/
























































Теги: химия химическая промышленность химические реакции
Год: 1961




Текст
Выпуск 1 вышел в свет под названием
.Химические реактивы и препараты*
(Методы получения).
СОДЕРЖАНИЕ
n-Фенилкоричная кислота. Р. С. Мильнер, М- И. Довгошея,
Е. А. Преображенская, В. Д. Безуглый.................. 5
₽-Индолкарбоновая кислота. Д. Ш. Розина, Н. Е. Кожевникова,
Н. Д. Сапожкова .................. 8
4-Нитро-З-оксибензойная кислота. А. М. Лукин, Е. Я. Яро-
венко................................................ 10
2,3,4,5-Тетраацетилслизевая кислота и её хлорангидрид.
Г. Н. Дорофеенко .................................... 13
Холиновые эфиры монокарбоновых кислот. Н. Е. Кожев-
никова, Т. И. Генералова, Н. П. Смирнова ....... 15
Индофенилацетат. Н. Е. Кожевникова, Т. И. Генералова,
Н. П. Смирнова............................’ ... . 19
о-Бромнитробензол. А. М. Лукин, Е. Я- Яровенко........... 21
5-Нитро-4-оксн-1,3-диметилбензол. В. М. Дзиомко, И. С. Мар-
кович ............................................... 23
5-Амино-4-окси-1,3-диметилбензол. В. М. Дзиомко, И. С. Мар-
кович .............................................. 24
о-Фениленмалонамид. В. М. Дзиомко, И. С. Маркович .... 25
Салицилоилхлорид. В. М. Дзиомко, И. С. Маркович........ 27
о-Нитробензоилхлорид В. М. Дзиомко, И. С. Маркович ... 28
n-mpern.-Амилфенол. С. П. Старков, В. М. Савоськин,
И. Г. Гах, Д. К. Захарова, Л. Г. Федосюк, Л. Г. Гах . . 29
Бутирофенон. С. П. Старков, М. Г. Сулейманова............ 32
н.-Капрофенон. С. П. Старков, М. Г. Сулейманова, В. Т. Бур-
мистров. И. П. Попова................................ 33
₽-Хлорпропиофенон. С. А. Кочеткова....................... 35
5-Ацетилаценафтен. Р. Д. Бондарчук, Г. Н. Дорофеенко,
Е. П. Бабин, Б. А. Розенберг......................... 37
я-Метоксибензофенон. Г. Н. Дорофеенко, Л. В. Полищук 39
гг-Метоксиацетофеион. Г. Н. Дорофеенко, Л. В. Полищук 40
Высшие первичные спирты. К. В. Вильшау, В. А. Гаврилова 42
н-Хлористые алкилы. В. Г. Брудзь, Д. А. Драпкина, Л. И. Гра-
чева ................................................ 45
N.N-Диметпл-т-нафтиламин. Е. П. Тупикова, В. И. Воронина 46
1,4-Нафтилендиамип. М. В. Березовская, А. А. Прянишников 48
Бромистый бутилгексаметилентетрамин. И. В. Хвостов,
В. В. Цодаков, Р. Т. Хлебородова..................... 50
3
Четвертичные аммониевые основания. В. В. Крохе,
А. И. Вольфсон............................................ 51
Кватерфенил. А. И, Греков.................................... 54
4,4'-Диизопропилдифенил. А. П. Греков.................... 55
2,5-Дифенил-1,3,4-оксадиазол. А. П. Греков................... 57
2-Фенил-5-(4-хлорфенил)-1,3,4-оксадиазол. А. П. Греков .... 58
2-Фенил-5-(4-метоксифенил)- 1,3,4-оксадиазол. А. П. Грекое 60
2,5-Ди-(4-метоксифенил)-1,3,4-оксадиазол. А. П. Греков .... 62
2-Фенил-5-(4-дифенилил)-1,3,4-оксадназол. А. П. Греков .... 63
2,5-Ди-(4-дифенилил)-1,3,4-оксадиазол. А. П. Греков....... 65
2-(1-Нафтил)-5-(4-дифенилил)-1,3,4-оксадиазол. А. П. Греков 67
2,5-Ди-(2-нафтил)- 1,3,4-оксадиазол. А. П. Греков............ 68
2-(2-Фурил)-5-(4-дифенилил)-1,3,4-оксадиазол. А. П. Греков,
Е. 11. Несынов ..................... 70
2-Ацетилфуран. Г. Н. Дорофеенко, В. И. Дуленко, Л. В. По-
лищук . ... .............................................73
2-Ацетотиенон. Г. Н. Дорофеенко, В. И. Дуленко............... 75
Нитрат хннолиния. В. М. Дзиомко, И. А. Красавин.............. 77
8-Нитрохияолин и 5-нитрохинолин. И. А. Красавин,
В. М. Дзиомко . . .................................... 80
8-Аминохинолин. В. М. Дзиомко, И. А. Красавин............. 85
5-Аминохиполин. И. А. Красавин, В. .14. Дзиомко........... 89
5-Хлорхинолин и нитрат 5-хлорхинолиния. И. А. Красавин,
В. 44. Дзиомко................. •....................... 92
5-Хлор-8-нитрохинолин. И. А. Красавин, В. М. Дзиомко ... 96
5-Хлор-8-аминохинолип. В. М. Дзиомко, И. А. Красавин ... 98
4-Этилпиридин. Н. Ф. Казаринова, М. И. Котеленеи,, К. А. Со-
ломко..................................... ........ ] 00
4-(/1-Диметиламинофенил)-пиридии. А. К. Шейнкман, Н. Ф. Ка-
заринова, Е. П. Бабин.................................. 102
Грамин. Н. А. Дзбановский, Е. М. Уринович .................. 104
4,4'-Диоксидифенилсульфон. В. Г. Брудзь, Д. А. Драпкина 107
Бис-(4-окси-3-аминофенил)-сульфон. Д. А. Драпкина,
В. Г. Брудзь............................................. НО
4,4'-Диоксидифенилдиэтилметан. И. М. Билик, А. М. Сереб-
ряный, Н. 44. Бондарец. Р. Л. Глобус, В. Г. Брудзь ... 113
4,4'-Диокси-3,3'-диметилдифенил-2,2-пропан. И. 44. Билик,
Н. М. Бондарец, А. М.Серебряный, Р. Л. Глобус, В. Г. Брудзь 114
л-ФЕ НИЛ КОРИЧНАЯ КИСЛОТА
Р. С. МИЛЬНЕР, М. И. ДОВГОШЕЯ, Е. А. ПРЕОБРАЖЕНСКАЯ,
В. Д. БЕЗУГЛЫЙ
(ВНИИ монокристаллов)
У у у—сн=сн—соон
С^Н^О, М. в. 224, 25
В литературе описано несколько способов получения д-фе-
нилкоричной кислоты. Впервые она получена Хеем [1], кото-
рый, использовав общий метод Перкина для получения кислот,
выделил ее после 8-часового нагревания /г-дифенилальдегида
с уксусным ангидридом и уксуснокислым натрием. Хей также
упоминает об образовании и-'фенилкоричной кислоты при ча-
стичном декарбоксилировании бензилиденмалоновой кислоты,
полученной им из уксуснокислого раствора дифенил альдегида
и малоновой кислоты.
Весьма сложный многостадийный синтез и-фенилкоричной
кислоты был предложен Брауном и Нельеом [2].
При действии натрмалонового эфира на га-фенилбензил-
хлорид образуется эфир л-фенилбензилмалоновой кислоты;
после его омыления из свободной кислоты получают моно-
бромпроизводное, которое сначала частично декарбоксилиру-
ют, а затем дебромируют.
Форлендер [3] получил этиловый эфир л-фенилкоричной
кислоты, выдерживая в течение двух дней при 20° смесь из
л-дифенилальдегида, этилацетата и металлического натрия, а
затем для выделения свободной кислоты провел омыление
эфира спиртовой щелочью.
Все упомянутые авторы нс приводят данных о количест-
венных результатах реакции. Лишь Локк и Байер [4] указали,
что л-фенилкоричная кислота получается по методу Перкина с
выходом 0,3%. Кроме отмеченного, упомянутые способы гро-
моздки и длительны. Заслуживающим внимания оказалось
5
описание «коричного синтеза» по Кновенагелю [4]. Методика
заключается в двухчасовом нагревании равномолярной смеси
ароматического альдегида, малоновой кислоты и пиридина.
Реакция катализируется присутствием небольшого количества
пиперидина. Выход коричных кислот колеблется в широком
интервале; так, выход незамещенной коричной кислоты, по
данным Кновенагеля, достигает 94%, а 2,4,6-триметилкорич-
ная кислота получается с выходом 16%.
Указанный метод мы решили использовать также и для
получения фенилкоричной кислоты.
Для выбора оптимального режима получения п-фенилко-
ричной кислоты мы провели изучение кинетики ее образования
по указанному выше способу. О степени протекания реакции
судили по убыли дифенилальдегида в реакционной массе.
Для этого была разработана методика полярографического
определения дифенилальдегида, которая состоит в следующем.
Навеску пробы реакционной массы в количестве 0,02—0,1 г
помещают в мерную колбу на 10 мл и заливают 1 мл диокса-
па. Раствор доводят до метки метиловым спиртом.
В электролизер помещают 4 мл фона (буферного раствора
с pH 8—12) и добавляют 1 мл приготовленного, как указано
выше, раствора. После пропускания азота в течение 10 минут
проводят полярографирование при подходящей чувствительно-
сти гальванометра. Потенциал полуволны дифенилальдегида
при pH 8 равен 1,45 в против насыщенного каломельного
электрода. Затем производят добавку стандартного раствора
фенилальдегида (0,01 М в метаноле) и вторично проводят
снятие полярограммы. При этом берут такое количество стан-
дартного раствора, чтобы высота волны возросла примерно в
2,5 раза. В этом случае наблюдается минимальная ошибка
определения [5].
Таким образом, расчет концентрации дифенилальдегида
производят по методу добавок.
Получающиеся результаты определения дифенилальдеги-
да вполне воспроизводимы, что подтверждается представлен-
ными в таблице 1 данными.
Таблица 1
Результаты проверки воспроизводимости полярографических определений
дифенилальдегида в реакционной смеси в %
№ образцов № определений
1 2 | 3 4 5
1 2 3 5,9400 1,5215 1,4500 6,1000 | 6,4000 1,5215 1 1,5250 1,3500 । 1,4900 1 ,5235 1,1900 1,6000
li
Результаты кинетических опытов сведены в таблицу 2.
Таблица 2
Кинетика образования я-фенилкоричной кислоты
Время Равномолярные количества я-ди- фепилальдегида, малоновой кислоты и пиридина 15-молярный избыток малоновой кислоты
выдерж- ки в час Количество остаточного я-дифенилальдегида к %
80° 90э 100° 60° 80°
0,25 5,47 5,2] 3,27
0,5 44,87 4,56 1 ,55 4,19 2,з<)
0,75 4,70 — 3,14 2,01
1,0 19,5-1 4.0 0.75 4J1 1,87
2,0 4,98 3,84 0,79 2,00 1,50
3,() 4,73 4,00 0,56 1,92 1,50
4,0 2,40 3,84 — 1,50 —
5,0 2,10 — о.зе — 1,15
6,0 1,70 3,43 0,46 1,40 —
7,0 — — — — 1,14
8,0 0,80 5,00 0,56 1,11 —
9,0 — — 1,06
10,0 0,80 3,76 0,42 0,98
Как видно из таблицы 2. температура оказывает сущест-
венное влияние на скорость реакции образования фенилкорич-
иой кислоты.
На основании полученных кинетических данных предложен
вполне обоснованный эффективный метод получения л-фенил-
коричной кислоты, отличающийся от всех ранее известных
своей простотой. Он заключается в следующем.
В круглодонную колбу, снабженную обратным холодильни-
ком и мешалкой, загружают 18,2 г дифенилальдегида, 10,4 г
малоновой кислоты, 7,9 г пиридина (всех компонентов по
0,1 /И) и несколько капель пиперидина и при перемешивании
нагревают в кипящей водяной бане в течение 2 часов. Реакци-
онная масса, сначала имеющая вид раствора, в конце нагре-
вания представляет собой твердое кристаллическое вещество
светло-коричневого цвета. Эту массу обрабатывают 500 мл
кипяшего толуола (порциями по 100 мл), из которого фенил-
коричная кислота кристаллизуется в виде белых длинных во-
локон.
Выход составляет 20—22 г (90—92% теории), т. пл. 221 —
223°. После повторной кристаллизации из толуола т. пл. 223—
224°, что совпадает с литературными данными.
ЛИТЕР АТУ РА
1. D. Н. Неу, J. Chem. Soc., 1931. 2476.
2. J. И. Braun, I. Nelles. Ber., 66, 1464 (1933).
3. D. Vorldnder, Ber., 68, 45.3 (1935).
4. O. Lock, E. Haver, Ber.. 72, 1064 (1939).
5. Xp. /Нейтанов. Годишник на хим. технол. ин-т т. IV. кн. I. 19—22
(1957)
р-ИНДОЛ КАРБОНОВАЯ КИСЛОТА
Д. Ш. РОЗИНА, Н. Е. КОЖЕВНИКОВА, Н. Д. САПОЖКОВА
(ВНИИ химических реактивов и особо чистых химических веществ)
/Х;—— СООН
N
И
C,H7OsN
М. в. 161, 16
По литературным данным, p-индолкарбоновую кислоту по-
лучают сплавлением fj-мегнлиидола с едким кали [1], окисле-
нием и идол-(3-альдегида щелочным раствором перманганата
калия [2, 3], действием двуокиси углерода на йодистый индо-
лил-магнин [4, 5, 6, 7], карбоксилированием индола ['8, 9, 10].
Согласно прописи [8], индол нагревают с металлическим
натрием в течение 2 часов до 190—200° под током сухого угле-
кислого газа.
При проверке указанной прописи [8] установлено, что ме-
тод трудно воспроизводим и не безопасен.
Нами предложено проводить синтез [3-индолкарбоновой
кислоты в среде диизопропилбензола, что обеспечивает более
гладкое течение реакции и безопасность процесса.
СИНТЕЗ ^-ИНДОЛКАРБОНОВОЙ кислоты
| I | 4- со2 + Na
COONa
-—-COONa
4-НС1--->j I
N
Н
N
H
8
N
H
Характеристика основного сырья
Диизопропилбензол, т. кип. 205—2103, уд. в. 0,857.
Индол, ч„ ВТУ 1064—54.
Натрий металлический, ГОСТ 3273—55.
Соляная кислота, ч., ГОСТ 3118-46.
Условия получения
В круглодонную четырехгорлую колбу емкостью 500 м ?,
снабженную мешалкой, термометром, трубкой для пропуска-
ния углекислоты и обратным .холодильником, помещают 90 -•
индола и 300 мл диизопропилбензола, нагревают на масляной
бане до 70° и загружают 15 г мелко нарезанного металличе-
ского натрия. Перемешивание продолжают при 190—200° г.
течение 3 часов, после чего при этой же температуре 2 часа
пропускают через реакционную смесь углекислый газ, осушен-
ный серной кислотой и фосфорным ангидридом. Содержимое
колбы (см. примечание 1) переносят в фарфоровый стакан
с 800 л л. воды и при перемешивании нагревают в течение 30
минут при 90°. Затем реакционную массу переносят в дели-
тельную воронку, отстаивают и отделяют нижний слой, содер-
жащий натриевую соль p-ипдолкарбоновой кислоты.
Раствор натриевой соли [3-индолкарбоновой кислоты ней-
трализуют до pH 8 разбавленной (1:1) соляной кислотой с
таким расчетом, чтобы кислота не выпала в осадок, прибав-
ляют 5 г активированного угля, нагревают при 70° 15 минут
и фильтруют. Из фильтрата выделяют подкислением соляной
кислотой до pH ~4 |3-индолкарбоновую кислоту. Выпавшие
кристаллы отсасывают, тщательно отжимают, дважды промы-
вают дистиллированной водой (по 20 мл) и сушат в эксикато-
ре над едким натром. Выход 29 г (23,5% теории), т. пл. 208—
210°.
Перекристаллизацию проводят в токе азота в колбе с об-
ратным холодильником и стеклянной трубкой, доходящей до
дна колбы и служащей для ввода азота. 29 г р-индолкарбоно-
вой кислоты растворяют в 700 мл нагретого до 70° 50%-ного
этилового спирта. К полученному раствору розового цвета до-
бавяют 4 г активированного угля и нагревают на кипящей водя-
ной бане в течение 30 минут. Раствор фильтруют горячим,
звн [ вн и HdXiBdauwai ионтвныоя ой" ютвДжвггхо твйтшшф
помещают в лед. р-Индолкарбоновая кислота кристаллизует-
ся в виде белых пластинок. Продукт отсасывают, промывают
50%-ным спиртом (по 20 мл), отжимают и сушат в эксика-
торе над едким натром.
Получают 22.8 г (18,5% теории) (см. примечание 2'1
3-индолкарбоновой кислоты, т. пл. 2)7—218°, что соответствует
литературным данным [8].
9
Примечания.
I. Необходимо удалить кусочки металлического натрия, не вошедшего
в реакцию.
2. Регенерация индола из раствора диизопропилбензола через бисуль-
фитное соединение позволяет увеличить выход fi-индолкарбоновой кисло-
ты до 24% теории, считая на израсходованный индол, и производится сле-
дующим образом:
В аппарат для встряхивания помещают склянку, содержащую
250 мл отработанного диизопропилбензола и 250 мл бисульфита нат-
рия, и взбалтывают в течение 1 часа. Выделившиеся бесцветные
кристаллы бисульфитного соединения индола отфильтровывают, про-
мывают 100 мл метилового спирта (по~- 33 мл) и разлагают 5%-иым
раствором углекислого калия или 5%-ным раствором аммиака при
нагревании до 60°. При охлаждении выпадает индол в количестве
22.3 г.
ЛИТЕРАТУРА
1. Ciamician, Zatti, Вег., 21, 1933, (1888).
2. Ellinger. Вег., 39, 2519 (1906).
3. Pschorr, Hoppe, Вег., 43, 254J (1910),
4. Majiema, Kotake, Вег., 55, 3859 (1922).
5. Allessandry. Gazz. chem. ital., 51, 272 (1921).
6. Oddo, Sessa, Gazz. chim. ital., 41, 1. 234 (1911).
7. Oddo, Gazz. chim Ital., 42, 1. 361 (1912).
8. Weissgerber. Ber., 43, 3526 (1910).
9. Weissgerber, Ber., 46, 651 (1913).
10. Zatti, Ferratinl, Ber., 23, 2296 (1890).
4-НИТРО-З-ОКСИБЕНЗОИНАЯ КИСЛОТА
A. Al. ЛУКИН, E. Я. ЯРОВЕНКО
(ВНИИ химических реактивов и особо чистых химических веществ)
СООН
\ хон
NO.,
C,H5O5N
М. в. 179,07
По литературным данным, 4-нитро-З-оксибензойную кисло-
ту можно получить из 4-нитро-З-оксибензальдегида при воз-
действии на него едкого кали [1] пли из З-оксибензойной кисло-
10
ты нитрованием (в смеси с изомерами) [2]. В свою очередь
3-оксибензойную кислоту получают щелочным плавлением
натриевой соли 3-сул ьфобецзой нон кислоты [3]. Во всех слу-
чаях авторы не приводят подробных условий синтеза.
Для получения 4-нигро-З-оксибензойной кислоты мы оста-
новились на методе синтеза из 3-сульфобензойной кислоты.
Во всех стадиях синтеза были уточнены условия проведения
реакции и выделения получающихся продуктов.
4-Нитро-З-оксибензойная кислота была предложена для
качественного определения нона бария [4], однако широкого
применения этот метод не получил.
СИНТЕЗ 4-НИТРО-З-ОКСИБЕНЗОйНОЙ КИСЛОТЫ
СООН СООН
.1 i
| ч-нгзо, —> | | -J-H2O
SO3H
СООН СООН
I i
I -NaCl-------- j I HCI
\o3H SO3Na
COOH COOH
/\ KOH /\
| I + NaOH —Э- I i _ Na.,so3
X/XSO3Na ' OH
COOH COOH
I I +HNO3 —> I I + H.,0
OH : XOH
NO,
Характеристика основного сырья
Азотная кислота, ГОСТ 701—41.
Бензойная кислота, ч. СТ ГОХП 27/1810.
Олеум 20%, ч. ГОСТ 2184—43.
И
Условия получения
Получение натриевой соли м-сульфобензоиной кислоты.
В трехгорлой колбе, снабженной мешалкой, термометром к
обратным холодильником, нагревают 100 г (0,82 М) бензой-
ной кислоты с 200 г 20%-ного олеума в течение 5 часов при
температуре 210°. По окончании сульфирования (см. приме-
чание 1) массу охлаждают до комнатной температуры и осто-
рожно растворяют в 400 мл холодной воды. Раствор выливают
в 1 литр кипящего, насыщенного при температуре кипения
раствора хлористого натрия; при охлаждении выкристалли-
зовывается натриевая соль лпсульфобензойпой кислоты, кото-
рую перекристаллизовывают из концентрированного раствора
хлористого натрия. Получают 150 г натриевой соли .и-сульфо-
бензойной кислоты.
Получение м-оксибензойной кислоты. В никелевом тигле
(см. примечание 2), снабженном мешалкой, смешивают 50 г
натриевой соли ж-сульфобензойнон кислоты с 20 мл 40°/)-кого
раствора едкого натра; полученную смесь нагревают на водя-
ной бане и добавляют еще 50 г твердого едкого натра. Плав-
ление н размешивание проводят до тех пор, пока смесь не ста-
нет однородной. Плав выливают па противень, по охлаждении
его растирают и вносят отдельными порциями в рас-
плавленную при 200—210° смесь, находящуюся в тигле и со-
стоящую из 30 г едкого кали и 20 г едкого натра. Расплав-
ленную массу выдерживают при хорошем размешивании в
течение 2 часов при той же температуре. Охлажденный плав
растворяют в 400 мл воды; к полученному раствору добавляют
соляную кислоту до прекращения выделения .и-оксибензойной
кислоты; последнюю отфильтровывают и промывают холод-
ной водой. Получают 20 г. Вещество имеет т. пл. 200е—201°.
Получение 4-нитро-З-оксибензойной кислоты. В стакане
емкостью 50 мл растворяют в возможно меньшем количестве
кипящей воды 10 г .и-оксибензойной кислоты и прибавляют
100 мл азотной кислоты уд. в. 1,045. Смесь, окрашенную в ин-
тенсивно красный цвет, кипятят в течение 15 минут. После
охлаждения выпадает оранжевый осадок 4-нитро-З-оксибен-
зойной кислоты в количестве 5 г. Полученную 4-нитро-З-окси-
бензойную кислоту перекристаллизовывают из воды с углем.
Получают 2,2 г, т. пл. 234,5—235е (см. примечание 3).
Найдено %: N 7,52, 7,62.
C7H5O5N Вычислено %: N 7,61.
Примечания.
1. Конец реакции определяют по полному растворению пробы реак-
ционной массы в воде.
2. На результаты щелочного плавления сильно влияет материал аппа-
рата. По нашим данным, эту операцию целесообразно проводить в нике-
левом тигле, причем температурный перепад не должен превышать 15°.
3. По литературным данным, т. пл. равна 230 [1], 234° [2], 234—234'
(51, 238’ [6].
12
ЛИТЕРАТУРА
1. G. Lock, Ber„ 20, 406 (1887).
2. P. Griess, Ber., 62. 1177-88 (1929).
3. H. Offermann, Liebigs Ann. Chetn., 280, 6 (1894).
4. Л. M. Кульберг, P. Б. Лиокуяович, Ж- ана.чит. химии, 4,
255 (1949).
5. U. Froelicher. J. Cohen, J. Client. Soc., 119. 1425-32 (1927).
6. IF. Hodgson. H. Smith, J. Clieni. Soc., 76—78 (1937).
2,3,4,5-ТЕТРААЦЕТИЛСЛИЗЕВАЯ КИСЛОТА И ЕЕ
ХЛОРАНГИДРИД
Г. Н. ДОРОФЕЕНКО
(Институт органической химии АН УССР, Донецкое отделение)
Н ОСОСН, OCOCHS н
I I i I
ноос-с-----с-с---с-соон
1111
ОСОСНз Н Н ОСОСНз
О Н ОСОСНз ОСОСНз н о
II I I ; I II
С1-С-С---С------С------С-С-С1
11^1
ОСОСН3 Н Н ОСОСНз
2. 3, 4, 5-Тетраацетилслизевая кислота получается при аце-
тилировании слизевой кислоты хлористым ацетилом [1] или ук-
сусным ангидридом в присутствии серной кислоты [2].
Ди.хлорангидрид этой кислоты был получен при обработке
тетраацетилслизевой кислоты пятихлористым фосфором в ра-
створе хлористого ацетила [3].
Нами предлагается простой препаративный способ получе-
ния 2,3,4,5-тетрацетилслизевой кислоты, заключающийся в
ацетилировании слизевой кислоты уксусным ангидридом в
присутствии каталитических количеств НСЮ4.
Дихлорангидрид тетраацегилслизевой кислоты образуется
с высоким выходом при кипячении хлороформенного раствора
кислоты с пятихлористым фосфором.
13
СИНТЕЗ 2,3,4,5-ТЕТРААЦЕТИЛСЛИЗЕВОЙ КИСЛОТЫ
И ЕЕ ХЛОРАНГИДРИДА
н он онн
; I I ;
НООС-С-С-С-С-СООН
I I !
онн н он
4(СН5СО)2О
нею.
Н ОСОСНз ососн3 н
। ; I ।
-> HOOC—С------С------С-----C-COOH
I I I !
ОСОСНз Н Н ОСОСНз
Н ОСОСНз ОСОСНз Н
: i i I 2PCL
НООС--С—-----С-----С------С-СООН------->
I , . СНС15
ОСОСНз Н Н ососн3
О H ОСОСНз ососн3 н о
I I I I II
_ С1-С-С-------С-----с-------C-C-CI
i I : i
ОСОСНз Н Н ОСОСНз
траацетилслизевой кислоты отфильтровывают, промывают
водой до полного удаления уксусной кислоты и высушивают
в сушильном шкафу при 100—110°. Выход тетраацетилелпзевой
кислоты 154—160 г (81,5—85% теории), т. пл. 238°. После пе-
рекристаллизации из спирта продукт имеет т. пл. 243°.
Получение хлорангидрида 2,3,4,5-тетраацетилслизевой кис-
лоты. В трехгорлую колбу емкостью 1 л, снабженную обрат-
ным холодильником и механической мешалкой с ртутным за-
твором, помещают 75,6 г (0,2 М) тетраацетилслизевой кисло-
ты, 300 мл сухого хлороформа и 93,6 г (0,45 М} пятихлористо-
го фосфора (вытяжной шкаф’). Реакционную смесь перемеши-
вают и нагревают при слабом кипячении з течение 45 минут.
Смесь охлаждают льдом до 0° и хлорангидрид отфильтровы-
вают на воронке Бюхнера (вытяжной шкаф!), промывают хо-
лодным хлороформом и петролейным эфиром и высушивают
в вакуум-эксикаторе. Выход хлорангидрида 77,1 г (94%
теории), т. пл. 178—179°.
ЛИТЕРАТУРА
1. М. Adelman, J. G. Breckenri^e. Canad. Chem. Research, 24B, 297
(1946).
2. Z. H. Skraup, Monatsh. 14, 488 (1893).
3 O. Diels, E. LOflund, Ber.. 47, 2351 (1914).
4. И. Губен, «Методы органической химии», 3, вып .1, стр. 189, Гос-
химтехиздат, 1934.
Характеристика основного сырья
Слизевая кислота получена окислением D-галактозы азот-
ной кислотой [4], т. пл. 214—215 °.
Уксусный ангидрид, ч.д.а.
Хлорная кислота 57%-ная, х. ч.
Пя гихлористый фосфор, ч.
Условия получения
Получение 2, 3, 4, 5-тетраацетилслизевоЛ кислоты. В трех-
горлую круглодонную колбу емкостью 1 л, снабженную меха-
нической мешалкой и обратным холодильником, помещают
105 г (0,5 М) слизевой кислоты п 420 мл уксусного ангидрида.
К полученной суспензии при перемешивании через отверстие
холодильника из капельной воронки по каплям добавляют
2 мл 57%-ной хлорной кислоты и затем реакционную смесь
•перемешивают в течение полутора часов на водяной бане, на-
гретой до температуры 75—80°. Смесь выливают в 3 л ледяной
воды и оставляют стоять в течение нескольких часов для раз-
ложения избытка уксусного ангидрида. Бесцветный осадок те-
14
ХОЛИНОВЫЕ ЭФИРЫ монокарбоновых кислот
(Ацетилхолинйодид, пропионилхолинйодид,
н-бутирилхолинйодид)
Н. Е. КОЖЕВНИКОВА, Т. И. ГЕНЕРАЛОВА, Н. П. СМИРНОВА
(ВНИИ химических реактивов и особо чистых химических веществ)
R--COO-CH,-CH2 N- |СН3|з,
J-
где R —СН3; С2Н5; н-С,Н,.
По литературным данным, хлориды и йодиды холиновых
эфиров монокарбоновых кислот можно получать нагреванием
холинхлорида с галоидангидридами соответствующих кислот
[1], взаимодействием диметиламиноэтиловых эфиров с йоди-
г>
стым метилом [2] из йодангидридов кислот с алкиленоксндамн
с последующим воздействием триметиламина [3].
Фурно и Пейдж [4] пытались получить холиновые эфиры
карбоновых кислот взаимодействием соответствующих |3-хлор-
этиловых эфиров с триметиламином в бензоле при нагревании
до 100° в запаянных трубках. Однако индивидуальных про-
дуктов авторам выделить не удалось.
Описанные в литературе способы получения холиновых
эфиров карбоновых кислот являются, как правило, многоста-
дийными.
Холиновые эфиры уксусной, пропионовой и масляной кис-
лот широко применяются в качестве субстратов для истинной
и псевдохолинэстераз.
Ацетилхолинйодид наряду с ацетилхолинхлоридом приме-
няется в качестве лечебного препарата [5] и имеет обширную
область применения в медицине.
Нами предложен [8] двустадийный метод получения холи-
новых эфиров уксусной, пропионовой и н-масляной кислот из
Р-йодэтиловых эфиров этих кислот взаимодействием их с три-
метиламином на холоду.
Весьма существенным является то, что 0-йодэтиловые эфи-
ры кислот были получены в одну стадию взаимодействием га-
зообразного йодистого водорода со смесью этиленгликоля с
соответствующими кислотами [6].
Описание метода получения холиновых эфиров дано на
примере н-бутирилхолинйодида.
Температуры кипения полученных р-йодэтиловых эфиров
и температуры плавления холиновых эфиров приведены в
таблице.
СИНТЕЗ н-БУТИРИЛХОЛИНИОДИДА
н-С8Н7СООН-НСН,ОН)2-г-Н.1 —
-> н-С3Н--СОО—CH,-CH2-J4-2H2O
h-C,H-COOCH,-CH,-J+N(CH.,)3 —
н-С3Н7-—COO—СН5—СН, —iN+(CH3)j
J-
Характеристика основного сырья
Йод металлический, ч., ГОСТ 4159- 48.
Фосфор красный, технический, ТУ МХП 518—45.
Подистоводородная кислота, ГОСТ 4200—48.
н-Масляная кислота, ч, ТУ 41—47.
Этиленгликоль, ч, ТУ МХП 2789—55.
Триметиламин, ТУ МХП 2624—51, 20%-ный спиртовый ра-
створ.
16
Условия получения
Получение $-йодэтилового эфира н-масляной кислоты. В
трехгорлую колбу, помещенную в баню с охладительной
смесью (лед с водой), снабженную обратным холодильником,
термометром и стеклянной трубкой, доходящей до дна колбы и
служащей для пропускания газообразного йодистого водоро-
да, помещают смесь 176 г (2 34) н-масляной кислоты и 124 г
(2 М) этиленгликоля. Через смесь в течение 6—7 часов при
температуре не выше 20° пропускают 492 г (3,75 34) йодистого
водорода, что соответствует расходу 24 г красного фосфора
(см. примечания 1 и 2).
Реакционную смесь переливают в делительную воронку н
дважды промывают (по 200 мл) 2%-ным раствором едкого
натра. Слой масла отделяют и трижды промывают водой (по
100 мл) до слабокислой реакции по универсальной индикатор-
ной бумажке. Далее, масло дважды обрабатывают (по
175 .ил) 10%-ным раствором гипосульфита натрия и трижды
(по 100 мл) водой. Масляный слой отделяют и сушат прока-
ленным серпокислым натрием. Получают 290 г масла, которое
подвергают разгонке в вакууме. Собирают фракцию, кипящую
в пределах 99—102715 мм рт. ст. Выход ₽-йодэтилового эфира
н-масляной кислоты 160—165 а (30—33% теории, считая на
масляную кислоту). ,
Получение н-бутирилхолинйодида. В склянку с притертой
пробкой помещают 162 г (0,67 М) fl-йодэтилового эфира
н-масляной кислоты, 200 мл абсолютного серного эфира и
прибавляют 255 е (0,86 34) 20%-ного спиртового раствора
триметиламина. Через сутки наблюдается выделение большого
количества белых кристаллов (примечание 3), которые от-
фильтровывают, промывают 50 мл смеси абсолютного этило-
вого спирта и абсолютного серного эфира (1:1), а затем 50 мл
серного эфира и сушат в эксикаторе над смесью парафина с
хлористым кальцием (1 : I).
