислоты до нек-рой величины, харак-
терной для каждого АгН, нитрование может практи-
чески протекать только через промежуточную реакцию
с двуокисью азота, т. е. каталитически. В конц.
смесях серной и азотной к-т значительная или даже
большая часть HONO2 находится в форме NO* в со-
ответствии с константой равновесия ур-ния (16),
равной ~30—40.
HNO3 в смеси, % . . 5-10 15 30 40 60 80 100
Превращение HNO3 в NO2+,% .... 100 80 63 29 17 10 2
При содержании 5—8% Н2О в самой азотной к-те
NO2 нельзя определить спектроскопически, но его
присутствие ясно обнаруживается химически даже
при дальнейшем разбавлении. Концентрацию NO2
понижают также анионы (NO~ и т. п.) и основные
растворители (эфир и т. п.).
Нитрование, в противоположность сульфированию
АгН, является необратимой реакцией и с термодина-
мич. точки зрения его можно было бы осуществить
даже очень разб. HNOs; каталитич. Н. может служить
примером преодоления кинетич. затруднений в реа-
лизации термодинамич. выводов. Дальнейшим расши-
рением каталитич. Н. было его проведение в присут-
ствии солей ртути с менее активными АгН —бензолом,
С6Н5СООН и т. п. Оно протекает через превращение
АгН в ртутьароматич. соединение ArHgNOa и переход
последнего в нитрозосоединение по реакции с NO +
или N2O4:
+ “Н+ NO+ +
CeHe + HgNO3--> CeH5HgNO3---> CeH5-NO + HgNOa (23)
В условиях нитрования ArNO может перейти в ArNO2
по ур-нию (19) или в ряд других соединений, напр.
бензол по этому консекутивно-каталитич. методу мож-
но на 80% превратить в 2,4-динитрофенол (окис-
лительное нитрование).
и in
IV V VI
Возникающий нитрозобензол в среде 40—60%-ной
HNOs, присоединяя Н + , переходит в фенил-нитрозо-
511
НИТРОВАНИЕ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
512
ний-катион I, в к-ром атомы углерода ядра в пара-
и отчасти в орто-положениях приобретают, благодаря
сопряжению, заряд -|-6, что позволяет катиону атако-
вать неподеленную пару электронов НоО. В резуль-
тате этого с отщеплением атома водорода воды в виде
Н+ возникает хинол оксим И. Прототропная изомери-
зация II приводит к образованию арилгидроксила-
мина III, последовательное окисление к-рого NO2
дает нитрозофенол IV, нитрофенол V, а нитрование —
2,4-динитрофенол VI. Возможно также превращение
ArNO по реакции с NO в диазосоединения и т. д.
Реакционноспособность АгН может сильно изме-
няться в зависимости от соотношения между его фор-
мами, напр. ArSOsH при нормальном нитровании
может реагировать как более активный анион ArSO3 ,
фенолы при каталитич. нитровании— в форме АгО-,
бензол при консекутивно-каталитич. нитровании —
в виде C6H5HgNOs и т. п. Превращение АгН в катион
АгН* и т. п. электрофильные комплексы при увели-
чении кислотности среды очень сильно понижает
скорость нитрования после достижения наиболее
благоприятной кислотности ок. 90% H2SO4 (т. н. оп-
тимум Мартинсена). Ориентация также может зависеть
от формы АгН, природы нитрующего или нитрозирую-
щего агента, степени его сольватации, стерич. факто-
ров и т. д. Так, нитрование свободного ArNH2 дает
орто- и пара-нитропроизводные, а в форме ArNH* —
лета-изомер. Более электрофильный NO* реагирует
менее избирательно, чем NO + ; повышение темп-ры
также выравнивает реакционноспособность во всех
положениях ядра; в том же направлении должно дей-
ствовать уменьшение сольватации NO*. Объемистые
заместители затрудняют нитрование в орто-поло-
жение и могут дезактивировать в этом положении
влияние других групп, например—N(CHs)2, вслед-
ствие нарушения их копланарности и сопряжения с
ядром.
При нитровании АгН выход ArNO2 обычно достигает
95—98%. Побочное образование нитрофенолов при
нитровании АгН объясняется частичной реакцией
+ +д' через кислород и нитрованием возникших
O=NjO фенолов Аг—ОН:
. _[O=N = O]+ Н +
Аг —H + NO- —> Аг(-------► Аг —ONO----*АгОН(25)
2 'и -Н+ -NO+
Возможностей для побочных реакций при каталитич.
нитровании больше. Для устранения нежелательных
процессов при нитровании ArNHo их часто переводят
в анилиды ArNH—СО—В и после нитрования послед-
них и омыления нитроанилидов получают нитроанили-
ны. В тех же целях фенолы предварительно сульфи-
руют, причем в процессе нитрования SOsH-rpynna
замещается —NO2; эфиры фенолов иногда нитруют
при —30°, —50°.
Особым видом понно-комплексной реакции является
внутримолекулярное нитрование, напр. превращение
фенилнитрамина СвН5—NH—NO2 при действии Н +
в смесь орто- и пара-нитроанилинов. Оно характери-
зуется почти исключительным образованием орто-
нитропроизводного. Ход этого процесса схематично
можно представить так:
Н+ . Е . NH2 -Н +
CeHs-NH-no2—s-C6H3-NH2-NoT —>C„hZ ------------->
I
nNO2+
nh2
Нитрамин, присоединяя H + , переходит в катион I.
При небольшом значении энергии активации Е
начинается перемещение NO* в поле л-электронов
молекулы, напр. в форме комплекса II. Естественно,
что при этом NO* имеет большую возможность уста-
новить ковалентную связь с ближайшим орто-атомом
углерода, и комплекс с отщеплением Н* переходит
на 95% в opmo-нитроанилин. В ряде методов получе-
ния opmo-нитропроизводных эта перегруппировка
имеет решающее значение (нитрование в уксусном
ангидриде, СНзСООН, нитрование бензоилнитратом
и т. п.).
Бензол можно пронитровать NOo по радикальному
механизму при нагревании с N2O4 или с 10—20%-ной
HNOs, под давлением. Первой стадией нитрования
является медленное присоединение NO2 к л-связи
ядра; возникший радикал I подвергается весьма раз-
нообразным быстрым превращениям, напр. присоеди-
няет последовательно еще 5 молекул NO2, а затем ад-
дукт И, отщепляя 3 молекулы HNO2, дает симм-
тринитробензол;
В этой реакции образуется также нитробензол, мета-
и napa-динитробензолы и нитрофенолы. Нитробензол
возникает, напр., в результате дегидрирования ради-
кала I (см. ур. 27):
/Н
•С6Н< +'NO2—oCJIsNOj + HNOj (28)
XNO,
Более энергичными инициирующими агентами ради-
кального нитрования являются NOs- и -ОН, к-рые
вовлекают в реакцию даже нитробензол. В качестве
источника NOs- применяют раствор ХгО5 в ССП
при нагревании (см. ур-ние 9). В этих случаях наблю-
дается относительно высокий выход нитрофенолов
(ок. 40%) и образование аномальных нитросоединений,
напр. лета-нитрохлорбензола при нитровании хлор-
бензола.
В научных и промышленных целях уже более 100 лет
широко применяется нитрование АгН по ионному
типу, чаще всего смесью серной и азотной к-т. Обычно
нитрующую смесь или HNOs постепенно прибавляют
к АгН при непрерывном энергичном размешивании и
строгом соблюдении температурного режима. В техни-
ке для нитрования применяют чугунные нитраторы
с пропеллерной быстроходной мешалкой, рубашкой
и змеевиком для охлаждения и нагревания. Описано
много аппаратов непрерывного нитрования АгН, из
к-рых нашли применение нитраторы с прямым током
реагентов. После отделения из смеси кислот часто
регенерируют H2SO4, а иногда и HNOs. Нитрование
водной HNOs требует обычно применения большого
избытка кислоты и аппаратов из эмалированной или
специальной стали. Предложено нитрование HNOs
с. отгонкой воды или с добавлением конц. HNOs для
поддержания ее крепости.
Нитрование и последующие об-
работки требуют тщательного со-
блюдения всех указаний по без-
опасности работы, особенно в слу-
чае образования полинитросоеди-
нений. Следует устранить возможности возникно-
НИТРОГЛИЦЕРИН
51.3
веиия легко взрывающихся нитрофенолятов, перегре-
ва остатков при перегонке, попадания ртути в ре-
акционную смесь и т. п. Принимают все меры про-
тив отравления парами и возможными поражения-
ми кожи, напр. динитробензолом, динитрохлорбен-
золом.
Нитрование гетероциклических соединений. Ус-
ловия и результаты нитрования гетероциклич. соеди-
нений сильно зависят от их степени ароматичности,
устойчивости ониевых катионов, чувствительности
к кислотам, возможности перевода в анионы. Так,
фуран и его производные, занимающие промежуточное
положение между ароматич. и ненасыщенными соеди-
нениями, при нитровании азотной к-той в среде уксус-
ного ангидрида часто дают (как и 1,3-диолефины)
продукты присоединения NO*— ООС—СН3 —2,5-нит-
роацетаты, к-рые при действии пиридина переходят
в 2-нитрозамещенные. Пиррол и его аналоги, вслед-
ствие их крайней чувствительности к кислотам, нит-
руют RONOa в виде аниона CiHiN-. Условия нитро-
вания имидазола и особенно пиразола приближаются
к условиям нитрования бензола. Тиофеновые соедине-
ния сравнительно гладко нитруются в среде уксус-
ного ангидрида. Пиридин, дающий в смеси серной и
азотной к-т устойчивый катион C5H5NH + , нитруется
только при темп-ре ок. 300°. Нитрование хинолина
происходит в 5- и 8-положения. Вступление электро-
фильных заместителей в ядра фурана и пиррола, а
NH2- и ОН-групп в ядро пиридина приближает усло-
вия Н. этих производных к Н. бензола.
О- и N-нитрование. Н. к атому азота аминов,
амидов, гуанидина и т. п., а также к атому кисло-
рода спиртов и углеводов весьма сходно по своей
природе с Н. ароматич. углеводородов; роль ароматич.
секстета играют неноделенные пары электронов N и О:
+ н+ .. NOz + ,no2 -н + .. н26 ..
ВОН2~ПОН R-О( BONO, ROH + HNO,
, хн
(29)
Однако Н. к азоту, в отличие от Н. к углероду, приоб-
ретает в сильно кислой среде равновесный характер,
а для этерификации спиртовой группы (нитрование
ROH) обратимость общеизвестна. Это изменение
степени обратимости зависит от уменьшения свободной
энергии нитрования в связи с увеличением сродства
к электрону в ряду С, N, О. При накоплении высоко-
электрофильных заместителей у атома углерода, в
особенности карбонилов и нптрогрупп, способность к
отщеплению—NO2 в виде NO^ появляется и у связи
С—NO2, напр. в тетранитрометане (NC^sC—NO2,
CeH5C(NO2)3, нитроиндандионе С6н4/ \сн—NO2 и т.п.
О механизме нитрования и реакциониоспособности. Пара-
фины, вследствие отсутствия у них пространственно доступных
я-электронов, не способны к комплексообразованию с NO2 ,
NO + , N2O4, C(NO2)4,N2Oe и т. п. реагентами и поэтому они
не вступают в Н. по ионному типу, напр. смесью серной и азот-
ной к-т. Парафиновую цепь можно нитровать только по ради-
кальному механизму, начальной стадией к-рого является от-
щепление периферии, водорода и возникновение свободного
радикала R ; скорость нитрования в основном пропорциональ-
на степени стабилизации R.сопряжением.
Олефины благодаря пространственной доступности я-элек-
тронов, сравнительно невысокому перекрыванию их орбит,
большому индексу свободной валентности F—0,7 весьма легко
присоединяют .NOa с разрывом я-связи. Нанр., в случае сти-
рола С„Н5—СН=СН2, когда возникающий свободный р-нит-
роалкил СвН5—СН—СН2—NO2 дополнительно стабилизован
сопряжением, Н. крайне быстро протекает даже при охлаж-
дении. Полагают, что бутадиен присоединяет 2 молекулы NO2
при —20° к атомам углерода в положении 1,4, т. е. с бблыпим
индексом F (0,84). При действии нитрующих агентов типа
NO* или NO2—ONOj ненасыщенные соединения, вследствие
пространственной доступности я-электронов и высокой поля-
ризуемости я-связи, нитруются по ионно-комплексному меха-
514
низму, обычно по типу присоединения с легкостью, пропорци-
ональной степени их электродонорного характера.
Соединения ряда циклопропана способны к нитрованию по
типу нитрования парафиновой цепи, по крайней мере при
400°. Так как характер перекрывания орбит С—С-связей ядра
циклопропановых соединений приближается к конфигурации
обычной л-связи, то можно ожидать от них вступления в нит-
рование по типу реакций с олефинами, т. е. с разрывом трех-
членного кольца, что подтверждается рядом наблюдений.
Углеводороды ряда циклопентана и циклогексана нитруются
подобно парафинам, причем вторые отличаются повышенной
реакционной способностью [благодаря их т. н. криптоароматич.
(скрытоароматич.) состоянию]. При Н. циклогексена по ион-
ному типу HNO3 легко возникают изомерные продукты при-
соединения и замещения.
л-Электроны ароматич. соединений типа бензола благодаря
двухстороннему перекрыванию их орбит, меньшему значению
Т?=0,40 характеризуются сравнительной трудностью их раз-
межевания, и вследствие этого они реагируют с *NO2 практи-
чески только при нагревании; быстро вступает в эту реакцию
нафталин в а-положенпи (F=0.45) и особенно легко антрацен
в .мезо-положениях благодаря пониженному перекрыванию
соответствующих л-электронов и высокому значению F (0,52).
Для типичных ароматич. соединений характерны легкость и
гладкость нитрования по ионному механизму с замещением
атомов водорода ядра. В переходных состояниях этой реакции
большую роль играет высокая поляризуемость системы л-элект-
ронов АгН с приближением структур реакционных комплексов
к хиноидным формам без полного нарушения ароматич. со-
пряжения. На основе учета хиноидного характера переходных
комплексов АгН с NO2 и сопоставления сущности этого типа
нитрования и окисления ароматич. диоксипроизводных в хи-
ноны возможно определение относительных потенциалов элект-
рона АгН. Если потенциал бензола принять равным нулю, то
потенциал нафталина в a-положении равен 0,231, в 0 = 0,139,
антрацена в .мезо-положении 0,561, в а=0,314, в 0 = 0,225 и т.д.
Эти значения потенциала пропорциональны величине log /,
где / — относительная скорость нитрования при / для бензола,
принимаемой за 1. АгН с потенциалом >0,1, в частности фе-
нолы и амины легко нитруются водной HNO3, через промежу-
точную реакцию с различными формами NO2 (см. ур-ние 17).
Напротив, нитробензол с потенциалом 0,16 реагирует толь-
ко с высокоэлектрофильным NOg .
Трополон и его производные в их отношении к нитрованию
сходны с АгОН. Очень легко нитруются азулены и циклопен-
тадиен в форме аниона С5Н5 , даже посредством зтилнитрата
C2H5ONO2; очень малая реакционная способность этого нит-
рующего агента перекрывается электродонорной активностью
карбаниона, т. е. высоким значением потенциала. Ферроцен
(С5Н5)2Ге медленно дает в сходных условиях продукт деструк-
тивного нитрования — нитроциклопентадиен С5Н5—NO2, а
обычными нитрующими и нитрозирующими средствами он
окисляется в феррициний-катион (С5Нб)2Ге+ и разлагается;
нитроферроцен все же может быть с низким выходом получен
при действии C8H7ONO2 на литий ферроцен (C5H4Li)Fe(C5Hsr
Лит.: Топчиев А. В., Нитрование углеводородов и
других органических соединений, 2 изд., М., 1956; В о р о ж-
ц о в Н. Н., Основы синтеза промежуточных продуктов и кра-
сителей, 4 изд., М.—Л., 1955; Орлова Е. Ю., Химия и тех-
нология бризантных взрывчатых веществ, М., 1960; А з и н-
гер Ф., Химия и технология парафиновых углеводородов,
пер. с нем., М., 1959; X у б е н И., Методы органической хи-
мии, пер. с нем., т. 4, М.—Л., 1949; Титова. И., Усп. химии,
1952, 21, вып. 8; ДАН СССР, 1951, 81, Кг 6, 1085; Сборник ста-
тей по общей химии, т. 1, М.—Л., 1953, 242; Т и т с в А. И.,
Усп. химии, 1958, 27, вып. 7, 845; ДАН СССР, 1953, 91, Кг 5,
1099; Ж. общ. химии, 1952, 22, вып. 8, 1329; ДАН СССР,
1957, 114, Кг 4, 777; ТитовА. И., Л а п т е в Н. Г., Успехи
и проблемы исследования окислительного нитрования арома-
тических соединений, в кн.: Органические полупродукты и
красители. Сб. статей, вып. 1, М., 1959, с. 5; Т и т о в А. И.,
Вопросы реакционной способности и ориентации в теории
нитрования ароматических соединений по ионно-комплексному
типу, там же, вып. 2, М., 1961; Реакции и методы исследования
органических соединений, кн. 7, М., 1958; Titov А. I.,
Tetrahedron, 1963, 19, Кг 4, 557; ДАН СССР, 1963, 149, Кг 2;
De la Mare Р. Б. D., R idd J. Н., Aromatic substitu-
tion. Nitration and Halogenation, L., 1959. А. И. Титов.
НИТРОГЛИЦЕРИН (глицеринтринитрат), мол. вес
227,1, CHaONOsCHONOaCHaONOa —полный эфир гли-
церина и азотной кислоты; маслянистая бесцветная
(технич. Н.— обычно желтая) жидкость без запаха
(выше 50° — со слабым сладковатым запахом), слад-
кого жгучего вкуса. Н. кристаллизуется в двух мо-
дификациях: стабильной (бипирамидально-ромбиче-
ской), т. пл. 13,5°, и лабильной (триклинной), т. пл.
2,8°; djj 1,596; 1,4732; вязкость 36 спуаз (20°);
давление паров (мм рт. ст.) : 0,0002 (20е), 0,003
(40°); 0,019 (60°); —2,0 (125°); чрезвычайно склонен
017 К.Х.Э. т. 3
515
НИТРОГЛИЦЕРИН— N-НИТРОЗОАМИНЫ
51©
к переохлаждению и стеклованию. Плотность кристал-
лич. Н. при 10° 1,735. Н. смешивается во всех отно-
шениях с ацетоном, эфиром, метиловым спиртом,
амилацетатом, ледяной уксусной к-той, бензолом,
толуолом, ксилолом, нитробензолом, метилнитратом,
нитрогликолем и др. нитроэфирами', ограниченно раст-
ворим в этиловом, пропиловом и амиловом спиртах;
трудно растворим в сероуглероде, глицерине, бен-
зине, лигроине, керосине. В воде Н. растворяется:
0,14% (25°); 0,24% (50°). Н. хорошо желатинирует
нитроцеллюлозу.
Н.— одно из самых мощных взрывчатых веществ',
теплота взрыва 1500 ккал/кг (в 1,5 раза больше, чем,
напр., тротила); скорость детонации 7700 м/сек (спо-
собен в нек-рых условиях детонировать с т. н. «малой»
скоростью 1500—2000 м/сек)', объем газообразных
продуктов взрыва 713 л/кг; темп-ра вспышки ок. 200°.
Н. способен взрываться при очень слабом ударе и яв-
ляется одним из самых опасных в обращении взрывча-
тых веществ.
Конц, серная к-та растворяет и разлагает Н. с обра-
зованием глицеринсерной и азотной к-т; спиртовые
р-ры едких щелочей и растворы сульфида натрия
энергично разлагают Н. Скорость гидролиза Н. водой
невелика; в присутствии щелочей и кислот она очень
сильно возрастает. При нагревании Н. происходит
его разложение, сопровождаемое окислением и гидро-
лизом. При умеренно повышенных темп-рах (~ 150°)
изотермич. разложение резко самоускоряется в ре-
зультате накопления в жидкости продуктов распада —
воды, кислот и окислов азота. При известных усло-
виях это ускорение может привести к саморазогреву
и взрыву. Время полного разложения Н. в присутствии
продуктов распада составляет: 10 час. (100°), 2—3 дня
(80°), 24—25 дней (60°). При добавлении к чистому
Н. кислот или воды это время существенно умень-
шается. Большая скорость разложения и резкое его
самоускорение делают Н. самым нестойким из обычных
вторичных взрывчатых веществ.
Получают Н. по реакции:
CH2OH-CHOH-CH2OH + 3HNO3±=J
CH2ONO2CHONO2CH2ONO2 + 3H2O
В качестве этерифицирующего агента применяют т. н.
нитрующую смесь — безводную или почти безводную
смесь серной и азотной к-т с содержанием 50—60%
последней. Этерификацию (нитрование) ироводят в ап-
паратах — нитраторах периодического или непре-
рывного действия, добавляя глицерин к тщательно
перемешиваемой и охлаждаемой кислотной смеси.
Производство Н. чрезвычайно взрывоопасно, его
транспортировка на сколько-нибудь значительные
расстояния запрещена.
Н.— первое вторичное взрывчатое вещество, при-
мененное для взрывных работ. Массовое произ-во и ши-
рокое применение Н., начавшееся с 60-х гг. 19 в.,
связано с именем шведского инженера и предприни-
мателя А. Нобеля. Почти весь Н. (произ-во его в круп-
нейших странах измеряется десятками тысяч тонн
в год) идет на изготовление динамитов и других
взрывчатых смесей и баллиститных порохов. Неболь-
шие количества Н. в виде очень разб. р-ров приме-
няются в медицине (в малых дозах он является хоро-
шим сосудорасширяющим средством). В больших
дозах Н. ядовит, может всасываться в кровь через
кожу, вызывает сильную головную боль.
При действии анилина и серной к-ты на Н. появляет-
ся пурпурно-красное окрашивание, переходящее в зе-
леное при добавлении воды. Для колич. определения
Н. применяют нитрометр Лунге, измеряя объем окиси
азота, выделяющейся в нем при разложении Н. сер-
ной к-той в присутствии ртути.
Лит.: Наум Ф., Нитроглицерин и нитроглицериновые
взрывчатые вещества, пер. с нем., М.—Л., 1943; S t е 11 Ь а-
cher A., Exploslv stoffe, 1959, 7, Ks 4, 9,10; Kirk,v. 6,
N. Y., 1951. Б. H. Контриков.
НИТРОЗИРОВАНИЕ — реакция замещения атома
водорода в органич. соединениях нитрозогруппой
с образованием нитрозосоединений или оксимов:
R2CHNO2—*R2C —NO.; NOH
I II
NO RCOCH.COOR—>RCOCCOOR
Впервые H. наблюдал В. Мейер в 1873 при
подкислении щелочных р-ров нитропарафинов и солей
азотистой к-ты. Н. алифатич. соединений требует, за
немногими исключениями, присутствия по соседству
с нитрозируемой группой электрофильных замести-
телей (ацильной, карбонильной, нитро-, карбоксиль-
ной, карбалкоксильной, циано-, имино- и арильной
групп). Среди ароматич. соединений Н. подвергаются
соединения, имеющие в ядре группы ОН, NR2, NHAr
и иногда NH2-rpynny. При Н. нитрозогруппа стано-
вится вой «-положения по отношению к имеющемуся
заместителю. Н. подвергаются также вторичные-
амины как жирные, так и ароматические (см. Нитро-
зоамины),
Нитрозирующие агенты: азотистая к-та, хлористый
нитрозил, нитрозилсерная к-та, окислы азота и эфиры
азотистой к-ты. В случае двух последних реагентов,
обычно добавляют кислоты или основания (RONa)
в качестве катализаторов. Активным агентом в реак-
циях Н. является нитрозоний-катион NO + , обра-
зующийся при действии сильных к-т на нитрозирую-
щие соединения:
HNO, + H + yzi h2no2+ ^no++h2o
Существование нитрозоний-катиона доказано исследо-
ванием электропроводности нитрозилсульфата и нит-
розилперхлората.
Активность Н. агентов убывает в ряду
NO +>NOC1—NO—ONO2>NO—ONO>O=N—OH>O=N—OR
К H. относятся и другие методы введения в орга-
нич. соединения нитрозогруппы. Такими методами
являются присоединение хлористого нитрозила
к кратным связям, получение нитрозосоединений,
основанное на способности окиси азота захватывать,
свободные радикалы, и др.:
250”
(CH3)2CHHgCI + NO --------► (CH3)2CHNO
2 CF,J + 2 NO 2CF..NO + l2
° ng
Последняя реакция используется в пром-сти для
получения циклогексаноноксима. Н. широко исполь-
зуется для синтеза а-дикетонов, а-аминокислот и их
эфиров.
Лит.: Тоустер О., в кн.: Органические реакции, сб. 1,.
пер. с англ., М., 1956, с. 409. Э. Е. Быховская.
N-НИТРОЗОАМИНЫ общей формулы R2N—NO,
где R=Alk или Аг,— продукты замещения атома
водорода во вторичных аминах нитрозогруппой. В таб-
лице приведены свойства нек-рых Н.
Соединение Т. кип °C;'AtAt и20 nD
Диметилнитрозамин .... 152-153 1,0049е 1,4374е-
Диэтилнитрозамин 176,9 93.5/18 0.9431 1,43864
Дипропилнитрозамин .... 0.9163 1.44458
Дифенилнитроэамина .... разл. — —
Этилфенилнитрозамин . . . 119-120/15 1,0814 1,55977
а Т. пл. 67°. 6 При 18°.
517
НИТРОЗОБЕНЗОЛ—N- НИТРОЗОДИФЕНИЛАМИН
518
Н. обладают слабоосиовиым характером. Слабые
восстановители превращают Н. в соответствующие
гидразины, а сильные — в исходные вторичные
амины:
Я,
Ar3N-NH2-<— Ar2N-NO + H2--»-Ar2NH + NHs
НС1
При действии газообразного НС1 на спиртовые р-ры
жирноароматич. Н. последние изомеризуются в п-нит-
розанилины, причем нитрозогруппа вступает всегда
в пара-положение к аминогруппе:
XR
• Аг-n; —>NOCSH,NHR
XNO
Нек-рые жирноароматич. нитрозамины при действии
разб. щелочи превращаются в антидиазотаты:
ZR
Ar-Nf +КОН—> ArN = N-OK +ROH
XNO
H. получают нитрозированием вторичных аминов
(действием подкисленных водных р-ров NaNO2 на
соль вторичного амина) или действием аминнитрита
(или другого нитрозирующего агента) на р-р вторич-
ного амина в органич. растворителе:
R2NH + HNO2—>R2N-NO + H2O
С помощью этой реакции можно отличить и отделить
вторичные амины от третичных и первичных. Арома-
тич. Н. получают также действием окиси азота на
тетраарилгидразины:
Ar2N-N-Ar2 + 2NO —>2 Ar2N-NO
Все Н. дают положительный результат при испытании
на нитрозогруппу по Либерману. При нагревании
с неорганич. к-тами Н. расщепляются на исходный
амин и азотистую к-ту, что используется для качест-
венного определения Н.
Известны также ацилнитрозамиды общей ф-лы
.NO
RN(
XCOR'
Применяют Н. для выделения и очистки вторичных
аминов из их смесей с первичными и третичными. Н.
используются для получения несимм. дизамещенных
гидразинов.
Лит.: Губен И., Методы органической химии, пер. с
нем., т. 4, вып. 1, кн. 1, М.— Л., 1949. Э. Е. Быховепая.
НИТРОЗОБЕНЗОЛ C8H5NO, мол. в. 107,11—крис-
таллы, бесцветные иглы; т. пл. 67,5—68°; т. кип.
57—59°/18 мм; нерастворим в воде, хорошо растворим
в органич. растворителях с образованием зеленых
растворов; летуч. Н. типичный представитель арома-
тич. нитрозосоединений. При окислении Н. превра-
щается в нитробензол, при восстановлении — в ани-
лин; с первичными ароматич. аминами реагирует
с образованием азосоединений, с Р-арилгидроксилами-
нами — азоксисоединений:
ТЛ NJ _ А г
.—----> CeH5N = N-Ar+H2O
CeHsNO---( HONHAr
х------C„H3N-N-Аг+Н3О
\/
О
В присутствии вторичных алифатич. аминов Н. пре-
вращается в азобензол. С арилгидразинами Н. реаги-
рует с образованием азогидроксиламинов:
он
I
C0H5NO + H2N-NHAr—► C»HSN - N = N- Ar + H2
Аналогично из Ar(R)N—NH2 получают окиви азо-
иминов (напр., C,H5N(O)=N—N—(R) Ar]; с NHzOH
в щелочной среде Н. образует антидигзотаты, с диазо-
метаном — производное глиоксима
C»H3N-CH-CH-N-C„Hs
\ / \ /
О о
Н. реагирует с соединениями, содержащими двой-
ные и тройные связи, с образованием продуктов,
строение к-рых строго не доказано. С дифенилкете-
ном образуется смесь двух веществ (см. Нитрозосо-
единения).
При действии конц. H2SO4 Н. превращается в п-нит-
розодифенилгидроксиламин. Реакцией Н. с окисью
азота получается азотнокислый фенилдиазоний, хло-
рированием Н.— 4-хлор-1-нитрозобензол, действием!
пятиокисн азота—4-нитро-1-нитрозобензол. Н. обычна
получают из нитробензола или окислением 0-фенил-
гидроксиламина хроматом калия в присутствии серной
к-ты. Н. можно также получить действием NOBr
(или N2O4) на дифенилртуть и реакцией NOC1 с фенил-
магнийбромидом. Э. Е. Быховская.
п-НИТРОЗОДИМЕТИЛАНИЛИН (N,N-димети л-4-
нитрозоанилин) (СНз)2И—С6Н4—-NO, мол. в. 150, 18 —
зеленые кристаллы; т. пл. 85°; хорошо растворяет-
ся в органич. растворителях и в разб. минеральных
к-тах с образованием желтых растворов; плохо рас-
творяется в воде; константа диссоциацииК~ 1,95- 1О~10
(25°); в сухом виде способен к самовозгоранию.
Н. легко восстанавливается до Ы,И-диметил-гг-фени-
лендиамина (напр., Sn в НС1); перманганатом окис-
ляется в Ы,Ы-диметил-4-нитроанилин; при нагревании
до 190—-195° превращается в 4,4-бисдиметиламино-
азоксибензол; в присутствии щелочных агентов или
сплавлением конденсируется с соединениями, содер-
жащими активную метиленовую группу:
При действии щелочей Н.расщепляется с образова-
нием диметиламина и паранитрозофенолята. Это
свойство используется для получения чистого диметил-
амина. Н. обычно получают нитрозированием NjN-ди-
метиланилина. Н. является важным промежуточным
продуктом синтеза многих красителей (напр., метиле-
нового голубого), применяется для осаждения фенил-
гликозидов и для различных препаративных целей.
При попадании на кожу Н. может вызвать экзему.
N -НИТРОЗОДИФЕНИЛАМИН (дифенилнитроз-
амин) (С8Н5)2 N—NO, мол. вес 198, 23 — желто-зеле-
ные кристаллы, пластинки. Т. пл. 66,5°; мало раство-
рим в воде; легко растворим в теплом бензоле и теп-
лом спирте с коричневым окрашиванием; растворим
в конц. р-рах щелочей и H2SO4. Спиртовым р-ром хло-
ристого водорода Н. превращается в 4-нитрозодифе-
ниламин; азотной к-той нитруется, образуя смесь
.\хнитрозо-2-нитродифениламина и N-miTpo3o-4-HHT-
родифениламина; сухим хлористым водородом при 0°
разлагается на дифениламин (C8H5)2NH и нитрозил-
хлорид NOC1; с гидразином NH2NH2 дает дифенил-
амин и ^^дифенилгидразин (С6Н5)2 NNHz", нагрева-
нием с гидразингидратом NH2NH2-H2O и спиртовым
р-ром едкого натра — дифениламин и азид натрия;
обработка фенилмагнийбромидом при —15° приводит
к образованию дифенила, фенола и трифенилгидра-
зина; нагревание с n-толуидином при 100° — к об-
разованию дифениламина и п-диазоаминотолуола.
Н. получают, добавляя водный р-р нитрита натрия
17
519
НИТРОЗОКРАСИТЕЛИ—а-НПТРОЗО-0-НАФТОЛ
520
связями.
он
NO
II
о
N
О 1
О
NOH
III
в орто-положении
вряд ли способны к
к холодному спиртовому р-ру солянокислого дифенил-
амина. Н.— полупродукт в синтезе органич. красите-
лей; применяют также для колориметрич. определе-
ния Pd. Э. Быховская.
НИТРОЗОКРАСИТЕЛИ (хиноноксимиминные кра-
сители) — небольшая группа нитрозопроизводных фе-
нолов, нафтолов и их аналогов. Гидроксил аромати-
ческих нитрозосоединений обладает повышенными
кислотными свойствами, что делает Н. кислотами,
образующими соли с металлами. В этих солях анион
имеет строение частицы с выравненными
Юн является общим ио-
ном (I) для двух тауто-
мерных кислот: нитро-
зофенола (II) и хино-
ноксима (III).
Практическое значе-
ние имеют только та-
кие Н., в к-рых окси-
и нитрозогруппы расположены
друг к другу. Эти соединения
таутомерии, так как взаимное расположение указан-
ных групп делает возможным возникновение внутри-
молекулярной симметричной водород-
ной связи, замыкающей 6-членное коль- R -
цо, связи в котором выравнены. ] Г •
Способность к образованию водородных 1 %, ,-н
связей определяет, по-видимому, воз- О
можность возникновения комплексных соединений
с металлами: железом, хромом, никелем, кобальтом
и др. Эти комплексные соединения применяются как
красители для печати, а также в аналитической
химии. Некоторые из них содержат сульфогруппы
и тогда лаки этих соединений (комплексные соеди-
нения с металлами) применяют для окраски шерсти
и шелка как кислотные красители. Практич. значе-
ние для получения Н. имеет только нитрозирование
фенолов.
Динитрозорезорцин образует при действии серно-
кислой железо-аммонийной соли интенсивно зеленый
лак, применяющийся в ситцепечатании; с кобальто-
выми солями получается лак оранжевого цвета. Боль-
шое значение имеют производные 2-нафтола, напр.
1-нитрозо-2-нафтол, образующий с солями 2-валент-
ного железа лак, известный как Пигмент зеленый В.
Нитрозирование 2-нафтол-6-сульфокислоты и образо-
вание аналогичного лака приводит к более ценному
Кислотному зеленому ЗЖ (Нафтоловому зеленому В).
Строение лаков очень сложно и не может считаться
окончательно установленным. Подобным же образом
получают ценные красители из арилидов 2-нафтол-
3-карбоновой к-ты (азотолов) или же из арилидов
ацетоуксусной к-ты.
Н. иногда служат промежуточными продуктами для
синтеза более сложных красителей, лекарственных
соединений или фотореагентов.
Лит..: Коган И. М., Химия красителей, 3 изд., М.,
1956, с. 96; Венкатараман К. Химия синтетических
Красителей, пер. с англ., т. 1, Л., 1956, с. 449.
Б. А. Порай-Коиыц.
НИТР030МЕТИЛМ0ЧЕВИНА СН3—N(NO)CONH2,
мол. в. 103,09 — белые кристаллы; т. пл. 123—124°;
легко растворяется в воде, спирте, эфире. Н. ус-
тойчива только на холоду; при повышенной
темп-ре (—30°) иногда без видимой причины разла-
гается с образованием метилизоцианата CH3NCO,
азота и воды. При хранении в течение нескольких
месяцев заметное разложение может наблюдаться уже
при 20°. При этом вместо метилизоцианата образуется
•его тример — N-триметилциануровая к-та. Щелочной
.гидролиз Н. приводит к образованию диазометана:
ON.
^NCONH2+KOH —>CH2N3+KCNO+2 н2о
H.CZ
Эта реакция благодаря доступности исходных реаген-
тов лежит в основе самого удобного способа получения
диазометана. При восстановлении цинком в СН3СООН
образуется метилгидразин CH3NHNH2. Н. обычно
получают нитрозированием метилмочевины.
Э. Е. Быховская.
НИТРОЗОМЕТИЛУРЕТАН (этиловый эфир N-ни-
трозометилкарбаминовой кислоты), мол. в. 132,12,
CH3N(NO)COOC2H5 — жидкость желтоватого цве-
та; раздражает слизистые оболочки; т. кип. 65—
65,5°/13 мм; d’°l,1224; п™ 1,4363. При хранении Н.
не изменяется, однако иногда после длительного хра-
нения начинается самопроизвольное разложений. Ре-
комендуется хранить при темп-ре ниже 20° в темноте
и в сосудах из стекла, не содержащего, по возмож-
ности, щелочных солей. При взаимодействии Н. с вод-
ным аммиаком образуется уретан, метиловый спирт
и азот:
CH3N (NO) COOC2H3 + NH3-->- H2NCOOC2H5 + CH3OH + N-
При действии на Н. конц. водного р-ра КОН при 0°
образуется нестойкая калиевая соль CH3N=NOK,
к-рая даже влагой воздуха разлагается с образованием
диазометана. Со спиртовыми р-рами щелочей Н.
образует с почти колич. выходом диазометан:
спирт
CH3N (NO) СООС,Н3 + КОН---->CH3N3+ КНСО3 + С2Н5ОН
Вместо щелочей могут быть использованы углекислые
соли щелочных металлов:
Na2CO,
C1ISN (NO) СООС2Н3 + С2Н5ОН--->CH,N2 + O = C (ОС2Н3)2
Нитрозометилуретан получается действием азоти-
стой к-ты на этиловый эфир N-метилкарбаминовой
к-ты:
CH3NHCOOC3H3 + HNO2 —*- CH3N (NO) СООС2Н,
Э. Е. Быховская.
а-НИТРОЗОЩ-НАФТОЛ (1
актив Ильинского) CJ0H7O2N,
-нитрозо-2-нафтол, ре-
мол. в. 173,17—тем-
но-оранжевые кристаллы; т. пл. 108—
110°; мало растворим в воде (при 20° в
100 г Н2О растворяется 0,1 г реактива)
и холодном спирте, хорошо растворим
в горячем спирте, бензоле, эфире, ле-
NO
дяной уксусной к-те и в р-рах щелочей; летуч с па-
рами воды. При взаимодействии уксуснокислого р-ра
реагента с нейтральным р-ром соли кобальта выпадает
красно-бурый осадок [C,0H6(NO)O]3 Со. Получают а-Н.
взаимодействием fi-нафтола и NaNQ2. Реагент об-
с некоторыми катиона-
75%-ном диоксане. Кон-
разует малорастворимые окрашенные внутрикомплек-
сные соединения (хелаты) с иона- n = О
ми нек-рых металлов. ''Me}
Этим свойством пользуются для ОХ 3
качественных реакций, а также
для гравиметрия, и фотометрич.
определения Со, Си, Fe, Pd и др.; можно применять
для разделения элементов экстракцией.
Ниже приведены константы диссоциации (ступен-
чатые) комплексов а-Н.
ми, определенные при 30° в
станта диссоциации реаген-
та, определенная при тех же
условиях рА (НА), равна
11,60.
Впервые как аналитич. ре-
агент был применен в 1885
М. А. Ильинским.
₽к2
Катион
+ + + 6,2 11,50 9,50 4,40 10.10 7,80
Лит.: Welcher F.J., Organic analytical reagents, v.3,
N. Y., 1947; Моррисон Дж.,Фрейзер Г., Экстрак-
ция в аналитической химии, пер. с англ., Л., 1960.
И. П. Ефимов.
521
Р-НИТРОЗО-а-НАФТОЛ— НИТРОЗОСОЕДИНЕНИЯ
522
р-НИТР030-а-НАФТ0Л (2-нитрозо-1-нафтол), мол.
вес 173,17, C10H7O2N — желто-зеленые иглы, т. пл.
162—164° (с разл.), легко растворяется в
ОН метиловом и этиловом спиртах, ацетове,
Nо ледяной уксусной к-те, мало растворим в
I Г бензоле, хлороформе,эфире, почти нера-
створим в холодной воде. Растворы в ор-
ганич. растворителях окрашены в оранжевый цвет. Ле-
туч с парами воды. Натриевая соль — красно-коричне-
вые иглы, хорошо растворяется в воде и спирте. При
взаимодействии р-ра соли кобальта и спиртового р-ра
реагента с последующим подогреванием образуется
красный осадок. Получают Р-Н. взаимодействием
а-нафтола с NaNO2. Реагент образует с рядом катионов
окрашенные внутрикомплексные соединения, исполь-
зуемые для качественных реакций, а также для гра-
виметрия. и фотометрия, определений Со, Си, Fe,
Pd для разделения нек-рых элементов, напр. с исполь-
зованием экстракции.
Ниже приведены константы диссоциации (ступенча-
тые) комплексов Р-Н. с нек-рыми катионами, опреде-
ленные при 30° в 75%-ном диоксане. Константа дис-
социации реагента, определенная при тех же условиях
рК (НА), равна 11,2.
Катион рК, рК, Катион рК1 рК2
Mg2+ . . 5,80 3,80 Ni2+ . . 10,70 9,20
Mh2+ . . 7,10 5,50 Zn2+ . . 8,70 7,00
Лит. см. при ст. а-Нитрозо-^-нафтол. И. П. Ефимов.
НИТР030-К-С0ЛЬ (динатриевая соль 1-нитрозо-
2-нафтол-3,6-дисульфокислоты) C10H5O8NS2Na2, мол. в.
377,26— золотисто-желтые кри-
NO сталлы. Н. легко растворима в
ОН' метиловом и этиловом спиртах,
III ограниченно — в воде (2,5 г в
NaO3s'44^x^A'SO3Na 100 2 воды); водный р-р имеет
светло-желтую окраску, при
подщелачивании переходящую в оранжевую. При на-
гревании 1 лл 0,5% водного р-ра Н. с 1 мл нейтраль-
ного 0,1% р-ра соли кобальта появляется красное
окрашивание. Применяется для колориметрич. опре-
деления Со и для косвенного колориметрич. опреде-
ления калия. Э. Е. Быховская.
НИТРОЗОСОЕДИНЕНИЯ — органич. соединения,
в к-рых нитрозогруппа—N=O непосредственно свя-
зана с атомом углерода. Различают первичные, вто-
ричные и третичные Н., в зависимости от характера
атома углерода (первичного, вторичного или третич-
ного), к к-рому присоединена нитрозогруппа. Первич-
ные и вторичные Н. изомеризуются в оксимы (изо-
нитрозосоединения):
ч zH ч
;с;--------------<-)C = N-OH
7 4 NO 7
Долгое время считалось, что в форме истинных Н.
существуют только третичные Н. Однако позднее было
найдено, что в нек-рых случаях можно выделить и
вторичные Н., напр. 2-нитрозо-З-хлор-З-метилбутан,
к-рый только при нагревании переходит в соответ-
ствующий оксим. Большинство С-нитрозосоединений
существует в димерной форме, легко переходящей при
плавлении или растворении в мономерную форму
(I). Димерной форме приписывают строение биполярно
ионизированного соединения (II):
(R3CNO)2—>R3CNO
I
о-
+ I
R-N—>N-R
II
О
II
Сполна галогенированные Н., такие как CClaNO,
CF3NO, (CF3)3 CNO, и другие не образуют димеров.
Это объясняется сильным индукционным эффектом
пергалогенированных групп, что препятствует обра-
зованию связи за счет свободной электронной пары
азота.
В мономерной форме Н. большей частью газы или
перегоняющиеся жидкости, в димерной форме —бес-
цветные твердые тела (см. таблицу).
Соединение T. кип. мономе- ров,0С/лии T. шг. димеров, °C
(CH3)3CNO CH3CBr(NO)CH3 29/74 76—76,5
CC1HNO — CH, — 65
CF,NO —86,6 —
cf2= cfno -23,7 4
(CF3),C- NOa 24/760 —
CC13NO6 5/70 —
сн3
a d° 1,6630. 6 d 20 1,506.
4 4
При действии сильных окислителей Н. могут быть,
превращены в соответствующие нитросоединения:
RSCNO —<-R3CNO2
Конечным продуктом восстановления Н. являются
амины, однако в нек-рых случаях восстановление
останавливается на стадии гидроксиламина:
Н2
R3CNO —► R3CNHOH —<- RaCNH2
Известны реакции, в к-рых нитрозогруппа ведет себя
аналогично карбонильной. Напр., с первичвыми ами-
нами нитрозобензол реагирует с отщеплением воды
и образованием азосоединений:
CoHjNO+RNHj —> C,H5N=N- R +Н2О
Ароматич. Н. при действии концентрированной сер-
ной кислоты вступают в конденсацию типа аль-
дольной:
.он
C«H3NO + CBH.,NO —>C3H3-N(
XCSH,NO
С дифенилкетеном Н. дают продукты присоединения,
аналогичные продуктам присоединения к дифенилке-
тену альдегидов и кетонов:
(C»H5)2C = C = O + CeH5NO—>(С»НГ,)2С-С = О 1С8Н3)2С-С = О
II + II
o-n-c„h5 c»h3-n-o
Известны и другие случаи присоединения по двойной
азот-кислородной связи. Так, трифторнитрозометан
легко присоединяет фторолефины, давая наряду с со-
ответствующими продуктами присоединения (оксазети-
динами) и высокополимерные продукты, используемые
523
НИТРОЗОСОЕДИНЕНИЯ—2-НИТРОИНДАНДИОН
524
для получения термостойких каучуков:
CFSNO + CF2=CF2—»-CF,-N-O4-
CF2-CF2
+ _N-O-GFa — CF2-[O-N-CF2-CF2]ft-O-N —
I I I
CF3 GF3 GF,
При действии на H. азотистого ангидрида образуются
нитрозонитриты (нитрозиты). Эта реакция исполь-
зуется для качественного определения терпенов. При-
соединяясь к диеновым углеводородам в положение
1,4, Н. образуют производные 3,6-дигидро-орто-
ксазина:
zcn2
сн
II
сн
сн2
Аг /СН2х
А СН N-Ar
и II I
---- сн о
'сн/
Н. жирного ряда получают: 1) Окислением жирных
аминов и N-алкилгидроксиламинов к-той Каро:
(CH3),CNH2—>-(CH3)3CNHOH—4CH3).,CNO
2) Восстановлением с помощью амальгамы алюминия
третичных нитросоединений до соответствующих ги-
дроксиламинов с последующим окислением их в Н.:
Н2 О2
R3CNO2—«-RjCNHOH—>R3CXO
3) Действием на оксимы галогенов (реакция Пилоти):
NO
R2C = N- ОН + Вг2—>R,cf
хВг
4) Действием на олефины хлористого, бромистого и
фтористого нитрозила или азотистого ангидрида;
в последнем случае образуются нитрозонитриты (нит-
розиты):
ZR NOCI, NOBr R2C-CR2
R2 “ XR NOF NO Cl (Br; F)
N 2O з .R2
R2G = GR2--->R,C — C.
| OXO
NO
5) Взаимодействием окиси азота и перфторалкилиоди-
дов под влиянием УФ-освещения:
CF3J + 2 NO—>- 2 CF3NO + J2
Специфич. методы получения ароматич. Н.: 1) Дей-
ствие окислов азота на диарилртуть:
Ar2Hg + N2O,—>ArHgNO3+ ArNO
2) Действие хлористого нитрозила на арилмагний-
бромиды или на ароматич. ртутьорганич. соединения:
ArMgX+NOCl—>ArNO + MgXCl
ArHgX+NOCl—>Ar.NO + HgXCl
3) Действие азотистой к-ты на N-диалкиламины, содер-
жащие в о-и м- или только в л-положении алкильную
или алкоксильную группу:
Нитрозогрупиа является ориентантом первого рода.
Нитрозокислоты, нитрозокетоны, нитрозофенолы и
др. получают аналогичными методами. Для количест-
венного определения нитрозогруппы используют ее
окислительную способность (выделение иода из под-
кисленных р-ров йодистого калия) или реакцию с фе-
нилгидроксиламином :
2 RNO + 2 GeH5NHNH2—>.RN = NR + 2C6H<, + 2H2O + 2 N2
Последний метод применяют гл. обр. в случае арома-
тич. Н.
В зарубежной литературе Н. иногда называют
также вещества, имеющие состав R—N=N=O и
R—О—N=O.
Лит.: Губен И., Методы органической химии, пер. с
нем., т. 4, вып. 1, кн. 1, М.— Л., 1949; И aszel di ne R. N.
[a. о.], J. Chem. Soc.,’ 1953, 2075; 1954, 912; 1955 , 1881;
Арбузов Ю. А., Федюкина H. Л., ДАН СССР, 1948,
60, .Xs 7, 1173. Э. E. Быховская.
n-НИТРОЗОФЕНОЛ (n-хинонмоноксим), мол. в.
123,11, НО—С6Н4—NO, или O=CeH4=NOH, — бес-
цветные кристаллы; т. пл. 133°; разлагается при
144°; теплота образования при постоянном объеме
715,4 ккал/моль, при постоянном давлении константа
диссоциации А = 3,3-IO-7 (25°); трудно растворим
в холодной воде, лучше в горячей, нерастворим в угле-
водородах; легко растворим в спирте, эфире, ацетоне,
щелочах. Растворы Н. в полярных растворителях
зеленого цвета. Получают действием азотистой к-ты
на фенол или расщеплением щелочью п-нитрозоди-
метиланилина, а также взаимодействием хинона
с гидроксиламином. Н. является таутомерным соеди-
нением (хиноноксимная таутомерия):
Н. вступает в реакции, характерные как для фено-
лов, так и для оксимов. Так, Н. окисляется в п-нитро-
фенол, восстанавливается в n-аминофенол; при дей-
ствии двуокиси азота образует 2,4-динитрофенол.
При пропускании газообразного НС1 в эфирный р-р
Н. образуется дихлор-4-аминофенол, в спирто-
вой р-р — дихлор-4-аминоанизол. В щелочных р-рах
с двумя молями брома Н. образует 2,6-дибромбензо-
хиноноксим; с солянокислым гидроксиламином —
хинондиоксим, с солянокислым семикарбазидом —
n-хиноноксимсемикарбазон. Качественной реакцией
на Н. служит появление красного окрашивания при
добавлении конц. H2SO4 к р-ру Н. в феноле. Количест-
венно Н. можно определить по объему выделившегося
азота при действии на него уксуснокислого р-ра фе-
нилгидразина.
Н.— полупродукт в синтезе органич. красителей.
2-НИТРОИНДАНДИОН, мол. в. 191Д4Б- безвод-
ного, 227,17—с2Н2О — желтые кристаллы; растворим
/СО
в воде; c6h,^coxchno2.2h2O легко образует хорошо
кристаллизующиеся соли. Соли Н. и первичных или
вторичных аминов мало растворимы, что можно
использовать для выделения и идентификации этих
аминов. Н. и его бромпроизводное дают характерные
цветные реакции на гидразин, формальдегид, антипи-
рин, пирролы, индолы, карбазол и др. в спиртовом,
водном или уксуснокислом р-ре (табл. 1) или в ледя-
ной уксусной к-те (табл. 2).
2-Нитрозоиндандион-1,3 (Р-оксим индантриона) дает
с л-аминофенолом и n-аминосалициловой к-той
(ПАСК) в ледяной уксусной к-те при нагревании зеле-
ную флуоресценцию; предельное разбавление для
л-аминофено ла 1 : 40000, для ПАСК — 1 : 28000.
При нагревании 2-бром-2-нитроиндандиона в высо-
ко кипящих растворителях образуется индантрион.
Это наиболее простой способ быстрого приготовления
нингидрина в лабораторных условиях. Нитроиндан-
дион легко конденсируется с бензгидролом и другими
гидролами. Продукты конденсации при действии ще-
лочи расщепляются на фталевую к-ту и соответствую-
щее нитрометильное соединение. Т. обр., нитроиндан-
525
НИТРОКРАСИТЕЛИ—НИТРОМЕТАН
526
дионом можно «нитрометилировать» (вводить группу
€H2NO2):
/С0\ /Н НОСНАг2 СО СНАгэ
На С
'coz \о2 „ COONa 2 NaOH * СеЩ 'со КО2
+ Ar2CH-CH2NOg
COONa
Таблица 1. Колориметрические реакции
2-нитроиндандиона-1,3
Соединение Окраска раствора Предельное раз- бавлениеа
Пиррол Красная 1:200 000; 1:3 333 000
N-М ети л пирр ол '2,4-Диметилпиррол То же « « 1:333 000; 1:10 000 000 1:83 000; 1:1 000 000
Индол .2-Метилиндол •З-Мети линдол 7-Метилиндол « « Фиолетовая Оранжевая Красная 1:333 0006 1:100 0006 1:50 0006 1:500 000; 1:5 000 000
N-Метилиндол Красная 1:125 000; 1:250 000
.Антипирин (при нагре- вании) Гидразин (s кислом р-ре при нагревании) 'Формальдегид (в конц. HoSO<; оставляют иа 15 мин, потом нагре- вают) Оранжевая Оранжево-крас- ная Зеленая флуорес- ценция 1:10 000в 1:100 000г 1:33 000
а Первое число — без нагревания, второе—после нагревания
раствора. 6 Чувствительность не увеличивается от нагрева-
ния в Окраска из водного слоя переходит в хлороформен-
ный. г Открытию гидразина не мешает NH2OH (р-р не ок-
рашивает).
Таблица 2. Колориметрические реакции 2-бром-2-ннтроин-
данд иона-1,3
Соединение Окраска раствора Предельное разбавление
Карбазол Красная 1:500 000
Индол Красная 1:100 000
2-Метилиндол Красно-фиолетовая 1:40 000
З-Метилиндол Синяя 1:4000
7-Метилиндол Красная Г.40 000
2,3-Дифенилин- ДОЛ Фиолетово-красная 1:100 000
Триптофан Розовая с коричнево-зеле- ной или зеленой флуорес- ценцией (и черным осад- ком до разбавления 1:20 000) 1:100 000
Тетероауксин Красная 1:1000
Пиррол Красно-фиолетовая 1:10 000
Лит.: В а наг Г., Нитроиндандион, Рига, 1954; Цикли-
ческие р-дикетоны. Сб. статей, под ред. Г. Ванага, Рига, 1961.
Г. Я. Ванаг.
НИТРОКРАСИТЕЛИ — амины и фенолы бензоль-
ного и нафталинового рядов, содержащие одну или
несколько нитрогрупп. По крайней мере одна нитро-
группа должна быть расположена в орто-положении
к амино-или оксигруппе.Нитрогруппы обусловливают
кислый характер этой группы красителей. Благодаря
этому они способны образовать щелочные соли. Эти
соли содержат ароматич. анионы с выравненными
связями, чем и обусловлена их стойкость и окраска.
Н. способны существовать в виде нитро- и ацинитро-
соединений, напр.:
NOg NOg
no2
нитроиедтние ион нитрокрасителя аиинктросоединекие
Ранее предполагали, что окрашенными частицами
являются две последние, т. е. ион Н. и ацинитросо-
единение в силу их возможного хинонного строения,
однако в настоящее время причину цветности припи-
сывают наличию иона с выравненными связями.
Орторасположение одной из нитрогрупп по отношению
к окси- или аминогруппе в Н. делает возможным воз-
никновение водородной или хелатной связи между
кислородом нитрогруппы и водородом, напр. окси-
группы, или к.-н. атомом металла. С этим связана
способность Н. давать комплексные соли с металлами.
Получают Н. чаще всего нитрованием окси- или
аминосоединений. Иногда удобнее нитровать галоге-
нопроизводные и затем заменять атом галогена амино-
или оксигруппой. Так, первый синтезированный кра-
ситель — пикриновая кислота (I) получается нитро-
ванием фенола или гидроксилированием динитрохлор-
бензола с последующим нитрованием образующегося
динитрофенола. Другой старинный краситель для
кожи — Ауранция, представляющий
собой аммонийную соль гексанитро-ди- q n Nq
фениламина (II) •— получается взаимо- 2 2
действием динитрохлорбензола с ани- L
лином — с последующим нитрованием
образовавшегося динитродифенилами- t NOa
на до гексанитропроизводного, и за-
ключительным образованием аммонийной соли. Мно-
гие Н. содержат сульфогруппы, что делает их проч-
ными к воздействию тепла (напр., при глажении).
Нафтоловый желтый S (III), содержащий сульфо-
группу, применяют для окраски шерсти и шелка;
он является также пищевым красителем:
NOg Ш
\ Vcf3 Представляют интерес также не-
)=( растворимые Н., применяющиеся
NO2 N(CH3)2 для окраски ацетатного шелка.
Примером может служить димети-
lv ламинодинитротрифтортолуол (IV),
обладающий хорошей прочностью и высоким срод-
ством к ацетатному шелку. Производные нитродифеннл-
амина, содержащие карбоксильную или сульфогруп-
пу, дают лаки с металлами, обладающие повышенной
прочностью. Многие Н., нерастворимые в воде, при-
меняются в качестве пигментов в лакокрасочной
пром-сти.
Лит.: К о г а н И. М., Химия красителей, 3 изд., М., 1956,
с. 91; Венкатараман К., Химия синтетических краси-
телей, пер. с англ., т. 1, Л., 1956, с. 453.
Б. А. Порай-Бошиц.
НИТРОМЕТАН CH3N0g, мол. в. 61,042 — бесцвет-
ная жидкость с своеобразным запахом, напоминающим
горький миндаль; 1,13816; Яр 1,38188; т. пл.
—28,55°; т. кип.: ‘ 101,1867760 juju; 20727,34 .и.и;
136, 40472026 ». Т. кип. азеотропной смеси Н. —
вода 83,6° (76,4% Н.). Теплота парообразования
527
НИТРОМЕТАН—НИТРОНАФТАЛИНЫ
528
8,225 ккал/моль (т. кип.); теплота плавления
2,319 ккал/моль', теплоемкость 25,76 ккал/моль -град
(30°); Т. крит. 315°; эбуллиоскопия, константа 1,86°;
диэлектрич. проницаемость 35,87 (30°); дипольный
момент 3,17 D; вязкость 0,646 спуаз (20°); поверхност-
ное натяжение 36,98 дин/см (20°); т. воспл. 44,4°;
растворимость (при 20°): Н. в воде 9,5 .ил/100 .мл;
воды в Н. 2,2 .и л/100 мл. Н. очень хорошо растворим
в большинстве органич. растворителей; исключение
составляют парафины и циклопарафины.
Н. является простейшим представителем класса
нитропарафинов (см. Нитросоединения алифатиче-
ские). Как все нитросоединения, имеющие а-атом водо-
рода, Н. проявляет свойства псевдокислоты. При
действии конц. щелочей реакция не останавливается
на образовании метаннитроната щелочного металла —
дальнейшая конденсация приводит к получению
соли метазоновой к-ты, к-рая затем превращается
в соль нитроуксусной к-ты (1). Сухие щелочные соли
аци-И. обладают взрывчатыми свойствами, вероятно,
в результате превращения их в соли гремучей кислоты.
При взаимодействии с солями ртути они дают грему-
чую ртуть (2):
„о
2 CH, = N( ->HON = CHCH = NC ->KOCOCH = N< (1)
4 ок 4 ок 4 ок
,О HgCl2 / ло\
CH„ = Nf -------►. CH2 = N< Hg—*Hg (ONC), (2)
XOK \ хО/г
Конц, к-ты разлагают Н. с образованием аммиака,
гидроксиламина СОг, СО и т. п. Нитрометан легко
нитрозируется, восстанавливается, галогенируется,
карбоксилируется. Благодаря наличию подвижного
водорода Н. может участвовать в азосочетании,
в Манниха реакции и Михаэля реакции.
Наибольшее значение имеет взаимодействие Н.
с карбонильными соединениями. При этом в зависи-
мости от исходных продуктов и условий получаются
нитроспирты, нитрогликоли, нитроолефины, динитро-
парафины и т. д.:
кон
-------------------->о2хс (СН2ОН)3
ch3no2+ch2o—
----»-CH2 = CHNO2
РЬ2 +
C2H5ONa „О
ClI3NO2 + CeH5CHO---> CeH5CHOHCH = N<
I xONa
H2SO, I
CeH5CH = CHNO2
CH,ONa
CH 3NO3 + (CH3)2CO--
(C,HS),NH
I CH3COOH
CaH5CHOHCH2NO2
(CH3)3CCH2NO3
I
OH
(CH3)2C (CH2NO2)2
Промышленные методы получения H. основаны на
деструктивном нитровании парафиновых углеводо-
родов. Обычно для нитрования используется пропан,
к-рый дает 4 нитропарафина, применяющихся
в пром-сти: Н., нитроэтан и два изомерных нитро-
пропана. Пропан нитруют в паровой фазе при
400—450°. В поток пропана впрыскивают под давле-
нием азотную к-ту в количестве 0,1—0,2 моля на
1 моль пропана, и полученную смесь пропускают через
реактор — хромоникелевую трубку. Затем смесь ох-
лаждают, нитропарафины промывают водой и подвер-
гают разгонке. Непрореагировавший пропан промы-
вают р-ром солянокислого гидроксиламина (для уда-
ления побочно образующихся альдегидов и кетонов)
и вновь возвращают на нитрование. Окислы азота,
оэрузающиеся при нитровании, снова превращают
в азотную к-ту. В лаборатории Н. получают ме-
тилированием солей азотистой к-ты (2, а и б) и по спо-
собу Кольбе — из монохлоруксусной к-ты (3); Н.
образуется также при перегруппировке метилнит-
рита (4):
420°
CSHS + HNO3 (пар) —> CH3CH3CH2NO2 (32 %) +
+ (CH3)2CHNO2 (33 %) + CH3CH2NO3 (26 %) + CH3NO2 (9 %) (1)
(CH3)2SO,+NaN02—>-CH3NO3 (2a>
CH3J + AgNO2—>-CH3NO3 (26>
C1CH2COOK + KN02—>- CH3NO3 (3)
120—130°
CH3ONO---------►СНзКЮ, (4>
H. применяют как растворитель, напр., для эфиро-
целлюлозных лаков, виниловых смол, для экстракции
ароматич. углеводородов из смесей с алифатич. и
циклоалифатическими; важное значение имеет приме-
нение Н. как полупродукта для синтеза хлорпикрина,
нитроспиртов, взрывчатых веществ типа трихлорнит-
рометана 2-нитро-2-оксиметил-1,3-пропандиола и т. п.
Н. можно применять так же, как добавку к дизельно-
му топливу и как горючее для реактивных двигателей.
Н. является сильным ядом для центральной нервной
системы. Допустимая концентрация Н. в воздуха
0,01%. При нагревании Н. выше 100° под давлением,
особенно в присутствии окисляющихся веществ, сле-
дует соблюдать осторожность, т. к. иногда при этом
происходит взрыв.
Л.ит.: Азингер Ф., Химия и технология парафиновых
углеводородов, пер. с нем., М., 1959, с. 265—355; Новиков.
С. С., Б е л и к о в В. М., Справочник физических и физико-
химических свойств нитросоединений, содержащих нитрогруп-
пу в жирной цепи. Приложение к «Новостям спецхимии»,
М 4/224, 1958; Вайсбергер А. [и др.], Органические ра-
створители, пер. с англ., М., 1958, с. 194,413. Е.М.Рохлин.
НИТРОН [1,4-дифенил-(3,5-энданил)-дигидро-
1,2,4-триазол] C20H13N4, мол. в. 312,38 — желтые-
кристаллы; т. пл. 189—190° (с разл.); нерастворим
в воде; плохо растворяется в эфире, (
лучше в горячем спирте и ацетоне; c»Hs-N
легко растворим в бензоле, хлоро-
форме, этилацетате и разб. к-тах (кро-
ме HNOs и НСЮ4). Спиртовой р-р Н.
на свету разлагается и краснеет. Н.—
сильное основание, образующее устой-
чивые соли, даже с угольной к-той. Получают Н. из.
карбодифенилдиимида (I), взаимодействием с фенил-
гидразином (II); при этом образуется трифениламино-
гуанидин (III), дающий Н. при нагревании с муравьи-
ной к-той:
нн н
I I I
C,H5N = C = NCeH5 NH2-NH-C„H5 C5H5-N N-GsHsN-С.Щ
4 1 J-
C--- ~N
I II III
Применяют H. для весового определения NO, s
CIO 4“, WO’“ и ReO^. И' n' E&tMOe'
НИТРОН — см. П олиакриловые волокна.
НИТРОНАФТАЛИНЫ Ci0H,O2N, мол. в. 173,14—
два изомера, отличающиеся положением нитрогруппы:
° 0
а-Н итронафталин — кристаллы, светло-
желтые иглы, без запаха; т. пл. 52° (метастабильвая
форма), т. пл. 57,8° (стабильная форма); технич. про-
дукт плавится при 58—61°; т. кип. 3047750 -м.ч. ок.
169712 .и.и; возгоняется при 30—4070,01 мм; с водя-
ным паром не перегоняется, очень хорошо растворим
в большинстве органич. растворителей, лучше всего
в ацетоне (в 100 г растворяется: 131,6 г при 18° и
529
НИТРОНИЙ-ИОН—НИТРОНОВЫЕ КИСЛОТЫ
530
327,6 г при 32°). Р-Н итр онафт а л ин — кри-
сталлы, бесцветные пластинки или иглы; т. пл. 78,7°;
т. кип. 312,5°/734,4 .и.«; не возгоняется; перегоняется
с водяным паром. При нитровании и галогенирова-
нии а-Н. образуется смесь, соответственно, 1,5- и
1,8-динитронафталинов или 1,5- и 1,8-галогенонитро-
нафталинов. При сульфировании а-Н. получается
только 1-нитронафталин-5-сульфокислота. При нит-
ровании и сульфировании Р-Н. заместитель вступает
преимущественно в 5- и 8-положения.
Восстановление а-Н. в кислой среде приводит к об-
разованию а-нафтиламина (см. Нафтиламины). При
восстановлении Н. в щелочной среде образуются
азокси-, азо- и гидразонафталины. Окисление а-Н.
хромовой к-той дает нитрофталевую к-ту, в то время
как при окислении КМпОа в щелочной среде образует-
ся фталоновая к-та.
а-Н. получают непосредственным нитрованием наф-
талина (HNOs при 95° или смесью HNO3+H2SO4 при
60°); при этом образуется 2—4% Р-Н. Последний от-
деляют перегонкой с водяным паром и последующей
кристаллизацией из лигроина и спирта. Если нитро-
ванию подвергается аддукт нафталина с 2 молями ге-
ксахлорпентадиена, атакуется исключительно (на
99%) P-положение в ядре нафталина; при нагревании
до 200° продукт нитрования диссоциирует с образова-
нием Р-Н. Последний не имеет технич. значения
вследствие малой доступности. а-Н.—технически
важный продукт, применяется в синтезе красителей
различных классов.
Л.ит. см. при ст. Нафталин. В. А. Пучков.
+
НИТРОНИЙ-ИОН — O=N=O— однозарядный по-
ложительный ион. Н.-и. входит в состав ряда солей
нитрония, к-рые представляют собой кристаллы, легко
разлагающиеся при действии влаги воздуха. Темп-ры
плавления нек-рых солей нитрония приведены в таб-
лице.
Соль нит- рония NO,* СЮ , 2 4 NO* HS.0 ; NO * FSO s NO* BF4
Т пл., °C > 135а 100-105 200а 170а
а Плавятся с разложением.
Соли нитрония образуются при действии сильных
к-т на азотную к-ту или азотный ангидрид:
HNO3 + HC1O, —> no2* сю; +Н3О + С1О4+
N2O5 + HF + PF5—»-NO* PF" +HNO3
* 8
Двуокись азота в присутствии окислителей также
может явиться источником Н.-и. при получении солей
нитрония: N2O4 + B2O3 + BrF3 —>n о* BF~
Br2
Чистые соли нитрония, содержащие комплексный
анион, удобно получать из фтористого нитрила:
NO3F+SbF6 —> NO2+ SbF6"
Н.-и. образуется в маловодных смесях азотной и
серной к-т:
HNO3+2 H2SO4 5=± NO2+ ч-Н3О + +2 HSO;
Его присутствие доказывается физич. методами (крио-
скопия, эбулиоскопия, определение электропровод-
ности, спектры комбинационного рассеяния). Кон-
станта равновесия этой реакции К == 30— 40.
Содержание HNO3 в смеси с H2SO, (в %) 5 10 20 60 90 100
Степень превращения HNO3 в NO * (в %) 100 100 62,5 16,7 5,9 1
100%-ная азотная к-та содержит ок. 1% Н.-и., обра-
зующегося по ур-нию:
HNO3 + HNO3 7Z* NO2+ +NOs" + H2O
Н.-и. имеет линейное симметричное строение: оба
атома кислорода в нем эквивалентны.
Н.?и. является основным нитрующим агентом при
нитровании ароматич. соединений с помощью конц.
азотной к-ты или нитрующих смесей. Для нитрования
применяют также индивидуальные соли нитрония.
Взаимодействие олефинов с азотным ангидридом и
сопряженное фторнитрование олефинов также осуще-
ствляются с промежуточным образованием Н.-и. См.
также Нитрование органических соединений.
Лит.: Ворожцов Н. Н., Основы промежуточных про-
дуктов и красителей, 4 изд., М., 1955 с. 125; Kuhn 8. J.,
О 1 a h G. A., J. Amer. Chem. Soc., 1961, 83, № 22, 4564.
Е. М. Рохлин.
НИТРОНОВЫЕ КИСЛОТЫ (ацинитросоединения,
изонитросоединения) — кислотная (аци) форма жир-
ных или жирноароматич. первичных и вторичных
нитросоединений. Н. к. находятся в таутомерном рав-
новесии с соответствующими нормальными нитросое-
динениями:
z-O /°
CH3CH2N< ch,ch=n/
4 он
сн3.
>СН—N
СН/
/°
'о
сн3. ,о
)C = N/
СН/ 4 он
Для ряда нитропарафинов определено количество Н. к.,
находящейся в таутомерном равновесии. Для этой цели кон-
дуктометрически устанавливают значение константы кислот-
ности ациформы (Каци), это удается вследствие того, что ско-
рость превращения данной формы достаточно мала. Затем
находят константу кислотности нормальной формы (КнорМ),
к-рую выводят из конечного постоянного значения первого,
определения. Отношение -Кнорм к каци Дает содержание И.к.
Таким путем были найдены следующие результаты (при 25“
разб. водных р-рах):
^аци ^норм Количество ациформы, % CH3NO2 C2H5NO. u30-C3H?NO2
5,6-10-* 6,1-10-” 0,00001 1 3.9-10-5 3,5-10-» 0,0089 7,7-10-» 2,1-10-» 0,275
Наиболее известными производными Н. к. являются
их соли со щелочными металлами, образующиеся при
взаимодействии первичных или вторичных нитросоеди-
нений со щелочами или алкоголятами, твердые про-
дукты, устойчивые при комнатной темп-ре, но взры-
вающиеся при нагревании. Первичные и вторичные
нитросоединения при —(30— 40°) в жидком аммиаке
образуют аммониевые соли, к-рые при 0—10° разла-
гаются с выделением аммиака и регенерацией нитро-
соединений. Известны и другие соли Н. к., напр.
ртутные. Натриевая соль нитрометана с сулемой,
образует неустойчивую ртутную соль, превращаю-
щуюся в гремучую ртуть:
2 CH2 = NO2Na + HgCl2 —>-(CH2=NO3) Hg —> Hg (ONC)2
Соли H. к. легко вступают в реакцию с галогенами:
R' R'
I zO |
R-C = N" +Br2 —>R-C-NO2 + NaBr
'ONa |
Br
Эта реакция используется для получения а-гало-
генонитросоединений, т. к. сами нитропарафины непо-
средственно не галогенируются. В случае первичных
нитросоединений при реакции с избытком галогена
образуются дигалогенные соединения ИСВггМОг,.
в случае вторичных — моногалогенные соединения;
531
НИТРОНОВЫЕ КИСЛОТЫ—НИТРОНЫ
532
реакция используется для различия первичных
нитросоединений от вторичных.
Соли Н. к., реагируя с карбонильными соедине-
ниями, образуют fJ-нитроспирты, из к-рых далее по-
лучают а, P-непредельные нитросоединения:
R-C
Н
?
R-C—CH2-N
I *
н О
Na +
ОН
I
—- r-chch2-no2 u ~ r-ch=ch-no,
-П2О
Н. к. получают при подкислении их щелочных солей
вычисленным количеством минеральной к-ты. Чисто
жирные Н. к. не выделены в индивидуальном виде,
т. к. они легко превращаются в нитроалканы. Жирно-
яроматич. Н. к., напр. ациформу фенилнитрометана,
удалось получить в индивидуальном виде (III):
ш
Соединение III — кристаллы, т. пл. 84°, а его тауто-
мерная нитроформа (I) — жидкость, т. кип. 225—227°.
Н. к., в отличие от таутомерных им нитроалканов,
электропроводны, дают интенсивную окраску с хлор-
ным железом и выделяют углекислый газ из водных
р-ров бикарбоната натрия. Н. к. при взаимодействии
с холодными минеральными к-тами образуют альде-
гиды и соответственно кетоны (Нефа реакция).
В последнее время эта реакция часто применяется
для наращивания цепи в моносахаридах (Саудена —
Фишера реакция), напр. для синтеза гексоз из араби-
нозы:
ОНО CHaNO„
но- -ОН
— — он chsno2 но—
-он » - -ОН +
-он
1Н20Н Ш20Н
zO
г IH2NO2- с H = N" 'ONa 2Н0
но— — -ОН — — ОН
но — но— - 110-
—он -ОН — — ОН
— -он — -ОН — —он
СНгОН- СН2ОН СН2ОН
Аммониевые соли H. к. восстанавливаются в ок-
симы. Так, нитроциклогексан с выходом 85—90% гид-
рируется в циклогексаноноксим в присутствии жид-
кого аммиака над медным катализатором. Этот непре-
рывный процесс имеет технич. значение при получении
капролактама.
С диазометаном Н. к. образуют метиловые эфиры
(метилнитронаты), представляющие собой жидкости,
перегоняющиеся в вакууме:
//О ‘ /О
R-CH = N< +CH2N,—>RCH=N<; +N2
'OH ‘ XOCH3
Аналогичные эфиры получаются при алкилировании
натриевых солей Н. к. галогенными алкилами или
диалкилсульфатами; наряду с ними образуются гомо-
логи нитросоединений (С- и О-алкилирование):
R'
-RCH-N^°
7/О 7 'О
RCH=NZ +R'X(
ONa \ .0
*R-CH = N"
Ч-R'
Алкплнитронаты распадаются при нагревании на ок-
симы и альдегиды:
,0 t°
R-CH = N<' —>R-CH=N-0H + 0 = CH2
осн,
Н. к. с нитропарафинами образуют диоксимы нитро-
Р-дикетонов, превращающиеся далее в триалкилизок-
сазолы:
о NOH NOH
Z -2Н..0 II II
2₽CH = Nx + RCH2NO2 с C-R —
ХОН RX V
О 'NO2
NOH NOH R-r---------rR
‘-HOSOZ A I — “ Lr
R CH XR
z
R
Э. E. Нифантъев.
НИТРОНЫ — окиси иминов, содержащие группи-
ровку R
х I
^C = N—>0
Нитроны (I) — кристаллы; обладают слабыми основ-
ными свойствами, с кислотами образуют непрочные
соли. Первоначально им приписывалось неправильное
циклич. строение (II), в последние годы получен и
подробно исследован ряд соединений типа II (изонит-
ронов), к-рые оказались совершенно отличными от Н.
R . t U'N-R"
N Г = X- — П" R'Z
При обработке сильными к-тами (напр., 85%-ной сер-
ной к-той) первичные ацинитросоединения превра-
щаются в гидроксамовые к-ты, к-рые далее гидроли-
зуются до карбоновых к-т, что является одним из
промышленных методов получения карбоновых к-т и
гидроксиламина:
,0 н+ ,0 ,0
В-CH = N<y —>R-C^' -->R-C<f + NH20H
'OH 'NHOH H20 'ОН
Действие безводного НС1 на Н. к. приводит к а-
тслорнитрозосоединениям часто с хорошими выходами:
R\ z°
CH-N
R'Z 40
Rx
------ XC-N = O
' 1
R Cl
Изонитроны — жидкости с темп-рами кипения гораздо
более низкими, чем у соответствующих Н.; ИК-спект-
ры этих классов соединений также резко различаются.
Алифатич. и алициклич. Н. существуют обычно в
виде линейных или циклич. димеров:
Исключение составляют устойчивые разветвленные
циклич. Н. типа I:
(СН3)2С CH2 Zn,H2o
СН2 СО NH4CI
I I
NO2 CH3
(СН3)2С—сн2
^2 С-х. ~ СН з
4 о
I
533
НИТРОНЫ—НИТРОПРОПАН
534
Ароматич. Н. — устойчивые соединения.
Н. реагируют со многими соединениями как бипо-
лярные ионы с зарядами в положениях 1,3. Примера-
ми являются реакции с гриньяровскжми соединениями
(1), цианистым калием (2), изоцианатами (3), олефи-
нами (4); особенно легко, уже при комнатной темп-ре,
взаимодействуют с Н. соединения, содержащие акти-
вированную двойную связь (5):
(С6Н5)2СН
евН5СН-N-СН2С6Н5 -I- C6H5MgBr —- JN-OMgBr(I)
О" С6Н5СН2
CN CN
RCH-N-С6Н5 + KCN—- RCH-N-C6H5—*R-C=NCeH5 (2)
О- ок
свн5-£н + N-свн5 С6Н5-СН—N-C6H5 (3)
R-N сх r-n^c=o
'о- ''о
С8Н,-CH CH2 CeH5~CH—CH (4)
5 I + II —»- I I
c6h5-n ch- c6h5 c8h5-n^ch-c6h5
xcr
н3с г j+ itH2 ~(5)
HoC^r CHCOOC2H5 H3C^N^
3 1 | [
A о—l-cooc2h5
Изонитроны содержат активный атом кислорода, бла-
годаря чему они количественно выделяют иод из K.J;
Н. не окисляют K.J. Изонитроны легко восстанавли-
ваются в амины или в анилы (6); Н.восстанавливаются
гораздо труднее, причемобразуются замещенные гидро-
ксиламины (7). Изонитроны очень легко, иногда уже
при комнатной темп-ре, перегруппировываются в ами-
ды к-т (8); в случае Н. подобная реакция имеет место
лишь при действии катализаторов, обычных для пе-
регруппировки Бекмана (9). Изонитроны устойчивы
ж водным щелочам, но при действии кислот они быстро
разлагаются на сложную смесь продуктов; Н. гидро-
лизуются щелочами и кислотами, причем образуются
карбонильное соединение и замещенный гидроксил-
амин (см. Крёнке реакция) (10). Н. окисляются легче,
чем изонитроны, в обоих случаях образуются карбо-
нильное соединение и нитрозосоединение (И). Н. и
изонитроны способны превращаться друг в друга:
Н. изомеризуются в изонитроны при облучении УФ-
светом, обратный процесс протекает при нагревании
до 60-100° (12).
о Н
/ \ ,-----> RCH2NHR'
R-CH-NR'A (СеН5)3Р
х------> RCH = NR'
О
1 Н ZOH
RCH = N-R' —> RCH2N<
R'
R . /° R'
)C-N-R"—>RC—Nf
R'Z\Z 4R"
О
R 4 (CH,CO)2O ,R'
)C = NR''---► RCONf
R'Z | XR"
О
(6)
(9)
Н к Н2О R ,
)C = NR" _rz/ ; н+ о о R ч t )G = N-R" - R'z ► ;c=O4-r"nhoh ,2(b или НО- R'z uu-’ О ^R
О В ч /\ >G—N-R"- R'z ZC = O + R" —NO (И) °
R v hv R.
)C = N-R"TZ± JC-N-R" (12)
R'z 1 Д R'z \Z
О О
Основные методы синтеза Н. — конденсация карбо-
нильных соединений с N-замещенными гидроксил-
аминами: R'
R2G = 0 + R'NH0H —>-r2c = n/
*0
действие алкилирующих агентов на нитрозосоеди-
нения:
RNO + R2CN2
O-N
R-< ||
zc-n
r'z V
C6H5NO 4- O2N-C6H4-CH2C1---- O2NCbH4-CH=N-CeHb (6)
о
(CH3),,NC8H4NO + N+-CH2R — RCH = N-C6H/,N(CH3)2 (b)
.0
дегидрирование и окисление N, N -дизамещенных
гидроксиламинов:
он о
I HgO t
С„Н5СН2-N —CH (C»H5)2--->C,H5CH = N-CH (CeH5)2
он о
I (СН3)з ссоон t
rch2-n-ch2r------------>rch=n-ch2r
Меньшее значение имеют синтезы Н. N-алкилированием
оксимов, конденсацией ацетиленов с нитрозосоединениями,
действием азотистой к-ты на эфиры фенолов, восстановлением
нитробензальдегидов.
Основной метод получения изонитронов — окисле-
ние иминов надкислотами:
о
R R s RCOOOH R ч /\
vC = 0 + HoNR"—► )С= NR"--------► ;G-N-RW
R'z ' R" R'z
Благодаря доступности и многообразию превраще-
ний Н. изонитроны находят все более широкое при-
менение в органич. синтезе.
Лит.: Smith L. I., Chem. Revs, 1938, 23, 224; К г i m m
H Chem. Вег., 1958, 91, № 5, 1057; G r a s h e у R., H u 1-
s g’e n R., Leitermani) H., Tertrahedron Letters, 1960,
№ 12, 9; Mare N. E., Coppinger G. M., J. Org. Chem.,
1963, 28, 1068. H. П. Гамбарян.
НИТРОПАР АФИНЫ — см. Нитросоединения али-
фатические.
НИТРОПРОПАН C,H,NOS, мол. в. 89,09. Извест-
ны 2 изомера: 1-нитропропан CH,CH2CH2NO2 и
2-нитропропан (CH3)2CHNO2. Н. — бесцветные жид-
кости с характерным запахом. Ниже приведены их
физич. свойства:
(7)
(8)
1-нитропропан 2-нитропропан
T. пл., °C — 103,99 -91.32
Т. кип., °С;мм 131 ,18/760 20/7,52 120,25/760
20/12,99
1,00144 0,98839
Теплота парообразования 1,40160 1,39439
(25°), ккал/моль 10.37 9.88
Крит, темп-ра, °C Диэлектрич. проницаемость 402,0 344,7
(30°) 23,24 25,52
Дипольный момент, D . . 3,57 3,73
Вязкость (25°), спуаз . . . Поверхностное натяжение, 0,798 0,750
дин iCM ......... 29,28 (25°) 29,08 (21°)
Т. восп., °C Растворимость (20°), об. °/с 48.9 39,4
Н. в воде 1,4 0,5 • 1,7
Воды в Н 0,6
535
НИТРОСОЕДИНЕНИЯ АЛИФАТИЧЕСКИЕ
536
Н. дают минимальные азеотропные смеси с водой,
Н. хорошо растворимы в большинстве органич. ра-
створителей. Химич, свойства Н. — обычные для нит-
ропарафинов (см. Нитросоединения алифатические).
Смесь Н. и нитропарафинов с меньшим числом атомов
углерода получают в пром-сти нитрованием пропана
в паровой фазе и разделяют на компоненты ректифи-
кацией. В лаборатории Н. можно синтезировать обыч-
ными методами получения нитропарафинов. Н. при-
меняют в качестве растворителей, для синтеза нитро-
спиртов. Допустимая концентрация паров Н. в воз-
духе 0,005%.
Лит. см. при ст. Нитрометан. Е. М. Рохлин.
НИТРОСОЕДИНЕНИЯ АЛИФАТИЧЕСКИЕ — со-
единения, содержащие нитрогруппу — NZ°npn атоме
углерода алифатич. цепи. Сюда относятся нитропара-
фины, нитроолефины и различные их функциональные
производные, содержащие, помимо нитрогруппы, дру-
гие заместители. В зависимости от количества нитро-
групп в молекуле различают моно-, ди- и полинитро-
соединения. Основным и наиболее важным классом
среди Н. а. являются нитропарафины. Физич. свой-
ства основных моно-, ди- и полинитропарафинов при-
ведены в табл. 1 и 2.
Таблица 1
Мононитропарафины Т. пл.,°C Т. кип., °C “Г п20 nD
Нитрометан CH3NO2 —28.55 101,186 1,13816 1,38188
Нитроэтан C2H5NO2 -89,52 114,07 1,05057 1,39193
1-Нитропропан CH3CH2CH2NO2 — 103,99 131,18 1.00144 1,40160
2-Нитропропан (СН3)2 chno2 — 91 ,32 120,25 0,98839 1,39439
1-Нитробутан CH3CH2CH,CH2NO2 — 81.33 152,77 0,97344 1,41019
2-Нитробутан СН3 . >chno2 С2 н5 — 132 139,50 0,96535 1,40407
1-Нитроизобутан СН3. )CHCH2NO2 сн,х — 76,85 141,72 0,96349 1,40642
2-Нитроизобутан (CH,)8CNO2 26,23 127,16 0,95028 (30°) 1,39715 (30°)
1-Нитропентан СН3 (СН2). no2 — 172,5 0,9525 1,41751
1-Нитрогексан СН3 (СН2)5 no2 — 81 , 5/15 0,9390 1,42346
Таблица 2
Полинитропара- фины3 Т. пл.,°C Т. кип., °C 1мм rf23 20 nD
Нитроформ HC(NO2)3 25 45—47/22 1,615
Тетранитрометан . С (NO2). 13,9 125,7/760 1,6306 1,4384
1,1-Динитроэтан СН3СН (NO2)2 — 185 — 186/760 1,35 1,4346
1,2-Динитроэтан O2NCH2CH2NO2 39-40 135/6 1,45 1,4488
1,1,1 -Тринитроэтан СН3С (NO2)9 57 68/17 — —
1,1 -Динитропропан СН3СН2СН (NO2)2 -42 100/30 1.2610 1,4339
2,2-Динитропропан (СН3)2 С (NO2)2 53 185,5/760 — —
1,3-Динитропропан O2NCH2CH2CH2NO2 -20 — 1,354 1,4654
а Динитрометан в свободном виде неустойчив.
Для введения нитрогруппы в алифатич. соединения
чаще всего пользуются азотной к-той, окислами азота
и солями азотистой к-ты. Для нитрования парафинов
и других алифатич. соединений обычно применяют
разб. азотную к-ту. Для достижения необходимой
темп-ры при нитровании, особенно в случае низших
парафинов, реакцию проводят под давлением (Ко-
новалова реакция):
СН3. ,СН3
>СН-СН2-СН2-СН(
СН3Х ^СН,
13"/0HNO,
110°
сн3ч
сн/
.сн.
-СН2-СН2-СН(
хсн,
с------>-
no2
Иногда для введения нитрогруппы в легко нитрую-
щиеся соединения их обрабатывают конц. азотиой
к-той при комнатной темп-ре:
zCO-NHx
н2/ 'со
4 СО-NHZ
HNO3 Ю0%-»ая
аыход 85-907.
со-кн^
o2nhcz 'со
CO-NH
Наиболее важный технич. метод получения низших
нитропарафинов — нитрование в паровой фазе пра
400—450° (по X. Б. Хэссу):
HNO3
СН3СН2СН2СН3 —> СН3СНСН2СН3 (50%) +
420° |
NO2
+ СН3СН2СН.СН2 (27%) + CH3CH2CH2NO2 (5%) +
I
NO.
+ CH3CH2NO2 (12%) + CH3NO2 (6%)
Вместо паров азотной к-ты при парофазном нитро-
вании можно применять также окислы азота:
NO3
СН3СН2СН3------>- СН3СНСН3 (55»/„)+CH3CH2CH2NO2 (15%)+-
510° I
NO2
+ CH3CH2NO2 (15%) + CH3NO2 (15%)
H. а. можно получить также присоединением азот-
ной к-ты, окислов азота, хлористого нитрила к нена-
сыщенным соединениям:
С»Н3. HNO3 С»Н5.
)С=СН2------>C-CH2NO2
С3Н3 H2SO. С»Н3 I
он
no2 no2
C12C = CC12--->C12C - CC12; C,H5C sCH —>C»H3C = CH-
800; p I I II
NO2 NO2 NO3 Ж
NO2C1 Br
BrCH = CH2——-+ >ch-ch2no2
cr
Фторсодержащие H. а. получают из олефинов взаимо-
действием с азотной к-той в безводном фтористо!»
водороде (сопряженное фторнитрование):
HNO3
СС12 = СН2--->-CCI2F —CH2NO2
HF
Широко распространен в лабораторной практике’
метод получения Н. а. алкилированием солей азоти-
стой к-ты. Впервые нитропарафины получены взаимо-
действием алкилиодидов с нитритом серебра (В. Мейер,
1872). Побочно при этом образуются алкилнитриты:
RJ + AgNO2 —> RNO2 + RONO
Подобным образом можно получать не только нитро-
парафины, но и другие Н. а.:
CH2 = CHCH2J + AgNO2 —»CH2 = CHCH2NO2
HOCH2CH,J + AgNO2 —> HOCH2CH2NO2
В качестве алкилирующих средств могут применяться
другие галогениды, а также алкилсульфаты, причем
537
НИТРОСОЕДПНЕНИЯ АЛИФАТИЧЕСКИЕ
538
вместо нитрата серебра можно брать нитриты щелоч-
ных металлов:
HCON (СН,)3
CH3CHCOOC,H5 + NaNO3----------* СН.СН-СООС2Н5
I NH.CONHj I
Вг NO3
(CH3O)2SO2 + KNO2 —>CH3NO2
Особенно легко нитриты калия и натрия алкили-
руются солями а-галогенкарбоно вых к-т. При этом од-
новременно происходит декарбоксилирование (способ
Кольбе):
к NO.,
С1СН2СООК —> CH3NO2
Иногда нитрогруппа в молекуле алифатич, соеди-
нения может образоваться в результате окисления
других азотсодержащих группировок, напр.:
(СН3)3 CNH,-----!--► (СН,)з CN03
Н2О; 50-60° выход S3°,o
Характерная реакционная способность Н. а. обус-
ловливается как склонностью нитрогруппы к различ-
ным превращениям, так и влиянием ее на другие реак-
ционные центры в молекуле. Нитрогруппу в Н, а.
можно восстановить в нитрозо-, гидрокснлампнную
или аминогруппу (1); нитрогруппа может отщеплять-
ся в виде нитрит-иона (2), в нек-рых случаях может
происходить присоединение по двойной связи Х = О
(3):
I (Н) I (Н) I (Н) I
~C-NO3 —> -C-NO —> -C-NHOH —> -C-NHa (1)
i 1 I
I I
-C-NO,—>-C++NO2 (2)
i Z° RMgX I 7 /OMgX RMgX
— C-N\------ -C-N\ ---->
I No I NO
R R
II II
—>—C-N-OMgX —->-C-N-OH (3)
I I
Влияние нитрогруппы на другие реакционные цент-
ры связано гл, обр. с ее большим электронооттягиваю-
щим эффектом. В связи с этим нитрогруипа активи-
рует сопряженную с ней олефиновую связь (4), сооб-
щает подвижность а-атомам водорода (5) и галогена
(6), обусловливает легкость декарбоксилирования
а-нитрокарбоновых к-т (7):
х«+ z ~ +:в- . ।
-С=А (4)
\ В О
а
I С° -н+ X +х0’
---------- ZC = N (5)
Н О '(Г
|^+ -’В- :
ci о в о
^Д| N / С® -н+ s, +/Q
Н~О—C-tCtN —л- C=nC (,)
II О \ -со2 / n
0 0
Н. а., содержащие атомы водорода в а-положении
к нитрогруппе (напр., первичные и вторичные нитро-
парафины), являются псевдокислотами; они раство-
ряются в щелочах с образованием солей нитроновых
кислот;
огг
-нао
* н о
Rx +/0
C=N
r'
При подкислении полученных щелочных р-ров обра-
зуются более или менее устойчивые свободные нитро-
новые к-ты (аци-нитросоединения, изонитросоедине-
ния), к-рые затем перегруппировываются обратно в
Н. а. Таким образом можно отделить первичные и
вторичные нитропарафины от третичных, к-рые не
реагируют со щелочами. Однако при подкислении
р-ров щелочных солей нитроновых к-т часто наблюда-
ются побочные реакции, к-рые при определенных ус-
ловиях могут стать основными. Так, при действии теп-
лых разб. минеральных к-т эти соли превращаются
в альдегиды и кетоны (Нефа реакция):
Rx лО Н+ Rx
>C = N< —» ^C=0+N,0
R xONa R
При продолжительном действии конц. щелочей на
Н. а. происходят разнообразные превращения, вклю-
чающие конденсацию с образованием новой С—С-
связи, отщепление нитрогруппы, окислительно-вос-
становительные реакции:
о о
кон л л
2CH3NO2—>HON=CHCH=N —> KOCOCH = N
\ N
OK OK
CH3CH2NO2
CH3 CH3 CH3
ch-c-ch
> NO NO2 NO
CH3
CH3zCH zch3
xcz 'c
II II
HON О
CH3 CH3 CH3
—— c-ch-c^
HON NOH
CH3
CH3-C'C4-CH3
N------О
+ NaOH
CH3CH — CH -----—> CH3CH=CHNO3
I I -NaNO3
NOj NO, - ll2O
zNO2 +2KO1I /°
CH3C-NO3 —----' CH3C=N
\N-n -KNO, I \K
4№3 _щО no, 0K
Rx ZH IIO- Rv R.
-----► C--O+ 'C-NOH+
Rz '\NO3 Rz R'
R4 R
+ 'ZC — c-
Rz I i ll
NOj NO,
При действии конц. соляной, серной или фосфор-
ной к-т на первичные Н. а. образуются гидроксамо-
вые к-ты, к-рые затем опыляются до карбоновых к-т
и солей гидроксиламина:
z/O /011 +11ч-
R-CH3-N< J±R-CH=Nx------------->
NO NO -H2O
4- +HX + H 2 о
—>-R-CH = N = O---> R-CH-NO----► R-CI1-NO —>
-HT I -HX I
X OH
+ H2O /О
—>-R-C = NOH----»-R-C<’ +H.N0II
I 'OK
OH
По этой реакции из нитроэтана в пром-сти получают
уксусную к-ту и гидроксилампн. Галогенированием
первичных и вторичных Н. а. в присутствии осно-
ваний получают соответствующие а-галогенопроиз-
водные:
NaClO
CH3N03 —г CCljNOj
/° Вг,
CH3CH = Nx —> CH3CHBrNOa—> CH3CBraNOa
NQK
539
НИТРОСОЕДИНЕНИЯ АЛИФАТИЧЕСКИЕ
540
Хлорирование нитропарафинов в условиях обра-
зования радикалов (при облучении УФ-светом, в
отсутствие оснований) приводит к замене хлором ато-
мов водорода, более удаленных от нитрогруппы (на-
чиная с fj-атомов):
С12
ch3ch2ch2ch2no2 —» C1CH2CH2CH2CH2NO2 +
hv
+ch3chch2ch2no2+ch3ch2chch2no2
Cl Cl
Атомы галогена, занимающие а-положение по от-
ношению к нитрогруппе в молекуле Н. а., обладают
повышенной реакционной способностью:
сн3. ,ci сн3. ло сн3ч zcir3
'IY и, Г'=\/ --> -- C.z
сн/ xno2 сн/ ХО№ СН/>1о, 1 4 С н. no2
,С1 сн3сн/ KNO2 .NO2 >сн3сн;
,С1 [ОН-]
СН3СН(---------»-СН3С = ССН3
4N0, | |
N02 no2
Характерной реакцией, позволяющей различить
первичные, вторичные и третичные Н. а., является
нитрозирование. При действии HNO2 на первичные
Н. а. образуются нптроловые к-ты,*растворимые в
щелочах с кроваво-красным окрашиванием; вторичные
Н. а. превращаются в нейтральные псевдонитролы,
бесцветные в твердом виде, но синие в жидком или
растворенном состоянии; третичные Н. а. вообще не
реагируют с HNO2:
rch2no2
r2chno2
r3cno2
NO HO- z/NO-
—>-НСН/ ---------->R — C<
xno2 ^no2
NaNO3 NO
-----► —>R2C<
H2SO, xN03
—>- не реагирует
При окислении первичных и вторичных Н. а. обра-
зуются, помимо других соединений, альдегиды и ке-
тоны:
R ч ,н н3О2 R .
;с<---------» >с=0
r,z ^no2 r,z
Восстановление Н. а. при сохранении атома азота
в молекуле в конечном счете приводит к получению
алифатич. аминов. При этом применяют самые различ-
ные восстановительные агенты: в пром-сти — железо
в присутствии соляной к-ты и водород над никелем
Ренея, в лаборатории — алюмогидрид лития, металлы
(олово, цинк, свинец, железо) в присутствии кислот
(соляной, серной, уксусной), амальгамы натрия и
алюминия, водород над платиной или палладием,
гидросульфит натрия, треххлористый титан:
[HJ
rno2 —>rnh2
Иногда восстановление Н. а. можно остановить на
промежуточной стадии, при этом образуются оксимы
либо алкилгидроксиламины:
Zn
RCH2NO2---------->-RCH = N0H
СН3СООН
электрическое
RN02-------------► RNHOH
восстановление
Алкилирование солей нитроновых к-т приводит как
к С-, так и к 0-алкилпроизводным; последние обычно
легко распадаются, образуя оксимы и альдегиды!
,N03
CH<N/°
^OAg
CH3J сн,х znto2
сн/ 4no2
CH,. п-СН3-С«НЧ — СН2Вг
)C = N< —-------————►
СН/ \ONa
—► n-CH3-C6H1-CH2ON = C/ —►
4- ЧСН3
о
снзх
—>n-CH3-CsH,-CHO + ;c = noh
сн/
При взаимодействии Н. а. с металлоорганич. сое-
динениями образуются производные гидроксиламина:
C2H5MgJC2H5. ,с,н5
CH3CH,NO2--------► ;NOH + CH3CH;
C2H5Z )N0H
С2Н5Х
Среди реакций Н. а., связанных с подвижностью,
а-атома водорода, следует упомянуть Манниха реак-
цию (8), азосочетание с ароматич. диазосоединениями
(9), конденсации типа Михаэля реакции с ненасыщен-
ными соединениями, молекулы к-рых содержат акти-
вированную двойную связь (10), а также карбокси-
лирование Н. а. (11):
.СН2СН2.
CH3NO2 + 2HOCH,N< /н2—>-
ХСН2СН/
.СН.СН-.
,ch2n; >сн2
п nctt СН2СЫ3'
O2NCH СН,СН2.
СП,./ )СН2
\ch,chZ
СН2 =n/"0 +C,H,N3X—»-C3H-,N = N-C = NNHC,eH.
\ONa I
(8>
(9>
NO;
CH3NO3 + CH3 = CHCN —>O2NC (CH2CH2CN)3 (10>
OCOOCH3 RCHCOOH
RCH3NO3 + Mg( —► I
XOCOOCH3 NO,
(11>
Конденсация H. а. с альдегидами или кетонами при-
водит к получению нитроспиртов. В присутствии ще-
лочных (иногда кислых) катализаторов в зависимости
от исходных продуктов и условий реакции образуются
нитроспирты, динитропарафины и т. д.
сн2о
CH3NO3-----> HOCH2CHaNO3
(СН3),СО
CH3NO2------->(СН3)2 С (CH2NO2)2
Дигидратацией нитроспиртов получают нитроолефиныг
r-ch-ch2no2 —>rgh=chno2
I
он
Алпфатич. нитроолефины представляют собой зе-
леновато-желтые жидкости; низшие члены этого ря-
да имеют резкий запах и являются лакриматорами.
Физич. свойства нек-рых нитроолефинов приведены
в табл. 3.
Сопряженные нитроолефины присоединяют различ-
ные нуклеофильные реагенты, образуя соответствую-
щие Р-нитронроизводные:
R. ,R R. ,R
)С = С< +НХ —► )с— с/
RZ XNO2 RZ I I H
X NO,
X=R'O, R'S, SO3Na, CN, r'n, SH, C (NO3)3 и т. д.
541
НИТРОСОЕДИНЕНИЯ АРОМАТИЧЕСКИЕ
542:
Таблица 3
Нитроолефин Т. кип., °С/ЛШ d (t° С)
Нитроэтилен ch2=chno2 98,5/760 38—39/80 1,073(13,8) —
1-Нитропропен CH3CH = CHNO3 37/10 1,0650(20) 1 , 4539 (20°)
2-Нитропропен СН3С=СН3 4no2 58/90 1,0660(20) 1,4292 (23°)
3-Нитропропен O2NCH2CH=CH2 125—130/760 87—89/1 80 1,051(21) —
со-Нитростирола C6H5CH = CHNO2 149,5/14 — —
а Т. пл. 58°.
Активированная двойная связь делает возможным
участие нитроолефинов в Дильса —Альдера реакции в
качестве диенофилов:
Сн-R
Ch-no2
Алифатич. нитроолефины легко полимеризуются в
присутствии влаги и оснований. Нек-рые нитроолефи-
ны обладают свойствами инсектицидов, фунгицидов,
дезинфицирующих и консервирующих средств. Нитро-
олефины используют как полупродукты для синтеза
различных нитро- и аминосоединенпй. Мононитропа-
рафины имеют большое значение как промежуточные
продукты в синтезе разнообразных веществ, их также
применяют в качестве растворителей для красителей,
смол, каучуков и т. д. Нек-рые полинитропарафины
применяют в качестве окислителей в ракетном топ-
ливе. Галогеносодержащие Н. а., напр. трихлорнит-
рометан, применяют в качестве инсектицидов и фун-
гицидов.
Н. а. приобретают все большее значение как сами
по себе, так и в качестве промежуточных продуктов
для получения аминов, кетонов, альдегидов, карбо-
новых к-т и т. д.
См. также Нитрование органических соединений,
Коновалова реакция, Нефа реакция, Нитроспирты
алифатические, Нитрометан, Нитропропан, Нит-
роциклогексан, Нитроэтан, Нитроэтилен, Тетранит-
рометан, Трихлорнитрометан.
Лит.: А з и н г е р Ф., Химия и технология парафиновых
углеводородов, пер. с нем., М., 1959, с. 265; Перекал ин
В. В., Непредельные нитросоединения, Л., 1961.
Е. М. Рохлин.
НИТРОСОЕДИНЕНИЯ АРОМАТИЧЕСКИЕ — со-
единения, содержащие нитрогруппу в ядре ArNO2—
собственно ароматич. Н., или же в боковой цепи —
жирноароматические. Примером первых могут слу-
жить нитробензол C6H5NO2 или пара-нитротолуол
CH3C6H4NO2; примером вторых — фенилнитрометан
CeH5CH2NO2, изомерный пара-нитротолуолу. Немно-
гие Н. а. имеют смешанный характер, как, напр., пара-
нитрофенилнитрометан 1,4-O2N—С6Н4—СН2—NO2.
Чисто ароматич. Н. относятся к третичным, а жирно-
ароматич. Н. могут быть различных типов. По числу
нитрогрупп различают моно-, ди-, тринитросоедине-
ния и т. д.
Н. а. обычно получают нитрованием ароматич.
углеводородов АгН (см. Нитрование органических
соединений). Нафталин, антрацен и подобные им мно-
гоядерные углеводороды легко нитруются водной
HNOs в присутствии двуокиси азота. Бензол и алкил-
бензолы обычно нитруют смесью серной и азотной к-т.
Иногда, в частности при получении полинитросоеди-
нений, нитрогруппу вводят в ядро посредством окис-
ления— NH2 и —NH—ОН-групп. Алкилбензолы в
зависимости от условий нитрования дают нитрозамс-
щенные в ядре или в a-положении парафиновой цепи.
Так, нитрование толуола HNOs в отсутствие окислов.
азота дает смесь нитротолуолов, а той же кислотой
в присутствии NO2 при 20° — фенилдинитроме-
тан CeH5CH(NO2)2, при 100° — фенилнитрометан
C5H5CH2NO2.
Мононитропроизводные бензола и его гомологов в.
чистом виде — бесцветные (обычно желтоватые) жид-
кости с миндальным запахом и сладким вкусом; мно-
гие пара-нитрозамещенные — твердые продукты.
Н. а. кипят более чем на 100° выше соответствующих
углеводородов; плотность > 1. Н. а. почти нераство-
римы в воде, являются хорошими, умеренно поляр-
ными растворителями для нек-рых неорганич. (А1С13,
H2SO4 и т. п.) и многих органич. веществ, особенно-
при нагревании, даже в тех случаях, когда обычные
растворители не применимы (индиго и т. п.). Мононит-
росоединения горят, но не взрывают, нек-рые из
них, в частности нитробензол, токсичны; нитротолуо-
лы в этом отношении, по-видимому, безопасны. Поли-
нитросоединения — кристалличны и обычно слабо-
окрашены вследствие присутствия примесей; они взры-
вают при ударе и детонации, нек-рые из них (динитро-
бензол, 2,4-динитрохлорбензол) вызывают дерматиты.
В Н. а. имеются обычная поляризация связи С—N02
и индуктивный эффект ее, резко уменьшающийся
по цепи атомов. Еще большую роль играет сопряже-
ние л:-электронов (I) или детальнее — на основе фор-
мулы Кекуле (II); оно несколько приближает струк-
туру ArNO2 к их пара и орто-хиноидным формам типа
III. Вследствие большей стабильности пара-хиноид-
Аг—N
хо
ных форм 6пара > 6орто и этот т.н. х иногенный
эффект сильнее проявляется в пара-положении.
Заместители в орто-положениях, напр. в 2-нитро-
мета-ксилоле, нитродуроле и их производных, нару-
шают копланарность и сопряжение нитрогруппы с яд-
ром, и в таких соединениях хиногенный эффект отсут-
ствует; напротив, обычная поляризация связи С—N02
в этих случаях даже усиливается.
Повышение электронной плотности у атомов кисло-
-S
рода a2-NO2 вызывает появление у Н. а. основных
свойств сравнительно с алифатич. нитросоединения-
ми. Так, Аг—NO2 растворяются в 70%-ной НС1О4илц
Г / о л +
конц. H2SO4 с образованием катионов Аг—N<f у
L xohj
с А1С1з дают комплексы типа Аг—NO2-----*-А1С13и т. п.
Хиногенный эффект в такого рода катионах и комплек-
сах еще более усиливается. Напротив, в полинитро-
производных ясно выражена склонность к образова-
нию аддуктов с электродонорными соединениями —
ароматич. углеводородами, аминами, эфирами фенолов
+6
по типу (ХОг)з С,Н,-*—-СвН5—N(CH3)2. Чем выше
вероятная степень смещения 6, тем глубже окрашен
комплекс. Эти аддукты находят применение для
очистки и идентификации АгН и в исследовании
проблем стереохимии. Продуктам присоединения ще-
лочей и алкоголятов к полинитросоединениям при-
дают пара-хиноидное строение, напр.
сн3о.
;CeH2(NO3)2 = NOOK
Hz
543
НИТРОСОЕДИНЕНИЯ АРОМАТИЧЕСКИЕ
544
Иногда в такого рода реакциях происходит вос-
становление группы = NOOH в оксимную = NOH, в
частности в реакциях СвН5—NO?.
Восстановление ArNO? в амины ArNH2 обычно про-
изводят посредством чугунной стружки в присутствии
слабого р-ра FeCl?, Н2 в газовой фазе с Си в ка-
честве катализатора или в жидкой фазе в присут-
ствии Ni под большим давлением. В нейтральной
среде реакция протекает через стадии возникновения
ArNO и Аг—NHOH; в щелочной среде последние
быстро образуют азоксисоединения, к-рые далее вое-
ArNO + HONH—Аг->Аг—N = N—Аг,
I
О
станавливаются в азо-Аг—N=N—Аг и гидразосое-
динения Аг—NH—NH—Аг и, наконец, в амины.
Полинитросоединения можно восстановить в нит-
роанилины действием сульфидов. Н. а. играют
роль окислителя в реакциях Скраупа, при получении
фуксина и др. Известно много случаев окисления нит-
рогруппой заместителей, чаще в орто-положении,
напр. превращение орто-нитротолуола при дейст-
вии КОН в антраниловую к-ту H2N—СвЩ—СООН, с
Вг2 при 170° — в дибромантраниловую к-ту, образо-
вание производных стильбена при нагревании щелоч-
ного р-ра 4-нитротолуола-2-сульфокислоты и т. и.
Общая поляризация ядра и хиногенный эффект нит-
рогруппы сильно затрудняют электрофильное замеще-
ние в Н. а. и приводят к ориентации преимущественно
в мета- и отчасти в орто-положения. Переход
ArNO2 в катионы или комплексы с А1С1з иногда не поз-
воляет его реализовать вовсе. Так, СвН5—NO2 инер-
тен к действию N2O4, даже при нагревании, и нитрует-
ся только крепкой HNO3 или ее смесью с H2SO4,
образуя мета-динитробензол с примесью 7—12% орто-
изомера. Для сульфирования применяют олеум или
HOSO2CI. При действии С1г или SOC12 на динитробен-
зол при нагревании происходит замещение нитрогруп-
пы галогеном. Реакция Фриделя-Крафтса, как пра-
вило, не приложима, т. к. образование ArNCh-AlCb
дезактивирует как ArNO2,TaK и А1С1з; хлорметилиро-
вание (СНгО и НС1) возможно в присутствии H2SO4
и т. п.
Поляризация ядра, возникающая от всех эффектов,
благоприятствует реакциям Н. а. с электродонорными
анионами (RO-,НО", CN-, SO2-H т.п.) и молекулами
(RNH2, NH3, Н2О и т. п.), при этом обычно пре-
обладает влияние хиногенного эффекта в орто- и
пара-положениях. В случае орто- и пара-производных
X—Аг—NO2 это т. н. нуклеофильное замещение на-
чинается с присоединения, напр.НО", возникновения
переходного комплекса
НО"
и заканчи-
вается отщеплением X- с образованием нитрофенола
НО—Аг—NO2. Нуклеофильное замещение легко про-
текает в случаях X, имеющих большое сродство к элек-
трону и способных играть роль аниона: —NO2, —С1,
—OR,—NH2 и т. п. Так, в орто- и пара-динитробензо-
лах при 100° одна из нитрогрупп может быть заме-
щена НО—, H2N—, СН3О — соответственно при реак-
циях с NaOH, NH3 и CHsONa, а тетранитробензолы
гидролизуются даже водой. В случае водорода (Х=Н)
отщепление в виде гидрид-иона Н- возможно только
в присутствии его акцепторов, т. е. окислителей, роль
к-рых может играть Аг—NO2. Так, 1,3,5-тринитро-
бензол окисляется в щелочном р-ре в пикриновую
к-ту, а-нитронафталин с NH2OH дает 1,4-аминонитро-
нафталин; при действии сухого КОН на CeH5NO2
образуется смесь орто- и пара-нитрофенолов, при
этом часть CeH5NO2 восстанавливается до азоксисое-
динения. При накоплении нитрогрупп обычная поля-
ризация связи С—NO2 и индуктивный эффект других
групп также могут привести к замещению, напри-
мер 1,3,5-тринитробензол при кипячении с CHsONa
в СНзОН превращается в 3,5-динитроанизол
СвНз^О2)?ОСНз. Н. а. в форме катионов [ArNO2H] +
или комплексов типа ArNO—О—*-SOs,благодаря очень
сильному проявлению в них хиногенного эффекта,
могут претерпевать в растворе H2SO4 перегруппиров-
ку в 1,4- и 1,2-нитрозофенолы или продукты их прев-
ращений (см., напр., динитронафталины); эта реак-
ция по своему механизму является своеобразным
нуклеофильным замещением и вместе с тем внутри-
молекулярным окислением-восстановлением.
Реакции ArNO2 со свободными R , Аг-, НО-, полу-
чаемыми, напр., при разложении перекисей ацилов,
протекают быстрее, чем с бензолом, и приводят к об-
разованию продуктов замещения водорода на эти
радикалы, гл. обр. в пара- и орто-положениях. Ори-
ентация при этом, т. н. радикальном замещении,
объясняется на основе учета стабильности промежу-
точных аддуктов. По-видимому, эти реакции проте-
кают не гладко; так, при действии R • обнаружено вос-
становление ArNO2 в Аг—N=O, к-рое особенно легко
реализуется внутримолекулярно при возникновении
радикалов типа орто-нитробензилаГ^О2—СвН4—СН2-.
В орто- и пара-нитроалкилбензолах
no3-c„h,-ch2r, no2-свн<-сн3
и особенно их полинитропроизводных в результате
сопряжения
а-атомы водорода приобретают в щелочной среде
способность отщепляться в виде протонов, а анио-
ны нитросоединений вступают в реакции конденса-
ции с электрофильными компонентами: альдегидами
СвН5СН=О, нитрозосоединениями Аг—N=O, щавеле-
вым эфиром и т. п. Особенно легко эти реакции идут
для 2,4,6-тринитротолуола. а-Нитропроизводные ал-
килбензолов типа С,Н5—СН2—NO2 отличаются от
жирных нитросоединений R—СН2—NO2 благодаря
сопряжению связи С= N с ядром, большей стабильно-
стью их ациформ Аг—CH=NOOH и анионов; поэтому
они могут быть выделены в кристаллич. виде. По той
же причине Аг—CH2NO2 обладают повышенными
кислотностью и реакционноспособностью и менее ус-
тойчивы при нагревании.
Общеизвестно влияние нитрогруппы, особенно в хи-
ногенных положениях, на химич. свойства других за-
местителей — повышение подвижности галогенов, ос-
лабление основных свойств аминогрупп, усиление
кислотного характера — ОН, —СООН и т. и. Наруше-
ния копланарности ослабляют это влияние. В орто-
нитропроизводных фенолов и аминов имеет место об-
разование внутримолекулярной водородной связи и
отсутствие межмолекулярной ассоциации по этому
типу; по этой причине орто-изомеры отличаются от
пара- и мета-нитропроизводных низкими темп-рами
плавления и кипения, лучшей растворимостью в ма-
лополярных жидкостях, интенсивной окраской и т. п.
Н. а. широко применяют для получения аминов, яв-
ляющихся исходными веществами в произ-ве кра-
сителей, ускорителей вулканизации и т. п., а также
для получения сернистых красителей, нигрозинов и ин-
дулинов. Многие полинитросоединения используются
как взрывчатые бризантные вещества, другие как
душистые (тринитро-третично-бутил-мета-ксилол —
ксилольный мускус и т. п., нитробензол — суррогат
миндального масла). В природе найдены нек-рые про-
изводные 9-нитрофенантрена и известный антибиотик
хлоромицетин — производное пара-нитрофенилпро-
545
НИТРОСОРБИД—НИТРОТОЛУОЛЫ
546
пана, один из стереоизомеров, идентичный синтетич.
левомицетину.
NO2 - С„Н, - СН(ОН) - CH(NHCOCHC12) - сн2он
Качественно Н. а. определяют восстановлением в
амины, диазотированием и азосочетанием. Для ко-
личественного определения используют восстанови-
тельное титрование Т1С1з и т. п. В отличие от мононит-
росоединений, ди- и трннитросоединения обычно да-
ют в растворе ацетона при действии щелочи соответ-
ственно фиолетово- и кроваво-красные окраски. Ана-
лиз смесей изомерных Н. а. удобно производить спект-
роскопии. методами.
Лит. см. при ст. Нитрование. А. И. Титов.
НИТРОСОРБИД (2,5-динитрат-1.4 : 3,6-диангидро-
D-сорбита) C8H8O8N2, мол. вес 236,14 — бесцвет-
ные кристаллы (из этанола), т.пл. 69—
71°, [а]р=134—139° (этанол), Н. почти
Н2С------1
HCONO2 О нераствОрим в воде, хорошо растворим
,--сн в большинстве органич. растворителей,
| ______I устойчив при длительном хранении; при
О HCONO,
i 1
1—сн2
быстром нагревании до 200° и выше Н.
взрывает. Н. получают нитрованием сор-
бида конц. HNO3 или смесью HNOa и
H2SO4. Н. применяют для лечения сер-
дечно-сосудистых заболеваний (стенокардии, гиперто-
нии, перемежающейся хромоты).
Лит.: Krantz J. С. [а. о.], J. Pharmacol, and Exptl.
Tlierap., 1939, 67, 187; К о ч e p г и н П. M., Титкова
Р. М., Мед. пром-сть, 1959, № 8, 18. П. М. Кочергин.
НИТРОСПИРТЫ АЛИФАТИЧЕСКИЕ — органи-
ческие соединения, содержащие нитрогруппы и окси-
группы при атомах углерода, алифатич. цепи; из-
вестны моно-, ди- и тринитроспирты, нитрогликоли
и т. д.
Одноатомные мононитроспирты — обычно жидкости,
ди- и тринитроспирты, а также нитрогликоли часто
представляют собой кристаллич. продукты. Физич.
свойства нек-рых одноатомных нитроспиртов представ-
лены в табл. 1:
Таблица 1
Мононитроспирты Т. кип., ° С 1мм d' 4 п20 nD
2-Нитроэтанол 02NCH2CH2OH 194°/7б5 102°/11 1,296 (20°) 1,443
2-Нитропропанол-1 ,no2 СН3СН< ХСН2ОН 99°/10 1 ,1841 (25°) 1,4379
1-Нитропропанол-2 .ОН СН3СН< xCH2NO2 86—89°/8 1,192 (20°) 1.441
2-Нитробутанол-1а .NO3 C2HSCH( СН2ОН 105°/10 1,1332 (25°) 1,4390
1-Нитробутанол-2 .ОН С2Н3СН( xCH2NO2 92°/10 — —
2-Нитро-2-метилпро- панол-1 б снзх zno2 CH3Z ''CHjOH 94,5— 95,5°/10
а Т. пл.—47—48°. 6 Т. пл. 90—91°.
В табл. 2 приведены темп-ры плавления некоторых
ди- и тринитроспиртов и нитрогликолей.
Нитроспирты можно ацилировать органич. или ми-
неральными к-тами. Так, напр., получают тринитрат
Таблица 2
Нитроспирты Т. пл., °C Нитроспирты Т. пл., °C
2,2-Динитроэтанол (O2N)2 СНСН20Н 2,2,2-Тринитроэта- нол (O2N)3CCH2OH 2-Нитро-1, 3-про- пандиол .СН2ОН O2NCH< ХСН2ОН 2-Нитро-2-метил-1, 3-пропандиол СН3. .СН.ОН )С< O2Nz хСН2ОН 2-3 36-37 56-58 147-149 2-Нитро-2-этил-1, 3-пропандиол С2НЗХ .СН2ОН O2Nz хСН2ОН 2,2-Динитро-1, 3-пропандиол 02N, 'СН2ОН )С< O2Nz хСН2ОН 2-Нитро-2-оксиме- тил-1,3-пропандиол хсн2он O2NC—СН2ОН ЧСН2ОН 56—57 142 165-170
2-нитро-2-оксиметил-1,3-пропандиола (взрывчатое ве-
щество):
/СНзОН HN0 zch2ono2
o2n—с—сн2он------+ o2n—с—ch2ono3
чсн2он H=so* 4ch2ono2
Нитроспирты или их сложные эфиры сравнительно
легко отщепляют воду либо кислоту, превращаясь в
сопряженные нитроолефины:
R—сн—ch2no2------► rch=chno2
I -R'OH
OR'
R'=H, R"CO, NO2
Нек-рые нитроспирты применяют как растворите-
ли. Восстановлением нитроспиртов получают амино-
спирты и аминогликоли, применяемые для приготов-
ления эмульгаторов, фармацевтич. препаратов, смол
типа глифталей и т. д.:
и ,
O2N—С (СН.ОН)3-----------> H2N—С (СН2ОН).
N1, 7 ат., 50°
Н. а. получают конденсацией алифатич. нитросое-
динений с альдегидами и кетонами, обычно в при-
сутствии щелочных, иногда в присутствии кислых
катализаторов. В зависимости от исходных продук-
тов и от условий реакции образуются нитроспирты,
нитрогликоли, динитропарафины и т. д.:
сн2о сн.о сн2о
CH3NO2----->HOCH2CH2NO2---->(НОСН2)2 chno2--->
—>(HOCH2)3CNO2
3COCH3CH34 ,CH2N0:
; NO2-------->
сн/ xch2no.
ch3-ch-ch-no2
I 1 CH3CHO
OH Cl----------->
/СН,
CH3-CH-C--CH
OH Cl\o24°H
Лит. см. при ст. Нитросоединения алифатические.
E. M. Рохлин.
НИТРОТОЛУОЛЫ — нитропроизводные толуола,
содержащие одну или несколько нитрогрупп у С-ато-
мов ароматич. ядра. Известны следующие изомеры:
3 мононитротолуола, 6 динитротолуолов, 6 тринитро-
толуолов и тетранитротолуол. Н., имеющие технич.
значение, приведены в таблице.
Мононитротолуолы получают нитрованием толуола
смесью азотной и серной к-т при темп-ре не выше 40°.
При этом образуется ок. 60% о-Н., 35—36% и-Н.
и 4—5% .ч-Н. Изомеры разделяют вымораживанием
части n-Н. и дистилляцией в вакууме. При произ-ве
2,4-динитротолуола темп-ру нитрования повышают до
70—80°. В зависимости от состава нитруемого моно-
О 18 К.Х.Э. т. 3
547
НИТРОТОЛУОЛЫ — НИТРОФЕНОЛЫ
548
Свойства Мононитротолуолы CH3CeHtNO2, мол. в. 137,13 Динитротолуолы CHSC»H3 (NO2)2, мол. в. 18 2,13
орто мета парав 2,4Г>Д 2,6
Темп-ра пл.у °C . . —3,17а 16,1 51,6—52,1 69,5—70,5 64,За
-9,27° 65,5°
48°
Темп-ра кип.4 °С[мм 221,7/760; 120/31 232,6/760; 113-114/15 104,5/9 — —
4 1,1742* 4 1 ,16181® 4 1,122655 4 1,521’® 4 1,538—1,540’® 4
nD Давление паров, мм/°С 1,5474 (20,4°) 81,8/5°; 94,8/10°; 109,6/20°; 119,2/30° 1,5475 (21°) 1.5554 (21°) 92,3/5°; 105,6/10°; 117,8/18° — —
— —
Растворимость
(г/100 мл', для п-Н. г/100 г) в воде . . 0,0652 (30°) 0,0498 (30°) — 0,027 (22°) —
в этаноле | в эфире f ’ ’ * ’ смешиваются во всех отношениях — 3,046 (15°) 9,4 (22°) —
Вязкость т), пуаз Теплота сгорания при 0,0262 (15°) 0,0256 (15°) 0,01204 (60°) — —
постоянном объеме, ккал/моль .... 897 893 889 848,7 854,4
Криоскопическая по- 7,18а 6,78—6,84
стоянная, °C 7,80 — —
5,08б
а Устойчивая форма. 6 Неустойчивая форма. в Т. всп. 103° С. г Т. всп. 150 °C. ДТ. самовоспл. 330° С.
нитротолуола получают след, количества (в %) ди-
нитротолуолов:
Сырье 2,4-Н. 2,6-Н. Остальные изомеры
Техническая смесь моно- нитротолуолов . . . 75 20 5
о-Нитротолуол .... 67 33 —
п- Нитротолуол . . . . 100 — —
Тетранитротолуол (т. пл. 1°) получают действием
окислов азота на трипитро-.ч-толуидин.
Н. применяются для синтеза красителей и промежу-
точных продуктов. Так, окисление Н. двуокисью мар-
ганца в серной к-те, хромовой к-той и другими окис-
лителями приводит, в зависимости от условий, к нит-
робензальдегидам или нитробензойным кислотам.
Восстановлением мононитротолуолов металлами в
кислой среде или в присутствии электролитов полу-
чают аминотолуолы (толуидины), а в щелочной среде—
гидразотолуолы (диметилгидразобензолы, см. Гидра-
зосоединения). Бензидиновой перегруппировкой гид-
разотолуолов, полученных из о- и .и-нитротолуолов,
синтезируют о- и .ч-толидины. Восстановлением ди-
нитротолуолов получают диаминотолуолы (толуилен-
диамины); 2,4-динитротолуол с NaHS в мягких усло-
виях дает 4-нитро-2-аминотолуол. n-Н. при нагрева-
нии со спиртовым р-ром КОН образует наряду с
другими веществами динитростильбен. Сульфиро-
ванием и-Н. получают 4-нитротолуол-2-сульфокисло-
ту, окислением к-рой синтезируют 4,4'-динитростиль-
бен-2, 2'-дисульфокислоту. Хлорированием Н. полу-
чают нитрохлортолуолы. Н., содержащие две и более
нитрогрупп, взрывчаты. 2,4-Динитротолуол входит в
состав нек-рых взрывчатых веществ.
Н. ядовиты, окисляют гемоглобин в метгемоглобин.
Предельно допустимые концентрации в воздухе для
мононитротолуолов 0,005 мг/л, для динитротолуолов—
0,001 мг!л. Для количественного определения восста-
навливают Н. цинковой пылью в кислой среде и тит-
руют полученные толуидины нитритом натрия (ди-
азотирование), а .ч-толуилепдиамипы — бензолдиазо-
нием (азосочетание). Эти же реакции пригодны для
качественного распознавания. Н. можно определять
также титрованием сернокислым ванадием. См. также
Тринитротолуол.
Лит.: Ворожцов Н. Н., Основы синтеза промежуточ-
ных продуктов и красителей, 4 изд., М., 1955; Орлова
Е. Ю., Химия и технология бризантных взрывчатых веществ,
М., I960. М. А. Чекалин.
НИТРОФЕНОЛЫ — замещенные фенолы, содер-
жащие одну или несколько нитрогрупп; ниже рассмат-
риваются только мононитрофенолы HOCjHaNO,,
мол. в. 139,11, для к-рых известны 3 изомера Н.
Свойства Орто- Мета- Пара-
Т. пл., °C 45,1 97 114
Т. кип., °С/.м..ча . . . d. (тверд.) 214/760 194/70 —
1,447 1 ,48542° t , 47920
di (жидк.) 1,2945*= —- 1,2809"*
Константа диссоциации (25°) 7,5-10-» 1-10-» 6,6-10-8
Растворимость г/100 г | 0,32 (38°) 1,35 (25°) 0,804 (1 5°)
11,08 (100°) 13,3 (90°) 29,1 (90°)
в этаноле 10,16 (0°) 116,9 (1°) 115.7 (0°) 119 (1°)
в эфире 37,76 (1°) 51 ,4 (0,2°)
а При темп-ре кипения о-Н. частично разлагается.
Н. образуют с металлами соли. о-Н. желтого цвета,
Na-соль красно-оранжевого цвета; м- и n-Н. бес-
цветны, соли желтые. о-Н., в отличие от м- и п-Н.,
летуч с водяным паром. о-Н. отличается по физич.
свойствам от м- и n-изомеров вследствие внутримоле-
кулярной водородной связи между атомами водорода
оксигруппы и кислорода нитрогруппы, что препятст-
вует ассоциации молекул. Восстановление Н. приво-
дит к аминофенолам, о-алкилирование дает простые
эфиры Н. Практич. значение имеют о- и и-Н., получае-
мые нагреванием о- и n-нитрохлорбензолов с водным
едким натром (10—15%) при 130—160° и давлении
2—6 ат; их можно получать также нитрованием
фенола. Н. — промежуточные продукты в синтезе
аминофенолов; применяют также непосредственно
при получении сернистых красителей.
и-Н. является фунгицидом, применяют также как
индикатор в алкалиметрии и ацидиметрии. Н. токсич-
ны, наиболее токсичен n-изомер. Для количествен-
549
НИТРОФОРМ—НИТРОХЛОРБЕНЗОЛЫ
550
ного определения Н. проще всего восстанавливать их
в кислой среде цинковой пылью до аминофенолов и
титровать последние нитритом натрия (диазотирова-
ние). См. также Динитрофенола, Тринитрофенол.
Лит.: Воронцов И. И., Полупродукты анилинокра-
сочной промышленности, М., 1955; Фирц-ДавидГ. Э.,
Б л а н ж е Л., Основные процессы синтеза красителей, пер.
с нем., М., 1957. М. А. Чекалин.
НИТРОФОРМ (тринитрометан) CH(NO2)s, мол. в.
151,04—низкоплавкое вещество с характерным запа-
хом;т. пл.22—23° (по др.данным 25°); т. кип.45—47°/22
мм', d™ 1,479 (по др. данным 1,5967); 1,4451;
поверхностное натяжение дин/см: 33,98(20°); 33,6(25°);
теплота сгорания при постоянном объеме746 ккал/молъ',
дипольный момент 2,7D; легко поглощает влагу;
хорошо растворим в воде и органич. растворителях.
Н. — сильная к-та, константа диссоциации в воде
Х=6,8.10_I(20°). В полярных растворителях диссо-
циирует по схеме:
+ ,о-
НС (NO2)s (NO2)2C = N(
хон
Н. неустойчив, начинает разлагаться при 14,3° (по
др. данным выше 25°), взрывает при ударе, детонации,
быстром нагревании. Н. образует соли с металлами
и органич. основаниями. Соли металлов постепенно
разлагаются уже на холоду, напр.
2(NO2)2C=NOOK—>2KNO3-|-2COs-|-2NO-|-N2
При хлорировании (бромировании) Н. образует хлор-
нитроформ (бромнитроформ), при нитровании—тетр -
нитрометан. При взаимодействии Н. с формальдегидом
получается тринитроэтиловый спирт (NOsb ССН2ОН.
Н. легко вступает в Михаэля реакцию, напр.:
(N03)3 CH + CH2 = CHN02 —> (N02)3 cch2ch2no2
Н. получают действием конц. HNO3 на ацетилен в
присутствии катализатора — нитрата ртути при 30—
50°. Н. токсичен.
Лит.: О р л о в а Е. Ю., Химия и технология бризантных
взрывчатых веществ, М., 1960; Паушнин Я. М., Химия
реактивных топлив, М., 1962. М. А. Чекалин.
НИТРОХЛОРБЕНЗОЛЫ (хлорнитробензолы) —
производные бензола, содержащие атомы хлора и нит-
рогруппы, общей ф-лы CeHe_n_mCln(NO2)m. По числу
атомов хлора и нитрогрупп различают мононитрохлор-
бензолы (п=1, т=1), динитрохлорбензолы (n= 1,
zn=2), нитродихлорбеизолы (п=2, т = 1) и Н., со-
держащие более 3 заместителей (п-/-т>3). Наиболь-
шее технич. значение имеют Н., приведенные в таб-
лице.
Положение за- местителей Мол. вес Т. пл., °C Т. нип., °С[мм Плотность, di т. всп., °C т. воспл., °C
С1 no2
1 2 157,56 32,5 119/8 1 ,368 (22°)
245,5/753 1,305 (80°)
1 3 157,56 44,5а 23,7® 234/756 1 ,534 (20°) — —
1 4 157,56 83,5 113/8 1 , 520 (18°) 127 127
238,5/753 1,272 (110°)
1 2,4 202,56 53,4а 315/760 1.697 (22°) 187 194
43 6 (с разл.) 1,471 7 (99,4°)
27 в
1 3.5 202,56 59 — —
1,2 4 192,01 43 255—6/756 1.4558 (75°) — —
1,4 2 192,01 56 266/760 1,669 (22°) —
1,2,4 5 226,46 57—58 288/760 1 , 790 (22°) — —
1,2,4 3,5 271,46 102,5 —
103,5
а Устойчивая форма. ® Неустойчивая форма. в 2-я неустойчивая форма.
H.— кристаллы, светло-желтого цвета (некоторые не
окрашены), с характерными запахами; почти нерас-
творимы в воде, растворимы в этаноле, бензоле, эфи-
ре и других органических растворителях. Н., осо-
бенно содержащие 2 и более нитрогруппы, взрыв-
чаты.
При действии восстановителей Н. образуют обыч-
но соответствующие аминохлорбензолы, но в ще-
лочной среде металлы (Zn, Fe) восстанавливают Н.
в гидразосоединения', так, из о-нитрохлорбензола
получают 2,2'-дихлоргидразобензол, который в кис-
лой среде перегруппировывается в 3,3'-дихлорбен-
зидин.
Н., содержащие нитрогруппы в о- или п-положе-
ниях к атому хлора, обменивают последний на «нук-
леофильные» заместители (OH.OCsOs, NH2 и др.)
значительно легче, чем хлорбензол. Особенно легко
протекает замещение, если две и более нитрогруппы
находятся в о- и n-положениях к атому хлора. Влия-
ние нитрогруппы на подвижность ароматически свя-
занного галогена в реакциях нуклеофильного заме-
щения объясняется смещением электронного облака
к нитрогруппам. В результате на атоме углерода, при
к-ром находится галоген, появляется положитель-
ный заряд. Вследствие этого облегчается атака нук-
леофильного реагента, несущего отрицательный за-
ряд. В процессе реакции нитрогруппы, оттягивая от-
рицательный заряд, понижают энергию переходного
состояния и, следовательно, энергию активации, напр.:
Мононитрохлорбензолы получают нитрованием хлор-
бензола смесью азотной и серной кислот при 40—
70°. При этом образуется ок. 34% о-, 65% п- и
1% .w-изомера, которые разделяют сначала дроб-
ной кристаллизацией, а затем дистилляцией. Другие
Н. получают нитрованием: 2,4-динитрохлорбензол —
n-нитрохлорбензола, 2-нитро-1,4-дихлорбензол — и-ди-
хлорбензола, 5-нитро-1,2,4- и 3,5-динитро-1,2, 4-три-
хЗГорбензолы — 1,2,4-трихлорбензола; а также хло-
рированием: 3,5-динитрохлорбензол — зг-динитро-
хлорбензола; 2-нитро-1,4-дихлорбензол—п-нитрохлор-
бензола.
Н. применяют гл. обр. для получения промежуточ-
ных продуктов синтеза органич. красителей: 2,4-ди-
нитрофенола, о- и п-нитроанилинов,
.м-хлоранилина, о- и п-нитроанизолов
и др. 3,5-Динитро-1,2,4-трихлорбен-
зол и его изомеры являются фунгици-
дами.
Н. ядовиты, действуют на кровь,
печень, кожные покровы. Наиболее
изучены токсичные свойства монони-
трохлорбензолов, к-рые окисляют ге-
моглобин в метгемоглобин; сильнее
других действует мета-изомер. Дини-
трохлорбензолы очень сильно раздра-
жают кожу, вызывая дерматиты и эк-
земы. Предельно допустимая концен-
трация нитрохлорбензола в воздухе
0,001 мг/л.
Лит.: Ворожцов Н. Н., Основы
синтеза промежуточных продуктов и кра-
сителей, 4 изд., М., 1955; Фирц-Да-
в и д Г. Э., Б л а н ж е Л., Основные про-
цессы синтеза красителей, пер. с нем., м.,
1957; Ullmann, 3 Aufl., Bd 12, В.—
[u.a.], 1960. М. А. Чекалин.
18*
551
НИТРОЦЕЛЛЮЛОЗА—НИТРОЦИКЛОГЕКСАН
552
НИТРОЦЕЛЛЮЛОЗА (нитрат целлюлозы) — слож-
ный эфир целлюлозы и азотной к-ты. Этерификацию
целлюлозы можно схематично выразить ур-нием:
[C«H7O2(OH)3]„+3nHNO3^zt [С,Н7О2 (ONO2)3]„+3nH2O
В зависимости от числа гидроксильных групп эле-
ментарного звена целлюлозы, участвующих в реак-
ции, степень замещения (С. з.) может колебаться от
О до 3; ее можно вычислить по ф-ле:
С. B.=3,6N/(3t,t-N)
где JV — содержание азота в %. Часто С. з. рассчи-
тывают не на один глюкозный остаток СвН,О2(ОН)3,
а на 100. В этом случае степень замещения, обозна-
чаемая у, для тринитрата целлюлозы равна 300.
Степень полимеризации Н. (число этерифициро-
ванных глюкозных остатков в макромолекуле Н.)
колеблется в широких пределах: 150—300 (лаковые
сорта), 500—800 (целлулоидные сорта), 1000—2000
(технич. высоковязкие пироксилиновые сорта). Н.
неоднородна полиол, весу, что объясняется как неод-
нородностью исходной целлюлозы, так и ее гидроли-
тич. распадом в процессе нитрации. Предельное
содержание азота, соответствующее этерификации
трех гидроксильных групп, равно 14,14%; при эте-
рификации двух ОН-групп 11,1%, одной ОН-группы
6,8%.
Различают след, основные виды технич. Н. (в
скобках содержание азота в%): коллоксилин
(10,7—12,2), пироколлодий, особый вид Н.,
открытый Д. И. Менделеевым (12,6+0,1), пирок-
силин № 2 (12,2—12,5) и пироксилин
№ 1 (13,0—13,5). Растворимость Н. зависит от содер-
жания в ней азота. Низкоазотные Н. (0,5—2%) раст-
воримы в 6%-ном NaOH при низкой темп-ре, при со-
держании азота 9—11% растворимы в этаноле; при
содержании азота до 13%—в смеси этанола и эфира,
метилэтилкетоне, этил-, бутил- и амилацетате. Уни-
версальным растворителем для всех видов техничес-
кой Н. является ацетон. Многие органич. в-ва вызы-
вают набухание (пластификацию) Н. с образованием
гелей, имеющих консистенцию от жидкого студня до
твердой термопластич. массы. Важнейшими пласти-
фикаторами являются: нитроглицерин и др. эфиры
азотной к-ты, камфара, дибутилфталат и др. сложные
эфиры.
Н.—• рыхлая, белая или желтоватая волокнистая
масса, сохраняющая структуру исходной целлюлозы.
Н. немного гигроскопична, с уменьшением степени
этерификации гигроскопичность увеличивается. При
действии разб. минеральных к-т происходит медленная
денитрация и гидролитич. расщепление целлюлозы.
Конц, серная к-та растворяет Н. В присутствии ртути
образовавшаяся при этом HNO3 восстанавливается
до NO:
2HNO3+ 3H2SO. + 6Hg —> 2NO + 3Hg2SO, + 4H2O
На этой реакции основано определение содержания
азота в Н. (по Лунге). Щелочи энергично действуют
да Н., омыляя ее и разрушая до низкомолекулярных
продуктов. Поэтому регенерировать целлюлозу из
ее нитрата можно только мягкодействующими реаген-
тами, напр. сульфгидратами натрия или аммония.
Нитраты целлюлозы обладают двойным лучепрелом-
лением, величина к-рого зависит от степени нитрации.
В поляризованном свете Н. с большим содержанием
азота окрашены в синий цвет, благодаря чему их
можно отличить от целлюлозы, окраска к-рой изме-
няется от желтой до оранжевой.
Н. уже при умеренных темп-рах самопроизвольно
разлагается, скорость разложения быстро растет с
тёмп-рой. Начальная скорость распада резко возрас-
тает в присутствии следов к-т, щелочей и примесей,
образующихся при получении Н.; в связи с этим Н.
интенсивно промывают (для стабилизации), после
чего контролируют ее химич. стойкость. Разложе-
ние Н. является самоускоряющимся. Особенно ве-
лико самоускорение в присутствии кислорода (возду-
ха) и следов влаги. При неблагоприятных условиях
самоускоряющийся распад может закончиться вспыш-
кой и взрывом. В изделия из Н., особенно высоко-
азотной, вводят стабилизаторы (дифениламин, произ-
водные мочевины и др.), к-рые увеличивают химич.
стойкость Н., связывая продукты ее разложения,
ускоряющие распад.
Н.— вторичное взрывчатое вещество, легко вос-
пламеняющееся и чувствительное в сухом состоянии
к удару и трению. Т. всп. 180—190°. После превраще-
ния путем пластификации в твердую, непористую мас-
су Н. приобретает свойства метательного вещества
(см. Пороха}.
Для получения Н. обычно применяют коротково-
локнистую хлопковую целлюлозу (линтер), к-рую об-
рабатывают смесью конц. азотной и серной к-т в ап-
парате из нержавеющей стали с механическими ме-
шалками. В лабораторных условиях нитрование про-
изводят также смесью азотной и фосфорной к-т или
смесью азотной, уксусной к-т и уксусного ангидрида.
По окончании нитрования Н. отделяют от отрабо-
танной к-ты на центрифуге и многократно промы-
вают разб. серной к-той, водой при нагревании ос-
трым паром, р-ром соды и опять водой (для удаления
к-т, низкомолекулярных примесей и сульфоэфиров).
Для лучшей стабилизации Н. в процессе промывания
дополнительно измельчают. Стабилизированную Н.
обезвоживают, в случае необходимости промывая ее
этанолом на центрифуге.
Н. применяют в зависимости от содержания в ней
азота: коллоксилин с высоким содержанием азота
(11,5—12,2%) применяют для произ-ва пороха (твер-
дого ракетного топлива) типа баллистит, а также для
желатинизации жидких нитроэфиров в произ-ве ди-
намитов и др. взрывчатых веществ промышленного
назначения (колоксилин с меньшим содержанием
азота применяют для произ-ва целлулоида,
искусственной кожи и др. материалов): пироксилины—
для произ-ва различных бездымных порохов (т. наз.
пироксилиновый порох состоит в основ-
ном из пироксилина, пластифицированного летучим
растворителем, удаляемым из пороха в процессе
произ-ва). Из р-ров низкоазотной Н. в летучем раство-
рителе, содержащей пластификатор, производят кино-
и фотопленку, нитролаки и нитроклеи различного
назначения. Основным недостатком пленок, лаков,
пластич. масс и т. п, продуктов, получаемых на осно-
ве Н., является горючесть. В связи с этим, а также
вследствие интенсивного развития произ-ва синтетич.
полимеров других видов, Н. вытесняется из произ-ва
фото- и кинопленки и др. материалов промышленного
и бытового назначения.
Лит.; Закощиков А. П., Нитроцеллюлоза, М., 1950;
Роговин 3. А., Шорыгина Н. Н., Химия целлюлозы
и ее спутников, М.—Л., 1953; Никитины. И., Химия дре-
весины и целлюлозы, М.—Л., 1962; Ott Е., Cellulose and
cellulose derivatives, pt 2, N. Y.— L., 1954, p. 746.
Г. А. Петропавловский.
НИТРОЦИКЛОГЕКСАН C6HnNO2, молекулярный
вес 129,16 — бесцветная жидкость; т. заст. —34°; т.
кип. 205,5—206°/7б8 мм (с частичным разложением),
109,5°/40 мм; d*’ 1,0605; 1,4612, нерастворим в
воде, хорошо растворим в щелочах (с образованием
солей) и в органич. растворителях.
Н. может быть восстановлен до циклогекса-
н оноксима хлористым оловом в соляной к-те,
цинковой пылью и амальгамой натрия в соляной и
уксусной к-тах, а также водородом в присутствии
смешанных катализаторов (Са—Zn—Сг—Ag) при
140—160° и 100—120 ати. В пром-сти Н. восставав-
553
НИТРОЭТАН —НИТРОЭТИЛЕН
554
ливают водородом при повышенных темп-ре и давле-
нии в присутствии металлической меди как катали-
затора и жидкого аммиака. Обработка щелочных
солей нитроциклогексана серной к-той и водным
р-ром гидроксиламинсульфата, к-рый может быть за-
менен р-ром NaNO2 и NaHSOs, насыщенным SO2,
также приводит к циклогексаноноксиму. При обра-
ботке хлорной водой Н. превращается в 1-хлор-1-
нитроциклогексан, к-рый восстанавливают до цикло-
гексаноноксима водородом в присутствии Pd на угле.
Циклогексаноноксим превращают Бекмана перегруп-
пировкой в капролактам.
Контактирование Н. борфосфатным катализатором
при 200—400° и атмосферном давлении в присутствии
азота и различных добавок (метанол, ацетон и др.)
сопровождается непосредственным образованием ка-
пролактама с выходом до 40%. Такие же результаты
получаются при взаимодействии аммониевой соли Н.
с олеумом или нитрозилсерной к-той в растворителях
(циклогексан, ССП). Увеличение выхода капролакта-
ма до 70—75% достигается в случае обработки ще-
лочных солей Н. гидроксиламинсульфатом или нитри-
том натрия и гидразинсульфатом в сернокислой среде
(или олеума), а также при действии серной к-ты в
присутствии первичных нитроалканов. При электро-
химии. восстановлении Н. в кислой среде получают с
хорошим выходом циклогексилгидроксиламин. С азо-
тистой к-той Н. образует циклогексилпсевдонитрол,
с ацетальдегидом — 1,1-нитроциклогексилэтанол.
Н. вступает в реакцию азосочетания, например с
n-нитробензолдиазонием в щелочной среде.
Н. токсичен; вызывает функциональные изменения
нервной системы и дерматиты, понижает кровяное
давление. Предельно допустимая концентрация Н. в
воздухе 0,05л1г/л. Н. может быть получен нитрованием
циклогексана разб. азотной к-той или двуокисью
азота. Процесс проводят как в жидкой фазе, так и в
парах. В пром-сти Н. получают путем жидкофазного
нитрования циклогексана азотной к-той при повышен-
ных темп-ре (125—150°) и давлении. В оптимальных
условиях выход Н. достигает 65—70%, при степени
превращения циклогексана 28—30%, одновременно
получаются дикарбоновые к-ты (адипиновая, глутаро-
вая, янтарная), суммарный выход к-рых составляет
22—25%. Побочными продуктами, получаемыми при
нитровании гексана, являются: изомерные моно-,
ди- и тринитрофенолы, циклогексанон, циклогекса-
нол, mpem-нитроциклогексан, изомерные нитро-
спирты, 1,1-динитроциклогексан и др. Тонкая очист-
ка Н. достигается, ректификацией.
Лит.: Современные методы синтеза мономеров для гете-
ро цепных волокнообразующих полимеров, под ред, Л. И.
Кнунянца, М., 1961; Хопфф Г., Мюллер А., Вен-
гер Ф., Полиамиды, пер. с нем., М., 1958. М. А. Чекалин.
НИТРОЭТАН CH3CH2NO2, мол. в. 75,07— бесцвет-
ная жидкость с характерным запахом; т. пл.— 89,52°;
т. кип. 114,077760 мм, 20715,65 мм; 1,05057;
«д 1,39193. Н. с водой образует азеотропную смесь,
т. кип.<100°. Растворимость при 20°: Н. в воде
4,5 л1л/Ю0л1л;воды в Н.0,9.ил/100л1л. Н. хорошо рас-
творим в большинстве органич. растворителей. Тепло-
та парообразования 9,94 ккал/моль (25°); теплоемкость
33,8 кал/моль-град (23—95°); (крит_ 388,6°; диэлектрич.
проницаемость 28,Оо (30°); дипольный момент 3,19 D.
Вязкость 0,661 спуаз (25°); поверхностное натяжение
31,31 дин/см (25°). Т. воспл. 41,1°.
Н. обладает обычными свойствами нитропарафинов
(см. Нитросоединения алифатические). Продолжи-
тельное действие концентрированных щелочей на Н.
приводит к получению триметилизоксазола (1);
взаимодействием Н. с 85%-ной серной к-той получают-
ся уксусная к-та и гидроксиламин (2), тогда как при
обработке солей аци-Н, холодной разб. серной к-той
по Нефа реакции образуется ацетальдегид (3). При
взаимодействии с альдегидами образуются нитроспир-
ты, нитрогликоли и нитроолефины (4а и 46):
ОН" C2H-,NO2 Н3С-п- “ГСНз O'N (i)
н3сА
c2h5no2 сн3( :оон + nh2oh (2)
о CH3CH = N Нг5О, ► CH3CHO + n2o (3)
4ONa
CH3CH2NO2 + СН2О КгС°3— СН3СНСН2ОН + СН3С(СНаОН)2(4а)
no2 no2
ch3ch2no2
c2h5nh2
сн=с-сн3
NO2
(46)
Нитрозирование, галогенирование, восстановление
и т. н. реакции производят обычным для нитропара-
финов образом. В пром-сти Н. вместе с другими нитро-
парафинами получают деструктивным нитрованием
парафиновых углеводородов, в лаборатории — эти-
лированием солей азотистой к-ты, по способу Кольбе,
перегруппировкой этилнитрита (см. Нитрометан).
Н. ядовит. Максимально допустимая концентрация
в воздухе 0,01%. Применяют Н. как растворитель,
исходный продукт в синтезе уксусной к-ты, 2-нитро-
2-метил-1,3-пропандиола и т. д.
Лит. см. при ст. Нитрометан. Е. М. Рохлин.
НИТРОЭТИЛЕН CH2 = CHNO2, мол. в. 73,06—
зеленовато-желтая жидкость с резким запахом (лак-
риматор); т. кип. 98,57760 мм, 38—39°/80 мм;
d13’8 1,073; растворим в органич. растворителях; при
растворении в воде осаждается полимер Н. Бесцветный
аморфный полимер получается также при действии
щелочей (реакция со взрывом) или при продолжитель-
ном хранении Н. Реакции Н. характерны для
сопряженных нитроолефинов — ненасыщенных со-
единений с двойной связью, активированной элект-
ронооттягивающим заместителем. Соответствующие
p-производные нитроэтана образуются при взаимодей-
ствии Н. с водой (1), сероводородом (2), аминами (3),
бисульфитом (4), сульфиновыми к-тами (5):
[Н + ]
CH2 = CHNO2 + H2O--<-HOCH2CH2NO2 (1)
CH2 = CHNO2 + H2S—>-S (CH2CH2NO2)2 + HSCH2CH2NO2 (2)
CH2 = CHNO2 + C6H5NH2 —>-C«H5NHCH2CH2NO2 (3)
CH2 = CHNO2 + NaHSO3 —>- HOSO2CH2CH2NO2 (4)
CH2 = CHNO, + n-CHa-CjH,-SO2Na —>-
—>.n-GH3-C»H1S02CH2CH2NO2 (5)
H. способен восстанавливаться (6), вступает в Дильса-
Альдера реакцию как диенофил (7):
CH2=CHNO2 + Zn > CH2CH2NH2
CH
CH=CH H
ch2=chno2 + I CH2 — X (7)
Ц1-Ш XI/
CH
H. получают конденсацией нитрометана с формаль-
дегидом при 250—300° в присутствии ацетата свинца
ch3no2+ch2o —>ch2=chno2
а также из соответствующих [J-производных нитро-
этана отщеплением от них при нагревании воды, НС1,
555
НИТРОЭФИРЫ—НОВОБИОЦИН
556
существенного различия.
СН3
1.4JA
о
125
О
NOa (~40 ккал/моль)
в одной плоско-
углерода распо-
плоскости, пер-
уксусной к-ты, HNOs или HNOs, напр.:
hoch2ch2no2-------------------> ch2=chno2
NaHSO, и др.
C1CH2CH2NO2 —> CH2 = CHNO2
240°
ch,cooch2ch2no2-------->ch2=chno2
Al2 (SO4)3
E. M. Рохлин.
НИТРОЭФИРЫ (органические нитраты) — соеди-
нения, содержащие связанную с атомом углерода
О
группу —ONO2 (—О—N<^ ), сложные эфиры азот-
ной к-ты и спиртов, углеводов и др. Наименования
Н. обычно составляются из названия органич. части
молекулы Н. (или соединения, из к-рого Н. полу-
чается) с добавлением слова «нитрат»— напр., ме-
тилнитрат, диэтиленгликольдинитрат, нитраты
целлюлозы и т. д. В технике до сих пор широко
употребляются названия Н., содержащие приставку
«нитро» (напр., нитроглицерин, нитроцеллюлоза),
сохранившиеся с тех времен, когда между Н. и нитро-
соединениями не делали
Детально известна струк-
тура лишьпростейшего Н.—
метилнитрата. Она м. б.
представлена в виде:
Все атомы нитратной груп-
пы лежат--------- -----
сти, а атом
ложен в
пендикулярной ей. Наиме-
нее прочна в Н. связь О—NO2 (~40 ккал/моль). Ди-
польный момент первых мононитратов жирного ряда
(от метил- до «-бутил-) составляет ~2,9 D.
Основной способ получения Н.— этерификация
соответствующего исходного соединения (спирта, уг-
левода) азотной к-той, обычно в при-
сутствии серной к-ты, уксусного ангид-
рида и т. п.:
ROH + HNO3T=i RONO2 + H2O (1)
На этой реакции основаны все мето-
ды промышленного производства Н. В
лабораторной практике для получения
Н. используют также реакцию галоге-
ноорганич. соединений с нитратом се-
ребра:
RJ+AgNO3—>R0NO2 + AgJ (2)
Свойства пек рых Н. жирного ряда
приведены в таблице.
Одной из наиболее характерных ре-
акций Н. является гидролиз, к-рый
идет либо с образованием спирта и HNOs
(см. ф-лу 1), либо иными путями (давая
HNO2 и альдегид, HNOs и алкилен и
т. д.). В присутствии кислот и особенно
щелочей скорость гидролиза сильно возрастает. Спир-
товые р-ры едких щелочей разлагают Н. почти мгно-
венно. При взаимодействии конц. серной к-ты с Н.
образуются сульфоэфиры:
rono2 + h2so4 roso3h+hnos (з)
Эта реакция используется, в частности, для ко-
личественного определения Н. по методу Лунге,
при к-ром Н. переводится конц. НгЗО4 в сульфат, а
выделившаяся HNOs со ртутью в сернокислом р-ре
дает окись азота, измеряемую волюмометрически.
Склонность Н. к гидролизу и незначительная проч-
ность связи О—NO2 обусловливают весьма малую
стойкость этих соединений даже при обычной темп-ре.
При 150—200° скорость разложения Н. так велика,
что многие из них самовоспламеняются. Если коли-
чество вещества достаточно и имеется возможность
накопления катализирующих разложение кислых
продуктов, самовоспламенение может произойти и при
более низкой темп-ре — вплоть до обычных темп-р
хранения. Многие Н. обладают способностью к горе-
нию (без участия кислорода воздуха) и взрыву, выра-
женную тем сильнее, чем больше содержится в них
активного кислорода (групп ONOj). Нек-рые Н. яв-
ляются мощными взрывчатыми веществами. Одни
из них — пентаэритриттетранитрат (тэн) и диэтанол-
N-нитроаминдинитрат (дина) — непосредственно при-
меняются в качестве вторичных взрывчатых веществ
(гл. обр. военного назначения), другие — в основ-
ном нитроглицерин, диэтиленгликольдинитрат и
нитрогликоль — используются в качестве составной
части промышленных взрывчатых веществ (см. Дина-
миты, Аммониты, Предохранительные взрывчатые
вещества)', нитроглицерин, диэтиленгликольдинитрат
и нитроцеллюлоза широко применяются для изготов-
ления порохов. Произ-во Н. для этих целей достигает
сотен тысяч т в год. Другие области применения Н.
весьма ограничены.
Производство Н. является одним из самых взрыво-
опасных химич. производств. Весьма велика опас-
ность взрыва также при хранении, перевозке и
применении Н. Поэтому обращение с ними строго
регламентируется, а транспортировка таких Н., как
нитроглицерин и нитрогликоль, в чистом виде в боль-
шинстве стран вообще запрещена.
Многие Н. обладают значительным физиологии,
действием. Вдыхание паров или попадание Н. на ко-
жу, а тем более в.пищеварительный аппарат вызывает
учащенное сердцебиение, головную боль и может при-
вести к сильному отравлению. Нитроглицерин в виде
очень разб. р-ров применяется в медицине в качестве
сосудорасширяющего средства.
Нитроэфир rf20 4 T. пл., °C T. кип., °C Теплота взрыва, ккал 1кг
Метилнитрат CH3ONO2 1,21 -83 65 1400
Этилнитрат CH3CH2ONO2 1,11 -94 89 700
н-Пропилнитрат CH3CH2CH20N02 1,05 — 110 550
Этиленгликольдинитрат CH20NO2 (нитрогликоль) | CH20NO2 1,50 -22,3 ~ 200 (с разл.) 1600
Глицеринтринитрат CH2ONO2 (нитроглицерин) | CHONOj 1 ch2ono2 1,60 13,5 (2,8) 1500
Маннитгексанитрат (нитроманнит) O2N0CH2 (CHONO2)4 ch2ono2 1 ,73 112—113 — 1450
Диэтанол- N-нитроаминдинитрат (дина) ,CH2CH,ON02 O„NN( xCH2CH2ONO2 1 .67 52,5 1200
Лит.: Наум Ф., Нитроглицерин и нитроглицериновые
взрывчатые вещества (динамиты), пер. с нем., М.—Л., 1934;
Boschan R., Merrow R. Т., Dolah R. W. van,
Chem. Revs, 1955, 55, № 3, 485; Андреев К. К., Терми-
ческое разложение и горение взрывчатых веществ, М.—Л.,
1957 Б. Н. Кондриков.
НОВАРСЕНОЛ - см. Сальварсана препараты.
НОВОБИОЦИН C„HS6N2On, мол. в. 612,3 — ан-
тибиотик, продуцируемый актиномицетами Str. sphe-
roides и Str. niveus. H. (I) — бесцветные оптически
активные кристаллы.
ОН
557
НОВОКАИН—НОВЭМБИХИН
558
Н. существует в двух формах, темп-ра плавления
соответственно 170—172° (или 174—178°) и 152—156°;
[а]р=—63° (С=1%, в этаноле); [а]р= —27°(С=1%,
1 н. р-р NaOH). Н. образует соли с неорганич.
и органич. основаниями. В виде свободной к-ты
Н. плохо растворим в воде (при pH не ниже 7,5);
хорошо — в метиловом и этиловом спиртах, этил-
ацетате, бутил- и амилацетате и пиридине; в виде
солей хорошо растворим в воде. УФ-спектр Н.
сильно меняется в зависимости от значения pH. По-
тенциометрия. титрованием обнаружены две кислые
группы в молекуле Н.: рХ 4,3, рЛ^ 9,1 (в воде).
В щелочной среде Н. изомеризуется в изоновобиоцин,
не обладающий биологич. активностью. При ката-
литич. гидрировании Н. образуется биологически
активный дигидроновобиоцин C3iHS8N20ii, т. пл.
163—165°. Агликоновой частью Н. и дигидроново-
биоцина являются новобиоцевая (II) и дигидро-
новобиоцевая к-ты (III). Осуществлен полный син-
тез Н. Количественно Й. можно определить микро-
биологически, спектроскопически и колориметрически
(по окрашиванию с N-2,6, трихлорхинонимином), а
также хроматографией на бумаге.
Н. образуется в условиях глубинной ферментации.
После отделения биомассы при pH 8,0 Н. выделяют из
культуральной жидкости экстракцией амилацетатом
при рЙ 6,0, с последующим переводом антибиотика
в воду при pH 10,0, повторной экстракцией органич.
растворителем и осаждением ацетоном или петролей-
ным эфиром. Н. может быть очищен перекристаллиза-
цией из водного ацетона или этанола или из смесей
петролейного эфира с этими растворителями.
Н. подавляет в основном грамположительные бак-
терии, в том числе их штаммы, устойчивые к боль-
шинству известных антибиотиков: пенициллинам,
стрептомицину, тетрациклинам, эритромицину и др.
Н. биологически активен, обладает умеренной ток-
сичностью; поэтому он нашел практич. применение
для лечения пневмоний и других инфекций, а главное
для борьбы с заболеваниями, вызванными стафило-
кокками, устойчивыми к другим антибиотикам.
Н. первоначально был описан под названиями стрептони-
вицин, катомицин, кардельмицин. Позднее было показано, что
гризеофлавин также идентичен Н. За рубежом Н. выпускают
под названиями: катомицин, кардельмицин, вулкамицин,
инамицин и альбамицин.
Лит.: Шемякин М. М. [и др.], Химия антибиотиков,
3 изд., М., 1961; Walton Е., Spencer С., пат. США,
2966484 (Chem. Abs. 1961, 55, № И, 10473); Stammer
С. Н., пат. США 2 925 411 (Chem. Abs. 1960, 54, № 18, 18555).
Д. М. Трахтенберг.
НОВОКАИН (прокаин, хлоргидрат диэтиламино-
этилового эфира n-аминобензойной к-ты), мол. в.272,77,
Ci3H2o02N2-HCl -
t \ бесцветные кри-
H2N-\ )-COO(CH2)2N(C2H5)2-НС1 СТаллы, без запаха;
т. пл. 154—156°;
хорошо растворим в
воде (1 : 1), спирте (1:8), плохо — в неполярных
органич. растворителях. В водных р-рах щелочей Н.
легко гидролизуется с образованием диэтиламиноэта-
нола и n-аминобензойной к-ты (витамина Hi), явля-
ющейся антагонистом сульфаниламидных препаратов,
чем обусловлено заметное антисульфаниламидное
действие Н.; в кислой среде Н. более устойчив. При
взаимодействии Н. с нитритом натрия образуется соль
диазония, к-рая со щелочным р-ром [J-нафтола дает
карминово-красный осадок азокрасителя; с нитратом
серебра Н. дает характерную реакцию на С1~. Коли-
чественное определение Н. основано на алкалиметрич.
титровании в спиртово-хлороформном р-ре по фенол-
фталеину. Н. можно получать, напр., из этилового
эфира n-аминобензойной к-ты и диэтиламиноэтанола
n-H,NCBH1COOC2H5 + HOCH.CH..N (С2Н5)2 —»- Н
перэтерифинацией в присутствии небольшого коли-
чества алкоголята натрия.
Н.— эффективное местноанестезирующее средство,
широко применяемое для инфильтрационной и спин-
номозговой анестезии (часто в сочетании с адренали-
ном), для анестезии слизистых оболочек, для раство-
рения пенициллина в целях удлинения его действия,
для новокаиновой блокады и т. п. Наряду с Н. в ме-
дицинской практике применяется новокаин-основание,
мол. в. 236,31, бесцветные кристаллы, т. пл. 60—63°
(из органич. растворителей) или 51° (с двумя молеку-
лами кристаллизационной воды — из воды или вод-
ного спирта). Это основание хорошо растворимо в
обычных органич. растворителях, растворимо в пер-
сиковом и абрикосовом маслах (10%) и плохо — в
воде (0,25%); применяют в виде масляного р-ра под-
кожно или внутримышечно как местноанестезирую-
щее средство при длительных болях в случаях огра-
ниченных патологич. процессов.
Лит.: G г а у Т. С., Gedde! I. С., J. Pharmacy and
pharmacol., 1954, 6, № 2, 89; A d г 1 a n 1 J., Klin. Wochenschr.,
1958, 36, H. И. Л. H. Яхонтов.
НОВОКАИНАМИД (прокаинамид, пронестил, хлор-
гидрат диэтиламиноэтиламида n-аминобензойной кис-
лоты) Ci3H21ON3-HC1, мол. в. 271,79 — белый кри-
сталлич. поро- г,—\
шок; т. пл. 165— h2n-( У-CONH(CH2)2N(C2H5)2 НС1
170°; хорошо ра- \=/
створим в воде, растворим в спирте, нерастворим в
эфире и дихлорэтане. Н. с нитритом натрия образует
соль диазония, к-рая со щелочным р-ром [J-нафтола
дает осадок оранжево-красного цвета. С нитратом се-
ребра Н. дает характерную реакцию на ион С1_. Ко-
личественное определение Н. основано на потенцио-
метрии. титровании нитритом натрия с платиновым
электродом. Н. получают из хлорангидрида п-нитро-
бензойной к-ты и диэтиламиноэтиламина с последую-
щим восстановлением диэтиламиноэтиламида п-нит-
робензойной к-ты в присутствии скелетного никелевого
катализатора или при взаимодействии с диэтилами-
ноэтиламином этилового эфира п-аминобензойной
к-ты. Н. более стоек, чем новокаин, значительно
медленнее разлагается плазмой крови. Н. применяют
при различных расстройствах сердечного ритма.
Лит.: С ы р н е в а Ю. И., Фармакология и токсикология,
1955, 18, № 4, 27. Л. Н. Яхонтов.
НОВЭМБИХИН [хлоргидрат 2-хлорпропил-М,М-бис-
(Р-хлорэтил)амина] CH3CHC1CH2N(CH2CH2C1)2 -НС1,
мол. в. 255,02 — бесцветные кристаллы, т. пл. 69—
71°; легко растворим в воде и спирте; нерастворим в
эфире и бензоле. Количественно Н. определяют арген-
тометрически. Получают Н. конденсацией диэтанол-
амина с окисью пропилена с последующим взаи-
модействием хлоргидрата 2-oKcHnponnn-N,N-6«c-([J-
оксиэтил) амина с хлористым тионилом. Н. относится
к группе азотистых ипритов, обладающих алкили-
рующими свойствами по отношению к нуклеопротеи-
дам клеточных ядер и в связи с этим применяющихся
для лечения опухолевых заболеваний кроветворной
системы. Подобно другим препаратам группы азотис-
тых ипритов (эмбихину, трихлортриэтиламину и
559
НОМЕНКЛАТУРА И КЛАССИФИКАЦИЯ ФЕРМЕНТОВ
560
др.), Н. при попадании на кожу оказывает раздражаю-
щее И нарывное действие. р- Г. Глушков.
НОМЕНКЛАТУРА И КЛАССИФИКАЦИЯ ФЕР-
МЕНТОВ — современная классификация и номенк-
латура ферментов, разработанная Комиссией по фер-
ментам и одобренная Ассамблеей Международного
биохимия, союза (Москва, август 1961).
В основу научной классификации и номенклатуры
ферментов положена выражаемая суммарным урав-
нением реакции природа превращения, катализируе-
мого тем или иным ферментом. На основе этого при-
знака все ферменты разделены на 6 главных классов:
1. Оксидоредуктазы, катализирующие окислитель-
но-восстановительные реакции.
2. Трансферазы, катализирующие межмолекуляр-
ный перенос различных групп и остатков.
3. Гидролазы, катализирующие реакции гидроли-
тич. расщепления внутримолекулярных связей.
4. Лиазы, катализирующие реакции негидролитич.
отщепления тех или иных групп с образованием двой-
ных связей и обратные реакции присоединения групп
по двойным связям.
5. Изомеразы, катализирующие различные реак-
ции изомеризации.
6. Лигазы (синтетазы), катализирующие реакции
присоединения друг к другу двух молекул, сопряжен-
ные с расщеплением пирофосфатной связи в молекуле
аденозинтрифосфорной к-ты (АТФ) или аналогичного
трифосфата (напр., гуанозинтрифосфата).
Внутри каждого класса ферменты разделяются на
подклассы. В свою очередь каждый подкласс делится
на несколько подподклассов на основе дальнейшего
уточнения природы ферментативной реакции. Все
подклассы в пределах каждого класса и все подпод-
классы в пределах подкласса имеют свою порядко-
вую нумерацию. Аналогичным образом нумеруются
ферменты в пределах каждого подподкласса. Такой
принцип нумерации позволяет при введении в систему
классификации новых подразделений (классов, под-
классов и подподклассов), а также при внесении в
список новых ферментов присваивать им номера, не
меняя всей предшествующей нумерации. Ферменты,
отнесение к-рых к тому или иному подклассу или под-
подклассу в настоящее время затруднительно из-за
отсутствия точных сведений о деталях катализируе-
мых ими реакций, включают в специальные разделы.
Эти разделы обозначены номером 99, чтобы оставить
место для новых подразделений. В такие подподклас-
сы, например, включены оксидоредуктазы, для ко-
торых известны только реакции с искусственны-
ми акцепторами, а истинный акцептор не установлен
(см. 1.1.99 и др.).
Ключ к классификации и нумерации (индексации)
ферментов
1. ОКСИДОРЕДУКТАЗЫ
1.1 Действуют на СН—ОН группу доноров
1.1.1 Акцептором служит НАДа или НАДФ^
1.1.2 Акцептором служит цитохром
1.1.3 Акцептором служит О2
1.1.99 Используют другие акцепторы
1.2 Действуют на альдегидную или кетонную группу доноров
1.2.1 Акцептором служит НАД или НАДФ
1.2.2 Акцептором служит цитохром
1.2.3 Акцептором служит О2
1.2.4 Акцептором служит липоевая к-та
1.2.99 Используют другие, акцепторы
1.3 Действуют на СН—СН группу доноров
1.3.1 Акцептором служит НАД или НАДФ
1.3.2 Акцептором служит цитохром
1.3.3 Акцептором служит О3
1.3.99 Используют другие акцепторы
1.4 Действуют на СН—NH2 группу доноров
1.4.1 Акцептором служит НАД или НАДФ
1.4.3 Акцептором служит О2
а НАД — никотинамид-аденин-динуклеотид.
6 НАДФ—никотинамид-аденин-динуклеотид-фосфат. См.
также Никотпинамиднуклеотидные коферменты.
1.5 Действуют на С—NH группу доноров
1.5.1 Акцептором служит НАД или НАДФ
1.5.3 Акцептором служит О2
1.6 Действуют на НАДН2 или НАДФН2 в качестве донора
1.6.1 Акцептором служит НАД или НАДФ
1.6.2 Акцептором служит цитохром
1,6.4 Акцептором служит дисульфидное соединение
1.6.5 Акцепторами служат хиноны или родственные им
соединения
1.6.6 Акцептором служит азотистая группировка
1.6.99 Используют другие акцепторы
1.7 Действуют на другие азотистые соединения в качестве
доноров
1.7.3 Акцептором служит О2
1.7.99 Используют другие акцепторы
1.8 Действуют иа серусодержащие группы доноров
1.8.1 Акцептором служит НАД или НАДФ
1.8,3 Акцептором служит О2
1.8.4 Акцептором служит дисульфидное соединение
1.8.5 Акцепторами служат хиноны и родственные им
соединения
1.8.6 Акцептором служит азотистая группировка
1.9 Действуют иа группы гема доноров
1.9.3 Акцептором служит О2
1.9.6 Акцептором служит азотистая группировка
1.10 Действуют на дифенолы и родственные им соединения
в качестве доноров
1.10.3 Акцептором служит О2
1.11 Действуют на Н2О2 в качестве акцепторов
1.98 Ферменты, использующие Н2 в качестве восстановителя
1.99 Прочие ферменты, использующие О2 в качестве окис-
лителя
1.99.1 Гидроксилазы
1.99.2 Оксигеназы
2. ТРАНСФЕРАЗЫ
2.1 Переносят одноуглеродные остатки
2.1.1 Метилтрансферазы
2.1.2 Трансферазы оксиметильных, формильных и род-
ственных им групп
2.1.3 Карбоксил- и карбамоилтрансферазы
2.2 Переносят альдегидные или кетонные остатки
2.3 Переносят ацильные остатки
2.3.1 Ацилтрансферазы
2.3.2 Аминоацилтрансферазы
2.4 Переносят гликозильные остатки
2.4.1 Гексозилтрансферазы
2.4.2 Пентозилтрансферазы
2.5 Переносят алкильные или родственные им остатки
2.6 Переносят азотистые группы
2.6.1 Аминотрансферазы
2.6.2 Амидинотрансферазы
2.6.3 Оксиминотрансферазы
2.7 Переносят группы, содержащие фосфор
2.7.1 Фосфотрансферазы со спиртовой группой в роли
акцептора
2.7.2 Фосфотрансферазы с карбоксильной группой в роли
акцептора
2.7.3 Фосфотрансферазы с азотистой группировкой в роли
акцептора
2.7.4 Фосфотрансферазы с фосфатной группировкой в роли
акцептора
2.7.5 Фосфотрансферазы с кажущимся внутримолекуляр-
ным переносом
2.7.6 Пирофосфотрансферазы
2.7.7 Нуклеотидилтрансферазы
2.7.8 Переносят другие замещенные фосфатные груп-
пировки
2.8 Переносят группы, содержащие серу
2.8.1 Сульфидтрансферазы
2.8.2 Сульфотрансферазы
2.8.3 КоА-трансферазы
3. ГИДРОЛАЗЫ
3.1 Действуют на сложиоэфириые связи
3.1.1 Гидролазы эфиров карбоновых кислот
3.1.2 Гидролазы тиоловых эфиров
3.1.3 Гидролазы фосфомонозфиров
3.1.4 Гидролазы фосфодиэфиров
3.1.5 Гидролазы трифосфомоноэфиров
3.1.6 Гидролазы сульфоэфиров
3.2 Действуют на гликозильные соединения
3.2.1 Гидролазы гликозидов
3.2.2 Гидролазы N-гликозильных соединений
3.2,3 Гидролазы S-гликозильных соединений
3.3 Действуют на эфирные связи
3.3.1 Гидролазы тиоэфиров
3.4 Действуют на пептидные связи (пептидгидролазы)
3.4.1 а-Аминопептид-аминоацидогидролазы
3.4.2 а-Карбоксипептид-аминоацидогидролазы
3.4.3 Дипептидгидролазы
3.4.4 Иминопептидгидролазы
3.4.5 Пролинпептидгидролазы
3.4.6 Пептид-пептидогидролазы
3.5 Действуют на С—N связи, отличающиеся от пептидных
связей
3.5.1 В ливейных амидах
561
НОМЕНКЛАТУРА И КЛАССИФИКАЦИЯ ФЕРМЕНТОВ
562
3.5.2 В циклических амидах
3.5.3 В линейных амидинах
3.5.4 В циклических амидинах
3.5.99 В прочих соединениях
3.G Действуют на кислотноангидридные связи
3.6.1 В фосфорилсодержащих ангидридах
3.7 Действуют на С—С связи
3.7.1 В кетосоединениях
3.8 Действуют на галоидные связи
3.8.1 В С-галоидных соединениях
3.8.2 В Р-галоидных соединениях
3.9 Действуют на Р—N связи
4. ЛИАЗЫ
4.1 Углерод — углерод-лиазы
4.1.1 Карбокси-лиазы
4.1.2 Альдегид-лиазы
4.1.3 Лиазы кетокислот
4.2 Углерод — кислород-лиазы
4.2.1 Гидро-лиазы
4.2.99 Прочие углерод — кислород-лиазы
4.3 Углерод — аэот-лиаэы
4.3,1 Аммоно-лиазы
4.3.2 Амидин-лиазы
4.4 Углерод — сера-лиазы
4.5 Углерод — галогеи-лиазы
5. ИЗОМЕРАЗЫ
5.1 Рацемазы и эпимеразы
5.1.1 Действуют на аминокислоты и их производные
5.1.2 Действуют на оксикислоты и их производные
5.1.3 Действуют на углеводы и их производные
5.2 Дис-траяс-изомеразы
5.3 Внутримолекулярные оксидоредуктазы
5.3.1 Взаимопревращают альдозы и кетозы
5.3.2 Взаимопревращают кетонные и енольные группы
5.3.3 Перемещают С=С связи
5.4 Внутримолекулярные трансферазы
5.4.1 Переносят ацильные группы
5.4.2 Переносят фосфорильные группы
5.4.99 Переносят другие группы
5.5 Внутримолекулярные лиазы
6. ЛИГАЗЫ (СИНТЕТАЗЫ)
6.1 Образуют С—О связи
6.1.1 Лигазы аминокислот — РНК
6.2 Образуют С—S связи
6.2.1 Кислото-тиоловые лигазы
6.3 Образуют С—N связи
6.3.1 Кислото-амиачные лигазы (амид-синтетазы)
6.3.2 Кислото-аминокислотные лигазы (пептид-синтетазы)
6.3.3 Циклолигазы
6.3.4 Прочие С—N-лигазы
6.3.5 С—N-лигазы с глутамином в роли N-донора
6.4 Образуют С—С связи
Новая система классификации ферментов имеет
ряд существенных отличий от систем, предлагав-
шихся ранее, в частности от классификации Диксона
и Уэбба (см. М. Диксон и Э. Уэбб, Ферменты, М., 1961)
или Гофмана — Остенгофа (1953), хотя и значительно
более сходна с последней, нежели с первой. В част-
ности, в новой системе классификации так же, как и в
системе Гофмана—Остенгофа, выделены в самостоя-
тельный класс оксидоредуктазы, отнесенные Диксо-
ном и Уэббом в общую группу трансфераз (группа А).
Лиазы и синтетазы (лигазы), входившие в системе
Гофмана — Остенгофа в один класс, по новой классифи-
кации разделены на два самостоятельных класса. Вы-
делены в самостоятельный класс также изомеразы,
входившие в системе Диксона и Уэбба частично
в общую группу В (т. наз. «прочие ферменты»),
а частично в группу трансфераз. При этом к классу
изомераз отнесены ферменты, катализирующие внут-
римолекулярный перенос не только водорода, но и
других групп, в частности метильной. К их числу от-
носится метиласпартат-мутаза, катализирующая ре-
акцию изомеризации: L-znpco-3-метиласпартат
^L-глутамат, а также метилмалонил-КоА-мутаза, осу-
ществляющая превращение; 2-метилмалонил-КоА1;
^сукцинил-КоА. Кофакторами этих ферментов яв-
ляются производные витамина В12. В этот же класс
включена фосфомутаза, катализирующая внутримо-
лекулярную изомеризацию 2-фосфо-П-глицериновой
к-ты в З-фосфо-Б-глицериновую к-ту. В то же время
такие фосфомутазы, как фосфоглюкомутаза или фос-
фоглицеромутаза по новой классификации выделены
в подподкласс 2. 7. 5 (фосфотрансферазы с кажущим-
ся внутримолекулярным переносом) и отнесены к
главному классу трансфераз, поскольку истинный ме-
ханизм реакций, катализируемых этими фермента-
ми (подробнее см. Коферменты}, состоит не во внутри-
молекулярной изомеризации, а в межмолекулярном
переносе остатка фосфорной к-ты.
В целях идентификации каждому ферменту при-
сваивается шифр (номер) по четырехзначному коду,
тесно связанному с системой классификации (см. вы-
ше «Ключ к классификации. . . »). По этой системе
первая цифра шифра указывает главный класс, вто-
рая — подкласс, а третья — подподкласс, определяя
тем самым тип катализируемой ферментом реакции.
Четвертая цифра означает порядковый номер фермен-
та в пределах данного подподкласса. Так, напр.,
ксантиноксидаза имеет шифр 1.2. 3. 2, к-рый иденти-
фицирует ее как оксидоредуктазу (главный класс 1),
действующую на альдегид в качестве донора (под-
класс 2) и использующую О2 в качестве акцептора
(подподкласс 3), порядковый номер фермента внутри
данного подподкласса 2. Вновь открытым ферментам
шифры присваиваются не отдельными исследователя-
ми, а специально уполномоченными на то органами
Международного биохимического союза: Постоянным
комитетом и периодически формируемыми Комиссия-
ми по ферментам. Однажды присвоенный ферменту
шифр сохраняется за ним как постоянный идентифи-
кационный признак и может быть изменен лишь в слу-
чае выявления ошибочности классификации и пере-
мещения фермента вследствие этого в другую группу.
В соответствии с рекомендациями Комиссии по фер-
ментам, одобренными Ассамблеей Международного
биохимического союза, приняты две номенклатуры
ферментов: систематическая и тривиальная (рабочая).
Систематич. наименование идентифицирует фермент
и по возможности точно определяет характер катали-
зируемой им реакции. Согласно «Правилам система-
тической и тривиальной номенклатуры», систематич.
наименование фермента составляется из двух частей.
Первая часть указывает название субстрата, а при би-
молекулярной реакции — названия двух субстратов,
разделенные знаком деления (двоеточие без интер-
вала). Вторая часть названия с окончанием «—аза»
указывает природу реакции. На русском языке часть
систематич. наименования, к-рая включает названия
субстратов, пишется, в отличие от английской номен-
клатуры, не раздельно от второй части наименования,
а через длинную соединительную черточку (знак свя-
зи), напр.: «Ацетил-КоА—гидролаза» 3. 1.2.1; «D-глю-
козо-6-фосфат:НАДФ — оксидоредуктаза», 1.1. 1.49.
Если суммарная реакция включает два различных
превращения, напр. окислительное дезаминирование,
то характер второй реакции следует указывать, до-
бавляя в круглых скобках соответствующее причас-
тие, напр.: «П-аспартат:О2 — оксидоредуктаза (де-
заминирующая)» 1.4.3.1. Входящие в название фер-
мента химич. наименования субстратов должны быть
сформулированы, следуя официальным правилам
Международного союза чистой и прикладной хи-
мии (см. Handbook for chemical society authors. Che-
mical Society, L., 1960).
Систематич. наименование может быть присвоено
ферменту лишь после того, как выяснена природа ка-
тализируемой им реакции. В ряде случаев, напр-
для пептидгидро лаз, разработать систематич. наиме-
нования оказалось невозможным ввиду того, что их
специфичности широко перекрываются, а раздельное
существование нек-рых из них как индивидуальных
ферментов не совсем достоверно. Эти ферменты иден-
тифицируются только шифрами и не имеют система-
тич. наименований (но имеют тривиальные названия—
см. ниже).
563
НОМЕНКЛАТУРА И КЛАССИФИКАЦИЯ ФЕРМЕНТОВ
564
Помимо систематич. наименования, каждый фермент
имеет тривиальное (рабочее) название, используемое
в повседневной практике. Это название должно быть
достаточно простым и кратким, но не обязательно
очень точным и строго систематичным. Так, напр., фер-
мент под шифром 2. 7. 5. 3 имеет систематич. наи-
менов ание «D-2,3-дифосфоглицерат: D-2-фосфоглице-
рат — фосфотрансфераза», и тривиальное — «фосфо-
глицеромутаза». Если систематич. наименование дос-
таточно кратко и просто, то в нек-рых случаях оно
используется и в качестве тривиального, напр. «Аце-
тил-КоА — гидролаза». В большинстве случаев в ка-
честве тривиальных наименований сохранены наз-
вания, принятые ранее до введения систематич. но-
менклатуры. В то же время ряд старых наименований,
идущих вразрез с «Правилами» или дезориентирую-
щих относительно истинного субстрата или природы
катализируемой реакции, признаны подлежащими
исключению из употребления и замене. Перечень
нек-рых подлежащих замене наименований приве-
ден в таблице.
Старое наиме- нование фер- мента Новое наименование фермента Шифр фермента
систематическое тривиальное
Аконитаза Цитрат (изоцит- Аконитатгид-
рат) — гидро-лиаза ратаза 4.2.1.3
Диафораза НАД-На1 липоамид — Липоамидде-
оксидоредуктаза гидрогеназа 1.6.4.3
Гиалуронида- Гиалуронат — лиаза Гиалуронат-
за лиаза 4.2.99.1
Енолаза D-2-фосфоглицерат — гидро-лиаза Фосфопиру- ват - гидра- таза 4.2.1.11
Инвертаза P-D-фруктофурано- р-фруктофу-
(сахараза) зид — фруктогидро- лаза ранозидаза 3.2.1.26
Карбоксилаза Карбокси-лиаза 2-ок- сокислоты (а-кето- Пируватде- карбоксила-
кислоты) за 4.1.1.1
Коллагеназа Нет Клостридио- пептидаза 3.4.4.19
Кротоназа L-3-оксиацил-КоА — Еноил-КоА —
гидро-лиаза гидратаза 4.2.1.17
Лизоцим N -ацетилмурамид — гликаногидролаза Мурамидаза 3.2.1.17
Липоксидаза Нет Липоксигена- за 1.99.2.1
В соответствии с «Правилами систематической и
тривиальной номенклатуры» исключается из употреб-
ления термин «дегидразы», к-рый ранее применялся
как в отношении дегидрогенизирующих (отщепляю-
щих водород), так и дегидратирующих (отщепляю-
щих воду) ферментов. Для первого типа ферментов
официально принят термин «дегидрогеназы», а для
второго •— «дегидратазы». Признано нежелательным
употребление в качестве названия ферментов описа-
тельных терминов, таких, как «конденсирующий фер-
мент», «промежуточный фермент», «яблочный фермент»
и др. В частности, термин «конденсирующий фермент»
заменяется тривиальным наименованием цитрат-син-
таза [систематич. название и шифр: цитрат—оксало-
ацетат-лиаза (ацетилирующая КоА), 4. 1. 3. 7]. В тех
случаях, когда субстрат в нормальных условиях на-
ходится в форме аниона, то в наименовании фермента
указывается название аниона с окончанием «—ат», а
не свободной к-ты с окончанием «—ико», напр.,
лактатдегидрогеназа, а не лактикодегидрогеназа.
Окончание «—аза» в систематич. наименованиях
не должны присоединяться непосредственно к наз-
ванию субстрата; в тривиальной номенклатуре пря-
мое присоединение окончания «—аза» к названию суб-
страта обозначает гидролитич. фермент (напр.,
уреаза). Названия, предназначенные для обозначения
ферментов, особенно термины с окончанием «—аза»,
могут употребляться только для индивидуальных
ферментов; их не следует применять для обозначения
систем, содержащих более одного фермента. Если
желательно присвоить такой системе название, осно-
ванное на катализируемой ею суммарной реакции, то
такое название должно включать слово «система».
Так, систему, катализирующую окисление сукцината
кислородом и состоящую из сукцинатдегидрогеназы,
цитохромоксидазы и нескольких промежуточных пе-
реносчиков, не следует называть сукцинатоксидазой,
но ее можно обозначить как «сукцинатоксидазная сис-
тема».
В соответствии с классификацией ферментов и пра-
вилами их номенклатуры разработан официальный
список ферментов. В список включено более 700 из-
вестных к настоящему времени ферментов. В списке
указаны шифр (номер) фермента, его систематич. наи-
менование, тривиальное название или названия (в
порядке предпочтения), а также суммарное уравнение
катализируемой реакции. При открытии нового фер-
мента, еще не фигурирующего в «Списке», автор может
предложить новое систематическое и (или) тривиаль-
ное название, составленное обязательно в соответст-
вии с рекомендованными «Правилами». Поддержание
списка ферментов на уровне текущего дня, внесение
в него дополнений и изменений, апробация назва-
ний новых ферментов и присвоение ферментам шиф-
ров возлагается на специальные органы Междуна-
родного биохимического Союза — Постоянный Коми-
тет или Комиссию по ферментам.
Классификация ферментов, а также их системати-
ческая и тривиальная номенклатуры вступили в силу
после их одобрения Ассамблеей Международного био-
химического союза. Редакциям журналов и неперио-
дич. научных изданий, а также отдельным авторам
рекомендуется пользоваться систематич. и тривиаль-
ными названиями, приведенными в списке ферментов,
и руководствоваться «Правилами систематической и
тривиальной номенклатуры». Если основным пред-
метом научного труда или реферата является тот или
иной фермент, то при первом упоминании его в тексте
должен быть указан его шифр, систематич. название и
биологич. источник, после чего может употребляться
тривиальное название. Ферменты, не составляющие
основной предмет работы, при первом упоминании
идентифицируются путем указания их шифра.
Помимо классификации и номенклатуры ферментов,
Ассамблеей Международного союза утверждены также
рекомендации, касающиеся определения единиц актив-
ности ферментов, символов кинетики ферментов (см.
Ферменты), номенклатуры коферментов (см. Нико-
тинамиднуклеотидные коферменты), а также класси-
фикации и номенклатуры цитохромов.
Лит.: Классификация и номенклатура ферментов. Отчет
Комиссии по ферментам Международного биохимического
союза, 1961, пер. с англ., М., 1962; Report of the Commission
on enzymes of the International Union of biochemistry, 1961,
Oxf.—L.—N. Y.—P., 1961. Б. В. Спиричев.
НОМЕНКЛАТУРА КОМПЛЕКСНЫХ СОЕДИНЕ-
НИЙ — см. Комплексные соединения.
НОМЕНКЛАТУРА НЕОРГАНИЧЕСКИХ СОЕДИ-
НЕНИЙ. Содержание:
Общие вопросы химической номенклатуры. Номен-
клатура химических элементов ................. 564
Возникновение и развитие русской Н. н. с. в 19 в. . . 565
Русская Н. н. с. в первой половине 20 в..........566
Новые правила Н. н. с............................568
Общие вопросы химической номенклатуры. Номен-
клатура химических элементов. Химич, номенклату-
ра — система наименований химич. элементов и сое-
динений, к-рая должна обеспечивать ясное обозна-
чение состава веществ как в письменном тексте, так
и в разговорной речи. Главное требование, предъяв-
ляемое к рациональной номенклатуре химич. соедине-
565
НОМЕНКЛАТУРА НЕОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
566
ний,— однозначность определения того или иного
соединения, исключающая возможность спутать од-
но соединение с другим. Рациональная номенклатура
должна быть достаточно универсальна и проста по
структуре, а также, по возможности, увязана с вопро-
сами классификации и строения соответствующих
соединений. При удовлетворении последним требо-
ваниям номенклатура не только отображает, но и
дополняет химич. символику, т. е. систему формул,
однозначно определяющих состав, но часто не дающих
представления о характере химич. соединения.
Первую номенклатуру химич. соединений разра-
ботали (1787) Лавуазье, Гитон де Морво, Бертолле и
Фуркруа. Основные принципы этой номенклатуры бы-
ли использованы при разработке номенклатуры химич.
соединений в других странах, в том числе и русской
химич. номенклатуры. Номенклатура химич. элемен-
тов, сложившаяся в России в течение 19 в., в боль-
шинстве случаев исходила из латинских названий,
приспособленных к строю русского языка (отбрасы-
вание лат. окончания «ium», напр. titanium — титан,
или замена его на русский «ий», напр. natrium — нат-
рий). Названия нек-рых элементов явились переводом
латинских названий, напр. oxygenium—кислород.
У элементов, с давних пор известных в России, были
оставлены русские традиционные названия: железо,
золото, медь, мышьяк, олово, ртуть, свинец, сера,
серебро, сурьма.
Возникновение и развитие русской Н. и. с. в 19 в.
Русская Н. н. с. создавалась постепенно в течение 19 в.
трудами А. И. Шерера, Я. Д. Захарова, В. М. Север-
гина и особенно Г.И.Гесса и Д. И. Менделеева. К кон-
цу 19 в. она приобрела определенную систематич-
ность и стройность. Эта стройность объяснялась тем,
что рамки систематич. Н. н. с. того времени охваты-
вали лишь немногочисленные группы соединений,
а именно, простые соединения элементов с водородом,
галогенами, кислородом (а также группой ОН) и се-
рой, кислородные кислоты, соли кислородных кислот.
Соединения, относящиеся к иным группам, были мало
известны, не играли существенной роли и, как пра-
вило, не получали систематич. названий.
По русской Н. н. с. того времени в случае соеди-
нений двух элементов название электроположитель-
ного элемента обычно давалось в форме существитель-
ного, а электроотрицательного — прилагательного,
напр. NaCl — хлористый натрий. Однако при образо-
вании данными элементами двух или нескольких со-
единений было необходимо отмечать в названии ва-
лентность (точнее степень окисления) элемента и свя-
занный с этим количественный состав соединений.
В этих случаях для простых соединений элемента с
галогенами, к-рые в названии соединения фигуриро-
вали в виде прилагательных, последним придавались
окончания «истый» и «ный», напр. FeCl2— хлористое
железо, FeCls — хлорное железо и т. п. Для обозна-
чения окислов элемента, в к-рых он имеет различную
валентность, служили названия «закись», «окись» и
«перекись», напр.: МпО — закись марганца, МщО.з—
окись марганца, МпО2 — перекись марганца. Кислот-
ные окислы назывались ангидридами, напр.: SOs —
серный ангидрид, SO2—сернистый ангидрид, Мп2О,—
марганцовый ангидрид.
При такой системе не было полного соответствия
названий количественному составу, что создавало
большие неудобства. Однако использование в наз-
ваниях соединений обозначения числа атомов элемен-
та в молекуле хотя и вводилось, но с рядом оговорок.
Так, Д. И. Менделеев писал, что окислам нередко при-
дают названия по числу атомов содержащегося кис-
лорода, напр.: SO2 — двуокись серы, SO3 — трех-
окись серы, МпО — одноокись марганца, МП2О3 —
полуторная окись, МпО2 — двуокись и т.п.; а такая
номенклатура игнорирует качества, к-рые в химии
стоят на первом плане. Поэтому Менделеев предлагал
номенклатуру количественного состава использовать
только для окислов, не обладающих явно качествами
оснований и кислот. В связи с тем, что выявилась
необходимость отличать названием «перекись» соеди-
нения, содержащие в своих молекулах группировку
—О—О—, Менделеев указал, чтоМпО2иРЬО2 лучше
называть двуокисями, чем перекисями.
В названиях кислородных кислот использовались
в виде прилагательных названия кислотообразующего
элемента с суффиксами, характеризующими степень
его окисления (по мере ее повышения): . . .оватис-
тая, . . .истая, . . . оватая и.ная или . . . овая (пос-
леднее для максимальной степени окисления), напр.:
НС1О— хлорноватистая кислота, НС10г— хлористая,
НСЮз — хлорноватая, НСЮа — хлорная кислота.
Прилагательными к слову ангидрид с подобными же
суффиксами обозначались и соответствующие кисло-
там окислы элементов (кислотные окислы или ангид-
риды к-т).
Названия солей составлялись из названий соответ-
ствующей кислоты и металла, к-рые объединялись в
прилагательное к существительному «соль», напр.:
Na2SO4 — сернонатриевая соль, КС10з—хлорно-
ватокалиевая соль и т. п. Позднее эти названия были
вытеснены названиями, в к-рых прилагательным слу-
жило название кислоты с суффиксом «кислый», напр.:
NaNOs — азотнокислый натрий, K.NO2 — азотисто-
кислый калий. Подобным же образом составлялись
еще мало распространенные в то время названия солей
комплексных кислот,напр.:Хаг81Гв—кремнефтористый
натрий, K3Fe(CN)6— железосинеродистый (железо-
цианистый) калий, К.гРЮ16— платинохлористый ка-
лий. Наряду с систематич. названиями в русской хи-
мич. литературе широко применялись названия три-
виальные.
Русская Н. н. с. в первой половине 20 в. В связи
с недостатками русской Н. н. с. в конце 19 в. и начале
20 в. делались попытки ее улучшить, а также сбли-
зить с международной Н. н. с. Так, Ф. М. Флавицкий
предложил номенклатуру, к-рую он назвал «рацио-
нальной числительно-элементной». Однако эти по-
пытки не были реализованы, а согласно постановле-
нию Русской комиссии при РФХО, подписанному
Н. С. Курнаковым, А. И. Горбовым и Л. А. Чугаевым
(1912), нормой признавалась та номенклатура, к-рая
была предложена Гессом и разработана Менделеевым.
В 1-й половине 20 в. в русскую Н. н. с. постепенно
вводятся обозначения, заимствованные из иностран-
ной номенклатуры. Это прежде всего обозначения
электроотрицательной части простых соединений
элементов (напр., двойных, или бинарных,соединений)
и анионов солей кислородных к-т. Для первых из них
используются корни латинских названий элементов,
играющих роль электроотрицательных в двойном сое-
динении, с суффиксом «ид». Для вторых — корни
латинских названий элементов, образующих кислород-
ные кислоты, с суффиксами «ат» — при проявлении эле-
ментом высшей валентности, и «ит» — низшей валент-
ности. Названия соответствующих соединений состав-
ляются из двух слов, первым из к-рых является ука-
занное обозначение электроотрицательного элемента
или аниона в именительном падеже, а вторым — обыч-
ное русское название металла или вообще электропо-
ложительного элемента (или группы) в родительном
падеже. Т. обр., хлористые соединения получили наз-
вание хлоридов, окислы — оксидов, сернистые соеди-
нения — сульфидов, азотистые — нитридов и т. п.
Азотнокислые соли получили название нитратов, азо-
тистокислые — нитритов, сернокислые — сульфатов,
сернистокислые — сульфитов, углекислые — карбо-
натов, и т. д.
567
НОМЕНКЛАТУРА НЕОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
568
Использование в русской Н. н. с. этой системы обо-
значений оказалось довольно плодотворным. Оно спо-
собствовало приданию Н. н. с. большей систематич-
ности, давало возможность кратко и выразительно
называть целые ряды многочисленных производных, а
также сближало русскую Н. н. с. с номенклатурой
иностранной. Трудности и осложнения появились и
здесь в связи с необходимостью, в случае образования
данным элементом нескольких однотипных соедине-
ний, отмечать в названии степень окисления эле-
мента и связанный с этим количественный состав сое-
динений. Использование числительных (греческих)
в качестве префикса в обозначении электроотрица-
тельной части соединений оказалось возможным толь-
ко в названиях простых соединений элементов, напр.:
ЕеС1г — дихлорид железа, FeCl3 — трихлорид же-
леза. В названиях же солей кислородных кислот
этот способ не был применен; напр., Sn(SO4)2 не
было названо, в отличие от SnSO4, дисульфатом олова.
Для названий различных соединений практиковалось
также использование числовых обозначений степени
окисления, предложенных Штоком в 1919 и принятых
в немецкой Н. н. с. с 1925. По этому способу FeCh
соответствует название хлорид железа (II), FeCl3—
хлорид железа (III), SnSO4 — сульфат олова (II),
Sn(SO4)2 — сульфат олова (IV).
Исключения и непоследовательности при использо-
вании в русской Н. н. с. указанной системы наимено-
ваний проявились также при образовании названий
солей в случае многочисленности кислородных кис-
лот данного элемента, напр. аниону С104 присвоено
название перхлорат, C1OS — хлорат, в то время как
манганатом назван анион Мп04~, а перманганатом —
анион Мп04-. Далее, не было однотипности в назва-
ниях анионов кислых солей. Так, напр., кислые
анионы двухосновных кислот обозначались с при-
ставкой «би»: HS04 — бисульфат, HSOs — бисуль-
фит, НСО3 — бикарбонат; однако бихроматом обоз-
начался анион Сг2О* ,а не НСгО4 Кислые анионы
трехосновных кислот получили названия: Н2РО~—
однозамещенный фосфат, HPOJ —двузамещенный фос-
фат. Были попытки называть также различные со-
ли многоосновных кислот, обозначая число атомов
металла греч. числительными, напр., NaH2PO4 —
мононатрийфосфат, Na2HPO4 — динатрийфосфат,
Na3PO4 — тринатрийфосфат. Однако эти названия
не получили широкого распространения.
Для комплексных соединений сис-
тематич. номенклатура была предложена А. Верне-
ром. Принцип этой номенклатуры в адаптированном к
русскому языку виде сводится к следующему. Сначала
называется собственно комплекс (комплексный ион), а
затем внешнесферный ион. В названии комплекса
сначала обозначаются внутрисферно связанные атомы
или группы, т. е. лиганды (или иначе адденды), а за-
тем центральный атом. Обозначение последнего сос-
тавляется из латинского названия соответствующего
элемента с суффиксом, характеризующим степень его
окисления (т. е. положительную валентность) таким
образом: 1 — «а», 2 — «о», 3 — «и», 4 — «е», 5 — «ан»,
6 — «он», 7 — «ин», 8 — «ен». Если комплекс является
анионом, то его название оканчивается на «ат». Отри-
цательно заряженные лиганды обозначаются сокра-
щенными латинскими названиями с соединительной
гласной «о», нейтральные лиганды — их обычными
названиями, аммиак — названием «аммин», вода —
«акво». Число лигандов данного рода обозначается
приставкой греч. числительного — ди-, три-, тетра-...
и т. д. Если комплекс в целом нейтрален и, т. обр., не
связан с внешнесферными ионами, то его название
заканчивается обычным русским обозначением цент-
рального атома без всяких указаний степени окисле-
ния. Примеры названий соединений с комплексными
анионами: Na2[SiFe] — гексафторосилицеат натрия,
K3[Fe(CN)s]— гексацианоферриат калия, К2 [PtClJ—
гексахлороплатеат калия. Примеры названий соеди-
нений с комплексными катионами: [Co(NH3)e]Cl3 —
гексамминкобальтихлорид, [Pt(OH)2(NH3)4]SO4 — ди-
гидроксотетрамминплатесульфат. Примеры названий
нейтральных комплексных соединений: [Со(НОг)з
(NH3)3] — тринитротриамминкобальт, [PtCl4(NH3)2]—
тетрах л op одиамминпл атина.
Эта номенклатура комплексных соединений была
широко использована в русской химич. литературе.
Однако для комплексных анионов наряду с этой сис-
темой используется и другая система с обратным
порядком перечисления обозначений центрального
атома и лигандов, напр. [Fe(CN)e]3- — феррицианид.
Подробнее см. в ст. Комплексные соединения.
Здесь были приведены краткие сведения о приме-
няемых в русской химич. литературе номенклатурных
системах, охватывающих лишь главные типовые
Н. н. с. Помимо этого, в русской Н. н. с. применяются
многочисленные термины для обозначения атомных
групп, или радикалов, а также названия соединений,
относящихся к специальным областям неорганич.
химии (региональная Н. н. с.).
Из названий, применяемых для обозначения атом-
ных групп, или радикалов, следует указать на наз-
вания электроположительных групп, состоящих из
положительно многовалентного атома элемента и
атомов кислорода, соответственно уменьшающих по-
ложительную валентность группы в целом. Названия
этих групп составляются из корня латинского назва-
ния элемента и суффикса «ил», например сульфу-
рил— SO2, карбонил — СО, нитрозил — NO, нитро-
ил — NO2 и т. п.
В связи с развитием химии и расширением круга
соединений отдельных элементов появилась необходи-
мость в региональной Н. н. с. Придание последней
систематичности проводилось на основе аналогий.Так,
региональная номенклатура соединений кремния соз-
давалась посредством аналогии с номенклатурой ор-
ганич. соединений. Соединения кремния с водородом,
аналогичные по составу и структуре предельным угле-
водородам — алканам, были названы силанами, их гид-
роксильные производные — силанолами и т. д. По-
скольку кремний является аналогом углерода, ана-
логия между номенклатурами кремниевых и органич.
соединений оказалась вполне уместной и плодо-
творной.
К середине 20 в. в русской химич. литературе —
учебной, научной, справочной, одновременно приме-
няются различные Н. н. с«, но ни одна из них не охва-
тывает всего многообразия химич. соединений. Кроме
того, каждая из применяемых систем имеет свои част-
ные недостатки, греша в основном непоследовательно-
стью. Приблизительно такое же положение сложилось
и в иностранной химич. литературе.
Новые правила Н. н. с. Попытки упорядочения рус-
ской Н. н. с., к-рая создавалась и дополнялась посте-
пенно, по мере расширения круга известных соедине-
ний, делались неоднократно. Наиболее серьезными из
них следует считать проекты, разработанные номен-
клатурными комиссиями Всесоюзного Менделеевского
съезда в 1932 и Всесоюзного Менделеевского общества
в 1937. Однако эти проекты не были реализованы.
В целях упорядочения Н. н. с. в международном
масштабе в 1959 Номенклатурной Комиссией Между-
народного союза чистой и прикладной химии (JUPAC)
были разработаны правила номенклатуры неорганич.
569
НОМЕНКЛАТУРА ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
570
химии. Работы по подготовке проекта русской номен-
клатуры неорганич. соединений, удовлетворяющей
современным требованиям, ведутся номенклатурной
комиссией Отделения химич. наук АН СССР. В СССР
ведется также разработка правил Н. н. с. на украин-
ском, азербайджанском, литовском, латышском язы-
ках. Разработаны правила Н. н. с. на молдавском и
эстонском языках.
Лит.: Савченков Ф., ЖРХО, 1870, 2, вып. 5—6,
205; Менделеев Д. И., Основы химии, т. 1, 9 изд., М.—Л.,
1927; Флавицк и й Ф. М., ЖРФХО, 1920, 50, вып. 3—4,
25; ФрицманЭ., там же, 1929, 61, 3; Г р и н б е р г А. А.,
Введение в химию комплексных соединений, 2 изд., Л.—М.,
1951; Труды VI Всесоюзного Менделеевского съезда по теоре-
тической и прикладной химии, т. 1, М., 1933; Усп. химии, 1933,
2, вып. 2, 249; Бори А. X., там же, 1937, 6, вып. 4, 605, 615;
VIII Менделеевский съезд по общей и прикладной химии.
Проект номенклатуры неорганических соединений, М., 1959;
International Union of pure and applied chemistry. Inorganic
chemistry section. Report of the Commission on the nomencla-
ture of inorganic chemistry. Definitive rules for nomenclature
of inorganic chemistry, L., 1959; Л учинский Г. П., Проект
правил номенклатуры неорганических соединений, М., 1962;
А б л о в А. В., Б а т ы р Д. Г., Номенклатура кимией неор-
ганиче, Кишинэу, 1962; Номенклатура неорганических со-
единений. (Проект правил), М., 1 963. Г. П. Лучинскии.
НОМЕНКЛАТУРА ОРГАНИЧЕСКИХ СОЕДИНЕ-
НИЙ. Содержание:
Введение .................................569
Рациональнан номенклатура ............... 570
Женевская номенклатура .................. 571
Льежские правила и номенклатура .TUPAC-1957 . .574
Номенклатура циклических соединений ..... 575
Стереохимические обозначения..............577
Введение. Номенклатура органических соединений—
система названий органич. соединений. Развитие
Н. о. с. тесно связано с историей открытия отдельных
соединений и с развитием теоретич. представлений в
органич. химии. В первоначальный период развития
органич. химии вновь открытые соединения получали
тривиальные (несистематические) названия. В та-
кого рода названиях отражаются, напр., природные
источники веществ (муравьиная к-та, винная к-та,
мочевина, молочный сахар, кофеин, бензойная к-та),
особенно заметные свойства вещества (пикриновая
к-та, какодил, гремучая к-та, свинцовый сахар), спо-
собы получения (пирогаллол, серный эфир), имя от-
крывшего ученого ( кетон Михлера, углеводород Чи-
чибабина, комплекс Иоцича).
В начале 19-го столетия, когда в органич. химии
были распространены дуалистич. представления Бер-
целиуса (теория радикалов), были введены такие на-
звания, как хлористый бензоил, бромистый этил, этил-
ацетат, окись мезитила и т. д. В дальнейшем, в сере-
дине прошлого столетия, появились названия, от-
ражающие представления унитарной теории Жера-
ра (теории типов). Названия такого рода составили
первую обширную систему научной номенклатуры в
органич. химии, т. н. рациональную номенклатуру.
В 1892 на международном химич. конгрессе в Же-
неве была принята т. н. Женевская номенклатура, где
в основу названия было положено химич. строение и,
прежде всего, характер углеродного скелета молеку-
лы. Официальные Женевские правила были разработа-
ны лишь для сравнительно простых соединений.Более
полными явились Льежские правила, утвержденные
на X конференции Международного Союза чистой и
прикладной химии (JUPAC) в 1930 в г. Льеже и в
нек-рой части противоречащие правилам женевской
номенклатуры. В последующие годы эти правила неод-
нократно дополнялись. Эту работу и в настоящее
время продолжает комиссия по Н. о. с. при Между-
народном Союзе чистой и прикладной химии.
Тривиальные названия особенно час-
то применяются в технике, а также для обозначения
сложных веществ и вновь открытых веществ неуста-
новленного строения. Тривиальные названия не от-
ражают химич. строения, а число их весьма велико и
продолжает расти, поэтому для ориентировки в по-
добных названиях имеются соответствующие справоч-
ники.
Рациональная номенклатура. При составлении ра-
ционального названия соединения рассматриваются
как продукты усложнения простейшего представителя
данного гомология, ряда. Т. обр., основу рациональ-
ных названий насыщенных углеводородов составляет
метан, ненасыщенных — этилен, ацетилен; спиртов—
метиловый спирт и т. д. Для указания положения ра-
дикалов атомы углеродной цепи обозначают обычно
буквами грея, алфавита, начиная счет от функциональ-
ной группы. Приводимые ниже примеры иллюстриру-
ют способы составления рациональных названий
(в ф-лах основа названия обведена пунктиром):
СНЧ
СН3гС+СН2—сн3
СН3
триметил-этил-метан
СН3уСН=СН4-СН3
a,ft - диметилэтилен
(диметилзтилен сими.)
СН3
СН3
СН3
\ /Г'3
zCH+CH-t-CH-CH2-CH3
сн3
мети л-иэопропил - аторичнойутил - метан
СН
СН
а, а-диметилзтилен
(диметилзтилен несимм)
СН3
n’t**
CHo-t-C-IOH
•Т 44./
сн3
гриметилнарбинол
сн3-------------х
;сн-соон'
с2н5а----------
метил-згил-уксусная кислота
с6н54сн2-со-сн3)
фенилацетон
В дальнейшем аналогичный принцип составления
названий стал применяться в более гибкой форме:
за основу часто выбирают не обязательно простейший,
а наиболее удобный для данного конкретного случая
представитель ряда. Примерами таких названий яв-
ляются:
(CH3-CH-CHjr соон')
он"
^-онсимаслякд кислота
С6Н5 /СбН
СбН5+с-с^с6н
^6^5 С6Н
генсафенилэтан
5
5
5
свн5/сн-сн3 \ (ноос-сн2-сн-сн2-сн2-соо"н';
сн;
«-фенилэтиламии
В - метиладипииовая кислота
Принципы рациональной номенклатуры применяют-
ся также в ряде «региональных номенклатур», спе-
циально приспособленных для нужд определенных раз-
делов органич. химии. Так, напр., углеводород ментан
является основой ра- сн
циональных названий 71 3
моноциклич. терпе- ^СН
нов, напр. ментан (I), Н2С® 29На
ментен-1(П) и мента- Н2С5.4^СН2
диен-1,4(8) (III). СН
В основу рациональ- асн
ных названий стерои- н3СХ СН
дов положено триви- 3
альное обозначение 1
in
полициклич. углеродного скелета. При этом, поскольку
в стероидных системах особое значение имеет простран-
ственное строение, вводят специальные названия для
571
НОМЕНКЛАТУРА ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
572
важнейших стереоизомеров,в частности андростан(1У),
этиохолан (V). Один из гормонов, производных андро-
стана — тестостерон (VI) — получает, напр., назва-
ние андростен-4-ол-17Р-он-3.
iv v Vi
В последнем из названий греч. буква 0 относится
к конфигурации асимметрич. центра, у к-рого стоит
гидроксильная группа; буквой 0 обозначают про-
странственное положение тех заместителей, к-рые
в условной ф-ле оказываются расположенными над
плоскостью кольца (сплошная линия связи), буквой
а — заместители под плоскостью кольца (пунктирная
линия связи).
Женевская номенклатура. В отличие от рациональ-
ной номенклатуры, допускающей построение несколь-
ких равноправных названий для одного и того же ве-
щества, Женевская номенклатура была задумана
как номенклатура официальная для словарей, спра-
вочников, указателей, причем определенной струк-
турной ф-ле должно соответствовать одно и только
одно официальное название.
В проекте, послужившем основой для дискуссии на
Женевском конгрессе, были четко сформулированы че-
тыре принципа, положенные в основу создаваемой
номенклатуры: а) номенклатура основана на принципе
замещения; б) названия всех представителей к.-л. се-
мейства строятся на основе названия углеводорода,
от к-рого путем замещения могут быть произведены
эти представители (т. н. генетич. принцип); в) нали-
чие заместителей выражается префиксами и суффик-
сами; г) названия образуются на основе ф-лы строения
вещества.
Женевские постановления состоят из 62 правил,
к-рые устанавливают порядок составления официаль-
ных названий для углеводородов ациклич. строения
и их производных с одним или несколькими одинако-
выми заместителями. Женевская система была поло-
жена в основу номенклатуры справочника Бейлып-
тейна, причем практика потребовала дальнейшего раз-
вития и дополнения женевских правил.
Для построения женевского названия любого
ациклич. соединения надо прежде всего выбрать в нем
главную, наиболее длинную цепь углеродных атомов
(пример 1, главная цепь выделена жирным шриф-
том). Если же строение соединения таково, что
можно выбрать несколько цепей максимальной дли-
ны, главной из них считается та, к к-рой примыка-
ет наибольшее число боковых цепей и заместителей
(пример 2). При равном числе боковых цепей или
заместителей вопрос решается на основании их стар-
шинства (пример 3). (О старшинстве см. ниже).
с с с
I I I
1. С-С-С-С-С-С 2. С-С-С-С-С-С-С
II II
с с с с-с-с
I I I
с с с
с
I
3. с-с-с-с.
ХС-С-С-С-С-С
c-c-c-cz
I
с
I
с
Углеродные атомы главной цепи нумеруют, начиная
счет с того конца цепи, к к-рому ближе стоит к.-л.
из боковых цепей (пример 4). Если же с обоих кон-
цов имеются боковые цепи, неодинаковые по слож-
ности, то нумерацию начинают с того конца, к к-рому
ближе простейшая цепь (пример 5). Если, наконец,
на одинаковом расстоянии от обоих концов главной
цепи расположены одинаковые разветвления, а кроме
них имеются еще и другие боковые цепи, то нумера-
цию начинают с того конца, где разветвлений больше
(пример 6).
1 2 2 4 S в 7 12S4S878
4. С-С-С-С-С-С-С 5. С-С-С-С-С-С-С-С
с
7 8 S 4 S 2 1
в. С-С-С-С-С-С-С
I I I
с с с
В простейшем случае для насыщенных углеводо-
родов женевское название строится из обозначения
главной цепи с принятым для парафинов окончанием
«ан» и примыкающих к ней боковых цепей, причем
цифрами указываются места расположения последних.
Разветвления в боковых цепях — боковые цепи вто-
рого порядка, обозначаются префиксами: мето, это,
пропо и т. д. Так, насыщенные углеводороды со ске-
летом, отвечающим примерам 1—6, получат названия:
1. 3,4,5-триметил-гептан;
2. 2,2,6-триметил-5-этил-З-метоэтил-гептан;
3. 4-метил-5(51-этобутил)-декан;
4. 2,5-диметил-гептан;
5. 2-метил-6-этил-октан;
6. 2,3,6-триметил-гептан.
Если имеется более сложное органич. соединение,
содержащее, напр., кратные связи, функциональные
группы и нефункциональные заместители, то в наз-
вание дополнительно вводятся суффиксы и префиксы,
отражающие наличие соответствующих структурных
элементов, и цифры, обозначающие их положение.
При этом правила выбора главной цепи и ее нумерации
сохраняют свою силу: в соответствии с § 17 Женев-
ских правил «для продуктов замещения углеводородов
сохраняется нумерация углеводородов». Практически
это означает, что последующие изменения (введение
кратных связей, различного рода заместителей) не
изменяют той части слова-названия, к-рая отражает
строение углеродного скелета. Все части слова-наз-
вания располагаются всегда в строго определенной
последовательности, что позволяет дать наглядную
графич. схему, облегчающую построение и расшиф-
ровку женевских названий. Разделы схемы перенуме-
рованы в порядке падающего старшинства (самый
старший раздел — I, самый младший—VI). По вер-
тикали внутри разделов старшинство возрастает свер-
ху вниз, за исключением раздела II, где порядок воз-
растания старшинства обратный:' самым старшим яв-
ляется метил. Старшим структурным элементам от-
дается предпочтение при выборе главной цепи и опре-
делении порядка нумерации. Ниже приведена схема
построения женевских названий ациклических соеди-
нений (стр. 573).
Практич. применение схемы показано на приведен-
ных ниже примерах. Для иллюстрации применения
параграфа 17 выбраны соединения, имеющие одина-
ковый углеродный скелет (отвечающий углеводороду 5)
и отличающиеся лишь характером и расположением
кратных связей и заместителей.
он
1 2 3 4 5 | 6 7 8
СН3-СН.-СН-СН = СН-С-СН2-СН2С1
I I
сн3 сн2
сн3
8-хлор-|3-метил-6-этил-окт|ен-4-ол-6
573
НОМЕНКЛАТУРА ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
574
VI V 11 I III IV
Нефункцио- нальные заместители Азотсодер- жащие функции Боковые цепи Главная цепь Связи Кислородные функ- ции и их сернистые аналогии
F фтор С1 хлор Вг бром J иод NO нитрозо NO3 нитро NH2 амино NH0H гид- роксиламино NHNH2 гид- разино СН, метил С2Н5 этил С.Н, пропил С,Н9 бутил CjHn амил С,Н13 гексил С7Н16 гептил СЬН17 октил С.Нг, нонил С,„Н21 децил С„Н23 ундецил С1 мет С2 эт С, проп С, бут С, пент С, гекс С7 гепт С8 окт Cs нон С,о дек С1( ундек С-С ан С = С ен С 55 С ин —ОН ол — SH тиол ^)С] = О* он \C]=S тион ,0 — C]v ал ХН /° — C]<z тиал ХН /° — С]" оновая ^ОН к-та ,S — С]" тионовая ХОН к-та /° — Civ тиоловая ^SH к-та — С]^ дитионовая ^SH к-та — SO3H сульфоновая к-та
Префиксы Корень Суффиксы
* Углеродный атом отделен квадратной скобкой от собственно функции в знак
того, что он входит в счет атомов углеродного скелета молекулы.
1 2 3 4 5 6 7 S
СН.-СН-СН-СН2-СН,-СН-СН2-СООН
I I I
NHa СН, СН2
сн,
2-амино-:3-метил-6-этил-окт1ановая-8-кислота
1 2 3 4 5 6 7 8
СН3-СО-СН-СНОН-СН2-СН-СН2-СН2ОН
СН, СН2
I
СН,
;3-метил-.6-этил-окт
андиол-4, 8-ОН-2
В области ароматич. соединений женевским поста-
новлением предлагалось прежде всего заменить наз-
вание «бензол» на «бензен», однако это изменение
привилось лишь в английском языке, в то время как
в русской и немецкой номенклатурах сохранилось
традиционное название «бензол». Нумерацию в бен-
зольном ядре по женевским правилам следует начи-
нать с атома, несущего заместитель, связанный с яд-
ром при посредстве атома с наименьшим атомным но-
мером. Направление нумерации выбирают таким об-
разом, чтобы наименьшие номера получили те атомы
ядра, к-рые непосредственно связаны с атомами-
заместителями наименьшего атомного веса, напр.
2-амино-1-метил-бензолсульфоновая-5-кислота (VII).
Если имеется несколько боковых цепей, наименьшие
номера надо придавать более простым из них, напр.
1-метил-З-этил-бензол (VIII). Когда одна и та же за-
(HO3S
снэ
nh2
VII
мещающая группа повторяется не-
сколько раз, нумерацию начинают
от одной из повторяющихся групп
и ведут так, чтобы остальные груп-
пы получили возможно меньшие
номера, напр.4-амино-1,3-диметил-
бензол (IX).
Женевские правила не регламен-
тировали названий соединений
жирного ряда со смешаннымифунк-
циями, а также полициклич. кон-
денсированных структур и других
сложных соединений.
Льежские правила и номенкла-
тура JUPAC-1957. По льежским
правилам название ациклич. сое-
динения строится из тех же состав-
ных частей (корня, префиксов и
суффиксов), что и женевские назва-
ния. Однако в целевых установках
обеих номенклатур, а также в ряде
существенных деталей (выборе
главной цепи, ее нумерации, по-
рядке расположения элементов сло-
ва-названия) имеются существен-
ные различия.
Для Льежской номенклатуры ха-
рактерен прежде всего отказ от
однозначности названий, допуще-
ние возможности употребления для
одного и того же соединения не-
скольких равноправных названий.
Так, допускается выбрать за осно-
ву не самую длинную, а какую-либо
иную цепь, допустимы изменения в
порядке перечисления заместителей, отсутствует стро-
гая регламентация, какие функции (в соединениях со
смешанными функциями) выражаются префиксами, а
какие суффиксами. Правила JUPAC-1957 развива-
ют льежские, но отличаются от них допущением и в
корне отличных от женевских названий, напр. по-
строенных по принципу рациональных (см. также
ниже о циклич. соединениях). Сами правила чрезвы-
чайно обширны, составляя книгу объемом до 200 стр.
В 1957 Международной комиссией по номенклатуре
органич. соединений JUPAC утвержден новый вари-
ант правил номенклатуры углеводородов и гетеро-
циклич. ядер, однако по номенклатуре прочих соеди-
нений (т. е. всех органич. веществ, кроме углеводо-
родов) комиссия еще не приняла официального ре-
шения. В 1962 опубликованы лишь временные правила,
регламентирующие номенклатуру соединений с функ-
циональными группами, содержащими О, N, Hal, S, Se
Те. В основном эти правила являются развитием и
уточнением льежских с широким допущением назва-
ний типа рациональных (см. выше).
Названия парафиновых углеводородов по правилам
JUPAC-1957 в основном сходные женевскими; главной
считается самая длинная цепь, нумерацию ее ведут
с того конца, к к-рому ближе боковая цепь, напр.:
1234 5 6 789 10
СН,-СН-СН,-СН2-СН2-СН,-СН-СН-СН2-СН,
I I I
СН, сн, сн,
2, 7, 8-триметилдекан
При равенстве этого признака нумерацию ведут с
того конца, у к-рого больше боковых цепей, наир.:
1 2 3 4 5 6
сн,-сн-сн-сн2-сн-сн,
I I I
сн, сн, сн,
2, 3, 5-триметилгексан
Если две или большее число различных боковых це-
пей находятся на равном расстоянии от концов главной
575
НОМЕНКЛАТУРА ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
576
цепи, то для выбора начала нумерации по правилам
1957 можно применить один из двух приемов: а) наи-
меньший номер получает та из боковых цепей, к-рая
перечисляется первой в алфавитном порядке; б) наи-
меньший номер получает более простая боковая
цепь. Напр., приведенный ниже углеводород должен
быть назван по (а) 4-пропил-5-этил-октан, по (б)
5-пропил-4-этил-октан.
СН,-СН2-СН2-СН-СН-СН2-СН2-СН2
сн2 сн2
I I
сн3 сн2
I
сн,
В случае ненасыщенных углеводородов вводятся
новые правила, отдаляющие современную номенкла-
туру JUPAC от женевской. В ненасыщенных углеводо-
родах главной считается цепь с наибольшим числом
кратных связей, причем она может быть и не самой
длинной. Нумеруют ее (вне зависимости от положения
боковых цепей) так, чтобы кратные связи получили
наименьшие номера, напр.:
С,Н7 сн,
в 5 4 \ S 2 1 в S | 4 S 2 1
СН S3 с-с=с-СН = СН2 СН,-С —сн2-сн2—сн=сн2
с,н, сн,
3,4-дипропил-гексади- 5,5'-диметилгексан-1
ен-1, З-ин-5
Названия монофункциональных производных по
Льежским правилам внешне совпадают с женевскими
названиями, но отличаются порядком выбора главной
цепи и нумерации; предпочтение отдается функции, а
не углеродному скелету, напр.:
5 4 S 2 1
СН,-СН-СН2-СН-СН2ОН
I I
сн, с,н7
4-метил-2-пропил-пентанол
Для наименования соединений со смешанными функ-
циями § 51 Льежских правил предлагает оставлять в
суффиксе обозначение только одной функции (глав-
ной), а обозначения остальных выносить в префикс.
Для обозначения функций применяют приведенные
ниже префиксы и суффиксы
Функциональная группа Префикс Суффикс
- С] ООН свая к-та или оил
— соон карбокси- карбоновая к-та
— CN циано- нитрил (карбонитрил)
- ОН окси(гидрокси)- ол
- С-]<^ хн оксо, альдо- ал
— СНО формил- —
)с]=о оксо(кето) он
— OR алкокси- —
— О — эпокси-
-SH- меркапто- тиол
— SO,H сульфо- сульфоновая к-та
— NH, амино- амин
- NH — NH2 гидразино- гидразин
—N=N — азо- —
-NO, нитро- —
соли аммония — аммоний
— NHCONH, уреидо- мочевина
Примеры:
S4321 6 5 4 3 2 J
СН,-СН-СН2-СН2-СООН сн,-сн-сн3-с-сн2-сно
I I «
он сн, о
4-оксипентановая к-та, или з-кето-5-метил-гексанал-1
4-оксибута-карбоновая к-та
Номенклатура циклических соединений. В прави-
лах JUPAC-1957 имеется несколько вариантов номен-
клатуры циклич. соединений.
Для бициклических мостиковых структур правила
JUPAC-1957 рекомендуют пользоваться системой
Байера, согласно к-рой название строится из префикса
«бицикло» и греч. числительного, отвечающего общему
числу атомов в циклич. системе. Между обеими со-
ставными частями названия помещается в квадратных
скобках шифр, указывающий длину мостиков, свя-
зывающих «узловые» углеродные атомы: напр., би-
цикло[1, 1, 0]бутан (X), бицикло[3, 2, 1]октан (XI),
бицикло[2,2,1]гептен-2 (XII).
X XI ’ XII
В примере X цифры шифра [1,1,0] указывают длину
мостиков А, Б, В, связывающих СН-группы (2 мостика
с одним промежуточным атомом в каждом, один мо-
стик без промежуточных атомов). Аналогично строятся
и названия в примерах XI и XII.
Система Паттерсона. Для большой
группы более сложных карбо- и гетероциклич. ядер
(25 срощенных, конденсированных ароматич. угле-
водородов и 60 гетероциклич. ядер) в правилах
JUPAC-1957 зафиксированы рекомендуемые три-
виальные названия и принятая нумерация атомов.
Названия более сложных полициклич. ядер предла-
гается строить по системе Паттерсона, существо
к-рой заключается в следующем: сложная конден-
сированная система расчленяется на 2 составные
части, и названия строятся из тривиальных названий
составных частей. Для обозначения типа сращения
составных частей периферич. связи одной из них
(основной структуры) обозначаются буквами латин-
ского алфавита, а в заключаемом в квадратные скобки
шифре указывается, к какой из связей прпчленена
вторая структура. В случае необходимости указы-
вается также, за счет каких атомов причлененной
структуры произошло сращение, напр. бенз [а] ант-
рацен (XIII), нафто [2,1-а] пирен (XIV), бензо [h]
изохинолин (XV), пиразоло [3,2-а] пиримидин (XVI).
XV XVI
Структура в целом получает свою нумерацию,
отличную от нумерации ее составных частей. Для
установления порядка нумерации ф-лу определенным
образом ориентируют на плоскости, а именно так,
чтобы наибольшее число колец образовало горизон-
тальный ряд, а из не вошедших в этот ряд максимально
возможное число колец располагалось бы справа и
сверху:
правильно неправильно
577 НОМЕНКЛАТУРА ОРГАНИЧЕСКИХ СОЕ ДИНЕНИЙ—НОНИЛОВЫЙ СПИРТ
578
с первого
3
XVII
В ориентированной т. обр. формуле нумерацию не-
свободного угла правого верхнего
2 кольца и ведут ее по ча-
совой стрелке. Узловые
uaJ^jcJsa 4 атомы не нумеруются;
>г в случае необходимости
они обозначаются но-
7а 7 6а в мером предыдущего ато-
ма с добавлением бук-
XVIN венного индекса, напр.
пирен (XVII), 4-аза-бензантрацен (XVIII).
а-С и с т е м а. Последний из приведенных приме-
ров одновременно иллюстрирует применение т. н.
«a-системы» (также рекомендуемой правилами JUPAG-
1957 в качестве одного из возможных вариантов),
согласно к-рой названия гетероциклич. соединений
строятся из названий соответствующих карбоциклич.
систем с добавлением префикса, характеризующего
тип и положение гетероатома. При этом атом азота
обозначают «аза», кислорода — «окса», серы — «тиа»
и т. д.
Для гетероциклич. соединений в правилах JUPAC-
1957 предусмотрена также возможность применения
названий, построенных на принципах, выдвинутых
в свое время Ганчем и Видманом. При этом моноци-
клич. соединения, содержащие один или несколько
гетероатомов в составе 3—10-членных колец, получают
названия, составленные из обозначения гетероатома
по «a-системе» и окончания, характеризующего число
членов и степень насыщенности цикла в соответствии
с приводимой ниже таблицей.
и
Азотсодержащие циклы | Безазотистые циклы
3
4
5
6
7
8
9
10
ол
ин
епин
один
онин
ецин
ирин
етин
олин
иридии
етидин
олидин
ол
ин
епин
один
онин
ецин
ирен
етен
олен
*
Иран
етан
олан
ан
епан
оцан
онан
ецан
Степени гидрирования, обозначенные звездочкой,
называются путем добавления префикса дигидро,
тетрагидро и т. д. к названию низшей степени гидри-
рования. Примерами применения системы Ганча и
Видмана являются такие названия, как оксиран
(XIX), азиридин (XX), 1,2-оксатиолан (XXI).
О
Н2С — СН2
NH
Н2С S
Н2С—СН2
XIX XX XXI
Стереохимические обозначения. Отдельной и до-
вольно сложной областью номенклатуры органич.
соединений являются стереохимии, обозначения. Воп-
рос об обозначении пространственной конфигурации
в названиях остается и поныне весьма запутанным.
Более или менее общепринятым стало в последнее
время употреблять для обозначения стереохимии,
рядов прописные буквы DhL, в отличие от d и 1, за
к-рыми должна быть сохранена их первоначальная
функция как синонимов знаков (+) и (—), относя-
щихся к направлению вращения. Наиболее общие
предложения в области стереохимических обозначе-
ний внесены Каном, Ингольдом и Прелогом, а так-
же Терентьевым с сотрудниками. Последние пред-
ложили также систему обозначений для поворотных
изомеров.
Лит.: Терентьев А. П. [и др.], Номенклатура орга-
нических соединений, М., 1955; М е н ш у т к и н Н. А.
ЖРХО, ч. хим., 1892, 24, вып. 8, приложение; Ворож-
цов Н.,Уч. зап. Казахского гос. ун-та, 1941, 10, 82; Т е ге п-
р,-. Potapov V. М., Tetrahedron, 1957, 1, К5 Ч,,
119; Chemical nomenclature. American chemical society,
Washington, 1953 (Advances in chemistry, Ser. 8); Patter-
s о n A M., C a p e 11 L. T.,The ring index, N. Y., 1940; Cahn
R. S., I n g о 1 <fc. К., P r e 1 о g V., Experientia, 1956, 12, 81;
trade names index, N.Y., 1941 (Special libraries association);
< п?оГ ° n e r W.,Chemical synonyms and trade names, 5 ed., L.,
1948; Nomenclature of organic chemistry. Definitive rules for
section A. Hydrocarbons, section B, Fundamental heterocyclic
systems, L., 1958; International Union of pure and applied
chemistry. Tentative rules for nomenclature of organic chemi-
stry. 1961. Sec. C., L., 1962; Cahn R. S., An introduction
to chemical nomenclature, L., 1959. В. M. Потапов.
НОНАН СНз(СН2)7 СНз, мол. в. 128,26 — бесцвет-
ная жидкость; т. пл.—53,519°; т. кип. 150,798°;
d™ 0,71763; zip 1,40542; плотность пара при 20° по
отношению к воздуху 4,424; теплота испарения
68,8 кал/г (при т. кип.); Ркрит. 22,5 ат, /крит. 322°,
^крит. 0,236; теплота сгорания 1463,80 ккал/моль;
теплоемкость 0,3945 кал/г-град (25°); вязкость 0,7160
спуаз (20°); поверхностное натяжение 22,92 дин!см
(2б°); октановое число 45 (по моторному методу).
Содержится в нефтях и отогнанных из них бензинах,
продуктах синтеза углеводородов из СО и Н2 (син-
типе); может быть получен в чистом виде химической
очисткой и последующей четкой ректификацией
синтина, а также через магнийорганич. соединения
из соответствующих нормальных альдегидов и 1-хлор-
или 1-бромалканов, напр.:
C<H,O + C;.H1,MgCl —»-C»H„OMgCl —>С9Н1ВОН —►
---------------► С9Н,8 >- СдН20
Н. содержится также в эфирных маслах, выделенных
из Pittosporum engenoils (до 60% Н.) и из Sarothra
gentianoids. М. И. Розенгарт.
НОНИЛОВЫЙ АЛЬДЕГИД (нонаналь, пеларго-
новый альдегид) СНз(СНг)7 СНО, мол. в. 142,23 —
бесцветная жидкость; т. кип. 1927760 мм, 80,6712 мм;
0,8269, пр 1,4274; с водой не смешивается, рас-
творим в спирте, эфире, хлороформе.
Н. а. обладает всеми свойствами альдегидов; иден-
тифицируют его по семикарбазону, т. пл. 101,5°,
и 2,4-динитрофенилгидразону, т. пл. 106,5°; количе-
ственно определяют оксимированием. Н. а. находится
в нек-рых эфирных маслах; может быть получен
обычными способами, напр. из нонилового спирта
и из олеиновой к-ты:
/О Оз
СН3 (СН2)7СН = СН (СН2),С? —<-
'он
/°\ .о н2о
—► СН, (СН2)7 СН СН(СН3)7С<--------->
\(Э-О/ 'ОН
zzo
—>СН,(СН2)7С<
'Н
Н. а. при сильном разбавлении обладает приятным
запахом розы и мандаринов; применяют в парфюмерии
как душистое вещество и для синтетич. целей.
НОНИЛОВЫЙ СПИРТ (нонанол-i, 1-оксинонан)
СНз(СН2)7СН2ОН, мол. в. 144,25 — бесцветная жид-
кость; т. пл.— 5,0°; т. кип. 2127760 мм, 100,0710 мм,
86°/2 мм; ds° 0,8305, 1,4310. Н. с. не смешивается
с водой, растворим в хлороформе, эфире, спирте.
Н. с. обладает всеми свойствами одноатомных спир-
тов, идентифицируют его по фенилуретану, т. пл. 64°;
019 К.Х.Э. т. 3
579
НОНЛАКТОН—НОРВАЛИН
580
количественно определяют ацетилированием в среде
пиридина. Н. с. находится как в свободном виде,
так и в виде сложных эфиров в нек-рых эфирных мас-
лах; получают каталитич. гидрированием пеларгоно-
вого альдегида, пеларгоновой к-ты или ее эфиров.
Н. с. обладает сильным запахом цитронеллола и роз;
применяют в парфюмерии как душистое вещество,
для получения моно-, ди- и тринониламина, пеларго-
нового альдегида и разнообразных сложных эфиров
(карбоновых, фосфорной, фосфиновых, серной к-т),
применяемых в качестве синтетич. моющих средств
и экстрагентов ряда металлов. Е- Нифантъев.
НОНЛАКТОН (у-амилбутиролактон, у-пеларгоилак-
тон, нонанолид-4,1) C9HleO2 (I), мол. вес 156,22 —
бесцветная сильнопреломляющая жидкость; т. кип.
134—134,5712 », J24° 0,9662, «д 1,4468; растворим
в большинстве органич. растворителей, нерастворим
в воде; Н. (I) получают при нагревании £-оксипелар-
гоновой к-ты с серной к-той; из натриймалонового
эфира и окиси гексилена:
NaCH(COOC2H5)2 + C6HUCH-CH2 --—
хо
, н+
---- С5НПСНСН2СН(СООС2Н5)2—-
он
Ряд синтезов Н. основан на использовании фурфу-
рола, напр.:
Ц--[1 СН3СОС2Н5) в-----а Нао
7oJ-cho LoJ-ch=chcoc2h5 НО
хСН2СН2СООН z./Hg + HCI
----► СО —~------*- нонлактон
ЧСН2СН2СОС2Н5
или
C^HgMgBr
С2Н5ОН
СН(ОН)С4Н9
CH2COOC2HS DNaOH
СН2СОС5Н„ 2)[Н]:3)НС1
NH2, тогда как в адреналине
ровка — NHCHa:
НО
s—сн
I II
SO.NH-C^C-R
CHOHCHoNHR
(IV) имеется группп-
I. R=H II. R = CH3
III. R = H IV. R = CH3
Обозначением H. часто пользуются и в ряду терпе-
нов: так, норборнеол содержит одну группу у мости-
кового атома углерода, тогда как в борнеоле такой
атом углерода связан с двумя метильными группами.
Иногда, в особенности в случае аминокислот, в при-
ставку Н. вкладывается другой смысл, и она приме-
няется для обозначения изомерных соединений, отли-
чающихся отсутствием разветвленного углеродного
скелета, напр. валин СНзСН(СН3)СНЛ'Н2СООН и нор-
валин CH3CH2CH2CHNH2COOH. Я. Ф. Комиссаров.
НОРАДРЕНАЛИН [норлеворин, артеренол, 1-(.3,4—
диоксифенил)-2-аминоэтанол] С8НцО3К,мол.в.169,18—
вещество (метаболит), содержа-
щееся в мозговом слое надпочеч- НО~+|Г-СНСН ,nh2
ников и являющееся медиато- ho-AJ он
ром в передаче возбуждений сим-
патической нервной системы в мышечные ткани.
В качестве лекарственного препарата применяют
L-(—)Н. в виде битартрата (моногидрат), мол. в. 359,23
CjHnOsN -CeHjOs-Н2О — белый кристаллич. по-
рошок, т. пл. 100—106°, растворим в воде, pH вод-
ного р-ра 3,0—4,0; на воздухе и под действием света
темнеет. FeCls окрашивает в изумрудно-зеленый цвет,
переходящий при добавлении NH3 в вишнево-красный.
Основание (—)Н.— твердый продукт, т. пл. 216—218°(с
разл.), [а]“ =—37,3° (вода); его хлоргидрат, т. пл.
145,2—146,4°, [а]” =—40° (вода); основание рацеми-
ческого Н., т. пл. 191° (с разл.). Количественно Н.
определяют спектрофотометрически.
Рацемат Н. получают конденсацией пирокатехина
с хлорацетилхлоридом, последующим аминированием
и восстановлением:
ПО -+А С1СН2СОС1 HO-+A[-COCH2CI NH3
ho-^J ho-kJ ---------------
Н. обладает всеми свойствами бутиролактонов, напр.
легко омыляется щелочами; эта реакция используется
для его количественного определения. Для идентифи-
кации Н. применяют взаимодействие его с гидразином
(5-гидразидо-5-окси-2-амилтетрагидрофуран, т. пл.
83—84°). Н. обладает приятным запахом; используют
как компонент в парфюмерных композициях.
Э. Е. Нифантъев.
Н0Н03Ы—моносахариды, содержащие девять
атомов углерода в углеродной цепи; в зависимости от
характера карбонильной группы делятся на альдоно-
нозы (содержат альдегидную группу) и кетононозы
или нонулозы (содержат кетонную группу). Нонозы
получены синтетически, в природе же найдены про-
изводные нонулоз — нонулозаминовые к-ты, важней-
шими представителями к-рых являются нейраминовая
кислота и сиаловые кислоты.
Лит. см. при ст. Октозы. Л. И. Линевич,
НОР— приставка впереди названия химич. соеди-
нения, содержащего вместо метильной группы атом
водорода. Приставкой Н. обычно пользуются в случае
сравнительно сложно построенных природных или
физиологически активных соединений. Типичным
примером может служить норсульфазол (I), отлича-
ющийся от сульфазола (II) отсутствием метильной
группы в боковой цепи. Аналогичным образом норад-
реналин (III) содержит первичную аминогруппу —
h°-AA-COCH2NH2 н2
но-чА - н-
Известны и др. методы получения Н., в частности вза-
имодействием протокатехового альдегида с нитромета-
ном и гидрирования образующего нитросппрта.
Н. применяют для повышения артериального давле-
ния; отличается от адреналина более сильным
сосудосуживающим действием; не оказывает возбуж-
дающего действия на центральную нервную систему.
Лит.: М а ш к о в с к и й М. Д., Лекарственные средства,
4 изд., М., 1960, с. 148; В u d ё s i ns к у Z., Pro! Iva М.,
Syntlietische Arznelmlttel, В., 1961, S. 9. В. Г. Яшунский.
НОРВАЛНН (а-аминовалериановая кислота)
СНз—(СН2)2—CH(NH2)—СООН, мол. в. 111,1—бес-
цветные кристаллы, существует в виде двух оптически
активных D- и L-форм и рацемической D, L-формы;
т. пл.: D-H. 305°; L-H. 307°; D,L-H. 303°. Н. умерен-
но растворим в воде, из воды хорошо кристаллизуется,
плохо растворим в спирте, нерастворим в эфире.
Для L-Н. [а]д= + 24,9° (в 5 н. НС1) и+35° (в уксус-
ной к-те); рАа, 2,36; рКа 9,72; pl 6,04. Н. дает об-
щие реакции аминокислот (нингидриновую и др.);
в составе белков не обнаружен. Получают Н. из
а-бромвалериановой к-ты и др. общими методами син-
теза аминокислот. Н. в организме животного является
антагонистом валина и лейцина.
Лит. см. при ст. Норлейцин» Л. И. Линевич.
581
НОРДИГИДРОГВАЯРЕТОВАЯ КИСЛОТА —НОРМАЛЬНЫЕ ЭЛЕМЕНТЫ
582
НОРДИГИДРОГВАЯРЕТОВАЯ КИСЛОТА [2,3-
бис-(3,4-диоксибензил)-бутан] С18Н22О4, мол. вес
302,37 — кристаллы кремового цвета; т. пл. 184—
185°; растворима в спирте, эфире и в р-рах щелочей.
Природную кислоту выделяют из произрастающего
в США кустарника Larrea divaricate; выпускается
в Канаде под названием «NDGA». Ценным свойством
NDGA является способность задерживать окисление
природных пищевых жиров и нек-рых витаминов.
Н. к. (I) впервые синтезировали в СССР из эвге-
нола (II) в 1957:
Н3СО-г^>|-СНг-СН=СН2
НВ г
СНзСООН _
HO-kk СН3 СН3 к/ он НВт
В ряде стран NDGA широко применяют для защиты
жиров, молока, лярда, бекона, рыбы, кондитерских
изделий от прогоркания. NDGA вводят в количестве
0,001—0,1% от веса жира, что увеличивает стойкость
жиров и жиросодержащих продуктов к окислению
от 3 до 17 раз (см. Антиокислители пищевых про-
дуктов). В применяемых дозах NDGA безвредна
для человека. Синтетическая Н. к. по физико-химич.
и антиокислительным свойствам идентична природной
кислоте. Совместное введение 0,1% кислоты и 0,02%
каротина в молочный жир увеличивает стойкость по-
следнего к окислению в 16 раз. Кислота является
хорошим стабилизатором кондитерских изделий.
Лит.: Higgins J. W., Black Н. С., Oil and
Soap, 1944, 21, 277; Г е р ч у к М. П., Иванова В. М.,
Хим. наука и пром-сть, 1958, 3,№ 5; Герчук М. П., Ж.
Всес. хим. о-ва, 1960, 5, № 4. М. П. Герчук.
НОРКАР АН [бицикло-(0,1,4)-гептан] C7Hi2, мол.
в. 96,17—насыщенный бициклич. углеводород, про-
стейший аналог терпеновых углеводо-
родов ряда карана; жидкость, т. кип.
2 I. ксн_> 110°. И. обладает всеми свойствами на-
Н2(Х zC4 сыщенных циклич. углеводородов. При
^'2 н действии на Н. галогеноводородов, га-
логеноводородных к-т, минеральных и сильных орга-
нич. к-т происходит расщепление трехчленного цикла и
образуются метиленциклогексан, метилциклогексен или
их соответствующие производные. При взаимодействии
бензола с диазоуксусным эфиром образуется эфир
норкарадиенкарбоновой к-ты, восстановлением к-рого
можно получить производное Н.— метилбицикло-
-(0,1,4)-гептан:
+ n2ch-coor—-
|^^£сн-сн3
Лит. см^ при ст. Каран. И. И. Бардыгиев.
НОРЛЕИЦМН (а-аминокапроновая кислота), мол.
вес 131,2, СНз—(СН2)з—CH(NH2)—СООН — бесцвет-
ные кристаллы; существует в виде двух оптических
активных D- и L-форм и рацемической DjL-формы;
т. пл.: D-Н. и L-H. 301°, D,L-H. 297—300°. Н.
умеренно растворим в воде, плохо растворим в холод-
ном спирте, нерастворим в эфире; растворим в к-тах
и щелочах. Для L-Н. [o.]D =+24,5° (в 5н. НС1) и
+ 35° (в уксусной к-те); рКа 2,39; рКа 9,76; р! 6,08.
Н. дает нингидриновую реакцию и др. реакции амино-
кислот. Определение Н. производят общими спосо-
бами определения аминокислот. В составе белков Н.
не обнаружен. Получают Н. синтетически из а-бром-
капроновой к-ты и др. общими способами синтеза
аминокислот. Н. в организме животных является
антагонистом лейцина и метионина.
Лит.: Белки, под ред. Г. Нейрата и К. Бейли, пер. с англ.,
т. 1—2, М., 1956; Майстер А., Биохимия аминокислот,
пер. с англ., М., 1961; G г е е n s t е 1 n J. Ph.,Winitz М.,
Chemistry of the ainino acids, v. 1—3, N. Y.—L., [1961J; Gre-
enberg D. M., Metabolic pathways, v. 2, 2 ed., N. Y.—L.,
1961. См. также лит. Прист. Аминокислоты. Л. И. Линевич.
НОРМАЛЬНОЕ ДАВЛЕНИЕ — абсолютное дав-
ление, равное одной нормальной атмосфере, т. е.,
согласно решению десятой генеральной конференции
по мерам и весам (1954 г.), равное 1013250 дин-см~2 и,
следовательно, равное 101325 Н'М~2 (ньютон-м~2),
т. е. 1,01325 бар. Н. д. является давлением, условно
принимаемым в качестве стандартного при определе-
нии многих величин, зависящих от давления. Напр.,
темп-ры кипения при Н. д. и темп-ры плавления при
Н. д. являются характерными индивидуальными по-
стоянными различных веществ. Темп-ра кипения воды
при Н. д. служит одной из основных реперных точек
практич. температурной шкалы. Н. д. практически
равно давлению столба ртути в 760 мм при плотности
ее 13,5951 г-см~> (плотность ртути при 0° С и атмо-
сферном давлении) при ускорении силы тяжести
£=980,665 см-сек~2\ раньше (до 1954) это соотношение
включалось в определение Н. д. Т. обр., нормальному
давлению более или менее близко атмосферное давле-
ние на уровне моря для широт в области 45°.
В. А. Киреев.
НОРМАЛЬНОСТЬ раствора — концентрация
р-ра, выраженная числом грамм-эквивалентов раство-
ренного вещества, содержащегося в 1 л раствора. Спо-
соб выражения концентрации р-ров через Н. широко
используется в аналитич. химии. См. также Концент-
рация.
НОРМАЛЬНЫЕ ЭЛЕМЕНТЫ — гальванические
элементы с устойчивой электродвижущей силой (эдс)
при постоянных темп-ре и давлении, точно воспроиз-
водимые по строго определенным спецификациям мет-
рологии. лабораторий. Йаиболее полно этим условиям
удовлетворяют кадмиевые элементы Вестона со сле-
дующей электрохимии, системой и токообразующии
процессом:
(-) Cd I CdSO,:Hg2SOt | Hg (+)
Cd+Hg2 + 5=±Cd2++2Hg
(подробнее см. Вестона нормальный элемент).
В практич. электрометрии применяют: Н. э. насы-
щенного типа, содержащие насыщенный р-р электро-
лита, и ненасыщенного типа, в к-рых концентрация
электролита может изменяться под действием элек-
трич. тока.
Нормальный элемент насыщен-
ного типа изображен на рис. 1. Положительным
электродом служит ртуть с пастой, состоящей из
Hg2SOa с размельченными кристаллами CdSCH и
металлич. ртути. Отрицательным электродом являет-
ся амальгама, состоящая из 10% (по весу) кадмия
и 90% ртути. Электролитом служит нейтральный на-
сыщенный р-р сульфата кадмия CdSO4-’/sH2O с из-
бытком его кристаллов или же раствор CdSOi ‘8/»Н2О
в 0,03 н. H2SOa. Кроме обычно применяемой Н-образ-
ной формы стеклянной оболочки Н. э., разработаны
и применяются Н. э. с концентрически расположен-
ными электродами (рис. 2). Н. э. такой формы имеет
известные преимущества перед элементом Н-образиой
формы в смысле портативности, создания более бла-
гоприятных температурных условий для эксплуатации
19*
583
НОРМАЛЬНЫЙ ПОТЕНЦИАЛ
584
элемента и уменьшения числа спаянных мест у
стеклянной оболочки.
Н. э., в зависимости от точности определения их
эдс и устойчивости ее, разделяются на Н. э. I и II
классов. Для Н. э. принимается нормальная темп-ра
20°. Для вычисления эдс Н. э. в промежутке от 10 до
40° принимается следующая ф-ла:
Et-=Ei<s — 0,0000406 •(«—20) — 0,00000095 •((—20)2 +
+0,00000001 -(t—20)®
где t — темп-ра элемента, Et — эдс элемента при
данной темп-ре, Егп — эдс элемента при 20°.
Нормальный элемент ненасыщен-
ного типа изображен на рис. 3. Температурный
Рис. 1. Рис. 2. Рис. 3.
Рис. 1. Схема нормального элемента насыщенного типа
(I и II классов): 1 — раствор сульфата кадмия; 2 —кри-
сталлы сульфата кадмия; з — амальгама кадмия; 4 — суль-
фат ртути (I); 5 — ртуть.
Рис. 2. Схема нормального элемента насыщенного типа
с концентрическим расположением электродов: 1 — раствор
сульфата кадмия; 2—кристаллы сульфата кадмия; з —
амальгама кадмия; 4 — сульфат ртути (I); 5 — ртуть.
Рис. 3. Схема ненасыщенного нормального элемента (III
класса): 1 — раствор сульфата кадмия; 2—корковая проб-
ка; з — амальгама кадмия; 4 — сульфат ртути (I); 5 — ртуть.
коэфф, эдс этого элемента в пределах от 10 до 40° прак-
тически может быть принят равным нулю. Кадмиевые
элементы ненасыщенного типа находят большое при-
менение в различных электроизмерительных схемах,
и, в частности, в самописцах.
По своему назначению Н. э. подразделяются на об-
разцовые (эталонные) и рабочие. Образцовый Н. э.
изготовляется с особой тщательностью и служит для
поверки рабочих элементов. Кадмиевый элемент не-
насыщенного типа не может применяться в качестве
образцового для проверки элементов, т. к. от действия
электрич. тока изменяется концентрация электролита,
а следовательно, и эдс. Поэтому элемент этого типа,
строго говоря, не является Н. э. Пригодность вновь
изготовленного элемента по строго определенной
спецификации определяется эдс, величина к-рой
должна находиться в определенных пределах. Вели-
чина эдс (при 20°) у Н. э. I класса должна быть в пре-
делах от 1,01855 до 1,01875 в (абсолютных), а у Н. э.
II класса в пределах от 1,0185 до 1,0187 в (абсолютных).
Значение эдс (при 20°) кадмиевого элемента ненасы-
щенного типа должно быть в пределах от 1,0187 до
1,0196 в (абсолютных).
Лит.: Сочеванов В. Г., Гальванические элементы,
М.—Л., 1951; ГОСТ 1954—55. Нормальные элементы (меры
электродвижущей силы), в кн.: Электроизмерительные при-
боры, М., 1959, с. 275; Cihalik J., Potenciometrie, Praha,
1961, s. 770.
НОРМАЛЬНЫЙ ПОТЕНЦИАЛ (стандартный по
тенциал) — равновесная разность потенциалов между
электродом и раствором, при условии, что вещества,
участвующие в электродной реакции, находятся в
стандартном состоянии, т. е. что их активности равны
единице. Если потенциал электрода ф (см. Электрод-
ный потенциал) устанавливается в результате равно-
весия между металлом и ионами этого металла в ра-
створе, то его величина определяется ур-нием Нернста:
Ф=Фо+А? 1а а
nF
Нормальные окислительные потенциалы (<р0) по отношению
к потенциалу нормального водородного электрода прн 25°
Высшая степень окисления n Низшая степень окисления Фо» ®
F+2H + 2 2HF + 3,06
f2 2 2F~ + 2,87
О+2Н + 2 U2-f-HaU + 2,07
s2o28 2 2SO' + 2,01
NaB103-f-4H + 2 BiO+-(-Na + -|-2H,O ( + 1,8)
Н2О2+2Н + 2 2H2O + 1,77
MnO4 +4Н + 3 MnO2+2H2O +1,695
PbO2+4H + -)SO2- 2 PbSO,4'2H2O + 1,685
H5JOe+H + 2 JO~ +3H2O + 1,6
2BrO~+12H + 10 Br2-f-6H2O + 1,52
MnO~+8H+ 5 Mn2+4-4HaO + 1.51
Au3 + 3 Au + 1,50
2СЮа +12H + 10 C12-|-6H2O + 1,47
PbO2-p4H + 2 Pb2 + 4-2H2O + 1,456
ciOj 4-6H+ 6 С1~4-ЗН2О + 1,45
BrO“-f-6H + 6 Br~+3H2O + 1,44
Cl2 2 2C1- + 1,359
Cr302 +14H + 6 2CP++7H2O + 1,33
HBrO-f-H + 2 Br~ + H2O + 1,33
МШМ-4Н + 2 MiP + 4-2H2O + 1,23
O,+4H + 4 2H3O +1,229
2JO3 4-12H + 10 J2+6H2O + 1,195
JO~ + 6H + 6 J~ + 3H2O + 1,09
Br2 2 2Br- +1,0652
VO2++2H + 1 vo2+4-h2o + 1,00
HNO-+H + 1 NO-(-H2O + 1,00
NO3 4-4H + 3 NO + 2H2O + 0,96
NO3-+3H + 2 HNO,+H2O + 0,94
2Hg2 + 2 + 0,92
C10--f-H20 2 C1-4-2OH- + 0,89
HO2 +H2O 2 зон- + 0,88
Cu= + + J" CuJ + 0.86
Hg2 + Hg + 0,854
O2-|-4H+ (10-’M) 4 2H3O +0,815
Ag + 1 Ag +0,799
NO3 4-2H + 1 no2+h2o + 0,79
Fe3 + Fe2 + + 0,771
B10-+H.0 2 ВГ-4-2ОН- + 0.76
C6H,O2-|-2H+" (хинон) 2 c 6 н4 (О H)a (гидро- хинон) + 0,6990
O2 + 2H + 2 H2O2 + 0,682
М11О2 4-2H.O 2 MnO24-4OH- + 0,60
MnO~4-2HjO 3 MnO2 + 4OH- +0,588
MnO4 MnO^ +0,564
H3AsO,+2H + 2 HAsO3-f-2H3O + 0,559
Js 2 3J~ + 0,536
J, 2J- +0,5355
H2SO3+4H + S-f-3H2O + 0,45
O,+2H3O 4 4OH- + 0,401
H2MoO4-f-2H + MoO2++2H3O + 0,4
VO2 + -|-2H + 1 Va + -|-H2O + 0,361
SO' +8H + 6 S-j-4H2O + 0,36
Fe(CN)“- 1 Fe(CN)'- + 0,36
Cu2 + 2 Cu + 0,34
2SO'“+10H + 8 S2O3 +5H2O + 0,29
AgCl 1 Ag + Cl- + 0,222
SbO + -f-2H + 3 Sb+H3O + 0,212
so' +4H + 2 h2so3+h2o + 0,17
Gu2 + 1 Cu + + 0,153
Sn‘ + 2 Snq + + 0,15
585
НОРМАЛЬНЫЙ ПОТЕНЦИАЛ — НОРСТЕРОИДЫ
586
Продолжение
Высшая степень окисления n Низшая степень окисления Фо, e
S + 2H + 2 H.S + 0,141
Т1б2 ++2Н + 1 Ti3 + +H2O + 0,1
s«°e 2 2s,o; + 0,08
2Н + 2 H2 0,0
о2+н2о 2 OH--J-HO —0,076
РЬ2 + 2 Pb — 0 12
СгО“-+4Н2О 3 Cr (OH)34-5OH- — 0,
Sn2+ 2 Sn — 0,136
N2-f-5H + 4 N2H^(hoh гидразо- 3
НИЯ)
Ni2 + 2 Ni —0,250
уз + 1 v2 + — 0,255
Н.,Р0<4-2Н + 2 H,PO3-t-H2O —0,276
Со2 + 2 Go — 0,277
PbSO, 2 Pb+S04 — 0,356
Cd2 + 2 Cd —0,403
Gr3 + 1 Cr2 + —0,41
2Н+ (10-7Af) 2 H2 —0,414
Fe2 + 2 Fe —0,440
2CO2+2H + 2 H2C20. — 0,49
H.,P0,+ 2H + 2 H,PO2-f-H2O —0,50
Fe (OH), 1 Fe (OH).-t-OH- —0 ,56
As+3H + 3 AsHa — 0,60
Gr3 + 3 Gr — 0 ,74
Zn2 + 2 Zn —0,763
Sn(OH)g~ 2 HSnO2 +зон-+н2о — 0,90
S04 +H20 2 SOa +20H- — 0,93
OCN--f-H2O 2 CN- + 20H- — 0,97
N2+4H.O 4 N2H.+ 4OH- (гид- — 1,16
разин)
Mn2+ 2 M n — ',18
Zn0;-4-2H2O 2 Zn4~4OH“ — 1 .216
Al’ + 3 Al — 1 ,70
N.,+2H2O+4H + 2 2NH2OH-H + (гид- — 1 , 87
роксиламмоний)
A102 +2H2O 3 A1 + 40H- — 2,35
M»2 + 2 Mg —2,37
Na + 1 Na — 2,714
Ga2 + 2 Ga — 2,87
Ba2 + 2 Ba — 2,90
Gs + 1 Cs — 2,923
K + 1 К — 2,925
Rb + 1 Rb —2,925
N2+4H2O 2 2NH2OH-f-2OH- —3,04
(гидроксиламин)
L1 + 1 Li — 3,045
где а — активность иона металла в растворе, R —
универсальная газовая постоянная, Т — абс. темп-ра,
F — число Фарадея (см. Нернста уравнение), п —
валентность (заряд) иона.
При а=1, ф=ф0, следовательно ф0 —• величина Н.п.
Если на инертном электроде происходит реакция:
окислитель-J-е~ = восстановитель,то потенциал элект-
рода
RT , (окисл.).
m = mn = ——- 1П-7-
nF (восст.)
где (окисл.)/(восст.) — отношение активностей окис-
лителя и восстановителя. Когда это отношение равно
единице, то ф=фо. В общем случае, при электродной
реакции
аА+а'А' + ...+пе~ = ЬВ + Ь'В' + ...
применимо ур-ние
RT ,
Ф=Ф->+^1“
(А)« (А') °'
(В)6 (В'?'
где (Л) — активность вещества А и т. д. Поскольку
абс. значения отдельных разностей потенциалов элект-
род — раствор недоступны измерению, пользуются
величинами, характеризующими потенциалы различ-
ных электродов относительно нек-рого электрода срав-
нения. Обычно электродом сравнения служит нормаль-
ный водородный электрод, потенциал к-рого, следова-
тельно, принят равным нулю при всех темп-рах.
Отметим, что, хотя формулы для электродных потен-
циалов могут содержать активности отдельных ионов,
для определения Н. п. нахождение активностей от-
дельных ионов не требуется, а достаточно знание
средних активностей ионов в электролите. Согласно
приведенному выше определению, потенциал элект-
рода, заряжающегося отрицательно относительно
нормального водородного электрода, имеет знак
минус, а потенциал электрода, заряжающегося поло-
жительно,— знак плюс.
