/
































































































































































































































































































Текст
Л. Н. АЛЕКСАНДРОВ
3J ***** I '4 Ы I У\М \
ВЕЩЕСТВО
ПОЧВЫ
И ПРОЦЕССЫ
ЕГО
ТРАНСФОРМАЦИИ
АКАДЕМИЯ НАУК СССР
ВСЕСОЮЗНОЕ ОБЩЕСТВО ПОЧВОВЕДОВ
ИНСТИТУТ АГРОХИМИИ И ПОЧВОВЕДЕНИЯ
Л. Н. АЛЕКСАНДРОВА
ОРГАНИЧЕСКОЕ
ВЕЩЕСТВО ПОЧВЫ
И ПРОЦЕССЫ
ЕГО ТРАНСФОРМАЦИИ
ЛЕНИНГРАД
«НАУК А»
ЛЕНИНГРАДСКОЕ
ОТДЕЛЕНИЕ
1980
УДИ 631.417.2
Александрова Л. Н. Органическое вещество почвы и процессы его
трансформации. — Л.: Наука, 1980.— 288 с.
В монографии рассматриваются источники органического вещества в почвах,
их состав, процессы трансформации, продукты взаимодействия с
минеральной частью почвы. Основное внимание уделено характеристике гумусовых
кислот — процессам их образования, природе и свойствам, ибо они являются
главнейшим компонентом гумуса и имеют огромное значение в
почвообразовании и плодородии почвы. Обобщена основная мировая литература по
гумусовым веществам и приведены многолетние исследования автора, позволившие
предложить ряд новых гипотез о механизме гумификации и процессах
взаимодействия с компонентами минеральной части почвы. Освещена проблема
формирования гумусового горизонта в пахотных дерново-подзолистых
почвах — основном земельном фонде в Нечерноземной зоне, показано значение
органических удобрений в формировании гумуса. Описан ряд оригинальных
методов исследования состава гумуса, природы гумусовых кислот и их ор-
гано-минеральных производных, а также методов моделирования процесса
гумификации и миграции органических веществ из почвы. Лит. — 565 назв.,
ил. — 56, табл. — 74*
ОТВЕТСТВЕННЫЙ РЕДАКТОР
В. А. КОВДА
k '- 676-80 3802020000 © Издательство «Наука», 1980 г.
055(02)-80
ПРЕДИСЛОВИЕ
Значение органического вещества почвы трудно переоценить,
ибо уже сам процесс превращения материнской породы в новое
тело природы — в почву — произошел, по образному выражению
М. В. Ломоносова, «от согнития животных и растущих тел со
временем». Все звенья почвообразовательного процесса связаны
с ее органическим веществом. В форме органических и органо-
минеральных соединений аккумулируются в почве огромные
запасы элементов питания и энергии, в составе органо-минеральных
компонентов осуществляется миграция многих элементов как
в пределах почвенного профиля, так и за его толщу в лито- и
гидросферу. Органическое вещество почвы и его главнейший и
специфический компонент — гумус — являются не только запасными
источниками всех элементов питания растений, но и
регуляторами главнейших физико-химических и биологических свойств
почвы, обусловливающих водно-воздушный и питательный режим
растений в любой природной зоне на Земле.
Огромно значение органического вещества почвы в
регулировании и повышении плодородия почвы, в ее коренной переделке.
Дальнейшая интенсификация земледелия в Нечерноземной зоне
РСФСР неразрывно связана со все более широким применением
органических удобрений. Не менее велико значение органического
вещества в образовании и накоплении многих органогенных
природных тел (торфа, сапропелей, углей, нефти) в литосфере и
огромных запасов органических веществ в гидросфере, в составе
которых неизменно господствуют гумусовые вещества, являющиеся
продуктом особого процесса трансформации органических
остатков на Земле — их гумификации. Совершенно естествен поэтому
интерес к исследованию природы и свойств этих соединений и
механизму их образования, который характерен для почвоведения
и ряда сопряженных с ним дисциплин на протяжении текущего
столетия.
Публикуемая книга — итог более чем 40-летнего изучения
природы и свойств органической части почвы, и прежде всего
ее основного компонента — гумусовых веществ, начатого по
инициативе проф. Сергея Павловича Кравкова, выдающегося
ученого и талантливого педагога, раскрывшего автору огромную
3
1*
значимость гумуса в почвообразовании и указавшего на
наиболее сложные и неясные стороны учения об органическом веществе
почвы.
Автор выражает сердечную благодарность своим многолетним
сотрудникам И. М. Андреевой, В. Н. Ефимову, А. А. Короткову,
М. Ф. Дюжину, О. А. Найденовой, М. В. Новицкому, А. М. Пуп-
кову, М. П. Филатову, О. В. Юрловой, а также аспирантам
В. Ф. Аршавской, И. Н. Барановскому, М. Г. Васильковой,
В. В. Вячкилеву, Э. М. Дорфман, 3. С. Ефимовой, А. С. Кащенко,
В. П. Колодка, С. Р. Корюшкиной, Г. П. Крупиной, Л. В. Ла-
бицкой, Г. П. Ландсберг, М. Надю, А. В. Назаровой, И. Проху,
Э. А. Румянцевой и Ю. И. Фомину, оказавшим огромную помощь
в экспериментальной разработке отдельных гипотез, а также
в накоплении фактического материала по характеристике состава
и свойств гумусовых веществ.
Глава I
ИСТОЧНИКИ ОРГАНИЧЕСКОЙ ЧАСТИ ПОЧВЫ
Источники органической части почвы — органические остатки,
поступающие в нее. В целинных почвах это растительные остатки,
отмирающие микроорганизмы и почвенная фауна, являющиеся
как исходным материалом для образования гумуса, так и
возбудителем самого процесса гумусообразования. В пахотных почвах
существенное значение в качестве источника гумуса имеют
органические удобрения, состав которых очень разнообразен.
Биомасса растительного, микробного
и животного происхождения, поступающая в почву
Значение высших зеленых растений как гумусообразователей
общеизвестно благодаря их специфической способности
синтезировать органические вещества из минеральных соединений.
Большинство низших организмов и представители животного
мира не вносят в почву новых запасов органических веществ
и в этом аспекте являются вторичными формами. Роль автотроф-
ных бактерий и водорослей в первичном синтезе органических
веществ очень невелика. Более сложно оценить фактическое
соотношение этих групп в годичном цикле почвообразовательного
процесса, а следовательно, и в гумусообразовании.
К настоящему времени накоплен и обобщен значительный
материал о биомассе зеленых растений и их химическом составе
для всех природных зон и ассоциаций. Как показывают данные
Л. Е. Родина и Н, И. Базилевич (табл. 1), максимальная общая
биомасса характерна для лесной растительности; под хвойными
и лиственными лесами умеренных широт она колеблется от 100
до 400 т сухого вещества на 1 га, но основная часть этой биомассы
многолетняя, и в почву ежегодно поступает 3.5—9.0 т сухого
вещества в виде наземного опада, образующего подстилку.
Последняя и является основным источником гумуса в лесных почвах.
Общий запас подстилки колеблется в широком диапазоне в
зависимости от состава, возраста и густоты насаждений, а также от
условий минерализации ежегодного опада. В среднем, по данным
Л. Е. Родина и Н. И. Базилевич [265], общий запас подстилки
5
Таблица 1
Биомасса растительности в различных природных зонах,
ц на 1 га сухого вещества (по: [265])
Природная зона
Тундра
арктическая
кустарниковая
Ельники тайги
северной
центральной
южной
Сосняки южной тайги
Леса
березняки
буковые
дубовые
Болота сфагновые
Степи
луговые
сухие
Пустыни
полукустарниковые
эфемерно полукустарниковые
субтропические
Саванна
смешанная
сухая
Леса
субтропические лиственные
тропические дождевые
Биомасса
общая
<50
280
1000
2600
3300
2800
2200
3700
4000
370
250
100
43
125
60
666
268
4100
>5000
корни
<35
231
220
600
735
636
505
950
960
40
205
85
38
104
35
39
ИЗ
'820
900
Ежегодный
прирост
<10
24
45
л 70
85
61
120
130
90
34
112
42
12
95
25
(120)
73
245
325
Ежегодный
опад
<10
9
35
50
55
47
70
90
65
25
112
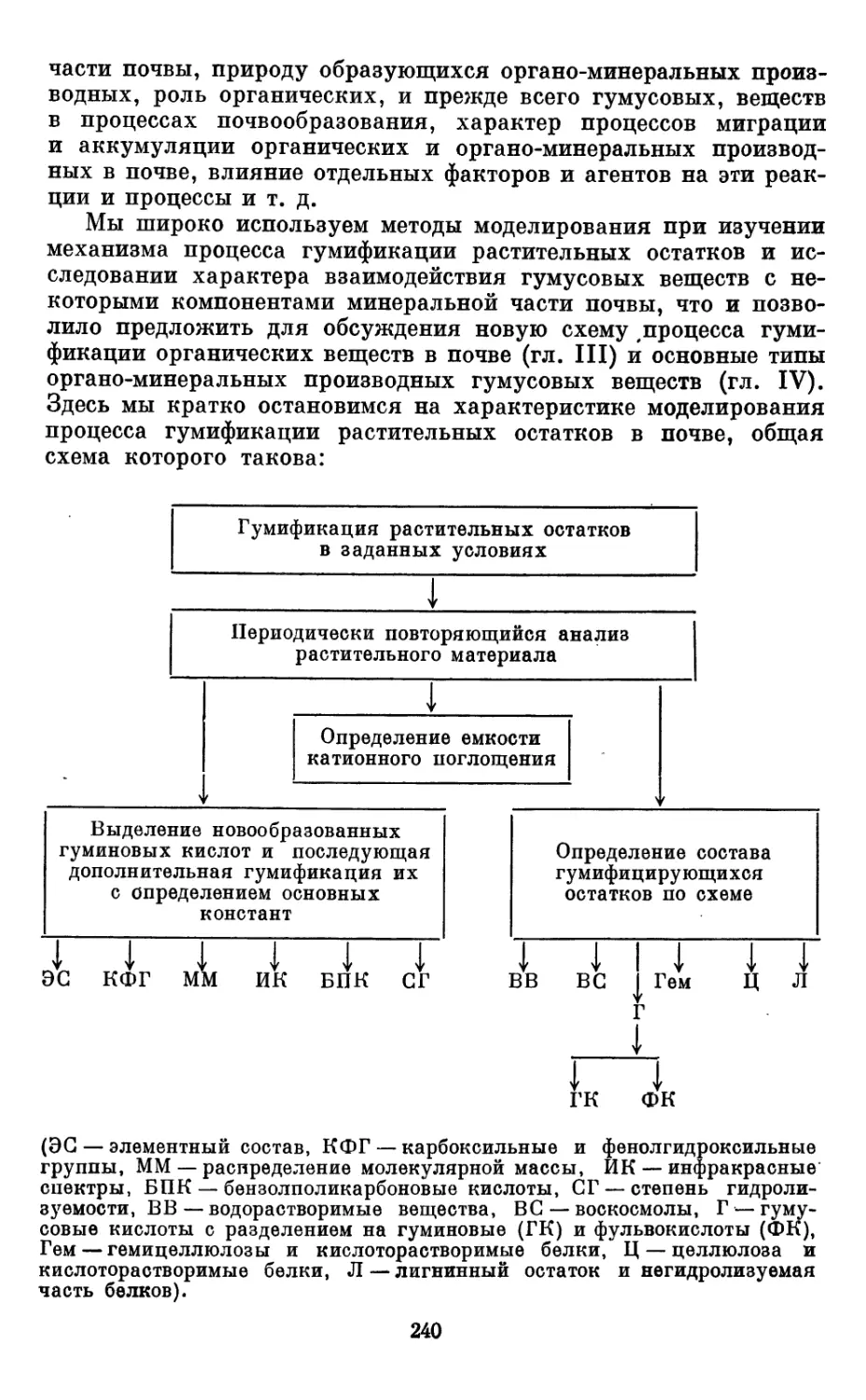
42
12
94
24
(115)
72
210
250
Запасы
органических
остатков
в
подстилке
и степном
войлоке
<35
835
300
450
350
448
300
150
150
>1000
62
15
13
100
20
колеблется от 15 до 45 т сухого вещества на 1 га. Он минимален
в широколиственных лесах, ибо условия минерализации подстилки
наиболее благоприятны.
Общая масса корней в лесах таежно-лесной зоны jb среднем
составляет 25% наземной биомассы, корни в основном многолетние
и обычно не рассматриваются как главный источник гумуса
в лесных почвах. Следует отметить, что некоторая часть корней
представлена тонкими, ежегодно отмирающими волосками,
равная, по данным Н. А. Качинского [1321 и А. П. Малянова [193],
6
приблизительно 30% общей массы корней, и исчисляется,
следовательно, величинами порядка 3—5 т сухого вещества на 1 га,
приближаясь к количеству корней под многими культурными
растениями. В лесах влажных субтропиков и тропиков общая
биомасса резко возрастает, достигая 400—500 т и более на 1 га,
а количество подстилки уменьшается, составляя 2—10 т,
вследствие интенсивной минерализации опада.
Природная травянистая растительность образует значительно
меньшую общую биомассу, но она всегда однолетняя, и вся
неотчуждаемая человеком часть ее полностью участвует в
ежегодном цикле почвообразовательного процесса, в том числе и в гуму-
сообразовании. Общая биомасса степной травянистой
растительности колеблется от 10 до 25 т сухого вещества на 1 га,
а луговая травянистая растительность суходольных лугов,
хвойных и смешанных лесов таежно-лесной зоны не превышает 10—
13 т. Характерная особенность травянистой растительности —
ежегодное отмирание не только надземной, но и корневой системы,
составляющей обычно в этих зонах не менее половины всей
биомассы. Их количество и глубина проникновения очень разные.
Общая биомасса корней под природной луговой и степной
растительностью составляет в среднем от 8.5 до 20.5 т сухого вещества
на 1 га. Эти данные, по-видимому, неоптимальны вследствие
известных методических трудностей отбора и полного учета корней
для всей почвенной толщи. Масса корней в метровой толще
черноземов и темно-каштановых почв достигает 30 т сухого вещества
на 1 га [269, 323]. В толще почвы она образует очень густую сетку,
пронизывая по существу весь почвенный профиль. Достаточно
указать, что общее количество корневых волосков у одного
растения составляет 14 млрд., а их длина — свыше 10 000 км [161].
Ежегодно отмирая, корни доставляют, таким образом, материал
для гумусообразования практически во все участки почвенной
толщи, в результате чего и образуется столь однородный по
степени гумусированности гумусово-аккумулятивный горизонт. В
области пустынь общая биомасса растений снижается до 4—6 т
сухого вещества на 1 га и лишь в эфемерно-кустарниковых
полупустынях равна в среднем 12.5 т. Корни здесь, глубоко проникая
в почву, образуют биомассу, почти равную наземной части
растений. Очень специфична болотная растительность, которая при
общей средней биомассе живых растений, составляющей лишь
десятки тонн сухого вещества на 1 га, и относительно невысоком
ежегодном приросте ее образует огромные запасы органических
остатков («очес») вследствие длительной консервации
значительной части ежегодного прироста.
Наименьшую биомассу для гумусообразования дают
культурные растения, наземная часть которых почти полностью
отчуждается человеком. К сожалению, до настоящего времени данные,
характеризующие биомассу культурных растений, остающуюся
в почве в виде пожнивных и корневых остатков, относительно
7
немногочисленны и разрозненны (табл. 2). Как видим, несмотря
на колеблющиеся урожаи культур в зависимости от их вида,
уровня агротехники и природных условий, пожнивные и
корневые остатки для зерновых, картофеля и овощей дают не более
Таблица 2
Биомасса культурной растительности, ц на 1 га сухого вещества
Культура
общая
Биомасса
пожнивные
остатки
корни
Источник
Дерно в о -подзо л и ста я суглинистая почва
Рожь озимая
Ячмень
Пшеница
озимая
яровая
Овес
Картофель
Клевер+тимофеевка (2-летние)
84.3
127.2
97.0
109.1
75.3
96.4
88.2
103.1
134.9
Дерново-подзолистая
Рожь озимая
Овес
Капуста кормовая
Подсолнечник
Картофель
Клевер+тимофеевка+ежа
-(-овсяница
1-летние
2-летние
3-летние
90.8
61.8
149.6
224.6
16.3
115.7
226.5
338.3
Чернозем выще
Пшеница яровая
78—122 |
7.4
18.6
10.0
14.8
10.9
12.4
12.2
2.8
18.8
п е с ч а н а
14.9
14.3
12.4.
15.4
34.5
37.8
39.4
81.9
> л о чен н ы
15-25 |
49.0
27.6
44.0
25.2
12.8
37.0
15.1
19.6
62.5
194]
[179]
[94]
[179]
[179]
[94]
[179]
[179]
[179]
я почва
13.5
11.4
57,.8
71.6
26.8
84.0
104.3
173.4
й
8-13 |
[155]
[155]
[180]
[180]
[180]
[180]
[180:
[331
[59]
50% наземной массы, что при средних урожаях составляет от 2
до 5—7 т сухого вещества на 1 га. Исключением являются
многолетние травы, под которыми в почве образуется значительное
количество исходного для гумусообразования материала в виде
поукосных и особенно корневых остатков. В зависимости от урожая
он колеблется от 7—10 до 15—25 т сухого вещества на 1 га.
Значительно сложнее решить вопрос о биомассе
микроорганизмов как гумусообразователей. По справедливому замечанию
8
Т. В. Аристовской [42 J, размер сезонной или годичной продукции
их биомассы до сих пор не решен из-за недостаточной точности
методов ее определения. Действительно, численность
микроорганизмов чрезвычайно изменчива и зависит от многих факторов:
типа почвы, характера растительности, гидротермического
режима; поэтому в годичном цикле почвообразования наблюдается
резко выраженная пульсация их количества. Так, например,
количество бактерий колеблется в почвах от 0.3 до 47 млрд./г.
Эти колебания максимальны в северных районах и минимальны
в средних широтах (табл. 3).
Таблица 3
Численность бактерий в разных почвах, млрд на 1 г сухой почвы
(по: [42])
Почва, растительность
Дерново-подзолистая
суглинистая, луг
Дерново-подзолистая
супесчаная
окультуренная v
Темно-серая оподзолен-
ная, дубовый лес
Чернозем обыкновенный,
пашня -
Луговая
тяжелосуглинистая
Местоположение
почвы
Карельский перешеек
ЛитССР
Харьковская обл.
» »
» »
Месяц
наблюдений
VII—VIII
V-IX
V
VI
XI
V
V
IV
Пределы
колебаний численности
2.0—15.0
0.7—3.3
4.76-32.65
1.05—3.58
1.05—8.41
0.191-0.931
0.452—3.328
0.304—2.232
И. В. Тюрин [299] считал, что масса сухого вещества
микроорганизмов в годичном цикле почвообразования не превышает
1 т/га, коррелируя с запасами гумуса; в почвах, бедных гумусом,
биомасса сухого вещества микроорганизмов колеблется около
1—2% от общего запаса гумуса; в почвах, содержащих
значительное количество гумуса, она составляет лишь десятые доли
процента. Н. А. Красильников [161] также отмечает трудность
подсчета количества микроорганизмов в почве и в качестве
ориентировочных данных называет величины порядка 7—10 т на 1 га
бактериальной массы, что при среднем содержании воды около
70—80% также составит не более 2 т сухого вещества на 1 га.
Аналогичные материалы приводят и другие авторы [56, 135].
Количество органических остатков почвенной фауны (фитофаги,
сапрофаги, некрофаги, крупные животные-хищники), по данным
В. А. Ковды [135], может достигать 1.0—1.5 т сухого вещества
на 1 га. Следует отметить, что все подсчеты биомассы почвенной
фауны и оценка ее значения как материала для гумусообразования
очень условны вследствие не только неточности методов подсчета
9
и определения массы, но и сложности взаимоотношений между
микроорганизмами и почвенной фауной. Как указывает Т. В. Ари-
стовская [42], значительная часть биомассы микроорганизмов
уничтожается почвенной фауной; аналогична судьба многих
представителей мезофауны. Основными источниками гумуса в почвах
являются, конечно, растительные остатки, масса которых во много
раз превышает биомассу всех вторичных форм организмов в почве.
Химический состав органических остатков
Химический состав органических остатков очень
разнообразен.1 Большую часть их массы составляет вода, содержание
которой минимально в древесине, грубых одревесневших корнях и
отмирающих стеблях. Основная масса сухого вещества состоит
из белков, углеводов, лигнина, липидов и липопротеидов,
дубильных веществ, смол, восков Стабл. 4), а также многих других
Таблица 4
; Химический состав органических остатков, % на сухую беззольную массу
Организмы
Бактерии
Водоросли
Лишайники
(кустистые и пластинчатые)
Мхи
Папоротникообразные
Хвойные
древесина
хвоя
Лиственные
древесина
листья
Многолетние травы
злаки
бобовые
Зола,
%
2-10
20—30
2-6
3-10
6-7
0.1-1
2-5
0.1-1
3-8
5—10
5—10
Белки-и
родствен-
,,ные им
[вещества
40-70
10-15
3-5
5-10
4-5
0.5—1
3-8
0.5—1
4-10
5—12
10-20
Углеводы
гемицел-
люлозы,
пектиновые
вещества
Есть
50-60
60-80
30-60
20—30
15-25
15—20
20-30
10-20
25-35
15-25
целлюлоза
Нет
5-10
5—10
15-25
20—30
45—50
15-20
40-50
15-25
25—40
25—30
Лигнин
0
0
8—10
20-30
25-30
20-30
20—25
20-30
15—20
15-20
Липиды,
дубильные
вещества
1—40
1-3
1-3
5—10
2—10
2—12
5-20
5-15
5-15
2—10
2—10
* Раздел составлен по материалам ряда монографий [58, 62, 103, 163, 182,
325J.
10
органических соединений (органические кислоты, глюкозы,
алкалоиды, витамины, стимуляторы роста и др.). В живом организме
весь этот комплекс соединений образует сложную открытую
изотермическую систему, обеспечивающую саморегуляцию и
самовоспроизведение за счет ферментативных реакций внутри живой
клетки. При отмирании живых организмов на поверхности или
в толще почвы вся эта система органических соединений
подвергается сложным процессам трансформации, превращаясь в новые,
уже неживые формы накопления органических веществ в
биосфере. Следует иметь в виду, что при отмирании живых организмов
в почву поступает масса различных, преимущественно
высокомолекулярных, соединений, что можно проиллюстрировать
схемой молекулярной организации живой клетки по А. Ленинд-
жеру [182]:2
клетка
t
ядро,
митохондрии,
хлоропласты
ферментные комплексы,
рибосомы,
I ^сократительные си- < л
стемы
органеллы
надмолекулярные
комплексы
(ММ 10е—109)
макромолекулы
(ММ 10»—10»)
строительные блоки
(ММ 100—350)
промежуточные
соединения (ММ 50—250)
предшественники,
поступающие из среды
(ММ 18-44)
нуклеиновые
кислоты
мононуклео-
тиды
i
белки
аминокислоты
рибоза,
карбамоил-
фосфат
полисахариды
простыв
сахара
а-кето- фосфо-
кислоты пируват,
малат
липиды
жирные
кислоты,
глицерин
t
ацетат,
малонат
— со,,—
Н20, N2
Соотношение между различными группами органических
соединений, образующих биомассу растительных, микробных и
животных остатков, конечно, варьирует, что предопределяет
различную скорость их трансформации в почве. Основная масса сухого
вещества растительных остатков состоит из углеводов и лигнина,
г Здесь и дадьще ММ — относительная молекулярная масса,
11
которые образуют клеточные стенки растения. Особенно богаты
ими остатки древесины и корни, относительно медленно поэтому
разлагающиеся в почве. Листья древесной растительности богаты
дубильными веществами, в хвое много восков и смол, также
тормозящих процессы разложения. Очень специфичен вещественный
состав мхов, масса которых состоит в основном из углеводов,
среди которых преобладают камеди, пектиновые вещества и поли-
урониды. Азотсодержащие компоненты представлены в основном
белками, количество которых максимально в остатках бобовых
трав, а также сложной системой нуклендов и полинуклеидов
(в том числе РНК и ДНК), функционирующих в живых
клетках как коферменты, информационные (матричные) транспортные
и рибосомные нуклеиновые кислоты. Их масса в составе
поступающих растительных остатков невелика.
Остатки микроорганизмов резко отличаются по составу
органических веществ от остатков растительного происхождения.
В составе сухого вещества резко доминируют белки и
нуклеиновые кислоты, отсутствует лигнин, что обусловливает наиболее
интенсивную трансформацию этих остатков в почве. Не менее
специфичен и состав остатков почвенной фауны. Наряду с белками
и углеводами в них содержатся хитин, липиды, а также
нуклеиновые кислоты и нуклеотиды. По элементному составу органические
остатки резко отличаются от компонентов литосферы (табл. 5).
В биомассе организмов органогены — С, N, О, Н — составляют
до 98—99%, причем основная масса органических соединений
Таблица 5
Средний химический элементный состав организмов суши,
% к массе живого вещества, почв и земной коры, % к массе
(по А. П. Виноградову; цит. по: [76])
Элемент
О
С
Н
N
Са
Si
Р
К
S
С1
Na
А1
Fe
Средняя масса
в
организмах
70.00
18.00
10.50
0.30
0.50
0.15
0.07
0.07
0.05
0.04
0.02
0.02
0.02
в почве
(на сырую
почву)
55.00
5.00
5.00
0.10
2.00
20.00
0.08
1.00
0.04
0.10
1.00
7.00
2.00
в земной
коре
47.20
0.10
0.15
2.3-10~2
3.50
27.60
7.8-10~2
2.50
0.05
4.8.10-2
2.64
8.80
5.00
Элемент
Ra
Mn
Sr
В
Ti
Ba
Li
Cu
Zn
Co
Mo
I
F
Ni
Средняя масса
в
организмах
2
7«
1
1
8
1.
1
1
3
1.
2
1«
8
'5«
¦ ИГ1
10~3
10"3
10~3
10~4
10~4
ю-4
10~4
10~4
10~б
10~б
10~б
кг*
иг*
в почве
(на сырую
почву)
1 -КГ"
0.06
0.02
8-Ю-4
0.40
0.01
ыо-8
5-Ю-4
МО"3
З-НГ4
мо-5
МО-4
0.01
з.нг8
в земной
коре
1.10-ю
0.09
0.04
з-ю-4
0.60
3.9.10~2
6.5-Ю-3
0.01
5. Ю-3
МО"8
1.5- Ю-3
з.ю-5
2.7.1СГ2
0.01
12
углерода находится в живых организмах в сильно восстановленной
и гидрированной форме и обладает, следовательно, высоким
запасом энергии. Это необходимо учитывать при оценке характера
их наиболее вероятной трансформации в почве. Помимо
органических соединений растительные остатки всегда содержат
некоторое количество зольных элементов, колеблющееся в зависимости
от видовой принадлежности и условий обитания в широких
пределах (1—10%). В составе золы доминируют калий, кальций,
кремний; в меньших относительных количествах содержатся
фосфор, сера, хлор. Минимальная зольность характерна для
древесины, максимально богаты зольными элементами травы
и водоросли.
Таким образом, в почву попадает чрезвычайно сложная смесь
органических соединений различной природы, что до сих пор
затрудняет точное решение вопроса о реальных источниках (вернее,
реальных строительных единицах) гумусовых, и прежде всего
гуминовых, кислот. Для более точных суждений о
вероятных источниках гумусовых кислот рассмотрим кратко
современные представления о природе основных компонентов, слагающих
массу сухого вещества организмов.
Белки и полинуклеотиды. Белки — типичные
высокомолекулярные соединения и в живой клетке играют первостепенную роль.
ММ белков колеблется в широких пределах: от 6000 до 1 000 000
и выше, но, несмотря на большое разнообразие отдельных
представителей, элементный состав их относительно постоянен, %:
С 48—55, Н 6—7, О 19—27, N 16—20, S— 2. Молекула белка
состоит из одной или нескольких полипептидных цепей, каждая
из которых содержит не менее 100 аминокислотных остатков,
ковалентно соединенных между собой пептидными связями. Все
белки независимо от их функций и видовой принадлежности
построены из остатков 20 а-аминокислот и обладают кислотно-
основными функциями благодаря наличию свободных концевых
а-аминогрупп и карбоксильной группы. Аминокислоты,
входящие в состав белков, следующие: глицин, аланин, валин, лейцин,
изолейцин, серии, треонин, аспарагиновая кислота, глутами-
новая кислота, аспарагин, глутамин, лизин, аргинин, гистидин,
цистин, метионин, фенилаланин, тирозин, пролин и триптофан.
По составу белки делятся на два класса: простые (протеины)
и сложные (протеиды). Простые белки состоят только из
аминокислот, сложные — из протеинов и каких-либо других
органических или неорганических продуктов, образующих простети-
ческую группу. Сложные белки конституционные и осуществляют
в клетке различные функции: регулируют процессы обмена,
участвуют в ферментативных реакциях, образуют мембранные
системы. Протеиды классифицируются по характеру веществ
простетической группы. Различают липопротеиды, содержащие
липиды, и глюкопротеиды, в которых роль простетической группы
играет какой-либо высокомолекулярный углевод; наиболее важна
13
Рис. 1. Конформация белковых
молекул.
а — глобулярные белки: 1 — единичная
цепь, г — олигомерный белок, б —
фибриллярные белки.
группа нуклеопротеидов, в
которых белок связан с нуклеотидом,
в частности с нуклеиновыми
кислотами. Белки классифицируют
также по их конфирмации
(пространственной структуре).
Выделяют фибриллярные белки,
молекулы которых состоят из
параллельных, сильно вытянутых
пептидных цепей (они выполняют
функции структурных элементов
в молекуле), и глобулярные белки,
молекулы которых представляют собой свернутые
полипептидные цепи, форма этих белков близка к сферической (рис. 1).
В настоящее время для нативных белков изучены различные
уровни структуры. Первичная структура характеризует
последовательность аминокислотных остатков в полипептидной цепи,
соединенных ковалентными связями; вторичная структура
определяет тип укладки полипептидных цепей; термин «третичная
структура» относится к способу укладки с образованием
компактной, плотно упакованной структуры, характерной для глобул-
лярных белков. Четвертичная структура характеризует способ
объединения (расположения в пространстве) отдельных
полипептидных цепей в молекуле.
Полинуклеотиды представлены в основном дезоксирибонукле-
иновыми (ДНК) и рибонуклеиновыми (РНК) кислотами,
содержание которых в живых организмах колеблется в очень широких
пределах, достигая 50% в рибосомах и снижаясь до 0.1—1.0%
в листьях и стеблях растений. Нуклеиновые кислоты
характеризуются огромной ММ, достигающей 2 000 000—8 000 000.
Основные компоненты их молекулы — пуриновые и пиримидиновые
основания, пентоза и фосфорная кислота:
он
I
но-р-о-сно
II I Л
о
Основание
«4$
он он
рибонуклеотид
ОН
I
Н0-Р-0-СН2
II |^0
о
Основание
ОН Н
феюпсинукюотид
14
Основаниями являются аденин (7), гуанин (2), цитозин (5),
урацил (4) и тимин (5); они соединены с пентозой гликозидной
связью, образуя нуклеозиды:
NH2
|
N С \
1 II СН
НС С /
^n/\n/
1
н
1
]
0=
H2N-
0
Л
HN GH
\N/
i
4
0
А\ /n\
н/ V \
J г /сн
А
2
0
Л
HN С-СНз
о=с &
\N/
1
н
5
NH2
1
t
/ Чсн
1 II
0=С СН
\N/
1
н
3
Фосфорнокислые эфиры нуклеозидов носят название нуклеоти-
дов. Они содержатся в значительных количествах во всех клетках
в виде сложной системы рибонуклеозидов и дезоксирибонуклео-
зидов. Молекулы ДНК состоят из ковалентно связанных между
собой дезоксирибонуклеотидов, а молекулы РНК — из рибо-
нуклеотидов (см. с. 16).
Варьирование оснований (1—5) и нуклеотидной
последовательности обусловливает большое разнообразие деталей строения
и ММ как нуклеиновых кислот, так и нуклеотидов.
К классу белков принадлежат и ферменты, осуществляющие
все процессы превращения органических веществ в природе как
в живой клбтке, так и вне ее. К настоящему времени
идентифицировано около тысячи различных ферментов, многие получены в
кристаллическом виде. Сущность действия ферментов сводится к катализу
химических реакций. Класоифицируют ферменты в зависимости от
природы катализируемых ими реакций. Различают: оксиредуктазы,
гидролазы, трансферазы, лиазы, лигазы и изомеразы [182].
Белковые вещества при отмирании организмов быстро
подвергаются биохимическим превращениям. Под влиянием
ферментативной деятельности микроорганизмов они расщепляются на менее
сложные компоненты, легко гумифицируются и минерализуются".
Разрушение в почве растительных первичных белков всегда
сопровождается синтезом новых вторичных белков, образующих
плазму микроорганизмов.
15
ДНК
.1
сн
1
РНК
I
сн,
Ш Й
о н
НО-Р=0
I
о
<Чск
о он
НО-Р = 0
I
о
сн9
hW нСГн
о н
НО-Р = 0
6
о он
НО-Р=0
о
СН2 О СНг О
ОН О ОН'
НО-Р=0
I
О
сн.
Углеводы. Углеводы —
полиоксиальдегиды или]по-
лиоксикетоны с
'эмпирической формулой (СН20) „;
они делятся на три группы:
моносахариды, олигосаха-
риды и полисахариды.
Последние составляют
главную массу углеводов во
всех органических остатках.
Моносахариды очень
разнообразны; известно
несколько сотен различных
по структуре и
стереохимии моносахаридов.
Простейшие из них — пира-
нозы (гексозы) и фуранозы
(пентозы) — содержат в
молекуле только
альдегидную или кетонную
группировку. Кроме этих групп
в молекулу моносахарида
могут входить
карбоксильные группы (сахарные
кислоты) и аминогруппы (ами-
носахара). Среди сахарных
кислот наибольшее
значение имеют уроновые
кислоты (глюкуроновая, га-
лактуроновая, маннуро-
вая) —компоненты многих
полисахаридов;
главнейшими аминосахарами
являются глюкозамин и га-
лактозамин, которые также
входят в состав
полисахаридов.
К олигосахаридам, широко распространенным в растительных
остатках, относятся сахароза, целлобиоза, лактоза, арабиноза,
мальтоза. Все они играют роль резервных углеводов в растениях.
Их молекула состоит из 2—10 моносахаридных единиц,
соединенных гликозидной связью. Главнейшими полисахаридами
растительного происхождения являются крахмал, целлюлоза, ге-
мицеллюлозы, пектин, а также камеди, хитин, агар; стенки
бактериальных клеток представляют собой сложный полисахарид-
пептид, называемый пептидогликаном, или муреином.
Все полисахариды — высокомолекулярные соединения с ММ
до 104—107; в клетках их молекулы образуют крупные гранулы
Н0-Р = 0
I
0
I
сн2
он о он
о н
Н0-Р=0
о
сн2
о
НО—f = 0
о
И Ш
о н
I
16
—•«. \ \ « *
j Ь
Рис. 2. Схема строения амилозы (а) и амилопектина (б).
диаметром 10—40 нм (крахмал) или длинные волокна, состоящие
из ряда параллельных цепей, соединенных поперечными
водородными связями (целлюлоза). Крахмал состоит из двух
компонентов: амилопектина с разветвленным типом строения молекулы
и спиралевидных неразветвленных молекул а-амилозы (рис. 2).
Цепи эти полидисперсны, их ММ варьирует от нескольких тысяч
до 500 000-1 000 000.
Целлюлоза — наиболее распространенный структурный
компонент клеточных стенок растений, ее молекулы состоят из
остатков D-глюкозы. Отдельные молекулы соединяются в пучки или
волокна диаметром около 20 нм. В стенках растительных клеток
целлюлозные волокна имеют правильную, почти кристаллическую
упаковку и цементируются рядом других полисахаридов (в
основном гемицеллюлозами).
Гемицел л ю лозы представлены комплексом полисахаридов,
главнейшие компоненты которых (ксиланы, глюкоманнаны, га-
лактаны, а также полиурониды) характеризуются относительно
невысокой ММ: 10 000—40 000. Пектин, по современным
представлениям, — полимер метил-Б-галактуроната, в растительных
и микробных клетках он пропитывает клеточные стенки.
Компонентами клеточных стенок являются также камеди, содержащие
остатки D-галактозы и D-глюкуроновой кислоты, а также агар
морских водорослей, состоящий из остатков D- и L-галактозы,
и хитин — главный структурный элемент наружного скелета
насекомых, также высокомолекулярный углевод, дающий при
гидролизе глюкозамин. Полисахариды бактерий изучены слабо;
основной их компонент муропептид является дисахаридом, к которому
присоединена тетрапептидная боковая цепь. У ряда бактерий
к этой сложной муреиновой цепи присоединяются и другие
полисахариды, липосахариды, липопротеиды.
17
Скорость трансформации углеводов в почве различна и
обусловлена прежде всего химическим составом. Моносахариды и оли-
госахариды разлагаются наиболее интенсивно. Скорость
разложения полисахаридов ниже; при этом существенное значение имеют
не только химический состав самих полисахаридов, но и общее
строение фибрилл, которые они образуют. Наличие восковых
оболочек, которыми часто покрыты целлюлозные волокна, а также
их связь с лигнином тормозят процессы разложения последних.
Большинство исследователей признают участие углеводов в
процессах гумификации, хотя характер его не представляется
достаточно ясным.
Лигнин. Обязательным компонентом растительных остатков
является лигнин, составляющий в высших растениях от 15 до 30%
общей массы растительных тканей. Он относится к классу
ароматических соединений, хотя, к сожалению, несмотря на
значительное число капитальных исследований, посвященных раскрытию
строения молекулы, ее природа до настоящего времени не вполне
ясна [58, 62, 335, 404]. По-видимому, это положение объясняется
большим разнообразием представителей лигнина в разных
растениях, а также отсутствием простого повторения какой-либо
структурной единицы, подобно углеводам или белкам.
Препараты лигнина, выделенные из различных растений,
представляют аморфный порошок желтого или коричневого цвета
с колеблющимся элементным составом, %: С 60—64, Н~6,
О 29—33; имеются данные о небольшом количестве азота: 0.6—
1.5%. Наибольшим признанием пользуются представления
К. Фрейденберга [404] о том, что основной структурной единицей
лигнинов древесины хвойных растений является остаток кони-
ферилового спирта, в лигнине лиственных древесных пород
присутствуют остатки синапилового спирта, а у травянистых
растений доминируют остатки мономеров тг-кумарилового спирта:
СН20Н
СН2ОН
1
1«
СН2ОН
Ч)СН8 НзСО/ у Х)СН3
)н он он
конифериловый синапиловый п-кумариловый
спирт спирт спирту
На схеме (см. с. 19) видно, что с бензольным ядром лигнина
связаны его основные функциональные группы: метоксильные,
гидроксильные и пропановая боковая цепь. Содержание метокси-
лов колеблется от 15 до 21%, общее количество гидрокси-
18
Схема полимерной структуры кониферилового лигнина
н2сон
нс-
I
со
MeoL Jo
н,со
nh
H2GOH HG
i^/'OMe H2GOH
I I
О 1
CH
H<!_
1
HC-
HC-
H2COH
-CH
I
HC
I
-X
I
о
H2GOH
-CH
I
HG
H2COH
HC-
I
HGOH
/\
2 2
HGO H2COH
I I
OH HG-
HC GO
I I
ft
ILjoMe
Ц>'ОМе ОН
У ОС GHa
H2COH I | I
ОЖе
H2GOH
-сн
//
Y
OH
!OMe
I HG CH
1С- ! I
Ц/]'(ОМе)о.5
MeO:
HCOH
I
HC HGOH
HC-
H2GOH
HG-
I
l^JoMe
/0\l
H2C CH
\y о CH V' V Y
бн o-
HCO(CeH10Oe)wH
A
\/ H2<
I
O-
MeO'
>сон
I
CH
HCOH
I
и
I
о—
Mei
A
I
-o
(Me — метильная группа— CH3)
лов — от 1 до 8%; они находятся как при бензольном кольце
(фенольные), так и в пропановом остатке (спиртовые). В
боковой пропановой цепи содержатся карбонильные группы, которые
19
представлены лактонными, кетонными и альдегидными
группировками.
ММ лигнинов точно неизвестна. Ф. Э. и Д. А. Брауне считают,
что лигнин — полимер с ММ от 3650 до 58 000 [62]; по сводным
материалам А. Блажея и Л. Шутого [58], использование
различных методов определения ММ лигнина привело к очень разным
величинам: от 200 до 50 000 000. В одревесневших тканях
растений лигнин находится в форме лигноуглеводного комплекса,
характер этой связи неясен. Лигнин — один из наиболее
устойчивых против разложения компонентов растительных тканей,
но он хорошо гумифицируется, вследствие чего многие
исследователи считают его основным гумусообразователем [65, 195,
281, 282, 393, 396].
Липиды. Общим термином «липиды» называют обширную
группу жиров и жироподобных веществ, обладающих гидрофоб-
ностью и нерастворимостью в воде. Они очень разнообразны
в живых клетках по составу и функциям: являются структурными
компонентами мембран, выполняют защитную роль, служат
дополнительными источниками энергии. Основные представители
класса липидов в растительных и микробных клетках —
нейтральные жиры (ацилглицерины), фосфоглицериды, гликолипиды,
воски, фитостерины, терпены и липопротеиды.
Главными строительными блоками в большинстве групп
этого класса соединений служат жирные кислоты, молекулы
которых представляют собой длинные углеводородные цепи
с концевой карбоксильной группой (см. с. 21).
К настоящему времени известно свыше 70 различных жирных
кислот с числом атомов углерода от 14 до 22, причем
преобладающее значение имеют ненасыщенные жирные кислоты.
Глицериновые эфиры жирных кислот называются
нейтральными жирами, или ацилглицеринами; они составляют главный
запас липидов в растениях. В фосфоглицеридах в отличие от ацил-
глицеринов одна из первичных гидроксильных групп глицерина
этерифицирована фосфорной кислотой, эту группу липидов часто
называют фосфатидами; многие из них содержат аминоспирты
(этаноламин, холин) и полярны. Гликолипиды — также полярные
соединения, неполярная часть их молекулы представлена
остатком жирной кислоты, а полярная группа — углеводными
компонентами, например гликозилдиацилглицеринами. Все эти липиды
входят в состав клеточных мембран.
Воски являются эфирами высших жирных кислот и
одноатомных спиртов с длинной цепью и содержат некоторое количество
углеводородов парафинового ряда. Покрывая тонким налетом
поверхность листьев и плодов, воски предохраняют ткани от
поражения микроорганизмами. Фитостерины представляют собой
сложные эфиры жирных кислот и высокомолекулярных
циклических спиртов (стеролов), в основе которых лежит циклическая
структура циклопентанопергидрофенантрена.
20
НО О
1
сна
1
сн2
1
сн2
1
сн2
1
сн2
1
сн2
1
сн2
1
сн2
1
сн2
1
сн2
1
сн2
1
сн2
1
сн2
1
сн2
1
сн,
льмитиновая
кислота
НО О |
1
сн2
1
сн2
1
сн2
1
сн2
1
сн2
1
сн2
1
сн2
1
GH
к
1
сн2
1
сн2
1
сн2
1
сн2
1
сн2
1
сн2
1
сн2
1
сн,
олеиновая
кислота
Н-
но-Ц
н-
н
1
н-с—
1
6
и
1
сн2
1
сн2
1
сн2
1
сн2
1
сн2
1
сн2
1
сн2
1 .
сн2
сн2
сн2
СН2
1
сн2
сн2
1
сн2
1
!_ сн,
-
онУсн2он
J
]
О
1
сн2
1
-С-Н
1
О
1
с=о
1
сн2
сн2
1
сн2
1
сн2
сн2
1
сн2
1
сн2
сн
сн2
1
сн2
1
сн2
1
сн2
сн2
1
СНз
моногалактозил-
диацилгли церин
Терпены образуют эфирные масла в растениях, и их молекулы
содержат две или более изопреновые группировки линейного или
циклического строения. К ним относятся ментол, лимонен,
камфора и др. Эта группа включает природный каучук, а также фитол
и каротиноиды — компоненты пигментов. Липопротеиды
представляют собой полярные липиды, ассоциированные с белками,
и входят в состав протоплазмы.
21
Н,С=сн снч (али-сно) Пигменты. Вещества, принадле-
1 I6 жащие к этой группе, хорошо
сн сн растворимы в органических рас-
1 3 творителях и обусловливают
окраску многих частей растений,
а также микроорганизмов.
Главнейшими растительными
пигментами являются каротин и ксан-
тофил, относящиеся к группе ка-
ротиноидов, и хлорофиллы,
окрашивающие клетки в зеленый цвет.
Каротиноиды — производные ли-
копина, эмпирическая формула
которого G40H6e; они содержат
значительное число сопряженных
двойных связей, обусловливающих
их участие в
окислительно-восстановительных реакциях.
Хлорофиллы состоят из длинной
гидрофобной терпеноидной цепи (остатка
фитола), соединенной сложно-
эфирной связью с пиррольными
кольцами (1—4), одно из
которых находится в восстановленной
форме (5).
В зеленых растениях
хлорофиллы составляют около 1 %
сухого вещества и образуют
комплекс с липидами и белками. Очень
разнообразны и многочисленны
С н - с нг пигменты микроорганизмов, в
частей н ности черные пигменты грибов [109].
3 Растительные вещества вторич-
хлорофилл а ного происхождения.8 Наряду
с белками, углеводами, липидами и лигнином в растениях
присутствует большая группа веществ, называемых обычно веществами
вторичного происхождения. Все эти вещества В. Л. Кретович [163]
делит на восемь групп: органические кислоты алифатического
ряда; ароматические и гидроароматические соединения; глюко-
зиды; дубильные вещества; эфирные масла и смолы; каучук
и гуттаперча; алкалоиды; регуляторы роста растений и
микроорганизмов, антибиотики. Некоторые из перечисленных групп
соединений содержатся в растениях и микроорганизмах в
заметных количествах, поступают в почву и ок&задвдют вдщщие на
состав органических веществ почвы.
8 Эта часть раздела написана М. В. Новицким.
Органические кислоты алифатического ряда очень
разнообразны, наиболее распространены щавелевая, многоосновные
оксикислоты, а также глюкуроновая и галактуроновая кислоты:
Алифатические
одноосновные
муравьиная
уксусная
масляная
многоосновные
щавелевая
фумаровая
янтарная
малоновая
цис-аконитовая
HGOOH .
СНдСООН
СН3-(СН2)2-СООН
СООН-СООН
НООС—СН=СН—GOOH
ноос—сн2—сн2—соон
ноос-сн2-соон
ноос-сн2—с=сн-соон
соон
Оксикислоты
одноосновные
гликолевая
молочная
глюконовая
многоосновные
яблочная
винная
лимонная
Альдегидо- и кетокислоты
глиоксилевая
глюкуроновая
галактуроновая
пировиноградная
а-кетоглютаровая
СН2(ОН)СООН
СН3СН(ОН)СООН
СН2(ОН)(СНОН)4СООН
СООН-СНОН-СН2—СООН
СООН-СНОН-СНОН-СООН
СООН-СН2-С(СООН)(ОН)—СН2-С00Н
GHO-GOOH
СООН
НО-С-Н
но-с-н
Н-С-ОН
но—с—н
GHO
СООН
но-с-н
н—с-он
I
н—с—он
но-с-н
I
сно
СНз-СО-СООН
СООН-СО—СН2-СН2- СООН
23
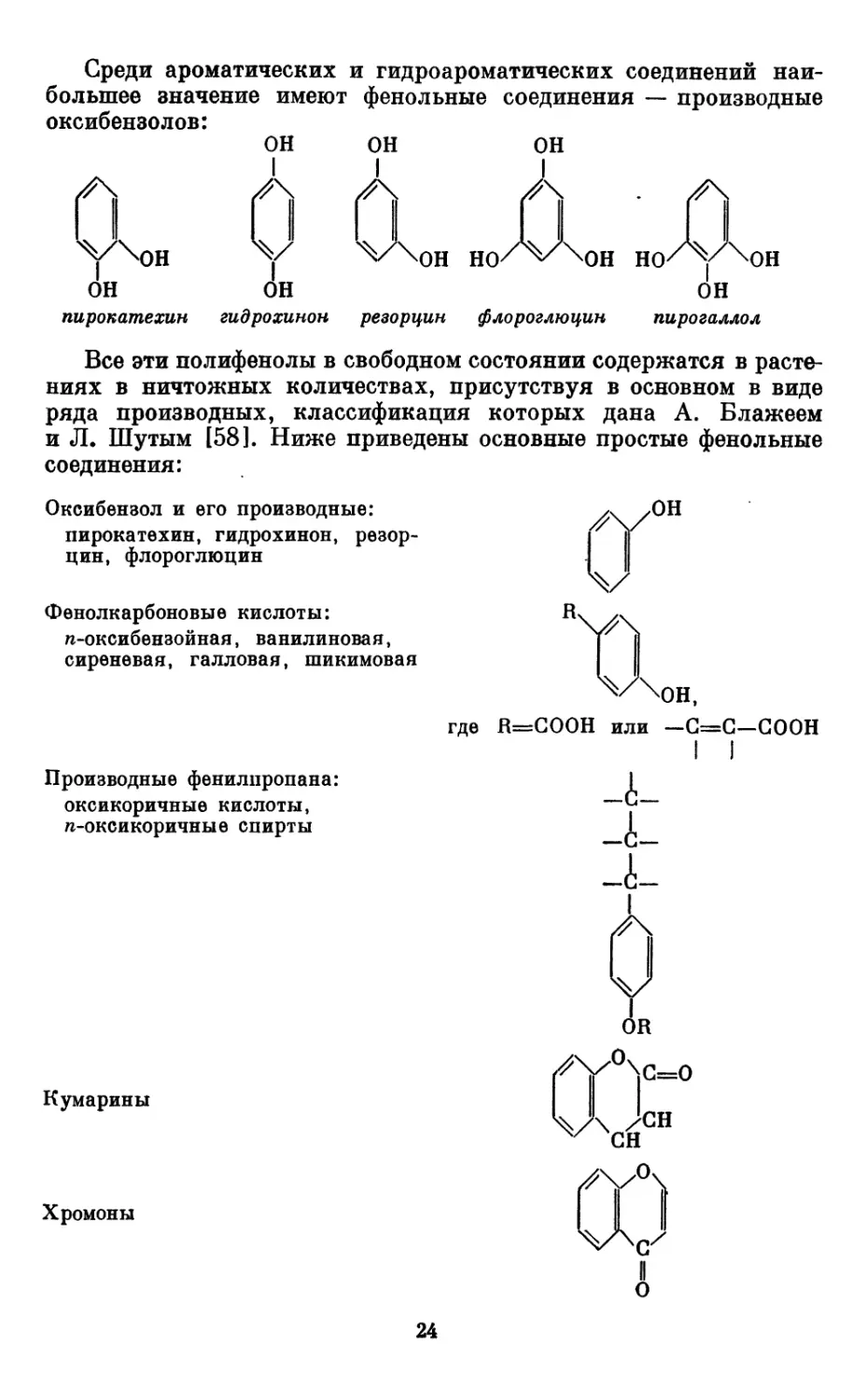
Среди ароматических и гидроароматических соединений
наибольшее значение имеют фенольные соединения — производные
оксибензолов:
ОН ОН ОН
I I
^\он но/^/\он
гидрохинон резорцин флороглюцин
НО' уЧ)Н
ОН
пирогаллол
Все эти полифенолы в свободном состоянии содержатся в
растениях в ничтожных количествах, присутствуя в основном в виде
ряда производных, классификация которых дана А. Блажеем
и Л. Шутым [58]. Ниже приведены основные простые фенольные
соединения:
Оксибензол и его производные:
пирокатехин, гидрохинон,
резорцин, флороглюцин
Фенолкарбоновые кислоты:
га-оксибензойная, ванилиновая,
сиреневая, галловая, шикимовая
/\/
ОН
где R=GOOH или — С=С—GOOH
Производные фенилиропана:
оксикоричные кислоты,
тг-оксикоричные спирты
Кумарины
Хромоны
АЛс=о
24
Бензофеноны
Ксантоны
Стильбены
Ауроны
Хал коны
<_^
vi<3
Os
/Ч
//
У-сн=сн-<С ^
//\/°
%/\
С=СН-!^'
¦с/
А _
>
\=
\/\сУ
СН
Флавоноиды
Хиноны
бензохиноны
нафтохиноны
антрахиноны
А
О
А
/
о
•Л
V\
о
^хА/хх
*
/V
25
Лигнаны
Трополоны
-С-т—С-
Все эти производные широко распространены в растениях;
они входят в состав различных глюкозидов, эфирных масел,
смол, дубильных веществ, лигнина [108].
Глюкозиды, являющиеся ди- и трисахаридами, соединенными
глюкозидной связью с каким-либо спиртом неуглеводной природы,
содержатся во многих растениях, придавая им горький вкус
и специфический запах, а также ту или иную окраску, хотя
количество их обычно ничтожно.
Дубильные вещества представляют собой гетерогенную группу
полиоксифенольных соединений и по химической природе
делятся на гидролизуемые и конденсированные. Первые —
сложные эфиры сахаридов и фенолкарбоновых кислот (галловой и
эллаговой); вторые (по гипотезе К. Фрейденберга) являются
производными катехинов, которые, по современным
представлениям, относятся к группе флаваноидов:
О
\/\У
флаванол-З
Все дубильные вещества легко окисляются под действием
окислительных ферментов с образованием высокомолекулярных
продуктов окисления и полимеризации.
Смолы представляют собой кислородсодержащие производные
дитерпенов, их молекула состоит из смоляной кислоты (левопи-
маровой или абетиновой) и смоляных спиртов, фенолов и
углеводородов.
К алкалоидам относится особая группа циклических азотистых
соединений основного характера. В зависимости от химической
26
природы азотистого гетероцикла различают производные
пиридина, пирролидина, хинолина, изохинолина, индола, пурина.
Они обнаружены только в высших растениях и накапливаются
в незначительных количествах, будучи физиологически
активными веществами. В растениях они присутствуют в виде солей
с органическими кислотами. Главнейшие алкалоиды — кокаин,
морфин, кураре, кофеин, никотин, теобромин.
Наряду с перечисленными группами органических соединений
вторичного происхождения в растительных и микробных остатках
присутствуют различные стимуляторы роста, среди которых
значительная роль принадлежит ауксинам и гиббереллинам, а также „
гербициды, антибиотики, фитонциды, витамины — сравнительно
низкомолекулярные соединения различного химического строения.
Таким образом, с органическими остатками в почву поступает
очень сложный комплекс различных по ММ, химическому
строению, разной степени устойчивости и реактивоспособности
органических соединений. Доминируют среди них высокомолекулярные
соединения, подвергающиеся в почве сложным процессам
превращения, главнейшими из которых являются гумификация и
минерализация.
Органические удобрения
Органические удобрения — существенный (а в ряде случаев
и главнейший) источник органических веществ в пахотных
почвах. Особенно большое значение они имеют для всех
минеральных почв Нечерноземной зоны, и прежде всего для подзолистых
и дерново-подзолистых, составляющих здесь основной фонд
пахотных земель. Количество органических удобрений, вносимых
в почву в этой зоне, различно и определяется как степенью окуль-
туренности почв, так и уровнем интенсификации земледелия в
хозяйстве. Оно колеблется в настоящее время в среднем от 2—5 до
10—12 т/га ежегодно. В ряде передовых хозяйств под овощные
культуры и картофель вносится до 90—150 т/га, а иногда эти
дозы поднимаются до 200—300 т/га. Естественно, что такие вы- ,
сокие дозы следует рассматривать как основной источник гумуса
в почве, резко изменяющий весь ход гумусообразования и
гумусовый профиль.
Работы по оценке органических удобрений как гумусообразо-
вателей, к сожалению, до настоящего времени очень
немногочисленны и все еще недостаточно полноценны, так как аналитические
материалы очень ограниченны [61, 167, 197, 243, 277, 294]. Для
оценки роли органических удобрений как гумусообразователей
необходимы прежде всего данные по их химическому составу не
только в плане валового содержания элементов питания, но и
состава их органической части. Если учесть, что виды
органических удобрений, используемые в сельском хозяйстве, очень
различны и не менее разнообразны дозы, способы внесения и глубина
27
заделки, следует признать настоятельную необходимость
детальных длительных исследований для оценки органических
удобрений как гумусообразователей.
Основными видами органических удобрений, поступающих
в почвы, служат стойловый навоз на торфяной (реже на
соломистой) подстилке, компосты на торфяной основе, торф, птичий
помет, зеленые удобрения, а также все шире используемые за
последние годы различные отходы народного хозяйства, также
содержащие значительное количество органических веществ.
Перевод животноводства на промышленную основу в корне меняет
природу ряда органических удобрений, так как вместо стойлового
навоза сельское хозяйство получает огромные количества жидкого
навоза, технология внесения которого в* почву еще практически
не разработана.
Основными параметрами органических удобрений как
гумусообразователей должны быть содержание в нем сухого вещества,
азота, зольность и химический состав их органической части.
Приведем некоторые показатели по этим параметрам (табл. 6).
При всем разнообразии этих данных органическая масса
удобрений при внесении их в почву составляет от 15 до 30%, за
исключением жидкого навоза, содержание сухого вещества в котором
ничтожно. В среднем можно считать, что при внесении каждых
Таблица 6
Основные компоненты органических удобрений, %
Виды удобрений
Навоз
на соломистой
подстилке
на торфяной
подстилке
жидкий
ТАУ
ТМАУ
Торф
верховой
переходный
низинный
Птичий помет
Бытовой мусор
Сырая масса
вода
65-85
67—80
90-98
60-75
55—65
1000—1200 *
350—950 *
460—900 *
5—20
40-65
сухое
вещество
15-35
20—33
2-10
25-40
35-45
80-95
35-60
Сухое вещество
сырая
зола
10-25
10-30
8—12
10-15
30-40
1-6
5-15
7-20
15-30
28-68
органическое
вещество
75-90
70-90
88-92
85—90
60-70
94—99
85—95
80—93
70—85
32—72
азот
0.5-20
1.0-2.0
3.0-5.0
1.5-3.0
1.0—3.0
0.5—2.0
1.0-2.5
1.5—4.0
3.0-5.0
0.5-1.5
Примечание. Звездочкой отмечена полная влагоемкость.
28
Таблица 7
Химический состав органической части удобрений, % на беззольную сухую массу
Виды удобрений
Навоз на подстилке
соломистой
торфяной
ТМАУ
1
2
3
4
ТАУ
1
2
Торф
верховой
переходный
низинный
Бытовой мусор
Водорастворимые
вещества
4
3
2
3
7
8
8
13
1-3
0.5-1.5
1
3-8
Липиды
(экстракция
спирто-
бензолом)
5
3
5
3
6
6
5
6
1
1.5—3.0
0.5—0.7
3—14
Гумусовые вещества,
экстракция 0.1 М
Na4P207
гуминовые
кислоты
16
28
39
43
34
33
34
32
3-5
20-30
40-55
0-4
«фульво-
кислоты»
22
16
12
23
15
14
14
13
5-7
10—18
10—18
4—12
Гемицеллюлозы
и белки,
гидролиз
1н. H2S04
8
12
6
3
2
4
5
6
7-8
10—12
3-5
3-6
Целлюлоза и
белки,
гидролиз
80%-ной
H2S04
12
10
6
5
3
3
3
4
33-37
7-15
3-5
6-23
Лигнино-
подоб-
ный
остаток
33
28
30
20
33
32
31
26
25-30
20—25
13—20
47-73
Примечание. «Фульвокислоты» — неосаждаемая при рН 1 часть веществ, извлекаемых
Na4P207.
10 т органических удобрений в почву поступает менее 0.3 т
органических веществ. Эти данные необходимо принимать во внимание
при оценке органических удобрений как источника гумуса.
Очевидно, что реальное значение в качестве гумусообразователей
органические удобрения могут иметь лишь в случае применения
значительных доз: порядка нескольких десятков тонн на 1 га.
Не менее существенно знание химического состава органической
части удобрений при оценке их как гумусообразователей. Эти
данные крайне ограниченны (табл. 7). Специфическая особенность
химического состава навоза и торфяных компостов по сравнению
с корневыми и пожнивными остатками в том, что в их составе
содержатся «готовые» гуминовые кислоты. Количество этих
соединений, конечно, различно и обусловлено прежде всего
содержанием в органическом наполнителе, а частично и степенью развития
процессов гумификации при приготовлении компоста; но эта
принципиальная особенность должна учитываться при
рассмотрении характера трансформации органических удобрений в почве.
Кроме гуминовых кислот органические удобрения содержат очень
29
разное количество легко и трудно разлагающихся компонентов.
В верховом торфе, который часто используют для подстилки,
много углеводов, но мало гуминовых кислот; низинные торфы
характеризуются высоким содержанием гуминовых кислот и
лигнина, но в них мало углеводов. Заметно выделяется навоз на
соломистой подстилке пониженным содержанием гуминовых
кислот и повышенным количеством углеводов и лигнина.
Все шире применяются в настоящее время различные компосты,
приготовленные из отходов народного хозяйства. Для них
характерен очень разный и колеблющийся по сезонам химический
состав органических веществ. Органическая масса в бытовых
отходах, переработанных на специальных заводах, колеблется от
32 до 72% на сухое вещество; в ней доминируют углеводы,
нередко значительно содержание таких трудно разлагаемых
веществ, как синтетические пластмассовые материалы типа
различных пленок, емкостей, пробок. Получают распространение и
донные осадки — различные илы, сапропели, которые также могут
быть дополнительными гумусообразователями в пахотных
почвах, хотя их реальное значение и химический состав органической
части исследованы еще недостаточно.
Зеленые удобрения (донник, люпин, сераделла, чина и др.)
по химическому составу органической части не отличаются от
всех остальных природных травянистых растительных остатков
семейства бобовых. Они содержат значительное количество
белковых веществ и углеводов и относительно легко разлагаются
и гумифицируются в почве.
Таковы некоторые, все еще ограниченные данные о
химическом составе различных органических удобрений, которые
необходимо учитывать для правильной оценки их роли в гумусообра-
зовании.
Глава II
СОСТАВ ГУМУСА, ПРИРОДА И СВОЙСТВА
ГЛАВНЕЙШИХ КОМПОНЕНТОВ
Современные представления о почвенном гумусе
формировались в течение длительного времени. Уже в работах классиков
естествознания XIX в. К. Шпренгеля, Я. Берцелиуса и Г. Муль-
дера было установлено, что гумус почвы является сложным телом
и главнейшие составные части его представлены гумусовыми
кислотами — гуминовой, ульминовой, креновой, апокреновой и
индифферентными модификациями первых двух — гумином и уль-
мином. Вопрос об азоте оставался открытым, и большинство
ученых считали гумусовые кислоты безазотистыми соединениями.
Процесс гумификации не исследовали, господствовала
умозрительная теория Мульдера, согласно которой гумусообразование
рассматривалось как цикл химических реакций дегидратации и
окисления целлюлозы. Следует отметить, что уже в то время
Я. Липовский [184], анализируя общее состояние вопроса об
условиях плодородия почвы, приходит к выводу о непрерывности
процессов гумусообразования в почве и неизбежности в силу
этого изменения гумусовых кислот.
Наиболее целенаправленными на рубеже XIX и XX вв. были
работы классиков почвоведения В. В. Докучаева и П. А. Косты-
чева, развивавшиеся на фоне бурного расчленения естествознания
на ряд смежных, но самостоятельных дисциплин и проникновения
в среду естествоиспытателей материалистического мировоззрения.
Работы В. В. Докучаева [93] и его учеников С. Козловского [1361
и С. Лесневского [183] положили начало исследованию
географических закономерностей гумусообразования и изменения
состава гумуса в различных почвах. Дальнейшее наиболее яркое
развитие это направление получило в трудах И. В. Тюрина [297],
М. М. Кононовой [142, 143] и В. В. Пономаревой [247].
П. А. Костычева [158] по праву считают основоположником
учения о процессе гумусообразования, ибо, проведя классические
исследования о характере и скорости разложения растительных
остатков, он впервые показал решающую роль микроорганизмов
в этом процессе и решил проблему накопления азота в гумусе.
После исследований Костычева появился ряд публикаций по
изучению скорости и характера процессов разложения
растительных остатков [429, 521, 561 ], но выводы из этих работ были раз-
31
норечивы, как и условия проведения лабораторных
экспериментов.
В XX в. изучение природы гумусовых веществ и путей их
образования в природе привлекает внимание не только
почвоведов, агрохимиков и микробиологов, но и химиков, исследующих
каменный уголь и торф [281, 282]. Широко развертываются
работы по изучению состава, строения и свойств гумусовых веществ;
выявляются условия, влияющие на характер и скорость гумусо-
образования; рассматриваются природа вероятных гумусообразо-
вателей, роль и значение гумуса в почвообразовании и плодородии
почв, в питании растений, миграции и аккумуляции некоторых
элементов в литосфере. Создаются крупные мировые центры по
изучению гумуса как в нашей стране, так и за рубежом. Их
возглавили И. В. Тюрин и М. М. Кононова (СССР), В. Фляйг (ФРГ),
С. Ваксман (США), М. Шнитцер (Канада), Ф. Дюшофур
(Франция). Детальный обзор основных направлений, разрабатываемых
в настоящее время, можно найти в ряде крупных монографий по
гумусу [65, 143, 230, 297, 385, 399, 500].
Общие представления о гумусе и вопросы номенклатуры
Анализируя современную литературу, посвященную
характеристике самого понятия гумус, следует отметить, что до
настоящего времени нет единой трактовки этого термина, как нет и
единой номенклатуры для характеристики органической части
почвы. Термины «гумус», «перегной», «органическое вещество
почвы», «органические вещества почвы», «органическая часть
почвы» часто употребляют как синонимы, что недопустимо,
особенно в учебной литературе. Часто термин «гумус» используют
как для характеристики органической части почвы, так и для
любых других скоплений органических веществ в результате
частичной трансформации отмирающих растительных остатков:
торфа, компостов, подстилок и т. д. Следует отметить, что вопросы
номенклатуры были предметом специального обсуждения на
VIII Международном конгрессе почвоведов, на котором с
обстоятельным обзором существующей терминологии выступил В. Фляйг
[394]. Однако и здесь не была принята единая номенклатура.
Достаточно указать, что М. М. Кононова [143а] предложила
расчленить понятия «гумус» и «органические вещества», тогда как
В. Фляйг отождествляет эти понятия, употребляя последний
термин в единственном числе.
Необходимо отчетливо представлять, что формы нахождения
остатков живых организмов и продуктов их трансформации в
биосфере очень разнообразны в различных экологических условиях
их формирования. При отмирании и последующих процессах
превращений живые организмы образуют очень различные типы
скоплений органических веществ в зависимости от местообитания
32
и условий их трансформации. При отмирании организмов в
минеральных почвах на поверхности накапливается подстилка или
дернина, а в верхней части почвенного профиля — гумус.
Отмирая на болотах, растения образуют массу торфа; на дне водных
бассейнов организмы при отмирании участвуют в образовании
сапропеля или различных илов, содержащих то или иное
количество органических веществ. Человек готовит различные
органические удобрения — компосты, используя для .этой цели
разнообразные органические материалы.
Развернутую классификацию форм гумуса и его составных
частей дали Ф. Шеффер и П. Шахтшабель [491 ]:
Органическая субстанция
I
живая (эдафон)
отмершая (гумус)
I
составные части
гумуса (вещественные)
1. Негуминовые
вещества: углеводы, смолы,
воски,
низкомолекулярные соединения
2. Гуминовые
вещества: фульвокислоты, ги-
матомелановые кислоты,
гуминовые кислоты, гу-
мины, гумусовый уголь
формы гумуса
(морфологические)
1. Йодводные формы,
гиттия, сапропель,
болотный ил, торф
2. Полуназемные
формы — торф: верховой,
переходный, низинный
3. Наземные формы:
сырой гумус, мо дер,
мулль
роды гумуса
(по функциональному
действию)
1. Питательный и
консервативный гуму-
сы
2. Различия по:
а) химическому
взаимодействию,
б) физиологическому
действию на растения,
в) физическому
воздействию на почву
Эта схема интересна своим построением. Характеристика
органической субстанции по нескольким показателям позволяет более
точно определить ее вещественный состав и функции. Но нельзя
согласиться с отождествлением понятия «гумус» со всей массой
отмирающих в биосфере организмов независимо от их
местообитания. Термины «гиттия», «торф», «сапропель» так же правомочны,
как и «гумус», ибо они генетически самостоятельны и формируются
в определенных условиях в биосфере.
Еще в 1970 г. мы предложили схему, отражающую основные
типы накопления органических веществ в биосфере [19]. Ниже
она приведена в несколько измененной и дополненной форме
(см. с. 34).
Нельзя отождествлять термин «гумус» с любыми другими
типами скопления продуктов трансформации растительных
остатков, накапливающимися в очень разных условиях их образования.
Термин «гумус» следует считать сугубо почвенным, он включает
лишь ту часть органических веществ почвы, которая потеряла
анатомическое строение исходных растительных остатков, под-
33
Формы продуктов трансформации организмов в биосфере
и их основные компоненты:
живые организмы
У
ткани отмирающих организмов, не потерявшие анатомического
строения
у
формы накопления органических веществ в зависимости от
местообитания
1
подстилка
I
\
дернина
по генезису
У
гумус
У
торф
компосты
сапропель
по химическому составу
1. Вещества исходных органических остатков:
углеводы, протеины, лигнин, липиды, дубильные вещества, смолы
и др.
2. Промежуточные продукты трансформации этих соединений:
протеиды, аминокислоты, поли- и монофенолы, моносахариды,
сахарные кислоты, азотсодержащие гетероциклы и др.
3. Гумусовые кислоты и их органо-минеральные производные:
группы гуминовых кислот и фульвокислот, их соли и сорбционные
комплексы.
верглась в почве процессам гумификации и формирует гумусовые
горизонты, равномерно прокрашивая минеральную массу их
в темный цвет. Его содержание в профиле педона любой целинной
почвы относительно постоянно, так как обусловлено самим ходом
почвообразовательного процесса. В то же время термин «гумус»,
не исчерпывает понятия об органической части почвы, содержание
которой колеблется в течение даже одного вегетационного периода
более значительно. В состав органической части почвы в каждый
данный момент входят не только гумус — главный ее компонент,
но и некоторое количество остатков зеленых растений, почвенной
фауны, органических удобрений. Все эти компоненты могут быть
отделены от массы почвы механическим путем, и лишь постепенно,
подвергаясь сложным процессам трансформации, они
превращаются в гумус.
Значительно сложнее решить вопрос о номенклатурном
месте живых микроорганизмов,, количество которых, по данным
И. В. Тюрина [299], составляет 0.5—0.7 т/га по сухому веществу,
т. е. около 1 % к массе гумуса в почве. Строго говоря, живые
микроорганизмы не могут включаться в понятие «гумус», так как
являются для него источником органического вещества и агентом
процесса гумусообразования, но практическое отделение их от
массы гумуса невозможно.