Получают 112 г препарата с т. пл. 89—90° (56% теории,
считая на |3-йодэтиловый эфир).
После кристаллизации (см. примечание 4) из абсолютного
этилового спирта выделением абсолютным серным эфиром
получают 100 г препарата (48% теории, считая на |3-йодэти-
ловый эфир), т. пл. 90,1—90,4° (см. примечание 5).
Примечания.
1. Газообразный йодистый водород мы рекомендуем получать по ме-
тоду Филлипса [7] гидролизом йодистых соединений фосфора. Для безо-
пасности следует брать не более 120 г красного фосфора на 2,8 кг йода.
Избыток йода препятствует образованию чрезвычайно взрывчатых соеди-
нений фосфора с водородом, образующихся в результате восстановления
фосфора йодистым водородом.
2. Пропускание йодистого водорода прекращают после того, как при
добавлении к 5 мл реакционной смеси 5 мл воды наблюдается образова-
ние не менее 4,5 мл масла.
17
о
ч
X
sS
с
X
к
св
X
X
X
о
еа
о
X
о
X
с
о.
X
е
л
0)
3
еа
©
х
о
и
X
18
3. При образовании недостаточного количества осадка или выпадении
масла в склянку прибавляют еще 150—200 мл абсолютного серного эфира;
при этом наблюдается мгновенное выделение кристаллического осадка.
4. Анетилхолинйодид кристаллизуется из абсолютного этилового спир
та.
5. Приведенные в литературе [2] температуры плавления йодидов ук-
сусной, пропионовой п н-масляион кислот даны для неперекристаллизо-
ванных препаратов и являются заниженными па 1—2°.
ЛИТЕРАТУРА
1. М. Kauffmann, D. VorlUnder, Вег., 43, 2740 (1910);
Levy-В. Schubar, С. г. Acad. Sci.. 231, 1262 (1950).
'2. L. Tammelin, Acta chem. scand., 10, № 1, 145 (1956).
3. С. И. Ивин, Ж- общ. химии, 22, 267, (1952); 28, 1332 (1958).
4. F. Faurneau, J. Page, Bull. Soc. Chim., 15, 544 (1914)
5. «Государственная Фармакопея», под ред. М. Д, Машковского, 1960.
6. A. Simpson, Liebigs Ann. Chem., 113, 115 (1860).
7. M. A. Phy Hips, Industr. chemist, 38, 759 (1946).
8. H. E. Кожевникова, T. И.Генерилова. Авт. свид. СССР 127663, 1960.
ИНДОФЕНИЛАЦЕТАТ
(Ы-(4'-ацетоксифенил)-п-хинонимин)
Н. Е. КОЖЕВНИКОВА, Т. И. ГЕНЕРАЛОВА, И. П. СМИРНОВА
(ВНИИ химических реактивов и особо чистых химических веществ)
О=ф \-N- / \-ОСОСН3
CuHnOjN — М. в. 241, 21
По литературным данным, индофенил ацетат может быть
получен методом Хирша [1, 2] сочетанием N-хлорхинонимина с
фенолом в щелочной среде с последующим ацилированием;
методом Хеллера [3, 4], окислением гипохлоритом натрия сме-
си фенола и солянокислого и-аминофенола с последующим
ацилированием получающейся натриевой соли индофенола.
Некоторыми исследователями отмечены трудности, возни-
кающие при попытке воспроизвести данные методы [5].
Х-(4'-ацетоксифенил)-п-хинонимин и различные его произ-
водные представляют интерес в качестве хромогенных субст-
ратов, служащих для определения ацетилхолинэстеразной ак-
тивности [4, 6], а также для препаративного синтеза.
В основу метода получения N-(4/-ацетоксифенил)-н-хино-
нимина положен метод Хеллера [3].
19
СИНТЕЗ М-(4‘-АЦЕТ0КСИФЕНИЛ)-я ХИНОНИМИНА
ОН
/\ 2NaC1° /~\ /— \
I |ОН , । .-------> О — , = N — ч /ONa
1 I I NaOH ' —' 4—z
NH3-HC1
0=^ /=N-( ^-ONa-h(CH3CO)2 0 -
-> 0=( \=xN-<^ 2~OCOCH3'
Характеристика основного сырья
Фенол, ч., ГОСТ 6417-52.
п-Аминофенол солянокислый, ТУ МХП 2787—51.
Хлорная известь, ГОСТ 210—53.
Едкий натр, ГОСТ 4328—48.
Уксусный ангидрид, ч., ГОСТ 5815—52.
Условйя получения
Получение натриевой соли индофенола. В 30 мл воды раст-
воряют 9,4 г (0,1 М) фенола, прибавляют 33 г 35%-го раство-
ра едкого натра. Отдельно растворяют 14,7 г (0,1 М) соляно-
кислого /г-аминофенола в 95 мл воды. Затем сливают оба рас-
твора вместе и охлаждают до нуля.
В фарфоровый стакан емкостью 1 л, снабженный мешал-
кой, помещают 90 г хлористого натрия и 300 мл концентриро-
ванного раствора гипохлорита натрия (см. примечание 1) и
добавлением мелкоизмельчснного льда температуру снижают
до минус 10°. При этой температуре и сильном размешивании
к раствору гипохлорита натрия быстро приливают смесь ра-
створов фенола с п-амипофеполом.
Содержимое стакана окрашивается в синий цвет, и вскоре
наблюдается выделение взвешенного осадка натриевой соли
индофенола. Через 30 минут осадок натриевой соли индофено-
ла отфильтровывают и промывают (дважды по 25 мл) насы-
щенным раствором поваренной соли.
Препарат сушат вначале на воздухе на пористой пластин-
ке, затем в вакуум-эксикаторе над фосфорным ангидридом.
Полученная натриевая соль индофенола представляет собой
темно-зеленый блестящий порошок, пригодный для дальней-
шего использования.
Выход сухой натриевой соли индофенола 16 г (71,7% тео-
рии, считая на n-аминофенол солянокислый) (см. примеча-
ние 2).
20
Получение П-(4'-ацетоксифенил)-п-хинонимина. В кругло-
донную колбу, снабженную обратным холодильником, поме-
щают 24 г уксусного ангидрида, смешанного с 2 г хорошо рас-
тертого безводного углекислого натрия. Через 5 минут при
взбалтывании вносят 4 г натриевой соли индофенола, предва-
рительно высушенной (см. примечание 3). При этом реакци
онная смесь окрашивается в оранжево-красный цвет. Смесь
нагревают до 58—60° и при этой температуре выдерживают
15 минут. Затем содержимое колбы охлаждают до комнатной
температуры и выливают на 200 мл ледяной воды. Мгновенно
выпадает осадок оранжевого цвета, который отфильтровывают
и промывают дистиллированной водой (дважды по 25 мл).
Препарат сушат в вакуум-эксикаторе над едким натром.
Выход сухого продукта 2,95 г (67,1% теории, считая на на-
триевую соль индофенола). Препарат кристаллизуют из ли-
гроина или н-октана. Выход сухого препарата 2,5 г (56,8%
теории), т. пл. 115—116°, что соответствует литературным дан-
ным [3].
Примечания.
1. Гипохлорит натрия удобнее всего готовить по .методу, описанному
Карякиным и Ангеловым [7]. Расход гипохлорита должен точно соответст
г.овать 2 грамм-атомам активного кислорода.
2. Натриевая соль индофенола может быть перекристаллизована из
смеси этилового спирта и серного эфира.
3. Натриевая соль индофенола должна быть обезвожена высушиваии-
ем в вакуум-эксикаторе над фосфорным ангидридом.
ЛИТЕРАТУР А
1. A. Hlrsch, Вег., 13, 1903 (1880).
2. В. Cohen, Public Health Reports, № 74 (1929).
3. G. Heller, Ann., 392, 28 (1912).
4. D. Kramer, R. Gamson, J. Organ. Chem., 24. 11, 1742 (1959).
5. M. M. Brooks, J. Amer. Chem. Soc., 53, 1826 (1931).
6. Nachlas. Joung, Seligman, Hystocliem and Cytochem, 5 565 (1957).
7. Ю. В. Карякин, И. И. Ангелов. «Чистые химические реактивы». Го<>
хнмиздат, 1953. стр. 421.
О -БРОМНИТРОБЕНЗОЛ
А. М. ЛУКИН. Е. Я. ЯРОВЕНКО
(ВНИИ химических реактивов и особо чистых химических веществ)
C,H4O,NBr М. в. 202,02
По литературным данным, о-бромнитробензол получают из
бромбензола нитрованием [1] или из о-нитроанилина разложе-
21
нием бромистого о-нитробензолдиазония [2]. В основу нашей
работы был положен последний метод. При проверке были
внесены некоторые уточнения в условия синтеза.
СИНТЕЗ о-БРОМНИТРОБЕНЗОЛА
I | NH2+NaNO2 + 2HBr - I I N2Br-%NaBr + 2H.,O
'x/l-NO2 \J~NO2
Си -J- Cu2Br2
----------
+ NT2
Характеристика основного сырья
о-Нитроанилин, ТУ 2639—51.
Кислота бромистоводородная, ГОСТ 2062—43.
Медь бромистая, ТУ МХП 86—47.
Медь в порошке, не ниже 98%-ой.
Условия получения
Диазотирование о-нитроанилина. В тре.хлитровый фарфо-
ровый стакан, помещенный в баню для охладительной смеси и
снабженный мешалкой, термометром и капельной воронкой,
загружают при размешивании 400 мл воды, 650 мл (3,2 Л4)
бромистоводородной кислоты и 207,3 г (1,5 М) о-нитроанили-
на. Затем в баню помещают охладительную смесь (лед с пова-
ренной солью), а к реакционной массе добавляют 300 г мелко
толченного чистого льда, чтобы температура реакционной сме-
си была не выше +5°. При этой температуре при размешива-
нии добавляют по каплям раствор 114 г (1,6 А4) нитрита нат-
рия в 264 мл воды, контролируя реакцию диазотирования по
йодкрахмальной бумаге. По окончании диазотирования раст-
вор выдерживают при той же температуре в течение 30 минут.
Полученное диазосоединение немедленно во избежание ос-
моления используют для синтеза о-бромнитробензола.
Получение о-бромнитробензола. В трехлитровый фарфоро-
вый стакан, снабженный мешалкой, загружают 350 мл (1,4 А4)
бромистоводородной кислоты, 112 г (0,78 А4) бромистой меди
и 7,5 г меди в порошке. К полученной реакционной смеси при,
размешивании в течение 1 часа добавляют вышеописанный
раствор диазосоединения. После загрузки реакционную массу
перемешивают еше в течение 1 часа и оставляют до следую-
щего дня для полного разложения. Выпавший о-бромнитро-
бензол отфильтровывают и сушат на воздухе. Получают
302,5 г технического продукта (99,7% теории). Полученное
22
вещество подвергают перекристаллизации, для этого в круг-
лодонную колбу емкостью 250 мл, снабженную обратным хо-
лодильником, загружают 100 г технического о-бромнитробен-
зола, 100 мл 90%-ой уксусной кислоты и 6 г активирован-
ного угля. Реакционную смесь выдерживают при температуре
кипения в'течение 30 минул, отфильтровывают в горячем со-
стоянии, фильтрат охлаждают, выпавший осадок вновь от-
фильтровывают и сушат на воздухе. Получают 75 г чистого
о-бромнитробензола, что составляет 75%, считая на техниче-
ский о-бромнитробепзол. Вещество имеет т. пл. 38—39° (см.
примечание).
Примечание.
По литературным данным, температура плавления о-бромнитробензо/и
38° [3], 41-41,5° [4], 41-42° [5], 43,1° [6].’
ЛИТЕРАТУРА
1. J. Dabble, D. Marsden, J. Chem. Soc., 73, 254 (1898).
2. Л Ullmann, Ber., 29, 1880 (1896).
3. В. И. Меишуткин, Ж.Р.Ф.Х.О., 41, 1071 (1909).
4. Tittig, Mager, Ber., 7, 1179 (1874).
5. Holleman, Recneil trav. chim., 19, 365 (1901).
6. Korner, Gazz. chim. ital., 4, 333 (1874).
5-НИТРО-4-ОКСИ-1,3-ДИМЕТИЛ БЕНЗОЛ
В. M. ДЗИОМКО, И. С. МАРКОВИЧ
(ВНИИ химических реактивов и особо чистых химических веществ)
сн,
H,C/'4'/X'NO2
С.чН^О, М. в. 167,16
По литературным данным, 5-нитро-4-окси-1,3-диметилбен-
зол получают нитрованием асил.л-ксиленола [1, 2]; диазоти-
рованием лг-ксилидина в среде серной кислоты с последующим
разложением диазосоединения в среде серной и азотной кис-
лот [31; действием азотистой кислоты на 2-окси-3,5-диметил-
бензиловый спирт [4].
Нами осуществлен синтез 5-нитро-4-окси-1,3-диметилбензо-
ла путем диазотирования асилмьксилидина в среде азотиой
23
кислоты с последующим разложением диазосоединения.
сн3 сн3
NaN0* Л/0Н
I I HNO, I I
Н:;С/Х/ H3C/XXXNO3
Условия получения
Получение 5-нитро-4-окси-1,3-димеп1лбензола. К 12,1
(0,1 М) аспл.лг-ксилидина в смеси с 30 мл азотной кис-
лоты (уд. в. 1,34) и 60 мл воды быстро добавляют
раствор 6,9 г (0,1 (И) нитрита натрия в 20 мл воды при темпе-
ратуре 0—5J. После окончания диазотирования диазораствор
отфильтровывают и оставляют при комнатной температуре
на 2 часа, затем его нагревают при 50° в течение 3 часов на
водяной бане. Выпавший светло-желтый осадок отделяют о г
тонкой смолистой пленки (см. примечание 1), отфильтровыва-
ют и сушат при 40°. Выход 10,5 г (63% теор.). Желтые иго-
лочки, т. пл. 69°. После 3-х последовательных перекристалли-
заций из спирта т. пл. 69—70° (см. примечание 2).
Примечания.
1. Из смолистого продукта после перегонки с паром можно получить
дополнительное количество 5-нитро-4-окси-1,3-диметнлбензола с т. пл. 69°.
2. По литературным данным, температура плавления 68,5° [1]: 72° [2, 31:
78° 14]. '
ЛИТЕРАТУРА
1. S. Ltiko, Liebigs Ann. Chem., 182, 32 (1876).
2. N. R. Hodgkinson, L. Limpach, J. Chem. Soc... 63, 105 (1893).
3. Br. Francke. Liebigs Ann. Chem., 296, 199 (1897).
4. K. Fries, K. Kann,Liebigs Ann. Chem., 353, 354 (1907).
5-АМИН0-4-0КСИ-1,3-ДИМЕТИЛБЕН30Л
В. M. ДЗИОМКО, И. С. МАРКОВИЧ
(ВНИИ химических реактивов и особо чистых химических веществ)
СИ,
/\ /он
1 А
H3CZ X NHa
CsH^NO М. в. 137,18
5-Амино-4-окси-1,3-диметилбензол получают восстановле-
нием 5-нитро-4-окси-1,3-диметилбензола цинком в соляной
кислоте с последующим выделением из его солянокислой соли
[1] или восстановлением 2-окси-3,4-диметилазобензола гидро-
сульфитом натрия [2].
24
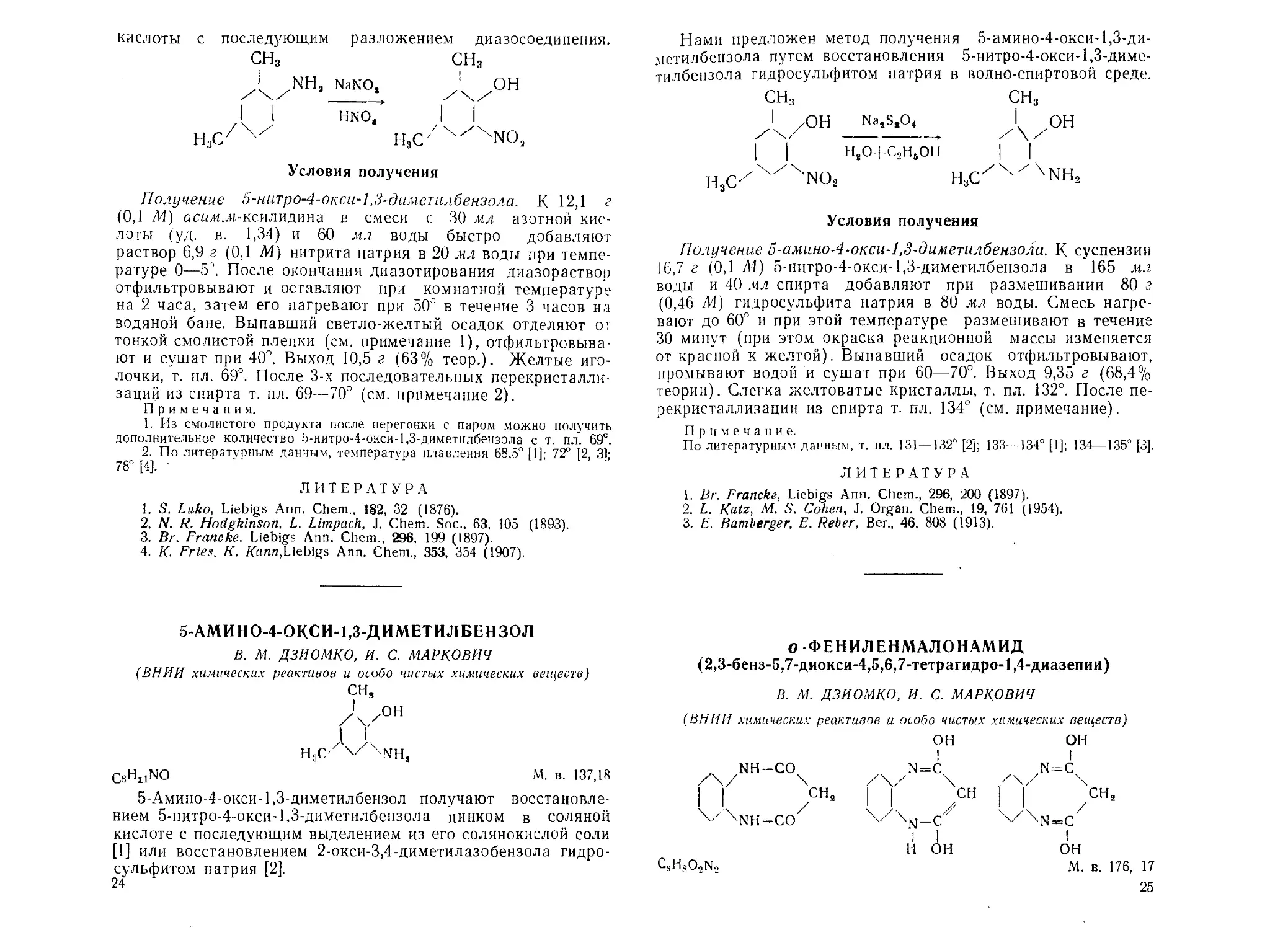
Нами предложен метод получения 5-амино-4-окси-1,3-ди-
мстилбензола путем восстановления 5-нитро-4-окси-1,3-диме-
тилбензола гидросульфитом натрия в водно-спиртовой среде.
СН3
Na2SsO4
I I H2O+C2HSO1I
H3C/X/4NOa
СН3
х0Н
h3c/x/xnh2
Условия получения
Получение 5-амино-4‘Окси-1,3-диметилбензола. К суспензии
16,7 г (0,1 /И) 5-нитро-4-окси-1,3-диметилбензола в 165 мл
воды и 40 мл спирта добавляют при размешивании 80 г
(0,46 М) гидросульфита натрия в 80 мл воды. Смесь нагре-
вают до 60° и при этой температуре размешивают в течение
30 минут (при этом окраска реакционной массы изменяется
от красной к желтой). Выпавший осадок отфильтровывают,
промывают водой и сушат при 60—70°. Выход 9,35 г (68,4%
теории). Слегка желтоватые кристаллы, т. пл. 132°. После пе-
рекристаллизации из спирта т. пл. 134° (см. примечание).
Примечание.
По литературным данным, т. пл. 131—132° [2], 133—134° [1]; 134—135° [3].
ЛИТЕРАТУРА
1. Br. Francke, Liebigs Ann. Chem., 296, 200 (1897).
2. L. Katz, M. S. Cohen, J. Organ. Chem., 19, 761 (1954).
3. E. Bamberger, E. Reber, Ber., 46, 808 (1913).
О ФЕНИЛЕНМАЛОНАМИД
(2,3-бенз-5,7-диокси-4,5,6,7-тетрагидро-1,4-диазепии)
В. M. ДЗИОМКО, И. С. МАРКОВИЧ
(ВНИИ химические: реактивов и особо чистых химических веществ)
/4/NH-COx
| I сн2
X-/XNH-CO/
C9HsO2N2
I I
и он
он
М. в. 176, 17
25
По литературным данным, о-фениленмалонамид получаюч
взаимодействием о-фенилендиамина с малоновым эфиром [1|
или малоновой кислотой [1, 2].
Нами проверен первый из указанных методов получения.
СИНТЕЗ о- ФЕНИЛ ЕНМАЛОНАМИДА
О
с2н5о-сх
+ СН2 -
с2н5о-сх
о
/О
NH—-С/
СН2 + 2С2Н,ОН.
nh~cC
Условия получения
В трехгорлую колбу емкостью 100 мл, снабженную мешал-
кой, обратным холодильником и термометром, загружают
10,80 г (0,1 М) о-фенилендиамина, 8,0 г (0,05 М) малонового
эфира, 20 мл концентрированной соляной кислоты и 30 мл во-
ды. Смесь нагревают до 103—106° и при этой температуре раз-
мешивают в течение 2,5 часов. Затем реакционную массу ох-
лаждают до комнатной температуры, отфильтровывают оса-
док, промывают его водой и сушат при 70и. Получают 2,4 г
Препарат представляет собой бесцветные кристаллы, субли-
мируется при 360° (литературные данные: температура субли-
мации 360° [1], 240° (0,68 мм рт. ст.) [2].
Растворим в 20—30% NaOH, диметилформамиде, не раст-
ворим в обычных органических растворителях, воде. Вступает'
в реакцию азосочетания.
ЛИТЕРАТУРА
1. R. L. Shriner. Р. G. Boermans, J. Amer. Chem. Soc.. 66, 1810(1944).
2. J. Bitchi, H. Dietrich, E. Eichenberger. Helv. Chim. acta, 39, 957 (1956).
САЛИЦИЛОИЛХЛОРИД
(Салициловой кислоты хлорангидрид)
В. М. ДЗИОМКО, И. С. МАРКОВИЧ
(ВНИИ химических реактивов и особо чистых химических веществ)
/s^/COCl
С,Н5ОЙС1 М. в. 156,16
По литературным данным, салицилоилхлорид получают
взаимодействием тионилхлорида с салициловой кислотой [1]
или салициловокислым натрием [2]. Реакцию рекомендуется
проводить в присутствии третичных аминов [3]. В последнем
случае реакцию проводят в среде насыщенных алифатических
углеводородов с т. кип. 30—60°. '
Проверка описанного метода получения салицилоилхлори-
да взаимодействием салициловой кислоты с тионилхлоридом
не дала положительных результатов. Проведение же этой ре-
акции в присутствии третичного амина позволило получить
салицилоилхлорид. В отличие от литературных данных нами
в качестве катализатора использован триэтиламин.
СИНТЕЗ САЛИЦИЛОИЛХЛОРИДА
/Ч/СООН СОС1
I | + SOC12 — | | + НС1 + so.
х/хон хон
Условия получения
В круглодонную колбу, снабженную мешалкой с ртутным
затвором, обратным холодильником, капельной воронкой и
термометром, загружают 28 г (0,21 М ) салициловой кислоты,
150 мл сухого петролейного эфира и 2 мл (0,014 М) триэтил-
амина. К суспензии при размешивании прибавляют в течение
15 минут 25 г (0,22 А4) хлористого тионила, смесь нагревают
до 80°, размешивают при этой температуре 2,5 часа, после че-
го обратный холодильник заменяют нисходящим и отгоняют
петролейный эфир. В колбе остается 21—21,5 г (66,5% тео-
рии) салицилоилхлорида, т. зам. 14,5°—19,0°.
Примечав и е.
По литературным данным, т. пл. 14—173 [3], 19—19,5° [4].
ЛИТЕРАТУРА
1. DRP 284161; Frdl., 12, 667.
2. DRP 262883; Frdl., 11, 211.
3. Пат. США 2 899 458; РЖХим, 1960, 85795 П.
4. Словарь органич. соед. III, 613.
27
О-НИТРОБЕНЗОИЛХЛОРИД
(о- Нитробензойной кислоты хлорангидрид)
В. М. ДЗИОМКО, И. С. МАРКОВИЧ
(ВНИИ химических реактивов и особо чистых химических веществ)
/Х/СОС1
\/ЧчМО2
C-H4OjNC1 ‘ М. в. 185, 57
По литературным данным, о-нитробензоилхлорид получа-
ют взаимодействием о-нитробензойной кислоты с тионилхло-
ридом [1] или пятихлористым фосфором [2].
Проверен второй метод получения.
СИНТЕЗ о-НИТРОБЕНЗОИЛХЛОРИДА
+ РС16
СОС1
| | +РОС13 + НС1
Характеристика основного сырья
о-Нитробензойная кислота, ч., ГОСТ 1192—55.
Пятихлористый фосфор, ч.
Условия получения
В трехгорлую колбу емкостью 150 мл, снабженную мешал-
кой с ртутным затвором, обратным холодильником и термо-
метром, загружают 37,5 г (0,2 Л4) о-нитробензойной кислоты,
нагревают ее до 90° и в течение 20 минут добавляют 50 г
(0,24 М) пятихлористого фосфора; температура при этом не
должна превышать 110°. Реакционную массу' размешивают
еще 1,5 часа при 90—95°, после чего обратный холодильник
заменяют нисходящим и отгоняют хлорокись фосфора в ваку-
уме, создаваемом водоструйным насосом. Остаток переносят
в колбу Кляйзена и перегоняют в вакууме (см. примечание).
Собирают фракцию с т. кип. 150° (12 мм рт. ст.) или 140—146°
(5—6 мм рт. ст.). Выход 35 г (84% теории).
Примечание.
Перегонку о-нитробензоилхлорида следует вести при равномерном на
гревании на масляной бане. При перегреве возможно разложение о-нитро
бензонлхлорида, проходящее со взрывом.
28
ЛИТЕРАТУРА
1. К. Auwers, W. Daniel, J. prakt. Chem., (2), 110, 259.
2. G. Lockemann, H. Rein, Chem, Ben, 80, 488 (1947).
n-mpem. АМИЛ ФЕНОЛ
С. П. СТАРКОВ, В. М. САВОСЬКИН, И. Г. ГАХ, Д. К. ЗАХАРОВА,
Л. Г. ФЕДОСЮК, Л. Г. ГАХ
(ВНИИ химических реактивов а особо чистых химических веществ,
Донецкий филиал)
СпН1вО
М. в. 164, 24
пд'рег.Амилфенол получают алкилированием фенола
изоамиловым и третичным амиловым спиртами в присутствии
хлористого цинка [1], хлористого алюминия [2], хлорного желе-
за [3], молекулярных соединений фтористого бора [4] и серной
кислоты [5].
Для получения /г-трет.амилфенола используют также и
изоамилены, применяя в качестве катализаторов серную кис-
лоту [6] и ее смесь с уксусной кислотой [7], фтористый бор и
его молекулярные соединения [8].
Мы применили метод алкилирования фенола третичными
амиленами в присутствии катионита КУ-2, описанный Исагу-
лянцем и Славской р9], с целью выделения их из пентан-ами-
леновой фракции, и метод алкилирования на том же катали-
заторе изоамиловым спиртом, описанный для изобутилового
спирта [10].
Преимуществами катионита КУ-2 перед другими катали-
заторами являются чистота получаемого продукта, легкость
отделения алкилата от катализатора и возможность исполь-
зования его без регенерации несколько раз.
29
СИНТЕЗ п-трет. АМИЛФЕНОЛА
ОН
Ы+С5н1о
он
КУ-2
Н3С-С-СН3
С2Н5
ОН он
I i
/Х| КУ-2
| | +СН3-СН-СН2-СН3ОН--------* | 1+н3о
Х/ сн5 V
Н3С-С-СН3
С2115
Характеристика основного сырья
Фенол, т. пл. 40,3°, т. кип. 177,5—178° (740 щм).
Третичные амилены, т. кип. 31—38° (750 мм), п20о 1,3876
d420 0,6623.
Изоамиловый спирт, ч.д.а., т. кип. 128—132°.
Катионит КУ-2 в Н-форме с обменной емкостью 3,86
мг-экв/г, обезвоженный кипячением с толуолом.
Условия получения
Получение п-трет.амилфенола из фенола и третичные
амиленов. В четырехгорлую колбу емкостью 0,75 л, снабжен-
ную механической мешалкой, термометром, барботером для
подачи амиленов и обратным холодильником (см. примеча-
ние 1), помещают 25 г катионита КУ-2 и 70,5 г (0,75 М) фено-
ла. Колбу нагревают на масляной бане до 135° и вводят в те-
чение 25—30 минут 35 г (0,5 .И) газообразных амиленов (см.
примечание 2). После десяти-пятнадцатиминутной выдержки
при 135—145° (см. примечание 3) реакционную смесь охлаж-
дают до 90°, алкилат осторожно декантируют и растворяют В|
1,5 г 1,5%-ного водного раствора едкого натра. Выделившееся
масло отделяют, а щелочной раствор амилфенола промывают
несколько раз небольшими порциями четыреххлористого угле-
рода и обрабатывают углекислым газом. Выпавшие белые
кристаллы п-трет. амилфенола отфильтровывают, промы-
30
вают водой и сушат в вакуум-эксикаторе над пятиокисыо фос-
фора (см. примечание 4). Выход 48,6 г (59% теории). Т. пл.
86___89°. После однократной перекристаллизации из н-октана
получают продукт с т. пл. 92—93°.
Получение п-трет.амилфенола из фенола и изоамило-
вого спирта. В трехгорлую колбу емкостью 0,75 л, снабжен-
ную механической мешалкой и обратным холодильником с
ловушкой типа Дина и Старка, помещают 10 г катионита
КУ-2, 47 г (0,5 /И) фенола и 44 г (0,5 Л4) изоамилового спир-
та. Перед началом опыта ловушку заполняют изоамиловым
спиртом. Затем колбу нагревают на масляной бане. При тем-
пературе 130° реакционная смесь закипает, и в ловушке начи-
нает отделяться вода. Через 6—8 часов нагревания выделение
воды практически заканчивается.
В горячем состоянии алкилат осторожно декантируют с
катионита и растворяют в 2 л 1,5%-ного водного раствора ед-
кого натра. Нерастворившийся масляный слой отделяют, а ще-
лочной раствор обрабатывают описанным выше способом.
Выход п-трет. амилфенола 35 г (43% теории). Т. пл. 86—
88°. После однократной перекристаллизации из н-октана
92—93°.
/г-трет.Лмилфецол представляет большой практический
интерес для синтеза алкил- и арилформальдегидных смол,
полностью растворяющихся в углеводородах и высыхающих
маслах.
Примечания.
1. Обратный холодильник должен охлаждаться водой с температурой
пе выше 5—10°.
2. В качестве алкилирующего реагента может быть использована и
смесь амиленов, полученная при дегидратации изоамилового спирта. Вы-
ход’ п-трет. амилфенола в этом случае не превышает 52% теории,
Т. пл. 81,6°.
3. Необходимо следить за тем, чтобы темпера гура в реакционной кол-
бе не поднималась выше 150—155°.
4. Потери /1-трет.амилфенола с маточником не превышает 1 г.
ЛИТЕРАТУРА
1. Ad. Liebman, 15, 150, 1990 (1882); 14, 1844 (1881); В. Fischer,
В. Griltzner, Вег., 26, 1646 (1893).
2. И. П. Цукерваник, 3. Н. Назарова, Ж. общ. химии, 5,767 (19351:
7, 623, (1937).
3. 3. Н. Назарова, И. П. Ну кер в аник, Ж. общ. химии, 10, 1151 (1940).
4. И. А. Ромадан, Г. Т. Стипниек, Ж. общ. химии, 30, 2193 (1960).
5. А. Г. Белороссова, М. И. Фарберов, Уч. зап. Ярославского техноло-
гии. ин-та, 2, 21 (1957); Пат. ФРГ 943707; РЖХим, 1957, № 8, 28189 п.