Любая электродная реакция является окислительно-
восстановительным процессом. В приведенной ниже
таблице Н. п. графа «высшая степень окисления» со-
держит исходные вещества электродной реакции aA-J-
-j-a'A'-J-..., графа «низшая степень окисления» —
продукты ЬВ+Ь'В'4-.... Окислительно-восстанови-
тельные системы приведены в табл, в порядке убы-
вающих значений ф0, поэтому системы, расположен-
ные в начале таблицы, способны окислить расположен-
ные ниже, при активностях, равных 1.
Совокупность Н. п. металлов и водорода, располо-
женных в порядке их возрастания, наз. рядом напря-
жений. Металлы с менее положительным Н. п. вытес-
няют металлы с более положительным Н. п. из р-ра их
сол°й (при активностях ионов металлов, равных 1).
II. п. вычисляются из результатов измерений элект-
родвижущих сил гальванич. элементов, а также из
стандартных значений изменения свободной энергии
Гиббса AG° при реакции. Обратно, Н. и. могут быть
использованы для вычисления AG0 и константы равно-
весия реакции. Для окислительно-восстановитель-
ной реакции А .°=—J?TlnK= — пЕ(ф0окисл—ф^осст.),
где ф0окисл. и ф„восст. — н. п. окисляющей и восста-
навливающей систем.
Лит.: Л у р ь е Ю. Ю., Справочник по аналитической хи-
мии, М., 1962; см. также при ст. Электродный потенциал.
НОРСТЕРОИДЫ — стероиды, отличающиеся от
обычных отсутствием метильной группы в месте со-
членения колец или СНг-группы в боковой цепи, а
также стероиды с суженными кольцами. За соедине-
ниями типа эстрона и эквиленина (см. Эстрогенные
гормоны) по традиции остаются, однако, прежние
названия. Буква, стоящая перед приставкой «нор»,
указывает на кольцо, в к-ром произошло сужение,
а цифра, предшествующая этой приставке, показывает
номер отсутствующего углеродного атома. Нумерация
скелета при этом не меняется, а номер выпавшего
углеродного атома опускается, напр.:
Наиболее интересными в биологич. отношении являют-
ся 19-норстероиды, к-рые недавно были обнаружены и
выделены из природных объектов.
587
НОРСТЕРОИДЫ — НОРСУЛЬФАЗОЛ
588
19-Н. общей формулы (I) — кристаллич. продукты,
растворимые в обычных органич. растворителях.
Физич. свойства наиболее важных 19-Н. приведены
в табл. 1.
Таблица 1
Стероид Заместители в формуле 1 Т. пл.,°C [«Id
R X
19- Нортестосте- рон /ОН '•Н /II чн 124—125 + 55° (хлоро- форм)
19-Норпрогесте- рон /СОСН3 '•Н и и 144—145 + 147°
19-Норгидрокор- тизон /СОСН2ОН ‘он /011 "н 255—257 + 112° (метанол)
19-Норкортизон /СОСН2ОН 'он = 0 230—232 —
19-Норкортико- стерон /СОСН2ОИ ' н /ОН 'н 195—197 + 155° (этанол)
О механизме действия 19-Н. пока мало известно,
однако почти доказано, что их начальное действие вы-
ражается в стимулировании
СН3 биосинтеза ферментов. Нек-рые
производные 19-нортестостеро-
Н J. 1 Г на обладают способностью сти-
мулировать синтез белка в ор-
qJ I J ганизме (анаболическая актив-
ность). Производные 19-норте-
I стостерона обладают и андро-
генной активностью, свойствен-
ной самому тестостерону (см. Андрогены, Андросте-
рон, Лекарственные вещества). Поэтому их применение
в терапии приводит иногда к расстройствам половой
сферы и нарушению вторичных половых признаков
у женщин (явление гермафродитизма). В последние
годы синтезирован ряд соединений, где андрогенное
действие сведено к минимуму, а анаболическое сильно
повышено. Отношение анаболич. активности к андро-
генной для данного соединения наз. терапевти-
ческим индексом (К). Нек-рые из наиболее
широко применяемых 19-иорстероидных анаболич.
соединений и их К приведены в табл. 2.
Таблица 2
Соединение Анаболи- ческая ак- тивность* Андроген- ная ак- тивность* К
Пропионат тестостерона (эта- лон) 1 1 1
Фенилпропионат 19-нортесто- стерона 1.5 0,15 10
Аоетат 19-нор-4-хлортестосте- роиа 0,57 0,04 14,2
Циклопентилпропионат 19-нор- тестостерона 2 0.2 10
* Подкожное введение.
19-Н. можно получить различными способами,
напр. восстановлением производных эстрона щелоч-
ными металлами в жидком аммиаке в присутствии
спирта с последующим кислым гидролизом промежу-
точного енольного эфира.
19-Н. применяют для лечения остеопороза печени,
последствий тяжелых травм, рака грудной железы,
полиомиелита, надпочечной недостаточности и др. При
старческой дряхлости введение анаболич. агентов по-
вышает общий тонус.
18-Норстероиды, 18,19-бис-норстероиды, А-норсте-
роиды и В-норстероиды обладают пониженной физио-
логии. активностью и в связи с этим пока значи-
тельного интереса не представляют. Класс С-норсте-
роидов почти не известен, за исключением нескольких
соединений, полученных синтетически из стероидных
сапогенинов. Видоизмененный скелет С-норстероидов
лежит в основе нек-рых сложных алкалоидов (жервин,
вератрамин, цевин, жермин и др.). Первые D-норсте-
роиды (четырехчленное кольцо D) получены только
в 1962 и еще не изучены.
Ли.т,: Назаров И. Н., Бергельсон Л. Д., Химия
стероидных гормонов, М., 1955; Кочетков Н. К., Тор-
ге в И. В., Б от в и н и к М. М., Химия природных соедине-
ний, М., 1961; FieserL. F., Fieser М., Steroids, N. Y.—
L., 1959; Camerlno B., Sala G., Fortsclir. Arzneimlttel-
forsch., 1960, 2, 71. В. H. Леонов.
НОРСУЛЬФАЗОЛ (сульфатиазол, 2-п-аминобен-
золсульфамидотиазол) C9H9O2N3S2, мол. в. 255,31 —
белый кристаллич. порошок, без N____
запаха, т.пл. 198—202°; раство- j j]
рим в ацетоне; плохо растворим H2NCtiH4SO2NH-lxs^
в воде (0,1 : 200) и спирте, нера-
створим в эфире; хорошо растворим в водных р-рах
едких и углекислых щелочей с образованием солей.
Н. легко подвергается диазотированию. Образующая-
ся соль диазония с щелочным р-ром Р-нафтола дает
азокраситель вишнево-красного цвета. С сульфатом
меди Н. дает осадок грязно-фиолетового цвета. Ко-
личественное определение Н. основано на алкали-
метрии. титровании в водно-ацетоновом р-ре по тимол-
фталеину.
Н. может быть получен конденсацией 2-аминоти-
азола с n-ациламинобензолсульфохлоридом или п-фе-
2 CH3OCONH
SO2C1 + H2N
пилуретилаисульфамидом. Принципиально иной путь
синтеза Н. основан на превращении ц-фенилуретилан-
сульфохлорида через п-фенилуретилансульфоцианамид
в карбометоксисульфанилтиомочевину с обработкой
последней монохлорацетальдегидом или его произ-
водными.
CH3OCONH/~ySO2CI--- CH3OCONH-^^-SO2NHCN—
Н,— эффективный сульфаниламидный препарат,
применяется как антимикробное средство при инфек-
циях, вызванных гемолитич. стрептококком, пневмо-
кокком, гонококком, стафилококком, кишечной па-
лочкой и др. Н. показан при пневмонии, цереброспи-
нальном менингите, гоноррее, стафилококковом и
стрептококковом сепсисе и других инфекционных за-
болеваниях. Наряду с Н. в медицинской практике
применяют также натриевую соль Н,— норсульфазол
589
НОСИТЕЛИ—НУКЛЕАЗЫ
590
растворимый (для перорального и парэнтерального
введения) и серебряную соль Н. в виде коллоидно-
дисперсного препарата для лечения верхних дыхатель-
ных путей, а также острой и хронич. дизентерии.
Лит.: Засосов В. А., Мед. пром-сть СССР, 1957, М 4,
6; М а ш к о в с к и й М. Д., Лекарственные средства, 4 изд.,
М., 1960. Л. В. Яхонтов.
НОСИТЕЛИ в катализе (трегеры, подкладки)—
вещества (сами по себе каталитически неактивные или
малоактивные), нанесение на к-рые катализатора зна-
чительно повышает его активность. В простейшем
случае действие Н. заключается в увеличении поверх-
ности работающего катализатора. Поэтому в качестве
Н. часто применяют твердые тела с высоко развитой
поверхностью и пористостью — как искусственные
(активные угли, силикагель, алюмогель, алюмоси-
ликагель, MgO), так и естественные (природные глины,
пемза, диатомиты, асбест и др.). Однако в случае мно-
гостадийных реакций высокая пористость Н. может
оказаться невыгодной вследствие возможности пе-
рехода реакции во внутридиффузионную область
(см. Катализ, Кинетика химическая). Напр., в реак-
ции окисления пропилена в акролеин на СпгО высокая
пористость Н. будет способствовать нежелательной
реакции — дожиганию акролеина до СОг и Н2О.
В этих случаях применяют непористые Н., напр.
карборунд, и роль их заключается в создании большей
механич. прочности и большей термостойкости ка-
тализатора. В нек-рых случаях Н. способствует
стабилизации определенной химич. формы катализа-
тора, напр. S1O2 способствует стабилизации окисно-
хромового катализатора полимеризации этилена в
состоянии Сг5+. В случае дорогих катализаторов
применение Н. обеспечивает их экономию, напр.
в платиновых катализаторах окисления SO2 в SOa
платину наносили на асбест, MgSCU, силикагель. Наи-
более распространенные методы приготовления ката-
лизаторов на Н.: пропитка Н. раствором соли ката-
лизатора с последующим ее разложением, соосаждение
или совместное прокаливание солей Н. и катализа-
тора. Напр., прокаливанием смешанных формиатов
Ni и Mg можно получить очень активный никелевый
катализатор гидрирования на носителе MgO. Н. ши-
роко применяют в промышленном катализе, напр.
Со— катализатор синтеза углеводородов из СО+Н2
по Фишеру — Тропшу, наносят на ТйОг’, Ni —
катализатор гидрирования органич. соединений — на
кизельгур или АНОз и т. д.
Лит. см. при ст. Катализ, Катализ гетерогенный.
О. В. Крылов.
НОСИТЕЛИ в радиохимии — макрокомпо-
ненты, за к-рыми при химич. операциях следуют
микроколичества радиоактивного изотопа. При ра-
диохимии. анализе Н. добавляют в раствор в целях
предотвращения потерь микроколичеств радиоактив-
ного изотопа вследствие адсорбции на стенках посуды,
захвата посторонними осадками, образования микро-
коллоидов и т. д. Поведение микрокомпонента при
выделении с различными Н. позволяет судить о его
химич. природе. В качестве Н., как правило, исполь-
зуется естественная смесь стабильных изотопов изу-
чаемого элемента (изотопный носитель).
В ряде случаев (если элемент не имеет стабильных
изотопов или если ставится задача выделения изотопа
без изотопного носителя) добавляют соль к.-л. дру-
гого элемента, чаще всего химич. аналога выделяемого
(неизотопный носитель). Так, радиоак-
тивные изотопы бария могут быть выделены из рас-
твора с изотопным носителем барием (путем осаждения
BaSCH, ВаСО,, ВаСгСН ит. д.) или с неизотопным носи-
телем — свинцом [осаждением PbSCU, Pb(NO3)2 и т.п.].
См. также Адсорбция радиоактивных элементов,
Осколки деления.
Лит.: Радиохимия и химия ядерных процессов, под ред.
А. Н. Мурина, В. Д. Нефедова и В. П. Шведова, Л., 1960;
Радиохимический анализ продуктов деления. Сб. статей, под
ред. Ю. М. Толмачева, М.—Л., 1960. В. И. Барановский.
НОТАТИН (пенатин, пенициллин В, глюкозоокси-
даза) — фермент, к-рому присвоено систематич. на-
звание Р-П-глюкоза:О2 — оксидоредуктаза, класси-
фикационный шифр 1.1.3.4 (см. Номенклатура и
классификация ферментов). Н. катализирует окисле-
ние P-D-глюкозы в D-глюконовую к-ту:
нотатин
Р-П-глюкопираноза+02------>-б-П-глюконолактон+Н202
самопроизвольно
б-D глюконолактоя+Н20-------------->- D-глюконовая к-та
суммарно:
|3-Р-глюкопираноза+02 +Н2О —> D-глюконовая к-та + Н2О2
Образующаяся Н2О2 действует подавляюще на
рост бактерий, поэтому Н. обладает антибиотич.
свойствами; однако в организме человека или живот-
ных Н. неактивен, вероятно, вследствие быстрого раз-
ложения Н2О2 тканевыми каталазами. Аналогичными
антибиотич. свойствами обладают и другие оксидазы,
образующие Н2О2, в частности ксантиноксидаза.
Н. присутствует, по-видимому, только в плесневых
грибах и, в частности, продуцируется в культураль-
ную жидкость Penicillium notatum и Р. amagasakiense.
Чистый Н., выделенный в виде желтых кристаллов из
культуральной жидкости Р. amagasakiense, пред-
ставляет собой флавопротеид, т. е. белок с флавин-
адениндинуклеотидом (ФАД) в качестве простетич.
группы (см. Флавиновые коферменты). 1 моль Н. со-
держит 2 моля ФАД; мол. в. Н. 154 000, его УФ спектр
характеризуется наличием двух максимумов погло-
щения при 377 и 455 ммк. Активность Н. может быть
измерена по количеству образующейся Н2О2 или же
манометрически — по потреблению О2.
Н.— высокоактивный фермент: 1 мг Н. катализи-
рует при 15° поглощение 48 600 мкл О2 в час. Скорость
реакции увеличивается при повышении концентрации
Ог в р-ре. Оптимум pH при 30—40° составляет 5,5—
5,8. Н. весьма устойчив при pH 4,0—6,0 и быстро
утрачивает активность при pH выше 9,0. Михаэлиса
константа при 38° и pH 5,6 составляет 0,0019 М
(концентрация О2 5%). Активность Н. угнетают:
8-оксихинолин, семикарбазид, димедон, бисульфит,
фенилгидразин, гидразин, гидроксиламин, а также
п-хлорртутьбензоат. Н.— высоко специфичный фер-
мент: любое изменение в молекуле P-D-глюкозы при-
водит к резкому уменьшению скорости катализируе-
мой Н. реакции (даже аномер P-D-глюкозы— a-D-
глюкоза — окисляется в 150 раз медленнее). Н. ис-
пользуется для количественного определения P-D-
глюкозы в биология, материале путем сочетания его
действия с действием пероксидазы, к-рая использует
Н2О2 для окисления бензидина или о-дианизидина с
образованием окрашенных соединений.
Лит.: III е м я к и н М. М. [и др.], Химия антибиотиков,
3 изд., М., 1961; Диксон М., Уэбб Э., Ферменты, пер.
с англ., М., 1961. В. Б. Спиричев.
НУКЛЕАЗЫ — групповое название ферментов,
катализирующих расщепление (деполимеризацию) ну-
клеиновых к-т на олиго- и мононуклеотиды. По суб-
стратной специфичности Н. разделяют на рибону-
клеазы (РНК-азы), расщепляющие рибонуклеиновую
к-ту и дезоксирибонуклеазы (ДНК-азы), действующие
на дезоксирибонуклеиновую к-ту. ДНК-азы, выделен-
ные из различных тканей, отличаются по своим физи-
ко-химич. свойствам и по специфичности действия.
В поджелудочной железе животных содержится
ДНК-аза I, системат. наименование: олигонуклеоти-
догидролаза дезоксирибонуклеиновых к-т, шифр
3.1.4.5. (см. Номенклатура и классификация фермен-
тов). При действии ДНК-азы I на молекулу ДНК
591
НУКЛЕИНОВЫЕ КИСЛОТЫ
592
гидролитич. расщеплению подвергаются эфирные
связи между третьим углеродным атомом дезоксирибо-
зы и остатком фосфорной к-ты (см. схему). В резуль-
тате этого
ДНК-аза I
образующиеся моно- и олигонуклеотиды
содержат концевой остаток фос-
форной к-ты у пятого углерод-
ного атома дезоксирибозы.
ДНК-аза II (систематич. наи-
менование: 3-нуклеотидгидрола-
за дезоксирибонуклеиновых к-т,
шифр 3.1.4.6) широко распрост-
ранена в различных животных
тканях (селезенка, зобная желе-
за) и у микроорганизмов. Дей-
ствие ДНК-азы II, в отличие от
ДНК-азы I, специфически на-
правлено на связь между пятым
углеродным атомом дезоксирибо-
зы и остатком фосфорной к-ты
(см. схему) и приводит к обра-
зованию смеси моно- и олигону-
клеотидов с концевым остатком фосфорной к-ты у
х
X
г?1
I с2
о к
,зЧох I
НО
£'
С5 О
А?
о.
д^°Л4
° Гс5
ДКК-аза II,
нуклеаза микрококков
Х-азотистое основание
Специфичность дейст-
вия нуклеаз.
третьего углеродного атома пентозы.
В отличие от РНК-аз и ДНК-аз, действие к-рых
специфически направлено только на РНК или только
на ДНК, фермент нуклеаза микрококков (шифр
3.1.4.7) расщепляет как ту, так и другую нуклеино-
вые к-ты. Оптимум pH Н. микрококков, выделенной из
Staphylococcus aureus, находится в щелочной области
(pH 8,6). Для максимальной активности фермента
необходимо присутствие ионов Са2+ в концентрации
0,01 М. Н. микрококков подобно ДНК-азе II расщеп-
ляет связь между пятым углеродным атомом пентозы
и остатком фосфорной к-ты.
Н. широко распространены в животном и расти-
тельном мире, а также у микроорганизмов. Они играют
важную роль в процессах расщепления нуклеиновых
к-т и, в частности, их переваривания в желудочно-
кишечном тракте. Ряд авторов считает, что внутри-
клеточные ДНК-азы могут играть важную роль в за-
щите клетки от трансформирующего действия чуже-
родных ДНК.
Иногда к Н. относят не только ферменты, катали-
зирующие деполимеризацию нуклеиновых к-т, но
вообще все ферменты, действующие как на нуклеино-
вые к-ты, так и на продукты их распада (олиго- и
мононуклеотиды и нуклеозиды), в частности фосфоди-
эстеразы, нуклеотидазы, нуклеозидазы, нуклеозид-
фосфорилазы и даже неспецифич. фосфомоноэстеразы
(фосфатазы). Такое объединение в одну группу раз-
личных ферментов вряд ли может считаться оправдан-
ным. См. также Рибонуклеазы.
Лит.: Шмидт Г., Нуклеазы и ферменты, действующие
на компоненты нуклеиновых кислот, в кн.: Нуклеиновые кис-
лоты, пер. с англ., М., 1957; The enzymes, ed. Р. D. Boyer,
H. Lardy, К. Myrbiick, 2 ed., v. 5, N.Y.- L., 1961, pt 3, 123.
Б. В. Спиричев.
НУКЛЕИНОВЫЕ КИСЛОТЫ (полинуклеотиды) —
сложные полимеры биологич. происхождения. Моно-
мерными единицами Н. к. являются нуклеотиды,
к-рые состоят из фосфатного остатка, углеводного
компонента и какого-либо пуринового основания или
пиримидинового основания. В зависимости от природы
нуклеотидов все природные Н. к. разделяются на
2 химически различных типа — дезоксирибонукле-
иновую к-ту (ДНК) и рибонуклеиновую к-ту (РНК).
Главным химич. различием нуклеотидов ДНК и РНК
является природа их углеводного компонента (соот-
ветственно дезоксирибоза или рибоза). Оба типа Н. к.
обязательно входят в состав всех живых организмов
и лишь вирусы могут содержать только одну из них,
Н. к.— оптически активные вещества, вращают
плоскость поляризации света вправо; максимум по-
глощения света в УФ-области при 260 ммк. Высоко-
полимерные Н. к. и олигонуклеотиды обладают гипо-
хромизмом (имеют при данной длине волны
меньшее поглощение, чем смесь составляющих их
мононуклеотидов).При денатурации и деградации Н. к.
степень гипохромизма уменьшается и, следовательно,
УФ-поглощение возрастает (г иперхр омиче-
ский эффект).
Современные методы выделевия нативных препара-
тов Н. к. основаны на экстракции биологич. матери-
ала водными р-рами в сочетании с применением ве-
ществ, денатурирующих белок. Обычно для этой
цели применяют р-ры анионных детергентов типа
додецилсульфата (лаурилсульфата) натрия либо смесь
равных объемов фенола и воды. Белок в этих усло-
виях денатурирует и в значител! н >й »ере раство-
ряется в феноле, а Н. к. переходит в водный р-р. Из
полученного экстракта Н. к. осаждается обычно
этанолом.
Обычно принятые количественные определения Н.к.
основаны на определении фосфора после озоления
препаратов, определении сахарного компонента с
помощью цветных реакций с дифениламином и орци-
ном или на количественном определении азотистых
оснований (пуриновых или пиримидиновых) с по-
мощью спектрофотометрии в УФ-зоне.
Ниже приводятся данные о составе и строении обеих
Н. к.— ДНК и РНК — в отдельности.
Дезоксирибонуклеиновая кислота (ДНК). Мономер-
ной единицей ДНК является дезоксирибонуклеотид;
в качестве углеводного компонента дезоксирибону-
клеотидов природных ДНК присутствует 2-дезокси-1Э-
рибоза в Р-фуранозной форме (I). В дезоксирибонукле-
отидах глюкозидный гидроксил (при С1) дезоксирибозы
занят пуриновым или пиримидиновым основанием, а
гидроксилы при С3 (или при С5) этерифицированы
фосфатным остатком. В качестве оснований, представ-
ляющих собой агликоны, в подавляющем большинстве
нуклеотидов ДНК присутствуют 4 универсально рас-
пространенных производных: производные пурина —
аденин (II) и гуанин (III), и производные пиримидина—
цитозин (IV) и тимин (V). Помимо этих оснований, в
дезоксирибонуклеотиды могут входить в качестве
агликона и нек-рые другие производные пурина
или пиримидина. Так, в составе ДНК многоклеточных
животных и высших растений обнаружен 5-метил-
цитозин, к-рый отсутствует в ДНК всех низших форм
(бактерий, актиномицетов, грибов, водорослей и про-
стейших). Наоборот, в составе ДНК нек-рых бактерий
обнаружен 6-метиладенин, отсутствующий в ДНК
высших форм. Обособленно стоит ДНК нек-рых
бактериофагов — Т2, Т4, Тб, в к-рой вместо цитозина
присутствует 5-оксиметилцитозин. Во всех случаях
в природных ДНК азотистые основания связаны с
глюкозидным гидроксилом дезоксирибозного остатка
через свой атом азота в положении 9 пуринового коль-
ца или в положении 3 пиримидинового кольца.
Соединение основания с дезоксирибозой носит название
дезоксирибонуклеозид (нуклеозид дезоксирибозной природы),
или дезоксирибозид — адениловый, гуаниловый, цитидило-
вый, тимидиловый, метилцитидиловый. Присоединение фос-
фатного остатка к 3-му или 5-му гидроксилам дезоксирибозного
593
НУКЛЕИНОВЫЕ КИСЛОТЫ
594
компонента дезоксирибонуклеозида дает соответствующий
дезоксирибонуклеотид (нуклеотид дезоксирибозной природы,
нуклеотид ДНК) или дезоксириботид.Если зтерифицированный
фосфат находится в виде остатка фосфорной к-ты, то соответ-
ствующие дезоксирибонуклеотиды обозначают как дезок-
сиадениловая к-та, дезоксигуаниловая к-та и т. д. В зависи-
мости от положения фосфата могут быть: З'-дезоксиадениловая
к-та и 5'-дезоксиадениловая к-та, З'-дезоксигуаниловая к-та и
5'-дезоксигуаниловая к-та и т. д. Соответствующие дезоксири-
бонуклеотилы с нейтрализованным фосфатным остатком
(обычно в виде натриевой соли) называют часто дезоксиаде-
нозин-З'-монофосфат (VI) (З'-дАМФ), дезоксицитидин-5'-мо-
нофосфат (VII) (5'-дЦМФ) и т. д. Указанные Дезоксирибону-
клеотиды и являются мономерными единицами природных
ДНК. Эти мономеры разнотипны, различаясь природой своего
азотистого компонента. Т. обр., в состав каждой природной
ДНК входит 4—5 различных типов мономерных единиц —
различных типов нуклеотидов дезоксирибозной природы.
Изучение межнуклеотидных связей в ДНК показало
строгую их однотипность. Во всех случаях связь
между дезоксирибонуклеотидными остатками в цепи
главных валентностей ДНК осуществляется за счет
ХО
Н2С^ NC4H4ON2
СН Н С (цитозин)
н" V-------(/ ' н
о~ о н
\ /
Н2С^ NC5H4N4
ch н CZ (аденин)
hz V—— С^'н
I I
0-0 н
\ /
рк
Н2Сч /NC5H4ON4
С Н н С (гуанин)
hz У,..Н
I I
О" о н
х /
Н2СХ zNC5H4Nt
с"н HCZ (аденин)
HZ V—-</" н
I I
образования фосфатного
мостика между 3-м и 5-м
гидроксилами дезоксири-
бозных компонентов двух
соседних дезоксирибонук-
леотидных остатков. Фос-
фатный компонент дезок-
сирибонуклеотидов в цепи
ДНК всегда (за исключе-
нием концевых нуклеоти-
дов) этерифицирует одно-
временно два дезоксири-
бозных остатка: один —
при З'-гидрокспле и сосед-
ний— при 5'- гидрокси-
ле. Образуется последова-
тельность «—С5 дезокси-
рибозид С’ — фосфат — С5
дезоксирибозид С’ — фос-
фат — С5 дезоксирибозид
С’—» и т. д. (рис. 1).Связь
С3 — Р — С5 является уни-
версальным и единствен-
ным типом межнуклеотид-
ной связи в ДНК. Фосфат
всюду (кроме концов) ди-
этерифицирован. Никаких
разветвлений в цепи ДНК
нет. Все природные ДНК
представляют собой части-
цы с очень большим мол.
весом (4—16 млн.,а в слу-
чае ДНК из фагов до
150 млн.) и состоят из де-
сятков тысяч нуклеоти-
дов.
ДНК хорошо растворя-
ется в воде, в слабых и креп-
Рис. I. Фрагмент цепи ДНК. ких водносолевых р-рах,
образуя вязкую жидкость;
осаждается 2—3 объемами 96%-ного этанола, образуя
студнеобразный или волокнистый осадок. ДНК дена-
турирует при нагревании водных р-ров до 100°, при
их подкислении (до pH 1—2) или подщелачивании (до
pH 10—12). При кислотном гидролизе происходит
отщепление пуринов и частичный гидролиз эфирных
связей в молекуле ДНК; при мягком кислотном гид-
ролизе ДНК удается получить апуриновую
ДНК. При нагревании с гидразином расщепляются
пиримидиновые основания — образуется а п и р и-
мидиновая ДНК. При действии на ДНК дезок-
сирибонуклеазы происходит гидролиз основных фос-
фоэфирных связей ДНК.
Изучение природных ДНК из различных организ-
мов показало, что они неодинаковы как биологически,
так и химически, и каждый вид организмов характе-
ризуется, по-видимому, своей специфич. ДНК. Коли-
чественные молярные соотношения дезоксирибонукле-
отидов разных типов в ДНК из разных видов могут
быть различны (табл. 1).
Таблица 1
Вид Содержание нуклео- тидов, мол. % (Г+Ц)/(А+Т)
Г А Ц Т
Животные
Крыса 22,2 28,3 22,0 27,5 0,79
Морской еж .... 19,1 31 ,2 19,2 30,5 0,62
Тутовый шелкопряд 22,5 28,6 21,9 27,2 0,79
Высшие растения
Фасоль 20,6 29,7 20,1 29,6 0,69
Лук 18,4 31,8 18,2 31,3 0,58
Сосна 19,3 30,1 20,1 30,5 0,65
Грибы
Шампиньон .... 22,6 28,2 21,8- 27,4 0,80
Аспергилл 25,1 25,0 25,0 24,9 1,00
Бактерии Туберкулезная бак-
терия 34,2 16,5 33,0 16,0 2 08
Тифозная бактерия 26,7 17,3 23,5 26,4 23,4 1,13
Стафилококк . . . 32,3 17,4 33,0 0,53
Вирусы
Бактериофаг Т2 . . 18,2 32,5 16,8 (окси- метил) 32,5 0,54
Обозначения нуклеотидов: Г — гуаниловый, А — адени-
ловый, Ц — цитидиловый, Т — тимидиловый.
При рассмотрении аналитич. данных обращает на
себя внимание существование определенных общих
закономерностей (Чаргафф) в молярных соотношениях
оснований, к-рые заключаются в следующем: 1) мо-
лярная сумма пуриновых оснований (аденин и гуанин)
равна молярной сумме пиримидиновых оснований
(цитозин и тимин); 2) молярные содержания аденина
и тимина равны; 3) молярные содержания гуанина
и цитозина равны. Фактически от вида к виду варь-
ирует лишь соотношение между суммой гуанина и
цитозина (Г+Ц) и суммой аденина и тимина (А+Т).
Т. обр., отношение (Г4~Ц)/(А+Т) является показате-
лем специфичности состава ДНК, суммирующим все
возможные различия в нуклеотидном составе ДНК.
Макромолекулярная организация полимерных це-
пей ДНК, несмотря на их химич. специфичность,
является однотипной и универсальной у самых раз-
личных представителей живого мира. Согласно совре-
менным представлениям, в основе к-рых лежит мо-
дель Уотсона и Крика, нативная молекула ДНК
представляет собой две спирально идущих полидезок-
сирибонуклеотидных цепи, правильно закрученных
вокруг общей оси. Эти две цепи связаны водородными и
гидрофобными связями между азотистыми основани-
ями цепей. При этом основания обращены внутрь
спирали, к центру, располагаясь своей плоскости»
приблизительно перпендикулярно длинной оси спи-
рали. Между ними образованы водородные связи»
595
НУКЛЕИНОВЫЕ КИСЛОТЫ
596
причем пуриновому основанию одной цепи всегда
соответствует пиримидиновое основание в другой
цепи, а пиримидиновому основанию первой цепи —
пуриновое. Водородные мостики существуют между
N1 пуринового кольца и № пиримидинового кольца
и между азотом или кислородом при 6-м атоме
углерода пуринового кольца и кислородом или азо-
том при 6-м атоме углерода пиримидинового кольца.
Т. обр., в молекуле ДНК существуют только специ-
фические пары оснований: аденин с тимином и гуанин
Рис. 2. Водородные связи в структуре ДНК.
с цитозином (рис. 2). В паре гуанин —цитозин есть так-
же третий водородный мостик между азотом при 2-м
атоме углерода гуанина и кислородом при 2-м атоме
.углерода цитозина. На протяжении всей длины моле-
кулы создается жесткая система ре-
гулярных водородных связей между
двумя цепями ДНК. Расстояние меж-
ду плоскостями пар оснований вдоль
оси. молекулы составляет при этом
3,4А; 1 виток включает 10 пар осно-
ваний и имеет длину 34 А вдоль оси.
Схема фрагмента нативной структуры
ДНК изображена на рис. 3. Последо-
вательность оснований одной цепи
полностью детерминирована последо-
вательностью оснований в другой це-
пи, так как всегда аденину первой
цепи должен соответствовать тимин,
гуанину — цитозин, тимину — аденин
и цитозину — гуанин. Следовательно,
если допустить расхождение двух це-
пей молекулы ДНК, то на каждой об-
Рис.З. Схема двух- разевавшейся половинке может быть
спиральной струк- синтезирована только точно допол-
дель УотсонаМ<и няющая ее комплементарная цепь с
Крика). заданной последовательностью. Если
это произойдет, то вместо одной мо-
лекулы ДНК образуются две, в точности идентич-
ные исходной. Это послужило основой для ряда
предположений о возможном механизме самоудвое-
ния («редупликации», «ауторепродукции») молекул
ДНК. Экспериментальные данные последних лет
подтверждают гипотезу самоудвоения молекул ДНК
путем построения на каждой из двух исходных це-
пей комплементарных к ним новых цепей. В отно-
шении макромолекулярной структуры ДНК имеется
исключение в виде ДНК бактериофага фХ174 и
родственных ему фагов, к-рая представляет собой
простую однотяжную полидезоксирибонуклеотидную
цепь с мол. весом ок. 2 млн.; однако немедленно после
заражения клеток этими вирусами их ДНК достра-
ивает на себе комплементарную цепь и превращается
в обычную двухтяжную ДНК.
В клетках живых организмов ДНК не распределена
диффузно, а организована в особые структуры, свя-
занные с передачей наследственных свойств. Эти
структуры составляют основу т. наз. ядерного вещест-
ва клеток — хроматина; они представлены в
виде хромосом и у высших организмов локализованы
в клеточном ядре, а у большинства бактерий и нек-рых
других низших организмов — в протоплазме, в виде
т. наз. нуклеоидов. Хромосома представляет
собой единый гигантский комплекс, состоящий по
преимуществу из молекул ДНК и белка.
В отношении биологич. функций ДНК в живой клетке давно
уже стала несомненной прямая связь специфич. структуры
ДНК с явлениями наследственности и с регуляцией белкового
синтеза. Генетич. роль ДНК впервые была экспериментально
установлена в опытах с бактериями в 1944. Оказалось, что если
выделить чистую ДНК из капсулированной разновидности
бактерий (пневмококков) и ввести ее в клетки некапсулирован-
ной разновидности, то потомство последней начинает образо-
вывать капсулу; при этом капсула этих клеток состояла из
того же полисахарида, к-рый был характерен для бактерий
донаторов ДНК. Следовательно, под действием ДНК произо-
шло превращение одной разновидности в другую. Эти опыты
получили название трансформации бактерий. В последующем
они были подтверждены на различных видах бактерий по линии
разнообразных наследственных признаков. Т. обр., была осу-
ществлена передача наследственных свойств у бактерий с
помощью химически чистой ДНК.
Генетич. роль ДНК связана с двумя процессами; 1) точным
воспроизведением самой себя, т. е. способностью к редупли-
кации (самоудвоению); это необходимо для того, чтобы у потом-
ков была такая же ДНК, что и у родительских клеток или ор-
ганизмов; 2) определяющим влиянием на обмен веществ клетки
и, следовательно, на ее биологич. свойства. Это влияние ДНК
не является прямым и непосредственным, а идет различными
путями через белковый синтез.
Рибонуклеиновая кислота (РНК). Мономерной еди-
ницей РНК, в отличие от ДНК, является рибонукле-
отид. Все природные РНК имеют в качестве углевод-
ного (пентозного) компонента рибонуклеотидов D-ри-
бозу в Р-фуранозной форме (VIII). В остальном строе-
ние рибонуклеотидов целиком подобно строению
дезоксирибонуклеотидов. Азотистый компонент ну-
клеотидов РНК всюду представлен одним из обяза-
тельных для каждой РНК оснований: аденином (II),
гуанином (III), цитозином (IV) и урацилом (IX), пред-
ставляющим собой деметилированное производное
тимина (V). Помимо этих оснований, в составе ряда
РНК (особенно бактериальных) обнаружены нек-рые
другие, представляющие собой, как правило, мети-
лированные производные пуриновых и пиримидино-
вых оснований. Так, в составе нуклеотидов РНК были
обнаружены тимин и 5-метилцитозин, ранее известные
только как составные части ДНК, а также различные
метилированные производные аденина и гуанина.
ОН ОН
VIII IX х
Так же как и в дезоксирибонуклеотидах, азотистые
основания РНК связаны с глюкозидным гидроксилом
рибозы, как правило, через свой С8 пуринового кольца
или С’ пиримидинового кольца, образуя соответствую-
щий рибонуклеозид или рибозид. Однако в составе
597
НУКЛЕИНОВЫЕ КИСЛОТЫ
598
различных РНК был обнаружен уридиловый нуклео-
зид иного типа — т. наз. «псевдоуридин» (X), у к-рого
связь между урацилом и рибозой идет через С° пири-
мидинового кольца и С1 рибозы с образованием угле-
род _ углеродной связи. Фосфат, этерифицируя один
из свободных гидроксилов рибозного компонента ну-
клеозида, дает соответствующий рибонуклеотид. В
зависимости от положения фосфата различают 2 -, 3 -
и 5'-монофосфаты соответствующих риоонуклеозидов
{или 2'-, 3'- и 5'-рибонуклеотиды). 3-' и 5'-Рибонуклео-
тиды могут рассматриваться как мономеры природных
РНК.
Межнуклеотидные связи в РНК полностью идентич-
ны таковым в ДНК и столь же строго однотипны. Во
всех случаях связь между
ных валентностей идет за
Рис. 4. Цепь «растворимой»
РНК.
нуклеотидами в цепи глав-
счет фосфатного мостика
между 3-м п 5-м гидрок-
силами рибозных компо-
нентов двух смежных ну-
клеотидных остатков,т. е.
по типу С3—Р—С5 (рис. 4).
Эта связь является един-
ственным типом связи в
РНК: цепь РНК, как и
ДНК, является неразвет-
вленной.
РНК хорошо растворя-
ется в воде й слабых вод-
носолевых р-рах; высоко-
полимерная РНК (мол.
вес 0,5— 2 млн.) медлен-
но выпадает в осадок в
1 —1,5 молярных солевых
р-рах, чтоиспользуют для
ее отделения от низкопо-
лимерной РНК. РНК оса-
ждается спиртом: тремя
объемами спирта с добав-
лением ацетатного буфера
(pH 5) или при осаждении
спиртом из 0,1 молярного
р-ра NaCI; при осаждении
выпадает мелкий хлопье-
видный осадок. Фосфоди-
эфирнаясвязь РНК легко
гидролизуется в кислой
(рН<2) или щелочной
(рН>13) средах. В ре-
зультате гидролиза обра-
зуется смесь рибонукле-
озид-2'- и З'-фосфатов.
Гидролиз РНК до нукле-
отидов может быть про-
изведен при помощи раз-
личных рибонуклеаз и
фосфодиэстераз. С по-
мощью ряда химич. аген-
тов можно видоизменить
азотистые основания цепи РНК; напр., обработка
РНК HNO2 приводит к превращению цитозина (IV)
в урацил (IX).
РНК из различных организмов не одинаковы как
биологически, так и химически; каждый вид организ-
мов характеризуется, по-видимому, своей специфич.
РНК. Количественные молярные соотношения нукле-
отидов разных типов в РНК из разных видов могут
быть различными (табл. 2).
У РНК соотношения нуклеотидов, как правило,
подчиняются след, закономерности: молярная сумма
гуанина и урацила равна молярной сумме аденина и
цитозина (т. наз. «правило Чаргаффа»), Этому правилу
не подчиняются лишь вирусные РНК. При переходе
от вида к виду наиболее изменчивым является соот-
Таблица 2
Вид Содержание нуклеотидов (Г+Ц)/(А + У)
г А Ц У
Животные Крыса 32,8 18,7 29,6 18,9 1,66
Морской еж . . . . 29,4 22,6 27,2 20,8 1 ,30
Тутовый шелкопряд 26,1 26,1 23,9 23,8 0,99
Муха дрозофила . . 24,2 29,6 20,8 25,4 0.81
Высшие растения Фасоль 31,4 24,9 24,1 19,6 1 , 25
Лук 29,8 24,9 24,7 20,6 1,20
Сосна 27,6 25,3 23,4 23,7 1 , 04
Грибы Шампиньоны . . . 28,8 27,2 21,6 22,4 1,01
Аспергилл 30,1 25,0 25,0 19,9 1 ,23
Бактерии Туберкулезная бак- терия 33,0 22,6 26,1 18, 3 1,45
Тифозная бактерия 30,8 26,1 24,0 19,1 1,21
Стафилококк . . . 28,7 26,9 22,4 22,0 1,05
Вирусы Вирус табачной моза- ики 25,4 29,9 18,5 26,3 0,78
Вирус желтой мозаи- ки репы 17,2 22,6 38,0 22,2 1 ,23
Обозначения нуклеотидов: Г — гуаниловый, А — аденило-
вый, Ц — цитидиловый, У—уридиловый.
ношение между суммой гуанина и цитозина и суммой
аденина и урацила—(Г-|-Ц):(А+У). В подавляющем
большинстве случаев Г-|-Ц>А+У, т. е. состав РНК
относится к ГЦ-типу. АУ-тип РНК встречается
гораздо реже.
Рассматриваемый специфич. состав РНК из раз-
личных организмов выражает их суммарный состав.
Однако любая живая клетка содержит, ио-впдимому,
сложный набор различных макромолекул РНК с раз-
личной химич. и биологич. специфичностью. Уста-
новлено существование нескольких функционально
различных типов РНК внутри каждой клетки —
рибосомальная РНК, «растворимая» РНК, «информа-
ционная» РНК; нуклеотидный состав этих РНК
различен.
Основную массу (80%) от всей суммарной РНК
клеток составляет рибосомальная РНК, схо-
дящая в состав клеточных рибонуклеопротеидов, или
рибосом (см. Нуклеопротеиды). Эта РНК характери-
зуется большим мол. весом (0,5—1,5 млн.); ее нукле-
отидный состав у разных организмов очень близок,
что, по-видимому, в основном и обусловливает срав-
нительно малые вариации в составе суммарной РНК
у разных видов. Другой тип клеточной РНК — РНК
клеточного сока или т. наз. «раство-
рима я», или адапторная, РНК — составляет обыч-
но 10—15% от всей РНК клетки; характеризуется
мол. весом ок. 20 000—30 000 и не связана в какие-
либо фиксированные нуклеопротеидные комплексы.
Около 1—10% всей клеточной РНК составляет откры-
тая сравнительно недавно «информационна я»,
или матричная, РНК, молекулы к-рой по соотношению
своих нуклеотидов и нуклеотидной последователь-
ности представляют собой полирибонуклеотидные
копии различных участков одной из двух цепей моле-
кул клеточной ДНК. По мол. весу молекулы «инфор-
мационной» РНК, по-видимому, очень гетерогенны,
но значительную часть составляют большие частицы
с мол. весом до 2 млн. (и более). В клетке эта РНК
может присутствовать во временной связи с ядерными
компонентами, с рибосомами, а также в свободном
виде. В состав вирусов рибонуклеопротеидной при-
роды входит только один тип молекул РНК — высо-
кополимерная РНК с мол. весом ок. 2 млн.;
599
НУКЛЕИНОВЫЕ КИСЛОТЫ
600
по-видимому, она аналогична «информационной» РНК
клеточных организмов. Химически все эти РНК
(рибосомальная, «растворимая», «информационная»
и вирусная) полностью гомологичны, обладая иден-
тичным набором азотистых оснований, строением ну-
клеотидов и характером межнуклеотидных связей.
Различаются эти РНК по степени полимерности их
цепей, соотношению нуклеотидов и нуклеотидной
последовательности.
Молекулы «растворимой» РНК, независимо от источника
выделения, имеют идентичные концевые группы: с одного кон-
ца — гуаниловый нуклеотид с моноэтерифицированным конце-
вым 5'-фосфатом; с другого конца — аденозин со свободными
2'- и З'-гидроксилами. Вслед за аденозином на этом конце
всегда следует цитидиловый нуклеотид, а за ним — еще один
цитидиловый нуклеотид. Т. обр., все растворимые РНК имеют
универсальную концевую последовательность — Ц-Ц-А (ем.
рис. 4). Однако дальнейшая последовательность нуклеотидов
в цепях различных молекул «растворимой» РНК оказывается
различной.
Каждая молекула РНК представляет собой гибкую
неразветвленную нить, состоящую, по-видимому, из
одной полинуклеотидной цепи. В обычных услови-
ях (в растворе и, очевидно, в живой клетке) эта нить
свернута в компактную частицу под влиянием сильно-
го внутримолекулярного взаимодействия (водород-
ные связи) между основаниями отдельных ее уча-
стков. Отдельные короткие участки цепи, взаимодей-
ствуя друг с другом посредством таких водородных
связей, закручиваются (спирализуются) друг вокруг
друга и тем самым формируют внутримолекулярные
двуспиральные участки. Свыше 60% нуклеотидов
в цепи РНК образуют при этом поддерживаемые во-
дородными связями спиральные участки, к-рые чере-
дуются с неспирализованными. Двуспиральные ко-
роткие участки могут быть не столь совершенны, как
двойная спираль ДНК; возможно, что они могут
включать известное число неспаренных оснований,
дающих одно- или двунуклеотидные петли.
Можно считать установленным, что биологич. функции раз-
личных РНК в живой клетке непосредственно связаны с осу-
ществлением синтеза белков. Процесс биосинтеза белков начи-
нается с активации свободных аминокислот при помощи спе-
циальных ферментных систем, катализирующих образование
(аденин) U II
^^N>^2c-o-ro-c-cH-R
Xi мЧ/| о" о nh2
н \о< н
^..растворимая" РНКJ
ох
I
\ NC5H4N4
\н нХ, <а“виУ
н\| ✓ Н
о он
C-CH-R XII
II I
о nh2
активированной формы аминокислот — аминоациладенилатов
(XI) — из аминокислот и аденозинтрифосфорной к-ты (АТФ).
На следующей стадии вступает в реакцию «растворимая» РНК.
Она взаимодействует с активированной аминокислотой так,
что аминокислотный остаток присоединяется своим карбокси-
лом к свободному 2'-или З'-гидроксилу концевого аденозина
«растворимой» РНК с образованием сложноэфирной связи
(XII). Для такого акцептирования аминокислоты необходима
концевая гоуппировка — Ц-Ц-А. В ферментативном взаимо-
действии активированных аминокис-
лот с «растворимой» РНК каждому
виду аминокислоты соответствуют
определенные, специфич. для дан-
ной аминокислоты, молекулы «раст-
воримой» РНК. Молекула «раство-
римой» РНК с присоединенной к ней
аминокислотой, строго определен-
ного вида, поступает вслед за этим
в рибосомы, к-рые являются места-
ми собственно белкового синтеза;
здесь происходит полимеризация
аминокислот в белковые цепи. Эта
полимеризация активированных
ахминокислот происходит при уча-
стии специальных ферментных си-
стем в рибосоме, рибосомальной РНК, заключенной в рибо-
соме и «информационной» РНК, приходящей в рибосому извне,
после ее образования на ДНК. При этом «информационная»
РНК, по-видимому, играет роль основной матрицы для син-
теза белковой цепи; специфич. нуклеотидная последователь-
ность данной молекулы «информационной» РНК является
основным фактором, определяющим соответствующую после-
довательность расположения аминокислотных остатков в стро-
ящейся полипептидной цепи белка.
По общепринятым представлениям происходит это, по-
видимому, вследствие того, что вдоль цепи «информационной»
РНК чередуются различные группировки нуклеотидов (в на-
стоящее время считается, что основную роль играют сочетания
по три нуклеотидных остатка—триплеты), причем каждой груп-
пировке соответствует дополнительная ей (комплементарная)
группировка нуклеотидных остатков в цепи той или иной мо-
лекулы «растворимой» РНК. Этим достигается определенная,
специфич. для каждой матрицы, последовательность располо-
жения различных молекул «растворимой» РНК с присоединен-
ными к ним соответствующими аминокислотами, а в результа-
те — столь же однозначная аминокислотная последователь-
ность в полипептидной цепи. Таким образом, в цепи «информа-
ционной» РНК в виде определенных сочетаний различных
нуклеотидных остатков как бы закодированы определенные
аминокислотные остатки, и последовательность указанных
сочетаний нуклеотидов в цепи РНК определяет тем самым по-
следовательность аминокислот в белках.
Молекулы «растворимой» РНК выступают в роли высоко-
специфич. переносчиков активированных аминокислот к месту
белкового синтеза и, что самое главное, в роли расшифровщи-
ков того кода, к-рым записана в цепи «информационной» РНК
аминокислотная последовательность строящегося белка. Спе-
цифичность белка, зависящая в конечном счете гл. обр. от его
аминокислотной последовательности, определяется в процессе
биосинтеза соответствующей ему полирибонуклеотидной мат-
рицей — молекулой «информационной» РНК. В то же время
показано, что молекулы «информационной» РНК образуются
на соответствующих участках молекул ДНК. Это образование
полирибонуклеотидных цепей на дезоксиполирибонуклеотидах
происходит по тому же принципу комплвхментарности, что и
образование одной полидезоксирибонуклеотидной цепи на
другой при самоудвоении (репликации) ДНК. В результате
нуклеотидная последовательность соответствующей цепи «ин-
формационной» РНК будет комплементарна к нуклеотидной
последовательности одной из цепей двутяжной ДНК и, следо-
вательно, точной копией другой цепи. Таким образом, если
«информационная» РНК является матрицей для синтеза бел-
ков, то ДНК служит матрицей для синтеза «информационной»
РНК, и тем самым ответственна за специфич. структуру бел-
ков. Так как белки определяют все остальные биологич.
синтезы и звенья обмена, а последние в конечном счете опре-
деляют биологич. признаки и свойства живых тел, то таким
путем действительно осуществляется влияние ДНК на эти
признаки и свойства. Точное воспроизведение ДНК на молеку-
лярном уровне, возможность к-рого прямо заложена уже в са-
мой ее структуре, и последующая передача воспроизведенной
ДНК из поколения в поколение рассматриваются как основные
факторы, обусловливающие У потомков синтез тех же белков,
а следовательно, и проявление тех же специфич. видовых био-
логич. свойств, что и у родителей.
Синтетические полинуклеотиды. Синтетич. поли-
рибонуклеотиды могут быть получены с помощью
ферментов — полинуклеотидфосфорилаз — из рибонук-
леозид-5'-дифосфатов (С. Очоа). Синтез идет по
схеме: пХРР-^(ХР)п-\-пР, где X — любой из рибо-
нуклеозидов и Р — остаток фосфорной к-ты. Если в
качестве исходных взята смесь двух или более нукле-
озиддифосфатов, образуются гетерополимеры, если
только один — гомополимеры.
Искусственный синтез ДНК проводится с помощью
фермента ДНК-полимеразы (Корнберг). Исходными
веществами служат только дезоксирибонуклеозид-5'-
трифосфаты. Синтез идет по схеме:
пХРРР —> (ХР)п+пРР
где X — любой из дезоксирибонуклеозидов, Р —
остаток фосфорной к-ты (РР — свободная пирофосфор-
ная к-та). Для этого требуется наличие в реакционной
смеси всех четырех дезоксинуклеозидтрифосфатов и
затравки в виде ДНК. Нуклеотидный состав образо-
вавшегося полимера совпадает с составом затравки,
к-рая, по-видимому, служит матрицей.
Биосинтетич. полинуклеотиды, как и природные
Н. к., содержат одни тип фосфодиэфирной связи
С3—р—Q5 и неразветвлены. Мол. вес их, как пра-
вило, достаточно велик для использования их в ка-
честве моделей природных Н. к. при физико-химич.,
химич. и ферментативных исследованиях.
Лит.: Белозерский А. Н., Нуклеопротеиды и ну-
клеиновые кислоты растений и их биологическое значение,
М., 1959; его же, Нуклеиновые кислоты и их биологиче-
ское значение, М., 1961; Белозерский А. Н., С п и-
р и н А. G., Усп. соврем, биологии, 1956, 41, № 2; и х же,
Изв. АН СССР, Серия биологическая, 1960, № Г, Б р е с л е р
G.E., Введение в молекулярную биологию, М., 1963; Д о т и П.,
Вестник АН СССР, 1960, № 9; Спирин А. С., Усп. биол.
химии, 1962, 4, 93; СпиринА. С., Гаврилова Л. П.,
Изв. АН СССР, Серия биологическая, 1961, № 4; Спирин
А. С. [и др.], Усп. биол. химии, 1963, 5, 3; Хесин Р. Б.,
601
НУКЛЕОЗИДАЗЫ—НУКЛЕОЗИДЫ
602
Усп. соврем, биологии, 1958, 46, № 2(5); Нуклеиновые
кислоты. Сб. статей, М., 1957; М., 1962; Проблемы цитофизио-
логии. Сб. [переводных] статей, М., 1957; Биологическое вос-
произведение макромолекул. Сб. статей, М., 1960; Химиче-
ские основы наследственности. [Тр. Международн. симпо-
зиума], пер. с англ., М., 1960; The nucleic acids, ed. E. Char-
gaff, J. N. Davidson, v. 1—3, N. Y.—L., 1955—60; Progress
in nucleic acid research, ed. J. N. Davidson, W. E. Cohn, v. 1,
N. Y.—L., 1963. A. C. Спирин.
НУКЛЕОЗИДАЗЫ — тривиальное название груп-
пы ферментов, катализирующих гидролиз нуклеози-
дов на пуриновое или пиримидиновое основание и
углевод. По современной номенклатуре Н. входят
в подподкласс гидролазы N-гликозильных соединений,
шифр 3.2.2 (см. Номенклатура и классификация фер-
ментов). К их числу, в частности, относятся: нуклео-
зидаза (систематич. наименование N-рибозил-
пурин — рибогидролаза, шифр 3.2.2.1), катализиру-
ющая гидролиз N-рибозилпуринов (аденозин, гуано-
зин) на соответствующее основание и D-рибозу; ино-
зиназа (инозин—рибогидролаза, 3.2.2.2), гидроли-
зующая инозин, и уридин-нуклеозидаза (уридин —
рибогидролаза, 3.2.2.3), катализирующая гидролиз
уридина на урацил и D-рибозу. Все известные Н.
действуют только на рибонуклеозиды и неактивны
в отношении дезоксирибонуклеозидов.
Н. присутствуют гл. обр. в микроорганизмах (дрож-
жи, различные бактерии), где, по-видимому, прини-
мают участие в расщеплении нуклеотидов на их со-
ставные элементы. В животных тканях Н. практически
отсутствуют и нуклеозиды в них распадаются, гл. обр.
не гидролитич., а фосфоролитич. путем (см. Нуклео-
зидфосфор илазы).
Лит.: Fermente, Hormone, Vitamlne und die Beziehungen
dieser Wirkstoffe zueinander, Bd 1, 3 Aufl., Stuttgart, 1959,
S. 242. В. Б. Спиричев.
НУКЛЕ ОЗИДФОСФОРИЛ АЗЫ — тривиальное
название группы ферментов, катализирующих обра-
тимую реакцию фосфоролитич. расщепления нуклео-
зидов'.
Нуклеозид + Н3РО, основание+D-рибозо-!-фосфат.
По современной классификации Н. входят в под-
подкласс пентозилтрансфераз (шифр 2.4.2), поскольку
механизм катализируемой ими реакции состоит в пе-
реносе остатка пентозы нуклеозида на фосфорную к-ту
(см. Номенклатура и классификация ферментов).
К Н. относятся: пуриннуклеозидфосфорилаза (си-
стематич. наименование пуриннуклеозид: ортофос-
фат — рибозилтрансфераза, шифр 2.4.2.1), катализи-
рующая расщепление ряда пуриннуклеозидов; адено-
зинфосфорилаза (аденозин: ортофосфат — рибозил-
трансфераза, 2.4.2.2), осуществляющая фосфоролиз
аденозина; уридинфосфорилаза (уридишортофосфат—
рибозилтрансфераза, 2.4.2.3) и др. По субстратной
специфичности Н. можно подразделить на пуриннукле-
озидфосфорилазы, активные только в отношении пу-
риннуклеозидов, и пиримидиннуклеозидфосфорилазы,
расщепляющие только нуклеозиды пиримидина.
Большинство Н. способно расщеплять как рибо-, так и
дезоксирибонуклеозиды.
Н. широко распространены в животных тканях
(печень, мозг, эритроциты, мышцы), а также у микро-
организмов (дрожжи, Escherichia coli). Н. играют
важную роль в процессах расщепления нуклеозидов,
к-рые в животных тканях осуществляются преим. фос-
форолитич., а не гидролитич. путем. Катализируемая
Н. реакция фосфоролиза обратима, причем равновесие
в этой реакции сдвинуто в сторону синтеза нуклеози-
дов. На основе этого делались выводы о возможной
роли Н. не только в распаде, но и в синтезе нуклеоти-
дов, однако такая роль Н. сомнительна, поскольку
установлено, что мононуклеотиды in vivo синтезиру-
ются, минуя промежуточное образование нуклеозидов.
Пуриннуклеозидфосфорилаза из эритроцитов чело-
века выделена в высокоочищенном состоянии с по-
мощью хроматографии на геле Саз(РО4)2 и фракцио-
нирования (NH4)2SO4; pH оптимум фермента 7,0,
константа Михаэлиса (по инозину) 8-10~4М, уд. актив-
ность 2,1-10’ единиц. Ферментативная активность по-
давляется солями тяжелых металлов и п-хлорртуть-
бензоатом (реагенты на SH-группу) и восстанавли-
вается при добавлении этилендиаминтетраацетата или
цистеина.
Активность Н. определяют колориметрически по
убыли (прямая реакция) или образованию (обратная
реакция) неорганич. фосфата.
Лит.: The enzymes, ed. by P. D. Boyer, H. Lardy, K. Myr-
back, 2 ed., v. 5, N. Y., 1961, p. 235. В. Б. Спиричев.
НУКЛЕОЗИДЫ — природные N-P-D-гликозиды
общей ф-лы (I), содержащие в качестве агликона
(R) азотистое гетероциклич. осно-
вание, где R—пуриновое основание НОН2С
или пиримидиновое основание', аде-
нин, гипоксантин, гуанин, тимин,
урацил или цитозин, к-рым coot- I I
ветствуют Н.: аденозин, инозин, - он
гуанозин, тимидин, уридин и ци- !• (или Н>
тидин. В составе нуклеиновых к-т
в значительно меньших количествах обнаружены ме-
тилированные основания: 5-метилцитозин, 2-(или 6)-
метиладенин, 1-(или 2)-метилгуанин, 1-метилгипо-
ксантин, 5-оксиметилцитозин и др. Углеводным ком-
понентом обычно является D-p и б о з а в рибонуклео-
зидах и 2-д е з о к с и -D-p и б о з а в дезоксири-
бонуклеозидах. Аномерный атом углерода пентозы,
к-рая находится в фуранозной форме, присоединен
к азоту в кольце остатка пуринового (N9) или пири-
мидинового (№) основания.
Свободные Н. содержатся в небольших количествах
в различных биологич. тканях. Основная масса Н.
входит в состав нуклеиновых кислот и нуклеотидов',
впервые выделены из нуклеиновых к-т, откуда и по-
лучили свое название.
В последнее время к Н. стали относить также аналогично
построенные соединения, не входящие в состав нуклеиновых
к-т и содержащие другие углеводные компоненты и азотистые
основания, напр. антибиотик пуромицин, рибозид оротовой
кислоты — оротидин, рибозид мочевой кислоты, вИцин —
глюкозид 2,6-диамино-4-окси-пиримидина, арабинозиды ура-
цила и тимина и др.
Н.— бесцветные кристаллы, оптически активны,
имеют высокую темп-ру плавления. Большинство Н.
растворимы в горячей, хуже — в холодной воде, хо-
рошо растворимы в кислых или щелочных р-рах.
Пиримидиновые Н. растворимы в воде лучше пурино-
вых. Н., содержащие первичную NH2-rpynny, осаж-
даются из растворов ацетатом ртути или фосфороволь-
фрамовой к-той и образуют хорошо кристаллизирую-
щиеся пикраты. Осаждение рибонуклеозидов произво-
дят ацетатом свинца в аммиачной среде, большая
часть дезоксирибонуклеозидов остается при этом
в растворе. Характерные спектры поглощения У Ф-
света используются при идентификации Н.
Химич, свойства Н. определяются природой азо-
тистого основания, сахара и прочностью N-гликозид-
ной связи. Н., содержащие NH2-rpynny в гетероцикл,
ядре, имеют свойства слабых оснований, содержащие
ОН-группу—свойства слабых к-т. N-гликозидная
связь пуриновых Н. легко гидролизуется разб. к-тами
(1н. H2SO4 при 100° за 1 час) с освобождением осно-
вания и сахара. Пиримидиновые Н. устойчивы к кис-
лотному гидролизу; их N-гликозидная связь гидроли-
зуется лишь в жестких условиях, при к-рых происхо-
дит разрушение сахара. Устойчивость к гидролизу
теряется при восстановлении или бромировании пи-
римидинового основания, что дает возможность выде-
лить сахар и хроматографически его идентифициро-
вать. Н. алкилируются и ацилируются по азоту и
аминогруппам азотистого основания. Пиримидиновые
Н. легко присоединяют галоген-, нитро- и нитрозо-
603
НУКЛЕОЗИДЫ
604
Соединение pH ^макс.» мм к Вмакс.’Ю 3 8260 *Ю 3 -Ь350 ^280 рк
^ЗйО Е3ао основания сахара
Аденозин 1-2 7—12 257 259,5 14,6 14,9 14,3 14,9 0,84 0,78 0,215 0,144 3,63 12,5
Адениндезоксирибозид 2 258 14,1 — 0,8 3 0,24 — —
Гуанозин 1 7 И 256,5 252,5 258— 266 12,2 13,65 11,3 11,75 11,7 11,3 0,94 1,15 0,89 0,695 0,67 0,61 (1,6) (9.2) 9,5 2.2 12,3
Гипоксантиндезоксирибозид 2 249 13,2 1,63 0,23
Инозин 2н. НС1 3-6 11,2 251 248,5 253 10,9 12,25 13,1 9,0 7, 1 11.7 1 ,21 1 ,68 1,05 0,11 0,25 0,18 1 .2 8.9 12,3
Уридин 1 — 7 11-12 262 262 10,1 7,45 9,95 7,35 0,74 0,83 0,35 0,29 9,25 12,5
Урацилдезоксирибозид 1—7 11-12 262 262 10,2 7,63 10,1 7,55 0,72 0,81 0,375 0,31 9,3
Оротидин 2 12 267 267 9,57 7,6 — 0,65 0,82 0,79 0,72
Цитидин 1-2 7—1 2 280 271 13,4 9,1 6,4 7,55 0,45 0,86 2,10 0,94 4,11 12,3
Цитозиндезоксирибозид 1-2 7—1 2 280 271 13,2 9,0 6.15 7,35 0,42 0,83 2,14 0,965 4,25
Тимидин (тимидиндезоксирибо- зид) 1—7 12-13 267 267 9,65 7,38 8,75 6,65 0,65 0,75 0,72 0,67 9,8
Ксантозин 2 263 235 8,95 8,4 8,7 0,75 0,28 — 0
7 277 248 — 7,9 1,29 1,10 5,5 -13
8-11 278 248,5 8,9 10,2 7,65 1,30 1,13
5-Метилцитозин-дезоксирибозид 2 6 — 13 286 277 И ,6 8,81 3,2 6.0 0,45 0,95 3,4 1,45 4,5 — 13
группы к Cs основания. При действии HNO2 на Н. с
NH2-rpynnon (аденозин, гуанозин, цитидин) последние
дезаминируются с образованием соответствующих
ОН-производных (инозина, ксантозина, уридина).
Пиримидиновые Н. разрушаются гидразином и гидро-
ксиламином; эта реакция лежит в основе получения
апиримидиновых нуклеиновых кис-
лот (см. Нуклеиновые кислоты). Гликольная груп-
пировка сахара рибозидов подвергается перйодатному
окислению.
Методы качественного и количественного опреде-
ления Н. основаны на поглощении Н. УФ-света (ха-
рактеристика основания), а также на цветных реак-
циях пентоз и на хроматографии, и электрофоретич.
исследовании Н. и продуктов их химич. и фермента-
тивного гидролиза. Нек-рые физич. и спектральные
свойства Н. приведены в таблице.
Получают Н. химич. или ферментативным гидро-
лизом нуклеиновых к-т. Рибонуклеозиды получают
щелочным гидролизом рибонуклеиновой к-ты, напр.
конц. NH4OH или Ва (ОН)2. Дезоксирибонуклеозиды
получают ферментативным гидролизом дезоксирибо-
нуклеиновой к-ты. Образующаяся при гидролизе
смесь Н. разделяется методом ионообменной хромато-
графии. Синтетич. методы играют большую роль при
получении неприродных аналогов Н., используемых
в биологии и медицине. Наибольшее практич. значе-
ние имеют синтезы, связанные с присоединением
остатка моносахарида к пуриновому или пиримидино-
вому основанию, что достигается взаимодействием
галогенацилсахаров с металлич. (чаще ртутными)
производными азотистого основания. Известны также
методы получения Н., связанные с синтезом гетеро-
циклич. ядра. Новым методом синтеза пиримидино-
вых дезоксирибонуклеозидов является получение
их из природных рибонуклеозидов путем удаления
ОН-группы у С2 рибозного остатка, что достигается
получением 2-алкилмеркаптопроизводного и после-
дующей обработкой натрийиодидом, каталитич. гид-
рированием и гидролизом.
Н. входят в состав биологически важных соедине-
ний— нуклеиновых к-т и нуклеотидов; в последнее
время открыты Н., обладающие антибиотич. свойства-
ми, напр. кордицепин, пуромицин, амицетин. Синте-
зировано также большое число веществ, близких по
структуре Н., к-рые как антиметаболиты привлекают
внимание исследователей в связи с поисками лечебных
средств для борьбы с инфекционными заболеваниями
и с злокачественными образованиями.
Лит.: Шабарова 3. А., Усп. химии, 1959, 28, вып. 4,
369; Кочетков Н. К., Торгов И. В., Ботвиник
М. М., Химия природных соединений, М., 1961; F о х J. J.,
We треп I., Advances Carbohydr. Chem., 1959, 14; Hof-
f e г M. [a. о.], J. Amer. Chem. Soo., 1959, 81, № 15, 4112;
В г о w n D. M. [a. o.J, J. Chem. Soc., 1958, 80, 3035; The
nucleic acids, v. 1, N. Y., 1955, v. 3, N. Y., I960; S t а с e у M.,
Proceeding of the Fourth International Congress of biochemistry,
Vienna, 1958, v. 1, N. Y.—[a. o.J, 1959, p. 107; Jordan D.O.,
The chemistry of nucleic acids, L., 1960; Methods in enzymology,
ed. S. P. Colowik, N. O. Kaplan, v. 3, N. Y., 1957; Advances In
Carbohydr. Chem., 1962, 17, 301. Л. H. Боброва.
605
НУКЛЕОПРОТЕИДЫ
60S
НУКЛЕ ОПРОТЕИДЫ — высокомолекулярные при- I
родные вещества, представляющие собой комплек-
сы белков и нуклеиновых кислот. Й. относятся к слож-
ным белкам и присутствуют во всех без исключения
живых организмах, где они входят в состав клеточных
ядер и структурных элементов цитоплазмы; Н. явля-
ются также главными компонентами различных ви-
русов.
Несмотря на большое значение Н., они изучены сравни-
тельно мало. Это объясняется трудностями получения неиз-
мененных (нативных) препаратов Н. Кроме того, нуклеиновые
к-ты, подобно многим другим высокомолекулярным полианио-
нам, могут реагировать с белками с образованием солеобразных
соединений. Это обстоятельство долгое время вызывало затруд-
нения при ответе на вопрос, действительно ли существуют те
или иные Н. в клетках или же они образуются в процессе вы-
деления, как результат вторичного взаимодействия белков и
нуклеиновых к-т.
В зависимости от природы нуклеиновой к-ты,
входящей в состав Н., последние делят на дезоксири-
бонуклеопротеиды (ДНП), содержащие дезоксирибо-
нуклеиновую к-ту (ДНК) и рибонуклеопротеиды
(РНП), содержащие рибонуклеиновую к-ту (РНК).
Йаряду с естественными ДНП и РНП существуют Н.,
получаемые искусственно путем соединения различ-
ных белков с нуклеиновыми к-тами. О типах связей
между белком и нуклеиновой к-той в Н. известно
очень мало. Предполагают, что большую роль здесь
играют электростатические (ионные),координационные
и водородные связи. Как правило, частица Н. состоит
из одной пли нескольких молекул высокомолекуляр-
ной нуклеиновой к-ты (ДНК или РНК) и многочис-
ленных (несколько сотен или даже тысяч) белковых
молекул — субъединиц. Поэтому мол. веса как ДНП,
так и РНП обычно очень велики — порядка несколь-
ких миллионов. ДНП входят в состав структурных
элементов ядер клеток — хромосом, обусловливаю-
щих передачу наследственных признаков. ДНП
составляет также главную часть многих вирусов жи-
вотных, напр. осповакцины, желтухи шелкопряда,
папиломы кролика и др., а также вирусов бактерий
(т. н. бактериофагов).
Наиболее изученными представителями клеточных
ДНП являются нуклеопротамины и нуклеогистоны,
обычно приводимые как классич. примеры Н. Нук-
леопротамины были выделены из ядер спер-
матозоидов нек-рых рыб. Эти ДНП почти полностью
состоят из ДНК и протамина (белка с сильно основ-
ными свойствами, содержащего большое количество
остатков аргинина). Йуклеопротамин в ядре сперма-
тозоидов форели содержит, напр., 64% ДЙК и 36%
протамина. Н уклеогистоны находятся в яд-
рах многих клеток животных, а также высших расте-
ний. Удобным объектом для выделения нуклеогистона
является зобная железа. Количество ДНК в этих ДНП
составляет ок. 50%, остальная масса приходится на
гистон (белок с основными свойствами), а также на
небольшое количество негистонового белка, содержа-
щего остатки триптофана. Мол. вес этого ДНП 18,5
млн., он содержит всего одну макромолекулу ДНК
с мол. весом ок. 8 млн.
Морфологически эти дезоксирибонуклеопротеид-
ные частицы в р-ре представляют собой очень длинные,
довольно жесткие нитевидные макромолекулы диа-
метром ок. 30 А. В целом это, по-видимому, нить
макромолекулы ДНК (диаметр 20 А), более или менее
равномерно окруженная белком, гл. обр. гистонового
типа. Гистон в макромолекуле ДНП не является одно-
родным, а состоит из двух четко различающихся фрак-
ций: фракции А, составляющей 20—30% всего ги-
стона и представляющей т. наз. богатый лизином ги-
стон («лизиновый гистон»), мол. в. ок. 10 000; фракции
Б, составляющей преобладающую часть гистона и
иногда обозначаемой как богатый аргинином гистон
(«аргининовый гистон») с мол. в. ок. 20 000 и более |
сложным набором аминокислот. В одну указанную
выше дезоксирибонуклеопротеидную частицу входит
ок. 200—300 молекул лизинового гистона и ок. 350—
400 молекул аргининового гистона. Негистоновая
фракция ДЙП изучена слабо. То же можно сказать и о
всех других ДНП ненуклеогистонного типа.
Рибонуклеопротеиды входят в состав всех струк-
турных образований цитоплазмы и ядра клеток живых
организмов. Это — т. н. клеточные РНП. Принципи-
ально другую группу образуют вирусные РНП,
представителями к-рых являются почти все изученные
до сих пор вирусы растений, а также ряд вирусов жи-
вотных и человека (напр., вирусы полиомиелита,
различных энцефалитов, ящура и т. д.). Нек-рые
более сложные вирусы (напр., вирус гриппа) хотя
и не являются чистыми РНП, но содержат их в каче-
стве главного компонента.
Основным типом клеточных РНП являются т. н.
рибосомы, или рибонуклеопротеидные частицы. Они
являются специализированными внутриклеточными
структурами, осуществляющими биосинтез белков.
Эти частицы, или рибосомы, могут быть получены в
химически чистом виде путем препаративного уль-
трацентрифугирования гомогенатов клеток. Они со-
держат 40—65% высокополимерной РНК (в зависи-
мости от типа клеток), а остальное приходится на
структурный рибосомальный белок. Физико-химич.
изучение рибонуклеопротеидных частиц показало,
что они представляют собой довольно однообразные
по строению макромолекулы, примерно сферич.
формы с несколько неправильными очертаниями.
Мол. вес преобладающего типа нативных РНП-частиц
из различных тканей и организмов колеблется от
2,8 млн. (у бактерий кишечной палочки) до 4,5 млн.
(в проростках гороха), что соответствует линейным
размерам (диаметру) от 170—200 А до 300—400 А
и константам седиментации от 70 до 85 единиц Свед-
берга. Помимо этого преобладающего типа частиц,
могут быть получены, с одной стороны, более мелкие
РНП-частицы, являющиеся, по-видимому, продуктами
обратимой диссоциации первых на две субъединицы
неравной величины, и, с другой стороны, более круп-
ные частицы, представляющие собой ассоциаты пер-
вых. Диссоциация и ассоциация рибонуклеопротеид-
ных частиц в растворе в значительной степени опре-
деляется концентрацией ионов двухвалентных ме-
таллов и особенно Mg2 + . Внутренняя пространствен-
ная организация и структура рибосом до сих пор не-
ясна. О белке рибосом также имеются весьма скудные
сведения. Показано, что он состоит из молекул с мол.
в. ок. 20 000 и имеет слабо основную природу.
Что касается вирусных РНП, то все они характе-
ризуются правильными геометрия, формами и регу-
лярной периодичной внутренней структурой. Многие
вирусные РНП могут быть получены в виде кристал-
лов. Различают два основных типа вирусных РНП:
палочкообразные и сферические. Типичный предста-
витель первых — вирус табачной моза-
ики (ВТМ). Он представляет собой палочкообразную
частицу с мол. в. ок.. 40 млн., длиной ок. 3000 А и
диаметром 150—170 А; внутри сквозь всю палочку
проходит полый канал с диаметром 40 А. В каждую
такую частицу входит лишь одна макромолекула вы-
сокополимерной РНК с мол. в. 2 млн. и свыше 2000
белковых молекул — субъединиц с мол. в. 17 500
каждая; по весу РНК составляет 5,5% от всей массы
ВТМ. Белковые молекулы в частице ВТМ. располага-
ются по правильной спирали с шагом 23 А; по той же
спирали, между рядами белковых молекул, проходит
цепь РНК. Представители сферич. вирусов, такие
как вирус желтой мозаики турнепса или вирус полио-
миелита, имеют форму шара с диаметром в 300 А и
мол. в. 5—7 млн. Указанные представители содержат
607
НУКЛЕОТИДАЗЫ—НУКЛЕОТИДИЛТРАНСФЕРАЗЫ
608
30—35% РНК на сухой вес, что соответствует одной
макромолекуле РНК с мол. в. ок. 2 млн.; остальную
массу составляют белковые молекулы — субъединицы,
уложенные по типу кубич. симметрии по периферии
частиц.
Лит.: Белки, под ред. Г. Нейрата и К. Бейли, пер. с англ.,
т. 3, ч. 1, М., 1958, с. 54; Белозерский А. Н., Нуклео-
протеиды и нуклеиновые кислоты растений и их биологическое
значение, М., 1959; Збарекий И. Б., Усп. биол. химии,
1960, № 1, 91; Спирин А. С., Усп. соврем, биол., 1960, 50,
№ 3—6, 261; его же, Усп. биол. химии, 1962, № 4, 93;
Спитковский Д.М.,Тонгур В. С.,Д иекина Б.С.,
Биофизика, 1958, 3, № 2, 129; ZubayG., Do t у Р., J. Mole-
cular Biol., 1959, 1, № 1, 1; Nucleoproteins, N. Y., [1960],
(Inst. International de ehlmie Solvay. XI conseil de ehimie);
Microsomal particles and protein synthesis, ed. R. B. Roberts,
N. Y.—L., 1958; The nucleic acids, ed. E. Ghargaff, J. N. David-
son, V. 1, N. Y., 1955, v. 3, 1960. В. О. Шпикитер.
НУКЛЕОТИДАЗЫ — тривиальное название груп-
пы ферментов, катализирующих гидролиз монону-
клеотидов на нуклеозид и фосфорную к-ту. По сов-
ременной классификации Н. входят в подподкласс
гидролаз фосфомоноэфиров, шифр 3.1.3 (см. Номен-
клатура и классификация ферментов). К ним отно-
сится 5-нуклеотидаза (систематич. наименование:
5'-рибонуклеотид—фосфогидролаза, шифр 3.1.3.5) и
3-нуклеотидаза (З'-рибонуклеотид — фосфогидролаза,
3.1.3.6). 5-Нуклеотидаза гидролизует только нуклео-
зид-5'-фосфаты и неактивна в отношении нуклеотидов
с иным расположением остатка фосфорной к-ты, в
частности в отношении нуклеозид-З'-фосфатов. Этот
фермент присутствует во многих животных тканях,
где он, по-видимому, принимает известное участие в
процессах распада нуклеотидов на составные элемен-
ты. 5-Нуклеотидаза из спермы быка гидролизует
5'-монофосфаты аденозина, инозина, уридина и цити-
дина, а также никотинамидмононуклеотид. рН-оп-
тимум фермента 8,5; Михаэлиса константа (по 5'-
аденозимонофосфату) менее 10-4Л7. Mg2+ стимулирует,
а Са2+ тормозит активность фермента; 0,1М NaF пол-
ностью подавляет фермент. Zn2+ и Ni2+ также явля-
ются сильными ингибиторами 5-нуклеотидазы. Ак-
тивная 5-нуклеотидаза содержится также в яде змей,
являясь, по-видимому, одним из действующих начал
этого яда.
З-Нуклеотидаза специфически гидролизует только
нуклеозид-З'-фосфаты и неактивна в отношении нук-
леозид-5'-фосфатов или нуклеозид 3',5'-дифосфатов.
З-Нуклеотидаза присутствует гл. обр. в растениях
(плоды картофеля, проросшие семена ячменя). Она
с большой скоростью гидролизует З'-монофосфаты аде-
нозина, инозина, уридина и цитидина, а также кофер-
мент А; pH-оптимум фермента 7,5.
В связи с высокой субстратной специфичностью
5-нуклеотидаза и 3-нуклеотидаза широко применя-
ются в научных исследованиях для идентификации
и количественного определения соответственно нукле-
озид-5'-фосфатов и нуклеозид-З'-фосфатов. Актив-
ность Н. измеряют определением количества неорга-
нич. фосфата, освобождающегося при гидролизе
нуклеотидов.
Лит.: The enzymes, ed. Р. D. Boyer, H. Lardy, К. Myr-
back, 2 ed., v. 5, N.Y—L., 1961, p. 49; Fermente, Hormone,
Vitamlne und die Bezlehungen dieser Wlrkstoffe zuelnander,
hrsg. R. Ammon, W. Dlrscherl, 3 Aufl., Bd 1, Stuttgart, 1959,
S. 171. В. Б. Спиричев.
НУКЛЕОТИДИЛТРАНСФЕРАЗЫ — ферменты, ка-
тализирующие перенос нуклеотида от нуклеозидтри-
или дифосфата на тот или иной акцептор. По совре-
менной классификации ферментов Н. относятся к
трансферазам, переносящим группы, содержащие
фосфор, и образуют подподкласс 2.7.7 (см. Номенкла-
тура и классификация ферментов). Реакции переноса
нуклеотида от нуклеозидтрифосфатов идут с образова-
нием неорганич. пирофосфата и легко обратимы,
в связи с чем до введения современной номенклатуры
эти ферменты носили название пирофосфорилаз.
Н. широко распространены в животном и расти-
тельном мире, а также у микроорганизмов и играют
исключительно важную роль в различных процессах
обмена веществ, в частности в биосинтезе нек-рых ко-
ферментов, ди- и полисахаридов, фосфолипидов и
нуклеиновых к-т. По своему участию в тех или иных
процессах обмена Н. можно подразделить на след,
группы:
1. Н., принимающие участие в синтезе кофермен-
тов-динуклеотидов, в частности никотинамид-аденин-
динуклеотида (НАД) и флавин-аденин-динуклеотида
(ФАД). К ним относится АТФ: НМН —аденилилтранс-
фераза (шифр 2.7.7.1), катализирующая обратимую
реакцию образования НАД из никотинамид-моно-
нуклеотида (НМН) и АТФ:
АТФ + НМН 7—>- НАД + пирофосфат
а также АТФ:ФМН—аденилилтрансфераза (шифр
2.7.7.2), катализирующая образование ФАД из фла-
винмононуклеотида (ФМН):
АТФ + ФМН 7 ФАД + пирофосфат
АТФ: НМН — аденилилтрансфераза, выделенная в
высокоочищенном состоянии из дрожжей, обладает
оптимумом активности при pH 7,6 и характеризуется
величиной Михаэлиса константы (К м) для НМН и
АТФ 1,5-10-4 и 4,6-10-4 М. Для проявления актив-
ности фермента необходимо присутствие ионов Mg2 + ,
что характерно для многих Н.; Fe2+, Cu2+ и Zn2 +
подавляют ферментативную активность.
2. Н., участвующие в синтезе нуклеозиддифосфат-
сахаров. Последние представляют собой активирован-
ные реакционноспособные производные моносахари-
дов, в виде к-рых они вступают в различные реакции
взаимопревращения (превращение галактозы в глю-
козу), а также биосинтеза дисахаридов (сахароза,
лактоза) и полисахаридов (гликоген, целлюлоза,
различные мукополисахариды). Из числа ферментов
этой группы можно назвать УТФ: а-П-глюкозо-1-фос-
фат — уридилилтрансферазу (шифр 2.7.7.9), катали-
зирующую обратимую реакцию образования уридин-
дифосфатглюкозы (УДФ-глюкозы) из уридинтрифос-
фата (УТФ) и а-П-глюкозо-1-фосфата:
УТФ+ а-В-глюкозо-1-фосфат т—> УДФ-глюкоза + пирофосфат
а также У ТФ:а-П-галактозо-1-фосфат — уридилил-
трансферазу (шифр 2.7.7.10), катализирующую ана-
логичную реакцию образования уридиндифосфога-
лактозы (УДФ-галактозы). Первый фермент, получен-
ный в очищенном состоянии из дрожжей, обладает
оптимальной активностью при pH 7,0 и характери-
зуется величиной К для УТФ- и УДФ-глюкозы
4-10~6 и 5-10~5 М. Mg2+ в концентрации 2-10~2М
активирует фермент; анионы (сульфат, фосфат) в
концентрации 0,03 М и п-хлорртутьбензоат 10~7М
подавляют его активность. Оба фермента присут-
ствуют в клетках печени и др. животных тканей.
3. Н., осуществляющие синтез цитидин-дифосфо-
холина (ЦДФ-холина) и цитидин-дифосфоэтаноламина
(ЦДФ-этаноламина), принимающих участие в биосин-
тезе фосфолипидов (лецитина и коламинфосфатидов).
Синтез ЦДФ-холина из цитидин-трифосфата (ЦТФ)
и фосфохолина катализирует ЦТФ : холинфосфат —
цитидилтрансфераза (шифр 2.7.7.15). Синтез ЦДФ-
этаноламина осуществляет ЦТФ:этаноламинфосфат—
цитидилилтрансфераза (шифр 2.7.7.14). Оба фермента
выделены из печени и активируются Mg24".
4. Н., участвующие в синтезе нуклеиновых кислот.
К ним относится выделенный из микроорганиз-
ма Esherichia coli и изученный А. Корнбергом фер-
мент дезоксинуклеозидтрифосфат: ДНК — нуклеоти-
дилтрансфераза (шифр 2.7.7.7) (тривиальное название
ДНК-нуклеотидилтрансфераза), осуществляющийсин-
609
НУКЛЕОТИДЫ
610
тез ДНК из нуклеозидтрифосфатов. Необходимым
условием синтеза является присутствие 4 дезоксину-
клеозидтрифосфатов (аденозин-, гуанозин-, цитидин-
и тимидинтрифосфата), а также «затравки» моноспи-
ральной ДНК, играющей роль матрицы, на к-рой
осуществляется синтез новой ДНК. Количество син-
тезируемой ДНК в десятки раз превосходит количе-
ство «затравки», а ее природа определяется природой
ДНК-матрицы. ДНК— нуклеотидилтрансфераза яв-
ляется первым изученным ферментом, осуществля-
ющим синтез ДНК. В связи с особенностями меха-
низма действия к подподклассу Н. относится также
рибонуклеаза поджелудочной железы.
Лит.: Современные проблемы биохимии. Сб. статей, М.,
1961, с. 236; The enzymes, ed. Р. D. Boyer, H. Lardy, К. Myr-
back, v. 5, 2 ed., N. Y.—L., 1961, p. 282. В. В. Спиричев.
НУКЛЕОТИДЫ — фосфорные эфиры нуклеозидов,
природные биологически активные соединения, ши-
роко распространенные в животных и растительных
тканях и микроорганизмах как в свободном состоя-
нии, так и в составе различных соединений (нуклеи-
новых к-т, нек-рых коферментов и витаминов).
Термин «Н.» первоначально был предложен для обозначе-
ния продуктов разрушения нуклеиновых кислот — фосфори-
лированных нуклеозидов, имеющих структуру: пуриновое осно-
вание или пиримидиновое основание — пентоза — фосфорная
к-та. Затем Н. стали называть и соединения, к-рые не входят
в состав нуклеиновых к-т, но имеют аналогичную структуру.
Вместо пуриновых и пиримидиновых оснований они могут
содержать другое циклич. азотистое основание, напр пиридин
пли изоаллоксазин. Позже название Н. было распространено
также на соединения с большей степенью фосфорилирования,
т. е. на нуклеозидполифосфаты, к-рые вместе с нуклеозидмо-
нофосфатами и многочисленными низкомолекулярными произ-
водными образуют т. наз. кислоторастворимые Н. (в отличие от
высокомолекулярных полинуклеотидов, не растворимых на
холоду в разб. к-тах). Нек-рые исследователи включают
в группу Н. и их полимеры.
Соединения, состоящие из одной молекулы азотистого ос-
нования, сахара и фосфорной к-ты, относят к мононуклеоти-
дам, или собственно Н. Мононуклеотиды, соединяясь между
собой, образуют динуклеотиды (см. Никотинамиднуклеотид-
ные коферменты, Флавиновые коферменты), тринуклеотиды
и т. д. Комбинации из нескольких связанных между собой мо-
нонуклеотидов по аналогии с олигосахаридами
носят название олигонуклеотидов, в то время
как макромолекулы называют полинуклеотидами (см. Нукле-
иновые кислоты). По количеству остатков фосфорной к-ты
различают нуклеозидмонофосфорные к-ты и нуклеозидполифос-
форпые к-ты. Нуклеозидфосфаты сокращенно обозначают на-
чальными буквами, указывающими на входящий в Н. нуклео-
зид и количество фосфора, напр. аденозин-З'-монофосфат —
АМФ-3', уридин-5'-дифосфат — УДФ-5', цитидин-5'-трифос-
фат — ЦТФ-5'. Цифра указывает на порядковый номер угле-
рода пентозы, к к-рому присоединен остаток фосфорной к-ты
(в обозначениях нуклеозид-5‘-фосфатов она часто опускается).
Перед сокращенным названием дезоксинуклеотидов ставят
слово Дезокси- или букву «д» (напр., дезокси-ГМФ, д-ГМФ)
Наиболее широко распространенными пуриновыми
и пиримидиновыми основаниями природных Н. яв-
ляются аденин, гуанин, гипоксантин, цитозин, ура-
цил и тимин, к-рым соответствуют Н.: аденозин-,
гуанозин-, инозин-, цитидин-, уридин- и тимидин-
фосфорные к-ты. Сахарный компонент представлен
обычно D-рибозой в рибонуклеотидах и 2-дезокси-
D-рибозой в дезоксирибонуклеотидах.
Идентификацию Н. и определение чистоты их
препаратов проводят хроматографии., электрофоре-
тич. и спектральными методами. Н. дают реакции,
характерные для компонентов, входящих в их со-
став, — азотистых оснований (см. Пуриновые основания
и Пиримидиновые основания), пентоз и органически
связанного фосфора. Пентозы определяются цветными
реакциями, напр. реакцией с орцином или дифенил-
амином соответственно для рибо- и дезоксирибонукле-
отидов. Пентоза пиримидиновых Н. определяется
полностью только после обработки, ослабляющей
N-гликозидную связь. Положение фосфата в нукле-
озид-2'-, З'-и 5'- фосфатах устанавливается химич.
методами, основанными на реакциях определения
свободных соседних ОН-групп в пентозном остатке Н.:
перйодатным методом, реакцией образования раство-
020 к. X. Э. т. 3
римого медного комплекса, а также методами, осно-
ванными на скорости гидролиза фосфора. Для этой
цели используют специфич. ферменты 3- и 5-нук-
леотидазы, отщепляющие фосфат от соответствую-
щих нуклеозид-3'- и 5'-фосфатов. Для спектраль-
ной характеристики Н. часто применяют величины
отношений оптич. плотностей, измеренных в УФ-свете
при разных длинах волн (в ммк) и pH (см. таблицу).
Концентрация Н. может быть определена по оптич.
плотности (Е) раствора Н. и коэфф, молярной экс-
тинкции (е): молярность =Е/е.
Нуклеозидмонофосфорные кис-
лоты (нуклеозидмонофосфаты, мононуклеотиды)
представляют собой N-гликозидфосфаты, т. е. орто-
фосфорные эфиры нуклеозидов. По положению фос-
фатной группы возможно существование трех изомеров
для рибонуклеотидов (нуклеозид-2'-, 3'- и 5'-фосфа-
тов) и двух изомеров для дезоксирибонуклеотпдов
(нуклеозид-3'- и 5'-фосфатов). В природе в свободном
состоянии преобладают нуклеозид-5'-фосфаты (I), где
R — аденин, гуанин, цитозин, тимин, урацил пли
гипоксантин. Нуклеозид-2'- и З'-фосфаты образуются
при химич. и ферментативном гидролизе нуклеиновых
к-т. Число изомеров увеличивается образованием
пиклич. нуклеозид-2':3'-(П) и 3':5'-фосфатов.
Нуклеозидмонофосфаты — бесцветные оптически
активные вещества с высокой темп-рой плавления (ча-
сто с разл.); растворимы в воде, нерастворимы в обыч-
ных органич. растворителях; обладают свойствами
кислот, по кислотности превосходят минеральный
ортофосфат; образуют соли с ионами металлов и осно-
ваний. Соли нуклеозидмонофосфатов с бруционом и
циклогексиламином хорошо кристаллизуются; соли
щелочных металлов хорошо растворимы в воде (за
исключением калиевых и натриевых солей гуанозин-
фосфатов, выпадающих уже из 5%-ного р-ра при хра-
нении на холоду), соли щелочноземельных металлов
хуже растворимы в воде и нерастворимы в спирте.
Нуклеозидмонофосфаты, содержащие первичную NH2-
группу, осаждаются р-рами Hg (СН3СОО)г или фосфо-
ровольфрамовой к-ты. Осаждение нуклеозидмонофос-
фатов в виде свинцовых солей используют для отделе-
ния их от нуклеозидов, аминокислот, сахаров.
Характерные спектры поглощения нуклеозидмонофос-
фатов в УФ-области определяются в основном приро-
дой основания и зависят от pH. Спектры дезоксирибо-
нуклеотидов и рибонуклеотидов различаются лишь
при высоких pH, вызывающих ионизацию ОН-групп
сахара. При длительном воздействии УФ-лучей может
наступить фотолиз пуриновых и пиримидиновых
Н., что проявляется в сильных изменениях спектра
поглощения. Нек-рые спектрофотометрия, константы
нуклеозидмонофосфатов приведены в таблице.
Химич, свойства нуклеозидмонофосфатов опреде-
ляются свойствами соответствующего азотистого осно-
вания, углеводного остатка, а также стабильностью
N-гликозидной и фосфоэфирной связей (см. Пурино-
вые основания, Пиримидиновые основания, Нуклеозиды).
Фосфоэфирная связь пуриновых нуклеозидмонофос-
фатов более лабильна в кислой и щелочной среде, чем
пиримидиновых. Пуриновые нуклеозидмонофосфаты
полностью гидролизуются в кислой среде (1 н. НС1 за
1 час при 100°) до азотистого основания, пентозы и фос-
611
НУКЛЕОТИДЫ
612
Соединение pH ^макс, ммк е-10-3 макс. е-10-3 260 ^250 РК
^260 £<260 основания фосфата
Аденозин-5 '-монофосфа! 2 7—12 257 259 15,0 15,3 14,5 15,3 0,84 0,79 0,22 0,15 3,74 6,2-6,4; 0,9
Аденозин- 5' - дифосфат 2 7-12 257 259 15,0 15,4 14,5 15,4 0,85 0,78 0,21 0,16 3,95 6,1—6,7
Аденозин-5'-трифосфат 2 7—11 257 259 14,7 15,4 14,3 15,4 0,85 0,80 0,22 0,15 4,00 6,0—6,95
Аденозин-2'- и 3'-фосфат (смесь изомеров) 2 7—12 257 259 15,0 15,3 14.5 15,3 0,85 0,80 0,23—0,22 0,15 3.81 (2') 3,74 (3') 6,17 (2') 5,92 (3'>
Гуанозин- 5 '-фосфат 1 7 И 256 252 258 12,2 13.7 11,6 11,6 11,7 11,5 0.96 1,16 0,90 0,67 0,66 0,61 2,4 9,4 6,1; 0,7
Гуанозин- 5' - дифосфат 1 7 11 256 253 257 12,3 13,7 11,7 11,8 11,8 11,7 0,95 1,15 0,91 0,67 0,66 0,61 2.9 9,6 6,3
Гуанозин-5'-трифосфат 1 7 И 256 253 257 12.4 13,7 11,9 11.8 11,7 11.8 . 0,96 1,17 0,92 0,67 0,66 0,59 3,3 9.3 6,5
Гуанозин-2'- и З'-фосфат (смесь изомеров) 1 12 257 253 12,5 13,7 12,3 11,8 0,90—0,93 1.15 0.89 0,68 0.68 0.60 2,3 9,36 5,9; 0,7
У ридин-5'-монофосфат 2-7 11 12 262 261 10,0 7,8 9.9 7 , 7 7,3 0,73 0,83 0,82 0,39 0,31 0,33 9,5 6,4; 1,0
Уридин-5 '-дифосфат 2-7 11 262 261 10,0 7,9 9,9 7,8 0,73 0,80 0,39 0,32 9,4 6.5
Уридин-5 '-трифосфат 2-7 11 262 26f 10,0 8,1 9,9 8,0 0,75 0,81 0.38 0,31 9,5 6,6
Уридин-2'- и З'-фосфат (смесь изо- меров) 2 7 12 26 2 262 261 10,0 10,0 7,29 9,9 9 , 9 7,29 0,80—0.76 0,7 8—0,73 0,85-0,83 0.28—0,32 0,3 0—0,3 5 0,25 9,43 5,88; 1,02
Цитидин- 5 '-монофосфат 1-2,5 6-12 280 271 13,2 9.1 6.3 7,4 0,45 0,84 2,10 0,99 4,5 6,3; 0,8
Цитидин-5 '-дифосфат 1—2,5 6—12 280 271 12,8 9,1 6,2 7,4 0,46 0.83 2,07 0,98 4,6 6,4
Цитидин-5'-трифосфат 2 7—И 280 271 12,8 9.0 6,0 7.4 0.45 0,84 2.12 0,97 4.8 6,6
Цнтидин- 2 '-монофосфат 1-2,5 6—12 278 270 12,75 8,89 6,9 7,75 0,48 0.90 1 ,83 0.86 4,30 4,44 6,19
Ц итидин- 3' -монофосфат 1-2,5 6—12 279 271 13,2 9,2 6,6 7,6 0,45 0,86 2.00 0,93 4, 16 4,31 6,04
Тимидин- 5' -монофосфат 1-7 12 267 9,6 8,4 6,7 0,65 0,74 0,72 0,67 10,0 6, 5; 1,6
форной к-ты; пиримидиновые — в этих условиях прак-
тически не гидролизуются. Для отщепления фосфора
от пиримидиновых нуклеозидмонофосфатов требуются
более жесткие условия, в к-рых происходит разруше-
ние органич. части молекулы; связь фосфата и пентозы
значительно ослабляется в соединениях с фосфоди-
эфирной связью, как, напр., в нуклеиновых к-тах.
Фосфорная связь в нуклеозид-2'-и З'-фосфатах более
кислотолабильна, чем в нуклеозид-5'-фосфатах. Дефос-
форилирование нуклеозидмонофосфатов происходит
также при действии щелочи, что используется при
получении рибонуклеозидов (см. Нуклеозиды). Цик-
лич. нуклеозид-2':3'-фосфаты гидролизуются в разб.
щелочах и слабых к-тах с образованием равновесной
смеси нуклеозид-2'- и З'-фосфатов. Циклич. нуклео-
зид-3':5'-фосфаты более устойчивы, особенно в щелоч-
ной среде. В присутствии конденсирующих агентов,
напр. карбодиимидов, нуклеозидмонофосфаты вступа-
ют в различные реакции конденсации (с фосфорной
к-той, с первичными и вторичными аминами).
N-гликозидная связь пуриновых нуклеозидмоно-
фосфатов менее прочна в кислой среде, чем фосфоэфир-
ная связь; она разрывается при мягком кислотном
гидролизе с освобождением пентозофосфата и основа-
ния. Нестойкость N-гликозидной связи пуриновых
нуклеозидмонофосфатов в кислой среде используют
при получении апури новых нуклеино-
вых кислот, т. е. лишенных пуриновых компо-
нентов без деполимеризации (см. Нуклеиновые кисло-
ты). N-Гликозидная связь пиримидиновых нуклеозид-
монофосфатов очень прочпая; она гидролизуется без
разрушения пентозы лишь после восстановления или
бромирования пиримидинового кольца.
Получают нуклеозидмонофосфаты химич. илп фер-
ментативным гидролизом нуклеиновых к-т. Рибону-
клеозид-2'- и З'-фосфаты получают мягким щелочным
613
НУКЛЕОТИДЫ
614
гидролизом рибонуклеиновой к-ты; при этом в конеч-
ном итоге образуется равновесная смесь нуклеозид-2'-
(40%) и З'-фосфатов (60%), к-рые обозначались как
а- и б-изомеры. Пиримидиновые нуклеозид-2'- и З'-фос-
фаты образуются также при кислотном гидролизе
рибонуклеиновой к-ты. Пуриновые нуклеозидмоно-
фосфаты в условиях кислотной деполимеризации раз-
рушаются. Рибонуклеозид-5'-фосфаты получают фер-
ментативным гидролизом рибонуклеиновой кислоты,
дезоксирибонуклеозидмонофосфаты — ферментатив-
ным гидролизом дезоксирибонуклеиновой к-ты. Смесь
нуклеозидмонофосфатов в гидролизатах нуклеиновых
кислот разделяют с помощью ионообменной хромато-
графии.
Синтетически нуклеозидмонофосфаты получают
фосфорилированием нуклеозидов. В качестве фосфо-
рилирующих агентов используют различные произ-
водные фосфорной и пирофосфорной к-т, напр. ди-
бензил- и дифенилхлорфосфат, тетрахлорпирофосфат
и др. Для этих целей применяют также фосфорную
к-ту и Р-цианоэтилфосфат в присутствии конденси-
рующего агента, напр. дицикл огексилкарбодиимида.
Нуклеозидмонофосфаты являются составными ча-
стями нуклеиновых к-т, нуклеотидных коферментов,
нек-рых витаминов и др. Они играют важную роль в
обмене веществ', в виде производных с аминокислотами
(процесс активирования аминокислот) участвуют в
биосинтезе белка.
Нуклеозидполифосфорные кис-
лоты (нуклеозидполифосфаты) образуются в био-
логич. тканях путем фосфорилирования свободных
нуклеозидмонофосфатов; являются производными ли-
нейных полифосфатов (исключение составляют нукле-
озид-2',3'- и 3',5'-дифосфаты, образующиеся при фер-
ментативном расщеплении нуклеиновых к-т, а также
аденозин-3'-фосфат-5'-фосфосульфат, выделенный из
биологич. тканей). Общая ф-ла нуклеозид-5'-ди- и
трифосфатов, наиболее распространенных в природе,
приведена ниже (IIГ).
ООО
и „ н и п
НО—Р—О—р—О— Р—О—Н2С / основание
I I I I I L/ \|
I ОН I ОН ОН \Н н/
; ! н\|—|<н
I ! он он
1 1 , 1
1 (---нуклеоэид-5—дифосфат-1
I-------нунлеозид-5-трифосфат-1
До недавнего времени (1950) были известны лишь адено-
зинди- и трифосфат. Позже в составе биологич. тканей в сво-
бодном состоянии об-
наружены аналогич-
но построенные ди- и
трифосфаты уридина,
гуанозина, инозина,
тимидина, цитидина.
Выделены также аде-
нозинтетра-и пента-
фосфаты, атакже раз-
нообразные производ-
ные нуклеозидполифосфатов, большинство к-рых имеет общую
ф-лу (IV), где X — остаток гексозы, пентозы, аминосахара,
уроновой к-ты, аминокислоты и др. соединений.
Нуклеозидполифосфаты — бесцветные оптически
активные гигроскопичные вещества без четкой темпе-
ратуры плавления, поглощают УФ-свет; раство-
римы в воде, нерастворимы в обычных органических
растворителях; проявляют сильные кислотные свой-
ства. Соли щелочных металлов нуклеозидполифосфа-
тов хорошо растворимы в воде, соли щелочноземельных
и тяжелых металлов плохо растворимы. С увеличением
количества фосфатных остатков растворимость в воде
солей щелочноземельных металлов уменьшается.
Лучше растворимы соли пиримидиновых нуклеозид-
полифосфатов. Присоединение остатка углевода к ну-
клеозидполифосфату повышает растворимость в воде
солей щелочноземельных металлов. Наиболее часто
получают соли Са, Ba, Pb, Hg. Спектры в УФ-свете
нуклеозидполифосфатов с разной степенью фосфори-
лирования практически одинаковы (см. таблицу).
По химич. свойствам нуклеозидполифосфаты напо-
минают нуклеозидмонофосфаты с той разницей, что
они имеют дополнительно кислотолабильную пиро-
фосфорную связь, гидролизующуюся уже за 10 мин.
в 1 н. к-те при 100°; значительное дефосфорилирование
происходит в щелочной среде. В соединениях типа
УДФ-глюкозы пирофосфорная связь имеет повышен-
ную щелочелабильность, однако действие щелочи
на эти соединения меняется с природой сахарного
остатка. При наличии свободной ОН-группы в цис-
положении к полуацетальному гидроксилу (напр.,
в глюкозе) вещество очень чувствительно к щелочи.
Стабильность повышается, если свободная ОН-группа
у Сг второго углеводного остатка отсутствует, как,
напр., в УДФ-ацетилгексозамине.
Нуклеозидполифосфаты могут быть выделены из
биологич. материала экстракцией их разб. НСЮа,
СС1зСООН, кипящей водой или водным спиртом; при
последующей обработке экстрактов (адсорбция на угле,
осаждение в виде солей щелочноземельных и тяжелых
металлов) можно выделить лишь группы веществ со
сходными физич. и химич. свойствами. На индивиду-
альные вещества нуклеозидполифосфаты разделяются
с помощью ионообменной хроматографии и ионофореза.
Синтетич. методы получения нуклеозидполифосфа-
тов связаны с фосфорилированием нуклеозидов химич.
или биологич. путем. В биологич. синтезе применяют
ферменты, к-рые фосфорилируют нуклеозиды до нукле-
озидмоно- и полифосфатов. Из химич. методов синтеза
нуклеозидполифосфатов наиболее широко распростра-
нен карбодиимидный метод, в к-ром нуклеозид конден-
сируется последовательно с несколькими молекулами
фосфорной к-ты с образованием пирофосфорной связи.
Удобны и перспективны в препаративном отношении
методы получения нуклеозидполифосфатов в мягких
условиях с использованием нуклеозид-5'-фосфоами-
датов
ОН
нуклеозид—О. . |
)P-NH, +НО-Р-ОН —>
- ох|| 3 II
о о
нуклеозид—О. .ОН
—>- Xp-0-Pf
-ОХИ ПХОН
О о
и нуклеозид-5'-фосфоморфолидатов. Последними ме-
тодами (карбодиимидным и фосфоамидатным) полу-
чают в мягких условиях и более ложные производные,
напр. УДФ-глюкозу, ГДФ-маннозу и др. В этом
случае вместо фосфорной к-ты используют фосфорные
эфиры сахаров.
Нуклеозидполифосфаты — одна из наиболее актив-
ных групп соединений, встречающихся в природе;
в качестве субстратов они участвуют в биосинтезе
нуклеиновых к-т. Коферментная функция связывает
их со всеми видами обмена веществ (углеводным, бел-
ковым, липидным). Нуклеозидполифосфаты участвуют
в биологич. окислительно-восстановительных реак-
циях, в процессах эпимеризации, фосфорилирования,
переноса гликозильных остатков на углеводные и др.
акцепторы и др.; как макроэргич. соединения они
играют важную роль в накоплении химич. энергии и в
превращении ее в механич. работу при мышечном сок-
ращении; нек-рые из них являются стимуляторами
сердечной деятельности. См. также А денозинфосфор-
ные кислоты, Инозинфосфорные кислоты, Уридинфос-
форные кислоты, Тимидинфосфорные кислоты, Цити-
динфосфорные кислоты.
20*
615
НУКЛЕОФИЛЬНЫЕ И ЭЛЕКТРОФИЛЬНЫЕ РЕАГЕНТЫ —НУКЛОНЫ
616
Лит.: Белозерский А. Н.,Спирин А. С., Успехи
соврем, биол., 1956, 41, вып. 2, 144; Боброва Л. Н., Сте-
паненко Б. Н., Успехи биол. химии, 1962, 4, 134; Ко-
те л ь н и к о в а А. В., там же, 1958, 3, 206; е е ж е, Успехи
соврем, биол., 1957, 43, вып. 2, 133; Кочетков Н. К.,
Торгов И. В., БотвиникМ. М., Химия природных со-
единений, М., 1961; Березовский В. М., Артемкина
Р. В., Усп. химии, 1962, 31, 724; СпиринА. С., Успехи
биол. химии, 1962, 4, 93; The nucleic acids. Chemistry and
biology, ed. E. Chargaff, J. N. Davidson, v. 1—3, N. Y., 1955—
1960; Methods in enzymology, ed. S. P. Colowlck, N. O. Kaplan,
v. 3, N. Y., 1957; Henderson .1. F.,L e Page G. A., Chem.
Revs, 1958, 58, № 4L 645; Baddl ley J., Buchanan
J. G., Quart. Revs, 1958, 12, № 2, 152; S t e 1 n e r R. F., Be-
ers H. F., Polynucleotides, Amst.—L.—N. Y., 1961; J о r-
d a n D. O., The chemistry of nucleic acids, L., 1960; H e p p e 1
L. A., Nucleotides and nucleosides, в кн.: Chemical pathways of
metabolism, ed. D. M. Greenberg, v. 2, N. Y., 1954, p. 263;
Baddlley J., Hughes N. A., Advances In enzymology,
1960, 22, 157; Strominger J. L., Phys. Revs., 1960, 40,
№ F, 55; К h о r a n a H. G., Some recent development in the
chemistry of phosphate esters of biological interest, N. Y.—L.,
1961; Annual Rev. Biochem., 1961, 30, 133; J a v I 1 1 i e г M.
[e. a.], Tralte de biochimie gdndrale, t. 1, P., 1959.
НУКЛЕОФИЛЬНЫЕ И ЭЛЕКТРОФИЛЬНЫЕ
РЕАГЕНТЫ — два типа реагентов в ионных органич.
реакциях, различающихся по характеру участия в об-
разовании новой или разрыве имеющейся связи. К
нуклеофильным (электронодонорным или анионоид-
ным) реагентам относятся ионы или молекулы, к-рые,
участвуя в реакции, отдают свои электроны или раз-
деляют их с другими атомными ядрами:
Z-+R ]:Х >R-Z + X-
Y |:Z + R |:Х —>R-Z+YX
Y |:Z + ^C = C^—► ^Z-C-C-J ——>Z —С —C—Y
Z"h YZ — нуклеофильные реагенты по отношению к
RX и \>С=с/ . Нуклеофильными реагентами явля-
ются все анионы, а также соединения, несущие не-
поделенную пару электронов (амины, спирты, мер-
каптаны, фосфиты, фосфины, сульфиды и др.). Ну-
клеофильная активность повышается параллельно
основности, напр. СгН5О ~ >СвН5О->С$Н5ОН, однако
основность и нуклеофильность не являются тожде-
ственными характеристиками. Основность служит
мерой сродства иона или молекулы к протону; нуклео-
фильная активность характеризует сродство частицы
к атому углерода. Так, CeHsO ~ и Вт - обладают почти
одинаковой нуклеофильностью, однако основность
фенолят-иона значительно выше.
Реагенты, действие к-рых связано с приобретением
электронов либо частичным приобретением электрон-
ной пары, наз. электрофильными (электроноакцеп-
торными, или катионоидными):
Z++R:| X--->RZ + X +
Z+ и YZ — электрофильные реагенты по отношению
к RX и
Электрофильными являются все
катионы, а также соединения, стремящиеся достроить
свою электронную оболочку до устойчивой конфигу-
рации, как, напр., апротонные к-ты (BF3, FeCl3,
А1С13 и др.). Кислотность, т. е. степень сродства к
электрону, является частным случаем электрофиль-
ности, т. е. сродства к внешним электронам.
Степень нуклеофильности или электрофильности
в общем случае определяется величиной энергии дан-
ной частицы. Чем больше энергия данной системы,
т. е. чем эта система неустойчивее, тем выше ее соот-
ветственно нуклеофильная либо электрофильная ре-
акционная способность. Нуклеофильный или электро-
фильный характер молекулы, способной к диссоциа-
ции, определяется характером менее устойчивого
иона, образующегося при диссоциации. Так, если
молекула Вгг в условиях реакции образует ионы
Вг+ и Вг~, то катион брома, обладающий значительно
большей энергией, чем соответствующий анион,опре-
деляет поведение молекулы брома в целом, и она реа-
гирует как электрофильная частица.
В молекуле органич. соединения часто можно раз-
личить четко выраженные нуклеофильные и электро-
фильные центры. Так, карбонильная группа в альде-
гидах или кетонах имеет электрофильный центр на
углероде, а нуклеофильный центр — на кислороде; в
большинстве случаев электрофильный центр более
активен и соединения, содержащие эту группировку,
носят электрофильный характер. Электрофильный
характер карбонильной группы доказывается кине-
тич. исследованиями циангидринной реакции. Уста-
новлено, что реакция не осуществляется в кислотах
и скорость процесса определяется концентрацией
цианид-иона, из чего следует, что первичным актом
является атака электрофильного атома углерода:
R' медленно
'С = О + C = N ..— ХСХ
R-'Z R" C=N
о. О" быстро R' он
'< + н* S—- - хсх
'c=N R" C=N
Нуклеофильная или электрофильная реакционная
способность молекулы часто зависит от условий про-
цесса. Так, если в нейтральной или щелочной среде
альдегиды или кетоны характеризуются электрофиль-
вой активностью и легко подвергаются атаке аниона-
ми, то в присутствии сильных к-т первоначальным
актом реакции является присоединение протона, т. е.
карбонильное соединение проявляет нуклеофильный
характер.
Переход от нуклеофильной к электрофильной ак-
тивности наблюдается при переходе от олефинов к
фторолефинам. Для этилена типичными являются
реакции, начинающиеся с атаки катионом, а анионоид-
ная атака резко затруднена: в случае тетрафторэти-
лена специфическими являются реакции, начинаю-
щиеся с атаки анионом либо анионоидным реагентом,
а реакции электрофильного присоединения осущест-
вляются с трудом. Электрофильность и нуклеофиль-
ность являются понятиями относительными. Одно
и то же соединение, в зависимости от реакционной
способности другого реагента, может проявлять нук-
леофильную или электрофильную активность. Так,
напр., тетрафторэтилеи является типичным электро-
фильным реагентом, однако при взаимодействии с
серным ангидридом, электрофильность к-рого выра-
жена более ярко, тетрафторэтилеи выступает как ну-
клеофильная частица:
л-0
CF2-S<
cf2=cf2+so3—>| I'O
cf2-o
Лит.: ИнгольдК., Механизм реакций и строение орга-
нических соединений, пер. с англ., М., 1959, о. 166—168, 289.
Л. С. Герман.
НУКЛОНЫ (нуклеоны) — общее название частиц
(протонов и нейтронов), входящих в состав всех атом-
ных ядер. Названы от лат. nucleus — ядро. Масса
нейтрона всего на 0,14% больше массы протона, спины
их одинаковы. Обе эти частицы могут взаимопревра-
щаться — как внутри ядер в процессах бета (Р~ и
Р + )-распада, так и в свободном состоянии — в про-
цессах, происходящих с переносом т. наз. л-мезонов
от одного Н. к другому. Во взаимодействиях, обус-
ловленных ядерными силами, протон и нейтрон
617
НУЛЕВАЯ ЭНЕРГИЯ—НУТЧ-ФИЛЪТР
618
проявляют себя одинаковым образом, и различия в их
поведении связаны лишь с наличием у протона
электрич. заряда. Поэтому в ряде случаев оказывается
целесообразно характеризовать протон и нейтрон как
два различных «зарядовых состояния» единой ядерной
частицы — Н.
См. также Элементарные частицы.
НУЛЕВАЯ ЭНЕРГИЯ — низший энергетич. уро-
вень системы, отсчитанный от наименьшего возможно-
го по классич. механике значения энергии; энергия,
сохраняющаяся при абс. нуле темп-ры. Существова-
ние Н. э., не равной нулю, обусловлено квантово-
механич. законами. Ее происхождение и величина за-
висят от вида энергии и характера системы.
Для отдельной частицы, свободно движущейся на
отрезке длиной а (в т. н. одномерном потенциальном
ящике), квантовая механика дает следующие возмож-
ные уровни энергии Е, к-рая в данном случае сводится
к кинетической:
Е = n2h2/8ma
где п=1, 2, 3. . . квантовое число, h — Планка по-
стоянная, т — масса частицы. Низший уровень энер-
гии, т. е. Н. э. (Еа), соответствует п=1; при этом им-
пульс частицы равен:
р — V 2тЕ^ — ± Л/2а
а неопределенность в значении импульса составляет
Ap = hl2a— (— Л/2а) --hja
что соответствует принципу неопределенности Гай-
зенберга: Az-Ap=/i, где Ах—а— неопределенность
в местонахождении (координате) частицы. Если бы
Н. э. такой частицы равнялась нулю, то тогда: Ар—О
и Ах -Ар—О, что противоречило бы принципу неопре-
деленности.
Для поступательного движения частицы, ничем не
ограниченной в пространстве, Н. э. равна нулю.
Полной определенности импульса частицы (наимень-
шее значение равно нулю) соответствует полная нео-
пределенность координаты, в соответствии с прин-
ципом неопределенности. Вращение жестко связанных
частиц также имеет Н. э., равную нулю, т. к. в этом
случае у системы на низшем энергетич. уровне полной
определенности углового момента (равного нулю),
играющего роль импульса, соответствует полная
неопределенность угловых координат, характеризую-
щих вращение. Напр., вращательная энергия двух-
атомной молекулы (если считать атомы жестко связан-
ными) равна:
Е = h2J (J + 1)/8л27
где 7 — момент инерции, .7=0, 1, 2, 3... — враща-
тельное квантовое число (см. Молекулярные спектры).
В низшем состоянии при .7=0: Е=Ео=0.
В отличие от поступательного и вращательного
движений, колебание частиц предполагает нек-рую
локализацию колеблющихся частиц друг относительно
друга. Поэтому в соответствии с принципом неопре-
деленности нулевая энергия колебаний не равна нулю.
В отличие от Н. э. частицы в потенциальном ящике,
она складывается из кинетич. и потенциальной энер-
гий. Напр., колебательная энергия двухатомной мо-
лекулы (если считать колебания гармоническими)
выражается величиной:
E = kca>e (а+'/г)
где со,, — частота колебаний в см~1 (равновесное зна-
чение), с— скорость света, у=0, 1, 2, 3...— колеба-
тельное квантовое число (см. Молекулярные спектры).
Н. э. (7?0) соответствует у=0: 7?о=7гссое/2. Ангармо-
ничность колебаний вносит лишь относительно не-
большую поправку в эту величину. Т. обр., колеба-
ния в молекуле в противоположность вращению и по-
ступательному движению, не исчезают при абс. нуле
темп-ры. Величина энергии, необходимая для пере-
хода молекулы с нулевого колебательного уровня на
следующий, зависит от колебательных частот, к-рые
имеют значения нескольких сотен, а иногда тысяч
см ~ *; энергия такого перехода соответствует обычно
темп-ре порядка нескольких сотен (иногда тысяч)
градусов (°C) и, следовательно, ниже таких темп-р
большинство молекул имеет колебательную энергию,
равную Н. э.
Н. э. может обладать не только отдельная частица
или система связанных частиц, но также и совокуп-
ность нек-рых свободных невзаимодействующих ча-
стиц. В этом случае Н. э. имеет несколько другое,
а именно квантово-статистич. происхождение. Такой
Н. э. обладает т. н. ферми-газ (напр., электронный
газ в металле), т. е. совокупность частиц, подчиняю-
щихся статистике Ферми — Дирака (см. Квантовая
статистика). Эти частицы, имея полуцелый спин и
подчиняясь Паули принципу, могут лишь попарно
(с противоположными спинами в каждой паре) зани-
мать определенное энергетич. состояние. Т. обр.,
на нулевом уровне с энергией 8=0 будут находиться
только две частицы, а остальные будут занимать более
высокие уровни, вплоть до нек-рого максимального
с энергией е0. Такое распределение энергий сохраняет-
ся при абс. нуле темп-ры; при этом средняя энергия,
составляющая по статистике величину Зе0/5, является
Н. э. ферми-газа. Электронный газ в металле сохраня-
ет энергию, равную Н. э., вплоть до темп-ры поряд-
ка 10000°.
Лит.: К о з м а н У., Введение в квантовую химию, пер.
о англ., М., 1960; Г леестонС., Теоретическая химия, пер.
о англ., М., 1950; ГерцбергГ., Атомные спектры и строе-
ние атомов, пер. о англ., М., 1948; егож е, Спектры и строе-
ние двухатомных молекул, пер. с англ., М., 1949; его же,
Колебательные и вращательные спектры многоатомных моле-
кул, пер. с англ., М., 1949. С. Э. Вайсберг.
НУТЧ-ФИЛЬТР — см. Фильтрация.
о
ОБЕПИН (анисовый альдегид, п-метоксибензаль-
дегид) СНзО — С„Н4— СНО, мол. в. 136,14— бес-
цветная жидкость; т. пл. 2,5°; т. кип. 248°/760 мм,
134—135712 мм, 106—10775 мм; d™ 1,1260;
1,5730; в воде нерастворим, растворим в большинстве
органич. растворителей и в жидких, сернистом анги-
дриде и аммиаке. Производные: п-нитрофенилгидра-
зон, т. пл. 160—161°; 2,4-динитрофепилгидразон, т. пл.
250°; n-бромфенилгидразон, т. пл. 150°.
О. обладает всеми свойствами ароматич. альдеги-
дов. Качественно определяют О. электрофорезом на
бумаге, пропитанной бисульфитом натрия, или при
действии резорцина в 80 %-ной серной к-те (желто-
оранжевое окрашивание). Количественное опреде-
ление производят оксимированием. О. находится в
эфирных маслах аниса, акации, укропа и др.; обладает
запахом цветов боярышника; получают при окисле-
нии анетола или метилового эфира п-ксилола:
метилированием n-оксибензойного альдегида, карбо-
нилированием анизола, по Гаттерману, и взаимодей-
ствием n-хлорметиланизола с уротропином, по Сом-
мле. О. применяют как компонент парфюмерных ком-
позиций и в синтетич. целях. Э. Е. Нщрантьев.
ОБЕСФЕНОЛИВАНИЕ СТОЧНЫХ ВОД — см.
Воды сточные.
ОБЕСФТОРЕННЫИ ФОСФАТ — см. Фосфаты
кормовые, Фосфорные удобрения.
ОБМЕН ВЕЩЕСТВ — комплекс химич. процессов,
специфически присущий живым организмам и состав-
ляющий, по определению Ф. Энгельса, характерный
отличительный признак жизни. Суть О. в. состоит в
непрерывном потреблении живым организмом из
внешнего мира определенных веществ, их химич. пре-
вращениях в организме и выделении конечных про-
дуктов в окружающую среду. О. в.— единственный
источник энергии, к-рую организм постоянно расхо-
дует в различной форме (механич. работа мышц,
электрич. работа нервных элементов, осмотич. ра-
бота ультрафильтрации и т. п.). В результате О. в.
потенциальная химич. энергия пищевых веществ ос-
вобождается при их постепенном распаде в организме
и либо непосредственно расходуется им, либо запа-
сается в легко мобилизуемой форме, удобной для ре-
гулируемого расходования. Не менее важным значе-
нием О. в. является образование из пищевых продук-
тов таких веществ, к-рые используются организмом
для непрерывного обновления и построения заново его
структурных элементов посредством биосинтеза. Та-
ким образом, О. в. имеет две стороны — энергетиче-
скую и пластическую, к-рые между собой тесно
связаны.
Нередко в понятие О. в. включают не только не-
обходимые для поддержания жизни процессы прев-
ращения пищевых веществ, но также превращения
посторонних веществ (лекарств, ядов и т. п.), попа-
дающих в организм. Такого рода процессы часто назы-
вают метаболизмом. Термин «метаболизм» час-
то применяется и для обозначения химич. превраще-
ний в организме любых веществ, в том числе пищевых.
Ниже рассмотрены пути обмена основных пищевых
веществ в организмах животных.
Энергетика обмена веществ. Характерной для каж-
дого организма является величина основного
обмена, под которым подразумевают то количе-
ство энергии, которое необходимо при полном мышеч-
ном покое. Средние величины основного обмена за
сутки, рассчитанные в килокалориях на 1 кг живого
веса (ккал/кг сутки) и на 1 мг поверхности тела
(ккал/м2-сутки), составляют, соответственно: лошадь
11,3 и 948, свинья 19,1 и 1078; человек 32,1 и 1042;
собака 51,5 и 1039; мышь 75,1 и 917. Интенсивность
О. в. в зависимости от физиологии, состояния челове-
ка может быть охарактеризована след, величинами
(ккал/час): состояние сна 65; бодрствования 105; мы-
шечная работа — слабая 170, умеренная 290, тяже-
лая 450, очень тяжелая 600. Общая энергетич. потреб-
ность организма складывается из величины основного
обмена и энергетич. трат, зависящих от рода деятель-
ности.
С точки зрения термодинамики организм представ-
ляет собой систему, в к-рой происходит изотермич.
процесс превращения пищевых веществ в конечные
продукты О. в. Энергетич. ценность этого процесса
может быть определена, как обычно, через величину
термодинамич. потенциала:
AF=AH-TAS
где ДЕ — изменение свободной энергии, Д/7 — изме-
нение теплосодержания; Т — темп-ра °К; ДА — изме-
нение энтропии.
Для превращений в организме нек-рых пищевых
веществ ДЕ может быть достаточно точно подсчитано
по табличным данным, исходя из величин энергии об-
разования из элементов этих веществ и конечных
продуктов О. в. Для сложных по химич. строению
пищевых веществ (напр., белков, нек-рых сложных
липидов) такие расчеты затруднительны. Однако в
связи со сравнительно небольшими величинами изме-
нения энтропии в ходе окислительных превращений,
к-рые являются основными в О. в., изменения свобод-
ной энергии мало отличаются от изменений теплосо-
держания начальных и конечных продуктов обмена.
На этом основании для практич. оценки энергетич.
ценности пищевых продуктов широко пользуются ка-
лориметрия. измерениями теплоты их сгорания. При
этом для таких веществ, как углеводы и жиры (липи-
621
ОБМЕН ВЕЩЕСТВ
622
ды), окисляющихся в организме до СО г и НгО, полу-
чаются достаточно точные данные. Для белков (и др.
азотистых соединений) данные калориметрия, изме-
рений отличаются от результатов непосредственных
измерений энергии обмена вследствие того, что в ор-
ганизме основным конечным продуктом превращений
азота белков является мочевина (частично также мо-
чевая к-та и аммиак, см. ниже). Количество энергии,
освобождающейся при окислении основных пищевых
продуктов в организме, характеризуется след, вели-
чинами (ккал/г): белки 4,1, жиры 9,3, углеводы 4,1.
Приведенные значения являются средними, поскольку
они могут изменяться в нек-рой степени в зависимости
от конкретной химич. природы пищевых веществ. При
оценке энергетич. эффективности пищевых веществ
необходимо иметь в виду, помимо прямой калорийной
ценности, и т. наз. специфически динамич. действие,
состоящее в увеличении интенсивности О. в. при по-
ступлении в организм. При всасывании белков это
действие равно 30% потенциальной энергетич. цен-
ности пищевых веществ, при всасывании углеводов
6% и жиров 4% .
Промежуточный обмен веществ. Все поступающие
из внешней среды вещества подвергаются в организме
химич. превращениям, суммарная схема к-рых отно-
сительно несложна. Так, простые липиды и угле-
воды в конечном итоге превращаются в углекислый
газ и воду:
С6Н12О» + 6О2 —> 6СО2 + 6Н2О .
сн,осос„н31
I
С.НОСО(:1-П31 + 72,5О2 —»51СО2 + 49П2О
I
СН2ОСОС15Н31
В результате О. в. белки, распадаясь, образуют
СОг, Н2О и NHs.
Однако в действительности эти превращения, иду-
щие при участии ферментов, весьма сложны. При О.в.
организм не только расщепляет пищевые вещества и
последующие полупродукты их распада (эти процессы
называют диссимиляцией, или к а т а-
болизмом), но непрерывно синтезирует свои со-
ставные части, постоянно обновляя их (эти процессы
носят название ассимиляции, или анабо-
лизма). Для биосинтеза специфичных веществ
каждого организма необходимы полупродукты, обра-
зующиеся в организме в результате химич. модифика-
ции пищевых веществ.
Постепенный, ступенчатый распад пищевых веществ
позволяет с наибольшей эффективностью использовать
освобождающуюся энергию путем ее аккумулирования
в форме универсального источника — макроэргич. со-
единений (см. Макроэргические связи). Механизм внут-
риклеточных и внеклеточных превращений пищевых
веществ в организме носит наименование проме-
жуточного О. в. Еще не все процессы проме-
жуточного О.в. расшифрованы с исчерпывающей пол-
нотой. Однако обмен основных веществ может быть
охарактеризован достаточно точно.
Обмен белков1. Поступающие в организм
белки подвергаются постепенному ферментативному
гидролитич. расщеплению первоначально в желудке
(с помощью пепсина) и далее в кишечнике (с помощью
трипсина, химотрипсина, карбоксипептидазы, амино-
пептидазы, дипептидаз). При последовательном и
частично совместном действии этих ферментов в бел-
ках гидролитически разрываются пептидные связи,
в результате чего вначале образуются крупные по-
липептиды, к-рые постепенно гидролизуются до сво-
бодных аминокислот. Аминокислоты всасываются че-
1 Вследствие того, что белки и аминокислоты являются
главным источником различных азотистых веществ организма,
их метаболизм часто называют азотистым обменом.
рез стенки кишечника и транспортируются кровью
в органы и ткани организма; в результате образуется
подвижной фонд свободных аминокислот, необходи-
мый для синтеза белков организма. Этот фонд характе-
ризуется общим содержанием свободных аминокислот
в крови (35—55 мг на 100 мл плазмы). Содержание
свободных аминокислот в тканях организма в 5—10
раз выше, чем в крови. Свободные аминокислоты
используются в различных внутриклеточных процес-
сах, наиболее важным и существенным из к-рых яв-
ляется биосинтез органоспецифичных белков — струк-
турных белков клеток, ферментов и т. п. (пластич.
функция О.в.). В этом процессе необходимым звеном
является активирование аминокислот, осуществляе-
мое путем ферментативного образования смешанных
ангидридов при взаимодействии аминокислот и адено-
зинтрифосфата (АТФ):
R-СН-СООН + АТФ
I
nh2
R - СН - С О - О - РО - О —рибозо-адении + пирофосфат
I I
nh2 о-
аминоациладепилат
В дальнейшем аминоациладенилаты при участии
рибонуклеиновых к-т (РНК) клетки вовлекаются в
процесс биосинтеза белков, структура к-рых задает-
ся строением т. наз. «информационной» РНК, слу-
жащей переносчиком генетич. информации, заложен-
ной в ДНК хромосом (см. Нуклеиновые кислоты,
Молекулярная биология).
Неиспользованные для биосинтеза белков амино-
кислоты подвергаются в клетке разнообразным химич.
превращениям, общим для многих аминокислот:
а) Окислительное дезаминирова-
н и е:
- 2н +н2О
RCH-COOH----->-R —С-СООН---> R-CO-COOH + NH.
I il
NH2 NH
Этот процесс катализируется оксидазами аминокис-
лот.В результате образуются кетокислоты и аммиак,
включающиеся в дальнейшие превращения (см. ниже).
В организме животных этот процесс имеет ограничен-
ное значение; большую роль он играет в О. в. микро-
организмов.
б) Переаминирование:
ноос-сн.—сн2-сн-соон+R-со-соон —►
I
nh2
—s-HOOC-CH2-CH2-CO-COOH + R-CH-COOH
I
nh2
Перенос аминогруппы от аминокислоты (как правило,
дикарбоновой) к кетокислоте без промежуточного от-
щепления аминогруппы в виде аммиака осуществляет-
ся при участии ферментов трансаминаз (аминофераз),
простетич. группой к-рых является фосфопиридок-
саль. Переаминирование играет важную роль в био-
синтезе аминокислот из кетокислот, образующихся
при обмене углеводов и жиров, являясь, т. обр., зве-
ном, связующим эти виды обмена с обменом белков.
Помимо того, переаминирование играет важную роль
в образовании аммиака из аминокислот. Это осуществ-
ляется посредством переаминирования их с а-оксо-
глутаровой к-той, в результате чего образуется глута-
миновая к-та, к-рая при действии фермента глутамат-
дегидрогеназы окисляется с образованием а-кетоглу-
таровой к-ты и аммиака.
в) Декарбоксилирование:
R-CH-COOH
I —► RCH2NH3 + С Од
nh2
623
ОБМЕН ВЕЩЕСТВ
624
Декарбоксилирование происходит под влиянием фер-
ментов декарбоксилаз (интенсивно у бактерий и в не-
значительной мере у животных). Образующиеся при
этом амины, как правило, обладают биология, актив-
ностью (см. Кадаверин, Путресцин, Гистамин,
Триптамин, Серотонин). В организме животных опи,
благодаря ферментативному окислению, быстро обез-
вреживаются, превращаясь сначала в альдегиды:
- 2Н +Н2О
RCH2NH2--------->-КСН = №1------>-RCH = O + NH3
и далее в СО2 и Н2О. Образующийся аммиак является
физиологически активным веществом;
низмы в ходе эволюции выработали
механизмы его связывания и превра-
щения в конечные продукты, к-рые
выводятся в окружающую среду. У
млекопитающих, рыб и амфибий та-
ким конечным продуктом является
мочевина. Синтез мочевины осущест-
вляется с помощью т. наз. орнити-
нового цикла. Необходимая
энергия черпается из макро- гли"оген™нтетаза
эргич. соединения аденозин-
трифосфорной к-ты (АТФ), об-
разующейся при окислитель-
ном распаде углеводов (см.
ниже). Источником СО2 для
биосинтеза мочевины являете я удф-:
также обмен углеводов и ли-
пидов (см. ниже). Для отдель-
ных аминокислот характерны, помимо опи-
санных общих путей обмена, специфич.
превращения, ведущие к биосинтезу био-
логически активных веществ, витаминов,
коферментов, а также нек-рых конечных
СН2ОН
поэтому орга-
СН2ОН
о
он
н
н
он
Н,РО.
фосфорилаза
—о
+ УТФ
(+„затравиа‘
гликогена)
глюкоза -«--------------
глюкоза-1-фифэт-
Урилилтрансфефаза'
продуктов, выводимых из организма (см.
Аминокислоты, статьи об индивидуальных Начальные стадии обмена углеаодое
аминокислотах, а также Креатинин, Го-
могентизиновая кислота, Кинуренин, Никотиновая
кислота, Мочевая кислота п др.).
Обмен углеводов. Основным углеводом,
используемым в питании человека и животных, яв-
ляется крахмал. Нек-рую роль в питании человека
играют дисахариды (сахароза и в раннем возрасте
лактоза). Эти углеводы гидролитич. расщепляются
в желудочно-кишечном тракте (частично в ротовой
полости) под воздействием ферментов (амилазы, маль-
тазы, сахаразы и лактазы). Образующиеся при этом
моносахариды (гл. обр. глюкоза, фруктоза и галак-
тоза) всасываются в кровеносное русло, разносятся
кровью по органам и тканям, причем в крови здоро-
вого животного постоянно поддерживается определен-
ный уровень содержания сахара; у человека он сос-
тавляет 80—120 мг глюкозы в 100 мл крови.
Галактоза в печени при участии уридинтрифосфата
(УТФ) ферментативно превращается в глюкозо-1-
фосфат. Фруктоза ферментативно фосфорилируется
при участии АТФ с образованием 1,6-дифосфата фрук-
тозы. Образующиеся из галактозы и фруктозы фос-
форилированные моносахариды включаются в обмен-
ные реакции, характерные для глюкозы. Значитель-
ная часть глюкозы из крови поглощается печенью,
где путем последовательных превращений глюкозы в
глюкозо-6-фосфат, глюкозо-1-фосфат и далее в ури-
диндифосфат-глюкозу происходит ферментативное об-
разование запасного полисахарида — гликогена (см.
схему).
При недостатке глюкозы в крови происходит повы-
шенный ферментативный распад гликогена печени с
образованием и поступлением в кровь необходимого
количества глюкозы. При действии фосфорилазы об-
разуется глюкозо-1-фосфат, превращающийся под
влиянием фосфоглюкомутазы в глюкозо-6-фосфат,
к-рый, в свою очередь, при воздействии фосфатазы
образует глюкозу (см. схему).
Во внутриклеточном углеводном обмене централь-
ное место занимают превращения глюкозы, к-рая по-
степенно окисляется, в результате чего образуются
СО2 и Н2О, а освобождающаяся энергия аккумули-
руется в форме АТФ. Окисление глюкозы может про-
исходить как с участием кислорода (аэробно),
так и без его участия (анаэробно). Двоякий меха-
низм имеет физиология, смысл, поскольку реально
могут возникать условия дефицита кислорода, при
к-рых жизнеспособность
клеток поддерживается
фосфоглюноМутаза
СН2ОН
о
н
н
н
н
н
н
глюкоза
он
он
Н2О
фосфатаза
генсо-
киназа
♦АТФ
Н
НО
СН2ОН
о
н
н
он
глюкозо-1-фосфат
ОН
О-РО3Н2
н ОН
глюкозо-6-фосфат
гликолиз и аэробные
окислительные реакции
нек-рое время за счет анаэробного окисления глюкозы.
Анаэробное окисление глюкозы, конечными продук-
тами к-рого являются молочная к-та и Н2О, носит
название гликолиза; ему, как правило, предшествуют
те же реакции, вплоть до стадии образования пирови-
ноградной к-ты (см. схему к ст. Гликолиз), но в этом
случае последняя не превращается в молочную к-ту,
а подвергается окислению по схеме цикла лимонной
к-ты (см. Цикл трикарбоновых кислот.) с участием
кофермента А и липоевой кислоты. Первоначально
пировиноградная к-та с участием липоевой к-ты (I)
превращается в ацетилкофермент А (II)
S ---- S
CH3COCOOJI + CH2CH,CH(CH2),COOH------->
! ' -со2
+ KOASH
—> изс:п2с:и2с:п(с:н.,),с:оон------>-
I
S-COCH.,
—>-CH3COSKoA + HSCH2CH..CH(CH.)4COOH
к-рый конденсируется в ферментативной реакции с
щавелевоуксусной к-той, образуя лимонную к-ту.
Последняя через ряд последовательных реакций дегид-
рирования и декарбоксилирования снова дает щаве-
левоуксусную к-ту, готовую к взаимодействию с но-
вой молекулой ацетилкофермента А.
При сопоставлении энергетич. эффективности ана-
эробного гликолиза и аэробного окисления глюкозы
видно, что последний процесс гораздо выгоднее пер-
вого, т. к. при этом выше не только изменение свобод-
ной энергии (в случае образования СО2 и Н2О ДЕ° =
=688,1 ккал/моль, в случае образования молочной
625
ОБМЕН ВЕЩЕСТВ
626
к-ты AF°= 30 ккал/моль), но и относительная доля по-
лезной энергии при полном окислении глюкозы. При
аэробном окислении молекулы глюкозы образуется 36
молекул АТФ, энергию к-рой можно полезно исполь-
зовать для выполнения работы, в то время как при
гликолизе образуется соответственно лишь 2 молеку-
лы АТФ. Другим путем аэробного окисления глю-
козы является образование 6-фосфоглюконовой к-ты
из глюкозо-6-фосфата и далее ее ступенчатое дегид-
рирование и декарбоксилирование до фосфотриозы,
к-рая далее превращается обычным путем через пиро-
виноградную к-ту.
Обмен жиров (липидов). Основную
массу липидов, употребляемых в пищу, представляют
собой триглицериды высших карбоновых к-т(нейтраль-
ные жиры). Нек-рое значение имеют также фосфоли-
пиды, и стероиды. Расщепление липидов происходит
в основном в кишечнике при участии ферментов ли-
пазы, лецитиназы, коламин-(холин)-фосфатазы, гли-
церофосфатазы; при этом существенную роль играют
эмульгаторы — желчные кислоты. Нейтральные жиры
в кишечнике гидролизуются с образованием ди- и
моноглицеридов и карбоновых к-т:
ch2ocor ch2ocor
I + Н2О 1
CHOCOR----------> RCOOH + C1I-0H
| липаза I
CH2OCOR CH2OCOR
В результате гидролиза лецитина образуются жирные
к-ты, холин и фосфорная к-та. При гидролизе кефа-
лина вместо холина образуется коламин. Эфиры холе-
стерина гидролизуются с образованием свободного
холестерина и карбоновых к-т.
Продукты гидролитич. разложения липидов, а так-
же коллоидные частицы эмульгированного желчными
к-тами жира всасываются из кишечника в кровенос-
ные сосуды (частично прямо, частично через лимфа-
тич. систему). Значительная часть этих продуктов
претерпевает в клетках организма окислительное
разложение. Нек-рое количество их подвергается не-
посредственно в стенке кишечника и др. тканях ре-
синтетич. превращениям с образованием специфич.
для данного организма жиров. Наконец, известное
количество всосавшегося и ресинтезированного жира
откладывается в специальных жировых депо (жи-
ровая клетчатка) про запас.
В результате уравновешенного притока и оттока
липидов в крови создаются их относительно постоян-
ные концентрации, характеризующиеся, напр., для
человека следующими средними величинами (мг %):
Суммарное содержание...........500
Нейтральные жиры...............150
Фосфолипиды ...................200
Холестерин свободный .......... 50
Эфиры холестерина .............100
Глицерин, являющийся продуктом гидролиза липи-
дов, претерпевает внутриклеточно ферментативное
превращение в монофосфорный эфир и далее в фос-
фоглицериновый альдегид, последующее окисление
к-рого идет по схеме гликолиза. Высшие карбоновые
к-ты окисляются по следующей схеме:
ацил-КоА-синтетаза
RCH2CH2COOH +K0ASH + АТФ-___—
RCH2CH2COSKoA +АМФ+пирофосфат
ацил-КоА-дегидрогеназа и
цитохромоксидаза
RCH2CH2COSKoA------------------------->
кротоназа + Н20
—► RCH = CHCOSKoA--------------->-
Р-оксиацил-КоА-дегидрогеназа+ НАД
-> RCHOHCH2COSKoA -— ..................
-НАДН2
ацил-КоА-транс-ацилаза + KoASH
RCOCH2SKoA ~ — Т
RCOSKoA + CH3COSKoA
В результате молекула жирной к-ты уменьшается
на два углеродных атома и образуется ацетилкофер-
мент А, к-рый окисляется по схеме цикла трикарбо-
новых к-т. R—СО—S—КоА претерпевает повторное
превращение до тех пор, пока полностью не превра-
тится в CHaCOSKoA. Освобождающаяся при окис-
лении жирных кислот энергия аккумулируется в
форме АТФ.
Биосинтез жирных к-т происходит гл. обр. в цито-
плазме клеток, причем исходным веществом для него
является ацетил-кофермент А. Последний при учас-
тии биотинсодержащего фермента, СОг, АТФ и Мпг +
образует малонил-кофермент А, к-рый, конденси-
руясь с ацетил-коферментом А, дает а-карбоксиаце-
то-ацетилкофермент А. Далее реакциями дегидриро-
вания и декарбоксилирования происходит образова-
ние бутирилкофермеита А, т. е. наращивание двух-
углеродного фрагмента к ацетилкоферменту А. После-
довательное наращивание углеродной цепи приводит
к образованию жирных к-т необходимого мол. веса.
Биосинтез жиров (триглицеридов) осуществляется
из соответствующих ацилпроизводных кофермента А
и фосфата глицерина при участии ферментов. Обра-
зующийся при окислении жирных кислот, а также
углеводов (см. выше) ацетил-кофермент А (называе-
мый иногда активной формой ацетата) может служить
для биосинтеза разнообразных биологически важных
соединений (стероиды, ацетилхолин, ацетилглюкоз-
амин и т. п.). Через это соединение, а также через пи-
ровиноградную к-ту, являющиеся общими в ходе
метаболизма аминокислот, глюкозы и карбоновых
к-т, осуществляется связь и единство белкового, уг-
леводного и жирового обмена. Именно наличие таких
общих соединений в обмене позволяет организмам осу-
ществлять биосинтезы почти всех необходимых био-
субстратов даже при недостатке каких-либо компо-
нентов в пищевом рационе. Нек-рые компоненты,
к-рые не могут синтезироваться в организме, носят
название незаменимых (см. Незаменимые аминокис-
лоты, Незаменимые жирные кислоты).
Другие виды обмена. Выше были рас-
смотрены пути обмена основных веществ, являющихся
источником энергии и источником структурных ком-
понентов тканей живых организмов. Однако этих
основных веществ далеко не достаточно для возник-
новения и поддержания жизнедеятельности, а также
для воспроизведения жизни (размножения).
Большое значение в О.в. имеет минеральный
обмен — совокупность процессов всасывания в же-
лудочно-кишечном канале, превращений в органах
и тканях и выделения из организма животных неор-
ганич. соединений, основную часть к-рых составляют
хлористые, сернокислые, углекислые и фосфорнокис-
лые соли натрия, калия, кальция и магния. Общее
содержание этих соединений в тканях и органах, за
исключением костной системы и нек-рых опорных
тканей у низших животных, обычно не превышает
1% по отношению к весу органа (только в костях
скелета и в эмали и дентине зубов неорганич. соеди-
нения, гл. обр. фосфорнокислые и углекислые соли
кальция, составляют значительную часть их общей
массы). Особенно мало содержание в организме жи-
вотных т. н. микроэлементов (солей меди, марганца,
молибдена, цинка, кобальта, алюминия, титана, бро-
ма, фтора и т. д.), количество к-рых в большинстве
тканей и органов выражается долями мг%.
Несмотря на то, что минеральный обмен в энерге-
тич. отношении не играет роли в организме живот-
ных и имеет лишь ограниченное значение в пластич.
отношении, его влияние на процессы жизнедеятель-
ности организма животных чрезвычайно велико. При
лишении организма животных таких элементов, как
Na, К, Са, Р, Си, Zn, J, Сои др., у них быстро разви-
€27
ОБМЕННОЕ ВЗАИМОДЕЙСТВИЕ
628
ваются различные специфич. заболевания, и они часто
погибают быстрее, чем при лишении их органич. со-
единений или при полном голодании. Это объясняется
тем, что неорганич. соединения являются основными
регуляторами ряда физико-химич. процессов в орга-
низме (поддержание на определенном, специфич. для
каждого вида животных уровне осмотич. давления
крови, спинномозговой жидкости, лимфы, внутри- и
внеклеточной тканевой жидкости, сохранение кислот-
но-щелочного равновесия в организме и поддержание
постоянства pH крови и тканей, что в значительной
степени определяет проницаемость мембран клеток
для тех или иных органич. веществ, влияет на способ-
ность белков связывать воду и т. д.). Очень большое
значение для почти всех процессов О. в. в организме
имеет функция различных неорганич. катионов и ани-
онов как стимуляторов и ингибиторов различных
ферментативных реакций. Нек-рые катионы (напр.,
Zn2 + , Mg2 + , Fe’ + , Мов+ и др.) входят в состав метал-
ло-ферментов, где выполняют коферментную функцию.
От концентрации ионов кальция и магния в клетках
мышечных волокон в очень сильной степени зависит
способность этих волокон к сокращению и расслабле-
нию. Минеральный обмен в организме регулируется
нервной и гормональной системой, а также рядом вита-
минов. Так, для регулирования содержания натрия и
калия в тканях и жидкостях организма особое зна-
чение имеет гормон коры надпочечников — альдосте-
рон, а для регуляции содержания кальция— гормон
паращитовидных желез и витамин D.
Огромную роль в О. в. играют витамины, нуклеи-
новые кислоты и многочисленные другие компоненты,
часто присутствующие в незначительных количест-
вах, но выполняющие ответственную роль. Их обмен
протекает особым образом и в настоящей статье не
рассматривается. В ней также не рассмотрены про-
цессы О. в. у растений, в частности важнейший про-
цесс фотосинтеза, а также процессы обмена у микро-
организмов, к-рые часто более разнообразны, чем
аналогичные процессы у животных.
Нормальный О. в. нарушается при воздействии на
организм различных факторов, напр. патогенных ми-
кробов, вирусов, отравляющих веществ и др.
Лит.: Збарский Б. И., Иванов И. И., М а р-
д а ш е в С. Р., Биологическая химия, 3 изд., М., 1960; Дик-
сон М., Уэбб Э. С., Ферменты, пер. с англ., М., 1961;
Капланский С. Я., Минеральный обмен, М.— Л., 1938;
Chemical pathways ot metabolism, ed. D. M. Greenberg, v.
1 — 2, N. Y., 1960; U m b г e i t W. W-, Metabolic maps, Ml-
neapolis, [1952); Principles of biochemistry, 2 ed., N. Y., 1959;
West E. S., Todd W. R., Textbook of biochemistry,
3 ed., N. Y., 1961; Mineral metabolism, ed. C. L. Comar, F.
Bronner, v. 1, pt A—B, N. Y.— L., 1960—61; Straub F. B.,
Biochemie, Bdpst, i960. В. А. Яковлев.
ОБМЕННОЕ ВЗАИМОДЕЙСТВИЕ — специфич. тип
взаимодействия частиц, определяемый квантовыми за-
конами движения рассматриваемых объектов; иг-
рает фундаментальную роль в образовании ковалент-
ной химич. связи, в явлениях ферромагнетизма, во
внутриядерных взаимодействиях. Согласно законам
квантовой механики, существует корреляция между
направлениями спинов взаимодействующих частиц и
относительным пространственным расположением час-
тиц. Это обусловливает зависимость энергии системы
от взаимной ориентации спинов. Наличие такой за-
висимости эквивалентно существованию дополнитель-
ного взаимодействия между частицами, к-рое и полу-
чило название О. в. Следует подчеркнуть, что О. в.
не является непосредственным взаимодействием спинов
друг с другом, т. е. не является взаимодействием спи-
новых магнитных моментов, оно гораздо больше по
величине и для электронов имеет чисто электростатич.
происхождение (для нуклонов в ядре определяющим
является поле ядерных сил, природа к-рого в настоя-
щее время недостаточно ясна).
Понятие «О. в.» было введено впервые при расчете
возбужденных состояний атома гелия (В. Гайзенберг,
1926) и основного состояния молекулы водорода
(В. Гейтлер, Ф. Лондон, 1927). Метод расчета Гейт-
лера — Лондона явился основополагающим для всего
дальнейшего построения теории химической связи.
Особенно нагляден в этом отношении пример вычис-
ления энергии взаимодействия двух электронов. В
нулевом приближении кулоновское взаимодействие
электронов не учитывается; тогда можно считать, что
один из электронов находится в квантовом состоя-
нии, характеризуемом волновой функцией Ф1(г1), дру-
гой — в состоянии с волновой функцией Ф2(г2), где
И и г2— радиусы-векторы, характеризующие поло-
жение электронов в пространстве по отношению к на-
чалу координат. Волновая функция системы двух
электронов в этом приближении равна просто произве-
дению Ф1(г1)Ф2(г2). Вследствие неразличимости элек-
тронов такой же энергии будет отвечать волновая
функция Ф1(г2) Фг(т,х), соответствующая обмену элек-
тронов местами, т. е. имеет место т. наз. обменное
вырождение. Шредингера уравнению в нулевом при-
ближении удовлетворяет любая линейная комбина-
ция приведенных функций. Для вычисления энергии
взаимодействия электронов (в первом приближении),
как следует из теории возмущений, пригодны,однако,
лишь следующие две линейные комбинации:
= Йг [Ф1 °2 (+ Ф>°2
= ~ !Ф, (П) Ф2 (г2) -Ф, (гг) Ф2 (г,)J
Согласно Паули принципу, полная волновая функ-
ция, включающая, в отличие от волновой функции’Г,
наряду с пространственными координатами также спи-
новые координаты электронов, должна быть анти-
симметричной относительно перестановок электронов.
Нетрудно установить, что это условие выполняется,
если отвечает противоположно направленным
спинам (т. н. синглетное состояние), а параллель-
но направленным спинам (триплетное состояние).
Энергия взаимодействия электронов в этих состояниях
равна интегралу
I' -Ч^ (г,, rJrfF.rfF,
J » 12
где е — заряд электрона, ri2— расстояние между элек-
тронами (волновые функции считаем действительны-
ми), т. е. эта энергия равна среднему значению c2/<i2.
В результате подстановки сюда функций и Чгц
получаем:
АП = А+А, Вп = К-А
где
* = J (г>)Ф2’ (r^dV.dV,
А = J' Ф. (г,) Ф2 (г2) Ф, (г2) Ф2 (г.) dV,dV2
Интеграл К имеет простой физич. смысл — это энер-
гия кулоновского взаимодействия двух зарядов,
размазанных в пространстве с плотностями еФ2(г) и
еФ2(г). Интеграл А наз. обменным интегралом и харак-
теризует энергию О. в., он не имеет классич. аналога.
Обменный интеграл появляется вследствие того, что
каждый электрон, как это следует из вида волновых
функций фЦ и t|)ff, с равной вероятностью находится
в состояниях Ф1 и Ф2. При этом в случае симметрич-
ной волновой функции ФЦ вероятность найти элек-
троны близко расположенными велика, в случае анти-
симметричной — мала, т. е. появляются силы, имею-
щие квантово-механич. происхождение, к-рые воз-
действуют на электроны так, что изменяют вероят-
ность их нахождения друг около друга. Эти силы и
629
ОБОГАЩЕНИЕ ПОЛЕЗНЫХ ИСКОПАЕМЫХ
630
являются причиной возникновения О. в. Т. обр.,
«обменная энергия» является частью обычной электро-
статич. энергии взаимодействия электронов, обуслов-
ленной квантово-механич. корреляцией между отно-
сительными положениями электронов в пространстве.
Описанный расчет применим к электронам в атоме
(напр., к возбужденным состояниям Не), т. к. в отсут-
ствии взаимодействия между электронами волновые
функции могут быть легко найдены: Фх и Ф2 отвечают
водородоподобному атому. В расчете молекулы Н2
п более сложных молекул, по Гейтлеру и Лондону,
при написании волновых функций нулевого прибли-
жения пренебрегают не только взаимодействием ва-
лентных электронов друг с другом, но и их взаимо-
действием с «чужими» ядрами или атомными остовами
(атомами, лишенными валентных электронов). Эти
взаимодействия далее учитываются в первом прибли-
жении с помощью волновых функций нулевого при-
ближения. Таким образом, обменный интеграл в за-
дачах химич. связи имеет более сложный вид, чем в
задаче о взаимодействии электронов в атоме.
Величина обменного интеграла А определяется сте-
пенью перекрытия волновых функций. Обменный ин-
теграл вносит наибольший вклад в энергию ковалент-
ной химич. связи. Так, для молекулы Н2 на долю А
приходится ок. 85% энергии связи, а на долю К —
ок. 15%. Если волновые функции Ф1 и Ф2 отличны
от нуля в различных областях пространства, то А =0.
Напр., в случае взаимодействия электронов двух раз-
личных атомов О. в. проявляется лишь при непосред-
ственном сближении атомов. Отсюда вытекает, что
обменные силы принадлежат к близкодействующим
силам (известные в класспч. физике электромагнитные
и гравитационные силы являются дальнодействую-
щими). Особенностью обменных сил является прису-
щее им свойство насыщения. Напр., два электрона
с антипараллельными спинами в двух атомах Н соз-
дают их притяжение, но в результате О. в. с третьим
электроном отталкивают третий атом Н.
В зависимости от знака обменного интеграла низ-
шее энергетич. состояние может быть как синглетным,
2?t)(A <0), так и триплетным, ЕЦ(А>0). Для боль-
шинства молекул обменный интеграл отрицателен
(при расстояниях между атомами, отвечающих длинам
связей), поэтому их низшее энергетич. состояние яв-
ляется синглетным; в атомах обменный интеграл по-
ложителен, поэтому, напр., Не в основном состоянии
триплетен (различие в знаке А связано с различием
природы этого интеграла в случае молекул и атомов,
отмеченным выше). О. в. являются причиной возник-
новения ферромагнетизма (см. Магнитные свойства).
Состояние с параллельными спинами может быть
термодинамически устойчивее состояния с беспоря-
дочно ориентированными спинами лишь в случае,
если оно обладает меньшей энергией. Для этого не-
обходимо, чтобы обменный интеграл для данного
вещества был положителен. Лишь вещества, удовлет-
воряющие этому условию, могут быть ферромагнети-
ками. Характерная для ферромагнетиков точка Кю-
ри — темп-ра, выше к-рой у вещества исчезают фер-
ромагнитные свойства, может быть грубо охарактери-
зована как темп-ра TQ, при к-рой энергия, характе-
ризующая тепловое движение, кТ0, становится равной
выигрышу энергии при параллельной ориентации
спинов.
Лит.: Левич В. Г., Вдовин Ю. А., Мямлин
В. А., Курс теоретической физики, т. 2, М., 1962, § 67; В о н-
совский С. В., Шур Я. С., Ферромагнетизм, М., 1948;
Блохинцев Д. И., Основы квантовой механики, 3 изд.,
М., 1961, § 123; Бом Д., Квантовая теория, пер. с англ.,
М., 1961. II. Г. Каплан.
ОБОГАЩЕНИЕ ПОЛЕЗНЫХ ИСКОПАЕМЫХ —
совокупность физич. и физико-химич. методов обработ-
ки минерального сырья для разделения его на обога-
щенные продукты (концентраты) и отходы
(хвосты). Простейшие виды обогащения, такие,
напр., как ручная промывка в ковшах и на наклонных
столах, измельчение руды в толчеях, ручная разбор-
ка кусков породы, были известны давно. В конце
19 в. открыто магнитное обогащение железных и
нек-рых других руд. Флотацию стали применять с
начала 20 в.
В соответствии с требованиями последующего
технологии, передела концентрат должен иметь доста-
точно высокое (кондиционное) содержание
ценного компонента и низкое — вредных примесей.
Обычно добываемые руды, а часто и уголь не м< гут
быть непосредственно использованы в пром-сти. Напр.,
содержание свинца в руде достигает 1,5—4,0%, в то
время как для металлургич. передела оно должно
составлять 30—70%. Содержание редких металлов в
рудах часто составляет десятые и сотые доли про-
цента, тогда как для металлургич. переработки оно
должно быть, напр. в случае молибдена, 48—50%.
Необходима также высокая степень удаления приме-
сей, как, напр., фосфора и мышьяка — в случаях воль-
фрамового и молибденового концентратов. Обогащение
имеет большое значение для переработки горнохимич.
сырья (фосфаты, сера, пирит, минеральные соли и др.).
Весьма большое экономии, и технич. значение имеет по-
лучение богатых концентратов; напр., железный кон-
центрат, содержащий 68—71% Fe, дает возможность
значительно интенсифицировать доменный процесс и
организовать переход на внедоменное произ-во метал-
ла высокой чистоты. В нек-рых случаях сразу после
обогащения получают готовую продукцию (асбест,
графит, энергетич. уголь, алмазы и др.). При обога-
щении руд иногда в результате первичного процесса
обогащения неизбежно получение промежуточных про-
дуктов вследствие тонкого взаимного прорастания
минералов. Промежуточные продукты можно допол-
нительно перерабатывать на обогатительной фабрике
или, если это более выгодно, отправлять на металлур-
гич. или химич. передел. Отходы также подвергают
обогащению с целью доизвлечения из них ценных
компонентов.
Наивысшее содержание ценного компонента (в
расчете на данный элемент), достигаемое в концентра-
те, зависит от того, в виде какого химич. соединения
этот компонент входит в состав обогащаемого сырья;
напр., медные концентраты можно получить более
богатыми медью, если они содержат халькозин Cu2S
(79,7% Си), чем в случае халькопирита CuFeS2 (34%
Си). При обогащении руды физико-химич. и физич.
методами не происходит изменения состава разделяе-
мых зерен. Поэтому важной задачей является рас-
крытие минеральных зерен измель-
чением руды до крупности, допускающей по возмож-
ности полное механич. разделение ее на отдельные
минералы. В ряде случаев обогащение дополняет
извлечение или разделение компонентов на одной из
стадий переработки химич. или металлургич. метода-
ми (напр., разделение флотацией сплава сульфидов
меди и никеля, выплавляемого при переработке мед-
но-никелевых руд). Получение концентратов дости-
гается также в ряде случаев комбинированием обога-
щения с химич. переработкой (см., напр., Гидроме-
таллургия).
Методы обогащения используют также и в различ-
ных химико-технологич. процессах, как, напр., для
разделения смеси кристаллов солей в их насыщенном
р-ре; обогащение (флотация) может быть применено
для извлечения анионитов или катионитов после сорб-
ции на них веществ из раствора; для извлечения и
разделения ионов в р-рах низких концентраций (ме-
нее 10“’%) перспективен новый метод ионной флота-
ции. Методы обогащения применяют также в пищевой
631
ОБОГАЩЕНИЕ ПОЛЕЗНЫХ ИСКОПАЕМЫХ
632
пром-сти, в технич. микробиологии (напр., флотация
для выделения микроорганизмов) и т. д.
Методы О. п. и. основаны на использовании разли-
чий в физич. и физико-химич. свойствах разделяемых
минералов или других компонентов.
Физические методы обогащения основаны на раз-
делении компонентов по таким признакам, как цвет,
блеск, плотность, магнитная проницаемость, диэлек-
трические, полупроводниковые и др. свойства. Ниже
перечислены главные из этих методов.
Рудоразборка (сортировка) применяется
для отделения кусков руды или угля от породы бла-
годаря их различному цвету и блеску. В простейшей
форме производится ручным отбором кусков на ленте
конвейера. Применение такой сортировки целесооб-
разно для кусков размером не ниже 50 мм, редко —
до 25 мм. Возможно разделение кусков и по форме.
Для радиоактивных руд применяется автоматизиро-
ванная механич. рудоразборка, основанная на дей-
ствии приборов, реагирующих на излучения.
Гравитационное обогащение ис-
пользует воздействие силы тяжести или центробеж-
ной силы на частицы различного веса. Вес частиц
может отличаться как вследствие различий в их раз-
мерах, так и вследствие отличий в плотности минера-
лов. При гравитационном обогащении руды, измель-
ченной до частиц, не очень сильно отличающихся
размером, основным влияющим фактором служит раз-
личие в плотностях частиц разных минералов. Так,
напр. кварцевая порода имеет уд. вес 2,65, а многие
самородные металлы (в форме природных сплавов)
от 17 до 19. Также значительна разница уд. весов
кварца (или силикатов) и минералов олова, вольфрама
и др. Разделение минералов обычно производят в
воде, иногда в тяжелой жидкости (напр., в тетра-
бромэтане) или в тяжелой суспензии, реже — в воз-
душной среде (большой уд. вес жидкости или сус-
пензии особенно способствует разделению в тех слу-
чаях, когда уд. вес жидкости или суспензии находится
между уд. весами разделяемых минералов или мине-
рала и пустой породы). Один из простейших видов
гравитационного обогащения — отсадка в вос-
ходящих и нисходящих струях воды на решете, на
к-ром помещен слой минеральных частиц или кусков
металла («п остель»). Вибрации создаются гид-
равлически за счет пульсаций воды (или за счет дви-
жения решета, поршня или диафрагмы). Легкая
фракция разделяемых минералов сносится потоком
воды, тяжелая собирается (в случае мелких час-
тиц) в камере машины, проходя под решетом, или (в
случае крупного материала) непосредственно на ре-
шете (создается естественная постель из тяжелого
обогащаемого минерала). Отсадка применяется при
обогащении руд и россыпей песков редких и благо-
родных металлов, угля, алмазсодержащих пород
и др. К тонкой фракции (менее 0,3 мм в случае угля
и менее 100—70 .ик в случае руд) отсадка неприменима.
Более простым методом является обогащение в
потоке воды, в желобах (уголь) или на наклонных
столах-шлюзах (благородные металлы, руды редких
металлов, олова), покрытых трафаретом, рифленой
резиной или ворсистой тканью. Для обогащения мел-
кого (6—5 мм и мельче) материала более эффективно
обогащение в слое воды на сотрясающихся столах.
Поверхность столов покрывается линолеумом, пласти-
катом и др. материалом и набивается параллельными
деревянными планками; применяется и рифленый по-
кров. Обогащение на сотрясающихся столах приме-
няется к вольфрамовым, оловянным, иногда к золо-
тым, редкоземельным и нек-рым др. рудам.
Новым направлением гравитационного обогащения
является применение тяжелых суспензий: магнети-
товой — для угля, ферросилициевой — для полиме-
таллических, железных, алмазных, урановых и др.
руд. Иногда в качестве утяжелителей суспензии при-
меняют и другие вещества, напр. такие суспензии,
как глинисто-баритовая (для угля), арсенопиритовая
(для оловянной руды), галенитовая (для свинцово-
цинковой сульфидной) ит. п. Крупную фракцию руды
обогащают в почти неподвижной среде, иногда пере-
мешиваемой перегребающими (транспортирующими)
гребками. Мелкую фракцию обогащают в суспензии,
быстро вращающейся по спирали в сильно вытянутых
по оси конусах, в которые пульпа под давлением
вводится в тангенциальном направлении (гидро-
циклоны).
В последнее время достигнуты хорошие результаты
обогащения в гомогенной среде — в тяжелой жид-
кости, напр. в тетрабромэтане, что применимо для руд
редких металлов, вольфрамовых и оловянных. Обо-
гащение в тяжелой жидкости в циклонном аппарате
позволяет разделить весьма мелкие частицы (до 70 мк).
Иногда обогащение в тяжелой среде крупных классов
руды производят в сотрясающемся желобе. Это по-
зволяет компактно и достаточно эффективно обога-
щать железные, оловянные и другие руды. Значитель-
но реже гравитационное обогащение ведут в воздуш-
ной среде, что может иметь значение для безводных
или весьма холодных районов, а также при нежела-
тельности увлажнения продукта обогащения. Так,
воздушное обогащение (пневматич. столы и отсадоч-
ные машины) применяют для асбеста, иногда — для
энергетич. угля. Воздушное обогащение применяют
также в электрохимия, металлургии нек-рых туго-
плавких металлов (тантал и др.), выделяемых на
катоде в форме металлич. порошка, смешанного с рас-
плавленными солями электролита. После остывания
и измельчения катодного осадка соли электролита
отделяются от более тяжелых частиц металла воздуш-
ной струей.
Магнитное обогащение основано на
различии воздействия магнитного поля на частицы
минералов неодинаковой магнитной восприимчивос-
ти. Минералы подразделяются на магнитные (магнетит,
титаномагнетит и др.), обогащение руд к-рых произ-
водится в слабом магнитном поле, и немагнитные (кас-
ситерит, кальцит, кварц и др.), вообще неизвлекае-
мые в магнитном поле. Частицы с большой магнитной
восприимчивостью сильнее притягиваются к полюсам
магнитного сепаратора, вследствие чего отделяются
от других. Магнитным методом обогащают или сухую
измельченную руду или ее же в смеси с водой (п у л ь-
п а). Мокрое магнитное обогащение позволяет избе-
жать распыливания, для тонких классов руды иногда
оказываясь эффективнее сухого.
Нек-рые новые фабрики начинают применять сухое
магнитное обогащение, что требует повышенного вни-
мания к технике безопасности. Магнитному обогаще-
нию железных руд иногда предшествует предвари-
тельный магнетизирующий обжиг слабомагнитных
минералов в восстановительной газовой среде с при-
менением высокопроизводительного метода — в к и-
пящем слое; при этом гематит Ре20з, сидерит
ЕеСОз и бурый железняк Ре20з -пН2О переводятся
в более магнитную закись-окись FesOj.
Электрическое обогащение осно-
вано на разделении частиц, заряжающихся различно
(по величине и знаку заряда) при действии электрич.
поля, создаваемого электродами с высокой разностью
потенциалов. Кроме электрич. сепараторов, можно
применить т. наз. коронные, к-рые удобны также для
обеспыливания при сухом обогащении (напр., желез-
ных руд), а также трибоадгезионные, основанные на
неодинаковой прилипаемости частиц, различно заря-
жающихся при контакте с поверхностью металлич.
барабана. Электрич. обогащение применяют для
633
ОБОГАЩЕНИЕ ПОЛЕЗНЫХ ИСКОПАЕМЫХ
634
доводки разделения и для разделения коллективных
концентратов на мономинеральные (напр., титано-
циркониевых, оловянно-вольфрамовых и др. концент-
ратов), а также в нек-рых случаях для обогащения
руд (напр., тонкой фракции хризотил-асбестовых) или
в сочетании с другими процессами (напр., магнит-
ным — для обогащения железных руд и отходов раз-
личных произ-в).
К новым методам физич. обогащения относится
радиометрическая сортировка, ос-
нованная на воздействии естественного или искусст-
венно созданного радиоактивного излучения минера-
лов на устройство (датчик), воспринимающее сигнал
излучения и передающее его через усилитель испол-
нительному механизму, к-рый и отделяет радиоактив-
ную часть руды (куски или порции) от нерадиоактив-
ной. В зависимости от естественной радиоактивности
руды делятся на контрастные и неконтрастные. На
основе естественной радиоактивности таким образом
выделяются куски руды, содержащие уран. Искусст-
венная радиоактивность может быть создана у-облу-
чением материала; напр., при облучении бериллиевой
руды вследствие ядерной реакции (у, п) возникает
поток нейтронов, воздействие к-рого на датчик и по-
зволяет выделить куски руды, содержащие бериллий.
Радиосортировку применяют для кусков не ниже
50—25 мм. Иногда сортируют не куски, а отдельные
порции руды.
Физико-химические методы обогащения основаны
на различии физико-химич. свойств разделяемых ми-
нералов.
Флотационное обогащение (фло-
тация) — наиболее распространенный метод обо-
гащения. В случае пенной флотации использует-
ся способность частиц избирательно прилипать к
пузырькам воздуха, продуваемого через суспензию
измельченной руды в воде (пульпу). Гидрофобизиро-
ванные действием флотореагентов частицы минералов
(несмачиваемые или плохо смачиваемые водой) при-
липают к поднимающимся воздушным пузырькам и
выносятся с ними на поверхность, образуя слой мине-
рализованной пены. Частицы других минералов с гид-
рофильной поверхностью к пузырькам воздуха не
прилипают, остаются взвешенными в пульпе и выносят-
ся ее потоком, проходящим последовательно через
камеры флотационной машины.
Регулирование флотации производится подбором
и дозированием реагентов, к-рые делятся на 3 класса:
собиратели (коллекторы), избирательно гидрофоби-
зирующие частицы минералов, извлекаемые в пену;
пенообразователи (вспениватели), создающие пену;
регуляторы (активаторы, депрессоры). Большое зна-
чение для проведения флотации имеет водородный
показатель среды (pH), регулируемый в случае
pH > 7 известью, едким натром, содой, а в случае
рН<7 — минеральными кислотами, в частности сер-
ной. Существенное значение в процессах обогаще-
ния (флотация, сгущение, фильтрация) имеют пеп-
тизация, флоккуляция и коагуляция частиц пуль-
пы путем введения неорганич. и органич. соединений
(силикат и карбонат натрия, гидроокись кальция и
др.). Флотация — наиболее распространенный метод
разделения тонко измельченных компонентов различ-
ного состава. Ограничение области ее применения дру-
гими, ранее перечисленными, методами определяется
экономии, соображениями.
Флотация особенно необходима для разделения
тонко измельченных компонентов размером менее
0,15—0,3 мм, иногда 70 мк и даже 40 мк, т. к.
другие методы почти неприменимы для разделения час-
тиц порядка менее 40 л и. Иногда применяют комбини-
рованные методы О. п. и. Так, для разделения частиц
(более 0,2 мм), недоступных для пенной флотации, при-
меняют флотогравитацию, также основанную
на избирательном прилипании к частицам пузырьков
воздуха; при этом образующиеся легкие агрегаты от-
деляются от частиц, не прилипших к пузырькам, на
рифленой поверхности сотрясающегося стола и легко
сносятся потоком воды. Такого рода флотогравитация
(в частном случае — флотация на столе)
применяется в конечной стадии обогащения для очист-
ки концентратов руд олова, вольфрама и титана от
примесей. Флотация применяется для обогащения руд
цветных и благородных металлов, сплава сульфидов
никеля и меди в никелевом производстве (с сульф-
гидрильными собирателями), руд редких и черных ме-
таллов, неметаллич. полезных ископаемых (преиму-
щественно с оксигидрильными реагентами, особенно
с карбоновыми к-тами, иногда со спиртами Св—С8, с
аминами); для обогащения угольных шламов. Кроме
того, возможно применение флотации для разделения
смеси солей в их насыщенном р-ре, для обогащения
агрономических (апатиты, фосфориты) и серных руд,
для переработки отходов ряда производств, выделе-
ния нек-рых микроорганизмов и т. д. (см. Флотация).
Обогащение на липких (жировых)
поверхностях также основано на избирательном
прилипании к ним частиц пульпы, напр. алмазов.
Особым видом физико-химич. обогащения, основан-
ного на избирательном смачивании ртутью, является
амальгамация, применяемая преим. для из-
влечения благородных металлов (см. Амальгамы).
Подготовка сырья для обогащения. Наряду с ос-
новными операциями сепарирования и концентрации
при обогащении большое значение имеют подготови-
тельные и вспомогательные операции. При подготовке
руды к обогащению производится дробление крупных
кусков в щековых, гирационных, конусных и валковых
дробилках с доведением размера максимального кус-
ка до 18—12 мм, а иногда — до 4—2 мм, а затем из-
мельчение в мельницах (шаровых, стержневых, а
также самоизмельчеиие), при к-ром крупность мак-
симальных частиц доводится до 0,07 мм и даже
0,04 мм, но иногда ограничивается 1,0 мм. Дробление
и измельчение при подготовке к сепарированию конт-
ролируется для более крупного материала грохо-
чением и для более мелкого — классифи-
кацией, чтобы «не дробить лишнего» и преследовать
лишь задачу раскрытия структурных компонентов
(минералов), подлежащих разделению (подробнее см.
Измельчение).
Путем измельчения получают необходимое раскры-
тие зерен компонентов, для чего необходимо разбить
взаимно сросшиеся зерна. Для получения про-
дукта 0,15—0,3 мм обычно применяют измельчение в
замкнутом цикле мельница — классификатор. Руда,
выходящая после измельчения из шаровой мельницы,
поступает в классификатор, представляющий аппарат,
из к-рого вода выносит частицы достаточно измельчен-
ной руды. Более крупные частицы оседают на дно клас-
сификатора и возвращаются обратно для доизмельче-
ния в мельницу.
Вспомогательные операции при обогащении сво-
дятся в основном к обезвоживанию, к-рое
производят отстаиванием в сгустителях (до40—50%
влаги), на фильтрах (до 10—15%. влаги) и, наконец,
путем сушки (до 1—5% влаги). Скорость отстаивания
при сгущении и фильтруемость могут быть весьма
интенсифицированы введением реагентов-флокулян-
тов (полиакриламид и ряд др.) как синтетических, так
иногда и природных. Современные обогатительные фаб-
рики имеют суточную производительность, достигаю-
щую 30—50 тыс. т руды в сутки и 600—800 т угля
в час. Предприятия используют системы дистанцион-
ного управления, автоматизации и всестороннего
контроля.
635
ОБРАБОТКА РЕЗУЛЬТАТОВ АНАЛИЗА
636
Главными методами контроля являются: грануло-
метрический (ситовой и седиментационный) контроль
крупности; определение плотности пульпы (отноше-
ние веса твердой части к жидкой в суспензии); весовой
учет количества обогащаемого вещества в данной вет-
ви потока; непрерывное автоматич. или порционное
определение состава исходного вещества и продуктов
обогащения; контроль за подачей, распределением и
остаточной концентрацией реагентов. С целью конт-
роля в настоящее время широко вводится пьезомет-
рич. контроль плотности пульпы (иногда — радио-
метрический), аналитический, инструментальный и
радиометрический (активационный, ядерные реак-
ции) контроль состава; определение ряда физич. и
физико-химич. характеристик рудного вещества и
пульпы на разных стадиях обогащения (сгущаемость,
фильтруемость, сорбционная способность, коллоидно-
химич. особенности и др.).
Лит.: Ясюкевич С. М., Обогащение руд, М., 1953;
Плаксин И. Н. [и др.], Технологическое оборудование
обогатительных фабрик, М., 1955; Атлас технологического
оборудования обогатительных фабрик, М., 1959; Г л е м б о ц-
кий В. А., Классен В. И., Плаксин И. Н.,
Флотация, М., 1961; Классен В. И., Мокроусов
В. А., Введение в теорию флотации, 2 изд., М., 1959; Дер-
кач В. Г., Колычев П. А., Специальные методы обо-
гащения полезных ископаемых, М., 1956; Олофинский
Н. Ф., Электрические методы обогащения, 2 изд., М., 1962;
Полькин С. И., Флотация руд редких металлов и олова,
М., 1960; Горное дело. Энциклопедический справочник, т. 11,
Обогащение и брикетирование углей, М., 1960; Митро-
фанов С. И., Исследование полезных ископаемых на обо-
гатимость, 3 изд., М., 1962; Плаксин И. Н. (и др.],
Применение радиоактивных изотопов для исследования про-
цессов флотации, М., 1963; Справочник по обогащению полез-
ных ископаемых, пер. с англ., т. 1—4, М., 1949—52; [Обога-
щение полезных ископаемых], М., 1960; Обогащение руд и
углей. Сб. статей, М., 1963; 5 Международный конгресс по
обогащению полезных ископаемых, Лондон, 1960. [Доклады],
пер. с англ., М., 1962. Л. Д. Плаксина, И. Н. Плаксин.
ОБРАБОТКА РЕЗУЛЬТАТОВ АНАЛИЗА (гра-
фическая) — подсчет результатов анализа, при
к-рых для установления значения неизвестной вели-
чины пользуются заранее составленным градуировоч-
ным (калибровочным) графиком (а не вычислением).
Градуировочный график может иметь любой вид, но
наиболее точным является прямолинейный, когда за-
висимость между величинами х и у линейна. При
аналитич. определениях часто имеют дело с построе-
нием прямолинейных градуировочных графиков,
связывающих измеряемую величину у (напр., силу
диффузионного тока Id при полярография, определе-
ниях или оптич. плотность D при фотоколориметрич.
и спектрофотометрия, определениях) с искомым содер-
жанием (концентрацией) определяемого вещества х(С).
Прямолинейный график описывается ур-ннем:
У = а Ьх
Для вычисления параметров а и Ь, оценки их точно-
сти, интервального значения и для вычисления с по-
мощью этого уравнения неизвестных концентраций
в исследуемых объектах применяют приводимые ниже
формулы.
1. Вычисление параметров а и Ь.
Чтобы найти значения параметров а и Ь в уравнении
прямой У = а+6г, анализируют серию эталонов. По-
лученные данные могут быть представлены системой
ур-ний:
>/, = а + Ьхг
У г = (I + Ъх2
.........I (1)
yj^a + bx; I
Уп = а + Ъхп
где п — число опытов; г,— известная концентрация
определяемого вещества в г-том эталоне; у; — резуль-
тат прямых измерений, связанных с анализом i-того
эталона; а и Ь — неизвестные величины.
При построении градуировочных графиков обычно
считают, что величины xj являются точными резуль-
татами или что ошибки в измерении Х[ значительно
меньше, чем в измерении у;. Так как значения yt
отягчены ошибками измерений, то, строго говоря,
ни одна система значений а и ft не может удовлетворять
всем п уравнениям и задача сводится к отысканию
таких значений параметров a nb, к-рые бы наилучшим
образом удовлетворяли всем уравнениям. Они вы-
числяются по формулам, полученным по методу
наименьших квадратов:
п п п п
2 xi 2 xi 2хт
'-Г1- (2)
(3)
Когда значение а мало отличается от нуля, проверяют
значимость этой величины. Для этого вычисляют зна-
чение 1Д= |а|Аа (где Sa—дисперсия параметра а) и
находят его вероятность Р(|/[>:1а) при к = п — 2
по таблице распределения Стьюдента либо по формуле
Р=([6>и = [1—Sk(ta}]-2. Если окажется, что по-
явление величины ta за счет одних случайных погреш-
ностей маловероятно (Р<0,05), то считают,что метод
включает систематическую ошибку а. Если же Р>
>0,05, т. е. появление величины а за счет одних
случайных погрешностей окажется вероятным, то ме-
тод не включает систематич. ошибку и величину а в
уравнении У = а-}-Ьх можно приравнять нулю и за-
ново рассчитать угловой коэфф, прямой, проходящей
через начало координат:
п
Ь = ~~
1=1
2. Вычисление дисперсии № и № па-
, а Ь
раметров а и ft.
Установление параметров уравнения а и ft по фор-
мулам (2) и (3) можно рассматривать как результаты
косвенных измерений. Поэтому дисперсии А® и А&
вычисляются по формулам:
4-(£)’4 + (й)М+-.. + (£)Ч
+ 161
Полагая, что А’= А“= ... = А* = А’, после пре-
образования получают:
Дисперсию ,$ характеризующую рассеяние значений
В37
ОБРАБОТКА РЕЗУЛЬТАТОВ АНАЛИЗА
638-
у относительно прямой, вычисляют по формуле:
п
у (ш-У;)2
гДе Vi— результат прямых измерений для i-того эта-
лона, Yj— результат для итого эталона, полученный
расчетным путем по ур-нию У,-=а+ Ьх,, по известному
х; и значениям а и 6, найденным по вышеприведенным
формулам.
Если метод не включает систематич. ошибку а и
прямая проходит через начало координат, то диспер-
сию параметра Ь вычисляют по формуле:
п
2^’ где "-1"- (Яа)
3. Точность значений параметров а и Ь:
Sa , , Sh
а = ta, к-^, a b = la,
V п У и
где ia,t — коэфф, нормированных отклонений (к-рый
находят из табл. 1 статьи Ошибки аналитического
определения при А=п—2 и надежности а. Если
а=0, то при оценке точности значения параметра b
значения ta,k находят при h:--n—1.
4. Интервальные значения параметров
а и 6:
(10); (“)
Число значащих цифр, сохраняемое в конечном ре-
зультате при вычислении параметров а и 6, опреде-
,,
ляется точностью их значении: t , • —zz и , -77=
u, К v п u ’ к _» п
(см. пример па прямые измерения в ст. Ошибки анали-
тического определения).
Пример. Рассчитать уравнение линейного калибровочного
графика для полярографии, определения кадмия на фоне
аммонийно-аммиачного р-ра. В табл. 1 приведены экспери-
ментальные данные.
9093,75
1 5-9093,75 — (262.5)2
Sa = 0,745-
= 0,274
Sf, 0,745-j/^ 1 5-9093,75 —262,52 °-011
; ~L = 2,478±2,16-CXiZA = 2,5 + 0.16
Н — 13 у п 3,6
a+ t
~ 0 = 0,95,
;а=о,,5;й=1,Й-- = 2Л42±2’,6-^Т-=2’742±0’0°7
Ур-ние линейного калибровочного графика имеет вид:
У = 2,5 + 2,742х (12)
или
C = (h — 2,5)/2,742 мкг/мл (12')
5. Вычисление концентрации вещества.
После того как рассчитаны параметры а и 6 и
их интервальные значения, для установления неиз-
вестной концентрации вещества (она должна быть
заключена в интервале концентраций, для к-рых со-
ставлен график) производят несколько раз измерение
величины у (у,, у2,..., уп), по ур-нию х = (У—а)/6
вычисляют значения xi, х2,..., хп, а затем, как в слу-
чае прямых измерений (см. ст. Ошибки аналитического-
определения), находят среднее значение х, дисперсию
№, точность измерений и относительную ошибку
измерений^- - 100%.
Пример. Рассчитать среднее и интервальные значения
концентрации кадмия (С^д) и относительную ошибку с надеж-
ностью 0,95, если при повторных полярография, измерениях
растворов, содержащих кадмий, сила диффузионного тока
(в условных единицах) равнялась h, = 140,0; h2 — 138,5;
h3 = 139,5. Концентрации кадмия (в мкг/мл), вычисленные
для hi, h2, h3 по ур-нию (12'), равны: С1 = 50,15; С, = 49,60;
С3 = 49,96. Окончательные данные, рассчитанные по" форму-
лам для прямых измерений (ст. Ошибки аналитического опре-
деления), приведены в табл. 2.
Таблица 2
Число оп- ределе- ний, п Среднее ариф- метич. найден- ных значений Ccd’ мг1мл Диспер- сия, S2-10* Точ- ность, ± Еа = 0,Я5 Относитель- ная ошибка,% Бо , '15 Ccd
3 49,9 797 0,52 1,0
Таблица 1. Расчет параметров линейного калибровочного графи для определения кадмия ка Если для расчетов пользуются не уравнением, а построенным калибровоч- ным графиком, то следует принимать во
Ко опыта Введено Cd2 + , мкг 1мл (ч) Сила диф- фузионно- го тока измерен- ная (у,) х~ 1 Xi У! Сила диф- фузионно- го тока расчетная (У;) (Ю-У/)Х хю внимание: 1) Графики, к-рые служат измеритель- (У1—¥г)‘Х ным целям, необходимо вычерчивать с ХЮ2 большой точностью и в достаточно боль- шом масштабе (во многих случаях при- мерно 20 см х 25 см или 25 см х 30 см).
1 2 3 4 5 6 7 8 9 10 И 12 13 14 15 2,5 2,5 2,5 5 5 5 10 10 10 20 20 20 50 50 50 9.0 10,0 9,0 16,0 15,5 15,0 30,0 29,5 31,0 57,5 58,0 58,5 140,0 138,0 139,5 6,25 6,25 6,25 25 25 25 100 too 100 400 400 400 2500 2500 2500 22,5 25,0 22,50 80,0 77,5 75,0 300,0 295,0 310,0 1150,0 1(60,0 1170,0 7000,0 6925,0 6975.0 9.26 3 9.263 9,26 3 16,188 16,188 16,188 29,898 29,898 29,898 57,318 57,318 57,318 139,578 139,578 139,578 — 2,63 + 7.37 — 2.63 — 1,88 —6.88 -11,88 + 1,02 —3,98 +11,02 + 1,82 + 6,82 +11,82 + 4,22 —10,78 -0,78 2) Значение аргумента (независимой 6 Q2 переменной х) откладывают, как правило, 53 ’ 00 по оси абсцисс, а функции (у) — по оси 6’92 ординат. 3*5 3 3) Если данные разбиты на небольшие 47 * 30 группы, то примерно половина точек каж- 141*40 дой группы должна лежать по однусто- 1*0 4 рону кривой, а половина— по другую 15*80 сторону. 121*40 Если необходимо, чтобы точность 3* 31 отсчетов по графику отвечала точности 46 *51 измерений, то наименьшее расстояние, 139 *40 к-рое можно отсчитать на графике, дол- 17 ’ 80 жно соответствовать величине ошибки из- 116*40 мерений. Так, напр., если ошибка отсче- 0'61 тОв на графике достигает ±0,5 мм, то абсолютную ошибку измерений и следует
Сум- ма 262,5 757,0 9093,75 25587,5 — — 722 2х принять равной этой величине (в мас- Х1б~2 штабе). Однако рассмотренное требование далеко не во всех случаях можно выпол-
_ 9093,5-757,0-26 2,5.25587.5 а 1 5-9093,75 —(262,5)2 ~ ‘ 1 5-25587,5 -262,5-757,0, „ 15-9093,75 -(268.5)2 -“'7 него отступать обычно в сторону умень- шения масштаба. Следовательно, отсчеты по графику могут быть менее точными, чем измерения, по к-рым он составлен. Лит.: Комар ь Н. П., Ж. аналит. химии, 1952, 7. вып. 6, 325; Налимов В. В., Применение математиче- ской статистики при анализе вещества, М., 1960; Методы ана- лиза веществ особой чистоты и монокристаллов, вып. 1, Харь- ков, 1962; Романовский В. И., Применение математи- ческой статистики в опытном деле, М.— Л., 1947; Брод-
639
ОБРАТИМЫЕ РЕАКЦИИ —ОГНЕОПАСНЫЕ ВЕЩЕСТВА
640
с к и й А. Д., Каи В. Л., Краткий справочник по мате-
матической обработке результатов измерений, М., 1960,
с. 147—48; Яковлев К. П.; Математическая обработка
результатов измерений, М.— Л., 1950. И. П. Калинкин.
ОБРАТИМЫЕ РЕАКЦИИ — химич. реакции, про-
текающие при данной темп-ре одновременно в двух
противоположных направлениях (прямом и обратном).
Примером может служить реакция:
H2 + J2 2HJ
к-рая в определенной области темп-р является обра-
тимой. (Знак употребляется, когда нужно под-
черкнуть, что реакция в данных условиях обратима).
Преобладание одного из направлений и, соответствен-
но, изменение состава реакционной системы зависят
от соотношения скоростей прямой и обратной реак-
ций. В общем случае это соотношение может быть раз-
личным, т. к. оно зависит, в свою очередь, от концент-
раций реагирующих веществ в данный момент време-
ни, а во многих реакциях — также от давления.
Однако при отсутствии затраты работы извне большей
скоростью всегда обладает то из направлений, к-рое
приближает реакционную систему к состоянию рав-
новесия. По мере приближения к этому состоянию
скорость преобладающей реакции уменьшается, а
противоположной — возрастает; поэтому разница в
скоростях уменьшается,
в обоих направлениях
(рис.). Эти соотношения
Время
Зависимость скорости прямой и об-
ратной реакций от времени.
и при равновесии скорости
становятся одинаковыми
между скоростями теорети-
чески могут быть обо-
снованы, исходя нехо-
рошо известной зави-
симости скорости хи-
мич. реакций от кон-
центраций реагирую-
щих веществ, выра-
жаемой действующих
масс законом.
На рисунке пред-
ставлен частный слу-
чай, когда в начальном
состоянии в реакцион-
ной системе имеются
только вещества, указанные в левой части ур-ния
реакции, напр. Н2 и J2 в приведенной реакции. По
мере израсходования водорода и иода уменьшается
скорость Vi прямой реакции. Скорость V2 обратной
реакции, будучи равной нулю в начальный момент,
возрастает с повышением концентрации Н J в системе.
При V, = V2 достигается равновесие и концентрации
каждого из этих трех веществ сохраняются во времени
постоянными. Так как химич. взаимодействие между
молекулами при этом не прекращается, то и в состоя-
нии равновесия реакция является обратимой. При
прибавлении к такой равновесной системе веществ,
указанных в левой части уравнения (Н2 или j2), в
результате химич. взаимодействия повысятся и кон-
центрации веществ, указанных в правой части урав-
нения (HJ). И наоборот — прибавление HJ вызовет
повышение концентрации Н2 и J2. Прп этом в обоих
случаях в системе вновь будет достигнуто равновесие,
причем концентрации каждого из веществ претерпят
изменения в ту или другую сторону (см. Равновесие
химическое).
Когда равновесие в данной реакции сильно смещено
в одну сторону, то по внешним признакам она часто
может быть принята за необратимую реакцию (напр.,
реакция нейтрализации сильного основания сильной
кислотой). Однако истинное химич. равновесие всегда
сохраняет динамич. характер, т. е. химич. взаимодей-
ствие не прекращается и реакция происходит в обоих
направлениях (с одинаковой скоростью) и,следователь-
но, является обратимой. К необратимым ре-
акциям принято относить такие, в к-рых вещества,
получаемые при реакции, или не вступают во взаимо-
действие между собой в данных условиях, или обра-
зуют при взаимодействии вещества, отличные от ис-
ходных веществ прямой реакции. Примером необра-
тимых реакций могут служить разложение взрывча-
тых веществ, реакции сгорания углеводородов в обыч-
ных условиях И др. В. А. Киреев.
ОБЪЕМНЫЙ анализ — совокупность методов
химич. анализа, основанных на измерении объемов
для установления количества (содержания) определя-
емого вещества. К объемным методам анализа относят
широко применяемые на практике различные вариан-
ты титриметрического анализа, основанного на изме-
рении объема израсходованного р-ра реагента извест-
ной концентрации, необходимого для достижения точ-
ки эквивалентности. Иногда титриметрич. методы не
совсем точно отождествляют с объемными методами.
К О.а.относят также многие методы газового анализа,
когда при выполнении определения измеряют объем
какого-либо поглотившегося или выделившегося газа.
Имеются методы О. а., основанные на измерении объе-
ма осадков, напр. количество серы в чугуне можно
определять по объему осадка сульфата бария в граду-
ированной центрифужной пробирке. Количество ве-
щества определяют по объему полученного осадка
в ультрамикроанализе, когда взвешивание затруднено
пли невозможно.
Лит. см. при ст. Аналитическая химия. И. А. Бусев.
ОБЪЕМНЫХ ОТНОШЕНИИ ЗАКОН - при оди-
наковых условиях темп-ры и давления газы вступают
в химич. реакцию друг с другом в простых объемных
отношениях, и объем газообразного продукта реакции
также находится в простом отношении к объемам
каждого из исходных газов. См. Гей-Люссака законы.
ОВОМУКОИД — глюкопротеид, выделенный из
белка яиц. Константа седиментации О. равна 2,8
единиц Сведберга; мол. в. 27 000; изоэлектрич. точка
при pH 3,9; хорошо растворяется в воде, растворы
О. при нагревании не коагулируют; содержит 21%
углеводного компонента, состоящего из 1% D-галак-
тозы, 4,7% D-маннозы, 14,5% D-глюкозамина и
0,5% N-ацетилнейраминовой к-ты. Аминокислотный
состав О. (в %): аланин 2,3; глицин 3,8; лейции 5,1;
валин 6,0; изолейцин 1,43; пролин 2,72; фенилаланин
2,91; тирозин 3,18; триптофан 0,3; серин 4,2; треонин
5,5; цистин/2 6,7; метионин 0,95; аргинин 3,7; гисти-
дин 2,15; лизин 6,0; аспарагиновая к-та 13,0; глута-
миновая к-та 6,5. Получают О. из белка яиц, для
чего последний коагулируют нагреванием или осаж-
дают 10%-ной трихлоруксусной к-той; из фильтрата
О. осаждают сернокислым аммонием, этанолом или
ацетоном. О. известен как один из ингибиторов трип-
сина. Механизм его тормозящего действия не выяс-
нен. При замещении карбоксильных, аминных или
гуанидиновых групп в О. теряется его активность как
ингибитора.
Лит.: Белки, пер. с англ., под ред. Г. Нейрата, К. Бейли,
т. 3, ч. 1, М., с. 764, 1958. В. О. Шпикитер.
ОГНЕОПАСНЫЕ ВЕЩЕСТВА — вещества, спо-
собные под действием источника воспламенения
(огонь, накаленное тело, электрич. искра, теплота
окружающей среды, химич. реакции и т- д.) к само-
распространяющемуся химич. превращению (горению,
взрыву). К огнеопасным относятся все горючие веще-
ства и материалы, в том числе взрывоопасные (см.
Взрывоопасные вещества) и взрывчатые (см. Взрывча-
тые вещества). В настоящей статье огнеопасность
взрывчатых веществ не рассматривается. Все О. в. по
степени огнеопасности можно подразделить на 3 груп-
пы: самовозгорающиеся, легковоспламеняющиеся и
трудновоспламеняющиеся. При этом за показатель
огнеопасности веществ приняты: темп-ра самовоспла-
менения, темп-ра вспышки (см. Вспышки темпера-
641
ОГНЕПРОВОДНЫЙ ШНУР —ОГНЕУПОРНЫЕ МАТЕРИАЛЫ
642
тура) и способность веществ образовывать с воздухом
взрывоопасные смеси.
Самовозгорающимися наз. вещества,
способные воспламеняться под действием теплоты,
выделяющейся при химия., физич. и биологич. про-
цессах. Наибольшую огнеопасность представляют ве-
щества, самовозгорающиеся при соприкосновении с
кислородом воздуха: белый фосфора карбиды щелоч-
ных металлов, сульфиды железа, порошки алюминия
и железа, фосфористый и кремнистый водороды, а
также растительные масла, олифы и скипидар (рас-
пределенные тонким слоем на поверхности волокнис-
тых или измельченных материалов), фрезерный торф,
влажные сено и листья и др. При соприкосновении с
галогенами (хлор, бром) самовозгораются на свету:
ацетилен, красный фосфор, водород, метан, этилен
и др. При контакте с азотной к-той самовозгораются:
целлюлозные материалы (солома, хлопок, древесные
стружки, опилки, бумага), скипидар, этиловый спирт,
керосин, олифа, льняное масло, триэтилампн, дре-
весный уголь и др.; при соприкосновении с перекисью
натрия самовозгораются: метиловый, этиловый, про-
пиловый, бутиловый спирты, анилин, диэтиловый
эфир и уксусная к-та. Хромовый ангидрид вызывает
самовозгорание метилового, этилового, бутилового и
изоамилового спиртов, уксусного, масляного, бен-
зойного и пропионового альдегидов, уксусной к-ты,
ацетона и др.; перманганат калия вызывает самовозго-
рание глицерина и этиленгликоля. Щелочные метал-
лы: калий, натрий, рубидий, цезий при соприкосно-
вении с водой самовозгораются и поджигают выделя-
ющийся при реакции водород.
Легковоспламеняющимися наз. ве-
щества, способные при темп-рах производственных
помещений быстро воспламеняться даже от малока-
лорийных источников воспламенения (электрические и
другие искры). К ним относятся: горючие газы и
аэрозоли, жидкости, имеющие темп-ру вспышки до
45°, и твердые вещества с темп-рой самовоспламене-
ния до 150° или находящиеся в виде отдельных тонких
листов и волокон. Горючие газы (СНц С2Н2, Н2, СО
и др.) и аэрозоли (сахарная, древесная, мучная и др.
пыли) способны образовывать взрывоопасные смеси
с воздухом при любой темп-ре. Такими же свойствами
обладают легковоспламеняющиеся жидкости (темп-ра
вспышки ниже 45°), когда они нагреты выше темп-ры
вспышки. К таким жидкостям относятся: бензин,
тракторный керосин, бензол, ацетон, диэтиловый эфир,
метиловый, этиловый, пропиловый и бутиловый спир-
ты и др. К твердым легковоспламеняющимся вещест-
вам относятся: целлулоид, горючая кинопленка,
спички, фосфор сернистый, ледерин, листы тонкой
бумаги, аэрогели (хлопка, льна, джута и др. волок-
нистых веществ).
Т рудновоспламеняющимися наз.
горючие вещества, имеющие темп-ру вспышки (для
твердых веществ темп-ру воспламенения) выше 45°,
к-рые при обычных темп-рах производственных поме-
щений не способны быстро воспламеняться даже от
высококалорийных источников воспламенения. К та-
ким веществам относятся минеральные и растительные
масла, глицерин, этиленгликоль, мазут, дизельное
топливо, анилин, ксилол и др., а также древесина,
торф, бумага, пластмассы, сахар, целлюлоза, ткани,
парафин, нафталин, смолы и др.
Лит.: Легковоспламеняющиеся и горючие жидкости.
Справочник, под общ. ред. Н. А. Тарасова-Агалакова,
М., 1956; Нормы и технические условия проектирования склад-
ских предприятий и хозяйств для хранения легко воспламе-
няющихся и горючих жидкостей (Н и ТУ 108—56), М., 1956.
П. Г. Демидов.
ОГНЕПРОВОДНЫЙ ШНУР (бикфордов шнур)-
трубочка из нескольких слоев нитяных оплеток,
внутренний канал к-рой, диаметром 1—2 мм, заполнен
4 з
Огнепроводныйшнур: 1—
центральная направляю-
щая нить; 2 — пороховая
сердцевина; 3 — нитяные
оплетки; 4 — пластикато-
вая оболочка.
дымным (черным) порохом (см. рис.). Применяется
для возбуждения взрыва капсюлей-детонаторов и за-
рядов дымного пороха при т. наз. огневом взрывании,
а также для других целей. При
поджигании пороховая сердце-
вина О. ш. горит с постоянной
скоростью ~1 см!сек (реже при-
меняется т. наз. медленно горя-
щий О. ш., скорость горения
к-рого ок. 0,5 см/сек) и, догорев
до конца, дает сноп искр и пла-
мя, к-рые и воспламеняют взры-
ваемый заряд. Для предохране-
ния О. ш. от увлажнения нитя-
ная оплетка пропитывается вла-
го-изолирующим составом или
заключается в пластикатовую оболочку. Оболочка нор-
мально горящего О. ш. белая, серая или черная, мед-
ленно горящего — желтая.
Лит.: Яременко Н. Е., Светлов Б. Я., Теория
и технология промышленных взрывчатых веществ, М., 1957,
с. 233—35. Б. Н. Кондриков.
ОГНЕУПОРНЫЕ МАТЕРИАЛЫ (огнеупоры) —
изделия, изготовляемые на основе минерального сырья
и применяемые для строительства различных промыш-
ленных агрегатов, работающих в условиях высоко-
темп-рного нагрева.
О. м. должны отвечать след, требованиям: обладать
высокой огнеупорностью (не расплавляться под дей-
ствием высоких темп-р); не деформироваться под дей-
ствием строительной нагрузки при высоких темп-рах
(сохранять в этих условиях постоянство объема);
обладать термич. стойкостью (не растрескиваться при
резких колебаниях темп-р) и шлакоустойчивостью
(противостоять воздействию шлаков и расплавленных
стекол и др. агрессивных сред при высоких темп-рах).
О. м. делятся на след, группы: кремнеземистые;
алюмосиликатные; магнезиальные; хромо-магнезито-
вые и хромитовые; циркониевые; углеродистые; кар-
бидные; нитридные; окисные.
В каждой из указанных в классификации групп
объединены О. м. с определенным физико-минерало-
гич. составом и свойствами.
Кроме того, О. м. подразделяются по след, признакам:
а) по степени огнеупорности — на огнеупорные (1580—
1770°), высокоогнеупорные (1770—2000°) и высшей огнеупор-
ности (выше 2000°);
б) по форме и размерам — на нормальный кирпич, фасон-
ные изделия и крупноблочные;
в) по способу изготовления — на изделия, полученные
пластичным формованием, полусухим прессованием или трам-
бованием из порошкообразных масс, литьем из шликера,
литьем из расплава;
г) по характеру термич. обработки — на безобжиговые,
обожженные и плавленые;
д) по характеру строения и пористости — на спекшиеся
с пористостью менее 1%; плотные с. пористостью 10—30%;
легковесные (теплоизоляционные) с пористостью выше 50%.
Кремнеземистые О. м.
а) Д и н а с о в ы е О. м. (табл. 1) изготовляют
из кварцевых пород с известковым или иным связую-
щим материалом, обжиг ведут при темп-ре, обеспечи-
вающей полиморфное превращение кремнезема (квар-
ца) в тридимит и кристобалит. Основным сырьем для
производства динаса служат цементные и кристаллич.
кварциты, содержащие не менее 95% 8Юг. Для изго-
товления динаса кварциты измельчают, зерна необ-
ходимых размеров смешивают в бегунах с известко-
вым молоком, полученную массу с влажностью 5,5—
9% формуют в изделия на механич. прессах. Высу-
шенные изделия обжигают при 1430—1450° в течение
нескольких суток. Образующаяся в результате об-
жига кристаллич. структура обусловливает высокую
темп-ру деформации изделий под нагрузкой при на-
гревании. Динас — кислый огнеупор, характеризует-
ся невысокой термич. стойкостью; пригоден в усло-
виях воздействия кислых шлаков, расплавленных
О 21 К.х.э. т. 3
643
ОГНЕУПОРНЫЕ МАТЕРИАЛЫ
644
стекол ит. п.; применяются гл. обр. для футеровки
коксовых и стекловаренных печей, а также для кладки
мартеновских и электросталеплавильных печей. Для
получения динасо-хромитовых и динасо-карборун-
довых О. м. в шихту вводят 30% молотого хромита
или до 30% тонкоизмельченного карборунда. Указан-
ные О. м. характеризуются повышенной термич.
стойкостью по сравнению с динасом.
б) Кварцевые О. м. изготовляют из плав-
леного кварца (непрозрачного кварцевого стекла),
содержащего ок. 99% S1O2. Сырьем служат кварце-
вые пески, содержащие не менее 99,5% SiOs- Изделия
формуют в горячем состоянии прессованием на мощ-
ных прессах, прокаткой или другими приемами.
Кварцевые О. м. имеют низкий коэфф, термич. рас-
ширения (0,5Х10~6) и поэтому отличаются высокой
термич. стойкостью; применяют для кладки бассей-
нов и протоков печей, в к-рых варят малощелочные
тугоплавкие стекла. Освоено производство О. м. из
непрозрачного кварцевого стекла керамич. методами.
Формование из водного шликера позволяет получать
крупноразмерные изделия сложной формы, характе-
ризующиеся высокой термич. стойкостью.
Алюмосиликатные О. м.
а) Полукислые О. м. (табл. 1) содержат в
обожженном виде 17—30% А120з и 65—80% SiO2.
Высококачественные полукислые О. м. изготовляют
из первичных каолинов с естественной примесью квар-
ца. В массу вводят шамот (глину, предваритель-
но обожженную до спекания) из того же каолина.
О.м. менее высокого качества изготовляют из глин с
примесью песка или др. кварцевых пород. Полукислые
О. м. обычно рассматриваются как экономически це-
лесообразный местный заменитель шамотных изделий.
Положительным свойством полукислых О. м. являет-
ся постоянство их объема при обжиге, обусловленное
расширением кварца, к-рое компенсирует усадку спе-
кающейся глины. Полукислые О. м. применяют для
кладки некоторых частей коксовых печей, работаю-
щих при температурах до 1450°, для насадки каупе-
ров, футеровки сталеразливочных ковшей, а так-
же в стенах, сводах, регенераторах и т. п. в условиях,
когда отсутствует непосредственное воздействие
шлака.
б) Ш а м о т н ы е О.м. (табл. 1) изготовляют из
огнеупорных глин и каолинов с добавлением 50—
85% шамота. Шамотные изделия являются наи-
более распространенными О. м.; по огнеупорности де-
лятся на 4 класса (от 1580 до 1750°). Свойства шамот-
ных О. м. (плотность, шлакоустойчивость, термич.
стойкость и др.) регулируются качеством исходного
сырья и зерновым составом. Шамотные О. м. изготов-
ляют гл. обр. полусухим прессованием масс, содержа-
щих шамотные зерна и высокодисперсную глину.
Пластич. формование применяют для получения особо
сложных фасонных изделий. Высушенные изделия
обжигают при темп-рах, обеспечивающих спекание
связующей глины (обычно 1300—1450°). На совре-
менных заводах обжиг шамотных огнеупоров произ-
водят в туннельных печах. Шамотные О. м. характери-
зуются универсальностью свойств, могут успешно при-
меняться в агрегатах с резкими сменами температур,
весьма устойчивы против воздействия кислых шлаков
и расплава стекла (до 1400—1500°), а также основных
шлаков (до 1250—1350°); применяются в коксовых,
доменных, мартеновских, нагревательных, стеклова-
ренных и туннельных печах, в вагранках, в топках
котлов, а также при разливке стали. В отдельных
случаях изготовляют также бесшамотные изделия,
изделия на низкожженом шамоте и безобжиговые
шамотные О. м.
в) Высокоглиноземистые О. м.
(табл. 1) изготовляют из природного и искусственно
получаемого высокоглиноземистого сырья — киа-
нита, андалузита и др., прокаленного гидрата глино-
зема и электроплавленого корунда. Основными кри-
сталлич. фазами высокоглиноземистых огнеупоров
являются муллит и корунд, содержание к-рых, на-
ряду с зерновым составом наполнителя, определяет
термич. и химич. стойкость и строительную прочность
изделий при высоких темп-рах. Увеличение содержа-
ния А120з повышает химич. стойкость изделий, что
обусловлено большой химич. инертностью корунда.
Производство высокоглиноземистых огнеупоров ана-
логично производству шамотных изделий; различие
заключается в применении шамота с более высоким
содержанием А120з и введением в качестве связующего
наряду с огнеупорной глиной технич. глинозема.
Высокоглиноземистые О. м. имеют высокую темп-ру
начала размягчения под нагрузкой и повышенную
огнеупорность; применяются преим. в сводах метал-
лургич. печей, в электропечах, в высокотемп-рных
стекловаренных и туннельных печах.
Для нужд стекловаренной промышленности изго-
товляют плавленые высокоглиноземистые (муллито-
вые) О. м., к-рые характеризуются повышенной плот-
ностью и прочностью, высокими темп-рами начала раз-
мягчения под нагрузкой.
Корундовые О.м. (табл. 1) — изделия, сос-
тоящие в основном из А120з (корунда); характеризу-
ются наиболее высокими для алюмосиликатных огне-
упорных материалов свойствами. Различают полно-
стью спекшиеся корундовые изделия и корундовые
О. м. зернистого строения. Первые производят из
предварительно обожженного тонко измельченного
технич. глинозема, вторые из масс, в состав к-рых вхо-
дит ок. 70% корундового наполнителя (электрокорунда
или корундового шамота определенного зернового
состава) и ок. 30% связующего тонкомолотого глино-
зема. Корундовые О. м. зернистого строения отли-
чаются от спекшихся корундовых О. м. повышенной
термич. стойкостью. Температура обжига корундовых
огнеупорных материалов без добавок, снижающих
темп-ру спекания, составляет ок. 1700°. При добавле-
нии 1—2% TiO2 эта температура может быть снижена
до 1500—1600°.
Таблица 1
Содержание и свойства Динасовые* О. м. Полукислые О. м. Шамотные О. м. Высокогли- ноземистые О. м. Корундовые О. М.
SiO2, % 93—98 65—80 55—60 <55
А12О3, % Огнеупорность, °C Темп-ра начала размягчения под нагрузкой 1-1,5 17—30 30—45 45—95 >95
1690—1720 1610—1710 1690—1740 1750—1950 >1950
2 кг/см2» °C Пористость, % 1620-1660 1300—1400 1300 — 1450 1400—1700 1700—1900
13—25 27—30 14-30 12—34 10-25
Предел прочности при сжатии, кг1см2 .... Коэфф, термич. расширения а-10“ в интерва- 150—600 100-150 100—650 150-1 500 550—2500
ле 20—1000° 11,5—13 7—9 4,5—6 5,5—7,5 8-8,5
В динасовых О. м,, кроме того, содержится 2—3,5% СаО.
645
ОГНЕУПОРНЫЕ МАТЕРИАЛЫ
646
Магнезиальные О. м.
а) Магнезитовые (периклазовые) О. м.
(табл. 2) состоят в основном из периклаза (содержание
MgO 80—85% и выше); сырьем служит природный
магнезит MgCOa, брусит Mg(OH)2 и окись магния.
Для производства магнезитовых О. м. применяют
спекшийся и плавленый магнезит. Спекшийся маг-
незит (металлургия, порошок) получают обжигом
природного обогащенного магнезита в шахтных или
вращающихся печах при 1550—1600°. Наиболее чис-
тые разновидности спекшегося магнезита (>91% MgO)
используются для изготовления изделий, менее чис-
тые — в качестве металлургия, порошка для наварки
и набивки подин мартеновских и основных электро-
сталеплавильных печей. Плавленый магнезит отли-
чается большой чистотой, но вследствие высокой стои-
мости применяется лишь для особо ответственных из-
делий. Изделия из магнезитовых О. м. изготовляют
прессованием смеси спекшегося магнезита различной
зернистости (50—55% крупных фракций размером
0,8—2 мм); иногда вводят добавки (окислы железа
или титана), способствующие спеканию, а также крем-
незем, серпентин, тальк, повышающие темп-ру дефор-
мации под нагрузкой и термич. стойкость (глинозем).
Обжиг готовых изделий производят при 1600° (не
выше). Изготовляют также безобжиговые магнезито-
вые О. м. Магнезитовые изделия характеризуются
высокой огнеупорностью (>2000°), началом деформа-
ции под нагрузкой при 1550—1600° и невысокой тер-
мич. стойкостью. Последняя может быть повышена
укрупнением зерен наполнителя и введением добавки
А120з (до 10%).
Магнезитовые О. м. применяют гл. обр. в стале-
плавильном произ-ве (мартеновские и электростале-
плавильные печи); термостойкие — используются гл.
обр. для выкладки сводов мартеновских печей и зоны
спекания вращающихся печей цементной пром-сти.
б) Д о л о м ит о в ы е О. м. (табл. 2) изготовля-
ют в основном из минерала доломита CaMg(CO3)2- Раз-
личают два вида доломитовых О. м.: содержащие сво-
бодную СаО и подвергающиеся гидратации при хра-
нении на воздухе; водоустойчивые, содержащие СаО
только в связанном виде. Обжиг металлургия, доло-
мита в кусках или в виде шлама производят во вра-
щающихся или шахтных печах при 1600—1700°. Во-
доустойчивые доломитовые О. м. изготовляют введе-
нием в обжигаемый доломит кварцита, трепела и др.;
при этом окись кальция связывается кремнеземом в
трехкальциевый и двухкальциевый силикаты. Пос-
ледний стабилизируется введением фосфорита; обжиг
ведется при 1450—1550°. Доломитовые О. м., содер-
жащие свободную СаО, изготовляют как без обжига,
так и путем обжига; связующим веществом является
смола или битум. Доломитовые О. м. устойчивы
против воздействия основных шлаков, применяются
аналогично магнезитовым. Разновидностью доломи-
товых О. м. являются магнезито-доломитовые О. м.,
изготовляемые по технологии доломитовых О. м. и
отличающиеся от них лишь большим содержанием
MgO. Для изготовления магнезито-доломитового
клинкера применяют природные магнезито-доломиты,
смеси доломитов с магнезитом или каустич. магнези-
том и хромитом.
г) Форстеритовые О. м. (табл. 2): их ос-
новой является минерал форстерит (Mg2SiO4); изго-
товляются из магнезиально-силикатных пород (ду-
нита, серпентинита, оливинита и др.) с добавкой магне-
зита (для связывания SiO2 в форстерит, а полутор-
ных окислов в шпинелиды). Изделия обжигают при
1600—1700°. Изготовляют также безобжиговые фор-
стеритовые О. м. (при этом исходный дунит предва-
рительно обжигают при 1450°). Форстеритовые О. м.
шлакоустойчивы к основным шлакам и могут служить
заменителем магнезитовых огнеупоров в мартеновских
печах, во вращающихся цементно-обжигательных
печах и в насадке регенератора мартеновских печей.
Таблица 2
Содержание и свойства Магне- зитовые (перик- лазовые) Доломи- товые Форсте- ритовые
MgO, % 89—95 -30 35—55
СаО, % 1 — 3 —'46 0,2—1.5
S1O,, % 1—3 -16 21 — 38
Огнеупорности °C Темп-ра начала размягчения >2000 1800— 1950 >1750
под нагрузкой 2 кг/см2, °C 1500— 1630 1550— 1610 1 550— 1670
Объемный вес, г.с.и3 2,6— 3,0 2,7— 2,9 2.40 — 2,9
Пористость, % Предел прочности при сжатии, 13—25 10 — 14 11 — 28
кг[см2 Коэфф, термич. расширения 350— 700 175—650
а-10е в интервале 20—1000° 14—15 — -И
д) Шпинельные О. м. изготовляют из мате-
риалов, в к-рых главной составной частью являются
различные шпинели с общей формулой R2 + R’ + O4.
Типичными представителями шпинелей являются
магнезиальная шпинель MgAl2O4, магнезиоферрит
MgFe2O4, магнезиохромит MgCr2O4 и др. Промыш-
ленное значение в качестве О. м. имеют шпинелиды
на основе хромита FeCr2O4 и магнезиальной шпинели
MgAl2O4 (т. пл. 2135°). Магнезиальную шпинель по-
лучают спеканием или сплавлением смеси окиси алю-
миния (или боксита) и обожженного магнезита, взя-
тых в стехеометрич. соотношениях. Шпинельные О. м.
характеризуются высокими темп-рами деформации
под нагрузкой и шлакоустойчивостью.
Хромо-магнезитовые и хромитовые О. м.
а) Хромитовые О. м. изготовляют на ос-
нове хромистого железняка (не менее 85% FeCr2O4,
огнеупорность 2100—2200°); характеризуются посто-
янством объема в обжиге (в нек-рых случаях могут
Таблица 3
Содержание и свойства Хромо-магнезитовые Магнезито-хромитовые Периклазо- шпинелид- ные
обожженные безобжиговые обожженные безобжиговые
MgO, % Сг2О3, % Огнеупорность, °C Темп-ра начала размягчения под нагрузкой 2 кг/см2, °C Объемный вес, г/ел3 Пористость, % Предел прочности при сжатии, кг/см2 .... Коэфф, термич. расширения а-106 в интервале 20—1000° 42-55 15-17 >2000 1450—1520 2,8 23-24 200—400 33—42 20 >2000 2,90—2,98 18—24 100—200 В 55—75 8—18 >2000 1450—1600 2,7—2,8 12-32 250—1000 пределах 8,5— 45—78 8-15 >2000 1350—1450 2,85—3,15 9-25 200—750 10 57—70 11 — 18 >2000 1580—1630 3,3 10-20 400—850
21
647
ОГНЕУПОРНЫЕ МАТЕРИАЛЫ
648
быть использованы без предварительного обжига),
низкой плотностью и прочностью, низкой темп-рой
начала размягчения под нагрузкой (1400—1450°) и
коэфф, термич. расширения 9— И -10“6 (в интервале
20—1000°).
б) Хромо-магнезитовые и магне-
зито-хромитовые О.м. (табл. 3) изготовля-
ют из материалов на основе хромита и спекшегося маг-
незита, содержание к-рого колеблется от 30 до 80%.
Спекшийся магнезит применяют преимущественно в
виде тонкомолотого порошка с зернами размером
<0,088 .и.и, хромит — в виде зерен размером 0,5—
2 мм и частично <0,09 л«л1.
В СССР освоено производство пе р и к л азо-
шпине л ид ных О.м. (табл. 3), в к-рых магне-
зит представлен крупными зернами, а хромит — тон-
кими; они характеризуются термич. стойкостью, вы-
сокой плотностью и прочностью.
Для изготовления хромо-магнезитовых О. м. массу
прессуют на мощных гидравлич. прессах, обжиг ведут
в туннельных высокотемп-рных печах при 1600—
1700°. Изготовляют также безобжиговые хромо-маг-
незитовые и магнезито-хромитовые О. м. Хромо-маг-
незитовые О. м. применяют для кладки сводов мар-
теновских печей, в сталеплавильных печах, в конвер-
торах, для футеровки цементно-обжи-
гательных печей (в зоне спекания).
Циркониевые О. м. Эти О. м. изго-
товляют из материалов на основе дву-
окиси циркония ZrO2 (цирконие-
вые) и ортосиликата циркония ZrSiOi
(цирконовые), характеризующие-
ся повышенной огнеупорностью (т. пл.
ZrO2 ок. 2700°); стойки к расплавам
металлов, шлакам, кислотам. Трудно-
сти производства О. м. из ZrO2 обус-
ловлены его полиморфным превраще-
нием при темп-ре ок. 1000°, связанным
с объемными изменениями (для перево-
да в устойчивые твердые р-ры поэтому необходима ста-
билизация добавками СаО и MgO). Широкое примене-
ние циркониевых О. м. ограничено высокой стоимостью
исходного сырья. Для нужд гл. обр. стекловаренной
пром-сти изготовляют также плавленые цирконо-мул-
литовые огнеупорные брусья, характеризующиеся вы-
сокой стойкостью к расплавленной стекломассе.
Углеродистые О. м.
а) Коксовые О.м. изготовляют из смеси кокса
со смолой. Смешение ведут в обогреваемых паром ме-
шалках, формование производят протягиванием в лен-
точных прессах, трамбованием или прессованием на
мощных гидравлич. прессах. Обжиг ведется в газо-
камерных печах с шамотными муфелями при 1400—
1500°. Изделия характеризуются высокой огнеупор-
ностью и отличной термич. стойкостью. Коксовые
О. м. применяют для кладки лещади и горна доменных
печей, а также в печах для плавки цветных металлов.
б) Графитовые О.м. изготовляют из смеси
графита и огнеупорной глины, характеризуются вы-
сокой огнеупорностью, шлакоустойчпвостью, высо-
кой тепло- и электропроводностью. Графитовые О. м.
применяют для изготовления тиглей и реторт для
плавки стали и цветных металлов, а также пробок
и стаканов для разливочных ковшей. Изделия из
химически чистого графита применяют в реакторо-
строении.
Недостатком коксовых и графитовых О.м. являет-
ся их способность к окислению кислородом уже
при 700°.
Карбидные, нитридные и др. О. м. К этой группе
О. м. в первую очередь относятся карборундо-
вые, изготовляемые из карбида кремния (SiC), ис-
Карбиды
TIC 3140
ZrC. 3500
HfC 3900
VC 2830
NbC 3500
TaC 3880
Cr3C2 1895
Mo2C 2690
wc 2600
кусственно получаемого из смеси кварцевого песка
и кокса. Различают след, карборундовые изделия:
рекристаллизованные, на глиняном связующем и на
др. минеральных связующих, в частности нитридном
(SiaNi), характеризующиеся высокой плотностью и
хорошей коррозионной стойкостью. В зависимости от
назначения О. м. содержание SiC колеблется от 20
до 90% и выше. При высоком содержании SiC, во из-
бежание его окисления, обжиг ведут в капселях (огне-
упорных коробах) в засыпке, содержащей кварцевый
песок, кокс или графит; темп-ра обжига 1400—1500°.
Карборундовые изделия применяют для футеровки
пода коксовых печей, для произ-ва капселей, муфелей
и пр. Карборундовые О. м. характеризуются высо-
кой термич. стойкостью, теплопроводностью, высоки-
ми темп-рами начала размягчения под нагрузкой.
Широкое внимание привлекают и нек-рые другие
высокоогнеупорные бескислородные соединения туго-
плавких металлов — карбиды, бориды, силициды и
нитриды (табл. 4), а также керамико-металлич. из-
делия (см. Керметы), характеризующиеся в ряде слу-
чаев исключительной твердостью, очень высокой
темп-рой плавления и др. Нек-рые из них (напр.,
карбиды кремния SiC и бора В4С, нитрид кремния
SiaN4, дисилицид молибдена Мо312идр.) широко при-
меняют, другие находятся в стадии исследования.
Таблица 4
Нит- риды T. пл., °C Бориды T. пл., °C Силициды T. ПЛ., °C
TIN 3200 T1B2 2980 TiSi2 1 540
ZrN 2980 ZrB2 30 40 ZrSi2 1700
HfN 3380 ШВ. 3250 HfSl, 1750
VN 2360 VB. 2400 VSi2 1660
NbN 2300 NbB- 3000 NbSi2 1950
TaN 3087 TaB2 3100 TaSi2 2200
CrN 1500 CrB2 2200 CrSi2 1550
— MO,Bs 2100 MoSij 2050
— — W2B5 2300 WS1, 2165
Окисные О. м. Эти О. м. изготовляют на основе чис-
тых высокоогнеупорпых окислов. Наибольшее рас-
пространение получили материалы из А120з, ВеО,
MgO, СаО, ZrO2, ThO2 и UO2 (табл. 5). Огнеупорность
указанных материалов выше 2000°. Как правило,
технология изготовления изделий из чистых окислов
предусматривает предварительную термич. обработку
материала, тонкое измельчение, производимое в ша-
ровых или вибрационных мельницах, обработку со-
ляной к-той, формование (литьем из водных суспен-
зий, горячим литьем под давлением, протягиванием
пластифицированной массы, прессованием полусухих
масс, гидростатпч. прессованием, горячим прессова-
нием, отливкой из расплавленной массы). Отформо-
ванные изделия обжигают при темп-ре ок. 1700° (и
выше) до спекания.
Окисные О. м. применяют для изготовления футе-
ровки высокоогнеупорных печей, тиглей для плавки
металлов, деталей для авиационных и ракетных дви-
гателей, ядерных реакторов, пирометрия, трубок для
термопар и др.Одним из ценных свойств окисных О. м.
является их стойкость в окислительных средах; не-
достатком — относительно невысокая термич. стой-
кость. Для ее повышения к окислам добавляют на-
полнитель (спекшиеся зерна из того же материала,
связываемого высокодпсперсным порошком). Таким
способом, в частности, получают материалы из А120з,
MgO, ZrO21 используемые для футеровки высоко-
огнеупорных печей.
Легковесные О. м. Наряду с плотными О. м. широко
производятся пористые (легковесные) О. м., приме-
няемые в тепловых агрегатах для уменьшения рас-
хода топлива за счет снижения потерь тепла на ак-
кумуляцию массивом кладки и на излучение поверх-
649
ОГНЕУПОРНЫЕ МАТЕРИАЛЫ
650
Таблица 5
Окислы
Свойства А1.,О3 ВеО MgO СаО ZrO, Th02 ио2
Плотность, г/сл13 . . . Т. пл., °C 3,97 3,02 3,60 3,35 5,60 9.69 10,97
2050 2530 2800 2585 2700 3050 2800
Коэфф, термич. расшире- 20 — 1 700° 20 — 1700°
ния а-10“ 20 — 1000° 25—1700° 20 — 1000° 20—1700° —
8,4 10,6 15,6 13,8 7,7 10,2
Коэфф, теплопроводно- сти, ккал/л1-час-град
100° 26,0 187,0 29.62 13,10 1,68 8,82 8,41
1600° 5,22 13,03 5,65 — 2,10 2,11
Удельное сопротивле- 1000°—80-Ю6 1000°—1-Ю7 763°— 70 • 10е
ние, ом-см 1 4°— 1 -10'“ 1000°—10* 20°—4-10” 22°—10 . 7-Ю2
800°—3 ,5-10-' 2000°— 1 , 6-Ю3 2000°— 5-1 О2 1 235°—10-103 1700°—6—7 500°—1,2-Ю12 3 27°—0,42-102
Предел прочности при 20°—14000 20°—21000
сжатии, кг/см2 . . . 20°—30000 20°—8000 — 20°—1 5000 —
1 600°—500 1 600°—500 1000°—1 1500 — 1500°—200 1500°—100 —
Поперечное сечение рас- 0 , 42 0,24 0,46
сенния, барн .... Поперечное сечение зах- 0,34 0,68 0,40 —
0,182
вата нейтронов, барн 0,010 0,0007 0,0033 0,010 0,132 —•
Таблица 6
Свойства Шамотный,легковес с выгораю- щими добавками Пенолег- ковесные БЛ-08 Ультра- легковес- ные Б’Л-0,4 Динасо- вый легковес
А Л-1 ,3 БЛ-1 , 3 БЛ-1 ,0
Огнеупорность, °C (не ниже) 1750 1670 1700 1670 1690 1670
Объемный вес, г/см3 (не более) 1 ,3 1,3 1 ,0 0,8 0,4 1.2
Пористость, % — — 59 — 82 54
Предел прочности при сжатии, хг.'см2 (не ниже). . . 45 30 — 20 10 30
Дополнительная усадка при темп-ре, °C 1 400° 1350° 1 350° 1250° 1250°
в % 1 , о 2,0 0,5 1,0 0,4
Коэфф, теплопроводности при средней темп-ре, °C 780° 720° 760° 700° 515° 550°
в ккпл/м-час-гра9 0,64 0,53 0,48 0,40 0,18 0,68
Предельная темп-ра применения, °C 1400 1 300 1200 1200 1200 1500
ностыо печи в окружающую среду. Характеристика
нек-рых легковесных О. м. приведена в табл. 6. Лег-
ковесные О. м. изготовляют пенометодом (смешением
суспензии О. м. с отдельно приготовленной устойчи-
вой пеной), методом химич. газообразования, введе-
нием выгорающих добавок и др. способами из тех же
исходных материалов, что и плотные огнеупорные
изделия. По сравнению с обычными О. м. легковес-
ные характеризуются малым объемным весом (0,4—
1,3 г/см!), высокой пористостью (60—8,5%), низкой
теплопроводностью (0,15—0,60 ккал/м-час-град), низ-
кой теплоемкостью, пониженной механической проч-
ностью, высокой газопроницаемостью и т. п. Глав-
ные недостатки, ограничивающие их применение,—
низкая шлакоустойчивость, низкая термич. стой-
кость, малая прочность.
Легковесные О. м. применяются: для незащищен-
ной футеровки (непосредственно у огня) печей, когда
исключено воздействие расплавленных шлаков, ме-
таллов, стекол и т. п.; для защищенной изоляции,
если темп-ра в рабочем пространстве превышает до-
пустимые для данного легковесного О. м. или при воз-
действии расплавленных агентов.
Для высокотемп-рной теплоизоляции в ограничен-
ном масштабе производятся легковесные материалы
высокоглиноземистые, корундовые, циркониевые и др.
Печп для обжига О. м. Подразделяются: по виду
тепловой энергии, применяемой для обогрева, на пла-
менные и электрические; по периодичности производ-
ственного цикла — на периодического и непрерыв-
ного действия; по конструктивному оформлению —
на камерные, кольцевые, туннельные, вращающиеся,
шахтные и др. Печи периодич. действия характери-
зуются большим расходом топлива, связанным с боль-
шими потерями тепла с отходящими газами и на ак-
кумуляцию тепла стенками печи, а также использова-
нием ручного труда. Поэтому основными видами печей
Рис. 1. Схема кольцевой печи: 1 —
футеровка печи; 2 — кольцевой ка-
нал; з —отверстия для загрузки и
выгрузки; 4 — дымовая труба; 5—
подача топлива.
Рис. 2. Схема многокамерной печи:
7 — футеровка; 2 — камера; з —
дымовая труба.
для массового обжига О. м. являются пламенные
печи непрерывного действия — кольцевые, много-
камерные, туннельные (для обжига формованных ма-
териалов), вращаю-
щиеся, шахтные (для
обжига кусковых ма-
териалов — шамота,
магнезита и др.). В
кольцевых пе-
чах (рис. 1) рабочая
камера представляет
собой сплошной коль-
цевой канал, запол-
няемый обжигаемыми
изделиями. Канал
кольцевой печи обыч-
но футеруют шамотным кирпичом. Высота канала 2—
3,5 м, ширина 2,5—6 м, длина 60—200 .и. Кольце-
вые печи весьма экономичны, применяются гл. обр.
для обжига рядовых шамотных и легковесных О. м.
В многокамерных печах (рис. 2)
рабочее пространство разделено на ряд камер, каж-
дая из к-рых представ-
ляет собой периодиче-
ски действующую ка-
мерную печь. Камеры
разделены стенками и
каналами для прохода
воздуха и горячих га-
зов. Печь обычно отап-
ливается генераторным
газом. Изделия обжи-
гают в одной из камер,
продукты горения проходят из нее в камеры, располо-
женные впереди, подогревая загруженные в них изде-
лия, затем удаляются через дымовую трубу или дымо-
сос. Число камер в печи колеблется от 16 до 32, емкость
651
ОГНЕУПОРНЫЕ МАТЕРИАЛЫ —ОДНОКОМПОНЕНТНЫЕ СИСТЕМЫ
652
однойкамеры.14—45 м!(длина 4—6м, ширина 2—4,5 м,
высота 2—3 л»). Одним из основных недостатков мно-
гокамерных печей является неравномерное распреде-
ление температур по высоте камер.
Туннельные печи (рис. 3), являющиеся
наиболее современными печами для массового обжига
О. м., имеют длинный узкий канал, по к-рому навстре-
чу газовому или воздушному потоку движутся ваго-
нетки с обжигаемыми изделиями. Туннель условно
Рис. 3. Простейшая схема туннельной печи прямого дей-
ствия: 1 — продукты горения; 2 — загрузка; 3 — подача
топлива; 4 — воздух для охлаждения и горения.
2
разделяется на зоны подогрева, обжига и охлаждения.
Вагонетки с изделиями подогреваются отходящими
из зоны обжига продуктами горения, затем проходят
через зону обжига, после чего поступают в зону ох-
лаждения, отдавая свое тепло омывающему их воз-
духу. Туннельные печи могут работать на твердом,
жидком и газообразном топливе; наиболее целесооб-
разны печи, работающие на газовом и жидком топливе;
они экономичны в отношении расхода топлива и пот-
ребности в рабочей силе. В туннельных печах наибо-
лее полно осуществляется автоматизация и точное
регулирование процесса обжига. В огнеупорной
пром-сти применяются мощные туннельные печи дли-
ной 120—150 м и сечением рабочего канала до 3 X
Х2,3 мг, производительность достигает 200 пг/сутки
и более. Указанные печи работают преимущественно
на газе и мазуте. Для кладки топок, свода и внутрен-
них стен рабочего канала в зоне обжига применя-
ются магнезитовые, хромомагнезитовые, высокогли-
ноземистые динасовые и шамотные О. м.
Вращающиеся печи (рис. 4) представляют
собой длинный барабан, установленный с уклоном
3—5° и вращающийся со скоростью 0,5—1,2 об/мин.
В верхний холодный конец барабана подается обжи-
гаемый материал, в нижний горячий конец — топ-
ливо. Обожженный материал поступает в холодиль-
ник длиной 15—25 м. Длина барабана 40—185 м,
диаметр 2—5 м", внутренняя поверхность печи футе-
руется огнеупорным кирпичом. Воздух, проходя по
холодильнику, охлаждает материал и нагретым по-
ступает в зону горения. Топливом для вращающихся
печей служит природный или коксовый газ, мазут,
угольная пыль. Преимуществами вращающихся пе-
чей является их высокая производительность и меха-
низация всех процессов, недостатками — высокий
удельный расход топлива и большой унос сырья.
Рис. 4. Схема вращающейся печи: 1 — головка; 2 — печь;
3 — холодильник; 4 — горячая камера; 5 — холодная ка-
мера.
В шахтных печах (рис. 5) материал непре-
рывно загружается в верхнюю часть шахты и непре-
рывно или периодически выгружается внизу. Мате-
риал передвигается по шахте под действием силы тя-
жести, подвергаясь при этом термич. обработке.
Движение газов и материала противоточное. Воздух
поступает снизу, охлаждает обожженный материал,
затем направляется в зону горения. Продукты горе-
ния движутся вверх, отдают тепло необожженному
Рис. 5. Схема круглой шахт-
ной печи: 1 — горелки; 2 —
отверстия для подачи воз-
духа.
материалу и удаляются в ат-
мосферу. Достоинства шахт-
ных печей — малые потери
материала, низкий удельный
расход топлива, невысокая
стоимость; недостатки — не-
равномерное качество про-
дукции, невысокая произво-
дительность печей (см. также
Печи технологические).
Наряду с пламенными пе-
чами для обжига О. м. при-
меняются электриче-
ские печи, к-рые по срав-
нению с пламенными обла-
дают такими преимущества-
ми, как возможность точного
контроля температурного ре-
жима, автоматич. регулиро-
вания обжига, чистота обжи-
га, снижение количества бра-
ка, малая продолжительность
обжига, простота обслужива-
ния и др. К важнейшим ти-
пам электрич. печей относят-
ся печи сопротивления, ин-
дукционные и дуговые; наи-
более распространены печи
сопротивления, конструкция
к-рых позволяет регулиро-
вать темп-ру в узких пре-
делах. Электрич. печи могут
быть периодически действу-
ющими (камерными) и непрерывно действующими
(туннельными); применяются гл. обр. при обжиге
изделий небольшого разме ра, а также в лабораторной
и исследовательской практике.
Лит.: Технология керамики и огнеупоров, под общ. ред.
П. П. Будникова, 3 изд., М., 1962; Техника высоких темпе-
ратур, под общ. ред. и. Э. Кэмпбелла, пер. с англ., М., 1959;
Справочник на огнеупорные изделия, материалы и сырье,
М., 1'961; Новые виды огнеупорных изделий. Каталог, М.,
1958; Тресвятский С. Г., Черепанов А. М.,
Высокоогнеупорные материалы и изделия из окислов, М.,
1957; Г и н з б у р г Д. Б. [и др.], Печи и сушила силикатной
промышленности, 2 изд., М., 1 956; Мамыкин П. С. |и
др.], Печи и сушила огнеупорных заводов, Свердловск, 1963.
И. Я. Гузман.
ОДНОКОМПОНЕНТНЫЕ СИСТЕМЫ — физико-
химические системы, образованные одним компонен-
том. Состояние О. с. определяется двумя параметрами
состояния, в качестве к-рых обычно берут темп-ру
и давление (реже темп-ру и объем). Согласно фаз
правилу, вариантность (т. е. число параметров состоя-
ния, к-рым можно задавать произвольные значения
без изменения числа фаз) О. с. равна двум — если
система состоит из одной фазы, одному — если в си-
стеме сосуществуют две фазы, ну-
лю— в случае трех фаз. Общий
вид диаграммы состояния О. с. изо- §
бражен на рисунке, где по гори- ®
зонтальной оси отложены величи- <g
ны темп-ры, а по вертикальной— |
давления; А — тройная точка, со-
ответствует равновесию трех фаз;
твердой, жидкой и парообразной. Е точке А пере-
секаются три кривые, каждая из к-рых отвечает
равновесию каких-либо двух фаз: АВ — твердой и
пара (кривая сублимации), АС — жидкой и пара
(кривая испарения), AD — твердой и жидкой фаз
(кривая плавления). Области между этими кривыми,
т. и. поля, изображают состояния лишь одной фазы.
температура
653
ОЖИЖЕНИЕ ГАЗОВ —ОЗОН
654
Для веществ типа серы, а таких большинство, кривая
AD при подъеме отклоняется вправо так, как это
изображено на рисунке. Для веществ же типа воды
(висмут и др.) кривая при подъеме уклоняется влево
(диаграмму состояния воды см. в ст. Диаграмма со-
стояния), т. к. их темп-ры плавления при повышении
давления понижаются (а не повышаются, как в пер-
вом случае). Равновесие двух фаз О. с. (линии АВ,
AC, AD) описывается Клайпейрона — Клаузиуса урав-
нением. Если вещество в твердом состоянии имеет
полиморфные модификации (см. Полиморфизм), то
диаграмма состояния усложняется; появляются по-
ля, отвечающие полиморфным модификациям, новые
линии двухфазных равновесий и новые тройные точки,
причем нек-рые из этих равновесий могут быть неу-
стойчивыми — метастабильными (см., напр., области
существования различных модификаций льда на рис. 2
в ст. Вода).
Лит.: Аносов В. Я. и Погодин С. А., Основ-
ные начала физико-химического анализа, М.— Л., 1947.
В. Я. Аносов.
ОЖИЖЕНИЕ ГАЗОВ — см. Охлаждение.
ОЗАЗОНЫ — продукты конденсации а-оксикарбо-
нильных соединений с замещенным гидразином, со-
держащие два остатка последнего. О. об- R'
разуют а-оксиальдегиды, а-оксикетоны, 1_м „
альдозы, кетозы и восстанавливающие оли-
госахариды. Гидразин может быть пред- c=N-NHR
ставлен монозамещенным производным, I
напр. фенилгидразином, 2,4-динитрофе- R
нилгидразином, n-бромфенилгидразином, или несим-
метричным двузамещенным гидразином, напр. метил-
фенилгидразином. Две эпимерные альдозы и кетоза,
имеющая одинаковое с ними строение в части молеку-
лы, не вовлекаемой в реакцию, дают один и тот же
О. Напр., О. D-маннозы, D-глюкозы и фруктозы иден-
тичны; аллоза, альтроза и псикоза образуют один и
тот же О. и т. д.
О.— желтые или оранжевые кристаллич. продукты,
плавящиеся обычно с разложением. Большинство О.
плохо растворимы в воде; О. лактозы, мальтозы и
нек-рых олигосахаридов растворимы в горячей воде.
В спирте О. растворимы плохо.
Получают О. нагреванием а-оксикарбонильных сое-
динений с избытком замещенного гидразина. Вначале
в конденсацию вовлекается только карбонильная груп-
па и одна молекула гидразина; при этом образуется
гидразон. Далее в реакцию вступают еще две моле-
кулы гидразина и получается О., напр.:
сно ch=n-nhc„h5
I I
н-с-он н-с-он
I I
НО-С-Н H2N-NHCeH5 НО-С-Н
Н-Ь-ОН -Н2О * Н-С-ОН
Н-С-ОН Н-Ь-ОН
сн2он <!;н2он
Образованием О. пользуются для характеристики са-
харов. Для О. каждого сахара характерна определен-
ная скорость образования, так О. фруктозы выде-
ляется из раствора за 2 мин; глюкозы — за 4—5 мин,
ксилозы — за 7 мин; арабинозы — за 10 мин; галак-
тозы — за 15—19 мин; рафинозы — за 60 мин. О. са-
харов имеют специфич. форму кристаллов.
Лит.: Шемякин М. М. [и др.], ДАН СССР, 1959,
128, № 3, 564; Simon Н., Kell К. D., Weygand F.,
Chem. Вег., 1962, 95, Л 1, 17; Percival Е. G. V., Ad-
vances Carbohyd. Chem., 1948, 3, 23. Л. И. Линевич.
ОЗОКЕРИТ—минерал из группы нефтяных биту--
мов, генетически связан с месторождениями парафи-
нистой нефти; встречается в виде жильных, пластовых
и смешанных жильно-пластовых месторождений. Со-
2H2N-NHC«Hs
-нао
держание его в песках и известняках от 4 до 16%.
О.— смесь высокомолекулярных твердых насыщен-
ных углеводородов, по виду напоминает пчелиный
воск (плотн. 0,85—1,0, т. пл. 52—85°), окрашен в жел-
тый или бурый цвет, жирный на ощупь, пахнет ке-
росином; элементарный состав О. 84—86%С, 13,5—
15% Н.
О. получают экстракцией из содержащей его руды
тяжелым бензином (т. кип. 100—200°) под давлением
1—3,5 ат. После отгонки растворителя получают
сырец, к-рый расплавляют перегретым паром, фильт-
руют от механич. примесей и отгоняют легкие фрак-
ции под вакуумом при темп-ре до 300°. Полученный
т. обр. стандартный О. применяют как товарный про-
дукт или перерабатывают в церезин, для чего
стандартный О. обрабатывают 95—98%-ной H2SO4
при 220° с последующей нейтрализацией известью и
очисткой отбеливающей глиной. Церезин может быть
также получен из различных фракций нефти (напр.,
петролатума), из продуктов восстановления СО во-
дородом по Фишеру — Тропшу; заменители церезина
можно получить из битумов твердого топлива (напр.,
гидрированием т. наз. горного или монтанного воска).
Церезин выпускают след, марок: 80, 75, 67, 57 (белый)
и 57 (желтый). Индексы марок соответствуют темп-ре
каплеиадения, к-рую определяют как темп-ру падении
первой капли испытуемого продукта при расплавле-
нии его в строго определенных условиях. Глубина
проникновения иглы под определенной нагрузкой
при 25° для церезинов лежит в пределах 16—30. Син-
тетич. церезин выпускается четырех марок: конден-
саторный, 100, 93 и 90 (см. также ст. Парафин).
Церезин — хороший диэлектрик; обладает способ-
ностью прочно удерживать различные масла; приме-
няется в качестве электроизоляционного материала,
для изготовления пропиточных и протектирующих
составов, для приготовления различных смазок и ма-
зей для технич. и медицинских нужд, для пропитки
бумаги, при приготовлении лаков, политур и др.
Основные промышленные месторождения О. в СССР
имеются в районе г. Борислава (УССР), на полуост-
рове Челекен (Туркмен. ССР), в Фергане (Узбек. ССР).
Лит.: Барановский Н. Ф., Сухарев М. Ф.,
Озокерит, М., 1959. В. В. Щекин.
ОЗОН — простое вещество, аллотропия, видоизме-
нение кислорода. В отличие от двухатомной молеку-
лы обычного кислорода (Ог) молекула О. трехатомна
(Оз). При обычных условиях О.— резко пахнущий
взрывчатый газ синего цвета, обладающий силь-
г, „ тг ным окислительным дейст-
СН—N-NHCaH3 вием.
C=N-NCeH5 Впервые О. был обнару-
I жен в 1785 Ван-Марумом
но-с-н +CsH5NH+NH1 по характерному запаху и
Н-С-ОН окислительным свойствам,
I к-рые приобретает воздух
н-с-он после пропускания через
СНгОН него электрич. искр. Ван-
Марум приписал эти свой-
ства «электрической материи». В 1840 Шёнбейн со-
поставил изменение свойств кислорода при пропуска-
нии через электрич. разряд и при электролитич.
выделении и объяснил эти изменения образованием
особого газа, к-рый он назвал «О.» (от греч. б£ш —
пахну). Позже Мариньяк и де ля Рив показали, что О.
является видоизменением кислорода.
Физические и химические свойства. Наиболее ве-
роятным считается строение молекулы О. в виде рав-
нобедренного треугольника ос двумя равной длины
связями О—0 1,278±0,003 А и углом между ними
116°49'±30'. Моменты инерции (г-см2): /а=7,8749-
.10-«°; 7в=62,844-107е= 70,888• 10Диполь-
ный момент 0,53 ±0,02 D. При нормальных условиях
655
ОЗОН
656
О.— газ синего цвета (синий цвет становится заметным
при 15—20 об.%). Жидкий О.—темно-синяя жидкость.
Твердый О.— темно-фиолетовые призматич. кристал-
лы. Т. пл.— 192,7±0,2° (приводимая до последнего
времени во многих справочниках т. пл. —251° оши-
бочна, т. к. при ее определении не учитывалась боль-
шая способность О. к переохлаждению. Т. о., свойства
жидкого О., определенные при темп-рах ниже —193°,
относятся к переохлажденному состоянию). Т. кип.
—111,9°; £крит.—12°; Ркрит. 54,6 атм} t/крит. 0,437
г/мл.. Постоянные в уравнении Ван-дер-Ваальса: а=
= 3,545 л/моль; 6=0,4903 атм-лг/моль2. Плотность га-
за 2,144 г/л (при 20° и 1 атм)} жидкости (г/мл): 1,46
(—112°), 1,571 (—183°), 1,614 (—196°). Плотность
твердого О. (как и другие его свойства) не определена.
Теплоемкость Ср (кал/моль-град): 7,95 (—173°), 9,10
(0°), 9,37 (25°), 10,44 (127°). Теплота испарения
75,6 кал/г (—112°). Стандартная теплота образования
ЛЯ°298 = 4-34,5±0,2 ккал/моль. Давление пара О.
в интервале от —183° до —30° выражается уравне-
нием: 1g Р (мм рт. ст.) = 8,25313 — 0,001966943 Т—
—814, 941587/7’. Вязкость (спуаз): 1,55±0,02 (—183°),
4,20 ±0,01 (—195°). Поверхностное натяжение
38,4±0,7 дин/см (—183°). Уд. магнитная восприим-
чивость (CGSM): газ 0,002-10-в; жидкость 0,15-10-в.
Нормальный потенциал по отношению к стандартно-
му водородному электроду — 2,07 в (25°).
Выше —180 ±0,5° жидкий О. смешивается с жидким
кислородом во всех отношениях. Ниже этой темп-ры
происходит расслоение на две фазы. Состав фаз (в вес.
% О.): 72,4—29,8 (при —183°), 90,8—9,0 (при — 195,4°).
Плотность жидких смесей О. с кислородом аддитивна
в отношении состава. О. растворяется в воде, образуя
нестабильные р-ры, лучше, чем кислород. Раствори-
мость О. сильно зависит от примесей (в газе и жидко-
сти), могущих ускорить разложение О. Даже в чистой
воде О. разлагается быстрее, чем в газовой фазе.
Растворимость О. в воде, в пересчете на 100% О.
при 1 атм:
°C 0 10 20 30 40 50 60
г/л 1,09 0,78 0,57 0,40 0,27 0,19 0,14
Теплота растворения 3,9 ккал/молъ. В водных р-рах
солей О. растворяется слабее, чем в воде, хотя ско-
рость разложения О. увеличивается. Растворимость
О. в СС14 примерно в 5 раз выше, чем в воде. О. раство-
ряется также в уксусной и пропионовой к-тах, их
ангидридах и хлоропроизводных и др. О. хорошо
адсорбируется силикагелем и алюмогелем, причем ад-
сорбированный О. намного стабильнее газообразного
или жидкого О. Это позволяет использовать адсорбцию
О. для его извлечения из р-ров или газовых смесей,
а также для безопасного обращения с концентрирован-
ным О.
Химич, свойства О. характеризуются двумя основ-
ными чертами: нестойкостью и сильным окислитель-
ным действием. При небольших концентрациях (без
посторонних примесей) при нормальных условиях О.
разлагается довольно медленно. При повышении
темп-ры до 100—150° скорость разложения О. значи-
тельно возрастает. Механизм распада О., в к-ром
участвуют гомогенные и гетерогенные процессы, сло-
жен и изменяется с изменением внешних условий.
Разложение О. ускоряется в гомогенных системах га-
зообразными добавками (NO, С12 и др.), а в гетероген-
ных системах металлами (Pt и др.) и окислами металлов
(Ag, Си, Fe, Ni и др.).
При больших концентрациях разложение О. про-
исходит со взрывом. Взрываемость О. в сильной сте-
пени зависит от примесей, особенно органич. проис-
хождения. Взрывные свойства чистых газообразных
смесей О. с кислородом по отношению к локальным им-
пульсам можно разбить на три области: при концент-
рациях меньше 20 вес. % разложение О. происхо-
дит лишь в месте локального импульса и не распро-
страняется на весь объем; при 20—48 вес. % разложе-
ние идет по всему объему, однако происходит лишь
слабый взрыв, к-рый можно проводить в стеклянных
сосудах, что используется для анализа О.; выше
48 вес.% происходит мощный взрыв, переходящий
в детонацию. При мощных подрывах могут детониро-
вать и более разбавленные смеси О. В жидком и твер-
дом состояниях О. также является инициирующим
взрывчатым веществом, причем примеси повышают
его взрывную чувствительность.
О.— один из наиболее сильных окислителей. Он
окисляет все металлы (за исключением Аи и группы
Pt), а также большинство других элементов. Разла-
гает галогеноводороды (кроме HF) и нек-рые их соли;
переводит низшие окислы в высшие и т. п. Однако
в реакции с Н2О2 выступает как восстановитель
(Н2О2-±Оз= Н2О-±2О2). При действии на нек-рые неор-
ганич. и органич. соединения О. образует озониды (см.
Перекиси и перекисные соединения).
О. очень токсичен. Смертельная доза для мышей
(JID5o после 4-часового пребывания) соответствует
содержанию О. всего в 4 -10~4 %, что примерно в 12 раз
меньше соответствующей величины для фосфина. Ха-
рактерной чертой токсичности О. является способ-
ность животных «привыкать» к О., выработка своеоб-
разного иммунитета. Так, после 4—6 часов пребыва-
ния в атмосфере, содержащей 10 ~4 % О., мыши приоб-
ретают способность в течение 4—6 недель переносить
дозы О., во много раз больше смертельных. У людей
длительное пребывание в атмосфере, содержащей
ок. 10-5% О., может вызвать головные боли, а также
раздражение дыхательных путей и глаз. Концентра-
ция О. в 10~5 % может считаться предельно допусти-
мой. При этой концентрации О. обладает резким разд-
ражающим запахом. В пределе запах О. различается
при его содержании в несколько миллионных долей
%, т. е. много ниже допустимой нормы, что снижает,
но не устраняет опасность отравления при работе с О.
Атмосферный О. В приземной атмосфере концентра-
ция О. очень мала (порядка 10“’—10~6%). Однако
с увеличением высоты она возрастает, проходя через
максимум на высоте 20—30 км. Общее содержание О.
в атмосфере можно характеризовать слоем О., приве-
денным к нормальным условиям (0°С,1 атм). Этот
слой достигает величины 0,4—0,6 см. Содержание О.
в атмосфере зависит от география, широты и времени
года. Как правило, оно больше в высоких широтах,
максимально весной и минимально осенью. Атмосфер-
ный О. играет большую роль в обеспечении условий
жизни на земле. О. задерживает вредное для жизни
ультрафиолетовое излучение солнца. С другой сто-
роны, он поглощает инфракрасное излучение земли,
препятствуя ее охлаждению. Содержание и переме-
щение О. в атмосфере влияет также на метеорология,
обстановку.
Получение. О. образуется в процессах, сопровож-
дающихся выделением атомарного кислорода, напр.
при разложении перекисей, окислении фосфора, элект-
ролизе кислородсодержащих кислот и др. О. обра-
зуется также при действии, на кислород коротковол-
новых излучений (Х<2000А) и потоков быстрых ча-
стиц (электронов, протонов и др.). В промышленности
О. получают при электрич. разряде в озонато-
рах. Разрядный промежуток озонатора ограничен
одним или двумя диэлектрич. электродами, к-рые ста-
билизуют ток разряда и обеспечивают равномерное
распределение разряда по поверхности электродов.
Озонатор питается переменным током, причем мощ-
657
ОЗОНИДЫ—ОКИСЛЕНИЕ БИОЛОГИЧЕСКОЕ
658
ность озонатора пропорциональна частоте тока. Ос-
новным фактором, определяющим работу озонатора,
является отношение активной мощности озонатора к
расходу газа (u/v). При одинаковых u/v получаются
равные концентрации О. При повышении u/v концент-
рация О. сначала растет, а затем достигает стационар-
ного значения, зависящего от величины разрядного
промежутка и темп-ры электродов. Как правило, с ро-
стом концентрации получающегося О. энергетич. вы-
ход О. и производительность озонатора падают. Для
эффективной работы озонатора требуется тщательная
осушка исходного газа (точка росы ок. —50°). При син-
тезе О. из воздуха выход О. в 2—3 раза меньше, чем
при синтезе из кислорода. Энергетич. затраты на син-
тез О. в современных озонаторах составляют 10—
15 кеч на 1 кг О.
Применение О. обусловлено его окислительным,
а также дезинфицирующим и бактериоцидным дейст-
вием. Наиболее широко О. применяется для очистки
питьевой воды. Озонированная вода обладает хоро-
шими гигиенич. и вкусовыми качествами. Возможно
также использование О, для доочистки сточных про-
мышленных вод. В пищевой пром-сти О. применяется
как бактериоцидное средство на холодильниках, кон-
сервных, пивоваренных и др. заводах. Как окислитель
О. используется для отбеливания бумаги, соломы,
масел и др., а также в производстве нек-рых органич.
химич. продуктов (ванилин, камфара, жирные кис-
лоты и др.). В очень небольших концентрациях О.
применяется для дезодорации воздуха.
Лит. см. при ст. Кислород, а также: Прокофьева
И. А., Атмосферный озон, М.— Л., 1951; Кожинов В. Ф.,
Озонирование питьевой воды, М., 1961; Филиппов Ю. В.,
Электросинтез озона, Вести. МГУ. Сер. математики..., 1959,
№ 4, с. 153; Т h о г р С. Е., Bibliography of ozone technology,
v. 1—2, Chicago, 1954—55; Ozone chemistry and technology.
Advances in chemistry, ser. 21, Washington, 1959 (Amer. Chem.
Soc.). Ю. В. Филиппов.
ОЗОНИДЫ — см. Перекиси и перекисные соединения,
ОКИСЛЕНИЕ БИОЛОГИЧЕСКОЕ — совокупность
окислительно-восстановительных реакций в живых
организмах, катализируемых ферментами оксидоредук-
тазами. Сущность окислительно-восстановительных
реакций сводится к переносу водорода или электронов
от окисляемого вещества — донора — на восстанав-
ливаемое вещество — акцептор.
Учение об О. б. развивалось на основе двух раз-
личных концепций — теории активирования кисло-
рода при О. б. и теории активирования водорода, т. е.
окисления путем дегидрирования. Первая гипотеза
была развита А. Н. Бахом, предложившим «перекис-
ную теорию окисления» (1897), а также О. Варбургом,
развившим представление о роли связанного с белками
железа в активировании кислорода (1927). Вторая
теория о роли активирования и переноса водорода
в реакциях О. б. была обоснована В. И. Палладиным
(1908), а затем Г. Виландом (1912). В процессе даль-
нейшего развития обе эти гипотезы слились в единую
теорию О. б., включающую как активирование водо-
рода, так и кислорода.
В реакциях О. б. принимают участие низкомолеку-
лярные и высокомолекулярные переносчики, способ-
ные к обратимому окислению и восстановлению. Эти
переносчики сами по себе не являются оксидоредук-
тазами, но в одних случаях они входят в состав окси-
доредуктаз в качестве их простетич. групп, способных
к диссоциации в большей или меньшей степени, в дру-
гих случаях играют самостоятельную роль. Для О. б.
характерно,'что весьма ограниченный набор переносчи-
ков осуществляет перенос водорода или электронов
от множества метаболитов разного строения. К числу
важнейших переносчиков О. б. относятся никотин-
амидадениндинуклеотиды, флавиновые нуклеотиды,
хиноны, железосодержащие белки — цитохромы, свя-
занная с белками медь и тиоловые соединения.
Никотинамидадениндинуклеотиды — НАД, НАДФ
(см. также Кодегидрогеназы и Никотинамиднуклео-
тидные коферменты) являются первичными акцепто-
рами водорода при окислении большинства субстратов
в реакциях, катализируемых различными дегидроге-
назами. При этом НАД играет роль переносчика во-
дорода преим. при реакциях окисления субстратов,
тогда как НАДФ участвует в реакциях переноса во-
дорода, гл. обр. в реакциях восстановительного
синтеза.
Флавиновые нуклеотиды (см. Флавиновые кофермен-
ты) выполняют роль переносчиков водорода, входя
в состав флавопротеидов — дегидрогеназ и оксидаз.
Для ряда флавиновых ферментов доказано, что они
катализируют перенос водорода через промежуточное
образование семихиноидной формы флавина.
Активную роль в качестве переносчиков водорода
играют хиноны — бензохиноны и нафтохиноны. Из
них наиболее важным переносчиком является убихи-
нон (УХ), или кофермент Q, представляющий 2,3-ди-
метокси-5-метил-6-алкилбензохинон. Убихиноны ши-
роко распространены в природе.
К железосодержащим белкам принадлежат гемо-
протеиды — цитохромы, осуществляющие перенос
электронов путем обратимого изменения валентности
железа, входящего в состав гема.
Ряд оксидоредуктаз содержит в качестве просте-
тич. группы медь—полифенолоксидазы, аскорбатокси-
даза, цитохромоксидаза и др. Последняя содержит
медь в простетич. группе наряду с цитохромом типа а.
Изменение валентности меди.при окислительно-вос-
становительной реакции установлено для нек-рых мед-
ных протеидов методом электронно-парамагнитного
резонанса, напр. для полифенолоксидазы и цитохром-
оксидазы.
Важными окислительно-восстановительными пере-
носчиками у живых организмов являются также тио-
ловые соединения, перенос водорода к-рыми осуществ-
ляется по реакции
— 2Н
2R-SH Z^±R-S-S-R
+ 2Н
К тиоловым соединениям относятся цистеин, глюта-
тион, липоевая кислота, кофермент А, играющие
весьма ответственные роли в О. б.
О. б. играет огромную роль в жизнедеятельности
организмов. На первом месте стоит энергетич. роль.
О. б. является у живых организмов источником энер-
гии, к-рая аккумулируется с образованием макроэр-
гич. соединений. Не менее важной функцией О. б.
является образование разнообразных метаболитов —
субстратов, коферментов, гормонов, запасных и дру-
гих веществ, необходимых организму для осуществле-
ния многочисленных реакций обмена веществ, пост-
роения клеточных структур, процессов роста и т. и.
Важным назначением О. б. является устранение
токсич. продуктов обмена или проникающих извне
ядов путем их окисления.
Большинство живых организмов, существующих в
аэробных условиях, получают необходимую энергию
за счет процесса дыхания, в результате к-рого пита-
тельные вещества в клетках окисляются до воды, дву-
окиси углерода и аммиака. В первой стадии, идущей
с освобождением не более 1% общей энергии, белки,
сложные углеводы, жиры подвергаются гидролизу,
превращаясь, соответственно, в аминокислоты, про-
стые сахара, жирные к-ты и глицерин. Часть полу-
ченных продуктов организм использует как строи-
тельный или запасной материал, другая же часть под-
вергается во второй фазе превращений неполному
окислению, образуя, кроме воды, углекислого газа и
аммиака, три основных продукта: ацетил-кофермент А
(ацетил-КоА), а-кетоглутаровую к-ту и щавелево-
659
ОКИСЛЕНИЕ БИОЛОГИЧЕСКОЕ
660
уксусную к-ту. На этой стадии О. б. выделяется ок.
общей энергии сгорания питательных веществ,
причем эта часть энергии не превращается в энергию
макроэргич. связей.
В третьей фазе происходит полное окисление ве-
ществ, образовавшихся во 2-й фазе, до Н2О и СО2.
Это окисление осуществляется в клетках по схеме
цикла трикарбоновых кислот. В результате этого
сложного цикла, состоящего из 10 отдельных фермен-
тативных реакций, молекула уксусной к-ты в форме
ацетил-КоА окисляется до Н2О и СО2. Окисление ор-
ганич. к-т — компонентов цикла — происходит пу-
тем отщепления водорода, к-рый далее окисляется
кислородом до воды. Углекислый газ — второй про-
дукт реакции — не образуется при О. б. непосредст-
венным присоединением кислорода к углероду, как
это указывал уже В. И. Палладии, но возникает лишь
при реакциях декарбоксилирования из атомов угле-
рода, окисленных до карбоксила в результате чередо-
вания процессов дегидрирования с реакциями при-
соединения воды. Поэтому кислород углекислого газа
возникает непосредственно не из поглощаемого при
дыхании кислорода, а из воды.