34
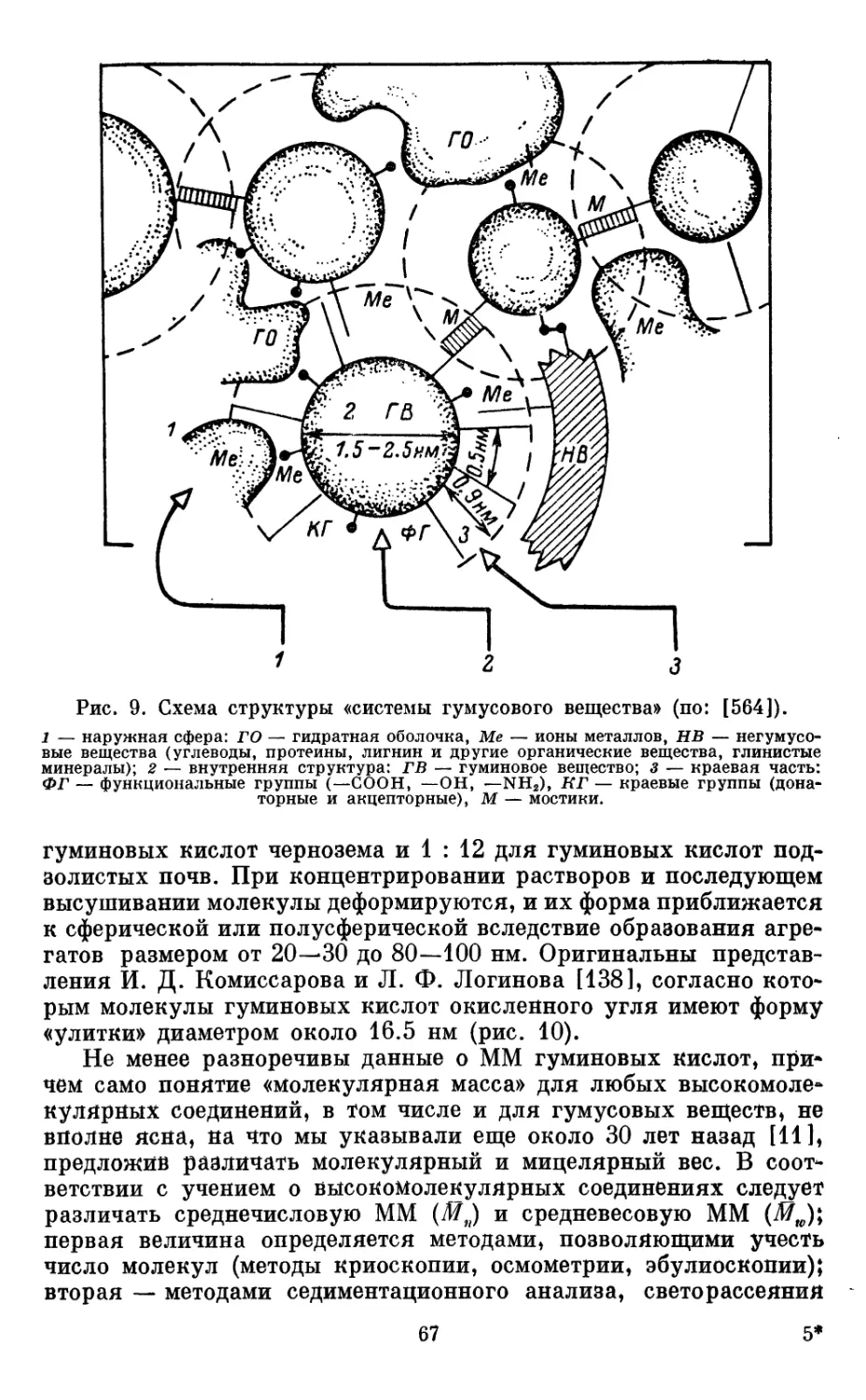
Из приведенной схемы видно, что гумус — всегда сложная
многокомпонентная система, включающая три группы веществ:
органические соединения исходных органических (в основном
растительных) остатков, постепенно подвергающихся
трансформации (их состав рассмотрен в гл. I); промежуточные продукты
трансформации, образующиеся в процессе разложения первой
группы соединений (эта группа очень разнообразна, эфемерна и
все еще плохо изучена, рассматривается в специальном разделе
данной главы); гумусовые кислоты и их производные — особый
класс органических соединений, образующихся в процессе
гумификации органических остатков.
Схематический состав гумуса можно представить следующим
образом:
ГУМУСОВЫЕ ВЕЩЕСТВА
Следует отметить, что эти три группы продутов
трансформации мертвых органических остатков универсальны и присутствуют
в любых типах скоплений органических веществ в биосфере, но
35
соотношение между ними различно в зависимости от условий
образования и местообитания (табл. 8).
Таблица 8
Соотношение основных компонентов органических веществ в различных
формах их накопления в биосфере, % к массе сухого беззольного вещества
Компоненты
органической части
Вещества исходных
органических остатков
Промежуточные
продукты разложения
Гумусовые вещества
Гумус
5-20
1
80-95
Подстилка,
дернина
65-90
1-5
5-30
Торф
35-85
1—10
10—60
Органические
удобрения
ТМАУ
30—45
2-5
50-65
навоз
55-80
2-5
15-40
Только в составе гумуса доминируют гумусовые вещества,
составляя до 90—95% всей его массы. В подстилках, торфе, ком-
постах количество гумусовых веществ резко снижается, хотя и
колеблется в очень широком интервале, всегда сохраняясь
благодаря наличию процесса гумификации. Доминируют здесь
исходные, еще не гумифицированные компоненты органических
остатков. Своеобразен состав органических веществ в
органических удобрениях — различных компостах и навозе. В них,
так же как и в торфе, присутствуют все три группы
компонентов, в том числе гумусовые вещества, причем в зависимости от
вида торфа содержание последних может быть значительным.
Совершенно справедливо указывает Ф. Дюшофур [103] на
необходимость отличать понятие «гумус» от органической части
почвы в целом. В зависимости от интенсивности процесса
гумификации растительных остатков автор выделяет мюллевый гумус
(мюлль), формирующийся в лесных почвах при благоприятных
для биологической активности условиях, и грубый гумус (мор),
характерный для тех почв, где биологический цикл освобождения
элементов питания замедляется и на минеральной части почвы
аккумулируется мощный слой из полуразложившихся
растительных остатков. Эта терминология широко используется среди
лесоводов. На сельскохозяйственных угодиях автор различает
свежее органическое вещество и собственно гумус.
В то же время недопустимо отождествление понятий «гумус»
и «гумусовые вещества», так как последние всегда являются лишь
компонентом (хотя и наиболее существенным) гумуса. Вместе
с тем гумусовые вещества присутствуют не только в гумусе, но
и в торфе, сапропеле, компосте, т. е. везде, где развивается
процесс гумификации.
36
Гумусовые кислоты как специфический класс
органических соединений
Исследование гумусовых кислот имеет более чем вековую
историю с естественной и закономерной эволюцией взглядов на
процессы их образования, состав и свойства. Если в первый период
их изучения, в конце XIX—первой четверти XX в., исследователи
стремились определить элементный состав этих соединений,
способность к реакциям солеобразования, то современный период
характеризуется очень широкой программой работ. Изучаются
механизмы и условия их образования, исследуются природа и
свойства этого класса соединений, их роль не только в процессах
почвообразования и плодородия почв, но и в общей биогеохимии
ландшафтов. За последние 15—20 лет опубликовано много
монографий и обзорных работ, в которых детально проанализированы
в историческом аспекте все основные направления в изучении
этого класса соединений [19, 143, 230]. В настоящей главе
рассматриваются лишь основные современные представления о
природе и свойствах этих соединений и дискуссионные вопросы
проблемы.
Первый существенный вопрос — отнесение этой категории
соединений к тому или иному классу органических веществ. Как мы
уже отмечали [8, 19], гумусовые соединения независимо от их
происхождения и местообитания являются высокомолекулярными
азотсодержащими органическими кислотами. Это определение не
вызывает каких-либо серьезно аргументированных возражений,
ибо кислотная природа этих соединений известна со времен
К. Шпренгеля и Я. Берцелиуса. Приходится лишь удивляться
тому, что даже в наиболее капитальных монографиях по
органической химии гумусовые кислоты как определенная группа
природных органических соединений не только не рассматриваются,
но и не упоминаются. Если у А. Е. Чичибабина [321] можно
найти упоминание об образовании в почве «сложных, богатых
углеродом черных гуминовых веществ», формирующихся в
процессе гниения или тления, то в новейшей капитальной монографии
по органической химии [306] исчезло даже упоминание об этой
категории соединений. Вместе с тем следует отметить, что
недостаточная изученность деталей строения и свойств вещества еще не
может быть причиной того, что обширная группа природных
органических веществ не находит в настоящее время места в недрах
органической химии. Реальность существования в почве, торфе,
компостах и других местообитаниях нативных гумусовых кислот
и принадлежность их к особому классу органических соединений
можно вновь подтвердить сводными данными элементного состава
различных групп природных органических соединений (табл. 9).
Не менее многочисленны к настоящему времени и данные о
количестве карбоксильных и фенолгидроксильных групп, о
существенной разнице в емкости обменного поглощения катионов
37
Таблица 9
Элементный состав органических соединений различного происхождения,
% на сухую беззольную массу
Соединения
Белки
Углеводы
Лигнин
Жиры
Гуминовые кислоты
Фульвокислоты
С
50—53
40—45
60-66
76—79
52—62
41—49
Н
6-7
6
6
11-13
3-6
4-5
о
19—24
49—54
28—34
10—12
31—39
47-52
N
16-18
Нет
1
Нет
2—5
2-4
Источник
[163, 297]
Тот же
[62]
[163, 297]
Наши данные
» »
в почвах до и после удаления из них гумусовых веществ.
Наиболее убедительные современные данные о реальности
существования гумусовых кислот как особого класса органических
соединений приводит Д. С. Орлов [230]. Основными параметрами
гумусовых кислот, подтверждающими реальность их
существования, следует в настоящее время считать элементный состав,
обязательное наличие азота и многообразие его форм, наличие
кислых функциональных групп, ароматических и
гетероциклических группировок, специфичность электронного и ИК спектров.
С удивительным для своего времени научным предвидением
И. В. Тюрин писал: «. . .термин „гуминовая кислота" является
групповым понятием, под которым следует подразумевать целую
группу высокомолекулярных соединений, имеющих несколько
различный состав, но обладающих рядом общих свойств и, как
будет видно далее, известным общим типом строения. Поэтому
правильнее говорить не „гуминовая кислота", а „гуминовые
кислоты", подобно тому как принято говорить о белках, лигнинах
и т. д. Указанный вывод, однако, не может служить основанием
для отрицания самостоятельности группы соединений,
объединяемых названием гуминовые кислоты. . .» [297, с. 116].
Номенклатура гумусовых кислот
Современная номенклатура ведет начало от классических
исследовании и фундаментальных обобщений И. В. Тюрийа.
В своих последних работах по этим вопросам И. В. Тюрин [302],
четко разграничив понятия «гумус» и «гумусовые вещества»,
предложил для последних следующую номенклатуру: гумусовые
вещества: I — группа гуминовых (и ульминовых) кислот: бурые
(т. е. ульминовые) кислоты, черные гуминовые кислоты; II —
фульвокислоты; III — «гумины».
Достоинства этой номенклатуры: использование термина
«гумусовые» как более широкого по сравнению с понятием
«гуминовые», в то же время подчеркивающего неоднозначность понятий
38
«гумус» и «гумусовые вещества»; объединение гуминовых и уль-
миновых кислот в одну группу; употребление этого термина во
множественном числе, чем уже в то время подчеркивалась их
гетерогенность на фоне принципиально однотипного строения;
четкая идентификация фульвокислот как особой группы
гумусовых кислот.
И. В. Тюрин, по-видимому, понимал уже в то время условность
термина «гумины», брал его в кавычки и относил к ним все
вещества, нерастворимые в щелочах после многократного
гидролиза. Не выделил И. В. Тюрин в составе гумусовых веществ и
особой группы гиматомелановых кислот, считая имеющиеся
данные очень противоречивыми [297].
Эта номенклатура И. В. Тюрина, подвергавшаяся при
дальнейших исследованиях лишь некоторым изменениям, выдержала
проверку времени, и термины «гуминовые кислоты» и «фульво-
кислоты», употребляемые во множественном числе, прочно вошли
в литературу. Так, например, на основе классификации И. В.
Тюрина Ф. Дюшофур [104] предложил выделять следующие
гумусовые кислоты: креновые кислоты — растворимую в воде часть
гумусовых веществ; фульвокислоты — фракцию гумусовых
веществ, растворимую в щелочах и не осаждаемую кислотами;
гумусо-лигниновые комплексы или гумусо-фульватные вещества —
промежуточные продукты гумификации, осаждаемые только
достаточно концентрированной серной кислотой; гуминовые
кислоты — часть гумусовых веществ, осаждаемая серной кислотой
при ее более слабой концентрации; в их составе выделяются:
бурые гуминовые кислоты — слабо полимеризованные и слабо
связанные с минеральной частью почвы; серые гуминовые
кислоты — сильно полимеризованные и прочно связанные с
минеральной частью почвы гуминовые кислоты; гиматомелановые
кислоты — часть гуминовых кислот, растворимые в спирте, слабо
полимеризованные, бурые; гумины — масса гумусовых веществ,
нерастворимых в щелочных растворах; комплексы, частично
гидролизуемые 6 н. НС1.
За последние годы вопросы номенклатуры гумусовых кислот
широко обсуждались в литературе [17, 230, 231, 394], в
большинстве работ вслед за терминологией И. В. Тюрина предложено
пользоваться термином «гумусовые вещества» как наиболее
широким, отражающим весь специфический класс
высокомолекулярных азотсодержащих органических кислот, образующихся
в процессе гумификации. Лишь Ф. Шеффер и Р. Шахтшабель
[491 ], а также большинство специалистов в области химии
угля и торфа [185] продолжают пользоваться термином
«гуминовые вещества» как более широким, включая в него
фульвокислоты.
М. М. Кононова [143а] и В. Фляйг [394] в последних
номенклатурных схемах в составе гумусовых веществ выделяют
гуминовые кислоты, фульвокислоты, гиматомелановые кислоты, гу-
39
мины. Новую схему для составления номенклатуры гумусовых
веществ почвы предложил в последнее время Д. С. Орлов [231]:
-Почва-
Неорганическая
часть почвы
Органическая часть
почвы
Органическое вещество
почвы
Живые организмы
(эдафон)
I
i
Гумус
Остатки, не утратившие
анатомического строения
I
Специфические
гумусовые вещества
Промежуточные
продукты распада
и гумификации
Небпецифические
соединения
Прогуминовые
вещества
Гумусовые
кислоты
Гумин
1
Пигменты
Гуминовые кислоты
Фульвокислоты
{
1 1
Серые
Бурые
Гиматоме-
лановые
1 1
I II
Эта схема представляет несомненный интерес. Автор вводит
понятие о прогумусовых веществах, включает гиматомелановые
кислоты в группу гуминовых кислот. Не вполне ясно только
положение гуминов как особой группы гумусовых веществ.
На основании наших многолетних исследований и критического
анализа литературных материалов мы предложили [19] различать
в классе гумусовых кислот две основные группы: группу
гуминовых кислот и группу фульвокислот. Это деление достаточно
хорошо обосновано всем комплексом данных по их основным
параметрам (разной растворимостью и осаждаемостью, разли-
40
чиями в элементном составе, в деталях строения молекулы),
а главное — в существенных различиях их роли в
почвообразовании вследствие неодинаковой растворимости как самих кислот,
так и их органо-минеральных производных. Если группа гумино-
вых кислот аккумулируется в почве на месте образования,
формируя аккумулятивный тип гумусового профиля, то фульвокис-
лоты легко мигрируют по профилю как в форме свободных
кислот, так и большинства солей, формируя иллювиально-гумусовые
типы профиля. Конечно, каждая группа множественна в
количестве отдельных фракций (подгрупп) с присущими ей
отличительными свойствами. Так, исторически сложился термин «гиматоме-
лановые» кислоты, которые по всем основным параметрам
(элементному составу, количеству кислых функциональных групп
и т. д.) не выходят за рамки группы гуминовых кислот. В таком
положении находятся более растворимые в воде и менее темно -
окрашенные ульминовые (бурые гуминовые) кислоты, хотя
исследования В. В. Пономаревой и Т. А. Плотниковой [253]
свидетельствуют о более высокой миграционной способности черных
гуминовых кислот.
Применение различных растворителей показало близость, но
не идентичность гуминовых кислот, выделяемых из одного и
того же объекта. К настоящему времени имеются материалы,
подтверждающие возможность расчленения гуминовых кислот на
много фракций по ряду показателей: по растворимости [139, 547],
по варьирующей устойчивости к осаждающему действию катионов
и скорости передвижения в электрическом поле и при
ультрацентрифугировании [8, 479, 492], по степени сорбции и
фильтрации на ионообменных смолах, агаре и сефадексах [285, 451, 462,
536, 556]. Применение сефадексов позволило расчленить любой
препарат гуминовых кислот на большое число фракций,
варьирующих по химическому составу и молекулярной дисперсности.
Не менее гетерогенна группа фульвокислот [72, 96, 98, 214].
Все эти материалы лишь подтверждают необходимость
установления новых, более точных критериев для расчленения каждой из
основных групп гумусовых кислот на подгруппы и фракции и их
номенклатуры. Очень неопределенна и группа «гумина», остатка
органических веществ после экстракции гумусовых веществ
кислотами и щелочами. Термин, вошедший в литературу со времен
Ёерцелиуса, сохранился до настоящего времени для
характеристики той части гумуса, которая не выделяется из почвы.
Современные исследования показали, что в «гумине» содержатся
две принципиально различные группы соединений: наиболее
устойчивая против щелочного и кислотного гидролиза часть
веществ растительного происхождения (главным образом лигнин и
частично целлюлоза) и наиболее прочно закрепленная на
поверхности минеральных частиц часть гумусовых веществ.
Оригинальные работы И. В. Тюрина и Е. Л. Гуткиной [303], О. А.
Найденовой [206], Н. Г. Зырина [113], а также современные исследова-
41
ния С. Кана и М. Шнитцера [437] показали, что принципиальных
отличий в составе гумусовых кислот, выделяемых и не выделяемых
обычными экстрагентами, нет. В «гумине» обнаруживаются гуми-
новые кислоты и фульвокислоты со всеми характерными для них
показателями. Более детально природа этой группы соединений
будет рассмотрена ниже.
Гуминовые кислоты
Гуминовые кислоты представляют группу высокомолекулярных
азотсодержащих органических кислот, молекула которых
содержит ароматические группировки. Высокомолекулярная природа
их подтверждается многочисленными определениями
молекулярной массы, кислотная природа — наличием ряда
кислородсодержащих функциональных групп, водород которых способен к
обменным реакциям с катионами оснований; обязательный выход
бензолкарбоновых кислот при частичном окислении их
свидетельствует о наличии ароматических группировок. Характерные
особенности гуминовых кислот — их полидисперсность и
химическая гетерогенность, вследствие чего гуминовые кислоты любой
почвы можно расчленить на ряд фракций с близкими, но не вполне
однородными параметрами. Высокая поглотительная способность
гуминовых кислот и образование ряда нерастворимых в воде
органо-минеральных производных, а также клеящая способность
их новообразованных фракций позволяют рассматривать эту группу
гумусовых кислот как наиболее существенный аккумулятор
энергии и элементов питания растений.
Элементный состав. Элементный состав гуминовых кислот
хорошо изучен, достаточно специфичен и заметно отличается от
любых других природных органических соединений (табл. 10, 11).
Из приведенных статистически обработанных данных Д. С.
Орлова, а также ряда других литературных источников [19, 29]
следует лишь один основной вывод: элементный состав гуминовых
кислот колеблется в некоторых, относительно узких пределах
(С от 52 до 62%, Н от 2.8 до 5.8, О от 31 до 39, N от 1.7 до 5%).
Причины этих колебаний до настоящего времени не вполне
ясны, хотя на протяжении последних 30—40 лет высказывались
разные гипотезы. И. В. Тюрин [297 ] на основании данных В. В. Ти-
щенко и М. Д. Рыдалевской [291 ] считал, что эти колебания
закономерны и обусловлены изменениями степени окисленности и
обводненности гуминовых кислот от северных почв к черноземам.
М. М. Кононова [142] также считала эти колебания
закономерными, носящими зональный характер. Однако Д. С. Орлов [230],
обработав статистически большой материал по элементному
составу гуминовых кислот различных почв, пришел к иным выводам.
Вычислив элементный состав не только в весовых, но и в атомных
процентах (что дает более четкое представление об истинном
42
Таблица 10
Средний элементный состав гуминовых кислот различных типов почв,
% на сухое беззольное вещество (по: [230])
Почвы
Торфяно-болотные, торфяники
Пойменные, луговые
Темноцветные, рендзины
Дерново-подзолистые, подзолы
Бурые лесные, буроземы
Серые лесные
Черноземы
Каштановые
Солонцы, солоди
Сероземы
Красноземы, красноцветные
Горно-коричневые
Горно-луговые
Гумусо-аллофановые, пепловые
Глеевые, псевдоглеевые
Гуминовые кислоты из
растительных остатков
Состав
растительных остатков
торфов
С
58.7 (45)
55.5 (14)
54.1 (9)
53.4 (53)
55.1 (32)
54.5 (18)
57.9 (75)
55.9 (9)
54.5 (3)
56.0 (18)
54.8 (3)
55.2 (10)
54.5 (2)
58.3 (17)
55.3 (10)
56.1(44)
49.6 (6)
58.4 (18)
н
5.0 (44)
4.1 (14)
5.0 (7)
4.8 (47)
5.2 (10)
4.8 (18)
4.0 (71)
5.2 (9)
4.1 (2) .
4.8 (18)
3.6 (3)
5.6 (10)
3.4(2)
4.2(15)
Не
5.5(44)
6.3(6)
5.6(18)
о
32.9
36.8
36.8
37.7
35.4
36.7
36.3
34.2
36.4
34.4
37.5
33.9
37.6
34.8
N
3.4 (41)
3.6 (14)
4.1 (7)
4.1 (48)
4.3 (10)
4.0 (18)
3.8 (75)
4.7 (9)
5.0 (3)
4.8 (18)
4.1 (3)
5.3 (10)
4.5 (2)
2.7 (17)
определялись
33.5
41.6
33.4
4.9 (40)
2.5 (6)
2.6 (18)
Примечание. В скобках — численность выборки.
соотношении атомов в молекуле гуминовых кислот), автор
приходит к заключению, что «четкого зонального ряда по степени
окисления гуминовых кислот обнаружить не удается» [230, с. 52].
Для выявления специфических различий элементного состава
гуминовых кислот автор предложил использовать степень окислен-
ности
2<?0-ен
где Qq — число атомов кислорода, (?н — число атомов водорода
в молекуле гуминовых кислот, a Qq — число грамм-атомов
углерода. По атомным отношениям Н : С, О : С и степени окисленности
гуминовые кислоты разделены Д. С. Орловым на две группы:
серые (группа I) и бурые (группа II); причем группа I по
степени окисленности делится на две подгруппы: окисленную (1а)
и восстановленную (16). К группе I автор отнес по этим
показателям гуминовые кислоты черноземов, пойменных, луговых, крас-
ноцветных, горно-луговых гумусо-аллофановых почв, солонцов и
солодей (подгруппа 1а — с пониженным отношением Н : С и
положительной ведичиной о>), а также торфяяо-болотшде цочш,
43
Таблица 11
Средний элементный состав гуминовых кислот
(вычислено по средним данным), ат.% (по: [230])
Почвы
Торфяно-бо л отные,
торфяники
Пойменные, луговые
Темноцветные, ренд-
зины
Дерново-подзолистые,
подзолы
Бурые лесные,
буроземы
Серые лесные
Черноземы
Каштановые
Солонцы, солоди
Сероземы
Красноземы, красно-
цветные
Горно-коричневые
Горно-луговые
Гумусо-а л лофановые,
пепловые
Гуминовые кислоты
из растительных
остатков
Состав
растительных
остатков
торфов
С
40.2
41.1
37.4
37.5
37.4
38.1
42.5
37.7
40.5
39.0
42.1
36.2
42.7
42.5
37.0
31.3
38.1
н
41.0
36.4
41.5
39.8
42.2
40.3
35.2
42.1
36.4
40.1
33.4
44.1
32.0
36.7
43.5
47.6
43.9
О
16.8
20.2
19.8
20.3
17.9
19.2
19.9
17.4
19.9
18.1
21.8
16.7
22.3
19.1
16.7
19.7
16.5
N
2.0
2.3
2.2
2.4
2.5
2.4
2.4
2.8
3.2
2.8
2.7
3.0
3.0
1.7
2.8
1.4
1.5
н :С
1.02
0.89
1.11
1.06
1.13
1.06
0.83
1.12
0.90
1.03
0.79
1.22
0.75
0.86
1.18
1.52
1.15
О : С
0.42
0.49
0.53
0.54
0.48
0.50
0.47
0.46
0.49
0.46
0.52
0.48
0.52
0.45
0.45
0.63
0.43
G:N
20.1
17.9
17.0
15.6
14.9
15.9
17.7
13.5
12.7
13.9
15.6
12.1
14.2
25.0
13.2
22.4
25.4
Окислен-
ность
А (О, Н),
г • экв./lOO г
-0.8
+0.5
-0.4
0
-0.8
-0.2
+0.6
-1.0
+0.5
-0.4
+1.0
—1.4
+1.4
+0.2
-1.3
-1.1
-1.4
со
-0.16
+0.11
—0.09
0
-0.17
—0.004
+0.13
—0.21
+0.11
-0.09
+0.22
-0.30
+0.31
+0.04
-0.28
—0.27
—0.29
для которых тоже характерна повышенная обуглерожвнность, но
отрицательная величина со (подгруппа 16). Группа II объединяет
рендзины, темноцветные* дерново-подзолистые, бурые и серые
лесные, каштановые почвы и сероземы. Как видим, в каждую
группу и подгруппу попадают почвы с очень различными
условиями и характером почвообразования (например, черноземы и
пойменные или дерново-подзолистые и каштановые), и пока трудно
судить о причинах, которые обусловливают эти закономерности.
Таким образом, установление причин колебаний элементного
состава гуминовых кислот требует дополнительных
экспериментальных исследований, и в первую очередь нужны работы по
изучению самого механизма процесса гумификации. В настоящее
время следует признать гетерогенность элементного состава
гуминовых кислот их характерным свойством.
Кислые функциональные группы. Носителем кислотных
свойств гуминовых кислот являются функциональные группы
44
полярной природы, водород которых способен замещаться на
другие катионы с образованием солей. В молекулах гуминовых
кислот установлены различные по природе функциональные группы:
карбоксильные, фенольные и спиртовые гидроксильные,
карбонильные, хинонные и сульфоновые. Основными полярными
кислотными группами являются карбоксилы (—СООН) и фенольные
гидроксилы (—ОН); они предопределяют реакционную
способность гуминовых кислот и их участие во всех реакциях
взаимодействия с минеральной частью почвы. Предложено много
методов как для суммарного определения количества кислых
функциональных групп, так и для раздельного учета количества
карбоксилов и гидроксилов. Детальный обзор этих методов дан
О. Б. Максимовым с сотрудниками [81, 82].
Данные о количестве кислых функциональных групп в
гуминовых кислотах, полученные нами баритным и ацетатным хемо-
сорбционными методами (табл. 12), показывают, что как общее
Таблица 12
Содержание карбоксильных и фенолгидроксильных групп в гуминовых
кислотах, мг -экв./ЮО г
Объект, из которого выделены
гуминовые кислоты.
Чернозем
типичный
обыкновенный
выщелоченный
Дерново-подзолистые суглинистые
почвы
слабоокультуренные
хорошо окультуренные
под многолетними травами
Торфяная почва
переходная
низинная
Растительные остатки
листья клевера, гумифицирован-
ные, сут
45
300
листья дуба, гумифицированные,
сут
30
360
Горизонт
А
А
пах
А
А
А
^пах
А
лпах
А
Т
Т
Т
—
—
— ;
—
COOH-KOH)
746
736
721
713
815
664
807
731
642
752
576
576
573 |
672 1
соон
530
375
516
461
320
327
331
287
309
252
270
331
250
327 |
(ОН)
216
301
207
252
495
337
476
434
333
500
306
245
323
350
Примечание, Количество COOH+(OH) определено баритным, GO0H — ацетатным
методом.
45
рН
[ I I I i
1.0 2.0 3.0 мл 0.1н.К0Н
Рис. 3. Кривые потенциометрического титрования гуминовых кислот
чернозема.
1—111 — концентрация Н: 1 — 1.6-Ю-3, II — 2.6-10-», III — 5.0-10-*.
количество активных кислых функциональных групп, водород
которых способен замещаться на металл, так и содержание
карбоксилов и фенольных гидроксилов в различных препаратах
гуминовых кислот заметно колеблются. Максимальное содержание
карбоксилов характерно для наиболее зрелых гуминовых кислот
черноземов (375—530 мг-экв./100 г), минимально оно в гуминовых
кислотах торфяных почв и молодых гуминовых кислотах,
образующихся в процессе гумификации растительных остатков. Еще
более значительные колебания характерны для количества
фенольных гидроксилов. Обычно их содержание максимально
в молодых гуминовых кислотах и препаратах, выделенных из
дерново-подзолистых и торфяных почв: 300—500 мг-экв./100 г.
Эти колебания, фиксируемые всеми исследователями, не находят
еще однозначного достаточно обоснованного объяснения.
По-видимому, существенное значение имеют степень зрелости гуминовых
кислот (т. е. стадия их гумификации), природные условия и
характер почвообразовательного процесса, а также химический
состав исходных гумусообразователей.
Широкое распространение получил также метод
потенциометрического титрования в водных и неводных средах, детальный
обзор которых дан Л. И. Глебко и соавторами [82].
Большинство исследователей полагают, что при рН от 3 до 7
определяются карбоксильные группы, а в интервале рН от 7 до 10 —
фенольные гидроксилы (рис. 3). Однако и этот метод дает очень
колеблющиеся результаты в зависимости от концентрации
титруемого раствора гуматов натрия, исходных объектов, из которых
выделены гуминовые кислоты, и методов их выделения и очистки.
В последние годы для идентификации кислых функциональных
групп в гуминовых кислотах широко щщользуется метод Щ
48
Рис. 4. ИК спектры (см"*1) гуминовых кислот (1—3) и монтмориллонита (4)
(по: [399]).
1 — исходная, 2 — серая, 3 — бурая фракция.
спектроскопии, который позволяет получить ряд
характеристических полос поглощения, по интенсивности которых судят о
приблизительном количестве тех или иных кислых
функциональных групп (рис. 4). Детальный обзор современных данных
о ИК спектрах гуминовых кислот и возможностях их
использования для характеристики кислых функциональных групп
гуминовых кислот дан Д. С. Орловым [230]; автор указывает на ряд
серьезных затруднений при расшифровке спектров для
диагностики кислых функциональных групп.
Таким образом, следует признать, что наряду с несомненным
присутствием значительного количества активных кислых
функциональных групп точное раздельное определение их количества
все еще затруднено. Если учесть, что до настоящего времени нам
неизвестно строение молекулы этой категории
высокомолекулярных кислот, но установлена их способность образовывать
надмолекулярные структуры типа ассоциатов и агрегатов,
становятся ясными все методические затруднения при определении
количества кислых групп в молекулах гуминовых кислот.
47
Таблица 13
Влияние рН на количество групп СООН, способных к обменному
поглощению катионов в гуминовых кислота^
Почва
Чернозем
типичный
выщелоченный
Дерново-подзолистая суглинистая
-' -
СООН, мг • экв.ДОО г, при разных
значениях рН
4.6
236
227
211
7.0
384
404
341
8.2
531
560
488
Примечание. Гуминовые кислоты выделены из горизонта А.
Для гуминовых кислот характерна многоступенчатая
диссоциация карбоксильных и фенолгидроксильных групп; при
разных значениях рН их реакционная способность очень различна
(табл. 13). Ионизация как карбоксилов, так и фенольных гидро-
ксилов протекает также многоступенчато, поэтому реакционная
способность гуминовых кислот в зависимости от реакции среды
различна. Для практических целей при оценке этих функций
гуминовых кислот в почвообразовании достаточно определить
кажущуюся константу диссоциации при заданной величине рН
на основании потенциометрического титрования выделенного из
почвы препарата. Величины кажущихся констант диссоциации
кислых функциональных групп гуминовых кислот обычно
вычисляются с помощью эмпирического уравнения Гендерсона—
Гассельбаха
PK = pH + lg^-,
где рК — кажущаяся константа диссоциации активной группы,
а — степень диссоциации активных групп при данном значении
рН. Следовательно, величину рК для гуминовых кислот можно
рассчитать по любой точке кривой титрования.
Не менее убедительные данные о разнокачественности кислых
функциональных групп гуминовых кислот приводят А. Шиге-
мицу и К. Кумада [514]. По кривым кондуктометрического
титрования они выделяют три группы кислых функциональных групп
в гуминовых кислотах: очень слабые —- фенольные гидроксилы.
карбоксилы и азотистые основания с очень высокими величинами
рК; слабые — алифатические и ароматические карбоксилы,
азотистые основания со средними величинами рК; сильные —
карбоксилы с малыми величинами рК. В гуминовых кислотах, по
данным этих авторов [514], доминирует вторая группа кислых
функциональных групп.
48
Другие функциональные группы. В последние годы появилось
значительное количество работ, свидетельствующих о наличии
ряда спиртовых гидроксилов и карбонильных групп, хотя
данные о природе этих групп и о количестве их очень противоречивы.
Некоторые исследователи вновь подтверждают существование
карбонильных группировок алифатической природы [497, 546, 554].
Отмечается наличие хиноидных группировок, количество
которых, по данным Т. А. Кухаренко и Л. Н. Екатерининой [174],
колеблется в пределах 62—75 мг-экв./100 г. Б. Чанг и А. Познер
[546], отрицая существование этих группировок, указывают на
присутствие кетонных карбонильных группировок, соединенных
водородными связями. Очень плохо исследованы свободные
аминогруппы, которые в основном служат мостиками между
отдельными компонентами молекул; определения количества этих групп
дают очень противоречивые результаты.
Природа и строение молекулы гуминовых кислот. Накоплен
большой аналитический материал по характеристике составных
компонентов молекулы гуминовых кислот, полученный|
различными методами.
Первая группа методов предусматривает постепенную
деструкцию молекулы гуминовых кислот с последующей идентификацией
продуктов деструкции. Широко применяются кислотный и
щелочной гидролиз, постепенное окисление или восстановление с
использованием широкого набора окислителей и восстановителей,
а также методы глубокой деструкции путем щелочной плавки или
пиролиза с одновременным снятием дериватограмм. Методы
химической деструкции с последующей идентификацией образующихся
продуктов позволили обнаружить разнообразные органические
вещества, принадлежащие ко многим классам органических
соединений (табл. 14).
Кислотный гидролиз, разрушающий, как известно, глюкозид-
ные и пептидные связи, установил наличие в продуктах
частичной деструкции почти всех алифатических и ароматических
аминокислот, пуриновых и пиримидиновых оснований, поли- и
моносахаридов, уроновых кислот и ряда фенолов. Методы
щелочного гидролиза приводят обычно к более энергичной
окислительной деструкции гуминовых кислот, затрагивающей ароматическую
часть молекулы. Продукты гидролиза в этом случае более
разнообразны в зависимости от степени жесткости метода. Это прежде
всего ряд ароматических производных — альдегиды, фенолы, кар-
боновые кислоты, а также аминокислоты, нуклеозиды и много
неидентифицированных продуктов. Различные продукты
деструкции обычно получают методами окисления, причем в
зависимости от интенсивности воздействия образуется сложная система
ароматических, гетероциклических и алифатических производных.
Наиболее распространен метод окисления гуминовых кислот
щелочным раствором перманганата с последующей более или менее
детальной идентификацией образующихся продуктов при помощи
4 Л. Н. Александрова 49
Таблица 14
Продукты и методы частичной деструкции гуминовых кислот по данным
разных авторов
Продукты деструкции
Соединения
органических остатков,
содержащие
эти продукты
Метод деструкции
Аминокислоты: 17
аминокислот алифатического и
ароматического ряда
Азотсодержащие
гетероциклические соединения
нуклеозиды
пуриновые и пирими-
дановые основания
Углеводы:
полисахариды, гексозы, пентозы,
уроновые кислоты, ами-
носахара
Фенолы и их
производные: пирокатехин,
резорцин, гидрохинон,
нитрофенол
Ароматические
альдегиды: бензойный,
коричный, ванилин, сиреневый
Ароматические карбоно-
вые кислоты: фталевая,
протокатеховая,
ванилиновая, сиреневая, феру-
ловая, кумаровая, окси-
бензойная, моно-, ди-,
три-, тетра- и пентабен-
золкарбоновые; феноль-
ные и оксифенольные
кислоты
Хиноны: бензохиноны,
нафтохиноны, антрахи-
ноны
Углеводороды от Сц до
Алифатические кислоты:
малоновая, уксусная,
малеиновая, яблочная,
щавелевая, муравьиная
и др.
Протеины,
протеиды
Нуклеиновые
кислоты, нуклеопро-
теиды
Те же
Углеводы
Лигнин,
дубильные вещества,
глюкозиды,
эфирные масла, смолы
Те же
Пигменты
растений и
микроорганизмов
Воски, терпены,
каротиноиды
Продукты обмена
низших—высших
организмов
Кислотный и
щелочной гидролиз
Щелочной
гидролиз
Кислотный
ролиз
Тот же
гид-
Щелочной
гидролиз, кислотный
гидролиз,
окисление НСЮ3,
щелочная плавка,
восстановление с
амальгамой
натрия
Тот же
Окисление в
щелочной среде,
щелочная плавка
Экстракция
спиртом
Гидрогенолиз
Окисление СЮ2,
Н202, HN03,
КМп04 в
щелочной среде
50
химических методов, газожидкостной хроматографии и их
спектроскопии. Основными продуктами деструкции в этих случаях
являются ароматические карбоновые и фенольные кислоты, в
меньших количествах обнаружены производные фурана, алканы и
многочисленные полиядерные и гетероциклические соединения,
перечень которых дает Д. С. Орлов [230].
Продуктами глубокой деструкции при окислении гуминовых
кислот являются алифатические низкомолекулярные кислоты
(щавелевая, малоновая, малеиновая и др.)- Большой
аналитический материал по составу продуктов окислительной деструкции
гуминовых кислот щелочным раствором пермангапата получен
М. Шнитцером и его соавторами [437, 497, 509]. Они
идентифицировали 50 индивидуальных соединений, среди которых
преобладали три-, тетра-, пента- и даже гексакарбоновые кислоты, ряд
фенольных кислот и низкомолекулярные алифатические кислоты
(табл. 15). Полученные данные позволили подтвердить представ-
Таблица 15
Продукты деструкции гуминовых кислот при окислении щелочным раствором
КМп04, мг из 1 г гуминовых кислот (по: [437])
Продукты деструкции
Идентифицированные
кислоты
бензолкарбоновые
фенольные
алифатические
Неидентифицированные
Всего
Чернозем
177
83
10
69
339
Солонец
170
75
4
46
295
Солодь
197
80
4
51
332
ление о том, что основными компонентами ароматической части
молекулы гуминовых кислот являются бензольные и фенольные
производные, составляющие, однако, лишь 25—30% массы
молекулы. К сожалению, М. Шнитцер и его школа не исследуют
природу азотсодержащих компонентов молекулы в пределах ее
ароматической части. Этот пробел восполняет Д. С. Орлов [230];
по его данным, в составе бензолкарбоновых кислот всегда
присутствует значительное количество азота, что свидетельствует о
наличии в продуктах окислительной деструкции каких-то
ароматических азотсодержащих группировок, природа которых пока
не уточнена. Содержание азота в бензолкарбоновых кислотах
гуминовых кислот чернозема, дерново-подзолистой почвы
практически одинаково: 9%. Д. С. Орлов предполагает наличие
риридид- и диримидинкдрбоновых кислот, образующихся в ос-
51
4*
новном из нуклеозидов. По данным М. Ф. Овчинниковой [225],
полученные бензолкарбоновые кислоты гетерогенны и методами
хроматографии могут быть расчленены на ряд фракций, в которых
по ИК спектрам обнаружены группы СООН и ароматические
группировки —С=С—. Наличие бензолкарбоновых кислот в
продуктах окисления каменного угля показано в исследованиях Т. А. Ку-
харенко с сотрудниками [171—г173]; для гуминовых кислот
почв — в ряде работ [445, 447, 474, 509].
Использование методов восстановления гуминовых кислот
амальгамой натрия в разбавленных щелочных растворах, а также
методов гидрирования позволяет идентифицировать
ароматические спирты и углеводороды [360, 385, 386, 466, 530].
Таким образом, методы химической деструкции позволили
обнаружить в качестве компонентов молекулы разные
многочленные и одночленные ароматические производные,
гетероциклические кислород- и азотсодержащие группировки, углеводные и
аминокислотные остатки. Соотношение между ними остается до
сих пор гипотетичным.
Вторая группа методов основана на использовании приемов
исследования без применения химического расщепления молекул
и лишь подтверждает выводы, полученные при помощи
химической идентификации вещества. Широко применяются
спектроскопические методы исследования. Электронные спектры
поглощения как в видимой, так и в УФ части спектра не дают
специфических полос поглощения и отражают лишь зависимость
цветности раствора от длины волны. Все спектры носят монотонный
характер и вскрывают лишь различную степень цветности
разных препаратов гуминовых кислот. Некоторые авторы по этим
данным считают возможным судить о степени конденсированности
молекул гуминовых кислот [122, 143], о содержании
свободных радикалов [133] или о наличии фракции Рд [484]. Однако,
как показал детальный анализ природы этих спектров и
имеющихся в литературе материалов, проведенный Д. С. Орловым [230],
ни одно из суждений не может считаться строго доказанным, так
как природа взаимосвязи между цветностью и структурой
вещества и тем более гуминовых кислот неясна.
Дж. Чен с соавторами [365] экспериментально показали
отсутствие прямой корреляции между 2?4 : Ев и содержанием
конденсированных ароматических колец. Следует отметить, что
электронные спектры поглощения (оптическая плотность) очень
широко используются исследователями для характеристики
гуминовых "кислот и «степени их конденсированности», хотя и
природа поглощения света гуминовыми кислотами, и тем более
«степень их конденсированности» остаются неясными. Вряд ли
поэтому данный показатель может быть использован для
изучения природы молекулы гуминовых кислот.
Значительно более четкую картину дают ИК спектры (см.
рис. 4), на которых, как известно, отчетливо выделяется ряд
52
полос поглощения в различных участках спектра, позволяющих
идентифицировать некоторые группировки и типы связей в гуми-
новых кислотах. Однако точная количественная характеристика
особенностей молекулы и соотношения компонентов по ИК
спектрам все еще невозможна [54]. ИК спектры подтвердили
принадлежность гуминовых кислот к особому классу органических кислот
и наличие в молекуле ряда функциональных групп, расширили
наши представления о вероятных типах связи и некоторых
структурных единицах. ИК спектры показали, что при общности
спектральных кривых гуминовых кислот, выделенных из
различных почв, четко выявляются и некоторые особенности:
различное участие пептидных связей, ароматических колец, карбо-
нилов, влияние типа гумусообразователёй на детали структуры
молекулы.
Сводную таблицу полос поглощения гумусовых кислот и
некоторых сопутствующих компонентов, составленную на
основании очень большого аналитического материала и глубокого
анализа его, приводит Д. С. Орлов [230].
Основными, наиболее существенными для характеристики
гуминовых кислот полосами поглощения на ИК спектрах
являются: полоса в области 3400—3200 см"1, обусловленная
валентными колебаниями гидроксильных (фенольных и спиртовых)
групп, соединенных в основном водородными связями (в этой же
области могут обладать заметным поглощением группа NH,
а также группа NH3+ аминокислот или пептидов); полосы
в области 2950—2860 см""1, обусловленные валентными
колебаниями С—Н в СН2-группах (в этой области фиксируются одна
или две полосы различной, чаще слабой интенсивности,
свидетельствующие о наличии метильной и метиленовой групп); полоса
в области 1720—1700 см"1 — одна из наиболее характерных,
свидетельствующая о наличии карбоксильных групп в молекуле
(при образовании солей полоса исчезает почти полностью и
появляются две полосы, характерные для карбоксилат-ионов,
для которых интенсивность изменчива; поэтому, как указывает
Д. С. Орлов [230, 240], работать необходимо со строго
стандартизированными препаратами); полосы в области 1650—1640 и
1550—1540 см"1 (полосы Амид I и Амид II), характерные для амид-
ной группы, имеющие очень существенное значение для
идентификации азотсодержащих группировок (к сожалению, как
отмечает Д. С. Орлов [230], эта полоса не всегда обнаруживается
в спектрах вследствие наложения ряда более сильных полос);
полоса в области 1625—1610 см"1, достаточно интенсивная;
исследователи трактуют ее по-разному: связывают с наличием
ароматических связей С=С, колебаниями С—Н в
гетероциклических группировках, Н и СО в первичных и вторичных амидах
[54, 177, 240], а также наличием карбонилов С=0, соединенных
водородной связью, и хинонов, также сочлененных этой связью
с группами ОН; полоса в области 1510—1500 см"1, в случае со-
53
ZOO Ш 600 800 1000° С
Рис. 5. Термограммы кривых ТВ (1) и
ДТА (2) гумусовых кислот
обыкновенного чернозема (/) и подзолистой почвы
северной тайги (77) (по: [328]).
а — гуминовые кислоты, б — фульвокислоты.
четания с полосой около 1610 см"1
свидетельствующая о наличии
ароматических группировок (ее
интенсивность обычно невелика);
полосы в области 1470—1370 см"1,
недостаточно интенсивные и часто
трудно расшифровывающиеся
(большинство исследователей
объясняют их наличие
деформационными колебаниями С—Н, хотя эти
полосы могут быть обусловлены
группировками О—Н, С—О, а
также ОН); полосы в
длинноволновой части ИК спектра
(области 1260-1200, 1100-1000 и
менее 1000 см"1) выражены не так
четко, для них характерна
переменная интенсивность
поглощения, и они обычно не используются для идентификации
компонентов молекулы гуминовых кислот из-за влияния
многочисленных минеральных веществ, присутствующих в препаратах
гуминовых кислот в виде примесей или сорбционных комплексов.
Несомненные достоинства ИК спектроскопии по сравнению
с классическими методами аналитической химии — быстрота и
простота определения, а также возможность снятия спектров
непосредственно в нативных гуминовых кислотах почвы без их
экстракции [238], что позволяет высказывать более
обоснованные гипотезы о структуре молекулы гуминовых кислот. Вместе
с тем точная идентификация компонентов молекулы, их
количественное соотношение все еще невозможны по данным ИК
спектроскопии. Необходимо их сочетание с иными приемами
исследования.
Метод дериватографии позволяет в своем наиболее простом
варианте получать сопряженные кривые дифференциального
термического (ДТА) и термовесового (ТВ) анализов (рис. 5), на
основании которых судят о наличии ряда структурных элементов
в молекуле гуминовых кислот [310, 328, 332]. Термическая
деструкция гумусовых кислот обусловливает ряд экзо- и
эндотермических эффектов, свидетельствующих о постепенном
разрушении молекулы. В ее составе отчетливо выделяются две резко
различные по термической устойчивости части: ядерная аромата*
54
ческая (более устойчивая) и боковые алифатические цепи, для
которых характерна значительно меньшая термоустойчивость.
Высокая интенсивность экзотермических реакций между 500 и
600°С обусловлена деструкцией ядерной части,
эндотермические и экзотермические эффекты в области низких температур
(60—400°) связаны с изменениями и постепенным разрушением
периферической части. Гуминовые кислоты чернозема и
подзолистой почвы заметно отличаются по потере массы в этих двух
областях, первые характеризуются большей относительной долей
ароматических структур. Сопряженное же применение
термографического и ИК спектрального методов или газожидкостной
хроматографии позволяет судить более точно об особенностях
структуры молекулы, хотя и эта группа методов не в состоянии
пока установить строение молекулы с точной идентификацией
всех компонентов [174, 320, 514].
В последние годы мы провели сравнительное исследование
природы гуминовых кислот различного происхождения с целью
изучить их природу. Для исследования использовали три группы
препаратов: гуминовые кислоты черноземов, дерново-подзолистых
суглинистых почв и новообразованные гуминовые кислоты,
полученные из растительных остатков, гумифицирующихся в
лабораторных условиях в течение 1—2 лет при оптимальных
условиях увлажнения и температуре 20—25°. Методики выделения
препаратов гуминовых кислот и определения отдельных констант
детально описаны в работах А. В. Назаровой [202, 204].
Приведенные данные об элементном составе гуминовых кислот
(табл. 16) показывают, что группа молодых гуминовых кислот,
образующихся на первых стадиях процесса гумификации
растительных остатков, наименее обуглерожена, содержит
максимальное количество водорода, отношение С : Н наиболее узкое, эти
препараты являются восстановленными соединениями.
Гуминовые кислоты дерново-подзолистых почв по элементному составу
близки к предыдущей группе. Это также восстановленные
соединения, но характеризуются они более широким отношением
С : Н, свидетельствующим, по-видимому, о некотором
возрастании доли ароматических структур в молекуле.
Гуминовые кислоты чернозема, как и следовало ожидать,
характеризуются максимальной обуглероженностью, широким
отношением С : Н, свидетельствующим о высокой степени
ароматизации молекул; для них же характерна наиболее высокая степень
окисленности. Необходимо отметить также, что молодые
гуминовые кислоты характеризуются наиболее заметными
колебаниями элементного состава, что обусловлено различиями в
химическом составе самих гумусообразователей. Заметны эти
колебания и в препаратах гуминовых кислот, выделенных из очень
близких в генетическом плане суглинистых, дерново-подзолистых
пахотных почв, хотя достоверность данных, выполненных в 3—5-
кратной повторности, была подтверждена математически [205].
55
Таблица 16
Элементный состав гуминовых кислот, ат.%, и отношение числа грамм-атомов
(на сухое беззольное вещество; по: [205])
f Объект, из которого
г выделены гуминовые
кислоты
Наземная масса
клевера, инкубированная
1 год
Подстилка,
инкубированная 2 года
осинового
насаждения
елового насаждения
Дерново-подзолистая
почва (Апах)
слабоокультуренная
тяжелосуглинистая
среднесуглинистая
хорошо
окультуренная,
среднесуглинистая
Чернозем типичный
(Апах)
С
34.9
37.0
37.0
36.4
38.6
38.5
43.6
42.9
Н
45.6
53.6
41.4
41.6
40.1
41.3
33.7
35.3
о
17.1
17.3
19.9
19.4
18.7
18.2
20.2
19.6
N
2.4
2.1
1.7
2.6
2.6
2.0
2.5
2.2
с
н
0.77
0.85
0.89
0.87
0.96
0.93
1.29
1.22
с
о
2.04
2.13
1.86
1.87
2.06
2.12
2.16
2.18
с
N
14.7
17.5
16.5
14.1
14.6
19.7
17.7
19.2
ш
-0.33
0.24
-0.04
—0.08
—0.07
-0.13
+0.15
+0.09
с
ароматических
структур, %
61.6
61.6
66.2
64.0
72.2
69.2
91.7
88.7
Если использовать графостатический метод Ван-Кревелена для
расчета содержания ароматической и алифатической частей
(рис. 6), то при всей относительности и условности абсолютных
величин минимальная доля ароматических структур характерна
для молодых гуминовых кислот, максимальная — для гуминовых
кислот черноземов. Отчетливо выделяется эта группа гуминовых
кислот и по диаграмме Ван-Кревелена, характеризующей
атомные отношения.
Не менее показательны материалы по содержанию
функциональных групп и выходу бензолполикарбоновых кислот в'
препаратах (табл. 17). Минимальное количество карбоксилов
содержится в новообразованных гуминовых кислотах
растительных остатков, для них же типично повышенное содержание фе-
нольных гидроксилов. В гуминовых кислотах
дерново-подзолистых почв количество карбоксильных групп несколько возрастает
при одновременном снижении содержания групп (ОН).
Максимальное количество карбоксильных групп типично для гуминовых
кислот черноземов, где количество фенольных гидроксилов
минимально. По-видимому, эти различия в составе кислых
функциональных групп играют существенную роль в формировании
разных по формам связи органо-минеральных производных в дер-
56
Рис. 6. Атомные отношения в гу-
миновых кислотах (по: [205]).
а — дегидрогенизация, б —
окисление, в — гидрогенизация. 1—5 —
новообразованные гуминовые кислоты:
I, 2 — из подстилки осинового
насаждения; 3, 4 — из подстилки
елового насаждения; б — из наземной
массы клевера. 6* 7 — из типичного
чернозема. 8—11 — из
дерново-подзолистых суглинистых почв.
ново-подзолистых почвах и
черноземах. Не менее
существенны различия и в
степени ароматизации молекул
гуминовых кислот,
характеризуемой по выходу бензол-
карбоновых кислот при
окислении препаратов щелочным раствором перманганата. При
сопоставлении гуминовых кислот дерново-подзолистых почв и черноземов
становится очевидным более высокий выход этих продуктов при
окислении гуминовых кислот черноземов, что согласуется и
с литературными данными [230]. Несколько неожиданным
оказался относительно высокий выход бензолполикарбоновых кислот
из препаратов новообразованных «молодых» гуминовых кислот,
Таблица 17
Функциональные группы, мг»экв./100 г, и продукты окисления, % гуминовых
кислот (на сухое беззольное вещество; по: [205])
Объект, из которого выделены
гуминовые кислоты
Наземная масса клевера,
инкубированная 1 год
Подстилка, инкубированная
2 года
осинового насаждения
елового »
Дерново-подзолистая почва
(АпахЛ
слабоокультуренная
тяжелосуглинистая
среднесуглинистая
хорошо окультуренная,
среднесуглинистая
Чернозем типичный (А„ах)
HOOD
233
243
1 287
313
320
327
392
375
(ОН)
444
636
| 607
384
495
337
332
361
Продукты окисления
КМп04 в щелочной среде
бензол-
поликар-
боновые
кислоты
18.7
13.4
25.4
8.8
| 10.9
11.6
19.4
J 19.5
низко-
молекулярные
кислоты
51.8
52.2
53.4
48.4
50.7
49.7
40.5
43.2
С02
29.5
34.4
21.2
42.8
38.4
38.5
40.1
37.3
^0.01%
1 см
0.027
0.023
0.031
0.048
0.052
i 0.053
0.103
Е4
Ее
9.0
7.7
6.2
6.0
5.8
5.3
3.2
57
1 I I I 1 1 1 1 1 I I »
3400 3200 3000 2800 1700 1500 1300 1100 900 см'1
Рис. 7. И К спектры гуминовых кислот различного происхождения.
1—11 — то же, что и на рис. 6.
выделенных из гумифицирующихся растительных остатков; для
этой группы кислот характерны наибольшие колебания: от 13.4
до 25.4%. Приведенные данные еще раз свидетельствуют о
сложности и недостаточной изученности самого механизма
гумификации (подробно будет рассмотрено в гл. III). Отчетливо выражены
некоторые различия в величине коэффициентов экстинкции
(i^o.01%/1 см) и цветности (Z?465/i?605), хотя в целом они однозначны.
Наибольшим поглощением, постепенно снижающимся в
гуминовых кислотах дерново-подзолистых почв и группе молодых
новообразованных кислот гумифицирующихся растительных
остатков (в последних оно минимально), характеризуются гумино-
вые кислоты чернозема. Более глубокая интерпретация этих
данных, как мы уже отмечали ранее, затруднена из-за неясности
самой природы цветности гуминовых кислот. Более четкую
картину дают ИК спектры (рис. 7). Все они имеют одинаковый
принцип строения, и (по установленной Д. С. Орловым
классификации) на них отчетливо выделяются полосы поглощения
спиртовых и фенольных гидроксилов, соединенные
межмолекулярной водородной связью (3400—3300, 1400, 1270 и ИЗО см""1),
метальные и метиленовые группы СН3 и СН2 (2935, 2860, 1460—
1380 см-1), карбоксильные группы как ароматических, так и
алифатических группировок (1720—1700 и 1220 см"1), амидные группы
58
(3200, 1650 и 1510 см""1), различного рода карбонилы — СО
(1625—1600 см""1), сопряженные связи С=С ароматических ядер
(1600 и 1510 см"1). Степень выраженности различных полос
в одном препарате, как одной и той же полосы в разных препаратах,
неодинакова. Наиболее сложные спектры по набору полос
поглощения дают препараты новообразованных гуминовых кислот
растительных остатков, что свидетельствует о их резко выраженной
гетерогенности. Следует отметить при этом, что они заметно
отличаются друг от друга по набору полос поглощения. Наиболее
гомогенен спектр гуминовых кислот чернозема. Если признать
справедливыми суждения К. Наканиси [216], что полоса
поглощения в области 1270 см"1 свидетельствует о хорошо выраженном
углеродном скелете с открытой цепью, нетрудно заметить, что
в максимальной степени она выражена в новообразованных
гуминовых кислотах гумифицированных растительных остатков,
в меньшей степени заметна в препаратах гуминовых кислот
дерново-подзолистых почв и практически незаметна в гуминовых
кислотах черноземов. Меньшая степень зрелости «молодых»
гуминовых кислот по сравнению с более сформированными гуми-
новыми кислотами дерново-подзолистых почв и наиболее «зрелыми»
кислотами черноземов может быть подтверждена наличием в
первой группе препаратов полосы в области ИЗО см"1,
обусловленной колебаниями спиртовых гидроксилов третичных спиртов.
В гуминовых кислотах чернозема эта полоса отсутствует. Таким
образом, ИК спектры гуминовых кислот различного
происхождения, свидетельствуя об общности химического строения молекул
гуминовых кислот, позволяют вскрыть и их различия, что может
быть использовано при исследовании самого механизма процесса
гумификации.
Азот гуминовых кислот.1 Изучение форм азота в гуминовых
кислотах различного происхождения показало их многообразие
(табл. 18). Прежде всего необходимо отметить различия в
валовом содержании азота в «молодых» гуминовых кислотах
гумифицированных растительных остатков, обусловленное главным
образом его содержанием в исходных гумусообразователях.
Достаточно показательны в этом плане данные о количестве азота
в гуминовых кислотах, выделенных из гумифицированной массы
клевера и еловой подстилки. Минимальное количество азота
характерно для гуминовых кислот чернозема. Очень разнообразны
формы азота; во всех препаратах они идентичны и представлены
аммонийной, аминной фракциями, фракцией аминосахаров и
азотсодержащих гетероциклов, причем их относительная доля в
гуминовых кислотах различного происхождения колеблется нерезко.
Наиболее значительно по степени подвижности азотистой части
молекулы выделяются гуминовые кислоты чернозема, для
которых характерны минимальная гидролизуемость 6 н. раствором
Эта часть раздела написана совместно с А. В. Назаровой [205].
59
Таблица 18
Содержание, степень гидролизуемости и формы азота в гуминовых кислотах
различного происхождения (по: [205])
Объект, из которого
выделены гуминовые
кислоты
Наземная масса клевера,
инкубированная 1 год
Подстилка елового
насаждения, инкубированная
2 года
Дерново-подзолистая
почва (Апах)
слабоокультуренная
тяжелосуглинистая
среднесуглинистая
хорошо окультуренная,
среднесуглинистая
Чернозем типичный (АпаХ)
Валовой N, %
(определен методом
Дюма)
6.35
2.96
4.37
4.40
3.33
3.76
3.49
Гидроли-
вуемый N,
% к
валовому
а
оГсо
14.9
11.7
12.7
11.9
11.3
10.4
и
К
щ
CD
53.5
51.0
59.4
52.5
49.5
44.2
45.8
Формы N, % к валовому
аминокислот
а
26.5
28.4
26.1
25.2
29.8
28.2
28.1
<*>
я
к a
я К
Но
6.0
6.1
8.3
8.2
5.6
3.3
5.1
я
Я
S
я
о
S
S
еб
18.3
13.8
21.3
16.6
12.0
10.6
9.7
¦
а
X
еб
о
о
я
Я Я
2.7
2.7
3.7
2.5
2.1
2.1
2.9
i
Я
Я
о*
ftg
ф К
н о
14.0
19.3
8.3
10.0
13.5
25.0
27.7
К*
s a
32.5
29.7
32.3
37.5
37.0
31.8
26.5
НС1 (44—46%) и максимальное содержание гетероциклических
группировок (25—27%). Одновременно гуминовые кислоты чер-
нозема\содержат пониженное количество аммонийных
группировок (около 10% от общего содержания азота).
Новообразованные гуминовые кислоты и выделенные из дерново-подзолистых
почв близки по соотношению азотсодержащих компонентов:
V8 всей массы азота представлена аминокислотами,
преимущественно их а-формами; колеблющиеся данные получены для
аммонийных группировок, составляющих от 12 до 21% азота
гуминовых кислот; содержание азота аминосахаров во всех препаратах
очень незначительно. Аналогичные данные о формах азота в
гуминовых кислотах различных почв приведены во многих работах
[53, 226, 230, 352, 354, 407, 559]. Но по-прежнему гипотетичны
наши представления о реальных формах нахождения различных
азотсодержащих группировок в молекуле гуминовых кислот.
Ранее [19] мы указывали на различные суждения о реальных
формах нахождения азота в молекуле гуминовых кислот. Одни
исследователи [143, 195] считают, что аминокислоты являются
компонентом молекулы гуминовых кислот в виде продуктов
конденсации с фенолами и хинонами, другие [425, 516, 559] полагают,
что аминокислоты — составные части протеинов, образующих
60
с гуминовыми кислотами единый комплекс. Можно высказать
еще одно предположение: аминокислоты, так же как и другие
идентифицированные азотсодержащие группировки, следует
рассматривать как продукты гидролиза гуминовых кислот,
образовавшиеся из протеидов (подробно эта гипотеза будет рассмотрена
в гл. III).
Структурные схемы, форма и ММ. Каковы же реальные
компоненты молекулы гуминовых кислот и насколько последние
однотипны? Окончательный ответ на этот вопрос дать нельзя.
Можно считать лишь доказанным, что основные компоненты
молекулы гуминовых кислот — ароматические и гетероциклические
безазотистые и азотсодержащие кольца, алифатические цепи и
ряд функциональных групп, среди которых главенствуют
карбоксилы и фенольные гидроксилы, обусловливающие кислотные
свойства молекулы. Достаточно хорошо изучены, как мы отмечалп
выше, состав и количество кислых функциональных группк но
нет ясности о их пространственном положении в молекуле.
Значительны успехи в определении природы алифатических цепей,
легко поддающихся гидролитическому отщеплению при
кислотном гидролизе. Всеобщим признанием пользуются представления
о наличии аминокислотных и углеводных остатков, причем четко
идентифицированы их индивидуальные представители. Но все
еще неясно структурное положение этих компонентов, ибо не
существует до сих пор общепризнанной структурной формулы
молекулы гуминовых кислот. Наиболее сложен и противоречив
состав реальных компонентов циклической, в том числе и
ароматической, части молекулы. Список ароматических и
гетероциклических компонентов, найденных в продуктах частичной деструк-
Таблица 19
Компоненты молекулы гуминовых кислот (по: [3&7])
Фрагменты молекулы
Аминокислоты
Гексозамины
Многочисленные ароматические кольца
Функциональные группы
Алифатические группы
Одночленные ароматические кольца
Предполагаемая
структура
CH3CHNH2COOH
C6H1306N
(глюкозамин)
С14Н10
(антрацен)
СООН
ОН
СО
сн3о
—с6н12—
с6н5о-
(фенол)
Всего
Доля
молекулы, %
10.5
2.5
10.0
11.2
11.1
1.5
2.0
3.6
2.5
54.9
61
Таблица 20
Баланс структурных единиц гуминовых кислот, % к веществу (по: [230])
Структурные единицы
Чернозем
Гидро л изуемая часть молекулы
Аминокислоты, всего
В том числе
основные
циклические
Аминосахара
NH3
Вещества типа фульвокислот
Углеводы, всего
В том числе
пентозы
метилпентозы
гексозы
Бензолкарбоновые кислоты
Сумма (округленно)
1 5.8
0.8
0.6
1.9
0.8
6
29
6
15
0.5
44
8.4
1.2
0.8
1.9
1.4
7
27
6
7
14
0.4
46
Негидро л изуемая часть
Ароматические продукты окисления перман-
ганатом, всего X 2.5
В том числе
бензолкарбоновые кислоты
феноксикислоты
Аминокислоты (вероятное содержание —1.3
от гидролизуемых аминокислот)
Сумма (округленно)
Общая сумма гидролизуемой и негидроли-
зуемой частей
1 42
25
17
1.9
44
88
17
10
7
2.8
20
66
ции препаратов, очень велик (см. табл. 14). Наиболее вероятными
компонентами ароматической части молекулы считаются
одноядерные и многоядерные ароматические остатки бензольного и
фенольного рядов; гетероциклическая часть ее представлена, по-
видимому, азотсодержащими основаниями пуринового и пири-
мидинового (пиридинового) рядов и остатками аминокислот,
содержащих гетероциклический азот (пролин, оксипролин, трипто-
62
q>;
&¦'¦
^
-d)
Рис. 8. Компоненты молекулы гуминовой кислоты (по: [425]).
j — пептиды, 2 — углеводы, 3 — фенольные кислоты, 4 — металлы.
фан, гистидин). Рассматриваются гуминовые кислоты и как
радикалы [493].
Предприняты попытки установить реальные фрагменты
молекулы гуминовых кислот и их ориентировочное соотношение.
Наиболее известны и обоснованы материалы Г. Фелбека и Д. С.
Орлова. В схеме Г. Фелбека (табл. 19) определяется 55% массы;
в качестве фрагментов выделяются аминокислоты, гексозамины,
многочленные и одночленные ароматические кольца,
алифатические и функциональные группы [387]. Несомненным
достоинством ее является выделение кислых функциональных групп
как обязательной составной части молекулы, но приводимая
в схеме «предполагаемая структура», практически исключающая
гетероциклические группировки азота, не может быть принята.
Д. С. Орлов [230] дает более полный баланс вероятных
структурных единиц (табл. 20), отводя большую роль ароматическим
компонентам, но не выделяет отдельно кислых функциональных
групп — одной из наиболее специфических частей молекулы
гуминовых кислот; неясно также положение гетероциклических
азотсодержащих группировок. Кроме того, в схемах Г. Фелбека
и Д. С. Орлова не вполне четко выдержана терминология. Вряд ли
можно пользоваться терминами «аминокислоты», «аминосахара»,
«углеводы», «бензолкарбоновые кислоты» как компонентами
молекулы гуминовых кислот; в последней должны присутствовать
лишь типичные группировки этих соединений, в противном случае
эти индивидуальные соединения не могут быть основными
структурными единицами молекулы. Р. Хэуорс [425], используя
ряд этих терминов, рассматривал их как компоненты,
сорбированные к основному ядру молекулы, не содержащему азота
(рис. 8).
На основании материалов по характеристике основных
компонентов молекулы гуминовых кислот можно составить
фрагментарную схему ее основных компонентов:
63
Компонент
Доля к массе, %
Ароматическая и гетероциклическая часть
азотсодержащие группировки типа пуриновых и
пиримидиновых оснований и ароматических
аминокислот и феноксазонов i 30—50
одночленные и многочленные ароматические коль- '
ца различных степеней замещения типа фенолов
и хинонов
Алифатическая и алициклическая часть
аминокислотные группировки 1 25—40
углеводные » I
Функциональные группы: —СООН; (—ОН), —ОН, 10—25
=СО, -СОН
Относительное содержание каждой группы компонентов,
конечно, очень условно, особенно на фоне достаточно хорошо
выраженной гетерогенности молекул гуминовых кислот, но в первом
приближении она отражает эти основные компоненты.
Не менее разнообразны и структурные схемы молекулы
гуминовых кислот. По данным С. С. Драгунова и сотрудников [97],
молекула гуминовых кислот имеет линейную форму и состоит
из ароматического ядра двух- и трехатомных фенольных и
сочлененных с ними азотистых гетероциклов, алифатических
углеводных и аминокислотных цепей, ряда периферических
функциональных групп:
с6н„о5
СООН о ОН
ОН СНг СН-СНг ОН СН-СНг Л)
ъ нг осн3 н О н2 сн2 н 0х н2 он
C0-NH-CgH1803N
Схема Г, Фелбека [385] предусматривает в качестве основных
компонентов пироновые кольца, связанные через метиленовые
мостики, к которым присоединены фенолы и аминокислоты (см.
с. 65). Кроме того, предложено еще несколько схем [120,138, 392].
Сложной и в значительной степени условной является схема
Д. Кляйнхемпеля [439], в которой предусмотрены, по-видимому,
все возможные группировки в молекуле гуминовых кислот и
способы их сочленения. Наиболее обоснована теоретически и
подтверждена экспериментально схема Д. С. Орлова [232] для
структурной ячейки гуминовой кислоты, в которой выделены
негидролизуемая и гидролизуемая части этой элементарной
ячейки. Негидролизуемая часть состоит из ряда шестичленных
циклов с трех- и четырехзамещенными структурами, азот
представлен в ней феноксазоновыми группировками; гидроли-
64
Фенолы
I
О
I
Фульвокислота i
Центральная
частица
пирона
N
Аминокислота,
пептид
Гуминовая
кислота
+
О
НзСО\А/ОСНз
I
о
н~
I
н—о
I L» «L. I!
(1 }-GH2-\J-Cn2-\ ;-CH2 ->
N N
I I
Н HG-R
I
с=о
I
NH
HC-R'
и
зуемая часть — в основном из углеводных и аминокислотных
остатков:
Н Н Н ^СНз Н Н Н ~~
-L->v<U(U^Y0V4--o-c!-cU(!-
¦с—„A-g=gVY yv°-c-
о1 Ay *
l I
н сн
н н f (%
i A [J Ун
сн2
<jo
неон
I
ядро
H JR3 H
(j_G_0~G-
I II I
но н
i-
(CH2)W—
(CH3)2CHCH2-CHNH2COOH
(CeH10O5)2
-(COOH)„,
-(0Н)„,
-(NH2)W,
-(CH2)W—
!U
он
d
_J перидберичбекая часть
Эта элементарная ячейка с массой около 1500 образует
молекулу гуминовых кислот с массой, достигающей 60 000—
70 000. Д. С. Орлов указывает, что данную схему нужно
рассматривать как первое приближение и «было бы неверным
представлять все структурные ячейки гуминовой кислоты
идентичными по составу и строению» [232, с. 16]. Следует отметить также
ряд блок-схем, в которых авторы стремятся отразить реальное
состояние всей системы гумусовых веществ в почве [199, 564].
В схеме В. Цихмана [564] выделены три основных компонента
системы: внутренняя (ядерная) структура, краевая часть и
наружная сфера (рис. 9, 1—3). Ядерная структура образована за
5 Л. Н. Александрова
65
счет сил главных валентностей и представлена единичными ч-дСОДц-
цами гуминовых кислот диаметром 1.5—2.5 нм; краевая част<ь¦—-
системой функциональных групп (СООН), (ОН), $Н2, С—б)
и т. д.; наружная сфера — системой негумусовых веществ:" (угле-'
водов, протеинов, лигнина, энзимов и т. д.), а также гидратиро-
ванных ионов металлов и их органо-минеральных производных,*
сорбированных в основном за счет межмолекулярных форм связи;
[565]. В этой сложной блок-схеме автор пытается отразить сумму
наших знаний об органических веществах почвы, но не
приближает нас к познанию структуры молекул гуминовых кислот.
Характер межмолекулярных форм связи автор рассматривает как
Донорно-акцепторное взаимодействие между ароматическими и
йинойднымй компонентами в молекуле. Наличие этого
взаимодействия й энергия связи определяются по изменению УФ и
ЙК Спектров, электропроводности, а также методом
колоночной и тонкослойной хроматографии [565].
Следует отметить, что аналогичные блок-схемы,
иллюстрирующие строение молекулы гуминовых кислот, приводились,
как мы уже отмечали, рядом авторов. В. Мистерски и В.
Логинов [199] рассматривают гуминовые кислоты как органо-мине-
ральные соединения, частицы которых включают три зоны:
ароматическое ядро, периферические части, способные к гидролизу,
и минеральные компоненты. Основной компонент
периферических частей, которые содержат почти 10% азота, — аминб^
кислоты; минеральные элементы представлены полуторными1
окислами, кремнеземом и фосфатами. Последние соединены с
массой молекулы эфирной связью по типу фосфоропротеидов.
Д. Кляйнхемпель [439] также выделяет ароматическую часть
молекулы, представленную фенолами, хинонами и бензолкарбо-
новыми кислотами, периферические линейные группировки,
состоящие из полисахаридов и пептидов, а также мостики,
соединяющие эти компоненты. По справедливому указанию
Д, С. Орлова [230], эта схема носит в значительной степени
условный характер, так как последовательность структурных
единиц и их набор произвольны.