6. М. Г. Имаев, Изв. вузов, Нефть и газ, № 6, 77 (1960).
7. W. Koenigs. Вег., 23, 3145 (1890), /?. Anschutz, Н. Beckerhoff,
Liebigs Ann. Chem., 327, 218 (1903).
8. А. В. Топчиев, Б. М. Тумерман, В. Н. Андронов, Л. И. Коршунова,
Нефт. хоз., 7, 65 (1954); А. В. Топчиев, Я. М. Паушкин и М. В. Курашев,
Докл. АН СССР, 30, № 3, 559 (I960).
31
9 В И Исагулянц, Н. А. Славская, Ж. прикл. химии, 33, № 4, 95Э1
(I960).
10. В. Я. Исагулянц, Авт. свид. СССР 110953, 1.957.
БУТИРОФЕНОН
С. П. СТАРКОВ, М. Г. СУЛЕЙМАНОВА
(ВНИИ химических реактивов и особо чистых химических веществ,
Донецкий филиал)
/\-CO-Clh- СН.- СН.
1| lit
C„HnO '' м. в. 118,20
Бутирофенон получают ацилированием бензола бутирил-
хлоридом [1] и масляной кислотой [2] в присутствии хлористо
го алюминия. Кроме прямого ацилирования, бутирофенон по
лучают также и косвенными способами, к числу которых отно^
сятся пропускание смеси паров масляной и бензойной кислот1
над закисью марганца при температуре 400—450° [3], дезамш
нирование 2-амино-1-фенил-бутанола-1 [4], присоединение
фенилмагнийбромида к пропилнитрилу с последующим разлоэ
жением полученного продукта [5].
СИНТЕЗ БУТИРОФЕНОНА
С3Н7СООН PSOCI2 C3H7COC1+SO2+HC1S
Al Cl,
С3Н;СОС1-4-С8Н6-----> C3H7COC6H5 |-HCI
Характеристика основного сырья
Масляная кислота, т. кип. 161° (748 мм), dd'‘ 0,957,
1,3990.
Тионил хлористый, т. кип. 75° (745 мм).
Бензол, ч.д.а, т. кип. 79,5—79,7° (745 мм). d>2!' 0,8751.
Бутирилхлорид, т. кип. 99—102° (750 л.ч).
Алюминий хлористый, технический.
Условия получения
Получение бутирофенона. В четырехгорлую колбу ем-
костью 1 л, снабженную механической мешалкой с ртутным
* См. примечание 2 к методике получения н-капрофенона (стр. 35)
32
затвором, обратным холодильником, капельной воронкой и
термометром, вносят 160 г (1,2 /И) хлористого алюминия, к
которому при температуре не выше 6—8° сначала прибавляют
106,5 г (1 Л1) бутирилхлорпда, а затем порциями в течение ча-
са 195 г (2,5 Л1) высушенного бензола (см. примечание 1). По
окончании прибавления бензола реакционную смесь медленно
нагревают до 30° и выдерживают при этой температуре 0,5
часа. Затем охлажденную массу выливают на смесь 1 —1,2 кг
измельченного льда с 8—10 мл концентрированной соляной
кислоты. Выделившийся бутирофенон экстрагируют 2—3 раза
небольшими порциями эфира, экстракт промывают водой, сла-
бым раствором карбоната натрия и сушат сульфатом натрия.
После этого экстракт фильтруют в колбу Кляйзена, из кото-
рой сначала отгоняют эфир и избыток бензола, а затем при
уменьшенном давлении бутирофенон, кипящий в пределах
114—115° (18 мм). Выход 126 г (85% теории) (см. примеча-
ние 2). Содержание основного вещества 98—99,5% (см. при-
мечание 4 к методике получения н-капрофенона, стр. 35).
Бутирофенон представляет интерес как продукт для синте-
за третичных ароматических спиртов, арилкетоноспиртов и
других соединений.
Примечания.
1. Холодильник и капельную воронку закрываю! хлоркальциевыми
трубками.
2. Пользуясь этой методикой, но прибавляя к 160 г (1,2 А1) хлористо-
го алюминия 106,5 г (1 Л!) изобутнрилхлорида и 195 г (2,5 .<И) высушен-
ного бензола при температуре не выше 0—5° и выдерживая реакционную
смесь в течение 0,5 часа при 10°, можно получить изобутирофенон с выхо-
дом 84% теории с содержанием основного вещества 98—99,5%.
ЛИТЕ Р А Т У Р А
1. Е. Burcher, Ann. Chim. Phys., 5, (26), 468 (1882),
2. M. С. Малиновский, А. А. Ляпина, Ж. общ. химии, 11, 168 (1941).
3. Sabatier, Mellhe. С. г. Acad. Sci., 158. 830 (Ю14).
4. М. Т. Tlffenlau, Levy, С. г. Acad. Sci. 183, 969 (1926).
5. R. Z. Schriner, T. A. Turner. J. Amer. Cliem. Soc., 52. № 3, 1627
(1930).
н-КАПРОФЕНОН
С. П. СТАРКОВ, M. Г. СУЛЕЙМАНОВА, В. T. БУРМИСТРОВ,
И. П. ПОПОВА
(ВНИИ химических реактивов t: особо чистьо химических веществ.
Донецкий филиал)
|/’\-СО-СН.-СН.-СН.-СН, - СН.
С1аН16О М. в. 176,25
Основным методом получения н-капрофенона является ре-
акция конденсации н-капроилхлорида с бензолом в присутст-
33
вии хлористого алюминия [1]. Кроме прямого ацилирования,
н-капрофенон может быть получен пропусканием смеси паров
капроновой и бензойной кислот над МпО при температуре
400—450° [2], дезаминированием 2-амино-1-фенилгексанола-1
[3], каталитическим гидрированием сорбофеноиа [4], присоеди-
нением н-амилмагнийбромида к бензонитрилу или фенилмаг-
нийбромида к амилнитрилу с последующим разложением по-
лученных продуктов [5].
СИНТЕЗ Н-КАПРОФЕНОНА
C5HUCOOH + SOC12 - C5HnCOCl+HCl+SO2
А1С1,
С5НиСОС1+С6Нв--------> C5HUCOC6H5+HC1
Характеристика основного сырья
н-Капроновая кислота, т. кип. 125° (40 мм), d^ 0,9260,
л20о 1,4160.
Тионил хлористый, т. кип. 75° (745 мм). -
Бензол, ч.д.а., т. кип. 79,5—79,7“ (745 мм), 0,8751.
н-Капроилхлорид, т. кип. 148—153° (740 мм).
Алюминий хлористый, технический.
Условия получения
Получение н-капроилхлорида. В сухую четырехгорлую кощ
бу емкостью 1 л, снабженную механической мешалкой с ртут-
ным затвором, капельной воронкой, термометром и обратным
холодильником, помещают 116,16 г (1 М) н-капроновой кис-
лоты (см. примечание 1). Затем нагревают ее на водяной бане
до 95° и постепенно при хорошем размешивании прибавляют
в течение 20—25 минут 154,7 г (1,3 М) хлористого тионила. По
окончании прибавления реакционную смесь выдерживают в
течение часа при 95°, охлаждают, переносят в колбу Фаворско-
го и перегоняют. н-Капроилхлорид собирают в интервале
148—153° (740 мм). Выход 108,9 г (82,0% теории) (см. приме-
чание 2).
Получение н-капрофенона. В четырехгорлую колбу ем-
костью 1 л, снабженную механической мешалкой с ртутным
затвором, обратным холодильником, капельной воронкой и
термометром, вносят 160 г (1,2 М) хлористого алюминия, к ко-
торому при температуре не выше 20—25° сначала прибавляют
сразу 134,5 г (1 41) капроил.хлорида, а затем порциями 117 г
(1,5 М) высушенного бензола (см. примечание 1). По оконча-
нии прибавления бензола реакционную смесь медленно нагре-
вают до 50° и выдерживают при этой температуре в течение
часа. Затем охлажденную массу выливают на смесь 1—1,2 кг
34
измельченного льда с 8—10 мл концентрированной соляной
кислоты. Выделившийся н-капрофенон экстрагируют 2—3 ра-
за эфиром. Экстракт промывают водой, слабым раствором
карбоната натрия и сушат сульфатом натрия. После этого
экстракт фильтруют в колбу Кляйзена, из которой сначала
отгоняют эфир и избыток бензола, а затем при уменьшенном
давлении н-капрофенон, кипящий в пределах 107,8—108,Зэ
(3 мм). Выход 146 г (85% теории) (см. примечание 3). Содер-
жание основного вещества 98—99,5% (см. примечание 4).
н-Капрофенон представляет интерес как промежуточный
продукт для синтеза инсектицидов, третичных ароматических
спиртов, арилкетоноспиртов и других соединений.
Примечания.
1. Холодильник и капельную воронку закрывают хлоркальциевыми
трубками. '
2. При аналогичных условиях, но с уменьшенным на 0,1 М количест-
вом хлористого тионила, были получены u-валерил-, изовалсрил,- бути
рил- и изобутирилхлориды с выходами 62,5—93,8% теории.
3. Пользуясь этой методикой, можно получить н-валерофенон (выход
81% теории, т. кип. 107—107,5° (6 леи) из 120,5 г н-валерилхлорида и 117 г
бензола; изовалерофепон (выход 81 % теории, т. кип. 107,2—107,5° (10 мм)
из 120,5 г изовалерилхлорида и 117 г бензола. Содержание основного ве-
щества 98—99,5% (см. примечание 4).
4. Количественное определение н-капро-, н-валеро-, изовалеро-, бутиро-
и изобутирофенона проводилось так же, как это было описано Новиковой
и Петровой [6], только вместо 0,5 н. раствора солянокислого гидрокси-
ламина, с которым оьсимированпе идет не полностью, применялся 1 н. рд-
створ этого реагента, а реакционная смесь при нагревании на кипящей во-
дяной бане периодически встряхивалась через каждые 10 минут.
ЛИТЕРАТУРА
1. Schroeter, Вег., 40, 1603 (1907); РЖХим, № 7, 21392, 1958.
Р. L. Breusch, М. Ogazer, Вег., 87, 1225 (1954).
2. Sabatier, Meilhe, С. г. Acad. Sci, 158, 830 (1914).
3. М. Т. llffenlau, Levy, С. г. Acad. Sci., 183, 969 (1926).
4. Kuhn Richard, H. A. Staab, Angew. Chemie, 65, 371 (1953).
5. R. L Schrlner, T. A. Turner, J. Amer. Chem. Soc., 52 (193u);
РЖХим, 1957, № 20, 65981,
6. E. H. Новикова, Л. H. Петрова, Ж- прикл. химии 23, 1336 (1950).
р-ХЛОРПРОПИОФЕНОН
С. А. КОЧЕТКОВА
(ВНИИ химических реактивов и особо чистых химических веществ)
СОСН,СН,С1
I
С9Н,ОС1 М. в. 168, 63
Р-Хлорпропиофенон получают ацилированием бензола
хлорангидридом p-хлорпропионовой кислоты в присутствии
3 _ 35
хлористого алюминия в среде сероуглерода [1] или. в избытке
бензола [2, 3].
Проверен и подтвержден последний метод получения.
СИНТЕЗ £ -ХЛОРПРОПИОФЕНОНА
ЗС1СН2СН2СООН + РС13 — ЗС1СН2СН,СОС1+Н3РОз
СОСН.2СН2С1
I
| |ч-С1СН2СН2СОС1 Ж’- I I +НС1
'-‘6 Г1 в \ /
Характеристика основного сырья
Алюминий хлористый безводный, ГОСТ 4452—48.
Бензол, ч.д.а., ГОСТ 5955—51.
н-Гексан, МХ-ТУ-271—59.
Кальций хлористый плавленный, ГОСТ 4460—48.
Фосфор треххлористый, ГОСТ 91—41, уд. в. 1,58.
Р-Хлорпропионовая кислота, т. затв. 39°.
Условия получения
Получение Q-хлорпропиофенона. В круглодонную колбу ем-
костью 250 мл. снабженную обратным холодильником, загру-
жают 75,0 г (0,7 Л4) p-хлорпропионовой кислоты и добавля-
ют 67,5 г или 42,3 мл (0,5 М) треххлористого фосфора. Содер -
жимое колбы нагревают на кипящей водяной бане в течение
1,5 часа, охлаждают до комнатной температуры, декантируют
и разбавляют 90 мл абсолютного бензола (см. примечание 1(.
В трехгорлую литровую колбу, снабженную мешалкой с
ртутным затвором, обратным холодильником и капельной -во-
ронкой, защищенными хлоркальциевыми трубками, помещают
112,5 г безводного хлористого алюминия и 210 мл абсолютно-
го бензола, перемешивают и постепенно из капельной воронки
за ~2 часа прибавляют бензольный раствор хлорангидрида
[З-хлорпропионовой кислоты. Затем реакционную массу нагре-
вают при размешивании на водяной бане (80—90°) 3 часа,
охлаждают до комнатной температуры и постепенно при раз-
мешивании выливают на лед (---500 г).
В делительной воронке емкостью 1,0 л отделяют водный
слой, бензольный несколько раз промывают сначала водопро-
водной, потом дистиллированной водой (по 150 мл) и сушат
над прокаленным хлористым кальцием. Избыток бензола ис-
паряют током нагретого воздуха (см. примечание 2).
36
Высушенный бензольный раствор загружают в колбу Вюр-
ца, снабженную термометром, погруженным в раствор, барбо-
тером для пропускания горячего воздуха, холодильником Ли-
биха с приемником, соединенным с вакуум-системой, и отго-
няют избыток бензола при температуре 6—10°. Остаток после
отгонки бензола охлаждают до 6°, выпавшие светло-желтова-
тые кристаллы отфильтровывают и сушат над безводной сер-
нокислой медью. Выхад 68,0 г (70% теории), т. пл. 46—48°.
После перекристаллизации из н-гексана т. пл. 47—48°, что со-
ответствует литературным данным [2].
При перекристаллизации fj-хлорпропиофенон растворяют в
н-гексане при комнатной температуре (соотношение (3-хлор-
пропиофенона к н-гексану 1:30), нерастворившийся осадок
отфильтровывают, фильтрат охлаждают в ледяной бане, вы-
павшие белые кристаллы p-хлорпропиофенона сушат над без-
водной сернокислой медью. Выход после перекристаллизации
60% теории.
Примечания.
1. Бензол абсолютируют сушкой над прокаленным хлористым каль.
цисм с последующей перегонкой и сушкой над металлическим натрием.
2. Воздух подогревают до 70—90° в змеевике, погруженном в кипя-
щую водяную баню.
ЛИТЕРАТУРА
1. I. Kennez, S. Statham, J. Chem. Soc., 299 (1935).
2. Campbell, La Forge, J. Organ. Chem., 14, 1, 348 (1949).
3. 5. Searles, K. Pollart, E. F. Lutz, J. Amer. Chem. Soc., 79, 950 (1957).
5-АЦЕТИЛАЦЕНАФТЕН
P. Д. БОНДАРЧУК, Г. H. ДОРОФЕЕНКО, Е. П. БАБИН,
Б. А. РОЗЕНБЕРГ
IИнститут органической химии АН УССР, Донецкое отделение)
Н2С—сн2
сосн3
Основным методом получения ацетилаценафтена является
ацетилирование аценафтена хлористым ацетилом в сероугле-
37
роде [1] или нитробензоле [2] в присутствии безводного хлори-
стого алюминия; иногда в качестве ацетилирующего агента
применяют уксусную кислоту. При этом используют в качест-
ве катализатора и одновременно растворителя безводный фто-
ристый водород [3].
Во всех описанных случаях образуется смесь изомеров 3-
и 5-ацетнлаценафтена, причем 5-ацетилаценафтен существует
в двух взаимнопревращаемых формах, плавящихся при 57э и
70°; в присутствии же безводного фтористого водорода обра-
зуется исключительно 3-ацетилаценафтеи.
По предлагаемой методике ацетилирование проводят ук-
сусным ангидридом в присутствии незначительных каталити-
ческих количеств безводного хлорнокислого магния (ангидро-
на).
По данному способу получается 5-ацстилаценафтен с тем-
пературой плавления 57°. Выход 5-ацегилаценафтена состав-
ляет 75—82%.
5-Ацетилаценафтен представляет интерес как промежуточ-
ный продукт для синтеза 5-вииилаценафтена.
СИНТЕЗ 5-АЦЕТИЛАЦЕНАФТЕ'НА
Н2С—сн2 Н2С —сн2
I
| I ! — (СН3СО)2О . | V ; +сн3соон
г
СОСНз
Характеристика основного сырья
Аценафтен, т. пл. 94°.
Уксусный ангидрид, ч.д.а., г. кип. 140'.
Магний хлорнокислый, безводный, ч.
Метанол, х.ч., т. кип. 64,7°.
Раствор карбоната натрия, насыщенный.
Бензол, ч.д.а., т. кип. 79,5—79,7° (745 мм).
Условия получения
В круглодонную колбу емкостью 500 мл с обратным холо-
дильником загружают 46,2 (0,3 М) аценафтена, 42,4 лгл‘
(0,45 .М) уксусного ангидрида и 3,3 г (0,015 М) ангидрона.
Реакционную смесь нагревают при кипении в течение 30 минут,'
после чего для разложения избытка уксусного ангидрида осто-
рожно добавляют ЮО мл воды и кипятят еще 2—3 минуты;
Охлажденную реакционную смесь экстрагируют 120 мл бен-’
38
зола. Органический слой промывают двумя порциями (по
50 мл) насыщенного раствора карбоната натрия, водой, а за-
тем раствор высушивают безводным сульфатом натрия. Раст-
воритель отгоняют на водяной бане, оставшуюся вязкую тем-
ную массу перегоняют в вакууме. При температуре 216—220е
и давлении 20 мм рт. ст. собирают 5-ацетилаценафтен. Выход
неочищенного продукта 47,5 г (81,6% теории).
Продукт достаточно чист для большинства работ. Для
окончательной очистки кетона его перекристаллизовывают из
метанола и получают продукт с температурой плавления 57°.
Примечав и е.
Ангидрон может быть заменен 57—60%-нои хлорной кислотой. При
ат ом расход хлорной кислоты составляет 0,01 М на 1 М аценафтена Реак-
цию проводят при нагревании в течение 30 минут на водяной бане при тем-
пературе 60—70".
ЛИТЕРА ТУ Р А
1. С. Graebe, Р. Haas, Liebigs Ann. Chem,, 327, 91 (1903).
2. L. F. Fieser, E. B. Hersberg, J. Amer. Chem, Soc.. 61, 1272. (19391.
3. D. T. Mowrv, M. Renoll, W. F. Huber, ,1 Amer. Chem. Soc.. 68, 1105
(1946).
л-МЕТОКСИБЕНЗОФЕНОН
Г. H. ДОРОФЕЕНКО, Л. В. ПОЛИЩУК
(Институт органической химии АН УССР, Донецкое отделение)
СН,0 -/ СО—У
/7-Метоксибензофенон получают по реакции Густавсона-
Фриделя-Крафтса при действии бензоилхлорида на анизол в
присутствии хлористого алюминия в растворе сероуглерода
[1], или взаимодействием бензола с га-анизоилхлоридом с этим
же катализатором [2]. Описано получение п-метоксибензофе-
нона при реакции анизола с бензойным ангидридом в присут-
ствии молярных количеств хлорной кислоты [3]. Хорошие ре-
зультаты дает метод бензоилирования анизола хлористым
бензоилом в присутствии алюминия [4] или йода [5].
СИНТЕЗ п-МЕТОКСИБЕНЗОФЕНОНА
, —, —. нею,
сн3о-/ \ л- / \-СОС1----------
СН3О- -СО — у: НС
39
Характеристика основного сырья
Анизол, ч.
. Хлористый бензоил, технический, перегнанный.
Хлорная кислота, 57%-ная, х. ч.
Условия получения
В круглодонную колбу емкостью 250 мл, снабженную об-
ратным холодильником с хлоркальциевой трубкой, помещают
54 г (0,5 Л4) анизола и 105 г (87 мл, 0,75 /И) хлористого бен-
зоила. К полученной смеси постепенно, при перемешивании,
добавляют 4,38 г (0,025 Д4) хлорной кислоты и нагревают со-
держимое колбы при кипении в течение 3 часов. В течение
всего времени кипения наблюдается выделение паров хлори-
стого водорода. После охлаждения реакционной смеси ее пе-
реносят в колбу Кляйзена и перегоняют при пониженном дав-
лении (60—80 мм) в вакууме водоструйного насоса (т. кип.
240°—280°). При этом отгоняется немного бензойной кислоты и
кетон, которые собирают в один приёмник. Отогнанный сырой
продукт промывают равным объемом 10%-ного раствора ще-
лочи, сушат над хлористым кальцием, фильтруют, помещают
в колбу Кляйзена и перегоняют в вакууме. При разгонке по-
лучают 84,5 г (79,7% теории) /г-метоксибензофенона с т. кип.
190—195° (11 мм). Кетон затвердевает в светло-желтую мас-
су с т. пл. 59—60°.
Примечание.
В аналогичных условиях был получен п-этоксибснзофепон с выходи г
80% теории.
ЛИТЕРАТУРА
1. L. Gattermann, R. Ehrhardt, Н. Maisch. Ber., 22, 1129 (1889).
2. А. Орехов, Bull. Soc. chim. France (4), 47, 621 (1930).
3. H. Burton, P. F. G. Prail, J. Chem. Soc., 1951, 529.
4. Докл. АН Арм. ССР, 29, № 3, 111 (1959)
5. I. A Kave, H. C. Klein, W. J. Burlant, J. Amer. Chem. Soc., 75,
№ 3, 745 (1953).
и-МЕТОКСИАЦЕТОФЕНОН
Г. И. ДОРОФЕЕНКО, Л. В. ПОЛИЩУК
(Институт органической химии АН УССР, Донецкое отделение)
СН30-<'~>-С0 СН,
/г-Метоксиацетофенон получают ацетилированием анизола
хлористым ацетилом [1, 2], уксусным ангидридом [3, 4] или ке-
40
теном [5] в присутствии обычных катализаторов реакции
Густавсона-Фриделя-Крафтса: хлористого алюминия [1, 2],
хлористого цинка [6], фтористого бора и его комплексов [3],
хлорной кислоты [7, 8] и асканита [9].
Предлагаемый способ получения н-метоксиацетофенона за-
ключается в ацетилировании анизола уксусным ангидридом в
присутствии незначительных количеств хлорнйй кислоты. Спо-
соб прост, удобен и позволяет быстро приготавливать п-мето-
ксиацетофенон и другие п-алкоксиацетофенопы с хорошими
выходами.
СИНТЕЗ л-МЕГОКСИАЦЕТОФЕНОНА
z —, НС1О4
СН,О-( \ + (СН3СО)2О
СН3О-/“ ^-СОСН3+СП3СООН
Характеристика основного сырья
Анизол, ч.
Уксусный ангидрид, ч.д.а.
Хлорная кислота, 57%-ная, х. ч.
Условия получения
В круглодонную колбу емкостью 250 мл, снабженную об-
ратным холодильником, помещают 54 г (0,5 Л1) анизола и
76,5 мл (0,75 Л1) уксусного ангидрида. К полученному раст-
вору по каплям, при перемешивании, прибавляют 2,5 г 60 % -
ной хлорной кислоты.
Добавление хлорной кислоты вызывает разогревание реак-
ционной смеси до 60—65° и окрашивание раствора в оранже-
во-коричневый цвет. Реакционную смесь выдерживают при
комнатной температуре в течение. 30 минут, а затем выливают
в 500 мл ледяной воды. При этом выделяется маслообразный
продукт и немного кристаллического вещества темно-оранже-
вого цвета, которое отделяется фильтрованием. Маслообраз-
ный.продукт реакции экстрагируют бензолом. Бензольную вы-
тяжку промывают разбавленным раствором едкого натра, во-
дой и сушат над обезвоженным сульфатом натрия. Бензоль-
ный раствор фильтруют с целью освобождения от сульфата
натрия, помещают в колбу Вюрца и перегоняют. При разгон-
ке получают 45,5 г (60%) п-метоксиацетофенона с т. кип.
254—258°. При охлаждении продукт затвердевает в кристалли-
ческую массу с т. пл. 35—37°.
Примечание.
При ацетилировании фенетола и бутилового эфира фенола уксусным
ангидридом в присутствии хлорной кислоты на кипящей водяной бане в те-
41
чение 30 минут могут быть получены ч-этоксиацетофенон и п-бутоксиаие-
тофенон с выходом 50% и 50% соответственно. Температура' кипения
п-этоксиацетофенона 143—144" (15 мм); температура кипения л-бутоксиаце-
тофенона 159—161’ (12 л.и).
ЛИТЕРАТУРА
1. Р. М. Baranger, Bull. Soc. chiin., 49. 1213 (1931).
2. A. F. Holleman, Recueil trav. chim., 10, 215 (1891).
3. H. Meerwein, D. Vossen. J. Amer. Chem. Soc., 67, 284 (1945).
4. C. R. Holler, R. Adams, J. Amer. Chem. Soc., 46, 1889 (1924).
5. C. D. W. Hurd, J. Amer. Chem. Soc., 47, 2777 (1925).
6. H. Button, P. F. G. Prail, J. Chem. Soc., 1951, 726.
7. H. Burton, P. F. G. Prill. J. Chem. Soc., 1950, 1203.
8. Г. И. Дорофеенко, X. общ. химии, 31, 994 (1961).
9. В. И. Белов, Л. А. Хейфиц, Авт. свид. СССР № 416562, 1950 г.
ВЫСШИЕ ПЕРВИЧНЫЕ СПИРТЫ
нормального строения с числом углеродных атомов
более десяти
К. В. ВИЛЬШАУ. В. А. ГАВРИЛОВА
(ВНИИ химических реактивов и особо чистых химических веществ)
Индивидуальные высшие спирты находят широкое приме-
нение в исследованиях по химии поверхностноактивных ве-
ществ, химии высокомолекулярных соединений, как препара-
ты для газожидкостной хроматографии, а также как исходные
соединения для получения чистых альдегидов, кетонов, кислот,
аминов и представителей других классов органических
веществ.
Синтез индивидуальных высших спиртов очень сложен, ис-
ходные продукты трудно доступны.
В то же время отечественная промышленность освоила про-
изводство спиртов гидрирования—смесей спиртов, получаю-
щихся в результате гидрирования соответствующих фракций
синтетических жирных кислот.
Спирты гидрирования представляют собой смесь первич-
ных спиртов нормального строения с небольшой примесью уг-
леводородов (до 8%), воды (2—3%) и спиртов изомеров
(5-10%).
. Индивидуальные высшие спирты были получены нами раз-
делением смеси спиртов гидрирования при помощи ректифика-
ции в вакууме.
42
Характеристика нормальных первичных спиртов от Си до
проект ТУ * со ' О о оо СМ ч'_-х 00 0,8145-55 (4) 0,8165-75 ( rfs°3 \ "20 ) 0,8195-205 (4) 0,8210-20 (4°) 0,8150—60 при 60° 0,8165-75 при 60°
Удельный вес литер. rf50 м20 сТ ОС с О LO QO i 0,8157(17] 0,8162[8] । 0,8176] 14] U1106l8‘0
эксперим. ( ) • 6858‘0 №) О518‘О 0,8170—79 0,8197 -2011 (4) 0,8216 0,8206 ( (I50 1 ( "20 ) 0,8157 0,8154 при 60° 0.8167 при 60° 1
i. ф со проект ТУ . не ниже . 14,5 23—24 29-31 35,5 -.39,0 СО 47,5-50 51,5-54
лпература девання литер, (т. пл.) 5 о 23,95[4] 3 38,26[4] 39,5[7 ] 44|11] 46(9] 49,5112] 50,0|13] 53,5(16] 54[11]
£ (Зэпэяб 116,0 14,7 23.0 30,5 ,30,7 чС= со 44.2 44,1 ю о СО Т" CS ГО ю ю
ЯОНО1В 'trod -Э1Л1Л огонь СМ 2 23 О
43
Для этой цели была применена насадочная колонка диа-
метром 22 мм и высотой ректифицирующей части 1,200 м.
Насадка—трехгранные спиральки из стальной проволоки
d = 0,2 мм. Диаметр элемента насадки 2,5 мм. Число витков
3—4. Сталь марки 1Х18Н9Т.
Ректифицирующая часть колонки снабжена шестисекцион-
ным обогревом, регулируемым латрами. Куб обогревается ба-
ней со сплавом Вуда.
Режим работы: 1. Время работы «на себя» 90—100 минут.
2. Скорость отбора 25-—35 мл/час.
3. Флегмовое число 11 —12.
В этих условиях производят отбор фракций, руководст-
вуясь, главным образом,характером изменения температуры
затвердевания погона. Пока идет основная фракция,температу-
ра затвердевания держится постоянной или изменяется по по-
логой кривой, имеющей максимум. Заметное понижение темпе-
ратуры затвердевания погона до минимальной точки с после-
дующим повышением характеризует отбор промежуточной
фракции. Температура затвердевания каждой промежуточной
фракции меньше температуры затвердевания предыдущей и
последующей основных.
Отобранные в результате нескольких операций первичной
ректификации фракции соответственно объединяются и под-
вергаются повторной ректификации, в результате которой по-
лучаются спирты, имеющие константы, приведенные в таб-
лице.
Рассмотрение данных таблицы позволяет квалифицировать
полученные спирты как «очищенные» (содержание ’основно-
го вещества от 90 до 95%).
ЛИТЕРАТУРА
1. Annales de chimie 19, 490 (1944).
2. .1. Chem. Soc., 1818 (1948).
3. Fette. Seifen., 49, 572 (1942).
4. J. Organ. Chem., 9. 268 (1944).
5. J. Amer. Chem. Soc., 70, 1637 (1948).
6. J. Amer. Chem. Soc.. 55. 1577 (1933).
7. Слов. орг. соед., И. Л.. 1949, 11, 824.
8. Докл. АН СССР, 68, 52 (1949).
9. К. Бауэр, Анализ орг. соед. И. Л. 1953.
10. Trans. Faraday Soc.. 44, 464 (1948).
11. J. Chem. Soc., 1660 (1934):
12. Beilstein, Hl, E Ill, 1816.
13. Angew. Chem. 68, 641 (1956).
14. J. Amer. Chem. Soc., 60, 1230 (1938).
15. Beilstein. Hl, Ell, 466.
16. Chem. Abstrs., 50, 9993 f (1956).
17. Fette. Seifen, 53, 680 (1951).
44
н ХЛОРИСТЫЕ АЛКИЛЫ
(типовой метод)
В. Г. БРУДЗЬ, Д. А. ДРАПКИНА, Л. И. ГРАЧЕВА
(ВНИИ химических реактивов и особо чистых химических веществ)
Сп Нзл-1 С1
В промышленности н-хлористые алкилы получают взаимо-
действием хлористого тионила с-соответствующими спиртами.
Недавно [1] было опубликовано сообщение о каталитическом
действии диметилформамида на реакцию кислот с хлористым
тионилом. Применение нами диметилформамида в синтезе
хлористых алкилов также дало значительный эффект, а имен-
но: позволило снизить температуру реакции с 60—70° до 20—
25°; сократить продолжительность синтеза с 7 часов (для С3
и С4), 12—14 часов,(для Се и С8) и 40 часов (для С5 и С?) до
двух часов для любого из спиртов от Сз до С8; повысить выхо-
да на 20—35%.
СИНТЕЗ ХЛОРИСТЫХ АЛКИЛОВ
C^Hj^OH 4- SOC12 --------------> СЛН,Я_!С1 + SO2 + НС1
Характеристика основного сырья
Диметилформамид, техн., ВТУ ЕУ 111—57.
н-Спирт (см. таблицу).
Тионил хлористый, ч., II сорт, ВТУ № АУ 84—56.
Условия получения
Получение н-хлористых алкилов. В колбу, снабженную ме-
шалкой с ртутным затвором, обратным холодильником, ка-
пельной воронкой и термометром, загружают 135 г 96%-ного
(1,1 ^И) хлористого тионила и 0,08 г (6,01 Л4) диметилформа-
мида. При 20—25° и размешивании прибавляют в течение 1,5
часов 1 моль соответствующего спирта, после чего размешива-
ют еще 30 минут при той же температуре. Затем отсоединяют
обратный холодильник и ртутный затвор с мешалкой, встав-
ляют насадку типа Вюрца, соединенную с нисходящим холо-
дильником, и постепенно, по мере ослабления выделения хло-
ристого водорода, повышают температуру до кипения и отго-
няют образовавшийся хлористый алкил. Дистиллат промыва-
ют 5%-ным раствором едкого натра до нейтральной реакции
45
на бумажку конго. Продукт, высушенный над хлористым каль-
цием, фракционируют.
В таблице приведены данные, характеризующие реакцию
получения отдельных хлористых алкилов.
Исходный спирт Загрузка спир- та на 135 г 96%-ного хлорист. тиони- ла и 0.08 г диме- тил формамида Темпер, отбора фракций чистого продукта Выход хлорист. алкилов Соответ- ствует требова- ниям ВТУ №
г % тео- рии
г [ °/0 содерж.