В цикле освобождаются остальные 2/3 общей энер-
гии, заключенной в питательных веществах. Освобож-
дение энергии происходит, однако, не в самом цикле,
а при переносе на кислород обезличенных атомов во-
дорода (электронов), отщепившихся от субстратов
цикла трикарбоновых к-т. Этот процесс осуществляет-
ся в клетке через ряд переносчиков, составляющих
«дыхательную цепь». Сложность процесса переноса
электронов требует его пространственной организа-
ции. Поэтому дыхательная цепь неразрывно связана
с белково-липидными структурными элементами клет-
ки и локализована в клеточных тельцах, наз. мито-
хондриями. Последние представляют собой ча-
стицы длиной 3—4 мк и диаметром ок. 1 мк. Их обо-
лочка состоит из двухслойной липопротеидной мем-
браны диаметром ок. 180 А. Внутренняя мембрана
образует в митохондрии многочисленные гребни,
или кристы, окруженные жидкостью, заполняющей
митохондрию. Дыхательные цепи находятся в мито-
хондриальных мембранах, а ферменты цикла трикар-
боновых квслот и некоторые другие содержатся в
жидкости, окружающей кристы. Переносчики элек-
тронов расположены в дыхательной цепи в такой
последовательности, что их нормальные редокспо-
тенциалы возрастают от отрицательных величин до
значений, приближающихся к потенциалу нормаль-
ного кислородного электрода. В таблице приведены
значения нормальных редокспотенциалов (EJ для
некоторых субстратов и компонентов дыхательной
цепи.
Реакция
Ео при pH 7, в
а-Кетоглутарат+Н2О 7—* сукцинат + СО2 +
+ 2Н + + 2е —0,67
Н2 ;=± 2Н+ + 2е . .. . —0.42
НАД-Н. TZA НАД+ 2Н++2е — 0,32
Лактат tlz? пируват + 2Н+ + 2е —0,19
Флавопротеин-Н2^г± флавопротеин + 2Н+ + 2е — 0.07
Сукцинат фумарат + 2Н + + 2е + 0,03
УХ-Н2 jzt УХ + 2Н + + 2е + 0,12
Цитохром b (Fe2 + 7—* Fe3 + +е) ....... + 0,08
Цитохроме (и cj (Fe2 + Fe3 + +е) + 0,25
Цитохром a (Fe2 + Т"*’ Fe3 + +e) + 0,29
Н2О ^2.‘/2 О2 + 2Н+ + 2е + 0,815
Перенос водорода и электронов по дыхательной
цепи показан на схеме 1.
Схема 1. S — субстрат; фа.-пр.— флавопротеин;
е — электрон.
Первым переносчиком водорода от различных суб-
стратов цикла трикарбоновых к-т через посредство
специфич. дегидрогеназ является НАД. С НАД водо-
род переносится на флавиновые ферменты, в состав
простетич. группы к-рых, кроме флавина, входит
также железо, не относящееся к гемопротеидам; вслед
за флавинами водород (электроны) акцептирует, по-
видимому альтернативно, цитохром Ь или убихинон;
с них электроны переносятся последовательно на
цитохром с, и цитохром с. Последним звеном классич.
дыхательной цепи является цитохром а, или цитохром-
оксидаза, к-рая переносит электроны непосредствен-
но на кислород. Перенос электронов по дыхательной
цепи при переходе от двухэлектронных переносчи-
ков, к к-рым относятся НА Д, флавопротеины и убихи-
нон, к одноэлектронным переносчикам — цитохро-
мам — происходит, по-видимому, через образование
семихинонов при окислительно-восстановительной
реакции флавопротеинов и убихинона. При переносе
электронов с флавопротеинов (или убихинона) на
цитохромы протоны уходят в раствор, образуя на
кислородном конце цепи воду с ионом О2-. Ответвле-
ние дыхательной цепи составляет цепь, окисляющая
сукцинат. Последний, согласно значению своего нор-
мального редокспотенциала, окисляется не через
НАД, но непосредственно через специфич. флаво-
протеин.
Работами лаборатории Д. Грина удалось полностью
восстановить дыхательную цепь митохондрий из от-
дельных ферментных комплексов.
Реконструированная дыхательная цепь представ-
лена на схеме 2.
Схема 2. УХ — убихинон; фл.-пр.— флавопротеин; цит.—
цитохром; FeHr — негемовое железо.
Она состоит из четырех нерастворимых фермента-
тивных комплексов и двух растворимых компонен-
тов — убихинона и цитохрома с. Комплекс I состо-
ит из специфич. флавопротеина и негемового железа;
он переносит водород сНАД-Н2 на убихинон; ком-
плекс II содержит другой флавопротеин, перенося-
щий водород с сукцината на убихинон, негемовое же-
лезо и цитохром Ь; комплекс III содержит цитохромы
Ь и Cj и негемовое железо; он переносит электроны с
убихинона на цитохром с; комплекс IV является ци-
тохромоксидазой. Убихинон и цитохром с связывают
между собой нерастворимые комплексы.
В точках наибольших перепадов редокспотенциалов
в дыхательной цепи прохождение водорода (электро-
нов) сопряжено с образованием макроэргич. фосфата
АТФ (см. Макроэргические связи) в процессе наз
окислительным фосфорилирова-
нием. Этот процесс является основным источником
661
ОКИСЛЕНИЕ БИОЛОГИЧЕСКОЕ
662
образования макроэргич. фосфата АТФ (~Р) для
всех живых организмов, за исключением фотосинте-
зирующих. Для субстратов, окисление к-рых проис-
ходит через НАД, перенос водорода сопряжен с фос-
форилированием в трех точках дыхательной цепи —
между НАД и флавопротеином,между цитохромами Ь
и с и в области цитохромоксидазы. Баланс реакций
окислительного фосфорилирования выражается
уравнением:
АН2 + В + Рн + АДФ А + ВН2 + АТФ
где А и В — соседние переносчики в дыхательной
цепи, а Рн— неорганич. фосфат. Действительный
механизм окислительного фосфорилирования гораздо
более сложен и, по-видимому, включает образование
нескольких промежуточных макроэргов. Заключи-
тельной стадией реакции окислительного фосфорили-
рования является перенос ~ Р с фосфорилированного
макроэрга неизвестной природы на АДФ с образова-
нием АТФ. На схеме 3 представлена дыхательная
цепь и сопряженное с дыханием окислительное фос-
форилирование.
Сукцинат-----— фл. — пр.
малат
цитрат 2Н
пируват -—*-НАД-«-(рл.—пр
—► цит. с —► цит. а —►
О2
ЦИТ- с
н Лр-
субстраты.
АТФ АТФ
ри
<р
АДФ
АТФ
Схема 3. фл.-пр.— флавопротеин; цит.— цитохром; А,, А2,
А3 — переносчики дыхательной цепи, образующие первич-
ные макроэрги (А, — х,, А2 — х2, А3 ~ .х3) о соединения-
ми xt, х2, х3; xi~P, х2~Р, х., — Р — фосфорилированные
макроэрги; УХ —убихинон; Рн—неорганич. фосфат.
Перенос водорода по цепи тормозится дыхательными
ядами. Антибиотик антимицин А действует между ци-
тохромами Ь и ci, цианид угнетает цитохромоксидазу.
Другие ингибиторы, т. наз. «разобщающие яды», не
тормозят дыхание в малых концентрациях, но подав-
ляют фосфорилирование. К ним относятся динитро-
фенол, грамицидин и др. Третий тип ингибиторов,
представителем которых является антибиотик олиго-
мицин, тормозит окислительное фосфорилирование
и сопряженное с ним дыхание, не влияя на дыха-
ние, не связанное с окислительным фосфорилиро-
ванием.
Превращение энергии окислительно-восстанови-
тельных реакций в энергию макроэргич. связей
АТФ при О. б. происходит не только при аэроб-
ных процессах окислительного фосфорилирования,
но и при анаэробных реакциях гликолиза или бро-
жения, к-рые имеют место в случае недостаточного
поступления кислорода. Однако эти способы образо-
вания АТФ с точки зрения расходования организмом
питательных веществ являются значительно менее
экономными, чем процесс окислительного фосфорили-
рования.
Ниже представлен механизм образования АТФ при
гликолитич. оксидоредукции:
1. R R
I I /ОН
С = О+ 8Н-фермент С:
I I XS-фермент
Н Н
где R: СН2ОРО,Н2
I
снон
I
I /ОН I
С( +НАДЩ±С=О + НАД-Н2
I S-фермент $
Н S-фермент
ацилмеркаптан
3. R R
I I
С=О + H.P0,^z± 0 = 0 +Н8-фермент
> (
S-фермент О—РО2Н2
1,3-дифосфоглпцеринов. к-та
4. R R
I I
0 = 0 + АДФ ЦгАСООН + АТФ
0-Р0,Н2
5. Н АД-Н2 +пиру ват НАД + лактат
В реакции 1, 3-фосфоглицериновый альдегид обра-
зует комплекс с специфич. дегидрогеназой. В реакции
2 комплекс окисляется с образованием макроэрга ацил-
меркаптана, водород переносится на НАД. В реакции
3 ацилмеркаптан подвергается фосфоролизу с образо-
ванием 1,3-дифосфоглицериновой к-ты. В реакции
4 происходит перенос~Р на АДФ с образованием АТФ.
В реакции 5 регенерируется НАД за счет восстановле-
ния пирувата, образующегося на более поздней ста-
дии гликолиза.
Необходимо подчеркнуть коренное различие в про-
странственной организации реакций О. б., связанных
с переносом электронов по дыхательной цепи, от тако-
вых при анаэробных оксидоредукциях. Первые обя-
зательно связаны со структурами, тогда как вторые
протекают в растворимой части протоплазмы — гиа-
лоплазме.
О. б. имеет важное значение в образовании различ-
ных метаболитов. Косвенным путем О. б. является
движущей силой всех реакций биосинтеза, в к-рых
принимает участие АТФ, поскольку АТФ образуется
за счет энергии реакций О. б. почти во всех случа-
ях, за исключением фотосинтезирующих организмов.
В качестве примера прямого участия реакций О. б. в
синтезе метаболитов может быть рассмотрен синтез
и окисление жирных к-т в организме, к-рый происхо-
дит с образованием ацилпроизводных, представляю-
щих тиоэфиры кофермента А (КоА).
В митохондриях имеется ферментная система, ката-
лизирующая окисление жирных к-т путем последо-
вательного отщепления двух углеродных остатков
в виде ацетил-КоА.
Суммарно такое превращение выражается уравне-
нием:
Сп+з-ацил-КоА + НАД+ фл.-пр.+ КоА дД: Сп-ацпл-КоА +
+Н АД • Н2 + фл.-пр. -Н2 + ацетил-КоА
Реакция является обратимой. За счет обратной реак-
ции в митохондриях осуществляется, напр., синтез
стеариновой к-ты из пальмитиновой. Иначе идет син-
тез жирных кислот в растворимой части клетки —
гиалоплазме. В этой системе пальмитиновая к-та син-
тезируется при участии ацетил-КоА, малонил-КоА
и НАДФ -Нг по ур-нию:
О
II
СН,-СО-SKoA+ 7Н00С-СН2-С-SKoA <- 14НАДФ-Н, —►
О
II
—> СН3-(СН2),4-С-S -КоА+ 14НАДФ + 7СО- + 7КоА + 7На0
пальмитил-КоА
Это типичная реакция восстановительного синтеза,
в к-рой восстановительные эквиваленты поставляет
НАДФ.
Другой пример можно привести из области обмена
углеводов. Помимо основного пути окисления глюкозы
расщеплением на две молекулы триозы с последую-
щим окислением образующихся продуктов,у многих
663 ОКИСЛЕНИЕ БИОЛОГИЧЕСКОЕ— ОКИСЛЕНИЕ НЕФТЕПРОДУКТОВ 664
организмов имеется другой путь окисления глюкозы,
т. наз. пентозофосфатный цикл (см.
Пентозный цикл), в к-ром превращение глюкозы идет
через образование пентоз. Этот сложный путь окисле-
ния глюкозы через глюкозо-6-фосфат выражается сум-
марным ур-нием:
глюкозо-6-фосфат + 6НАДФ + ЗН2О —>
—> фосфоглпцериновый альдегид + ЗСО2 + 6 НАДФ Н2
Функция цикла — не энергетическая, т. к. он не свя-
зан с образованием ~Р. Одной из основных его функ-
ций является образование пентозофосфатов, являю-
щихся промежуточными продуктами цикла, к-рые не-
обходимы для синтеза нуклеиновых кислот и нуклео-
тидов.
Образование пространственных структур молекул
белка также связано с реакциями О. б. В инсулине,
напр., две полипептидные цепи соединены между
собой двумя дисульфидными мостиками, образован-
ными SH-группамп цистеина, входящего в обе цепи
инсулина. Дисульфидные мостики, получившиеся в ре-
зультате окисления SH-групп, тем самым определяют
третичную конфигурацию молекулы инсулина, как и
других белков.
Важной функцией реакций О. б. является устране-
ние вредных продуктов обмена путем их окисления.
Можно привести ряд примеров этой функции О. б.
Так, при окислении ряда субстратов кислородом воз-
духа при участии флавопротеиновых оксидаз (ксан-
тинокспдазы, глюкозооксидазы, оксидазы аминокис-
лот ит. д.) продуктом реакции является токсич. про-
дукт — Н2Ог : R+H2O+O2=RO+H2O2. Накопление
П8О2 предотвращается действием фермента каталазы,
расщепляющей Н2О2 по уравнению: 2Н2О2—г2Н2О-|-
+ О2. В значительной мере защитную роль выполняют
ферменты оксидазы и гидроксилазы. Так, накопление
в организме мощного фармакология, агента гистами-
на, образующегося при декарбоксилировании гисти-
дина, предотвращается действием аминооксидазы, оки-
сляющей гистамин в альдегид.
Особенностью реакций О. б. в растениях является
широкое распространение оксидоредуктаз полифенол-
оксидаз и пероксидаз, окисляющих полифенолы, пи-
рокатехин, флороглюцин, пирогаллол и другие сое-
динения, относящиеся к растительным веществам
вторичного происхождения.
Большое разнообразие реакций О. б. по сравнению с
животными тканями наблюдается у микроорганизмов.
Так, среди микробов имеются представители как аэро-
бов (азотобактер), так и анаэробов (Clostridium),
способных превращать молекулярный атмосферный
азот в белковый азот в случае недостатка в окружаю-
щей среде аммониевого азота или азота нитратов. Этот
процесс имеет большое значение для обогащения почвы
легко усваиваемыми формами азота. Восстановление
молекулярного азота достигается сопряжением этой
реакции с реакциями анаэробной оксидоредукцип
промежуточных продуктов обмена углеводов таких,
как пируват. Установлено, что первым продуктом
фиксации азота является аммиак, однако механизм
этого сложного процесса до настоящего времени еще
не выяснен.
Обратной реакцией по отношению к фиксации азота
является реакция денитрпфикацииу микроорганизмов,
представляющая собой восстановление нитратов до
молекулярного азота. Эта реакция используется мик-
роорганизмами для получения энергии в анаэробных
условиях за счет переноса электронов от окисляю-
щихся субстратов, как лактат, по дыхательной цепи,
в принципе сходной с дыхательной цепью митохондрий,
конечным акцептором электронов в к-рой является
не кислород, а нитрат. Прохождение электронов поцепи
при этом сопровождается, как и в случае реак-
ций окислительного фосфорилирования, образованием
АТФ.
Лит.: Грин Д., Труды V Международного Био-
химического конгресса, том «Пленарные заседания», М.,
1962; то же, Симпозиум V, М., 1962; Кребс Г. А., Корн-
берг Г. Л., Превращения энергии в живых системах, пер.
с англ., М., 1959; Классификация и номенклатура ферментов,
пер. с англ., М., 1962; Михлин Д. М., Биохимия клеточ-
ного дыхания, М., 1960; Котельникова А. В., Усп.
биол. химии, 1962, 4, 173; Скулачев В. П., Соотношение
окисления и фосфорилирования в дыхательной цепи, М.,
1962; Северин С. Е., в кн.: Химические основы процессов
жизнедеятельности, М., 1962; Н а у а 1 s h 1 О., Annual Rev.
Biochem., 1962, 31, 25; М а з s е у V., V е е g е г С., там же,
1963, 32, 579. А. /1, Котельникова.
ОКИСЛЕНИЕ НЕФТЕПРОДУКТОВ — процесс
окисления нефтепродуктов молекулярным кислоро-
дом (автоокисление). При окислении нефтепродуктов,
представляющих собой в основном смесь углеводоро-
дов различного строения, образуются различные кис-
лородсодержащие соединения. В одних случаях эти сое-
динения являются целевыми продуктами, исполь-
зуемыми в различных областях народного хозяйства
(см., напр., Высшие жирные кислоты), в других — на-
копление кислородсодержащих соединений в нефте-
продуктах влечет за собой резкое ухудшение их эк-
сплуатационных свойств.
Механизм окисления углеводородов молекулярным
кислородом раскрыт А. Н. Бахом и Энглером, а
затем развит и дополнен Н. Н. Семеновым (цепной
механизм окисления). В соответствии с современными
взглядами автоокисление протекает по цепному ме-
ханизму: окислительная цепь развивается благодаря
накоплению в реакционной среде свободных атомов
пли радикалов, образующихся за счет термич. (или
любой иной) активации исходного углеводорода (ос-
новная цепь) или за счет распада образовавшихся
промежуточных продуктов — перекисей (вырожден-
ное разветвление). Первичным продуктом автоокис-
ления углеводородов являются перекиси (гидропере-
киси) .
В общем виде окисление углеводородов молекуляр-
ным кислородом может быть выражено след, схемой:
rh + O2 —>R +НО2
r + o2 —>ro2
ro2 + rh —»ROOH+R
И т. д.
Разложение перекисей идет по схеме:
ROOH —> RO + OH
RO + RH—»ROH + R
OH + RH —>H2O + R
Развитие реакционных цепей может прекратиться—
обрыв окислительной цепи — при непосредственном
взаимодействии радикалов, приводящем к образо-
ванию устойчивых димеров, и при любых других реак-
циях, дающих устойчивые соединения, в том числе
спирты, кислоты и т. п., не способные продолжить
окислительную цепь. Особенно интенсивно проис-
ходит «стабилизация» активных молекул (радикалов)
на стенке реакционного сосуда или в более общем
случае при соударении с к.-л. третьей частицей, спо-
собствующей отводу энергии от активных молекул.
Все вещества, способствующие образованию радика-
лов — носителей цепей окисления, являются ката-
лизаторами (ускорителями) окисления, а все вещества,
реагирующие с радикалами и приводящие пх в неак-
тивное состояние, будут тормозить окисление (инги-
биторы окисления, или антиокислители).
Одним из основных факторов, ускоряющих окисле-
ние, является темп-ра. При невысоких темп-рах, напр.
в условиях хранения, нефтепродукты (топлива, масла)
изменяются незначительно. При 50° и выше их окис-
ление достаточно заметно, особенно при длительном
665
ОКИСЛЕНИЕ НЕФТЕПРОДУКТОВ
666
соприкосновении нефтепродуктов с воздухом. Напр.,
в турбинах или трансформаторах нефтяные масла
редко нагреваются выше 45—50°, однако вследствие
длительности работы (1—2 года в турбине, 5—7 лет
в трансформаторе) в маслах происходят заметные из-
менения — накапливаются кислые продукты и оса-
док, и масла теряют необходимые эксплуатационные
качества. С увеличением темп-ры скорость окисления
резко возрастает. Так, при окислении масла до выде-
ления одного и того же количества осадка при 50“
требуется 10 200 час, а при 150°— всего 6 час. При
повышении темп-ры от 150° до 200° и выше процесс
окисления еще больше ускоряется, возрастает коли-
чество летучих продуктов окисления и продуктов
глубокой окислительной конденсации. В начальной
стадии процесса в окисляемом нефтепродукте ие наб-
людается видимых изменений — индукционный пе-
риод окисления; его продолжительность зависит от
химического состава нефтепродуктов, температу-
ры процесса, наличия катализирующих веществ и т. п.
По окончании индукционного периода взаимодей-
ствие нефтепродуктов с кислородом ускоряется, а
затем скорость реакции замедляется, что, по-види-
мому, связано с накоплением веществ, тормозящих
окисление.
Окисление нефтепродуктов ускоряется в присутст-
вии металлов и их солей. Особенно эффективно уско-
ряют окисление медь, свинец и их сплавы, менее
активны железо и марганец. Относительно слабо ка-
тализируют окисление цинк и алюминий. В присут-
ствии двух катализаторов (напр., железа и меди)
процесс окисления ускоряется в большей степени,
чем при использовании тех же катализаторов в от-
дельности. Это обстоятельство использовано в нек-рых
лабораторных методах искусственного старения неф-
тепродуктов (ГОСТ 981—55). Наиболее активно уско-
ряют окисление соли жирных и нафтеновых кислот
металлов с переменной валентностью (РЬ, Мп, Си).
Соли марганца используются, напр., при окислении
парафинов для получения жирных к-т. О. н. уско-
ряется также в присутствии газообразных инициато-
ров (НВг, СЬ, NO2); напр., в отсутствии инициато-
ров парафин при 127° не удавалось окислить даже
за 370 час, а после добавления к кислороду в течение
1 мин 10% NO2 окисление (до кислотного числа
70 мг КОН) проходит менее чем за 26 час. При воз-
действии у-излучений О. н. ускоряется. В этих усло-
виях происходит более интенсивное образование
свободных радикалов, что приводит к резкому ускоре-
нию развития окислительных цепей. Чем больше доза
облучения, тем интенсивнее О. н. Добавление инги-
биторов окисления не оказывает заметного влияния,
т. к. под действием ядерного излучения они теряют
свою активность. Скорость окисления в большой сте-
пени зависит от скорости диффузии кислорода в нефте-
продукт. Чем больше поверхность соприкосновения
нефтепродукта с кислородом, тем интенсивнее идет
процесс. Поэтому окисление, проводимое в условиях
продувания воздуха или кислорода через нефтепро-
дукт, всегда проходит быстрее, чем при пропускании
воздуха или кислорода над его поверхностью. Очень
резкое увеличение скорости окисления, вплоть до
взрыва, происходит при распылении нефтепродукта
в среде кислорода. Диффузия кислорода в окисляе-
мой среде увеличивается также и при повышении
давления. Окисление одного и того же нефтепро-
дукта в одном случае чистым кислородом, в дру-
гом — воздухом, но при увеличении в пять раз дав-
ления дает чрезвычайно близкие результаты как
по степени окисления, так и но составу продуктов
реакции.
Склонность нефтепродуктов к изменениям при воз-
действии кислорода воздуха в значительной мере
зависит от строения входящих в их состав компо-
нентов.
Ароматич. углеводороды, не имеющие боковых
цепей (бензол, нафталин, антрацен, фенантрен, дифе-
нил и т. п.), б. ч. пассивно относятся к воздействию
кислорода или воздуха. Только под воздействием та-
ких окислителей, как озон, пли при окислении в усло-
виях высоких темп-р и давлений в присутствии ката-
лизаторов эти соединения претерпевают те или иные
химич. превращения. Основные реакции при этом на-
правлены в сторону образования продуктов окисли-
тельной конденсации. Лишь незначительная часть
ароматич. углеводородов (при жестких условиях
окисления) окисляется с разрывом ароматич. ядра и
образованием кислых продуктов. Наиболее устойчи-
вы ароматич. углеводороды с конденсированными яд-
рами. Ароматич. углеводороды с боковыми парафино-
выми или изопарафиновыми цепями или имеющие
нафтеновые циклы окисляются значительно легче.
Чем больше боковых алифатпч. цепей у ароматич.
циклов, чем цепи длиннее и разветвленнее, чем больше
третичных атомов углерода в молекулах углеводо-
родов, тем они легче окисляются и тем больше обра-
зуют продуктов реакции. Наличие четвертичных ато-
мов углерода, особенно в конце боковой цепи, обыч-
но увеличивает стабильность углеводорода к окис-
лению.
Нафтеновые углеводороды легче окисляются, чем
ароматич.; боковые цепи уменьшают стойкость наф-
теновых углеводородов к окислению (как и в случае
ароматич. углеводородов). Окисление обычно сопро-
вождается разрывом полиметиленовых колец и обра-
зованием большого числа кислых продуктов. Основ-
ными продуктами окисления нафтеновых углеводоро-
дов являются кислоты и оксикислоты. При добавлении
ароматических углеводородов к нафтеновым послед-
ние увеличивают свою стабильность к окислению.
Наиболее эффективно тормозят окисление нафтенов
ароматич. углеводороды, лишенные боковых цепей
и способные при окислении образовывать фенолы.
Добавление к нафтеновым .углеводородам 5% нафта-
лина, антрацена или фенантрена снижает скорость
окисления нафтенов в несколько раз.
Парафиновые углеводороды по своей окисляемости
близки к нафтеновым. Окисляемость парафиновых
углеводородов возрастает с увеличением мол. веса.
Первичными продуктами окисления являются гидро-
перекиси, разложение к-рых приводит к образованию
соединений с меньшим содержанием углеродных ато-
мов в молекуле, нежели в исходном углеводороде.
В продуктах окисления парафиновых углеводородов
преобладают кислоты, спирты, соединения с карбо-
нильной группой и др. Образование продуктов окис-
лительной конденсации — смол, относительно неве-
лико. Изопарафиновые углеводороды, содержащие
третичные атомы углерода, окисляются легче, чем
парафиновые с нормальной цепью; наличие четвертич-
ных атомов углерода, как правило, увеличивает ус-
тойчивость углеводородов к окислению.
Наименее устойчивы при окислении молекулярным
кислородом непредельные углеводороды с активными
двойными связями. Легче всего окисляются диены
(особенно с сопряженными двойными связями) и аро-
матич. углеводороды с диеновой боковой цепью, не-
сколько хуже — ароматич. углеводороды с одной
двойной связью в боковой цепи. Ненасыщенные али-
циклич. соединения более склонны к реакциям окис-
ления, чем олефины. Окисление непредельных угле-
водородов обычно приводит к накоплению продуктов
уплотнения и окислительной конденсации. При окис-
лении непредельных углеводородов кислород присое-
диняется не по месту двойной связи, а в р-положение
к двойной связи; при этом в качестве первичных
667
ОКИСЛЕНИЕ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
668
продуктов окисления образуются соответствующие
гидроперекиси:
RCH = CHCH2R'-->RCH = CHCHR’
I
ООН
Чем больше непредельных углеводородов в нефтепро-
дуктах (обычно это продукты термич. крекинга), тем
больше склонность их к смоло- и осадкообразованию.
Для повышения стабильности топлив, содержащих
в своем составе продукты термич. крекинга, при-
меняют специальные антиокислительные присадки
(см. Ингибиторы). Содержание в нефтепродуктах
кислородных и сернистых компонентов б. ч. является
нежелательным, т. к. эти вещества ухудшают каче-
ство нефтепродуктов. Однако в случае, напр., мине-
ральных масел содержание небольшого количества
смол и нек-рых сернистых соединений может оказать-
ся полезным, т. к. эти продукты являются естествен-
ными ингибиторами в маслах и предохраняют их от
окисления.
Лит.: Черножуков Н. И., Крейн С. Э., Окис-
ляемость минеральных масел, 3 изд., М., 1 955; Черно-
жу ков И. И., Крейн С. Э., Л о с и к о в Б. В., Химия
минеральных масел, 2 изд., М., 1959; Иванов К. И.,
Промежуточные продукты и промежуточные реакции авто-
окисления углеводородов, М.— Л., 1949; Семенов Н. Н.,
О некоторых проблемах химической кинетики и реакционной
способности, М., 1954; Моторные и реактивные масла и
жидкости, под ред. К. К. Папок и Е. Г. Семенидо.М., 1964;
Заславский Ю. С., Радиационная стойкость смазочных
материалов, М., 1961; Сборник работ по радиационной химии
[отв. ред. Н. А. Бах], М., 1955. С. Э. Крейн.
ОКИСЛЕНИЕ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ—
введение в молекулу органич. соединения атомов
кислорода или отщепление водорода.
О. о. с. осуществляется при помощи окислителей,
мерой активности к-рых является их окислительный
электрохимич. потенциал. Действие окислителя на
органич. соединение зависит от характера окисляе-
мого вещества, темп-ры, катализатора, концентрации
реагентов, кислотности среды и т. д. Напр., при окис-
лении анилина (I) хромовой к-той образуется хинон
(II), перманганатом калия в кислой среде — анили-
новый черный (III), перманганатом калия в нейтраль-
ной или щелочной среде — азобензол (IV) и нитробен-
зол (V), хлорноватистой кислотой—ге-аминофенол (VI).
N-N
IV
В качестве окислителя чаще всего применяют: кисло-
род воздуха, перманганат калия, хромовый ангидрид
и хромовую смесь, азотную к-ту, висмутат натрия,
озон, двуокись свинца, двуокись селена, трет-бу-
тилхромат, окись серебра, перекись осмия, ацетон
в присутствии znpezn-бутилата алюминия, хлоранил и
др. Большое значение имеет окисление углеводородов
кислородом воздуха. Установление режима окисления,
регулирование соотношения между образующимися
продуктами с целью придания реакции необходимой
селективности, подавление глубокого выгорания уг-
леводорода, стимулирование процесса и управление
их становятся возможными в результате исследования
элементарных и макроскопич. стадий, изучения кине-
тики и механизмов реакций.
Современное понимание механизма О. о. с. основано
на перекисной теории, согласно к-рой ор-
ганич. вещества реагируют с кислородом, предвари-
тельно переходящим в активное состояние (см. Пе-
рекиси и гидроперекиси органические). При взаимо-
действии молекулы окисляемого вещества с возбуж-
денной молекулой кислорода получается «мольокись»
(мольоксид), к-рая превращается в более устойчивую
молекулу перекиси:
R4 R. R.
)сн,+ -о-о----> )СН,-О2—>- )СН-О-ОН
R'z R'z ' R'z
Т. обр., первичный акт окисления не требует разрыва
связи между атомами О в молекуле кислорода, энергия
диссоциации к-рой очень велика (117 ккал). Энергия
диссоциации связиО—О в перекисях значительно ниже
(30—40 ккал), чем в молекуле Ог, поэтому перекиси
являются неустойчивыми веществами, легко претерпе-
вают превращения, приводящие к разрыву связи О—О
и к образованию устойчивых продуктов окисления.
Образование перекисей в качестве практически един-
ственного продукта на начальной стадии окисления
экспериментально установлено на примере большого
числа углеводородов. В начальный период окисления
количество поглощенного кислорода эквивалентно
количеству образовавшихся гидроперекисей, в даль-
нейшем рост концентрации перекисей отстает от коли-
чества поглощенного кислорода и затем проходит через
максимум; в системе начинается разрушение пере-
кисей, приводящее к образованию продуктов более
глубокого окисления.
Начальная стадия окисления — образование гид-
роперекисей — является цепной реакцией:
R+O, —>RO2-
RO2+RH —^ROOH + R-
Основным признаком, указывающим на цепной ха-
рактер этой стадии окисления, является ускоряющее
(инициирующее) действие веществ, способных гене-
рировать свободные радикалы (перекись бензоила,
тетраацетат свинца, динптрил азоизомасляной к-ты,
фенилдиазоацетат и нек-рые др. соединения). Цепной
характер окисления может быть вызван также и ка-
тализаторами, в качестве к-рых широко используют
соединения переходных металлов; каталитич. действие
связано с их способностью либо присоединять,либо от-
давать один электрон валентноненасыщенной системе,
что приводит к образованию свободных радикалов и
ускорению реакции:
с,,н,с^ 0+Со3+—> С„Н3С=О+Со2 ++н+
хн
/СН, 2 + /СИз
C„H=C(-OOH+Fe + —^С0Н5С<-О. +Fe +OH +
\сн2 \сн2
Цепной характер доказывается также тем, что О. о. с.
может быть резко заторможено небольшим количест-
вом антиокислителей, к-рые вступают в ре-
акцию с ведущими цепь свободными радикалами и об-
рывают цепи окисления.
Основным отличием современного цепного меха-
низма окисления от «классической» перекисной тео-
рии является то, что реакция начинается не с акти-
вации молекулы Ог, а с активации молекулы окисляю-
щегося вещества.
Скорость реакции в первую очередь определяется
величиной константы скорости продолжения цепи; ве-
личина энергии активации этого процесса в основном
определяется энергией разрыва атакуемой связи
С—Н. В таблице приведены соответствующие данные
для ряда углеводородов, согласно к-рым труднее
всего должны окисляться ароматич. углеводороды и
парафины по связи Сцервич.—II; легче должны окис-
ляться разветвленные парафиновые. Резкое повышение
склонности к окислению надо ожидать у углеводо-
родов, имеющих в a-положении к окисляемой группе
669
ОКИСЛЕНИЕ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
670
двойную связь или ароматич. ядро, т. е. олефины и
жирноароматич. углеводороды, причем реакция дол-
жна практически затрагивать только углеродный
атом, находящийся в a-положении к двойной связи
или ароматич. ядру. Снижение энергии С—Н-связи
в двух последних случаях объясняется образованием
единой сопряженной системы их л-электронов двойной
связи (или ароматич. ядра) и неспаренного p-электро-
на по месту свободной валентности:
сн 2=сн-сн2«-—► • сн2-сн=сн2
Энергия С—Н-связей в углеводородах
Углеводород Связь Энергия связи, ккал
Бензол СВН5—Н 102
Метан СН3—Н 101
Пропан С3Н,— Н 100
Бутан ..... м-С.Н.—Н 102
Изобутан mpem-CiHn—H 89
Толуол .... С»Н,СН2—н 77,5
Этилбензол С0НБСН—н 75
сн,
Кумол С„Н,С (СН2)2—н 74
Пропилен ...... сн2=сн-сн2—н 77
В соответствии с данными таблицы бензол и метан —
углеводороды, стойкие к окислению; окисление их
с заметной скоростью начинается при темп-рах выше
соответственно 500° и 400° в присутствии катализа-
тора НВг. Значительной стойкостью к окислению
обладают парафины с нормальной цепью. При их
окислении в первую очередь реакция затрагивает
вторичные СН2-группы. При окислении н-декана было
установлено, что с почти равной вероятностью обра-
зуются все возможные вторичные гидроперекиси
и почти не образуется первичных гидроперекисей.
Разветвленные парафины окисляются значительно
легче, например 2- и 3-метилгептаны реагируют с за-
метной скоростью в условиях, когда окисление изо-
мерного им октана не имеет места. В случае раз-
ветвленных углеводородов атаке кислородом в пер-
вую очередь подвергается третичный атом углеро-
да, например 2-й и 5-й при окислении 2,5-диметилге-
ксана.
Гидроперекиси, образующиеся при окислении пара-
финов, превращаются в спирты, карбонильные соеди-
нения, кислоты, сложные эфиры и соединения со сме-
шанными функциями. При распаде первичных гид-
роперекисей получаются вода и спирт с тем же
углеродным скелетом, что и у исходного соединения. В
случае гидроперекиси метила и этила в продуктах
распада найдены также соответствующие альдегиды.
При распаде вторичных гидроперекисей главными
продуктами являются спирты и кетоны с тем же чис-
лом углеродных атомов, что и у исходных гидропере-
кисей. Третичные гидроперекиси без разрушения
скелета образуют только спирты, а с разрушением
скелета — кетоны.
Образование спирта из гидроперекиси должно со-
провождаться либо выделением кислорода, либо па-
раллельным окислением компонентов системы, наир,
спиртов в кетоны. Действительно, карбонильные
соединения образуются параллельно: при распаде
гидроперекисей и при окислении спиртов.
Окисление углеводородов может привести к син-
тезу карбоновых к-т, к-рые получаются пз промежу-
точно образующихся кетонов путем их окисления
до а-кетогидроперекисей, далее распадающихся на
альдегиды и кислоты:
о 00
, ,О /° || ||
R -Се —>R'-C-CHR—> R'-С-OH + R-С-Н
xch2r /
ООН
Общая схема и механизм реакции окисления опи-
сываются след, стадиями:
лепи рты
•-гидроперекись^^/ III
О кетоны —
углеводород
I
кислоты
IV V
RH —>- R -; R- + О2 •—>- RO.-; RO2- +RH —>- ROOH + R-ит. д
I II
RO2-+RO2. —>ROOR + O2
ROOH—>RO- + OH-; RO-+RH—>ROH + R-
II III
RH + OH---.R- + HaO; ROOH + RH—> 2ROH
I II
ROOH —>R'COR" + H3O; ROH + O, —>R'COR" + HaO
II IV III
R'COR" —> кислоты
IV V
Многие крупные произ-ва основного органич. син-
теза основаны на окислении углеводородов. Наиболее
перспективным промышленным методом получения
высших карбоновых к-т является окисление кислоро-
дом воздуха алканов (см. Высшие жирные кислоты).
В пром-сти используется жидкофазное окисление
кумола кислородом воздуха в присутствии медных
или марганцовых катализаторов. Первоначально об-
разующуюся гидроперекись кумола разлагают сер-
ной к-той до фенола и ацетона, к-рый при дистилля-
ции легко выделяется из реакционной смеси. Эта
реакция протекает по гетеролитич. механизму через
образование ионов оксения:
сн. сн. сн.
I н+ I -Н2о ----------------- |
с„нБ-с-сн3 —> СвНБ-С-СН3----» |С.,Н-| -с-сн„—►
ООН о-он Ч
I О +
н
СНз СН,
1 Н2О 1
—>сн3-с-ос»нБ------> снз-с-оед,—>.
+ Н+ |
он
о
п
—> СНз-С-СНз + С.НзОН
При разложении гидроперекиси в щелочной среде
с высоким выходом образуется диметилфенилкарби-
нол, к-рый далее дегидратируется до а-метилстирола:
СНз
,СН /СНз 140° /
С»НБС—OONa —> С„НБС(-ОН + О+ +NaOH--► СБНБС = СН2
\СН3 \СН3
При окислении олефинов слабым (~1%) водным р-ром
перманганата калия образуются а-гликоли (Вагнера
реакция):
К МпО,
r-ch=ch2--------*.-r-ch-ch2
он <Ьн
Промышленным методом синтеза ацетилена являет-
ся термоокислительный крекинг метана (850—1500°);
6СН, + 4О3 —>СН = СН4-8Н2 + ЗСО + СО2 + ЗН2О
Окисление этилена кислородом воздуха над мелко-
раздробленным серебром при 270—290° является наи-
671
ОКИСЛЕНИЕ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
672
более распространенным методом получения этилена
окиси'.
Ag
СН2 = СН2 + ’/2Ог —> СН2-СН2
4OZ
Пропилена окись получают аналогично или же окисле-
нием пропилена надуксусной к-той {Прилежаева реак-
ция). Большое технпч. значение имеет окисление
пропилена до акролеина кислородом воздуха над мед-
ным катализатором:
О2 хО
сн.-сн=сн2 —> сн2=снс/
Си ХН
Пропилен и другие олефины окисляются в водной
среде до кетонов в присутствии хлористого палладия:
СН3-СН = СН2 + О2 -> СН3СОСН3
MCI,
Промышленное значение имеет синтез циклогексано-
на и циклогексанола при окислении циклогексана
кислородом воздуха (145—150°, 10—34 ат, катализа-
тор — нафтенат кобальта). Одновременно получается
адипиновая к-та.
Перспективным методом синтеза фенола является
окисление толуола кислородом воздуха при 220—
250° в присутствии медного катализатора (1). Боль-
шое значение в пром-сти красителей имеет окисле-
ние антрацена до антрахинона (2). В качестве окис-
лителя в этой реакции чаще всего используют р-р
бихромата натрия в 25%-ной серной к-те, гипохлорит
натрия и кислород воздуха (жидкофазное окисление
при 170—175°, парофазное при 380—425°). Парофазное
окисление нафталина кислородом воздуха является
лучшим промышленным методом синтеза фталевого
ангидрида (3):
Фталевый ангидрид получают при аналогичном окис-
лении о-ксилола (540°). При окислении бензола ки-
слородом воздуха над ванадиевым катализатором
(400— 500°) с высоким выходом образуется малеиновый
ангидрид', вместо бензола можно использовать фур-
фурол, бутан, бутены, бутадиен. Наиболее известным
методом синтеза формальдегида является окисление
метанола кислородом воздуха:
сн.,он +1 /2о2 —> сн2о + н,о
Промышленным методом получения водорода перекиси
с выходом 90% является окисление 2-этилоксиантрона
кислородом воздуха.
Важный промышленный метод синтеза первичных
спиртов— окисление кислородом воздуха алюминий-
органич. соединений:
nCH2 = CH2 /R' /OR'
А1(С2Н5)3------->A1<-R"------>A1-OR" —>
\r'" XOR"'
[H2SO«]
-------->R'OH + R"OH + R"'OH
В пром-сти реализовано несколько вариантов окис-
ления ацетальдегида. При использовании воздуха
в присутствии окислов металлов (марганец, железо,
ванадий, уран, серебро) образуется уксусная кислота.
Промежуточным продуктом в этой реакции является
перуксусная к-та, к-рая в реакторе распадается на
уксусную к-ту и кислород:
/О /О /О
СНз-С^ + О2—>СН-С" —>CH3C<Z +’/2О2
хн ‘ хоон хон
При более мягком окислении и активации УФ-облу-
чением перуксусная к-та связывается в комплекс
с ацетальдегидом: 0 0
сн,-с<^ .сн/
хн хоон
Этот комплекс (кристаллич. продукт) разделяют на
компоненты в ректификационной колонне, причем аце-
тальдегид снова возвращается в процесс. Окисление
ацетальдегида кислородом воздуха в жидкой фазе со
взвешенным в ней катализатором, состоящим из ацета-
тов кобальта и меди (1 : 2), используют для пром, по-
лучения уксусного ангидрида и уксусной к-ты.
При нагревании углеводородов, аммиака и кислоро-
да получаются нитрилы. Простейшим вариантом этой
реакции является окислительный аммонолиз метана,
используемый для массового произ-ва синильной к-ты:
1000°
CH< + NH3 + O--cHCN + H.O
Pt
В пром-сть внедрен (1961) новый экономически выгод-
ный метод синтеза акрилонитрила совместным окис-
лением пропилена и аммиака кислородом воздуха:
ch3=ch-ch3+nh2+o2 —>ch2=ch-cn+h2o
Аналогично из толуола, аммиака и кислорода полу-
чают бензонитрил, а из пиколинов — нитрилы пи-
ридинкарбоновых к-т.
Окисление широко применяют для получения дру-
гих типов органич. соединений. При совместном окис-
лении кислородом предельных и непредельных угле-
водородов и треххлористого фосфора (хлорфосфпнов)
получают фосфорорганич. соединения:
/О
СН3-СН3 + РС13 + О. —>СН3-СН2РХ
/6
CH,-CH, + RPC12 + O2 —>-СН3-СН2Р<'-С1
XR
Большое значение как окислитель приобрел тетрахлор-
п-хинон С^СБОг; в мягких условиях с его помощью
количественно дегидрируют дибеизпл в стильбен.
Насыщенные и ненасыщенные спирты можно с хо-
рошим выходом окислить до соответствующих кето-
нов, применяя znpczn-бутилат алюминия в присутст-
вии большого избытка ацетона (Оппенауера реакция):
R, СН,. ((CH,),COJ3AI R. СН3.
ХСН-ОН+ ХС = О------------> ХС = О + ХСНОН
R'z СН/ R'z СН/
Реакция имеет очень большое значение в химии сте-
роидных гормонов и др. областях тонкого органич.
синтеза. а-Гликоли и а-аминосппрты легко окисляют-
ся иодной к-той (реактив Малапрада)с об-
разованием двух молекул карбониальных соединений:
/° /°
RCHOHCHOHR'+HJO,—>RC/ + R'C<' +H,O + HJO3
ХН ХН
RCHOHCHNH,R' + HJO,—> RCZ +R'CX + NH, + HJO,
H XH
Наиболее широко эта реакция используется в химии
углеводов. При окислении целлюлозы иодной к той
получается т. наз. диальдегидцеллюлоза:
О О
СН2ОН СН2ОН
673
ОКИСЛЕНИЕ-ВОССТАНОВЛЕНИЕ
674
Лит.: Губен-Вейль, Методы органической химии, т.
2, вып. 1, М.— Л., 1941; Препаративная органическая химия,
пер. с польск., М., 1959, с. 655; Джексон Э., в кн.: Орга-
нические реакции, сб. 2, пер. с англ., М., 1950, с. 363; Д ве-
ра с с и К., там же, сб. 6, М., 1953, с. 235; С в е р н Д.,
там же, сб. 7 М., 1956, с. 476; Хасселл Ч. X., там же,
сб. 9, М., 1959, с. 82; М е л ь н и к о в Н. Н., в кн.: Реакции
и методы исследования органических соединений, кн. 1, М.—
Л., 1951, с. 99; Лаптев Н. Г., там же, кн. 7, М., 1958,
с. 223; Белов В. Н., Хейфиц Л. А., Вирезуб
С. И., там же, кн. 10, М., 1961, с. 7; Березил И. В.,
Денисов Е. Т., Эмануэль Н. М., Окисление
циклогексана, М., 1962; Иванов К. И., Промежуточные
продукты и промежуточные реакции автоокисления углеводо-
родов, М.—Л., 1949; Бах А., /КРФХО, 1897, 29, вып. 6,
373; Engler С., Вег., 1897, 30, 1669; Rieche А.,
Angew. Chem., 1937, 50, 520; К н о р р е Д. Г. [и др.], Усп.
химии, 1957, 26, вып. 4, 416; Марголис Л. Я., там же,
1959, 28, вып. 5, 615; Сы ркинЯ. К., Моисеев И. И.,
там же, 1960, 29, вып. 4, 425. Э. Е. Нифантьев.
ОКИСЛЕНИЕ-ВОССТАНОВЛЕНИЕ — широкий
класс химич. реакций, включающих полный или ча-
стичный переход электронов от одних атомов к дру-
гим. Отдача электронев наз. окислением, при-
соединение электронов — восстановлением.
Эти термины объясняются их происхождением; пер-
воначально они применялись к реакциям образования
и разложения окислов металлов: окисление металла,
напр. Cu+1/2O2=CuO, и восстановление металла из
окисла, напр. СиО+Н2=Си+Н2О. При схематич.
описании строения окислов металлов принимают, что
они содержат положительные ионы металла и что,
следовательно, присоединяя кислород, металл те-
ряет электроны; с этим согласуется электронная трак-
товка понятия окисления. Пример окислительно-вос-
становительной реакции в водном р-ре:
Sn2+ + 2Fe’+=Sn‘ + + 2Fe2 +
в этой реакции ионы Sn2 + окисляются, ионы Fe’ +
восстанавливаются; ион Fe’+ является окислите-
лем, ион Sn2+ — восстановителем. Одним
из первых сформулировал и в дальнейшем широко
пропагандировал определение О.-в. как перехода
электронов Л. В. Писаржевский.
Изменение свободной энергии при окислительно-
восстановительных процессах может служить источ-
ником электродвижущей силы гальванич. элементов.
Напр., в элементе Даниеля анодом служит цинк, он
окисляется: Zn=Zn2+-j-2e_; на катоде восстанавлива-
ются ионы меди: Си2 + +2е~ = Си.Эти процессы проис-
ходят и тогда, когда потенциал медного электрода по-
ложительнее потенциала медного провода, присоеди-
ненного к цинковому электроду, поэтому во внешней
цепи идет ток от меди к цинку (электроны движутся
от цинка к меди). При электролизе также происходит
электрохимическое окисление на
аноде и электрохимическое восста-
новление на катоде, но под действием электро-
движущей силы во внешней цепи. Напр., при элект-
ролизе водного р-ра NaCl с ртутным катодом образу-
ются хлор и амальгама натрия: С1_ = 1/2С12+е~,
Na+-f-e_=Na.
Наряду с ионными соединениями имеются много-
численные соединения с ковалентными связями, об-
ладающими частично ионным характером. Распростра-
нение на реакции таких соединений концепции О.-в.
облегчается при использовании понятия электро-
отрицательности, к-рое, хотя и не является
строгим, все же полезно при общей систематизации
химич. явлений. Электроотрицательность показывает
стремление атома удержать имеющиеся электроны
и присоединить новые; для одновалент-
ного атома она пропорциональна сумме
ионизационного потенциала и сродства
к электрону (Малликен). Электроотрица-
тельность некоторых элементов цо По-
лингу (см. табл.).
Приведенные величины отвечают эмпирия, прибли-
женной ф-ле Полинга дляэнергии связи/) атомовАиВ:
Р(А-В) =4 {D (А-А) 4-.D (В-В)} + (zA — хв)2
где хд и — электроотрицательности А и В, причем
энергии связей А—В, А—А и В—В выражены в элект-
рон-вольтах. Значение х= 2 является приблизительной
границей, металлов (х<2) и неметаллов (х>2).
Электроны, образующие связь, смещены к более
электроотрицательному атому. В соответствии с этим
представлением дается определение величины «сте-
пень окисления» (английский термин oxidation num-
ber, буквально «окислительное число»; иногда упо-
требляют синоним «электрохимическая валентность»).
Для атомного иона степень окисления атома п равна
его заряду (считая за единицу элементарный элект-
рич. заряд), напр. в Fe’ + , n(Fe)=+3. Степень окисле-
ния атома, связанного ковалентно, определяется так:
если электроны находятся в общем обладании неоди-
наковых атомов, мысленно отдают их более электро-
отрицательному атому; если электроны связывают
атомы одного и того же элемента, делят их между
этими атомами поровну. Заряд атома после такого
распределения электронов есть его степень окисле-
ния. В согласии с этим правилом степень окисления
элемента в свободном состоянии равна 0.
В молекуле Н2О п((Т)=—2, п(Н)=4-1; в молекуле
перекиси водорода НО—ОН n(O) = — 1, n(H) = -j-l,
в обоих случаях валентность кислорода равна 2. Сте-
пень окисления в ряде случаев определяется без
написания структурной ф-лы соединения. Напр., в
H2SOj n(H)= + l, n(O) =—2 (молекула не имеет ха-
рактера перекиси); т. к. сумма п должна равняться
общему заряду частицы, в данном случае — нулю, то
п (S)=+6. Степень окисления может быть и дробным
числом, напр. в КО2 п(О)= — %, а в КОз (озонид ка-
лия) п (О)= — ’/,.
Для комплексных соединений обычно указывают
степень окисления центрального атома. Напр., ион
[PtCl,]2-, n(Pt)=-|-4. В этом примере ив более слож-
ных, чтобы определить п центрального атома, доста-
точно мысленно отделить от него лиганды в их устой-
чивой форме, такой, как Cl-, CN-, NH3, Н2О и т. п.
Напр., в ионе [Fe(CN)6]4~ n(Fe) = -|-2.
Обобщенное определение окис-
ления и восстановления: увеличение
степени окисления наз. окислением, уменьшение сте-
пени окисления — восстановлением. Всякое окисле-
ние сопровождается восстановлением, потому что сум-
ма степеней окисления атомов при химич. реакции
постоянна. В реакции Н2-|-С12==2НС1 га(Н) изменяет-
ся от 0 до +1, следовательно, водород окисляется;
п(С1) изменяется от 0 до —1, следовательно, хлор
восстанавливается. Согласно вышеприведенной ф- ле
Полинга, если хА = хв, то тепловой эффект реакции
А2-|-В2=2АВ равен нулю; поэтому можно считать,
что освобождение значительного количества энергии
при реакции Н2+С12=2НС1 обусловлено ее окисли-
тельно-восстановительным характером (такой простой
способ оценки теплового эффекта неприменим, если
в реакции участвуют кратные связи).
В ряду СН4, СН3ОН,СН2О, НСООН, СО2 п(С) при-
нимает значения —4, —2, 0, +2, +4, т. е. С ступен-
чато окисляется. Обычно говорят об окислении моле-
кулы, а не атома в ней, напр. «СЩ окисляется в
СНзОН»,«СН3ОН окисляется в СН2О», и т. п. Реакцию
F О Cl, N Вг C,S,j| Pt Н, Р В Геш Fen,Si Al Na Cs
4.0 3,5 3,0 2,8 2,5 2,2 2,1 2,0 1,9 1 ,8 1.5 0,9 0,7
22 к.х.э. т. 3
675 ОКИСЛЕНИЯ СТЕПЕНЬ—ОКИСЛЕНИЯ-ВОССТАНОВЛЕНИЯ МЕТОДЫ 676
4ННз+5О2=4НО+6Н2О наз. окислением аммиака,
хотя, согласно приведенному определению, окисляет-
ся азот: ra(N) увеличивается от —3 до +2.
В отличие от окислительно-восстановительных реак-
ций, в реакциях кислотно-основного взаимодействия
степень окисления участвующих атомов не изменяет-
ся, потому что пара электронов для образования свя-
зи поставляется более электроотрицательным атомом.
Примеры: Н+-J-OH _ = НгО, NH3+H+ = NH4 + . Следуя
Ингольду, обозначают окислители, т. е. молекулы
или ионы, стремящиеся присоединить электрон, и
кислоты, пли акцепторы электронной пары, общим
термином электрофильные реагенты, а
восстановители и основания, или доноры электронной
пары,— общим термином нуклеофильные
реагенты. Это объединение понятий отвечает за-
кономерностям ориентации при действии реагентов
на органич. вещества.
В явлениях катализа отчетливо проявляется раз-
личие указанных классов реакций. Окислительно-
восстановительные реакции, напр. окисление NHs,
SO2, гидрирование непредельных соединений, ката-
лизируются твердыми веществами, поверхность к-рых
может окисляться или восстанавливаться метал-
лами и окислами металлов переменной валентности:
Pt, Ni, V2O5, СО3О4 и др. Кислотно-основные реак-
ции, такие как присоединение Н2О, НС1, NH3, алки-
лирование олефинами или спиртами, катализируют-
ся кислотами (бренстедовскими или льюисовскими)
и основаниями: H2SO4, Н3РО4, А1С1з, HgCl2, NaNH2
и др.
Концепция О.-в., подобно другим классификацион-
ным понятиям, теряет определенность в пограничных
случаях; если полярность связей незначительна
(электроотрицательности атомов близки), обобщенное
определение О.-в. малопродуктивно.
Типичными окислительно-восстановительными реак-
циями являются реакции клеточного дыхания, реак-
ции горения и другие важные природные и технич.
процессы.
Лит.: Pauling L., General chemistry, S. Francisco,
1947; Латимер В. M., Окислительные состояния эле-
ментов и их потенциалы в водных растворах, пер- с англ.,
М., 1954; Кудрявцев А. А., Составление химических
уравнений, 3 изд., М., 1962. М. И. Темкин.
ОКИСЛЕНИЯ СТЕПЕНЬ — см. Окисление-восста-
новление.
ОКИСЛЕНИЯ-ВОССТАНОВЛЕНИЯ МЕТОДЫ в
аналитической химии — титриметриче-
ские методы, оспованные на окислительно-восстанови-
тельных реакциях. В процессе титрования изменяется
окислительно-восстановительный потенциал системы.
Если окислительно-восстановительную реакцию пред-
ставить ур-нием:
а Ок. + пе + с Вос.
1 11
(I — окисленная форма; II — восстановленная фор-
ма), то количественная зависимость окислительно-
восстаповительного потенциала от концентраций
(точнее активностей) реагирующих веществ данной
частной пары может быть выражена ур-нием (для
25° С):
77_ г» । 0.069 , [Ок.]а
о“Г пе g [Вос.]с
где Ео — нормальный окислительно-восстановитель-
ный потенциал данной пары, измеряемый в вольтах
по отношению к нормальному водородному электроду;
при [Ок.] — [Вос.] потенциал системы Е = Еп (см.
Нормальный потенциал). Изменяя концентрации (ак-
тивности) веществ, участвующих в окислительно-вос-
становительной реакции (напр., с помощью комплек-
сообразования, осаждения, экстрагирования и др.),
можно добиться количественного осуществления реак-
ции в необходимом направлении.
Так, связывание Fe (Ill) в комплекс фосфорной кислотой
или ЭДТА фторидом понижает окислительный потенциал в
системе Fe3 + /Fe2 + . Добавление фосфорной кислоты к системе
FeJ + + J ' I'e2 ++ , J. предотвращает сдвиг реакции
слева направо. Образование малорастворимого соединения
может изменить направление реакции окисления-восстанов-
ления. Так, реакция Cu2 + +2J~ , 3 CuJ4. + 1/2J., протекает
практически количественно вследствие образования малорас-
творимого иодида одновалентной меди.
Если в окислительно-восстановительном процессе
принимают участие ионы водорода, то величина
окислительно-восстановительного потенциала Е за-
висит не только от концентрации окисленной и вос-
становленной форм, но также и от концентрации
ионов водорода:
£ = £ I О.ОбЭ, [Ок.]«[Н + Г
0 ' пе ® [Вос.]с
Для обратимой окислительно-восстановительной ре-
акции между определяемым веществом и титрантом
можно найти величину константы равновесия:
а Ок., + Ъ Вос., = о Boc.j -f-d Ок.2
К _[Вос.,]° 10K.2]d , „ ^(Е'0~Е"0)
лравн.-[Ок 1]О ’[Вос.,]* и lg Л₽авн- -оТО59--
где Е„ и Ео—нормальные окислительно-восстанови-
тельные потенциалы частных реакций:
1) a Ok.j + пе = с Boc.j
2) Ь Вос.2 — пе тЛ d Ок.2
Из этой ф-лы видно, что константа равновесия будет
тем большей, чем больше разность нормальных оки-
слительных потенциалов обеих пар.
Возможность осуществления окислительно-восста-
новительной реакции обусловлена различием в нор-
мальных потенциалах более ~ 0,2 в. Окислительно-
восстановительные реакции часто проходят с измери-
мой скоростью, что затрудняет использование их
в титриметрич. анализе. На скорость окислительно-
восстановительных реакций большое влияние оказы-
вают темп-ра, катализаторы. Наряду с основной
окислительно-восстановительной реакцией часто про-
ходят побочные (индуцированные, сопряженные) ре-
акции, к-рые вызывают дополнительное расходование
основного (рабочего) вещества и приводят к искажен-
ным результатам (см. Индукция химическая). Прп
титровании в окислительно-восстановительных мето-
дах концентрации участвующих в реакции веществ
или ионов все время изменяются и, следовательно,
изменяется потенциал. Если величины окислительно-
восстановительпого потенциала, соответствующие раз-
личным моментам титрования, наносить на график,
то получаются кривые титрования, аналогичные кри-
вым, получаемым но методу нейтрализации. Папр.,
прп титровании р-ра Fe (II) сульфатом Се (IV) в соот-
ветствии с ур-нием реакции:
CeIV + FeII^CeIII + Feln
в р-ре будут находиться как исходные, так и образо-
вавшиеся ионы. В любой из моментов титрования
в р-ре будут две окислительно-восстановительные
пары: CeIV/CeIII[ и Fe^VFe11. Следовательно, окисли-
тельно-восстановительный потенциал можно рас-
считать по ур-нию:
£FeUI/FeII =0,77 + 0,0591g И
ИЛИ
£CeiV/Ceni = 1.45+0,0591g^
В точке эквивалентности потенциал системы может
быть вычислен по ф-ле:
аЕ0+ЬЕ"
677
ОКИСЛЕНИЯ-ВОССТАНОВЛЕНИЯ МЕТОДЫ
678
где а и Ь — стехиометрия, коэфф, окислительно-вос-
становительной реакции. На рисунке представлена
90 100 ю
Окислено F е, % Избыток Се, %
кривая титрования, из
к-рой видно, что вбли-
зи точки эквивалент-
ности наблюдается
резкое изменение по-
тенциала.
В ел ичина скачка по -
тенциала зависит от
разности нормальных
окислительно-восста-
новительных потен-
циалов обеих пар: чем больше эта разность, тем
больше и скачок потенциала. По скачку на кривой
титрования устанавливают точку эквивалентности.
Определение «конца титрования» при оксидиметрич.
титровании может быть проведено различными путями.
Если окраска титрующего р-ра достаточно резко
изменяется в точке эквивалентности при появлении
избытка титранта (напр., при титровании КМпО4
в кислой среде избыток его дает малиново-фиолетовую
окраску), может быть проведено безиндикаторное
титрование.
В качестве индикаторов в О.-в. м. могут применяться
различные вещества, не реагирующие на окисли-
тельно-восстановительный потенциал системы, но даю-
щие характерное окрашивание с определяемым веще-
ством или титрантом. Напр., при титровании с исполь-
зованием реакции .(2Z2J- в качестве индикатора
применяют крахмал, дающий адсорбционное соеди-
нение с иодом синего цвета. Широко применяют
в О.-в. м. для определения конца титрования органич.
вещества, изменяющие окраску при определенном
значении окислительно-восстановительного потенци-
ала титруемого раствора, т. наз. редокс-индикаторы
(см. Индикаторы). Применение окислительно-восста-
новительных индикаторов основано на способности их
окисляться или восстанавливаться, причем окислен-
ная и восстановленная формы имеют различную
окраску. Изменение окраски редокс-индикаторов свя-
зано с областью окислительно-восстановительных
потенциалов раствора, наз. интервалом перехода
индикатора. Чтобы окраска ред оке-индикатора из-
менялась при титровании резко и индикаторная
ошибка титрования была незначительной, необхо-
димо, чтобы область потенциалов перехода индикатора
находилась в пределах скачка потенциала анализи-
руемой системы. Известно большое число редокс-ин-
дикаторов. К важнейшим индикаторам для титрова-
ния сильными окислителями относятся индикаторы
группы о-фенантролин— Fe (II) (ферроин), фенилантра-
ниловая к-та и др. Широко используются индикаторы
группы дифениламина, триарилметановые красители
и др. Иногда в О.-в. м. применяют индикаторы, изме-
нение окраски к-рых необратимо. Напр., метиловый
красный или метиловый оранжевый обесцвечиваются
под действием сильных окислителей вследствие раз-
рушения молекулы индикатора. Окончание титрова-
ния в О.-в. м. может быть установлено также с по-
мощью инструментальных методов анализа (потенцио-
метрии, амперометрии, спектрофотометрии и др.).
Методы окисления-восстановления классифицируют
в зависимости от применяемого в данной реакции ос-
новного (рабочего) вещества — окислителя или восста-
новителя. К методам окисления-восстановления от-
носятся перманганатометрия, цериметрия, хромато-
метрия, иодометрия, иодатометрия, броматомет-
рия, ванадатометрия, титанометрия, аскорбиномет-
рия, хромометрия и т. д.
При помощи титрованных р-ров окислителей можно
количественно определять различные восстановители:
Sn (II), Fe (II), Мп (II), иодиды, сульфиты, сульфиды,
нитриты, арсениты, перекись водорода, ферроциа-
ниды, оксалаты и др. вещества. С помощью р-ров вос-
становителей можно определять разнообразные окис-
лители: Fe (III), хроматы и бихроматы, хлор, бром,
иод, гипохлориты, хлораты, броматы, йодаты, ванада-
ты, феррицианиды, перекись водорода и т. д. О.-в. м.
можно определять также вещества, не являющиеся
окислителями или восстановителями в условиях опы-
тов, но реагирующие с окислителем или восстановите-
лем в стехиометрия, соотношениях с образованием
осадков или комплексных соединений. В этих слу-
чаях определение проводят по количеству окислителя
или восстановителя, пошедшего на реакцию. Так,
напр., можно определять ряд элементов, образующих
осадки с оксалат-ионом методом перманганатометрии
или ряда катионов, образующих с 8-окспхинолииом
внутрикомплексные малорастворимые соединения,
с использованием метода броматометрии. Прежде чем
проводить оксидиметрич. титрование, необходимо
элемент, к-рый может существовать в различных сте-
пенях окисления, перевести количественно в опреде-
ленную высшую степень окисления и затем титровать
восстановителем или в определенную низшую степень
окисления и титровать раствором более сильного оки-
слителя. Во всех случаях применяемое для окисления
или восстановления вещество должно быть ущалено.
В качестве окисляющих агентов используются КМпСЧ,
(NH4)2S2O8 (с Ag), озон, Н2О2, KJO4, PbO2, NaBiOa,
КСЮз, HCIO4 и т. д. Восстановление может быть
проведено SnCl2, SO2, H2S, электролизом на ртутном
катоде, различными металлами или амальгамами, часто
для восстановления применяют различного рода
редукторы пли др. специальные приспособления.
Применение оксидиметрии для определения орга-
нпч. веществ основано на различной степени их окис-
ляемости в зависимости от силы применяемого окисли-
теля, pH среды, темп-ры и т. д. При использовании
очень сильных окислителей (£0>1,Зв при pH О
и £0>0,3 в при pH 14) ряд органич. веществ оки-
сляется полностью до СО2 и Н2О. При использовании
более умеренных окислителей окисление проходит
неполностью с образованием различных продуктов.
Неполное окисление менее желательно, так как оно
обычно протекает не стехиометрически. Однако неко-
торые даже сильные окислители не дают полного
окисления, но реагируют стехиометрически. Приме-
ром может служить «реакция Малапрада» — селектив-
ное определение при помощи иодной кислоты или
перйодата органич. соединений, в которых гидроксиль-
ные группы присоединены к двум рядом стоящим ато-
мам углерода. Окисление органич. веществ обычно
протекает с незначительной скоростью п сильно за-
висит от pH и темп-ры. Поэтому часто добавляют
окислитель в избытке и после прохождения реакции
избыток окислителя оттитровывают раствором под-
ходящего восстановителя.
О.-в. м. широко применяют на практике. С их
помощью возможно проводить определения огромно-
го количества неорганич. объектов; определение ме-
таллов в различных сплавах и сталях, часто при со-
вместном присутствии; определение всевозможных
органич. объектов, напр. с целью определения их чи-
стоты; определение воды с использованием реактива
Фишера (см. Акваметрия) и т. д.
Лит.: Кольтгоф И. М. [и др.], Объемный анализ,
пер. с англ., т. 3, М., 1961; Кольтгоф И. М., Стен-
гер В. А., Объемный анализ, пер. с англ., т. 1, М.— Л.,
1950; Крешков А. П., Основы аналитической химии,
кн. 1—2, М., 1961; Латимер В. М., Окислительные со-
стояния элементов и их потенциалы в водных растворах,
пер. с англ., М., 1954; Г и л л е б р а н д т В. Ф. [и др.],
Практическое руководство по неорганическому анализу, пер.
с англ., М., 1957; Ф а й г л ь Ф., Капельный анализ органи-
ческих веществ, пер. с англ., М., 1962; Berka A., V u 1-
t е г i n J., Z у k a J., УуЬгапё охуйабпё reduktni ойтёгпё
22*
679
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЙ ПОТЕНЦИАЛ—ОКИСЬ МЕЗИТИЛА
680
metody, Praha, 1961; Boef G. den, Polak N. L.,
Taianta, 1962, 9, 271; Treatise of analytical chemistry, v. 1,
N. Y-, 1959, p. 1. См. также литературу при статьях Анали-
тическая химия, Количественный анализ, а также при
соответствующих методах окисления-восстановления.
А. И. Бусев, К. П. Ефимов.
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЙ ПО-
ТЕНЦИАЛ — СМ. Нормальный потенциал, Элект-
родный потенциал.
ОКИСЛЫ (оксиды) — соединения элементов
с кислородом. В молекулах нормальных О.
все атомы кислорода непосредственно связаны с ато-
мами других элементов и не связаны друг с другом.
Иногда к О. относят и соединения, в молекулах к-рых
атомы кислорода связаны также друге другом,— п е-
р е к и с и (пероксиды), над пер екиси (супер-
оксиды) п озониды (см. Перекиси и перекисные
соединения).
Нормальные О. элементов образуются обычно при
непосредственном окислении кислородом простых
веществ. Образование О. происходит также при тер-
мич. разложении соответствующих гидроокисей, кар-
бонатов, нитратов, сульфатов и других солей кисло-
родных к-т.
Отдельные О. носят различные названия. Если
элемент образует с кислородом только одно соедине-
ние, то оно называется окисью. Так, Li2O, MgO,
АЦОз представляют собой соответственно окись лития,
окись магния, окись алюминия. Если для элемента
известны два различных О., то тот, в к-ром кислорода
относительно меньше, наз. обычно закисью,
а тот, в к-ром кислорода больше,— окисью.
Напр., Сп2О — закись меди, СпО — окись меди,
FeO — закись железа, Fe2O3 — окись железа и т. д.
О., в к-рых на один атом элемента приходится два
или три атома кислорода, часто наз. двуоки-
сями или трехокисями, напр.: NO2 — дву-
окись азота, СгОз — трехокись хрома п т. д. Если,
наконец, элемент образует большее число О., то осталь-
ные наз. обычно ангидридами тех кислот,
к-рые получаются при действии на них воды. Приме-
ром может служить азот, для к-рого известны пять
О.: N2O — закись азота, NO — окись азота, N2O3 —
азотистый ангидрид, NO2 — двуокись азота, N2O5 —
азотный ангидрид. От указанной номенклатуры встре-
чаются и отклонения. Напр., соединения состава
Э20з (где Э — общее обозначение элемента) иногда
наз. «полуторными» окислами; в тех случаях, когда
элемент образует два окисла типов ЭО и ЭО2, первый
наз. обычно окисью, второй — двуокисью (вместо
закиси и окиси). Пример: СО — окись углерода,
СО2 — двуокись углерода.
По химич. свойствам нормальные О. подразде-
ляются на основные, кислотные и проме-
жуточные, или амфотерные. Изменение их
химич. характера удобно проследить на высших О.
элементов III периода системы Менделеева: Na2O,
MgO, А120з, SiO2, РзО5, SOs, С12О7. Здесь ясно виден
постепенный переход от типично основного О. натрия
к типично кислотным О. серы и хлора. Этому соответ-
ствует и повышение степени окисления элементов,
образующих высший О. Такое же соответствие можно
наблюдать прн рассмотрении свойств О. одного и того
же элемента в разной степени окисления. Так, напр.,
в ряду МпО, Мп20з, МпО2, МпОз, Мп2О7 также виден
постепенный переход от основного О. марганца (II)
к кислотному О. марганца (VII). Подобные изменения
свойств наблюдаются у О. гл. обр. металлич. элемен-
тов с переменной валентностью. Низшие же О.
неметаллич. элементов часто не обладают свойствами
основных О. Нек-рые из них вообще не способны
к образованию солей и потому носят название и н-
дифферентных, т. е. безразличных
О., как, напр., СО и NO.
Многие О. встречаются в природе. Наиболее распро-
странен О. водорода — вода, образующая гидросфе-
ру. Главной составной частью горных пород является
О. кремвия. Нек-рые природные О. (железа, алюми-
ния и др.) служат главными источниками получения
металлов. Важную роль для жизни играет О. углеро-
да СО2, находящийся в атмосфере в количестве 0,03% .
ОКИСЬ МЕЗИТИЛА (2-метилпентен-2-он-4)
(СНз)2 С=СН—СО—СНз, мол. в. 98,14 — бесцвет-
ная жидкость; т. пл. —46,4°; т. кип. 128,3°; d*°
0,8548; п^ 1,4475; теплота испарения 87,9 кал/г\
давление паров 9,1 .«.« рт. ст. (20°); т. воспл. (откры-
тый тигель) 32,2°. При 20° в воде растворяется 2,8%
О. м., в О. м. — соответственно 3,4% воды. О. м.
смешивается со спиртом, эфиром, кетонами и многими
другими органич. растворителями. С водой, тетра-
хлорэтаном, диэтилкарбонатом и дибромэтаном О. м.
образует азеотропные смеси. Олефиновая двойная
связь в О. м. активирована карбонильной группой;
поэтому во многих реакциях предпочтительно полу-
чаются продукты присоединения по двойной связи,
а не по кетогруппе. Так, с аммиаком и первичными
аминами образуются Р-аминокетоны (1), с гидроксил-
амином — смесь оксима и Р-гидроксиламинокетона,
с семикарбазидом в кислой среде — семикарбазон
Р-семикарбазидокетона, с гидразином — замещенный
пиразолин, превращающийся при нагревании в про-
изводное циклопропана (2); взаимодействие с 1%-ной
НС1 приводит к диацетоновому спирту, с HCN обра-
зуется Р-цианкетон, с бисульфитом натрия — Р-сульфо-
кетон, с бромом—а, Р-дибромкетон; Фриделя —
Крафтса реакция приводит к Р-фенилкетону (3), а
взаимодействие с малоновым эфиром — к Р-дикарб-
этоксиметилкетону (4). Фторированный аналог О. м.,
напротив, присоединяет нуклеофильные реагевты
с образованием а-замещенных кетонов (5).
(СН3)2С—CHCOCH3 + RNH2 —>• (СН3)2С-СН2СОСН3 (1)
NHR
(СН3)2 C = CHCOCH3 + NH3NH, —>.
СНзХ/\/СН3 1° СН3\/\
—II (2)
снз NH-N снз снз
А1С1,
(СН3)2С=СНСОСН3 + С3Н,-^-(СН3)2С-СН2СОСН3 (3)
С6Н5
C2H3ONa
(CH3)..C;-CRCOCH3 + CH,(COOC.H-1),->
—>(СН3)2С-СН2СОСН3 (4)
СН(СООС2Н,)2
CF34 CF3.
>C=CHCOCH, + NH,—>• >CH-CH-COCII3 (5)
CF/ CF3z I
nh2
При восстановлении О. м. в зависимости от условий
могут быть получены ненасыщенный спирт, насыщен-
ный кетон, насыщенный спирт; возможно также обра-
зование гидродимера (6). При окислении О. м. полу-
чается кетоокись (7) или продукты деструктивного
окисления по двойной связи;
Na/Hg
(СН3)2С = СНСОСН3--> (СН3)2С-СН2СОСН3 (6)
(СН3)2С-СН2СОСН3
н.о2
(СН3)2С = СНСОСН3--►(СН1)3С - СНСОСН3 (7)
В пром-сти О. м. получают дегидратацией диацето-
нового спирта в присутствии минеральных к-т. С худ-
шими выходами О. м. образуется при действии водо-
отнимающих средств (хлористый водород, серная
681
ОККЛЮЗИЯ— ОКСАЗИНЫ
682
кислота, хлористый цинк, окись алюминия и др.) на
ацетон. Образование О. м. из ацетона обратимо: под
действием кислот она превращается в ацетон.
О. м. действует раздражающе на кожу и обладает
более сильным наркотич. действием, чем насыщенные
кетоны.
Применяют О. м. как растворитель для нитроцел-
люлозы, этилцеллюлозы, поливинилхлорида и сопо-
лимеров винилхлорида, каучука и природных смол;
используют также в качестве инсектицида.
Лит.: U 1 1 m а п п, 3 Aufl., Bd 9, Munchen — В., 1957,
S. 555. Н. П. Гамбарян,
окклюзия — поглощение (растворение) газов
расплавленными или твердыми металлами и нек-рыми
другими веществами. Окклюдированные газы распре-
деляются по всему объему, чем О. отличается от
адсорбции.
ОКСАЗИЛ {бис-[хлор(о-хлор)бензилат-Р, P'-(N,-N'-
тетраэтил) диаминодиэтил оксамида]', молекулярный
вес 608,49, С28Н42 О2 N4CI4, — бесцветные кристаллы,
т. пл. 188—192°; легко растворим в воде, раство-
рим в спирте 1 : 50 (20°), нерастворим в бензоле, хло-
роформе, ацетоне. Из спирта О. кристаллизуется
с двумя молекулами спирта. При нагревании О. раз-
лагается с выделением о-хлорбензилхлорида. Коли-
чественно О. определяют титрованием 0,1 н. НСЮ4
в ледяной уксусной к-те в присутствии индикатора
кристаллич. фиолетового. Получают О. конденсацией
М,]\-диэтилэтилендиамина с диэтиловым эфиром
щавелевой к-ты н взаимодействием образовавшегося
P,P',N,N'- тетраэтилдиаминодиэтилоксамида с о-хлор-
бензилхлоридом. Применяют О. для лечения миасте-
нии и последствий полиомиелита.
Лит.: Phillips А. Р., J. Amer. Chem. Sec., 1955.
77, № 9, 2400. Р. Г. Глушков,
ОКСАЗИНОВЫЕ КРАСИТЕЛИ — производные
феноксазина, рассматриваемые как мезомерные формы
оксониевых и аммониевых солей орто- и гаара-хиноид-
ного строения. Первый О. к.— основной темно-си-
ний 2К (I) — получен Мельдола в 1879 нагреванием
солянокислого п-нитрозодиметиланилина с [3-нафто-
лом:
орю-мномш строение (I)
пара-хиноидиое строение (Г)
Заменяя (3-нафтол галловой к-той, пирогаллолом или
резорцином, в тех же условиях получают другие
марки красителей, окрашивающих обработанный та-
нином хлопок.
Нек-рые полициклич. оксазины приобрели значение
в качестве пигментов, а их сульфокислоты, как прямые
красители, отличающиеся яркими, гл. обр. голубыми
и синими оттенками, хорошей прочностью к свету.
Получают полициклич. О. к. взаимодействием хлор-
анила с ароматич. аминами. В частности, с З-амп-
HO-N-этилкарбазолом образуется очень прочный пиг-
мент фиолетовый (II)
С2Н5 С1
с 4-амино-дифениламино-2-сульфокислотой хлоранил
образует прямой ярко-голубой светопрочный (III)
а
H5C6-HNYS^r°Y\^N>|'/S-SO3Na
... NaO3S^4JkNA^oJ<xJ-NH-C6H5
Группировку, характерную для О. к., содержат
антибиотики — актиномицины.
Лит.: Коган И. М., Химия красителей, 3 изд., М.,
1956, с. 324; Венкатараман К., Химия синтетических
красителей, пер. с англ., т. 2, Л., 1957, с. 894.
М. А. Чекалин.
ОКСАЗИНЫ — производные 6-членных гетероци-
клич. соединений, содержащих 4 атома углерода,
атом кислорода и атом азота, связанные между собой
четырьмя простыми и двумя двойными связями. В за-
2Н-1.2
4Н-1.2
бН-1,2
0 о
бН-1,3
1,3
висимости от взаимного расположения гетероатомов
различают 1,2-, 1,3- и 1,4-оксазины. Кроме
личные типы О. отли-
чаются расположением
двойных связей. Ниже
приведены всевозмож-
ные изомерные О.
Напр., в бН-1,3-0. при
6-м атоме С нет двой-
ных связей (группа
СН2), атом Онаходится
в положении 1, а атом
N — в положении 3.
1,2-0 к с азины
изучены плохо. Они,
по существу, являют-
ся циклич. О, N-дизаме-
щенными гидроксил-
аминами или О-заме-
щенными оксимами (1).
Дигидро-1,2-оксазины получены диеновым синтезом (2),
а тетрагидро-1,2-оксазины — алкилированием N-ok-
сиуретана тетраметилендибромидом (3):
того, раз-
4Н-1.3
2Н-1.3
N,
4 Н -1,4
2Н -1,4
СН
CeHs~C С—свн5
CeH5-C=O NOH
(1)
СН2=СнсН=СН2 + CgHgNo —-
кон
(2)
НОМНСООС2Н5 + Вг(сН2)4Вг
1,3-0 к с а з и н ы являются 6-членными аналога-
ми оксазолов. 4Н-1,3-оксазины получают циклизацией
N-ацил-р-аминокарбонильных соединений (4). Они
683
ОКСАЗОЛОНЫ
684
представляют собой слабые основания, менее устой-
чивые, чем оксазолы; гидролиз 4Н-1,3-О. приводит
к исходному (3-ациламинокарбонильному соединению.
Дигпдро-1,3-О. образуются при циклизации у-гало-
генопропплампдов карбоновых к-т (5) и при конденса-
ции 1,3-диолов с нитрилами (6). Тетрагидро-1,3-окса-
зины получены конденсацией 1,3-аминоспиртов с аль-
дегидами (7):
CeH5CONH(CH2)2COOC2H5
Л
100" с2н5оАоЛсвн5
сн2
н2с nh2
сн3-сн-он
1,4-0 к с а з и н ы изучены мало; гораздо лучше
изучены гидрированные 1,4-0., особенно тетрагидро-
1,4-0. (морфолины). Эти соединения, в том числе
незамещенный морфолин, получают гл. обр. циклиза-
цией диэтаноламинов (8) либо из ди-Р-галогенэтило-
вых эфиров (9);
NH
HN(CH2CH2OH)2
H2SO4
180“
(8)
NH
•NH3 < >
O(CH2CH2C1)2 — 122a> Ц
Морфолин — очень устойчивое соединение, не реа-
гирует с водой при 200°, с 10%-ным едким натром или
конц. соляной к-той при 160°, с фенилгидразином при
200°, но вступает почти во все реакции, характерные
для вторичных аминов. Он широко используется в ор-
ганич. синтезе как реагент и растворитель. Многие
полициклпч. соединения, содержащие кольцо О.
(обычно конденсированное с другими кольцами),
являются красителями, лекарственными веществами
и инсектицидами.
Лит.: Chemistry of carbon compounds, ed. E. H. Rodd,
v.4, Amst., 1959; Кромуэлл H., в кн.: Гетероцикличе-
ские соединения, под ред. Р. Эльдерфилда, пер. с англ., т. 6,
М., 1960, с. 406. Е. М. Рохлин.
ОКСАЗОЛОНЫ — кетодигидрооксазолы. Обычно О.
(или азлактонами) называют 5-кето-4,5-дигидроокса-
золы, к-рые являются внутренними ангидридами
а-ацилированных а-аминокислот:
.R'
/R
Ny-дС D.
II Э « I '-R
(1).
О.— активные ацилирующие агенты; при взаимо-
действии их с различными нуклеофильными соеди-
нениями обычно легко образуются соответствующие
производные а-ациламинокислот за счет разрыва
связи О1—С5 (2). Однако известны случаи, когда реак-
ции О. осуществляются с разрывом связи О1—С2 (3)
или №—С4 (4):
/R'
СУ *pR" +НВ> RCONHC-СОВ
R-C^C=O R'Z V (2)
В=ОН. OR,NR2, SR и т.п.
N C = CHR'
R-^/q^C-O
N---CH-R
II I
CF§-Cxo_C=O
+ HN3 R-C—N-C = CHR'
N-x^N COOH
N—C-R
CF„— CH ,6=0
з xo.
f'SH
I Hr
/R'
CF3CH^ • HBr
+ NH2 (4)
R-C-COOH
Z,\ z
SR' SR'
Иногда при превращениях О. атаке подвергаются другие
реакционные центры, а азлактонный цикл сохраняется (5,6).
Известно также 1,4-присоединение к сопряженной системе
ненасыщенного О. с переходом оксазолонового цикла в окса-
зольный (7):
xR'
N---С=С_ нг ,
|| । р ,г
R-CxoX=O к
.R'
N---СН-СН
II I хг>"
R-CxOxC=o R
(5)
N—С = СНС1 с6н6? у—С=снсвн5 (б)
С6Н5-Схо,С=О А1С13 СвН5-СхохС=О
CF ЬзСР
2 1/ 3 СН2=СО к,_Г/Схгн
у « (7)
С6Н5—Cxq^C=O CFg c6H5 Cxq^Cxq/C—о
О. являются слабыми основаниями — при взаимо-
действии с сильными к-тами в инертных растворите-
лях образуются соответствующие соли. Ненасыщенные
О. более устойчивы, чем насыщенные, и поэтому шире
применяются в различных синтезах. Насыщенные О.
часто используют в дальнейших реакциях без выде-
ления их в чистом виде.
Получение О. дегидратацией а-ациламинокислот
(1) достигается как при простом нагревании, так и
при действии дегидратирующих агентов: уксусного
ангидрида, хлористого бензоила, трехбромистого фос-
фора и т. п. Наиболее распространенный метод полу-
чения ненасыщенных О. — взаимодействие карбо-
нильных соединений с ацилглпцинами (метод Эр-
ленмейера) (8). Ненасыщенные О. получаются также
из а-ациламино-|3-оксикислот (метод Картера) (9) и
из Г4-(а'-галогеиацпл)-а-аминокислот (метод Бергма-
на) (10):
R'
/R< N С=СХ
RCONHCH2COOH + О=С ----- II I (8)
V R-CxoX=O R
Гх
RCONHCH-C
СООН ОН
N--С=С
r-CxqX-o R
(9)
Rx X
CHCONHCH-CHx ----*-
xz соон R"
zR'
R N CH-CH
x II I x
CH-CxOxC=O R
R'
N---C=CZ
RCH2-CxOxC“O R,
(10)
685
ОКСАЗОЛЫ—ОКСАЛАТЫ
686
Насыщенные О. обычно получают восстановлением
ненасыщенных (5). О. широко применяются в лабора-
тории для получения аминокислот, пептидов, а-кето-
кпслот и других соединений. См. также Оксазолы.
Лит.: Корнфорт Дж., в кн.: Гетероциклические
соединения, под ред. Р. Эльдерфилда, пер. с англ., т. 5. М.,
1961, с. 269; Лурье С. И., Ч а м а н Е. С., в кн.: Реакции
и методы исследования органических соединений, кн. 9, М.,
1959, с. 155. Е. М. Рохлин.
ОКСАЗОЛЫ — производные 5-членного гетеро-
цпклич. соединения, содержащего 3 атома углерода,
N______c-r' атом кислорода и атом азота (послед-
ц| *ц г ние в положении 1,3), связанные меж-
R-C^O'-C-K Ду собой тремя простыми п двумя двой-
ными связями.
О. обычно представляют собой жидкости, перегоняю-
щиеся при атмосферном давлении без разложения;
обладают «ароматическим» характером (общий секстет
образуется из двух р-электронов атома кислорода и
четырех л-электронов). Являясь слабыми основани-
ями, О. образуют неустойчивые солп с хлористоводо-
родной, пикриновой и другими к-тами. О. обычно
очень устойчивы к действию щелочей; при действии
к-т в довольно жестких условиях происходит гидролиз
(1). Окисление О. осуществляется легко и ведет к
более или менее полному расщеплению молекулы.
О. восстанавливаются лишь в жестких условиях с рас-
щеплением цикла (2). При действии аммиака или
аминов О. превращаются в имидазолы (3). О., содер-
жащие различные функциональные группы, часто
проявляют склонность к перегруппировкам (4,5):
Удобный метод получения О.— восстановление со-
ответствующих N-окисей, к-рые образуются из альде-
гидов и монооксимов 1,2-дикетонов (И). О. обра-
зуются также в результате некоторых перегруппиро-
вок (12). Незамещенный оксазол получен декарбок-
силированием соответствующей карбоновой к-ты;
эфир этой к-ты синтезирован циклизацией калиевой
соли а(а'-алкокспметиленамино)-|3-оксиакрилового
эфира (13):
О
HOM=C-RrHC1 N-------С—R zn N-------C-R'
RCHO + I ----------•- В । ---II । (11)
О=С—R R-CxqzC-R" R-CxozC-R"
N---С=СНОК
II I
R-CxozC=O
N---C-COOK
II II
R-C^ozCH
N—C-COOR^H3COOH J—J COORoh
R'OCH CHOK HCxqzCH ——»
N---‘CH
hLozCH<I3i
Я---СН
II II
Cxq^C С6Н5
конц.НС!
H2NCH2COC6H5 + C6H5COOH(1)
Na
1н5ОН.^СвН*СН^НСН2СНС6Н5 (2)
ОН
N---С-СеН.
II II ь 5
Н3С-СхохСН
NH3 N---С-С6н5
С2Н6ОН; 220-230* Н3С^С\0.ХН (3)
О. флуоресцируют при освещении дпевным или
УФ-светом. 2,5-Диарилоксазолы используют в ка-
честве сцинтилляционных добавок. Нек-рые О. обла-
дают физиологич. действием. Дигидропроизводные О.
называют оксазолинами, тетрагидропроизводные —
оксазолидинами. Эти соединения получают самостоя-
тельными путями, а не из О. См. также Гетероцикли-
ческие соединения, Оксазолоны.
Лит.: Корнфорт Д ж., в кн.: Гетероциклические
соединения, под ред. Р. Эльдерфилда, пер. с англ., т. 5, М.,
1961, с. 242. Е. М. Рохлин.
ОКСАЛАТЫ — соли щавелевой к-ты. Существуют
два ряда О. — кислые НООССООМе и средние
(СООМе)з, где Me — эквивалент металла. О. —
кристаллы, разлагающиеся без плавления; за исклю-
чением средних О. щелочных металлов, плохо рас-
творим в воде. Физич. свойства нек-рых О. приведены
в табл.
N--С-СОС1 N---С-СООС2Н5
11 11 ----II II
R-С'.0х-С~ОС2Н5 R—C-xqxC-Cl
N---С-СНО
Л II
R-CXqX-CI
R
(5)
О. могут быть синтезированы: дегидратацией а-ацил-
аминокетонов (6) и производных а-ациламинокислот
(7); нагреванием а-галогенкарбонильных соединений
с амидами (8); взаимодействием а-оксинитрилов с аль-
дегидами (9) или 1,2-дикетонов с N-алкилгидроксила-
мипами (10).
н on N сн
hconhch2coc6h5 Jis—А» i II (6)
POCI3 N-----СН
C6H5CONHCH2COOC2H5-----CgH5—С^охС—ОС2Н5
(7)
N---С-С6на
С6Н5СОСНаВг + C6H5CONH2 — 1 II н (8)
С6М5 ОхдАП
zCN
CeH5CHZ + С6Н5СНО
хон
HCI
Л и
С6Н5-^0.С-СвН5
(9)
СеН5СОСОСвН5 + HONHCH2C6H5
N----С-С6Н5
I! II ПО)
г г—г м
Океалат Плотность Растворимость, г/10 0 мл воды
холодной горячей
ВаС2О4 2,658 0.0093 (18°) 0,0228 (100°)
СаС-,0, 2,2 0,00067 (13°) 0,0014 (95°)
СаС204-Н20 2,2 нерастворим
FeC2O4-2H2O 2,28 0,022 0,026
Fes (С2О,)3-5Н2О к2с,о,-н)о КНС2О, — растворим
2,127 33 (16°)
2,0 2,5 16, 7 (100°)
КНСо04.0,5Н20 — 2,2 51 , 5 (100°)
K2[Fe(C2O,)2]-2,5H2O — 92 (21°) разл.
MgCoOi 2,45 (2,5 HoO) 0,0 7 (16°) 0,08 (100°)
(NH,)2C2O4-H2O 1,50 2,54 (0°) 11,8 (50°)
Na2C2O4 2,34 3,7 (20°) 6,33 (100°)
NaHC204-H,0 — 1 . 7 (15°) 21 (100°)
Na2 [Fe.. (C2O4)3]-xH2O 1,973 32,5 182 (100°)
Pbc..O4 5,28 0,00016 (18°) ——
ZnC,O4-2H2O 2,562 0,00079 —
О. широко распространены в природе. Так,
Са (СОО)з содержится во всех растениях в оболочках и
внутри клеток, особенно богаты ею водоросли, грибы,
лишайники и папоротники; кислая калиевая соль
КООССООН-(СООН)з-2Н2О (кисличная соль) —
в разновидностях щавеля и кислицы; в различных
растениях находятся О. натрия и магния. Моча чело-
века и животных всегда содержит немного О. кальция.
О. двухвалентного железа и кальция встречаются
в виде минералов. Синтетич. способы получения щаве-
левой к-ты основаны на образовании оксалата Na при
быстром нагревании до 400° формиата Na или при про-
пускании СОз над металлич. Na при 360°. Нераство-
римые в воде О. тяжелых металлов легко растворяются
687
ОКСАЛИЛХЛОРИД —ОКСАНТРОЛ И ОКСАНТРОН
688
в растворах О. щелочных металлов. При этом обра-
зуются комплексные оксало-соли, напр.
Fe2(C2O4)s+ ЗК2(С2О4) + 6Н2О —2KS [Fe*n(C2O4)e]-6H2O
Комплексные оксало-соли с участием иона трехвалент-
ного металла имеют октаэдрич. пространственную
структуру (координационное число 6) и могут суще-
ствовать в виде оптич. изомеров:
О. аммония применяют в аналитич. химии для опре-
деления Са2+; Кз [Fell (СзОНз] используют в фотогра-
фии как проявитель; О. алюминия и сурьмы приме-
няют В крашении. А. Е. Васильев.
ОКСАЛИЛХЛОРИД (хлорангидрид щавелевой
кислоты) Cl— С— С— С1, мол. вес 126,93 — бес-
цветная дымящая на воздухе жидкость с резким запа-
хом; т. пл. —10°; т. кип. 647763 мм; d™ 1,4785; п
1,4316; растворим в обычных органич. растворителях.
О. обладает типичными свойствами галогенангидри-
дов карбоновых к-т — легко реагирует со спиртами,
меркаптанами и аминами с образованием соответст-
вующих производных щавелевой к-ты. О. в обычных
условиях гидролизуется с образованием смеси СО2,
СО и НС1; гидролиз в газовой фазе приводит к ща-
велевой к-те и НС1. При повышенной температуре
О. разлагается на СО и фосген. В условиях Фриде-
ля — Крафтса реакции О., в зависимости от усло-
вий, образует либо хлорангидрид ароматич. кислоты,
либо дикетон:
(СОС1)а, А1С13, CS,
С«Н5-С„Н5----------------->. С„Н5-С„Н(СОС1
75%
(С0С1)з, А1С13, cs2
СН3О-С6Н.,
CH3O-CtH4-CO-CO-CcH4-OCH3
90% от теоретич.
Конденсация О. с полициклич. соединениями арома-
тич. ряда приводит к соответствующим орто-хиноид-
ным структурам:
N-алкиланилины с О. образуют N-алкилизатины:
(СОС1)2; А1С|3
С2Н5
При нагревании с мочевиной О. дает парабановую
кислоту:
o_cxNH2 (СОС1)2
>н2
NH-С=О
о=сх |
''5(Н-С=О.
Под влиянием УФ-лучей О. легко распадается на ради-
калы: С1—С—С—С1 —>- 2 С—С1. Этот процесс был
П II II
0 0 о
использован для свободнорадикального карбоксили-
рования органич. соединений.
хСН2 СН2/Н
Н2С сн2 (С0С1)а; ПУ Н2С с|
н2сх хсн2 Н2С сн2*0
СНг сн2
выход 60 %
Получают О. действием РС15 на безводную щавелевую
к-ту.
Лит..: Томас Ч., Безводный хлористый алюминий в ор-
ганической химии, пер. с англ., М., 1949; Runge F.,
Z. Electrochem., 1952, 56, № 8, 779. Л. С. Герман.
ОКСАМИД (диамид щавелевой кислоты), мол.
вес 88,07, H2NCOCONH2 — моноклинно-призматиче-
ские кристаллы, при медленном нагревании ча-
стично сублимируются с образованием неплавкого
остатка; при быстром нагревании в заплавленпом
капилляре т. пл. 419“ (с разл.); слабо растворим в воде
(1 : 2700 при 7,3°), в 100 мл кипящей воды, раство-
ряется 0,6 г; еще хуже О. растворим в спирте. В конц.
серной к-те растворяется с образованием сольвата
(C2H4O2N2 -ЗИгЗОа), аналогичный сольват образуется
с винной к-той (2C2H4O2N2-C4HeOe). О. проявляет об-
щие свойства амидов карбоновых кислот. Особые
свойства О. связаны с наличием двух соседних амид-
ных групп. С оксалилхлоридом О. образует параба-
новую кислоту, при восстановлении Zn в СНзСООН
дает гликолевую кислоту, а при электролизе в 80%-пой
серной к-те переходит в OHCCONH2. Обработка О.
муравьиной к-той дает оксалат аммония. О. обра-
зует окрашенные металлич. производные, напри-
мер K2Cu (C2H2O2N2)2-1,5H2O и K2Ni(C2H2O2N2)2.
Получают О. обычными методами — нагреванием окса-
лата аммония, аммонолизом диэтилоксалата, омыле-
нием дИциана И Т. Д. а. Е. Васильев.
ОКСАНТРОЛ И ОКСАНТРОН Ci4HI(lO2, мол. в.
210,23 — таутомерные формы антрагидрохинона (бен-
зоидно-хиноидная таутомерия). Бензоидная форма,
оксантрол (I) — коричневые кристаллы, т. пл. 180°;
водные р-ры обладают сильной зеленой флуоресцен-
цией. При подщелачивании р-ры оксантрола стано-
вятся темно-красными, на воздухе, окисляясь, обес-
цвечиваются вследств
хинона, которая раз-
лагается с выделе-
нием перекиси водо-
рода и антрахинона.
Получают оксантрол
восстановлением ан-
трахинона цинковой
пылью или гидросуль-
фитом вщелочнойсре-
де (образуется соль
оксантрола). Превра-
щения антрахинона в
оксантрол (или его
соль) и обратно испо-
льзуются в кубовом
крашении и для качественного определения антрахино-
на. Оксантрол—промежуточный продукт привосстанов-
лении антрахинона в технически важные антрон (III) и
антранол(1У). В холодном спиртовом р-ре в присутствии
НС1 оксантрол находится в равновесии с оксантроном
(II), причем равновесие сдвинуто в сторону первого
(97 : 3). Оксантрон может быть получен гидролизом
.чезо-бромантрона водой. Оксантрон — бесцветные
кристаллы, т. пл. 167°; в р-рах не флуоресцирует,
689
ОКСАФЕНАМИД —ОКСИАЛЬДЕГИДЫ И ОКСИКЕТОНЫ
690
в щелочи не растворяется, на воздухе не окисляется,
в оксантрол превращается при действии кипящего
спиртового р-ра щелочи. При действии цинка в уксус-
ной к-те оксантрон дает антранол. А- Е- Васильев.
ОКСАФЕНАМИД (дриол, дренамид, 4-оксифенил-
салнциламид) C13H11NO3, мол. в. 229,21 — белый
—. ___ или с фиолетовым оттенком
/ у.conh —( У-ОН мелкокристаллич. порошок;
\т. пл. 175—178°; практически
ОН нерастворим в воде, раство-
рим ₽ р-рах едких щелочей,
хорошо растворим в спирте и эфире. Водные фильт-
раты дают с р-ром FeCla фиолетовое окрашивание.
О. получают сплавлением п-аминофенола с фенило-
вым эфиром салициловой к-ты (салолом) при 200°. О.
применяют при холециститах, гепато-холециститах,
холангитах, желчекаменной болезни, в. А. Засосов.
ОКСИ АЛЬДЕГИДЫ И ОКСИКЕТОНЫ — альде-
гиды и кетоны, содержащие группу ОН. Различают
алифатические О. и о. (a-, J3-, у- и т. п. альдегида- и
кетоспирты) и ароматич. О. и о. (о-, м-, n-альдегидо- и
кетофенолы):
I ( I \
но—с— —с— —с—н
I \ I /п II
О
I ( I \
НО-С- —С— —С—R
I \ I /« II
о
но-с,н4-сно
НО—С„Н,—COR
Существуют также смешанные (жирноароматические)
О. и о.
Общие методы синтеза О. и о. — гидролиз соответ-
ствующих галогеналъдегидов и галогенокетонов (1),
озонирование непредельных спиртов (2), частичное
дегидрирование диолов (3), восстановление дикарбо-
нильных соединений или взаимодействие их с реак-
тивами Гриньяра (4), окисление трехатомных спиртов
(5), замена аминогруппы оксигруппой (применяется
гл. обр. для синтеза ароматич. О. и о.) (6):
.R' .R'
R-CO-(CH2)n-CH< —>R-CO-(CH,)„-CH< —>
'X XOCOR"
,R
—>R-CO-(CH2)n-CH( (1)
ЮН
I/O» I
R—CH-(CH3)„- C = C( —>R-CH-(CH2)„-C = O (2)
I 4 I
OH OH
-H2
R—®H—(CH2)n—CH2OH------>R—C—(CH2)„—CH2OH (3)
он о
+H2
RCO(CH2)nCOR'---------->-
(или R"MgX)
R" 4
—> RCO(CH2)nCHR' ( ИЛИ RCO(CHj)nC!-R' ) (4)
OH ' OH '
I | Pb(OCOCHs)4 I
RCH(CH2)n-C — C-----------cRCH(CH,)„-C=O (5)
OH OH OH OH
NaNO2
H2N—C»H.—COR-------> HO—C„H4—COR (6)
HC1
Реакции, характерные для большинства О. п о.,—
восстановление в диолы, спирты или кетоны, окисле-
ние в оксикислоты и дикарбонильные соединения.
Помимо общих методов получения и общих химич.
свойств, отдельные типы О. и о., в зависимости от
взаимного расположения гидроксильной и карбониль-
ной групп, обладают специфич. свойствами и полу-
чаются по-разному. Чем дальше функциональные
группы отделены одна от другой, тем меньше их взаим-
ное влияние и тем чаще О. и о. проявляют обычные
свойства спиртов (пли фенолов) и альдегидов (или ке-
тонов), вступая во все характерные для них реакции.
а-Окспальдегпды па-окспкетоны взаимно превращают-
ся под действием кислот или щелочей (7). Третичные
а-оксикетоны изомеризуются с перестройкой углерод-
ного скелета (8).
С6Н3СН2СНСНО — с,н3сн2с=сн у-*
<i>H он он
СДГ.-СП-С-СН,;// с„н5—сн—с=сн2 yzi
I : II
он о он он
•It с
5=iC0H3CH2C—СН.ПО — 1 СеН5СН=С—СН2ОН
II I
о он
+ /ОН /ОН
R—С—С—R"------>-R—С—С—R' (8)
I \R' -Н+ || \R"
ОН о
Простейший а-оксиальдегид— гликолевый альдегид —
образуется при конденсации формальдегида; а-О.
и а-о. получают также присоединением реактивов
Гриньяра к циангидринам (9):
,CN R'MgX
RCH<---------->RCH—COR' (9}
XOH I
ОН
Характерной особенностью а-О. и а-о. является
димеризация в диоксидиоксаны (10) и образование
озазонов при действии феннлгидразина (И):
/0Н
RCH-C-R' RCH-C-R' (10)
ОН О °\ /°
С — CHR
/\
НО R'
R'
C„H5NHNH3 |
RCH-C-R'-----------> RC—C=NNHCeH3 (11)
14 I!
ОН О NNHC.Hj
Третичные а-окснальдегиды вступают в Канниццаро
реакцию (12), а в присутствии гидрата окиси меди
претерпевают внутримолекулярную реакцию окисле-
ния-восстановления (13):
R. НО- R R.
;С—СНО-----> >С-СООН+ )С—СН2ОН (12)
R'z I R'z I R- I
OH OH OH
R. Cu(OH), R.
)C-CHO-----—7 )CH—COOH + R—C—CH—R' (13)
R'z I H2O R'z || |
ОН О OH
Основной метод синтеза р-оксиальдегидов и fi-окси-
кетонов — альдольная конденсация. Характерная реак-
ция |3-О. и |3-о. — отщепление воды с превраще-
нием в а-, Р-непредельные карбонильные соединения
(реакция, обратная синтез}' р-О. и P-о. гидратацией
последних).
H2O
I I Д , I
—С—C—COR ;C=C—COR
II H2O z
OHOH
Исключение составляют фторированные Р-оксикетоны
и нек-рые 6-О. и о. с третичным атомом углерода,
к-рые распадаются с разрывом С—С-связи:
CF»\ CFS.
)С—CHjCH.COR—» )C=O+CH»COR
CF/ I ' CF»
OH
691
ОКСИАНТРАХИНОНОВЫЕ КРАСИТЕЛИ —ОКСИАИЕТОН
692
у- и 6-Окспалъдегиды получают восстановлением соот-
ветствующих лактонов алюмогидридом лития в тет-
рагидрофуране (14), частичным восстановлением ал-
киндиолов (15), оксосинтезом (16) и конденсацией аце-
тоуксусного эфира с окисями (см. у-Ацетопропило-
еый спирт):
СНг-(СН2)н
I /
О — с = о
L1A1H,
-----> HOCH2(CH2)„CHO
(14)
Н2
иос1гд:-ссн,он —> носн2сн=снсн2он —->
Ni
—» НОСН2СН2СН,СНО (15)
СО(СО),
НО(СН,)ПСН=СН, + СО+Н2 -—>-
—>НО(СН3)„ + 2-СНО (16)
у-и 6-Оксиальдегиды и оксикетоны находятся в коль-
чато-цепном таутомерном равновесии с циклич. полу-
ацетальной формой; последняя со спиртом в присут-
ствии хлористого водорода дает О-алкильное произ-
водное, а при нагревании превращается в циклич.
виниловый эфир;
ZCH2 (СН2)л
к-f CHR-
° t!°H
R CH2-(CH2)n
I “
HOZ О------CHR'
R .СН2-(СН2)Г
Л । ,
R"-O О-----CHR
zCH-(CH2)n
R-cz |
О---CHR'
Ароматич. О. и о. получают по реакциям Реймера —
Тимана, Гаттермана, Гаттермана — Коха, Геша,
Фриса, Даффа. Они в основном обладают обычными
свойствами фенолов и альдегидов (пли кетонов). Од-
нако в орто-О. и о. образование клешневидных струк-
тур за счет внутримолекулярной водородной связи
вызывает характерное изменение физич. и химич.
свойств. Напр., ароматич. о-оксиальдегиды не всту-
пают в реакцию Канниццаро.
О. и о. широко используют в синтезах, особенно
лекарственных препаратов и природных веществ.
Важную роль в живой природе играют полиоксиаль-
дегиды и полиокснкетоны, относящиеся к группе уг-
леводов (см. Сахара). Многие ароматич. О. ио. входят
в состав природных веществ, нек-рые из них обуслов-
ливают окраску цветов и древесины. См. также
Ванилин, Оксиацетон, Маклурин.
Лит.: Chemistry of carbon compounds, ed, E, H. Rodd,
v. 4, pt C., Amst,— [a.o.J, 1960, p. 1465; Кромуэлл H.,
в кн,: Гетероциклические соединения, под ред. Р. Эльдерфил-
да, пер. с англ., т. 6, М., 1960, с. 406. Н. П. Гамбарян.
ОКСИАНТРАХИНОНОВЫЕ КРАСИТЕЛИ — при
родные и синтетич. оксипроизводные антрахинона,
образующие с солями алюминия, хрома, железа, оло-
ва и др. металлов интенсивно окрашенные прочные
комплексы — лаки. О. к. характеризуются наличием,
как минимум, двух оксигрупп в соседних а- и |3-поло-
жениях молекулы антрахинона. По методу примене-
ния эти красители относятся к протравным (см. Кра-
сители синтетические).
Основным представителем О. к. является 1,2-ди-
оксиантрахинонализарин [I]. По алюминиевой про-
траве ализарин дает на волокне
очень прочную яркую окраску — ку-
мач. Отдельно полученный алюми-
ниевый лак—крапплак применяют
в качестве пигмента, напр. в поли-
графии. В виде гликозида — рубе-
ритриновой к-ты — ализарин содер-
жится в корнях красильной маре-
ны, культура к-рой с древности
занимала большие площади во многих странах ми-
ра. Произ-во синтетич. ализарина, начатое еще в
1871, было первым примером промышленного изго-
товленпя природного красителя. В технике ализа-
рин получают щелочным плавлением соли 2-антра-
хинонсульфокислоты пли 2-хлорантрахинона. При-
сутствие в технпч. сульфокислоте 2,6- и 2,7-антрахн-
нондисульфокислот определяет наличие в ализарине
1,3,6- п 1, 3,7-триоксиантрахпнона флавопурпурпна
(II) и антрапурпурина (III).
Эти триоксисоединения с алюминием дают лаки более
желтого оттенка и содержание их определяет марки
продажного ализарина. В настоящее время ализарин,
ввиду сложности применения, практически полностью
вытеснен пз текстильной практики другими красите-
лями, прежде всего холодными. В качестве протрав-
ного красителя для шерсти сохраняет нек-рое значе-
ние ализариновый красный С —1,2-диокспаптрахи-
нон-3-сульфокислота. Нитрованием ализарина, вос-
становлением нптросоединения и последующей кон-
денсацией с глицерином получают краситель (IV).
В форме бисульфитного соединения этот краситель
под названием ализариновый синий ВС широко при-
меняли в ситцепечатании. Хинализарин (V), образую-
щийся из ализарина нагреванием в олеуме в присут-
ствии борной к-ты, по алюминиевой протраве окраши-
вает ткани в цвет бордо.
В качестве протравных красителей применялись
также 1,2,4,5,8-пентаокси-, 1,2,3,5,6,7-гексаокси- и
др. полпоксппроизводные антрахинона. Природные
красители кермес и кошениль также представляют
иолиоксипроизводные антрахинона.
Лит.: Коган И. М., Химия красителей, 3 изд., М.,
1956; Венкатараман К., Химия синтетических кра-
сителей, пер. с англ., т. 2, Л., 1957. И. С. Докунихин.
ОКСИАЦЕТОН (ацетилкарбпнол, ацетол, пиро-
виноградный спирт) СНзСОСН2ОН, мол. в. 74,08 —
бесцветная жидкость со сладким, жгучим вкусом:
т. п.т. ок. —17°; т. кип. 54718 ллс d2aa 1,0824;
1,4295; смешивается с водой, спиртом и эфиром. О.
получают действием муравьинокислого натрия на
галогенацетоны (1) или на а-бромпропионовый
альдегид (2) с последующим гидролизом химич. или
микробиологии, окислением 1,2-пропнленгликоля (3),
окислением ацетона, нагреванием глюкозы с угле-
кислым цинком, пиролизом глицерина и омылением
О-ацетилмолочного альдегида. Эфиры О. легко полу-
чают взаимодействием а-алкоксиацетонитрилов с ме-
тилмагнийгалогенидом (4):
HCOONa Н.о +
CH3COCH2X ------> СН3СОСН2ОСНО —>-
—>СН3СОСН2ОН (1)
СНО HCOONa Н3О +
СН.СН<------------>СН3СНСНО------^CHjCHCHO —►
4 Вг | I
осно он
.—>СН3С=СН—>СН3СОСП2ОН (2)
оной
693
ОКСИАЦЕТОФЕНОНЫ —ОКСИДОРЕДУКТАЗЫ
694
[О]
СН3СНСН2ОН—>СН3СОСН2ОН (3)
он
ROCH2CN + CH3MgX—<-ROCH2COCH3 (4)
В отличие от гликолевого и молочного альдегидов,
О. мономолекулярен и, по-видимому, таутомерен
с циклоацеталъной формой:
сн3—с—сн2он сн3—с—сн2
II /\/
о
но о
При нагревании О. с метиловым спиртом в присутст-
вии соляной к-ты получается димерный эфир:
СНЗХ ,CL
СН3ОН (-и q/Q сн2
СН3СОСН2ОН снзО I I хОСНз
2 хсн3
О., подобно а-оксиальдегидам, восстанавливает
феллингову жидкость, а с фенилгидразином обра-
зует озазон. При окислении О. дает пировиноградный
альдегид СН3СОСНО, к-рый превращается в молоч-
ную к-ту СН3СН(ОН)СООН, а при восстановлении,
в зависимости от условий, образуется 1,2-пропилен-
гликоль, изопропиловый спирт или ацетон. Для обна-
ружения О. служит характерная реакция с о-амино-
бензальдегидом, к-рая приводит к 3-оксихинальдину;
последний дает темно-красную окраску с хлорным
железом в спиртовом р-ре:
Х\г№Н2 О=С—СН3
1 + I "
сн.>—ОН
О. находит ограниченное применение в органич.
синтезе.
Лит.: Chemistry of carbon compounds. Ed. E. H. Rodd,
v. 1, pt A, Amst.— [a. o.j, 1951, p. 709. И. П. Гамбарян.
ОКСИАЦЕТОФЕНОНЫ — производные ацетофе-
нона, содержащие ОН-группу в фенильном ядре.
ОН О. — жидкости или твердые про-
СН3-СО-«2 'X' дукты; плохо растворимы в воде,
\в_§/ хорошо — в спирте.
Название Положе- ние ОН-груип Мол. в. Т. пл., °C Т. кип.,
о-Окспацетофенон а 9 136,14 213'717
91—92/1 3
п-Оксиацетофенон 4 136, 14 109° 147 — 148/3
Резацетофенон . . . 2,4 152,14 14 7° .—
Флорацетофенон. . . 2, 4, 6 168,14 285° —
Галлоацетофенон . . 2, 3, 4 168,14 173° —
2,5-Диоксиацетофе-
нон 2,5 152,14 202° —
а d21 1,131.
Многие О., встречающиеся в природе, являются
красящими веществами, обусловливающими окраску
нек-рых видов цветов и древесины. Так, 2-0. (о-окси-
ацетофенон) содержится в масле древесины Chione glab-
ra, 4-0. (n-оксиацетофенон) — в иглах пихты и в
коре ивы; некоторые производные 2,4-диоксиацет-
офенона, т. наз. резацетофенопа, содержатся в Asper-
gillus Clavatus (клаватол — диметильное производ-
ное) и корнях пионов (пеонол — 4-монометильное про-
изводное); диметиловый (2,4-) эфир (2,4,6-) триоксиаце-
тофенона, т. наз. флорацетофенона, найден в нек-рых
эфирных маслах (напр., в масле Blumea balsamifera)
и т. д. О. могут быть легко получены синтетически.
Так, 4-0. получают из анизола и СНзСОС! Фриделя—
Крафтса реакцией, с последующим омылением обра-
зующегося метилового эфира; 2-0. — нагреванием
фенилацетата с А1С1з, резацетофеноп — из резорцина
и СНзСООН в присутствии ZnCl2 и т. д. Получаемый
из пирогаллола и ледяной уксусной к-ты, 2,3,4-трп-
оксиацетофеноп, т. наз. галлоацетофенон, является
простейшим оксикетоновым красителем бледно-жел-
того цвета (ализариновый желтый С); его алюминие-
вый лак интенсивно желтого цвета, выкраска по
хромовой протраве — оливково-зеленого цвета. Близ-
ким по структуре к галлоацетофенону является галло-
бензофенон (2, 3, 4-триоксибензофенон) — протравной
краситель желтого цвета (ализариновый желтый А).
В. Н. Фросин.
ОКСИГЕМОГЛОБИН — см. Гемоглобины.
ОКСИДАЗЫ — ферменты, катализирующие оки-
слительно-восстановительные реакции с переносом
водорода или электронов от окисляемого вещества не-
посредственно на молекулярный кислород; входят
в класс оксидоредуктаз. К О. относятся многие важ-
нейшие ферменты, действующие на завершающих
этапах окисления биологического, в частности цитохро-
моксидаза, катализирующая перенос электронов с
цитохрома с на кислород, а также различные флаво-
протеиновые ферменты, простетич. группой к-рых
является флавинадениндинуклеотид (ФАД). К числу
последних принадлежат оксидазы L- и D-аминокислот,
осуществляющие окислительное дезаминирование,
а также глюкозооксидаза (нотатин), ксантинокси-
даза и др. Подробнее см. Оксидоредуктазы.
В. Б. Спиричее.
ОКСИДОРЕДУКТАЗЫ — ферменты, катализирую-
щие окислительно-восстановительные реакции. Сущ-
ность катализируемых О. реакций состоит в перено-
се атомов водорода и электронов от окисляемого
соединения (донора) на восстанавливаемое вещество
(акцептор). О. играют важную роль в обмене веществ
и энергии любой живой клетки, принимая участие
в биологическом окислении, обеспечивающем клетку
необходимой для ее жизнедеятельности энергией, а
также в реакциях, связанных с фотосинтезом, ив био-
синтезе углеводов, липидов, белков и др. соединений.
О. — высокомолекулярные вещества белковой
природы. Молекула О. состоит из белковой части, т. н.
апофермента, и низкомолекулярного небелкового ком-
понента (кофермента или простетич. группы), связан-
ного с белком более или менее прочной связью. В за-
висимости от природы небелкового компонента О.
можно разделить на след, группы: 1. О., исполь-
зующие в качестве кофермента никотинамид-аденпн-
динуклеотид (НАД) или никотинамид-аденин-днну-
клеотид-фосфат (НАДФ) (см. Кодегидрогеназы и Нико-
тинамиднуклеотидные коферменты). 2. Флавопротеи-
новые О., простетич. группой к-рых являются флавип-
аденин-мононуклеотид(ФМН) илифлавин-аденин-дину-
клеотид (ФАД) (см. Флавиновые коферменты). 3. Гемо-
протеиновые О., простетич. группа к-рых имеет геми-
новую природу, родственную гему гемоглобина.
Коферменты илп простетич. группы, входящие в со-
став О., являются непосредственными переносчиками
водорода и электронов в катализируемых О. окис-
лительно-восстановительных реакциях. В качестве
примера можно рассмотреть механизм действия флаво-
протеиновых О. Входящие в состав этих О. ФМН пли
ФАД обладают способностью обратимо присоединять
2 атома водорода:
В процессе катализируемых флавопротеиновыми О.
реакций атомы водорода, отщепляемые от окисляв-
695
ОКСИДОРЕДУКТАЗЫ
696
мого вещества (донора), в начале переходят на ФАД,
восстанавливая его. Затем атомы водорода с восста-
новленного ФАД-Н2 перено-
сятся на восстанавливаемое -*-^rRH2
вещество (акцептор): Y Y
Механизм действия нпко- *-фад-H2>x.r
тинамидадениндинук л е от и д-
ных О. более подробно рассмотрен в статье кодегидро-
геназы, а гемопротеиновых О. — на примере ката-
лазы или пероксидазы. Наиболее прочно связаны
с белками простетич. группы в гемопротеиновых О.,
где эти группы далеко не всегда поддаются обратимому
отщеплению. Значительно менее прочна связь между
коферментом и белковой частью молекулы в О., свя-
занных с НАД или НАДФ. Последние образуют с апо-
ферментом легко диссоциирующие комплексы.
Флавопротеиновые О. по прочности связи между апо-
ферментом и небелковым компонентом занимают
промежуточное положение между гемопротеиновыми
и никотинамидадениндинуклеотидными О.
Помимо коферментов и простетич. групп органич.
природы, в состав многих О. входят ионы металлов,
к-рые обычно принимают непосредственное участие
в механизме катализируемого О. переноса электронов.
Так, в состав многих флавопротеиновых О. входят
ионы Fe, Мо или Си. В состав гемопротеиновых О.
входит ион Fe, связанный с гемом. Кроме того, в со-
став цитохромоксидазы, помимо геминового Fe, входит
поп Си. С помощью метода электронного парамагнит-
ного резонанса показано, что в процессе многих
окислительно-восстановительных реакций, катализи-
руемых О., происходит обратимое изменение валент-
ности входящих в их состав ионов металлов. Мол. вес
различных О. изменяется в довольно широких преде-
лах (от нескольких десятков тысяч до миллиона).
Оксидазы отличаются друг от друга не только
физико-химич. свойствами, но и специфичностью
действия, а также местом, к-рое та или иная О. зани-
мает в общей цепи окислительных процессов. При
биологии, окислении водород и электроны, отщепляе-
мые от того или иного субстрата, как правило, пере-
носятся на кислород не в один, а в несколько этапов
путем последовательного перемещения по т. н. дыха-
тельной цепи, или цепи переноса электронов к кисло-
роду, образуемой промежуточными переносчиками
водорода, и электронов, к к-рым относятся упомяну-
тые выше НАД и НАДФ, ФАД и ФМН, а также раз-
личные цитохромы. Упрощенно эту цепь можно пред-
ставить след, образом:
RH2x s' НАД(Ф) *. жФАД-Нг^ окисленный» у Н2О
субстрат Y Y Y “ИТ0,’““ Y
окисления Л Л
R ^НАД(ф) Н2 'ч- ФАД ж восстановленный*^ /2О2
цитотрон
1 II III
Примечание: Символ НАД(Ф) означает, что фермент
реагирует как с НАД, так и с НАДФ.
В отдельных случаях эта цепь может быть короче.
При окислении нек-рых субстратов, напр. сукцината
пли жирных к-т, водород сразу переносится на ФАД,
минуя НАД(Ф). В ряде других случаев флавопротеи-
новые О. переносят водород непосредственно на кисло-
род, минуя систему цитохромов. Окислительно-вос-
становительные реакции, относящиеся к различным
этапам дыхательной цепи, помеченным на схеме циф-
рами I—III, катализируются различными О.
О., осуществляющие перенос водорода и электронов
на промежуточные акцепторы (НАД, ФАД, цитохро-
мы), обычно называют дегидрогеназами', О., перенося-
щие водород или электроны на кислород,— оксидаза-
ми. О., действующие на том или ином этапе дыхатель-
ной цепи, различаются субстратной специфичностью,
т. е. способностью взаимодействовать только со строго
определенным типом субстрата. Так, напр., алкоголь-
дегидрогеназа, осуществляющая окисление спиртов,
не окисляет молочную к-ту, с к-рой взаимодействует
другой специфич. фермент — лактатдегидрогеназа.
О., функционирующие на I этапе биологич. окисле-
ния, осуществляют многочисленные реакции деги-
дрирования самых различных субстратов с переносом
отщепляемых от них атомов водорода и электронов
на НАД(Ф), являющихся коферментами этих О. К их
числу относятся важнейшие ферменты гликолиза:
триозофосфатдегидрогеназа, катализирующая оки-
слительное фосфорилирование фосфоглицеринового
альдегида, в результате чего образуется новая богатая
энергией макроэргическая связь; лактатдегидроге-
наза, катализирующая перенос водорода от НАД -1]2
на пируват с образованием НАД, что обеспечивает
возможность непрерывного гликолиза в анаэробных
условиях. Сюда же относятся ферменты дегидрирова-
ния в цикле трикарбоновых кислот, в частности изо-
цитратдегидрогеназа и малатдегидрогеназа, окисляю-
щие соответственно изолимонную и яблочную к-ты.
В эту группу входят также глюкозо-6-фосфат-дегидро-
геназа и фосфоглюконатдегидрогенава — ферменты
т. н. пентозного цикла, или пентозофосфатного пути
окисления углеводов, обеспечивающего клетку рибо-
зо-5-фосфатом, идущим на синтез рибонуклеиновых
к-т, и НАДФ -Н2, играющего роль восстановительного
эквивалента в многочисленных процессах биосинтеза,
в частности при биосинтезе жирных к-т.
Из других О. этого типа следует упомянуть 0-окси-
ацил-КоА-дегидрогеназу, принимающую участие в
окислении жирных к-т (см. Обмен веществ, Жиры), a
также глутаматдегидрогеназу, играющую важную
роль в обмене аминокислот.
О., действующие на II этапе биологич. окисления
и осуществляющие перенос электронов от НАД(Ф) -Н2
на цитохромы, являются флавопротеидами. К ним от-
носятся НАД -Н2-цптохром с-редуктаза и НАДФ -Н2-
цитохром с-редуктаза.
Нек-рые флавопротеиновые О. катализируют пере-
нос электронов на цитохромы непосредственно от
окисляемого субстрата, минуя НАД(Ф). К таким фер-
ментам относится ацил-КоА-дегидрогеназа, принимаю-
щая участие в окислении жирных к-т. К числу флаво-
протеиновых О., способных взаимодействовать непо-
средственно с окисляемым субстратом, относится
также сукцинатдегидрогеназа — важнейший фермент
цикла трикарбоновых к-т, катализирующий дегидри-
рование янтарной к-ты. Акцептор, на к-рый этот
фермент переносит электроны и водород, отщепляемые
от сукцината, пока еще не установлен. Среди О., дей-
ствующих на III этапе биологич. окисления, важней-
шим является цитохромоксидаза — гемопротеиновый
фермент, катализирующий перенос электронов с цито-
хрома с на кислород.
К О. относятся также: а) Нек-рые флавопротеиновые
ферменты, переносящие водород и электроны непо-
средственно на кислород, минуя, т. о., как НАД(Ф),
так и цитохромы. Характерной чертой окислительных
реакций, катализируемых этими ферментами, является
образование при этом перекиси водорода. Такими
ферментами являются оксидазы L- и D-аминокислот,
осуществляющие окислительное дезаминирование по-
следних, а также ксантиноксидаза, глюкозооксидаза
(нотатин) и др. ферменты, б) Гемопротеиновые фер-
менты, использующие в качестве акцептора водорода
и электронов перекись водорода; такими ферментами
являются каталаза и различные пероксидазы, в) Гидро-
ксилазы, осуществляющие гидроксилирование раз-
личных органич. соединений с использованием для
этой цели молекулярного кислорода, по схеме:
RH + О2 + НАД(Ф) Н„ =ROH + Н20 + НА Д(Ф)
697
698
ОКСИДЫ — ОКСИКИСЛОТЫ
Некоторые важнейшие оксидоредуктазы и катализируемые ими реакции
Систематическое название, тривиальное (рабочее) название и номер (шифр) фермента Катализируемая реакция Кофермент или простсти- ческан группа
Оксидоредуктазы, осуществляющие деги- дрирование субстрата с переносом элек- тронов и водорода на НАД(Ф): В-глицеральдегид-3-фосфат;НАД — оксидоредуктаза (фос- форилирующая), триозофосфатдегидрогеназа, 1.2.1.12. Ь-лактат:НАД — оксидоредуктаза, лактатдегидрогеназа, 1.1.1.27. Ь-пзоцитрат:НАДФ — оксидоредуктаза (декарбоксилирую- щая), изоцитратдегидрогеназа, 1.1.1.42. Ь-малат:НАД — оксидоредуктаза, малатдегидрогеназа, 1.1.1.37. В-глюкозо-6-фосфат:НАДФ — оксидоредуктаза, глюкозо- б-фосфатдегидрогеназа, 1.1.1.49. 6-фосфо-D-глюконат: НАДФ — оксидоредуктаза (декарбок- силирующая), фосфоглюконатдегидрогеназа, 1.1.1.44. Ь-3-оксиацил-КоА:НАД — оксидоредуктаза, ₽-оксиацил- КоА-дегидрогеназа, 1.1.1.35. L-глутаматгНАД (Ф) — оксидоредуктаза (дезаминирующая), глутаматдегидрогеназа, 1. 4.1.3. D-глицеральдегид-3-фосфат + 113РО.+ + НАД = 1,3-дифосфоглицериновая к-та + + НАД-Н; L-лактат + НАд = пируват + НАд-Н2 L-изоцитрзт + НАДФ=2-кетоглутарат + + С02 + НАДф.Щ L-малат + НАД = оксалоацетат + НАД-Н, D-глюкозо-б-фосфат + НАДФ=Ь-глюконо- -б-лактон-6-фосфат + НАДФН2 6-фосфо-П-глюконат + НАДФ-П-рибулозо- -5-фосфат + СО. + НАДФ11. L-3-оксиацил-КоА + НАД = З-кетоацпл- -КОА + НАД Н2 L-глутамат + Н20 + НАД (Ф) = 2-кетоглу- тарат + NH3 + НАД (Ф)-Н. НАД, глута- тион НАД НАДФ НАД НАДФ НАДФ НАД НАД (Ф)
Оксидоредуктазы, переносящие элект- роны между НАД (Ф) и цитохромами: НАД-Н2;цитохром с — оксидоредуктаза, НАД-Н2: цито- хром с-редуктаза, 1.6.2.1. НАДф.Н2:цитохром с — оксидоредуктаза, НАДФН2- цятохром с-редуктаза, 1.6.2.3. НАД-Н2 + феррицитохром с = НАД + ч-ферроцитохром с НАДФ.Н2 + феррицитохром с = НАДФч- ферроцитохром с ФАД ФАД
Флавопротеиновые оксидоредуктазы, вза- имо д е йст в у ю щпе непосредственно с суб- стратом, минуя НАД (Ф) Ацил-КоА: цитохром с — оксидоредуктаза, зцил-КоА-де- гидрогеназа, 1.3.2.2. Сукцинат: (акцептор) — оксидоредуктаза, сукиинатдегидро- геназа, 1.3.99.1. Ацил-КоА + феррицитохром с = 2,3-де- гидроацил-КоА + ферроцитохром с Сукцинат + акцептор = фумарат + восста- новленный акцептор ФАД ФАД
Оксидоредуктазы, переносящие элект- роны на кислород: Цитохром с: 02 — оксидоредуктаза, цитохромоксидаза, 1.9.3.1. L-аминокислота: 02 — оксидоредуктаза (дезаминирующая), оксидаза L-аминокислота, 1. 4.3. 2. 4-ферроцитохром с + 02 = 4 феррицито- хром с + 2Н2О L-аминокислота + Н30 + 02 = а-кетокис- лота ч- NH, + Нг02 Железопор- фирин ФАД или ФМН
Сюда относятся ферменты, катализирующие гидрокси-
лирование стероидов, а также ряда аминокислот, напр.
превращение фенилаланина в тирозин, г) Оксигеназы,
осуществляющие окисление субстрата непосредствен-
ным присоединением к нему молекулярного кисло-
рода. К их числу относится липоксигеназа,
ранее называвшаяся липоксидазой, катализирующая
образование перекисей ненасыщенных жирных к-т
путем присоединения по двойной связи молекуляр-
ного 0-2.
В соответствии с современными правилами номен-
клатуры и классификации ферментов систематич.
наименования О. составляются по форме: «донор:
акцептор — оксидоредуктаза». В качестве тривиаль-
ных (рабочих) наименований используются многие
старые названия ферментов с сохранением терминов
«дегидрогеназа», «редуктаза» и «оксидаза». Нек-рые
важнейшие О. и катализируемые ими реакции при-
ведены в таблице.
Лит.: Северин С. Е., Внутриклеточный обмен угле-
водов и биологическое окисление, в кн.: Химические основы
процессов жизнедеятельности, М., 1962, с. 156; Нейландс
Дж., Штумпф П., Очерки по химии ферментов, пер.
с англ., М., 1958; Классификация и номенклатура ферментов,
пер. с англ., М,, 1962. В. Б. Спиричев.
ОКСИДЫ — см. Окислы.
ОКСИКИСЛОТЫ — соединения, содержащие од-
новременно карбоксильную и гидроксильную группы.
О. различаются по числу карбоксильных групп (ос-
новность) и общему числу гидроксильных групп,
включая и карбоксильные (атомность) в молекуле и по
принадлежности к предельному, непредельному, ци-
клич., ароматич. илп гетероциклич. рядам. О. очень
распространены в растительном мире. Молекулы мно-
гих О. содержат один или более асимметрич. атомов
углерода и могут существовать в оптически изомерных
формах (оптич. изомерия). Простейшими являются
предельные О., содержащие одну гидроксильную и
одну карбоксильную группы. В зависимости от того,
к какому атому углерода по отношению к карбоксиль-
ной группе присоединена гидроксильная группа, раз-
личают а-, Р-, у-, 6-оксикислоты и т. д. Простейшие
О.— вязкие, жидкие или кристаллич. продукты,
растворимые в воде. Кроме общих свойств спиртов и
кислот, присущих О., они обладают специфич. свой-
ствами. Так, они более сильные кислоты, чем соответ-
ствующие карбоновые; способны отщеплять воду, при
этом а-0. образуют циклпч. сложные эфиры-лактиды:
zOH нох о
R-CH + с=о __________в-нсх \=О
о=сх /CH-R О=С CH-R
ОН но
P-О. образуют а, P-ненасыщенные к-ты, у- и 6-0.
лактоны:
R—сн—сн.соон
I
он
1;-сп-сн,-с=о
I-----o-J
При большем удалении ОН-группы от СООН, начи-
ная с НО(СН2)7 СООН, происходит межмолекулярная
этерификация с выделением воды и образованием
699
ОКСИЛИЗИН —ОКСИМЫ
700
линейных макромолекул. При кипячении с разб. ми-
неральными к-тами О. расщепляются на альдегиды
(кетоны) и муравьиную к-ту.
О. можно получить: из галогенозамещенных к-т;
присоединением воды к ненасыщенным карбоновым
к-там; действием HNOs на аминокислоты; окислением
кислот с третичным атомом углерода; окислением
гликолей, имеющих хотя бы одну первичную спирто-
вую группу; окислением оксиальдегпдов; омылением
окспнитрилов; при помощи металлоорганич. соеди-
нений чаще всего Реформатского реакцией.
Среди одноосновных О. предельного ряда, содержащих
две и более ОН-группы, наибольший интерес представляют
глицериновая и оповые к-ты. В растениях широко распро-
странены О : двухосновные — яблочная (I) и винные (II),
трехосновная — лимонная (III), циклическая — хинная.
НООССН(ОН)СН3СООН НООССН(ОН)СН(ОН)СООН
I II
.он
НООССН3С(ОН)СН2СООН НО /Г \ОН
I /н \
СООН \Н /
III н\ <соон
он
К непредельным О. относится рицинолеиновая кислота
СН((СН2)-СН(ОН)СН2 — СН = СН(СН2)7СООН, глицерид к-рой
составляет основу касторового масла. К ароматическим О.
относятся фенолокислоты, содержащие обе функциональные
группы в ароматич. ядре, и О., у к-рых одна или обе функцио-
нальные группы находятся в боковой цепи. Основные способы
получения фенолокислот: действие СО2 на феноляты (реакция
Кольбе):
.ОН
CbH.,ONa + CO2 —>-CbHZ
xCOONa
действие HNO2 на аминокислоты; сплавление со щелочами
сульфированных ароматич. к-т; окисление гсмологов фенола;
действие СС14 на фенолы в щелочных р-рах:
ZOH
СбН5ОК +CCU+ КОН —> С6Н<
ХСООК
Специфич. свойством фенолокислот является их способ-
ность взаимодействовать с галогенангидридами фенолокислот
с образованием соединения типа сложных эфиров, наз. по-
ли де пс и да м и. Фенолокислоты находятся в родстве
с важными природными продуктами — смолами, красящими
веществами цветов и плодов. Важнейшие фенолокислоты:
салициловая (о-сксибензойная к-та) и галловая (о, о-диокси-
бензойная к-та).
Представителем ароматич. О., содержащих функциональ-
ные группы в боковой цепи, является миндальная кислота
СбН,СН(ОН)СООН. К ненасыщенным ароматическим О. от-
носятся цис- и транс-о-оксикоричпые к-ты, лактон к-рых —
кумарин, распространен в растительном мире. 3. Н. Парнес.
ОКСИЛИЗИН (д-оксилизин, а,8-диамино-6-оксика-
проновая кислота, 5-окси-2,6-диаминогексановая кис-
лота) H2NCH2CH(OH)CH2CH2CH(NH2)COOH, мол. в.
162,19—основная оксиаминокислота. Известны четы-
ре оптически активных стереоизомера: L-оксилпзин
(I), D-оксилизин (II), L-ялло-оксилизин (III), D-алло-
оксилизин (IV), и два оптически неактивных рацема-
та — И,Ь-оксилизин и 1Э,Ъ-алло-оксилизин.
СООН COOH COOH COOH
1 H—c—nh2 1 nh2—c—h 1 h-c-nh,
1 (CH,), (CH2)2 (CH2)2 (CH2)2
H-C-OH HO—C—H HO—C—H H-C-OH
1 ch2nh2 ch2nh3 ch2nh2 CHjNH,
I II III IV
В природе обнаружен L-O. (I), который входит в
состав коллагена и фосфатида, выделенного из Myco-
bacterium phlei; в виде фосфорного соединения L-O.
обнаружен в экстракте мышц эмбриона теленка.
Ъ-алло-О. (Ш) содержится в дентине зубов человека.
Остальные стереоизомеры получены разделением син-
тетич. рацематов. L-О, в кристаллич. виде получен
в виде монохлоргидрата (т. пл. 221 — 225°), моно-
пикрата (т. пл. 224—227°) и некоторых производ-
ных. L-О. хорошо растворим в воде и кислотах;
[а]д5° =4-14,5° (2% в 6 я. НС1); рА'«г = 2,13;
рА7г2=8,62; р7Г<г3 = 9,67; р! =9,15.
О. дает общие реакции аминокислот; осаждается
пикриновой и фосфорновольфрамовой к-тами. Методы
количественного определения О. основаны на окис-
лении его иодной к-той:
HJO,
CH2(NH2)CH(OH)CH2CII2CH(NH2)COOH--->
—>HCHO4-N’H34-HJO34-CHOCH,CH3CH[NH21COOH
О количестве О. судят, определяя образовавшийся
формальдегид, аммиак или израсходованную иодную
к-ту.
Получают L-О. из гидролизатов желатины. Синте-
тически смесь рацемич. форм О. получают различными
методами, напр. из глутаминовой к-ты через а,е-
дифталимидо-6-кето-Н,Е-капроат или из диэтил-
ацетамидомалоната и акролеина.
Лит.: Майстер А., Биохимия аминокислот, пер. с
англ., М., 1961; Biochemical preparations, v. 8, N, Y.—L.,
1961, p. 55—62; Greenstein J. P.,Winitz M., Chemi-
stry of the amino acids, v. 3, N. Y.—L., 1961, p. 1996.
Л. И. Линевич.
ОКСИЛИКВИТЫ — взрывчатые смеси жидкого
кислорода с твердыми органическими пористыми ве-
ществами, называемыми поглотителями. В качестве
поглотителей применяют преимущественно волок-
нистые целлюлозные материалы (измельченный мох,
солому и т. п.), иногда в виде прессованных брикетов;
поглотители, богатые углеродом (сажа, уголь), обра-
зуют О., опасные в обращении. Из порошкообразного
поглотителя готовят патроны в пористой оболочке
(напр., мешковина), к-рые пропитывают жидким кис-
лородом непосредственно на месте произ-ва взрывных
работ. Время от окончания пропитки до момента, когда
в патроне О. еще сохраняется количество кислорода,
достаточное для получения нужной энергии взрыва за
счет реакции окисления поглотителя, наз. време-
нем жизненности О.; это время зависит от
характера поглотителя, размеров и конструкции пат-
рона, условии взрывных работ и составляет от не-
скольких минут (патроны малого диаметра) до не-
скольких часов.
О. весьма мощное вторичное взрывчатое вещество.
По силе взрывного действия О., в зависимости от
свойств поглотителя и содержания кислорода в мо-
мент взрыва, могут резко отличаться: от ВВ, подобных
черному пороху, с четко выраженными метатель-
ными свойствами, до мощных ВВ, обладающих силь-
ным бризантным действием. Преимущества О.— дос-
тупность исходных компонентов и безопасность в слу-
чаях отказов (через нек-рое время после заряжания
патрон О. становится невзрывчатым из-за испарения
кислорода). Недостатки О.— сложность организа-
ции взрывных работ. О. находят применение при
взрывных работах, гл. обр. в районах, куда затрудне-
на доставка обычных взрывчатых веществ.
Лит.: Марченко Л. Н., Оксиликвиты па открытых
горных разработках, М., 1955; Яхонтов А. Д. [и др.],
Оксиликвиты, их производство и применение, М., 1958.
Л. II. Марченко.
ОКСИМЕТИЛФОСФОНОВАЯ КИСЛОТА (оке и-
м е т и л ф о с ф и и о в а я кислота) —см. Фос-
фоновые кислоты.
ОКСИМИРОВАНПЕ — см. Оксимы.
ОКСИМЫ — функциональные производные аль-
дегидов и кетонов, образующиеся из них при дейст-
вии гидроксиламина:
R . R ч
>С-0 -}-H2NOH—> )C = NOH
R'z
701
ОКСИМЫ — ОКСИНАФТОЙНЫЕ кислоты
702
О.— жидкости или сравнительно низкоплавкпе кри-
сталлы; хорошо растворимы в органич. растворителях
и плохо — в воде. Для получения О. карбонильное
соединение обычно нагревают с солянокислым гидро-
ксиламином в присутствии эквивалентного количест-
ва или избытка щелочи в водном или спиртовом
р-ре; О. получают также в среде пиридина в отсутст-
вие щелочи, а иногда и в кислой среде.
О. можно получать также нитрозированием нек-рых
углеводородов (1) или соединений, содержащих ак-
тивную метильную или метиленовую группу (2),
окислением первичных аминов (3), восстановлением
нитросоединений (4):
хсн2 — сн2 ХОС1 СНз
СН2 У СН2 —> СН2 ^C=NOH (1)
хсн2-сн2 hv хсн2-сн2
C,HtlONO
СН.СОСбН,------->C„H5COCH = NOH (2)
C2H5ONa
R . Н2О2 R.
)CHNH3------> )C = NOH (3)
Rz/ сольМоН'7
H2
C2H,CH2NO2-----zC2H3CH = NOH (4)
Cu; NH,
При нагревании с водными р-рами минеральных
к-т О. гидролизуются, регенерируя карбонильное co-
о. используют для выделения, идентификации и
количественного определения карбонильных соеди-
нений, а также в качестве промежуточных продуктов
в нек-рых синтезах. Оксим циклогексанона исполь-
зуется в производстве капрона; оксим дпацетила (ди-
метилглиоксим) — для определения никеля и др.
Лит.: Неницеску К. Д., Органическая химия, т. 1,
М., 1962, с. 695; Вейганд К., Методы эксперимента в
органической химии, пер. с нем., ч. 2, М., 1950, с. 288.
Е. М. Рохлин.
ОКСИНАФТОЙНЫЕ КИСЛОТЫ СцН80з, мол. в.
188,18 — замещенные нафталина, содержащие окси-и
карбоксильную группы. Константы нек-рых О. к. и
их производных приведены в таблице.
Технич. значение имеют 1-окси (I) и З-окси-2-нафтой-
ные (II) к-ты:
II
О. к. вступают в реакции электрофильного замеще-
ния. При хлорировании и бромировании 1-окси-2-
нафтойной к-ты в CIISCOOH атом галогена вступает
в положение 4; бромирование З-окси-2-нафтойной
к-ты двумя атомами брома в присутствии SO3 дает
единение. С безводными минераль-
ными к-тами происходит-Беклаиа пе-
регруппировка. При дегидратации
альдоксимов образуются нитрилы
(5). Взаимодействие О. со щелочами
или с алкоголятами щелочных ме-
таллов приводит к соответствующим
солям. Последние можно алкилиро-
вать, причем образуются О-алкиль-
ные производные; эти же производ-
ные получаются из О-алкилгидрок-
силаминов и карбонильных соеди-
нений (6). При алкилировании О. в
отсутствие оснований образуются
Положение заместителей Свойства Т. пл. °C производных О. к.
о.
раствори- Ае я ° «S ЕГ S е- ное
ОН соон °C ' мость в воде, % g=s с 3 s s я S ЕС S S S Б Я л еб g
2 « © © И с- еб еб О Е-
1 2 200 0,058 (17°) 0,55 (100°) 78 49 86 — 156 —
9 1 1 57-9 80 55 —. 186—8 172 131,5
3 9 222—3 0,1 04 (25°)а 75 85 99 217 243—4 186
6 2 245-8 — — — — 1 98 224
а Хорошо растворим в горячей воде.
нитроны (7). Галогенированием О.
получают галогенонитрозопроизвод-
ные (8) (см. Пилотти реакция), восстановлением —
первичные амины
лы (10):
RCH=NOH
(9), нитрованием — псевдонитро-
(СН3СО)2О
-------4—RCN
(5)
R R Rx
C=NONa + R"J —»- XC = NpR"-« C=O + H2NOR" (6)
R' R< R' О
Rx Na R"1 Rx I
(9) zCHNHa ” zC=N-R (7)
* \ / R'
C=NOH
О. обычно существуют в виде двух стереоизомерных
форм (син- и а.нти-):
R-C-R' R -C-R'
II И
НО — N N-OH
Методы анализа О. обычно связаны с гидролизом их
до карбонильного соединения и гидроксиламина;
иногда для количественного определения О. исполь-
зуют их восстановительные свойства.
4,7-дибром-3-окси-2-нафтойную к-ту. Сульфирование
1-окси-2-нафтойной к-ты в конц. H2SO4 при 30° при-
водит к смеси 5- и 7-сульфопроизводных; нагревание
с олеумом при 100° дает 5,7-дисульфо-3-оксп-2-наф-
тойную кислоту. При нагревании З-окси-2-нафтойной
кислоты в водном р-ре с Na2SO3 и МпО образуется
4-сульфопроизводное.
1-окси-2-нафтойная к-та сочетается с диазосоеди-
нениями в положение 4(a); в случае избытка диазо-
соединения (2 моля) происходит вытеснение СООН-
груипы с образованием 2,4-бис-бензолазо-1-нафтола.
В случае З-окси-2-нафтойной к-ты азосочетание идет
также в положение 4(a). Практич. интерес представля-
ет взаимодействие арилидов З-окси-2-нафтойной к-ты
(азотолы) с формальдегидом с образованием произ-
водного (III), содержащего метилольную группу и
обладающего еще большими субстантивными свойст-
вами, чем азотолы. При азосочетанип метилольная
группа вытесняется азогруппой:
СООН
он
СН2ОН
III
RNgX(
N=N-R
О. к. получают по Колъбе-Шмидта реакции карбо-
ксилированием а- и |3-нафтолов. При карбоксилиро-
вании |3-нафтола в зависимости от условий образуется
703
оксиндол
704
либо 2-окси-2-нафтойная к-та (IV), либо З-окси-2-
нафтойная к-та (V):
СООН
АА-°н
L^JL^J-COOH
Арилиды (амиды) 1-окси-2-нафтойной к-ты приме-
няют как цветообразующие (голубые) компоненты в
цветной фотографии, а арилиды З-окси-2-нафтойной
к-ты — как азосоставляющие для получения нераст-
воримых азокрасителей методом холодного (ледяного)
крашения. Арилиды О. к. получают взаимодействием
хлорангидридов оксинафтойных к-т с ароматич. ами-
нами. Часто (в случае 2,3-оксинафтойной к-ты) для
получения арилидов смесь О. к. и амина обрабатыва-
ют в подходящем растворителе РС1з или SOC12 при на-
гревании; в этих случаях полезно добавление третич-
ного амина или ZnCl2. Образование красителя из
арилида 1-окси-2-нафтойной к-ты происходит по ре-
акции:
проявитель
голубой краситель
4,4'-Мстилен-бис-3-окси-2-нафтойнан к-та, С2.Н1ВО6, мол. в‘
388,05—почти бесцветные кристаллы с темп-рой разложения
выше 285°; не растворяются в воде, спир-
те’ эФи₽е и бензоле, трудно растворяют-
\ | CUUH оя в хлороформе, хорошо — в нитробен-
L JL ОН золе и пиридине; образует трудно раство-
римые в воде устойчивые на воздухе неги-
Р и гроскопичные соли с хинином, стрихнином
|Пг и др алкалоидами, соли с акрихином, ви-
Ю тамином В, и др. Соли не имеют горького
ОН вкуса, под действием к-т распадаются на
СООН исходные компоненты. Эти факторы обу-
словливают их применение в фармако-
логии. Получают 4,4’-метилен-бис-3-окси-
2-нафтойную к-ту при нагревании формальдегида с р-ром 3-ок-
си-2-нафтойной к-ты в разбавленной щелочи.
Лит.: Ворожцов Н.Н., Основы синтеза промежуточ-
ных продуктов и красителей, 4 изд., М., 1955; Венката-
раман К., Химия синтетических красителей, пер. с англ.,
т. 1—2, М., 1956—57; Donaldson N., The chemistry and
technology of naphthalene compounds, L., [1958]. В.А.Пучков.
ОКСИНДОЛ (лактам 2-аминофепилуксусной кис-
лоты) C8H7NO, мол. в. 133,14—бесцветные иглы;
т. пл. 126—127°; т. кип. 195°/17.ил1, 227°/73 мм', раст-
ворим в горячей воде и обычных органич. раствори-
телях; образует легкорастворимый в воде хлоргид-
рат. При действии этилата натрия или амальгамы
натрия в бензоле образуется натриевая соль, при
действии AgNOa — серебряная соль О. Известны про-
изводные лактамной (I), енольной (II) и лактимной
(III) форм О.
I и
Действие CH3J на Na-соль О. приводит к N-метил-
производным О., а на Ag-соль — к О-метилпроизвод-
ным. У незамепдевного О. преобладает лактамная фор-
ма и обнаружить равновесие лактама с енольной и
лактимной формами методами спектроскопии не уда-
лось. Нагревание О. с Ва(ОН)2 до 150° приводит к
Ba-соли о-ампнофенилуксусной к-ты, подкисление
к-рой дает О. Восстановление О. литийалюминийгид-
ридом приводит к индолу.
О. легко вступает в реакции электрофильного заме-
щения в бензольном ядре, заместители вступают гл.
обр. в положения 5 и 7. В то же время в пиррольном
кольце активным местом является положение 3. Бро-
мирование О. в СС1< дает 3,3-дибромоксиндол, а в
воде 5-бром-или 5,7-дибромоксиндол. К аналогичным
результатам приводит хлорирование и иодирование.
При действии РС15 на О. образуется 2,3-дихлориндол.
При нитровании и сульфировании получаются 5-или
5,7-производные. Действие HNO2 приводит к изатин-
2-оксиму.
О. вступает в реакции типа альдольной конденсации
и Клайзена конденсации с образованием соответствую-
щих 3-производных. Конденсация О. с нитрозобензо-
лом приводит к (IV), с изатином— к изоиндиго (V), с
бензальдегидом—к 3-бензальоксиндолу, со сложными
эфирами — к 3 ацилоксиндолу.
О. получают восстановлением изатина (с промежу-
точным образованием диоксиндола) с помощью раз-
личных восстановителей. Прямое восстановление иза-
тина в О. может быть достигнуто амальгамой натрия
или электролитически. Применяют и другие методы,
напр. переводят изатин действием РС15 в 3,3-дихло-
роксиндол и восстанавливают последний. О. может
быть также получен щелочным плавлением индол-2-
сульфокислоты, образующейся при сульфировании
индола пиридинсульфотриоксидом. Препаративным
способом получения О. является восстановительная
циклизация о-нитрофенилуксусной к-ты (Байер),
получаемой из о-нитротолуола по Рейссерту (1).
Другими методами являются синтезы Гинсберга (2),
Бруннера (3) и Штолле (4):
(СООС2Н5)2
Ссн2сосос2н5
г
no2
Многие производные О. были изучены при исследова-
нии химии алкалоида физостигмина (эзерина) и дру-
гих индольных алкалоидов и являются промежуточ-
ными продуктами синтеза этих алкалоидов.
705
ОКСИПИРИДИНЫ
706
Диоксиндол (3-оксиоксиндол, лактам о-ами-
номиндальной кислоты), мол. в. 149,14— кристаллы;
___________ т. пл. 180°; разлагаются при 195°; ра-
\ j i-ОН створим при нагревании в воде и эти-
ловом спирте. Диоксиндол — амфотер-
NH ное соединение, растворимое в кисло-
Vi тах и щелочах; в водных р-рах легко
окисляется кислородом воздуха с об-
разованием изатида. Более энергичное окисление при-
водит к изатину. При ацетилировании образуются
моно- (VII) и диацетильные производные (VIII); при
галогенировании, как и в случае изатина, образуются
5- и 5,7-дигалогенопроизводные.
руются в положения 2 и 4); сочетается с солями диа-
зония с образованием азокрасителей; реагирует с
СН2О; алкилируется (диазометаном и галогеналки-
лами) по атому кислорода, напр.:
2- и 4-О., каки соответствующие алинопиридины, про-
являют двойственную реакционную способность; они
реагируют с образованием производных двух форм —
оксипиридина и пиридона:
Способы получения диоксиндола и его производных
см. Изатин. Диоксиндол получен также восстанов-
лением о-нитроминдальной к-ты:
СНОН
I
СООН
'2
Sn
нсГ
Следует также отметить реакцию Мартине:
IX
Промежуточный продукт в синтезе Мартине может
быть получен из циангидрина изатина. Диоксиндол
образуется в смеси с изатином при действии разб.
к-т на дигидроиндиго:
Свойства диоксиндола широко изучались в связи с
тем, что он является одним из важнейших производ-
ных ИНДИГО. М. Н. Преображенская.
ОКСИПИРИДИНЫ (пиридолы) — замещенные пи-
ридина, содержащие одну или несколько гидроксиль-
вых групп; а- и у-О. и их нек-рые произ-
водные часто нзз. п ир и донам и. Моно-
• Ijs4з] ₽ оксипиридивы C5H5ON, мол. в. 95,10—бес-
аг чА “ цветные кристаллы; легко растворимы в во-
де и спирте, огравиченно — в эфире, бен-
золе, лигроине. О. образуют соли с кислотами (на-
пример, CSHSON-HC1) и едкими щелочами (так называ-
емые пиридоляты); последние, подобно фенолятам,
разлагаются СО2:
СО;
Н2О
ОН + NaHCOj
3-0.— типичный фенол (рКа 4,86); дает пурпурное
окрашивание с водными р-рами FeCls; легко брони-
руется, сульфируется и нитруется с образованием
моно- и дипроизводных (замещающие группы ориенти-
2-оксипиридия
2-лиридоя 4-оисипиридии 4-пиридов
Так, алкилирование 2- и 4-О. галогеналкилами или
окисями приводит к образованию N-алкилпиридонов:
снг-снг
soz
снгснгон
CH3J
В то же время при действии на 2- и 4-0. диазометана
образуются соответственно 2-метоксипиридин и смесь
4-метоксипиридина с М-метил-4-пиридоном. Как по-
казывают результаты исследований УФ- и ИК-спект-
ров, в нейтральных водных р-рах 2- и 4-0. существуют
в пиридонной форме, а их соли имеют оксипиридино-
вую структуру:
сг
2- и 4-0. вступают так же, как и 3-0., в реакции элек-
трофильного замещения (ориентация происходит в
положения 3 и 5), сочетаются с солями диазония и т. д.
«Фенольные» свойства 2- и 4-0. несколько ослаблены:
окрашивание с FeCl3 они дают более слабое, чем 3-0.,
не реагируют с СН2О и т. д. О. могут быть получены
декарбоксилированием оксиниридинкарбоновых к-т,
взаимодействием аминопиридинов с азотистой к-той,
сплавлением пиридинсульфокислот с едкими щелоча-
ми и др. способами. 2-0. обычно получают из 2-амино-
пиридина или непосредственным гидроксилированием
пиридина по Чичибабину (пропусканием паров пири-
дина над КОН при 300—320°). 3-0. получают сплав-
лением легкодоступной пиридин-3-сульфокислоты с
КОН (при 170° в течение 1—2 час.) или из 3-амино-
пиридина. 4-0. обычно получают гидролизом хлор-
гидрата М-(4-пиридил)пиридиниумхлорида (1), ре-
же — пиролизом хелидамовой к-ты (II), образующей-
ся при взаимодействии NH3 с хелидоновой
к-той (I) (2):
Нг°-> HO-Z^N-HCl t
150е,8час- \/
+ NHtCl + ОСНСН-СНСНгСНО . О)
© 23 к.х.э. т. з
707
ОКСИПРОЛИН—3-ОКСИТИОНАФТЕН
708
Нек-рые замещенные О. нашли применение в меди-
цине: пиридоксин и др. витамины группы Вв; пири-
доновые аналоги сальварсана (напр., 3,3'-диамино-5,5'-
арсено-2,2'-пиридон); рентгеноконтрастные препа-
раты (напр., 5-иод-2-О. и натриевая соль N-ацетил-
5-иод-2-О. и т. д.). К производным 2-0. относится ал-
калоид рипинин. Известны также диоксипиридины
C5H5O2N, мол. в. 111,10 (2,4-, 2,5-, 2,6-, 3,4- и 3,5-),
с темп-рой плавления соответственно 265°, 248°, 195°,
< 250° (разл.), 237—239°, а также триоксипиридины
C6H5O3N, мол. в. 127,10 (напр., 2, 4, 6, т.пл. 218—220°).
Лит.: Мошер Г., в кн.: Гетероциклические соединения,
под ред. Г. Эльдерфилда, пер. с англ., т. 1, М., 1953, с. 337—42,
408—19. В. Н. Фросин.
ОКСИПРОЛИН (у-оксипролин, 4-оксипирроли-
дин-2-карбоновая кислота) C5HeNOs, мол. в. 131,14 —
_________ оксиаминокислота. Известно 4 опти-
I 1 чески активных стереоизомера: L-ок-
сипролин(Т), алло-Ь-оксипролип (II),
D-оксипролин (III) иалло-Й-оксипро-
лин (IV) и два рацемата — И,Ь-оксипролин и D, L-ал-
ло-оксипролин.
В природе обнаружен L-O. (I), к-рый входит преим-
в состав белков соединительной ткани — коллагена
(до 13%) и эластина. Алло-L-O. (II) встречается в сво-
бодном состоянии в цветах сандалового дерева и вхо-
дит в состав ядовитых пептидов бледной поганки —
фаллоидина и а- и 0-аманитинов. Остальные стерео-
изомеры получены разделением смеси синтетич. ра-
цематов. L-O.— кристаллы, т. пл. 274° (с разл.),
хорошо растворим в воде; [а]д = — 72,5° (1% в воде
при 22,5°), —0,5° (1% в 5 н. НС1) и —77° (0,25% в ук-
сусной к-те при 24—26°); рАа] 1,92; рКа> 9,74; р7 5,83.
Для «лло-1.-О. [а]д = —59,5° (2% в’воде), —18,8°
(2% в 5 н. НС1) и —30° (2% в уксусной к-те). О. дает
общие реакции аминокислот. С нингидрином О. дает
красное окрашивание, к-рое быстро переходит в жел-
тое, с изатином — синее. С рейнекатом аммония О. об-
разует нерастворимую соль C5H.,OSN-(C,H7N6S1Cr2) H2O.
Количественно О. определяют колориметрически,
используя нингидриновую реакцию или реакцию с
п-диметиламинобензальдегидом пиррол-2-карбоновой
к-ты, образующейся при окислении О. перекисью
водорода.
Получают L-О. из гидролизатов коллагена и неко-
торых др. белков. Синтетически смесь рацемич.
форм О. получают различными методами, напр. исходя
из 1,2-эпоксифталимидопропана или из 0-, у-дибром-
пропилацетамидомалонового эфира. В организме О.
образуется из пролина. Продуктами его превращения
в организме являются пиррол-2-карбоновая и глута-
миновая к-ты. О. является антагонистом пролина.
Лит.: Майстер А., Биохимия аминокислот, пер. с
англ., М., 1961; G г е е n s t е 1 n J. P.,Winitz М., Che-
mistry of the amino acids, v 3, N.Y.—L., 1961, p. 2018; Bioche-
mical preparations, v. 8, N.Y.—L., 1961, p. 114. См. также лит.
при ст. Аминокислоты. Л. И. Линевич.
ОКСИТЕТРАЦИКЛИН (террамицин) — антибиотик
группы тетрациклинов. О. кристаллизуется из воды в.
виде дигидрата, теряющего „ он Он N(CH3),
воду при нагревании до 7 3 \/ н н L пи
100° в вакууме;т.пл.184,5—
J J П И г l6aD |4аА j
185,5° (с разл.), [а]^=
= -199° (С=0,5% в 0,03 ‘%н Д ПаТ*2П£ CONH2
н. НС1); умеренно раство- он °
рим в низших спиртах и кетонах, несколько хуже —
в воде, в кислой и щелочной среде растворимость О.
в воде значительно повышается, что связано с его
способностью образовывать соли как с кислотами,
так и с основаниями (рКа 3,5; 7,6; 9,2). Хлоргид-
рат О. имеет т. пл. 204°; ди-О-ацетат, т. пл. 208—
213° (с разл.), +214° (С = 0,4 в СНзОН).
Молекула О. отличается полифункциональностью-
и способна к разнообразным превращениям; та-
ковы, например, дегидратация СОМН2-группы в CN
(террамицинонитрил) или превращение ее в резуль-
тате Манниха реакции в группу CONHCH2N(R)21
эпимеризация (СНз)2М-группы (кватримицин), ее
восстановительное отщепление или превращение в
четвертичную аммониевую группу; ацилирование
ОН-групп при С5, С,?) и Ci2a, кислотная дегидратация
при С5а—С6 (ангидротеррамицин), гидрогенолиз груп-
пы ОН при С6 с одновременной эпимеризацией, вос-
становительное дегидроксилирование при Ci2a. Даже
в довольно мягких гидролитич. условиях происходит
расщепление Сц—Сца, Сца—С12, Ci2—С12а-связей,
приводящее к образованию продуктов глубокого
распада молекулы О.
Наиболее характерным для молекулы О. является
фенолполи-0-карбонильная система С10—Сз, обладаю-
щая сильными комплексообразующими свойствами и,
вероятно, играющая основную роль в проявлении О.
антибиотич. активности (предполагается, что в основе
последней лежит подавление О. процессов белкового
синтеза у микроорганизмов путем связывания в проч-
ные хелаты многовалентных катионов, необходимых
для нормального функционирования нек-рых энзима-
тич. систем).
О. образуется при аэробной ферментации Strepto-
myces rimosus и S. flavus (наряду с римоцидином),
S. gilvus, S. platensis, S. vendargensis (наряду с вен-
гицидом), S. armillatus, S. aureofaciens var. oxytetra-
cyclini var. nov. и ряда других актиномицетов. О.
относится к антибиотикам широкого спектра дейст-
вия: он высоко активен in vitro и in vivo против боль-
шого числа грамположительных, грамотрицательных
и кислотоустойчивых бактерий, а также в отношении
ряда риккетсий, крупных вирусов и простейших. Всо-
четании с устойчивостью и довольно низкой токсич-
ностью это обусловливает широкое применение О.
в медицине, особенно при лечении заболеваний, воз-
будители к-рых устойчивы к менее токсичным лекар-
ственным веществам, напр. пенициллинам.
Лит.: Шемякин М. М. [ид р.], Химия антибиотиков,
т. 1, 3 изд., М., 1961, с. 181; Hochstein F. А. [а. о.],
J. Amer. Chem. Soe., 1953, 75, № 22, 5455. Ю. А. Берлин.
3-ОКСИТИОНАФТЕН (3-окситионафтен, тиоин-
доксил) C8H6OS, мол. в. 150,20 — кристаллы, бесцвет-
ные иглы (из воды); т. пл. 71°; по запаху напоминает
1-нафтол; перегоняется с водяным паром; легко раст-
ворим в большинстве органич. веществ. О. реагирует
709
ОКСИТОЦИН —8-ОКСИХИНАЛЬД ин
710
в таутомерных формах, повторяя многие свойства
1-нафтола и индоксила:
О. сочетается с диазосоединениями; при окислении
дает тиоиндиго; конденсируется по активной метиле-
новой группе, что широко используется в технике для
синтеза несимметричных индигоидных красителей. О.
получают циклизацией фенилтиогликоль-о-карбоно-
вой к-ты щелочным плавлением или нагреванием в
уксусном ангидриде. Образующаяся в качестве про-
межуточного продукта З-окситионафтен-2-карбоно-
вая к-та легко декарбоксилируется:
G СООН
SCH2COOH
-Н2О; -со2
Для синтеза кубовых красителей находят применение
хлор-, метил-, алкоксизамещенные 3-0. и 3-оксибенз-
тионафтена.
Лит.: Коган И. М., Химия красителей, 3 изд., М.,
1956; Венкатараман К., Химия синтетических краси-
телей, т. 2, Л., 1957. Н. С. Докунихин.
ОКСИТОЦИН — гормон задней доли гипофиза,
циклич. октапептид; кристаллич. продукт, хорошо
прол-лейц-глиц- NH2
I
цистеин
асл- NH,
I
глут- NH2
иеол
I
дир
цистеин
растворим в воде, мол. в. 1007, изо-
электрич. точка при pH 7,7. О. встре-
чается всегда совместно с другим гор-
моном — вазопрессином, имеющим
очень близкое строение. Исследова-
ние О. и вазопрессина является пер-
вым в истории химии природных со-
единений примером установления
строения полипептидных гормонов,
подтвержденного полным синтезом. За эти исследова-
ния их основной автор В. Дю-Виньо удостоен Но-
белевской премии.
О. был выделен из задней доли гипофиза крупного
рогатого скота и свиньи и очищен методом противо-
точного распределения. Индивидуальность получен-
ного гормона была подтверждена хроматографией
и др. методами.
На основании совокупности всех данных изучения
строения О. была постулирована его структура. При
действии на О. металлич. натрия в жидком аммиаке
происходит размыкание цикла с образованием дитио-
ла, к-рый, взаимодействуя с бензилхлоридом (также
в жидком аммиаке), дает ди-8-бензильное производ-
ное, не обладающее биологич. активностью О. Однако
дебензилирование этого производного (Na в жидком
NH3) с последующим окислением кислородом воздуха
приводит к веществу, полностью идентичному по хи-
мич. и биологич. свойствам исходному О. Таким обра-
зом, ключевым соединением в синтезе О. является
К-карбобензокси-ди-8-бензил-нонапептид,
Бз
I
Кбз-цист-Э-тир-изолейц-глут (NH3)
I
(КН2)асп-цист-8-прои-лейц-глиц (NH2)
I
Бз
где Кбз — карбобензокси, Бз — беизокси
к-рый и был синтезирован из соответствующих L-ами-
нокислот общепринятыми методами синтеза пептидов.
Дебензилирование синтетич. а-пептида с последующим
окислением и снятием защитных групп привело к
образованию О., полностью идентичного природ-
ному гормону. Этот синтез подтвердил постулирован-
ную структуру О.
Основное биологич. действие О. состоит в том, что
он при секреции из гипофиза в кровяное русло вызы-
вает усиление сокращений гладкой мускулатуры бе-
ременной матки. Это усиление имеет существенное зна-
чение в стимуляции родовой деятельности. Помимо
того, О. играет важную роль в качестве фактора, оп-
ределяющего отдачу молока (но не его образование)
грудной железой. Об активности О. говорят такие
данные: при внутривенном введении кормящим жен-
щинам 0,1 мкг О. уже через 20—30 сек. наблюдается
существенное усиление отделения молока грудной
железой; стимуляция сокращений беременной матки
также наблюдается при малых дозах гормона. О. об-
ладает частично биологич. действием вазопрессина:
способностью повышать кровяное давление (вазопрес-
сорный эффект) и снижать образование мочи (антиди-
уретический эффект), что объясняется близостью их
строения.
Лит..: Дю-Виньо В., в сб.: Современные проблемы
биохимии, М., 1957; Straub F., Biochemie, Praha, 1962.
См. также лит. при ст. Гормоны. В. А. Яковлев.
5-ОКСИТРИПТАМИН — См. Серотонин.
5-ОКСИТРИПТОФАН [|3-(5-оксииндолил-3)-аланин]
C11H12N2O3, мол. в. 220,22 — аминокислота; природ-
ный L-5-окситриптофан —
бесцветные кристаллы, т. ---л-СН2СНСООН
пл, 273°, [а]” = - 32,5° J NH2
(вода); рацемич. 5-окси-
триптофан — бесцветные кристаллы, т. разл. 293—295°.
О. образуется в организме животных в результате
гидроксилирования триптофана под влиянием трип-
тофан-5-гидроксилазы. Микроорганизм Chromobac-
terium violaceum также превращает триптофан в О.
При действии 5-окситриптофандекарбоксилазы, со-
держащейся, напр., в почках, О. превращается в 5-ок-
ситриптамин (серотонин). Введение О. в организм,
минуя желудочно-кишечный тракт, вызывает галю-
циногенный эффект, подобный действию диэтиламида
лизергиновой кислоты.
Синтетически О. можно получить из 5-бензилокси-
индола (I); действием на него формальдегида и диме-
тиламина получают (II); обрабатывая последний
Н2
СН2СНСООН
nh2
НССЖНСН(СООС2Н6)2 в щелочной среде с последую-
щим гидролизом и декарбоксилированием в щелочной
среде, получают (III), при восстановлении к-рого в
присутствии Pd образуется О. Оптически активный
О. получен расщеплением Н-карбобензокси-5-бен-
зилокситриптофана хинином с последующим гидро-
лизом и гидрогенолизом.
Лит.: Суворов Н. Н., Морозовская Л. М.
Сорокина Г. М., Ж. общ. химии, 1961, 31, вып. 3 1937’
U tlenfriend S. [а. о.], J. Biol. Chem., 1956 219’ № 1’
335; Clark С. Т., Weissbach Н., Udenfriend s’
там же, 1954, 210, № 1, 139; Morris A. J., Armstrong
М. D., J. Organ. Chem., 1957, 22, 306. М. Н. Преображенская.
8-ОКСИХИНАЛЬДИН (о-оксихинальдин, 2-метил-
8-оксихинолин) C10H9ON, мол. в. 159,18—бесцветные
кристаллы с запахом фенола; т. пл. ..
74°; т. кип. 266— 267°; нерастворим в p’Y'ji
воде, хорошо растворим в спирте, аце-
тоне, хлороформе и бензоле. О. обра- ' N
зует соли с кислотами и щелочами; он
сублимируется при нагревании до 100°; летуч с пара-
ми воды; окрашивает водные р-ры FeCla в зеленый
23*
711
8-ОКСИХИНОЛИН
712
цвет. Получают О. сплавлением 8-сульфохинальдина
со щелочью, а также нагреванием смеси о-аминофе-
нола, паральдегида и конц. НС1.
О., подобно 8-оксихинолину, образует с катионами
многих металлов малорастворимые внутрикомплекс-
ные соединения, различающиеся по
^*1 своей устойчивости.
L J-CH3 Ниже приведены константы устой-
। чивости (ступенчатые) комплексов 8-
Q—Ме/Я оксихинальдина с нек-рыми катиона-
ми, определенные при 25° в 50%-ном
диоксане. Константа диссоциации, определенная при
тех же условиях, равна рХ'(НА) = — 11,71.
Катион 1$ К. 1g к2 Катион 1g К, 1g к2
Cd2 + 9,00 Мп2 + 7,44 6,55
Се3+ 7,71 Ni2 + 9,41 8,35
Со2+ 9,63 8,87 Pb2 + 10,30 8,20
Си2 + Mg2 + 12,48 '5,24 11,52 4,48 Zn2+ 9,82 —
О. осаждает ионы металлов менее полно, чем 8-ок-
сихинолин; с алюминием О. не взаимодействует. При-
меняют О. для гравиметрия, определения ряда эле-
ментов, напр. в уксуснокислом р-ре, содержащем
ацетат, для определения Zn, Cd, Mo(VI), W(VI) и др. О.
используется для разделения элементов методами экст-
ракции, напр. при анализе сплавов для удаления ме-
шающих ионов металлов перед экстракцией алюми-
ния с 8-оксихинолином. О. может быть обнаружен
так же, как 8-оксихинолин, по возникновению флуо-
ресценции при хемосорбции на гидроокиси Mg.
Лит.: Hollingshead R. G., Oxine and i ts deriva-
tives, V. 1—4, L., 1954—56. В. H. Фросин.
8-ОКСИХИНОЛИН (о-оксихинолин, оксин), мол. в.
145,15, C9H7ON —светло-желтые кристаллы с харак-
терным запахом; т. пл. 75—76°; т. кип.
266,6°/752 мм; растворим в спирте, аце-
|т, I ! J тоне, хлороформе, бензоле, кислотах и
Iff щелочах, весьма трудно — в холодной во-
ОН де и эфире; легко летуч с парами воды;
7Сосч.= 2 'Ю-10; дает положительную ре-
акцию Бейлыптейна; окрашивает водный р-р FeCi3 в
зеленый цвет; окисляется КМпО« и др. окислителями
в хинолиновую к-ту; с бромом дает 5-бром- и 5,7-дп-
бромпроизводные; сочетается с солями диазония (в по-
ложение 5) и т. д.
О.— один из наиболее широко применяемых орга-
нич. реагентов. Взаимодействие О. изучено более чем
с половиной элементов периодич. системы (рис.). В ос-
н L1 Na К Be Mg| Ca Sc Ti V Cr В C N |a7|Si P Mn Fe Co Nl Cu Zn Ga|Ge As He 0 F Ne S Cl Ar Se Br Kr
Rb Cs Sr Y Zr Nb Mo Ba La Hf Ta W Tc Ru Rb|pd Ag Cd In Sn Sb Re Os Jr Pt]Au Нг TI Pb Bl Те J Xe Po At Rn
Fr Ra Ac|Th|Pa|U
новном это ионы металлов, к-рые осаждаются из вод-
ных р-ров аммиаком. О. образует малорастворимые
в водных р-рах кристаллич. внутрикомплексные сое-
динения (хелаты), напр. типа Me(II) (C9HeON)2,
Me=7,Mg, '/,Al и т. д.
Me (III) (C9HeON)3, со многими катионами метал-
лов. Составы соединений многовалентных металлов
с О. могут несколько различаться: Се(С9НвОМ)«,
Th(C9HeON)4-C9H7ON, MoO2(C9HeON)2, WO2(C9H,ON)2,
UO2(C9HeON)2. В табл. 1 приведены рТС диссоциа-
ции (ступенчатые) комплексов О. с нек-рыми ка-
тионами, определенные при 25° в 50 %-ном диоксане;
при тех же условиях рТС (НА) = 11,54.
Таблица 1
Катион pAi ptf2 Катион p*. pA,
Cd2 + 9,43 7.68 Mg3 + 6,38 5,43
Ce3 + 9,15 7,98 Mn2+ 8,28 7,17
Co2 + 10,55 9,11 Ni2 + 11,44 9,94
Cu2 + 13,49 12,73 Pb2 + 10,61 8,09
La3+ 8,66 7,74 Zn2 + 9,96 8,90
Для Th*+ при ионной силе р-ра ц = 0,1 (СЮ ) найдено:
рА, рК2 Р-Кз РК,
10,45 9,95 9,45 8,95
Количественное осаждение О. различных металлов
происходит при определенных значениях pH так же,
как и экстракция (табл. 2).
Таблица 2
Металл .Значения pH
для полного осаждения для максимальной экстракции
Алюминий 4,4—9,8 4,8—6,7; 8,2—11,5
Ванадий 2.7—6,1 3,3-4,5
Висмут 4,8—9,4 4,0—5,2
Вольфрам 4,9 —5,6 2,4—4,За
Галлий 3,1—> 3,0—6,2
Железо (III) .... 2,8—11 ,2 1 ,9—12,9°
Индий 2, 5—3,0 >3,0
Кадмий 5,4—13,3 8,0 неполное извлечение
Кальций 9,2—12,7 13,0
Кобальт 4,2 — 11 ,6 9.4—12,6 >6,80
Магний 10.20
Марганец 5,9—9,5 7,2-12,5 в присутствии тартрата и ферроцианида
Медь 5,3—14,5 2,8—14 в области высоких зна- чений pH — тартратная среда
Молибден 3,3—7,6 1 ,6—5,6 в присутствии ЭДТА
Никель 4,6—10,0 4,5—9,5
Ниобий — 1 н. NHiOH цитрат- ная среда
Плутоний ~5,0 4,0—8,0 амилацетат Pu (IV), Ри (VI)
Свинеп 8,4 — 12,3 6,5—8,5 8,4—12,3
Скандий 6,5—8,5
Таллий (III) .... 4,0—8,0 6,5—7.0 экстрагируется на 85-89%
Титан 4,8—8,6 3,8—5,0 в присутствии Н3О*
Торий 4,4—8,8 4,9
Уран 4,1—8,8 4,7-8,0
Церий (III) .... — 9,9—10,5
Цинк 4,6—13,4 4,6—13,4 неполное извлечение
Цирконий — уксусная к-та — ацетат
d Б присутствии этилендиаминтетрауксусной к-ты.
& В области высоких значений pH применяют тартрат-
ною среду.
В уксуснокислом р-ре О. можно использовать для
определения Al, Zn, Cd, Си, Mn, Fe, Ti, Со, Ni, In,
Мо, W, Zr, Nb, Th, U и др., в аммиачном р-ре — для
определения Mg, Cd, Al, Ga. Ряд металлов можно
определить фотометрии, или флуорометрия, методами;
для разделения металлов с использованием О. приме-
няют экстракцию. О., имеющий атом азота с основ-
ными свойствами и фенольную гидроксильную группу,
в водной среде проявляет амфотерные свойства. Вслед-
713
ОКСОНИЕВЫЕ СОЕДИНЕНИЯ
714
ствие этого О. экстрагируется неполностью при рН<5
и рН>9. Коэфф, распределения нейтрального соеди-
нения между хлороформом и водой (18°) равен 720,
если величина pH водного р-ра находится в пределах
5—9. Для экстракции он употребляется обычно в ви-
де 1%-ного р-ра в хлороформе. Возможности приме-
нения О. для разделения значительно расширяются
при использовании различных маскирующих веществ
(ЭДТА, цианиды, ацетаты и др.). Реагент чувствителен
к свету; р-р его необходимо хранить в темной посуде
в прохладном месте. Сам реагент так же, как и его
производные, может быть обнаружен по флуоресценции
его р-ров или после сорбции на гидроокиси магния.
О. получают действием едкой щелочи на 8-сульфо-
хинолин; последний образуется на холоду из хино-
лина и дымящей серной к-ты; О. может быть также
получен щелочным гидролизом 8-хлорхинолина и
Скраупа реакцией (из о-аминофенола и глицерина в
присутствии серной к-ты). Кроме использования для
определения металлов, О. и нек-рые его производные
применяют в качестве фунгицидов [напр., медную соль
(C8HeNO)2 Си] и антисептиков амебоцидного и наруж-
ного действия [ятрен, хинозол, виоформ (5-хлор-7-
иод-8-оксихинолин), 7-иодо-7-оксихинолин-5-сульфо-
кислота и др.]. Бензоат О. применяют для борьбы
с нек-рыми болезнями растений.
Лит.: Hollingshead R. G., Oxine and Its derivati-
ves, v. 1—4, L., 1954—56; Моррисон Дж., Фрейзер
Г., Экстракция в аналитической химии, пер. с англ., Л.,
i960, с. 168; Кольтгоф И. М. [ид р. ], Объемный анализ,
пер. с англ., т. 3, М., 1961; Берг Р., Применение о-оксихи-
нолина в аналитической химии, пер. с нем., М., 1937; W е 1-
с h е г F. J., Organic analytical reagents, v. 1, N. Y.—[a. o.],
1947. И. 11. Ефимов, В. H. Фросин.
ОКСОНИЕВЫЕ СОЕДИНЕНИЯ — соединения, со-
держащие положительно заряженный атом кисло-
рода, связанный тремя ковалентными связями с ор-
ганич. радикалами и ионной связью с анионом, напр.
+ __
[(С2Н6)зО] BFi . О. с. принадлежат к ониевым соедине-
ниям. О. с.— твердые солеобразные соединения с до-
вольно высокой темп-рой плавления (см. таблицу),
растворимые в полярных растворителях (ацетон,
CH3NO2, CeH6NO2, CH3CN, жидкий SO2, СН2С1),
нерастворимые в эфире и углеводородах.
Анионы и т. пл. О. с., °C
Катионы SbCl6“ BF, 4 FeCl, 4
(СН3)3О + 156—158° 141° 81°
(С2Н5)3О+ 131° 92° 74°
(СН,)2ОН' 116° — —
с.н6-о; хсна 168° — —
(СанБ)ас5а — 226° —
,н с»на-с; + хон 181° — —
.0—н C.Hs-C'f \осна 112-114° —
с„н6сн=6сна 166° — —
(CHaO)2C = dcH, 130—131° 138° —
а Т. пл. [<CSH5),OJ +ВГ- 182», [(C»Hs)aOJ + J" 177-178°.
Алифатич. О. с. устойчивы только при наличии
комплексных анионов (BF , SbCl , FeCl ит. п.).
Соли с простыми анионами легко распадаются. Аро-
матич. О. с. очень устойчивы, с водой не реагируют
и разлагаются лишь при сильном нагревании:
+ _185°
[(С.н5)ао ]J —> C.HJ 4- (СаН8)2О
Все О. с. обладают электропроводностью в жидком
SO2. По электропроводности [(СНз)зО]ВР4 близок к
KJ. Очень нестойкая гидроокись триэтилоксония, по-
добно [(C2H5)3SO]H и [(C2H5)4N]OH, является сильным
основанием.
Известны О. с. след, типов: 1) [К3О] + Х_, где R—
алкил или арил; 2) карбоксониевые соединения, про-
изводные карбонильных соединений, напр. (I) или (II):
bf4-
3) О. с., в к-рых оксониевый атом кислорода входит
в ароматич. цикл, напр. соли пирилия (см. Пири-
лиевые соли), ксантилия, флавилия.
О. с. первых двух типов получают: а) взаимодейст-
вием спиртов, простых эфиров, альдегидов и кетонов
с комплексными к-тами H4[Fe(CN)a], Нз[Ре(СМ)в],
Нз[Со(С1М)в], H2 [PtCl,]; б) при действии НС1 на аддук-
ты спиртов, простых эфиров или к-т с галогенидами
металлов или при действии SbCl5 на смесь кислород-
содержащего соединения и жидкого НС1. Реакции
ведут при минус 70—80° и полном отсутствии влаги:
(C2H6)2O4-SbCl6-[-HCl —. [(C2H„)2OHjSbCl6
ceH5OH+HCi+sbci5—> [свн5он2]8ЬС1Г
Вместо SbCl5 можно брать FeCla, А1С13, SnCh, ZnCl2
и ВРз и HF.
Триалкилоксониевые соли впервые были получены
Меервейном в 1937 след, образом:
С1СНг-СН С2н5 С2Н5
3 I /О + 4 ZO—BF3 + 2 ХО-----------
снг с2н6 с2н5
выход 90 7,
СНгС1-СН-СН2ОС2Н5 в
о
Триарилоксониевые соединения образуются при раз-
ложении борфторидов арилдиазониев в дифениловом
эфире:
ArN2BF, + (CsHs)2O —► [<С3Н5)2О - ArJBF?
или внутримолекулярным замыканием:
Известны и др. способы получения О. с., напр. про-
стой и удобный способ получения [А1кзО] с помощью
безводного AgBF4 или действие галогеиалкилов на
эфираты BF3 или SbCl5:
(С2Н5)2О + C2HsBr + AgBF, ► [(С2Н5)аО ] + BF, + AgBr
(С2Н5)2О SbCl5 + C2HSC1 —> [(С2Нь)а0 ]+ SbCla
Триалкилоксониевые соединения, ведущие себя в ре-
акциях как свободные алкил-катионы, являются са-
мыми сильными из известных в настоящее время
715
ОКСОСИНТЕЗ
716
алкилирующих средств, алкилируют анионы всех
слабых к-т:
[R3O] + BF~ 4-CN- —> RCN4-R2O4-BF“
Спирты, фенолы, органич. к-ты, амины алкилируются
ими в очень мягких условиях—при комнатной темп-ре
или при слабом нагревании. Третичные О. с. алки-
лируют соединения, к-рые другими алкилирующими
средствами алкилируются плохо или вовсе не алкили-
руются (мочевина, ацетамид, диэтилсульфоксид).
О. с. можно использовать для получения других
ониевых соединений, напр. сульфониевых соединений
из сульфидов. О. с. применяют для получения недо-
ступных ранее ацеталей амидов к-т и лактамов, ока-
завшихся чрезвычайно реакционноспособными со-
единениями:
Н
I
(CH3)2N — С = O-|-[RaO]BF4 ——>
—► [(CH3)aN-
С помощью триалкилоксониевых соединений изучен
механизм нек-рых катализируемых к-тами реакций,
в к-рых предполагалось промежуточное образование
оксониевых ионов. Первичные и вторичные О. с.
очень неустойчивы, легко гидролизуются уже влагой
воздуха. Устойчивость их в сильной степени зависит
от аниона:
Fe(CN)’-, PtCl®-» SbCl” > FeCl~ > A1C1” > BFj>
> SnCr“> ZnCl“
H"! H
I RONa I ,OR
C....ORJ BF,-HCH,),N-Cf
'OR
Карбоксоииевые соединения могут реагировать как
оксониевые соли и как карбониевые соли. С сильными
нуклеофильными реагентами они реагируют след,
образом:
СН2-0 +
| хс-свн5
сн2-ох
NaCN ^Нг °'-c^'CN
СН2— OZ SCeH5
BF
+ NaBF
СН2-0 ,ОС2Н5
I с
C2HsONa CH2-OZ SC6H6
«
С более слабыми нуклеофильными реагентами они
взаимодействуют как оксониевые соли. Триарилок-
сониевые соединения, в отличие от триалкилоксоние-
вых, представляют собой чрезвычайно прочные со-
единения с очень высокой темп-рой разложения, хи-
мически очень инертные.
При действии хлористого ацетила на эфират SbCl6
в отсутствие влаги при —75° образуется оксониевая
соль, способная ацилировать ароматич. соединения
без катализатора и при темп-рах ниже 0°:
,0 SbCl5 г о "I
СН3-С^ +0Ra-------->- II + SbClJ-
ХС1 -75° [CH3-C-OR2J
Лит.: Губен И., Методы органической химии, пер. с
нем., т. 3, вып. 3, М., 1935, с. 109; И е е г we i n Н. [и. а.],
J. prakt. Chem. [21, 1937, 147, 257; 1939, 154, 83; Meerwein
Н. [и. a.], Liebigs Ann. Chem., 1960, 632, Н. 1—3, 38; К 1а-
g е s F. [и. а.], там же, 1953, 592, Н. 2, 81; К 1 a g е s F.
[и. a.], Chem. Вег., 1959, 92, № 8, 1819, 1828; Meerwein
Н. [и. а.], там же, 1956, 89, 2060; Несмеянов А. Н.,
Избранные труды, т. 2, М., 19.59, с. 458, 467; Хю кнель
В., Теоретические основы органической химии, пер. с нем.,
т. 1, М., 1955, с. 103. Т.П. Толстая.
ОКСОСИНТЕЗ (гидроформилирование) — присо-
единение окиси углерода и водорода к олефинам с обра-
зованием альдегидов:
ХС = С^ + СО4-Н3—> XCH-t-CHO
х 7 I
Реакция происходит при обработке олефина эквимоле-
кулярной смесью СО и Н2 (напр., водяным газом)
при 50—250° и 100—700 ат, обычно же при 75—200®
и 100—300 ат, в присутствии Со-содержащих катали-
заторов. Активным началом катализаторов является
гидрокарбонил кобальта НСо(СО)4, образующийся в
условиях О. как из металлич. Со, так и из его соеди-
нений, через промежуточную стадию дикобальтокта-
карбонила Со2(СО)в. В присутствии катализаторов,
содержащих Со в виде карбонилов, скорость О. выше,
чем в присутствии металлич. Со или его солей, так
как в этом случае медленной стадией реакции является
образование карбонилов. Заранее приготовленный
раствор НСо(СО)4 катализирует О. даже при низкой
темп-ре и атмосферном давлении. Темп-ра и давле-
ние СО и Н2 определяют концентрацию Co(CO)s и
НСо(СО)4, и, чем выше темп-ра, тем большее давление
требуется для предотвращения разложения карбо-
нилов.
О. представляет собой гомогенный каталитич. про-
цесс, действительный механизм к-рого не установлен,
хотя схематически его можно представить как раскры-
тие водородом первоначально образующегося цикло-
пропанонового кольца:
\/
сн
I
хс-сно
Характер продуктов О. определяется строением
олефина и условиями реакции. а-Олефины нормаль-
ного строения дают смесь примерно равных количеств
альдегидов с прямой и разветвленной углеродной
цепью. Изомерные олефины с неконцевой двойной
связью образуют смеси того же состава, что объяс-
няется равновесием, устанавливающимся между оле-
финами под влиянием сильной к-ты — НСо(СО)41
н +
сн3сн2сн2сн = сн2 jzi сн3сн2сн=снсн3
I \
CH3CH2CH2CH2CH2CHO i (СН,СН2)2СНСНО
45% СН3 10%
СН3СН2СН,СНСН0
45%
Аналогичная картина наблюдается для разветвлен-
ных олефинов:
сн3
сн3сн2с=сн3
сн3сн—сн=сн2
сн3
сн3-с =сн-сн3
сн3
I I
CH3—CH—СН-СНО 5%
СН3—СН-СН2СН2СНО
СН3 50%
СН3СН2СНСН2СНО
I
СН, 45%
Изобутилен образует почти исключительно продуй
соответствующий присоединению СО по метиленов
группе:
сн3. сн3. СН’\
ХС=СН2—> хсн-сн2сно+сн3—с—сно
Снз СН“ 90 - 95% СН3Х 5-10%
Тетразамещенные этилены вступают в О. только
предварительным перемещением двойной связи;
с
СН3
сн3 I
ХСН-С = СН2
СН/
сн3. ,сн3 н+
;C —С'
СН/ ХСН3
сн3
СН3. I
хсн-снсн2сно
сн/
Известны примеры вовлечения в О. олефинов с
функциональными заместителями (винилацетат, ал-
лилацетат, аллилцианид, эфиры кротоновой, фумаро-
717
ОКТАМЕТИЛ —ОКТАНОВОЕ ЧИСЛО
718
вой, ундециленовой к-т), а, Р-Иепредельные альдеги-
ды и кетоны в условиях О. обычно восстанавливаются
в соответствующие насыщенные соединения. Гидро-
формилирование стирола, а также фторолефинов тоже
сопровождается значительным гидрированием. Для
диолефинов наблюдается гидрирование одной двойной
связи и гидроформилирование другой; так, из бута-
диена получается смесь С5-альдегидов. Помимо гид-
рирования олефинов, побочными реакциями при О.
являются восстановление альдегидов до спиртов, их
конденсация по альдольному илп кротоновому типу,
ацетализация, полимеризация и т. п., а также обра-
зование кетонов, напр.:
2СН2=СН2-|-Н2-|-СО —> (СН3СН2)2 с = о
Нек-рые из этих реакций в известных случаях могут
быть превращены в целевые. Так, гидроформилиро-
вание высших олефинов обычно проводят так, чтобы
получать спирты. О. в присутствии аммиака дает
соответствующие амины (восстановительное аминиро-
вание альдегидов).
Продукты О. находят широкое практич. примене-
ние. В особенности это относится к высшим спиртам,
используемым для получения пластификаторов, ла-
ков, смазочных масел, синтетич. моющих средств.
Непосредственно используют и нек-рые альдегиды.
Так, и-масляный альдегид применяют для получения
поливииилбутираля, и-масляной к-ты, и-бутилового
спирта. О. в промышленных условиях осуществляют
по разным технология, схемам, к-рые в основном раз-
личаются формой вводимого в реакционную зону
катализатора и способом выделения кобальта из
получаемого продукта.
Катализатор (Со на кизельгуре) может быть введен
в реакционную зону в форме взвеси в жидкости, где
и образуются карбонилы кобальта. После разложе-
ния карбонилов в стадии декобальтизации металлич.
Со осаждают на кизельгуре, катализатор отфильтро-
вывают из полученного продукта и возвращают в
реактор. В схеме с неподвижным катализатором кар-
бонилы кобальта получают в специальном аппарате
(кобальтизере), заполненном пемзой с осажденным
на ней Со. Карбонилы растворяют в сырье или рас-
творителе и этот р-р направляют в реактор. Разруше-
ние карбонилов кобальта происходит в другом аппа-
рате, также заполненном пемзой, на к-рой осаждается
выделяющийся металлич. Со. В процессе работы коли-
чество Со в кобальтизере уменьшается, а в декобаль-
тизере увеличивается. После уменьшения количества
Со на пемзе в кобальтизере до определенного предела
направление потоков жидкости и газов изменяют и
карбонилы кобальта получают во втором аппарате,
а разрушают в первом. При проведении процесса с со-
лями кобальта катализатор в реакционную зону вво-
дят в виде р-ра солей жирных к-т или же его подают
в виде суспензии в сырье или растворителе. Карбо-
нилы кобальта разлагаются при нагревании продукта
или при обработке его растворами к-т. Однотипность
технология, схемы для произ-ва различных альде-
гидов и спиртов является важным преимуществом О.
Оксосинтез открыт О. Реленом в 1938.
Лит.: Орчин М., в кн.: Химия углеводородов нефти,
под ред. Б. Т. Брукса и др., пер. с англ., т. 3, М., 1959, с. 286;
Гольдштейн Р., Химическая переработка нефти, пер.
с англ., М„ 1952, с. 179; Н о u b е n-W е у 1, 4 Aufl., Bd 4,
TI 2, Stuttgart, 1955, S. 375; [Рудковский Д. M.J, в
кн.: Основы технологии нефтехимического синтеза, под ред.
А. И. Динцеса и Л. А. Потоловского, М., 1960, с. 325;
Силич М. И., Получение альдегидов и спиртов методом ок-
сосинтеза, М., 1960. В. Л. Дяткин, М. И. Силич.
ОКТАМЕТИЛ — СМ. Фосфорорганические инсекти-
циды.
ОКТАН (и-октан) СН3(СН2)6СН3, мол. в. 114,224—
бесцветная жидкость со слабым запахом; т. пл.
—56,795°; т. кип. 125,665°; <7*0,70252 (20°), 0,69849
(25°); 1,39743; Ркрит. 24,64 ат, 7крит. 296,2°, <7крит.
0,235; теплота сгорания жидкого О. до жидкой во-
ды и СОг (25°) 1307,53 ккал/моль', теплота образования
газообразного О.— 49,82 ккал/моль (25°); вязкость
0,47 спуаз (20°); поверхностное натяжение 21,76 дин/см\
т. воспл. 240°; т. всп. 13°; теплоемкость Ср 0,3952
кал/г-град (25°);концентрационные пределы взрываемо-
сти паров 0,8—6,0%. Содержится в нефтях, бен-
зинах, продуктах синтеза из СО и Но по Фише-
ру— Тропшу. В пром-сти рекомендуется выделять
О. ректификацией или в более чистом виде — при
сочетании ректификации с выделением нормальных
парафиновых углеводородов мочевиной или молеку-
лярными ситами. В лаборатории О. синтезируют гид-
рированием октиленов, полученных дегидратацией
и-октанолов, реакцией Вюрца из 1-хлорбутана или
другими реакциями, общими для получения предель-
ных нормальных углеводородов. Химич, свойства О.
аналогичны свойствам других гомологов ряда нор-
мальных парафинов. При пиролизе О. получают в
основном метан, этилен, этан. При крекинге на алю-
мосиликатном катализаторе возрастает выход угле-
водородов С3—С5. В присутствии ароматизирующих
катализаторов при 450— 500° из О. получают с высо-
ким выходом смесь о-ксилола и этилбензола (см. Аро-
матизация). При дегидрировании О., напр. при по-
вышенных темп-рах на хромовых катализаторах, об-
разуются октены. Под действием водорода в присут-
ствии никелевых или кобальтовых катализаторов при
повышенных темп-рах и давлениях протекает гидро-
генолиз (гидрокрекинг О.) с образованием метана и
других предельных нормальных углеводородов. При
мягком окислении О. получаются октанол, октаналь-
дегид, перекиси и гидроперекиси О., а также продук-
ты деструкции, альдегиды, кислоты и др. Под дей-
ствием С12 и SO2 в присутствии перекисей образуются
сульфохлориды. Наряду с н-октаном существуют еще
17 изомерных октанов общей ф-лы С8Н18, из них три-
метилпентаны и тетраметилбутан обладают высокими
октановыми числами (97—105) и являются желатель-
ными компонентами моторных топлив (см. Изооктан).
Сам н-октан имеет низкое октановое число (19).
М. И. Розепгарт.
ОКТАНОВОЕ ЧИСЛО — показатель, характеризу-
ющий детонационную стойкость (антидетонационные
свойства) бензинов и керосинов; определяется на спе-
циальных установках, к-рые представляют собой мало-
размерный стенд, состоящий из одноцилиндрового дви-
гателя с переменной степенью сжатия, электротор-
моза, соединенного с двигателем, пульта управления
с приборами, а также аппаратуры для замера детона-
ции. Двигатель при определении О. ч. работает прп
строго постоянных условиях. Определение О. ч. за-
ключается в сравнении неизвестного (испытуемого)
бензина с эталонными топливами. Для этого одноци-
линдровый двигатель, работающий на испытуемом
бензине, изменением степени сжатия доводят до опре-
деленной степени детонации, а затем подбирают такую
смесь эталонных топлив, к-рая по интенсивности дето-
нации соответствует этому бензину. В качестве эта-
лонных топлив используют индивидуальные угле-
водороды — изооктан (2,2,4-триметилпентан, С8Н18) и
нормальный гептан (С7Н16). Для изооктана, облада-
ющего высокой детонационной стойкостью, этот пока-
затель условно принят за 100 единиц, а для «-гептана,
обладающего низкой детонационной стойкостью, за
нуль. Процентное (по объему) содержание изооктана
в смеси с и-гептаном принято называть О. ч. эталон-
ной смеси.
О. ч. бензина называется процент-
ное содержание изооктана в та-
кой его смеси с и-гептаном, к-рая
при стандартных условиях йены-
719
ОКТАНОВОЕ ЧИСЛО — ОКТИЛОВЫЙ АЛЬДЕГИД
720
т а н и я на специальном одноцилин-
дровом двигателе детонирует так
же, как испытуемый бензин.
Применяются три метода определения О. ч.: мотор-
ный, исследовательский и температурный. Для всех
этих методов используются однотипные одноцилинд-
ровые двигатели. В основу методов положен общий
принцип определения О. ч., т. е. сравнение испы-
туемого бензина с эталонными топливами. Отличи-
тельными особенностями являются основные пара-
метры работы двигателей при проведении испыта-
ний (табл. 1), некоторое специальное оборудование
и аппаратура, а, главное, различное назначение ме-
тодов.
Таблица 1. Условия определения октанового числа
Методы
Параметры моторный исследов ате ль- ский температурный
Число оборотов двигателя, об/мин ......... Темп-ра охлаждающей жид- кости, °C Темп-ра рабочей смеси, °C Угол опережения зажига- ния, градусы Назначение 900 100 149 26 до ВМТ * при степени сжатия 5,0. Переменный Авиа- и авто- бензины 600 100 Не контроли- руется 13 до ВМТ * для всех степе- ней сжатия. Постоянный Автобензины 1200 190,5 104,5 35 до ВМТ для всех степеней сжатия. Постоянный Авиабензины
* Верхняя мертвая точка.
Моторный метод разработан для опре-
деления О. ч, бензинов и обеспечивает надежную оцен-
ку детонационной стойкости бензинов с О. ч. от 65
до 100 единиц. О. ч., определяемые с помощью этого
метода, лучше всего отвечают требованиям авиацион-
ных поршневых двигателей.
Исследовательский метод разрабо-
тан для определения О. ч. автомобильных бензинов;
получаемые результаты определения О. ч. являются
лучшей характеристикой детонационной стойкости
для автобензинов.
Температурный метод разработан для
определения О. ч. высокооктановых авиабензинов;
служит для оценки детонационной стойкости авиа-
Таблица 2. Октановые числа некоторых углеводородов
(по Герну и Смиту)
Углеводороды Октановое число
моторный метод исследоват. метод
Парафиновые
Изобутан 97,6 101,1
Изопентан 90,3 92,3
Изооктан 100,0 100,0
н-Пентан 61,9 61 , 7
н-Гексан 26,0 24,8
Олефиновые
Пропилен 84,9 101,4
Пентен-1 77,1 90,9
Гексен-1 63,4 76,4
Н афтеновые
Метилциклопентан 80,0 91,3
Этилциклопентан 61,2 67,2
Метилциклогексан 40,8 46,5
Ароматические
Бензол 108,0 >100,0
Толуол 103,0 115,0
Этилбензол 97,9 106,0
бензинов и компонентов с октановыми числами от 90
до 115. Авиационные и автомобильные двигатели, в
зависимости от снимаемой мощности и условий экс-
плуатации, большее время работают на двух режи-
мах: максимальной (взлетной) и эксплуатационной
мощностях. На максимальной мощности двигатели
работают на богатых рабочих смесях (коэфф, избытка
воздуха а==0,6—0,7), а на эксплуатационной (крей-
серской) мощности — на бедных рабочих смесях
(c«rl,0—1,1). О. ч. характеризует детонационную
стойкость бензинов на бедной рабочей смеси, т. к.
оно определяется при а=ь0,9—1,1.
Углеводороды, входящие в состав бензинов, обла-
дают различной детонационной стойкостью. Наиболь-
шие О. ч. имеют изопарафиновые и
ароматич. углеводороды, наимень-
шие — нормальные парафиновые;
нафтеновые и олефиновые углеводо-
роды по детонационной стойкости за-
нимают промежуточное положение.
Чем выше О. ч., тем лучше детона-
ционная стойкость бензина. Бензины
в чистом виде имеют сравнительно
низкие О. ч. Для повышения О. ч. к
бензинам добавляют специальные
присадки, наз. антидетонаторами (см.
Антидетонаторы моторных топ-
лив}.
При определении О. ч. различны-
ми методами получают разные ре-
зультаты (табл. 2 и 3); поэтому каж-
дый метод определения О. ч. имеет
свое назначение, определяемое стан-
дартами на бензины. Результаты оп-
ределения О. ч. в зависимости от метода обозна-
чаются: по моторному методу — О. ч./М; по исследо-
вательскому методу — О. ч./И; по температурному
методу — О. ч./Т.
Таблица 3. Октановые числа некоторых топлив
Топливо Октановое число с 4 мл1кг Р-9
моторный метод температур- ный метод
Пиробензол 95,0 92,0
Бензол 101 ,0 98,0
Толуол 104,0 100,0
Изооктан 110,0 111,0
Алкилат 105,0 106,0
Бензин прямой гонки 95,0 96,0
Бензин прямой гонки + 30% пи- робензола 95,0 93,0
Бензин прямой гонки + 30% бензола 94,0 93,0
Бензин прямой гонки + 30% толуола 95,0 94,0
Бензин прямой гонки + 30% изооктана 99,0 100,0
Бензин прямой гонки + 30% алкилата 98,0 99,0
Лит.: Нефтепродукты. Методы испытаний, М., 1961;
Забрянский Е. И., Зарубин А. П., Детонационная
стойкость и воспламеняемость моторных топлив, М., 1958;
Лосиков Б В., Пучков Н. Г., Энглин Б. А.,
Основы применения нефтепродуктов, 2 изд., М., 1959; Мотор-
ные, реактивные и ракетные топлива, под ред. К. К. Папок
и Е. Г. Семенидо, 4 изд., М., 1962. А. П. Зарубин.
ОКТИЛОВЫЙ АЛЬДЕГИД (октаналь, каприло-
вый альдегид) СН3(СН2)вСНО, мол. в. 128,21— бес-
цветная жидкость; т. кип. 172°/766 мм; 85°/35 мм,
65°/11 мм; d,™ 0,8211, Пд 4,4217; с водой не смешивает-
ся, растворяется в спирте, эфире, хлороформе. О. а.
обладает всеми свойствами альдегидов; идентифици-
руют его по семикарбазону, т. пл. 101°, и 2,4-динит-
рофенилгидразону, т. пл. 106°. Количественно опре-
721
ОКТИЛОВЫЙ СПИРТ—ОЛЕАНДОМИЦИН
722
деляют оксимированием и др. методами определения
альдегидов. При.сильном разбавлении обладает при-
ятным апельсиновым запахом; находится в нек-рых
эфирных маслах, может быть получен окислением
или каталитич. дегидрированием октилового спирта.
Применяют О. а. в парфюмерии как душистое вещество
и для синтетич. целей. Э. Е- Нифаптьев.
ОКТИЛОВЫЙ СПИРТ (октанол-1,1-оксиоктан)
СН3(СН2)6 СН2ОН, мол. в. 130,23—бесцветная жид-
кость с ароматным запахом; т. пл.— 16,3°; т. кип.
195,27760 мм, 1.357Ю0 мм, 100,7720 мм; 0,8270;
Пд 1,4304; 4° крит. 384,6°, вязкость 10,640 спуаз (15°),
т. всп. 81°; образует азеотроп с водой, т. кип. 99,4°
(содержит 90% воды); с водой не смешивается, раство-
рим в хлороформе, эфире, спирте. О. с. обладает всеми
свойствами одноатомных спиртов, идентифицируют
его по 3,5-динитробензоату, т. пл. 62°; количествен-
но определяют ацетилированием в среде пиридина.
О. с. находится в свободном виде и в виде сложных
эфиров в нек-рых эфирных маслах; получают ката-
литич. гидрированием каприлового альдегида, кап-
риловой к-ты или ее эфиров. В пром-сти О. с. исполь-
зуют при эмульсионной полимеризации этилена, при
синтезе моно-, ди- и триоктиламинов, каприлового
альдегида и сложных эфиров (карбоновых, фосфор-
ной, фосфиновых, серной к-т), применяемых в ка-
честве синтетич. моющих средств, экстрагентов ряда
металлов, пластификаторов и косметич. средств.
Э. Е. Нифантьев.
ОКТОГЕН(циклотетраметилентетранитрамин,1,3,5,7-
тетранитро-1,3,5,7-тетразоциклооктан) С4Н8К8О8,мол.
вес 296,17 — бесцветные кри-
сталлы моноклинной системы; т.
<,пл. 281—282°, т. всп. ок. 290°;
плотп. 1,96. О. существует в четы-
рех кристаллич. модификациях,
стабильных при темп-рах < 115°,
115—156°, ок. 156° и >156—280°.
О. негигроскопичен, нерастворим в воде, спиртах, бен-
золе, толуоле, эфире; плохо растворим в дихлорэта-
не, анилине, нитробензоле, диоксане; лучше в ацето-
не (4,13% при 56°), нитропарафинах, HNO3. В силь-
ных к-тах и щелочах О. гидролизуется. На разнице
в энергиях активации щелочного гидролиза О. и гек-
согена основан количественный анализ их смесей.
Получают О. нитролизом динитропентаметилентетр-
амина в присутствии NH4NO3 и при взаимодейст-
вии гексаметилентетрамина (уротропина) с HNO3 и
NH4NO3 в присутствии уксусного ангидрида. Очищают
О. перекристаллизацией из ацетона, нитрометана,
HNO3. О.— мощное вторичное взрывчатое вещество,
практически равное по взрывчатым характеристикам
гексогену. Высокая термич. стойкость и высокая
т. пл. в сочетании с большой мощностью позволяют
применять О. для взрывных работ при повышенных
темп-рах, напр. в глубоких и сверхглубоких сква-
жинах.
Лот.: Орлова Е. Ю., Химия и технология бризантных
взрывчатых веществ, М., 1960; Urbanski Т., Chemia I
technologia materiatow wybuchowych, t. 3, Warszawa, 1955.
В. Ф. Жилин.
ОКТОЗЫ C8H16O8 — моносахариды, содержащие
восемь атомов углерода в цепи. В зависимости от ха-
рактера карбонильной группы О. делят на альдо-
октозы СН2ОН(СНОН)8СНО и кетооктозы, или ок-
тулозы СН2ОН(СНОН)5СОСН2ОН. О. дают общие ре-
акции моносахаридов. Получают О. синтетически об-
щими методами удлинения цепей в моносахаридах.
В природе обнаружена одна октулоза — D-глицеро-
D-манно-октулоза (в растении авокадо).
Лит.: Webber I. М., Adv. Carbohydr. Chem., 1962, 17,
49; см. также ст. Моносахариды. Л. И. Линевич.
ОКТЭСТРОЛ [бензэстрол, октофоллин, 2,4-ди- (п-
оксифенил)-3-этилгексан] С20Н2вО2, мол. в. 298,43—
ХСН2
o2n-nz xn-no2
сн2 сн2
o2n-n n-no2
Хсн2х
белый кристаллич. порошок; т. пл. 160—162°; трудно
растворим в воде, хлороформе, бензине, бензоле,
растворим в растительных маслах, хорошо растворим
в спирте, эфире и в разб. р-рах щелочей. При добав-
С2Н5 СН
СН-СН-СН
I
С2Н5
он
лении к уксуснокислому р-ру О. бромной воды обра-
зуется желтый осадок. При взаимодействии О. с ще-
лочным р-ром диазосульфаниловой к-ты образуется
интенсивно окрашенное соединение, что используется
для количественного определения. О. получают, ис-
ходя из анисового альдегида (I) и п-метоксибутиро-
фенона (II). Полученный при их конденсации а-этил-
п, п'-диметоксихалкон (III) с бромистым этилмагнием
образует 1,3-дианизил-2-этилпентанон-1 (IV). При его
взаимодействии с бромистым метилмагнием, дегидра-
тации полученного продукта, гидрировании и деме-
токсилировании получают О.
Н3СО
О. проявляет высокую физиологич. активность, при-
меняется при лечении болезней, вызываемых эстро-
генной недостаточностью.
Лит.: Машковский М. Д., Лекарственные средства,
4 изд., М., 1960; Государственная фармакопея СССР, 9 изд.,
М„ 1961; Киприанов Г. И., Куценко Л. М., Ж.
прикл. химии, 1958, 31, № 4, 665; Stuart А. Н. [а. о.],
J. Amer. Chem. Soc., 1945, 67, № 9, 1475; 1946, 68, № 5, 729;
Burger A., Walter R., J. Amer. Chem. Soc., 1950, 72,
M 5, 1988. Г. M. Кадатский.
ОЛЕАНДОМИЦИН (матромицин) C35H61NO12, мот.
в. 687,5—антибиотик из группы антибиотиков-мак-
ролидов.
СН3 СН3 СН3 СН3 СН3
сн3—сн—сн-сн-сн-со-с-сн2-сн-сн-сн-сн-сн-со
1,3 12 11 10 9 /8\7 6 5 4 3 2.
I / \ I I
ОН О —СН2 OR OR'
I--------:-------о-------------------
R-Дезозаминил
R'-олеандрозил
В молекуле О. содержится макроциклич. лактонное
кольцо. Агликоновая часть молекулы представ-
ляет собой полиоксиэпоксилактон, гликозидно свя-
занный с аминосахаром дезозамином (при С(6)) и
нейтральным сахаром
L-олеандрозой (приС(3)).
О.-основание— кристал-
лы горького вкуса, т. пл.
110°; [«]“= — 65° (2%,
метанол); дает устойчи-
N(CH3)2
.Дезозвмин
L - олеандроза
вые соли с минеральными к-тами: хлоргидрат, т. пл.
134—135°, [а]д= — 54° (1%, метанол); фосфат, т.
пл. 150°, [а]д= — 52° (1%, метанол). Основание
723 олеиловый спирт — оливомицин 724
хорошо растворимо в большинстве органич. растворите-
лей, нерастворимо в воде и углеводородах, хорошо рас-
творимо в разб. водных р-рах кислот и щелочей.
Основание и хлоргидрат О. имеют пик малой интенсив-
ности в УФ-области спектра при 286—289 ммк.
Основание, соли О. и хлоргидрат-хлоргидрин О.
[т. пл. 152°, [а]р= — 79° (1%, метанол)] легко обра-
зуют сольваты с различными органич. растворителя-
ми, напр. сложными эфирами или кетонами. О. ста-
билен при комнатной темп-ре в водном р-ре в течение
24 час. в пределах pH от 2,0 до 9,0; нестоек при на-
гревании в кислой среде. При действии на основание
О. уксусным ангидридом в присутствии пиридина
образуется триацетилолеандомицин, темп-ра пл. 176°
[с разл., [cc]q= — 23° (1%, метанол)], рА’а=6,6. Это
соединение биологически активно и не обладает горь-
ким вкусом, создает более высокую концентрацию
в крови и моче (по сравнению с основанием О.).
О. образуется при глубинной ферментации штамма
Str. antibioticus. Определяют О. обычными микробио-
логии. методами, а также колориметрически. Для иден-
тификации используют нерастворимый в воде кристал-
лич. комплекс с тетрафенилбором, т. пл. 157—161°.
О. активен в отношении грамположительных микро-
организмов, микобактерий, риккетсий, крупных ви-
русов и простейших. На грамотрицательные организ-
мы, за исключением Neisseria, Hemophilis и Brucella,
О. не действует. О. активен по отношению к микроор-
ганизмам, устойчивым к пенициллину, стрептомици-
ну, тетрациклинам, хлорамфениколу и полимиксину
В. Для хлоргидрата О. ЛД50 при внутривенном вве-
дении мышам составляет 550 мг/кг. Применение О.
в клинике в виде его хлоргидрата и фосфата дало
положительный результат. Весьма эффективно при-
менение триацетилолеандомицина, а также комбина-
ций О. с тетрациклинами (сигмамицин; алететрин).
Лит.: Шемякин М. М. [и др.], Химия антибиотиков,
т. 1, 3 изд., М., 1961, с. 641. Д. М. Трахтенберг.
ОЛЕИЛОВЫЙ СПИРТ (tyuc-9-октадеценол-!)
СН3(СН2),—СН=СН(СН2)8ОН, мол. в. 268,47—бесцвет-
ная жидкость; растворим в спирте и эфире; т. пл. 5,5—
7,5°; т. кип. 158°/2.и.и; 0,8489, Пр 1,4607; диполь-
ный момент 1,72 D. О. с. при каталитич. гидрировании
образует октадециловый спирт, при окислении пере-
кисью водорода — 9,10-эпоксиоктадециловый спирт,
при взаимодействии с бромом — 9,10-дибромоктаде-
канол, озонолизе — пеларгоновый и ш-оксипеларго-
новый альдегиды:
о,
СН3(СН2)7СН = СН(СН2)8ОН —
/О ,О
—>СН3(СН2)7С^ +НОСН3(СН2), С"
Н ХН
При алкилировании бензола О. с. в присутствии хло-
ристого алюминия получается смесь 9 и 10-фенилокта-
деканолов:
О. с. изомеризуется при нагревании до 220° в элаиди-
новый спирт, являющийся тра«с-9-октадеценолом-1;
при взаимодействии с серной к-той он превращается
в дисульфат октадеканола -1,10, а с ацетиленом — в
октадециленвиниловый эфир; последнее соединение
рекомендуется как мономер и пластификатор. Окись
этилена присоединяется к О. с. с образованием водо-
растворимых полигликолевых эфиров (перегаль О):
о
СН3(СН2),СН = СН(СН2)„ОН + /\ —>
сн2-сн2
—»-СН3(СН2)7СН = СН(СН2)8—О—(СН2СН2О)—ОСН2СН2ОН
Эти соединения применяют как диспергирующие,
эмульгирующие и эгализирующие средства.
О. с. получают восстановлением простейших эфи-
ров олеиновой к-ты:
Н2
СН3(СН2)7СН = СН(СН2)7СООС2Н5 —►
—»-СН3(СН2)7СН = СН-(СН2)7СН2ОН
В виде эфиров высших жирных к-т О. с. входит в
состав спермацетового и нек-рых др. природных ма-
сел. Э. Е. Нифантьев.
ОЛЕИНОВАЯ КИСЛОТА (^ис-октадецен-9-овая-1
кислота) СН3(СН2)7СН=СН(СН2)7СООН, мол. вес
282,46— кристаллы, существует в виде двух кристал-
лич. модификаций: а-, т. пл. 13,4° и 0-, т. пл. 16,°3;
т. кип. 225-226710 мм; d™0,8906; п2'1 1,45823; кислот-
ное число 199; иодное число 89,87. О. к.— одна из
наиболее распространенных в природе кислот; встре-
чается практически во всех растительных и животных
жирах в виде триглицерида. В оливковом масле ее
содержится 70—85%, в миндальном 75%, в пальмо-
вом 41—48%, в подсолнечном и соевом 33%. Отделе-
ние от сопутствующих линолевой, линоленовой и
стеариновой к-т представляет значительные трудно-
сти. Для выделения более или менее чистой 0. к.
смесь к-т (после гидролиза жира) фракционируют
и затем подвергают неоднократной кристаллизации
из 90%-ного метанола. Иногда предварительно про-
изводят селективную гидрогенизацию жира так, что
линолевая и линоленовая к-ты восстанавливаются до
О. к., дальнейшую обработку ведут аналогично.
О. к. обладает свойствами ненасыщенной к-ты с
изолированной двойной связью. Последняя разрывает-
ся нормальным образом при окислении, озонирова-
нии, действии надкислот, галогенировании и т. д.
Из О. к. могут быть получены ее функциональные про-
изводные обычным образом. При расщепительном
окислении О. к. дает смесь азелаиновой и пеларгоно-
вой к-т. О. к. может быть легко изомеризована в эла-
идиновую к-ту (транс-изомер О. к.), катализаторами
являются HNO2, Hg(NOa)2, NaHSOa и др. В виде
эфиров и глицеридов (часто в смеси с глицеридами
других кислот). О. к. применяют в качестве основы
для получения лаков, покрытий, эмалей, олиф, кра-
сок; нек-рые эфиры применяют 'также в качестве
пластификаторов. Соли О. к. являются мылами.
Н. А. Несмеянов.
ОЛИВКОВОЕ МАСЛО — см. Жиры растительные.
ОЛИВОМИЦИН — антибиотик, выделенный из
культуральной жидкости Actinomyces olivoreticuli
экстракцией бутилацетатом при pH 5,0. Полученный
препарат представляет собой смесь, состоящую, по
крайней мере, из 3 компонентов с различной биоло-
гич. активностью. Основной компонент, получаемый
при разделении исходного препарата методом хромато-
графии на А120з или на кремневой к-те, составляет
70% суммарного препарата, кристаллизуется из смеси
СНС1з с петролейным эфиром в виде желтых игл с
т. пл. 164—166°, мол. в. ок. 1000. Максимум поглоще-
ния в УФ-области при 227, 275 и 405 ммк, в ИК-об-
ласти (с КВг) при 770, 815, 912, 1060—1070, 1125,
1155, 1175, 1200, 1245, 1260, 1325, 1370—1400, 1450,
1520, 1585, 1640, 1740, 2920, 2950, 2990 см"1, широкая
полоса при 3450 см~'. О. обладает кислыми свойст-
вами, растворим в воде в виде натриевой соли, [а] “=
=—41,7° (метанол, С=0,5%); содержит: 57,2% С;
725
ОЛИГО—ОЛИГОСАХАРИДЫ
726
7,31%И; 7%—ОСНз; 6,6% ацетильных групп. О.
реагирует с солями тетразолия, диазосульфаниловой
к-той и FeCl3. При гидрировании в присутствии А дамса
катализатора поглощает 2 моля водорода, теряя при
этом биологич. активность. При действии на О.
уксусного ангидрида в пиридине в течение 96 часов
образуется белый кристаллич. неактивный ацетат
с т. пл. 204—205°, максимум поглощения в УФ-об-
ласти при 220, 265 и 320 ммк. О. образует соединения
с Al3+, Fe3+, Ni2+, Со2+, Mg2+, при этом наблю-
дается батохромный сдвиг спектра (см. Батохромный
аффект). Комплексы О. с А13+ и Fe’+ не кристалли-
зуются.
О. обладает противораковыми свойствами (в опытах
на животных), относится к липофильным антибиоти-
кам кислого характера, к-рые химически плохо оха-
рактеризованы. К этой группе относятся такие анти-
биотики, как абурамицин, ауреловая кислота,
М5-18903, NSCA-649, LA-7017, 24-10, хромомицин,
митрамицин. О. хроматографически отличается от
указанных антибиотиков.
Лит.: Шемякин М. М. [и др.], Химия антибиотиков,
т. 1—2, М., 1961; Бражникова М. Г. [и др.], Антибиоти-
ки, 1962, № 3; 1 9 64, № 2; Кру г л я к Е.Б. [и др.], там же, 1963,
Л 12; Nlshimora Н. га. о.], J. Antibiot., 1957, 10, М 5,
205; Philip J. Е. [а. о.т, Antibiot. a. Chemother., 1953, 3,
№ 12, 1218; Gale R. М. [а. о.], Antibiot. Ann., 1958—1959,
489; Schmitz Н. [а. о.], Antibiot. a. Chemother., 1960,
10, № 12, 740; S е n s i Р. [а. о.], там же, 1958, 8, № 5, 241;
Bring! N. V. [а. о.], Hindust. Antiblot. Bullet., 1960, 2,
Гй 4, 120; Mizano К., J. Antibiot. A., 1963, № 1, 22; К о p-
paka V. [a. o.], Antibiot. a. Chemother., 1962, 12, № 3.
M. Г. Бражникова.
ОЛИГО — приставка в названиях полимерных со-
единений, указывающая на то, что молекула полимера
построена из небольшого числа мономеров. Так,
олигосахаридами называют сложные угле-
воды, построенные не более чем из 10 остатков мо-
носахаридов, олигоуронидами —• полимеры,
в состав к-рых входит несколько остатков уроновых
к-т, олигонуклеотидами — полинуклео-
тиды, содержащие только 8—10 остатков нуклеоти-
дов, олигопептидами — пептиды, в состав
к-рых входит ок. 10 остатков аминокислот (последний
термин широкого распространения не имеет). Резкой
разницы в свойствах олигосоединений и низших пред-
ставителей соответствующих им полимерных соедине-
ний нет, и граница между ними проводится условно.
О. в названиях сложных слов указывает на малое
количество чего-либо, на отклонение от нормы в сто-
рону уменьшения, напр. олигоцитемия — уменьше-
ние количества форменных элементов в крови, олигу-
рия — уменьшение выделения количества мочи, оли-
годинамич. действие металлов — действие на организм
небольших количеств металлов. л. И. Линевич.
ОЛИГОМЕР — полимер низкого мол. веса (обычно
не более нескольких тысяч). В химии высокомолеку-
лярных соединений О. часто называют полимеры со
степенью полимеризации меньше той, при к-рой начи-
нают проявляться специфические свойства полимера,
связанные с гибкостью его макромолекул. Важное зна-
чение имеют О., к-рые содержат функциональные
группы, обусловливающие способность молекул О.
соединяться друг с другом с образованием длинных
молекулярных цепей или трехмерных сетчатых струк-
тур. Примерами таких О. могут служить смолы феноло-
формальдегидные в стадии резола, смолы эпоксидные,
полиэфиракрилаты, к-рые полимеризуются за счет
концевых двойных связей, и др.
Лит.: Лосев И. П., Тростянская Е. Б., Химия
синтетических полимеров, М., 1964; Берлин А. А. |и др.],
Хим. пром-сть, 1962, № 12. В. А. Кабанов.
ОЛИГОСАХАРИДЫ (олигозиды, голозиды) — по-
лимерные углеводы, построенные из небольшого числа
(2—10) остатков моносахаридов и не содержащие ни-
каких компонентов неуглеводного характера. В зави-
симости от числа остатков моносахаридов, входящих
в состав молекулы О., различают: дисахариды, или
биозы; трисахариды; тетрасахариды, или тетраозы;
пентасахариды, или пентаозы; гексасахариды, или
гексаозы; гептасахариды, или гептаозы; октасахариды,
или октаозы; нонасахариды, или нонаозы и декаса-
хариды, или декаозы. Полимеры с большим содержа-
нием моносахаридов относят уже к полисахаридам.
Резкой разницы в свойствах олигосахаридов и низ-
ших полисахаридов нет, и граница между ними прове-
дена условно. О. называют также сахароподобными
кристаллизующимися полисахаридами, т. к. эти ве-
щества, подобно простым сахарам, легко растворимы
в воде и, в отличие от высших полисахаридов, до-
вольно легко получаются в кристаллич. виде. В за-
висимости от моносахаридных компонентов О. делятся
на гомоолигосахариды, построенные из
остатков одного сахара, и гетероолигосаха-
риды, в состав к-рых входят остатки различных
сахаров. О. обеих групп, гл. обр. в виде ди- и триса-
харидов, широко распространены в природе, встре-
чаясь в свободном виде и в виде структурных компо-
нентов сложных белков, мукополисахаридов, глико-
липидов, гликозидов и других веществ, имеющих
большое биологич. значение. Остатки моносахаридов
в О. связанна- или р-гликозидными связями. Если в
гликозидных связях принимают участие все полу-
ацетальные гидроксилы моносахаридных компонентов,
то О. не содержат свободной полуацетальной (потен-
---------------группы, не дают реакций мо-
СН2ОН Н ОН
O^H_J^C>A нон^он
н
циальной альдегидной)
носахаридов и не об-
ладают восстанавли-
вающими свойствами.
Эту группу О. назы-
вают невосста-
навливающими
О., или О. типа трега-
лозы (I). К О. этой
группы относят саха-
розу (I). Восстанавли-
вающими О., или О.
типа мальтозы(П), на-
зывают О., содержа-
щие свободную полу-
ацетальную группу.
О. этой группы по свойствам
НО
О
н ОН
н2он
н
О
ОН
очень
СН.ОН
Н
Н
ОН
ОН
U
близки моносаха-
ридам: они восстанавливают Фелинга реактив, мута-
ротируют в водных р-рах, дают гидразоны, озазоны
и другие характерные для простых сахаров производ-
ные. О. обеих групп гидролизуются под действием
кислот и специфич. ферментов (см. Гликозидазы).
Легкость гидролиза в значительной степени зависит
от компонентов О.; так О., построенные из остатков
глюкозы, гидролизуются очень легко, тогда как О.,
в состав к-рых входят уроновые к-ты, гидролизуются
с трудом (см. Альдобиуроновые кислоты). Для опре-
деления О. идентифицируют их структурные моноса-
харидные компоненты, образующиеся при гидролизе,
устанавливают характер связи между компонентами,
используя преим. ферментативные методы, и пользу-
ются хроматография, характеристиками О.
О., встречающиеся в природе в свободном виде,
выделяют из природных источников. Напр., сахарозу
получают в промышленных масштабах из свеклы и
сахарного тростника, лактозу выделяют из молока.
Многие О. получают, отщепляя простетич. труппу от
сложных белков, напр. расщеплением мукопротеинов
подчелюстной железы получают дисахариды, в состав
к-рых входит сиаловая к-та. Ряд О. получен при не-
полном гидролизе сложных гликозидов. Синтетически
различные О. получают тремя общими методами:
1) изомеризацией уже существующих О.; 2) расщеп-
лением О. и 3) конденсацией моносахаридов.
727
ОЛИФЫ
728
Лит.: Кочетков Н. К., Торгов И. В., В о т-
виник М. М., Химия природных соединений, М., 1961;
Hassid W. Z., В а 1 1 о и С. Е., в кн.: The carbohydrates,
ed. W. Pigman, N. Y., 1957; S h a 11 z a d e h F., Wolf-
rom M. L., в кн.: Handbuch der pflanzenphyslologie, Bd 6,
B., 1958; Stanek J., Cerny M., Paca kJ., Oligosa-
charldy, Praha, 1962; Kuhn R., Les oligosaccharides du lalt,
P., 1962; Comprehensive biochemistry, v. 5, ed. M. Florkin,
E. H. Stotz, L.—N. Y., 1963; Bailey R. W. [a. o.],
Advances Carbohydr. Chem., 1962, 17, 121. Л. И. Линевич.
ОЛИФЫ — жидкие пленкообразующие вещества, по-
лучаемые переработкой растительных масел, жиров
и др. органич. веществ; в состав О. вводят сиккативы
(ускорители высыхания) и летучие растворители (для
разжижения густых продуктов до малярной консис-
тенции). При нанесении на поверхность О. высы-
хают с образованием более или менее прочной, элас-
тичной, хорошо прилипающей к поверхности и не-
растворимой в воде пленки, к-рая способна прочно
связывать введенные в О. пигменты и наполнители.
О. применяют для изготовления и разведения густо-
тертых красок (см. Краски), шпаклевок и для грун-
тования окрашиваемой поверхности. Краски, изго-
товленные на О., являются одним из основных средств
защиты металлов от атмосферной коррозии, а дерева
от гниения; они используются также для декоратив-
ной отделки внешней поверхности объектов и из-
делий.
Масла и жиры, применяемые для приготовления О.,
должны быть прозрачными и очищенными от сопут-
ствующих нежировых веществ (фосфатидов, восков,
углеводов, красящих и др.). Очистку масел произ-
водят гидратацией, а в случае необходимости — ще-
лочной рафинацией, обработкой адсорбентами, тер-
мич. обработкой и др. способами. Очищенное масло
не должно содержать мыла; содержание фосфатидов
не должно превышать 0,2—0,3%, золы 0,02%, не-
омыляемых веществ 1 %.
Типы олиф, нек-рые их свойства и применение (числа даны по ГОСТ, в скобках приведены цифры,
взятые из др. источников)
Типы олиф Содержа- ние пленко- образо- вателя, % (не менее) Цвет по иодометрич. шкале, мг (не темнее) Вязкость3 по вискози- метру ВУ при 20° Кислот- ное число, мг КОН (не более) Применение для
изготовле- ния красок разведения красок внутренних покрытий наружных | покрытий
Льняная полимеризованная — Н а т у рал ьны е олифь/*
ГОСТ 7931-56 100 300 12—16° 7 + +
Льняная окисленная— ГОСТ 7931—56 100 400 (300) 12-16° 6 + + +
Конопляная окисленная — ГОСТ 7931—56 100 1600 (600) 12—16° 7 + + +
Льняно-тунговая быстро высыхающая. . . 100 (300) 8-12° ' 8 — + +
Полунатуральные (п о л у с и н т е т и ч е с к и е) олифы
Полимеризованная (ИМС) — ОСТ НКТП 7476/583 54в 686 6—8° 6 — + +
Оксоль6—ОСТ НКТП 74 74/581 55 1 200 (400) 6—8° 6 +
Оксоль — смесь — ГОСТ 190—41 .... 55 827 7-10° 6 __
Оксоль из полувысыхающих масел с тун- говым маслом 55 (300) 6—1 2° 8
Оксиполимеризованная из рыжикового и соевого масел —ВТУМПП № 258 . . . 50 —
Оксиполимеризованная из полу высыхаю- щих масел с тунговым маслом 50 (600) 7-12° 10 +
Сульфооксоль — ОСТ НКТП 7475/582 . . 55 1820 6—8° 6 — +
Касторовая — ОСТ НКПП 538 50-55 489 7-10° 10 — + ——
Комбинированные Kn К2, Ks и К* — ТУ МХП 482-45 70 827 6—12° 7 + +
Комбинированная К5 — ТУ МХП 482—45 73 827 6—12° 7 + — + +
Глифталевая^—ГОСТ 8040—56 .... 50 636 (200) 8—14° 12 — +
Глифталевая малеинизированная . . . 50 — 8 — 14° 12 — +
Глифтале-тунговая 50 (130) 8 — 14° 6-10 —— + +
Глифтале-тунговая быстровысыхаюьиая . . 50 (250) 8-14° 1 2 — + 4-
Пентафталевая хлопковая 50 (170) 8 — 1 2° 12 —— + + +
Пентоль X хлопковая 68,5 (14 сек — + —— +
Пентоль Хр хлопковая 49,5 по НИИЛК) 8—10° + 4-
Ксифтале-тунговая 50 (300) (22—30 сек 12 — —
Стиролизованная соево-тунговая .... 50 по ВЗ-4) 6-12° + + —
Сланцевая дизельная — ГОСТ 5596—50 С и н т 40 этические темная олифы 10° _j_ + + +
Сланцевая генераторная — ГОСТ 5596—50 45 темная 10° — -1- 4-
Нафтеноль — ОСТ 8'713'НКТП 2019 . . . 50 827 (6—1 0 сек ней- 4- + + —
Синтол Ц 40 4000 по НИИЛК) (8 — 12 се,г тральн. +
Карбоноль — ОСТ 8714/НКТП 2020. . . 50 3076 по НИИЛК) (6—8 сек ней- + Т +
Лакойль — ГОСТ 3540—47 40 коричнев. по НИИЛК) 6—10° тральн. ней- + 4- 4- —
Полидиеновая-углеводородная — светлая — тральн. + + —
Вязкость препарированных масел и олиф, помимо вискозиметра ВУ (ГОСТ 1 532—54 и 6258—52), измеряют вискози-
метрами ВЗ-4 и ВЗ-1 (ГОСТ 9070—59 и 8420—57) и воронкой НИИЛК. Для определения вязкости в процессе варки О.
применяют упрощенный прибор Осноса и Головистикова. 6 Срок службы натуральных О. в покрытиях с Железным суриком
2,5—4 года, оксоли 2—3 года и глифталевых О. 3—4 года. в В том числе 12% жидкого сиккатива 64. г Только для ре-
цептуры Ка.
729
ОЛИФЫ
730
Общими для всех О. являются следующие требова-
ния: полная прозрачность после отстаивания в тече-
ние 24 час. при 20°; отстой за то же время не более
1 об.%; высыхание в тонком слое при 18—22° «от
пыли» не более 12 час., полное или практическое не
более 24 часов (синтетических О.— не более 48 и
72 часов), с образованием пленки соответствующего
качества; через 5 суток после нанесения пленки дол-
жны выдерживать изгиб на 180° вокруг стержня
диаметром в 1 мм (ГОСТ 6806—53).
Технич. требования к О. приведены в таблице. По
происхождению исходного сырья и химич. составу О.
делятся на натуральные, полунатуральные (полусин-
тетические) и синтетические.
Натуральные О. готовят из высыхающих масел
типа льняного и их смесей с др. маслами типа тун-
гового, макового и др. (см. Масла высыхающие и
Жиры растительные) с введением в них сиккативов
преим. в виде маслорастворимых линолеатов, нафте-
натов, резинатов и рицинолеатов или в виде окислов
(из расчета содержания в О. активных металлов:
РЬ 0,06—0,10%, Мп 0,06—0,08%, Со 0,01—0,06%
и др.). Натуральные О. применяются для приго-
товления и разведения густотертых красок, а также
в качестве самостоятельного материала при ответст-
венных наружных покрасках (железных крыш и т.п.).
Преимуществами натуральных О. и красок на их
основе перед другими лакокрасочными материалами
являются их относительная негорючесть, отсутствие
запаха и физиологич. действия, легкость растира-
ния и возможность нанесения на поверхность различ-
ными методами. В зависимости от способа обработки
масла и вида исходных материалов приготовляют
след, виды натуральных О.:
Полимеризованная, к-рую получают на-
греванием льняного масла до 140—150° при непре-
рывном перемешивании до исчезновения пены и уда-
ления влаги; после этого проводят его полимериза-
цию при 260—280° в присутствии катализатора с
последующим добавлением сиккатива при 220°; по-
лимеризацию заканчивают, когда плотность масла
достигает 0,935 e!cMz и вязкость по вискозиметру ВУ
10—14° при темп-ре 20°.
Окисленная — из препарированного льня-
ного или конопляного масла, содержащая сиккатив.
Масло нагревают при 150—160° с одновременным
перемешиванием и продуванием через него воздуха
со скоростью 0,5—0,7 м’Чмин на 1 т масла.
Быстровысыхающая — смесь препари-
рованных льняного и тунгового (2,3:1) масел, содер-
жащая железный или кобальтовый сиккатив и орга-
нич. активатор (1—1,5% фуроина или диэтилового
эфира диоксималеиновой к-ты). В нормальных усло-
виях (20°, относит, влажность воздуха 60—70%)
время высыхания этой О. не превышает 2 час.; хра-
нится в заполненной и плотно закрытой таре, защи-
щенной от солнечных лучей.
Полунатуральные (полусинтетические) О. приго-
товляют на основе препарированных (сгущенных)
растительных масел и жиров, их сополимеров и поли-
эфиров (см. Масла препарированные и Смолы алкид-
ные) с последующим добавлением сиккативов (0,06—
0,12% РЬ, 0,03—0,06% Мп, 0,01—0,02% Со или Са)
и растворителей (27—50% уайт-спирита, сольвента,
скипидара и др.). Применяются след, типы полуна-
туральных (полусинтетических) О.:
Полимеризованная (ИМС) — из сгу-
щенного нагреванием льняного масла; полимериза-
цию проводят в закрытом котле под вакуумом и в
атмосфере инертного газа при 300°.
Оксоль и оксоль-смесь — из окислен-
ного при 130—150° льняного масла или смесей льня-
ного или конопляного масел с подсолнечным путем
продувания через них воздуха (2—2,5 м’/мин на 1 т
масла) в присутствии катализатора (0,02% Мп).
Масла при окислении сгущают до условной вязкости
50—60 единиц (вязкость исходного масла равна 1)
по прибору Осноса и Головистикова. Процесс сопро-
вождается выделением большого количества тепла,
а также большого количества летучих продуктов и
сопровождается значительным увеличением вяз-
кости реакционной среды. Продолжительность про-
цесса и характер образующихся продуктов зависят
от вида масла и режима окисления. Кроме того, О.
оксоль получают смешением окисленных полу-
высыхающих масел (рыжикового, соевого или под-
солнечного) до условной вязкости 60—80 единиц с
нагретым и обезвоженным при 140—150° тунговым
маслом (3,5:1).
Оксиполимеризованная — из поли-
меризованных при 270° (до условной вязкости 80—
90 единиц), предварительно окисленных воздухом
при 150—160° в присутствии катализатора (0,02% РЬ,
0,02% Мп и 0,01% Со) полувысыхающих масел или
из совместно полимеризованных при 260° тех же окис-
ленных масел с обезвоженным тунговым маслом
(3,9:1) — до условной вязкости 60— 70 единиц.
Сульфооксоль — из окисленного возду-
хом льняного масла при 140—145° и обработанного
2,5—3% серного цвета, растертого с льняным маслом
в виде пасты. О. должна содержать не более 1,5%
серы и не должна содержать минеральных к-т.
Касторовая — из термически обработанного
(до 280°) касторового масла в присутствии катализа-
тора (0,06% РЬ или др.).
Комбинированные, предназначенные для
растирания пигментов.— из смесей оксидированных
и термообработанных растительных масел; они могут
быть след, типов:
Тип комбини- рованной олифы Состав масляной части
оксидированное масло термообработанное масло
Ki полувысыхающее касторовое
К2 то же льняное или др. высыхающее
Кз высыхающее высыхающее
К4 полувысыхающее полувысыхающее
к5 то же тунговое
Глифталевая — из глифталевой смолы, мо-
дифицированной высыхающими или полувысыхаю-
щими маслами (термически обработанными или с до-
бавлением 10% тунгового масла). При замене фтале-
вого ангидрида малеиновым получают глифталевую
малеинизированную О.; при этом сокращается техно-
логич. процесс, повышается механич. прочность плен-
ки и снижается ее растворимость.
Глифтале - тунговая О. отличается от
глифталевой О. большим содержанием тунгового мас-
ла (до 50%); в нормальных условиях высыхает за
12—16 час.
Глифтале-тунговая быстровысы-
хающая — из жирной глифталевой смолы, мо-
дифицированной смесью полувысыхающего и тунго-
вого масла (1,36:1) с введением органич. активатора
и железного сиккатива; время высыхания в нормаль-
ных условиях не более 2 часов.
Пентафталевая — из жирной пентафта-
левой смолы, модифицированной полувысыхающими
маслами; от глифталевой отличается большей ско-
ростью высыхания (16 часов) и образованием более
твердой и атмосферостойкой пленки.
731
ОЛИФЫ — ОЛОВА ГАЛОГЕНИДЫ
732
ПентольХ и пентоль Хр — из хлопко-
вого масла, переэтерифицированного пентаэритри-
том и сгущенного до вязкости, соответственно, 8—
12 минут или 20—25 минут по воронке НИИЛК при
20°. Пентоли могут также изготовляться этерифика-
цией пентаэритритом свободных жирных к-т различ-
ных растительных масел или жирных к-т таллового
масла. Пентоль X предназначена для применения в
качестве связующего при изготовлении густотертых
красок, а пентоль Хр — для разведения красок.
Ксифталевая — из ксилита, фталевого ан-
гидрида и предпочтительно из свободных жирных
к-т, высыхающих и полувысыхающих масел, а также
из фракций кислот, получаемых из отходов масло-
жировой пром-сти, из таллового масла и из рыбьих
жиров. По качеству пленка несколько уступает гли-
фталевой и пентафталевой. Добавление тунгового
масла (до 30—50%) улучшает цвет, скорость высы-
хания и механич. свойства пленки. Из ксилита и жир-
ных к-т получают ксилитовую О. Все О. на основе
ксилита по мере высыхания образуют неплавкие и
водостойкие пленки.
Стиролизованная — из сополимеров нена-
сыщенных растительных масел (80% окисленного
соевого и 20% тунгового масел) и стирола (до 35%).
Сополимеризацию ведут обычно в растворителе в при-
сутствии инициаторов (перекиси бензоила и др.).
Пленки О. обладают хорошей водо- и светостойко-
стью, а также электроизоляционными свойствами.
При введении в натуральные и полунатуральные
О. водного р-ра эмульгатора (СаО и др.) в качестве
диснерсионной среды получают эмульсионные
О. молочно-белого цвета (допускается кремоватый
оттенок), время высыхания не более 20 часов; упот-
ребляются для разведения густотертых красок и для
непосредственного приготовления красок малярной
консистенции; краски на эмульсионной натуральной
О. применяют для неответственных внутренних и
наружных покрытий (но не в качестве противокор-
розионных покрытий по металлу), а на полунатураль-
ной — только для неответственных внутренних по-
крытий по дереву, штукатурке и т. п.
Синтетические О. получают на основе продуктов
переработки нефти, угля, сланцев, отходов производ-
ства синтетич. каучука и др.; содержат 45—60% лету-
чих органич. растворителей (уайт-спирит, сольвент,
ксилол). К синтетич. О. относятся:
Сланцевые — из препарированных дизель-
ного и генераторного сланцевых масел, получаемых
окислением кислородом воздуха в присутствии ката-
лизатора при 140—150° с последующей дегидратацией
и полимеризацией их при 240—250°. Сланцевые О.
имеют темный цвет, высыхают «от пыли» через 12 ча-
сов, полностью — через 24 часа; используются для
приготовления красок с темными пигментами или с
алюминиевой пудрой; применяются для наружных
покрытий по железу, гл. обр. для окраски кровли.
Нафтеноль'— р-р алюминиевых и кальциевых
солей природных нафтеновых кислот (асидола) в уайт-
спирите с добавлением скипидара или сольвент-нафты;
светлая жидкость, срок высыхания до 72 часов; при-
меняют для приготовления густотертых паст, к-рые
можно использовать для малярных работ внутри
помещения (по дереву, железу и штукатурке, при
темп-ре не выше 50°). Цинковые и свинцовые белила
на этой О. сильно загустевают при хранении.
Синтол — из продуктов окисления уайт-спирита
или очищенного керосина. Краски, получаемые на
этих О., имеют светлые тона; пигменты, растираемые
на этой О., должны содержать окись цинка, к-рая
после окраски и испарения растворителей взаимодей-
ствует с карбоксильными группами к-т, образуя цин-
ковые мыла, за счет к-рых и происходит твердение
пленки краски. Синтоловые краски имеют стойкий
очень резкий запах. Пленки на основе синтола не-
прочны (стареют) и обладают плохой атмосферостой-
костью; применяются для окраски дерева и штука-
турки внутри помещений (при наличии хорошей вен-
тиляции).
Карбоноль — из продуктов окисления соляро-
вого масла, окрашена в темный цвет; применяется
для приготовления темных красок; употребляется
для разбавления красок при работах внутри помеще-
ний (для окраски дерева, металла и штукатурки);
высыхает не позднее чем через 48 час.
Лакойль — р-р сгущенных (полимеризованных)
углеводородов, получаемых из отходов пиролиза
нефти в сольвент-нафте; имеет темный цвет, высыхает
«от пыли» в течение 6—8 часов, полностью — не
более 24 часов. Лакойль пригодна лишь для неответ-
ственных работ гл. обр. внутри помещения.
Полидиеновая-углеводородна я—
из отходов произ-ва бутадиенового каучука; имеет
светлый цвет, высыхает в тонких слоях с образова-
нием светлой, глянцевой и достаточно твердой пленки;
из-за недостаточной водо- и атмосферостойкости ис-
пользуется только для неответственных покрытий
внутри помещения; иногда ее применяют в смеси с О.
оксоль (не более 25% от веса смеси).
Э т и н о л ь (лак этиноль) — см. Лаки.
Дальнейшее усовершенствование методов получе-
ния синтетич. О. вероятно пойдет по пути расшире-
ния произ-ва высококачественных полиэфирных О.
типа глифталевой, пентафталевой и др. При этом
значительно сократится расход растительных масел
за счет применения синтетич. сырья (фталевого ангид-
рида, малеинового ангидрида, пентаэритрита, кси-
лита, стирола и. др.), а также отходов масложиро-
вой пром-сти, таллового масла, рыбьих жиров, мно-
гоатомных спиртов и синтетич. эфирокислот, полу-
чаемых из продуктов переработки нефти, угля, слан-
цевого масла и др.
Лит.: Руководство по технологии получения и переработки
растительных масел и жиров, т. 3, Л., 1961; Киселев В. С.,
Абашкина А. Ф., Производство лаков, олиф и красок,
2 изд., М., 1961; Пэйн Г. Ф., Технология органических по-
крытий, пер. с англ., т. 1, Л., 19 59; т. 2, Л. ,1963; Варла-
мов В. С., Производство олиф и сиккативов, М., 1957;
Дринберг А. Я., Технология пленкообразующих веществ,
2 изд., Л., 1955; Лакокрасочные материалы. Сырье и полупро-
дукты. Справочник, под ред. И. Н. Сапгира, М., 1961; Бодя-
ж и н а 3. И., Маслобойно-жировая пром-сть, 1959, № 7, 12;
Субботин А. А., Лакокрасочные материалы и их приме-
нение, 1962, JM5 5, 27. И. И. Головистиков.
ОЛОВА ГАЛОГЕНИДЫ — соединения олова с га-
логенами состава SnX2 и SnXa. Наибольшее практич.
значение имеют хлориды олова. Олово хло-
ристое SnCla— кристаллы белого цвета ромбич.
структуры, а = 6,623 А, b = 9,359 А, с = 10,000 А;
плотн. 3,95 (25°); т. пл. 247°, т. кип. 652°; теплота
образования ДЯ°298 = —83,6 ккал/моль, молярная
теплоемкость 19,26 кал/молъ • град (20—99°); давление
насыщенного пара (мм рт. ст.): 0,76 (302°), 7,60
(382°), 760 (652°). Растворимость безводного SnCl2
в воде (в г на 100 г Н2О): 83,9 (0°), 269,8 (15°); в разб.
р-рах гидролизуется. С соляной к-той SnCl2 дает
комплексные хлорокислоты Н2[8нСЦ] и Н[8нС1з].
Получают SnCl2 растворением олова в конц. соляной
к-те, из растворов кристаллизуются бесцветные крис-
таллы SnCl2-2Н2О; плотн. 2,71, т. пл. 37,7°. Хлорис-
тое олово растворимо в спирте, эфире, ацетоне и др.
органич. растворителях. SnCl2 в р-рах — сильный вос-
становитель, оно быстро окисляется кислородом воз-
духа, особенно на свету. Применяют SnCl2 при син-
тезе органич. красителей и как протраву при кра-
шении.
Олово четыреххлористое SnCU—
бесцветная жидкость, дымящая на воздухе вследст-
вие гидролиза парами воды; плотн. 2,23; т. пл.—
733
ОЛОВА ОКИСЛЫ—ОЛОВА СПЛАВЫ
734
33°, т. кип. 113,9°; теплота образования ДЯ°298=
= —130,3 ккал/моль', молярная теплоемкость 38,45
кал/моль -град (14—98°); давление насыщенного пара
(мм рт. ст.)'. 5,53 (0°), 122,2 (60°), 760 (113,9°). SnCl4
легко растворяется в воде, но подвергается гидролизу.
Из р-ров кристаллизуется в виде кристаллогидратов,
при 19—56°— SnCl4-5H2O. Четыреххлористое олово
легко растворяется в нек-рых органич. растворите-
лях (неполярных) и само растворяет неэлектролиты,
напр. S, Р, J2. Получают SnCl4 действием сухого
хлора на жидкое олово, реакция экзотермична,
в пром-сти она служит для снятия олова с отходов
белой жести. Применяется SnCl4 как аппретура для
отяжеления шелковых тканей и в ситцепечатании.
Другие галогениды Sn2+ и Sn4+ но способам полу-
чения и многим свойствам сходны с хлоридами;
нек-рые данные о них приводятся ниже:
Соединение SnF2 SnF, SnBr. SnBr, Sn J2 SnJ4
—^Н°,ккал/моль 3,6 7. 1 4,4
Цвет бес- — желт. бес- красн. желт.
цветн. цветн.
Т. пл., °C . . — 215,5 31,0 320 143,5
Т. кип., °C . . 705 620 202 720 341
Плотн., г(смА — 4,78 5,12 3,34 5,28 4,70
Галогениды Sn4+ образуют многие комплексные
соединения с Н2О, NH3, РС1В, спиртами, эфирами
и др. органич. веществами. Весьма прочны комплекс-
ные галогениды Sn (IV) с галогеноводородными к-тами
типа H2[SnXe] и их соли, хорошо растворимые в воде
и мало подверженные гидролизу, напр. (NH4)2 [SnCle],
Двойные галогениды Sn4+ по свойствам сходны с
обычными, примерами служат SnBr3Cl — желтая
жидкость с плотн. 3,12, т. пл. 1°, т. кип. 73°; SnBrCl3—
бесцветная жидкость с плотн. 2,51, т. пл.— 1°, т.
кип. 50°; SnBr2J2— оранжевые кристаллы, плотн.
3,63, разл. при 50°.
Лит. см. при ст. Олово. Н. Н Севрюков.
ОЛОВА ОКИСЛЫ — соединения олова с кислоро-
дом, SnO и SnO2. Олова окись (часто называе-
мая закисью олова) SnO черного цвета, структура
тетрагональная, с периодами решетки а = 3,804 А
и с = 4,826 А; плотн. 6,446; т. пл. 1040°, т. кип.
1425°. Стандартная теплота образования ДЯ°298=
= —68,4 ккал/моль', молярная теплоемкость 10,52
кал/моль -град (19,5°). На воздухе SnO быстро оки-
сляется до SnO2. Без доступа воздуха при 400°
диспропорционирует: 2SnO—>SnO2-|-Sn. В кислотах
SnO легко растворяется с образованием ионов Sn2+.
Окись олова растворима в жидком олове, причем
выше 1040° SnO и Sn образуют два несмешивающихся
взаимно насыщенных жидких слоя. SnO и SiO2 дают
эвтектику с темп-рой пл. ок. 900°, состав индивиду-
альных силикатов не установлен. В природе SnO не
встречается. Ее получают термич. разложением SnC2O4
или действием щелочей и соды на растворы Sn2+,
выпадающие при этом гидраты обезвоживают на-
греванием. Промышленного применения SnO не имеет.
Олова двуокись SnO2 белого цвета, струк-
тура тетрагональная, с периодами решетки а=4,737А,
с=3,185 А; плотн. 7,0096, т. пл. ок. 2000°. Стандарт-
ная теплота образования ДЯ°298= —138,8 ккал/моль.
Молярная теплоемкость 14,03 кал/моль -град
(0—100°). В природе встречается в виде минерала
касситерита (см. Олово). Двуокись олова, особенно
в виде касситерита, исключительно стойка к дейст-
вию водных р-ров кислот, щелочей, солей и восстано-
вителей. С, Н2идр. восстановители легко восстанавли-
вают SnO2 до металла уже при красном калении.
При тех же темп-рах смесь SnO2 с углем реагирует
с С1г или с СС14, образуя пары хлоридов олова. Полу-
чают SnO2 обезвоживанием гидроокиси, выпадающей
в осадок при действии NH3 на р-ры солей Sn4+, либо
обезвоживанием метаоловянной к-ты, образующейся
при действии HNOs на Sn. В пром-сти SnO2 получают
сжиганием олова в кислороде; применяют для при-
готовления жаростойких эмалей и свинцово-оловя-
нистых глазурей.
Лит. см. при ст. Олово. Н. Н. Севрюков.
ОЛОВА СПЛАВЫ — сплавы олова с другими ме-
таллами, в к-рых олово преобладает. Для олова весьма
характерно образование химич. соединений с др.
металлами, нередко имеющих высокие темп-ры плавле-
ния. Примеры: Zr3Sn2 (1985°), Ti3Sn (1663°), Pt3Sn
(-1420°), La2Sn (-1420°), Pr2Sn (-1415°),Ce2Sn(1400°),
Ni3Sn2 (1264°), Co2Sn (1161°), Ca2Sn (1122°), SeSn
(860°), Mg2Sn (778°). Способность Sn к образованию
твердых р-ров невелика. В О. с. с тяжелыми металлами
наблюдаются лишь небольшие области твердых р-ров.
Со многими легкоплавкими тяжелыми металлами
олово образует эвтектики, напр. Bi —Sn (43% Sn,
139°), Cd—Sn (67,75% Sn, 177°), Pb—Sn (61,9% Sn,
183°), TI—Sn (56,5% Sn, 170°), Zn—Sn (91% Sn, 198°).
Частичная несмешиваемость в жидком состоянии
наблюдается относительно редко, напр. в системах
Сг—Sn, Fe—Sn, Se—Sn.
Вследствие низкой механич. прочности О. с. не при-
меняют как конструкционные материалы. Наиболь-
шее значение в технике имеют сплавы систем Sn—Sb—
Си и Sn—Sb—РЬ, применяемые в качестве антифрик-
ционных сплавов (баббитов), припоев и для изготов-
ления фольги (см. таблицу). Оловянные баббиты об-
ладают наилучшими антифрикционными свойствами
по сравнению с другими подшипниковыми сплавами.
Их структура состоит из твердого р-ра на основе
олова и равномерно распределенных в нем кристаллов
соединения, близкого по составу к SnSb. Твердый
р-р служит пластичной основой, обеспечивающей
прирабатывание вкладыша к валу, а более твердые
кристаллы SnSb выносят на себе трение. Двойные
сплавы Sn—Sb при медленном охлаждении сильно
ликвируют: кристаллы SnSb всплывают. Добавка
Си предотвращает ликвацию благодаря первичному
выделению из расплава кристаллов Cu3Sn, образую-
щих скелет, на к-ром нарастают кристаллы SnSb.
Оловянно-свинцовые припои (ПОС) отличаются лег-
коплавкостью, малой твердостью (поэтому они полу-
чили название мягких) и сравнительно низкой меха-
нич. прочностью. Они хорошо смачивают поверхность
большинства металлов, напр. железа, меди и их спла-
вов, обладают высокой пластичностью, не хрупки
и хорошо сопротивляются усталости. Это позволяет
применять их для пайки изделий, испытывающих
небольшие ударные и вибрационные нагрузки. Однако
область их применения ограничивается только пайкой
деталей, от к-рых не требуется большая механич.
прочность и к-рые работают при невысоких темп-рах.
Создание шва при пайке меди, латуни и стали обеспе-
чивается образованием оловом твердого р-ра или
интермета л лич. соединения с металлом изделия.
Свинец добавляют в припои для снижения их стои-
мости, а небольшие присадки сурьмы повышают
прочность. В технике применяют припои, содержащие
от 90% до 3—4% Sn. ПОС 90, плавящийся при 220—
230°, отличается благодаря большому содержанию Sn
высокой стойкостью к коррозии. Поэтому его исполь-
зуют наряду с чистым оловом, гл. обр. для пайки
пищевой посуды и консервной тары. Припои ПОС 61
и ПОС 50 кристаллизуются в весьма малом интервале
темп-р (0—25°). ПОС 61 по составу примерно соответ-
ствует эвтектике, плавящейся при 183°. Эти сплавы
отличаются очень высокой жидкотекучестью и при-
меняются в тех случаях, когда при пайке не допу-
скается высокий нагрев изделия и требуется хорошее
735
ОЛОВА СУЛЬФИДЫ—ОЛОВО
736
Состав и свойства оловянвых сплавов
Назначение сплава Марка сплава Состав, вес. % Механические свойства
Sn Sb Си РЬ предел прочности, кГ(мм2 относи- тельное удлине- ние, % твердость по Бринел- лю, кГ/мм*
Антифрикцион- ные сплавы Б89 остальное 7,25—8,25 2.5—3,5 8,0 10,6 24,3
Б83 > 10—12 5,5—6,5 9,0 6,0 30,0
Припои * ПОС90 89 — 90 не более 0,15 остальное 4.3 12,8
ПОС61 59 — 61 не более 0,8 — 6—7 8 — 4 —
ПОС50 49 — 50 не более 0,8 — > — — —
Сплавы для фоль- ги оловянный 95,9 — 97.1 1,9—3,1 0,5 0,5
оловянно-свинцо- вый 82—85 1,75 — 3,25 — 12 — 15 — — —
* Припои, содержащие менее 50% Sn, описаны в ст. Свинца сплавы.
заполнение узких зазоров, гл. обр. в электротехник,
пром-сти и в приборостроении.
О. с., применяемые для литья под давлением и для
центробежного литья посуды и деталей измерительных
приборов, содержат 64—92% Sn, 5,5—36% Sb,
1—8% Си и иногда 5—14% РЬ. Для литья украше-
ний из О. с. применяют формы из синтетич. каучука.
Примеси алюминия и цинка в О. с. образуют тончай-
шие пленки окислов, затрудняющие пайку и лужение.
Железо и мышьяк ухудшают пластич. свойства спла-
вов, повышают их твердость и хрупкость. Медь сни-
жает коррозионную стойкость припоев. В связи с де-
фицитностью олова в технике широко применяют заме-
нители О. с.— антифрикционные сплавы на основе
свинца (см. Свинца сплавы), текстолит, углеграфито-
металлич. материалы и т. п., а также свинцовооло-
вянные припои, в к-рых содержание Sn не превышает
40%. Заменителем фольги из олова и О. с. служит
алюминиевая фольга.
Лит.: Хансен М., А н д е р к о К., Структуры двой-
ных сплавов, пер. с англ., т. 1—2, 2 изд., М., 1962; Б о ч в а р
А. А., Металловедение, 5 изд., М., 1956; Шпагин А. И.,
Антифрикционные сплавы, М., 1956; Основы металлургии, под
ред. Н. С. Грейвера [и др.], т. 1, ч. 1, М., 1961; Лакеде-
монский А. В., X р я п и н В. Е., Паяние и припои, М.,
1961; М a n t е 1 1 С. L., Tin, Its mining, production, technology
and applications, 2 ed., N. Y., 1949; Tin and its alloys, ed. E. S.
Hedges [a. o.J, L., 1960. А. Я. Фишер.
ОЛОВА СУЛЬФИДЫ — соединения олова с серой:
SnS, SnS2 и Sn2S3. Олово сернистое SnS —
кристаллы синевато-черного цвета ромбич. струк-
туры, а = 4,339 А, Ь =11,202 А, с=3,988 А; плотн.
5,08; т. пл. 880°; т. кип. 1230°; теплота образования
АЯ°298= —25,1 ккал/моль', молярная теплоемкость
17,90 кал!моль-град (80—952°); давление насыщенного
пара (мм рт. ст.)'. 2,43 (811°), 760 (1230°). В природе
встречается в виде редкого минерала герценбергита.
Получается нагреванием олова с серой и углем до
темп-ры ок. 900°; для очистки возгоняется в инертном
газе или вакууме. Из водных р-ров осаждается в кис-
лой среде сероводородом аморфное сернистое олово.
В разб. минеральных к-тах SnS, особенно кристалли-
ческое, трудно растворимо, в конц. соляной к-те при
нагревании растворяется с образованием комплексных
хлорокислот Н[8пС1з] и H2[SnClJ, а азотной к-той
окисляется в нерастворимую метаоловянную кислоту
H2SnO3. Летучесть SnS вызывает потери олова при его
выплавке. Она используется для извлечения О. из
бедного сырья. Обжигом при доступе воздуха SnS
количественно превращают в SnO2.