Не менее разноречивы современные данные о форме молекул
ц ее размерах. Электронно-микроскопические исследования,
проведенные еще в 50-х гг. В. Фляйгом и Г. Байтельспахером [398],
свидетельствовали о сферической форме частиц гуминовых кислот
размером от 3 до 8 нм; Г. Тиле и Г. Кеттнер [548] отмечали
несколько большие размеры: 8—10 нм. Детальные электронно-
микроскопические исследования провели Д. С. Орлов с
сотрудниками [230, 233]; они показали, что истинные размеры молекул
гуминовых кислот очень трудно определять из-за их
полидисперсности, В итоге электронно-микроскопических исследований и
последующих расчетов авторы считают, что в разбавленных
растворах частицы гуматов имеют диаметр около 3 нм и
характеризуются вытянутой формой при отношении осей 1 : 2—1 : 6 для
66
Рис. 9. Схема структуры «системы гумусового вещества» (по: [564]).
1 — наружная сфера: ГО — гидратная оболочка, Me — ионы металлов, НВ —
негумусовые вещества (углеводы, протеины, лигнин и другие органические вещества, глинистые
минералы); 2 — внутренняя структура: ГВ — гуминовое вещество; з — краевая часть:
ФГ — функциональные группы (—СООН, —ОН, —NH2), КГ — краевые группы (дона-
торные и акцепторные), М — мостики.
гуминовых кислот чернозема и 1 : 12 для гуминовых кислот
подзолистых почв. При концентрировании растворов и последующем
высушивании молекулы деформируются, и их форма приближается
к сферической или полусферической вследствие образования
агрегатов размером от 20—30 до 80—100 нм. Оригинальны
представления И. Д. Комиссарова и Л. Ф. Логинова [138], согласно
которым молекулы гуминовых кислот окисленного угля имеют форму
«улитки» диаметром около 16.5 нм (рис. 10).
Не менее разноречивы данные о ММ гуминовых кислот, при*
чем само понятие «молекулярная масса» для любых
высокомолекулярных соединений, в том числе и для гумусовых веществ, не
вполне ясна, на что мы указывали еще около 30 лет назад [11],
предложив различать молекулярный и мицелярный вес. В
соответствии с учением о высокомолекулярных соединениях следует
различать среднечисловую ММ (Л/я) и средневесовую ММ (Мю)\
первая величина определяется методами, позволяющими учесть
число молекул (методы криоскопии, осмометрии, эбулиоскопии);
вторая — методами седиментационного анализа, светорассеяния
67
5*
Рис. 10. Условная модель
макромолекулы гуминовой кислоты (по:
[138]).
или гельфильтрации [19].
Для полидисперсных
высокомолекулярных соединений
та и другая величины
относительны^ причем Мю всегда
больше Мп. В тех случаях,
когда в растворах
присутствуют агрегаты молекул (ас-
социаты, по терминологии
Д. С. Орлова), следует
говорить о мицеллярной массе.
Д. С. Орлов предложил
более дробную номенклатуру
для характеристики ММ гумусовых веществ [230]. Он различает
минимальную ММ, характеризующую элементарную ячейку
молекулы гуминовых кислот; структурные единицы (или
структурный элемент) — часть молекулы, отщепляемой в ходе деструкции;
молекулу гумусовой кислоты — индивидуальную частицу,
образованную за счет сил главных валентностей; простые ассоциаты
молекул (димеры) и сложные ассоциаты (мицеллы или агрегаты),
образующиеся за счет межмолекулярных сил. Эта детальная
характеристика будет безусловно полезна после установления
структуры молекулы гуминовых кислот.
В настоящее время наиболее распространенным способом
определения и характеристики ММ гумусовых веществ является метод
гельфильтрации на системе сефадексов или молселектов, который
позволяет составить кривые распределения массы в зависимости
от размеров частиц (молекул) в растворе. Данные, полученные
этим методом, очень варьируют: от 4000—6000 до 50 000—
70 000 и даже 100 000 и более. На кривых распределения ММ
обычно выделяется несколько максимумов, что свидетельствует
о резко выраженной полидисперсности Гуминовых кислот [137,
233, 363]. Для иллюстрации приводим кривые, полученные в
нашей лаборатории для гуминовых кислот различного
происхождения (рис. 11). Наибольшая ММ, достигающая 100 000, у
новообразованных гуминовых кислот, минимальная ММ характерна для
гуминовых кислот черноземов (менее 70 000), причем любой
препарат легко расчленяется на ряд фракций по размерам частиц.
Очень обстоятельные материалы приводит М. П.
Колесников [137], определив параметры распределения ММ гуминовых
кислот для главнейших типов почв СССР. Исследования показали,
что гуминовые кислоты всех изученных типов почв полидисперсны
и у них очень широкий диапазон колебаний для Mw: от <500
68
/о
MMZ700000t
** I
Рис. 11. Распределение ММ гумусовых кислот на сефадексе Г-75.
По оси абсцисс — объем элюата; по оси ордипат — доля препаратов от их исходного
количества. 1 — новообразованные гуминовые кислоты, 2 — гуминовые кислоты чернозема,
3 — фульвокислоты (гор. В^).
до >200 000, причем основную массу составляют фракции от 5000
до 150 000, тогда как наиболее высокомолекулярные фракции
с Mw ^>200 000 составляют лишь 4—9% и характерны для
некоторых типов почв (рис. 12). Трудно уловить закономерности,
которым подчиняются ММ гуминовых кислот ряда зональных
почв. Достаточно указать, что минимальные величины Мп 35 000—
50 000 характерны для черноземов, тундрово-глеевых и
горнолуговых почв, а более высокие показатели, лежащие в пределах
60 000—80 000, отмечены для серо-бурых и бурых почв: пустынно-
степных, сероземов, дерново-сильноподзолистых, красноземов,
горно-луговых субальпийских.
Причины столь значительных колебаний не вполне ясны.
Д. С. Орлов указывает, что эти колебания обусловлены в
основном методом исследования и состоянием препарата: его
концентрацией, величиной рН, присутствием поливалентных катионов и
склонностью гуминовых кислот к образованию ассоциатов
(надмолекулярных структур). Кроме того, существенное значение
имеет полидисперсность самих молекул гуминовых кислот как
следствие обязательных и специфических особенностей этого
класса соединений.
Гетерогенность гуминовых кислот. Характерной особенностью
всего класса гумусовых кислот, и в частности гуминовых, является
их гетерогенность — «некоторая неоднородность состава и свойств
69
Рис. 12. Молекулярно-весовое
распределение фракций (%) гуминовых
кислот некоторых почв СССР (по: [137]).
1 — дерново-палевоподзолистая, 2 — мощный
чернозем, 3 — темно-каштановая, 4 — серозем.
отдельных фракций в пределах
каждой группы перегнойных
кислот при сохранении общего типа
строения, характерного для груп-
лы в целом» [8, с. 5].
Эти представления,
высказанные нами еще в 1955 г.,
подтвердились в настоящее время по
ряду параметров. Прежде всего
гуминовые кислоты гетерогенны по ММ. Особенно ярко это
свойство выявляется при применении метода распределительной
хроматографии на сефадексах, агаре, декстранах и молселектах,
который позволил расчленить любые препараты гуминовых
кислот на ряд фракций, различных по ММ [70, 100, 121, 285, 446,
451, 533, 536, 556]. В. Дубин и В. Фильков [100] разделили
гуминовые кислоты черноземов, серых и бурых лесных почв на
две фракции с различными ММ: для первой фракции она
колебалась от 50 000 до 100 000, для второй — около 5000. С. Кан и
Д. Фризен [435] также выделили методом гельфильтрации на
сефадексах Г-75, Г-100 и Г-150 две фракции из препаратов
гуминовых кислот чернозема и солонца с резко различными ММ.
Исследования Д. С. Орлова [230] показали, что методом гель-
фильтрации препараты гуминовых кислот расчленяются на ряд
фракций с различными ММ. Автор указывает, что наименее
гетерогенны гуминовые кислоты черноземов. Г. Филэп и Л. Татар [390]
получили пять фракций гуминовых кислот чернозема, причем ММ
наиболее высокомолекулярной фракции более 100000, а
наименее высокомолекулярной — около 27 000. Ф. Поспишил [480]
расчленил препарат на четыре фракции. Аналогичные
данные можно умножить, и представления о неоднородности
(полидисперсности) гуминовых кислот по ММ общепризнанны.
Значительно менее ясен, как мы уже отмечали, вопрос о причинах этой
:полидисперсности.
Для установления причин полидисперсности А. В. Назаровой
исследован методом гельфильтрации ряд препаратов гуминовых
>кислот, выделенных из чернозема, дерново-подзолистых почв и
мумифицирующихся растительных остатков [203]. Методика
получения препаратов описана в одной из работ А. В. Назаровой [205],
а основные параметры их приведены в табл. 16 и 17. Автором
использованы сефадексы Г-25, Г-50 и Г-100, в качестве элюента
выбрана дистиллированная вода. Калибровка колонок проведена
70
по голубому декстрану (ММ 2 000 000), сывороточному бычьему
альбумину (ММ 70 000—68 000), дезоксирибонуклеазе (ММ 40 000)
и липазе (ММ 30 000—25 000). Данные о кривых элюации
(рис. 13, а) показывают, что все гуминовые кислоты
полидисперсны, их ММ более 5000, так как они полностью выходят во
внешнем объеме геля Г-25 и четко расчленяются на ряд фракций.
Новообразованные гуминовые кислоты и гуминовые кислоты
дерново-подзолистых почв отчетливо расчленяются на три фракции,
а гуминовые кислоты чернозема — на две фракции; при этом
гуминовые кислоты дерново-подзолистых почв и гумифицирую-
щихся растительных остатков более высокомолекулярны, чем
гуминовые кислоты чернозема. Использование сефадекса Г-100
показало (рис. 13, б), что ММ одной из фракций гуминовых кислот
дерново-подзолистых почв и гумифицирующихся растительных
остатков более 100 000, тогда как максимальная ММ гуминовых
кислот чернозема не превышает 70 000 (фракция I
дерново-подзолистых почв и гумифицированных растительных остатков
выходит в свободном объеме при фильтрации через Г-100, а гуминовые
кислоты чернозема распределяются внутри его). Учитывая
калибровку колонок, можно считать, что ММ фракции II гуминовых
кислот из дерново-подзолистой почвы и гумифицированных
растительных остатков колеблется около 40 000, а фракции III —
в пределах 14 000—10 000. Очень показательны отношения
площадей пиков фракций гуминовых кислот: SISlu (табл. 21).
Сравнивая отношения SUI/Sr и Sn/Sv можно выявить существен-
Таблица 21
Соотношение площадей пиков фракций гуминовых кислот (по: [203])
Объект, из которого выделены
гуминовые кислоты
Еловая подстилка,
инкубированная
6 мес
2 года
Наземная масса клевера,
инкубированная 1 год
Дерново-подзолистая сред-
несуглинистая почва (Апах)
плохо окультуренная
хорошо »
Чернозем типичный (Апах)
s
?
Г-50
0.37
0.47
0.88
1.31
1.29
—
пг
i
Г-100
I
\
1.29
—
8
&
Г-50
Г
г
0.88
1.11
1.54
0.65
0.90
2.60
гт
i
Г-100
—
¦ —
0.90
2.60
su +-?пг
sT
i
1.25
1.58
2.42
1.96
2.19
2.60
Примечание. S1f 8и и Sm — площади пиков фракций Т, II и III с ММ >
> 100 000, ~ 40 000 и 14 000—10 000 соответственно.
71
140 мл
Рис. 13. Распределение ММ гуминовых кислот различного происхождения
на сефадексах Г-50 (а) и Г-100 (б).
По оси абсцисс — объем элюата. 2 — новообразованные гуминовые кислоты из гумифици-
рованной наземной массы клевера, 2 — из подстилки елового насаждения, з — из под
стилки оринового насаждения, 4—6 — из дерново-подзолистых цочв,^7 — из чернозема
i
s
120 ш
Ъ V0
Рис. 13 (продолжение).
Таблица 22
Элементный состав, ат.% па сухое беззольпое вещество,
и отношение числа грамм-атомов гуминовых кислот,
фракционированных через сефадексы Г-50 и Г-100 (по: [203])
Фракция
G
Н
С
О
с
N
НСРо,
Еловая подстилка, гумифицированная 6 мес
I
II
III
I
II
III
37.0
37.3
36.2
44.9
43.5
38.7
15.3
17.4
23.4
2.8
1.8
1.7
0.82
0.86
0.93
2.42 1
2.15
1.54 |
13.47 I
21.32
21.46
0.25
3.37
3.30
Та же подстилка, гумифицированная 24 мес
1 37.1
38.2
1 36.6
45.8
42.5
38.2
14.5
17.7
23.9
2.6
1.6
1.3
0.81
0.90
0.96
2.55 1
2.16
1.53
14.40 I
24.02
26.68
0.25
4.40
Наземная масса клевера,
I
II
III
1 32.6
37.4
37.5
49.3
44.6
42.0
14.3
15.1
18.0
3.8
2.9
2.5
0.66
0.84
0.90
2.28 1
2.49
2.09
гумифицированная1
12.89 | \ 0.20 | 2.90
14.91
Дерново-подзолистая почва (Ах)
I
II
III
34.0
37.2
35.3
46.9
42.4
39.9
16.0
17.7
22.7
3.1
2.7
2.2
0.73
0.88
0.89
2.12
2.11
1.57
11.10 1
13.42
16.89
0.26
3.4
3.90
год
1.9
2.7а
Примечание,
и 10 000-14 000.
ММ фракций I, II и III — соответственно > 100 000, ~ 40 00CP
ные различия между наиболее высокомолекулярными и менее
высокомолекулярными фракциями. Максимальная
полидисперсность и наиболее высокие числа для ММ получены в препаратах
новообразованных гуминовых кислот и выделенных из дерново-
подзолистых почв. Гуминовые кислоты чернозема более
гомогенны и характеризуются менее высокой ММ; отношение Sjj/Sj^ и
{Su-\- Sj^/Si значительно шире на сефадексах Г-50 и Г-100.
Гетерогенность по размерам молекул проявляется и в
неоднородности химического состава выделенных фракций. Следует
отметить при этом, что если полидисперсность гуминовых кислот
иллюстрируется в настоящее время значительным объемом
информации, то данных о химической гетерогенности отдельных фракций
гуминовых кислот значительно меньше. Можно отметить лишь
работы, в которых показаны различия в оптической плотности
фракций с разной ММ [362, 450, 542], а также исследования,
вскрывшие существенные различия в элементном составе и
количестве функциональных групп в разных фракциях гуминовых
кислот, выделенных из серой лесной почвы и чернозема [276].
Интересна статья Р. Свифта с соавторами [536], разделивших
74
Рис. 14. Взаимосвязь между
коэффициентом оптической плотности (#сгк,
1 г""1'См"1) и ММ (Х10_3) различных
фракций (показаны крестиками) гу-
миновых кислот (по: [536]).
j 2 и з — светофильтры при длине волны
230, 278 и 400 нм соответственно.
гуминовые кислоты гельфильтра-
цией на агаре и диализом на
50 фракций с ММ от 1360 до 62 000
и показавших резко различную Y% ?
оптическую плотность и разную |^х- * * х—
степень ароматизации их (арома- ^ ^ 15ZMM
тизация возрастала параллельно
уменьшению величины ММ —
рис. 14).
Полученные в нашей лаборатории данные позволяют дать
дополнительную характеристику химической гетерогенности гу-
миновых кислот различного происхождения и высказать
предположение о вероятных причинах ее.
Достаточно отчетливо заметны колебания как по элементному
составу, так и по соотношению числа грамм-атомов трех фракций
гуминовых кислот, выделенных на сефадексах Г-50 и Г-100 из
дерново-подзолистой почвы и гумифицирующихся
растительных остатков в нашей лаборатории (табл. 22). Менее
высокомолекулярные фракции во всех препаратах характеризуются
минимальным отношением С : О, т. е. наиболее окисленны. Они же
содержат минимальное количество азота и характеризуются
возрастающим отношением С : Н, свидетельствующим о заметном
увеличении относительной доли ароматической части в молекуле.
В гуминовых кислотах дерново-подзолистой почвы содержание Н
максимально в наиболее высокомолекулярной фракции и
постепенно снижается с уменьшением ММ. Аналогичные
закономерности можно наблюдать и в препаратах новообразованных
гуминовых кислот. Очень четкие изменения характерны для
азотсодержащей части молекулы. Содержание азота уменьшается от
наиболее высокомолекулярной фракции I к фракции III. Все
приведенные данные статистически достоверны, что видно из
результатов их математической обработки. Аналогичные
колебания в содержании азота приводят Р. Свифт и А. Познер [534]; по
их данным, количество азота уменьшается в основном за счет
снижения содержания аминокислот в продуктах гидролиза
отдельных фракций.
Наиболее ярко химическая гетерогенность отдельных фракций
выявляется на ИК спектрах (рис. 15). Так, например, для
фракции III гуминовых кислот дерново-подзолистой почвы и
гуминовых кислот, выделенных из гумифицированной подстилки,
характерна более высокая степень ароматизации (полосы в области
75
-;
3800 3600 ЗЩ}0 32003000 2800 1900 1700 1500 1300 1100 900 700 см
Рис. 15. И К спектры гуминовых кислот после фракционирования на сефа-
дексе Г-50.
а — из подстилки елового насаждения, инкубированной 2 года; б — из дерново-подзо»
листой почвы. 1 — исходные, 2 — фракция 1,3 — фракция II, 4 •— фракция III.
1610—1600, 860 и 800—760 см"1). Подтверждается также меньшее
содержание азота в наименее высокомолекулярной фракции, так
как нет смещения максимума при 1610 см'1.
Каковы же причины гетерогенности, которая характерна для
всего класса гумусовых веществ? Дать однозначный ответ на этот
вопрос в настоящее время трудно: еще мало экспериментальных
данных. По-видимому, основными причинами полидисперсности
и гетерогенности по химическому составу (вероятно, и по деталям
строения молекул) являются неоднородность и сложность
химического состава гумусообразователей, стадийность самого
процесса гумификации и варьирование условий, в которых протекает
этот процесс, а также реакции взаимодействия образующихся
гуминовых кислот с компонентами минеральной части почвы.
Некоторые данные, полученные для гуминовых кислот
различных по составу гумифицирующихся растительных остатков
(табл. 23), показывают, что препараты заметно различны по
элементному составу. Это проявляется в варьировании отношения
числа грамм-атомов, в различиях по степени окисленности,
количеству реактивоспособных карбоксильных и фенолгидроксильных
76
Таблица 23
Отношение числа грамм-атомов и функциональные группы,
мг -экв./ЮО г, гуминовых кислот различного происхождения
Объект, из которого
выделены гуминовые
кислоты
Подстилка
осинового
насаждения
елового
насаждения
Наземная масса
клевера
Листья дуба
Корни
разнотравно-злаковой
растительности
Дерново-подзолистая почва (AjJ
Чернозем (А)
Срок
гумификации, мес
6
24
6
24
24
24
24
с
н
0.77
0.85
0.88
0.89
1.10
0.83
0.88
0.96
1.29
С
о
2.04
2.13
1.85
1.86
2.20
1.03
1.99
2.06
2.16
С
N
14.7
17.5
16.4
16.5
10.7
11.2
19.3
14.6
17.7
н
с
1.30
1.18
1.14
1.12
0.90
1.15
1.18
0.78
1.04
и)
-0.33
-0.24
-0.04
—0.22
-0.22
-0.25
—0.20
—0,07
+0.15
соон
297
243
307
287
342
420
266
320
392
(НО)
722
736
657
507
424
497
513
495
332
Бензол-
поликар-
боновые
кислоты,
%
12.5
13.4
25.4
18.1
22.2
14.1
19.4
10.9
групп, выходу бензолполикарбоновых кислот. Действительно,
гуминовые кислоты, выделенные из гумифицированных листьев
клевера, отличаются минимальным отношением С : N вследствие
высокого содержания азота в наземной массе клевера, а также
более широким отношением С : Н и по выходу
бензолполикарбоновых кислот; в гуминовых кислотах, выделенных из
гумифицированных листьев дуба, высокое содержание карбоксильных
и фенолгидроксильных групп, что обусловлено повышенным
содержанием таннинов, богатых фенольными гидроксилами,
в листьях дуба. Различен выход бензолполикарбоновых кислот:
максимален в молодых гуминовых кислотах, выделенных из
подстилки елового насаждения; в хвое ели, как известно,
повышено количество смол, восков, дубильных веществ, лигнина.
Следует отметить, что эти различия наиболее заметны в менее
гумифицированных препаратах гуминовых кислот; они постепенно
сглаживаются в ходе дальнейшего развития процесса гумификации.
Не менее существенное влияние на химическую
гетерогенность гуминовых кислот должна оказывать стадийность
процессов гумификации растительных остатков (детально
рассматривается в гл. III). Процесс гумификации длительный, и
новообразованные гуминовые кислоты подвергаются в почве непрерыв^
ной трансформации, в течение которой изменяются детали
строения молекулы. Совершенно естественно, что в каждый данный
момент в почве присутствуют разновозрастные гуминовые кислоты,
77
заметно гетерогенные и по размерам молекул, и по деталям их
структуры.
Существенное влияние на фракционирование гуминовых кис*
лот в почве оказывают реакции взаимодействия их с минеральной
частью почвы. Действительно, исследования по дробному
осаждению гуминовых кислот, проведенные еще 30 лет назад [5],
показали, что различные катионы осаждают отдельные фракции
гуминовых кислот, заметно отличающиеся по элементному составу.
Варьирует элементный состав гуминовых кислот в различных
частях гумусовой пленки, покрывающей поверхность
гранулометрических фракций, причем в разных почвах степень этой
вариабельности различна (табл. 24). Поверхностная часть пленки
(фракция I) гуминовых кислот дерново-подзолистой почвы меньше
обуглерожена и ароматизирована, содержит пониженное
количество карбоксильных групп. Различные по степени закрепления
фракции гуминовых кислот чернозема менее гетерогенны.
Очень четко выявляется влияние процессов взаимодействия
с минеральной частью почвы на степень гетерогенности
гуминовых кислот при использовании метода гельфильтрации на сефа-
дексах [27 ]. Если до взаимодействия гуминовые кислоты отчетливо
расчленялись на три фракции, то после взаимодействия в центри-
фугате полностью сохраняется наиболее высокомолекулярная
Таблица 24
Элементный состав, % к сухому беззольному веществу,
и функциональные группы, мг-экв.ДОО г,
отдельных фракций гуминовых кислот
Фракция
Зола, %
N
Функциональные группы
при разных значениях рН
4.6
7.0
8.2
ё
Дерново-подзолистая почва
1 3.79
3.78
1 4.75
54.3
55.4
57.0
5.0
4.8
4.4
36.5
35.7
34.3
4.2
4.1
4.3
171
193
189
332 1
332
348
482
495
487
I
II
III
Чернозем
I 2.04
2.16
[ 2.41
60.2
60.1
60.2
3.9
3.7
3.7
32.6
32.9
32.7
3.3
3.3
3.4
236
236
241
365 1
384
375 |
531
531
522
фракция, исчезает фракция III и значительно уменьшается выход
фракции II, которые прочно сорбируются бентонитом (рис. 16).
Иными словами, в самой почве происходит своеобразное
фракционирование гуминовых кислот по степени их сорбции на
поверхности глинистых минералов, что неизбежно оказывает
влияние на состав экстрагируемой из почвы части гуминовых кислот*
7S
0.2
0.1
0.2
0.1
140 мл
Рис. 16. Распределение ММ исходных (1) и не сорбированных бентонитом (2)
гуминовых кислот на сефадексе Г-50.
По оси абсцисс — объем элюата. а — гуминовые кислоты из дерново-подзолистой почвы,
б — из гумифицированной наземной массы клевера.
Наблюдается, по-видимому, фракционирование гуминовых
кислот в ходе почвообразовательного процесса и его
многообразных элементарных звеньев, но эти явления еще не подвергались
экспериментальному исследованию.
Таким образом, охарактеризовав основные современные
представления о природе гуминовых кислот, необходимо отметить,
что все еще недостаточно разработана теория структуры и
строения гуминовых кислот как основной группы особого природного
класса органических соединений — гумусовых кислот. Чем же
можно объяснить столь длительные и все еще недостаточно
результативные поиски в области исследования их природы? И не
79
являются ли стремления найти однотипное строение в настоящее
время тормозом для дальнейших исследований? Как высокомоле-
кулярныелпродукты трансформации различных компонентов
растительных, микробных и животных остатков гуминовые кислоты
могут и не иметь однотипного строения из-за различий в строении
тех исходных веществ, которые подвергаются гумификации. Для
познания природы образующихся продуктов настоятельно
необходима дальнейшая разработка теории гумификации.
«Гиматомелановые» кислоты
Термин «гиматомелановые кислоты» был введен Ф. Гоппе-
Зейлером в 1889 г. для части гуминовых кислот, растворимых
в спирте [429]. С тех пор этот термин широко используется, хотя
разные исследователи отводят данной группе кислот различное
номенклатурное место. Некоторые выделяют эту группу гумусовых
кислот отдельно наравне с гуминовыми кислотами и фульвокисло-
тами [143а, 394, 513]. В большинстве же номенклатурных схем
гиматомелановые кислоты включаются в состав группы гуминовых
кислот как их фракция, растворимая в органических
растворителях [19, 231, 491].
Частичная растворимость гуминовых кислот в органических
растворителях — в смеси этилового спирта и бензола, в
метиловом и этиловом спиртах, ацетоне, диоксане и др. — в настоящее
время установлена многими авторами [139, 192, 500]. Определены
и основные параметры этой фракции. Наиболее обстоятельные
исследования в последние годы проведены Т. А. Кухаренко и
Л. Н. Екатерининой [172, 173] для гиматомелановых кислот
торфа и ископаемых углей, Л. Н. Александровой [8], а также
С. И. Шурухиной и А. И. Чебаевским [327] для препаратов,
полученных из почвенных гуминовых кислот. По всем основным
параметрам эта фракция принципиально не отличается от
показателей, характерных для группы гуминовых кислот в целом, но
содержит несколько повышенное количество углерода и водорода
и характеризуется пониженным содержанием карбоксилов и
повышенным содержанием метоксильных групп (табл. 25).
Аналогичные данные получены для гиматомелановых кислот,
выделенных из гуминовых кислот торфяной и дерново-подзолистой
почв, чернозема [327], а также для спирторастворимой фракции
гуминовых кислот торфа и ископаемых углей [172]. Следует
отметить, что повышенное содержание метоксильных групп и
снижение количества карбоксилов — результат частичной этерифика-
ции карбоксильных групп в процессе выделения гиматомелановых
кислот спиртом. На эту особенность обращали внимание еще
в 40-е годы [71, 490], а в последнее время Т. А. Кухаренко и
Л. Н. Екатеринина [172], С. И. Шурухина и А. И. Чебаев-
ский [327]. Немногочисленные, к сожалению, данные по изучению
80
Таблица 25
Элементный состав, % к сухой беззольыой массб*
и кислые функциональные группы, мг -экв./ЮО г, в гуминовых кислотах
Фракция
Растворимая
в спиртобензоле
в спирте
Нерастворимая
Растворимая
в спиртобензоле
в спирте
Нерастворимая
Выход фрак-jl
ЦИИ, %
к массе
гуминовых
кислот 1
с
н
о
N
Функциональные
группы, способные
к обмену при
разных значениях рН
4.5
.7.0
'8.2
Подзолистая почва
9.2
4.8
86.0
61.0
60.1
54.1
6.3
6.0
4.8
' 31.1
31.1
36.9
1.6
2.8
4.2
103
182
203
321
307
495
Чернозем
8.9
2.4
88.7
60.7
62.6
61.1
5.3
5.7
2.8
31.1
28.6
32.7
2.9
3.5
3.4
152
270
280
364
387
601
OGHs,
%
1.4
1.8
0.9
0.5
0.5
0.3
природы этой фракции гуминовых кислот приведены Т. А. Ку-
харенко и Л. Н. Екатерининой в другой статье [173]. Окисление
препаратов в щелочной среде парманганатом не обнаружило
принципиальной разницы в продуктах окисления (табл. 26).
Таблица 26
Продукты окисления щелочным перманганатом калия
гуминовых и гиматомелановых кислот углей (по: [173])
Исходный уголь
Тощий
Бурый
Бурый
выветрившийся
Кислоты
Гуминовые
Гиматомелановые
Гуминовые
Гиматомелановые
Гуминовые
Гиматомелановые
Выход С, % от исходного
ароматических кислот
53.0
42.6
24.8
16.3
18.1
14.2
низкомолекулярных кислот
18.0
20.6
37.0
39.1
21.9
22.7
СО,
29.0
36.8
38.2
44.6
60.0
63.1
Найдены ароматические кислоты и продукты глубокой
деструкции, представленные низкомолекулярными органическими
кислотами, но содержание ароматических кислот в гиматомелановых
кислотах ниже, чем в гуминовых. ИК спектры дают аналогичные
с гуминовыми кислотами кривые [173, 448, 538]. Гиматомелано-
6 Л. Н, Александрова
81
вые кислоты также гетерогенны. Гетерогенность этой фракций
отмечена в работе Дж. Трояновского [551], она заметно
проявляется при применении различных органических
растворителей [444]. Все эти данные свидетельствуют о принадлежности
гиматомелановых кислот к группе гуминовых кислот и позволяют
рассматривать их как одну из фракций последней. Сам термин
«гиматомелановые кислоты» имеет, по-видимому, сейчас лишь
историческое значение, так как химическая гетерогенность
гуминовых кислот, являющаяся одним из специфических свойств
этой группы гумусовых кислот, и выделение большого количества
варьирующих по составу фракций не дают оснований для
сохранения только для одной из них специального наименования.
Ульминовые кислоты
Этот термин был введен также в прошлом столетии Г. Мульде-
ром для бурых органических веществ и используется в ряде работ
для характеристики более растворимой части гуминовых кислот.
По предложению И. В. Тюрина [301] он был заменен на «бурые»
гуминовые кислоты, которые отличаются от «собственно»
гуминовых (черных) кислот меньшим содержанием углерода и большим
содержанием элементов воды, меньшей емкостью обменной
сорбции оснований, пониженной степенью окисленности и более
высокой степенью подвижности. Многие дсследователи вслед за
предложением Д. С. Орлова [230] используют для характеристики
«собственно» гуминовых кислот термин «серые», сохраняя для
менее окисленной фракции гуминовых кислот, которую
именовали «ульминовой кислотой», термин «бурые» гуминовые кислоты,
характерные, по данным Д. С. Орлова, для большинства почв
широкого географического ареала: от подзолистых до
горнокоричневых.
Гумины
Термин «гумины», также предложенный еще в прошлом
столетии Я. Берцелиусом и Г. Мульдером для гумусовых веществ,
нерастворимых в щелочах, широко используется в настоящее
время для характеристики органических веществ, не
экстрагируемых из почвы при определении группового состава гумуса*
Сохранение этого термина в номенклатуре органических веществ
почвы нельзя признать целесообразным. Я. Берцелиус и Г. Муль-
дер, а затем и другие исследователи конца XIX—начала XX в,
рассматривали «гумины» (и «ульмины») как «изомерные»
разновидности гуминовых (и ульминовых) кислот, причем эта изомерия
проявлялась лишь в том, что гуминовые кислоты растворяются
в щелочах, а гумины — не растворяются, имея одинаковый эле^
ментный состав. В настоящее время такое определение не может
82
считаться удовлетворительным, а работы ряда ученых 1113, 206,
303, 316, 437, 511, 512] показали, что основными компонентами
гуминов являются гуминовые и фульвокислоты, выделяющиеся
после предварительной дополнительной обработки почвы
относительна крепкими растворами HN03 или смеси HC1 и HF.
Действительно, приведенные данные о характеристике
гуминов, выделенных из чернозема, подзолистой и торфяной почв
многократной обработкой 5 н. раствором HN03 и последующей
экстракцией 0.1 н. раствором NaOH (табл. 27), показывают, что
Таблица 27
Характеристика гуминов почвы (по: [206])
Показатели
Гумины, % от общего содержания
гумуса
Компоненты гуминов, %
гуминовые кислоты *
фульвокислоты *
целлюлоза
лигнин
Не идентифицировано *
Элементный состав, %
гуминовые кислоты
С
Н
О
N
СО ОН по емкости обмена при
рН6.5
фульвокислоты
с
н
О
N
Чернозем
23.7
47.0
18.0
6.0
7.0
19.0
59.7
3.3
34.0
3.0
460.0
52.0
5.2
38.7
4.1
Подзолистая
почва
26.8
31.0
10.0
9.0
10.0
43.0
57.8
5.0
34.7
2.5
370.3
50.0
6.5
38.9
4.6
Торф
22.8
9
3
10.0
17.0
61.0
57.3
5.5
34.8
2.4
300.0
49.6
6.3
39.8
4.3
Примечание. Звездочкой отмечены данные, вычисленные нами по материалам
О. А. Найденовой,
количество гуминов в различных почвах практически почти
одинаково и составляет 23—27%. Их состав оказался
многокомпонентным. Наряду с гуминовыми кислотами, составляющими в
различных почвах очень разную часть гуминов, обнаружены
фульвокислоты, целлюлоза и лигнин. Если в черноземе половина массы
гумина представлена гуминовыми кислотами, то в подзолистой
почве они составляют только 31%, а в торфяной — 9%. От
чернозема к торфяной почве также резко уменьшается содержание фуль-
вокйсдот и возрастает количество целлюлозы, лигнина и ряда
83
6*
других неидентифицированных веществ, находящихся,
по-видимому, на начальных стадиях гумификации. В подзолистой почве
негумусовым веществам в составе гуминов принадлежит 62%, что
также свидетельствует об условности этого термина. Гуминовые
кислоты и фульвокислоты гуминов по элементному составу
практически не отличаются от собственно гуминовых и фульвокислот,
выделяемых обычными методами; достаточно близки также
данные по содержанию групп СООН в гуминовых кислотах,
выделенных из гуминов и из самих гуминовых кислот.
Не менее показательны данные о характеристике продуктов
деструкции гуминов, выделенных из чернозема, солонца и солоди
(табл. 28). По элементному составу и продуктам деструкции гу-
мины не отличаются от гуминовых кислот, но общий выход
продуктов деструкции во фракции гуминов значительно ниже (на 50%)
по сравнению с фракцией гуминовых кислот. Наиболее вероятная
Таблица 28
Элементный состав и продукты деструкции гуминов
при обработке щелочным раствором
перманганата калия (по: [437])
Показатели
Элементный состав, %
С
Н
N
0+S
Продукты деструкции из 1 г,
мг
бензолкарбоновые кислоты
фенольные »
алифатические »
не идентифицировано
Чернозем
55.4
5.5
4.6
34.5
161
ИЗ
30
6
12
Солонец
56.3
6.0
5.1
32.6
186
132
42
2
10
Солодь
53.8
6.0
4.9
35.3
138
96
26
4
12
причина этого явления — наличие во фракции гуминов
значительного количества полугумифицированных веществ,
относительно легко окисляющихся перманганатом до конечных
продуктов минерализации. Исследования А. Перрода с соавторами
[477] показали сложность и неоднородность фракции гумина.
В их составе авторы выделяют три различные категории веществ:
высокополимеризованные гумусовые вещества, связанные с
глиной и железом, частично гумифицированные вещества и свежие
органические вещества, окруженные минеральными частицами
почвы.
Таким образом, термин «гумины» следует признать
неопределенным, включающим наряду с гуминовыми и фульвокислотами,
прочно связанными с минеральной частью почвы, а потому и
84
неэкстрагируемыми при обычных обработках, полугумифициро-
ванные продукты разложения растительных остатков, не
растворяющиеся в разбавленных кислотах и щелочах. К ним, как
известно, относятся целлюлоза, лигнин, воски, смолы, часть
гетероциклических соединений азота. На присутствие комплекса
полисахаридов в составе гуминов указывают Ф. Жакэн с
сотрудниками [430]. Особенно значительно содержание негумифицирован-
ных растительных остатков в пахотных почвах, получающих
значительное количество органических удобрений в виде навоза
и торфяных компостов. Наши исследования показывают, что после
внесения высоких доз органических удобрений (порядка 80—
100 т/га) всегда заметно возрастает величина нерастворимого
в кислотах и щелочах остатка органических веществ, который
в большей части состоит из полугумифицированных остатков
торфа. Называть этот остаток гумином нет никаких оснований.
Не менее убедительны многочисленные данные по групповому
составу гумуса, выполненные различными методами. В одном
и том же образце количество гуминов, т. е. неэкстрагированного
остатка органических веществ, в зависимости от способа
экстракции может колебаться от 30 до 60%.
Таким образом, следует отказаться от термина «гумины»;
считаем целесообразным заменить его термином «нерастворимый
остаток» с последующим изучением состава этого нерастворимого
остатка¦
Фульвокислоты
Термин «фульвокислоты» ввел С. Оден [472] вместо терминов
«креновые» и «апокреновые» кислоты Я. Берцелиуса, выделившего
их из железных охр и болотных руд [350]. Из почвы и торфа
эти кислоты впервые выделили Р. Герман [77] и Г. Мульдер [464].
С. Оден относил к этой категории гумусовых кислот желтоокра-
шенные вещества, остающиеся в растворе после осаждения гу-
миновых кислот; описывая эти кислоты, автор отмечал их
неоднородность, а также растворимость в воде, спирте и щелочах.
Интерес к фульвокислотам был восстановлен классическими
работами И. В. Тюрина [297, 298, 300, 302], в которых даны
новые методы их выделения, исследован состав, описаны
существенные свойства и показана роль в почвообразовании. И. В.
Тюрин определил группу фульвокислот как высокомолекулярные
оксикарбоновые (и содержащие азот) кислоты с эквивалентной
массой (по отношению к NH3) около 300, отличающиеся от группы
гуминовых кислот светлой окраской, более низким содержанием
углерода, растворимостью в воде и минеральных кислотах и более
значительной способностью к кислому гидролизу. В дальнейшем
суждения И. В. Тюрина о фульвокислотах как обязательной и
специфической части гумусовых кислот были развиты В. В.
Пономаревой [245—248], разработавшей оригинальные методы выде-
85
ления, изучившей их состав и свойства в основных типах почв
и показавшей их роль в подзолообразовании. После работ
Й. В. Тюрина и В. В. Пономаревой накоплен значительный
материал по характеристике этой группы гумусовых кислот; обзор
основных исследований можно найти в ряде публикаций [19,
143, 230]. Современные данные подтвердили выводы И. В.
Тюрина и В. В. Пономаревой о том, что фульвокислоты, так же как и
гуминовые кислоты, являются специфическими органическими
кислотами, образующимися в процессе гумификации
органических остатков в природе. Они обязательные компоненты
почвенного гумуса, входят в состав органической части торфа, сапропеля,
органических компостов, а также встречаются в водах многих
болот, рек и озер благодаря хорошей растворимости.
Элементный состав и функциональные группы. Выделенные
из почвы препараты фульвокислот окрашены в светло-бурые
тона, а растворы их в зависимости от концентрации и степени
фракционирования имеют соломенно-желтую, светло-бурую и
оранжево-вишневую окраску. Они хорошо растворимы в воде,
кислотах и многих разбавленных щелочных растворах, а также
в ряде органических растворителей, причем водные растворы их
характеризуются резко кислой реакцией (рН 2.8—3.5).
Элементный состав их заметно отличается от элементного состава гуми-
новых кислот и колеблется в следующих пределах, %: С 40—52,
Н 4-6, N 2-6, О 42-52.
Следует отметить, что точная характеристика элементного
состава фульвокислот до настоящего времени затруднена
вследствие очень разных методов их выделения и очистки. Достаточно
указать, что если М. Шнитцер [495] для фульвокислот горизонтов
Вь ряда подзолистых почв приводит колебания в содержании С
от 44 до 49%, то Ф. Мартин с сотрудниками [459] для этого же
типа почв — лишь 37—40%.
Наиболее полные сводные данные Д. С. Орлова по
элементному составу фульвокислот верхних горизонтов различных почв
1230] подтверждают более ранние заключения И. В. Тюрина
о пониженном по сравнению с гуминовыми кислотами
содержании С и повышенном количестве Н, хотя возможные колебания
элементного состава в этой группе гумусовых кислот даже в
пределах одного типа почв могут быть значительны (табл. 29 и 30).
Д. С. Орлов отмечает возможность выделения двух групп
фульвокислот по элементному составу: для первой группы характерно
повышенное содержание С (45—47%), для второй — среднее
(41—43%). При этом как в первую, так и во вторую труппу
попадают фульвокислоты почв различного генезиса, но,
по-видимому, элементный состав фульвокислот достаточно четко
коррелирует (в обратной связи) с относительным содержанием гуми-
новых кислот. В почвах первой группы (подзолистые/
красноземы, луговые), для которых характерно меньшее количество
гуминовых кислот в составе гумуса, содержание С в фульвокисло-
86
Таблица 29
'Средний элементный состав фульвокислот, % на сухое беззольное'
вещество (по: [230])
Почвы
Подзолистые и дерново-
подзолистые
Бурые лесные, буроземы
Серые лесные
Черноземы, каштановые
Сероземы
Коричневые
Красноземы, красноцвет-
ные
Горно-луговые
Луговые, пойменные
Темноцветные, рендзины
Гумусо-аллофановые
С
45.8
42.3
44.9
42.9
41.4
41.1
46.3
43.0
44.5
45.0
48.9
н
4.5
4.8
4.8
4.7
5.9
3.7
3.5
5.0
4.7
4.5
4.3
О
46.5
49.3
47.2
48.9
50.0
52.3
47.1
48.2
46.7
47.7
44.5
N
3.2
3.6
3.1
3.5
3.7
2.9
3.1
3.8
4.1
2.8
2.3
н
с
0.10
0.11
0.11
0.11
0.12
0.09
0.08
0.12
0.11
0.10
0.09
о
с
1.02
1.16
1.05
1.14
1.21
1.27
1.02
1.12
1.05
1.06
0.91
с
N
14.3
11.8
14.5
12.3
11.2
14.2
14.9
11.3
10.9
16.1
21.2
Пределы
колебаний
С
39.0-52.5
35.7—48.9
39.3-50.5
34.6-51.3
35.5-47.4
39.1—50.7
42.6-55.1
Таблица 30
Средний элементный состав фульвокислот, ат.% (по: [230])
Почвы
Подзолистые и
дерново-подзолистые
Бурые лесные, буроземы
Серые лесные
Черноземы, каштановые
Сероземы
Коричневые
Красноземы, красноцветные
Горно-луговые
Луговые, пойменные
Темноцветные, рендзины
Гумусо-аллофановые
с
33.4
30.8
31.9
30.9
29.4
32.2
36.6
30.2
31.9
32.9
! 36.0
н
39.3
40.9
40.9
40.6
41.7
34.9
33.4
42.1
41.2
39.4
37.9
о
25.3
26.1
25.3
26.3
26.7
30.9
27.9
25.4
24.4
26.0
34.7
N
2.0
2.2
1.9
2.2
2.2
2.0
2.1
2.3
2.5
1.7
1.4
н
с
1.18
1.33
1.28
1.31
1.42
1.08
0.91
1.39
1.27
1.20
1.05
о
G
0.76
0.85
0.79
0.85
0.91
0.96
0.76
0.84
0.79
0.79
0.68
с
N
16.6
13.9
17.0
14.3
13.3
16.3
17.6
13.2
12.8
18.8
25.5
со
+0.34
+0.36
+0.32
+0.34
+0.38
+0.73
+0.65
+0.28
+0.30
+0.67
+0.32
тах заметно повышено. В почвах второй группы с высоким
относительным содержанием гуминовых кислот (черноземы, сероземы,
каштановые, коричневые, горно-луговые) фульвокислоты
содержат меньшее количество С и наиболее окислены.
Природа фульвокислот исследована в нашей лабораторий
Н56, 157, 207, 209—213, 215]. Показано, что элементный состав
фульвокислот варьирует как в различных типах почв, так и
в разных горизонтах одного профиля (табл. 31). Фульвокислоты
дерново-подзолистой почвы характеризуются повышенным
содержанием С, минимально оно в фульвокислотах серозема. Для
87
н
о
ч
о
я
о
а
ч
Я
к
рцю
ф
Я о
if
II
Я ?
^ яя
Я"
Я
ч
vo
ев
Н
н л
к *>
я 8
Л со
ф Ф
НЮ
Я о
Sl
г «
я я
««о
та
«а
я
I
0>
(ОН)
СООН
3
о|й
ою
up
К|и
U №
о
Й
и
и
Горизонт
Почва
СО Ю О* 00 СО СО СО !>. Ю
СО CD 00 С5ЮЮ 00 СО G5
t>- t>- CD Ю CD 00 С-~ 1> 1>
00 00 -И ЮГ^Ю CDCSIUO
00 ОЭ СО СОСЧКМ ЮОЫЪ
(MCOsf* <МСО<М ^^ЧГ
СО СО -чН t^SflO 00 00-О5
СЧ-*ч СО С^СОЮ СОООСО
odd ооо ооо
+++ +++ +++
ОЭ00Ю SPOOS СООЭхч
OsF<M СОСЧСО OrHOi
CM •*¦» тН -ч-l "*Н тН тН -^Н "ЧН
00COl>- COt^UO (МОСТ»
cD t^- о* оо <л о ем-чр ю
ООО О О -чн «?н -чн -чн
t^«*4<N Ю<МчР 0505СО
*tf st< О *r<<Z>OS I>» CO CO
T^Hri <НгнО ООО
Ю 05 sf< СМ-тчсО sfrHO
"чн са со thcdlo oqoio
тНтН-ri гНтН«н -чН CM CM
|>.Ю"чН OOCsl-tf sfQO
00l>CD CDCDCD LQNlW
odd odd odd
^rHsf ЧГСОЬ» -чч stf CO
со со d iodod о«чн"о
<M<M<M <NCSKN COCOCO
NT CSJ CM C4J0505 СО-чН-чП
odd счсч^н cxicMeM
40.1
42.2
44.4
42.9
43.9
42.3
43.8
44.2
48.1
oocdo ^^"^ oqcocsi
vjicvit^ dt^i> со см d
COCOCM CM<MC\1 CXICM^
<<& <JpQPQ <?CQPQ
Дерново-подзолистая
Чернозем типичный
Серозем »
OS
фульвокислот чернозема получены средние данные по
элементному составу. Степень окисленности также возрастает от
фульвокислот дерново-подзолистых почв через черноземы к сероземам.
Достаточно четко выявляются различия и в пределах одного
профиля по генетическим горизонтам, вскрывая особенности
генезиса каждого типа почвы. Очень заметно и закономерно
изменяется элементный состав фульвокислот в профиле дерново-
подзолистой почвы: вниз мигрирует все более окисленная и менее
обуглероженная фракция. В профиле чернозема и серозема
элементный состав фульвокислот более однороден, что обусловлено,
по-видимому, особенностями гумусообразования в этих типах
почв. В черноземе и сероземе гумификация идет во всей мощной
корнеобитаемой части; следовательно, фульвокислоты в условиях
непромывного типа водного режима и недостаточной влажности
в значительной степени фиксируются на месте образования.
В профиле дерново-подзолистой почвы в условиях промывного
водного режима условия для миграции фульвокислот наиболее
благоприятны, причем в процессе миграции происходит своеобразное
фракционирование этой категории гумусовых кислот, и в нижнюю
часть профиля поступают наиболее окисленные и менее обугле-
роженные фракции.
Фульвокислоты, так же как и гуминовые кислоты, содержат
ряд функциональных групп, среди которых наибольшее значение
имеют кислые группы, определяющие реактивную способность
этой группы гумусовых кислот. Кислые функциональные группы
в основном представлены карбоксильными и фенолгидроксиль-
ными, водород которых при определенных условиях замещается
на металл. Еще в работах И. В. Тюрина [298] и В. В.
Пономаревой [245] было установлено наличие этих групп. Следует, однако,
заметить, что определение количества кислых групп, активных
в тех или иных интервалах реакции, затруднено по ряду причин.
Невозможно определять их количество буферными растворами
хлоридов кальция или бария вследствие растворимости этих
солей фульвокислот. Наиболее пригодными оказались методы
потенциометрического и обратного высокочастотного титрования
в неводных средах [81, 82, 207, 212], а также новый метод с бор-
гидридом натрия, использованный О. А. Найденовой и С. Р. Ко-
рюшкиной [213]. Полученные в нашей лаборатории в последние
годы данные о количестве карбоксильных и фенолгидроксильных
групп методом обратного высокочастотного титрования в водно-
ацетоновой среде свидетельствуют о том, что их сумма выше
таковой в гуминовых кислотах и колеблется от 800 до
1250 мг.экв./100 г (табл. 31). Она минимальна для фульвокислот
черноземов и максимальна в фульвокислотах серозема, при этом
в составе кислых групп доминируют фенольные гидроксилы,
на долю карбоксилов приходится от 225 мг-экв. в фульвокислотах
черноземов до 450—500 мг-экв./ЮО г в сероземе. Лишь в пределах
профиля дерново-подзолистой почвы отчетливо выражено увелц-
89
230 232 242 254 264
296 300
333 340
360 нм
Рис. 17. Дифференциальные кривые УФ спектров фульвокислот из разных
почв.
1 — дерново-подзолистая, 2 — краснозем, 3 — чернозем, 4 — каштановая, 5 и 6 —
фульвокислоты фракции I (ММ > 4000) и фракции II (ММ < 4000) из
дерново-подзолистой почвы.
чение количества групп СООН при одновременном снижении
содержания (ОН) с глубиной.
Очень разные данные по содержанию кислых групп в фульво-
кислотах из одного типа (подзола) можно найти в работах многих
авторов [309, 455, 459, 529, 562]. Следует иметь в виду, что в
зависимости от жесткости обработки в фульвокислотах, так же как
и в гуминовых кислотах, обнаруживается неодинаковое
содержание кислых групп в результате различий в величине рК. Так,
например, методом потенциометрического титрования
фульвокислот 0.1 н. растворами КС1 и НС1 в 0.1 М КС1 обнаружены три
категории кислых групп [524]: очень сильные (ионизированы
при рН < 2) — сульфоновые (—S02OH), кислые (ионизированы
при рН ~ 7) — карбоксильные (—СООН) и очень слабые
(ионизированы при рН > Ю) — группы —(ОН) и —SH.
Очень четко разнокачественность фенольных гидроксилов
показана О. А. Найденовой [210] при применении метода УФ
спектроскопии в модификации Г. Аулин Эрдтмана в сочетании
с методом гельхроматографии (рис. 17). На дифференциальных
кривых УФ спектров отчетливо видно несколько полос
поглощения, свидетельствующих о наличии групп (ОН) различной сте-
90
пени кислотности (т. е. с разными значениями рК). Для
фракции I фульвокислот дерново-подзолистой почвы (ММ > 4000)
обнаруживается пять характерных полос поглощения: в области
234, 280, 300, 316 и 338 нм; для фракции II (ММ < 4000) — шесть
полос: в области 240, 256, 262, 280, 286 и 300 нм. Аналогичные
данные были получены для фульвокислот чернозема,
каштановой почвы и краснозема.
Необходима поэтому строгая стандартизация как метода
получения и очистки препаратов, так и способов определения
кислых групп. Наряду с кислыми функциональными группами
в фульвокислотах обнаружены метоксильные группы в
значительно больших количествах, чем в гуминовых кислотах,
показано наличие карбонильных группировок, спиртовых гидрокси-
лов, хинонных групп [441, 455, 456, 494]. Все эти, к сожалению
единичные, определения не дают еще возможности
характеризовать все функциональные группы^ их количество,
местоположение в молекуле и функции.
Природа и строение молекулы. За последние годы получен
ряд данных о природе молекулы фульвокислот методами частичной
деструкции с последующей идентификацией продуктов
тонкослойной или газожидкостной хроматографией, а также при
помощи УФ и ИК спектроскопии, рентгенографии и дериватогра-
фии. Данные о продуктах частичной деструкции фульвокислот
(табл. 32) показывают, что в них, так же как и в гуминовых
кислотах, найдены разнообразные компоненты: аминокислоты,
углеводы, фенольные производные, ароматические соединения, —
что свидетельствует о принципиальной однотипности природы
молекулы фульвокислот и гуминовых кислот. Указывая на эту
однотипность, авторы в то же время отмечают иные соотношения
между ароматической и алифатической частями молекулы [343,
437, 509]. Д. С. Орлов [230], проанализировав литературу
последних лет и дополнив ее своими материалами, показал, что
в молекуле фульвокислот доминирует алифатическая часть,
представленная аминокислотными и углеводными компонентами.
Аналогичные мнения высказывают и исследователи,
базирующиеся в основном на частичной деструкции препаратов
фульвокислот при окислении их щелочным раствором перманганата
калия [343, 423, 496, 498].
Лишь в работе Р. Хансена и М. Шнитцера [424] отмечается
большая ароматичность ядра молекулы фульвокислот по
сравнению с гуминовыми кислотами. Данные о продуктах частичной
деструкции фульвокислот при окислении КМп04 в щелочной
среде (табл. 33) также подтверждают значительно более низкий
выход бензолкарбоновых кислот из препаратов различных почв.
Если гуминовые кислоты, как мы отмечали ранее, дают при
окислении до 20% бензолполикарбоновых кислот, то в фульвокислотах
их выход не превышает 8%, причем максимален он для
фульвокислот дерново-подзолистой почвы. Следует отметить обратную
91
Таблица $2
Продукты и методы частичной деструкции фульвокислот
по данным разных авторов
Продукты деструкции
Метод деструкции
Источник
Аминокислоты: 17 аминокислот
алифатического и ароматического ряда
Азотсодержащие гетероциклические
соединения: ароматические амиды
Углеводы: полисахариды, гексозы,
пентозы, уроновые кислоты, амино-
сахара
Фенолы и их производные:
полифенолы, пирокатехин, фенолальдегид
Ароматические соединения: три-, тет-
ра-, пента-, гексабензолкарбоновые
кислоты, феноксикарбоновые кислоты,
фенольные кислоты, конденсированные
ароматические кольца
Углеводороды от С4 до С24
Алифатические кислоты дикарбоновые,
жирные (Св—С18)
Кислотный гидролиз
Щелочной »
Кислотный »
Щелочной »
Щелочной гидролиз,
окисление КМп04
в щелочной среде,
щелочная плавка
Масс-спектрометрия,
окисление в
хлороформе
Окисление КМп04
в щелочной среде
[225, 226, 343,
407, 419, 420,
563]
[407, 468]
[157, 230, 376,
407, 419, 427]
[469, 496, 519]
[230, 343, 423,
424, 436, 437,
467, 496, 499]
[436, 474]
[436, 437, 467,
474, 496]
Таблица 33
Продукты частичной деструкции фульвокислот при окислении
КМп04 в щелочной среде (по: [156])
Почвы
Дерново-подзолистая
Чернозем
Серозем
Горизонт
А,
А?
в
А
в.
в2
А
Продукты окисления, %
к сухой беззольной
массе
бензол-
поликар-
боновые
кислоты
8.4
7.2
5.4
5.2
5.0
5.3
1.4
лекулярные
кислоты
48.6
50.7
53.8
52.2
53.5
52.9
55.4
со2
43.0
42.1
40.8
42.6
41.5
41.8
43.1
Углеводные
компоненты, %
13.8
14.0
15.2
16.5
15.6
14.9
18.0
?0.001% С
465
0.015
0.017
0.005
0.004
0.003
0.005
0.002
92
Рис. 18. ЙК спектры (см^1) фульвокислот.
1—3 — из дерново-подзолистой почвы (1 — гор. Аь 2 — гор. А2, 3 — гор. В), 4 и 5 —
из чернозема (4 — гор. А, 5 — гор. АВ).
корреляцию между выходом бензолкарбоновых кислот и содержанием
углеводных компонентов в молекуле. Содержание углеводных
компонентов максимально в фульвокислотах серозема и
чернозема, где, по-видимому, гумификация и минерализация
растительных остатков проходят наиболее полно. В дерново-подзолистых
почвах фульвокислоты по составу и свойствам приближаются
к гуминовым кислотам.
ИК спектры в целом аналогичны таковым для гуминовых
кислот, но характеризуются большей сложностью (рис. 18).
В них подтверждаются участие ароматических структур (полоса
1625—1610 см"1), большое количество алифатических цепей
(полоса 1720—1700 см"1), а также ряда полос, характеризующих
93
наличие амидных, спиртовых и карбонильных группировок. Все
это свидетельствует о наличии ароматических и алифатических
компонентов в молекуле фульвокислот и позволяет
идентифицировать функциональные группы и некоторые детали строения
молекулы. Применение рентгеноструктурного анализа позволило
исследователям также высказать предположение о меньшей роли
ароматических группировок в молекуле фульвокислот по
сравнению с гуминовыми кислотами [230, 343, 409, 495, 529]. Многие
авторы [122, 320, 442, 540] используют ДТА и ТВ для
исследования структуры молекул фульвокислот, что также свидетельствует
о доминирующей роли алифатических группировок в молекуле,
хотя выявить отдельные группировки в ней этими методами не
удается.
Азот фульвокислот. Материалов по формам азота в фульво-
кислотах все еще недостаточно. За последние годы опубликовано
лишь несколько работ, позволяющих дать сжатую характеристику
азотсодержащей части молекулы [225, 226, 407, 438].
Азотсодержащая часть молекулы фульвокислот значительно
легче гидролизуется при действии минеральных кислот по
сравнению с азотистой частью гуминовых кислот, что подтверждает
доминирующее значение аминокислотных остатков (а также,
вероятно, и пептидных группировок) в молекуле* Действительно,
если азотсодержащая часть гуминовых кислот в зависимости
от типа почвы гидролизуется на 40—60%, то азот фульвокислот —
на 70-75%.
В составе азотсодержащих компонентов обнаружены формы,
аналогичные таковым в гуминовых кислотах. Найдены аминные,
аммонийные, гетероциклические формы, а также азот аминоса-
харов. Некоторая часть азота остается неидентифицированной.
Соотношение между этими компонентами иное по сравнению
с гуминовыми кислотами. В молекуле фульвокислот доминируют
аминокислотные и аммонийные формы азота, составляющие до
70% всей массы азота [407] (см. с. 95).
На долю гетероциклических компонентов и
неидентифицированной части азота приходится меньшая, но все же значительная
доля азота: ~40%. По набору аминокислот фульвокислоты ана-
.логичны гуминовым кислотам. В их составе найдены аспараги-
шовая, глютаминовая кислоты, серии, глицин, треонин, р-аланин,
явалин, метионин, лейцин, лизин, гистидин, аргинин, пролин,
'тирозин, фенилаланин, цистин [225, 420]. На аминокислоты
^циклического ряда в фульвокислотах приходится только 3—4%
ют всей суммы их, тогда как в гуминовых кислотах их
относительная роль достигает 10%.
ММ и гетерогенность фульвокислот. Данные о ММ молекул
(фульвокислот очень разноречивы даже при применении одного
метода определения. Все исследователи, используя метод гель-
«фильтрации, отмечают большую полидисперсность фульвокислот.
1Из любого препарата выделяется несколько фракций с очень
94
Основные формы азота в гуминовых кислотах и фульвокислотах
ГК
N -аминов
N -нерастворимый
N -гетероциклический
ФК
N «аминокислотный
/ 45.5% / ДО -Аминов
N -гетеро- f к. 16.6: I
циклическийЛ 22.5 / ^v J
N. /l5.4^X
N -нерастворимый
различными ММ.. Таек, например, Н. Роуль и соавторы [481],
йейбльзуя сефадексы Г-50 ж Г-75 для фульвокислот горизонта
h-., подзола, называют величины от 4500 до 100 000. Аналогичные
даййше приведены в работе А. А. Юхнина и Д. С. Орлова [330],
получивших две фракции фульвокислот дерново-подзолистой,
луговой и каштановой почв, а также чернозема с ММ для
фракции I 10 000—42 000 и для фракции II 5000—6000. Фракция II,
по данным авторе неоднородна и при дополнительной обработке
делится еще на 2—3 шодфракции. О. А. Найденова с
сотрудниками [211], фракционируя препараты фульвокислот чернозема,
краснозема и дерново-подзолистой почвы на системе сефадексов
Г-25 Г-50 и Г-75, получили из каждого препарата по три фракции
с колебанием ММ 5000-30 000 (фракция I), 30 000-70 000
(фракция II) и более 70 000 (фракция III). Кроме этих фракций иа
каждого препарата выделяется некоторое количество
низкомолекулярной фракции с ММ меньше 1000. Следует отметить, что
все фульвокислоты были выделены с использованием
активированного угля по несколько измененному О. А. Найденовой
методу Форсита, но установить четкие закономерности в
колебаниях ММ не удалось. Наименьшее количество
низкомолекулярных фракций получено для фульвокислот подзолистых почв*
N
-аминокислотный
95
максимальное содержание их типично для серозема [211]. Иной
порядок величин приводят К. Гох и М. Райд [408]; по их данным,
в фульвокислотах, выделенных из песчаных почв Новой
Зеландии, доминируют фракции с ММ более 100 000, причем эти
величины получены также методом гельфильтрации. Очень высокая
степень полидисперсности фульвокислот показана в тех работах,
где авторы, используя систему сефадексов, выделили несколько
фракций в препаратах фульвокислот подзолистой почвы,
чернозема и краснозема с ММ от 170 до 11 250 [117, 119, 164, 228].
При этом если в фульвокислотах чернозема и краснозема почти
половина массы фульвокислот представлена крайне
низкомолекулярной фракцией (ММ 170), то в подзолистой почве основная
масса фульвокислот характеризуется значительно более
высокой ММ: 2000—11 000. Исследование ИК спектров не обнаружило
существенных различий, что, по мнению А. И. Карпухина [116],
свидетельствует о близкой химической природе выделенных
фракций. Многие исследователи, отмечая резко выраженную
полидисперсность фульвокислот, указывают на существенные
различия в химической природе отдельных фракций, т. е.
подтверждают их химическую гетерогенность. Так, С. С. Драгунов
и Б. Г. Мурзаков [98] выделили 13 фракций из фульвокислот
чернозема с очень различным элементным составом и не менее
различным содержанием карбоксильных групп (табл. 34). По данным
Таблица 34
Элементный состав, % на беззольное вещество,
и функциональные группы, мг«экв./100 г,
фракций фульвокислот чернозема (по: [98])
Фракция
1
2
4
5
6
7
9
10
11
12
13
с
47.3
66.7
36.5
34.2
45.1
51.6
41.2
40.6
31.1
42.7
25.6
н
4.8
6.8
6.3
4.6
6.9
6.5
5.0
5.1
9.0
6.2
4.4
N
3.6
2.9
0.9
1.7
5.5
1.5
4.3
2.0
5.8
4.6
1.4
о
44.2
23.6
56.3
59.6
42.5
40.4
49.6
52.2
54.1
46.8
68.6
Зола, %
0.9
—
1.1
1.7
4.8
5.2
2.9
[2.5
2.8
3.2
3.1
соон
240
—
—
586
326
615
—
761
402
534
634
(ОН)
80
—
—
128
54
412
—
234
56
130
81
осн3
67
-,-
—
98
464
217
—
719
115
405
47
Выход
фракции,
мг на 100 г
почвы
70
30
36
106
286
28
20
70
120
250
44
О. А. Найденовой и С. Р. Корюшкиной [210, 213, 214], разные
фракции фульвокислот характеризуются различными по
кислотной силе фенолгидроксильными группами (рис. 17). Л. Турчиник
р Дж. Оадес [553], выделившие с помощью ультразвука из фульво-
96
кислот шесть фракций, указывают на неоднородность их по ряду
параметров. Неоднородность элементного состава и ИК спектров
отдельных фракций фульвокислот, выделенных из одного и того же
препарата, отмечают многие исследователи [164, 338, 436, 453,
455, 542, 563].
Причины столь резко выраженной полидисперсности по ММ
и химической гетерогенности фульвокислот все еще неясны.
Д. С. Орлов [230] считает, что одна из них — наличие
низкомолекулярных примесей в препаратах, а также различия в
методах выделения и очистки отдельных фракций. По-видимому,
фракции с ММ примерно 150—700 нельзя относить к категории
фульвокислот, так как они не являются высокомолекулярными
соединениями и представляют собой, вероятнее всего,
низкомолекулярные оксикарбоновые кислоты типа полиуроновых.
Практически отсутствуют данные о форме и размерах молекул
фульвокислот. Лишь М. Шнитцер и X. Кодама [501] на основании
электронно-микроскопических наблюдений указывают, что фульво-
кислоты при рН около 2.5 дают сфероиды с диаметром 1.5—2.0 нм,
а при рН 3.5 образуют губчатые структуры толщиной в 10—30 нм.
Все эти, все еще крайне ограниченные материалы по
характеристике фульвокислот свидетельствуют о необходимости
дальнейшего детального изучения их основных параметров, и прежде
всего вещественного состава, при условии четкой
стандартизации как методов выделения и фракционирования препаратов,
так и приемов исследования природы и структуры молекулы.
Промежуточные продукты разложения 2
В процессе разложения и гумификации различных
органических остатков в почве образуется сложная система промежуточных
продуктов, частично расходуемая на процессы гумификации,
частично усваиваемая гетеротрофными микроорганизмами или
постепенно минерализующаяся до конечных продуктов. Запас этих
соединений в почве непрерывно пополняется за счет микробного
синтеза и корневых выделений растений, а также трансформации
и взаимодействия этих веществ между собой. Все это
обусловливает чрезвычайно сложный химический состав системы, несомненно
участвующей в процессах почвообразования.
К сожалению, вещественный состав промежуточных продуктов
разложения изучен значительно слабее, чем природа гумусовых
веществ. В первом приближении эта система промежуточных
продуктов разложения может быть разделена на две категории
веществ: группу высокомолекулярных продуктов разложения и
группу низкомолекулярных соединений; при этом имеющиеся
5 Раздел написан М. В. Новицким.
7 Л. Н. Александрова 97
в литературе данные в основном относятся к характеристике
низкомолекулярных соединений. О природе высокомолекулярных
соединений мы практически не имеем достоверных материалов.
Следует отметить, что точно установить наличие в почве тех или
иных групп свободных индивидуальных соединений очень трудно.
Исследователи обычно вынуждены прибегать к общепринятым
методам кислотного или щелочного гидролиза с последующим
определением в гидролизате тех или иных индивидуальных
соединений. Поэтому остается неизвестным, какая часть веществ
действительно находится в почве в свободном состоянии, а какая
входит в состав более сложных органических соединений.
Азотсодержащие соединения. Среди азотсодержащих
соединений — промежуточных продуктов разложения белков и нуклео-
протеидов — в почве обнаружены аминокислоты, аминосахара,
амины, нуклеиновые кислоты [45, 146, 230, 353, 397, 475, 523].
Наибольшее количество данных получено по составу
азотсодержащих соединений в различных гидролизатах из почвы и гумусовых
кислот, а также из плазменных белков и гумусоподобных
продуктов жизнедеятельности грибов. Материалы о содержании и составе
свободных азотсодержащих соединений в почве ограниченны и
относятся лишь к категории низкомолекулярных соединений.
Известны работы о наличии свободных аминокислот в почвах
Белоруссии [168, 169], о выделении свободных аминокислот из
почв Украины [194] и в других регионах [115, 305, 476]. Несмотря
на разные методы выделения и идентификации, авторы этих
исследований отмечают качественно однотипный состав аминокислот
в различных почвах. В свободном состоянии обнаружены аспараги-
новая, глютаминовая кислоты, аланин, серии, валин, лейцин,
лизин, пролин, глицин, треонин, гистидин, тирозин и фенилаланин,
т. е. практически почти все аминокислотные компоненты белков.
Данные о количественном содержании свободных аминокислот
в почвах очень разноречивы: их сумма в минеральных почвах
колеблется от 1—3 до 50—70 мг/кг. Лишь в болотных почвах
содержание свободных аминокислот резко возрастает, достигая
ЮО—400 мг/кг [169, 194, 305]. Эти расхождения обусловлены, по-
видимому, не только разным содержанием свободных аминокислот
в почвах, но и различиями в методах их выделения, очистки и
идентификации.
В последнее время появились работы о количестве и составе
индивидуальных азотсодержащих веществ в природных водах,
куда эти соединения в значительной степени мигрируют из почвы.
А. Д. Семенов с сотрудниками [274] провели цикл исследований
в водах главнейших рек, озер и водохранилищ СССР. Свободные
аминокислоты найдены в природных водах; общее содержание
их в водах рек в среднем составляет 3—12 мкг, озер — 2—7.5,
водохранилищ — 3.5—19 мкг аминного азота в 1 л воды.
Содержание белковых веществ в этих водах, как правило, в 10—20 раз
больше, чем свободных аминокислот. Концентрация свободных
98
аминокислот в грунтовых и родниковых водах также невелика:
в среднем 2—7 мкг на 1 г аминного азота [319].
Очень ограниченны данные по содержанию аминокислот в
почвенных растворах. В. Ф. Купревич и Т. И. Щербакова [169]
обнаружили в водах, насыщающих торфяно-болотную почву, от 4 до
220 мг аминного азота в 1 л воды; Р. И. Царева [318] в
аналогичных водах -^ от 6 до 12.5 мкг аминокислот в 1 л.Шо данным М.В; Но^-
вицкого [221 ], в лизиметрических водах, мигрирующих в
профиле дерново-подзолистых суглинистых почв, содержится в
различных горизонтах и в разные периоды наблюдений от 7 до
100 мкг на 1 л аминного азота. По данным И. EL Барановского
[48], концентрация аминокислот в лизиметрических водах из
пахотного горизонта дерново-подзолистой суглинистой почвы
составила в зависимости от доз вносимых удобрений 57—
278 мкг/л.
Такие же данные можно обнаружить и в других работах, что
свидетельствует о наличии в почвах очень незначительных
количеств свободных аминокислот, так как они интенсивно
используются почвенной микрофлорой как источник питания и энергии
или минерализуются, образуя аммиак. Это подтверждают и
данные, полученные в последнее время в нашей лаборатории (табл. 35):
на всем протяжении процесса гумификации растительных остатков
различного химического состава содержание свободных
аминокислот в водной вытяжке из этих образцов очень невелико и
составляет около 0.5—0.01%; лишь из свежих растительных
остатков водная вытяжка извлекает до 5% аминокислот. При этом
очень большое влияние оказывает химический состав
разлагающихся остатков: в максимальных количествах свободные
аминокислоты экстрагируются водой из наземной массы клевера и
листьев дуба; абсолютное количество аминокислот непрерывно
уменьшается и через год составляет лишь тысячные доли процента
к сухой беззольной массе гумифицирующихся растительных
остатков. Можно полагать, что незначительное количество
аминокислот в конце срока гумификации обязано своим
происхождением разложению плазмы отмирающих микроорганизмов»
Аминокислоты, внесенные в почву, довольно быстро разлагались,
образуя аммиак и нитраты [191].
Углеводы. Наличие разных углеводов в почвах отмечается
многими исследователями, но в большинстве работ изучен
углеводный состав кислотных гидролизатов из почв. В продуктах
кислотного гидролиза обнаружено значительное количество
углеводов: от 5 до 30% от общей массы органических веществ.
Обстоятельная сводка по углеводам почв дана в работах как советских,
так и зарубежных авторов [239, 397, 414, 421, 465, 471, 541].
Идентификация углеводов в кислотных гидролизатах ряда почв
позволила обнаружить практически все группы этого класса
соединений: разнообразные моносахариды, дисахариды, амино-
сахара, сахарные спирты, уроновые кислоты и полиурониды, пенто-
99
7*
о
и
В
8
>>
s
И"
к
н
Я
»
gzi
© н
со ^и
О Сб
VO
сб
Н
N О
* Ь4
И 3
В №
ев Ч
ф
а н
Kg
°&
vo "
о
»
и
S
В
&
п
cd
Рн
о
и
ф
№
сб
о,
CD
о
и
1 и
1 а >>
«о
S В
нольной
гумифи]
ния фе
сроки
ф ф
3»
•J
Л
ные
со
cd bt
-> о
w
as
1*
|8
5 я
si
Я&!
бодные
сроки
Сво
360
о
со
с-
©
о
8
8
с—
о
PS
cd
о
«
ф
1 о
Я
§
о
О
°9
со
\°°.