н-Пропиловый спирт очищ. ГОСТ 15/23-54 63,3 95 45,5 47° 45,6 58 3398-52
н-Бутиловый спирт техн. ГОСТ 5208-50 80,0 92,7 77—78° 69,5 75 2640—51
н-Амиловый спирт очищенный ВТУ № РУ 1298-57 90,0 98,0 106-109° 82,1 77 1366 - 57
н-Гексиловый спирт очищенный ВТУ № РУ 1299—57 106,0 96,0 132-137° 96,5 80 970 - 53
н-Гептиловый спирт ч. ВТУ № РУ 1324-57 120,0 98,0 154-160° 111,5 83 1365-57
н-Октиловый спирт очищенный ВТУ № РУ 130-57 133,0 98,0 81-83° при 20 мм рт. ст. 132 89 132-57
ЛИТЕРАТУРА
Н. Н. Bosshard, К- Могу, М. Schmid, И. Zollinger, Helv. chim. acta,
42, 1653 (1959).
N,N-flHMETHJI а НАФТИЛАМИН
E. П. ТУПИКОВА, В- И. ВОРОНИНА'
(ВНИИ химических реактивов и особо чистых химических веществ
N(CH3),
CKH13N М. в. 171,23
По литературным данным, NN-диметил-а-нафтиламин мо-
жно получить нагреванием u-нафтиламина с йодистым мети-
46
лом и метиловым спиртом [1, 2], нагреванием а-нафтиламина
с метиловым эфиром п-толуолсульфокислоты [3] или метили-
рованием а-нафтиламина с помощью диметилсульфата [4, 5].
Нами для получения NN-диметил-а-нафтиламина был вы-
бран метод, основанный на метилировании а-пафтиламина ди-
метилсульфатом [5], как наиболее простой и дающий хороший
выход препарата.
СИНТЕЗ N.N-ДИМЕТИЛ- а -НАФТИЛАМИНА
nh2
| I I + (CH3)aSO4 +
2NaHCO3
------->
N(CH3)2
4- Na2SO44-2CO2+2H,O
Характеристика основного сырья
Диметилсульфат, ч., ГОСТ 3180—52.
а-Нафтиламин, ч., ГОСТ 8827—58.
Условия получения
В трехгорлую колбу, снабженную мешалкой, термометром
и газоотводной трубкой, загружают 151,2 г (1,8 М) двуугле-
кислого натрия, 300 мл воды и 87 г (0,6 М) а-нафтиламина.
Смесь перемешивают, охлаждают и при температуре 5—73
прибавляют 144 мл (1,5 М) диметилеульфата.
После 1,5-часового размешивания реакционной смеси при
указанной температуре выделение углекислого газа прекра-
щается.
Избыток диметилеульфата разлагают нагреванием в тече-
ние 45 минут при температуре 55—60°.
К полученной смеси аминов приливают 200 мл (2 М) хло-
роформа и переносят в делительную воронку, тщательно
встряхивают и отделяют хлороформенный слой, который про-
мывают водой (3 раза по 50 мл), отделяют промывные воды,
отгоняют хлороформ. К остатку прибавляют 30 мл (0,3 М)
уксусного ангидрида (см. примечание), смесь нагревают на
кипящей водяной бане в течение 20 минут, охлаждают и при-
ливают 75 мл петролейного эфира. Выпавший осадок удаля-
ют фильтрованием, а фильтрат, содержащий раствор М,М-ди-
47
метил-а-нафтиламина в петролейном эфире, промывают 5%-
ным раствором едкого натра и высушивают над едким нат-
ром. NN-Диметил-а-нафтиламин разгоняют в вакууме при
13 мм остаточного давления, отбирают фракцию, кипящую в
пределах 139—140°.
Выход чистого продукта -—73 г (70% теории). Получен- '
пый NN-диметил-а-нафтиламин имеет т. кип. 275,2°/760 мм
рт. ст.; d420 1,040; я20о 1,6200.
Найдено %: С 85,1; Н 8,2.
Ci2Hi3N Вычислено: % С 84,17; Н 7,65.
Примечание.
Смесь амилов подвергают обработке уксусным ангидридом для удале-
ния не вступившего в реакцию а -нафтиламипа и образовавшихся следов
метил-а -нафтиламина.
ЛИТЕРАТУРА
1. Z. Landshoff, Вег., 11, 643 (1878).
2. A. Hantzsch, Вег.. 13, 1348 (1880).
3. В. М. Родионов, В. Е. Введенский, Хим. пром-сть, 7, I, 11 (1930).
4. F. Germuth. Chem. Zbl., 1, 2338 (1929).
5. S. Httnlg, Chem. Ber., 85, 1056 (1952).
1,4-НАФТИЛ ЕНДИАМИН
Al. В. БЕРЕЗОВСКАЯ, А. А. ПРЯНИШНИКОВ
(ВНИИ химических реактивов и особо чистых химических веществ)
C10H10N2
М. в. 150,20
В литературе описан ряд методов получения 1,4-нафтилен-
диамина: восстановлением нитро-амипо-ацетонафталина оло-
вом в соляной кислоте [1]; восстановлением бензол-азо-а-на-
фтиламина гидросульфитом натрия [2] или цинковой пылью
[3]; восстановлением азо-динафтилдиамина оловом в соляной
кислоте [4]; декарбоксилированием нафтилендиамин-1,4-карбо-
повой-(2)-кислоты [5] без указания на выход 1,4-нафтиленди-
амина.
48
Нами в основу получения 1,4-нафтилендиамина был поло-
жен метод Бамбергера [3], заключающийся в восстановлении
бензол-азо-альфа-нафтиламина (нафтиловый красный) цинко-
вой пылью в уксусной кислоте.
Характеристика основного сырья
Бензол-азо-альфа-нафтиламин (нафтиловый красный),
инд., т. пл. 125—126°, ТУ МХП 1660—47.
Кислота уксусная 98%-ная, х. ч., ГОСТ 61—65.
Условия получения
В круглодонную колбу (или стакан) емкостью 1 л, снаб-
женную мешалкой, загружают 24,8 г нафтилового красного,
120 мл концентрированной ускусной кислоты и 630 мл воды.
К полученному раствору при размешивании порциями по 10—•
15 г прибавляют цинковую пыль при температуре 60—65°
(всего 70 г) до тех пор, пока окраска раствора перейдет из
темно-малиновой в слабо-розовую (4—5 часов). По оконча-
нии реакции и охлаждении раствор фильтруют на воронке
Бюхнера в смесь, состоящую из 50 мл концентрированной сер-
ной кислоты и 250 мл воды.’Выпавший осадок сульфата 1,4-
нафтилендиамина отфильтровывают на воронке Бюхнера и
сушат при температуре 50°. Выход 21,5 г (63% теории). Для
получения основания загружают в стакан емкостью 1 л 21,5 г
сернокислой соли 1,4-нафтилендиамина, раствор 20 г углекис-
лого натрия в 500 мл воды и 2—3 г активированного угля.
Полученный раствор кипятят 10—20 минут, фильтруют го-
рячим на воронке Бюхнера и прибавляют к фильтрату 1 —
1,5 г гидросульфита натрия. При стоянии 1,4-нафтилендиамип
выкристаллизовывается в виде игл темно-песочного цвета
(з—6 по шкале Бондарцева), которые отфильтровывают и су-
шат на воздухе. Выход 7,5 г (~50%, считая на нафтиловый
красный). Вещество имеет т. пл. 119—120°, что соответствует
литературным данным [3].
Найдено %: С 75,66; 75,70; Н 6,70; 6,46. .
CmHioNa Вычислено %: С 75,85; Н 6,37.
4 Зак. 1020 , 49
Л 14 Т Е Р А Т У Р А
1. С. Liebermann, Liebigs. Ann. Chem., 183, 241 (1876).
2. E. Qrandmougin. Ber.. 39, 3562 (1906).
3. E. Bamberger, IT. J. Schiffelin, Ber,, 22, 1381 (1889).
4. W. H. Perkin, Liebigs Ann. Chem., 137, 359 (1886).
5. J. F. Thorpe, J. Chem. Soc., 91, 1009 (1907).
БРОМИСТЫЙ БУТИЛ ГЕКСАМЕТИЛЕНТЕТРАМИН
И. В. ХВОСТОВ, В. В. ДОДИКОВ, Р. Т. ХЛЕБОРОДОВА
(ВНИИ химических оеактивое и особо чистых химических веществ)
[(CH2)eN4-С4Н9] + Вг~
C19HslN4Br М. в. 277, 22
Бромистый бутилгексаметилентетрамин был получен Шуль-
те и Гоесомпри действии бромистого бутила на гексаметилен-
тетрамин в среде хлороформа. Авторы не приводят данных
элементарного анализа для данного продукта, но указывают
температуру плавления (152°) [1].
При проверке метода нами внесены следующие уточнения:
1. Установлено, что наличие следов влаги сказывается от-
рицательно на качестве и выходе продукта.
2. Одно-двухкратная промывка готового продукта горячим
хлороформом позволяет получать более чистый продукт.
(CH2)6N4 + CH3CH2CH2-CHaBr —
— [(CH2)6N4.CH3CHaCH2CH3]+Br-
Характеристика основного сырья
Гексаметилентетрамин фармакопейный.
Бромистый бутил, перегнанный п™ 1,4400.
Условия получения
В трехгорлую колбу, снабженную обратным холодильни-
ком, помещают 77 г (0.55 Л1) гексаметилентетрамина (см.
примечание 1), 600 мл свежеперегнанного хлороформа (см.
примечание 2) и 68 г (0,5 М) бромистого бутила. Нагревают
10 часов на водяной бане, поддерживая спокойное кипение
смеси. Выпавший белый кристаллический осадок отфильтро-
вывают и промывают на воронке горячим хлороформом (1—2
раза, всего 100—150 мл). Продукт высушивают на воздухе в
50
течение двух суток. Выход 65 г (46% теоретического, в пере-
счете на бромистый бутил), т. пл. 138,5—141°.
Найдено ' %: С 43,4; Н 7,85; Вг 28,1.
CI0H2iN4Br Вычислено %: С 43,3; Н 7,6; Вг 28,8.
Примечания.
1. Гексаметилентетрамин был предварительно просушен в течение су-
ток над безводным сульфатом натрия.
2. В начале добавляют 450 мл хлороформа, а в течение реакции дог
бавляют еще 150 мл.
ЛИТЕРАТУРА
К. Schulte, Н. Goes, Arch. Pharm., 290, 118 (1957).
ЧЕТВЕРТИЧНЫЕ АММОНИЕВЫЕ ОСНОВАНИЯ
(Ионообменный хроматографический синтез на анионите)
В. В. КРОХВ, А. И. ВОЛЬФСОН .
(ВНИИ химических реактивов и особо чистых химических веществ)
Общая формула*): [R4N]+OH_,
где R—органический радикал.
Четвертичные аммониевые основания находят широкое
применение в полярографий в качестве фонового электролита.
В литературе описаны способы получения на анионитах
гидрата окиси тетраэтиламмония [1] и гидроокиси триметил-
бензиламмония [2].
Нами предложен типовой метод получения гидратов оки-
сей тетраэтиламмония из йодистого тетраэтиламмония [3] и
тетраметиламмония из йодистого тетраметиламмония [4].
В настоящем сообщении приводятся методики получения
неконцентрированных водных растворов гидратов окисей тет-
раметиламмония, тетраэтиламмония, тетра-н-бутиламмония,
диметилфенилбензиламмония и н-бутилгексаметилентетрами-
на с помощью анионообменной смолы промежуточной основ-
ности ЭДЭ-10П в ОН-форме.
Ионообменный хроматографический синтез четвертичных
аммониевых оснований протекает по схеме:
A+OH--|-r-R+ 71 A+r-+R+OH~
где А — скелет анионита ЭДЭ-10П;
R — радикал четвертичного аммония;
Г — галоид.
* За исключением гидрата окиси н-бутилгексаметилентетрамина.
4* ’ 51
Для синтеза применяется стеклянная колонка высотой
750 мм и внутренним диаметром 9,5 мм, на верхнем конце ко
торон имеется расширенное горло для заливки исходного ра-
створа, а на нижнем капилляр. На выходе нз колонки поме-
щается слой стеклянной ваты, назначение которого-—удержи-
вать на себе столб анионита.
Зарядка колонки анионитом и переведение его в ОН-форму
динамическим методом производится следующим образом:
10,1 г воздушно-сухого анионита ЭДЭ-10П, помещенного в
колбу, заливают дистиллированной водой и оставляют на сут~
ки для набухания. Затем взбалтывают содержимое колбы и
постепенно заливают им колонку. После этого через заряжен-
ную колонку, не содержащую воздушных пузырей в столбе
анионита, пропускают 1 л 4%-ного водного раствора едкого
натра со скоростью фильтрования 2,8 мл/мин.
Раствор едкого натра подают на колонку из делительной
воронки.
Затем на колонку подают дистиллированную воду для от-
мывки анионита от едкого натра. Степень отмывки контроли-
руют по универсальному индикатору. Расход дистиллирован-
ной воды при отмывке составляет 400 мл. Скорость фильтро-
вания остается прежней.
Отмывку анионита производят до достижения pH промыв-
ных вод 6, после чего капилляр закрывают резиновой Проб-
кой и считают колонку подготовленной для хроматографиче-
ского синтеза.
СИНТЕЗ ЧЕТВЕРТИЧНЫХ АММОНИЕВЫХ ОСНОВАНИИ
Через подготовленную\вышеописанным способом колонку
пропускают при 18п100,«.г исходного раствора соответствую-
щей галоидной соли четвертичного аммония. Затем через ко-
лонку пропускают 50—60 мл дистиллированной воды до до-
стижения значения pH 6. Скорость фильтрования 2,8 мл/мин.
Содержание гидрата окиси четвертичного аммония определя-
ется титрованием 0,1 н. раствором серной кислоты в присутст-
вии метилоранжа.
Степень чистоты полученного гидрата окиси четвертично-
го аммония определяется полярографически по потенциалу
разложения.
Характеристика исходного сырья для хроматографическо-
го синтеза, условия проведения этого синтеза и данные, отно-
сящиеся к полученному продукту, приведены в таблице.
Методика проведения синтеза описана выше.
Концентрация всех синтезированных оснований может
быть повышена в 10 раз путем упаривания их растворов при
30° под вакуумом 30—40 мм рт. ст.
52
Потенциал разложения синтезиро- ванного основания в в —2,0 -1,8 ~2,0 -1,8 1,8
Выход основания В % теории 97,3 - 78,4 97,5 95,7 99,6
Концентра- ция основа- ния в филь- трате в вес. % 0,3 > 0,46 0,23 0,33 0,45
Объем филь- трата О Ф © о о Ф «Э Ю Г- - г-
Концентра- ция исход- ной соли н вес. % 1,30 1,25 1 0,50 0,62 1,00
Мол. вес основания 91 147 - 259 109 214
гг та х S т s S м С CXQ S О X m-e-g С О о О О -'г г z: г х £ £ т I - £ * СО « а о и jГ и
№ ВТУ У 243 -51 ру 864-53 Синтезирован по методике [5] Синтезирован на заводе Синтезирован по методике |6] (см. также стр. 50)
Наименование исходной соли Тетраметилам- моиий йодистый Тетраэтиламмо- ний бромистый Тетра-н-бутил- аммоний йодистый Диметилфенил- бензиламмоиий хлористый н-Бутилгекса- метилентетрамин бромистый
53
Л И Т Е Р А Т У Р А
1. М. Н. Платонова, Ж. общ. химии, 27, 264 (1957).
2. J. Stuchlik, . М. Tickhv И. Prochazka, Chem. listy, 50 (80), 4, 662,
(1956).
2. А. И. Вольфсон, В. В. Крохе, «Вещества высокой чистоты и реакти-
вы», Вып. 24, Госхимиздат, 1961.
4. А. И. Вольфсон, В. В. Крохе, Ж. прикл. химии, 34, 223 (1961).
5. Г. И. .Михайлов, Ж. прикл. химии, 27, 217 (19521.
6. К. Schulze, М. Goes, Arch. Phann., 290, 1 IS (1957).
КВАТЕРФЕНИЛ
С„н
18
А. П. ГРЕКОВ
(ВНИИ монокристаллов)
М. в. 306, 8
Кватерфенил получают при нагревании 4-йоддифенила с
медным порошком до температуры 270° [1], при взаимодейст-
вии 4-бромдифенила с активированным магнием в безводно?^
эфире [2], при действии лития на эфирный раствор 4-бромдн-
фенила [3], путем пропускания паров дифенила над раскален-
ной проволокой [2, 4], при действии на 4-бром- и 4-йоддифенит
спиртового раствора КОН в присутствии палладий-кальций
карбоната [5, 6], при каталитическом восстановлении 4-бром-
дифенила [7]. Кватерфенил получается при обработке бензол-
диазонийсульфата медным порошком и концентрированной
муравьиной кислотой [8], а также при продолжительном кипя-
чении перекиси бензоила с бензолом [9].
Нами разработана методика получения кватерфенила из
4-йоддифенила в присутствии металлической меди по реакции:
2
— / — — \ + CuJ,
Условия получения
В термостойкой пробирке смешивают 12 г 4-йоддифенила
с 12 г медного порошка, активированного раствором йода в
ацетоне (см. примечание 1). Полученную смесь нагревают на
металлической бане до температуры 270° и при этой темпера-
туре дают выдержку 30 минут. Реакционная масса постепенно
54
густеет и под конец прогрева становится твердой. Затем полу-
ченный плав охлаждают, измельчают в порошок и обрабаты-
вают 200 мл кипящего нитробензола. Полученный раствор от-
деляют от нерастворимого осадка фильтрованием в горячем
виде, из которого при охлаждении выпадает бесцветный мел-
кокристаллический осадок, который отфильтровывают, промы-
вают на фильтре холодным бензолом и сушат, т. пл. 317—318°
выход 5 г (75,8% теории) (см. примечание 2). .
Кватерфенил — бесцветный кристаллический порошок, не-
растворим в спирте, эфире, хлороформе, лигроине. Трудно ра-
створяется в ледяной ускусной кислоте, легче в кипящих ни-
тробензоле, анилине, ксилоле, пиридине, хинолине. Кватерфе-
нил' растворим в 500 частях кипящего бензола. При растворе-
нии кватерфенила в дымяшей серной кислоте раствор окра-
шивается в зеленый цвет.
Примечания.
1. 200 г медиого порошка обрабатывают в течение 5 — 10 минут 2 л
2%-иого раствора йсда в ацетоне. Затем отфильтровывают на воронке
Бюхнера (или удаляют раствор декантацией), вынимают из воронки, раз-
мешивают с 1 л раствора концентрированной соляной кислоты в ацетоне
(1:1) и раствор сливают. Затем медный порошок промывают ацетоном и
сушат в вакуум-эксикаторе. Применять его следует немедленно.
2. Для дальнейшей очистки кватерфенил перекристаллизовывают и>
чистого нитробензола, затем опять из нитробензола с углем, после чет о
следуют 2 перекристаллизации с фильтрованием из чистого толуола. Чи-
стый кватерфенил имеет т. пл. 320°, т. кип. 428718 мм рт. ст., т. субл.
310—320°/50 мм рт. ст.
ЛИТЕРАТУРА
1. F. Ullmann, G. Meyer. Ann., 332, 51 (1904).
2. S. Bowden, j. Chem. Soc., Ill (1931).
3. E. MUller, T. Г Opel. Ber., 72, 273 (1939).
4. G. Meyer, A. Hoffmann, Monatsli, 37, 722 (1916).
5. M. Busch, W. Schmidt, Ber., 62, 2618 (1929).
6. M. Busch, W. Weber, J. pr. Ch.. 146, 1 (1936).
7. F. Mago. M. Harwitz, J. amer. Chem. Soc., 71, 776 (1949).
8. O. Gemgross, Dunkel, Ber.. 57, 742 (1924).
9. H. Gelissen, P. Hermans, Ber., 58, 290 (1925).
4,4 -ДИИЗОПРОПИЛДИФЕНИЛ
А. П. ГРЕКОВ
(ВНИИ монокристаллов)
(CH3),HC—/ CH(CH,)S
CjqH jo
М. в. 238, 37
55
л Изойропилдифенйл получают при действии изопропилового
спирта на дифенил с применением B'F3 или BF3 Н3РО4 [1, 2].
а также взаимодействием дифенила с пропиленом или хлори-
стым изопропилом в присутствии А1С13 [3].
Поскольку первые два метода получения изопропилдифе-
нила технологически трудно осуществимы, а для последнего
имеются лишь патентные указания, нами разработан простой
метод синтеза изопропилдифенила, * который выражается сле-
дующей схемой:
/ + 2(СН3)2СНС1 (А1С1,)
(СН3),НС~/ )
^-СН(С11з)г4-2НС1
Условия получения
Получение 4,4'-<Зни:юпропилдифенила. В трехгорлую круг-
лодонную колбу емкостью 1 л, снабженную механической ме-
шалкой, обратным водяным холодильником и капельной во-
ронкой (см. примечание 1), помещают 474 г (3 М) дифенила
и 99,2 г безводного хлористого алюминия (см. примечание 2).
Полученную массу нагревают на водяной бане до темпе-
ратуры 90—95° и к ней при хорошем перемешивании в течение
двух часов прибавляют 518,4 г (6,6 М) хлористого изопропи-
ла (см. примечание 3). После прекращения выделения хлори-
стого водорода реакционную массу охлаждают и выливают в
равное по объему количество 15%-ной соляной кислоты. Обра-
зовавшийся темно-коричневый маслянистый слой 'разделяют
на делительной воронке, несколько раз промывают водой (по
100 мл) и высушивают безводным хлористым кальцием. За-
тем полученный продукт перегоняют над ,твердой щелочью
(15 г) (см. примечание 4), собирают фракцию, кипящую в
пределах' температур 291—316°. . 1
Для получения бесцветного продукта его, пофве^тают дву-
кратной перегонке в вакууме над металлическим натрием (см.
примечание 5), выход 440 г (92% теории) (см. примечание 6).
Примечания.
1. Следует применять капельную воронку с длинным капиллярным
концом.
2. Работу необходимо проводить в вытяжном шкафу.
3. Для синтеза можно использовать реактивы квалификации «чистый».
4. Можно употреблять NaOH или КОН.
5. Необходимо применять прибор для вакуум-перегонки на шлифах.
6. Обычно получается смесь д-изопро'пил- и я.л’-диизопропилдифенила.
которая вполие пригодна как растворитель для жидкостных сцинтиллято-
ров и для органического синтеза. При необходимости получать индивиду-
альные вещества смесь подвергают эффективной ректификации.
* В разработке данной методики принимали участие Д. М. Александ-
оова и Г. Д. Тицкий.
56
ЛИТЕРАТУРА
1. И. А. Ромадин, Т. И. Рендель, Ж. общ. химии, 26, 202 (1956).
2. И. А. Ромадан, С. Э. Берги, УК. общ. химии, 28, 413 (1958).
3. A. Levine, О. Cass, Анг. пат. 497284 (19о9); Chem. Zbl„ 1939, 1, 3961.
2,5-ДИФЕН ИЛ-1,3,4-ОКСАДИАЗОЛ
А. П. ГРЕКОВ
(ВНИИ монокристаллов)
С„Н10ОМ2
М. в. 222, 26
2,5- Дифенил-1,3,4-окса диазол получают из 1,2-дибензоил-
гидразина при нагревании до 250—300° или под влиянием во-
доотнимающих средств [1], а также при действии на 1,2-дибен-
зоилгидразин хлорокиси фосфора [2].
Нами разработан препаративный метод получения 2,5-ди-
фенил-1,3,4-оксадиазола из гидразингидрата и бензоилхлорида
по схеме:
/~^-С0С1 + n2h4
• ( \-CONHNHCO—f + 2НС1
^~^-CONHNHCO—
N — N
(~)-с с-<2/ + Н2°
ъ
Условия получения
Получение 1,2-дибензоилгидразина. В колбу емкостью
0,5 л, снабженную механической мешалкой и обратным холо-
дильником, помещают раствор 10 г 98%-го гидразингидрата в
200 мл сухого пиридина и к нему медленно, по каплям добав-
57
ляют 56 г бензоилхлорида. После окончания прибавления все-
го бензоилхлорида смесь кипятят 20 минут, а затем содержи-
мое колбы выливают в холодную воду (500—600 мл). Выпав-
ший бесцветный кристаллический продукт отфильтровывают,
промывают на фильтре водой и сушат. Выход количественный,
т. пл. 237° (см. примечание).
Получение 2,5-дифенил-1,3,4-оксадиазола. Смешивают 24 г
1,2-дибензоилгидразина с 80 мл хлорокиси фосфора и кипятят
в колбе емкостью 250 мл с обратным холодильником в течение
1 часа, причем находящееся в виде суспензии вещество цели-
ком переходит в раствор. После этого отгоняют 40 мл хлор-
окиси фосфора, а остаток осторожно выливают в пятикратное
количество холодной воды. Выпавший бесцветный кристалли-
ческий осадок отфильтровывают, промывают водой и сушат,
выход 19 г (91% теории), т. пл. 137°.
2,5-Дифенил-1,3,4-оксадиазол — бесцветное кристалличе-
ское вещество, хорошо растворимое в большинстве органиче-
ских растворителей. Оптические и сцинтилляционные свойст-
ва приведены в работах [3, 4].
Примечание.
Для получения чистого препарата с т. пл. 241е необходимо его пере-
кристаллизовать.
Л ИТ Е Р А Т У Р А
1. R. Stolle, J. pr. Chem., [21, 69 157 (1904).
2. N. Hayes, В. Rogers, D. Ott, J. Am. Chetn. Soc., 77, 1850 (1955).
3. И. П. Шиманская, А. П. Килимов, А. П. Греков, Л. M. Егупова,
P. С. Авен, Оптика и спектроскопия, 7, 366 (1959).
4. О. П. Швайка, А. П. Греков, Оптика и спектроскопия, 7, 824 (1959).
2-ФЕНИЛ-5-(4-ХЛОРФЕНИЛ)-1,3,4-0КСАДИАЗОЛ
А. И. ГРЕКОВ
(ВНИИ монокристаллов)
N N
CHH,ONSC1 м. в. 256, 71
2-Фенил-5-(4-хлорфенил)-1,3,4-оксадиазол впервые опи-
сан нами [1]. Его синтез осуществляется по следующей схеме;
^-CONHNH» + С1СО-( )-С1---------
\-CONHNHCO-\—^-С1<НС1
58
/—\ z“ ?0С]»
/ \-CONHNHCO-f \/“cl --
N - N
Z—х I! II Z-.
- ; ^-с с-/ ;-ci + H2o
о
Условия получения
Получение 1-бензоил-2-(4-хлорбензоил)-гидразина. В трех-
горлую колбу емкостью 0,5 л. снабженную механической ме-
шалкой и обратны^ холодильником, помещают 250 мл сухого
пиридина и 45 г хлорангидрида 4-хлорбепзойной кислоты.
К полученному раствору постепенно, небольшими порциями до-
бавляют 34 г бензгидразида. После прибавления всего бенз-
гидразида реакционную массу кипятят 20—30 минут на воз-
душной бане, а затем после охлаждения выливают в 500—
800 мл холодной воды. Выпавший бесцветный кристалличе-
ский 1-бензоил-2-(4-хлорбензоил)-гидразин отфильтровывают,
промывают водой и сушат, выход 22 г, т. пл. 216° (см. приме-
чание 1).
Получение 2-фенил-5-(4-хлорфенил)-1,3,4-оксадиазола. В,
колбу емкостью 250 мл, снабженную обратным холодильни-
ком, помещают 22 г 1-бензоил-2-(4-хлорбензоил)-гидразина
и 70 мл хлорокиси фосфора. Полученную смесь кипятят на
воздушной бане 90 минут, причем все вещество переходит в
раствор. Затем из реакционной массы отгоняют 30 мл хлор-
окиси фосфора, а остаток осторожно при перемешивании вы-
ливают в 200—300»мл холодной воды. После того как масса
полностью закристаллизуется, ее отфильтровывают, хорошо
промывают на фильтре водой (5—6 раз по 25 мл) и сушат, вы-
ход 19,5 г, т. пл. 147—148°. Полученный продукт перекристал-
лизовывают из этанола (200—300 мл), выход 17 г, т. пл. 155°.
Для дальнейшей очистки оксадиазол пропускают через хро-
матографическую колонку, наполненную окисью алюминия, с
применением толуола в качестве растворителя. Выход 12 г
(50,8% теории), т. пл. 162° (см. примечание 2).
2-Фенил-5-(4-хлорфенил)-1,3,4-оксадиазол — бесцветное
кристаллическое вещество, хорошо растворимое в органиче-
ских растворителях; его сцинтилляционные свойства изучены
в работе [2].
Примечания.
1. Соединение пригодно для дальнейшего синтеза. Для получения чи-
стого препарата с т. пл. 222—223° необходимо его подвергнуть дополни
тельной очистке.
2. Хроматографическую очистку можно заменить перекристаллизацией
из толуола.
59
ЛИТЕРАТУРА
1. А. П. Греков, Л. Н. Кулакова, О. П. Швайка, УК- общ. химии, 29,
3054 (1959).
2. О. П. Швайка, А. П. Греков, Оптика и спектроскопия, 7, 824 (1959).
2-ФЕНИЛ-5-(4-МЕТОКСИФЕНИЛ)-1Д4-ОКСАДИАЗОЛ
А. П. ГРЕКОВ
( ВНИИ монокристаллов )
N—N
х II II-------X
J>-C С-ф \ осн,
'о'
С-1»Н1гОг1Чг
М. в. 252,28
Получение 2-фенил-5-(4-метоксифенил) -1,3,4-оксадиазола
из бензгидразида и 4-метоксибензоилхлорида впервые описа-
но нами [1.]
Процесс протекает в две стадии:
/ \-CONHNH24-ClCO-^ ^-ОСН3---------------
/ ^-CONHNHCO- ( ^-~ОСНз+НС1
х РОС),
/ CONHNHCO-' ^-ОСН3 >
N-N
х-0СНз + Н,0
Условия получения
Получение 1-бензоил-2-(4-метоксибензоил)-гидразина. В
трехгорлую колбу емкостью 250 мл, снабженную механиче-
ской мешалкой и обратным холодильником, помещают 100 мл
сухого пиридина (см. примечание 1) и 42 г 4-метоксибензоил-
60
хлорида. В полученный раствор в течение 40—60 минут не-
большими порциями добавляют 34 г бензгидразида, по мере
прибавления которого реакционная масса постепенно разогре-
вается и окраска раствора меняется от светло-желтой до тем-
но-оранжевой. После прибавления всего количества бензгид-
разида полученный раствор кипятят 20—30 минут на воздуш-
ной бане (см. примечание 2).
Затем реакционную массу охлаждают и выливают в 250—
300 мл холодной воды. Выпавший бесцветный осадок филь-
труют, промывают на' фильтре водой (пятью порциями по
20 мл), хорошо отжимают и сушат. Выход 50 г (75% теории),
т. пл. 178—184° (см. примечание 3).
Получение 2-фенил-5-(4-меток.сифенил)-1,3,4-ок.садиазола.
В колбу емкостью 2э0 мл, снабженную обратным холодильни-
ком, помещают 50 г 1-бензоил-2-(4-метоксибензоил)-гидразина
и 150 мл хлорокиси фосфора. Полученную смесь нагревают на
воздушной бане до полного растворения осадка, на что требу-
ется 45—60 минут, после чего ее прогревают еще один час.
Затем реакционную массу охлаждают и осторожно при пере-
мешивании в течение 30—60 минут выливают в смесь 1600 г
льда и воды (см. примечание 4). После того как масса пол-
ностью закристаллизуется, ее отфильтровывают, хорошо про-
мывают на фильтре водой (5—6 раз по 50 мл) и сушат. Полу-
ченный продукт растворяют при кипячении в 500 мл этанола,
к раствору добавляют 2—3 г активированного угля и фильтру-
ют. При охлаждении из раствора выкристаллизовывается ве-
щество с т. пл. 143—144°, выход 39 г. Полученный оксадиазол
Перекристаллизовывают из 180—200 мл толуола, выход 28 г
(см. примечание 5). После повторной перекристаллизации из
150 мл толуола получается 23 г (49% теории) оксадиазола
для сцинтилляционных целей с т. пл. 144,5—146° (см. примеча-
ние 6).
2-Фенил-5-(4-метоксифенил) -1,3,4-оксадиазол — бесцветное
кристаллическое вещество, хорошо растворимое в большинст-
ве органических растворителей. Оптические и сцинтилляцион-
ные свойства описаны в работах [2—4].
Примечая и я.
1. Пиридин сушат твердым КОН с последующей его перегонкой.
2. Если получился гомогенный раствор, то его кипячение можно исклю-
чить.
3. Продукт пригоден для дальнейшего синтеза. Для получения чистого
препарата с т. пл. 186—187е его подвергают пере.кристаллизации из этано-
ла.
4. Реакцию необходимо проводить в вытяжном шкафу.
5. При наличии нерастворимого осадка следует провести- горячее филь-
трование.