Олово двусернистое SnS2 — кристаллы
золотисто-желтого цвета гексагональной структуры,
а = 3,646 А, с=5,880 А, плотн. 4,51; теплота образо-
вания А77°298=—40 ккал/моль', молярная теплоем-
кость 21,85 кал/моль -град (12—85°). Двусернистое
олово нелетуче, при темп-рах выше 520° оно разла-
гается на Sn2S3 и серу. В природе SnS2 не встречается.
Кристаллич. SnS2 получают нагреванием амальгамы
олова с серным цветом и хлористым аммонием до
темп-ры ок. 300° либо нагреванием аморфного SnS
с серой ок. 450°. Аморфное двусернистое олово выде-
ляется также сероводородом из кислых р-ров солей
Sn (IV) в виде SnS2-2Н2О. В конц. соляной к-те SnS2
растворяется с выделением H2S и S, в азотной к-те —
с образованием нерастворимой метаоловянной к-ты.
Двусернистое олово, особенно аморфное, легко раст-
воряется в р-рах сернистых щелочей, напр. Na2S, об-
разуя растворимые в воде тиостаннаты Na2SnSa
и NaaSnSi. Растворимость NaaSnSa в воде (вес %):
32,4 (20,1°), 54,8 (89,9°). В щелочах SnS2 растворяется
с образованием Na2SnS3 и Na2Sn(OH)e. Тиостаннаты
железа Fe2SnSa, меди Cu2SnSa и большинства др.
металлов нерастворимы в воде. Кристаллич. двусер-
нистое олово, наз. сусальным золотом,
входит в состав красок для золочения деревянных,
гипсовых и др. изделий.
Олово полуторасернистое Sn2S3 —
синевато-черного цвета. Теплота образования А//°298 -=
= —85,5 ккал/моль. При темп-рах выше 640° оно
разлагается на SnS и S. По строению Sn2S3, по-види-
мому, представляет собой тиостаннат Sn (II), т. е.
SnSnS3. Получают Sn2S3 длительным нагреванием
SnS2 в инертном газе ок. 600°.
Лит. см. при ст. Олово. Н. Н. Севрюков.
ОЛОВО (Stannum) Sn — химич. элемент IV гр.
периодич. системы Менделеева; п. н. 50, ат. в. 118,69.
Природное олово состоит из десяти изотопов:
Sn112 (0,95%), Sn114 (0,65%), Sn115 (0,34%), Sn11’
(14,24%), Sn117 (7,57%), Sn118 (24,01%), Sn119 (8,58%),
Sn120 (32,97%), Sn122 (4,71%), Sn124 (5,98%). Последний
изотоп слабо радиоактивен (двойной 0“-распад с пе-
риодом, большим 1016—10” лет). Сечение захвата
тепловых нейтронов атомом О. 0,625 барн. Получено
более 10 искусственных радиоактивных изотопов О.
В качестве радиоактивных индикаторов применимы
Sn11’ (Г>/,= 118 дней), Sn119 (Г1/а=175 дней) и Sn12’
(7'1/2=136 дней). Конфигурация внешних электронов
атома 0.5s2 5 р2. Э нергии ионизации (в эв): Sn°—*Sn+—>•
—>Sn2+—>Sn’+—>-Sn4+—>-Sn5 + соответственно 7,342;
14,628; 30,49; 39,4; 80,7.
Сплавы О. с медью—бронзы—были известны более
чем за 4000 лет до н. э. В древности чистое О. применя-
лось также для изготовления украшений. Промышлен-
ное применение О. нашло в 14—15 вв. Происхождение
названия металла не установлено.
737
ОЛОВО
738
Содержание О. в земной коре 0,04 вес. %. Самород-
ное О. не встречается. Известны 16 минералов О., из
к-рых промышленное значение имеют: касситерит
(оловянный камень) SnO2 и в меньшей степени стан-
нин (оловянный колчедан) Cu2FeSnS4. Оба минерала
встречаются в коренных рудах, а касситерит — также
в россыпях, где он концентрируется благодаря высо-
кой плотности. Касситерит образует кристаллы
разной крупности (обычно мелкие, изредка более
10 см), окрашенные примесями (окислы Fe, Nb, Та,
Мн) в желтый или буро-черный цвет; структура тетра-
гональная, плотн. 6,8—7,0; твердость 6—7. Стан-
нин — минерал серо-стального цвета с зеленоватым
оттенком, обычно наблюдается в виде неправильных
зерен и зернистых масс; структура кристаллов тетра-
гональная (аналогична структуре халькопирита
CuFeS2); плотн. 4,3—4,5; твердость по Моосу 3—4.
В СССР месторождения О. находятся в Восточной
Сибири (Читинский район и др.), на Дальнем Востоке
(Колыма, Чукотка), а также в Якутской АССР и Ка-
захской ССР. В зарубежных странах крупные место-
рождения О. известны в Китайской Народной Респуб-
лике, в Малайе, Индонезии, Таиланде, Конго, Ниге-
рии, Боливии и др.
Физические и химические свойства. О.— серебристо-
белый блестящий металл, медленно тускнеющий на
воздухе. О. полиморфно, обычная 0-модификация
(«белое» О.) устойчива выше 13,2°. Кристаллич. ре-
шетка 0-0.,тетрагональная с периодами, а = 5,831 А,
с = 3,176 А; плотн. 7,2984. При охлаждении 0-0.
переходит в a-модификацию кубич. структуры,
а = 6,4912 А, плотн. 5,8466. В связи с резким увели-
чением уд. объема (на 25,6°/0) при превращении 0^а
металл рассыпается в серый порошок («серое» О.).
Наиболее быстро превращение протекает при —33°;
оно резко ускоряется в присутствии зародышей а-О.
ц поэтому быстро распространяется по слиткам ме-
талла (в связи с этим явление наз. «оловяннойчумой»).
Для предохранения от разрушения оловянные пред-
меты следует хранить в отапливаемых помещениях.
Образование ромбич. y-О., устойчивого от 161°
до темп-ры плавления, не доказано. Ат. радиус О.
1,58 А, ионный радиус Sn4+ 0,67 A, Sn2+ 1,02 А. Т. пл.
231,9°, т. кип. 2270°. Теплоты плавления и испарения
соответственно равны (в кал/г-атом): 1710 и 64 700
(2750°). Уд. теплоемкость в кал/г-град: 0,0536 (0°);
0,0589 (230°). Теплопроводность 0,157 кал/см -сек -град
(0°). Термич. коэфф, линейного расширения 23 •IO-’
(0—100°). Уд. электрич. сопротивление (99,9885%
Sn) 11,5-10_® ом-см (20°). Темп-ра перехода 0-О.
в сверхпроводящее состояние 3,73° К (а-О. в сверх-
проводящее состояние не переходит). Давление насы-
щенного пара О. (в мм рт. ст.): 1 (1492°); 10 (1703°);
60 (2169°); 100 (1968°); 760 (2270°). Белое О. слабо
парамагнитно с атомной магнитной восприимчивостью
5(дТ=4,5-Ю-’ (30°), но при темп-ре плавления ста-
новится диамагнитным с ХАТ =—5,1 -10-6; это изме-
нение обратимо. Для серого О. %АТ=----37 -10“6 (20°)-
О.— весьма мягкий и пластичный металл, его механич.
свойства значительно зависят от чистоты, обработки
и темп-ры. Модуль нормальной упругости 4150 кГ/мм2;
модуль сдвига 1640 кГ/мм2; предел прочности
1,7 кГ/мм2; относительное удлинение 80—90%; твер-
дость по Бринеллю 3,9—4,2 кГ/мм2.
В соединениях О. проявляет валентности 4 (наибо-
лее характерная) и 2. Производные 2-валентного О.
легко окисляются и поэтому используются как восста-
новители. При обычных условиях О. весьма устой-
чиво к химич. воздействиям. На воздухе при комнат-
ной темп-ре окисляется очень медленно даже в при-
сутствии влаги. Образующаяся на поверхности ме-
талла тонкая прочная окисная пленка предохраняет
его от дальнейшего окисления. Заметное окисление О.
наблюдается ок. 150°, а в очень влажном воздухе —
при более низкой темп-ре. О., сильно перегретое выше
темп-ры плавления, начинает с поверхности тлеть,
а при действии струи кислорода на расплавленный
металл горит, образуя наиболее устойчивый окисел
SnO2. Продукты коррозии О. состоят гл. обр. из
SnO2 и содержат также SnO (см. Олова окислы). С фто-
ром и хлором О. легко взаимодействует, образуя
SnF4 и SnCl4 (см. Олова галогениды). При нагревании
О. с серой образуется низший сульфид SnS; при
нагревании SnS под давлением идет образование SnS2
(см. Олова сульфиды). Гидрид О. (станно-
метан) SnH4 получается в незначительных коли-
чествах как примесь к водороду при разложении кис-
лотами сплавов О. с магнием (т. е. при действии
водорода в момент выделения). SnH4 — бесцветный
ядовитый газ, т. пл.—150°, т. кип.—52°. По устой-
чивости SnH4 превосходит РЬН4, но уступает GeH4;
при хранении постепенно разлагается на свободное О.
и водород; такой распад идет быстро при 150°. С азотом
и аммиаком О. не реагирует. Косвенным путем полу-
чен твердый коричневый нитрид Sn3N4, распадаю-
щийся на элементы при 360°. С углеродом и кремнием
О. не взаимодействует. С многими металлами дает
соединения (см. Олова сплавы).
По отношению к воде О. устойчиво. Стандартный
электродный потенциал О. в кислой среде равен
—0,136 в, поэтому О. медленно растворяется в разб.
сильных к-тах. В горячей конц. соляной к-те О.
растворяется быстро с образованием комплексных
хлорокислот H2[SnCl4] и H[SnCl3]. Горячую конц.
серную к-ту О. восстанавливает до SO2, образуя
хорошо растворимый сульфат Sn (SO4)2. Разб. азотную
к-ту О. медленно восстанавливает до NH3, переходя
в раствор в виде Sn2+, а концентрированную— до
NO2, превращаясь в нерастворимую мета оло-
вянную кислоту H2SnO3. В царской водке О.
растворяется, переходя в SnCl4. В щелочной среде
потенциал системы HSnO2 /Sn приблизительно равен
0,3 в, поэтому в конц. щелочах О. растворяется с вы-
делением водорода, образуя соли оловянистой к-ты
H2SnO2 (станниты), а в присутствии окислителя-
соли оловянной к-ты H2SnO3 (с т а н н а т ы). Стан-
ниты и станнаты при подкислении гидролизуются,
выделяя осадки гидратов окислов О. Последние полу-
чаются также при действии аммиака или щелочей на
р-ры солей О. Гелеобразный гидрат окиси
nSnO -mH20 хорошо растворим в кислотах с образо-
ванием солей Sn (II), при старении переходит в чер-
ную SnO. Гелеобразный гидрат двуокиси
iSnO2 -г/Н2О — так наз. a-о ловя иная к-та,
растворима в щелочах и кислотах. При стоянии
а-оловянная к-та стареет; полимеризуясь и теряя
воду, она постепенно приобретает кристаллич. струк-
туру и переходит в 0-о ловянную (метаоловян-
ную) к-ту, трудно растворимую в щелочах и кислотах.
Большинство солей Sn2+ бесцветно и хорошо рас-
творимо в воде. Таковы нитрат Sn(NO3)2, ацетат
Sn(C2H3O2)2 и др. Практически нерастворимы фос-
фаты Sn3(PO4)2 и Sn(H2PO4)2. Растворы солей Sn (II)
постепенно окисляются кислородом воздуха, быст-
рее — на свету. Из кислородных кислот 2-валентного
О. наибольшее практич. значение имеет сульфат
SnSO4 — бесцветные кристаллы, разлагающиеся выше
360° с выделением SO2; растворимость в воде: 15,9%
(19°) и 15,3% (100°).
Для 4-валентного О. соли кислородных кислот мало-
характерны. Получены, вчастности, Sn(N О3)4, Sn(SO4)2,
Sn(CH3COO)4 и др. Все они легко гидролизуются.
Аналитическое определение. О. от-
носится к аналитич. группе элементов, осаждаемых
О 24 к. X. Э. т. 3
739
ОЛОВО — ОЛОВООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
740
сероводородом в кислой среде и образующих с суль-
фидами натрия и аммония растворимые тиосоли. Ка-
чественно О. открывают, восстанавливая его цинком
из кислого р-ра до металла или SnHa. Все соединения
О. при сплавлении с NajOg дают растворимые стан-
наты, а после подкисления р-ра — соли Sn4+, к-рые
восстанавливают Fe, Al или РЬ и титруют иодометри-
чески. При растворении сплавов в HNOs часто удается
количественно выделить О. в виде осадка НгЗпОз,
к-рую прокаливают и взвешивают в виде SnOj. Метод
отделения О. перегонкой основан на летучести S11CI4.
Колориметрически малые количества О. определяют
по синей окраске с силикомолибденовой к-той или по
красной окраске с дитиолом.
Получение. Руды О. обычно содержат не более 1%
Sn, а россыпи — иногда только 0,01—0,02% Sn. Руды
часто комплексны, они содержат вольфрам, титан,
редкоземельные и др. редкие металлы. Руды обога-
щают гравитацией, магнитной сепарацией, флотацией
и флотогравитацией, а россыпи — преим. гравита-
цией, получая 40—70%-ные концентраты. Для уда-
ления примесей S и As концентраты обжигают при
темп-ре 600—700°, для удаления Fe, Bi, ЬЬ и др. при-
месей выщелачивают конц. НС1, а затем плавят с углем
и флюсами в отражательных или электрич. печах.
Шлаки богаты О., они подвергаются доработке, т. е.
повторной плавке в электрич. печах и фьюмингованию
с углем и пиритом. Черновое О., содержащее 97—99%
Sn, рафинируют от примесей в обогреваемых стальных
полусферич. котлах. Железо и медь удаляют вмеши-
ванием в жидкое О. угля и серы; мышьяк и сурьму
отделяют в виде соединений и сплавов с алюминием;
свинец — действием SnCl2, а висмут — в виде соеди-
нений с кальцием и магнием. Рафинированный металл
разливают в изложницы, первые марки его содержат
99,56—99,90% Sn. Электролитическое рафинирова-
ние О. не нашло широкого применения. Особо чистое
О. для полупроводниковой техники (99.99985% Sn)
получают дополнительной очисткой способами элект-
ролиза и зонной плавки. Вторичное О. извлекается
из отходов белой жести и отходов сплавов, например
бронз.
Техника безопасности. При выплавке
и рафинировании О. возможно выделение вредных
газов: SO2, СО, As2O3, AsH3 и топкой пыли, вызываю-
щей легочные заболевания — пневмокониоз и сили-
коз. Опасность отравлений и заболеваний предупреж-
дается работой плавильных печей под разрежением,
приточно-вытяжной вентиляцией цеховых помещений
и устройством местных отсосов. Для предупреждения
ожогов рабочим выдаются суконные костюмы, шляпы,
рукавицы и валенки.
Применение. Около 40% выплавляемого О. расхо-
дуется на произ-во белой жести для консервной
пром-сти. Эю обусловлено стойкостью О. против кор-
розии, легкостью покрытия им железа и безвред-
ностью продуктов коррозии. О. применяют для
пайки, лужения, а также изготовления припоев,
бронз, типографских, подшипниковых и др. сплавов
(см. Олова сплавы). Ок. 7% металла расходуется в виде
химич. соединений, гл. обр. двуокиси, хлоридов,
станнатов и др.
Лит.: Мурат Н. Н. [и др.], Металлургия олова, М.,
1964; Tin and its alloys, ed. E. S. Hedges [a. 0.], L., 1960;
M a n t e 1 1 C. L , Tin, its mil Ing, production, technology and
applications, 2 ed., N. Y., 1949; Севрюков H. H.. Кузь-
мин Б. A„ Чеаищев E. В , Общая металлу!гия, 2 изд.,
М.,1962; Герасимов Я. И.,КрестовниковА. Н.,
Шахов А. С., Химическая термодинамика в цветной метал-
лургии, т. 2, М., 1961; Основы металлургии, т. 2, М., 1962;
Tafel V., Lehrbuch der Metallhiittenkunde, Bd 2, Lpz., 1953;
M e 1 1 0 r, v. 7, L.—N. Y.—Toronto, 1947; то же, v. 7, 1952;
Sneed M. C., Brasted R. C., Comprehensive Inorganic
chemistry, v. 7 — The elements and compounds ot group
IVa, N. Y.—L.— Toronto, 1958; Kirk, v. 14, N. Y., 1955,
p. 136—64. H. Н. Севрюков.
ОЛОВООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ — орга-
нич. соединения, содержащие по меньшей мере
одну связь С—Sn. Число известных О. с. весьма
велико, а свойства их существенно различны. Подав-
ляющее большинство О. с. образовано 4-валентным,
оловом.
Соединения четырехвалентного олова. В эту группу
включаются: симметричные О. с., в молекулы к-рых
входят 4 одинаковых радикала R; несимметричные
О. с., содержащие разные радикалы; оловоорганич.
галогениды; кислород- и серусодержащие О. с., гид-
риды алкилолова и другие.
Симметричные О. с. RiSn. Тетраалкиль-
ные соединения, б. ч. гомеополярные жидкости, рас-
творимые в обычных растворителях и нерастворимые
в воде. Тетраарильные соединения — твердые веще-
ства, с т. пл. > 170°. Низшие О. с. могут быть пере-
гнаны без разложения. Соединения R$Sn получают
в основном двумя методами — действием на хлорное
олово органич. соединений Mg, Li, Al и Zn, напр.:
SnCli + 4RMgX —>R,Sn + 4MgXCl
и обработкой сплавов Sn/Na или Sn/Mg галоидными
алкилами, напр.:
Mg2Sn + 4C2H.X —> (C2Hs),Sn + 2MgX2
Алкилированием оловоорганических галогенидов
R„SnX4_„ могут быть получены оловотетраалкилы,
содержащие различные радикалы. Соединения R4Sn
устойчивы по отношению к воде и кислороду воздуха;
в отличие от более реакционноспособных металлоор-
ганич. соединений не реагируют с карбонильными
соединениями, спиртами и т. п., однако связь Sn—С
относительно легко расщепляется действием галоге-
нов, кислот и нек-рых неорганич. галогенидов.
Несимметричные О. с. В несимметрич.
О. с. группы R могут быть алкильными, арильными
или же эти группы могут содержаться одновременно.
Несимметрич. О. с., как правило, более низкоплавки,
лучше растворимы в органич. растворителях. Обыч-
ным методом получения несимметрич. О. с. является
взаимодействие реактивов Гриньяра с оловоорганич,
галогенидами, напр.:
R2SnX2 + 2R'MgX —► R2SnR' + 2MgX2
Несимметрич. О. с. по химич. свойствам не отличаются
от соответствующих симметрии. О. с., но большей
частью несколько более реакционноспособны.
А л кил ол ов ог а л оген иды. При взаимо-
действии соединений ряда R4Sn с галогенидами олова
в зависимости от соотношения реагентов образуются
различные алкилоловогалогениды:
3RtSn+SnX4 —► 4R3SnX; R^n + SnX,—>+R2SnX2
R4Sn + 3SnX4—>- 4RSnX3
Эти соединения являются важными полупродуктами в
синтезе разнообразных О. с. Алифатич. алкилоловога-
логениды, за исключением твердых и труднораство-
римых фторидов, являются либо жидкостями, либо
легкоплавкими твердыми веществами; ароматич. сое-
динения — твердые вещества (ArSnXa часто бывают
жидкими). Алкилоловогалогениды получаются также
при отщеплении групп R от тетраалкильных О. с.
действием галогенов; первые две группы отщепляются
ступенчато, две оставшиеся — одновременно:
R4Sn + X2—> R3SnX+RX; R3SnX+X2—>-R2SnX2 + RX
R2SnX2 + 2x2'—- SnX, + 2RX
Важными синтетич. методами являются также не-
посредственное взаимодействие металлич. Sn с гало-
геналкилами;
нагревание
Sn+2RX--------► R2SnX2
741
ОЛОВООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ — ОМЫЛЕНИЕ
742
и частичное алкилирование галогенидов олова при
помощи, напр., реактивов Гриньяра:
SnXj + nRMgX —> RBSnX4_и + nMgXa
Диарилоловодигалогениды могут быть получены раз-
ложением двойных солей арилдиазониев с хлорным
оловом:
(Ar2N2Cl)2SnCl1 + 2Sn —> Ar2SnCl2 + 2N2 + 2SnCl2
Атомы галогена в соединениях R„SnX4n являются
весьма подвижными и легко замещаются алкилами
(прп действии RMgX и т. п.), алкоксилами (при дей-
ствии алкоголятов), водородом (LiAlH4), натрием
(в жидком аммиаке) и т. д.
Важнейшие реакции алкилоловогалогенидов —
гидролиз (щелочи, аммиак) и образование оловоорга-
нич. сульфидов (H2S). Гидролиз приводит к оловоор-
ганич. гидроксидам и оксидам:
R3SnX +ОН-—► R3SnOH + X~
R2SnX2+2OH“ —> R3SnO + H2O + 2X ~
RSnX3+ЗОН-—* RSnOOH+ H2O +3X _
Кислородные соединения. Гидро-
окиси триалкилолова при дегидратации образуют
окиси типа (R3Sn)2O, к-рые при щелочном гидролизе
вновь дают R3SnOH. В отличие от алифатич. анало-
гов, (CeH5)3SnOH обладает кислыми свойствами.
Гидроокиси R3SnOH — обычно твердые вещества,
растворимые в органич. растворителях (низшие члены
растворимы и в воде); обладают резким запахом и
в растворе несколько ассоциированы. (R3Sn)2O —
жидкости, перегоняющиеся в вакууме; (Ar3Sn)2O —
твердые нерастворимые вещества. Окиси R2SnO —
твердые, нерастворимые и неплавкие вещества, поли-
мерные при не слишком больших R (R<Ci2H25);
соответствующие гидраты R2Sn (ОН)2 нестойки.
Станноновые кислоты — амфотерные неплавкие ве-
щества с т. разл. > 300°, обычно плохо растворимые
в органич. растворителях и нерастворимые в воде.
Алифатич. RSnOOH получают также взаимодействием
SnCl2 с галогеналкилами в щелочной среде. СО2
вытесняет RSnOOH из их солей; с другой стороны,
действие галогеноводородов приводит к тригалоге-
нидам аЛкилолова:
RSnOOH + 3HX —>RSnX3+2H3O
Реакция последнего типа является общей для всех
оловоорганич. оксидов и служит способом разделения
И очистки соответствующих галогенидов. Аналогично
получаются алкилоловянные соли других к-т, в том
числе и многих органич. к-т, напр.
2RCOOH + R2SnO —> R2Sn(OCOR)2
Сульфиды. Сульфиды типа R2SnS — обычно
твердые вещества с четкими темп-рами плавления,
имеющие, по-видимому, структуру 6-членных триме-
ров; сульфиды (R3Sn)2S сходны с окисями триалкил-
олова. Известны также нерастворимые трисульфиды
(RSnS)2S, к-рые разлагаются, не плавясь, при высо-
ких темп-рах.
Гидриды. Оловоорганические гидриды легче
всего можно получить восстановлением галогенидов
А1Н
алюмогидридом лития: R„SnX4_n —,’RnSriH4n
(n= 1—3). Они мономерны, не ионизированы и раст-
воримы в инертных растворителях. Большая часть
этих гидридов может быть перегнана (CH3SnH3 газо-
образен), однако медленное разложение имеет место
уже при комнатной темп-ре; устойчивость гидридов
повышается с увеличением числа алкильных групп.
Соединения R„SnH4n обладают восстановительными
свойствами и присоединяются к непредельным соеди-
нениям, напр.:
R3SnH + R'CH = CHR"—> RsSnCHR'CHaR"
(R' и R" могут содержать функциональные группы).
Производные щелочных металлов.
Соединения типа R3SnMe и R2SnMe2 (Me=Na, Li) —
нестабильные вещества, обладающие в растворах
в жидком аммиаке свойствами сильных электролитов.
Обычный способ их получения — действие щелочного
металла на галогениды алкилолова в жидком аммиаке
или в среде тетрагидрофурана. Эти соединения силь-
но нуклеофильны (остаток R3Sn аналогичен карба-
ниону); щелочной металл легко замещается алкилами,
галогенами, остатками R3Sn и т. п.
Гексаалкил- и гексаарилдистан-
наны R3Sn—SnR3. Низшие алифатич. соединения
этого типа — устойчивые в отсутствие кислорода
перегоняющиеся жидкости; высшие и ароматиче-
ские — твердые, часто весьма высокоплавкие тела.
Гексаалкил дистаннаны нередко образуются в качестве
побочных продуктов при получении других О. с.;
препаративно могут быть получены при действии
натрия на триалкилоловогалогениды:
Na R3SnX
R3SnX —> RjSnNa------> R3Sn -SnR3
Связь Sn—Sn легко расщепляется галогенами,
серой, кислородом, галогенидами ряда металлов
(Hg, Bi), щелочными металлами и их амидами, некото-
рыми органич. галогенидами ит. п. Во всех этих слу-
чаях образуются соответствующие производные ряда
R3SnY (Y — галоген или металл) или (R3Sn)2Z
(Z=O, S).
Соединения двухвалентного олова. О. с., образован-
ные двухвалентным оловом (R2Sn), немногочисленны.
Для этих соединений характерна склонность к легкой
полимеризации уже при стоянии; мономерные или
низкомолекулярные оловодиалкилы с сильным окра-
шиванием растворяются в органич. растворителях
(кроме спирта), но в полимерном состоянии они нера-
створимы. Темп-ры плавления также зависят от
«возраста» препарата, т. е. от степени полимерности.
Соединения R2Sn получаются при действии реактивов
Гриньяра или литийорганич. соединений на двухло-
ристое олово:
SnClt+2RMgX —> RaSn + MgX2+MgCl2
или при дегалогенировании диалкилоловодигалогени-
дов цинком, амальгамой натрия или натрием в жидком
аммиаке, напр.:
(C2H5)aSnCI2 + Zn —> (C2H5)2Sn + ZnCI2
Оловодиалкилы обладают сильными восстановитель-
ными свойствами (окисляются кислородом воздуха
в окиси R2SnO, выделяют серебро из AgNO3 и ртуть
из сулемы); присоединяют многие реагенты, давая
производные Sn(IV) (галогены, алкилгалогениды, ли-
тийалкилы). При нагревании R2Sn выделяют метал-
лич. олово и переходят в соединения четырехвалент-
ного олова.
О. с., особенно производные триалкилолова, очень
токсичны. Многие О. с., в частности дилаурат дибутил-
олова и сернистые О. с., применяют в качестве эф-
фективных стабилизаторов поливинилхлорида, анти-
оксидантов для каучуков и катализаторов образова-
ния полиуретанов. Соли диалкилолова используют
также в ветеринарной практике (антигельментики);
соединения ряда триалкилолова — в качестве фунги-
цидов и антисептиков в произ-ве бумаги.
Лит.: И н г а м Р. [ид р. J, Оловоорганические и герма-
нийорганические соединения, М., 1962; Кочешков К. А.,
Синтетические методы в области металлоорганических соеди-
нений элементов IV группы, М.—Л., 1947; Рохов Ю. Д ж.,
Херд Д.,Льюис Р., Химия-металлоорганических соеди-
нений, пер. с англ., М., 1963; GoatesG. Е., Organo-metalliC
compounds, 2 ed., L.—N. Y., 1960. О. IO. Охлобыстин.
ОМЫЛЕНИЕ — гидролиз сложного эфира с обра-
зованием спирта и кислоты (или ее соли):
/О /,°
R—Су +НОН —>-R—C<f +R'OH
4OR' ХОН
24*
743
ОМЫЛЕНИЯ ЧИСЛО — ОНИЕВЫЕ СОЕДИНЕНИЯ
744
Происхождение термина «О.» связано с методом
произ-ва мыла из жиров. О. часто называют гидро-
литич. превращения любых производных к-т, приво-
дящие к кислотам или их солям (О. нитрилов, амидов,
хлорангидридов и т. и.):
R-C = N + H3O —>RCOOH+NH,
R-CONH3 + H2O —>rcooh + nh8
R—COC1 + H2O —>RCOOH + HC1
Поскольку галогенозамещенные углеводороды можно
рассматривать как эфиры галогеноводородных к-т,
их гидролиз также часто называют О.:
R—С1 + н2о —>R—0Н + НС1
Иногда термин «О.» применяют к другим случаям
гидролиза органических соединений, напр. О. простых
эфиров. Е. М. Рохлин.
ОМЫЛЕНИЯ ЧИСЛО — количество КОН, необ-
ходимое для омыления 1 г сложного эфира, численно
выраженное в миллиграммах. Омыление ведут спирто-
вым р-ром КОН при темп-ре кипения. Одновременно
проводят в таких же условиях контрольный опыт.
О. ч. вычисляют по формуле: О. ч.=28,055х(о—б)!в,
где а — расход 0,5 н. р-ра НС1 на титрование конт-
рольного опыта, мл', б — то же на титрование избыт-
ка щелочи после омыления, мл', в—навеска эфира.
Для определения О. ч. трудно омыляемых эфиров
вместо этилового спирта применяют пропиловый
спирт, в среде к-рого реакция идет значительно быст-
рее. О. ч. является важной технич. характеристикой
жиров, т. к. оно характеризует мол. вес кислот, вхо-
дящих в их состав в виде глицеридов. О. ч. жира соот-
ветствует количеству миллиграммов КОН, необхо-
димому для омыления глицеридов жирных к-т и ней-
трализации свободных к-т, содержащихся в жире,
т. е. сумме эфирного и кислотного чисел жира.
Лит.: Губен-Вейль, Методы органической химии,
т. 2, Методы анализа, пер. снем., 4 изд., М., 1963; Бауэр К.,
Анализ органических соединений, пер. с нем., 2 изд., М.,
1953. А. А. Черкасский.
ОНЗАГЕРА УРАВНЕНИЕ ЭЛЕКТРОПРОВОДНО-
СТИ — связывает эквивалентную электропровод-
ность электролита с основными величинами, характе-
ризующими электролит и растворитель: концентра-
цией и валентностью ионов электролита, диэлектрич.
проницаемостью и вязкостью растворителя, а также
темп-рой р-ра. Модель, к-рую описывает О. у. э.,
рассматривает движение иона в р-ре электролита под
действием электрич. поля и содержит след, допуще-
ния: растворитель представляет собой однородную
изотропную среду с диэлектрич. проницаемостью D
и вязкостью т|; концентрация электролита столь мала,
что размеры ионов не учитываются (т. е. они предпо-
лагаются точечными), а электролит полностью диссо-
циирован; каждый ион окружен ионной атмосферой,
состоящей из ионов противоположного знака.
В простейшем случае для одно-одновалентного
электролита (тип 1—1, напр. NaCl) О. у. э. имеет вид:
A=AO-(A + BAO)KC
где А — эквивалентная электропроводность, Ао —
эквивалентная электропроводность при бесконечном
разбавлении, С — концентрация в молях на литр, а
А = 82,4/(Р7’),'2т] и В = 8,20-105/(РП3'2 — кон-
станты, зависящие только от природы растворителя
и темп-ры. Для несимметричных электролитов (типы
1—2, напр., СаС12, 1—3 и т. д.) константы А и В при-
обретают значительно более сложный вид и вычисление
их весьма затруднительно.
О. у. э. объясняет падение эквивалентной электро-
проводности при увеличении концентрации электро-
лита тем, что подвижность ионов уменьшается под
действием сил электростатич. взаимодействия ионов.
Первый член в скобках отражает т. наз. электрофоре-
тич. эффект, а второй — т. наз. релаксационный эф-
фект (см. Электропроводность электролитов). Экспе-
риментальная проверка подтверждает О. у. э. в доста-
точно разб. р-рах сильных электролитов.
О. у. э. используется для определения степени
диссоциации а электролитов. Последняя может быть
найдена из ур-ния:
Ур-ние позволяет проверить применимость представ-
лений о р-рах электролитов, к-рые следуют из теории
Дебая и Хюккеля, не только по термодинамич., но и по
кинетич. свойствам р-ров (см. Электролиты).
Лит.: Глесстон С., Введение в электрохимию, пер.
с англ., М., 1951. Л. И. Богуславский.
ОНИЕВЫЕ СОЕДИНЕНИЯ — вещества ионного
характера общей ф-лы (Rn+1M"+)X_, где R—водород,
органич. радикал или, реже, другой заместитель, М —
атом N, Р, As, Sb, О, S, Se, Те, J, Вг, С1, п — низшая
валентность М в органич; соединениях, X-— анион.
Термин «О. с.» образован как обобщение от названия
аммония соединения для веществ типа R^N + X-. Ана-
логичные производные других элементов наз. фосфо-
ниевые соединения RiP + .X~, арсониевые соединения
RiAs + X ~,стибопиевыесоединения RiSb + Х ~, оксоние-
вые соединения RsO + X-, сулъфониевые соединения
RsS + X ~, селенониевые соединения Вз8е+Х~,теллуро-
ниевые соединения RaTe + X ~, галогенониевые соедине-
ния (иодониевые R2J+X~, бромониевые R2Br+X~,
хлорониевые R2C1+X_), а соответствующие катионы—
аммоний, фосфоний, стибоний и т. д. Частными слу-
чаями аммониевых соединений являются соли иммония
R2N=-, гидроксиламмония R2N + (ОН), гидразония
R2N — N + R2, гуанидония R2NC(= NR)N + R2. Аммоние-
вое состояние одного из атомов азота характерно для
солей диазония R—N + =NX~(cm. Диазониевая струк-
тура). Ониевыйатом мо-
жет входить в состав re- s—л
тероцикла. Так, пири- 1^+J L 1^+J l^+JI
дин образует соли пи- 7 X' I* X' ? X'
ридиния (I), ХИНОЛИН— R R R
хинолиния соединения I 11 ш
(II), тиазол—тиазолия
(III) и т. и. В ониевом состоянии находятся ато-
мы, несущие положительный заряд и участвую-
щие в образовании семиполярной связи, напр. азот
в комплексах аминов с BFs (IV), окисях аминов (V),
нитросоединениях (VI), сера в сульфоксидах (VII),
сульфонах (VIII) и сульфокислотах (IX) ит. д. Та-
ким образом, переход к соединениям азота, серы и
т. и. высшей валентности связан с переходом цент-
рального атома в ониевое состояние.
R3NT—BF3 R3fj—0-
IV V
R. + /О
Rz ^Cl-
VIII
+/0 R. +
R—NC )S—0
X0- Rz
VI VII
+ /°
R-S-OH
o-
IX
В состоянии низшей валентности атомы элементов,
способных давать О. с., пмеют неподелеиную пару
электронов. Формально процесс образования О. с.
всегда можно представить как обобщение свободной
пары в результате взаимодействия с веществами,
имеющими вакантную орбиту или способными образо-
вать вакантную орбиту путем перестройки молекулы
в ходе реакции. Примерами соответствующих реакций
(1) могут служить образование аддукта триметил-
амина с BFs:
(CH,)3N: + BF3 —» (СН3)3Й -BF,
(1)
745
ОНИЕВЫЕ СОЕДИНЕНИЯ —ОПИЙ
746
и хорошо известное образование солей аммония из
аминов и галогеналкилов:
(CH3)3N:+JCH3 —HCH,)4NJ- (2)
В результате обобщения свободной пары электронов
атом-донор приобретает один положительный заряд
и одну дополнительную ковалентную связь. Так,
азот в аминах нейтрален и трехвалентен, в солях ам-
мония — положительно заряжен и четырехковален-
тен; сера в органич. сульфидах нейтральна и двух-
валентна, в солях сульфонил — положительна и трех-
валентна; иод в органич. иодидах нейтрален и одноко-
валентен, в солях иодония — положителен и двух-
ковалентен.
Реакция (2) — наиболее общая схема образования
ониевых солей. Аналогично аммониевым соединениям
так могут быть получены соли из фосфинов, арсинов,
стибинов, сульфидов, теллуридов. Известны отдель-
ные примеры получения по этой реакции также и со-
лей оксония, напр.:
(С2Н.,)20 BF8+C2H5F —> (C,H,),OBf7
Соли галогенониев так получить нельзя. Таким обра-
зом, реакция (2) типична для получения О. с. элемен-
тов V группы, менее характерна для VI и не имеет
места для VII группы периодич. системы. Легкость
образования О. с. по этой схеме падает в V группе
в последовательности P>As>N>Sb, а в VI группе —
Te>Se>S>0. Во многих случаях для получения
О. с. требуются другие, специфич. методы. В особен-
ности это относится к О. с. галогенов и кислорода,
в меньшей степени — других элементов.
Стабильность О. с. для данной группы периодич.
системы возрастает сверху вниз — в V группе от N
к Sb, в VI — от О к Те, в VII — от С1 к J.
Стабильные О. с. представляют собой вещества,
растворимые вследствие своего ионного характера
в воде, причем водные р-ры обладают большой электро-
проводностью. Продуктами термич. разложения солей
четвертичных аммониевых оснований являются тре-
тичные амины и галогеналкилы. Разложение первич-
ных, вторичных и третичных солей аммония приводит
к продуктам алкилирования аммиака или аминов
(см. Алкилирование). Действием влажной окиси
серебра ониевые соли могут быть переведены в соответ-
ствующие гидроокиси, являющиеся довольно силь-
ными основаниями, выделяющими NHa из NH4CI,
связывающими СО2 и реагирующими с металлич.
алюминием с выделением Н2. Гидроокиси менее устой-
чивы, чем соли; порядок же изменения их стабиль-
ности тот же, что и для солей. Гидроокиси четвер-
тичных аммониевых оснований вполне устойчивы,
гидроокиси сульфониев, хотя и неустойчивы, все же
могут быть выделены; гидроокиси оксониев совер-
шенно неустойчивы. Термич. разложение гидрооки-
сей четвертичных аммониевых оснований, а также
третичных сульфониевых, селенониевых и теллуро-
ниевых оснований происходит с образованием олефи-
нов по схемам:
(C2H5)4N + ОН - —>( C2H5)3N + сн2=сн2 + Н2О
(C2Hs)3S + ОН - —> (C2H6)2S + СН2=СН2 + Н2О
Гидроокиси же фосфониевых, арсониевых и стибоние-
вых оснований разлагаются с образованием насыщен-
ных углеводородов и соответствующих окисей фосфи-
нов, арсинов, стибинов:
(С2Н3)4Р + ОН- —>СН3-СН3 + (С2Н;)3РО
Центральный атом О.с. имеет тетраэдрич. конфигу-
рацию, что показано разделением на оптические анти-
поды аммониевых, фосфониевых И арсониевых солей,
имеющих четыре различных заместителя. Могут быть
разделены на антиподы также соли сульфония, ссле-
нония и теллурония, содержащие три различных заме-
стителя: роль четвертого заместителя играет в этом
случае неподеленная пара электронов S, Se или Те.
Многие О. с. обладают физиологич. активностью,
действуя гл. обр. на вегетативную нервную систему
аналогично курарину, мускарину или никотину;
ониевые структуры входят в состав многих природных
и биологически важных соединений (см., напр., Лекар-
ственные вещества. Алкалоиды, Холин, Холинэстераза
и т. д.), находят применение в качестве гербицидов
и фунгицидов. Ониевые соли, содержащие радикалы
с длинной углеродной цепью, обладают поверхностной
активностью и применяются как нейтральные мыла
и закрепляющие вещества в текстильной пром-сти.
К О. с. относятся многие классы органич. красителей
(см. Цветности теории, Красители синтетические,
Красящие вещества природные). О. с. азота, фосфора
и серы применяют в качестве стабилизаторов для ви-
нильных смол. О. с. являются промежуточными про-
дуктами во многих органич. реакциях и перегруппи-
ровках (см., напр., Арбузова реакция, Брауна реакция,
Виттига реакция, Гофмана реакция, Кренке реакция,
Ладенбурга реакция, Соммле реакция, Стивенса пере-
группировка, Илиды). Термин «ониевые катионы»
применяют иногда также по отношению к неорганич.
и органич. катионам, имеющим секстет электронов
на внешней оболочке. К частицам такого рода, играю-
щим важную роль в органич. реакциях, относятся
катионы карбония ВлС + , нитрония ОгК + , нитрозокия
O=N + , сульфония HOSO2+, хлорония Cl+, бромония
Вг+, иодония Js+ и J , иодозония O=J +. В отличие от
О. с., рассмотренных выше, эту группу частиц назы-
вают также «нестабильными ониевыми катионами»,
чтобы в какой-то мере избежать путаницы, вызываемой
совпадением терминологии. Следует отметить, что если
названия ионов карбония, нитрония, нитрозонпя и
иодозония прочно укоренились, то вместо «хлоровин»,
«бромония» и «иодония» для ионов галогенов говорят
обычно «катион хлора, брома, иода» (см. Карбония ион,
Галогенозамещенные углеводороды). Б.Л.Дяткин.
ОНОВЫЕ КИСЛОТЫ — см. А лъдоновые кислоты.
ОПИЙ — высохший на воздухе млечный сок несо-
зревших плодов мака Papaver somniferum L. сем.
маковых (Papaveraceae). Комки или масса темно-бу-
рого цвета с характерным запахом. Частично раство-
рим в воде с образованием бурого р-ра с кислой реак-
цией. Состав О. весьма сложен. Он содержит воду
(до 40— 50%), алкалоиды, белки, углеводы, смолы,
воск, жиры, пигменты. Из О. выделено св. 20 алкалои-
дов. Содержание главных алкалоидов в воздушно-
сухом отечественном О. составляет: 12—16% морфина,
10—18% наркотина, 0,5—1,5% папаверина, 1—3%
кодеина. Алкалоиды, за исключением слабых осно-
ваний (наркотина и папаверина), находятся в виде
солей различных кислот. Качество О. в основном опре-
деляется содержанием в нем морфина и кодеина.
Обычно соли алкалоидов экстрагируют из О. горя-
чей водой, а затем из остатка минеральной к-той извле-
кают наркотин. Водный экстракт упаривают; алка-
лоиды осаждают подщелачиванием и разделяют, ис-
пользуя различие их силы основности и разную раст-
воримость в различных растворителях.
В медицине применяется как вся сумма алкалоидов
О. (пантопон, или омнопон, содержит ок. 50% морфи-
на), так и отдельные алкалоиды — морфин, кодеин,
папаверин. Физиологич. действие самого О. то же,
что и у морфина.
Лит.: Преображенский И. А.,Генки и Э. И. ,
Химия органических лекарственных веществ, М., 1953, с. 208;
Атлас лекарственных растений СССР, м., 1962; Машков-
ский М. Д., Лекарственные средства, 4 изд., М., I960.
А. Е. Васильев,
747
ОПОПАНАКС —ОПРОБОВАНИЕ МАТЕРИАЛОВ
748
ОПОПАНАКС — сок или каучукообразная часть
выделений растения Opopanax chironium. В настоя-
щее время коммерч. О. также называют высушенный
на солнце экст р шт коры дерева Commiphora erythraea,
произрастаюп его гл. обр. в Сомали. Масло О. —
жидкость зеленоватого цвета, обладающая характер-
ным бальзамным запахом смолы, частично раствори-
мая в спирте; спиртовой р-р после фильтрования и
концентрирования в вакууме дает т. наз. смолистый
О. Типичный запах О. определяется наличием летучих
сн масел, к-рые могут быть выделены
I 3 при перегонке с водяным паром. Со-
став эфирного масла О. мало изучен,
I I 1ГН из него выделен ряд сесквитерпенов
3 и терпеновых спиртов. Первым иден-
« тифицированным сесквитерпеном
хСх масла О. является бисаболен(т.кип.
нзС Сн3 268°, а?20 0,8725).
Масло О. используется самостоятельно или как
компонент в парфюмерных композициях в смеси
с другими компонентами, напр. с маслом сандала.
Э. Е. Нифамтьев.
ОППЕНАУЭРА РЕАКЦИЯ — окисление вторич-
ных спиртов в кетоны действием ацетона в присутст-
вии трет-бутилата алюминия:
R. [(СН3)3СО]3А1 R.
>СНОН + СН3СОСН3( _ —> )С = О + (СН3)2СНОН
R'z R'Z
О. р. является реакцией, обратной восстановлению
по Меервейну — Понндорфу (см. Меервейна —Понн-
дорфа — Берлея реакция). Применением большого
избытка ацетона равновесие сдвигается в сторону
образования кетона. Трет-бутилат алюминия может
быть заменен изопропилатом или фенолятом алюми-
ния. Обычно спирт кипятят с алкоголятом в бензольно-
ацетоновом растворе. В случае трудно окисляемых
спиртов иногда в качестве акцепторов водорода при-
меняют кетоны, кипящие при более высокой темп-ре,
чем ацетон: метилэтилкетон, циклогексанон, бензо-
хинон в толуольном или ксилольном р-ре.
Мягкие условия и селективность О. р. обуслов-
ливают ее широкое применение для получения ла-
бильных соединений, гл. обр. стероидов, гормонов,
алкалоидов и пр. При О. р. (3,у-ненасыщенных спир-
тов двойная связь мигрирует в а, Р-положение (1).
Насыщенные и ненасыщенные диолы окисляются в ди-
кетоны (2). Специфичность О. р. проявляется в успеш-
ном окислении ряда соединений, содержащих аллиль-
ную, винильную, этинильную, бензилиденовую, диа-
зокетонную, галогенокетонную, ацетальную, лактон-
ную и т. п. группировки. Алкалоиды типа хинина не
окисляются в обычных условиях, но использование
трет-бутилата калия и бензофенона в бензольном р-ре
привело к количественному окислению хинина в хинн-
ой (3). Первичные спирты, как правило, не окисляются
в условиях О. р., однако превращение спирта в алко-
голят и использование высококипящих альдегидов
в качестве акцепторов водорода приводят к желаемому
результату (4):
ОН О
I _ II х / Q 4
R-CH-R ------ R-C-R'
(RCH2O)3A1 + с6н5сн=снсно —•-
---- RCHO + (С6Н5СН=СНСН2О)3А1 (4)
Механизм О. р. представляется след, образом:
сначала образуется алкоголят окисляемого спирта,
затем молекула алкоголята отдает гидрид-ион моле-
куле ацетона (подобно тому как нек-рые реактивы
Гриньяра восстанавливают карбонильное соединение,
а не присоединяются к нему):
ГН
RXH + cof3
R'Z ЧОН \н3 R/ Ч0А1-^-0
з
R4 Нч СН3
---- хС=О 4- С
R' A£OZ ЧСН3
Реакция открыта в 1925—26 А. Берлеем и В. Понн-
дорфом; в 1937 Р. В. Оппенауэр продемонстрировал
широкое применение этого метода в основном для син-
теза стероидов.
Лит.: Surrey A. R., Name reactions in organic chemist-
ry, N. Y., 1954, p. 129; Д ь e p а с с и К., в кн.: Органические
реакции, пер. с англ., сб. 6, М., 1953, с. 235. Н. 11. Гамбарян.
ОПРОБОВАНИЕ МАТЕРИАЛОВ
I. Введение................................. 743
II. Опробование газов........................750
III. Опробование жидкостей.................... 750
IV. Опробование полужидких (вязких) масс .... 751
V. Опробование твердых материалов........... 752
I. Введение. О. м. — процесс отбора и обработки
проб, взятых пз месторождений, добытых ископаемых,
полупродуктов и продуктов производства. Целью
О. м. является определение физич. ц химич. свойств
материала.
Для опробования отбирают среднюю про-
бу — часть материала, небольшую по сравнению
со всей массой его, средний состав и свойства ко-
торой идентичны во всех отношениях (минералогия.,
химич. п т. д.) среднему составу исследуемого
продукта.
В зависимости от назначения пробы разделяют на: 1) мине-
ралогия.— дает возможность выявить структурные особенно-
сти и состав полезных ископаемых и т. д., применяется для со-
ставления вероятных схем переработки сырья; 2) химич.—
имеет целью установление химич. состава сырья и продуктов
переработки; 3) технология,— служит для установления
вещественного состава материала и применяется для разработ-
ки технологической схемы производства; 4) пробы для опре-
деления влаги — имеет значение для контроля процессов
сушки и обезвоживания, содержания воды в твердом теле;
5) пробы для ситового и седиментационного анализа — при-
меняется для установления гранулометрического состава сы-
пучей массы.
Различие между пробами заключается преимущественно в
количестве материала, отбираемого от исходной средней про-
бы материала для проведения того или иного исследования.
Одна и та же проба может служить одновременно для разных
целей, напр. для установления химич. и минералогия, состава
или влажности и т. п.
Для получения средней пробы правильного состава
решающее значение имеет методика отбора пробы.
О. м. заключается в отборе средней пробы и ее обра-
ботке. Средняя проба (так наз. генеральная
проба) составляется из нескольких ч а с т и ч-
н ы х, отбираемых в разных местах опробуемой
партии материала. От генеральной пробы путем
749
ОПРОБОВАНИЕ МАТЕРИАЛОВ
750
«сокращения и, если необходимо, измельчения отби-
рают лабораторную пробу, из к-рой не-
посредственно берут навеску для анализа. Места
{точки) отбора проб распределяют так, чтобы в сред-
ней пробе был равномерно представлен весь исследуе-
мый материал. Для материала, расположенного пря-
моугольником, точки отбора проб располагают часто
так наз. «конвертом» — по углам и в середине
или аналогичными способами. От материалов, распо-
ложенных в бочках, чанах и т. п., пробы отбирают из
точек, расположенных на концентрических окруж-
ностях на разной глубине. Число частичных проб
устанавливается специальными правилами для каж-
дого опробуемого материала. Для сыпучих мате-
риалов число частичных проб (п) часто вычисляют по
формуле:
n = (rlm-R)2
где г и R — соответственно вероятная погрешность
частичной и средней проб, т — коэффициент, завися-
щий от степени гарантии:
степень гарантии, % . . . 82 96 99
т................ . 0.50 0,33 0,25
Минимальный вес средней пробы зависит от опробуе-
мого материала. Для однородных материалов вес
пробы меньше, чем для неоднородных. Минимальный
вес частичной пробы для твердых сыпучих материалов
часто вычисляют по формуле:
W=k-d*
где W — вес пробы в кг, к — коэффициент пропорцио-
нальности, зависящий от свойств материала (плот-
ность, форма зерен, крупность вкрапления определяе-
мого компонента и др.), d — размер наибольших кус-
ков или частиц смеси, лл, а — показатель степени
{для минералов колеблется в пределах 1,5—2,7).
Значение величин к и а устанавливают эмпирически.
Во всех случаях различают отбор пробы от непод-
вижно лежащего материала или из потока его. В пер-
вом случае частичные пробы отбирают в разных точках
опробуемой массы материала, пользуясь специаль-
ными правилами. Во втором
случае для отбора проб при-
меняют методы продольных
струй или поперечных сече-
ний. Метод продол ь-
Проба ных струй заключается
в делении движущейся струи
материала на ряд полос вдоль
потока (рис. 1) и непрерыв-
₽ис> 1. ном отборе пробы. Отбирают
одну или несколько чередую-
щихся полос. Метод пригоден для отбора пробы одно-
родного материала. Для неоднородного материала ста-
новится заметным неоднородное расположение частиц
по крупности. Пробы из неоднородного материала
предпочитают отбирать методом
поперечных сечений. В этом
В пробу СЛуЧае периодически отсекают в пробу
по всему сечению равные порции мате-
риала через равные промежутки време-
ни (рис. 2). Для правильного отбора
в пробу ПрОбы большое значение имеет частота
отсекания. Последняядолжна быть свя-
зана с переменным содержанием опре-
В пробу Деляемого компонента.
При отборе пробы следует обращать
внимание на однородность материала.
Рис. 2. Неоднородный кусковой материал при
транспортировке и укладке может рас-
слаиваться (явление сегрегации). При этом крупные
куски располагаются в нижней части кучи, наир,
конуса; размер частиц уменьшается по высоте. Рас-
слоение объясняется тем, что при движении материала
по наклонной плоскости крупные куски и куски
с большей плотностью движутся быстрее (большая
кинетическая энергия) и скапливаются гл. обр. в ос-
новании, Поэтому пробу необходимо брать по всей
толщине кучи опробуемого материала. Генеральная
средняя проба, в зависимости от величины партии,
может достигать величины в сотни килограммов или
несколько тонн. В дальнейшем пробу сокращают,
уменьшая ее вес до необходимого для анализа; сокра-
щение генеральной пробы может быть в 100 и более
раз. Эту операцию производят по правилам, позво-
ляющим сохранить один и тот же состав отобранной
и сокращенной проб. Для твердых тел сокращение
обычно сопровождается дроблением пробы.
II. Опробование газов. Пробу газов отбирают в гер-
метически закрывающиеся сосуды, из к-рых вытес-
няют воздух пропусканием через них длительное
время испытуемого газа. В нек-рых случаях пробу
газа отбирают в эвакуированные сосуды, к-рые после
отбора пробы герметизируют. Для отбора пробы из
потока газа заборную трубку располагают по оси
трубопровода.
III. Опробование жидкостей. Различают опробо-
вание однородных или расслаивающихся жидкостей,
а также жидкостей, находящихся в бассейнах, резер-
вуарах, цистернах и т. п. или же текущих по тру-
бам и желобам. Пробы однородных неподвижных
жидкостей берут после перемешивания специальными
пробоотборниками или стеклянными трубками в раз-
ных точках сосуда; если перемешивание невозможно,
частичные пробы отбирают из разных слоев жидкости
по высоте. Пробу маловязких жидкостей, напр.
нефтепродуктов, находящихся в резервуаре, можно
отбирать, пользуясь спец, пробоотборником, к-рый
представляет собой вертикальную трубу, помещен-
ную внутрь резервуара и закрепленную в нем. По
высоте всей трубы просверлены калиброванные отвер-
стия. При отборе пробы содержимое трубы сливают
в промежуточную емкость, из к-рой через кран отби-
рают пробы для анализа. Жидкость, оставшуюся
в емкости, инжектором возвращают обратно в резерву-
ар. При этом резко сокращается продолжительность
отбора. От расслаивающихся жидкостей пробы отби-
рают из каждого слоя пропорционально его содержа-
нию. Пробу жидкости, текущей по трубопроводу,
отбирают через кран, вмонтированный в линию. Ча-
стичные пробы отбирают методом сечения непрерывной
струи черпаком или другим приспособлением и соби-
рают в общий бак с закрытой крышкой. Количество
опробуемых мест в партии и минимальный вес средней
пробы определяют специальными правилами. Напр.,
для серной и соляной к-т берут 5% мест, минималь-
ный вес пробы 1,0 кг; азотную к-ту берут из каждой
цистерны или контейнера в 3 местах, минимальный
вес пробы 0,1 кг.
Приводим как пример условия отбора проб нек-рых жид-
костей. Вода. Пробу с намеченной точки открытого водоема
отбирают батометром. Допускается отбор проб воды бутылью
емкостью 2—5 л, к-рую закрывают пробкой. Бутыль устанав-
ливают на намеченной глубине, пробку вынимают при помоши
шнура. Пробу воды с небольшой глубины (особенно зимой)
отбирают шестом с прикрепленной к нему бутылью. Бутыль
заполняют водой до верха. Перед закрытием бутыли пробкой
верхний слой воды сливают так, чтобы под пробкой остался не-
большой слой воздуха. Моторный бензол. Пробы от-
бирают от каждой цистерны в 3 местах по ее высоте. При по-
грузке моторного бензола в бидоны или бочки отбирают 10%
всех мест. От каждой отобранной бочки, пользуясь пробной
трубкой, отбирают частичную пробу через наливное отверстие
примерно из середины. Отбор проб производится для автомо-
бильного бензола зимнего при темп-ре не ниже —16°, для авто-
мобильного бензола летнего — не ниже —7°. Отобранные про-
бы, в общем количестве не менее 2 л, соединяют вместе, тща-
тельно смешивают и помещают в 3—4 сухие склянки с при-
тертыми пробками, емкостью каждая 0,75 л. Склянки перед на-
полнением ополаскивают моторным бензолом из отобранной
пробы.
Мазут. В зависимости от тары, в к-рой хранится мазут,
применяют разные способы отбора проб. Для этого пользуются
751
ОПРОБОВАНИЕ МАТЕРИАЛОВ
752
пробоотборником, представляющим собой сосуд с крышкой,
открываемой на заданной глубине. Среднюю генеральную про-
бу от мазута, хранящегося в вертикальном резервуаре, а также
в горизонтальном диаметре больше 2,5 м составляют из частич-
ных проб, отбираемых с 3 уровней: на 200 мм ниже поверхно-
сти, с середины высоты и на 100 мм ниже приемно-раздаточной
трубы; если трубы нет или если она расположена менее 250 мм
от дна резервуара, пробу отбирают на уровне, отстоящем от дна
на 250 мм. Для пробы, взятой из вертикального сосуда, берут
в среднюю пробу: с верхнего и нижнего уровней 1 часть, со
среднего уровня 3 части; для проб, взятых из горизонталь-
ного резервуара, соответственно 1 часть и 6 частей. Из налив-
ных судов пробы отбирают не менее чем из 25% трюмов (тан-
ков) в разных местах судна; частичные пробы отбирают с уров-
ней: 200 мм ниже поверхности (1 часть), с середины высоты
слоя мазута (3 части), с нижнего слоя, доступного для отбора
(1 часть). Из двухосной цистерны отбирают одну пробу с уров-
ня, расположенного па высоте 1 от ее дна. От четырехосной
цистерны отбирают 2 частичных пробы: одну на расстоянии
200 мм от дна ее и одну на высоте диаметра, считая от ее дна.
Среднюю пробу мазута, перекачиваемого по нефтепроводу, со-
ставляют из частичных проб, отбираемых через пробоотборный
кран в конце трубопровода. Пробы отбирают через разные про-
межутки времени в зависимости от продолжительности пере-
качки. Напр., если перекачка продолжается до 1 часа —в на-
чале и в конце перекачки, при продолжительности от 2 до
24 часов — в начале перекачки и через каждый час.
IV. Опробование полужидких (вязких) масс. В за-
висимости от условий опробования и физич. состояния
полужидких масс (пульп, лаков и красок, паст) при-
меняют ручной или автоматический отбор проб. Руч-
ное опробование производят специальными пробоот-
борниками, имеющими вид ковшей разной конструк-
ции (рис. 3). Пробоотборник 1 на
длинной ручке 3 опускают на нуж-
ную глубину в опробуемую массу
и открывают крышку 2, пользуясь
привязанным к ней шнуром. Важ-
ное значение имеет место отбора
пробы. Автоматические опробовате-
ли включают в поток движущейся
полужидкой массы. Для отбора про-
бы применяют периодически вклю-
чающийся механич. отсекатель ча-
сти потока или периодически напол-
няющийся ящик; известны и другие
конструкции. Из неподвижных па-
стообразных масс (масляныекраски,
нефтепродукты ит. п.) пробу отбира-
ют пробоотборниками, представляющими собой полу-
цилиндрические желобки с ручкой. Пробоотборник вво-
дят в исследуемую массу, поворачивают и вынимают,
при этом желобок остается заполненным материалом.
Применяют также пробоотборник и более сложной
конструкции. Обработка проб полужидких продуктов
заключается в сокращении, обезвоживании, измель-
чении и отборе аналитической пробы. Для сокращения
применяют делитель, в котором масса из воронки 1
стекает по наклонным желобкам, расположенным
так, что из части их масса выводится
наружу, напр, в карманы 2 и 3, часть
же делится дальше на других желоб-
ках 4 и выводится наружу через отвер-
стие (рис. 4). Обезвоживание (напр.,
пульпы) производят декантацией или
фильтрованием и сушкой. Для филь-
трования применяют вакуум- или
прессфильтры, а также гравитацион-
ные фильтры. Сушку производят при
атмосферном давлении, при комнат-
ной или повышенной темп-ре. Сухую
пробу обрабатывают далее по общим
правилам опробования твердых тел.
В качестве примера приводим условия
отбора проб нек-рых материалов. Лако-
красочные материалы. При
упаковке лакокрасочного материала в бочки и барабаны пробу
отбирают из 10% бочек и барабанов, при упаковке в бутыли,
фляги—из 5% бутылей или фляг, при упаковке в банки (более
1 кг)—из 3% банок. В случае упаковки материала в цистерны
или в специальные контейнеры пробу отбирают от каждого
места: одну на расстоянии 200 мм от дна цистерны, вторую —
Рис. 4.
из середины цистерны или контейнера, третью — на расстоя-
нии 200 мм. от верхнего уровня материала От жидких и пасто-
образных продуктов пробы отбирают стеклянной трубкой,
пробоотборником или деревянным веслом. Перед взятием про-
бы жидкие продукты тщательно перемешивают (кроме масля-
ных лаков); если на поверхности материала есть пленка, ее
аккуратно удаляют перед взятием пробы.
Консистентные смазки. Пробу отбирают от
2% или от 5% общего количества мест (бочки, бидоны и т. п.).
Для взятия пробы снимают с бочек или барабанов днища, с
ящиков крышки, после чего снимают слой смазки на глубину
5 мм с поверхности диаметром до 200 мм. Пробу отбирают вин-
тообразным или поршневым щупом. Щуп вводят в смазку до
дна тары, вынимают и снимают с него смазку. На длине 5 лм|
от конца щупа смазку в пробу не включают. Частичные пробы
складывают в чистый сухой сосуд и перемешивают не рас-
плавляя.
V. Опробование твердых материалов. Опробуемый
твердый материал может находиться в виде крупных
кусков, порошкообразной сыпучей массы, волокни-
стой массы, слитков или чушек. В первых двух слу-
чаях отбор пробы производят одинаково с учетом се-
грегации в случае крупнокусковых материалов; это'
достигается отбором проб по специальным правилам.
В зависимости от хрупкости материала, находящегося,
в слитках или чушках (металл, сплав, сера и т. п.)г
пробу отбирают по-разному. Слиток, еслй это воз-
можно, дробят и далее отбирают среднюю пробу, как
это делают для крупнокусковых материалов. Из слит-
ков, не поддающихся дроблению (чугунные, стальные,
медные и др., вязкие нефтепродукты и т. п.), пробу
отбирают сверлением или распиливанием, выполняе-
мым по специальным правилам. От неподвижных сы-
пучих материалов, находящихся в мелкой таре (моло-
тая сера, пигменты, бура и борная к-та, синтетич.
красители и т. п.), пробы берут щупом сверху, из
середины и со дна тары от небольшого числа мест
(2—10%) опробуемой партии. В нек-рых случаях
пробу берут щупом, проходящим через всю толщу
массы или до половины толщины. Пробу сыпучих
материалов, находящихся в вагонах, напр. соды и
т. п., отбирают обычно по схеме
«двойного конверта»
(рис. 5). Кружками на рис. от-
мечены точки отбора частичных
проб. Для неподвижных сыпу-
чих материалов, находящихся в
кучах, напр. песка, наиболее ра-
спространен метод «в ы ч е р п ы-
в а н и я», заключающийся в отборе лопатой небольших
порций материала из разных точек его поверхности.
Вес порции должен быть пропорционален весу опро-
буемой партии. Применяется также отбор проб
щупом, бурением, прокладкой шурфа и др. Для опро-
бования движущихся масс пользуются описанным
выше способом продольных или поперечных сечений-
Отбор проб крупнокускового материала производят
так же, как и однородного. При отборе проб крупно-
кускового материала необходимо следить за тем, чтобы
в пробе отношение кусков разной величины было таким
же, как и во всей массе материала. Обработка пробы
твердых материалов состоит из дробления, перемеши-
вания и сокращения. Дробление материала
производят в несколько приемов. Для грубого дроб-
ления применяют или дробление вручную или, при
больших размерах генеральной пробы, измельчение
в щековых дробилках. В щековых дробилках полу-
чающиеся куски в 4—5 раз меньше поступающих
(в нек-рых случаях в 8—9 раз больше). Далее следует
измельчение до частиц размером 1—2 ял на дробиль-
ных вальцах или аппаратах типа кофейной мельницы.
Тонкое дробление осуществляют в дисковом истира-
теле, молотковой дробилке или шаровой мельнице.
Измельченные частицы имеют размеры долей милли-
метра. Дальнейшее измельчение производят в меха-
нич. агатовой ступке. Перемешивание имеет
целью устранить неравномерность состава пробы в раз-
Рис. 5.
753
ОПРОБОВАНИЕ МАТЕРИАЛОВ
754
ных ее точках. В зависимости от крупности кусков
и веса пробы применяют: 1) способ «кольца и
конуса», заключающийся в том, что исходную
пробу рассыпают попеременно в конусообразную
кучу и окружающее ее кольцо
(рис. 6). Затем конусообразную
кучу рассыпают на кольцо и по-
лученное кольцо большого объ-
ема снова ссыпают в конус. В
вершипу последнего вставляют
доску и «развертывают» конус.
Снова раскидывают материал
в кольцо и повторяют все сна-
чала. Во избежание сегрегации
перелопачивание производят по
специальным правилам. Этот
способ применим для пробы ве-
сом 200—2500 кг и крупности
кусков не выше 50— 60 мм. 2)
«П ерекатывание», при
к-ром пробу высыпают на бре-
зент или клеенку и попеременно
поднимают и сближают углы брезента или клеенки, рас-
положенные по диагонали. Операцию повторяют много-
кратно, меняя поднимаемые углы. Способ применим для
проб весом не более 20—25 кг, в пробе не должно быть
крупных кусков. 3) Просеивание, при к-ром
пробу пропускают через сито, размер отверстий к-рого
в 2—3 раза больше размера самых крупных кусков.
Комки порошкообразного материала на сите расти-
рают резиновой пробкой. Обычно просеивание пов-
торяют 2—3 раза. Способ применяют для пробы
весом в несколько килограммов. 4) Механич.
перемешивание в спец, аппаратах, напр.
во вращающихся четырехугольных ящиках, через
к-рые проходит по диагонали вал, в шаровых мельни-
цах и др. Способ применим для проб весом 1—2 кг.
Сокращение в зависимости от крупности и веса мате-
риала производят разными способами: 1) Фрак-
ционный отбор, заключается в том, что при
погрузке, разгрузке или перемещении материала
через определенное число лопат отбирается одна
лопата. Отобранную таким образом пробу, если нуж-
но, сокращают далее. 2) Прокладка канав
через материал, к-рому придан вид прямоугольного
параллелепипеда. В сокращенную пробу отбирают
материал, вынутый из канав. 3) Квартование.
Описанный выше конус материала делят на 4 части
проходящими через центр перпендикулярными ли-
ниями. Отбирают две противоположные четверти,
укладывают их в конус и снова повторяют всю опера-
цию до тех нор, пока проба не достиг-
нет нужного веса. 4) Деление в
делителях разных конструкций. Для
сокращения пробы больших размеров
применяют механические делители с
приводом от электромотора. Все дели-
тели или сократителн устроены так, что
часть пробы отводится в приемники.
Для этого в них устраивают специаль-
ные направляющие или различно рас-
положенные приемные камеры для ча-
сти пробы и основного потока ее. В ла-
бораториях широко применяют желоб-
чатый рифленый делитель, состоящий
из ряда желобков шириной не менее
трех диаметров наибольших зерен в
пробе. Половина общего числа желоб-
ков разгружает материал в одну сто-
Рис. 7. рону, а другая половина в другую сто-
рону (рис. 7). Сокращение пробы в ри-
фленом делителе может сопровождаться потерями
вследствие пылеобразования. Эти потери исключаются
Рис. 9.
при пользовании совратителями соответствующей кон-
струкции. Простейший из них (рис. 8) состоит из ци-
линдра со съемным дном, в bi ж-
нюю часть к-рого входит сектор- тХ
ный приемник. Приемник пред- - Ха.
ставляет собой цилиндр, разде-
ленный на секторы одинакового
размера. Секторы с дном чере-
дуются с секторами без дна.
Все секторы соединены между со-
бой стержнем, заканчивающим- II таИк "Д
ся вверху конусом. Над конусом
вставляют воронку с отверстием
у вершины и с крышкой. Пробу хиВаа
засыпают в воронку, закрывают ^8»
крышкой и приподнимают ворон- Рис 8
ку. В образовавшийся зазор про-
сыпается сокращаемая проба. При этом проба сокра-
щается в 2 раза. В случае необходимости сокращение
повторяют.
В аппаратах для опробования, применяемых в ла-
боратории, нередко совмещают операции усреднения
(перемешивания) пробы и ее сокращения. Ниже опи-
саны 2 аппарата такого типа. Один из них (рис. 9)
представляет собой вра-
щающийся барабан 1, по-
ловина к-рого работает
как смесил ель, половина
2 — в качестве делителя.
Смеситель состоит из не-
скольких рядов переме-
шивающих элементов,
расположенных в шах-
матном порядке. Элемент
представляет собой пря-
мую ромбическую приз-
му, поставленную на ос-
нование или же смежные
боковые грани такой
призмы. В первом случае
барабан может вращать-
ся в обе стороны, во втором—только в направлении уг-
ла, образованного гранями элемента. Делитель разде-
лен перегородками на ряд отсеков, количество к-рых,
как и количество элементов в каждом ряду смесителя,
равно или кратно отношению общего количества мате-
риала к количеству его, составляющему среднюю
пробу. Например, если проба составляет ’/10 часть
материала, то аппарат имеет 10 отсеков. Барабан
открывается со стороны центральной стенки дели-
теля. Имеются съемная крышка 3, закрывающая все
отверстия целиком, и малые съемные крышки 4,
каждая из к-рых закрывает только один отсек дели-
теля. Барабан закрепляют отверстием вверх, загру-
жают его опробуемым материалом, закрывают крыш-
кой 3 и заставляют вращаться нужное число раз.
Поток частиц при прохождении через смеситель на
ребро очередного элемента делится примерно ионо-
лам, в результате происходит перемешивание мате-
риала и равномерное его распределение по длине
барабана. Затем барабан снова закрепляют отвер-
стием вверх, снимают большую крышку 3 и закры-
вают малыми крышками 4 такое число отсеков, чтобы
попадающее в них количество материала было бы рав-
но средней пробе. После этого барабан поворачивают
на полоборота, основная часть материала просыпается
из барабана в бункера, в барабане остается средняя
проба. Можно рассчитать число оборотов барабана,
необходимых для хорошего перемешивания.
Для одновременного усреднения и сокращения
пробы сыпучих материалов применяют механич. де-
литель. В универсальном делителе для материалов
с частицами диаметром до 10 мм или длиной до 15 мм
755
ОПРОБОВАНИЕ МАТЕРИАЛОВ
756
(рис. 10) опробуемый материал поступает из бункера 1,
Снабженного клапаном 2, н корпус аппарата 3, в к-ром
Закреплен конус
Рис. 10.
4. Острие конуса находится точно
против отверстия воронки. В кор-
пусе расположена также съемная
гребенка 5. На валу червячного ре-
дуктора 7 расположена тарелка, на
к-рую устанавливают обойму 6, за-
к репленную 4 или 8 объемными сек-
торами, служащими тарой. Обойму
вращают от электромотора 8 через
редуктор. ~
П одсушенный образец
опробуемого материа-
ла засыпают в ворон-
ку, откуда оп посту-
пает, на острие усред-
нительного конуса и
растекается со всех его
сторон во вращающие-
ся секторы сборника.
Усреднение происхо-
дит вследствие равно-
мерного распределе-
ния различных по раз-
меру частиц по поверх-
ности конуса во время
вращения сборника. Усредняемая проба делится при
этом на 4 или на 8 частей. Повторением этой опера-
ции достигается дальнейшее сокращение пробы.
Ниже приводятся примеры отбора проб различных твердых
материалов. Описания отбора проб расположены по степени
грануляции. Сода, хлорная известь и т. п.— по-
рошкообразные материалы. Из материалов, находящихся в
вагонах, бочках, мешках и т. п., частичную пробу отбирают
щупом сверху, из середины и со дна бочки или мешка, снимая
каждый раз со щупа нижний слой высотой 10—15 см. Отоб-
ранные пробы соединяют вместе, тщательно перемешивают и
квартованием доводят вес пробы до 0,5 кг. Порошкооб-
разные нефтепродукты (сажа, нефтяной кокс).
Опробуют 2% от общего числа мешков и 1% от общего числа
пакетов, по не менее 2 мешков или пакетов. Продукты, храня-
щиеся в мешках и пакетах, опробуют винтовым или поршневым
щупом длиной около 400 мм при отборе из банок, бидонов и
мешков, и около 800 мм—при отборе из бочек и барабанов. При
опробовании щуп погружают во всю толщу опробуемого мате-
риала. Частичные пробы из отдельных мест, взятые в равных
количествах, собирают в один сухой чистый сосуд, перемеши-
вают и после этого отбирают лабораторную пробу. Молотая
сера. В мешок, содержащий опробуемый продукт, погружают
щуп на 1 высоты мешка. Вес частичной пробы должен быть
не менее 50 г. Пробы берут не менее чем от 5% мешков всей
партии. Все частичные пробы смешивают и сокращают до 1 кг.
Сокращенную пробу делят на 2 равные части, помещают в 2
чистые сухие банки, закрывают корковой пробкой и заливают
ее сургучом или парафином. Песок. Среднюю (генераль-
ную) пробу составляют из отдельных частичных проб, отоб-
ранных с различной глубины от каждого вагона, баржи, плат-
формы, автомашины в точках, равномерно отстоящих друг от
друга, но не ближе 333 мм от края и не ближе 200 мм от по-
верхности материала. Число частичных проб от каждого ваго-
на и т. п. должно быть не менее 8. При наличии комьев они
должны быть включены в состав средней пробы в количестве,
примерно пропорциональном содержанию их в партии. Вес
частичной пробы должен быть не менее 1 кг. Для отбора проб
применяют лопату, совок или пробоотборник, представляющий
собой тонкостенную цилиндрическую трубу с закрытым дном,
с продольной щелью, через к-рую отбираемый материал по-
падает в пробоотборник. Среднюю пробу сокращают квартова-
нием до 6 кг и сохраняют в таре, предохраняющей ее от загряз-
нения, высыхания и увлажнения. Пигменты. Пробу от-
бирают от 10% общего количества мест в партии, но не менее
трех при малых партиях. Минимальный вес средней пробы от
0,5 до 1,2 кг. Частичную пробу отбирают щупом, проходящим
до 3/< глубины тары; количество отбираемых порций пропорцио-
нально объему тары, в днище бочки или в середине крышки
ящика просверливают отверстие, через к-рое проходит щуп.
Минералы и горные породы. Среднюю пробу
обычно отбирают в 2 стадии: 1) взятие генеральной пробы на
месте; минимальный вес ее может колебаться от 25 до 100 кг;
2) взятие лабораторной пробы весом от 25 до 50 г. Для этого
пробу, поступившую в лабораторию, измельчают и сокращают.
Отдельные куски породы дробят на специальной стальной
плите отвесно направленными ударами хорошо закаленного
стального молотка до кусочков диаметром 0,5—0,75 см. Затем
пробу измельчают до порошка раздавливанием в стальной
ступке небольшими порциями. Измельченную пробу помеща-
ют на лист глянцевой бумаги, тщательно перемешивают и рас-
пределяют ровным тонким слоем при помощи шпателя в виде
прямоугольника. Прямоугольник делят на 20—26 квадрати-
ков, из к-рых отбирают шпателем около 25 г пробы и просеива-
ют ее через сито 100 меш. Пробу сохраняют в бюксе или склян-
ке с пробкой. Едкий натр технический. Пробу
твердого едкого натра отбирают от 2% всех барабанов партии;
при малых партиях не менее чем из трех барабанов. Барабан
вскрывают по продольному шву и быстро отбирают одинаковые
количества продукта из трех мест: вблизи дна, из середины и из
верхней части барабана. Частичные пробы быстро перемешива-
ют, полученную генеральную среднюю пробу общим весом не
менее 1 кг помещают в 2 чистые сухие банки и плотно закупори-
вают. Для анализа передают одну банку, другая хранится как
арбитражная проба. Аналогично отбирают пробы технич. хло-
ристого кальция и хлористого магния, плавленного технич.
сернистого натрия и других солей и щелочей.
Стеклянная вата. Стеклянную вату упаковывают
в трех-, четырехслойные бумажные непропитанные мешки, в
мешки из паковочной хлопчатобумажной ткани или в рогожные
мешки; вес одного мешка не должен превышать 60 кг. От каж-
дой партии отбирают 5% упакованных мешков, но не менее
пяти. Из каждого мешка отбирают по 1 кг ваты. Полученные
пробы перемешивают вручную и из общего количества отвеши-
вают 4 кг.
Особо остановимся на анализе руды, металлов и сплавов.
Руда. Если руда или другой подобный материал прибы-
вает к месту потребления водным путем, то при выгрузке из
каждого десятого, двадцатого или пятидесятого сосуда, в за-
висимости от величины всей партии, однородности крупных
кусков и мелочи берут ручной лопатой (длиной —30 сии ши-
риной — 15 см) в различных местах по небольшому количеству
руды с таким расчетом, чтобы набрать полную лопатку. Отоб-
ранную пробу (генеральную) ссыпают на специально подго-
товленную площадку. Затем пробу сокращают (квартуют)
для уменьшения ее количества.
Если руда доставляется в железнодорожных вагонах, то
пробу берут из каждого вагона не менее чем в пяти его точках—
по углам и в середине (обязательно со дна), после чего отобран-
ное количество материала тоже сокращают.
Чугун и сталь (проба для спектрального анализа).
При анализе чугунов отбор пробы должен обеспечить сплошной
отбел и однородную мелкозернистую структуру, для чего мож-
но применять: а) кокиль для пальчиков диаметром 5,5—6,0 мм;
б) кокиль для получения отливки в виде кольца толщиной
не более 10 мм и диаметром 70—80 мм; в) кокиль для получе-
ния отливки в виде усеченной призмы, где нижняя площадка
равна 100x10 мм и верхняя 100x15 мм; высотой до 10 мм и
г) чугунную сковородку для отливки лепешки толщиной не
более 10 мм.
Чугун. При отборе проб чугуна используемая для за-
ливки изложница должна быть холодной, чтобы обеспечить
необходимую закалку всего образца. Изложница должна быть
чистой без следов загрязнений песком, глиной и др. Рабочий,
отбирающий пробу, хорошо прогревает металлическую ложку
в чугуне, прогретой ложкой зачерпывает чугун и наполняет
изложницу доверху. Пробы литейного чугуна отливают в
стальную изложницу с отверстием для пробы диаметром 30 и
высотой 120 мм. При разливке чугуна на разливочных машинах
от каждого ковша отбирается по 3 пробы (в начале, середине и
конце разливки ковша). Пробы отливаются в песок, в котором
сформированы прямоугольные углубления, соответствующие
размеру пробы 40x50x100 мм. Миксерный чугун отбирается
в форме кольца, если анализ выполняется спектрографически,
и прямоугольной формы, если анализ выполняется химиче-
ским путем. Пробу жидкого чугуна (из доменной печи) берут у
разливочной машины там, где нет этой машины, проба берется
специальной ложкой в середине каждой трети данного выпуска.
Каждая из трех порций жидкого чугуна наливается в специ-
альную чугунную изложницу. Пробу жидкого чугуна из мик-
сера берут во время выпуска его в ковш, когда последний на-
полнен почти наполовину. Пробу жидкого чугуна из вагранки
берут в середине выпуска при помощи ложки из-под струи ме-
талла, поступающего в спускной желоб, обычно берется одна
проба.
Сталь. Среднюю пробу стали по ходу плавки берут
ошлакованной ложкой. Отобранный металл выливают в проб-
ницу, в которую предварительно насыпается алюминиевая
стружка, взятая сверлением из чушкового алюминия. При
сливе металла из ложки в пробницу не должен попадать шлак.
В зависимости от химического состава металла и его свойств
пробу по ходу плавки отбирают или в виде небольших слитков
весом от 0,5 до 1 кг или в виде «скрапипы», т. е. тонкого слоя
металла, налитого па железную лопату. Пробу стали в мар-
теновском цехе берут непосредственно на рабочей площадке
из ванны (для экспресс-анализов) или на литейгой канаве из
ковша (для маркировочных анализов). Перед отбором пробы
металлическую ванну тщательно перемешивают (в мартенов-
ской печи — через два — три окна), т. к. распределение эле-
ментов в жидком металле неоднородно.
Верхние слои ванны богаче углеродом и серой, а нижние —
марганцем. Поэтому необходимо брать пробу металла ближе
к середине глубины ванны. Проба стали для химического ана-
лиза поступает в лабораторию в виде стружек или скрапины,
как только металл несколько затвердеет, лопату опускают в
сосуд с холодной водой. Полученную пластинку (скрапину) от-
деляют от лопаты и высушивают (у печи). Выбирают тонкую(пе
толще 0,5 мм) и лишенную шлаковых загрязнений часть
757
ОПТИЧЕСКИЕ СВОЙСТВА КОЛЛОИДНЫХ СИСТЕМ
758
скрапины, разбивают и затем измельчают ее в ступке ударного
действия, затем просеивают через железную или медную сетку
с отверстиями 1 мм2. Для анализа берут фракцию, которая ос-
тается на сетке. Пробу листовой стали отрезают от нескольких
листов по всей их длине полоски шириной —5 см-, каждую по-
лоску складывают в несколько слоев и строгают по торцовой
поверхности. Полученную от всех полосок стружку переме-
шивают в общем сосуде.
Ферросплавы. Опробование ферросплавов обычно
производят при погрузке или выгрузке из вагона, особенно в
тех случаях, когда данный ферросплав транспортируют без
тары, навалом. Количество ферросплава, отбираемое для пер-
вичной средней пробы, не должно превышать 1 % от общего веса
партии. Для средней пробы берут от каждого сотого кус’»*а
1 ка сплава (по всему поперечному сечению). От мелочи доста-
точно отбирать каждую сотую лопату. При транспортировке
ферросплавов в та| е пробу берут с таким расчетом, чтобы вес
первичной пробы не превышал 2—3% от общего веса партии (в
зависимости от однородности и ценности данного ферроспла-
ва). Практически достаточно взять 1—2 куска от каждой плав-
ки илп по одному куску из каждой тары. Отобранную сред-
нюю пробу измельчают в дробилках со щеками из особо твердой
стали или кувалдами на плите из марганцовистой стали.
Баббит. Отбирают в различных местах штабеля, со-
блюдая симметрию относительно контура последнего, 1—3%
баббитовых чушек и разламывают их, чтобы убедиться в одно-
родности излома. Затем сверлом или ручной пилой берут
стружку от каждой чушки, сверля или распиливая чушку на-
сквозь в поперечном направлении в нескольких местах. Полу-
ченную стружку сплавляют в железном (или шамотовом) тигле
конической или цилиндрической формы под слоем поваренной
соли, королек очищают от поверхностных загрязнений, за-
тем сверлят его или распиливают во многих местах. Полу-
ченная стружка является средней пробой.
Лит.: Дымов А. М., Технический анализ руд и метал-
лов, 5 изд., М., 1949; Яковлев П. Я., Яковлева
Е.Ф., Технический анализ в металлургии, М., 19 6 3; Бабаев
М. В., Опробование ферросплавов, М., 1954; Б а р ы ш е в Н.
В., Заводск. лаборатория, 1947, 13, № 5; Соколов В. А.,
Анализ газоз, М.—Л., 1950; ГОСТ 5424—50. Железные руды
и агломерат; ГОСТ 3594—62. Глины формовочные; ГОСТ
2189—62 Пески и смеси формовочные; Пономарев А. И.,
Методы химического анализа минералов и горных пород, т. 1,
М., 1951; Хан Г. А., Анфимова Е. А., Опробование
сырья и продуктов промышленности, М.—Л., 1953; П о ж а-
р и цк ий К. Л., Опробование месторождений цветных,
редких металлов и золота, М., 1947; ГОСТ 2517—52. Неф-
тепродукты. Отбор проб; Касимов Б. С., Заводск. лабор.,
1963, 29, № 4, 500; Алексеев Р. И., там же, 1961, 27,
№ 4, 474; Р у д а н о в с к и й С. Н., там же, 1963, 29, № 5, 629.
1£. Я. Яковлев, А. И. Оржеховская, Д. Н. Васкевич.
ОПТИЧЕСКИЕ СВОЙСТВА КОЛЛОИДНЫХ СИ-
СТЕМ — эффекты, связанные с рассеянием и погло-
щением света коллоидными системами. Формально
оба эффекта можно описать одинаковым образом,
характеризуя ослабление светового пучка прп про-
хождении нек-рого слоя толщины I, заполненного
коллоидными частицами. Интенсивность I пучка ио
выходе из этого слоя равна 1 = 10е~'1, где /0— ис-
ходная интенсивность, а у — коэфф, экстинкции,
связанный ст. наз. оптич. плотностью!) соотношением
у = 2,303Р (т. е. D = Z-1lg/0//). Величина D или у,
вообще говоря, спектрально зависимая, легко может
быть определена методом пропускания света с по-
мощью любого стандартного спектрофотометра.
На самом деле поглощение и рассеяние света —
различные явления, хотя во многих «классических»
коллоидных системах (золи золота, графита и т. и.)
они наблюдаются одновременно. Поглощение света
обусловлено затратой части электромагнитной энергии
падающего пучка на возбуждение электрич. тока в про-
водящих коллоидных частицах. Преобразуясь в джо-
улево тепло, эта энергия не «возвращается» в виде
света. Коэфф, экстинкции в этом случае именуется
коэфф, истинного поглощения. (По указанной при-
чине проводники и полупроводники всегда непро-
зрачны, если не говорить об очень тонких слоях).
При рассеянии (от диэлектрич. частиц) свет не
«исчезает», но меняет свое направление. Если кол-
лоидные частицы достаточно велики, они становятся
видимы в направлении, перпендикулярном проходя-
щему пучку. На этом, основана улътрамикроскопия.
В более общем случае опалесценция коллоидных
р-ров и аэрозолей, их «мутность», обусловлена рассея-
нием. Даже в случае очень мелкодисперсных золей
и истинных полимерных р-ров проходящий пучок
становится видимым сбоку (эффект Тиндаля).
На этом явлении основан ряд методов определения
размеров и формы коллоидных частиц и макромолекул.
Коэфф, у в этом случае именуется коэфф, экстинкции
за счет рассеяния, или мутностью, п обозначается т.
Ослабление проходящего пучка теперь обусловлено
не поглощением, а тем, что часть электромагнитной
энергии распространяется в направлениях, отличных
от исходного. Количественно рассеяние света описы-
вается на оспове двух теорий. Первая, связанная
с именами Рэлея и Ми, оперирует представлением
о возбуждении на коллоидных частицах или их участ-
ках вторичных источников излучения. Эта теория
позволяет почерпнуть информацию о размере и форме
частиц из анализа углового распределения интенсив-
ности рассеянного света. Вторая, связанная с именами
Эйнштейна, Смолуховского и Дебая, оперирует пред-
ставлением о флуктуациях концентрации. Коллоид-
ная система при этом становится оптически неодно-
родной, по аналогии с оптич. стеклом, содержащим
включения с др. показателем преломления. Эта теория
связывает абс. интенсивность рассеяния с мол. весом
и силами взаимодействий между частицами или
макромолекулами.
Мутность входит в выражение для абс. интенсив-
ности рассеяния под углом 90° к падающему пучку
(т. наз. константы Рэлея) и может быть измерена не
только по пропусканию, но и нефелометрически (см.
Нефелометрия и турбидиметрия}. Можно показать,
что для частиц, значительно меньших длины световой
волны в рассеивающей среде
128л1 2
т=-----ПХ2
Кг
где п — число частиц в I с.л3. а и — их поляризуе-
мость. Этот закон Рэлея объясняет, почему рассеянный
свет кажется голубоватым, а проходящий краснова-
тым: благодаря повышенной интенсивности рассеяния
в коротковолновой области проходящий свет как бы
обогащается длинноволновыми компонентами. В самом
общем виде теория рассеяния света изотропными
сферич. частицами была развита Ми. Согласно этой
теории, х = К*лггп, где К* — так наз. коэфф, рас-
сеяния, а г—радиус частицы. Критич. параметрами
теории являются относительный показатель пре-
ломления т и величина а=2лг/Л. Рассмотрим три
случая:
1) 2г< 0,(истинные р-ры полимеров; мицеллы в
водных р-рах мылообразующих веществ). Здесь рас-
пределение интенсивно-
сти рассеяния симметрии- Zj' i°' в'
но относительно 90°; из-
мерения х позволяют оп-
К
в кото-
Схематическое изображение
рассеяния света от большой
частицы. Стрелкой указано
направление падающего пуч-
ка. О—центр тяжести ча-
стицы. Отрезки АО и ОВ рав-
ны. Точки А' и В',
рых регистрируется интен-
сивность рассеяния от уча-
стков частицы А и В, расположены симметрично относитель-
но перпендикуляра к АВ, 00'. Интерференция в точке А'
меньше, чем в В', а следовательно, интенсивность больше.
Полная интенсивность в любой паре точек определится де-
тальной формой частицы; на этом основано определение фор-
мы из индикатрисы рассеяния.
ределить мол. в. полимеров или вес и степень агрегации
мицелл, а также критич. концентрацию мицеллообра-
зования. 2) 2г~-/.т. Теперь, вследствие интерференции
волн, рассеянных участками одной и той же частицы,
свет рассеивается вперед сильнее, чем назад (см. рис.).
759
ОПТИЧЕСКИЕ СВОЙСТВА КОЛЛОИДНЫХ СИСТЕМ — ОРГАНИЧЕСКАЯ ХИМИЯ
760
По угловому распределению интенсивности можно
определить не только мол. в., но и размер (радиус
инерции) и форму частицы, если она отлична
от сферич. Этот метод приложим для умеренно дисперс-
ных коллоидов и р-ров полимеров с мол. в. выше 10е.
3) Этот диапазон соответствует латексам,
крупнодисиерсным аэрозолям, туманам. Важным
отличием этого случая от предыдущих является то,
что теперь коэфф, рассеяния К* становится пе-
риодич. функцией а, а интенсивность — периодич.
функцией угла рассеяния. В этом диапазоне а
можно получить однотипную информацию, измеряя
угловое распределение интенсивности рассеяния или
волновую зависимость пропускания. Для расчетов
размеров здесь приходится пользоваться протабули-
рованными функциями К* при различных т или
номограммами. Периодичность К* делает возмож-
ными методы определения размеров, основанные на
анализе гало, счете дифракционных колец, регистра-
ции тиндалевских спектров высших порядков и т. д.
Разумеется, в случае полидисперсных коллоидов все
эти методы дают средние размеры частиц. 3-му
диапазону а соответствует также рассеяние рентге-
новских лучей под малыми углами от коллоидных
р-ров и пористых систем.
Теория Ми учитывает и собственное поглощение (это
достигается автоматически введением комплексного
показателя преломления). Спектральной зависимостью
собственного поглощения объясняется цвет золей
металлов. Так, высокодисперсные золи золота погло-
щают в основном в желто-зеленой части спектра; этим
объясняется их ярко-красный свет (который «остается»
и рассеивается). Однако положение максимума погло-
щения в таких системах зависит от формы частиц:
напр., золь золота с вытянутыми частицами уже не
красный, а голубой. Теория Ми распространена на
случай несферич. частиц Гансом. Поглощением света
пользуются в физич. химии и биохимии при анализе
подвижных границ, напр. при седиментации в ультра-
центрифуге или электрофорезе.
Дополнительные сведения о размерах и форме ча-
стиц могут быть получены при измерениях депо-
ляризации рассеянного света. Рассеянный под
углом 90° от системы малых оптич. изотропных частиц
свет всегда поляризован вертикально. Появление го-
ризонтально поляризованной компоненты легко может
быть зарегистрировано; отношение интенсивностей
горизонтально и вертикально поляризованной компо-
ненты наз. фактором деполяризации и обозначается
Qu, Qu или Qa (индексы соответствуют естественному,
вертикально и горизонтально поляризованному па-
дающему свету). Факторы деполяризации связаны
между собой соотношением взаимности Кришнана,
т. е. лишь два из трех являются взаимонезависимымп:
Можно показать, что фактор Qv чувствителен к собст-
венной анизотропии частиц, тогда как фактор оа бо-
лее чувствителен к размерам. Метод деполяризации
весьма удобен при наблюдении созревания и старе-
ния гелей, так как эти процессы сопровождаются
изменениями размеров и формы образующих гели
частиц.
Следует еще упомянуть о специальном случае рас-
сеяния света — критич. опалесценции в р-рах полиме-
ров вблизи темп-ры осаждения. Этот эффект связан
с гетерофазными флуктуациями —
возникновением неустойчивых зародышей истинной
коллоидной фазы. Метод критич. опалесценции может
быть использован для изучения сил, действующих
между молекулами в р-рах полимеров.
Ориентация анизометрич. коллоидных частиц под
влиянием электрич. (эффект Керра), магнитного или
гидродинамич. (эффект Максвелла) поля приводит
к возникновению двойного лучепрелом-
ления. Оно может быть измерено, и по его вели-
чине и углу ориентации (в случае гидродинамич. поля)
можно наиболее прямым образом получить сведения
о форме (размерах и степени асимметрии) и жесткости
частиц (в потоке, наряду с ориентацией, возможна
и деформация). Измерения двойного лучепреломления
позволяют также наблюдать за созреванием гелей,
для наблюдения кинетики этого процесса достаточно
регистрировать нарастание интенсивности проходя-
щего света при скрещенных поляроидах.
Лит.: Шифрин К. С., Рассеяние света в мутной среде,
М.—Л., 1951; Наука о коллоидах, под ред. Г. Кройта, пер. с
англ., т. 1, М., 1955, гл. 3; Шелудко А., Коллоидная
химия, пер. с болг М., 1960, гл. 2; S t а с е у К. А., Light-
scattering in physical chemistry, L., 1956; Цветков В. H.,
Эскин В. E.,Френкель С. Я., Структура макромоле-
кул в растворе, Л., 1964, гл. 3, 4, 7, 8; Н u 1 s t Н. С.
van de, Light scattering by small particles, N. Y.— L.,
1957. С. Я. Френкель.
ОРГАНИЧЕСКАЯ ХИМИЯ — естественнонаучная
дисциплина, предметом изучения к-рой являются
соединения углерода с другими элементами, называе-
мые органич. соединениями, а также законы, к-рым
подчиняются превращения этих веществ.
В последние десятилетия многие разделы О. х.
развивались столь интенсивно, что выросли в боль-
шие специализированные разделы, называемые по
научному или прикладному признаку. Так, часто
говорят как о самостоятельных видах химич. науки
о стереохимии, химии элементоорганич. соединений,
химии высокомолекулярных веществ, полимеров,
природных веществ, антибиотиков, витаминов, гор-
монов, химии фторорганпч. соединений, красителей
и др. Однако естественно, что все эти разделы осно-
вываются на общих законах О. х.
В последние годы О. х. тесно переплелась со смеж-
ными областями знания естественных наук — био-
химией, медициной, биологией и др. Выдающееся зна-
чение приобрели методы О. х. в современной техно-
логии произ-ва каучука, пластмасс, синтетич. волокна,
красителей, медикаментов, пром-сти кино-фотомате-
риалов, стимуляторов роста, средств борьбы с вреди-
телями сельского хозяйства и мн. др. Благодаря
успехам О. х. в установлении зависимостей между
строением и свойствами органич. соединений стано-
вится возможным создание новых материалов различ-
ных назначений с заранее заданными свойствами.
О. х. оказалась в состоянии удовлетворить все воз-
растающие потребности науки и техники и вследствие
этого тесно с ними связана. Успехи О. х. в области
тяжелого органич. синтеза не только изменили техно-
логию ряда произ-в, но и повели к созданию новых
видов продукции. За последние 2 десятилетия созданы
новые виды произ-в на основе химич. переработки
природных газов и нек-рых узких фракций нефти.
Особое место О. х. заняла в биологии, медицине
и с. х-ве. Именно с помощью методов О. х. удалось
установить структуру нуклеиновых к-т и важнейших
белков, создать огромную новую пром-сть антибиоти-
ков, новые ростовые вещества и мощные средства
борьбы с вредителями сельского хозяйства. О. х.
достигла такого состояния, к-рое позволяет говорить
о ее ведущей роли в создании материальной культуры
современного общества.
О. х. сложилась и оформилась в самостоятельную
химич. науку во второй половине 19 в. Причинами вы-
деления О. х. в самостоятельную область науки яви-
лись особенности углерода и его соединений. Углерод
образует соединения с большинством элементов и об-
ладает наиболее выраженной способностью по срав-
нению с другими элементами к образованию молекул
761
ОРГАНИЧЕСКАЯ ХИМИЯ
762
цепного и кольчатого строения, скелет к-рых обра-
зуется как непосредственным соединением практи-
чески неограниченного числа атомов углерода друг
с другом, так и включением в цепь или кольцо атомов
других элементов. Для соединений углерода также
наиболее характерно явление изомерии, заключаю-
щееся в том, что при одном и том же составе и мол.
весе возможно существование ряда веществ, отличаю-
щихся друг от друга химич. и физич. свойствами,
это различие является следствием различной последо-
вательности сцепления атомов или расположения их
в пространстве в молекулах этих соединений. В ре-
зультате этих особенностей число органич. веществ
чрезвычайно велико и к настоящему времени состав-
ляет более 1 млн., в то время как соединений всех
остальных элементов насчитывается немногим более
50 тыс.
Органич. соединения способны к сложным и много-
образным превращениям, существенно отличным от
превращений веществ неорганич. Соединения угле-
рода играют особо важную роль в построении и жизне-
деятельности растительных и животных организмов.
К органич. соединениям относятся, напр., белки,
с к-рыми связан обмен веществ, нуклеиновые кислоты,
являющиеся материальными носителями наследствен-
ных признаков и обусловливающие тот или иной
вид организма, витамины, гормоны, регулирующие
его обмен, и др. закономерности. О. х. широко исполь-
зуется биохимией, биологией, медициной. Таким
образом, О. х. можно рассматривать как науку,
представляющую собой как бы своеобразный мост от
неживой природы к ее высшей форме — жизни.
Многие представления и закономерности химич. науки
такие, как, напр., валентность, изомерия и мн. др.,
впервые были открыты при изучении именно ор-
ганич. соединений. Перечисленные особенности угле-
рода и его соединений, своеобразие законов превраще-
ний органич. веществ и их огромное практич. и тео-
ретич. значение и явились причиной выделения О. х.
в самостоятельную область химич. науки.
Классификация органических соединений. Все органич.
соединения подразделяются на три основных ряда, или клас-
са: ациклических, изоциклических и гетероциклических соеди-
нений. К классу ациклических соединений (жирных соедине-
ний) относят углеводороды и их производные с открытыми це-
пями: гомологии, ряд метановых углеводородов наз. также ря-
дом насыщенных парафиновых углеводородов, или алканов,
общей ф-лы С.,гомологии, ряды углеводородов, изоло-
гичных углеводородам ряда метана: гомологии, ряд этиленовых
углеводородов С„Н2„ и ряды других углеводородов с открытыми
цепями. К классу изоциклических соединений (карбоциклич.
соединений) относят углеводороды и их производные, в моле-
кулах к-рых имеются циклы или кольца из атомов углерода:
углеводороды и их производные циклопарафинового или
полиметиленового ряда С„Н.,„, циклич. ненасыщенные соеди-
нения этого же ряда, а также ароматич. углеводороды ряда
С„Н2„_в и их Производные, содержащие бензольные ядра. К
этому же ряду относят и многоядерные ароматич. соединения,
напр. нафталин, антрацен и др. К классу гетероциклических
соединений относят органич. в-ва, молекулы к-рых содержат
циклы или кольца, в к-рые, кроме атомов углерода, входят
также и атомы других элементов (О, N, S, Р, As и др.).
От каждого углеводорода можно произвести отдельный ге-
нетич. ряд, представители к-рого формально производятся пу-
тем замены атома водорода в углеводороде той или иной функ-
циональной группой, определяющей химич. свойства соедине-
ния. Так, в генетич. ряд метана СН, входят: хлористый метил
СН,С1, метиловый спирт СН3ОН, метиламин CH,NH2, нитро-
метан CH,N03 и др. Аналогично представителями генетич. ря-
да бензола СвНв являются: хлорбензол СвН,С1, фенол С,,Н,ОН,
нитробензол СцН.Д’О, и др. Сумма одноименно замещенных
представителей различных генетич. рядов составляет гомоло-
гия. ряд производных: галогенных соединений, спиртов, ами-
нов, нитросоединений и др.
Истоки О. х. лежат в глубокой древности. Так,
спиртовое и уксуснокислое брожение было известно
в древности, финикияне знали крашение индиго и
ализарином. Однако еще в средние века (алхимии)
число известных индивидуальных органич. веществ бы-
ло очень ограничено. Начиная с 16 в., в период т. наз.
ятрохимии, исследования под влиянием идей Парацель-
са были направлены гл. обр. на использование различ-
ных веществ в медицине. В это время был выделен из
растений ряд эфирных масел, получен простой эфир
(Кадар), перегонкой дерева были получены древесный
спирт и уксусная к-та, из винного камня — винная
к-та, перегонкой свинцового сахара — уксусная и
перегонкой янтаря — янтарная к-та. Однако все
исследования этого периода сводились гл. обр. к опе-
рациям, при помощи к-рых, как тогда думали, можно
одни простые вещества превращать в другие. Огром-
ную роль в развитии О. х. сыграли аналитич. методы
А. Лавуазье, к-рый разработал основные принципы
количественных методов определения состава химич.
соединений. Эти методы (сожжение веществ в атмо-
сфере кислорода и весовой анализ продуктов юрения)
были последовательно улучшены Л. Тенаром, Я. Бер-
целиусом, затем Ю. Либихом, Ж. Дюма. Принципы
этих методов лежат и в основе современного элемен-
тарного анализа, в том числе и микроанализа,
разработанного Прэглем. В результате анализа огром-
ного числа различных веществ ранее доминирующее
представление о принципиальном различии веществ
растительного и животного происхождения постепенно
отпало. Впервые название «органические соединения»
встречается в конце 18 в. Термин «О. х.» впервые был
введен Берцелиусом в 1827, когда им было написано
первое руководство по органич. химии. Явление
изомерии впервые было открыто Ф. Вёлером и Ю. Ли-
бихом в 1822—23. Первый синтез органич. вещества
был осуществлен Вёлером, к-рый получил щавелевую
к-ту из дициана и в 1828 нагреванием циановокислого
аммония получил мочевину. Начиная с середины 19 в.
количество искусственно получаемых органич. ве-
ществ быстро увеличивается. Так, в 1845 Г. Кольбе
получил уксусную к-ту, в 1854 П. Бертло синтезиро-
вал вещества типа жиров, а в 1861 А. М. Бутлеров
синтезировал первое искусственное сахаристое ве-
щество, названное им метиленитаном и оказавшееся,
как впоследствии было установлено, акрозой. Син-
тетич. направление в О. х. приобретает все большее
значение. В результате успехов синтеза идеалистич.
учение о необходимости «жизненной силы» для созда-
ния органич. веществ было отвергнуто. Теоретич.
представления О. х. начали развиваться со второй
четверти 19 в., когда была создана радикалов теория
(Либих, Вёлер, Франкленд, Бунзен и др.). Основное
положение теории радикалов о переходе группы ато-
мов-радикалов из одного соединения в другое в не-
изменном виде остается в огромном числе случаев
справедливым и в настоящее время; и на этом пред-
ставлении основаны многие методы исследования ве-
ществ неизвестной структуры. Эта теория, в значитель-
ной степени основывающаяся на дуалистич. гипотезе
Берцелиуса, пала в результате работ Дюма (1834—39),
показавшего возможность замещения положительно
заряженных атомов в радикале на электроотрица-
тельные без серьезных изменений электрохимия,
характера радикала, что до работ Дюма считалось
невозможным.
На смену теории радикалов пришла типов теория,
созданная Ш. Жераром и О. Лораном. Последним
удалось расклассифицировать органич. вещества по
типам простейших неорганич. соединений. Так,
спирты по Жерару и Лорану считались соединениями
типа воды, амины — типом аммиака, галогенал-
килы — типом хлористого водорода. Позднее А. Ке-
куле установил четвертый тип — тип метана, от
к-рого он производил все углеводороды. Теория типов
позволила создать четкую классификацию органич.
соединений, основа к-рой лежит в современной клас-
сификации органич. веществ. Однако теория типов,
стремившаяся лишь к объяснению реакционной спо-
собности органич. в-в, не могла долго продержаться
763
ОРГАНИЧЕСКАЯ ХИМИЯ
764
вследствие декретированной ею принципиальной
невозможности познания их строения. В 1852 Э. Фран-
кленд при изучении металлоорганич. соединений
устанавливает правило валентности. В 1857 Кекуле
высказывает впервые мысль о возможности сцепления
атомов углерода друг с другом и доказывает четырех-
валентность углерода. В 1858 А. Купер, используя
правило валентности и положение Кекуле о сцепле-
нии атомов углерода, впервые отходит от теории типов
и пишет формулы органич. веществ, к-рые являются
очень близкими современным. Однако идеи теории
типов остаются еще очень сильны и создание теории
продолжает отставать от темпов эксперимента.
В 1861 Бутлеров создает теорию строения органич.
веществ. Он ввел в О. х. идею о строении органич.
веществ и ряд новых понятий о химич. связи, порядке
связей атомов в молекуле, о взаимном влиянии атомов
посредственно или непосредственно связанных и др.
представления. Теория строения Бутлерова блестяще
объяснила до него остававшееся непонятным явление
изомерии. В 1864 Бутлеров предсказывает возмож-
ность изомерии углеводородов и вскоре подтверждает
это синтезом изобутана (1867). В целом Бутлеровым
было создано стройное учение о химич. строении ор-
ганич. веществ, к-рое является основой современных
представлений. Одним из важнейших элементов тео-
рии строения явилось положение о взаимном влиянии
атомов, впоследствии развитое В. В. Марковниковым.
Детальное изучение этого влияния послужило основой
для дальнейшего развития теории строения и пред-
ставлений о распределении электроввсй плотности и
реакционной способности органич. соединений.
В 1869 Вислиценус показал, что плоскостные струк-
турные ф-лы недостаточны для объяснения всех слу-
чаев изомерии, к-рая наблюдается и при совершенно
одинаковой последовательности сцепления атомов в
молекуле. Он доказал идентичность строения обыч-
ной молочной и мясо-молочной к-т и пришел к выводу,
что тонкие различия в свойствах молекул с одинако-
вой структурой следует искать в различном располо-
жении их атомов в пространстве. Вскоре (1874)
Я. Вант-Гофф и Ж. Ле Бель создают теорию простран-
ственного расположения атомов в молекуле — стерео-
химию, в основе к-рой по Вант-Гоффу лежит представ-
ление о тетраэдрич. модели четырехвалентного атома
углерода и о том, что оптич. изомерия является след-
ствием пространственной асимметрии молекулы,
в к-рой атом углерода соединен с четырьмя раз-
личными заместителями (см. Асимметрический атом
углерода). Вант-Гофф же высказал предположение
о возможности и другого вида пространственной изо-
мерии и при отсутствии в молекуле асимметрия,
атома углерода. Вскоре Вислиценус доказал, что
фумаровая к-та, ранее считавшаяся полимером мале-
иновой, представляет ее геометрия, изомер (цис-,
транс-изомерия). Естественно, что стереохимия, уче-
ние могло быть создано только на оснопе представле-
ний о строении (структуре) молекулы в Бутлеровском
понимании.
К концу 19 в. накопился огромный фактич. мате-
риал, в том числе и по ароматич. соединениям; в част-
ности, широко была изучена химия бензола, откры-
того впервые М. Фарадеем в 1825. Первая т. н. «бен-
зольная теория» строения ароматич. соединений была
создана Кекуле. В этой теории впервые высказывается
мысль о том, что атомы углерода в органич. соедине-
ниях могут сцепляться, образуя кольцо. Согласно
этой теории, бензол обладает симметрия, структурой
вследствие кольцеобразного строения попеременно
сцепленных простыми и двойными связями шести
метиновых групп. Однако вскоре возникли серьезные
сомнения в правильности ф-лы Кекуле. Исходя из
строения бензола по Кекуле, следовало допустить
наличие двух ортозамещенных гомологов или произ-
водных бензола, чего на самом деле не наблюдалось.
Устойчивость бензола по отношению к сильным
окислителям и нек-рые другие т. наз. ароматич.
свойства бензола и его производных также проти-
воречили этому. Поэтому Кекуле ввел представле-
ние об осцилляции (быстром перемещении) двойных
связей и устранил формальные различия между двумя
ортоположениями. Несмотря на то, что строение бен-
зола по Кекуле находилось в противоречии с рядом
его физич. и химич. свойств, оно долгое время без
всяких изменений принималось подавляющим числом
химиков. Таким образом, возникла группа вопросов,
являвшихся неразрешимыми с точки зрения «клас-
сической» теории строения. К этой же группе вопросов
относится и своеобразие свойств многих других соеди-
нений с сопряженными системами связей. Вопрос о
строении бензола и других ароматич. систем мог быть
решен лишь с появлением физич. методов исследова-
ния строения и в результате развития квантовохими-
ческих представлений о строении органических
веществ.
Впервые электронные представления о химической
связи были введены В. Косселем (1915) и Г. Льюисом
(1916). Эти представления явились шагом вперед и
придали физич. содержание понятию связи (пара обоб-
щенных электронов); однако в том виде, как они были
сформулированы, они не смогли отразить тонких пере-
ходов от гомеополярной к электровалентной связи
и в области О. х. оставались в значительной степени
формальными. Только с помощью квантовохимич.
учения было вложено принципиально новое содержа-
ние в правильные в основном представления элект-
ронной теории.
Как было показано, представления Льюиса о паре
электронов, образующих связь и всегда строго лока-
лизованных на этой связи, оказались приближенными
и не могли быть приняты для всех случаев. Учет
квантовых свойств электрона и отсюда новое пред-
ставление об электронной плотности, с помощью к-рой
и реализуется химич. связь, представление о взаимо-
действии электронов в сопряженных системах открыли
новые возможности рассмотрения вопросов строения
взаимного влияния и реакционной способности орга-
нических веществ. В симметрично построенных на-
сыщеьных углеводородах ординарвые С—С о-свя-
зи действительно реализуются парой электронов,
причем электронная плотность в пространстве между
ядрами соединившихся С—С атомов больше суммы
соответствующих электронных плотностей тех же
изолированных атомов, и она симметрично рас-
пределена относительно оси, соединяющей центры
атомов.
Увеличение электронной плотности есть результат
перекрывания электронных облаков атомов по прямой,
соединяющих их центры. В несимметрич. парафинах
появляется возможность неполной симметрии в ра-
спределении электронной плотности; однаьо эта асим-
метрия столь незначительна, что не может быть прак-
тически установлена при помощи соьременных мето-
дов, и у всех парафиновых углеводородах дипольный
момент равен нулю. То же касается и симметрично
построенных непредельных углеводородов, где атомы
углерода соединены друг с другом л-связио, образую-
щейся за счет добавочного заполнения р-о{бит; так,
этилен, бутадиен и т. п. непредельные углеводороды
дипольным моментом также не обладают. Однако вве-
дение в молекулы этих веществ электродонорной ме-
тильной группы вследствие высокой поляризуемости
л-связи привело к смещению электронной плотности
к крайнему атому углерода и пропилен (I) уже имеет
дипольный момент в 0,35 D, а 1-метилбутидиен —•
0,68 D. Распределение электронной плотности в этих
765
ОРГАНИЧЕСКАЯ ХИМИЯ
766
случаях
схем:
принято изображать
одной из следующих
сн3 -ch=Qh2 или н^с-сн=О:н2
! Нг8" 8+ 8"
причем значки 6+ и б~ показывают возникающие
частичные заряды на атомах углерода.
В представления о распределении электронной
плотности хорошо укладывается ряд эмпирич. правил
О. х. Так, напр., из вышеприведенной ф-лы пропилена
вытекает, что при гетеролитич. присоединении к нему
галогеноводородов протон должен фиксироваться
в месте наибольшей электронной плотности, т. е.
у наиболее «гидрогенизированного» атома углерода
(см. Марковникова правило). Естественно, что гораздо
сильнее сказывается введение в молекулы углеводо-
родов атомов или радикалов сильно разнящихся по
электроотрицательности атомов углерода или водо-
рода. Введение электрофильного заместителя в моле-
кулу углеводородов, как известно, ведет к изме-
нению подвижности атомов водорода в С—Н, О—Н
и др. связях. Подобного рода взаимное влияние атомов,
также объяснимое изменением распределения элект-
ронной плотности, быстро затухает в случае насы-
щенных соединений и почти без затухания пере-
дается по цепи сопряженных связей.
Принято различать два вида взаимного влияния:
индукционное — по цепи о-связей, и влияние сопря-
жения — по цепи имеющих сопряженные я-связи.
Так, увеличение кислотности хлоруксуспых к-т по
сравнению с уксусной к-той объясняется индукци-
онным влиянием атомов хлора: С1*-СН2*-С*-О*-Н,
о
а подвижность атомов водорода метильных групп
уксусного (II) или сорбинового (III) альдегида рас-
сматривается как влияние сопряжения. То же ка-
сается, напр., нитрометана (IV) и нитротолуола (V) и
ряда других случаев:
томерии. Так, алкилирование натрийенолятов к атому
углерода также является результатом перемещения
реакционного центра благодаря сопряжению связей;
~С=сн—
62)3-
Na
RJ
-с-сн-
II
о
I
R
+ NaJ
Взаимное влияние
связей проявляется
в ароматич. соединениях.
Так, напр., при электро-
фильном замещении элек-
трод опорные (нуклеофиль-
ные) заместители (VI) ори-
ентируют в орто- и пара-
положения, а электроно-
акцепторные (VII) элек-
трофильные в .чета-поло-
атомов в результате
также в правилах
сопряжения
ориентации
жение.
VII
Таким образом, на основании современных кванто-
вохимич. представлений разнообразные процессы.
О. х. нашли естественное объяснение. Теоретич.
представления О. х. окрепли и получили предсказа-
тельные возможности. В результате развития тео-
ретич. и физич. методов исследования был оконча-
тельно решен и вопрос о строении ароматич. систем,,
в том числе и бензола; строение последнего описы-
вается след, образом: шесть атомов углерода бен-
зольного кольца находятся в одной плоскости и сое-
динены о-связями, бр-электронов составляют единую
подвижную электронную систему и заполняют кван-
товые энергетич. уровни в соответствии с принципом,
исключения. Таким образом, все С — С связи тож-
Перераспределение электронной плотности и осо-
бенно в момент реакции происходит не только в свя-
зях, к-рые затрагиваются реакцией, но и в других
частях молекулы. Кажущаяся ненормальность соле-
образования парадиметиламино шобензола с фикса-
цией протона одним из слабоосновных атомов азота
азогруппы находит объяснение в перемещении реак-
ционного центра молекулы вследствие сдвига элек-
тронной плотности в момент реакции в направлении,
указанном стрелками:
дественны, все атомы углерода и водорода соответ-
ственно идентичны. Следствием этого является полная
наблюдающаяся на опыте равноценность С — С.
связей (1,39 А) и высокая симметрия бензола с осью
шестого порядка. Из этих положений следует то, что
бензол не полярен и обладает анизотропией диамаг-
нитной восприимчивости. Аналогичными свойствами:
обладают все ароматич. системы, если в них число,
p-электронов равно 4п + 2 (правило Хюккеля). Бен-
зол является далеко не единичным примером соеди-
нений с выровненными двойными и простыми связя-
ми, аналогичная картина наблюдается у трополона*
тропилийбромида, ферроцена, дпфенилполиенов и ря-
да др. (см. А рематические системы). Вполне удачного
графич. изображения строения бензола и др. арома-
тич. систем выработать не удалось. Для описания их
строения используют набор валентных схем, впервые
предложенных Л. Полингом в его резонанса теории'
Это же явление (сопряжения связей) лежит в основе
тех случаев, когда двойственная реакционная способ-
ность органич. веществ не является следствием тау-
или систему обозначения, где изогнутые стрелки по-
казывают также выровненность связей (впервые при-
менена в теории изомерии). Этим же обозначением
пользуются в целях графич. интерпретации равно-
мерного распределения электронной плотности в
симметричных ионах (карбоксилатаниоп, анион
угольной к-ты, гуанидиний катион и т. п.), в амидах
кислот — при объяснении их слабо основных свойств
Со
R—с и в других многочисленных случаях.
В последнее десятилетие О. х. вошла в новую фазу
своего развития. Успехи теории и развитие физиче-
ских методов исследования: рентгеноскопии, УФ- и
ИК-спектроскопии и в особенности ядерно-магнитнога
767
ОРГАНИЧЕСКИЕ РЕАГЕНТЫ
768
и протонного резонанса, а также методы разделения
сложнейших веществ с помощью хроматографии
сделали возможным быстрый структурный анализ
сложнейших соединений и быстрое решение тех
проблем, на к-рые ранее уходили десятилетия.
Огромные сдвиги произошли также в направлении
изучения кинетики и катализа; на основе последнего
вырос ряд новых произ-в. Наиболее выдающихся
результатов О. х. достигла в познании жизненных
процессов.
Лит : Ч и ч и б а б и н А. Е., Основные начала органиче-
ской химии, 5 изд , т. 1—2, М., 1953—57; Каррер П., Курс
органической химии, пер. с нем., Л., 1960; Неиицеску
К. Д., Органическая химия, пер. с рум., т. 1, 1962, т. 2, 1963;
Хюк ке ль В., Теоретические основы органической химии,
пер. с нем., т. 1—2, М., 1955—58; Темникова Т. И.,
Курс теоретических основ органической химии, 2 изд., Л.,
1962; РеутовО. А., Теоретические проблемы органической
химии, М., 1956; Ингольд К. К., Механизм реакций и
строение органических соединений, пер. с англ., М., 1959;
Перспективы развитии органической химии, ред. А. Тодда,
пер. с англ , М., 1959; Мюллер Е., Новые воззрения в ор-
ганической химии, пер. с нем., М., 1960; Пространственные эф-
фекты в органической химии, пер. с англ., под ред. А. Н. Не-
смеянова, М., 1960. И. Л. Кнунянц.
ОРГАНИЧЕСКИЕ РЕАГЕНТЫ — органич. сое-
динения, применяемые в аналитич. химии для обна-
ружения, отделения или количественного определе-
ния как неорганич., так и органич. соединений. Пер-
вым сложным синтетич. соединением, примененным
в качестве аналитич. реагента, был, по-видимому,
л-фенилендиамин, рекомендованный для определе-
ния окислов азота (П. Грисс, 1878). Далее были ре-
комендованы не потерявшие своего значения и ныне
а-нитрозо-Р-нафтол для Со (М. А. Ильинский, 1885)
и диметилглиоксим для Ni (Л. А. Чугаев, 1905).
В настоящее время находят применение несколько
сот органич. реагентов, описано же было более 3000
представителей, к-рым посвящены тысячи публикаций.
О. р. могут обеспечивать высокую чувствитель-
ность, избирательность, скорость и разнообразные
другие удобства выполнения аналитич. определений,
часто превосходя в этом отношении неорганич. реа-
генты. Характерной особенностью О. р. по сравнению
с неорганическими является возможность их «модер-
низации», т. е. создание аналогов известных реаген-
тов, превосходящих по своим достоинствам исходные.
Так, напр., хромотроповая к-та (I) является инте-
ресным реагентом на титан, но нестойкость ее раство-
ров на воздухе (буреет) ограничивает применение
этого реагента. Предложенная позднее дихлорхромо-
троповая к-та (II) дает с титаном еще более яркую цвет-
он он
он он
III
N-ONH,
NO
£
N-ONH,
NO
ную реакцию и при этом стойка в растворах. Возможно
также аналогичными способами «улучшение» эффек-
тивности действия
О. р. Так, органич.
осадители в общем
осаждают элементы
« гем полнее, чем
больше гидрофоб-
ных частей утяже-
ляет молекулу оса-
дителя. Напр., нео-
купферрон (IV) оса-
ждает те же са-
мые элементы пол-
нее, чем купферрон
(III), аантрахинон-
фениларсоновая (V).
о
AsOjHj
AsO3Hj
V
IV
VI
О
(V1) полнее, чем
к-та
а-арсоновая
Общепринятой классификации О. р. не существует.
Предлагавшиеся системы в основном распределяют
О. р. на группы по их назначению или по строению
образуемых ими продуктов реакции. Имеются и
смешанные системы. Так, говорят об органич. осади-
телях, соосадителях, титрантах (реагентах для ти-
трования), реагентах для цветных реакций, для эк-
стракции и др. Наряду с этим прибегают к делению
реагентов на группы: реагенты, образующие нормаль-
ные соли, внутрикомилексные соли, реагенты, обра-
зующие характерные продукты окисления или вос-
становления, и др.
Реагенты, применяемые в неорга-
ническом анализе. Аналитич. действие мно-
гих О. р. основано на комплексообразовании. Ре-
зультат действия таких О. р. в общем виде опреде-
ляется двумя особенностями строения их молекул.
Одна часть молекул, являющаяся функциональной
или характерной атомной группировкой, обусловли-
вает сам факт протекания реакции с ионами данного
элемента, а другая — наблюдаемый аналитич. эф-
фект.
В табл. 1 приведены важнейшие характерные атом-
ные группировки О. р.
Если, кроме характерной атомной группировки,
молекула реагента содержит, напр., группы, придаю-
щие растворимость в воде или окраску, то и реагент
и продукты реакции непременно будут растворимыми
в воде или окрашенными. С другой стороны, харак-
терная атомная группировка определяет возможность
и условия протекания реакции с данным элементом.
Вне зависимости от того, образуются окрашенные или
неокрашенные продукты реакции, растворимые или
нерастворимые. При отсутствии рассматриваемых
ниже особенностей строения молекул О. р. все эти
реагенты будут взаимодействовать с атомами данного
элемента и даже при приблизительно одинаковых
значениях pH. В табл. 2 представлена схема взаимо-
связи О. р. различного аналитич. назначения.
Важным положением, облегчающим понимание
влияния pH на действие комплексообразующих О. р.,
является наличие аналогии между их действием и
лежащими в основе этого действия простейшими
неорганич. реакциями. Так, имеется аналогия
между реакциями катионов с водой и реакциями,
протекающими при действии О. р., содержащих
группы —ОН, т. е. О. р. вида R — ОН:
Ме+ -|-Н — ОН = Me—ОН +Н +
Ме+ 4-Н- OR = Me — OR-f-H +
В обоих случаях образуется связь Me — О, определя-
ющая химизм реакции. При отсутствии образования
полидентатных комплексов, т. е. сое-
динений, в к-рых один и тот же атом металла связан
со многими (3—4 и более) точками той же самой
молекулы реагента, влияние pH на течение обеих
этих реакций приблизительно одинаково. Поэтому
катионы элементов, реагирующих с водой уже в
сильно кислых растворах (Ge, Zr), взаимодей-
ствуют с О. р. этого вида также в сильно кислых
р-рах; гидролизующиеся труднее (Th, Al) реагируют
при меньшей кислотности, а гидролизующиеся сов-
сем трудно (Mg, Са, Li) требуют щелочной среды.
Полидентатные комплексы образуются в более ки-
слых р-рах, но очередность расположения элементов
по их способности давать реакции в зависимости от
кислотности обычно сохраняется. Подобная же ана-
логия имеет место между условиями действия П»8 и
реагентов вида R—SH, образованием аммиакатов
и действием реагентов с аминным азотом и др.
Реагенты, применяемые для раз-
деления элементов. Органич. осадители
могут осаждать элементы в виде нормальных или
769
ОРГАНИЧЕСКИЕ РЕАГЕНТЫ
770
Таблица 1
Атомная группировка Примеры элементов, для к-рых группировка характерна Атомная группировка Примеры элементов, для к-рых группировка характерна Атомная группировка Примеры элементов, для к-рых группи- ровка ха- рактерна
- с = сн Ag, Си (0 = ) с-с=с- 1 1 НО N(H2) Си, Zn, Cd, Hg, Pb, Co, Ni /0 —As-OH \он Zr, Hf, Nb, Та, Ti, Sn
-с=с-он 1 FeIII
— С = С —С(Н) 1 II НО N- ОН Cu, Zn, Cd, Pb, Fe, Ni —N=N-+-SO4H или —СООН Zr, Hf
-с=с- 1 1 но он Al, In, Ga, Ge, Tl, VIV, Sb, Bi, Mo- re, ва, Pb
(HN =) C-N-C ( = NH) 1 । । НО N (Н) Ag, Cu, Ni, Pd
__ZH hov N=N— Coin Сгш, Pd
-с—С—С- или 1 1 но он -С=С-С(=О) но он Lt, Си, Ca, Be, Mg. Al, Ga, Ti, U, Fe
-C-SH Hg
_y0H HOX_ ^-N=N-\ Co, Mg, Zn, Cd, Cu и многие другие
-s/0 'ОН Fem, Ti, Zr
-с=с-с- i II НО О B, Al, Ga, Ge
-N —C-SH 1 (-NH-C = S) 1 Hg, Ag, Си и др. элементы группы H2S и (NH4)2S
-с=с-с- | 11 + CS. НО О Tl _/COOH HOX__ /-N=N— Al, Cu и другие
-N = Си и др. элементы, образующие амми- акаты ( = N-)-C-NH, II S Bi, Ru, Os
_/SO.,H H0\_ /—N=N—\ Ba, Sr, РЬ
- NO2 и подвижн. Н К, Rb, Cs, NH,+ -S-C-SH II N — Cu, Cd, Hg, Ti, Pb, Bi
- N - ОН 1 NO Многие элементы, катионы к-рых лег- ко гидролизуются
_/AsO3H2 HOx_ У N=N \ Al, редк. земли, Zr, Th, U
(0 = ) c-c- 1 1 HO SH Fe, U и другие эле- менты группы H2S или (NH,)2S
= С - N О + (Alk)3N - С = 1 1 Pd
(0=) c-c = 1 1 —(H)N SH Элементы группы H2S или (NH4)2S _/AsO3H2 HO, / N = CH — \ Sc, Th, U
-С^С- 1 1 НО NO Со
S=C-SH 1 Си и другие элемен- ты группы H2S или (NH,)2S
—N=C—С—N- Fe11
1 1 Й Z, II z 1 z z\z Z. 1 Cu
(0 = ) С-С^ НО N (Н2) Си, Ni \ /S PZ / 4SH Мо, Bi, Ru, Os
1 Z 1 z II к z о 1 1 Cu, Ni
(0 = ) С-С- 1 II НО N - Си, Zn, Cd и многие другие -c=c- 1 1 HS SH Sn, Bi, Mo, W
-с-с - II II 0 N —ОН Fe11, Со, N1 —C—S—c— 1 II HS S —C—N—C— И | II HS s Bi, Sb /СООН N^ “\ \ — ;-n=n z z \ Cu, NI
-С-С- 1 II НО N-0H Си, Мо
_/SO3H N-^ N=N_Z- 7 \ Cu, Ni
-С-С- II II HO-N N-0H -С-С- ИЛИ II II N N Ni, Pd
-Se/° 'OH Zr, Fe111
комплексных солей определенного состава. Получае-
мые осадки чисты. После высушивания они нередко
могут быть взвешены как таковые, имея хороший
фактор пересчета на элемент. Иногда возможны
объемные или фотометрич. определения, после раст-
ворения осадков или определения остатка реагента в
фильтрате. Применяются в амперометрическом тит-
ровании.
Вследствие сложности работы применение органич.
осадителей рассматриваемого вида для собственного
определения элементов практикуется нечасто. Бла-
годаря избирательности действия, легкости филь-
трования, способности многих осадков экстрагировать-
ся и другим причинам органич. осадители чаще ис-
пользуются для разделений или предварительных от-
делений элементов. Для осаждения алюминия и мно-
гих других элементов применяют 8-оксихинолин, цен-
ный О. р. для никеля и палладия — диметил гли-
нке им, для рения— нитрон. Цругие органич. осадите-
ли (купферрон, фениларсоновая кислота, таннин и др.)
не дают весовых форм и перед взвешиванием требуют
озоления осадков. Кремневая к-та осаждается жела-
тиной. Известны также осадители, выделяющие эле-
менты в виде неорганич. соединений. Это или рН-регу-
ляторы — беззольные буферные смеси (ацетаты, пири-
дин, уротропин и др.), или восстановители, как,
напр., гидрохинон. Осадители этих групп использу-
ются для отделения или выделения элементов.
Органические осадители способны выделять эле-
менты из растворов с концентрацией не ниже
— 0,1—1 лг/100 мл. Экстракцией, где это возможно,
можно выделять элементы и из более разб. р-ров.
Для последней цели используются также органич.
соосадители, способные практически полностью выде-
лять элементы из необычайно разб. р-ров. Так, в
виде комплекса с арсеназо (с осадком, образованным
С25 к. X. Э. т. 3
771
ОРГАНИЧЕСКИЕ РЕАГЕНТЫ
772
Таблица 2
Тип реагента Схема строения а Труп хромо- форная пы в молен придаю- щие раст- воримость (SO3H и др.) уле б гидрофоб- ные (С6Н5, СН3, С1, NO2 и др.)
1. Неокрашенный осади-
тель
2. Неокрашенный утяже-
ленный осадитель; О. р.
для экстракции
3. Окрашенный осадитель
4. Окрашенный утяжелен-
ный осадитель; О. р. для
экстракции
5. Неокрашенный маскиру-
ющий комплексообразо-
ватель (титрант); О. р.
для цветных реакций
6. Окрашенный комплексо-
образователь; О. р. для
цветных реакций, инди-
катор для комплексоно-
метрии
7. Ингредиенты органич.
соосадителей:
а) окрашенные для экст-
ракции
б) бесцветные
в) индифферентные
а Четырехугольником обозначена молекула О.р.; М характерная атомная труп-
1 ZSO3H
пировка, —(N = N)— хромофорная группа, ' означает, что для хорошего
J 4so3H
действия реагента требуется наличие нескольких групп — SOaH. ° + и-присут-
ствие или отсутствие той или иной группировки в составе О. р.
арсеназо с кристаллвиолетом) америций еще хорошо
выделяется из растворов с разбавлением 1 : 1 • 1015,
т. е. содержащих 2-10-’л1Кг Ат в 2 л объема. Для
улучшения избирательности осаждения, чтобы пред-
отвратить осаждение нежелаемых элементов, при
осаждении вводят маскирующие комплексообразова-
тели (маскирующие реагенты): винную, лимонную,
сульфосалициловую к-ты, комплексон III и др.
Для отыскания улучшенных осаждающих О. р. и
частично для экстрагирующих О. р. имеет значение
т. наз. «эффект утяжеления». Чтобы О. р. полнее
осаждал или экстрагировал металлы, молекула реа-
гента должна быть «утяжелена» введением замести-
телей. Более тяжелые заместители приводят в общем
к менее растворимым в воде продуктам реакции.
Заместители должны быть гидрофобными. Введение
солеобразующих групп, напр.—SOsH, наоборот, улуч-
шает растворимость продуктов реакции в воде и
затрудняет их экстракцию. Наиболее отчетливо дей-
ствует введение арильных или алкильных групп:
Св115, С2Н5, СНз и др., замена бензольных ядер
нафталиновыми или антрахиноновыми. Выше были
приведены примеры, относящиеся к эффекту утяже-
VII VIII
ления в виде пар реагентов: куп-
ферон (III), неокупферон (IV), фе-
ниларсоновая кислота (V), антра-
хинон-а-арсоновая к-та (VI). Бато-
фенантролин (VIII) дает продукты
реакции, экстрагирующиеся лучше,
чем фенантролин (VII). Этот реа-
гент обеспечивает более чувстви-
тельное определение железа, чем
фенантролин. Используются также
заместители Cl, Вг, J, NO2, к-рые
одновременно могут добавочно вли-
ять на кислотность солеобразую-
щих групп реагента.
Механизм эффекта утяжеления
заключается в уменьшении плот-
ности заряда, обусловленного нали-
чием в молекуле реагента солеобра-
зующих групп (последние придают
реагенту растворимость). Это ослаб-
ляет сольватацию молекулы О. р.
молекулами.воды, от чего зависит
растворимость. Иногда гидрофоб-
ные заместители экранируют соле-
образующие группы, что также'
уменьшает растворимость в воде.
Реагенты для экстракции приме-
няют в тех случаях, когда хотят пе-
ревести элемент в способное экстра-
гироваться гидрофобное соедине-
ние, содержащее большую органич.
составную часть. Экстрагирующим-
ся соединением чаще всего являет-
ся: 1) нормальная соль, содержащая
металл-комплексный анион; 2) нор-
мальная соль, содержащая металл-
комплексный катион; 3) внутриком-
плексная соль (органич. адденд и
органич. анион заключаются в ос-
татке одной и той же молекулы О.
р.). В нек-рых случаях применяемый
(«активный») экстрагент является
поставщиком требующихся органич. катионов или слу-
жит аддендом и одновременно сольватирует экстраги-
рующиеся соединения. В таких случаях для экстракции
практически достаточно только одного экстрагента.
В других случаях, когда используется «пассивный»
экстрагент, к-рый только растворяет экстрагируемые
соединения, производя их слабую сольватацию, необ-
ходимо обеспечить образование экстрагирующихся
соединений добавлением других органич. реагентов.
Для экстрагирования железа из солянокислотных
р-ров применяют активный экстрагент — изоамил-
ацетат, заменивший употреблявшийся ранее огнеопас-
ный диэтиловый эфир. Уран из азотнокислотных
р-ров экстрагируется смесями, содержащими трибу-
тилфосфат. Для многих целей применяются также
метилэтилкетон, циклогексанон, бутиловый и ами-
ловый спирты и др. Пассивными растворителями,
используемыми для экстрагирования, напр., куп-
ферронатов, 8-оксихинолинатов и других соединений
служат СНС1з. СС1«, СвНв. В качестве аддендов исполь-
зуют пиридин, о-фенантролин. Экстрагировать можно
и растворимые в воде окрашенные соединения, напр.
комплексы многих элементов, образованные содер-
жащими сульфогруппы реагентами арсеназо, торон,
нитрозо-И-соль и многими другими. В этом случае
вводят соли органич. катионов-дифенилгуанидиния,
S-бензилтиурония, тетрафенил арсония и экстраги-
руют бутиловым или амиловым спиртом.
Реагенты, применяемые для опре-
деления элементов. Реагенты для фото-
метрии. определений используются очень широко,
773
ОРГАНИЧЕСКИЕ РЕАГЕНТЫ
774
т. к. в современном неорганич. анализе фотометрии,
определения выполняются чаще других. Многие оп-
ределения выполняются в экстракционно-фотометрич.
вариантах, в к-рых окрашенный продукт реакции
экстрагируется. Это приводит к улучшению изби-
рательности и одновременно за счет уменьшения
объема и других причин может повысить чувстви-
тельность. Реагенты дитизон, 8-меркаптохинолин,
применяемые для определения следов тяжелых ме-
таллов, используются только в экстракционно-фото-
метрич. вариантах. Фотометрии, методы, основанные
на образовании комплексных соединений, выполня-
ются при использовании неокрашенных или окрашен-
ных О. р. Неокрашенные О. р. дают окрашенные ком-
плексы только с элементами, являющимися носите-
лями цветности, т. е. образующими хромофорные
сочетания (Fe, Со, Ni, Mn, Ti, U, V и др.). Элементы,
такими свойствами не обладающие (Al, Са, Cd, Ga,
In, Mg, Zn, Zr и мн. др.), образуют неокрашенные
комплексы и поэтому определениям не мешают.
Вследствие хорошей избирательности неокрашенные
реагенты применяются особенно часто, но чувстви-
тельность реакций относительно мала. Для опреде-
ления железа применяются сульфосалициловая к-та,
о-фенантролин, а,а'-дипиридил; для титана — хромо-
троповая и дихлорхромотроповая к-ты, для меди
и ряда прочих элементов — диэтилдитиокарбаминат
натрия и др.
Более высокой чувствительностью, но худшей изби-
рательностью обладают образующие комплексы ок-
рашенные реагенты. Чувствительность достигает
0,05—0,5 мкг/мл элемента. Для улучшения избира-
тельности и обеспечения надежности определений
здесь прибегают к работе при точных значениях pH,
применению маскирующих комплексообразователей
и другим приемам. Химизм цветных реакций этого
вида заключается в том, что под влиянием атома эле-
мента остаток окрашенной молекулы реагента в
комплексе переходит в такое ионное состояние, к-рое
при данном значении pH, не связанному в ком-
плексе реагенту, не свойственно. Поэтому при дан-
ном значении pH реагент и комплекс обладают раз-
личными окрасками, но комплексы разных элементов
могут иметь близкую или даже одинаковую окраску.
Было описано несколько сот окрашенных комплексо-
образующих О. р. Для определения Al, Th, суммы
редкоземельных элементов, Zr, U и ряда других эле-
ментов применяют арсеназо, торон, арсеназо III, пиро-
катехиновый фиолетовый, ксиленоловый оранжевый
и др.
В значительно меньшем количестве описаны ком-
плексообразующие реагенты, дающие люминесцирую-
щие продукты реакции. Чувствительность люминес-
центных реакций в 10—50 и более раз выше чувстви-
тельности фотометрии, реакций. Для определения
алюминия применяют 8-оксихинолин и морин, для
магния — биссалицилальэтилендиамин и др.
Элементы, образующие комплексные анионы, та-
кие, как8ЬС16 , Т1С14 , Zn(SCN)4 , HgJ4 и др., опре-
деляются в виде солей этих анионов с окрашенными
органич. катионами. Применяются основные краси-
тели: метилвиолет, кристаллвиолет, родамин Б,
бутилродамин и др. Определения выполняются в виде
т. наз. цветных твердофазных реакций, где возникаю-
щая суспензия малорастворимой соли отличается по
окраске от р-ра реагента или, чаще, в виде экстрак-
ционно-фотометрич. методов. Последние основаны на
неодинаковой пригодности длн экстракции ионов с
различной степенью гидрофобности. В применяемых
реагентах один (окрашенный) ион хорошо пригоден
длн экстракции, а другой (неокрашенный) — нет.
Так, напр., в случае бутилродамина положительно
заряженный окрашенный катион пригоден для экс-
тракции, а неокрашенный хлорлд-ион непригоден:
При определении, напр., таллия, присутствующего
в виде Т1С14 , толуол или бензол экстрагируют соль,
образованную катионом бутилродамина и анионом
Т1С14 . Интенсивность окраски пропорциональна кон-
центрации таллия. Избыток реагента, длн окрашен-
ных катионов к-рого не хватило пригодных для эк-
стракции анионов, остается в водной фазе в виде прак-
тически не экстрагирующегосн хлорида. При исполь-
зовании люминесцирующих основных красителей
(родамин Б и др.) экстракционно-фотометрич. прие-
мами можно определять 0,07 мкг элементов.
Ионы окислители и восстановители, к-рые обра-
III IV VI IV—VII V
зуют Au , Се , Сг , Mn , V и др. элементы,
определяются при использовании бесцветных О. р.,
образующих окрашенные продукты при действии
окислителей или восстановителей. Для хрома приме-
няется высокоизбирательная реакция с дифенилкарба-
зидом, для многих окислителей пригодны: бензидин,
толидин, лейкомалахитовый зеленый и др.
Реагенты, применяемые для объ-
емных определений. Растворы нек-рых
органич. реагентов пригодны в качестве титрантов для
комплексонометрич., оксидиметрич. и редуктометрич.
титрований. Особенно широко применяются первые,
причем на практике используется почти исключи-
тельно этилендиаминтетрауксуснан к-та в виде дина-
триевой соли (ЭДТА, комплексон Ш, хелатон III,
трилон Б, версен). Титрованием ЭДТА определяют
более 20 элементов, в их числе Са, Mg, Al (при нагрева-
нии), тяжелые и цветные металлы, многие редкие эле-
менты. Титрование проводят в присутствии металл-
индикаторов, изменяющих окраску при достижении
точки эквивалентности. В качестве металл-инди-
каторов пригодны многие О. р., дающие цветные ре-
акции. Часто используются эриохром черный Т, кси-
леноловый оранжевый, арсеназо и др. Значительно
менее распространено комплексонометрич. титро-
вание при использовании иных комплексообразующих
реагентов: нитрилотриацетата (трплон А), цитрата,
цианидов и др. В качестве слабого окислителя, за-
менителя иода, используетсн р-р хлорамина Т. Р р
гидрохинона применяется как восстановитель при
объемном определении золота с потенциометрия,
определением достижения точки эквивалентности.
Индикаторы, применяемые в аналитической химии,
практически все являются органич. соединениями.
Прочие случаи применения О. р.
в неорганическом анализе. Нек-рые
О. р., легко получаемые во вполне чистом виде и легко
определяемые объемными методами, используются
в качестве первичных стандартов. В алкали- и аци-
диметрии применяются щавелевая и п-нитробензой-
ная к-ты, дифенилгуанидин и др., в комплексоно-
метрии — динатриевая соль этплендиаминтетраук-
сусной к-ты. Желатина и агар-агар используются в
качестве стабилизаторов различных суспензий так
же, как и глицерин, придающий растворам вязкость.
В полярографии органич. комилексообразователи
используются для смещения и раздвигании волн
25*
775
органические реагенты—ориентации правило
776
нек-рых элементов. При спектральных и поляро-
графии. определениях органич. соосадители приме-
няются для предварительного концентрирования
определяемых элементов.
Применение О. р. в органическом
анализе. Для целей идентификации многие орга-
нич. соединения, при употреблении органич. реа-
гентов, переводятся в соответствующие производные,
идентифицируемые по их физич. свойствам, обычно
по темп-ре плавления или, при окрашенных соеди-
нениях, по положению максимума светопоглощения.
Напр., разлагающиеся без плавления сульфокислоты
четко плавятся в виде S-бензилтиурониевых и других
солей, сахара в виде гидразонов и др. Многие амины
и их сульфокислоты могут быть идентифицированы
по положению максимума светопоглощения азосое-
динений, получаемых сочетанием диазотированных
аминов с Р-нафтолом.
Цветные качественные реакции и количественные
методы определения разнообразнейших органич. сое-
динений различных классов основаны на реакциях
их функциональных группировок и других свойствах.
Многие неокрашенные органич. в-ва при простом сме-
шении с другими органич. в-вами дают окрашенные
молекулярные соединения, образование к-рых исполь-
зуется для аналитич. целей. Реагентами на амины слу-
жат полинитросоединения и особенно тетрахлор-
хинон (хлоранил). Напр., светло-желтый хлоранил с
бесцветным диметиланилином образует интенсивно
синее молекулярное соединение. Обратно, амины
(диметиланилин и др.) служат реагентами на нитро-
соединения, особенно на полинитросоединения. Мно-
гие взрывчатые в-ва могут быть идентифицированы по
окраске их растворов в аминах.
Органич. соединения, способные при подходящих
значениях pH образовывать ионы — катионы или
анионы,— определяют экстракционно-фотометрич. ме-
тодами, аналогичными используемым в неорганич.
анализе. Напр., бутилродамин, содержащий окрашен-
ный катион, применяют для экстракционно-фотомет-
рич. определения 2,4-дихлорфеноксиуксусной к-ты.
Такими же способами определяют многие поверхно-
стно-активные в-ва.
При выполнении реакций на функциональные груп-
пировки используют многие органич. и неорганич.
реагенты. Таких реакций известно очень много и
они охватывают очень многие группы органич. соеди-
нений. Напр., неокрашенные карбонильные соедине-
ния при использовании динитрофенилгидразина пере-
водятся в удобные для фотометрирования желтые
динитрофенилгидразоны. Для аминокислот широко
применяется нингидрин. Аскорбиновая к-та титруется
синим раствором 2,6-дихлориндофенолята натрия
(реагент Тильманса), к-рый обесцвечивается.
Многие О. р., кроме аналитич. химии, находят боль-
шое применение в различных отраслях пром-сти и
при научных исследованиях. Для отделения и очистки
редких элементов широко используют органич. оса-
дители и соосадители и реагенты для экстракции.
Трилон Б используется во многих случаях для свя-
зывания присутствующих в природной воде кальцие-
вых и магниевых солей (смягчения воды), для предот-
вращения образования накипи. Комплексы различ-
ных металлов с О. р. являются широко применяе-
мыми объектами при разнообразных научных иссле-
дованиях.
Лит.: Кульберг Л. М., Органические реактивы в
аналитической химии, М.—Л., 1950; W е 1 с h е г F. J.
Organic analytical reagents, v. 1—4, N. Y., 1947—48; F e 1 g i
F., Chemistry of specilic, selective and sensitive reactions, N. Y.,
1 949; ProdingerW., Organische Fallungsmittel in der quan-
titativen Analyse, 4 Aufl., Stuttg., 1957; Сонг ин а О. A.,
Амперометрическое (полярометрич.) титрование в анализе
минерального сырья, М., 1957; Моррисон Д ж., Фрей-
зер Г., Экстракция в аналитической химии, пер. с англ.,
Л., 1960; Кузнецов В. И., Химические основы экстрак-
ционно-фотометрических методов анализа, М., 1963; S a n d е 11
Е. В., Colorimetric determination of traces of metals, 3 ed., N.
Y., 1959; И в а н ч e в Г., Дитизон и его применение, пер. с
нем., М., 1961; Кузнецов В. И., Усп. химии, 1952, 21,
вып. 2, 175; его ж е, Ж. общ. химии, 1950, 20, вып. 5, 816;
Органические реагенты в аналитической химии, М., 1960 (Тр.
комиссии по аналитической химии, т. И); Труды по химии и
химической технологии (г. Горький), 1960, М 3, 559; Куз-
нецов В. И., Успехи химии, 1949, 18, вып. 1, 75; П р ш и-
бил Р., Комплексоны в химическом анализе, пер. с чеш.,
2 изд., М., 1960; Д ж о н с о н В. С., Ш е н н а н Р., Р и д Р.,
Органические реактивы для органического анализа, пер. с
англ., М., 1948; Ф а й г л ь Ф Капельный анализ органиче-
ских веществ, пер. с англ., М., 1962; S 1 g g i a S., Quantitative
organic analysis via functional groups, 2 ed., N. Y., 1954; Аса-
тиани В. С., Биохимическая фотометрия, М. 1957.
В. И. Кузнецов.
ОРГАНИЧЕСКОЕ СТЕКЛО — см. Полиметакри-
латы.
ОРГАНОГЕНЫ — исторически сложившееся наз-
вание углерода, водорода, кислорода и азота как
основных элементов состава растительных и живых
организмов. В последующее время было выяснено, что
в состав многих природных соединений наряду с
элементами-органогенами входят фосфор, сера и
нек-рые металлы. Т. обр., понятие О. имеет лишь
история, значение. д. в. Ншрашпъев
ОРЕХОВОЕ МАСЛО — см. Жиры растительные.
ОРИЕНТАЦИИ ПРАВИЛО — совокупность за-
кономерностей, определяющих строение соединений,
образующихся при реакции замещения в ароматич.
ряду. В замещенных бензолах, конденсированных
ароматич. системах, а также гетероциклич. соедине-
ниях ароматич. природы электронная плотность рас-
пределена в цикле неравномерно. Вследствие этого
вступающий заместитель направляется в положение,
определяющееся как характером имеющегося заме-
стителя (ориентанта), так и природой атакующе-
го реагента. По характеру влияния на ароматиче-
ское ядро различают 2 типа ориентантов. Ориентан-
тами 1-го рода являются электронодонорные груп-
пировки (ОН, OR, -O-C-R, SH,—SR, -NH3, NHR, NR3,
О
алкилы, атомы галогена и др.); ориентантами
2-го рода — электроноакцепторные группировки
о
(SO3H, NO3, СООН, COOR, Ce=N, CF3, NR3, cf ,С1О3идр.).
H
В реакциях электрофильного замещения (атакующим
является либо катион, либо катионоидный реагент)
ориентанты 1-го рода направляют вступающий заме-
ститель в орто- и пара-положения; в случае ориентан-
тов 2-го рода вступающий заместитель направляется в
.иеша-положение. Существуют заместители, которые
обусловливают смешанную ориентацию (например,
+ /R
NO, С-СН,, СНС12, CH3NO„ CH2CH2N“R и др.).
Асимметрия электронного облака моно.замещен-
ного бензола, содержащего заместитель 1-го рода,
характеризуется общим увеличением электронной
плотности по сравнению с незамещенным бензолом.
Вследствие этого в реакциях электрофильного замеще-
ния подобные соединения более активны, чем бензол.
Характер распределения электронного облака обу-
словливает повышение электронной плотности осо-
бенно в орто- и пара-положениях. След., приближение
электрофильной частицы к орто- и пара-положениям
является энергетически более выгодным по сравнению
с щгта-положением (1, а). Кроме того, переходное
состояние при атаке орто- и пара-положений катионом
или катионоидным реагентом характеризуется боль-
шим рассредоточением положительного заряда (1, б),
чем в случае атаки лщта-положения (1, в). Т. обр.,
777
ОРИЕНТАЦИИ ПРАВИЛО — ОРНИТИН
778
в этом случае статич. и динамич. факторы действуют
согласованно (см. Сопряжение).
та-двузамещенные бензолы, содержащие ориентанты
одного рода, либо орто- или пара-двузамещенные
бензолы с заместителями различного ориентирую-
щего действия) реакция осуществляется в соответст-
вии с влиянием обоих заместителей. В случае
Особое положение в ряду ориентаитов 1-го рода
занимают атомы галогена, к-рые, как и другие ориен-
танты 1-го рода, направляют вступающую электро-
фильную группу в орто- и пара-положения, однако
реакция осуществляется труднее, чем в незамещенном
бензоле. Этот факт объясняется несогласованностью
статич. и динамич. факторов. В нереагирующей моле-
куле галогенобензола атом галогена является отри-
цательным концом диполя, в результате чего арома-
тич. ядро в общем несет частичный положительный
заряд (II, а). Следствием этого является затруднен-
ное, по сравнению с бензолом, электрофильное заме-
щение галогенобензола в ядро. Вместе с тем в пере-
ходном комплексе р-электроны галогена-заместителя
участвуют в рассредоточении положительного заряда
вступающей группы лишь в том случае, если она
направляется в орто- и napa-иоложения (II, б). При
атаке жста-положения атом галогена в этом рассре-
II,а
На1
доточении не участвует
(II, в). Т. обр., дина-
мич. фактор способству-
ет ориентации атакую-
* щей электрофильной ча-
стицы в орто- и пара-
положения.
В монозамещенных
бензолах, содержащих
заместители 2-го рода, ароматич. ядро в статпч. состоя-
нии обеднено электронами и электрофильная атака зат-
руднена. Однако в мет a-положениях это обеднение ме-
нее ярко выражено и катионоидный заместитель направ-
ляется именно в эти положения (III, а). В переходных
состояниях в любом случае рассредоточение положи-
тельного заряда происходит без участия заместителя
(III, б и III, в). В согласии с теми же электрон-
ными схемами закономерности становятся обратными
в реакциях нуклеофильного замещения. Ориентанты
2-го рода направляют анион либо анионоидный реа-
гент в орто- и пара-положения, причем замещение
происходит легче, чем в незамещенном бензоле (IV).
Ориентанты 1-го рода направляют вступающий заме-
ститель в лета-положение, причем атака значительно
затруднена по сравнению с незамещенным бензолом.
В реакциях радикального замещения наличие за-
местителя вне зависимости от его ориентирующего
действия определяет замещение в орто- и пара-поло-
жения. Участвуя в рассредоточении облака неспарен-
ного электрона, любой заместитель (V, а; V, б) облег-
чает реакцию по сравнению с незамещенным бензо-
лом (V, в). Наличие двух и более заместителей в
бензольном кольце влияет на направление реакции
по-разному. В случае согласованной ориентации (ме-
конкурирующей ориентации направление реакции
определяется влиянием ориентанта 1-го рода. При
несогласованном действии двух ориентаитов одного
рода ориентация определяется заместителем более
активным (правило Бейльштейна). Относительная
активность заместителей определяется положением в
так наз. рядах Голлемана: ориентанты 1-го рода:
OH>NH2>C1>J>Вг>СН3; ориентанты 2-го рода:
С00Н>80зН>]\02. Непременным условием ориен-
тирующего влияния на ароматич. кольцо какого-
либо заместителя является параллельность осей л-
или р-электронов заместителя и л-электронов ядра.
В случае нарушения этого условия ориентирующее
действие не имеет места. Так, напр., М,И-диметил-
анилин легко нитрозируется в пара-положение, т. к.
ничто не мешает параллельности осей р-электронного
облака азота и л-электронов арома-
тич. ядра, однако в диметилксилиди- нзСх ZCH3
не (VI) такая параллельность невоз- N:
можна из-за стерич. препятствий, со-
здаваемых двумя метильными группа- Y |
ми в орто-положении по отношению
к диметиламиногруппе. В этом случае V|
диметиламиногруппа выключена из
сопряжения с ядром и нитрозирование не может быть
осуществлено. См. также Ароматические системы.
Лит.: Реутов О. А., Теоретические проблемы органи-
ческой химии, М., 1956, с. 314; II н г о л ь д К., Механизм
реакций и строение органических соединений, пер. с англ.,
М., 1959, с. 184. Л. С. Герман.
ОРНИТИН (а,6-диаминовалериановая кислота)
Н2М(СН2)зСН(МН2)СООН, мол. вес 132,2— белые
кристаллы; т. пл. 140° (D-орнптина), [а]” = + 11,5“
(вода); легко растворим в воде и спирте, трудно в
эфире; раствор О. в воде обладает щелочной реакцией.
О. образует производные D-0. — дихлоргидрат,
[«1д=+16,8° (вода); хлороплатинат, т. разл. 208°; D.L-0.—
хлоргидрат, т. пл. 215°; нитрат, т. пл. 185°; оксалат, т. пл. 218°;
пикролонат, т. пл. 220—221° и др.
При действии на О. NH3 и С02 образуется цитрул-
лин, при декарбоксилировании — путресцин', при
взаимодействии H2NC(= ]\Н)ОСНз с медной солью
О. последний может быть переведен в аргинин.
Синтетически О. может быть получен по схеме
(выход 85%):
C,H,0Na
NCCH=CH2 + HC(COOC2H;)2 ------->
I
NHC0CH3
H,.Ni
—<-mcch2ch2c(cooc3h5)2 —:—>
NHCOCH,
COOC2HS
—>-| I ' NHCOCH, —> H2N(CH2)SCH(NH2)COOH-HC1
\z = 0
N
I
H
779
ОРНИТИНОВЫЙ ЦИКЛ-ОРОТОВАЯ КИСЛОТА
780
а также из 1,1,5-трихлорпентена (продукта теломери-
зации СН2=СН2 и СС14).
В организме О. образуется при гидролизе арги-
нина, восстановительном аминировании а-амино-
у-формилмасляной кислоты (продукта восстановле-
ния глутаминовой кислоты); при окислительном от-
щеплении d-аминогруппы О. может переходить в
пролин. В незначительных количествах О. содержит-
ся в тканях человека, плазме крови. В нек-рых
растениях О. присутствует в значительных количе-
ствах.
О.— заменимая аминокислота, играет весьма важ-
ную роль в организме, особенно в биосинтезе моче-
вины (см. Орнитиновый цикл); биосинтетически тесно
связан с пролином и оксипролином, входит в состав
антибиотиков-полипептидов (бацитрацина, грами-
цидина С).
Лит.: Фердман Д. Л., Биохимия, 2 изд., М., 1962;
Кочетков Н.К., Торгов И. В., БотвиникМ. М.,
Химия природных соединений, М., 1961; Substances natu-
relies de synthese, v. 3, P , 1951, p. 52. A. E. Васильев.
ОРНИТИНОВЫЙ цикл — цепь реакций превра-
щения орнитина в процессе биологич. синтеза моче-
вины, являющейся у многих видов животных конеч-
ным продуктом азотистого обмена (см. Обмен ве-
ществ); предложен Кребсом в 1932. О. ц. состоит из
трех основных реакций: превращения орнитина в
цитруллин (I); превращения цитруллина в аргинин
(II) и расщепления аргинина на мочевину и орни-
тин (III):
+ CO, + NH,
---------—► цитруллин
Г П | +NH°
| III | аргинин
орнитин -*—•-------
мочевина
Образовавшийся орнитин вновь вступает в этот цикл
превращений и выполняет, таким образом, роль ката-
лизатора (что подтверждено в опытах с орнитином,
меченным N15 в а- и d-положении). Орнитиновый цикл,
изображенный на схеме, имеет сложный фермента-
тивный механизм. Реакции I и II требуют затраты
энергии и протекают только в аэробных услови-
ях при наличии субстратов клеточного дыхания
и при сохранности клеточной структуры. В гомогена-
тах и экстрактах, где клеточная структура наруше-
на, эти реакции осуществляются только при доба-
влении аденозинтрифосфорной кислоты (АТФ) и ио-
нов Mg.
Реакция I осуществляется ферментной системой
митохондрий и состоит из двух этапов: 1) образова-
ния из NH3, СО2 и АТФ карбамилфосфата, обладаю-
щего высокоэргической фосфатной связью; актива-
тором этой реакции является N-ацетилглутаминовая
кислота:
nh3+co3+ato —>нн2со-о~ро-он+адф
он
карбамилфэсфат
2) образования цитруллина в результате взаимодей-
ствия карбамилфосфата с орнитином:
NH2CO-O~PO-OH + NH2CH2CH2CH(NH2)COOH —►
он
—i-N H2CONHCH2CH2CH(NH2)COOH + Н3РО,
Реакция II О. ц. осуществляется растворимой фер-
ментной системой за счет энергии окислительно-вос-
становительных процессов в митохондриях. Реакция
II состоит из двух этапов: 1) конденсации цитрул-
лина с L-аспарагиновой к-той с образованием арги-
нинянтарной к-ты (реакция требует затраты энергии
и протекает при участии АТФ):
NH II с-он соон NH СООН 11 1 С—NH—СН
NH NH2—СН | + j +АТФ—> (СН2)з СН2 1 1 NH СН2 | | +АДФ + Н,РО. (СН2)3 СООН
chnh2 соон CHNH,
1 соон цитруллин аспарагиновая к-та 1 соон аргининянтарная к-та
2) ферментативного расщепления аргининянтарной
к-ты на аргинин и фумаровую к-ту:
NH II C-NH- 1 соон -Ан NH II С—NHj соон 1
кн 1 (СН2)3 1 сн2 1 соон I NH ^~(СН2)3 сн + II сн
1 chnh2 CHNHa I соон
соон аргининянтарная к-та । СООН аргинин фумаровая к-та
Реакция III представляет собой гидролитич. расщеп-
ление аргинина аргиназой на мочевину и орнитин.
Реакция О. ц. способствует синтезу цитруллина и
аргинина в организме животных. Цитруллин, синте-
зирующийся в печени, переносится током крови к
почкам, где, как и в печени, происходит синтез и ча-
стично гидролиз аргинина. Биологич. значение реак-
ций О. ц. определяется тем, что они являются важней-
шим механизмом обезвреживания NH3 в организме
животных. Установлено, что реакции О. ц. имеют
место не только у высокоорганизованных животных,
но и у грибка Neurospora, Penicillium, бактерий
Escherichia coli и некоторых молочнокислых бакте-
рий. У бактерий аргинин гидролизуется с образова-
нием NH3, СО2 и орнитина не аргиназой, а другой
ферментной системой с промежуточным образованием
цитруллина. Реакции О. ц. лежат в основе био-
синтеза аргинина у дрожжей. Перечисленные факты
являются доказательством общебиологического значе-
ния О. ц.
Лит.: Збарский Б. И., И в а н о в И. И., Мард а-
ш е в С. Р., Биологическая химия, 3 изд., М., 1960, с. 340;
Майстер А., Биохимия аминокислот, пер. с англ., М.,
1961, с. 1 76, 338. И.В.Цветкова.
ОРОТОВАЯ КИСЛОТА (2,6-диоксипиримидин-4-
карбоновая кислота, 4-я карбокси-урацил) C5H4N2O4,
мол. в. 156,1 — слегка кислые на вкус бесцветные
кристаллы, т. пл. 323—345° (с разл.); кристалли-
зуется из воды в виде пластинок ромбич. формы с 1
молекулой воды; при 130° теряет воду. О. к. (I)
растворима в воде (1:550/18°, 1:70/100°), трудно рас-
творима в эфире и др. органич. растворителях, имеет
характерный максимум поглощения УФ-света при
278 ммк (Е * = 420). Карбоксильная группа О. к.
не отщепляется при длительном кипячении с 20 %-ной
H2SO4 даже при 200°; легко образует сложные
эфиры. Количественное определение О. к. производят
микробиологии, методом по ее влиянию на рост
Lactobacillus vulgaris. Строение О. к. подтверждено
ее синтезом из аспарагиновой к-ты.
Основное биологич. значение О. к. и ее производ-
ных состоит в том, что они являются предшественни-
ками при синтезе нуклеозидов пиримидиновых осно-
ваний (цитозина, урацила и тимина). О. к. взаимодей-
ствует в присутствии оротидилпирофосфорилазы с ри-
бозил-5'-пирофосфатом (II), образуя оротидин-5-фос-
фат (III), к-рый под влиянием оротидил-5-фосфат-
781
ОРСИН—ОРТОНА ПЕРЕГРУППИРОВКА
782
декарбоксилазы декарбоксилируется с образованием
уридин-5'-фосфата (IV):
О. к. стимулирует рост животных и растений, при-
меняется при болезнях печени.
Лит.: Чхиквадзе К. А., Магидсон О. Ю.,
Мед. пром-сть СССР, 1960, Ks 10, 24; N у с J. F., М 1 t с h е 1 1
Н. К., J. Amer. Chem. Soc., 1947, 69, 1382; Lieberman I.,
там же, 1955, 77, Л: 9, 2661; Schwi etzer C., Blochem.
Z., 1956, 328, H. 4, 291; Kosti r J. V., Chemie (Praha),
1958, 10 304. Л. А. Вакулова, Г. И. Самохвалов.
ОРСИН (орцин, орсинол, 3,5-диокситолуол,
5-метилрезорцин) C7HSO2, мол. в. 124,13 — бесцветные
кристаллы; т. пл. 107—108°; т. кип. 287—290°; хорошо
растворим в воде, спирте, эфире, ограниченно — в
СвНв; плохо — в СНС1з, лигроине, петролейном
эфире; образует гидрат C7HSO2 -Н2О, т. пл. 56°;
пикрат, т. пл. 92°; дибензоильное производное, т. пл.
88°; окрашивает водные р-ры FeCls в глубокий фиоле-
товый цвет; с СНС1з и КОН дает красное окрашива-
ние, переходящее при разбавлении водой в желтое
с зеленой флуоресценцией. О. (I) может быть получен
из ацетил-п-толуидина рядом последовательных ре-
акций или сухой перегонкой орселлиновой к-ты
(т. пл. 176°); последнюю обычно получают гидролизом
I
нек-рых видов лишайников (напр., Roccella, Lecanora
и др.). Специальной обработкой тех же лишайников
получают орсейль — непрочный краситель, применяв-
шийся ранее для крашения шерсти и шелка, и лак-
мус — широко распространенный индикатор. О.
применяют для качественного определения нитрит-
и нитрат-ионов, нитрозилсерной к-ты, хроматов, для
колориметрич. определения пентоз, пентозанов и
фурфурола; в микроскопии — для открытия инули-
на; для синтеза реактива на Be и др. В. и. Фросин.
ОРТО-, МЕТА-, ПАРА—обозначение трех возмож-
ных изомеров положения в дизамещенных бензола.
Нередко пользуются более краткими обозначениями
о-, л-, п-. В орто-изомерах оба заместителя (одина-
ковые или различные) расположены у соседних атомов
углерода ядра (положения 1,2), в случае мета-соеди-
нений зшестители находятся в положениях 1,3, в
пара-изомерах заместители наиболее удалены друг
от друга (положения 1,4). Примерами могут служить
3 диоксизамещенных бензола:
ОН
пирокатехин
о- диоксибензол
ОН
резорцин гидрохинон
м- диоксибензол п-диоксибензол
Другой смысл имеют приставки орто- и мета-
перед названием кислот, их солей и производных.
Ортоформы кислот содержат максимальное число
ОН-групп при данном центральном атоме. Так, кисло-
ту НзРО4, или ОР(ОН)з, наз. ортофосфор ной, кислоту
НзВОз, или (ОН)зВ,— ортоборной и т. д. Эта же при-
ставка применяется и в случае производных нек-рых
органич. к-т: так, к-ту НС(ОН)з (гидратную форму
муравьиной к-ты НСООН -Н2О) наз. ортомуравьи-
ной к-той, С(ОН)4 (гидратированная угольная к-та)—
ортоугольной к-той; обе эти кислоты устойчивы в
виде эфиров. Метаформы кислот — это дегидратиро-
ванные ортокислоты, напр. метафосфорная к-та
НРОз(НзРО4 — НгО), метаборная к-та НВОг и т. д.
Приставка пара- иногда применяется для обозначе-
ния продуктов полимеризации, например парациан
(CN)n, паральдегид — полимерный уксусный альде-
гид (СНзСН0)„.
Приставки орто- и пара- применяются также для
обозначения двух модификаций водорода. Орто- и
параводород обладают одинаковыми химич. свойства-
ми, но их физич. свойства (уд. теплоемкость, темп-ры
плавления и кипения и т. д.) несколько различаются.
ОРТОНА ПЕРЕГРУППИРОВКА ^-Ф’превращенйе
М-галоген-И-ацилариламинов в о- и п-галогеноза-
мещенные ариламиды карбоновых к-т:
COR
NHCOR
О. п. проводят при освещении, нагревании (лучше
в полярных растворителях) и наиболее часто при
действии кислот на И-галоген-И-ацилариламины.
Помимо наиболее изученных N-галогенацетанилидов,
О. п. описана для N-галогеноформанилидов, N-гало-
генобензанилидов, замещенных в ядре N-галоген-
ацетанилидов, N-хлорнафтостирила и т. п. Накопле-
ние атомов галогена в ядре Н-галоген-И-ациларил-
амина затрудняет О. п. Н,Н-Дихлорариламины, на-
против, перегруппировываются очень легко уже при
комнатной темп-ре (в эфирном р-ре).
О. п. в большинстве случаев не является истинной
внутримолекулярной перегруппировкой и протекает
в две стадии: а) ацидолиз Н-галоген-И-ацилариламина
в N-ацилариламин и галоген (при катализе галогено-
водородными к-тами); б) обычное галогенирование
N-ацилариламина в ядро:
+ НС1
CeH3NClCOCH3----ь CeHr.NHCOCH3 + Cl3 —ь
(а) (б)
—>CiCoH<NHCOCH3 + HCI
Такое направление реакции подтверждается образо-
ванием бром- или иодацетанилида при обработке
N-хлорацетанилида бромистоводородной или иодисто-
водородной к-той, а также получением радиоактив-
ного хлорацетанилида из N-хлорацетанилида и мече-
ного хлористого водорода:
НВг НС1*
BrCeH,NHCOCH34-----C„H5NClCOCHa-----►
—bCICsH.NHCOCHj
783
ОРТОЭФИРЫ —ОСАЖДЕНИЕ
784
Межмолекулярный характер О. п. подтверждается
также образованием галогенанизола при перегруппи-
ровке N-галогенацетанилидов в присутствии анизола.
При О. п. в неводных растворителях в присутствии
карбоновых к-т (хлор- или дихлоруксусная, фенилук-
сусная, о- или .и-нитробензойная) галогенирующими
агентами являются первоначально образующиеся
аци лгипогалогениты:
C»H5NXCOCH3 + RCOOH —<-C6HsNHC0CH3 + RC00X —>
—XCeHjNHCOCHj + RCOOH
О. п. открыта Г. Бендером в 1886 и подробно изу-
чена К. Ортоном начиная с 1899.
Лит.: Houben-Weyl, 4 Aufl., Bd И, TI i, Stutt-
gart, 1957, S. 827; Ингольд К. К., Механизм реакций п
строение органических соединений, пер. с англ., М., 1959,
С. 488. Н. II. Гамбарян.
ОРТОЭФИРЫ карбоновых кислот —
класс производных гидратной формы карбоновых к-т
ИС(ОН)з; известны как в жирном, так и в арома-
тич. ряду. В отличие от ортокарбоновых к-т, не из-
вестных в свободном виде, О.— вполне устойчивые сое-
динения; простейшие из них — легко перегоняющие-
ся жидкости с эфирным запахом, растворимые в орга-
нич. растворителях; в воде практически нерастворимы.
Физич. свойства нек-рых О. приведены в таблице.
Формула Т. кип., °C 20 di 20 Примечание
первичный пропило-
вый
вторичный пропило-
вый
т. пл. 76—7°
D. ; nD
» »
20
D
4
НС (OCHA, НС (OC2H5)S НС (0С3Н,)3 10 2—3 145-7 196—8 0,9676 0,8909 0,8805 1,3793 1,3922 1,4072
НС (ОС3Н7)а 106—8 0,8621 1,4000
НС (0С6Н3)3 269—70/50 (с разл.) — —
СН3С (0СН,)3 107—9 0,94375 1 , 38585
СН.С (ОС.Нг,)з 14 4—6 0,8847 1,39465
СН,СОС2НГ, (ОС3Н7)2 190—4 0,87129 1,40635
С2Н3С (ОС2Н..)3 159—60 — —
С.,Н5С (0С2Н3)3 238—40/7 47 .м.ч 0,9902 —
Вода и водные р-ры щелочей не омыляют О., разб.
к-ты легко гидролизуют (аналогия с ацеталями и
кеталями)',
рН<7
RC (OR')3 + 2H2O---> RC00H + 3R'0H
Самым существенным свойством О., широко исполь-
зуемым в препаративной химии, является их склон-
ность к реакциям обмена эфирных групп; так, различ-
ными реакциями с пх помощью можно получать аце-
тали альдегидов и кетонов (а следовательно, и сами
альдегиды и кетоны). 1) Реакцией с магнийгалогенор-
ганич. соединениями (Чичибабин, Бодру):
НС (OC2H.,)s + CH3MgJ —>СН3СН (OC2He)2 + C2H3OMgJ
2) Восстановлением:
0,25MLiAlH.
СН3С (ОСН3)3-----------^СН3СН (ОСН3)2 + СН3ОН
3) Действием на альдегиды и в особенности кетоны,
для к-рых этот способ является главным (Клайзен,
Арбузов):
(СН3)2С=.О+НС (ОС2Н,)3—> (СН3)2 С (ОС2Н;)2 + НСООС2Н3
О. легко конденсируются с соединениями, содержа-
щими реакционноспособные метиленовые группы,
напр.:
НС (ОС2Н-)3 + СН2 (СООС2Н,.)2 —>
--*С2Н3ОСН = С (С00С2Н5)2+ 2C2HsOH
Способы получения О. 1) Реакция между алкоголя-
тами и хлороформом или аналогичными по строению
тригалогенопроизводными;
CHCl3 + 3NaOC2H5 —>СН (ОС2Н3)3 +3NaCl
2) Взаимодействие между хлористоводородными со-
лями иминоэфиров карбоновых к-т и спиртами:
+
,nh2ci~
2С2Н>ОН + СН3С<^ —>СН3С (OC2H5)3+NH,C1
хОСоН5
3) Действие магнийгалогенорганич. соединений на
ортоугольные эфиры (Чичибабин):
С (OC2H3), + C2H3MgJ —>С2Н5С (OC2Hs)3 + C2H3OMgJ
Чаще всего в лабораторных синтезах используют
этиловый эфир ортомуравьиной к-ты, т. наз. орто-
муравьиный эфир. Р. Р- Галле.
ОСАЖДЕНИЕ — выделение одного или нескольких
компонентов в виде малорастворимого соединения. О.
принадлежит к наиболее распространенным методам
разделения элементов в анализе и в химич. техноло-
гии. На образовании осадков основано много мето-
дов качественного анализа как для обнаружения от-
дельных элементов, так и для разделения групп
элементов. О. является основой большей части мето-
дов весового анализа. В физич. и химич. методах
анализа также нередко применяют О. для отделения
от мешающих элементов или для получения «ана-
литических концентратов». Кроме того, процессы О.
важны для получения чистых ве-
ществ, для процессов образования
накипи, в геохимии и др. областях.
Катионы металлов обычно осаждают
различными анионами и наоборот.
Реже используют реакции окисле-
ния (напр., О. двуокиси марганца)
или комплексообразования. Ряд ме-
таллов и нек-рые анионы выделяют
также электроосаждением.
В химич. отношении осадки мож-
но разделить на след, группы: 1.
Соли сильных к-т. Как пра-
вило, соли простых сильных к-т ра-
створимы в воде; известны исключе-
ния изэтого правила. Нерастворимые
соединения широко применяют для
О.: сульфаты бария, стронция, свинца, одновалентной
ртути, хлориды серебра, одновалентной ртути и нек-рые
другие. О. этих веществ обычно ведут в кислой сре-
де, т. к. Н + -ионы пе связывают осаждающий анион
в молекулу, тогда как присутствующие анионы слабых
к-т при этом обычно не реагируют с ионами металлов.
Соли многих сильных к-т комплексного характера,
напр. ферроцианиды, принадлежат к этой же группе,
т. е. малорастворимы в воде и в кислотах. 2. Соли
слабых к-т, как правило, малорастворимы в
воде; поэтому наибольшее количество осадков принад-
лежит к данной группе. Однако по этой же причине
реакции менее избирательны и для разделения элемен-
тов требуются определенные условия, прежде всего
pH среды. Большинство солей слабых к-т не осажда-
ется в сильнокислой среде, т. к. Н + -ион связывает
осаждающий анион в молекулу малодиссоциирующей
к-ты. Нек-рые соли слабых к-т осаждаются также и в
сильнокислой среде, напр. сульфиды меди, ртути и
др. Последнее обусловлено тем, что иногда связь
Ме"+— А"1-более прочна, чем связь Н + — А"1-. Ши-
роко применяют О. сульфидов металлов, карбонатов,
фосфатов, а также О. металлов многочисленными
органич. реагентами. Последние применяют также
для О. нек-рых анионов, напр. О. сульфатов бензиди-
ном и др. Ряд органич. осадителей, имеющих характер
слабых к-т, образует со многими катионами внутри-
комплексные соединения. Для последних, а также для
сульфидов металлов и нек-рых других соединений
характерны значптельные различия растворимости
солей разных металлов. Эти случаи особенно удобны
785
ОСАЖДЕНИЕ —ОСВЕТИТЕЛЬНЫЕ СОСТАВЫ
786
для разделения сложных смесей, т. к. О. легко регу-
лировать, напр., создавая определенное значение pH
раствора. Кроме того, мешающие компоненты можно
связать в растворимые в воде комплексные соедине-
ния (маскировка). 3. Свободные к-ты такие,
как кремневая, оловянная, вольфрамовая, а также
гидроокиси многих металлов. 4. Осадки, обра-
зующиеся при взаимодействии трех компонентов.
Для О. металлов их переводят иногда в комплек-
сные анионы, напр. BiJ4 , а затем вводят катион
органич. основания R+, причем осаждается RBi.T4.
Аналогично осаждают, нанр., фосфат в виде солей
фосфорномолибденовой к-ты и др.
Разделение элементов путем О. нередко осложняет-
ся соосаждением. В зависимости от формы частиц
различают два типа осадков: кристаллич. и аморфные.
Процесс образования частиц осадка довольно сложен.
Прибавление первых порций осадителя после достиже-
ния предела растворимости приводит к образованию
центров кристаллизации. Расчеты на основании кине-
тич. исследований приводят к выводу, что эти центры
состоят из 5—8 ионов. Затем образуются первичные
кристаллы размером 10~4— 10-5 см. Дальнейший
процесс в зависимости от условий идет различным
путем для разных веществ. Для более растворимых
соединений обычно характерно образование пересы-
щенных р-ров; новые центры кристаллизации обра-
зуются медленно, поэтому дальнейшее прибавление
осадителя приводит к росту ранее образовавшихся
центров, без увеличения числа частиц. Электронно-
микроскопич. исследования показывают, что при этом
сильно разветвленные первичные кристаллы (тип
«снежинки») приобретают более совершенную форму.
Медленное приливание осадителя, а также нек-рые
специальные приемы (см. Возникающие реагенты)
позволяют еще более замедлить процесс и получить
более крупные кристаллы. Для менее растворимых
соединений часто центры кристаллизации образуются
быстро, поэтому прибавление каждой порции осади-
теля приводит к образованию новых частиц. Однако,
особенно в случае гидрофобных осадков (как сульфи-
ды),новые частицы соединяются с ранее образованными
в более крупные, хотя и не имеющие кристаллич.
формы, получается аморфный осадок. Общее число
частиц (при том же общем количестве вещества) при-
близительно одинаково для кристаллич. и для аморф-
ных осадков. Аморфные осадки характеризуются
сильно развитой поверхностью.
Аморфные и кристаллич. осадки требуют различ-
ной техники О.,фильтрования и промывания.Кристал-
лич. осадки обычно получают в форме более крупных
частиц при медленном О. из горячих разб. р-ров;
многие кристаллич. осадки заметно растворимы в
воде, поэтому их обычно промывают не водой, а раство-
ром избытка осадителя. Кристаллич. осадки часто
дают пересыщенные р-ры, поэтому для полного осаж-
дения их необходимо нек-рое время. Аморфные осад-
ки бывают гидрофобные (напр., сульфиды металлов)
и гидрофильные (напр., кремнекислота, гидроокись
алюминия и др.). Для последних получаются более
плотные и чистые осадки, если О. ведут из конц. и
горячих р-ров. Аморфные осадки при промывании
склонны переходить в раствор вследствие образова-
ния коллоидной суспензии (пептизация). Для устра-
нения этого осадки необходимо промывать р-ром элек-
тролита, напр. нитрата аммония.
Зная произведение растворимости (ПР) во многих
случаях можно рассчитывать оптимальные условия
О.: необходимый избыток осадителя, pH раствора,
допустимые количества посторонних ионов и др.
Принцип ПР применим для многих осадков, исполь-
зуемых в количественном анализе. Однако в общем
AsO3H3
СуХмнСОСНз
он
получают 4-ок-
для малорастворимых соединений процессы нередко
значительно усложняются. Наиболее часто усложне-
ния обусловлены образованием растворимых комплек-
сов при действии избытка осадителя. При этом, вме-
сто понижения растворимости (как это следует из
принципа ПР), наблюдается увеличение раствори-
мости. Так, в избытке осадителя заметно раство-
ряется хлорид серебра, оксалаты лантанидов, легко
растворяется и поэтому вовсе не применим для оса-
ждения иодид висмута и др.
Кроме обычных процессов разделения или выделе-
ния ионов фильтрованием или центрифугированием,
О. применяется в аналитич. химии в других формах.
Многие осадки, образованные катионами металлов
с органич. реагентами, способны растворяться в не-
смсшивающихся с водой органич. жидкостях. В этих
случаях О. иногда совмещают с экстракцией. На
основе образования осадков и измерения интенсив-
ности помутнения или рассеивания света основаны
методы нефелометрии и турбидиметрии, а также
метод «гетерометрического» титрования, заключаю-
щийся в установлении точки эквивалентности по
изменению помутнения системы при постепенном
прибавлении титрующего р-ра.
Лит.: Бабко А. К., П я т н и ц к и й И. В., Количест-
венный анализ, 2 изд., М., 1962. А. К. Бабко.
ОСАРСОЛ (стоварсол, З-ацетамино-4-оксифенил-
арсиновая кислота) C8Hi0NO6As, мол. в. 275,09 —
бесцветные кристаллы; плохо раство-
рим в воде и органич. растворителях,
легко растворяется в щелочах и р-ре
соды с образованием солей. Наиболее
простой способ получения О. основан
на реакции Барта, исходя из 4-амино-
фенола: действием солей 4-оксифенил-
диазония на соли мышьяковистой к-ты
сифениларсиновую к-ту, из к-рой нитрованием с пос-
ледующим восстановлением получают З-амино-4-окси-
фениларсиновую к-ту. Ацетилирование этой к-ты
лучше всего производить уксусным ангидридом в вод-
ном р-ре КааБгОз; при этом сразу получается чистый
О. (выход 90%). О. применяют при лечении сифилиса,
трихомонадных кальцитов и др., а также в ветери-
нарии. М. я. Крафт.
ОСВЕТИТЕЛЬНЫЕ СОСТАВЫ — пиротехнич. со-
ставы (см. Пиротехника), применяемые для снаряже-
ния изделий, предназначенных для освещения мест-
ности (осветительные авиабомбы, ракеты, снаряды
и др.). О. с. состоят из горючего (обычно порошок
магния или алюминия, а также их сплавы или смеси),
окислителя (чаще всего нитраты Na или Ва) и нек-рых
органич. веществ, используемых в качестве связую-
щего, для увеличения пластичности, замедления
горения и др. (смолы, парафин, стеарин и др.).
Примером могут служить след, рецептуры О. с.:
1) 36% Ba(NO3)2, 6% Sr(NO3)2, 54% Mg и 4% пара-
фина; 2) 48% NaNOs, 45% Mg и 7% связующих орга-
нич. веществ; в нек-рых О. с. может содержаться до
70% Mg. Теплота горения современных О. с. 1500
ккал/кг, темп-ра горения ~2500—3000°.
О. с. изготовляют в специальных смесителях;
перед смешением исходные материалы измельчают,
сушат, просеивают; готовую смесь прессуют. При
этом получаются цилиндрич. изделия, запрессован-
ные в картонные или металлич. оболочки (факелы)
диаметром 20—500 мм. При горении изделий обра-
зуется яркое (тысячи стильбов) белое или желтовато-
белое пламя. Сила света пламени зависит от диаметра
изделия, темп-ры и скорости горения О. с., а также
от свойств продуктов горения; она колеблется в пре-
делах 104—107 международных свечей; время
горения факелов колеблется от 8 сек до 8 мин. Ско-
рость горения спрессованных О. с. 0,5—10 мм/сек}
787
ОСКОЛКИ ДЕЛЕНИЯ
788
при понижении давления (горение на больших высотах)
скорость горения и свечение пламени О. с. умень-
шаются. О. с. часто характеризуют значением уд. свето-
суммы (Lo)', для современных О. с. Lo 40-10’
сек-свеч/г.
Излучение пламени О. с. гл. обр. температурное и
частично люминесцентное. Температурное излучение
обусловлено наличием в пламени О. с. накаленных
твердых и жидких частиц (MgO, А120з, ВаО и др.)
и быстро возрастает с повышением темп-ры пламени.
На О. с. по своим свойствам весьма похожи трасси-
рующие составы и фотосмеси; последние используют
для снаряжения авиафотобомб (фотаб), применяемых
при ночных аэрофотосъемках; сила света фотаб до-
стигает сотен миллионов свечей, время свечения до
0,1 сек.
Лит.: Шидловский А. А., Основы пиротехники, 2
изд., М., 1954; Y Symposium on combustion, N. Y.—L., 1955, p.
267; Tavernier P., Mem. poudres, 1949, 31, 309; E 1 1 e r n
H., Modern pyrotechnics, N. Y., 1961; Cassel H. M., L i-
e b m a n I., Combustion and ’flame, 1962, 6, № 3, 153; 1963,
№ 1, 79. А. А. Шидловский.
ОСКОЛКИ ДЕЛЕНИЯ — радиоактивные изотопы,
образующиеся при делении тяжелых элементов (см.
Ядро атомное). Целью анализа таких изотопов являет-
ся их идентификация, изучение их ядерных свойств
и определение их выходов. Выяснение того, какие
именно ядра и в каких количествах образуются в
ходе реакции деления, имеет существенное значение
как для теории деления атомных ядер, так и для
ядерной технологии. Эта проблема решается в основ-
ном методами радиохимии. Приемы радиохимия,
анализа О. д. были разработаны в 40-х гг. 20 в. неза-
висимо в СССР и США.
При делении урана и плутония нейтронами не
слитком высоких энергий (до 15 М&в) образуется
сложная смесь радиоактивных изотопов элементов
периодич. системы от Zn до Gd с материалом мишени
(U или Ри). Большинство О. д. представляет собой
нейтроноизбыточные изотопы, распадающиеся путем
испускания Р “-частиц, образуя довольно длинные изо-
барные цепочки, напр.:
ВГ9°------*К1Гв--------------------” Sr3 8-----
35 16 сек. 36 33 сек. 37 2,74 мин. 38 27,7лет 39
________Zr9U
64,2 часа 10
(стаб.)
Изменение изотопного состава смеси во времени, а
также непрерывное накопление дочерних веществ
в выделяемых фракциях должно самым тщательным
образом учитываться при планировании и проведе-
нии эксперимента.
При изучении выхода какого-либо изотопа — про-
дукта деления — определяют количество его атомов
в смеси О. д., для чего измеряют его абс. активность.
Т. обр., необходимо произвести количественное выде-
ление исследуемого изотопа из смеси О. д., содержа-
щейся в растворе, полученном при растворении облу-
ченной мишени, или, по крайней мере, знать, какая
доля атомов этого изотопа выделена и попала в пре-
парат, поступивший для измерений активности.
В обычных опытах образуются ничтожные количества
О. д. (10“12—10“14г), находящихся в растворе веще-
ства мишени. Работа с такими малыми количествами
осколочных элементов весьма сложна. Во-первых,
нельзя производить выделение исследуемых элемен-
тов путем осаждения их труднорастворимых соеди-
нений, как это обычно делается в аналитич. практике.
Далее, в ходе работы возможны неконтролируемые
потери радиоактивных веществ за счет адсорбции на
стенках посуды и пылевых загрязнениях, образова-
ния микроколлоидов и т. д. Поэтому радиохимии,
анализ обычно проводится с применением носителей
(как правило, изотопных).
Главное условие, к-рое необходимо соблюдать при
использовании метода носителей,— это достижение
полного изотопного обмена между введенным носи-
телем и данным изотопом в р-ре мишени. При соблю-
дении этого условия радиоактивные атомы и атомы
носителя при всех химич. операциях будут вести себя
одинаковым образом и их потери в ходе выделения бу-
дут одинаковы, так что если в осадок перешло, напр.,
80% введенного носителя, то в осадке будет нахо-
диться и 80% радиоактивного изотопа (изотопов)
того же элемента. Поэтому, если соблюдено условие
достижения полного изотопного обмена, то отпадает
необходимость 100%-го выделения изучаемого эле-
мента. Это весьма существенно, т. к. необходимость
тщательной очистки препаратов осколочных элемен-
тов от всех других радиоактивных изотопов и связан-
ные с этим многочисленные очистительные операции
часто приводят к большим потерям вещества.
В большинстве случаев полнота изотопного обмена
легко достигается, если носители добавляются в виде
азотнокислых или солянокислых солей соответствую-
щих элементов (большинство осколочных элементов
является металлами). При выделении циркония можно
достигнуть полного изотопного обмена добавлением
к раствору плавиковой к-ты (при этом все соедине-
ния циркония переходят в ZrF2-), а при выделении
молибдена — кипячением раствора с азотной к-той
(что сопровождается образованием молибденовой
к-ты). Сложнее обстоит дело, если выделяемый эле-
мент может находиться в нескольких валентных
состояниях (J, Вг, Ru). В этих случаях после добавле-
ния носителя проводят несколько окислительно-
восстановительных циклов. Так, при выделении иода
носитель, добавленный в виде иона J", окисляется
с помощью NaClOj. Образовавшийся при этом J04
восстанавливается сульфитом до .(“-иона, к-рый оки-
сляется до элементарного иода с помощью NaNO2.
Элементарный иод экстрагируют СС14.
Обычно раствор облученной мишени разделяют на
несколько аликвотных частей, и из каждой части
производят выделение только одного элемента. В соот-
ветствии с этим к каждой аликвоте добавляют изве-
стное количество (от 2 до 100 мг) носителя выделяемого
элемента. После этого производится первичное выде-
ление этого элемента из исходной смеси путем оса-
ждения его труднорастворимого соединения (напр.,
сульфата — при выделении бария, или фосфата —
при выделении циркония, хлористого серебра —
при изучении серебра и т. д.) либо путем экстракции
(так, иод экстрагируется СС14, железо и галлий —
эфиром из солянокислых растворов), или возгонки
(рутений отгоняют после окисления в виде RuO4,
при этом происходит полный изотопный обмен радио-
активных изотопов рутения с добавленным перед
окислением носителем), а также путем электролитич.
выделения (Ag, Ро).
Однако одна такая операция, какой бы специфич-
ной она ни была, еще не дает требуемой степени
очистки вследствие захвата маточного р-ра осадком,
явлений соосаждения и адсорбции на поверхности
осадка, неполного разделения фаз при экстракции
и других причин. Поэтому приходится применять
очистку выделенного элемента от всех возможных
радиоактивных загрязнений. Очистка состоит из
многократно повторяющихся операций. Во-первых,
можно снова производить отделение интересующего
элемента с помощью различных специфич. реакций,
в т. ч. и той, к-рая применялась для первичного
выделения. Далее, широко используются операции,
при к-рых примеси захватываются к.-л. осадком,
а очищаемый элемент остается в растворе. При этом
используются такие реакции, к-рые дают возможность
789
ОСКОЛКИ ДЕЛЕНИЯ —ОСЛАБЛЕНИЕ ФОТОГРАФИЧЕСКОЕ
790
сразу освободиться не от одной, а от большого числа
примесей. Так, широко применяют осаждение гидро-
окиси железа (к-рую предварительно добавляют к
раствору в пробирке в количестве 5—10 мг) аммиа-
ком или щелочью, осаждение сульфидов серебра
или сурьмы в кислой среде и т. д. При этом происхо-
дит захват осадками микроколичеств не только тех
элементов, к-рые образуют нерастворимые гидрооки-
си, сульфиды и т. д., но и многих других. Так, при
осаждении аммиаком осадок гидроокиси железа почти
нацело захватывает микроколичества молибдена. Для
предотвращения соосаждения микроколичеств при-
месей с осадком анализируемого элемента часто перед
осаждением в раствор добавляются макроколичества
этих примесей (удерживающие носители, см. А дсорб-
ция радиоактивных элементов). Напр., при очистке
бария от стронция в раствор добавляют небольшое
количество Sr и производят осаждение хромата бария
в уксуснокислой среде, причем Sr остается в растворе.
Обычно вся работа проводится в центрифужных
пробирках на 15—100 мл с использованием мини-
мальных объемов растворов. Осадки отделяются от
раствора центрифугированием. Конечной стадией
радиохимии, анализа является выделение изучаемого
элемента в форме, удобной для взвешивания (или
легко переводимой в эту форму путем, напр., про-
каливания). Этот препарат поступает для измерения
активности содержащихся в нем радиоактивных изо-
топов (см. Радиоактивности измерения). Отношение
количества выделенного к количеству добавленного
носителя наз. в радиохимии химическим вы-
ходом. Разделив абсолютную активность изотопа
в полученном препарате на химич. выход, мы узнаем
его активность в исходном р-ре.
Для количественного выражения степени очистки
вводится понятие о коэфф, очистки, к-рый показы-
вает, во сколько раз активность загрязнений в выде-
ленном препарате меньше, чем в исходной смеси.
Опыт показывает, что химич. выход должен быть не
менее 50—70%, а коэфф, очистки — не менее 105.
С учетом этих требований и разрабатываются мето-
дики выделения отдельных осколочных элементов.
При отработке методик поступают следующим обра-
зом. Если, напр., разрабатывается методика выделе-
ния Ва14“, то к раствору неактивного нитрата бария
добавляются радиоактивные изотопы тех элементов,
к-рые присутствуют в реальной смеси О. д., а также
и другие необходимые компоненты (соли урана и т. д.).
После проведения первичного выделения и намечен-
ных очистительных операций измеряют активность
полученного препарата бария. Операции повторяются
(с необходимыми видоизменениями) до тех пор, пока
не будет достигнута желаемая степень очистки.
В ряде случаев (напр., при анализе образцов почв)
в исследуемом растворе могут присутствовать в макро-
количествах разнообразные неактивные загрязне-
ния (Si, Са, Fe, Ва и т. д.), что необходимо иметь в
виду при разработке хода анализа. Нужно также
тщательно контролировать возможность загрязне-
ния выделенных препаратов неактивными примеся-
ми. Присутствие макропримесей необходимо также
учитывать при определении химич. выхода. Так,
если в растворе присутствует неактивный барий, его
количество должно быть определено с достаточной
точностью, чтобы в дальнейшем можно было ввести
соответствующую поправку при расчете химич. вы-
хода бария. Если активность исследуемого образца
невелика, то не производят разделения раствора про-
бы на аликвотные части. Методика строится на после-
довательном выделении отдельных элементов из всего
объема раствора. Все последующие очистительные
операции производят обычным образом, как описано
выше.
Для выделения О. д. все шире применяют хрома-
тография. и экстракционные методы, позволяющие
ускорить процесс и повысить степень очистки. Все
описанные приемы с успехом используются при изу-
чении и других ядерных реакций, в ходе к-рых возни-
кают сложные смеси радиоактивных элементов (ре-
акции высокоэнергетич. деления, глубокого отщеп-
ления под действием частиц больших энергий, реак-
ции с многозарядными ионами и др.).
Лит.: Радиохимический анализ продуктов деления. Сб.
статей, [под ред. Ю. М. Толмачева], М., i960; Радиохимия и
химия ядерных процессов, под ред. А. Н. Мурина [и др.],
Л., 1960, гл. 15, 16, 17; Radiochemical studies: Ttie fission pro-
ducts, book 1—3, N, Y., 1951. В. И. Барановский.
ОСЛАБЛЕНИЕ ФОТОГРАФИЧЕСКОЕ—понижение
оптич. плотностей (D) проявленных негативных или
позитивных фотография, изображений; применяется
для исправления преимущественно перепроявленных
и (менее успешно) передержанных материалов. По-
нижение D достигается удалением части металлич.
серебра изображения из фотография, слоя путем его
окисления и перевода в растворимые соединения,
легко вымываемые из фотография, слоя. При О. ф.
понижение D возможно: 1) без изменения коэфф, кон-
трастности у; 2) с уменьшением у; 3) с повышением у.
Ослабляющие растворы (ослабители) в зависимо-
сти от их действия разделяют на: ^пропорцио-
нальные, к-рые удаляют серебро в количестве,
пропорциональном его содержанию в отдельных уча-
стках слоя, и, следовательно, помимо общего умень-
шения D, отчасти снижают также и у; 2) с в е р х-
пропорциональные — удаляют серебро с
участков высокой плотности в большей степени, чем
с участков малой плотности и используются для силь-
ного понижения у; 3) субпропорциональ-
ные — действуют гл. обр. на участки малой D и
повышают у.
Эффект действия ослабителя зависит не только от
природы окисляющего вещества, но и от условий
применения: в первую очередь от концентрации оки-
слителя и pH среды. Один из наиболее распростра-
ненных ослабителей — феррицианид тиосульфатный
(ослабитель Фармера) — в сильно разбавленном виде
действует как пропорциональный ослабитель (осо-
бенно при использовании феррицианида калия и
тиосульфата натрия в раздельных р-рах); обычные же
его составы имеют субпропорциональное действие.
Крупность и распределение зерен серебра также влия-
ют на характер действия ослабителя. Так, при широ-
ком диапазоне размеров зерен ослабитель Фармера
действует гл. обр. на участки малой D. Примеси
нек-рых ионов влияют не только на эффект О. ф., но
и на скорость процесса. Так, типичный сверхпропор-
цпональный ослабитель — персульфат аммония — в
присутствии серной к-ты при повышении концентра-
ции ионов серебра действует как пропорциональный.
Химические реакции О. ф. еще не вполне изучены.
Перманганат калия в слабокислой среде является
одним из распространенных пропорциональных осла-
бителей. В присутствии серной к-ты КМпО4 превра-
щает серебро изображения в его растворимый суль-
фат:
10Ag + 2KMnO, + 8H2SO, = 5Ag2SO, +K2SO, + 2MnSO, + 8Н2О
В случае ослабителя Фармера образующийся сна-
чала нерастворимый ферроцианид серебра перево-
дится с помощью тиосульфата натрия в раствор:
a) 4Ag + 4K,Fe(CN)«=Ag,Fe (CN)» + 3K,Fe (CN)»
б) Ag.Fe (CN)» + 8Na2S2O3=4Na3Ag (S2O3)2 + Na,Fe (CN),
При персульфатном ослаблении происходит след,
реакция:
2Ag + (NH,)2S2Oa = Ag2SO, + (NH,)2 SO,
791
ОСМИЙ
792
Однако при этом наблюдается промежуточное обра-
зование окислов серебра, участие к-рых не отражено
в приведенном ур-нии; ионы водорода и серебра
ускоряют реакцию.
Известно много ослабителей, в состав к-рых вхо-
дят и другие окислители (иод, парабензохинон, соли
четырехвалентного церия и др.). Нек-рые из них
(напр., бихромат калия) могут быть использованы
также и прп усилении фотографическом, несмотря
на то, что этот процесс имеет прямо противоположное
назначение (повышение D). Это связано с тем, что
оба процесса (ослабление и усиление) проводятся
каждый в две стадии: 1) окисление металлич. серебра
с образованием нерастворимой его хромовой соли
(в фотографии, практике эта стадия называется
отбеливанием); 2) в случае ослабления —
удаление серебра в фиксирующем р-ре (иногда после
предварительного частичного проявления); в случае
усиления — полное проявление (чернение) отбелен-
ного изображения, в результате чего выделяется
металлич. серебро, а также его нерастворимые хро-
мовые соединения, что повышает D.
В цветной фотографии ослабляющие р-ры применя-
ют в стадии обработки экспонированной пленки для
полного удаления серебра из фотография, слоя (изо-
бражение создается только за счет красителей).
Специальный вид О. ф. представляет собой частич-
ное обесцвечивание красителей в многослойных плен-
ках, используемых в цветной фотографии, что приме-
няется для частичного ослабления нежелатель-
ного оттенка или понижения общей плотности цвет-
ного изображения (подробнее см. Цветная фото-
гра фия).
Ни один из способов О. ф. не может дать резуль-
татов, вполне равноценных тем, к-рые достигаются
правильным экспонированием и проявлением.
Лит.: Н е б л и т К, Б., Фотография, ее материалы и про-
цессы, пер. с англ., М., 1958; Миз К., Теория фотографиче-
ского процесса, пер. с англ., М.—Л., 1949. Р. Р. Галле.
ОСМИЙ (Osminin) Os — химич. элемент VIII гр.
периодич. системы Менделеева, принадлежит к пла-
тиновым металлам; п. н. 76, ат. в. 190,2. Природный
О. состоит из семи стабильных изотопов: Os184
(0,018%), Os186 (1,592%), Os’8’ (1,64%), Os'88 (13,3%),
Os189 (16,1%), Os190 ( 26,4%), Os192 (41,0%). Из искус-
ственно радиоактивных изотопов О. наиболее долго-
живущий Os194 700 дней). Поперечное сечение
поглощения тепловых нейтронов атомом 15,3 ±0,7
барн. Конфигурация внешних электронов атома
5d’6№. Энергии ионизации (в эв): Os° Os+ -+ Os2 +
соответственно равны 8,7; 17.
О. был открыт в 1804 С. Теннантом в остатке
после растворения платины в царской водке. Назва-
ние «осмий» происходит от греч. ocrpiv; — запах,
и было дано из-за характерного запаха летучего осмие-
вого ангидрида OsO4, напоминающего запах хлора;
этим запахом обладает также измельченный О. при
окислении на воздухе. Содержание О. в земной коре
5 -10 -6 вес. % .О. встречается в виде минералов группы
осмистого иридия, иногда вместе с самородной пла-
тиной (о минералах этой группы см. Иридий). Мине-
ралы группы осмистого иридия встречаются в корен-
ных и россыпных месторождениях платины и золота—
на Урале и в Сибири (СССР), в Колумбии, Калифор-
нии, на Аляске (США), в Южной Африке, Канаде.
Физические и химические свойства. О.— оловянно-
белый с серо-голубым оттенком металл, самый тяже-
лый из всех известных металлов, очень твердый, хруп-
кий, поддающийся растиранию в порошок. Имеет гек-
сагональную плотноупакованную решетку с периодами
а = 2,7353 А, с = 4,3188 А; плотн. 22,5 (20°). Дру-
гие аллотропные модификации О. неизвестны. Ат.
радиус 1,35 А, ионные радиусы Os4+ 0,75 A, Os8 +
0,539 А. Т. пл. 3000°, т. кип. 5500° (вероятно). Уд.
теплоемкость 0,0309 кал/г-град (0°); термич. коэфф,
линейного расширения 4,6 •10-6 (50°); уд. электро-
сопротивление 9,5 мком -см (0°); термич. коэфф,
электросопротивления 0,0042 (0—100°); поверхно-
стное натяжение в точке плавления 2450 эрг/смг.
О. наименее парамагнитен из металлов платиновой
группы; его атомная магнитная восприимчивость
при комнатной темп-ре равна 9,5-10-в. Модуль нор-
мальной упругости для отожженного металла 58 000
кГ/мм2 (при комнатной темп-ре); твердость по Моосу
7,0, по Бринеллю 400 кГ/мм2.
По химич. свойствам О. резко отличается от дру-
гих платиновых металлов, проявляя иногда свойства
неметалла. При нагревании на воздухе О. сгорает
в OsO4. О. устойчив к действию бескислородных
кислот при комнатной темп-ре, но уступает рутению
по коррозионной стойкости. Конц. HNOs окисляет О.
до OsO4, что также нехарактерно для других плати-
новых металлов. Порошок О. растворяется в царской
водке, в растворах гипохлоритов щелочных металлов.
При сплавлении О. с Na2O2 пли со смесью KNO3+
4- КОН образуются соответствующие соли неустой-
чивой в свободном состоянии осмиевой к-ты H2OsO4—
о с м иа ты, к-рые после обработки хлором или HNO3
переходят в Os04. О. поглощает водород. По разно-
образию валентных состояний (0, 2, 3, 4, 6 и 8) О.
напоминает рутений, но для него более устойчивы
соединения с высокой валентностью. Наиболее часто
проявляются валентности +4 и -±6. Все соединения
О. восстанавливаются до металла при нагревании в
токе водорода.
Известны о к и с л ы 2-, 4- и 8-валентного О.;
моноокись осмия OsO — черный порошок, нераство-
римый в воде, имеет структуру пирита. Получается
при нагревании О. в ограниченном количестве кисло-
рода в закрытом сосуде. Двуокись осмия OsO2 —
коричневый или черный порошок, нерастворимый в
воде, имеет тетрагональную структуру типа SnO2;
плотн. 7,91 (22°); разлагается при 650°. С соляной
к-той OsO2 реагирует по обратимой реакции: OsO2+
-±6НС1Д;Н2 [OsClgJ -± 2Н2О. Получают OsO2 при
нагревании О. в ограниченном количестве кислорода,
в NO или в парах OsO4, а также при всс<тановлении
OsO4, при обработке K2[OsCle] карбонатом натрия.
Черная гидратированная двуокись OsO2 -2Н2О оса-
ждаетсяпривосстановлениисернокислого р-ра Na2OsO4
этиловым спиртом. Дегидратировать ее следует в
отсутствии кислорода, т. к. получающийся продукт
обладает пирофорными свойствами.
Наиболее характерное соединение О. четырех-
окись осмия, или осмиевый ангид-
р и д, OsO4 — почти бесцветные кристаллы, плотн.
4,96 (22°), т. пл. 39,5° и 41,0° (две модификации),
т. кип. 130°; уд. электропроводность в жидком со-
стоянии<10 "" мком; растворим в СС14 (5,00 вес. %
при 0°; 6,56 вес. % при 25°) и воде (79,0 вес. % при
25°). OsO4 получается при окислении О. азотной к-той
или при нагревании О. на воздухе и в кислороде и
служит промежуточным продуктом при выделении О.
из природных объектов. OsO4 очень ядовита, раздра-
жает глаза, слизистые оболочки, кожу. При растворе-
нии в воде 0s04 дает слабую к-ту H2[OsO4(OH)2].
Фториды OsF4, OsFe, OsF8 образуются из эле-
ментов при 250—300°. Тетрафторид OsF4 — черный
порошок; растворяется в воде с разложением, частич-
но образуя H2[OsFe], Гексафторид OsF6— зеленые
кристаллы, разлагающиеся водой на OsO2, OsO4 и
HF; т. кип. ок. 200°. Октафторид OsF8 (единственное
соединение типа АВ8) — желтые кристаллы, реагиру-
ющие с водой; OsF8 — самый летучий из всех фто-
ридов О., т. кип. 47,5°. Дает соединения с фторидами
щелочных металлов. При нагревании О. в токе хлора
793
ОСМИЙ
образуются хлориды: OsCl2, OsCla, OsCl4. Дихло-
рид OsCl2 — темно-коричневое вещество, нераствори-
мое в холодной воде и слаборастворимое в горячей.
Трихлорид OsC1.3 — гигроскопичные коричневые ку-
бич. кристаллы, растворимые в воде, сублимирует
выше 350°, при 560—600° разлагается на OsCl4 и OsCl2.
Спиртовой и водный р-ры OsClg устойчивы, последний
имеет кислую реакцию. Тетрахлорид OsCl4 — красно-
коричневое вещество, нерастворимое в воде, но мед-
ленно разлагаемое ею, нерастворимое в бескислород-
ных к-тах. Из других галогенидов известен тетраиодид
0sJ4. Мелкораздробленный О. загорается в парах се-
ры, давая OsS2 — черное вещество со структурой
пирита, окисляющееся горячей азотной к-той до
OsO4. Известны соединения OsSe2 и OsTe2, также со
структурой пирита. С фосфором О. дает серо-черный
порошок OsP2. Получены соединения О. с азотом,
углеродом, водородом и многочисленные сплавы с
платиной, иридием, кобальтом, вольфрамом, молиб-
деном и т. д.
Как и для других платиновых металлов, для О.
во всех валентных состояниях характерно образова-
ние многочисленных комплексных соеди-
нений, гл. обр. анионного типа. Преобладающее
координационное число О. равно 6. Для 2-валент-
ного О. известны комплексы типов Me4[Os(CN)e] и
Ме4[Ов(8Оз)з]. Для 3-валентного О. известны: 1) Оран-
жевые комплексные нитриты Me2[Os(NO2)6], большин-
ство к-рых хорошо растворимо в воде (кроме производ-
ного Ag); пентанитроосмиат (III) калия K2[Os(NO2)6]
получают обработкой раствора K2[OsCle] избытком
KNO2. 2) Комплексные галогениды Мез[ОвХв], на-
пример гексахлороосмиат (III) калия K3[OsCls]—
красно-коричневое вещество, растворимое в воде и
спирте; его получают при действии С12 на смесь хло-
ридов О. и калия или из KJOsOsN] при добавлении
НС1. Гексабромоосмиат (III) калия Кз[ОвВгв] полу-
чают в р-ре при электролитич. восстановлении гекса-
бромоосмиата (IV) калия K2[OsBre],
Для 4-валентного осмия известны: 1) Гексамины
[Ов(амин)в]С14; гексатиомочевинотетрахлорид ос-
мия jOs[(NH2)2CS]ejCl4-4Н2О получают при нагре-
вании Na2[OsCle] с тиомочевиной. 2) Сульфитокомп-
лексы Ме8[Ов(8Оз)в], Ме8[ОвС14(8Оз)4], Ме6[О8(8Оз)6],
Me7[OsCl(SO3)6], Ме6[О8С12(8Оз)4]. 3) Комплексные
галогениды Me2[OsXe], где Xe— Cle, Fe, Bre, J6,Cl6Br,
С1зВгз, (ОН)С16, (ОН)С13Вг2, (NH2)CI6. Гексахлориды и
гексабромиды О. изоморфны с платинохлоридами.
Хлороосмиевая к-та H2[OsCle] образуется при на-
гревании с обратным холодильником OsO4 с НС1 и
спиртом.
Для 6-валентного О. известны: 1) Осмиаты, или соли
осмиевой к-ты H2OsO4. Осмиат (VI) калия K2OsO4-
•2Н2О образуется при восстановлении щелочного р-ра
OsO4 спиртом или KNO2 и выделяется в виде окта-
эдрич. фиолетовых кристаллов. Осмиаты менее устой-
чивы, чем рутенаты. 2) Комплексы осмила (OsOj+):
аммины [ОзО2(ХНз)4]Х4, цианиды Me2[OsO2(CN)4]
и замещенные осмиаты Me2[OsO2X4], где X — С1,
Br, NO2, '/2С2О4, SOsNa. 3) Соли оксиосмила (ОзОз)—
Ме2[ОвОзХ2], где X—Cl, Br, NO2, ’/2С2О4. 4) Нитрил-
галогениды Me[OsNX4], Me2[OsNX6], к-рые могут
быть получены при обработке осмиаматов Me[OsO3N]
соответствующим галогеноводородом:
K[OsO,N] .4- КС1 + 6НС1^ K2[OsNCI5| + С12 + Н2О.
Для 8-валентного О. характерно образование осмиа-
матов Ме[ОвОзХ] при действии конц. NH4OH на конц.
р-р OsO4 в щелочи. Известны также перосматы
Me2[OsO4X2], где X — ОН, F.
О. образует два карбонила: Os(CO)6, т. пл.— 15°
и Os2(CO)9, т. пл. 22,4°, возгоняющийся выше 130°.
794
При действии СО на OsCl2 образуется соединение
состава Ов(СО)зС12, т. пл. 269—273°, т. разл. 280°,
нерастворимое в воде и большинстве кислот. Известны
также соединения типа 0s(C0)4X2 и [Os(CO)4X]2,
где X — Cl, Br, J.
Аналитическое определение. При
нагревании О. или его соединений на воздухе всегда
появляется характерный запах OsO4. Сероводород
выделяет коричневый или черный осадок из кислых
р-ров солей О. и из щелочных р-ров осмиатов и пер-
осматов. При сплавлении соединений О. с Na2O2 при
подщелачивании водной вытяжки появляется корич-
невое окрашивание (образование перосмата), перехо-
дящее в фиолетовое при добавлении спирта (рутений
дает черный осадок). При добавлении к этому раствору
КОН выделяются фиолетовые кристаллы K20s04,
а при добавлении NH4C1 — желтый кристаллич. оса-
док [OsO2(NH3)4]Cl2, нерастворимый в НС1 и разла-
гающийся при кипячении с выделением OsO4. Раствор
OsO4 обесцвечивает индиго, окисляет С2Н6ОН до аль-
дегида и уксусной к-ты, причем раствор окрашивает-
ся в голубой цвет. В присутствии эфира (1 —10) -10_6г
О. можно открыть по зеленому цвету эфирного слоя.
FeSO4 выделяет из растворов соединений О. черный
осадок OsO2. Для отделения О. от остальных метал-
лов платиновой группы его отгоняют в виде OsO4 из
растворов, содержащих HNO3, причем для количе-
ственного отделения от рутения необходимо, чтобы
в растворе содержалась серная к-та, а концентрация
азотной к-ты не превышала 40% по объему.
Методами пробирного анализа О. не определяется
ввиду летучести образующейся OsO4. Количественно
О. определяют весовым методом в виде металла, полу-
чаемого при восстановлении различных соединений
О. (OsO2, OsS2) водородом. Для определения малых
количеств О. в свинцовых сплавах применяют осаж-
дение его тионалидом и колориметрич. или спектро-
фотометрии. определение с тиомочевиной (реакция
Чугаева). Образующееся соединение окрашивает
раствор в красный или розовый цвет. Т. обр. может
быть определено от 4 -10 ~4 г до 5 -10“’г О.в 1 л с ошиб-
кой, не превышающей 5% . Определение О. может быть
проведено также потенциометрии., полярографии.,
спектральным и рентгеноспектральным методами.
Получение. О. добывается совместно с платиной и
другими платиновыми металлами. Отличительной осо-
бенностью аффинажа О. является выделение его после
отгонки летучей OsO4, тогда как другие платиновые
металлы (кроме Ru) выделяют осаждением. Выделение
О. проводится из различных концентратов, шламов
от электролиза никеля и меди, вторичного металла
(лома). При растворении платины в царской водке
О. остается в осадке в виде осмистого иридия, осмие-
вого концентрата и OsO4. Осадок сплавляют с 6—
8-кратным количеством Zn по весу для получения мел-
кого порошка. Порошок спекают с ВаО2. Массу после
спекания обрабатывают смесью НС1 и HNO3 в перегон-
ном аппарате для отгонки OsO4. В результате этой
обработки О., содержащийся в опеке в виде двуокиси,
образует четырехокись, отгоняющуюся из раствора.
OsO4 улавливается щелочным р-ром с образованием
Na2OsO4. Получающийся раствор восстанавливают
гипосульфитом, после чего осаждают хлористым
аммонием соль Фреми [ОвО2(ХНз)4]С12. Осадок про-
мывают, фильтруют и прокаливают в восстановитель-
ном пламени. После этого губчатый О. обрабатывают
HF и НС1 и восстанавливают в электропечи в струе
водорода; получают продукт, достигающий по чистоте
99,9% Os.
Металлический О. может быть получен в различных
формах: а) кристаллический О. — при нагревании
смеси 1 ч. О. с 7—8 ч. Zn или Sn до красного каления,
медленном охлаждении расплава и обработке соляной
795
ОСМОЛ —ОСМОТИЧЕСКОЕ ДАВЛЕНИЕ
796
к-той; б) «чернь» — при восстановлении р-ров осмиа-
тов пли OsO4 формиатами щелочных металлов; в) плен-
ка — при термич. разложении карбонилов О.; г) кол-
лоидный О.— при восстановлении р-ра K2OSO4 гид-
разином в присутствии гуммиарабика как защитного
коллоида.
Применение. Большая твердость и высокая кор-
розионная устойчивость О., а также его искусствен-
ных и природных сплавов с другими металлами плати-
новой группы позволяют использовать их в различных
изнашивающихся деталях точных измерительных при-
боров. Сплавы О. с другими платиновыми металлами
пли с W и Со используют для наконечников перьев в
авторучках; в различных нагревательных элементах;
для изготовления нитей радиоламп. В фарфоровой
пром-сти О. применяют для получения яркой и сочной
черной краски. О. и его соединения — хорошие ката-
лизаторы, для гидрогенизации более эффективны, чем
платина. О. используют как катализатор в синтезе
NH3. Из соединений О. наиболее часто применяется
OsO4. Ее используют при микроскопия, исследовании
животных и растительных тканей как окрашивающий
и фиксирующий препарат; применяют в качестве
мягкого окислителя в органич. реакциях. OsO4, как
и металлич. О., обладает каталитич. свойствами, что
также находит применение в лабораторной практике
и пром-сти, иапр. при синтезе кортизона. Хлоро-
осмиаты применяют в фотографии вместо солей золота.
Лит.: Звягинцев О. Е., Аффинаж золота, серебра и
металлов платиновой группы, 3 изд., М., 1945; Бетехтин
А. Г., Курс минералогии, 3 изд., М., 1961; Плаксин
И. Н., Металлургия благородных металлов, М., 1958; G m е-
11 п, 8 АиП., Syst.-Num. 66'—Osmium, В., 1939; Mellor,
v. 15, L., 1947, р. 686; К 1 г k, v. 10, N. Y., 1953, p. 849—51;
v. 7, N. Y., 1951, p. 509; Rare metals handbook, ed. C. Hampel,
2 ed., L., 1961, p. 304; Handbook of chemistry, comp, and ed.
N. A. Lanse, 10 ed., N. Y.— [a. o.], 1961; Pascal, t. 19, P.,
1958; Гиллебранд В. Ф. [и д p.], Практическое руко-
водство по неорганическому анализу, пер. с англ., М., 1957;
Рутений и осмий. Библиогр. указатель лит-ры (1804—1960),
М., 1962; Методы анализа платиновых металлов, золота и се-
ребра. Сб. научи, трудов, М., 1960. Т. Н. Леонова.
ОСМОЛ — просмоленная древесина сосны (иногда
кедра сибирского и корейского), служащая сырьем для
смоло-скипидарного (смолокуренного) и канифольно-
экстракциопного произ-ва. Наибольшее значение име-
ет пневый О. (естественно просмолившаяся ядровая
часть пней, включая крупные корни); используется
также стволовый О. (стволовая древесина сосны,
просмолившаяся в результате осмолоподсочки). О.
пневый, считая на 20%-ную влажность, содержит
13—21% канифоли и 3—5% летучих (скипидара,
терпеновых спиртов и др.). При переработке О. пнево-
го соснового на канифольно-экстракционных пред-
приятиях из 1 складочного м3 получают 40—45 кг
канифоли, 12—14 кг скипидара, 2—2,5 кг масла сос-
нового флотационного (содержащего 50—70% терпе-
новых спиртов) и 1 кг смолы абиетиновой. Экстракци-
онная канифоль темнее живичной; опа может быть
осветлена обработкой отбельными глинами или фур-
фуролом. Заготовка О. пневого сосцового произво-
дится преим. взрывным способом. Л. В. Гордон.
ОСМОС — диффузия веществ (обычно из растворов)
через перегородку, разделяющую раствор и чистый
растворитель или два раствора различной концентра-
ции. О. приближает систему к равновесию в результа-
те выравнивания концентраций по обе стороны пере-
городки. Важнейшим случаем О., позволяющим изме-
рять осмотическое давление, является О. через т. н.
полупроницаемую перегородку, т. е. перегородку,
проницаемую только для молекул одного из веществ,
составляющих раствор, обычно — для молекул раство-
рителя. Перегородки, проницаемые только для воды
илидругого растворителя и непроницаемые для раство-
ренных веществ как низкомолекулярных, напр.
сахара и солей, так и высокомолекулярных, могут
быть изготовлены из полимерных плейок (коллодия)
или гелеобразных осадков, напр. ферроцианида меди
Cu2[Fe(CN)e]; такой осадок образуется в порах пере-
городки стеклянного фильтра или керамич. пластин-
ки при погружении пористого материала сначала в
раствор медного купороса, а затем желтой кровяной
соли.
При О. через полупроницаемые перегородки раство-
ритель, напр. вода, диффундирует в направлении вы-
равнивания осмотич. давлений, т. е. от чистого раство-
рителя или разбавленного р-ра к более концентри-
рованному р-ру, налитому в осмотич. ячейку (см.
Изотонические растворы). Осмотич. равновесие и
действие разностей осмотич. давлений весьма важ-
ны в жизнедеятельности живых организмов. Фи-
зиологии. р-р в медицине и хирургии, как и раз-
личные заменители крови, пзотоничен по отношению
к плазме крови. Даже когда чистый растворитель или
более разбавленный р-р отделен от концентрирован-
ного р-ра перегородкой, в нек-рой степени проницае-
мой для всех компонентов р-ра, О. может приводить
к отсасыванию растворителя, если в данных условиях
он диффундирует с большей скоростью. Поэтому
увлажненные пористые тела или гели могут обезвожи-
ваться при помещении в концентрированные водные
р-ры солей или в жидкости, растворяющие воду, если
для выравнивания концентраций вода диффундирует из
них быстрее, чем проходит обратная ее диффузия в
микропоры, заполненные водой. Легко осуществимы
мембраны, проницаемые для растворителя (воды) и
низкомолекулярных растворенных веществ (обычных
электролитов) и непроницаемые для высокомолеку-
лярных веществ — полимеров и дисперсных (колло-
идных) фаз. О. через такие мембраны используется в
виде так наз. диализа, электродиализа и ультрафиль-
трации для очистки высокомолекулярных соединений
(напр., белков) и коллоидов от низкомолекулярных
примесей — солей и др. О. имеет особо важное значе-
ние в жизнедеятельности растительных и животных
организмов.
Лит. см. при ст. Осмотическое давление. П. А. Ребиндер.
ОСМОТИЧЕСКОЕ ДАВЛЕНИЕ (диффузионное
давление) — мера стремления растворенного вещества,
вследствие теплового движения, перейти в процессе
диффузии из раствора в чистый растворитель и равно-
мерно распределиться по всему объему растворителя,
предельно понизив первоначальную
концентрацию раствора. Если раст-
вор отделен от чистого растворителя
полупроницаемой пере-
городкой (проницаемой только
для растворителя), О. д. вызывает
осмотич. всасывание растворителя в
раствор для предельного уменьше-
ния концентрации растворенного
вещества в растворе (см. Осмос).
О. д. измеряется уравновешиваю-
щим его противодавлением, т. е. из-
бытком гидростатич. давления со сто-
роны раствора, препятствующим
диффузионному потоку чистого раст-
ворителя в раствор. Этим обеспечп-
Рис. 1. Схема осмометра: Н„—равновес-
ная высота столба, соответствующая ос-
мотич. давлению П; Н'— высота столба,
при к-рой идет всасывание растворителя
в раствор Н'—высота столба,
ритель выдавливается из раствора через
перегородку.
вается термодинамич. равновесие раствора и раство-
рителя, разделенных полупроницаемой перегородкой
А (см. рис. 1). Всасывание воды извне через перего-
797
ОСМОТИЧЕСКОЕ ДАВЛЕНИЕ
798
родку А постепенно замедляется возрастающим избыт-
ком гпдростатич. давления со стороны раствора, и
скорость осмоса спадает до нуля. Уравновешивающее
избыточное давление, т. е. О. д., (П) определяется
наибольшей высотой избыточного манометрич. столба
Но, медленно достигающейся в самом процессе осмоса;
(И = H0Qcg, где о,. — плотность раствора, g — напря-
жение поля тяжести). Таков статич. метод измерения
О. д. в обычных осмометрах. Для высоких О. д.
осмометры снабжаются ртутными затворами, мано-
метрами и источником высокого давления — балло-
ном со сжатым газом. Для достижения равновесия
в осмометрах требуется длительное время — 5—10
часов, и потому для ускорения и уточнения измере-
ний О. д. применяют динамич. метод — постепенно
увеличивают избыточное противодавление Р и наблю-
дают скорость всасывания V за короткое время. При
Р<П эта скорость положительна (Е>0) и убывает с
уменьшением разности (П — Р) пропорционально
ей. При Р>П скорость становится отрицательной
(Е<0), т. е. растворитель выжимается избыточным
давлением из раствора через полупроницаемую иере-
городку (этот процесс, обратный осмосу, использует-
V
V<0
Рис. 2. Определение осмотич. дав-
ления по динамич. методу.
ся в ультрафильтрации для концентрирования кол-
лоидных р-ров и р-ров высокомолекулярных веществ).
Откладывая на графике (рис. 2) зависимость V от Р,
легко найти интерполяцией О. д. (Р =П),
т. е. значение Р, при к-ром V меняет знак,
переходя через К --= 0. 1 %-ный р-р сахара
в воде обладает О. д. ок. 0,7 атм. В конц.
р-рах сахара и других
низкомолекулярных ве-
ществ (напр., солей) О. д.
достигают сотен атмо-
сфер.
Термодинамически на-
личие О. д. связано с по-
нижением химич. потен-
циала растворителя в
растворе (или давления
насыщенного пара рас-
творителя) под действи-
о
P-TUV-0!
ч—1—I—\-Р
ем растворенного вещества. Для идеальных или пре-
дельно разбавленных р-ров всегда справедлив закон
Рауля — относительное понижение давления насы-
щенного пара растворителя не зависит от темп-ры и
равно молярной доле растворенного вещества х2:
(Ро— Ре)' Ро =
(Л
здесь р0 — давление насыщенного пара растворителя,
рс — то же для р-ра; х2 = пг.Длп+иг) == п21щ, где
«1, «2 — числа молей растворителя и растворенного
вещества соответственно. В схеме осмометра на рис. 1
равновесие в жидкой фазе обеспечено разностью уров-
ней в обеих трубках, т. е. О. д.; для равновесия же
в газовой фазе необходимо, чтобы на уровне Но пар-
циальное давление пара растворителя над раство-
ром в трубке С(рс) и над чистым растворителем в труб-
ке О (рн„) было одинаково; но на этом уровне над
чистым растворителем в трубке О давление пара по-
нижено, т. к. оно убывает с высотой в поле земного
тяготения:
Pua^Pe=--P^M'gH'‘!RT (2)
Здесь М — мол. вес растворителя, R — универсаль-
ная газовая постоянная, Т — абс. темп-ра. По закону
Рауля рс!ра = 1 — х2, заменяя На через n/Q,g и
подставляя vi = M/Qi, где vi — молярный объем
растворителя, Qi — его плотность, находим связь
между О. д. и понижением давления пара раствори-
теля:
(Ро—Рс)'Ро=1 —е“1,,П/ЯТ
Отсюда для достаточно разбавленных р-ров полу-
чаем: П — — 1п (рс/р0). Это устанавливает связь
О. д. с молярной долей х2 или молярной концентра-
цией с растворенного вещества:
n = -^ln(l-z2) (3)
или, разлагая в ряд In (1—х2), имеем
п=^^о+?+4+-)
при Z2<C1, т. е. в разбавленных р-рах этот ряд—
быстро сходящийся, в этом случае, ограничиваясь
первым членом, находим: П= RTx2/vi, или, с до-
статочной степенью точности:
П=ЯТс (5)
Из этого ур-ния (уравнения состояния
Ван т-Г о ф ф а) следует аналогия между О. д. и
парциальным давлением газа (пара) в идеально-газо-
вой смеси: О. д. численно равно давлению, к-рое
оказывало бы растворенное вещество в газовом состоя-
нии, занимая тот же объем, что и объем разбавленного
р-ра, в к-ром оно находится. Из ур-ния (5) видно, что,
как и все подобные энтропийные термодинамич.
эффекты, О. д. в идеальных и предельно разбавленных
р-рах не зависит от природы растворителя и раство-
ренных веществ, а определяется при Г = const только
общим числом «кинетических элементов» в единице
объема раствора — ионов, молекул, их ассоциатов
или коллоидных частиц. Это позволяет измерять
среднечисленные молекулярные веса М растворенных
веществ (или истинные М в случае одного вида раство-
ренного вещества) по ур-нию (5), зная навеску веще-
ства, растворенную в 1 см3 раствора (т г/см3)'.
M=RT
, О. д. при данной весовой концентрации раство-
' ра обратно пропорционально М, т. е. относительно
велико для р-ров низкомолекулярных веществ и очень
мало для р-ров полимеров. О. д. в предельно разбав-
ленном р-ре равно сумме О. д. оiдельных раство-
ренных веществ, в полной аналогии с газовыми сме-
сями. В смесях газов О. д. равно парциальному дав-
лению того компонента, для к-рого перегородка не-
проницаема. Так, напр., можно определить осмомет-
рически парциальное давление азота в смеси с
водородом, если отделить эту смесь от чистого водо-
рода тонким листочком металлич. палладия, прони-
цаемого только для водорода.
Если молекулы растворенного вещества в растворе
диссоциированы на ионы, то П = iRTc, где i (>1)—
коэффициент Ван т-Г о ф ф а, дающий уве-
личение числа частиц вследствие диссоциации на v
ионов; если степень диссоциации а, то i — av+(l—а);
при v= 2 (бинарные электролиты) i = 1 + а, при
полной диссоциации при предельном разбавлении
i = V. В случае ассоциации растворенного вещества
«<1. Уравнения (1)—(5) справедливы для идеальных
жидких смесей, напр. смесей углеводородов, а для
полимеров — в случае растворов в идеальном так наз.
0-растворителе. Для любых растворенных веществ
п растворителей эти ур-ния верны только при предель-
ном разбавлении — до х2 = 0,01 для неэлектроли-
тов, а для электролитов — только до х2 = 10’в.
В более конц. р-рах в формулы для О. д. вводится по-
правочный множитель — осмотический ко-
эффициент растворителя: G = П/П;,
где Пг — реальное, а П, — идеальное О. д., вычис-
ленное по (3). Коэфф. G связан с коэфф, активности
799
ОСНОВАНИЯ—ОТБЕЛИВАНИЕ ТЕКСТИЛЬНЫХ МАТЕРИАЛОВ
800
растворителя Д и растворенного вещества /2 уравнени-
ями:
In ft = —(1 — G) In х,
ln/2=- C
Х2 = 0
при х2—>0, G, /] и /2—>0
Вместо прямых измерений О. д., иногда затрудни-
тельных и ненадежных как из-за необходимости под-
бирать полупроницаемые перегородки, так и из-за
медленного установления равновесия, для нахожде-
ния мол. веса данного вещества обычно измеряют
величину одного из т, наз. осмотических
свойств его раствора в соответствующем раствори-
теле, пропорциональных О. д. (в разбавленных р-рах).
Таковы понижение точки замерзания растворителя,
вызываемое растворенным веществом \TS=TOS-—ТcS
(криометрия), повышение точки кипения растворителя
Д7'г = Tc[-\-Toi (эбуллиометрия) и относительное
понижение давления насыщенного пара (тонометрия).
Термодинамич. теория показывает, что
йт’», RTolv'
^s = G-^-c-, \Tl=G—~—c
где Qs и Qt — теплоты плавления и испарения при
ToS и То[ — темп-рах замерзания и кипения чистого
растворителя (Tcs и ТсГ— то же для р-ра). Отклонения
от идеальных соотношений, характеризующиеся G
или /, определяются взаимодействием частиц — моле-
кул или ионов растворенных веществ и таким образом
зависят от химич. природы — состава и строения ком-
понентов раствора.
Лига.: Гуггенгейм Э. А., Современная термодинами-
ка, изложенная по методу У. Гиббса, пер. с англ., Л,—М.,
1941; Жуховицкий А. А., Шварцман Л. А., Фи-
зическая химия, М., 1963; Гильдебранд Д. Г., Раство-
римость неэлектролитов, пер. с англ., М., 1938; Цянь
Ж э н ь-ю а н ь, Определение молекулярных весов полимеров,
пер. с кит., М., 1962; Вагнер Р. Г., в кн.: Вайсбергер
А. [и др.), Физические методы органической химии, пер. с
англ., т. 1, М., 1950; Тагер А., Растворы высокомолеку-
лярных соединений, М.—Л., 1951. II. А. Ребгтдер.
ОСНОВАНИЯ — см. Кислоты и основания.
ОСТВАЛЬДА ЗАКОН РАЗБАВЛЕНИЯ устанав
ливает зависимость эквивалентной электропроводности
р-ра электролита от концентрации р-ра (или его раз-
бавления, т. е. величины, обратной концентрации).
Для электролита, диссоциирующего по уравнению ви-
да АВ^А++В_, закон выражается равенством;
СА2/А (Ах —Ах)-- А
где А и А со — эквивалентные электропроводности
(о.ч-1 -г-экв-' -см2) р-ра при концентрации С (г-экв/л) и
соответственно при бесконечном разбавлении (С-+0 и
«-<!, а — степень диссоциации), К — константа дис-
социации (г-экв;л), определяемая действующих масс
законом для приведенной реакции диссоциации:
А = Сд+Сg-/ССа2/ (1 а), Слв= С (1 —а),
С д + — С q — = С а
Подстановка в выражение закона действующих масс
значения степени диссоциации по Аррениусу: а=
= А/Асо, непосредственно приводит к О. з. р.
О. з. р. позволяет определять весьма важные в тео-
рии р-ров электролитов константы К и А® (см. Элек-
тролитическая диссоциация, Подвижность ионов, Коль-
рауша закон). Их определяют по наклону эксперимен-
тально полученной прямой О. з. р.
CA = /(1/A)=-AAoo+XA2k/A
и отрезку, отсекаемому этой прямой на оси ординат.
Для электролитов с дву- или многовалентными катио-
нами или анионами О. з. р. вытекает из выражений
для нескольких констант диссоциации и имеет более
сложный вид, а также более ограниченные возмож-
ности использования,
О. з. р. применим лишь в той мере, в какой справед-
ливо использование в законе действующих масс кон-
центраций вместо активностей и предположение о
независимости подвижности ионов от концентрации
раствора, лежащее в основе равенства a=A/A®. Поэ-
тому О. з. р. справедлив лишь в области сравнительно
малых концентраций (в особенности для сильных
электролитов). Напр., в случае соляной к-ты он при-
меним до концентраций порядка 5-10~4 М/л. Для сла-
бых электролитов применимость О. з. р. распростра-
няется и на более высокие концентрации.
Лит.: М е л в и п-Х ь ю з Э. А., Физическая химия, пер.
с англ., кп. 2, М., 1962; Раковский А. В., Введение в
физическую химию, М., 1938; Ostwald W., Z. phys.
Chem., 1888, 2, № 4, 270. С. Э. Вайсберг.
ОСТРОМЫСЛЕНСКОГО РЕАКЦИЯ — каталитич.
конденсация ацетальдегида и этанола в бутадиен-1,3:
/°
СН3СН2ОН+CH3Cf------->СН2 = СН—СН.:=СН,
II -Н2О
Вместо ацетальдегида с равным успехом можно исполь-
зовать паральдегид. Синтез имеет общий характер,
в него можно вводить высшие альдегиды и спирты.
Напр., привзаимодействии ацетальдегида с пропиловым
(или изопропиловым) спиртом при400°с хорошим выхо-
дом образуется пиперилен СН3— СН=СН— СН=СН2,
этот же диен получается из ацетона и этилового
спирта. О. р. протекает обычно при 360—440°; катали-
затор — красный фосфор, сульфаниловая к-та, хло-
ристый барий, окись алюминия и окись циркония
или тантала на силикагеле, последний катализатор
используется при промышленном осуществлении реак-
ции в США. Выходы реакции подсчитывают в мо-
лях: 1) на пропущенный спирт; 2) на разложенную
смесь; 3) на разложившийся спирт; 4) на прореагиро-
вавший альдегид. При использовании О. р. получен
бутадиен 97—98%-ной чистоты в количестве 0,35—
0,36 кг на 1 кг абс. спирта.
Механизм О. р. окончательно не установлен; на
основе экспериментальных данных может быть пред-
ставлен след, схемой:
,О ,О
2СН.С^ —>сн.сн=снсс
хн чт
C2H5OH+CH3CH=CHC<f°—>СН3с/° +СН3СН=СН-СН2ОН
хн хн
СН3СН=СН—СН2ОН —> сн2=сн—сн=сн2
Реакция открыта И. И. Остромысленским в 1911.
Лит.: Остромысленский И. И., ЖРФХО. Часть
химич., 1915, 47, вып. 6, 1494; Остромысленский
И. И., К е л б а с и н с к и й С. С., там же, 1915, 47, вып. 6,
1509; Е g 1 о 1 f G„ Hulla G., Chem. Rews, 1945, 36, 73;
Смирнов Н. И., Синтетические каучуки, 2 изд., Л., 1954;
Синтетический каучук, под ред. Г. С. Уитби, пер. с англ., Л.,
1957. Э. Е. Нщрантьев.
ОСУШКА ГАЗОВ — см. Газов осушка.
ОТБЕЛИВАНИЕ ТЕКСТИЛЬНЫХ МАТЕРИА-
ЛОВ — удаление из волокнистых материалов различ-
ных примесей, придающих волокнам и изделиям из
них плохую смачиваемость, неприятный оттенок и
др. Суровые ткани (не отбеленные) не имеют нужной
степени белизны, плохо окрашиваются и не обладают
надлежащими санитарно-гигиенич. свойствами. Опе-
рации, связанные с удалением примесей и приданием
волокнам, пряже и тканям требуемой белизны и гигро-
скопичности, и составляют сущность беления.
О. т. м. состоит из трех основных операций: 1) рас-
шлихтовки (удаления шлихты); 2) отварки, при к-рой
удаляют лигнин, пектиновые в-ва, замасливатели и
др. примеси; 3) собственно отбелки, при к-рой разру-
801
ОТБЕЛИВАНИЕ ТЕКСТИЛЬНЫХ МАТЕРИАЛОВ
802
шаются естественные красящие вещества волокна.
Первые две операции применяют прп отбеливании
тканей из растительных волокон.
Для отбеливания ткани обрабатывают р-рами
белящих веществ (хлорная известь, гипохлорит нат-
рия, перекись водорода и др.). В разрушении естест-
венных красящих веществ хлопка прп белении хлор-
ной известью и гипохлоритом натрия большое значе-
ние имеет хлорноватистая к-та, образующаяся при
их гидролизе. По современным представлениям НОС1
присоединяется по месту двойной связи окрашенных
примесей, а затем, в зависимости от pH среды, полу-
чаются бесцветные продукты их окисления или хло-
рирования. Беление перекисью водорода дает лучшие
результаты как по степени белизны, так и по меньшему
снижению прочности ткани.
Продолжительность беления можно значительно
сократить за счет совмещения щелочной отварки и
собственно беления в одну операцию. По современным
представлениям белящее действие перекиси водорода
обусловливается ионом иергидроксила, к-рый обра-
зуется в результате диссоциации перекиси: Н2О2—
=Н + 4-НО2 .При повышении щелочности р-ра снижает-
ся концентрация водородных ионов и увеличивается
концентрация пергидроксильных ионов. С ростом pH
среды скорость беления возрастает, но вместе с тем
падает устойчивость перекисных р-ров и происходит
потеря активного кислорода. Последняя реакция не-
желательна по двум причинам: 1) повышается непро-
изводительный расход перекиси водорода и 2) имеется
опасность окислительной деструкции целлюлозы (в
случае отбеливания растительных волокон). Поэтому
на практике применяют стабилизаторы, повышающие
устойчивость перекисных р-ров. Наибольшее распро-
странение в качестве стабилизаторов перекиси имеет
силикат натрия. Для этой цели можно применять
также фосфаты и иолифосфаты.
В качестве белящих веществ получают распростра-
нение хлорит натрия и нек-рые надкислоты, в част-
ности надуксусная к-та СНзСОООН. Эти вещества,
имея сравнительно низкий окислительный потенциал,
достаточный, однако, для разрушения естественных
красящих веществ хлопка и льна, совершенно не де-
структируют целлюлозу. Прп белении хлоритом нат-
рия действующим началом является СЮ2, выделяю-
щаяся в кислой среде. Режим хлоритного беления:
концентрация активного хлора 0,5—1,5 г/л, pH 3—4,
темн-ра 85—90°, продолжительность беления 45—60
минут. Существенным недостатком хлоритного беле-
ния является сильная коррозия аппаратуры, изготов-
ленной даже из нержавеющих сталей. Титан лучше
всего выдерживает действие СЮ2. Большой интерес
заслуживает способ беления хлоритом с добавкой эфи-
ров органич. к-т почти в нейтральной среде. При гид-
ролизе эфиров, напр. этплформиата, выделяется к-та,
активирующая беление вследствие разложения хло-
рита с выделением СЮг’, последняя расходуется не-
посредственно на беление. Коррозия аппаратуры при
этом резко снижается. Надуксусная к-та применяется
для беления в слабокислой и нейтральной среде при
pH (>—7. Хлорит натрия и надкислоты рекомен-
дуются для беления синтетич. волокон.
Качество отбеленных тканей оценивают по степени
белизны, капиллярности ткани, ее прочности на
разрыв и вязкости медно-аммпачных р-ров.
Отбелка тканей пз растительных волокон. Беле-
ние хлопчатобумажных тканей. Рас-
шлихтовка сводится к удалению шлихты, к-рую на-
носят на нити основы, в процессе ткачества с целью
снижения обрывности и повышения производительно-
сти ткацких станков. Для приготовления шлихты ши-
роко применяют крахмал, а также синтетич. материа-
лы (карбоксиметилцеллюлозу, поливиниловый спирт
и др.). Последние сравнительно легко удаляются с
ткани, благодаря их хорошей растворимости в воде.
Гораздо труднее удалить с ткани крахмальную шлих-
ту, нерастворимую в воде. В этом случае применяют
различные расщепители крахмала, к-рые гидролизуют
его, переводя в растворимое состояние. Этим процессам
предшествует набухание крахмала. В качестве расще-
пителей крахмала применяют кислоты (обычно серную,
концентрацией ок. 5 г/л). На специальных машинах
ткань обрабатывают этим раствором при комнатной
темп-ре илп прп 40—50°. Расщепителями крахмала мо-
гут служить окислители: хлорамины, хлорная известь
пли гипохлорит натрия и перекись водорода. Обра-
ботку окислителями проводят в условиях, достаточных
для разложения крахмала и не вызывающих деструк-
цию целлюлозы. Применение окислителей особенно
желательно прп расшлихтовке тканей из низких сор-
тов хлопка. В этом случае при последующей щелоч-
ной отварке легче удаляется лигнин, содержащийся
на ткани в виде «галочек» (остатки коробочек и скор-
лупы семян хлопкового волокна). Обработка окисли-
телями важна также и потому, что щелочная отварка
может быть произведена при более низкой темп-ре и
меньшей продолжительности; прп этом наблюдается
меньшая потеря прочности ткани. Широкое примене-
ние при расшлихтовке тканей имеют различные фер-
менты солода (диастафор, диафорин), панкреаза,
содержащаяся в панкреатине (получается из сока под-
желудочной железы животных), и биолаза —фермент
бактериального происхождения. Действующим нача-
лом этих ферментов являются а- и 0-амилазы, обладаю-
щие свойством переводить крахмал в растворимые в
воде декстрины и сахаристые вещества. Особо эффек-
тивными расщепителями крахмала являются биолаза
и супербполаза, позволяющие вести расшлихтовку
непрерывным методом на агрегатах, состоящих из
ряда машин, обеспечивающих пропитку ткани р-ром
расщепителя и вылеживание ее в течение 15—20 мин.
Непрерывный метод расшлихтовки можно использо-
вать, применяя и другие расщепители крахмала. При
всех способах расшлихтовки процесс заканчивают
промывкой ткани для удаления продуктов гидролиза
крахмала и оставшегося неизрасходованным расще-
пителя.
Щелочная отварка — одна из наиболее важных ста-
дий беления хлопчатобумажных тканей. В этом про-
цессе удаляется большая часть естественных примесей
хлопкового волокна: воскообразные, белковые и пек-
тиновые вещества, лигнин. Воскообразные вещества
удаляют гл. обр. эмульгированием; поэтому в со-
став варочной жидкости, помимо едкого натра (10—
12 г/л), вводят неионогенные эмульгаторы, напр.
ОП-7, ОП-Ю и др. Белковые вещества под действием
щелочи при высокой темп-ре гидролизуются и перехо-
дят в растворимые в щелочной среде аминокислоты.
Также путем распада и гидролиза разрушаются в ус-
ловиях щелочной отварки пектиновые вещества, да-
вая растворимые натриевые соли уроновых к-т. Лиг-
нин в условиях щелочной варки расщепляется на ком-
поненты, растворимые в щелочах. Расщепление про-
исходит особенно легко после предварительного хло-
рирования лигнина или в присутствии сульфитов ще-
лочных металлов. В состав варочной жидкости вво-
дят также силикат натрия, способствующий предот-
вращению образования ржавых пятен на ткани и
лучшей очистке ее от примесей. Щелочную отварку
тканей при периодич. способе беления производят в
варочных котлах различных систем при 130—135°
в течение 6—8 часов. Весь же процесс отварки, вклю-
чая загрузку и выгрузку ткани, промывку ее в котле
и другие подсобные операции, продолжается до 20
часов. В последние годы разработана технология и
° 26 к.х.э. т. з
803
ОТБЕЛИВАНИЕ ТЕКСТИЛЬНЫХ МАТЕРИАЛОВ — ОТБЕЛИВАЮЩИЕ ВЕЩЕСТВА
804
аппаратура, позволяющие щелочную отварку про-
водить непрерывным способом, при этом продолжи-
тельность операции сокращается до 1—2 часов.
Условия беления должны быть такими, чтобы
окисление самой целлюлозы было сведено к минимуму.
Существенную роль при этом играют концентрация
раствора, темп-ра, pH среды и продолжительность
воздействия белящих р-ров на ткань. Исходя из тео-
ретич. представлений и экспериментальных исследо-
ваний установлены примерно следующие условия про-
цесса: концентрация активного хлора 0,5—1 г/л,
темп-ра 20—35°, pH среды 9—9,5, продолжительность
от 20—30 минут до 2 часов. Беление целесообразно
вести непрерывным методом на агрегатах, подобных
тем, к-рые применяют при расшлихтовке тканей.
В этом случае темп-ра белящих р-ров должна быть по-
вышена до 30—35°, а время беления сокращено до
30 минут. Беление хлопкового волокна и пряжи прин-
ципиально не отличается от беления хлопчатобумаж-
ных тканей, однако аппаратурное оформление процес-
сов будет иным. Естественно, что в данном случае
расшлихтовка исключается.
Наиболее прогрессивным является щелочно-пере-
кисный запарной метод беления, способ‘двухстадий-
ный: первая стадия — обработка ткани щелочным
р-ром при концентрации едкого натра ок. 25 г!л и обра-
ботка паром (запаривание) в течение 1 часа; вторая
стадия — обработка ткани перекисным р-ром при
концентрации перекиси водорода 3—5 г/л, запарива-
ние в течение ~ 1 часа. После каждой обработки ткань
последовательно промывают горячей и холодной во-
дой. Время, необходимое для беления ио непрерывно-
му способу, 3—4 часа. В целях дальнейшего ускоре-
ния процесса разработан одностадийный способ беле-
ния, к-рый может быть осуществлен на малогабарит-
ном агрегате.
Беление льняных тканей сложнее,
чем хлопчатобумажных, что обусловлено особен-
ностью строения льняного волокна и большим содер-
жанием в нем примесей. Значительная часть льняных
тканей отбеливается периодич. методом с многократ-
ной щелочной отваркой и гипохлорптной (илп пшо-
хлорптно-перекисной) отбелкой; разработан непре-
рывный процесс беления предварительно отваренных
в котлах льняных тканей.
Беление вискозных тканей про-
текает значительно легче, чем хлопчатобумажных,
т. к. эти ткани лишены примесей, имеющихся в при-
родных волокнах хлопка и льна. Преимущество имеет
перекисный способ беления, осуществляемый на спе-
циальных агрегатах, в лучших конструкциях к-рых
беление проводится в расправленном состоянии.
Существенное значение для повышения качества
тканей имеет мерсеризация. Этот процесс особенно
необходим для нек-рых видов хлопчатобумажных
тканей (сатин, батист, маркизет и др.). Сущность про-
цесса состоит в обработке ткани при натяжении на
холоду конц. р-рами едкого натра (-250 г/л) с после-
дующей отмывкой щелочи с ткани. Мерсеризация со-
общает тканям блеск, повышенную прочность и луч-
шую сорбционную способность ио отношению к кра-
сителям.
Беление тканей пз животных и синтетических воло-
кон. Отбелка шерсти. Природноокрашеи-
ную шерсть отбелить практически невозможно без
значительного ослабления волокна. Отбеливают шер-
стяные ткани, имеющие лишь светлые оттенки, и
только в случае, если они идут для крашения в свет-
лые яркие окраски или когда необходимо получить
чисто белую ткань. Окраску шерстяного волокна
разрушают, обрабатывая шерсть восстановителями
(SO2, NaHSOs, Na2S2O4) или окислителями (Н2Ог,
КМпОа и др.). Наибольшее применение имеет перекись
водорода в слабощелочной среде при темп-ре ок. 50°.
Окрашенную регенерированную шерсть обычно обес-
цвечивают гидросульфитом или ронгалитом в слабо-
кислой среде при 90°. После беления тщательно про-
мывают водой.
Отбелка шелка. Натуральный шелк пред-
варительно обесклеивают (для удаления серицина)
обычно мыльными растворами в слабощелочной среде
(рН10) при концентрации мыла 10—15 г/л и темп-ре,
близкой к кипению (95°), в течение 1—3 часов. Процесс
проводят периодически в механич. жгутовых барках.
Если шелк не предназначен для крашения, то после
промывки дается «оживка» р-ром уксусной к-ты с
целью придания шелковому волокну особого грифа.
Разработан и непрерывный способ обесклеиванмн
шелка. Беление шелка для получения чисто белых тка-
ней и тканей, идущих для крашения в светлые яркие
оттенки, производят в настоящее время перекисью
водорода в слабощелочной среде (pH 8—8,5) при
70—75° с последующей промывкой теплой и холодной
водой.
Синтетические волокна перед краше-
нием освобождают от замасливателей с помощью мою-
щих препаратов (1—2 г/л) при 50—70° в течение
30—60 минут. Если требуется отбелка, то ее предпоч-
тительно производить хлоритом натрия или надкис-
лотами с целью сохранения физико-механич. свойств
синтетич. волокон.
Лит.: Садов Ф. И., Корчагин М. В., Мате ц-
кий А. И., Химическая технология волокнистых материа-
лов, 2 изд., М., 1956; Г о т о в ц е в а Л. А., Ш и х е р М. Г.,
Суровая А. В., Текстильная пром-сть, 1959, .V» 5; Ф е д о-
р о в а Н. Е., М о р ы г а н о в П. В., там же, 1960, № 12,
1961, № 6; Известия ВУЗов. Технология текстильной пром-' тп,
1960, -У 4; 1961, № 2; 1962, № 4; Каталог текстильных ма-
шин, аппаратов и агрегатов, М., 1961.
II. Г>. Морыганов, Б. Н. Мельников.
ОТБЕЛИВАЮЩИЕ ВЕЩЕСТВА (о пт ич е с-
к и) — бесцветные или слабоокрашенные органич.
соединения, способные поглощать УФ-лучи в области
300—400 ммк и преобразовывать их в синий илп фио-
летовый свет с длиной волны 400—500 ммк. Основ-
ные максимумы поглощения О. в. находятся в области
335—380 ммк, а максимумы флуоресценции сдвинуты
в длинноволновую область на 70—130 ммк (правило
Стокса).
Неокрашенные растительные, животные и синтетич.
материалы даже после отбеливания их окислителями
или восстановителями имеют желтоватый оттенок,
вызванный более сильным поглощением дополнитель-
ных синих и фиолетовых лучей. Отбеливающее дейст-
вие О. в. основано на том, что излучаемый ими свет
компенсирует недостаток синих лучей в отражаемом
материалом свете. Материалы, обработанные незначи-
тельными количествами О. в. (обычно 0,01 — 0,1% от
веса материала), приобретают не только высокую
степень белизны, но и повышенную яркость. Передо-
зировка О. в. может привести к снижению степени
белизны благодаря концентрационному тушению флу-
оресценции пли нек-рой окрас-
ки материала самим отбелива-
телем.
На рисунке схематически по-
казаны кривые отражения све-
та хлопком: отбеленным обыч-
ным методом (я), а затем отдель-
но обработанным синим ультра-
Длины волн, ммк
марином («синькой») (б) и оптически отбеливающим ве-
ществом (в). Отражение света окисью магния принято
за 100%. При устранении желтизны подсиниванием
ультрамарином происходит частичное поглощение
последним света в зелено-желтой и оранжево-красной
областях спектра, что естественно приводит к сни-
жению яркости материала. Подсиненный материал
выглядит белее, но менее ярким. При отбеливании с
805
ОТБЕЛИВАЮЩИЕ ВЕЩЕСТВА —ОТВЕРЖДЕНИЕ
806
помощью О. в. увеличивается количество отражае-
мого света благодаря флуоресценции в синей части
спектра.
В 1929 П. Крайс впервые наблюдал отбеливающее
действие на льняную ткань водного р-ра глюкозида
эскулетпна (Ша), флуоресцирующего синим цветом.
Однако отбеливающий эффект оказался нестойким к
свету и стирке. В 1940 было выпущено (Германия)
первое О. в. на основе п, п-диаминостильбен-о,
о'-дисульфокислоты и хлористого цианура (бланкофор
Б) (при R'=C6H5NH и R"=OH), обладающий хорошим
сродством к целлюлозным материалам. В дальнейшем
синтезировано большое число О. в., принадлежащих
к различным классам ароматич. и гетероциклич. со-
единений с развитой системой сопряженных двойных
связей. К 1963 мировой ассортимент О. в. включает
ок. 200 марок веществ с общим выпуском св. 7000 т
в год.
Ниже приведены нек-рые О. в., относящиеся к
производным стильбена (I, II; R' и R"обычно разные,
R% R"=NHAlk, N(Alk)2, NHC2H4OH, N(C2H4OH)2, NHAr,
OCH3, NHC6H4SO3Na, NH2, ОН и др.
N—N
R = SO2OAr, SOjNHAlk, SO2N(Alk)2, SO3Na и др.
R = H, SO3Na
реже они бывают одинаковые), кумарина (III), ими-
дазола (IV, при X = N—R') и оксазола (IV, ipnX=O).
x=n-r; о
R = H, Aik, CH2CH2OH, CH2CH2N(CH3)2 и др.
R=H, Aik, OCH3 и др,
Большая часть О. в., выпускаемых для целлюлозных
материалов, принадлежит к стпльбен-триазпновым
производным (I). Соединения (11), не содержащие
свободных сульфогрупп, применяют для отбеливания
полимерных материалов, а содержащие — SOaNa —
для натуральных волокон. Производные 7-аминоку-
марина (III) отбеливают протеиновые волокна, кап-
рон и ацетатный шелк. Соединения типа (IV) исполь-
зуют для отбеливания натуральных и синтетич. ма-
териалов.
О. в. применяют для оптич. отбеливания хлопка,
бумаги, лубяных волокон, шерсти, натурального шел-
ка, кожи, меха, искусственных и синтетич. волокон,
мыла, пластич. масс, лаков, воска, жиров и др. За-
патентованы О. в. для отбеливания пищевых продук-
тов (сахар, мука, рис, рыба и др.). Окрашенным из-
делиям О. в. придают яркость, а набивным тканям —
яркостьи контрастность; при этом увеличивается све-
топрочность окрасок. С помощью О. в. удается повы-
сить контрастность черно-белых фотобумаг. Эти
вещества применяют также прп приготовлении косме-
тпч. средств, прп химчистке, «несминаемой» отделке
белых тканей, изготовлеипп светозащитной оберточ-
ной бумаги для пищевых продуктов, для нанесения
невидимых при обычном свете меток и в люминесцент-
ной дефектоскопии. Бведенпе нек-рых О. в. в поли-
мерные материалы повышает устойчивость последних
к разрушающему действию УФ-радиацпи.
Применяемые О. в., кроме интенсивной флуорес-
ценции в синей части спектра, должны быть доста-
точно устойчивыми к свету п не давать окрашенных
продуктов деструкции. Б зависимости от назначения
они должны обладать сродством к волокну, устойчи-
востью к стирке, поту, повышенной темп-ре, к дейст-
вию кислот, щелочей, окислителей и восстановителей,
совмещаться с компонентами, входящими в состав
пластич. масс и т. д.
Основное количество выпускаемых О. в. потребляет-
ся в произ-вах моющих средств, бумаги и текстиля.
О. в. применяют на различных стадиях изготовления
и обработки натуральных и синтетич. материалов.
Методы оптич. отбеливания в основном аналогичны
способам крашения материалов органич. красящими
веществами, в частности отбеливание целлюлозных и
протеиновых материалов сходно с крашением послед-
них соответственно прямыми и кислотными красите-
лями. Однако для достижения нужного эффекта тре-
буется значительно меньшее количество О. в., чем
красителей. Водонерастворпмые О. в. можно приме-
нять в высокодисперсной форме пли в виде раствора
в органич. растворителях. Отбеливающий эффект
обычно оценивают визуально, хотя практически важ-
но измерять также интенсивность флуоресценции. См.
также Фотолюминесценция.
Лит.: (Ihleln Е., Optisclie Auflieller, 1957; Адамс Д.,
Химия и химическая технология. Органическая химия, 1959,
А1 6, 144; ШВА Kuiidscliau, 1960, 13, № 152; Петро-
вич П. И., Б о р и с е в и ч П. А., Изв. АН СССР. Сер. фи-
зическая, 1963, 27, Л» 5, 703. II. И. Петрович.
ОТБОР ПРОБ — см. Опробование материалов.
ОТВЕРЖДЕНИЕ — образование полимеров трех-
мерного строения из олигомера или полимера линей-
ной или разветвленной структуры. О. можно проводить
с участием отвердителя и без него.
О. с участием отвердителей приме-
няется для олигомеров пли полимеров, содержащих
функциональные группы, по не способных к межмо-
лекулярному взаимодействию. В качестве отверди-
телей используют иолпфхнкцпональные соединения,
к-рые могут реагировать с исходным олигомером или
полимером, соединяя их молекулы в полимер трех-
мерного строения. Закономерности такого типа О. оп-
ределяются характером функциональных групп. Так,
О. иолиэфпрмалеппатов впнильнымн или аллильными
производными подчиняется закономерностям ради-
кально-цепной полимеризации, эпоксидных смол по
диаминами — ступенчатой полимеризации, феполо-
формальдегидиых смол новолачпого типа гексамети-
лентетрамином — поликонденсации.
Процесс получения трехмерного полимера можно
искусственно остановить на стадии образования тер-
мореактпвиого полимера; в случае поликонденсации
26*
807
ОТДЕЛКА ТЕКСТИЛЬНЫХ МАТЕРИАЛОВ
808
это достигается снижением темп-ры и изменением pH
среды, в случае полимеризации — введением инги-
биторов. О. такого термореактивного полимера прово-
дят без отвердителя, повышая лишь темп-ру
процесса и изменяя pH среды (для поликонденса-
ции) или вводя инициаторы и активируя их распад (для
полимеризации). О. в этом случае подчиняется тем
закономерностям, к-рые лежали в основе образования
термореактивиого полимера.
В зависимости от температурных режимов разли-
чают холодное (при комнатной темп-ре) и г о-
р я ч е е (при повышенной теми-ре) О. Холодное О.
феноло-формальдегпдных смол резольного типа про-
водят в кислой среде, карбамидных смол — в кислой
среде или в присутствии солей сильных к-т, эпоксид-
ных смол — с участием полиаминов и т. д. О.— важ-
ная технология, операция в произ-ве пластич. масс,
при использовании заливочных компаундов, клеев
на основе термореактпвных полимеров, нанесении ла-
кокрасочных покрытий и др.
Лит.: Барг Э. И., Технология синтетических пластиче-
ских масс, Л., 1954; Лосев И. П., Тростянская
Е. Б., Химия синтетических полимеров, М., 1964; Бай-
те й д т А. А., Кузнецова Н. Н., Ж. прикл. химии,
1957, 30, вып. 12, 1850; Барановская Н. Б. [и д р. J,
ДАН СССР, 1958, 122, № 4, 603; Королев Г. В., М а X о-
нина Л. И., Берлин А. А., Высокомолекулярные со-
единения, 1961, 3, К» 2, 198; П а к е н А. М., Эпоксидные со-
единения и эпоксидные смолы, пер. с нем., Л., 1962.
Е. Б. Тростянская.
ОТДЕЛКА ТЕКСТИЛЬНЫХ МАТЕРИАЛОВ —
операции, цель к-рых— улучшение внешнего вида и
повышение добротности и износоустойчивости тканей.
В результате отделки ткани приобретают ряд ценных
свойств — несминаемость, безусадочность, повышен-
ную устойчивость к истиранию, противогнилостные
свойства и т. д. Отделка тканей из различных воло-
кон имеет свои особенности. Помимо тканей, отделы-
вают также различные трикотажные изделия и в от-
дельных случаях пряжу, когда она выпускается в ви-
де товарной продукции (ниточные изделия). Сущест-
вуют различные способы отделки тканей в связи с их
целевым назначением и волокнистым материалом, из
к-рого они изготовлены.