об
см
t^-
со
о
ю
чг
«*
5
ю
об
ю
•ч-н
ю
ОЭ
о
чн
LO
OS
со
in
о
со
**
о
00
ч-н
1
ф
ч
н
ч
ф
«
о
«
>
<
**
со
LO
со
ю
ю
см
ю
со
со
ю
ю
со
чН
чР
чн
t^
о
см
со
со
чН
р
о
-*ч
о
см
1С
ч—|
|>
о
со
об
-чН
ю
00
о
ч-ц
еб
О,
CD
я
CD
ч
к
Сб
о
о
Сб
S
ее
сб
и
S
CD
со
Сб
р
и
см
со
IO
СО
ЧР
о
00
¦чЧ"
00
о
со
со
со
^н
см
-чН
d
•*н
d
Ч*<
см
-чН
со
d
со
о
чрН
со
о
со
о
см
со
4F
t-
сб
%
t=C
«
л
н
о
в
t
н
ю
ю
с*
см
с-
ю
SJ*
4ji
4i«
00
ю
см
¦чН
чг
чеН
со
ю
о
о
ю
со
ЧИ
см
1
а
CD
ч
CJ
со
о
ел
чр
со
чн
СО
о
CQ
о
(-1
>>
ч
а
К
0
CD
CD
>&•
о
а
н
а
и
о*
о
&
m
Б
i
S
О
«
О
и
«а
cog
ф й
я«
& о
сб л
*3
заны, гексозаны и целлюлозу. В. Фляйг 1397 ] приводит следующие
углеводы, обнаруженные в гидролизатах разных почв:
Группа углеводов Идентифицированные углеводы
Моносахариды Гексозы: глюкоза, галактоза, манноза,
фруктоза; пентозы: арабиноза, ксилоза, рибоза, рам-
ноза, фу коза
Дисахариды Сахароза, целлобиоза
Олигосахариды Целлотриоза
Полисахариды Целлюлоза, гемицеллюлоза
Уроновые кислоты Глюкуроновая, галактуроновая, алигино-
вая кислоты
Сахароспирты Маннит, инозит
Амипосахара Глюкозамин, галактозамин, N-ацетил-О-
глюкозамин
Метилированные сахара 2-0-метил-Б-ксилоза, 2-0-метил-Б-араби-
ноза, 2-О-метилрамноза, 4-О-метилгалак-
тоза
Совершенно очевидно, однако, что основная масса углеводов
является компонентом гумусовых веществ. Свободные углеводы
в почве содержатся в значительно меньших количествах, их доля
в среднем, как указывают Д. С. Орлов с сотрудниками [232],
составляет около 1% всей органической части минеральных почв.
В составе свободных углеводов, переходящих в водные или
спиртовые вытяжки, найдены преимущественно моносахариды,
составляющие лишь сотые доли процента от всей массы
органических веществ [102, 338, 416, 422]. В некоторых работах [349,
383 ] отмечено наличие в почве полисахаридных фракций,
содержание которых в случае применения мягких методов выделения также
не превышает 1—2%. Природа и состояние в почве этих фракций
неясны, но имеющиеся в литературе данные свидетельствуют о их
гетерогенности по ММ, которые колеблются от 4000 до 450 000,
а также химической гетерогенности, обнаруживаемой методами
гельфильтрации, ультрацентрифугирования и хроматографии.
В составе полисахаридной фракции найдены остатки моносахаров,
полиуронидов, аминосахаров, целлюлозы, гемицеллюлозы;
методами ИК спектроскопии в этих полисахаридах обнаружены
карбоксильные, альдегидные, гидроксильные и метильные группы
[239]. Но состояние этой полисахаридной фракции в почве все
еще не представляется ясным. Присутствие ее в свободном
состоянии определяется наличием микробной плазмы, состоящей, как
известно, в значительной степени из полисахаридного комплекса,
но реальные количества свободных полисахаридов в почве,
по-видимому, очень невелики.
Следует отметить возможность сорбции углеводов глинистыми
минералами почвы, способность образовывать простые соли и
101
сложные комплексные соединения с полуторными окислами,
а также вступать во взаимодействие и сорбироваться гумусовыми
кислотами и полифенолами [410, 548]. Следует отметить также
незначительное количество моносахаридов в природных водах.
По данным Гидрохимического института, количество
моносахаридов в речных водах составляет 0.82—1.50 мг/л, в водах озер и
водохранилищ — 0.1—4.6 мг/л. Несколько выше содержание в водах
водорастворимых полисахаридов [273, 274, 304]. Таким образом,
количество свободных углеводов в почвах, по-видимому,
незначительно и достаточно эфемерно вследствие быстрого
использования их микроорганизмами.
Соединения фенольной природы. В составе промежуточных
продуктов разложения органических остатков в почве
присутствуют вещества фенольной природы. Большинство исследований
посвящено изучению веществ фенольной природы в гидролизатах
лочв или гумусовых кислот. Наиболее обстоятельный обзор
этих работ дан в монографии Д. С. Орлова [230], где приведены
три основные группы этих соединений, найденные в составе
продуктов расщепления: фенолы (пирокатехин, резорцин, флоро-
глюцин, нитрофенол, 2,4,6-тринитрофенол, тринитродиоксибензол),
альдегиды (тг-гидроксибензальдегид, ванилин, сиреневый
альдегид) и кислоты (м- и тг-гидрооксибензойные, ванилиновая,
сиреневая, вератровая, протокахетовая, 2,4- и 3,5-дигидрооксибензой-
ные, 3,4,5-тригидрооксибензойная, изофталиевая, терефталиевая,
бензолтрикарбоновые, меллофановая, пирромеллитовая, бензол-
пентакарбоновая и меллитовая). В зависимости от методики
получения и идентификации эти кислоты были выделены в виде
различных производных хинонов, полициклических соединений и
азотсодержащих гетероциклов.
В последние годы появились данные о том, что в почве
встречаются свободные соединения фенольной природы. Для их
выделения из почвы применяют обычно щелочные или щелочно-спир-
товые растворы с последующей идентификацией экстрагированных
продуктов хроматографическими методами. В ряде публикаций
[239, 358, 359, 361, 369, 417, 418, 458, 558] отмечено наличие в
почвах ванилиновой, тг-оксибензойной, я-кумаровой, сиреневой, феру-
ловой кислот, а также ароматических альдегидов,
индивидуальных флавонов, флавонолов, катехинов и других соединений.
Содержание каждого из этих соединений очень незначительно и
исчисляется тысячными и сотыми долями процента органической части
почвы. Так, например, содержание фенолкарбоновых кислот в
низинной торфяной почве составляет 0.75—1.50 мг, в
дерново-подзолистой — 0.03—0.09 мг/100 г [239].
Доля фенольных соединений типа таннинов в водорастворимом
органическом веществе лесных подстилок и почвенном растворе
составляет 30—80% от общего содержания его [130]. В
органическом веществе лизиметрических вод из дерново-подзолистой
почвы их содержание, выраженное в С, колеблется от 0.26 до
102
2.16 мг/л, что составляет лишь 1.5—8.0% всей массы
водорастворимых органических веществ [221 ].
Следует указать, что низкомолекулярные соединения феноль-
ной природы несомненно присутствуют в продуктах разложения и
гумификации растительных остатков, о чем свидетельствуют
данные, полученные в последнее время в нашей лаборатории (табл. 35).
Общее количество фенольных соединений, образующихся при
разложении различных по химическому составу растительных
остатков, колеблется от 3—5 до 41 % ко всей сумме водорастворимых
органических соединений; при этом в максимальных количествах
они появляются при гумификации листьев дуба и хвои ели;
минимальное содержание их характерно для разлагающихся корней
тимофеевки. В абсолютных единицах их содержание колеблется
от 0.01—0.03 до 1.1% к сухой беззольной массе растительных
остатков.
Таким образом, абсолютные количества свободных
соединений фенольной природы в почве в каждый данный момент,
вероятно, относительно невелики, но они длительно присутствуют
в почве, прежде всего, по-видимому, вследствие непрерывного
появления в процессе разложения растительных остатков и
относительно медленной минерализации.
Низкомолекулярные органические кислоты. Наряду с другими
группами низкомолекулярных продуктов разложения
органических остатков обычными компонентами являются
низкомолекулярные органические кислоты алифатического ряда. Наиболее
распространенные из них щавелевая, янтарная, фумаровая, лимонная,
малоновая, яблочная, а также масляная, уксусная и муравьиная.
Эти кислоты образуются в почве при разложении растительных
остатков, а также в результате синтетической деятельности многих
бактерий и грибов; поступают они в почву и из корневых выделений
растений. Максимальные количества обнаруживаются в водных
вытяжках из лесных подстилок и торфяных почв, где содержание
этих кислот достигает 30—60 мг/л. В минеральных почвах они
составляют 3—15 мг/л [130, 221 ]. В большинстве случаев
органические кислоты присутствуют в почвах в виде солей, так как
активно мобилизуют основания почвы. Относительная доля их в составе
водорастворимых органических соединений достаточно высока:
до 30—44% [221 ]. Их содержание в почве, по-видимому, очень
динамично (табл. 36). Количество солей органических кислот
различно в зависимости от состава разлагающихся растительных
остатков и заметно изменяется в течение периода разложения.
Оно колеблется от 1—2 до 6% к общей массе водорастворимых
органических веществ, лишь продукты гумификации хвои образуют
постепенно более значительное количество органических кислот.
Очень существенное влияние на количество образующихся
низкомолекулярных органических кислот оказывают условия аэрации.
При разложении лесных подстилок в анаэробных условиях
содержание низкомолекулярных органических кислот воз-
103
Таблица 36
рН и содержание водорастворимых солей органических кислот
в гумифицирующихся растительных остатках (по: [165])
Объект
исследования
Хвоя ели
Наземная масса
клевера
Листья дуба
Корни тимофеевки
луговой
рН и соли органических кислот в разные сроки гумификации,
сут
0
рН
6.6
6.4
6.2
6.6
соли
органических
кислот
1.9
0.9
1.5
66.2
1.5
53.0
3.1
11.6
7
рН
7.9
6.0
6.7
соли
органических
кислот
19.1
275.8
3.6
37.8
5.3
87.9
5.8
13.8
30
РН
6.4
7.2
5.9
6.6
соли
органических
кислот
14.0
44.4
2.1
24.3
5.1
36.8
5.7
14.7
360
рН
6.2
7.7
7.3
6.3
соли
органических
кислот
6.2
20.6
1.3
10.7
2.1
11.9
5.5
11.1
растает, а качественный состав их становится более
разнообразным.
Органические кислоты, очевидно, менее эфемерны в почве
по сравнению с аминокислотами и моносахаридами; благодаря
своей активности они вступают во взаимодействие с компонентами
минеральной части почвы, участвуют в разрушении
кристаллической решетки минералов и, образуя с продуктами разрушения
соли и комплексные соединения, мигрируют по профилю почвы [127,
129].
Прочие промежуточные продукты разложения. В. Фляйг [397]
указывает на наличие в почвах незначительного количества
органических фосфатов — продуктов разложения нуклеиновых
кислот и фосфатидов, а также синтезирующихся
микроорганизмами. Единичные работы [341, 366] посвящены
идентификации инозитфосфатов; найдены в ничтожных количествах
пуриновые и пиримидиновые основания (гуанин, аденин,
цитозин), образующиеся при разложении нуклеотидов. Все эти
продукты обнаружены при помощи хроматографических методов,
обычно в следовых количествах, преимущественно в органогенных
горизонтах — подстилках или торфе. Длительность их сохранения
в почве и доля участия в гумификации и формировании органо-
минеральных продуктов практически не изучены.
Экстракция почвы спиртобензолом с последующей
идентификацией полученных продуктов показала наличие ряда
промежуточных цродуктов разложения липидов и родственных им
104
веществ. Эти соединения рассматривались как в работах 40-х гг.
[65], так и более поздних [351, 527]. Общее содержание липидной
фракции в почвах колеблется от 2—3 до 10—21 % к общему
содержанию органических веществ в почве; минимальное количество
их характерно для черноземов и каштановых почв, максимальное —
для тундровых почв и подбуров [315].
При использовании современных методов идентификации в
составе липидной фракции найдены в ничтожных количествах многие
индивидуальные жирные кислоты, фталиевые эфиры, стероиды,
каротиноиды, порфирины, воски, глицериды, нормальные алканы;
обзор этих исследований дан В. Фляйгом [397]. Однако как
количество этих индивидуальных представителей класса липидов, так
и их роль в почвообразовании все еще не изучены.
Глава III
ПРОЦЕССЫ ТРАНСФОРМАЦИИ
ОРГАНИЧЕСКИХ ВЕЩЕСТВ В ПОЧВЕ
Учение о гумусообразовании имеет длительную историю
развития с закономерной сменой представлений о характере этого
процесса. Еще у Я. Валериуса и Т. Соссюра можно найти указания
на то, что гумус является продуктом частичного разложения расти-
тельйых остатков, хотя, конечно, сам процесс разложения
рассматривался с химических позиций, а агентами его считали влагу,
тепло и кислород воздуха. Длительное время господствовала
теория Г. Мульдера, согласно которой процесс гумусообразования
рассматривался как ряд химических реакций дегидратации и
окисления целлюлозы. Блестящие работы Л. Пастера и последующее
бурное развитие микробиологии и биохимии оказало огромное
влияние на дальнейшее изучение процессов образования гумуса
в почве.
Основоположником современного учения о гумусообразовании
следует считать П. А. Костычева, четко сформулировавшего
основные положения этого учения: 1) разложение органических
веществ в почве происходит под влиянием бактерий, грибов и
вследствие химического взаимодействия составных частей
разлагающегося вещества; 2) только относительно грибов несомненно, что
они способны образовывать продукты разложения, окрашенные
в темный цвет; бактерии этой способностью не обладают; что
касается чисто химического разложения, то роль его в этом
неизвестна; 3) образование аморфных продуктов разложения
обусловливается главным образом деятельностью животных; без их участия
разлагающиеся растительные остатки долгое время сохраняют
анатомическое строение [160].
П. А. Костычев считал, что гумус (перегной, по его]
терминологии, — Л. А.) образуется в процессе разложения растительных
остатков микроорганизмами, но не расшифровал характера этого
процесса. Огромная историческая заслуга П. А. Костычева
в совершенно новой трактовке причин обогащения гумуса азотом.
В серии тщательно выполненных экспериментов автор показал, что
«при всяком процессе разложения (растительных остатков, —
Л- А.) с участием низших организмов распадение белковых веществ
несомненно сопровождается синтезом их» [158, с. 356]. Но этот
микробиологический процесс синтеза и накопления вторичных
106
белков в составе плазмы микроорганизмов, развивающихся в
разлагающемся материале, П. А. Костычев не отождествлял с
процессом образования гумуса. Он подчеркивал, что перегнойные
кислоты — это продукты разложения растительных остатков,
а обогащение гумуса азотом обусловлено ресинтезом микробной
плазмы, входящей в состав гумуса [ИЗ, 113а, 158, 159]. Таким
образом, П. А. Костычев с удивительной для того времени
прозорливостью четко сформулировал все, что было впоследствии
доказано экспериментально.
В дальнейшем, на протяжении всего XX в., проблема гумусо-
образования находилась и находится в центре внимания
исследователей [19,142,143, 230, 399], и этот интерес обусловлен огромной
значимостью процессов трансформации органических остатков
в биосфере не только для почвоведения, но и для агрохимии, торфо-
ведения, биогеохимии и ряда других смежных дисциплин.
Общая схема процессов трансформации
К настоящему времени накопился огромный
экспериментальный материал, позволяющий в общих чертах составить
представление о процессах трансформации. Любые органические остатки,
поступающие в почву, подвергаются в ней процессам разложения под
воздействием микроорганизмов и мезофауны, использующих эти
остатки как строительный и энергетический материал. Этот
процесс расчленяется на два основных звена:
разложение
I I
минерализация гумификация
Конечный результат первого — постепенное исчезновение
органических компонентов и образование минеральных соединений,
используемых в биологическом круговороте; итог второго —
консервация органического вещества в форме новых устойчивых к
разложению продуктов — гумусовых кислот, являющихся, таким
образом, аккумуляторами огромных запасов элементов питания и
энергии. По данным В. А. Ковды [135], в гумусе почвы содержится
до п • 1019 ккал связанной энергии.
Эта общая схема не вызывает в настоящее время дискуссий
и общепризнанна. Следует иметь в виду, что характер
трансформации органических остатков универсален в пределах
биосферы и осуществляется не только в почвах, но и в любых
других скоплениях мертвого органического вещества,
доступного для микроорганизмов. Интенсивность этих двух звеньев,
соотношение между ними, процессы взаимодействия
продуктов трансформации с компонентами окружающей среды, и
107
прежде всего с минеральной частью почвы, конечно, очень
разнообразны и обусловлены всем комплексом условий, в которых
развивается этот процесс.
Процессы разложения—минерализации
органических остатков в почве
Все процессы разложения органических остатков носят
биокаталитический характер и протекают при непосредственном участии
^ферментов микроорганизмов как вне живых клеток последних,
так и внутри их. Вследствие высокомолекулярной природы
большинства компонентов органических остатков процессы трансформации
начинаются вне живых клеток микроорганизмов и сводятся к
гидролитическому расщеплению экзоферментами, после чего продукты
расщепления благодаря уменьшению ММ постепенно проникают
через клеточные мембраны и подвергаются дальнейшим
превращениям при участии различных оксиредуктаз. Следовательно, в почве
вне живых клеток микроорганизмов постоянно образуется очень
сложная система высокомолекулярных продуктов
гидролитического расщепления, непрерывно пополняемая низкомолекулярными
органическими веществами и продуктами их полной
минерализации вследствие завершения жизненного цикла клетки
микроорганизма или в процессе осмоса. Такова общая картина
постепенной минерализации органических остатков, которая описана в
современных монографиях по микробиологии [256, 325].
Процессы разложения—минерализации органических
остатков можно представить схемой, где R—Ca2+, Mg2+, K+, Na+,
NHt (см. с. 109).
Если низкомолекулярные продукты разложения
органических остатков изучены достаточно хорошо, то количество, состав
и свойства первой категории веществ — системы
высокомолекулярных соединений, образующиеся в процессе гидролитического
расщепления, — практически неизвестны, ибо невозможно
выделить их из почвы без нарушения живых функционирующих клеток
микроорганизмов. И в то же время именно эта группа продуктов
первичного расщепления, по-видимому, играет существенную роль
в формировании молекул гумусовых кислот, так как она
находится в непосредственном контакте с массой4 почвы.
Белки. Как известно, белки вначале, вне живых клеток
микроорганизмов, при участии протеаз распадаются на сложную
систему пептидов, нуклеотидов, нуклеиновых кислот и ряд других
компонентов. Постепенно при дальнейшем гидролизе образуются
менее высокомолекулярные продукты, проникающие через
мембранные оболочки непосредственно в живые клетки, где и
происходит их дальнейшая трансформация под действием обширной
группы внутриклеточных ферментов — пептидаз — до
аминокислот, пуриновых и пиримидиновых оснований, моносахаридов
и других относительно простых органических соединений. Конеч-
108
Органические остатки
белки и поли-
нуклеотиды
углеводы
лигнин,
дубильные вещества
1
липиды, смолы
Гидролитическое расщепление
Система высокомолекулярных продуктов разложения,
накапливающаяся в почве вне живых клеток микроорганизмов
1
высокомолекулярные продукты
разложения
лигнина (типа
полифенолов), окси-
поликарбоновые
кислоты
пептиды, нуклеотиды,
нуклеиновые кислоты
олигосахара,
целлобиоза,
полиуроновые
кислоты
J Гидролиз и оксиредукции
Система низкомолекулярных продуктов разложения,
накапливающаяся в почве и в живых клетках микроорганизмов
-j—
глицерин,
смоляные
кислоты,
жирные
кислоты, спирты
алифатические и
ароматические
аминокислоты,
пуриновые и пири-
мидиновые
основания
моносахариды,
аминосахара,
уроновые
кислоты
1
низкомолекулярные продукты
расщепления
лигнина и
дубильных веществ
I
Окисление, восстановление, дезаминирование, декарбоксилирование
1 1
S н Ч^
? s и я
g ез Я Я
4 « « Н
(б Sou
I
1
1
1 I
I О
о га
И ф~
К ЧД
а»?
Soo
а
о
ф
я^
г я ч
н с, Я
,^_ О Ф Н
>§<Ч «Он
а о ^ а
4SRB
ЛИЛО
н
се
° ф «g<
ф Н о §.
И И О СГ
>>2 3 ?
н Р* н ч ~
ф н о 2 Я
ф
Ом
*о^ я
S о Я и
н щ н Н
се о о И
Svo Ч
OftOJ
Рчсе а 3
се к К Ч
Н Д
ф
я
и
1st №
К Ч
О К
Я
и ^
tc н Я
ФОН
ЛЧ Оч
нон
ф а а
н к о
Я
о
*=с
о
га
ф
Ч~
i I
i
i
J 1
Полная минерализация
R3P04, R2S04, RN02, RNOg, NH8l H20, C02, CH4, H2, N2, H2S, PH3
аэробиозис (I)
анаэробиозис (II)
109
ными процессами разложения белков являются, как известно,
Декарбоксилирование, аммонификация, нитрификация,
осуществляемые обширной группой оксиредуктаз [по: 325]:
Бепки
Протеочитические
ферменты
(экзоформс^нты)
Почипептпды
О 1ИГОПРПТМДЫ
ПоптидазЫ
А м инокиспоты
дезамшшровлше
и распад С-скечета
докарбокси шрованио
непосредственное .
испопьзование
в качестве
строительных бпоков
д 1я синтеза бепка
переаминирование
и распад С-скепета
Менее изучены процессы разложения безазотистых
органических соединений, которые (как указано в гл. I) очень
разнообразны. Ж. Пошон и Г. де Баржак [256] приводят общую схему
процессов превращения этих соединений в почве (см. с. 111).
Скорость разложения протеинов в различных растительных
остатках неодинакова (табл. 37). Протеины наземной массы
клевера разлагаются быстро, и через 6 мес остается лишь V3 их
первоначального количества, причем степень аэробиозиса
оказывает незначительное влияние на скорость их минерализации.
Заметно больше воздействуют химический состав и состояние
растительных остатков. Белки корней клевера разлагаются
медленнее как в аэробных, так и в анаэробных условиях из-за
особенностей морфологической структуры тканей в корнях.
По-видимому, белки здесь находятся в комплексе с лигнином,
что и тормозит скорость их минерализации.
Расщепление углеводов также Начинается с гидролиза вне
живых клеток микроорганизмов, катализируемого обширной
группой гидролаз, в результате чего вначале образуется сложная
смесь достаточно высокомолекулярных продуктов разложения —
олигосахаридов, полиуроновых кислот, а затем — система
низкомолекулярных продуктов гидролиза. В аэробных условиях
доминируют моносахариды и уроновые кислоты, в условиях анаэробио-
зиса — низкомолекулярные органические кислоты и спирты.
Целлюлоза и гемицеллюлозы. Расщепление целлюлозы,
вызываемое обширной группой бактерий, грибов и актиномицетов,
обусловленное действием «целлюлаз»,1 происходит в природных
1 До сих пор исследователи не пришли к единому мнению о природе фермента
«целлюлаза»: является ли он действительно одним ферментом или
представляет собой комплекс ферментов.
110
Автотрофные
микроорганизмы
СО.
Растения
содержащие
хлорофилл
Клетчатка,
гемицеллюлоза,
крахмал,
пектин
Лигнин
Фиксаторы
азота
условиях медленнее и труднее по сравнению с другими
углеводами. Основная причина этой особенности — в специфике
строения природных волокон целлюлозы, которые обычно
пропитаны лигнинами, восками, смолами. Этим объясняется
медленное разложение в почве относительно крупных корней
травянистой растительности. Ж. Пошон и Г. де Баржак [256] приводят
такую схему расщепления целлюлозы в аэробных условиях:
длинная цепочка глюкозидаза
клетчатка —> р-1—4-ангидроглюкозы-» целлобиоза —> глюкоза
вне клетки
в клетке
Эти сахара, однако, обнаруживаются в ничтожных коли*
чествах, так как трансформируются в слизистое вещество —
111
co
cej
К
a
H
о
I
0s»
Рн
Ф
И
<D
И
К
«
и
О)
I
о
Рч
а
по
005
11
X >>
is
ев
Рн
pa
о
н
я
§
И
§
В
8
О
И
S
и
8
CD
й
я
Сумма
фракций, %
Протеины
Лигнинный
остаток
Целлюлоза
Геми целлюлоза
Растворимые в сйир-
тобензоле
1 *
О
я ».
т
§
Срок
разложе-
*-<
1—1
м
1-1
t-H
ни
•—<
м
м
Н4
&
ее
S
8
ОООО
00 <ЯО>
ООС
05 05С
)00
>00
о
00 ¦
а-
юсо^оо
00 ОООО 00
•00 00 СО СМ
о
Осмоэсмсо
^ t-~Xt<COCM
ООЭООчРСО
о
Ооо г>со
"^00 СО** СМ
Ocmiovfco
^4 оо i> см «ч1
?i
о
о
¦чН
©СОСМ"^
t»*tf СМчН
00 00 00 00
8
vjh COOOO
о
о
О00»*СМ-чН
•чг! t^ со СО СО
^IOnFOOCO
СМ00 1> !>• t^
о ю со см см
о t^ со со со
>^сою****
о.
о
со со со со
«
сб
И
Я
О)
со
сб
и
00 СОЮ 00 СМ
СЧ С» СО -ч-н *«•«
«чЧ
о
о
«чН
©«ч-чОЭСО
CONf-чН^Н
PL,
о
Осмючрсм
о'сО-ч^^ч^
о.
о
©l>t^CO
со со со со
СОЮ-чн-чН
?ГЧ>СОСМ«-Н
СООЯЛчЧ
СО СО СО СМ
о
о
о
о
oot-^o
Ю СО «чН<«Н
О5 00т-1СО
соооэоо
о
о
о
о
S
00005
Од 00 О)
Oit-чн t^
о*-*юсо
ot^ooo
СО 00 CM
-чЧ t-ч
оюооо
«ччООСОСМ
¦ччСО t-
«цитофаговое желе» с высокой ММ, которое является йолиуронй-
дом. В анаэробных условиях целлюлоза разлагается бактериями
рода Cytophaga, Cellvibrio, Cellulomans с образованием серии
низкомолекулярных органических кислот (уксусной, молочной,
масляной, муравьиной) и спиртов.
Более интенсивно разлагаются в почве гемицеллюлозы и
пектиновые вещества также при участии различных гидролаз,
выделяемых бактериями, грибами и актиномицетами во внешнюю по
отношению к клетке среду — в почву. В процессе разложения
в зависимости от степени аэробиозиса и компонентов, слагающих
массу гемицеллюлоз, образуется система плохо изученных
продуктов гидролитического расщепления, а затем фиксируются и
наиболее простые компоненты — гексозы, пентозы, уроновые
кислоты (в условиях аэробиозиса) и низкомолекулярные
органические кислоты и спирты (в условиях анаэробиозиса).
Лигнин. До настоящего времени неясен характер процессов,
обусловливающих биологическое расщепление лигнина, хотя
постепенное использование этого компонента растительных
остатков не только грибами, но также бактериями и актиномицетами
несомненно. Происходит оно медленно и длительное время
развивается при участии экзоферментов вне живой клетки
микроорганизмов. По данным Дж. Трояновского с сотрудниками [452,
552], расщепление лигнина начинается при участии
экзоферментов грибов с деметилирования и окисления, но реальные продукты
этой частичной деградации неизвестны. Г. Шлегель [325]
считает, что расщепление лигнина осуществляется полифенолазами
грибов и сопровождается разрывом эфирных связей и связей С—С,
вследствие чего должны образовываться различные димеры кони-
ферилового спирта и другие олигомеры. Если учесть, что, по
современным представлениям, молекулы лигнина неоднородны
по деталям строения, станут очевидными трудности исследования
продуктов частичной трансформации этого вещества. В
последние годы показано участие ряда анаэробных и особенно аэробных
бактерий в расщеплении лигнина, но механизм этого расщепления
остается неясным [256, 473]. Следует отметить, что большинство
данных получено путем определения «лигнина» как
негидролизу емого кислотами остатка разлагающихся материалов, в составе
которого всегда значительна доля образовавшихся гумусовых
веществ. При более четкой в методическом плане схеме анализа
скорость исчезновения лигнина достаточно заметна, хотя и
замедлена по сравнению с интенсивностью разложения углеводов.
Интересные данные Ф. Броадбента [355] о скорости
разложения лигнина, выделенного из овсяной соломы по методу
Нормана, приводят В. Фляйг и соавторы [399]. Инкубацию
препаратов проводили в течение полутора лет без добавления и с
добавлением азота (табл. 38). Как видно из приведенных данных,
разложение препаратов лигнина, замедленное в первый период,
происходит длительное время и за полтора года минерализуется
8 Л. Н. Александрова ИЗ
Емкость обменного
поглощения, мг«экв./100 г
СООН, мг • экв.ДООг
о
и
о
S2
Е
о
*-2
И
о
2
С)
С)
*
«°
Р.
Период
•-•
м
Н-1
н
H-t
1-1
"
1-1
w
Ь-1
1-Н
1-1
м
разложения, сут
^^ООО^ОО'чнСО
СТ>е<1СЧ1<М-*-4©СОСО^!<
С^^СООООО<М<^С<1
0000OOT-lCSI«4H^Sf
ОЭОЭОтЧС^-чНСОСЧСО
7.84
7.24
6.63
5.32
6.77
6.70
6.31
5.77
5.20
6.82
7.53
7.77
5.05
4.13
4.01
5.83
6.37
6.05
11.48
11.42
11.23
10.37
8.66
8.96
9.00
7.71
7.99
ocsic<icivt<ooco.iom
Nfeoioeocvjcoiooqoo
6.25
5.88
6.12
6.04
9.24
5.65
5.74
5.85
5.67
6.67
6.30
6.21
6.35
6.30
6.34
5.81
6.23
5.93
63.0
63.0
63.9
63.1
61.7
60.7
61.1
62.0
60.5
61.5
61.9
61.6
61.2
59.7
60.2
60.7
59.8
59.4
0
22.7
37.7
47.6
56.4
58.0
59.3
61.3
63.9
0
27.4
38.8
44.6
58.0
60.1
67.2 (?)
66.0
65.5
-чи ¦*¦« CM CO чП
8
»
и
a
s
«ев
OS
О «й
•rtg
I Ш
~J
is-
-I
*2
2/3 первоначальной массы, причем добавление азота практически
не изменяет скорости разложения. Отчетливо выражен процесс
гумификации массы лигнина, не подвергшейся полной
минерализации, так как количество карбоксильных групп и общая
емкость обмена возрастают в процессе инкубации в 2 раза.
Увеличение количества карбоксильных групп сопровождается
заметным уменьшением количества гидроксилов. Аналогичные
данные получены в нашей лаборатории М. Ф. Люжиным по
изменению химического состава растительных остатков в аэробных
и анаэробных условиях. В схему анализа было включено
определение новообразующихся гумусовых веществ, экстрагируемых
пирофосфатом натрия при рН 9.5 (табл. 37).2
Таким образом, реальное количество лигнинного остатка
после удаления веществ, растворимых в 0.1 М растворе Na4P207
и спиртобензоле, гемицеллюлоз, целлюлозы и протеинов, также
относительно быстро уменьшается в процессе разложения. Через
полгода сохраняется лишь 47—63% от первоначального
количества лигнина при разложении в аэробных условиях и 45—74%
при разложении в условиях анаэробиозиса. Через два года остается
лишь 26—30% исходного лигнина, содержавшегося в листьях
клевера, в то время как лигнинный остаток разлагающихся
корней сохраняется высоким: 62—73%. На интенсивность
разложения лигнина (и целлюлозы) большое влияние оказывает его со-
стояние в растительных остатках, в первую очередь, по-видимому,
его связь с другими компонентами тканей. Очень медленно
разлагается лигнин корней травянистой растительности, для
которых характерно наличие целлюлозно-лигнинных волокон.
Липиды и прочие компоненты. Недостаточно исследован
механизм расщепления в почве липидов. Общая схема их
трансформации не вызывает сомнений. Жиры подвергаются
гидролизу, осуществляемому липазой с последующим окислением
в аэробных условиях и процессами брожений, развивающимися
при анаэробиозисе; менее исследован процесс трансформации
восков, фосфатидов и гликолротеидов, хотя и для этой категории
соединений несомненны гидролиз и постепенное окисление с
последующей полной минерализацией продуктов окисления.
Продуктами гидролитического расщепления являются
многочисленные высокомолекулярные жирные кислоты, глицерин и другие
спирты жирного и ароматического рядов, а также углеводороды.
Получены данные о микробиологическом окислении спиртов
жирного и ароматического рядов, при котором образуется
сложная система альдегидов, кетонов и главным образом кислот под
влиянием бактерий и грибов, а также о процессах трансформации
углеводородов парафинового ряда, окисление которых бакте-
Детальная методика работы и вычисление количества отдельных
компонентов приведены в работе М. Ф. Люжина [188].
115
8*
риями и грибами также идет через их гидроксилирование и кар-
боксилирование (кислотообразование).
Вероятные схемы процессов трансформации нафталина, фе-
нантрена и антрацена таковы [313, 325]:
НО
^ч/v
v\^«
нафталин 1,2-диоксинафта-
лин
у\
ноч
он
,w
W
фенантрен 3,4-диоксифе- 1-окси-2-нафтил-
нантрен метиленпируват
антрацен
S\/\/
ОН
+ пируват
салициловый альдегид
/у\Ум
О
салициловая кислота
1
•
соон
пирокатехин
Продукты начальных стадий гидролитического распада этой
категории соединений практически не изучены.
Расщепление различных фенольных соединений, а также
алкалоидов, пигментов и многих других органических соединений,
входящих в состав гумифицирующихся в почве органических
остатков, также, по-видимому, происходит по общей схеме гидролиза
и оксиредукций, хотя реальные материалы по характеристике
образующихся продуктов очень ограниченны. Сводку основных
типов микробиологического окисления различных органических
соединений дают Г. Фонкен и Р. Джонсон [313]. Процессы
окисления обусловливают гидроксилирование ароматического кольца
с последующим раскрытием его по типу орто- и метарасщепления.
Осуществляются эти процессы обширной группой оксигеназ,
катализирующих включение молекулярного кислорода в субстрат,
116
что и приводит к образованию сложной системы алифатических
кислот (по: [325]):
Тип I — орторасщепление
НООС
ч
ОН
Тип II — метарасщепление
НООС
пирокатехин
о2
он
\Lo
//
Vc^h
цис% цис-
му коновая
кислота
протокатеховая пирокатехин протокатеховая
кислота
кислота
02
ноос он
V\c^o
3-карбокси-цис,
цис-муконовая
кислота
о2
о
с^он
Нч /О Н
с^он
семиалъдегид семиальдегид
2-оксимуконо-
2-окси-4-карбоксивой кислоты муконовой кислоты
Окисление углеводородов также приводит к их гидроксилиро-
ванию, а затем и образованию органических кислот, хотя детали
этих процессов исследованы еще недостаточно. Конечными
продуктами трансформации любых компонентов органических
остатков в почве являются продукты полной минерализации их — С02,
Н20, NH3, а также (в зависимости от степени аэробиозиса) ряд
окисленных или восстановленных продуктов (см. схему на
с. 109).
Таким образом, в процессе разложения растительных остатков
в почве образуется очень сложная система высоко- и
низкомолекулярных продуктов разложения, соотношение между которыми
практически неизвестно. Можно привести лишь некоторые, не
законченные еще исследования М. В. Новицкого и Г. И. Крупи-
ной об относительном содержании водорастворимых
высокомолекулярных продуктов разложения различных растительных
остатков в течение годичного цикла их разложения в аэробных условиях
(табл. 39). Используя оригинальный метод гельфильтрации в
сочетании с центрифугированием, авторы показали, что на
протяжении годичного цикла разложения относительное содержание
высокомолекулярных продуктов (с ММ > 1500) высокое. 'Эти
дродукты практически доминируют начиная с 7—30-суточного
срока инкубации в течение всего года [223]. К сожалению,
подобные исследования только начаты и более детально обсудить их
можно будет значительно позднее. Сейчас лишь с достаточной
долей уверенности следует отметить, что на протяжении всего
периода разложения любых растительных остатков в почве доля высоко-
117
Таблица 39
Содержание водорастворимых органических веществ
в разлагающейся растительной массе, мг С на 1 л (по [223])
Объект исследования
Хвоя ели
Листья
клевера
Дуба
~
Корни тимофеевки
луговой
Срок
гумификации, сут
0
7
30
360
0
7
30
360
0
7
30
360
0
7
30
360
Общее
количество
1.04
30.87
6.80
4.28
64.43
20.79
20.79
11.12
52.47
25.11
16.13
12.56
6.44
5.72
4.50
3.56
Высокомолекулярные соединения
мг/л
0.77
14.72
4.94
2.92
50.57
12.11
18.92
9.64
27.90
19.33
12.71
7.39
3.21
3.81
2.78
Не опр.
% от общего
содержания
74.63
47.68
68.17
68.22
77.29
58.25
81.76
86.70
53.17
76.98
78.80'
89.11
55.27
66.61
61.78
Не опр.
молекулярных продуктов разложения достаточно высока и несом-
ненно не менее значительна их роль в процессах гумификации.
Скорость разложения—минерализации органических остатков
Скорость разложения органических остатков в почве, конечно,
различна и регулируется комплексом причин, среди которых
наибольшее значение имеют химический состав и анатомическое
строение растительных остатков, степень увлажнения и аэробиозиса,
а также гранулометрический и химический состав почвы, в
которой развивается этот процесс. Наиболее интенсивно разложение
большинства растительных остатков идет в первый период (в
течение первых 3 мес) в аэробных условиях при оптимальной
влажности и достаточно оптимальной для микроорганизмов
температуре. Скорость разложения лимитируется содержанием
легкоусвояемых микроорганизмами органических соединений, и прежде
всего белков. Довольно интенсивно с относительно одинаковой
скоростью в аэробных условиях разлагаются углеводы, протеины
и вещества, экстрагируемые спиртобензолом, содержащиеся в
наземной массе клевера (см. табл. 37; рис. 19). Значительно медленнее
в первый период разложения снижается количество лигнинного
остатка, но за два года наблюдений и его содержание уменьшается
118
/о
О 90 180 360 720 сут
Рис. 20. Скорость минерализации органических остатков различного
химического состава.
1 — наземная масса клевера, 2 — корни клевера, 3 — наземная масса люцерны, 4 —
хвоя, 5 — навоз на соломистой подстилке, в — навоз на торфяной подстилке, 7 — ТМАУ,
8 — опилки.
на 70—80%. Кажущееся замедление процессов разложения
протеинов в конце опыта обусловлено, по-видимому, ресинтезом их
вторичных форм в массе микроорганизмов. Анаэробиозис, как и
следовало ожидать, тормозит процессы разложения всех
компонентов растительных остатков, но наиболее ясно это торможение
выражено в первый период инкубации. К концу инкубационного
срока (2 года) влияние анаэробиозиса сказывается слабее.
Значительно медленнее разлагаются все компоненты корневых
остатков. Особенно резко они замедляются для углеводно-лигнинного
комплекса и протеинов, что подчеркивает существенное влияние
особенностей анатомического строения корней.
Данные об интенсивности разложения органических остатков
различного химического состава (рис_. 20) показывают, что
корневые остатки всегда минерализуются медленнее наземного опада
вследствие значительного содержания в них одревесневших
тканей. Интенсивность минерализации органических удобрений
определяется природой наполнителя, которым в последнее время чаще
всего бывает торф. Чем выше степень разложения торфа, тем ниже
интенсивность минерализации компоста.
Очень большое влияние на скорость минерализации оказывает
гранулометрический и химический состав почвы, в частности
степень ее гумусированности. При всех оптимальных условиях
разложения тяжелый гранулометрический состав и низкое
содержание гумуса тормозят минерализацию органических остатков
(рис. 21), так как значительная свободная минеральная
поверхность обусловливает процессы сорбции промежуточных продуктов
разложения. Наши исследования показали, что коэффициент
гумификации растительных остатков в значительной степени ре-
120
1
2
'3
Of 15 90 120 Ш) Tub 720 cyrn
Рис. 21. Влияние гранулометрического состава почвы на скорость
минерализации растительных остатков (по: [190]).
1,2 — наземная масса клевера; з, 4 — корни клевера. 1,3 — в песке; 2,4 — в суглинке.
гулируется именно этим фактором. Если в песчаном и супесчаном
субстрате он составляет около 25%, то в суглинистых и особенно
глинистых почвах может достигать 60—70% даже в условиях
оптимального увлажнения. Поэтому приемы глинования песчаных
почв всегда приводят к заметному накоплению в них гумуса.
Следует иметь в виду, что гранулометрический состав регулирует
физические свойства почвы, а следовательно, и водно-воздушный
режим, при котором протекают процессы разложения. Усиление
анаэробиозиса всегда задерживает эти процессы и обусловливает
торфообразование.
Очень разноречивыми были до недавнего времени материалы,
характеризующие влияние реакции почвы на скорость разложения
растительных остатков. Детальный критический обзор этих
исследований дает И. В. Тюрин [297]; автор показал, что
нейтральная реакция в почвах, насыщенных кальцием, ускоряет процессы
разложения растительных остатков, причем эти процессы
приобретают более резко выраженный окислительный характер.
Одновременно усиливаются процессы гумификации компонентов,
содержащих циклические группировки, и накопления гуминовых веществ
в форме нерастворимых гуматов кальция.
В последнее время эти выводы И. В. Тюрина нашли
достаточно убедительное подтверждение: В. В. Рассохина [261] в серии
оригинальных модельных опытов показала, что кислая реакция
тормозит процессы разложения растительных остатков вследствие
угнетения бактериальной микрофлоры и стимуляции развития
грибов, тогда как насыщение почвы обменным кальцием
усиливает эти процессы и полную минерализацию. Существенное
влияние на скорость разложения органических остатков
оказывает, конечно, уровень питательного для микроорганизмов
режима [65].
Таким образом, физико-химические свойства всегда прямо и
косвенно влияют на скорость разложения растительных остатков.
Прямое влияние проявляется в степени развития процессов взаимо-
121
действия с компонентами почвы (реакции солеобразования,
сорбции), косвенное — в регулировании интенсивности
жизнедеятельности микрофлоры и ее группового состава (доминировании
грибной или бактериальной микрофлоры).
Деятельность почвенной фауны
Наряду с микроорганизмами, обусловливающими процесс
разложения органических остатков, огромна роль почвенной фауны,
которая обусловливает как измельчение и механическое
перемешивание органических остатков с массой почвы, так и их
химическую трансформацию. Представители макро-, мезо- и микрофауны
не только перераспределяют и измельчают все органические
остатки в пределах профиля, но и сами участвуют в процессах
разложения [142]. М. М. Кононова выявила роль личинок мушки
Scaria и тироглифоидных клещей в превращении разлагающихся
растительных остатков в относительно однородную и
измельченную массу, а также показала те гидролитические процессы,
которые протекают в кишечнике животных и обусловливают
трансформацию всех компонентов растительных тканей (лигнина,
углеводов, белков и т. д.),
Обстоятельный обзор работ последних лет и личные
наблюдения приводит крупнейший зоолог-почвовед современности
М. С. Гиляров [78, 79]. Почвенная фауна, очень разнообразная
в видовом составе, должна рассматриваться в настоящее время
как активный и обязательный агент разложения и гумификации
любых органических остатков, поступающих в почву, и все звенья
динамического комплекса: корневые системы—беспозвоночные—
гумус — взаимосвязаны [79]. Детально изучена роль дождевых
червей в формировании капролитов и специфике их органической
массы [255, 278], не менее существенна деятельность позвоночных,
обусловливающих прежде всего измельчение растительных
остатков и перемешивание их массы с почвой. Общий обзор роли
почвенной фауны в гумусообразовании можно найти в работе Д. Кева-
на [434].
Процессы гумификации органических остатков
в почве и вероятные компоненты ее
Современные представления о процессе гумификации. Если
характер процессов разложения—минерализации органических
остатков в почве недостаточно изучен только в деталях, а общая
схема его не вызывает дискуссий, то механизм гумификации,
несмотря на длительное изучение, до сих пор неясен. Существует
ряд гипотез, иногда совершенно по-разному трактующих сущность
этого процесса.
Прежде всего необходимо уточнить само понятие «гумифика-
122
ция» органических остатков. В современной литературе нет четкого
разграничения между понятиями «гумификация» и «гумусообра-
зование», хотя они нетождественны [17]. Мы уже отмечали и
неправомочность пользования этими понятиями как синонимами.
Гумусообразование, т. е. процесс формирования в толще почвы
особой системы органических веществ, — частный случай
биохимических преобразований органических остатков в биосфере.
Этот процесс развивается только в толще почвы, причем
гумификация — лишь звено этого процесса. В биосфере наряду с
процессом гумусообразования, являющимся неотъемлемой частью
почвообразования, развиваются и другие процессы трансформации
растительных остатков с формированием иных природных тел.
Идет процесс торфообразования, сапропелеобразования,
формирования органических компостов, в результате которых
образуются массы торфа, сапропеля, компостов и т. д. Для каждого из
этих процессов также характерна гумификация органических
остатков.
Следовательно, гумификация — универсальное звено
трансформации любых скоплений органических остатков в природе.
Она развивается при обязательном участии микроорганизмов,
всегда носит биохимический характер, но не является синонимом
процесса гумусообразования. Общая схема процесса
гумусообразования в почве такова (см. с. 124).
Как видим, гумификация является лишь звеном процесса
гумусообразования, для которого характерны и иные звенья:
синтез микробной плазмы, взаимодействие с компонентами
минеральной части почвы и т. д. Гумификацией следует называть лишь
процесс образования особого класса органических веществ —
гумусовых кислот, которые накапливаются при трансформации мертвых
растительных, микробных и животных остатков в биосфере,
в почве, торфе, сапропеле и других органогенных телах
природы.
Проблемой механизма гумификации исследователи занимались
на протяжении всей истории развития учения о почвенном гумусе,
генезисе торфа и угля [18, 19, 65, 142—144, 147, 230, 473]. Не
останавливаясь на представлениях В. Р. Вильямса о внутриклеточном
образовании гумусовых кислот как экзоферментах
микроорганизмов, так как эта гипотеза не получила экспериментального
подтверждения и противоречит современным представлениям о
природе ферментов [66, 66а, 67], отметим только вторую гипотезу
внутриклеточного происхождения гумусовых кислот [186, 260].
По этой гипотезе Е. С. Лукошко и В. Е. Раковского синтез
молекулы гуминовой кислоты протекает в две стадии, первая из
которых развивается внутри клеток зеленого растения, где происходит
циклизация глюкозы, пентозы и уроновой кислоты с образованием
мономера, являющегося дифуранбензолом. Вторая стадия —
дальнейшая конденсация мономеров, развивающаяся уже вне живой
клетки микроорганизмов, — обусловливает формирование много-
123
ф
в
о
к
в
в
в
Сб
в
о
со
сб
Рн
\о
о
о
о
>>
S
>>
Сч
Сб
О
О
ф
Я"
о
Он
в
Сб
3
ф
И
и
я
в
о
в
в
1=2
ф
Я Сб
в Ч д
S Ч °
? в g.
Я
в
ф
Ч
В
Ч
ф
И
я
в
Ч
ф
В
в
о
в
сб
Н
, О
^ о
Ф °^^
|->Я ? В
я о в
» и»
О Ч сб
ч фф
сб В
№ Ч
в
о
-со"
в
ч
о
ее
В
я
н
и
>>
1 **
о
?"•«
в в
В 1
ф ф
Я *
и 2
F Ч
о со
Н Сб
>>Оч
*
Ф
2
о
Он
а
в
в
я*
сб
--»»
Я—>
>&
я
«в
—
е кислоты
Я
В
о
о
>»
Я
>>
Сн
^—^
В
в
В"
йствие
адсорб
ф .
ее ф
~~*S
5 к
Я сб—>
->Вза
эразов
"з:
§
(сол
н
ьвокисло
? Ч
я >>
в>&«
ч в
[инера
овых
s в
в я
Сб ^»
С Сн
В
ч:
О
В
произ
ф
в
в
Сб
в
——. ч ——
— д —
IS
я
в
ф
в
в
ф
*в
ф ф
Сб О
со в
81
В
Я
=г
сб
со
Я
^•еб-
&
ф
Я
я
5 «
в о
в (-.
ф >>
?*
&§
Сб w
sS
IS
gvo
5 в
в
в
ф
Ч
Сб
ч:
В °*
Я 2
Я 8
Я *
PQ в
йдерной макромолекулы гуминовых кислот, которая может быть
представлена 160—200 мономерами с ММ около 40 000 [186]:
2С6Н1гОГОН^
,снгон
чсоон
+ н2о
аяьЭольная
конЭенсощия
гуминоВые
Вещества
Эта оригинальная гипотеза, принимаемая за основу многими
исследователями гуминовых веществ торфа [185], не подтверждена,
однако, экспериментальными данными по модельному
воспроизведению этой реакции и носит умозрительный характер, а потому
ее и нельзя считать доказанной. Наличие в продуктах гидролиза
производных уроновых кислот и моносахаров, конечно, не
подтверждает этого механизма гумификации. На вероятность
внутриклеточного синтеза гуминовых кислот, но уже в отмирающих
клетках микроорганизмов указывают Р. Свэби и Дж. Лэд [532],
считая, что микроорганизмы только разрушают клеточную оболочку
и освобождают образовавшиеся гуминовые кислоты. Внимательный
анализ материалов заставляет признать, что нет сколько-нибудь
убедительных доказательств о внутриклеточном синтезе гумусовых
веществ.
Большинство исследователей рассматривают образование
гумусовых веществ как процесс, осуществляющийся вне живой
клетки организмов из продуктов их частичного разложения,
признавая в разных вариантах биохимическую природу этого процесса.
125
Родоначальником современных гипотез о гумификации является
А. Г. Трусов [293J. По его мнению, гуминовые кислоты
образуются в процессе окисления и конденсации продуктов частичного
разложения белков, лигнина и дубильных веществ, причем
непосредственными «строительными единицами» являются пирроль-
ные, бензольные, фенольные и хинонные соединения — продукты
гидролитического расщепления указанных выше компонентов.
Первая стадия гумификации, по Трусову, — гидролитический
распад исходных органических соединений, второй — окисление и
конденсация продуктов гидролиза. Агентами гумификации
являются кислород воздуха, аммиак и оксидазы микроорганизмов.
Природные гуминовые кислоты А. Г. Трусов рассматривал как
«очень сложную смесь органических соединений, в которую входят
продукты гумификации дубильных, белковых и инкрустирующих
веществ, некоторых жирных и эфирных масел, вероятно
хлорофилла и смол» [293, с. 209]. Сам термин «гуминовая кислота»
А. Г. Трусов брал в кавычки, способ образования и природу фуль-
вокислот не обсуждал.
Обоснованием этой новой и очень оригинальной для того времени
гипотезы были многочисленные модельные опыты по гумификации
ряда органических соединений и различных растительных
остатков с последующим определением количества образующейся гу-
миновой кислоты. Эта гипотеза, выдвинутая в 1917 г., т. е. тогда,
когда учения о высокомолекулярных соединениях не
существовало, а по представлениям органической химии природные
органические соединения рассматривались как относительно простые
низкомолекулярные соединения, была безусловно очень
прогрессивной и оказала огромное влияние на все последующие работы по
изучению процесса гумификации.
После работ А. Г. Трусова наиболее обстоятельные
экспериментальные исследования по проблеме гумусообразования провел
С. Ваксман [65]. Его гипотеза о происхождении, составе и
свойствах гумуса, подтвержденная большим количеством
экспериментальных данных, очень быстро получила всеобщее признание,
а методика — широкое применение при исследовании f состава
гумуса. Процесс гумусообразования С. Ваксман рисует
следующим образом. При разложении растительных остатков микро-
юрганизмами в почве накапливаются лигнин, воски, смолы и
некоторые другие относительно устойчивые вещества.
Микроорганизмы синтезируют в почве новые органические комплексы —
^клеточное вещество, состоящее главным образом из белков.
Последние, а также часть первичных протеинов образуют гумусовое
:ядро — лигнино-протеиновый комплекс, составляющий, по
данным автора, 60—70% гумуса. Взаимодействие лигнина с
протеидом осуществляется в результате химической конденсации по типу
шиффовских оснований. Гумус и состоит из многочисленных
««дублеров», входивших в состав растений. Гумусовых кислот
жак специфических продуктов процесса гумификации в почве нет.
126
Несмотря на то что эта гипотеза подтверждалась большим
количеством аналитических данных, она была подвергнута критике
из-за недостатков методики исследований, рекомендованной самим
Ваксманом. Применявшийся им метод растительного анализа для
исследования природы гумуса почвы оказался непригодным [2,
3, 181, 297]. Вследствие несовершенства методики С. Ваксман
«прошел мимо» гумусовых кислот, реально существующих в почве*
Наиболее ценная часть концепции Ваксмана — та, что
свидетельствует об обязательном участии лигнина в образовании гумусовых
кислот.
В дальнейшее развитие представлений А. Г. Трусова о
сущности процесса гумификации крупный вклад внесли работы
М. М. Кононовой [140, 142, 143, 149], в которых также
подчеркнуто, что специфической реакцией гумификации является
конденсация ароматических соединений фенольного типа с
аминокислотами и протеинами; при этом если в первых работах М. М.
Кононова считала обязательной реакцию конденсации мономеров, то
по мере накопления новых данных автор расширяет
представления о механизме гумификации процессами поликонденсации
(полимеризации). Конденсация основных структурных единиц
осуществляется путем окисления фенолов ферментами типа фенол-
оксидаз через семихиноны до хинонов и взаимодействия
последних с аминокислотами. Поликонденсация, по мнению автора, —
химический процесс. Источником структурных единиц являются
продукты частичного распада лигнинов, таннинов, фенольные
соединения продуктов метаболизма микроорганизмов, продукты
частичного распада и ресинтеза белковых соединений —
аминокислоты и пептиды. Схема процессов гумификации М. М.
Кононовой такова (см. с. 128).
Эта гипотеза, сложившаяся у автора на протяжении
многолетних исследований с применением оригинального
микроскопического метода изучения хода гумификации растительных
остатков и многочисленных модельных опытов по взаимодействию
фенолов с различными аминокислотами и другими продуктами
частичного расщепления белков, пользуется широким признанием,
хотя и не вскрывает основной характерной черты процесса
гумификации — образования гумусовых кислот. Все реакции
конденсации неизбежно приводят к уменьшению количества
карбоксильных групп в образующихся продуктах, а не к их возрастанию,
тогда как гумусовые кислоты всегда содержат значительно больше
карбоксильных групп по сравнению с любыми гумусообразова^
телями. Не обсуждается также и механизм образования
различных групп гумусовых кислот, и прежде всего фульвокислот,
которые рассматриваются автором как наименее зрелые представители
группы гуминовых кислот [143].
К числу так называемых конденсационных гипотез следует
отнести меланоидиновую гипотезу С. М. Манской и Т. В.
Дроздовой [195 J, согласно которой основным звеном гумификации яв-
127
Схема процессов гумификации (по: [145])
Ў
3
Целлюлоза и прочие
углеводы
Растительные остатш
1
Белки
i
У
Лигнины, таннины
и АР- ч
1
Микроорг а низмы
со2, н2о
и др.
1
со2, н2о
и др.
Фенольные
соединения (продукты
метаболизма)
Аминокислоты,
пептиды (продукты
распада и ресинтеза)
Фенольные
соединения (продукты
распада)
ОН
-2е
-2Н+
V*
/М
;
H2N
Н-С-СООН
I
R
Конденсация
О.
-2е
2Н+
НО
но!
/
ляется конденсация хинонов с аминосоединениями типа меланои-
дов — продуктов взаимодействия хинонов с сахарами, пептидами,
аминокислотами, аминами и аммиаком.
Много лет механизм гумификации исследовал В. Фляйг —
наиболее яркий представитель так называемых полимериза-
ционных гипотез гумификации [393, 395, 396, 401—403].
Основным звеном гумификации, по Фляйгу, является
окислительная полимеризация соединений фенольной природы с
азотсодержащими органическими веществами, образующимися при
распаде белков, сопровождающаяся дегидратацией и деме-
тилированием. Основными катализаторами этих реакций
являются ферменты микроорганизмов, и прежде всего фенолокси-
дазы, а поставщиками структурных единиц молекулы — лигнин
и белки [396]. Следует отметить постепенную эволюцию
представлений В. Фляйга как на источники гумусовых кислот, так и на
детали самого механизма гумификации. Если в первых работах [393]
автор рассматривал гумификацию как полимеризацию мономеров
(фенолов и аминокислот), то в дальнейшем он рисует более слож-
128
ную картину, считая, что стадия «мономера» необязательна для
образования молекулы гуминовых кислот, и придавая все
большее значение реакциям окисления. В одной из работ [396] он
указывал, что непосредственными структурными единицами для
гуминовых кислот являются не «мономеры», а более крупные
продукты частичного расщепления лигнина растительного
происхождения, а также фенолы микробного происхождения с
разнообразными азотсодержащими компонентами (пептидами, аминокислотами,
аммонием). В более поздней публикации В. Фляйг и
соавторы [399] рисуют достаточно сложную схему образования
гуминовых кислот, указывая на многообразие источников,
участвующих в построении молекулы (см. с. 130).
При этом авторы все же остаются на позициях гипотезы
полимеризации как основного процесса гумификации. В формировании
молекулы, по Фляйгу, участвуют продукты распада лигнина и
азотсодержащие вещества микроорганизмов и растений, а также
фенолы, синтезированные микроорганизмами или содержащиеся
в растении. К сожалению, и в этой схеме не показан механизм
образования карбоксильных групп в гуминовых кислотах и
формирования центральной и периферической частей молекулы.
Различные варианты этой принципиальной схемы Фляйга
о механизме гумификации как окислительной полимеризации
можно найти в ряде работ, но, к сожалению, ни в одной из них
не обсуждается вопрос о механизме формирования кислотных
свойств гумусовых веществ. Так, например, своеобразна гипотеза
Дж. Финкла [391 ], в соответствии с которой характерная
реакция гумификации — сополимеризация гидрополистиренов как
продуктов деполимеризации лигнина с ароматическими
аминокислотами. Не менее оригинальна гипотеза Г. Фелбека [385, 387],
согласно которой гуминовые кислоты образуются в процессе
полимеризации пироновых колец с аминокислотами и фенолами,
причем основные источники этих структурных единиц — продукты
метаболизма грибов и плазма микроорганизмов. В серии работ
Л. Батистика, Дж. Майодона и Р. Симонара [348, 461, 516]
высказывается ряд предположений о характере связи протеинов
с основной частью молекулы гуминовых кислот; в исследованиях
М. Хайеса с соавторами [428] обсуждаются пути включения
полисахаридов в молекулу гуминовых кислот.
Несколько схем образования гуминовых кислот
исследователями немецкой школы (В. Фляйг и В. Цихман, Ф. Шеффер и
С. Шахтшабель, Ф. Шеффер и Г. Ульрих) приводят Г. Фидлер
и Г. Рейсиг [388]; они предусматривают возможность образования
гуминовых кислот из хинонов и аминокислот, из моносахаридов
и аминосоединений, из производных фенолов путем полимеризации
этих компонентов. В схеме Дж. Мартина и К. Хайдера [457]
также основной реакцией гумификации является полимеризация
фенольных радикалов и гидроксибензохинонов с пептидами и
аминокислотами.
9 Л. Н. Александрова 129
со
о
О
ф
а
га
о
К
Я
3
«
т
сб
«
о
00
ев
сц
VO
о
cd
I
о
*SeO"?
№
-0~5=5"s*°~0"
Необходимо указать на ряд работ Ф. Дюшофура с
сотрудниками [104, 378, 379, 381, 382], посвященных исследованию и
обсуждению механизма гумификации растительных остатков в почве.
Если в первых публикациях авторы понимали процесс
гумификации как полимеризацию мономеров лигнина с азотсодержащими
компонентами, то в последующих отмечают возможность
различных путей гумификации в зависимости от экологических условий,
в которых протекает этот процесс. Ф. Дюшофур в сводной работе,
опубликованной на русском языке, рассматривая органическое
вещество почв и его эволюцию, детально излагает свои взгляды
на процесс гумификации как на «сумму процессов синтеза,
приводящих к новообразованию коллоидных компонентов гумуса,
которые появляются за счет продуктов разрушения свежего
органического вещества» [104, с. 142]. Автор различает два основных
типа гумификации: абиотическую и биохимическую.
Абиотическая гумификация развивается в биологически малоактивной
среде (кислые торфяники, подстилки типа «мор»); характерная
черта ее — разложение свежего органического вещества при почти
полном отсутствии процессов синтеза, вследствие чего образуются
слабополимеризованные полифенолы из продуктов деградации
таннинов и лигнина. Эти полифенолы дают с протеинами
комплексы типа фульвокислот и бурых гуминовых кислот. В
биологически активной среде имеет место биохимическая гумификация,
которая в зависимости от химического состава исходных
растительных остатков возможна в двух вариантах. При гумификации
растительных остатков, богатых лигнином — основным
источником гумусовых веществ, — происходят его окисление, потеря ме-
токсильных групп, увеличение количества групп СООН и
взаимодействие образующихся полифенолов с продуктами разложения
белков (аминокислотами и аммонием). Растительные остатки,
богатые целлюлозой и гемицеллюлозами, в процессе гумификации
подвергаются разложению с образованием глюцидов и уронидов,
которые через глиоксаль подвергаются циклизации.
Образующиеся таким путем хинонные ядра фиксируют азотистые вещества
с образованием веществ, которые «сходны с серыми гуминовыми
кислотами» [104].
Таким образом, по представлениям Ф. Дюшофура, для
гумификации характерны реакции полимеризации относительно простых
продуктов разложения лигнина и белков. Оригинальная черта
его концепции — стремление объяснить происхождение различных
гумусовых кислот, хотя, конечно, эти суждения нуждаются еще в
дополнительной экспериментальной проверке. Аналогичные
представления развивает О. Даннеберг [371 ].
Таковы современные гипотезы сущности процесса
гумификации, основными реакциями которых являются конденсация
и полимеризация. Не рассматривая механизмов конденсации или
полимеризации, которые авторы трактуют по-разному, следует
отметить, что конденсацию или полимеризацию нельзя признать
131
9*
основной реакцией гумификации, так как она не доказана
экспериментально и недостаточно обоснована с позиций химической
кинетики.
Действительно, ни одна из этих гипотез не объясняет причин
развития процессов разложения только до относительно простых
промежуточных продуктов разложения (аминокислот, фенолов)
без их дальнейшей минерализации и причину смены этих
процессов реакциями конденсации или полимеризации, развивающимися
вне живых клеток микроорганизмов. Вместе с тем хорошо известно,
что реакции синтеза сложных органических соединений из простых
в природе осуществляются внутри живых клеток организмов и эта
особенность — универсальная принадлежность живой клетки.
Ее неизбежность во внешней по отношению к клетке среде требует
реальных доказательств. Хорошо известны многочисленные
модельные опыты с самыми различными мономерами, способными
к реакциям конденсации в условиях, близких к природным под
влиянием процессов окисления, но, к сожалению, все они
начинаются со своеобразного «постулата»: низкомолекулярных продуктов
распада — мономеров типа фенолов и аминокислот. Следует
отметить, что если свободные фенолы можно обнаружить в продуктах
разложения растительных остатков, то количество свободных
аминокислот обычно ничтожно, ибо они по мере образования
используются клетками микроорганизмов для построения плазмы или
процессов нитрификации.
В последнее время для объяснения причин реакций
конденсации и полимеризации используют теорию свободных
радикалов [440, 493, 526], которыми, как известно, называются атом или
группа атомов с одним или несколькими неспаренными
электронами, вследствие чего они способны участвовать в химической
связи [244]. В силу этих особенностей свободные радикалы
обладают высокой реактивной способностью и парамагнитными
свойствами. К числу свободных радикалов, образующихся при
окислении фенолов и их производных, относят, например, анион семихи-
нона
О 6
I
I J "^
он
V
\^
Наличие парамагнитных свойств у гуминовых кислот
установлено в ряде исследований. Большинство авторов предполагают,
что источником этих свойств являются радикалы семихинонного
типа [35, 275]. Но до настоящего времени нет каких-либо
экспериментальных данных, подтверждающих участие свободных
радикалов в реакциях полимеризации при образовании гумусовых
132
кислот. Неясна также природа линий в спектрах электронного
парамагнитного резонанса гумусовых кислот [47].
Не получает объяснения и сам механизм образования
высокомолекулярных кислот (процесс кислотообразования), хотя
гумификация — прежде всего образование особого класса кислот,
которые со времен К. Шпренгеля и Я. Берцелиуса называют
гумусовыми кислотами. Хорошо известно, что большинство реакций
конденсации сопровождается уменьшением количества
карбоксилов в процессе их взаимодействия с гидроксилами или
аминогруппами, в то же время не менее известно, чтр наличие большого
количества групп СООН присуще только гумусовым кислотам и
нехарактерно для любых других компонентов растительных остатков.
Остается неясным также механизм образования различных
групп гумусовых кислот: группы гуминовых кислот и группы фуль-
вокислот. По этому вопросу мнения диаметрально
противоположны: одни исследователи [143] считают фульвокислоты
предшественниками гуминовых кислот, другие [14, 37 ] — что
фульвокислоты представляют собой продукты разрушения гуминовых кислот.
Экспериментальные исследования и новая схема процесса
гумификации органических веществ. На протяжении последних
15 лет мы детально изучали динамику процесса гумификации
различных растительных остатков в условиях модельных
экспериментов [11-13, 15, 16,19, 20, 22, 22а, 24, 29, 30, 36-38, 43, 188-
190, 204, 221, 257, 267, 268]. Основные итоги исследований
позволили высказать ряд принципиально новых суждений о механизме
гумификации. Гумификация представляет собой сложный
биофизико-химический процесс трансформации промежуточных
высокомолекулярных продуктов разложения органических остатков
в особый класс органических соединений — гумусовых кислот.
Основными элементарными звеньями ее являются окислительное
кислотообразование, формирование азотистой части молекулы,
фракционирование и дальнейшая трансформация новообразованных
гумусовых кислот (их дальнейшая ароматизация и
гидролитическое расщепление, сорбция, конденсация), а также процессы
взаимодействия с минеральной частью почвы.
Необходимо подчеркнуть, что гумификация — длительный
процесс и трансформация новообразованных гуминовых кислот
продолжается в течение всего периода их существования, хотя
подтверждением этих суждений могут быть в настоящее время
лишь данные об абсолютном возрасте гуминовых кислот, который
исчисляется тысячелетиями [266, 487].
Первый элементарный процесс гумификации — биохимическое
окислительное кислотообразование, т. е. формирование системы
гумусовых кислот, подвергающихся в дальнейшем длительным и
сложным процессам трансформации. Данный процесс
осуществляется при непосредственном участии оксидаз
микроорганизмов, роль которых показана в исследованиях М. М.
Кононовой [140—142, 148]. Этот первый принципиальный тезис о началь-
133
153090 180 360 720 1530 30 180
Рис. 22. Изменение емкости обменного поглощения катионов в
разлагающихся растительных остатках (а) и количество групп СО ОН в
новообразованных гуминовых кислотах (б), образующихся из этих остатков в процессе
гумификации в разное время (по: [188]).
1 — наземная масса клевера, 2 — корни клевера, з — листья дуба, 4 — корни
разнотравной злаковой растительности.
ном этапе гумификации как кислотообразовании, впервые
опубликованный нами [19] на основании исследований тогда еще
аспирантов М. Ф. Люжина [188], В. Ф. Аршавской [43] и Э. А.
Румянцевой [268], можно иллюстрировать данными о количестве
кислых функциональных групп как в гумифицирующейся массе
растительных остатков, так и в образующихся в процессе
гумификации гуминовых кислотах (рис. 22). Емкость обменного
поглощения катионов в растительных остатках определена буферным
раствором СаС12+Н3В03 при рН 7, количество групп СООН —
ацетатным методом. Эти данные однозначно свидетельствуют о том,
что действительно в процессе гумификации формируются кислые
функциональные группы, т. е. происходит формирование
органических кислот из каких-то промежуточных продуктов разложения
органических остатков. В ряде случаев можно наблюдать уменьшение
количества фенольных гидроксилов, но эта закономерность
выражена нечетко и характерна лишь для растительных остатков
с их высоким содержанием.
Можно полагать, что это окислительное кислотообразование
идет по типу С ->СОН ->СНО ->СООН, хотя, конечно,
необходимы дальнейшие исследования для выявления тех группировок,
которые подвергаются окислению. Эти представления о начальном
звене гумификации как кислотообразовании, т. е. формировании
кислых функциональных групп, и в первую очередь карбоксилов,
нашли в последние годы подтверждение в работах ряда
исследователей [47, 336, 373, 377, 515, 520, 557]. Особый интерес
представляют исследования Р. Свифта и А. Познера [535], в условиях
лабораторного эксперимента инкубировавших в течение
длительного времени различные растительные остатки и выделявших
из них экстракцией 0.1 М Na4P207 новообразованные гуминовые
кислоты. Определение количества карбоксильных и фенолгидро-
ксильных групп методом потенциометрического титрования пока-
134
Рис. 23. Изменение количества
кислых функциональных групп в
образующихся гуминовых кислотах
при рН 7 (по: [535]).
1 и з — наземная масса клевера и пшеницы,
2. и 4 — корни клевера и пшеницы.
300
200
•1
'3
•2
360
720 сут
зало резкое возрастание количе- g ЮО
ства СООН на первом этапе
гумификации (рис. 23). Авторы
приходят к справедливому выводу
о том, что в процессе
гумификации формируются карбоксильные
группы, и подтверждают его ИК спектрами, на которых
интенсивность полосы 1720 см'1 в процессе гумификации возрастает во
всех объектах.
Высказываются опасения по поводу возможного
искусственного самоокисления молодых гуминовых кислот при экстракции
их щелочными растворами [145, 148]. Следует указать, однако,
что (и это отмечают сами авторы) данные опасения правомочны
лишь для свежих растительных остатков и самых
кратковременных сроков гумификации. Кроме того, водорастворимые
новообразованные гуминовые кислоты по содержанию кислых
функциональных групп, реактивоспособных при кислой, нейтральной
и слабощелочной реакциях, мало отличаются от
новообразованных гуминовых кислот, экстрагируемых слабыми растворами
щелочей (табл. 40) [257]. Наиболее интенсивно процесс кислото-
образования развивается в первый период гумификации, в
дальнейшем он затухает и сменяется иными процессами. На
аналогичные закономерности указывают Р. Свифт и А. Познер [535].
Таблица 40
в
Препарат
1
2
3
Содержание кислых функциональных групп
новообразованных гуминовых кислотах, мг-экв./100 г
Метод извлечения гуминовых кислот при разных значениях рН
водой
7
135
157
186
8
190
218
234
0.02 н. NaOH
7
188
210
8
208
272
Процессу окислительного кислотообразования подвергаются
не мономеры, а высокомолекулярные продукты разложения
растительных остатков, вследствие чего образующиеся гуминовые
кислоты с первых этапов своего существования представляют собой
135
Рис. 24. Распределение ММ новообразованных гуминовых кислот из гуми-
фицирующейся наземной массы клевера на сефадексе Г-75.
По оси абсцисс — объем элюата, по оси ординат — доля препаратов от их исходного
количества. I—3 — сроки гумификации, сут 1 — ол " 'сл ° ОЛА
36
32
28
24
20
16
12
6
Ч
г
г
""
-
Ц—_
3
г
1
—.—-д
-v—¦*
0
13 5 7 9 1113151719
13 5 7 9 1113151719
Рис. 25. Распределение ММ гуминовых кислот из гумифицирующихся
растительных остатков на сефадексе Г-75.
По оси абсцисс — фракции элюата; по оси ординат — содержание гуминовой кислоты а —
наземная масса клевера, б — листья дуба 1—5 — сроки гумификации, сут. J, 4 — 30;
2Х 5 — 180, 3 — 360.