6. Две последние перекристаллизации из толуола можно заменить хро-
матографической очисткой толуольного раствора вещества на окиси алюми-
ния.
61
ЛИТЕРАТУРА
1. А. П. Греков, Л. Н. Кулакова, О, П. ШваЦка, /К. общ химии, 29.
3059 (1959).
2. О. П. Швайка, А. П. Греков, Оптика и спектроскопия, 7, 824 (1959).
3. Н. П. Шиманская, А. П. Килимов, А. П. Греков, Л. М. Егупова,
Р. С. Авен, Оптика и спектроскопия, 7, 366 (1959).
4. Н. П. Шиманская, А. П. Килимов, А. П. Греков, Оптика и спектро-
скопия, 6, 194 (1959).
2,5-ДИ’-(4-МЕТОКСИФЕНИЛ)-1,3,4-ОКСАДИАЗОЛ
А. П. ГРЕКОВ
(ВНИИ монокристаллов)
N — N
/— - II I! /— \
Н,СО-<^ ^-С С-/ \-ОСН3
с/
C„H14O,N, М. в. 282, 32
По литературным данным, 2,5-ди-(4-ме,токсифенил)-1,3,4-
оксадиазол можно получать из 1,2-ди-(4-метоксибензоил)-гид-
разина при нагревании с пятихлористым фосфором [1] или
хлорокисью фосфора [2]. Нами разработан метод получения
этого соединения из хлорангидрида 4-метоксибензойной кисло-
ты и гидразингидрата по схеме:
2Н3СО— / \ — СОС1 + N2H4
т Н9СО-/ \-CONHNHCO-/ \-ОСН3 + 2НС1
H3CO-/~\-CONHNHCO —х \-ОСН3
N - N
II II
Н3СО-^ ^-С С-^. ^-ОСНз-4-Н.О
о
Условия получения
Получение 1,2-ди-(4-метоксибензоил) -гидразина. В трех-
горлую колбу емкостью 0,5 л, снабженную капельной ворон-
62
кой, обратным холодильником и механической мешалкой, по-
мещают 150 мл сухого перегнанного пиридина, 10,5 г 98%-но-
го гидразингидрата и к полученному раствору медленно, при
перемешивании в течение одного часа прибавляют из капель-
ной воронки 70 г хлорангидрида 4-метоксибензойной кислоты.
Затем полученную смесь нагревают до полного растворения
осадка, после чего через' 15 минут раствор охлаждают и выли-
вают в 300 мл холодной воды. Выпавший продукт отфильтро-
вывают, промывают на фильтре водой (50 .ил) и сушат. Вы-
ход 38 г (61,7% теории), т. пл. 222°.
Получение 2.5-ди-(4-метоксифенил)-1,3,4-оксадиазола. В
колбе емкостью 100 мл. осторожно смешивают 38 г 1,2-ди-(4-
метоксцбензоил)-гидразина с 55 мл хлорокиси фосфора. Полу-
ченную смесь нагревают на водяной бане с обратным холо-
дильником до полного растворения осадка, на что требуется
обычно 1—1,5 часа. Затем реакционную массу охлаждают и
очень осторожно, медленно при перемешивании выливают в
300 г льда (см. примечание 1). Выпавший продукт отфильтро-
вывают, промывают на фильтре водой (50 мл) и сушат. После
перекристаллизации из 500 мл этанола с применением 2—3 г
активированного угля выход 30 г, т. пл. 158—159°. После вто-
рой перекристаллизации из 500 мл этанола выход 25 г (70%
теории), т. пл. 164—165° (см. примечание 2).
2,5-Ди-(4-метоксифенил)-1,3,4-оксадиазол растворим в
большинстве органических растворителей. Оптические и сцин-
тилляционные свойства описаны в работе [3].
Примечания.
1. При очень энергичном ходе реакции следует применять охлаждение.
2. Все перекристаллизации можно заменить хроматографической очи-
сткой толуольного раствора вещества на окиси алюминия.
ЛИТЕ Р А Т У Р А
1. /?. Stolle, A. Bombach, J. pr. Ch., [2], 74, 15 (1906).
2. N. Hayes, В. Rogers. D. Ott. J. Amer. Chem. Soc., 77. 1850 (1955).
3. И. П. Шиманская, А. П. Килимов, А. П. Греков. Л. A-f. Егупова,
P. С. Авен, Оптика и спектроскопия, 7, 366 (1959).
2-ФЕНИЛ-5-(4-ДИФЕНИЛ ИЛ )-1,3,4-ОКСАДИАЗОЛ
.4. П. ГРЕКОВ
(ВНИИ монокристаллов)
N— N
/—\ И И /—' л—\
\ /~С с~\ у \ /
О
C!0H14ONs М. в. 298, 36
2-Фенил-5-(4-дифенилил)-1,3,4-оксадиазол впервые описан -
з работе [I]. Нами разработан препаративный метод его полу-
63
чения из бензгидразида и хлорангидрида 4-дифенилкарбоно-
вой кислоты по схеме:
. c5h5n
CONHNHH-C1CO --->
Условия получения
Получение 1 -бензоил-2-(4-дифенилоил)-гидразина. В колбу
емкостью 1 л, снабженную механической мешалкой и обрат-
ным холодильником, помещают 500 мл- сухого свежепе-
регнанного пиридина, 50 г бензгидразида и 50 г хлорангидри-
да 4-дифенилкарбоновой кислоты. Полученную смесь медлен-
но нагревают до полного растворения осадка, после чего ра-
створ доводят до кипения. Через 20 минут нагревание прекра-
щают, а реакционную массу выливают в холодную'воду (2 л).
Выпавший бесцветный кристаллический осадок фильтруют,
промывают водой и сушат. После перекристаллизации 'из ле-
дяной уксусной кислоты выход 85 г (73% теории), т. пл. 23Р—
222°.
Получение 2-фенил-5-(4-дифенилил)-1,3,4-оксадиазола. 85г
1-фенил-2-(4-дифенилоил)-гидразина и 335 мл хлорокиси фос-
фора помещают в колбу емкостью 0,75 л, снабженную обрат-
ным холодильником, и полученную смесь кипятят в течение
четырех часов. Затем отгоняют половину первоначально взя-
того количества хлорокиси фосфора, а остаток осторожно вы-
ливают в холодную воду (см. примечание 1). Выпавший кри-
сталлический осадок фильтруют, промывают водой и сушат.
После перекристаллизации из ледяной уксусной кислоты вы-
ход 69 г (86,5% теории), т. пл. 167—167,5°.
2-Фенил-5-(4-дифенилил)-1,3,4-оксадиазол растворим в ор-
ганических растворипелях, оптические и сцинтилляционные
свойства описаны в работах [2, 3].
Примечания.
1. Реакцию необходимо проводить в вытяжном шкафу.
2. Для окончательной очистки полученный оксадиазол подвергают хро-
64
матографической очистке на окиси алюминия с применением толуола и ка-
честве растворителя.
ЛИТЕРАТУРА
1. W. Hayes, В. Rogers, D. Ott. J. Amer. Chem. Soc.. 77, 1850 (1955).
2. H. П. Шиманская, А. П. Килимов, А. П. Греков, Оптика и спектро-
скопия, 6, 194 (1959).
3. А. П. Греков, О. П. Швайка, сб. «Сцинтилляторы и сцинтилляцион-
ные материалы». М., 1960 г., стр. 105. •
2,5-ДИ-(4-ДИФЕНИЛ ИЛ )-1,3,4-ОКСАДИАЗОЛ
А. П. ГРЕКОВ
(ВНИИ монокристаллов)
CjeHjgON,
М. в. 374, 45
Ввиду того, что для 2,5-дн-(4-дифенилил)-1,3,4-оксадиазола
в литературе [1] имеется лишь одно упоминание, без каких бы
то ни было указаний о подробностях синтеза, была разработа-
на методика его получения из хлорангидрида дифенилкарбо-
новой кислоты и гидразингидрата, которая излагается ниже.
Синтез осуществляется по следующей схеме:
/—V X — ч CSHSN
2 / < \-COCl + N,H4--------
\ / \ /
>-CONHNHCO \
Условия получения
Получение 1,2-ди-(4-дифенилоил)-гидразина. В литровую
колбу, снабженную механической мешалкой, обратным холо-
дильником и капельной воронкой, помещают 20 мл сухого,
свежеперегнанного пиридина, 124 г хлорангидрида 4-дифенил-
карбоновой кислоты (см. примечание 1) и полученную смесь
слегка нагревают до полного растворения твердого продукта.
К содержимому колбы при интенсивном перемешивании через
капельную воронку прибавляют по каплям раствор 17 мл
98%-ного гидразингидрата в 50 мл сухого пиридина, на что
требуется около 30 минут. Затем реакционную массу нагрева-
ют до кипения, выдерживают при этой температуре 30 минут,
после чего ее выливают в литровый стакан и медленно охлаж-
дают до комнатной температуры. Выпавший продукт реакции
отфильтровывают и промывают на фильтре большим количест-
вом воды до отсутствия запаха пиридина. После перекристал-
лизации 1,2-ди-4- (дифенилоил) -гидразина из ледяной уксус-
ной кислоты выход 96а (85% теоретического), т. пл. 280—295°
(с разложением) (см. примечание 2).
Получение 2,5-ди-(4-дифенилил)-1,3,4-оксадиазола. В круг-
лодонную колбу емкостью 0,5 л, снабженную обратным холо-
дильником, помещают 60 г 1,2-ди-(4-дифенилоил)-гидразина и
150 мл хлорокиси фосфора. Полученную смесь кипятят в тече-
ние 8 часов, причем осадок не растворяется, а приобретает
лишь вид блестящих зеленовато-желтых лепестков. После
прекращения нагревания содержимое колбы охлаждают до
комнатной температуры, а затем осторожно при перемешива-
нии выливают в 2 л холодной воды. Образовавшийся бесцвет-
ный кристаллический 2,5-ди-(4-дифеншшл)-1,3,4-оксадиазол
фильтруют, хорошо промывают на фильтре водвй и сушат,
выход 55 г, т. пл. 228—231°, После перекристаллизации из
1,2 л ледяной уксусной кислоты и затем из 0,7 л диоксана вы-
ход 30,6 г (52% теоретического), т. пл. 234° (см. примеча-
ние 3).
2,5-Ди-(4-дифенилил)-1,3,4-оксадиазол — бесцветный кри-
сталлический порошок, плохо растворимый в эфире, спирте,
бензоле, толуоле, лучше — в ледяной уксусной кислоте и ди-
оксане. Оптические и сцинтилляционные свойства описаны в
работе [2].
Примечания.
1. Необходимо применять чистый хлорангндрид 4-дифеиилкарбоновой
кислоты с т. пл. 115°.
2. Без дополнительной очистки продукт пригоден для дальнейшего син-
теза.
3. Последние перекристаллизации можно заменить хроматографической
очисткой толуольного раствора вещества на окиси алюминия.
66
ЛИТЕРАТУРА
1. N. Hayes, В. Rogers, D. Ott, J. Amer. Chem. Soc., 77, 1850 (1955).
2. И. П. Шиманская, А. П. Килимов, А. П. Греков, Л. M. Егупова,
P. С. Авен. Оптика и спектроскопия, 7, 366 (1959).
2-(1-НАФТИЛ)-5-(4-ДИФЕНИЛИЛ)-1,3,4-ОКСАДИАЗОЛ
QuHieQN,
А. П. ГРЕКОВ
(ВНИИ монокристаллов)
М. в. 348, 42
Это соединение впервые описано нами [1]. Его синтез осу-
ществляется из гидразида 1-нафтойной кислоты и хлорангид-
рида 4-дифенилкарбоновой кислоты по схеме:
^Ч-НС!
Условия получения
Получение 1-(1-нафтоил)-2-(4-дифенилоил)-гидразина. В
колбу емкостью 0,5 л, снабженную обратным холодильником
и механической мешалкой, помещают 48 г 1-нафтилгидрази-
: • 67
да, 310 мл сухого перегнанного пиридина и 50 г хлорангидри-
да 4-дифенилкарбоновой кислоты. Полученную смесь кипятят
при перемешивании 1,5—2 часа, после чего реакционную мас-
су охлаждают до комнатной температуры и выливают в 3—4 л
холодной воды. Выпавший бесцветный кристаллический осадок
фильтруют, промывают на фильтре водой и сушат. Получают
продукт с т. пл. 211—214°. После двух перекристаллизаций из
ледяной уксусной кислоты т. пл. 219—220°, выход 58 г (69%
теории).
Получение 2-(1-нафтил ) -5-(4-дифенилил )-1,3,4-оксадиазс-
ла. Смешивают 58 а 1-(1-нафтоил)-2-(4-дифенилоил)-гидрази-
на с 150 мл хлорокиси фосфора и кипятят в колбе с обратным
холодильником в течение 5 часов. По мере нагревания вещест-
во постепенно растворяется и к концу кипячения образуется
темно-коричневый раствор, из которого отгоняют половину
первоначально взятого количества хлорокиси фосфора. Оста-
ток осторожно выливают в 1 л холодной воды и оставляют
стоять при комнатной температуре в течение 3—4 часов. Вы-
павший бесцветный кристаллический продукт фильтруют, про-
мывают водой и сушат. После двух перекристаллизаций из
ледяной уксусной кислоты получают 34 г (62% теории) окса-
диазола с т. пл. 156—157". Для дальнейшей очистки получен-
ное вещество подвергают хроматографической очистке на оки-
си алюминия с применением толуола в качестве растворителя.
2- (1 -Нафтил) -5- (4-дифенилил) -1,3,4-оксадиазол хорошо ра-
створим в большинстве органических растворителей. Оптиче-
ские и сцинтилляционные свойства приведены в работе [2, 3].
ЛИТЕРАТУРА
1. А. П. Греков, Р. С. Авен, Ж. общ, Химин, 29, 1995 (1959).
2. Н. П. Шиманская. А. П. Килимов, .4. П. Грисов, Оптика и спектро-
скопия, 6, 194 (1959).
3. А. П. Греков, О. П. Швайка, сб. «Сцинтилляторы и сцинтилляцион-
ные материалы», М„ I960., стр. 105.
2,5-ДИ-(2-НАФТИЛ)-1,3,4-ОКСАДИАЗОЛ
А. Я. ГРЕКОВ
(ВНИИ монокристаллов)
N — N
/\/\ /\/\
I I Г \/ I I 1
ч/\/ О \/\/
CbH14ONt М. в. 322, 38
Для этого соединения в литературе [1] имеется лишь одно
упоминание без указаний о подробностях синтеза. Ниже при-
68
водится методика его получения из хлорангидрида 2-нафтой-
ной кислоты и гидразингидрата по схеме:
2 ( Х|7 '| C0C1 %- N2H4 Csl-5-—>
Г Y XrC0NHNHC°^ Y) + 2НС1
/ 7 \ —CONHNHCO — / yZ POCI,
Условия получения
Получение 1,2-ди-(2-нафтоил)-гадразина. В трехгорлую
круглодонную колбу емкостью 2 л, снабженную механической
мешалкой, обратным холодильником и капельной воронкой,
помещают 1 л свежеперегнанного сухого пиридина и к нему
при перемешивании добавляют 168 г хлорангидрида 2-нафтой-
ной кислоты (см. примечание Г). Затем из капельной воронки
при энергичном перемешивании медленно прибавляют 25 мл
98%-го гидразингидрата (см. примечание 2), после чего через
30 минут содержимое колбы охлаждают до комнатной темпе-
ратуры и выливают в 4 л холодной воды. Выпавший бесцвет-
ный кристаллический продукт фильтруют, промывают водой и
сушат при температуре 100—110°, выход 70 г, т. пл. 215°. Пос-
ле перекристаллизации из ледяной уксусной кислоты (1,0—
1,5 л) получается 1,2-ди-(2-нафтоил)-гидразин с т. пл. 242—
244°, выход 45 г (32% теории).
Получение 2,5-ди-(2-нафтил)-1,3,4-оксадиазола. В колбу
емкостью 250 мл помещают 45 г 1,2-ди-(2-нафтоил)-гидразина,
к нему добавляют 100 мл хлорокиси фосфора и полученную
смесь кипятят с обратным холодильником в течение трех ча-
сов, причем вещество растворяется за 30—40 минут. Затем из
реакционной массы отгоняют 60 мл хлорокиси фосфора, а
остаток медленно при хорошем перемешивании выливают в
300 мл холодной воды со льдом (см. примечание 3). Вначале
вещество выпадает в виде желтовато-серой массы, которая
при растирании и охлаждении переходит в сероватый кристал-
69
лический осадок. Полученное вещество фильтруют, промывают
на фильтре водой и сушат при температуре 80—90°, выход
42,7 г, т. пл. 182—184°. После перекристаллизации из толуола
(300 мл) с активированным углем (1—2 г) выпадает бесцвет-
ный кристаллический 2,5-ди-(2-нафтил)-1,3,4-оксадиазол, ко-
торый отфильтровывают и сушат, выход 34 г (81% теории),
т. пл. 187—189° (см. примечание 4).
• 2,5-Ди-(2-нафтил)-1,3,4-оксадиазол— бесцветное кристал-
лическое вещество, хорошо растворимое в органических рас-
творителях. Его оптические и сцинтилляционные свойства опи-
саны в работах [2, 3].
Примечания.
1. Происходит сильное разогревание, причем большая часть хлор-
ангидрида 2-нафтойной кислоты переводит в» раствор.
2. После прибавления половины гидразингидрата раствор становится
прозрачным, а затем начинает выпадать осадок.
3. Происходит сильное разогревание, поэтому реакционную массу необ-
ходимо выливать в воду небольшими порциями при энергичном перемеши-
вании.
4. Если оксадиазол не будет соответствовать указанным характеристи-
кам, то его следует дополнительно перекристаллизовать из толуола.
ЛИТЕРАТУРА
1. N. Hayes, В. Rogers, D. Ott, J. Amer. Chem. "Soc, 77, 1850 (1955).
2. А. П. Греков, О. П. Швайна, сб. «Сцинтилляторы и сцинтилляцион-
ные материалы», М„ 1960 г., стр. 105.
3. Н. П. Шаманская, А. П. Килимов, А. П. Греков, Л. М. Егупова,
Р. С. Авен, Оптика и спектроскопия, 7, 366 (1959).
2-(2-ФУР ИЛ)-5-(4-ДИФЕНИЛ ИЛ )-1,3,4-ОКСАДИАЗОЛ
А. П. ГРЕКОВ, Е. П. НЕСЫНОВ
(ВНИИ монокристаллов)
М. в. 288, 31
2-(2-Фурил)-5-(4-дифенилил)-1,3,4-оксадиазол впервые
описан нами в статье [1], где приведены общие указания о его
синтезе и свойствах. Ниже приводится препаративный метод
70
его получения из гидразида пирослизевой кислоты и хлоран-
гидрида 4-дифенилкарбоновой кислоты.
Процесс проходит в две стадии:
J—CONHNH, + СЮС—
О
k J-CONHNHCO-( )+НС1
о
/ \ \ РОС!
4/-CONHNHCO-^ ')
6 \_/ \—
На первой стадии образуется 1-(2-фуроил)-2-(4-дифенило-
ил)-гидразин в среде пиридина [2], который связывает выде-
ляющийся в результате реакции хлористый водород, что бла-
гоприятствует более полному использованию исходных реа-
гентов. На второй стадии с помощью хлорокиси фосфора про-
исходит циклизация полученного гидразина в 2-(2-фурил)-5-
(4-дифенил ил) -1,3,4-оксадиазол.
Условия получения
Получение 1-(2-фуроил)-2-(4-дифенилоил)-гидразина. В
трехгорлую круглодонную колбу емкостью 250 мл, снабжен-
ную обратным холодильником с хлоркальциевой трубкой и
механической мешалкой, помещают 140 мл безводного пири-
дина (см. примечание 1) и 18 г (0,13 М) гидразида пирослизе-
вой кислоты. В полученную смесь при хорошем перемешива-
ний. добавляют в течение 10 минут 30 г 0,14 Л4 хлорангидри-
да 4-дифенилкарбоновой кислоты, при этом реакционная мас-
са разогревается до температуры 45—50°. Перемешивание
продолжают еще 2 часа, после чего содержимое колбы выли-
вают в стакан емкостью 1 л и в него прибавляют 750 мл ди-
стиллированной воды. После 12-часового стояния выпавший
осадок отфильтровывают, промывают на фильтре дистилли-
рованной водой (100 мл) и Сушат вначале при комнатной
температуре, а затем при 80°, выход 34,7 г (79% теории), т. пл,
213,5—215° (см. примечание 2).
71
Получение 2- (2-фурил)-5-(4-дифенилил)-1,3,4-оксадиазоли.
В круглодонную колбу емкостью 250 мл, снабженную обрат-
ным холодильником и хлоркальциевой трубкой, помещают 30 г
(0,1 Л4) сухого 1-(2-фуроил)-2-(4-дифенилоил)-гидразина (см.
примечание 3) и 100 мл хлорокиси фосфора. Полученную
смесь кипятят 2 часа на воздушной бане, причем через 15-
20 минут после начала Кипения осадок полностью переходи:
в раствор. Затем нагрев прекращают и дают 3-часовую выдерж
ку, после чего обратный холодильник заменяют насадкой
Кляйзена с нисходящим холодильником и отгоняют в вакуугп
55 мл хлорокиси фосфора. Оставшуюся в колбе массу медлен
но при перемешивании выливают в 250 мл воды (см. примеча-
ние 4) и выдерживают 12 часов. Выпавший осадок отфильтро-
вывают, промывают дистиллированной водой (50 мл) и сушат
при 80°, выход 27,6 г, т. пл. 125—127°. Полученный продукт
при слабом нагреве (30—40°) растворяют в смеси диоксана
(100 мл) и бензола (200 лы), образовавшийся раствор обра-
батывают активированным углем (7 г) и пропускают через
хроматографическую колонку, наполненную окисью алюми-'
ния (диаметр 2 см; высота 14 см). К очищенному' раствору;
добавляют 900 мл петролейного эфира. Через 10—12 часов
выпавший осадок отфильтровывают, промывают на фильтре
петролейным эфиром (50 мл) и сушат при 70°. Выход 15,7 г,
т. пл. 133,5—134,5°. Затем вещество снова растворяют в 500 лл
бензола и повторно подвергают хроматографической очи-
стке на окиси алюминия (диаметр 2 см; высота 8 см), после
чего продукт выделяют добавлением к его бензольному раст-
вору 700 мл петролейного эфира. Выпавший бесцветный
2-(2-фурил)-5-(4-дифенилил) -1,3,4-окс а диазол отфильтровыва-
ют на стеклянном фильтре, промывают петролейным эфиром
(40 мл) и сушат при 70°. Выход 9,7 г (34,4% теории), т. пл.
135—135,5°.'
Из маточных растворов можно выделить еще 2,5—3 г ме-
нее чистого оксадиазола.
2- (2-Фурил) -5- (4-дифенилил) -1,3,4-оксадиазол растворим в
бензоле, толуоле, ксилоле, фенилциклогексане, диоксане, аце-
тоне, пиридине. Не растворим в воде и холодном петролейном
эфире. Гидролизуется водными растворами минеральных кис-
лот и щелочей. Люминесцирует в ультрафиолете.
Примечания.
1. Пиридин сушат твердым КОН с последующей его перегонкой.
2. Препарат пригоден для дальнейшего синтеза. В случае необхо щ-
мости его очищают путем кристаллизации из смеси диоксана с бензолом
(1 :2). Очищенный продукт имеет т. пл. 216,5—218°.
3. В случае применения влажного 1-(2-фуроил)-2-(4-дифенилопл)-гид-
разина при прибавлении хлорокиси фосфора происходит энергичная реак-1
ция, которая иногда сопровождается выбросом реакционной массы.
4. Реакция экзотермичная. поэтому реакционную массу необходимо
выливать в воду небольшими порциями при энергичном перемешивании
72
Л И Т Е Р А Т У Р А
1. А. П. Греков, Г. II. Несынов, /К. общ. .химии. 30, 3240 (I960).
2. А. Л. Греков, Е. П. Несынов, Ж. общ. химии, 30, 3237 (I960).
2-АЦЕТИЛ ФУРАН
Г. Н. ДОРОФЕЕНКО, В. И. ДУЛЕНКО, Л. В. ПОЛИЩУК
(Институт органической химии АН УССР, Донецкое отделение)
НС - сн
Ii !1
нс с сосн3
о
Основными методами получения 2-ацетилфурана являют-
ся методы, основанные на каталитическом ацетилировании фу-
рана по реакции Гусгавсона-Фриделя-Крафтса. Для этой цели
в качестве ацилирующих средств описано применение уксус-
ной кислоты [1], хлористого ацетила [2], кетена [3], уксусного
ангидрида [4—10] и тетраацетилоксисилана [11].
Наилучшие результаты получаются при ацетилировании
фурана уксусным ангидридом в присутствии следующих ка-
тализаторов: йода и йодистоводородной кислоты [4], хлори-
стого цинка [5], фтористого бора и его комплексов [6,], фосфоп-
ного ангидрида [7], хлорной кислоты [8, 9] и других минераль-
ных кислот [10].
Из других методов, не имеющих препаративного интереса,
следует упомянуть о взаимодействии нитрила пирослизевой
кислоты с метилмагниййодидом [12], хлорангидрида этой же
кислоты с диметилкадмием [13], диазсметана с фурфуролом
[14], также приводящем к образованию 2-ацетилфурана.
Нами Предлагается способ ацетилирования фурана уксус-
ным ангидридом в присутствии незначительных каталитиче-
ских количеств безводного хлорнокислого магния (ангидрона).
Описываемый здесь способ отличается простотой, удобством и
быстротой выполнения и позволяет получать 2-ацетилфуран и
Другие алкил-2-фурил'кетоны с высокими выходами.
73
СИНТЕЗ 2-АЦЕТИЛФУРАНА
НС—СН Mg(C104)« НС—СН
|| II +(СН,СО)„О---------II [| +СНЭСООН
НЕ 2Н ' НС ССОСН,
о о
Характеристика основного сырья
Фуран, чистый, перегнанный при температуре 31—34°.
Уксусный ангидрид, ч. д. а.
Магний хлорнокислый безводный, ч.
Условия получения
В круглодонную колбу емкостью 500 мл, снабженную об-
ратным холодильником, помещают 68 г (1 Л1) фурана, 117 мл
(1,2 М) уксусного ангидрида и 2,23 г (0,01 М) ангидрона, ко-
торый вскоре растворяется в реакционной смеси. Колбу погру-
жают на 40 минут в водяную баню с температурой воды 45—
50° (при этом реакционная смесь находится в состоянии сла-
бого кипения), затем баню убирают и реакционную смесь на-
гревают при сильном кипении в течение 5 минут. Продукт ре-
акции, окрашенный в темно-фиолетовый цвет, выливают в
500 мл холодной воды, тщательно перемешивают и через 10—
15 минут органический слой отделяют от водного. Водный ра-
створ нейтрализуют карбонатом калия, выделившийся продукт
дважды экстрагируют бензолом (каждый раз по 50 мл). Бен-
зольные вытяжки и органический слой соединяют, промывают
5%-ным раствором соды, водой и в течение 30 минут сушат
над обезвоженным сульфатом натрия. Высушенный от следов
воды раствор 2-ацетилфурана в бензоле декантируют с осад-
ка в колбу Вюрца, из которой отгоняют бензол, а затем про-
дукт, кипящий при температуре 170—175°. Выход 2-ацетилфу-
рана 82,5 г (75% теории). При охлаждении в ледяной воде
2-ацётилфуран затвердевает в кристаллическую массу с темпе-
ратурой плавления 28—29°.
Примечания.
1. Пользуясь вышеописанной методикой, можно получить 2-пропиони.т-
фуран с выходом 51—56%, . 2-ацстилс.ильван (48,5—50%) и 2-пропионил-
сильван (40%).
2. В качестве катализатора ацилирования фурана вместо перхлората
магния может быть применена 57—70%-пая хлорная кислота [9], взятая в
очень небольших количествах (0,001—0,005 М на 1 М фурана). В этом
случае катализатор предварительно растворяют в части уксусного анги 1-
рида, а раствор при охлаждении осторожно добавляют к реакционной
смеси. Выход 2-ацетилфурана и 2-пропиопилфураиа составляет 67—70%.
74
ЛИТЕРАТУРА
1. Е. I. Bourne, Г. Stacey, I. С. Tdtlow, I. Л1. Tedder, J. Chem. Soc.
1951, 718.
2. 7". Reichstein, Helv. chim. acta 13, 356 (1930); fl. Л. Гольдфарб,
JI. M. Сморгонский, Ж. общ химии, 8. 1523 (1938).
3. Пат. США 2.460.822, 2.460.82г> 1949; Chem. abstrs, 43. 8465а (1949).
4. Н. D. Hurtough, A. I. Kosak, J. Amer. Chem. Soc., 68, 2639 (1946).
5. H. D. Hurtough, A. I. Kosak, J. Amer. Chem. Soc., 69, 1012 (1947),
6. J. V. Held, R. Leuine, J. Organ. Chem., 13, 409 (1948).
7. H. D. Hartough, A. I. Kosak, J. Amer. Chem. Soc.. 69, 3098 (1927).
8. Пат. США 2.515. 123 (1950), Chem. Abstrs, 44, 8955 (1950).
9, Г. H. Дорофеенко, Ж- общ. химии, 31, 994 (1961).
10. - Н. D. Hurtough, A.-I. Kosak, J. Amer. Chem. Soc., 69, 3093 (1947).
И. Ю. К. Юрьев, Г. Б. Еляков, Док.т. АН СССР, 86, 337 (1952).
12. G. Asahina, I. Muroyana, Arch. Pharm., 252. 443 (1914).
13. F. Kipnis, I. Onitelt. J. Amer. Chem. Soc., 70, 3948 (1948).
14. T. Gabuta. T. Tamura, J. Arg. Chem. Soc. Japan, 19, 546 (1943).
2-АЦЕТОТИЕНОН
Г. II. ДОРОФЕЕНКО, В. И. ДУЛЕНКО
(Институт органической химии АН УССР, Донецкое отделение)
нс-сн
II II
НС с со СНз
\ /
S
2-Ацетотиенон получают главным образом по реакции
Густавсона-Фриделя-Крафтса. Для этой цели используются
различные ацилирующие средства: уксусная кислота, хлори-
стый ацетил, уксусный ангидрид, кетен и тетраацетилоксиси-
лан [1]. 13 качестве катализаторов ацилирования применяют
хлористый алюминий [2—4], хлорное олово [5], четыреххлори-
стый титан [6], йод и йодистоводородную кислоту [7], эфираты
фтористого бора [8], ортофосфорную [9] и хлорную кислоту
[10, 11]. Другие некаталитические способы получения тиенил-2-
алкилкетонов [12] существенного интереса не представляют.
Предлагаемые ниже две методики получения 2-ацетотиено-
на и других кетонов тиофенового ряда состоят в ацилировании
тиофена уксусным ангидридом и хлористым ацетилом в при-
сутствии каталитических количеств перхлората магния (ан-
75
гидрона). Описываемый способ прост, удобен в выполнении и
требует очень мало времени для приготовления кетонов.
СИНТЕЗ 2-АЦЕТОТИЕНОНА
1) НС-СН Mg(C104)s НС-СН
|| II +(СН3СО),О--------- || II ЧСНзСООН
НС СН НС ССОСНз
\/ V/
S S
Характеристика основного сырья
Тиофен технический, перегнанный, с т. кип. 82—84°.
Уксусный ангидрид, ч.д.а.
Магний хлорнокислый безводный, ч.
Условия получения
В круглодонную колбу емкостью 250 мл, соединенную с
обратным холодильником, помещают 42 г (0,5 М) тиофена,
61,2 г (57 мл 0,6 М) уксусного ангидрида и 2,23 г (0,01 Л4) ан-
гидрона. По мере растворения ангидрона реакционная смесь
окрашивается в фиолетовый цвет, изменяющийся при нагрева-
нии до зеленого. Нагревание раствора проводят на кипящей
водяной бане в течение 25—30 минут, после чего реакционную
смесь выливают в холодную воду (300 мл) и после гидролиза
непрореагировавшего уксусного ангидрида отделяют органи-
ческий продукт, выделившийся в виде маслянистой жидкости
тяжелее воды, а водный слой дважды экстрагируют бензолом.
Полученный продукт, соединенный с бензольным экстрактом,
промывают раствором бикарбоната натрия, водой и высуши-
вают безводным сульфатом натрия. Бензольный раствор сли-
вают с осадка в перегонную колбу, растворитель отгоняют, а
2-ацетотиенон перегоняют, собирая фракцию с т. кип. 209—
213°. Выход кетона составляет 56,7—59.8 г (90—95% теории)
2) НС-СН Mg(ClO4)„ НС-СН
|| И +СН3СОС1-------I! || +НС1
НС СН НС ССОСНз
\ 's
Характеристика основного сырья
Тиофен технический перегнанный, с т. кип. 82—84“.
Хлористый ацетил, ч., свежеперегнаннып, с т. кип. 53—55
Магний хлорнокислый безводный, ч.