Для повышения белпзны фона пли фигуры рисунка
ткань обрабатывают слабым р-ром гипохлорита с до-
бавкой ультрамарина. Однако эта обработка вызывает
нек-рое ослабление ткани. Для повышения белпзны
ткани без снижения прочности ее используют также
оптические отбеливающие в-ва, к-рые адсорбируются
целлюлозой и флуоресцируют сине-фиолетовым цве-
том. При этом экранируется желтизна ткани, значи-
тельно повышается белизна и придается свежесть бе-
лому фону и фигурам ненапечатанной ткани; концент-
рация применяемых р-ров 0,0033—0,00166 г/л [см.
Отбеливающие вещества (оптически)].
Целлюлоза при действии света и атмосферных усло-
вий подвергается деструкции, при этом ткани из рас-
тительных волокон теряют устойчивость к изнаши-
ванию. Защитить целлюлозу от воздействия этих фак-
торов можно с помощью аппретов, наносимых на ткань
при ее заключительной отделке. Хлопчатобумаж-
ные и льняные ткани до сих пор аппретируют крахма-
лом. Крахмал окисляется иод действием света раньше
целлюлозы, и, т. обр., предотвращает ее окисление.
Однако крахмал удерживается на ткапи очень непроч-
но и смывается при первой же стирке. Кроме того,
это пищевой продукт и его применение для аппрети-
рования нежелательно. В заграничной практике для
этой цели пользуются различными эфирами целлю-
лозы, и в частности оксиэтиловым эфиром. Пленка
эфиров целлюлозы прочно фиксируется на ткани и
выдерживает многократные стирки.
Сравнительно широко применяют синтетич. высо-
комолекулярные соединения, получаемые в виде высо-
кодисперсных суспензий (т. наз. латексов). В нашей
пром-сти используются латексы: сополимер хлорис-
того винила и хлористого винилидена, полиметилме-
такриловый, иолистирольный и другие. Ткань про-
питывают на аппаратах непрерывного действия (плю-
совках) латексами, разбавленными водой до опреде-
ленной концентрации, и высушивают на сушильных
барабанах при темп-ре выше 100°. В этих условиях
латекс коагулирует и образует на поверхности эле-
ментарного волокна тонкую пленку полимера. Такая
обработка повышает прочность ткани на разрыв и
истирание и улучшает внешний вид.
Несмываемые аппреты на ткани мож-
но получить, используя также смеси предконденсатов
термореактивных смол и полиоксисоединений. Одна-
ко такое совмещение вызывает резкое падение проч-
ности волокна. Если же иредконденсат смолы, напри-
мер карбамидной (т. наз. карбамол), предварительно
модифицировать аммиаком, то его можно совмещать
с полиоксисоединениями, не опасаясь деструкции
ткани:
-NHCHjOH
СО (ХНСН20Н)2 + ХН3 — +Н2о
ХХНСН2ХН2
Технология нанесения этого аппрета весьма проста,
экономически выгодна, аппрет совершенно не вымы-
вается с ткани при многократных стирках.
Большое значение имеет придание хлопчатобумаж-
ным, льняным тканям и тканям из вискозного штапеля
свойств н е с м и н а е м о с т и. Известно, что сми-
наемость волокнистых материалов зависит от упруго-
эластич. свойств волокна; чем они выше, тем меньше
волокно подвергается смятию. Упруго-эластические
свойства волокна проявляются при наличии прочных
поперечных связей между макромолекулами данного
волокна. Наир., макромолекулы кератина шерсти
связаны прочными химич. цистиновыми связями и
шерстяное волокно обладает хорошо выраженными
упруго-эластическими свойствами; при смятии оно
легко расправляется. В целлюлозных волокнах мак-
ромолекулы целлюлозы не имеют прочных химич.
поперечных связей, а силы водородной связи и Ван-
дер-Ваальса пе обеспечивают проявления упругих
свойств этих волокон, п при смятии они ие выпрям-
ляются.
Для придания хлопчатобумажным и штапельным
тканям свойств несмпнаемостп применяют гл. обр.
продукты начальной конденсации мочевины или ме-
ламина с формальдегидом (соответственно, «карба-
мол» и «метазпн»). Оба продукта хорошо растворимы
в воде. Ткань пропитывают раствором, высушивают
и обрабатывают термически. Основной реакцией, ве-
дущей к образованию поперечных связей, является,
наир., в случае карбамола:
—ОН НО-СН—НХ-С—ХН-СН2-ОН НО-
11
о
—ОН НО—CHo-IIN-С—ХН-СН2—ОН НО-
11
о
Н—X—CH.OH
I
с = о +
I
H-N-CH.OH
Н—X—СН-ОН
<!. = о -н’°
I *
Н—N—СН20Н
H-x-cHj-----n—с:н2он
Z^o (Lo
I '
Н—X—СН20Н Н—N—СН20Н
809
ОТДЕЛКА ТЕКСТИЛЬНЫХ МАТЕРИАЛОВ
810
3. H-N-CH20H НОСНг-N-H
С = О + С. = О ~На°
I I *
h-n-ch2oh hoch2-n-h
H-N-CHj--О--CH2-N-H
-Н20 (1 = 0 С-0
* I I
H—N—CH,0H HOCH,—N—H
При этом образуются также циклич. соединения пз
2, 3 и более молекул диметилолмочевины:
4- HOH2CNH сн2 nhch2oh
znh-ch2oh o=c-nz 4n-c=o
C = ° ----- H2C 6h2
NH-CHaOH N
I
c=o
nhch2oh
В реакции 1 могут принимать участие не только
продукты начальной конденсации, но также и более
сложные соединения, к-рые получаются по реакциям
2—4. В нек-рых случаях для предотвращения излиш-
ней потери прочности волокна в процессе его отделки
выгодно уменьшить склонность продукта начальной
конденсации к смолообразованию н, наоборот, жела-
тельно, чтобы преимущественно шла реакция 1. Сде-
лать это можно, применяя вместо карбамола димети-
лолэтиленмочевину (карбамол ЦЭМ, I) или тетраме-
тилолацетилендимочевину (II).
СН2ОН
zNx
/ сн2
О=С I
\ _сн2
N
I
СН2ОН
НОН2С н СН2ОН
N-C-N
°=С\ I /С=°
N-C-N
Z I \
нон2с н сн2он
1 II
У этих соединений отсутствует очень активный
атом водорода у азота, связанного с метилольной
группой, поэтому реакции смолообразования и цик-
лизации подавляются, реакция же 1, наоборот, уси-
ливается. К числу т. наз. реактивных смол, помимо
карбамола ЦЭМ и ацетилендимочевины, относят еще
диуретан и эпоксидные соединения.
Для отделки тканей из гидратцеллюлозных волокон
пользуются, напр., след, рецептурой пропиточных
ванн: 200—300 г/л иредконденсата аминоформальде-
гидной смолы, 20 — 40 г/л смягчающего вещества (пре-
парат AM пли ОС-2), 4—5 г/л катализатора, напр.
MgCl2. Раствором ткань пропитывают на плюсовке,
высушивают и термически обрабатывают при теми-ре
ок. 150° в течение 3—5 мин. В результате такой об-
работки сминаемость ткани резко уменьшается. В це-
лях повышения эффекта несминаемости в пропиточную
ванну иногда вводят гидрофобизирующие вещества.
Применение приведенной рецептуры для отделки
хлошгатобумажных тканей сопряжено с резким паде-
нием их прочности (до 30—40%). Этот недостаток мо-
жет быть частично устранен предварительным (перед
отделкой) разрыхлением природного волокна мерсе-
ризацией, или комбинированной отделкой. Последняя
состоит из обработки ткани термопластич. смолами, а
также нредконденсатами термореактпвных амипоформ-
альдегпдных смол, напр. с карбамолом ЦЭМ. Назна-
чение первого компонента — компенсация падения
прочности ткани, на разрыв и истирание в процессе ее
отделки, роль второго компонента — придание ткани
несминаемости. Пример отделки: 70 г/л карбамола
ЦЭМ, 17,5 г/л акриловой эмульсии, 8 г/л силиконовой
эмульсии, 5 г/л катализатора MgCL или Zn(NOs)2.
Режим обработки такой же, как и ранее. Прп этом
способе обработки ткань приобретает настолько вы-
сокую несминаемость, что не требует глажения после
стирки. Потери прочности не превышают 10%, эффект
несминаемости не изменяется после 24 стирок.
Пользуясь спнтетич. смолами, можно получать
на тканях эффекты тиснения, к-рые делают их похо-
жими на ткани жаккардового переплетения, а также
эффекты лощения, муаровый и др. Для этого приме-
няют специальные типы каландров. Несминаемые
ткани можно изготовлять также из смесей волокон
натуральных и синтетических, напр. из смеси хлоп-
ка и лавсана (до 60%).
Важной задачей для нек-рого ассортимента тка-
ней, напр. плащевых, является придание им водо-
отталкивающих свойств. Для этого по-
верхность таких тканей подвергают гидрофобизации.
Используя комплексные соединения хрома со стеари-
новой к-той или силиконы, удается получать ткани с
водоотталкивающими свойствами и в то же время мяг-
кие па ощупь. Эти соединения можно также комбини-
ровать со смолами, придающими тканям несмипае-
мость. Реагентами могут быть: алкилизоцианат (III),
алкилэтиленимпномочевина (IV), веланоподобные
соединения (V), хромолаи (VI), силикон (VII):
zch2
R-N = CO R-.NH-CO— N( |
<Н.
IV
Ш
R-O-CH2-N^ СГ СГ\ОН11+4С1-
V VI
- СН3 СН3 СН3 -
—Si—О—Si—О—Si—О—
I I I
L Н Н Н -1Н
VII
Соединения III, IV nV, реагируя с гидроксильными
группами целлюлозы, образуют химич. связи. О ха-
рактере взаимодействия соединений VI и VII с волок-
нистыми материалами нет единого мнения.
Большинство текстильных материалов, прежде все-
го целлюлозные волокна, подвержено разрушению
микроорганизмами: бактериями и плесневыми гриб-
ками. Для продления срока службы изделий, особен-
но подвергающихся длительному воздействию тепла
и влаги, их обрабатывают антисептическими вещест-
вами (противогнилостная обработка). С этой целью
широко применяют дубильные экстракты, к-рые
фиксируются на ткани солями меди и хрома. Сущест-
вуют и другие способы защиты волокон от действия
микроорганизмов. Химич, превращение целлюлозы
дает наиболее устойчивый противогнилостный эффект.
Одним из таких способов является цианэтилирование
целлюлозы:
цел. -ОН + СН2 = СН—>цел.-O-CHj-CHj
I I
CN CN
С целью придания текстильным материалам свойств
негорючести их подвергают огнезащитным
пропиткам. В ранних способах огнезащитной пропит-
ки ткань обрабатывали растворами различных мине-
ральных соединений, напр. смесью буры, борной к-ты
и фосфата аммония. Однако после промывки ткани пол-
ностью теряют огнестойкость. Более эффективно нане-
сение на тканьили изделия из нее солей сурьмы, тита-
на, олова и др. и затем переведение их в нерастворимые
в воде соединения, а также хлорированных углеводо-
родов, наир, хлорнафталина. Одним из новых эффек-
тивных способов огнезащитной обработки ткани яв-
811
ОТДЕЛКА ТКАНЕЙ СПЕЦИАЛЬНАЯ —ОТПУГИВАЮЩИЕ СРЕДСТВА
812
ляется образование эфиров целлюлозы при обработке
фосфорной к-той в присутствии дициандиамида, а
также препаратом ТНРС (VIII):
НОН2С. „СН20Н1
>Р( С1
нон2сх xch2ohJ
VIII
Потерю прочности ткани устраняют введением в про-
питочный р-р мочевины, к-рая связывает выделяющий-
ся НС1.
При отделке шерстяных тканей особое внимание
обращается на безусадочность их. С этой
целью предусматривают тепловую обработку ткани в
специальной аппаратуре, к-рая позволяет получить
стабильные линейные размеры. Для изделий из син-
тетич. полимеров особое значение приобретает ста-
билизация заданных размеров и формы материала.
Это достигается прогревом изделий в заданной форме
прп темп-рах более высоких, чем те, к-рым они будут
подвергаться в условиях эксплуатации. Для прогре-
вания пользуются горячим воздухом, перегретым па-
ром п др. теплоносителями.
Для защиты шерстяных тканей от моли их обраба-
тывают, напр., препаратом ДДТ и др.
Лит.: Садов Ф. И.. Корчагин М. В., М а т е ц-
к и Й А. И., Химическая технология волокнистых материалов,
2 изд,, М.. 1956; Гордон Н. Б., Б о р и с о в Н. А., От-
делка льняных тканей, М., 1956; М а р ш Д ж., Заключитель-
ная отделка текстильных материалов, пер. с англ., М., 1956;
Ф р о т ш е р Г., Химия и физическая химия текстильных
вспомогательных материалов, пер. с нем., т. 2, М., 1958;
Морыганов П. В. [и др.], вкн.: Труды Всесоюзной
Межвузовской конференции по вопросам синтеза и примене-
ния органических красителей, Иваново, 1962, с. 125; Архи-
пова Т. Н. |и др.], Сб. рефератов трудов ЦНИХБИ,
сер. Отделка, М., 1955—60; СимигинП. А., 3 усман
М. Н.. Р а й X л и н Ф. И., Защитные пропитки текстильных
материалов, М., 1957; Морыганов П. В. [и др.], Тек-
стильная пром-сть, 1961, № 4; Вацлав Ф., За-
ключительная отделка текстильных материалов, пер. с
чеш.., М., 1964. П. В. Морыганов, Б. Н. Мельников.
ОТДЕЛКА ТКАНЕЙ СПЕЦИАЛЬНАЯ — обра-
ботка тканей с целью придания им свойств несминае-
мости, отсутствия усадки, способности не воспламе-
няться и др. Подробнее см. Отделка текстильных ма-
териалов.
ОТКРЫВАЕМЫЙ МИНИМУМ — наименьшее ко-
личество вещества, достоверно открываемое аналитич.
методами. Подробнее см. Чувствительность реакции.
ОТНОСИТЕЛЬНАЯ ВЛАЖНОСТЬ — отношение
весового количества водяного пара в каком-нибудь
объеме газовой смеси (в частности, в воздухе) к весо-
вому количеству насыщенного водяного пара, содер-
жащегося в том же объеме прп той же темп-ре, выра-
женное в процентах. О. в. характеризует, т. обр.,
степень насыщенности водяного пара в данной газо-
вой смеси. Эта величина используется при различных
технич. (и других) расчетах.' Она дает возможность,
напр., легко определить, при каком понижении темп-ры
данной газовой смесп в ней начнется конденсация
водяного njpa в жидкость. Темп-ра начала конден-
сации наз. точкой росы. Измерение ее позво-
ляет с помощью таблицы давления водяного пара в
зависимости от темп-ры определить О. в. Для опре-
деления О. в. воздуха используется также психрометр.
Этот прибор содержит 2 термометра, шарик одного
из них окружен тканью, смоченной водой. Вследствие
испарения воды темп-ра смоченного термометра ниже
темп-ры окружающего воздуха. Это понижение темп-ры
термометра прп прочих равных условиях тем меньше,
чем выше О. в. воздуха. Из показаний сухого и смо-
ченного термометра находят влажность или О. в.
воздуха с помощью специальных психометрических
таблиц.
ОТПУГИВАЮЩИЕ СРЕДСТВА (репелленты) —
вещества, применяемые для предохранения человека
и животных от укусов насекомых и клещей. К О. с.
предъявляют след, требования: малая токсичность;
отсутствие резорбтивного действия и раздражающего
действия на кожу; возможно большая длительность
отпугивающего действия; отсутствие неприятного за-
паха и др. Важнейшие О. с. и нек-рые их свойства
приведены в таблице.
СООСНд
СООСНз
Г^ГСООСНз
%>СООСН3
II
СООС4Н9
С3Н7--СН--СН--СН2ОН
он С2Н5
IV V
С4Н9ОСО(СН2)4СООС4Н9
Ml
сн2-сн2-сн2ч
I ncoc6h5 .
сн2-сн2-сн2
VII
Свойства некоторых наиболее распространенных отпугивающих препаратов
Название Т. кип., °C jtAt рт. ст. 20 d 4 Продолжительность действия на коже, в ча- сах Способ получения
Дпмстилфталат (I) 280/734 1 ,192 2 — 4 Этерификацией фталевого ангидрида ме- танол ом
дпметилкарбат (II) 114 — 115/3,8 1.173 2 — 4 Конденсацией дпметилмалеата с цинло- пеитадиеном
2-Этплгександиол- 1 .3 (III) 244/760 0,979 2-4 Альдольной конденсацией масляного альдегида, с Последующим восстановле- нием полученного окснальдегида до 2-этил- гександиола
Индалон (IV) 175/20 1 ,051 5 — 6 Конденсацией окиси мезитила с дибутил- оксалатом в присутствии бутилата нат- рия
ДЭТА (диэтилмета- толуамид) (V) 111/1 1 ,009 10 — 1 2 Взаимодействием хлорангпдрпда .п-то- луиловон к-ты с диэтпламином
дпбутиладипат (VI) 18 3/14 0,965 24-48; только против клещей Этерификацией адипиновой к-ты бута- нолом
Гексаметаленбенз- амид (VII) 190 — 190,5/760 1 6; при опрыскивании С.-Х. ЖИВОТНЫХ 2%-НЫ- МИ водными эмульсия- ми — до 3 суток- Бензоилированием 4 о—50%-пой азео- тропной смеси гексаметиленимида с водой
813
ОТРАВЛЕНИЕ КАТАЛИЗАТОРОВ — ОТРАВЛЯЮЩИЕ ВЕЩЕСТВА
814
Таблица 1
Довольно широкое применение получили также
смеси нескольких О. с., напр. препарат рют-
гер 622 (60% диметилфталата, 20% индалона и
20% 2-этплгександиола-1,3), препарат ДИД
(75% диметилфталата, 20% индалона и 5% диметнл-
карбата) и др. Кроме того, в качестве О. с. предложе-
ны эфироамиды янтарной к-ты, алкилацетанилиды и
др. Для предохранения посевов от птиц применяют
препарат м о р к и т, изготовляемый на основе
антрахинона.
Обычно О. с. наносят на открытые участки кожи;
от кровососущих можно также защищаться путем
пропитки О. с. одежды, специальных защитных се-
ток и т. п.
Лит.: Мандельбаум Я. А,,Сафьянова В.М.,
Ломакина В. И., Ж. Всес. хим. об-ва, i960, 5, № 3, 307;
Advances in pest control research, ed. R, L. Metcalf, v. 1, N. Y.,
1957; Dethier V. G., Chemical Insect attractants and repel-
lents, L., 1948. H. H. Мельников.
ОТРАВЛЕНИЕ КАТАЛИЗАТОРОВ — см. Ката-
литические яды.
ОТРАВЛЯЮЩИЕ ВЕЩЕСТВА (О В) — высокото-
ксичные соединения, пригодные для военного приме-
нения с целью поражения живой силы противника.
ОВ в больших масштабах были применены в первую
мировую войну.
Массированное применение ОВ впервые было осуществлено
Германией в апреле 1915; на фронте шириной 6 км было выпу-
щено!80 тхлораиз 6000 баллонов. В результате этого внезап-
ного нападения было выведено из строя ок. 15000 человек, из
к-рых 5000 погибло; фронт на протяжении 8 км был прорван.
В дальнейшем немцами были применены и другие ОВ: в конце
1915 — фосген, его смесь с хлором, дифосген; в 1916 —хлор-
пикрин, к-рый действует пе только на органы дыхания, но и на
слизистые оболочки глаз.
Вслед за применением ОВ появились и средства защиты от
них. Вначале это были влажные повязки, пропитанные р-ром
тиосульфата (гипосульфита) натрия, соды, уротропина. Затем
начали применять противогазы с активным углем в качестве
универсального поглотителя газообразных и парообразных
О В. Дальнейшие поиски новых ОВ и способов их применения
были направлены на преодоление средств противохимич. за-
щиты. В июле 1917 Германия впервые применила дихлорди-
этилсулъфид (иприт), обладающий в жидком и парообразном
состояниях наряду с общетоксическим и сильным кожно-на-
рывным действием. Применение иприта, к-рый сыграл большую
роль в 1-й мировой войне, потребовало разработки средств за-
щиты не только органовдыхания, но и кожных покровов (см.
Защитная одежда и Противохимическая защита). Позднее, с
целью преодоления противогаза, немецкие войска путем тер-
мич. возгонки (ядовито-дымные шашки) или механич. распыле-
ния (артиллерийские химич. снаряды) применили ОВ раздра-
жающего действия (дифенилхлорарсин, дифенилцианарсин).
Для защиты от ядовитых дымов в противогаз был введен про-
тиводымный фильтр (вначале из ваты, затем из пуха и, наконец,
из тонковолокнистого асбеста).
Во время 1-й мировой войны широко применялись также
хлор- и бромацетоны, эфир бромуксусной к-ты, эфиры хлор-
уюльноЙ к-ты, алкилдихлорарсины, фенилдихлорарсин и ряд
др. ОВ. Всеми воюющими державами было использовано
125 000 т ОВ, из к-рых 47 000 — Германией. Из большого чис-
ла разнообразных примененных ОВ только нек-рые сохранили
в дальнейшем свое значение.
Общие потери от применения ОВ за время первой мировой
войны составили 700—800 тыс. человек. В армии США,
•вступившей в войну лишь в 1917 г., т. е. в период наиболее ин-
тенсивного применения ОВ, потери от них составили ок. 25%.
Накануне окончания 1-й мировой войны были оценены в
качестве ОВ. но не были применены: люизит C1CH=CHAsC12—
ОВ быстрого кожно-нарывного действия, хлорацетофенон
С«Н GOGHoCl (мощный лакриматор), адамсит и др.
В период между 1-й и 2-й мировыми войнами про-
должались интенсивные поиски новых ОВ. Так, в
С1\ .
1929 г. были получены фосгеноксим q]/^= ХОН —
ОВ многостороннего физиологии, действия; три (|3-
хлорэтил)-ампн NfCH.CH.Cl^ (см. Азотистые ип-
риты) — мощное кожно-нарывное ОВ и ряд других.
В 1932 появились первые работы в области веществ
пефвно-пара.чптич. действия из класса диалкп.чфтор-
фосфатов (RO2)POF. О масштабах подготовки к веде-
нию химич. войны свидетельствует качественный и
количественный рост производства ОВ, к-рого достиг-
ла Германия ко второй мировой войне (табл. 1).
Производственные мощности, тыс. m
в 1914—1918 гг.
Фосген.................. 11
Дифосген................ 15,6
Мышья ^содержащие
ОВ..................... 1,1
Иприт ............. 9
Лакриматоры .... 1,6
Хлорпикрин......... 4
Трихлортриэтиламин —
Зарин.............. —
Табун.............. —
Прочие О В............... 1,4
4
7
7
125
15
(хлорацетофенон)
1 . 4
7,2
12
ок. 45
ок. 180
После поражения фашистской Германии стало из-
вестно, что она имела на вооружении высокотоксич-
ные быстродействующие О В нервно-паралитич. дейст-
вия. К ним относятся: табун, зарин и зоман (см. табл.
2). Одного вдоха воздуха, содержащего 2—.3 мг/л
этих ОВ, достаточно для смертельного отравления.
Современные средства защиты дыхания (противогаз)
надежно защищают от этих сильно действующих О В.
Эти О В сильно токсичны и при попадании на кожу,
что ведет к необходимости ее общей защиты, как и в
случае иприта. Наибольшей кожнорезорбтивной ток-
сичностью из веществ этого типа обладают фосфорил-
тиохолины общей формулы
О
II ХОВ
СН.-Рб
XS—СН2—СН2—NR,
Попадание 2—3 мг веществ этого типа на обнаженную
кожу может привести к смертельному исходу. Физич.
свойства и константы нек-рых современных О В при-
ведены в табл. 2.
Во время второй мировой войны О В не были при-
менены, несмотря на то, что фашистская Германия бы-
ла подготовлена к применению новых высокотоксич-
ных ОВ. Это может быть объяснено тем, что в начале
войны фашистская Германия делала ставку на молние-
носное окончание войны, а после потери стратегии,
инициативы и нанесения советскими армиями ударов
по фашистским захватчикам фашистское командова-
ние уже не решалось начать химич. войну, т. к. при-
менение химич. оружия в условиях, когда военные
действия переместились к границам Германии постави-
ло бы густо населенную территорию Германии под от-
ветный удар советских и союзных войск.
Токсичность ОВ. В основе токсич. действия боль-
шинства О В лежат реакции ингибирования фермент-
ных систем организма. Так, синильная к-та чрезвы-
чайно быстро ингибирует железосодержащие окисли-
тельные системы Варбурга, вызывая вначале внутри-
клеточное кислородное голодание, а затем паралич
дыхательных центров. Люизит быстро реагирует с
сульфидрильнымп группами различных ферментов,
выводя пх из строя. Аналогичным действием облада-
ют все лакриматоры. Иприт, помимо ингибирования
гексокпназы, действует на ядерный аппарат клеток.
Действие трихлортрпэтиламина аналогично иприту.
Фосфоросодержащие ОВ—табун, зарин, зомаи и др.
очень быстро фосфорилируют холинэстеразу, инги-
бируя ее функции в передаче нервного импульса.
Действие фосгена и его аналогов (дифосген, эфиры
хлоругольноп к-ты, фосгеноксим и др.) наряду с реак-
цией со всевозможными белками также, по-видимому,
направлено на различные ферментные системы. В ос-
нове взаимодействия ряда О В с ферментами лежат
реакции с пх нуклеофильными группировками: NH2,
815
ОТРАВЛЯЮЩИЕ ВЕЩЕСТВА
816
Таблица 2
Название ОВ Формула 20 d 4 20 nD т. пл., °C т. кип., °C Давление па- ра при 20°, мм рт. ст. Летучесть при 20°(макс, кон- центр.), мг/л Растворимость в воде при 20°, %
Иприт Азотистый иприт Диизопро- пилфтор- фосфат S (СН2СН2С1)2 N (СН2СН2С1)3 0 (С3Н7О)2р( F 1 ,28 1,23 1,07 1,529 1 ,496 1 , 383 14,5 —4 -82 217 (760 мл» с разл.) 117/26 мм 1 25/1 0 мм 94/1 мм 67,5/12 мм 0,055/1 0° 0,007 0,9 0,6—0,7 70 9,2 0,05 0,02 1,5
Зарин Зоман Табун С3Н7О 0 СН/ XF (СН3)3С—СН—О—Р— О 1 СН3 Н3С F 0 II NC—Р—N (СН3)2 1,09 1,013 1,08 1,383 1,408 1,424 -54 -80 -50 151,5; 50/12 мм 42/0,2 мм 220; 108/1 2 мм 1,43 0,073 11,3 3 0,60—0,65 Смешивается во всех отноше- ниях Ограниченно растворим 12
ОС2Н,
ОН, SH и др. Начальной ступенью взаимодействия
нек-рых OB (HCN, AsHa, РНз и др.) является комплек-
сообразование с атомом железа гемоглобина крови
или железосодержащих ферментов, в ряде случаев с
последующим изменением образующегося комплек-
са. Многие ОВ обладают сильным мутагенным дейст-
вием, благодаря к-рому нек-рые из них применяются
н качестве канцеролитич. веществ (иприт, азотистый
иприт и их аналоги). В последнее время констатиро-
вано явное мутагенное действие и веществ типа за-
рина. Степень токсичности ОВ зависит от избиратель-
ности их действия. Так, токсичность фосгена сравни-
тельно невелика, в связи с тем, что он, помимо фермен-
тов, реагирует с многочисленными веществами орга-
низма. Наоборот, чрезвычайно сильная токсичность
фосфорилирующих ОВ объясняется высокой избира-
тельностью их взаимодействия с холинэстеразой. Взаи-
модействие ОВ с ферментами в ряде случаев приводит
к весьма прочным соединениям, что при полном инги-
бировании ферментов делает терапевтии, вмешательст-
во практически безрезультатным. В нек-рых случаях
возникает возможность регенерации фермента. Так,
введение в организм антидотов, напр. димеркаптопро-
панола («ВАЛ»), снимает токсич. действие люизита,
даже в случаях далеко зашедших отравлений. Дп-
меркаптоиропанол реагирует с продуктами взаимо-
действия ферментов с люизитом, освобождая фермент:
HS-CH2
.S—ферм. I
ClCH = CHAs( + HS—СН—>
4 S—ферм. |
HS—сн2
S—сн3
—»-С1СН = СН—As, | +2 ферм.—SH
XS-CH
I
но—сн2
Введение в организм иодметилата а-пиридилальд-
оксима («ПАМ») предотвращает смертельный исход
при отравлении зарином, зомапом и др. фосфорили-
рующими ОВ. При этом ПАМ фосфорилируется про-
дуктом фосфорилирования холинэстеразы зарином и
освобождает последнюю. Прп отравлении синильной
к-той смертельный исход может быть предотвращен
введением в организм мощного окислителя гемоглоби-
на (амилнитрит и др.). Гемоглобин при этом окисляет-
ся в метгемоглобин и последний, вследствие большего
сродства к HCN нежели к ней имеют ферменты, «сни-
мает» синильную к-ту, и ферменты освобождаются.
Из образующегося при этом циаиметгемоглобина
гемоглобин медленно регенерируется за счет углевод-
ного и серного обмена (окисление HCN до HCNO и
HCNS). Естественно, что создание антидотов возмож-
но лишь при глубоком познании природы и механизма
действия ОВ; отсутствие антидотов против ряда ОВ
(фосген, иприт, азотистые пириты и др.) в значитель-
ной степени объясняется именно слабым знанием ме-
ханизма действия этих ОВ.
Классификация отравляющих веществ. О В отно-
сятся к разнообразным классам неорганич. и особенно
органич. соединений. Обычно ОВ обладают разносто-
ронним токсич. действием на организм; одно и то же
вещество может применяться в различных средствах
химического нападения в зависимости от поставленной
задачи. Вещества, принадлежащие к различ. классам
соединений, могут быть близкими по характеру дейст-
вия. Напр., слезоточивым действием обладают хлор-
пикрин, хлорацетофенон, бромбензилцпанпд, акро-
леин и др. Поэтому строгая, последовательная клас-
сификация ОВ чрезвычайно затруднительна. Наиболь-
шее практич. значение имеют физиологич. и тактич.
классификации О В.
Физиологич. классификация основа-
на на преобладающем характере действия О В на орга-
низм. По этой классификации все ОВ можно разбить
на 5 групп: нервно-паралитические, общеядовитые,
кожно-нарывные, удушающие и раздражающие
(табл. 3).
Условность физиологич. классификации видна хотя
бы из того, что все представители кожно-нарывной
группы О В обладают не меныпим удушающим дейст-
вием, чем ОВ, к-рые составляют самостоятельную
группу удушающих ОВ. Совершенно так же ряд
лакриматоров обладает побочным — кожным (хлор-
ацетофенон) , либо удушающим (хлорпикрин) действием.
Тактическая классификация груп-
пирует О В по тактическому назначению и поведению
их на местности в условиях боевого применения. Сог-
ласно этой классификации все ОВ разделяются на:
нестойкие отравляющие вещества (НОВ); стойкие от-
равляющие вещества (СОВ); ядовитые дымообразую-
щие вещества (ЯДВ).
К группе нестойких относятся вещества с высоким
давлением пара п низкими темп-рами кипения (при-
мерно до 140°). Эти вещества, попадая в атмосферу в
виде паров, образуют «газовое облако», к-рое быстра
распространяется в атмосфере и рассеивается. Ско-
рость рассеивания в сильной степени зависит от ме-
теорология. условий и от рельефа местности. Нестой-
кие ОВ применялись гл. обр. для поражения живой
силы противника через органы дыхания.
817
ОТРАВЛЯЮЩИЕ ВЕЩЕСТВА — ОТСТАИВАНИЕ
818
Таблица 3
Нервно-парали- тические Ов Общеядовитые ОВ ОВ, действующие на ткани легких (уду- шающие) ОВ, поражающие кожу и действу- ющие через кожу (кожно-нарыв- ные) ОВ, раздражающие слизистые оболочки глаз (лакриматоры) ОВ, раздражающие верхние дыхательные пути, вызывающие чи- ханье, кашель (стерни- ты)
диизопропил- фторфосфат Зарин Зоман Табун Фосфорилтио- XОЛИНЫ Синильная к-та Хлорциан Окись углерода Мышьяковистый водород Фосфористый водо- род Хлор Фосген Дифосген Алкилхлоркарбо- наты Фосгеноксим Иприт Трихлортри- этиламин Фосгеноксим Бромбензилцианид Хлорацетофенон Хлорпикрин Акролеин Галогенацетаты дифенилхлорарсин Дифеннлцианарсин Адамсит Капсаицин и его ана- логи
К группе стойких ОВ относятся вещества, имеющие
сравнительно высокую темп-ру кипения (выше 140°)
и незначительное давление пара. Эти ОВ обладают
значительной химич. стойкостью: от нескольких
часов (летом) до нескольких дней и даже месяцев (зи-
мой). Стойкие ОВ могут применяться для заражения
местности, а также для поражения живой силы против-
ника; в виде тумана эти ОВ могут применяться и для
заражения атмосферы. Между группами нестойких
и стойких ОВ существует постепенный переход, п
ряд веществ может быть с одинаковым правом отне-
сен к любой из них (дифосген, фосгеноксим и др.); поэ-
тому часто говорят о группе (<полустойких» ОВ.
К группе ядовитых дымообразующих веществ отно-
сятся соединения, обладающие почти всегда очень вы-
сокой темп-рой кипения и весьма малым давлением
пара. Они могут быть применены в виде аэрозолей
для заражения атмосферы.
Индикация ОВ. Большое значение при защи-
те от ОВ имеет быстрое определение вачала химич.
нападения, установление характера и концентрации
применяемого ОВ. Наиболее надежным методом инди-
кации ОВ является инструментальная разведка, ос-
нованная на использовании физич. и химич. свойств
ОВ. В основе большинства технич. средств индикации
лежат те пли иные реакции ОВ со специальными реак-
тивами, приводящие к образованию окрашенных сое-
динений. Возникающая окраска определяется визуаль-
но либо колориметрически. Так, для определения
фосгена и дифосгена используется их способность к
образованию с п-дпметилбензальдегидом красителей
ди- и трифенплметанового ряда. При взаимодействии
синильной к-ты с уксуснокислой медью и бензидином
образуется окрашенная бензидиновая синь. Опреде-
ление иприта, хлорцпана, фосгеноксима основано
на их способности образовывать с пиридиновыми ос-
нованиями соответствующие солп пиридиния, к-рые
при последующем взаимодействии с ароматич. амина-
ми или метплциклоамониевыми солями дают окра-
шенные производные глютакондиальдегида. Галоге-
нокетоны индицируют по окрашенным в красный цвет
комплексам, образующимся при их взаимодействии с
метадинитробензолом в щелочной среде. Люизит вна-
чале разлагают щелочью до ацетилена, к-рый далее
обнаруживают по образованию красного ацетиленида
меди и т. д.
Индикацию ОВ чаще всего проводят в индикатор-
ных трубках или на индикаторных бумажках. Инди-
каторные трубки обычно наполняют сорбентом (си-
ликагелем, стеклянной ватой, асбестом и др.), про-
питанным тем пли иным реактивом, дающим окрас-
ку с ОВ. Если требуется применить несколько реак-
тивов, свойства к-рых не позволяют их совместить на
сорбенте, то второй реактив запаивается в ампулу,
к-рый вводится в индикаторную трубку. Перед опреде-
лением у трубки обламывают концы; трубка вставляет-
ся в насос и через нее просасывается исследуемый
воздух. После просасывания ампула разбивается и ее
содержимое смачивает сорбент, к-рый окрашивается
в соответствующий цвет. В современных индикатор-
ных приборах (напр., в приборе химич. разведки —
ПХР) содержится набор таких индикаторных трубок
на важнейшие ОВ. Чувствительность трубок находит-
ся в соответствии с требованиями обнаружения безо-
пасных концентраций ОВ. Так, в трубке на фосген
при его концентрации 0,005 мг/л воздуха сорбент ок-
рашивается в синий цвет; на синильную кислоту (0,005
мг/л) — в вишнево-красный цвет; на иприт и люизит
при концентрациях 0,003 и 0,002 мг/л — в красный
цвет; на зарин (0,0003 мг/л) — в светло-желтый до
светло-оранжевого цвета и т.д. В случаях, когда хи-
мич. методы индикации оказываются недостаточно
чувствительными, прибегают к биохимии, методам
определения. Напр., о наличии фосфорфторсодержа-
щих ОВ нервно-паралитич. действия можно судить
по разнице во времени омыления субстрата (ацетил-
холина), расщепляемого р-ром чистой холинэстеразы
(контрольный опыт), и холинэстеразой, подвергшей-
ся воздействию ОВ. В последнем случае омыление
происходит медленнее, и окраска, появляющаяся в
результате изменения индикатора под влиянием выде-
ляющей уксусной к-ты, наступает позднее, чем в
контрольном опыте. Чувствительность этого метода
10"’ мг/л.
Одним из важнейших физич. методов индикации О В
является метод, основанный на изменении иониза-
ционного тока в специально сконструированных
ионизационных камерах, в к-рые непрерывно по-
дается исследуемый воздух.
Дегазация ОВ производится различными физич.
и химич. методами (см. Дегазация).
Лит.: Степанов А. А., Попов В. Н., Химическое
оружие и основы противохими>1еской защиты, М., 1962; Н е-
м е ц В. Г., С о ч и л и н Е. Г., Химин отравляющих веществ,
М.—Л., 1941; СоборовскипЛ. 3., Эпштейн Г. Ю.,
Химин и технология боевых химических веществ, М.—Л.,
1938; Сондерс Б., Химин и токсикология органических
соединений фосфора и фтора, пер. с англ., М., 1961; Сар-
тори М., Усп. химии, 1954, 23, № 1, 62; О’Б райан Р.,
Токсичные фосфоросодержащие эфиры, пер. с англ., М., 1964.
А. А. Степанов.
ОТСТАИВАНИЕ (сгущение, седиментация) — про-
цесс механич. отделения взвешенных в суспензии
или эмульсии частиц под действием силы тяжести.
Условием осаждения частицы, находящейся в жид-
кости, является неравенство силы ее тяжести и архи-
медовой силы. Постоянная скорость свободного осаж-
дения, к-рую приобретает сферич. частица при урав-
новешивании действия силы тяжести сопротивлением
среды, может быть определена для наиболее распро-
страненного случая ламинарного осаждения по за-
кону Стокса:
.d- (0-0„)g ,n
819
ОТСТАИВАНИЕ
820
где d — диаметр частицы, Q — плотность частицы,
р0— плотность среды, g — ускорение силы тяжести,
р, — динамич. вязкость.
Применимость закона Стокса ограничена нижним
пределом размера частиц, когда их дисперсность до-
стигает коллоидных величин (1-10-5—5-10“5 с.и),
подверженных броуновскому движению. Верхний
предел применимости этого закона зависит как от
размеров и плотности частиц, так и от физич. свойств
среды, в к-рой они осаждаются. Этот предел харак-
теризуется числовым значением критерия (числа)
Рейнольдса:
Re=v-^ (2)
Для случая ламинарного осаждения 7?е<1,85. Прп
любых значениях Re процесс осаждения описывается
критериальным уравнением, полученным приемами
теории подобия:
Ле=а(фЛг)” (3)
где А г критерий Архимеда:
А gi3 (Q-Qo) On
ц2 ' ’
Is — линейный размер частицы; в случае сферы l=d;
ф— коэфф, формы (для сферич. частицы ф =1), а и п—
экспериментально определенные коэфф., зависящие
от величины Re'.
Re а п
<1,85 1/8 1
>1,851 0,1 52 0,71 5
<500 f
>500 1,74 0,5
После подстановки в ур-ние (3) значения Re а Аг
из ур-ний (2) и (4) может быть получено выражение
для скорости осаждения частицы более общее, чем
«формула Стокса.
Приведенные расчетные ф-лы применимы, как
отмечено выше, лишь прп свободном падении частиц,
т. е. в случае неконцентрированных суспензий, когда
осветление жидкости происходит постепенно; сначала
выпадают более крупные частицы, а более мелкие
образуют мутный раствор. При О. концентрированных
суспензий нельзя уже говорить о свободном падении
частиц; более крупные частицы при падении увлекают
за собой также и более мелкие, наблюдается т. н.
солидарное О.
Приведенные ур-ния позволяют судить о влиянии
лишь основных физич. параметров на скорость осаж-
дения. Они не учитывают влияния коагуляции, флок-
куляции, поверхностных явлений, а также влияния
изменения концентрации твердой фазы в процессе
ее осаждения, стенок отстойника, перегородок и
.др. факторов.
Процесс О., протекающий во времени, может быть
охарактеризован след, образом: вначале, в зоне сво-
бодного падения частиц, скорость их оседания пос-
тоянна, затем оседание замедляется (переходная зона)
и, наконец, резко уменьшается, падая до нуля (зона
уплотнения). Образующиеся прп О. осадки по своему
характеру можно подразделить на два типа. В случае
крупнозернистых суспензий взвешенные частицы осе-
дают на дно в виде плотного слоя. Мелкозернистые тон-
кие суспензии не образуют плотного осадка, в них
происходит лишь постепенное повышение концентра-
ции частиц сверху вниз п жидкость не осветляется.
В случае полпдисперсных суспензий при невысоких
концентрациях, когда имеет место свободное оседа-
ние частиц, осадки образуются в виде слоев; в ниж-
нем слое самые крупные, а затем более мелкие и т. д.
Это свойство полпдисперсных систем используется
в процессах о т м у ч и в а н п я — классификации
твердых веществ по их плотности пли но величине
частиц.
Практически О. производится пропусканием сус-
пензии (эмульсии) через камеру (отстойник), на дне
к-рой осаждаются взвешенные частицы. При этом
продолжительность пребывания элемента потока в
камере должна быть равна или больше времени осаж-
дения частицы.
Средняя продолжительность (т) пребывания сус-
пензии в отстойнике с площадью поперечного сечения
F и рабочей высотой h равна, очевидно, Fh/V, где V —
часовая производительность (м3/час). Но, с другой
стороны, т = h/v, поэтому
V = Fv (5)
Отношение V/v наз. нагрузкой поверх-
ности отстаивания. Из ур-ния (5) следует,
что производительность отстойника пропорциональна
поверхности осаждения; для увеличения последней
применяют горизонтальные или наклонные перего-
родки. Для практич. расчетов в случае концентри-
рованных пульп величина и в ур-нии (5) принимается
равной скорости «свободного осаждения твердой фазы»
(иногда скорости уплотнения осадка, определяемой
опытным путем).
Представления о процессе отстаивания, рассмот-
ренные выше, требуют корректировки в зависимости
от конструкции отстойника и характера обрабатывае-
мой жидкости. В расчетных уравнениях скорость
потока в сечении длинного прямоугольного гори-
зонтального отстойника можно принимать постоян-
ной. В вертикальных отстойниках условия отстаива-
ния суспензий менее благоприятны, чем в горизон-
тальных (в смысле равномерности распределения
струй).
На процесс отстаивания искажающее влияние ока-
зывает турбулентность потока разделяемой жидко-
сти, к-рая в большей степени наблюдается в верти-
кальных отстойниках. Турбулентный режим в гори-
зонтальных отстойниках возникает при Ле>500.
При турбулентном потоке траектории движения час-
тиц жидкости искривляются, возникает перемешива-
ние жидкости, способствующее переносу твердых
частиц и их транспортированию во взвешенном со-
стоянии на значительные расстояния.
Эффективность отстаивания суспензий существенно
повышается при ламинарном режиме течения потока
в отстойнике. Последний обеспечивается как надлежа-
щей скоростью иодачи обрабатываемой жидкости, так
п применением гори-
зонтальных, наклон-
ных и вертикальных
перегородок. Несмо-
тря на более благо-
приятные условия для 6
осаждения суспензии
в горизонтальных от-
стойниках, они более
громоздки, и в пром-
сти чаще применяют
вертикальные.
Рис. 1. Непрерывно дей-
ствующий отстойник с
гребками; I— резервуар;
2 — гребки; — желоб д.т
провод для удаления осадка; 6 — насос; 6 — сливной желоб
для осветленной жидкости.
11 химич. пром-сти отстойники применяют для от-
деления большей части жидкой фазы суспензий перед
фильтрацией, для промывки осадков методом декан-
тации, улавливания из сточных вод ценных или вред-
ных продуктов, для разделения по крупности зерен
твердой фазы суспензий при мокром помоле в замк-
нутом цикле, для отделения примесей или крупных
821
ОТСТАИВАНИЕ —ОХЛАЖДЕНИЕ
822
Рис. .2. Схема двухъярусного
отстойника непрерывного дей-
ствия.
зерен при «отмучивании» земель и других материалов.
На рис. 1 изображен гребковып отстойник непрерыв-
ного действия. Пульпа непрерывно подводится в
аппарат по желобу. Оседающая твердая фаза транс-
портируется гребками к
трубопроводу для удале-
ния осадка. Слив удаляет-
ся через кольцевой желоб,
расположенный вверху.
На рис. 2 показан двухъ-
ярусный отстойник непре-
рывного действия. Оба от-
деления отстойника сое-
динены друг с другом тру-
бой, нижний конец к-рой
находится ниже уровня
сгущенной суспензии в
нижнем отделении. Обра-
батываемая суспензия
подводится раздельно в
оба отделения отстойни-
ка. Сгущенный продукт
отводится из нижнего от-
деления, а осветленная жидкость — из верхней части
каждого отделения.
На рис. 3. показан многоярусный отстойник с про-
межуточной промывкой осадка. Суспензия непрерыв-
но поступает в верхний ярус отстойника, из к-рого
также непрерывно отводится по желобу и осветлен-
ная жидкость. Осадок накапливается в ловушке,
расположенной у днища яруса. Здесь он промывается
Рис. 3. Многоярусный отстойник с промывкой осадка:
1 — корпус; 2 — бачок для свежей промывной жидко-
сти; з — ловушки; 4 — трубопровод для отвода осветленной
жидкости; 5 — бачки для промывной жидкости; 6 — трубо-
проводы для осветленной жидкости, поступающей для
промывки.
жидкостью, поступающей пз яруса, находящегося
ниже, и смывается ею на нижележащий ярус, где
происходят отстаивание, промывка и т. д.
В конечном итоге осадок выводится через
патрубок, находящийся в днище аппарата,
а промывная жидкость отводится из верх-
него яруса.
Преимуществом отстойников являются:
простота конструкции, незначительность
энергетич. затрат и надежность в эксплуа-
тации. Недостатками их являются: малая
производительность на единицу объема и
получение сгущенной суспензии в виде те-
кучего шлама, а не осадка, как на филь-
трах и центрифугах.
Лит.: Касаткин А. Г., Основное про-
цессы и аппараты химической технологии, 7 изд.,
М.—Л., 1961; П л а н о в с к и и А. Н., Н и к о-
л а е в П. II., Процессы и аппараты химической
и нефтехимической технологии. Л., 106'1; Е р к о-
ва Л. Н., Смирнов Н. II., Ж. прикл. хи-
мии, 1956, 29, вып. 5; П а в л у ш е н к о И. С., там же, 1956,
29, вып. 6; С о к о л о в В. И., Хим. пром-сть, 1948, .N, 5;
Шифр и и С. М..Современные способы механической очистки
сточных вод, Л.—М., 1956. В. И. Соколов.
ОХЛАЖДАЮЩИЕ СМЕСИ (холодильные сме-
си) — системы из двух или нескольких твердых (илп
твердых п жидких) веществ, прп смешении к-рых
происходит понижение темп-ры вследствие поглоще-
ния теплоты при плавлении или растворении. Состав
О. с. из льда и соли определяется по диаграммам
растворимости, изображающим зависимость темп-ры
начала кристаллизации раствора от его концентрации.
Чтобы получить наиболее низкую темп-ру, веще-
ства, входящие в О. с., берут в количествах, отвеча-
ющих криогидратной точке (см. Эвтектика). Чаще
всего пользуются О. с. из снега (или мелкого льда)
и NaCl; криогидратная точка системы Н2О—NaCl
лежит при темп-ре'—21,2° и 22,4 вес. % NaCl. Еще
большее понижение темп-ры О. с. достигается введе-
нием в ее состав двух солей, а также использованием
твердой СО2 в смеси со спиртом (—77°), эфиром (—72°)
и другими жидкостями. Чтобы О. с. скорее достигла
своей предельной темп-ры, твердые компоненты смеси
следует измельчить и охладить.
Наиболее употребительны О. с. следующих составов
(состав указан в вес. частях полужирным шрифтом; в
скобках отмечены предельные понижения темп-ры в °C):
1) вола 16, NH.C1 5, KNO, 5 (-12°);
2) вода 1, NH. NO3 1 (—50°);
3) вода I, Na2CO3-10H2O 1, NH,NO3 1 (—22°);
4) соляная кислота 9, Na2SO410H2O 8 (—18°);
5) азотная кислота 4, Na2SO,10H2O 6, NH,NO, 5 (—40°);
6) лед 2, NaCl I (—20°>-
7) лед 5, NaCl 2, NH.NO, 1 (—25°);
8) лед 4, СаС1.-6Н2О 5 (—40°);
9) CO2+SO2 (до -82°).
О. с. применяют гл. обр. в лабораторной технике
для получения и поддержания низких темп-р, при-
мерно до —40°.
Лит.: Комаров Н.С., Справочник холодильщика,2 изд.,
М., 1962; Справочник химика, т. 3 — Химическое равнове-
сие и кинетика. Растворы..., Л.—М., 1952; Аносов В. Я.,
Погодин С. А., Основные начала физико-химического
анализа, М.—Л., 1947, С. 271, 306. С. А. Погодин.
ОХЛАЖДЕНИЕ — процесс отвода тепла от ка-
кого-либо вещества с целью понижения его темп-ры.
О. веществ ниже темп-ры окружающей среды, если
оно не может быть достигнуто применением естест-
венных источников холода — воды, льда и их смесей
с солями, осуществляется, согласно второму началу
термодинамики, путем затраты подводимой извне ме-
ханич. или тепловой энергии. О. до темл-р выше минус
100° принято называть умеренным, а ниже минус
100° — глубоким.
Охлаждение умеренное достигается применением
преим. жидких холодильных агентов, гл. обр. ам-
миака, пропан-пропиленовых смесей и фреонов. Фп-
зич. свойства важнейших холодильных агентов при-
ведены в таблице.
Физические свойства некоторых холодильных агентов
Холодильный агент Т. кип. (при 0° и 760 мм), °C Критические Т. затв., °C Показа- тель адиа- баты, К
темп-ра, °C давление, ат
Вода + 100 +374,15 218,53 0.0 1 , 33
Сернистый ангидрид — 10,08 — 33,4 + 157.5 77,8 — 75,2 1,26
Аммиак +132,40 111 ,5 —7 7,7 1 . 3
Углекислый газ . . -78,5 + 31,0 72,9 —56,6 1 . 3
Фреоны: Ф-12 (CFoCL) . . —29,8 +112,0 41.96 —155,0 1.14
Ф-14 (CF,) .... -127,8 —45,45 38,13 —190,92 —
Ф-23 (CHFS) . . . -82,2 — — — 163,0 1 ,21 0,644
Ф-143 (C,HSF3) . . —47.3 + 71,40 + 96,80 42,0 —111.3
Пропан -42.17 42,0 —187,1 1.13
Этан ....... —88,6 + 32,3 48,2 — 183, ,2 1,25
Этилен —103,9 + 9.90 50.50 -181.0 1,25
823
ОХЛАЖДЕНИЕ
824
Для переноса тепла из среды с низкой в среду с
более высокой темп-рой используются обратные тер-
модинамич. (холодильные) циклы, осуществляемые
холодильными машинами или установками. На рис. 1
приведен замкнутый обратный холодильный цикл.
При расширении по кривой АаВ темп-ра холодиль-
ного агента понижается, чем вызывается приток
тепла из охлаждаемой среды. Последующее сжатие
холодильного агента по кривой ВЬА вызывает повы-
шение его темп-ры и возможность отдачи тепла в
окружающую среду. Расходуемая при этом механич.
работа изображается в Р—Г-диаграмме площадью
АаВЬА. Тепло А1, эквивалентное затраченной работе,
а также тепло q0, отнятое холодильным агентом у
охлаждаемых веществ, передаются им окружающей
среде: q=q0A~Al ккал.
По рассмотренному простейшему холодильному
циклу можно судить лишь о принципе работы хо-
лодильных машин, использующих для получения
Рис. 2.
холода механич. пли тепловую энергию. Действи-
тельные холодильные циклы значительно сложнее.
В качестве идеального цикла для холодильных машин
принимается обратный цикл Карно (см. Карно цикл),
изображенный на рис. 2. Отвод тепла от охлаждаемых
веществ при темп-ре 7’0(°К) совершается по изо-
терме АВ, а передача тепла среде с более высокой
темп-рой 7'(°К) — по изотерме CD. Эти количества
тепла, отнесенные к 1 кг холодильного агента, соот-
ветственно равны:
qa = Ta(S2-Sx) п q = T(S2-Si)
Сжатие холодильного агента по адиабате ВС при-
водит к повышению темп-ры с То до Т, а расширение
по адиабате DA — к понижению его темп-ры до ТГ:.
Следовательно, расходуемая работа на сжатие хо-
лодильного агента частично компенсируется работой,
получаемой при его расширении. В Т—А-диаграмме
тепло, эквивалентное этой затраченной работе, выра-
жается площадью BCD А, т. е. А 1=(Т —Ta)(S2—Si).
Отношение количества тепла q0, отведенного из охла-
ждаемой среды при низкой темп-ре То, к затраченной
работе АI называется холодильным коэф-
фициентом ек. Для обратного цикла Карно:
р Со ТО (^2 ~ 81) То
‘’«“АПТ-Т,) (S2-S0 ~Т-Т0
Значение е^, максимальное в случае цикла Карно,
не зависит от природы холодильного агента и опре-
деляется только граничными температурами цик-
ла и Т.
В зависимости от физич. состояния холодильных
агентов холодильные машины делятся на воздушные и
паровые, а по методам отсасывания паров холодиль-
ного агента из охлаждаемой среды и их последующего
сжатия различают компрессионные, абсорбционные
и пароэжекторные паровые холодильные машины.
Воздушная холодильная машина
(рис. 3) состоит из компрессора 1, к-рый отсасывает
воздух из охлаждае-
мой среды при давле-
нии Pi, близком к ат-
мосферному, сжимает
его до давления Р2
(обычно 4—5 ат), пос-
ле чего воздух охлаж-
дается водой в холо-
дильнике 2 до темп-ры
1\, расширяется в де-
тандере 3 и с темп-рой
Тг (минус 75 — минус
70°) направляется в
качестве холодильного Рис. з.
агента в камеру 4.
В Т—S-диаграмме прямые АВ и CD соответствуют процес-
сам адпабатич. сжатия и расширения воздуха, а кривые СА
и DB — нагреву воздуха в камере при P,=const и охлаждению
в холодильнике 2 при Р,—const (в действительном цикле сжа-
тие и расширение воздуха совершаются политропически).
Воздушные холодильные машины пе нашли широ-
кого применения из-за их громоздкости, малого кпд,
вымерзания водяных паров в детандере, переменной
темп-ры охлаждения и др. недостатков.
Паровая компрессионная холо-
дильная машина (рис. 4). Пары холодиль-
ного агента отсасываются из охлаждаемой среды ком-
прессором 1 и после сжатия ожижаются в конденса-
торе 2. Жидкость дросселируется до давления Pi,
при к-ром, испаряясь, она отводит тепло от охлаж-
даемых ею веществ, а образующиеся пары снова заса-
сываются компрессором.
В Г—S-диаграмме испарение жидкого холодильного аген-
та изобразится прямой АВ, адиабатич. сжатие образовавшихся
паров — прямой ВС, конденсация их — прямой CD, а дрос-
селирование — кривой DA. Тепло q„, отводимое из охлаждае-
мой среды 1 кг холодильного агента, выражается площадью
АаЬВ, а работа Alt,, расходуемая на сжатие паров,— площадью
BCDdB. В действительном цикле паровой холодильной машины
пары, засасываемые компрессором, перегреты (точка В,) и в
конденсаторе, помимо скрытой теплоты испарения, охлаждаю-
щей водой отводится еще тепло перегрева (отрезок СС,). Для
снижения количества испаряющейся жидкости при дроссели-
ровании она часто после
Рис. 4.
Рис. 5.
В случаях, когда отношение давления конденсации
Р2 к давлению испарения Pi велико, сжатие паров
производится в многоступенчатом компрессоре. Прин-
ципиальная схема холодильной установки с двух-
ступенчатым сжатием паров приведена на рис. 5.
Абсорбционные холодильные ма-
шины. В абсорбционных машинах пары холодиль-
ного агента поглощаются жидким абсорбентом, из
к-рого они затем десорбируются и ожижаются. В
качестве холодильного агента в абсорбционных маши-
нах применяют преим. жидкий аммиак, а в качестве
825
ОХЛАЖДЕНИЕ
826
поглотителя его паров — воду. Принципиальная схе-
ма абсорбционной холодильной машины приведена
на рис. 6. Пары аммиака из
камеры (испарителя) 1
направляются в абсор-
бер 2, где поглощаются
слабым р-ром аммиач-
ной воды. Тепло, выде-
ляющееся при абсорб-
ции, отводится водой,
проходящей по змееви-
ку 3. Обогащенный р-р
аммиачной воды насо-
сом 4 подается в подо-
греватель 5, где отгоня-
ются пары аммиака,
ожижаемые в конденса-
торе 6. С помощью ре-
гулирующего вентиля
в подогревателе и кон-
денсаторе поддерживается одинаковое давление, опре-
деляемое темп-рой ожижения аммиака, а посредст-
вом другого вентиля — одинаковое давление в испа-
рителе и в абсорбере, определяемое темп-рой испа-
рения аммиака в камере. Абсорбционные машины
часто дополняют ректификационными колоннами для
уменьшения количества водяных паров, уносимых
парами аммиака из подогревателя и отрицательно
влияющих на процесс охлаждения.
Тепловой баланс абсорбционной холодильной машины опи-
сывается следующим образом;
Фкил.-*’ Qo + QHac,— Фконд. + Qa6c. ккал/час
гдеРКип — тепл0> подводимое в кипятильник; Qo— холодо*
производительность; QHac — тепло, эквивалентное работе насо-
са; QK0H1 — тепло, отводимое в конденсаторе; Ра6с — тепло,
отводимое* в абсорбере. Холодильный коэфф.
_____Ор_____
Ркип. + Рнас.
Значения холодильного коэфф, абсорбционных машин при
одинаковых темп-рах кипения и конденсации холодильного
агента теоретически равны холодильным коэфф, компрессион-
ных машин. Однако при низких темп-рах более эффективны
компрессионные холодильные машины. Преимущество абсорб-
ционных холодильных машин — возможность использования
в них низкопотенциальных тепловых источников; их недоста-
ток — громоздкость и повышенный расход воды (помимо тепла
конденсации, приходится отводить тепло абсорбции).
Адсорбционные холодильные ма-
шины. Эти машины используют в качестве холо-
дильного агента аммиак, а адсорбентом
служит СаС1з илп SrCh. Принципиаль-
ная схема такой машины периодич. дей-
ствия приведена на рис. 7. Жидкий ам-
миак из ресивера 1 поступает в камеру
2, где, испаряясь, охлаждает загружен-
ные в нее вещества. Пары аммиака по
трубе 3 и конденсатору 4 непрерывно от-
водятся для поглощения в адсорбер 5.
После окончания цикла охлаждения
включают подогреватель 6 и десорбиру-
ют аммиак, к-рый ожижается в конден-
саторе 4. Теплота адсорбции и конден-
сации отводится в окружающую среду
через ребристые поверхности конденса-
торов и адсорбера. Период регенерации
адсорбента обычно длится 1,5 часа, пе-
риод охлаждения—6,5 часов.
Паро эжекторные холо-
д и л ь н ы е м а ш и н ы (рис. 8). Во-
дяной пар сдавлением 8—10 ат подво-
дится к соплу эжектора 1, где расширяется; его ки-
нетич. энергия расходуется на создание вакуума
в испарителе 2, смешение с отсасываемым из него
паром и сжатие образовавшейся смеси в диффузоре 3
до давления Р^. В испарителе 2 за счет частичного
самопспаренпя понижается темп-ра циркулирующего
хладоагента. Водяные пары ожижаются в конден-
саторе 4, откуда конденсат частично подается для
восполнения потерь через регулирующий вентиль в
испаритель, а основная его масса насосом 5 возвра-
щается в котельную. Величина холодильного коэфф,
пароэжекторных машин значительно ниже, чем у
компрессионных, а в нек-рых случаях и абсорбцион-
ных машин.
Охлаждение глубокое достигается: 1. Изоэнталь-
пийным расширением сжатого газа (г= const), т. е.
дросселированием, используя эффект Джоуля — Том-
сона (изменение темп-ры — положительное или от-
рицательное — прп изоэнтальпийном расширении га-
зов). 2. Изоэнтропийным расширением сжатого газа
(A=const) в детандере (машине для получения холода
за счет расширения газов с отдачей внешней работы),
т. е. с совершением внешней работы. В данном случае
получают дополнительное количество холода (поми-
мо обусловленного эффектом Джоуля — Томсона),
т. к. работа расширения газа совершается за счет его
внутренней энергии. Первый из этих методов исполь-
зуется только в ожижителях малой производитель-
ности,где в целях упрощения их аппаратурного оформ-
ления пренебрегают нек-рым перерасходом энергии.
Ниже рассматриваются основные холодильные цик-
лы, нашедшие наиболее широкое применение в про-
цессах глубокого охлаждения, причем приведенные
ниже ф-лы для определения холодопроизводитель-
ности q и коэфф, ожижения х не учитывают потерь
холода на недокуперацию и в окружа-
ющую среду. В действительных холо-
дильных циклах значения q и х значи-
тельно ниже. Термин «недокуперация»
означает потери тепла, связанные с соз-
данием разности темп-р между потоками,
входящими в теплообменные аппараты
и выходящими пз них.
Цикл Линде. На рис. 9 приведена
схема цикла Линде с однократным дрос-
селированием газа высокого давления.
После сжатия в компрессоре 1 до 140—
220 ат газ проходит теплообменник 2
и дросселируется в сосуд 3. Ожиженная
часть газа отводится через вентиль, а не-
ожиженная, отдав свой холод в теплооб-
меннике 2, снова всасывается компрес-
сором. Весовую долю ожиженного газа х
принято называть коэффициентом
ожижения, к-рый легко может быть найден из
ур-ния теплового баланса:
Откуда
Ц = Чк +(1— х] it
где t2 и (4 — энтальпия газа на входе и выходе из
теплообменника, — энтальпия ожиженного газа.
827
ОХЛАЖДЕНИЕ
828
Это ур-ние составлено без учета потерь холода вслед-
ствие недокуперации и в окружающую среду.
Очевидно, что рассмотренный цикл может быть осуществлен
только при условии i, > 12, т. е. когда давление и темп-ра сжа-
того газа на входе в теплообменник имеет значения, соответ-
ствующие положительному эффекту Джоуля—Томсона. Хо-
лодопроизводительность рассматриваемого цикла q опреде-
ляется изотермич. дроссельным эффектом Д1Г:
?=-Дгг=х =
Разность энтальпий (Ц—г2) сжатого и несжатого
газа увеличивается с уменьшением темп-ры и ростом
давления сжатия. Так, в случае сжатия воздуха от
1 до 60 ат при 30° Ц—i2=2,96 ккал1кг, а при минус
45° и конечном давлении 200 ат Д—i2= 16,34 ккал!кг,
т. е. холодопроизводительность увеличивается почти
в 5,5 раза. Ниже приводится описание холодильного
цикла с предварительным охлаждением дросселируе-
мого газа.
Цикл с однократным дроссели-
рованием и предварительным (ам-
миачным) охлаждением сжатого
газа (рис. 10). Сжатый газ из компрессора 1 про-
ходит предварительный теплообменник 2, аммиачный
холодильник 3, в к-ром охлаждается обычно до —45°,
а затем основной теплообменник 4, после чего дрос-
селируется. Неожиженная часть газа отдает свой
холод в теплообменниках 2 и 4.
Точки А, и А, на изобаре Р2 (см. Т—S-диаграмму на рис. 10)
соответствуют темп-рам сжатого газа на выходе из предвари-
тельного теплообменника и аммиачного холодильника. В слу-
чае совершенного теплообмена для предварительного теплооб-
менника справедливо равенство:
i,-in = (l-x) (i.-ij
Расход <аммиачного» холода q =i( —г или после подста-
новки значения г : ам* * 11 а
а =г -г = ( 1 -г ) - (г± - М + х (I,-i )
*амм. п а \ 4 а/ ' 4 \ 1 4/
Где г4—га— холодопроизводительность при темп-ре Т2; К—Ъ—
холодопроизводительность при темп-ре 1\ изотермич. сжатия
газа; х (г4—г )—расход холода на охлаждение О/ьтькениой
части газа до темп-ры выхода его из основного теплообменника.
Общая холодопроизводительность установки в случае, если
ожиженный газ не выводится из системы:
Цикл с двумя давлениями газа
схематически представлен на рис. 11. Исходный
газ первоначально сжимается в компрессоре 1 до
промежуточного давления Р,, после чего часть его —
Мкг — отводится, а остаток (1 — М) кг дожимается
в компрессоре 2 до высокого давления Р2. После ох-
лаждения обратными потоками н теплообменниках
3 и 4 газ высокого давления дросселируется венти-
лем. Если обратные потоки отводятся под начальным
давлением Р\, то холодопроиз-
водительность рассматривае-
мого цикла выразится:
9 = М (Ц—ir)-|-(l— M)(it—i2)
Из ур-ния теплового баланса
без учета потерь холода на не-
докуперацию и в окружаю-
щую среду следует, что коли-
чество сжиженного газа х со-
ставит:
г4-г2—М (г~-г2)
х=----------------
Эффект Джоуля — Томсона с по-
вышением давления дросселируе-
мого газа растет весьма значитель-
но. В то же время расход работы
на сжатие газа до более высокого
давления увеличивается в значи- Рис. И.
тельно меньшей степени. Так, при
дросселировании воздуха с давлений 100 и 200 ат расход энер-
гии на получение 1000 ккал холода в действительном цикле
составляет соответственно 40 квт-ч и 27 квт-ч.
Таким образом, при переработке больших количеств газов
целесообразно для экономии энергии сжимать только неболь-
шую часть газа до высокого давления, а основную массу — до
промежуточного более низкого давления, определяемого тех-
нология. схемой данного процесса. Количество газа высокого
давления в этом случае определяется потерями холода от
недокуперации и в окружающую среду.
Циклы с двумя давлениями газа нашли широкое
применение, напр. в крупных установках разделения
воздуха для получения газообразного кислорода, где
доля воздуха высокого давления колеблется в преде-
лах от 5 до 10%; в установках для получения азота
она достигает 20%.
Цикл с двойным дросселирова-
ние м. Схема такого цикла и его изображение в.
Т — S-диаграмме приведены на рис. 12. В компрессо-
ре 1 Мкг газа с давлением Рг сжимают до давления
циркулирующего потока PIV смешивают с (1—М) кг
циркулирующего газа и сжимают в компрессоре 2
до давления В теплообменнике 3 газ высокого
давления охлаждается отходящими газами среднего
и низкого давления, после чего он последовательно
дросселируется вентилями в отделители 4 и 5.
В 71 — S-диаграмме линия АВ соответствует сжатию газа
в компрессоре 1 от давления Pi до давления Рц, а линия ВС —
сжатию всего количества газа от Рц до Р2. Если в компрессоре ?
сжимается 1 кг газа, а при дросселировании его до давления
Рч (линия DE) ожижается М кг, то из отделителя 4 в теплооб-
менник .7 отводятся (1 — .W) кг паров. Дальнейшее дросселиро-
вание жидкости (линия EG) в отделитель .5 до давления р,
приводит к образованию паров низкого давления гв количестве-
(-W— л) кг.
Коэфф, ожижения л- и холодопроизводительность цикла q
легко определить, исходя из того, что энтальпия газа высокого
829
ОХЛАЖДЕНИЕ
830
давления i_> равна сумме энтальпий, выведенных из системы
количеств газа:
% = (11-г2) + М (ij-in)
q—x (ij -1ж) = (г1-г2) + Л/(г4-гц)
Охлаждение газа высокого давления перед его дросселирова-
нием за счет подвода холода извне применяется на практике и в
рассматриваемом цикле. При этом в связи с большим увеличе-
нием эффекта Джоуля— Томсона уд. расход энергии на еди-
ницу получаемого холода значительно снижается.
Такие холодильные циклы с двойным дросселиро-
ванием (или с циркуляцией, газа высокого давления)
нашли применение в агрегатах разделения коксового
газа, промывки конвертированного газа жидким азо-
том, в установках небольшой производительности —
для получения жидкого воздуха или кислорода и др.
Экономичность этих циклов объясняется тем, что эффект
Джоуля — Томсона при одной и той же темп-ре определяет-
ся только разностью давлений до и после дросселирова-
ния (Р2—Р|), а работа, затрачиваемая на сжатие исходного
газа, пропорциональна 1п (Р2!Р{). Напр., разность энтальпий
при дросселировании воздуха с темп-рой 30° от 200 до 20 ат
составляет 7,25 ккал^кг, а при дросселировании от 20 до 1 ат
(абс.) всего 0,92 ккалiкг. В то же время, на изотермич. сжатие
1 кг воздуха с 20 до 200 ат расходуется ART — 46,9 ккал,
20
а с 1 до 20 am ART In — — 60 ккал.Следовательно,расход энер-
гии на получение 1000 ккал холода в первом случае составит
1000-46,9 „ гл 1000.60 __ о
7~25 8 60 а во втором случае^ =/э,8кбт-ч.
Таким образом, значительная экономия может быть достигну-
та, если часть газа высокого давления дросселировать не до
1 ат (абсолютной), а до нек-рого промежуточного давления,
определяемого условиями данного технология, процесса.
Холодильные циклы с детанде-
ром. Известны след, циклы подобного рода, разли-
чающиеся давлением и темп-рой газа перед его адиа-
батич. расширением:
а) В цикле высокого давления Гейландта (рис. 13)
газ сжимается в компрессоре 1 от начального давле-
ния Pi до требуемого давления Р?, после чего одна
его часть М направляется в теплообменники 2 и 3,
а другая (1 — М) кг — в детандер 4. Охлажденный в
теплообменниках газ далее дросселируется вентилем
в отделитель 5, из к-рого в случае необходимости
отводится ожиженная его часть х кг. Неожиженные
пары в количестве (М—т) кг отдают сначала свой
холод в теплообменнике 3, а после смешения с коли-
чеством газа(1 — М), выходящего из детандера, про-
ходят теплообменник 2 и могут снова всасываться
компрессором 7. ,
б) В цикле среднего давления Клода (рис. 14) газ
после сжатия в компрессоре 1 проходит теплообмен-
ник 2 и разделяется на две части. Одна часть (М кг)
проходит теплообменники 3 и За и дросселируется в
отделитель о. Другая часть сжатого газа (1 — Л7) кг—
направляется в детандер 4, где расширяется до дав-
ления Р.
в) В цикле низкого давления П. Л. Капицы (рис.
15), осуществленном благодаря созданному им эффек-
тивному турбодетандеру, газ сжимается в турбокомп-
рессоре 7, охлаждается в теплообменнике 2, после-
чего большая его часть (1—М) кг расширяется!
в турбодетандере 4 и направляется в трубное про-
странство конденсатора 3. Меньшая часть газа (М кг)
поступает в межтрубное пространство конденсатора
3, где ожижается за счет охлаждения основной массой
газа, прошедшей турбодетандер. Ожиженный газ.
дросселируется вентилем в нижнюю камеру конден-
сатора 3.
Холодопроизводительность циклов, с детандером слагается
из холода, получаемого за счет изотермич. дросселирования
всего количества исходного газа Д1г и холода, получаемого за
счет расширения части газа в детандере Дгд, т. е.
? = Д1Т + Д1д
Применительно к рассмотренным холодильным циклам е
детандером и принятым обозначениям эту формулу можно пе-
реписать след, образом:
цикл высокого давления с детандером:
q = i4 -i2 + (l— М) (i2-i3)
цикл среднего и низкого давления с детандером:
где — энтальпия газа на входе в детандер, a i3 — энталь
пня на выходе из детандера. Количество ожиженного газа я
выразится: x=q'l(i\—гк). В отличие от циклов, использующих:
эффект Джоуля — Томсона, холодопроизводительность цик-
лов с детандером определяется количеством (1—М), темн-pofr
и давлением газа, поступающего в детандер. Тепловой пе-
репад растет с повышением темп-ры газа перед детан-
дером, но при этом снижается его количество (1—М) кг, что-
может привести к ухудшению теплообмена и большим по-
терям холода. Выбор этой темп-ры производится с учетом
возможности полной рекуперации холода расширяемой части
газа для охлаждения М кг газа высокого давления.
Цикл низкого давления с детандером применяется
во всех крупных воздухоразделительных установках,
а циклы высокого и среднего давления с детандером —
в установках для получения жидкого кислорода и.
разделения газовых смесей (коксового газа, конвер-
тированного газа и т. д.). Давление сжатого газа и
цикле высокого давления с детандером достигает
200 ат, среднего давления 40—60 ат и низкого дав-
ления — 7 ат.
Каскадный холодильный ц и к л..
Принципиальная схема такого цикла (предложенного
Кеезомом) приведена на рис. 16. Сжатые в компрес-
соре 7 пары аммиа-
ка с давлением 10 ат
ожижаются водой и
дросселируются в
конденсатор - испа-
ритель 2, где испа-
ряются, ожижаяпри
этом сжатый в комп-
рессоре 3 этилен с
давлением 19 ат.
Ожиженный этилен
в свою очередь дрос-
селируется до 1 ат
в конденсатор-испа-
ритель 4 для ожи-
жения сжатого в
компрессоре 5 мета-
на с давл ением 2 5 а т.
Последний дроссе-
лируется до 1 ат в конденсатор-испаритель 6, в к-ром,
испаряясь, ожижает сжатый в компрессоре 7 азот
с давлением 18,6 ат. Жидкий азот дросселируется в
отделитель 8, пз к-рого он отводится как целевой про-
дукт. Теплообменники 9, 10, 11 служат для рекупера-
ции холода испаряющихся хладоагентов перед пх
повторным сжатием в компрессорах. Каскадный хо-
лодильный цикл выгоден в случаях, когда возможно
использование хладоагентов в состоянии, близком к
насыщению. Для сравнения ниже приводятся расходы
831
ОХЛАЖДЕНИЕ —ОЧИСТКА НЕФТЕПРОДУКТОВ
832
энергии (в квт-ч) на ожижение 1 кг воздуха в зависи-
мости от применяемого холодильного цикла.
Идеальный обратимый цикл...............0,11
Цикл высокого давления с однократным дрос-
селированием ...........................2,86
То же, с предварительным охлаждением до
—45°....................................1,54
Цикл с двумя давлениями газа ...........1,76
То же, с предварительным охлаждением до
—45°....................................0,99
Цикл высокого давления с детандером . . . 0,925
То же, среднего давления................0,99
Каскадный цикл Кеезома (рассчитано для
азота) ................................ 0,60
Sa Si
Рис. 17.
Из-за громоздкости оборудования каскадные холо-
дильные циклы не нашли пока применения в пром-сти.
Частично принцип каскадного цикла использует-
ся при ожижении водорода, когда газ предвари-
тельно охлаждается жидким азотом, и при ожижении
гелия, к-рый предварительно охлаждается жидким
азотом и водородом или неоном.
Ожижение газов. Любой газ при соответствующем
его охлаждении или сжатии может быть ожижен.
Темп-ра ожижения определяется давлением, под
к-рым находится газ, но она всегда ниже критической.
Для определения минимального теоретич. расхода
энергии на ожижение газообразного вещества рас-
смотрим в диаграмме Т — S идеальный термодинамич.
цикл ожижения (рис. 17). Газ с начальным давлением
Pi, темп-рой Ti и энталь-
пией И (точка Л) изотерми-
чески сжимается до давления
Рг, к-рому соответствует эн-
тальпия г'г (точка В). Тепло-
та, эквивалентная работе
сжатия газа и изотермич.
дроссельному эффекту, при
этом непрерывно отводится в
окружающую среду. Сжатый
газ далее подвергается изоэн-
тропийному расширению до
давления Рг (точка С), при-
чем совершаемая работа ис-
пользуется для уменьшения
подводимой извне энергии
присжатиигаза. Давление Р2
выбирается таким, чтобы в
результате изоэнтропийного расширения газ был пол-
ностью ожижен (точка С).
Очевидно, что за вычетом отведенного тепла Q
повышение энтальпии газа после сжатия должно быть
равно затраченной работе Nc, т. e.i2—ii=Nc—Q =
= Ус-Т(А1-А2).
Работа А'и, произведенная сжатым газом при изо-
энтроцийном его расширении, равна N„=i2—is.
Следовательно, работа, расходуемая в идеальном
термодинамич.цикле на ожижение 1 кг газа, выразится:
7V = TVe—2V„= Т (б’. —^2) —(i. —i3)
Величина N характерна для обратимого процесса
ожижения газа и зависит только от свойств послед-
него. Ниже приведены теоретич. значения N (дж/кг
________________________— и квт-ч/кг) для ожижения
нек-рых технич. газов.
Применяемые на прак-
тике циклы ожижения зна-
чительно отличаются от
рассмотренного. Это объяс-
няется не только неосуще-
ствимостью изотермическо-
дж1кг квт-ч > кг
Воздух 723 0,2
Азот . . 715 0,21
Кислород 6.35 0,18
Водород 11 900 3,30
Гелий - . 6 800 1 .89
го сжатия и последующего
изоэнтропийного расширения ожижаемого газа, но
также требуемым огромным конечным давлением сжа-
тия (в случае воздуха — 458 000 ат).
Лит.: Герш С. Я., Глубокое охлаждение, ч. 1, 3 изд.,
М.~ Л., 1957, Розеифел ьдЛ. М.» ТкачевА. Г.,
Холодильные машины и аппараты, 2 изд., М., 1960; Гель-»
перин И. И. [и др.], Справочник по разделению газовых
смесей методом глубокого охлаждения, 2 изд., М., 1963.
ОХРА — железоокисный
Пигменты.
ОЦИМЕН С10Н16, мол.
ациклических терпеновых
(2,6-диметилоктатриена-1,5,
метил октатриена-2,5,7):
Н,С.
°>с-сн2-сн2
н3сх
а
Н3С.
)С = СН-СН2-
H3CZ
II. II. Гельперин.
пигмент. Подробнее см.
вес 136,23 — смесь двух
углеводородов: а-оцимена
7) и Р-оцимена (2,6-ди-
сн3
-сн=с-сн=сн3
сн3
I
-сн=с-сн=сн2
О.— бесцветная, приятно пахнущая жидкость;
легко окисляется и полимеризуется; т. кип. 176—
178° (с изомеризацией); 73—74°/21 льи; 0,7982;
Ид 1,4860. При нагревании легко изомеризуется в
аллооцимен. Действием Na в спирте О. превращается
в дигидрооцимен, идентичный дигидромирцену (см.
Мирцен). Смесь водного р-ра H2SOi и СНзСООН
превращают О. в спирт оцименол Сц,Н17ОН,т. кип.
97°/10 ллГ, с/” 0,901; и’д 1,4900. В природе О. встре-
чается во многих эфирных маслах; выделяют фрак-
ционированной перегонкой масла из Ocimum basi-
licum L. О. получается в качестве промежуточного
продукта при термич. изомеризации а-пинена в алло-
оцимен, откуда его можно выделить.
Лит. см. При ст. Мирцен. II. II. Бардышев.
ОЧИСТКА ГАЗОВ — см. Газов очистка.
ОЧИСТКА НЕФТЕПРОДУКТОВ — совокупность
процессов, имеющих целью освобождение нефтепро-
дуктов от нежелательных или, чаще всего, недопус-
тимых в товарном продукте компонентов. К таким
компонентам относятся сернистые, кислородные и
азотистые соединения, а также высокомолекулярные
соединения сложного химич. состава, называемые
смолами. В нек-рых случаях в задачу очистки входит
выделение из нефтепродуктов, содержащихся в них
твердых углеводородов (депарафинизация).
К числу нефтепродуктов, подвергаемых очистке,
относятся бензиновые, лигроиновые, керосиновые,
дизельные (газойлевые), соляровые и более высоко-
кипящие масляные дистилляты, а также высоковязкие
остатки от перегонки нек-рых нефтей. В соответствии
с характером применяемых реагентов и аппаратур-
ным оформлением процесса принято различать очи-
стку светлых нефтепродуктов (бензиновые, лигрои-
новые, керосиновые и дизельные фракции) и очистку
масляных дистиллятов и остатков от перегонки.
Очистка светлых нефтепродуктов. Простейшим спо-
собом очистки является щелочная, при к-рой дис-
тиллят обрабатывается водным р-ром едкого натра или
кальцинированной соды крепостью 10—30% при
темп-ре 20—90°. Расход едкого натра составляет
0,02% для бензина и лигроина прямой перегонки,
0,09—0,1% для керосина и крекпнг-бензина и 0,2—
0,3% для бензинов пз сернистых нефтей. При этом
из дистиллятов удаляются нафтеновые к-ты, фенолы,
сероводород п меркаптаны в соответствии с реак-
циями:
HCOOH NaOH —>RCOONa + H3O
ROH+NaOH —>RONa+H3O
H2S + 2NaOH —5-Na3S + 2H,O
RSH + NaOH —> RSNa + H3O
Удаление меркаптанов связано с нек-рыми трудно-
стями, обусловленными гидролизом образующихся
833
ОЧИСТКА НЕФТЕПРОДУКТОВ
834
меркаптидов. Для избежания этого очистку бензи-
новых фракций от меркаптанов производят 43%-ным
р-ром щелочи, в к-рую, в целях повышения раство-
римости меркаптидов и уменьшения гидролиза, до-
бавляют таннин, пропионовую или масляную к-ту.
При этом полнота извлечения меркаптанов, содержа-
щихся в бензиновых фракциях, достигает 95—97%.
Полнота удаления меркаптанов резко убывает с
повышением их мол. веса; поэтому для очистки от
меркаптанов керосиновых и дизельных фракций
щелочной метод не эффективен. Более совершенной
является очистка серной к-той (до 92%), проводимая
при 0—30° и расходе кислоты 1—8% (в зависимости
от свойств дистиллята). Помимо сернистых соедине-
ний, серная к-та удаляет из дистиллятов смолистые
и азотистые соединения, а также непредельные угле-
водороды; с последними серная к-та образует кислые
или средние эфиры, к-рые со щелочью дают соответ-
ствующие соли.
Оптимальные условия очистки серной к-той зависят
от природы дистиллята. Иногда, в целях экономии
к-ты, последнюю заменяют кислым гудроном, остаю-
щимся после очистки к-той бензинов прямой пере-
гонки и содержащим 60—75% H2SOi. Очищенный
серной к-той дистиллят для нейтрализации обрабаты-
вают щелочью и промывают водой. В случае очистки
крекинг-дистиллятов обработке к-той предшествует
иногда обработка щелочью для удаления сероводоро-
да, а очищенный кислотой и нейтрализованный про-
дукт подвергают вторичной перегонке, а затем повтор-
но обрабатывают щелочью и промывают.
Основным методом удаления сернистых соедине-
ний из светлых нефтепродуктов является каталитич.
гидроочистка, проводимая для бензинов в паровой
фазе над катализатором (смесь сульфидов никеля и
вольфрама) при 50—55 ат и 230—370° в присутствии
водорода. Выбор темп-ры зависит от фракционного
состава сырья, желаемой глубины обессеривания и
активности катализатора. Процесс гидроочистки
дизельных фракций ведут обычно в жидкой фазе над
неподвижным алюмо-кобальт-молибденовым катали-
затором при 40—50 ат и 390—420° в присутствии
вводимого извне водорода. Разновидностью гидро-
очистки является автогидроочистка, при
к-рой необходимый для гидрирования водород полу-
чают дегидрогенизацией нафтеновых углеводородов,
содержащихся в сырье. Автогидроочистку ведут на
том же катализаторе при 5—15 ат и темп-ре 400°
и выше. Выход очищенного продукта: при гидро-
очистке ок. 100%, степень обессеривания 95—98%;
при автогидроочистке бензинов 95—97%, дизельных
фракций 50— 85%.
Крекинг-бензины очищают также в, паровой фазе
отбеливающими землями, способствующими поли-
меризации непредельных, что повышает стабильность
бензина. Процесс ведут при 4,0—4,5 ат и 230—240°.
Крекинг-бензины, богатые сернистыми соединениями,
подвергают очистке ZnClj. При этом удаляется 30—
40% содержащихся в них сернистых соединений.
Дистилляты, содержащие меркаптаны, очищают р-ром
плумбита натрия, получаемым прямым смешением
6% свинцового глета, 10% едкого натра и 84% воды.
Сущность плумбитной очистки сводится к переводу
меркаптанов в нейтральные дисульфиды, не действу-
ющие на металлы. Процесс очистки проводится в
две стадии:
2RSH+Na2PbO2 —>-R—S—Pb—S—R+2NaOH
R-S-Pb-S-R + S —R-S-S—R + PbS
Отработанный p-p NajPbOa по мере падения его ак-
тивности подвергается регенерации путем продувки
воздухом:
4NaOH + PbS + 2O2=Na2PbO2+Na2SO, + H2O
топлив с низкой темп-рой
депарафинизация соответ-
Наиболее современным и
депарафинизации светлых
очистка с помощью кар-
При этом к р-ру добавляются едкий натр для по-
полнения его потерь.
При получении из парафинистых нефтей керосинов,
реактивных и дизельных---- ~ ------------
застывания необходима
ствующих дистиллятов,
прогрессивным методом
нефтепродуктов является
бамида (мочевины); последний образует с парафино-
выми углеводородами нормального строения нераст-
воримые в углеводородах комплексы, легко отделя-
емые от основной массы нефтепродукта декантацией
или фильтрованием. После разложения комплекса
горячей водой карбамид пригоден для повторного
использования.
Очистка масляных фракций. Основным способом
очистки масляных фракций являлась обработка их
серной к-той. При этом способе дистиллят обрабаты-
вают 92 %-ной H2SO4 (4—16% по весу), дают отсто-
яться образовавшемуся кислому гудрону, нейтрали-
зуют кислые масла водным р-ром едкого натра, от-
стаивают щелока и промывают масла водой. Для
дистиллятов, содержащих значительное количество
нафтеновых к-т, напр. нек-рых дистиллятов эмбен-
ских нефтей, кислотной очистке предшествует обра-
ботка щелочью. Для нек-рых специальных масел,
где требуется особо глубокая очистка, применяют
серную к-ту повышенной крепости, олеум или газо-
образный SOs. Многие виды масел (автолы, дизель-
ные, масла для авиационных поршневых двигателей)
нейтрализуют после очистки к-той отбеливающими
землями при 60—300° (расход земли от 2 до 10—15%).
В целях улучшения стабильности и деэмульсирующей
способности нек-рых масел, подвергшихся кислотно-
щелочной очистке (напр., турбинных), их дополни-
тельно очищают отбеливающими землями. Частным
случаем очистки масел с помощью отбеливающих
земель является перколяционная очист-
к а. Этот метод, применяемый и для других нефте-
продуктов, заключается в фильтрации их в чистом
виде или в р-ре легкого лигроина через слой зернен-
ного адсорбента. Для очистки применяются специаль-
ные фильтры, представляющие собой вертикальные
цилиндры, снабженные перфорированным внутренним
днищем или горловиной с ситом и задвижкой для
выгрузки отработанного адсорбента.
В последнее время серная к-та в процессах очистки •
масел все более вытесняется селективными (избира-
тельными) растворителями — фенолом, фурфуролом,
нитробензолом, смесью фенола с крезолом и пропаном
(дуосол-процесс). Смесь фенола с крезолом
и пропаном применяют гл. обр. для очистки остаточ-
ных масел. Метод основан на способности раствори-
телей селективно извлекать из дистиллятов смолис-
тые вещества, нек-рые ароматич. и сернистые соеди-
нения. При обработке дистиллятов селективными
растворителями образуются два слоя: рафинатный
(очищенное масло) и экстрактный, представляющий
собой р-р извлеченных из сырья компонентов в се-
лективном растворителе. Селективно очищенные
масла часто подвергают дополнительной очистке от-
беливающими землями. Иногда селективная очистка
сочетается с сернокислотной или гидрогенизаци-
онной.
Для удаления из остаточных масел асфальто-смо-
листых веществ применяется обработка масел пропа-
ном (деасфальтизация), Этот способ основан
на понижении растворимости асфальто-смолистых
веществ в пропановом р-ре, в результате чего они
осаждаются из р-ра в виде пластичной фазы, кото-
рая легко может быть отделена от р-ра. Деасфальти-
зацию проводят жидким, пропаном при 25—30 ат
и 60—85°.
° 27 к.х.Э. т. 3
835
ОШИБКИ АНАЛИТИЧЕСКОГО ОПРЕДЕЛЕНИЯ
836
При получении дистиллятных или остаточных ма-
сел из парафинистых нефтей необходимо удалить из
них твердые парафиновые и циклич. углеводороды.
Депарафинизацию проводят глубоким охлаждением
р-ра депарафинируемого масла в к.-л. растворителе
(бензин, смеси бензола с ацетоном или метилэтилке-
тоном, смесь дихлорэтана с бензолом) с отделением
выделяющихся при этом кристаллов твердого пара-
фина на центрифугах или фильтрах. Кроме перечис-
ленных методов очистки, нек-рые масла (трансфор-
маторные, моторные и др.), получаемые из сернистых
нефтей или несернистых низкокачественных нефтей,
очищают каталитич. гидрированием или адсорбци-
онным способом с помощью силикагеля, алюмосили-
катной крошки и др.
Для современного технология, оформления про-
цессов очистки нефтепродуктов характерно широкое
применение аппаратов, обеспечивающих возможность
непрерывного автоматич. управления процессами.
Это достигается применением противоточных экстрак-
торов, трубчатых печей, непрерывно разгружающих-
ся центрифуг и фильтров, инжекторных и дисперси-
онных смесителей для обработки сырья серной к-той
и др. реагентами.
Лит.: Технология нефти, ч. 3, 3 изд., М.—Л., 1952; Мо-
торные, реактивные и ракетные топлива, 4 изд., М., 1962; Со-
вершенствование производства масел и парафина из сернистых
нефтей, М., 1961. Б. В. Логиков.
ОШИБКИ АНАЛИТИЧЕСКОГО ОПРЕДЕЛЕНИЯ.
Результаты, полученные при аналитич. определе-
нии, должны быть воспроизводимыми и правиль-
ными. Под воспроизводимостью пони-
мают малое отклонение параллельных определений
от среднего результата. Воспроизводимость характе-
ризует метод анализа и технику его выполнения.
Правильность аналитич. определения —
разность между полученным результатом и истинным
содержанием определяемого вещества. Воспроизво-
димость сама по себе недостаточна, чтобы судить о
пригодности метода анализа. Последнее можно сде-
лать при сопоставлении воспроизводимости и правиль-
ности. Для определения правильности результатов
анализа основное значение имеет оценка ошибок ана-
лиза. Различают след, виды ошибок измерения: гру-
бые, систематические и случайные.
Грубые ошибки связаны с неверными от-
счетами или с недостаточной точностью в работе.
При обработке экспериментальных данных резуль-
таты с грубыми ошибками отбрасывают.
Систематические ошибки зависят от
постоянных причин и повторяются при всех наблю-
дениях. К числу систематич. ошибок относят: индиви-
дуальные, напр. связанные с особенностями зрения
наблюдателя, инструментальные ошибки, зависящие
от точности изготовления приборов, напр. точности
калибрования разновесок или мерной посуды, ошибки
метода, напр. индикаторная ошибка при титриметрич.
анализе или ошибки, связанные с гигроскопичностью
осадков при прокаливании в весовом анализе и др.
Систематич. ошибки можно либо исключить, либо
ввести на них поправку.
Случайные ошибки зависят от неопреде-
ленных причин, помех, несовершенства приборов и
органов чувств наблюдателя. Исключить эти ошибки
невозможно, но, пользуясь теорией ошибок, можно
уменьшить их влияние на окончательный результат
измерений и установить величину возможной ошибки.
Выводы теории ошибок применимы только к случай-
ным ошибкам, т. е. предполагается, что грубые и си-
стематич. ошибки исключены.
При определении величины ошибки различают
абсолютную и относительную ошибки. Абсолют-
ная ошибка — разница в абсолютных цифрах
между истинным или наиболее достоверным значением
определяемой величины и полученным результатом.
Напр.: истинный вес осадка 5,3 мг, полученный при
анализе — 5,1 мг, абсолютная ошибка 0,2 мг. О т-
носительная ошибка — отношение абсо-
лютной ошибки к истинному или среднему значению.
В приведенном примере относительная ошибка равна
0,2-100/5,35=3,7% . Ниже рассмотрены возможные слу-
чаи учета влияния случайных ошибок при оценке точ-
ности аналитич. определения. В метрологии измерения
делят на прямые, косвенные и совокупные.
Ошибки прямых равноточных измерений. При пря=
мых (непосредственных) измерениях числовое значе-
ние измеряемой величины (ж) сразу получается из
показаний прибора, при помощи которого выполняет-
ся данное измерение (например, объем при отсчете
по шкале бюретки; значение оптич. плотности при
отсчете по шкале барабана на фотоколориметре и т. д.).
Результат каждого прямого измерения может вклю-
чать случайную ошибку, которая зависит от большого
числа случайных факторов. Если отклонения, вызы-
ваемые этими факторами по абсолютной величине,
значительно меньше чувствительности регистрирую-
щего прибора, то они не обнаруживаются; при много-
кратных измерениях одной и той же величины резуль-
таты получаются одинаковые, хотя ошибка и не равна
нулю. В этом случае (как и при однократных измере-
ниях) критерием точности является цена наимень-
шего деления шкалы прибора или десятые доли наи-
меньшего деления. Если же отклонения, вызванные
случайными факторами, сравнимы по абсолютной
величине с чувствительностью прибора, то их обна-
руживают приборами, и при п измерениях одной и той
же величины получаются результаты ц, х2, ..., хп,
к-рые могут отличаться друг от друга в пределах
точности данных измерений. Когда число измерений
невелико (щ>=2), то для расчета точности полученного
ряда прямых измерений пользуются методами совре-
менной математич. статистики, разработанной для
малого числа наблюдений. В таких случаях получен-
ную систему наблюдений над измеряемой величиной
рассматривают как случайную выборку из нек-рой
гипотетической генеральной совокупности, к-рая
представляет собой совокупность всех мыслимых наб-
людений над измеряемой величиной при данных усло-
виях эксперимента. Задача математич. статистики
заключается в том, чтобы оценить параметры гене-*
ральной совокупности по результатам данной случай-
ной выборки с учетом элемента неопределенности,
к-рый вносится ограниченностью опытного материала.
Поэтому в статистическом анализе следует четко раз-
граничивать параметры генеральной совокупности,
к-рые обычно обозначают греческими буквами, и выбо-
рочные параметры, обозначающиеся латинскими бук-
вами.
Для оценки точности прямых равноточных изме-
рений используются след, критерии:
1. Среднее значение случайной
величины (среднее арифметическое). Пусть xi,
х2, ..., хп обозначают п результатов измерений величи-
ны, истинное значение к-рой а. На основании закона
нормального распределения случайных ошибок до-
казывается, что при п измерениях одинаковой точ-
ности среднее арифметич. из результатов, полученных
при всех измерениях, является наиболее вероятным
и наилучшим значением измеряемой величины:
-=х,+хг±...+х, + ...+хя = _£У (1)
п п 1 ' '
1 = 1
х принимают за приближенное значение а и пишут
а^х.
837
ОШИБКИ АНАЛИТИЧЕСКОГО ОПРЕДЕЛЕНИЯ
838
2. Остаточные погрешности (вероят-
нейшие). Отклонение отдельных измерений от ариф-
метич. среднего наз. остаточной погрешностью vf.
vi — xi — х (2)
Алгебраическая сумма остаточных погрешностей
2г;,-=0.
3. Дисперсия и «средняя» квад-
ратичная ошибка. Рассеяние случайной
величины относительно среднего значения принято
характеризовать дисперсией. Чем меньше точность
измерений, тем больше дисперсия. Для п найденных
значенийц, гг, хп случайной величины выборочная
дисперсия определяется выражением (3). Положи-
тельное значение корня квадратного
из дисперсии наз. средней квадратич. ошибкой от-
дельного определения или выборочным стандартным
отклонением (4)
При оценке точности полученных результатов вычис-
ляют также выборочную дисперсию средней квадра-
тичной (5). Корень квадратный из этой величины
наз.
Таблица 1. Значения t , для разных значений k и а
k а
0,95 0,99 0,999
1 12,706 63,657 636,619
2 4,303 9,925 31,598
3 3,182 5,841 12,941
4 2,776 4,604 8,610
5 2, 571 4,0 32 6,859
6 2,447 3,707 5,959
7 2, 365 3,499 5,40 5
8 2,306 3,355 5,041
9 2,262 3,250 4,781
10 2,228 3,169 4,587
11 2,201 3,106 4,487
12 2,179 3,055 4,318
13 2,160 3.012 4,221
14 2,145 2,977 4,140
15 2,131 2,947 4,073
16 2,120 2,921 4,015
17 2,110 2,898 3,965
18 2,103 2,878 3.922
19 2,093 2,861 3,883
20 2,086 2,845 3,850
21 2,080 2,831 3,819
22 2,074 2,819 3,792
23 2,069 2,807 3,767
24 2,064 2, 797 3,745
25 2,060 2,787 3,725
26 2,056 2,779 3,707
27 2,052 2,771 3,690
28 2,048 2,763 3,674
29 2,045 2,756 3,659
30 2,042 2, 750 3,646
40 2,021 2,704 3,551
60 2,000 2,660 3,460
120 1,980 2,617 3,373
00 1,960 2,576 3,291
2
S- =-----тт-
х п(п-1)
средней квадратичной ошибкой среднего арифмети-
ческого (6)
г —п
—--------- (6)
п (п- 1) ' '
Генеральные (общие) дисперсии обозначают соответ-
ственно через а2 и а—, а стандартные отклонения
X
генеральной совокупности — через а и а—.
4. Точность прямого измерения
(вероятное квадратичное отклонение среднего ариф-
метического). Точностью прямого измерения наз.
абсолютную величину разности между х и а, т. е.
ea=|J— а|; e,° = t^k-s- (7)
где а — доверительная вероятность или надежность
(доля случаев, в к-рых среднее арифметич. при данном
числе определений будет лежать в определенных пре-
делах, обычно пользуются надежностью 0,95 и реже
0,99; 0,999); t 4—коэфф, нормированных отклонений
при малом числе отсчетов (малой выборке), к-рый
зависит от и и а. Числовые значения величины д,
для различных значений а и к=п—1 (где п — число
измерений, а к — число степеней свободы) приведены
в табл. 1.
5. Вероятная относительная по-
грешность. Для характеристики точности мето-
да недостаточно найти только ва, но необходимо рас-
считать и вероятную относительную погрешность
метода по ур-нию:
±-^-100% (8)
X
6. Интервальное значение изме-
ряемой величины (доверительный интервал).
Значение этой величины определяется выражением
(9): а=ж±еа или_ _
х—ea<a<i-f-ea (9)
х — t ks- <a<x-\-t ks-
Равенство (9) разрешает совершенно точно задачу об
оценке приближенного равенства а^~х.
При обработке результатов небольшого числа из-
мерений очень важно не только найти интервальные
значения измеряемой величины, но решить и другую
задачу теории ошибок, найти и оценить приближен-
ное значение стандартного отклонения а (для гене-
ральной совокупности). Считают a=s=s и интервальное
значение а находят в виде a=y±dtz или
s—Sa<a<s + Sa (Ю)
где Sa — точность стандартного отклонения, опреде-
ленная с надежностью а при к=п—1, 6a=?aifeS, где
s — средняя квадратич. ошибка отдельного измере-
ния [стандартное отклонение, см. ур-ние (3)], fe-
множитель, к-рый находят по табл. 2. Для значе’ний
д > 1 левая доверительная граница интервального
значения а приравнивается нулю.
Если величина а известна и |'5—а | > еа, то при из-
мерениях допущена систематич. ошибка (погрешность)
Е, интервальное значение к-рой заключено в пре-
делах _
х — а — еа<Е<х—а+еа (11)
Таким образом, значения х, х±ва", s и пол-
ностью определяют точность, правильность и воспро-
изводимость измерения.
Пример. При повторных взвешиваниях стеклянного
фильтра были получены следующие результаты (в г): 10,2375;
10,2374; 10,2378; 10,2375. Определить среднее и интервальное
значения веса фильтра и стандартного отклонения, а также
27
839
ОШИБКИ АНАЛИТИЧЕСКОГО ОПРЕДЕЛЕНИЯ
840
Таблица 2. Значения & для разных значений ft и а
k a
0,95 0,99 0,999
1 14,947 77,786 397,947
2 3,415 9,978 30,802
3 1,914 4,111 10,112
4 1,372 2,670 5,637
5 1,091 2,006 3,877
6 0,915 1,624 2,977
7 0,797 1 ,377 2,424
8 0,711 1,203 2,055
9 0,645 1,076 1,796
Ю 0,593 0,977 1 ,599
12 0,515 0,833 1,327
14 0,460 0,733 1,145
16 0,418 0,659 1,013
18 0.385 0,602 0,916
20 0,358 0,556 0,838
25 0,310 0,473 0,700
30 0,276 0,416 0,609
35 0,253 0,375 0,544
40 0,234 0,343 0,494
45 0,219 0,318 0,455
50 0,209 0,297 0,424
60 0,186 0,266 0,375
70 0.172 0,242 0,339
80 0,160 0,224 0.311
90 0,150 0,209 0,289
100 0,142 0,196 0.271
150 0,115 0,159 0,220
200 0,099 0,135 0,184
250 0,089 0,120 0,162
300 0,081 0,109 0,146
400 0,075 0,100 0,134
450 0,070 0,093 0,124
500 0,066 0,088 0,116
0,062 0,083 0,110
600 0,057 0,076 0,099
700 0,053 0,070 0,091
800 0,049 0,065 0,085
900 0,046 0,061 0,080
1000 0,044 0,058 0,076
5000 0,020 0,026 0,037
10000 0,014 0,018 0,023
относительную ошибку взвешивания с надежностью а=0,95.
Записываем данные в след, таблицу:
JM5 опыта Вес фильтра, г(хО (х; — х)-10* (х;-х)-10а
1 10,2375 —1 1
2 10,2374 -2 4
3 10,2378 + 2 4
4 10,2375 -1 1
Суммы: 40,9502 10-10-8
п= 4
й = 4— 1 = 3
тт - ~ 40,9502
Применяя вышеприведенные формулы, находят: х——-----=
= 10,2376 (деление суммы результатов отдельных измерений на
число этих намерений п надо продолжать до тех пор, пока част-
ное станет иметь на одну цифру больше, чем их имеется в ре-
зультатах отдельных измерений. Затем в полученном частном х
отбрасывают последнюю цифру по правилу округления и после
вычисляют остаточные погрешности v,=xi—я).
10- IO-8
3
= 3,3-10-8
s=V3,3-10-’ = l,8-10-‘
S—
X
8
Vn
1,8-10-4
2'
0,9.10-<
Из таблицы 1 находят: t _ = 3,182 и ел . =
а—о и,уэ
= 1 -s_ =3,182-0,9-10-4:=310_4. При оформлении оконча-
ас
тельного результата обычно придерживаются следующего пра-
вила: погрешность должна иметь одну или две значащие циф-
ры, а число, выражающее среднее значение измеряемой вели-
чины, должно оканчиваться разрядом, к-рым начинается по-
грешность. Тогда: а=(10,2376±0,0003) г при п=4 и а=0,95,
т. е. истинное значение веса фильтра лежит в пределах:
Ю,2373< а< 10,2379 с вероятностью 95%.
Для определения интервального значения стандартного от-
клонения из табл, 2 находят:
д . =1,914 и
a=o,9s; ft=s
в =1,914-1,8 • 10 “4 = 3,4 • 10 ~4
0,95
0«Т<5,2-10-4
Относительная ошибка взвешивания, вычисленная с надежно-
3 • 1 0 — 4
стью а=0,95, равняется: q . --7.-100=0,003%.
Отдельные результаты анализа не должны отли-
чаться друг от друга на величину, превышающую
V 2-ea. Эта величина может служить критерием для
выявления данных, к-рые должны быть исключены
из обработки.
Ошибки косвенных измерений. Косвенным назы-
вается измерение, при к-ром вначале проводят прямые
измерения нек-рых величин (xi, хг, ..., хь ..., хп),
а затем по определенным формулам, связывающим
эти величины с измеряемой величиной (у), вычисляют
ее значение у — функция от непосредственно изме-
ряемых величин xi, xt, ..., х,-, хп:
xt, ..., xb ..., х„)
Каждая из величин ц, хг, ..., хь ... , хп имеет свою
ошибку и в зависимости от формулы, по к-рой вычис-
ляют результат анализа, эти ошибки различно влия-
ют на ошибку результата.
При оценке точности косвенных измерений приме-
няют следующие формулы:
1. Среднее значение косвенно определяемой вели-
чины получают подстановкой в расчетную формулу
средних арифметических значений непосредственно
измеренных величин: y=/(xi, х2, ..., хь ... хп} и счи-
тают yz^a, где а — истинное значение косвенно опре-
деляемой величины.
2. Дисперсию косвенно измеряемой величины s*
вычисляют по формуле:
df .
где — частная производная функции/(xj, х», ...,
хь ..., хп) по х,- — прямо измеренной величине, а
s’ — дисперсия прямо измеренной величины ц.
Иногда при обработке результатов косвенных измере-
ний ограничиваются только расчетом дисперсии s’
или дают расчет т. наз. предельной абсолютной и
предельной относительной ошибок, вычислив пред-
варительно предельные абсолютные ошибки прямых
измерений по формуле: X=3s_; строго говоря, при
малом числе прямых измерений предельная абсолют-
ная ошибка
3. Точность значения косвенно измеренной величи-
ны (е ) равняется: е =taft-^=x , а точность стандарт-
’ V п
ного отклонения (S7) : f>x=q,,it'sy (значения t^ k и
ya ft находят соответственно из табл. 1 и 2).
А. Интервальное значение косвенно измеряемой
величины (а): у—e0<a<y-f-ea, а интервальные зна-
чения стандартного отклонения (a): sy—Sa<<T<sv-f-6a.
Пример. Для определения концентрации железа в ис-
следуемом растворе (Сх) был применен фотоколориметрнч. ме-
тод сравнения и из 3 измерений были получены следующие
средние значения оптич. плотности исследуемого и стандартного
растворов: Пт=0,216; ОСт.= 0,148. Выборочные дисперсии
соответственно равнялись: =4-10_“;s2tj =4-10_“;концент-
х ист.
рация стандартного раствора Ссг. =1,20 мкг1мл. Определить
среднее и интервальное значения концентрации Сх и стандарт-
841
ОШИБКИ АНАЛИТИЧЕСКОГО ОПРЕДЕЛЕНИЯ
842
ною отклонения, с надежностью ц=0,95. Сст. примем за точ-
ное число. Тогда:
('сст.^-') с а(сст^') д
0 \_____^ст. / _ист. . \ 17ст, /_
dDx Ост. ’ SDct.
2 _^ст. 2 Сст. °х 2
8С,=О*т W р^/Пст.
С ст V D2 s‘D +D‘s‘D
. С J , * •L'CT.
»ся= =
CT.
1,20-2-IO-’ /219-10-' + 467-10-> _
= 219-10- - U ’J J
При a = 0,95 и fe=n-l=2 го,ж=4,30-!руу = 0,072
Ca.=(l,75± 0,07) мкг/мл
^O,9S=®a=o,»s; ft = 2’Sc^. = 3,41 5-0,0 29 = 0,099
0«J<0,13
При многих косвенных определениях (при установ-
лении нормальности растворов, титра и т. д.) прихо-
дится брать несколько навесок. Получить одинаковые
навески, к-рые различались бы только за счет случай-
ных погрешностей взвешивания, трудно, но если
навески приблизительно одинаковы, то результаты,
полученные по расчетной формуле для каждой навески
отдельно, можно считать за прямые измерения и вы-
числить средние и интервальные значения измеряемой
величины и ее стандартного отклонения, как описано
для прямых измерений.
Совокупные измерения. Совокупными наз. измере-
ния, при к-рых непосредственно наблюдаемая вели-
чина является функцией от определяемых неизвест-
ных величин. Напр., при спектрофотометрич. опре-
делениях непосредственно измеряемое значение оп-
тич. плотности (D) смеси окрашенных веществ явля-
ется функцией концентраций этих веществ (Ci, Cz,
..., Сп), значения к-рых необходимо найти. При обра-
ботке результатов совокупных измерений применяют
метод наименьших квадратов.
При совокупном измерении все т измеряемых
величин Xj, ..., хт вычисляют одновременно с помощью
р. линейных ур-ний: у=/(х„ х2, ... , хт, Л,, Аг...Ат),
где у — прямо измеряемая величина, А[ — заранее най-
денные постоянные. Число р измерений величины у
должно превышать число совокупно измеряемых ве-
личин пг, т. к. только в этом случае можно рассчиты-
вать на компенсацию случайных погрешностей при
измерении величины у. Согласно принципу суммы
квадратов наименьших отклонений для получения
наивероятнейших значений совокупно измеряемых
величин Xi, х2, ..., хт проводят р измерений величи-
ны у и получают р (р>т) «условных» уравнений
AiiXi-\-Aizxz-\-... -\-Aimxm=yi. Из них получают си-
стему т «нормальных» уравнений:
лпх1 + Aizx> + • • • + А'1тхт= Ri
Azixi 4" Аггхг 4* • • • 4* Агтхт =
Amixi + Атгхе~^ • • • 4* Аттхт ~ Rm
где
f i=U ( i=u г=ц
Ап= 5 Л11- А'гг= 5 Aiz и вообще A'kk= 2 АЛ
i = i i = i i = i
г=ц
<=<=2 АМ,-г
г = 1
г = ц 1 = ц
4,= 4i = 2 И вообще A'jt= А' = АцА-л
i=i i=i
г=ц г=ц г=ц
= 2 = 2 и вообп*е Rk= 2 AikVt
1=1 1=1 1=1
Систему нормальных уравнений решают методом
обращения матрицы ее коэффициентов, эквивалентным
методу решения с помощью неопределенных множи-
телей. Решение имеет вид:
х, = MnRt + MltRt + ... + MlmRm
хг =M21Rj -f-Af22R2 . 4-MimRm
xm—A^miRi 4* • • •
где x^—наивероятнейшие точечные значения совокупно
измеряемых величин. Дисперсию величины xi вы-
числяют по формуле “ MuSy, дисперсию xz по
формуле $£з=М22$у и вообще для Xj по формуле
slx где з’= 2 (у«—Уг)2/*,' А=Р—т и у,- вы-
г=1 о
числяют по формуле 4,-ixi+4I-2a:24-•A-Aimxm=yi,
то есть подстановкой найденных точечных значений ху
в каждое из условных уравнений. Интервальные зна-
чения ху и aXj находят по формулам, приведенным для
прямых измерений.
При повторных совокупных измерениях проводят
ц прямых измерений величины у, вычисляют величи-
ны Ri,Rz, ..., Rm, вычисляют с помощью готовой об-
ращенной матрицы коэффициентов (решения) тачеч-
ные значения ху , подставляют их в систему исходных
уравнений, находят s’, затем , ..., и интерваль-
ные значения всех совокупно измеряемых величин.
Пример. Определить спектрофотометрически концент-
рацию орто- (xt), мета- (х2) и пара- (х.) крезола в их смеси. Оп-
ределение, проведенное в спиртовом р-ре при толщине слоя
1,00 см, основано на прямом измерении оптических плотностей
(D>) при различных длинах волн (А.,), к-рым отвечают след,
миллимолярные козфф. поглощения: е^у —для орто-, —
для мета- и для пара-ксилола. Значения ; и О, приве-
дены в табл. 3, последние 4 столбца к-рой составляют матрицу
системы условных уравнений (ц = 11).
Таблица 3. Определение смеси крезолов
А.,, А % =Л" е, . =А/а ek. =Ais Dt=yt
2685 0,185 0,257 0,480 0.525
2697 0,178 0,200 0,432 0,420
2703 0,195 0,168 0,316 0,382
2709 0,204 0,168 0,257 0,379
2721 0,178 0,313 0,209 0,500
2727 0,148 0,311 0,309 0,530
2733 0,112 0,234 0,355 0,432
2739 0,080 0,210 0,562 0,412
2745 0,071 0,20 0 0,676 0,410
2752 0,042 0,126 0,501 0,280
2758 0,031 0,071 0,355 0 , 174
При вычислении козфф. Afeft, Ajfe и величин Rj считают
значения eAiy и D/ абсолютно точными и сохраняют в резуль-
ОШИБКИ АНАЛИТИЧЕСКОГО ОПРЕДЕЛЕНИЯ
843
тагах все значащие цифры. Полученная система уравнений
включает три нормальных уравнения (т=3): 0,225848 х,+
+ 0,316620х2 + 0.527441Х. = 0,618739 = й( 0,316620х, +-
+0,516960х2 + 0,892161xs= 0,985345 = й2 0,527441х1Ч-0,892161
х24 1,997962 хя=1,783939=й,. Решая эту систему путем обра-
щения матрицы, находят точечные значения:
X, = 32,109416 й,—21,960465 й2+1,329579 й3=0,600602
х2=—21,960455 Й! + 23,452561 й2-4,675072 й3=1,181031
4=1,329574 Й!-4,675070 й3+2,237101 й3 = 0,206954
Подставляя найденные значения х,, х2 и х„ в исходную систему
условных уравнений, вычисляют значения £>;, находят, что
i = ii -з/
(Dj-£>j)2=l,105-Ю-3, 3^ = 1,105-10 /s=l,381-10-‘
г = 1
так как А=11—3=8
В2Х =32,109416-1,381-10-*=4,434-10-’
в2 = 23,452961-1,381 • 10-*=3,239-10-3
х2
S2 =2,237101 -1,381 -10-‘=3,089-10
844
Используя i . „=2,31, находят, что при а=0,95
Ct — v, У Э, И —• о
еХ1 = 2,31 /4,434 • 10 ~ 3/11 =0,0464
еЖа=2,31 /3,239-10 -»/11 =0,0396
еЖа=2,31 /3,089-10“ */11 =0,0122
и искомые интервальные значения концентраций (милли-
моли, л) равны:
Х! = 0,60 +0,05; х2=1,18±0,04 и х3=0,21 + 0,01
Определить границы доверительных интервалов с большей
точностью в этом случае нельзя, т. к. при А=8 дО1!П₽=0,71,
а это значит, что с заданной надежностью а=0,95 стандартные
отклонения ах. определяются с точностью до i0,71 sX;.
При последующих измерениях концентраций крезолов в
спиртовом р-ре измеряют D; при тех же 11 длинах волн, вы-
числяют Ri и подставляют их в готовые расчетные формулы.
Лит.: КрыловА. Н., Лекции о приближенных вычис-
лениях, Л., 1933; Линник Ю. В., Метод наименьших квад-
ратов и основы математико-статистической теории обработки
наблюдений, М., 1962; Романовский В. И., Основные
задачи теории ошибок, М.—Л., 1947; Комарь Н. П., Ж.
аналит. химии, 1952, 7, вып. 6; Бродский А. Д., К а н
В. Л., Краткий справочник по математической обработке ре-
зультатов измерений, М., 1960; Налимов В. В., Приме-
нение математической статистики при анализе вещества, М.,
1960; Яковлев К. П., Математическая обработка резуль-
татов измерений, М.—Л., 1950; Дымов А. М. (и др.], За-
водск. лаборатория, 1955, № 4, 504. И. П. Калинкин.
ПАБК, см. _ п-Аминобензойная кислота.
ПАЛЛАДИИ (Palladium) Pd — химич. элемент
VIII гр. периодич. системы Менделеева, принадлежит
к платиновым металлам; и. н. 46, ат. в. 106,4. При-
родный П. состоит из 6 стабильных изотопов: Ра102
(0,96%); Pd101(10,97%); Pdlos(22,23%); Pd106(27,33%);
Pd108 (26,71%) и Pd110 (11,81%). Поперечное сечение
поглощения тепловых нейтронов атомом П. 8,0 ±
±1,5 барн. При делении U, Th, Pu образуются радио-
активные изотопы П.; важнейший из них Pd1<”(T>;2=
=13,6 час.) используется в качестве радиоактивного
индикатора. Конфигурация внешних электронов атома
П. 4р’4<710. Энергии ионизации (в эв): Pd°->-Pd+->-Pd2+
соответственно равны 8,33 и 19,8.
П. открыт в 1803 Волластоном в неочищенной пла-
тйне и назван в честь астероида Паллады, открытого
в 1802. Химия П. и методы отделения его от других
металлов изучены К. К. Клаусом, Л. А. Чугаевым,
И. И. Черняевым. Содержание П. в земной коре
1-10_’вес.%. П. находится в природе в самородном
виде, в виде сплавов с другими благородными метал-
лами и химич. соединений.
Ниже приводятся важнейшие минералы П, А л-
лопалладий (в основе металлич. П.; гексаго-
нальная структура, содержит примеси Hg, Pt, Ru,
Си); палладистая платина (до 37% Pd,
остальное Pt); палладит PdO, образующийся
в результате окисления П. или палладистой платины
и встречающийся в виде бурой массы; п о т а р и т
PdHg (34,9—35,9% Pd, остальное Hg); станно-
палладинит Pd3Sn2 (с примесью Си и Pt);
арсенопалладинит Pd3As; стибиопал-
л а д и н и т Pd3Sb, встречающийся совместно со
спериллитом PtAs2; майченерит PdBi2; пор-
пецит (85,98% Аи, 4,17% Ag, 0,1% Си, 8,2—11,6%
Pd); брэггит (Pt, Pd, Ni)S (18—20% Pd, 58—60%
Pt, 2,8—4,7% Ni, 16,8—19%S) — первый минерал,
идентифицированный методом рентгеновского ана-
лиза и названный в честь основателей кристаллогра-
фии Брэггов. П. совместно с другими платиновыми
металлами и золотом встречается в месторождениях,
содержащих сульфидные минералы никеля и меди,
в СССР — на Урале и в Сибири, в Канаде и США,
Южной Америке и Южной Африке. Содержание П.
в самородной платине различных месторождений
составляет от 0,3 до 1,0%.
Физические и химические свойства. П.— серебристо-
белый металл, по внешнему виду напоминающий
платину, с наименьшими уд. весом и темп-рой плав-
ления из всех платиновых металлов; в чистом виде
мягок, пластичен и легко поддается обработке.
Кристаллизуется в гранецентрированной кубич. ре-
шетке а=3,8824 А., Атомный радиус 1,37 А; ионные
радиусы Pd2+0,88 A, Pd4+0,73 А, Плотн. 12,02 (20°),
11,0 (1550°). Т, пл. 1552°; т. кип. 3980° (вероятно);
теплота плавления 38,6 кал)г', теплота испарения
88,Зк ал/г. Уд. теплоемкость 0,0584 кал/г-град (0°);
термич. коэфф, линейного расширения 11,67-10-6
(0°). Теплопроводность 0,17 кал/см-град-сек. Уд.
электросопротивление (.ико.и-с.и): 10,0 (0°), 10,8
(20°); термич. коэфф, электросопротивления 37,7-10-1
(0—100°). Магнитный момент атома П. 8,01 магнето-
нов Бора; П. парамагнитен, атомная магнитная
восприимчивость 6,42-10-’ (20°). Механич. свойства
П. (при комнатной темп-ре): модуль нормальной
упругости 12600 кГ/мм2', предел прочности при рас-
тяжении 18,5 кГ/мм2', относительное удлинение 24—
30%; твердость по Бринеллю 49 кГ/мм2 (отожженного).
Механич. свойства П. изменяются в зависимости
от примесей и способа обработки. При холодной
прокатке твердость П. увеличивается в 2—2,5 раза.
Прочность при растяжении увеличивается при хо-
лодной обработке и при добавлении нек-рых элемен-
тов, особенно Ru и Rh.
П. легче, чем другие платиновые металлы, подвер-
гается химич. воздействиям. Он хорошо растворим
в царской водке; в отличие от других платиновых
металлов, растворяется в конц, горячей азотной
к-те и в конц. серной к-те при 300°. П. может быть
также переведен в раствор электролитически, анодным
растворением в соляной к-те. П. окисляется с поверх-
ности при нагревании на воздухе до 700°, реагирует
с влажными хлором и бромом при комнатной темп-ре,
с сухими фтором и хлором, а также с серой — при
темп-ре красного каления. Характерным свойством
П. является поглощение большого количества во-
дорода с увеличением объема металла (до 900 объемов
Н2 на 1 объем П.). Предполагается, что при опре-
деленном насыщении образуется соединение состава
Pd2H, но существующие литературные данные раз-
норечивы. Водород легко диффундирует через пал-
ладиевые пленки; благодаря достаточно высокой
скорости диффузии при 400° это свойство П. исполь-
зуют для получения очень чистого водорода; весь
поглощенный водород может быть удален при нагре-
вании П. в вакууме при 100°. При повышенных
темп-рах П, растворяет небольшие количества кис-
лорода и, если после этого поместить образец в ат-
мосферу водорода, образуются пары воды, разрыва-
ющие кристаллич. решетку.
В соединениях П. обычно 2- и 4-валентен, реже
3-валентен. Наиболее характерная валентность 2.
По химич. свойствам П. наиболее близок к платине,
хотя является более активным. Известны окислы
2-, 3- и 4-валентного П. 3 а к и с ь П. PdO — чер-
ный порошок; разлагается на элементы выше 870°;
сильный окислитель (окисляет СО до СО2 при 100°).
Образуется при нагревании П. в кислороде при 600—
700° или при сплавлении дихлорида PdCl2 с NaNO3
при 600°. В воде PdO нерастворима, соответствующая
гидроокись Pd(OH)2 может быть получена при гид-
ролизе Pd(NO3)2 в слабощелочном р-ре. Pd(OH)2
легко восстанавливается до металлич. П. О к и с ь П.
Pd2O3 получена в гидратированной форме Pd2O3-xH2O;
847
ПАЛЛАДИЙ
848
шоколадно-черный порошок. Соединение эндотер-
мично и разлагается при обычной темп-ре; при 0°
переходит в PdO. Почти не растворяется в HNO3
и H2SO4, но легко растворяется в НС1 с выделением
хлора. Образуется при осторожном анодном окисле-
нии р-ра Pd(NO3)2 при —8°. Двуокись П. PdO2
темно-красного цвета; сильный окислитель, медленно
теряющий кислород уже при комнатной темп-ре,
а выше 200° переходящий в PdO. Образуется при
обработке хлоропалладатов Me2[PdClJ щелочами.
Из соединений П. с фтором известны PdF2 и PdF3.
Дифторид PdF2— коричневые кристаллы, быст-
ро разлагается на воздухе. Очень хорошо растворим
в воде. Может быть получен при восстановлении PdF3
водородом, сернистым газом или иодом. Наиболее
устойчив трифторид PdF3— черные кристаллы,
изоморфные с трифторидами Fe, Со, Rh. При нагре-
вании на воздухе переходит в PdO и Pd. PdF3 очень
гигроскопичен, реагирует с водой с образованием PdO
или Pd(OH)2. Может быть получен из элементов во
флюоритовой трубке при 500° или нагреванием PdCl2
с фтором при 200—250°.
Из соединений П. с хлором хорошо изучен д и-
хлорид PdCl2— красные кристаллы, гигроско-
пичные и легко растворимые в воде. Из раствора
выделяется коричневато-красный осадок PdCl2-xH2O.
Установлено, что безводные кристаллы PdCl2 состоят
из цепочек атомов
Расстояние Pd—Cl 2,31 А (теоретически 2,36 А).
PdCl2— пример типичного мостикового соединения:
в полимерной цепочке атом Pd связан с 4 атомами С1,
а каждый атом С1 является мостиком между 2 атомами
Pd. При нагревании PdCl2 сублимируется при 600°,
диссоциируя на элементы; давление хлора достигает
1 апгм при 738°; т. пл. под давлением 936°. В растворе
PdCl2 легко восстанавливается до металла — водоро-
дом на холоду, а этиленом при нагревании (эта ре-
акция специфична для П. и позволяет отделять его
от др. платиновых металлов). PdCl2 может быть по-
лучен из элементов при темп-ре красного каления,
при анрдном растворении П. в соляной к-те и при
растворении П. в царской водке.
При растворении П. в смеси бромистоводородной
к-ты и брома образуется дибромид PdBr2
коричневого цвета, растворимый в НВг и почти не-
растворимый в воде. При добавлении KJ к р-ру PdCl2
образуется д и и о д и д PdJ2 черного цвета; в воде
нерастворим, слегка растворим в р-ре KJ с образо-
ванием комплексного иодида K2[PdJ4], Дииодид
применяется для количественного определения П.
Из соединений П. с серой известны PdS и PdS2.
М оносульфид PdS образуется при нагревании
П. с S или H2S в виде голубоватых кристаллов либо
при действии H2S на р-ры солей Pd*+ в виде коричне-
вого порошка; т. пл. 950°; в воде нерастворим. Д и-
сульфид PdS2— шоколадно-серое или черно-
серое вещество, растворимое в царской водке, нераст-
воримое в воде и кислотах. Может быть получен
при нагревании PdS с S при 400°, PdCl2 с S при 400—
500° и др. способами. Известны селениды PdSe, PdSe2
и Pd4Se и теллурид PdTe2. При пропускании СО
через суспензию PdCl2 в абс. спирте при 0° или при
пропускании смеси СО и паров СН3ОН над сухим
PdCl2 образуется светло-желтый карбонил
Pd(CO)Cl2, устойчивый в сухом состоянии, но раз-
лагаемый водой; р-р PdCl2 служит очень чувствитель-
ным реактивом на СО. При пропускании NO в смеси
с парами СН3ОН через сухой PdCl2 на холоду обра-
зуется темно-коричневый нитрозил Pd(NO)2Cl2,
разлагаемый водой.
П. дает сплавы с благородными и другими
металлами, широко применяющиеся в технике. Из-
вестны сплавы П. с Pt, Rh, Au, Ag, Ru, Cu, Ni, Mo,
W, Zn. Сплавы П. c Ag характеризуются коррозион-
ной стойкостью, постоянством и низким темпера-
турным коэфф, электросопротивления. Сплавы П.
с Au и Pt обладают высокой и стабильной термич.
эдс; сплавы П. с Ni, Си, Ru пластичны, хорошо поли-
руются. Как и сам П., его сплавы обладают способ-
ностью поглощать водород. Так, напр., сплав Pd —
Pt (23,97% Pt) поглощает 702 объема Н2 на 1 объем
сплава, причем объем сплава увеличивается на 8,423 %.
Из простых солей П. наиболее изучены следующие.
Нитрат Pd(NO3)2 — коричневато-желтые кри-
сталлы, растворимые в воде и гидролизующиеся при
кипячении р-ра с o6pa3OBainieMPd(OH)2 или PdO -Н2О.
Получается при растворении порошка П. в теплой
азотной к-те. Сульфат PdSO4-2H2O — красно-
коричневые кристаллы, растворимые в воде и легко
гидролизующиеся при нагревании с образованием
Pd(OH)2 и основных сульфатов. Получается при
добавлении небольшого количества воды к р-ру П.
в горячей конц. серной к-те и охлаждении р-ра.
С е л е н а т PdSeCh— мелкие коричневатые гигро-
скопичные призматич. кристаллы, восстанавлива-
ющиеся до SeO2 горячей конц. соляной к-той. PdSeO4
дает двойные соли с сульфатом и селенатом аммония.
Получается при растворении порошка П. в смеси
селеновой и азотной к-т. Дицианид Pd(CN)2—
желтое желатиноподобное вещество, разлагающееся
при 210°; образуется при добавлении р-ров KCN
или Hg(CN)2 по каплям к р-ру PdCl2. Т етрациа-
н и д Pd(CN)4— светло-розового цвета, менее ус-
тойчив, чем Pd(CN)2, медленно разлагается с выде-
лением HCN. Получается при добавлении водного
р-ра K2[PdClJ к Hg(CN)2.
Аналитическое определение. П.—
единственный металл платиновой группы, для к-рого
существует избирательный реактив — диметилглиок-
сим в кислом р-ре, выделяющий желтый объемистый
осадок диметилглиоксимина П. Качественно П. можно
также обнаружить либо осаждением черного PdJ2
при добавлении KJ к р-рам солей Pd2+, либо осаж-
дением желтого Pd(CN)2 при действии Hg(CN)2
на р-р PdCl2. Количественно П. определяют весовым
методом в виде металла после прокаливания раз-
личных его соединений последовательно в токе Н2
и СО2. Кроме того, существуют весовые методы опре-
деления П. в виде соединений с KJ, диметилглиокси-
мом, нитро-6-хинолином, о-фенантролином, циклогек-
сандиондиоксимом. Колориметрич. метод определе-
ния П. основан на образовании коричневато-желтой
окраски при добавлении SnCl2, усиливаемой введе-
нием следов Hg2Cl2. П. определяют полярографи-
чески, на фоне различных веществ: KCN, NH3, C.HSN,
NH2CH2CH2NH2. Предел концентрации П. 510-3—
2-10-1 моль! л.
П. совместно с Rh и 1г может быть отделен от Pt
гидролитич. осаждением в присутствии бромата при
pH 8; Pt остается в р-ре, а вышеуказанные металлы
переходят в осадок. Затем П. количественно отде-
ляется от Rh и 1г из кислого р-ра однократным осаж-
дением диметилглиоксимом.
Получение. П. добывается совместно с платиной
преимущественно из сульфидных медно-никелевых
РУД! а также из россыпных и коренных месторож-
дений и из золотых россыпей и руд. Аффинаж П.
основан на разделении химич. соединений платино-
вых металлов. П., переведенный в 2-валентное со-
стояние, осаждают в виде хлоропалладозаммина
[Pd(NH3)2Cl2] следующим образом: р-р нейтрализуют
849
ПАЛЛАДИЙ —ПАЛЛАДИЯ КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ
850
водным р-ром аммиака и доводят до сильнощелочной
реакции (при большом избытке кислоты сначала до-
бавляют р-р NaOH до слабокислой реакции). При
действии NH3 П. образует аммиакаты: сначала крас-
ный осадок [Pd(NH3)4][PdC14], а затем при растворении
в избытке аммиака тетрамминхлорид [Рб(ХНз)4]С12.
Из р-ра в осадок выпадают гидроокиси Fe, Rh и др.
металлов. Их отфильтровывают; фильтрат подкисляют
соляной к-той до слабокислой реакции (избыток
к-ты не должен превышать 2—3%). Выпадает рыхлый
осадок хлоропалладозаммина [Pd(NH3)2C12], к-рый
промывают холодной водой, подкисленной соляной
к-той. Осадок прокаливают при 800—900°, полученный
П. измельчают и восстанавливают в струе водорода.
Металлич. П, может быть получен в нескольких
видах: а) крупнокристаллический м е-
т а л л — при прокаливании комплексных соединений
или при восстановлении солей водородом; б) пал-
ладиевая чернь — при восстановлении вод-
ных р-ров солей Pd различными восстановителями:
формальдегидом, гидразингидратом, окисью угле-
рода и т. д.; в) коллоидный П.— при восста-
новлении водных р-ров солей П. вышеуказанными
восстановителями в присутствии защитного коллоида.
Иногда для восстановления применяют T1CI3. В этом
случае защитным коллоидом служит образующаяся
ТЮа-яНзО.
Применение. П. наряду с платиной, благодаря
своей пластичности и сравнительной дешевизне,
применяется в технике гораздо чаще, чем другие
металлы платиновой группы. В чистом виде П.
применяют для изготовления реторт при перегонке
плавиковой к-ты, для изготовления сосудов-коллек-
торов водорода и сосудов, применяющихся при
разделении изотопов водорода.
Сплавы П. с серебром (40—80% Ag) широко
применяют в аппаратуре связи. Так, сплав, состоящий
из 60% Pd и 40% Ag, является в настоящее время
основным в телефонной индустрии США для изготов-
ления контактов. Сплавы П. с Au, Pt, Rh применяют
в терморегуляторах и термопарах; сплавы с Ag, Au,
Ni, Ru, Rh — в ювелирном и зубоврачебном деле.
Соли П. используют в фотографии.
Палладий как катализатор. П., на-
несенный на носители, а также палладиевую чернь
и коллоидный П. широко используют в органич.
катализе как в лабораторных условиях, так и в
производственных масштабах. Реакции гидрогени-
зации на П. легко протекают при комнатной темп-ре
(и даже при более низких темп-рах). Дегидрогени-
зация циклопарафинов на П., нанесенном на уголь,
асбест, BaSO4, протекает при 300—400°. При этом
П., в отличие, напр., от Ni-катализатора, проводит
реакцию дегидрирования избирательно до ароматич.
соединений без деструктивного расщепления. П.
катализирует также реакции окисления: П. на ас-
бесте или фарфоре применяют для очистки Н2 от
примесей О2 (и наоборот Оз от примесей Н2). Из
соединений П. в качестве катализатора в последнее
время применяют PdCl2. Водные р-ры PdCl2 с добавкой
окислителей (CuCl2, FeCls и др.) легко и избирательно
окисляют этилен до ацетальдегида.
Лит.: Звягинцев О. Е., Аффинаж золота, серебра и
металлов платиновой группы, 3 изд., М., 1945; Плаксин И.
Н., Металлургия благородных металлов, М., 1958; Минералы.
Справочник, т. 1, М., 1960; Методы анализа платиновых ме-
таллов, золота и серебра. Сб. научных трудов, М., I960; Г и л-
лебрандВ. Ф. [ид р.], Практическое руководство по не-
органическому анализу, пер. с англ., М., 1957, с. 393; G m е-
1 i п, 8 Aufl., Syst.-Num. 65-Palladium, В., 1942; Kirk, v.
10, N. Y., 1S53, p. 819; v. 4, N. Y., 1949; M e 1 1 о r, v. 15, L.—
N. Y. — Toronto, 1947; то же, v. 15, 1953; Rare metals hand-
book, ed. C. Hampel, 2 ed., L., 1961, p. 304—36; Handbook
of chemistry, ed. N. A. Lange, N. Y., 1961; P a s с a 1, v. 19,
P., 1958, p. 577; U 11 m a n n, 3 Aufl., Bd 14, Mtlnch.—в.,
1963, S. 14—43. T. H. Леонова.
ПАЛЛАДИЯ КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ.
Палладий — один из наиболее характерных ме-
таллов-комплексообразователей. Он образует ком-
плексные соединения в 2-, 4- и нульвалентном со-
стояниях. Сравнение аналогичных комплексов пал-
ладия, никеля и платины показывает, что в ряду
Ni—Pd—Pt увеличивается устойчивость высшего
валентного состояния. Особенно многочисленны и
прочны комплексы Pd (II), комплексы же Pd (IV)
сравнительно малочисленны; соединений, содержащих
формально нульвалентный Pd, известно всего не-
сколько. Для Pd (II) характерно координационное
число 4 (комплексы плоского строения); для Pd
(IV) — координационное число 6 (октаэдрич. строе-
ние); соединения Pd (0), напр. К4[Р<1(СХ)4], вероятно,
имеют плоское строение. Описано также несколько
соединений формально 3-валентного Pd, напр. состава
K2PdCls и Pd(NH3)2C13. По аналогии с однотипными
соединениями Pt считают, что эти комплексы содержат
одновременно Pd (II) и Pd (IV) и их строение таково:
К2 [РаС14] • К2 [PdClJ и [Pd(NH3)2Cl2J-[Pd(NH3)2C14].
Для Pd характерно образование комплексных
соединений следующих типов:
1) Комплексы, к-рые формально можно предста-
вить как продукты сочетания простых сол:ей Pd
(напр., PdCl2) с соединениями, молекулы к-рых
содержат N„S, О и др. атомы со свободной электрон-
ной парой [напр., NH3, (NH3)2CS и т. п.]. Таковы
производные Pd(II): а) тетраминового типа [PdA4]X2;
6) триаминового [PdAsXJY; в) диаминового [PdA2X2]
и г) моноаминового [PdAX3]Me (А — нейтральная
молекула, X и Y — однозарядные анионы, Me —
однозарядный катион, простой или комплексный).
Примеры указанных производных: а) [Рй(КНз)4]С12,
б) [Pa(NHs)sCl]2-[PdcL], в) [Pd(NH3)2(NO2)2],
г) [PdNH3C13h-[Pd(NH3)4].
Среди комплексов Pd (IV) известны только диа-
мины, напр. [Pd(NH3)2C14].
2) Комплексы типа двойных солей, напр. K2[PdCl4]
или K2[PdClJ.
3) Внутрикомплексные соединения Pd (II), напр.
ОНО
/ \
CH3-C=N N=C-CH3
I 4 Pd 7 j
CH,-C = N N = C—CH3
\ /
OHO
4) Многоядерные комплексные соединения Pd (II),
напр.
(CH,).,As Cl Cl
\ / \ /
Pd Pd
/ \ / \
Cl Cl As(CH3)3
5) Сверхкомплексные соединения, напр, описанное
выше [Pd(NH3)2Cl2]- [Pd(NH3)2C14],
Для комплексных соединений Pd характерно яв-
ление изомерии. Вследствие меньшей прочности
связи Pd — адденд, по сравнению со связью Pt—
адденд, соединения Pd более склонны к изомеризации,
причем более устойчив более симметричный транс-
изомер. Ниже перечислены типы изомерии комплек-
сных соединений Pd.
1) Геометрич. изомерия
NH3 NO3 NIL N03
Pd и Pd
NH3 NOa NO2 NH,
Геометрич. изомерия Pd (IV) не изучена.
2) Оптич. изомерия для Pd (II) не характерна
вследствие плоского строения комплексов. Она по-
является, однако, если во внутреннюю сферу комплек-
са входит оптически активный адденд. Оптич. изо-
мерия комплексов Pd (IV) не наблюдалась.
851
ПАЛЛАДИЯ КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ—ПАЛЮСТРОВАЯ КИСЛОТА
852
3) Координационная полимерия, напр.
[Pd(NH,)J-fPdCl4], [PdNH,Cls],-[Pd(NH,)H и [Pd(NH,),C],J
4) Координационная изомерия, напр.
[Pd(NHs)3Cl],-[PdCM и [PdNH,Cl,],-[Pd(NH,)J,
[Pd(NHs),)-[PdCle] и rPd(NH3)3Cl2]*Pdf(NH1)2Ci,J
5) Сольватная изомерия, напр.
[PdBr,| AsC2H-,(CeH5)2}2]AsC2HB(CeIJ5)2
и [Pd{ AsC,H6(C.H5),}3 Вг]Вг
Сольватные изомеры Pd (IV) неизвестны.
Комплексные соединения Pd и Pt имеют много
общего. Так, как и в случае Pt (II), высокой проч-
ностью отличаются связи: Pd (II) — N, Pd (II) — S
и Pd (II) — Р и Pd(II) — Hal. Связь Pd(II) — О, как
и Pt(II)—О, малоустойчива. Однотипные производ-
ные Pd и Pt изоморфны, напр. [Pt(NH,)4]Cl2-Н2О и
[Pd(NH,)JCl2.H2O или K2[PtCl,] и K2[PdCl,].
Комплексы того и другого элемента подчиняются
закономерности трансвлияния, причем последова-
тельность изменения силы трансвлияния координи-
рованных групп, по-видимому, одинакова для обоих
элементов. Ниже перечислены нек-рые правильности
в химии комплексных соединений Pd(II), наблюда-
ющиеся и в случае Pt(II) и объяснимые с позиций
закономерности трансвлияния, а) При взаимодействии
ацидосоединений Pd(II) со свободными аминами об-
разуются тетрамины:
2K2[PdClJ + 4А —> [PdAJ • [PdClJ + 4КС1
где A=NH2, C,HsN и t. п.
Однако при проведении реакции в слабокислой
среде образуются, в соответствии с закономерностью
трансвлияния, как и в случае соединений Pt(II),
цис- диамины:
Если же ацидосоединения Pd(II) взаимодействуют
с соединениями с повышенной трансактивностью,
напр. с алкильными производными фосфора, то
образуются транс-изомеры, напр.:
R3P С1
Pd
Cl PR3
где R — органич. радикал, б) При расщеплении тет-
раминов Pd(II) кислотами (или их солями) в боль-
шинстве случаев сначала образуются тпранс-диамины,
к-рые при дальнейшем нагревании в кислой среде
переходят в ацидопроизводные:
[Pd(NH3)4] С13 + 2НС1 —> трстс= [Pd(NH3)2Cl2] + 2NH,C1
mpanc = [Pd(NH3)2Cl2] + 2HCl —> (NH1)3[PdCli]
У производных Pd(II) эта реакция протекает быстрее
и в более мягких условиях, чем в случае Pt(II).
Отклонения от этого правила у производных Pd(II)
наблюдаются чаще, чем у Pt(II). Напр., [Pd(NH,)4]Cl2
при взаимодействии с KNO2 образует смесь цис-
и шранс-динитропроизводных [Pd(NH,)2(NO2)2J, а
не только транс-изомер, как это имеет место при
взаимодействии [Pt(NHs)4]Cl2 с KNO2.
в) При действии тиомочевины как на цис-, так и
на тпранс-диамины Pd(II) образуются [Pd(Thio)4]Cl2,
где
NH,
/
Thio означает S = C
\
NH,,
тогда как в случае платиновых комплексов эту ре-
акцию применяют для того, чтобы отличить цис-
от транс-диамина.
г) Совместная кристаллизация двух к.-л. транс-
диаминов Pd(II), как и в случае соответствующих
производных Pt(II), приводит к образованию смешан-
ных диаминодиацидосоединений:
NH, Cl NH, NO, NH, NO,
Pd + Pd —>2 Pd
Cl NH, NO, NH, Cl NH,
что также объясняется закономерностью трансвлия-
ния.
д) При окислении производных Pd(II), как и Pt(II),
образуются производные 4-валентных элементов, от-
личающихся от исходных — плоскопостроенных, толь-
ко наличием третьей координаты;
Вг
Ру,------_,С1 Ру,—I_____ CI
/ Pd / окис л. / lpd /
ciZ——” ci^-—|—
Вг
Комплексные соединения Pd(IV) мало изучены.
Описаны K2[PdCle], K2[PdBre], mpaHC-[Pd(NH2)2C14],
тпранс-[РЙРу2С14], тпранс-[Р(1Ру2С12Вг2], транс-
[PdPy2Cl2J2] и т. д., получающиеся в результате
окисления хлором, бромом или иодом р-ров K2[PdC14],
K2[PdBr4], mpaHC-[Pd(NH2)2Cl2], транс-[Р<1Ру2С12]
соответственно. Все производные Pd(IV) являются
сильными окислителями и легко переходят в соеди-
нения Pd(II). Производные нульвалентного Pd полу-
чают восстановлением производных Pd(II). Напр.,
K.4[Pd(CN)4] — при восстановлении K2[Pd(CN)4] ме-
таллич. калием в жидком NH,. Все комплексы Pd(O)—
сильные восстановители. Напр., тетрацианопалладаты
(О) Me4[Pd(CN)4] восстанавливают ионы Ag+ и Hg22+
до металлов и выделяют водород из воды.
Н. Н. Желиговская.
ПАЛЬМИТИНОВАЯ КИСЛОТА (гексадекановая
кислота) С15Н21СООН, мол. в. 256,42 — кристаллы;
т. пл. 63,1; т. кип. 351,5; d’20,8534; п™ 1,4309. П. к.—
наиболее распространенная в природе жирная кис-
лота. Находится практически во всех природных
жирах (в виде триглицерида); главная составная
часть свиного сала, кокосового масла; пальмовое
масло содержит 35—40%. П. к. отделяют от сопут-
ствующих жирных к-т (после расщепления жира)
либо дробной кристаллизацией из ацетона или мета-
нола, либо фракционной перегонкой. П. к. обладает
общими свойствами жирных к-т. Соли П. к. в смеси
с солями нек-рых других кислот используют для
получения мыла; нек-рые эфиры — в приготовлении
красок и покрытий, а также для получения синте-
тич. детергентов. В смеси со стеариновой к-той П. к.
употребляется в виде т. наз. стеарина для произ-ва
свечей И пр. Н. А. Несмеянов.
ПАЛЬМОВОЕ МАСЛО —• см. Жиры растительные.
ПАЛЬМОЯДРОВОЕ МАСЛО — см. Жиры расти-
тельные.
ПАЛЮСТРОВАЯ КИСЛОТА С20Н20О2, мол. в.
302,45 — бесцветные кристаллы без запаха; раство-
рима в большинстве органич. растворителей, нераст-
ворима в воде. Т. пл. 162—167°; [о]д=+71,6°; X макс.
266 .и.ик (спирт). Соли щелочных металлов и аммония
растворимы в воде, лучше в этаноле, но нерастворимы
в неполярных растворителях. Положение двух двой-
ных сопряженных связей в П. к. пока точно не уста-
новлено; предполагают два возможных положения их:
5,7 или 7,13 (см. I). П. к. изомеризуется минераль-
ными или органич. к-тами и при нагревании (см.
Кислоты смоляные); конечным продуктом является
абиетиновая кислота. При дальнейшем нагревании
происходит диспропорционирование водорода в абие-
853
ПАНГАМОВАЯ КИСЛОТА—ПАНТОТЕНОВАЯ КИСЛОТА
854
типовой к-те. Гидрогенизацией П. к. получают ди-
и тетрагидрокислоты, к-рые пока мало изучены.
При нагревании с серой, селеном и палладированным
углем П. к. образует ретен (II). УФ-спектр П. к. в сме-
сях кислот маскируется УФ-спектром левопимаровой
к-ты. Поэтому для качественного и количественного
определения П. к. лучше всего подвергнуть смесь
к-т вначале хроматографич., а затем спектроскопич.
и химич. анализу. П. к. присутствует почти во всех
хвойных бальзамах и полученных из них различных
видах канифоли. Присутствует она (10—15%) в
живице отечественных сосен и ели обыкновенных.
Синтетически ее можно получить кислотной или
тепловой изомеризацией левопимаровой и неоабие-
тиновой к-т с последующим разделением продуктов
реакции хроматограф., или химич. методом или со-
четанием обоих методов. В смеси с другими смоляными
к-тами П. к. (см. Канифоль) применяется в пром-сти.
И. И. Бардышев.
ПАНГАМОВАЯ КИСЛОТА (витамин В15, панга-
метин) Ci0HI9O8N, мол. в. 281,27 — эфир D-глюко-
новой к-ты и диметиламиноуксусной к-ты (D-глюко-
но дим етил аминоацетат).
н он н н
Н ООО- С-С---С - С-СН3-О-СО—СН3—N(CH,)3
II II
онн он он
В кристаллич. виде выделена из рисовых отрубей,
дрожжей, крови, печени. Специфич. качественные
реакции и методы количественного определения не
описаны.
П. к. активирует перенос кислорода в клетках
тканей и участвует в реакциях метилирования раз-
личных биосубстратов; применяется в виде натриевой
соли — бесцветные кристаллы, весьма гигроскопич-
ны, т. пл. 186°, растворимы в воде, нерастворимы в
ацетоне, спирте, хлороформе. Применяется также
кальциевая соль П. к., к-рая почти негигроскопична.
П. к. получают взаимодействием глюконовой к-ты
и диметиламиноуксусной к-ты или D-глюконолактона
с монохлоруксусной к-той с последующей обработкой
полученного хлорацетата диметиламином; высшие
гомологи П. к. (с 4 и 8 метильными группами) обла-
дают более высокой биологич. активностью. П. к.
применяют при лечении хронич. гепатита и цирроза
печени, кардиосклероза, ревмокардита, при явлениях
аноксии, коронарном тромбозе и др. нарушениях
коронарного кровообращения. При применении П. к.
в лечебных целях отмечено прекращение болей при
стенокардии, устранение цианоза, одышки, повы-
шение использования кислорода при острых и хро-
нич. отравлениях алкоголем и др. ядами. П. к.
токсич. действием не обладает.
Лит.: Гаркина И. Н., Вопросы мед. химии, 1962, 8,
вып. 3, 236; Удалов Ю. Ф., ДАН СССР, 1962, 143, № 3,
734; Krebs Е. Т. [а. о.]. Internal. Rec. Med., 1951, 164;
Beard H. H., Wofford G., Exptl. Med. and Surg.,
1956,14,169. B.H. Букин.
ПАНКРЕАТИН — высушенный экстракт подже-
лудочной железы свиней и крупного рогатого скота,
подвергшейся аутолизу. Он содержит ферменты под-
желудочной железы: панкреатич. амилазу, липазу,
карбоксипептидазу, панкреатич. протеиназы (трип-
син и химотрипсин). Медицинский П.— желтоватый
порошок с характерным запахом, плохо растворяется
в воде и не растворяется в этиловом спирте. В каче-
стве наполнителя добавляют сахарозу или лактозу.
Оптимум действия П. находится в нейтральной и
слабощелочной среде.
Для получения П. смешивают измельченную под-
желудочную железу с водой, подкисляют до pH 4—5,
прибавляют хлороформ и выдерживают при комнат-
ной темп-ре в течение суток, затем ткань отфильтро-
вывают и фильтрат выпаривают в вакууме до порош-
ковидного состояния. Медицинский П. выпускается
в виде порошков и таблеток и назначается для приема
внутрь при расстройствах пищеварения, связанных
с недостаточностью секреции поджелудочной железы
или нарушением функции печени. Технич. препараты
П.— высушенная и измельченная поджелудочная
железа — применяются в кожевенной пром-сти, в
произ-ве желатины, бактериальных сред и др.
Лит.: Машковский М. Д., Лекарственные средства,
4 изд., М., 1960, с. 385; Northrop J. Н., К uni tz М.,
Herriott R. М., Crystalline enzymes, 2 ed., N. Y.,
1948. __ Г. А. Левдикова.
ПАНТОТЕНОВАЯ КИСЛОТА[витамин В„ М-(а,у-ди-
окси-0, Р~диметилбутирил)-р-аланин]
CH3OH-C(CH,)3-CHOH-CO-NH-(CH)3-COOH
мол. в. 219,24 — распространенный водораствори-
мый витамин, встречающийся в природе только в
D-форме, светло-желтый, вязкий, маслообразный
продукт; [а]”=-|-37,5о (вода). П. к. легко растворима
в воде, этиловом спирте, этилацетате, диоксане,
ледяной уксусной к-те; слабо растворима в эфире и
высших спиртах, нерастворима в бензоле и хлорофор-
ме. П. к. образует хорошо кристаллизующиеся
соли: пантотенат натрия — бесцветные сильно гиг-
роскопич. иглы, т. пл. 121—122° (с разл.), [а]р=4-27°
(вода); пантотенат кальция, т. пл. 193,5—195°, [а]^=
= 4-24,3° (вода).
П. к. устойчива в нейтральных р-рах; при нагре-
вании с к-тами и щелочами гидролизуется по амидной
связи на 0-аланин и пантоевую (а,у-диокси-0,0-
диметилмасляную) к-ту. П. к. не окисляется кисло-
родом воздуха, разрушается КМпО4, способна обра-
зовывать простые и сложные эфиры, хлорангидриды,
азиды; с холином П. к. образует соль, к-рая обладает
биологич. активностью.
Синтез П. к. сводится к конденсации пантоевой
к-ты или ее лактона с 0-аланином, его эфирами или
солями:
сн, сн, сн,
\ \ / KCN
сн-сно-------> носн3- с-сно ---->
/ К3СО, СаС13
СН,
СН, СН, сн, сн,
\ / \ /
—>-НОСН2 С —СН—CN —>- СН3-С-СН(ОН)-СО—►
нс11—°—1
H3NCH3CH3CO3Na
-------------->- П. к.
СаСО,
П. к. широко распространена в природе; в наиболь-
ших количествах присутствует (в мкг/г сухого веса):
в печени крупного рогатого скота (80—180), яичном
желтке (125), пивных дрожжах (200), мясе (38),
картофеле (28), молоке (22/1 лм). Определение П. к.
производят микробиология, методами анализа. По-
требность в П. к. для человека составляет не менее
10 мг в сутки; лечебная доза может доходить до 500 мг.
В клетках животных и растительных тканей П. к.
находится преим. в связанной форме, являясь со-
ставной частью кофермента А.
Лит.: The vitamins chemistry, physiology, pathology, ed.
W. H. Sebrell, R. S. Harris, v. 2, N. Y., 1954; Березов-
ский В. M., Химия витаминов, М., 1959, с. 91; П р е о б-
855
ПАНТОЦИД—ПАРАЛЛЕЛЬНЫЕ РЕАКЦИИ
856
раженский Н. А., Генкин Э. И., Химия органиче-
ских лекарственных веществ, М.— Л., 1953, с. 153.
Л. А. Вакулова. Г. И. Самохвалов.
ПАНТОЦИД (пантосепт, халазон, N, N-дихлор-п-
карбоксибензолсульфамид) C,HsNO4SC12, мол. в.
соон 270,11— белый пдрошок со слабым запа-
с ° хом хлора, почти нерастворим в воде и
разб. к-тах, легко растворим в р-рах ед-
ких и углекислых Щелочей. П. содержит
не менее 50% активного хлора.
SOg-NCiB При прибавлении к р-ру П. р-ра KJ и
хлороформа хлороформный слой окра-
шивается выделившимся иодом в фиолетовый цвет.
При добавлении к разб. водному р-ру П. щелочного
р-ра метилового красного жидкость окрашивается в
красный цвет и далее обесцвечивается. Для количест-
венногоопределения П. растворяют в разб. NaOH, при-
бавляют р-р KJ и разбавленную HjSOi; выделившийся
иод титруют Na2S2O, в присутствии крахмала.
П. получают, исходя из хлорангидрида п-толуол-
сульфокислоты, к-рый действием водного NH, пре-
вращают в n-толуолсульфамид, его окисляют К2Сг2О7
в среде H2SO4 до n-карбоксибензолсульфамида и по-
следний обрабатывают водным р-ром NaOCl. П.
применяют для обеззараживания воды, обработки
ран, дезинфекции рук и др.
Лит.: Машковский М. Д., Лекарственные средства,
4 изд., М., 1960; Государственная фармакопея СССР, 9 изд.,
М., 1961. В. А. Засосов.
ПАПАВЕРИН C20H21O4N, мол. в. 332,39 — один
из алкалоидов мака снотворного (Papaver somnife-
ОСН3
П,— весьма
rum L.), содержится в опии (получаемом
из опийных сортов мака) и в масличном
маке; кристаллы, т. пл. 147°; оптически
неактивен; нерастворим в воде; раство-
рим в горячем спирте и хлороформе; об-
разует слабо растворимый в воде (1 : 40)
хлоргидрат с т. пл. 225—226°; сульфат
несколько лучше растворим в воде; пик-
рат, т. пл. 186°.
слабое основание; он экстрагируется
органич. растворителями из кислых р-ров. Строение
П. подтверждено многочисленными синтезами. Боль-
шую часть потребляемого в медицинской практике
П. получают синтетически из доступных исходных
веществ, напр. из ванилина или вератрола. В расте-
ниях биосинтез П., по-видимому, происходит из двух
молекул тирозина. П. (в виде хлоргидрата) широко
применяется в медицине как спазмолитич. средство
при спазмах кровеносных сосудов (при гипертонии,
стенокардии, мигрени), спазмах гладкой мускулатуры
органов брюшной полости (холецистит, спастич. ко-
лики, спазмы мочевых путей). П. расслабляет глад-
кую мускулатуру.
Лит.: Budeiinsky Z., Protiva М., Syntheti-
sche Arzneimittel, В., 1961, S. 87; Преображенский
H. А., Г e н к и н Э. И., Химия органических лекарственных
веществ, М.— Л., 1953, с. 254; М а й о ф и с Л. С., Техноло-
гия химико-фармацевтических препаратов, Л., 1958, с. 515;
Машковский М. Д., Лекарственные средства, 4 изд.,
М., 1960. А. Е. Васильев.
ПАПАИН (папайотин) — протеолитич. фермент, со-
держащийся в млечном соке дынного дерева Carica
papaya. IL— кристаллич. продукт, растворим в
воде, водных солевых р-рах, а также в 70%-ном
этиловом и метиловом спиртах. Изоэлектрич. точка
П. находится при pH 8,75, константа седиментации
2,42 единицы Сведберга, константа диффузии D—
= 10,23 см/сек*, мол. в. 20700. Молекула П. пред-
ставляет собой одиночную полипептидную цепь из
187 аминокислотных остатков, из ,к-рых 13 принад-
лежит аланину, 10 аргинину, 17 аспарагиновой к-те,
8 цистеину, 17 глутаминовой к-те, 23 глицину,
2 гистидину, 10 изолейцину, 10 лейцину, 9 лизину,
4 фенилаланину, 9 пролину, 11 серину, 7 треонину,
5 триптофану; 17 тирозину, 15 валину; в П. имеется
19 амидных групп. Установлено расположение 165
аминокислотных остатков П., N-концевой амино-
кислотой является изолейцин, С-концевой — аспа-
рагин. П. катализирует гидролиз белков и имеет
оптимум действия при pH 5—8, обладает сравни-
тельно широкой специфичностью. При pH 5—7,5 П.
гидролизует пептиды, амиды и эфиры основных
аминокислот, имеющих незамещенную аминогруппу
в боковой цепи. П. активируется цистеином, циа-
нидами, восстановленным глютатионом, сульфи-
дами. Максимальная активность П. наблюдается в
присутствии цистеина и этилендиаминтетраацетата.
П. не теряет активности при нагревании до 50—60°.
Ионы тяжелых металлов, нек-рые окислители и п-
хлормеркурибензоат обратимо инактивирует П., иод-
ацетамид инактивирует П. необратимо. Предпола-
гают, что в состав активного центра П. входит сульф-
гидрильная группа, образующая с ионизированной
карбоксильной группой тиоэфир. Выделяют П. из
млечного сока дынного дерева или из продажных
препаратов высушенного сока фракционированием
сульфатом аммония, хлористым натрием и последу-
ющей кристаллизацией. П. применяют в пищевой и
легкой пром-сти (для обработки кож, осветления
напитков, размягчения мяса и др.).
Лит.: Современные проблемы биохимии. Сб. статей, пер.
[с англ.], под ред. В. А. Энгельгардта, М., 1961, с. 67; The
enzymes, ed. Р. Boyer, Н. Lardy, К. MyrbHck, v. 4, N. Y.,
1960, p. 133; Смит Э. Л., Киммель Дж. P., Лайт A.,
Структура папаина, в кн.: Труды V Международного биохи-
мического конгресса. Москва. 10—16 августа, 1961. Симпо-
зиум IV, М., 1962. 3. Ф. Евтихина.
ПАРА — см. Орто-, Мета-, Пара.
ПАРАБАНОВАЯ КИСЛОТА (N, N'-оксалилмоче-
вина, 2,4,5-триоксоимидазолидин) C3H2O3N2, мол. в.
114,05 — кристаллы, т. пл. 243—245° (с
разл.), константа диссоциации K=l,5- HN______с=о
•10-’(25°); растворимость в воде 1 : 21,2 I 1
(8°); с нингидрином в аммиаке дает синюю O-CxNpc
окраску.
П. к. можно рассматривать как производное ими-
дазолидина или как смешанный имид угольной и
щавелевой к-т. П. к. обладает слабыми кислыми
свойствами; при действии оснований водород заме-
щается металлом. Аммониевая соль при 100° раз-
лагается на аммиак и П. к.; серебряная соль при дей-
ствии галогеналкилов дает N-алкильные производ-
ные. П. к. легко омыляется щелочами:
NaOH NaOH
О = С —NH —COCONH------*- NH3CONHCOCOONa-----►
I___________J
—> (COONa)2 + NH2CONH2
При электролитич. восстановлении одна из СО-групп
превращается в метиленовую группу, образуется
гидантоин. Содержится П. к. в моче животных.
Она может быть получена окислением мочевой к-ты
азотной к-той и ацилированием мочевины оксалил-
хлоридом или же диэтилоксалатом в присутствии
этилата натрия:
NHjCONHj + С1СОСОС1 —>- OCNHCOCONH
I J
C2H,ONa
NH2CONH2 + (COOC2H,)2 ---► OCNHCOCONH
I J
H. А. Несмеянов.
ПАРАЛЛЕЛЬНЫЕ РЕАКЦИИ. Совместно проте-
кающие реакции наз. параллельными, если по край-
ней мере одно исходное вещество этих реакций яв-
ляется общим (реже говорят о П. р. в случае разных
исходных веществ и общего продукта). Примеры:
нитрование фенола с образованием орто-, мета-,
и паранитрофенола (одни и те же исходные веще-
ства), нитрование смеси бензола и толуола (одно из
исходных веществ — азотная к-та, является общим).
Если необратимые П. р. с участием одних и тех
857
ПАРАМАГНЕТИЗМ — ПАРАФИН
858
же исходных веществ протекают по одинаковому
порядку, с константами скорости к1, к%, к3 ... и на-
чальные концентрации продуктов Х1, Хз, Хз ...
равны нулю, то в любой момент времени концент-
рации продуктов удовлетворяют соотношению
[X,] : [Х2] : [Х3] ...=А, : Аа:Л3...
Лит.: Раковский А. В., Введение в физическую хи-
мию, М., 1938, § 457; Эмануэль Н. М., Кнорре Д. Г.,
Курс химической кинетики, М., 1962, гл. 6, § 2.
М. И. Темкин.
ПАРАМАГНЕТИЗМ — см. Магнитные свойства.
ПАРАМЕТРЫ СОСТОЯНИЯ — величины, харак-
теризующие состояние физико-химич. системы. Для
однокомпонентных систем обычно за П. с. принимают
темп-ру и давление; для систем из двух и более ком-
понентов указывают, кроме того, и концентрации
последних. Если система находится в силовом поле,
указывают и величину, характеризующую это поле,
напр. его напряженность. Если система состоит из
частей с сильно развитой поверхностью раздела,
добавляют к указанным выше П. с. поверхностное
натяжение.
ПАРАМИОН (дииодметилат мезо-п,п'-бис-диме-
тиламино-3,4-дифенилгексана) CaiHjjNaJa, мол. в.
608,39 — белые кристаллы, т. разл. 220—230°; хо-
рошо растворим в горячей воде, при 20° в воде раст-
воряются ок. 0,5%. П. (I) нерастворим в бензоле,
эфире и ацетоне; при нагревании разлагается с
образованием мезо-п,п'-бис-диметиламино-3,4-дифе-
нилгексана (П), т. пл. 210—212°. Количественно П.
определяют в горячем водном р-ре титрованными
р-рами AgNOa и NHiSCN в присутствии железоам-
мониевых квасцов. П. получают взаимодействием
мезо-n,п'-диамино-3,4-дифенилгексана (III) с йоди-
стым метилом в водном метиловом спирте в присут-
ствии углекислого кальция.
Г(С(^)3Й-/~Д-сн-СН-^^-Й(СНз)з J • 2J-
L Н6С2 С2Н5
П. обладает курареподобным действием; применяет-
ся как средство, расслабляющее скелетную муску-
латуру, а также для выключения самостоятельного
дыхания при операциях с управляемым дыханием.
Лит.: Машковский М. Д., Лекарственные средства,
4 иад., М., 1960; Торф С. Ф. [и др.], Фармакология и токси-
кология, 1952, т. 15, 6, с. 12; Торф С. Ф., Хромов-
Борисов Н. В., Мед. пром-сть СССР, 1961, № 6, с. 18.
И. В. Хромов-Борисов.
ПАРАТИРЕОКРИН (паратиреоидин) — гормо-
нальный белковый препарат из околощитовидных
желез млекопитающих, регулирующий соотношение
кальция и фосфора в крови. П. растворим при ком-
натной темп-ре в разб. солевых р-рах, разб. этаноле,
уксусной к-те, при нагревании — в феноле и в гли-
церине; нерастворим в абс. спирте, эфире и др. ор-
ганич. растворителях; изоэлектрич. точка П. 4,8—
6,0. При кислотном и щелочном гидролизе, а также
под влиянием пепсина и трипсина П. инактивирует-
ся. П. содержит два компонента с мол. в. 500 000—
1000 000 и 15 000-25 000.
П. получают из свежезамороженных, консервиро-
ванных или высушенных ацетоном околощитовидных
желез экстрагированием слабой соляной к-той при
100°. После выпадения из экстракта балластных
белков фильтрат высаливают, растворяют его в
слабощелочном р-ре, центрифугируют и многократно
осаждают.
П. повышает содержание кальция в крови, устра-
няет явления, связанные с недостаточной секретор-
ной деятельностью околощитовидных желез. Биоло-
гич. активность П. определяется в единицах действия.
В зарубежной литературе 1 единица соответствует
0,01 количества препарата, вызывающего повышение
содержания кальция в сыворотке крови собак на
5 мг%. В советской фармакопее за единицу прини-
мается 0,01 количества препарата, вызывающего
повышение содержания кальция в сыворотке крови
собак на 30%. П. применяют для лечения заболе-
ваний, связанных с пониженной функцией около-
щитовидных желез.
Лит..- Камерон А. Т., Достижения современной эн-
докринологии, пер. с англ., М., 1948; Руководство по клини-
ческой эндокринологии, под ред. Е. А. Васюковой, М., 1958;
Hani О., Hormone, 2 Aufl., Jena, 1959; Bersin Th.,
Biochemle der Hormone, B., 1959. И. A. Sckuh.
ПАРАФИН — смесь твердых высокомолекулярных
углеводородов предельного характера, нормального
и изостроения, с незначительной примесью циклич.
углеводородов, получаемая гл. обр. из нефти, озоке-
рита, а также синтетически — восстановлением СО
водородом. Углеводороды, входящие в состав П.,
делят на парафины и церезины.
К парафинам, характеризующимся пластинчатой
или ленточной структурой кристаллов, относятся
смеси твердых углеводородов, являющиеся малораз-
ветвленными гомологами метана с т. пл. 50—70°,
состава С18Н40—С35Н72 и мол. в. 300—500; выкипают
в интервале 40—500°. Химически парафины очень
устойчивы.
Церезины обладают мелкокристаллич. строением,
т. пл. 65—88°, мол. в. 500—700, что соответствует
составу С37Н7в—C53HI0s, бблыпими по сравнению
с парафинами плотностью, показателем преломления,
вязкостью и более высокой реакционной способно-
стью. Церезины являются изопарафинами с беспо-
рядочно расположенными боковыми цепями, содер-
жащими в среднем 3 атома углерода каждая.
Очищенный П.— бесцветный продукт, без запаха
и вкуса, жирный на ощупь, нерастворим в воде и
спирте, хорошо растворим в большинстве органич.
растворителей и минеральных маслах; при нагре-
вании растворим во многих растительных маслах.
Плотность твердого П. при 15° в зависимости от его
чистоты колеблется от 0,881—0,905 (неочищенный
П.) до 0,907—0,915 (очищенный П.). Вследствие
неоднородности состава П. темп-ры начала и конца
его плавления могут различаться на 10—12°. Плохо
очищенный П. имеет желтый или бурый цвет и тем-
неет на свету.
Химич, свойства П. соответствуют характеру вхо-
дящих в его состав метановых углеводородов. При
обычной темп-ре П. очень устойчив к действию кис-
лот и оснований, щелочных металлов, окислителей
и галогенов. При нагревании П. вступает во взаимо-
действие с галогенами, при окислении, напр., HNO2
образуются карбоновые к-ты, при действии кисло-
рода — высшие жирные к-ты, спирты и другие кис-
лородсодержащие продукты.
П. получают в основном (— 90%) из парафинистых
нефтей (парафинистый дистиллят, фракция, кипящая
при 300—500°). Наиболее выгодны для переработки
т. наз. высокопарафинистые нефти с темп-рой за-
стывания парафинистой фракции 21° и выше и с
содержанием П. выше 2%. Из нефтей, добываемых
в СССР, наибольшим содержанием П. (до 14%)
отличаются нефти Западной Украины (бориславская,
859
ПАРАФИНЫ—ПАРАХОР
860
долинская), а также грозненские парафинистые (до
9%), туймазинская и мухановская нефти (до 7%).
Парафинистый дистиллят из нефти получают на
атмосферно-вакуумных трубчатых установках, после
чего дистиллят перерабатывают обычно след, мето-
дами: а) фильтрпрессованием с последующим «поте-
нием»; б) депарафинизацией с избирательными рас-
творителями. Фильтрпрессование слагается из кри-
сталлизации, фильтрования и «потения». Кристал-
лизацию проводят в кристаллизаторах различного
типа, предварительно охлаждая дистиллят и доби-
ваясь наиболее полного отделения твердого П. от
масла и получения его в крупнокристаллич. форме.
Выкристаллизовавшийся П. в смеси с маслом фильт-
руют в охлажденном состоянии для отделения от
масла на фильтрпрессах под давлением до 25 ат,
или же на барабанных вакуум-фильтрах. Получа-
емая при фильтровании лепешка твердого П. содер-
жит до 30% масла и наз. парафиновым гачем.
Для отделения от масла гач подвергают «потению»,
выдерживая его в специальных камерах при опре-
деленном температурном режиме. При этом масло,
заключенное между кристаллами П., вытекает. Очист-
ку полученного сырца производят 102—103%-ным
олеумом при темп-ре ~ 100° с последующей нейтра-
лизацией щелочью и очисткой отбеливающими гли-
нами.
Депарафинизацию производят с помощью селек-
тивных растворителей, при этом получают не только
П., но и низкозастывающие смазочные масла. Сырьем
служит масляный парафинистый дистиллят с т. кип.
350—500°, смешиваемый при 70—90° в отношении
1:3 с селективным растворителем (30% ацетона,
40% бензола, 30% толуола). Смесь последовательно
проходит через ряд кристаллизаторов с прогрессивно
понижающейся темп-рой. Выкристаллизовавшееся сы-
рье поступает на вакуум-фильтр, где отделяется
частично обезмасленный П., к-рый затем смешивают
с охлажденным до +5° растворителем для отмывки
от масла и вновь фильтруют. Получаемый парафин-
сырец очищают перколляцией (см. Очист-
ка нефтепродуктов).
Нек-рые сорта низкоплавких П., состоящих из
и-парафинов, получают при карбамидной депарафи-
низации парафинистых газойлей, однако основное
назначение этого процесса — получение низкозасты-
вающих дизельных топлив. Синтетический П. полу-
чают восстановлением СО водородом при 160—300°
и давлении 10—20 ат над железным или кобальт-
ториевым катализатором. Получаемый П. не нуждает-
ся в дополнительной очистке.
Выпускаемые пром-стью нефтяные П. подразде-
ляются на технич. высокоочищенные, марок А и Б,
медицинский, технич. очищенные, марок Г и Д и
неочищенный (спичечный). Важнейшими характе-
ристиками П. являются: темп-ра плавления — не
ниже 50—54° (спичечный не ниже 42°) и содержание
масла — не более 0,6—2,3% (спичечный 5%).
П. применяют в бумажной, текстильной, полигра-
фической, кожевенной и лакокрасочной промышлен-
ности, в электротехнике, для медицинских целей
и др. Как химич. сырье П. используют для получения
высших жирных кислот и спиртов, моющих и поверх-
ностно-активных веществ, присадок к смазочным мас-
лам и т. д.
Лит.: Наметкин С. С., Химин нефти, [3 изд.], М.,
1955; Рудакова Н. Я.,Тимошина А. В., Череп-
н е в а Е. И., Производство парафина, М., 1960; Технические
нормы на нефтепродукты, под ред. Н. Г. Пучкова, М., 1957;
Нефтепродукты. Методы испытаний, М., 1961. В. В. Щекин.
ПАРАФИНЫ — см. Предельные углеводороды.
ПАРАФОРМАЛЬДЕГИД (параформ) — см. Фор-
мальдегид.
ПАРАФУКСИН — краситель группы трифенилме-
тановых, трудно растворимый в холодной воде,
лучше — в горячей. П. — соль слабого основания:
рК—7,57 (25°); максимумы поглощения 543,5 и 486,0
i— _,+ ммк. При восстановле-
нии П. превращается в
4,4',4"-триаминотрифе-
нилметан. В П. благо-
даря сопряжению с цен-
тральным углеродным
с атомом основность ами-
ногрупп сильно подав-
лена, а кислотность, в
NH2
свою очередь, увеличе-
на. При добавлении ще-
лочи к раствору П. мгновенно образуется иминоосно-
вание, получившее название основания Гомолки. За-
тем, постепенно, в результате «медленной нейтрали-
зации» раствор обесцвечивается и равновесие сдвигает-
ся в сторону образования карбинольного основания.
Последнее при обработке кислотами превращается в
краситель.
Получают П. нагреванием п-диаминодифенилме-
тана с анилином, солянокислым анилином, нитробен-
золом в присутствии хлорного железа. П. окрашивает
непосредственно шерсть, шелк и кожу, а также
хлопок, подготовленный по прописям основного
крашения. П.— один из первых синтетич. красителей
(Я. Натансон, 1855).
Лит.: Коган И. М., Химин красителей, 3 изд., М.,
1956, с. 275; Венкатараман К., Химин синтетических
красителей, пер. с англ., т. 2, Л., 1957, с. 819.
О. Ф. Гинзбург.
ПАРАХОР — эмпирическая функция, выражаю-
щая связь между мол. весом, плотностью и поверх-
ностным натяжением неассоциированной индивиду-
альной жидкости:
р — м . у
D-d
(1)
где Р — парахор; М — молекулярный вес; Dud —
плотности жидкости и ее насыщенного пара соот-
ветственно в г/см3 при темп-ре измерения; у — по-
верхностное натяжение жидкости на границе с ее
насыщенным паром при той же темп-ре, дин/см.
П. практически не зависит от темп-ры вплоть до
темп-р, на 30—40° ниже критической.
Происхождение П. связано с наблюдением, что для
неассоциированных жидкостей (углеводородов, эфи-
ров и т. д.) зависимость у от (D—d) в логарифмич.
координатах представляет семейство параллельных
прямых, отвечающих ур-нию 2:
1g у = А + n lg (Д — d) (2)
Для веществ, в заметной степени ассоциированных,
напр. для воды, эта зависимость представляется
уже кривыми линиями. Среднее из различных из-
мерений значение коэфф, пропорциональности п
прямых в ур-нии (2) равно 4. Величина А характерна
для каждого вещества и не зависит от темп-ры. По-
тенцирование ур-ния (2) приводит к выражению
закона Мак-Леода (1923):
у = 10^(Д — d)n — C(D — d)1 (3)
где С — константа, зависящая от природы жидкости.
Появление четвертой степени плотности в формуле
(3) связано, по Мак-Леоду, с тем, что обусловли-
вающее поверхностное натяжение когезионное взаи-
модействие между молекулами зависит от четвертой
степени расстояния между ними.
Сегден (1924) ввел в выражение закона Мак-Леода
множитель М (мол. вес), сформулировав тем самым
функцию Р, к-рая связывает свойство совокупности
861
ПАРАХОР
862
молекул с особенностями отдельных молекул. При
темп-рах измерения, обычно достаточно далеких от
критической, можно пренебречь d по сравнению с D.
Тогда для определения П. пользуются выражением
(4) и учитывают d только при более точных измере-
ниях:
Р=^у’/4 (4)
Для расчета d при невысоких темп-рах пользуются
ур-нием состояния идеального газа или значениями
d, взятыми из литературы. При повышенных темп-рах
d рассчитывают по ур-нию (5):
1g (5)
\ кип. J \ кип. /
0122М/Ткяп.
где d — плотность пара жидкости при Т’°К, в г/с.и3;
<7КИП,— плотность пара при Ткип °К. Множитель M/D
в формуле (4) имеет размерность [слг3] и представляет
собой молярный объем, т. е. объем, занимаемый
одним молем вещества при темп-ре измерения. По-
этому отмечалось, что сравнение П. двух веществ оз-
начает сравнение их молярных объемов при темп-рах,
при к-рых у!=у2. Другими словами, П. численно
равен молярному объему при у=1. По Сегдену,
выполняется постоянное отношение П. к критич.
объему жидкости: Р=0,78Гкрит.. Такое сравнение
является слишком приближенным, т. к. сравнивать
молярные объемы можно только в соответственных
состояниях, т. е. при 7’=0°К, Ткип, или Ткрит.. Срав-
нение же при произвольных темп-рах является
физически неопределенным. Таким образом, функ-
ция П. не имеет строгого физич. смысла, т. к. неясно,
что ею измеряют.
Тем не менее П. сыграл положительную роль
при решении ряда вопросов строения. Это объясняет-
ся тем, что П. в общих чертах аддитивен. Это оз-
начает, что значение П. целой молекулы можно
представить в виде суммы констант, относящихся к
структурным элементам. Такое отнесение можно
выполнить, напр., по атомам:
Р = хс -фyh -фzo -ф wn -ф ...
по группам:
Р=х (ch,) +y(ch,) + z(ch) + w(oh) + . . .
или по связям:
Р =х (с—с) -фу (с — h) -фг (о — п) -ф ...
где х, у, г....— число атомов, групп или связей соот-
ветственно указанного типа (в скобках); с, h, о, п—
значения П., отнесенные к атомам С, Н, О, N и т. д.
Полученные константы, напр. «атомные П.», не
имеют ничего общего с объемом атома и являются
чисто расчетными величинами, лишенными физич.
смысла. Отклонения от аддитивности, обусловленные
наличием в молекуле кратных связей, циклов и т. д.,
учитываются введением соответствующих инкремен-
тов. В этом случае разложение П. по Сегдену при-
нимает вид (6):
Р = хсуh-)-zown21 (6)
Где 21 — сумма инкрементов.
Кроме ур-ния 6, известно несколько отличное раз-
ложение П. по Зиппелю и разложение по группам
(Гибблинг, 1943). Ниже приведены значения атомных
и структурных П. по Сегдену (в скобках) и исправ-
ленные позже Мемфордом и Филлипсом на основании
более тщательных расчетов.
Н 15,4 (17,1) N 17,5 (12,5) В 21,5 Сложные эфи- ры — (—3,2)
С 9,2 (4,8) Р 40,5 (37,7) Al 55,0 Одноэлект- роннан связь (И.6)
СН2 40 As 54 Tl 62,0 Трехчленный цикл + 12,5 (16,7)
S1 31 Sb 68 Hg 69 Четырехчлен- ный цикл + 6 (+11,6)
Sn 64,5 Bl 80 Be 42 Пнтичлен- ный цикл + 3 (8,5)
F 25,5 (25,7) О 20,0 (20) Простая связь — 9,5(—) Шестичлен- ный цикл + 0,8 (6,1)
Cl 55 (54,3) S 50 (48,2) Двойная связь + 19 (23,2) Семичлен- ный цикл -4 (-)
Вг 69 (68) Se 63 Тройная связь + 38 (46,6)
J 90 (91) Сг 58,0 Семиполяр- ная связь ±0(1,6)
Обращают на себя внимание нек-рые особенности
П. и его отличия от рефракции молекулярной, к-рые
позволяют рассматривать эти методы как взаимодо-
полняющие. Эти различия следующие:
1. П. имеет только один инкремент двойной связи,
не зависимо от вида атомов, ею связанных. Так, П.
связей С=С, С=О, C=N, N=O — одинаков. То же
относится и к инкременту тройной связи, который
одинаков для связей С=С, С Л' и т. д.; 2. Сопряжение
связей не влияет на П.; 3. Инкремент тройной связи
вдвое больше инкремента двойной связи; 4. Семипо-
лярные связи не вносят никакого инкремента, что
позволяет легко отличить эти связи от двойных.
Так, при расчете П. нитросоединений необходимо
вводить только один инкремент двойной связи, а
при расчете П. окисей фосфинов ни одного, что соот-
ветствует изображенным структурам I и II этих
соединений:
О
R-N^ (I) R3P—>-0(11)
Однако выводы о более тонком различии в электрон-
ном строении на основании величины П. следует
делать с большой осторожностью, т. к. зависимость
П. от электронного строения слишком сложная и
неявная. Попытки связать П. с электронным строе-
нием делались неоднократно. Отмечалось, напр.,
что можно рассматривать П. аминов и оксисоедине-
ний, если допустить, что эффективный объем атома
водорода уменьшается с увеличением электроотрица-
тельности атома, с к-рым он связан, и т. д. К подоб-
ным же попыткам относится и введение Копли (1941)
изостерических П. При этом считалось, что атомы
в одинаковом изостерич. состоянии содержат одина-
ковое число и тип заполненных электронных обо-
лочек, свободных пар и связывающих электронов.
Далее предполагалось, что изменения в изостерич.
состоянии, напр. замена —С— на —N:, —N: на
I III
—О: и т. д., сопровождаются равным изменением П.
Однако эти попытки не привели к заметному успеху.
Возможность использования П. для предсказания
поверхностного натяжения смесей показана Катцем
(1943) для трех бинарных смесей (CHi+CaHj, Na-ф
-f-CaHj, Na+H—С8Н12) при темп-рах до 90° и давле-
863
ПАРОМОМИЦИН—ПАРЦИАЛЬНЫХ ВАЛЕНТНОСТЕЙ ТЕОРИЯ
864
НИЯХ
Y
до 100 ат, при этом выполняется соотношение
D d \ .
м м j "i" (
, жидк. пар J
D d \
М х* М Уг
жидк. пар )
где х — молярная доля компонента в жидкой фазе,
у — молярная доля компонента в газовой фазе,
[Р] — «эффективные» П.
Большой вклад для развития метода П. в органич.
химии сделан работами Б. А. Арбузова.
Лит.: Хюккель В., Теоретические основы органиче-
ской химии, пер. с нем., т. 2, М., 1958, с. 221; Оно С., К о н-
д о С., Молекулярная теория поверхностного натяжения
в жидкостях, пер. с англ., М., 1963, с. 81; Физические методы
в органической химии, под ред. А. Вайсбергера, пер. с англ.,
т. 1, М., 1950. Ю. Д. Корешков.
ПАРОМОМИЦИН (паромомицин I, гуматин, ан-
тибиотик С-1488, катенулин, оксимицин, аминоси-
дин) — антибиотик группы стрептомицинов. Ему изо-
мерен паромомицин II.
П.— аморфное основание с т. разл. >200°, [а]”=
=+64° (С 1, вода), устойчивое при обычной темп-ре
в широком интервале pH (1,5—10) и растворимое в
воде и метаноле; описаны его сульфат с |а]р=+50,5°
(С 1,5, вода); хлоргидрат с [а]р=+56,5° (С 1, вода),
растворим в спирте, на холоду лучше, чем при на-
гревании; N-пентаацетат, [а]р=+64о (С 1,5, вода)
и ряд других производных.
Молекулы П. I и П. II состоят из углеводных ос-
татков, соединенных с полностью трансоидным 4,5,
6-триокси-1,3-диаминоцикл огексаном (2-дезоксистрепт-
амином). П. I представляет собой 4-О-(ц-В-2'-амино-
-2-дезоксиглюкопиранозил)-5-О-(P-L-2", 6"-диамино-2",
6"-дидезоксиидопиранозидо-1", 3"'-0- D-рибофурано •
зил)-2'-дезоксистрептамин; П. II отличается конфигу-
рацией при Cs — асимметрия, центре, т. е. вместо
0-Ь-диаминоидозы — остаток a-D-диаминоглюкозы.
Невыясненной остается абсолютная конфигурация
остатка 2-дезоксистрептамина в П. I и П. II (хотя,
как таковой, этот фрагмент симметричен, замещение
гидроксилов в положениях 4 или 6 приводит к оп-
тич. антиподам).
П. I образуется (наряду с П. II) при ферментации
Streptomyces rimosus forma paromomycinus, S. pau-
cisporogenes и ряда других актиномицетов. П. высоко
активен против различных грамположительных, грам-
отрицательных и кислотоустойчивых бактерий, а
также нек-рых простейших, патогенных грибов и
актиномицетов. Наиболее интересна высокая ак-
тивность П. против Endamoeba histolytica, превы-
шающая активность большинства известных аме-
бицидных препаратов. Обладая невысокой токсич-
ностью, П. является эффективным средством в те-
рапии ряда заболеваний, в особенности различных
амебиазов, а также бациллярной дизентерии, местных
поверхностных инфекций и др.
Лит.: Шемякин М. М. [и др.], Химия антибиотиков,
т. 1, 3 изд., М., 1961, с. 750; Rinehart К. L. [а. о.],
J. Amer. Chem. Soc., 1962, 84, № 16, 3216, 3218; Schillings
R. T., Schaffner С. P., в кн.; Abstracts of the 1-st Inter-
science conference on antimicrobial agents and chemotherapy,
New York, oct. 31—nov. 2, 1961, N. Y., 1961, p. 274.
Ю. А. Берлин.
ПАРООБРАЗОВАНИЕ — см. Испарение.
ПАРЦИАЛЬНОЕ ДАВЛЕНИЕ — давление, ока-
зываемое ударами молекул рассматриваемого компо-
нента i газовой смеси. Общее давление смеси Р равно
сумме парциальных давлений всех ее компонентов:
P=Sp. П. д. i-ro компонента pi= Р, где п;— число
молей этого. компонента, ап — общее число молей
смеси. Для идеальных газов П. д. данного компо-
нента газовой смеси равно давлению, к-рым обладал
бы этот компонент, если бы он один при той же темп-ре
занимал весь объем, занимаемый смесью (см. Даль-
тона законы).
ПАРЦИАЛЬНЫХ ВАЛЕНТНОСТЕЙ ТЕОРИЯ —
гипотеза, предложенная в 1899 И. Тиле для объяс-
нения особенностей реакционной способности сопря-
женных и ароматич. соединений. П. в. т.'основана на
представлениях о химич. сродстве, согласно к-рым
образование химич. связи между атомами сопровож-
дается расходом сродства и пропорциональным этому
расходу выделением энергии. На базе более ранних
исследований, показавших, что теплота образова-
ния двойной связи меньше, чем удвоенная теплота
образования простой связи, Тиле предположил, что
при образовании кратных связей на создание каждой
последующей связи расходуется меньше сродства,
чем на создание предыдущей. Отсюда следовало, что
в этилене каждый из атомов углерода израсходовал
на образование двух простых и одной двойной связей
не весь запас своего химич. сродства и обладает
еще нек-рой величиной остаточного сродства, парци-
альной (остаточной) валентностью. Наличием оста-
точных валентностей и обусловлена, по Тиле, способ-
ность этилена к реакциям присоединения:
сн2... Вг СН2Вг
и + I —* I
СН2... Вг СН2Вг
Приложение этих представлений к бутадиену-1,3
позволило Тиле хорошо объяснить многочисленные
факты 1,4-присоединения, до тех пор не находившие
удовлетворительного истолкования.
Написав формулу бутадиена-1,3 с остаточными валентно-
стями (а), логично было предположить, что остаточные валент-
ности атомов 2 и 3, если не полностью, то в значительной мере
взаимно насыщаются, тогда как у атомов 1 и 4 избыток срод-
ства остается свободным (б). Понятно, что присоединение
реагентов, напр. Вг2, происходит в положение 1—4. При этом
предполагается, что вначале атомы брома связываются непроч-
но, только за счет сил остаточной валентности, а потом проис-
ходит перераспределение сродства и образование! ,4-дибромида:
Вг2
СН2=СН—СН=СН2 СН2 = СН—СН=СН2—►
а ' б
—► СН2ВгСН = СНСН2Вг
Рассмотрение формулы б показывает, что с точки зрения Тиле
сопряженное соединение является более насыщенным, чем
аналогичное соединение с несопряженными двойными связя-
ми. В дальнейшем это было подтверждено экспериментально,
т. к. было найдено, что вещества с сопряженными связями
обычно имеют меньшие теплоты сгорания.
Рассматривая ароматич. соединения как замкнутые
сопряженные системы, Тиле предложил для бензола
ф-лу с равномерным распределением связей (в).
865
ПАСК —ПАССИВНОСТЬ МЕТАЛЛОВ
866
Ф ормула Тиле объясняет симметрию бензола, изо-
мерию производных бензола, прочность бензольного
в
ядра и малую склон-
ность его к реакциям
присоединения. Из этой
ф-лы для нафталина сле-
дует активность а-поло-
жений нафталинового
цикла в реакциях замещения: остаточные валентности
атомов углерода в положениях 9 и 10 распределяются
каждая между двумя соседними а-атомами углерода,
и какая-то часть собственной парциальной валентно-
сти а-атома углерода остается свободной.
Механизм П. р. окончательно не установлен. На
основании кинетич. данных предложены две схемы
реакции (А и Б):
Rx
R~NC + С=О
z
R'
+ zR
R"N=C---С
н-6^сГо-К
о' V"
+ / + Л
r"n=c-c -*—R"N=C-C
i-R' oV
ZR A
R"N=C~C
। । zR
HO OCOR — R"NHCO-C
|V
OCOR"'
О HO R
—- r"-c^ V —
O-Cx R'
%NR"
Из формул Тиле вытекало существование связей
нецелочисленной кратности (связь между атомами
2 и 3 в бутадиене-1,3, ароматич. углерод —углерод-
ная связь). Слабым местом гипотезы Тиле была ее
неспособность объяснить присоединение к сопряжен-
ным системам в положение 1—2.
Наиболее плодотворной частью в И. в. т. является
сама идея о делимости валентности, т. е. то, что
соответствует современным представлениям о делока-
лизации валентных электронов, лежащим в основе
современных концепций реакционной способности
органич. соединений — резонанса, мезомерии, ре-
акционного центра переноса, см. также Ароматиче-
ские системы, Сопряжение.
Лат.: Мюллер Е., Новые воззрения в органической
химии, пер. С нем., М., 1960; Р е м и к А., Электронные пред-
ставления в органической химии, пер. с англ., М.,1950, с. 34.
Б: Л. Дяткин.
ПАСК (n-аминосалициловая кислота, 4-амино-2-
оксибензойная кислота) C,H7OsN, мол. в. 153,13—
МНг мелкие бесцветные кристаллы; т. пл. 146—
j. 147° (с разл.); растворима в большинстве
[ff органич. растворителей, трудно — вводе.
П. легко декарбоксилируется при нагре-
Д вании в разб. к-тах, устойчива при нагрева-
СООН нии в щелочных р-рах. При диазотировании
П. в соляной к-те и последующем разложении обра-
зовавшейся диазониевой соли получают 4-хлорсалици-
ловую к-ту. При вливании диазотированного П. в
щелочной р-р р-нафтола раствор окрашивается в
вишнево-красный цвет. Получают П. карбоксилиро-
ванием .и-аминофенола СОа под давлением в при-
сутствии КНСОз и др. щелочных агентов, а также
восстановлением 4-нитро-2-оксибензойной к-ты. В ме-
дицине под термином ПАСК подразумевают легко
растворимый в воде дигидрат Na-соли 4-амино-2-окси-
бензойной к-ты, обладающий выраженной бактерио-
статич. активностью в отношении микобактерий
туберкулеза; применяют для лечения туберкулеза.
Лит.: М а й о ф и с Л. С., Технология химико-фармацев-
тических препаратов. Л., 1958, с. 210. Р. Г. Глушков.
ПАССЕРИНИ РЕАКЦИЯ —• конденсация арилизо-
цианатов с карбонильными соединениями и карбоно-
выми к-тами, приводящая к анилидам а-ацилоксикар-
боновых к-т:
R
/С = О + R"NC + R'"COOH
R'
R
R
^C-CONHR"
Z I
OCOR'"
П. p. протекает в очень мягких условиях — смеше-
нием реагентов при —20° или при комнатной темп-ре
в инертных растворителях. Выходы а-ацилоксианили-
дов, как правило, близки к количественным. П. р.при-
менима к алифатич., ароматич. и циклич. карбониль-
ным соединениям, но не к а, fi-непредельным кетонам.
R ZR
—» R"N=C-Cx »- R"NHCO-C
I |XR' IV
HO OCOR'" OCOR"'
Б
В согласии, с обеими схемами, зависимость скорости
П. р. от строения альдегидов и кетонов почти такая
же, как при циангидринном синтезе; добавка соли
карбоновой к-ты не увеличивает скорости реакции.
В последние годы разрабатываются синтезы на
основе изонитрилов, аналогичные П. р., с вовлече-
нием все новых реагентов, напр.:
R'
RNH2 + ^C = O + R'"NC + HOCOR""—>-
R"
•—»-R'"'CONHC-CONHR"'
I z\
RR' R"
R
CeH,CH2NH2 + HCOCH(CH3)2 + C = NCH^ +
COOR'
+ HOCOCH3NHCOCH3 —>.
CH(CH3)2
—r CH3CONHCH2CONHCHCONHCHCOOR'
СНДД R
П. p. открыта M. Пассерини в 1921.
Лит.: Ugl D. D. I., Angew. Chem., 1962, 74, № 1, 9;
Baker R. H., S t a n о n 1 s D., J. Amer. Chem. Soc., 1951,
73, № 2, 699. H. П. Гамбарян.
ПАССИВНОСТЬ МЕТАЛЛОВ — состояние повы-
шенной коррозионней стойкости металлов в усло-
виях, когда с термодинамич. точки зрения они до-
статочно реакционноспособны. Пассивирование свя-
зано с образованием на поверхности металла адсорб-
ционных или фазовых защитных слоев, вызывающих
торможение процесса растворения. Свойством пас-
сивироваться обладают многие металлы: Fe, Сг, Ni,
Со, Cd, Zn, Мо, Mg, Си, Sn, Pb, Al, Ti, Ta, Nb, Zr,
Ag, Pd, Pt, Au и др.
Характерными признаками пассивации, т. е. пере-
хода металла из активного в пассивное состояние
являются резкое снижение скорости его растворения
и заметный сдвиг потенциала металла в положитель-
ном направлении. Пассивация активного металла
может быть вызвана либо пропусканием через него
анодного тока, либо введением в агрессивную среду
нек-рых химич. агентов, напр. окислителей. При
этом пассивность наступает только в тех случаях,
если плотность анодного тока или концентрация до-
бавки превосходит нек-рую критич. величину. После
наступления пассивности возможно снижение плот-
ности анодного тока или концентрации пассивиру-
ем 28 ц.х.э. т. 3
867
ПАССИВНОСТЬ МЕТАЛЛОВ
868
ющей добавки до значений ниже критических без
нарушения пассивного состояния. Активация пас-
сивного металла может наступить под действием
нек-рых агрессивных анионов, напр. галогенидов,
под действием катодной поляризации и при контакте
с более электроотрицательным металлом. Примером
проявления пассивности является резкое замедление
растворения железа при перенесении его из разб.
в конц. азотную к-ту.
В соответствии с представлениями электрохимич.
кинетики скорость растворения металла в агрессивной
среде, являющаяся функцией потенциала, в общем
случае должна возрастать при сдвиге потенциала в
положительном направлении. Пассивность характе-
ризуется отклонением от этой зависимости, выража-
ющимся в замедлении растворения металла при
смещении потенциала в положительном направлении.
На рис. 1 схематически представлена зависимость
скорости растворения металла i, выраженной в еди-
Рис. 1. Схематическая за-
висимость скорости раство-
рения металла от потен-
циала.
ницах плотности тока, от по-
тенциала Е. При потенциалах
отрицательнее £пас. происхо-
дит активное растворение ме-
талла: при сдвиге Е в положи-
тельном направлении i возра-
стает в соответствии с т. наз.
ур-нием Тафеля: E=a^-blgi
(а и Ъ — постоянные). В интер-
вале потенциалов от £’пас_ до
£акт. металл находится в пере-
ходном состоянии (от активно-
го к пассивному); при смеще-
нии потенциала в положитель-
ном направлении скорость ра-
створения металла в этой обла-
сти уменьшается. Интервал по-
тенциалов Еакт —Етр является областью существова-
ния металла в пассивном состоянии; скорость раство-
рения металла в этой области, как правило, мало зави-
сит (иногда вообще не зависит) от потенциала. Потен-
циал £’тр. характеризует нарушение пассивного состоя-
ния металла и переход его в транспассивное
состояние (иначе наз. состоянием п е-
репассивации); при Е>Етр. сдвиг потенциала
в положительном направлении вновь приводит к
росту скорости растворения.
Из теорий пассивности наибольшее развитие полу-
чили фазовая и адсорбционная теории. Фазовая
теория предполагает покрытие металлов сплошными
(обычно окисными или солевыми) пленками конечной
толщины и связывает растворение пассивного ме-
талла с химич. растворением самой пленки. При
этом обычно предполагается, что рост пленки про-
исходит по диффузионному механизму. Адсорбцион-
ная теория исходит из того, что причиной пассивности
является адсорбция компонентов электролита (напр.,
анионов или молекул воды, иногда в количестве
долей монослоя) на поверхности металла. В соот-
ветствии с адсорбционной теорией природа процесса
растворения пассивного металла остается электрохи-
мической, но в связи с изменением состояния поверх-
ности металла изменяется кинетика процесса, причем
состояние поверхности пассивного металла является
функцией потенциала. В зависимости от природы
металла пассивность может быть связана с образо-
ванием на его поверхности либо фазовых, либо ад-
сорбционных слоев и удовлетворительно объясняется
одной из двух указанных теорий. Возможны и такие
случаи, когда фазовые пористые пленки облегчают
наступление пассивности, к-рая сама по себе в этих
случаях может иметь адсорбционную природу. Яв-
ление транспассивности объясняется растворением
металла в виде ионов более высокой степени окис-
ления. Важнейшими характеристиками способности
металла пассивироваться и сохраняться в пассивном
состоянии являются потенциал пасси-
вации Е„ас. (рис. 1), критический ток
пассивации iKp, величина скорости раство-
рения металла в пассивном состоянии in, положение
и ширина области потенциалов пассивного состояния
металла (Еакт.—А’тр.), потенциал перепас-
с и в а ц и и £гр.. Важное значение имеет также
потенциал активации Аакт., часто наз.
Флад е-п отенциалом. Смысл Етъ заключа-
ется в том, что при Е<ЕЛК.[ пассивный металл акти-
вируется.
Кривые зависимости скорости растворения металла
от потенциала (рис. 1) являются осн®вными харак-
теристиками коррозионного поведения металла. Они
хорошо объясняют известные закономерности пас-
сивационных явлений, а также позволяют найти
условия, необходимые для пассивации металлов, и
предсказать их коррозионную стойкость в пассивном
состоянии. Очевидно, что условием пассивации ме-
талла является смещение его потенциала в область
значений Яакт—Етр-, а скорость растворения пассивно-
го металла определяется величиной in (рис. 1). В зави-
симости от способа смещения потенциала данного
металла в область потенциалов пассивного состояния
различают химическую пассивацию и
анодную защиту.
Если в отсутствие внешнего тока металл имеет
стационарный потенциал Е'ст- (рис. 1), то смещение
его в область потенциалов пассивного состояния
(до ) возможно, прежде всего, за счет повышения
скорости катодной деполяризующей реакции (сдвиг
катодной кривой 1 на рис. 1 до положения 2). Для
этой цели могут применяться пассиваторы типа
окислителей, ускоряющие катодный процесс за счет
повышения концентрации восстанавливаемой формы.
Введение в раствор окислителей лежит в основе
метода химич. пассивации. При анодной защите сме-
щение потенциала Аст. в область /?акт_—Етр. осущест-
вляется за счет пропускания через металл внешнего
анодного тока. Другими возможными путями сме-
щения потенциала Е'^. данного металла в область
потенциалов пассивного состояния является частич-
ное нанесение на его поверхность нек-рых других
металлов, снижающих перенапряжение катодного
деполяризующего процесса (напр., Pt, Au, Си и др.),
или контактирование с нек-рыми более электрополо-
жительными металлами.
Величины анодного тока или концентрации окис-
лителя, требуемые для перевода металла из актив-
ного в пассивное состояние, определяются значе-
ниями iKp ; чем выше £кр, тем при большем расходе
тока или окислителя удается занассивировать металл.
Расход тока или окислителя в стационарном режиме
защиты (для поддержания устойчивого пассивного
состояния металла) определяется величиной ia, к-рая,
как правило, значительно (часто в тысячи раз) ниже,
чем iKp..
Для подбора материалов, устойчивых в определен-
ной агрессивной среде, значительный интерес пред-
ставляет проблема выбора металлов с выгодными
в практич. отношении пассивационными характери-
стиками. В зависимости от природы металлов склон-
ность их к пассивации и коррозионная стойкость в
пассивном состоянии могут быть весьма различны
(см. таблицу). Так, критич. ток пассивации желез .
в серной к-те превышает соответствующую величину
для хрома более чем в 500 раз. Скорости растворе-
ния пассивных хрома и железа в той же кислоте раз-
личаются в 100 раз.
Одним из важных путей пассивации металлов и
повышения их стойкости в пассивном состоянии яв-
869
ПАССИВНОСТЬ МЕТАЛЛОВ —ПАУЛИ ПРИНЦИП
870
Пассивацпонные характеристики нек-рых .металлов
и сплавов для 1 н. H2SOt
Металл Е , 8* пас. г , а, см2 кр-’ 1 гп, а/см2
Железо + 0,460 17,0 7 • 1 0 — «
Хром —0,350 3,2 • 10 -2 6-10-»
Никель + 0,150 1 10“2 3-1 о-«
Сталь 1Х18Н12Т . . . —0,150 1 10 1 10-"
* Относительно нормального водородного электрода.
ляется легирование. Высокая коррозионная стойкссть
нержавеющих сталей, напр., основана па значи-
тельном улучшении пассивационных свойств железа
при введении в него хрома, никеля, молибдена и др.
легирующих элементов, обладающих большей склон-
ностью к пассивации. Для каждого сплава, как
правило, существует нек-рое оптимальное содержа-
ние пассивирующего компонента, повышение к-рого
существенно не улучшает пассивационных характе-
ристик сплава.
Необходимые для практич. использования зависи-
мости скорости растворения различных металлов и
сплавов от потенциала получают потенциостатич.
методом измерения, основанным на определении плот-
ностей анодного тока или скоростей растворения
при различных постоянных значениях потенциала до
достижения устойчивых значений i при каждом зна-
чении Е. Основные пассивационные характеристики
металлов и сплавов, как правило, зависят от их при-
роды, условий термообработки, pH раствора и темп-ры
и не зависят от наличия в растворе окислителей.
На реальных Е — Igi кривых, в отличие от приве-
денных на рис. 1, могут наблюдаться несколько по-
следовательных областей пассива-
Е,в(н.в.э.) ции и активации (рис. 2), в связи с
_______чем для практич. целейинтерес пред-
0 0-___°’ ставляет область потенциалов опти-
’ | мальвой запассивированности (АВ
+ 0,5- ВЧ_ на рис. 2). Иногда до начала транс-
пассивации, т. е. при Е<Етр., наб-
1,0' ' людается выделение ки-
+ 1,5- слорода, к-рое может
V также и сопровождать
+2’0^L-,—•—1——1—- -igitA/cM1) транспассивацию. Во
Рис. 2. Зависимость скорости ра- многих случаях транс-
створения стали Х22Т в 0,1 н. пассивация может во-
H2SO4ot потенциала. обще не иметь места. В
нек-рых р-рах, напр. га-
логенидов, возможно активирование пассивного метал-
ла при E<EtV' с возникновением питтинговой корро-
зии, в результате чего в таких растворах сокращается
область потенциалов пассивного состояния.
При выборе путей защиты металла, склонного к
транспассивации, всегда необходимо учитывать по-
ложение его стационарного потенциаиа на Е — Igi
кривой. В случае, если Ест<Епас, при анодной или
химич. пассивации недопустимо, чтобы величины
анодного тока или концентрации окислителя пре-
вышали нек-рый предел (во избежание возможного
сдвига потенциала металла в область потенциалов
транспассивного состояния). Если Ест.>Егр , для пас-
сивации металла можно применять катодную защиту.
Свойство металлов пассивироваться используется
для их защиты от коррозии (см. Коррозия металлов).
Лит.: Фрумкин А. Н. [и др.], Кинетика электродных
процессов, М., 1952; Томашов Н. Д., Теория коррозии и
защиты металлов, М., 1960; Эванс Ю. Р., Коррозия и окис-
ление металлов, пер.с англ., М., 1962;К олотыр кин Я.М.,
Проблемы физ. химии, 1958, вып. 1, 81; Новые проблемы сов-
ременной электрохимии, под ред. Дж. Бокриса, пер. с англ.,
М., 1962, с. 284; Коррозия металлов и сплавов. Сб. статей, М..
1963; Vetter К. J., Elektrochemische Kinetik, В. [и. а.],
1961; Труды IV совещания по электрохимии (1—6 окт. 1956 г.),
М., 1959; Bericht ilber das Internationale Kolloquium liber die
Passivitat der Metalle in Heiligenberg bet Darmstadt vom 2
bis 7 September 1957, Z. Elektrocliem., 1958, 62, № 6/7; К о ло-
ты p к и н Я. М., К п я ж е в а В. М., Хим. пром-сть, 1963,
•У 2; Е d е 1 е a n и С., J. Iron and Steel Inst., 1957, 185^
pt 4, 482. Г. M. Флорианович.
ПАСТЕРА ЭФФЕКТ — подавление реакций бро-
жения (у микроорганизмов) и гликолиза (у животных)
реакциями дыхания; открыто Л. Пастером. При изу-
чении брожения, обусловленного дрожжами, было
установлено, что образование спирта и ССЦ дрожжа-
ми, происходящее в отсутствие кислорода, прекра-
щается в аэробных условиях, при к-рых развиваются
процессы клеточного дыхания. Одновременно клет-
ки начинают экономнее расходовать питательный
материал. Впоследствии аналогичное явление было
обнаружено Мейергофом в клетках организмов жи-
вотных — подавление образования молочной к-ты,
наблюдающееся в ходе гликолиза в условиях покоя,
когда клетки снабжаются достаточным количеством;
кислорода. В клетках животного организма в состоя-
нии активной функции П. э. отсутствует: гликолиз
протекает наряду с дыханием.
Механизм П. э. до сих пор не установлен. Согласно
гипотезе Линена — Джонсона, общие для гликолиза
и дыхания кофакторы (неорганич. фосфат, аденозин-
фосфорные к-ты) имеют большее сродство к ферментам
реакций сопряженного фосфорилирования. В резуль-
тате этого кофакторы становятся недоступными для
ферментов гликолиза. Согласно «мембранной гипоте-
зе», П. э. объясняется изменениями проницаемости
мембран митохондрий в зависимости от уровня адено-
зинтрифосфата (АТФ) в клетке. При высоком уровне
АТФ в клетке мембраны митохондрий непроницаемы
и в гиалоплазму клетки пе поступают особые «уси-
ливающие факторы», без к-рых гликолиз не идет.
При низком уровне АТФ проницаемость мембран
митохондрий увеличивается, и в гиалоплазму посту-
пают «усиливающие факторы», стимулирующие про-
цессы гликолиза. Отсутствие П. э. при активной
функции клеток объясняется усиленными затратами
АТФ и снижением концентрации АТФ в работающей
клетке.
Лит.: Регуляция клеточного обмена, под ред. С. Я. Кап-
ланского, М., 1962; Нейфах С. А. [и др.], ДАН СССР,
1961,138, № 1, 227; Казакова Т. Б., там же, 1963, 152,
№ 2, 471. С. А. Нейфах.
ПАУЛИ ПРИНЦИП — фундаментальный принцип
квантовой механики, определяющий свойства систем;
тождественных частиц с полуцелым спином, иначе
называемый принципом исключения
(или принципом запрета). П. п. формулируется след,
образом: в каждом квантовом состоянии системы ча-
стиц с полуцелым спином может находиться не более
одной частицы.
Паули сформулировал свой принцип в январе 1925, т. е.
до создания квантовой механики (1925—26). Для объяснения
ряда закономерностей электронных спектров атомов и их свй-
зи с т. наз. аномальным эффектом Зеемана Паули постули-
ровал, что в атоме не может существовать двух или больше эк-
вивалентных электронов, для к-рых значения всех четырех
квантовых чисел в магнитном поле одинаковы. В то время по-
нятие спина еще не существовало, поэтому четвертое кванто-
вое число не описывалось у Паули никакой моделью. В даль-
нейшем выяснилось, что П. п. является следствием вытекаю-
щих из законов квантовой механики свойств симметрии вол-
новых функций тождественных частиц с полуцелым спином.
Связь значения спина и симметрии волновой функции была
теоретически обоснована Паули уже в рамках релятивистской
квантовой механики (1940).
Из принципиальной неразличимости одинаковых
частиц в квантовой механике следует, что квадрат
модуля волновой функции, дающий вероятность на-
хождения системы в определенном состоянии, не дол-
жен меняться при перестановках частиц. Отсюда для
волновой функции имеются две возможности: 1) вол-
новая функция системы симметрична относительно
перестановок одинаковых частиц; 2) волновая функ-
ция — антисимметрична. Первый тип симметрии осу-
ществляется для частиц с целым значением спина, вто-
28*
871
ПАУЛИ РЕАКЦИЯ —ПЕКИ
872
рой — для частиц с полуцелым спином.. Требование
антисимметричности волновой функции системы час-
тиц с полуцелым спином является наиболее общей
квантово-механич. формулировкой П. п. Связь П.п.
с условием антисимметричности волновой функции
была показана Дираком (1926) на системе А7 невзаи-
модействующих частиц. Антисимметричную волновую
функцию такой системы можно представить в виде
детерминанта:
Фр,^) Фл(^2) ••• ФрЛ.у)
Фр3(5>) фц2(^) ... фц2(1у)
ф(£„ ?2, ... ,
Фр (S1) Фр (£2). • - Фр.. &v)
г П г N г N
где фр^ (В/) — волновая функция i-той частицы, —
совокупность пространственных и спиновой коорди-
нат частицы, — номер квантового состояния (см.
Квантовая механика). Если какие-либо две строки де-
терминанта совпадают, то он тождественно обращает-
ся в нуль, отсюда следует, что все Pk должны быть
разные, т. е. не может быть двух частиц в одном и том
же состоянии.
Определение П. п., включающее понятие квантового
состояния частиц, справедливо в тех случаях, когда
взаимодействие между частицами можно заменить
нек-рым эффективным полем, вследствие чего каждую
частицу можно характеризовать индивидуальным на-
бором квантовых чисел, тогда как при строгом учете
взаимодействия должны существовать только состоя-
ния всей системы в целом. Такой способ рассмотрения
обычно дает хорошее приближение и является поэтому
основным в большинстве конкретных применений
квантовой механики (теория атомных и молекулярных
спектров, оболочечная модель ядра, квантовая теория
химич. связи и т. д.), чем и объясняется практич.
важность П. п. Этот принцип является определяющим
также и при изучении статистич. поведения систем
тождественных частиц с полуцелым спином (см. Кван-
товая статистика).
Применение П. п. к классификации состояний тож-
дественных частиц может быть наглядно проиллюст-
рировано на примере атомных термов. Электрон в
атоме характеризуется четырьмя квантовыми числами,
к-рые обозначаются п, I, mt, ms (см. Атом). Макси-
мальное число электронов с данным п определится,
согласно П. п., числом различных троек I, пц, ms,
п—1
т. е. У, 2(21+1) = 2п2. Отсюда сразу получаются
г = о
числа заполнения оболочек 2,8,18,32,.... в полном
согласии с периодической системой элементов Менде-
леева. Для т. наз. эквивалентных электронов, т. е.
электронов с одинаковыми п, I, осуществляются не
все возможные состояния, а лишь те, к-рые удовлет-
воряют П. п. Так, для конфигурации (пр)2 правило
векторного сложения дает 6 термов ’’’А, Ъ’Р, I,3D
(см. Атомные спектры), а разрешены только три *А,
2Р, 'D, т. к. остальные три терма соответствуют сов-
падающим наборам квантовых чисел для двух элект-
ронов.
Лит.: Ландау Л.,ЛифшицЕ., Квантовая механика,
ч. 1, М., 1964 (Теор. физика, т. 3); Г лес стон С.,
Теоретическая химия, пер. с англ., М., 1950; Ван-дер-
Варден Б. Л., в сб.: Теоретическая физика 20 века, М.,
1962, с. 231; Паули В., там же, с. 357; его же, Phys.
Rev., 1940, 58, 716. И. Г. Каплан.
ПАУЛИ РЕАКЦИЯ (диазореакция) — ка-
чественная цветная реакция на белок, зависящая от
наличия в молекуле белка тирозина и гистидина. При
добавлении к щелочному р-ру белка свежеприготов-
ленного р-ра диазобензолсульфокислоты появляется
вишнево-красная окраска. П. р. основана на образо-
данди азокрасителя в результате сочетания диазо-
бензолсульфокислоты с фенольным кольцом тирозина
или имидазольным кольцом гистидина:
^-р-сн2-^н-соон N.,CO,
nh2
N---J -CH2-CH-COOH
so2
O"
П. p. применяют для открытия белков и количе-
ственного колориметрич. определения гистидина и
тирозина; реакция не специфична (аналогичную реак-
цию дают вещества, имеющие в своей структуре аро-
матич^ или гетероциклич. кольца). Описана Паули
Лит.: Блок Р., Боллинг Д., Аминокислотный сос-
тав белков и пищевых продуктов, пер. с англ., М., 1959, с. 54,
ИЗ. И. В. Цветкова.
ПАХИКАРПИН [(+)-спартеин] ClsH2eN2, мол. в.
238,37—алкалоид, выделенный из листьев и стеблей
растения софоры толстоплодной 5 7 8 л
(Sofora pachycarpa) сем. мотыль-
ковых (Papilionaceae). Найден I СНг 7 , 1
также в других растениях. Оптич.
антиподом алкалоида является 2 18 18 °
(—)-спартеин, с к-рым П. имеет одинаковые физич.
и химич. свойства. П.—почти бесцветное густое мас-
ло; т. кнп. 3257754 мм, 188—190718 мм; da 1,034;
[а]д= + 5,4° (без растворителя), +16,3° (в спирте).
Двутретичное насыщенное основание, дает моно- и
дисоли; дипикрат, т. пл. 206—208°; моноиодгидрат,
т. пл. 235—236°; дииодгидрат, т. пл. 257—258°. Обра-
зует только моноиодметилат, к-рый является основа-
нием (дает иодгидрат — иодметилат). Циклич. скелет
П. (спартеина) лежит в основе структуры ряда алка-
лоидов: лупанила, афиллина, афиллидина и др. Про-
стейший синтез П. осуществлен по след, схеме:
НСНО(ш CH;J,)
--(i)-спартеин ► пахииарпин
Известны другие пути синтеза П. По фармаколо-
гия. свойствам П.— ганглионарный яд; от никотина
и анабазина отличается тем, что угнетает ганглии без
заметного предварительного возбуждения. В малых
дозах успокаивает центральную нервную систему, в
больших — вызывает возбуждение и судороги. На-
ряду со снижением возбудимости узлов вегетативной
нервной системы уменьшает реактивность мозгового
слоя надпочечника и каротидного клубочка. Повыша-
ет тонус и усиливает сокращение мускулатуры матки.
Иодгидрат П. применяют в медицине для лечения
облитерирующего эндартериита, мышечной дистро-
фии и поражения симпатич. ганглиев, а также в ка-
честве родоускоряющего средства.
Лит.: Преображенский Н. А., Генкин Э. И.,
Химия органических лекарственных веществ, М.— Л., 1953;
Орехов А. П., Химия алкалоидов, 2 изд., М., 1955.
,, А. Е. Васильев.
ПЕК ДРЕВЕСНЫЙ — см. Пеки.
ПЕК НЕФТЯНОЙ — см. Пеки.
ПЕКИ — остаточные продукты перегонки дегтей
(смол), образующихся при термич. переработке твер-
873
ПЕКИ —ПЕКТИНОВЫЕ ВЕЩЕСТВА
874
дых топлив (каменного и бурого углей, торфа, горю-
чих сланцев, древесины). К П. относят также остаток
перегонки нефтяной пиролизной смолы. П.— твердые,
иногда густые, вязкие, черные продукты с раковис-
тым изломом. П. нерастворимы в воде (за исключе-
нием древесного и торфяного П., из к-рых вода вы-
щелачивает водорастворимые соединения); раствори-
мы во многих органич. растворителях (остаются не-
растворенными минеральные примеси, свободный уг-
лерод, карбоиды и, в нек-рых растворителях, асфаль-
тены). Свойства П. зависят от природы дегтя и мето-
да и режима его перегонки. Отгоном от дегтя большего
или меньшего количества масел получают П. с задан-
ной темп-рой плавления, нужной твердостью и дук-
тильностью (растяжимостью). Качество П. зависит
также от конечной темп-ры процесса перегонки и
длительности пребывания П. при этой темп-ре. П.
получают на смолоперегонных установках (трубча-
тых, кубовых непрерывного действия и периодиче-
ских); наиболее термически стойкие П. с наиболее
высокой темп-рой плавления получают на установках
периодич. действия. При выходе из перегонных уст-
ройств П. имеет темп-ру 250—350°. Во избежание
воспламенения на воздухе П. охлаждают в пекотуши-
телях до 150—180°, после чего разливают в формы,
пековые ямы или на охлаждающие транспортеры.
П. делятся на мягкие (т. пл. 40—50°), средние (т.
пл. 65—70°) и твердые (т. пл. 85—100° и выше). Для
получения П. с т. пл. 140—150° их подвергают до-
полнительной перегонке или окислению в кубах не-
прерывного действия при 300—340°.
Наибольшее значение имеет каменноуголь-
ный П., получаемый из высокотемпературной (кок-
совой) смолы. Выход П. 55—65% от перерабатываемой
смолы, плотн. 1,2—1,3 г/см”. В состав кам.-уг. П.
входят гл. обр. высокомолекулярные ароматич. угле-
водороды, высшие фенолы и органич. основания.
Установлено присутствие флуорантрена, пирена и
хризена. Кислые и основные компоненты изучены
мало. Каменноугольный П. используется гл. обр.
для получения кокса, идущего на изготовление элек-
тродов (электродный кокс). Такой кокс должен со-
держать не более 1% золы, до 0,7% серы, до 1% лету-
чих. Коксование П. производится в камерных печах
периодич. действия типа коксовых, а также в подовых
печах или в установках непрерывного действия с
твердым теплоносителем.
Торфяной П. получают из дегтей, образую-
щихся при газификации или полукоксовании торфа.
Состав и свойства его колеблются в зависимости от
условий получения и переработки дегтя. В состав
торфяных П. входят асфальтены, карбоиды, воски,
парафины, высокомолекулярные фенолы и углеводо-
роды. Преимущественным направлением использова-
ния торфяного П. является произ-во на его основе
крепителя литейного марки «КТ», к-рый представляет
собой смесь торфяного П. с сульфитным щелоком и
формовочной глиной. Для приготовления толя и при-
менения в дорожном строительстве торфяной П. мало
пригоден, вследствие содержания водорастворимых
продуктов и наличия парафинов и воска.
Древесный П. получают при термич. перера-
ботке древесины. Свойства древесного П. зависят от
породы древесины, метода его термич. переработки,
глубины и длительности перегонки древесного дегтя
при получении П. и др. факторов. В состав древесных
П. входят смоляные к-ты, асфальтены, карбоиды,
оксикислоты, жирные к-ты, ангидриды оксикислот,
высокомолекулярные фенолы, нейтральные дегтевые
продукты и др. Способом повышения твердости дре-
весного П., кроме вторичной перегонки и окисления,
является обработка крепкой серной к-той и удаление
растворимых в воде сульфопрод}ктов. Основное ис-
пользование древесного П.— получение древеснопе-
кового трехкомпонентного крепителя (50% тонко-
размолотых древесных П., 25% сухой сульфитной
барды и 25% дисперсной глины). На основе древесно-
го П. может быть получено связующее для корковых
оболочек (60% П., 30% пульвербакелпта и 10% урот-
ропина). Для вышеуказанных применений пригоден
П. с темп-рой размягчения не ниже 80°.
П. получают также при дистилляции буроугольных
и сланцевых смол. Пути их использования аналогич-
ны описанному. П. сланцевый иногда используют
как топливо. К П. относят Также продукты, получае-
мые при очистке нефтяных масел от смолисто-асфаль-
товой части. Эти продукты состоят преим. из осме-
лившихся .асфальтоподобных высокомолекулярных
ароматич. и нафтеновых углеводородов и продуктов
их окисления, а также сернистых органич. соедине-
ний, полинафтеновых к-т и карбоидов.
П. используют как связующее при брикетировании
твердых топлив, как сырье для произ-ва кокса пеко-
вого, в дорожном строительстве, для изготовления
толя и руберойда, как составную часть литейных
формовочных материалов, для приготовления пеко-
вого лака, применяемого для окраски металлокон-
струкций и др.
Лит.: Белов К. А., Переработка химических продук-
тов коксования, Харьков — М., 1949; Кор ото в С. Я. [и
др.], Энергохимическое использование древесины, Л., 1958;
Крейцер Г. Д., Асфальты, битумы и пеки, 3 изд., М.,
1952; Химия и технология искусственного жидкого топлива и
газа, сб. статей, М.— Л., 1951 (Труды ВНИГИ, вьГп. 3);
Раковский В. Е., Общая химическая технология тор-
фа, М.— Л., 1949. А. М. Кунин.
ПЕКТАЗЫ — см. Пектиновые ферменты.
ПЕКТИНАЗЫ — см. Пектиновые ферменты.
ПЕКТИНОВЫЕ ВЕЩЕСТВА — высокомолекуляр-
ные углеводы растительного происхождения, главным
структурным компонентом к-рых является D-галак-
туроновая к-та; встречаются в тканях наземных рас-
тений и в нек-рых водорослях. В тканях нек-рых
растений содержание П. в. достигает 30% сухого
веса (напр., в белой части кожуры цитрусов), в дру-
гих— их содержание не превышает долей процента.
К П. в. относятся: пектовая кислота, к-рая
построена из остатков D-галактуроновой к-ты, связан-
ных а-1,4-гликозидными связями в длинные цепи (яв-
ляется основой всех П. в.); пек та ты —соли
пектовой к-ты; пектины — продукты различной
степени метилирования пектовой к-ты по карбо-
ксильным группам, растворяются в воде с образовани-
ем плотных гелей; пектинаты — соли пектинов;
протопектины — нерастворимые в воде веще-
ства высокого мол. веса, в к-рых линейные молекулы
пектинов связаны поперечными мостиками, и др. про-
изводные пектина.
В растениях П. в. присутствуют преим. в виде не-
растворимого протопектина, к-рый содержится гл.
обр. в стенках растительной кдетки, в межклеточном
цементирующем материале, играя роль опорных эле-
ментов тканей; в клеточном соке содержатся пектины
и их соли пектинаты. При извлечении пектинов из
растительного материала протопектин разрушают го-
рячей соляной к-той, и образующийся пектин осаж-
дают спиртом. Пектины различных растений харак-
Н ОН СООСНз н он
соон н он соон
Участок цепи макромолекулы пектина.
теризуются неодинаковыми мол. весом, степенью эте-
рификации карбоксильных групп, распределением
метильных групп в молекуле. Полностью метилиро-
875
ПЕКТИНОВЫЕ ФЕРМЕНТЫ —ПЕЛЛОТИН
876
ванная по карбоксилам пектовая к-та содержит ок.
14% метоксилов, однаио природные пектины содер-
жат и свободные карбоксильные группы. В зависи-
мости от степени метилирования пектины делятся на
и-пектины, в к-рых метилировано более 50% всех
карбоксильных групп, и L-пектины, содержащие мень-
ше 50% этерифицированных карбоксильных групп.
Пектины растворимы в воде и способны в присутствии
сахара и органич. к-т образовывать плотные гели.
К действию кислот пектины довольно стойки, под
действием щелочей они разрушаются; легко подвер-
гаются окислительному расщеплению.
Ферменты, действующие на пектины, делятся на
пектинэстеразы (старое название пектазы),
гидролизующие сложноэфирные группировки, от-
щепляя метильные группы, и полигалакту-
роназы (старое название пектиназы), расщепля-
ющие полигалактуронидную цепь до олигоуронидов
и далее до D-галактуроповой к-ты. Эти пектиновые
ферменты, различные представители к-рых встреча-
ются в микроорганизмах и растениях, используют
для расщепления П. в. при осветлении фруктовых
соков.
Определение пектинов производят измерением ко-
личества СОг, образующегося при нагревании пекти-
нов с горячей к-той; спектрофотометрически — по
окрашенному комплексу, получаемому при их взаи-
модействии с карбазолом, и др. способами. Для ха-
рактеристики промышленных препаратов большое
значение имеет определение вязкости пектинов и их
способности образовать гели.
Промышленные препараты пектинов получают из
кожуры цитрусов, яблок и кормового арбуза. П. в.
применяют в пищевой пром-сти (гл. обр. при получе-
нии мармелада и джемов), а также в фармацевтич.
пром-сти.
Лит.: Гапоненков Т. К., Проценко 3. И.,
Ботанический журнал, 1962, 47, № 10; Хенглейн Ф,,
в кн.: Биохимические методы анализа растений, пер. [с нем.],
М., 1960; Comprehensive biochemistry, ed. М. Florin, Е. Н.
Stotz, v. 5, N. Y.—L., 1963; Industrial gums, ed. R. L. Whist-
ler, N. Y.— L., 1959; Handbuch der Pflanzphyslologle, Bd 6,
B.— [u. a.], 1958, S. 405. Л. И. Линевич.
ПЕКТИНОВЫЕ ФЕРМЕНТЫ — ферменты, ка-
тализирующие гидролитич. расщепление пектиновых
веществ. Наиболее известны два представителя П. ф.:
пектинэстераза и полигалактуропаза, относящиеся
к классу гидролаз.
Пектинэстераза (систематич. наименова-
ние: пектил-гидролаза пектинов, шифр 3.1.1.11; си-
нонимы: пектаза, пектинметилэстераза, пектиндемето-
ксилаза) катализирует гидролитич. расщепление
сложноэфирных связей в пектинах и протопектинах
с освобождением карбоксильных групп и отщепле-
нием метанола (см. схему); высоко специфична: рас-
полигалантуроназа
пектинэстераза
Схема действия пектиновых ферментов.
щепляет только метиловые эфиры полигалактуроно-
вой к-ты и не действует на ее этиловые эфиры или
на метиловые эфиры моно-, ди- и тригалактуроновых
к-т. Активность пектинэстеразы можно определить по
количеству отщепляемого метанола или же титрова-
нием освобождающихся СООП-групн. Пектинэстера-за
широко распространена в растительных тканях, а
также в нек-рых грибах и бактериях. Роль этого
фермента, по-видимому, состоит в превращении водо-
нерастворимых протопектинов в водорастворимые
пектиновые к-ты в процессе созревания плодов.
Полигалактуроназа (систематич. наи-
менование: полигалактуронид-гликаногидролаза,
шифр 3.2.1.15; синонимы: пектиназа, пектолаза, пек-
тингликозидаза, пектиндеполимераза) гидролизует
гликозидные (а-1,4'-П-галактуронидные) связи в
пектиновых к-тах (пектатах) и др. полигалактурони-
дах (см. схему). Активность полигалактуроназы мож-
но определять по уменьшению оптич. плотности или
вязкости р-ров гидролизуемых пектиновых веществ
или по возрастанию количества образующихся в про-
цессе гидролиза СНО-групп. Фермент инактивируется
иод действием таннина и формальдегида. Полигалак-
туроназы, выделенные из различных источников, от-
личаются по своему действию на пектиновые вещества:
одни ферменты, получившие название эндополига-
лактуроназ, гидролизуют внутренние гликозидные
связи в молекуле полигалактуроновой к-ты, расщеп-
ляя ее на более или менее крупные фрагменты; другие
полигалактуроназы, получившие название экзополи-
галактуроназ, ступенчато расщепляют цепочки поли-
галактуроиовой к-ты, начиная с одного из ее концов.
Полигалактуроназа встречается в растительных
тканях, в частности в семенах, где ее роль, по-види-
мому, связана с процессами прорастания. Особенно
широко полигалактуроназа представлена у различ-
ных микроорганизмов, плесеней и грибов, являющих-
ся сапрофитами или паразитирующих на раститель-
ных тканях.
Препараты полигалактуроназы применяют для
просветления фруктовых соков, содержащих нераст-
воримые пектиновые вещества.
Лит.: Классификация и номенклатура ферментов, пер.
с англ., М.,1962; Advances in enzymology, v. 20, N. Y.— L.,
1958, p. 341; Ullman n, 3 Aufl., Bd 7, Munch.— B.,1956,
S. 413. В. Б. Спиричев.
ПЕКТИНЭСТЕРАЗА — см. Пектиновые ферменты.
ПЕЛАРГОНОВАЯ КИСЛОТА (нонановая кис-
лота) СНз(СН2),СООН, мол. в. 158,23; т. пл. 12,5°;
т. кип. 253—2547760 мм; 186°/100 мм; «^1,4343;
d™ 0,9057; константа диссоциации 217=1,11 • 10~s (25°).
П. к. обладает общими свойствами предельных али-
фатич. к-т и может быть получена всеми обычными
способами. П. к. встречается в природе в виде эфиров
в летучем масле герани, в сивушном масле кормовой
свеклы и картофеля, японском воске и др. Ее можно
получить также окислением олеиновой к-ты.
Этиловый эфир П. к. применяют как душистое
вещество; он имеет запах фруктового характера с
оттенком розы. В таблице приведены основные свой-
ства нек-рых простейших эфиров П. к.
Эфир Мол, вес Т. кип., оС}мм ,2 0 d4 пго ГГ>
Метиловый 172,26 213,8/760 0,8748 1,4216
Этиловый 186,17 227/760, 75.5/3.5 0,8657 1,4220
н-Пропиловый 200,19 120 — 1 22/20 0,8641 1,4236е
и-Бутиловый 214,21 122-124/2') 0.87205 1,4262е
н-А милов ый 228.23 130/20 0,8506° 1,4318
а d°. 6 d”. в Пр 4 4 1 и 25°. Н. А. Несмеянов.
ПЕЛЛОТИН (I)C1SH19NO„ мол. в. 236,49-Н-мети-
льное производное ангалонидина (II), изохинолино-
вый алкалоид; кристаллы, т. пл. 111—112°;кристал-
лизуется из спирта в виде прозрачных табличек.
Основание алкалоида (ОА) образует хлоргидрат, т. пл.
877
ПЕЛЬТЬЕРИН — ПЕНИЦИЛЛИНЫ
878
4 47—148°, пикрат, т. пл. 166—168°, иодметилат ОА-
•CHSJ'H2O, т. пл. 198— 199°. Свободная гидроксиль-
ная группа у П. и ангалонидина находится, как
I
H3CO'^Yx7/NH
он сн3
II
было установлено Шпетом, при С8; строение этих
алкалоидов было установлено и синтетически. П.
синтезирован, исходя из бензолового эфира 2-ок-
си-3,4-диметоксиацетофенона, к-рый действием ами-
ноацеталя превращают в шиффово основание. При
обработке последнего 73%-ной серной к-той, а затем
горячей водой получается 8-окси-6,7-диметокси-1-ме-
тилизохинолин, иодметилат к-рого при восстанов-
лении образует П.
П.—судорожный яд, вызывает замедление сердеч-
ных сокращений и падение кровяного давления;
содержится в кактусах Anhalunium Williamsii
(ок. 3,5%), произрастающих в горах Мексики.
Лит.: Генри Т. А., Химия растительных алкалоидов,
пер. с англ., М., 1956; Орехов А. П., Химия алкалои-
дов, 2 изд., М., 1955.
ПЕЛЬТЬЕРИН — описанный ранее как главный
алкалоид коры гранатового дерева (Punica grana-
tum L.), сем. гранатовых (Punicaceae), к-рому при-
писывалось строение р-(а'-пиперидил)-пропионового
альдегида. Более поздние исследования как алка-
лоидов, выделяемых из коры Punica granatum, так и
препаратов, приготовляемых из нее и употребляемых
в медицинской практике, привели к заключению,
что в них не содержится П. и что зти вещества состоят
гл. обр. из псевдопелътъерина, изопельтьерина (I)
и метилизопельтьерина (II).
С^~со-снг-сн3
NH
С^Хсо-сн,
СН3 (1
Препараты алкалоидов из коры гранатового дерева
(т. наз. «пельтьерин») применяют против ленточных
глистов, хотя они и весьма токсичны.
Лит. см. при ст. Псевдопельтьерин. А. Е. Васильев.
ПЕНИЦИЛЛИНАЗА — тривиальное наименова-
ние фермента, катализирующего гидролиз пеницил-
лина с образованием пенициллоиновой к-ты:
s А
RCONH-СН-СИ С(СН3)г НгО RCONH—СН—СН хС(СН3)г
ос—N—СНСООН НООС HN—снсоон
Систематич. наименование пеницидлин-амидогидро-
лаза (шифр 3.5.2.6) относится к классу гидролаз (см.
Номенклатура и классификация ферментов).
11.— простой белок, не содержащий каких-либо
простетич. групп. Для П. характерно очень низкое
содержание серусодержащих аминокислот. П. выраба-
тывается многими видами грамположительных и
грамотрицательных бактерий, в частности различными
штаммами стафилококков и бацилл. Способность
этих штаммов продуцировать II., инактивирующую
пенициллин, лежит в основе их устойчивости к дей-
ствию этого антибиотика. Нек-рые бактерии синтези-
руют П. в виде внутриклеточного фермента, другие —
выделяют ее во внешнюю культуральную среду. У
нек-рых штаммов микробов П. является конститутив-
ным ферментом, к-рый постоянно образуется клеткой
независимо от наличия или отсутствия в среде пени-
циллина. В то же время у др. штаммов П. образует-
ся индуктивно при добавлении в среду пенициллина.
Физико-химич. свойства кристаллич. П. несколько
варьируют в зависимости от источника их получения
(см. таблицу).
Фпапко-химические свойства различных П.
Источник фермента Мол. вес Оптимум pH Константа Михаэлиса (по бензил- пенициллину)
Bacillus cereus 5/в * . . . 35200 7,0 4,3210-5 М
Bacillus cereus 569 .... 31500 6,0—6,5 4,87-Ю-5 М
Bacillus cereus 569/Н . . . 30800 6,0—6,5 —
* Изоэлектрич. точка рГ 5,0.
П.— высокоспецифичный фермент, гидролизующий
как природные, так и синтетич. пенициллины с раз-
личными заместителями (В) в боковой цепи. С наи-
большей скоростью П. гидролизует феноксиметилпе-
нициллин (В = НОС8Н4СН2), несколько медленнее
бензил- и еще медленнее н-гептилпенициллин. Обя-
зательным условием ее активности является наличие
в молекуле субстрата неизменной, характерной для
пенициллина, бициклич. структуры, образованной
конденсированными тиазолидиновым и р-лактамным
кольцами. П. совершенно неактивна в отношении
аналогов пенициллина, у к-рых эта структура хотя
бы частично изменена, в частности в отношении дез-
тиобензилпенициллина или N-фенацетил-З-цикло-
-(L-цистеинил-П-валина).
Молекулярная активность кристаллич. П. из Ba-
cillus cereus 569 составляет 1,6 • 10s единиц при 30°
и pH 7,0; 1 мг П. способен разрушить за час 77 г
пенициллина. 2-Бензимидазол и бензилпенициллине •
вая к-та в концентрации 10~s М подавляют активность
П. соответственно на 74% и 26%. Гистамин, D-
и L-пеницилламин, 2-метилимидазол тормозят II.
в концентрации 10~4 М. Са2 + и Fe2 + в концентрации
10”3 М подавляют активность П. на 50%. п-Хлор-
ртуть-бензоат практически не влияет на активность
П. Активность П. не меняется также при диализе.
Энергия активации гидролиза бензилпенициллина П.
из Bacillus cereus 569 составляет 5,3 ±0,3 ккал/моль.
П. довольно стабильна в водных р-рах при комнат-
ной темп-ре в интервале pH 3,0—10,0. Нагревание
при 50° и pH 7,0 инактивирует П. в течение 30 мин.
Присутствие в р-рах высокополимерных соединений,
в частности желатины и др. белков, защищает П. от
инактивации. В их присутствии П. выдерживает на-
гревание при 100° в течение 20 мин без потери актив-
ности. Поскольку в ходе катализируемой П. реакции
на каждый моль превращенного субстрата образует-
ся 1 эквивалент кислоты (новая СООН-группа), то
активность П. определяют титриметрич. методом или
же манометрически — по количеству СОг, вытесняемой
из бикарбонатного буфера. Для определения активно-
сти П. предложен также иодометрич. метод, основан-
ный на способности пенициллоиновой к-ты присоеди-
нять J2.
Лит.: The enzymes, ed. Р. D. Boyer, H. Lardy, К. Муг-
back, v. 4, 2 cd., N. Y.— L., 1960. В. В. Спиричев.
ПЕНИЦИЛЛИНЫ — антибиотики, действующие
на многие грамположительпые, нек-рые грамотрица-
тельные микроорганизмы и отдельные грибки. Для
П. характерна бициклич. система из сконденсирован-
ных между собой ^-лактамного и тиазолидинового
колец. Центральной частью молекулы П. является
6-аминопеницилланован к-та (6-АПК):
° /А
H2N-CH-HC5 гС(СН3)г
СО — N*—"СНСООН
Один из атомов водорода в группе NH2 замещен
879
ПЕНИЦИЛЛИНЫ
880
Отроение радикала К Название (рациональ- ное и тривиальное общепринятое) Синонимы
Пенициллины, выделенные из приро д н ых материалов
СН3 (СН2)3 сн, П-Амилпепициллин Гигантсновая к-та, диги- дропенициллин F
СН3СН2СН = СНСН2 2-Пснтеннлпенициллин Пенициллин I, пеницил- лин F
СН3 (СН2)3 сн3 71-Гсптилпенициллин Пенициллин К, пеницил- лин IV
С„Н5СН2 Бензилпенициллин Пенициллин С, пеницил- лин II
п-НОС«Н,СН2 п-Оксибснзилпеницил- лин Пенициллин III
Пени ц и л л и н ы, полученные би осинтезом
СьН3ОСН2 Феноксиметилпсницил- лин Пенициллин V
СН2 = СНСН2 SCH2 Аллилмеркаптометил- пенициллин Пенициллин О
НООССН (NH2) (СП2)3 4-Амино-4-карбокси-п- Цефалоспорин N, синие-
бутилпенициллин матин В
Пенициллины, полученные сочетанием химических
и био логических метод о в
С„Н,ОСН (СН3) а-Феноксиэтилпеницил- Броксил, максипен, син-
лин, фенетициллин циллин
С„Н5ОСН (С2Н5) а-Фсноксипропилпсни- Броциллин, ультрапен,
циллин,левопропицил- лин байциллин
С»Н3ОСН (СЬН5) а-Фен оксибензилпени- циллин, фенбеницил- лин Пенспек
2,6 = (СН3О)2СоН3 2,6-Диметоксифенилпе- Селбенин, стафциллин
нициллин, метициллин
С6Н5-С—с 5-Метил-З-фенилизокса- Простафлип, микроне-
II II з оли л-4-п ени цил ли и, нин, стапенор, крипто-
N С—СНа оксациллин циллин
\/ О
С«Н(С1-С—с 3 -о-Х л ор ф они л- 5-метил - Орбенин
II II - 4-изоксазолилпени-
N С-СН3 циллин
4 Z О
С„Н5СН (NHa) Аминобензилпеницил- лин, ампициллин Певбритин, бинотал
группой RCO. Молекула П. строится из цистеина,
валина и замещенной уксусной к-ты, образующей
боковую цепь. В молекуле П. три асимметрия, атома
углерода — 3-й, 5-й, 6-й; пространственное располо-
жение заместителей установлено только у атомов 3
и 6. П. отличаются между собой радикалом R, от стро-
ения к-рого в значительной степени зависят их био-
логич. и физико-химич. снойства.
По способу получения П. разделяют на: 1) выделен-
ные из природных материалов; 2) полученные био-
синтезом; 3) полученные комбинацией методов химпч.
и биологич. синтеза. В табл. 1 приведены строение
и название наиболее известных П.
П. образуются многими микроорганизмами Peni-
cillium и Aspergillus. П. получают в процессе биосин-
теза, добавлением в питательную среду предшествен-
ников, к-рые используются плесневым грибком для
построения радикала, замещающего атом водорода в
аминогруппе молекулы П. Но этим путем может быть
введен в молекулу П. не всякий радикал. Получено
значительное число биосинтетич. П., из к-рых ши-
роко применяются в мед. практике бензилпеницил-
лин — в виде Na, К и новокаиновой солей, фен-
оксиметилпенициллин — в виде свободной к-ты и ре-
же аллилмеркаптометилпенициллин — в виде К-соли.
С открытием 6-АПК стало возможным получение
разнообразных П. определенного строения. В 6-АПК
Таблица 1 можно ввести ацильные остатки со
всевозможными алифатич., арома-
тич. и гетероциклич. системами, вклю-
чая и такие, которые сами по себе
могут обладать химиотерапевтич. дей-
ствием. Для получения П. из 6-АПК
рекомендуют несколько способов: 1)
ацилирование Na-соли 6-АПК хлоран-
гидридами различных карбоновых к-т;
2) ацилирование 6-АПК смешанными
ангидридами кислот; 3) действие кар-
бодиимидов на смесь 6-АПК с соответ-
ствующей к-той, в частности в качестве
конденсирующего агента применяют
дициклогексилкарбодиимид; 4) конден-
сация 6-АПК с хлорангидридами суль-
фокислот, изоцианатами и изотиоциана-
тами; этим синтезом пользуются редко.
Известно большое число П., получен-
ных сочетанием химич. и биохимич. ме-
тодов синтеза (см. табл. 1), из к-рых
наиболее известны фенетициллин, ме-
тициллин, оксациллин, ампициллин,
фенбепициллин. Характерной особенно-
стью таких П. является их устойчи-
вость к пенициллиназе и значительная
стабильность нек-рых из них в кислой
среде.
Полный синтез П. осуществлен впер-
вые Де Виньо в 1946. Наиболее изу-
ченным в химич. отношении является
бензилпенициллин, полный синтез к-ро-
го осуществлен Шиханом в 1959.
Для получения П. в промышленном ма-
сштабе применяют штамм плесени Peniclllium
chrysogenum № 194, к-рый при глубинном вы-
ращивании образует культуральные жидко-
сти с высоким содержанием антибиотика
(7000—9000 мкг/мл). Этот штамм был полу-
чен в результате селекционной работы проду-
центов П. с помощью хнмич. и физич. воздей-
ствий,он резко отличается от продуктивности
«диких» штаммов (см. табл. 2).
В процессе биосинтеза применяют различ-
ные питательные среды, к-рые позволяют по-
лучать высокие выходы П. Обычно в состав
этих сред входят лактоза (3—5%), глюкоза
(1—2%), кукурузный экстракт (1—4%), мел
(0,5—0,8%), животные или растительные жи-
ры (0,2—0,5%), сернокислый или серновати-
стокпслый натрий (0,2—0,3%), а также соответствующий
предшественник: в произ-ве бензилпенициллина — фенил-
уксусная к-та, феноксиметилпенпциллина — феноксиуксусная
к-та, а в случае аллилмеркаптометилпенициллина — аллпл-
меркаптоуксусная к-та.
Таблица 2
«Дикие» штаммы Активность, международ- ные единицы Селекционирован- ные штаммы Активность, 1 международ- ные единицы
Р. chrysogenum 248/1 0 Р. chrvsogenum Q № 176 1000
То же № 182 20—36 То же «Новый гибрид» 3970
То же № 189 50 Р. chrysfgenum Ns 194 7000 9000
Биосинтез П. осуществляют в ферментерах, при соблюде-
нии определенных условий: темп-ре ок. 24°, pH 7,0—7,4,
степени аэрации—1 объем воздуха в 1 минуту на 1 объем пи-
тательной среды, интенсивности размешивания и т. п. Фермен-
тация протекает в 2 стадии. На первой стадии происходит
рост мицелия, при к-ром усваиваются углеводы, достигает
максимума обмен органич. и нитратного азота, сильно повы-
шается значение pH среды.
На второй стадии происходит образование основного коли-
чества П. В зависимости от состава питательной среды и дру-
гих факторов ферментацию проводят в течение 3—5 дней. По-
сле окончания ферментации мицелий отделяют от Культураль-
ПЕНИЦИЛЛИНЫ
881
ной жидкости фильтрацией или центрифугированием. Для
сравнения активности различных препаратов П. и для оценки
общего содержания антибиотиков в различных культуральных
жидкостях применяется международная единица, равная
0,6 мкг чистой Na-соли бензилпенициллина.
Выделение П. из культуральных жидкостей и очистка их
производятся экстракцией органич. растворителями с примене-
нием сепараторов, суперцентрифуг или экстракторов разного
типа. Для повышения концентрации и чистоты П. экстракцию
проводят из органич. растворителя фосфатным буферным р-ром
при pH 7,0. После окончания экстракции растворы подкисляют
до pH 2,0 и экстрагируют бутилацетатом, амилацетатом или
хлороформом. К-соль бензилпенициллина обычно получают
непосредственно из этих растворов прибавлением ацетата ка-
лия. Выпавшие кристаллы К-соли отделяют, промывают бута-
нолом и сушат. Na-соль бензилпенициллина получают экстра-
гированием антибиотика из его раствора в бутилацетате вод-
ным р-ром бикарбоната натрия. Растворы солей П. сушатся
лиофильным способом или на распылительных сушилках.
Для определения П. в препаратах и в культураль-
ных жидкостях существуют различные микробиоло-
гия., химич. и физико-химич. методы. Наиболее рас-
пространенным методом для определения общего ко-
личества П. является йодометрический, основанный
на различном отношении П. и пенициллоиновых к-т
к иоду. Для быстрого и точного определения П. в
смеси пользуются методами распределительной хро-
матографии. Наиболее надежным методом качестве! -
ного определения П. является применение пеницил-
линазы.
При действии на П. (II) различных реагентов они
довольно легко расщепляются с образованием соедь-
нений, лишенных антибактериальной активности.
В большинстве случаев происходит раскрытие менее
устойчивого р-лактамного кольца. При действии на
П. водными р-рами щелочей, спиртами, аминами, пе-
нициллазой и др. образуются весьма неустойчивые
пенициллоиновые кислоты (I) и их
более стойкие производные: эфиры, соли, амиды и
И(4)-ацильные производные. Пенициллоиновые к-ты
можно сравнительно легко синтезировать. Так, кон-
денсацией пенальдовых к-т или их производных,
882
напр. ацеталей, с пеницилламином или его хлоргид-
ратом получают a-эфиры пенициллоиновых к-т;
HS
RCONHCHCHO ЧС(СНз)г _________
R'OOC + H2N-СНСООН *
RCONHCH-HC С(СН3)2
” R'OOC HN-------СНСООН
В молекулах пенициллоиновых к-т и их производных
имеются 3 асимметрия, атома углерода (3-й, 5-й, 6-й),
и поэтому они могут существовать в 8 стереоизомер-
ных формах. Построенные из D- или L-пеницилламина
обозначаются соответственно как D- или [.-пеницил-
лоиновые к-ты, а члены этих двух рядов стереоизо-
меров как а-, $-, у- и б-формы. По физич. свойства.,,
эти формы резко отличаются между собой; под влия-
нием различных реагентов они могут превращаться
друг в друга. Карбоксильные группы в пенициллои-
новых к-тах неравноценны. Сулемой пенициллоино-
вые к-ты расщепляются на пеницилламин
(XIII) и на пениллоальдегиды (XI). При
нагревании они превращаются в пениллоиновые к-ты
(IX); при каталитич. гидрировании их диэфиров об-
разуются диэфиры детиопенициллоиновых к-т (VI).
Пенициллоиновые к-ты могут циклизоваться в П.
Действием М,М'-дициклогексилкарбодиимида уда-
лось циклизовать D-a-феноксиметилпенициллоиновую
к-ту в природный феноксиметилпенициллин с выходом
10-12%.
П. расщепляются сулемой на пеницилламин (XIII)
и пениллоальдегиды (XI), к-рым предшествуют п е -
нальдовые (пенальдиновые) к-ты
(XII). Пенальдовыек-ты—весьма неустойчивые соеди-
RCONHCH-HC^ 1 ^С(СН3)2 RCONHCH-HC^ ЧС(СН3)2. СН3ОН
ХОС HN-—’СНСООН СО - N СНСООН
xs4
RCONHCH-HC С(СН3)2
СО - N---СНСООСН3
III
x-ОН, ONa, OR'
H2/(N1)
RCONHCH—
R'OOC
-СО2
CH2 HC(CH3)2
NH-CHCOOR'
VI
RCONHCHCH3 CH(CH3)2
CO—NH—СНСООН
IV
RCONHCH2CH2 CH(CH3)
RCONHCH- CH2
CO—N--------
CH(CH3)2
СНСООН
V H+,OH“
)2 RCONHCH-CH2 CH(CH3)2
hn — СНСООН -CO2 HOOC NH —СНСООН
H2/(N1)7 VII
HgCI2
VIII
HSCN
IX
RCONHCHjCHO
XI
s
rconhch2-hcx XC(CH3)2
RCONHCHCHO
I
HOOC
XII
s
RCONHCH-HCZ 4C(CH3)2
CO N-----CHCOOCH3
I I
HN----cs
HSC(CH3)2
HjNCHCOOH
XIII
883
ПЕНИЦИЛЛИНЫ
884
нения, обычно выделяемые в виде эфиров, амидов,
ацеталей и др. производных. При кипячении подкис-
ленных водных р-ров П. образуются пениллои-
новые к-ты (IX), к-рые расщепляются суле-
мой на пеницилламин (XIII) и пениллоальдегиды (XI);
при каталич. гидрировании образуются детиопенил-
лоиновые к-ты (VII). В молекулах пениллоиновых к-т
имеются 2 асимметрич. атома углерода; эти к-ты
могут существовать в 4 стереоизомерных формах.
Построенные из D- или L-пеницилламина обознача-
ются соответственно как D- или L-пениллоиновые
к-ты, а члены этих двух рядов как а- и (i-формы. Сте-
реоизомерные формы могут легко переходить друг в
друга. Описано значительное число пениллоиновых
к-т и их производных, полученных декарбоксилирова-
нием пенициллоиновых к-т (I) или синтетически.
Гидрогенолиз П. приводит к образованию ацилиро-
ванного L-аланил-П-валина (IV) и детиопенициллинов
(V); последние под влиянием кислот или щелочей
могут превращаться в детиопенициллоиновые к-ты
(VIII), при декарбоксилировании к-рых образуются
детиопениллоиновые к-ты (VII). При действии на П.
различными спиртами образуются эфиры (III), к-рые
в присутствии тиоциановой к-ты HSCN дают тиогид-
роурацильные производные (X) (см. стр. 881—882).
Несколько отлично от остальных ведет себя в кис-
лой среде феноксиметилпенициллин. Характерной
особенностью П. является способность к изомерным
превращениям. Известно пять типов соединений, изо-
мерных природным П.:
S
RCONHCH—НС С(СН3)2
СО—N---------СНСООН
RC-N-C = СН С(СНД
О—ОС NH----СНСООН
пеницилленовые кислоты
CH2CO-N---СНСООН
11
I
СООН
нс н S
f с' >(СН3)
RC----N---СНСООН
пеннлловые кислоты
i
соон
А н\
N СН С(СН3)2
RC----N СНСООН
пенилломовые кислоты изопеннлловые кислоты
+ *
RC-NH-СН-НСХ ЧС(СН3)2
О-------СО HN---СНСООН
псевлопеяицнллины
Пеницилленовые к-ты имеют интенсивную полосу
поглощения в УФ-области (320 ммк), отсутствующую
у П., а также у других изомеров и продуктов их
превращений. Кислоты и многие их производные труд-
но поддаются очистке и выделению в кристаллич.
состоянии. Пеницилленовые к-ты способны изомери-
зоваться в пенилловые, пениллоновые к-ты и с очень
малым выходом в П.
Пенилловые кислоты легко образуют-
ся непосредственно из П. в водных р-рах при pH 2.
С увеличением концентрации водородных ионов вы-
ход пенилловых к-т снижается, а в присутствии 2 н.
НС1 они совсем не образуются. Образование пенил-
лозых к-т характеризуется увеличением угла враще-
ния по сравнению с П., напр., бензилпенилловая
к-та имеет [а]д=+500; пенилловые к-ты имеют так-
же характерный УФ-спектр с максимумом ок. 240 ммк.
Пенилловые кислоты—довольно устойчивые соедине-
ния, могут быть получены несколькими синтетич.
методами, напр.:
СООСНд s
NH HC1 H2NCH-HCZ ЧС(СН3)2 ______
С|Н5СН2СОС2Н5 + HN---CHCOOCHj
соосн3
СН S
---- 1/ Hf/ ЧС(СН3)2
C|HSCH2C----N---CHCOOCHj
Под действием р-ра сулемы пенилловые к-ты рас-
щепляются в пенилламины; при кипячении водных
р-ров превращаются в пениллоиновые к ты, под дей-
ствием водных р-ров щелочей изомеризуются в и з о-
пенилловые к-ты.
В пениллоновых кислотах имида-
золидиновое кольцо сконденсировано с тиазолиди-
новым. Наиболее изучены бензилпениллоновая к-та
и ее метиловый эфир [т. пл. 152 —154°; [а]^ = + 318°
(метанол)]. Последний можно получить нагреванием
метилового эфира бензилпенициллина в вакууме в при-
сутствии следов иода. Известны способы синтеза этого
эфира. При омылении его образуется бензилпенилло-
вая к-та. Образование п с е в д о и е и и ц и Л л и-
н о в изучено на примере метилового эфира бензил-
пенициллина, к-рый в совершений безводных раство-
рителях в присутствии НС1 превращается в смесь
моно- и дихлоргидратов метилового эфира бензил-
псевдопенициллина .
Основной причиной нестабильности П. в водных
р-рах является неустойчивость их 0-лактамного коль-
ца. Скорость его расщепления зависит от pH среды и
темп-ры. П. довольно устойчивы в сухих неполярных
растворителях и быстро инактивируются в органич.
растворителях в присутствии НС1. Сухие соли П.
достаточно устойчивы даже при повышенной темп-ре.
П. применяют для лечения сепсиса, пневмонии,
скарлатины и др. заболеваний, а также при обработке
ран. Кислотоустойчивые П.— фенетициллин и фенок-
сибензилпенициллин — при введении per os эффектив-
ны при лечении инфекций, вызванных чувствитель-
ными к бензилпенициллину микроорганизмами. Фене-
тициллин всасывается лучше, чем феноксиметилпе-
нициллин. Метициллин и оксациллин эффективны при
лечении инфекций, вызванных резистентными к бен-
зилпенициллину стафилококками. Метициллин может
применяться только парентерально, а оксациллин —
и парентерально и per os, что является его преимуще-
ством. Ампициллин эффективен при инфекциях, вы-
званных грамположительными микроорганизмами,
т. е. является антибиотиком широкого спектра.
Наибольший интерес представляют П., действую-
щие на резистентные к бензилпенициллину стафило-
кокки (метициллин и оксациллин), а также П., дей-
ствующий на грамотрицательные микроорганизмы
(ампициллин). В табл. 3 (стр. 885—886) приведены
антибактериальные спектры различных П.
Лит.: Шемякин М. М. [и др.], Химия антибистиков,
3 изд., т. 2, М., 1961; Antimicrobial agents and chemotherapy.
1961. Proc, of the First conference on antimicrobial ....
[Detroit], 1962; Левитов M. M., Савицкая E. M.,
Товар ова И. И., Антибиотики, 1962, 7, № 5, 387; Knox
R., Nature, 1961, 192, № 4802, 492; Doyle F.P. [a. o.],
там же, 1961, 191, № 4793, 1091; Патент США, 2941995' (Chem.
Abst. 54, 20072c, I960); P e г г о n Y. G. [a. o.J, J Organ.
Chem., 1961, 26, № 9,3365; Левитов M. M. [и др.],
Антибиотики, 1962, 7, № 2, 104; Doyle F. P. [a. o.J,
Nature, 1961, 192, № 4808, 1183; Knudsen E. T.,
Brown D. M., Roll n son G. N., Lancet, 1962, 2, Л»
7257, 632; DoyleF. P. [a. o.], J. Chem. Soc., 1962,
1140: Some new penicillins, Brit. Med. J., 1962, № 5271;
885
ПЕНИЦИЛЛОВАЯ КИСЛОТА —ПЕНОПЛАСТЫ
886
Таблица 3
Минимальная бактериостатическая концентрация, мкгрлл
Тест-микробы бензил- пеницил- лина фенокси- метилпе- ницил- лина фенокси- этилпени- циллина фенокси- бенз илпе- ницил- лина оксацил- лина метицил- лина аминобен зилпени- циллина
S. typhosa 6.0 50 >100 50 100 >100 1,2
S. paratyphi В 12,5 100 >100 100 100 >100 2,5
S. typhi murium 3.0 50 100 100 100 >100 0,6
Sh. dysenteriae Flexheri 25 50 >100 25 100 >100 2,5
E. coli 25 >100 >100 100 100 >100 5,0
Pseud, aeruginosa 100 >100 >100 100 100 >100 >100
Prot. vulgaris 25 100 >100 100 100 >100 2,5
Klebs pneumoniae >100 100 100 25 100 >100 2.5
Shept. faecalis 0.5 1,0 0,25 1.25 1.25 50 0,5
Shept. pyogenes 0,03 0,015 0,012 0,03 0,006 1 , 25 0,015
Dipl, pneumoniae 0,012 0.015 0,012 0,015 0,12 0,3 0.03
C. diphfheriae 0,5 0,25 0,5 1.0 1.0 12,5 0,25
Staph, aureus 209 P 0,03 0,015 0.03 0,03 0,12 1, 5 0.06
Он же, адаптирован к бензилпенициллину . . 500 500 500 500 500 500 250
Staph, aureus № 8 0,03 0,06 0,12 0,25 0.25 3,0 0,06
Sheehan J. С., Henery-Logan К. R., J. Amer.
Chem. Soc., 1957, 79, № 5, 1262; Алиханян С. И., Изв.