высокомолекулярные соединения. Высокомолекулярная природа
образующихся гуминовых кислот доказана методом гельфильтра-
ции на сефадексах [43, 268]. По данным Э. А. Румянцевой [268],
все исследованные ею новообразованные гуминовые кислоты
характеризуются ММ 50 000—60 000, тогда как для гуминовых
кислот черноземов при использовании тех же марок сефадексов
характерны величины в пределах 40 000. Иными словами,
новообразованные гуминовые кислоты всегда более
высокомолекулярны, чем гуминовые кислоты, выделенные из почвы (рис. 24).
Следует отметить, что в этот первый этап процесса гумификации
новообразованных гуминовых кислот ММ их изменяется
незначительно (рис. 25). Некоторое возрастание величины ММ
относительно и обусловлено разложением наиболее низкомолекулярной
фракции микроорганизмами (табл. 41).
Таблица 41
Динамика ММ новообразованных гуминовых кислот
в процессе их дополнительной гумификации
Срок
дополнительной
гумификации, сут
0
150
300
Фракции по ММ
100 000—70 000
34.2
46.2
51.4
, % к сухому беззольному веществу
70 000—30 000
33.3
32.8
26.1
30 000-5000
32.5
21.0
22.5
Мер
51000
59000
60000
Примечание. Здесь и дальше МСр — условные средние ММ..
Этот вывод нашел подтверждение в работе А. В.
Назаровой [203], а также и в других исследованиях [46, 107, 336, 446,
520], где приведены данные о более высоких величинах для ММ
новообразованных гуминовых кислот по сравнению с гуминовыми
кислотами, выделенными из почв.
Новообразованные гуминовые кислоты очень гетерогенны как
вследствие большого разнообразия «структурных единиц», так
и благодаря непрерывности и длительности самого процесса
гумификации. Изучение' распределения ММ в новообразованных
гуминовых кислотах и выделенных из почв показало, что для
первых характерно более сложное распределение при гельфиль-
трации, чем для гуминовых кислот черноземов, и лишь постепенно
в процессе дальнейшего развития процесса гумификации степень
гетерогенности снижается (см. рис. 13, а). Дополнительным
подтверждением высокомолекулярной природы «структурных
единиц», которые подвергаются карбоксилированию, являются
материалы, полученные в нашей лаборатории в последнее время (см.
табл. 39); приведенные данные показывают, что на протяжении
137
12-месячной непрерывной гумификации растительных остатков
их водорастворимая фракция состоит в основном из веществ с ММ
более 1500. По-видимому, именно эти продукты неглубокого
расщепления лигнина, белков и других ароматических соединений —
непосредственный материал для образования системы гумусовых
кислот. Необходимы специальные исследования по их
идентификации.
Обязательным звеном этого начального этапа процесса
гумификации является расчленение системы образующихся гумусовых
кислот на ряд фракций, отличающихся друг от друга по составу и
степени подвижности. Эти процессы расчленения, химические
по своей природе, обусловлены реакциями фракционного
осаждения различных по миграционной способности компонентов
гумусовых кислот и их органо-минеральных производных.
Наши многолетние исследования показали, что основными
реакциями, определяющими эти процессы фракционирования,
являются реакции соле- и комплексообразования, формирующие
соли различной степени подвижности. Не менее существенно
значение процессов сорбции и адгезии, также обусловливающих
фракционирование системы гумусовых веществ.
Фракционирование системы гумусовых кислот при солеобразовании подтверждено
исследованиями по дробному осаждению отдельных фракций гуми-
новых кислот при осаждении их хлористыми солями натрия
различной концентрации [380] или при использовании катионов
разной валентности [5]. Прямым доказательством
фракционирования системы новообразованных гумусовых кислот при
взаимодействии с почвой являются данные, полученные в нашей
лаборатории [37, 188, 257]. И. Прох [257] фильтровал водорастворимые
гумусовые вещества через толщу почвы и наблюдал самопроиз-
Таблица 42
Расщепление водорастворимых гумусовых веществ
при фильтрации их через почву, мг С (по: [257])
Горизонт
Профильтровано
всего
в том числе
гк
ФК
Найдено в фильтрате
всего
в том числе
ГК
ФК
Дерно в о -подзо листа я суглинистая почва
Ai I 79 I 52 I 27 I 60 14 1 56
С | 79 I 52 I 27 | 51 | 6 | 45
Чернозем
1 79
79
52
52
27
27
52
43
6 1
5 j
Примечание. Здесь и далее ГК — гуминовые кислоты, ФК — фульвокислоты.
138
вольное отщепление фульвокислот от новообразованных гумино-
вых кислот (табл. 42), ибо в фильтрате во всех вариантах опыта
обнаружено больше фульвокислот, чем внесено, тогда как
количество гуминовых кислот резко уменьшилось частично при
поглощении почвой, а главным образом в результате трансформации
в фульвокислоты. Не менее убедительны опыты И. М.
Андреевой [37], инкубировавшей гуминовые кислоты в массе суглинка
и наблюдавшей в течение длительного времени отщепление
фульвокислот. В минимальных количествах фульвокислоты отщеплялись
от гуминовых кислот чернозема, в максимальных количествах —
от менее сформированных гуминовых кислот дерново-подзолистой
почвы и особенно от новообразованных гуминовых кислот,
выделенных из гумифицирующихся растительных остатков (табл. 43).
Таблица 43
Расщепление гуминовых кислот при их инкубации в суглинке
(внесено 100 мг С гуминовых кислот) (по: [37])
Объект, из которого выделены гуминовые
Гумифицированные листья клевера
Дерново-подзолистая почва
Чернозем
Найдено гумусовых кислот, мг С, после
инкубации, мес
1
68
32
70
30
90
10
6
67
33
68
32
87
13
12
69
33
60
40
80
20
Примечание. В числителе — гуминовые кислоты, в знаменателе —
фульвокислоты.
Приведенные данные достаточно убедительно подтверждают
нашу гипотезу о том, что одним из путей образования
фульвокислот является дифференциация образующейся при
гумификации системы гумусовых кислот при взаимодействии с минеральной
частью почвы. Конечно, велико значение направленности
процесса гумификации, обусловленной экологическими условиями,
в которых она протекает, или, пользуясь терминологией
Д. С. Орлова, глубиной гумификации. Компоненты системы
гумусовых кислот в зависимости от химического состава
гумифицирующихся растительных остатков, от гидротермических условий,
в которых протекает этот процесс, от состава самой минеральной
части почвы будут варьировать, но принципиальная схема
формирования гуминовых и фульвокислот, по-видимому, остается
единой.
Существенным звеном процесса гумификации является
формирование азотистой части молекулы гумусовых кислот. Действи-
139
тельно, каково происхождение столь разнообразных детально
рассмотренных выше форм азота в гумусовых кислотах? К
сожалению, этот вопрос исследован очень плохо, и в настоящее время
можно высказать лишь ряд предварительных суждений. Прежде
всего (как мы уже отмечали в гл. II) валовое содержание азота
в новообразованных гуминовых кислотах зависит от содержания
белков в исходном материале. Минимально содержание азота
в гуминовых кислотах, образующихся при разложении корневой
системы трав (2.4—4.0%), и максимально оно при гумификации
наземной массы клевера (6.4—7.8%; табл. 44). Эти данные также
косвенно подтверждают, что гуминовые кислоты образуются не
за счет реакций конденсации фенолов с аминокислотами, а путем
карбоксилирования высокомолекулярных продуктов разложения
различного химического состава, что подтверждается увеличением
количества групп СООН в препаратах. В процессе гумификации
содержание азота в гуминовых кислотах не остается постоянным.
При гумификации растительных остатков, богатых белками,
постепенно снижается содержание азота в молекуле образующихся
гуминовых кислот; в случае гумификации материалов, бедных
азотом, его количество заметно возрастает. Таким образом,
возможны как частичная потеря азота гуминовыми кислотами в
процессе их гумификации, так и присоединение каких-то форм его
к молекуле образующихся гуминовых кислот. Следует отметить,
что в процессе гумификации растительных остатков непрерывно
образуются новые молекулы гуминовых кислот с тем или иным
содержанием азота в молекуле, поэтому даже эти модельные
Таблица 44
Элементный состав, % на сухую беззольную массу,
функциональные группы, мг-экв./100 г, и гидролизуемость, %,
новообразованных гуминовых кислот (по: [43])
Объект исследования
Клевер, наземная
масса
Листья дуба
Корни
разнотравно-злаковой
растительности
Срок
гумификации,
сут
15
90
180
360
15
90
180
360
15
90
180
360
Зола,
%
2.26
0.60
1.42
2.30
2.68
1.30
1.65
1.00
2.35
1.45
2.49
2.60
С
57.1
57.8
58.4
59.1
56.2
50.7
51.1
49.3
57.5
55.3
55.6
55.3
н
5.0
5.0
4.5
3.5
5.7
4.8
5.0
5.1
5.3
5.5
5.1
5.5
N
7.8
6.4
6.4
6.4
5.5
6.4
6.9
6.6
2.4
3.1
4.0
3.3
О
30.1
30.8
30.0
31.0
32.6
38.1
37.0
39.0
34.8
36.1
35.3
35.9
соон
246
311
322
327
275
285
381
408
213
245
282
294
(ОН)
320
281
341
350
495
464
393
378
318
332
236
337
Гидролизуемость
С
40
40
34
18
34
36
26
26
Не
N
48
38
31
28
50
28
32
23
опр.
»
»
>
140
опыты дают лишь общую картину изменения содержания азота
в гуминовых кислотах в процессе их дальнейшей гумификации.
Для точного определения господствующих в течение гумификации
гуминовых кислот процессов трансформации ее азотистой части
Э. А. Румянцева провела серию специальных модельных опытов,
в которых препараты гуминовых кислот подвергались длительное
время дополнительной инкубации в оптимальных условиях
влажности и температуры при инокуляции их микрофлорой (табл. 45).
Таблица 45
Изменение элементного состава, % к сухой беззольной массе,
количества функциональных групп, мг-экв./100 г,
гумусовых кислот при их дополнительной гумификации (по: [268])
Объект, из которого
выделены гумусовые
кислоты
Гуминовые кислоты
клевер, гумифи-
цированный, сут
45
180
300
чернозем
Фульвокислоты
Bh подзола
Срок
дополнительной
гумификации, сут
0
300
0
300
0
300
0
300
0
300
с
55.6
57.2
56.6
53.1
54.4
51.5
57.9
57.8
46.4
41.1
н
4.4
4.3
5.0
4.1
4.2
5.7
3.4
5.0
3.0
3.9
о
31.5
32.1
30.0
35.9
35.2
38.5
34.2
33.8
47.5
54.1
N
8.5
6.4
8.4
6.9
6.2
5.3
4.5
3.4
3.1
0.9
Атомное
отношение
С
N
7.6
10.4
7.9
9.0
10.2
11.3
15.0
19.8
17.5
53.4
соон
270
382
300
388
331
400
461
461
(ОН)
306
158
276
248
245*
211
252
231
Полученные данные показывают, что в процессе дальнейшей
гумификации гуминовых кислот любого «возраста» господствуют
процессы постепенного отщепления азота, тем более интенсивные,
чем выше его содержание в гуминовых кислотах. В максимальных
количествах теряется азот из гуминовых кислот, образующихся
при гумификации материалов, богатых азотом; в минимальных
количествах обедняются азотом гуминовые кислоты, выделенные
из чернозема, но и в них содержание азота постепенно снижается.
Изменяется, по-видимому, и соотношение между алифатическими
(гидролизуемыми) и гетероциклическими (негидролизуемыми)
формами азота. В процессе постепенной гумификации доля
гетероциклических форм азота в молекуле гуминовых кислот
возрастает (табл. 44). Все эти данные свидетельствуют о том, что
в процессе гумификации непрерывно трансформируется
азотсодержащая часть молекул гуминовых кислот, причем прежде всего
141
вследствие реакций постепенного отщепления наименее
устойчивых алифатических группировок. Возможны, конечно, и реакции
противоположного порядка, при которых молекулы обогащаются
азотом. Некоторые авторы предполагают сорбцию аминокислот
молекулой гуминовых кислот [333, 508, 518], а также аммония
с последующей трансформацией его в формулу амидов [370, 560].
Не исследован экспериментально вопрос о происхождении
гетероциклических форм азота в молекуле гуминовых кислот.
Можно полагать, что в основном эти формы сохраняются в
молекуле от исходных структурных единиц, хотя не исключена,
конечно, и частичная трансформация компонентов самой
молекулы гуминовых кислот в процессе ее дальнейшей гумификации.
Такова в общих чертах характеристика начального этапа процесса
гумификации, который развивается относительно быстро, причем
интенсивность его регулируется прежде всего напряженностью
деятельности микроорганизмов. Именно этот этап следует
считать «биологической гумификацией», по терминологии Ф. Дюшо-
фура.
Существенным является вопрос о том, в какой фазе гумифици-
рующейся растительной массы развивается процесс гумификации.
Иными словами: обязательна ли для гумификации «водорастворимая
стадия»? Возможно ли развитие гумификации в пределах
поверхностной, достаточно гидратированной пленки тканей,
подвергающихся воздействию микроорганизмов? Для ответа на эти вопросы
необходимы специальные исследования, но косвенным
подтверждением возможности развития процесса гумификации вне ясно
выраженной «водорастворимой стадии» может служить хорошо
известный факт о необходимости использования щелочных
растворителей для выделения некоторой части новообразующихся
гумусовых кислот даже в том случае, когда гумификация
растительных остатков протекает вне какого-либо минерального
субстрата.
Очень слабо изучены процессы дальнейшей трансформации
новообразованных гумусовых кислот — этапы гумификации, но
они несомненно имеют место, о чем свидетельствуют пока еще
немногочисленные публикации о модельных опытах, проведенных
в нашей лаборатории [43, 268]. Данные А. Ф. Аршавской о
характеристике некоторых основных параметров гуминовых кислот,
образующихся при гумификации различных растительных
остатков (см. табл. 44), показывают, что по мере увеличения срока
гумификации эти кислоты обогащаются карбоксильными
группами, в них возрастает содержание кислорода, отчетливо
снижается гидролизуемость как по углероду, так и по азоту.
Использование метода гельфильтрации показало, что на протяжении
всего периода гумификации новообразованные гуминовые кислоты
остаются высокомолекулярными веществами с резко выраженной
полидисперсностью (рис. 25). В то же время по этим данным еще
нельзя достаточно обоснованно установить дзмецеция струк-
142
Таблица 46
Продукты окисления щелочным раствором КМп04 препаратов
гуминовых кислот до и после их дополнительной гумификации,
% к сухому беззольному препарату (по: [268])
Объект, из которого
выделены гуминовые
кислоты
Клевер, гумифициро-
ванный, сут
45
180
300
Чернозем
Срок
дополнительной
гумификации, сут
о
30
150
300
0
30
150
360
0
30
150
360
0
150
360
Бензолполи-
карбоновые
кислоты
12.8
15.3
18.5
18.3
11.8
16.3
18.4
18.4
16.7
17.9
18.2
15.0
22.1
20.0
14.6
Кислоты типа
щавелевой
68.7
66.2
63.0
63.7
78.0
70.8
68.9
69.1
69.6
70.9
66.8
52.1
41.8
43.8
31.1
Продукты
полного
окисления
18.5
18.5
18.5
18.0
10.2
12.9
! 12.7
12.5
12.7
11.2
15.0
22.9
36.1
36.2
54.3
туры самих молекул гуминовых кислот, так как в ходе
гумификации растительных остатков постоянно образуются новые
молекулы, а определяемые в опыте параметры гуминовых кислот дают
какие-то средние величины. Для более точного определения
характера изменения гуминовых кислот в процессе их
дальнейшей гумификации были поставлены специальные модельные опыты
по дополнительной гумификации препаратов новообразованных
гуминовых кислот (табл. 46, 47). Они показали, что наряду с
обеднением азотом и возрастанием количества карбоксилов постепенно
изменяется соотношение между ароматической и алифатической
частями в молекуле. Во всех «молодых» гуминовых кислотах
в процессе дальнейшей гумификации возрастает выход бензол-
карбоновых кислот при заметном снижении количества
низкомолекулярных кислот, что свидетельствует, по-видимому, о некотором
разрушении алифатической части молекулы и относительном
возрастании степени ароматизации; ММ гуминовых кислот при
этом изменяется мало, препараты остаются высокомолекулярными.
Определение массы фракций, различных по молекулярной
полидисперсности, показало, что содержание наименее
высокомолекулярной фракции в процессе дополнительной гумификации
препаратов новообразованных гуминовых кислот заметно
снижается, а количество фракции более 70 000 возрастает (табл. 47).
143
Таблица 47
Молекулярная полидисперсность и Жср препаратов
новообразованных гуминовых кислот до и после
их дополнительной гумификации (по: [268])
Препарат из
клевера, гумифици-
рованного
в различные
сроки, сут
45
180
300
Срок
дополнительной
гумификации,
сут
со со со
о о о
оооооо
Полидисперсность, % к сухой беззольной
массе, фракции с разной ММ
100 000-70 000
34
51
38
46
39
43
70 000-30 000'
33
26
32
33
31
32
30 000-5000
33
23
30
31
30
25
Afcp
51000
60 000
53 000
53 000
54 000
57 000
С повышением срока гумификации (т. е. с увеличением возраста
гуминовых кислот) прирост Л7ср уменьшается. Но в целом в
процессе дополнительной гумификации характер молекулярной
полидисперсности новообразованных гуминовых кислот заметно не
меняется (рис. 26, б). Вероятно, лишь длительное развитие
процессов трансформации гуминовых кислот в почве приводит к
постепенному снижению полидисперсности и уменьшению Жср,
что видно при сопоставлении этих величин в новообразованных
гуминовых кислотах и гуминовых кислотах, выделенных из
чернозема. Не исключена, конечно, частичная взаимная
конденсация или полимеризация молекул.
Иные закономерности дальнейшей гумификации
(трансформации) выражены в гуминовых кислотах чернозема и фульвокисло-
тах подзола. Гуминовые кислоты чернозема наиболее «инертны»,
хотя и для них характерны заметное обеднение азотом и изменение
отношения С : Н. Содержание карбоксильных и фенолгидроксиль-
ных групп практически не изменяется, а выход бензолкарбоновых
кислот к концу опыта уменьшается (см. табл. 45, 46). Не менее
своеобразны и изменения в структуре фульвокислот при их
дополнительной гумификации: в них резко снижается содержание
азота и не менее резко возрастает степень окисленности, но
количество карбоксилов остается практически прежним. Мало
изменяется и Мср. Он колеблется в пределах 36 000—42 000, хотя
до и после дополнительной гумификации фульвокислоты
становятся более полидисперсными (рис. 26, а): появляются как
наиболее высокомолекулярная фракция (Л/ср > 70 000), так и
более низкомолекулярные компоненты в пределах фракции
Л/ср от 70 000 до 30 000.
Для более глубокого анализа тех изменений, которые
наблюдаются в молекулах новообразованных гуминовых кислот при их
дальнейшей гумификации, был использован графостатический
144
34 44
MMZ70000 v0
MM* 70000
MM $3000
Рис. 26. Распределение MM фульвокислот (а) и новообразованных гуминовых
кислот (б) до (1) и после 300 сут (2) дополнительной гумификации (по: [268]).
По оси абсцисс — объем элюата, по оси ординат — доля препаратов от их исходного
количества
метод Ван-Кревелена [162]. Этот метод (рис. 27) дает, конечно,
лишь приближенное представление о характере процессов,
протекающих при дальнейшей гумификации новообразованных
гуминовых кислот, так как мы не знаем числа углеродных атомов в
расчете на одну молекулу и нам неизвестны все кислородные функции
ее. Но в первом приближении, по-видимому, отношение Н : С можно
принять за показатель относительной доли ароматических
группировок в молекуле, а отношение (N+0) : С — как степень
развития процессов окисления. Из приведенных условных расчетов
соотношения ароматической и алифатической частей молекулы,
которые позволяет сделать метод Ван-Кревелена (табл. 48),
Ю Л. Н. Александрова 145
В 0.5 10 (A/+0)/C
С
Рис. 27.|Зависимость между атомными отношениями H/G и (N+0)/G в гу-
миновых кислотах после их дополнительной гумификации в течение 300 сут
(по: [162]).
а — дегидрогенизация, б — окисление, в — гидрогенизация. 1—3 — гуминовые кислоты
из наземной массы клевера, гумифицирующегося 45 (I), 180 (2) и 360 сут (5); 4 — из
чернозема; 5 — из подзола (гор. Вь).
видно, что при дальнейшей гумификации новообразованных гумино-
вых кислот развивается комплекс реакций: реакции дегидратации,
деметилирования и окисления. В наиболее молодых гуминовых
кислотах отчетливо выражены частичное разрушение
алифатической части молекулы и возрастание относительного содержания
ароматических группировок, ибо отношение Н : С становится
менее широким. Этот процесс подтверждается, как мы уже
отмечали, и увеличением выхода бензолполикарбоновых кислот.
Постепенно процессы окисления и дегидрогенизации затухают и
сменяются реакциями частичного разрушения ароматического ядра,
что заметно для новообразованных гуминовых кислот,
выделенных после 300 сут гумификации клевера, и особенно ярко
выражено в препарате гуминовых кислот чернозема. Для фульвокислот
характерны процессы окисления и разрушения ароматической
части молекулы.
Одновременно с процессами трансформации молекул
гумусовых кислот в почве развиваются процессы их частичной
минерализации, причем наиболее интенсивны они для молодых
гуминовых кислот. К сожалению, данные о минерализации гуминовых
кислот все еще ограниченны [1, 41, 64, 166, 200, 219, 405, 449,
510], что побудило нас провести специальные исследования.
Скорость минерализации гуминовых кислот детально изучена
146
Рис. 28. Скорость минерализации qq мг
ГуМИНОВЫХ КИСЛОТ. j2Qr
1 и 2 — из наземной массы клевера, гуми-
фицированного 45 (1) и 360 сут (2); 3 и
4 •— из чернозема при добавлении (3) и
без добавления (4) глюкозы; 5 —
минерализация глюкозы.
Э. А. Румянцевой на препаратах,
описанных выше. Основные
показатели скорости
минерализации — динамика выделения С02
при дополнительной
гумификации (инкубации) гуминовых
кислот и определение их
минерализующейся массы. В связи с тем
что для наиболее интенсивной
минерализации гуминовых
кислот необходимо добавление
глюкозы [405], ее вносили во все
препараты (рис. 28).
Прежде всего необходимо отметить, что минерализация
гуминовых кислот — процесс очень медленный по сравнению с
гумификацией и минерализацией свежих растительных остатков, что
и приводит к накоплению гумуса в процессе преобразования любой
горной породы в почву. По нашим наблюдениям, за 300 сут в за-
Таблица 48
Соотношение ароматической и алифатической частей молекулы
гумусовых кислот до и после их дополнительной гумификации
по Ван-Кревелену (по: [268])
WU1
во
60
?0
20
-
-
/
~ 1/1
рС-* 1
/ ; \
/rW
1 ь \\Ч
5- **\
1 _....*1 Г 1
4 6 3 10 сут
Объект, из которого выделены
гумусовые кислоты
Гуминовые кислоты
клевер, гумифицирован-
ный, сут
45
180
300
чернозем i
Фульвокисяоты Вь
подзола
Срок
дополнительной
гумификации,
сут
о
300
0
300
0
300
0
300
0
300
Алифатическая
часть, %
Ароматическая
часть, %
31.0
24.8
31.0
24.8
24.8
36.6
11.4
31.0
15.2
38.6
69.0
75.2
69.0
75.2
75.2
61.4
88.6
69.0
84.8
81.4
147
10*
висимости от «возраста» гуминовых кислот минерализовалось от
7.6 до 9.0% первоначальной массы их (табл. 49). Длительное
изучение процесса минерализации гуминовых кислот в лабораторных
условиях затруднено из-за неизбежного затухания этого процесса
при образовании токсических для микрофлоры продуктов и
уменьшении коэффициента использования энергии с возрастом
культуры. Приведенные материалы позволяют высказать ряд новых
предположений о вероятном характере процессов гумификации
растительных остатков в почве и представить для обсуждения
новую схему ее (см. с. 149).
Таблица 49
Гуминовые кислоты, минерализованные в присутствии глюкозы,
% к исходной массе (по: [268])
Объект, из которого выделены гуми-
Клевер, гумифицированный, сут
45
180
360
Чернозем
Минерализация при разном сроке инкубации,
сут
30
6.5
6.0
5.8
4.6
150
8.6
6.9
6.3
4.8
300
9.0
7.6
Не опр.
»
Прежде всего необходимо подчеркнуть длительность самого
процесса гумификации и многообразие отдельных звеньев. В
настоящее время следует уже выделять несколько стадий (этапов)
его. На первой стадии гумификации ведущими являются процесс
кислотообразования как результат биохимического окисления
высокомолекулярных продуктов разложения органических
остатков и одновременно развивающееся фракционирование системы
образующихся гумусовых кислот по степени растворимости на
группу гуминовых кислот и группу фульвокислот. В зависимости
от реакции среды в почве сохраняются свободные гумусовые
кислоты или их соли как результат взаимодействия с минеральной
частью почвы. Таким путем в почве образуется сложная система
свободных гумусовых кислот и их органо-минеральных
производных, которые находятся в виде гидратированных пленок на
поверхности различных по размерам гранулометрических
фракций, а также в виде агрегатов (в той или иной степени
гидратированных) в порах между ними. Одновременно формируется
азотистая часть молекулы образующихся гумусовых кислот путем
малоизученных реакций сорбции и десорбции аммиака, последующей
трансформации сорбированного аммиака в состав азотсодержащих
группировок, поглощения продуктов разложения белков, прежде
148
иийееик^онии
ионнэкУем HiHiCtfodn
ф
и
v
о
в
n
н
о
ф
S
и
а
в
о
о
>>
Я"
со
S
Си
о
>©•
о
»
О,
Н
Ф
В
Ч
ев
В
В
еб
«
В
>8<
В
3
>>
(-1
ей
ф
И
и
в в
В 3
еб еб
Я ?
пользе
эганиз
§о
ф
в
в
ф
азлож
си
.
ф
в
в
ф
ч
о
В
SCJ
Q
< А
еб
ОЛЬЦ
ние
v ф
и о
ч В
Й4 о
со
еб
4
В
В
tf
^—s
/
S
S
ф
в
в
ф
в
л
в
ф
>>
в
в
Эй 2-
еб еб
В о
| и 9
еб и
~ о
леяие
ГК
образ
н —-
о
>>
Г Ч
I °
1*
об
Ч
о
»
CQ
Я
р.
со
еб
Рн
к
в °
в
ф
ч
ф ад
1-е
еб
Рн
ф
о
й е
ф
н
В
Ч
§—>55
§™е
е
t
101ГЭИН ХНЯОО^И^Л
ХИШВЖ^вЙОО-^ BW9X0H0
¦ -
еб
ю
О
со
еб
О,
\о
о
о
н
§
о
в
^
г
+1
ф
в
в
винэжоке^ niHiCWodn
> 9HHdBirXH9ifowoHO0Ha
i
Ф
В
В
ф
ч
в
1
о
еб
Рч
„
ф
окислвни
к
2
S
ф
в
в
ф
а
^
в
ф
2
>»,
| ф ф
о в
в в
! да Ф
2 В
остег
азру
i я р*
I 1 2
1 о
Н в
ьн В
ф
1 §а
Н Ф
со 3
[ i
еб
! И
1 В
в
1 >>«
шая г
рваци
зВ Ф
ф О
в и
л о
ч к
и «
в в
V И"
1
1—1
1—»
в
8
со
ф
в
азован
ислот
л. т
новооб]
СОВЫХ ]
1 >>
еб
со
В
еб со
РнВ
^ |JH Р*
VD Ф
В
В
2
всего аминокислот, а также, возможно, и внутримолекулярных
перегруппировок, обусловливающих динамическое соотношение
гетероциклических и алифатических компонентов.
Для второй стадии гумификации характерны процессы
дальнейшей трансформации молекул гуминовых кислот и фульвокис-
лот. В гуминовых кислотах постепенно возрастает степень
ароматизации вследствие частичной деструкции — отщепления
белковых алифатических цепей, а также дезаминирования и
внутримолекулярных перегруппировок. Механизм этой стадии
гумификации еще не изучен; по-видимому, и она совершается при участии
ферментативных систем микроорганизмов и, развиваясь вне
живых клеток под воздействием гидролитических ферментов и окси-
даз, приводит к уменьшению ММ гуминовых кислот. Д. И.
Никитин [219] указывает на участие пероксидазы в этой деструкции
молекулы; Ф. Соуден [522] описывает частичный гидролиз
гуминовых кислот с помощью лейцин-аминопептидазы и папаина.
Отмечая возрастание степени ароматизации молекулы гуминовых
кислот в процессе их дальнейшей гумификации путем
постепенного отщепления ее алифатических частей, мы хотим лишь
подчеркнуть принципиальную черту этой стадии, имеющей не
синтетическую, а гидролитическую и окислительную направленность,
но осложненную спектром сопутствующих реакций. Несомненны
реакции сорбции на гуминовых кислотах и их органо-минераль-
ных производных продуктов разложения органических остатков,
показанные в исследованиях ряда авторов [118, 286, 311, 333,
508]. Не исключено, что эти реакции сорбции сопровождаются
последующими процессами конденсации, внутримолекулярными
перегруппировками, обусловленными как биологическими, так
и абиотическими факторами. Эта стадия гумификации,
по-видимому, очень длительная, а если учесть, что одновременно в почву
поступают новые массы органических остатков, разлагающихся
с образованием сложной системы высоко- и низкомолекулярных
продуктов разложения, становятся очевидными сложность и
многообразие процесса дальнейшей трансформации
новообразованных гуминовых кислот.
Трансформация молекул фульвокислот в соответствии с
единичными, к сожалению, данными происходит, по-видимому,
преимущественно по типу их дальнейшего гидролитического
расщепления и окисления при непосредственном участии
микроорганизмов. Известна относительно высокая доступность
фульвокислот микроорганизмам [37, 268]. Эти процессы также приводят
к постепенному уменьшению ММ. И этот ход трансформации
показан выше лишь принципиальной схемой, осложненной в
природных условиях реакциями взаимодействия как с промежуточными
продуктами разложения, так и с компонентами минеральной части
почвы.
Неизбежна и следующая стадия трансформации гумусовых
веществ — их постепенная минерализация. К сожалению, и этот
150
процесс практически не изучен, хотя сам факт минерализации
гумусовых веществ подтверждается многими авторами [1, 41, 64,
200, 219, 267, 449]. Несомненно, что эти процессы биохимические
и совершаются они при участии многообразной системы экзофер-
ментов микроорганизмов. Основными реакциями должны быть
гидролиз и оксиредукции, которые приводят к гидролитическому
расщеплению молекулы, разрушению гетероциклических и
ароматических группировок и постепенному полному окислению до
конечных продуктов: аммиака, воды и углекислого газа.
Многообразие типов микробиологического окисления самых
различных по степени устойчивости органических соединений
показано в трудах ряда исследователей [289, 292, 326]; сводка
современных работ этого плана дана Г. Фонкеном и Р.
Джонсоном [313]. Типы микробиологического окисления гумусовых
веществ и образующиеся при этом продукты еще не изучены.
Такова общая принципиальная схема процесса гумификации
органических остатков в почве, обусловливающая наличие в
каждый данный момент сложной и динамической системы гумусовых
кислот и их производных, находящихся на разных стадиях этого
процесса. В зависимости от конкретного сочетания факторов
гумификации скорость этого процесса и характер образующихся
продуктов варьируют.
Факторы гумификации
Если факторы, регулирующие скорость и характер процессов
разложения—минерализации растительных остатков, хорошо
исследованы совместными трудами микробиологов, биохимиков и
почвоведов, то факторы, регулирующие скорость и характер
процессов гумификации, изучены все еще недостаточно, и до
настоящего времени фактические материалы по этой проблеме
ограниченны.
К числу основных факторов, регулирующих скорость и
характер гумификации, следует отнести количество и характер
поступления растительных остатков в почву, их химический состав,
режим влажности и аэрации, реакцию среды и
окислительно-восстановительные условия, интенсивность микробиологической
деятельности и групповой состав микроорганизмов, а также
гранулометрический, минералогический и химический состав
минеральной части почвы.
Масса растительных остатков, поступающая в почву в
различных природных условиях, очень разная (см. табл. 1), а потому
несомненно при прочих равных условиях регулирует
интенсивность гумификации. Иллюстрацией могут служить данные о
среднем ежегодном поступлении в почву растительных остатков и
запасов гумуса в метровом слое большинства почв (рис. 29).
Уничтожение природной растительности в любой зоне приводит к посте-
151
т/га
Рис. 29. Запасы гумуса (1) и среднее количество растительных остатков (2)
в почвах СССР (в слое 0—100 см).
а — тундровые, б — подзолистые, в — серые лесные, г — черноземы, д — каштановые,
е — бурые пустынно-степные, ж — серо-бурые пустынные.
пенному обеднению почвы гумусом. Не менее существенное
значение имеет характер поступления растительных остатков в почву.
Поступление их на поверхность почвы всегда снижает темп
гумификации, обусловливая при благоприятных для
жизнедеятельности микроорганизмов условиях господство процессов
разложения—минерализации или консервации этих остатков в случае
господства постоянного избыточного увлажнеция.
Химический состав гумифицирующихся растительных остатков
регулирует при прочих равных условиях как скорость
гумификации, а следовательно, и количество образующихся: гумусовых
веществ, так и ее характер, от которого зависит групповой состав
гумусовых веществ. Хорошо известно, что органические остатки,
богатые белками, дают максимальный выход гумусовых кислот,
в составе которых доминируют гуминовые кислоты, тогда как
растительные остатки, богатые лигнино-целлюлозными
комплексами с ярко выраженным волокнистым строением тканей, гуми-
фицируются медленно и образуют значительно меньше гумусовых
кислот. Примером могут служить наши данные о скорости и
характере гумификации различных по химическому составу
растительных остатков. При гумификации древесных опилок и корней
разнотравно-злаковой растительности образуется в 2—3 раза
меньше гуминовых кислот, в составе которых доминируют фульво-
кислоты (рис. 30). Аналогичные данные получены и другими
исследователями [218, 482].
Не менее существенное значение для процесса гумификации
имеет режим влажности и аэрации, что также было отмечено
ранее. Постоянное избыточное увлажнение, тормозя процессы
152
мг/10г
2000
1750
1500
1250
1000
750
500
250
Г °
Г г-П 3
г б и
L 2
Nil Nil UN г-гП
3 6 12 3 6 12 3 6 12 3 612 ' 3 В 12мес
Рис. 30. Влияние химического состава растительных остатков на количество
образующихся гуминовых кислот.
По оси абсцисс — длительность гумификации, а — наземная масса клевера, б — корни
клевера, в — листья дуба, г — хвоя ели, д — опилки.
разложения, одновременно обусловливает преимущественное
накопление группы фульвокислот. Впервые эта закономерность была
показана еще Б. Одинцовым [227] и подтверждена современными
исследованиями [188, 517].
Наиболее благоприятными для гумификации и закрепления
в почве образующихся гумусовых веществ считаются чередование
оптимальных условий увлажнения с недостатком влаги,
вследствие чего периоды интенсивной деятельности микроорганизмов
сменяются их депрессией и фиксацией в почве образовавшихся
гумусовых веществ [143, 218]. Постоянный аэробиозис при
оптимальном увлажнении усиливает минерализацию образующихся
гумусовых веществ, постоянный недостаток влаги приводит
к слабоизученным процессам тления и полной минерализации
органических остатков.
Не менее существенное влияние на скорость и характер
гумификации оказывают реакция среды и
окислительно-восстановительные условия. По данным исследователей [143, 431], кислая
реакция тормозит развитие процесса гумификации прежде всего
вследствие подавления деятельности бактерий. Данные,
полученные в последнее время В. В. Рассохиной [261 ] в условиях
модельного опыта, четко показали, что кислая реакция не только
тормозит новообразование гумусовых веществ, но существенно влияет
и на состав образующихся продуктов. В кислой среде резко
снижалось количество новообразованных гуминовых кислот и
возрастало содержание фульвокислот, что свидетельствует об
усилении реакций гидролиза в процессе гумификации.
М. М. Кононова указывает [143], что
окислительно-восстановительные условия, регулирующие степень развития
окислительных процессов, — тоже существенный фактор гумификации,
хотя первые фазы разложения растительных остатков обусло-
153
426
Щ7\ ШбЗ_ 7050
а.
Рис. 31. Микроорганизмы (А, тыс.) и гумус (Б, т/га) в различных
почвах СССР (по: [143]).
1 — на 1 г почвы, 2 — на 1 г гумуса, 3 — 0—100 см, 4 — 0—20 см. а — бурые и сероземы,
б — каштановые, в — черноземы обыкновенные, г — подзолистые, д — тундрово-глеевые.
вливают значительное снижение
окислительно-восстановительного потенциала. Оптимальными величинами Eh, благоприятными
для гумификации, являются средние величины: 250—300 мВ.
При резком возрастании Eh до 500—600 мВ резко усиливается
окислительная минерализация продуктов разложения.
К числу ведущих факторов гумификации относится безусловно
интенсивность деятельности микроорганизмов, так как весь
процесс гумификации биохимический. Как отмечает М. М.
Кононова, высокая биогенность почвы не способствует накоплению
значительного количества гумуса вследствие интенсификации
процессов полной минерализации, слабо развивается
гумификация при низкой биохимической активности микроорганизмов,
обусловленной как малым количеством их, так и снижением их
ферментативной активности. Наиболее интенсивно процесс
гумификации (и консервации гумусовых веществ) развивается при
некоторой средней биохимической активности в почве (рис. 31).
Очень существенно влияние минеральной части почвы на
процесс гумификации, причем оно многообразно. Наши
исследования, проводившиеся в течение ряда лет, показали, что скорость
и интенсивность гумификации, а также количество
накапливающихся в почве гумусовых кислот регулируются прежде всего
гранулометрическим составом, т. е. величиной общей и удельной
поверхности почвы. Чем тяжелее гранулометрический состав,
тем больше гумусовых веществ накапливается в почве при
разложении растительных остатков (рис. 32). Механизм этого
накопления детально рассматривается в следующей главе; здесь
отметим только, что это накопление обусловлено сорбцией
промежуточных продуктов разложения, образованием ряда нерастворимых
в воде органо-минеральных производных и их сорбцией на поверх-
154
С,мг
7 30 90 180 360 540 720 Сцт
Рис. 32. Влияние гранулометрического состава на накопление гумусовых
веществ, извлекаемых 0.1 М раствором Na4P207 при гумификации корней
клевера, в песке (1, 2) и суглинке (3, 4) (по: [189]).
По оси абсцисс -— длительность гумификации. 1,3 — общая сумма; 2, 4 — гуминовые
кислоты.
ности минеральных частиц почвы. Кроме того, наличие гумуси-
рованных пленок на поверхности частиц уменьшает сорбционную
способность поверхности, и накопление новообразованных
гумусовых веществ снижается вследствие их быстрой минерализации.
Из приведенных данных (рис. 32) видно, что суглинистая масса
почвы с большой свободной поверхностью оказывает существенное
влияние на ход гумификации и свойства образующихся гумусовых
веществ. Если гумификация растительных остатков в массе песка
длительное время сопровождается образованием значительного
количества водорастворимых органических веществ, то наличие
суглинистой массы резко снижает содержание всей
водорастворимой фракции, которая, вероятно, интенсивно поглощается на
свободной поверхности гранулометрических фракций. Как было
показано исследованиями А. В. Назаровой [203], эти процессы
сорбции приводят к фракционированию гуминовых кислот, наи-.
более энергично сорбируются фракции менее
высокомолекулярные, т. е. наиболее сформированные как гуминовые кислоты.
Не менее велико влияние минералогического и химического
состава минеральной части почвы на гумификацию. Как показали
исследования, проведенные в нашей лаборатории [31, 312],
минералы группы каолинита сорбируют значительно меньше
гумусовых веществ, так как меньше их удельная поверхность. Каолинит
и бентонит с зернами размером 1—10, 1.0—0.2 и меньше 0.2 мкм,
а также лимонит настаивали с водорастворимыми продуктами
гумификации наземной массы клевера, а также с гуминовыми
кислотами дерново-подзолистой почвы и после удаления
центрифугированием избытка органических веществ и высушивания фракций
с сорбированными органическими веществами в них определяли
удельную поверхность БЭТ методом [63] и снимали микрофото-
155
а
а
If
о
H
§
со
P
1
Kg
?-©
о
в
111 11
0)
в
ег
ев 3
К о
Я
tc
s
я
о
§
о
о
ю
л
1=1
n
о
§
5
1
CO
§
о
s
к
H"
8
s
s
>»
5
в
1
ff
В
И
о
s
a
etf
CD
s
s
A
R
графии шлифов. Детально эти материалы рассматриваются в
следующей главе; здесь же отметим, что сорбция органических
веществ, в том числе и гуминовых кислот, бентонитом была
значительно выше, чем сорбция этих же продуктов на каолините.
Основная причина — в резком различии величины удельной
поверхности этих минералов.
Очень велико влияние состава обменных катионов, количества
и степени гидратации несиликатных полуторных окислов на
формирование в почве системы гумусовых веществ. Присутствие
в почве способных к обмену катионов кальция, магния, натрия,
калия обусловливает образование различных по растворимости,
а следовательно, и способности к аккумуляции гуматов и фульва-
тов [19, 73, 143]. Несиликатные полуторные окислы участвуют
в формировании сложных железо- и алюмогумусовых
производных, которые также, характеризуясь неодинаковой подвижностью,
обусловливают разную миграционную способность образующихся
гумусовых веществ [19, 95, 178].
Таким образом, конкретное проявление процесса гумификации
растительных остатков в почве всегда очень многообразно и
обусловлено комплексом факторов, благодаря чему в почвах
формируются различные по составу и миграционной способности
гумусовые вещества. Первая современная классификация характера
гумификации (точнее, типов накопления органического вещества)
принадлежит, по-видимому, И. В. Тюрину, предложившему
выделять четыре главных типа накопления органического вещества
в почвах: торфяно-болотный, гумидный кислый, семигумидный
нейтральный и аридный щелочной [297]. Она хорошо отражает
основные характерные особенности процесса гумификации в
зависимости от конкретного сочетания факторов этого процесса.
Несколько модернизируя эти представления, предлагаем свою
группировку основных типов гумификации (табл. 5(3).
Глава IV
ОРГАНО-МИНЕРАЛЬНЫЕ ПРОИЗВОДНЫЕ
ГУМУСОВЫХ КИСЛОТ И ИХ СОСТАВ
Формирование органо-минеральных производных гумусовых
кислот — обязательное основное звено любого типа
почвообразовательного процесса, обусловливающее развитие почвенного
профиля. Еще в конце прошлого и начале текущего столетия Ф. Зенфт,
К. Тархов, П. Лященко, И. Белецкий и ряд других исследователей
изучали роль гумусовых кислот в процессах выветривания.
К настоящему времени накоплен большой материал по
характеристике процессов взаимодействия органических веществ с
минеральной частью почвы при формировании всех основных типов
почв [60, 69, 80, 83].
Минеральная часть почвы является очень сложной и
гетерогенной по составу и дисперсности системой.
Главнейшие''компоненты ее, участвующие в процессах взаимодействия с
органическими веществами почвы, представлены первичными и вторичными
силикатами, несиликатными окислами кремния, железа и
алюминия, поглощенными катионами почвенных коллоидов и
минералами простых кислородсодержащих солей, среди которых
главнейшее значение имеют карбонаты. Все эти компоненты, находясь
в основной массе в составе твердой фазы почвы, вступают в
различные реакции взаимодействия с гумусовыми кислотами и
промежуточными продуктами разложения органических остатков.
Во всем многообразии этих реакций можно выделить две
основные категории их: реакции, обусловливающие мобилизацию
компонентов минеральной части почвы с образованием различных
органо-минеральных соединений, мигрирующих по профилю почвы,
и реакции образования и аккумуляции в почве
органо-минеральных коллоидов и других более крупных органо-минеральных
гранулометрических фракций, обусловливающих накопление в
профиле этих продуктов.
Не рассматривая процессов мобилизации минеральных
компонентов гумусовыми веществами (обзор ранних исследований
можно найти в монографии К. Д. Глинки [83]), отметим только,
что как фульвокислоты и гуминовые кислоты, так и
водорастворимые промежуточные продукты разложения органических
остатков при взаимодействии с минералами переводят в раствор
все их компоненты. Интенсивность мобилизации и скорость ее
158
очень различны в зависимости от природы минерала и типа
органических веществ. Роль низкомолекулярных продуктов раскрыта
в основном И. С. Кауричевым с сотрудниками 1123, 125—128],
а также в работах других исследователей [389, 432]. Процессы
мобилизации элементов из минералов гумусовыми кислотами и их
солями показаны в исследованиях В. В. Пономаревой с
сотрудниками [105, 247, 254, 279, 290]. Достаточно интенсивно разлагают
минералы не только гумусовые кислоты, но и их растворимые
в воде соли в результате господства в этих процессах обменных
реакций.
Наряду с мобилизацией элементов из кристаллической решетки
минералов в почве образуются органо-минеральные коллоиды
и другие более крупные по степени дисперсности
гранулометрические фракции, представляющие собой минеральные зерна,
покрытые пленками органических веществ. Естественно поэтому,
что природа, свойства и механизм образования всех этих очень
различных органо-минеральных производных привлекают
внимание исследователей на протяжении всей истории изучения
почвенного гумуса.
Общие представления и номенклатура
Первые современные классификации для органо-минеральных
производных гумусовых кислот были описаны в 30-х гг. [296,
297, 525]. У. Шпрингер [525] предложил различать гумусовые
вещества в свободном состоянии, в рыхлой связи с
двухвалентными катионами Са и Mg, в прочной связи с этими же катионами,
в рыхлой или прочной связи с ионами Fe и А1, гумусовые вещества
в комплексной связи с минеральной частью почвы. Эта
классификация подводила итог знаний о природе продуктов
взаимодействия с компонентами минеральной части почвы, накопленных
к тому времени. Слабым звеном классификации являлись очень
схематичные представления о природе продуктов взаимодействия
гумусовых кислот с полутораокисями и глинистыми минералами,
но экспериментальные данные по этим группам
органо-минеральных производных были в то время очень относительными. Наиболее
ценной частью этой классификации, оставшейся незамеченной
в течение длительного времени, были предложения автора
различать прочность связи, хотя детально эти понятия не
рассматривались.
Схема А. Ф. Тюлина [296] составлена по формам связи, т. е.
по наиболее рациональному принципу, но, к сожалению, в основу
ее были положены не доказанные экспериментально схемы Е. Н. Га-
пона [74], по которым гумусовые кислоты связаны с
кристаллической решеткой глинистых минералов непосредственно через
мостики с обменными катионами Са и А1. Эти схемы Гапона
пользовались в 30-х гг. всеобщим признанием, хотя в последующих
159
работах [26, 85, 358, 374, 375] показано, что основная реакция
между этими компонентами носит ионный характер.
Наиболее точной в тот период была схема И. В. Тюрина [297],
в которой автор выделил пять категорий соединений: гумусовые
вещества в свободном состоянии, гуматы сильных оснований,
«гуматы» и смешанные гели с гидроокисями алюминия и железа,
гумусовые вещества, прочно связанные с глиной («аргиллогу-
мины»), и гумусовые вещества в форме комплексных органо-мине-
ральных соединений (с Al, Fe, Р, Si). Эта классификация в
наибольшей степени созвучна современным представлениям, в ней
выделены в особую группу продукты взаимодействия гумусовых
кислот с полуторными окислами, хотя еще нет достаточно четких
различий по типам связей между органическими и минеральными
компонентами почвы. Следует отметить также классификацию
И. Н. Антипова-Каратаева с сотрудниками [39], в которой
предложена более детальная группировка органо-минеральных
производных гумусовых веществ и предусмотрена возможность
формирования как собственно органо-минеральных соединений, так
и адсорбционных комплексов.
В настоящее время изучение органо-минеральных
производных гумусовых кислот и низкомолекулярных продуктов
разложения органических остатков многопланово. Разрабатываются и
изучаются методы выделения органо-минеральных производных,
исследуется их состав; все шире используются методы
моделирования для раскрытия природы и свойств органо-минеральных
производных; большое количество работ посвящено изучению
механизма взаимодействия гумусовых кислот и
низкомолекулярных продуктов разложения растительных остатков с катионами
сильных оснований, полутораокисями, глинистыми минералами,
микроэлементами; обсуждаются вероятные формы связи между
компонентами, предлагаются различные классификационные
схемы для этой категории соединений [127, 129, 237, 287, 322,
337, 406, 412, 413, 443, 503, 544].
Исследования, осуществляемые нами совместно с
сотрудниками на протяжении последних 20 лет, позволили выдвинуть для
обсуждения группировку наиболее вероятных форм связи между
гумусовыми кислотами и основными компонентами минеральной
части почвы, а также рассмотреть природу и свойства этих
продуктов, предложив для них наиболее рациональную номенклатуру
и классификацию.
Как мы уже отмечали [13], наиболее рациональная
номенклатура органо-минеральных производных гумусовых кислот
должна базироваться на типах связи между органическими
минеральными компонентами. Еще в 1967 г. мы предложили различать три
основных класса органо-минеральных производных гумусовых
кислот с компонентами минеральной части почвы в соответствии
с тремя основными типами связи между ними: гетерополярные
соли, образующиеся за счет ионной (гетерополярной) связи, комп-
160
лексно-гетерополярные соли, формирующиеся при участии
координационной (донорно-акцепторной) формы связи, и
адсорбционные комплексы — в случае участия межмолекулярных форм
связи.1 Проявление гомеополярной (ковалентной) связи, широко
распространенной при формировании основного скелета молекул
органических и минеральных соединений, при взаимодействии
гумусовых кислот с компонентами минеральной части почвы до
сих пор не получило экспериментального подтверждения и
теоретически неясно.
_ Гетерополярная связь обусловлена, как известно,
электростатическим притяжением двух разноименно заряженных ионов:
А+ + В- +± А+В-.
Характерные особенности такой молекулы — резко
выраженный дипольный момент и полярнорть, обусловливающие ее
диссоциацию. Реакция взаимодействия между ионными молекулами
носит обменный характер:
А+В- + X+Y- 5± A+Y- + Х+В-,
Реакции этого типа являются типичным солеобразованием
и направлены в сторону малодиссоциированного соединения.
Координационная связь, в соответствии с представлениями
Люиса и Сиджвика, характеризуется сочетанием гетерополярной
и гомеополярной связи и обусловливает образование комплексных
соединений:
К + А+В- ^> [КВ]~А+ или [КА]+В-.
В этих соединениях часть атомов связана гомеополярной связью
и не проявляет себя как ион, так как втягивается в анионную или
катионную часть молекулы [52, 86]. Комплексообразователями
в системе органо-минеральных производных гумусовых кислот
служат металлы переменной валентности или легко образующие
амфотерные соединения (Си, Ni, Co, Zn, Cd, Fe, Mn, Al). Они
являются акцепторами электронов, тогда как доноры электронов
представляют собой молекулы или ионы, присоединяющиеся к
металлу. Часто суммарный заряд внутренней сферы не компенсирует
полностью заряда центрального иона, и в этом случае комплекс —
комплексный ион того или иного знака заряда, а ионы
противоположного знака заряда образуют внешнюю сферу данного
соединения и способны к обменным реакциям, что приводит к
образованию комплексно-гетерополярных солей:
[КА]+В- + X+Y- —> [KAJ+Y- + Х+В-.
1 Несмотря на некоторую условность ионной связи в соответствии с волновой
природой электрона, этот тип связи широко используется химиками при
характеристике валентной связи [217, 244].
Ц Л. Н. Александрова J6J
Металлы-комплексообразователи присутствуют в почве
преимущественно в виде аквакомплекса
Н2Ох /ОНз
H20-Fe—OH2
нао/ чон2
При реакции комплексообразования в этом случае
координационно связанные молекулы воды замещаются молекулами йЛй
ионами лиганда.2 Одним из типов комплексных соедийенйй явЛя--
ются хелаты.3 Они имеют два типа связей с металлом: кбордйна--
ционную и ионную. За счет ионной связи осуществляются ббмёвк
ные реакции, за счет донорной связи формируется комплексный ион.
Третий тип связи — межмолекулярная, природа которой сложна
и многообразна. Она обусловливает образование агрегатов
(мицелл), состоящих из минеральных частиц с сорбированными на
ее поверхности гумусовыми кислотами и их солями, а также
агрегатов (ассоциатов) самих гумусовых веществ. К этому типу
относится водородная связь, образующаяся между атомами
водорода, химически связанными в одной молекуле и достаточно
электроотрицательными атомами другой молекулы (ОН. . .0,
NH, , .0, NH. , .N). Ее природу объясняют электростатическим
взаимодействием между водородом и наиболее
электроотрицательными элементами, на которое накладываются резонансная
структура лигандов и изменение ионных расстояний. К этому же типу
относится и поляризационная связь, возникающая между
постоянными диполями за счет электростатических сил. За счет
межмолекулярного типа связи в почве осуществляются очень
распространенные явления адгезии (склеивание разнородных по химической
природе тел) и когезии {склеивание однородных веществ),
обусловливающие формирование сложных и неоднородных по составу
микро- и макроагрегатов. Аналогичные типы возможных связей
описали Н. И. Горбунов и Д. С. Орлов [85].
Выделенные нами три класса органо-минеральных
производных гумусовых кислот (гетерополярные соли, комплексно-гете-
рополярные и адсорбционные комплексы) образуются в любой
почве, формируя массу органо-минеральных коллоидов, и в
зависимости от минералогического и химического состава обладают
различной степенью устойчивости, вследствие чего способны к
аккумуляции или миграции в почвенном профиле.
Предложенная нами классификация не претендует на полноту
характеристики всех органо-минеральных производных гумусовых
2 Лигандами, как известно, называются молекулы или ионы органических
или неорганических соединений, непосредственно присоединяющиеся
к комплексообразователю и образующие внутреннюю сферу комплексного
соединения.
3 Хелаты — соединения, в состав которых входят две или более донорные
группы, вследствие чего образуется циклическая структура, замкнутая
координационными связями концевых атомов.
162
кислот, но отражает, как нам представляется, основные, наиболее
характерные типы взаимодействия. В различных экологических
условиях сочетание тех или иных типов связей, а следовательно,
и свойства органо-минеральных производных очень разнообразны.
Гетерополярные соли
Типичными гетерополярными солями гумусовых кислот
являются гуматы и фульваты щелочных и щелочноземельных осно*
ваний. Как показали наши исследования [26], механизм их
образования сводится к обменной реакции между водородом кислых
функциональных групп гумусовых кислот, способных к обменным
реакциям при заданной величине рН, и соответствующими
катионами щелочных и щелочноземельных оснований:
минеральный! л „/ллл„ —> минеральный! л„. пЛ '2
v Caa+4-R(C00H„ F 2H+ + IT
коллоид J <— коллоид J \(СООШ
Эта обменная реакция протекает в строго эквивалентных
количествах и обратима, что подтверждено в ряде работ [85, 185*
358]» Обменный характер реакции снимает возможность
образования мостиков из катионов кальция между гумусовыми
кислотами и глинистыми минералами по известной схеме Гапона
-_Si—О—Са—СОО
,СОО—Са—О—Sift
—Si—О-Са-СОСК \G00-Ca-0-Si-
ГуматЫ кальция лишь выпадают в осадок и остаются «свобод*
ными», образуя на поверхности тех или иных гранулометрических
фракций йленку* Механизм прочного закрепления гуматОв каль*
ция имеет другую природу. Он обусловлен процессами склеивания
за счет сил адгезии при дегидратации пленки гумата. Поэтому
невозможно прочно закрепить гуминовые кислоты известкованием.
Хорошо известно, что и после известкования гуминовые кислоты
подзсщистых почв легко снимаются непосредственной обработкой
щелочными растворами без предварительного декальцированйя
почвы.
Емкость обменной сорбции оснований гумусовых кислот (т. ек
максимальное связывание основания) определяется реакцией
среды й природой гумусовых кислот (табл, 51). Для гуминовых
кислот она колеблется от 300 до 700 и в условиях резко щелочной
реакций достигает 1000 мг*экв./100 г. Причина этих различий
в зависимости от реакции среды заключается» как известно>
в разной степени диссоциации кислых функциональных групп
163
11*
Таблица 51
Ёмкость обменной сорбции оснований
гумусовыми кислотами, мгэкв. на 100 г сухого
беззольного препарата, при разных значениях рН
РН
4.5-6.5
6.6—7.5
7.6—8.5
8.5
300-
400-
600-
700-
гк
-400
-600
-700
-1000
ФК
200-
500-
600-
800-
-500
-600
-800
-1200
при разных значениях рН. Большинство исследователей считают,
что в слабокислой среде, вплоть до рН 7, постепенно возрастает
степень диссоциации карбоксильных групп, в щелочном
интервале в реакцию обменного солеобразования вступают фенольные
гидроксилы. Однако в условиях сильнощелочной реакции соле-
образование осложняется молекулярной сорбцией гидроокисей,
на что указывали еще Г. Л. Стадников и соавторы [283]. Емкость
обменного поглощения оснований фульвокислотами колеблется
примерно в таких же пределах, хотя, как мы отмечали выше,
количество кислых функциональных групп в фульвокислотах очень
изменчиво. Следует иметь в виду, что некоторые колебания
в сорбции оснований зависят также от радиуса иона основания,
но они незначительны.
Гетерополярные соли гумусовых кислот в зависимости от
состава катионов и природы самих кислот характеризуются
различной растворимостью, а следовательно, и разной миграционной
способностью. Гуматы кальция очень плохо растворимы в воде
при любых встречающихся в почве значениях рН (до 12). Гуматы
магния более подвижны и, находясь в виде достаточно гидратиро-
ванного геля, пептизируются водой и мигрируют по профилю.
Следует отметить, что таковы лишь общие закономерности,
известные со времен Шпренгеля и Берцелиуса. Гетерогенность гуми-
новых кислот, установленная в последнее время, требует
дополнительных исследований по выявлению растворимости отдельных
фракций гуматов кальция и магния [253]. Гуматы калия, натрия
и аммония при замещении всех карбоксильных групп хорошо
растворимы в воде и легко передвигаются по толще почвенного
профиля. Фульваты всех щелочных и щелочноземельных оснований
хорошо растворимы в воде при кислой, нейтральной и щелочной
реакциях, и только при рН более 10, по данным В. В.
Пономаревой [245], фульваты кальция и магния выпадают в осадок.
Вследствие этих особенностей миграция вниз по профилю как свободных
фульвокислот, так и всех фульватов оснований ярко выражена
во всех почвах.
Возможно ли образование комплексно-гетерополярных солей
гумусовых кислот со щелочными и щелочноземельными основа-
164
ОМ 1.2 2.0 2.8 3.6
Рис. 33. График функции [Mg]/[Ca]=/(Mg)/(Ca) (по: [1 5]).
ниями? Этот вопрос нельзя считать достаточно хорошо изученным.
Большинство исследователей отрицают эту возможность из-за
резко выраженного полярного характера как гумусовых кислот,
так и сильных оснований. Но в ряде работ [176, 507, 550] отмечается
возможность образования хелатов и определяются константы
устойчивости для них.
Однако тщательно выполненные модельные опыты по
исследованию ионного обмена для гуминовых кислот торфа с растворами
постоянной ионной силы опровергают эти выводы [185] (рис. 33).
Не подчиняются эквивалентности реакции взаимодействия
гумусовых кислот с ионом аммония. Численные константы обмена
NH4+ на Са2+ постепенно уменьшаются с увеличением
концентрации Са2 + в исходном растворе, что свидетельствует о более сложном
механизме взаимодействия и частичной трансформации иона NH4
в необменную форму.
Как показали наши исследования, в почвах чистые гуматы или
фульваты сильных оснований встречаются редко, обычно
формируются более сложные органо-минеральные производные, которые
следует относить к группе комплексно-гетерополярных соединений,
хотя степень насыщенности кальцием этих соединений в
различных почвах разная: максимальна в черноземах и сероземах и
минимальна в подзолистых почвах [11].
Источниками щелочных и щелочноземельных оснований в почве
для образования гуматов и фульватов являются элементы,
входящие в состав золы растительных остатков, остающихся в почве
и подвергающихся разложению: простые соли их
(преимущественно карбонаты), находящиеся в твердой или жидкой фазе почвы,
обменные катионы диффузного слоя почвенных коллоидов,
способные к реакциям обмена при различных значениях рН, а также
основания, входящие в состав кристаллической решетки
первичных и вторичных силикатов, которые мобилизуются гумусовыми
кислотами [105, 150, 280]. В пахотных почвах существенным
источником этих оснований являются известь, минеральные,
органические и органо-минеральные удобрения, вносимые в почву.
165
Комплексно -гетерополярные соли
Если природа и механизм образования гуматов и фульватов
сильных оснований исследованы относительно хорошо, то продукты
взаимодействия гумусовых кислот с поливалентными катионами
изучены все еще недостаточно, хотя наличие в почве значительных
количеств железа, алюминия, марганца, меди, цинка, никеля
и образование «гуматов» и «фульватов» этих металлов были
известны основоположникам учения о почвенном гумусе. Еще в конце
прошлого столетия Г. Густавсон [90] обратил внимание на
своеобразие свойств «гуматов» железа и алюминия, но до 50-х гг.
природа этих соединений практически не изучалась, хотя И. В.
Тюрин [297] брал в кавычки термины «гумат железа» и «гумат
алюминия». Лишь во второй половине текущего столетия были
показаны природа и свойства этих соединений [6, 95, 246, 503—506].
К настоящему времени можно считать экспериментально
доказанным, что некоторые металлы (железо, алюминий, медь, цинк,
никель) при взаимодействии с гумусовыми кислотами образуют
комплексные соли, в которых металл входит в состав анионной
части молекулы и не способен к обменным реакциям [284, 334,
364, 367, 372, 470, 504, 505, 528]. Наиболее детально изучены
реакции взаимодействия гумусовых кислот с железом и алюминием.
Наши исследования показали, что металл, вступая во
взаимодействие с гумусовыми кислотами, вытесняет водород части
кислых функциональных групп и образует комплексную соль, входя
в анионную часть молекулы [6]. Схематически эту реакцию можно
представить так:
ЛСООН). Г уСООч Т(СООН)м
Г /соо\ Т
ч(ОН)ш L \он^ J(°H>
т-у
где М - Fe(OH)?, Fe (ОН)2+, А1 (ОН)?, А1 (ОН)2*.
Участие карбоксильных групп в образовании этих соединений
подтверждено прямым определением их количества в серии
модельных препаратов до и после взаимодействия с солями железа и
алюминия (рис. 34), а в дальнейшем — в ряде работ [95, 504—507].
Характерной особенностью этих соединений является остаточная
«емкость обменного поглощения катионов щелочных и
щелочноземельных металлов, которой они всегда обладают, что
свидетельствует о сохранении некоторой части карбоксильных групп
свободными и способными к обменным реакциям с катионами
щелочных и щелочноземельных оснований:
[уСООч -1СОО-Н+ Г уСООч IGOOMx
К ХМ + Ыг —> \К ХМ + 2Н,
Хон^ J°" н+ L Хон^ J°Mi
где М - Fe(OH)+, Fe(OH)2+, А1(ОН)+, А1(ОН)2+; Мг - Са, Mg,
Na, К, А1.
166
2 b 6 8 10 12 П 16 Fe
1 2 3 4 5 6 7 8 Al
Рис. 34. Емкость обменного поглощения Са при рН 6.8 Fe- и А1-гуминовыми
соединениями с различным содержанием Fe (1) и А1 (2).
По оси абсцисс — содержание Fe и А1, г на 100 г препарата гуминовой кислоты.
Эта особенность и послужила основанием для отнесения
данных соединений к категории комплексно-гетерогенных солей.
Называть эти соединения «гуматами» или «фульватами» нельзя,
так как металл, входя во внутреннюю сферу молекулы, не способен
к реакциям солеобразования. Если оставшиеся свободными кислые
функциональные группы не замещены на основания, эти
соединения проявляют себя как своеобразные металлогумусовые
кислоты (железо- и алюмогумусовые). Следует отметить двойственную
природу многих металлов, входящих в анионную часть молекулы,
в составе этих комплексно-гетерополярных соединений. Нами
доказано это на примере алюмогуминовых соединений (табл. 52).
Часть алюминия, входящего в их состав, не способна к обменным
реакциям; часть его вытесняется катионом нейтральной соли
(например, СаС12).
Алюминий вытесняется в раствор постепенно, но полностью
вытеснить его невозможно даже при очень длительной обработке,
продолжающейся несколько недель. Электродиализ также приводит
Таблица 52
Вытеснение Fe и А1 кальцием (из 0.2 н. раствора СаС12)
из железо- и алюмогуминовых солей, г на 100 г препарата
Препарат
1
2
3
4
5
6
7
8
Содержание Fe
исходное
1.5
3.0
6.3
7.4
8.5
12.4
14.0
15.4
после обработки
1.4
2.9
5.9
7.0
8.3
12.10
13.6
15.0
Содержание А1
исходное
2.7
3.3
4.3
5.6
6.4
7.2
после обработки
1.8
2.0
2.7
3.9
4.6
5.3
167
к частичному вытеснению алюминия в раствор. Аналогичные
особенности меди, никеля и марганца отметили А. Бейзу и Д. Му-
кереджи [346]. Железо не вытесняется другими катионами,
оставаясь полностью в анионной части молекулы.
Емкость связывания металлов в комплекс с гумусовыми
кислотами очень различна и, по данным ряда исследователей,
применяется в широких пределах. Так, содержание железа в железо-
гуминовых солях колеблется от 50 до 150 мг, а А1 — от 27 до 55 мг
на 1 г гуминовых кислот [6, 101, 116, 134, 178, 290, 504—506].
Фульвокислоты связывают до 250 мг Fe и до 140 мг А1 на 1 г
кислот. Эти данные о емкости связывания в комплексе очень условны,
так как получены в модельных опытах; в природных условиях
в почвах и почвенно-грунтовых водах степень насыщенности
гумусовых кислот железом и алюминием заметно ниже.
Емкость связывания металлов в комплексе зависит от ряда
причин: природы самих металлов, состава гумусовых кислот,
реакции среды, общих условий и характера почвообразовательного
процесса. Большинство исследователей отмечают меньшее
количество железа, связываемого гуминовыми кислотами и фульво-
кислотами, по сравнению с алюминием [95, 134, 504, 505].
Общепринято и представление о том, что фульвокислоты
связывают в комплекс больше железа и алюминия по сравнению с
гуминовыми кислотами [95, 134]. Существенное значение имеют
степень и характер гетерогенности самих гумусовых кислот, что
очень четко было показано на примере фульвокислот [116].
Разные по ММ фракции фульвокислот обладают различной способностью
к комплексообразованию, причем образующиеся комплексы
характеризуются неодинаковой степенью устойчивости, которая
максимальна для наиболее высокомолекулярных фракций
фульвокислот. Не менее существенна величина рН, при которой
осуществляется реакция комплексообразования. Детальное исследование
Э. М. Дорфман [95] показало, что в зависимости от величины рН
и концентрации Fe и А1 гумусовые кислоты образуют растворимые
и нерастворимые комплексные железо- и алюмогумусовые
соединения переменного состава. Чем ниже концентрация металла,
тем при более низких значениях рН образуются растворимые
комплексы; при этом фульвокислоты на единицу углерода
связывают больше железа и алюминия по сравнению с гуминовыми
кислотами, что имеет огромное значение для оценки роли
гуминовых кислот и фульвокислот в мобилизации и миграции полуторных
окислов в почве (рис. 35).
Для других металлов-комплексообразователей данных о
емкости их связывания в комплекс гумусовыми кислотами
значительно меньше, но все же и для них она исчисляется несколькими,
а иногда и десятками процентов на 100 г гумусовых кислот. Так,
например, в зависимости от рН количество меди, сорбированной
гуминовыми кислотами и фульвокислотами, выделенными из
торфяной почвы, колеблется от 8—9 до 60—75% [196]. Столь
J68
§uKi '««Гц '|
al'sJI-H
O^ S ев со t
о о -Я д п t
SS<jMSE?fe<Jc5g
высокие величины свидетельствуют о многообразий реакций
взаимодействия; по-видимому, при высоких количествах
сорбированной меди имеют место какие-то побочные реакции.
Какова же реальная структура этих комплексных алюмо-
й железогумусовых соединений? К сожалению, до настоящего
времени она не представляется достаточно ясной. На основании
исследования корреляции между изменением емкости обменного
поглощения кальция в препаратах алюмо- и железогуминовых
соединений с различным содержанием в них железа и алюминия
еще в 50-е гг. нами [6] было высказано предположение о том, что
эти металлы присутствуют в форме основных ионов: Fe(OH)2+,
Fe(OH)2+, Al(OH)2+, Al(OH)2+.
Это предположение подтвердили М. Шнитцер и С. Скиннер
[504, 505]. Следовательно, схематически строение этих
соединений можно представить таким образом:
[уСООч ПСОО-Н+ Г уСООч 1СОО-Н+
R( ;Fe(OH) ; R( ;А1(0Н)
Следует обратить еще раз внимание на то, что в молекуле этих
соединений всегда остается свободным некоторое количество
карбоксильных и фенолгидроксильных групп, водород которых может
замещаться на любой обменный катион почвы. Комплексный
характер этих соединений (наличие металла во внутренней анионной
части молекулы) подтвержден в последнее время многими
авторами, применяющими разнообразные методы: метод
электрофореза [153], метод горизонтального электрофореза на бумаге в
сочетании с определением кинетических констант изотопного обмена,,
констант нестойкости и ИК спектроскопии [116], методику потен-
циометрического титрования и определение рН эффекта [95, 126,,
433, 505]. Проведенные исследования подтверждают комплексную1
природу этой категории соединений, но еще не решают, по нашему
мнению, вопроса о принадлежности их к хелатам.
Необходимо отметить, что рН-эффект резко снижается при
титровании А1- и Fe-гуминовых солей, отдиализованных до нейтральной
реакции (рис. 36), и сохраняется в этих условиях лишь в щелочном
интервале. Можно полагать поэтому, что в комплексообразовании
участвуют фенольные гидроксилы, а резкое смещение кривой
титрования в кислом интервале рН в неотдиализованных
препаратах обусловлено гидролизом сернокислых или хлористых солей
железа и алюминия, используемых обычно в аналитической
практике. Если понимать под хелатами внутрикомплексные
соединения, в которых комплексообразователь одновременно связан
с двумя и более атомами одного и того же лиганда с помощью как
валентной, так и донорной связи, необходимо уточнить характер
связи металла со всеми реактивоспособными группами молекулы
гумусовых кислот. Наряду с карбоксильными и фенолгидроксиль-
170
12
10
8
6
Ч
2
\
3
12
10
8
6
t
Z
/s*
I
4?
в
?2?^
<^-
1 ,_ _r i_
1
i
/ 2 3
мл 0 1 н
Рис. 36. Кривые потенциометрического титрования гумусовых кислот и их
Fe- и Al-гумусовых солей после (а, в) и до (б, г) диализа (по: [95]).
а, б. 1 и 4 — исходные гуминовые кислоты (2 6-10-3 Н+), 2и5- Fe-гуминовые соли
(3 3 -Ю-8 мМ Fe*+), 3 и 6 — АЬгуминовые соли (3 7 -Ю-2 мМ А13+), виг: 1 и 4— исходные
фульвокислоты (8 1 -Ю-3 Н+), ги5- Fe-фульватные соли (3 3-Ю-2 мМ Fe3+), 3 и 6 —
Al-фульватные соли (3.7-10-2 мМ А13+).
ными группами в реакциях могут участвовать аминогруппы
[356, 357].
Э. М. Дорфман [95] на основании модельных опытов и
определения констант устойчивости железогумусовых комплексов
предложила три схемы их строения.
I. При замещении водорода одной карбоксильной группы и
координировании одного фенольного гидроксила в присутствии ионов
Ме(ОН)+:
Н,Оч °\П /ООСч /(СООН^.х
Ч/
н2о/1 \
он
ч>'
(ОН),,
(Н — ионы, способные к обмену; Me — Fe3+ или А13+)4
171
II. При замещении водорода двух карбоксильных групп
и координировании одного фенольного гидроксила ионом Ме(ОН)2+:
ОН
Нг°\1 /оос\ /(СООН)„_г
Me R
На0 он оос/| Noh)^
III. При участии только одной карбоксильной группы,
способной занимать два координационных места:
он /(СООН)^
НгОч | ,0—С—R(
)Ме( / \(ОН)т
Э. М. Дорфман полагает, что схемы I и III вероятны для гуми-
новых кислот, а схема II — для фульвокислот.