76
Условия получения
В круглодонную колбу емкостью 250 мл с обратным холо-
дильником помещают 42 г (0,5 М) тиофена, 58,8 г (0,75 Л1)
хлористого ацетила и 2,23 г (0,01 М) ангидрона. Реакционную
смесь нагревают в течение 15 минут на кипящей водяной ба-
не и затем выливают в 200 мл холодной воды. Выделившееся
1емное масло отделяют в делительной воронке, водный раст-
вор дважды экстрагируют бензолом, бензольный экстракт сое-
диняют с отделенным продуктом, промывают раствором би-
карбоната натрия, водой и высушивают сульфатом натрия.
При разгонке бензольного раствора получают 2-ацетотиенон
с т. кип. 210—214°. Выход кетона 37,8—41 г (60—65% тео-
рии) .
Примечания.
I. По методу 1 из тиофена и пропионового ангидрида может быть по-
лучен 2-пропиотиенон с выходом 64—69%.
2. Из тиофена и хлористого пропионила или бутирила по методике
2 получают 2-пропиотиенон (55%) и 2-бутиротиенон (65%).
3. Вместо перхлората магния (в тех же. каталитических количествах)
при ацилировании тиофена по методике 1 или 2 может быть использована
продажная 57—60%-ная хлорная кислота [И]. Выход кетонов с этим ка-
тализатором почти такой же. как при реакции с ангидроном.
ЛИТЕРАТУРА
1. Ю. К. Юрьев, Г. Б. Еляков. Докл. АН СССР, 86, 337 (1952).
2. Steinkopf, Ann., 543, 128 (1940).
3. Peter, Ber., 17, 2643 (1884).
4. L. Qattermann, Ber., 18, 3013 (1885).
5. Г. Стадников, Л. Каштанов, ЖРФХО, 60, 1117 (1928).
6. «Синтезы органических препаратов» сб. 2, стр. 76, ИЛ, 1949; Г. Стад-
ников, Л. Каштанов, Вег., 61, 1389 (1928).
7. И. D. Hartou.gh, A. I. Kosak, J. Amer. Chem. Soc., 68, 2639 (1946).
8. I. N. Heid, R. Levine, J. Oran. Chem., 13, 109 (1948).
9. H. D. Hrtough, A. I. Kosak, J. Amer. Chem. Soc., 69, 3093 (1947).
10. Пат. США 2.575.123 (1950); Chem. Abstrs. 44, 8955rf (1950).
11. Г. H. Дорофеенко, Ж. общ. химии, 31, 994 (1961).
12. «Гетероциклические соединения», т. I, стр. 181, ИЛ, 1953.
НИТРАТ хинолиния
В. М. ДЗИОМКО, И. А. КРАСАВИН
(ВНИИ химических реактивов и особо чистых химических веществ)
C9HsN.,O3 м. В. 192, 18
Нитрат хинолиния является промежуточным продуктом
при нитровании хинолина в мягких условиях по методу, опи-
санному на стр. 82.
77
В литературе имеется только одно сообщение, опубликован-
ное более 100 лет назад [1], о попытке получения этой соли пу-
тем упаривания раствора хинолина в слегка разбавленной
азотной кислоте, взятой в небольшом избытке. Образовавшая-
ся густая масса после охлаждения и застывания была пере-
кристаллизована из спирта. Автор охарактеризовал получен-
ное им вещество как белые иглы, устойчивые на воздухе и
«не плавящиеся при 100°».
Никаких других указаний па получение нитрата хиполиния
в литературе обнаружить не удалось, если не считать того,
что это вещество могло образоваться при некоторых вариан-
тах проведения реакции нитрования хиполнпа [2, 3], хотя и не
было выделено в чистом виде.
Нами был приготовлен химически чистый образец нитра-
та хинолиния, представляющего собой (после перекристалли-
зации из спирта или из смеси метанол—ацетон) белоснежные
игольчатые кристаллы с т. пл. 123—123,5°. Вещество хорошо
растворяется в воде и кипящих метиловом и этиловом спир-
тах, мало растворимо в этих же спиртах на холоду и в ацетоне.
Нами разработаны два варианта получения нитрата хинс-
линия— с применением слабой (54—60%-ной) азотной кисло-
ты и упариванием в вакууме [4] и из 80%-ной кислоты. Второй
способ дает худшие результаты, но более прост, т. к. не тре-
бует применения кислотостойкой вакуумной аппаратуры. Оба
метода допускают использование хинолина коксохимического
происхождения, который содержит, обычно, значительные ко-
личества примесей. Путем повторной кристаллизации легко
получить азотнокислую соль желаемой степени чистоты, тогда
как очистка хинолина-основания представляет, как известно,
трудную задачу.
Условия получения
Получение нитрата хинолиния с применением разбавлен-
ной азотной кислоты. В круглодонную короткогорлую колбу
емкостью 2—3 л со шлифом помещают 387,6 г (3 М) хинолина
(см. примечание 1) и, охлаждая се на водяной бане, медленно
добавляют 256 мл (3,15 Л4) 57%-ной азотной кислоты (см.
примечание 2). Присоединяют на шлифе насадку с капилля-
ром, доходящим до дна колбы, и отводом, ведущим к нисходя-
щему холодильнику и приемнику. Отвод приемника соединя-
ют с вакуум-насосом (см. примечание 3) и. по достижении ос-
таточного давления около 15—20 мм рт. ст., начинают мед-
ленно нагревать колбу на водяной бане. Отгоняют воду при
температуре бани не выше 80° (см. примечание 4). Продолжи-
тельность отгонки 2—3 часа, отгоняется около 140—150 мл
жидкости (см. примечание 5). Когда отгонка полностью пре-
кращается, прибор разбирают и еще теплую массу продукта
78
в колбе грубо измельчают, смачивая ее 100—150 мл ацетона.
Куски продукта извлекают из колбы (см. примечание 6), дро-
бят в ступке и перекладывают в воронку Бюхнера, отжимают
возможно более полно и отсасывают. Промывают на фильтре
150—200 мл ацетона, добавляя его небольшими порциями, ка-
ждый раз сильно отжимая и отсасывая (см. примечание 7).
Полученное вещество помещают в колбу емкостью 1,5 л и ра-
створяют в 200—300 мл метилового спирта при нагревании
на водяной бане. Прозрачный раствор слегка охлаждают и
добавляют к нему через обратный холодильник равный объем
ацетона, после чего вновь разогревают до полного растворения
и оставляют на ночь. Выпавшие кристаллы отсасывают, отжи-
мают, высушивают при 95—100° и растирают. Выход 430—
460 г (75—80% теоретического), т. пл. 120—121° (см. приме-
чания 8—-10).
Получение нитрата хинолиния с применением 80%-ной
азотной кислоты. Охлаждают 129,2 г (1 М) хинолина, поме-
щенного в фарфоровый стакан емкостью 300—400 мл, на ле-
дяной бане до 0—5° (см. примечания 4 и 11) и медленно при-
ливают при размешивании 57 мл (1,06 ЛГ) 80%-ной азотной
кислоты, предварительно освобожденной от окислов азота (см.
примечание 12). Оставляют на 3 часа на ледяной бане, затем
размешивают осадок с 30 мл ацетона, отсасывают, промывают
еще 30—50 мл ацетона, отжимают и перекристаллизовывают
из смеси 60 мл метилового спирта и 60 мл ацетона, как описа-
но выше. Выход сухого вещества 135—144 г (70—75% теоре-
тического). т. пл. 120—120.5°.
Примечания.
1. Применялся продажный хинолин квалификации «чистый», выпуска-
емый коксохимической промышленностью. Дополнительная перегонка его
перед употреблением не дала существенных преимуществ.
2. Пригодна продажная «слабая» азотная кислота с содержанием ос-
новного вещества 54—60%, если ее брать в количестве, соответствующем
198,5 г (3,15 М) 100%-ной.
3. Удобнее воспользоваться хорошим водоструйным насосом. При
использовании масляного вакуум-насоса его следует защитить от попада-
ния паров воды и кислоты при помощи ловушек с твердым едким натром.
4. При более высокой температуре усиливаются окислительные про-
цессы и продукт приобретает темно-красную окраску.
5. Для получения хорошего выхода важно отогнать воду возможно
более полно. Желательно, чтобы масса в колбе не застывала как можно
дольше. Для этого колба должна быть погружена в воду бани по самое
горло; не следует прерывать процесс или добавлять в баню холодную воду.
6. Иногда вещество в колбе застывает в твердую массу, извлечь кото-
рую оказывается трудно. Все же не рекомендуется перекристаллизовы-
вать продукт непосредственно в реакционной колбе без предварительного
измельчения и промывки, т. к. остатки азотной кислоты могут вызвать са-
мопроизвольную реакцию окисления метилового спирта.
7. Промывка ацетоном позволяет удалить остатки воды и азотной кис-
лоты, а также окрашенные в красный цвет примеси. Последние представ-
ляют собой, вероятно, продукты окисления гомологов хинолина, т. к. при
79
использовании чистого хинолина (полученного по реакции Скраупа) такой
окраски не наблюдается.
8. Продукт пригоден для нитрования. Качество его зависит от чистоты
применяемого хинолина. Вещество, полученное из хинолина коксохимиче-
ского происхождения, обычно сохраняет розовую или кремовую окраск},
которую можно устранить путем еще 1—2 кристаллизаций. Некоторые
сорта коксохимического хинолина дают соль, окрашенную в желтый цвет,
избавиться от которого значительно труднее.
9. Если использовать синтетический хинолин, то получается чисто-бе-
лое вещество с т. пл. 122—123°. При таком сырье нитрат можно и не пе-
рекристаллизовывать, а лишь промыть ацетоном. Выход в этом случае
увеличивается до 90--95% теоретического.
10. Маточные растворы от кристаллизации нитрата нельзя упаривать
по причинам, указанным в примечании 6.
11. Процесс сопровождается сильным разогревом. При недостаточном
охлаждении или применении более концентрированной кислоты вещество
сильно окисляется. Использование более разбавленной кислоты снижает
выход.
12. Присутствие окислов азота также способствует окислению вещест-
ва. Их удаляют, продувая кислоту быстрым током азота в течение 20—•
30 мин.
ЛИТЕРАТУРА
1. С. G. Williams, Jahresbcricht der Cliemie. 1856, 533; Trans. Roy. Soc.
Edinburgh, XXI (3), 377 (1856); Chem Gaz., 1856. 261, 283; Chem, Zbl, 1856,
817.
2. S. F. Dufton, J. Chem. Soc., 61, 782 (1892).
3. C. G. Le Fevre, R. J. W. Le Fevre, J. Cheni. Soc., 1935, 1470.
4. В. Л1. Дзиомко, И. А. Красавин, Ю. П. Радин, Удостов. о регистра-
ции, № 18267 от 10 мая 1959 г.
8-НИТРОХИНОЛИН И 5-НИТРОХИНОЛИН
И. А. КРАСАВИН, В. А/. ДЗИОМКО
(ВНИИ химических реактивов и особо чистых химических веществ)
Cc>HeN2CC
М, в. 174, 16
Наиболее часто для получения 8- и 5-нитрохинолинов при-
меняется нитрование хинолина смесью азотной и серной кис-
80
лот [1—16], при котором оба нитросоединения образуются од-
новременно в примерно равных количествах.
Для приготовления свободного от изомеров 8-нитрохиноли-
на иногда используется синтез Скраупа с о-нитроанилином
[10, 17—21]. Однако хорошие результаты (выход 80%) [20]
этот способ дает лишь при употреблении в качестве окисли-
теля соединений мышьяка [10, 18—21]. Ядовитость последних,
так же как и трудность регулирования хода реакции, ограни-
чивает применение такого метода. Еще менее удобно исполь-
зование его для синтеза 5-нитрохинолина, т. к. при циклизации
.и-нитроанилина по Скраупу [18, 22, 23] образуется смесь 5- и
7-нитрохинолинов (в соотношении 3,5:1) [23].
8-Нитрохинолин был получен также при нагревании 2-ме-
токсн-3-нитрокоричного альдегида со спиртовым аммиаком в
запаянной трубке при 130—140° [24], при нагревании 8-хино-
линсульфокислоты с дымящей азотной кислотой в запаянной
трубке при 160° [25], при декарбоксилировании 8-нитрохиналь-
диновой [26] п 8-нитроцннхонпновой [27] кислот.
Образование 5-нитрохинолина наблюдалось при декарбо-
ксилировании 5-нитрохинальдиновой [26], 5-нитропинхониновой
[28] и 5-нитрохинолин-8-карбоновой [23] кислот, а также при
нагревании диазотированного 5-нитро-8-аминохинолина со
спиртом [29].
Однако все эти методы имеют лишь теоретическое значе-
ние из-за малой доступности исходных веществ.
Все авторы, осуществлявшие нитрование хинолина смесью
азотной и серной кислот [1 —16] использовали для этой цели
«дымящую» азотную кислоту уд. веса 1,5—1,52 (94—100%-
ную) .
Почти во всех случаях применялась также и «дымящая»
серная кислота (слабый олеум) или олеум с содержанием
свободного серного ангидрида 20% [14], 40% [6—9, 11] и даже
65% [12]. Лишь в двух случаях указано применение «концен-
трированной» серной кислоты, наряду с «дымящей» азотной
кислотой [4, 13].
Суммарный выход нитрохинолинов достигает 86,4% [5].
Мы разработали метод нитрования хинолина в форме ни-
трата хинолиния, не требующий применения ни концентриро-
ванной азотной кислоты, ни олеума. Выход 8-нитрохинолина
составляет 35—38%, 5-нитрохинолина — около 30%, причем
потери возникают лишь при очистке и могут быть значительно
уменьшены путем выделения вещества из маточных растворов.
Нитрат хинолиния может быть приготовлен из хинолина и
50—60%-ной азотной кислоты (см. стр. 77).
Важным преимуществом нашего метода является возмож-
ность использования хинолина коксохимического происхожде
6 Зак. 1020 Ю
ния, который содержит обычно большое количество примесей.
Путем перекристаллизации нитрата легко осуществить пред-
варительную очистку исходного вещества, тогда как очистка
хинолина-основания весьма затруднительна. Степень очистки
удобно контролировать по т. пл. нитрата.
Благодаря применению высушенного нитрата хинолиния
процесс нитрования проходит легко и быстро в 100%-ной сер-
ной кислоте, и даже при использовании 95%-пой кислоты бы-
ли получены удовлетворительные результаты и не было обна-
ружено следов непрореагировавшего хинолина.
Следует отметить, что ранее уже применялся сходный ме-
тод нитрования хинолина [5, 10], но авторы этих работ исполь-
зовали дымящую азотную кислоту (уд. веса 1,5) и высоко-
концентрированный олеум уд. веса 2,0.
Нитрование хинолина в форме нитрата хинолиния
и разделение смеси изомеров
HNO,
• HNO3
В трехторлую колбу емкостью 1 я, снабженную мешалкой
и термометром, помещают 400 мл (7,5 Л4) 100%-ной серной
кислоты (примечание 1) и охлаждают до —3° (примечание 2).
Продолжая размешивание и охлаждение, вносят небольшими
порциями в течение 1 —1,5 часа 192,2 г (1,0 Л4) сухого тонко
измельченного нитрата хинолиния с такой скоростью, чтобы
температура не поднималась выше 5°, а твердые частицы ни-
трата полностью растворялись (примечание 3). Перемешива-
ют еще 1,5—2 часа при 2—5и, после чего добавляют 10 г
(0,1 Л4) сухого растертого азотнокислого калия за 10—15 ми-
нут, размешивают при 5° в течение 1—2 часов и оставляют па
ночь при комнатной температуре. На другой день прозрачный
желтый раствор медленно выливают при размешивании на
2 кг толченого льда, помещенного в колбу емкостью 5 л с ме-
82
шалкой. Колбу охлаждают снаружи льдом и постепенно при-
бавляют 1100—1200 .ил 25%-го раствора аммиака до щелоч-
ной реакции (примечание 4). Образовавшуюся суспензию раз-
мешивают при охлаждении еще 1,5—2 часа, затем осадок от-
сасывают, промывают несколько раз водой и отжимают.
Влажный осадок—смесь 5- и 8-нитрохинолинов—растворя-
ют в 400 мл 57%-ной азотной кислоты и 2,5 л воды при нагре-
вании. Раствор кипятят с 10—15 г активированного угля и
фильтруют в горячем виде. Фильтрату дают охладиться до на-
чала кристаллизации, перемешивают его в течение 4—5 часов,
пока он примет комнатную температуру, и оставляют до сле-
дующего дня (примечание 5).
Выпавшие желтые кристаллы нитрата 5-нитрохинолиния
отсасывают и промывают на фильтре водой два раза. Промыв-
ную жидкость собирают отдельно и отбрасывают, а фильтрат,
содержащий 8-нитрохинолин, сохраняют для дальнейшей пе-
реработки.
Получение 8-нитрохинолина.
I I I • HNOS 4- NH4OH — I I l-%NH4NO3 + H2O
IN IN
no2 no2
Фильтрат помещают в колбу емкостью 5 л и охлаждают
до 0—5°. При размешивании медленно добавляют 420—450 мл
25%-ного раствора аммиака до щелочной реакции (примеча-
ние 4). Образовавшуюся суспензию размешивают 1,5—2 часа
при охлаждении, осадок отсасывают, промывают 2 раза водой
и отжимают. Полученный 8-нитрохинолин, не высушивая, рас-
творяют в 400 мл метилового спирта (примечание 6) при на-
гревании с обратным холодильником. Раствор обрабатывают
активированным углем и фильтруют. К фильтрату добавляют
100 мл воды, вновь разогревают до растворения выделивше-
гося вещества и оставляют для медленной кристаллизации на
12—15 часов. Выпавшие игольчатые кристаллы отсасывают,
отжимают и сушат на воздухе или в эксикаторе. Выход 8-нит-
рохинолина 60—66 г (34,5—37,9%, теории), т. пл. 88—88,5°.
После повторной кристаллизации т. пл. 89—90° (примечания
7, 8).
Получение 5-нитрохинолина
NO, NO2
I I
II I • HNO3 + NH4OH — I I I % NH4NO3 % H2O
\/\/
N N
83
Кристаллический осадок нитрата 5-нитрохинолиния, полу-
ченный после отделения 8-изомера, перекристаллизовывают из
350—375 мл 10% азотной кислоты с применением активи-
рованного угля. Выход очищенного нитрата 79—80 г (33,3—
33,7% теории), т. пл. 187 -187,5° (примечание 8). Его раство-
ряют в 350 мл 10% азотной кислоты при нагревании и горячий
раствор при энергичном размешивании медленно прибавляют
ь колбу, содержащую 75 мл 25%-ного раствора аммиака. Колбу
нагревают на водяной бане в течение 30—40 минут (примеча-
ние 9), после чего охлаждают на ледяной бане до полного за-
твердевания 5-нитрохинолина. Осадок отфильтровывают, про-
мывают водой, отжимают и высушивают на воздухе. Получен-
ное вещество перекристаллизовывают из 150 мл метилового
спирта и высушивают в вакуум-эксикаторе над серной кис-
лотой (примечание 10). Выход 5-нитрохинолина 50—53 г
(28,7—30,4% теор.), т. пл. 70,5—71,5° (примечания 8,11).
Примечания.
1. При проведении синтеза в меньшем масштабе (0,2—^-0,5 М нитрата
хинолиния) удовлетворительные результаты можно получить с 95—97%-нон
серной кислотой.
2. При этом серная кислота затвердевает, по вскоре после начала
нитрования она вновь становится жидкой.
3. Если останутся (на стенках колбы п т. п.) нерастворнвшиеся ча-
стицы нитрата, то конечный продукт будет загрязнен хинолином.
4. В начале нейтрализацию можно вести значительно быстро, при уме-
ренном охлаждении. Когда появится не исчезающее при размешивании по-
мутнение, жидкость следует охладить до 2—5° и дальнейшее прибавление
раствора аммиака производить при хорошем охлаждении и размешивании
с такой скоростью, чтобы вещество выделялось в твердом виде, а не в виде
масла.
5. Для наиболее полного разделения изомеров необходимо, чтобы ра-
створ охлаждался медленно и перемешивался до тех пор, пока выделится
большая часть нитрата 5-нитрохинолинпя.
G. Метиловый спирт может быть заменен на этиловый.
7. По литературным данным, т. пл. 8-нитрохинолина 87° [8]; 87—88" [21];
88—89° [10, 12], 89° [2, 17, 26]; 89—90° [20]; 90-91,5° [27]; 91—92° [24].
8. Общий выход нитрохинолинов может быть значительно повышен,
если вещество выделить из маточных растворов и присоединить к реакци-
онной массе при повторении синтеза.
9. Необходимо полностью нейтрализовать азотнокислую соль.
10. 5-Нитрохиполии кристаллизуется в форме моногидрата. Для получе-
ния безводного вещества приходится применять длительное высушивание в
вакуум-эксикаторе.
11. По литературным данным, т. пл. 5-нитрохинолина (безводного) 70’
[23], 70—71° [12], 71" [23, 26], 71—72° [13], 72е [2, 5, 8, 10, 29].
ЛИТЕРАТУРА
1. W. Koenigs, Вег., 12, 448 (1879)
2. A. Claus, Т. Kramer, Вег., 18, 1243 (1885).
3. Е. Noelting, Е. Trautmann, Вег., 23, 3654 (1890).
4. 5. F. Dufton, J. Chem. Soc., 59, 756 (1891).
5. S. F. Dufton, J. Chem. Soc., 61, 782 (1892).
6. A. Claus, E. Setzer, J. prakt. Chem.. [2], 53, 390 (1896).
7. W. Meigen, J. prakt. Chem., [2] 77, 472 (1908).
84
8. R. P. Dikshoorn, Recueil trav. chim., 48. 147 (1'129).
9. IT. Langenbeck, R. Jnttemann, F. Hellrung, Liebigs Ann. Chem.,
499, 201 (1932).
10. C. G. Le Fevre, R. J. W. Le Fevre, J. Chem. Soc., 1935, 1470,
II. W. Borsche, Л1. Wagner-Roemmich, Liebigs Ann. Chem., 544, 280
(1940).
12. L. F. Fleser, E. B. Hershberg. J. Amer. Chem. Soc.. 62, 1640(1940).
13. J. W. Wetzel, D.E. Weldton, J.E. Christian, G.L. Jenkins, G.B. Ba-
chman, J. Amer. Pharm. Assoc., 35, 331 (1946).
14. F. II. S. Curd, W. Graham, D. N. Richardson, F. L. Rose. .1. Cliein.
Soc.. 1947, 1613.
15. E. A. Steck, Q. W. Ewing, .1. Amer. Chem. Soc., 70, 3397 (194-8).
16. M. J. S. Dewar, P. M. Malllis, J. Chem. Soc., 1957, 2521.
17. W. La Caste, Ber., 16. 669 (1883).
18. C. A. Knueppel, Ber., 29, 703 (1896).
19. 0, F. Smith, C. A. Getz, Chem. Reviews, 16, 113 (1935).
20. F. Misani, M. T. Bogert, J. Organ. Chem., 10, 347 (1945).
21. H. L. Yale, J. Bernstein, J. Amer. Chem. Soc.. 70 254 (1948).
22. H. Decker, J. prakt. Chem., [2] 63, 573 (1901).
23. L. Bradford, t. J. Elliott, F. M Rowe, .1. Chem Soc., 1947, 437.
24. W. V. Miller, F. Kinkelin, Ber., 22, 1716 (1889).
25. A. Claus, Kttttner, Ber. 19, 2886 (1886).
26. E. Besthorn. J. Ibele, Ber., 59, 23^9 (1906).
27. M. Ishikawa, J. Kikkawa, J. Pharm. Soc. Japan, 75. 33 (1955); Chem.
Abstrs., 50, 10071 (1956).
28. W. Koenigs, E. Lossow, Ber.. 32, 717 (1899).
29. R. P. Dikshoorn, Recueil trav. chim., 48. 517 (1929).
8-АМИНОХИНОЛИН
В. M. ДЗИОМКО, Я. А. КРАСАВИН
(ВНИИ химических реактивов и особо чистых химических веществ)
c,h8n2
М. в. 144,18
Практическое значение имеют два способа получения
8-аминохинолина: аминирование 8-оксихинолина [1—7] и вос-
становление 8-нитрохинолина [8—25].
Аминирование 8-оксихинолина осуществлялось при дейст-
вии водного аммиака под давлением (20—22 атм) в присутст-
вии сульфита аммония [1—3,5] или бисульфита натрия [4].
Выход достигает 93% [5]. Существенным недостатком этого
метода является необходимость применения автоклава.
85
Другой способ аминирования 8-оксихинолина состоит в на-
гревании его до высоких температур с аммиачным комплексом
хлористого цинка [6] или хлористого кальция [7]. Выход не
превышает 50% [7].
Значительно чаше для получения 8-аминохинолина приме-
нялось восстановление 8-нитрохинолина (или его смеси с 5-
нитрохинолином, с последующим разделением аминов). В ка-
честве восстановителя были использованы: металлическое
[8] или хлористое олово [9—15] в солянокислом растворе, по-
рошкообразное железо в уксусной кислоте [12, 13, 15, 16], в
очень слабо-кислой среде [17] или в спирте в присутствии без-
водного хлористого кальция [18]. Однако тщательные иссле-
дования показали [12, 14, 15, 19], что применение хлоридов и
соляной кислоты приводит к получению загрязненного, частич-
но хлорированного продукта и лишь восстановление железом
в разбавленной уксусной кислоте дает хорошие результаты
(Выход 75%) [12].
В более поздних работах описано восстановление 8-нитро-
хинолина сульфатом гидразина в жидком аммиаке под давле-
нием (выход около 80%) [20] и каталитические методы: гидри-
рование водородом на никеле Ренея [21, 22] или на окиси пла-
тины [23], а также восстановление гидратом гидразина в при-
сутствии палладия на угле [24] или никеля Ренея [25]. Выход
составляет соответственно 69% [21], 96% [23], 65% [24], 90—
95% [25].
Известны и другие реакции, ведущие к образованию 8-ами-
нохинолина [26—28], но они имеют только теоретическое зна-
чение.
Мы проверили несколько методов восстановления 8-нитро-
хинолина в амин. При использовании гидразин-гидрата в при-
сутствии никеля Ренея [25] нами были получены неудовлетво-
рительные результаты.
Простой и удобный способ восстановления порошкообраз-
ным железом в разбавленной уксусной кислоте [12] дал не-
устойчивый выход амина (от 30 до 63%), зависящий от ка-
чества железного порошка.
Нами был также разработан не применявшийся ранее ме-
тод восстановления 8-нитрохинолина сульфидом аммония в
среде метилового спирта с выходом амина 69—79%.
Получение 8-аминохинолина.
| | | + 3(Na,S-9HsO)+ 6NH4C1 —
V
NO,
86
-> | | | +6NaC14-3S4-6NH3-f-29H.2O
w
nh2
В трехгорлую колбу емкостью 1—1,5 л, снабженную ме-
шалкой, обратным холодильником и капельной воронкой, по-
мещают 26,1 г (0,15 Л1) 8-нитрохинолина и 400 мл метилового
спирта и нагревают на водяной бане до растворения. Добав-
ляют 63,5 г (1,17 Л4) тонко измельченного хлористого аммония
и размешивают при кипении растворителя 15—20 минут. Одно-
временно приготавливают раствор 152,7 г (0,585 Л4) кристал-
лического сернистого натрия (см. примечание 1) в 200 мл ме-
тилового спирта при нагревании на водяной бане с обратным
холодильником. Полученный горячий раствор сульфида нат-
рия прибавляют через обратный холодильник к хорошо разме-
шиваемой суспензии, находящейся в трехгорлой колбе, от ко-
торой временно отставляют баню с горячей водой. Прибавле-
ние производят в течение 5—10 минут с такой скоростью, что-
бы метиловый спирт спокойно кипел. Затем возобновляют на-
грев, кипятят в течение 30 минут и слегка охлаждают. Добав-
ляют через капельную воронку 260 мл разбавленной (1:1)
соляной кислоты до кислой реакции на бумажку конго, отводя
выделяющийся сероводород через холодильник в ловушку
(см. примечание 2). Снова кипятят 30 минут. Горячую жид-
кость фильтруют от осадка серы, осадок промывают горячей
водой до исчезновения красного окрашивания промывных вод
и отбрасывают его. Фильтрат и промывные воды соединяют,
добавляют 8—10 г активированного угля и метиловый спирт
полностью отгоняют. Оставшийся водный раствор фильтруют,
фильтрат охлаждают на ледяной бане и подщелачивают 20%-
ным раствором едкого натра. Выделившийся и затвердевший
при охлаждении 8-аминохинолин отфильтровывают, фильтрат
экстрагируют эфиром (2 раза по 150 мл) и в экстракте раст-
воряют осадок, Раствор промывают водой, высушивают без-
водным сульфатом натрия, добавляют 5—7 г активированного
угля и оставляют на 8—10 часов (см. примечание 3). Затем
уголь отфильтровывают и эфир отгоняют. Остаток перегоняют
в вакууме при 120—123° (5 мм) (см. примечания 4 и 5). Ве-
щество застывает в приемнике в светло-желтую массу. Выход
16—17 г (74—78,6% теор.), т. пл. 64—65° (см. примечания
6—9).
Примечания.
1. Применялся кристаллический девятиводпый сернистый натрий ква
лификации «чистый» с содержанием основного вещества не ниже 92%. Не
пригоден сернистый натрий, хранившийся длительное время при доступе
воздуха. Раствор его в метиловом спирте должен быть почти прозрачным.
2. Под конец добавления соляной кислоты происходит энергичное вы-
87
деление сероводорода. Згу операцию, так же как и последующее фильтро-
вание следует проводить под тягой.
3. Отработка эфирного раствора активированным углем позволяет
устранить примесь серусодержащих соединений, придающих продукту не-
приятный запах.
4. По литературным данным, т. кип. 12375 мм [30]; 157—158719 мм
[29]; 157—162720—24 мм [13].
5. Перегонку можно произвести из колбы Клайзена с елочным дефлег-
матором, используя is качестве холодильника короткий форштосс.
6. По литературным данным т. п. 62,5—64° [24]; 63—64° [5, 25]; 64—65'
[23]; 64,5 [26, 27]; 64,5—65° [4]; 65 [12, 13, 28]; 65—65,5° [1, 3]; 65—66° [22];
66-67° [6]; 67° [9]; 67—70’ [17]; 70° [30].
7. Для окончательной очистки 8-аминехинолина (иногда очень неболь-
ших количеств его) применялась перегонка с водным паром [4, 9, 11, 12], пе-
регонка в вакууме [5, 7, 13, 29, 30], сублимация [13] и перекристаллизация
из воды [1, 3], спирта [17, 20] петролейного эфира [24, 26], лигроина [6, 27],
бензина [28], октана [22] или из смеси эфир-гекеан [23].
8. Вещество следует хранить в плотно закрытых банках из темного
стекла.
9. При проведении синтеза в увеличенном в 4 раза масштабе и,
104,5 г (0,6 М) 8-нитрохинолина было получено 59,6—61,9 г (69—71,7%
теор.) амина с т. пл. 63,5—65°.
Л И I Е Р А Т У Р А
1. Н. Н. Ворожцов, И. М. Коган, Вег., 65, 112 (1932).
2. И. И. Ворожцов, И. Л(. Коган, Авт. свпд. 28215 (1932).
3. И. М. Коган, Труды МХТИ, вып. Ill—IV, 55 (1938).
4. Г. И. Михайлов, Труды НЕВА, вып. 20, Гос.химиздат, 1951, стр. 264
5. Е. С. Hardts, J Organ Chem., 23, 891 (1958).
6. С. Bedall, О. Fischer, Ber., 14, 2570 (1881).
7. IF. Borsche, M. Wagner-Roemmich, Liebigs Ann. Chem. 544,280(1940).
8. W. Koenigs, Ber., 12, 448 (187 9).
9. A. Claus, T. Kramer, Ber., 18, 1243 (1885).
10. S. F. Dufton, J. Chem. Soc., 59, 756 (1891).
11. W. Melgen, J. prakt. Chem., (2) 77, 472 (1908).
12. R. P. Dikshoorn, Recueil trav. chim., 48, 147 (1929).
13. A. Kaufmann, O. Zeller, Ber., 50, 1626 (1917).
14. R. H. Baker, C. J. Albisetti jr„ R. M.. Dodson, О R. Lappin,. B. Rie-
gel, J. Amer. Chem. Soc., 68, 1532 (1946).
15. J. Howitz, H, Fraenkel, E. Schroeder, Liebigs Ann. Cliem., 396, 53
(1913).
16 F. H. S. Curd, W. Graham, D. N. Richardson, F. L. Rose, .1. Chem.
Soc., 1947, 1613.
17. A. Claus, E. Setzer, J. prakt. Chem., (2) 53, 390 (1896).
18. R. Seka, Monatsh. Chem., 45, 287 (1924).
19. С. C. Price, D. B. Guthrie, J. Arner. Chem. Soc., 68, 1592 (1946),
20. K. Takeda, M. Tokuyama, J. Pharm. Soc. Japan, 74, 1274 (1954);
Chem. Abstrs., 49, 3192e (1955).