АН СССР. Сер. биол., 1962, М 4, 544; Левитов М. М.,
Лурье Л. М., Завилейская Г. Ф., Антибиотики,
1961, 6, № 12, 1058; Навашин С. М., там же, 1963, 8, № 6,
564; Р а б и н о в и ч М. С. [ид р.], Ж. общ. хим., 1963, 33,
вып. 10, 3135; Струков И. Т., Антибиотики, 1963, 8, № 11,
963; Германова К. И., там же, 1963, 8, № 11, 976; П а-
н и н а М. А. [и д р.], там же, 1963, 8, № 11, 989; J. Amer.
Chem. Soc., 1959, 81, № 21, 5838. Е. В. Качалина.
ПЕНИЦИЛЛОВАЯ КИСЛОТА С8Н10О4, мол. в.
170,16—антибиотик, продуцируемый несколькими
пгн видами грибков Penicillium и Asper-
х0СНз gillus, а также Paecilomyces Ehrlichii.
[ [_/СНз П- к. — бесцветные иглы (из бензина),
(У^ОЛсн т' пл' м°ногидрат (из воды), т. пл.
ОН С”2 64— 65°; максимум поглощения в вод-
ном р-ре при 225 ммк (е 12 750), в 0,02
н. НС1 при 228 ммк (е 11 500); хорошо растворима
в горячей воде, этаноле, эфире, бензоле и хлоро-
форме, умеренно— в холодной воде (2%) и плохо —
в холодном беизине. П. к. в кристаллич. состоянии и
в растворах существует в лактольной форме, она лег-
к о образует нейтральный ацетат (R=COCH3) и ме-
тиловый эфир (R=CH3) (с подкисленным метанолом).
П. к. синтезирована в 1947, исходя из метилаллило-
вого спирта. Биосинтез П. к. протекает конденсацией
прп участии коэнзима А) 4 молекул уксусной к-ты
или 1 молекулы уксусной и 3 молекул малоновой
/СНз SeO2; HC = CNa
HOCHjC ------------------------—
VH co2; сн3он(н+)
СН3
CHjOOCCSCCHC
I ч
ОН снг
СН3ОН
BF3: НгО; СС13СООН
о
4 СН3СООН
0СНз NH
сн3----
сн
О 0
осн,
cн33^
пн3 с
ОСН
СН.
сн
НО
сн3
соон
О
ОСН3
сн3
он сн’
1. N —бромсукцинимид
2.N(CH3)3
3. Mg(OH)2
,ОСН3
СН3
0 01?сн'
OR
II
R=COCH3; СН3
к-т) с образованием орселлиновой к-ты (II), биохи-
мии. метилирование, окислительно-гидролитич. рас-
щепление и декарбоксилирование к-рой приводят к
П. к. Выше приведен синтез П. к. и связанной с ней
дигидропеницилловой к-ты (I).
Лучшим продуцентом П. к. является Р. cyclopium,
к-рый на среде Раулина — Тома образует до 2,5 г/л
П. к., выделяют П. к. из культурального фильтрата
адсорбцией на активном угле при pH 2 с последующим
вымыванием метанолом. Количественно П. к. опреде-
ляют колориметрич. (по цветной реакции с гидрок-
силамином) или полярографич. методом.
П. к. подавляет многие грамположительные, грам-
отрицательные и кислотоустойчивые микроорга-
низмы (напр,, Staphylococcus aureus при концентра-
ции 20—100 мкг/мл, Escherichia coli и Vibrio comma
при концентрации 10 мкг/мл), а также ряд патогенных
грибков и фагов. Токсичность П. к. невысока: для
мышей и кроликов переносимая доза при подкожном
введении составляет 100—200 мг/кг.
Лит.: Шемякин М. М. [и др.], Химия антибиотиков,
т. 1, 3 иад., М., 1961, с. 471; М о s b а с h К., Actachem. stand.,
1960, 14, 457; BentleyR.,Keil J. G., Proc. Chem. Soc.,
1961, 111. В. В. Оноприенко.
ПЕНОПЛАСТЫ (ячеистые полимерные материа-
лы) — газонаполненные пластмассы, в к-рых газ за-
полняет несообщающиеся между собой полости (ячей-
ки). Наряду с несообщающимися в П. обычно имеется
и нек-рое количество сообщающихся ячеек. П. харак-
теризуются малым объемным весом, плавучестью,
высокими тепло-, звуко- и электроизоляционными по-
казателями. Свойства П. определяются степенью
вспенивания, строением ячеек и химич. природой
полимера. Физич. свойства нек-рых П. приведены в
таблице. Наиболее распространены след, способы
произ-ва П.
Вспенивание полимеров в высо-
коэластическом состоянии. Твердый
термопластичный полимер, содержащий вспениваю-
щие вещества (твердые или жидкие), нагревается до
темп-ры, на несколько градусов превышающей темп-ру
стеклования. При этом газ, образующийся вследствие
разложения или испарения газообразователя, вспе-
нивает полимер. Наибольшее распространение полу-
чил прессовый метод произ-ва П., заключающийся
в прессовании смеси порошкообразного термопластич-
ного полимера, напр. полистирола, с твердым газооб-
разователем. При темп-ре 140—160° и давлении 100—
300 кг/см‘ газообразователь разлагается с выделением
газообразных продуктов, к-рые распределяются или
растворяются в размягченном полимере. После ох-
лаждения запрессовка извлекается из прессформы
и нагревается до 100—105°. При этом вследствие
887
ПЕНОПЛАСТЫ — ПЕНТАЛАН
888
Пенопласт на основе эпоксидной смолы 0.3 50 70 1.5 * При t 0ft ги
0.2 20 35 0,8 0.038 130-165 1.6* 8 *
0.1 7 14 480 0.6 0,029 1.2* 6.3 *
1 Пенополиуретан жесткий 0.2 29-39 15-22 4 2-60 1300 1.3-1 .6 0.04 1 ’.5 1,25 4-8
0.1 5.5-13 4.5-12 11 -24 300 0,8-0,9 0,032 1.1 1 .5-3.7
0.05 1 .7-4.0 2.5-3.5 4.5-12 too 0.5-0,6 0.029 120 t . 1 1 .2
Пенополи- стирол бесп расо- вый 0,02-0,04 1 .1—3.0 t . 8-2,0 t .3-4,0 40 — 100 O.t—0.4 0.027-0,031 60
Пенополистирол 0.2 30 30 65 tooo 1.9 0,044 70 1 .28—1,36 2,4
0.1 10 550 1 , t 0,033 60 1,1 1.2
0,06 3,0-4,3 400 0,94 0,22 60
Пенополивинилхлорид 0,2 26 45 40 1800 1.5 0.045 60 2,4 16,6 *
O.t 9 20 20 900 1,0 0.037 60 1 .6 3,6 *
0,06 2,3 0.7 0,022 60
Показатели Объемный вес, г/см? .... Предел прочности, кг{см2 при сжатии при растяжении при статич. изгибе .... Модуль упругости при сжа- тии, кг см2 Уд. ударная вязкость, кг-см^см2 ........ Коэфф, теплопроводности, ккал Дм • час • гр ад Теплостойкость, °C .... Диэлектрич. проницаемость при 1010 гц Тангенс угла диэлектрич. потерь при 1010 гц (tgft-lO—3)
размягчения полимера и повышения давления газа
запрессовка расширяется, образуя П. К этому же
способу относится произ-во пенополистирола из
гранульного или блочного полимера. В этом слу.
чае блочную или суспензионную полимеризацию
стирола проводят в присутствии низкокипягцих жид-
костей (изопентан, бутан, петролейный эфир и др.)
или же насыщают ими готовый полимер. Вспенивают
последний обычно в перфорированных формах паром
или кипящей водой в течение нескольких минут.
Вспенивание вязко-жидких по-
лимерных композиций в процессе
образования полимера. При изго-
товлении по этому способу П. на основе феволо-
формальдегидных смол вспенивание производится
газообразными продуктами, выделяющимися при
взаимодействии кислых отвердителей с металлич.
порошками (водород) или с солями угольной к-ты
(двуокись углерода). При получении пенополиуре-
тана исходная смесь вспенивается вследствие об-
разования двуокиси углерода при взаимодействии
диизоцианата с добавляемой в композицию водой.
Полимер отверждается при комнатной или повышен-
ной темп-ре в зависимости от типа взятых компонен-
тов. Полиуретановые композиции вспенивают также
низкокипящими жидкостями, испаряющимися за счет
тепла экзотермич. реакции образования полимера.
Этот способ позволяет изготовить П. в виде готовых
формованных изделий или получать их непосредствен-
но в полостях изделий; можно также наносить исход-
ную композицию методом пульверизации на поверх-
ность изделий, где и образуется П.
Лит.: Берлин А. А., Основы производства газонапол-
ненных пластмасс и эластомеров, М., 1954; Пенопластмассы.
Сб. статей, М., 1960. Н. А. Напалков.
ПЕНТАЛАН (бициклооктан-[0.3,3]) С8Н14, мол. в.
110,20 — бесцветная жидкость со слабым характерным
запахом, нерастворимая в воде, растворимая в эфи-
ре, спирте, петролейвом эфире. П. — бициклич. угле-
водород; существует в виде двух геометрич. изомеров:
цис- (I) и трене- (II). Транс- _ ,
форма П. является напря- I Т I I Т I
жевной и жесткой системой,
для нее возможна только од- । и
на конформация (III). Для
цис-формы П. возможны две конформации (IV а и IV б),
из к-рых IV а почти также напряжена, как и транс-
форма; IV б почти свободна от напряжения. Транс-
форма П. как система напряженная имеет теплоту
сгоргния, на 6 ккал превышающую теплоту сгорания
цис-формы.
Транс-ТТ., т. пл.—30°; т. кип. 1327755 мм; Т"
0,8626; Нд 1,4625; теплота сгорания 1203,8 ккал/моль
(20°, v — const). Транс-П. получают восстановлением
по Клемменсену или Кижверу траис-бицикло-[0,3,3]-
октанона-2. Дис-П. не кристаллизуется при охлаж-
дении до —80°; т. кип. 1357765 мм; 0,8719, u'J
1,4629; теплота сгорания 1197,7 ккал/моль (20°,
=const). Дис-П. получают восстановлением по Клем-
мевсеву или Кижнеру цис-бицикло-[0,3,3]-октано-
на-1 или цис-бицикло-[0,3,3]-октанона-2. Дис-П. при
88'9
ПЕНТАМИН—ПЕНТАЭРИТРИТ
890
действии хлористого алюминия превращается в транс-
П. Дие-П. найден в нек-рых нефтях.
Э. Е. Нифантьев.
ПЕНТАМИН [пендиомид, 3-метил-1,5-бис-(Х,М-ди-
метил-Ат-этиламмоний)-3-азапентан- дибромид], мол.
“Н,С СН3 -|
Н3С -N-(CH2)2-N- (CH2)2 -n-ch3
-Н,С2 сн3 С2Н3_
2Вг-
вес 391,25, С]3Н33ХзВг2, — белый или слегка желтова-
тый кристаллич. порошок со слабым запахом, т. пл.
210—215° (с разл.), гигроскопичен, легко растворим
в воде и спирте, нерастворим в эфире; при прибавле-
нии конц. H2SO4 выделяется бром. При смешивании
р-ра П. с насыщенным р-ром пикриновой к-ты выпа-
дает желтый осадок. Количественно П. определяют
аргентометрически.
Исходным продуктом для получения П. служит
смесь полиэтиленполиаминов. Из этой смеси дву-
кратной ректификацией выделяют диэтилентриамин
H2NCH2CH2NHCH2CH2NH2, к-рый обрабатывают му-
равьиной к-той и формалином, и получают пентаметил-
диэтилентриамин (CH3)2NCH2CH2N(CH3)CH3CH2N(CH3)2;
при его нагревании с бромистым этилом получают П.
Применяют П. при гипертонич. болезни, гипертонич.
кризах, спазмах периферии, сосудов, спазмах кишеч-
ника и желчных путей, почечных коликах, при за-
держке мочеиспускания, связанного с циститами и
камнями мочевого пузыря, и бронхиальной астме.
Jl'im.: Государственная фармакопея СССР, 9 изд., М.,
1961; Машковский М. Д., Лекарственные средства,
4 изд., М., 1960. В. А. Засосов.
ПЕНТАН (и-пентан) СНз(СНа)зСНз, мол. в. 72,146—
бесцветная, подвижная, горючая со слабым запахом
жидкость; т. пл.— 129,721°; т. кип. 36,074°; <7* 0,62624
(20°); 0,62139 (25°); ng 1,35748; С°р 0,3982 кал/г-град
(25°); 1крит.196,62°; Ркрит. 33,31 am; dKpi)T. 0,232; т. воспл.
285°; т. всп. <— 40°; пределы взрываемости в смеси па-
ров с воздухом 1,45—7,5 об. %; теплота сгорания
жидкого П. при 25° до жидкой воды и СО2 838,80
ккал/молъ; теплота образования газообразногоП.—35,00
ккал/молъ. П . в значительных количествах содержится
в бензинах, конденсатах, выделяемых из природного
газа, в нефтях и легких погонах сланцевой смолы, а
также в углеводородах, синтезируемых по Фишеру-
Тропшу из СО и Нг. П. легко выделяется ректифика-
цией или при сочетании ее с селективной адсорбцией
парафиновых углеводородов молекулярными ситами.
В лаборатории П. синтезируют б. ч. гидрированием
и-амиленов, получаемых дегидратацией и-амиловых
спиртов или реакциями, общими для получения
нормальных предельных углеводородов. П. образует-
ся как единственный продукт гидрогенолиза цикло-
пентана на платинированном угле при 300°. В свою
очередь на этом же катализаторе П. циклизуется с об-
разованием циклопентана. Химич, свойства П. по-
добны свойствам других гомологов ряда «-парафино-
вых углеводородов. При пиролизе П. получаются
этилен и пропилен — реакция протекает в промышлен-
ном процессе пиролиза легких низкооктановых бен-
зинов. В присутствии А1С1з, алюмоплатинового ката-
лизатора П. изомеризуют в основном в изопентан —
сырье для синтеза изопрена. Дегидрированием П.
при высокой темп-ре в присутствии хромовых катали-
заторов или кислорода и иода получают смесь амиле-
нов. Хлорирование П. осуществляют в пром-сти с
целью превращения получающихся хлорпентанов в
амиловые спирты и эфиры, получения амилфенола и
многих других веществ, а также в произ-ве гексахлор-
циклопентадиена. При пропускании П. в токе азота
над C0F3 при повышенной темп-ре образуется пер-
фторпентан. С HNO3(68%) при 400° наряду с продук-
тами деструкции получаются 1-, 2- и 3-нитропен-
таны. В присутствии А1С1з при повышенных темп-ре
и давлении П. с СО образует кетоны, кислоты, а с
SO2— пентансульфоновую к-ту. П. окисляется бак-
териями Bact. aliphaticum liquefaciens. П. имеет два
изомера общей ф-лы С3Н12: изопентан и неопентан.
М. И. Розенгарт.
ПЕНТАФЕНИЛФОСФОР (С6Н3)3Р, мол. в. 416,47—
бесцветные кристаллы, призмы; т. пл. 124°
(с разл.); в воде нерастворим, растворим в горячем
циклогексане. П. имеет строение тригональной пира-
миды, в центре к-рой находится атом фосфора. Для
установления характера связи фенильных ядер с цен-
тральным атомом проведено специальное исследова-
ние. Двумя различными способами (I и II) получен
П., одно из фенильных ядер к-рого содержит дейте-
рий вместо атомов водорода:
I. (C»H,)(PJ + CeD5Li\/„
IT (С ВМС Н^РЫГНЫ >»HS).P(C.D5)
При взаимодействии П., содержащего дейтерий вместо
атомов водорода, со спиртами и хлороформом отрыв
фенильных радикалов происходит с одинаковой легко-
стью из полюсных и экваториальных положений. Это
объясняется тем, что, несмотря на геометрич. неравно-
значность расположения фенильных ядер в П., все
пять фенильных групп энергетически равноценно свя-
заны с фосфором.
Для П. особенно характерны радикальные реакции.
При нагревании П. выделяется бензол и получает-
ся Р-фенилдифениленфосфин (I), в небольшом количе-
стве образуется также дифенил (II) и трифеннлфос-
фин (III):
III
П. реагирует с хлороформом с образованием тетра-
фенилфосфонийхлорида:
(С.Н5)зР —с(С.Н,)4Р.+С.Н5.
С3Н3- +СНС13 —>-С.Нв + СС13-
(С.н5),р. +СНС1, —> (СвНг,),РС1 + СНС12-
Аналогично П. вступает во взаимодействие с СС1«.
При стоянии во влажном спирте П. превращается в
окись трифенилфосфина и бензол:
(СвН3)3Р + 2R0H —► (CeH3)3P(OR). + 2С„Н«
(C„HJ3P(OR)2 + H2O —>- (C»HS)3P=O + 2ROH
П. может реагировать также и по гетеролитич. ме-
ханизму, напр. с иодистым водородом:
(CeH.)5P+HJ—»(C4H5)4PJ + C3H,
Для синтеза П. используют реакцию между тетра-
фенилфосфонийиодидом и фениллитием:
(C6H;),PJ + CeH5Lt —>-(CsH5).-)P + L1J
Аналогично получают тетрафенилтолилфосфор и тет-
рафенилтритилфосфор:
(C.H.O.PJ
СН3С,Н4Ы
----------->- (C.HshPCaH.CH,
(СвН5)3СЫ
-----------►(C.Hb),PtC(C.Hs),]
Э. Е. Нифантъев.
ПЕНТАЭРИТРИТ (2,2-диметилолпропандиол, тет-
раметилолметан) (СНгОН^С, мол. в. 136,15—четырех-
атомный спирт с разветвленной углеродной цепью;
891
ПЕНТОЗАНЫ— ПЕНТОЗНЫЙ ЦИКЛ
892
бесцветные кристаллы, т. пл. 263,5°, d’°l»397; раство-
римость (%): в воде 7,1 (25°), 19,3 (55°), 76,6 (100°),
этиленгликоле 12,9 (100°), глицерине 10,3 (100°),
формамиде 21 (100°), пиридине 3,7 (100°); в других
растворителях — ацетоне, эфире, бензоле и др. мало
растворим.
П. обладает всеми свойствами полиатомных спир-
тов: при действии ацилирующих средств образует
моно-, ди-, три- и тетраацильные производные; с азот-
ной к-той дает нитраты, с хлорсульфоновой к-той —
сульфаты, с хлористым тионилом — пентаэритрит-
дисульфит:
носн2 СН2ОН о-сн2х сн2-о
zcz -S°2C'»- o=sz zcz \=о
носн2 ХСН2ОН XO-CH2Z чсн2-о'
или хлориды (моно-, ди-, три- и тетра-); метилируется
диметилсульфатом, иодистым метилом в присутствии
окиси серебра и диазометапом (неполно). При нагре-
вании с полиоксиметиленом и соляной к-той П. обра-
зует 5,5-бисхлорметоксиметил-1,3-диоксан и пента-
эритритдиметиленовый эфир:
носн2 zch2oh
zcz + СН2О + HCI —-
носня хсн2он
С1СН2ОСН2 хСН2Ох хО-СН2ч zCH2-Ox
—- ZCH2 + СН2 Сх ZCH2
С1СН2ОСн/ XCH2OZ XO-CH2Z XCH2-OZ
П. дает бициклич. ацетали с бромалем, фурфуролом,
циклопентаноном, циклогексаноном и другими альде-
гидами и кетонами; с ацеталями образует ацетали
пентаэритрита и соответствующий спирт;
ZOR' НОСН2 СН2ОН
2 RCH 9- XCZ ---►
OR' HOCH2Z ХСН2ОН
zoch2x сн2оч
---- RCH V zCH2R + 4 R'OH
OCH2 ch2oz
При нагревании до 270—280° с активированным
алюминием или медью П. превращается в 2-метил-
акролеин, метанол и формальдегид; при гидрирова-
нии над CuO—СгаОз-ВаО-катализатором при 250° и
175 ат образует изобутиловый спирт и метанол.
П. получают при действии формальдегида на аце-
тальдегид или акролеин в присутствии гидрата окиси
кальция или др. щелочных агентов (промышленный
продукт обычно загрязнен 10—15% ди- и трипента-
эритрита). В пром-сти П. используется для получения
высокомолекулярных соединений при действии на
него ангидридов и др. производных двух- и полиос-
новных к-т или окиси этилена и в синтезе тетра-
нитропентаэритрита — мощного взрывчатого ве-
щества.
Лит: Marr i an S. F., Cbem. Revs, 1 948, 43, Ks 1,
149. Э. E. Нифантъев.
ПЕНТОЗАНЫ — полисахариды, построенные из
остатков пентоз. В природе распространены II. двух
типов — арабаны, построенные из остатков L-араби-
позы, и к с и л а н ы, построенные из остатков
D-ксилозы. Нек-рые ксиланы, помимо ксилозы, содер-
жат незначительные количества L-арабинозы, D-inio-
козы, D-глюкуроновой к-ты и ее 4-О-метильного про-
изводного; ксиланы с высоким содержанием араби-
нозы называют арабоксиланами. Арабаны
в растениях сопутствуют пектиновым веществам',
ксиланы входят в состав гемицеллюлоз, чрезвычайно
широко распространены в наземных растениях,
встречаются и в водорослях.
Арабаны (I) — полисахариды, мол. в. 5000—6000,
остатки арабинозы в виде фуранозной формы связаны
в арабанах преим. а-1,3- и а-1,5-связями. Ксиланы
(II) объединяют группу полисахаридов, значительно
отличающихся по строению; мол. в. порядка 10000—
30000. Молекулы пек-рых ксиланов представляют
линейные или слабо разветвленные цепи остатков кси-
лозы в виде пиранозной формы, соединенных Р-1,4-
связями; остатки других моносахаридов образуют в
ксиланах обычно боковые цепи.
П,— бесцветные аморфные вещества. Арабаны хо-
рошо растворимы в воде, ксиланы в воде не раство-
ряются. Восстанавливающими свойствами П. не обла-
дают. П. оптически активны, вращают плоскость
поляризованного света влево; [а]в арабанов минус
60—180°, [а]в ксиланов минус 90—110°. Получают
П. из растительного сырья. Удобным сырьем для по-
лучения ксиланов является солома, кукурузные по-
чатки, подсолнечная шелуха. Пентозансодержащее
сырье используется для получения фурфурола и про-
изводных фурана. См. также Гемицеллюлозы.
Лит.: Никитин Н. И., Химия древесины и целлюлозы,
М.— Л., 1962; Роговин 3. А., Ш о р ы г и н а Н. Н., Хи-
мия целлюлозы и ее спутников, М.— Л., 1953; Cellulose and
cellulose derivatives, ed. E. Ott [a. o.J, 2 ed., N. Y.— L., 1954—
1955; Neumuller G., в кн.; Handbuch der Pflanzen-
physiologle, Bd 6, B.— [u a.], 1958, S. 252; Whistler R. L.,
Smart C. L., Polysaccharide chemistry, N. Y., 1953.
Л. И. Линевич.
ПЕНТОЗНЫЙ ЦИКЛ (пентозофосфатный цикл,
гексозомонофосфатный шунт, апотомический, или
прямой, путь окисления углеводов)— сложный цик-
лич. ферментативный процесс, обеспечивающий аэроб-
ное окисление углеводов, минуя гликолиз и цикл
трикарбоновых кислот-, сопровождается образованием
восстановленного никотинамид-а денин-динуклеотид-
фосфата (НАДФ-На) — см. Никотинамиднуклеотид-
ные коферменты. Исходным соединением цикла яв-
ляется D-глюкозо-б-фосфат (I), к-рый образуется
в клетке в процессе фосфорилирования глюкозы и
является ключевым соединением, общим как для П. ц.,
так и для гликолитич. пути расщепления углеводов.
Последовательность реакций, входящих в П. ц.,.
представлена на схеме.
В реакции (1), являющейся начальной реакцией цикла,
D-глюкозо-б-фосфат (I) дегидрируется под действием глюкозо-
6-фосфат-дегидрогеназы (систематич. номер 1.1.1.49, см.
Номенклатура и классификация ферментов). Акцептором во-
дорода, отщепляемого отглюкозо-6-фосфата, является никотин-
амид-аденин-динуклеотгд-фосфат (НАДФ). В результате об-
разуется D-глюконо-б-лактон-б-фосфат (II) и НАДФ-Н,.
Глюконолактон может спонтанно гидролизоваться до 6-фосфо-
D-глюконовой к-ты (III); в клетке этот процесс ускоряется
специальным ферментом D-глюконо-б-Лактон — гидролазой
(3.1.1.17). Образующаяся 6-фосфо-В-глюконовая к-та подвер-
893
ПЕНТОЗНЫЙ цикл
894
(9)
СНО
2 неон
<!:н2о®
VII
СНО
IX
носн
СНО
2 HCOiH
неон
снгон
со
2
неон
СН2ОН
СНО
неон
неон
СН2О©
VIII
со
2 НОС Н
неон
I Z-X
СН2О ©
неон
+н2о
H3FO4 неон
6
(10)
носн
носн
неон
СН2О®
нос----
о
НС
6 над®
6 над®-н2
о
со
неон
о
НС
сн
СН2ОН
нос----
, носн
5 I
неон
неон
НА_____
СН2О®
XI
неон
неон
СН2О®
(П)
носн
неон
I
НС------
СН2О®
II
6 н2о
(2)
соон
H<j:OH
„ носн
6 I
неон
неон
СН2О®
соон
бнад®-н2
6 над®
неон
со
6 I
неон
(ЗБ)
неон
-6С0
неон
неон
I
СН2О ©
IV А
неон
(jiHgOH
со
неон
I Z-4
СН2О®
неон
®-остаток -РО(ОН)2 СН2О®
IV Б
гается окислительному декарбоксилированию (реакции 3 4 и ЗБ)
с помошью фермента фосфоглюкопатдегидрогеназы (1 1 1.44>,
что приводит к образованию рибулозо-5-фосфата (IA Б). Акцеп-
тором водорода в этой реакции также ярлрстся НАДФ.
Пока еще не установлено, катализирует ли фосфоглюконатде-
гидрогеназа одновременно и окисление и декарбоксилирование
или реакция протекает с участием двух различных ферментов.
Имеются указании, что промежуточным продуктом реакции яв-
ляется З-кето-6-фосфоглюконовая к-та (111 А). Т. обр., на этом
этапе П. ц. молекула глюкозо-6-фосфата подвергается частич-
ному окислительному расщеплению и укорачивается на один
углеродный атом,превращаясь в молекулу рибулозо-5-фосфата.
Это превращение сопровождается образованием двух эквива-
лентов НАДФ-Н2 и выделением одной молекулы СО2. После-
дующие этапы П. ц. не связаны с процессами окисления, а
представляют собой различные превращения фосфорных эфи-
ров сахаров. Из 6 молекул образовавшегося рибулозо-5-фос-
фата (1VB) две с помощью фермен-
та рибозофосфат-изомеразы (5. 3. l.G>
изомеризуются (реакция 4) в рибозо-
5-фосфат (V). Оставшиеся 4 молекулы
рибулозо-5-фосфата превращаются в
ксилулозо-5-фосфат (VI) (реакция 5>
с помощью фермента П-рибулозо-5-
фосфат — 3-эпимеразы (5.1.3.1). По-
ловина образовавшегося ксилулозо-
5-фосфата взаимодействует с рибозо-
б-фосфатом (реакция 6). Эта реакция
осуществляется с помощью транс-
кетолазы (2.2.1.1) — переносчиком
«активного гликольальдегида» (обо-
значенного на схеме пунктиром) от
молекулы кетосахара к различным
альдегидным акцепторам. В резуль-
тате реакции образуются 3-фосфо-
глицериновый альдегид (VII) и D-ce-
догептулозо-7-фосфат VHI), которые
вступают во взаимодействие друг с
другом, катализируемое трансалъ-
долазой (2.2.1.2), осуществляющей
перенос трехуглеродного остатка от
молекулы седогептулозо - 7 - фосфата
на фосфоглицериновый альдегид
(реакция 7). В результате образует-
ся Г)-эритрозо-4-фосфат (X) и один
из конечных продуктов П. ц. — D-
фруктозо-6-фосфат (IX). В свою оче-
редь П-эритрозо-4-фосфат взаимо-
действует с оставшимся ксилулозо-
5-фосфатом (реакция 8), в результа-
те чего также образуются D-фрукто-
зо-6-фосфат (IX) и фосфоглицерино-
вый альдегид (VII). Эту реакцию,
так же как и реакцию (6), катализи-
рует транскетолаза, с тем отличием,
чго в данном случае двухуглеродныи
остаток от ксилулозо-5-фосфата пе-
реносится не на рибозо-5-фосфат, а
на эритрозо-4-фосфат. Образовав-
шийся в реакции (8) фосфоглицери-
новый альдегид в результате двух,
последовательных превращений, ка-
тализируемых ферментами триозо-
фосфат-изомеразой (5.3.1.1) и аль-
долазой (4.1.2.7), конденсируется с
образованием фруктозо -1,6- дифос-
фата (XI) (реакция 9). Последний
под действием гексозодифосфатазы
<3.1.3.11) превращается в D-фрукто-
зо-6-фосфат (IX) (реакция 10). Обра-
зовавшийся фруктозо-6-фосфат с по-
мощью фермента глюкозофосфат-изо-
меразы (5.3.1.9) превращается в ис-
ходное соединение цикла глюкозо-
6-фосфат (I) (реакция 11). в резуль-
тате чего цикл замыкается. Таким
образом, если исходить из 6 молекул
глюкозо-6-фосфата, то суммарное
ур-ние реакций П. ц. за один его
оборот можно представить в след,
виде:
6 молекул глюкозо-6-фосфата-|-
+ 12НАДФ->6 СО2+12НАДФ-Н24-
+ Н3РО4-|- бмолекул глюкозо-
6-фосфата
или
глккэзо-6-фосфат+12 НАДФ->
->6СО2-Н2 надф-н2-ин#ро4
В результате однократного
оборота через П.ц. шести моле-
кул глюкозо-6-фосфата одна мо-
лекула полностью окисляется до СО2, а 5 молекул реге-
нерируются и вновь возвращаются в цикл. Указанная
последовательность реакций представляет собой лишь
идеализированную схему основных превращений П. ц.
В реальных условиях П. ц. значительно сложнее,
поскольку многие промежуточные соединения цикла
могут образовываться и использоваться не только
в реакциях самого цикла, но также и в других прев-
ращениях. Это, в первую очередь, относится к исход-
ному соединению П. ц.—глюкозо-6-фосфату, а также
к таким промежуточным соединениям, как фрукто-о-
6-фосфат, фруктозо-1,6-дифосфат и фосфоглицерипо-
вый альдегид, к-рые одновременно являются субстра-
895
ПЕНТОЗЫ—ПЕНТОН
896
тами гликолитич. пути расщепления углеводов.
В результате наличия общих субстратов гликолиз и
П. ц. оказываются тесно связанными друг с другом.
П. ц. представляет собой альтернативный путь
окисления углеводов, идущий в обход гликолиза и
цикла трикарбоновых кислот, в связи с чем его иног-
да называют шунтом. П. ц.— широко распространен-
ный биохимия, процесс, осуществляемый во многих
животных и растительных тканях, а также у микроор-
ганизмов. Хотя доля участия П. ц. в общем обмене
углеводов значительно уступает гликолитич. пути
расщепления, тем не менее его роль в обеспечении жи-
знедеятельности клетки и целого организма исклю-
чительно велика. П. ц. является универсальным по-
ставщиком пентоз, к-рые входят в состав нуклеино-
вых кислот и нуклеотидов, в том числе важнейших
коферментов— нуклеотидов (НАД, НАДФ, кофер-
мент А). Важную биологич. роль играет другой про-
межуточный продукт П. ц.— эритроза, к-рая являет-
ся предшественником ароматич. кольца, входящего
в состав нек-рых аминокислот, фенолов, лигнина и
многих других соединений. Не менее важным продук-
том П. ц. является НАДФ-Н2, широко используемый
в качестве донора электронов и водорода в различных
синтетич. процессах, происходящих в клетке, в част-
ности при биосинтезе жирных к-т, холестерина, сте-
роидных гормонов, пуринов, а также при образова-
нии гидрированных форм фолиевой к-ты. Т. обр.
роль П. ц. в обмене веществ состоит в обеспечении
тканей гл. обр. пластич. материалами и НАДФ-На,
являющегося донором водорода и электронов для вос-
становительных реакций и процессов биосинтеза.
Исключительно велико значение П. ц. в обмене
веществ у растений, где реакции П. ц., идущие в об-
ратном направлении, принимают участие в процессах
ассимиляции СО2 (подробнее см. Фотосинтез).
Лит.-. Химические основы процессов жизнедеятельности,
М., 1962, с. 156; Диксон М., Уэбб Э., Ферменты, пер.
с англ., М., 1961, с. 542, 555; Кребс Г. А., Корнберг
Г. Л., Превращение энергии в живых системах, пер. с англ.,
М.. 1959; Шонка И., Ермоленко Р., Вопр. мед. хи-
мии, 1961, 7, вып. 2, 115; Кальвин М., Углеродный цикл
при фотосинтезе, в кн.: Современные проблемы биохимии. Сб.
статей, перевод, М., 1957; Metabolic pathways, ed. by D. M.
Greenberg, v. 1, N. Y.— L., 1960. В. Б. Спиричев.
ПЕНТОЗЫ — моносахариды, содержащие в моле-
куле пять атомов углерода. П. в зависимости от харак-
тера карбонильной группы делят на альдопентозы и
кетопентозы (пентулозы). Группа альдопентоз с нор-
мальной углеродной цепью включает 8 стереоизомер-
ных сахаров: 4 пары D-и L-изомеров, D-формы к-рых
представлены след, рядом:
СНО СНО СНО СНО
НСОН 1 НОСН 1 НСОН НОСН
1 НСОН 1 неон носн носн
НСОН 1 1 неон 1 НСОН 1 НСОН 1
СН2ОН В -рибоза снаон D-арабиноза СН2ОН D-ксилоза СН2ОН Г>-ликсоза
В кетопентозах карбонильная группа может зани-
мать положение 2 или 3 в углеродной цепи, но в при-
роде встречаются только 2 кетопентозы, D-форма
к-рых представлена ф-лами:
сн.он
неон
I
неон
I
сн2он
1)-эритро-пентулоза
(D-рибулоза)
СН2ОН
I
СО
I
носн
I
неон
I
сн2он
D -шрео-пентулоза
(D-ксилулоза, D-ликсулоза
П.— кристаллы, хорошо растворимые в воде; в вод-
ных р-рах П.муторатируют. П.дают всереакциивосста-
навливающих сахаров. При окислении альдопентоз об-
разуются одноосновные пентоновые кисло-
ты и двухосновные пентаровые кислоты,
что используется для идентификации пентоз.При окис-
лении первичной спиртовой группы (при защите аль-
дегидной группы) образуются пентуроновые
кислоты. Кетопентозы окисляются с расщепле-
нием цепи. При восстановлении П. образуются пя-
тиатомные спирты — пентиты.
П. широко распространены в природе. Они встре-
чаются в свободном виде, входят в состав гликози-
дов, полисахаридов (арабанов, ксиланов). D-рибоза и
ее дезоксипроизводное — D-дезоксирибоза — являются
структурными компонентами нуклеиновых кислот. Фос-
ферные производные П. являются важными проме-
жуточными продуктами в обмене углеводов. Получают
П. из природных источников, гл. обр. гидролизом по-
лисахаридов, напр. ксилозу — из ксиланов, арабино-
зу — из арабанов. Синтетически П. получают гл. обр.
из гексоз, используя общие методы укорачивания
цепи.
Лит. см. прист. Моносахариды. Л. И. Линевич.
ПЕНТОКСИЛ (2,6-диокси-4-метил-5- оксиметил-пи-
римидин) CeH80sN2, мол. в. 156,14 — белый кри-
сталлич. порошок; т. пл. 303—304°
(с разлож.); плохо растворим в во-
де, нерастворим в спирте и эфире,
растворяется в щелочах. При наг-
ревании с водой П. разлагается с
выделением формальдегида; водный
р-р П. обесцвечивает бромную воду,
при прибавлении
водного р-ра П. к смеси салициловой и серной к-т
появляется красное окрашивание. Количественно П.
определяют по содержанию в нем азота по Кьельдалю.
Для получения П. конденсируют 4-метилурацил с
формальдегидом.
П.— оригинальный советский препарат, способный
стимулировать различные регенеративные процессы,
в том числе при воспалении. П. применяется как сти-
мулятор лейкопоэза при алейкиях различного проис-
хождения (лучевой, лекарственной, токсической лей-
копении), агранулоцитозах различной этиологии,
а также для лечения язвы желудка.
Лит.: Синтезы органических препаратов, пер. с англ.,
сб. 2, М., 1949, с. 335; Хромов-Борисов Н. В., Кар-
линская Р. С., Ж. общ. химии, 1956, 26, вып. 6, 1728;
Лазарев Н. В., Фелистович Г. И., Пентоксил
и его применение при алейкиях, [Л.], 1954; Конференция по
проблеме медицинского применения пиримидиновых производ-
ных. Ростов-на-Дону. 1961. Материалы, Ростов н/Дону, 1961;
Материалы Конференции по применению пиримидиновых про-
изводных в онкологии и других областях медицины. 22-24
мая. 1963, Л., 1963. И. В. Хромов-Борисов.
ПЕНТОН [-СН2С (СН2С1)2 СН2О-]П— гетероцепной
простой полиэфир; темп-ра плавления достигает 180°.
П. устойчив к гидролизу в слабокислых и щелочных
средах, к действию конц. водных р-ров аммиака, пе-
рекиси водорода, дымящей азотной к-ты, 96 %-ной сер-
ной к-ты, анилина, толуола, этанола и др. соединений.
Ниже приведены нек-рые физич. свойства П.:
Плотн., г'см'1 .............. 1,4
Предел прочности, кг/см1
при разрыве.................. 420 (23°); 245 (100°)
при изгибе....................... 775 (23°)
Относительное удлинение, %
при разрыве...................... 35 (23°);
200-250 (100
Уд. объемное электрич. сопро-
тивление, ом см................... 5-10’3
Диэлектрич. проницаемость . . 3,1 (60 гц);
2,8 (10» гц)
Тангенс угла диэлектрич. по-
терь ........................... 0,016 (60 ги);
0,01 (10» гц)
Электрич, прочность, кв/мм . . 16
897
ПЕПТОНОВЫЕ КИСЛОТЫ—ПЕНЫ
898
П. получают полимеризацией 3,3-бис-(хлорметил)-
оксациклобутана в растворителе (хлористый метил,
сернистый ангидрид и др.) при низкой темп-ре;
в качестве катализатора чаще всего используют фто-
ристый бор и пятифтористый фосфор.
П. легко перерабатывают литьем под давлением и
шприцеванием. Он может быть использован для изго-
товления пленок, волокна, химически стойких труб,
вентилей, клапанов, деталей часовых механизмов и др.
Лит.: Химия и технология синтетических высокомолеку-
лярных соединений, М.,1961 (Итоги науки. Хим. науки, вып. 7);
Лосев И. П., Тростянская Е. Б., Химия синтети-
ческих полимеров, М., 1964; J. Appl. Chem., 1958, 8, pt 3, 186;
Кронин Е., в кн.; Химия и технология полимеров, сб.
1, №.. 1958. С. В. Виноградова.
ПЕПТОНОВЫЕ КИСЛОТЫ (альдопентоновые кис-
лоты) НООС(СНОН)зСН2ОН, мол. в. 166,13 — сте-
реоизомерные альдоновые кислоты с 5 углеродными
атомами в углеродной цепи молекулы. П. к. содержат
по 3 асимметрия, атома углерода в молекуле и обра-
зуют 8 стереоизомеров (4 пары D- и L-антиподов)
соон СООН соон соон
Z \ \ \
Н-С-ОН НО-С-Н Н-С-ОН НО-С-Н
н-с-он 1 н-с-он 1 НО-С-Н 1 НО-С-Н
н-с-он 1 н-с-он 1 н-с-он Н-С-ОН
СН20Н 1 СН2ОН 1 СН2ОН 1 СН2ОН
D-рибоновая D-арабоновая D -нейлоновая D-лик соко-
к-та к-та к-та вая к-та
Названия П. к. даны по альдопентозам, продуктом
окисления к-рых они являются. П. к. хорошо раство-
римы в воде, в р-рах едких щелочей, плохо — в боль-
шинстве органич. растворителей; дают химич. реак-
ции альдоновых к-т.
Т. пл. L-рибоновой к-ты 104—105°, L-арабоновой
И8—119°; [а]д (в воде); D-рибоновой к-ты-|-8,420; L-рибоновой
4-17,6°-*—4,6° (через 30 дней); D-арабоновой 4-18,65°-*4-48,6°
(через 2 дня); L-арабоновой—9,5°-*—41,7° (через 21 день);
L-ксилоновой —2,9° -* 4-20,1° (через 36 дней).
Для характеристики П. к. получают их бруциновые
соли и двойные соли с CdBr2. П. к. получают общими
методами, принятыми для альдоновых к-т, напр. окис-
лением пентоз. Образованием П. к. при окислении
пентоз пользуются для идентификации последних.
Лит.: Methods in carbohydrate chemistry, ed. R. L. Whist-
ler, M. L. Wolfrom, V. t, N. Y —L., 1962. Л. И. Линевич.
ПЕНЫ — дисперсные системы, образованные мно-
жеством ячеек — пузырьков газа (пара), разделенных
тонкими пленками жидкости. Обычно газ рассматри-
вается в П. как дисперсная фаза (2), а жидкость —
как дисперсионная непрерывная среда (1). Однако
дисперсность, определяемая удельной поверхностью
П., может быть выше либо у жидкой фазы (1), ук-рой
она равна 51г/уп либо у газовой фазы, где она состав-
ляет S12/v2 (здесь г, и г2 — объемы фаз, 512 — общая
поверхность раздела). Это зависит от отношения vi/vi
или кратности (т. е. «обильности») П.— объема П., по-
лучаемого из единицы объема жидкости; P=(f2+yi)/yi-
Когда крупные ячейки газа (в очень устойчивых П.)
разделены тенчайшими пленками жидкости, т. е.
т0 значение очень велико и может до-
стигать 200—600 и даже 1000. В таких «сухих» П. уд.
поверхность соответственно значительно выше у жид-
кой, чем у газовой фазы. Ячейки в таких П. представ-
ляют собой многогранники. Наоборот, в случае
«влажных» П. величина Р имеет порядок 1—10, а
ячейки П. — круглые пузырьки, разделенные про-
слойками жидкости значительной толщины. Такие П.
можно считать эмульсиями газа в жидкости. Следует
отметить, что предельно концентрированные эмульсии
жидкости (2) в другой жидкости (1), напр.углеводоро-
да в воде или воды в нефти, в к-рых эмульгированная
жидкость образует ячейки, разделенные сравнитель-
но тонкими пленками дисперсионной среды, надо рас-
сматривать как пенообразные системы (с п у м о и д ы).
Как и все вообще лиофобные дисперсные системы,
обладающие избытком свободной энергии, П. термоди-
намически неустойчивы (см. Коллоидные системы).
Для характеристики П. важны следующие их свойст-
ва: 1) т. наз.кинетич. устойчивость, т. е. время само-
произвольного разрушения столба П. на половину
начальной высоты (или объема); 2) наибольший дости-
жимый объем (высота столба) П.; 3) дисперсность.
Для получения П. используют один из двух про-
цессов: 1) диспергирование газа (воздуха) встряхи-
ванием сосуда, частично заполненного жидкостью,
либо интенсивным перемешиванием вращающимися
мешалками или, наконец, пропусканием газа через по-
ристую перегородку в слой жидкости (барботирова-
ние) — оба последних способа используют для обо-
гащения полезных ископаемых методом пенной фло-
тации; 2) выделение газа или пара в виде новой
дисперсной фазы при кипении жидкости либо из пере-
сыщенного р-ра, напр. в результате реакции, один из
продуктов к-рой газ:
1
NaHCO3+HCl=NaCl + H2O + CO2
1
2А1 + 2Са(ОН)2 + 2Н2О = 2СаНА1О3+ЗН3
1
СаН2 + 2Н,_ О = Са(О Н)2 + 2Н2
1
Zn + 2HCl = ZnCl2 + H2
Первой из этих реакций пользуются в пенных («хи-
мических») огнетушителях, второй — при изготовле-
нии газобетона.
П. не возникают в обычных чистых жидкостях, т. к.
при любых значениях поверхностной энергии пузырь-
ки, соприкасаясь друг с другом, очень быстро коа-
лесцируют или же лопаются под действием отрица-
тельного капиллярного давления, а также силы тяже-
сти. Стабилизация пузырьков и П. в целом в этих
условиях достигается резким повышением вязкости
жидкости путем понижения ее темп-ры с переходом,
как в случае стекол, в область ниже интервала размяг-
чения; на этом основано стеклодувное искусство (вы-
дувание крупных пузырьков — колб и др. заготовок)
и произ-во пеностекла. Самопроизвольное разрушение
столба П. выражается в стекании жидкости из пленок
верхнего слоя пузырьков в нижний слой, а затем в слой
жидкости, из к-рой образовалась П. Этот процесс,
наз. синерезисом П., ведет к тому, что коа-
лесценция пузырьков начинается в верхних слоях
(пленки нижних слоев сохраняют свою толщину).
На начальной стадии синерезиса П. становится более
«сухой». В открытых сосудах П. разрушаются, кроме
того, и вследствие испарения пленок пузырьков верх-
него слоя. Пузырьки в П. укрупняются также и вслед-
ствие диффузии газа через пленки из меньших пузырь-
ков (радиусом га) в более крупные (радиусом rb>ra)
под влиянием избыточного капиллярного давления
Др=2а(1/га—1/гй).
Для образования устойчивых, обильных и высоко-
дисперсных П. в данную жидкость вводятся в сравни-
тельно небольших количествах пенообразова-
тели (вспениватели), облегчающие диспергирование
газа в виде мелких пузырьков и повышающие устой-
чивость тонких пленок между пузырьками. Типич-
ными пенообразователями являются поверхностно-
активные вещества. Поскольку поверхностная актив-
ность особенно резко выражена на границе вода —
газ, то и действие пенообразователей лучше всего
проявляется в водных р-рах. Пенообразователи де-
лятся па 2 группы: 1) слабые пенообразователи, не
образующие структуры ни в адсорбционных слоях,
ни в объеме раствора, а лишь изменяющие поверх-
ностное натяжение жидкости на границах двусторон-
0 29 к.х.Э. т. з
899
ПЕНЫ—ПЕПТИДАЗЫ
900
жих пленок. Такие пенообразователи, концентрируясь
в поверхностных слоях пленки, способствуют возник-
новению местных разностей поверхностного натяжения
и, в соответствии с принципом Марангони — Гиббса,
замедляют стекание жидкости в наиболее тонких ее
слоях из-за возникновения противодействующего сте-
канию двухмерного давления (разности поверхност-
ных натяжений) вследствие уменьшения адсорбции в
утончающейся средней части пленки. Благодаря
этому замедляются дальнейшее утончение и разрыв
пленки. В этом случае наблюдается ярко выражен-
ный максимум устойчивости П. (от нескольких се-
кунд до минуты) в зависимости от концентрации пено-
образователя в соответствии с общей теорией поверх-
ностных явлений. Слабыми пенообразователями мо-
гут быть не только поверхностно-активные вещества
(растворимые в воде спирты, жирные к-ты, фенолы),
но и поверхностно-инактивные электролиты, создаю-
щие местные разности поверхностных натяжений.
Это имеет практич. значение и, в частности, требует
тщательной очистки воды в паровых котлах высокого
давления (возможность вспенивания и переброса
жидкости при интенсивном кипении).
2) Сильные пенообразователи, образующие в адсорб-
ционных слоях высоковязкие и прочные пространст-
венные структуры, сильно замедляющие утончение
и разрыв пленок. Устойчивость П. при этом непре-
рывно растет с увеличением концентрации пенообразо-
вателя по мере насыщения адсорбционного слоя и
достигает нескольких часов, суток н более. К силь-
ным пенообразователям относятся полуколлоиды ти-
па мыл (анионактивных, катионактивных и неионо-
генных) и красителей, а также поверхностно-ак-
тивные гидрофильные высокомолекулярные веще-
ства, белковые вещества, гликозиды (сапонин), тан-
ин ды.
Для еще большего повышения устойчивости П. к пе-
нообразователю добавляют загустители, по-
вышающие структурную вязкость жидкости в плен-
ках П., либо студнеобразующие высокомолекуляр-
ные вещества (альгинаты, белки, карбоксиметилцел-
люлоза), либо полимеризующиеся кристаллизациоп-
нотвердеющие вещества, отверждающие пленки П.
Именно так получают твердые пеноматериалы — пе-
нопласты, пенобетоны, пенорезины.
В тех случаях, когда образование устойчивых П.
препятствует нормальному ходу технологич. процесса
(в нек-рых пищевых произ-вах, в произ-ве антибио-
тиков, в химич. пром-сти, при эксплуатации паровых
котлов и бурении глубоких скважин), вводят малые до-
бавки (10~3—10 _ 3 %) пеногасителей — сильно
поверхностно-активных веществ, очень мало раство-
римых в воде, но быстро растекающихся по ее поверх-
ности в виде мономолекулярного слоя. Вытесняя пено-
образователи из поверхностного слоя пленки, пенога-
сители (средние гомологи спиртов, напр. октиловый
спирт, полиамиды жирных к-т, жидкие жирные к-ты
и их глицериды, пропиленгликоли и их гидрофобные
производные, силиконовые масла) на несколько по-
рядков понижают устойчивость П.
Малоустойчивые П., стабилизованные пенообразо-
вателями типа спиртов, применяют в процессах пен-
ной флотации для выноса прилипших к пузырькам
частиц ценного дисперсного продукта (минерала).
Устойчивость таких минерализованных П. значи-
тельно увеличивается вследствие образования «бро-
ни» из твердых частиц, прилипших к поверхности пу-
зырьков. Высокоустойчивые и обильные П. служат
наиболее эффективным средством борьбы с нефтяными
пожарами (противопожарные П.); их применяют
в произ-ве тепло- и звукоизоляционных строительных
материалов и в пищевой (кондитерской) пром-сти
(взбитые сливки, «молочная пена», белки, кремы, па-
стила); качество таких напитков, как пиво, квас, ши-
пучие вина, в значительной степени также опреде-
ляется их способностью к пенообразованию.
Вследствие концентрирования пенообразователей
в поверхностных слоях пленок пенообразование ис-
пользуется для фракционирования смесей поверх-
ностно-активных веществ (белковых веществ, жирных
и холевых — желчных кислот) или для удаления по-
верхностно-активных веществ из жидкости (вспени-
вание). Пенообразование может служить чувстви-
тельным аналитич. признаком: при определении жест-
кости воды конец ее титрования р-ром олеата натрия
обнаруживается по устойчивому вспениванию после
выпадения всех ионов кальция и магния в виде соот-
ветствующих мыл, нерастворимых в воде.
В природных видах большие количества устойчивой
П. образуются экстрактивными веществами растений
(гликозиды, танниды). Морская П. вызвана присут-
ствием в воде солей и экстрактивных веществ водо-
рослей.
Лит.: Шварц А., Перри Д ж., Берч Д at., По-
верхностноактивные вещества и моющие средства, пер. с
англ., М., 1960; Р е б и н д е р П. А., П о с п е л о в а К. А.,
в кн.: Клейтон В., Эмульсии, пер. с англ., М., 1950;
Bikerman J. J., Foams, N. Y., 1953; его же, Surface
chemistry, 2 ed., N. Y., 1958. П. А. Ребиндер.
ПЕПСИН — основной протеолитич. фермент же-
лудочного сока; относится к группе протеипаз; от-
крыт в 1836 Т. Шванном. П. получен в кристаллич. ви-
де Д. Нортропом в 1930, мол. вес 35 000; изоэлектрич.
точка находится ок. pH 1, что обусловлено высоким
содержанием в П. дикарбоновых аминокислот и при-
сутствием остатка фосфосерина. Молекула П. пред-
ставляет собой одиночную полипептидпую цепь при-
мерно из 340 аминокислотных остатков. Последова-
тельность аминокислот на N-концевом участке П.
свиньи: изолейцил-глицил-аспарагинил-аспарагинил-
гистидил-глутаминил-...; С-концевом участке ...— ва-
лил-аланин. В молекуле П.— три дисульфидных
мостика.
П. наиболее устойчив при pH 5—5,5; в кислой среде
происходит самопереваривание (аутолиз) П., при pH
выше 6 П. быстро инактивируется. П. гидролизует
белки до пептидов, хотя среди продуктов гидролиза
встречаются и аминокислоты. Гидролизу подвергаются
пептидные связи, образованные различными амино-
кислотными остатками. Помимо белков, П. расщеп-
ляет нек-рые низкомолекулярные пептиды и способен
катализировать реакцию транспептидацпи
(перенос аминокислотных остатков с одного пептида,
на другой). Оптимум действия П. находится ок.pH 2;
при pH 5 П. вызывает створаживание молока. Неак-
тивный предшественник П.— пепсиноген, вы-
рабатываемый клетками слизистой оболочки желудка,,
превращается в П. в присутствии соляной к-ты, со-
держащейся в желудочном соке. Процесс активации
протекает автокаталитически. При этом от N-koh-
цевого участка молекулы пепсиногена отщепляется
несколько пептидов с общим мол. весом ок. 8000.
П. получают высаливанием из автолизатов слизистой
оболочки желудка с последующей кристаллизацией из.
20%-ного спирта или из водного р-ра. В лаборатор-
ных условиях для получения чистого П. используют
хроматографию на аниопообмешюй целлюлозе. Для
определения П. применяются методы, основанные па
измерении его активности при расщеплении различ-
ных белковых субстратов. П. применяют в сыроваре-
нии и при лечении нек-рых заболеваний желудочно-
кишечного тракта.
Лит.: Н о р т р о п Д.,Кунптц М., X е р р и о т т Р.,
Кристаллические ферменты, пер. с англ., М., 1950; Белки, под.
ред. Г. Нейрата и К. Бэйли, пер. с англ., т. 3, ч. 2, М.,
1959; The enzymes, ed. Р. D. Boyer [a. o.J, v. 4^ 2 ed., N. Y.,
1960, p. A. 62. Л. Д. Локшина.
ПЕПТИДАЗЫ — см. Пептидгидролазы.
901
ПЕПТИДГИДРОЛАЗЫ
902
ПЕПТИДГИДРОЛАЗЫ (протеолитические фер-
менты)— ферменты, катализирующие гидролитич. рас-
щепление пептидных связей в белках и пепти-
дах:
.._HNCHRC0-NHCHRC0... + H20 —>-
—» ...hnchrcooh + h2nchrco...
Классификация, номенклатура и специфичность.
В соответствии с современной системой классификации
и номенклатуры (см. Номенклатура и классификация
ферментов) все ферменты, катализирующие гидро-
лиз пептидной связи, входят в класс гидролаз, обра-
зуя подкласс пептидгидролаз (шифр 3.4), к-рый со-
стоит из след, подподклассов:
а-А минопептид-аминоацидогидро-
л а з ы (аминопептидазы по старой номенклатуре
Гофмана — Остенгофа), расщепляющие крайние свя-
зи в пептидной цепи, расположенные рядом со свобод-
ными концевыми а-аминогруппами и не действующие
па связи, рядом с к-рыми находятся свободные кар-
боксильные группы. Эти ферменты отщепляют конпе-
вые аминокислотные остатки с N-конца пептидной
цепи (см. схему).
Эндопептидазы,
пептид пептидогидролазы
I I
N-консц I I
NH2CHCO—NHCHCO—NHCHCO —.. .—NHCHCO—NHCHCO—NHCHCOOH
Illi I I I
R | R R R R R
аминопептидазы,
а-аминопептид-
аминоацидогидролазы
карбоксилептидазы,
а-карбоксипептид-
аминоацидогидролаэы
экзопептидазы
К ним относятся такие ферменты, как лейцинамино-
пептидаза (шифрЗ. 4.1.1.) и аминотрипептидаза, рас-
щепляющая в трипептидах пептидную связь, бли-
жайшую к свободной а-аминогруппе, с образованием
свободной аминокислоты и дипептида:
H2NCHCO-NHCHCO-NHCHCOOH + Н20 —>
1:1 I
R ' R R
—>-H2NCHCOOH + H2NCHCO-NHCHCOOH
а-К арбоксипептид-аминоацидоги-
д р о л а з ы (карбоксипептидазы по старой но-
менклатуре), расщепляющие крайние связи в пептид-
ной цепи, расположенные рядом со свободными кон-
цевыми а-карбоксильными группами. Эти ферменты
последовательно отщепляют аминокислотные остатки
с С-копца пептидной цепи.
Поскольку амино- и карбоксипептидазы катализи-
руют отщепление концевых аминокислотных остатков
пептидной цепи, то их иногда объединяют под общим
названием экзопептидаз.
Дипептидгидролазы, гидролизующие ди-
пеитиды (дипептидазы). Для многих дипептидгидролаз
характерна исключительно высокая субстратная спе-
цифичность, напр. глицил-L-лейцин—гидролаза (три-
виальное наименование глициллейции-дипептидаза,
шифр 3.4.3.2.), выделенная из животных тканей,
гидролизует только глицил-Ь-лейцин и не действует
на какие-либо др. пептиды.
Пептид-пептидгидролазы, дейст-
вие к-рых направлено гл. обр. на внутренние связи
в пептидной цепи (эндопептидазы). Под действием этих
ферментов молекула белка или полипептида распа-
дается на более или менее крупные фрагменты. К ним
относятся ферменты желудочно-кишечного тракта
(пепсин, трипсин и химотрипсин), растительные
активные
С-конец
протеолитич. ферменты (папаин, химопапаин, фицин),
протеолитич. ферменты животных тканей (катепсины,
плазмин, тромбин и клостридиопептидазы — пре-
жнее название коллагеназы) и др. Ферменты этой под-
группы характеризуются значительно более широ-
кой субстратной специфичностью, чем, напр., дипеп-
тидазы, однако в их действии наблюдается изве-
стная избирательность. Так, напр., пепсин преим.
гидролизует пептидные связи, образованные остатками
ароматич. или дикарбоновых а-аминокислот. Трип-
син и химотрипсин предпочтительно расщепляют пеп-
тидные связи, в к-рых участвуют карбоксильные
группы L-аргинина или L-лизина. Многие ферменты
этой группы, напр. трипсин, химотрипсин, папаин и
др., катализируют расщепление не только пептидных,
но и сложноэфирных связей.
Все П. стереоспецифичны: они расщепляют пеп-
тидные связи, образованные остатками только L-амп-
нокислот, и не действуют на пептиды, построенные из
D-аминокислотных остатков. В нек-рых микроорга-
низмах обнаружены т. н. D-пептидазы, гидролизую-
щие пептиды, образованные из D-аминокислот, и не
в отношении L-пептидов.
Физико-химические свойства. Все П.—вы-
сокомолекулярные вещества белковой при-
роды. Многие П. выделены в кристаллич.
состоянии и их физико-химич. свойства под-
робно охарактеризованы (см. Карбокси пеп -
тидаза А, Лейцинаминопептидаза, Плаз-
мин, Пепсин, Папаин, Трипсин, Химотрип-
син и др.). Нек-рые пептид-пептидогидрола-
зы резко отличаются друг от друга опти-
мумом pH. Например, пепсин, секретируе-
мый вместе с желудочным соком, имеет оп-
тимум активности в интервале pH 1,5—2,5,
а трипсин и химотрипсин, функционирующие в слабо-
щелочной среде кишечника, имеют оптимум pH, близ-
кий 8,0.
Активный центр и механизм действия. По строению
активного центра и возможному механизму действия
П. можно разделить на 3 типа.
К первому типу относятся металлов и зимы,
т. е. ферменты, в состав активного центра к-рых вхо-
дят ионы металлов. К ним относится большинство
экзо-и дипептидаз. Наиболее часто в построении ак-
тивного центра этих П. принимают участие ионы Mg2 + ,
Mn2 + , Со2 + , Zn2 + , Fe2+и др. Прочность связи металла
с белковой частью фермента (апоферментом) может
быть различной. В нек-рых случаях, как, напр.,
в карбоксипептидазе, ион металла (Zn2+) связан с апо-
ферментом очень прочно и не может быть отделен диа-
лизом. В других случаях (лейцинаминопептидаза,
пек-рые дипептидазы) эта связь значительно менее
прочна и может нарушиться в процессе выделения
или очистки фермента. Активность ферментов этой
группы обычно угнетается агентами, связывающими
ионы металлов: этилендиаминотетрауксусной кислоты
(ЭДТА), о-фенаптролином, цианидами. Входящие в со-
став активного центра ноны металлов, по-видпмо-
му, принимают участие в образовании фермент-суб-
стратных комплексов и облегчают активацию суб-
страта.
Ко второму типу относятся такие ферменты, как
трипсин, химотрипсин, тромбин и пек-рые бактериаль-
ные П.; они характеризуются отсутствием в составе
активного центра к.-л. небелковых простетич. групп
или коферментов. Установлено, что в состав активного
центра этих ферментов входит остаток серина, при-
чем последовательность аминокислотных остатков
в этом участке полипептидной цепи у всех перечислен-
вых ферментов одинакова: аспарагиновая кислота-се-
рин-глицин. Гидроксильная группа серина, входя-
щего в состав каталитич. центра, характеризуется
29*
903
ПЕПТИДНАЯ СВЯЗЬ—ПЕПТИДЫ
904
высокой реакционноспособностью. Второй функцио-
нальной группой, принимающей участие в каталитич.
процессе, является имидазол остатка гистидина, ак-
тивирующий гидроксил серина, возможно, в резуль-
тате образования водородной связи. Строение актив-
ного центра и механизм действия П. этой группы весь-
ма сходны со структурой каталитич. центра и меха-
низмом действия эстераз, в частности холинэстеразы,
а также фосфоглюкомутазы.
Третий тип П. образуют ферменты, в составе актив-
ного центра к-рых не обнаружены ни ионы металлов,
ни последовательность аминокислотных остатков, ха-
рактерная для П. рассмотренного выше типа. Эти
ферменты утрачивают свою активность под дейст-
вием реагентов на SH-группы: ионов тяжелых метал-
лов, иодацетамида, п-хлорртутьбензоата и др. Таким
ферментом является папаин, в состав активного цент-
ра к-рого входят сульфгидрильная и ионизированная
карбоксильная группы.
Распространение, физиологическая роль и приме-
нение. П. широко распространены в животных и
растительных тканях и в микроорганизмах. П. желу-
дочно-кишечного тракта (пепсин, трипсин, химотрип-
син и др.) играют важную роль в процессе перевари-
вания белков, поступающих с пищей. Нек-рые П.
(тромбин, плазмин) имеют большое значение в систе-
ме свертывания крови, обеспечивая, с одной стороны,
образование кровяного сгустка (тромба), а с другой—
его растворение (лизис).
Препараты различных П. широко применяют в на-
учных исследованиях, медицине и нек-рых отраслях
пром-сти. Кристаллич. препараты папаина, пепсина и
трипсина используют для частичного расщепления
белковых молекул в процессе изучения последователь-
ности расположения в них аминокислотных остатков.
Препараты П. применяют для лечения нек-рых желу-
дочных заболеваний (пепсин), очистки поверхности
ран от некрозирующей ткани (трипсин, химотрипсин),
а также для предотвращения тромбообразования при
операциях. Препараты П. из поджелудочной железы
(панкреатин) применяют как лечебное средство, а
также при дублении кож. Реннин (сычужный фер-
мент) используют в сыроварении.
Лит.: Современные проблемы биофизики, т. 1, М., 1961,
с. 238; Труды V Международного биохимического конгресса.
, Симпозиум IV. Молекулярные основы действия и торможения
ферментов, М., 1962; Классификация и номенклатура фер-
ментов, М., 1962; The Enzymes, 2 ed., v. 4, N. Y., 1960.
В. Б. Спиричев.
ПЕПТИДНАЯ СВЯЗЬ — связь между углеродом
карбонильной и азотом имидной групп аминокислот-
ных остатков в белках и пептидах. С помощью П. с.
происходит образование основной (первичной) по-
I
R
Рис. 1. Схематическое изображение двух полипептпдных
цепей и водородных связей между ними (указаны пунк-
тиром).
лчпептидной структуры белков (см. рис. 1). Теплота
образования П. с. составляет 5,7 ккал/моль. П. с.
характеризуется сопряжением л-электронов азота,
углерода и кислорода , в результате че-
го она имеет характер частично двойной связи, что про-
является в уменьшении ее длины (1,32 А) по срав-
нению с длиной ординарной связи С—N (1,47 А).
Такое сопряжение обусловливает и копланарность
заместителей при атомах азота и углерода: во всех
изученных пептидных структурах группа СО—NH
оказалась плоской с точностью до нескольких граду-
сов (см. рис. 2). В белках найдена шранс-конфигура-
Рис. 2. Схематическое изображение пептидной группы
(межатомные расстояния даны I ангстремах).
ция пептидной группы, изображенная на рис. 2.
В белковых веществах многие СО- и NH-группы пеп-
тидных группировок соединены между собой водород-
ными связями (см. рис. 1). Основные полосы поглоще-
ния в ИК-области спектра находятся для NH-группы
при 3300 и 3080 ел»-1, а для СО-группы при 1660 см~'.
В УФ-области спектра электронным переходам в пеп-
тидной группе соответствуют полосы поглощения
при 1800—2300 А. В присутствии конц. к-т и щело-
чей, а также под влиянием специфич. протеолитич.
ферментов происходит гидролиз П. с. В результате
гидролиза всех П. с. в белках или пептидах образуются
свободные аминокислоты. Вопрос о механизме фер-
ментативного образования П. с. остается пока не
выясненным. Известно, что П. с. образуются в рибо-
сомах клеток при участии различных рибонуклеино-
вых к-т (рибосомальной, информационной и транспорт-
ной РНК). См. также Белки, Пептиды, Молекулярная
биология, Нуклеиновые кислоты.
Лит.: Современные проблемы биохимии, Сб. статей, пере-
вод, под ред. В. А. Энгельгардта, М., 1957, с. 54; 3 б а р с к и й
И. В., Усп. соврем, биологии, 1962, 54, № 3(6), 265.
ПЕПТИД-ПЕПТИДОГИДРОЛАЗЫВ-см.П/7:г5-
гидролазы.
ПЕПТИДЫ (полипептиды)— соединения, состоящие
из двух или более остатков аминокислот, соединен-
ных друг с другом амидными (пептидными) связями.
П., состоящие из двух аминокислотных остатков, на-
зывают дипептидами, из трех — трипептидами, из
четырех—тетрапептидами и т. д.; П. с числом аминокис-
лотных остатков, превышающим 6—10, наз. полипеп-
тидами. Аминокислотный остаток пептида, а-амино-
группа к-рого свободна, наз. N-конпевым. Амино-
кислота со свободной а-карбоксильной группой носит
название С-концевой. Название пептида составляет-
ся из названий, входящих в его состав аминокислот,
перечисляемых последовательно, начиная с N-koh-
цевой. При этом суффикс «ин» в названиях всех ами-
нокислот, кроме С-копцевой, заменяется суффиксом
«ил», например «аланил-пролил-фенилаланин». П., в
905
ПЕПТИДЫ
906
к рых имеются сложноэфирные связи с оксигруппами
аминокислот или оксикислот, наз. депсипепти-
д а м и, или О-пептидами. П.. замкнутые в цикл за
счет взаимодействия концевых а-аминных и а-кар-
боксильных групп, называют гомод етными
циклопептидами, а замкнутые за счет боковых
гидроксильных, карбоксильных, аминных или сульф-
гидрильных групп — гетеродетными цик-
л о п е п т и д а м и.
Низшие П. по свойствам близки к аминокислотам,
увеличение длины пептидной цепи обусловливает воз-
никновение вторичной структуры (см. Белки), во
многом определяющей свойства П. Высшие П. по
свойствам приближаются к белкам. Низшие П., как
правило,— кристаллич. вещества, разлагающиеся
при нагревании до 200—300°. Они хорошо раствори-
мы в воде, разб. к-тах и щелочах, почти нерастворимы
в органич. растворителях. П. обладают амфотерными
свойствами. Химич, реакции П. определяются их сво-
бодными функциональными группами и во многом
напоминают соответствующие реакции аминокислот.
П. дают биуретовую реакцию и нингидриновую реак-
цию. Дипептиды и их эфиры легко циклизуются с об-
разованием дикетопиперазинов. Для разделения сме-
сей П. применяют различные виды хроматографии
(в особенности ионообменную), электрофорез, проти-
воточное распределение.
Для выяснения строения П. в первую очередь уста-
навливают количественный состав исследуемого П.
С этой целью П. гидролизуют до аминокислот, содер-
жание к-рых в гидролизате определяют хроматогра-
фически, часто с использованием автоматик, методов
анализа. Затем проводят частичный ферментативный
или (реже) кислотный гидролиз П.для получения более
мелких его фрагментов. Эти небольшие П. разделяют
и определяют последовательность расположения в них
аминокислотных остатков. Для определения N-koh-
цевых аминокислот по Сейнджеру П. обрабатывают
в щелочной среде 1-фтор-2,4-динитробензолом, к-рый
реагирует со свободными аминогруппами:
R' R" И'"
I I I
F + H2N—СН—СО—NH—СН—СО—NH—СН—СООН—
O2N_—/^°2 R' R" R'"
—X^Z-NH-CH-CO-NH-CH-CO-NH-CH-COOH
Полученное производное П. подвергают гидролизу,
после чего идентифицируют Х-2,4-динитрофениламино-
кислоту, соответствующую N-концевой аминокислоте
исследуемого П. Предложенный Эдманом фенилтио-
гидантоиновый метод позволяет устанавливать после-
довательность расположения нескольких аминокислот,
примыкающих к аминному концу пептида. П. обраба-
тывают фенилизотиоцианатом, превращая его в произ-
водное фенилтиокарбамила. Последнее легко распа-
дается в кислой среде, отщепляя N-концевую амино-
кислоту в виде соответствующего фенилтиогидан-
тоина:
R' R" R'"
СеН5—N = C = S + NH2-CH—СО—NH-CH—СО-NH—СН—СООН
S R' R" R'"
II I I I
—> CeH-NH—С—NH—СН—СО—NH — СН—СО—NH—СН—СООН
R" R'"
—>R'-CH----С = О | |
| | + NH2—СН-СО—NH—СН—СООН
NH N
\ / \
С с„н5
II
S
Образовавшийся в результате отщепления фенил-
тиогидантоина укороченный П. может быть подверг-
нут новому циклу превращений, что позволит иден-
тифицировать вторую аминокислоту и т. д.
Для определения С-концевой аминокислоты П.
нагревают с безводным гидразином (гидразипо-
л и з). В результате расщепления пептидных связей
все аминокислоты, кроме С-концевой, превращаются
в гидразиды. После отделения гидразидов идентифи-
цируют С-концевую аминокислоту. Установление
С-концевой последовательности аминокислот дости-
гается при помощи протеолитич. фермевтов — кар-
боксипептидаз А и В, к-рые последовательно, одну за
другой, отщепляют аминокислоты от карбоксильного
конца пептидной цепи. Точно так же для определения
N-концевой последовательности используют фермент
аминопептидазу. После того как определен порядок
расположения аминокислот во фрагментах исследуе-
мого П., устанавливают общую последовательность
аминокислот. Для этого сравнивают строение отдель-
ных коротких пептидных фрагментов и находят среди
них П. с «перекрывающимися» последовательностями
аминокислотных остатков. Выписав такие последо-
вательности одну под другой, можно установить и
строение изучаемого П.
Для синтеза П. прибегают к активации реагирую-
щих карбоксильных или (значительно реже) аминных
групп и временной защите других функциональных
групп аминокислот или П., участие к-рых в реакции
нежелательно. После окончания синтеза защитные
группы удаляют. Особенно трудным является предот-
вращение рацемизации аминокислот в ходе пептид-
ного синтеза и сопутствующих операций.
В качестве производных аминокислот и пептидов,
содержащих активированную карбоксильную группу,
чаще всего используют азиды, n-нитрофениловые эфи-
ры, реже — смешанные ангидриды угольной к-ты.
При синтезе П. с применением дициклогексилкарбо-
диимида, по-видимому, также происходит активация
карбоксильной группы. Аминогруппу чаще всего за-
щищают карбобепзокси-остатком (CSHSCH2O—СО—),
к-рый легко удаляется гидрогенолизом над палладием
или действием НВгв ледяной СНзСООН. Используют
также n-толуолсульфонильную и трет-бутоксикар-
бонильную защиту (удаляются, соответственно, дей-
ствием натрия в жидком аммиаке и слабых к-т). Для
защиты карбоксильной группы используют метило-
вые эфиры (для удаления прибегают к омылению)
или бензиловые эфиры (защита может быть снята гид-
рогенолизом). При синтезе П., содержащих допол-
нительные функциональные группы, применяют спе-
циальные способы их временного блокирования.
Современные методы синтеза П. позволили получить
почти все физиологически активные П. природного
происхождения, вплоть до 20-членных, и синтезиро-
вать их многочисленные аналоги.
Большое количество П. образуется при неполном
гидролизе белковых веществ. В организмах животных
П. образуются при ферментативном расщеплении
белков (автолиз тканей, действие бактерий, пищева-
рение). Однако вследствие быстрого последующего
гидролиза такие П. присутствуют в небольших
количествах. КП., выделенным из жидкостей и
—> тканей организмов, относятся глютатион, карно-
+ зин, ансерин. Среди биологически активных П.
н , встречаются гормоны, напр. вазопрессин, оксито-
цин, адренокортикотропный гормон, а- п 0-мела-
нотропные гормоны, глюкагон и др. Известно большое
число П.—антибиотиков-полипептидов, наиболее рас-
пространенными из к-рых являются циклопептиды,
построенные из L- и D-аминокислот (грамицидин Дю-
бо, тироцидины, грамицидины С и J, полимиксины,
бацитрацины, актиномицины и др.). П. или их произ-
водными являются нек-рые витамины и факторы роста
растений и микроорганизмов (пантотеновая кислота,
907
ПЕПТИЗАЦИЯ—ПЕРВЫЙ ЗАКОН ТЕРМОДИНАМИКИ
908
фолиевая кислота, стрепогенин и др.); см. также
Аминокислоты, Белки. Пептидная связь.
Пит.: Кочетков Н. К., Торгов И. В., Б о т в и -
и и к М. М., Химия природных соединений, М., 1961, с. 486;
Кнунянц И. Л., П е р в о в а Е. Я., Усп. химии, 1955,
24 вып. 6, 641; Шемякин М. М., там же, 1962, 31, вып. 3,
269; Шемякин М. М. [и др.], Химия антибиотиков, т. 2,
М., 1961, с. 1057; Goodman М., Kenner G. W., Ad-
vances in protein chemistry, 1957, 12, 465; Hofmann K.,
Ann. Rev. Biochemistry, 1962, 31, 213; Bamford С. H.,
Elliott A., H a n b у W. E., Synthetic polypeptides,
N. Y., 1956. В. О. Шпикитер.
ПЕПТИЗАЦИЯ — расщепление агрегатов частиц
в коллоидных осадках, гелях или суспензиях на пер-
вичные частицы. Й. возникает под действием жидкой
дисперсионной среды, напр. воды, или добавленных
к ней адсорбирующихся веществ — пептизато-
р о в; адсорбционные силы, связывающие коллоидные
частицы с молекулами или ионами среды или пепти-
затора, преодолевают силы сцепления частиц дисперс-
ной фазы друг с другом в агрегат (коагуляционные
силы); образующиеся на поверхности частиц адсорб-
ционно-сольватные оболочки или, диффузные двой-
ные слои ионов препятствуют сближению частиц, а сле-
довательно, и их коагуляции. В случае достаточной
лиофильности, т. е. адсорбционного сродства среды
и дисперсной фазы, для П. достаточно броуновского
движения самих частиц. Так, бентонитовые глины,
в к-рых ионы катионпо-обменного комплекса в доста-
точной степени замещены натрием, самопроизвольно
распускаются в воде — растворяются до коллоидной
дисперсности. Классич. примером П. в присутствии
пептизатора является П. коагулята гидроокиси же-
леза в воде малыми добавками ионов Fe3+ (напр.,
в виде FeClg), к-рые и способствуют развитию диффуз-
ного двойного слоя на поверхностях первичных ча-
стиц. Если не вся поверхность частиц, образующихся
при П., стабилизована адсорбционно-сольватными обо-
лочками, то П. благоприятствует развитию коагуля-
ционной пространственной сетки геля из частиц,
свободно участвующих в броуновском движении;
при этом наблюдается переход от компактной коагу-
ляции с образованием осадка к сплошному структу-
рированию — гелеобразованию. Зависимость прочно-
сти Рт образующейся пространственной структуры от
концентрации С пептизатора выражается в общем
виде кривой с максимумом. Начальный участок подъе-
ма прочности до максимума соответствует резкому
увеличению числа частиц при П.; дальнейшее падение
прочности соответствует «разжижению» структуры
геля — переходу геля в золь.
П. — один из методов получения коллоидных р-ров;
имеет большое значение в технике при получении вы-
сокодисперспых суспензий глин и других материалов.
Лит.: Песков Н. П., Физико-химические основы кол-
лоидной науки, 2 изд., М.— Л., 1934; Ребиндер П. А.,
Физико-химические исследования процессов деформации твер-
дых тел, в кн.: Юбилейный сборник посвященный тридцатиле-
тию Великой Октябрьской социалистической революции,
ч. 1 , М.— Л., 1947. П. А. Ребиндер.
ПЕР — приставка к наименованию хим. соедине-
ния, означающая: 1) наличие в молекуле данного со-
единения атома, находящегося в состоянии высшей
степени окисления (перманганат КМпО4, перхлорат
КС1О4); 2) наличие перекисной группировки в мо-
лекуле (персульфат Кг82О8, пербензойная кислота
С6НбСО—О—ОН); 3) полное замещение атомов водоро-
да в органич. соединении какими-либо другими атома-
ми (наир., перхлорбензол С6С16, перфторгептан C,F16);
4) исчерпывающее гидрирование кратных связей в
ароматич. соединении (пергидрофенантрен).
Л. С. Герман.
ПЕРБОРАТЫ — см. Пероксобораты.
ПЕРВИТИН (адипекс, амфедроксин, дезамин, дезок-
сиэфедрин, гидрохлорид П-1-фенил-2-метиламинопро-
пана) C8H5CH2CH(CH3)NHCH3-HC1, мол. в. 185,70-
белый кристаллический порошок горького вкуса; |
т. пл. 170—172°; хорошо растворим в воде и спирте,
нерастворим в эфире. При добавлении к подкислен-
ному HNO3 водному р-ру П. AgNO3 выпадает творо-
жистый осадок AgCl; при нагревании до кипения
водного р-ра П., смешанного с конц. р-ром NaOH,
выделяются пары, к-рые окрашивают лакмусовую бу-
мажку в синий цвет. Для количественного определе-
ния навеску П. растворяют в НгО, прибавляют р-р
бромфенолового синего и СНзСООН до перехода фио-
летовой окраски р-ра в зеленовато-желтую; получен-
ный р-р титруют р-ром AgNO3, пока осадок AgCl и
жидкость не окрасятся в фиолетовый цвет.
П. может быть получен из L-эфедрина, к-рый вос-
станавливают HJ в присутствии красного фосфора:
HJ+P NaOH
СвН3-СН-СНСН3------->- C6H3-CH2CHCH.HJ----->
I I -Н2О I
ОН NHCH3 NHCH„
НС1
—>-СбН3-СНа-СНСН3—> п
NHCH3
П„ являясь N-метильным производным фенамина,—
активный стимулятор центральной нервной системы,
по действию близок к фенамину, но обладает более
выраженной фармакология, активностью и более ток-
сичен. П. применяют при депрессивных состояниях,
нарколепсии, отравлении наркотиками, для повыше-
ния физич. и умственной работоспособности.
Лит.: Машковский М. Д., Лекарственные средства,
4 изд., М., 1960. В. А. Засосов.
ПЕРВЫЙ ЗАКОН ТЕРМОДИНАМИКИ •— закон
сохранения энергии в применении к тепловым явле-
ниям (см. Сохранения энергии закон). Его иначе на-
зывают первым началом (или первым принципом)
термодинамики. П. з. т. был открыт в результате уста-
новления эквивалентности теплоты и работы теорети-
чески Ю. Р. Майером (1842) и экспериментально Дж.
Джоулем (1843). По современным данным, одна
15-градусная калория равна 4,1855 джоулей (между-
народная калория равна, по определению, 4,1868 дж,
термохимия, калория, также по определению, равна
4,1840 дж).
Согласно П. з. т., при изменении состояния систе-
мы тел сумма количеств сообщенного системе тепла
и затраченной работы, выраженных в одинаковых
единицах (напр., в джоулях), зависит только от на-
чального и конечного состояний системы, но не от пу-
ти перехода от начального к конечному состоянию.
Эта сумма определяет увеличение внутренней энергии
системы U. Обозначим сообщенное тепло через Q,
полученную работу (равную затраченной работе с
противоположным знаком) через А, тогда:
ДС7 = (?-А (1)
Здесь Д?/=£Л,—Ц\, причем Б, и U,— внутренняя
энергия в конечном и в начальном состояниях, соот-
ветственно.
В отличие от величины U, являющейся функцией
состояния системы, количество работы и количество
теплоты, каждое в отдельности, зависят не только от
начального и конечного состояний, но также и от
процесса, т. е. от той последовательности состояний,
в результате прохождения к-рой система приходит
в конечное состояние.
Для элементарного (т. е. связанного с бесконечно
малыми изменениями термодипамич. параметров) про-
цесса математич. формулировка П. з. т. принимает
вид:
6Q = dl7 + 6A (2)
Здесь dU — полный дифференциал внутренней энер-
гии U, а величины 6Q и 6А — лишь бесконечно малые
количества сообщенной теплоты и выполненной сис-
темой работы. Эти две величины нельзя рассматривать
909
ПЕРВЫЙ ЗАКОН ТЕРМОДИНАМИКИ—ПЕРЕАМИНИРОВАНИЕ
910
как дифференциалы функций состояния, но разность
их 6Q — bA=dU является полным дифференциалом.
Справедливость П. з. т., подтвержденная опытной
проверкой многочисленных его следствий, приводит
к выводу о неосуществимости вечного двигателя пер-
вого рода, т. е. такой машины, которая увеличивала
бы количество энергии в изолированной системе.
Понятие о работе, выполняемой той или иной
системой, возникло раньше всего в механике. С точки
зрения закона сохранения энергии, работа является
формой передачи энергии от той системы, к-рая вы-
полняет работу, той системе, над к-рой эта работа
выполнена. Для бесконечно малого изменения состоя-
ния тела количество работы 6А может быть представ-
лено дифференциальным выражением:
k
6A-p'dv + '^j Xjdxi (3)
где р'— внешнее давление, v — объем тела, X,-— обоб-
щенные силы и dx. — бесконечно малые изменения
параметров, определяющих состояние. Уравнения та-
кого вида наз. уравнениями Пфаффа.
В отличие от работы, другая форма передачи
энергии от одной системы к другой — теплота — не
может быть представлена в виде суммы произведений
обобщенных сил на малые изменения соответствую-
щих термодинамич. параметров. С точки зрения ста-
тистич. физики, теплота проявляется лишь в виде пере-
носа энергии от одного тела к другому в результате
беспорядочных молекулярных столкновений на гра-
нице соприкосновения тел или испускания и погло-
щения светового излучения. Т. обр., под количеством
теплоты Q следует понимать ту часть энергии, к-рая
передана от одного тела к другому в форме теплоты.
Для многих процессов количество теплоты может быть
представлено с помощью теплоемкости С тела:
6Q = Cdi
где dt — изменение темп-ры рассматриваемого тела.
Наряду с пониманием теплоты как формы переда-
чи энергии встречается представление о теплоте как
об особой форме энергии, именно тепловой энергии.
Подобное толкование теплоты как особой формы
энергии противоречит установленной в термодинами-
ке эквивалентности теплоты и работы, коль скоро
последнюю нельзя рассматривать в виде особой формы
энергии.
Для элементарного равновесного (обратимого)
процесса внешнее давление равно внутреннему дав-
лению р, и П. з. т. можно придать вид дифференци-
ального соотношения:
k
6Q=dU + pdi; + 2 X;dXi (t)
i=l
Из теории дифференциальных соотношений Пфаффа
следует, что в общем случае при наличии трех или
большего числа слагаемых они не допускают интег-
рирующего делителя, т. е. такой величины, при деле-
нии на к-рую правая часть равенства Пфаффа стала
бы дифференциалом яек-рой функции, вообще говоря,
всех аргументов. Однако и в этом случае на основа-
нии одного лишь П. з. т. нельзя утверждать, что ин-
тегрирующий делитель будет функцией только темпе-
ратуры тела, а ие какой-либо иной функцией U и v.
Согласно второму закону термодинамики, для правой
части равенства (4) всегда существует интегрирующий
делитель, являющийся функцией только темп-ры.
По определению абсолютной темп-ры Т, этот дели-
тель равен Т. Следовательно, для равновесного про-
цесса 6QjT=^dS, где S — функция состояния, назы-
ваемая энтропией. Первый и второй законы являют-
ся основой термодинамики.
Одной из первых задач, решаемых при помощи
П. з. т., является разбор свойств равновесных поли-
тропных процессов, выполняемых тем или иным веще-
ством. Процесс наз. политропным, если система, его
выполняющая, во все время процесса подчиняется
соотношению py"=const, в к-ром п — показатель
политропы. Напр., для идеального газа политропными
являются процессы: изохорный (r=const), изобарный
(р=const), адиабатный (дQ=0), изотермический (Т=
= const) и др. Главные области применения П.з. т. объ-
единяются в термохимии, к к-рой относится изучение
теплот различных фпзико-химич. и химич. процессов
(см. Теплота сгорания, Теплота образования и др.),
тепловых эффектов реакций и их зависимости от
темп-ры (см. также Гесса закон).
В отличие от закона сохранения энергии, имеюще-
го всеобщее значение, П. з. т. как основной закон тер-
модинамики относится только к макроскопия, систе-
мам, т. е. системам, содержащим достаточно большое
количество вещества, для к-рых имеют смысл понятия
темп-ры, давления и отдаваемой или поглощаемой
теплоты.
Лит.: Карапетьянц М. X., Химическая термодина-
мика, 2 изд., М.— Л., 1953; А к о п я II А. А., Общая термо-
динамика, М.— Л., 1955; Киреев В. А. Курс физической
химии, 2 изд., М., 1956; Гуггеигейм Э. А., Современная
термодинамика, изложенная по методу У. Гиббса, пер. с англ.,
М.— Л., 1941; Ван-дер Ваальс И. Д., К о н с-
т а м м Ф., Курс термостатики, пер. с нем., ч. 1—2, М., 1936;
Кричевский И. Р., Понятия и основы термодинамики,
М., 1962; Эпштейн П, С., Курс термодинамики, пер.
с англ., М.— Л., 1948. К. В. Астахов.
ПЕРЕАМИНИРОВАНИЕ (трансаминирование) —
ферментативная реакция обратимого переноса ами-
ногрупп между амино- и кетокислотами без выделения
свободного аммиака:
R R' R
I I I
h2n—сн—СООН + О=С—СООН Т~» О=С—СООН+
R'
I
+ h2n—сн—соон
II. в значительной мере определяет направление
ряда реакций азотистого обмена и обмена углеводов
и жиров, являясь поэтому одним из основных факто-
ров химич. интеграции процессов обмена веществ
на уровне клетки. П. играет важнейшую роль в про-
цессе обмена азотистых соединений в тканях живот-
ных, растений, микроорганизмов, будучи основным
путем как биосинтеза, так и распада аминокислот
в биологич. системах.
Ферментативное П.было открыто в 1937 А. Е. Браун-
штейном и М. Г. Крицман, обнаружившими перенос
азота а-аминогрупп многих аминокислот на а-оксо-
глутаровую к-ту с образованием L-глутаминовой к-ты
или на щавелевоуксусную к-ту с образованием L-ac-
парагиновой к-ты в сердечной мышце голубя.
Наибольшее значение в процессах превращения
азотистых соединений имеют:
1. L-a-амипокислота +а-оксоглутаровая к-та izta-кетокис-
лота-|-Ь-глутаминовая к-та.
2. у-аминомасляная к-та-|-а-оксоглутаровая к-тз^пепол-
пый альдегид янтарной к-ты -pL-глутаминоаая к-та.
L-орнитин -|-а-кетокислота * у-пеполный альдегид L-глу-
таминовой к-ты + L-a-аминокислота.
3. L-глутамин + a-кетокислота —- а-кетоглутарамовая
к-та L L-a-аминокислота.
L-аспарагин 4- а-кетокислота^з? a-оксосукцинамовая к-та 4-
+L-a-аминокислота.
4. L-a-аминомонокарбоновая к-та-ра-кетомонокарбоповая
к-та а-кеТомонокарбоновая к-та+L-a-аминомонокарбоно-
вая к-та.
У нек-рых микроорганизмов обнаружены анало-
гичные реакциям типа (1) ферментативные процессы
обратимого переноса аминогрупп между D-a-амино-
кислотами и a-кетоглутаровой к-той. По современным
представлениям, реакции типа (1) в сочетании с реак-
циями типа (4) играют центральную роль в биосипте-
911
ПЕРЕ АМИНИРОВАНИЕ—ПЕРЕГОНКА НЕФТИ
912
зе аминокислот в биологич. системах (процесс транс-
реаминирования).
Нек-рые исследователи допускают также возмож-
ность прямого аминирования кетокислот с образова-
нием аминокислот (напр., аланина из пировиноград-
ной к-ты) в биологич. системах, но биохимич. меха-
низм этих реакций еще недостаточно изучен. Дезами-
нирование аминокислот с образованием конечного
продукта обмена белков — аммиака—происходит в два
этапа (трансдезаминирование), путем сочетания реак-
ций П, типа (1) и дальнейшего окислительного дез-
аминирования глутаминовой к-ты под влиянием спе-
цифич. глутаматдегидрогеназы. Реакции П. типов
(1—4) имеют важнейшее значение в процессах обез-
вреживания аммиака и биосинтеза конечных продук-
тов азотистого обмена (мочевины, мочевой к-ты)
в организме животных и человека. Реакциям П. типа
(2) принадлежит важная роль в регуляции функций
центральной нервной системы.
Реакции П. катализируются специфич. фермен-
тами — трансаминазами (аминоферазами), содержа-
щими в качестве кофермента пиридоксалъ-5-фосфат
(см. Пиридоксалевые ферменты). В биологич. химии,
помимо П., известны след, ферментативные реакции:
а) переамидирования, т. е. обратимого
переноса амидной группы между молекулами, напри-
мер биосинтез аспарагина (IV) из глутамина (I) и ас-
парагиновой к-ты (II) с образованием глутаминовой
к-ты (III):
и
С—NH3 СООН
I I
(СН2)2 + СН2
I /
ноос-сн—nh2 hooc-ch-nh2
(I) (II)
о
II
соон с—nh2
< (СН2)2 4- (!'Н2
ноос-сн—nh2 НООС—СН—NH3
(III) (IV)
б) переамидинирования, т. е. обратимого
переноса амидиновой группировки между молекула-
ми, напр. биосинтез гуанидинуксусной к-ты (VII)
из аргинина (V) и глицина (VI) с освобождением ор-
нитина (VIII):
NH
и
nh-c-nh2
(СН2)3 + H2N-CH2-COOHj=t
I (VI)
H2N—сн-соон ' 1
(V)
NH3
I
C = NH +
I
HN—CH2—COOH
(VII)
nh2
(CH2)3
1
H2N—CH—COOH
(VIII)
Значение этих реакций в обмене веществ мало по
сравнению с П.
Нарушения нормального течения реакций П. обна-
ружены при ряде патология, состояний. Результаты
определений содержания трансаминаз в крови исполь-
зуются для диагностики нек-рых заболеваний (ин-
фаркт миокарда, заболевания печени).
Лит.: Браунштейн А. Е., Биохимия аминокислот-
ного обмена, М., 1949; Майстер А., Обмен аминокислот,
пер. с англ., М., 1961; Браунштейн А. Е., Шемя-
кин М. М., Биохимия, 1953, 18, вып. 4, 393; Браун-
штейн А. Е., Главные пути ассимиляции и диссимиляции
азота у животных, М., 1957. В. 3. Горкин.
ПЕРЕГОНКА НЕФТИ — процесс термич. разде-
ления нефти на ее составные части или фракции с по-
лучением при этом бензина, лигроина, керосина, реак-
тивного и дизельного топлив, топочного мазута и ко-
тельного топлива. Остаток после такой перегонки неф-
ти — мазут — используют так же, как сырье для
произ-ва смазочных масел (дистиллятных и остаточ-
ных), парафина, гудрона, кокса и др. нефтепродуктов.
Дистилляты и остатки перегонки нефтей применяют
также в качестве сырья для вторичных процессов пе-
реработки нефти — термич.
и каталитич. крекинга, ри-
форминга, пиролиза, дегид-
рогенизации и др.
В лабораторных условиях
П. н. производят в специ-
альных аппаратах, снабжен-
ных р ектификационными ко-
лоннами , напр. Ц И АТ ИМ-58
(рис. 1). Фракции,выкипаю-
щие до 200°, отбираются
при атмосферном давлении,
от 200 до 320°— при оста-
точном давлении 10 мм рт.
ст., остальные при 1 мм
рт. ст.
Разделение нефти на фрак-
ции (дистилляты) в промыш-
ленных условиях осуществ-
ляется на непрерывно дейст-
вующих трубчатых установ-
Рис. 1. Схема аппарата
ЦИАТИМ-58; 1 —колонка; 2 —
перегонный куб; 3 — конденса-
тор; 4 — электропечь; 5 — ва-
куумный приемник; 6 — буфер-
ная емкость; 7 — ловушки; S — ртутные вакуумметры; 9 —
дифференциальный манометр; 10 — вакуумный масляный на-
сос; 11 —термопары.
ках,прототипом к-рых являются лабораторные аппара-
ты однократного испарения ОII (рис. 2). Основными
частями этого аппарата являются
трубчатый нагреватель, испари-
тель, конденсатор и холодильники.
Однократное испарение нефти про-
водится при нарастающих темп-рах
через каждые 25°, начиная с 250° и
кончая 400°. Определение доли от-
гона при заданной темп-ре равно-
холодильник; 6 — ртутный вакуумметр; 7 — подогреватель ос-
татка; 8 — отвод дистиллята; 9 — отвод остатка; 10 — ваку-
умный приемник; II —ловушка; 12 — буферная емкость; 13 —
термопары.
веского испарения ведется после того,как будут достиг-
нуты адиабатич. условия, т. е. когда темп-ры жидкой
и паровой фаз сравняются. У дистиллятов и остатков
913
ПЕРЕГОНКА НЕФТИ
914
•с
420
380
340
300
260
220
180
140
’°°0 10 20 30 40 50 60 70 80 90 100
Отгон, вое %
d 4
1-0,960
~ 0,940
0,920
0,900
0,880
- 0,860
0,840
- 0,820
10,800
Мол. вес
> - & ,
, 400 Л
0,948
Рис. 3. График для определения
качества отгона и остатка в усло-
виях однократного испарения (ОИ).
определяют плотность, мол. вес и др. свойства. По най-
денным величинам строят кривые разгонки методом 011
(рис. 3). По числу ступеней испарения сырья разли-
чают трубчатые уста-
новки одно-, двух-, трех-
и многократного испа-
рения нефтей. Произво-
дительность современ-
ных трубчатых устано-
вок для разделения неф-
ти на фракции превы-
шает 6 млн. т в год. На
одноступенчатой уста-
новке нефть нагревает-
ся дотакой темп-ры, при
к-рой однократным ис-
парением и ректифика-
цией паров из нефти
получают всю гамму ди-
стиллятов, от легчайшего бензинового до высоковяз-
кого цилиндрового. Остатком перегонки является
гудрон. Установки двукратного испарения, именуе-
мые атмосферными трубчатками (АТ), состоят из 2
ректификационных колонн: предварительного испа-
рения и основной. Нефть после подогрева в теплооб-
менниках и отстоя в водогрязеотделителях поступает
в испарительную колонну. Головным продуктом (рек-
тификатом) являются легкий бензин и пары воды.
Отбензиненная нефть снизу колонны насосом подается
через трубчатую печь в основную колонну, к-рая
обычно снабжена отпарными секциями: внутренними
или внешними — боковыми. В них дистилляты (бо-
ковые погоны) обрабатываются водяным паром, ос-
вобождаясь таким образом от легкокипящих фракций,
понижающих темп-ру вспышки нефтепродуктов.
Наряду с АТ, остатком перегонки к-рых является
мазут, существуют атмосферно-вакуумные трубчатые
установки АВТ. Их отличительной особенностью яв-
ляется наличие вакуумной ступени, где осуществляет-
ся вакуумная перегонка мазута до гудрона и отбор
масляных дистиллятов. В зависимости от путей исполь-
зования масляных дистиллятов различают АВТ ма-
сляные или топлив-
ные. Технология, ре-
жим АВТ в основном
зависит от природы пе-
регоняемых нефтей и
ассортимента получае-
мых нефтяных фрак-
ций. На рис. 4 приве-
дена принципиальная
схема АВТ, перераба-
тывающей 6 млн. т
нефти в год. Установка
Рис. 4. Схема атмосфер-
но-вакуумной трубчатой
установки АВТ: 1 — элек-
тродегидраторы; 2 — От-
бензинивающая колонна;
3 — стабилизатор; t —
очистка легкого бензина;
5 — печь атмосферной колонны; 6 — атмосферная колонна;
7 — секция тяжелого лигроина; 8 — секция керосина; 9 — сек-
ция легкого газойля; 10 — очистка тяжелого лигроина;
11 — печь вакуумной колонны; 12 — вакуумная колонна.
работает в комбинации с электрообезвоживающей и
обессоливающей установкой (ЭлОУ), где нефть подвер-
гается обезвоживанию и обессоливанию в 4 горизон-
тальных электродегидраторах 1. Далее нефть подается
в отбензинивающую колонну 2. Ее головным продук-
том является бензин, направляемый на стабилизацию.
Отбензиненная нефть через атмосферную печь 5 по-
дается в атмосферную колонну 6, оснащенную трех-
секционной отпарной колонной (7, 8, 9). Газообразные
углеводороды со стабилизационной колонны 3 на-
правляются на газофракционирующую установку
(ГФУ), где смесь газов разделяется на индивидуаль-
ные углеводороды. Головным продуктом атмосферной
колонны является легкий лигроин (105—150°), ис-
пользуемый после гидроочистки в качестве сырья для
каталитич. риформинга. Боковыми погонами атмо-
сферной колонны, отбираемыми через отпарную ко-
лонну, являются тяжелый лигроин (фракция 150—
205°), керосиновый дистиллят и легкий газойль, ис-
пользуемые после гидроочистки соответственно как
компонент реактивного топлива, осветительный ке-
росин и дизельное топливо. Мазут снизу атмосфер-
ной колонны, пройдя вакуумную печь 11, поступает
в вакуумную колонну 12. Ее головным продуктом яв-
ляются перманентные газы (продукты разложения),
отсасываемые вакуумсоздающей аппаратурой. Бо-
ковым погоном вакуумной колонны является тяжелый
вакуумный газойль, используемый как сырье для ка-
талитич. крекинга. Остаток снизу колонны — гуд-
роп —направляется на установку коксования. Вверху
вакуумной колонны поддерживается темп-ра 70—75°
(за счет циркуляционного орошения), благодаря чему
основная масса углеводородных паров конденсирует-
ся. Колонна снабжена высокоэффективной вакуум-
создающей аппаратурой, обеспечивающей в эвапо-
рационной зоне колонны остаточное давление 15—
17 мм рт. ст., а в верхней части 5 мм рт. ст. Осо-
бенностью вакуумсоздающей аппаратуры является
система поверхностных конденсаторов и эжекторов
(вместо барометрич. конденсатора). Несконденсиро-
ванные газы отсасываются параллельно работающими
паровыми трехступенчатыми эжекторами, а конден-
сат — мощными вакуум-насосами. Применение та-
кой системы способствует созданию более высокого
вакуума в колонне и избавляет от огромных коли-
честв загрязняющих водоемы стоков воды. Суммар-
ная тепловая мощность печей 90 млн. ккал/час. В топ-
ках и форсунках печей можно сжигать жидкое, газо-
образное и твердое (порошковый кокс) топлива. В со-
став АВТ входит также секция очистки легкого бензи-
на и тяжелого лигроина от меркаптанов; очистка про-
изводится смесью, состоящей из гидроокиси калия и
изомаслянокислого калия (раствор «солютайзер»).
Почти полное отсутствие воды и солей в нефти
(после ЭЛОУ), применение антикоррозионных добавок
к сырью и аммиака (в верхней секции атмосферной ко-
лонны), коррозионноустойчивых металлов и сплавов,
большой запас прочности в аппаратуре обеспечивают
высокую надежность работы АВТ. Расходные показа-
тели на перегонку 1 т нефти на АВТ: воды охлаждаю-
щей 7,85 т/час; пара давлением 2,7 атм 0,059 т/час;
пара давлением 12 атм 0,013 т/час; электроэнергии
2,0 квт/час; топлива (кг/час): 16,3 газа, 16,5 жидкого-
топлива и 10 кокса (50%). Выходы основных продук-
тов при переработке тяжелой (плотн. 0,92) сернистой
(3,5% серы) нефти составляют (вес. %): газ сухой 1;
бензин легкий 8; лигроин легкий 8; лигроин тяжелый
2,5; керосин 8,5; газойль легкий 9; вакуумный тя-
желый газойль 30,5 и гудрон 32,5.
Экономия, показатели работы АВТ зависят от ее
производительности: чем она больше, тем ниже уд.
капитальные и эксплуатационные затраты, а также
удельный расход металла на тонну перегоняемой
нефти; ниже приводятся нек-рые показатели рабо-
ты АВТ:
Производительность, млн-т!год .
Капит. затраты, тыс. рублей . . .
Затраты металла, кг на 1 т нефти
Эксплуатационные расходы, рублей
на 1 т нефти ..................
Занимаемая площадь, м1.........
3
4 060
1,17
0,743
28 800
2 507
1,51
0,775
21 376
1
1 721
1 .85
0,841
19 320
915
ПЕРЕГРУППИРОВКА АЛЛИЛЬНАЯ
916
При одноступенчатой вакуумной перегонке мазута в
гудроне остается до 40% и выше вязких масляных дис-
тиллятов, к-рые с успехом могут быть применены для
произ-ва высоковязких смазочных масел или для ката-
литич. крекинга. В таких случаях пользуются двух-
ступенчатой вакуумной перегонкой мазута по схеме,
изображенной на рис. 5. Особенностью схемы являет-
Рис. 5. Трубчатая установка с двухступенчатой вакуумной
перегонкой мазута: 1 — колонна предварительного испаре-
ния; 2 — атмосферная колонна; з — печь; 4 — первая ва-
куумная колонна; 5 — вторая вакуумная колонна; 6 —
испаряющий агент.
•ся последоват. перегонка мазута в 2 вакуумных ко-
лоннах. Во 2-й вакуумной ступени пользуются также
испаряющим агентом, к-рым служит легкий газойль.
Лит.: Павлова С. Н., Д р и а ц к а я 3. В., Типовая
методика исследования состава нефтей, в ки.: Состав и свой-
•ства нефтей и бензино-керосиновых фракций. Сборник работ
по изучению состава и свойств нефтей и нефтепродуктов, М.,
1957; Гуревич И. Л., Технология нефти, ч. 1, 2 изд.,
М.— Л., 1952; Казьмин Г. И. [и др.], Нефтеперерабаты-
вающие заводы США, М., 1962; Багиров И. Т., Современ-
ная атмосферно-вакуумная установка, М., 1957. И.Л.Гуревич.
ПЕРЕГРУППИРОВКА АЛЛИЛЬНАЯ — перегруп-
пировка аллильных систем, связанная с переме-
щением атома (или группировки) из положения 3
в положение 1, с одновременным сдвигом двойной
связи в противоположном направлении:
R . 3 2 1 R" R . 3 2 1 R"
C = C' —> J>C=C —
R' 1 R'" R' 1 j. R'"
В зависимости от природы мигрирующей группы
различают прототропные П. а. (миграция протона) и
анионотропные П. а. (миграция аниона). Прототроп-
ные П. а. особенно характерны для а, р и р, у-нспре-
дельных карбонильных соединений (1) или нитри-
лов, для аллиловых эфиров (2), аллиламинов и ари-
лироваиных пропиленов (3); многие из них изоме-
ризуются уже при комнатной темп-ре в присутствии
следов оснований. Основания являются универсаль-
ными катализаторами прототропной П. а., причем си-
ла катализатора характеризуется его основностью;
в нек-рых случаях прототропную П. а. катализируют
п кислоты (4, 5). Олефины изомеризуются значительно
труднее, при нагревании до 200—300°, обычно в при-
сутствии сильных оснований (6); в диметилсульфок-
сиде в присутствии трет-бутилата калия олефины изо-
меризуются при низкой темп-ре (7). Большинство
прототропных П. а. обратимо, и равновесие сдвинуто
в сторону устойчивой формы, т. е. в сторону изомера,
у к-рого при кратной связи расположено наибольшее
число арильных, алкильных (лучше метильных), ви-
нильных, карбонильных и других группировок,обес-
печивающих создание сопряженной цепи (8, 9).
RCH2—СН = СН—С—R' 72 RCH = CH—СН2—С—R' (1)
II II
о о
АгОСН2—СН = СН2 АгОСН = СН—СН3 (2)
АгСН2СН = СНАг' АгСН = СНСН2Аг' (3)
+Н+ -н+
(СН3)3С-СН = С(СН3)2 72 (СН3)3С-СН2-С + (СН3)2 72
-Н+ +Н+
сн3
±2(СН3)3С-СН2—С = СН2 (4)
+ Н +
СН.. = СН—СН..ОН —>-СН3—СН—СН..ОН—»-
- н +
—^СН3—СН = СНОН—^СН3СН2СНО (5)
300»
СН3—СН2-СН = СН2 222 СН3—СН = СН—СН3 (6)
А12О3
СН3 сн3
СН3СН2СН2С = СН2 2 СН3СН2СН =С СН3 т2
сн3
СН3СН = 1 снснсн, (7)
О СН2 = ССН2С 2± 1 \ R OR' О СН3С = СН—с 1 \ R OR' (8)
R О 1 / С„Н3СН = ССН2С — \ о- R О 1 / I С3Н3СН2С = СНС \ о- О)
Механизм прототропной П. а. включает в себя от-
рыв протона основанием с образованием мезомерпого
аниона, к-рый затем снова присоединяет к себе про-
тон (10). Однако иногда возможен синхронный ме-
ханизм перегруппировки:
-вн
П+вн
R-C-C=C-R'_- R—С=С—С—R'
III III
н
RC=C—C—R +
(10)
(И)
н
R-C-C=^C-R
r-c=c-c-r'+ нв,+в2
H
Н
Прототропные П. а. характерны также для ацетиле-
нов (ацетилен-алленовая перегруппировка, 12), алле-
нов (аллен-диеповая перегруппировка, 13) и азо-
метинов (14); кетоенольная таутомерия, по существу,
также является прототропной П. а. (15):
РСН2—С -С—R' 2 7 RCH = C = CHR' (12)
сн3 сн3
I I
СН3-С=С=СН2—► СН2 = С—СН = СН3 (13)
ArCH=N—СН3Лг'72 АгСНг—N=CHAr' (14)
)С-2ОСС/’ = Г-О (15)
7 I I 7 I I
н н
Очень часто под П. а. подразумевают исключительно
анионотропные П. а., широко изученные па большом
числе примеров. Анионотропные II. а. обычно катали-
зируются кислотами, в том числе апротонными (ВБ3,
917
ПЕРЕГРУППИРОВКА АЛЛИЛЬНАЯ
918
ZnCl2, ионы Ag, Fe’+ипр.). Многие аллильные соеди-
нения претерпевают анионотропную П. а. самопро-
извольно или при слабом нагревании. Легкость пере-
группировки зависит в основном от строения моле-
кулы и от природы мигрирующей группы: спирты
перегруппировываются труднее, чем сложные эфиры;
легче всего перегруппировываются галогенопроиз-
водные. Прп наличии нескольких сопряженных крат-
ных связен в соединении анион мигрирует к концу
сопряженной цепи (16), даже в тех случаях, когда
кратные связи входят в цикл (17). Из производных
ацетилена, в зависимости от мигрирующей группи-
ровки, образуются аллены (18) или а, р-иепредельные
кетоны (19). Аллиловые спирты с атомами галогена
в винильном положении (и эфиры таких спиртов)
перегруппировываются в а, Р-непредсльпые карбо-
нильные соединения (20, 21, 22). В отличие от обычных
анионотропных П. а., перегруппировка а-галогено-
кетопов (22) и производных а-моногидронерфторви-
нилуксуспой к-ты (23) катализируется основаниями;
°Н
, I „ 3Z H2SO4
(сн3)2с-сн=сн-сн=сн2-----------
(сн3)2с=сн--сн=снсн2он
ЦЦ(С6Н6)2
СН\ I Си2С12 СН»
'С-С = СН------» )С = С = СНС1
„„ z NH,C1 z
Xl g KJ XI 2
(16)
(17)
(18)
H +
RCHC=CR' —>RCH=C = CR' —>RCH = CH—C—R' (19)
I I II
ОН он о
H
RCHCH—CR'—>.RCH = CH—C—R' + HX (20)
I I II
OH X о
о
н± /
АгСНСН = СС1,—>.ArCH = CHC (21)
I \
ОН он
РЬО
СН3СН,ССН201 —>- СН30Н = ССН2С1—хСН3СН-С = СН
100° | I
ОН С1
—>СН3СН—с—сн3
I II
он о
о о
/ (C2H-)3N /
CF2 = CF-CHF—с---------► CF3—CF = CH—С
\ \
NR2 NRa
Многие аномальные реакции замещения в аллиль-
ных системах объясняются переносом реакционного
центра по типу П. а. (24) (см. Анионотропия). В цик-
лопропановых аналогах аллильных систем наблю-
дается гомоаллильная перегруппировка, аналогичная
П. а.(25). х3 ^7^ +у- ч3 2
/С~С = Сх zC=c-(jk (24)
XJ Y
\ I z
с=с-с
с
Y-<
(25)
Механизм анионотропной П. а. зависит от строения
исходного соединения и от условий перегруппировки.
В сильно полярных растворителях, в условиях, спо-
собствующих сольволизу, образуется мезомериый ион.
к-рый далее присоединяет анион к одному из двух
концевых атомов аллильной системы (26). В случае
соединений, менее склонных к сольволизу, перегруп-
пировка осуществляется через стадию образования
псевдоциклич. соединения (27), иногда с участием
катализатора (28). Сложные эфиры аллиловых спир-
RJ
с-сн=сн2
R'
R R
к z
С
НС \
чсн2
R X
к z
С +
чсн=сн2
о R
\+ \ +
с-сн=сн2 - с=сн-сн2
R' R'
(26)
R
чс=сн-сн2х
R'
R R'
к /
С.
НС.' X
X-
сн2
R R
Л
НС X
X z
сн2
(27)
R .X.
\ Н
R" СН ,х
^'сн2
R
—«~хс + НХ
/II
R СН X
хсн2
(28)
тов изомеризуются через шестичленный псевдопиклич.
комплекс (29);таков же механизм Клайаена перегру ппи-
ровки, перегруппировки аллилвиниловых эфиров (30),
О-аллилимипоэфиров (31), аллилтиоцианатов (32),
диаллилов (33) и солей винилаллиламмония (34):
R z°x ZRZ Rk °Ч
\ ЧС \ С-R' ....
I II ------► „zll I (29)
н СН О н СН о
чс чсх
z к / к
R R R R
о
сн^с-с6н5
1-х /II
сн) <N-c6Hs
чсн
I
R
СН3
СН
сн.-сн сн
сн2
R СН2
^N^CH
r/CH)^CH2
V ••
сн2
О
%
сн2 СН
II I
сн сн2
сн2
(23)
210-220°
---------
о
сн2 чс-с6н.
сн n-c6h5
сн
I
R
S
сн2 с
II II
СН N
хсн
I
R
сн3
сн
сн3-сн сн
сн сн2
чсн2
Rx+ Сч2
ЧСН
R СН zCH2
ЧСН2
(30)
(31)
(32)
(33)
(34)
919
ПЕРЕДАЧА ЭНЕРГИИ ЛИНЕЙНАЯ —ПЕРЕКАЧИВАНИЕ ЖИДКОСТЕЙ
920
П. а. очень распространена; изучение ее сыгра-
ло важную роль в развитии теоретич. органической
химии.
Лит.: Темникова Т. И., Курс теоретических основ
органической химии, 2 изд., Л., 1962; Ингольд К. К.,
Механизмы реакций и строение органических соединений,
пер, с англ., М., 1959; DeWolfe R. Н., J о u n g W. G.,
Chem. Revs., 1956, 56, 753. _ Н. П. Гамбарян.
ПЕРЕДАЧА ЭНЕРГИИ ЛИНЕЙНАЯ (сокр. ЛПЭ,
в иностр, лит. LET) — количество энергии ионизи-
рующего излучения, передаваемое молекулам среды
на единице длины пути частиц излучения. Понятие
ЛПЭ используется в радиационной химии и радиобио-
логии. В физике более употребительно равнозначное
понятие тормозная способность. Вели-
чина ЛПЭ излучения имеет существенное влияние
на характер радиационнохимич. процесса и на выходы
продуктов в этом процессе. Это обусловлено тем, что
с изменением ЛПЭ изменяется строение трека, а сле-
довательно, и относительная роль процессов диффузии
и рекомбинации, протекающих внутри трека. Величи-
ны ЛПЭ пек-рых часто применяющихся на практике
типов излучения для воды и разб. водных р-ров при-
ведены в таблице.
Излучение
Начальная
ЛПЭ, эв/ц
•у-Лучи Со™.................
Электроны, Е=1 —2 Mas . . .
Рентгеновские лучи, £ = 250 Лэв
Рентгеновские лучи, £ = 8 Лэв .
(1-Лучи Н3(ЕС =5,5 Лэв) . . .
Дейтоны, Е=20 Mas...........
Протоны, Е = 8,4 Мэе........
Дейтоны, Е = 5,2 Мэв........
Протоны, Е=2,0 Мэв..........
Гелионы, £ = 38 Мэв.........
0,02
0,02
0,10
0,28
0.36
0,45
0,55
1.3
1 , 7
2,2
3,0
5,0
8,8
12
700
Тритоны, Е = 2,1 Мэв.......................
Гелионы, £ = 12 Мэе . . . ...............
а-Лучи Ро210 (£=5,3 Мэв)..................
а-Лучи, Е = 3,4 Мэв ......................
Осколки деления (масса 138, Е = 65 Мэв) . . . .
Расчет ЛПЭ для пучков высокоэнергетич. заряжен-
ных частиц проводится с помощью уравнений, выве-
денных для модели одноатомною газа. Однако в случае
многоатомных газов и конденсированных сред сущест-
венное значение могут иметь межатомные и межмо-
лекулярные взаимодействия, к-рые не были учтены
при выводе уравнений.
Уравнение потери энергии для протонов, дейтонов,
гелионов и других тяжелых ионов имеет вид:
dE 2ne'z2A’Z Af ,_ 4£ т
die ~ Е т 111 ~Г ’ М
где Е — энергия падающей частицы, е и т— заряд и
масса электрона, Muz — масса и число единичных
зарядов движущегося иона, NZ — количество элект-
ронов в единице объема облучаемого вещества и I —
средний потенциал возбуждения вещества, опреде-
ляемый эксперпментально. Согласно этому уравне-
нию, при очень малых энергиях ЛПЭ должна стать
бесконечно большой. Обнаружено, однако, что ЛПЭ
достигает максимума при. нек-рой энергии и при даль-
нейшем ее снижении начинает уменьшаться. Причи-
ной снижения является то, что медленная заряженная
частица может захватывать электроны из окружаю-
щей среды и образовывать частицу с меньшим заря-
дом или нейтральный атом. Просуществовав нек-рое
время в таком состоянии, частица потеряет электрон
и вновь вернется в первоначальное состояние. Так как
нейтральный атом или частица с меньшим зарядом
теряют энергии много меньше, результатом будет сни-
жение наблюдаемой величины ЛПЭ. Очевидно, что
для электронов и электромагнитных квантов подоб-
ные явления не могут иметь места.
Ур-ние для ЛПЭ нерелятивистских электронов в
основном подобно ур-нию для тяжелых ионов. Не-
сколько изменяется лишь выражение под логариф-
мом вследствие неразличимости электронов и другого
значения приведенной массы. При энергиях больше
50 Кэв ур-ние видоизменяется в связи с необходимо-
стью учета релятивистского увеличения массы элект-
рона с ростом его скорости, что приводит к более вы-
соким ЛПЭ при высоких энергиях. Вследствие этого
с ростом энергии падающих электронов ЛПЭ сначала
уменьшается, достигает минимального значения
0,02 эв/А при энергиях от 1 до. 2 Мэв и далее начинает
медленно возрастать. При энергиях выше области
минимума при расчете ЛПЭ следует принимать во вни-
мание плотность среды (поляризационный эффект),
а также потери на тормозное излучение. Потеря энер-
гии при взаимодействии со средой приводит к умень-
шению скорости частицы и одновременному увеличе-
нию ЛПЭ вдоль трека движущейся частицы. Поэтому
следует различать мгновенное и усредненное по длине
пути значения ЛПЭ.
Лит.: Гайсинский М. Н., Ядерная химия и ее при-
ложения, пер. с франц., М., 1961, гл. XI; Экспериментальная
ядерная физика, под ред. Э. Сегре, пер. с англ., т. 1, М., 1955,
ч. 2; Gordon S., Hart Е.. Radiation Res., 1961, 15, № 4,
440; Hochanadel C. J., в кн.: Comparative effects of
radiation, N. Y.-L., 1960, 151. в. н. Шубин.
ПЕРЕКАЧИВАНИЕ ЖИДКОСТЕЙ — непрерыв-
ное или периодич. перемещение жидкостей из одного
резервуара в другой, находящийся на большей высо-
те или под повышенным давлением. Для перекачива-
ния жидкостей б. ч. при-
меняются насосы, т. е.
машины, сообщающие
жидкости энергию, не-
обходимую для ее пере-
мещения. Работа насоса
характеризуется пода-
чей (производительно-
стью) Q, измеряемой в
м3/час, л/сек или л/мин
(режев кг!час, т/час), и
напоромТ/, измеряемым
в метрах столба перека-
чиваемой жидкости. Н а-
пор насоса равен коли-
честву энергии, сооб-
щаемой единице пере-
3 — нагнетательный резервуар; 4—
трубопроводы.
качиваемой жидкости, и определяется условиями уста-
новки насоса (рис. 1):
я="ст.+'ЦгНи
где Нст — разность уровней в резервуарах (статич. напор), м;
Р,—абс. давление в заборном резервуаре, хгс/.ч2; Р2— абс.
давление в нагнетательном резервуаре, кгс/м2; у— объемный
вес перекачиваемой жидкости, кгс/м3; hx—гидравлич. потери
во всасывающем трубопроводе, м; h2— гидравлич. потери в
нагнетательном трубопроводе, м.
Напор действующего насоса контролируется по
показаниям манометров, установленных до и после
насоса. Высоконапорные насосы часто характеризу-
ются давлением после насоса (давлением нагнетания),
выраженным в кгс/см2, или в атм. Вакуумметрич.
высота всасывания йвак., измеряемая в метрах стол-
ба перекачиваемой жидкости, составляет:
ГГ ____ IT I 7, _ 1
л вак. — 27 вак.ст. “г ,li у
где Нвак сг — расстояние от уровня жидкости в заборном
резервуаре до оси насоса (статич. высота всасывания), м; Ри—
атмосферное давление, кгс/м2.
Если насос расположен ниже уровня жидкости в
заборном резервуаре или давление Рх достаточно ве-
лико, то величина 7/вак. может быть отрицательной
(работа насоса с подпором).
Вакуумметрич. высота всасывания действующего
насоса не должна превышать величины, допустимой
921
ПЕРЕКАЧИВАНИЕ ЖИДКОСТЕЙ
922
для данного режима работы насоса. Несоблюдение это-
го требования приводит к возникновению в рабочих
органах насоса т. наз. к а в и т а ц и и, т. е. наруше-
ния сплошности потока с образованием паровых пу-
зырей, что сопровождается снижением подачи, шу-
мом, вибрацией и эрозионным разрушением деталей
проточной части насоса. Допускаемая вакуумметрич.
высота всасывания указывается в каталоге; она зави-
сит также от давления паров перекачиваемой жидко-
сти при заданной темп-ре и от атмосферного давления
в месте установки насоса. От допускаемой вакууммет-
рич. высоты всасывания следует отличать самовса-
сывающую способность, т. е. способность насоса при
его включении в работу отсасывать воздух из неза-
полненного всасывающего трубопровода, что необ-
ходимо для подъема жидкости из заборного резер-
вуара.
Коэфф, полезного действия (кпд) насоса определяет-
ся как отношение полезной (гидравлической) мощ-
ности насоса к мощности, затрачиваемой на его привод
(мощности на валу), и существенно зависит от типа
насоса и режима его работы. В качестве привода насо-
са используются гл. обр. электродвигатели с непо-
средственным соединением (через муфту) валов насо-
са и двигателя. Реже применяются двигатели внут-
реннего сгорания, гидродвигатели, паровые и газо-
вые турбины и др.
Насосы делятся на лопастные и объемные. К лопа-
стным насосам относятся центробежные, осевые и
вихревые насосы, к объемным — поршневые привод-
ные, поршневые паровые и роторные.
Насосы центробежные — наиболее
распространенный тип насосов. Рабочими органами
центробежного насоса являются подвод, рабочее
колесо и спиральный отвод. Рабочее колесо оказы-
вает динамич. воздействие на поступающую в насос
жидкость, повышая ее энергетич. уровень за счет
увеличения в основном кинетич. энергии. В спираль-
ном отводе скорость жидкости снижается, а давление
повышается, т. е. кинетич. энергия жидкости преобра-
зуется в потенциальную.
Подача и напор центробежного насоса при постоян-
ном числе оборотов в минуту взаимно связаны и соот-
ветствуют одной из точек
рабочей характеристики
насоса (рис. 2), на к-рой
обычно приводятся так-
же мощность на валу и
кпд насоса. Т. к. напор
выражен в метрах стол-
ба перекачиваемой жид-
кости, то он не зависит
от объемного веса послед-
ней, тогда как мощность
на валу пропорциональ-
на объемному весу. Ра-
бочая зона, в к-рой целе-
сообразно эксплуатиро-
вать насос, определяется
областью подач, в к-рой
кпд насоса близок к мак-
симальному. Рабочая точ-
зона подач
Рис. 2. Характеристика центро-
бежного насоса: 1 — характери-
стика трубопровода; 2—рабочая
•точка характеристики насоса;
3 — мощность; 4 — кпд; 5 — на-
пор.
ка насоса (а следовательно, его подача и напор) опреде-
ляется пересечением рабочей характеристики насоса
и суммарной гидравлич. характеристики всасывающе-
го и нагнетательного трубопроводов. Изменение ха-
рактеристики трубопровода приводит к смещению
рабочей точки и, следовательно, к изменению подачи
(рис. 2) и напора. Аналогично влияет изменение уров-
ней и давлений в резервуарах, смещающее харак-
теристику трубопровода по вертикали. Мощность
центробежного насоса растет с увеличением подачи,
поэтому пуск центробежных насосов производят при
закрытой задвижке на нагнетательной линии. Центро-
бежные насосы изготовляются как с горизонтальным,
так и с вертикальным валом.
Наибольшее распространение получили горизонталь-
ные центробежные насосы консольного типа (рис. 3).
Рис. 3. Схема центробежного насоса консольного типа:
1 — рабочее колесо; 2 — спиральный отвод; 3 — всасы-
вающий патрубок; 4 — вал; 5 — опорная стойка; 6 — саль-
никовое устройство с мягкой набивкой.
Перекачиваемая жидкость поступает через всасываю-
щий патрубок 3 на рабочее колесо 7, а затем через
спиральный отвод 2 в корпусе подается в нагнетатель-
ный трубопровод. Рабочее колесо расположено кон-
сольно на валу 4, вращающемся в подшипниках, за-
крепленных в опорной стойке 5. Для предохранения
от утечки жидкости через зазор между валом и корпу-
сом служит сальниковое устройство 6. При больших
подачах применяются насосы с колесом двухсторон-
него входа, при больших напорах — многоступенча-
тые насосы, в к-рых перекачиваемая жидкость после-
довательно проходит через 2 или большее число рабо-
чих колес. Конструктивное исполнение центробежных
насосов определяется не только его рабочими парамет-
рами, но и назначением. Изготовляются насосы для
чистой воды, химически агрессивных жидкостей,
шламов и суспензий, для откачки из артезианских
скважин и др. Центробежные насосы применяют при
подачах от 3 до 65000 м3!час, напорах до 2000 м,
их мощность достигает 8000 л. с., а кпд — 90%.
К центробежным насосам, применяемым для пере-
качивания химически агрессивных жидкостей, предъ-
являются специфич. требования: коррозионная стой-
кость материалов гидравлич. части, отсутствие утечек
перекачиваемой жидкости через уплотнение, макси-
мальная автоматизация работы и т. п. Для изготовле-
ния рабочих колес и корпуса таких насосов применя-
ют нержавеющие стали, кислотоупорные чугуны,
пластмассы, фарфор, а также различные покрытия
(эмали, лаки, резина). В насосах для химически аг-
рессивных жидкостей применяют торцовые уплотне-
ния, основными деталями к-рых являются подвижное
металлич. и неподвижное графитовое или фарфоро-
вое кольца, поверхности к-рых тщательно взаимно
притерты, что практически исключает утечку жидко-
сти. При всех типах уплотнений у насосов для хими-
чески агрессивных жидкостей предусмотрен отвод
возможной утечки во всасывающий трубопровод, в
дренаж или в специальную емкость. Полным отсут-
ствием утечек перекачиваемой жидкости и ее паров
отличаются герметичные (бессальниковые) насосы
(рис. 4). Рабочее колесо 1 такого насоса и ротор элек-
тропривода 5 вращаются на общем валу 3 в подшип-
никах 4, помещенных в перекачиваемую жидкость.
Статор электропривода 6 отделен от ротора металлич.
или пластмассовой гильзой 7 и заполнен нейтральной
жидкостью. Толщина гильзы не более 0,3—0,5 мм.
Для охлаждения и смазки подшипников перекачивае-
мая жидкость подается к ним от спирального отвода 2
923
ПЕРЕКАЧИВАНИЕ ЖИДКОСТЕЙ
924
(на рис. 4 не показано). Если перекачиваемая жид-
кость содержит взвеси, то охлаждение и смазка под-
шипников осуществляются за счет автономной цирку-
ляции той части жидкости, к-рая заполняет ротор
Рис. 4. Схема герметичного (бессальникового) наеоса:
1 — рабочее колесо; 2 — спиральный отвод; 3 — вал; 4—
подшипники; 5 — ротор электропривода; 6 — статор элек-
тропривода; 7 — гильза.
Рис. 5. Схема верти-
кального осевого на-
соса: 1 — рабочее ко-
лесо; 2 — лопасть; 3—
направляющий аппа-
рат.
электропривода; в этом случае перекачиваемая жид-
кость в полость ротора практически не проникает.
Герметичные насосы применяют при перекачивании
наиболее агрессивных, взрывоопасных, ядовитых и
других жидкостей, утечка к-рых (и проникновение в
атмосферу их паров) совершенно недопустима.
Насосы осевые (пропеллерные) отличаются
большой подачей (от 750 до 65 000 л’час) при отно-
сительно низких напорах (от 2 до
22 .и). Рабочими органами осевых
насосов (рис. 5) являются рабочее
колесо 1 пропеллерного типа с же-
стко закрепленными или поворот-
ными лопастями.2 и направляющий
аппарат 3. Рабочие характеристи-
ки аналогичны характеристикам
центробежных насосов; кпд дости-
гает 90% и выше.
Насосы вихревые (рис.
6) близки по конструкции к цент-
J робежным, но отличаются от пос-
/ дедних рабочим процессом. При
> вращении рабочего колеса вихре-
‘ вого насоса, имеющего радиальные
или наклонные лопатки, возника-
ющие между лопатками вихри ув-
лекают находящуюся в кольцевых
каналах корпуса жидкость и пере-
мещают ее от полости всасывания
к полости нагнетания. Спираль-
ные отводы или направляющие ап-
параты у вихревых насосов отсутствуют. По сравнению
с одноступенчатыми центробежными вихревые насосы
Рис. 6. Схема вихревого насоса: 1 — рабочее колесо;
2 — корпус.
являются более высоконапорными (до 90 м), но при-
меняются обычно при относительно малых подачах
кости, и поршневых
с а х (рис. 7) при ходе скал-
(от 1 до 35 м’/час). К их недостаткам относятся низ-
кий кпд (не выше 40%) и относительно низкая всасы-
вающая способность.
Насосы поршневые относятся к насосам
объемным, т. е. к насосам, у к-рых подача жидкости
осуществляется за счет периодич. изменения объема
рабочей полости (или рабочих полостей) и вытесне-
ния перекачиваемой жи ~
приводных н а с <
ки I вправо освобожда-
ющийся объем цилиндра
заполняется перекачи-
ваемой жидкостью, по-
ступающей через всасы-
вающий клапан 7. При
ходе скалки влево жид-
Рис. 7. Схема поршневого
приводного насоса: 1—скал-
ка или поршень; 2 —шток;
3 — ползун; 4 — шатун; 5—
кривошип или коленчатый вал; в — напорный клапан; 7 — вса-
сывающий клапан; з — напорный колпак; 9— всасывающий
колпак; 10 — цилиндр; 11 — уплотнение.
кость вытесняется через напорный клапан 6. Кол-
паки 8, 9, верхняя часть к-рых заполнена возду-
хом, служат для смягчения толчков жидкости и по-
вышения степени равномерности подачи. Этой же
цели служит увеличение числа цилиндров (до 2—3),
что в ряде случаев позволяет отказаться от колпаков.
Подача поршневого приводного насоса при постоянном
числе оборотов приводного вала с изменением напора
практически пе изменяется. Предельный напор (или
давление нагнетания) определяется максимально до-
пустимым усилием на скалке и прочностью механизма
движения. Насос обычно снабжается предохранитель-
ным клапаном, отрегулированным на заданную ве-
личину максимального давления.
Привод насоса осуществляется гл. обр. от электро-
двигателя через понижающий редуктор. Наибольшая
компактность достигается при использовании встро-
енного в насос редуктора (шестеренного или червяч-
ного). Регулирование подачи осуществляется или за
счет сбрасывания части жидкости из напорного трубо-
провода во всасывающий через байпас (что не эконо-
мично) или за счет изменения числа оборотов привод-
ного вала. У регулируемых насосов подача может
изменяться за счет изменения длины хода скалки с
помощью специального механизма или за счет выклю-
чения всасывающих клапанов на определенной части
хода нагнетания. Наиболее точное регулирование по-
дачи обеспечивают дозировочные насосы.
Поршневые приводные насосы в основном применя-
ют для подачи от 0,3 до 160 лР/час при относительно
высоких давлениях нагнетания (до 400 кгс/см2 и выше)
и отличаются высоким кпд (до 95%) и хорошей вса-
сывающей способностью. Большинство поршневых
насосов обладает также самовсасывающей способ-
ностью. В зависимости от материала гидравлич.
части насоса его можно использовать для подачи раз-
личных (в том числе химически агрессивных) жидко-
стей. Небольшая утечка через уплотнение обычно от-
водится в дренаж или возвращается во всасывающий
трубопровод. При недопустимости утечки иногда
применяют т. наз. гидравлич. затвор, т. е. подачу
в уплотнение нейтральной жидкости под давлением,
превышающим давление нагнетания, или используют
диафрагмовые (мембранные) насосы.
Для дозирования нескольких жидкостей, подавае-
мых в различные аппараты, применяют многоплун-
жерные насосы-дозаторы, у к-рых каждый плунжер
предназначен для подачи определенной жидкости.
Величина подачи каждой жидкости регулируется соот-
ветствующей установкой длины хода плунжера.
925
ПЕРЕКАЧИВАНИЕ ЖИДКОСТЕЙ — ПЕРЕКИСИ И ГИДРОПЕРЕКИСИ
926-
Поршневые паровые (прямодействую-
щие) насосы работают за счет давления пара
(реже — воздуха, масла) на поршень, жестко связан-
ный (через шток) с гидравлич. поршнем. Прямодей-
ствующие насосы изготовляют большей частью с двумя
гидравлич. и соответственно двумя паровыми поршня-
ми (дупл е к с - н а с о с ы), реже — с одним гидрав-
лич. и одним паровым поршнем (симплекс-на-
сос ы). Каждый гидравлич. поршень — двойного
действия: если в левой полости цилиндра происходит
всасывание, то в правой в это же время — нагнета-
ние, и наоборот. Паровые прямодействующие насосы
применяют гл. обр. для перекачивания воды и взрыво-
опасных жидкостей при подачах от 1 до 500 м3[час
и давлениях нагнетания до 50 кгс/см2. К недостаткам
этих насосов относится весьма низкий кпд (порядка
1%); к положительным качествам — отсутствие эле-
ктропривода, редуктора, коленчатого вала и шатун-
ной группы и простота регулирования подачи путем
дросселирования подводимого пара и достигаемого
при этом снижения числа ходов поршней. Дуплекс-
пасосы, кроме того, отличаются высокой равномер-
ностью подачи, ввиду чего эти пасосы б. ч. пе снабжа-
ются напорными колпаками.
Насосы роторные (рис. 8) также относятся
К объемным насосам и отличаются большим коп-
Рис. 8. Схема роторных насосов: а — шестеренный; б —
пластинчатый; в — винтовой; г — поршеньковый.
структивным разнообразием. Наибольшее распро-
странение получили насосы шестеренные, пластин-
чатые (шиберные), винтовые, поршеньковые (с акси-
альным или радиальным расположением поршней).
Общим для роторных насосов является наличие трех
основных элементов: вращающегося ротора, б. ч.
неподвижного статора и замыкателя (или замыка-
телей), совершающего вращательное (коловратные
пасосы) или возвратно-поступательное движение. Ро-
торные насосы отличаются от поршневых отсутствием
всасывающих и нагнетательных клапанов, повышен-
ными числами оборотов (1000—3000 в мин) и более
высокой степенью равномерности подачи.
Рабочие органы роторных насосов непрерывно об-
разуют замкнутые объемы, заполняемые перекачивае-
мой жидкостью, и перемещают их от всасывающего
патрубка насоса к нагнетательному. Из-за больших
поверхностей трения роторные насосы применяют
гл. обр. для перекачивания жидкостей с хорошей сма-
зывающей способностью (напр., минеральных ма-
сел). Роторные насосы применяют при подаче от 0,1
до 200 м31час и давлениях нагнетания от 2 до
320 кгс{см2. Наибольшие подачи имеют место у насо-
сов винтовых, давления — у насосов поршеньковых.
Нек-рые типы роторных насосов имеют механизмы
регулирования подачи (напр., аксиально-плунжерные-
насосы). С увеличением давления подача роторных на-
сосов несколько уменьшается ввиду роста внутренних
утечек. Большим преимуществом роторных насосов
перед поршневыми являются их компактность и ма-
лый вес.
Для перекачивания жидкостей, кроме насосов,
применяют аппараты, в к-рых для создания напора
перекачиваемой жидкости использу-
ется энергия другой (рабочей) жид-
кости, газа или пара. На рис. 9 пред-
ставлена схема струйного аппарата.
Перекачиваемая жидкость поступает
через патрубок 1. Через сопло 2 по-
дается под давлением рабочая жид-
кость, газ или пар, образующие в
корпусе 3 струю, к-рая постепенно
передает свою энергию перекачивае-
мой жидкости, полностью смешиваясь
с пей.
Несмотря на низкие кпд (не выше
25—30%), подобные устройства ши-
роко применяют в различных конст-
руктивных модификациях ввиду про-
стоты и отсутствия движущихся ча-
стей, в особенности при кратковре-
менной работе, когда величина кпд
несущественна. Струйные аппараты
могут применяться как для отсасыва-
ния жидкости (эжекторные насосы),
так и для ее нагнетания (инжектор-
ные пасосы) с напором до 10 м.
Для периодич. подачи жидкостей (в
том числе весьма агрессивных) при-
меняют мопжусы (монтежю) — аппа-
I
Рис. 9. Схема*
струйного аппара-
та: 1 — патрубок;
2— сопло; ,3 — кор-
пус.
раты, из к-рых перекачиваемая жидкость выдавли-
вается в нагнетательный трубопровод с помощью
подаваемого под давлением инертного газа (напр.,
азота). Известны также конструкции автоматич.
монжусов, т. наз. пульсометров.
Лит.: Ломакин А. А., Центробежные и пропеллерные-
насосы, М.—Л., 1950; Степанов А. И., Центробежные
и осевые насосы, пер. с англ., М., 1960; Черкасский
В. М., Романова Т. М., Каул ь Р. А., Насосы, ком-
прессоры, вентиляторы, М.—Л., 1962; Касаткин А. Г.,
Основные процессы и аппараты химической технологии, 7 изд.,.
М., 1960; Насосы. Каталог — справочник, 3 изд., М., 1959.
А. М. Васильев.
ПЕРЕКИСИ И ГИДРОПЕРЕКИСИ органи-
ческие — соединения, в состав к-рых входит
группа —О—О —, связанная с атомом углерода. Они
разделяются на два класса: 1) гидроперекиси, в к-рых
I
перекисная группа соединена с водородом —С—О—ОН
I I I
и 2) перекиси, имеющие группировку —С—О—О—С—.
Гидроперекиси и перекиси углеводородов делятся на
первичные, вторичные и третичные в зависимости ог
атома углерода, связанного с группой —О—О —;
атом углерода перекисной группы может быть со-
единен с алкильными, арильными, аралкильными
радикалами, входить в состав циклоалканов, циклоал-
кенов и гетероциклич. соединений. Гидроперекиси,
в к-рых углерод перекисной группы входит в арома-
тич. цикл, неизвестны, но перекиси такого рода полу-
чены.
Гидроперекиси (Г.). Наиболее важные методы полу-
чения Г.: 1) окисление углеводородов в газовой или
жидкой фазе. Окисление направляется на третичный
атом углерода или при его отсутствии на вторичный.
У олефинов идет окисление СН-группы, связанной с
927
ПЕРЕКИСИ И ГИДРОПЕРЕКИСИ
928
атомом углерода, находящимся у двойной связи. Од-
нако при этом часто получаются продукты распада Г.
Этот метод имеет промышленное значение для получе-
ния третичных Г. В газовой фазе автоокисление уско-
ряется добавками, напр. НВг. При окислении жидких
веществ применяют соли металлов переменной валент-
ности (Со, Мп и др.), щелочи, соли Г. и др. 2) Синтезы
с использованием Н2О2; эти способы дают широкие
возможности получения Г. Реакции проводят в при-
сутствии к-т или щелочей, напр. для спиртов:
[Н+]
roh + h2o2---->rooh + h2o
В кислой среде Г. могут быть получены из галогенал-
килов и из олефинов:
[Н+]
R2C = CHR' +Н2О2-->. R2C(OOH)CH2R'
В щелочной среде Г. образуются из эфиров серной
к-ты, алкилметансульфонатов и Н2О2. 3) Окисление
при пониженных темп-рах нек-рых элементоорганич.
соединений (В, Mg, Zn, Cd и др.) с последующим гидро-
лизом:
н2о
RMgX + O2—>ROOMgX------>ROOHfMg(OH)X
Г. в зависимости от строения могут быть жидкостя-
ми. перегоняющимися в вакууме, или твердыми веще-
ствами. Последними бывают сильно разветвленные
алкильные Г. от С8, аралкильные, а также Г., содержа-
щие конденсированные циклич. системы. Г. раство-
ряются в органич. растворителях, Г. метила и этила
растворяются в воде. Многие Г. взрывчаты. Г. обла-
дают слабокислыми свойствами и дают соли со ще-
лочными и щелочноземельными металлами, что часто
используется для их выделения.
При действии HJ, SnCl2, LiAlH4, (СвН5)3Р и др.
восстановителей Г. восстанавливаются до спиртов;
при нагревании распадаются на свободные радикалы
по —О—О-связи:
ROOH —>RO- +но-
Образовавшиеся радикалы могут вызывать распад Г.
(цепной распад). Радикалы RO часто разлагаются да-
лее, напр.:
(СН3)3СО —> сн3- +сн3сосн3
При действии кислот происходит гетеролитич. распад
Г.; это используется в практике получения фенола
и ацетона из гидроперекиси кумила:
[Н + ]
С„Н5(СН3)2СООН----> слон + СН3СОСН3
При действии на Г. солей металлов переменной ва-
лентности легко образуются свободные радикалы RO:
ROOH+Fe11—>RO+Fen,+HO-
При разложении циклич. Г. наблюдается перегруп-
пировка О-радикала в С-радикал:
---- \-(СН2)ю-с'
Н Н
При гомолитич. распаде Г. (см. Гомолитические
реакции) в растворителях первичные радикалы могут
реагировать с растворителем, отрывая от него водо-
род с образованием вторичных радикалов из раство-
рителя. Могут идти реакции присоединения первич-
ных радикалов к кратным связям и др. процессы.
Перекиси R—О—О—R' (П.). Эти соединения могут
быть получены: 1) При автоокислении углеводородов
в определенных условиях П. образуются в числе дру-
гих продуктов, напр. при окислении изобутилена
в присутствии НВт и паров воды получается до 40%
перекиси третичного бутила. П. триарилметилов ко-
личественно образуются при действии воздуха на
р-ры гексаарил этанов (1):
О2
Аг3ССАг3 ;—> 2Аг3С-»-Ar3COOCAr3 (1)
2) Прямым алкилированием перекиси водорода спир-
тами или эфирами в присутствии кислот, а также диал-
килсульфатами или алкилметансульфонатами в ще-
лочной среде (2):
2 (AlkO)2SO2 + H2O2—> AlkOOAlk + 2AIkOSO3H (2)
3) Из Г. и спиртов в присутствии кислот (3):
ROOH + R'OH—>R0OR' + H20 (3)
из Г. и галогеиалкилов или диалкилсульфатов в ще-
лочной среде (4):
ROONa + R'X—>ROOR'+NaX (4)
присоединением Г. к соединениям с неактивной двой-
ной связью в кислой среде (5):
(СН3)3СО О Н + (СН3)2С = СНСН3—>(СН3)3СО ОС(СН3)2СН2СН3 (5)
и к двойной связи, активированной полярными груп-
пами в щелочной среде (6):
(CHs)3COOH + CHa = CHCN —> (CH3)3COOCH2CH2CN (6)
из Г. и циклич. соединений типа окисей алкенов или
алкенимипов (7):
ROOH + CH2-CH2—>ROOCH,CH2OH (7)
\ /
о
Методом (3) можно получить несимметричные П.
Перекись диметила СН3—О—О—СН3— газ (т. кип.
13°), большая часть П.— перегоняющиеся в вакууме
жидкости. П. с нормальной цепью атомов углеро-
да, у которых радикал содержит 14 атомов С и бо-
лее, а также содержащие несколько циклов — твер-
дые вещества. Перекись диметила крайне взрывчата.
С увеличением мол. веса П. их взрывчатые свойства
уменьшаются. Наиболее устойчивы дитретичные П.
На практике используются перекиси дитретичного
бутила и дикумила. При нагревании или при облуче-
нии П. распадаются по связи —О—О—; ROOR^2RO •.
Как и в случае Г., могут проходить инициирован-
ные распады П., а также распад первичных RO-pa-
дикалов. При разложении П. в растворах свобод-
ные радикалы реагируют с растворителем. Восстанов-
ление П. (HJ, TiClj, SnCl2 и др.) идет с трудом и не-
полно.
Перекисная группа может входить в цикл. Такие П.
образуются при фотоокислении диенов с конъюгиро-
ванными связями: 1,4-эпипероксиды — из диенов с
открытой цепью (8):
При фотоокислепии непредельных циклич. соедине-
ний получаются трансанулярные П. (9). К таким П.
относится природная перекись — аскари-
дол (I). Известен ряд трансанулярных П.
стеринов.
Ароматич. П. АтООАт известны для
о,о,n-три- замещенных бензолов и про-
изводных нафталина; они легко получа-
ются окислением стерически затруднен-
ных фенолов и диссоциируют на свободные радикалы.
В интенсивноокрашенных парамагнитных радикалах
неспаренный электроп кислорода «размазан» обычно
сщснА
।
929
ПЕРЕКИСИ И ГИДРОПЕРЕКИСИ
930
на о- и n-положениях ядра. Радикал реагирует в хи-
ноидной форме С-радикала (10):
Эти формы проявляются при окислении р-ров ради-
кала, к-рое ведет к образованию П. (11).
Перекисные соединения, в кото-
рых углерод группы С—0—О —-сое-
динен еще с одним атомом кисло-
рода. Эта группа перекисных соединений весьма
многочисленна и разнообразна. Такие производные,
содержащие гидроксил, эфирную группу, а также вто-
рую группу —О—О—, могут быть получены несколь-
кими способами: 1. Окислением алифатич. или циклич.
спиртов, простых эфиров, диоксана, тетрагидрофурана
и др. Автоокисление ускоряется светом или добав-
ками катализаторов. Такие П. образуются при хра-
нении соответствующих органич. соединений при дос-
тупе воздуха. 2. Взаимодействием перекиси водорода
с альдегидами или кетонами образуются смеси пере-
кисных соединений. Вначале перекись водорода присо-
единяется к карбонильной группе, давая а-оксигидро-
перекиси: RCH(OH)OOH (из альдегида), R2C(OH)OOH
(из кетона). Они могут реагировать с избытком карбо-
нильного соединения, напр. для кетонов (12):
R он R R R
''(/ +О = С/ —>R - i-О - О - С-R (12)
RZ ХООН XR (!>Н ОН
или друг с другом с отщеплением воды и образованием
перекиси оксигидроперекиси (13); и при более глу-
боком процессе дегидратации получаются циклич.
диперекиси:
R ОН НОО R R ОН НОО R
ХС/ + ХС/ >- ХС/ Z \ Z \ -Н2о Z \ R ООН НО R R 0 XG/ —► Z \ —0 R
R 0-0 R \ Z \ Z
>- С С -Н2О Z \ Z \ R 0-0 R (13)
3. Присоединением перекиси водорода в кислой среде
к виниловым эфирам (14):
RO-CH = CH2 + H2O2—>ROCH(OOH) СН3 (14)
4. Присоединением Г. к альдегидам (15) или кето-
нам (16):
RCHO + R'OOH—> RCH(OH) OOR' (15)
R2C = O + R'OOH—>RaC(OH)OOR' (16)
Получающиеся а-окси-перекиси могут реагировать с
отщеплением воды с избытком Г., давая диперекиси
(17):
RjC(OH) OOR'+JR'OOH--->R3C(OOR')1 (17)
- Н2О
5. Действием озона на олефины в спиртовых р-рах.
Реакция проходит через биполярный ион (18).В инерт-
ных растворителях биполярный ион дает димерную
или полимерную П., а также может реагировать с
образовавшимся карбонильным соединением, давая
озонид (19):
о—о-о
I I +
RaC = CRa + 03 —»RaC-CRj—>R2C = O+R2C-OO- (18)
RaC-00~+R'0H —<-RaC(0R')00H
+ _ z°-\
2 RjC-OO -- R2C CRj
o-o
о---о
+ - II
r2c—оо + r2c=o—- R2C zCR2 (19)
о
П. этого класса взрывчаты. В присутствии следов во-
ды происходит распад с выделением перекиси водорода.
Такое разложение а-оксигидроперекиси, полученной
окислением изопропилового спирта, может служить
методом получения ацетона и Н2О2 (20):
(СН3)2С(ОН)ООН —> СН3СОСН3 + НаО2 (20)
а-Алкоксигидроперекиси при нагревании с водой
разлагаются (21):
СН3СН(ОС2Н3) 00Н + Н20 —>СН3СН0+С2НБ0Н + На0а (21)
Циклич. а-оксигидроперекиси при действии солей
Fe11 распадаются с образованием О-радикала, далее
идет раскрытие цикла с изомеризацией в С-радикал
и димеризация в дикарбоновую к-ту (22):
% , S
С-(СН2)8-С
Н(/ ХОН
(22)
П еркислоты. Особо важное значение имеют П.,
в к-рых перекисная группа соединена с карбонильной:
перкислоты RCOO2H и их эфиры и ацильные перекиси
RCOOOCOR'. Перкислоты получают: 1) окислением
альдегидов при низкой темп-ре; 2) действием на ки-
слоты или на ангидриды 98 %-ной Н2О2 в присутствии
1% H2SO4 и 3) гидролизом алкоголятами ацильных
перекисей (23):
(RCO2)2+R'ONa —> RC002Na + RC00R' (23)
Перкислоты, содержащие до 6 атомов углерода,—
жидкости; обладающие более высоким мол. весом,—
твердые вещества, растворимые в органич. растворите-
лях.Первые три кислоты гомологич. ряда смешиваются
с водой. Перкислоты — сильные окислители: окисля-
ют амины, сульфиды и др. При их взаимодействии с
олефинами образуются окиси алкенов — П риле Исаева
реакция (24):
RGO3H^C=C'C--> RCOaH + -(!-С —
z \ \z <24>
о
Реакция имеет широкое практич. применение. Эфиры
перкислот получаются из хлорангидридов и Na-солей
Г. (обычно третичных) (25). Из фосгена образуются
эфиры перугольной к-ты. При нагревании эфиры
перкислот распадаются на радикалы (26):
RCOCI + NaOOC(CH3)3 —>- RCO -00С(СНа)3 (25)
о
II
RG —О — О —С(СН3)3 —>. R- + СО2 + -ОС(СН3)3 (26)
Перекиси диацилов. Основным методом
синтеза этих соединений является реакция хлоран-
гидридов к-т с перекисью водорода в щелочном р-ре
или в присутствии пиридина (27):
2RCOCl + NaaOa—>RC0-00-C0R (27)
Смешанные ацильные П. получают взаимодействием
перкислот или их солей с хлорангидридами и ангид-
ридами к-т. Перекись ацетилбензоила и нек-рые
О 30 к.Х.Э. т. 3
931 ПЕРЕКИСИ И ГИДРОПЕРЕКИСИ—ПЕРЕКИСИ И ПЕРЕКИСНЫЕ СОЕДИНЕНИЯ 932
другие образуются при окислении бензальдегида в
присутствии ангидридов к-т. Ацильные П.— твердые
вещества, взрывчаты, особенно низшие члены гомо-
логии. ряда. Они легко восстанавливаются до кислот,
количественно выделяют иод из растворов KJ.
Ацильные П. распадаются на радикалы (28):
RCO-OO-COR >• 2RCO, —>-2R-+2CO2 (28)
При R=CHS, С2Н5, С,Н, не образуется RCO2, а сра-
зу происходит декарбоксилирование. Термический
распад ацильных П. является удобным методом полу-
чения свободных радикалов, что широко используется
в практике и в научных исследованиях. Такого рода
реакций крайне много. Напр., радикалы из П. могут
вырывать атом водорода из молекулы растворителя,
при этом вторичные радикалы, полученные из раство-
рителя, димеризуются. Такие процессы наблюдаются
для углеводородов, напр. для перекиси ацетила (29),
для кислот (30), для кетонов (31), полигалогенных
соединений (32):
RH+CH,---->CHt + R-; 2R---->R-R (29)
СН3- + СН3СО2Н —> CHt + CH2CO2H;
2СН2СО2Н —> НО2ССН2СН2СО2Н (30)
сн,-+сн3сосн3 —► сн.+сн3сосн2-:
2СНаСОСН,- —>СН3СОСН2СН2СОСН, (31)
СН,-+СНС1,—>СН, + СС13-; 2СС13-->- С3С1„ (32)
Такие реакции могут проходить далее между П. и
димерными продуктами, т. о. образуются из раство-
рителя дегидрополимеры. При нагревании П. (АтСО2)2
в ароматич. растворителях легко проходит арилиро-
вание (33):
(С,Н5СО2)2 + АгН —> СвН,Аг + СО2 + С,Н5СО2Н (33)
Бензоатные радикалы из перекиси бензоила могут
быть введены в нек-рые соединения (напр., эфиры,
диоксан и др.) при нагревании их с П. бензоила (34):
(С«Н5СО,)2 + С2Н5ОС2Н5 >
--► С,Н,ОСН (02СС,НБ) СН, + СеН,СО3Н (34)
Особенно важны процессы между ацильными П. и не-
предельными соединениями: радикалы, образовав-
шиеся из П. (RCO2- или R-), могут присоединяться
по двойной связи (35):
rco2-+с=с^-—>-RCO2-i-i- или
/ \ 1 I
\ / II
R- + С = С —>R-C-C- (35)
/ \ II
Новые радикалы могут реагировать далее с олефина-
ми — развитие цепной реакции полимеризации. При
взаимодействии П. со смесью олефинов и насыщенных
соединений к олефинам могут присоединяться сво-
бодные радикалы, образовавшиеся при взаимодей-
ствии П. с насыщенным производным. Т. обр. П. ини-
циируют реакции теломеризации и многочисленные
процессы присоединения к олефинам.
Элементоорганические переки-
с и. Известен ряд перекисных органич. соединений,
в к-рых группа —О—О— связана с гетероатомами
S, Р, Si, Ви др., а тажже с металлами металлоорганич.
соединений. Такие производные могут быть получены:
1) автоокислением, напр. (36):
RL1 + O2—s-ROOLi; R3SlLi + O2—>R3S1OOL1 (36)
Автоокисление, напр. соединений бора (37), прохо-
дит следующим образом:
R3B+O,—>-R373-OO+—>R,B-OO-R (37)
2) Исходя из Н2О2 или гидроперекиси (Г.), напр. (38):
ROOH+NaH—>ROONa + H2 1
ROOH + R2Zn—> R'ZnOOR+R'H J> (38)
H2O2+ 2(CH3)aCd —► CHaCd-00-CdCH, + 2CH. )
Для Si, Ge, Sn, Pb (в присутствии пиридина)
ROOH + R'siCl—► r's100R + HC1 (39)
П., получающиеся из перкислот, неустойчивы и
сразу перегруппировываются (40):
о OR
R3SI — 00 —ССеН5—►RjSi'' (40)
\
О-СОС„Н,
Практическое применение пере-
кисей. В практике П. используют весьма разно-
образно: 1) как добавки к моторному топливу,
отбеливающие материалы и др.; 2) как промежуточ-
ные продукты синтеза: получение фенолов и кетонов
разложением Г. алкилбензолов; получение перекиси
водорода распадом Г., образующейся при окислении
изопропилового спирта, или при разложении Г. алкил-
антрахинона; 3) для синтезов при помощи П.: полу-
чение дегидрополимеров при взаимодействии П. с
различными органич. соединениями (перекись отры-
вает водород), синтез эпоксипроизводных из олефи-
нов и перкислот (пербензойная к-та), использование
П. как окислителей (амины, сульфиды и др.). В этих
синтезах расход П. эквимолекулярен целевым про-
дуктам реакции; 4) особенно для разнообразных син-
тезов, инициируемых П. Сюда относятся многочис-
ленные реакции окисления кислородом, галогениро-
вания, присоединения к олефинам. Присоединение
галогеноводородов в присутствии П. проходит против
правила Марковникова. Особенно важны вызываемые
П. цепные процессы полимеризации и теломеризации.
В пром-сти широко используются перекиси бензоила
и третичного бутила для получения высокомолекуляр-
ных соединений. П. применяют в произ-ве привитых
и блок-сополимеров, при вулканизации каучуков
и других полимерных материалов. В практике часто
бывает необходимо предупредить образование П. или
их удалить. Образовавшиеся при автоокислении П. в
мономерах могут вызывать их несвоевременную поли-
меризацию. Очень опасны из-за взрывчатых свойств
П., получающиеся при хранении на воздухе орга-
нич. растворителей. Предложены методы удаления
перекисных соединений обработкой щелочами, про-
мыванием р-рами сульфата железа, пропусканием
через колонну с активным глиноземом или ионооб-
менными смолами и др. Для предотвращения обра-
зования П. к органич. продуктам добавляют ингиби-
торы — полиатомные фенолы, соли меди и др.
Лит.: Карножицкий В., Органические перекиси,
пер. с франц., М., 1961; Davies A. G., Organic peroxides,
L., 1961; Н a wk i ns E. G. E., Organic peroxides, L., 1961;
Toboisky A. V., Mesrobi an R. B., Organic pero-
xides, N.Y., 1954; H о u b e n—W e у 1, 4 Aufl., Bd 8, Stuttgart,
1952. Г. А. Разуваев.
ПЕРЕКИСИ И ПЕРЕКИСНЫЕ СОЕДИНЕНИЯ
неорганические — неорганич. соединения,
характеризующиеся наличием химически связанного
кислорода, легко выделяющегося в активной форме:
делятся на простые и комплексные. Кристаллич.
решетки простых неорганич. перекисных соединений
состоят из ионов металлов и из молекулярных анионов
кислорода: О2“, О2 иО, . К комплексным перекисным
соединениям относятся соединения, в к-рых п е р -
оксогруппа —О—О—(кислородныймостик), как
таковая или в виде Н2О2 и НО2, соединена с элемен-
том, образующим перекисное соединение, не ионной,
а координативной связью.
Простые неорганические перекисные соединения.
Эту группу соединений образуют преим. элементы I а
и II а подгрупп периодич. системы — щелочные и
щелочноземельные металлы. Эти соединения могут
быть разделены на следующие три подгруппы: пере-
киси, надперекиси, озониды.
933
ПЕРЕКИСИ И ПЕРЕКИСНЫЕ СОЕДИНЕНИЯ
934
Перекиси характеризуются наличием пере-
кисного иона О2 , со строением [:О:О:]2 .Длина
связи О—О в ионе О2 1,49 А.
Наиболее важные свойства перекисей щелочных и
щелочноземельных металлов приведены в табл. 1.
Фор- мула Содержание активного кислорода, вес. % Плот- ность, г/см3 Т. пл., °C Т. разл., °C Теплота образо- вания, ДНО298, ккал/моль Тип решетки
Li 2O2 34,8 2,363 (425)* 315-342 —152 + 4,0 гексаг.
Na2O2 20,5 2,60 > 59'6 (675)* -122+1,2 гексаг.
К2О2 14,5 2,40 (490)» — -118+10 ромбич.
Rb2O2 7.9 3,65 (567)* —- -101,5+10 ромбич.
Cs2O2 5.4 4,47 (597)» — - 96±15 ромбич.
СаО, 22,2 2,92 — 375-425 -156,4±1 тетраг.
SrOa 13,4 4,7 410-450 -153,2+4 тетраг.
ВаО2 9,4 5,43 — 790—900 -151,8^0,25 тетраг.
* Не вполне достоверные зиачеиия.
Высокая реакционная способность ще-
лочных металлов (за исключением лития)
по отношению к кислороду позволяет син-
тезировать их перекиси непосредственным
окислением металла кислородом при атмо-
сферном давлении. Эта способность обус-
ловлена тем, что Na, К, Rb и Cs, в отли-
чие от других металлов, обладают наиболь-
шими значениями атомного радиуса и на-
именьшими значениями энергии иониза-
ции. Литий же этими свойствами не обла-
дает, и синтез перекиси лития осуществим
лишь взаимодействием гидроокисис р-рами
Н2О2. Все перекиси щелочных металлов в
чистом виде бесцветны и диамагнитны, ак-
тивно взаимодействуют с влагой и угле-
кислым газом воздуха и поэтому их хранят в гер-
метически закрытой таре. Единственным соединением
этой группы, широко применяемым в народном хо-
зяйстве, является натрия перекись. Элементы Ij,
группы периодич. системы—Cu, Ag, Аи — не образуют
перекисей Me* О2-(Me — металл) и устойчивых пере-
кисей, в к-рых металл находится в более высоком
состоянии валентности, ни при непосредственном
окислении их кислородом, ни с помощью р-ров Н2О2.
Последнее обстоятельство объясняется тем, что Н2О2
проявляет как окислительные, так и восстановитель-
ные свойства (см. Водорода перекись), a Cu, Ag и Аи
обладают переменной валентностью. При попытке
синтеза перекисей Cu, Ag и Аи из соединений этих
металлов и растворов Н2О2 ионы этих металлов пе-
реходят в более высокое валентное состояние. При
этом может образоваться неустойчивая перекись, ко-
торая, однако, быстро восстанавливается до окиси.
Высшие кислородные соединения Си20з, Аи20з и AgO
являются окислами Cu’+, Аи3+ и Ag2 + , а не пере-
кисями.
Все элементы II гр. периодич. системы, за исклю-
чением Be, образуют перекисные соединения.Перекиси
Са, Sr, Ва принадлежат к типу Ме2+О2~. Перекисные
соединения Mg, Zn, Cd состава МепО2'хН2О, по-
видимому, относятся к типу НО—Me—ООН, с
ковалентной связью между пероксогрушюй и атомом
металла. Перекись бария ВаО2 получают окислением
ВаО в токе кислорода при 500—520°, перекисные со-
единения других элементов этой группы — взаимодей-
ствием соответствующих гидроокисей с р-рами Н2О2.
Перекиси Са, Sr, Ва и гидратные формы перекисей
Mg, Zn и Cd в чистом виде бесцветны и диамагнитны;
перекись ртути HgO2 желтого цвета. Практич. приме-
нение имеет в основном ВаО2 (для получения Н2О2, в
органич. синтезе, в пиротехнике, для покрытия термо-
ионных катодов). В меньшей степени применяют пе-
рекись кальция (в хлебопечении, вулканизации бутил-
каучука), перекись стронция (в пиротехнике), гидрат-
ные формы перекисей магния и цинка (в медицине).
Надперекиси характеризуются
наличием надперекисного иона 02 , со
строением р0210:]-. Длина связи О—О в
ионеО2 1,32—1,35 А. Известны лишь над-
перекиси щелочных металлов Na —Cs. Их
свойства представлены в табл. 2.
Надперекиси калия КОа, рубидия ВЬОа
и цезия CsOa образуются преим. при сго-
рании этих металлов в атмосфере кислоро-
да или воздуха, обогащенного кислородом,
при атмосферном давлении. Их получают
также окислением кислородом растворов
этих металлов в жидком аммиаке при
температуре ниже —50°. Надперекись
Таблица 2
Фор- мула Содержание, активного кислорода, вес. % Плот- ность, г/см3 Т. пл., °C Т. разл., ’С Теплота образо- вания, дн°298, ккал/моль Тип решетки
NaO2(I)* 43,6 2,21 — >540 —62.1 + 0,7 кубич. граней.
ц-КО2** 33,8 2,158 (535) 543 —67,6±0,8 тетраг.
RbO2 20,4 3,06 412 >567 —63.0± 10 тетраг.
CsO2 14,5 3,80 450 >597 —62,0± 1 0 тетраг.
* У NaO2 имеются две низкотемпературные модификации Na02 (II) — ку-
бическая, и NaOs (III) — ромбическая.
** У К02 имеется еще g-модификация — кубич. гранецентрированная, ус-
тойчивая при (>100°.
натрия NaO2 (впервые полученная И. А. Казарнов-
ским) образуется при окислении кислородом перекиси
натрия Na2O2 при сравнительно высокой температуре
и повышенном давлении. Надперекиси щелочных
металлов окрашены и парамагнитны, что обусло-
влено наличием неспаренного электрона в ионе
О”. Они активно взаимодействуют с влагой и угле-
кислым газом, и поэтому их хранят в герметически
закрытой таре; легко окисляют органич. вещества и
могут вызывать их воспламенение. Из всех надпереки-
сей NaO2 и КО 2 имеют практич. значение и произво-
дятся в промышленном масштабе. Их используют
для регенерации выдыхаемого воздуха в дыхательных
аппаратах изолирующего типа и в генераторах кисло-
рода типа аппарата Киппа.
Озониды характеризуются наличием озонид-
ного иона О, со строением
Длина связи О—О в ионе О2 1,34 ± 0,05 А. Извест-
ны озониды щелочных металлов и аммония. Они ок-
рашены в красный цвет и парамагнитны. Ион О,
с нечетным числом электронов является носителем
огромной химич. активности озонидов, к-рые можно
рассматривать как свободные радикалы с необычно
большой продолжительностью жизни. Среди озо-
нидов наиболее изученным является озонид калия
КО3. Кристаллич. решетка КОз тетрагональная с
параметрами: я = 8,597 ± 0,002 А и с = 7,080 £
30*
935
ПЕРЕКИСИ И ПЕРЕКИСНЫЕ СОЕДИНЕНИЯ
936
±0,002 А, ДЯ°88 =—62,1 ± 0,9 ккал/моль. Предел
термич. устойчивости 60 ± 2°. Содержание активного
кислорода 46,0 вес. %. Озониды щелочных металлов
получают взаимодействием озонированного кислорода
с гидроокисями с последующей экстракцией жидким
аммиаком.
Различие характера связи между атомами кисло-
рода в «мостике» для перекисей, надперекисей и
озонидов отчетливо проявляется при гидролизе.
Перекиси гидролизуются по ур-нию Ме2О2 + 2Н2О^
->2 МеОН + Н2О2, с образованием перекиси водоро-
да, количественно титруемой перманганатометрически
в кислой среде. Надперекиси гидролизуются с обра-
зованием промежуточного радикала НО2 — гидро-
пероксила — по ур-ниям:
2МеО3 + 2Н3О —> 2МеОН + 2НО3
2Н.Оа —> Н2О2 + О2
2МеО3 + 2Н3О —>- 2МеОН + Н2О2 + О2
Вследствие этого 2/, активного кислорода, к-рые
принято наз. «надперекисным кислородом», выделя-
ются в виде О2 и определяются газометрически, а
’/» активного кислорода, к-рую принято наз. «пе-
рекисным кислородом», выделяется в виде Н2О2 и
титруется перманганатометрически в кислой среде.
Озониды гидролизуются без заметного образования
H2O2, по ур-нию МеОз + Н2О^-МеОН + ОН + О2.
К простым перекисным соединениям можно отнести
и гидроперекиси, общей ф-лы ЭООН (где
Э— элемент). Они характеризуются наличием г и д-
ропероксильного иона НО~. В этих соеди-
нениях, к к-рым следует отнести и перекись водоро-
да, пероксогруппа соединена ионной связью с эле-
ментом, образующим гидроперекись, и ковалентной
связью с водородом. Ионная природа гидроперекисей
экспериментально доказана лишь для соединения
NH4OOH и тем, что Н2О2 является слабой к-той.
Необходимо отметить, что нек-рые гидроперекиси
могут быть отнесены к пероксигидратам, другие —к
пероксокислотам. Напр., соединение NaOOH может
быть представлено в виде димера МагОг-НгОг, а со-
единение (ОН)з TIOOH можно рассматривать как пер-
оксокислоту.
Комплексные перекисные соединения. Все эти
соединения могут быть разделены на след, пять групп:
пероксокислоты, перекисные комплексы, гидраты пе-
рекисей, пероксигидраты, гидраты пероксигидратов.
Пероксокислоты и их соли, в к-рых пер-
оксогруппа —О—О—входит в состав комплексного
аниона. Известны одноядерные и многоядерные пер-
оксокислоты и их соли. В этих соединениях координи-
рующими являются, в большинстве случаев, атомы
элементов IV—VI групп периодич. системы — неме-
таллов С, N, Р, S или металлов Ti, V, Cr, Мо, W и др.
Примером одноядерной пероксокислоты служит иерок-
сомоносерная к-та
го о-олн
V
-</ хо Jh
примером многоядерной — пероксодисерная к-та
го о-о олн
(см. Пероксосерные кислоты). Пероксокислоты и соли
пероксокислот типич. элементов периодич. системы
синтезируют преим. электролитич. путем; соответст-
вующие соединения переходных металлов—взаимодей-
ствием солей их кислородных к-т с Н2О2. Сохранив-
шиеся в химич. литературе названия «персульфаты»,
«перкарбонаты», «пербораты» и т. д. для обозначения 1
солей пероксокислот являются весьма неудачными
и приводят к неправильному представлению об их
сходстве с солями нек-рых кислородных кислот, как,
напр., перхроматами, перманганатами, перренатами
и т. д. В последних соединениях префикс «пер» озна-
чает лишь высшую степень окисления элемента, а не
наличие пероксогруппы. Для солей пероксокислот
правильными являются термины: пероксобораты, пер-
оксокарбонаты, пероксосульфаты, пероксофосфаты и
т. д. Количественное определение активного кисло-
рода в пероксокислотах и их солях проводят обыч-
но иодометрически, так как их гидролиз до Н2О2
проходит ступенчато, через образование промежуточ-
ных перекисных соединений и иногда со значительным
выделением молекулярного кислорода.
Перекисные комплексы, не являю-
щиеся пероксокислотами или их производными, также
делятся на одноядерные и многоядерные. Одноядерные
могут быть представлены соединениями, включаю-
щими во внутренней сфере пероксогруппу, напр.
[(UO2)2 (О2)г (Н2О)8]. Соединения такого типа могут
быть объединены в подгруппу пероксоком-
п л е к с о в. Одноядерные перекисные комплексы
могут быть также представлены соединениями, вклю-
чающими во внутренней сфере молекулы H2O2, как,
напр., [Fe(H2O)5 (НгОг)]’4-. Соединения такого типа
составляют подгруппу пергидрокомплексов.
Наконец, во внутренней сфере могут быть координи-
рованы пергидроксильные радикалы НО2, как, напр.,
в соединении K4[(UO2)2 (О2)з (НО2) (ОН) (Н2О)].
Соединения такого типа составляют подгруппу
гидропероксокомплексов. Примером
многоядерных перекисных комплексов, не являю-
щихся производными пероксокислот, могут служить
соединения типа:
о-о
/ \
O2Zr ZrO3
4 so/
Н, [Cl5Re-O-O-ReCl5]-2H3O и
[(NH,), Co-O-O-Co(NH,)5] (NO,)s
Перекисные комплексы, не являющиеся пероксоки-
слотами или производными пероксокислот, в большин-
стве случаев гидролизуются с образованием H2O2,
но нек-рые многоядерные перекисные комплексы,
как, напр., [(NHs)5Co—О—О—Co(NH3)i]5 + , весьма
устойчивы по отношению к воде.
Гидраты перекисей — аддитивные со-
единения перекисей с кристаллизационной водой.
Пероксигидраты — молекулярные соеди-
нения, аналогичные кристаллогидратам, содержащие
кристаллизационную перекись водорода. Известны
пероксигидраты перекисей, напр. СаОг^НгОг, и
пероксигидраты солей, напр. КгСО-ЗНгОг. При вза-
имодействии пероксигидратов с водой кристаллиза-
ционная перекись водорода переходит в раствор и
может быть определена количественно перманганато-
метрически.
Гидраты пероксигидратов — моле-
кулярные соединения, содержащие и кристаллиза-
ционную воду и кристаллизационную перекись водо-
рода, напр. ВаОг-НгОг^НгО и МагЭСП-О^НгОг-НгО.
В табл. 3 приведена схема классификации и но-
менклатуры неорганич. перекисных соединений.
Различие в строении неорганич. перекисных соеди-
нений обусловливает различие их физич. свойств и
реакционной способности и, следовательно, возмож-
ность их применения в самых разнообразных услови-
ях. Перекись водорода, ее производные или предста-
вители других вышеуказанных групп перекисных со-
единений с каждым годом завоевывают новые и новые
области применения: текстильную и целлюлозно-бу-
937
ПЕРЕКИСНАЯ ТЕОРИЯ БАХА—ЭНГЛЕРА —ПЕРЕКИСНЫЙ ЭФФЕКТ
938
Таблица 3
Тип Группа Формула
/ Гидроперекиси I МеООН
Про- стые Перекиси Надперекиси I Ме3О, I МеО,
Озониды ' Пероксо- кислоты и их соли Перекис- ные комп- лексы одноядерные V многоядерные z / пероксо ядер- < пеР™Д₽о < ные гидропер- 1 оксо I МеО, [90.]»-; [ЭОЕ]’-; [ЭОЕ]»~ [9,0,]»-; [Э2О8]‘-; [Э,О,]»- [Ме (О3)в (A)j,]"+ [Me (НаО,)я (А)у]"+ [Me (НО2)„(А)у]п+
Комп- \ многоядерные [(А)уМе - О - О -Me (A),]" +
ле к— сные Гидраты перекисей I Ме2Оа-хНаО
Пероксигидраты перекисей солей I МеаОа«х НаОа I МелЭОш.хН2О2
Гидраты перокси- J гидратов | перекисей солей I Me3O2xH2O3- yH2O I МепЭОт-хН2О3-уН3О
Ме-металл; Э-элемент; А-адденд .
мажную пром-сть (в процессах отбеливания и краше-
ния естественных и искусственных волокон и от-
беливания древесной массы), медицину и косметич.
пром-сть, строительное дело (для произ-ва пористого
бетона), пищевую пром-сть (для отбеливания жиров
и масел, консервирования ряда продуктов и в хлебо-
печении), с. х-во (для ускорения созревания семян и
в борьбе с вредителями), пиротехнику, извлечение
нек-рых металлов из рудных концентратов, травление
металлич. поверхностей, тонкую неорганич. и орга-
нич. технологию, произ-во стиральных порошков и
синтетич. моющих средств и др. Кроме того, неорга-
нич. перекисные соединения играют важную роль в
процессах радиолиза, протекающих в гомогенных атом-
ных реакторах; они служат компонентами жидкостных
реактивных двигателей и смесей, используются для
регенерации воздуха в закрытых помещениях.
Лит.: Перекись водорода и перекисные соединения,
под ред. М. Е. Поаина, Л,—М., 1951; Шамб У., Сеттер-
филд Ч., Вентворс Р., Перекись водорода, пер. с
англ., М., 1958; Химия перекисных соединений. Сб. статей,
М., 1963; Писаржевский Л. В., Избранные труды. [Сб.
работ в честь 40-летнего юбилея], Киев, 1936. И. И. Вольнов.
ПЕРЕКИСНАЯ ТЕОРИЯ БАХА — ЭНГЛЕРА —
теория, представляющая окисление как процесс, пер-
вой стадией к-рого является присоединение молеку-
лы кислорода О2 к активной молекуле окисляемого
вещества А' с образованием промежуточного пере-
кисного соединения АО2, а второй стадией — окисле-
ние этой перекисью обычных молекул вещества А.
I стадия А' + О2=АО2
II стадия АОа+А=2АО
Теория была сформулирована А. Н. Бахом (1893—
1897) и К. Энглером (1897). Современные схемы цеп-
ного окисления органич. веществ показывают боль-
шую роль промежуточных перекисных соединений
в этих процессах (см. Цепные реакции).
ПЕРЕКИСНЫЙ ЭФФЕКТ (пероксидный эффект) —
изменение порядка присоединения бромистого водо-
рода к несимметричным олефинам в присутствии пере-
кисей. Так, напр., свежеприготовленный бромистый
аллил в отсутствии кислорода воздуха и в темноте
очень медленно присоединяет НВт в соответствии
с Марковникова правилом с образованием
только СНзСНВгСН2Вт. Такой порядок при-
соединения объясняется ионным характе-
ром реакции и соответствует полярности
реагентов:
Н л- л-
Н^С-СН-СИ, + НВт" ----- СН3СН-СНЭ
Н Вг
При применении же бромистого аллила, хра-
нившегося нек-рое время в присутствии воз-
духа, НВт присоединяется оченьбыстрои об-
разует 1,3-дибромпропан ВтСН2СН2СН2Вт.
К образованию этого изомера приводит так-
же добавление готовых перекисей, напр. пе-
рекиси бензоила, или же облучение. Изме-
нение порядка присоединения объясняется
изменением характера реакции и переходом
от ионного механизма к радикально-цеп-
ному:
НВг + О2'—>НОО- + -Вг
ВгСН2СН=СН2 + • Вг—> ВгСН2СНСН2Вг
ВгСН2СНСН2Вг + НВг —>• ВгСН2СН2СН2Вг + • Вг
и т. д.
Присоединение атомарного брома к мети-
леновой, а не к метиновой группе объяс-
няется тем, что образующийся радикал
ВтСН2СНСН2Вг (радикал А) более устой-
чив, чем радикал ВтСН2СНВтСН2’ (Ради-
H. в радикале А неспаренный электрон
кал Б), т.
атома С может находиться в сопряжении с а-элект-
ронами четырех С—Н-связей, а в радикале Б воз-
можно сопряжение лишь с одной С—Н-связью:
В. -CHt-C — СН,
в>
Такое объяснение (а не атака со стороны -Вг,
обладающего электрофильным характером, места с
наибольшей электронной плотностью) вытекает пз
многих опытных данных, напр. при облучении смеси
ВСН=СН2 и CFsJ группа CFs всегда присоединяется
исключительно к метиленовой группе, даже при
R = CFs, СООСНз, CN, когда поляризация двойной
связи противоположна поляризации несимметричных
незамещенных олефинов.
Фтористый водород всегда присоединяется по ион-
ному механизму, т. к. перекиси не могут вызвать го-
молитич. разрыв очень прочной связи Н — F. Ио-
дистый водород, обладающий восстановительными
свойствами, раскисляет перекись и поэтому также
может присоединяться лишь по ионному механизму.
П. э. в нек-рой мере сказывается и щ>и присоединении
НС1. Так, в присутствии перекиси бензоила НС1 при-
соединяется к трет-бутилэтилену и хлористому ал-
лилу с образованием смесей, содержащих незначи-
тельную примесь продуктов радикально-цепной ре-
акции. Прибавление антиоксидантов (гидрохинона,
тиокрезола, дифениламина и др.) снимает П. э., так
как они легко реагируют с образующимися активными
радикалами (СНзСОО-, Вт- и т. д.) с образованием
неактивных радикалов, могущих лишь димеризо-
ваться, но не способных продолжить цепную реакцию:
R- +H-S-CBH5 •—>RH + C„H;S-
2C„HES —> C,HESSC,H.
П. э. наблюдается при присоединении НВт не толь-
ко к этиленовым, но и к ацетиленовым соединениям
и диолефинам.
Лит.: Kharasch М. S., Mayo F. R., J. Amer.
Chem. Soc., 1933, 55, Ks 6, 2468, 1938, 60, Ks 12, 3097; Mayo
939
ПЕРЕМЕТИЛИРОВАНИЕ—ПЕРЕМЕШИВАНИЕ
940
F. R., Walling Ch., Chem. Revs, 1940, 27, № 2, 351;
MayoF. R., J. Amer. Chem. Soc., 1954, 76, Ks 5, 5392; T e M-
u и к о в а Т. И., Курс теоретических основ органической
химии, 2 изд., Л., 1962. Я. Ф. Комиссаров.
ПЕРЕМЕТИЛИРОВАНИЕ (трансметилирование)—
ферментативная реакция межмолекулярного пере-
носа СНз-групп. Играет важную роль при био-
синтезе многих веществ биологич. значения (напр.,
креагина, адреналина, ансерина, N-метилникотин-
амида, алкалоидов и др.). Основным донором метиль-
ных групп при реакции П. является S-аденозил-
L-метионин (I), образующийся ферментативно из
метионина и аденозин-5-трифосфата:
N = C-NH3
I I H3N-СН-СООН
НС С-N |
II II /СН (СНз)»
N —С —N —СН(СН3ОН)2 -СН—СН3—S —СН,
1-----о-----1
I
(СНа)„Т?-СН3СН3ОН (СН3),Й-СН3-СОО-
II III
Возможен обратимый ферментативный перенос ме-
тильных групп холина (II) или бетаина (III) на го-
моцистеин (а-амино-у-тиомасляную к-ту) с образо-
ванием метионина, к-рый затем превращается в (I).
Необратимым является катализируемый специфич.
ферментами перенос метильных групп (I) на атомы
азота, серы (но не углерода) таких акцепторов, как
гуанидинуксусная к-та, норадреналин, карнозин,
никотинамид и др. Нарушения процессов П. (напр.,
при недостаточном поступлении в организм метиони-
на в пищевыми продуктами) приводят к возникнове-
нию тяжелых функциональных расстройств (жировая
дегенерация печени). Поэтому метионин широко при-
меняется для подкормки животных. Строение ката-
литич. центров многочисленных ферментов, участ-
вующих в П. (в частности, роль витамина В12 в их
действии), еще не достаточно изучено. Эти ферменты
интенсивно исследуются в связи с проблемой их
участия в превращениях ряда биогенных аминов
(норадреналин, адреналин, дофамин, гистамин, се-
ротонин) в организме животных и человека в норме
и при патологии, состояниях (болезнь Паркинсона,
стенокардия).
Лит.: Браунштейн А. Е., Биохимия аминокислот-
ного обмена, М., 1949; Майстер А., Биохимия аминокис-
лот, пер. с англ., М., 1961; Смит Л., Витамин Б13, пер.
с англ., М., 1962; Черкес Л. А., Холин как пищевой фак-
тор и патология холинового обмена, М., 1953; Axelrod J.,
J. Pharmacol, and Exptl Therap., 1962, 138, Ай 1, 28.
ПЕРЕМЕШИВАНИЕ — процесс, применяемый в
химич. произ-вах для получения эмульсий, суспен-
зий и смесей твердых веществ, а также для интен-
сификации процессов массообмена и теплообмена.
В зависимости от того как проводятся химич. реакция
и П. (раздельно или они совпадают во времени) про-
цесс проводят в смесителях или непосредственно в
реакционных аппаратах, снабженных мешалками.
Выбор метода П. и аппаратуры для его осуществления
обусловливается, в первую очередь, агрегатным со-
стоянием перемешиваемых материалов. Соответствен-
но подразделяют: П. в жидкой и П. в твердой сыпу-
чей средах.
Перемешивание в жидких средах. Этот вид П. при-
меняют в химич. технологии для ускорения реакций
в гетерогенных средах, получения стойких эмуль-
сий и суспензий, а также интенсификации тепловых
и диффузионных процессов. Наибольшее распро-
странение получило механич. П. посредством разно-
образных лопастных мешалок, вращающихся в жид-
кости. Реже применяют П. путем барботажа газов
пли воздуха через жидкость, а также дросселировани-
ем или циркуляцией жидкости.
Механическое перемешивание.
Различают след, группы механич. мешалок: лопа-
стные (двух-, трех- и многолопастные с горизонталь-
ными лопастями, рамные), якорные, пропеллерные,
турбинные открытого и закрытого типов и специаль-
ные. Работа мешалок характеризуется интенсивно-
стью и эффективностью перемешивания, а также по-
требляемой энергией.
Лопастные мешалки состоят из прямо-
угольных пластин, укрепленных на валу в плоско-
сти его оси в один или несколько рядов (рис. 1, а
и б); обычно применяют для перемешивания маловяз-
ких и легко смешиваю-
щихся жидкостей, а так-
же в случае смешива-
ния хорошо раствори-
мых порошкообразных
сыпучих материалов с
жидкостями. Для пере-
мешивания густых жид-
костей к стенкам аппа-
рата прикрепляют до-
полнительные горизон-
тальные неподвижные
перегородки (рис. 1, в),
препятствующие вра-
щению массы вместе С Рис. 1. Лопастные мешалки: а —
лопастями мешалки И двухлопастная многоярусная; б—
мттмчшяюптиопплпосс П трехлопастная; в — двухлопастная
улучшающиепроцесс и. многоярусная с неподвижными пе-
Разновидностью лопа- регородками; г—рамная; 1—со-
стных мешалок являют- суд; 2 — лопасти; з — неподвиж-
ся рамные мешалки ные перегородки.
(рис. 1, г), применяемые
для П. больших объемов жидкостей малой и средней
вязкости. Экспериментально установлено, что различ-
ные типы лопастных мешалок при разных скоростях
вращения и прочих равных условиях (одинаковых
отношениях радиуса лопастей к радиусу сосуда 7?Л/Я,
вязкости, расходуемой энергии) приводят поток
жидкости в сосуде в одинаковое гидродинамич. со-
стояние, характеризуемое определенной деформа-
цией свободной поверхности жидкости (профилем
воронки). Таким образом, при П. маловязких жидко-
стей (р. от 1 до 100 спуаз) целесообразно применять
самые простые конструктивные формы радиально-
лопастных мешалок; число их должно быть сведено
до рационального минимума, поскольку при одина-
ковом расходе энергии их эффективность не уступает
более сложным. С точки зрения правильного распре-
деления силовых нагрузок наиболее целесообразными
являются трехлопастные мешалки с расположением
лопастей под углом 120°.
Отношение радиуса лопастей мешалок к радиусу
сосуда R^/R при работе их в маловязких жидких
средах следует принимать равным 0,5, т. к. в этом
случае мощность, необходимая для перемешивания,
будет наименьшей. При П. более вязких жидкостей
(р, > 100 спуаз) указанное соотношение следует при-
нимать равным 0,66 (учитывая уменьшение сечения
вихревого потока с повышением вязкости).
Высоту уровня жидкости в сосуде следует прини-
мать равной (0,75—1,0) D (где Л—диаметр аппарата),
скорость вращения конца лопастей мешалки (по внеш-
ней окружности) 2—5 м/сек. П. жидкости более эко-
номично производить в сосудах без дополнительных
устройств и перегородок, лопасти мешалок целесо-
образно располагать по оси сосуда.
Мощность, потребляемую трехлопастной мешалкой (рис.
1, б) при установившемся движении при перемешивании мало-
вязких жидкостей, можно определить по ф-ле:
Г^л+мС (0>25+in5i)l
34 L 4 \ 2 Л / \ Hq / J
941
ПЕРЕМЕШИВАНИЕ
942
где N — мощность, кет: ю — угловая скорость лопасти,
1/сек; h — высота лопасти, м; — радиус лопасти, м; Ro —
радиус вихря, м; С — интенсивность циркуляции, м2/сек;
— плотность жидкости, кг/м3.
Интенсивность циркуляции определяют по ф-ле: С=ю17?0,
где Wj— угловая скорость вихревого потока. При работе
трехлопастных мешалок в цилиндрич. сосудах диаметром от
600 хн и выше, наполненных жидкостями до уровня Hy=D,
угловую скорость вихревого потока при 7?л/7?=0,5 можно
определить по ф-ле:
где 7?э — радиус эталонного сосуда, равный 0,12 м; цв— абс.
вязкость воды при 20°, спуаз; — абс. вязкость перемеши-
ваемой жидкости при 20°, спуаз.
Радиус вихря определяют по ф-ле:
Яе = Лехр
л е °*25
0,5+---г-т—
l+tgiy
R3 -
2Яу
где пг в большинстве случаев равно 2; R — радиус сосуда;
7?У—радиус сечения воронки при работе мешалки по статич.
уровню жидкости; tgy== 0,527 (Л/Л^о»73— экспериментальная
зависимость между окружной и радиальной скоростями по-
токов для различных радиально-лопастных мешалок (7?д—
радиус лопастной мешалки); R? определяют по ф-ле:
Яу=д [ 0,508 + 0,215 (^_о,3
Якорные мешалки (рис. 2, а и б) отли-
чаются тем, что лопасти строго следуют контуру
Рис. 2. Якорные мешалки: а — с
прямолинейными лопастями; б — с
криволинейными лопастями.
днища и стенок аппара-
та, образуя с ними нез-
начительный зазор (5—
20 мм). Мешалки этого
типа применяются для
перемешивания густых
жидкостей (вязкостью
10—20 тыс. спуаз), ког-
да требуется предупре-
дить пригорание и обра-
зование накипей и осад-
ков на стенках сосу-
дов.
Пропеллерные мешалки (рис. 3) пред-
ставляют собой гребной винт с 2—4 лопастями (чаще
всего 3). При вращении винта происходит интенсив-
ная циркуляция жидкости с сильными
вихреобразованиями, обеспечивающи-
ми энергичное перемешивание. Мешал-
ки этого типа применяются для пере-
мешивания жидкостей с вязкостью до
4000 спуаз, получения суспензий и
эмульсий, поддержания тяжелых ча-
стиц во взвешенном состоянии, интен-
сивного охлаждения жидкости в про-
цессе нитрации и т. д. Диаметр про-
Рис. з. Пропел- пеллера обычно колеблется от */4 до
лерная мешалка. диаметра аппарата, окружная ско-
рость от 6—7 до 11—12 м/сек.
Необходимая для перемешивания жидкости мощность на
валу пропеллерной мешалки, потребляемая при установив-
шемся режиме работы, может быть определена по ф-ле:
^ = 0,845Q0^95 p.o,os П2,»з d4>9 (кгм[сек)
где d — диаметр пропеллерной мешалки, м; Q — плотность
жидкости, г/см3; р. — абс. вязкость, пуаз; п — число об/сек.
Этому ур-нию отвечает прямая в турбулентной области
(50<Яем<10’, где Re^—модифицированный критерий Рей-
нольдса).
Турбинные мешалки работают на прин-
ципе всасывания и выброса жидкости рабочим коле-
сом; эти мешалки бывают двух типов: открытые, име-
ющие рабочее колесо с плоскими (рис. 4, а) или криво-
линейными лопатками (рис. 4, б), и закрытые (рис.4,в),
имеющие рабочее колесо с каналами. Диаметр тур-
бомешалки d выбирается в зависимости от диаметра
Рис. 4. Турбинные ме-
шалки: а — с плоскими
лопатками; б — с кри-
волинейными лопатка-
ми; в — турбинная ме-
шалка закрытого типа.
аппарата D; при D = 1,6 м (для аппаратов емкостью
меньше 0,75 л?) d = 0,33—0,5 D; для сосудов с большим
диаметром d = 0,25—0,33 D, причем большие значения
берутся для более .вязких жидкостей. Окружная ско-
рость конца лопаток турбинных мешалок колеблется
в пределах 3—9 м/сек, причем ско-
рость мешалок диаметром меньше
300 л.н составляет 6—9 м/сек, а ме-
шалок диаметром больше 300 мм —
3—6 м/сек. Турбинные мешалки
применяют для перемешивания
жидкостей вязкостью до 20 тыс.
спуаз, интенсивного перемешива-
ния и диспергирования жидкостей
в больших объемах (до 4,5—6 м*)
и для перемешивания суспензий с
крупными твердыми частицами.
Дисковая мешалка
(рис. 5) применяется для перемеши-
вания жидкостей с разными плот-
ностями. Мешалка состоит из 2 дис-
ков, укрепленных на небольшом
расстоянии один от другого на вер-
тикальном валу и вращающихся с
большой скоростью в направляю-
щих цилиндрах. Для увеличения
интенсивности перемешивания к
цилиндрам прикреплены три кры-
ла 3, не позволяющие жидкости
вращаться с перфорированными Рис. 5. Дисковая ме-
дисками. шалка: 1 -цилиндры;
Диафрагменный смеси- ®
тел ь (рис. 6) представляет собой ные крылья.
цилиндр, по оси к-рого помещен
стержень, несущий 4—6 дисков (диафрагм) с несколь-
кими отверстиями в каждом. Смешиваемые жидкости
Рис. 6. Диафрагменный смеситель: а—схема аппарата;
б — схема движения жидкости через отверстия в диа-
фрагмах.
проходят последовательно через отверстия в дисках.
При подходе к отверстиям струйки жидкости сталки-
ваются, что способствует равномерному смешению
обеих жидкостей.
943
ПЕРЕМЕШИВАНИЕ — ПЕРЕНАПРЯЖЕНИЕ
944
Переммппвяние сыпучих материалов. Это П. приме-
няю! с целью получения из двух или более компонен-
тов, взятых в определенных соотношениях, однород-
ной в любом объеме сыпучей массы. Для этого поль-
зуются след, смесителями.
Шнековые смесители (рис. 7) представ-
ляют собой корыто 1, в к-ром вращаются два парал-
лельных горизонтальных вала 2 с плоскими лопас-
тями 3, насаженными на валы по винтовой линии через
Рис. 7. Шнековый барабан: 1 — корыто; 2 — валы;
3 — лопасти.
определенные промежутки, образуя как бы преры-
вистую винтовую поверхность. При вращении ва-
лов материал, находящийся в смесителе, перемеши-
вается по направлению их вращения и вдоль оси
шнека.
Смесовые барабаны (рис. 8) представля-
ют собой горизонтальный стальной барабан 1, на
Рис. 8. Смесовой барабан: 1 — барабан; 2 — винтообразные лопасти; 3 — пе-
регородки; 4 — шнек; 5 — загрузочный патрубок; 6 — разгрузочный патрубок.
внутренних стенках к-рого укреплены винтообразные
лопасти 2 и тангенциальные перегородки 3, не дохо-
дящие до оси барабана. Для загрузки и выгрузки
материала служит шнек 4. Материал поступает в ба-
рабан через патрубок 5. После окончания перемеши-
вания направление вращения барабана меняется на
обратное и материал тем же шнеком выгружается из
барабана через патрубок 6.
Центробежный смеситель (рис. 9).
Его действие основано на смешении материала в рас-
пыленном состоянии, осуществляемом при вращении
конуса 1 с бурно движущимся потоком материала. Ча-
стицы под действием центробежной силы поднимаются
по конусу и образуют параболоид вращения, к-рый
разрушается при помощи ножей 8; частицы сбрасы-
ваются с конуса, после чего через окна 6 они снова
поступают в конус. Цикл многократно повторяется.
В нижней части конуса имеются скребки 7, к-рые при
вращении устраняют мертвые зоны и усиливают дви-
жение материала. Лопасти - приводятся в движение
самим материалом, способствуют разделению потоков
массы и продвижению ее в окна конуса. Для регули-
рования скорости вращения лопастей имеется тормоз-
ное устройство 4. Смеси-
тель предназначен для тон-
кого смешения легко под-
вижных сыпучих материа-
лов, для смешивания су-
хих красок, приготовле-
ния пудры и других сме-
сей. На крупных предпри-
ятиях химической промыш-
ленности такие смесите-
ли часто устанавливаются
батареями.
Рис. 9. Центробежный смеси-
тель: 1 — конус; 2 — корпус;
3 — лопасти; 4 — шкив; 5 —•
тормозное устройство; в — окно; 7 — скребок; 8 — нож;
9 _вал; 10 — загрузочный люк; 11 — разгрузочный люк.
Лит.: Касаткин А. Г., Основные процессы и аппа-
раты химической технологии, 7 изд., М., 1961; Канторо-
вич 3 Б., Машины химической промышленности, т. 1, М.,
1957; П л а н о в с к и й А. Н., Н и к о л а е в П. И., Про-
цессы и аппараты химической и нефтехимической технологии,
М., 1960; С к о б л о А.И., Трегубова И. А., Егоров
Н. Н., Процессы и аппараты нефтеперерабатывающей и неф-
техимической промышленности, М., 1962; Гзовский С. Я.,
Плановский А. Н., Вестник технической и экономиче-
ской информации, 1959, № 5. С. Я. Гзовский.
ПЕРЕНАПРЯЖЕНИЕ — часть общего сдвига по-
тенциала электрода от его равновесного значения,
обусловленное электрохимия, поляриза-
цией. Различают катодное и анодное
цд перенапряжение:
Т)к = (ро_(рк Лд^фд_фо
где ср' — равновесный потенциал катода
или анода; <рк и <рд — потенциалы соот-
ветственно катода и анода при прохожде-
нии тока (см. Поляризация электрохими-
ческая).
Величина П. зависит от природы элек-
тродной реакции. Наиболее высокие зна-
чения П. наблюдаются в случае электро-
литич. выделения водорода (пер е н а п-
ряжение водорода) и кислоро-
да (перенапряжение кисло-
рода). Наиболее изучено П. водорода
на металлич. катодах. П. водорода воз-
растает с увеличением плотности тока,
зависит от природы катода, состава рас-
твора и других факторов. При малых
плотностях тока, когда значения П. суще-
ственно меньше R Т/F (при комнатной темп-ре 0,025 в),
наблюдается линейная зависимость перенапряжения ц
от плотности тока г, т. е. ц = coi, где со — константа,
к-рую часто наз. «сопротивлением» электрохимии, про-
цесса. При более высоких плотностях тока, когда
для зависимости П. от плотности тока спра-
ведливо ур-ние Тафеля:
ц = a -j- Ъ lg i
где а и Ь — константы.
Константы со и а зависят от природы металла элек-
трода, от состояния его поверхности, состава раствора
и темп-ры. Для металлов с высоким П. водорода
(Hg, РЬ, Zn, Cd, Sn, Т1 и др.) значение константы а.
(П. водорода при плотности тока ia/см2) колеблется
в пределах от 1,2 до 1,6 в. Для других металлов эта
величина меньше. Напр., для никеля а= 0,6 в, а
для металлов платиновой группы колеблется от 0,1
до 0,3 в. В отличие от константы а,константа Ь мало
зависит от природы металла—катода. Для многих ме-
таллов Ь имеет при 25° значения, близкие к 0,12 в. Во-
945
ПЕРЕНОС ЭНЕРГИИ
946
лее низкие значения Ъ наблюдались на Pt,Pd и нек-рых
других металлах. С повышением темп-ры при средних
плотностях тока П. водорода снижается примерно
на 3—4 мв на 1°, величина константы b при этом воз-
растает пропорционально абсолютной темп-ре. При
увеличении концентрации кислоты или щелочи в ряде
случаев наблюдается уменьшение П. водорода. Однако
в общем зависимость П. от состава раствора являет-
ся достаточно сложной. Возникновение П. при выде-
лении водорода обычно связывают с замедленностью
к.-л. одной или нескольких из следующих трех ста-
дий этого процесса: 1) разряда ионов водорода: Н+4-
-|-Ме-|-е-^МеН(Ме—металл, МеН—атом водорода,
хемосорбированный на металле); 2) рекомбинации ад-
сорбированных атомов: 2МеН^ Н2-|-2Ме; 3) электро-
химия. десорбции: Н++МеН + е Н2 + Me. Стадии
1—2, 1—3 являются последовательными, а стадии 2—
3 — параллельными.
Для металлов с высоким П. водорода (Hg, РЬ и т. п.)
наибольшее подтверждение на опыте получила теория
замедленного разряда ионов водорода, т. е. теория,
объясняющая П. замедленностью стадии 1. Для ме-
таллов с низким П. водорода еще нет единой точки
зрения на механизм этого явления. В ряде случаев
было показано, что механизм П. изменяется при пере-
ходе от малых к большим плотностям тока.
Изложенные выше основные закономерности для П.
водорода в общем справедливы для П. при других
электрохимия, процессах. Следует отметить, что для
П. кислорода имеет место зависимость от природы
металла, обратная указанной зависимости для П.
водорода. А именно, на металлах с высоким П. водо-
рода П. кислорода, как правило, ниже, чем на ме-
таллах с низким П. водорода. Перенапряжению, как
одному из видов поляризации, уделяется большое
внимание в теоретической и прикладной электрохимии
(см. Поляризация электрохимическая).
Лит.: Фрумкин А. Н. 1и д р.]„ Кинетика электрод-
ных процессов, М., 1952. П. Д. Лукавцев.
ПЕРЕНОС ЭНЕРГИИ (на молекулярном
уровне) — широко распространенное явление,
заключающееся в том, что энергия, первоначально
поглощенная одним участком системы, безызлуча-
тельно передается другому. В зависимости от того,
происходит ли П. э. в пределах одной молекулы,
либо же между молекулами, различают внутримо-
лекулярный П. э. и межмолекулярный П. э. Явление
П. э., состоящее в многократном перескоке энергии
от одной молекулы (или атома) к другой, обычно наз.
миграцией энергии.
Впервые явление П. э. наблюдалось Карио в 1922. При
облучении смеси паров ртути и таллия резонансным светом,
поглощаемым ртутью, Карио наблюдал флуоресценцию таллия.
Поскольку таллий не поглощает в области излучения ртути,
Д. Франк и Карио (1922) интерпретировали наблюденное
явление как безызлучательный перенос энергии от возбуж-
денных атомов ртути к атомам таллия.
Для того, чтобы П. э. стал возможным, необходимо,
чтобы для энергий возбуждаемых уровней донора D
и акцептора А выполнялось соотношение Eit ЕА .
Явление П. э. отличается от реабсорбции
(т. е. излучения кванта энергии донором с последую-
щим поглощением его акцептором) тем, что при П. э.
происходит бузызлучательная передача, прежде чем
имеет место излучение донора. В результате время
жизни возбужденного состояния донора сокращается
по сравнению с временем жизни в отсутствии пере-
носа. П. э. происходит вследствие взаимодействия до-
нора и акцептора, к-рое может осуществляться как
при непосредственном контакте, так и на расстоянии.
Расстояние между донором и акцептором не должно
быть велико, обычно оно не превышает 100 А, т. е.
величина взаимодействия должна быть достаточной,
чтобы вызвать перенос прежде, чем произойдет дезак-
тивация возбужденного состояния. П. э. в системе
наблюдается как при облучении светом, так и при
воздействии ионизирующих излучений. В послед-
нем случае, в отличие от резонансного поглощения
света, уже нельзя четко зафиксировать поглощение
энергии одним сортом молекул и передачу ее другому
сорту. Эффект П. э. проявляется здесь в том, что лю-
минесценция, либо же химич. изменение компонента
становится непропорциональным доле поглощенной
им энергии (нарушается так наз. правило аддитив-
ности). При действии ионизирующих излучений,
кроме возбужденных молекул, образуются ионы и
свободные электроны. Поэтому отклонение от правила
аддитивности может быть связано также с переносом
заряда от одного компонента системы к другому.
Внутримолекулярный П. э. наблюдал-
ся на множестве молекул, в к-рых адсорбирующая
часть не идентична с излучающей. Вайссман (1942)
исследовал впервые это явление в хелатных соедине-
ниях европия. Свет поглощался органич. частью мо-
лекулы, после чего энергия возбуждения передава-
лась иону Еи’ + , к-рый флуоресцировал. Впоследствии
явление внутримолекулярного П. э. было исследовано
на хелатных комплексах других редкоземельных
металлов (Гросби и др.). В этом случае возбуждение
передается по синглетным уровням молекулы (см.
Молекулярные спектры). А. Н. Теренин и В. Л. Ермо-
лаев (1958) показали, что в нафтилкарбонильных
молекулах возбуждение карбонильной группы вызы-
вает фосфоресценцию нафтильной группы. Энергия
с триплетного уровня карбонильной группы пере-
дается на триплетный уровень нафтильной группы.
В настоящее время накоплено большое количество
экспериментальных данных, указывающих на внут-
римолекулярный П. э. к ароматич. кольцу. Ароматич.
соединения обладают повышенной радиационной
стойкостью по отношению к воздействию ионизирую-
щих излучений. Наличие внутримолекулярного П. э.
к ароматич. части молекулы приводит к повышению
общей радиационной устойчивости молекулы, т. к.
энергия возбуждения диссипирует в кольце, не при-
водя к химич. изменениям. Показателен такой эк-
сперимент (Р. Александер, А. Чарльзби, 1954).
Додекан облучался в реакторе до состояния полной
нерастворимости, т. е. до появления пространствен-
ной сетки вследствие образования поперечных химич.
связей. Замена в мономере атома Н циклогексильным
кольцом почти не меняла радиационную дозу, необ-
ходимую для образования пространственной сетки,
замена нафтильным кольцом увеличивала дозу почти
вдвое. При этом максимальный эффект получался,
когда нафтильное кольцо присоединялось к середине
цепи додекана, минимальный — к концу цепи. Эти
результаты получают естественное объяснение при
допущении миграции энергии к нафтильному кольцу.
Межмолекулярный П. э. может проис-
ходить как в газовой, так и в конденсированной фазе.
Типичным проявлением является сенсибилизирован-
ная флуоресценция: находящаяся в возбужденном
синглетном состоянии молекула донора безызлуча-
тельно переходит в основное состояние и передает
свою энергию молекуле акцептора, переводя ее из
основного в возбужденное синглетное состояние с
последующей флуоресценцией (т. наз. перенос синг-
лет — синглет). Облучение раствора люминофора
(вещества с высоким квантовым выходом флуорес-
ценции) светом, поглощающимся только растворите-
лем, при соответствующем подборе растворителя вы-
зывает люминесценцию люминофора уже при концен-
трациях 10-6 — 10-1 М/л. Сенсибилизированная лю-
минесценция происходит также при воздействии иони-
зирующего излучения на раствор. Необходимым усло-
вием для П. э. на расстояния, превышающие кинетич.
радиусы молекул, является наличие резонанса энер-
947
ПЕРЕНОС ЭНЕРГИИ
948
гетич. уровней молекул донора и акцептора, другими
словами, наличие перекрытия спектра излучения до-
нора со спектром поглощения акцептора. Этому
условию лучше всего удовлетворяют пары, подобран-
ные из ароматич. соединений, либо из красителей.
П. э. происходит не только между молекулами раз-
ного сорта, но и между одинаковыми молекулами.
В последнем случае сдвиг спектра флуоресценции по
отношению к спектру поглощения ухудшает перекры-
тие спектров донора и акцептора, тем не менее П. э.
может быть достаточно эффективным. Проявляется
такой перенос, напр., в явлении т. наз. «концентра-
ционной деполяризации», когда, вследствие переско-
ка энергии между различным образом ориентирован-
ными молекулами, первоначально поляризованный
свет деполяризуется. Для растворов красителей явле-
ние концентрационной деполяризации наблюдается
при средних расстояниях между молекулами до 70 А.
В кристаллах наличие упорядоченной структуры
одинаковых молекул способствует П. э. между ними
посредством экситонного механизма (см. ниже).
П. э. по триплетным уровням впервые наблюдался
А. Н. Терениным и В. А. Ермолаевым (1952) при изу-
чении фосфоресценции в твердых р-рах при низких
темп-рах. Среднее расстояние между молэкулами
донора и акцептора составляло 12—14 А. Ответ-
ственными за П. э. в данном случае являются обмен-
ные взаимодействия (на этих расстояниях возможно
перекрытие периферия, электронных оболочек моле-
кул донора и акцептора). В дальнейшем рядом ис-
следователей (X. Бэкстрем и К. Сандрос, 1958, Г. Пор-
тер и Ф. Уилкинсон, 1961) был обнаружен П. э. по
триплетным уровням в жидких р-рах при комнатной
темп-ре. Оказалось, что перенос тем эффективнее,
чем больше разность энергий триплетных уровней
донора и акцептора. В наиболее благоприятных слу-
чаях скорость переноса приближалась к числу диф-
фузионных встреч молекул за 1 сек.
Если при П. э. к акцептору последний претерпе-
вает химич. изменение, то имеет место сенсибилизи-
рованный фотолиз или радиолиз, в зависимости
от рода облучения. Примером сенсибилизированного
радиолиза является у-сенсибилизированный распад
в бензоле ряда перекисей, ароматич. азосоединений
и дисульфидов (В. А. Кронгауз и X. С. Багдсарьян,
1957—60). Выход разложения этих веществ резко
возрастает с ростом их концентрации и более чем на
порядок превосходит выход, соответствующий прямо-
му действию излучения на растворенное веществе.
П. э. от донора к относительно более стабильному
акцептору повышает радиационную устойчивость
системы. Для того, чтобы защита была эффективной,
П. э. должен успеть произойти за время жизни со-
стояния, с к-рого происходит распад молекулы (обыч-
но 10_” — 10-13 сек). Наиболее изученной является
радиационная защита циклогексана добавками бен-
зола. Заметное уменьшение распада циклогексана
при у-облучении начинается уже при концентрациях
бензола 10_’ М/л (М. Бэртон с сотр., 1957). Однако
накладывающиеся в данной системе побочные химич.
эффекты мешают точно определить относительный вес
П. э. в защите циклогексана от распада.
Механизм переноса. В зависимости от
того, какой процесс определяет скорость П. э., разли-
чают три основных механизма переноса: диффузион-
ный, индуктивно-резонансный и экситонный.
1. Диффузионный механизм П. э. наблюдается при
сближении молекулы донора с молекулой акцептора
до расстояний порядка кинетич. радиусов. Скорость
процесса не превышает скорости диффузии. Для мо-
лекул с одинаковым поперечником константа скорости
диффузии равна . _ srt л
диф. 3000т) М -сек
где г) — вязкость среды, R — газовая постоянная,
Т — абсолютная темп-ра. Число встреч Z за 1 сек
возбужденной молекулы D* с молекулами А при кон-
центрации последних С а равно/ГдИф. Са, средний про-
межуток времени между встречами Дг = 1/А:дИф Са.
Для того, чтобы процесс переноса был эффективен,
необходимо, чтобы соблюдалось неравенство т > Дг,
где т — время жизни возбужденного состояния.
Большое время жизни триплетных состояний благо-
приятствует переносу по этому механизму.
2. Индуктивно-резонансный механизм (механизм
Ферстера — Галанина — Декстера) П. э. обусловлен
кулоновым взаимодействием электронных оболочек
молекул и имеет место лишь при наличии резонанса
энергетич. уровней донора и акцептора. Этот механизм
ответствен за большинство случаев сенсибилизиро-
ванной люминесценции в конденсированной фазе.
Вероятность переноса пропорциональна шестой сте-
пени расстояния R между молекулами донора и ак-
цептора и выражается формулой
где т — среднее время жизни возбужденного состоя-
ния донора, — характеристич. расстояние, на
к-ром вероятность П. э. равна вероятности спонтан-
ной дезактивации возбужденного состояния донора.
Величина тем больше, чем больше перекрытие
спектра излучения донора со спектром поглощения
акцептора. Вышеприведенная формула выведена в
приближении диполь-дипольного взаимодействия при
условии неподвижности молекул и справедливости
распределения Больцмана. В тех случаях, когда дйф-
фузионное смещение молекул становится сравнимым
с Ra, необходим совместный учет диффузии и индук-
тивно-резонансного переноса.
3. Экситонный механизм. В молекулярных кристал-
лах возбуждение какой-либо молекулы вследствие
эквивалентности молекул кристалла будет переме-
щаться от молекулы к молекуле в виде волны элек-
тронного возбуждения. Возбужденное состояние по-
добного типа наз. экситоном (Я. И. Френкель, 1931).
Время П. э. тпер_ обратно пропорционально вели-
чине межмолекулярного взаимодействия и, именно:
Тпер. = h/iu, где h — Планка постоянная. Различа-
ют два случая: а) Возбуждение переходит от одной
молекулы к другой быстрее, чем успевает произойти
колебание ядер (период колебания 10~14 — 10-1’ сек).
Движение возбуждения не сопровождается локальной
деформацией решетки. Такое состояние называется
свободным экситоном, б) Ядра успевают совершить
несколько колебаний, прежде чем происходит пере-
дача возбуждения. Возникает местная деформация,
к-рая перемещается вместе с возбуждением по кри-
сталлу. Такое состояние наз. локализованным экси-
тоном. Скорость перемещения локализованного экси-
тона значительно меньше, чем свободного. В ионных
кристаллах возбуждение можно рассматривать как
нейтральную пару электрон — положительная дырка,
притягивающиеся друг к другу в силу кулонова
взаимодействия. Экситон такого типа также перемеща-
ется по кристаллу от ячейки к ячейке (Г. Ванье, 1937).
Значение явления П. э. По мнению не-
которых исследователей, явление П. э. играет суще-
ственную роль в механизме фотосинтеза растений;
предполагается, что энергия солнечных лучей, погло-
щаемая хлорофиллом, мигрирует через 100—200 моле-
кул хлорофилла, пока не захватывается молекулой
фермента, что дает начало цепи биохимич. реакций,
составляющих вторичный механизм фотосинтеза.
Явление П. э. получило широкое применение в иссле-
довании различных вопросов спектроскопии и радиа-
ционной химии. П. э. используется в практич. вопро-
949
ПЕРЕСУЛЬФИРОВАНИЕ —ПЕРЕХОДНОГО СОСТОЯНИЯ МЕТОД
950
сах создания люминесцирующих материалов, лежит
в основе работы сцинцилляционных счетчиков, реги-
стрирующих ионизирующие излучения. В последние
годы П. э. получил применение для создания новых
типов квантевых генераторов света. Защита облучае-
мых систем от радиационных повреждений малыми
добавками нек-рых веществ — антирадов, в значи-
тельной степени, видимо, также связана с П. э. или
переносом заряда.
Лит.: Теренин А. Н., Ермолаев В. Л., Усп.
физ. наук-, 1956, 58, вып. 1, 36; Усп. физ. наук, 1963, 80,
вып. 1, 3; Кронгау а В. А., Усп. химии, 1962, 31, вып. 2,
222; Рид С., Возбужденные электронные состояния в химии
и биологии, пер. с англ., М., 1960; FSrster Th., Fluo-
reszenz organlscher Verbindungen, GOttlngen, 1951; Energy
transfer with special reference to biological systems, Disc.
Faraday Soc., 1959, № 27; Comparative effects of radiation, ed.
M. Burton [a. o.J, N. Y., 1960. И. Г. Каплан.
ПЕРЕСУЛЬФИРОВАНИЕ* — ферментативная ре-
акция переноса серы L-гомоцистеина (а-амино-у-мер-
каптомасляной к-ты, I) на остаток L-серина (а-амино-
-Р-оксипропионовой к-ты, II) с образованиемL-цистеина
(а-амино-р-меркаптопропионовой к-ты, IV) и L-гомо-
серина (а-амино-у-оксимасляной к-ты, V). Значение П.
в организме животных обусловлено его ролью в био-
синтезе цистеина из синтезирующейся в организме
аминокислоты (серина) и незаменимой аминокислоты—
метионина, являющегося источником гомоцистеина
в тканях. У микроорганизмов возможен и обратный
процесс — биосинтез гомоцистеина (и метионина)
из цистеина и гомосерина. Промежуточным продуктом
П. является L-цистатионин [S — (Р-амино-р-карбок-
сиэтил)-Б-гомоцистеин — III]:
CH2-SH
I сн2он
сн2 + । —>.
I hooc-ch-nh2
hooc-ch-nh2
I II
сн2— s — сн2
Г I +н2о
СН2 HOOC-CH-NH3---->
hooc-ch-nh2
III
СН3ОН
сн2
HOOC-in-NH.
CH2-SH
—* I
hooc-ch-nh2
IV
Биосинтез и распад цистатионина катализируются
специфич. ферментами, в каталитич. центрах к-рых
участвует пиридоксальфосфат.
Лит..: Браунштейн А. Е., Биохимия аминокислот-
ного обмена, М., 1949; Майстер А., Биохимия аминокис-
лот, пер. с англ., М., 1961; Янг Л., М о у Д ж., Метаболизм
соединений серы, пер. с англ., М., 1961. В. 3. Горкин.
ПЕРЕСЫЩЕННЫЙ РАСТВОР — см. Раствори-
мость.
ПЕРЕФОСФОРИЛИРОВАНИЕ (трансфосфорилиро-
вание) — ферментативная реакция преимущественно
межмолекулярного переноса остатка ортофосфор-
ной к-ты
R-P0sH2 + R'0H^± r'-po3h2+roh
R-PO3H2 + R'NH2 RH + R'NH-POaH2
Основным переносчиком фосфата в биологич. системах
является аденозин-5'-пирофосфат, к к-рому в резуль-
тате ферментативных реакций (в частности сопряжен-
ных с тканевым дыханием) присоединяется остаток
ортофосфата с образованием аденозин -5'-трифосфата
(АТФ). Концевая фосфатная группа АТФ легко может
быть использована при биосинтезе ряда фосфорили-
рованных соединений (напр., пиридоксальфосфата,
креатинфосфата, пиридиновых и флавиновых нуклео-
* Термин «П.» не является точным, поскольку речь идет
о реакциях переноса — SH-групп, а не — SOsH-rpynn.
тидов и др.), необходимых для протекания биохимия,
процессов в клетках животных, растений и микроорга-
низмов. В роли акцептора переносимых специфич.
ферментными системами групп в большинстве случаев
(53% всех известных реакций П.) выступают спиртовые
группы сахаров, нуклеотидов, аминокислот и азоти-
стых оснований. Однако большой интерес представля-
ют и такие реакции, в к-рых акцепторами фосфатных
групп являются, напр., карбоксильные (ацетат, аспар-
тат) или азотистые группировки, а также процессы
внутримолекулярного переноса фосфорсодержащих
групп с регенерацией донора.
Ферменты, катализирующие реакции П., акти-
визируются ионами Mg’+, а также нек-рых других
двухвалентных металлов. Широко распространены
ферменты, катализирующие обратимый перенос фос-
фата между указанными нуклеозиддифосфатами и АТФ.
Обратимость реакций П. определяется соотношением
энергетич. уровней вступающих в реакцию соединений
и ее продуктов. Так, перенос фосфата АТФ на креа-
тин, не сопровождающийся потерей свободной энер-
гии, является легко обратимой реакцией; геКсоки-
назная реакция:
АТФ+глюкоза АДФ+глюкозо-6-фосфат+Н +
при к-рой имеет место рассеивание энергии, характе-
ризуется резко сдвинутым вправо положением равно-
весия.
Лит.: Котельпинова А. В., Успехи соврем, биол.,
1957, 4 3, вып. 2, 133; X е с и н Р. Б., Биохимия цитоплазмы,
М., 1960; Диксон М., Уэбб Э., Ферменты, пер. с англ.,
М., 1961. В. 3. Горкин.
ПЕРЕХОДНОГО СОСТОЯНИЯ МЕТОД (метод
активированного комплекса, теория абсолютных ско-
ростей реакций) служит для теоретич. вычисления
скоростей процессов, в к-рых преодолевается энерге-
тич. потенциальный барьер за счет энергии теплового
движения. Основная область применения П. с. м.—
вычисление скоростей химич. реакций.
В ходе элементарного акта химич. реакции проис-
ходит перемещение атомов, входящих в состав реаги-
рующих молекул. Так как электроны много легче
атомных ядер и движутся много быстрее их, можно
принять, что в каждый момент движения ядер со-
стояние электронов будет совпадать со стационарным
состоянием при так же расположенных неподвижных
ядрах (приближение Борна — Оппенгеймера). Ос-
новываясь на этом, называют часть энергии атомной
системы, определяемую взаимным расположением
атомных ядер, потенциальной энергией, хотя она
включает, наряду с истинной потенциальной энер-
гией также и кинетич. энергию электронов. Часть
энергии, определяемая скоростями ядер, обозна-
чается соответственно, как кинетич. энергия. Поль-
зуясь наглядным геометрич. языком, говорят, что
зависимость потенциальной энергии от конфигурации
ядер задается нек-рой потенциальной по-
верхностью. При построении такой поверхно-
сти по одной оси откладывается потенциальная
энергия, по другим — координаты ядер. Такое по-
строение не осуществимо в трехмерном пространстве,
если число параметров, определяющих взаимное
расположение ядер, больше 2; это, однако, не препят-
ствует использованию геометрич. терминологии.
Энергия атомной системы в принципе доступна
вычислению методами квантовой химии, фактически,
однако, такое вычисление удается выполнить лишь
приближенно для простейших систем. Получаемые
результаты иллюстрируются схематич. потенциаль-
ной поверхностью для реакции X-|-YZ = XY+Z,
где X, Y и Z — одновалентные атомы. Если ограни-
читься конфигурациями, в к-рых атомы расположены
на одной прямой, как показывает расчет — наиболее
выгодными энергетически, то потенциальная поверх-
951
ПЕРЕХОДНОГО СОСТОЯНИЯ МЕТОД
952
ность будет трехмерной (рис. 1). Она изображается,
подобно горной местности, с помощью горизонталей:
высота местности показыва-
ет потенциальную энергию,
как функцию координат —
расстояний Y—Z и X—Y.
Изменения взаимного рас-
положения атомов переда-
ются движением по потен-
циальной поверхности точ-
ки, показывающей значения
координат, т. н. к он фи-
гуративной точки.
На поверхности имеются
Рис. 1. потенциальная по- Дпе «Долины», одна из них
верхность для реакции отвечает начальному состо-
X+YZ=XY+Z янию X-f-YZ, другая—ко-
нечному XY + Z. Наивы-
годнейший, т. е. требующий наименьшего подъема,
путь из одной «долины» в другую (показанный на
рис. 1 пунктиром) проходит через «перевал» (знак X
на рис. 1). Точка «перевала» является седловой точкой
потенциальной поверхности: в ней находится мак-
симум потенциальной поверхности при движении
вдоль наивыгоднейшего пути и минимум — при дви-
жении в перпендикулярном направлении.
Расстояние вдоль наивыгоднейшего пути, отсчи-
танное от какой-либо начальной точки, наз. реак-
ционной координатой; обозначим его х.
Разрез потенциальной поверхности по наивыгод-
нейшему пути, развернутый
<с У £о\ в плоскость (рис. 2), нагляд-
Д X+YT—\. н0 показывает энергети-
1 XY+Z ческий барьер EQ, пре-
।одолеваемый реагирующей
системой при движении кон-
фигуративной точки по наи-
выгоднейшему пути; соответ-
ствующее вершине барьера
Реакционная координата
Рис. 2. Энергетический
профиль для реакции
X+YZ=XY+Z
значение реакционной коор-
динаты, х0, будем называть критическим.
Состояние реагирующей системы, в к-ром реак-
ционная координата имеет критич. значение (или
отличающееся от последнего на бесконечно малую
величину), наз. переходным состоянием
или активированным комплексом
(рис. 3). Это состояние реализуется в момент пере-
сечения конфигуративной точкой плоскости, перпен-
дикулярной реакционной координате в седловой
точке. Согласно закону Максвелла — Больцмана,
вероятность состояния пропорциональна е~Е^Т, где
Е — энергия состояния, к — постоянная Больцмана,
Т — темп-ра, так что с ростом Е вероятность со-
стояния резко падает. Поэтому в переходном со-
стоянии конфигуративная точка практически всегда
Начальное
состояние
Переходное
состояние
конечное
состояние
Рис. 3. Изменения относительного расположения атомов
в реакции X4-YZ=XY+Z
проходит вблизи седловой точки потенциальной
поверхности; ее отклонение от наивыгоднейшего пути
можно рассматривать, как малое колебание в на-
правлении, перпендикулярном реакционной коор-
динате.
Подобные соотношения имеют место и в более слож-
ных системах, когда потенциальная поверхность яв-
ляется многомерной. Поэтому переходное состояние,
или активированный комплекс, является образова-
нием, аналогичным молекуле и отличающимся от
устойчивой молекулы лишь тем, что в то время как
малые изменения координат атомов в молекуле
всегда повышают потенциальную энергию (колеба-
тельные степени свободы) или оставляют ее без изме-
нения (поступательные и вращательные степени
свободы), активированный комплекс обладает одной
координатой, изменение к-рой приводит к уменьшению
потенциальной энергии — реакционной координатой;
остальные степени свободы активированного ком-
плекса вполне подобны таковым стабильной молекулы.
При движении конфигуративной точки по потенци-
альной поверхности в направлении к вершине энер-
гетич. барьера происходит переход кинетич. энергии
в потенциальную; в зависимости от величины перво-
начального запаса кинетич. энергии возникает или
не возникает переходное состояние. Примем вначале,
в соответствии с классич. механикой, что если реак-
ционная координата не достигает критич. значения,
реакция не происходит, а если это значение дости-
гается, реакция происходит с неизбежностью, т. к.
атомы продолжают движение по инерции в прежнем
направлении. Тогда для вычисления скорости ре-
акции нет надобности прослеживать движение кон-
фигуративных точек на всем пути от начального
состояния к конечному; достаточно подсчитать число
этих точек, проходящих через переходное состояние
в нужном направлении за единицу времени.
Выберем произвольный весьма малый интервал
значений реакционной координаты dx, такой чтобы
ее критич. значение х0 лежало внутри интервала.
Будем считать активированным комплексом прямой
реакции каждую группу атомов реагирующих мо-
лекул, у к-рых х лежит внутри указанного интервала,
а направление движения конфигуративной точки
отвечает протеканию элементарного акта реакции
в прямом направлении. При х, лежащем внутри dx
и противоположном направлении движения, имеем
активированный комплекс обратной реакции. По-
скольку интервал dx задан, приобретает определенное
значение концентрация активированных комплексов,
т. е. их число в единичном объеме — для гомогенной
реакции, или на единичной поверхности — для ге-
терогенной реакции. Очевидно, что эта величина
должна быть пропорциональна dx, поэтому запишем
ее так: Са dx — для прямой реакции и Са dx — для
обратной реакции. Величины Са и Са являются кон-
центрациями активированных комплексов прямой
и обратной реакций, соответственно, на единицу
длины реакционной координаты.
Обозначим через v скорость изменения реакцион-
ной координаты при x=xS)\ v = dx/dt, где t — время.
Продолжительность жизни активированного комплек-
са dt = dx/v. Будем называть скоростью реакции в
прямом направлении со, или в обратном направлении
со, число элементарных актов реакции соответствую-
щего направления, происходящих в единичном объеме
или на единичной поверхности за единицу времени.
Если бы величина v (а следовательно и dt) была оди-
накова для всех активированных комплексов, кон-
центрация активированных комплексов Cadx опре-
деляла бы число элементарных актов за общее для
—7- —э-
всех них время dt, и со равнялась бы-^- =Cav. В дей-
ствительности скорость v неодинакова для разных
активированных комплексов, поэтому w=Cav, где
v — среднее значение v. При вычислении скорости
953
ПЕРЕХОДНОГО СОСТОЯНИЯ МЕТОД
954
реакции должна быть учтена возможность, что при
нек-рых условиях, хотя конфигуративная точка
проходит над вершиной барьера, реакция не про-
исходит, или напротив, реакция происходит без
достижения вершины барьера. Поэтому вводится
коэффициент прохождения (трансмис-
сионный коэффициент) х и уравнение для скорости
реакции принимает вид
со=хСдр (1)
Если движение подчиняется законам классич.
механики, факторы, вызывающие отклонение х от 1
(напр., кривизна реакционного пути), мало сущест-
венны, так что х ЭИ. Квантово-механич. туннельный
эффект ядер, создающий возможность перехода си-
стемы с одной стороны энергетич. барьера на другую,
при значении кинетич. энергии, недостаточном для
достижения вершины барьера, мало проявляется в
химич. реакциях, хотя его учет может быть сущест-
венен при обсуждении кинетич. изотопных эффектов,
т. е. различий в скорости реакций молекул, разли-
чающихся изотопным составом. Большие отклонения
х от 1 встречаются лишь как исключение, а именно —
в случае неадиабатических реакций.
Движение происходит неадиабатически (в квантово-
механич. смысле), если элементарный акт реакции
сопровождается изменением общего спина атомной
системы, а также в нек-рых др. случаях. При этом
к потенциальной кривой основного состояния, рас-
сматривавшейся выше (рис. 2), в окрестности вершины
барьера близко подходит кривая возбужденного
состояния. Квантово-механич. перескок конфигура-
тивной точки на эту кривую предотвращает реакцию,
т. к. за ним следует возвращение системы в исходное
состояние. Вероятность о осуществления реакции
при достижении конфигурации переходного состояния
определяется ур-нием Ландау — Зинера (1932):
р = 1 — ехр ( — 4л2еАВ//гг | Fa — /’в | ) (2)
Здесь h — постоянная Планка, еАВ— половина наи-
меньшего расстояния между потенциальными кри-
выми, |Fa—Fb| —абсолютная величина разности на-
клонов потенциальной кривой до и после барьера.
Согласно ур-нию (2), если еАВ мала, то при обычных V,
отвечающих скорости теплового движения, о может
быть много меньше 1, тогда и х, равный среднему
значению о, много меньше 1 (типичная для неадиа-
батич. реакций величина х = 10-5).
Нахождение Са и v весьма облегчается предполо-
жением, являющимся основным для П. с. м., согласно
к-рому протекание химич. реакции (или другого
процесса, скорость к-рого рассматривается) не на-
рушает существенно максвелл-больцмановское рас-
пределение по состояниям, предшествующим пере-
ходному состоянию. Начнем с вычисления v. При
движении конфигуративной точки в пределах отрезка
dx, включающего вершину барьера, потенциальная
энергия постоянна, поэтому движение можно рас-
сматривать как поступательное, с нек-рой эффек-
тивной массой т*. Скорости v соответствует кинетич.
энергия Yz т*v1, поэтому
® m*D2 а m*D2
С ve dVK e~~^dv = ’ (3)
J / J (urn* j
о о
Для вычисления Ca рассматриваем систему в со-
стоянии химич. равновесия; это допустимо, т. к.,
согласно вышеприведенному основному предполо-
жению П. с. м., удаление из системы продуктов
реакции, влекущее за собой обращение со в 0, не ска-
залось бы на со. При равновесии со = со, и т. к. х и v
одинаковы для прямой и обратной реакции, Са = Са.
Следовательно, суммарная концентрация активиро-
ванных комплексов независимо от направления дви-
жения, т. е. (Ca-\-Ca)dx, при равновесии равна 2Cadx.
Она пропорциональна сумме по состояниям (стати-
стич. сумме) координаты х:
2Сadx — fх dx (4)
где jx — сумма по состояниям реакционной коорди-
наты на единицу длины, С72— равновесная по отноше-
нию к исходным веществам концентрация воображае-
мых молекул, во всем подобным активированным ком-
плексам, но имеющим вместо реакционной координаты
полностью «замороженную» колебательную степень
свободы, т. е. такую, для к-рой сумма по состояниям
равна 1 (у величины и других величин с этим
индексом для упрощения обозначений не помечаем,
что они относятся к прямой реакции). Т. к. движение
в пределах dx является поступательным, то согласно
статистич. механике (см. Квантовая статистика)
fx = (2am*kT)'l'[h
Поэтому ур-ния (1), (3) и (4) дают
ш = хС^ (5)
Это ур-ние сводит задачу вычисления скорости
реакции к вычислению равновесной концентрации С^.
Входящая в него величина kT/h имеет размерность
частоты; по порядку величины она близка к частоте
молекулярных колебаний, например при Т’ = 300°К
ЛТ//г=6-1012сек-1.
Если реакция
IL + mM-f- ... = qQ -f-rR -f- ... (6)
(Z молекул вещества L реагируют с т молекулами
вещества Мит. д.) происходит в идеальной газовой
смеси или в идеальном растворе, то
c^'c[c™...=z^ (7)
где Cl, См,...— концентрации веществ L, М и т. д.,
— константа равновесия молекул, подобных ак-
тивированным комплексам (в разъясненном выше
смысле) с исходными молекулами. Согласно ур-ниям
(5) и (7)
ш = х^^с£с«... (8)
Величина
Г = (9)
постоянна при данной темп-ре, следовательно, ур-ние
(8) выражает закон действующих масс для скорости
реакции, к — константа скорости.
По аналогии с известным термодинамич. уравне-
нием, введем стандартное изменение
свободной энергии Гиббса при ак-
тивации с помощью ур-ния
Дб'с = — кТ In К"Р (10)
Знак Д обозначает здесь и в дальнейшем величину
в N раз меньшую, чем в химич. термодинамике (N—
число Авогадро), т. е. ДС^ относится к 1 активиро-
ванному комплексу, а не к 1 молю активированных
комплексов. Обычно ведут расчет на 1 моль, тогда к
заменяется на газовую постоянную R. Отметим,
кроме того, что в соответствии с ур-нием (7) ур-ние
955
ПЕРЕХОДНОГО СОСТОЯНИЯ МЕТОД
956
(10) подразумевает в качестве стандартных состояний
газов состояние с концентрацией, равной 1, а не с
давлением, равным 1, как принято в химич. термоди-
намике. Т. к. G=H—TS, где Н — энтальпия, S—
энтропия, пишут —T&S^, тогда ур-ния
(9) и (10) дают
Д5^ ДН^
Л = е hT (11)
Это ур-ние близко по форме к ур-нию Аррениуса (см.
Кинетика химическая)
~Е
'k=Ae~hT (12)
Поэтому приближенно можно отождествить с
энергией активации Е и считать, что предэкспонен-
циальный фактор А определяется стандартным
изменением энтропии при активации
(а также коэффициентом прохождения х).
Более строгая трактовка величины Е может быть прове-
дена следующим образом. Применяя термодинамич. ур-ние
Вант-Гоффа к для случая реакции в идеальной газовой
смеси, получаем, что
<ПпК*_ДС7*
dT kT2 ( '
где ДУ^— среднее изменение внутренней энергии при обра-
вввании из исходных молекул молекулы, подобной активиро-
ванному комплексу. Если х=1, можно считать х не зависящим
от Т. Тогда получаем из ур-ния (9):
dlnft ДУ^+йТ
“йт-=^тз----- (14)
Условимся называть энергией активации Е средний из-
быток энергии активированных комплексов по сравнению
с исходными молекулами. Тогда
E = &.U^ + EX (15)
где Ех— слагаемое в средней энергии активированных ком-
плексов, обязанное реакционной координате. При вычислении
Ех следует учесть, что вклад в скорость реакции конфигура-
тивных точек, имеющих скорость v вдоль реакционной коор-
динаты, пропорционален v, поэтому
со m*va оо m*vJ
2hTdvl^ve 2hT d'c=hT (16)
о 0
Таким образом получаем ур-ние Аррениуса
При х«1, согласно ур-нию (2), коэфф, х пропорционален v”1,
следовательно он пропорционален Т~ и вместо (14) получаем
^2=(дсг%-| kr^kT2
При этом произведение ос не зависит от v, следовательно при
вычислении Е„ вводить весовой множитель г не нужно, и
kT. Мы вновь получаем ур-ние (17).
В случае реакции в идеальном жидком р-ре, находящемся
под давлением Р, ур-ние (13) заменяется следующим
(18)
\ дТ 'Р kT2 '
Отсюда приходим к ур-нию Аррениуса для реакции в растворе
причем энергия активации Е=ДН7± + Е1 и Еш, как и выше,
равна ЙТ или ЙТ при х=1 или при х«1 соответственно.
4
Чтобы ур-ния (18) и (19) были строго применимы, кон-
центрации и скорость реакции должны вычисляться так,
чтобы на них не сказывалось тепловое расширение раствора;
напр., можно рассчитывать концентрации и скорость реакции
на единичный объем раствора при какой-то одной темп-ре.
Константа скорости реакций в жидких растворах
зависит от давления, под к-рым находится раствор;
при повышении давления от 1 до 1000 атм, к в ти-
пичных случаях изменяется в 2—3 раза. Согласно
термодинамич. ур-нию Планка, изменение константы
равновесия в растворе К с давлением определяется
величиной изменения объема при реакции AV:
Zflln ______ду
\ дР )т~ п'
Концентрации должны вычисляться так, чтобы для
данного раствора они не менялись с давлением,
несмотря на изменение объема раствора. Применяя
ур-ние Планка к и учитывая ур-ние (9), получаем
ур-ние Эванса и Поляни (1935)
ДУ*
kT
(20)
Здесь —изменение объема при акти-
вации, т. е. разность парциального молекулярного
объема активированных комплексов и суммы пар-
циальных молекулярных объемов исходных веществ
(под парциальными молекулярными величинами по-
нимаются величины в N раз меньше, чем парциальные
мольные величины). Ур-ние (20) позволяет определить
объем активированного комплекса из опытных данных
по зависимости к от Р, подобно тому, как ур-ния (17)
и (19) позволяют определить энергию активирован-
ного комплекса по зависимости к от Т.
Если реакция происходит в неидеальной системе,
ур-ние (7) заменяется следующим:
---V с---= К^
J с1 vm cm
YLCL yMCM-
(21)
Здесь у5*— коэфф, активности активированного комп-
лекса (одинаковый для прямой и обратной реакции)
yL , Ym> —коэфф, активности исходных молекул.
Поэтому получаем для скорости реакции в неиде-
альной системе ур-ние Бренстеда
, , J vm
a = k -L ” ClLe™... (22)
Y
где к по-прежнему определяется ур-нием (9). Множи-
Ybs х
тель называется кинетическим ф а к-
VA
тором активности.
Если идеальная газовая смесь находится в равно-
весии с идеальным разбавленным жидким р-ром,
то для каждого компонента газовой смеси выпол-
няется закон Генри
Cg=VCs (23)
где Г — коэфф. Генри, Cg— концентрация в газовой
фазе, Cs — то же в растворе. Условимся измерять ак-
тивность вещества в растворе его концентрацией в
сосуществующей идеальной газовой фазе Cg, тогда Г
приобретает смысл коэфф, активности в растворе.
Ур-ние (22) показывает теперь, что константа ско-
рости в растворе
. . гг гт
= L ” - (24)
Г>“
957
ПЕРЕХОДНОГО СОСТОЯНИЯ МЕТОД —ПЕРЕХОДНЫЕ ЭЛЕМЕНТЫ
958
где kg — константа скорости реакции в газовой фазе,
Г А — коэфф. Генри активированных комплексов. Т.о.,
влияние растворителя на константу скорости опре-
деляется растворимостью в нем исходных веществ
и активированных комплексов.
Статистич. механика дает возможность выразить
в идеальной газовой смеси через суммы по состоя-
ниям молекул. Обозначим через/L, /м и т. д., а также
Л6, суммы по состояниям молекул L, М и т. д. и мо-
лекулы подобной активированному комплексу, но
с замороженной координатой х. Эти суммы будем
рассчитывать на единичный объем, принимая для
каждого сорта молекул за нуль энергии наименьшую
возможную энергию этих молекул (их энергию при
Т=0). Концентрации в ур-нии (7), определяющем
будем считать выраженными числом молекул в
единице объема. Тогда
kT (25)
7l 'М'"
где Еа — высота энергетич. барьера, т. е. разность
наименьших энергий активированного комплекса и
исходных молекул. Е отличается от Ел так же, как
тепловой эффект реакции при какой-либо темп-ре
Т^О отличается от теплового эффекта при Т=0.
Подстановка в ур-ние (9) дает ур-ние Эйринга (1935)
для константы скорости гомогенной газовой реакции:
к = х^--7-1^----е kT (26)
п /I хГП ' >
ZM
Это уравнение позволяет определить абсолютную
величину к, если известна высота барьера Ео; от-
сюда термин «теория абсолютных скоростей ре-
акций».
Суммы по состояниям вычисляются с достаточным при-
ближением, как произведение след, множителей: статистич.
вес ge основного электронного состояния (для нелинейной
молекулы всегда g„= 1), сумма по состояниям поступательного
движения для единицы объема f пост = (2лтйТ) ,2/h’ (т —
масса молекулы), сумма по состояниям вращения f Вращ
=8л2 IkT/oh1 в случае линейной молекулы (7 — момент инер-
ции, о — число симметрии) или/ =8л2(8л37д/д7с^2Х
3/ Р Щ*
X(feT) /ой3 в случае нелинейной молекулы (7д, 1В, 1С — глав-
ные моменты инерции), сумма по состояниям колебаний
/ колер = П [1— exp(hv;/AT)]-1 (V;—частоты нормальных
колебаний). Число нормальных колебаний при п атомах равно
Зп — 5 или Зп —6 для линейной и нелинейной молекулы, со-
ответственно, и Зп — 6 или Зп — 7 для линейного и нелиней-
ного активированного комплекса.
Необходимые для вычисления величины, харак-
теризующие активированный комплекс — моменты
инерции и частоты нормальных колебаний, могут,
в принципе, быть найдены из соответствующей по-
тенциальной поверхности. Приближенная оценка
во многих случаях осуществима и без знания потен-
циальной поверхности. Вычисление абсолютной ско-
рости гомогенных газовых реакций по ур-нию (26)
дает результаты, хорошо согласующиеся с опытными
данными.
Методы статистич. механики могут быть также
применены для вычисления на основе ур-ния (5)
абсолютной величины скорости реакций на поверх-
ностях твердых тел, таких как гетерогенные ката-
литич. реакции и процессы хемосорбции газов.
Поверхностная концентрация молекул, подобных
активированным комплексам, С^, может вычисляться
как поверхностная концентрация адсорбированных
частиц; однако даже при простейшей трактовке,
опирающейся на модель идеального адсорбированного
слоя (см. Кинетика химическая), необходимо принять,
что активированный комплекс может занимать не-
сколько мест поверхности; напр., при реакции между
двумя адсорбированными частицами активированный
комплекс занимает 2 соседних места (рис. 4). Число
Рис. 4. Кружки О и заштрихованные — реагирующие
молекулы; пунктирные кружки — другие равноценные по-
ложения молекулы О- Переходное состояние занимает 2 ме-
ста на поверхности.
мест поверхности, занимаемое активированным ком-
плексом, входит в ур-ние для скорости реакции
[ур-ние (15) статьи Кинетика химическая]. Константа
скорости в этом ур-нии определяется равенством
—►
Г=х A (gf^l 4 . /L /S •) (27)
где I, J, ... вещества, молекулы к-рых вступают в
реакцию из адсорбированного состояния в числе
i, /, ..., L, М , ...— вещества, молекулы к-рых всту-
пают в реакцию из газового состояния в числе I, т,
..., Д — число мест для адсорбции на поверхности
единичной площади, g — число возможных поло-
жений активированного комплекса, когда одно из
Занятых им мест фиксировано (рис. 4). При вычис-
лении сумм по состояниям адсорбированных частиц
и активированного комплекса, строго говоря, должно
бы учитываться производимое ими изменение частот
колебаний твердого тела. Поскольку в адсорбирован-
ном состоянии все степени свободы являются колеба-
тельными, суммы по состояниям легких адсорбиро-
ванных частиц близки к 1 (потому что для BHxhvj>kT),
это позволяет просто оценить абсолютную скорость
реакции на поверхности. В ряде случаев подобные
оценки подтверждены сравнением с опытом.
Метод П. Ci применяется также для вычисления
абсолютных скоростей электродных процессов, про-
цессов диффузии и др.
Лит.: Г л ест он С., Лейдлер К., Эйринг Г.,
Теория абсолютных скоростей реакций, пер. с англ., М.,
1948; Кондратьев В. Н., Кинетика химических газовых
реакций, М., 1958; Семенов Н. Н.. О некоторых пробле-
мах химической кинетики и реакционной способности, 2 изд.,
М.—Л., 1958, приложение 1; Эйринг Г., Уолтер Д.,
Кимбалл Д., Квантовая химия, пер. с англ., М., 1948,
гл. 16; Гоникберг М. Г., Химическое равновесие и
скорость реакций при высоких давлениях, 2 изд., М.—Л., 1960;
Темкин М. И., Переходное состояние в поверхностных
реакциях, Ж. физ. химии, 1938, 11, вып. 2, 169; его же.
Энергия активации и предэкспоненциальный фактор при
электрохимическом процессе, в кн.: Труды совещания по
электрохимии (1950), М., 1953, с. 181. М. И. Темкин.
ПЕРЕХОДНОЕ СОСТОЯНИЕ (активированный
комплекс) — совокупность атомов, участвующих в
элементарном акте химич. реакции, в момент преодо-
ления системой энергетич. барьера, разделяющего
начальное и конечное состояния. Понятие «П. с.»
широко используется в химич, кинетике (см. Пере-
ходного состояния метод). М. И. Темкин.
ПЕРЕХОДНЫЕ ЭЛЕМЕНТЫ — встречающееся в
литературе название химич. элементов (расположен-
ных в периодич. системе Менделеева друг за другом),
959 ПЕРЕЭТЕРИФИКАЦИЯ—ПЕРИОДИЧЕСКАЯ СИСТЕМА ЭЛЕМЕНТОВ МЕНДЕЛЕЕВА 960
в атомах к-рых происходит заполнение внутренних
электронных d-оболочек, после того как уже произош-
ло заполнение более высших (с большим главным кван-
товым числом п) s-оболочек. Напр., при переходе от
кальция, имеющего конфигурацию внешних электро-
нов 4s2, к скандию с конфигурацией 3d4s2 новый элек-
трон попадает не на 4р-уровень, к-рый должен был
бы следовать за 4я-уровнем, а на Зй-уровень, соответ-
ствующий более низкому значению главного кван-
тового числа, равному 3 (см. Атом). Скандий и
следующие за ним элементы до цинка (конфигурация
3d”4s2) составляют первую группу П. э. Вторую
группу П. э. составляют элементы от иттрия до
серебра, третью — от лютеция до ртути. В приве-
денной в ст. Атом периодич. системе элементов Мен-
делеева группы П. э. расположены друг под другом.
С заполнением внутренних d-оболочек связан ряд
закономерностей в свойствах П. э. (см. Периодиче-
ская система элементов Менделеева).
ПЕРЕЭТЕРИФИКАЦИЯ — общее название для
алкоголиза (1), ацидолиза (2) и двойного обмена (3)
сложных эфиров:
RCOOR' + R"OH RCOOR" + R'OH (1)
RCOOR' + R"COOH^R"COOR' + RCOOH (2)
RCOOR' + R"COOR'" RCOOR"' + R"COOR' (3)
Катализаторами П. являются кислоты, напр. НС1,
НВг, В Fa, нек-рые соли металлов и в особенности
основания, напр. алкоголяты и гидроокиси щелочных
металлов. В первом случае механизм П. сходен с
механизмом прямой этерификации:
HOR
+ R"OH |
R-C-OR' + H + R-C-OR' R-C-OR"?ZZt:
II I I
О он он
RCOOR" + R'OH2
во втором случае — с механизмом реакции Sn2:
0-
R-C-0R'+R''0~ zzt R-C-OR' zzzt R-C-0R" + R'0~
II I II
0 OR" О
Смещение равновесия в П. обычно достигается от-
гонкой более летучего спирта, кислоты или сложного
эфира. При помощи П. могут быть получены сложные
эфиры спиртов, не вступающих в прямую этерифи-
кацию, напр. ментола. Обменом с винилацетатом
или изопропенилацетатом могут быть получены со-
ответственно виниловые и изопропениловые эфиры
высших к-т. Частным случаем П. является алкоголиз
лактонов:
сн2-с=о
I I +ROH—>hoch2ch2coor
сн2-о
П. во многих случаях является удобным методом
получения сложных эфиров и находит широкое
применение в препаративной органич. химии и химич.
пром-сти. Так, П. диметилтерефталата этиленглико-
лем в присутствии ацетатов Zn и Со получают бис-р-
оксиэтилтерефталат, нагревание к-рого в вакууме
при 230—290° дает полиэтилентерефталат (терилен,
дакрон).
Лит.: Н о и Ь е n-W е у 1, 4 Aufl., Bd 8, Stuttgart, 1952,
S. 526; Bd 4, TI 2, Stuttgart, 1955, S. 9. Б. Л. Дятпин.
ПЕРИ — обозначение расположения двух заме-
стителей в молекуле нафталина в по-
ложениях 1 и 8. Типичными приме- ноэ? j*"2
рами являются нафталиндикарбоно-
вая-1,8 кислота (нафталевая кислота), u I з|
1-нафталамино-8-сульфокислота(пери-
кислота, I) и Т. д. я. Ф. Комиссаров. 1
ПЕРИЛЕН С20Н12, мол. в. 252,32 — кристаллы,
золотисто-желтые пластинки; т. пл. 273—274°, при
350—400° возгоняется; хорошо раство-
рим в сероуглероде и хлороформе, лпл /ГаХ
менее растворим в бензоле, умеренно A VyZ sj)
растворяется в уксусвой к-те, очень itX=/i5 iX=Z0
мало — в этиловом спирте, этиловом /з iw___оз к\
эфире, ацетоне, нерастворим в лиг- \2_1/
роине; сильно разб. р-ры обладают го-
лубой флуоресценцией; растворяется в конц. серной
к-те, давая зеленый р-р, к-рый быстро меняет окраску
на сине-зеленую, синюю и в конечном счете красно-
фиолетовую; теплота сгорания при постоянном объеме
Q.a = 2333,0 ккал/моль. П. образует моно- и дипикрат,
комплекс со стифниновой к-той и тринитробензолом,
а также с неорганич. хлоридами, напр. с SbCl5.
Наиболее реакционноспособными центрами в П.
являются положения 3, 4, 9 и 10. При введении двух
заместителей образуются в основном 3,10-изомеры,
наряду с 3,9-изомерами. Напр., при кипячении
П. с водным р-ром СгО, образуется периленхинон-3,10.
Восстановление П. с помощью HJ и красного фосфора
дает гексагидроперилен, с помощью Na в амиловом
спирте — октагидро- и декагидроперилены; ката-
литич. гидрогенизацией над паладием получены
гексагидро- и тетрадекагидроперилены; при пропу-
скании хлора в раствор П.— гексахлорперилен. При
бромировании выделены 3,9- и 3,10-дибромперилены;
нитрование азотной к-той в среде ССЦ приводит к
3,10-динитроперилену; нагревание с конц. серной
к-той и СНзСООН дает 3,9- и 3,10-перилендисульфо-
кислоту. В присутствии А1С1, П. с СНзСОС1 дает 3,9-
диацетилперилен; аналогично протекает реакция и
с С,Н5СОС1. При нагревании П. с СвН5СОС1 и А1С1з
при 150° с последующей обработкой двуокисью мар-
ганца образуется изодибензантрон'.
С малеиновым ангидридом П. образует ангидрид
бензперилендикарбоновой к-ты.
Получают П. нагреванием ди-р-нафтола с РС15
и НзРО, (препаративный метод, выход 86%); дей-
ствием А1С1з на динафтил-1,1 или на диметиловый
эфир ди-Р-нафтола; при нагревании 1,8-дииоднафта-
лина с порошкообразной медью и др. П. был выделен
также из кам.-уг. смолы и американской нефти.
Лит.: Chemistry of carbon compounds, ed. E. H. Rodd,
v. 3, pt B, Amst.—[a. 0.], 1954, p. 1478; Физер Л., Ф и-
зер M., Органическая химия, пер. с англ., М., 1949, с.
715. Л. С. Поваров.
ПЕРИЛЛОВОЕ МАСЛО — СМ. Жиры раститель-
ные.
ПЕРИОД ПОЛУРАСПАДА — см. Полураспада пе-
риод.
ПЕРИОДИЧЕСКАЯ СИСТЕМА ЭЛЕМЕНТОВ
МЕНДЕЛЕЕВА. Содержание:
История открытия периодического закона и пе-
риодической системы ...........................961
Периодическая система и строение атома...........962
Принципы построения периодической системы . . . 964
Описание периодической системы..................967
Периодичность свойств в системе Менделеева . . 972
Периодическая система элементов Д. И. Менде-
леева — естественная система химич. элементов, со-
зданная Д. И. Менделеевым на основе открытого им
периодического закона. В 1869 Менделеев впервые
сформулировал сущность периодич. закона, а в 1871
предложил более развернутую его формулировку:
«физические и химические свойства элементов, про-
являющиеся в свойствах простых и сложных тел,
ими образуемых, стоят в периодической зависимости...
961
ПЕРИОДИЧЕСКАЯ СИСТЕМА ЭЛЕМЕНТОВ МЕНДЕЛЕЕВА
962
от их атомного веса» (Соч., т. 14, 1949, стр. 907).
Современная, более точная и глубокая формулировка
периодич. закона отражает периодич. зависимость
свойств элементов от числа электронов в атоме, опре-
деляемого зарядом атомного ядра; это число равно
порядковому (атомному) номеру элемента в системе
Менделеева. Поскольку, однако, атомные веса эле-
ментов, как правило, возрастают в той же последо-
вательности, что и заряды атомных ядер, современ-
ная табличная форма периодич. системы принципи-
ально совпадает с менделеевской. Периодич. система
отображает объективно существующую взаимосвязь
между химич. элементами. Поэтому она и была наз-
вана Менделеевым «естественной» системой эле-
ментов.
Периодич. закон и периодич. система Менделеева
служат ключом к глубокому познанию строения ве-
щества и являются одним из наиболее широких обоб-
щений в области физики и химии.
История открытия периодического закона и перио-
дической системы. Первые попытки классификации
элементов относятся к концу 18 — началу 19 вв.
Особенно же богат работами в этой области 19 век,
что связано с открытием и исследованием многих
новых элементов. (Если в середине 18 в. было из-
вестно не более 15 элементов, а в конце 18 в.— ок. 30,
то к 1869 их число возросло до 63). Первоначально
классификацию основывали лишь иа резко выражен-
ных физич. или химич. свойствах. Так, в конце 18—
начале 19 вв. возникло деление элементов на металлы
и неметаллы (А. Лавуазье, Я. Берцелиус). В начале
19 в., с развитием идеи химич. атомистики и методов
химич. анализа, появились первые попытки систе-
матизации элементов ио их атомным весам, признан-
ным основной количественной характеристикой эле-
мента. Предшественники Менделеева — И. Дёберей-
нер, Ж. Дюма, А. Шанкуртуа, У. Одлинг, Дж. Нью-
ленде и др.— установили, что имеются группы эле-
ментов, сходных по химич. свойствам,— т. наз. «ес-
тественные группы» («триады» Дёберейнера, «октавы»
Ньюлендса и др.), причем разница между атомными
весами родственных элементов в группе равна нек-рой
постоянной величине. Авторы этих работ, однако, не
шли дальше установления частных закономерностей
внутри групп.
В 1864 Лотар Мейер изложил в монографии «Со-
временные теории химии и их значение для химиче-
ской статики» имевшиеся литературные данные по
вопросу о соотношении атомных весов родственных
элементов и привел таблицу, где показал такие соот-
ношения для нескольких характерных групп. Каких-
либо теоретич. обобщений из своей таблицы Мейер
не вывел. Таким образом, ни в одной из работ по
классификации химич. элементов, предшествовавших
трудам Менделеева, не была обнаружена взаимосвязь
всех химич. элементов. Вместе с тем работы пред-
шественников подготовили почву для открытия Мен-
делеева. Важным событием, подготовившим это от-
крытие, явился международный химич. съезд в
г. Карлсруэ (1860), где были разграничены понятия
«атомный вес» и «химич. эквивалент», до тех пор не-
редко смешиваемые. Это позволило создать единую
систему атомных весов, и рассмотрение соотношении
между атомными весами элементов получило проч-
ную основу.
Менделеев открыл периодич. закон в 1869 (перво-
начальный набросок периодич. системы помечен
1 марта 1869). Это открытие было подготовлено и
предшествующей 15-летней научной деятельностью
самого Менделеева, нашедшего отдельные важные
соотношения в свойствах элементов; непосредствен-
ным же поводом к поискам послужило составление
систематич. курса химии, названного впоследствии
«Основы химии». Как и его предшественники, Мен-
делеев в качестве основной характеристики, одно-
значно определяющей химич. элемент, выбрал атом-
ный вес. Но, в отличие от них, Менделеев руковод-
ствовался твердой уверенностью в существовании об-
щего закона природы, определяющего сходства и раз-
личия между всеми элементами, и искал закономерно-
сти в изменении атомных весов не только у химически
сходных элементов, внутри одной естественной груп-
пы, но и между несходными элементами. Сопоставив
такие крайне противоположные в химич. отношении,
но близкие по атомным весам их членов группы,
как щелочные металлы и галогены, и написав первые
под вторыми, Менделеев расположил под и над ними и
другие группы сходных элементов в порядке изме-
нения атомных весов. Оказалось, что члены этих
естественных групп образуют общий закономерный
ряд, причем химич. свойства элементов периодически
повторяются.
При размещении элементов в периодич. системе
Менделеев руководствовался не только правилом
постепенного возрастания атомного веса, по и прин-
ципом периодичности химич. свойств; среди послед-
них в качестве непосредственного и основного кри-
терия он выбрал формы кислородных и водородных
соединений элементов, соответствующие их высшей
валентности. Соблюдение принципа периодичности
позволило Менделееву в нескольких местах системы
правильно расположить элементы не в порядке воз-
растания атомных весов, а с нарушением этого по-
рядка, как требовали химич. аналогии (Со—Ni,
Те—J), а для нек-рых элементов изменить обще-
принятые в то время атомные веса даже в 1,5—2 ра-
за (Be, In, Се, U и др.). Одновременно Менделеев
предсказал многие неизвестные тогда элементы, для
к-рых в периодич. системе обнаружились незаполнен-
ные места, а для трех из них — так наз. экаалюмипия,
экабора и экасицилия, подробно описал ожидаемые
свойства. Вскоре эти элементы были открыты: аналог
алюминия — галлий, Лекоком де Буабодраном в
1875; аналог бора — скандий, Л. Нильсоном в 1879;
аналог кремния — германий, А. Винклером в 1886.
Поразительное совпадение их свойств с предсказа-
ниями Менделеева привлекло внимание ученых всего
мира; периодич. закон получил всеобщее признавие
и лег в основу всего последующего развития химии.
Над уточнением и развитием своей системы Менделеев
работал ок. 40 лет. Но особенно больших успехов
достигла система Менделеева после его смерти, с
открытием самой причины периодичности, заключен-
ной в сложном строении атомов.
Периодическая система и строение атома. Замеча-
тельные достижения физики в конце 19 и начале
20 вв. позволили раскрыть сложную структуру
атомов и выяснить физич. смысл построения перио-
дич. системы. В свете этих новых результатов стало
ясно, сколь велико значение периодич. системы
для науки и какой гениальной прозорливостью
обладал ее автор — Д. И. Менделеев. К числу основ-
ных достижений физики этого времени относятся
открытие радиоактивного распада (1896) и рентге-
новских лучей (1895), создание квантовой теории
излучения М. Планком (1900), планетарной модели
атома Э. Резерфордом (1911) и, наконец, теории
строения атома Н. Бором (1913).
Опыты Резерфорда в области рассеяния а-частиц
дали следующие фундаментальные результаты: вся
масса атома практически сосредоточена в его ядре,
имеющем размеры порядка 10_,3с.и; ядро атома несет
положительный заряд, вокруг ядра обращаются
электроны. Так как атом в целом нейтрален, то
положительный заряд ядра Е должен быть целым
кратным от заряда электрона е, т. е. E — Ze, где Z—
031 к.х.э, т. з
963
ПЕРИОДИЧЕСКАЯ СИСТЕМА ЭЛЕМЕНТОВ МЕНДЕЛЕЕВА
964
число электронов в атоме. Измерения Резерфорда
показали, что величина Z различных элементов
равна приблизительно половине их атомного веса.
С другой стороны, уже давно было известно, что
атомные веса элементов, грубо говоря, равны удвоен-
ному значению их порядкового номера в системе
Менделеева. Отсюда возникла гипотеза, высказанная
в 1913 Ван-дер-Бруком, что число электронов в
атоме Z равно порядковому номеру элемента.
Модель атома Резерфорда в сочетании с классич.
электродинамикой, господствовавшей тогда в физике,
приводила к выводу о крайней неустойчивости атома,
что противоречило опыту. В 1913 Бор, связав плане-
тарную модель атома с новой квантовой теорией
излучения, сформулировал свои постулаты о стацио-
нарных (устойчивых) состояниях атомов и о возмож-
ности перехода атома из одного стационарного состоя-
ния в другое; Бор предположил, что стационарные
состояния соответствуют движению электронов по
вполне определенным орбитам (см. Атом). Для атома
водорода расчет Бора получил превосходное согласие
с опытом.