Различна степень миграционной способности железо- и алю-
могумусовых соединений, изучение которой необходимо для
расшифровки роли этих соединений в элементарных звеньях
процесса почвообразования. Она определяется не только природой
гумусовых кислот, но и составом обменных катионов, замещающих
оставшиеся свободными кислые функциональные группы этих
соединений, а также степенью насыщенности их металлом.
Максимальной миграционной способностью обладают соли
фульвокислот [95, 178, 248], и только при очень высокой степени
насыщения металлом они переходят в твердую фазу, формируя
многочисленные конкреции. Алюмо- и железогуминовые соли менее
подвижны, причем степень их миграционной способности
определяется не только степенью насыщенности их этими металлами, но
также степенью гидратации и составом катионов свободных кислых
функциональных групп. Гидратированные алюмо- и
железогуминовые соли с относительно невысоким количеством А1 и Fe в них,
содержащие в качестве обменных катионов натрий и калий,
обладают ярко выраженной миграционной способностью. Эти же
соединения, свободные кислые функциональные группы которых
замещены кальцием, а также свободные А1- и Fe-гуминовые
кислоты не способны к миграции и выпадают в твердую фазу на месте
своего образования (табл. 53). Все эти особенности А1- и Fe-ry-
мусовых солей в последнее время нашли подтверждение в ряде
исследований [116, 311 и др.].
Для выделения алюмо- и железогумусовых соединений из
почвы еще 20 лет тому назад нами [9] был предложен 0.1 М раствор
пирофосфата натрия с рН 9.5—10. В настоящее время этот метод
рекомендуется М. М. Кононовой и Н. П. Бельчиковой [152],
широко применяется С. В. Зонном [111, 112]. Используя этот
метод и осаждая гуминовые кислоты кальцием для предотвращения
172
Таблица 53
Пептизация в воде Fe- и Al-гуматов калия
Препарат
Fe-гумат калия
гидратированный
дегидратированный
Al-гумат калия
гидратированный
дегидратированный
Содержание
Fe (A1), о/0
8.5
12.4
15.4
7.2
5.4
2.3
Пептизация, %
ГК
100
72
47
14
100
22
Fe (A1)
95
49
27
9
100
36
разрушения комплексов при действии кислоты, мы показали
наличие этих соединений в ряде основных типов почв (табл. 54).
Максимальная насыщенность железом и алюминием как гумино-
вых кислот, так и фульвокислот характерна для краснозема,
значительна для дерново-подзолистой почвы и минимальна для
черноземов, что соответствует особенностям почвообразовательного
процесса в этих почвах. Впоследствии эти выводы были
подтверждены исследованиями М. М. Кононовой и Н. П. Бельчиковой[152].
Следует отметить, что применение кислоты для осаждения гумино-
Таблица 54
Содержание Fe и А1 в железо- и алюмогумусовых солях,
выделенных 0.1 М Na4P207 из почв,
мг на 1 г гумусовых кислот
Гумусовые кислоты
Осаждение CaS04
А1
Fe
Осаждение Н2804
А1
Fe
Дерно в о - подзо л и ста я почва
Гуминовые
Фульвокислоты
Гуминовые
Фульвокислоты
Гуминовые
Фульвокислоты
133
7
127
27
Краснозем
117 1 79
.42 1 7
Чернозем
6
38
27
69
S
1 23
| 115
5
41
72
65
1 63
| 22
9
86
173
вых кислот резко искажает фактическую степень насыщенности
гумусовых кислот железом и алюминием.
Источники железа и алюминия, а также других металлов
переменной валентности для образования этих комплексно-
гетерополярных солей очень многообразны. Для железа и
алюминия, как известно, характерны следующие основные типы
соединений: силикатные формы в составе первичных и вторичных
силикатов, где железо и алюминий входят в состав кристаллической
решетки; аморфные и окристаллизованные гидраты окислов
с различной степенью гидратированности; свободные основные
ионы этих металлов, освобождающиеся из органических остатков
при разложении последних; плохо исследованные соединения этих
металлов, вносимые в почву с минеральными, органическими и ор-
гано-минеральными удобрениями.
Кроме того, некоторая часть алюминия и ряда других металлов,
за исключением Fe3+, может находиться в виде обменных катионов,
В почвах болотного ряда присутствуют закисные соединения
железа в виде различных солей (преимущественно бикарбонатов
и сульфидов).
При всем многообразии соединений железа и алюминия в почве
их мобилизация гумусовыми кислотами с образованием ком-
плексно-гетерополярных солей возможна только в форме
основных ионов. В противном случае формируются иные категории
органо-минеральных производных, относящиеся к типу
адсорбционных комплексов.
Адсорбционные комплексы 4
Способность гумусовых веществ сорбироваться на поверхности
твердых частиц почвы общеизвестна и подтверждается
многочисленными аналитическими и микроморфологическими
исследованиями. Накоплено большое количество данных о наличии
органических веществ в отдельных гранулометрических фракциях,
приведены микрофотографии шлифов, на которых отчетливо видны
скопления гумусовых агрегатов в почвенных порах, пленки
органических и органо-минеральных веществ на поверхности
различных гранулометрических фракций и полуразложившихся
корневых остатков [23, 31, 32 и др.]. Минеральная часть почвы,
очень гетерогенная по составу и размерам элементарных частиц,
характеризуется огромной суммарной поверхностью, на которой
и развиваются поверхностные явления, все еще недостаточно
изученные в аспекте почвообразования.
Мы сохраняем наиболее распространенный в мировой литературе термин
«адсорбционные комплексы», понимая под ним органо-минеральные
продукты поглощения гумусовых веществ на минералах и не вкладывая в этет
термин химического представления о комплексных соединенияхi
174
Рис. 37. Состав железо- и алюмогуминовых комплексов и емкость их
обменного поглощения.
а—в — содержание Fe203: a — при взаимной коагуляции золей гуминовых кислот или
гуматов с золями Fe(OH)3, б — при поглощении золя Fe(OH)3 гелем гуминовой кислоты,
в — при осаждении комплексного золя кальцием; г—е — содержание А1403: г — при
взаимной коагуляции золя-гумата и золя А1(ОН)3, д — при поглощении золя А1(ОН)3
гелем гуминовых кислот, е — при осаждении комплексного золя кальцием.
За последние десятилетия накоплено значительное количество
^модельных опытов и теоретических суждений о вероятных типах
«связи между органическими и минеральными компонентами почвы
[19, 85, 287, 317, 412, 486]. Анализ литературных данных и много*
летние исследования позволили нам выделить три основные
труппы адсорбционных комплексов, формирующихся в процессе
«сорбции гумусовых веществ на компонентах минеральной части
почвы: алюмо- и железогумусовые, кремнегумусовые и глиногуму-
совые. Экспериментально исследованы алюмо-, железо- и глино*
гумусовые комплексы. Алюмо- и железогумусовые комплексы
детально изучены методом моделирования [6, 178]. Это типичные
хемосорбционные продукты, образующиеся за счет химической
реакции между компонентами на цоверхности раздела твердая
фаза—раствор, сопровождаемой межмолекулярными формами
связи. Схематическое строение их, по-видимому, таково:
Fe203 • /гН20
FeO—ООСч у(СООН)я
NOH)^
jFeO О/
ШО-ООС
А1203 • лН20
1А10-
AIO-
4r/
(СООН)^!
-О/ \ОЩт.г
175
Соотношение между .компонентами в этих продуктах может
быть очень различно, т. е. они являются типичными мутабильными
веществами, по терминологии А. Е. Ферсмана. В отличие от А1-
и Fe-гумусовых соединений, описанных выше, в этих комплексах
преобладает минеральная основа, состоящая из полуторных
окислов, причем единица массы гидроокиси алюминия или железа
может сорбировать до 100% массы гумусовых кислот (рис. 37).
Эти хемосорбционные комплексы, которые мы назвали
комплексными алюмо- и железогумусовыми гелями [6], могут
образовываться в почве различными путями. Гумусовые кислоты могут
сорбироваться гелями полуторных окислов, образующими
многочисленные пленки и конкреции в толще почвы; может иметь
место обратный процесс — сорбция золей полуторных окислов на
осадках гуминовых кислот; возможна взаимная коагуляция золей
полуторных окислов и золей гумусовых кислот или их солей.
В зависимости от соотношения компонентов различен и состав
образующихся алюмо- и железогумусовых гелей (рис. 37). Резко
снижает хемосорбцию дегидратация пленки полуторных окислов
(табл. 55)*
Таблица 55
Сорбция гумата натрия на Fe(OH)3 и А1(ОН)3,
мг гумата на 100 мг R203
Препарат
• 1
2
рН
| 5.0
6.5
J
Fe(OH)3
гидратиро-
ванный
13
8
сухой
3
2
А1(ОН)3
гидратиро-
ванный
192
169
сухой
3
2
Реакция хемосорбции подтверждается заметным уменьшением
количества кислых функциональных групп. Величина емкости
обменного поглощения катионов кальция в железо- и алюмогуми-
новых гелях, вычисленная на единицу массы гуминовых кислот,
всегда значительно ниже исходной (рис. 37). Следует отметить,
что в условиях модельного опыта не удается полностью заместить
все кислые^функциональные группы гумусовых кислог в
^образующихся комплексных ^ гелях, вследствие Хчего..они ^обладают
некоторой .^остаточной, емкостью обменного поглощения катионов
и ведут себя как ацидоиды.
Очень разная степень подвижности, а следовательно, и
миграционная способность этих комплексных гелей. Наши
исследования показали, что гидратированные комплексные железогумино-
вые гели с относительно невысоким содержанием железа частично
* пептизируются водой после насыщения свободных
функциональных групп гумусовой части их щелочными катионами. Алюмогу-
176
Таблица 56
Пептизадил Fe- и Al-гуминовых гелей,
% к исходному содержанию компонентов
Препарат
Fe-гуминовый
гидратированный
сухой
гидратированный
А1-гуминовый
гидратированный
сухой ц*
гидратированный
Содержание
R203, г на
100 г ГК |
39
39
297
35
35
169
Пептизация Н20 .
после насыщения
препарата К+
ГК
37
22
0
100
34
74
R203
24
12
0
100
29
27
Пептизация 0.1 н,
NaOH
ГК
85
43
44
100
62 t
100
R2o3
38
22
12
100
51
72
миновые гели еще более подвижны и могут в этих условиях пепти-
зироваться полностью, образуя комплексные золи (табл. 56).
При поступлении в такую систему кальция комплексные золи
«коагулируют и переходят в состояние геля, который вновь
проявляется как ацидоид и обладает остаточной емкостью обменного
поглощения катионов. Таким путем осуществляются,
по-видимому, миграция железогуминовых и особенно алюмогуминовых
комплексных золей из верхней части профиля солонцов и
солонцеватых почв и переход их в твердую фазу в иллювиальной части
профиля,
Алюмо- и железофульватные комплексы, исследованные
Г. А. Левашкевич [178], более подвижны; автор отмечает еще одно
характерное свойство этих комплексных гелей: при взаимодействии
гидроокисей с избытком гумусовых кислот последние переводят
в раствор некоторое количество^железа и алюминия в форме
комплексных золей. Фульвокислоты обладают этой способностью
в наибольшей степени. Следовательно, как гуминовые кислоты,
так и особенно фульвокислоты приj наличии нисходящих токов
влаги в^ любом типе почв могут мобилизовать некоторое
количество полуторных окислов и образовать с ними растворимые
комплексные золи, мигрирующие по профилю^и выпадающие
в осадок при повышении концентрации кальция.
Природа кремнегумусовых комплексов практически не
изучена. Можно полагать лишь, что их образование возможно за
счет адгезионных сил в процессе дегидратации пленок гумусовых
кислот или их солей на поверхности кристаллических шиаГ
аморфных форм кремнезема. Количество сорбированных таким путем
органических веществ должно определяться величиной удельной
поверхности частиц кремнезема.
Природа глиногумусовых, и прежде всего глиногуминовых,
комплексов изучается многими исследователями [4, 7, 10, 10а, 11,
12 Л. Н. Александрова 177
!23, 25, 31, 32, В8,242, 317, 384, 411, 412, 485, 486, 539, 545, 555].
В настоящее время общепризнанно, что гумусовые кислоты
сорбируются минералами только экстраламеллярно, т. е. на
поверхности кристаллической решетки. Этот вывод, сформулированный
нами более 20 лет назад на основании оригинальных модельных
исследований М. Надя [26], в настоящее время подтвержден во
многих работах. Так, И. А. Пивоварова [242] в серии модельных
опытов со многими силикатными минералами (каолинитовыми,
монтмориллонитовыми глинами, вермикулитом, биотитом,
мусковитом) показала, что гуминовые кислоты и фульвокислоты,
выделенные из ряда почв, сорбируются этими минералами в соот-
1ветствии с уравнением Лэнгмюра. Общая масса сорбированных
:гуминовых кислот составляла от 0.2 до 3.5% к навеске минерала.
Аналогичные данные для каолинита и асканита, а также
некоторых почв получены нами ранее [26, 31].
Значительно Сложнее решить вопрос о механизме этого
поглощения. По этому вопросу единого мнения нет. Одни
исследователи допускают непосредственную химическую связь через
функциональные группы гумусовых кислот и гидроксилы алюмокисло-
]родных октаэдров [486, 489, 531 ] или через мостики из обменных
^катионов или алюминия [412]; другие [237] отмечают сложность
>и многообразие форм связи в зависимости от. конкретных условий
^почвообразования и указывают на существенное значение
азотсодержащих групп гумусовых кислот в явлениях их сорбции на
минералах. В одной из наших публикаций [26] убедительно
показано, что взаимодействие гумусовых кислот или их
растворимых гуматов с обменными катионами Са и Mg глинистых минералов
всегда является обменной реакцией, в результате которой
образуется соответствующий гумат, не связанный с кристаллической
решеткой минерала и лишь выпадающий в осадок на ее
поверхности:
глинистый! п ^,ллл+* глинистый"! „ _ ллл л
Са2+ + R СООН 2 —» 2Н+ + R(COO)2Ca.
минерал J минерал J
Глиногумусовые комплексы (сорбция гумусовых веществ на
кристаллической решетке глинистых минералов) образуются не
через главные валентности, а в процессе склеивания поверхностей
при дегидратации компонентов. Процесс склеивания (адгезия)
осуществляется за счет межмолекулярных сил (поляризационной
и водородной). Этот вывод удалось доказать при постановке
специальных модельных опытов по изучению влияния обменных
катионов и возможности обменных реакций. Для этого получены
модельные препараты из Н-форм асканита или минеральной массы
коллоидов и гуминовой кислоты, определена в них емкость
обменного поглощения катионов кальция при трех значениях рН и
сняты изотермы сорбции паров воды (табл. 57; рис. 38). Во всех
препаратах независимо от реакции и степени дегидратации наблю-
178
Рис. 38. Изотермы сорбции водяных паров модельным препаратом {а) и
коллоидами чернозема (б).
1 — исходный асканит, 2 — асканит-f-гуминовая кислота (опыт), з — асканит+гумино*
вая кислота (расчет), 4 — гуминовая кислота, 5 — исходные коллоиды, 6 — коллоиды
после удаления гумуса.
далась полная аддитивность емкости катионного обмена, что
подтвердило сохранение свободными всех кислых реакционно-
способных функциональных групп. Аддитивности сорбции по
отношению к парам воды нет, фактически полученная величина
ниже вычисленной на всем отрезке сорбционной кривой.
Следовательно, наблюдалось уменьшение суммарной поверхности за счет '
склеивания поверхностей минерала и гуминовой кислоты [26],
что подтвердили более поздние исследования [31]. Во всех
модельных препаратах бентонита и гумата калия на шлифах резко
выделялась четко выраженная пленка последнего на поверхности
минерала без сколько-нибудь выраженного прокрашивания всей
минеральной массы. Определение удельной поверхности в полу-
Таблица 57
Емкость катионного обмена адсорбционных комплексов
глинистых минералов и гуминовой кислоты,
мгэкв. на 100 г препарата
Препарат
Асканит
То же-)-гуминовая кислота
опыт
расчет
Минеральные коллоиды чернозема
То же+гуминовая кислота
опыт
расчет
Емкость катионного обмена при разных
значениях рН
4.5
85.8
92.2
93.6
71.6
85.7
83.5-
6.6
96.5
114.0
113.9
88.6
111.3
112.4
8.2
99.6
127.3
121.6
90.3
126.6
120.4
179 12*
со
со
00
05
Я
я
«
а
Н
9
ft
>е<
м
н
&
6
е-
ffi
Я
о
R
О)
Оч
к
3
в
1 *
§
К
**
о
к
1 к
1 ф
1 я
1 о
К
*S
1 о
1 '*н
а
В^ч
0, Г,
мкм
В"
• количес
фракций
§<*
1 фф
s»
1 се
ч|Й
1 ° w>
«Г
1 о
уму
©
S3
и
ев
*
а
ф
**
°
ф
о
п
2
§
А
о
V
*ч
i
cq
о
7
о
о
7
S
<м
О и
я
Я
sg
О о
>»
S
-ч
(ч
1 ~-2
1 °
и
~-2 1
° 1
и
•-2 1
о 1
и
о
и
о 1
о 1
Я
Ф
СО
о
W
ft
ф
F
»н
3
W
и
ф
о
«
ф
а
в
i
о
чН
о
4F
00
ю
чН
|
О
чН
О
4F
О
О
О
чН
г»
1
О
ч-н
СО
СО
СО
05
чН
г»
О
чг»
333
00
СО
<м
I
О
чН
О
1 139
ю
t^
чН
ю
00
о
чН
1
1
о
о
чН
ю
см
а
i
о
чн
05
СО
ю
со
00
7
о
•"Н
со
СМ
см
ю
о
ЧвН
Г"
о
чН
161
00
00
ю
*г
о
чН
со
см
г^
со
о
¦чН
*ф
о
1
1
о
sf
1
о
•чН
СМ
•чН
«чН
ю
VH
1
о
ч-Ч
о
•ч-»
со
см
**
г»
1
о
¦чН
см
СМ
чН
чН
со
см
1^
о
4-4
646
ч-Ц
со
см
1*
1
о
чН
ю
t-
о
¦чН
о
о
чН
о
чН
ЧН
1
1
о
о
о
и
тая
о
а
Д 3 0 Л
' о
и
о
W
А
ч
N
о
1
о
И
о
н
ft
ф
п
ь
чН
о
ю
г;
ел
см
г»
о
¦чг»
о
|>
<М
00
ю
чН
•чН
чН
о
00
¦чН
г»
о
чН
см
ч-t
см
00
1
о
•чН
а>
124
ю
ю
чН
о
г—
Nt*
vt<
тЧ
1
1
о
чН
о
*tf
со
со
¦чН
г*
о
чН
о
V}*
чН
о
05
г»
1
о
чН
-ч-Ч
-ЧН
ю
ю
г»
о
чН
со
см
¦чН
00
о
г»
1
о
чН
Oi
СО
о>
о
о
со
о
ю
см
1
1
ю
чН
i
чН
СО
о
38
•4-4
г*
о
-чН
1^
о
00
о
•чН
г-
1
о
чН
со
о
СЛ
о
1^
о
чН
^
ч!<
чН
о
1
о
чН
г-
см
v-t^
о
о
ю
см
о
о
со
1
1
о
ю
о
чН
ю
о
о
чН
г»
о
чН
ю
о
оэ
оа
о.
г»
4»
чН
см
00
со
о
с»
о
чН
VP
чН
о
г»
1
о
ч-Ч
ю
см
4J*
о
о
00
чН
о
о
ю
ЧГ^
1
1
о
VF
ф
со
о
W
о
ей
ft
Й
1
о
чН
ю
со
со
см
чНа
г»
1
о
чН
ч-Ч
vt<
со
00
1
о
чН
см
чН
чН
ч-Н
„
f
о
чН
348-
чН
со
г»
1
о
чН
7889-
00
00
ч-t
чН
о
чН
см
чН
СО
чН
1
1
СО
1
о
чН
со
о
чН
см
t*
1
о
ч-4
о
ю
см
о
чН
чН
СО
о
см
Ч-»
г»
1
о
чН
со
см
с*
00
о
г-
1
о
ч-Н
со
чН
со
о*
о
о
чН
on
1
1
00
со
1
о
чН
t^
о
S
см
!>.
1
о
чН
ю
о
О)
о
т
о
чН
см
см
о
г>
\
о
ч-t
см
чН
о
t^
1
о
чН
315-
о
о
СО
о
о
со
ч-Н
1
1
о
см
ченных органо-минеральных комплексах БЭТ методом показало ее
резкое снижение по сравнению с исходными компонентами.
Следует отметить поэтому, что наблюдающаяся сорбция являлась
по существу коагуляцией соответствующих гуматов на
поверхности минералов, из которых предварительно не были удалены
обменные катионы кальция и магния. И. И. Лиштван с
сотрудниками [185] также отмечают обменный характер сорбции гуми-
новых кислот на глине за счет реакции замещения водорода
карбоксильных и фенолгидроксильных групп при
соответствующих значениях рН; при этом ни гуминовые кислоты, ни гуматы
не проникают в межпакетное пространство кристаллической
решетки. Обменный характер взаимодействия отмечают и другие
исследователи [84, 237, 375].
Косвенным, но достаточно убедительным подтверждением
описанного механизма формирования глиногумусовых комплексов
являются данные о распределении гумуса в различных
гранулометрических фракциях ряда почв (табл. 58).
Весовое содержание гумуса во фракциях вскрывает хорошо
известную картину. Максимальное количество гумуса характерно
для илистой и коллоидной фракций. Совершенно другие
закономерности выявляются при вычислении количества гумуса на
единицу поверхности фракции. Наименьшее и примерно одинаковое
количество гумуса распределено в единице поверхности
коллоидной фракции. В аккумулятивных горизонтах всех почв оно равно
3.5—5.0«10~7 г/см2. Если бы гумусовые вещества были связаны
с поверхностью кристаллической решетки минералов через
мостики с обменными катионами, количество которых в этих почвах
очень различно, следовало бы ожидать, что максимальное
содержание гумуса будет в единице поверхности чернозема. Таким
образом, закрепление гумуса на илистой фракции обусловлено
величиной поверхности, на которой может распределиться масса
гумусовых веществ.
Интересно отметить, что аналогичные данные получены
И. А. Пивоваровой [242], количество поглощенной асканитом и
каолинитом гуминовой кислоты на 1 м2 поверхности оказалось
примерно одинаковым, несмотря на очень разную емкость
обменного поглощения катионов в них (61.8 мг*экв. в асканите и
16.5 мг«экв./100 г в каолините).
Дискуссионным является также вопрос о возможности
проникновения молекул гумусовых кислот или их растворимых солей
в межслоевое пространство глинистых минералов. Наши
исследования со многими модельными препаратами, полученными на
различных глинистых минералах, дают отрицательный ответ.
Снятые дифрактограммы неизменно свидетельствуют о сохранении
межплоскостного расстояния d001, характерного для данного
препарата (рис. 39). Аналогичные материалы получены в нашей
лаборатории А. В. Назаровой (рис. 40), а также другими
исследователями [242]. В тоже время Д. С. Орлов с сотрудниками [237],
181
14 9
Рис. 39. Рентгенодифрактометрические кривые (d0oi) асканита,
компостировавшегося 1 год с гуминовой кислотой при 60 (/) и 150%-ной (//) влажности
(от полной влагоемкости).
а — контроль, б — обработка 0.1 н раствором NaOH, в — обработка Н20.
подтвердив поверхностную сорбцию гуматов и фульватов на
глинистых минералах, указывают на возможность
проникновения фульвокцслот в межплоскостное пространство
монтмориллонита в условиях резко кислой реакции (рН 2.5).
В последнее время появились данные, свидетельствующие об
избирательном характере поглощения гумусовых веществ
минералами в зависимости от их ММ, что обусловливает своеобразное
фракционирование их в почве. Некоторые исследователи считают,
что избирательно сорбируется наиболее высокомолекулярная
182
3 80
9 80
Рис. 40. Рентгенодифрактометрические кривые (d001) бентонита,
насыщенного гуматом Na и гуминовой кислотой.
а — Na-бентонит, б — Na-бентонит+гумат Na, в — Na-бентонит+гуминовая кислота,
г — Са-бентонит+гумаг Na
фракция гуминовых кислот. По данным И. А. Пивоваровой [242],
наиболее интенсивно поглощается наименее высокомолекулярная
фракция как гуминовых кислот, так и фульвокислот. К таким же
выводам приходит А. В. Назарова [205]. Приведенные кривые
молекулярной полидисперсности гуминовых кислот на сефадексе
Г-50 до и после их взаимодействия с бентонитом (см. рис. 16)
показывают, что если гуминовые кислоты до взаимодействия
с бентонитом делятся на три фракции, то после взаимодействия
на хроматографических кривых сохраняются лишь две фракции.
Таким образом, фракционирование гумусовых веществ при их
взаимодействии с минеральной частью почвы можно считать
установленным фактом, хотя его характер требует дальнейшего
изучения.
Возможность сорбции и внедрения низкомолекулярных
продуктов разложения в межплоскостное пространство глинистых
минералов не вызывает сомнений и отмечается многими
исследованиями. Хорошо поглощаются минералами аминокислоты, амиды,
нуклеиновые кислоты, низкомолекулярные органические кислоты,
спирты, альдегиды, кетоны, многие пестициды. Механизм их
поглощения, по-видимому, различен и детально рассматривается
в ряде работ [110, 224, 406, 411, 454, 537].
183
Природа органо-минеральных коллоидов и их состояние
Материалы о характере взаимодействия гумусовых веществ
с минералами почвы позволяют дать общую характеристику
основных компонентов органо-минеральных коллоидов (табл. 59).
Органо-минеральные коллоиды — комплекс переменного состава
из высокодисперсных минералов, покрытых пленками гумусовых
кислот, гу матов, фульватов, алюмо- и железогумусовых
производных. Это типичные мутабильные соединения, образующиеся
в процессе почвообразования и трансформирующиеся в
дальнейшем ходе его. Основными минералами, входящими в состав
коллоидов, являются монтмориллонитовая и гидрослюдистая группы
минералов, вермикулит, а также всегда сопутствующие им
кристаллические и аморфные полуторные окислы и кремнезем;
несколько меньше распространены каолинитовые минералы.
Формирование органо-минеральных коллоидов в почве —
результат комплекса реакций, среди которых главнейшее значение
имеют обменные реакции солеобразования при взаимодействии
гумусовых кислот с обменными катионами породы и почвы,
мобилизация гумусовыми кислотами и их солями элементов
кристаллической решетки глинистых минералов и сопутствующих
им полуторных окислов с образованием системы гетерополярных
и комплексно-гетерополярных солей, а также склеивание всех
этих веществ в процессе дегидратации в основном за счет
межмолекулярных сил. Химический и минералогический состав органо-
минеральных коллоидов в разных почвах достаточно
разнообразен и определяется характером материнской породы и
растительности, участвующих в почвообразовании, природными условиями
и типом почвообразовательного процесса.
В любой почве коллоиды неоднородны по своему состоянию
(степени подвижности). Часть коллоидов находится в почве
в свободном состоянии, не связана прочно с поверхностью более
крупных гранулометрических фракций, и степень их подвижности,
а следовательно, и миграционной способности определяется
составом обменных катионов. Эти коллоиды легко выделяются из почвы
при пептизации их по методу Гедройца насыщением почвы натрием
и при последующей пептизации водой. Некоторая часть
коллоидов образует^ пленки на поверхности более крупных
гранулометрических фракций почвы, дегидратирована и неподвижна.
Для их выделения необходимы дополнительные приемы обработки
почвы: разминание для снятия пленок вследствие
расклинивающего действия воды и диссолюционная пептизация почвы слабыми
щелочными растворами, при которой происходит добавочная
пептизация наиболее застаревших и дегидратированных коллоидов.
Эти свойства, детально исследованные М. Надем [201], можно
проиллюстрировать данными о количестве коллоидов, выделяемых
из почвы при различных способах их пептизации (табл. 60).
Особенно значительно количество прочно связанных в почве коллоидов
184
«с
3 ca
S
о*
S
з ?
Н ф
О Ои
о Ы
<* Я
Я
«и Я
is
5 и
и g Р
и Я Я
Я ?о
о «
_ _ tr
ч « а
ч ч н
о о о
Я5
fcf ф
о а
§ ьс
В м И
яваяя
fa ч и © g
® И и eq 2
о-, сьн ^
Й й « и и
о в о ч "
еь о. ?: Я н
И о
Я п И ?*
¦*ю о
>>ев
©
m
о
И
о ,
«
к
о
ч
¦§
«
и
я
я
О) ?
ю Ч
ев .
« §Н
Й* И
5 S
ев ев
о
Я
н
и
ф
W
о
к
a
о
к
ф
Я
и
в
о
Я
g s 3
2 и 5
ft О W ы
и § 2'3
2 «вЮ Я*
д к о к
О. „- н *
« 5 ф 2
^§св
А2
И Я"4'4
11 Ли
№~*
?М^»Ы ев
SgA/я л
^.ч
ы ф ТО
2'§К
2 ч &
0-1 в
ев Я tT
^ а ев
2 н о
>>Ч 2.
со 2
ев в g
к 2
о о ?
« S
CQ ф В
О Н-Н"
ft" к
« w Ф
ев О
РнЧ
И
+"
. +*
ев
I
+
I
о
I
о
о
и
ф
\/
ее
ев
I О
I
о
о
U
\/
С*
/\
О
о - о
о
\/
+
и
I
о
о
и
I
к
I
о
\/
ее
/\
и
о о
о
о
о
и
о
- О О
ф
? О ь
о
^ *Й Q
о~5Г
ее
о
ф
ее;
о
/\
о,
ф
й
\/
t •
°
in —
/
+ аГ *Х ¦
S 'о ,Х
i \/ j
?__3_g
/ \
-о^-^О-
/\
2
О о
Ч/
2
,Qg,
о
~"'<л
\
2 »
S се
о л
as
ф &
PQO
W
ее
Я
Я
• О
S
Я ее
N Я
Ы я
РчО
2 S
° Я*
ю «
Лев
Я И
Я
о
к
Я"
с
a
ф
Ол.
Я—'
Ф Я
t- ев
«И?
0,cd
fcf сб
8 Н & и
о S и
я S^°
ф S I S
§1
с
се Я
^?
ф ч
Я >»
я *©*
ч: „
о в
ю
а
6о »
00 Ч м со
2 н Ч a о
g g о g к
м ° 2 2
2 и § °9g
ООн«бЙ
g
<J С, О
I
Ф
Ч ф
I я я
« CQ о
о к
в о ф
о Я И
я h^
* Q к
Ч го
. я
о о
° к
>~> ф
Я Ч
>^Я
ф s
н§
ф ф
^2
Я >>
я 5
ч §
ев
Я . В
и ч: —
5 ч в
в 2 и
г о a
&&Ч
н г 2
я ^ и
о
Я
я
о
О
Я со
2 о
се *
И Я
в ч
с
>>
§33
ев Я В
°?|
О
РнЯ
2 Я в
ф Й*я
» РнЯ
2 ф и
я^чЭ
о о о §
й я И ср
Л Я
^ ч
vo н
Рн2 S
° я §
U Я g
Рис. 41. Схематическое строение органо-минеральной частицы.
фракция I, свободная; 2 — фракция II, рыхло связанная; з — фракция III, прочно
связанная с поверхностью минеральной частицы почвы.
при малом содержании их. Так, в бурой лесной почве основная
масса коллоидов выделяется лишь при дополнительной диссолю-
ционной пептизации.
Современные представления о природе органо-минеральных
коллоидов заставляют внести ряд поправок в толкование данных
Таблица 60
Содержание коллоидов, выделяемых при различных способах
пептизации (по: [201])
Способ выделения
Количество коллоидов, % к сухой почве
(размер фракции в мкм)
10—1
1-0.2
0.2-0.1
<0.1
суммарно
<0.2
<1
Чернозем
1 25.6
18.7
18.2
15.2
21.9
15.2
5.7
7.1
6.0
18.3
21.8
30.1
24.0 1
28.9
36.2
По Гедройцу
Дополнительное разминание частиц|
>1 мкм
Дополнительная обработка 0.02 н.
раствором NaOH частиц >0.1 мкм
Бурая лесная почва
По Гедройцу
Дополнительное разминание
частиц >1 мкм
Дополнительная обработка 0.02 н.
раствором NaOH частиц >0.1 мкм
Примечание. Образцы взяты из горизонта А
№
20.4
18.6
18.1
6.7
18.0
15.4
0.8
3.2
3.8
1.9
4.5
7.0
1.7 I
7.7
10.8
39.2
50.8
51.3
8.4
25.7
26.3
фракционного анализа гумуса, выполненного по любой схеме, на
что мы неоднократно указывали ранее [10, 19]. Постепенное
фракционирование гумусовых веществ и выделение фракций I —
III обусловлено не различиями в формах связи их с
компонентами минеральной части почвы, а степенью прочности связи
с поверхностью тех или иных гранулометрических фракций
(рис. 41). Фракция I, выделяемая при непосредственной обработке
почвы 0.1 н. раствором NaOH, наиболее подвижна, практически
свободна и не связана с поверхностью минералов. Последующие
обработки почвы 0.1 н. растворами H2S04 и NaOH обусловливают
выделение фракций гумусовых веществ, связанных с
поверхностью рещетки и лишь постепенно отделяющихся от нее.
Поэтому-то при фракционном анализе состава гумуса солонцов,
в которых доминируют обменные катионы натрия, а не кальция,
•фракции II и III гумусовых веществ значительны. Они прочно
связаны с поверхностью минеральных частиц.
Глава V
ГУМУС ПАХОТНЫХ ДЕРНОВО-ПОДЗОЛИСТЫХ ПОЧВ
И ВЛИЯНИЕ ОРГАНИЧЕСКИХ УДОБРЕНИЙ
НА ЕГО СОДЕРЖАНИЕ И СОСТАВ
Дальнейшая интенсификация земледелия и преобразование
сельского хозяйства Нечерноземной зоны Российской федерации
невозможны без все более широкого использования органических
удобрений, роль которых в повышении плодородия и
преобразовании дерново-подзолистых почв огромна. Органические удобрения
являются не только источником элементов питания культурных
растений,-но и регулятором всех главнейших физико-химических
и биологических свойств пахотного слоя: запасов и состава гумуса,
поглотительной способности, биологической активности, водно-
физических свойств.
Вместе с тем если влияние разных видов и доз органических
удобрений на питательный режим дерново-подзолистых почв и
урожай всех основных культур изучается широко, то их влияние
на гумусовый режим, количество и состав гумуса в
дерново-подзолистых почвах исследованы все еще недостаточно. Не
обсуждаются также сами понятия «гумусовый профиль», «гумусовый
режим», не рассматриваются их основные константы. Без
изучения этих констант и процесса их формирования нельзя дать
научно обоснованные рекомендации о наиболее рациональных
способах оптимизации количества гумуса и его состава в дерново-
подзолистых почвах, составляющих основной земельный фонд
в Нечерноземной зоне.
Гумусовый профиль и гумусовый режим,
их основные параметры
Количество и состав гумуса в почвах колеблются в очень
широких пределах и представляют собой в настоящее время один из
существенных диагностических признаков. Для каждого типа
почв характерен свой гумусовый профиль, сохраняющийся в
условиях того или иного гумусового режима. Основные показатели
гумусового профиля: содержание и запасы гумуса в пределах
педона, характер его распределения, а также групповой и
фракционный состав гумусовых веществ, свидетельствующий о
соотношении между группами гумусовых кислот и прочностью связи
188
7 и гп
100 100 - 100
N V
8642021+68 8642 0 2468%
100 100
Рис. 42. Типы гумусового профиля.
/ — грубогумусовый, II — гумусово-иллювиальный, III — аккумулятивный неполно-
развитый, IV — аккумулятивный полноразвитый, V — неразвитый.
их с минеральной частью почвы. Все многообразие типов
гумусового профиля по характеру распределения гумуса в педоне
минеральных почв можно в первом приближении свести к пяти
основным типам: грубогумусовый, гумусово-иллювиальный,
аккумулятивный неполноразвитый, аккумулятивный полноразвитый и
неразвитый (рис. 42).
Грубогумусовый тип профиля формируется в лесных почвах
бореального гумидного климата при отсутствии или слабо
выраженном травянистом растительном покрове. Он характеризуется
накоплением на поверхности почвы значительной массы подстилки
с содержанием в ней более 30% органических веществ. В составе
органических веществ значительна доля негумифицированных и
полугумифицированных растительных остатков, количество
гумусовых веществ не превышает 30%. Гумусовый аккумулятивный
горизонт здесь практически не формируется, и содержание
органических веществ под подстилкой резко падает, а гумус в
минеральной части профиля носит потечный характер. Этот тип гуму-
189
Совогчэ профиля характерен для целинных подзолистых, глеепод-
золистых, таежно-мерзлотных почв и подбуров.
Гумусово-иллювиальный тип гумусового профиля формируется
в пределах полярного и бореального поясов почвообразования и
характеризуется наличием иллювиально-гумусового горизонта
в пределах педона, что является результатом непрерывной
миграции части гумусовых веществ в нижние горизонты профиля.
Фульвокислотный состав гумусовых веществ и постоянно
господствующие нисходящие и боковые токи влаги при наличии на
некоторой глубине водонепроницаемых прослоек или их
формирование в ходе почвообразовательного процесса обусловливают
эти шотечные» формы гумусовых веществ, выпадающие в виде
алюмо- и железофульватных комплексовав. профиле. Этот тип
гумусового профиля характерен для многих тундровых и
подзолистых почв, а также подбуров.
Аккумулятивный неполноразвитый тип гумусового профиля
очень распространен во многих типах почв, Он формируется при
обязательном участии корневых систем травянистой
растительности, основная масса которых распределяется лишь в верхней
15—20-сантиметровой части педона, что обусловлено как
климатическим режимом, так и особенностями строения самого
почвенного профиля — его гранулометрическим составом и плотностью*
Для этого типа гумусового профиля характерно образование
небольшого по мощности аккумулятивного гумусового горизонта,
органическая часть которого представлена в основном
гумусовыми веществами (они составляют 80—90% всей массы
органических веществ). Мощность его невелика и всегда коррелирует
с глубиной проникновения основной массы корней травянистой
растительности. Он типичен для дерново-подзолистых целинных
и залежных почв, для дерновых почв бореального пояса,
каштановых и бурых полупустынных почв, а также многих почв
влажных и ксерофитных субтропиков и тропиков.
Аккумулятивный полноразвитый тип гумусового профиля по*
лучает яркое развитие в почвах, где благодаря благоприятному
гидротермическому режиму и не менее благоприятным физическим
свойствам самой почвы развивается глубокопроникающая
корневая система травянистой растительности. Содержание гумуса
с глубиной снижается очень постепенно, в его составе доминируют
гумусовые вещества, а гумусовый профиль практически совпадает
^с профилем всей почвы. Типичными представителями этого типа
^гумусового профиля являются черноземы, различные луговые
jriogBbi и темные сероземы.
^развитый тип гумусового профиля характерен для почв
^пуст^ндого ряда, где практически нет аккумуляции гумуса
j;B верхней части педона из-за крайней бедности растительного
^покрова, фн типичен для серо-бурых, примитивных песчаных
(Почв, такыров. ^Особое положение занимают торфяные почвы, весь
профиль которых имеет органогенную природу.
190
. Для пахотных дерново-подзолистых почв, которые по
зональному положению имели аккумулятивный неполноразвитый тиш
гумусового профиля, необходима более дробная группировка
типов гумусового профиля вследствие его антропогенного
происхождения, обусловленного использованием этих почв в качестве^
пахотных угодий с очень различным уровнем агротехники.
Характеристику гумусового профиля в пахотных дерново-подзолистых
почвах целесообразно дополнить показателями мощности
гумусового горизонта и общими запасами гумуса в нем. В зависимости
от этих дополнительных показателей пахотные
дерново-подзолистые почвы характеризуются как неполноразвитым, так и
полноразвитым аккумулятивным типом гумусового профиля (табл. 61).
Таблица 61
Основные показатели разных типов гумусового профиля
пахотных дерново-подзолистых почв
Тия гумусового профиля
Маломощный
Неполноразвитый
Полноразвитый
Мощность
Аиах, см
1
! <22
22—27
; 27 и более
Содержание
гумуса, %
1-3
3-4
4-6
Запас гумуса4
в Агав?, т/га
<100
100—160
160—250 и более-
По доминирующему типу гумусовых веществ мы выделяем
четыре градации с разным отношением СГк * Сок
фульватный <0.6
гуматно-фульватный . . . 0.6—0.8
фульватно-гуматный 0.8—1.2
гуматный >1.2
Аналогичную группировку с несколько иными константами
предложили Л. А. Гришина и Д. С. Орлов [87]. Описаны и более
дробные группировки типов гумусовых веществ: В. Р. Волобуев;
[69] в дополнение к основной характеристике по составу
гумусовых кислот включает солефракционный состав и обменные^
катионы; С. А. Алиев [34] использует в качестве диагностического *
признака содержание «гуминов», хотя сам термин, как мы уже*
отмечали, очень неопределенен. Особенно сборным он является
при анализе пахотных почв, систематически получающих
значительное количество органических^удобрений. Существенны
показатели, характеризующие-прочность связи тумусовых веществ
с минеральной частью почвы. Вряд ли тольшГцелесообразно
выделять фракции, связанные с кальцием, ибо,*какТмы уже
отмечали, фракция II, выделяемая после декалыщрования серной
кислотой, представлена той частью гумусовых веществ, которая
191
более прочно связана с поверхностью кристаллической решетки
Минералов и не всегда насыщена кальцием. Для характеристики
с1епени подвижности и прочности связи мы предлагаем
использовать относительное содержание подвижных и прочносвязанных
^Уминовых кислот (отношение фракции I к фракции II гуми-
**овых кислот, выделяемых по методу И. В. Тюрина).
Существенным показателем является, конечно, относительное
содержание негидролизуемого остатка, предложенное Л. А. Гришиной
** Д. С. Орловым [87], но для этого показателя необходима
дополнительная расшифровка его природы, так как он состоит по
крайней мере из двух резко различных в генетическом плане групп
компонентов: очень прочно связанных с поверхностью
минеральных частиц гумусовых веществ, а также полугумифицированных
И слабоэкстрагируемых компонентов органических удобрений.
Наряду с фактической характеристикой гумусового профиля
Необходима характеристика гумусового режима почвы, т. е.
потенциальной способности органической части почвы к процессам
Гумификации, накоплению гумуса и его минерализации. К
сожалению, эти показатели не разработаны, хотя крайне необходимы
Нак для оценки роли гумуса в почвообразовании, так и для
определения уровня плодородия.
Наши исследования показали, что для характеристики
гумусового режима пахотных дерново-подзолистых почв (по-видимому,
И других типов почв) можно использовать следующие основные
Параметры: потенциальную способность к гумусообразованию,
°иределяемую по разности между общим содержанием органических
веществ (Сорг) и количеством гумуса (Сгум) в заданном горизонте;
интенсивность гумификации органических остатков,
учитываемую по динамике количества гумуса с введением поправки на
Степень его минерализации; интенсивность минерализации как
Всей массы органических веществ, так и гумуса в заданном
горизонте; степень миграции гумусовых и низкомолекулярных
органических веществ из заданного горизонта за тот или иной период
Наблюдений.
Определение всех этих показателей требует предварительного
Изучения пространственной пестроты территории исследования
И уточнения необходимого количества повторностей для
получения достоверных результатов, в противном случае получаемые
результаты не будут отражать реальную характеристику
гумусового режима.
Особенности формирования гумусового горизонта
в пахотных дерново-подзолистых почвах
Гумусовый горизонт в пахотных дерново-подзолистых почвах
формируется в период их сельскохозяйственного использования.
Целинные дерново-подзолистые почвы, как справедливо
указывают исследователи [187, 220, 241, 263], характеризуются мало-
193
vn
чо
30
20
70
, /о
1 1 1 1 1 1
/ 1 1 1 1 1 1
Ч
1 Л L 1
10
20
30
40 ГК}%
рис. 43. Типы гумусовых веществ
в дерново-подзолистых почвах.
2 — фульватный, 2 — гуматно-фуль-
ватный, 3 — фульватно-гуматный, 4 —
гуматный.
мощным гумусово-аккумуля-
тивным горизонтом,
недостаточным для развития
корневой системы большинства
сельскохозяйственных
растений и обеспечения для них
благоприятного
водно-воздушного и питательного
режимов. Гумусовый горизонт
маломощен, бесструктурен,
обеднен илистой фракцией,
имеет кислую реакцию
вследствие многовекового развития подзолообразовательного
процесса. Даже тяжелосуглинистые и глинистые разновидности
целинных дерново-подзолистых почв обеднены не только
коллоидами, но и илом. Эти особенности и обусловили
необходимость формирования более мощного гумусово-аккумулятив-
ного горизонта, совпадающего в большинстве случаев с глубиной
вспашки.
Содержание гумуса в пахотном слое дерново-подзолистых почв
и его состав формируются как за счет сохранения остаточных
показателей, типичных для данной фации целинных вариантов
гумусовых веществ, так и за счет производственной деятельности
человека. Общее содержание гумуса в пахотном слое дерново-
подзолистых почв разных регионов Нечерноземной зоны и состав
гумусовых веществ очень различны в зависимости от степени окуль-
туренности этих почв. Анализ многочисленных данных по
количеству и составу гумуса в пахотном слое дерново-подзолистых
почв Псковской, Новгородской, Ленинградской, Вологодской,
Кировской, Горьковской, Московской и ряда других областей
показал, что количество гумуса колеблется в зависимости от
гранулометрического состава, степени окультуренности и общих
биоклиматических условий от 1—2 до 4—5%, причем решающее
значение имеют гранулометрический состав и степень
окультуренности (вернее, степень и длительность применения
органических удобрений). Не менее значительно варьирует и состав
гумуса (рис. 43). В зоне все еще доминируют почвы с фульватным
и гуматно-фульватным типами гумусовых веществ, а следовательно,
и с сохранением агрессивной роли свободных фульвокислот.
Современный гумусово-аккумулятивный горизонт в пахотных
дерново-подзолистых почвах формируется за счет остаточных
13 Л. Н. Александрова 193
гумусовых веществ целинной почвы, наиболее прочно связанных
с минеральной частью почвы, пожнивных и корневых остатков
культурной растительности и вносимых органических удобрений.
Мощность этого горизонта определяется, как мы уже
указывали, глубиной ежегодной вспашки.
К настоящему времени у большинства пахотных
дерново-подзолистых почв удовлетворительный, а в ряде случаев — хороший
по мощности пахотный слой: 22—27 см; лишь вновь осваиваемые
дерново-подзолистые почвы, а также некоторые старопахотные
почвы восточных районов Нечерноземья (Вологодская и
Кировская обл.) имеют пахотный слой менее 22 см; многие хорошо
окультуренные почвы характеризуются мощным пахотным слоем: 27—
30 см. Следовательно, увеличение мощности пахотного слоя
путем углубления вспашки или припахивания подпахотного,
в большинстве случаев подзолистого, слоя или верхней части
горизонта А2В необходимо лишь на вновь осваиваемых или очень
плохо окультуренных почвах для формирования мощного кор-
необитаемого слоя с благоприятными для корневой системы
растений физико-химическими свойствами. Менее благоприятными
следует считать содержание и состав гумуса, а также его динамику
в пахотном слое этих почв. Как справедливо указывают А. М.
Лыков [187], В. К. Пестряков [241] и другие исследователи [48,
154, 197], в большинстве пахотных дерново-подзолистых почв
любого гранулометрического состава постепенно, но постоянно
снижается количество гумуса и ухудшается его состав. Для
иллюстрации можно привести данные о содержании и составе гумуса
в дерново-подзолистых почвах Северо-Запада (табл. 62). Лишь
в почвах, систематически получающих высокие дозы
органических удобрений порядка 80—100 т/га ежегодно, или на
огородных участках можно наблюдать высокое, систематически
поддерживаемое на этом уровне содержание гумуса и относительно
благоприятный (фульватно-гуматный) его состав.
Каковы же причины, обусловливающие неудовлетворительное
состояние гумусового режима, количества и состава гумуса
в пахотных дерново-подзолистых почвах? Основными из них,
по нашему мнению, являются недостаточное количество исходных
гумусообразователей, ежегодно поступающее в пахотный слой,
обедненность пахотного слоя высокодисперсными глинистыми
минералами, кислая реакция его, небольшое количество
обменного кальция и неблагоприятный для гумификации органических
остатков биоклиматический режим. Действительно (как мы уже
отмечали в гл. I), источниками гумуса для пахотного слоя
дерново-подзолистых почв служат пожнивные и корневые остатки
и вносимые в него органические удобрения. Количество корневых
и пожнивных остатков, ежегодно поступающее в пахотный слой,
в зависимости от видового состава возделываемых культур и
урожая колеблется от 2—3 до 8—10 т сухого вещества на 1 га
и только под многолетними травами (2—4-летнего пользования)
194
Таблица 62
Содержание и состав гумуса в дерново-подзолистых почвах
Северо-Запада (по: [241])
Угодье
Глубина,
см
С, о/о
Состав гумуса,
% к валовому С
СГК
СФК
СГК
Сфк
Дерново-сильноподзолистая суглинистая
почва на покровном суглинке (Вологодская обл.)
Лес
-
Пашня
освоенная
окультуренная
сильноокульту-
ренная
| 1—5
5-10
10-20
21—28
30—40
0-10
10—22
22—30
30—40
0—10
10-20
20—30
30—40
0-10
10—20
20—25
25—32
32—40
50—60
1 2.3
1.2
0.6
0.3
0.3
1.3
1.3
0.4
0.4
1.2
1.2
1.2
0.6
5.4
4.9
3.4
2.1
0.7
0.3
24
27
19
5
3
30
25
21
14
20
19
20
4
34
35
30
24
20
6
44
41
40
41
34
34
34
29
33
34
33
24
16
23
24
27
35
35
45
0.5
0.6
0.5
0.1
0.1
0.9
0.7
0.7
0.4
0.6
0.6
0.8
0.2
1.5
1.5
1.1
0.7
0.5
0.1
Дерново-подзолистая глинистая почва (Валдай)
Лес
Пашня
3—12
12-17
17—26
26—36
0-10
20—29
29-35
40-50
1 3.8
2.1
0.7
0.3
1.7
| 1.4
1 0.4
! о.з
29
30
23 1
7
23
24
20
13
43
50
45
55
34
40
38
34
0.7
0.6
0.5
0.1
0.7
0.6
0.5
0.4
поднимается до 15 т/га. Количество вносимых органических
удобрений варьирует в очень широких пределах: от 5—10 до 80—
—100 т/га. В зависимости от мощности пахотного слоя и плотности
его сложения, а также влажности и зольности их количество
углерода, поступающего в пахотный слой в составе органической
массы на каждые Ют гумусообразователей, не превышает 0.1%
к массе сухой почвы (табл. 63). Если учесть, что основная масса
пожнивных и корневых остатков ежегодно минерализуется и
195
13*
коэффициент их гумификации не превышает обычно 25%,
становится очевидным, что использование пахотных
дерново-подзолистых почв на фоне одних минеральных удобрений
неизбежно приводит к постепенному снижению запаса гумуса
вследствие недостаточного ежегодного поступления гумусообразователей.
Лишь под многолетними травами при условии их высоких
урожаев можно наблюдать достаточно интенсивное
новообразование гумусовых веществ, в составе которых возрастают
относительная роль гуминовых кислот и накопление гумуса.
Однако распашка пласта неизбежно приводит к интенсификации
процессов минерализации новообразованных гумусовых веществ
и резкому снижению количества гумуса вследствие слабого
закрепления их в пахотном слое.
Таблица 63
Содержание С в 10 т на 1 га органических остатков
и удобрений, внесенных в пахотный слой
Мощность
пахотного слоя,
см
0—20
0-25
0-30
С, % к сухой почве, при различной плотности
сложения
1.2
0.06—0.10*
0.05-0.08
0.04-0.07
1.3
0.06-0.10
0.05-0.08
0.04-0.06
1.4
0.05—0.09
0.04—0.07
0.04-0.06
Примечание. Звездочкой отмечен интервал колебаний в зависимости от зольности
и влажности удобрений.
Не менее существенное значение для формирования гумусово-
аккумулятивного горизонта в пахотных дерново-подзолистых
почвах имеют его неблагоприятные физико-химические
показатели, являющиеся результатом многовекового развития подзоло-
образовательного процесса во всей верхней части профиля
целинных дерново-подзолистых почв. Пахотный слой дерново-
подзолистых почв любого гранулометрического состава обеднен
илистой и особенно коллоидной фракцией. Заслуживают
внимания обширные аналитические данные В. К. Пестрякова [241] по
содержанию не только ила, но и коллоидов в пахотных дерново-
подзолистых почвах различного гранулометрического состава
и разной степени окультуренности (рис. 44). Даже
тяжелосуглинистые и глинистые относительно хорошо окультуренные
пахотные дерново-подзолистые почвы содержат в пахотном слое крайне
незначительное количество минеральных коллоидов, не
превышающее 2—3%, хотя общее содержание ила здесь может
достигать 8—10%. Произведенные автором подсчеты убедительно
свидетельствуют о непрерывно продолжающемся выносе ила и
особенно коллоидов из пахотного слоя. Если учесть, что незна-
196
25%
Рис. 44. Содержание ила (i, < 1 мкм) и коллоидов (2, < 0.2 мкм) в профиле
дерново-подзолистых глинистых почв (по: [241]).
/ — лес, // — луг, III — пашня окультуренная.
чительное количество минеральных коллоидов обусловливает
низкую общую и удельную поверхность пахотного слоя, то
становятся очевидными крайне неблагоприятные условия для
закрепления образующихся гумусовых веществ в нем.
Незначительное количество минеральных коллоидов в
пахотном слое — основная причина низкой емкости обменного
поглощения и ненасыщенности основаниями большинства
пахотных дерново-подзолистых почв [154, 187, 220, 241 ]. Если в нижней
части профиля в случае суглинистой или глинистой материнской
породы емкость обменного поглощения катионов достигает 25—
30 мг-экв./100 г при достаточно высокой степени насыщенности
основаниями (до 85—95%), то пахотный слой, как правило,
характеризуется резко пониженной общей емкостью поглощения
и ненасыщенностью основаниями, несмотря на систематически
повторяющееся известкование. Исследование состава
мигрирующих из пахотного слоя лизиметрических вод всегда фиксирует
значительное вымывание кальция в виде водорастворимых солей
низкомолекулярных кислот и фульвокислот. Многочисленные
эксперименты свидетельствуют о том, что в условиях
избыточной влажности при разложении и гумификации органических
остатков образуется много фульвокислот, которые усиливают
миграцию кальция из пахотного слоя вследствие растворимости
фульватов кальция.
Таковы основные причины формирования недостаточно гуму-
сированного пахотного горизонта в дерново-подзолистых поч-
197
вах и обоснование огромной роли органических удобрений в
частичной ликвидации этих недостатков и обеспечении
положительного баланса гумуса в этих почвах.
Трансформация органических удобрений
в пахотном слое дерново-подзолистых почв
и пути оптимизации гумусового режима в нем х
Дальнейшая интенсификация земледелия и коренное
повышение плодородия вновь осваиваемых и недостаточно
окультуренных дерново-подзолистых почв Нечерноземной зоны невозможны
без устойчивого улучшения гумусового режима в них: увеличения
запасов гумуса, улучшения его состава, усиления процессов
гумификации органических остатков и органических удобрений.
В условиях интенсивного земледелия гумус является не только
источником питания растений, хотя в нем сосредоточены почти
все запасы азота и значительная часть фосфора и серы, но и
основным условием для создания благоприятного для растений
водно-воздушного, питательного и биологического режимов.
Поэтому роль органических удобрений в повышении
производительности дерново-подзолистых почв очень велика.
Без применения органических удобрений формирование
положительного баланса гумуса в пахотном слое почвы невозможно
[187, 220, 241]. В то же время вопрос о роли этих удобрений
в поддержании бездефицитного баланса гумуса в пахотных
дерново-подзолистых почвах дискуссионный. Одна группа
исследователей, а их большинство [187, 197, 241], считает обязательным
использование органических удобрений для создания
бездефицитного баланса по гумусу, а тем более для увеличения его
содержания в пахотном слое, без которых неизбежно снижение
количества гумуса в почве. Наиболее убедительные данные приводит
А. М. Лыков (табл. 64). Как при бессменной культуре ржи, так
и в севообороте минеральные удобрения постепенно
обусловливают некоторое, незначительное возрастание содержания гумуса
вследствие большего количества остающихся в почве пожнивных
и корневых остатков, но оптимального содержания гумуса
таким путем создать не удается. Теоретически это подтверждается
прямыми расчетами количества поступающих в пахотный слой
растительных остатков и коэффициентом их гумификации на фоне
достаточно интенсивной минерализации как новообразованных*
так и сохранившихся от целинного периода гумусовых веществ.
Другая группа исследователей [198] считает возможным
сохранить бездефицитный баланс гумуса в пахотном слое дерново-
подзолистых почв без применения органических удобрений. На
основании экспериментов, проведенных на Ротамстедской опыт-
Раздел написан совместно с А. М. Пупковым.
198
Таблица 64
Влияние длительного (60 лет) систематического применения удобрений
на содержание и запасы углерода в пахотном слое
дерново-подзолистой легкосуглинистой почвы
Тимирязевской сельскохозяйственной академии (по: [187])
Вариант опыта
Рожь бессменная
- контроль
NPK
навоз
Севооборот, 132-е поле
контроль
NPK
навоз+NPK
С, %, в слое 0—20 см
без извести
0.97
1.07
1.56
0.77
0.95
1.16
по извести
0.96
1.04
1.48
0.76
0.97
1.04
Запас С, т/га, в слое
0—40 см
без извести
31.7
36.9
46.3
31.4
34.6
41.6
по извести
29.4
34.6
46.5
29.4
35.7
38.7
ной станции, а также опытов в Галле В. Г. Минеев и Л. К.
Шевцова [198] считают, что за 50—80 лет использования почвы
только по фону минеральных удобрений содержание гумуса не
уменьшилось.
Многолетние опыты А. М. Лыкова на дерново-подзолистых
почвах различного гранулометрического состава показали, что
систематическое внесение невысоких доз органических удобрений
обеспечивает как бездефицитный баланс гумуса, так и
увеличение его количества [187]. Аналогичные материалы приводят в
обзорной работу В. Г. Минеев и Л. К. Шевцова [198]. Следует
отметить, что все эти данные получены в условиях опытных
станций с полным соблюдением всех агротехнических требований и
на достаточно хорошо окультуренных почвах, не требующих
коренной переделки. В то же время при освоении закустаренных
или использовании плохо окультуренных дерново-подзолистых почв
необходима существенная переделка гумусового состояния
пахотного слоя, что возможно только при внесении в почву
значительного количества органического материала в виде органических
удобрений. К сожалению, данные, характеризующие влияние
высоких доз органических удобрений на гумусовый режим
пахотных дерново-подзолистых почв, очень ограниченны, хотя многие
хозяйства вносят ежегодно достаточно высокие дозы этих
удобрений.
Имеющиеся в нашем распоряжении материалы свидетельствуют
о том, что однократное внесение высоких доз органических
удобрений (навоза или торфяных компостов) порядка 80—150 т/га
коренным образом переделывает гумусово-аккумулятивный
горизонт пахотных дерново-подзолистых почв, резко улучшая все
основные физико-химические и биологические показатели в нем.
В песчаных и супесчаных почвах возрастает влагоемкость, сни-
199
жается водопроницаемость, повышается емкость обменного
поглощения катионов, а в глинистых почвах заметно увеличивается
диапазон активной влаги, улучшается питательный режим, и,
конечно, во всех случаях значительно возрастают количество
гумуса и относительное содержание в нем гуминовых кислот.
Эти высокие дозы резко увеличивают урожай и в ряде случаев
оправдывают себя экономически уже в первые годы, что
позволяет хозяйствам, особенно расположенным в пригородной зоне,
ежегодно вносить по 100—200 т и более этих удобрений.
Вместе с тем если влияние различных видов и доз органических
удобрений на питательный режим дерново-подзолистых почв и
урожай всех основных культур исследуется достаточно широко,
то их влияние на элементарные звенья почвообразовательного
процесса, именуемого часто «культурным», а также на процессы
гумусообразования изучено все еще недостаточно. А именно эти
вопросы должны находиться в настоящее время в центре внимания
почвоведов для разработки теоретических основ любой системы
мероприятий по коренному улучшению вновь осваиваемых и
плохо окультуренных дерново-подзолистых почв Нечерноземья.
Исследования, проводимые под нашим руководством на
протяжении последних 15 лет в условиях производственных полевых
опытов на полях совхозов Ленинградской и Калининской областей,
а также лабораторно-полевые и лабораторные наблюдения
позволили изучить основные закономерности трансформации
органических удобрений в пахотном слое дерново-подзолистых почв
различного гранулометрического состава [89].
Внесение органических удобрений (навоза и ТМАУ) повышает
запас органических веществ в пахотном слое в соответствии с
поступающей дозой; содержание же гумуса при этом повышается
в значительно меньшей степени и коррелирует со степенью
гумификации используемых удобрений. Иллюстрацией могут служить
данные одного из полевых производственных опытов, проведенных
в совхозе «Губинский» Калининской обл. с 1974 по 1977 г. А. М.
Пупковым (табл. 65). Изучалось влияние двух доз навоза и ТМАУ на
общее содержание органического вещества, количество гумуса, их
динамику в течение трех лет, а также групповой состав гумуса и
миграцию водорастворимых органических и минеральных веществ
из пахотного слоя песчаной и легкосуглинистой
дерново-подзолистой почвы. Повторность опытных делянок 3-кратная, детальная
методика работы и физико-химическая характеристика почв
приведены в работах А. М. Пупкова и В. П. Колодка [258, 259].
Следует отметить только, что для получения достоверных
результатов все определения содержания углерода проведены в 12-
кратной повторности с каждой делянки. Каждый образец,
поступающий в анализ, отбирали, кроме того, из 10 точек. Результаты
математической обработки показали, что такого количества по-
вторностей достаточно для получения достоверных результатов (s$
не превышает 7%).
200
ю
СО
о
1=1
tsi
I
9
О
m
I
Рн
а
I
IS
s S
и tsi
о
и
о
Рн
Рн
О
«
К
и
Рч
а
о
/-ч
р"
977
1 ""^
О
В
я
се
одерж
и
1 2«
R н
пос
рен
Содержание С
запашки удоб
(1975 г.)
ее
^
3
Б
о
а
акладк
СО Рн
С до
(1974
со
а
И
се
?
а
Сод
• >» 1
5 о
*5
+!§
о
14
со
-
S°
>»
Ен
А>»
5 о
«а
4HS
о
>Н 1
о
о
р<
CD
«->
m
'Н
о
Р*
®
03
о
'Й
с«
t-c
s?
fi
*
9 о
8
8*?* 1
«5ан 1
2
ag
в S
»5
ев О |
и
1
ев
П
F
О
Н
«
сб
И
е с ч а
К
СЛ ЮСО О СО
ООО'гН'^н
ооооо
1 1 +++
F-* <Л СО СО О
lOst" *tf СМ СО
COCO** O00
Ю СО СО OS 00
ооооо
^ Osf 00O
-54 СО СО С- 00
ооооо
1 1 1 1 1
0*^«54СМ 00
со сою со <м
ЧРОСОЮСО
СО СО !>• <Л <Л
ооооо
см** оо coo
СО СО СО Ю СО
00 Г» ОСОСО
t>- 05 «чН I>- t^
О* О «54 «54 ч-н
!>OMt>0
Ю СО *«tf "* СО
Ю00-54ОСМ
со со со оо t-
ооооо
ЮСО О СО Ь-
St1 Ю СО «<Р СО
оочр о* с- со
NI>COC5t*
ооооо
| ОООО
1 СО СО OS 05
л
ч
Р< СО >& СО >5
йМнКн
w
PQ
о
н
«
л
н
о
и
к
Рч
о
Рн
О)
1=3
«?4 00 «5-4 СМ СО
ОО «*н СМ СМ
ооооо
1 ++++
ООЮОтнЮ
COCO CO«tf СО
CNI О СО 05 СТ>
ОСМ CM«<f СО
s^h v-jh сО СМ СМ
ОСМ «54 LOCO
ооооо
1 1 1 1 1
-НОЮСОС5
со ю ю со со
^СООч^О
СМ СО СОСОО
«?н «*н -чн т-1 СМ
00 00 СО Ю СМ
со ююсо со
ооосо со см
СМ СО 1> -54 СО
«54 ч-Н -54 СМ* СМ*
or» as-* оою
СОЮ1СЮЮ
-*4<М Ю1> СО
О -5Н -54 CM «54
-54 -54 -«4 «54 «54
СМ СО <М СМ СО
СО ЮСО 00^
оо см ооюсм
CM COCO Vf СО
ч-Н -54 т4 -54 -54
1 ОООО
1 00 00 05 05
А
Ч
Рн СО р>5 СО ^>
ОСЙЙсб^
tfffiHffiH
у
s
СИ
кже орг
н
и"
о
н
се
н
о
о
3
m
а
о
В
шнивных
о
G
к
?
1=3
ей
>>
со
CD
о
ев
«
з углерс
S
в
Я СО
ад
«§¦
о а
Ос?
gee
2 «
и о
о к
» се
мече
ельь
й от
осит
§s
{н О
m со
со •
6 §
a S
се о
CD . .
рим
ских
в
Внесение органических удобрений обусловливает прежде всего
возрастающую пестроту в содержании органического вещества
в пахотном слое, коэффициент вариабельности достигает 30—
45% и сохраняется длительное время. Непосредственно после
внесения удобрений общий запас органического вещества в
пахотном слое возрастает в строгом соответствии с внесенной дозой.
При внесении 90 т/га общее количество органических веществ
удваивается, что соответствует в первом приближении массе
внесенных удобрений. Резко возрастает вследствие этого
потенциальная способность к гумусонакоплению, так как Сорг — Сгум
становится больше 0.5%. Вариабельность в содержании гумуса
значительно ниже, причем его количество менее резко, но также
увеличивается непосредственно после внесения удобрений. При
этом в суглинистой почве на протяжении трех лет наблюдений
сохраняется бездефицитный баланс гумуса даже без внесения
органических удобрений в результате включения в севооборот
многолетних трав. При применении органических удобрений
баланс гумуса становится положительным, причем наибольшие
прибавки в содержании гумуса получены при внесении 90 т ТМАУ
на 1 га. По-видимому, эти «прибавки» в количестве гумуса
непосредственно после внесения органических удобрений
обусловлены его «антропогенным» происхождением. Аналогичные данные
получил И. Н. Барановский при изучении влияния высоких доз
навоза и ТМАУ в модельно-полевом опыте на суглинистой
дерново-подзолистой почве совхоза «Ленсоветовский» (табл. 66).
Таблица 66
Динамика содержания гумуса и его состава в пахотном слое
тяжелосуглинистой дерново-подзолистой почвы в октябре 1971
и 1972 гг. (полевой опыт
в лизиметрических сосудах; по: [50])
Вид
удобрений
Контроль
Навоз
ТМАУ
Доза
удобрений, га
30
150
30
150
Глубина, см
0-10
10-20
0-10
10-20
0—10
10—20
0—10
10-20
0-10
10—20
±с
1971
-0.03
—0.01
+0.06
+0.07
+0.18
+0.23
+0.06
+0.09
+0.31
+0.38
1972
-0.05
-0.03
+0.03
+0.02
+0.08
+0.13
+0.03
+0.08
+0.16
+0.30
сгк: сфк
1971
0.74
0.76
0.81
0.79
0.92
0.88
0.80
0.81
0.96
0.91
1972
0.77
0.73
0.78
0.78
0.90
0.85
0.75
0.82
0.95
0.94
Примечание. Исходное содержание углерода 2.66%; весна 1971 г.
202
Таблица 67
Состав органического вещества навоза и ТМАУ,
% к органической массе
Вид
удобрений
Навоз
1
-2
ТМАУ
1
2
Собщ» /о к 1
сухому
веществу
35
30
43
32
Зольность,
%
16
26
18
30
Водорастворимые
вещества
4
—
2
—*
Битумы
5
3
5
3
Гумусовые
вещества
41
44
51
65
Гуминовые
кислоты
20
28
39
42
Гемицеллю-
лозы
8
8
5
5
Целлюлоза
12
17
6
6
Негидроли-
зуемый
остаток
30
28
31
21
Источник
[259]
[50]
[259]
[50]
Как в навозе, так и особенно в ТМАУ содержится
значительное количество гумифицированных и полугумифицированных
материалов, которые невозможно механически отделить от массы
почвы. Определение состава органической части вносимых
удобрений подтвердило наши предположения (табл. 67). Как видим,
основная часть удобрений — органическое вещество. Органическая
масса навоза и ТМАУ хорошо гумифицирована, причем в ТМАУ
на долю гумусовых веществ приходится больше половины ее.
В составе гумусовых веществ этих удобрений преобладают
гуминовые кислоты, содержание которых в ТМАУ также выше. Не-
гидролизуемый остаток, представленный в основном лигнинопо-
добными соединениями, также составляет значительную часть
органических веществ. Навоз богаче, чем ТМАУ, гемицеллюлозами
и целлюлозой, вероятно, во-первых, благодаря различному
биохимическому составу растений и наличию в экскрементах
животных большого количества этих соединений, а во-вторых, из-за
потери части углеводов в процессе торфообразования.
Водорастворимые вещества составляют наименьшую фракцию, причем в навозе
их в 2.6 раза больше, чем в ТМАУ. Таким образом, вследствие
большего содержания в навозе водорастворимых веществ,
битумов, гемицеллюлоз и целлюлоз, а также меньшей степени его
гумификации можно предположить, что минерализация этого
удобрения пойдет интенсивнее, чем ТМАУ Г
Изучение динамики содержания как общего количества
органических остатков в пахотном слое, так и количества гумуса
показало, что основным звеном процесса трансформации
органических удобрений в пахотном слое дерново-подзолистых почв
является их постепенная минерализация, достаточно
интенсивная для всей массы органических остатков, хотя темпы ее очень
разные, прежде всего в зависимости от гранулометрического
состава почвы и состава удобрений (рис. 45). Суглинистый грануло-
203
V1975 XW75 Х1976 Х1977
Рис. 45. Изменение содержания органического вещества (/) и гумуса(/7)
в пахотном слое дерново-подзолистой песчаной (а) и легкосуглинистои (о)
почвы при внесении различных доз навоза и ТМАУ.
1 — контроль, 2 и 3 — навоз и ТМАУ по 30 т/га, 4 и о — навоз и ТМАУ по 90 т/га.
метрический состав резко тормозит скорость минерализации
органических удобрений, причем, чем выше степень гумификации
самих удобрений, тем ниже темпы их минерализации.
Содержание гумуса, как и следовало ожидать, оставалось во все годы
наблюдений более стабильным и по высоким дозам удобрений
возросло на 20—25%, или (в абсолютных цифрах) на 0.20—0.26 /о,
что примерно соответствует количеству гумифицированныхвеществ,
содержащихся во вносимых удобрениях.
Органические удобрения, вносимые в пахотный слой дерново-
подзолистой почвы, изменяют групповой состав гумуса, причем
204
V1975 Х1975 Х1976
Рис. 45 (продолжение).
Х1977
степень его изменения обусловлена количеством удобрений,
поступивших в почву, и содержанием в них гумусовых, и прежде
всего гуминовых, кислот или их солей. Этот вывод
подтверждается и другими исследованиями [48, 187]. При длительном
применении средних доз органических удобрений достигается тот же
эффект, что и при однократно используемых высоких дозах.
Действительно, состав гумуса в пахотном слое
дерново-подзолистых почв различного гранулометрического состава изменяется
при длительном применении средних доз навоза, но все же
остается в большинстве случаев фульватным или гуматно-фуль-
ватным с резким доминированием в составе гумуса фульвокислот
(табл. 68).