21. R. Winterbottom. J. Amer. Chem, Soc., 62, 160 (1940).
22. E. A. Steck, G. W. Ewing, J. Amer. Chem. Soc.- 70. 3397 (1948).
23. L. F. Fieser, E. В Hershberg. J. Amer. Chem. Soc., 62, 1640 (1940).
24. M. J. S. Dewar, T. Mole, J. Chem. Soc, 1956, 2556.
25. T. L. Fletcher, Al. J. Namkung, J. Organ. Chem., 23, 680 (1958).
26. H. H. Ворожцов мл.. С. П. Миценгендлер, Ж. общ. химии, 6, 681
(1936).
27. С. П. Миценгендлер, Н. Н. Ворожцов мл., Авт. евид.. 47691 (1936).
28. L. Kochanska, В Bobranski, Ber., 69В, 1807 (1936).
29. R. Р. Dikshoorn, Recueil tiav. chim., 48, 517 (1929).
30. F. Misani, M. T. Bogert. J. Organ. Chem., 10, 347 (1945)
88
5-АМИ НОХИ НОЛ И Н
И. А. КРАСАВИН, В. М ДЗИОМКО
(ВНИИ химических реактивов и особо чистых химических веществ)
М1г
Х/ V
C9H8N2 м. в. 144, 18
Единственным способом получения 5-аминохинолина, имею-
щим практическое значение, является восстановление 5-нитро-
хинолина [1 —13]. Известен лишь один пример, когда 5-амино-
хинолин был получен иначе, а именно путем нагревания 5-ок-
сихинолина с аммиачным соединением хлористого цинка при
300° [14].
Восстановление 5-нитрохинолина в амин было осуществле-
но при помощи хлористого олова в соляной кислоте [1—3] с
выходом до 64% [3], порошкообразного железа в солянокислом
[2] или уксуснокислом [3—6] растворе с выходом до 60% [3] и
сульфата гидразина в жидком аммиаке под давлением при
100° с выходом около 80% [7].
Описано также каталитическое гидрирование 5-нитрохино-
лина водородом в присутствии никеля Ренея [8—10], палладия
на угле [11, 12] или окиси платины [13]. Выход от 75% [9] до
95% [13], считая на неочищенное вещество, или 82% [13] — на
очищенное.
Следует отметить, что в недавно опубликованной работе
[16] сообщается о неудачных попытках восстановления 5-нит-
рохинолина в 5-аминохинолин: при проведении процесса в
различных условиях всегда имело место значительное осмоле-
пие' и выход амина не превышал 15—20%.
Мы неоднократно проводили восстановление 5-нитрохино-
лина хлористым оловом в солянокислом растворе по литера-
турным данным [3] и не встретили при этом каких-либо за-
труднений. Амин неизменно получался с выходом 55—60% в
виде зеленоватых хлопьев, которые после очистки приобрета-
ли вид светло-желтых игл с т. пл. 105°, как и у автора ориги-
нальной работы [3].
В дальнейшем нам удалось существенно усовершенство-
вать методику восстановления, благодаря чему 5-аминохино-
лин получается (с выходом 80—82%) непосредственно после
выделения из реакционной массы в виде светло-желтых кри-
сталлов с т. пл. 105—106,5°. Наиболее важные изменения заклю-
чаются в употреблении избытка хлористого олова, проведе-
нии реакции при 60°, разбавлении реакционной массы водой
89
сразу по окончании восстановления и в применении экстрак-
ции продукта бензолом. Мы обнаружили также, что бензол
является значительно более удобным растворителем для пе-
рекристаллизации 5-аминохинолина, чем применявшиеся ра-
нее эфир [1, 13], спирт [7, 8] или разбавленный спирт [3].
Получение 5-аминохинолина.
NO„ NH2 NH2
I ! I
SnCl2 /\/\ NaOH
| | |----> ] | |.SnCl4.2HCl —* | | |
NN N
Растворяют при нагревании 43,5 г (0,25 М) 5-нитрохиноли-
на в 125 мл 35%-ной соляной кислоты, помещенной в стакан
емкостью 1 л с мешалкой, термометром и капельной воронкой,
и раствор охлаждают до 30°. К нему прибавляют по каплям
раствор 200 г (0,886 /И) кристаллического хлористого олова
(см. примечание 1) в 50 мл 35%-ной соляной кислоты при ин-
тенсивном размешивании. Реакционная масса приобретает
вначале оранжевый, а после добавления ’А—7з части раство-
ра ярко-красный цвет, причем температура быстро возрастает
до 60—65°. Стакан немедленно погружают в глубокую баню с
ледяной водой (см. примечание 2) и остальную часть раствора
хлористого олова прибавляют с такой скоростью, чтобы под-
держивалась температура 55—60°. Сразу же по окончании
прибавления, которое длится около 1 часа, образовавшуюся
ярко-красную густую пасту разбавляют 200 мл воды.
Для завершения реакции нагревают стакан на водяной
бане за 30 минут до 100° и выдерживают на кипящей бане
1 час. Охладив содержимое стакана до загустевания, медлен-
но выливают его при хорошем размешивании в сосуд с раст-
вором 240 г едкого натра в 760 мл воды, погруженный в баню
со льдом. Полученную светло-желтую суспензию перемешива-
ют до полного исчезновения в ней частиц красного цвета (см.
примечание 3), осадок отсасывают, промывают водой и отжи-
мают. Затем его растворяют при нагревании на водяной бане
в 700 мл бензола и образовавшийся водный слой отделяют.
Бензольный раствор кипятят с активированным углем, филь-
труют и фильтрат упаривают на водяной бане до начала
кристаллизации. Выделившиеся при стоянии светло-желтые
кристаллы 5-аминохинолина отделяют и сушат в вакуум-экси-
каторе, защищая от действия света (см. примечание 4). Выход
22—24 г (61—67% теории) т. пл. 104,5—106,5°. Маточный ра-
створ еще раз упаривают и получают дополнительно 6—7 г
вещества с т. пл. 103—104,5°. Общий выход 5-аминохинолина
29—29,7 г (80,5—82,4% теории). После перекристаллизации
90
из 250—300 мл бензола получают 24,1—24,7 г (67—68,6% тео-
рии) продукта с т. пл. 107—108° (см. примечания 5 и 6).
Примечания.
1. Применялось кристаллическое двухводное хлористое олово квали-
фикации «чистое».
2. Подъем температуры начинается внезапно, и в этот момент тре-
буется интенсивное охлаждение. Когда температура установится при 55—
60°, эффективность охлаждения снижают, чтобы не замедлять течение ре-
акции.
3. Необходимо растереть стеклянным шпателем все крупные комки и
частицы красного цвета до образования однородной желтой суспензии.
4. Продукт чувствителен к действию света и воздуха.
5. По литературным данным, т. пл. 105—106° [3], 108—110° [13], 109—
110° [4, 5, 14], 110° [2, 3], 110—11 Г [8]. Имеются указания [3] на го, что при
восстановлении 5-нитрохинолина хлористым оловом в солянокислом раст-
воре образуется 5-аминохинолин с т. пл. 105—106°, которая не повышается
даже после двухкратной перекристаллизации. Предположение о возмож-
ности частичного хлорирования продукта в процессе восстановления и о
присутствии хлорсодержащих соединений не оправдалось, т. к. чувстви-
тельная проба не показала наличия хлора в образцах амина, полученного
этим способом [3].
6. Вещество следует хранить в плотно закрытых банках из темного
стекла.
ЛИТЕРАТУРА
1. 5. F. Dufton, J. Chem. Soc., 61, 782 (1892).
2. A Claus, Е. Setzer, J. prakt. Chem., (2) 53, 390 (1896).
3. R. P. Dikshoorn, Recueil trav. chim., 48, 147 (1929).
4. A. Kaufmann, O. Zeller, Ber., 50, 1626 (1917).
5. M. J. S. Dewar, P. M. Maitlis, J. Chem. Soc., 1957, 944.
6. F. H. S. Curd, W. Graham, D. N. Richardson, F. L. Rose, .1. Chem.
Soc., 1947, 1613.
7. K. Takeda. M. Tokuyama, J. Pharm. Soc. Japan, 74, 1274 (1954);
Chem. Abstrs., 49, 3192 e (1955).
8. E. A. Steck, G. W. Ewing, J. Amer. Chem. Soc., 70, 3397 (1948).
9. R. Winterbottom, J. Amer. Chem. Soc., 62, 160 (1940).
10. A. Roe, G. F. Hawkins, J. Amer. Chem. Soc., 71, 1785 (1949).
II. W. Langenbeck, R. Jtlttemann, F. Hellrung, Liebigs Ann. Chem., 499,
201 (1932).
12. W. Porsche, M. Wagner-Roemmich, Liebigs Ann. Chem., 544, 280
(1940).
13. L. F. Fieser, E. B. Hershberg, J. Amer. Chem. Soc., 62, 1640 (1940).
14. C. Riemerschmied, Ber., 16, 721 (1883).
15. Ю. А. Банковский, Л. А. Федотова, Д. Э. Зарума, Ж- общ. химии,
30, 1614 (1960).
5-ХЛОРХИНОЛИН И НИТРАТ 5-ХЛОРХЙНОЛЙНЙЯ
И. А. КРАСАВИН. В. М. ДЗИОМКО
(ВНИИ химических реактивов и особо чистых химических веществ)
С1 С1
I I
II I [Д I-HNO,
N ч/ N
C,H6C1N М. в. 163, 61 C9H,C1N2O3 М. в. 226, 63
В литературе имеются сведения о получении 5-хлорхиноли-
на путем циклизации л-хлоранилина по Скраупу [1—6], по ре-
акции Зандмейера из 5-аминохинолина [7—10], при декарбо-
ксилировании 5-хлорхинальдиновой кислоты [11], при восста-
новлении 4,5-дихлорхинолииа оловом в солянокислом раство-
ре [12], а также в результате побочной реакции при взаимодей-
ствии 1-(5-хинолил)-3,3-диметилтриазена с бензолом в токе
хлористого водорода [13]. Три последних способа нс имеют
препаративного значения.
Серьезным недостатком синтеза по Скраупу является то,
что реакция с л-хлоранилином приводит к образованию смеси
изомерных 5- и 7-.хлорхинолинов. Соотношение их зависит от
условий реакции, но 7-изомера образуется всегда больше (в
1,4—3,9 раза) [10]. Удовлетворительного метода разделения
этой смеси не описано.
Напротив, замена аминогруппы в 5-аминохинолине на хлор
по реакции Зандмейера позволила получить свободный от
изомеров 5-хлорхинолин с выходом до 59% [10].
В литературе имеется упоминание о получении нитрата
5-хлорхинолиния в виде бесцветных призматических игл с
т. пл. 159—161° (из воды) [10].
Мы проверили и уточнили метод [10] получения 5-хлорхи-
нолина из 5-аминохинолина. Благодаря введению некоторых
усовершенствований и использованию свежеприготовленной
хлористой меди удалось повысить выход до 65—68%. Кромер
того, мы разработали способ получения 5-хлорхинолина с вы-
ходом 57% непосредственно из 5-нитрохинолина без выделе-
ния промежуточно образующегося амина.
В обоих случаях продукт реакции может быть выделен как
в виде основания, так и в виде азотнокислой соли. Последнее
целесообразно в тех случаях, когда предполагается нитрова-
ние 5-хлорхинолина. Перевод основания в нитрат осуществля-
ется количественно, выход соли после перекристаллизации
90—92% (с учетом продукта, выделенного из маточного ра-
створа) .
92
Получение 5-хлорхинолина из 5-нитрохинолина.
no2 nh2
I I
/\/\ SnCl2 /XXX NaNOj
I I I НСГ~* | |' | HC1
X/ V
N=N-'C1- Cl
В стакане емкостью 1 л, снабженном мешалкой, термомет-
ром и капельной воронкой, растворяют при нагревании 43,5 г
(0,25 М) 5-нитрохинолина в 125 мл концентрированной соля-
ной кислоты и охлаждают до 8—10°. При хорошем размеши-
вании прибавляют к образовавшейся желтой суспензии раст-
вор 200 г (0,886 Л4) кристаллического хлористого олова (см.
примечание 1) в 50 мл концентрированной соляной кислоты.
ААасса приобретает ярко-красную окраску и разогревается.
Продолжительность прибавления 1 час, температуру поддер-
живают в пределах 55—60°. Затем охлаждают образовавшую-
ся ярко-красную пасту до —8° и через капельную воронку,
конец которой опущен под поверхность жидкости, прибавляют
при хорошем размешивании раствор 17,2 г (0,25 М) нитрита
натрия в 50 мл воды. Температуру поддерживают в пределах
от —5° до —2°, продолжительность прибавления 1 час, конец
диазотирования контролируют по йодкрахмальной бумажке.
После 30-минутной выдержки при этой же температуре сни-
мают избыток азотистой кислоты добавлением сульфаминовой
кислоты
Одновременно с диазотированием приготавливают в кру-
глодонной колбе емкостью 3 л (см. примечание 2) раствор
свежеосажденной хлористой меди (см. примечание 3) в 112мл
концентрированной соляной кислоты и охлаждают его до 0°.
Диазораствор прибавляют за 15 минут к раствору катали-
затора (см. примечание 4), регулируя охлаждение таким об-
разом, чтобы температура смеси не превышала 6—8°. Остав-
ляют па 30 минут на холоду, затем за 30 минут нагревают на
водяной бане до 90° и выдерживают еще 30—40 минут. Реак-
ционную массу подщелачивают раствором едкого натра и от-
гоняют 5-хлорхинолин с водяным паром, собирая 3—3,5 л ди-
стиллата (см. примечание 5). Последний экстрагируют эфи-
ром (3 раза по 300 мл), экстракт высушивают безводным
сульфатом натрия и эфир отгоняют. Остаток перегоняют в
93
вакууме при 94—9572 мм рг. ст. (см. примечание 6). Продукт
закристаллизовывается в приемнике. Выход 5-хлорхинолина
23,4 г (57,2% теоретического, считая на 5-нитрохинолин),
т. пл. 43°.
Получение нитрата 5-хлорхинолиния из 5-аминохинолина.
NH3 N=N+C1-
NaNO2 Cu.Cls
I I | нсг~7 | | *
N N
С1 С1
I I
| | [ • HNO3.
N N
В стакане емкостью 300—400 мл, снабженном мешалкой
и термометром, растворяют 14,4 г (0,1 Л4) 5-аминохинолина
(см. примечание 7) в смеси 60 мл концентрированной соляной
кислоты и 50 мл воды при нагревании и охлаждают при сильном
размешивании до —5°. К выделившейся суспензии розового
цвета добавляют за 20—25 минут раствор 7 г (0,1 М) нитрита
натрия в 25 мл воды из капельной воронки, конец которой по-
гружен под поверхность жидкости. Во время прибавления
поддерживают температуру от —4 до —2°, а затем размеши-
вают при этой же температуре еще 15 минут.
Одновременно с диазотированием готовят в круглодонной
колбе, емкостью 1 —1,5 л (см. примечание 2) раствор свеже-
осажденной хлористой меди (см. примечание 8) в 45 мл кон-
центрированной соляной кислоты и охлаждают его до 0°.
Раствор диазосоединсния добавляют за 10—15 мин. к охла-
жденному раствору хлористой меди, поддерживая температу-
ру 6—8°, оставляют на 15 минут на холоду, затем — на 45 ми-
нут при комнатной температуре. Потом нагревают на кипящей
водяной бане в течение 1 часа. Подщелачивают раствором ед-
кого натра и перегоняют с водяным паром, собирая 500--
600 мл дистиллата (см. примечание 5). Охладив дистиллат.
экстрагируют его эфиром (3 раза по 100 мл) (см. примеча-
ние 9), эфирный раствор взбалтывают в конической колбе со
смесью 8 мл 57%-ной азотной кислоты и 32 мл воды, причем
на стенках колбы выделяется белый осадок нитрата 5-хлор-
хинолиния. Эфир сливают в делительную воронку и встряхи-
вают со смесью 12 мл такой же кислоты и 168 мл воды в 2—3
приема. Азотнокислые экстракты возвращают в колбу с осад-
ком, осторожным нагреванием удаляют остатки эфира, а за
94
тем добиваются растворения всего осадка. Раствор обраба-
тывают активированным углем, фильтруют в горячем виде и
оставляют для медленного охлаждения. Выделившиеся круп-
ные бесцветные кристаллы собирают и сушат. Выход 12,4 г,
т. пл. 159—159,5°. Из маточника путем подщелачивания, экст-
ракции эфиром, реэкстракции небольшим количеством раз-
бавленной азотной кислоты и перекристаллизации получают
еще около 2,8 г вещества с т. пл. 158,5—159,5°. Общий выход
нитрата 5-хлорхинолиния 15,2г (67,1% теоретического).
Примечания.
1. Применялось кристаллическое двухводное хлористое олово квали-
фикации «чистое». Качество его существенно влияет на выход конечного
продукта.
2. Колбу заранее, приспосабливают для последующей перегонки с па-
ром.
3. Катализатор — хлористую медь приготавливают заблаговременно в
тот же день. К нагретому до 60—70° раствору 51,2 г (0,3 Л-1) кристалличе-
ской двухводной хлорной меди и 7,3 г (0,125 Л1) хлористого натрия в
250 мл воды добавляют 150 г свежего раствора бисульфита натрия (полу-
ченного путем насыщения сернистым ангидридом раствора 6,6 г безводно-
го углекислого натрия в 125 мл воды до привеса 18—20 г). Выпавший бе-
лый осадок хлористой меди отсасывают, сильно отжимают и немедленно
растворяют в соляной кислоте.
4. Выделение азота происходит немедленно, масса вспенивается и ра-
зогревается.
5. Перегонку продолжают до получения прозрачного погона.
6. Температура кипения 5-хлорхиполина при атмосферном давлении
256—257° [10].
7. Вместо 5-аминохинолина можно применить его дигидрохлорнд в ко-
личестве 21,7 г (0,1 А1).
8. Катализатор приготавливают, как описано в примечании 3, но коли-
чества реагентов уменьшают в 2,5 раза.
9. При желании получить 5-хлорхинолин-основание эфирные экстракты
перерабатывают, как описано выше.
ЛИТЕРАТУРА
1. W. La Costs, J. Bodewig, Ber., 17, 926 (1884).
2. W. La Costs, Ber., 18, 2940 (1885).
3. A. Claus, R. Kayser, J. prakt. Chem., [2] 48, 270 (1893).
4. E. Fourneau, T. st. T-me Trefouel, A. Wancolle, Bull. Sec. chim.
France, (4) 47, 738 (1930).
5. С. C. Price, D. B. Guthrie, J. Amer. Chem. Soc., 68, 1592 (1946).
6. A. K. Sen, N. K. Ray, U. P. Basu, J. Sci. Ind. Research, 1 IB, 325
(1952); Chem. Abstrs, 47, 10 534i.
7. J. Freydl, Monatsh. Chem., 8, 580 (1888).
8. A. Claus, C. Nassau, J. prakt. Chem., [2] 48, 170 (1893).
9. J. Howitz, H. Fraenkel, E. Schroeder, Liebigs Ann. Chem., 396, 53
(1913).
10. L. Bradford, T. J. Elliott, F. M. Rowe, J. Chem. Soc., 1947, 437.
11. A. M. Spivey, F. H. S. Curd, J. Chem. Soc., 1949, 2656.
12. H. Andersag, Chem. Ber., 81, 499 (1948).
13. J. Elks, D. H. Hey, J. Chem. Soc., 1943, 441.
5-ХЛОР-8-НИТРОХИНОЛ ИН
И. А. КРАСАВИН, В. М. ДЗИОМКО
(ВНИИ химических реактивов и особо чистых химических веществ)
С1
I
\ \/
: N
NO.,
C,H6C1N2O2 ’ М. в. 208, 61
Для получения 5-хлор-8-нитрохинолина применялась реак-
ция Скраупа с 2-нитро-5-хлоранилином [1—3] или его ацетиль-
ным производным [4], нитрование смеси 5- и 7-хлорхинолинов
(полученной также по Скраупу из ж-хлоранилина) с после-
дующим разделением их 8-нитропроизводных [1, 5—7] и нитро-
вание 5-хлорхинолииа [8—12]
Данные первой работы |1] по циклизации 2-нитро-5-хлор-
анилина методом Скраупа в присутствии мышьяковой кислоты
(выход 80,3%) не удалось воспроизвести при проведении это-
го синтеза в большем масштабе: даже после введения некото-
рых усовершенствований и замены мышьяковой кислоты на се
ангидрид выход составил всего 50% 12]. В более поздней ра-
боте [4] оба эти варианта признаны неудовлетворительными и
предложено вместо 2-нитро-5-хлоранилина применять 2-нитро-
5-хлорацетальдегид (выход 50—70%). Два последних вещест
ва, необходимые для синтезов этого типа, в свою очередь были
получены [1—4] путем нитрования ж-хлорацетанилида при низ-
ких температурах (до —50°). Выход 2-нитро-5-хлорацетанили-
да не превышал 60% [3]. т. к. при этом образуются два изоме-
ра, которые приходится затем разделять. При нитровании м-
хлорацетанилида в увеличенном масштабе существует опас-
ность взрыва [4].
Нитрование полученной скраупированием ж-хлоранилина
смеси 5- и 7-хлорхинолинов при помощи дымящей азотной
кислоты [1, 5, 6] или нитрата калия [7] в присутствии концен-
трированной серной кислоты приводило к образованию смеси
5- и 7-хлор-8-нитрохинолинов. Разделение их было основано
на различной растворимости в абсолютном спирте [1] или в
кислотах [5—7].
При нитровании 5-хлорхинолина [8—12] получается чистый
5-хлор-8-нитрохинолин, свободный от примеси изомеров и ди-
нитропроизводных. Даже в жестких условиях, при нагревании
5-хлорхинолина в течение 2 часов при 100° со смесью дымя-
щих азотной и серной кислот не наблюдалось образования
динитропроизводных [И].
96
Нами разработан оригинальный метод нитрования 5-хлор-
хинолина в форме его азотнокислой соли, не требующий при-
менения концентрированной азотной кислоты и позволяю-
щий полупить 5-хлор-8-нитрохинолин с количественным выхо-
дом, Перевод 5-хлорхинолина в его азотнокислую соль также
осуществляется почти без потерь (см. стр. 92). Нитрование
проходит очень гладко, в мягких условиях.
Получение 5-хлор-8-нитрохинолина.
С1
С! z1
(H.SO4) 1 | |
| I | -HNO3----------► \/Х/
I N
N NOo
В трехгорлой колбе емкостью 250 мл с мешалкой и термо-
метром охлаждают 80 мл 100%-ной серной кислоты до —3—0е.
За 40 минут добавляют небольшими порциями 22,7 г (0,1 Л1)
сухого тонко растертого нитрата 5-хлорхинолиния (см. приме-
чание 1) при хорошем размешивании, поддерживая темпера-
туру 4—5°. После 20 минут выдержки охлаждающую баню
удаляют, вносят 1 г (0,01 М) сухого растертого азотнокислого
калия и размешивают до полного растворения твердых частиц.
Затем нагревают колбу на кипящей водяной бане в течение
1 часа. Охладив реакционную массу, выливают ее на 1 кг во-
ды со льдом, выпавший осадок отфильтровывают, промывают
водой и сушат. Выход 17,3 г (82,9%) 5-хлор-8-иитрохинолина
с т. пл. 136—136,5° (см. примечания 2 и 3).
Фильтрат нейтрализуют крепким раствором едкого натра,
причем осаждается дополнительное количество более загряз-
ненного вещества. После фильтрации, промывки и сушки по-
лучают около 3,3 г продукта с т. пл. 133—134° (см. примеча-
ние 3).
Общий выход 20,6 а (98,7% теоретического).
Примечания.
1. Применялся нитрат 5-хлорхинолиния с т. пл. 159—159,5°, предвари-
тельно высушенный при 105—108° и растертый.
2. По литератуоным данным, т. пл. 134—135° [2], 135° [7, 11], 135—136°
[4, 6], 136° [1, 12], 138° [9].
3. Продукт пригоден для восстановления в 5-хлор-8-аминохинолии.
При желании его можно перекристаллизовать из спирта или разбавленной
уксусной кислоты.
ЛИТЕРАТУРА
1. Е. Fotirneau, М. et. M-me Trefouel, A. Wancolle, Bull. Soc. chim.
France, (4) 47, 738 (1930).
2. R. E. Lutz, P. S. Bailey, T. A. Martin, J. M. Salsbury. J. Amer.
Chem. Soc., 68, 1324 (1946).
7 Зак. 1020 97
3. R. C. Fuson, R. A. Bauman, E. Howard jr„ E.N. Marvell, J. Organ.
Chem., 12. 799 (3947).
4. J. H. Burckhalter, W.H. Edgerton, J. A. Darden jr„ J. Amer. Chem.
Soc., 76, 6089 (1954).
. 5. IT. La Caste, J. Bodewig, Ber., 17. 926 (1884).
6. С. C. Price, D. B. Guthrie, J. Amer. Chem. Soc., 68, 1592 (1946).
7. A K. Sen, N K. Ray, U. P. Basu, J. Sci. Ind. Research, 1 IB. 325
(1952); Chem. Abstrs, 47. 105341.
8. W. La Coste, Ber,. 18, 2940 (1885).
9. A. Claus, R. Kayser, J. prakt. Chem. |2], 48, 270 (1893).
10. J. Howitt, H. Fraenkel, E. Schroeder, Liebigs Ann. Chem.. 396, 53
(1913).
11. G. M. Bennett, J. F. Grove, J. Chem. Soc.,'1945, 378.
12. H. Andersag, Chem. Ber.. 81, 499 (1948).
5-ХЛОР-8-АМИНОХИНОЛ ИН
В. M. ДЗИОМКО, И. А. КРАСАВИН
(ВНИИ химических реактивов u особо чистых химических вещее/в)
C,H7C1N,
I N
NH2
М. в. 178, 63
5-Хлор-8-аминохинолин был получен при восстановлении
5-хлор-8-нитрохинолина хлористым оловом в соляной кислоте
[1—3] или железной пылью в уксуснокислом растворе [2—4].
Было, однако, замечено, что применение хлористого олова и
соляной кислоты приводит к образованию, наряду с ожидаем
мым монохлораминохинолином, значительных количеств 5,7-
дихлор-8-аминохинолина [3].
Аналогичный процесс хлорирования наблюдался и при
восстановлении хлористым оловом 8-нитрохинолина: кроме
8-аминохинолина образуются два монохлорпроизводных пос-
леднего [5]. Позднее их строение было уточнено, и оказалось,
что они являются 5- и 7-хлор-8-аминохинолинами [3, 4]. Была
также сделана попытка использовать это явление для получе-
ния 5-хлор-8-аминохинолина непосредственно из 8-нитрохино-
лина (4]. Получение чистого продукта по этому методу затруд-
нено необходимостью выделять его из сложной смеси веществ.
98
В противоположность -этому, восстановление 5-хлор-8-ии-
трохинолина железной пылью в уксусной кислоте [2—4] проте-
кает без каких-либо осложнений; выход достигает 79,4% [3].
Последний способ восстановления был нами проверен и
дал удовлетворительные результаты, хотя выход амина коле-
бался в зависимости от качества применяемой железной пыли,
в пределах от 55 до 72%.
Получение 5-хлор-8-аминохинолина.
С1 С1
/\/\ Fe4 CHSCOOH
В круглодонной колбе емкостью 1 л (см. примечание 1) с
мешалкой растворяют 14,0 г (0,067 А4) 5-хлор-8-нитрохиноли-
ва в смеси 120 мл уксусной кислоты и 80 мл воды при нагре-
вании. Продолжая нагревать колбу на кипящей водяной бане
и размешивая, добавляют небольшими порциями 11,2 г
(0,2 г-ат) железа в порошке (см. примечание 2) за 30 минут н
нагревают еще 30 минут. Затем разбавляют массу водой, под-
щелачивают раствором едкого натра и перегоняют продукт с
водяным паром. По охлаждении отфильтровывают выпавшие
светло-желтые волокнистые кристаллы амина (см. примеча-
ние 3), а фильтрат экстрагируют эфиром (2 раза по 250 мл).
В эфирном экстракте растворяют осадок, раствор сушат без-
водным сульфатом натрия, эфир отгоняют и остаток перего-
няют в вакууме при 125—13171 мм рт. ст. (см. примечание 4).
Выход 5-хлор-8-аминохинолина 8,6 г (71,7% теоретического)..
Т. пл. 86,5—87° (см. примечание 5).
Примечания.
1. Колбу следует заранее приспособить для последующей перегонки с
водяным паром.
2, Применялось «железо восстановленное водородом» (Ferrum redu-
ctum р. а.) объединения «Reanal» (Венгерская Народная Республика). При
использовании «железа восстановленного реактивного» производства Сов-
нархоза Латвийской ССР выход был на 10—15% ниже
3. Если не стремиться к получению максимального выхода, то можно
исключить экстракцию фильтрата эфиром, а также перегонку в вакууме.
Продукт, перегнанный с паром, достаточно чист.
4. Вместо перегонки в вакууме вещество можно перекристаллизовывать
из разбавленного спирта.
5. По литературным данным, т. пл. 86,5— 87,5 [3, 4].
ЛИТЕРАТУРА
1. A. Claus, R. Kayser, J. prakt. Chem., [2] 48 , 270 (1893).
2. J. Howitz, H. Fraenkel, E. Schroeder, Liebigs Ann. Chem, 396, 53
(1913).
7* 99
3. С. С. Price. D. В. Guthrie, .1. Amer. Chem. Soc., 68, 1592 (1946).
4. R. H. Baker, C. J. Albisetti jr., R. M. Dodson. G. R. Lappin
B. Riegel, J. Amer. Chem. Soc., 68, 1532 (1946).
5. R. P. Dikshoorn, Recueil trav. chem., 48, 147 (1929).
4-ЭТИЛПИРИДИН
H. Ф. КАЗАРИНОВА, M. И. КОТЕЛЕНЕЦ, К. А. COJIOMKO
(Институт органической химии АН УССР, Донецкое отделение)
С2Н6
Метод получения 4-этилпиридина путем алкилирования пи-
ридина уксусным ангидридом в присутствии порошка цинка
опубликован в сборнике «Синтезы органических препара-
тов» [1].
Предлагаемый способ представляет собой видоизменение
методик, описанных [2—3] и является более простым, не тре-
бует большого расхода уксусного ангидрида.
4-Этилпиридин является исходным сырьем для получения
4-винилпиридина — ценного мономера промышленности пла-
стических масс.
СИНТЕЗ 4-ЭТИЛПИРИДИНА
сн;1-с<
)о Fe
сосн3
c2Hs
NCOCHS
сн,соон
Fe
100
Характеристика основного сырья
Пиридин, технический.
Уксусный ангидрид, ч.д.а., т. кип. 140".
Хлорбензол, ч., т. кип. 130—132°.
Железный порошок (270 меш).
Условия получения
К смеси 50 г пиридина (0,63 Л1), 130 г (0,126 М) уксусно-
го ангидрида и 150 мл хлорбензола добавляют при интенсив-
ном перемешивании (см. примечание 1) 101 г железного по-
рошка. Реакционную массу нагревают до 80° и перемешива-
ют при этой температуре в течение 1,5 часа. Затем к реакци-
онной смеси добавляют 15 мл уксусной кислоты и нагревают
до кипения (см. примечание 2). После 1,5 часового кипения
снова добавляют 30 мл уксусной кислоты и кипячение про-
должают еще в течение часа (см. примечание 3). По оконча-
нии кипячения реакционную смесь разбавляют водой прч
одновременном охлаждении (см. примечание 4). Затем при
охлаждении смесь подщелачивают 50%-ным раствором едкого
натра (см. примечание 5) до pH 10 и перегоняют с водяным
паром до объема 1,5—2 литра. Дистиллят подкисляют концен-
трированной соляной кислотой (60 мл) до кислой реакции но
бумажке конго и упаривают в вакууме до объема 300 мл. Из
отгона выделяют хлорбензол в количестве 95% от загружен-
ного (см. примечание 6). Остаток подщелачивают 50%-ным
раствором едкого натра до pH 10. Выделившееся масло от-
деляют, сушат безводным поташом и фракционируют.' Выход
этилпиридина составляет 60—65% теории, т. кип. 163—166°.
Примечания.
1. При недостаточно интенсивном перемешивании железо осаждается
на дно колбы, образуя твердую корку, которая мешает дальнейшему про-
ведению реакции.
2. Во время кипячения иногда наблюдается загустевание реакционной
массы, затрудняющее перемешивание, в этом случае следует добавляш
еще некоторое количество хлорбензола.
3. На этой стадии процесса также может наблюдаться загустевание
реакционной массы. В этом случае следует прекратить нагревание и при-
ступить к выделению продукта.
4. Воду необходимо добавлять постепенно, небольшими порциями, в
противном случае может произойти сильное разогревание реакционной
массы и осмоление продукта.
5. Подщелачивание следует проводить постепенно, при непрерывном
охлаждении реакционной смеси во избежание осмоления продукта.
6. Хлорбензол может быть снова использован в качестве растворителя.