Теория Бора явилась основой для работ Г. Мозли
(1913—14) в области линейчатых (характеристиче-
ских) рентгеновских спектров, возникающих при
бомбардировке вещества быстрыми электронами. Эти
спектры обладают весьма важной особенностью:
их общая структура (число, яркость и относительное
расположение линий) сходна для различных элемен-
тов; однако, чем дальше в системе Менделеева
расположен элемент, тем в большей мере весь его
рентгеновский спектр отодвинут в область коротких
волн. Мозли открыл, что при переходе от одного
элемента с п. н. 2 к другому с п. н. Z-|-1 спектральные
серии рентгеновских лучей смещаются на одну и ту
же величину. Следовательно, положение линейчатого
рентгеновского спектра на спектральной шкале од-
нозначно определяет номер данного элемента в таб-
лице Менделеева. Систематич. исследование периодич.
системы Мозли начал с А1, приняв для него Z = 13,
т. к. от Н до А1 все места таблицы Менделеева были
давно заняты и не было оснований предполагать су-
ществование неизвестных элементов в этом проме-
жутке. Затем, переходя от элемента к элементу, он
определял их номера по величине смещения спектра,
показав, в частности, что п. н. Со действительно 27,
a Ni — 28 (хотя атомный вес Со больше, чем у Ni),
нашел порядковые номера еще не известных в то
время элементов и установил, что в периодич. системе
от Н до U должно находиться 92 элемента. Т. обр.,
фундаментальным параметром, определяющим химич.
свойства элементов, является не атомный вес, как
это предполагал Менделеев, а порядковый номер
элемента в перисдич. системе Z, или, что то же,
число электронов, окружающих ядро.
Теория Бора, развитая в дальнейшем А. Зоммер-
фельдом, качественно объяснила и саму причину
периодичности в системе Менделеева. Согласно этой
теории, электроны движутся вокруг ядра лишь
определенным образом, но «разрешенным» орбитам.
Иными словами, энергия электрона в атоме может
иметь ряд определенных дискретных значений (уров-
ней энергии). В многоэлектронном атоме электроны,
заполняя «разрешенные» орбиты, располагаются сло-
ями и оболочками. (Слоистое расположение элек-
тронов в атоме было подтверждено экспериментально;
исследования оптич. спектров единожды, дважды
и т. д. ионизированных атомов свидетельствовали
о том, что разные группы электронов в атоме — груп-
пы К, L, М, N и т. д., обладают различной энергией
связи). Чем дальше от ядра находится оболочка,
тем слабее связаны с ядром входящие в ее состав
электроны. Эти периферия, (внешние) электроны и
определяют способность элементов к химич. взаи-
модействию. Причина периодичности свойств эле-
ментов, открытая Менделеевым, заключается, следо-
вательно, в том, что по мере возрастания числа
электронов, окружающих ядро, наступает такая
стадия, когда заканчивается заполнение данного
электронного слоя и начинается заполнение сле-
дующего. При этом элементы с одним, двумя, тремя
и т. д. электронами в этом новом наружном слое
воспроизводят химич. свойства элементов, имевших
также один, два, три и т. д. электронов в предшест-
вовавшем, теперь уже глубинном, слое. Следует
указать, что периодически меняются не только химич.
свойства элементов, но и многие их физич. свойства,
такие, напр., как атомный объем, козфф. объемного
сжатия, коэфф, теплового расширения, электропро-
водность, темп-ра плавления и т. п., т. е. именно те
свойства, к-рые связаны гл. обр. с наружными элек-
тронными слоями, тогда как свойства, связанные с
глубинными слоями, меняются монотонно, без какой-
либо периодичности. Такого рода свойствами яв-
ляются атомный вес и характеристич. рентгеновский
спектр элементов. Причина послойного расположения
электронов в атоме стала ясна в 1925, когда Паули,
используя представления о спине электрона, сформу-
лировал «принцип запрета», являющийся одним из
фундаментальных законов физики (см. Паули прин-
цип). Согласно этому принципу, на одном энергетич.
уровне (в атоме, молекуле) может находиться не более
двух электронов, причем эти электроны должны иметь
противоположно ориентированные спины.
Квантовая механика, созданная в 1924—28, ра-
дикально изменившая и углубившая теорию Бора —
Зоммерфельда и, в частности, отказавшаяся от рас-
смотрения электронных орбит, оставила, однако,
без изменения основные принципы заполнения элект-
ронных оболочек в атоме. Точное решение задачи о
движении электронов в атоме по законам квантовой
механики требует одновременного рассмотрения дви-
жения всей совокупности электронов, взаимодейст-
вующих электрич. силами с ядром атома и друг с дру-
гом, что крайне сложно. Однако удовлетворительное
приближенное решение может быть получено при рас-
смотрении для каждого электрона в отдельности его
движения в неизменном электрич. ноле, создаваемом
ядром и усредненным воздействием остальных элект-
ронов; результат уточняется дополнительным учетом
тождественности электронов (метод Хартри — Фока).
В рамках этого приближения можно говорить об ор-
битах отдельных электронов в атоме, понимая под
орбитой одноэлектронную волновую функцию. При
этом квантово-механич. формулировка принципа Пау-
ли сводится к приведенной выше первоначальной
формулировке. Основное (негозбужденное) состояние
атома отвечает последовательному заполнению атом-
ных орбит электронами, начиная с орбиты, к-рой отве-
чает наинизшая энергия, и далее переходя к все более
высоким энергетич. уровням.
Принципы построения периодической системы. Каж-
дый электрон в атоме, в соответствии с квантовой
механикой, характеризуется четырьмя квантовыми
числами: главным квантовым числом п, принимающим
значения и=1, 2, 3, 4 ..., азимутальным I, прини-
мающим значения Z=0, 1, 2..., п—1, магнитным mlt
имеющим (2Z+1) значений, и спиновым ms, прини-
мающим значения +% и —%. Состояния с 1 = 0, 1,
2, 3, 4... принято обозначать буквами s, р, d, f, g ...
и соответственно называть s-, р-, d-, f-, g- ... состоя-
ниями (см. Атом). Электроны с данным п образуют
электронный слой, к-рый состоит из п оболочек с
1 = 0, 1, 2 ... п—1 и в соответствии с Паули принципом
содержит 2 я2 электронов. Отсюда получаем след,
распределение электронов по слоям и оболочкам.
965
ПЕРИОДИЧЕСКАЯ СИСТЕМА ЭЛЕМЕНТОВ МЕНДЕЛЕЕВА
966
Таблица 1. Распределение электронов по слоям и оболочкам
п 1 1 0 2 о 3 1 2 0 1 2 3
0 1
Обозначение слоев и оболочек . . . 1s 2s 2р 3s Зр 3d 4s 4р 4d 4/
Число электронов в слое 2 2 + 6 = 8 2+6+10 = 18 2+6+10+14 = 32
Отсюда видно, что максимально возможное число
электронов в первом слое (и=1) равно 2 (ls-со-
стояния); во втором слое — 8 (2s- и 2р-состояния);
в третьем и четвертом слоях — соответственно 18
и 32 электрона. Тем самым устанавливается связь
между слоистым расположением электронов в атоме
и периодич. системой: периоды таблицы Менделеева
содержат 2, 8, 18 и 32 элемента. Остается выяснить,
почему в таблице Менделеева встречаются по два раза
периоды из 8 и 18 элементов. Это объясняется тем,
что в табл. 1 указано возможное максимальное число
электронов в различных слоях, в действительности
же электроны в атомах располагаются в тех состоя-
ниях (из числа возможных), к-рые соответствуют
наименьшей энергии. Энергия электрона на орбите
тем выше, чем больше п, а при данном п тем выше, чем
больше I. Поэтому последовательность энергетич. уров-
ней отдельных состояний электрона не всег-
да совпадает с последовательностью главных
квантовых чисел п.
В табл. 2 представлена схема заполнения
электронных оболочек последовательно для
мех элементов периодич. системы по отдель-
ным периодам. Первый период содержит лишь
два элемента, что соответствует максималь-
ному числу электронов в ls-состоянии пер-
вого слоя. Начиная с Li идет постепенное за-
полнение второго слоя вплоть до Ne (Z=10).
С одиннадцатого электрона Na начинается
заполнение третьего слоя — третьего периода
системы Менделеева. В элементах, следую-
щих за Na, идет последовательное заполне-
ние электронами состояний 3s и Зр третьего
слоя. У Аг восемь электронов составляют
симметричную группу и обусловливают сход-
ство его физике-химич. свойств с неоном.
Этим завершается третий период, содержа-
щий, как и второй, 8 элементов. 19-й эле-
мент К начинает новый, четвертый, слой и
четвертый период таблицы. Однако число сво-
бодных мест третьего слоя далеко не исчер-
пано. После Аг остаются еще свободными 10
мест Зй-состояния. Здесь впервые последова-
тельный порядок заполнения нарушается из-
за энергетич. соображений. Состояние 4s ока-
зывается энергетически более выгодным, чем
состояние 3d. Но начиная со Sc и вплоть до
Ni вновь становится энергетически более вы-
годным заполнение состояния 3d. Все места
этого состояния у Ni оказываются заполнен-
ными и с Си начинается нормальное запол-
нение состояния 4s четвертого слоя. Это об-
стоятельство обусловливает нек-рое сходство
меди со щелочными металлами. Кг с восемью
электронами в этом слое, заканчивающий чет-
вертый период, оказывается сходным с Nen
Аг. С последующего за Кг 37-го элемента Rb
начинается застройка пятого слоя, хотя в
четвертом слое еще остаются 24 свободных
места — 10 мест в состоянии 4d и 14 мест в
состоянии 4/. Здесь, как и у К, состояние
5s оказывается энергетически болеевыгодным.
Достройка четвертого слоя возобновляется с
ся
Y, начиная с к-рого идет заполнение 4с?-оболочки.
Состояния 4/ остаются незаполненными вплоть до
58-го элемента. Лишь с Се они начинают заполнять-
ся. Застройка 4/-состояний охватывает 14 элемен-
тов, образующих своеобразную группу лантанидов,
весьма сходных между собой по своим физико-хи-
мич. свойствам. Такая же группа элементов с дост-
раивающимися Бу-состояниями, называемая акти-
нидами, начинается с Th (Z = 90).
Т. обр.,каждый период (кроме первого) начинает-
со щелочного металла с одним валентным электро-
ном и кончается инертным газом с восемью валентны-
ми электронами, образующими замкнутую оболочку.
Второй и третий периоды, где нормально застраи-
ваются s- и p-состояния, содержат в соответствии с
этим по 8 элементов. Периоды же четвертый и пятый,
в к-рые вклиниваются группы элементов с достраи-
вающимися ^-состояниями, содержат по 18 элементов.
Наконец, последний полный период — шестой —
содержит 32 элемента, т. к. в нем появляется новая
группа из 14 элементов с достраивающимися 4/-
состояниями. Тем самым вся сложная периодичность,
открытая Менделеевым, полностью объясняется рас-
положением электронов по группам, характеризу-
емым определенными квантовыми числами.
Интересно отметить, что 72-й элемент в то время,
когда Н. Бор строил табл. 2, не был открыт. Было
Таблица 2. Схема заполнения электронных оболочек атомов
Пери- оды z Эле- мент Оболочки
К L M N О P Q Is 2s 2p 3s 3p 3d 4s 4p 4d 4/ 5s 5p 5d 5/ 6s 6p 6d 7s
I II III 1 2 Н Не 1 2
3 4-9 10 Li Be—F Ne 2 1 заполнение состояний 2s и 2 p 2 2 6
11 12-17 18 Na Mg—Cl Ar 2 2 6 1 заполнение состояний 3s и 3 p 2 2 6 2 6
IV V VI VII 19 20 21 22-28 29 30-35 36 К Ca Sc Ti—Ni Cu Zn—Br Kr 2 2 6 2 6 1 2 2 6 2 6 2 2 2 6 2 6 1 2 заполнение состояний 3 d 22 62610 1 заполнение состояний 4s и 4 р 2 2 6 26 10 26
37 38 39 40-46 47 48-53 54 Rb Sr Y Zr—Pd Ag Cd—J Xe 22626 10 26 1 22626 10 26 2 22626 10 261 2 заполнение состояний 4 d 22626 10 26 10 1 заполнение состояний 5 s и 5 р 22626 10 26 10 56
55 56 57 58 59—71 72 73—78 79 80—85 86 Cs Ba La Ce Pr—Lu Hf Ta—Pt Au Hg—At Rn 22626102610 26 1 22626102610 26 2 22626 10 26 10 1261 2 22626 10 26 10 226 2 заполнение состояний 4 f 2 2 6 26 10 26 10 1426 2 2 заполнение состояний 5 d 2 2 6 2 6 10 2 6 10 14 2 6 10 1 заполнение состояний 6 s и 6 р 2 2 6 26 10 2610 14 26 10 2 6
87 88 89 90- ЮЗ Fr Ra Ac Th—Lw 2262610 2 610142610 26 1 22626 10 26 10 14 26 10 26 2 22226102610 14 2610 261 2 заполнение состояний 5/
31*
967
ПЕРИОДИЧЕСКАЯ СИСТЕМА ЭЛЕМЕНТОВ МЕНДЕЛЕЕВА
968
Пери- оды I а b ПЕРИОДИЧЕСКАЯ СИСТЕМА ХИМИЧЕСКИХ ЭЛЕМЕНТОВ Д.И. МЕНДЕЛЕЕВА VIII a b
1 1Н 1,00797 s’ 2 He 4,0026 s2
п а b III a b IV a b V a j b VI a b VII a b
2 3.Li 6,939 s’ 4 Be 9,0122 S2 5B 10,811 $2p1 6C 12,01115 s2p2 7N 14,0067 s2p3 80 15,9994 s2p‘ 9F 18,9984 S2p’ io Ne 20,183 S2p’
3 11 Na 22,9898 . s1 12 Mg 24,312 S2 13 Al 26,9815 s2p1 14 Si 28,086 s2p2 15 P 30,9738 S 2p3 16 s 32,064 s2p* 17 Cl 35,453 s2p’ 18 Ar 39,948 s 2p’
4 19 К 39,102 s’ 20 Са 40,08 S2 21 Sc 44,956 d’s2 22 Ti 47,90 d2 S2 23 V 50,942 d3s2 24 Cf 51,996 d’s’ 25 Mn 54,9381 d5 s2 26 Fe 55,847 d’ s2 27 Co 58,9332 d’s2 28 Ni 58,71 dBs2
29 Cu 63,54 d»s’ зо Zn 65,37 d'° S2 31 Ga 69,72 S2p’ 32 Ge 72,59 S2p2 зз As 74,9216 s2p3 34 Se 78,96 S2p4 35 Br 79,906 s2p’ 36 Kr 83,80 S2p’
5 37Rb 85,47 s’ 38 Sr 87,62 s2 39 Y 88,905 d’s2 40 Zr 91,22 d2 s2 41 Nb 92,906 d‘s’ 42 Mo 95,94 d’s’ 43 Tc [9?] d’s2 44 Ru 101,07 d’s1 45 Rh 102,905 46 Pd 106,4 dio
47 Ag 107,870 tfIOS1 48 Cd 112,40 d10 s2 49 In 114,82 S2p’ 50 Sn 118,69 s2 p2 51 Sb 121,75 s2p3 52 Те 127,60 s 2p4 53 J 1269044 s*p’ 54 Xe 131,30 s 2p’
6 55 Cs 132,905 s’ 56 Ba 137,34 s2 57 La 138,91 d’s2 ланта- ниды 58-71 72 Hf 178,49 d2s2 73 Ta 180,948 d3 s2 74 W 183,85 d‘s2 75 Re 186,2 d5$2 760s 190,2 d’s2 77 lr 192,2 d7s2 78 Pt 195,09 d’s’
79 Au 196,967 d IDs’ 80 Hg 200,59 tflO s2 81 Tl 204,37 s2p’ 8?Pb 207,19 s2p2 83 Bi 208,980 s2p3 84 Po [209] *s2p‘ 85 At [210 ] s2p’ 86 Rn [ 222 1 s2pB
7 87Fr [223] s1 88 Ra [226] s2 89 Ac [227] d’s2 акти- ниды 90-103 (104) (105) (106) (107) (108) (109) (110)
(111) (112) (113) (114) (115) (116) (117) (118)
Лантаниды
58 Ce 140,12 f2S2 59 Pr 140,907 /3S2 60 Nd 144,24 Л S2 61 Pm [1Л] f5 s2 62 Sm 150,35 /’s2 63 Eu 151,96 /’ s2 64 Gd 157,25 /’d’s2 65 Tb 158,924 (/’d’s2) 66 Dy 162,50 /’’ s2 67 Ho 164,930 /”s2 68 Er 167,26 . /«S2 69 Tu 168,934 /’’'s2 70 Yb 173,04 M*s2 71 Lu 174,97 /’‘d’s2
Актиниды
90 Th 232,038 d2 s2 91 Pa [231] /2d’s2 92 U 238,03 /3d’s2 93 Np [237 j /‘d’s2 94 Pu [2644]2 /О s2 95 Am [2«]2 f7 s2 96 Cm [247] (/’d’s2) 97 Bk [247] (/’d’s2) 98 Cf [251] (/”s2) 99 ES [254] (/”s2) 100 Fm [253] (/’2s2) 101 Md [254] (/” s2) 102(NO) [255] (/’‘s2) 103 Lw [257] (/'‘d's2)
неясно, сколько должно быть лантанидов. Полагая,
что число их равно пятнадцати, 72-й элемент искали
среди минералов, содержащих редкие земли. Т. к.
число 4/-электронов равно 14, то этот элемент должен
иметь близкую к Zr внешнюю электронную оболочку.
Поэтому Бор предложил искать 72-й элемент в цирко-
ниевых рудах. Этот элемент, наз. гафнием, к торжеству
теории Бора, и был обнаружен в циркониевых рудах.
Что же касается аналогов лантанидов — актини-
дов, у к-рых заполняются 5/-состояния, то здесь сле-
дует иметь в виду, что 6rf- и 5/-состояния энергети-
чески весьма близки, что обусловливает легкость
переходов электронов между ними и приводит к
большим трудностям при установлении истинного
расположения электронов по этим состояниям.
С момента открытия системы Менделеева было
опубликовано неск. сот различных вариантов ее
изображения на плоскости и н пространстве. Наи-
более употребительна т. н. короткая таблица, один
из возможных вариантов к-рой помещен выше. Перио-
дич. система составлена по данным, опубликованным
до середины 1963 г.
Описание периодической системы. В таблице по-
мещены символы элементов, принятые в 1961 Меж-
дународным съездом Союза чистой и прикладной
химии. Для ряда элементов в литературе употребля-
ются различные названия; напр., 86-й элемент наз.
радоном (Rn), или эманацией (Em), 71-й элемент —
лютецием (Lu), или кассиопеем (Ср). Для нек-рых
элементов употребляются различные символы, напр.
наряду с символами Ar, J, Tu, Md, применяются
также символы A, I, Tm, Mv. Возможно, что назва-
ние 102-го элемента будет изменено, поэтому его
символ помещен в скобках.
Атомные веса даны по таблице международных
атомных весов на 1962 в углеродной шкале (за еди-
ницу принята ‘/12 массы изотопа углерода С12).
Для радиоактивных элементов, не имеющих стабиль-
ных или долгоживущих (f'/^lO8 лет) радиоактивных
изотопов, понятие атомного веса, как среднего зна-
чения для природной смеси изотопов, теряет смысл.
Поэтому для таких элементов приведено массовое
число наиболее долгоживущего изотопа (в прямых
скобках).
Для каждого элемента приведена конфигурация
внешних электронных оболочек. Номера электронных
слоев, к к-рым принадлежат оболочки, даны в первой
колонке слева от таблицы. Напр., для Вг (2=35)
969
ПЕРИОДИЧЕСКАЯ СИСТЕМА ЭЛЕМЕНТОВ МЕНДЕЛЕЕВА
970
конфигурация внешних оболочек s2p5 означает на-
личие 2 электронов в 4<;-оболочке и 5 электронов в
4р-оболочке, т. е. 4s24p5; в атоме Вг, кроме внешних
оболочек, заполнены и внутренние; полная электрон-
ная конфигурация атома ls22s22pe3s23p63d104s24p5.
Таблица состоит из 7 горизонтальных периодов,
в каждом из к-рых начинается заполнение нового
электронного слоя. В 1-й колонке приведены номера
периодов п, совпадающие с величиной главного
квантового числа электронного слоя, наиболее уда-
ленного от атомного ядра. Как видно из таблицы,
в 1-м периоде застраивается ls-оболочка; период со-
держит 2 элемента. Во 2-м и 3-м периодах, содержащих
по 8 элементов, сначала застраиваются s-, а затем
р-оболочки. 4-й и 5-й периоды состоят каждый из 18
элементов, у к-рых застраиваются сначала соответст-
вующие s-оболочки (4s и 5s), затем d-
оболочки предыдущих слоев (3d и 4d)
и, наконец, р-оболочки (4р и5р). Эле-
менты, у к-рых происходит застройка
d-оболочек, паз. переходными. В 6-м
периоде, содержащем32 элемента, пос-
ле заполнения бя-оболочки у С8 иВа
начинает застраиваться 5й-оболочка у
La, но у следующих 14 элементов —
лантанидов (Се—Lu) происходит заст-
ройка более глубокой 4/-оболочки и
только после ее заполнения достраива-
ются 5d- и бр-оболочки (Hf—Hg и Т1—
Ви). В 7-м периоде заполнение оболо-
чек происходит аналогично заполне-
нию в 6-м периоде; последовательно
застраиваются 7з-оболочка (Fr, Ra),
затем замещается 1-е место в 6<7-оболо-
чке (Ас), после чего происходит запол-
нение 5/-оболочки у 14 актинидов (Th—
Lw). У следующих, еще неизвестных
элементов (их порядковые номера на-
печатаны в скобках), должно происхо-
дить заполнение 6й-оболочки (элемен-
ты с п. н. от 104 до 112) и 7р-оболочки
(113 — 118).
Исходя из аналогии в строении 6-го
и 7-го периодов, можно лишь указать
места еще неизвестных элементов с
2>103 в 7-м периоде. Однако устано-
вить, на каком элементе кончается пе-
риодич. система, т. е. с какого элемен-
та все изотопы имеют настолько малое
время жизни, что практически распа-
даются в момент образования, еще
нельзя. Поэтому помещение в скобках
элементов с 2=104 до 2=118' отнюдь
не следует понимать как гипотезу о
существовании в природе именно 118
элементов. Общий порядок заполнения
оболочек с ростом 2 в неск. случаях
нарушается, т. к. у нек-рых элементов
энергии связи s-, d- и /-электронов
весьма близки, и происходит «переска-
кивание» электронов между оболочка-
ми. Напр., при переходе от Zr(2=40)
к Nb(Z=41) строение внешних оболо-
чек меняется от <?s2 к di s', а при пе-
реходе от Np(Z = 93) к Ри(2=94) —от
fd's3 к s2/6.
Элементы в периодич. системе раз-
деляются на 8 вертикальных групп
(обозначены римскими цифрами I —
VIII). В короткой форме таблицы группы состо-
ят из подгрупп — а (основная) и Ъ (побочная).
У элементов 1а подгруппы (щелочных металлов)
каждый раз начинается образование новой электрон-
ж
1
?
i
ной оболочки, а у элементов Villa подгруппы (инерт-
ных газов) она заканчивается. Первый по порядку
переходный элемент — скандий, впервые начинает
формирование подгруппы b (в Ш гр. сдвинут вправо).
Т. обр., у элементов главных подгрупп застраива-
ются s- и р-оболочки, у элементов побочных подгрупп
d- и /-оболочки.
Лантаниды и актиниды, принадлежащие к III гр.
системы, помещены для компактности внизу таб-
лицы, хотя правильнее было бы располагать все
элементы периодич. системы друг за другом, по
мере возрастания 2. В соответствующих местах
таблицы, после La и Ас, действительное положение
лантанидов и актинидов помечено их порядковыми
номерами в двух квадратах (58—71 и 90—103). Вопрос
о месте Th, Ра и U долго обсуждался в литературе в
«тем эжйжйк во здпмш» и ряджь.
ГРУППЫ ЗЛ t KIHTO В-tl
¥ Yt j V’i
м» *
Me ill
в '
в
А
Sc
ИВ
й®
till
вв'
в
ill
Ай»»
S7,t
iff
II
ИИИ
MB
€1».
s
S.‘
sa,«.
. Ji!!
Ж1И
Bl
liilih
жи
МИ
S
«.8
A
ЯшЯ
ИМИ
Zb
sm
eie
TI -
w
Iff
h
МВЙ
Ю
V,
Рис.
ft
Mfilj
liliih
“jti
...
и
I
MM
i
, .ЯИИГ
A
8»
t
1
Willi
L»
Ж®
.ы
И
fir
Кжяи
V
Mmsss.-
ЙУЛ J
Xpsxb.
Cr
ft
; i
Ж «миг ч»ч
Ш Fe ’
w
I
i
ШЯ
*>.
Sn
J
МИ1
Ж8ЯЯ
п
I
М.Й
Sb
£9 Ж
Вг"
is»
1Ш
Т»
S«<-:
i
IT
jljjl
ОШ
12?
m i
I i
E»*t« Sts*
fc ft M
ot.t HSA
Г «- Им -
! 6s Jr pt (Jtajj
: 8
»««»!♦ < > < t 14 p > « < « » « ♦ л Ы:
| 8 ГО ! И I F* I W! i»s
1, Периодическая
химии» (1906 г.,
Ж :
система элементов, приложенная к 8-му изданию «Основ
последнее издание при жизни Д. Л. Менделеева).
связи с тем, что эти элементы имеют также сходство
с элементами IV, V и VI групп соответственно (в
частности, у Th электронная конфигурация ближе к
элементам IV гр.). В связи с этим во многих таблицах
971
ПЕРИОДИЧЕСКАЯ СИСТЕМА ЭЛЕМЕНТОВ МЕНДЕЛЕЕВА
972
эти три элемента расположены (иногда в скобках)
в клетках, к-рые в действительности должны занять
неизвестные еще элементы с п. н. 104, 105 и 106.
Как видно из таблицы, номер группы периодич.
системы совпадает с общим числом наружных s-
и р-электронов у элементов главной подгруппы,
т. е. с максимальным числом их валентных электро-
нов. В побочных подгруппах эта правильность имеет
ряд исключений. Тем не менее можно в общем
сказать, что номер группы в периодич. системе
совпадает с общим числом валентных электронов
у атомов элементов, расположенных в этой группе.
Подтверждается таким образом и общее определение
групп, данное Менделеевым: номер группы равен
максимальной валентности элемента по кислороду.
гл. обр. короткой системой (на рис. 1 дана периодич.
система, опубликованная ее автором в 1906). В длин-
ной форме периодич. системы каждый элемент с ана-
логичной электронной структурой помещен в отдель-
ной группе, поэтому длинная форма таблицы более
строго соответствует электронной структуре и химич.
аналогиям. На рис. 2 приведена длинная форма
системы Менделеева по Бору—Томсену.
7H
2 He
5Li
4 Be
5В
SC
7N
SO
9F
70 Ne
. 77Na
72M^X
74 Si
/5 Р
16 S
17С1
18 Аг
Рис. 2. Периодическая система элементов
по Томсену и Бору (длинная форма таб-
лицы Менделеева).
19 К. -
20Са-
21 Sc
22 Ti
23 V
24 Сг
25Мп
26Fe
27Со
28 Ni
'29Си-
30Zn-
31Ga-
32Ge-
33As-
34Se-
35Br-
36Kr-
. 37Ць
.58 Sr
59 V
40Zr
47 Nb
42Mo
45Tc
44RU
45Rh
46Pd
47Аг
4SCd
49In
50Sn
51 Sb
52Te
531
54 Xe
В помещенной на стр. 967 таблице все главные
подгруппы (а) находятся слева, а побочные (6)—спра-
ва. Такое расположение удобно для сравнения у- и
p-элементов, с одной стороны, и переходных — с дру-
гой. Во многих других вариантах таблицы 6 элемен-
тов 2-го периода, начиная с бора, сдвинуты вправо;
этим отмечается переход от типичных металлов — Li
и Be, к элементам, образующим неметаллич. тела —
В, С, N, О, F, Ne. Подгруппы в этом варианте оста-
ются теми же, но меняется их относительное рас-
положение, начиная с III гр.; напр., подгруппа В,
Al, Ga, In, Т1 расположена правее, чем подгруппа
Sc, Y, La, Ас. Такое расположение дает ряд преиму-
ществ прп рассмотрении химич. свойств (напр., в
одной и той же группе элементы, образующие основа-
ния, оказываются левее, чем элементы, образующие
кислоты).
Не общепринято и объединение в одну группу
инертных газов и элементов триад VIII группы.
К моменту создания периодич. системы инертные
газы еще не были известны. После их открытия (в
конце 19 в.) Менделеев предложил объединить их
в особую нулевую группу (что подчеркивало их
«нулевую валентность», т. е. химич. недеятельность).
Однако в последнее время многие авторы рассмат-
ривают прежние разобщенные 0-группу и триады
как две подгруппы VIII гр. Это обосновывается не
столько тем, что, как оказалось, инертные газы спо-
собны при определенных условиях к химич. взаимо-
действию, но гл. обр.— для придания большей стро-
гости и единообразия самой системе.
Периодич. система была опубликована Менделее-
вым в 2 формах — короткой, в к-рой элементы боль-
ших периодов подразделяются на два ряда, и длинной,
где элементы больших периодов располагаются в
один непрерывный ряд. Сам Менделеев пользовался
55Cs
56 Ba-
57 La
58 Ce
59 Pr
60 Nd
61 Pm
62 Sm
63 Eu
64 Gd
S5Tb
66 Dy
67 Ho
68 Er
69 Tu
70 Vb
71 Lu
72 Hi
75 Ta
74 W
75 Re
76 Os
77 Ir
78 Pt
79 Au
80 Hg
81 TI
82 Pb
85 Bi
84 Po
85 At
86 Rn
II
II
Ф
'/
-87 Fr
-88 R a
89 Ac
90 Th
91 Pa
92 U
95 Np
94 Pu
95 Am
96 Cm
97 Bk
98 Cf
99 Es
100 Fm
101 Md
702(No)
103 Lw
Периодичность свойств в системе Менделеева.
В итоге многочисленных экспериментальных иссле-
дований была установлена связь самых разнообраз-
ных химич. и многих физич. свойств элементов с
их положением в периодич. системе. Наиболее простое
выражение принцип периодичности имеет в приложе-
нии к свойствам самих элементов (отдельных атомов).
При образовании химич. соединений (молекул) или
простых и сложных тел (конденсированных фаз)
закономерности значительно усложняются, будучи
обусловлены не только свойствами отдельных атомов,
но и их взаимодействием при различных условиях;
эти закономерности сохраняют, однако, ярко выра-
женный периодический характер. Как подчеркивал
сам Менделеев, периодический закон «...выра-
жает свойства элементов, а не прос-
тых тел. Свойства простых и сложных тел находят-
ся в периодической зависимости от атомного веса
элементов только потому, что ...сами составляют
результат свойства... элементов, их образующих»
(«Основы химии», СПБ, 1899, 5-е издание, с. 463,
прим. И).
Одним из важнейших свойств химич. элемента,
непосредственно связанным со структурой электрон-
ной оболочки, является ионизационный потенциал /»
характеризующий энергию связи электрона в атоме1.
На рис. 3 представлена зависимость первого иониза-
ционного потенциала Ц от порядкового номера эле-
мента Z. Элементы-аналоги на рис. соединены пунк-
973
ПЕРИОДИЧЕСКАЯ СИСТЕМА ЭЛЕМЕНТОВ МЕНДЕЛЕЕВА
974
4 8 12 16 20 24 28 52 36 40 44 48 52 56 74 78 82 86
Порядковый номер
Рис. 3. Периодичность потенциалов ионизации.
тарными линиями. Резкие максимумы наблюдаются
у атомов инертных газов, к-рые обладают наиболее
устойчивой электронной конфигурацией s’-p6. В мини-
мумах кривой находятся щелочные металлы, атомы
к-рых, отдавая свой единственный валентный 4-элект-
рон, приобретают конфигурацию предшествующего
инертного газа, Т. обр., длины периодов на кривой
полностью соответствуют длинам периодов системы
Менделеева. В пределах одного периода/, изменяет-
ся не монотонно. На кривой наблюдаются вторичные,
менее резкие максимумы, соответствующие запол-
нению 4-оболочкн у элементов II гр.—Be, Mg, Zn, Cd
и Hg. У следующих за ними элементов III гр.—В, А1,
Ga, 1п,Т1, появление 1-го р-электрона снова снижает
потенциал ионизации. Следующие максимумы на-
блюдаются у элементов V гр.—N, Р, As, что соот-
ветствует энергетически выгодному половинному за-
полнению р-оболочки, содержащей три неспаренных
электрона. У расположенных за ними элементов VI
гр.—O,S,Se, потенциалы ионизации снова снижаются.
Во II гр. периодич. системы более резкие вторичные
максимумы у Zn, Cd и Hg, по сравнению с Be и Mg,
объясняются влиянием застроенной d-электронной
прокладки. В пределах одной группы с увеличением
Z значения I, в общем, убывают, что связано с увели-
чением расстояния от ядра внешней электронной
оболочки (см. также Ионизации потенциал). Перио-
дич. закономерности можно проследить и на изме-
нении вторых, третьих и т. д. потенциалов иони-
зации. Периодически изменяется и сродство к элек-
трону, выражающее работу присоединения электрона
к нейтральному атому (см. Сродство к электрону).
Положение элемента в нериодич. системе опреде-
ляет и его валентность — число электронов, участ-
вующих в образовании химич. связи. Наиболее
четко периодич. зависимость выявляется на примере
высшей положительной валентности, равной числу
электронов в наружной электронной оболочке атома.
В малых периодах она возрастает от 1 до 7 при пере-
ходе от I к VII гр. и равна 0 в VIII гр. (у инертных
газов). У элементов больших периодов высшая
положительная валентность изменяется более сложно.
Как правило, в 1-й половине периода она возрастает
от 1 до 8 (в побочной подгруппе VIII гр.), затем
при переходе к побочной подгруппе II гр. (Zn, Cd, Hg)
уменьшается до 2 и вновь возрастает к концу периода
(VII гр.—галогены) до 7. Нек-рые элементы прояв-
ляют аномально высокую валентность, объясняемую
особенностями строения их атомов. Так, валентность
элементов подгруппы 1 Ь (Cu, Ag, Аи) доходит до 3.
Это, по-видимому, связано с непрочностью 18-элект-
рониой оболочки, формирование к-рой заканчивается
в середине большого периода; часть входящих в
ее состав электронов проявляет свойства электронов
валентности. Интересные примеры аномально вы-
сокой положительной валентности наблюдаются в
семействе лантанидов. Характерная для этого семей-
ства высшая положительная валентность 3 соот-
ветствует номеру группы в системе Менделеева.
Однако при начале формирования 4/-оболочки об-
разующие ее электроны связаны не очень прочно.
Вследствие этого С.е, Рг и отчасти Nd дают соедине-
ния, в которых они 4-валентны. Рассматриваемая
оболочка заполняется в два приема, и электронная
конфигурация седьмого элемента Gd (/7d’ss) ока-
зывается наиболее устойчивой; валентность 3 — для
Gd наивысшая. ТЬ, следующий за Gd, снова
проявляет высшую положительную валентность 4.
Еще более резкие отклонения наблюдаются в семей-
стве актинидов (см. также Валентность, Химическая
связь).
Периодич. закономерность проявляется в атомных
оптич. спектрах, возникающих при электронных
переходах в валентной оболочке. Спектры атомов
щелочных металлов, обладающих одним валентным
электроном, весьма сходны между7 собой. Так же
сходны между собой и спектры атомов с большим
числом электронов па внешней оболочке, но строение
этих спектров значительно более сложно. Особенно
сложны спектры атомов с достраивающимися cl- и
/-оболочками, состоящие из сотен и тысяч линий.
Истолкование спектров многоэлектронных атомов и
периодичности их строения производится с доста-
точной полнотой на основе квантовой механики
(см. Атомные спектры).
Для рассмотрения свойств химич. элементов и их
соединений необходимо учитывать наряду с элект-
ронной структурой и размеры атомов, в общем харак-
теризуемые эффективными атомными и ионными
радиусами. Как видно из рис. 4, эффективные атомные
радиусы Ra периодически изменяются в зависимости
от порядкового номера элемента Z. В малых периодах
с возрастанием Z атомные радиусы сначала умень-
шаются, что обусловлено повышением заряда ядра,
стягивающего электроны ближе к центру атома;
Рис. 4. Периодичность атомных радиусов.
затем, с увеличением числа электронов в наружной
оболочке атома, благодаря их взаимному отталки-
ванию, значения Ra возрастают. Конкуренция этих
двух факторов при увеличении Z обусловливает более
сложный характер кривой, чем в случае ионизацион-
ных потенциалов. Еще более сложны соотношения
в больших периодах. Тем не менее очередной резкий
975
ПЕРИОДИЧЕСКАЯ СИСТЕМА ЭЛЕМЕНТОВ МЕНДЕЛЕЕВА
976
максимум всякий раз приходится на щелочной эле-
мент — первый в новом периоде. Примерно такая
же закономерность характерна для ионных радиусов
(см. также Ионные радиусы, Межъядерные расстояния).
Важная особенность наблюдается в семействе ланта-
нидов; при переходе от La к Lu действие возраста-
ющего заряда ядра преобладает над взаимным оттал-
киванием электронов наружных оболочек, в связи
с чем атомные и ионные радиусы уменьшаются (так
называемое лантанидное сжатие). В результате раз-
меры атомов элементов-аналогов, разделенных груп-
пировкой лантанидов, оказываются практически оди-
наковыми, что приводит к повышенному химическо-
му сходству этих элементов (Zr и Hf, Nb и Та, Мо
и W и др.).
С кривой эффективных атомных радиусов очень
сходна кривая атомных объемов соответствующих
простых тел, впервые данная Мейером в 1870 и имев-
шая большое значение в истории периодич. закона.
Как известно, атомный объем равен частному от де-
ления атомного веса па удельный вес. Поэтому кри-
вая уд. весов простых тел имеет в общем обрат-
ный характер по сравнению, с кривой атомных объ-
емов.
Положение элемента в периодич. системе опреде-
ляет внешний вид, кристаллич. структуру и многие
физич. свойства соответствующих простых тел. Эле-
менты начала периодов, с низкими ионизационными
потенциалами и большими атомными радиусами,
содержат в твердом и жидком состояниях часть элек-
тронов в особом «полусвободном» состоянии, в виде
так наз. электронного газа, распределенного по
всему объему тела. Такие тела — металлы, харак-
теризуются блеском, высокой электро- и теплопро-
водностью, пластичностью и др. Отчетливое ослаб-
ление металлич. характера наблюдается с увеличе-
нием ионизационного потенциала и уменьшением
атомного радиуса, т. е. при переходе слева направо
или снизу вверх в малых периодах системы (в боль-
ших периодах элементы, у к-рых происходит запол-
нение d- и /-оболочек, проявляют свойства метал-
лов). На рис. 5 представлены изменения темп-ры
Термодинамич. свойства веществ также явно за-
висят от положения элемента в периодич. системе.
В качестве примера на рис. 6 приведены теплоты
образования окислов и хлоридов для элементов III
500
450
400
550
500
250
/// группа
il I I— // группа I
*-300 ? | | - > J,
-250-f
I S
-200 g
2^3
Al??vfscko3
/5<
’-50
--------100
.^>LaCl3_
in,b, r
.InCi
BCI
-GaCfg-
вг°.
- ScCh-
z>ici
200
150
100
500 10 20 50 40 50 60 70 80
гСЦ
’ХЕПСЦГ I
,_______Si
:od
•пО
«*5пС1;'
Порядковый номер
"I ThCij
-ШО*.
РЪОа
>ССЦ
10 20 50 40 50 60 70 80 90
Порядковый номер
?РЬС1Л
Рис. 6. Зависимость тепл от
образования окислов и хло-
рпдов элементов III и IV гр.
от порядкового номера.
Рис. 5. Периодичность физич. свойств: обратные темп-ры
плавления -у- 10* (верхняя кривая), термич. коэфф, ли-
нейного расширения a-10s (средняя кривая), коэфф,
объемной сжимаемости и-10е (нижняя кривая); для участ-
ков кривых, обозначенных пунктиром, достоверных све-
дений нет.
плавления, коэфф, линейного расширения и коэфф,
объемной сжимаемости элементов в кристаллич.
состоянии. Здесь также совершенно явственно вы-
ступает периодичность этих свойств.
н IV групп. Кривые наглядно показывают, что эле-
менты как главных, так и побочных подгрупп обеих
групп проявляют (по сравнению с А1 и Si соответ-
ственно) закономерное изменение свойств.
Периодичность в свойствах химич. соединений
удобно проследить на примере высших окислов и
гидроокисей элементов. Элементы начала периодов
образуют окислы основного характера. С увеличе-
нием номера группы (в малых периодах) основной
характер окисла сменяется амфотерным, а затем
кислотным. У элементов одной подгруппы с увели-
чением Z, как правило, усиливаются основные (п
ослабляются кислотные) свойства окислов. Эти общие,
а также наблюдаемые в подгруппах и периодах более
сложные закономерности объясняются на основе
представлений о поляризации и деформации атомов
и ионов — явлениях, к-рые, в свою очередь, обуслов-
лены структурой электронной оболочки, валентностью
и размерами взаимодействующих частиц (см. Гидро-
окиси). Более сложным образом зависит от положения
в системе способность элементов к комплексообразова-
нию, особенно сильно развитая у переходных элементов
(см. Комплексные соединения). На основе периодич.
системы изучаются и взаимодействия металлов меж-
ду собой. Так, установлено, что элементы одной под-
группы, как правило, не образуют металлич. соедине-
ний; напротив, твердые р-ры дают либо металлы Одной
подгруппы, либо металлы, стоящие в одном периоде,
с небольшим различием в размере атомов. Чем даль-
ше друг от друга в периоде расположены метал-
лы, тем меньше тенденция к образованию твердых
растворов (см. Металлические соединения, Твердые
растворы).
Приведенные примеры лпшь иллюстрируют при-
ложение принципа периодичности. Их перечень
может быть далеко продолжен: на основании перио-
дич. системы изучаются и окислительно-восстанови-
тельные, и каталитические, и полупроводниковые, и
магнитные, и многие другие — самые разнообразные
свойства элементов и их соединений. Необходимо,
однако, отметить, что современная теория строения
атомов может быть использована для количественного
расчета пока еще очень ограниченного круга хими-
ческих и физических свойств (таковы оптические
и рентгеновские спектры и магнитная восприимчи-
вость) для сравнительно простых систем. Другие важ-
ные физические константы (такие, как температу-
ры плавления и кипения, теплоты сублимации, уд.
электросопротивление и т. д.) могут быть в настоя-
щее время лишь качественно связаны со строением
атомов.
977
ПЕРИОДИЧЕСКИЙ ЗАКОН МЕНДЕЛЕЕВА—ПЕРКИНА РЕАКЦИЯ
978
Развитие физики ядра показало, что периодич-
ность проявляется в ряде ядерных свойств и что в
ядре, как и в атоме, вероятно, существуют оболочки.
Однако сравнение периодичности в системе Менделе-
ева и периодичности в ядерных свойствах показыва-
ет, что они не совпадают друг с другом. Это обуслов-
лено тем, что, несмотря на наличие нек-рых общих
черт, электроны и частицы, составляющие ядро,— нук-
лоны (протоны и нейтроны) обладают различной мас-
сой, и между ними действуют силы различной приро-
ды — в случае электронов электрич. силы притяже-
ния и отталкивания, а в случае нуклонов, кроме сил
электрич. отталкивания (между протонами), играю-
щие главную роль, быстро убывающие с расстояни-
ем ядерные силы притяжения. Периодич. система
Менделеева является систематикой атомов химич.
элемептов на основе строения их электронных обо-
лочек, а систематика изотопов должна классифици-
ровать изотопы по ядерным свойствам, зависящим от
строения их атомных ядер (см. Изотопы, Радио-
активность, Ядро атомное, Ядерная химия).
Периодич. система элементов Менделеева, создан-
ная в то время, когда наука не только не располагала
сведениями о строении атомов, но исходила из пред-
ставления о их неделимости, тем не менее выдержала
самое серьезное испытание — испытание временем.
Все дальнейшее развитие физики и химии, особенно
в XX в., не изменило сколько-нибудь существенно
сделанное Менделеевым, но лишь развило и углубило
те основные принципы, к-рые были положены Мен-
делеевым в основу периодич. системы элементов. Мож-
но без преувеличения сказать, что вся современная
химич. наука имеет своим фундаментом периодич.
систему элементов, к-рая всегда служила и служит
основным критерием правильности возникающих но-
вых представлений в области строения вещества.
Однако значение периодич. системы не исчерпывает-
ся ее ролью в области физики и химии. Она оказала
огромное влияние на развитие таких наук, как гео-
химия, геология, ядерная физика, физико-химия
космоса и т. п. Периодич. закон химич. элементов
является одним из тех общих законов природы, к-рые
незыблемо остаются в науке, обогащаясь с ее разви-
тием и, в свою очередь, обогащая ее дальнейшее
разви тие.
Лит.: Менделеев Д., Соотношение свойств с атомным
весом элементов, ЖРХО, 1869, 1, вып. 2—3, 60—77; его
ж е, Новые материалы по истории открытия периодического
закона, М.—Л., 1950; Д. И. Менделеев. Научный архив, т. 1,
Периодический закон, М., 1953; его же, Основы химии,
т. 1—2, 13 изд., М.—Л., 1947; Рабинович Е., Т и л о 9.,
Периодическая система элементов, пер. с нем., М.—Л., 1933;
Некрасов Б. В., Курс общей химии, 14 изд., М., 1962;
Реми Г., Курс неорганической химии, пер. с нем., М.,1963;
Спицын В. И., Современное состояние периодического
закона Д. И. Менделеева, М., 1959; Кондратьев В. Н.,
Структура атомов и молекул,2 изд., М.,1959; СелиновИ. П.,
Усп. физ. наук, 1951, 44, вып. 4; С е м и ш и н В. И., Перио-
дический закон и периодическая система химических элемен-
тов Д. И. Менделеева в работах русских ученых, М., 1959
(библ. 898 работ). Н. П. Моаповенко.
ПЕРИОДИЧЕСКИЙ ЗАКОН МЕНДЕЛЕЕВА — см.
Периодическая система элементов Менделеева.
ПЕРИТЕКТИКА — см. Двойные системы.
ПЕРКАРБОНАТЫ — см. Пероксокарбонаты.
ПЕРКИНА РЕАКЦИЯ — метод синтеза коричных
к-т и их производных взаимодействием ароматич.
альдегидов с ангидридами карбоновых к-т в присут-
ствии щелочных катализаторов (солей, карбонатов
щелочных металлов, третичных аминов):
о
С»Н5-</ + R'-CH3-C-O-C-CH2-R' —>
\ n II
Н 0 0
—^сан5-сн=с-соон
I
R'
R' =Н, алкил, арил
Щелочной катализатор енолизирует ангидрид кисло -
ты, после чего анион енолята осуществляет нуклеоф иль-
ную атаку карбонильного атома углерода ароматич.
альдегида:
сн3-с-сг + сн3-с-о-с-сн, -
° II d II II 3
о о о
СН3—С—ОН + СН2=С-О-С-СН,
II I II °
О Q1-I о
н s'—''ч
ceH5-cz----сн2=с-о-с-сн3
II л 1л II °
СИ "О' о
__н+
--_ СвН5-СН-СН2-С-О-С-СН3 ==7
О" об
____ -н2о
-— свн5-сн-сн2-с-о-с-сн3—
он о о
н2о
—- с6н5-сн=сн-с-о-с-сн3—-
о б
свн5-сн=сн-соон + сн3соон
Промежуточное образование [5-оксикислоты доказа-
но реакцией бензальдегида с ангидридом изомасляной
к-ты:
о сн3
/, 100° |
С0Н6-С' + / изо = С.Н;-С \ О —^С6Н5-СН-С-СООН
\ II II
Н\ 0/2 о сн3
с=о
С3Н7
Выход 33% от теоретического. В том случае, когда
ангидрид карбоновой к-ты трудно доступен, П. р.
осуществляют с эквимолекулярным количеством соот-
ветствующей Na-соли в избытке уксусного ангидрида
(видоизменение Ольялоро). В условиях реакции соль
реагирует с уксусным ангидридом с образованием
смешанного либо полного ангидрида соответствующей
кислоты, к-рый затем реагирует с бензальдегидом;
CfiH5CH2COONa + (CH,CO)20 (C6HsCH2CO)2O + CH3COONa
О
С»Н5С^
н
(С6Н,СН2СО)2О-----—>сен5-сн = с-соон
CH.COONa |
С„Н3
Часто в условиях П. р. происходит декарбоксилиро-
вание образующейся a, f)-непредельной к-ты:
о
/ (С2Н,СО)2О; 200°
СН,О-С0Н,-С---------------->
\ C2H5COONa
н
-со2
—> [СН3ОСВН4СН = С-СООН]----->
сн,
—>СН3О-СаН1-СН = СН-СН3
Введение галогена или нитрогруппы в молекулу аро-
матич. альдегида, вне зависимости от положения, уве-
личивает выход в П. р.; алкильные заместители, как
правило, уменьшают выход. Производные бензаль-
дегида, содержащие оксигруппы в мета- и пара-поло-
жениях, образуют соответствующие оксикоричные
к-ты с удовлетворительными выходами. Салициловый
979
ПЕРКИНА РЕАКЦИЯ—ПЕРМАНГАНАТЫ
980
альдегид в условиях П. р. образует смесь кумарина и
ацетильного производвого о-кумаровой к-ты:
г
'н (СН3СО)2О
CH3COONa
ОН 3
GCH=CH-COOH
о-с-сна
II d
о
Бензальанилин реагирует с уксусным ангидридом с
образованием смешанного ангидрида [5-фсниламино-р-
фенилпропионовой к-ты:
(СН3С0),0
CH3COONa
c6h5-ch=n-c„h,
—>-CeH5NH-CH-CH2-C-O-C-CH3
I II II
с„н5 о о
Коричный альдегид, являющийся винилогом бензаль-
дегида, в условиях П. р. дает р-стирилакриловую
к-ту:
о
/ (СН3СО)2О; 165°
с»н3-сн=сн-с --------------->
\ CH3COONa
Н
—> С„НБ - СН = СН = СН = СН - СООН
Ароматич. диальдегиды— фталевый, изофталевый и
терефталевый — с уксусным ангидридом образуют со-
ответственно о-, м- и n-фениленбисакриловые к-ты.
В П. р. были вовлечены также нек-рые гетероцик-
лич. альдегиды:
|сн3соьо
CH3COONa
сн=сн-соон
Аналогично реагирует 2-тиофенальдегид. Алифатич.
альдегиды вступают в П. р. в редких случаях и соот-
ветствующие а,Р-ненасыщенные к-ты образуются с
малыми выходами. Несколько лучшие результаты
были получены с фенилацетатом натрия:
О
/ CsH,CH2COONa
СН3-С------------------► СН3-СН = С-СООН
\ (СН3СО)2О |
н с»н5
В качестве кислотного компонента в П. р. наряду с
уксусной и монозамещенными уксусными к-тами уда-
лось ввести кротоновый ангидрид:
О
/ (СН,-СН = СН-СО)2О; 125°
СД1С- с ----:---------------> свн3-сн=с-сн =сн2
\ B3N I
н соон
Взаимодействие янтарной к-ты с бензальдегидом при-
водит к у-фенилпараконовой к-те:
о
Нгс--СН2 Чн5с<н,100" с6нь-сн—сн-соон
О= С.. ^=0 сукцина! натоив О СН..
° V
6
В конденсацию с бензальдегидом легко вступает
также тиодиуксусная к-та:
CH2-COONa
/ с„н3сон /-с-соону
S ---------S п
\ (CH3CO)2O \ CHCtH5 /2
CH3COONa
О поведении ациламинокислот в П. р.— см. Эрлен-
мейера — Плохла синтезы. П. р. открыта Перкицым
в 1868.
Лит.: Джонсон Д ж., в кн.: Органические реакции,
об. 1, пер. с англ., М., 1948, с. 267; А и g е 1 H.S.,Day A.R.,
J. Amer. Chein. Soc., 1950, 72, № 9, 3874; Polya J. В.,
Spotswood T. M., Recuell trav. chini., 1951, 70, № 2,
146. JI. С. Герман.
ПЕРМАНГАНАТОМЕТРИЯ — титриметрич. ме-
тоды количественного анализа, основанные на при-
менении р-ров перманганата калия в качестве окисли-
теля. В кислых р-рах окислительный потенциал сис-
темы
МпО“+8Н + + 5е“ Мпз + + 4Н2О
„ 0,059 [Mn04][H+Js
сильно зависит от концентрации ионов водорода и
является высоким:
_ -f- -1,5 2 в
МпО + 8Н /Мп2 +
Обычно титрование перманганатом калия проводят в
кислой среде, т. к. в этих условиях в большинстве
случаев возможно прямое титрование до конечной
точки. Однако нек-рые вещества (сульфиты, сульфиды,
тиосульфаты, гидразин и др.) окисляются легче в
нейтральных или щелочных р-рах. Окисление КМпСЦ
в слабокислой, нейтральной или щелочной средах
может проходить по ур-нию:
МпО ~ + 4Н + + Зе~ ^МпОа + 2НгО
Прямое титрование в нейтральных и щелочных р-рах
осложняется образованием МпО2, к-рая должна осесть
на дно сосуда, чтобы можно было увидеть цвет раство-
ра над осадком. Основным стандартным (рабочим)
р-ром является 0,05 н. или 0,1 н. р-р КМпСЦ. Перман-
ганат калия нельзя использовать как исходное веще-
ство, т. е. титр его р-ров нельзя рассчитать по весу
растворенного КМпСЦ. Для приготовления титрован-
ного р-ра КМпСП в воде растворяют несколько боль-
шее количество перманганата, чем это требуется по
расчету, затем р-р кипятят, охлаждают и устанавли-
вают его титр. Титр р-ра КМпСЦ с течением времени
несколько изменяется (частичное разложение, особен-
но в присутствии Мп2 + , МпО2, света и др.),поэтому его
необходимо периодически проверять. Для установле-
ния титра перманганата применяют Н2С2О4-2Н2О,
Na2C2O4, As2Os, K.4[Fe(CN)6]-ЗН2О, металлич. желе-
зо и ряд других веществ. При титровании перманга-
натом не требуется индикатора, т. к. даже разб. р-ры
его интенсивно окрашены, и избыток КМпСЦ может
быть легко обнаружен визуально по появлению розо-
вого окрашивания. Методы П. широко используют для
определения восстановителей (Fe11, питритов, окса-
латов, арсенитов, фосфитов, сульфидов, сульфитов,
тиосульфатов, роданидов, селенитов, теллуритов,
иодидов, бромидов, перекисей в кислой среде, ряда
органич. веществ и др.). Перманганатометрически по-
сле восстановления солями Fe11 можно определять
многие окислители (персульфаты, бихроматы, хлора-
ты, нитраты, ванадаты, перманганаты и т. д.). Тит-
рованием КМпСЦ можно определять различные метал-
лы в их низших степенях валентности (V, Мо, W, U,
Ti, Sn, Сг, Nb, Re, Mn, Tl, Pt). Перманганатометри-
чески можно определять также ряд различных метал-
лов после их осаждения в виде оксалатов (Са, Mg, Zn,
La, Th). КМпСЦ может быть использован для опре-
деления многих органич. веществ.
Лит.: Кольтгоф И. М. [и др.], Объемный анализ,
пер. с англ., т. 3, М., 1961; Крешков А. П., Основы
аналитической химии, кн. 2, М., 1961. И. П. Ефимов.
ПЕРМАНГАНАТЫ (марганцовокислые соли) — со-
ли марганцовой к-ты НМпСЦ. П. окрашены, как пра-
вило, в красно-фиолетовый цвет иона Мп04“. В воде
П. натрия и 2-валецтпых металлов хорошо раствори-
мы, сравнительно мало растворим П. калия, трудно
растворимы соли Rb и Cs. По отношению к нагреванию
П. весьма неустойчивы; так КМпО4 распадается уже
выше 200° в основном по схеме: 2КМпО4=К2МпО4-(-
981
ПЕРМУТИТЫ —ПЕРОКСОБОРАТЫ
982
+М11О2+О2. П.— сильные окислители, причем харак-
тер их восстановления зависит от среды. В кислой
среде Мп’+ восстанавливается до Мп2 + (напр., до
MnSOi), в нейтральной и слабощелочной— до Мп4+
(МпОг), в сильнощелочной— до Мп5+ (К2МпО4).Т.
обр., наиболее сильно окислительные свойства П.
выражены в кислой среде. П. широко используют как
окислители в химич. практике; в медицине применяют
как дезинфицирующие средства, при ожогах и проч.
См. также Калия перманганат.
ПЕРМУТИТЫ — искусственно приготовленные
гидратированные алюмосиликаты, близкие по соста-
ву и свойствам цеолитам. Химический состав П.
определяется общей ф-лой КагО-А^Оз-иЗЮг-тНгО,
где и от 1 до 10 (в зависимости от условий приготовле-
нии П.), а т от 1 до 2. Так же, как и природные цео-
литы, П. обладают способностью к обмену ионов ме-
таллов в растворах (см. Иониты). Напр., при обработ-
ке натриевого П. р-ром соли кальция происходит обмен
ионов натрия на ионы кальция. При этом кальций
удаляется из р-ра, а натрий переходит в р-р. Реакция
обратима, обмен катионов происходит эквивалентно.
П. получают сплавлением каолина или полевого
шпата с кварцем и содой при 1000° или осаждением
алюмосиликата натрия жидким стеклом из р-ра сер-
нокислого алюминия. Наиболее часто в практике
применяется второй способ. По этому способу для
получения одной весовой части П. состава ХагО-
•Al2O3(SiO3)4-2SiO2-2H2O растворяют одну часть
сернокислого алюминия AlsfSO^s-lSEhO в 7—12 ча-
стях горячей воды и вливают этот раствор при непре-
рывном энергичном перемешивании в р-р 0,8 частей
растворимого стекла NasSiOs и 0,35 части кальциниро-
ванной соды Na2CO3 в 10 частях воды. Образующийся
гель отжимают на фильтрпрессе и высушивают в токе
горячего воздуха (100—105°) до влажности 10—15%.
Высушенный гель дробят до размера зерен 0,5—1,5 мм.
Обменная способность приготовленных осаждением
П. колеблется в пределах 1—1,5 мг-экв!г. П. применя-
ют гл. обр. для умягчения воды, к-рое достигается
фильтрованием ее через слой П. в открытом или на-
порном фильтре. В результате ионного обмена каль-
ций и магний поглощаются П., а в воду переходит
эквивалентное количество натрия. По истощении
обменной способности П. регенерируют; для этого его
обрабатывают 5—10%-ным раствором поваренной со-
ли. Умягченная вода дает значительную экономию
топлива (до 10%), устраняя необходимость специаль-
ных мероприятий по очистке котлов от накипи. П.
могут применять также для извлечения цветных и ред-
ких металлов из слабых р-ров их солей (напр., из
сбросных вод), для обезвоживания газов, обработки
нек-рых пищевых продуктов, для удаления из воды
железа и марганца, а также в качестве носителя при
изготовлении катализаторов (обработкой П. р-рами
солей никеля или кобальта получают никелевые или
кобальтовые катализаторы с весьма развитой поверх-
ностью) .
Лит.: Астафьев В. П., Технология пермутитового
водоумягчения, М.—Л,— Новосибирск, 1933; Гельфе-
р и х Ф., Иониты. Основы ионного обмена, пер. с нем., М.,
1962; Kunin R., Myers R. I., Ion exchange resins, 2 ed.,
N. Y., 1958.
ПЕРОКСИДАЗЫ — ферменты, катализирующие
окислительно-восстановительные реакции, в к-рых
участвует перекись водорода: АН2+Н2О2 JI А^ЗНгО.
В обычных условиях равновесие этой реакции сильно
сдвинуто вправо. Субстратами окисления (АНг)
для П. могут служить биогенные полифенолы (пиро-
катехин, пирогаллол, гваякол) и амины (бензидин,
адреналин, тирозин), а также нек-рые др. соединения.
По современной классификации ферментов (см.
Номенклатура и классификация ферментов) П. сов-
местно с каталазами входят в класс оксидоредуктаз.
П. содержатся гл. обр. в растительных тканях, в
частности они найдены в корнях хрена и соке фиго-
вых деревьев. В животных тканях и у микроорганиз-
мов П. встречаются реже и активность их значительно
ниже.
Растительная П., в частности П. хрена (систематич.
наименование: донор: Н2Ог— оксидоредуктаза, три-
виальное— пероксидаза, шифр 1.11.1.7), является
активным ферментом, отличающимся широкой специ-
фичностью по отношению к окисляемым субстратам
(фенолы, амины, аминофенолы и др.). Фермент пред-
ставляет собой сложный белок — гемопротеид с мол. в.
44100. Простетич. группой фермента является железо-
протопорфирин IX (см. Гемоглобины). Молекула
фермента содержит один атом железа, находящийся в
обычных условиях в трехвалентном состоянии. Про-
стетич. группа может быть обратимо отщеплена от бел-
ковой части фермента путем мягкой обработки П.
холодным ацетоном, подкисленным соляной к-той.
В спектре поглощения П. имеются характерные мак-
симумы при 645, 583, 548 и 498 ммк и ярко выражен-
ная полоса при 410—420 ммк (полоса Соре). Фермен-
тативную активность П. измеряют по скорости окис-
ления пирогаллола в пурпурогаллин, количество
к-рого определяют фотометрически.
В процессе катализируемой реакции П. образует
с Н2О2 промежуточный комплекс, окрашенный в
ярко-зеленый цвет, к-рый быстро переходит в светло-
красный комплекс, расщепляющийся затем с освобож-
дением свободной П. и продуктов реакции. В при-
сутствии избытка Н2О2 светло-красный комплекс пере-
ходит в темно-красный комплекс. П. термостабильна.
Циалид и сульфид в концентрации 10—10~6 М, а
также азид Na в концентрации 10-s М подавляют ак-
тивность П.; гидросульфит натрия, гидроксиламин и
окись азота также оказывают тормозящее действие
на активность П. Окись углерода не оказывает па П.
ингибирующего действия.
К П. животного происхождения относится лак-
топероксидаза, выделенная в кристаллич.
виде из молока и имеющая мол. в. 92 000, и миело-
пероксидаза, содержащаяся в лейкоцитах.
Из нек-рых видов бактерий и дрожжей выделена цито-
хром с:НгО2 — оксидоредуктаза (цитохром с-перок-
сидаза, шифр 1.11.1.4), катализирующая окисление
восстановленного цитохрома с перекисью водорода:
2-феррицитохром с + Н2О2 = 2-ферроцитохром с+2Н2О
Из бактерий выделена также НАД-Н2:Н2О2—
оксидоредуктаза (НАД-пероксидаза, шифр 1.И.1.1.),
катализирующая окисление восстановленного нико-
тинамид-аденин-динуклеотида (НАД-Нг) (см. Нико-
тинамиднуклеотидные коферменты):
НАД Н2 + Н2О2 J—Г НАД + 2Н2О
В отличие от других П., этот фермент является не
гемопротеидом, а флавопротеидом, т. е. сложным бел-
ком с флавин-аденин-динуклеотидом в качестве про-
стетич. группы.
Лит.: Нейла ндс Дж., Штумпф П., Очерки по
химии ферментов, пер. с англ., М., 1958, с. 279; Диксон М.,
Уэбб Э., Фермэнты, пер. с англ., М., 1961; Классификация
и номенклатура ферментов, пер. с англ., М., 1962.
В. Б. Спиричев.
ПЕРОКСОБОРАТЫ — соли не выделенной в сво-
бодном состоянии пероксометаборной кислоты НВОз,
напр. NH4BO3 и КВОз. В безводном состоянии
П. можно получить лишь в редких случаях, т. к. бла-
годаря гидролизу они весьма легко превращаются в
гидраты пероксигидратов метабората МеВОг-гНгОг-
• г/Н2О. К последним относятся технич. препараты,
известные под названием «пербората» натрия. Из-
вестны три соединения метабората натрия ИаВОг
с перекисью водорода и водой: ИаВОг-НгОг-ЗНгО;
983
ПЕРОКСОКАРБОНАТЫ —ПЕРОКСОФОСФАТЫ
984
NaBO2-H2O2-2H2O и NaBO2-H2O2. Это — белые
кристаллы, устойчивые при хранении в герметически
закрытой таре и растворяющиеся в воде с отщеплени-
ем кристаллизационной перекиси водорода. Содержа-
ние активного кислорода в этих соединениях соответ-
ственно составляет 10,4; 11,7 и 16 вес%. Т. пл. три-
гидрата 65,5±0,5°; плотн. 1,731; т. пл. дигидрата
81,7±0,2°; плотн. 1,860. Оба соединения кристалли-
зуются в триклинной системе.
Промышленный способ получения NaBO2-H2O2-
ЗН2О состоит из след, операций: приготовление р-ра
NaBO2 с pH 9,4—9,9 смешением р-ров буры и NaOH;
обработка р-ра метабората перекисью водорода при
30—60°; быстрое охлаждение полученного р-ра до
15—10° после внесения затравки «пербората», выса-
ливающего агента (NaCl) и стабилизатора (MgSiOs);
отделение кристаллов фильтрованием;сушка при ком-
натной темп-ре. Тригидрат NaBO2-H2O2-3H2O можно
получить также электролизом р-ра буры и карбоната
натрия, применяя платиновые аноды. Дигидрат
NaBO2-H2O2-2H2O получают, обрабатывая опреде-
ленное количество тригидрата одинаковым по весу
количеством воды при 50° с последующей сушкой
полученной массы в вакууме при 40°. Сушка тригид-
рата до NaBO2-H2O2 проводится в псевдоожиженном
слое в токе горячего воздуха (60—100°).
Пероксигидраты метабората натрия широко приме-
няются в народном хозяйстве. Преимущество в ис-
пользовании этих соединений и им подобных по срав-
нению с водорода перекисью состоит в том, что они
содержат Н2О2 в твердом виде, легче транспортируют-
ся и хранятся и могут быть смешаны, без заметного
разложения, с другими порошкообразными вещест-
вами. В основном их применяют в технике отбелива-
ния шерсти, льна, хлопка, искусственного волокна, в
качестве окислителя при крашении кубовыми краси-
телями и в печатании самых разнообразных видов
текстиля и полиамидных волокон. В смеси с мыльным
порошком или с поверхностно-активными веществами
пероксигидраты метабората натрия входят в состав
стиральных порошков и синтетич. моющих средств.
Находят применение также в косметич. и фармацевтич.
пром-сти и в органич. синтезе.
Лит. см. при ст. Перекисные соединения. И. И. Вольное.
ПЕРОКСОКАРБОНАТЫ — соли не выделенных в
свободном состоянии пероксомонокарбоновой Н2СО4
и пероксодикарбоновой Н2С2О6 кислот. Пероксомо-
нокарбоновая к-та способна образовывать довольно
устойчивые средние и кислые соли, из к-рых наиболее
изучены пероксокарбоновые соли щелочных метал-
лов. Кислые соли пероксомонокарбоновой к-ты
NaHCCU, КНСО4, RbHCOi образуются при взаимодей-
ствии спиртовых р-ров соответствующих гидропере-
кисей или щелочных р-ров Н2О2 с углекислым газом
по ур-нию: MeOOH4-CO2=MeHCOi, а также при
взаимодействии Н2О2 с этилкарбонатами при —10°
по ур-нию: МеС2Н5СОз+ Н2О2-^ MeHCOi-|- С2Н5ОН.
Средние соли получают карбонизацией гидратов пере-
кисей при 0°— 20° по ур-нию Ме2О2-хН2О-|-СО2=
= Ме2СО1-|-л:Н2О. Пероксодикарбоновая к-та обра-
зует лишь средние соли, напр. К2С2О6. Они получены
электролизом конц. р-ров карбонатов при—15°, а
также взаимодействием СО2 с гидратами перекисей.
Технич. препараты, известные под названием «пер-
карбонаты», являются продуктами присоединения пе-
рекиси водорода и воды к карбонатам, а не производ-
ными пероксокарбоновых к-т, и отвечают составу:
Ме2СОз хН2О2-уН2О. Среди них наибольшее значение
имеет соединение Na2CO3-l,5H2O2-H2O («персоль»),
выпускаемое в промышленном масштабе. Персоль
получают смешиванием р-ров карбоната натрия
и Н2О2, при pH —11, в присутствии стабилизатора, с
последующей сушкой кристаллич. осадка при темп-ре
ок. 100° (белый порошок с содержанием 14 вес.%
активного кислорода). Области применения «перкар-
боната» натрия в основном те же, что и «пербората»
(см. Пер оке обора ты); используется также для отбели-
вания муки, в хлебопечении и для обеззараживания
воды.
Лит. см. при ст. Перекисные соединения. И. И. Вольнов.
ПЕРОКСОСЕРНЫЕ КИСЛОТЫ — кислородные
к-ты серы, характеризующиеся наличием пероксо-
группы — О—О—. Известны три пероксокислоты се-
ры: пероксомоносерная H2SOs, пероксодисерная
H2S2O8 и пероксотрисерная H2SsOn. Наиболее
изучены и широко применяются пероксомоиосерная
(кислота Каро) и пероксодисерная (надсерная) к-ты
и их соли (см. Пероксосулъфаты). Ангидриды пероксо-
моно-, пероксоди- и пероксотрисерной к-т не являют-
ся, как ранее считалось, мономерами, соответствую-
щими ф-лам SO4, S2O7, SsO10, а являются полимерами,
отвечающими ф-лам (ЗОз^з)*— (SO39)X, и образуются
в тихом разряде из смесей SO2 и’О2 или SO3 и 02
с последующей конденсацией продуктов, выходящих
из зоны разряда в ловушке, охлажденной жидким
азотом. Пероксотетрасерная к-та H2S«O14 и ее ангид-
рид не выделены, но известны соответствующие соли.
Пероксомоносерная и пероксодисерная к-ты — бес-
цветные кристаллы, плавящиеся соответственно при
45°и 65°.Их водные р-ры получают электролизом р-ров
H2SO4. Поскольку H2SOs менее устойчива, нежели
H2S2O8, то образование ее при электролизе должно
быть, по возможности, подавлено для получения хо-
рошего выхода H2S2O8. Водные р-ры H2SOs выгодно
получать гидролизом H2S2O8 или пероксодисерных
солей, а также смешением р-ров Н2О2 и H2SO4 на хо-
лоду. Пероксодисерную к-ту получают электролизом
р-ра H2SO4 (обычно деся.тинормального); темп-ра
анолита не должна превышать 20—30°. Анод плати-
новый; плотность тока на аноде 0,86 а/см2. Пероксо-
моносерная к-та является сильным окислителем, при-
меняемым в органич. синтезе. Пероксодисерную к-ту
и ее соли также применяют как окислитель, они слу-
жат исходными продуктами при промышленном по-
лучении р-ров Н2О2 (см. Водорода перекись).
Лит. см. при ст. Перекисные соединения. И. И. Вольнов.
ПЕРОКСОСУЛЬФАТЫ — соли пероксосериых к-т.
Кислые соли пероксомоносерной к-ты MeHSOs (Me—
щелочной металл) получают нейтрализацией р-ров
H2SO3 р-рами соответствующих карбонатов или взаи-
модействием пиросульфатов с конц. Н2О2. Они обла-
дают хорошими отбеливающими свойствами. Средние
соли пероксомоносерной к-ты Me2SO5 образуются при
реакции перекисей щелочных металлов с SO3; практич.
применения они не нашли.
Пероксодисерная к-та образует лишь средние соли
Me2S2O8. Из них наиболее широкое применение нашли
(NH4)2S2O8 (см. Аммония персульфат) и K2S2Os. По-
следний получают прибавлением KHSO4 к раствору
(i\H4)2S2O8. Растворимость в воде при 15°: 44,2 вес. %
(NH4)2S2O8,3,8 вес.% K2S20g.Распад K2S2O8->-K2S2O74-
+’/2 Ог происходит экзотермически при 200°.K2S208 ис-
пользуется для получения р-ров Н2О2 (см. Водорода
перекись), а также в качестве окислителя, инициатора
реакций полимеризации, для отбеливания жиров и
мыла, в составе брикетов для инициирования термич.
разложения взрывчатых веществ и в качестве состав-
ной части нек-рых инсектофунгицидов. Сульфаты ще-
лочных металлов способны к присоединению Н2О2
и Н2О с образованием перокепгидратов сульфатов об-
щей ф-лы Me2SO4-гНгОг-у Н2О.
Лит. см. при ст. Перекисные соединения. И. И. Вольнов.
ПЕРОКСОФОСФАТЫ — соли пероксомонофосфор-
ной Н3РО5 и пероксодифосфорной Н4Р2О8 кислот.
НзРО5 и ее соли в чистом виде не получены. Пероксо-
дифосфорная к-та не выделена в свободном состоя-
985
ПЕРСИКОВОЕ МАСЛО—ПЕРФТОРБЕНЗОЛ
986
нии, но выделены ее калиевые, рубидиевые, цезиевые
и аммониевые соли, получаемые анодным окислением
водных р-ров фосфатов, при пониженной анодной плот-
ности и низкой темп-ре. Пероксофосфат калия КлР2О8
— бесцветные кристаллы с плотностью 2,388, устой-
чив до 340°. Растворимость в воде при 25° 45 вес. %.
Нормальные и двузамещенные фосфаты натрия и
калия, а также пирофосфат натрия, реагируя с Н2О2,
образуют продукты молекулярного присоединения.
Среди них практич. применение, в качестве мягкого
окислителя и в фармацевтике, имеет соль NajP2O7 •
• тН2О2, где х=1—3, известная под названием «пер-
фосфат»; эта соль не является производной пероксо-
дифосфорной к-ты. Получается растворением на холо-
ду одного моля Na4P2O7 в трех молях конц. Н2О2, с
последующей вакуум-выпаркой образующейся сиро-
пообразной жидкости.
Лит. см. при ст. Перекисные соединения. И.И.Вольнов.
ПЕРСИКОВОЕ МАСЛО — см. Жиры раститель-
ные.
ПЕРСУЛЬФАТЫ — см. ПерЬксосулъфаты и Аммо-
ния персульфат.
ПЕРФОСФАТЫ — см. Пероксофосфаты.
ПЕРФТОРАЛКАНЫ — см. Фторалифатические
с @ед инен ия
ПЕРФТОР АЛКИЛМАГНПЙГАЛОГЕНИД — см.
Фторалифатические соединения.
ПЕРФТОРАЦЕТИЛЕНЫ — см. Фторалифатиче-
ские соединения.
ПЕРФТОРАЦЕТОН (гексафторацетоп, перфторди-
метилкетон) CF3—СО—CF3 — бесцветный газ с рез-
ким запахом, т. кип. —28°, т. замерз. —129°. П. уме-
ренно растворяется в обычных нейтральных органич.
растворителях; с водой, спиртами и аминами образует
устойчивые аддукты (гидраты, полуацетали и соли
полуаминалей). Гидрат П.— гигроскопичные белые
кристаллы с т. пл. 38—40°, неограниченно растворим
в воде; при действии конц. H2SOi регенерирует П.
В связи с наличием двух электронооттягивающих три-
фторметильных групп П. гораздо активнее в реакциях
с нуклеофильными агентами, чем нефторированные
кетоны, и даже вступает в реакции, не свойственные
алифатич. кетонам:
(C.H5)3P=CHCCCH
(CFj)2C=CHCOCH3-«——----------:
(CF3)2C-NR
(C6H5)3P=NR \ / (CH.3)2C = CH
--------------- C F3-C О - C F,-----------
, , NyCHCOOCjHc
(CF3)jC-CHCOOC2H5—------—
V
X
ch/y
(CH3)2NCH2C6H,
В реакциях с электро-
фильными реагентами П., сн
напротив, менее активен, гнх+/ 3
чем ацетон. Так, например, (CF3)2CHON—СН2С6Н5
ацетон реагирует с фено- СН2
лом в безводном HF уже
при комнатной темп-ре, а П. — лишь при 100°:
CF3COCF3 + С6Н5ОН
CF3
Наиболее просто и с хорошим выходом П. полу-
чается окислением перфторизобутилена водным р-ром
перманганата калия в мягких условиях или кисло-
родом воздуха при высокой темп-ре. П. получают так-
же взаимодействием галогенангидрида или нитрила
трифторуксусной к-ты с CF3MgJ и фторированием
ацетона элементарным фтором:
CF3 CF3 О
\ О CF3MgJ /
C = CF2—>- С = О-<----CF3C (ИЛИ CF3CN)
/ / \
CF3 CF3 X
Для колич. определения П. титруют щелочью вод-
ный р-р гидрата П., а для идентификации получают
кристаллич. аддукт с семикарбазидом (т. пл. 161°,
с разл.) или дианилиновую соль гидрата П. (т. пл.
64—65°). Такие производные, как имин или оксим П.,
Удается получить лишь обходными путями:
NH,
(CF3)2C = NC„H3
А
nh2oh
(CF,)2C = NH
А
н
(CF3)2C = NOH-<--(CF3)3CFNO.
Интересным производным П. является тиоперфтор-
ацетон; в этом соединении, в отличие от П., наблюдает»
ся «обратная» поляризация двойной связи, так что
атом серы оказывается более электрофильным, чем
атом углерода:
II
Н3С CHj
-w
Н3Сч ZCH3
с
сч< Усн>
Сч сн2
cf/ чн
П. используется для синтеза самых разнообразных
фторорганич. соединений, в том числе и мономеров.
Лит.: Кнунянц И. Л. [и д р.], Изв. АН СССР. Отд.
хим. н., 1963, № 6, 1123; MlddletonW. J.[a. о.], J. Amer.
Chem. Soc., 1961, 83, Ks 11, 2589. II. П. Гамбарян.
ПЕРФТОРБЕНЗОЛ (гексафторбензол) C6F6, мол.
в. 188,06 — бзсцветная жидкость; т. пл. 5,29°; т. кип.
OH X
(cf3)2c-ch/
Y
(cf3)2c-ch2-c-ch2
OH CH3
(CFj)jC-OH
N3
80—81°; Пд 1,3777. Нераст-
ворим в воде, растворим в ор-
ганич. растворителях. П.при-
соединяет С1 при УФ-облуче-
нии с образованием C6F3Cle>
гидрируется над Pd/C (или
Pt/C) при 300°, давая смесь
фторбензолов с преоблада-
нием C3F5H (40%). Взаимо-
действие П. с нуклеофиль-
ными реагентами приводит к
замещению атомов F. Так,
обработка П. гидроокисями или алкоголятами щелоч-
ных металлов в пиридине или спирте дает пентафтор-
фенол или его эфиры:
сл+кон —>-c0F5oh
C6F0 + CH3OH —> C,.F;OCH3
При действии на П. NaNH2 в жидком NH3 при —70°
или водноспиртового NH3 в автоклаве при 160—170°
образуется пентафторанилин:
C.F«+nh3—>c»fsnh2
Аналогичные реакции происходят при действии ал-
киламинов, N2Hi, KSH. Ряд интересных производных
П. получен из пентафторбензола:
Вг, Mg
CbF5H----->C„HsBr >C1,F,MgBr
|so3 leu ICO,
C3FjSO3H C3F5-C3F5 CoFsCOOH
987
ПЕРФТОРБУТАДИЕН-1,3—ПЕРФТОРПРОПИЛЕН
988
Синтез П. осуществлен
несколькими методами:
630 - 640"
С F Вг, ----
*Pt—трубка
I
„г- ✓F-C
CsC'5 450-500'
F
I
>С
C-F
И
.C-F
Fe C°F3
------C5FI2 -------CtH,
l-cnn® 5 12---5 !
CoF3
CjHjFq C.H,
С
I
F
| Zn
C5BrCl4F,
BrF3 lSbF3
CgClg C5Br2CI4F6
ПЕРФТОРБУТАДИЕН-1,3 (гексафторбутадиен-1,3,
перфтордивинил) CF2=CF—CF=CF2, мол. в. 162,04—
газ без цвета и запаха; т. пл. —61°; т. кип. 0° (по
другим данным: 4-3°; 4-6,6°; 4-7,4------1-7,6°); d4 20
1,602; nD20 1,298. П. получают дехлорированием
1,2,3, 4-тетрахлорперфторбутана:
560°
CF2 = CFC1---->-CF2Cl-CF = CF-CF2Cl CFC1 = CFC1
Pcl Zn I |F’
cf2cicfcu--------->CF2C1CFC1CFC1CF2C1^—I
(CH3CO)2O I Zn/C2H-,OH
cf2=cf-cf=cf2
П. может быть получен действием Zn на трифториод-
этилен:
Zn
2CF2 = CFJ—>cf2=cf-cf=cf2
или пиролизом Na-соли перфторадипиновой к-ты:
NaOOC—CF2CF2CF2CF2-COONa —>
—>. CF2=CF - CF = CF3 + 2NaF + 2CO2
Как диен с сопряженными двойными связями, П. спо-
собен присоединять галогены и N2O4 в положение 1—4:
CF2 = CF — CF = CF2 + X2 —> CF2X - CF = CF - CF2X
X = C1, Br, NO,
К реакции присоединения в положение 1,4 относится
и внутримолекулярная термич. циклизация П. в
перфторцикл обутен:
CF = CF, CF-CF,
I I
CF = CF, CF-CF,
Эти превращения осуществляются по гомолитиче-
скому механизму. П. легко вступает в типичные
для фторолефинов реакции с нуклеофильными ре-
агентами — алкоголятами и аминами; при этом ос-
новным процессом является замещение атомов F, на-
пример:
CFa=CF-CF = CFj + HN(C2H;)2 —►CF2=CF-CF = CF-N(C2H5)j
Реакции с электрофильными реагентами для П. неиз-
вестны. П. не вступает в характерные для диеновых
углеводородов реакции диенового синтеза; в то же вре-
мя он сам является диенофилом:
/н2
СН
I
сн
\нг
CF-CF = CF,------
II
сн2
СН CF-CF=CF,
II I
СН CFS
чсн2
При термич. полимеризации П. образуются димер,
тример и низкомолекулярный полимер; механизм ее
сводится к циклодимеризации с образованием перфтор-
циклобутановых звеньев. При дальнейшем нагрева-
нии димера получается соединение, представляющее
собой, по-видимому, конденсированную трициклич.
структуру:
cf2=cf-cf=cf2 cf2-cf-cf=cf2
CF2 = CF — CF = CF2 * OF, — ir - CF = CF, ”
CF, —CF -CF-CF,
—► Illi
CF2-CF-CF-CF,
П. удалось заполимеризовать в присутствии перекиси
бензоила. В различных условиях могут быть получены
как низкомолекулярные, так и высокомолекулярные
полимеры П. Последние представляют собой резино-
образные материалы, приобретающие пластич. свой-
ства при 250—275° и деполимеризующиеся при 300°.
Интересные эластомеры получены на основе 1,1-
дигидр оперфторбута диена-1,3 С Н 2=С F— С F=С F2.
Лит.: Л о в л е й с А.М.,Роуч Д., П о с т е л ьи е к У.,
Алифатические фторсодержащие соединения, пер. с англ.,
М., 1961; Гудлицкий М., Химия органических соеди-
нений фтора, пер. с чеш., М., 1961; Кнунянц И. Л. [и
др.], ДАН СССР, 1959,124, № 5, 1065. Б. Л. Дяткин.
ПЕРФТОРИЗОБУТИЛЕН (ф-изобутилен, октафтор-
изобутилен) (CF3)2C=CF2, мол. в. 200,04 — бесцвет-
ный газ с фосгеноподобным запахом; т. кип. 4“7°; <7°
1,5922; растворим в эфире, бензоле, плохо растворим
в воде. Благодаря ярко выраженной полярности, яв-
ляющейся следствием сильного электро-
ностягивающего действия двух CF3-
групп, молекула П. проявляет сильные
электрофильные свойства. П.— самый
реакционноспособный по отношению к
нуклеофильным реагентам фторолефин;
он очень легко вступает в реакции со
спиртами, аминами и др. Так, в отличие
F4C
8 +
CF,
F-'
от перфторпропилена, П. реагирует с этиловым спир-
том па холоду в нейтральной и слабокислой среде;
медленно реагирует даже с таким слабым нуклеофиль-
ным соединением, как Н2О и др.:
н,о
(CF3),C = CF,--->[(CF,)2CH-CF,OH]---->
-HF
О ’
(CF3),CH-c/
\
F J
О
H,0 /
-----> (CF3), СН-С
-HF \
ОН
Окисление П. приводит к перфторацетону.
КМпО
(CF..),C=CF,------ CF..COCF..
35-40°
восстановление — к 1,2-дигидрооктафторизобутапу.
П. может быть получен пиролизом нек-рых фторугле-
родов— перфторциклобутана (700—725°), политет-
рафторэтилена (750°) и др. способами. П. токсичен,
по характеру действия сходен с фосгеном.
ПЕРФТОРКАРБОНОВЫЕ КИСЛОТЫ с^Фтор-
алифатические соединения.
ПЕРФТОРОЛЕФИНЫ — см. Фторалифатические
ПЕРФТОРПРОПИЛЕН (ф-пропилен, перфторпро-
пен, гексафторпропилен) CF3—CF=CF2, мол. в.
150,03 — бесцветный газ без запаха; т. кип. —29,4°,
т. пл. —156,2°; плохо растворим в эфире, нерас-
творим в воде. Благодаря сильному электроностяги-
вающему действию СБз-группы и эффекту сопряже-
ния П. является одним из реакционноспособных, по
отношению к нуклеофильным реагентам, фтороле-
финов.
989
ПЕРХЛОРАТЫ—ПЕРХЛОРЦИКЛОПЕНТАДИЕН
990
При взаимодействии со спиртами при 20—30° наря-
ду с насыщенными эфирами (I) образуются непредель-
ные соединения II и III:
cf3-cfh-cf2or cf3-cf=cf-or
+ HF
I II
CF2=CF-CF2OR
+ HF
III
Тенденция к образованию ненасыщенных соединений
растет с увеличением подвижности электронной пары
атакующего агента; так, в реакции П. с аминами
CF3 — CF = CF2 + HNR2 —>.
--> С F3 - CF H - CF2NR2 + CF3 - CF = CF - NR,
ненасыщенного продукта образуется больше, чем в
случае взаимодействия П. со спиртами. Также легко
П. реагирует с меркаптанами, гидразином, магнийор-
ганич. соединениями, гидроксиламином, азотисто-
водородной к-той, фосфитами и др. нуклеофильными
соединениями.
Гетеролитич. реакции П. с электрофильными реа-
гентами затруднены и часто носят радикальный ха-
рактер:
/--------------*CF3-CFBr-CF2Br
' FNO
CF3-CF = CF2 -----------> CF3 —CF(NO) —CF3
\--------------»- CF3-CF(NO2)-CF2NO2 +
+CF3-CF(ONO)-CF2NOj и др.
П. обычно получают пиролизом (600—700°) фторуг-
леродов— дифторхлорметана (фреона-22), тетрафтор-
этилена, перфторциклобутана и политетрафторэти-
лена. П. может быть получен также термич. разложе-
нием К- или Na-соли перфтормасляной к-ты:
290 — 300°
CF3-CF2-CF2-COOK-------> cf3-cf = cf2 + co2 + kf
Сополимеры П. с фторолефинами нашли примене-
ние в производстве некоторых фтрркаучуков («ви-
тон-А>> — сополимер с винилиденфторидом) и фторо-
пластов (сополимер с тетрафторэтиленом — «теф-
лоп-ЮОх»),
Лит. см. прп ст. Фторолефины. В. Н. Фросин.
ПЕРХЛОРАТЫ (хлорнокислые соли) — соли хлор-
ной к-ты HCIO4. Подобно самой кислоте, большинство
П. бесцветно. При нагревании П. устойчивы примерно
до 300—600°, выше этой темп-ры начинается разложе-
ние с выделением кислорода. Большинство П. хорошо
растворяется в воде и в органич. растворителях (ме-
тиловом спирте, ацетоне и др.); труднорастворим из
обычных П. KCIO4. Получают П. электролизом р-ров
хлоратов: КС1Оз-|-Н2О=Н2(катод) КСЮ4 (анод).
Они образуются также при растворении металлов
в хлорной к-те. П. калия получают, кроме того, при
осторожном нагревании чистого хлората калия:
4КС1Оз=ЗКС1О4+КС1. Применяют П. для изготов-
ления взрывчатых веществ и пиротехнич. составов.
См. также Калия перхлорат.
ПЕРХЛОРВУТАДИЕН (гексахлорбутадиен, пер-
хлордивинил, гексахлордивинил) cci2=cci—cci=cci2
мол. вес 260,79 — прозрачная бесцветная жидкость
с запахом скипидара; т. пл.—18,6°; т. кип. 210—
2127760 мм, 144,17100 лм»; d*’l,6794;n^° 1,5557; раст-
воряется в спирте, диоксане, практически нераство-
рим в воде. Вязкость П.: 2,446 спуаз (37,8°) и 1,31
спуаз (98,9°); поверхностное натяжение 36 дин/см
(20°); диэлектрич. проницаемость 2,55 (20°); т. воспл.
537°, воспламеняется при разбрызгивании в струе
кислорода, не имеет темп-ры вспышки. Максимальное
поглощение в УФ-области при Х=253 ммк, в ИК-
области при Х=792, 847 и 978 см~*.
При восстановлении П. цинком в кипящем этаноле
образуется бутадиен; при восстановлении цинком и
окисью дейтерия в диоксане — целиком дейтерирован-
ный дивинил. При хлорировании на свету при ком-
натной темп-ре или на диатомите при 450° образуется
гексахлорэтан. При взаимодействии П. и жидкого
хлора под давлением получается октахлорбутен и де-
кахлорбутан. П. устойчив к сильным минеральным
к-там, а также к водным р-рам щелочей и сульфгидра-
тов щелочных металлов при кипячении; в спиртовом
р-ре П. заметно реагирует со щелочью. Количествен-
ное определение П. (в отсутствие др. хлорсодержащих
соединений) может быть произведено известными ме-
тодами, напр. разложением металлич. натрием с по-
следующим определением хлор-иона.
П. получают в основном хлорированием и-бутана,
бутиленов с прямой цепью и дивинила. При этом обра-
зуется октахлорбутан, дегидрохлорированием к-рого
получают П.:
С.Ищ 1 +С]2 -2НС1
С.Н, V------>С,Н2С13---->п.
С.н» )
П. используют для уничтожения виноградной фи-
локсеры, для борьбы с к-рой П. оказался наиболее
эффективным из всех известных соединений. Водный
р-р П. (1—2 мг/л) подавляет или сильно угнетает раз-
витие зеленых, сине-зеленых и диатомовых водорос-
лей. П.— гербицид по отношению к узколистным сор-
някам (траве), развивающимся среди широколистных
огородных культур. П. может быть применен также в
качестве гидравлической, электроизоляционной и
смазочной жидкости. Известно применение П, в каче-
стве селективного растворителя для очистки НС1
от хлора, а также в качестве среды для проведения
реакций, особенно хлорирования.
Лит.: Коган Л. М., Усп. химии, 1964, 33, вып. 4.
Л. М. Коган.
ПЕРХЛОРЦИКЛОПЕНТ А ДИЕН (генсахлорцикло-
пентадиен) С5С16, мол. в. 273,00 — прозрачная жид-
кость, соломенно-желтого цвета с едким
неприятным запахом; т. пл. 12° и 0,8— С1-п-----ц-С1
0,2° (нестабильная форма); т. кип. 236— ci-L J-ci
2387760 мм, 164—165°/100л»л», 108°/10л1Л1, fi'ci
78-7971 мм; d201,7101; п д 1,5652; ра-
створяется в спирте, практически нерастворим в воде,
скрытая теплота плавления 10,0 кал/г; поверхностное
натяжение 37,5 дин/см (20°); поглощение в УФ-облас-
ти при Л 322 ммк, в ИК-области при Л 1603, 1572, 803,
704 и 678 см~'; спектр комбинационного рассеяния
показывает две линии в области 1606 и 1572 см~'.
Наличие системы сопряженных двойных связей и
двух «аллильных» подвижных атомов хлора в молеку-
ле П. определяют многообразие реакций этого соеди-
нения. Наибольшее значение имеет реакция диенового
синтеза (см. Хлорциклодиены). Следует также отме-
тить реакции восстановления П., присоединения по
двойной связи, окисления, замещения двух подвижных
991
ПЕСТИЦИДНЫЕ ПРЕПАРАТЫ
992
атомов хлора и конденсации с галогенопроиаводными
в присутствии А1С1.3. Молекулярный кислород при
взаимодействии с П. (90— 200°) при освещении
образует смесь двух изомеров гексахлорциклопенте-
она:
Качественной пробой на II. может служить образо-
вание осадка со спиртовым р-ром AgNOg; при кипяче-
нии в течение 30 минут с 1 н. спиртовым р-ром КОН
от молекулы П. количественно отщепляются два
атома хлора.
Основным способом получения П. является глубо-
кое хлорирование углеводородов с открытой цепью,
содержащих 5 атомов углерода, кроме неопентана, до
образования декахлорпентанов. Далее реакция про-
водится по схеме:
С5Н2С110---->СС1,-СС1^СС1-СС1=СС|2 —>.
-НС1
С12С---СС1
—> I II----------*п.
С12С ХСС1 -С12
СС12
Кроме того, П. можно получить из циклич. углеводо-
родов с 5 атомами углерода в молекуле (циклопен-
тана, циклопентепа, циклопентадиена), напр. хлори-
рованием циклопентадиена при 50—60° гипохлоритами
щелочных металлов, а также их полихлоралканов и
полихлоралкенов.
П. применяют для синтеза биологически активных
веществ, используемых в качестве хлорциклодиеновых
инсектицидов, особенно эффективных для борьбы с
почвенными вредителями, а также в качестве фунги-
цидов, гербицидов, регуляторов роста растений и
дефолиантов. Кроме того, П. применяют для получе-
ния жаропрочных и термостойких полимеров, в част-
ности синтеза полиэфирных и эпоксидных смол, элас-
томеров (фторсодержащих и силоксанового типа),
пластификаторов и присадок к маслам.
П. токсичен для теплокровных, ЛД5О=ЗОО мг/кг
(для мышей); его главная опасность состоит в кумуля-
тивном действии (накапливается в организме); хоро-
шо всасывается кожным покровом.
Лит.: Roberts С. W., Chem. Ind., 1958, № 5, 110;
Ungnade Н. Е., Мс В ее Е. Т., Chem. Revs., 1958, 58,
№ 2, 249; Коган Л. М., Хим. пром-сть, 1959, № 5, 448;
Вольфсон Л. Г., К о г а н Л. М., там же, 1961, № 10,
672; Мельников Н. Н. [и др ], в кн.: Реакции и методы
исследования органических соединений, кн. И, М., 1962.
С. Д. Боловкович, Л. М. Поган.
ПЕСТИЦИДНЫЕ ПРЕПАРАТЫ — композиции пес-
тицидов с другими ингредиентами, употребляемые для
борьбы с вредителями полезных растений, а также
для подавления или стимулирования роста растений.
Действие пестицидов в значительной степени зависит
от формы, в виде к-рой применяется препарат, и ус-
ловий его соприкосновения с растениями и возбуди-
телями их заболеваний. В нек-рых случаях форма
препарата влияет не только на продолжительность,
но и на характер действия пестицидов.
При выборе тех или иных П. п. существенное зна-
чение имеют физико-химич. свойства соединения и
характер действия его на вредителей растений и на
растения. В зависимости от применяемого объекта од-
но и то же соединение можно использовать в виде са-
мых различных препаратов. Так, гербицид 2,4-Д
можно применять в виде гранулированных препара-
тов, в виде р-ров в минеральных маслах и водных
эмульсий, а инсектицид ДДТ — в виде дустов, гра-
нул, смачивающихся порошков, эмульсий, растворов
в минеральных маслах, аэрозолей и т. д.
П. п. часто называют формами примене-
ния пестицидов; к ним относятся порош-
ки (дусты), гранулированные препараты, смачиваю-
щиеся порошки, концентраты эмульсий, аэрозоли,
растворы в воде или органич. растворителях и др.
Кроме того, в нек-рых случаях пестициды применяют
в виде кремов, восков, паст, в смеси с красками и т. п.
Порошки (дусты) — механич. смесь пести-
цида (или нескольких пестицидов) с инертным напол-
нителем, размолотая до величины частиц 2—30 мк.
Размол обычно производят в помольных агрегатах
(см. Измельчение') различных типов. Пестицидные по-
рошки можно также изготовлять смешением жидкого
(или расплавленного) действующего начала с напол-
нителем, а также растворением пестицидов в маслах
или других растворителях с последующим смешением
с наполнителем. В качестве наполнителей обычно
применяют гидрофобные минералы (тальк, пирофиллит,
сланцевая или шиферная мука и др.). В нек-рых слу-
чаях к порошкам добавляют вещества, способствующие
удерживанию порошков на поверхности растений.
Гранулированные препараты по
своему характеру близки к дустам, размер их обычно
от 0,1 до 0,5 мм. Чаще всего их готовят пропиткой пер-
лита, вермикулита и других аналогичных минералов
жидкими пестицидами. Гранулированные препараты
можно также получать путем агрегирования пести-
цидных порошков. Гранулированные препараты чаще
всего применяют для внесения гербицидов и инсекти-
цидов в почву; в нек-рых случаях гранулированные
препараты используют для обработки растений (напр.,
для борьбы с кукурузным мотыльком хорошие резуль-
таты дают гранулированные препараты ДДТ).
Смачивающиеся порошки — препа-
раты, к-рые при разбавлении водой дают достаточно
устойчивые суспензии. В виде смачивающихся порош-
ков чаще всего применяют пестициды, нерастворимые
в воде (в отдельных случаях в виде суспензий можно
применять и растворимые в воде химич. соединения).
В состав смачивающихся порошков, кроме действую-
щего начала, обычно входят: наполнитель, поверх-
ностно-активное вещество и вспомогательное вещест-
во. Для приготовления смачивающихся порошков с
высоким содержанием пестицида (более 50%) исполь-
зуют достаточно чистые твердые пестициды и сорбцион-
ноемкий наполнитель с малым насыпным весом (напр.,
силикагель). Известны, напр., препараты след, со-
става:
ДДТ...................... 50% 75%
Наполнитель...............40—41 % 15%
Смачивающие, стабилизирующие
и диспергирующие вещества . 9—10% 10 %
В качестве наполнителей для смачивающихся по-
рошков используют каолин, бентонит, силикат каль-
ция, опоку, силикагель и др. гидрофильные вещества,
не способные вступать во взаимодействие с пестици-
дом. В качестве поверхностно-активных веществ при-
меняют эфиры полиэтилен- и полипроииленгликолей,
алкилсульфонаты и алкиларилсульфонаты щелочных
металлов, алкилсульфаты щелочных металлов и др.
Из стабилизаторов чаще всего используют сульфитно-
спиртовую барду. Смачивающиеся порошки получа-
ют размолом смеси ингредиентов в мельницах. Чем
тоньше помол, тем более стойкие суспензии получа-
ются при разбавлении смачивающегося порошка во-
дой. Применение пестицидов в виде суспензии в боль-
шинстве случаев более эффективно, чем опыливание
растений пестицидами.
Концентраты эмульсий — одна из наи-
более удобных и распространенных форм П. п.; при
разбавлении водой образуют достаточно устойчивые
эмульсии. Известно два вида таких концентратов:
1) Конц, эмульсии пестицидов (или р-ров пестицидов
993
ПЕСТИЦИДЫ—ПЕХМАНА КОНДЕНСАЦИЯ
994
в гидрофобных органич. растворителях) в воде в при-
сутствии стабилизаторов; диспергирование обычно
производится в жидкостных коллоидных мельницах.
Такие эмульсии могут сохраняться длительное время,
выдерживают относительно низкие темп-ры и доста-
точно дешевы, однако для их приготовления непригод-
ны пестициды, легко гидролизуемые водой. 2) Конц,
эмульсии пестицидов в органич. растворителях,
известные под названием смешивающиеся
масла. В состав таких концентратов входят поверх-
ностно-активные вещества, пестицид и растворитель
(напр.: 25% ДДТ, 71% растворителя и 4% эмульга-
тора или 25% паратиона, 70% растворителя и 5%
эмульгатора). В качестве растворителей используют
гл. обр. ароматич. углеводороды и их смеси, а в каче-
стве эмульгаторов — эфиры полиэтилен- и полипро-
пиленгликолей, а также смеси нескольких поверх-
ностно-активных веществ.
Аэрозоли получают механич. диспергирова-
нием растворов пестицидов в маслах; нек-рое приме-
нение получили также дымовые шашки, в состав к-рых,
кроме пестицида, входят горючая смесь и флегматиза-
тор (напр., 54,9% ДДТ, 12% тиомочевины, 7,3% мо-
чевины, 25,8% хлората калия).
Растворы. Большое число органич. пестицидов
выпускают в виде р-ров (5—20%) в минеральных
маслах. Эти р-ры используют для тонкодисперсного
опрыскивания растений. Растворимые в воде препа-
раты чаще всего используют в виде водных р-ров.
В качестве П. п. применяют также нек-рые зооциды
путем их смешения с каким-либо питательным продук-
том (мука, крупа, зерно и т. п.).
Лит.: Дусты, эмульсии и смачивающиеся порошки орга-
нических инсектофунгисидов, М., 1959; Дусты, эмульсии и
суспензии ДДТ и ГХЦГ, М., 1955; Безуглый С. Ф.,
Ж. Всес. химич. об-ва, 1960, 5, № 3, 298; Ш о г а м С. М.[и
д р.], там же, 1960, 5, 3, 312; Мельников Н. Н.,
Баскаков Ю. А., Химия гербицидов и регуляторов роста
растений, М., 1962; Попов П. В., Справочник по ядохи-
микатам, М., 1956. Н. Н. Мельников.
ПЕСТИЦИДЫ — химич. средства борьбы с раз-
личными вредными организмами: клещами (см. Ака-
рициды), насекомыми (см. Инсектициды), бактериями
(см. Бактерициды), растениями (см. Гербициды),
низшими растениями — грибами (см. Фунгициды),
моллюсками (см. Лимациды), круглыми червями (см.
Нематоциды) и др. В эту же группу веществ обычно
включают и антисептики, применяемые для предо-
хранения неметаллич. материалов от разрушения
микроорганизмами, а также вещества, употребляемые
для предуборочного удаления листьев (см. Дефолиан-
ты) й подсушивания растений (см. Десиканты),
предпосевной обработки семян (см. Протравители
семян) и др. В зависимости от физико-химич. свойств
и назначения П. обычно применяют в виде препаратов
различных форм (см. Пестицидные препараты).
Н. Н. Мельников.
ПЕТРОЛАТУМ — смесь высокомолекулярных
твердых углеводородов (парафинов и церезинов) с
Остаточным маслом, получаемая при депарафинизации
масел сернокислотной или селективной очистки с
применением избирательных растворителей, напр.
тяжелого бензина. Остаточное масло, предварительно
подвергнутое деасфальтизации и очистке, подогревают
и смешивают с 2—2,5 частями растворителя-разбави-
теля, полученный раствор охлаждают до кристалли-
зации парафина и центрифугируют. Получаемый П.
содержит до 20% масла и наз. «масляным». Для даль-
нейшего отделения масла этот продукт повторно об-
рабатывают 5—6 частями растворителя и вновь цент-
рифугируют. Из получаемого при этом р-ра, т. н.
«сухого» П., отгоняют растворитель и П. подверга-
ют дальнейшей переработке, напр. в церезин. Вы-
ход масляного П. достигает 15—25% на исходное
сырье.
П.— продукт гелеобразной консистенции, светло-
или темно-коричневого цвета, содержит 1—2% воды,
нейтрален; его т. каплепад. 50—55°, т. всп. 240—250°.
П. применяют гл. обр. для получения нефтяного
церезина, для изготовления медицинского и технич.
вазелина, консервирующих и пластичных (консистент-
ных) смазок, а также как компонент изоляционных
и кабельных масс. П., получаемый из сернистых неф-
тей, применяют для сушки древесины.
Окислением П. воздухом при 140—160° в присут-
ствии перманганата калия получают окисленный П.—
однородный вязкий продукт темного цвета, с кислот-
ным числом не ниже 55, числом омыления не ниже 140,
полностью растворимый в тяжелом бензине (уайт-
спирите) и содержащий не более 2% воды. Окислен-
ный П. применяют в качестве эмульгатора (вместо жи-
ров и высокомолекулярных карбоновых к-т при изго-
товлении смазочно-охлаждающих жидкостей), как
компонент нек-рых консистентных смазок и как флото-
реагент. Омыляя окисленный П. каустической содой,
получают омыленный окисленный П., применяемый
для отделки шерстяных тканей. Этот продукт должен
содержать не более 0,25% свободной щелочи и не более
22% воды.
Лит.: Богданов Н. Ф., Переверзев А. Н.,
Депарафинизация нефтяных продуктов, М., 1961; Технические
нормы на нефтепродукты, под ред. Н. Г. Пучкова, 6 изд.,
М., 1957; Технические условия на нефтепродукты, М., 1960.
,. _ В. В. Щекин.
ПЕТРОЛЕЙНЫЙ ЭФИР — смесь легких углево-
дородов (преим. предельных с 5 и 6 атомами углеро-
да), получаемая из попутных нефтяных газов и легкпх
фракций нефти, а также каталитически (синтезом из
СО и Нг при среднем давлении). П. э.— бесцветная
жидкость, d=0,633—0,670 (при получении из нефти)
и 0,685 (синтетич. продукта); начало кипения П. э.,
полученного из нефти, не выше 40°, конец кипения не
выше 70°. Фракционный состав П. э., полученного
синтезом из СО и На: 10% перегоняется не ниже
75° и 95% не выше 100°. Нефтяные П. э. применяют в
качестве растворителя жиров, смол, нек-рых эфиров
целлюлозы, для экстракции эфирных масел и др.;
синтетич. П. э.— растворитель в лабораторных усло-
виях, а также сырье для произ-ва нек-рых химич.
реактивов.
Лит.: М е 1 1 a n I., Handbook of solvents, v. 1, N. Y.—L.,
1957; Технические нормы на нефтепродукты, под ред.
Н. Г. Пучкова, 6 изд., М., 1957. В. А. Гетлинг.
ПЕХМАНА КОНДЕНСАЦИЯ — метод синтеза ку-
марина и его производных конденсацией фенолов с
яблочной к-той или эфирами р-кетокислот в присут-
ствии кислотных катализаторов:
нооссн-сн,соон
Применение яблочной к-ты позволяет с успехом син-
тезировать кумарины, не замещенные в пироновом
ядре. В данном случае П. к. предшествует разложение
яблочной к-ты на муравьиную и формилуксусную
к-ты:
О%
ноос-сн-снг-соон—- нсоон + с-снг-соон
он Нх
0 32 к.х.э. т. з
995
ПЕХМАНА КОНДЕНСАЦИЯ—ПЕЧАТАНИЕ ТЕКСТИЛЬНЫХ МАТЕРИАЛОВ
996
Вместо яблочной в П. к. могут быть использованы
также малеиновая или фумаровая к-ты.
В качестве карбонильного компонента в П. к. всту-
пают различные кетоэфиры, содержащие алифатич.,
ароматич. и гетероциклич. заместители. Установлено,
что П. к. облегчается наличием заместителей, усилива-
ющих енолизацию или стабилизирующих енольную
форму. Эфиры а-алкил- и а-арилацетоуксусных к-т,
как правило, легко вступают в П. к.; с еще большей
легкостью реагируют эфиры ацетоянтарной и ацето-
глутаровых к-т:
сосн,
СаН5ООССНСНаСООС1Н5
н+
о
соосгн5
сн3
сн3 сн3
Ацетондикарбоновая к-та и ее эфиры реагируют с фе-
нолами с образованием соответствующих кумаринук-
сусных к-т и их эфиров;
СН3 CHjCOOH
СН
Лимонная к-та в зависимости от условий может да-
вать те же продукты, что и ацетоуксусный эфир или
ацетондикарбоновая к-та. Легкость осуществления
П. к. в значительной степени зависит от подвижности
водорода в opmo-положении к гидроксильной группе
фенола. Малая склонность к конденсации у самого
фенола повышается при введении в мета-положение
к оксигруппе электронодонорных заместителей и резко
понижается при введении электроноакцепторных
групп. Заместители в opmo-положении, как правило,
затрудняют П. к., пара-замещенные фенолы занимают
промежуточное положение.
а-Нафтол легко взаимодействует с яблочной к-той,
ацетоуксусным эфиром и его аналогами с образова-
нием 1,2-а-нафтопиронов;
(З-Нафтол вступает в П. к. несколько труднее.
Окситиофены в условиях П. к. реагируют с эфирами
(3-кетокислот, образуя производные тиофенопирона;
сн,ссн,соосгн5
<t 3 и 2 „
НзС-Ог0Н ——-НзС^
сн3
Катализаторами П. к. обычно являются H2SOi,
РОС1з, Н3РО4, Р2О3, Aids и др., причем в отдель-
ных случаях от характера катализатора зависит не
только осуществление реакции, но и общее ее на-
правление. Применение H2SC>4, Р2О3, РОС13 и ZnCl2
в случае активных фенолов гладко приводит к заме-
щенным кумаринам. Сам фенол и его малоактивные
аналоги в присутствии H2SO4 не конденсируются с
эфирамир-кетокислот вообще либо реагируют с боль-
шим трудом. В присутствии Р2О3 конденсация фено-
ла, о-крезола, Р-нафтола и других малоактивных фено-
лов с Р-кетоэфирами, как правило, приводит не к ку-
маринам, а к хромонам (реакция Симониса):
О
Безводный Aids в р-ре сухого эфира или нитробензо-
ла— один из наиболее активных катализаторов П. к.
СН3
Выход кумарина достигает 40—55% от теоретического.
В ряде случаев не только выход, но и структура
образующегося кумарина определяется характером
применяющегося катализатора:
Считается, что первоначальным актом П. к. яв-
ляется С-алкилирование фенола эфиром кетокислоты с
последующей циклизацией промежуточно образующе-
гося эфира оксикислоты (I), сопровождающейся от-
щеплением воды и спирта. Согласно другому предпо-
ложению, на первой стадии из фенола-и енольной фор-
мы р-кетокислоты образуется соответствующее про-
изводное коричной к-ты (II), к-рое затем претерпевает
циклизацию в кумарин:
П. к. открыта Пехманом в 1884.
Лит.: С е т и а С., Пхадке Р., в кн.: Органические
реакции, сб. 7, пер. с англ., М., 1956, с. 7; В а в з о н е к С.,
в сб.: Гетероциклические соединения, под ред. Р. Эльдерфилда,
пер. с англ., т. 2, М., 1954. Л. С. Герман.
ПЕЧАТАНИЕ ТЕКСТИЛЬНЫХ МАТЕРИАЛОВ
(узорчатая расцветка тканей) — окрашивание от-
дельных участков ткани, величина и форма к-рых за-
висят от наносимого рисунка. Различают грунтовые,
полугрунтовые и белоземельные рисунки. В грунто-
вых фон составляет более 60% площади ткани, в
полугрунтовых — от 40 до 60%, в белоземельных
тканях рисунок печатают по белому фону. По технике
печатания различают три способа получения окраски:
прямой (накладной), резервный и вытравной. Прямая
печать заключается в непосредственном получении
на ткани рисунка в нужных расцветках. Краски на-
носят на белую или слабоокрашенную ткань, к-рук>
можно также подкрашивать после печатания. Резерв-
ный способ заключается в печатании на ткани защит-
ного слоя (резерва) печатной краски, в состав к-рой
входят вещества, препятствующие ее закрашиванию
при последующей окраске ткани. После удаления
краски получается белый рисунок. Аналогично полу-
997
ПЕЧАТАНИЕ ТЕКСТИЛЬНЫХ МАТЕРИАЛОВ
998
чают цветные рисунки, но в этом случае в защитную
печатную краску вводят краситель. При вытравной
печати по предварительно окрашенной ткани печата-
ют краску, содержащую вещества, разрушающие кра-
ситель фона. Разрушение красителя может быть пол-
ным— на ткани получается белый рисунок,-или же
частичным, когда на окрашенной ткани получается ри-
сунок того же цвета, но более светлого оттепка. Воз-
можна также цветная вытравка. В этом случае одно-
временно происходит разрушение красителя фона и
нанесение па ткань невытравляемого красителя. Наи-
более часто вытравную расцветку осуществляют по
тканям, окрашенным прямыми и нерастворимыми
оксиазокрасителями. Основу хромофорной системы
этих красителей составляют одна или несколько азо-
групп, разрушая к-рые печатанием загущенного р-ра
ронгалита или кубовой печатной краски можно полу-
чить соответственно белый или цветной узор. Разру-
шение окраски фона и фиксация кубовых красителей
в местах нанесения рисунка происходят в среде на-
сыщенного водяного пара («запаривание») при 102—
105° в специальных аппаратах — восстановительных
зрельниках.
Техника печатания и состав кра-
сителей. Узорчатую расцветку текстильных ма-
териалов осуществляют: ручной набивкой, способом
аэрографии, сетчатыми шаблонами и на печатных ма-
шинах. Последние два способа являются главными.
При печатании сетчатыми шаблонами рисунок нано-
сят на ткань, протирая краску через шаблон, представ-
ляющий прямоугольную раму, на к-рую натянута
шелковая или капроновая ситовая ткань или частая
медная сетка. Ткань или сетку покрывают тонкой
непроницаемой для краски лаковой пленкой т. обр.,
чтобы участки, не закрытые пленкой, образовали ри-
сунок. В этих местах печатная краска при протира-
нии специальной резиновой теркой (раклей) свободно
проходит на ткань. Печатание сетчатыми шаблонами
производят на набивных столах или на спец, машинах.
Этот способ наиболее широко применяют при печата-
нии тканей из натурального шелка.
Основным способом узорчатой расцветки хлопчато-
бумажных тканей и тканей из искусственных волокон
является нанесение рисунка при помощи печатной
машины. Медные хромированные печатные валы ма-
шины имеют углубленную гравюру; процесс печата-
ния заключается в передавливании на ткань печатной
краски, предварительно нанесенной на гравюру вала.
Для многокрасочной печати служат машины с числом
валов от 1 до 16. Как правило, число печатных валов
соответствует числу печатных красок, переносимых
на ткань. При т. наз. «орбис»-печати вместо несколь-
ких печатных валов весь рисунок наносят на ткань с
помощью одного вала, покрытого слоем печатных кра-
сок разных цветов в соответствии с расцветкой рисун-
ка. Печатные краски имеют в данном случае консистен-
цию густых паст. По мере их израсходования на вал
вновь наносят многокрасочный рисунок. При способе
трехцветной печати минимальным числом красителей
можно воспроизводить на ткани тоновые цветные пе-
реходы в рисунке. В этом случае очень важен подбор
тканей, красителей и загустителей. Наиболее подходя-
щими являются гладкие ткани типа поплинов, репсов,
сатинов и красители, полностью фиксируемые волок-
ном. Помимо печатания тканей, иногда производят
расцветку чесальной лепты («фигуре») и пряжи как в
виде мотков, так и в форме основы («фламме») для по-
лучения впоследствии меланжевых эффектов. В неко-
торых случаях ткани расцвечивают приклеиванием
в электростатич. поле к поверхности текстильного
материала мелко нарезанного волокна-флока. Для
специальных целей применяют фотохимия, метод пе-
чатания.
В большинстве случаев состав печатных красок со-
ответствует составу красильных р-ров; разница за-
ключается лишь в консистенции. В целях получения
при печатании рисунка с четкими контурами в печат-
ные краски вводят загущающие средства. Наряду с
давно известными загустителями— крахмалом, про-
дуктами его гидролиза, трагантом, камедью — исполь-
зуют различные продукты их модификации. В качестве
хороших заменителей камеди успешно используют
альгиновую к-ту и альгинат натрия. Загустителями
животного происхождения являются альбумин, казеин
и хитозан. Кроме перечисленных природных загусти-
телей, применяют неорганич. и синтетич. загущающие
в-ва: нек-рые виды глин и гидроокисей, напр. гидро-
окись кремния, простые и сложные эфиры целлюлозы,
напр. карбоксиметилцеллюлозу и ацетилцеллюлозу,
а также синтетич. смолы — алкидные, акриловые,
мочевипо- п меламино-формальдегидные и др., исполь-
зуемые при печатании пигментами. Широкое распро-
странение получили также и т. наз. эмульсионные
загустки.
Печатание азокрасителями. При
образовании на текстильных материалах нераствори-
мых оксиазокрасителей методом печатания по предва-
рительно пропитанной р-ром азотола и высушенной
ткани печатается загущенное диазосоединение. Затем
следует сушка, во время к-рой происходит образова-
ние красителя из компонентов, и промывание напеча-
танной ткани. Наряду с этим существуют и специал ь-
ные составы, предназначенные преим. для печатания
текстильных материалов. К числу таких соединений
относятся, напр., диа.зотолы, диазоаминолы и рапидо-
золи. Этими препаратами печатают непосредственно
по белой ткани.
Диазотолы представляют собой смесь азотолов
и устойчивой формы диазосоединения в виде нитроза-
мина или антидиазотата:
R-N R-N-Na
II I
N-ONa N = O
При печатании к диазотолу добавляют едкий патр,
к-рый переводит азотол в раствор, а также ализарино-
вое масло и хромпик. Загущенную печатную краску
наносят па ткань и для образования окраски послед-
нюю пропускают через пары уксусной к-ты:
R-N-Na
I
N=O
Наибольшее распространение из диазаминолов по-
лучили такие, к-рые могут проявляться при запарива-
нии в нейтральной среде, т. наз. палогены— смесь
триазонов (I) с азотолами: ,
Для печатания к этой смеси добавляют едкий натр,
нек-рые вещества, улучшающие растворимость, и за-
густку. Печатную краску наносят на ткань и после вы-
32*
999
ПЕЧАТАНИЕ ТЕКСТИЛЬНЫХ МАТЕРИАЛОВ
1000
сл’шивапия подвергают действию насыщенного водяно-
го пара («запаривают»):
NaO3S
NaOH
NaO3S
NaO3S
запаривание
N-N-R
+ NaOH
Рапидозоли — смесь диазоарилсульфонатов и азо-
толов:
R—N = N—OSO2Na
наносят на ткань в форме щелочной печатной краски,
содержащей хромпик; ткани после высушивания за-
паривают в среде насыщенного водяного пара. При
этом происходит проявление окраски:
ОН- + NaHSO3
R-N НдО
N—OSO2Na запаРиа"
Вместо диазоарилсульфонатов можно применять так-
же соли гидразинсульфокислот
R—NH—NH-OSO.Na,
Печатание кубовыми красителя-
м и. Для печатания можно применять кубовые кра-
сители в виде загущенных щелочных р-ров лейкосоеди-
нений, к-рые получают предварительным восстанов-
лением кубовых красителей гидросульфитом натрия.
В этом случае в печатную краску, помимо гидросуль-
фита натрия, вносят еще и ронгалит, к-рый поддержи-
вает краситель в состоянии лейкосоединения в момент
протекания диффузионных процессов при запарива-
нии напечатанной ткани в восстановительном зрель-
нике. Более удобен т. наз. ронгалитно-поташный
метод печатания, основанный на применении специаль-
ных высокодисперсных паст кубовых красителей.
При этом предварительное восстановление красителя
гидросульфитом не требуется, а в состав печатной крас-
ки входит только краситель, поташ, ронгалит и за-
густка. Печатный состав наносят на ткань, к-рую за-
тем высушивают и обрабатывают в восстановительном
зрельнике; конечной операцией является окислитель-
ная промывка. Для окисления лейкосоединения на
волокне используют слабые р-ры хромпика (3 г/л)
и перекиси водорода (2—3 г/л). В последнее время при
расцветке тканей кубовыми красителями начали при-
менять двухфазное печатание, сущность к-poro заклю-
чается в том, что вначале на ткань наносится только
загущенная суспензия невосстановленного красителя,
а затем после высушивания ткань плюсуют р-ром гид-
росульфита и щелочи и обрабатывают в восстанови-
тельном зрельнике. При печатании тканей применяют
также и эфиры лейкосоединений кубовых красите-
лей — кубозоли; в этом случае почти всегда пользу-
ются запарным или нитритным способом проявления
окраски.
Печатание активными красите-
лями. При узорчатой расцветке текстильных
материалов широко используют т. наз. активные
красители. Чаще всего их применяют по запарному
способу. Для этого в состав печатной краски вводят,
помимо красителя, мочевину и бикарбонат натрия.
Последний при запаривании ткани переходит в карбо-
нат и создает необходимую для фиксации красителя
волокном концентрацию гидроксильных ионов. В ка-
честве загустителей необходимо применять альгинат
натрия или эмульсионные загустки. Эти загустители
выгодны тем, что позволяют свести к минимуму
реакцию активного красителя с веществом загустки и
способствуют наиболее полному образованию нового
химич. соединения красителя с субстратом волокнисто-
го материала.
Печатание пигментными красите-
лями. Красителями этой группы можно печатать но
любой ткани и даже по тканям из смешанных волокон.
Пигментная печатная краска обычно состоит из тонко-
дисперсного пигмента, синтетич. смолы, используемой
в качестве связующего вещества, эмульсионной за-
густки и катализатора. Пигмент на ткани фиксируют
пропусканием последней через пагретую до 140—150°
камеру. Наиболее часто применяют фталоцианиновые,
кубовые и азопигменты, а также окислы нек-рых ме-
таллов, напр. двуокись титана и металлич. порошки.
Пигменты должны состоять из высокодисперсных
частиц порядка 0,2—0,3 мк. В качестве связующего
вещества применяют предконденсаты термореактив-
ных смол и термопластич. смолы. Из первых наиболее
широко используют меламино- и мочевино-формальде-
гидные препараты, напр. мета-
зин или тетраметилольное про-
изводное ацетилендимочевины.
Из второй группы веществ ча-
ще применяют поливиниловый
спирт, поливинилхлорид, эфиры
акриловой и метакриловой к-т.
нонгс н снгон
N-С —N
О“\ I "оо
N-C-N
I I I
ЙОНгС н снгон
В качестве эмульсионной загустки употребляют вяз-
кие эмульсии, состоящие из органич. летучего раство-
рителя, напр. тяжелого бензина с т. кип. 150—160°.
Можно употреблять также керосин и даже нек-рыо
виды смазочных масел. Обычную эмульсионную за-
густку типа «масло в воде» можно получить, эмульги-
руя при сильном размешивании растворитель в вэдо
при наличии стабилизатора эмульсии — препарата
ОС-20. Для приготовления пигментной печатной
краски берут, напр., 75—100 г пасты пигмента, 140?
метилметакрилата в смеси 1:1с метазином, 8 г ни-
трата аммония (катализатор) и 702—747 г эмульсион-
ной загустки. После нанесения печатной краски на
ткань последнюю высушивают и прогревают при 140°
в течение 8 минут. Напечатанная ткань не требует
промывки. В этом заключается существенное преиму-
щество пигментов по сравнению с красителями дру-
гих классов.
Печатание черным анилином. Прп
образовании на текстильных материалах черного ани-
лина методом печатания загущенный р-р, содержащий
анилиновую соль, хлорат калия или натрия и ферри-
или ферроцианид калия, наносят па ткань, к-рую за-
тем высушивают и обрабатывают в окислительном
зрельнике при 95—97° в течение 1 минуты. Далее сле-
дуют окислительная обработка и промывка в мыльном
р-ре и воде. Часто пользуются получением белых или
цветных резервных узоров по фону из черного ани-
лина. Для этого па ткани, пропитанной названным
выше составом, но без загустки, печатают загущенный
раствор соды, ацетата натрия, бисульфита натрия или
। вносят готовую печатную краску, напр. кубовую.
При последующей обработке такой ткани паром в ме-
стах нанесения печатной краски черный анилин пе
образуется и они выглядят белыми или окрашенными
в цвет соответствующего кубового красителя. Резерв-
ный метод печатания широко используется и при об-
1001
ПЕЧАТНЫЕ КРАСКИ
1002
разовании на волокне нерастворимых оксиазокраси-
телей. В этом случае по ткани, пропитанной р-ром азо-
тола, печатают загущенные р-ры кислых солей, напр.
ацетата алюминия, цинка и др., к-рые способны пере-
вести азотолят натрия в недеятельную форму свобод-
ного азотола. В качестве резервирующих средств ис-
пользуют также загущенные восстановители типа би-
сульфита натрия, воздействующие, помимо азотолята,
еще и на диазосоединение. После высушивания напе-
чатанной ткани ее пропускают через р-р диазосоли.
При этом в местах нанесения «резерва» азосочетание
не происходит и пигмент не образуется.
Лит.: Г р ю н е р т А. [и д р.], Оборудование и техно-
логия мокрой обработки текстильных материалов из целлю-
лозных волокон, пер. с нем., М., 1959, с. 235—300; Садов
Ф. И., Корчагин М. В., Матецкий А. И., Хими-
ческая технология волокнистых материалов, 2 изд., М., 1956,
с. 599—748; Справочник по крашению и отделке шелковых
тканей, под ред. А. А. Копьева, М., 1953, с. 237—338; X а р-
харов А. А.,Калантаров И. Я., Активные красители
и их применение в текстильной промышленности, М., 1961,
с. 77—94; Knecht Е., Fothergill J. В., The prin-
ciples and practice of'textile printing, 4 ed., L., 1952; Мои-
сеев А. Г., П e т p о в В. M., Руководство по гравированию
текстильного рисунка, М., 1961; С и н е г у б-Л а в р е н-
к о А. А., А н и с и м о в В. И., Т а р а с о в а Л. А., Фото-
механические способы изготовления форм для печати на тканях,
М., 1961. 17. В. Моръианов, Б. И. Мельников.
ПЕЧАТНЫЕ КРАСКИ — краски, применяемые в
полиграфия, произ-ве для различных способов печата-
ния. К ним относятся: типографские, офсетные, фото-
типные, для глубокой печати, эластографские, этмо-
графские (шелко-трафаретные), электрографские и
нек-рые другие виды П. к., различающиеся в основном
вязкостью, скоростью закрепления на бумаге или на
другой подложке. П. к. в зависимости от характера
изданий, для к-рых они предназначены, разделяются
на газетные, книжные, журнальные, иллюстрацион-
ные, репродукционные (для трех- и четырехкрасоч-
ного печатания), картографические и др.; в зависи-
мости от скорости печатания — на П. к. для ротаци-
онных и для плоских печатных машин. П. к. наносят
обычно слоем ок. 2 мк, гл. обр. на поверхность отно-
сительно пористой бумаги. В этом их отличие от
обычных индустриальных красок, наносимых слоем
ок. 100 мк на хорошо прогрунтованную невпитываю-
щую подложку. Кроме того, цветные П. к., как пра-
вило, прозрачные, а индустриальные — кроющие
(см. Краски, Лаки, Пигменты).
П. к. — суспензии пигментов в связующем, иногда
подкрашенном маслорастворимым красителем. Ис-
ключение составляют нек-рые П. к., напр. эластограф-
ские, представляющие собой р-ры красителей в свя-
зующем; электрографские краски — порошкообраз-
ные смеси смол, пигментов и красителей. П. к. при-
надлежат к коллоидным системам, в к-рых молекулы
поверхностно-активных веществ связующих адсор-
бируются на поверхности частичек пигментов и об-
разуют защитные (сольватные) оболочки, препятст-
вующие агрегации пигмента. Таким образом, при де-
формации П. к. трение происходит в относительно
жидкой прослойке связующего, а не между поверх-
ностью частичек пигментов.
Для офсетных, типографских и нек-рых других
П. к. характерны вязкость структурная и ясно вы-
раженные тиксотропные свойства (см. Тиксотропия).
Маловязкие краски глубокой печати подчиняются за-
конам вязкого течения истинно вязких жидкостей
(см. Вязкость). Структурная вязкость и эластичность
(см. Вязкость полимеров) определяют печатные свой-
ства красок, т. е. их способность раскатываться вали-
ками, накатываться на поверхность печатной формы
и переноситься под давлением на поверхность бу-
маги.
Пигменты. Наибольшее значение имеют син-
тетич. органич. пигменты (желтые и красные азопиг-
менты; голубые, синие и зеленые фталоцианиновые
пигменты) и красочные лаки (красные бариевые и
кальциевые лаки из анионоидных азокрасителей и
лаки основные разных цветов из катионоидиых трифе-
нилметановых и ксантеновых красителей, осажден-
ных фосфорновольфрамовомолибденовой к-гой) (см.
Красители синтетические). В качестве черного
пигмента для П. к. применяется сажа, гл. обр. ка-
нальная и особенно ее окисленная разновидность.
Из неорганич. пигментов используют ферроцианиды
железа (милори), сульфохроматы свинца (хромовые
желтые), молибдаты свинца, двуокись титана и раз-
личные виды белил (см. Пигменты).
Связующие. В качестве связующего для ти-
пографских, офсетных и ряда других П. к. применяют
олифы: 1) алкидные, напр. из пентаэритрита, дифе-
новой к-ты и нек-рого количества растительных ма-
сел или высших жирных к-т; 2) эпоксидные, получае-
мые взаимодействием эпоксидных смол с высшими
жирными к-тами; 3) масляно-стирольные, получае-
мые сополимеризацией льняного и тунгового масел
со стиролом; 4) композиционные, состоящие из син-
тетич. смол, высоковязких олиф, масел (нефтяных и
растительных) и высококипящих органич. раствори-
телей (чаще всего это фракции алифатич. нефтепродук-
тов ст. кип. 220—290°); 5) фирнисы — простейшие свя-
зующие, не высыхающие на невпитывающей поверх-
ности; к таким связующим относятся р-ры смол (би-
тум, кумароновая смола, эфиры канифоли) в нефтя-
ных маслах; 6) эмульсионные типа «вода в масле» или
«масло в воде». Для красок глубокой печати в качест-
ве связующих применяют лаки из смол (фенол о-аль-
дегидных, резината цинка, эфиров канифоли) и соот-
ветствующих органич. растворителей (бензин, уайт-
спирит, толуол, ксилол).
Специфич. особенностью П. к. является их закреп-
ление, т. е. пленкообразование, на поверхности по-
ристой бумаги. Пленка образуется в результате вкис-
лительной полимеризации связующего, испарения
органич. растворителя или избирательного впитыва-
ния. В последнем случае в поры и капилляры бумаги
впитываются сравнительно низковязкие и низкомоле-
кулярные компоненты связующего — масла и раст-
ворители, а высокомолекулярные и высоковязкие
компоненты — смолы и высоковязкие олифы — остают-
ся на поверхности бумаги и закрепляют пигмент.
На этом принципе основано изготовление большин-
ства видов быстрозакрепляющихся П. к.
В и д ы П. к. Типографские черные ротационные
П. к. (газетные и книжные) состоят из канальной са-
жи, р-ра индулина в олеиновой к-те (подцветка) й
битумного фирниса; они закрепляются исключительно
избирательным впитыванием. Газетные цветные П. к.
состоят из пигмента или красочного лака, маслораст-
воримой подцветки и композиционной олифы. Чер-
ные иллюстрационные книжные П. к. для плоских
печатных машин содержат канальную сажу, синие и
фиолетовые пигменты и красочные лаки (подцветка),
алкидную или композиционную олифу и сиккатив.
Цветные типографские П. к. содержат пигменты или
красочные лаки, наполнители (А1(ОН), и BaS04],
алкидные или композиционные олифы и сиккатив.
Репродукционные типографские П. к. для трех- или
четырехкрасочного печатания (желтая, пурпурная и
голубая) отличаются от обычных цветных красок
спектральной характеристикой (см. рисунок) и полной
прозрачностью (желтая краска, наносимая первым
слоем, может быть непрозрачной). Быстрозакрепляю-
щиеся типографские П. к. приготовляются на компо-
зиционных олифах из интенсивных высокодисперсных
пигментов или красочных лаков с минимальным коли-
чеством наполнителя и нек-рого количества сиккати-
ва. Офсетные П. к. (черные и цветные) отличаются от
1003
ПЕЧИ
1004
соответствующих типографских красок несколько по-
вышенным содержанием пигмента и более вязким и
эластичным связующим.
Фототипные П. к. подоб-
ны офсетным, но приго-
товляются предельно вяз-
кими и содержат пигмен-
ты, обладающие высокой
стойкостью к воде. Эла-
стографские П. к. пред-
ставляют собой р-ры кра-
сителей в спиртах, они
применяются для печата-
ния преим. этикеток пище-
вых продуктов с эластич-
Спектры отражения триады
красок для трех- и четырех-
красочного печатания: 1 —
желтая из пигмента желтого
светопрочного 23; 2 — пурпурная из лака основного розо-
вого; 3 — голубая из пигмента зеленовато-голубого фталоци-
анинового.
пых печатных форм. Картографические П. к. — это
особый вид офсетных красок, предназначенных для
печатания география., топография., гидрография, и
т. и. карт. Этмографские П. к. приготовляют из свя-
зующих, представляющих собой, напр., продукт взаи-
модействия феноло-альдегидной смолы с тунговым
маслом, растворенный в алифатич. нефтяных раство-
рителях; предназначены они для печатания продав-
ливанием через форму-трафарет. Краски для глубо-
кой печати состоят из пигментов или красочных ла-
ков и р-ров смол в соответствующих органич. раство-
рителях.
Лит.: Apps Е. A., Printing ink technology, L., 1958;
Wolfe H. J., Printing and litlio inks, 5 ed., N. Y., 1957;
Березин Б. И., Печатные краски, М., 1961; его же,
Что должен знать печатник о красках, 2 изд., М., 1957.
Б. И. Березин.
ПЕЧИ технологические — тепловые устройства, в
к-рых получаемое тепло (в основной химия, пром-сти
за счет сжигания топлива или горючих компонентов
сырья) передается находящемуся в П. материалу, в
результате чего протекают химич. процессы с обра-
зованием заданного продукта. По способу обработки
сырья различают: шахтные печи, вращающиеся труб-
чатые печи—механич. многополочные печи и подовые
печи, печи с псевдоожиженным слоем, печи пылевид-
ного обжига — форсуночные и циклонные — и др.
Шахтные печи работают по принципу про-
тивотока; опускающийся слой материала омывается
встречным потоком газа. Обжигаемый материал про-
ходит последовательно зоны подсушки, подогрева, об-
жига и охлаждения. Зона обжига частично совпа-
дает с зоной горения топлива, в к-рой образуются
горячие топочные газы. Размеры кусков обрабаты-
ваемого в шахтных печах материала должны нахо-
диться. в соотношении 1 : 3—1 : 4 (обычно применяют
куски размером 40—120 лглг, иногда 80—240 и 20—
60 мм). При обжиге в шахтных печах кусковой мате-
риал не должен спекаться и сильно измельчаться
(средний размер кусков после обжига должен быть
не менее 0,5 среднего исходного размера). Шахтная
печь (рис. 1) имеет обычно цилиндрич. форму; сна-
ружи она заключена в стальной кожух, а изнутри
футерована огнеупорным кирпичом. Сырье подается
на верх печей скиповыми подъемниками или вагонет-
ками. Распределение сырья но поперечному сече-
нию печи производится устройствами различной кон-
струкции, напр. конусами-рассекателями. Из вы-
грузочных устройств наибольшее распространение
получили каретки Антонова, совершающие возвратно-
иоступадельное движение, вращающиеся или качаю-
щиеся поды, а также вибровыгружатели. Конструк-
тивное оформление шахтных печей зависит от вида
применяемого топлива
(твердое, жидкое, газо-
образное).
Шахтные печи, рабо-
тающие на твердом топ-
ливе (кокс, антрацит),
смешанном с сырьем («в
пересыпку»), наз. пере-
сыпными. Размеры
шахты таких печей: внут-
ренний диаметр 2—8 м,
высота 12—40 м; произ-
водительность 25—600 т
в сутки; съем обжигае-
мого материала, напр.
извести (в пересчете на
100% СаО), достигает
15—20т/л12в сутки. Наи-
меньший расход тепла
900—1000 ккал/кг СаО.
Количество топлива в
шихте составляет 8—10
вес. %. Обжигаемый кус-
ковой материал, филь-
трующий газовый поток,
является как бы колос-
никовой решеткой для
сжигаемого топлива, по-
этому размеры кусков
топлива для предотвра-
щения его просыпания в
промежутки между кус-
ками материала должны быть равны 0,5—1 диаметра
среднего куска обжигаемого материала. Пересыпные
печи применяют для обжига карбонатных пород: из-
вестняка, мела, мрамора, доломита (CaCO5-MgCO,).
Теплота сгорания топлива расходуется на эндотермич.
реакции:
СаСО3=СаО + СО2-42,5 кг.алхоль
MgCO, = MgO + CO2-23 ккал/лоль
Скорость термич. диссоциации зависит от темп-ры
окружающей среды, физич. свойств сырья, формы и
размеров кусков.
При обжиге карбонатных пород образуется СО2, ис-
пользуемый в ряде химич. производств (папр., содо-
вом). Концентрация СОа в печном газе зависит от объе-
ма и состава топочных газов, с к-рыми он смешивается.
Т. к. в шахтных печах расход топлива, а следователь-
но, и объем топочных газов минимальный, то концен-
трация СО* в печном газе получается наиболее высо-
кой. При обжиге известняка на коксе она достигает
41 %, а при обжиге влажного мела, содержащего 10—
12% влаги,— 34—36%. Во избежание разбавления
СОа воздухом шахтные печи работают под небольшим
избыточным давлением (до 700 мм вод. ст. внизу и до
2—Ь мм вод. ст. вверху).
Все процессы (приготовление шихты и ее загрузка,
поддержание постоянства уровня шихты и регулирова-
ние расхода воздуха) на современных шахтных печах
механизированы и в значительной степени автомати-
зированы. Созданы опытные регуляторы выгрузки
извести. К достоинствам шахтных печей относятся:
простота конструкции; сравнительно малые капитало-
вложения; низкие эксплуатационные расходы; высо-
кие теплотехнич. и технико-экопомич. показатели
(производительность, расходные коэффициенты, кон-
центрация СОа в печном газе). Недостатками этих пе-
чей являются: необходимость применения сравни-
тельно узкофракциопировапного по размерам кусков
сырья, что связано с большим (до 50%) количеством
отходов горной массы; трудность равномерного рас-
1005
ЛЕЧИ
1006
пределения сырья и топлива по сечению печи; боль-
шая инерционность печей.
Шахтные печи на газообразном топливе обору-
дуются специальными горелками, обеспечивающими
равномерное распределение газа по сечению печи.
Если внутренний диаметр печи не превышает 2 м,
то ограничиваются установкой периферийных одно-
сопловых горелок. При такой схеме ввода газа и воз-
духа коэфф, избытка воздуха (а), вводимого в П., под-
Йзвес/пь
тип IIIIIIIH
П I
;IHIllllWIIIIIUIIliiil
III 11ПЯН11Н |I 111 I
lli1 :
iimniiir
1ММ111»
111Ш1Ш1Н11.! д
IIHIIHIIIUIIIII :
LUUIHII II II <' ir
I >.>(«11111 и"
il IIIIIIIIIIIH'
111111111 1'1
11НПМ111НI >
mamnniiiiiiiiif
Газ
Рис. 2. Шахтная печь на газообразном топливе: а — ос-
нащенная балочными и периферийными горелками; б —
оснащенная периферийными горелками.
держивают ок. 2. Если диаметр шахты больше 2 м, то
газообразное топливо подают и в центральную часть
печи, для чего используют многосопловые балочные
горелки (рис. 2, а), охлаждаемые проточной водой, или
центральные горёлки, изготовляемые из жаропрочной
стали и охлаждаемые воздухом (рис. 2, б). В этом
случае козфф. избытка воздуха может быть снижен
до 1,1 — 1,3, расход тепла достигает 1050 ккал/кг
СаО, а съем извести до 30 т/м2 в сутки (и выше).
Скорость горения газообразного топлива, а следова-
тельно, и производительность печи лимитируются про-
цессом перемешивания газа с воздухом; поэтому при
применении специальных горелочных устройств с ча-
стичным предварительным смешением газа и воздуха
(балочные или центральные горелки) резко увеличи-
вается съем продукта с 1 м2 сечения печи.
Преимуществами применения газообразного топ-
лива в шахтных печах являются: низкая стоимость
топлива и легкость регулирования его расхода;
высокое качество получаемого продукта; меньшая
инерционность печи; отсутствие золы; отсутствие
отделения приготовления шихты; улучшение сани-
тарно-гигиенич. условий труда. К недостаткам этого
способа относятся: необходимость установки в пе-
чи горелочных устройств, охлаждаемых водой или
воздухом; снижение концентрации СОг в печном газе;
возможность образования взрывоопасных смесей при
неполном сгорании газа.
Шахтные печи на жидком топливе аналогичны по
конструкции печам на газообразном топливе, но имеют
дополнительные устройства по предварительной гази-
фикации жидкого топлива. Для этого используют цик-
лонные газификаторы, в к-рых за счет частичного
сжигания мазута поддерживается температура ок.
1000°. В этих условиях происходит испарение жид-
кого топлива и его термич. разложение; образующаяся
газовая смесь имеет теплотворную способность до
1900 ккал/нм2. В нек-рых случаях шахтные печи на
жидком топливе эксплуатируют и без предваритель-
ной газификации топлива. В таких печах топливо рас-
пыляется форсунками непосредственно в подбалоч-
ном пространстве.
Для обжига мелкокускового сырья (10—50 мм)
применяют шахтные печи с поперечным потоком газа;
они состоят из двух шахт прямоугольного сечения,
между к-рыми находится камера для сжигания топ-
ливного газа. С внешней стороны шахт имеются ка-
меры для отходящих газов. Нагретый доменный газ
вместе с нагретым воздухом подается в камеру для
сжигания. Образовавшиеся горячие газы поступают
в шахты и производят обжиг сырья. После этого то-
почные газы удаляются во внешние камеры; часть то-
почных газов используется для нагрева сырья.
Удельный расход тепла в такой печи при обжиге
известняка 1050 ккал/кг извести, расход электроэнер-
гии до 22 квт-ч/т извести. Положительной особен-
ностью описываемых печей является высокая степень
обжига известняка.
Вращающиеся трубчатые печи с
внутренним обогревом (за счет факельного сжига-
ния жидкого или газообразного топлива) работают
по принципу противотока. Передача тепла осущест-
вляется непосредственным излучением факела, кон-
векцией от газов к материалу и к стенке печи, свобод-
ной от материала. От нагретой стенки П. тепло откры-
той поверхности материала передается лучеиспуска-
нием, закрытой поверхности — теплопроводностью.
Материал проходит зоны: подсушки, нагрева и реак-
ции (напр., диссоциации, плавления и химич. взаи-
модействия); зона охлаждения в этих печах отсутст-
вует. Корпус печи (рис. 3) представляет собой опи-
рающийся на ролики металлич. цилиндр с внутрен-
ней огнеупорной футеровкой. С одного торца цилиндра
размещается форсунка для распыления топлива,
с противоположного — производится отвод топочных
газов и подача материала. Готовый продукт выгру-
жается через специальные окна в головке печи либо
пересыпается через порог, поддерживающий опреде-
ленный уровень заполнения печи. Для использова-
ния тепла выгружаемого продукта устанавливают
теплообменники-рекуператоры. Диаметр вращаю-
щихся трубчатых печей колеблется в пределах 1,5—
2,7 л, длина от 12 до 122 л. Для утилизации тепла от-
ходящих газов целесообразно устанавливать шахтные
или трубчатые теплообменники-рекуператоры или
цепные решетки для подогрева сырья. Вращающиеся
Рис. 3. Вращающаяся трубчатая печь для обжига
известняка.
печи применяются для обжига мелкокусковых мате-
риалов, к-рые не поддаются обработке в шахтных пе-
чах, а также в случаях особых требований к про-
дукту. Во вращающихся Л. обжигают карбонатные
породы (известняк, доломит, ракушечник), серный кол-
чедан, обесфторивают фосфаты, получают феррит ат-
рия и др.
Конструкция вращающихся трубчатых печей за-
висит от особенностей осуществляемого процесса.
1007
ПЕЧИ
1008
Так, при обжиге серного колчедана, протекающем по
реакции:
4FeS2+l lOa = 2FeaOa+8SOa + 203,8 ккал/моль
не требуется подвода тепла. Для предотвращения спе-
кания колчедана производится подача воздуха через
сопла, установленные по длине печи по винтовой ли-
нии. Образующийся при горении SO2 используется
для производства серной к-ты. Феррит натрия полу-
чают во вращающихся печах при 1000—1100° по след,
реакции:
Na2CO3+Fe2Oa=Na2O-Fe2O3+COa- 38,8 3 ккал/моль
Темп-ра в печи поддерживается за счет сжигания
мазута или газа; длина печи 12—15 м, темп-ра отходя-
щих газов в ней достигает 500°. Обесфторенные фос-
фаты получают при 1450° в присутствии водяного
пара по реакции:
Ca10(P0))eFa + 2H20 = Ca,a(PO4)e(0H)2 + 2HF-62,3 ккал/моль
Расход тепла во вращающихся трубчатых печах всегда
выше, чем в шахтных (при обжиге известняка 1300—
2000 ккал/кг СаО).
В случаях, когда не допускается смешение топоч-
ных газов с газообразными продуктами, выделяю-
щимися во время реакции, применяют вращающиеся
трубчатые печи с наружным обогревом, к-рые дейст-
вуют по принципу прямотока. Вращающийся ци-
линдр печи помещают в обмуровку, где он омывается
горячими топочными газами. Такие печи применяют,
напр., для кальцинации бикарбоната натрия:
2NaHCO3=NaaCO., + CO2 + H2O (пар)-29,95 ккал/моль .
Процесс протекает при 140—160°. В связи с тем, что
влажный NaHCO5 склонен к комкованию и образова-
нию корок на нагретых поверхностях печей, предус-
мотрена специальная цепь для очистки стенок ци-
линдра и измельчения образовавшихся комков. Для
использования газов, выделяющихся при полу-
чении соды, печи тщательно герметизируются. Га-
бариты цилиндра: диаметр 1,5—2,65 л, длина 17—
23 м, полезная поверхность 80—185 .и2; производи-
тельность 50—170 т в сутки, темп-ра факела в топке
достигает 1400°, темп-ра отходящих газов 380—470°.
Для обжига колчедана используют многопо-
лочные печи, к-рые имеют обычно 8 сводов, вы-
ложенных из огнеупорного кирпича. Принципиально
они не отличаются от аналогичных печей, описанных
в статье Пирометал-
лургия. Недостатка-
ми механич. многопо-
лочных печей явля-
ются большие затра-
ты ручного труда,
низкая производи-
тельность, большая
металлоемкость и
практически полная
потеря тепла, выде-
ляющегося при об-
жиге колчедана.
Печи с псев-
доожиженным
слоем разделяют-
ся на однокамерные
и многокамерные. Од-
нокамерные печи
(рис. 4) применяют
для обжига серного
колчедана (производ-
ство серной к-ты), а
Рис. 4. Схема печи с псевдоожижен-
ным слоем: 1— форсунка; 2— термо-
пара; 3— провальная решетка; 4—
беспровальная решетка.
также для сжигания серы в слое инертного материала
(напр., песка). Камеры печей имеют преимущественно
круглое сечение, подина камеры является распредели-
тельной решеткой для воздуха (газа) и состоит из про-
вальной и непровальной частей. Провальная часть ре-
шетки устанавливается под местом загрузки колчедана
в печь для вывода из печи попадающихся В сырье
крупных частиц. Наибольшее распространение полу-
чили трубные провальные решетки. Непровальная
часть решетки имеет отверстия, прикрытые грибками,
выполненными из жаропрочной стали. Скорость вы-
хода воздуха из отверстий трубной решетки 4—6 м/сек,
из колпачковой 10—12 м/сек. Для отвода тепла, выде-
ляющегося при сжигании колчедана, в зоне реакции
установлены трубчатые змеевиковые охлаждающие
элементы. Равномерная подача колчедана в печь до-
стигается установкой специальных тарельчатых пи-
тателей, отличительной чертой к-рых является вра-
щающийся стол и бункер. Высота псевдоожиженного
слоя поддерживается равной примерно 800 мм. Со-
держание серы в огарке не превышает 0,5% при ин-
тенсивности обжига 8—9 т/м1 в сутки. Концентра-
ция сернистого ангидрида в газах колеблется в преде-
лах от 11 до 14%. Обжиг ведется при 750—850°, при
этом в охлаждающих элементах образуется 1,0—1,1 т
пара на 1 т колчедана. Печи с псевдоожиженным слоем
снабжаются системой авторегулирования, поддер-
живающей постоянство высоты слоя, расхода воз-
духа и концентрации SO2 в заданных пределах.
Многокамерные печи, пока еще не получившие ши-
могут иметь различные фор-
Рис. 5. Схема многокамерной печи:
1 — камеры нагрева; 2 — камера
обжига; з — камера охлаждении;
4 — переточное устройство; & —
решетка с колпачками; 6 — фор-
сунка.
рокого распространения,
мыигабариты. Имеют-
ся печи (рис. 5), диа-
метр обжиговой ка-
меры которых равен
4000мм и высота 13,7 м\
печь по высоте разде-
лена распределитель-
ными решетками на 5
камер: 3 верхних
используются для под-
сушки и подогрева ма-
териала и по одной
камере для обжига и
охлаждения продук-
та. Обжигаемый мате-
риал, напр. известняк,
перемещаясь из каме-
ры в камеру с помощью
переточных труб, по-
степенно нагревается
до 500°, 750° и 850° за
счет тепла отходящих
газов. Обжиг произ-
водят при 1000°, а го-
товый продукт охлаж-
дается в последнейка-
мере до 370°. Произво-
дительность П., отап-
ливаемой нефтью, со-
ставляет 7—15 т/м‘ в
сутки при расходе
тепла ок. 1270 ккал/кг
СаО. Имеются образ-
цы печей, предназна-
ченные для работы на
твердом и газообразном топливе. Обжигу подвергается
фракция 1—5 .u.u (иногда 5—10 .и.и) с обязательным от-
делением мелкой фракции. Продукт, получаемый в
печах с псевдоожиженным слоем, отличается высокой
степенью обжига. Недостатками этих печей являются:
необходимость применения узкофракционироваипого
материала; возможность изменения производитель-
ности в узких пределах; большой унос пыли, требую-
щий сложной системы пылеулавливания (циклоны
и электрофильтры); повышенный расход электроэнер-
гии на переработку сырья (см. Псевдоожижение).
1009
ПЕЧИ
1010
Печи пылевидного обжига, где об-
работка материала происходит во всем объеме, имеют
камеру прямоугольного или круглого сечения, в к-рую
вместе с газом вдувается обрабатываемый порошкооб-
разный материал. Образующиеся газообразные про-
дукты реакции, содержащие значительные количества
пыли, отводятся обычно из верхней части камеры,
а твердые продукты реакции удаляются снизу в спе-
циальные бункеры. Печи такого типа применяют для
обжига флотационного колчедана. Камера печи из
огнеупорного кирпича заключена в стальной кожух.
Колчедан с воздухом вводится в печь сверху (при по-
мощи специальной форсунки) или снизу. Для повы-
шения интенсивности П. и более полного выгорания
серы в камеру над уровнем бункера для огарка по-
дается вторичный воздух. Интенсивность работы печи
600—1250 кг/м3 в сутки в пересчете на колчедан, со-
держащий 45% серы. Печи работают нормально при
равномерной подаче колчедана и равномерном его
распределении по сечению камеры. Поддерживае-
мый в П. температурный режим зависит от содержа-
ния серы в колчедане. Повышение температуры интен-
сифицирует процесс до тех пор, пока не начинается
оплавление частичек, препятствующее доступу кис-
лорода внутрь частиц и выгоранию серы. Недостат-
ками этого типа печей являются: большой унос пыли
с обжиговыми газами, сложность удаления огарка,
малая инерционность печей.
Форсуночные печи для сжигания рас-
плавленной серы аналогичны по конструкции печам
пылевидного обжига. Камера печи представляет со-
бой вертикально или горизонтально расположенный
цилиндр с устройствами для лучшего перемешивания
газов. Расплавленная сера самотеком или насосом
подается в распиливающую форсунку, откуда раз-
дробленная до мельчайших частиц' сера поступает в
камеру печи, смешивается с воздухом и при 1200°
интенсивно испаряется и сгорает с образованием сер-
нистого газа. Полученный SO2 используется для
производства серной к-ты. Наиболее полное сгорание
серы достигается в вертикальных печах, в к-рых обес-
печивается лучшее перемешивание паров серы с воз-
духом.
Печи циклонного
типа (рис. 6) представ-
ляют собой полый цилиндр (отношение длины к
диа-
Рис. 6. Схема циклонной
метру 1,5—2), в к-рый
тангенциально вводится
газовоздушная смесь. По-
ток получает вихревое
движение. Материал мо-
жет подаваться тангенци-
ально, вместе с горючей
смесью, или аксиально
(вдоль осишечи). Для по-
дачи материала приме-
няется пневмотранспорт.
Печи циклонного типа
еще не получили широко-
го распространения; при-
менение их перспективно
в процессах обработки
порошковых материалов
с образовавием плава
(жидкое шлакоудаление).
В некоторых случаях печи такого типа могут найти
применение для обжига карбонатных пород.
Другие разновидности печей см. в статьях Огнеупор-
ные материалы, Псевдоожижение, Пирометаллургия,
Стекло, Цементы, Кальция карбид.
Теоретически обоснованные методики расчета производи-
тельности П. еще не созданы. Обычно съемы принимаются по
практическим данным, достигнутым в производстве, а габа-
риты П. выбираются по заданной мощности и принятым удель-
ным съемам. Расчет пересыпных шахтных П. сводится к оп-
печи.
ределению расходных коэфф, по сырью и топливу из матери-
ально-теплового баланса, нахождению поперечного сечения
П. по принятому съему и заданной производительности и
определению потребной высоты П. Последняя определяется
след, образом:
Высота зоны подогрева:
ЙПд. = —0,418 1g/1—---------gc.de Укаж.сСс .
\ *г У й2Унас.с
Высота зоны охлаждения:
D d у /-»
и л г / / ии ’каж.и Си
лОхл- = 01544 --------------—
а у I \
S ’нас.и I----1
\Wr J
Высота участка зоны горения, совпадающего с зоной обжига:
Лобж. = 2,1В<<г 1g Reo>” к
где t"— темп-ра на входе и выходе из зоны, °C; D — расход
материала (или съем извести), кг/м2час', d — определяющий
размер куска, м\ уКаж. Унас.— кажущийся и насыпной вес.
кг/ж3; С — средняя теплоемкость, ккал/кг°С; W — водяное
число, ккал/час°С; Х'о, X — содержание кислорода в газе на
входе и на выходе из зоны обжига, %;
а = 0,106-Re при Re<200
а = 0,61 ReM’—5- при Re>200
( ^сух.с -Ксух.т \ -Ксух.т
\ ^нас.с ^нас.т / ^каж.т
X — коэфф, теплопроводности, ккал/м•час°С; ^Сух — удельный
расход сухой массы, кг/m; м — материал; с — сырье; и — из-
весть; г — газ; т — топливо.
Во вращающихся печах без подпорных колец скорость
осевого перемещения материала может быть подсчитана по
ур-нию:
т, л /В+ф cos а \
V = 2nrn *-—А---- ]
sin а J
где г — радиус барабана печи, м; п — число оборотов бара-
бана, мин'., 3— угол наклона барабана, град.; ф— угол наклона
поверхности материала к оси печи, град.; а—динамич. угол
естественного откоса материала, град. Для печей с подпорными
кольцами необходимо учитывать дополнительное торможение
материала. Расчет теплопередачи в печи с внутренним обо-
гревом ведется по ур-нию:
Q=ea[rJ-T‘]
Где е — приведенная степень черноты излучающей среды;
0=4,9,10“"—степень черноты абсолютно черного тела; Тг—
Темп-ра излучающей среды, “К; Тм—темп-ра материала, “К.
Степень черноты излучающей среды выбирается в зависимо-
сти от запыленности (и темп-ры) газового потока. Количе-
ство тепла, передаваемое конвекцией, может быть рассчитано
по ф-ле: £>коив.=“к (4г“4м) l\L, где ак — коэфф, теплоотдачи
конвекцией, ккал/м2град'час; ty — темп-ра газов, °C; tM —
темп-ра материала, °C; 1Х —длина хорды, ж; L — длина ба-
рабана, -ч. Темп-ра газов при факельном сжигании может
быть определена по ур-нию:
^г.х^теор.
где Тг х— температура газа в сечении х,°К; Ттеор.— теоретич.
темп-р’а сгорания, °К; х — расстояние от места ввода топ-
лива, м; п — показатель темп-рного режима, определяемый
размерами и режимом работы печи; ц— показатель, характе-
ризующий процесс выгорания топлива.
Показатели п и ц определяются на основе данных эксплуа-
тации промышленных печей. Для вращающихся печей с на-
ружным обогревом расчет топки производится по норматив-
ному методу (см. литературу в конце статьи «Тепловой
расчет котельных агрегатов»). Газоход рассчитывается как
конвективное теплообменное устройство.
По данным кинетики технологического процесса, скорости
осевого перемещения материала и теплообмена выбираются
размеры вращающейся печи.
Лит.: Производство кальцинированной соды, [под ред.
М. Б. Зеликина], М., 1959; Попандопуло Ю. Г., Хим.
пром-сть, 1945, № 7; М а л и н К. М. [и др.], Технология
серной кислоты, М.—Л., 1950; Применение в СССР процессов
1011
ПИГМЕНТЫ
1012
о7;кига в кипящем слое. Сб. докладов..., М-, 1960; Семе-
ненко Н. А., С и д е л ь к о в с к и й Л. Н., Шуры-
гин А. П., Хим. пром-сть, 1956, К» 3; Г у р в и ч А. М.,
Теплообмен в топках паровых котлов, Л.—М., 1950; X о д о
ров Е. И., Печи цементной пром-сти, ч. 1, М., 1950; Тепловой
расчет котельных агрегатов, под ред. А. М. Гурвича и Н. В.
Кузнецова, М.—Л., 1957; Sa ema n W. С., Chem. Engng.
Progr,, 1951, 47, AS 10. Н.П. Табунщиков.
ПИГМЕНТЫ. Содержание:
Введение.................................. 1011
Важнейшие свойства пигментов................1011
Пигменты белые..............................1015
Пигменты черные ............................1017
Пигменты желтые, оранжевые, красные и коричневые 1017
Пигменты синие, зеленые и фиолетовые.......1019
Пигменты антикоррозионные...................1022
Введение. Пигменты — тонкодисперсные окрашен-
ные (включая белый и черный цвета) порошки, нера-
створимые в воде (в отличие от красителей) и плен-
кообразуклцих веществах; образуют при растирании
с последними дисперсии, называемые красками.
В биологии П. наз. окрашенные вещества, входящие
в состав тканей животных и растительных организ-
мов [см. Пигменты (биологические), Красящие ве-
щества природные}.
По происхождению П. делят на природные и синте-
тические, по составу — на минеральные и органиче-
ские, по цвету — на ахроматические (белые, серые,
черные) и хроматические (все цветные), по примене-
нию — на грунтовочные, малярные, для художест-
венных красок, специального назначения и др. Наи-
более широко в лакокрасочной и смежных областях
пром-сти применяют минеральные П., представляю-
щие собой гл. обр. окислы и соли различных метал-
лов. Такие П. обычно дешевле органич. П. (см. Кра-
сители синтетические), отличаются более высокой
свето- и термостойкостью, лучшей кроющей способ-
ностью. Однако богатая гамма оттенков, высокая ин-
тенсивность, яркость и глубокий тон органич. П.
создают основу для более широкого их применения
в ближайшем будущем. Помимо органич. П., приме-
няемых обычно в смеси с белыми минеральными П.,
выделяют группу пигментных лаков, по-
лучаемых осаждением органич. красителей на мине-
ральных носителях (субстратах). Роль пигментных
лаков в лакокрасочной пром-сти невелика.
Большинство важнейших минеральных П. полу-
чают синтетически (цинковые белила, двуокись ти-
тана, свинцовые окислы, кроны, милори и др.). Раз-
витие химии и технологии этих П., создание непре-
рывных способов произ-ва, совершенствование ме-
тодов контроля и испытаний позволяют получать
продукты с более высокими, заранее заданными пиг-
ментными свойствами. Но и до настоящего времени не
утратили своего значения более дешевые природные
(земляные) П. (охры, сиены, железный сурик и др.),
отличающиеся химич. инертностью, атмосфере- и
светостойкостью.
Большую роль играют наполнители (некроющие П.);
их вводят в лакокрасочные композиции с целью сни-
жения стоимости и улучшения технич. свойств кра-
сок и покрытий. Основные требования, предъявляе-
мые к высококачественным наполнителям,— это вы-
сокая степень белизны (не ниже 90% по сравнению
с эталоном белого цвета — MgO или бланфиксом) и то-
нины помола. В качестве наполнителей в лакокрасоч-
ной пром-сти широко применяют барит, ангидрит, до-
ломит, мел, тальк, слюду и др.
Большое число соединений, относящихся к П.,
ТРУДНО классифицировать по какому-либо одному
признаку. Наиболее распространенвым и удобным
является, деление минеральных П. на группы по цвет-
ности, а внутри групп — по химич. общности.
Важнейшие свойства пигментов. Основными харак-
теристиками П. являются: цвет, укрывпстость, интен-
сивность (красящая сила), форма и размер частиц,
смачиваемость, маслоемкость, уд. и насыпвой веса,
антикоррозионные свойства, устойчивость к атмос-
ферным воздействиям, свету, теплу, химич. стойкость.
Цвет П. определяется их способностью к погло-
щению и отражению света в видимой части спектра
(380—760 ммк). Белые П. обладают высокой отража-
тельной способностью, черные сильно поглощают ви-
димый свет; цветные П. поглощают избирательно.
Основной цветовой тон П. зависит от
того, в какой части спектра находится максимум от-
раженного света. Цвет П. характеризуют также
яркостью и насыщенностью. Чем боль-
шую часть падающего света отражает П., тем он ярче.
Яркость характеризуют коэфф, отражения (в %).
Чем меньше в отраженном свете содержится лучей,
не соответствующих основному тону, тем цвет насы-
щенней, чище. Цвет П. обусловлен наличием в них
определенных атомов или их группировок—хромофо-
ров (напр., CrOj в кронах). Уменьшение размера ча-
стиц П. обычно повышает яркость и насыщенность его
цвета. Иногда от размера частиц зависит и основной
тон П. Напр., красный и оранжевый свинцовые кро-
ны имеют одинаковый химич. состав и кристаллизуют-
ся в одной и той же тетрагональной системе, но отли-
чаются размером частиц (у красного частицы круп-
нее). Цвет П. определяют визуально (сравнением
с эталонами) и фотоэлектрич. методами (с помощью
колориметров, рефлектометров и спектрофотометров).
Укрывистость (кроющая способность) —
свойство П. делать невидимым (перекрывать) цвет
закрашиваемой поверхности. Укрывистость опреде-
ляется гл. обр. диффузным отражением (рассеянием)
света в пленке; у темных П. заметную роль играет
также поглощение света. Поэтому укрывистость ра-
стет с увеличением разности показателей преломления
П. и среды, в к-рой они диспергированы. П. с очень
низкой укрывистостью («прозрачные») наз. лесси-
рующими. Они используются для произ-ва худо-
жественных и полиграфия, красок (см. Печатные
краски). Укрывистость увеличивается с уменьшением
размера частиц до определенного оптимума (0,25—
0,4 мк), ниже к-рого она понижается. С ростом кон-
центрации П. в пленке укрывистость также проходит
через максимум. Низкая укрывистость органич. П.
объясняется тем, что размеры их частиц значительно
ниже оптимальных. Методы определения укрывисто-
сти, принятые в СССР, основаны на определении ко-
личества П. (в г), необходимого для получения на сте-
клянной пластинке площадью 1 л2 непрозрачного
слоя краски.
Интенсивность (красящая сила) — спо-
собность П. придавать свой цвет смеси П. Чем больше
интенсивность П., тем больше требуется разбеливаю-
щего компонента для доведения смеси до стандартного
оттенка, полученного на эталонном П. На этом и ос-
нованы визуальные и объективные (рефлектометри-
ческие) методы измерения интенсивности. Высокая
интенсивность П. позволяет уменьшить их количе-
ство в краске, что особенно важно для таких дорогих
продуктов, как ультрамарин, милори и др. Интен-
сивность П. сильно зависит от их дисперсности. По-
лагают, что интенсивность цветных П. с уменьшением
размера частиц непрерывно повышается, а белых —
проходит через оптимум, близкий оптимуму для укры-
вистости.
Форма и размер частиц П., а также рас-
пределение частиц по размерам и плотность их упа-
ковки являются важными факторами, определяю-
щими реология, свойства красок и фпзпко-механич.
свойства лакокрасочных покрытий (выше уже отме-
чалось влияние размера частиц на оптич. свойства
красок). Многие свойства красочной пленки дости-
1013
ПИГМЕНТЫ
1014
гают максимума или минимума вблизи «критич е-
ской объемной концентрации пиг-
мент а». Последняя отвечает максимальной плот-
ности упаковки частиц П. в пленке и сильно зависит
от формы, размеров частиц и распределения их по
размерам. Размер частиц П. колеблется от 0,01 мк
для тонких сортов сажи до нескольких десятков мик-
рон у грубых природных П. Стандартами обычно не
допускается наличие очень грубых частиц (определяе-
мых ситовым методом), препятствующих получению
высококачественных покрытий. Размеры частиц П.
и распределение частиц по размерам определяют се-
диментационным, микроскопич. и электронномикрос-
копич. методами. Два последних метода позволяют
определять и форму частиц. Дисперсность П. характе-
ризуют также уд. поверхностью, т. е. поверхностью
единицы веса или объема П. Эта характеристика очень
важна, т. к. многие технич. свойства краски являются
функцией физич. и химич. взаимодействия П. со свя-
зующим на границе раздела фаз. Знание уд. поверх-
ности позволяет более правильно составлять рецеп-
туры красок. Для определения уд. поверхности при-
меняют адсорбционные и кинетич. методы.
П. обычно состоят из довольно прочных агрегатов,
включающих большое количество первичных частиц.
Сухой размол П. разрушает лишь самые крупные аг-
регаты. Дальнейшее разрушение происходит при из-
готовлении краски, т. е. при растирании П. со свя-
зующим. Качество краски в значительной степени за-
висит от смачиваемости П. связующим. Если
уд. поверхность характеризует площадь, на к-рой
происходит взаимодействие П. со связующим, то сма-
чиваемость определяет активность этого взаимодей-
ствия. От легкости смачивания зависят эффективность
работы аппаратов для приготовления красок и экс-
плуатационные свойства красок и пленок. Для улуч-
шения смачиваемости П. обрабатывают поверхностно-
активными веществами (стеаратами алюминия и каль-
ция, алифатич. четвертичными аммониевыми основа-
ниями и др.).
Количество масла (в г), необходимое для получения
пасты из 100 г П., наз. маслоемкостью 1-го
рода. Количество масла в краске, готовой к упот-
реблению, т. е. разбавленной олифой, наз. масло-
емкостью 2-го рода. Чем ниже маслоем-
кость П., тем экономически выгоднее его применение.
Маслоемкость является сложной функцией от смачи-
ваемости П., пористости и размера его частиц и рас-
пределения частиц еео размерам.
Свойства основных П. приведены в табл. 1.
Важнейшей функцией лакокрасочных покрытий
является защита металлических из-
делий от коррозии. Большую роль играют
П., способствующие замедлению электрохимия, кор-
розионных процессов. К наиболее эффективным
пассиваторам (в-вам, тормозящим анодный
процесс ионизации металла путем образования на его
поверхности адсорбгщонных или фазовых защитных
слоев) относят кроны (цинковые, свинцовые, барие-
вые, кальциевые, стронциевые), фосфат хрома, свин-
цовые П. (сурик, цианамид свинца, плюмбат каль-
ция). Для протекторной защиты широко
применяют покрытия, пигментированные цинковое!
пылью.
Весьма важной характеристикой П. является их
устойчивость к атмосферным воз-
действиям, свету, теплу, действию
химич. реагентов. Многие органич. П. на
свету быстро выцветают (обесцвечиваются), нек-рые
минеральные темнеют (желтый свинцовый крон),
Таблица 1
Пигменты
Плотн., Насыпной
г(см3 вес, г/л
Коэфф, отра-
жения (пре-
ломления)
Размер час- I Укрывис- ] °е“'
стиц, лек тость, г/ле3 „„А
I , | рида
Белые п.
Белила цинковые3 ...... 5,45—5,65 400—800 92.0-99,7 0,15—5,00 120-140 10-14
Двуокись титана (1,95—2.03)
рутил® 4,18—4,25 700—800 98 (2,55) 0,2 —0,8 35-40 17-26
анатаз8 3.82—3,95 600-700 99 (2,72) 0,2—0,8 35—45 18-23
Белила свинцовые 6,4 —6.8 — (1,94—2,09) 0,5 -1,2 130—200 9-12
Литопон 4,1 —4,3 750 95-97 1-3 120-160 11-15
(1,84—2,00)
Черные п
Железоокисный 1 4,73 700-800 3.0 0,2-0,5 28—35
Термостойкий железоокисный . . | 5,15—5,25 830 1,8-2,0 4,0—5,5 31-34
Желтые, оранжевые, красные и коричневые п.
Желтый кадмий 4,2 -4,7 1000—1400 1-5 65—75 3 2-3 5
Желтый кадмопон 4,0 -4.25 1 100—1200 — — 90—120 16—18
Желтая окись железа1, 3.8 5—3.9 0 420—550 0.2-0,6 10—15 40—60
Красный кадмий 4.5 —5,3 850 — 1100 —- 1—5 28-30 28—32
Красный кадмопон 4,2 -4,3 1 250—1 500 — — 30-50 18—23
Киноварь 8. 15 — (2,91—3.27) 8—13 2—20 — —
Красный железоокисный .... 4,8 —5,0 500—750 0,3-1, 5 5—8 20-40
Охры 2,7 -3,4 800-1 100 16—40 1-5 60—90 25-32
Сиены 3.0 —3.4 900-1000 25 — — 50-55
Мумия 2,5 770 7—12 —— 15-30 19-33
Железный сурик 3,66—4,46 1 500-1800 10-12 1,2-4,0 10-20 14-21
Умбра натуральная 2,5 —3,4 750 — 1 ,5—4,0 30-40 38—48
Спине, зеленые и фиолетовые П.
Ультрамарин
светлый темный 2,33—2,45 600—660 880—990 (1 .50 — 1 ,54) о; з—о, 5 2,0-3,0 — 33—34
Свинцовая зелень 3.4 -5,0 950-1250 — — 10 — 15 13-22
Окись хромад 4,64 1160 — —• 8 — 12 23-27
Изумрудная зелень 3,32 800-850 — 1-10 50 63—75
Синий кобальт 4.2 —4,4 630-740 — —— 75—80 31 — 37
«Зеленый кобальт 5,41—5,76 2100-2500 — — 50-60 —
Церулеум 6,1 -6,3 — — — 60—70 —
Марганцовая зеленая — — — — 3 0—3 5 12—16
а Уд. поверхность 4—6 м2/г. ® То же 4,8. в То же 8,8. Г То же 8—11. Д То же 6—7.
1015
ПИГМЕНТЫ
1016
нек-рые белые (литопон) желтеют при недостатке
света. Такие П., как анатаз, муфельные цинковые бе-
лила, обладают высокой фотохимии, активностью. Они
сенсибилизируют окислительно-восстановительные
процессы, ведущие к разрушению пленок (меление,
образование трещин и пр.). Эти процессы могут про-
текать весьма быстро при совместном действии света,
влаги, тепла и др. атмосферных факторов. Установ-
лено, что светостойкость и фотохимии, активность П.
зависят не столько от их дисперсности, сколько от
микроструктуры кристаллич. частиц, наличия де-
фектов структуры. Поэтому светостойкость и фото-
химии. активность П. в значительной степени оп-
ределяются условиями кристаллизации пигментных
частиц; свинцовые кроны, полученные нитратным
способом, более светостойки, чем полученные ацетат-
ным; витерилльиые цинковые белила более атмосферо-
стойки и имеют низкую фотохимии, активность по
сравнению с муфельными. Для повышения свето- и
атмосферостойкости П. применяют их поверхностную
обработку (гидроокисью алюминия, кремневой к-той
и др.).
Помимо лакокрасочной пром-сти, П. применяют
в произ-ве резины, бумаги, линолеума, керамики,
цемента, стекла, стеклянных эмалей, пластмасс и др.
В различных областях к П. предъявляют свои требо-
вания. Так, для резины требуются очень тонкоди-
сперсные, высокоактивные П., активирующие процесс
вулканизации. Для нек-рых печатных красок необхо-
димы П. с очень высокой укрывистостью и мягкой
текстурой; для керамики, стекла, эмалей — термо-
стойкие и способные хорошо диффундировать в рас-
плавах. Для произ-ва пластмасс используют термо-
стойкие П., способные совмещаться с полимерами.
Пигменты белые. Основными белыми П. являются
двуокись титана (титановые белила), цинковые и
свинцовые белила, литопон.
Двуокись титана (TiOs). Высокими пиг-
ментными свойствами обладают две кристаллич. мо-
дификации TiOs — рутил и анатаз. Двуокись титана
не ядовита, не образует мыла с жирными к-тами свя-
зующего, незначительно растворяется в щелочах, ор-
ганич. и минеральных к-тах, полностью растворяет-
ся при длительном кипячении в смеси конц. серной
к-ты с сульфатом аммония; обладает значительной
фотохпмич. активностью. В результате циклич. вос-
становления и окисления TiO2 под действием света,
воздуха и влаги связующее непрерывно окисляется
(разрушается); при этом покрытие теряет блеск и на-
чинает мелить.
Более светоустойчива и атмосферостойка рутиль-
ная форма. Высокая кроющая способность и интен-
сивность TiO-2 позволяют использовать ее в смеси с
большим количеством (от 25 до 70% от веса смеси)
наполнителя (тальк, бланфикс, ангидрит, каолин,
мел), к-рый сильно уменьшает меление покрытий.
Промышленное значение имеют два метода получе-
ния TiO2: 1) гидролиз р-ров сернокислого титана с
последующим прокаливанием метатитановой к-ты
(для получения рутильной формы применяют спе-
циальные добавки) и 2) гидролиз или сжигание
четыреххлористого титана в парообразном состоянии
(в этом случае всегда получается рутильная форма).
Исходные соли титана получают при обработке сер-
ной к-той или хлорировании концентрата ильменита
(FeTiOs) и реже — титансодержащих шлаков. Дру-
гие титансодержащие минералы (сфен, перовскит) не
нашли пока промышленного применения.
Тонкое измельчение, поверхностная обработка (гид-
ратом окиси алюминия, кремневой к-той, фосфатом
алюминия, органич. поверхностно-активными веще-
ствами) значительно повышают все пигментные
свойства двуокиси титана. Поверхностная обработка
особенно улучшает атмосферостойкость. Двуокись ти-
тана (гл. обр. в рутильной форме) очень широко ис-
пользуют для приготовления всех видов красок и эма-
лей, а также для окраски резины, линолеума, клеен-
ки, пластмасс, бумажной массы.
Цинковые белила — высокодисперсная
окись цинка (ZnO); по внешнему виду представляет
собой белый порошок; форма частиц — от зернистой
до игольчатой. Цинковые белила растворимы в к-тах
и щелочах, не ядовиты, устойчивы к действию света,
сероводорода; с жирными к-тами связующего обра-
зуют мыла. Благодаря активной поверхности цинко-
вые белила легко смачиваются связующим и диспер-
гируются в нем, придают покрытию долго сохраняю-
щийся блеск. Цинковые белила получают по сухому
и мокрому способам.
Сухой способ, в свою очередь, делится на две
группы; 1) косвенный способ — плавление металлич.
цинка, испарение его в муфельных (муфельные бе-
лила) или вращающихся (печные белила) печах и оки-
сление паров цинка кислородом воздуха; 2) прямой
способ — восстановление брикетированного цинк-
содержащего сырья (отходы, концентраты), испаре-
ние образующегося цинка в печах типа Ветерилля с
последующим окислением в специальных камерах. Про-
дукты окисления улавливают рукавными фильтрами.
Мокрый (гидрометаллургический) способ получения
цинковых белил, основанный на прокаливании легко
диссоциирующих тщательно очищенных соединений
цинка (гидроокись, основные карбонат и сульфат),
еще мало используют в пром-сти из-за недостаточной
белизны получаемого пигмента.
Пигментные свойства цинковых белил зависят от
условий их кристаллизации; темп-ры, концентрации
паров цинка и кислорода в процессе окисления. Ат-
мосферная устойчивость цинковых белил определяется
их фотохимии, активностью: чем выше активность,
тем ниже атмосферостойкость. Фотохимии, актив-
ность цинковых белил, полученных различными спо-
собами, понижается в следующем порядке: муфель-
ные, печные, ветерилльные, гидрометаллургические.
Муфельные белила имеют частицы зернистой формы,
обладают высокой степенью белизны и химич. чистоты.
Ветерилльные белила имеют частицы игольчатой фор-
мы; они уступают муфельным по белизне и чистоте
(гл. обр. вследствие примеси соединений свинца).
Цинковые белила широко используют для всех видов
красок и эмалей, в резиновой пром-сти, для окраски
пластич. масс, в парфюмерии, медицине.
Литопон — эквимолекулярная смесь ZnS и
BaSOt с примесью небольших количеств ZnO. Состав
литопона соответствует примерно ф-ле ZnS-BaSOi
(29,4% ZnS и 70,6% BaSO$). Известны марки с со-
держанием от 15 до 60% ZnS. Пигментные свойства
литоиопа определяет сернистый цинк; BaSOt является
наполнителем. Литопон не ядовит, устойчив к дейст-
вию щелочей, недостаточно устойчив к действию света
(темнеет), разлагается к-тами с выделением H2S. Лито-
пон получают совместным осаждением сернистого цин-
ка и сернокислого бария из р-ров тщательно очищенно-
го сернокислого цинка и сернистого бария. Промытый
осадок прокаливают при 700—750°. Для повышения
светостойкости в процессе синтеза вводят добавки
солей кобальта. Литопон широко используют для всех
видов белых и цветных масляных и эмалевых красок
для внутренних покрытий, в эмульсионных красках,
в произ-ве пластмасс, клеенки, линолеума и др.
Свинцовые белила — основной карбо-
нат свинца 2РЬСО3-РЬ(ОН)2. Свинцовые белила рас-
творяются в к-тах и щелочах, начинают разлагаться
ок. 150° и полностью разлагаются при 415°; устой-
чивы к воздействию света и влаги. Получают их обыч-
но карбонизацией р-ров основного ацетата свинца
1017
ПИГМЕНТЫ
1018
углекислым газом. Из-за ядовитости и высокой чув-
ствительности к H2S свинцовые белила применяют
только для окраски судов и в художественных
красках.
Пигменты черные. К черным П. относятся сажа
(имеет наибольшее значение), черни и черные железо-
окисные П.
Черни — продукты, полученные при прокали-
вании без доступа воздуха различных твердых орга-
нич. веществ растительного и животного происхожде-
ния. Черни растительного происхождения — «вино-
градная чернь» (из отходов виноделия) и «персиковая
чернь» (из скорлупы орехов) — используют только
для художественных красок: их коэфф, отражения
3,2—3,5%, маслоемкость 1-го рода 37—70. Черни из
костей животных — «жженая кость» (лучшие сорта—
«слоновая кость») —имеют коэфф, отражения 1,7—
2,0%, маслоемкость 1-го рода 50- 60.
Черный железоокисный П. по химич.
составу представляет собой закись-окпсь железа
Fe50i с примесью небольшого количества окиси же-
леза (общее содержание окиси железа 72—74%). Очень
интенсивный и укрывистый черный П., растворим
в к-тах, устойчив к слабым щелочам, свету, атмосфер-
ным воздействиям. При 150—170° начинает окислять-
ся, превращаясь в красную окись. Черный железо-
окисный П.получают окислением:!) свежеосажденного
гидрата закиси железа кислородом воздуха при 90°
и 2) металлич. железа ароматич. нитросоединениями
в р-ре солей железа, обычно хлористого. Применяют
черные железоокисные П. для нек-рых грунтовочных
и покровных красок.
В состав термостойкого (до 800°) чер-
ного ж ел езоок йен ого П. входят: 40%
FeO, 30% СнО и 30% МпО. Получают прокаливанием
смеси осажденных гидратов железа, меди и марганца
при 550—600°; используют для изготовления термо-
стойких эмалей с минимальным светоотражеиием.
Пигменты желтые, оранжевые, красные и коричне-
вые. Группу этих П. составляют: 1) кроны — свинцо-
вые (лимонный, желтый, оранжевый, красный), свин-
цово-молибдатные, цинковые и др.; 2) кадмиевые П.
(желтый и красный); 3) киноварь; 4) железоокисные
П. синтетич. и природные (желтые, красные, корич-
невые).
Желтый кадмий представляет собой серни-
стый кадмий CdS, встречающийся в природе в виде
минерала гринокита. Искусственный CdS имеет в за-
висимости от размера и формы частиц, наличия при-
месей и др. факторов насыщенный цвет от лимонно-
желтого до оранжевого. Наибольшее влияние на
цвет П. оказывает примесь ZnS. Отдельные сорта П.
имеют следующий состав: лимонный—CdS-0,4ZnS,
светло-желтый — CdS-0,2ZnS, золотисто-желтый —
CdS. Желтый кадмий неядовит, отличается высокой
интенсивностью, атмосферостойкостью, стойкостью
к действию света и высоких темп-р; нерастворим в ще-
лочах и разб. соляной и серной к-тах, хорошо раство-
ряется в конц. к-тах и в разб. HNOs- Желтый кадмий
получают взаимодействием в р-ре солей кадмия с
сульфидами, а также прокаливанием окиси или легко
диссоциирующих солей кадмия с серой при 500—600°.
Желтый кадмиевый П. выпускают также в виде
кадмопона — эквимолекулярной смеси желтого
кадмия с бланфиксом, получаемой совместным их
осаждением пли механич. смешением. Из-за дефицит-
ности и высокой стоимости желтые кадмиевые П.
применяют только в произ-ве художественных и по-
лиграфии. красок и для окраски стекол.
Красный кадмий — твердый раствор CdS
и CdSe, в к-ром соотношение между компонентами
может изменяться в широких пределах. Общую ф-лу
этого П. можно выразить в виде CdS nCdSe. Цвет
П. изменяется от оранжевого до пурпурно-красного.
Выпускают кадмий оранжевый (n=s:0,2), светло-крас-
ный (п==О,35), темно-красный (п==0,4) и пурпурный
(n=s0,5). Красный кадмий — самый насыщенный и
прочный красный минеральный П. В уксусной и ми-
неральных к-тах он не растворяется, устойчив к дей-
ствию света, нагреванию до 700°, атмосферостоек.
Получают его прокаливанием: 1) смеси серы и селена
с сульфатом или карбонатом кадмия и 2) осажден-
ной смеси CdS и CdSe. Красные кадмиевые П. выпус-
кают также в виде красных кадмопоно в—
эквимолекулярных смесей красного кадмия с блан-
фиксом. Чистый красный кадмий и кадмопон вслед-
ствие дороговизны применяют преимущественно для
произ-ва художественных красок и в произ-ве ру-
биновых стекол.
Киноварь по химич. составу представляет со-
бой HgS; встречается в природе в виде минерала. Ис-
кусственная киноварь отличается ярким красным
цветом (от желтоватого до синеватого оттенка). Раз-
личие оттенков определяется гл. обр. дисперсностью
П. (светлый имеет частицы размером 2—5 мк, темный—
5—20 мк), к-рая зависит от состава используемых при
синтезе сульфидов щелочных металлов, температур-
ного режима и др. Киноварь отличается высокой ин-
тенсивностью и укрывистостью. Устойчива к дейст-
вию к-т и щелочей, но недостаточно свето- и термостой-
ка. Киноварь получают возгонкой черной сернистой
ртути (сухой способ) или обработкой чистой ртути
полисульфидами щелочных металлов (мокрый спо-
соб); применяют ее исключительно для произ-ва ху-
дожественных красок.
Желтые железоокисные П. представ-
ляют собой гидраты окиси железа, к-рые в зависимо-
сти от условий образования имеют различные сте-
пень гидратации, дисперсность и, следовательно,
различные оттенки и пигментные свойства. Наиболь-
шее значение имеет желтая окись ж е л е-
з a (FeOOH, пли Ре2Оз-Н2О), кристаллизующаяся
в ромбич. системе (модификация гетита). Этот П. ра-
створим в минеральных к-тах, устойчив к действию
щелочей, света и атмосферных влияний. При 180—
200° начинает терять гидратную воду и переходит
в красную окись железа. Желтую окись железа полу-
чают окислением металлич. железа, погруженного
в р-р хлористого или сернокислого железа. В каче-
стве окислителей используют кислород воздуха или
ароматич. нитросоединения (напр., нитробензол).
Желтую окись железа применяют для приготовления
масляных, нитролаковых, клеевых и др. красок, для
окраски цемента и др. строительных материалов.
В очень больших количествах этот П. используют
для приготовления искусственной охры.
Красный железоокисный П. — чистая
окись железа Fe20s, кристаллизующаяся в гексаго-
нальной системе. В зависимости от размера и формы
частиц его цвет может быть от оранжевого до фиоле-
тово-красного. Частицы светлых оттенков имеют пла-
стинчатую форму и размер 0,3—0,5 мк, а темных то-
нов — зернистую форму и размер 0,8—1,5 мк. Крас-
ный железоокисный П. устойчив к действию щелочей,
слабых к-т, высоких темп-р, солнечного света и к ат-
мосферным влияниям. Получают его прокаливанием:
1) железного купороса при 700—750° и 2) осажденной
желтой окиси или закись-окиси железа при 600—700°.
Более светлые тона получают из предварительно обез-
воженного (до моногидрата) железного купороса в
присутствии углеродсодержащих добавок. Красный
железоокисный П. применяют для произ-ва всех ви-
дов красок и эмалей, окраски цемента и др. строитель-
ных материалов.
Коричневые железоокисные П.
представляют собой смеси красной (85—93%) и
1019
ПИГМЕНТЫ
1020
черной (6—14%) окиси железа. Их свойства близки к
вышеописанным железоокисным пигментам.
Охры — природный кристаллич. гидрат окиси
железа с примесыо большего или меньшего количе-
ства глины. По цвету охры делят на светло-желтые
(12—25% Ре20з), средне-желтые (25—40% Fe2O5)
и золотисто-желтые (40—75% Fe2O3). Охры ус-
тойчивы к действию слабых к-т, щелочей, солнеч-
ного света, атмосферных влияний; при нагревании до
150° начинают темнеть. Охры наиболее дешевые и
прочные П., вследствие чего их широко применяют
для всех видов окрасок (известковых, клеевых, мас-
ляных), а также для произ-ва шпаклевок и грунтовок.
Они имеют большое значение и в произ-ве художест-
венных красок.
Сиены — светло-коричневые природные П.,
отличающиеся от охр большим содержанием гидро-
окиси железа, почти полным отсутствием глины и на-
личием примесей кремнезема и окиси марганца. По
своим пигментным свойствам сиены близки к охрам,
за исключением повышенной лессирующей способ-
ности в масляных связующих, а также более темного
коричневого оттенка. Сиены применяют преим. в ка-
честве лессирующего П. в произ-ве художественных
и типолитографских красок, а также красок по де-
реву.
Мумия — красный природный П., содержащий
20—70% Ре20з. Мумии делят на светлые (20—35%
Fe20a) и темные (35—70% Fe20a). Мумии отличаются
очень высокой устойчивостью к действию света и
корродирующих агентов; атмосферостойки, с трудом
растворяются в азотной и серной к-тах; полностью
растворяются при кипячении в соляной к-те. Мумии
получают обжигом болотных руд, высокожелезистых
бокситов и охристых разновидностей гидрогематитов.
Применяют их для приготовления красок и эмалей
всех типов, а также грунтовок.
Железный сурик — темный вишнево-крас-
ный природный П. (нек-рые сорта обладают ярким
желтовато-красным цветом), по химич. составу он
представляет собой окись железа с примесью неболь-
ших количеств глинистых веществ и кварца. Содержа-
ние окиси железа в сурике находится в пределах 75—
95%, т. е. выше, чем у других природных П. Желез-
ный сурик стоек к действию света, корродирующих
агентов, щелочей и слабых к-т, атмосферостоек. Ра-
створяется при кипячении в конц. соляной к-те. В ка-
честве железного сурика используют железные руды,
богатые окисью железа, гл. обр. красные железняки.
Нек-рые гематитовые руды дают красные П. очень
большой чистоты. К таким П. относятся персид-
ская красная (75% Fe2O3) и испанская
красная (85% Fe20s). Железный сурик приме-
няют очень широко для всех видов окрасочных работ
(как покрывных, так и грунтовочных) и со всеми
связующими — водными, масляпымп, нитролаками
И др.
Умбра — коричневый природный П., образует-
ся в результате выветривания железных руд, содер-
жащих марганец. Различают умбру двух видов — на-
туральную и жженую. По составу натуральная умбра
близка охре, от к-рой отличается высоким содержа-
нием марганца (до 16% в пересчете на МпО2). Марга-
нец в умбре может находиться в виде окиси, гидрата
окиси или двуокиси. Умбра устойчива к действию ще-
лочей, света, прп нагревании приобретает более тем-
ный оттенок. Ее применяют для произ-ва полиграфия.,
масляных, фасадных, силикатных и художественных
красок.
Пигменты синие, зеленые п фиолетовые. Группу
этих П. составляют железная лазурь (милори), уль-
трамарин, смешанные зеленые, хромовые, кобальто-
вые, медные и марганцовые П.
Ультрамарин. Произ-во искусственного
ультрамарина было начато в 30-х гг. 19 в. До этого в
качестве синего П. применяли натуральный ультрама-
рин, к-рый получали из очень дорогого минерала —
ляпис-лазури. Искусственный ультрамарин представ-
ляет собой алюмосиликат натрия, содержащий серу;
в зависимости от условий термич. обработки ис-
ходных компонентов, а также соотношения в них
SiO2: А120з и Na:S состав и цвет ультрамарина могут
колебаться в значительных пределах. Различают виды
ультрамарина: 1) малосернистый и малокремнистый
примерного состава Na8Al,Si6S2O24 (зеленый) и
Na,AleSieS2O24 (синий) и 2) многосернистый и мно-
гокремнистый примерного состава NaeA14SieS4O24 (си-
ний), Na5Al4Si6S4O24 (фиолетовый) и Na3Al4Si6S4HsO2J
(красный). Известны условия перехода ультрамарина
из одного вида в другой. Окраска ультрамарина оп-
ределяется химич. составом, строением кристаллич.
решетки и характером связи в ней между атомами
Na и S. Ультрамарин неядовит, устойчив к действию
света, воздуха, высоких темп-р, щелочей, но легко
разлагается даже органич. к-тами с выделением H2S.
Ультрамарин отличается насыщенным цветом, обла-
дает лессирующей способностью и высокой ивтен-
сивностью. Последняя увеличивается с повышением
дисперсности П. В зависимости от качества и назна-
чения ультрамарин делят на пять марок: УХ К—для
произ-ва художественных красок, УС — для сахар-
ного произ-ва, УМ-1 — для произ-ва масляных
красок, УМ-2 и УМ-3 — для малярных работ.
Ультрамарин получают содово-серным способом.
Шихту, состоящую из каолина, соды, серы и угля или
другого восстановителя (кам.-уг. пека, канифоли и
т. п.), обжигают в шамотных тиглях вместимостью
3—7 кг или в муфелях емкостью ~1 т. К каолину ча-
сто добавляют кремневую кислоту в виде молотого
кварца или инфузорной земли. Практич. значение
имеет только синий ультрамарин примерного состава
NaeAl4SieS4O24. Его применяют для произ-ва разнооб-
разных колерных масляных красок и эмалей, окраски
линолеума, резины, цементных плиток, подсинивания
сахара, белья, бумаги и т. д.
Смешанные зеленые П. (зелени)
представляют собой механические смеси желтых и
синих П.
Свинцовая зелень — смесь желтого
свинцового крона с лазурью, получаемая механич.
смешением или (реже) совместным осаждением. Свин-
цовая зелень имеет яркий, насыщенный цвет, стойка
к атмосферным воздействиям, обладает хорошей ук-
рывистостью. Недостатки этого П.: невысокая свето-
стойкость, чувствительность к действию сернистых
соединений, склонность к расслаиванию в эмалях
(при хранении) и в пленке (при высыхании). Свин-
цовую зелень применяют обычно с большим количе-
ством наполнителя для произ-ва всех видов масляных
и эмалевых красок, в полиграфия, и др. отраслях
пром-сти.
Цинковая зелень — смесь цинкового кро-
на с железной лазурью. Цинковая зелень чище, ярче
и более светостойка, чем свинцовая зелень, но менее
укрывиста.
Окись хрома Сг20з — оливково-зеленый П.,
обладает высокой стойкостью к действию кислот,
щелочей, света, высокой темп-ры и агрессивных га-
зов (H2S,SO2), атмосферостоек. Получают окись хрсма
прокаливанием хромпика в присутствии восстанови-
теля (сера, древесный уголь и др.). Большое влияние
па малярно-технич. свойства П. оказывает темп-ра
прокаливания. Окись хрома применяют обычно в сме-
си с наполнителем (до 70—75% от веса смеси) для
изготовления художественных красок, для печати
денежных знаков, а также для живописи по фарфору.
1021
ПИГМЕНТЫ
1022
Изумрудная зелень — гидрат окиси хро-
ма Сг2О,-п112О (я=1—2), состоящий из частиц более
крупных (1—10 мк) и уплотненных, чем частицы обыч-
ного гидрата. Кроме того, в состав изумрудной зелени
входит борный ангидрид (в обычных сортах его
содержание составляет 4—9%, в хороших 1—2%).
Изумрудная зелень — красивый лессирующий П.,
отличается высокой атмосферной и химич. устойчи-
востью; однако он менее термостоек, чем окись хрома.
Изумрудную зелень получают прокаливанием смеси
хромпика с борной к-той при 500—600°; используют
для произ-ва художественных красок и при всех ви-
дах окрасочных работ — клеевых, масляных, изве-
стковых, фасадных.
Синий кобальт (с и и ь Т е н а р а) —
алюминат кобальта СоА12О4 (кристаллич. продукт
с решеткой шпинели); обычно, помимо алюмината, си-
ний кобальт содержит небольшие количества свобод-
ной окиси алюминия, а также зеленого кобальта
CoO-nZnO и фиолетового кобальта Со3(РО4)2. Синий
кобальт — термо- и атмосферостойкий П., устойчи-
вый к действию света, агрессивных газов; нераство-
рим в щелочах и к-тах (кроме горячей НС1). Полу-
чают синий кобальт при совместном прокаливании
Предварительно обезвоженной смеси солей алюмивия
и кобальта с добавкой (для улучшения цвета) солей
цинка и фосфорной к-ты при 1200—1250°. Приме-
няют его исключительно для произ-ва художествен-
ных красок.
Зеленый кобальт (зелень Ринма-
н а) — твердый р-р закиси кобальта и окиси цинка;
его химич. состав может быть представлен общей
ф-лой CoO-nZnO.
Цвет П. изменяется от светло- (п==50) до темно-зеле-
ного (п=^15). Зеленый кобальт атмосферостоек, устой-
чив к действию света, высоких темп-р; растворим
в кипящих минеральных к-тах; разлагается щелоча-
ми. Получают его при совместном прокаливании солей
кобальта с цинковыми белилами при 1000—1100°; при-
меняют преим. для производства художественных
красок.
Церулеум (небесно-голубой) —
ортостанпат кобальта Co2SnO4; обычно в его состав
входят также двуокись олова, алюминат кобальта (си-
ний кобальт), а иногда окиси алюминия и магния. Со-
став церулеума может быть представлен общей ф-лой
СоО-zSnOa-yAhOs-MgO. П. устойчив к действию ще-
лочей, света, растворяется в к-тах при нагревании, ат-
мосферостоек. Получают его совместным осаждением
оловянной к-ты и гидроокисей кобальта, алюминия и
магния и прокаливанием осадка при 1100—1250°.
Фиолетовый кобальт. Под этим назва-
нием известны два П.: темно-фиолетовый кобальт,
представляющий собой безводную соль фосфата ко-
бальта Со3(РО4)2, и светло-фиолетовый кобальт, яв-
ляющийся одноводной солью фосфата кобальт-аммо-
ния CoNH4PO4'H2O. П. темного оттенка стоек по отно-
шению к высоким темп-рам, атмосферным воздейст-
виям и свету; растворяется в к-тах н разлагается ще-
лочами. Получают его прокаливанием (900—1100°)
восьмиводного фосфата кобальта, образующегося при
взаимодействии соли кобальта с фосфатом натрия.
Темно-фиолетовый кобальт относится к полулесси-
рующим П. и применяется исключительно для произ-
водства художественных красок. Светло-фиолетовый
кобальт очень чувствителен к нагреванию (уже при
темп-ре до 100° заметно изменяет свой цвет); в к-тах
растворяется, щелочами разлагается. Получают свет-
ло-фиолетовый кобальт взаимодействием сульфата
кобальта с фосфатом аммония или смесью двузамещен-
ного фосфата аммония и аммиака. Полученный оса-
док промывают водой, а затем высушивают при 40—
50°. П. светлого оттевка обладает лессирующей спо-
собностью. Его применяют для произ-ва как масля-
ных, так и акварельных красок.
Медянка — яркий зеленовато-сипий П., по хи-
мич. составу представляющий собой основную уксус-
нокислую соль меди Cu(CH3COO)2-nCu(OH)2mH2O.
П. частично растворим в воде, разлагается при
нагревании (150—250°). Получают медянку обработ-
кой гидрата окиси меди уксусвой к-той. Медявка
имеет низкие пигментные свойства: ее укрывистость
и интенсивность незначительны, состоит она из мел-
ких кристаллич. игл, трудно поддающихся перетира-
нию. Применяют этот П. только в смеси со свинцо-
выми белилами для изготовления атмосферостойкой
масляной краски.
Зелень Шеелепо химич. составу представ-
ляет собой основную мышьяковистокислую соль меди
Cu(AsO2)2-«Cu(OH)2.mH2O(n = l—3). Цвет . П., в
зависимости от основности, колеблется от светло- до
темно-зеленого. Зелень Шееле — ядовитый П., имею-
щий очень ограниченное применение.
Швейнфуртская зелень — очень яркий
зеленый П. Cu(CH3COO)2-3Cu(AsO2)2, устойчивый к
атмосферным воздействиям; имеет низкие укрыви-
стость и интенсивность. Ее получают взаимодейст-
вием медного купороса, мышьяковистокислого нат-
рия и уксусной к-ты; применяют для произ-ва масля-
ных и акварельных красок.
К природным медным П. относят гор-
ную зелень CuCO3-nCu(OH)2 (этот П. мож-
но получать и искусственно), минералы мала-
хит CuCO3-Cu(OH)2 (зеленого пвета) и азурит
2CuCO3Cu(OH)2 (синего цвета). Природные медные П.
используют для отделочных работ.
Марганцовая голубая — изоморфная
смесь манганата и сульфата бария (10—16 молей суль-
фата на 1 моль манганата). Марганцовая голубая об-
ладает высокой свето- и атмосферостойкостью; ус-
тойчива к щелочам; сравнительно легко разлагается
к-тами и водными р-рами сульфатов (исключение со-
ставляет конц. соляная к-та). Марганцовая голубая—
лессирующий П. с низкой интенсивностью. Получают
прокаливанием смеси двуокиси марганца, нитрата и
сульфата бария при 750—800°; используют для
произ-ва художественных красок и для окраски
портландцемента.
Марганцовая фиолетовая — двой-
ная соль фосфатов марганца и аммония примерного
состава MnPO4-NH4H2PO4. ЭтотП. обладает красивым
красно-фиолетовым цветом, очень высокой светостой-
костью, лессирующей способностью. Получают мар-
ганцовую фиолетовую прокаливанием солей марган-
ца, фосфорной к-ты и аммония при 300—320"; приме-
няют для произ-ва художественных красок и для под-
цветки двуокиси титана.
Марганцовая зеленая — основной ман-
ганат бария ВаМпО4-пВа(ОН)2 (п=1—2,5) насыщен-
ного темно-зеленого цвета. П. устойчив к щелочам;
разрушается даже слабыми кислотами, напр. уголь-
ной. Получают марганцовую зеленую прокаливанием
карбоната марганца с нитратом бария при 650—
750°; применяют для произ-ва художественных красок.
Пигменты антикоррозионные. В группу антикорро-
зионных объединены П., для к-рых основной характе-
ристикой являются не цвет, а антикоррозионные
свойства. Эти П. применяют обычно в грунтах и по-
крытиях, имеющих гл. обр. защитное, а не декоратив-
ное назначение. Важнейшие антикоррозионные II.:
свинцовые (глет, сурик, основные карбонат, силикат
и сульфаты свинца, цианамид свинца, плюмбат каль-
ция), хромовые [хроматы цинка, свинца, бария,
стронция (см. Кроны), кальция, силикохромат свинца,
фосфат хрома] и металлич. порошки и пудры (алюми-
ний, цинк, свинец, медь, железо, никель).
1023
ПИГМЕНТЫ
1024
Свойства нек-рых антикоррозионных П. приве-
дены в табл. 2.
Таблица 2
Пигменты Плотн., г 1см3 Насыпной вес, г/л Укрыви- стость, г[М2 Маслоем- кость 1-го рода
Сурик свинцо- вый9 . . . ► 8,6 2050- 20—25
Основные суль- фаты свинца 6,3—6,4 2250 10-14
Основной сили- кат свинца . . 4,0 -15
Цианамид свин- ца^ 6,0—6,2 800—1 100 50—60 18-24
Плюмбат каль- ция 5,7 19
Основной си- ликохромат свинца . . . 4.1 900 55—65 30-45
Фосфат хрома® 2,37 500-700 100—120 70 — 100
Алюминиевая пудра .... 2,54 — 600 10
2.55
а Размер частиц 1,5—20 лог. ® Размер частиц
0,5—2 лк. в Уд. поверхность 5—10 м2/г.
Г лет— окись свинца РЬО. Встречается в при-
роде в виде двух модификаций: тетрагональной (крас-
ный цвет, плотн. 9,35) и ромбической (желтый цвет,
плотн. 9,63). Переход одной модификации в другую
происходит при 489°. Ниже этой темп-ры термодина-
мически устойчива тетрагональная модификация, но
перекристаллизация практически не идет. В водной
среде ромбич. форма глета более реакционноспособна.
Глет получают окислением расплавленного свинца:
тетрагональный — при 480°, ромбический — при 500°
и выше. Технич. глет (глет-сырец, зеленый глет), полу-
чаемый после первичного обжига, содержит до 20%
неокисленного РЬ. После вторичного обжига содержа-
ние свинца в глете составляет ок. 1%. В качестве П.
глет практически не применяют. Его используют гл.
обр. для произ-ва сурика, свинцовых кронов, белил,
сиккативов, хрусталя, в керамич. пром-сти для изго-
товления глазурей, для получения оптич. стекол,
различных замазок.
Сурик свинцовый — смешанный окисел
состава 2РЬ0-РЬО2(РЬзО4) или свинцовая соль орто-
свинцовой к-ты. П. окрашен в пурпурно-красный или
оранжевый цвет. При нагревании сурик разлагается
РЬзО4^?ЗРЬО-|-0,5 О2; под действием света и серни-
стых газов он сереет. Покрытия на сурике отли-
чаются стойкостью к атмосферным воздействиям,
высокой адгезией к стали. Практически сурик содер-
жит не 2 моля РЬО на 1 моль РЬО2, а несколько боль-
ше. Качество сурика тем выше, чем этот избыток
меньше, т. к. свободный глет, реагируя со свободны-
ми жирными к-тами олифы, вызывает ее загустевание.
Для малярных работ рекомендуется незагустеваю-
щий высокодисперсный сурик с большим содержа-
нием РЬО2 (30—33%). Получают сурик: 1) окислением
глета воздухом в печах с механич. перемешиванием
при 450—500° и давлении от 1 до 30 ат; 2) окислением
паров свинца, образующихся в электрич. дуге, чи-
стым кислородом или воздухом, обогащенным кисло-
родом. Применяют сурик для произ-ва грунтов и ан-
тикоррозионных красок по железу. Окраску ответст-
венных стальных конструкций, мостов, портовых соо-
ружений, подводной части судов и др., как правило,
производят по грунту, содержащему сурик. Широко
используют сурик в оптич. и радиотехнич. пром-сти.
Основные сульфаты свинца — П.
белого цвета следующего состава: PbSO4-PbO и
PbSO4-3PbO-H2O. Их используют для повышения ат-
мосферной устойчивости покрытий на основе свинцо-
вых или цинковых белил.
Основной силикат свинца 2РЬ0-
• SiO2 — П. белого цвета; используют в композиции
с другими антикоррозионными П.
Цианамид свинца PbCN2 — П. ярко-жел-
того цвета с частицами игольчатой формы; устойчив
к действию света; атмосферостоек. Высокие антикор-
розионные свойства цианамида свинца обусловлены
его способностью к мылообразованию и к щелочному
пассивированию. Цианамид свинца получают взаи-
модействием свинцовых солей с цианамидом Са или
Na. Применяют для грунтовочных и наружных пок-
рытий при окраске мостов, железнодорожного и мор-
ского транспорта и др.
Плюмбат кальция 2СаОРЬО2— П.
бледно-кремового цвета, обладающий высокой укры-
вистостью и стойкостью к атмосферным воздействиям.
Получают этот П. прокаливанием смеси гидроокиси
или карбоната кальция с глетом в окислительной ат-
мосфере при 700°. Плюмбат кальция применяют в чи-
стом виде или с наполнителями для производства
грунтов, антикоррозионных красок.
Основной силико-хромат свин-
ца — П. оранжевого цвета; содержит ядро двуокиси
кремния, покрытое активной оболочкой из хромата и
силикатов свинца различной основности. В состав П.
входят SiO2 (47%), РЬО (47%), СгОз (5,5%). Высокие
антикоррозионные свойства этого П. обусловлены
сочетанием стабилизирующих свойств основного си-
ликата свинца (связывает кислые продукты разложе-
ния связующего) и пассивирующих свойств хромата
свинца. Получают основной силико-хромат свинца
осаждением хромата свинца в присутствии глета и
измельченного диатомита или песка и последующим
прокаливанием осадка при 620—650° для образования
основных силикатов свинца. Применяют для произ-ва
грунтов и красок на различных связующих (масляных,
алкидных, виниловых, эпоксидных, полиамидных,
латексных), к-рые используют для защиты судовых
днищ, мостов, подводных опор и конструкций и др.
Хромат кальция СаСгО4 — желтый (с
зеленоватым оттенком), термостойкий (до 1000°) П.;
хорошо растворим в воде (22,3 г/л при 20°); его пасси-
вирующие свойства обусловлены большим содержа-
нием ионов СгО* в водной вытяжке и высоким значе-
нием ее pH (8,0—9,0). Получают хромат кальция
взаимодействием хромата натрия с хлористым каль-
цием или хромового ангидрида с окисью кальция с
последующим обезвоживанием полученного продукта
при — 600°. Применяют этот П. в грунтах для за-
щиты металлов, особенно магниевых сплавов.
Фосфат хрома СгРО4-ЗН2О — высокодис-
персный П., цвет к-рого в зависимости от дисперс-
ности изменяется от светло-зеленого до зеленого. Фос-
фат хрома "растворим при нагревании во всех мине-
ральных к-тах, кроме фосфорной, устойчив к дейст-
вию слабых щелочей, света, атмосферостоек. Полу-
чают взаимодействием р-ров бихромата Na и фос-
форной к-ты в присутствии восстановителя — смеси
бисульфита и сульфита Na. Продукт сушат при 105°.
Применяют фосфат хрома для изготовления фосфа-
тирующих грунтов на поливинилбутирале и одно-
упаковочного активного (с тетраоксихроматом цинка)
грунта для защиты корпусов кораблей, самолетов,
автомобилей, железнодорожного транспорта, мос-
тов и др.
Металлические П. используют в виде
порошков (частицы шарообразной или неправиль-
ной формы) и пудр (частицы формы чешуек).
Алюминиевая пудра — наиболее рас-
пространенный металлич. П., имеющий серебристо-
1025
ПИГМЕНТЫ—ПИ-КОМПЛЕКСЫ
1026
серый цвет. Размер частиц П.: диаметр 50—100 мк,
толщина 0,1—1,75 jmk. Алюминиевая пудра раствори-
ма в к-тах и щелочах, реагирует с водой, может взры-
ваться в смеси с воздухом. Высокие защитные свой-
ства алюминиевой пудры обусловлены: 1) способно-
стью к листованию — образованию чешуек, к-рые
всплывают в лакокрасочной пленке, образуя защит-
ный металлич. панцирь; 2) непроницаемостью для
ультрафиолетовых и инфракрасных лучей. В чешуй-
чатой форме широко используют также медные
и свинцовые металлич. П., характеризующие-
ся такими же размерами частиц, как у алюминиевой
пудры.
Современный способ получения чешуйчатых метал-
лических П. заключается в тонком размоле предва-
рительно измельченных металлов в органич. раство-
рителях в присутствии 3—4% стеариновой к-ты.
Последняя, смазывая поверхность, не только облег-
чает размол и предотвращает слипание чешуек, но
изменяет характер их поверхности, придает пудрам
способность к всплыванию в покрытиях. Далее пуд-
ры полируют (трением одной чешуйки о другую) в
специальных машинах и выдерживают до употребле-
ния! 5—20 дней для ориентации молекул смазки на
поверхности чешуек.
Порошки Al, Zn, Pb обычно получают рас-
пылением жидких металлов струей газа (метод «ато-
мизации»), а порошки Fe, Си, Ni — восстановлением
их окислов водородом, вытеснением из солей, измель-
чением катодных отложений при электролизе соответ-
ствующих солей.
Алюминиевую пудру используют для окраски же-
лезнодорожных цистерн, вагонов-холодильников,
оболочек дирижаблей и др. Цинковая, свинцовая
и др. пыль используются в протекторных грунтах и
покрытиях для судов, мостов и др.
Лит.: Беленький Е. Ф., Р и с к и н И. В., Химин
и технология пигментов, 3 изд., М., 1960; Якубович С. В.,
Испытания лакокрасочных материалов и покрытий, М.—Л.,
1952; Пэйн Г. Ф., Технология органических покрытий,
пер. с англ., т. 2, Л., 1963; Pigmente, 3 Aufl., hrsg. v. H. Kit-
tel, Stuttgart, 1960; Шампетье Г., Рабата Г., Химия
лаков, красок и пигментов, пер. с франц., т. 2, М., 1962;
Лакокрасочные материалы. Сырье и полупродукты. Справоч-
ник, М., 1961. М. А. Штерн, Г. Н. Горелик.
ПИГМЕНТЫ (биологические) — окрашен-
ные вещества, содержащиеся в тканях животных и
растений. Химич, природа и биологич. роль П. раз-
личны.
Обширную группу П. составляют хромопротеи-
ды — сложные белки, содержащие окрашенные про-
стетич. группы. Важнейшими из них являются ок-
рашенные в красный цвет пигменты крови — гемогло-
бины, осуществляющие доставку кислорода от орга-
нов дыхайия к тканям. Весьма близок к гемоглобину
хромопротеид мышц — миоглобин, обеспечи-
вающий накопление в мышцах резервных запасов
кислорода. К хромопротеидам, содержащим железо-
порфирин, относятся, кроме гемоглобина и миогло-
бина, такие важнейшие окислительно-восстанови-
тельные ферменты, как каталаза и пероксидазы, а
также различные цитохромы растительных и живот-
ных тканей, осуществляющие перенос электронов к
кислороду на завершающих этапах биологич. окисле-
ния. В крови и лимфе беспозвоночных, в частности
головоногих моллюсков и ракообразных, присутст-
вуют окрашенные в темно-голубой или зеленовато-
синий цвет гемоцианины, осуществляющие
у этих животных функцию, аналогичную функции
гемоглобина. В состав простетич. группы этих хромо-
протеидов входит медь. Кровь нек-рых кольчатых
червей содержит зеленый пигмент — хлорокру-
о р и н.
Особую группу П. животного происхождения сос-
тавляют желчные пигменты — гетероциклич. тетра-
пиррольные соединения, образующиеся в печени и
селезенке в результате распада гема гемоглобина.
Важнейшими из них являются зеленый пигмент
биливердин и окрашенный в желто-коричневый цвет
билирубин, выделяемый с желчью в пищеварительный
тракт. Из билирубина образуются пигменты кала—
стеркобилиноген и стеркобилин,
а также пигменты мочи — уробилиноген и
уробилин.
Обширную группу составляют П. растительного
происхождения. К ним относится зеленый пигмент
хлорофилл, принимающий непосредственное участие в
осуществлении процессов фотосинтеза. Помимо хло-
рофилла, в растительных тканях присутствуют мно-
гочисленные П., относящиеся к группе каротинои-
дов (а-, Р-, у-каротины, ксантофилл, криптоксантин
и др.). Нек-рые из них обладают способностью по-
глощать энергию солнечного света и передавать ее
на хлорофилл. Каротины имеют важное значение
также в связи с тем, что они являются источником
образования в организме внтамнпа А (см. Ретинол)
и зрительного пурпура — родопсина. Красные водо-
росли обязаны своей окраской пигменту фико-
эритрину, к-рый, подобно хлорофиллу, активно
участвует в фотосинтезе.
К растительным П. относятся также антоцианины,
обеспечивающие образование розовой, красной, си-
ней и фиолетовой окраски цветов и др. органов рас-
тений, а также флавоны, окрашивающие раститель-
ные ткани в желтые цвета. Окраска наружных по-
кровов (кожи, волос и перьев) у животных обуслов-
лена присутствием меланинов — пигментов темно-
коричневого или черного цвета. В состав чешуек
крыльев бабочек входят белые, желтые и красные
пигменты, являющиеся производными птерина', его
производным является также окрашенная в желтый
цвет фолиевая кислота. См. также Красящие вещества
природные.
Лит.: Фердман Д. Л., Биохимия, 2 изд., М., 1962;
Гудвин Т., Сравнительная биохимия каротиноидов, пер.
с англ., М., 1954; Lemberg R., Legge J. W., Hematin
compounds and bile pigments, N. Y.—L., 1949.
В. Б. Спиричев.
ПИГМЕНТЫ БЕЛЫЕ — двуокись титана, цин-
ковые и свинцовые белила, литопон и др. Подробнее
см. Пигменты.
ПИГМЕНТЫ ЖЕЛЕЗООКИСНЫЕ — П., окраска
к-рых обусловлена присутствием в них одного из
окислов железа. Между химич. составом и цветом
П. ж. существует следующая зависимость: желтые П.
окрашены гидратами окиси железа, красные —
окисью железа, черные — закисью-окисью железа.
Подробнее см. Пигменты.
ПИГМЕНТЫ КАДМИЕВЫЕ — неорганич. соеди-
нения кадмия, применяемые для приготовления кра-
сок, преимущественно художественных. Выпускаются
П. к. двух видов: 1) чистые пигменты желтого (CdS)
и красного (CdS-raCdSe) цветов и 2) кадмопоны (эк-
вимолекулярные смеси желтого или красного кадмие-
вого пигмента с BaSOa) тех же цветов. Подробнее см.
Пигменты.
ПИГМЕНТЫ ОРГАНИЧЕСКИЕ — см. Красители
синтетическ ие.
ПИКОЛИНОВАЯ КИСЛОТА — см. Пиридинкар-
боновые кислоты.
ПИКОЛИНЫ — см. Метилпиридины.
ПИ-КОМПЛЕКСЫ (л-комплексы) — комплексы
или соединения, в к-рых связь между акцептором
электронов (катионом, металлом, солью металла) и
донором электронов (непредельным или ароматич.
соединением) осуществляется за счет (или с участи-
ем) л-электронов донорной группы. К П.-к. относятся
разнообразные типы органич. соединений, многие из
к-рых стали известны в последнее время и интенсивно
О33 к.х.э. 1. з
1027
ПИ-КОМПЛЕКСЫ
1028
исследуются. Понятие «л-комплекс» не очень опре-
деленно; нет жесткой границы между обычными не-
органич. комплексными соединениями, П.-к. и орга-
нич. соединениями с чистой ковалентной связью.
К П.-к. можно отнести след, типы соединений:
1) комплексы солей металлов (Pt, Cu, Ag, Pd и др.)
с олефинами и алкинами, напр. [Р1(КНд)(СгН4)С12],
СиС1-С2Н4; 2) смешанные карбонильно-олефиновые
соединения переходных металлов (СО)Х-Me-олефин;
3) дициклопентадиенильные соединения переходных
металлов типа ферроцена (С5Н5)г Me и С6Н5Мп(СО)3;
4) соединения типа дибензолхрома (С6Н6)2Сг и С6Н8-
• Сг(СО)з; 5) комплексы AlClg, BFg и пр. с ароматич.
соединениями. Наиболее давно известны комплексные
соединения меди и платины с олефинами. Соединение
СиС1-С2Н4 образуется при поглощении этилена р-ром
СигС1г н соляной к-те; в чистом виде оно было полу-
чено при нагревании полухлористой меди с этиленом
под давлением. Бутадиен соединяется с двумя моле-
кулами однохлористой меди Си2С1г. Из хлорной пла-
тины и олефина образуются соединения типа
С1 ,С1.
% zPt’. '.Х ч
\ С1' С1
Известны также [Pt(NHg)(C2H4)(Cl2)J, K[Pt-C2H4-Cl3]
и др. Эти комплексы, как правило, легко элими-
нируют олефин; молекула олефина может быть вы-
теснена молекулой другого олефина, а также молеку-
лами аминов, пиридина и др. Такого рода соединения
известны для серебра, палладия, железа и нек-рых
других металлов. К ним близко примыкают смешан-
ные карбонильно-олефиновые соединения переход-
ных металлов, которые особенно характерны для же-
леза, кобальта и никеля; они образуются при нагрева-
нии или облучении смеси олефина с карбонилом метал-
ла; при этом одна СО-группа замещается молекулой
олефина или
две СО-группы—молекулой диена.
Особенно устойчивы соеди-
нения диеновых углеводо-
родов с карбонилами же-
леза; они представляют со-
бой кристаллич. вещества.
В бутадиен-железо-трикар-
бониле (I) молекула бута-
диена находится н плос-
кости, параллельной плоско-
" сти расположения углерод-
ных атомов СО-грулп. Аналогичное строение имеет
комплекс с циклооктатетраеном (II) с той разницей,
с—с
Fe
(СО),
(СО),
Fe
(СО),
что молекула углеводорода расположена в двух плос-
костях и каждая пара двойных связей способна об-
разовывать связь с железом. Связь между олефином
и атомом металла делокализована, т. е. металл свя-
зан не с отдельным атомом углерода, а с обоими в
равной мере; такая делокализация связи характерна
для 'Всех П.-к. Связь осуществляется, по-видимому,
за счет включения л-электронов олефина в вакантные
орбиты переходных металлов (или других тяжелых
металлов); не исключено, что нек-рый вклад в эту
связь вносят валентные электроны металла, вступая
н p-орбиты олефина.
Смешанные карбонильно-олефиновые соединения ме-
таллов являются переходной ступенью от карбонилов
металлов, с одной стороны, и от комплексов тяжелых
металлов с олефинами, с другой, к соединениям арома-
тич. углеводородов с переходными металлами. Еще
ближе к последним примыкает марганца метилцикло-
пентадиенилтрикарбонил (III) и родственные соедине-
ния. В соединении III между атомом металла и арома-
тизированным циклопентадиенильным кольцом осуще-
ствляется одна ковалентная делокализованная связь
(точно такая же, как в ферроцене)-, по дру-
гую сторону металла, н плоскости, парад- /,-Д
дельной кольцу, расположены три карбо- / (i ,'S
нильные группы. Это соединение весьма
устойчиво термически, а также к дейст- I
вию кислот, щелочей, кислорода; подобно Мп
ферроцену, легко вступает в реакции (СО)3
электрофильного замещения (сульфирова- ш
ние, Фриделя—Крафтса реакция и др.).
Развитию химии такого рода соединений и П.-к.
дало толчок открытие ферроцена (1951); в последую-
щие годы были получены дициклопентадиенильные
соединения почти для всех переходных металлов.
Большинство из них имеет ф-лу (CsHs)2Me и построено
подобно ферроцену, хотя они, как правило, менее
стабильны.
CsHsMgX + RuCL, —>- (CsH5)2Ru
СО2(СО),
(С5Н5)2СО
С5Н5СО(СО)а
ф
2C5H,Na + CrCl3 —>(С5Н5)2 СгС1
В соединениях нек-рых переходных металлов с арома-
тич. углеводородами типа дибензолхрома
(IV) связь между ароматич. кольцом и
атомом металла осуществляется, по-ниди-
мому, на тех же основаниях, что и в мо-
лекуле ферроцена. Катион дибензолхрома
был получен при нагревании смеси СгС13,
Al, AlClg и бензола и восстановлен бисуль-
фитом натрия до дибензолхрома:
ЗСгС13 + 2А1 + А1С13+6СбН«—►3[Cr(C,He)J]+ А1С14 IV
2 [Сг(СвН„)21 + +S2O*_ + 4OH- —>2Cr(C,He)2 + 2SOj~+2H2O
Соединения этого типа вполне стабильные кристаллич.
продукты; н отличие от ферроцена, они не вступают
в реакции электрофильного замещения. При на-
гревании или облучении нек-рых карбонилов метал-
лов (Сг, Мо, W) в присутствии ароматич. углеводоро-
дов образуются комплексы смешанного типа:
R-CeH5 + Cr(CO)„ —>. RCSHS-Cr (СО)3
В смешанных карбонильно-олефиновых и карбо-
нильно-ароматич. соединениях переходных металлов
часть СО-групп может быть замещена на нитрозильные
(NO) и нек-рые другие группы без того, чтобы свой-
ства соединения существенно менялись.
Наконец, к П.-к. можно отнести, по крайней мере,
значительную часть т. наз. комплексов «с переносом
заряда», напр. комплексы ароматич. соединений с
хлористым алюминием, трехфтористым бором и пр.
При растворении в конц. НгЗО4 ароматич. углеводо-
роды протонизируются; при этом образуется П.-к.
протона, напр. с бензолом С6Н6-|-Н+ [С8Н,] + .
По Дьюару при электрофильном замещении в арома-
тич. ряду вначале в качестве переходного состояния
образуются П.-к. между ароматич. углеводородом и
атакующим агентом (напр., нитроний катионом NO2+):
С6Н8 + NO2 —-
после чего отщепляется один из протонов и нитро-
группа мигрирует к атому углерода.
Электрофильное присоединение галогенов к двой-
ной связи также, вероятно, проходит через переходное
1029
ПИКРАМИД—ПИКРИЛХЛОРИД
1030
состояние П.-к. (Дьюар):
6+ 6-
СН2 = СН2 + Вг-Вг —> [СН2 гттттт СН2] + Вг~
4
Вг
ВгСН2СН2Вг
Как видно из сказанного, П.-к. представлены сое-
динениями и комплексами с широким диапазоном
свойств; часто они резко различаются по стабиль-
ности, способам образования, химич. поведению,
строению и даже типу связи. В последнее время тер-
мин «П.-к.» относят гл. обр. к соединениям переходных
металлов с непредельными и ароматич. углеводоро-
дами.
Олефиновые комплексы тяжелых металлов исполь-
зуются иногда для разделения смеси олефинов или
для разделения смеси солей металлов. Марганца цик-
лопентадиенилтрикарбонил предложено применять
в качестве антидетонатора, имеющего преимущества
перед тетраэтилсвинцом. Нек-рое применение нахо-
дит ферроцен. П.-к. являются промежуточными обра-
зованиями в ряде важнейших химич. реакций и про-
мышленных процессов. Так, в любых синтезах на
основе олефинов или ацетилена, в к-рых в качестве
катализаторов применяют карбонилы металлов или
металлы (Ni, Fe, Со) в присутст-
вии окиси углерода, промежуточ- 9Н
но образуются смешанные кар- сн CONH—rV NO2
бонильно-олефиновые соединения 3 । •
металлов (напр., Реппе синтезы).
Большое значение при полиме-
ризации и других реакциях име-
ют промежуточно образующиеся
П.-к. А1С13, BFs, SnCl4 и др. с
олефинами, ацетиленом и арома-
тич. соединениями.
Лит.: УилкинсонД ж., Кот-
тон Ф., Усп. химии, 1962, 31, вып. 7,
838; Несмеянов А. Н., Перева-
лова Э. Г., там же, 1958, 27, вып. 1, 3; П а р и и и В. П.,
там же, 1962, 31, вып. 7, 822; Химия координационных соеди-
нений, под ред. Дж. Бейлара, пер. с англ., М., 1960, гл. 15,
с. 411. Н. А. Несмеянов.
ПИКРАМИД (2,4,6-тринитроанилин, 2-амино-1,3,
5-тринитробензол) CaH4N4O6, мол. в. 228,12— кристал-
лы, темно-желтые пластинки; т. пл.
"J”2 190—191°; плохо растворим в спирте
О N-jX^r-NOa и эфире, легко —в горячем бензоле,
2 J ацетоне и уксусной к-те. Образует
комплексы с нафталином, т. пл.
NO3 168,8°; с антраценом, т. пл.158,8°; с
фенантреном, т. пл. 160,2°. П. легко
восстанавливается оловом в кислой среде до 1,3,5,6-те-
трааминобензола; в случае использования сульфида
аммония восстановление останавливается на стадии
образования 3,5-динитро-1,2-диаминобепзола. При
обработке щелочью П. переходит в пикриновую к-ту,
при действии HNOs в конц. H2SOa— в нитропикра-
мид;
no2
HNO3
NO2
N-Метильное производное этого соединения — тет-
рил — является важным взрывчатым веществом. П.
получают при аммонолизе пикрилхлорида, метилового
эфира пикриновой к-ты, метилпикрилнитрамина или
1,2,3,5-тетранитробензола; при взаимодействии 1,3,
5-тринитробензола с гидроксиламином в присутствии
ацетата натрия и при нитровании 2- или 4-нитроани-
линов. Используют П. для получения комплексов с по-
лициклич. ароматическими соединениями и в органич.
Синтезе. э. Е. Нифонтъев.
ПИКРАМИНОВАЯ КИСЛОТА (4,6-динитро-2 амп-
нофенол) CeHsOsN3, мол. в. 199,12— темно-красные
кристаллы; т. пл. 169,9°; плохо растворима в воде.,
эфире, хлороформе, растворяется в спирте, бензоле и
уксусной к-те. П. к. (I) — амфотерное соединение,
образующее соли с кислотами и основаниями; цинко-
вая и кадмиевая соли очень мало растворимы в воде;
константа диссоциации А=2,5-10-5 (0°) и lA-lO-4
(65°). Производные: N-ацетат, т. пл. 205—206°; O,N-
диацетат, т. пл. 190—191°. Вещество огне- и взрыво-
опасно. П. к. восстанавливается сероводородом до
4-нитро-2,6-диаминофенола, а треххлористым тита-
ном—до 2,4,6-триаминофенола; последнюю реакцию
используют для количественного определения П. к.
При ацетилировании П. к. уксусным ангидридом
(20—25°) образуется N-ацетильное производное (II);
в случае использования хлористого ацетила
(50—55°) — О, N-диацетилировапиьш продукт (III).
Фосгенирование П. к. при 130—140° приводит к 3,5-
динитро-2-оксифенилизоцианату (IV); метилирование
диметилсульфатом также сопровождается замещением
по аминогруппе с образованием 4,6-динитро-2-метил-
амино-(У1)и 4,6-дилитро-2-диметиламипофенола (V):
(сн3со)2о
И. к. реагирует с 2,4,6-тринитроанизолом с образо-
ванием 1, 3, 5, 7-тетранитрофеноксазина. Обработка
П. к. азотистой к-той приводит к 2-диазо-4,6-динитро-
фенолу:
NOj NO2
Получают П. к. восстановлением пикриновой кис-
лоты или гидролизом 4,6-динитро-2-ациламинофеио-
лов. П. к. используют при приготовлении нек-рых
красителей, в качестве цветного стандарта при коло-
риметрия. определении сахаров, для открытия бел-
ков и аминокислот (дает цветные реакции с протеи-
нами и аминокислотами, но не с их солями). П. к.
ТОКСИЧНа. э. Е. Нифантъев.
ПИКРИЛХЛОРИД (2-хлор-1,3,5-тринитробензол),
мол. в. 247,56—почти бесцветные кристаллы, иглы;
т. пл. 85°; образует комплексы: с нафталином, т. ил.
150—151°; с антраценом, т. пл. 160°.
П. обладает очень подвижным атомом хлора, легко
обменивающимся при действии нуклеофильных ре-
агентов. Так, с водой образуется пикриновая к-та
(1), с перекисью натрия — натриевая соль гидропе-
рекиси пикриновой к-ты (2), с гидроксиламином —
2,4,6-тринитрофенилгидроксиламип (3),с гидразином—
33*
1031
ПИКРИНОВАЯ КИСЛОТА—ПИКТЭ — ШПЕНГЛЕРА РЕАКЦИЯ
1032
Растворитель Растворимость, П. г/100г растворителя
при 17° при 50°
Хлороформ 12,36 233,4
Четыреххлористый углерод . . 0,557 2,45
Бензол 36,69 428,08
Толуол 89,435 321,05
Метанола 10,241 34,80
Этанол а 4,848 15,06
Ацетон 212,00 546,43
Уксусная кислота 91,52 238,35
Пиридин 120,79 173,38
Эфир 7,128 10,64 °, 0,95 0
Сероуглерод 0,499
Вода 0,0178 0.053
При частичном алкоголизе. 0 При 31°.
пикрилгидразид (4), с фенолятом натрия-— 2,4,6-
тринитродифениловый эфир (5), с аммиаком и амина-
ми— амиды пикриновой к-ты (6), с натриймалоновым
эфиром — 2,4,6-тринитрофенилмалоновый эфир (7):
(2)
Na.O.
no2
O,N
OONa
NO.
NO2
O2N
NHOH
NO.
NH2OH
HNRR1
„ C6HsONa
Cl ——2---
NO,
ПИКТЭ - ШПЕНГЛЕРА
o2n
NO.
(7)
(3)
NOj
При нагревании П.
с медью в нитробен-
золе образуется 2,4,
6,2'4',6*- гексанитро- ( ОСО)2СН
дифенил, в случае ис- г ’
пользования в качест- 0
ве растворителя влаж- 2
ного толуола получается 1,3,5-тринитробензол;
ного толуола получается 1,3,5-тринитробензол; при
восстановлении П. оловом в кислой среде— 1,3,5-
триаминобензол. П. реагирует с тиосульфатом нат-
рия с образованием 2,4,6,2',4',6'-гексанитродифепил-
сульфида.
Получают П. взаимодействием пикриновой к-ты
с пятихлористым фосфором или бензолсульфохлори-
дом в присутствии пиридина. П. образуется также при
действии на суспензию пикриновой к-ты в соляной
к-те гипохлоритом натрия, нитрованием хлорбензола
или 4-хлор - 1,3-динитробеизола. П. используют
для идентификации аминов и нек-рых других соеди-
нений. В пром-сти он применяется для получения кра-
сителя пинакриптола зеленого (N-фенил-! ,3-диами-
нофеназонийхлорида), являющегося сильным десен-
сибилизатором. э- Е- Нифантъев.
ПИКРИНОВАЯ КИСЛОТА (мелинит) — см. Три-
нитрофенол.
ПИКРОЛОНОВАЯ КИСЛОТА (1-га-нитрофенил-
3-метил-4-нитропиразолон-5) CI0H8OsN4, молекуляр-
O2n
СО-Сн-NO,
/ 5 41
N<2 з1
N=C-СН3
ОН
zC=C-NO2
o2n—/“n4 I
\=/ №С-Сн3
ный вес 264,20—кристаллы, желтые иглы, т. пл.
116,5°; т. разл. 125°. Растворимость П. к. приведена
в таблице:
Растворитель Растворимость 1 г П. к. в граммах растворителя
при 17° при т. кип.
Вода 116 108
Метанол . . 164,3 29,4
Этанол 96%-ный .... 21 9,6
Этиловый Эфир 198 112
П. к.— слабая к-та, образует соли с металлами
и органическими основаниями и устойчивые комплек-
сы со многими поликопденсированными ароматически-
ми соединениями; аммониевая соль, т. пл. 275°;
соль с гидроксиламипом, т. пл. 262°; соль с атропи-
ном, т. пл. 194°. Получают П. к. нитрованием 1-фе-
нил-З-метилпиразолона-5 или 1-п-нитрофенил-З-ме-
тилпиразолона. Для ка-
чественного определения
11. к. к водво-спиртовому
ее р-ру прибавляют по
каплям р-р метиленово-
го синего; выпадает оса-
док, растворимый в хло-
роформе.
П. к., также как и пи-
криновую кислоту, при-
меняют для идентифика-
ции алкалоидов и других
органич. аминов и поли-
циклических ароматич.
соединений, а также для
весового и микрокри-
сталлоскопич. определе-
ния Pb,Ca, Sr, Mg, Th.
Э. Е. Нифантпьее.
РЕАКЦИЯ — получение
Р-арил этил-
тетрагидроизохинолинов конденсацией
аминов с карбонильными соединениями:
В качестве аминного компонента можно использо-
вать незамещенный р -фенилэтиламин и (чаще) Р-фе-
нилэтиламины с алкоксильными или гидроксильными
заместителями в ароматич. кольце, причем, как пра-
вило, кольцо замыкается в пара-положение к акти-
вирующей группе (1). Гораздо реже используются
Р-фенилэтиламины, замещенные в боковой цепи,
напр. р-(о-метоксифенил)-р-оксиэтиламин, тирозин и
др. Вторичные р-арилэтиламины реагируют с карбо-
нильными соединениями аналогично первичным (2).
В качестве карбонильного компонента чаще всего и с
наибольшим успехом применяют формальдегид. Сре-
ди других альдегидов наибольший интерес представ-
ляют ацетальдегид и замещенные фенилацетальде-
гиды; вовлечение их в П.—Ш. р. приводит к синте-
зу алкалоидов сальсолина и тетрагидропапаверина.
а-Кетокислоты также легко вступают в реакцию (3).
П.— Ш.р.используется не только для синтеза производ-
ных изохинолина, но и для получения других конден-
сированных гетероциклич. систем, таких, как дибен-
зохинолизины и 2-карболины (4):
Г Т сн, + сн,о —
NH,
1033
ПИЛОКАРПИН —ПИМЕ ЛИНОВАН КИСЛОТА
1034
Обычно амин и альдегид нагревают до 100° с из-
бытком соляной к-ты; часто сначала получают основа-
ние Шиффа, к-рое затем циклизуется нагреванием с
к-тами. Вместо наиболее часто употребляемой НС1
иногда используют и другие конденсирующие аген-
ты— НВг, POClg, PC1S, (СНзСО)зО и др. В ряде слу-
чаев П.— Ш. р. удается осуществить в условиях, близ-
ких к физиологическим, что имеет большое значение
для выяснения механизма образования алкалоидов
в растениях. П.—Ш.р. является по существу частным
случаем Манниха реакции и включает в себя внутри-
молекулярное электрофильное замещение в ароматич.
ядре:
Реакция открыта А. Пиктэ и Т. Шпенглером в 1911.
Лит.: У э л л и В. М., Говиндачари Т. Р., в сб.:
Органические реакции, сб. 6, пер. с англ., М., 1953, с. 177;
Генслер В. Д ж., в сб.: Гетероциклические соединения^
под ред. Р. Эльдерфилда, пер. с англ., т. 4, М., 1955, с. 269.
Н. П. Гамбарян.
ПИЛОКАРПИН CuHuOaNa, мол. в. 203,26—алка-
лоид, выделенный из листьев африканского расте-
ния Pilocarpus Jalorandi
Hsc2-|---Гснг~И----N —СН Holms; П. (I) трудно
n=J J |l J кристаллизуется; т. пл.
340. г> кип 260о/5 л{л{.
[а]д= + 100,5° (СНС1з);
легко растворим в воде, спирте и хлороформе,
хуже — в бензоле, нерастворим в эфире; образует
соли с одним эквивалентом кислот; гигроскопич-
ный хлоргидрат, т. пл. 204—205°, [а]о= + 91,74°
(вода), легко растворим в воде и спирте, нерастворим
в эфире и хлороформе; нитрат, т. пл. 178°, [<x]d =
82,9° (вода), растворим в воде (1:6,4) и спирте
(1:146); бромгидрат, т. пл. 185°; пикрат, г. пл. 161—
162°. При действии щелочных агентов, а также час-
тично при перегонке П. изомеризуетси в изопилокар-
пин (транс-изомер П.).
Из растений П. выделяют экстракцией подкислен-
ным спиртом; очищают от сопутствующих алкалоидов ।
кристаллизацией его хлоргидрата или нитрата. Из I
1 т сухих листьев получают 5 кг хлоргидрата или |
нитрата И. Изомеры разделяют при получении смеси
пилоповой (II) и изопилоповой (III) к-т. Синтез И.
осуществлен Н. А.
Преображенским,
М. Н. Щукиной с
сотр.
П. — физиологи-
соон
чески активное ве- ц щ
щество, сужает зра-
чок, повышает тонус гладкой мускулатуры и увели-
чивает слюно- и потоотделение. В медицине П. при-
меняют (н виде хлоргидрата) для снижения внутри-
глазного давления при глаукоме.
Лит.: Преображенский Н. А., Генкин Э. И.,
Химия органических лекарственных веществ, М.—Л., 1953,
с. 62; М а й о ф и с Л. С., Технология химико-фармацевти-
ческих препаратов, Л., 1958, с. 523; Машковский М. Д.,
Лекарственные средства, 4 изд., М., 1960. А. Е. Васильев.
ПИЛОТИ РЕАКЦИЯ — синтез геминальных хлор-
и бромнитрозосоединений действием галогенов на
оксимы альдегидов и кетонов:
\ x2 C-N-0 ——н
C = N-OH—> / | -I.1
/ L X; X
-нх,
\
С-N = O
/I
X
Обычно обрабатывают хлором или бромом растнор
оксима в разбавленной соляной к-те, водном пиридине
или водном р-ре ацетата натрия, или же чистый ок-
сим, при 0—20°. П. р. служит также методом полу-
чения производных гидроксамоных к-т, поскольку
полученные из альдоксимов а-хлорнитрозоалканы лег-
ко изомеризуются в хлорангидриды гидроксамовых
кислот; последние, в сною очередь, вступают в П. р.
с образованием а, а-дихлорнитрозоалканов:
С12 ci,
R-CH = N- ОН—>R—СН—N—О —>R-C=N-OH—►
I I
Cl Cl
Cl2
—>R-CC13-N = O
Реакция открыта О. Пилоти в 1898.
Лит..- Р 1 1 о t у О., Вег., 1898, 31, 454; Aston J. G.,
Menard D. F., J. Amer. Chem. Soc., 1935, 57, № 10, 1920.
Б. Л. Дяткин.
ПИМЕЛИНОВАЯ КИСЛОТА (пентаметилендикар-
боновая кислота) НООС(СН2)5СООН, молекулярный
вес 160,17— кристаллы; т. пл. 105,5; т. кип.
272°/100 мм; константа диссоциации: Ki— 3,33-Ю-5,
Кг= 4,87-10_6; в 100 г воды растворяетси 2,5 г
П. к. (14°). П. к. обладает общими свойствами ди-
карбоновых к-т; образует производные как по од-
ной, так и по обеим карбоксильным группам. Полные
эфиры П. к. под действием алкоголята натрия или
натрия (в условиях сложноэфирной конденсации) за-
мыкаются с образованием эфиров циклогексанон-2-
карбоновой к-ты:
COOC,HS
(CH2)S(COOC2H5)2 ----►
о
П. к. может быть получена окислением касторового
масла; из пентаметиленхлорида и цианистого натрия
с последующим омылением образующегося нитрила;
при восстановлении салициловой к-ты натрием в ами-
ловом спирте:
ОН 2Н
СООН
а он r i/~'''r=o 0
соон ** соон
---НООС(СН2)5СООН
П. к. содержится в моче травоядных животных.
Я. А. Несмеянов.
О 34 К.Х.Э. т. 3
1035 ПИНАКОЛИН — ПИНАКОЛИНОВАЯ И РЕТРОПИНАКОЛИНОВАЯ ПЕРЕГРУППИРОВКИ Ю36
ПИНАКОЛИН (метил-трет-бутилкетон, 2,2-ди-
метилбутанон-3,а,а,а-триметилацетон) CeHi2O, мол,
вое 100,16 — бесцветная жидкость с характерным за-
пахом мяты, т. пл.— 49,8°; т. кип. 106,3°; 0,8114;
п™ 1,3944; теплота сгоолния 891,5 ккал/моль (v =
= const); растворим в эфпре, ацетоне, спирте, раство-
римость в воде 2,04% (20°); образует азеотропные сме-
си. Производные: 2,4-динитрофенилгидразон, т. пл.
125,5—126,5°; оксим, т. пл. 78,5—79,5°.
П. окисляется бихроматом калия и гипохлоритом
натрия до пивалоновой к-ты (1), перманганатом ка-
лия— до смеси пивалоновой (1) и диметилпировино-
градной к-т (2), двуокисью селена— до mpcm-бутил-
глиоксаля (3):
K2Cr20;(NaC10)
КМпО,
SeO3
(СН3)3ССООН
(СН3),С-СОСН3
(сн.хссоон (1)+
+ (СН,).,ССОСООН
о
(СН3)3СОС^
\
н
(1)
(2)
(3)
При восстановлении П. получается пинаколиновый
спирт. Он же наряду с ацетатом и карбонатом калия
образуется при нагревании П. со спиртовым р-ром
едкого кали. Магнийорганические соединения реаги-
руют с П. с образованием метил-трет-бутилалкил-
карбинолов или пинаколинового спирта:
R
RMgX---------НСНз)зС-с'-СН3
(СН3)3С-СОСН --------
СН3
-----►(СН3)3-СН/
\
он
Своеобразие П. заключается и в том, что он плохо
взаимодействует с реагентами типа бисульфита нат-
рия и не дает йодоформенную пробу, при взаимодей-
ствии с иодом и едким кали.
П. образуется при окислении пинаколинового
спирта, нагревании 2,2-диметил-З-нитробутана с хло-
ристым оловом и соляной к-той, взаимодействии пива-
лоилхлорида с метилмагнийхлоридом. Основным ме-
тодом синтеза П. является пинаколиновая перегруп-
пировка (см. Пинаколиновая и ретропинаколиновая
перегруппировки), заключающаяся в дегидратации
пинакона (пинаконгидрата) под действием серной
к-ты или хлористого цинка:
сн3 сн3 сн3
х / h3so< \
С С------------>-СН3-С-С-СН3
/I I \ /II
сн3 он онсн3 сн3 о
П. используется н органич. синтезе.
Э. Е. Нифантъев.
ПИНАКОЛИНОВАЯ И РЕТРОПИНАКОЛИНО-
ВАЯ ПЕРЕГРУППИРОВКИ — широко распростра-
ненные случаи изомеризации углеродного скелета при
дегидратации некоторых гликолей и спиртов изо-
строения:
R R R
\ / -Н3О \
С---С------->R-C-C-R
/I 1\ /II
R ОН ОН R R О
пцнаколииован перегруппировка
R Н R R
\ I -Н,0 \
R-C-C-R-------► С = С
/I / \
R ОН R R
ретропина ко линован перегруппировка
По характеру изменения скелета молекулы ретро-
пинаколиновая перегруппировка является процессом,
обратным пинаколиновой перегруппировке:
г г с
, пинаколиновая перегруппировка ,
С - С f *• С - С - С - С
ретропинаколиновая перегруппировка (-,/
Типичным примером пинаколиновой перегруппиров-
ки является превращение двутретичных спиртов (пи-
наконов) в кетоны (пинаколины), происходящее под
действием кислот, их галогенангидридов, ZnCl2 и др.
электрофильных реагентов. Аналогичная изомериза-
ция наблюдается при дегидрогалогенировании бром-
гидринов пинаконов и дезаминировании аминоспир-
тов пинаконового ряда:
HNO2
(СН3),С--С(СН,)2----->(СН3)3ССН3
нб nh2 <!>
В нек-рых случаях перегруппировке, подобной пина-
колиновой, подвергаются также вторично-третичные
гликоли и аминоспирты. Механизм пинаколиновой пе-
регруппировки предполагает следующий ряд проме-
жуточных стадий:
снэ СН3 + СН3 СН3 „ п СН, /СН.)
X / +н ' / ~н8о \ g м у
Zc-C С-с—сн3 с-С-сн3—-
СНзОНОНСНз СН3 о+ ОН СН3Х ОН
нх хн
СН3 —н+ СНз
СН3-С~С-СН3^^СН3-С-С-СНЭ
СН3 о сн/ о
н
Легкость отщепления гидроксила определяется ин-
дукционным влиянием соседних групп, которые по
активности располагаются в ряд л-анизил > фе-
нил > алкил > водород. При прочих равных услови-
ях подвижность мигрирующих групп определяется их
„ АгАг'С —САгАг’
электронодонорнои активностью. Так, ।
для пинакона подвижность заместителей изменяется
в порядке п-анизил > л-толил > л» толил > .«-ани-
зил > фенил > п-хлорфенил > о-анизил > .«-хлор-
фенил. Аномально низкая подвижность о-анизиль-
ной группы объясняется влиянием стерич. факторов.
Изучение стереохимии, природы пинаколиновой
перегруппировки на примере оптически активных
аминоспиртов показало, что в подавляющем большин-
стве случаев оптическая активность сохраняется, при-
чем наблюдается обращение конфигурации (см. Валь-
деновское обращение)'.
СвН5 И С„Н5
I I HNO, /
с,н5-с—»с-сн,-------> с»н5-с-»с
II -Na II 1\
ОН NH, О Н СН,
Обращение конфигурации подтверждается также ха-
рактером перегруппировки нек-рых пинаконов ряда
циклопентана:
Выход 87% в случае цис-гликоля; 7% в случае транс-
гликоля.
Сохранение оптич. активности молекулы в условиях
пинаколиновой перегруппировки, а также изучение
реакции дейтерообмена показывает, что механизм
пинаколиновой перегруппировки не включает стадию
1037
ПИНАКОНЫ
1038
образования иона карбония в виде кинетически неза-
висимой частицы. На примере дезаминирования диа-
стереомерных аминоспиртов показано, что в случае
конкуренции стереохимия, и электронных факторов
направление перегруппировки определяется в первую
очередь стереохимия, факторами. При благоприят-
ной конфигурации менее электронодонорная группа
мигрирует предпоятительно перед более электронодо-
норной.
Йинаколиновая перегруппировка используется для
полуяения разлияных кетонов алифатия., ароматия.
и гетероциклия. рядов. Особенно ценным оказалось
применение этой реакции для синтеза соединений спи-
ранового ряда:
Типияным примером ретропинаколиновой перегруп-
пировки является дегидратация пинаколинового спир-
та при действии безводной щавелевой к-ты:
-HZO
(СН3)3С-СН-СН3---->(СН3)2С = С(СН3)2
он
Подобное превращение претерпевает при дегидрога-
логенировании соответствующий галогенгидрин. Ме-
ханизм ретропинаколиновой перегруппировки апа-
логияен механизму пинаколиновой:
Таблица 1. Физические свойства пинаконов
Пинакон Т. пл., °C Т. кип., “С/.ч.ч
СН, СН3а /С—с;
сн/ 1 1 хсн, он он с2н. с,нБ )с— с. 43,4 1 74,35/760
С2н/ 1 | С2Н, он он 112/10
^О^оЙ^ 108 —
^ooiP 129-130 —
сн, сн, ц-форма 122
)с—с( СвН/ [ | чсвнб он он сен5 свн5 3-форма 117
)с—с( С«н, | 1 чс,н, он он 187 —
а Моногидрат, т. пл. 41,25°; гексагидрат, т. пл.
45,4; d” 0,9641; Пр 1,4430; продукт присоедине-
ния двух молекул НВг, т. пл. 52—54°.
Таблица 2. Азеотропные смеси пинакона
\ I
R-C-C-R
RZ ОН
R R
xc=cz
RZ 4R
R\+ zR
хс-сн
RZ \
Изомеризация углеродного скелета, подобная ретропи-
наколиновой перегруппировке, наблюдается также
в более простых системах:
сн-сн2он-
ZnCla
Компонент
Пентахлорэтан . . . .
Бромбензол ..........
ц-Лимонен............
а-Пинен .............
Мезитилен............
Фенол................
Диметилоксалат . . . .
Камфен ..............
сн,
-------->- СН3-СН = СН-СН,
сн,
\
с=сн2
Z
-------->-сн3
К перегруппировкам типа ретропинаколиновой от-
носятся многояисленные слуяаи камфеновых перегруп-
пировок.
Лит.: X ю к к е л ь В., Теоретические основы органиче-
ской химии, пер. с нем., т. 1, М., 1955, с. 306 исл.; И н г ольд
К., Механизм реакций и строение органических соединений,
пер. с англ., М., 1959, с. 385 и сл.; S t 1 1 е s М.,М а у е rR.P.,
J. Amer. Chem. Soc., 1959, 81, Ай 6, 1497; Scott R. В.,
Gayle J. В., J. Organ. Chem.. 1953, 18, № 6, 740.
Л. С. Герман.
ПИНАКОНЫ — вицинальные двухатомные двутре-
R R
тичные спирты с — с . Физия, свойства наи-
/I I \
R' ОН НО R'
более известных П. приведены в табл. 1.
Все П. плохо растворимы в холодной воде, хорошо в
спирте, хлороформе, они образуют азеотропные смеси
(см. табл. 2, для простейшего пинакона).
П. легко образуют продукты присоединения с во-
дой, галогеноводородными к-тами и др. соединениями.
Гидраты П. легко теряют воду, при выдерживании в
вакууме над едким кали, нагревании или луяше
всего при перегонке с бензолом. Й. вступают во многие
Т. кип.,
°C
Содержание
пинакона, %
1
1
25
28
3
33
1
28
реакции, характерные для простейших третичных
спиртов (алкилирование, дегидратация, восстановле-
ние иодисто-водородной к-той и т. д.). Высшие П. при
нагревании до темп-ры плавления распадаются с раз-
рушением центральной углерод — углеродной связи,
так, напр., бензпинакон превращается в бензофенон и
бензгидрол:
С,Н3 С«н5 СвНб С,Н,
\ Z 1° \ \
С С —ч- С = О + СН(ОН)
С,Н5 Ьн ОН С,. Н, С,Н, с,н.
Своеобразной реакцией й. является их перегруппи-
ровка при действии минеральных к-т в кетоны (т. наз.
пинаколиновая перегруппировка):
R R R
\ z H2SO4 \
с---С--------->R—С—C-R
Zl I \ Z JI
R OH OH R R 0
Для полуяения й. чаще всего применяют электро-
химия. восстановление кетонов или восстановление
их амальгамированными магнием или алюминием,
цинком, натрйем, нек-рыми гриньяровскими реакти-
вами (напр., трифенилметилмагнийбромидом); в эту
реакцию легко вступают алифатич., алициклич. и
ароматич. кетоны (I). Механизм образования П. за-
ключается в присоединении водорода к карбонильной
и
34*
1039
ПИНАН — ПИНЕНЫ
1040
группе кетона с последующей димеризацией проме-
жуточно образующихся радикалов:
R R R R
\=о+н—► Чс-он—► Чс— сх
RZ RZ RZ^H AhR
П. также синтезируют из диацетила и подобных
а-дикетонов и магнийорганич. соединений (II). Ана-
логично получают П. и из эфиров щавелевой к-ты
(III). Более ограниченное значение имеют методы
синтеза П. окислением тетраалкилэтиленов (IV) и
гидролизом тетраалкилдигалогенэтиленов (V):
ГН]
I. RCOR'------>RR'C(OH)C(OH)RR'
R'MgX
II. RCOCOR-------► RR'C(OH)C(OH)RR'
RMgX
III. R'OCOCOOR' ------->RaC(OH)C(OH)R2
fO]
IV. RR'C = CRR'---->RR'C(OH)C(OH)RR'
OH'
V. RR'BrC-CBrRR'----> RR'C(OH)C(OH)RR’
Промышленное значение имеет простейший П.—
тетраметилэтиленгликоль — для получения пинако-
лина; ранее тетраметилэтиленгликоль применялся в
синтезе 2,3-диметилбутадиена (дегидратация над
окисью алюминия), дающего при полимеризации кау-
чукоподобный продукт, наз. метилкаучуком.
Э. Е. Нифантъев.
ПИНАН (2,6,6-триметил-бицикло- [1,1,3]-гептан)
С10Н18, мол. в. 138,25—бесцветная жидкость, по внеш-
нему виду и растворимости похожая на пинен, сущест-
вует в виде цис- (I) и транс- (II) форм:
Транс-1-пинан, т. кип. 162— 47720 мм, 0,8519,
Пд7’5 1,4595, [a]D= — 16,1°; транс -d-пинан, т.
кип. 165—1667760 мм, d2* 0,8558, пг* 1,4618, [a]D=
= 20,4°; tyuc-1-пинан, т. кип. 164,8—165,87716
мм, d2’ 0,8562, п% 1,4624, [a]D = — 18,9°; цис-d-
пинан, т. кип. 163—47720 мм, d20 0,8566, п20 1,4624,
[а]д = 23,1°. По другим данным, tjuc-d-пинан, т.
кип. 167— 87760 мм, d™ 0,8590, п™ 1,4632, [a]D=
— + 0,35°. П. весьма устойчив по отношению к дей-
ствию окислителей и минеральных к-т. При действии
хлора на П. получается смесь хлорпинанов.
В природе П. пока не найден. Получается гидро-
генизацией пиненов. При гидрогенизации в мягких
условиях (Pt-чернь на холоду) получается в основном
цис-П., в жестких условиях (Ni при 220—230°),
в основном, транс-изомер. П. нашел практич. приме-
нение: при окислении кислородом воздуха образует
гидроперекись, к-рая служит инициатором полиме-
ризации.
Лит. см. при ст. Терпены. И. И. Бардышев.
ПИНЕНЫ С10Н1в, мол. в. 136,23. Известны три изо-
мера П., отличающихся положением двойной связи:
a-пинен (2,6,6-триметил-бицикло-[1,1,3]-гептен-2) (I);
Р-пинен, или нопинен (6,6-диметил-2-метилен-бицик-
ло-[1,1,3]-гептан) (II) и б-пинен (2,6,6-триметил-бицик-
ло -[1,1,3] - гептен -3)
(П1).
Все П.—бесцветные
жидкости со своеоб-
разным приятным за-
пахом (запах хвои
сосны), хорошо рас-
творимы в органич.,
особенно неполярных
растворителях, нера-
створимы в воде. Бы-
стро окисляются на
воздухе, особенно на
свету, превращаясь
при этом в желтое
вязкое масло (хранят
в заплавленных стек-
лянных сосудах в темноте). Существуют в виде d-,
I- и б,1-форм.
Пинены Т. кип., °С/760 мм „20 nD Г -1 2А [“Id
а-П.а Р-П. d, 1-6-П. 1-6-11. 155,9 166,0 157—9 156—7 0,8578 0,8712 0.8636 0,8590 1,4653 1,4790 1,4656 1,4667 ±50,6° —22,7° 0,0° — 6,2»
Т. кип. 52,2° (20 .ад): т. кристалл,—75.5°;
ц 4,40 спуаэ; ®Т. кип. 59,7° (20 мм); т. кристалл.
—61,5°; ц 1,70 спуаз.
П. весьма реакционноспособны. При нагревании
выше 300° a-П. изомеризуется в аллооцимен (1,6-ди-
метилоктатриен-2,4,6), [3-П.— в мирцен (2-метил-6-
метилен-октадиен-2,7). При нагревании до 250—300°
а- и р-пинены образуют гл. обр. дипентен. При
темп-ре красного каления (700°) все П. образуют
изопрен и ароматич. углеводороды, а при гидрогени-
зации П.— пинан. Под действием H2SO4, Н3РО4,
P2OS, TiO2, BF3, активных глин и др. П. полимери-
зуются. При действии разб. органич. и неорганич.
к-т а- ир-П. образуют дипентен, терпинолен, терпи-
нены, терпинеол, терпингидрат и др. ТЮг и активные
глины при 150° изомеризуют а- и р-П. в основном в
камфен. При действии влажного НС1 а- ир-П. обра-
зуют в основном дигидрохлорид дипентена, а при дей-
ствии сухого НС1 — борнилгидрохлорид и частично
фенхилгидрохлорид (см. Камфеновые перегруппиров-
ки). При действии смеси КМпО4 и НгО a-пинен обра-
зует ряд промежуточных продуктов и пиноновую (IV)
и норпиновую (V) к-ты;Р*П.— нопиновую к-ту (VI).
СООН
IV V VI
При действии бензойной, трихлоруксусной, абиети-
новой к-т, Pt-черни Р-П. изомеризуется в a-П. Те
же катализаторы частично изомеризуют a-П. в Р-П.
Характерное кристаллич. производное П.— нитро-
зохлорид d-a-П., т. пл. 81°, [а]д= -|- 322°, а для
d, l-a-П. т. пл. 103°.
a-П. является одним из самых широко распростра-
ненных терпенов в природе и встречается в составе
различных скипидаров, эфирных масел. р-П. встре-
чается обычно там же, где и a-П., но только в меньших
1041
ПИОЦИАНИН — ПИПЕРИДИН
1042
количествах. Хорошим источником (3-П. является
скипидар обыкновенной ели. б-П. в природе пока не
найден.
а- и Р-П. находят широкое практич. применение:
хорошие растворители лаков и красок, сырье для
произ-ва камфары, терпинеола, терпингидрата, фло-
тационных масел, двухосновных к-т, инсектицидов
(под названием: хлортен, хлортен-2, полихлорпинеп),
«-цимола, камфена, аллооцимена. |5-П. служит для
получения мирцена и его производных, из которых
наибольшее практич. значение имеют душистые ве-
щества.