205
Таблица 68;
Состав гумуса в пахотном слое дерново-подзолистой почвы
при длительном применении удобрений (по: [187])
Вариант опыта
С гумусовых веществ,
°/0 к валовому С
Сгк
СФК
СГК
СФК
Легко су г л инистая почва
(Тимирязевская сельскохозяйственная академия, 60 лет)
Рожь бессменная
NPK
навоз
Севооборот, 132-е поле
NPK
HaB03-[-NPK
17
20
16
16
27
23
30
28
Среднесуглинистая почва (Щапово, 10 лет)
Тяжел о с углиниста я почва
(Долгопрудная агрохимическая станция)
0.6
0.9
0.5
0.6
Пшеница озимая бессменная
NPK
навоз
навоз-fNPK
15
17
16
31
24
24
0.5
0.7
0.7
Севооборот
NPKCa
навоз
V2 NPK Ca-f-Va навоза
12
17
15
33
29
31
0.4
0.6
0.5
Иную картину можно наблюдать при единовременном
внесении в почву высокой дозы органических удобрений. Групповой
состав гумуса изменяется непосредственно после внесения
органических удобрений: в нем возрастает абсолютное содержание
гуминовых кислот, причем оно четко коррелирует с тем
содержанием гуминовых кислот, которое вносится с удобрением.
Результаты наших исследований подтверждают этот вывод (табл. 69).
При внесении высоких доз хорошо гумифицированных
удобрений гуматно-фульватный тип гумусовых веществ превращается
в фульватно-гуматный. Еще более разительные примеры резкого
изменения группового состава гумуса в пахотном слое дерново-
подзолистых почв приводит М. Н. Владимирова [68]; по ее данным,
на участке суглинистой дерново-подзолистой почвы на фоне
глубокой вспашки и многократно повторяющихся высоких доз
торфонавозных компостов количество гумуса возросло до
небывало высоких размеров, а его состав стал гуматным с
отношением Сгк : СФК 1.7—2.3 (табл. 70),
206
Таблица 69
Групповой состав гумуса в пахотном слое
дерново-подзолистых почв, % к валовому содержанию С
Вариант
опыта
Доля
удобрений,
т/га
1975 г.
С, %
к
сухой
почве
Сгк
СфК
Сгк
СфК
С негид-
ролиэуе-
мого
остатка
1977 г.
С, %
к
сухой
почве
сГк
СФК
Сгк
сФК
С негид-
ролизуе-
мого
остатка
Песчаная почва
Контроль
Навоз
ТМАУ
Навоз
ТМАУ
—
30
30
90
90
0.72
0.75
0.95
1.40
1.45
21.8
24.5
23.2
26.8
26.5
27.9
28.7
27.3
29.8
27.6
0.78
0.84
0.85
0.90
0.96
50.3
47.2
49.5
43.4
45.9
0.64
0.67
0.76
0.95
0.96
20.7
21.6
22.1
26.4
26.2
27.6
28.4
28.8
31.4
29.9
0.75 1
0.76
0.77?
0.84
0.88 1
51.7
50.0
49.1
42.2
43.9
Легкосуглинистая почва
Контроль
Навоз
ТМАУ
Навоз
ТМАУ
—
30
30
90
90
1.24
1.41
1.52
1.80
2.02
23.0
23.9
25.4
28.8
27.1
28.8
29.1
29.7
31.0
28.3
0.80
0.82
0.86
0.93
0.96
48.2
47.0
44.9
40.2
44.6
1.24
1.36
1.60
1.64
2.00
23.3
25.8
25.3
28.4
27.9
27.7
29.0
28.1
29.3
26.8
0.84
0.89
0.90
0.97
1.041
49.0
45.2
46.6
42.3
45.3
Примечание. В 1976 г. на легкосуглинистой почве были посеяны клевер и
тимофеевка.
Таблица 70
Содержание и состав гумуса в суглинистой
дерново-подзолистой почве (по: [68])
Глубина, см
Валовой С, %
С гумусовых веществ,
% к валовому С
сГк
СфК
сгк
СфК
0-10
10-22
22—32
32—39
Сл а б о ок ультуренная почва
2.6
2.4
0.7
0.4
30
30
27
17
53
56
37
0.6
0.6
0.5
0.5
0-10
10—20
20-30
30—37
Сильно о к ультуренна я почва
5.8
5.9
6.4
4.6
44
51
51
46
34
30
21
25
1.3
1.7
2.3
1.8
Примечание. В слабоокультуренную почву органические удобрения не вносились,
в сильноокультуренную 7 лет ежегодно вносилось по 100—150 т на 1 га торфона-
воаного компоста.
207
Столь быстрое изменение состава гумуса под влиянием
органических удобрений при применении высоких доз их обусловлено
в основном не интенсификацией процессов гумификации, а
спецификой химического состава последних: наличием
значительного количества гуминовых веществ. Если пожнивные и корневые
остатки являются только гумусообразователями и содержат лишь
постепенно гумифицирующиеся компоненты (белки, углеводы,
лигнин), то в составе органических удобрений, как показано
в гл. I, значительна доля гумифицированных продуктов типа
гуминовых кислот и фульвокислот. Естественно, что, внося эти
удобрения в пахотный слой, мы немедленно увеличиваем в нем
количество гумусовых веществ. Чем выше единовременно
вносимая доза навоза или различных видов торфяных удобрений, тем
больше гумусовых веществ вносится в них. Полугумифицирован-
ные компоненты органических удобрений, главным образом
лигнин, увеличивают количество негидролизуемого остатка, но
рассматривать его как гумусовые вещества, прочно связанные с
минеральной частью почвы и именуемые обычно термином «гумин»,
нельзя.
Дальнейшая трансформация группового состава гумуса в
пахотном слое дерново-подзолистых почв после внесения
органических удобрений идет в основном в сторону постепенного
возрастания относительной роли фульвокислот, и только в случае
использования навоза на соломистой подстилке, а также, по-
видимому, компостов на слабогумифицированном торфе можно
наблюдать некоторое абсолютное возрастание количества
гуминовых кислот. Несколько возрастает их содержание при посеве
и последующей распашке пласта многолетних трав, и в этом
незаменимая роль многолетних трав, содержащих бобовые
компоненты, в интенсификации процессов гумификации в пахотных
дерново-подзолистых почвах. Органические удобрения лишь в
небольшой степени усиливают процессы гумификации в случае
одновременного поступления в пахотный слой легкогумифици-
рующихся материалов в виде возрастающего количества корневых
и пожнивных остатков.-Иными словами, под влиянием
органических удобрений возрастает содержание гумуса в пахотном слое
и улучшается его групповой состав, но эти изменения адекватны
количеству вносимых с удобрениями гуминовых кислот.
Степень закрепления внесенных с удобрениями гумусовых
веществ обычно невелика благодаря недостаточной суммарной сорб-
ционной поверхности твердой фазы пахотного слоя. Большая
часть их экстрагируется при непосредственной обработке 0.1 н.
раствором NaOH. He имеет места, по-видимому, и
преобразование фульвокислот в гуминовые кислоты; постоянно можно
наблюдать обратный процесс — возрастание относительного
количества фульвокислот в составе гумуса пахотного слоя почвы.
Таким образом, следует признать, что ведущим процессом
трансформации органических удобрений в пахотном слое дерново-
208
подзолистых почв является, их постепенная минерализация с
освобождением элементов питания растений. Дополнительная
гумификация их, по-видимому, крайне ограниченна вследствие малого
количества легкогумифицирующихся компонентов. Замедленная
гумификация лигнина негидролизуемого остатка удобрений,
которая должна иметь место, невелика и не может оказать
существенного влияния на возрастание доли гуминовых кислот в составе
гумуса пахотного слоя.
Минерализация органических удобрений и гумуса в пахотном
слое всегда сопровождается образованием сложной системы
водорастворимых минеральных и органических веществ, мигрирующих
в4 нижнюю часть профиля, а частично выщелачивающихся из
почвы. Исследование И. Н. Барановским [49] состава
лизиметрических вод показало, что из пахотного слоя мигрирует
значительное количество водорастворимых веществ, при этом, оно
находится в прямой корреляции с дозами и видами вносимых
удобрений (табл. 71). На фоне высоких доз из пахотного слоя
выносятся в значительном количестве органические вещества,
представленные как фульвокислотами, так и низкомолекулярными
продуктами разложения: солями органических кислот и фенолами.
Миграция аминокислот невелика: 47—190 мкг/л, тогда как на
долю фульвокислот приходится около Vg всей массы
водорастворимых веществ. Не менее заметен и вынос оснований Са, Mg и К,
вследствие чего реакция лизиметрических вод близка к
нейтральной. Нужны, конечно, специальные балансовые опыты для
решения вопроса о степени выщелачивающего действия
водорастворимых органических кислот из пахотного горизонта, но несомненно,
Таблица 71
Количество водорастворимых веществ,
мигрирующих из пахотного слоя тяжелосуглинистой
дерново-подзолистой почвы (полевой лизиметрический опыт; по: [49])
Вид
удобрений
Контроль
Навоз
ТМАУ
Доза удобрений,
т/га
30
150
30
150
Рч
и
4.6
6.8
9.4
5.3
6.8
Водорастворимые
органические
вещества, %
&
в
35
33
33
36
39
о
8
И
И
10
И
я ,
К О
?§
R О Н
о <и о
о F РЗ
14
16
16
21
27
рГ
1.4
1.9
2,7
2.2
2.9
Формы
соединений
N, %
i
5
в _
С S
О О
18
21
24
14
14
сз
О»
82
79
76
86
86
Катионы,
Г/М2
+
ев
13.3
16.9
23.9
20.1
31.8
к
2.0
1.5
4.3
2.2
3.1
рН
7.3—6.5
7.6—6.6
7.5—6.9
7.0—6.5
6.9—6.4
Примечание. «ФК» — вещества типа фульвокислот.
14 Л. Н. Александрова 209
что высокие дозы органических удобрений могут вызвать
дополнительное оподзоливание подпахотной части профиля
дерново-подзолистых почв [154, 241 L Таковы некоторые
теоретические аспекты исследования процессов трансформации
органических удобрений в пахотных дерново-подзолистых почвах, которые
следует учитывать при обсуждении вопроса оптимизации.
Каковы же оптимальное для пахотных дерново-подзолистых
почв содержание гумуса и наиболее благоприятный его состав?
Прежде всего следует отметить, что однозначный ответ на эти
вопросы неправомочен вследствие многоплановой роли гумуса
в почвообразовании.
В первом приближении для решения вопроса об оптимальных
запасах гумуса все вновь осваиваемые и пахотные
дерново-подзолистые почвы можно разделить на две категории. К первой
следует отнести пахотные легко- и среднесуглинистые достаточно
хорошо окультуренные дерново-подзолистые почвы с
удовлетворительными для культурных растений физико-химическими
и водными свойствами. В этих почвах необходимо лишь
поддерживать характерное для данного региона количество гумуса, которое,
по-видимому, колеблется от 2—3% гумуса для легкосуглинистых
до 4—5% для среднесуглинистых почв при фульватно-гуматном
типе гумусовых веществ. Это можно осуществлять при
систематическом применении средних доз органических удобрений и
обязательном включении в севообороты посева многолетних трав,
при точном выполнении всей системы агротехнических и
агромелиоративных мероприятий, предусмотренных для этих почв
того или иного региона.
Вторая категория — вновь осваиваемые и плохо
окультуренные песчаные, супесчаные и особенно тяжелосуглинистые и
глинистые почвы, характеризующиеся неудовлетворительным водно-
воздушным режимом и (как следствие) неудовлетворительным
биологическим и питательным режимом. Для этих почв
органическая часть ее пахотного слоя должна служить прежде всего
мелиоративным целям: созданию мощного хорошо гумусирован-
ного пахотного слоя с оптимальными для культурных растений
параметрами водопроницаемости, влагоемкости, количества
продуктивной влаги. Для этих почв мощность гумусового горизонта
должна быть доведена до 28—30 см, а количество гумуса в нем —
до 3—4% в песчаных и супесчаных и до 5—6 % в тяжелосуглинистых
и глинистых почвах при фульватно-гуматном типе гумусовых
веществ.
Эти задачи можно решить, вероятно, лишь при
единовременном внесении больших доз органических удобрений на торфяной
основе, заделываемых непременно на всю глубину создаваемого
пахотного слоя, и обязательном возможно широком
использовании многолетних трав в севооборотах, при строгом соблюдении
всего комплекса мелиоративных и агротехнических
мероприятий. Некоторые результаты такого коренного улучшения гли-
210
нистой дерново-подзолисто-глеевой почвы показаны в табл. 72.
Опыт заложен в 1957 г., мощность гумусового горизонта доведена
до 38 см при глубокой заделке в него 100 т на 1 га торфонавозного
компоста. Весь вновь созданный гумусовый горизонт приобрел
и в течение 20 лет сохранил хорошую комковато-зернистую
структуру» в нижней части его скважность достигла 48% (вместо 38%
на контроле), содержание гумуса сохранилось на уровне 5%
при фульватно-гуматном типе гумусовых веществ.
Таблица 72
Влияние органических удобрений и глубокой вспашки
на порозность, содержание гумуса и его состав
в глинистой дерново-подзолисто-глеевой почве
Глубина,
см
0-10
10-20
20-30
30-40
40-50
Порозность общая,
% к объему
ее
еб
5
О
X!
О
К
57
46
35
34
35
через
48
52
49
45
34
1
и
со
52
50
47
43
35
1
50
49
45
42
34
Валовое содержание
гумуса, % к сухой массе
а>
о
a
о
X
о
S3
4.6
4.2
1.8
0.5
0.4
через
5.8
5.2
4.7
2.9
0.5
«
о
и
со
5.0
5.0
4.5
2.8
0.6
1
8
5.5
5.2
4.4
4.7
1.0
сгк
СфК
о
1
и
о
Я
1.3
1.3
1.2
0.8
0.7
через
о
и
1.7
1.8
1.4
0.9
0.8
i
ео
1.8
1.8
1.8
1.3
1.1
1.9
1.9
1.8
1.2
1.0
Примечание. На протяжении последующих 20 лет в верхнюю часть пахотного
слоя внесено еще 100 т на 1 га навоза и 2 раза были многолетние травы.
Наиболее сложные, но в то же время необходимые задачи —
обогащение пахотного слоя минеральными коллоидами и
регулирование самого процесса гумификации в сторону максимального
образования гуминовых кислот, а также использование для
приготовления органических компостов различных городских
отходов, количество которых непрерывно возрастает. Решение этих
вопросов еще ждет своего исследователя.
Глава VI
МЕТОДЫ ИЗУЧЕНИЯ
ОРГАНИЧЕСКОГО ВЕЩЕСТВА ПОЧВЫ г
Сложность состава органического вещества почвы и процессов
его трансформации требует от исследователя многообразия
приемов изучения как природы этого сложного комплекса веществ, так
и характера процессов его превращений. Естественно, что в
опубликованных руководствах, посвященных описанию методов изучения
органических веществ почвы, невозможно привести все методы
исследования. Это и побудило нас изложить в плане теоретической
оценки некоторые наиболее распространенные методы анализа
состава органических веществ почвы и дать прописи для наименее
распространенных и разработанных в нашей лаборатории.
Методы определения общего содержания углерода
и состава органических веществ в минеральных почвах
Определение общего содержания углерода органических
веществ. При определении количества органических веществ в
пахотных почвах необходимо знать содержание не только гумуса,
но и всей массы органических веществ, особенно в почвах,
систематически получающих органические удобрения, ибо общая
масса органических веществ в пахотном слое часто значительно
превышает количество гумуса. Для решения этой задачи
необходимо определять как Сорг, так и Сгум. Сорг определяют в почве без
отбора корешков, пожнивных остатков и органических удобрений.
Для определения Сгум из почвы очень тщательно при помощи лупы
и стеклянной палочки, наэлектризованной кусочком шерстяной
ткани, отбирают все заметные визуально органические включения.
Чистоту отбора следует проверить через лупу.
Наиболее сложной и ответственной операцией является
определение количества повторностей, необходимых для получения
достоверных результатов. Для аналитической работы обычно
достаточно 3—4-кратной повторности при условии расхождения данных
между параллельными определениями не более 5% (относительно
Глава написана совместно с О. А. Найденовой и М. В. Новицким.
212
м 1.5 3.0 Ь.5 6.0 7.59.0 1.5 3.0 ».5 6.0 7.5 9.0 1.5 3.0 4.5 6.0759.0%
U \ i I i r r~ I i i—ПТ—i 1 1 1 НГЛ—7"i 1 1
10
20
30
40
SOW-
Рис. 46. Содержание гумуса в профиле залежной и пахотной
тяжелосуглинистой почвы при внесении 100 т на 1 га торфонавозного компоста при
глубокой его заделке.
а — залежь, б и в — вспашка на 23 и 38 см соответственно.
средней цифры). Чтобы получить достоверные данные при
определении количества углерода в почве на какой-либо заданной площади
(опытной делянке, стационарной площадке), обязательно следует
предварительно определить пестроту в содержании углерода в
отдельных горизонтах почвы, которая всегда бывает значительной.
Это положение иллюстрируется данными о содержании гумуса
(после достаточно тщательного отбора корешков и видимых
органических остатков) на площадках по 1000 м2 одного из полевых
опытов с внесением 100 т на 1 га торфонавозного компоста на фоне
глубокой заделки его через 3 года после закладки опыта (рис. 46).
Полевая повторность при отборе образцов была 18-кратной,
количество гумуса колебалось очень существенно: от 3 до 7.5%.
Только большая повторность позволила получить достоверные
данные [307]. Наши исследования свидетельствуют о том, что
при определении Сорг и Сгум в дерново-подзолистых почвах для
гумусированной части профиля необходимо не менее 12-кратной
повторности на каждые 1000 м2, причем для каждой полевой повтор-
ности рекомендуется составлять смешанный образец из 8—10
точек. Для определения степени достоверности полученных данных
следует вычислять коэффициент вариабельности (F, %) и
относительную ошибку средней (%, %) в соответствии с указаниями
Е. А. Дмитриева [92].
В пахотных дерново-подзолистых почвах V обычно достаточно
высок (более 10—15%), особенно в пахотном слое после внесения
в него больших доз органических удобрений; $з не должна
превышать 5—7%. Не менее существенно приготовление средней пробы
213
в точном соответствии с указаниями, содержащимися в
руководствах по лабораторным работам [28, 40, 124].
Сорг и Сгум в минеральных почвах обычно определяют по методу
Тюрина в любом из вариантов. В образцах, где содержание
углерода более 15%, следует пользоваться методом Анстета в
модификации Пономаревой—Николаевой [249].
Определение группового и фракционного состава гумуса.
Изучение состава органических веществ в минеральных почвах
обычно ограничивается исследованием группового и фракционного
состава гумуса по одной из общепринятых схем. Наиболее
рациональными по опыту нашей работы являются схема анализа
И. В. Тюрина [302] и схемы В. В. Пономаревой [250—252].
В любой из схем фильтрация через бумажные фильтры должна быть
заменена центрифугированием, что не только заметно ускоряет
работу, но и сводит к минимуму неизбежный щелочной гидролиз
неспецифической части органических веществ почвы.
Метод М. М. Кононовой и Н. П. Бельчиковой с использованием
щелочного раствора пирофосфата натрия [151 ] может применяться
как экспресс-метод для приблизительной характеристики
группового состава гумуса, особенно для карбонатных почв, хотя и дает
результаты, несколько отличающиеся от общепринятой схемы
анализа И. В. Тюрина [251, 329]. Следует иметь в виду, что
любой метод определения группового и фракционного состава гумуса
в почве условен и при сравнительной характеристике заданных
объектов все анализы необходимо выполнять по строго одинаковой
прописи. Для иллюстрации приводим полученные В. Фляйгом
с соавторами [399 ] расхождения в групповом составе гумуса в
зависимости от метода выделения отдельных фракций (рис. 47).
Ранее мы уже отмечали, что толкование результатов
фракционного анализа по формам связи гумусовых веществ с компонентами
минеральной части почвы неправомочно, непосредственная
экстракция гумусовых веществ 0.1 н. раствором NaOH снимает
фракции, наименее прочно связанные с поверхностью
кристаллической решетки минералов, тогда как предварительное «де-
кальцирование» почвы 0.1 н. раствором H2S04 с последующей
щелочной экстракцией извлекает из почвы более прочно связанные
фракции [19].
Кратко опишем наиболее распространенный вариант метода
И. В. Тюрина в модификации кафедры почвоведения
Ленинградского сельскохозяйственного института [28]. В этой модификации
выделяются три фракции фульвокислот и две фракции гуминовых
кислот, а также количество гумуса, остающееся в виде
нерастворимого остатка. Фракция 1а — свободные или связанные
с подвижными несиликатными полутораокисями фульвокислоты,
экстрагируемые 0.5 н. раствором H2S04, а также вещества
неспецифической природы, выделяемые вместе с фульвокислотами.
Фракция I — гуминовые кислоты, фульвокислоты и их соли, свободные
или непрочно связанные с минеральной частью почвы, а потому
214
Ща Ш5 Ш6 Шг Шд
Рис. 47. Сравнительная оценка методов экстракции гумусовых веществ из
чернозема (по: [399]).
а — фульвокислоты, б — гиматомелановые кислоты, виг — гуминовые кислоты (в —
«коричневые», г — «серые»), д — неэкстрагируемое вещество I—VII — условия
экстракции. I — 2%-ный раствор HG1, 1%-ный раствор NaOH, II — 5%-ный раствор НС1 при
70°, 0 5%-ный раствор NaOH при кипячении; III — 5%-ный раствор HG1 при 70°,
1%-ный раствор NaOH; IV — 0.1 М Na4P207f 0.1 н NaOH, V — 0.1 н. NaJP,07, 0 1 н.
NaOH; VI — 0.1 н Na4P207 при кипячении; VII — 0 1 н Na4P207, 5%-ный раствор
гидразингидрата.
легко экстрагирующиеся в виде гуматов и фульватов натрия при
обработке почвы 0.1 н. раствором NaOH. Фракция II — гуминовые
кислоты, фульвокислоты и их соли, прочно связанные с
.минеральной частью почвы, вследствие чего необходима предварительная
обработка почвы минеральной кислотой для расшатывания этой
связи.
Для определения группового и фракционного состава гумуса
по этому варианту метода берут две параллельные навески почвы
от 2 до 20 г в зависимости от валового ^содержания гумуса.
Одновременно определяют количество гигроскопической воды и
валовое содержание гумуса. Первую навеску используют для выделения
фракций 1а и II гумусовых веществ, для чего ее помещают в стакан
емкостью 100—500 мл, заливают 20—200 мл 0.5 н. раствора H2S04
и оставляют на ночь. На следующий день почву декантацией
фильтруют через воронку с плотным фильтром и вновь заливают
небольшим объемом 0.5 н. раствора H2S04, перемешивают и
декантируют. Обработку кислотой повторяют до отрицательной реакции
на кальций (по хромогену черному). Все порции фильтрата соби-
215
рают в мерную колбу, почву промывают 3—4 раза
дистиллированной водой, чтобы удалить избыток серной кислоты. Во избежание
пептизации почву с первой порцией дистиллированной воды
переносят в центрифужный стакан и все дальнейшие операции
проводят в нем. Центрифугат переносят также в мерную колбу, раствор
доводят до метки и после тщательного перемешивания берут три
параллельные пробы по 50 мл для определения углерода
фракции 1а фульвокислот. Взятый для анализа объем сернокислой
вытяжки нейтрализуют 10%-ным раствором Na2C03 до появления
буроватого осадка полутораокисей и осторожно упаривают на
песочной бане до небольшого объема, после чего выпаривают досуха
на водяной бане. В сухом остатке определяют углерод по методу
Тюрина. При кипячении с двухромовокйслым калием необходимо
добавить прокаленный песок или суглинок для равномерного
кипения содержимого колбы.
Для выделения фракции II гуминовых кислот и фульвокислот
почву в центрифужном стакане обрабатывают 20—200 мл 0.1 н.
раствора NaOH, тщательно перемешивают и оставляют на ночь.
На следующий день раствор центрифугируют, центрифугат
собирают в мерную колбу, а оставшуюся почву повторно обрабатывают
0.02 н. раствором NaOH, настаивают в течение 1 ч и вновь
центрифугируют, повторяя эту операцию в зависимости от содержания
гуминовых кислот 2—3 раза. После последнего центрифугирования
центрифугат должен оставаться соломенно-желтым. Добиваться
полного осветления не следует, так как при длительной обработке
почвы раствором щелочи частичному гидролизу подвергаются
многие неспецифические органические соединения гумуса, и прежде
всего углеводы и лигнин. Весь центрифугат должен быть
прозрачным, в противном случае его необходимо дополнительно очистить
фильтрацией через пористые фарфоровые или глиняные фильтры
или повторным центрифугированием после высаливания пеп-
тизированных органо-минеральных коллоидов сухой солью
Na2S04.
Прозрачные центрифугаты доводят до определенного объема
водой и после тщательного перемешивания берут для определения
общего углерода, извлекаемого из почвы 0.1 н. раствором NaOH,
три параллельные пробы по 5—10—20 мл (в зависимости от
интенсивности окраски) в колбочки на 100 мл. К щелочному раствору
добавляют по каплям 1 н. раствор H2S04 до появления бурого осадка,
избыток кислоты нейтрализуют несколькими каплями 10%-ного
раствора Na2C03 до растворения осадка. Раствор упаривают
досуха на водяной бане, после чего в сухом остатке определяют
углерод методом Тюрина, что дает общий углерод гуминовых
кислот и фульвокислот фракции П.
Для определения углерода гуминовых кислот последние
осаждают серной кислотой, для чего к 25—50—100 мл щелочного центри-
фугата (в зависимости от интенсивности окраски) в стакан
прибавляют 1 н. раствор серной кислоты до рН 1 (проба с индикаторной
216
бумагой), нагревают на водяной бане приблизительно до 60—70° С
в течение 10—15 мин и для укрупнения выпавшего осадка гуми-
новых кислот оставляют на ночь. Осадок декантируют, переносят
0.02 н. раствором H2S04 на воронку с фильтром и на фильтре
несколько раз промывают тем же раствором серной кислоты для
удаления механической примеси фульвокислот. Затем осадок
растворяют горячим 0.02 н. раствором NaOH, собирая гумат натрия
в мерную колбу, раствор доводят до метки, тщательно
перемешивают и в 5—10—20 мл определяют углерод гуминовых кислот.
По разности между общим содержанием углерода в щелочном цен-
трифугате и содержанием его в растворе гумата натрия определяют
содержание углерода фульвокислот. Следует иметь в виду, что
при экстракции гумусовых веществ из почвы после ее обработки
0.5 н. H2S04 в раствор переходят гумусовые вещества фракции I,
что нужно учитывать при вычислении общих результатов анализа.
Остаток почвы после ее обработки 0.5 н. раствором H2S04 и
0.1 н. раствором NaOH 2—3 раза промывают водой (с
использованием центрифуги), переносят в заранее высушенную и
взвешенную фарфоровую чашку, высушивают до воздушно-сухого
состояния и взвешивают. Из воздушно-сухой почвы остатка
определяют углерод остатка по Тюрину (с последующим пересчетом на
исходную навеску).
Фракцию I гумусовых веществ выделяют при непосредственной
обработке почвы (второй навески) 0.1 н. раствором NaOH методом
центрифугирования с осаждением гуминовых кислот и определением
углерода в них аналогично вышеописанному. При этой обработке
в раствор переходят также фульвокислоты фракции 1а, что
необходимо учитывать при вычислении результатов анализа. Таким
образом, этим методом получают фракцию 1а фульвокислот,
извлекаемую при обработке почвы 0.5 н. раствором H2S04, фракцию
I гуминовых кислот по непосредственному определению С
гуминовых кислот в этой фракции, фракцию I фульвокислот по разности
между С фульвокислот фракции I и С фульвокислот фракции 1а,
фракцию II гуминовых кислот по разности между С гуминовых
кислот фракции II и С гуминовых кислот фракции I, фракцию II
фульвокислот также по разности между С фульвокислот фракции II и
С фульвокислот фракции I. Все результаты вычисляют в процентах
к сухой почве и к валовому содержанию углерода. В первом случае
целесообразно вычислять количество гуминовых кислот и
фульвокислот (умножая содержание С гуминовых кислот на 1.8, а С
фульвокислот — на 2.1), что позволит определить долю этих гумусовых
кислот в гумусе; второй пересчет дает все величины в углероде.
Определение С нерастворимого остатка необходимо для
контроля за точностью анализа. Сумма С всех фракций и нерастворимо-
мого остатка, вычисленная в процентах к валовому содержанию
углерода в почве, должна теоретически составлять 100. Вследствие
неизбежных погрешностей анализа она может быть меньше или
больше 100 на +3%. По общей сумме экстрагируемых гуминовых
217
кислот (фракция I+II) и сумме фульвокислот (фракции Ia+I-f-П)
вычисляют отношение СГк : Сфк, которое является важным
диагностическим признаком при оценке гумусового режима.
Следует иметь в виду, что описанный выше метод группового
и фракционного анализа органического вещества дает лишь
относительно верное представление о количестве гуминовых кислот
и фульвокислот в почве, так как, во-первых, значительная
часть гумусовых веществ, наиболее прочно связанная с
минеральной частью почвы, остается в составе нерастворимого остатка и,
во-вторых, вместе с гумусовыми кислотами или их солями серная
кислота л щелочь извлекают некоторое количество
неспецифических (негумусовых) веществ: белков, углеводов и других
соединений.
Методы определения состава негумусовых веществ в
минеральных почвах не разработаны. Применение методов кислотного
гидролиза почвы, широко практикуемых в последнее время для
определения количества углеводов [239, 414], не может считаться
приемлемым для минеральных почв, так как в гидролизат
одновременно переходят и фульвокислоты, количество которых необходимо
учитывать. По-видимому, может быть использован метод Форсита,
позволяющий выделять фульвокислоты из кислотного гидролизата
на активированном угле, но для массовых анализов этот метод не
опробован. Отсутствуют также методы определения количества
негумифицированных белков и лигнина. Любые варианты
кислотного или щелочного гидролиза переводят в раствор как азот
гумусовых кислот, так и азот находящихся в почве белков. В
нерастворимом остатке почвы после выделения всех фракций гумусовых
веществ всегда остается какая-то сложная смесь гумусовых кислот и
их солей, очень прочно связанных с минеральной частью почвы,
лигнина и азотсодержащих соединений, не подвергшихся
гумификации. Поэтому-то недопустимо определение каких-либо констант
(оптической плотности, элементного состава и т. д.) из кислых
гидролизатов, если принимать их за фракцию фульвокислот:
чистые препараты необходимо получать заранее.
Из состава неспецифических органических соединений в
минеральных почвах можно определить количество целлюлозы при
использовании реактива Швейцера [25] и количество воскосмол
экстракцией навески почвы в аппарате Сокслета смесью спиртобен-
зола (1 : 1).
Методы определения общего содержания углерода, азота
и состава органических веществ в органогенных материалах
(торфяных почвах, органических удобрениях,
гумифицирующейся растительной массе)
Для определения всех компонентов органической части в
торфяных почвах, органических удобрениях и других органогенных
материалах, состоящих в значительной части своей массы из орга-
218
нических веществ, методы анализа, разработанные для
минеральных почв, непригодны. Для определения общего содержания
углерода метод Тюрина не годится из-за трудности взятия очень
маленькой навески (0.01—0.02 г) для анализа, поэтому следует
использовать метод Кн,оппа, или сухого сжигания, с последующим
определением количества образовавшейся С02 или метод Анстета
[345] в модификации Пономаревой—Николаевой [249]. Последний
позволяет быстро и с достаточной точностью определить
содержание углерода в любом органогенном материале. Для
исследования состава органической части в этих материалах необходима
специальная схема анализа, так как в торфах, органических
удобрениях и гумифицирующихся растительных остатках имеются
гумусовые вещества и значительное количество исходных
компонентов: углеводов, белков, лигнина. На кафедре почвоведения
Ленинградского сельскохозяйственного института разработана
и многократно апробирована специальная схема анализа состава
органических веществ в этих органогенных материалах,
предусматривающая последовательный ход анализа одной навески
с экстракцией гумусовых веществ 0.1 М раствором Na4P207 и
0.02 н. раствором NaOH перед выделением углеводов и белков
(см. с. 220).
Исходный образец торфа, органического удобрения или
гумифицирующихся растительных остатков без предварительного
доведения его до воздушно-сухого состояния делят на две части:
одну высушивают на воздухе для последующего определения
количества гигроскопической воды, зольности, валового
содержания углерода, азота и воскосмол; другую берут в анализ без
высушивания, так как последнее дегидратирует и переводит в
нерастворимую форму некоторое количество водорастворимых
органических соединений. Высушенную часть образца измельчают
на мельнице.
Определение валового содержания углерода по методу Анстета
в модификации Пономаревой—Николаевой. Из высушенного
образца отбирают среднюю пробу, растирают и пропускают через
сито с диаметром отверстий 0.25 мм. Навеску воздушно-сухой
массы от 0.2 до 0.5 г, взятую на аналитических весах, помещают
в коническую колбу емкостью 200—250 мл из термостойкого
стекла. Для равномерности кипения в колбу на кончике ножа
прибавляют прокаленную пемзу или лёсс. Из бюретки со
стеклянным краном медленно приливают 30 мл сернохромовой смеси
и очень осторожно 20 мл концентрированной серной кислоты.
Колбу закрывают маленькой воронкой в качестве холодильника,
осторожно перевешивают содержимое и после окончания бурного
разложения органического вещества ставят на заранее
разогретую этернитовую плитку или песчаную баню, доводят до кипения
и кипятят точно 5 мин. После охлаждения содержимое осторожно
переносят в мерную колбу на 250 мл, затем, окончательно охладив,
доводят водой до метки. Полученный объем тщательно перемеши-
219
Схема анализа -состава органических веществ
в органогенных материалах
гигроскопическая
вода
Исходный материал
зольность
Воскосмолы
Водная вытяжка
С<-
Экстракция
гумусовых веществ
0.1 М Na4P207
осаждение гуминовых
кислот
Экстракция
гумусовых веществ
0.02 н. NaOH
осаждение гуминовых
кислот
идролиз гемицеллтолоз
и белков
углеводы
С<-
1
Гидролиз целлюлозы
и белков 80%-ной
H2S04
Валовое содержание
углерода и азота
N
->N
С и N в гуминовых
кислотах
->N
С и N в гуминовых
кислотах
углеводы
t
Остаток материала
->N
вают и пипеткой (с предохранителем!) берут 2—3 пробы
по 25 мл на титрование солью Мора. Одновременно в точно
таких же условиях проводят холостой опыт для установления
соотношения между сернохромовой смесью и солью Мора. Серно-
хромовую смесь приготавливают из двух объемов 12%-ного водного
раствора Сг03 и одного объема H2S04 (плотность 1.84).
Приготовлять смесь следует очень осторожно, прибавляя серную
кислоту к раствору хромового ангидрида. ?&
Одновременно из сухого образца берут пробу для определения
гигроскопической воды, зольности и количества воскосмол. Вос-
космолы определяют экстракцией воздушно-сухой навески
образца (3—5 г) бензолом в аппарате Сокслета, а после окончания
экстракции и отгонки избытка бензола высушивают при
температуре 70—80° и взвешивают. Валовое содержание азота
определяют методом Кьельдаля.
Определение состава органической части органогенного
материала. Для анализа используют свежий (не высушенный на
воздухе) образец, из которого тщательно берут среднюю пробу.
Одновременно в образце определяют влажность. Навеску
материала из расчета 3—5 г сухой массы в сыром состоянии помещают
в колбу и заливают 300—500 мл дистиллированной воды, уровень
которой отмечают восковым карандашом, настаивают в течение
10—12 ч и фильтруют через бумажный фильтр под слабым
разряжением при помощи водоструйного насоса.2 Остаток во влажном
состоянии переносят вновь в ту же колбу и повторно заливают
водой до метки. Экстракцию водорастворимых веществ проводят
2—3 раза, после чего весь фильтрат собирают вместе. После
учета его общего количества в фильтрате определяют С по Тюрину
и N по Кьельдалю. В случае необходимости можно определить
количество минеральных и органических соединений азота,
содержание углеводов и других низкомолекулярных органических
соединений. Остаток в сыром состоянии вновь переносят в колбу
и обрабатывают 300—500 мл 0.1 М горячего раствора Na4P207
для экстракции гумусовых веществ по методу, предложенному
В. Н. Ефимовым и М. Г. Васильковой [106]. Колбу с раствором
нагревают на водяной бане при температуре 80° в течение 2 ч,
горячий раствор фильтруют, а остаток торфа смывают горячим
раствором пирофосфата натрия обратно в колбу, доливают до 300—
500 мл горячим раствором пирофосфата натрия и вновь нагревают
при 80° 2 ч, а затем раствор фильтруют. Для полного выделения
гумусовых веществ необходимо проводить три экстракции. Все
растворы гумусовых веществ сливают вместе, очищают на
центрифуге и бактериологических свечах № 5—7 и в очищенном растворе
определяют С по Тюрину, N по Кьельдалю, а также С и N гуми-
новых кислот после их осаждения H2S04 при рН 1. Остаток во
? При наличии центрифуг с большими стаканами (на 600—800 мл)
фильтрацию везде можно заменить центрифугированием.
221
влажном состоянии смывают в колбу 0.02 н. раствором NaOH,
объем раствора доводят до 300—500 мл и нагревают на водяной
бане в течение 1 ч при 80°, после чего фильтруют горячим и
повторяют обработку 0.02 н. раствором NaOH еще раз. Обработка
материала 0.02 н. раствором NaOH экстрагирует еще некоторую
часть гумусовых веществ, находящихся, по-видимому, в виде
дегидратированной пленки на поверхности полугумифицирован-
ных остатков тканей и торфа. Гумусовые вещества, выделенные
0.02 н. раствором NaOH, очищают указанным выше способом,
в очищенном растворе определяют С, N и осаждают гуминовые
кислоты, в которых также определяют количество С и N.
Остаток материала с фильтра вновь смывают 300 мл 1.0 н.
раствора H2S04 в колбу, которую помещают на кипящую баню
на 2 ч для экстракции гемицеллюлоз и кислоторастворимых
белков. Вытяжку фильтруют через бумажный фильтр или
центрифугируют, остаток промывают несколько раз холодной водой,
а в растворе определяют количество С, N и углеводов. Для
выделения целлюлозы и трудногидролизуемых белков остаток с фильтра
смывают водой в фарфоровую чашку, воду удаляют
выпариванием, доводя остаток до слегка влажного состояния. После
охлаждения остаток в чашке обрабатывают 20 мл 80%-ного раствора
H2S04, часто перемешивая стеклянной палочкой, при комнатной
температуре в течение 1—2 ч. Затем содержимое чашки смывают
из промывалки в колбу 300 мл дистиллированной воды и помещают
на кипящую водяную баню на 3 ч. При обработке материала
80%-ным раствором H2S04 начинается гидролиз целлюлозы и
трудногидролизуемых белков, а в процессе дополнительной
обработки разбавленным раствором кислоты гидролиз целлюлозы
и белков заканчивается. Содержимое фильтруют или
центрифугируют, промывают 2—3 раза водой и в растворе определяют
С, N и количество углеводов. Негидролизуемый остаток переносят
в фарфоровую чашку, рысушивают и взвешивают, после чего в нем
определяют количество С и N.
Результаты анализа можно выражать в процентном
содержании углерода отдельных групп соединений к валовому количеству
его. Более наглядным способом, хотя и в какой-то степени
условным, является вычисление процентного содержания отдельных
групп соединений к общему количеству органических веществ.
Мы используем несколько приемов пересчета: для гуминовых
кислот — коэффициент 1.8, для фульвокислот — 2.1, для
определения содержания белков — коэффициент 6.25, на который
умножаем содержание азота в каждом гидролизате; количество
углеводов определяем непосредственно или по разности между
общим содержанием С в гидролизате и его количеством,
входящим в состав белков (в среднем в белках С составляет 55%). Если
есть данные по непосредственному определению углеводов,
разность между общим количеством С в гидролизате и суммой
углерода белков и углеводов (в среднем в углеводах С равно 45%)
222
Таблица 73
Состав органического вещества целинной верховой торфяной почвы,
%'к органической массе (по: [175])
Глубина, см
0-8
8-20
30—40
Воскосмолы
2.6
2.5
3.5
Водорастворимые
азотсодержащие
вещества
0.8
0.5
0.4
углеводы
1.4
1.0
0.2
прочие
0.9
0.2
0.8
сумма
3.1
1.7
1.4
Гумусовые вещества
12.1
15.0
17.4
Гидролизуемые
1 н. H2S04
азотсодержащие
вещества
0.3
0.3
0.2
геми целлюлозы
13.3
12.3
9.7
прочие
1.5
1.4
1.9
сумма
15.1
14.0
11.8
Гидролизуемые
80%-ной H2S04
азотсодержащие
вещества
0.7
0.6
0.8
целлюлоза
28.4
29.4
27.0
прочие
5.6
6.8
6.1
сумма
34.7
36.8
33.9
-Негидро-
лизуемый
остаток
азотсодержащие
вещества
1.5
1.5
1.3
лигнин
30.9
28.5
30.9
дает фракцию прочих «неидентифицированных» веществ,
количество которых обычно невелико. Для иллюстрации приводим
результаты анализа торфа (табл. 73).
Выделение и изучение состава
органо -минеральных производных гумусовых кислот
Методы выделения из почвы органо-минеральных производных
гумусовых веществ нельзя считать разработанными, они
находятся в стадии изучения преимущественно путем моделирования.
В последнее время появились интересные работы, посвященные
выделению из почвы и исследованию илистой фракции [85],
а также описывающие условия моделирования и выделения
из почвы этой фракции [237, 287, 317].
В соответствии с классификацией органо-минеральных
производных гумусовых кислот, предложенной нами [11], необходимо
выделять и исследовать вещественный состав, структуру и
свойства трех групп этих производных: гетерополярных солей, комп-
лексно-гетерополярных солей и адсорбционных комплексов
гумусовых кислот с компонентами минеральной части почвы.
Количество гетерополярных солей, а точнее, возможную степень
насыщенности гумусовых кислот минеральных почв катионами
сильных оснований, в настоящее время можно вычислить
расчетным путем при определении емкости обменного поглощения почвы
до и после удаления гумуса, используя метод Гедройца [75] —
обработку почвы перекисью водорода. Более легкий способ —
сжигание гумуса в муфеле даже при относительно низких
температурах (400°) — неизбежно изменяет емкость обменного
поглощения минеральной части почвы.
223
Определение возможной степени насыщенности гумусовых
кислот минеральных почв катионами сильных оснований.
Навеску почвы 3—10 г (в зависимости от содержания гумуса и
гранулометрического состава почвы) помещают в стакан емкостью
200—250 мл, смачивают 10 мл воды и приливают 20 мл 30%-ного
раствора Н202. Стакан прикрывают стеклом, взбалтывают и
оставляют на 30 мин при комнатной температуре, периодически
перемешивая. Затем стакан оставляют на ночь в теплом месте (при
температуре 40—50°) или помещают на кипящую баню для
ускорения окисления. После прекращения реакции (выделения
пузырьков С02) в стакан еще раз приливают 10 мл 30%-ного раствора
Н202 и оставляют на кипящей бане. При высоком содержании
гумуса обработку почвы перекисью водорода повторяют еще 2—
3 раза. Если почва карбонатна, необходимо предварительно
удалить соляной кислотой углесоли (0.1 н. раствор), а затем
фильтрацией — и образовавшийся хлористый кальций.
Емкость обменного поглощения катионов в дальнейшем
определяют в двух навесках почвы, для чего во второй стакан
помещают такую же навеску почвы без предварительной обработки
ее перекисью водорода. В связи с тем что в обработанной Н202
почве может содержаться заметное количество водорастворимых
солей кальция и магния, образовавшихся за счет окисления
органического вещества, емкость обменного поглощения катионов
в обоих навесках следует определять путем насыщения почвы
уксуснокислым аммонием с последующим учетом количества
поглощенного аммония. Можно использовать растворы хлоридов
натрия или калия и определять количество Na или К,
сорбированное почвой, вытеснением слабым раствором НС1 с последующим
анализом их на пламенном фотометре. Разность между величиной
емкости обменного поглощения исходной почвы и после ее
обработки перекисью водорода даст возможную емкость поглощения
оснований органической частью почвы.
Определение алюмо- и железогумусовых соединений. Метод
был предложен нами [9], а в дальнейшем проверен и
модифицирован М. М. Кононовой и Н. П. Бельчиковой [55, 152]. Данный
метод следует рассматривать лишь как первую попытку
выделения из почвы этой группы органо-минеральных производных для
изучения их состава и структуры.
Техника работы в нашей прописи такова. Навеску почвы в 3—
10 г помещают в коническую колбу и обрабатывают 150—300 мл
свежеприготовленного 0.1 М раствора Na4P207 при комнатной
температуре. Чем более гумусирована почва, тем меньше навеска
и шире отношение почва : раствор. Для коагуляции органо-
минеральных коллоидов (пирофосфат натрия — сильный пепти-
затор) прибавляют несколько кристалликов K2S04. Содержимое
колбы оставляют на 6—8 ч или на ночь, после чего отстоявшийся
раствор центрифугируют и дополнительно очищают от коллоидов
фильтрацией через мембранный фильтр или бактериальные свечи
224
Рис. 48. Установка для фильтрации через бактериальные свечи.
1 — к распределителю, 2 — колба толстостенная, 3 — бактериальный фильтр, 4 —
цилиндр вместимостью 0.5 л, 5 — шланги вакуумные, 6 — каплеуловитель, 7 —
вакуумметр.
(рис. 48). Наличие в центрифугате пептизированных органо-
минеральных коллоидов резко искажает результаты анализа,
так как увеличивает содержание А1 и Fe. В отдельных порциях
центрифугата определяют содержание С и после разрушения
комплексных соединений -— количество А1 и Fe. Если
необходимо разделить фульватные и гуминовые комплексы, последние
осаждают насыщенным раствором гипса и после диализа для
удаления фульватных комплексов осадок А1- и Fe-гуматных
комплексных солей вновь переводят в раствор с последующим
определением в нем количества С, А1 и Fe. Осаждение гуминовых
соединений серной кислотой заметно искажает результаты, так как
частично разрушает комплексные А1- и Fe-гуминовые соединения.
Углерод определяют по методу Тюрина. Для разрушения
комплексных соединений мы используем гидролиз определенного
объема центрифугата с соляной кислотой в течение 3 ч. Н. П. Бель-
чикова [55] рекомендует разрушать комплексы путем сжигания
органической части смесью серной и хлорной кислот в колбах
Кьельдаля. Для разрушения комплексных соединений 50—100 мл
центрифугата помещают в коническую колбочку, прибавляют
7—12 мл концентрированной HG1 (х. ч. не содержащей следов Fe)
с таким расчетом, чтобы конечная концентрация кислоты
составляла около 5%, и кипятят с обратным холодильником 1 ч. Во
избежание перегрева в колбу добавляют несколько стеклянных
капилляров. В процессе гидролиза Fe и А1 переходят в форму
хлоридов, а пирофосфат — в ортофосфат.
Для осаждения А1- и Fe-гуминовых солей 50—100 мл
центрифугата диализуют до отрицательной реакции на Р207, осаждают
насыщенным раствором CaS04, осадок отфильтровывают,
растворяют в 0.02 н. растворе NaOH и в аликвотном объеме определяют
15 Л. Н. Александрова 225
содержание углерода. В оставшейся части раствора разрушают
комплексные соединения, как описано выше.
Содержание Fe и А1 определяют любым колориметрическим
методом. Показателем степени насыщенности гумусовых веществ
А1 и Fe является отношение С гуминовых кислот или фульвокислот
(или их суммы) к сумме R203. М. М. Кононова и Н. П. Бель-
чикова [152] предлагают использовать обратное отношение, т. е.
(Fe+Al) : С.
Определение количества органо-минеральных коллоидов и
исследование их состава. Основную массу органо-минеральных
коллоидов следует выделять по методу Гедройца насыщением
почвы катионом натрия с последующей пептизацией коллоидов
водой. Некоторая часть коллоидов при этом остается на
поверхности различных гранулометрических фракций вследствие
прочного закрепления на поверхности более грубых частиц. Поэтому,
как показали исследования М. Надя [201 ] и А. К. Суворова [2881,
для полного выделения коллоидов необходимо обязательное
насыщение почвы обменным катионом натрия для образования на
поверхности первичных частиц подвижного ионогенного слоя с
последующим механическим разминанием почвы и диссолюционной
пептизацией. В любой почве следует различать три фракции
коллоидов: 1-я пептизируется по Гедройцу и наименее прочно
адгезионно склеена с поверхностью минеральных частиц; 2-я
снимается при дополнительном разминании остатка, т. е. путем
расклинивающего действия воды, и более прочно связана с
поверхностью минеральных частиц; 3-я может быть выделена
только методом диссолюционной пептизации при дополнительной
обработке остатка почвы 0.02 н. раствором NaOH. В зависимости
от задач исследования мы используем два основных приема для
выделения коллоидов из почвы. Если необходимо суммарно
выделить максимально возможное количество коллоидов, образец
почвы насыщают натрием из раствора NaCl или Na2S04 и длительно
разминают в тестообразном состоянии, после чего методом отмучи-
вания выделяют фракцию меньше 1 мкм, из которой
центрифугируют фракции коллоидов. Используя центрифугу С-100, можно
выделить последовательно предколлоидную фракцию (1—0.2 мкм),
грубые (0.2—0.1 мкм) и тонкие (.< 0.1 мкм) коллоиды. Остаток
почвы больше 10 мкм можно расчленить методом отмучивания
на ряд крупных фракций: 1—0.25, 0.25—0.05 и 0.05—0.01 мм.
Все полученные суспензии выпаривают на водяных банях и диали-
зируют до отрицательной реакции на С1~ или SO|~. Коагуляцию
суспензии НС1 с последующим сифонированием прозрачной
жидкости можно применять лишь в тех случаях, когда не
планируется детальное исследование органической части коллоидов,
так как НС1 вызывает заметное гидролитическое действие.
В полученных фракциях определяют в зависимости от
программы исследований количество углерода, состав гумусовых
веществ, валовой химический и минералогический состав колло-
226
идов, емкость обменного поглощения катионов и т. д.
Значительный интерес представляют определение суммарной поверхности
каждой фракции и вычисление количества гумуса не только
в процентах к массе фракции, но и на единицу поверхности. Все
определения проводят общепринятыми методами. Суммарную
поверхность можно вычислить по способу, предложенному
А. А. Роде [262], или путем непосредственного анализа:
определения величины сорбции паров воды или азота — БЭТ метод [63].
Количество сорбированной воды удобно определять также методом
Кутилика, сущность которого заключается в следующем: 1—5 г
фракции в стеклянном бюксе помещают в эксикатор над 58%-ным
раствором H2S04 или над насыщенным раствором ацетата калия
в темное место. Через 2—3 сут бюкс взвешивают, раствор в
эксикаторе меняют и вновь насыщают навеску парами воды. После
достижения постоянной массы количество сорбированной воды
определяют высушиванием. Общую удельную поверхность S
(м2/г) рассчитывают по формуле
5 = 3614 l2~~ll ,
ч у о
где V2 — масса почвы с бюксом после насыщения парами воды,
V1 — то же после сушки при 105°, V0 — масса бюкса; (V2—V-^j
(V1—V0) — количество воды (г), сорбированное 1 г почвы.
Коэффициент 3614 получают по формуле
NW0 1023 • 10 . 8 к2 • 10-2о
М — 18
где N — число Авогадро; W0 — площадь (м2=1(Г20), занимаемая
одной молекулой воды; М — молекулярная масса воды.
Если необходимо исследовать прочность закрепления
коллоидов на поверхности всех гранулометрических фракций, метод
выделения коллоидов усложняется, так как нужно снять коллоиды
с каждой гранулометрической фракции. При решении этой
задачи ход работы таков. Образцы почвы насыщают натрием и отму-
чиванием выделяют фракцию больше 10 мкм (группа I по Тю-
лину). Остаток почвы многократно разминают в тестообразном
состоянии и дополнительно отмучивают для выделения
микроагрегатов группы II (частиц больше 10 мкм). Из полученных
суспензий групп I и II микроагрегатов отмучиванием выделяют
фракцию меньше 1 мкм, в остатке — фракцию 10—1 мкм.
Суспензию частиц меньше 1 мкм на центрифуге С-100 расчленяют на
фракции 1—0.2, 0.2—0.1 и меньше 0.1 мкм. Кроме того, учитывая
возможность неполной пептизации коллоидов группы I
микроагрегатов во фракции 10—1 мкм, последнюю подвергают
многократному дополнительному разминанию, отмучиванию до полного
выделения фракции меньше 1 мкм и центрифугированию
последней на коллоидные фракции 0.2—0.1 и меньше 0.1 мкм. Для
максимально полного выделения коллоидов с поверхности круп-
227
15*
ных гранулометрических фракций последние следует
дополнительно обрабатывать 0.02 н. раствором NaOH, хотя этот прием
дает обычно уже незначительный выход коллоидов (пример
полного выделения коллоидов см. в табл. 60).
Наряду с методами выделения из почвы алюмо- и железогуму-
совых солей и органо-минеральных коллоидов и последующего
исследования их состава в настоящее время широко используют
различные приемы моделирования, методика которых очень
разнообразна в зависимости от целей исследования. Как справедливо
отмечает А. А. Роде [264], при моделировании любой процесс
изолируется жесткими условиями эксперимента, что позволяет
исследователю изучить данное явление в «чистом» виде, но в то же
время искажает его природное развитие, совершающееся на фоне
множества других элементарных процессов и реакций. Поэтому
методы моделирования в конечном итоге следует проверять
приемами выделения и аналитического исследования природного
аналога модели. Кроме того, необходимо очень четко вычленять
исследуемый процесс при постановке модельного опыта.
Так, например, при моделировании процессов формирования
железо- и алюмогумусовых солей и адсорбционных комплексов
с полутораокисями следует помнить о различиях в состоянии
(твердая фаза, раствор) компонентов, участвующих в модельной
реакции, степени их гидратации, реакции среды, влиянии
обменных катионов сильных оснований и т. д. Особенно четко нужно
ставить модельные опыты при исследовании характера
взаимодействия гумусовых кислот или их солей с кристаллической
решеткой минералов. Наличие на поверхности последней обменных
катионов кальция может резко исказить выводы, которые
надлежит сделать из модельного опыта. Для ликвидации побочного
влияния обменных катионов необходимо ставить модельный опыт
так, чтобы все компоненты (минерал, гумусовые кислоты) были
насыщены одним и тем же катионом (лучше всего натрием). Только
в этом случае можно избежать обменных реакций между
обменными катионами компонентов и установить характер
взаимодействия гумусовых веществ с кристаллической решеткой минерала.
Выделение чистых препаратов гуминовых кислот
и фульвокислот и определение некоторых основных констант
Для изучения природы гумусовых кислот различных почв
и других материалов, их содержащих, а также для исследования
изменений, происходящих в самих гуминовых кислотах в
процессе их дальнейшей трансформации, необходимо иметь чистые
препараты гуминовых и фульвокислот. Очень сложно выбрать
экстрагент, который позволил бы извлекать гумусовые кислоты
в относительно неизмененном виде и достаточно чистыми от
посторонних примесей. Чаще всего применяют три категории экстра-
228
гентов. Первый, самый мягкий экстрагент — дистиллированная
вода, которая используется для выделения водорастворимой
части новообразованных гуминовых кислот и фульвокислот.
Этот растворитель, однако, можно использовать крайне
ограниченно — только в модельных опытах при исследовании первых
этапов процесса гумификации растительных остатков, если опыты
проводить в условиях инертных наполнителей (чистый кварцевый
песок). Но даже в этих условиях вода извлекает лишь некоторую
часть образующихся гумусовых кислот, так как последние очень
быстро переходят в нерастворимое в воде состояние и не
экстрагируются в этих условиях.
Вторым, относительно мягким экстрагентом является 0.1 М
раствор Na4P207 с рН около 10. Этот экстрагент извлекает обычно
достаточно полно гумусовые кислоты и их соли из почв и из
различных гумифицированных органогенных материалов, но
основной недостаток его — трудность освобождения выделяемых
гуминовых и особенно фульвокислот от избытка пирофосфата
натрия.
Наиболее распространенным экстрагентом является третий —
0.1 н. раствор NaOH, также извлекающий основную массу
гуминовых веществ из любых материалов и почв. При использовании
его в качестве экстрагента гумусовых веществ из минеральных
почв в большинстве случаев необходима предварительная
обработка почвы 0.1 н. раствором H2S04. Основной недостаток этого
метода — возможное некоторое изменение в составе и структуре
молекулы гумусовых кислот и искусственная гумификация полу-
гумифицированных продуктов, содержащихся в анализируемых
образцах. Часть фульвокислот из почв можно экстрагировать
при помощи 0.1—0.5 н. раствора H2S04.
В последние годы применяют различные органические
растворители: диметилформамид, диметилсульфоксид [139, 192], —
но количество непосредственно экстрагируемых этими
растворителями гумусовых веществ значительно ниже, чем при
экстракции щелочными растворами, что не позволяет в настоящее время
заменить ими пирофосфат или гидроокись натрия.
Не менее сложным вопросом является способ получения
малозольных препаратов, так как при содержании золы более 5%
исследование природы и свойств гумусовых кислот невозможно.
Прием многократного растворения и переосаждения гуминовых
кислот для снижения зольности, очень распространенный в
практике работы, не дает обычно желаемых результатов, так как при
этом гуминовые кислоты не освобождаются от наиболее
высокодисперсной части органо-минеральных коллоидов; последние,
так же как и гуминовые кислоты, пептизируются под влиянием
щелочных растворов и вновь осаждаются при подкислении.
Наиболее достоверными приемами получения малозольных
препаратов с сохранением относительно неизменной природы
гуминовых кислот являются механическая очистка последних от высоко-
229
дисперсных органо-минеральных и минеральных частиц при
помощи центрифуг с достаточно большой центробежной силой
(примерно 20—30 тыс. об/мин) и фильтрации растворов через
тонкопористые фильтры (бактериальные свечи № 7) с диаметром
пор 0.9—1.2 мкм. В качестве вспомогательного приема очистки
можно рекомендовать высаливание части органо-минеральных
коллоидов сернокислым натрием. Осадки гуминовых кислот
следует переосаждать 1—2 раза для освобождения от фульво-
кислот, механически задерживающихся в составе геля гуминовых
кислот. Многократно повторяющееся растворение и
переосаждение гуминовых кислот может привести к частичному
гидролитическому расщеплению молекулы. Завершают этап очистки диализ
и электродиализ препаратов.
Многолетний опыт нашей работы свидетельствует о том, что
наиболее удобный технически экстрагент гумусовых кислот —
0.1 н. раствор NaOH, которым мы обычно пользуемся для
выделения гумусовых кислот и получения их препаратов из
минеральных и торфяных почв, а также органических удобрений и
достаточно хорошо гумифицированных растительных остатков. Лишь
в некоторых специальных модельных опытах по исследованию
процессов гумификации органических материалов в качестве
экстрагента используют воду и пирофосфат натрия с рН около 10.
При работе с минеральными почвами рекомендуем следующую
методику получения препаратов гуминовых и фульвокислот.
Навеску почвы от 300 до 1000 г обрабатывают многократно 0.1 н.
раствором H2S04 декантацией в большом сосуде (бутылки,
батарейные стаканы) до отрицательной реакции на Са, после чего 1 раз
остатки серной кислоты декантируют дистиллированной водой.
Гумусовые кислоты в форме гуматов и фульватов натрия
экстрагируют 3—6 л 0.1 н. раствора NaOH. Экстракцию не следует
проводить многократно, если не ставить перед собой задачу
исчерпывающе выделить всю массу гумусовых кислот, ибо
многократная обработка почвы щелочью пептизирует большое количество
коллоидов, которые загрязняют в дальнейшем препараты.
Целесообразно экстракцию проводить 2—3 раза (в зависимости от
содержания гумусовых кислот). Щелочные растворы собирают
вместе и очищают вначале центрифугированием, а затем
фильтрацией через бактериальные свечи. Гуминовые кислоты осаждают
1 н. раствором H2S04 при рН 1 и после отстаивания в течение 1 —
2 сут сливают сифоном сернокислый раствор, содержащий фульво-
кислоты. Осадок гуминовых кислот отделяют фильтрацией через
фильтр на бюхнеровской воронке или центрифугированием,
растворяют в 0.02 н. растворе NaOH и вновь осаждают H2S04
при рН 1—2 для освобождения от примеси фульвокислот. Осадок
в состоянии свежего геля переносят в среднюю камеру
электродиализатора и подвергают электродиализу до отрицательной
реакции на SOf~. Очищенный гель из средней камеры переносят
в большие чашки Петри и высушивают на воздухе под феном,
230
а затем в вакуумном сушильном шкафу при 25—30°. Зольность
гуминовых кислот не должна превышать 5%.
При использовании раствора пирофосфата натрия все
операции аналогичны вышеописанным, но повторно осадок гуминовых
кислот лучше растворять 0.1 н. раствором NaOH, а электродиализ
вести до отрицательной реакции на Р04 (пробу из анолита кипятят
несколько минут с небольшим объемом НС1 для перевода аниона
пирофосфата в ортофосфат).
При использовании в качестве экстрагента дистиллированной
воды отношение органогенного материала к воде должно быть
примерно 1 : 100, а экстракцию следует повторить 2—3 раза и
тщательно очистить центрифугированием и * фильтрацией через
бактериальные свечи полученную водную вытяжку. Гуминовые
кислоты осаждают также 1 н. раствором H2S04 при рН 1—2.
Для препаративного получения фульвокислот используют
принцип метода Форсита. Все фульвокислоты, оставшиеся в
фильтрате после осаждения гуминовых кислот или выделяемые при
непосредственной обработке почвы серной кислотой, прежде
всего очищают центрифугированием и фильтрованием через
бактериальные свечи. После этого растворы сгущают до
небольших объемов (500—800 мл) в кристаллизаторах под феном. При
выпаривании на водяной бане нельзя допускать выпадения
осадка. Полученные таким путем растворы фильтруют через
специально подготовленный активированный древесный уголь
марки БАУ-2 (на. этой марке угля наблюдается максимальная
десорбция фульвокислот). Перед работой уголь помолом 1—2 мм
настаивают в течение суток с 5%-ным раствором НС1, а затем
тщательно промывают тем же раствором до отрицательной
реакции на Са2+, SOf~, Fe3+ и дистиллированной водой при нагревании
до отрицательной реакции на С1". Уголь следует также обработать
0.1 н. раствором NaOH для растворения возможных примесей
органических веществ и вновь отмыть при кипячении
дистиллированной водой. Подготовленный таким образом уголь сушат
на фильтровальной бумаге под феном и хранят в банках с
притертыми пробками. Для работы уголь загружают в делительные
воронки и насыщают его раствором фульвокислот, настаивая
в течение суток. После настаивания элюат сливают и, если он
окрашен, повторно фильтруют через колонку с углем. Если элюат
не светлеет, в воронку прибавляют небольшую порцию угля.
По окончании сорбции уголь несколько раз промывают 0.1 н.
раствором НС1 для удаления простых органических соединений,
а затем дистиллированной водой — до отрицательной реакции
на С1~ и SOf~. Фульвокислоты с угля десорбируют путем
промывания 0.1 н. раствором NaOH до полного обесцвечивания
промывных вод. Д. С. Орлов с сотрудниками [235] рекомендуют
смачивать уголь ацетоном перед обработкой щелочью, что
обеспечивает более полную десорбцию фульвокислот. Полученные
растворы фульватов натрия нейтрализуют соляной кислотой до
231
рН 5.5—6.0, сгущают в вакууме до небольшого объема и
очищают диализом или фильтрацией через катионит, после чего
разливают тонким слоем в большие чашки Петри и высушивают под
феном. Можно использовать прием вымораживания как для
разделения гуминовых кислот и фульвокислот из первичных
щелочных экстрактов, так и для концентрирования фульвокислот.
В тех случаях, если описанными приемами не удается получить
малозольные препараты гуминовых и фульвокислот, прибегают
к дополнительному обеззоливанию препаратов смесью 5%-ных
растворов HF и НС1 до отрицательной реакции на Fe. Обработку
можно проводить в центрифужных стаканчиках.
Набор анализов для характеристики полученных препаратов
гуминовых кислот и фульвокислот очень велик и определяется
задачами исследования. Для относительно полной
характеристики гумусовых кислот минимально необходимы зольность,
элементный состав, количество кислых функциональных групп,
степень ароматизации и гидролизуемости, ИК спектры и характер
молекулярной полидисперсности. Целесообразно также
определить формы азота.
Элементный состав представляет собой первый
характеристический параметр, позволяющий установить принадлежность
вещества к классу гумусовых кислот и отличить гуминовые кислоты
от фульвокислот. Количество углерода и водорода определяют
методом сухого сжигания по полумикрометоду Коршун, а азот —
по микрометоду Дюма. Техника работы детально описана в
руководстве Д. С. Орлова с сотрудниками [235]. При определении
количества азота методом Кьельдаля получаются пониженные
результаты вследствие неполного гидролитического расщепления
пиррольных, пиримидиновых ядер, а также циклических систем,
содержащих два или более вицинально расположенных атомов
азота. Расхождения значительны и достигают 0.5% (табл. 74).
Для определения общего количества кислых
функциональных групп предложено несколько методов: метилирование, хемо-
Таблица 74
Валовое содержание азота в препаратах гуминовых кислот
Объекты, из которых выделены препараты
Подстилка елового насаждения
Гумифицированная масса клевера
Типичный чернозем (АпаУ)
Дерново-подзолистая суглинистая почва
(А пах)
хорошо окультуренная
среднеокультуренная
Валовое содержание азота,
% к беззольной массе
метод Дюма
2.96
6.35
3.76
3.33
4.46
метод Кьельдаля
2.39
5.46
2.82
2.88
4.02
232
сорбция, потенциометрическое титрование, восстановление ди-
бораном [19, 44, 82]. Наиболее распространены в настоящее
время хемосорбционные методы и методы потенциометрического
титрования, существующие во многих вариантах.
Общее количество кислых функциональных групп удобно
определять баритным методом Стадникова и сотрудников [283],
содержание карбоксильных групп — ацетатным методом Куха-
ренко [170]. Для суммарного определения количества кислых
функциональных групп мы используем 0.25 н. раствор Ва(ОН)2
с последующим автоматическим потенциометрическим титрованием
до рН 8.4 на рН-метрах с БАТ-15; для определения количества
карбоксильных групп — 0.2 н. раствор ацетата кальция с
последующим автоматическим титрованием до рН 9.8. Содержание
гидроксилов определяется по разности.
В последнее время все более широко применяются методы
потенциометрического и высокочастотного титрования в неводных
средах, позволяющие более точно определить количество очень
различных по кислотной силе функциональных групп,
характерных для гумусовых кислот [81,82]. В нашей лаборатории О. А.
Найденовой в последнее время разработан ряд методов для определения
кислых функциональных групп с использованием
потенциометрического и высокочастотного титрования в водно-ацетоновой
среде в сочетании с боргидридным методом и УФ спектроскопией
[207-210, 212, 213].
(г1 Определение суммы кислых групп прямым
потенциометрическим титрованием. При прямом потенциометрическом титровании
фульвокислот определяют суммарное количество фенолгидрок-
сильных и карбоксильных кислых групп. Для анализа
применяют малозольные препараты или очищенные растворы
фульвокислот. Если используется ряд растворов, то концентрация их
должна быть одинаковой и не ниже 600—800 мг/л (по
углероду).
При использовании сухих препаратов 10 мг (из расчета на
беззольное вещество) растворяют в 100 мл дистиллированной воды
при легком подогревании на водяной бане. Для
потенциометрического титрования берут 5—20 мл раствора фульвокислот в
стаканчик и измеряют рН на потенциометре, после чего из
микробюретки с ценой деления 0.01—0.02 мл добавляют порцию за
порцией титранта (0.02 н. раствор NaOH), одновременно
определяя рН. Вблизи эквивалентной точки наблюдается «скачок»
потенциала: рН резко меняется. Результаты титрования
изображают графически в виде дифференциальной кривой. Для этого
на оси абсцисс откладывают объем щелочи (мл), на оси ординат —
ДрН/ДУ, где ДрН — изменение рН, а ЬУ — изменение объема
между двумя последующими определениями. Дифференциальная
кривая имеет резкий максимум в точке эквивалентности.
Количество кислых групп определяют, опуская перпендикуляр на
ось абсцисс из точки максимума (см. рис. 17).
233
Сумму кислых групп х (мг'экв.) рассчитывают по формуле
x=Vh., где V — объем щелочи, найденный по графику (мл);
н. — нормальность щелочи.
Для пересчета суммы кислых групп (мг*экв.) на 100 г
препарата фульвокислот используют формулу
где хх —- количество кислых групп (мг-экв.), найденное по
графику; V — объем исходного раствора фульвокислот; Vx — объем
фульвокислот, взятый для титрования; К — поправка на
зольность; Кг — поправка на влажность.
Коэффициент К вычисляют по формуле 100/(100—q), где q —
зольность (или влажность) препарата (%).
Определение суммы кислых групп в фульвокислотах обратным
потенциометрическим титрованием. Обратное потенциометри-
ческое титрование дает возможность более полно учитывать
кислые группы по сравнению с прямым методом. Это можно
объяснить как дополнительной активацией кислых групп (СООН"
и ОН"), так и блокировкой части функциональных групп
(разрыв внутримолекулярных водородных связей) в щелочной
среде.
Для анализа берут 10 мг (в расчете на беззольное вещество)
сухого препарата и растворяют в 100 мл 0.1 н. раствора NaOH
путем настаивания в течение 24 ч. Можно использовать и фуль-
ваты натрия. Если сравниваются растворы нескольких образцов,
концентрация должна быть уравнена и не ниже 600—800 мг/л
(по углероду).
Для потенциометрического титрования берут 5—20 мл раствора
в стаканчик и определяют рН аналогично описанному выше.
В точке, соответствующей полной нейтрализации избытка
щелочи, наблюдается первый «скачок» потенциала. Затем идет
титрование кислых групп, и вновь наблюдается незначительное
изменение величины рН. Второй «скачок» потенциала
соответствует полной нейтрализации кислых групп, после чего рН опять
меняется мало. Результаты титрования изображают
дифференциальной кривой. Для этого на оси абсцисс откладывают объем
кислоты (мл), на оси ординат — величину рН. Дифференциальная
кривая обратного потенциометрического титрования имеет два
резких максимума: первый соответствует оттитрованию избытка
щелочи, второй — кислых групп фульвокислот.
Количество кислых групп определяют, опуская
перпендикуляр на ось абсцисс из точки второго максимума. Сумму кислых
групп х(мг*экв.) рассчитывают по формуле x=V1e., где V± —
объем кислоты, найденный по графику (мл); н. — нормальность
кислоты. Для пересчета суммы кислых групп на 100 г препарата
используют ту же формулу, что и при определении прямым
потенциометрическим титрованием.
234
Определение кислых групп методом обратного высокочастотного
титрования в водно-ацетоновой среде. Прямым и обратным по-
тенциометрическим титрованием нельзя раздельно учесть фенол-
гидроксильные и карбоксильные группы в фульвокислотах —
определяется лишь сумма кислых групп. Такое разделение,
возможно методом высокочастотного титрования, представляющего
собой разновидность кондуктометрического. Высокочастотное
титрование — более совершенный и точный метод, его
достоинство — в наличии очень четких перегибов кривых титрования,
в возможности определения различных групп, в отсутствии
контакта электродов с раствором. Применение данного метода
наиболее эффективно в сочетании с неводными растворителями, которые
обладают дифференцирующими свойствами по отношению к
солям, кислотам, основаниям. По отношению к слабым кислотам
наиболее дифференцирующим действием обладают кетоны, в
частности ацетон и водно-ацетоновые растворы.
Для титрования кислых групп этим методом навеску 30—
40 мг сухого препарата заливают 0.1 н. раствором NaOH
(лишенным С02) на 24 ч. Перед титрованием добавляют воду и ацетон
в таких количествах, чтобы к моменту полной нейтрализации
в растворе было около 50% объема ацетона (например, навеску
30 мг+5 мл щелочи+12 мл Н2О+20 мл ацетона).
Титрование проводят 0.1 н. раствором НС1 микробюреткой
с ценой деления 0.01 мл на титриметре ВУ-1А (или другой марки).