ЛИТЕРАТУРА
1. G. Wlllbert, G. Reich, L. Tenenbaum, J. Organ. Chem., 22, 6 (1957).
2. Янков Любен, Неделева Лиляна, Фармация, 9, № 1, 24 (1959).
3. «Синтезы органических препаратов», Сб. 4, ИЛ, 1953, стр. 599.
101
4-( п -ДИМЕТИЛАМИНОФЕН ИЛ )-ПИРИДИН
А. К. ШЕИНКМАН. Н. Ф. КАЗАРИНОВА, Е. П. БАБИН
(Институт органической химии АН УССР, Донецкое отделение)
/-N(CH3)2
Предлагаемая методика основана на способе, предложен-
ном Е. Кеннигом и Е. Руппельтом [1] и является наиболее
удобным синтезом 4-(/г-диметиламинофенил)-пиридина, поз-
воляющим использовать в качестве сырья различные хлоран-
гидриды кислот и дающим более высокие выхода продукта.
Без применения катализатора использованный метод дает не-
высокие выхода при проведении реакции в течение 7 недель [2].
Другим способом получения этого соединения является диазо-
сочетание пиридина и хлористого л-диметиламинофенилдиа-
зония [31.
4-я-(Диметиламинофеиил)-пиридин может быть использо-
ван для получения изоникотиновой кислоты и, как промежу-
точный продукт, для синтеза различных физиологически ак-
тивных препаратов и красителей.
СИНТЕЗ 4-(я-ДИМЕТИЛАМИНОФЕНИЛ)-ПИРИДИНА
4-С6Н5СОС1
+ (CH3),NC,HSA1C13
осс6нг,
НС1
—CjHjCHO
102
Характеристика основного сырья
Пиридин, тщательно высушенный кипячением над СаО,
т. кип. 113,5—114°; п™ 1,509.
Хлористый бензоил, технический, свежеперегнанный,
т. кип. 194—197° (см. примечание 1).
Диметиланилин, технический, перегнанный, т. кип. 190—
193°.
Хлористый алюминий, технический.
Условия получения
В трехгорлую круглодонную колбу, емкостью 500 мл, снаб-
женную механической мешалкой с затвором и обратным хо-
лодильником с хлоркальциевой трубкой, помещают 79 г (1 М)
пиридина и 70 г (0,5 А1) хлористого бензоила (см. примеча-
ние 2). Реакционную смесь выдерживают при перемешивании
1 час на кипящей водяной бане. После охлаждения прилива-
ют 72,5 г (0,6 А1) диметиланилина и при интенсивном переме-
шивании и охлаждении (на водяной бане со льдом) по частям
прибавляют 80 г (0,6 А1) хлористого алюминия за 1 час. Пос-
ле прибавления всего хлористого алюминия реакционную
смесь выдерживают на кипящей бане 2 часа при перемешива-
нии. Содержимое колбы при этом застывает в виде красно-бу-
рой густой массы.
Теплую реакционную смесь разлагают соляной кислотой
(150 мл) и отгоняют с водяным паром для удаления бензой-
ного альдегида и бензойной кислоты. Остаток подщелачивают
50%-ной щелочью до pH 9—10 (см. примечание 3) и вновь пе-
регоняют с водяным паром для освобождения от непрореаги-
ровавшего пиридина и диметиланилина.
Светло-коричневый аморфный осадок, оставшийся в кол-
бе, отсасывают, дважды промывают горячей водой для отмыв-
ки от неорганических солей. Промытый осадок выдерживают
в сушильном шкафу при 100° 4 часа. Высушенный светло-ко-
ричневый продукт кипятят в ацетоне (200—300 мл), фильтру-
ют и на фильтре промывают ацетоном.
Выход 4-(n-диметиламинофенил)-пиридина 83,5 г (84,3%
теории). Т. пл. 224—225°.
Для получения более чистого продукта вещество подверга-
ют сублимации. Сублимированный 4-(п-диметиламинофенил)-
пиридин перекристаллизовывают из спиртово-хлороформенной
смеси (10:1). Получают серебристо-белые пластинки. Т. пл.
233—234° (см. примечание 4).
Предлагаемый метод применим для синтеза других
4- (n-диалкиламинофенил) -пиридинов.
юз
Примечания,
1. Вместо хлористого бензоила можно использовать хлористый ацетил,
и-нитробензоилхлорид и другие хлораигидриды кислот.
2. При использовании других хлораигидридов прибавление их к пири-
дину следует производить по каплям при интенсивном перемешивании и
охлаждении.
3. Обрабатывать 50%-ной щелочью следует до тех пор. пока выпавшая
гидроокись алюминия не растворится.
4. Можно с менпшими потерями произвести очистку продукта путем
трехкратной перекристаллизации из большого количества спиртово-хлоро-
форменной смеси (10: 1) или из диоксана.
ЛИТЕРАТУРА
1. Е. Koennig, Е. Ruppelt, Liebigs Ann. Chem, 509, 142 (1934).
2. H. Adkins, Q. Thompson, J. Amer. Chem. Soc., 71, 2242 (1949).
3. MOhlan, Berger, Ber., 26, 2003 (1893),
ГРАМИН
(р-(диметиламинометил)-индол, получение в гетерогенной
среде)
Н. Л. ДЗБАНОВСКИИ, Е. М. УРИНОВИЧ
(ВНИИ химических реактивов и особо частых химических веществ)
H
Сц Нм N2
M. в. 174, 25
Простейший алкалоид группы индола — |3-(диметиламино-
метил)-индол, получивший название грамин, был впервые син-
тезирован Виландом и Тсингом [1] из индолилмагниййодида и
диметиламиноацетонитрила с 40%-ным выходом. Грамин
стал более доступен после того, как Кюн и Штейн [2, 3] осу-
ществили его синтез в уксуснокислой среде по реакции Ман-
ниха из индола, формальдегида и димстиламина с выходом
96%. технического продукта, не охарактеризованного по тем-
пературе плавления; после перекристаллизации т. пл. 134°
104
При проверке метода Кюна и Штейна в крупно-лаборатор-
ном масштабе выяснилось, что выходы грамина колебались
от 75% до 90%, а содержание грамина в техническом про-
дукте составляло от 80 до 95%, что было установлено титро-
ванием 0,1 н. НО с метиловым красным, по А. П. Орехову [4].
Соответственно т. пл. были различными: 116°, 120°; обычно не
выше 124°, даже если исходили из чистого индола с т. пл. 52°.
При очистке образцов грамина с т. пл. 116° мы находили за-
метное количество примеси, нерастворимой в разбавленной ук-
сусной кислоте. Природу этой примеси мы не исследовали, од-
нако Добенск и Мареш [5] нашли, что при получении грамина
по Кюну и Штейну всегда побочно образуется небольшое ко-
личество р,(У-дииндолилметана.
После того как было найдено, что аминогруппа в Грамине
довольно подвижна, он стал объектом многочисленных иссле-
дований, которые привели к синтезу таких физиологически
ценных соединений, как d, /-триптофан, гетероауксии и др.
В связи с этим, в целях дальнейшего упрощения и удешев-
ления технологического процесса был разработан принципи-
ально новый метод синтеза грамина, основанный на диметил-
аминометилировании индола водной смесью формалина и ди-
метиламина в гетерогенной слабо-щелочной среде (обуслов-
ленной наличием диметиламина).
Кюн и Штейн описали [2] аналогичный опыт, в котором
было получено масло, содержащее только 40% грамина.
Мы более подробно изучили эту реакцию и нашли, что
при комнатной температуре индол не реагирует с водной ами-
нометилирующей смесью; при нагревании до 60° в течение
40—60 минут и хорошем перемешивании, образуется жидкое
индольное основание, полностью растворяющееся в разбав-
ленной соляной кислоте и обратно выделяющееся при подще-
лачивании. Путем фракционирования в вакууме (при 1—2 мм)
мы установили, чго полученное нами индольное основание
(масло) состоит из грамина (около 10%, найденного в кубо-
вом остатке; по лит. данным, грамин возгоняется только в вы-
соком вакууме — порядка 0,01 мм) и масла, выкипающего в
пределах 100—105° при 1—2 мм (около 90%)- Изучение
свойств этой фракции показало, что она состоит преимущест-
венно из соединения, образующего пикрат и йодметилат с
теми же температурами плавления, что и грамин. При нагре-
вании с водой в тонкой эмульсии при 95—100° через несколь-
ко часов внезапно превращается в твердый продукт, оказав-
шийся чистым Грамином с т. пл. 126°.
Эти данные положены в основу разработанного нами син-
теза грамина.
105
СИНТЕЗ ГРАМИНА
। ч---г, । II---iiCH2N(CH3)2
| || II+CH2O + NH(CH3)2 -Ч I) II +н20
N 7 N
н н
К 20 г водного 48%-ного диметиламииа (0,22 Л1) при ох-
лаждении ледяной водой и хорошем перемешивании прибав-
ляют по каплям 40%-ный формалин в количестве 15 г (0,2 М),
с такой скоростью, чтобы температура смеси не поднималась
выше 10—15°. К этому раствору прибавляют 23 г индола
(0,2 Л4) с т. пл. 52° и 0,5 г эмульгатора (примечание 1) в 3 мл
воды. Реакционную смесь постепенно нагревают на водяной
бане при температуре не выше 60 и хорошем перемешивании
в течение часа. При этом уже при комнатной температуре на-
чинается образование эмульсии, а через 10—15 минут при тем-
пературе не выше 35—40°, смесь представляет собой сплош-
ную тонкую эмульсию. При дальнейшем нагревании при 95—
97°, через 5 часов, эмульсия внезапно превращается в твердый,
кристаллический грамин (в виде листочков). Для удобства пе-
ренесения из реакционной колбы кристаллической массы к го-
рячей реакционной смеси прибавляют 100 мл нагретой до ки-
пения воды и смесь перемешивают до полного охлаждения.
Грамин отфильтровывают, промывают и высушивают до по-
стоянного веса при 40°. Чешуйки желтоватого цвета со слабым
запахом индола. Т. пл. 126°. Выход 32 г или 91% теории. По-
сле очистки (растворением в разбавленной уксусной кислоте и
обратном выделении небольшим избытком щелочи) выход 31 с
или 90%. Очищенный грамин представляет собой белый мел-
ко-кристаллический порошок, без запаха, т. пл. 127°. По тит-
рованию 0,1 н. соляной кислотой содержал 98,4% основного
вещества (см. примечание 2).
Получение грамина в крупно-лабораторном масштабе.
В 30- литровый чугунный эмалированный аппарат, снаб-
женный пароводяной рубашкой, обратным холодильником,
мерником, механической мешалкой и термометром, загружа-
ют 8 кг 25%-ного водного диметиламина (основания). В ру-
башку аппарата пускают холодную воду, охлаждают аппарат,
включают мешалку и при непрерывном размешивании мед-
ленно спускают из мерника 3,1 кг 40%-ного формалина с та-
кой скоростью, чтобы температура смеси не превышала +10°.
После прибавления всего формалина смесь выдерживают при
этой температуре 30 минут. После выдержки в тот же аппарат
загружают 4,7 кг индола и раствор 40 г эмульгатора (сульфо-
ната) в 0,1 л воды. Смесь перемешивают и при размешивании
нагревают до 60° и выдерживают при этой температуре один
106
час. Затем температуру поднимают до 95—97° и выдерживают
при этой температуре и непрерывном размешивании 5—6 ча-
сов. После этого к реакционной массе приливают 5—6 литров
воды, содержимое аппарата доводят до кипения и дают двух-
часовую выдержку. Реакционную массу охлаждают, грамин
выгружают из аппарата, переносят на нутч-фильтр, промыва-
ют 5—6 раз холодной водой (по 2 литра на каждую промыв-
ку), хорошо отжимают и сушат в сушильном шкафу при 30—
40э около 6—8 часов. Грамин получают в виде желтовато-се-
рых чешуек со слабым запахом. Выход Грамина 5,8—6 кг. По
лученный грамин по анализу содержит 90—91 % основного ве-
щества.
Примечания.
1. В описываемом опыте в качестве эмульгатора применялся сульфо-
нат. В случае применения сульфонола (в том же количестве) получались
несколько лучшие результаты, выход грамина достигал 34 г или 98% тео-
рии, а после очистки (аналогичной предыдущему) 32 г или 93,5%. Полу-
ченный грамин имел такую же г. пл. (127г).
Применение эмульгаторов особенно важно потому, что дает возмож-
ность исходить из 25%-ного водного диметиламииа (вместо 50—55%). что
было нами использовано для серии полупроизводствепных опытов получе-
ния грамина.
2. Грамин с т. пл. 127° и содержанием 98,4—98,5% основного вещества
вполне пригоден для большинства препаративных целей; для получения
гетероакусииа ( /5-иидолилуксусной кислоты) пригоден даже неочищенный
грамин, с содержанием 90—91 % основного- вещества и с т. пл. 125—126°.
Для получения грамина с т. h.i. 133 и 134° рекомендуется двукратная
перекристаллизация из ацетона.
ЛИТЕРА ГУ Р А
1. Т. Wieland, С., I. Hsing, Liebigs Ann. Chem., 526, 188 (1936).
2. H. Kahn, О Stein, Ber., 70, 567 (1936).
3. О. Stein, H. Kahn, Герм, пат., 673949 (1939).
4. Л. П. Орехов, С С. Норкина, /К- общ. химии, 7, 673 (1937).
5. И. F. Dobeneck, G. Marech, Angew. Chemie, 63, 469 (1951).
4,4 ДИОКСИДИФЕНИЛ СУЛЬФОН
(Бис-4-оксифенилсульфон)
В. Г. БРУДЗЬ. Д. /1 ДРАП КН И А
(ВНИИ химических реактивов и особо чистых химических веществ)
НО / \-SOo-/ ^>-ОН
C1.,H10O4S м. в. 250, 20
Согласно литературным данным, основным методом полу-
чения Ф/Г-диоксидифенилсульфона является взаимодействие
107
фенола с серной кислотой: фенол сначала сульфируют олеу-
мом [1—6], моногидратом [7, 8] или купоросным маслом [9, 10]
при относительно низкой температуре, затем, не выделяя суль-
фокислоты, конденсируют ее со второй молекулой фенола при
150—200°. Выделяющуюся реакционную воду удаляют отгон-
кой: простой [1—7], азеотропной [8, 9, 11] или в вакууме [8]. В
результате получают плав, состоящий из смеси изомерных ди-
оксидифенилсульфонов, фенолсульфокислот, фенола, дифени-
лового эфира и смолистых примесей. Для выделения диокси-
дифенилсульфонов из плава их выщелачивают водой [7], или
же плав растворяют в растворе едкого натра, фильтруют и из
фильтрата диоксидифенилсульфон осаждают подкислением до
pH ~ 3,5 [8]. Полученный технический сульфон содержит от
10 до 30% 2,4'-изомера. Чтобы получить из него чистый 4,4'-
диоксидифенилсульфон с т. пл. 248—249° требуются особые
приемы [7, 12], т. к. обычная одно—двукратная перекристалли-
зация из воды или спирта дает продукт, плавящийся не выше
240° [1, 5, 6, 9, 10].
По нижеприведенной прописи нам удалось получить техни-
ческий 4,4/-диоксидифенилсульфон, практически не содержа-
щий других изомеров. Это достигнуто тем, что:
1. Сульфирование фенола начинают не при 50—60°, а при
температуре не ниже 100—110°, для сведения к минимуму об-
разования о-фенол сульфокислоты.
2. В реакцию вводят теоретическое количество фенола, т. к.
избыток его способствует образованию 2,4'-изомера.
3. Для выделения 4,4'-диоксидифенилсульфона из щелоч-
ного раствора нейтрализацию последнего производят только
до pH 7,5—7,8 при температуре 70 - 80°.
СИНТЕЗ 4,4 -ДИОКСИДИФЕНИЛСУЛЬФОНА
ОН
2 |/'4| + H2SO4 • НО-^ )-SO2-( /-OH+2H/J
Характеристика основного сырья
Фенол, ч. ГОСТ 6417—52.
Кислота серная, ч. ГОСТ 4204—48.
Едкий натр, ч. ГОСТ 4328—48.
Соляная кислота, ч. ГОСТ 3118—46.
Этиловый спирт, ч. ГОСТ 5962—51.
Условия получения
К 188 г (2 М) фенола, нагретого до 100—110°, прибавляют
при механическом размешивании 57 мл купоросного масла
108
(1,0 Л/ серной кислоты) в течение 15 минут. К концу прибав-
ления кислоты температура поднимается до 125—130°. Затем
размешивают еще в течение одного часа при 130—140°. По
окончании выдержки в системе создают вакуум (см. примеча-
ние 1) с помощью водоструйного насоса, снижая давление до
440—430 мм остаточного, и далее соблюдают приблизительно
следующий режим:
Время, часы Остаточное давление, мм, рт. ст. Температура, °C
2,5 410 430 140-170
1,5 400-380 170—175
1,0 360-340 170 - 175
1,0 постепенное сни- жение до 150 175-180
0,25 „ до 90 180- 185
0,25 . до 60 180-185
С момента подключения вакуума начинается отгонка воды
и частично фенола (нижний слой). Когда количество отогнав-
шегося фенола составит 25—30 мл, в реакционную смесь вво-
дят дополнительно 50 г фенола (см. примечание 2). По окон-
чании конденсации и прекращении отгонки избыточного фено-
ла плав охлаждают до 150—160е и растворяют добавлением
10%-ного раствора едкого натра. Раствор осветляют активи-
рованным углем. Розовый фильтрат нагревают до 70—80° н
при механическом размешивании нейтрализуют соляной кис-
лотой до pH 7,6—7,8 (pH определяют по советской индикатор-
ной бумаге «Рифан» с интервалом 7,2—8,8). Суспензию 4,4'-
диоксидифенилсульфона охлаждают до 20—25° и фильтруют.
Осадок многократно промывают водой. Получают около 200 а
(70—80% теории) белого или слегка розоватого цвета сульфо-
на. Температура плавления 237—245°. После однократной пе-
рекристаллизации из 15° этилового спирта при отношении 1 : 20
с обработкой углем получают 4,4'-диоксидифенилсульфон с
выходом 85%, считая на технический, т. пл. 246—247°; после
второй аналогичной кристаллизации т. пл. 248—249°.
Примечания.
1. При указанной величине загрузки перед снижением давления мож-
но прекратить размешивание, однако уже при увеличении масштаба в
5 раз размешивание необходимо.
109
2. Если в процессе конденсации отгоняется много фенола и в реакци-
онной смеси его остается меньше, чем требуется по теории, фенол вводит
дополнительно еще по 50 г один или два раза сверх указанного количества.
Конец конденсации (после 6-чи часов синтеза при пониженном давле-
нии) определяют следующим образом: в 15—20 мл дистиллированной водь:
суспензируют около 1 г (навеска с точностью до третьего знака) реакцион-
ной массы и нагревают до кипения; затем охлаждают до комнатной тем-
пературы и титруют 0,1 н. раствором едкого натра по метилоранжу. Оста-
точную кислотность, и пересчете да серную кислоту, рассчитывают по сле-
дующей формуле
«•49
1дс а —число мл точно 0,1 и. раствора щелочи, пошедшее на титропанн'.-.
б — навеска.
Если кислотность не превышает 3,0%, можно производить отгонку Ш-
быточного фенола, снижая давление до 90—60 мм остаточного. В против
пом случае добавляют еще 40—50 г фенола и выдерживают реакционную
массу при 170—175° и 250 300 мм остаточного давления в течение одного
часа; если после этого достигают требуемой величины кислотности, проит-
водят дальнейшее снижение давления.
ЛИТЕРАТУР А
1. J. Zehenter, Е. Fauser, J. prakr Chem., [2], 117, 233 (1927).
2. G. Machek, H. Haas, J. prakt. Chem., 160, 41 (1942).
3. F. Ullmann, J. Korselt, Ber., 40, 641 (1907).
4. J. Annaheim, Liebigs Ann. Chem., 172, 28 (1874),
5. P. Sah, J. Chinese. Chem. Soc., 15, 129 (1917).
6. fl. П. Беркман, H. Г. Гирджи. Ж. прикл. химии. 12, 451 (1939).
7. L. Е. Hinkel, G. H. Summers, J, Chem. Soc., 2854 (1949).
8. K. Okon, T. Urbanski, Roczn. Chem., 27, 348 (1953).
9. Нем. пат., 728276, 1940 г.
10. V. П. Беркман, А. Н. Сергеева, Научные зап. Львов, политехи, ин-1.1
Серия хим. техн., вып. 22, № 2, 121 (1956).
11. Франц, пат., 893369, 7.04.44 г. Client, abstrs, 47, 4112 (1953).
12. Пат. США, 2392137 1.01.46 г.
БИС-(4-ОКСИ-3-АМИНОФЕНИЛ)-СУЛЬФОН
(4,4/-Диокси-3,3/ диаминодифенилсульфон,
3,3'-диамино-ДДС)
Д. А. ДРАП РИНА, В. Г. БРУДЗЬ
(ВНИИ химических реактивов и особо чистых химических веществ)
HO-<f ^>-он
NH, \’Ha
CisH^N.OiS M. и. 280, 3
3,3'-Диамино-ДДС был получен восстановлением бис-(4
окси-3-нитрофенил)-сульфона оловом в солянокислой среде
110
[1, 2]; исходный нитропродукт—нитрованием 4,4'-диоксидифе-
нилсульфона азотной кислотой [1, 3, 4] или нитрующей
смесью [5] и гидролитической заменой хлора на гидроксил в
4,4'-дихлор-3,3'-динитродифенилсульфоне [2].
По нитрованию нами проверены все литературные данные.
В условиях [5] наряду с требуемым динитросоединением полу-
чается значительное количество тетранитропроцукта. Нитрова-
ние по [1, 3, 4] при загрузках свыше 50 г сульфона протекает
бурно, иногда с выбросом реакционной массы. Для осущест-
вления спокойного протекания процесса мы применили раз-
бавление реакционной массы уксусной кислотой.
По восстановлению бис- (4-окси-З-нитрофенил) -сульфона
памп разработан метод с использованием сернистого натрия в
качестве восстановителя.
СИНТЕЗ БИС-(4-ОКСИ-3-АМИНОФЕНИЛ)-СУЛЬФОНА
I. _>-ОН -k2HNO, сйда-от
\-ОН + 2Н2О
"no2
и. 2НО—\ ^-SO2-^ ^-OH4-6Na3S + 3H2O ->
no2 \о2
->2NaO—/-SO2—^-ONa + 3Na2S2O3 + 2NaOH
NH2 XNH2
III. NaO-
J^ — ONa I- 2NaHSO3 -
\h.
;-OH + 2Na2SO3
nh2
Характеристика основного сырья
4,4/-Диоксидифенилсульфон, неочищенный, с началом тем-
пературы плавления не ниже 235°.
Кислота азотная, ч., ГОСТ 4461—48.
Ill
Кислота уксусная, ч., 98%-ная, ГОСТ 61—51.
Натрий сернистый, ч., ГОСТ 2053—43.
Бисульфит натрия, ч., ТУ МХП 1944—49.
Спирт этиловый, ч., ГОСТ 5962—51.
Условия получения
Получение 4,4'-диокси-3,3'-динитродифенилсульфона.
175 г (0,7 7И) 4,4/-диоксидифенилсульфона и 540 г уксусной
кислоты нагревают при механическом размешивании до тем-
пературы 80° и затем к полученной суспензии приливают в те-
чение одного часа 280 мл азотной кислоты уд. в. 1,35, поддер-
живая температуру не выше 80—85°. После прибавления около
10% всего количества кислоты, обычно получается гомогенный
раствор, а после внесения около 25% кислоты начинает выпа-
дать желтоватый осадок динитропродукта. После прибавления
всего количества кислоты реакционную массу размешивают
еще в течение 10—15 минут при той же температуре и затем
охлаждают водопроводной водой до 20—25°. Осадок отфиль-
тровывают, промывают сначала ~ 100 мл 50%-ной уксусной
кислоты и далее водой до нейтральной реакции на бумажку
конго. Получают около 260 г пасты с содержанием влаги 30—
40%. Выход в пересчете на сухое вещество 162—173 г (68—
73% теории). Температура плавления высушенной при 100°
пробы — 228—233°,
Получение 4,4'-диокси-3,3'-диаминодифенилсульфона.
Растворяют 470 г кристаллического сернистого натрия в
370 мл дистиллированной воды при 50° и к полученному раст-
вору, не охлаждая, прибавляют 130 г — половину всей загруз-
ки пасты динитропродукта с содержанием влаги ~35%. Если
паста имеет другую влажность, количество воды и пасты соот-
ветственно пересчитывают.
Примерно через 10 минут после внесения первой части па-
сты температуру реакционной смеси поднимают до 80—85° ц
прибавляют остальное количество динитропродукта (всего
167 г в пересчете на сухой — 0,49 7И), и продолжают нагревать,
не закрывая загрузочного отверстия. Температура реакцион-
ной массы, по мере испарения воды, поднимается до 100—105°
(около двух часов). При этой температуре массу нагревают
еще в течение 1 часа. Для выделения диамина реакционную
массу, не охлаждая, разбавляют при размешивании 1 л ди-
стиллированной воды, нагревают снова до 80° и осаждают до-
бавлением 0,88 л бисульфита натрия; охлаждают до 40° и сра-
зу фильтруют, иначе фильтрование замедляется.
Осадок многократно промывают дистиллированной водой.
Выход 115—120 г (82—88% теории), т. пл. 228—232,5° с
разложением. Очищают перекристаллизацией из 30° этилового
спирта с добавлением активированного угля и нескольких мил-
112
лилитров бисульфита на!рия. Выход чистого продукта 100—
110 г (80—93%, считая на технический), т. пл. 228—232°.
ЛИТЕРАТУРА
1. G. J. Machek, Н. Buch. Monatsh. СЬеш., 80. 9 (1949).
2. J. Annaheim, Ber.. 8. 1060 (1875).
3. L. Glutz, Liebigs Ann. Chem.. 147, 52 (1868).
4. F. Ulltnann, I. Korselt. Ber.. 40, 641 (1907).
5. A’. Okon, T. Urbanski. Roczn. Chem., 27. 348 (1953).
4,4-ДИОКСИДИФЕН ИЛД ИЭТИЛМЕТАН
И. AL БИЛИК, A. Al. СЕРЕБРЯНЫЙ, H. Al. БОН ДАРЕЦ, P. Л. ГЛОБУС.
В. Г. БРУДЗЬ
(ВНИИ химических реактивов и особо чистых химических веществ)
с,н3
НО /-С-\ 011
С17Н20О3 С’‘Нэ м. В. 256,35
4,4'-Диоксидифенилдиэтилметан был впервые получен Диа-
ниным [1] из фенола и диэтилкетона в запаянных трубках с
применением соляной кислоты в качестве конденсирующего
средства и после перекристаллизации из водного спирта имел
т. пл. 198—200°.
Позднее этот продукт был получен рядом других авторов.
При применении в качестве конденсирующего средства хлори-
стого водорода [2] 4,4'-диоксидифенилдиэтилметан синтезиро-
ван с выходом 25% теории. Конденсацией фенола с диэтилке-
тоном в присутствии соляной кислоты при комнатной темпе-
ратуре в течение длительного времени (20 недель) получен
продукт с выходом 90% теории [3].
Учитывая, что при конденсации фенола с ацетоном в при-
сутствии серной и тиогликолевой кислот получают дифенилол-
пропан с высоким выходом [4] мы, по аналогии, конденсиро-
вали фенол с диэтилкегоном.
СИНТЕЗ 4,47-ДИОКСИД ИФЕНИЛДИЭТИЛМЕТАНА
С2Н5
%н5сос.,н5 — !
2/ ОН/-------------------—НО/ '—С—' '-ОН+Н„О
\---./ —х | \
С2Н-
8 Зак. 1020 113
К 8 .мл свежеприготовленного раствора катализатора (при-
мечание) при размешивании добавляют 4 .ил воды и при 20—
25° 47 г 92,5 %-ой серной кислоты. В смесь кислот в течение
15 минут при 24—26' вносит при размешивании раствор 13,3 с
(0,14 Л4) фенола в 1 мл воды, выдерживают при этой темпе-
ратуре 30 минут и, не прекращая размешивания, в течение
1 часа при 24—28° добавляют 6,5 г (0,076 А4) диэтилкетона.
Температуру повышают до 40°, размешивают массу в течение
14 часов, охлаждают до 20—25s и фильтруют через хлорино-
вую ткань ПЦ. Осадок промывают водой от кислоты и отго-
няют с паром не прореагировавший фенол и кетон. Продукт
отфильтровывают и сушат. Получают 10.7—13.0 г сухого
4,4 -диоксидифенилдиэтилметана (57,8—70.0% теории, считая
на фенол). После перекристаллизации из водного спирта
т. пл. 198—199°.
4.4/-Диоксидифепнлдиэтилметан представляет интерес как
препарат для проведения исследовательских работ.
Примечание.
Приготовление катализатора. Растворяют 1,76 г (0,022 А4) сернистого
натрия (в пересчете на 100%) в 55 .и.1 воды и при температуре 0— +2’
прибавляют при размешивании в течение 1 часа раствор 2,1 г (0,022 А1)
монохлоруксусной кислоты в 7 ли воды. После ЗО-мииутиой выдержки ра-
створ нагревают до 90—9о° н при этой темпера , \ ре размешивают в тече-
ние 1 часа.
Л И Т Е Р А Т У Р А
1. А. Дианин, Ж.Р.Ф.Х.О., 23, 499 (1891).
2. Hasson, Harrison. Swinew Pvtnan !. Pharmacv and Pharmacol. ",
509 (1934); Chem. Zbl. 1,3133 (1935). '
3. Reid, Wilson, J Amer. Chem. Soc.. 66. 967 (1944).
1. И. Al. Билик, P. Л. Глобус, В. Г. Брудзь, Пласт, массы. % 3, 69
(1961).
4,4/-ДИОКСИ-3,3/ ДИМЕТИЛДИФЕНИЛ-2.2-ПРОПАН
(Диортокрезилолпропан)
И М. БИЛИК, И. М. БОНДАРЕЦ, А. А1. СЕРЕБРЯНЫЙ, Р. . I. ГЛОБУС,
В. Г. БРУДЗЬ
(ВНИИ химических реактивов и особо чистых химических веществ)
СН3
С13Н20О2 М. в. 25S, 35
4,4,-Диокси-3.3/-диметил дифенил-2,2-пропан был получен
Цинке ’путем конденсации орго-крезола с апетоном в присут-
114
ствии концентрированной соляной кислоты с выходом 60—700-
теории, т. пл. 136° [I].
Учитывая, что при конденсации фенола с ацетоном в при-
сутствии серной и тногликолевой кислот получают дифенилол-
пропан с высоким выходом [2], мы по аналогии конденсирова-
ли орто-крезол с ацетоном.
СИНТЕЗ 4,4'-ДИОКСИ-3.3'-ДИМЕТИ.ЛДИФЕНИЛ-2,2-ПРСПАНА
_сн3
\он
+сн8сосн3
сн3
НО/ —
4—/ I 4
сн3
СПз
^он + н,о
К 32,5 .1/л свежеприготовленного раствора катализатора
(см. стр. 114) при 20—25е и при размешивании прибавляют
11,6 мл воды н при этой же температуре вносят 192 г 90%-ой
серной кислоты. В смесь кислот в течение 15 минут при 24—
26° и при размешивании вносят 58,4 г (0,54 М) орто-крезола
(чистого), выдерживают при этой температуре 30 минут и. нс
прекращая размешивания в течение 1 часа при 24—28°, до-
бавляют 17,5 г (0,3/И) ацетона. После 30-минутной выдержки
при размешивании и температуре 24—28°, вносят 3 мл толуо-
ла, повышают температуру до 39—40° и продолжают интен-
сивное размешивание еше в течение 8 часов. Массу охлаждают
до 20—25° и фильтруют через хлориновую ткань ПЦ. Отжа-
тый осадок размешивают со 100 мл воды и отфильтровывают.
Операцию повторяют несколько раз до исчезновения кислой
реакции по бумажке конго. Отмытый от кислоты отфильтро-
ванный продукт обрабатывают 100 мл аммиачной воды
(0,3%-ной), через 30 минут отфильтровывают и отмывают во-
дой до исчезновения щелочной реакции (проба на индикатор-
ную бумажку). Диортокрезилолпропан отжимают и сушат
сначала при 40—50° 5—6 часов, затем при 70—80°. Получают
26,6—32 г (77—92,5% теории, считая на загруженный орто-
кре.зол). После двухкратной перекристаллизации из 33%-ной
уксусной кислоты с применением активированного угля
(1 часть на 30 частей диортокрезилолпропана) т. пл. 134 —
138° (интервал т. пл. Г).
4,4/-Диокси-3,3/-димстилднфенил-2,2-пропан представляет
интерес как препарат для проведения исследовательских ра-
бот.
Л И Т Е Р А Т 5' Р . V
1. Th. Zincke, Liebigs Ann. Chem., 400. 33 (1913)..
2 И. Al. Билик, P. Л. Глобус, В. Г. Брудзь. Пласт, массы № 3 6'1
(1961).
8* 11.5