Лит.: Рудаков Г. А., Химин и технология камфары,
М., 1961, и при ст. Терпены. И. И. Бардышев.
ПИОЦИАНИН (10-метил-1-оксифеназин) С13Н10ОП^
мол. в. 210,23—бактериальный пигмент, образуемый
Pseudomonas aeruginosa (Р. руосуапеа), обладает
антибиотич. свойствами. П. кристаллизуется из сме-
си СНС13 с петролейным эфиром в виде синих игл,
т. пл. 133°; растворим в ацетоне, этилацетате, горя-
чей воде и нерастворим в эфире, бензоле и четырех-
хлористом углероде; в р-ре NaOH Хмакс. 326 и 660 ммк,
Igg соответственно 3,46 и 4,12. П. легко разлагается
до гемипиоцианина (I), а при гидрировании образует
бесцветное дигидропроизводное, к-рое с хлористым
оксалилом превращается в циклич. амидоэфир (III).
Эти данные позволили приписать П. строение (II).
На основании определения дипольного момента был
сделан вывод о заметной поляризации связей в П.,
что в предельном виде может быть выражено ф-лой
(IV). Синтез П. был осуществлен след, образом:
ОМе ОМе
П. извлекают из культуральной жидкости СНС13 и
отделяют от сопутствующего ему гемипиоцианина
хроматографированием на А12О3.
П. угнетает рост нек-рых грамположительных и
грамотрицательных микроорганизмов, а также ряда
патогенных грибов; однако он весьма токсичен. Име-
ются данные об участии П. в процессах бактериаль-
ного дыхания.
Лит.: Шемякин М. М. 1и др.1, Химин антибио-
тиков, т. 2, М., 1961, с. 825. В. В. Антонов.
ПИПЕКОЛИНЫ — см. Метилпиперидины.
ПИПЕРАЗИН CaHI0N2, мол. в. 86,14 — бесцветные
гигроскопич. кристаллы; т. пл. 104°; т. кип. 145—146°;
гексагидрат, т. пл. 44°, т. кип. 125—130°;
хорошо растворим в спирте, умеренно— в
Г j воде (15 г в 100 г воды), нерастворим в эфире;
''Nir П. — сильное органич. двукислотное основа-
ние, Ai=9-10"s, К2 == 4,6-10-’ (15°). П.
легко алкилируется по азоту галогеналкилами, с хлор-
новатистой к-той образует N, N'-дихлорпиперазин, с
азотистой к-той—N, N'-динитрозопиперазин. Над де-
гидрирующим катализатором П. превращается в пи-
разин, он легко присоединяется к акрилнитрилу и
а, р-непредельным карбонильным соединениям. При
взаимодействии с альдегидами П. образует полимеры:
О
-N N-CHR-
_ п
П. фосфорилируется по азоту хлорокисью фосфора и
тиоокисью фосфора, образуя тетрахлорангидрнды,
к-рые при реакции с этиленимином дают П,П'-бисди-
этилениминофосфопиперазины (дипин и тиодипин):
HN NH + POCI3(PSCl3)
?(s)r~\ ?(s>
CI2P- N N-PCla
О
применяемые в числе других аналогичных соединений
при лечении злокачественных опухолей. Качественной
реакцией на П. является взаимодействие его с
0,5%-ным р-ром нитропруссида натрия и уксусным
альдегидом (интенсивная синяя окраска). Для иденти-
фикации П. служит его пикрат, т. пл. 280°; комплекс
с ацетатом серебра, т. пл. 110—112°; комплекс с фе-
нолом, т. пл. 99—101°.
П. получают гидрированием пиразина, взаимодей-
ствием бромистого этилена с аммиаком, циклизацией
Р -оксиэтилдиаминоэтана и др. способами. Используют
П. в аналитич. химии для микрокристаллоскопич. ре-
акций на Мо, W, V; в качестве лекарственного сред-
ства при лечении аскаридоза и энтеробиоза и для
синтетич. целей.
Лит.: Пратт И., в сб.: Гетероциклические соединения,
под ред. Р. Эльдерфилда, пер. с англ., т. 6, М., 1960, с. 344.
Э. Е. Нифантьев.
ПИПЕРИДИН (гексагидропиридин, пентаметиле-
нимин) CsHnN, мол. в. 85,15— бесцветная жидкость
с резким аминным запахом; т. пл. — 9°; т.
кип. 106,3°/760 мм; 52,6°/170 мм; 36,7°/70 мм;
0,8606; п2д 1,4530. П. легко смешивается с fl
водой и большинством органич. растворите- NH
лей; с водой образует азеотропную смесь,
содержащую 35% воды с т. кип. 92,8°. П.— силь-
ное основание (Аосн = 1,6-10-®), обладает типич-
ными свойствами жирных аминов, легко образует
соли (хлоргидрат, т. пл. 244—245°; пикрат, т. пл.
151—152°). При действии галогенангидридов или
ангидридов кислот на П. образуются N-ацильные
производные (N-ацетилпиперидин, т. кип. 226°; N-бен-
зоилпиперидин, т. пл. 48°; N-n-нитробензоилпипери-
дин, т. пл. 121°). При действии слабых окислителей
П. переходит в N-окись (т. пл. 39°), при конденсации
с серой образует сульфид C5HI0N—S—NCSHIO. П.
относительно легко дегидрируется в пиридин, что
указывает на их генетич. родство. Типичными реак-
циями П. являются многочисленные случаи раскрытия
цикла. Расщепление по Гофману приводит к пипери-
лену (1). N-Бензоильное производное П. легко оки-
сляется до у-аминовалериановой к-ты, а в условиях
Брауна реакции образует дигалогенпентан (2):
1) СН3Л 2) А2дО
NH
СН3СН“СНСН=СНг (1)
КМпО«
НгЫ(СНг)4СООН
РС15
(2)
С1(СНг)5С1
1043
ПИПЕРИЛЕН — ПИРАЗИН
1044
Основные методы синтеза П.: 1) каталитич. гидриро-
вание пиридина (H2/Ni при 170—200°); электрохи-
мии. восстановление пиридина (последний способ при-
меняется в промышленности); 2) многочисленные слу-
чаи замыкания кольца:
Вг-(СН2)5— Вг
X-(CH2)5-NH2
№Br,OH,NH2
№C-(CH2)3-C=N
NH3
—НВг
-нх
Н г (каталитич.)
-NH3
Цикл П. входит в состав пиперина — алкалоида чер
ного перца, а также является основой многих алка-
лоидов лобелинового, тропанового и морфинового
рядов. В лабораторной практике П. используют в ка-
честве катализатора Михаэля реакции и Кневенагеля
реакции. Нек-рые производные П., в частности пен-
таметилендитиокарбамат, применяются как ускори-
тели вулканизации.
Каталитический метод гидрирования нашел при-
менение также и для восстановления гомологов пи-
ридина.
Лит.: Гетероциклические соединения, под ред. Р. Эльдер-
филда, пер с англ., т. 1, М., 1953, с. 490. Л. С. Герман.
ПИПЕРИЛЕН (1,3-пентадиен) С5Н8, мол. вес
68,11—бесцветная жидкость; хорошо растворим в
ацетоне, четыреххлористом углероде, бензоле, эфи-
ре, спирте; нерастворим в воде. Известен в цис- и
транс-формах:
сн2=сн-сн
I!
сн3-сн
цис-П.
сн-сн=сн2
II
сн3-сн
транс-П.
Т. пл., °C Т. кип., °C d20 4 20 nD
цис —140.82 44,07 0,6910 1,4363
транс .... -87,47 42,03 0,6760 1,4301
Цис- и транс-П. легко вступают во все реакции,
характерные для сопряженных диенов: гидрируются,
окисляются, присоединяют электрофильные реагенты
(напр., SO2) и т. д.:
СН3-СН = СН-СН=СН2 + SO2 —►
Последняя реакция иногда используется для отде-
ления П. от изопрена (образующийся сульфон при
нагревании легко разлагается на SO2 и транс-П.).
Транс-П. вступает в реакцию Дильса-
Альдера (отличие от цис-П.), напр.:
/Н2
СН
чсн
сн3
сн2
сн-со сн хсн-со
II >0 ---------- || I >0
сн-со сн сн-со
\ Z
сн
I
СН3 (т. пл 62"'
П. содержится в продуктах крекинга нефти; может
быть получен дегидратацией 2,3-пентандиола, ката-
литическим дегидрированием и-пентенов и из крото-
нового альдегида. Удобным методом получения П. яв-
ляется «исчерпывающее» метилирование пипери-
продукта метилирования по
дина и расщепление
Гофману:
Af2O,H2O CH3J
« H2C J
N
CH3 4CH3
CH2 ”1 CH
H(j Ch , HC^CH
H2c CH2 H2C CH3
П. легко полимеризуется и сополимеризуется (напр.,
с изопреном). Пленки полимера П., полученные в при-
сутствии стереоспецифич. катализатора [напр.,
(C2Hs),A1, VOC13 или CrOCl], отличаются высокой
прочностью И СТОЙКОСТЬЮ К окислению. В. Н. Фросин.
ПИПЕРИН С17Н190з N, мол. в. 285,35 — алкалоид,
выделенный из многих видов перца (Piper), где его
содержание может дости-
гать 9%. П.— кристаллы; ® \
т. пл.129—130°; оптически ^-\СН ^сн с-N >
неактивен; плохо раство- Г [ сн %СН 41—
рим в воде, лучше—в спир- 9~
те, эфире и хлороформе. I I
Спиртовые растворы П. снг~°
имеют жгучий вкус. П.
образует соли только с сильными к-тами; дает периодид
(•HJ • J2), т. пл. 145°; тринитробензоат, т. пл. 130°.
П. — слабое основание. При щелочном гидролизе он
распадается на пиперидин и пипериновую к-ту [ди-
транс-4- (3,4-метиленди оксифенил) -бутадиен -1,3-кар-
боновую кислоту]. Строение П. подтверждено син-
тезом. Для выделения П. спиртовый экстракт чер-
ного перца упаривают, промывают щелочью и из
остатка выкристаллизовывают П. Из экстракта выде-
лены также ди-^ис-изомер П. — хавицин, а также его
следующий высший винилог пипереттин.
Лит.: Генри Т. А., Химия растительных алкалоидов,
пер. с англ., М., 1956; Bolt Н. G., Ergebnisse der Alkaloid-
Chemie bis 1960, В., 1961. A. E. Васильев.
ПИПЕРОНАЛ — метиленовый эфир протокатехо-
вого альдегида; употребляется в парфюмерии; то же,
что гелиотропин.
ПИРАЗИН (1,4-диазин, пиазин) C4H4N2, мол. в.
80,09—бесцветные кристаллы с запахом, напоминаю-
щим запах гелиотропа; легко летуч с парами воды,
смешивается во всех отношениях с водой, спиртом,
эфиром; т.пл. 57°; т. кип. 118°; d’° 1,0254; Пр 1,4953.
Производные П.— пикрат, т. пл. 157°; сульфат,
т. пл. 137°; комплекс с сулемой, т. пл. 272°. П.
является электронным аналогом бензола, содержащим
систему 6л электронов («ароматический секстет»),
состоящую из 4л электронов четырех атомов углерода
и 2л электронов двух атомов азота. Строение П.
лучше всего передают ф-лы:
П.— очень слабое основание, с водой образует гидрат,
с галогеналкилами — моногалогеналкилаты (алкил
в галогеналкилатах при нагревании мигрирует к уг-
лероду). В условиях отсутствия воды П. может обра-
зовывать соль с двумя молекулами кислоты. II. легко
восстанавливается Na в спирте или др. способом в пи-
перазин, при окислении превращается в N-окись.
В реакции электрофильного замещения П. вступает
с большим трудом; так, хлорирование протекает лишь
при 400°; напротив, он сравнительно легко реагирует
1045
ПИРАЗОЛ - ПИРАЗОЛИН
1046
с нуклеофильными реагентами, например с амидом
натрия:
П. получают обработкой бромацетальдегида аммиаком,
гидролизом ацеталей аминоацетальдегида:
ВгСН2С^
'н
NH2CH2CH(OR)
о
nh2ch2c*
н
декарбоксилированием пиразинкарбоновых к-т, де-
гидрированием пиперазина и обработкой ацеталя
иминодиацетальдегида солянокислым гидроксилами-
ном:
NH
гы сн nh2oh-hci
СП2 ЬПз ------------:
<OR)2CH CH(OR)3
rNH^
нсх J2H
но'' '°" 'он
Лит.: Пратт И., в сб.: Гетероциклические соединения,
под ред. Р. Эльдерфилда, пер. с англ., т. 6,М., 1960, с. 312.
Э. Е. Нщрантьев.
ПИРАЗОЛ (1,2-диазол) C3H4N2, мол. в. 68,08 —
бесцветные кристаллы, горькие на вкус, с запахом
пиридина; т. пл. 70°; т. кип. 187°; хорошо
НС-—3СН растворим в воде (270 г в 100 г при 24,8°),
бензоле (38,6 г в 100 г), спирте, эфире, ху-
NH же — в циклогексане, плохо — в лигроине.
П.— слабое основание (рйГа= 2,53); соли П.
(с НС1, H2SO4, HNO3) полностью гидролизуются
водой. П. образует комплексы с солями тяжелых
металлов. Атом водорода у азота легко замещается
щелочными металлами, серебром, магнием. Металлич.
производные легко алкилируются и ацилируются.
Ядро П. обладает ароматич. характером. П. легко
галогенируется (легче, чем бензол, но труднее, чем
фенол и пиррол), нитруется, сульфируется; замести-
тель входит при этом в положение 4. П. устойчив
к окислению; восстановление приводит к пиразолину:
О.+ Нг ~~ о
NH NH
П. и его производные получают конденсацией гид-
разина и его производных (напр., семикарбазида и
др.) с 1,3-дикарбонильными и а, 0-ненасыщенными
соединениями:
RC=C-COR' + H2NNH2 ——
RCOCH2COR + R'NHNHj
или реакцией алифатич. диазосоединений с ацетиле-
нами:
нс=сн + ch2n2—-
NH
Известен также ряд менее общих методов. Широкое
применение получили красители и лекарственные пре-
параты на основе нек-рых производных П., т. наз.
пиразолонов (см. Пиразолом,, Пиразолоновые краси-
тели, Цветная фотография, Антипирин, Пирамидон,
Анальгин). П. является структурным изомером ими-
дазола.
Производные П.: пикрат, т. пл. 160°; оксалат, т.
пл. 192°.
Лит.: Джейкобс Т., в сб.: Гетероциклические сое-
динения, под ред. Р. Эльдерфилда, пер. с англ., т. 5, М.,
1961, с. 42. в. Н. Фросин.
ПИРАЗОЛИДИН— см. Пиразолин.
ПИРАЗОЛИДОН (дигидропиразолон, дигидроокси-
пиразол, оксопиразолидин) — циклическая система
пиразольного ряда.
НгС—С-0
Н2С^,’NH
NH
О=С СН2 Н2С--СН2
II I I
Н2сх ZNH О=СХ ZNH
NH NH
П. получают методами, аналогичными применяемым
для синтеза пиразолонов. Так, напр., взаимодействие
гидразина и замещенных гидразинов с а.0-ненасыщен-
ными к-тами приводит к образованию П.-5:
СН3СН»СНСООН + C6HSNH-NHa —
Н2С---СН-СНз
О=Сч NH
N—С6Н5
Для синтеза нек-рых замещенных П.-З используют
реакцию гидразинов с 0-галогенокислотами (или их
эфирами) и 0-пропиолактоном. П. обладают основ-
ными свойствами, легко окисляются в пиразолоны:
Н2С СН2 FeCl., Н2с СН
О=С NH О=СХ XN
'nh 'nh
П.-5—бесцветная жидкость; т. кип. 133—135°;
смешивается со спиртом и эфиром; плохо растворим
в воде.
Лит. см. при ст. Пиразол. В. Н Фросин.
ПИРАЗОЛИН (4,5-дигидропиразол) C3H,N2, мол. в.
70,09 —бесцветная жидкость; т. кип. 144°; 1,017;
” Не '1,^78; хорошо растворим в воде и н2с—5сн
спирте, ограниченно— в эфире. П.— 60- н qs
лее сильное основание, чем пиразол; об- ’ nh
разует неустойчивые комплексные соеди-
нения с солями тяжелых металлов (напр., с хлорной
платиной и др.); легко окисляется (даже кислородом
воздуха) в пиразол. С-производные П. известны в раз-
личных таутомерных формах, напр.:
Н2С----C-R
I II
Н2с^ /N
NH
НС=С- R
H2CkJlH иар'
NH
П., не замещенные у азота, сравнительно легко при
нагревании теряют азот, превращаясь в олефины или
производные циклопропана:
HjCOOC—НС С— СООСН3 150-200"
H2<k./N
NH
—>СНзООСС=СНСООСНз+ CHjOOCCH-СНСООСНз
СН3 65Х СН2 35%
Каталитич. восстановление П. (или действие цинка
и уксусной к-ты) приводит к образованию тетрагидро-
пиразола, т. наз. пиразолидина, бесцветной жидкости
с т. кип. 49—51°/25 мм.
П. и его производные получают методами, общими
для создания пиразольной системы, — взаимодейст-
1047
ПИРАЗОЛОН
1048
вием гидразина и замещенных гидразинов с а,р-нена-
сыщенными карбонильными соединениями, напр.:
ch2=chch=0 + h2nnh5 —- н2с—сн
-Н2О I И
,Н2С^ /N
NH
или реакцией алифатич. диазосоединений с олефи-
нами:
Сн.=Сн., + Ch2n,—- н2С—Сн
Н2С^
NH
Нек-рые производные П. и пиразолидина нашли при-
менение как лекарственные препараты жаропонижаю-
щего, болеутоляющего и анестезирующего действия
(напр., бутазолидин и др.).
Лит. см. при ст. Лиразол. В. Н. Фросин.
ПИРА30Л0Н (оксипиразол) C3H4N2O, мол. в.
84,08. Из трех теоретически возможных П. (3-, 4- и 5-)
Н0Т ’ll F~1 - |Г‘ зн он
4.1JN HO-^i^N \1_5n
NH NH NH
I II III
два П. (3- и 5-) и их производные 3- и 5- П., не имею-
щие заместителей у атома азота, являются таутоме-
рами; П.-4 (I) — т. пл. 118—118,5°; легко растворим
в воде и спирте, ограниченно— в эфире, плохо —
в СНС1з и бензоле; П.-5 (П.-З) — т. пл. 165°; рас-
творим в воде, спирте, плохо— в эфире и толуоле.
Для П. и их производных характерно равновесие
между таутомерными формами А, Б и В, к-рое, в за-
висимости от природы П. и его заместителей, может
быть смещено в ту или иную сторону:
в
Напр., для П.-5 и 1-фенил-3-метилпиразолона-5 (IV)
характерны реакции, отвечающие форме Б; антипирин
(V) и пирамидон (VI) существуют в форме В; П.-4 и
пикролоновая кислота (VII) — в форме А, и т. д.
Н3С
^N-r--]—СН3
HjC/o-k^N-CHj
I
С6Н5
VI
хлоралгидрата удается выделить промежуточный про-
дукт (IX). Конденсация П.-5 с нитрозосоединениями
приводит к азометиновым красителям, напр. (X):
(CH3)2n
№0
П.-5 вступают в сочетание также с солями диазония
с образованием светостойких пиразоло новых
красителей, нашедших весьма широкое приме-
нение в цветной фотографии, напр.:
С383 С6Н5
П., не замещенные у азота, при взаимодействии
с солями диазония образуют диазоаминосоединения.
Азотистая кислота превращает П.-5 в 4-изонитрозо-
соединения; при нитровании и сульфировании в пер-
вую очередь затрагивается пара-положение 1-фепиль-
ной группы; нитрование 1-фенил-З-метил-пиразоло-
на-5 в жестких условиях приводит к пикролоновой
кислоте. Галогенирование П.-5 идет также в поло-
жение 4;
С1
I---ГСн’ -С1^ СЙ---------гСНз
1 u 1
с6н5 с6н5
П. сравнительно устойчивы к действию восстанавли-
вающих агентов; так, 1-фенил-3-метил-пиразолон-5
не изменяется при обработке натрием в спирте, цин-
ком в соляной к-те, устойчив к действию HJ при 180°;
при перегонке над цинком или красным фосфором
образует 1-фенил-З-метилпиразол; каталитич. восста-
новление приводит к расщеплению кольца. При обра-
ботке хлорным железом и др. окислителями П. очень
легко окислиются, напр.:
Кипячением в ксилоле с PzSj П. гладко превращаются
в соответствующие тиопроизводные:
V
OjN-j----ir-CH,
hoAn,n
I
c6h4no2
VII
Наиболее широко изучены П.-5 и его многочисленные
производные. П.-5 и П.-З, содержащие активную
метиленовую группу в положении 4, вступают в реак-
ции с карбонильными соединениями, нитрозосоедине-
ниями и т. п. Так, П.-5 и его производные конденси-
руются с кетонами и альдегидами; напр., с ацетоном
при нагревании образуется соединение (VIII); в случае
(СН3)2С-г—рСН,
I
VIII СбН‘
СС13~СН(ОН)-Т---Ц-СН3
^N'N
I
IX сбН3
I
С6н5
CHj
Алкилирование II.-5 приводит, в зависимости от
природы реагента и условий реакции, к различным
продуктам: так, напр., 1-фенил-3-метил-пиразолон-5
с CH3J (при 100°) образует антипирин (V), с диазо-
метаном — 1-фенил-3-метил-5-метоксипиразол (XII),
а с диметилсульфатом либо V, либо XII:
1049
ПИРАЗОЛОНОВЫЕ КРАСИТЕЛИ — ПИРАН
1050
При ацилировании П. получаются как О-, так и
С-ацильные производные. Методы синтеза П. анало-
гичны методам синтеза пиразола и пиразолинов.
Наиболее общим методом является взаимодействие
гидразинов с р-кетоэфирами, напр.:
СН3СОСН2СООС2Н5 + c6h5nh-nh2
С6н5
о
NH
ОСН-СН2СООС2Н5 + nh2nh2
Нек-рые П. используют в произ-ве красителей и ле-
карственных препаратов болеутоляющего и жаропо-
нижающего действия (антипирин, пирамидон,
анальгин и др.).
Лит.: Джейкобс Т., в сб.: Гетероциклические сое-
динения, под ред. Р. Эльдерфилда, пер. с англ., т. 5, М., 1961,
с. 84. В. Н. Фросин.
ПИРАЗОЛОНОВЫЕ КРАСИТЕЛИ — группа кра-
сителей, получаемых с применением различных про-
изводных пиразолона и пиразола. П. к. чаще всего
являются азокрасителями и образуются сочетанием
солей диазония с замещенными 5-пиразолона, реже —
с производными 5-аминопиразола. П. к. принадлежат
к группам кислотных хромовых (протравных),
прямых, активных (проционовых), ацетонораствори-
мых красителей, а также пигментов и лаков. Для
П. к. характерны яркие и высокие цвета (желтые,
оранжевые, красные) и повышенная по сравнению
с другими азокрасителями светопрочность. Благо-
даря этим ценным свойствам П. к. широко применяют
в текстильной пром-сти, а также для окраски резины,
пластич. масс, пищевых продуктов (тартразин). Наи-
большее значение имеют кислотные и активные П. к.
ярко-желтого цвета, а также кислотные красители
оранжевого,
кобальт или
рых П. к.
алого, коричневого цветов, содержащие
хром. Ниже приведено строение некото-
r'-N-N-C С—R'
Н В
НО-С N
4NZ
R'"
ирвситель R' R" R"'
нсябтмый желтый светопрочный с6н6- -CH3 ^O3Na
емтмвиый яркожелтый SO3Na NH 1 Zx N N 1 H Cl-C4 ZC-CI N -CH3 Zrci ci-kj SO3Na
ТВрТрбЗНН NaO3S-^~~y~ —COONa ^O3Na
М. А. Чекалин.
Лит. см. к статье Азокрасители.
° 35 К.Х.Э. т. 3
-1==Гснз
<N>-CH3
ПИРАМИДОН (амидопирин, 1-фенил-2,3-диметил-
4-диметиламинопиразолон-5) С1зН17ОМз, мол. вес
231,30 — бесцветные кристаллы
слабогорького вкуса; т. пл. 107—
109°; легко растворим в больший- ZN
стве органич. растворителей, ра- Н3С о
створим в воде 1 : 18 (максимум ,
растворимости при 70°). Водные с,Н5
р-ры П. имеют слабощелочную ре-
акцию; с кислотами П. образует легко диссоции-
рующие соли; цветные реакции и комплексообра-
зование П. аналогичны антипирину. П. окисляет-
ся Н2Ог или KMnOi в диметиламид р-ацетил-р-
метилфенилгидразин-а-щавелевой к-ты (диоксипи-
рамидон) — бесцветные призмы, т. пл. 103—104°.
Количественно П. определяют титрованием 0,5 н.
НС1 в присутствии индикатора — смесь метило-
вого оранжевого и метиленового синего (3:2). По-
лучают П. из антипирина его нитрозированием до
нитрозоантипирина, восстановлением последнего и
метилированием образующегося аминоантипирина.
Применяют П. в качестве жаропонижающего, боле-
утоляющего и противовоспалительного средства.
Лит. см. при ст. Анальгин. Р. Г. Глушков.
ПИРАН С5Н6О, мол. в. 82,10 —шестичленное гете-
соединение, содержащее атом кислорода
в цикле. Двойные связи П.
могут быть либо сопряжены
Ч (а-пиран), либо изолированы
Ор (у-пиран). а-П. известен только
в виде производных (напр.,
“ 2-метил-а-пиран, т. кип. 106—
роциклич.
ос —пиран у-пнран Ш°; d|°0,907, nD 1,4542; 2,4,6-
(1,2-лиран) (1,4-пиран) Трифенил-а-пиран, т. пл. 225°
и др.). Многочисленные попыт-
ки получения а-П. окончились неудачей очевидно
вследствие крайней неустойчивости пиранового
кольца. у-П.— бесцветная жидкость, т. кип. 84°
(с разл.); быстро разлагается при комнатной темп-ре;
каталитически возбужденным водородом восстанавли-
вается в тетрагидропиран:
у-П. получен пиролизом (350°) 2-ацетокси-3,4-дигидро-
у-пирана:
_____1
ОСОСН3 -ch3csoh
Более доступными н устойчивыми являются дигидро-
и тетрагидропираны; так, напр., тетрагидропиран —
внутренний эфир пентадиола-1,5 —получают нагрева-
нием последнего с 60%-ной H2SO4. Наиболее важными
производными П. являются кетопираны, т. наз. а- и
у-пироны (см. 2,6-Диметилпирон, Кумарин, Хромоны,
Флавоны, Ксантон), и родственные им соединения
(хелидоновая кислота, меконовая кислота). Пирановое
кольцо широко распространено в природе: напр.,
гексозы (альдозы и кетозы) в большинстве своем яв-
ляются шестичленными пиранозами:
СНОН
НОСН СНОН
I I
НОСН СН"СН2ОН
чох
СНОН
НОСН СНОН
СН2 с^он
ХОХ ХСН2ОН
альлогеисозы четогеисоза
Лит.: Фред Д., в сб.: Гетероциклические соединения,
под ред. Р. Эльдерфилда, пер. с англ., т. 1.М., 1953, с. 269;
Chemistry of carbon compounds, ed. by E. H. Rodd, v. 4, pt
B., Amst.—L.—N. Y., 1959, p. 810—14. В. H. Фросин.
1051
ПИРЕН — ПИРИДАЗИН
1052
ПИРЕН С,вн,о, мол. в. 202,26 — бледно-желтые
пластинки; т. ил. 150°; т.кип. 392°;d* 1,277; сублими-
руется; в 100 г абс.
Р I 1 [э 10 [ 'sil спирта растворяется
AAJ или 1,37 г (16°) и 3,08 г (т.
Р9° Т J If Т Т кип.); в 100 г толуола
растворяется 16,54 г
(18°); очень легко раст-
воряется П. в сероуглероде, эфире, бензоле; как в
твердом виде, так и в растворе обладает голубой флу-
оресценцией; т. пл. пикрата 222° (красные иглы из
спирта); т. пл. комплекса с тринитробензолом 245°.
Хромовой смесью П. окисляется в пиренхинон-3,8
С1вН6О2 и пиреновую к-ту С13Н6О (СООН)2; гидриро-
вание дает различные продукты в зависимости от усло-
вий и катализатора: на сернисто-молибденовом
катализаторе были получены 1,2-дигидро- и 1,2,6,
7-тетрагидропирены (нумерация дана по левой ф-ле);
на ^медно-хромовом —• симметричный и несимметрич-
ный гексагидропирены, на никеле Ренея — гексаде-
капирен; П. присоединяет литий (вероятно, в поло-
жение 3,10), образуя производное, к-рое при обработке
СО2 дает дигидропирендикарбоновую к-ту; присоеди-
няет также натрий. Положения 3,5,8 и 10 в П. являют-
ся наиболее реакционноспособными. Легко образуют-
ся 3-хлор-, 3-бром-, 3-нитро-, 3-сульфо- и 3-альдегидо-
производные. Дальнейшее замещение дает 3,8-дизаме-
щенные, 3,5,8,10-тетразамещенные и более высоко-
замещенные П. При прямом хлорировании П. получен
1, 3, 4, 6, 8, 9-гексахлорпирен.
Прежде П. добывался из сажи, образующейся как
побочный продукт при выплавке ртути из битуминоз-
ных киноварных руд; разработан промышленный
способ получения из кам.-уг. смолы; образуется
при крекинге нефти и при гидрогенизации гудрона
под высоким давлением.
Лит.: Chemistiy of carbon compounds, ed. E. H. Rodd,
v. 3, pt B, Amst.— L.— N. Y., 195G, p. 1502; Фи'зер Л.,
Физер M., Органическая химия, пер. с англ., М., 1949,
С. 71.4. л. С. Поваров.
ПИРЕТРИНЫ (и их синтетические аналоги —
пиретроиды) — инсектициды, содержащиеся
в цветках нек-рых видов ромашки (рода хризантеми-
ум). П.—едва ли не первые инсектициды, получившие
практич. применение в борьбе с вредными насекомы-
ми. Инсектицид, выделяемый из цветков ромашки,
состоит в основном из четырех соединений: п и р е т-
рпна-1(1), пиретрина-2 (II), ц и н е р и н а-1 (III)
и цинерин а-2 (IV), к-рые представляют собой слож-
ные эфиры хризантемовой монокарбоновой и хри-
зантемовой дикарбоновой к-т и циклич. спиртов пи-
ретро лона и цинеролона.
н3с сн3 снэ
R\ /СС zK
С=СН—СН—СН —COO—СН c-ch2-cii-ch-r'
Н.С I I
"зС сн-со
I R=-CH3; R,= —СН = СН2
II R= —СНЭОСО , R'= —СН=СН2
111 R--CH3 : R'=-сн3
IV R=—CHjOCOi R'=-CH3
П. обычно используются в виде разб. р-ров
(2—10%-ных), получаемых экстракцией цветков ро-
машки органич. растворителями. За рубежом (США)
выпускаются препараты, содержащие до 90% П. Та-
кие препараты получают отгонкой растворителя от
экстрактов при низкой темп-ре.
Технич. П. представляют собой густую маслянистую
жидкость, не перегоняющуюся без разложения в обыч-
ном вакууме, плотность несколько больше единицы.
Токсичность для мышей ЛД50 = 1500 мг/кг, для мор-
ских свинок 150 мг/кг.
Инсектицидное действие П. достаточно высокое;
эти соединения довольно быстро действуют на насеко-
мых, однако в нек-рых случаях после действия II.
насекомые оживают. Действие П. усиливается в при-
сутствии различных веществ — синергистов. П. чаще
всего применяют для борьбы с синантропными насе-
комыми (постельные клопы, вши и т. п.), однако
вследствие малой стабильности действие их кратко-
временно.
Синтезирован ряд аналогов П. — аллитрин
(V), циклитрин (VI) и фуретрин (VII),
получивших практич. применение.
Н3С ZCH3 СН3
нс С С
н3с / \ / %
с=сн-сн-сн-СОО-СН C-R
u rz 11
нзС сн2-со
v r=-ch2-ch=ch2
/°х
VI R—сн-с сн
II II
сн—сн
ZCH=CH
VII R=-CH I
хсн2-сн2
Технич. аллитрин (V) — светлая жидкость, d’®
1,005—1,015, n’D’ 1,504, токсичность для эксперимен-
тальных животных 480—920 мг/кг. Циклитрин (VI)—
жидкость, d™ 1,02, п” 1,517, токсичность для экспе-
риментальных животных 280—600 мг/кг. Фуретрин
(VII) — жидкость (т. кип. 187—18870,4 мм рт. ст.),
d”l,Q25, га” 1,52, токсичность близка к токсичности
циклитрина (VI). Инсектицидное действие синтетич.
аналогов П. несколько слабее, чем природных II., но
в смеси с синергистами оно уравнивается.
Синтетич. аналоги П. получают из хлорангидрида
хризантемовой кислоты (VIII) и аналогов пиретро-
лона (IX),
Н3С
С = СН-СН-СОС1
h,cz 4cz
J \
HjC сн3
VIII
IX
к-рые в присутствии пиридина или другого третич-
ного амина образуют соответствующий эфир. Технич.
синтетич. П. обычно представляет собой смесь 8 изо-
меров, содержание к-рых в технич. препарате состав-
ляет 75—98%.
Лит.: Metcalf R. L., Organic insecticides. L.—N. Y.,
1961; Мельников H. H., Швецова-Шилов-
c к а я К. Д-, Хим. пром-сть, 1955, № 3, 178.
Н. Н. Мельников.
ПИРЕТРУМ — мелко размолотый тонкий порошок
цветка далматской ромашки, применяемый для борьбы
с вредными насекомыми. Обычно такой порошок со-
держит не менее 0,15—0,5% пиретринов.
ПИРИД'АЗИН (1,2-диазин, ойазин) C4H4N2,
мол. в. 80,09 — бесцветная жидкость со слабым запа-
хом белены; т. пл.—8°, т. кип. 208°; <3“°1,1054; «д
1,5231; легко растворим в бензоле, спирте, эфире и
воде, плохо — в петролейном эфире; пикрат, т. пл.
1053
ПИРИДИН
1054
169°, комплекс с четыреххлористой платиной, т. пл.
180°; поверхностное натяжение 46,9 дин/см (34°),
дипольный момент 4D. П. является электронным ана-
логом бензола, содержащим сопряженную систему
6л:-электронов («ароматический секстет»). Строение
П. лучше всего передают ф-лы:
П. — очень слабое однокислотное основание. Он
очень плохо вступает в реакции электрофильного
замещения. При восстановлении натрием в спирте
П. расщепляется до тетраметилендиамина:
<7, И
L Д —- nh2ch2ch2ch2ch2nh2
П. получают из малеинового альдегида и гидразина,
окислением дегидропиридазина, каталитич. восста-
новлением 3-хлор- и 3,6-дихлорпиридазина и декарбо-
ксилированием пиридазин-3- и пиридазин-4,5-дикар-
бон овых к-т.
Лит.: Джейкобс Т., в сб.: Гетероциклические со-
единения, под ред. Р. Эльдерфилда, пер. с англ., т. 6, М.,
1960, с. 88. Э. Е. Нщрантьев.
ПИРИДИН C6H6N, мол. в. 79,098 — "бесцветная
жидкость со специфич. запахом; т. пл. —41,8°;
_ т. кип. 115,587760 мм; 95,6°/600 мм;
s 75,07200 мм; 57,87100 мм; 13,2710 мм;
d™ 0,9819; 1,5100; теплоемкость 32,4
« кал/молъ-град (17°); т. воспл. 23,3°; тепло-
та сгорания 664 ккал/моль (при постоянном давлении);
уд. теплота парообразования 107,38кал/г (114,13°).
П. смешивается во всех отношениях с водой и боль-
шинством органич. растворителей. С водой П.
образует азеотропную смесь состава (на 1 моль П. 3 мо-
ля Н2О) с т. кип. 92—93°. Наличие неподеленной пары
электронов на атоме азота в П. обусловливает его ос-
новные свойства (ЙГПСН =2,3-10-9), к-рые,однако, более
слабо выражены, чем у аминов жирного ряда. Ослаб-
ление объясняется тем, что неподеленная пара элект-
ронов на атоме азота в П. находится на плоско-триго-
нальной орбите, имеющей больший S-характер по
сравнению с аминами жирного ряда, основность к-рых
обусловлена наличием неподеленной электронной пары
на тетраэдрич. орбите.
П. является типичным ароматич. соединением,
имеющим секстет л:-электронов, образующих единую
замкнутую электронную систему аналогично бензолу.
Реакции электрофильного замещения в П. протекают
несравненно труднее, чем в бензоле, т. к. в П. стяги-
вающее действие атома азота снижает общую плот-
ность электронов в ароматич. ядре, осо-
бенно в положениях 2,4 и 6. Кроме того,
эти реакции, как правило, проводят в кис- Ц |1/
лой среде, в к-рой П., реагируя в виде со- 8+°
ответствующей соли, несет на атоме азота
положительный заряд. П. нитруется лишь при 300°,
образуя 3-нитропиридин с небольшим выходом. Суль-
фирование П. также происходит только при высоких
темп-рах в присутствии катализаторов. Действием аце-
тата ртути на П. при 155° был получен 3-пиридилмер-
курацетат; при более высоких темп-рах образуются
ди- и полизамещенные производные. Бромирование П.
при 300° приводит к смеси 3-бромпиридина и 3,5-ди-
бромпиридина. При увеличении темп-ры до 500°ос-
новными продуктами реакции становятся 2-бром-
и 2,6-дибромпиридины. Изменение ориентации при
повышении температуры объясняется тем, что в этих
условиях процесс принимает радикальный характер.
35*
К реакциям радикального замещения в П. относится
фенилирование при действии фенилдиазогидрата (см.
Гомберга-Бахмана-Гея реакция), в результате к-рого
образуется смесь, содержащая 55% 2-фенил-, 30%
3-фенил- и 15% 4-фенилпиридина.
Нуклеофильное замещение в П. протекает легче, чем
в бензоле, что также определяется общим понижением
электронной плотности в ядре. Типичной реакцией
нуклеофильного замещения является синтез 2-амино-
пиридина при действии на П. амида натрия (см. Чи-
чибабина реакция и Аминопиридины). По нуклео-
фильному механизму также происходит гидроксили-
рование П. до а-оксипиридина при действии КОН
(см. Оксипиридины). П. является весьма эффективным
комплексообразователем и образует устойчивые соли
с большинством неорганич. к-т, а также с многими
солями — А1С13, ВеС12 и др.
Характерными производными П. являются хлоро-
платинат (C5H5N .HCl)2.PtC12, т. пл. 262—264°
(с разл.); хлормеркурат C5H5N-НС1-2Н§С12, т. пл.
177—178°. Комплекс П. с SOs— пиридинсульфотри-
оксид — является мягким сульфирующим агентом;
комплекс с бромом — C5H5NBr2-HBr— применяется
для введения брома в органич. соединения.
Четвертичные соли П. с органич. соединениями
играют существенную роль в химии П. При действии
алкилирующих агентов — галогеналкилов или диал-
килсулыфатов П. легко переходит в соли алкилпири-
диния C5H5N-RX, к-рые при нагревании образуют
пиридины, алкилированные в ядре (см. Ладенбурга.
реакция). Неустойчивые комплексы П. с галогенан-
гидридами к-т являются источниками ацилкатиона
в ацилировании по Шоттена — Баумана реакции.
Большинство реакций гидролитич. расщепления
ядра П. является реакциями его солей. Особенно
легкому расщеплению подвергаются комплексы П.,
несущие у атома азота электроностягивающие груп-
пировки. В частности, комплекс П. с 2,4-динитро-
хлорбензолом при действии щелочей разлагается с об-
разованием глутакондиальдегида, к-рый легко выде-
ляется в виде анила. Аналогично расщепляется
комплекс П. с бромцианом:
Н.О; C,H,NH,
(C5H5NCN) + Вг - —----->
—> CeHsN = CH-CH = CH-СН = СН-NHCeHs HBr
Расщепление комплекса П. с бромцианом было с ус-
пехом использовано для синтеза полиметиновых со-
единений. Образование анила глутакондиальдегида
при гидролитическом расщеплении солей пиридиния
в присутствии анилина является весьма чувстви-
тельной реакцией, которая положена в основу ряда
качественных и количественных методов определе-
ния П.
П., как правило, устойчив к окислителям, однако
при действии специфич. окислителей — кислоты Каро
или перекиси бензоила — легко образует N-окись.
При высокой темп-ре под действием БеС1з П. окис-
ляется в смесь изомеров дипиридила:
FeCl.; 300°
C3HSN--------^C.H4N-CsHtN
-н2
П. восстанавливается до пиперидина при действии Na
в спирте, каталитич. гидрированием над Ni либо
электролитически. Последний способ нашел примене-
ние в промышленности. Более жесткое восстановление
сопровождается расщеплением цикла и дезаминиро-
ванием:
HJ
cbh5n —> c5hI2+nh,j
Основным источником получения П. является
кам.-уг. смола, в к-рой содержание его достигает
1055
ПИРИДИНКАРБОНОВЫЕ КИСЛОТЫ
1056
0,08% (подробнее см. Пиридиновые основания). Син-
тетич. методы получения П. имеют лишь теоретич.
значение. Основные из них — каталитич. конденсация
бутадиена или ацетилена с HCN:
/Н,
сн
I + Н — C=N
СН
чсн2
Q 2НС = СН + HCN
действие гидроксиламина на 2-алкокси-3,4-дигидро-
пираны:
NH2OH -HCI
120”
(70-80Х)
каталитич. аминирование тетрагидрофурилового спир-
та над восстановленным молибдатом аммония:
NH3
катализатор
(45Z)
П. получают в больших количествах, применяют
в синтезе красителей, лекарственных веществ, инсек-
тицидов. П. — хороший растворитель, в том числе и
для многих неорганич. солей (AgBr, HgCl и др.), его
широко применяют для денатурации этилового спирта.
П. токсичен; максимально допустимая концентрация
паров в воздухе ~ 0,005 мг!л. П. впервые был выделен
Андерсеном в 1849 из костяного масла. Структура П.
установлена Дьюаром и Кернером.
Лит.: Pyridine and its derivatives, ed. E. Klingsberg, pt 1,
N.Y.'—L., 1960; Мошер Г., в кн.: Гетероциклические со-
единения, под ред. Р. Эльдерфилда, пер. с англ., т. 1. М., 1953;
Eerie в М., Jizba J., Chemie pyridinu, Praha, 1957.
Л. С. Герман.
ПИРИДИНКАРБОНОВЫЕ КИСЛОТЫ — заме-
щенные пиридина, содержащие одну или несколько
карбоксильных групп. Известны все 19 моно- и поли-
замещенных П. к. Свойства важнейших П. к. приве-
дены в таблице.
Кислота Т. пл., °C Производные
Пиридин-2-карбоно- вая (пиколиновая) 135-136 HCl-соль, т. пл. 47°; мети- ловый эфир, т. кип. 112°/13 мм; амид., т. пл. 107°
Пиридин-З-карбоно- __ См. Никотиновая кислота
вая
Пиридин-2,3-дикар- разл. Ангидрид, т. пл. 134°;
боновая (хинолино- вая) >190 имид, т. пл. 230°; диме- тиловый эфир, т. пл. 53°
Пиридин-3,4-дикар- боновая (цинхоме- роновая) 266—268 Ангидрид, т. пл. 67°; диме- тиловый эфир, т. кип. 160—171°/28 Л4.м; имид, т. пл. 230°
Пиридин-2.4-дикар- 248—250 Диметиловый эфир, т. пл.
боновая (лутидино- вая) (дигидрат) 58°; диамид, т. пл. 254— 255°
Пиридин-2,5-дикар- боновая (изоцинхо- 254 (моно- гидрат) Диамид, т. пл. 310—313°
мероновая)
Пиридин-2,6-дикар- 235—236 Диамид, т. пл. 302°
боновая (дипиколи- нов ая)
Пиридин-3,5-дикар- 322 Диамид, т. пл. 303—304°
боновая (диникоти- новая)
Пиридин-4-карбоно- вая (изоникотино- вая) 315 HCl-соль, т. пл. 16°; мети- ловый эфир, т. кип. 207— 20х9°/8,5 ллс; амид, т. пл. 155°
П. к. ведут себя как типичные ароматич. к-ты,
однако наличие пиридинового кольца придает им
слабоосновной характер. В П. к., содержащих не-
сколько карбоксильных групп, основные свойства
выражены еще слабее. Пиридин-2-карбоновая к-та и
ее гомологи, в отличие от 3- и 4-изомеров, способны
к образованию окрашенных комплексов с солями меди
и железа, что находит применение в аналитич. прак-
тике. Декарбоксилирование П. к. происходит легче,
чем в ароматич. ряду. Карбоксильная группа в 2-
и 4-положениях легко элиминируется, в то время как
3- и 5-замещенные П. к. относительно стойки.
СООН
СООН
СООН
180° _
-со/
соон
соон
Характерным свойством П. к. является образование
бетаинов при взаимодействии с галогеналкилами в ще-
лочных средах (1); бетаин образуется также при пере-
группировке эфиров изоникотиновой к-ты (2):
Многие П. к. относительно легко замещаются в ядре
(нитрование, галогенирование и др.), аналогично дру-
гим производным пиридина. Необычным является
галогенирование изоникотиновой к-ты при действии
хлористого тионила (3). Восстановление П. к. в зави-
симости от условий приводит к различным производ-
ным пиридинового и пиперидинового рядов (4):
Основными методами синтеза П. к. является окис-
ление замещенных пиридинов, а также конденсиро-
ванных систем, содержащих пиридиновое кольцо (хи-
нолин, изохинолин, хинин и др.). В ряде случаев
нитрилы П. к. получают действием смеси аммиака
и кислорода (при недостатке последнего) на соответ-
ствующие пиколины при повышенной темп-ре. Вы-
ходы достигают 80%. Гидролиз нитрилов приводит
к П. к.:
Частичным окислением полиалкилпиридинов полу-
чают алкилзамещенные П. к. Конденсациями различ-
1057
ПИРИДИННУКЛЕОТИДЫ — ПИРИДОКСАЛЕВЫЕ ФЕРМЕНТЫ
1058
вых соединений алифатич. ряда с успехом получают
замещенные П. к.:
-н,о
СН,-С-СН,-С-СООС2Н5 + СН,-С=СН-СООС,Н5—
3 л 2 II I 5
о о nh2
СООС2Н5
___~ ZAaS-COOC2H5
н3с—Ln#^—ch3
соон
Аналогично синтезированы к-ты, содержащие галоген,
окси- и аминогруппы. Нек-рые производные П. к.
встречаются в природных продуктах. Наиболее важ-
ное значение в ряду П. к. имеют никотиновая кислота
и ее производные. Другие П. к. находят применение
в аналитич. практике. Нек-рые эфиры пиридин-2,5-ди-
карбоновой к-ты представляют интерес в качестве
репеллентов.
Лит.: Гетероциклические соединения, под ред. Р. Эльдер-
филда, пер. с англ., т. 1, М., 1953, с. 437; Perles М., J 1 z-
b a J., Chemie pyrldinu, Praha, 1957, s. 382. Л. С. Герман.
ПИРИДИННУКЛЕОТИДЫ — группа нуклеоти-
дов, включающая в себя никотинамиднуклеотидные
коферменты, являющиеся коферментами многочис-
ленных оксидоредуктаз (см. также Кодегидрогеназы).
ПИРИДИНОВЫЕ ОСНОВАНИЯ — технич. смесь
гетероциклич. органич. оснований, содержащая пири-
дин и его гомологи (пиколины, лутидин ы—
см. Метилпиридины — и др.). П. о. образуются при
коксовании, полукоксовании и газификации каменных
и бурых углей, сланцев, торфа, обугливании древе-
сины, костей и др. При этом более 60% П. о. переходит
в коксовый газ (см.Газ коксовый); в кам.-уг. смоле со-
держится ок. 0,1% П. о. и ок. 2% хинолиновых осно-
ваний. Наиболее богаты П. о. продукты коксования
каменных углей, к-рые и служат основным источни-
ком их промышленного получения. При коксовании
углей общий выход П. о. колеблется от 70 до 140 г/т
угольной шихты и зависит от природы углей, режима
коксования и др. факторов. Основное количество
наиболее летучих П. о. извлекается из коксового газа
при произ-ве сульфата аммония. При этом часть маточ-
ного р-ра при накоплении в нем ок. 25 г/л П. о. отби-
рается в виде сульфатов оснований на пиридиновую
установку, где нейтрализуется парами аммиака из
аммиачной колонны. Образующаяся при этом гетеро-
азеотропная смесь вода — П. о. отгоняется и конден-
сируется, а затем в сепараторе происходит разделение
сырых П. о. и водного р-ра аммиачных солей. Сырые
П. о. содержат до 30% воды, до 10% нейтральных
масел и 60—70% оснований. Отделение сырых П. о.
производят с помощью конц. р-ра NaOH или азео-
тропной ректификацией с бензолом.
Эффективной ректификацией из сырых П. о. полу-
чают до 30% пиридина, до 15% а-пиколина, до 10%
Р-пиколиновой фракции, содержащей, в основном,
2,6-лутидин, р - и у-пиколины и др. фракции, состоящие
из высших гомологов пиридина. Дальнейшее выделе-
ние индивидуальных соединений — гомологов пири-
дина из фракций П. о.— производится химич. путем.
Разделение р-пиколиновой фракции с получением
2,6-лутидина и Р- и у-пиколинов можно производить
также и азеотропной ректификацией с водой или
уксусной к-той.
Значительное количество П. о., содержащихся
в сыром бензоле, а также в легкой, фенольной и,
в меньшей степени, в нафталиновой фракциях кам.-уг.
смолы, экстрагируется из этих продуктов слабой
серной к-той и затем перерабатывается совместно
с П. о. из коксового газа.
Наиболее ценными составляющими П. о. являются
пиридин и пиколины (см. Метилпиридины), к-рые
широко применяют в произ-ве фармацевтич. препара-
тов. Кроме того, пиридин, его гомологи, а также-
фракции П. о. широко применяют как растворители,
антисептич. средства, в произ-ве красителей, каучука,
при очистке антрацена, в произ-ве ионообменных
смол, в качестве ингибиторов коррозии, для денату-
рирования спирта и др.
Лит.: Гетероциклические соединения, Сб. статей, под
ред. Р. Эльдерфилда, пер. с англ., т. 1, М., 1953; Литви-
ненко М. С., Носалевич И: М., Химические про-
дукты коксования для производства полимерных материалов,
Харьков, 1962; Дирихс А., К у б и ч к а Р., Фенолы и
основания из углей, пер. с чеш., М., 1958. В. Е. Привалов.
ПИРИДОКСАЛЕВЫЕ ФЕРМЕНТЫ — ферменты,
прос топической группой к-рых является пиридок-
саль-5-фосфат. К П. ф. относятся след, ферменты,
катализирующие различные превращения амино-
кислот: трансаминазы, рацемазы, декарбоксилазы,
кинурениназа, цистатионаза (гомосериндезаминаза),
цистатионинсинтетаза, серин- и треониндегидразы,
триптофаназа, триптофансинтетаза, моноаминокси-
даза, серинтрансоксиметилаза и др. Пиридоксальфос-
фат, обнаруженный в фосфорилазе мышц, выполняет,
по-видимому, в ней, в отличие от других П. ф., струк-
турную, а не каталитич. функцию, участвуя в со-
единении 4 субъединиц или мономеров, образующих
молекулу фосфорилазы «а».
Общая теория действия П. ф. была разработана
в 1952 А. Е. Браунштейном и М. М. Шемякиным.
Согласно этой теории, каталитич. действие П. ф.
обусловлено способностью альдегидной группы пири-
доксальфосфата образовывать азометины I (шиффовы
основания) при взаимодействии с аминами, в том числе
с аминокислотами:
В возникающей при этом системе сопряженных
двойных связей в азометине происходит смеще-
ние электронов по направлению от а-углеродного
атома аминокислоты к электрофильному атому азота
гетероцикла. Это смещение может усиливаться под
влиянием различных дополнительных индуктивных
воздействий, напр. вследствие образования циклич.
структуры с участием фенольного гидроксила пирид-
оксальфосфата, атома азота шиффова основания и
внешней электроноакцепторной группы (иона металла
или полярной группы белка). Понижение электронной
плотности у а-углеродного атома аминокислоты в азо-
метинах типа (I) и (II) приводит к поляризации связей
этого атома. Направление и специфичность происхо-
дящих далее реакций определяются особенностями
структуры специфич. белка — апофермента — и ха-
рактером его связей с пиридоксальфосфатом. Связь
между пиридоксальфосфатом и белком в П. ф. осущест-
вляется через альдегидную и фосфатную группы и,
возможно, также через фенольный гидроксил и атом
азота гетероцикла пиридоксальфосфата. Альдегидная
1059
ПИРИДОКСАЛЬ-5-ФОСФАТ — ПИРИДОКСИН
1060
группа пиридоксальфосфата образует альдиминную,
или азометиновую, связь (внутримолекулярное осно-
вание Шиффа) с е-ГШ2-группой остатка лизина в моле-
куле белка.
Есть основания считать, что первым этапом реакций,
катализируемых И. ф., является взаимодействие
с аминокислотой с замещением или подстановкой
в альдиминной связи, в результате чего образуется
пиридоксилиденаминокислота (азометин I) и освобож-
дается NH2-rpynna белка (см. рисунок). В составе
фермент-субстратного комплекса азометин I подвер-
11 О
о °
о
I
"О-Р
N
II
СН.
,сн2. Т о-
R
+ I
ННЭ-С-СООН
Н
сн.
Н
I
N-C-COOH
I
R
О-
“О
. I
"О-Р
“'Vi-
ci 0
N^CH
й
гается далее различным превращениям, к-рые наи-
более хорошо изучены в случае реакции переамини-
рования.
Пиридоксилиденаминокислота, образующаяся на
первом этапе этой реакции, подвергается таутомер-
ному превращению в пиридоксилиминокислоту (азо-
метин II), к-рая легко гидролизуется с образованием
пиридоксаминфосфата и а-кетокислоты. Далее реак-
ция идет в обратном направлении между пиридок-
саминфосфат-протеидом и другой кетокислотой, что
приводит к образованию новой аминокислоты и пири-
доксальфосфат-протеида. Т. обр., в реакции переами-
нирования пиридоксальфосфат, прочно соединенный с
белком (апотрансаминазой), функционирует в качестве
промежуточного акцептора 1ЧН2-группы. Травсами-
назы отличаются от других П. ф. тем, что кофермент-
ные функции в них могут выполнять как пиридоксаль-
фосфат, так и пиридоксаминфосфат, подвергающиеся
взаимопревращению в ходе ферментативного переами-
нирования.
Рацемизация а-аминокислот под действием П. ф.,
подобно переаминированию, осуществляется путем
образования азометинов I и их обратимого превраще-
ния в азометины II. Это превращение, связанное
с диссоциацией атома водорода и перемещением свя-
зей, влечет за собой временное исчезновение центра
асимметрии и рацемизацию молекул. Механизм реак-
ций рацемизации аминокислот отличается, однако,
от механизма переаминирования тем, что возникаю-
щий в процессе рацемизации азометин типа (II) не
подвергается гидролизу, а вновь превращается в азо-
метин типа (I), к-рый в конечном итоге гидролизуется
с образованием D, L-аминокислот.
При декарбоксилировании аминокислот, катали-
зируемых П. ф., азометины типа (II), по-видимому,
вообще не образуются, т. к. не обнаружено диссоциа-
ции водорода, присоединенного к а-углеродному
атому.
Лит.: Браунштейн А. Е., Шемякин М. М.,
ДАН СССР, 1952, 85, № 5, 1115; Биохимия, 1953, 18, М 4,
393; The enzymes, ed. Р. D. Boyer, H. Lardy, К. MyrbSck,
v. 2, 2 ed., N. Y.—L., 1960, p. 113; Браунштейн A. E.,
в кн.: Труды V Международного биохимического конгресса.
Молекулярные основы действия и торможения ферментов.
Симпозиум IV, М., 1962; Metzler D. Е., Ikawa М.,
Snell Е. Е., J. Amer. Chem. Soc., 1954, 76, № 3, 648.
Ю. М. Торчинский.
ПИРИДОКСАЛЬ-5-ФОСФАТ (2-метил-3-окси-4-фор-
мил-5-пиридилметилфосфат) CsHI0NO6P, мол. вес
247,15 — основная биокаталитически активная форма
витамина В,; желтый кристаллич. порошок, раство-
рим в воде. В нейтральных и щелочных р-рах П. раз-
лагается под действием света. Спектры поглощения
СНО ОН
I
НО СНа-О-Р=О
Н3С он
сн
й
растворов И. обладают характерными максимумами
при 295 ммк (в 0,1 н. НС1), при 330 и 388 ммк (pH
7,0) и при 305 и 388 ммк (в 0,1
н. NaOH). Молярная экс-
тинкция моногидрата П. при
388 ммк (в 0,1 н. NaOH) рав-
на 6600; 5-фосфатная группа
П. гидролизуется с трудом;
при pH 1,5—2,0 ее гидролиз идет более быстро.
При освещении р-ров П. или сухих пятен его на
фильтровальной бумаге УФ-светом наблюдается ин-
тенсивная флуоресценция, особенно в
щелочной среде. П. легко взаимодей-
ствует с карбонильными реагентами
(гидроксиламином, гидразином, би-
сульфитом, цианидом). Альдегидная
группа П. легко вступает в реакцию
обратимой конденсации с NH2-rpyn-
пами первичных аминов и аминокислот
с образованием шиффовых оснований
(азометинов). Образование азометинов
сопровождается усилением желтой
окраски р-ров П. вследствие сдвига максимума погло-
щения от 388 ммк в зону 400—415 ммк; кроме того,
появляется новый максимум поглощения при 278 ммк.
Альдегидная группа П. реагирует с цистеином и дру-
гими 1-амино-2-тиолами с образованием производных
тиазол идина.
Наиболее надежным и чувствительным методом
идентификации и количественного определения П.
является ферментативный метод, основанный на акти-
вации аподекарбоксилазы тирозина из S. faecalis
или аспартат-апотрансаминазы дрожжей.
П. синтезируют конденсацией пиридоксаля с N-ди-
метилглицингидразидом; образующийся при этом
гидразон обрабатывают метафосфорной к-той и про-
дукт подвергают кислотному гидролизу при 100°. См.
также Пиридоксалевые ферменты и Пиридоксин.
Лит. см. при ст. Пиридоксалевые ферменты.
Ю. М. Тарчинский.
ПИРИДОКСИН (пиридоксол, 2-метил-3-окси-4,
5-диоксиметилпиридин) C8HUNO3, мол. в. 169,18 —
одно из производных витамина В,. П. (I) — бесцвет-
ный кристаллич. продукт, т. пл. 160° (с разл.); хорошо
растворим в воде и спирте, плохо растворим в ацетоне
и нерастворим в эфире. Спектры поглощения р-ров
П. обладают характерными максимумами при 290 ммк
(в 0,1 н. НС1), при 253 и 325 ммк (pH 7,0) и при 245
и 308 ммк (0,1 н. NaOH). Молярная экстинкция гид-
рохлорида П. при 308 ммк (0,1 н. NaOH) равна 7000.
но
Н3с
СН2ОН
"4YCH2°H
5Г
СНО
ch2nh2
НО-VX-CHoOH
H3C-kN>
П. устойчив к кислотам и щелочам, в нейтральных и
щелочных р-рах разрушается под действием света.
При нагревании до 120° в нейтральном р-ре П. подвер-
гается полимеризации. П., как и другие производные
витамина В, — пиридоксаль (II), п и р и д-
оксамин (III) — и их фосфорные эфиры, флуо-
ресцирует в УФ-свете, дает оранжевую окраску с ди-
азотированной сульфаниловой к-той при pH 10—И,
синюю окраску с реактивом Фолина — Дениса и с
2,6-дихлорхинонхлоримидом. Эти реакции исполь-
зуют для количественного определения П. В наиболь-
шем количестве П. содержится в рисовых отрубях,
зародышах пшеницы, бобах, дрожжах, почках, печени
и мышцах. В животных тканях П. легко фосфорили-
руется с образованием пиридоксин-5-фосфата; окисле-
1061
ПИРИДОНЫ— ПИРИЛИЕВЫЕ СОЛИ
1062
ние его 4-оксиметильной группы в альдегидную при-
водит к образованию пиридоксалъг-5-фосфата.
Лит. см. при ст. Пиридоксалевые ферменты.
Ю. М. Т арчинский.
ПИРИДОНЫ — гетероциклические соединения,
изомерные а- и у-оксипиридинам, соответственно
а-П. (I) и у-П. (II):
р -кетоацеталями или р -хлорвинилкетонами:
ОН о
I II
В нейтральных р-рах, по-видимому, преобладает пи-
ридонная форма.
ПИРИЛЕН (1, 2, 2, 6, 6-пентаметилпиперидин п-то-
луолсульфонат) C17H29O,NS, мол. в. 327,49— белый
П. с. в щелочной среде превращаются в основания
пирилия. При действии аммиака П. с. превращаются
в производные пиридина. При реакции с избытком
реактива Виттига П. с. претерпевают раскрытие пи-
рилиевого цикла с образованием непредельного фос-
форилированного кетона, к-рый далее циклизуется
в производное бензола:
СН3
n-CHaCjHjSOjH
кристаллич. порошок
со слабо-кремовым от-
тенком; т. пл. 158—
160°; легко растворим
в воде и спирте, ра-
створим в ацетоне,
почти нерастворим в эфире, бензоле; с р-ром ацеталь-
дегида и нитропруссида натрия образует темно-крас-
ное окрашивание.
Получают П. восстановлением триацетонамина по
Кижнеру гидразингидратом и едким кали в 2,2,6,6-
тетраметилпиперидин, к-рый при метилировании му-
равьиной к-той и формальдегидом, метиловым эфиром
«-толуолсульфокислоты или иодистым метилом пре-
вращается в 1, 2, 2,6, 6-пентаметилпиперидин; послед-
ний обрабатывают n-толуолсульфокислотой. По ган-
глиоблокирующему и гипотензивному действию П.
превосходит гексоний и бензогексоний, к тому же он
менее токсичен и хорошо всасывается в желудочно-
П. с. широко распространены в природе, они нахо-
дятся в основе большинства красных и синих крася-
щих веществ цветов и ягод. Эти вещества называются
антоцианинами, все они являются гликозидами, к-рые
при кислотном гидролизе распадаются на сахар и
соответствующий агликон (антоцианидин).
Гидроксиды, соответствующие солям пирилия, наз.
пирилия основаниями. Простейшие пирилия основа-
ния — бесцветные вещества, растворимые в эфире и
существующие, вероятно, в виде таутомерной смеси —
циклической (I) и открытой (II) форм, напр.:
кишечном тракте.
П. применяют при лечении заболеваний, связанных
с нарушением нервной регуляции: гипертония, бо-
лезни, спазмах периферия, сосудов (эндартериит
и др.), язвенной болезни желудка, токсикозах бере-
менности и т. д.
Лит.: Домбровская А. М.,Крементуло В. А.,
Черкес А. И., Врачебн. дело, 1960, № 12, 102; Дра-
чева 3. Н., Кузнецова Т. М., Клинич. мед., 1962,
40, № 9, 100; Тихоненко В. М., Фармацевтич. ж.,
1962, № 1, 31; Фармакол. и токсикология, 1962, № 6, 698;
Лебедев С. В., Врачебное дело, 1963, М 7, 21; Н о в и-
к о в а М. Н., там же, 1963, М 11; Симон И. Б., Вве-
денский В. П., Мед. пром-сть СССР. 1963, 5, 9; Н а 1 1
Н. К., J. Amer. Cliein. Soc., 1957, 79, А8 20, р. 5444; Leo-
nardN. J., Hauck F. Р., там же, № 19, р. 5279.
И. Б. Симон.
5 ОН
ь
Формула I лишь приблизительно отражает строение
циклич. формы вещества.
Пирилия основания образуются при осторожном
подщелачивании П. с. В случае использования П. с.,
содержащих в положениях 2 или 4 п- или о-оксифе-
нильные радикалы, образованию пирилиевых основа-
ний предшествует получение ангидрооснований:
ПИРИЛИЕВЫЕ СОЛИ (пироксония соли) —
ароматические кислородсодержащие гетероциклич.
соединения, в основе к-рых находится пи-
роксониевая система (см. формулу).
Приведенная ф-ла не совсем точно отобра-
жает строение П. с., т. к. положительный _
заряд пирилиевого иона не сосредоточен ис- X
ключительно на атоме кислорода цикла, а равномер-
но распределен по всему ядру.Для получения П. с.
обычно используют конденсацию а,р-непредельных
кетонов с кетонами в присутствии уксусного ангидри-
Металлич. производные пирилиевых оснований син-
тезированы при действии на замещенные пироны маг-
нийорганич. соединений:
да и хлорного железа:
С6Н5
СН
сн +
с6н5-с=о
СН3 (CHjCO)jO
O=c-C6H5 FeCl,
[FeCl4]~
Соли бензо- и нафтопирилия удобно синтезировать
взаимодействием фенолов с оксиметиленкетонами,
CH3MgJ
При взаимодействии пирилиевых оснований или
их металлич. производных с кислотами получаются
1063
ПИРИМИДИН — ПИРИМИДИНОВЫЕ ОСНОВАНИЯ
1064
соли пирилия; с аммиаком образуют соответствующие
производные пиридина.
Лит.: Фред Д., в сб.: Гетероциклические соединения,
под ред. Ф. Эльдерфилда, пер. с англ., т. 1, М., 1953, с. 269.
ПИРИМИДИН (1,3-или м-диазин, миазин)^ C4H4N2,
мол. в. 80,09— соединение класса диазинов. Строе-
ние П. лучше всего передается ф-лами:
П. — бесцветные кристаллы с наркотич. запахом;
т. пл. 21°; т. кип. 124°; дипольный момент 2,1 D-,
легко растворим в воде, спирте и эфире. Производные:
сульфат, т. пл. 95°; нитрат, т. пл. 135°; пикрат, т. пл.
156°; перхлорат, т. пл. 192°; соль с хлорным железом,
т. пл. 220°. П. — очень слабое однокислотное основа-
ние, образующее моногалогеналкилаты. При окисле-
нии П. перекисью водорода получается N-окись.
С большим трудом П. вступает в реакции электро-
фильного замещения, напр. галогенирование. По
реакционной способности к электрофильным реаген-
там атомы углерода П. располагаются в следующий
ряд CS>C4>C2.
С магний- и литийорганич. соединениями П. обра-
зует в мягких условиях 4-алкил (арпл)производные:
I) н,о
2) (о]
X-N
Синтезируют П. восстановлением 2,4-дихлор-,
2,4,6-трихлор и др. галогенированных П., а также
конденсацией производных малондиальдегида, напр.
fj-диэтиламиноакролеина, с формальдегидом и аммиа-
ком:
ох zo
zC-CH=CH-N(C?Hs)? + Н(/ + NH3----
н xnh2
Производные, содержащие систему П., широко
распространены в природе и играют важную роль во
многих ответственных биологич. процессах. Ядро П.
входит в состав витаминов (В,), коэнзимов (ури-
диндифосфатглюКоза), антибиотиков (амицетин) и
нуклеотидов, являющихся структурными единицами
нуклеиновых кислот.
Лит.: Кеннер Г., Т о д д А., в сб.: Гетероцикличе-
ские соединения, под ред. Р. Эльдерфилда, пер. с англ., т. 6,
М., 1960, с. 195; Blown D. J., The pyrimidines. N. Y.—L.,
1962; В re de reck H. [a. o.], Chem. Ber., 1958, 91,
№12, 2832. Э. E. Нифантьев.
ПИРИМИДИНОВЫЕ ОСНОВАНИЯ — группа
природных веществ, основой к-рых является пири-
мидин-, различаются характером и положением заме-
стителей в пиримидиновом ядре. П. о. широко рас-
пространены в животных, растительных тканях и
в микроорганизмах,
? I. R = OH; R'=H гл. образом в виде
„ о_|мц.о*_н N-гликозидных сое ди-
Г Г H-K-NHg, R-Н нений Биологическн
НС)4™ ш. R = OHi R' = Ch3 наиболее важными П.
о. являются урацил
(I), цитозин (II) и тимин (III), входящие в состав
нуклеиновых кислот, нуклеозидов и нуклеотидов.
Более ограниченное распространение в составе нуклеино-
вых к-т имеют метилированные основания — 5-метилцитозин,
5-оксиметилцитозин. Из других пиримидинов найден 2.4-диа-
мино-5,6-диоксипиримидин (д и в и ц и н) в составе гликозида
вики. Образование различных П. о. в организме идет через
4-карбоксипиримидин (см. Оротовая кислота).
П. о. — бесцветные кристаллич. вещества с высо-
кой темп-рой плавления (часто с разл.); плохо раст-
воримы в воде, но лучше пуриновых оснований. Раство-
римость в воде уменьшается в ряду пиримидин, цито-
зин, 5-метилцитозин, тимин, урацил, оротовая к-та.
П. о. растворимы в щелочах и к-тах с образованием
солей; хорошо кристаллизуются пикраты. Серебряные
соли П.о.,в отличие от пуриновых оснований, не осаж-
даются из кислых, но осаждаются из щелочных р-ров.
П. о. сильно поглощают УФ-свет. Характерные для
каждого П. о. спектры поглощения УФ-света опре-
деляются природой и положением заместителей в ге-
тероциклич. ядре, а также характером их ионизации.
Наибольшие различия в спектрах П. о. наблюдаются
при pH 1, 7 и 12, что используется при идентификации
П. о. Длительное действие УФ-света вызывает глубо-
кие изменения в структуре П. о., сопровождающиеся
потерей абсорбции. Характерный спектр П. о. исче-
зает также при обработке их бромной водой.
Анионные свойства оксипиримидинов связаны
с наличием карбонильных групп, обусловливающих
лактам-лактимную таутомерию:
О ОН
Hi I —-
HO-kJ
й
Окси- и оксотаутомеры образуют соответственно ме-
таллич. производные, в к-рых металл связан либо с
кислородом, либо с азотом. Катионные свойства П. о.
усиливаются наличием аминогруппы. Пиримидины,
содержащие ОН- или NH2-rpynny у С4 и Cs, нестабиль-
ны в щелочной среде даже при комнатной темп-ре.
Аминопиримидины дезаминируются HNO2, образуя
соответствующие оксипроизводные. П. о. способны
восстанавливаться (водородом в присутствии плати-
нового катализатора, амальгамой натрия и др.). На-
сыщение 4,5-двойной связи П. о. легко происходит
при обработке бромом. Образующиеся при восста-
новлении или бромировании насыщенные соединения
П. о. менее стабильны в кислой и особенно в щелоч-
ной среде, разрушаясь с разрывом кольца. Это свой-
ство используется при выделении и определении пен-
тоз пиримидиновых нуклеотидов и нуклеозидов. П. о.
разрушаются гидразином с образованием мочевины и
пиразолона, окисляются КМпО4, перекисью водорода,
озоном, поглощают иод из водных щелочных р-ров.
Металлич. производные П. о. легко алкилируются
при действии галогеналкилов; эта реакция исполь-
зуется при получении нуклеозидов. Аминогруппы II. о.
сравнительно легко реагируют с формальдегидом.
Качественно и количественно П. о. определяют
гл. обр. хроматографич., электрофоретич., спектро-
фотометрич. и полярографич. методами. Цветные реак-
ции П. о. основаны на восстановлении мышьяково-
вольфрамовой к-ты (бромированные урацил и цито-
зин) и на реакции с диазобензолсульфокислотой,
с к-рой в щелочной среде в присутствии восстанавли-
вающего агента образуется красное окрашивание
(тимин). Окраска в этих реакциях может быть изме-
рена количественно.
Из биологич. материала П. о. получают кислотным
гидролизом (12 н. НС1О4, конц. муравьиная к-та при
100°) нуклеиновых к-т, нуклеотидов и нуклеозидов.
Смесь П. о., полученную при гидролизе, препара-
тивно разделяют методом ионообменной хромато-
графии. Синтезируют П. о. конденсацией мочевины и
ее производных с производными ацетоуксусного и
циануксусного эфиров или конденсацией хлорангид-
рида N-алкоксиакриловой к-ты с цианатом серебра;
образовавшийся ацилизоцианат присоединением ами-
нов превращают в ацилмочевину, при циклизации
к-рой образуется П. о. При введении в организм
неприродных аналогов П. о. происходят серьезные
1065
ПИРО — ПИРОВИНОГРАДНАЯ КИСЛОТА
1066
нарушения обмена веществ, что объясняется их кон-
курентным действием с нормальными П. о.; напр.,
аллоксан, близкий по строению урацилу, вызывает
у животных экспериментальный диабет. Нек-рые
II. о. используются в качестве антиметаболитов,
напр. при лечении тиоурацилом повышенной функ-
ции щитовидной железы, при поисках противоопухо-
левых и бактериостатич. средств; азапиримидины
(6-азаурацил, 6-азатимин) и галогенопиримидины (осо-
бенно 5-фторурацил) используют в терапии лейкозов
и бластомной болезни; производные барбитуровой
к-ты, напр. веронал, мединал, люминал, применяют
в качестве снотворных и наркотич. средств.
Лит.: Кочетков Н. К., Торгов И. В., Б о т-
виник М. М., Химия природных соединений, М., 1961;
Кочетков Н. К., Будовский Э. И., G и м у к о-
в а Н. А., Биохимия, 1962, 27, вып. 3, 519; Bendlch А.,
в кн.: The nucleic acids, ed. E. Ghargaff [a. o.J, v. I, N. Y.,
1955, p. 8i; Beaven G. H., Holiday E. R., John-
son E. А., там же, p. 493; Jordan D. O., The-chemistry
of nucleic acids, L., 1960, p. 64, 99, 134; Steiner R. F,.,
В e e r s R. F., Polynucleotides, Amst.—L.—N. Y.. 1961, p. 21;
Methods In enzymology, ed. S. P. Colowlck, N. O. Kaplan, v. 3,
N. Y., 1957; J a villi er M. [e. a.J, Traits de biochimle
gengrale, t. 1, P., 1959. Л. H. Боброва.
ПИРО — приставка впереди названия исходного
соединения, превращающегося при высокой темп-ре
в другое соединение. Так, при нагревании ортофос-
форной кислоты Н2РО4 до 200° отщепляется вода и
образуется ангидрид (НО)2Р(О)О(О)Р (ОН)2 (Н4Р2О7],
т, наз. пирофосфорная к-та, для к-рой известны
также соли, эфиры и т. д. По аналогии с пирофосфор-
ной к-той ангидрид серной к-ты HOSO2OSO2OH
называют пиросерной к-той (другое название дисер-
ная к-та). Приставку пиро часто применяют для наз-
вания органич. соединений. Так, наиболее важная
простейшая а-кетокислота СН5СОСООН получила
название пировиноградной к-ты потому, что была
впервые получена нагреванием винной, или виноград-
ной, к-ты. Диоксибензол называют пирокатехином,
т. к. это соединение было впервые получено сухой
перегонкой различных видов катеху. Пирогаллол
(1,2,3-триоксибензол) образуется при отщеплении
О2 от галловой к-ты:
ОН. ОН
СООН
ПИРОБЕНЗОЛ — продукт пиролиза (при темп-ре
ок. 700°) керосино-газойлевой фракции нефти (фрак-
ции легкого масла), не содержащий толуола; выкипает
до 175°; содержит смесь бензола и его гомологов
(ксилола), а также 10—15% углеводородов других
классов. П. получают очисткой и ректификацией
легкого масла, при этом отбирается толуол. Получен-
ный обестолуоленный продукт представляет собой
прозрачную жидкость с темп-рой застывания не
выше —18°; начало перегонки не ниже 80°. Фракцион-
ный состав: 10% перегоняется не выше 95°; 50% —
не выше 120°; 90% — не выше 165° и 97,5% — не
выше 175°; содержание сульфируемых веществ не
менее 87%, серы не более 0,02%. П. не должен содер-
жать водорастворимых к-т, щелочей, воды и механич.
примесей. П. применяют как высокооктановую до-
бавку к легкому моторному топливу; в последнее
время применение П. для этих целей утратило свое
значение в связи с развитием риформинга.
Лит.: Технология нефти, ч. 2, 3 изд., М., 1952; Технические
нормы на нефтепродукты, под ред. Н. Г. Пучкова, 16 изд.,
М., 1957. В. А. Гетлинг.
ПИРОВИНОГРАДНАЯ КИСЛОТА (пирувино-
вая, а-кетопропионовая) СН3СОСООН, мол. вес
88,06 — бесцветная жидкость с запахом уксусной
кислоты; т. пл. 13,6°; т. кип.: 165°/760 мм(е частичным
разл.), 75—80725 мм, 65710 мм; </“’ 1,2668, n“’s
1,4303; во всех отношениях смешивается с водой,
спиртом и эфиром.
П. к. образует производные: семикарбазон, т. пл. —200°
(с разл., из воды); фенилгидразон, т. пл. 192° (из воды);
n-нитрофенилгидразон, т. пл. 220° (из спирта); 2,4-динитро-
фенилгидразон, т. пл. 218° (из спирта), амид, т. пл. 124—125°
(из спирта).
П. к. проявляет общие свойства а-кетокислот (см.
Алъдегидокислоты, и кетокислоты): при восстанов-
лении легко переходит в молочную к-ту, при окисле-
нии и декарбоксилировании дает ацетальдегид и
СО2; восстанавливает аммиачный р-р AgNOs, в конц.
H2SO4 выделяет СО и превращается в уксусную к-ту.
П. к. вступает во все реакции, характерные для
кетогруппы. С аммиаком она реагирует по схеме:
2GH3GOOOH + 2NH3—►
—> CO2 + H2O + GH3CONHCH(CH3)GOONH,
П. к. легко вступает в конденсации альдольного и
кротонового типа. Самоконденсация 2 молекул П. к.
в кислой среде дает а-кето-у-метил-у-карбокси-у-бу-
тиролактон (I), к-рый в более жестких условиях
сразу переходит в пировинную (метилянтарную) к-ту.
Самоконденсация 3 молекул П. к. в присутствии
Ва(ОН)2 приводит с отщеплением (СООН)2 к ме-
тилдигидротримезиновой (1-метилциклогексадиен-
2,4-трикарбоновой -1, 2, 5) к-те (II), к-рая при окис-
лении и декарбоксилировании дает 5-метилизофтале-
вую (увитиновую) к-ту (III). П. к. легко конденси-
руется с альдегидами;
СООН
НзСч^СООН
НООС
НООС
О
Н3С О
НООС w
III
н
GH3COGOOH + RGHO —► RGH = GHGOCOOH
с формальдегидом образует а-окси-р, Р-диметилол-у-бу-
тиролактон (IV), по реакции Дебнера (1) и Пфитцин-
гера реакции (2) образует соответственно 2-Б-4-карбо-
кси- и 2,4-дикарбоксихинолины:
СООН
I
со
I
сн3
OHCR
Получают П. к. перегонкой винной (или виноград-
ной) к-ты в присутствии KHSO4:
HOOGGH(OH)GH(OH)GOOH —> HOOGG(OH) = GHGOOH —►
—> HOOGGOGHjGOOH —► HOOGGOGH3
Другие способы — щелочной гидролиз а,а-дихлор-
пропионовой к-ты, кислый гидролиз ацетилцианида,
кетонное расщепление щавелевоуксусного эфира,
окисление молочной к-ты и т. д. — менее удобны.
Определяют П. к. колориметрически по реакции с ни-
тропруссидом или 2,4-динитрофенилгидразином.
1067
ПИРОГАЛЛОЛ — ПИРОКАТЕХИН
1068
П. к. играет весьма важную биохимии, роль. При
многих видах брожения углеводов П. к.— главный
продукт, дальнейшие превращения которого опре-
деляют конечные вещества процесса. При анаэроб-
ном обмене углеводов 2-фосфоглицериновая кислота
НОСН2СН(СООН)ОР(О)(ОН)2, образующаяся так же,
как и при брожении, под действием энолазы, дегид-
ратируется в богатую энергией 2-фосфоэнолпиро-
виноградную кислоту СН2=С(СООН)О ~ Р(О)(ОН)2,
к-рая при участии соответствующей фосфоферазы
отдает свою энергию аденозинтрифосфату, образуя
П. к. В организме П. к. восстанавливается далее
лактикодегидрогеназой в присутствии дипиридинну-
клеотида (Н2) в Ь-(Ц-)-молочную к-ту. При спиртовом
брожении П. к. в присутствии декарбоксилазы дает
ацетальдегид, к-рый восстанавливается в спирт. При
аэробном обмене (дыхании) П. к. участвует в образо-
вании ацетилкофермента А, к-рый далее участвует
в лимоннокислом цикле (см. Цикл трикарбоновых
кислот). При этом распад образующейся из углеводов
П. к. выражается суммарным уравнением:
С3Н4,О3 + 503—> ЗСО3 + 2Н3О
П. к. также принимает участие в переаминировапии
трехуглеродных аминокислот под действием амипо-
фераз. П. к. служит переходным веществом при био-
синтезе белков из углеводов и наоборот.
Метиловый эфир П. к., т. кип. 134—137°, образует оксим
с т. пл. 69° (из эфира) и т. кип. 122—123°/14 мм; 2,4-динптро-
фенилгидразон с т. пл. 186,5—187,5° и диметилацеталь с т. кип.
66—66,5°/16 мм. Этиловый эфир П. к., т. кип. 155° и 55°/14 мм,
дает оксим с т. пл. 97°, семикарбазон с т. пл. 206° (с разл.);
2,4-динитрофепилгидразои, т. пл. 154,5—155° (из водно-
го диоксана) и диэтилацеталь, т. кип. 190—191°/760 мм и
81,5— 82,5°15 мм.
П. к. содержится во всех тканях организма. Ави-
таминоз В j, а также ряд других заболеваний вызывает
накопление П. к. в организме. Нарушение обмена
П. к. приводит к ацетонурии.
Лит.: Колотилова А. И., Обмен веществ, Л., 1962.
А. Е. Васильев.
ПИРОГАЛЛОЛ (пирогалловая кислота, 1,2,3-
триоксибензол) С6Н3(ОН)3, мол. в. 126,11 — бесцвет-
ные легко сублимирующиеся кристаллы, пластинки
или иглы; без запаха, на воздухе темнеют; т. пл.
133—134°; т. кип.: 3097760 мм, 171,5712 мм; раство-
римость в 100 г воды: 40 г (13°), 62,5 г (25°), в 100 г
спирта 100 г (25°), в 100 г эфира 83,3 г (25°); П. почти
нерастворим в бензоле, хлороформе, четыреххлори-
стом углероде. Производные П.: триацетат, т. пл. 165°;
трибензоат, т. пл. 89—90°, трифенилуретан.т. пл. 173°.
Наиболее характерным свойством П. является спо-
собность легко окисляться; он мгновенно восстанав-
ливает соли золота и серебра, а его щелочные р-ры
настолько сильно связывают кислород, что их приме-
няют в газовом анализе для количественного опреде-
ления кислорода. При электрохимия, окислении или
при действии закисных солей железа П. (I) превра-
щается в пурпурогаллин, являющийся триоксибензо-
аД-трополоном (II):
рование, сульфирование и др. При действии органич.
к-т в присутствии хлористого цинка П. гладко аци-
лируется в (IV). При ацетилировании получается
галлоацетофенон (али-
зариновый желтый С)— О ОН
протравной краситель но-Х^7-0Н
бледно-желтого цвета; Г |ГПН \ Г „
при бензоилировании— \х'~он 4^/-COR
галлобепзофенон (али- ц |V
зариновый желтый А) —
протравной краситель желтого цвета. Для качествен-
ного определения П. используют реакцию с серно-
кислым железом (сине-фиолетовая окраска) или с
глицерином в сернокислой среде (красно-фиолетовая
окраска, усиливающаяся при нагревании).
Впервые П. синтезировал Шееле (1786) декарбокси-
лированием галловой к-ты; способ используется
в пром-сти и в настоящее время. Применение П.
разнообразно: используется как восстановитель, в фо-
тографии, в газовом анализе для поглощения кисло-
рода, при количественном и качественном определе-
нии Bi, Sb, Nb, Та, Ti, Fe, Al и для синтетич. и др.
целей. П. ядовит.
Лит.: Deichmann W. В., в кн.: Phenol and
phenolic compounds, v. 2, N. Y'., 1949, p. 1042; Critchlow
A., Haworth R. D., Pau son P. L., J. Chem. Soc.,
1951, May, 1318. Э. E. Нифантьев.
ПИРОГЕННЫЕ ПРОЦЕССЫ — см. Пиролиз, Тер-
мическая переработка топлива.
ПИРОКАТЕХИН (о-диоксибензол) СвН1(ОН)2,
мол. в. 110,11 — кристаллы, бесцветные иглы с фе-
нольным запахом, легко буреющие на воздухе; т. пл.
104°; т. кип.: 245,9°/760 мм; 1767100 мм; 1,371,
Цд 1,604; растворимость см. таблицу; средняя тепло-
емкость между 0° и 79° 0,3265 кал!г-град, константа
диссоциации К = 4,7-10-11 (25°). Производные П.:
моноацетат, т. пл. 57—58°; диацетат, т. пл. 63,5°.
Растворимость пирокатехина
Растворимость, вес. %
Растворитель 20° 60° 100°
Вода 31,2 80,8 98,8
Этиловый спирт 58,2 74,2 97,3
Диэтиловый эфир .... — 65,5 94,8
Ацетон 66,0 75,4 96.3
Бензол 0,8 7,5 93,1
Четыреххлористый углерод 0,1 0,9 86,0
Хлороформ 1 ,9 6.9 8У, 9
Для П. характерны все реакции фенолов: с основа-
ниями он образует моно- и дисоли, соли с тяжелыми
металлами нерастворимы в воде; легко окисляется
до о-бензохинона, на холоду восстанавливает аммиач-
ный р-р окиси серебра, при нагревании — реактив
Фелинга. При сплавлении с фталевым ангидридом П.
(I) дает два красителя: ализарин (П)и гистазарин (III):
При каталитич. восстановлении П. над никелем полу-
чен 2-оксициклогексадион (III), П. легко вступает
в реакции электрофильного замещения — галогени-
При взаимодействии с иодистым метиленом в щелоч-
ной среде П. превращается в метиленовый эфир П.
(IV), а с фосгеном и др. хлорангидридами двухоснов-
ных и полиосновных кислот — в циклич. сложные
1069
ПИРОКСИЛИН — ПИРОЛИЗ
1070
эфиры (V—VII), с о-аминофенолом образует дибензо-
ксазин (VIII):
П. легко вступает в Фриделя — Крафтса реакцию',
гак, при взаимодействии с изобутиленом он алкили-
руется до mpem-бутилпирокатехина, применяемого
в технике в качестве эффективного ингибитора поли-
меризации винильных соединений; с хлоруксусной
к-той превращается в хлорацетил П., являющийся
полупродуктом в синтезе адреналина.
Для качественного определения П. его нагревают
в щелочном р-ре с хлороформом до появления зеленой
окраски или используют реакцию с хлорным железом,
сопровождающуюся появлением зеленого окрашива-
ния, переходящего в красное при добавлении соды или
аммиака (в случае применения гидрохинона или резор-
цина образуется желто-бурый осадок гидрата окиси
железа). Количественно П. определяют весовым спо-
собом по образующейся свинцовой соли.
П. получают: сухой перегонкой протокатеховой
кислоты, лигнина, электрохимическим окислением
бензола и фенола, деалкилированием гваякола и
вератрола и др. способами. В пром-сти П. синтезируют
щелочным плавлением о-хлорфенола, о-фено л сульфо-
кислоты или фенолдисульфокислоты. В последнем
случае фенолдисульфокислоту сплавляют с едким
натром при 300°, при этом образуется пирокатехин-
моносульфокислота, сульфогруппа к-рой легко от-
щепляется при нагревании с разб. серной к-той.
П. используют в фотографии в качестве восстанови-
теля для колориметрич. определения Ti, Мо, W, V, Fe,
Ge и для синтетич. целей. П. токсичен, минимальная
смертельная доза 0,04 г на 1 кг живого веса. Впервые
П. открыт в 1839 Рейншем при сухой перегонке раз-
личных видов катеху, откуда и получил свое название.
Лит.: Harris Е. Е., Saeman J. F., Bergst-
rom С. В., Industr. and Engng Chem., 1949, 41, № 9, 2063;
H о ti b e n J., Fortschrltte der Hellstoffchemie Bd 1, B.,
1926, S. 201. Э. E. Нифантьев.
ПИРОКСИЛИН — см. Нитроцеллюлоза.
ПИРОЛИЗ. Содержание:
Введение................................1069
Механизм и кинетика пиролиза............1071
Пиролиз органических соединений основных классов 107 3
Техника пиролиза ...................... 1078
Введение. Пиролиз — превращение органич. со-
единений с одновременной деструкцией их под дейст-
вием высокой темп-ры. При П. не обязательно воздей-
ствие химич. реагентов и других, кроме теплоты, фи-
зич. факторов, напр. света, радиации и т. п. Поня-
тие П. уже понятия пирогенетич. превращений вооб-
ще, поскольку последние могут происходить и без
деструкции молекул исходного вещества, напр. изоме-
ризация, полимеризация, но, с другой стороны, шире
понятия термич. деструкции, т. к. при П., помимо
деструкции, происходят обычно вторичные реакции
уплотнения молекул, изомеризации их и т. п.
В применении к переработке углеводородного сырья,
напр. нефти и ее фракций, П. обычно наз. крекингом,
хотя, в свою очередь, един из видов крекинга — кре-
кинг тяжелого сырья под низким давлением — также
наз. П. Условным является понятие высокой темп-ры,
поскольку многие органич. соединения, напр. диазо-
соединения, стабильны только при темп-рах ниже 0°
и распадаются уже при обычной темп-ре.
Обычно под высокой темп-рой, при к-рой происходит
П., понимают темп-ру порядка нескольких сотен
градусов, при к-рой лабильными в органич. молекулах
становятся и прочные углерод — углеродные и угле-
род — водородные связи. Хотя воздействие химич. реа-
гентов и других физич. факторов при П. не обязатель-
но, однако на практике прибегают к воздействию хи-
мич. реагентов, напр. кислорода, водяного пара, ка-
тализаторов, а также и радиации. П., проводимый
в присутствии катализаторов, нельзя, однако, отож-
дествлять с термокаталитич. процессами вообще, т. к.
роль катализатора различна в обоих случаях. При-
менение катализатора при П. имеет целью ускорить
уже протекающий с заметной скоростью чисто термич.
процесс, тогда как применение катализатора в термо-
каталитич. процессах, напр. при присоединении во-
дорода к ароматич. соединениям, является необхо-
димым условием их осуществления; эти процессы
при данной темп-ре и в отсутствии катализаторов
не протекают с заметной скоростью.
Познавательное значение П. связано с определе-
нием влияния химич. состава и структурных особен-
ностей на термич. стабильность молекул органич.
веществ и на прочность образующих их связей. Тех-
нич. значение П. связано в первую очередь с перера-
боткой и использованием природных источников хи-
мич. сырья — нефти, газа, угля, торфа, сланцев, дре-
весины и т. п. Применение П. идет по двум основным
направлениям — топливному и «химическому». В пер-
вом случае основной целью является получение вы-
сококачественных (газообразных, твердых и жидких)
топлив и масел путем крекинга, коксования, полукок-
сования, гидрогенизации деструктивной, риформинга
сухой перегонки древесины. Получение химич. сырья
(олефины, ароматич. углеводороды, водород, сера
и т. п.) при этом не является самоцелью, хотя и су-
щественно в определении экономия, эффективности
процесса. Во втором случае целевым назначением П.
является получение химич. сырья, в первую очередь
олефинов и ароматич. углеводородов для пром-сти
органич. синтеза. В обоих случаях используется угле-
водородное сырье, поэтому наиболее изучен и разра-
ботан П. углеводородов. П. других классов соедине-
ний имеет несравнимо меньшее значение. Большое
практич. значение имеет деструкция полимеров при
термич. воздействии и процессы, происходящие в мо-
торных топливах и маслах при воздействии на них
высокой темп-ры. П. используется также и для ана-
литич. целей.
Основные процессы П.: а) расщепление углеродного
скелета молекул; б) полное или частичное отщепление
функциональных групп с образованием простейших
неорганич. соединений — воды, аммиака, сероводо-
рода, галогеноводорода, окиси и двуокиси углерода;
в) полимеризация или конденсация исходных молекул,
образующихся из них фрагментов и продуктов превра-
щения; в последнем случае образуются продукты уп-
лотнения и продукты деструкции последних; г) изо-
меризация, предшествующая или сопровождающая
процесс деструкции; д) отщепление водорода, вплоть
до выделения свободного углерода. .
Общий для всех случаев механизм процесса и пол-
ный анализ термодинамич. и кинетич. закономерно-
1071
ПИРОЛИЗ
1072
стей П. дать невозможно. Могут лишь быть показаны
общие пути подхода к изучению П. на примере наибо-
лее хорошо исследованной области П. углеводородов.
Механизм и кинетика пиролиза. Во многих случаях
при П. органич. соединений принимается, что распад
идет по радикально-цепному механизму. В случае П.
углеводородов считается, что первичной реакцией яв-
ляется образование свободных радикалов, напр.
метиленового или метильного, в результате разрыва
связи С—С. Простейшие радикалы СН,- ,C2HS- ,Н- реа-
гируют с молекулой исходного углеводорода, чем и
поддерживается реакция передачи цепи. Взаимодей-
ствие свободных радикалов между собой с образова-
нием стабильных продуктов ведет к обрыву цепи.
На основе радикально-цепного механизма П. могут
быть рассмотрены кинетич. закономерности процесса
и произведен расчет вероятного выхода продуктов П.
Во многих простейших случаях, напр. при П. низ-
ших углеводородов, получено удовлетворительное
совпадение между расчетными и опытными данными.
Во многих случаях процесс П. рассматривают как
реакцию молекулярного распада, несмотря на то,
что, напр., при П. углеводородов с этой точки зрения
нельзя объяснить более низкую величину энергии
активации, чем энергия разрыва связей С—С и С—Н.
Поэтому при истолковании кинетики распада реакции
П. часто рассматривают как гомогенные, некаталитич.
реакции первого порядка.
Представление о вероятном направлении П. может
быть получено при термодинамич. анализе процесса.
Точные термодинамич. расчеты П. в большинстве слу-
чаев невозможны, т. к. вследствие вторичных процес-
сов большая часть реакций не успевает дойти до
равновесия. Поэтому термодинамич. анализ дает
ответ только на вопрос о принципиальной возможно-
сти тех или иных реакций и, отчасти, об относитель-
ном преобладании тех или иных превращений.
Кинетич. особенности П. в каждом отдельном слу-
чае нуждаются в специальных исследованиях. Однако
для практич. целей, напр. для расчета реакционных
устройств, иногда оказывается возможным дать об-
щий метод описания кинетич. закономерностей, при-
ложимый к группе однотипных реакций.
В большинстве случаев реакции при П. являются
необратимыми; продукты первичного распада немед-
ленно превращаются дальше и неспособны вновь об-
разовать исходный продукт. Поэтому конечные ре-
зультаты П. в значительной мере определяются соот-
ношением скоростей отдельных реакций. Первичную
и основную реакцию П.— термич. распад — рассмат-
ривают обычно как гомогенный и мономолекулярный
процесс, скорость к-рого не зависит от давления и
является функцией только темп-ры:
W = Ce~E/RT (1)
где W — скорость реакции, Е — энергия активации
распада а С — константа, зависящая от природы ис-
ходного соединения. При П., напр. углеводородов,
можно во многих случаях рассматривать распад как
гомогенную некаталитич. реакцию первого порядка,
протекающую в газовой фазе.
В пром-сти П. обычно проводят в проточных усло-
виях при небольшом изменении давления, но при зна-
чительном изменении объема смеси. При этом объем
газовой смеси и концентрация ее компонентов изменя-
ются как по мере углубления процесса, так и в зави-
симости от изменения темп-ры. Скорость распада уг-
леводородов при П. обычно описывается ур-нием
первого порядка: 1ГА=А:ОСА, где П7А — скорость рас-
пада компонента A, kv—константа скорости реакция
при постоянном объеме и СА — концентрация компо-
нента А. Можно записать иначе: IFд=АуРА, где Рд —
парциальное давление компонента А и кр— константа
скорости реакции при постоянном давлении.
Для проточного реактора, работающего в стацио-
нарном режиме, применимо следующее ур-нпе мате-
риального баланса: W AdVp=FdxA, где Vp— объем
реактора, F — скорость подачи сырья (молъ\час),
dxA— количество компонента А, распавшегося в эле-
менте реактора dVp. После соответствующих преобразо-
ваний и интегрирования это ур-ние приводится к виду:
F Р MF dxA (2)
F ~ SF qf ~~ J WA
О
где MF— плотность сырья, qf— плотность сырья
при стандартных условиях, SF— объемная скорость
(отношение объема сырья, поступающего в реактор
в единицу времени, к объему реактора). Приведенное
выше ур-ние выражает связь между объемной скоро-
стью и степенью превращения, к-рые для данного ве-
щества при постоянной темп-ре и давлении не зависят
от скорости потока и формы реактора. Скорость реак-
ции, следовательно, может быть определена из экспе-
риментально найденных величин хА при изменении
скорости подачи, напр. по углу наклона касательной
из графика зависимости Vp[F от хА.Ур-ние (2) может
быть также представлено в виде:
F г п„ Г ( , nA„ \ I 1 ПАО 1
— = ш пл In -----------2---х Л + In ----— (3)
F hvp L \ A° па„-жа "Ao-^aJ v
где n0 — общее число молей сырья, подаваемого в
реактор в единицу времени, пАо—число молей компо-
нента А в том же количестве исходного продукта и
a>=6/n0, где 6—увеличение числа молей в результате
распада сырья при расчете на один моль прореагиро-
вавшего компонента. Это ур-ние позволяет рассчиты-
вать кр по результатам одного экспериментального оп-
ределения величин 7а и VpIF при условии соблю-
дения постоянства темп-ры и давления.
Очевидно, что подобный же метод описания кине-
тич. особенностей процесса может быть приложен и
к другим случаям П., скорость распада к-рых может
быть описана ур-нием первого порядка по распадаю-
щемуся компоненту. Применяя подобный метод рас-
чета для углеводородов, находят, что энергия акти-
вации их распада ок. 70 ккал/моль и не зависит от ве-
личины молекулы. Известный из опыта факт возраста-
ния скорости распада углеводородов с увеличением
мол. веса следует поэтому связывать с увеличением
значения предэкспоненциального члена в ур-нии (1).
Сопровождающие распад полимеризация и конден-
сация в обычных условиях П. в первом приближении
являются гомогенными бимолекулярными реакциями,
а поэтому, в отличие от процесса распада, в значи-
тельной мере зависят от давления. Скорость этих
процессов подчиняется ур-нию:
Wyns=kabexp(-EIRT) (4)
где а и Ъ— концентрации уплотняющихся веществ.
Поскольку а и b являются функциями давления, то
ур-ние (2) может быть представлено в виде:
Wynlt=kPaPbe-ElRT
тдерапрь— парциальные давления реагирующих ве-
ществ. Энергия активации реакций уплотнения обыч-
но значительно ниже, чем для реакций распада.
Влияние темп-ры, давления и времени реакции на
результаты П. может быть обрисовано только в общей
форме. С повышением темп-ры ускоряется распад не
только исходных молекул, но и первично образующих-
ся из них фрагментов. Поскольку верхнего предела
темп-ры П. не существует, то конечным итогом при
достаточно высокой темп-ре и продолжительности П.
1073
ПИРОЛИЗ
1074
является распад исходного соединения на молекулы
Н2, Nj, О2 и элементарный углерод или же на про-
стейшие термически стабильные молекулы СО, СО2,
СН4, Н2О, NH3, H2S п т. п. Влияние темп-ры на П.
до нек-рой степени можно компенсировать измене-
нием продолжительности реакции. Обратное положе-
ние справедливо только в известных пределах, т. к.
в силу термодинамич. ограничений увеличение вре-
мени реакции при низкой темп-ре может компенсиро-
вать влияние темп-ры только частично. Кроме того,
при высокой темп-ре увеличивается равновероятность
различных реакций, а следовательно уменьшается се-
лективность процесса. При практич. осуществлении П.
выбор оптимальных условий (давления, темп-ры и вре-
мени реакции) определяется требованием достижения
максимальной производительности реактора при за-
данном его объеме. Повышения темп-ры, ускоряющей
П., и давления, уменьшающего реакционный объем,
при прочих равных условиях способствуют повышению
производительности реактора.Верхний предел темп-ры
П., помимо термодинамич. соображений, определяется
в значительной мере также и возможностью подвода
необходимого количества тепла и наличием термостой-
ких конструкционных материалов.
Пиролиз органических соединений основных клас-
сов. Как правило, для соединений одного и того же
класса термич. стойкость понижается с увеличением
мол. веса и более сложные молекулы стремятся прев-
ратиться в более простые термически стабильные.
В большинстве случаев углеводороды, в особенности
простейшие, являются термически более стойкими,
чем соответствующие им производные. Наиболее
прочными углеводородами являются ароматич., про-
стейшие парафины (метан, этан) и олефины (этилен,
ацетилен). Образование их всегда имеет место при до-
статочно высокой темп-ре П. Поэтому распад и прев-
ращения различных органич. соединений с функцио-
нальными группами происходит легче, чем соответ-
ствующих им углеводородов, и при достаточно высо-
кой темп-ре наряду с продуктами расщепления и от-
щепления функциональных групп всегда образуются
также и молекулы углеводородов. С известными огра-
ничениями к П. применимо т. наз. «правило наимень-
шей молекулярной деформации», заключающееся
в утверждении, что термич. распад молекул идет
в первую очередь по направлениям, требующим наи-
меньшей деформации молекулы, и, следовательно,
строение продуктов П. будет, насколько возможно,
близко к структуре исходного соединения.
Однако во многих случаях, не прибегая к опыту,
трудно решить какое именно направление разрыва
связей приводит к наименьшей деформации молекулы.
Напр., при П. углеводородов трудно определить с по-
мощью этого правила в каком именно месте произой-
дет разрыв углеводородной цепи. Часто П. протекает
легче, если функциональные группы стоят при вто-
ричном или при третичном атоме углерода; большей
легкости П. часто способствует накопление функцио-
нальных групп в молекуле и возможность внутримо-
лекулярного их взаимодействия с отщеплением гете-
роатома, особенно если таким путем возможно обра-
зование пяти- или шестичленного циклов. Если же
внутримолекулярное взаимодействие затруднено или
же имеется возможность образования пяти- или шести-
членного циклов путем межмолекулярного взаимо-
действия, то такое направление процесса может быть
преобладающим. Такие процессы осуществляются ча-
сто очень гладко при невысоких темп-рах, иногда при
простой перегонке. Ниже описаны общие направления
превращений при П. главнейших классов органич.
соединений.
Углеводороды. П. углеводородов представ-
ляет наибольший практич. интерес и является наибо-
лее изученным. Легкость П. и его результаты зависят
от типа углеводорода, мол. веса и условий процесса.
С повышением мол. веса термич. стойкость всех угле-
водородов падает, и наиболее стабильными являются
низкомолекулярные углеводороды — метан, этан,
ацетилен, бензол, толуол и др. Основными направле-
ниями П. являются разрыв связей С—С с образова-
нием углеводородов меньшего мол. веса, и отщепле-
ние водорода с образованием олефинов и ароматич.
углеводородов, а при достаточно высоких темп-рах
приводящее к полному распаду на углерод (сажа, кокс)
и водород. Наряду с этим имеют место реакции поли-
меризации и конденсации, приводящие к получению
даже из низкомолекулярных углеводородов, напр.
пропана жидких продуктов.
Парафиновые углеводороды распадаются наиболее
легко. С повышением темп-ры скорость их распада
возрастает сильнее, чем углеводородов других клас-
сов. Преимущественным направлением распада яв-
ляется расщепление молекулы приблизительно посре-
дине с образованием парафинов и олефинов меньшего
мол. веса:
У парафинов изостроения отщепляются преим. более
короткие цепи, напр. метильные группы, а также во-
дород. Напр., из изобутана образуется до 50% изо-
бутилена. Изомеризации парафинов при П. практи-
чески не происходит. Термодинамически мало вероят-
на циклизация. Большой интерес с целью получения
олефинов представляет П. простейших парафинов —
метана, этана, пропана. Метан при темп-ре выше 1000°
разлагается с промежуточным образованием метильных
радикалов и отщеплением водорода. Взаимодействие
метильных радикалов приводит к образованию этана,
в свою очередь дегидрирующегося в этилен и далее
в ацетилен. Поскольку метан и ацетилен более устой-
чивы при высокой темп-ре, чем этилен, то получить
последний П. метана с хорошими выходами нельзя.
Этан при 1000° разлагается на этилен, водород и ме-
тан, являющиеся первичными продуктами его распада.
Вторичные продукты распада—ацетилен, углево-
дороды С3—С4, высшие олефины, ароматич. углево-
дороды, образующиеся в небольших количествах.
Реакция идет также с участием метильных радикалов
по цепному механизму. Этан — наилучшее сырье для
получения этилена.
Пропан при П. одновременно деметилируется и
дегидрируется, поэтому основными продуктами его
распада являются этилен и пропилен, а также метан
и водород. н-Бутан при П. одновременно деметили-
руется и претерпевает разрыв молекулы посредине.
Реакция дегидрирования имеет меньшее значение.
Поэтому в продуктах распада преобладают пропилен
и этилен. Соотношение пропилен: этилен выше, чем
при П. этана и для получения пропилена н-бутан яв-
ляется лучшим сырьем, чем этан. В продуктах П.
изобутана, благодаря сильному дегидрированию,
содержатся значительные количества изобутилена
наряду с пропиленом; этилен из него не образуется.
Высшие парафины при П. также дают большие коли-
чества газообразных и жидких олефинов.
Нафтеновые углеводороды более стабильны при П.,
чем парафины. Первоначально происходит отрыв бо-
ковых цепей, в особенности наиболее длинных, а в бо-
лее жестких условиях расщепляется кольцо с образо-
ванием олефина. Напр., при П. циклопентана обра-
зуется до 70 об. % олефинов; из циклогексана обра-
зуется до 40 об.% этилена, а также и бутадиен-1.4.
Характерной для нафтенов является изомеризация
цикла с изменением числа атомов углерода в нем.
Напр., гексаметилены переходят при П. в пентаме-
тилены, 7- и 8-членные нафтены — в 6-членные. Про-
исходит также и дегидрогенизация циклов, преим.
1075
ПИРОЛИЗ
1076
6-членных с образованием ароматич. углеводородов.
Наряду с этим могут образоваться ненасыщенные
циклич. углеводороды, склонные к уплотнению.
Напр., циклопентадиен дает при П. нафталин.
Непредельные углеводороды, особенно низкомоле-
кулярные, значительно устойчивее предельных, поэ-
тому они являются обязательным продуктом распада
парафинов. Двойная связь прочнее одинарной в усло-
виях П., поэтому у высокомолекулярных олефинов
преобладает распад по связи С—С. Возможно и отщеп-
ление водорода с образованием диолефинов, хотя эта
реакция проводится в практике как термокаталитич.
процесс. Наиболее характерны для олефинов полиме-
ризация и конденсация с последующим превраще-
нием образующихся продуктов. Реакции уплотнения
особенно выражены при одновременном наличии в
сырье П. ароматич. углеводородов. Может происхо-
дить и циклизация олефинов, особенно диолефинов,
с последующей дегидрогенизацией образующихся цик-
лич. углеводородов при достаточно жестких условиях,
приводящая к образованию ароматич. углеводородов.
Ароматические углеводороды при П. наиболее ус-
тойчивы. При не очень высоких темп-рах идет отщеп-
ление боковых цепей у алкилированных ароматич.
углеводородов, углеводороды с длинными цепями рас-
падаются наиболее легко, давая ароматич. углево-
дороды с более короткими боковыми цепями. Метиль-
ная группа отщепляется трудно, поэтому и сам бен-
зол и его метильные гомологи, особенно толуол, тер-
мически очень устойчивы. В отличие от парафинов,
энергия связи С—С в ароматич. кольце больше энер-
гии связи С—Н, поэтому бензол и его гомологи срав-
нительно легко распадаются до углерода и водорода.
Для ароматич. углеводородов характерна конденса-
ция, происходящая либо между ароматич. кольцами,
либо между ароматич. углеводородами и присутствую-
щими в реакционной смеси олефинами. Конденсация
первого типа идет с выделением водорода и в случае
бензола приводит к образованию дифенила, из наф-
талина образуется динафтил, последний переходит
далее в пирен и перилен. Конденсация осложняется
выделением углерода. Термич. стойкость и склонность
к конденсации сильно осложняют термич. переработку
нефтепродуктов, содержащих большое количество
ароматич. углеводородов. Технич. применение П. угле-
водородов описано в статьях крекинг, гидрогенизация
деструктивная, риформинг, ароматизация нефтепро-
дуктов, газы природные горючие.
Кислородные соединения. П. спир-
тов протекает обычно при темп-рах порядка 500—
700°; приболев низких темп-рах основным направлени-
ем реакции является отщепление воды с образованием
соответствующего олефина. При более высокой темп-ре
большую роль начинает играть дегидрирование гидро-
ксильной группы с образованием из первичных спир-
тов — альдегидов, а из вторичных и третичных спир-
тов — кетонов. Наряду с первичными продуктами
реакции — олефином и водой или альдегидом (кето-
ном) и водородом — реакционная смесь всегда со-
держит и продукты, образующиеся в результате вто-
ричных реакций — низшие олефины и предельные
углеводороды, окись углерода — причем роль вторич-
ных реакций возрастает с темп-рой. В нек-рых слу-
чаях дегидратация протекает черезвычайно легко уже
при низких темп-рах. Напр., этил-п-анизилкарбинол
превращается в анетол при перегонке в вакууме при
140°. Третичные спирты превращаются легче, чем вто-
ричные и первичные, хотя это различие выражено
слабее, чем при термокаталитич. реакциях. В отли-
чие от последних, при пиролитич. расщеплении ни-
когда не образуются эфиры. Исключением является
бензиловый спирт. Если есть возможность образова-
ния 5- или 6-членного цикла, то отщепление воды мо-
жет протекать с его образованием. Напр., при П. тер-
пингидрата при 260° отщепляется вода от первичного
гидроксила с образованием цис-терпина, тогда как
оба третичные гидроксила остаются нетронутыми.
П. двухатомных спиртов не отличается принципиально
от П. одноатомных, за тем исключением, что отщеп-
ляться может, в зависимости от условий, один или оба
гидроксила с образованием соответственно непре-
дельного спирта или диолефина. Могут образовываться
также непредельные альдегиды и оксиальдегиды.
Фенолы являются значительно более стабильными и
распад их происходит при темп-рах порядка 800—
900°. В результате образуются, напр., из фенола —
бензол, n-оксидифенил и дифениловый эфир.
Алифатич. эфиры в результате П. образуют основ-
ной продукт — соответствующий альдегид. При нали-
чии ароматич. заместителей распад идет до ароматич.
альдегида и ароматич. углеводорода, напр. из дибен-
зилового эфира образуются бензальдегид и толуол.
В смешанных алкилароматич. эфирах расщепление
приводит к соответствующему фенолу и олефину. Чи-
сто ароматич. эфиры, напр. окись дифенила, вещества
термически очень стойкие. Весьма сложные превра-
щения претерпевают при термич. воздействии ненасы-
щенные эфиры.
Общим для алифатич. альдегидов и кетонов при П.
является отщепление СО с образованием предельного
углеводорода. Наряду с этим образуются также оле-
фины и водород. Из простейших кетонов образуются
также кетены, из ацетона, напр., в значительных ко-
личествах. Ароматич. кетоны могут образовывать кон-
денсированные ароматич. углеводороды при умерен-
ном нагревании. Напр., диметилбензофенон при 320°
дает с отщеплением воды 2-метилантрацен.
Для П. алифатич. и ароматич. кислот характерно
декарбоксилирование с отщеплением СО2 и образова-
нием из радикала кислоты насыщенного углеводорода.
И в том и в другом случае для облегчения декарбокси-
лирования добавляют натронную известь. Дикарбо-
новые к-ты ведут себя более сложно, в зависимости
от взаимного расположения карбоксильных групп.
Малоновая к-та и ее гомологи, у к-рых обе карбоксиль-
ные>группы стоят при одном и том же атоме углерода,
легко отщепляют одну карбоксильную группу с вы-
делением СО2 и образованием соответствующей моно-
карбоновой к-ты. Янтарная к-та и ее гомологи дают
при П. циклич. 5-членные ангидриды; глутаровая к-та
и ее гомологи — 6-членные ангидриды, хотя и значи-
тельно труднее. 6-членные ангидриды образуются
и в том случае, когда серединная метиленовая группа
в кислоте замещена атомом кислорода, напр. дигли-
колевая и дилактоновая к-ты при перегонке образуют
циклич. ангидриды. Если же метиленовые группы
входят в состав цикла ядра, то часто наблюдается
не дегидратация с образованием ангидрида, а декарбо-
ксилирование. Напр., изофталевая к-та не дает ангид-
рида. Адипиновая и пимелиновая к-ты и их гомологи
дают обычно не ангидриды, а циклич. кетоны — цик-
лопентаноны и циклогексаноны. Соответствующие
ароматич. и гетероциклические двухосновные к-ты
очень часто не дают ни ангидридов, ни кетонов, но
просто декарбоксилируются.
Оксикислоты при П. могут вести себя различным
образом, давая ангидриды, лактиды, ненасыщенные
а, р-кислоты и альдегиды. Лактиды также при нагре-
вании, отщепляя СО, образуют альдегиды. Р-Окси-
кислоты склонны к образованию а, р-ненасыщенных
карбоновых к-т. у- и 6-Оксикислоты уже при низких
(ок. 100°) темп-рах отщепляют воду, образуя циклич.
лактоны. Соли алифатических кислот с одновалент-
ными металлами обычно декарбоксилируются с обра-
зованием насыщенного углеводорода из радикала кис-
лоты; соли двухвалентных металлов декарбоксили-
1077
ПИРОЛИЗ
1078
руются с образованием кетонов; эта реакция может
осуществляться как термокаталитич. процесс над оки-
слами двухвалентных металлов. Сложные эфиры
алифатич. к-т распадаются на исходную кислоту и
на олефиновый углеводород из эфирного радикала.
Поведение аминокислот при П. напоминает поведе-
ние оксикислот и фактором, определяющим результа-
ты процесса, является взаимное расположение кар-
боксильной и аминогруппы. Пиролиз а-аминокислот
происходит с отщеплением воды или СО2. В первом
случае аналогично образованию лактидов из а-ок-
сикислот образуется дикетопиперазин; во втором —
соответствующий амин. На преобладание того или ино-
го направления распада влияет характер нагревания.
При осторожном нагревании до более низкой темп-ры
отщепляется вода, при более быстром и сильном на-
гревании — СО2. Напр., при нагревании глицина
NH2CH2COOH в глицерине при 150—170° образуется
дикетопиперазин и отчасти полимерный ангидрид —
(C2H3ON)x. При сильном нагревании в глицерине или в
дифениламине (240°) образуется метиламин. Подобным
же образом ведет себя и фенилглицин, образуя при
130°дефинилдикетопиперазин, а при 190°метиланилин.
Отщепления СО от а-аминокислот не наблюдается.
Дипептиды образуют дикетопиперазины уже при
плавлении. Простейшие р-аминокислоты при П. от-
щепляют аммиак и образуют непредельную к-ту, напр.
из p-аминопроппоновой к-ты образуется акриловая
к-та, а, р-днаминокислоты декарбоксилируются с об-
разованием соответствующих диаминов. Если а, р-
атомы углерода являются частью ароматич. ядра, как,
напр., в антраниловой к-те, то происходит только
декарбоксилирование с образованием соответствую-
щего ароматич. амина; у-аминокислоты при П. отщеп-
ляют воду, образуя лактамы. Однако у-амипокислоты
более стойки,чем соответствующие им оксикислоты.
Напр., у-оксимасляная к-та устойчива только при
очень низких темп-рах, тогда как у-аминомасляная
образует пиролидон только ок. 200°, Подобно образо-
ванию 5-членных колец из у-аминокислот, 6-аминокис-
лоты образуют 6-членные кольца, отщепляя воду уже
при плавлении или при вакуумной перегонке. Напр.,
6-аминовалериановая к-та при плавлении образует
пиперидон. 7-членные кольца — лактамы е-аминокис-
лот — также образуются, но значительно труднее,
чем 5-и 6-членные, получаются гл. обр. продукты уп-
лотнения.
Азотистые соединения. Основными
продуктами распада аминов, образующимися в ре-
зультате отщепления аммиака или водорода, являют-
ся этиленовый углеводород и нитрил. При темп-рах
выше 700° распад идет глубже с образованием предель-
ного углеводорода, цианистого водорода и азота. Ди-
метпламин и трпметиламин при 1000° распадаются
на HCN, СН4 и Н2. Четвертичные аммониевые основа-
ния, кроме гидрата окиси тетраметиламмония, рас-
падающегося на метиловый спирт и триметиланилин,
образуют обычно с отщеплением молекулы воды соот-
ветствующий трпалкиламин и этиленовый углеводород.
Реакция протекает чрезвычайно легко. Галогенные
соли четвертичного аммония при П. образуют наряду
с триалкилампном не олефин, а соответствующий
галогеналкил.
Поведение амидов при П. напоминает поведение кар-
боновых к-т. Основными направлениями П. являются
отщепленпе аммиака с образованием предельного
углеводорода из радикала кислоты и дегидратация
с образованием соответствующего нитрила. Имидов,
однако, из амидов одноосновных к-т не образуется,
подобно тому, как не образуется и ангидридов из са-
мих одноосновных к-т. Амиды к-т типа янтарной и
глутаровой, наоборот, склонны образовывать циклич.
имиды.
Нитросоединения при П. разлагаются с различной
легкостью, иногда со взрывом, в зависимости от числа
и взаимного расположения нитрогрупп в молекуле.
В продуктах разложения в зависимости от условий
наряду с азотом и его окислами — NO, NO2 содержатся
также СО и СО2, образующиеся в результате окисляю-
щего действия окислов азота на углеводородную часть
молекулы.
Гетероциклические азотистые соединения часто тер-
мически очень устойчивы, напр. пиридин и его гомо-
логи, и поэтому образуются при П. различных азот-
содержащих веществ, напр. аминокислот, N-гете-
роциклич. карбоновых к-т и при сухой перегонке угля
и сланцев. Распад их, подобно распаду ароматич. сое-
динений, происходит только при высоких темп-рах.
П. сернистых соединений представ-
ляет большой практич. интерес, ввиду их содержания
в нефти и в различных нефтепродуктах. Разложение
их при различных процессах нефтепереработки вызы-
вает значительные осложнения, приводя к коррозии
аппаратуры и к необходимости очистки нефтепродук-
тов от сернистых соединений, образующихся в ре-
зультате П. более высокомолекулярных, содержащих
серу соединений (см. Гидроочистка, Очистка нефте-
продуктов). Разложение этих соединений идет гл.
обр. с отщеплением сероводорода и элементарной серы.
Меркаптаны распадаются с отщеплением сероводорода
и образованием сульфидов. Последние распадаются
дальше, отщепляя сероводород и бразуя парафино-
вые углеводороды и олефины. При этом часто образу-
ются смолистые остатки и сера. Дисульфиды, распа-
даясь, образуют сероводород, серу, меркаптаны,
алкилсульфнды, а также парафины и олефины и смоли-
стые продукты. Образуются иногда также и гетеро-
циклич. сернистые соединения, напр. трофеи и его
производные. Гетероциклич. сернистые соединения
значительно устойчивее термически, чем соединения
с открытой цепью, и поэтому могут образовываться
при темп-рах порядка 450—500° и выше из серово-
дорода и олефиновых углеводородов.
Галогенные соединения. Основным
направлением распада галогеиалкилов при П. яв-
ляется отщепление галогенводорода с образованием
олефина. Наряду с этим при умеренных темп-рах про-
исходит изомеризация с образованием из первичных
галогеиалкилов вторичных и из вторичных тре-
тичных. Третичные галогеиалкилы термически устой-
чивее первичных и вторичных; с повышением мол. веса
их термич. стойкость падает. Опа падает также и с по-
вышением ат. веса, входящего в молекулу галогена.
Дигалогенные соединения дают с отщеплением га-
логеноводорода непредельные галогеиалкилы. Наряду
с этим из соединений с атомами галогена при различ-
ных атомах углерода образуются галогеиалкилы
с двумя атомами галогена при одном и том же атоме
углерода. Галогеиалкилы с тремя и более атомами
галогена в молекуле также распадаются с образо-
ванием непредельных галогеиалкилов. Напр., из
четыреххлористого углерода и из хлороформа при
П. образуется тетрахлорэтилен, а также и гексахлор-
бутадиен. Непредельные галогеиалкилы значительно
стабильнее насыщенных, благодаря чему и могут быть
получены из дигалогенных насыщенных углеводо-
родов. Ароматич. соединения, содержащие атом га-
логена в ядре, очень устойчивы. Производные бензола
при этом переходят в производные дифенила, напр.
хлорбензол при длительном нагревании переходит
в 4,4-дпхлордифенил.
Техника пиролиза. Применение П. ограничивается
в основном переработкой различных видов углево-
дородного сырья с целью произ-ва, с одной стороны,
топлив и масел и, с другой — сырья для пром-сти
органич. синтеза, гл. обр. олефинов и ароматич. угле-
1079
ПИРОЛИЗ
1080
водородов (этилена, пропилена, высших олефинов,
бутадиена, ацетилена, бензола, толуола, ксилолов,
высших гомологов бензола и конденсированных аро-
матич. углеводородов).
Основным вопросом при П. является подвод доста-
точного количества тепла в реакционную зону. В ла-
бораторных условиях П. часто проводят в статич.
условиях, при постоянном объеме реактора, а источ-
ником тепла является раскаленная металлич. нить.
В промышленных условиях П. чаще всего осуществ-
ляют в проточных системах и реже в аппаратах пе-
риодич. действия. Все большее распространение полу-
чают непрерывно действующие реакционные устрой-
ства с внутренним обогревом, где тепло подводится
непосредственно
Рис. 1. Схема ре-
генеративной печи
Копперса—Хэша:
I — воздух; II —
продукты сгора-
ния; III — угле-
водородное сырье;
IV — газ пироли-
за; V — топлив-
ный газ.
ш
Рис. 2. Двухкамерная трубчатая
печь с экраном одностороннего об-
лучения: I — сырье; II — газ пиро-
лиза; III — топливо.
внутрь реактора, а не че-рез стенку.
Наряду с этим используются и ап-
параты периодич. действия и реак-
торы с подводом тепла извне, через
стенку реактора.
Наиболее проста конструкция а п-
паратов периодическо-
го действия — регенератив-
ных керамич. печей с неподвижной
насадкой (рис. 1). Печь представляет
собой камеру с огнеупорной насадкой,
нагреваемой топочными газами до
1000—1500°. После цикла разогрева
насадки на нее подается пиролизуе-
мое сырье, обычно разбавленное
водяным паром для улучшения те-
плопередачи и снижения коксообра-
зования. За счет аккумулированного насадкой теп-
ла происходит П. По охлаждении насадки прои.з-
водится новый цикл нагрева ее и т. д. Для лучшей
утилизации , тепла и обеспечения непрерывности про-
цесса при смене циклов регенеративные печи обычно
строятся многокамер-
ными. В то время, как
одни камеры исполь-
зуются для П. и за-
калки продуктов ре-
акции, в других каме-
рах подогревается по-
даваемый воздух или
топливо - воздушная
смесь или же проис-
ходит нагревание на-
садки. Иногда насад-
ка обладает катали-
тич. действием и в
этом случае представ-
ляет собой керами-
ку, пропитанную солями тяжелых металлов. Широ-
ко используются реакторы с внешним обогревом,
т. наз. трубчатые печи (рис. 2). П. в этом
случае протекает в трубах (змеевиках), через к-рые
прокачивается превращаемое сырье, находящихся
в радиантной камере печи и нагреваемых за счёт излу-
чения от факела сгорающего топлива до 800—900°.
Несмотря на преимущества, к-рые представляет П.
в реакторах с внутренним обогревом, трубчатые печи
в силу простоты конструкции и достаточно высоких
показателей работы не потеряли своего значения, осо-
бенно при пользовании т. наз. беспламенными (корот-
кофакельными) горелками.
Большие преимущества представляют проточ-
ные реакторы с внутренним обогревом
(рис. 3), в к-рых в реакционное пространство подает-
ся твердый или газообразный теплоноситель, нагре-
вающий превращаемое сырье до темп-ры П. Наибо-
лее распростравенными являются аппараты с по-
движным гранулированным или пылевидным твер-
дым теплоносителем, нагреваемым вне реактора ды-
мовыми газами и затем не-
прерывно вводимым в ре-
актор, где он отдает свое
тепло пиролизуемому про-
дукту и по охлаждении вы-
водится из реактора, при-
чем ввод и вывод теплоноси-
теля непрерывный и незави-
симый друг от друга. Преи-
муществом такого способа
подвода тепла является бо-
лее равномерный нагрев
сырья, большая гибкость ре-
гулирования темп-ры, а так-
же возможность выводить
из реактора вместе с тепло-
Рис. 3. Схема установки для пи-
ролиза с подвижным твердым
теплоносителем: 1 — загрузоч-
ный бункер; г — зона нагрева
теплоносителя; 3 — затвор; 4 —
трубопровод дымовых газов; .5 —
топливо; 6 — воздух; 7— топка;
8— газоотвод; 9— зона пироли-
за; 10—подача сырья; 11—подача
пара; 12— перегретый
пар;
— вывод теплоносителя; 14 — дозатор.
носителем осаждающиеся на нем продукты глубокого
распада и уплотнения, так называемый «кокс».
Новым и очень перспективным способом является приме-
нение газообразного теплоносителя (рис. 4).
Принцип метода заключается в том, что поток продуктов
сгорания к.-л. топлива в смеси с кислородом или с воздухом
из топочной камеры 2 при темп-ре до 2000° поступает в
смесительную зону, где смешивается с превращаемым сырь-
ем и водяным паром (для регулирования темп-ры процесса),
затем проходит в реактор 4, где сырье пиролизуется и, нако-
нец, поступает в закалочную зону, в качестве теплоносителя
можно применять также и перегретый до 1000—1200° водяной
пар, хотя при этой темп-ре он может действовать и окисля-
ющим образом (в данном случае пар играет роль теплоно-
сителя, а не разбавителя, как в других процессах, где практи-
куется его добавка). В стадии опытного применения находятся
реакторы с жидким теплоносителем, в качестве к-рого приме-
няется расплавленный и нагретый извне до заданной темп-ры
металл, напр. свинец, через к-рый пропускается превращаемое
сырье. Большой интерес представляет пиролиз в плаз-
ме, т. е. в потоке газообразного теплоносителя (метано-водо-
родная смесь, водород, аргон), нагреваемого до темп-ры по-
рядка 2000—3000° и выше (10 000—20 000°) электрич. тоном
и содержащего ионизированные частицы — ионы и электроны.
Разогрев теплоносителя и образование плазмы достигаются
в небольшом пространстве между катодом и анодом в так наз.
плазменной горелке. Мощность таких горелок доходит до
2000 кет. Этот метод особенно пригоден для получения'силь-
но эндотермичных продуктов, напр. ацетилена, HCN, чистых
кремния, бора путем разложения соответствующих кремний- и
Рис. 4. Реактор для пиролиза легких углеводоро-
дов в потоке газообразного теплоносителя: 1 —. го-
релка; 2 — топочная камера; з —смеситель; 4—
реактор; 5— смотровое окно; I — сырье; II — воздух;
III — топливо; IV — газы пиролиза; V — вода.
борорганич. соединений. Особым видом автотермич. П. яв-
ляется окислительный П., применяемый для превращения уг-
леводородов, при к-ром сырье до заданной темп-ры разогре-
вают за счет сжигания части самого сырья, смешиваемого
внутри реактора в нужном соотношении с кислородом или
кислородсодержащим газом (рис. 5). Для достижения высо-
кой темп-ры применяют также электродуговые печи, в к-рых
высокая темп-ра создается между электродами дуги постоян-
ного или переменного тока при напряжении порядка
7000 в и силе тока 850—900 а. Разновидностями этого метода
являются Д. в тлеющем разряде и в многократных микродугах.
1081
ПИРОЛИЗ
1082
Рис. 5. Реактор для окисли-
тельного пиролиза метана.
Получение химич. сырья
Пока только научный интерес представляет пиролиз
в ударных трубках, в к*рых нагрев разлагаемого
сырья до 1500—2000° и выше, вплоть до 1 000 000°, достигается
практически мгновенно адиабатич. его сжатием под действием
ударной волны.
Примером одновременного действия высокой темп-ры и
радиации является П. при облучении превращаемого сырья
у-излучением от Со60. Основ-
СН*+ 02 ными стадиями цепной реак-
-4 ции распада, напр. н-пара-
финов, являются иницииро-
вание, требующее энергии
активации 80 ккал/моль, и
рост цепи с энергией актива-
ции ок. 20 ккал'моль. Сум-
марная энергия активации
П. ок. 60 ккал/моль, т. е.
реакция может протекать с
заметной скоростью, начи-
ная с 600°. Если же реакция
инициируется свободными
радикалами, возникающими
под действием излучений,
удается при П. н-парафинов
при 650° (обычной темп-ре
термич. крекинга) увеличить
выход продуктов примерно
в 2 раза, или же проводить
крекинг с теми же скоростя-
ми, что и при чисто термич.
инициировании процесса, но
при темп-рах на 150—200°
более низких.
из углеводородов может
быть классифицировано, во-первых, по способу под-
вода тепла в соответствии с описанными выше мето-
дами, во-вторых, по характеру перерабатываемого
сырья (природный газ, попутный газ, нефтезаводские
газы, легкие, не находящие сбыта, бензиновые фрак-
ции, тяжелое, в том числе остаточное сырье и неразог-
нанная нефть) и, наконец, по целевому назначению.
Выбор способа подвода тепла обусловливается,
помимо экономии, соображений, также характером
наилучшим сырьем для получения этилена. Легкое
сырье вообще дает большие выходы газа, чем тяжелое.
Напр., из газового бензина при сопоставимых усло-
виях получается газа на 20—30% больше, чем из
керосино-газойлевых фракций и на 50% больше, чем
при применении тяжелых фракций нефти. Наоборот,
тяжелое сырье более пригодно для получения арома-
тич. углеводородов и жидких олефинов. Большое
влияние на выход газа оказывает групповой состав
сырья, в первом приближении характеризуемый от-
ношением С/Н. При большой глубине П. жидких угле-
водородов с одинаковым отношением С/Н, независимо
от их мол. веса и строения образуются примерно оди-
наковые количества газа. Кроме того, характер сырья
определяет скорость П., а следовательно и произво-
дительность реактора. Влияние природы сырья на
выход и состав газа пиролиза показан в табл. 1.
Таблица 1
Показатели и состав про- пан Сырье
газовый бензин мазут
Темп-ра, °C 816 816 843
Выход газа, вес % на сырье . . . 80,2 63,2 47,7
Жидкие продукты, вес. % на сырье Состав газа, об. %: 19,0 36,3 41 , 8
н2 24,2 22,5 25,6
СП, 48,3 48,3 43,0 2,7
с2на 2,6 2,8
С2Н. 21 ,7 23,5 23.7
С,н, 1,0 0,8 2,0
с.н10 0,1 0,2 —
Выход и состав газа при различных способах ве-
дения П. приведен в табл. 2.
Таблица 2
Условия П., выход и состав газа П. в трубчатых печах П. в регене- ративных печах П. с водяным пароли Окислительный П. П. с подвижным теплоносителем
этан газо- вый бензин этан этан тяжелый бензин этан легкий бензин этанпро- пиленовая фракция газовый бензин
Темп-ра, °C 850 835 950 1100 900 960 850 900
Выход газа, об. % на сырье . . Состав газа, об. %: 74 76,6 83,9 60 50-70 * 81,9 72,5 64,4 53,5*
н2 36 15,6 42,5 6,3 0,8 19,8 4,8 33,0 0,9
СП, 11,2 41,0 10,8 7,2 10,9 5,9 14,1 6,6 16,6
С2Н2 1,0 — 2,6 0,6 —— 0,3 —— ——
С2Н4 34,8 28,3 30,0 75,4 22,5 21 ,3 17,4 36,5 20,6
с2на 13,7 4,6 8,9 —— 2,4 6,6 3,1 22,5 10,9
с,Не 2,7 6,1 —— 12,0 —— 13,6 0,5 —
С3н„ — 0,6 3,0 8,6 0,4 0,6 3.3 0,3
с. 0,0 0,8 — —- — 0,4 13,7 0,2 —-
С О + СОд 0,62 —— 0,8 — 6,8 16,9 —. —
Выход жидких продуктов и ко- — — 1,4 — — 38,3 — — —*
кс а 2,1 23,4 — — — — 27,5 — —
♦ Вес. %
сырья и назначением процесса. Напр., для П. лег-
кого сырья применимы любые способы, тогда как П.
тяжелого сырья в трубчатых и регенеративных печах
создает ряд трудностей из-за легкой его коксуемости,
содержания золы и т. и. Если, напр., для получения
этилена практически пригодны все способы, то для
получения ацетилена, требующего высокой темп-ры
и быстрой закалки продуктов реакции, наиболее
пригодными будут электрокрекинг, окислительный
П., применение газообразного теплоносителя.
Выбор сырья обусловлен гл. обр. целевым назначе-
нием процесса. Напр., метан непригоден для получе-
ния этилена, т. к. он более стоек при высокой темп-ре,
чем этилен. В то же время метан является наиболее
дешевым сырьем для получения ацетилена, более
стойкого при темп-ре П., чем метан. Этан является
Сказанным далеко не исчерпываются потенциаль-
ные возможности П., области применения к-рого
в связи с развитием все расширяются.
Лит.: Херд Ч. Д., Пиролиз соединений углерода, пер.
с англ., М.—Л., 1938; Лукьянов П. И., Басистов
А. Г., Пиролиз нефтяного сырья, М., 1962; Введенский
А. А.. Термодинамические расчеты нефтехимических процес-
сов, Л., I960; Паушкин Я. М., Вишнякова Т. П.,
Производство олефиносодержащих и горючих газов из неф-
тяного сырья, М., 1960; Основы технологии нефтехимического
синтеза. Сб. статей, Под ред. А. И. Динцеса и Л. А. Потопов-
ского, М., 1960; Экспресс-информация. Химия и переработка
нефти и газа, Кг 18, 25, 28, 33, 37, М., 1962 (ВИНИТИ),
Радиолиз углеводородов, М., 1962; Клименко А. П.;
Получение этилена из нефти и газа, М., 1962; Гудков С.Ф.,
Переработка углеводородов Природных и попутных газов,
М., 1960; Артюхов И. М., Окислительная конверсия уг-
леводородов, М., 1961; Физико-химические свойства индиви-
дуальных углеводородов, под ред. В. М. Татевского, М.,
1960, гл. VII, с. 308. В. В. Щекин.
О 36 К.Х.Э. т. 3
АЛФАВИТНО-ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Абсолютных скоростей реакций теория —
см. Переходного состояния метод
Абсорбция, массообмен 52
Авертин — см. Нарколан
Автолы — см. Масла автотракторные
Адамсит 818 '
Адениндезоксирибозид 603
S-Ад еноз илметионин 198
Аденозин 603
Аденозин-Б-дифосфат 611
Аденозин-2'- и 3'-фосфат 611
Аденозин-5'-монофосфат 611
Аденозин-5'трифосфат 611
Адипекс — см. Первитин
Адонит 254
Адрианол — см. Мезатон
Азот
—. молекула, межъядерное расстояние
102
Азурит 67, 77, 1022
Аквамарин 239
Аквополимолибдаты 299
Акриловая кислота, метиловый эфир —
см. Метилакрилат
Акрилонитрил 494
Акролеин 818
Активированного комплекса метод —
см. Переходного состояния метод
Активированный комплекс — см. Пе-
реходное состояние
Актин 245
Акустические единицы 87
Алентин — см. Надизан
Ализарин 691
Алкил(арил)арсиновые кислоты 355
Алкил(арил)арсинтетрагалогениды 355
Алкиларилсульфонаты 335
Алкилат, октановые числа 18,720
Алкилбензолсульфонаты 335
Алкилнитраты 489
Алкилнитриты 498
Алкилоламиды 337
Алкилоловогалогениды 740
Алкилсульфаты 335
Алкилсульфонаты 336
Алкилхлоркарбонаты 817
Аллантоин 327
Аллилмеркаптометилпенициллин 879
N-Аллилнорморфин, хлоргидрат —см.
Налорфин
Аллильная перегруппировка 915
Аллит 255
Аллитрин 1052
Аллоксантин 327, 342
Аллопалладий 845
Алмаз 241, 242
Алхимия 420
Альдитолы 323
Альдозиды 323
Альдопентозы 895
Альдопентоновые кислоты — см. Пепто-
новые кислоты
Альтернативный запрет 288
Алюминиевая пудра 1024
Алюминиевые бронзы 71
Алюминий
— нейтронное облучение 165
— нитрид 491
— сплавы, микротвердость 141
— упругость 205
Алюминотермия 155
Амальгамация 634
Амблигонит 137
Амидопирин — см. Пирамидон
у-Амилбутиролактон — см. Нонлактон
п-Амилпенициллин 879
Аминобензилпенициллин 879, 885
n-Аминобензойная кислота
— диэтиламиноэтиламид, хлоргидрат —
см. Новокаинамид
—• диэтиламиноэтиловый эфир, хлоргид-
рат — см. Новокаин
— чувствительность микробиологии. ме-
тода химич. анализа 233
2-п-Аминобензолсульфамидотиазол — см.
Норсульфазол
М-(4-Аминобензолсульфонил)-1Ч'-н-бутил-
карбамид — см. Надизан
а-Аминовалериановая кислота— см. Нор-
валин
а-Аминокапроновая кислота — см. Нор-
лейцин
4-Амино-4-карбокси-п-бутилпенициллин
879
Аминокарбоксиоктулозы — см. Нейрами-
новые кислоты
Аминокислоты, чувствительность микро-
биологии. метода химич. анализа 233
— незаменимые 405
Аминомезитилен — см. Мезидин
а-Аминометилпиридины 196
а-Амино-у-метилтиомасляная кислота —
см. Метионин
о-Аминоминдальная кислота, лактам —
см. Диоксиндол
Аминонафталины — см. Нафтиламины
8-Амино-1-нафтойная кислота, лактам —•
см. Нафтостирил
4-Амино-2-оксибензойная кислота — см.
ПАСК
6-Аминопенициллановая кислота 878
а-Аминопептид-аминоацидогидролазы 901
Аминополисахариды кислые — см. Му-
кополисахариды
n-Аминосалициловая кислота — см.
ПАСК
Аминосидин — см. Паромомицин
2-Амино-1,3,5-тринитробензол —см. Пик-
рамид
2-Аминофенилуксусная кислота, лак-
там —. см. Оксиндол
Аммиачная селитра 245
Аммоний
— мономолибдат 299
— парамолибдат 299
— • пурпурат — см. Мурексид
— сульфат 24 5
Аммофос 245
Ампициллин — см. Аминобензилпени-
циллин
Амфедроксин — см. Первитин
Амфотерные соединения 336
Анаболизм 621
Анатаз 1013, 1015
Ангидриды кислот 679
Ангидрит 242
Англезит 135
Ангстрем (ед. измерения) 89
Анилин-З-сульфокислота — см. Метани-
ловая кислота
Анионоактивные вещества 335
Анисовый альдегид — см. Обепин
Анодная защита 868
Антибиотик С-1488 — см. Паромомицин
Антимонит 239, 241, 242
Антинейтрино 408
Антиокислители 668
Антипирин 525
Антироид — см. Мерказолил
Анторфин — см. Налорфин
Антранол 688
Антрацен 105
Антрон 688
Апатит 239, 241, 242
Апиоза 324
Апоморфин 325
Арабаны 891
Арабоксиланы 891
Арабоновая кислота 897
Арахидоновая кислота 406
Аргиродит 139
Ароматические соединения, нитрование
508
--- углеводороды, октановое число 719
Арсенаты и арсениты 352
Арсениды 349
Арсенолит 351
Арсенопалладинит 845
Арсеносоединения 354
Арсин — см. Водород мышьяковистый
Арсиноксиды 355
Арсины 354
Артеренол — см. Норадреналин
Асидол-мылонафт — см. Мылонафт
Ассимиляция 621
Атмосфера, масс-спектральное исследо-
вание верхних слоев 65
Атом
— строение и периодич. система 962
— электронные оболочки, схема запол-
нения 966
Атомные радиусы 974
Атомные спектры 281, 282
Атто (приставка) 89
Ауранция 526
Аурипигмент 348, 352
Ахондриты 174, 178
З-Ацетамино-4-оксифениларсиновая кис-
лота — см. Осарсол
6-Ацетил-1,1,2,3,3,5-гексаметилиндан —
см. Фантолид
Ацетилен 441
Ацетилкарбинол —• см. Оксиацетон
Ацетилсеротонин-метилтрансфераза 199
7-Ацетил-1, 1,4,4-тетраметил-6-этилтетра-
гидронафталин — см. Версалид
Ацетилцеллюлоза, механодеструкция 227
Ацетол — см. Оксиацетон
Ацетонитрил 494
Ациклические соединения 761
З-Ацилоксиндол 704
Ацинитросоединения — см. Нитроновые
кислоты
Б
Баббит, опробование 757
Бадделеит 139
Байциллин — см. а-Феноксипропилпени-
циллин
Бар (ед. измерения) 91
Барит 239, 241
Барн (ед. измерения) 89
Баха — Энглера перекисная теория 937
Беление вискозных тканей 803
---льняных тканей 803
--- тканей из животных и синтетич.
волокон 803
--- хлопчатобумажных тканей 801
Белила свинцовые 1013, 1016
--- цинковые 1013, 1016
Белки, биосинтез 278
— структурные особенности и функции
276
2,3-Бензантрацен —• см. Нафтацен
«Бензгидрилыюе правило» 129
Бензиловая кислота
—р-диметиламиноэтиловый эфир, иодме-
тилат — см. Метацин
—2-диэтиламинопропиловый эфир, хлор-
гидрат — см. Метамизил
Бензилпенициллин 879, 885
Бензины, октановое число 18, 720
— стабильность против окисления 436
Бензол 443
— октановое число 719
— энергия G—Н-связи 669
Бензол моторный, опробование 750
Бензолтрикарбоновая кислота — см. Бен-
золполикарбоновые кислоты (т. 1)
«Бензольная теория» 763
Бензонитрил 494
Бензэстрол —• см. Октэстрол
Берилл 137
Бериллиевые бронзы 71, 72
Бериллий 137
— • нитрид 491
Бетон, упругость 205
Бикфордов шнур — см. Огнепроводный
шнур
Бинотал— см. Аминобензилпенициллин
Биотин, чувствительность микробиоло-
гии. метода химич. анализа 233
Бисаболен 748
4,4'-Бис-(диметиламино)бензофенон —см.
Михлера кетон
2,3-Бис-(3,4-диоксибензил)-бутан — см.
Нордигидрогваяретовая кислота
Бис-(4-оксикумаринил-3 )уксус ная кисло-
та, этиловый эфир — см. Неодикума-
рин
Битумы 432
Бицикло-(0,1,4)-гептан —-см. Норкаран
Бициклооктан-[0,3,3] — см. Пенталан
Блокит 137
Бор
— молекула, межъядерное расстояние
102
— нитрид 491
Борацит 239
Бориды металлов, огнеупоры 648
Борнит 77, 135
Браунит 14, 133
Броксил — см. а-Феноксиэтилпеницил-
лин
Бром
— молекула, межъядерное расстояние
102
Бромбензилцианид 818
2-Бром-2-нитроиндандион-1,3 525
Бронзы 71
Броциллин — см. а-Феноксипропилпени-
циллин
Брошантит 74, 77
Брэггит 845
Буланжерит 135
Бура 367
1085 —1086
Бутадиен-1,2— см. Метилаллен
Бутамид 358
Бутан, энергия С—Н-связи 669
н-Бутан 439
Бутаналь — см. Масляный альдегид
Бутановая кислота — см. Масляная кис-
лота
Бутанон-2—• см. Метилэтилкетон
н-Бутилен 441
Бутиральдегид — см. Масляный альде-
гид
Бутиронитрил 494
Бутлерова теория строения 763
В
Валентные соединения 145
Ванадий 135
— нитриды 492
Ванадинит 135, 239
Вапор облегченный 36
Вар (ед. измерения) 87
Вебер (ед. измерения) 85
Велосит 36
Вернадскит 74
Версалид 343
Взаимная система 256
Видемана — Франца закон 162
Визопенилловые кислоты 884
5-Винил-а-пиколин — см. 2-Метил-5-ви-
нилпиридин
Винилтриметиламмоний, гидроокись —
см. Нейрин
Вирус табачной мозаики 606
Висмутол II — см. Меркаптофенилтио-
тиодиазолон
Витамин В3— см. Пантотеновая кислота
Витамин В15 — см. Пангамовая кислота
Витамин F — см. Жирные кислоты не-
заменимые
Вицин 602
Внутреннее трение 206
Внутримолекулярное движение 271
Вода, опробование 750
--- гигроскопическая 243
--- надсмольная 358
Водород
— молекула, межъядерное расстояние
102
Водород мышьяковистый 353, 817
--- фосфористый 817
Волокна (из полимеров)
— прочность, временная зависимость 223
Вольфрам 135
— упругость 205
Вольфрамит 135, 239
Восстановление — см. Окисление-вос-
становление
Время, единицы измерения 84, 85, 91
Высокомолекулярные соединения, моди-
фицирование 260
— молекулярный вес 292
Выход химический 789
Вязкое течение полимеров 220
Вязкость динамическая, единицы изме-
рения 93
--- кинематическая, единицы измере-
ния 93
,— масел синтетических 45, 47
--- нефтепродуктов 32
Г
Газовый разряд, масс-спектральное ис-
следование 65
Газы, ожижение 822, 831
— опробование 750
Галенит 135, 241, 242
Галит —• см. Каменная соль
Галлий 139
Галлоацетофенон 693
Галогенацетаты 818
Гамамелоза 324
Гаргойль 36
Гарниерит 133
Гаусманит 19, 133
Гафний 139
— нитрид 492
Гексаалкилдистаннаны 74 2
Гексаарилдистаннаны 742
Гексабромное число 27
Гексагидрометилпиперидины — см. Ме-
тилпиперидины
Гексагидропиридин — см. Пиперидин
Гексагидротолуол — см. Метилциклогек-
сан
Гексадекановая кислота — см. Пальми-
тиновая кислота
Гексадиен-2,4-дикарбоновая-1,6- кислота
— см. Муконовая кислота
Гексаметаленбензамид 812
н-Гексан, октановое число 719
Гексафторацетон — см. Перфторацетон
Гексафторбензол — см. Перфторбензол
Гексафторбутадиен-1,3 — см. Перфторбу-
тадиен-1,3
Гексафторпропилен —• см. Перфторпро-
пилен
Гексахлорбутадиен — см. Пенхлорбута-
диен
Гексахлордивинил—см.Перхлорбутадиен
Гексахлорциклопентадиен — см. Пер-
хлорциклопентадиен
Гексен-1, октановое число 719
Гекситы 254
Гексоген 499
Гексозомонофосфатный шунт — см. Пен-
тозный цикл
Гексозы 320
Гекто (приставка) 89
Гельвин 137
Гематит 133, 241, 242
Гемопротеиды 153
Гемоцианины 1025
Гепарин 339
Гептадекановая кислота — см. Маргари-
новая кислота
Гептандиовая-1,7-кислота — см. Пимели-
новая кислота
п-Гептилпенициллин 879
Гептиты 255
Гептозы 320
Германий 139
Германит 139
Герсдорфит 133
Гессит 239
Гетероауксин 525
Гетероолигосахариды 726
Гетерополимолибдаты 299
Гетероциклические соединения 761
— нитрование 513
Гетит 133, 239
Гиалуроновая кислота 339
Гига (приставка) 89
Гигантеновая кислота — см. п-Амилпе-
нициллин
Гидразин 525
Гидразинолиз 906
Гидроборацит 239
Гидрогалит 387
Гидролазы 560
Гидроочистка нефтепродуктов 833
Гидроперекиси
--- неорганические 935
--- органические — см. Перекиси и
гидроперекиси
Гидропероксил 935
Гидропероксильный ион 935
1-Гидроперокси-1-метилциклогексан 200
Гидропероксокомплексы 936
Гидроформилирование — см. Оксосин-
тез
Гидроциклоны 632
Гипоксантиндезоксирибозид 603
Гипосульфит — см. Натрий, тиосульфат
Гипс 239, 240 — 242
Гистерезис упругий 206
Гистон аргининовый 605
--- лизиновый 605
Глауберит 242
Глауберова соль 367, 382
Глаукодот 348
Глет 1023
Гликозы — см. Моносахариды
Глицеринтринитрат — см. Нитроглице-
рин
Глутаронитрил 494
4—(а-Д-глюкозидо)-Д-глюкоза — см.
Мальтоза
Глюкозооксидаза — см. Нотатин
Д-глюкопираноза 323
Д-глюкофураноза 323
Голозиды — см. Олигосахариды
Гомоолигосахариды 726
Гомоцистеин 202
Горные породы, опробование 755
Град (гон) (ед. измерения) 91
Градус Кельвина 84
Гранат 241
Гуанидинацетат-метилтрансфераза 198
Гуанозин 603
Гуанозин-5'-дифосфат 611
Гуанозин-2'- и З'-фосфат 611
Гуанозин-5'-трифосфат 611
Гуанозин-5'-фосфат 611
Гука закон 163, 205
Гуматии — см. Паромомицин
Гюбнерит 135
д
Давление, единицы измерения 91
--- нормальное 582
Дальтониды и бертолиды 14 2
Данаит 348
Даналит 137
Двойное лучепреломление 760
Двойные системы 142
Двуокиси 679
Дезамин — см. Первитин
Дезаминирование окислительное 622
2-Дезокси-Д-рибоза 60 2
Дезоксирибонуклеазы 590
Дезоксирибонуклеиновая кислота (ДНК)
592
— генетич. роль 277, 596
Дезоксирибонуклеиновая кислота апири-
мидиновая 594
--------апуриновая 594
Дезоксирибонуклеозиды 603
Дезоксирибонуклеопротеиды 605
Дезоксиэфедрин — см. Первитин
Дека (приставка) 89
Декарбоксилирование 622
Деклуазит 135
Декозы 320
Демаскировка (в химич. анализе) 27
Депсипептиды 905
Дерево, упругость 205
Дерматансульфат — см. Хондроитинсер-
ная кислота
Детонационная стойкость моторного топ-
лива 18
Деформационные свойства полимеров 214
Деформация остаточная 163
--- упругая 206
Деци (приставка) 89
Джемсонит 135
1,2-Диазин — см. Пиридазин
1,3- или At-Диазин — см. Пиримидин
1,4-Диазин — см. Пиразин
1,2-Диазол — см. Пиразол
Диазореакция — см. Реакция Паули
а.б-Диаминовалериановая кислота —см.
Орнитин
2, 4-Диамино-5,6-диоксипиримидин—см.
Дпвицин
а, е-Д иамино-б-оксикапроновая кислота —•
см. Оксилизин
Диаммофос 245
1,4 : 3,6-Диангидро-Д-сорбит —2,5-дини-
трат — см. Нитросорбид
1,2-Дибром-З-хлорпропан — см. Немагон
Дибутиладипат 812
Дибутилнафталинсульфокислота, натри-
евая соль — см. Некаль ВХ
Дивицин 1063
1,4-Дигидронафталин 390
Дигидрооксипиразол — см. Пиразоли-
дон
Дигидропенициллин F—-см. п-Амилпе-
нициллин
4,5-Дигидропиразол — см. Пиразолин
Дигидропиразолон — см. Пиразолидон
Диеновое число 27
Диизопропилфторфосфат 815, 817
1,4-Диметан-сульфонилоксибутан — см.
Миелосан
Ди-п-диметиламинобензофенон — см.Ми-
хлера кетон
Диметиламинодинитротрифтортолуол 526
2,2-Диметилбутан — см. Неогексан
2^2-Диметилбутанон-З —-см. Пинаколин
Диметилкарбат 812
Диметилнафталины — см. Метилнафта-
ЛИНЫ
Диметилнитрамин 499
Диметилнитрозамин 516
М,М-Диметил-4-нитрозоанилин—см. п-
Нитрозодиметиланилин
2,2-Диметилолпропандиол —• см. Пента-
эритрит
Диметилпиперидины — см. Лупетидины
Диметилпиридины —• см. Лутидины
2,4-Диметилпиррол 525
2,2-Диметилпропан — см. Неопентан
Диметилсульфат 186
Диметилуксусная кислота — см. Изо-
масляная кислота
Диметилфталат 812
2,6-Диметоксифенилпенициллин — см.
Метициллин
Динатрийарсенат 374
Дииикотиновая кислота — см. Пиридин*
карбоновые кислоты
36*
1087 —1088
4,6-Динитро-2-аминофенол — см. Пикра-
миновая кислота
2,4-Динитроанилин 500
3,5-Динитробензойная кислота 502
3,5-Динитро-2,6-диметил-4»трет-бутила-
цетофенон — см. Мускус-кетон
2,6-Д инитро-З-метокси-4-трет-бутилто-
луол — см. Мускус_амбровыи
1,1-Динитропропан 535
1.3-Динитропропан 535
2,2-Динитропропан 535
2,2-Динитро-1,3-пропандиол 546
Динитроспирты 545
Динитротолуолы 548
Динитрохлорбензолы 549
1.1-Динитроэтан 535
1,2-Динитроэтан 535
2,2-Динитроэтанол 54 6
1,2-Диоксиантрахинонализарин 691
2,5-Диоксиацетофенон 693
о-Диоксибенвол — см. Пирокатехин
N- (и, т-диокси-р, (З-диметилбутирил)-(З-
аланин — см. Пантотеновая кислота
2.2-Диокси-1,3-индандион —см. Нингид-
рин
2,6-Диокси-4-метил-5-оксиметил пири-
мидин — см. Пентоксил
5,8-Диоксинафтохинон-1,4 — см. Нафта-
зарин
Диоксиндол 705
Диоксипиридины 707
2,6-Диоксипиримидин-4-карбоновая кис-
лота — см. Оротовая кислота
3,5-Диокситолуол — см. Орсин
1-(3,4-Диоксифенил)-2-аминоэтанол—см.
Норадреналин
2,4-Ди-(п-оксифенил)-3-этилгексан —см.
Октэстрол
Диоксоаллен — см. Недоокись углерода
а, З-Диоксопропан — см. Метилглиок-
саль
Дипептидгидролазы 901
Дипиколиновая кислота — см. Пиридин-
карбоновые кислоты
Дипропилнитрозамин 516
Дисперсионные силы 97
Диссимиляция 621
Диурон — см. 1\-3,4-Дихлорфенил-Х',Х-
диметилмочевина
2.3-Дифенилиндол 525
Дифенилнитрозамин —• см. N-Нитрозо-
дифениламин
Дифенилхлорарсин 818
Дифенилцианарсин 818
1,4-Дифенил-(3,5-энданил)-дигидро-1,2.4-
триазол — см. Нитрон
Дифосген 814, 817
Дифракция нейтронов 409
Диффузионное давление —• см. Осмоти-
ческое давление
Диффузия вихревая 55
---• молекулярная 55
----- турбулентная — см. Диффузия вих-
ревая
N, N-Д ихлор-п-карбоксибензолсульфа-
мид — см. Пантоцид
Дихлорфенидим— см. М-3,4-Дихлорфе-
нил-1Ч',1Ч-диметилмочевина
1Ч-3,4-Дихлорфенил-М',.1Ч-диметилмочеви-
на 331
Г^-ЗЛ-Дихлорфенил-М'-метил-М'-бутил-
мочевина 331
N- 1,4-Д ихлорфенил-М'-метил-Х'-метокси-
мочевина 331
Дихлорхромотроповая кислота 767
Диэтанол-1Ч-нитроаминдинитрат 556
5,5-Диэтилбарбитуровая кислота, натрие-
вая соль — см. Мединал
Диэтилметатолуамид 812
Диэтилнитрозамин 516
Длина, единицы измерения 84, 89
— — связи 102
Доза, мощность (в радиац. химии) 331
— — облучения 331
— — поглощенная 331
Донорно-акцепторная связь 101
Древесный спирт — см. Метиловый
спирт
Дренамид — см. Оксафенамид
Дриол — см. Оксафенамид
Друзы 240
Дуалистическая система Берцелиуса
420
Дублетов метод (в масс-спектрометрии)
61
Дульцит 255
ДЭТА — см. Диэтилметатолуамид
Единицы измерения, международная си*
стема 84
--------английские 95
•т»
lit
Жаропрочные никелевые сплавы 468
Железо 133
— маскировка (в химич. анализе) 26
— молибдат 299
—• пассивационная характеристика 869
— упругость 205
Железоокисные пигменты 1013, 1017
Женевская номенклатура 571
Жидкости, перекачивание 920
Жидкость, метаетаб ильное состояние 171
---- перегретая 171
—— переохлажденная 171
Жирные кислоты незаменимые 406
Жиры, высыхание 27, 28
----- кухонные 23
—•—• растительные, высыхание 27, 28
3
Закиси 679
Запрета принцип — см. Паули принцип
Зарин 814 815, 817
Зеебека эффект 162
Зелень горная 1022
----- изумрудная 1013, 1021
---------- Ринмана — см. Кобальт зеленый
—— свинцовая 1013, 1020
—— цинковая 1020
---------- швейнфуртская 1022
----- Шееле 1022
Золото 137
Зоман 815
Зонная теория образования металлич.
соединений 144, 160
И
Идит 255
Известь натровая 388
—— натронная — см. Известь натровая
---- хлорная, опробование 755
Измерения, ошибки 835
----совокупные 841
Изобарные потенциалы образования сое-
динений металлов 153
Изоборнил-2-метилциклогексанон — см.
Мустерон
Изобутан
— октановое число 719
— энергия С—Н-связи 669
Изобутилен 441
(р-Изобутилен — см. Перфторизобутилен
Изомасляная кислота 50
Изоментол 115
Изоментон 117
Изомеразы 561
Изомеризация механохимичсспая поли-
меров 228
Изоморфины 325
Изоникотиновая кислота — см. Пиридин-
карбоновые кислоты
Изонитроны 533
Изонитросоединения — см. Нитроновые
кислоты
Изооктан, октановое число 719
Изопентан 439
— октановое число 719
Изополимолибдаты 299
Изотопный состав вещества, масс-спект-
ральный анализ 61
Изоциклические соединения 761
Изоцинхомероновая кислота — см. Пи-
ридинкарбоновые кислоты
Изумруд 240
Икс-единица (ед. длины) 89
Ильинского реактив — см. а-Нитрсзо-
р-нафтол
Индалон 812
Индол 525
Индукционные силы 97
Инконель 468
Инозин 603
Инозиназа 601
Инозит, чувствительность микробиоло-
гия. метода химич. анализа 233
Интермедин — см. Меланотропный гор-
мон
Интерметаллические соединения 139
Иод
— молекула, межъядерное расстояние
102
Ионизации потенциал 972
Ионизация в пламенах, масс-спектраль-
ное исследование 65
Ионизирующие излучения, единицы из-
мерения 89, 95
Ионные радиусы 974
Иприт 814, 815, 818
--- азотистый 815
Исключения принцип — см. Паули прин-
цип
Испанская красная 1019
Кавитация 921
Кадмий 139
---• желтый 1013, 1017
---красный 1013, 1017
Кадмопон желтый 1013, 1017
---красный 1013, 1018
Калаверит 137
Каламин 135
Калий
— гексабромоосмиат 793
— гексахлороосмиат 793
— манганат 22
— метаниобат 474
— молекула, межъядерное расстояние
102
— надперекись 934
— оксифторониобат 483
— пентанитроосмиат 793
— пероксофосфат 985
— сульфат 245
— фторониобат 483
— хлорид 245
Калийная соль 245
Кальциевая селитра 245
Кальциетермия 155
Кальций
— молибдат 299
— плюмбат 1024
— . хромат 1024
Кальцит 239, 241, 242
Каменная соль 239, 242, 367, 387
Канфильдит 139
Каолинит 241
Каприловый альдегид — см. Октило-
вый альдегид
Капсаицин 818
Карат (ед. измерения) 91
Карбамид — см. Мочевина
«Карбамол» 808
Карбиды, огнеупоры 648
Карбинол — см. Метиловый спирт
а-Карбоксипептид-аминоацидогидролазы
901
4-Карбокси-урацил — см. Оротовая кис-
лота
Карбоновые кислоты, нитрилы 494
—• ортоэфиры 783
Карбоноль 732
Карботермия 154
Карнотит 135, 137
Касситерит 135, 240, 241, 737
Катаболизм 621
Катализ, носители 589
Катенулин — см. Паромомицин
Катионактивные вещества 336
Кварц 241, 242
Кеммерерит 240
Кератосульфат 339
а-Кето-6-аминопентаоксикарбоновые кис-
лоты — см. Нейраминовые кислоты
Кетозиды 323
Кетопентозы 895
а-Кетопропионовая кислота—-см. Пи-
ровиноградная кислота
Кило (приставка) 89
Кинетический фактор активности 956
Киноварь 135, 241, 1013, 1018
Кислород
— молекула, межъядерное расстояние
102
Кислородное число 28
Кислотный зеленый ЖМ 310
--- хром синий 2К 310
Кладиноза 324
Клодетит 351
Кобальт зеленый 1013, 1021
--- синий 1013, 1021
---фиолетовый 1021
Ковалентная связь 158
Ковеллин 75, 77, 135
Колебательные спектры молекул 273,
282, 285
Колимицин 418
Коллидины 195
Коллоидные системы
— оптические свойства 757
— осмотическое давление, мембранное
равновесие ИЗ
Коллоксилин 551
Колумбит 137, 239
Колумбит-танталит 475
Комбижир 23
Коменовая кислота 108
Комплексные соединения меди 78
--- --- многоядерные 256
------- ------ осмия 793
------- - палладия 850
Комплексоны
— маскировка (в химич. анализе), при-
менение К. 26, 27
л-Комплексы — см. Пи-комплексы
Конкреции 240
Константа Михаэлиса 247
—• седиментации 296
Константан 73
Конфигуративная точка 951
Концентрат рудный 134
Копеллидины 194
Копель 73
Кордицепоза 324
Коррозия металлов, защита 1014
Корунд 239, 242
Коферменты никотинамиднуклеотидные
472
----пиридиннуклеотидные 472
Коэффициент Вант-Гоффа 798
----второй вириальный 100
----массопередачи 52
----ожижения 826
----прохождения 953
----растворителя осмотический 798
----теплообмена (теплоотдачи) и (те-
плопередачи), ед. измерения 93
----- теплопроводности, ед. измерения
---- упрочнения 207
----холодильный 823
----экстинкции 758
Красители
----моноазо- 310
---- нитро- 525
---- нитрозо- 519
----- оксазиновые 681
---------- оксиантрахиноновые 691
-----пиразолоновые 1049
-----хиноноксимиминные — см. Нитро-
зокрасители
Кремненикелевая бронза 71
Кремний, нитрид 491
Криолит 241, 367
Криптоциллин — см. Оксациллин
Кристаллическая структура металлов 158
Крокоит 239
Кронкит 74
Ксантозин 603
Ксиланы 891
Кейлоны 443
Ксилоновая кислота 897
Кумол, энергия С—Н-связи 669
Куприт 135, 241, 242
Купферрон 767
Курнакит — см. Марганец, окись
Курнакова соединения 143
Л
Лавеса фазы 144
Лазеры 314
Лазурит 240
Лаки пигментные 1011
Лакойль 732
Лакокрасочные материалы, опробование
751
Лакриматоры 814
Лактопероксидаза 982
Лакумин — см. Мепазин
Лангит 74
Латуни 70
Лаусон 403
Левопропициллин — см. а-Феноксипро-
пилпенициллин
Лепидолит 137
Летидрон — см. Налорфин
Лиазы 561
Ливингстонит 135
Лигазы 561
Ликсоновая кислота 897
Лимонит 133
Лимонная кислота 699
Линде цикл 826
Линоксин 28
Линолевая кислота 406
Линоленовая кислота 406
Липиды, обмен 625
Липополисахарид 338
Липопротеин 338
Литий 137
— молекула, межъядерное расстояние
102
— нитрид 491
Литопон 1013, 1016
Лопарит 137, 475
Лупетидины 194
Лутидиновая кислота — см. Пиридии-
карбоновые кислоты
Лутидины 195, 1057
Льежские правила 574
м
Магнетит 133, 239
Магниетермия 155
Магний, нитрид 491
Магнитные единицы 85
Магнитные никелевые сплавы 468
Магнитные свойства металлов 162
Мазеры 314
Мазут, опробование 750
Майченерит 845
Максипен — см. а-Феноксиэтилпеницил-
лин
Малапрада реактив 672
Малахит 67, 77, 1022
Малоновая кислота, ангидрид —см. Уг-
лерод, недоокись
Мальтаза 9
Мальтобионовая кислота 10
Мальтоза 9
Манганаты — см. Марганцовистая .кис-
лота и манганаты
Манганин 73
Манганиты — см. Марганец
Манганозит 19
Маннаровая кислота 13
Маннит 10, 254, 255
Маннитгексанитрат — см. Нитроманнит
Манниха реакция 11
Манноза 12
а-В-Маннозидо-^'-метил-а-Ь-глюкозами-
нидо-р-Ь-стрептозидострептидин — см.
Манноз пдострептомицин
Маннозидострептомицин 13
Маннозиды 12
Манноновая кислота 13
Маннуровая кислота —см. Гексуроновые
кислоты (т. 1)
Маннуроновая кислота 13
Марганец 13, 133
— аналитич. определение 16
— галогениды 15
— • гидроокись 19
— двуокись 20
— закись 19
— закись-окись 19
— карбиды 15
— • карбонат 17
— • карбонил 17
— • карбонилгалогениды 17
— метилциклопентадиенилтрикарб онил
18
— • нитрат 19
— • нитриды 15
— окислы 19
— окись 19
— • получение 16
— применение 17
— • силициды 15
— сульфаты 20
— сульфиды 21, 15
— • техника безопасности 17
— физич. и химич. свойства 14
— фосфиды 15
— хлориды 21
— циклопентадиенилтрикарбонил 18
Марганец двусернистый — см. Марганец,
сульфиды
---сернистый — см. Марганец, суль-
фиды
Марганцовая голубая 1022
Марганцовая зеленая 1013, 1022
Марганцовая кислота 22, 16
Марганцовая фиолетовая 1022
Марганцовистая кислота и манганаты
22, 16
Марганцовокислые соли — см. Перман-
ганаты
Марганцовые удобрения 235
Маргарин 22
Маргариновая кислота 23
Маргуселин 23
1089—1090
Марковникова правило 24
Маскировка (в химич. анализе) 26
---- буферная 27
Масла
— высыхание 2 7, 28
— дегидратация 43
— • изомеризация 43
— • моющие свойства 436
— переэтерификация 43
— рафинация 43
— регенерация 41
— старение 41
Масла авиационные 33
------ автотракторные 34
---- бакинские 32, 33
---- белые 40
----восточные (из восточных нефтей)
32, 33
------ высыхающие 27
------ гидравлические 39
----дизельные 30, 34
----для паровых машин 36, 37
----для производства сажи 30
-------- для технологических целей 40
------для фармакопеи и парфюмерии —
см. Масла белые
—•—• для флотореагентов 30
—•— для ядохимикатов 30
----древесно-смольные флотационные—
см. Смола древесная
----индустриальные 35
----кабельные 39
------ каменноугольные технические 30
---- катализированные 44
----компаундированные — см. Масла
смешанные
—•— компрессорные 38
----малеинизированные 43
----минеральные 30, 432
-----—. коррозионные свойства 436
-------------------— моющие свойства 436
----—•— — стабильность против оки-
сления 436
--------дистиллятные 31
--------остаточные 31
----моторные 32
----несмазочные 38
----нефтяные — см. Масла минераль-
ные
------ осевые 35
----поглотительные 30
------ препарированные 42
---- приборные 35
----синтетические 44, 45, 47
--------полиалкиленгликолевые 46
------ —•—полисилоксановые 47
----—.—. углеводородные 44
— •— --- фторуглеводородные 47
-------- ——• хлорфторуглеводородные 48
------------- эфирные 45
смазочные 33
-------- смешанные 31
— сополимеризованные 44
—— трансмиссионные 35
—•—• трансформаторные 39
---- турбинные 38
----цилиндровые —см. Масла для па-
ровых машин
----шпалопропиточные — см. Масла ка-
менноугольные технические
----электроизоляционные 38
Масло антраценовое— см. Каменноуголь-
ная смола (т. 2)
---- вазелиновое 36
—•— веретенное 36
----- газгольдерное 30
—•—• для подсвечивания пламени 30
------ древесно-смоляное флотационное 49
—•— касторовое, высыхание 28
----- конденсаторное 39
---- коровье 48
------ льняное, высыхание 28
----• маковое, высыхание 28
------ машинное 36
—•— медицинское вазелиновое 41
----• нафталиновое поглотительное 30
---- отопительное 30
— •—• парфюмерное 41
— — сепараторное 36
-------- сосновое флотационное 49
-------- —•— сухоперегонное 49
-------- -- экстракционное 49
-------- телеграфное 36
—•—• тунговое, высыхание 28
---- часовое 36
----швейное 36
Масляная кислота 50
1091—1092
Масляные фракции, очистка 834
Масляный альдегид 51
Масса, единицы измерения 84, 91
----ядра атома, прецизионные опреде-
ления 60
Массовое число 52
Массовый расход, единицы измерения 91
Массообмен 52
Массообменный аппарат 53
Масс-спектральные приборы 56
Масс-спектрография — см. Масс-спектро-
метрия
Масс-спектрографы 58
Масс-спектрометрия 60
Масс-спектрометры 58
Масс-спектроскопия —см. Масс-спектро-
метрия
Матромицин — см. Олеандомицин
Мевалоновая кислота 66
Мега (приставка) 89
Мединал 75
Медно-аммиачное волокно 75
Медноникелевые сплавы 73
Медные руды 77
Медные удобрения 235
Медный блеск — см. Халькозин
Медный колчедан — см. Халькопирит
Медный купорос — см. Медь, сульфат
Медь 76, 135
— аналитич. определение 79
— бромид 66, 67
— галогениды 66
— гидроокись 68
— закись 68
— ИОдИд 66
— карбиды 78
— карбонаты 67
— комплексные соединения 78
— молибдат 299
— нейтронное облучение 165
— нитрат 67
— нитрид 491
— окислы 68, 78
— окись 68
— перекись 68
— получение 79
— применение 81
— сплавы 69
— сульфат 74
— сульфиды 74
— техника безопасности 81
— упругость 20 5
— Феррит 68
— физич. и химич. свойства 77
— фторид 66, 67
— хлорид 66, 67
Медь азотнокислая —• см. Медь, нитрат
----полусернистая — см. Медь, суль-
фиды
----сернистая — см. Медь, сульфиды
----сернокислая — см. Медь, сульфат
Медьорганические соединения 81
Медянка 1022
Меервейна реакция 82
Меервейна—Понндорфа — Верлея реак-
ция 83
Международная система единиц 84
Межмолекулярное взаимодействие 96
Межмолекулярные силы — см. Межмоле-
кулярное взаимодействие
Межъядерные расстояния 101
Мезаконовая кислота — см. Цитрако-
новая кислота
Мезатон 103
Мезидин 103
Мезил 104
Мезилаты 104
Мезитил, окись 104
Мезитилен 104
Мезитиловая кислота 105
Мезо (приставка) 105
Мезовинная кислота — см. Винные кис-
лоты (т. 1)
Мезо-п,п'-бис-диметиламино-3,4-дифенил-
гексан, дииодметилат — см. Парамион
Мезоксалевая кислота 105
Мезомерия 105
Мезоны 107
Мезосидериты 174, 177
Мезоэритрит 254
Мейера реакция — см. Майера реакция
(т. 2)
Мейстера — Мишера реакция — см. Бар-
бье — Виланда расщепление (т. 1)
Меконин 364
Меконовая кислота 108
Мелаконит 68
Меламин 108
Меланиины 109
Меланотропин— см. Меланотропный гор-
мон
Меланотропный гормон НО
Меланофорный гормон — см. Мелано-
тропный гормон
Меланоцитостимулирующий гормон —
см. Меланотропный гормон
Мелатонин 111
Мелибиоэа 111
Мелинит — см. Тринитрофенол
Мелиссиловый спирт — см. Мирициловый
спирт
Меллитовая кислота — см. Бензолполи-
карбоновые кислоты (т. 1)
Мельхиоры 71, 73
Мембранное равновесие 112
Менделевий — см. Актиниды (т. 1)
Ментан 114
п-Ментанол-3 —см. Ментол
Ментены 114
Ментол 115
Ментон 116
Мепавлон — см. Мепротан
Мепазин 117
Мепанит 117
Мепробамат —• см. Мепротан
Мепробан — см. Мепротан
Мепросан — см. Мепротан
Мепротан 118
Меридил 118
Мерихиноидные соли 118
Мерказол — см. Мерказолил
Мерказолил 119
Меркамин 120
Меркаптали 120
Меркаптамин — см. Меркамин
Меркаптаны 121
Меркаптиды — см. Меркаптаны
Меркаптометан —• см. Метилмеркаптан
6-Меркаптопурин 123
Меркаптофенилтиотиодиазолон 123
Меркаптофос — см. Фосфорорганические
инсектициды
p-Меркаптоэтиламин, хлоргидрат —
см. Меркамин
Меркузал 123
Меркузаловая кислота 123
Меркуриметрия 123
Меркурометрия 124
Меромиозин 24 5
Меромиозины 245, 246
Мерпанит — см. Мепанит
Мёссбауэра эффект 124
Месторождения рудных минералов 132
Мета (приставка) 126—см. также Орто-,
Мета-, Пара-
Метаболизм 620
Метазид 126
Метакриловая кислота 127
— метиловый эфир — см. Метилмета-
крилат
Металепсия 128
Металлидные растворы 141
Металлирование 128
Металлическая связь—см. Металлы
Металлические руды 131
Металлические соединения 139
— классификация 142
— общие особенности 140
— • практич. значение 146
— • характеристика важнейших типов
142
Металлические соединения гетеродесми-
ческие 142
--------гомодесмические 14 2
Металлические фазы сложного состава
166
Металлкетилы 147
Металловедение 157
Металлоорганические соединения 148
--------непереходных металлов 148
------------------------- переходных металлов 151
Металлопротеиды 152
Металлотермия 153
-------------------- вакуумная 156
---------- внепечная 154
----------- печная 154
Металлоэнзимы 902
Металлургия 157
Металлы 157
— действие излучения 164 ;
— кристаллич. структура 158
— • магнитные свойства 162
— механич. свойства 163
— окисление 165
— пластичность 163
— • ползучесть 164
— полиморфизм 160 1
— прочность 164
— сверхпроводимость 162
— . тепловые свойства 161
— теплопроводность 161
— теплота сублимации 158
— термоэлектронная эмиссия 162
—• упругость 163
— • электрич. свойства 161
— электросопротивление 162
Метальдегид — см. Ацетальдегид (т. 1)
Метамарганцоватистая кислота 15
Метамерия 169
Метамизил 169
Метамин — см. Нитранол
Метамолибдаты 299
Метамышьяковая кислота 352
Метамышьяковистая кислота 352
Метан 170, 439
— энергия С—Н-связи 669
Метаниловая кислота 170
Метаниобаты 474
Метановое брожение — см. Брожение
(т. 1)
Метанол —• см. Метиловый спирт
Метантиол — см. Метилмеркаптан
Метаоловянная кислота 738
Метастабильное состояние 171
Метаторбернит 137
0-Метахромазия 173
у-Метахромазия 173
Метахроматическое окрашивание 173
Метацин 174
Метациннабарит 135
Метгемоглобин — см. Гемоглобины (т. 1)
Метеориты 174
------ железные 174
—•— железокаменные 174
—•— каменные 174
Метилакрилат 179
а-Метилакриловая кислота — см. Ме-
такриловая кислота
—- метиловый эфир — см. Метилметак-
рилат
Метилаллен 179
Метиламин 180
n-Метиламинофенол, сернокислая соль —
см. Метол
17 а-Метил-Д4-андростенол-170-он-3 —
см. Метилтестостерон
N-Метиланилин 180
2-Метилантрахинон 181
Метилацетат 182
Метилацетон — см. Метилэтилкетон
п-Метилацетофенон 182
М-(4-Метилбензолсульфонил)-М'-Н-бу-
тилкарбамид — см. Бутамид
З-Метил-1,5-6ис-(N.N-диметил -N-этилам-
моний)-3-азапентан-дибромид—см.Пен-
тамин
Метилбицикло-(0,1,4)-гептан 581
Метил-пгрет-бутилкетон — см. Пинако-
лин
2-Метил-5-винилпиридин 182
Метилвиолет — см. Метиловый фиолето-
вый
Метилглиоксаль 183
3-Метил-3,5-диокси - валериановая кисло-
та — см. Мевалоновая кислота
Метилдихлорарсин 183
Метилдихлорфосфин — см. Хлоралкил-
фосфиты
1, Г-Метилен-б ис-изоникотиноилгидра-
зон — см. Метазид
4,4'-Метилен-бис-3-окси-2-нафтойная кис-
лота 703
Метиленовая синь — см. Метиленовый
синий
Метиленовые основания 197
Метиленовый голубой — см. Метилено-
вый синий
Метиленовый синий 184
п-Метилизопропилбензол 184
1-Метил-4-изопропилциклогексанон-3 —
см. Ментон
З-Метил-6-изопропилциклогексанол —см.
Ментол
Метилиндолы 185, 525
Метилирование 186
Метилкаучук 187
Метилмеркаптан 187
1-Метил-2-меркаптоимидазол — см. Мер-
казолил
Метилмеркаптофос — см. Фосфороргани-
ческие инс ектицид ы
Метилметакрилат 188
Метилморфиметин 325
N-Метилморфинан 325
Метилнафталины 189
0-Метилнафтохинон — см. Витамины К
(т. 1)
Метилнитрамин 499
Метилнитрат 189, 556
Метиловый спирт 190
— получение 192
— применение 193
— • физич. свойства 190
— химич. свойства 191
Метиловый фиолетовый 194
2-Метил-3-окси-4,5-диоксиметилпиридин
— см. Пиридоксин
10-Метил-1-оксифеназин — см. Пиоциа-
нин
2-Метил-3-окси-4-формил-5-пиридилме-
тилфосфат — см. Пиридоксаль-5-фос-
фат
2-Метил-8-оксихинолин — см. 8-Оксихи-
нальдин
2-Метилпентен-2-он-4 — см. Окись ме-
зитила
Метилпентиты 254
Метилпиперидины 194
Метилпиридины 195
N-Метилпиррол 525
3"(К-Метил-а-пирролидило)-пиридин —•
см. Никотин
2-Метил-2-н-пропил-1,3-пропандиолди-
карбамат — см. Мепротан
5-Метилрезорцин — см. Орсин
Метилсерная кислота 186 — см. также
Алкилсульфаты (т. 1)
Метилсульфат — см. Алкилсульфаты (т.1)
Метилтестостерон 197
17а-Метилтестостерон — см. Метилтесто-
стерон
Метилтиоурацил 198
6-Метил-2-тиоурацил —см. Метилтиоура-
цил
Метил-тг-толилкетон — см. п-Метилацето-
фенон
Метилтрансферазы 198
Метилфенидат, хлоргидрат — см. Ме-
ридил
Метилфениламин — см. N-Метиланилин
5-Метил-3-фенилизоксазолил-4-пеницил-
лин — см. Оксациллин
N-Метилфенилнитрамин 499
О-Метилфенилнитрамин 499
Метилфосфин — см. Фосфины
Метилфоофпновая кислота — см. Хлор-
алкилфосфины
Метилфосфоновая кислота—см. Хлор-
ал килфосфины
Метилхолантрен 199
Метилцеллюлоза 199
Метилциклогексан 200
— октановое число 719
3-Метилциклопентадеканон —см. Мус-
кон
Метилциклопентадиенилтрикарбонил-
марганец — см. Марганец, метилцикло-
пентадиенилтрикарбонил
Метилциклопентан 201
— октановое число 719
5-Метилцитозин-дезоксирибозид 603
О-Метилэтилизонитрамин 499
Метилэтилкетон 202
2-Метил-5-зтилпиридин 197
Метионин 202
--- активный 198
Метициллин 879, 885
п-Метоксибензальдегид — см. Обепин
8-Метоксигидрастин — см. Наркотин
^-(5-Метокси-З-индолил) -N-ацетилэтил-
амин — см. Мелатонин
Метол 203
Метотирин — см. Мерказолил
Метрика химической диаграммы 203
Механические единицы 85
Механические свойства материалов 204
I 63
---------металлов 163
---------полимеров 214
Механохимия полимеров 225
Меченые атомы —• см. Изотопные инди-
каторы (т. 2)
Меченых соединений синтез 229
Мешалки — см. Перемешивание
Миазин — см. Пиримидин
Миарсенол — см. Сальварсана препара-
ты
Миграция химических элементов (гео-
химия.j 230
Миграция энергии — см. Перенос энер-
гии
Миелопероксидаза 982
Миелосан 231
Микароза 324
Микозамин 487
Микостатин — см. Нистатин
Микро (приставка) 89
Микроанализ — см. Микрохимический
анализ
Микробиологические методы химическо-
го анализа 232
Микрокристаллоскопия 233
Микропенин — см. Оксациллин
Микроудобрения 234
Микрохимический анализ 235
Миллерит 133, 242, 456
Милли (приставка) 89
Миллона реакция 237
Мильеран —. см. Миелосан
Мильтаун — см. Мепротан
Миндальная кислота 237
Миндальное масло — см. Жиры расти-
тельные (т. 2)
Минералы 238
— блеск 241
— габитус 239
— • генезис 243
— двойники 240
— классификация 238
— кристаллич. структура 242
— механич. свойства 241
— морфология 239
— окраска аллохроматич. 241
— идиохроматич. 240
— —— псевдохроматич. 241
— опробование 755
— оптические свойства 240
— парагенезис 243
— состав 242
— спайность 241
— • твердость 241
— удельные веса (плотности) 241
— физико-химич. природа 242
— физич. свойства 239
— цвет 240
Минеральные агрегаты 24о
---—-— зернистые 240
---—•— натечные 240
Минеральные удобрения 244
Минеральный обмен — см. Обмен веществ
Миоглобин 1025
Миозин 245
Мипора — см. Поропласты
Мирабилит — см. Глауберова соль
Мирбановое масло — см. Нитробензол
Миристиновая кислота 246
Мирициловый спирт 246
Мирцен 246
Митохондрии 659
Михаэлиса константа 247
Михаэлиса — Беккера реакция 249
Михаэля реакция 250
Михлера кетон 251
Мицеллообразование 333
— критич. концентрация 346
Мицеллы 252
Мицерин 418
Многоатомные спирты 253
Многокомпонентные системы 255
Многоядерные комплексные соединения
256
Модели атомные — см. Модели молеку-
лярные
Модели молекулярные 259
--------Дрейдинга 259
---—— Кекуле — Вант-Гоффа 259
--------Стюарта — Бриглеба 260
--------Хартли — Робинсона 260
Модифицирование высокомолекулярных
соединений 260
-------------методом макромолеку-
лярных реакций 261
-------------методом полимеранало-
гичных превращений 260
Модифицирование поверхности химиче-
ское 262
Мозли закон 264
Молекула 265
— внутренние движения в М. 271
— движения в М., виды 282
— дипольный момент 273
— история изучения 266
— магнитные свойства 273
— нормальные колебания 272, 285
— оптич. свойства 272
— парамагнетизм 274
— размеры и форма 269
— свойства 272
— спектр испускания 281
— комбинационного рассеяния 281
— —поглощения 281
— строение 267
— структура и энергетика, масс-спект-
ральное исследование 62
— уровни энергии 281
— химич. свойства 274
— электрич. свойства 273
1093 —1094
— энергетика 271
— энергия разрыва связей 272
Молекула ионная 269
—•— полярная 269
---гомеополярная 268
Молекулы двухатомные, межъядерные
расстояния 102
Молекулярная биология 275
Молекулярная дистилляция —• см. Дис-
тилляция молекулярная (т. 1)
Молекулярные силы — см. Межмолеку-
лярное взаимодействие
Молекулярные сита 280
Молекулярные спектры 281
—•— —•— колебательные 285, 282
-------полосатые 281
——-----электронные 288, 273, 282, 283
Молекулярный баланс моющих ве-
ществ 33 3
Молекулярный вес 290
Молекулярный вес высокомолекулярных
соединений 292
— измерения метод вискозиметрия. 297
— —•—------ осмотический 293
— --------- основанный на рассеянии
света 294
— ----- _— седиментационного равно-
весия 295
— -------скорости седиментации 296
— химический 294
Молекулярный масс-спектр 62
Молекулярный масс-спектральный ана-
лиз 63
Молекулярных орбит метод — см. Кван-
товая химия (т. 2)
Молибдаты 298
Молибден 300, 135
— аналитич. определение 302
— бромиды 304
— галогениды 303
— гексафторид 303
— гидроокиси 306
— двуокись 305
— дибромид 304
— дигидрат 306
— дииодид 304
— дисилицид 307
— иодиды 304
— карбонил 301
— кислоты 305
— моногидрат 306
— нейтронное облучение 165
— • нитрид 492
— окисел промежуточный 305
— окислы 305
— пентасульфид 309
— пентахлорид 3 04
— получение 302
— применение 303
— силицид 301
— сплавы 307
— • сульфиды 308
— тетрабромид 304
— тетраиодид 304
— • тетрахлорид 304
— трибромид 304
— трииодид 304
— трисульфид 309
— трифторид 303
— трихлорид 304
— трехокись 305
— физич. и химич. свойства 300
— фториды 303
— • хлориды 304
Молибденит 300, 135, 241
Молибденовая кислота — см. Молибден,
кислоты
Молибденовая синь 306
Молибденовые удобрения 235
Молочная кислота 309
Моль — см. Грамм-молекула (т. 1)
Молюскоциды — см. Лимациды (т. 2)
Моляльность раствора 310 — см. также
Концентрация (т. 2)
Молярность раствора 310 — см. также
Концентрация (т. 2)
Молярный коэффициент поглощения —•
см. Поглощение света
Монацит 137
Моноазокрасители 310
Моноалкилбензолсульфонаты 335
Моноалкилнитрамины 500
Монозы — см. Моносахариды
Монокристаллы 311
Мономеры 314
—•— образующие полимеры поликонден-
сацией 315
1095 — 1096
--- участвующие в цепной полимери-
зации 314
Монометилпиперидины — см. Пипеколи-
ны
Монометилпиридины — см. Пиколины
Мономинеральные агрегаты 240
Мономицин 316
Мономолекулярные реакции 316
Мономолекулярный слой 318
Мономолибдаты 298
Мононатрийарсенат 374
Мононитроанилин 500
Мононитробензойная кислота 502
Мононитробензол —• см. Нитробензол
Мононитропарафины 535
Мононитроспирты 545
Мононитротолуолы 547
Мононитрохлорбензолы 549
Мононуклеотиды — см. Нуклеотиды
Моносахариды 320
Монослой —см. Мономолекулярный слой
Монотектика — см. Двойные системы
(т. 1)
Монтанвоск — см. Воски (т. 1)
Монтанный воск — см. Воски (т. 1)
Монурон — см. М-4-Хлорфенил-М',М'-ди-
метилмочевина
Морин 324
Моркит 813
Морфин 325
Морфолин 683
Морфольный распад 325
Мостиковые группы (мостики) 256
Моторное топливо 326, детонационная
стойкость 18
Мочевая кислота 327
Мочевина 328, 245
— производные 331
Мощность, единицы измерения 93
— — дозы (в радиац. химии) 331
Моющее действие 332
Моющие вещества 332
Моющие свойства масел 436
Моющие средства 334
МСГ — см. Меланотропный гормон
Муковещества 338
Мукоидные вещества — см. Муковеще-
ства
Мукокомплексы —• см. Муковещества
Муколипоид 338
Муконовая кислота 339
Мукополисахариды 339, 338
Мукопротеиды 340
Мукопротеины — см. Мукопротеиды
Мукосоединенин — см. Муковещества
Мультиплетная теория катализа — см.
Катализ гетерогенный (т. 2)
Мультиротация — см. Мутаротация
Мумия (пигмент) 1013, 1019
Муравьиная кислота 340
Мурексид 342. 327
Мурексидная реакция 342, 327
Мускарин 342
Мусковит 241
Мускон 343
Мускус 343
— — амбровый 344
Мускус-амбретт — см. Мускус амбровый
Мускус-кетон 344
Мускус-ксилол 344
Мускусные препараты 343
Мустерон 344
Мутазы — см. Фосфомутазы
Мутаротация 344
Мутации 278
Мыла 345
— жидкие 347
---клеевые 346
— — порошкообразные 347
— — твердые 347
--- ндровые 346
Мыло сульфатное 347
Мылонафт 347
Мышьяк 348
— аналитич. определение 350
— бромид 351
— • галогениды 351
— иодиды 351
— кислоты 351
— окислы 351
— отравление М. 351
— получение 350
— применение 351
— сульфиды 352
— техника безопасности 350
— • физич. и химич. свойства 349
— фториды 351
— хлориды 351
Мышьяк желтый 349
---металлический 349
----- односернистый 352
—— пятисернистый 352
---серый — см. Мышьяк металличе-
ский
—— трехсернистый 352
Мышьяковая кислота 352
Мышьяковистый ангидрид 351
Мышьяковистый водород 353, 817
Мышьяковистый колчедан 348
«Мышьяковое стекло» 352
Мышьяковый ангидрид 352
Мышьяковый колчедан 348
Мышьякоорганические соединения 353
Мягчители 355
Мятное масло перечное — см. Эфирные
масла
н
Набухание полимеров 357
Надизан 358
Надкислоты — см. Перекиси и гидропе-
рекиси органические
Надперекиси 679, 934
Надсмольная вода 358
Найлон — см. Полиамидные волокна
Наллин — см. Налорфин
Налорфин 359
Намагниченность — см. Магнитике свой-
ства (т. 2)
Наметкина перегруппировка — см. Кам-
феновые перегруппировки (т. 2)
Напалм 360
Наполнители (для резин) 361
Напряжение разложения 361
Напряжений ряд — см. Ряд напряжений
Напряжения теория 362
Нарколан 363
Наркотин 363
Насадки 364
Наследственная информация 278
Насосы (для перекачивания жидкостей)
920
Насыщенный раствор — см. Раствори-
мость
Натр едкий — см. Натрий, гидроокись
Натриевая селитра — см. Натрий, нитрат
Натрий 367
— азид 368
— амальгамы 369
— амид — см. Натрийамин
— аналитич. определение 369
— арсенаты 374
— арсенит 374
— бикарбонат 378
— бисиликат 381
— бисульфат 383
— бисульфит 384
— бифторид 386
— • боргидрид 375
— бромид 375
— гидрид 375
— гидроокись 376, 756
— гидросульфид 383
— гидросульфит 377
— гипофосфит 377
— иодид 378
— карбиды 369
— карбонат нормальный 378
— карбонаты 378
—• кобальтинитрит 379
— метабисульфит — см. Натрий, пиро-
сульфит
— метаборат 982
— • метаниобат 474
— метасиликат 381
—• молекула, межъядерное расстояние
102
— мономолибдат 299
— надперекиси 934
— нитрат 379, 245
— нитрид 368, 491
— нитрит 379
—• нитрозопентациноферриат — см. Нат-
рий, нитропрусид
— нитропрусид 380
— окись 368
— • ортосиликат 381
— • парамолибдат 299
— пентобарбитал —• см. Нембутал
— • перекись 380
— • пиросиликат 3 81
— пиросульфат — см. Натрий, сульфаты
— пиросульфит 384
— полисульфиды 384
— получение 369
— применение 370
— родизонат 381
— сесквикарбонат 379
— силикаты 381
— сульфаты 382
— сульфид 383
— сульфит 384
— техника безопасности 370
— тиосульфат 385
— трисиликат 382
— физич. и химич. свойства 368
— фосфаты 385
— фосфовольфрамит 386
— фторид 386
— хлорид 386
— 5-этил-5-(1-метилбутил) -барбитурат-
ом. Нембутал
Натрий азотнокислый — см. Натрий,
нитрат
---бромистый — см. Натрий, бромид
— гидросернистокислый —см. Натрий,
гидросульфит
--- двууглекислый — см. Натрий, би-
карбонат
---иодистый — см. Натрий, иодид
---родизоновокислый — см. Натрий,
родизонат
---сернистокислый —см. Натрий, суль-
фит
---сернистый —• см. Натрий, сульфид
—•— серноватистокислый —• см. Натрий,
тиосульфат
---углекислый — см. Натрий, карбо-
нат нормальный
---фосфорноватистокислый — см. Нат-
рий, гипофосфит
---фосфорновольфрамовокислый — см.
Натрий, фосфовольфрамит
----- фтористый — см. Натрий, фторид
----- хлористый — см. Натрий, хлорид
Натрий — бутадиеновый каучук (СКБ>
371
Натрийамин 371, 368
Натрийорганические соединения 373
Натровая известь 388
Натронная известь — см. Натровая из-
весть
Науманнит 239
Нафтазарин 388
Нафталевая кислота 389
Нафталин 389
— сульфокислоты 391
Нафталин-1,8-дикарбоновая кислота —
см. Нафталевая кислота
а- и 0-Нафталинкарбоновые кислоты —
см. Нафтойные кислоты
Нафтацен 393
Нафтеновые кислоты 393
Нафтеноль 731
Нафтены 395
— октановое число 719
Нафтиламин, сульфокислоты 396
1-Нафтиламин-4-сульфокислота — см.
Нафтионовая кислота
Нафтиламины 397
Нафтилендиамины 397
Нафтионовая кислота 398
Нафтойные кислоты 398
Нафтол, сульфокислоты 399
Нафтол АС — см. Азотолы (т. 1)
0-Нафтол, метиловый эфир—см. Не-
ролин
Нафтолкарбоновые кислоты — см. Ок-
синафтойные кислоты
Нафтоловый желтый S 526
Нафтолы 400
Нафтостирил 402
0-Нафтохинолин 402
Нафтохиноны 402
Неадиабатические реакции 953
Неамин 417
Небензольные ароматические системы —
см. Ароматические системы (т. 1)
Небесно-голубой — см. Церулеум
Небурон — см. М-3,4-Дихлорфенил-К'-
метил-М'-бутилм оч евина
Негатив 404
Негативный процесс — см. Проявле-
ние фотографическое
Недоокись углерода 404
Незаменимые аминокислоты 405
Незаменимые жирные кислоты 406
Неионогенные вещества 336
Нейзильберы 71, 73
Нейраминидазы 406
Нейраминовые кислоты 407
Нейрин 408
Нейтрино 408
Нейтрон 409
— дифракция 409
— облучение металлов 165
Нейтронография 409
--- магнитная 413
Некаль ВХ 415
Немагон 415
Нематоциды 415
Нембутал 415
Ненасыщенные соединения — см. Не-
предельные соединения
Ненасыщенный раствор — см. Раство-
римость
Нсоабиетиновая кислота 416
Необиозамины 417
Необратимые реакции — см. Обратимые
реакции
Неогексан 416
Неодикумарин 417
Неодим — см. Лантаниды (т. 2)
Неоизоментол 115
Неозоны 417
Неокупферрон 767
Неоментол 115
Неомицин 417
Неон 418
Неопентан 418
Неопрен — см. Каучук хлоропреновый
(т. 2)
Неорганическая химия 419
Неорганические соединения, номенкла-
тура 564
Нео-синефрин — см. Мезатон
Неподеленная пара электронов 101
Непредельные соединения 425
— • нитрование 507
Непредельные углеводороды 425
— химич. переработка 440
Непрерывности принцип—см. Физико-
химический анализ
Нептуний — см. Актиниды (т. 1)
Нервон 426
Нержавеющая сталь — см. Железо,
сплавы (т. 2)
Нернста принцип— см. Третий закон
термодинамики
--- Уравнение 426
Нерол 427
Неролин 427
Несмеянова реакция 428
Несслера реактив 429
Нефа реакция 429
Нефелометрия и турбидиметрия 430
Нефтепродукты 432
— автогидроочистка 833
— высота некоптящего пламени 437
— вязкость 432
— гидроочистка 833
— давление паров Н. 433
— депарафинизация 832
— дуктильность (растяжимость) 434
— коксуемость 437
— коррозионные свойства 436
— окисление 664
— органич. кислоты, определение со-
держания в Н. 43 5
— очистка 832
— парафин твердый, определение со-
держания в Н. 435
— плотность 432
— сера, определение содержания в Н.
434
— сероводород, определение содержа-
ния в Н. 435
— смолы, определение содержания в
Н. 435
— стабильность против окисления 436
— температура воспламенения 434
— вспышки 434
— застывания 433
— плавления 434
— помутнения 433
— углеводородный состав, влияние на
свойства Н. 454
— физич. свойства, оценка 432
— фракционный состав 433
— химич. свойства, оценка 434
— цвет 434
Нефтепродукты порошкообразные, оп-
робование 755
Нефтехимический синтез 437
— основные продукты Н. с. 444
Нефть 446
— классификация 450
— обезвоживание 453
— обессоливание 453
— перегонка 912
— переработка 454, 453
— происхождение Н. 446
— стабилизация 454
— физич. свойства 447
— химич. состав 449
Нефть как источник химич. сырья 455
Ниацин — см. Никотиновая кислота
Нигрозины 455
Никелевые руды окисленные 456
Никелин 133, 239
Никель 455, 133
— аналитич. определение 459
— бромид 462
— галогениды 461
— гидрид 457
— гидроокиси 466
— диэтилдитиофосфат 462
— закись 466
— . иодид 462
— карбид 458
— • карбонат 463
— • карбонил 463
— комплексные соединения 464
— моносульфид 470
— нейтронное облучение 165
— нитрат 466
— нитрид 458
— окислы 466
—• пассивационная характеристика 869
— получение 459
— применение 460
— селениды 458
— сплавы 467
— ---жаропрочные 468
— магнитные 468
— —•— с особыми свойствами 469
— сульфат 469
— • сульфиды 470
— • теллуриды 458
— физич. и химич. свойства 456
— • фосфид 458
— • фторид 461
— хлорид 462
Никель как катализатор 460
---Ренея 461
Никель-арсенидные фазы 145
Никодин 471
Никотин 471
Никотинамид-метилтрансфераза 198
Никотинамиднуклеотидные коферменты
472
Никотиновая кислота 472
— амид 473
— N-оксиметиламид —. см. Никодин
— чувствительность микробиология, ме-
тода химич.анализа 233
Нимоник 468
Нингидрин 473
Нингидринная реакция —• см. Нингид-
риновая реакция
Нингидриновая реакция 474
Ниобаты 474
Ниобиевая кислота —• см. Ниобий, гид-
роокись
Ниобий 475, 137
— аналитич. определение 480
— взаимодействие с водородом 478
— • галогениды 482
— • гидрид 479
— • гидроокись 477
— • двуокись 478
— дииодид 484
— дихлорид 484
— нитриды 492
— • окись 477
— оксидииодид 485
— окситрибромид 484
— окситрииодид 485
— окситрифторид 483
— окситрихлорид 484
— оксихлорид 484
— пентабромид 484
— • пентаиодид 484
— пентафторид 482
— • пентахлорид 483
— получение 480
— применение 482
— пятиокись 478
— • сплавы 485
— тетраиодид 484
— • тетрахлорид 484
— • трибромид 484
— трииодид 484
— трихлорид 484
— физич. и химич. свойства 476
Ниобий — азот система 479
Ниобий — кислород система 477
Ниобий — углерод система 479
Ниренштейна реакция 486
Нистатин 487
Нитралетте — см. Нитранол
2-Нитраминопиридин 499
Нитрамины —• см. N-Нитроаминосоеди-
неиия
Нитранол 488
1097 — 1098
Нитраты металлов 488
----органические 489
Нитридные покрытия 493
Нитриды 490
— огнеупоры 648
— применение 493
Нитриды ионные 490
----ковалентные 491
----металлоподобные 492
Нитриллиевые соли 494
Нитрилы карбоновых кислот 494
Нитрильный каучук — см. Бутадиен-
нитрильный каучук (т. 1)
Нитриты металлов 497
------ органические 498
N-Нитроаминосоединения 499
Нитроанилины 500
Нитроантрахиноны 501
Нитробензойные кислоты 502
Нитробензол 503
1-Нитробутан 535
2-Нитробутан 535
1-Нитробутанол-2 545
2-Н итр об утанол-1 545
Нитрование, механизм 513
—— ароматических соединений 508
—— гетероциклических соединений 513
~ ненасыщенных соединений 507
------О и N- 513
—— окислительное 510
—•— органических соединений 505
------- парафиновых соединений 505
1-Нитрогексан 535
Нитрогликоль 556
Нитроглицерин 514, 556
Нитроцихлорбензолы 549
Нитрозирование 516
N-Нитрозоамины 516
Нитрозобензол 517
п-Нитрозодиметиланилин 518
N-Нитрозодифениламин 518
Нитрозокрасители 519
N-Нитрозометилкарбаминовая кислота,
этиловый эфир — см. Нитрозометил-
уретан
Нитрозометилмочевина 519
Нитрозометилуретан 520
а-Нитрозо-0-нафтол 520
р-Нитрозо-а-нафтол 521
1-Нитрозо-2-нафтол — см. а-Нитрозо-
6-нафтол
1-питрозо-2-нафто л-3,6-дисульфокислота
динатриевая соль — см. Нитрозо-В-
соль
2-Нитрозо-1-нафтол — см. 0-Нитрозо-
а-нафт о л
Нитрозосоединения 521
Нитрозо-Й-соль 521
п-Нитрозофенол 524
1-Нитроизобутан 535
2-Нитроизобутан 535
2-Нитроиндандион 524
Нитрокрасители 525
Нитроманнит 556
Нитрометан 526, 535
2-Нитро-2-метил-1,3-пропандиол 546
2-Нитро-2-метилпропанол-1 54 5
«Нитромускусы» 343
Нитрон 528
Нитрон (волокно) — см. Полиакриловые
волокна
Нитронафталины 528
Нитроний-ион 529
Нитроновые кислоты 530
Нитроны 532
2-Нитро-2-оксиметил-1,3-пропандиол 546
Нитроолефины 540, 541
Нитропарафины — см. Нитросоединения
алифатические
1-Нитропентан 535
Нитропропан 534
1-Нитропропан 535
2-Нитропропан 53 5
2-Нитро-1,3-пропандиол 546
1-Нитропропанол-2 54 5
2-Нитропропанол-1 545
1-Нитропропен 541
2-Нитропропен 541
3-Нитропропен 541
Нитросоединения алифатические 535
---- ароматические 541
Нитросорбид 545
Нитроспирты алифатические 545
(о-Нитростирол 541
Нитротолуолы 546
1-п-Нитрофенил - З-метил-4-нитропиразо-
лон-5 — см. Пикролоновая кислота
1099 — 1100
Нитрофенолы 548
Нитроформ 549, 535
Нитрофос 245
Нитрофоска 245
Нитрохлорбензолы 549
Нитроцеллюлоза 551
Нитроциклогексан 552
Нитроэтан 553, 535
2-Нитроэтанол 545
Нитроэтилен 554, 541
2-Нитро-2-этил-1,3-пропандиол 546
Нитроэфиры 555
Новарсенол—см. Сальварсана препараты
Новобиоцин 556
Новокаин 557
Новокаиномид 558
Новэмбихин 558
Номенклатура Женевская 571
----JUPAC-1957 574
—— комплексных соединений — см
Комплексные соединения (т. 2)
----Льежская 574
----неорганич. соединений 564
---- органич. соединений 569
---- рациональная 570
----ферментов 559
----химич. элементов 564
----• циклических соединений 575
Нона (приставка) 89
Нонан 578
Нонаналь — см. Нониловый альдегид
Нонановая кислота — см. Пеларгоновая
кислота
Нонанол-1 — см. Нониловый спирт
Нонанолид-4,1 — см. Нонлактон
Нониловый альдегид 578
Нониловый спирт 578
Нонлактон 579
Нонозы 579, 320
Ноноксы — см. Неозоны
Нонулозаминовые кислоты — см. Ней-
раминовые кислоты
Нор (приставка) 579
Норадреналин 580
Норвалин 580
19-Норгидрокортизон 587
Нордигидрогваяретовая кислота 581
Норкаран 581
19-Норкортизон 587
19-Норкортикостерон 587
Норлеворин — см. Норадреналин
Норлейцин 581
Нормальное давление 582
Нормальность раствора 582
Нормальные элементы 582
----—— насыщенного типа 582
--------ненасыщенного типа 583
Нормальный потенциал 583
19-Норпрогестерон 587
Норстероиды 586
Норсульфазол 588
19-Нортестостерон 587
— фенилпропионат 587
— циклопентилпропионат 587
19-Нор-4-хлортестостерон, ацетат 587
Носители в каталиэе 589
----в радиохимии 589
Нотатин 590
Нуклеазы 590
Нуклеиновые кислоты 591
----—— апиримидиновые 603
—•— —— апуриновые 612
—•— —•— синтетические 600
Нуклеогистоны 605
Нуклеозидазы 601
Нуклеозидмонофосфаты — см. Нуклео-
зидмонофосфорные кислоты
Нуклеозидмонофосфорные кислоты 610
Нуклеозидполифосфаты — см. Нуклео-
зидполифосфорные кислоты
Нуклеозидполифосфорные кислоты 613
Нуклеозидфосфорилазы 601
Нуклеозиды 602
Нуклеоиды 596
Нуклеоны — см. Нуклоны
Нуклеопротамины 605
Нуклеопротеиды 605
Нуклеотидазы 607
Нуклеотидилтрансферазы 607
Нуклеотиды 609
Нуклеофильные и электрофильные реа-
генты 615
Нуклеофильные реагенты 675
Нуклоны 616
Нулевая энергия 617
Нутч-фильтр — см. Фильтрация,
Ньютон (ед. измерения) 85
О
Обезвоживание руды 634
Обепин 619
Обесфеноливание сточных вод —. см. Во-
ды сточные (т. 1)
Обесфторенный фосфат — см. Фосфаты
кормовые, Фосфорные удобрения
Обмен азотистый 621
— — белкоп 621
— веществ 619
— —- —— — энергетика 620
— — > основной 620
— —- —- промежуточный 621
— — жиров (липидов) 625
• -- минеральный 626
— — углеводов 623
Обменное взаимодействие 627
Обменный интеграл 628
Обогащение полезных ископаемых 629
— подготовка сырья 634
— физико-химич. методы 633
— физич. методы 631
Обогащение полезных ископаемых гра-
витационное 631
— -—------магнитное 632
— —---—•— на липких (жировых) по-
верхностях 634
— — — ---— флотационное 633
— —---—— электрическое 632
Обработка результатов анализа (графи-
ческая) 635
Обратимые реакции 639
Объем, единицы измерения 91
Объемный анализ 640
Объемный расход, единицы измерения
91
Объемных отношений эакон 640
Овомукоид 640
Огнеопасные вещества 640
— —------ легковоспламеняющиеся 641
— -------- самовозгорающиеся 641
— трудновоспламеияющиеся 6'
Огнепроводный шнур 641
Огнеупорные материалы 642
— печи для обжига 649
Огнеупорные материалы алюмосиликат-
ные 643
— .—, —— высокоглиноземистые 644
— — —.—. графитовые 647
— — ——. динасовые 642
-----—.—. доломитовые 645
— — —— карбидные 647
----------- карборундовые 647
— — кварцевые 643
— — —.—. коксовые 647
— .—• —— корундовые 644
------ —.— кремнеземистые 642
— ---легковесные 648
— — —.— магнезиальные 645
— — —.— магнезитовые 645
— —. —— магнезито-хромитовые 647
— — —•—• нитридные 647
— —------ окисные 648
— — —•—• периклазовые —• см. Огне-
упорные материалы магнезитовые
— — —•—• периклазо-шпинелидные 647-
— — —— полукислые 643
— —------ углеродистые 647
— •—----- форстеритовые 646
——------хромитовые 6 46
— —------ хромо-магнезитовые 647 .
— .— —.— циркониевые 647
— ------- цирконовые 647
— ------- шамотные 643
— ,—--шпинельные 646
Однокомпонентные системы 652
Ожижение газов — см. Охлаждение
Озазоны 653
Озокерит 653
Озон 654
— - получение 656
— применение 657
— физич. и химич. свойства 654
Озон атмосферный 656
Озонатор 656
Озониды — см. Перекиси и перекисные
соединения
Ойазин — см. Пиридазин
Окисление, степень — см. Окисление-
восстановление
Окисление биологическое 657
— -— металлов 165
— —• нефтепродуктов 664
— •—► органич. соединений 667
— —- электрохимическое 673
Окисление-восстановление 673
—. методы в аналитич. химии 675
Окислительно-восстановительный по-
тенциал — см. Нормальный потенциал»
Электродный потенциал
Окислы 679
---амфотерные 679
---индиферентные 679
----- кислотные 679
---нормальные 679
---основные 679
Окись 679
—— мезитила 680
Окклюзия 681
Оксазил 681
Оксазиновые красители 681
Оксазины 682
Оксазолоны 683
Оксазолы 685
Оксалаты 686
N,N' -Оксалилмочевина — см. Парабано-
вая кислота
Оксалилхлорид 687
Оксамид 688
Оксантрол и оксантрон 688
Оксантрон — см. Оксантрол и оксан-
трон
Оксафенамид 689
Оксациллин 879, 885
Оксиальдегиды и оксикетоны 689
Оксиантрахиноновые красители 691
Оксиацетон 692
Оксиацетофеноны 693
тг-Оксибензилпенициллин 879
Оксигемоглобин — см. Гемоглобины
(т. 1)
Оксидазы 694
5-Окси-2,6-диаминогексановая кислота—
см. Оксилизин
Оксидоредуктазы 694, 559
Оксиды — см. Окислы
0-(5-Оксииндолил-3)-аланин — см. 5-Ок-
ситриптофан
Оксикетоны — см. Оксиальдегиды и ок-
сикетоны
Оксикислоты 697
Оксилизин 699
Оксиликвиты 700
Оксиметилфосфиновая кислота — см.
Фосфоновые кислоты
Оксиметилфосфоновая кислота — см.
Фосфоновые кислоты
Оксимирование — см. Оксимы
Оксимицин —• см. Паромомицин
Оксимы 700
ОКСИН —’СМ. 8-Оксихинолин
Оксинафталины — см. Нафтолы
Оксинафтойные кислоты 702
Оксиндол 703
1-Оксинонан —• см. Нониловый спирт
З-Оксиоксиндол — см. Диоксиндол
1-Оксиоктан — см. Октиловый спирт
Оксипиразол — см. Пиразолон
Оксипиридины 705
0-Окси-у-пирон 108
4-Оксипирролидин-2-карбоновая кисло-
та —• см. Оксипролин
Оксипролин 707
а-Оксипропионовая кислота —см. Молоч-
ная кислота
Окситетрациклин 708
З-Океитионафтен 708
Окситоцин 709
5-Окситриптамин — см. Серотонин
5-Окситриптофан 710
1-(3-Оксифенил)-2-метиламиноэтанол,
хлоргидрат — см. Мезатон
4-Оксифенилсалициламид — см. Окса-
фенамид
Оксифенилуксусная кислота — см.
Миндальная кислота
Оксифторониобиевая кислота 483
8-Оксихинальдин 710
8-Оксихинолин 711
Оксоль 729
Оксоль-смесь 729
Оксониевые соединения 713
Оксопиразолидин — см. Пиразолидон
Оксосинтез 715
Дис-Октадсцен-9-овая-1 кислота — см.
Олеиновая кислота
Дис-9-Октадеценол-1 —• см. Олеиловый
спирт
Октаметил — См. фосфорорганические
инсектициды
Октан 717
Октаналь — см. Октиловый альдегид
Октановое число 718
--------бензинов 18
Октанол-1 — см. Октиловый спирт
Октафторизобутилен — см. Перфтор-
изобутилен
Октиловый альдегид 720
Октиловый спирт 721
Октоген 721
Октозы 721, 320
Октофоллин — см. Октэсгрол
Октэстрол 722
Олеандомицин 722
Олеиловый спирт 723
Олеиновая кислота 724
Оливин 239
Оливковое масло — см. Жиры раститель-
ные (т. 2)
Оливомицин 724
Олиго (приставка) 725
Олигозиды — см. Олигосахариды
Олигомер 725
Олигонуклеотиды 609
Олигосахариды 725
Олифы 727
—.— быстровысыхающая 729
----глифталевая 730
----глифтале-тукговая 73 0
--------быстровысыхающая 730
---- касторовая 730
----комбинированные 730
---- ксифталевая 731
---- натуральные 729
---- окисленная 729
----оксиполимеризованная 730
---- пентафчалевая 730
----полидиеновая-углеводородная 732
— полимеризованная 729
----полунатуральные (полусинтетиче-
ие) 729
----синтетические 731
----сланцевые 731
---- стиролизованная 731
------ эмульсионные 731
Олово 736, 135
— аналитич. определение 738
— ацетат 738
— галогениды 732
— гидрид 738
—• двуокись 733
---- гидрат 738
— нитрат 738
— нитрид 738
— окислы 733
— окись 733
---- гидрат 738
— получение 739
— применение 739
— сплавы 734
— сульфат 738
— сульфиды 735
— техника безопасности 739
— физич. и химич. свойства 737
Олово двусернистое 735
----двухвалентное, оловоорганич. сое-
динения 742
----полуторасернистое 736
---- сернистое 735
---- хлористое 732
---- четырехвалентное, оловоорганич.
соединения 740
----четыреххлористое 732
Оловоорганические гидриды 741
----кислородные соединения 741
----производные щелочных металлов
742
----соединения 740
--------несимметричные 740
--------симметричные 740
----сульфиды 741
а-Оловянная кислота — см. Олово, гид-
рат двуокиси
р-Оловянная кислота — см. Метаоло-
вянная кислота
Оловянные бронзы 71
Омыление 74?
Омыления число 743
Онзагера уравнение электропроводности
743
Ониевые соединения 744
Оновые кислоты — см. Альдоновые кис-
лоты (т. 1)
Опий 746
Оппенауэра реакция 747
Оппонакс 748
Опробование газов 750
----жидкостей 750
—•— материалов 748
----полужидких (вязких) масс 751
—— твердых материалов 752
Оптическая плотность — см. Поглоще-
ние света
Оптические отбеливающие вещества —
см. Отбеливающие вещества (опти-
ческие)
Оптические свойства коллоидных систем
757
Оранил—см. Надизан
Орбенин — см. З-о-Хлорфенил-5-метел-
4-изоксазолил — пенициллин
Органическая химия 760
Органические реагенты 767
— применение в неорганич. анализе 774
— применение в органич. анализе 775
Органические соединения, классифика-
ция 761
— нитрование 505
— номенклатура 569
— окисление 667
— пиролиз 1073
— стереохимия, обозначения 577
Органическое стекло — см. Полимета-
крилаты
Органогены 776
Ореховое масло — см. Жиры раститель-
ные (т. 2)
Ориентации правило 776
Орнитин 778
Орнитиновый цикл 779
Оротидин 602, 603
Оротовая кислота 780
Орселлиновая кислота 886
Орсин 781
Орсинол — см. Орсин
Ортоклаз 242
Орто-, Мета-, Пара- 781
Ортомарганцоватистая кислота 15
Ортомышьяковая кислота — см. Мышья-
ковая кислота
Ортомышьяковистая кислота 352
Ортона перегруппировка 782
Ортониобаты 474
Ортонитробенэойная кислота 502
Ортоэфиры карбоновых кислот 783
Орцин — см. Орсин
Осаждение 784
Осарсол 786
Осветительные составы 786
Осколки деления 787
Ослабители фотографические 790
Ослабление фотографическое 790
Осмиаты 792
Осмиевая кислота 792
Осмиевый ангидрид — см. Осмий, че-
тырехокись
Осмий 791
— аналитич. определение 794
— гексафторид 792
— двуокись 792
— дихлорид 793
— комплексные соединения 793
— моноокись 792
— окислы 792
—• октафторид 792
— получение 794
— применение 795
— тетраиодид 793
— тетрафторид 792
—• тетрахлорид 793
— трихлорид 793
— физич. и химич. свойства 791
—. фториды 792
— хлориды 793
—• четырехокись 792
Осмол 795
—•— пневый 795
—— стволовый 795
Осмометр 796
Осмос 795
Осмотические свойства раствора 799
Осмотический коэффициент раствори-
теля 798
Осмотическое давление 796
--------коллоидных растворов, мем-
бранное равновесие 113
Основания — см. Кислоты и основания
(т. 2)
Оствальда закон разбавления 799
Остромысленского реакция 800
Осушка газов —см. Газов осушка (т. 1)
Отбеливание (в фотография, практике)
7 91
--- синтетических волокон 804
------- текстильных материалов 800
----- тканей из растительных волокон 8 01
----- шелка 804
---- шерсти 803
Отбеливающие вещества (оптически)
804
Отбор проб—см. Опробование материалов
Отверждение (при полимеризации) 806
Отделка текстильных материалов 807
— безусадочность 811
— водоотталкивающие свойства 810
— негорючесть 810
—. несминаемость 808
— несмываемые аппреты 808
1101 —1102
Отделка тканей специальная 811
Открываемый минимум 811
Отмучивание 819
Относительная влажность 811
Отпугивающие средства 812
Отравление катализаторов—см. Ка-
талические яды (т. 2)
Отравляющие вещества (ОВ) 813
— индикация 817
— классификация 816
— токсичность 814
Отравляющие вещества мышьяксодер-
жащие 814
Отстаивание 818
Отстойники 820
Отэнит 137, 239
Охлаждающие смеси 822
Охлаждение 822, 831
------ глубокое 826
--- умеренное 822
Охра — см. Пигменты
Оцимен 832
Очистка газов — см. Газов очистка (т. 1;
---масляных фракций 834
---• нефтепродуктов 832
Ошибки аналитического определения 8зь
---—•—-------абсолютная 835
-------------грубые 835
-------------относительная 836
-------------систематические 835
--------------- случайные 835
—•—------------ «средняя» квадратичная
837
---косвенных измерений 84 0
---• прямых равноточных измерений
836
ПАВК — см. n-Аминобензойная кис-
лота (т..1)
Пакатал — см. Мепазин
Палладий 845
— аналитич. определение 848
— двуокись 847
— дибромид 847
— дииодид 847
— дисульфид 847
— дифторид 847
— дихлорид 847
—• дицианид 848
— закись 846
— карбонил 847
— комплексные соединения 850
— моносульфид 847
— нитрат 848
—• нитрозил 848
— окислы 846
— окись 846
— получение 848
— применение 849
— селенат 848
— сплавы 848
— сульфат 848
—• тетрацианид 848
— трифторид 847
— физич. и химич. свойства 845
Палладий как катализатор 849
Палладит 845
Палласиты 174, 177
Пальмитиновая кислота 852
Пальмовое масло — см. Жиры расти-
тельные (т. 2)
Пальмоядровое масло — см. Жиры рас-
тительные (т. 2)
Палюстровая кислота 852
Пангаметин —• см. Пангамовая кислота
Пангамовая кислота 853
Панкреатин 853
Пантосепт — см. Пантоцид
Пантотеновая кислота 854
—. чувствительность микробиология, ме-
тода химич. анализа 233
Пантоцид 855
Папаверин 855
Папаин 855
Папайотин — см. Папаин
Пар пересыщенный 171
Пара- — см. Орто-, Мета-, Пара-
Парабановая кислота 856
Параллельные реакции 856
Парамагнетизм — см. Магнитные свой-
ства (т. 2)
Параметры состояния 857
Параминдальная кислота 238
1103 — 1104
Парамион 857
Паранитробензойная кислота 502
Паратиреоидин — см. Паратиреокрин
Паратиреокрин 857
Парафин 858
----в нефтепродуктах, определение со-
держания 435
Парафины —см. Предельные углеводороды
Парафины, нитрование 505
— октановые числа 719
— химич. переработка 438
Параформ — см. Формальдегид
Параформальдегид — см. Формальдегид
Парафуксин 859
Парахор 860
Паромомицин 863
Парообразование — см. Испарение (т. 2)
Парциальное давление 864
Парциальных валентностей теория 864
ПАСК 865
Пассерини реакция 865
Пассиваторы 1014
Пассивация, потенциал 868
—— химическая 868
Пассивность металлов 866
Пастера эффект 870
Патронит 135
Паттерсона система 576
Паули принцип 870
Паули реакция 871
Пахикарпин 872
Пек древесный 873
----каменноугольный 873
---- нефтяной 873
----торфяной 873
Пекаэин — см. Мепазин
Пеки 872
Пектазы — см. Пектиновые ферменты
Пектаты 874
Пектиназы — см. Пектиновые ферменты
Пектинаты 874
Пектингликоэидаза — см. Пектиновые
ферменты
Пектиндеметоксилаза — см. Пектиновые
ферменты
Пектиндеполимераза — см. Пектиновые
ферменты
Пектинметилэстераза — см. Пектиновые
ферменты
Пектиновые вещества 874
Пектиновые ферменты 875
Пектины 874
Пектинэстераза — см. Пектиновые фер-
менты
Пектовая кислота 874
Пектолаэа—см. Пектиновые ферменты
у-Пеларгонлактон — см. Нонлактон
Пеларгоновая кислота 876
Пелентан — см. Неодикумарин
Пеллотин 876
Пельтье эффект 162
Пельтьерин 877
Пенальдиновые кислоты — см. Пеналъ-
довые кислоты
Пенальдовые кислоты 882
Пенатин — см. Нотатин
Пенбритин—см. Аминобензилпенициллин
Пендиомид — см. Пентамин
Пениллоальдегиды 882
Пенилловые кислоты 883
Пениллоиновые кислоты 883
Пениллоновые кислоты 884
Пеницилламин 882
Пенициллин В — см. Нотатин
Пенициллиназа 877
Пенициллины 878
Пеницилловая кислота 885
Пенициллоиновые кислоты 881
Пеногасители 899
Пенообразователи 898
Пенопласты 886
Пенополивинилхлорид 887
Пенополистирол 88 7
Пенополиуретан 887
Пенспек — см. а-Феноксибензилпени-
циллин
Пентабромтолуол 201
Пентадеканолид — см. Тибеттолид
Пентадеканол-15-овая кислота, лактон —
см. Тибеттолид
1,3- Пентадиен — см. Пиперилен
Пенталан 888
Пентаметилендикарбоновая кислота —
см. Пимелиновая кислота
Пентаметиленамин — см. Пиперидин
1, 2, 2, 6, 6-Пентаметилпиперидин п-то-
луолсульфонат — см. Пирилен
Пентамин 889
Пентан 889, 439
— октановое число 719
3, 5, 7, 2', 4'-Пентаоксифлавон — см.
Морин
Пентаровые кислоты 896
Пентафен 117
Пентафенилмышьяк 354
Пентафенилфосфор 890
Пентаэритрит 890
Пентен-1
— октановое число 719
2-Пентенилпенициллин 879
Пентиты 254, 896
Пентландит 133, 456
Пентозаны 891
Пентозный цикл 892
Пентозофосфатный цикл 663 — см. так-
же Пентозный цикл
Пентозы 895, 320
Пентоксил 896
Пентоль X 731
Пентоль Хр 731
Пентон 896
Пентеновые кислоты 897, 896
Пентулоэы — см. Пентозы
Пентуроновые кислоты 896
Пены 897
Пепсин 900
Пепсиноген 900
Пептидазы — см. Пепгидгидролазы
Пептидгидролазы 901
Пептидная связь 903
Пептид-пепгидогидролазы — см. Пеп-
тидгидролазы
Пептиды 904
Пептизагоры 907
Пептизапия 907
Пер (приставка) 907
Пербораты — см. Пероксобораты
Первигин 907
Первый закон термодинамики 908
Пергидрокомплексы 936
Переамидинирование 911
Переамидирование 911
Переаминирование 910, 622
Перегонка нефти 912
Перегруппировка аллильная 915
---Ортона 782
--- пинаколиновая 1035
--- регропинаколиновая 1035
Передача энергии линейная 919
Перекачивание жидкостей 920
Перекиси, гидраты 936
--- диацилов 930
--- неорганические 932
--- органические 926
---элементоорганические 931
Перекисная теория окисления органич.
соединений 667
--------Баха — Энглера 937
Перекисные комплексы 936
Перекисные соединения 929
— —--комплексные 93 5
— —----неорганические — см. Пере-
киси и перекисные соединения неорга-
нические
Перекисный эффект 937
Переметилирование 939
Перемешивание 939
— •— в жидких средах 939
— •— сыпучих материалов 943
Перенапряжение 944
— — водорода 944
— .— кислорода 944
Перенос энергии (на молекулярном уров-
не) 945
— диффузионный механизм 947
— значение явления 948
— индуктивно-резонансный механизм
948
— экситонный механизм 948
Перенос энергии внутримолекулярный
946
--------межмолекулярный 946
Перепассивации состояние металлов
867
Пересульфирование 949
Пересыщенный раствор — см. Раство-
римость
Перефосфорилирование 949
Переходного состояния метод 950
Переходное состояние 958, 951
Переходные элементы 958
Переэтерификация 959
Пери (приставка) 959
Периклаз 242
Перилен 960
Перилловое масло — см. Жиры расти-।
тельные (т. 2) 1
Период полураспада — см. Полураспада
период
Периодическая система элементов Мен-
делеева 960
— история открытия 961
— описание 967
— • периодичность свойств 972
— принципы построения 964
Периодическая система элементов Мен-
делеева и строение атома 962
Периодический закон Менделеева — см.
Периодическая система элементов Мен-
делеева
Перитектика — см. Двойные системы
(т. 1)
Перкарбонаты — см. Пероксокарбонаты
Перкина реакция 977
Перкислоты — см. Перекиси и гидропе-
рекиси органические
Перманганатометрия 980
Перманганаты 980
Пермутиты 981
Пероксигидраты 936
— гидраты 936
Пероксидазы 981
Пероксидный эффект — см. Перекисный
эффект
Пероксиды — см. Перекиси
Пероксобораты 982
Пероксогруппа 932
Пероксодисерная кислота 984
Пероксодифосфорная кислота 984
Пероксокарбонаты 983
Пероксокислоты 935
Пероксокомплексы 936
Пероксомоносерная кислота 984
Пероксомонофосфорная кислота 984
Пероксосерные кислоты 984
Пероксосульфаты 984
Пероксофосфаты 984
Персидская красная 1019
Персиковое масло — см. Жиры расти-
тельные (т. 2)
Персоль 983
Персульфаты —• см. Пероксосульфаты и
Аммония персульфат (т. 1)
Перфосфаты — см. Пероксофосфаты
Перфторалканы — см. Фторалифати-
ческие соединения
Перфтора лкилмагнийгалогенид — см.
Фторалифатические соединения
Перфторацетилены — см. Фторалифа-
тические соединения
Перфторацетон 985
Перфторбензол 986
Перфторбутадиен-1,3 987
Перфтордивинил —• см. Перфторбута-
диен-1,3
Перфтордиметилкетон — см. Перфтора-
цетон
Перфторизобутилен 988
Перфторкарбоновые кислоты — см. Фто-
ралифатические соединения
Перфторолефины — см. Фторалифати-
ческие соединения
Перфторпропен — см. Перфторпропилен
Перфторпропилен 988
Перхлораты 989
Перхлорбутадиен 989
Перхлордивинил — см. Перхлорбутадиен
Перхлорциклопентадиен 990
Песок, опробование 755
Пестицидные препараты 991
Пестициды 993
Петалит 137
Петролатум 993
Петролейный эфир 994
Петцит 137
Пехмана конденсация 994
Печатание текстильных материалов 996
-------------азокрасителями 998
-------------активными красителями
999
--------—— кубовыми красителями
999
-------------пигментными красителя-
ми 1000
-------------черным анилином 1000
Печатные краски 1001
Печи для обжига огнеупорных материа-
лов 649
--- технологические (основной химич.
промышленности) 1003
Пиазин — см. Пиразин
Пигменты 1011
— интенсивность 1012
— хмаслоемкость 1014
— опробование 755
— смачиваемость 1013
— укрывистость 1012
— форма и размер частиц 1012
— цвет 1012
Пигменты антикоррозионные 1022
--- белые 1013, 1015
---биологические 1025
---для печатных красок 1001
---железоокисные 1026
-------- желтые 1018
--------- коричневые 1018
------- красный 1013
-------- черный 1013
----------- термостойкий 1013
— желтые 1013, 1017
---зеленые 1013, 1019
-------смешанные (зелени) 1020
---кадмиевые 1026
--- коричневые 1013, 1017
--- красные 1013, 1017
---лессирующие 1012
---медные природные 1022
—.—• металлические 1024
---оранжевые 1013, 1017
---органические — см. Красители син-
тетические (т. 2)
--- синие 1013, 1019
---фиолетовые 1013, 1019
---черные 1013, 1017
Пико (приставка) 89
Пиколиновая к г. слота — см. Пиридин-
карбоновые кислоты
Пиколины 1057 — см. также Метилпи-
ридины
Пи-комплексы 1026
Пикрамид 1029, 500
Пикраминовая кислота 1030
Пикрилхлорид 1030
Пикриновая кислота 526 — см. также
Тринитрофенол
Пикролоновая кислота 1031
Пикте — Шпенглера реакция 1032
Пилокарпин 1033
Пилоти реакция 1034
Пимелиновая кислота 1034
Пинаколин 1035
Пинаколиновая и ретропинаколиновая
перегруппировки 1035
Пинаконы 1037
Пинан 1039
Пинены 1039
Пиоцианин 1041
Пипеколины — см. Метилпиперидины
Пиперазин 1041
Пиперидин 1042
Пиперилен 1043
Пиперин 1044
Пиперонал 1044 — см. также Гелиотро-
пин (т. 1)
Пиразин 1044
Пиразол 1045
Пиразолидин — см. Пираэолин
Пираэолидон 1046
Пираэолин 1046
Пираэолон 1047
Пиразолоновые красители 1049
Пирамидон 1050
Пиран 1050
Пирен 1051
Пиреновая кислота 10 51
Пиренхинон-3,8 1051
Пиретрины 1051
Пиретроиды — см. Пиретрины
Пиретрум 1052
Пиридаэин 1052
Пиридин 1053
p-Пиридинкарбоновая кислота — см. Ни-
котиновая кислота
Пиридинкарбоновые кислоты 1055, 196
Пиридиннуклеотидные коферменты 472
Пиридиннуклеотиды 1057 — см. также
Кодегидрогеназы (т. 2)
Пиридиновые основания 1057
Пиридоксалевые ферменты 1058
Пиридоксаль 10 60
Пиридоксаль-5-фосфат 1059
Пиридоксамин 1060
Пиридоксин 1060
— чувствительность микробиология, ме-
тода химич. анализа 233
Пиридоксол — см. Пиридоксин
Пиридолы — см. Оксипиридины
Пиридоляты 705
Пиридоны 1061, 705
Пирилен 1061
Пирилиевые соли 1061
Пиримидин 1063
Пиримидиновые основания 1063
Пирит 239
Пиро (приставка) 1065
Пиробензол 1065
Пировиноградная кислота 1065
—альдегид — см. Метилглиоксаль
Пировиноградный спирт — см. Окси-
ацетон
Пирогалловая кислота — см. Пирогаллол
Пирогаллол 1067
Пирогенные процессы — см. Термичес-
кая переработка топлива
Пирокатехин 1068
Пироколлодий 551
Пироксилин — см. Нитроцеллюлоза
Пироксоний, соли—см. Пирилиевые соли
Пиролиз 1070
— механизм и кинетика 1071
— техника П. 1079
Пиролиз азотистых соединений 1077
— — в плазме 1081
--- в ударных трубках 1081
— — галогенных соединений 1078
—— кислородных соединений 1075
—.—• органич. соединений основных клас-
сов 1073
—•— сернистых соединений 1078
---углеводородов 1074
Пиролюзит 14, 20, 133
Пиромышьяковая кислота 352
Пироп 239
Пирофосфорилаэы 607
Пирохлор 137, 476
Пиррол 525
Пирувиновая кислота — см. Пировиног-
радная кислота
Пластификаторы — см. Мягчители
Пластичность материалов 207
---металлов 163
Пластмассы
— прочность, временная зависимость 223
Платина палладистая 84 5
Плотность, единицы измерения 91
Площадь, единицы измерения 89
Пневматическая химия 420
Поваренная соль — см. Натрий хлорид
Повеллит 239
Поверхность, модифицирование химич.262
Погрешности измерения 837
Подкладки — см. Носители в катализе
Подмыльный щелок 346
Полевые шпаты 239, 241
Поледентатные комплексы 768
Полезные ископаемые, обогащение 630
Ползучесть материалов 210
---металлов 164
---твердых полимеров 216
Полиалкиленгликоли 46
Поливинилацетат, механодеструкция 227
Поливиниловый спирт, механодеструк-
ция 227
Полигалактуронаэа — см. Пектиновые
ферменты
Полидепсиды 699
Полимеризация мономеров 314
Полимерные материалы ячеистые — см.
Пенопласты
Полимеры, высокая эластичность 218
— вязкое течение 220
— деформационные свойства 214
— механич. свойства 214
— механодеструкция 226
— механокрекинг 226
— механосинтеэ 227
— механохимич. изомеризация 228
— механохимия 225
— • мономолекулярный слой 319
— набухание 357
— ориентация и кристаллизация 261
— отверждение 806
— ползучесть 216
— прочность 222 _
— — временная зависимость 223
— релаксационные свойства 219
— реология. свойства 220
— фрикционные свойства 225
— хемомеханика 226
— химич. течение 228
Полимеры аморфные 215, 220, 223
---высокоэластические 215, 217, 222
--- вязкотекучие 215
---кристаллические 214, 220, 222
---стеклообразные 215
Полиметилметакрилат, механодеструк-
ция 227
Полиминеральные агрегаты 240
Полиморфизм металлов 160
Полинитропарафииы 535
Полинуклеотиды — см. Нуклеиновые
кислоты
Полипептиды — см. Пептиды
Полипропиленгликоли 46
Полисилоксаны 47
Полистирол, механодеструкция 227
Полиэтиленгликоли 46
1105 — 1106
Полуццит 137
Порох пироксилиновый 552
Порпецит 845
Потарит 84 5
Потенциальная поверхность 950
Предел вынужденной эластичности по-
лимеров 214
—— текучести 163, 207
— — упругости 163
— — усталости 209
Пренитрон •— см. Нитранол
Препарат ДИД 813
-----рютгер 622, 813
Преципитат 245
Приставки
—— атто 89
гекто 89
гига 89
дека 89
—— деци 89
—•— кило 89
—— мега 89
мезо 105
мета 126, 781
—— микро 89
—— милли 89
нона 89
—— нор 579
олиго 725
— орто 781
— пара 781
— пер 907
— пико 89
— — пиро 1065
— — Санти 89
— — тера 89
— — фемто 89
Провитамин РР —. см. Никотиновая
кислота
Прокаин — см. Новокаин
Прокаинамид — см. Новокаинамид
Пронестил — см. Новокаинамид
Пропан 439
— энергия С — Н-свнэи 669
Пропанол-2-ован-1 кислота — см. Мо-
лочная кислота
Пропилен 441
— октановое число 719
— энергия С—Н-связи 669
Ч>-Пропилен — см. Перфторпропилен
Пропилнитрамин 499
н-Пропилнитрат 556
Пропиолонитрил 494
Пропионитрил 494
Простафлин — см. Оксациллин
Пространство, единицы измерения 85
Протопектины 874
Прочность материалов 211
-----металлов 164
— — полимеров 222
«Псевдоуридин» 597
Псиломелан 14, 133
Пуаз (ед. измерения) 93
Пуриннуклеоэид 601
Работа, единицы измерения 93
Равновесие мембранное 112
Радиан (ед. измерения) 84
Радикалов теория 762
Радиометрическая сортировка руд 633
Радиохимия, носители 589
Рассеяние света антистоксово 281
— — —— коллоидными системами 757
--------стоксово 281
Раствор, моляльность 310
— молярность 310
— нормальность 582
Раствор ненасыщенный — см. Раство-
римость
— —• пересыщенный 171
Растворы металлидные 141
Рауля закон 797
Рациональная номенклатура 570
Реабсорбция 945
Реагенты органические 767
Реакции мономолекулярные 316
— — неадиабатические 953
— — необратимые — см. Реакции обра-
тимые
------ обратимые 639
— — параллельные 856
Реакционная координата 951
Реакция Меервейна 82
1107 — 1108
—— Меервейна — Понндорфа — Вер-
лен 83
---Мейера — см. Реакция Майера (т. 2)
---- Мейстера — Митера — см. Бар-
бье — Виланда расщепление (т. 1)
---Миллона 237
---Михаэлиса — Беккера 249
— Михаэля 250
---мурексидная 342, 327
—— Несмеянова 428
---Нефа 429
--- нингидринная — см. Реакция нин-
гидриновая
--- нингидриновая 474
---Ниренштейна 486
---Оппенау эра 747
--- Остромысленского 800
---Пассерини 865
------ Паули 871
---Перкина 977
---Пикте — Шпенглера 1032
— Пилоти 1034
— транспептидации 900
Реальгар 348, 352
Ревденскит 133
Регенерация масел 41
Регмаглипты 174
Резацетофенон 693
Резины, наполнители 361
Резонанса теория 766
Резонансное взаимодействие 101
Ректанол — см. Нарколан
Ректификация, массообмен 52
Релаксационные свойства полимеров 219
Рений 139
Репелленты — см. Отпугивающие сред-
ства
Ретропинаколиновая перегруппировка —
см. Пинаколиновая и ретропинаколи-
новая перегруппировки
D-Рибоза 602
Рибоновая кислота 897
Рибонуклеазы 590
Рибонуклеиновая кислота (РНК) 278,
596
——.------«информационная» (матрич-
ная) 598
----------- клеточного сока («раствори-
мая» или адапторная) 598
----- —•— рибосомальная 598
Рибонуклеозиды 603
Рибонуклеопротеиды 605
Рибосомы 278, 606
Рибофлавин, чувствительность микро-
биологии. метода химич. анализа 233
Риталин — см. Меридил
Родановое число 27
Родицит 137
Родонит 133
Родохрозит 133
Родохром 24 0
Роскоэлит 135
Ртуть 135
Рубидий 137
— надперекись 934
Рубин 240
Рудоразборка 631
Рутил 240, 1013, 1015
С
Сажи, модифицирование поверхности хи-
мич. 263
---как наполнители (для резин) 361
Сало растительное 23
Санти (приставка) 89
Сахар солодовый — см. Мальтозы
Сахара, чувствительность микробиоло-
гия. метода химич. анализа 233
---простые —см. Моносахариды
Сведберг (ед. седиментации) 296
Сверхпроводимость 162
Световые единицы 87
Свинец 135
— молибдат 299
— силикат основной 1024
— силико-хромат основной 1024
— сульфаты основные 1023
— цианамид 1024
Свинцовая бронза 71
Связь, длина 102
---гомеополярная (ковалентная) 158,
268
-------- донорно-акцепторная 101
---------- металлическая 157, 140
------ пептидная 903
—— химическая 267, 764
Сгущение — см. Отстаивание
Седиментация—см. Отстаивание
Селбенин — см. Метициллин
Селен 137
— молекула, межъядерное расстояние
. 102
Селитры 489
Семидурохинон 119
Семихиноны — см. Мерихиноидпые соли
Сера в нефтепродуктах
—• определение содержания 434
— молекула, межъядерное расстояние
102
Сера молотая, опробование 755
Сероводород в нефтепродуктах, опре-
деление содержания 435
Сиалидазы — см. Нейраминидазы
Сидерит 133, 241
Сиены 1013, 1019
Сила, единицы измерения 91
Силиконовые жидкости — см. Масла
синтетические полисилоксановые
Силикотермия 155
Силициды, огнеупоры 648
Сильванит 137
«Синий Вурстера» 119
Синильная кислота 817
Синнематин В—см. 4-Амино-4-карбок-
си-п-бутилпенициллин
Синтетазы —см. Лигазы
Синтол 731
Синциллин—см. а-Феноксиэтилпени-
циллин
Синь Тенара — см. Кобальт синий
Скандий 137
Скородит 239, 348
Скорость линейная, единицы измерения
91
----угловая, единицы измерения 91
Слюда 239, 241
Смазки консистентные, опробование 752
Смачиватель СВ-101 —см. Некаль ВХ
Смеси, масс-спектральный анализ хи-
мич. состава 63
Смесители 942
Смесь ДД 415
Смитсонит 135
Смолы в нефтепродуктах, определение
содержания 435
Сода, опробование 755
—.— кальцинированная —см. Натрий,
карбонат нормальный
—— питьевая — см. Натрий, бикар-
бонат
Соли сильных кислот, осаждение 784
—— слабых кислот, осаждение 784
Солодовый сахар — см. Мальтоза
Сорбит 254, 255
(+)-Спартеин — см. Пахикарпин
Спирты многоатомные 253
Сподумен 137
Спумоиды 897
Среднее арифметическое результатов ана-
литич. определения 836
Сталь
— нейтронное облучение 165
— опробование 756
Стандартный потенциал — см. Нормаль-
ный потенциал
Станнаты 738
Станнин 135, 737
Станниты 738
Станнометан — см. Олово, гидрид
Станнопалладинит 845
Стапенор — см. Оксациллин
Стафциллин — см. Метициллин
Стеаронитрил 494
Стекло, упругость 205
Стеклообразное состояние 172
Стеклянная вата, опробование 756
Стен (ед. измерения) 91
Стерадиан (ед. измерения) 84
Стереохимические обозначения 577
Стереохимия 763
Стеркобилин 1026
Стеркобилиноген 1026
Стероиды 586
Стибиопалладинит 845
Стоварсол—см. Осарсол
Стокс (ед. измерения) 93
Стрептоза 324
Стрептомицин В — см. Маннозидостреп-
томицин
Стронцианит 139
Стронций 139
Структура дефектная (дефицитная) ми-
нералов 243
Сульфатиазол—см. Норсульфазол
Сульфидные медноникелевые руды 456
Сульфонолы 335
Сульфооксоль 730
Супернапалм 360
Супероксиды — см. Надперекиси
Суперфосфат 245
Сурик железный 1013, 1019
---свинцовый 1023
Сусальное золото 736
Суспензия, отстаивание 818
Сфалерит 135, 241
т
Табун 814, 817
Тадексил — см. Мепротан
Талит 254, 255
Таллий 139
Тальк 241, 242
Тантал 137
— нитриды 492
Танталит 137
Твердость материалов 211
Твердые растворы замещения 166
Текстильные материалы, отделка 807
— печатание 996
Текстура руды 133
Тектиты 17 7
Теллур 137
— молекула, межъядерное расстояние
Температура, единицы измерения 84, 95
Температурный градиент, единицы изме-
рения 95
Тенардит 382
Тенорит 68
Тепловой поток, единицы измерения 93
— поверхностная плотность, единицы
измерения 93
Тепловые единицы 87
Тепловые свойства металлов 161
Теплоемкость, единицы измерения 93
Теплота, единицы измерения 93
Тера (приставка) 89
Терапевтический индекс стероидов 587
Термия (ед. измерения) 93
Термоэлектронная эмиссия 162
Террамицин—см. Окситетрациклин
Тесла (ед. измерения) 8 5
Тестостерон, пропионат 587
Тетраалкилдиарсины 3 54
Й-Тетрагидродициклопентадиенил-Х',
N '-дим етилм оче вина
Тетрагидронервон 426
Тетраметилдиаминобензофенон — см.
Михлера кетон
Тетраметилентионин, тригидрат хлори-
да — см. Метиленовый синий
Тетраметилолметан —см. Пентаэритрит
Тетранитрометан 535
1, 3, 5, 7-Тетранитро-1, 3, 5, 7-тетразо-
циклооктан — см. Октоген
Тетраэдрит 239
Тетриты 254
Тетрозы 320
Тиамин, чувствительность микробио-
логия. метода химич. анализа 233
Тибеттолид 343
Тимидин 603
Тимидин-5'-монофосфат 611
Тинделя эффект 758
Тинкал — см. Вура
Тиоиндоксил — см. З-Окситионафтен
Тиолы —• см. Меркаптаны
Тиоспирты — см. Меркаптаны
Типов теория 762
Титан, двуокись 1013, 1015
— нитриды 492
Толуол 443
— октановое число 719
— энергия С — Н-связи 669
Томасшлак 245
Топаз 242
Топливо, октановое число 720
Топливо моторное 326
— детонационная стойкость 18
Торбернит 137
Тормозная способность —см. Передача
энергии линейная
Точка росы 812
Транквил — см. Мепротан
Трансаминирование — см. Пе реамини-
рование
Трансметилазы — см. Метилтрансфера-
зы
Трансметилирование — см. Перемети-
лирование
Транспассивное состояние металлов —
см. Перепассивации состояние метал-
лов
Транспептидации реакция 900
Трансферазы 560
Трансферрин 152
Трансфосфорилирование —• см. Перефос-
форилирование
Трегеры — см. Носители в катализе
Трехокись 679
2, 4, 6-Триамино-з-триазин — см. Ме-
ламин
2, 4,6-Триамино-1, 3, 5-триазин — см.
Меламин
Трибромэтанол — см. Нарколан
Тривиальные названия 569
Трикетогидринденгидрат — см. Нингид-
рин
Тримезиновая кислота 105
2, 4, 6-Триметиланилин — см. Мезидин
а-Триметилацетон — см. Пинаколин
1,3, 5-Триметилбензол — см. Мезитилен
2,6, 6-Триметил-бицикло-[1,1,3]-гептан —
см. Пинан
Триметилпиперидины — см. Копелли-
дины
Триметилпиридины — см. Коллидины
Тримиристин 246
Тринатрийарсенат 374
2,4,6-Тринитроанилин — см. Пикрамид
2,4,6-Тринитробензойная кислота 502
2,4,6-Тринитро-1, З-диметил-5-mpem-
бутилбензол — см. Мускус-ксилол
Тринитрометан — см. Нитроформ
Тринитроспирты 545
2,4,6-Тринитрофенилнитрамин 499
1,1,1-Тринитроэтан 535
2,2,2-Тринитроэтанол 546
Триозы 320
1, 2, З-Триоксибензол — см. Пирогаллол
Триоксипиридины 707
2,6,8-Триоксипурин — см. Мочевая кис-
лота
2,4,5-Триоксоимидазолидин — см. Пара-
бановая кислота
Триптофан 525
Трихлортриэтиламин 814, 818
Триэтаноламин, дифосфат тринитрата —
см. Нитранол
Тромексан — см. Неодикумарин
Трона — см. Натрий, сесквикарбонат
Тунгусский метеорит 177
Турбидиметрия — см. Нефелометрия в
турбидиметрия
Турмалин 239
Тюямунит 137
У
Увитиновая кислота 105
Углеводороды, окисление 664
— октановое число 719
— пиролиз 1074
— энергия С — Н-связей 669
Углеводороды ароматические, алкили-
рование 443
— дегидрирование 444
— окисление 444
— химич. переработка 442
Углеводороды нафтеновые 395
— химич. переработка 444
---• ненасыщенные — см. Углеводо-
роды непредельные
— •— непредельные 425
—.— —.—. —. гидратация 441
---------- — окисление 442
----------—, полимеризация 442
---—.—. — химич. переработка 440
--------—• хлорирование 441
---парафиновые, химич. переработка
438
Углеводы, апотомический путь окис-
ления — см. Пентозный цикл
Углерод
— молекула, межъядерное расстояние
102
— недоокись 404
— окись 817
Угол плоский, единицы измерения 91
Удельная теплоемкость, единица измере-
ния 93
Удельная теплота, единицы измерения 93
Удельная энтропия, единица измеренияЭЗ
Удельный вес, единицы измерения 93
Удобрения минеральные 244
Узорчатая расцветка тканей — см. Пе-
чатание текстильных материалов
Уксусная кислота, метиловый эфир —
см. Метилацетат
Ультрамарин 1013, 1020
Ультрапен — см. а-Феноксипропилпе-
нициллин
Умбра 1013, 1019
Упругое последействие 206
Упругость материалов 205
——- металлов 163
Упрочнение материалов 207
—— металла 164
Уран 137
Уранинит 137
Ураты 327
Урацилдезоксирибозид 603
Уридин 603
Уридин-5'-дифосфат 611
Уридин-5'-монофосфат 611
Уридин-5'-трифосфат 611
Уридин-2'- и З'-фосфат 611
Уробилин 1026
Уробилиноген 1026
Усталость материалов 209
«Утяжеления эффект» 771
о-Фазы 143
----внедрения 167
----- Лавеса 144
—-— никель-арсенидные 145
Фантолид 343
Фемто (приставка) 89
Фенакит 137
Фенбенициллин — см. а-Феноксибен-
зилпенициллин
Фенетициллин — см. а-Феноксиэтилпе-
нициллин
Фенидим — см. Х-Фенил-Х', N'-диме-
тилмочевина
Фениларсоновая кислота 767
Фенилгликолевая кислота—см. Мин-
дальная кислота
1-Фенил-2,3-диметил-4-диметиламинопи-
разолон-5 — см. Пирамидон
Х-Фенил-Х', N'-диметилмочевина 331
В-1-Фенил-2-метиламинопропан, гидро-
хлорид— см. Первитин
1-Фенил-3-метилпиразолон-5 1047
Фенилнитрамин 499
1-Фенилциклопентан-1-карбоновая кис-
лота
— диэтиламиноэтиловый эфир, метил-
сульфометилат — см. Мепанит
а-Феноксибензилпеиициллин 879, 885
Феноксиметилпенициллин 879, 885
а-Феноксипропилпенициллин 879
а-Феноксиэтилпенициллин 879, 885
Фенолокислоты 699
Фенурон —см. Х-Фенил-Х', N'-диме-
тилмочевина
Ферберит 135
Ферменты, номенклатура и классифика-
ция 559
----пиридоксалевые 1058
----протеолитические — см. Пептид-
гидролазы
Ферромагнетизм 163
Ферросплавы, опробование 757
Фикоэритрин 1026
Фладе-потенциал 868
Флорацетофенон 693
Флотация — см. Обогащение полезных
ископаемых флотационное
Флотогравитация 634
Флуктуации гетерофазные 759
Флюорит 239, 241, 242
Фолиевая кислота, чувствительность
микробиологич. метода химич. анализа
233
Формальдегид 525
Форстерит 242
Фосген 814, 817
Фосгеноксим 817
Фосфат обесфторенный 245
Фосфор
— молекула, межъядерное расстоя-
ние 102
Фосфорилирование окислительное 660
Фосфорилтиохолииы 817
Фосфористый водород 817
Фосфоритная мука 245
Фотолиз нуклеотидов 610
Фотометрия энергетическая, единицы
измерения 87
Франка — Кондона принцип 289
Фреоны, холодильные агенты 822
Фтор
— молекула, межъядерное расстояние
102
Фторониобиевая кислота 483
Фторуглеводороды 48
Фуксит 240
Фунгицидин — см. Нистатин
Фуретрин 1052
1109 — 1110
Халазон — см. Пантоцид
Халькозин 75, 77 135
Хальконтит 74
Халькопирит 77, 135
Хасталлой 468
Хелидоновая кислота 706
Химическая связь 7 64
Хинализарин 692
Хинная кислота 699
Хиногенный эффект 542
Хинолиновая кислота — см. Пиридин-
карбоновые кислоты
п-Хинонмоноксим —• см. п-Нитрозофенол
Хладноломкость материалов 208
Хлор
— молекула, межъядерное расстояние
Хлорацетофенон 818
Х-Хлорбонил-Х', N'-диметилмочевина
331
Хлорнитробензолы — см. Нитрохлор-
бензолы
Хлорнокислые соли—-см. Перхлораты
Хлорокруорин 1025
Хлороосмиевая кислота 793
Хлорпикрин 814, 818
2-Xnopnponnn-N, ^бис-(р-хлорэтил)
амин, хлоргидрат — см. Новэмбихин
2-Хлор-1,3,5-тринитробензол — см. Пи-
крилхлорид
Хлорфенидим — см, Х-4-Хлорфенил-Х',
N'-диметилмочевина
№4-Хлорфенил-№, N'-диметилмочевина
331
3-о-Хлорфенил-5-метил-4-изоксазолил-
пенициллин 879
Хлорфторуглеводороды 48
Хлорциан 817
Холин, чувствительность микробиоло-
гии. метода химич. анализа 233
Холла эффект 162
Холодильные агенты 822
Холодильные машины 824
Холодильные смеси —• см. Охлаждаю-
щие смеси
Холодильный цикл 826
Хондриты 174, 177
Хондроитин 339
Хондроитинсерная кислота 339
Хондроитин-4-сульфат — см. Хондро-
итинсерная кислота
Хризоберилл 137
Хризоколла 77
Хром, нитриды 492
— окись 1013, 1020
— пассивационная характеристика 869
— фосфат 1024
Хроматин 596
Хромопротеиды 1025
Хромосомы 596
Хромотроповая кислота 767
Хромотропы 173
Хромофоры 240
Хрупкость материалов 207
ц
Цезий 137
— надперекись 934
Целестин 139
Целлюлоза, нитрат — см. Нитроцеллю-
лоза
Центендрин — см. Меридил
Церезины 858, 654
Церулеум 1013, 1021
Церулоплазмин 152
Церуссит 135
Цефалоспорин N—-см. 4-Амино-4-кар-
бокси-п-бутилпенициллин
Цианкобаламин, чувствительность мик-
робиологии. метода химич. анализа 233
Цианоформнитрил 494
Цианпиридины 196
Цибетон 343
Циклитрин 1052
Циклические соединения, номенклатура
575
Циклогексаноксим 553
Циклогептадецен-9-он—см. Цибетон
№Циклооктил-№, N'-диметилмочевина
331
Циклопентадеканон—см. Экзальтон
1111—1112
Циклопентилкарбоновая кислота 393
Циклопентилуксусная кислота 393
Циклопептиды 905
Циклотетраметилентетра нитрамин — см.
Октоген
•п-Цимол — см. п-Метилизопропилбен-
зол
Цинерины 1051
Цинк 135
— молибдат 299
— нитрид 491
Цинковые удобрения 235
Циннвальдит 137
Цинхомероновая кислота — см. Пири-
динкарбоновые кислоты
Циркон 139
Цирконий 139
— нейтронное облучение 165
— нитрид 492
Цистеамин — см. Меркамин
Цистроны 278
Цитидин 603
Цитидин-5'-дифосфат 611
Цитидин-2'-моноФосфат 611
Цитидин-З'-монофосфат 611
Цитидин-5'-монофосфат 611
Цитидин-5'-трифосфат 611
Цитозиндезоксирибозид 603
ч
Чаргаффа правило 597
Черни (пигменты) 1017
Чессилит 67
Четверные системы 255
Чилийская селитра 239, 367
Чугун, опробование 756
ш
Шеелит 13 5
Шпинели 239
щ
Щавельная кислота, диамид — см. Ок-
самид
— хлорангидрид — см. Оксалилхлорид
э
Эвдиалит 139
Эвкриты 177
Экзальтолид — см. Тибеттолид
Экзальтон 344
Экзопептидазы 901
Экстракция, массообмен 53
Электрические единицы 85
Электрический газовый разряд, масс-спек-
тральное исследование 65
Электронная концентрация 159
Электронные оболочки атомов, схема
заполнения 966
Электронные соединения металлич. 144
Электронные спектры молекул 288, 273,
282, 283
Электроны, неподеленная пара 101
— работа выхода 158
Электроотрицательность 673
Электропроводность, Онзагера уравне-
ние 743
Электростатические силы 97
Электрофильные реагенты 675 — см.
также Нуклеофильные и электрофиль-
ные реагенты
Элементарные процессы, масс-спектр а ль-
нов исследование 62
Элементы химические, миграция 230
Эмболит 239
Эмульсия, отстаивание 818
Энаргит 135
Энергетический барьер 951
Энергия, единицы измерения 93
Энтропия, единицы измерения 93
Эпидот 239
Эритрит 254
Этаминал-натрий — см. Нембутал
Этан 439
Этилбензол 443
— октановое число 719
— энергия С — Н-связи 669
2-Этилгександиол-1,3 812
Этилен 441
Этиленгликольдинитрат —• см. Нитро-
гликоль
Этилнитрамин 499
Этилнитрат 556
Этилуксусная кислота —• см. Масляная
кислота
Этилфенилнитрозамин 516
Этилциклопентан, октановое число 719
Этиноль —• см.' Лаки (т. 2)
Эфиры сложные алифатического ряда 45
ю
Юглон 403
Юнга модуль 163, 205
Яблочная кислота 699
Ядро атома
— масса, прецизионное определение 60
Яра-яра — см. Неролин
Ятрохимия 420, 761
Краткая химическая энциклопедия. Ред. кол. И. Л. Кнунянц
(отв. ред.) и др.т. 3 М., «Советская Энциклопедия»,
1964 (Энциклопедии. Словари. Справочники).
Т. 3. Мальтаза—Пиролиз. 1964. 1 112 стб. с илл.
Сдано в набор 18 октября 1963 г. Том подписан к печати 6 марта 1964 г.
Издательство «Советская Энциклопедия». Москва, Ж-28. Покровский бульвар, д. 8.
Т-00122. Тираж 81 тыс. экз. Заказ № 960. Формат 82Х1081,!.. Объем 343 , физич. п. л. 56,99 усл. п. л.
Уч.-изд. л. 95,85. Цена 1 экз. книги 3 р. 30 к.
Печать с матриц, изготовленных в Первой Образцовой типографии им. А. А. Жданова
Московская типография № 2 «Главполиграфпрома» Государственного комитета Совета Министров СССР по печати.
Москва, Проспект Мира, 10 5. Заказ № 3228