Предварительно необходимо познакомиться с конструкцией
прибора по прилагаемой инструкции. Перед началом титрования
снимают показание амперметра (мА), затем добавляют из
микробюретки порцию за порцией раствор титранта и после каждого
прибавления снимают показания амперметра. Моменту полной
нейтрализации избытка щелочи соответствует 1-й «скачок»
потенциала. После этого наблюдается медленное изменение силы тока
в период титрования суммы карбоксильных групп. В момент
полной нейтрализации карбоксильных групп происходит 2-й «скачок»
потенциала. Последующий ход титрования соответствует оттитро-
выванию фенолгидроксильных кислых групп. В момент их полной
нейтрализации намечается 3-й «скачок» потенциала, после чего
вновь постепенно изменяется величина силы тока.
Результаты титрования можно выразить графически. На оси
абсцисс откладывают (мл) титрант (0.1 н. раствор НС1), а на оси
ординат — показания амперметра (мА). На кривых титрования
всегда намечаются два отрезка, соответствующих титрованию
двух Кислых групп: отрезок АВ — карбоксильным группам,
отрезок ВС — фенолгидроксильным (рис. 49). Наиболее точные
результаты получают на высокочастотных титриметрах,
работающих в диапазоне 3—30 МГц. Для количественного
определения кислых групп опускают перпендикуляры из точек перегибов
кривой на ось абсцисс. Объем титранта, соответствующий В—А,
гз§
I » II I I 1 I I 1*1 I I I
1 2 3 <+ 5 6 1 Z 3 <t 5 6
мл 0.1 н. MCI
Рис. 49. Кривые обратного высокочастотного титрования гуминовых
кислот (/) и фульвокислот (77) дерново-подзолистой почвы в водно-ацетоновой
среде.
АВ _ участок, соответствующий группам СООН; ВС — участок, соответствующий
группам (ОН).
дает сумму карбоксильных групп; объем титранта ?— С
соответствует сумме фенольных гидроксилов.
Количество кислых групп х (мг-экв.) определяют по формуле
#=Fh., где V — объем титранта, найденный по графику; н. —
нормальность титранта.
Для перевода количества кислых групп (мг-экв.) на 100 г
сухого беззольного вещества расчет ведут по формуле
Xl • 100ККг
Где хг — количество кислых групп (мг-экв.), найденное по
графику; Ъ — навеска сухого препарата (г), К — коэффициент
пересчета на беззольное вещество; Кг — коэффициент пересчета на
сухое вещество.
Следует иметь в виду, что использование различных методов
и даже различных прописей одного метода определения
количества кислых функциональных групп дает колеблющиеся
показатели количества этих групп, что затрудняет сравнение
полученных данных с литературными материалами. Поэтому в работе
необходимо строго соблюдать используемую в анализе пропись
и сопоставлять результаты с данными, полученными по
аналогичной прописи.
Определение степени ароматизации, ИК спектров,
молекулярной полидисперсности и форм азота. Степень
ароматизации — очень существенный показатель при выявлении осо-
236
бенностей строения гумусовых кислот или их отдельных фракций.
Чаще всего используют два метода: определение количества
бензолкарбоновых кислот при окислении гумусовых кислот
щелочным раствором КМп04 и определение степени гидролизуе-
мости гумусовых кислот. Наиболее прямым методом следует
считать первый. Степень гидролизуемости дает только сравнительную
характеристику устойчивости гумусовых кислот или их фракций
к кислотному гидролизу и позволяет лишь косвенно судить об
относительной доле ароматических структур в молекуле.
Детальная пропись окисления гумусовых кислот перманганатом калия
и определение выхода бензолкарбоновых кислот даны Д. С.
Орловым с сотрудниками [235]; следует только рекомендовать
дополнительное определение количества азота в полученном при
анализе осадке бариевых солей бензол(пиридин)карбоновых
кислот, которое позволит получить прямые данные об этой части
азота.
Гумусовые кислоты гидролизуют на глицериновой бане при
температуре 120—130° в течение 20 ч с 20-кратным количеством
6 н. раствора HG1 в колбочках с обратным холодильником. Степень
гидролизуемости ^определяют путем непосредственного
взвешивания остатка навески после гидролиза, для чего остаток
высушивают при 50—60°.
ИК спектры поглощения гумусовых кислот
снимают на любых двулучевых ИК спектрофотометрах (ИКС-14,
UR-20 и многие зарубежные марки). Детальная пропись всей
техники работы и расшифровки полученных результатов дана
коллективом авторов [234]. Наибольшую сложность представляет
расшифровка полученной спектральной кривой из-за наложения
ряда полос поглощения друг на друга и наличия в препаратах
некоторого количества минеральных примесей в виде золы.
Поэтому препараты не должны содержать более 3—5% золы.
Молекулярная полидисперсность — также
существенный показатель, свидетельствующий о степени
гетерогенности гумусовых кислот. Полидисперсность определяют
обычно методом гельфильтрации на сефадексах или молселек-
тах. Детальная техника работы описана в ряде работ [91, 99,
137, 205, 233, 285]. Мы используем стеклянные колонки с
внутренним диаметром 22—24 мм и высотой рабочего слоя геля сефадекса
270—250 мм. Для хроматографирования на верхнюю часть
колонки (на поролоновый фильтр) наносят 7 мг гумусовой кислоты,
растворенной в 1 мл 0.02 н. раствора NaOH. В качестве элюиру-
ющего раствора используем дистиллированную воду, хотя
некоторые экспериментаторы применяют другие элюенты
(органические и минеральные растворители). Удобно работать на коллекторе
(рис. 50), который позволяет автоматизировать сбор фракций.
Кривые распределения ММ гумусовых кислот строят по величинам
оптической плотности, которую определяют для каждой фракции
на ФЭК-56 или непосредственно по углероду (рис. 51). Последний
237
Рис. 50. Автоматический коллектор фракций (тип 301В, ПНР).
1 _ хроматографическая колонка, 2 — счетчик капель, з — коллектор фракций.
40 I 60 80 100 120 ПО 160 180 200 220мл
Vn ММ*1500
Рис. 51. Кривая молекулярной полидисперсности водорастворимых орга-*-
нических веществ на сефадексе Г-15, составленная до углероду,
JJg дси абсцисс -*- объем эдюата.
прием необходим при работе с бесцветными или слабоокрашен-
ными растворами (например, водорастворимыми продуктами
гумификации растительных остатков). Для калибровки колонки
используют голубой декстран (Mw 2 000 000), альбумин бычий
сыворотный (Mw 70 000—68 000), дезоксирибонуклеазу из
поджелудочной железы свиньи (М^ 40 000), липазу (Mw 30 000—
25 000) и ряд других веществ, список которых опубликован
[91, 285]. По кривым распределения ММ можно подсчитать
площади, соответствующие отдельным фракциям (Sj, Sn, Sm), и
рассчитать соотношение выхода фракций с различной ММ (см.
рис. 13, а). Если необходимо получить значительное количество
фракций с разной ММ, их объединяют в соответствии с элюацион-
ной кривой, концентрируют и высушивают в вакуумном шкафу.
Путем многократного хроматографирования через колонку с
гелем сефадекса можно получить достаточное для анализа
количество фракций. В зависимости от целей хроматографирования
выбирают марку сефадекса (чаще всего Г-25, Г-50, Г-100) или
используют систему колонок от Г-10 до Г-100.
При исследовании форм азота рекомендуют
определение аммонийного азота, включающего в основном азот
амидов, летучих аминов, аминоспиртов, азот аминокислот
алифатического и ароматического рядов, азот аминосахаров (глюкоз-
аминаигалактозамина). Для определения этих соединений гидро-
лизуют гумусовые кислоты 6 н. раствором НС1 при температуре
120—130° так, как это описано выше. Общее содержание азота
в гидролизате определяют методом Кьельдаля, содержание
аммонийного азота — в чашках Конвея, азот аминокислот —
методом Белозерского и Проскурякова, сумму аммонийного азота и
азота аминосахаров — путем разложения амидов и аминосахаров
в щелочной среде с выделением аммиака. Азот циклических
группировок аминокислот вычисляют арифметически по разности
между общим содержанием азота в гидролизате и суммой всех
идентифицированных форм его. Азот гетероциклов можно
получить по разности между валовым содержанием азота,
определенным по методам Дюма и Кьельдаля. Детальный систематический
ход анализа описан А. В. Назаровой [205].
Методы моделирования процессов гумификации
органических остатков
Как мы уже отмечали, методы моделирования широко
применяют при исследовании механизма гумификации. Обзор [работ
с использованием метода моделирования для исследования
отдельных звеньев почвообразовательного процесса сделан
ранее [21]. Методом моделирования исследуют процессы разложения
и гумификации растительных остатков, реакции взаимодействия
различных органических веществ с компонентами минеральной
239
части почвы, природу образующихся органо-минеральных
производных, роль органических, и прежде всего гумусовых, веществ
в процессах почвообразования, характер процессов миграции
и аккумуляции органических и органо-минеральных
производных в почве, влияние отдельных факторов и агентов на эти
реакции и процессы и т. д.
Мы широко используем методы моделирования при изучении
механизма процесса гумификации растительных остатков и
исследовании характера взаимодействия гумусовых веществ с
некоторыми компонентами минеральной части почвы, что и
позволило предложить для обсуждения новую схему,процесса
гумификации органических веществ в почве (гл. III) и основные типы
органо-минеральных производных гумусовых веществ (гл. IV).
Здесь мы кратко остановимся на характеристике моделирования
процесса гумификации растительных остатков в почве, общая
схема которого такова:
Гумификация растительных остатков
в заданных условиях
Периодически повторяющийся анализ
растительного материала
Определение емкости
катионного поглощения
Выделение новообразованных
гуминовых кислот и последующая
дополнительная гумификация их
с определением основных
констант
W ф \h <Aj V V
эе кфг мм ик впк сг
Определение состава
гумифицирующихся
остатков по схеме
1
вв
1
ВС
:
гк
1
Гем
f
-л
1
ФК
1
ц
i
Л
(ЭС — элементный состав, КФГ— карбоксильные и фенолгидроксильные
группы, ММ — распределение молекулярной массы, ИК — инфракрасные
спектры, ВПК — бензолполикарбоновые кислоты, СГ — степень гидроли-
зуемости, ВВ—водорастворимые вещества, ВС — воскосмолы, Г
—гумусовые кислоты с разделением на гуминовые (ГК) и фульвокислоты (ФК),
Гем —гемицеллюлозы и кислоторастворимые белки, Ц —целлюлоза и
кислоторастворимые белки, Л — лигнинный остаток и негидролизуемая
часть белков).
240
Процесс гумификации растительных остатков мы моделируем
в зависимости от заданных условий в двух типах наполнителей:
в чистом кварцевом песке или суглинистой почвообразующей
породе. Чистый кварцевый песок, предварительно тщательно
отмытый 10%-ным раствором НС1 до отрицательной реакции
на Fe3+, PO^, Са2+, SOf*, а затем от следов СГ
дистиллированной водой, используют в тех случаях, когда исследователь
ставит перед собой задачу исключить влияние минеральных
компонентов на образующиеся гумусовые вещества и прежде всего
снять процессы взаимодействия этих веществ с компонентами
минеральной части почвы. Применение суглинистой материнской
породы того или иного химического состава необходимо для
имитации процесса гумификации в условиях, приближающихся
к природным. Мы уже отмечали, что наличие в наполнителе
веществ, с которыми образующиеся гуминовые кислоты дают
нерастворимые в воде соединения, снимает возможность выделять
водорастворимую часть этих соединений. При использовании
в качестве наполнителя суглинистой массы породы необходимо
определять в ней емкость обменного поглощения катионов, чтобы
внести поправки в модельном опыте.
Для изучения скорости и характера процесса гумификации
в почве (в гумусовом горизонте) необходима дополнительная
серия опытных сосудов с одним наполнителем, так как в процессе
инкубации растительных остатков происходят изменения ее
органической части. В зависимости от заданной программы
исследований модельные опыты закладывают с достаточно большим
количеством растительных остатков при отношении растительные
остатки : наполнитель как 1 : 40—50 в стеклянных или
полиэтиленовых сосудах при заданных условиях увлажнения. Чаще
всего изучают две категории увлажнения: аэробные при 60% и
анаэробные при 150% от полной влагоемкости исследуемого материала.
Опыты рекомендуется проводить в биологическом шкафу
при постоянной заданной температуре. Полив осуществляют по
массе после предварительного определения влагоемкости
наполнителя и растительных остатков и расчетной величины ее в
зависимости от соотношения между этими компонентами. Срок
инкубации материала зависит от программы; целесообразно
снимать сосуды для анализа через 7, 30, 90, 180, 360 и 720 сут
после начала опыта. Количество сроков, намеченных для
исследования, определяет необходимое число повторностей с учетом
не менее чем 4-кратной повторности для каждого срока. При этом
одну из повторностей каждого срока исследований употребляют
для определения влагоемкости и остаточной массы материала,
чтобы учесть степень минерализации его и корректировки
необходимого для полива количества воды. В процессе гумификации
масса растительных остатков уменьшается, изменяется их влаго-
емкость, и эту поправку необходимо вносить для сохранения
заданных условий опыта.
16 Л. Н. Александрова 241
В соответствии со схемой сосуд первой повторности (I)
расходуют полностью для выделения новообразованных гуминовых
кислот и исследования их дальнейшей трансформации. С этой
целью всю массу данного сосуда в одной повторности без
дополнительного высушивания на воздухе используют для выделения
новообразованных гуминовых кислот; массу второй
повторности (II) также без предварительного высушивания употребляют
для определения состава гумифицирующихся остатков; массу
третьей повторности (III) высушивают на воздухе и тщательно
растирают для дальнейшего использования.
Из образца повторности I новообразованные гуминовые
кислоты в случае инертного наполнителя выделяют двумя экстра-
гентами последовательно водой и 0.1 М раствором Na4P207,
а при использовании суглинистой массы в качестве наполнителя —
только 0.1 М раствором Na4P207 с последующим получением
чистых препаратов гуминовых кислот так, как это описано выше
(см. с. 228). Часть полученных препаратов подвергают
дополнительной гумификации в массе чистого кварцевого песка для
исследования тех изменений, которые имеют место в
последующем ходе гумификации новообразованных гуминовых кислот.
Добавочную гумификацию проводят в течение заданного периода
(30, 90, 180, 360, 720 сут), после чего в препарате определяют
намеченные программой параметры. Одновременно следует
определить степень минерализации заложенных в опыт гуминовых
кислот. Перечень анализов, необходимых для характеристики
гуминовых кислот, дан на схеме, а условия их выполнения
описаны выше (см. с. 233—239).
Состав гумифицирующихся растительных остатков определяют
из образца повторности II по прописи, изложенной на с. 221.
Для определения количества гигроскопической воды,
зольности и емкости обменного поглощения катионов, которую
рекомендуется проводить при помощи буферного раствора ВаС12
при рН 6.8, используют высушенный образец повторности III.
В процессе гумификации растительных остатков емкость
обменного поглощения постепенно возрастает вследствие увеличения
количества карбоксильных групп при формировании гумусовых
кислот.
При работе с суглинистым наполнителем необходимо вносить
поправку на величину емкости поглощения наполнителя. Работа
с гумусированной почвой очень осложняет исследование процесса
гумификации из-за наличия гумуса в самом наполнителе.
Необходимо вносить систему поправок, для того чтобы учесть
количество новообразованных гумусовых кислот; количество геми-
целлюлоз, белков и целлюлозы в этом случае практически
определить невозможно.
Аналогичные модельные опыты по гумификации растительных
остатков можно проводить непосредственно в природных условиях,
закладывая их в полиэтиленовых сосудах в поле.
242
Методы исследования процессов миграции
органических и органо-минеральных соединений
Процессы миграции водорастворимых органических и органо-
минеральных соединений из того или иного горизонта почвенного
профиля следует изучать лизиметрическим методом, так как метод
водных вытяжек дает искаженные результаты. В настоящее время
используют три основных типа лизиметрических установок,
позволяющих получать мигрирующую-часть почвенного раствора
и исследовать его состав: лизиметры конструкции Е. И.
Шиловой [324], при помощи которых получают растворы из любой
части профиля почвы; хроматографические воронки
конструкции И. С. Кауричева и Е. М. Ноздруновой, закладываемые под
любой генетический горизонт профиля почвы [131]; лизиметры
конструкции кафедры почвоведения Ленинградского
сельскохозяйственного института, позволяющие исследовать как
процессы миграции веществ, так и характер процессов разложения
и гумификации растительных остатков или органических
удобрений. Лизиметр третьего типа (рис. 52) представляет собой
полиэтиленовую бутыль с обрезанным дном емкостью 10 л (бутыли
меньшей емкости не рекомендуются, ибо в каждую из них можно
заложить очень небольшой объем исследуемого материала),
соединенную шлангом с бутылью — приемником почвенного
раствора. Раствор выкачивают при помощи насоса двойного действия
из приемника.
В бутыль, перевернутую вниз горловой частью, помещают
какой-либо инертный дренирующий материал (стеклянную вату)
и подготовленную массу исследуемого растительного материала
или органического удобрения, тщательно перемешанную с
пахотным слоем почвы, или кварцевым песком, или суглинистой
материнской породой в соотношении не уже чем 1 : 40.
Перемешанную массу утрамбовывают и устанавливают в заложенной
для этой цели траншее с таким расчетом, чтобы верхняя часть
перевернутой бутыли выступала на 2—3 см от поверхности после
закрытия траншеи. Если опыт закладывают с почвой, необходима
серия контрольных лизиметров без растительных остатков.
Количество повторностей определяют длительностью опыта.
Для каждого срока нужно закладывать лизиметрические
установки в трех повторностях. Целесообразно опыт проводить в
течение 3—5 лет, для чего при работе с одним видом органических
материалов и одним видом наполнителя необходимо заложить
не менее 15 лизиметров с исследуемым материалом и такое же
количество лизиметров с одним наполнителем, если им является
гумусовый горизонт почвы.
Поступающие в приемник лизиметрические воды периодически
откачивают, а сам лизиметр снимают в заданный программой срок
для изучения скорости разложения и характера гумификации
т
W
Рис. 52. Схема лизиметрической установки для исследования мигрирующих
из пахотного слоя почвы водорастворимых веществ.
1 — лизиметр, в который набивается почва; 2 — приемник мигрирующего раствора.
органических материалов, помещенных в лизиметр. Следует
наблюдать за скоростью наполнения приемника лизиметрическими
водами (в соответствии с выпадающими атмосферными осадками)
и не допускать переполнения приемника водой.
Лизиметрические воды перед анализом центрифугируют для
очистки от случайных загрязнений, после чего измеряют их объем,
а затем анализируют по составленной программе. Минимально
необходимыми видами анализа следует считать определение
общего количества водорастворимых веществ (плотный остаток),
рН, количества С и N, содержания фульвокислот и
низкомолекулярных органических веществ, состава А1- и Fe-органических
соединений, а для характеристики водорастворимых
минеральных соединений — определение количества анионов СО§-, HGO3,
NO3, SOf, СГ, POf и катионов NH+, Ca2+, Mg2+, K+, Na+, Fe3+,
Al3+.
Фульвокислоты определяют сорбцией их на активированном
угле, состав низкомолекулярных продуктов разложения — по
методике, изложенной в следующем разделе. Для исследования
количества А1- и Fe-органических соединений нужно определить
содержание А1 и Fe до кислотного гидролиза лизиметрических
вод и после такового с 5%-ным раствором НС1. Разность даст
содержание этих элементов в составе комплексных соединений.
Ежегодно в конце вегетационного периода можно снимать
три повторности для исследования процесса минерализации и
гумификации органических материалов. Эти исследования
проводят в соответствии с методикой, описанной выше (см. с, 239).
244
Методы изучения состава индивидуальных
(неспецифических) органических веществ в почве 3
В составе веществ, образующихся при разложении в почвах
остатков растительного и микробного происхождения, присутствуют
большое число индивидуальных соединений, а также вещества
гумусовой природы. Концентрация различных индивидуальных
соединений в почвах, как правило, невысока. Поэтому
определение количества и состава их возможно лишь после
предварительного концентрирования и фракционирования исходных
вытяжек или почвенных растворов.
Для концентрирования индивидуальных органических
веществ применяют обычно методы выпаривания, экстракции
различными растворителями, сорбции на различных сорбентах,
вымораживания и лиофильной сушки [270, 314].
Сложную смесь органических веществ фракционируют на
отдельные компоненты методами дробного осаждения из
растворов, ультрацентрифугирования, с помощью бумажной и
тонкослойной хроматографии. В последнее время все большее
распространение получают методы фракционирования органических веществ
на активированном угле, ионообменных смолах и сефадексах
[145, 235, 483]. Для определения общего содержания и состава
отдельных групп индивидуальных соединений и гумусовых
веществ применяют различные, главным образом
колориметрические и хроматографические, методы.
Приводим методику изучения состава индивидуальных
органических веществ в почве, применяемую на кафедре
почвоведения Ленинградского сельскохозяйственного института.
Органические вещества в вытяжках из почв или почвенных
растворах фракционируют по следующей схеме (см. с. 246).
Органические вещества в вытяжках предварительно
концентрируют методом вымораживания. Для этого в цилиндрические
полиэтиленовые стаканы диаметром 10 см и высотой 15 см
помещают по 500—700 мл исследуемой вытяжки, очищенной на
бактериальных свечах СФ-3. Стаканы с подставками из пенопласта
ставят в холодильник на 24 ч. В процессе вымораживания
образуются слой льда и концентрат органических веществ. После
нарастания слоя льда толщиной 4—5 см стаканы оставляют
на несколько минут при комнатной температуре и затем лед
отделяют от концентрата. При необходимости проводят
повторное вымораживание.
Для разделения сложной смеси органических веществ на
низко- и высокомолекулярную фракции применяют гельхрома-
тографию в сочетании с центрифугированием. Этот метод
представляется весьма эффективным для предварительного фракциони-
* Раздел написан М. В. Новицким.
245
м
н
о
ф
в
и
Н
К
о
ф
к
и
cd
и
Рн
о
«
н
и
cd
П
О
Рн
IS)
»
о
IS]
ST
К
cd
Рн
cd
Ф
И
a
cd
fc
ec
H
Я
П
дна
о
M
ф
И
н
cd
«
в
cd
Рн
о
Я
д
64
cd
Рн
н
нцен
в
ЕГ
cd
Рн
Н
Is)
ч
ф
и
и
cd
0
О
Рн
IS]
Сн
>>
>е<
в
Рн
н
я
—>
к
cd
*
ф
cd л
tt н
?§
о *
о
ф
И
о
Рн
1н
Он
S
ф
я
и
л
НОЛ
ф
Э
cd
И
н
щес
ф
«
II
о
И
CD
>>
cd
Рн §
о
л и
о н
И о
н ч
s S
и
Ф
Я
ш
Рн
S3 cd
Чи
>>н
И о
ф ф
« В"
о
»
со
М
->
-*< §« —>
Он Ог?
а g-°
^ tc
н
cd
Ч
>в<
<? go
g ^^
?? Рн^—>
«s&
и
^*s
^ i voO ^
Pf
Я cd
И М
ё 5
о ф
и В" !
ф ф j
в и
i
а
5*
>>
i
рганиче
ские
КИСЛОТЫ
. я
о н
Амин
КИСЛО
Рис. 53. Центрифужный стакан-вкладыш.
1 — цилиндр-вкладыш, 2 — вставной стакан с отверстиями
(сеткой), з — фильтр.
рования в мягких условиях, так как исключает
изменение химической природы и рН
растворов.
Методика разделения состоит в следующем.
К концентрированным растворам объемом 50 или
100 мл добавляют соответственно 20 или 40 г
сухого сефадекса Г-15. После 10-минутного
настаивания растворы с набухшими сефадек-
сами переносят в центрифужные стаканы со
специальными вкладышами-фильтрами и
центрифугируют при 3000 об/мин в течение 10 мин (рис. 53).
При этом набухающий гель поглощает воду и низкомолекулярные
вещества с ММ не более 1500. Высокомолекулярные соединения
(с ММ более 1500) остаются в свободным объеме и при
центрифугировании собираются на дне стакана. Низкомолекулярные
вещества из геля вытесняют путем обработки его
дистиллированной водой (3 раза по 50 мл) и последующим центрифугированием
в таких же стаканах.
Полученную таким образом низкомолекулярную фракцию
органических веществ разделяют на ионообменных смолах, выделяя
из смеси аминокислоты, низкомолекулярные органические
кислоты, углеводы, фенольные соединения и очищая их от
минеральных примесей [295].
Применение ионообменных смол. Для фракционирования
низкомолекулярных органических веществ применяют катио-
ниты КУ-2 (или Дауэкс-50Д?) в Н+-форме, аниониты
Дауэкс-3 (ЭДЭ-10П) в СО|-форме и ИРА-400 (АВ-17) в СНГ-
форме. Сильнокислотный катионит КУ-2, по данным А. Д.
Семенова [271], — наиболее эффективный сорбент для
аминокислот; вместе с тем он почти не сорбирует редуцирующих
Сахаров и органических кислот. На слабоосновных анионитах
Дауэкс-3 или ЭДЭ-10П сорбируется 90—95% органических
кислот и не более 6—10% Сахаров [271]. Кроме того, в отличие
от сильноосновных анионитов они не расщепляют Сахаров при
прохождении последних через колонку.
Все ионообменные смолы для колоночной хроматографии
необходимо тщательно подготовить к работе [483]. Используют
смолы с диаметром зерен 0.15—0.25 мм. Продажную
ионообменную смолу измельчают в шаровой мельнице или в ступке
и просеивают через сито с диаметром отверстий 0.25 мм. От более
мелких частиц (диаметром менее 0.10—0.15 мм) смолу
освобождают отмучиванием, для чего просеянную смолу суспендируют
в 8—10-кратном объеме воды и после 10—15-минутного отстаивания
247
Рис. 54. Колонка для ионообменных смол.
1 — стеклянная вата, 2 — смола.
сливают воду с неосевшими частицами смолы.
Операцию повторяют несколько раз, до обесцвечивания
промывной воды.
Отобранную фракцию катионита после
предварительного 24-часового набухания в насыщенном
растворе NaCl и промывания водой нагревают на
водяной бане с 2 н. раствором NaOH в течение 2 ч (для
удаления низкомолекулярных органических веществ).
Затем смолу переносят в большую делительную воронку
(емкостью 1—2 л) и отмывают от щелочи
дистиллированной водой до нейтральной реакции промывных вод.
После щелочной обработки смолу промывают 15, 10 и
1%-ным растворами НС1 до полного исчезновения
в фильтрате ионов железа и дистиллированной водой
до отрицательной реакции на С1~. Последнюю
промывку проводят 96%-ным этиловым спиртом до
исчезновения окраски в фильтрате. Промытый катионит
высушивают на воздухе и в воздушно-сухом состоянии хранят
в стеклянной банке с притертой пробкой.
Слабоосновной анионит Дауэкс-3 (фракцию 0.15—0.25) после
набухания в насыщенном растворе хлористого натрия промывают
5%-ным раствором соляной кислоты до полного удаления ионов
железа. Затем ионит обрабатывают 2 н. раствором соды,
отмывают водой до нейтральной реакции промывных вод,
промывают этиловым спиртом и после высушивания на воздухе хранят
в банке с притертой пробкой. Сильноосновные аниониты
(ИРА-400, АВ-17) готовят к работе так же, как Дауэкс-3, с той
лишь разницей, что вместо раствора соды применяют 1 н. раствор
NaOH.
Подготовленными ионитами заполняют хроматографические
колонки (можно использовать бюретки емкостью 25 мл с
расширенной частью вверху емкостью 40—50 мл). Внутренний диаметр
рабочей части колонки равен 1 см, высота слоя ионита в ней
20 см. На дно колонки помещают стеклянную вату и переносят
влажную смолу небольшими порциями с водой, утрамбовывая
стеклянной палочкой. Скорость пропускания растворов через
такую колонку можно регулировать с помощью хорошо
пришлифованного крана (рис. 54).
Общая схема очистки и разделения низкомолекулярных
органических веществ следующая. Пробу объемом 200—250 мл
пропускают последовательно через катионит Дауэкс-50 (КУ-2)
в Н+-форме и анионит Дауэкс-3 (или ЭДЭ-10П) в СО|~-форме.
На катионите сорбируются аминокислоты и неорганические
катионы, на анионите — все вещества кислотного характера
248
(органические и минеральные кислоты)» а вещества нейтральной
природы (углеводы) проходят через обе колонки.
Аминокислоты с катионита десорбируют 75 мл 2 н. раствора
NH4OH с последующей промывкой колонки 100 мл
дистиллированной воды. Элюат с промывными водами выпаривают в
фарфоровой чашке на водяной бане досуха, а сухой остаток
растворяют в 1—2 мл 30%-ного этанола. Органические кислоты
с анионита десорбируют 30 мл 2 н. раствора углекислого
аммония с последующей промывкой 50 мл катионированной
дистиллированной воды. Применение в качестве элюента углекислого
аммония имеет по сравнению с другими реактивами неоспоримое
преимущество, так как избыток (NH4)2C03 легко удаляется при
упаривании элюата. Кроме того, одновременно с элюацией кислот
происходит регенерация анионита. Десорбированные с анионита
органические кислоты также выпаривают в маленькой
фарфоровой чашке досуха на водяной бане. Сухой остаток растворяют
в 1—2 мл 30%-ного эталона с добавлением нескольких крупинок
тимола (в качестве антисептика).
Для разделения и идентификации отдельных
низкомолекулярных органических соединений используют методы бумажной,
тонкослойной и газожидкостной хроматографии. Ниже
приводится методика определения состава низкомолекулярных
органических кислот, аминокислот, Сахаров и фенольных соединений
с использованием метода хромотографии в тонком слое [222].
Аминокислоты. Аминокислоты разделяют на стеклянных
пластинках размером 13x18 см с закрепленным слоем ДЭАЭ-цел-
люлозы. Для нанесения сорбента применяют рамку из
плексигласа с регулируемой толщиной наносимого слоя или аппарат
Шталя. Пластинки после нанесения на них сорбента
высушивают на воздухе в течение 15—20 ч и хранят в эксикаторе над
силикагелем.
Подготовленную описанным выше способом вытяжку
аминокислот наносят (при помощи микропипетки на 0.1 мл с
вытянутым носиком) на хроматографическую пластинку по 5—20 мкл
в пятно. Стартовую линию помещают на расстоянии 1.5 см от
края пластинки; расстояние между пятнами на стартовой линии
1.5 см, диаметр пятна не более 2—3 мм. На каждую пластинку
наряду с исследуемыми вытяжками наносят не менее чем в двух
точках смесь аминокислот-«свидетелей» (рис. 55).
Разгонку производят в лотковой камере с насыщением парами
растворителя путем пропитки обклеивающей фильтровальной
бумаги или в С-камере по Шталю. Храмотографическая камера —
прямоугольный сосуд размером 14x20x20 см, закрытый сверху
стеклом. В сосуд наливают растворитель так, чтобы поставленная
вертикально (или с небольшим уклоном) пластинка погружалась
в растворитель на 5 мм. В качестве растворителей для
аминокислот применяют смесь w-бутанол—уксусная кислота—вода в
соотношении 4:1:5 (по объему) или н-бутанол—муравьиная кислота—
249
16
13\J 13 0q12 q1Z
On 11й
! 4
о " ю
Рис. 55. Хроматограмма аминокислот-<<свидетелей».
1 — аспарагиновая кислота, 2 — аспарагин, з — глутаминовая кислота, 4 — глутамин»
5 — пролин, 6 — серии, 7 — глицин, 8 — треонин, 9 — лизин, 10 — гистидин, 11 —
аргинин, 12 — аланин, 13 — метионин, 14 — валин, 15 — фенилаланин, 16 — лейцин.
вода в соотношении 5:1:1. Аминокислоты лучше делятся при
двукратной разгонке (длина пути растворителя 10 см, время
разгонки 2 ч) с просушиванием слоя после каждой
разгонки. Просушенные хроматограммы проявляют, опрыскивая
их 0.8%-ным раствором нингидрина в ацетоне с добавкой 0.1%-
ного уксуснокислого кадмия (по массе) и 5%-ной ледяной
уксусной кислоты (по объему). Для лучшего развития окраски хро-
матограмму помещают на 10—12 ч в эксикатор над
концентрированной серной кислотой.
Общее содержание свободных аминокислот определяют
колориметрическим методом с нингидрином после предварительной
очистки и концентрирования аминокислот на катионите [271 ].
Метод основан на реакции а-аминокислот с нингидрином с
образованием окрашенного в сине-фиолетовый цвет соединения.
Нингидриновую реакцию проводят в смеси изучаемого водного
раствора с бутиловым спиртом, добавление которого повышает
устойчивость окрашенного продукта реакции и позволяет
отделить его от посторонних окрашенных органических веществ
(гуминовых кислот и др.). Чувствительность метода составляет
1.5—2 мкг аминного азота в пробе. Точность определения 10—-15%.
Анализ проводят следующим образом. 150—200 мл вытяжки
пропускают через колонку с катионитом КУ-2 в Н+-форме. Затем
аминокислоты десорбируют 75 мл 2 н. раствора NH4OH, элюат
выпаривают в маленькой фарфоровой чашке на водяной бане.
Полученный осадок с помощью 2 мл дистиллированной воды
О
05
250
Рис. 56. Хроматограмма метчиков
органических кислот.
1 — щавелевая, г — винная, 3 — лимонная,
4 — яблочная, 5 — янтарная и аконитовая,
6 — фумаровая.
(в 3—4 приема) переносят в колбу
вместимостью 50 мл с притертой
пробкой. В колбу добавляют 1 мл
нингидринового реактива (75 мг
хлористого кадмия растворяют
в 6 мл воды, добавляют 0.3 мл
ледяной уксусной кислоты, 2 г нингид-
ринаиЮОмл ацетона), 1—2 капли
насыщенного раствора хлорида
кадмия и 5 мл бутилового спирта.
Колбу помещают на 15 мин в
кипящую воду. После охлаждения смесь
переносят в градуированную
центрифужную пробирку, содержащую 1 мл 25%-ного раствора сегне-
товой соли. Спиртовой слой доводят до 6 мл, содержимое пробирки
встряхивают и центрифугируют. Далее из спиртового слоя
отбирают 5 мл, смешивают их с 0.3 мл w-бутанола (для
разрушения водной эмульсии в спиртовом растворе) и колориметрируют
на фотоэлектроколориметре при длине волны 435 нм (красный
светофильтр). ¦>*••¦
Содержание аминокислот (в пересчете на аминный азот)
рассчитывают по колибровочной кривой, построенной по оптической
плотности стандартных растворов гликокола с 0—30 мкг амин-
ного азота в пробе.
Низкомолекулярные органические кислоты. Органические
кислоты разделяют и идентифицируют на хроматограммах с
закрепленным слоем силикагеля с добавкой 5% гипса. Разгонку ведут,
так же как и аминокислот, в лотковой камере с насыщением
парами растворителя. Наилучшие результаты достигаются при
применении в качестве растворителя смеси эфир—муравьиная
кислота—вода в соотношении 18 : 5 : 9 (разгонку осуществляют
при температуре 8—10° в течение 90 мин, длина пути
растворителя 10 см). Лучший проявитель — смесь ксилозы с анилином
в соотношении 1 : 1 (по массе). Удовлетворительные
результаты дает также применение 0.05%-ного раствора бромфенолового
синего (рис. 56). В остальном техника разделения
низкомолекулярных органических кислот аналогична разделению
аминокислот.
Общее содержание органических кислот можно определить
потенциометрическим титрованием растворов после
предварительного пропускания их через катионит КУ-2 (или Дауэкс-50)
О
02
! ¦
0<
6
О*
4
О 05
О
о
0*
0<
251
и кипячения в течение 5 мин. Для этой цели используют
потенциометр ЛПМ-60 с блоком автоматического титрования.
Углеводы. Удовлетворительное разделение ряда Сахаров —
глюкозы, сахарозы, ксилозы, галактозы, арабинозы, фруктозы —
возможно хроматографированием на тонком закрепленном слое
силикагеля с гипсом в лотковой камере с насыщением. В
качестве растворителя применяют смесь w-бутанол—^-уксусная
кислота—вода в соотношении 4:1:5 или смесь этилацетат—уксусная
кислота—вода (3:1 : 3). Разгонку производят при, комнатной
температуре (17—18°) двукратно, длина пути растворителя 10 см.
Наиболее чувствительным проявителем Сахаров является
свежеприготовленная смесь 0.5 мл анисового альдегида, 9 мл 95%-ного
этанола, D.5 мл концентрированной серной кислоты и 0.1 мл
ледяной уксусной кислоты (чувствительность реакции 0.5 мкг
сахара в пятне). Вполне удовлетворительные результаты дает
проявление хроматограмм свежеприготовленной смесью (2% -ный
водный раствор хлористого трифенилтетразолия и 1 н. раствор
NaOH 1 : 1) с нагреванием обработанной реактивом хромато-
граммы в течение 40 мин при 50—55°.
Для количественного определения общего содержания
углеводов применяют фенолсернокислотный метод Дюбуа в
модификации Э. С. Бикбулатова [57]. Метод основан на способности
углеводов давать фурфурол при взаимодействии с сильными
кислотами при нагревании и позволяет определить как свободные,
так и связанные углеводы, в том числе уроновые кислоты и
углеводные группирЪвки гумусовых веществ.
Углеводы определяют по следующей методике. К 5 мл
вытяжки, пропущенной через катионит КУ-2 и анионит Дауэкс-3,
взятой в колбу на 100 мл из тугоплавкого стекла, прибавляют
из бюретки 5 мл 5%-ного водного раствора фенола. Затем быстро
приливают 25 мл сернокислотного реактива (5 г гидразинсуль-
фата в 1 л концентрированной H2S04) при постоянном
перемешивании. Колбочки закрывают стеклянными пробками и охлаждают
при комнатной температуре. Затем измеряют оптическую
плотность D± раствора на спектрофотометре при длине волны 485 нм
или на фотоэлектроколориметре при соответствующем
светофильтре в 50-миллиметровых кюветах. Истинное значение
оптической плотности DacT находят по формуле Дтт=П1 — Z)2, где
D2 — оптическая плотность холостого раствора, который
готовят так же, как и рабочий, но вместо фенола берут 5 мл
дистиллированной воды. Образующаяся розовая окраска соединения
углеводов с фенолом устойчива в течение 24 ч. Концентрацию
углеводов определяют по калибровочному графику, построенному
по стандартным растворам, содержащим от 5 до 80 мкг глюкозы
в пробе.
Наряду с общим содержанием углеводов определяют и
свободные редуцирующие сахара. Наиболее приемлем для этой цели
спектрофотометрический метод [272], основанный на реакции
252
восстановления двухвалентной меди сахарами в щелочной среде.
Образующуюся при этом закись меди отделяют от избытка
щелочного медного реактива, окисляют вновь до двухвалентной
и определяют ее количество с диэтилдитиокарбаматом натрия.
Анализ проводят следующим образом. 30 мл анализируемой
вытяжки после пропускания через колонки с ионитами КУ-2
и Дауэкс-3 выпаривают досуха на водяной бане. Сухой остаток
переносят с помощью 1 мл дистиллированной воды в центрифужную
пробирку, добавляют 1 мл щелочного медного реактива,
нагревают смесь в кипящей воде в течение 5 мин и затем быстро
охлаждают холодной водой. (Щелочной медный реактив готовят из
двух растворов: 1-й — 12.5 г безводного карбоната натрия и
12.5 г сегнетовой соли растворяют в 300 мл дистиллированной
воды, содержащей 20 мл 1 н. раствора едкого натра; 2-й — 3 г
медного купороса растворяют в 50 мл дистиллированной воды;
перед определением растворы сливают и доводят объем смеси
водой до 500 мл). В пробирку с охлажденной смесью приливают
около 10 мл бидистиллята, раствор центрифугируют и сливают.
Промывку проводят трижды. После удаления последней порции
промывной воды окисляют закись меди в пробирке смесью
соляной и азотной концентрированных кислот (2 капли НС1 и 1 капля
HN03). Содержимое пробирки выпаривают почти досуха и с
помощью 5 мл катионированной дистиллированной воды переносят в
маленькую делительную воронку, куда приливают затем 1 мл
5%-ного раствора диэтилдитиокарбамата натрия и 5 мл смеси четы-
реххлористого углерода с изобутиловым спиртом (2 :1 по объему).
Смесь встряхивают 2 мин и отделяют окрашенный слой
органического растворителя. Тонкую водную эмульсшо в этом слое
разрушают прибавлением 0.5 мл изобутилового спирта, затем
измеряют оптическую плотность раствора при длине волны
433 нм.
Количество Сахаров определяют по калибровочной кривой,
построенной по стандартным растворам глюкозы, обработанным
аналогичным способом, в концентрациях от 0 до 30 мкг Сахаров
в пробе.
Одним из компонентов углеводов, анализируемых фенолсерно-
кислотным методом Дюбуа, являются уроновые кислоты. Для
определения их количества в вытяжках из почв или
растительных остатков применяют фотометрический метод с карбазолом [308].
Анализ проводят таким образом. К 2 мл исследуемого
раствора, помещенного в колбу из термостойкого стекла вместимостью
50—100 мл, приливают 1мл 0.5%-ного спиртового раствора кар-
базола. Колбу охлаждают в водяной бане со льдом и к смеси
добавляют 22 мл концентрированной серной кислоты, охлажденной
до 0°. Затем смесь нагревают в кипящей водяной бане в течение
15 мин, охлаждают до комнатной температуры и измеряют
оптическую плотность в сравнении с холостым раствором (вода,
подготовленная аналогичным способом). Наиболее точные результаты
253
получают при измерении разности оптической плотности при
длине волны 490 и 420 нм в кювете 1 см. Для устранения
действия моносахаридов добавляют водный раствор моноамида H2S04
(100 мг/мл) и Н3В03 (60 мг/мл).
Содержание уроновых кислот (%) вычисляют по формуле
где AJ9 — разность оптических плотностей при 490 и 420 нм,
V — объем реакционной смеси, I — толщина поглощающего
слоя (1 см), т — масса исследуемого вещества (мг) в объеме,
ATAD=68.6 мл • мг-1 • см"1.
Фенольные соединения. Для выделения фенольных
соединений из водных или спиртовых вытяжек применяют
активированный уголь, сильноосновные аниониты, а также полиамидные
смолы. В работе В. Т. Каплина с сотрудниками [114] показано,
что сорбция многоатомных фенолов на активированном угле БАУ
очень незначительна и не превышает 10—30% исходной
концентрации фенолов. Сильноосновные аниониты, содержащие
четвертичную аминогруппу АВ-17 или ИРА-400, обладают хорошей
сорбционной способностью по отношению к многоатомным
фенолам, сорбируя их практически полностью (95—100% исходной
концентрации). Для десорбции фенол ьных соединений,
сорбированных анионитом АВ-17 (или ИРА-400), применяют
хлороформ (в статических условиях) и этиловый спирт (в динамических
условиях). В динамических условиях используют колонку
1 : 15 см, скорость пропускания 10—12 капель/мин. Наиболее
селективными по отношению к различным фенольным
соединениям являются полиамидные смолы типа «капрон» или других
марок, которые успешно применяются в колоночной и
тонкослойной хроматографии фенолов [295].
Для разделения и идентификации отдельных фенольных
соединений применяют газожидкостную или тонкослойную
хроматографию. Многоатомные фенолы и фенолкарбоновые кислоты
разделяют на закрепленном флуоресцирующем слое силикагеля
с гипсом в лотковой камере или в С-камере. В качестве
растворителей применяют смеси бензол—диоксан—уксусная кислота
(95 : 25 : 4) или бензол—метанол—уксусная кислота (45 : 8 : 4).
Разгонку проводят двукратно при комнатной температуре с
просушкой хроматограмм после первой разгонки. Лучшим
проявителем являются смесь анисового альдегида с серной кислотой
в этаноле или бензидин (для проявления флавоноидов). Хорошее
разделение фенольных соединений достигается на
тонкослойных хроматограммах с закрепленным слоем полиамидного
порошка [295].
Суммарное количество фенольных соединений определяют
колориметрическим методом с использованием реактива Фолина —
Дениса [108] или бромид-броматным методом [51]. Для при-
254
готовления реактива Фолина—Дениса 100 г Na2W04 • 2НаО
смешивают с 20 г фосфорно-молибденовой кислоты, 50 мл 85% -ной
фосфорной кислоты и 750 мл дистиллированной воды. Смесь
кипятят с обратным холодильником 2 ч, фильтруют и
разбавляют водой до 1 л.
Методика определения фенольных соединений состоит в
следующем. К анализируемому раствору (5 мл) в мерной колбе
вместимостью 50 мл добавляют 30 мл дистиллированной воды, после
перемешивания — 2.5 мл реактива Фолина—Дениса. Смесь снова
перемешивают, через 3 мин добавляют 5 мл насыщенного
раствора Na2C03 и доводят водой до метки. Через 1 ч измеряют
оптическую плотность раствора при длине волны 725—730 нм. В
случае появления мути или осадка раствор перед колориметриро-
ванием центрифугируют. Калибровочную кривую строят по
стандартным растворам фенола.
Фенольные соединения бромид-броматным методом
определяют по такой методике. Пробу анализируемой вытяжки 5—50 мл,
содержащей не более 150 мг фенолов, переносят в коническую
колбу на 250 мл с притертой пробкой и разбавляют дважды
дистиллированной водой до 80 мл. Затем приливают 1 мл бромид-
броматной смеси (1.67 г бромата калия и 6 г бромида калия,
растворенные в 1 л дистиллированной воды), 5 мл разбавленной
(1 : 3) серной кислоты и оставляют стоять в темном прохладном
месте 30 мин. После этого добавляют 1 г иодида калия, закрывают
пробкой, перемешивают, ставят в темное место и через 5 мин
титруют выделившийся иод 0.002 н. раствором тиосульфата натрия
с добавлением 1—2 мл 0.05%-ного раствора крахмала в конце
титрования. Аналогичным образом проводят холостой опыт, где
вместо вытяжки берут 80 мл дистиллированной воды.
Результаты определения рассчитывают на фенольное соединение, которое
преобладает в анализируемой вытяжке, по формуле
(Ъ — а)к'И.-К- 1000
где х — содержание фенола в вытяжке (мг/л); Ъ — объем раствора
тиосульфата натрия, израсходованного на титрование в холостом
опыте (мл); а — объем раствора тиосульфата натрия,
израсходованного на титрование пробы (мл); к — поправочный коэффициент
для приведения к точной концентрации; н. — нормальность
раствора тиосульфата натрия; К — коэффициент, равный числу
миллиграммов соответствующего фенола, эквивалентному 1 мл
раствора тиосульфата натрия; V — объем вытяжки, взятой для бро-
мирования (мл). При содержании фенольных соединений более
150 мг в пробе бромирование проводят 5 мл бромид-броматной
смеси, а выделившийся в результате реакции иод оттитровывают
0.01 н. раствором тиосульфата натрия.
ЗАКЛЮЧЕНИЕ
В монографии изложены современные представления об
источниках, природе и свойствах органической части почвы на
основании литературных данных и многолетних исследований
автора. В составе компонентов, образующих основную массу
органических остатков в почве, главнейшая роль принадлежит
сложной системе азотсодержащих высокомолекулярных
соединений (белкам, углеводам, лигнину), формирующих биомассу
и являющихся гумусообразователями. В пахотных почвах
существенна и специфична роль органических удобрений,
содержащих всегда значительное количество «готовых» гумусовых
веществ.
Рассматривая состав и природу органической части
почвы, автор сосредоточил основное внимание на характеристике
гумусовых веществ, составляющих наиболее важную часть любых
скоплений органогенных тел в природе и являющихся основным
компонентом почвенного гумуса. Подчеркнута необходимость
четкой унификации терминологии при характеристике
органической части почвы, а также номенклатуры процессов, их
обусловливающих. Нельзя пользоваться как идентичными терминами «гумус»
и «органическое вещество почвы», «гумус» и «гумусовые вещества»,
«гумусообразование» и «гумификация». Гумус — основная
составная часть минеральных почв — состоит из гумусовых кислот
и их органо-минеральных производных, веществ исходных
органических остатков и продуктов трансформации этих двух групп
соединений.
Общепризнанны и несомненны специфичность и
самостоятельность гумусовых веществ как особого класса
высокомолекулярных азотсодержащих органических кислот циклического строения,
образующихся в процессе гумификации. Этот процесс
универсален везде, где создаются условия, благоприятные для
жизнедеятельности гетеротрофных микроорганизмов. Они
накапливаются в составе гумуса и лесной подстилки минеральных почв,
в торфе болот, в сапропеле и других илах на дне водоемов,
в различных компостах, приготовленных человеком.
К настоящему времени накоплен большой фактический
материал по характеристике гумусовых веществ, образующихся
256
в форме кислот, хотя все еще недостаточно унифицирована их
номенклатура. Наиболее рационально различать в их составе
две группы: группу гуминовых кислот и группу фульвокислот,
используя эти термины только во множественном числе как
групповые понятия. Эта группировка хорошо соответствует основным
отличиям в пределах каждой группы по составу и свойствам, а
следовательно, и по функциям в почвообразовании. Группа
гуминовых кислот в основном накапливается на месте своего
образования, фульвокислоты легко мигрируют благодаря хорошей
растворимости. Аккумуляция и миграция осуществляются в
большинстве случаев в форме различных органо-минеральных
производных. Термины «ульминовые», «гиматомелановые», «бурые
гуминовые», «серые гуминовые», «креповые», «апокреновые»,
«фульвановые», «фульвиновые» и другие следует рассматривать
как фракционные, исторически сложившиеся в период
развития учения о почвенном гумусе. Дальнейшее использование
термина «гумин» нецелесообразно из-за неоднородности его
состава и отсутствия реальных отличий от гуминовых кислот и
фульвокислот.
Характерная особенность класса гумусовых кислот,
отличающая их от любого другого класса природных органических
соединений, — специфичность элементного состава, обязательное
наличие кислых групп, разных по кислотной силе, присутствие
в молекуле ароматических, гетероциклических и алифатических
группировок, многообразие форм азота, гетерогенность по ММ
и химическому составу при сохранении однотипности строения.
Гуминовые кислоты более обуглерожены и ароматизированы
по сравнению с фульвокислотами, элементный состав их
колеблется в относительно узких пределах. Основными носителями
кислотных свойств являются карбоксильные и фенолгидрок-
сильные группы. Использование разнообразных методов частичной
деструкции позволило установить наличие одночленных и
многочленных ароматических колец различных степеней замещения
типа фенолов и хинонов, азотсодержащие группировки типа
пуриновых и пиримидиновых оснований, остатки ароматических
аминокислот и феноксазонов. Алифатическая часть представлена
аминокислотными и углеводными группировками. До настоящего
времени значительная часть молекулы не идентифицирована,
вследствие чего предложенные схемы строения молекулы
разнообразны и гипотетичны. Имеющиеся в литературе данные
свидетельствуют о резко выраженной полидисперсности ММ,
колеблющейся от 4000-6000 до 50000-70000 и даже 100 000.
Все еще не могут быть объяснены причины колебаний элементного
состава, количества кислых функциональных групп,
варьирования ММ и продуктов частичной деструкции молекулы.
Группа фульвокислот отличается от гуминовых кислот
пониженным содержанием углерода и более высоким количеством
кислорода, причем колебания в элементном составе выражены
17 Л. Н. Александрова 257
более резко. В продуктах частичной деструкции обнаружены
ароматические и алифатические группировки, но доминирующая
роль ^принадлежит последним. Данные о ММ очень разноречивы.
Гетерогенность гумусовых кислот — характерная особенность
этого класса природных соединений, но причины ее все еще не*
достаточно изучены. Гетерогенность гуминовых кислот резко
проявляется в колебаниях ММ, она максимальна в молодых
(новообразованных) и минимальна в наиболее сформированных
гуминовых кислотах (в черноземах). Химическая неоднородность
отдельных фракции изучена плохо; по-видимому, главнейшими
чертами ее являются различная степень окисленности, колебания
в содержании и формах азота, а также разная степень
ароматизации молекулы. Фульвокислоты более гетерогенны как по ММ,
так и по химическому составу и соотношению отдельных
компонентов в молекуле. Причинами гетерогенности гумусовых кислот
следует считать различия в химическом составе самих гумусо-
образователей, а также стадийность и длительность самого
процесса гумификации.
Основной причиной слабой изученности структуры и строения
гумусовых кислот и неясности причин их гетерогенности в пре-
делах каждой группы является отсутствие единой теории их
образования (механизма гумификации). В монографии описаны
главнейшие и очень противоречивые гипотезы, ни одна из которых
не может считаться общепризнанной и вполне достоверной.
Многолетние исследования автора позволили предложить для
обсуждения новую схему гумификации, согласно которой этот
процесс рассматривается как сложная биофизико-химическая
трансформация промежуточных высокомолекулярных продуктов раз-
ложения главнейших компонентов растительных остатков (бел-?
ков, лигнина, углеводов). Основными звеньями гумификации
являются окислительное кислотообразование, формирование азо-
тистой части молекулы, фракционирование системы образующихся
гумусовых кислот, сопровождающееся дальнейшей ароматизацией,
гидролитическим расщеплением, сорбцией. Гумификация -^
длительный процесс, вследствие чего происходит трансформация
самих гумусовых кислот и неизбежна гетерогенность
образующихся продуктов; в процессе дальнейшей гумификации ММ
гумусовых кислот постепенно уменьшается.
Детальное изучение органо-минеральных производных гу-
мусовых кислот, формирование которых — обязательное звено
любого типа почвообразования, позволило предложить их
классификацию, основанную на характере связи между
компонентами, и описать главнейшие условия их аккумуляции и миграции
в процессе почвообразования. Автор различает три основные
группы органо-минеральных производных: гетерополярные соли,
комплексно-гетерополярные соли и сорбционные комплексы.
Первая категория производных представлена гуматами и фульватами
сильных оснований. В зависимости от природы гумусовых кислот,
258
Состава их обменных катионов, а также степени проявления про^
цессов адгезии они накапливаются на месте образования или
мигрируют по профилю. Комплексноггетерополярные соли (в
основном это продукты взаимодействия гумусовых кислот с алюминием
и железом) образуются за счет координационной связи,
благодаря чему металл входит в анионную часть молекулы гумусовых
кислот. Максимальной миграционной способностью обладают
комплексно-гетерополярные соли фульвокислот, что обусловливает
формирование в почвах железисто(алюмо)гумусовых
иллювиальных горизонтов.
Типичные глиногумусовые сорбционные комплексы
формируются в результате межмолекулярного взаимодействия по типу
адгезии между молекулами или ассоциатами гумусовых кислот
или их солей с поверхностью глинистых минералов. Их
миграционная способность, обусловливающая лессиваж, регулируется
как типом водного режима и характером порозности, так и
степенью адгезионного закрепления на поверхности более грубых
скелетных частиц почвы. Вся совокупность этих органо-мине-
ральных производных образует массу органо-минеральных
коллоидов в почве.
До сих пор мало изучены промежуточные продукты
разложения и гумификации гумусообразователей. Лишь в последнее
время появляются работы, свидетельствующие о наличии на
протяжении всего периода разложения и гумификации растительных
остатков двух основных категорий продуктов:
высокомолекулярных, природа которых все еще не идентифицирована, и
низкомолекулярных, в составе которых доминирующее значение по массе
имеют углеводы, фенольные соединения и органические кислоты.
Содержание свободных аминокислот обычно очень незначительно.
Продукты частичной минерализации гумусовых кислот также не
исследованы.
Разнообразие органо-минеральных производных гумусовых
кислот свидетельствует о многогранной роли этого класса
органических соединений в почвообразовании, и прежде всего в
главнейшем итоге всякого почвообразовательного процесса —
развитии плодородия и формировании почвенного профиля.
Гумусовые кислоты и промежуточные продукты разложения
органических остатков участвуют в трансформации минералов в почве
(в синтезе органо-минеральных веществ) и в процессах
аккумуляции многих слагающих массу почвы элементов (в накоплении
макро- и микроэлементов, необходимых для жизни растений).
Образуя водорастворимые и пептизируемые органо-минеральные
продукты, гумусовые кислоты и различные продукты разложения
органических остатков участвуют почти во всех типах
передвижения (миграции) продуктов почвообразования по профилю: в опод-
золивании, выщелачивании, лессиваже, осолонцовывании и осо-
лодении почв. Аккумуляция органо-минеральных производных
как одно из основных звеньев почвообразования приводит к фор-
259
17*
мированию дерновых и серых лесных почв, черноземов,
каштановых и горно-луговых почв, а также многих других типов,
выделяемых в настоящее время.
Трудно переоценить роль органических веществ, и прежде
всего гумусовых кислот и их органо-минеральных производных,
в регулировании уровня плодородия почвы и ее основных
режимов, определяющих это свойство. Наличие органо-минеральных
коллоидов обусловливает поглотительную способность почвы;
участие в структурообразоваиии является причиной изменения
водно-воздушного режима; аккумуляция в форме
органо-минеральных производных всех главнейших элементов питания
регулирует биологическую жизнь почвы и уровень ее питательного
режима. Совершенно естественно поэтому, что сохранение почвы
как производительной силы возможно только при определенном
уровне равновесия между ее органической и минеральной час-
стями. Не менее значительна биогеохимическая роль органо-
минеральных соединений в миграции и концентрации многих
элементов в земной коре, приводящая к формированию тех или
иных геохимических ландшафтов.
В дальнейшем первоочередными задачами являются
следующие.
1. Необходимы исследования природы всех компонентов,
составляющих органическую часть почвы. По-видимому, будет
рационально использовать максимально дробное, но достаточно
мягкое фракционирование каждой группы гумусовых веществ и
промежуточных продуктов их трансформации с последующим
изучением всех основных параметров каждой фракции, а также
моделирование процесса их образования из различных компонентов
растительной и микробной массы.
2. Крайне нужна разработка теории гумификации, что должно
осуществляться путем широкого моделирования этого процесса
как при длительном стационарном исследовании в природных
условиях, так и при четко поставленных модельных лабораторных
экспериментах с учетом химического состава тех или иных
возможных гумусообразователей на фоне заданного биологического
и гидротермического режимов и учета роли минерального
субстрата. При этом особенно необходимы идентификация
высокомолекулярных продуктов разложения и выявление степени
развития окислительных, конденсационных, полимеризационных и
сорбционных процессов,- а также изучение всех этапов самого
процесса гумификации.
3. Исследование природы органо-минеральных производных
целесообразно сосредоточить на уточнении природы алюмо- и
железогумусовых производных, характера их дальнейшей
трансформации в почве. Крайне необходимо изучить условия
образования и природу кремнегумусовых комплексов, а также характер
взаимодействия гумусовых кислот и их солей с минералами почвы
при обязательном исследовании физико-химических поверхности
260
ных явлений на границе раздела этих компонентов. Обязательным
звеном при дальнейшем развитии учения о почвенном гумусе
является также изучение условий образования и природы органо-
минеральных производных промежуточных продуктов
трансформации гумусообразователей и новообразованных гумусовых
кислот.
4. Очень существенно детальное исследование характера
процессов превращения органических удобрений в почвах и их роли
в формировании гумуса в профиле пахотных почв, а также
влияния на почвообразование. Материалы по этой проблеме крайне
ограниченны, что мы показали на примере пахотных дерново-
подзолистых почв Нечерноземья, в которых органические
удобрения будут играть решающую роль как фактор почвообразования.
5. Не менее важна разработка методов изучения органических
веществ почвы и особенно методов выделения и идентификации как
всех основных промежуточных (неспецифических) продуктов
трансформации, так и веществ, образующихся при постепенной
минерализации самих гумусовых веществ.
Автор не освещает в монографии роли гумуса в
почвообразовании и плодородии, хотя эти вопросы имеют первостепенное
значение для теории почвообразования и системы мероприятий
по наиболее рациональному использованию в народном
хозяйстве. Эта проблема очень обширна и требует специального
исследования.
ЛИТЕРАТУРА
1. Александрова Я. В. Об использовании гуминовых кислот
микроорганизмами. —* Почвоведение, 1953, №6.
2. Александрова Л. Я. Применение ацетилбромида для изучения гумифи-
цированных веществ почвы. — Учен. зап. Ленингр. ун-та. Сер. ГПГ,
1937, № 16.
3. Александрова Л. Я. К характеристике углеводов в органическом
веществе чернозема. — Учен. зап. Ленингр. ун-та. Сер. геол.-почв. наук, 1939,
№ 34, вып. 7.
4. Александрова Л. Я. К характеристике органической и минеральной
части почвенных коллоидов. — Учен. зап. Ленингр. ун-та. Сер. геол.-
почв. наук, 1944, № 71.
5. Александрова Л, Я. Гумус как система полимерных соединений. —
В кн.: Тр. юбил. сессии, посвящ. 100-летию со дня рождения В. В.
Докучаева. М., Изд-во АН СССР, 1949.
6. Александрова Л. Я. О природе и свойствах продуктов взаимодействия
гуминовых кислот и гуматов с полутораокисями. — Почвоведение, 1954,
№1.
7. Александрова Л. Я. Процессы взаимодействия гуминовых веществ с
минеральной частью почвы. — Почвоведение, 1954, № 9.
8. Александрова Л. Я. О природе перегноя. — Зап. Ленингр. с.-х. ин-та,
1955, вып. 9.
9. Александрова Л. Я. О применении пирофосфата натрия для выделения из
почвы свободных гумусовых веществ и их органо-минеральных
соединений. — Почвоведение, 1960, №2.
10. Александрова Л. Я. Органо-минеральные соединения и органо-минераль-
ные коллоиды в почве. — В кн.: Докл. сов. почвоведов к VII Междунар.
конгр. почвоведов в США. М., Изд-во АН СССР, 1960.
-10а. (Александрова Л. Я.) Alexandrova L. N. On the composition of humus
substances and the Nature of organo-mineral colloids in soil. -* In: Trans.
7th Intern, congr. soil sci. Madison, USA, 1960, vol. II.
'И. Александрова Л. Я. Современные представления о природе гумусовых
веществ почвы и их органо-минеральных производных. — В кн.: Проблемы
почвоведения. М., Изд-во АН СССР, 1962.
12. Александрова Л. Я. О механизме образования гумусовых веществ и
процессах превращения их в почве. — Зап. Ленингр. с.-х. ин-та, 1966, т. 105,
вып. 1.
13. Александрова Л. Я. Органо-минеральные производные гумусовых кислот
и методы их изучения. — Почвоведение, 1967, № 7.
14. Александрова Л. Я. Вероятные пути образования двухкомпонентной
системы гумусовых веществ в почве. — В кн.: Химия, генезис и
картография почв. М., «Наука», 1968.
15. Александрова Л. Я. Изучение процессов гумификации растительных остат-
• ков и природы гумусовых веществ. — Почвоведение, 1972, № 7.
16. Александрова Л. Я. О природе новообразованных гуминовых кислот и
вероятном механизме их образования. — В кн.: Тр. I Нац. конгр. по
почвоведению. София, 1972.
262
17. Александрова Л. Я. О номенклатуре, применяемой в учении о почвенном
гумусе. — Почвоведение, 1975, № 2.
18. Александрова Л. Я. Некоторые дискуссионные вопросы механизма
гумификации органических остатков в почве. — Зап. Ленингр. с.-х. ин-та,
1975, т. 269.
19. Александрова Л. Я. Гумусовые вещества почвы (их образование, состав,
свойства и значение в почвообразовании и плодородии). — Зап. Ленингр.
с.-х. ин-та, 1970, т. 142.
20. Александрова Л. II., Андреева И. М. О процессах превращения
гумусовых веществ в почве. — Почвоведение, 1963, № 7.
21. Александрова Л. Я., Аристовская Т. В., Кауричев И. С, Пономарева В. В.
Моделирование почвенных процессов. — В кн.: Тез. докл. на IV Все-
союз. делегатском съезде почвоведов. Алма-Ата, 1970, кн. 1.
22. Александрова Л. Я., Аршавская В. Ф. Изменение состава гумусовых
кислот в процессе гумификации растительных остатков. — Зап. Ленингр.
с.-х. ин-та, 1968, т. 117, вып. 1.
22а. (Александрова Л. Я., Аршавская Я. Ф., Дорфман Э. М., Люжин М. Ф.„
Юрлова О. В.) Alexandrova L. N., Arshavskay V. F., Dorfman E. M.y
Lyuzin M. F., Yurloua 0. V. Humus acids and their organo-mineral
derivatives in soil. — In: Trans. 9th Intern, congr. soil sci. Australia, 1968*
vol. III.
23. Александрова Л. Я., Зверева Т. С, Фомин Ю. И. О формировании ор~
гано-минеральных каллоидов. — В кн.: Тр. X Междунар. конгр.
почвоведов. М., «Наука», 1974, т. II.
24. Александрова Л. Я., Люжин М. Ф. Влияние условий разложения на
соотношение процессов минерализации и гумификации растительных
остатков. — Зап. Ленингр. с.-х. ин-та, 1966, т. 105, вып. 1.
25. Александрова Л. Я., Мясникова А. М. К методике определения
целлюлозы в растительных материалах и почве. — Почвоведение, 1940, № 9.
26. Александрова Л. Я., Надь М. О природе органо-минеральных коллоидов
и методах их изучения. — Почвоведение, 1958, № 10.
27. Александрова Л. Я., Назарова А. В. Гетерогенность гуминовых кислот и
ее происхождение. — В кн.: Проблемы почвоведения. М., «Наука», 1978.
28. Александрова Л. Я., Найденова О. А. Лабораторно-практические занятия
по почвоведению. 3-е изд. Л., «Колос», 1976.
29. Александрова Л. Я., Румянцева Э. А. Изменение новообразованных
гуминовых кислот в процессе их дальнейшей гумификации. — Зап.
Ленингр. с.-х. ин-та, 1970, т. 137, вып. 4.
30. Александрова Л. Я., Румянцева Э. А. О процессах дальнейших
превращений новообразованных гуминовых кислот. — Зап. Ленингр. с.-х. ин-та,
1974, т. 237.
31. Александрова Л. Я., Фомин Ю. Я. О механизме взаимодействия глинистых
минералов с гуминовыми кислотами и продуктами разложения
растительных остатков. — Зап. Ленингр. с.-х. ин-та, 1972, т. 165, вып. 2.
32. Александрова Л. Я., Фомин Ю. И. Термографические исследования форм
связи монтмориллонита с продуктами гумификации растительных
остатков. — Зап. Ленингр. с.-х. ин-та, 1973, т. 206.
33. Александрова Л. Я., Юрлова О. В., Лобицкая Л. В. Распределение и
состав гумусовых веществ и их органо-минеральных производных в
гранулометрических фракциях некоторых типов почв. — Зап. Ленингр.
с.-х. ин-та, 1966, т. 105, вып. 1.
34. Алиев С. А. Классификация группового состава и физико-химических
свойств гумуса почв. — В кн.: Тез. докл. на V делегатском съезде Все-
союз. о-ва почвоведов. Минск, 1977, т. 2.
35. А лиев (•. А., Касимов Р. М. Парамагнитные свойства гуминовых кислот
Азербайджанской ССР. — Почвоведение, 1971, № 1.
36. Андреева Я. М. О процессах превращения воднорастворимых гумусовых
веществ. — Зап. Ленингр. с.-х. ин-та, 1966, т. 105, вып. 1.
37. Андреева Я. М. Процессы превращения гумусовых веществ в почве..
Канд. дис. Ленингр. с.-х. ин-т, 1966.
263
38. Андреева И. М. О процессах минерализации гумусовых веществ. —
Зап. Ленингр. с.-х. ин-та, 1968, т. 117, вып. 1.
39. Антипов-Каратаев И. Н., Келлерман В. В., Хан Д. В. О почвенном
агрегате и методах его исследования. М., Изд-во АН СССР, 1948.
40. Аринушкина Е. В. Руководство по химическому анализу. М., Изд-во
Моск. ун-та, 1961.
41. Аристовская Г. В. О разложении органо-минеральных соединений в
подзолистых почвах. — Почвоведение, 1963, № 1.
42. Аристовская Т. В. Численность, биомасса и продуктивность почвенных
бактерий. -— В кн.: Ресурсы биосферы. М., «Наука», 1975, вып. 1.
43. Аршавская В. Ф. Изменение состава и свойств новообразованных гуми-
новых кислот в процессе их дальнейшей гумификации. Канд. дис.
Ленингр. с.-х. ин-т, 1968.
44. Аршавская В. Ф. Сравнительная оценка методов определения
функциональных групп в гумусовых кислотах. — Зап. Ленингр. с.-х. ин-та,
1968, т. 117, вып. 1.
45. Асеева И. В., Паников Н. С, Чурсина О. Т. Содержание и состав
нуклеиновых кислот в дерново-подзолистой почве. — Вестн. Моск. ун-та.
Почвоведение, 1977, № 1.
46. Б амбалов Н. Н. Биохимические изменения гуминовых кислот при
гумификации торфяной залежи. — В кн.: Тез. науч. докл. 1-й конф. Моск.
ун-та. М., 1967.
47. Бабанин В. Ф., Ильин Н. 77., Орлов Д. С, Федотова Т. В,, Яблонский О. 77.
О природе линий в спектрах ЭПР гумусовых кислот. — Почвоведение,
1977, № 1.
48. Барановский И. Н. Процессы превращения органических удобрений
в дерново-подзолистых суглинистых почвах. Канд. дис. Ленингр. с.-х.
ин-т, 1974.
49. Барановский И. Н. Состав воднорастворимых продуктов, мигрирующих
из пахотного слоя дерново-подзолистых суглинистых почв. — Зап.
Ленингр. с.-х. ин-та, 1974, т. 237.
50. Барановский И. Н. Влияние равновеликих доз навоза и ТМАУ на
содержание и состав гумуса в дерново-подзолистой суглинистой почве. —
Науч. тр. Ленингр. с.-х. ин-та, 1976, т. 296.
51. Барс Е. А., Коган С. С. Органическое вещество подземных вод
нефтегазоносных областей. М., «Недра», 1965.
52. Басоло Ф., Джонсон Р. Химия координационных соединений. Пер.
с англ. М., «Мир», 1966.
53. Бацула А. А., Крупский Я. К. Формы азота гумусовых веществ
некоторых целинных и освоенных почв левобережной Украины. —
Почвоведение, 1974, № 8.
54. Беллами Л. Инфракрасные спектры сложных молекул. М., ИЛ, 1963.
55. Белъчикова Н. П. Определение органического углерода, Fe203 и А1203
в вытяжках 0.1 н. Na4P207. — Почвоведение, 1970, № 6.
56. Берестецкий О. А., Торжевский В. И. Численность и биомасса
микроорганизмов в основных типах почв Украинской СССР. — В кн.:
Закономерности развития почвенных микроорганизмов. М., Изд-во АН СССР,
1975.
57. Бикбулатов Э. С, Скопинцев Б. А. Определение общего содержания
растворенных углеводов в природных водах в присутствии гумусовых
веществ. — Гидрохим. матер., 1974, т. 60.
58. Блажей А., Шутый Л. Фенольные соединения растительного
происхождения. Пер. со словац. М., «Мир», 1977.
59. Бобрицкая Т. А. Роль однолетних культурных растений в балансе
органических и минеральных веществ в почве. — Почвоведение, 1958, № 1.
60. Боул С, Хоул Ф., Мак-Крекен Р. Генезис и классификация почв. Пер.
с англ. М., «Прогресс», 1977.
61. Брагин А. М., Романенкова Т. М., Шугля 3. М., Кондрыко В. Д.
Длительное применение удобрений и плодородие почвы. — В кн.: Проблемы
накопления и использования органических удобрений. Минск, 1976,
264
62. Брауне Ф. Э., Брауне Д. А. Химия лигнина. Пер. с англ. М., «Лесная
пром-сть», 1964.
63. Брунауэр С. Адсорбция газов и паров. М., ИЛ, 1948.
64. Былинкина В. Я. Микроорганизмы, минерализация гумусовых веществ
почв. — В кн.: Тр. Всесоюз. науч.-исслед. ин-та с.-х. микробиол. за
1941—1945 гг. Л., Сельхозгиз, 1949.
65. Ваксман С. Гумус. М., Сельхозгиз, 1937.
66. Вильяме В. Р. Почвоведение. Вып. 1. М., Кн. изд-во студентов Моск.
с.-х. ин-та, 1914.
66а. Вильяме В. Р. Почвоведение. Собр. соч., М., 1950, т. 5.
67. Вильяме В, Р. Земледелие с основами почвоведения. 4-е изд. М.,
Сельхозгиз, 1938.
68. Владимирова М. II. Изменение содержания и состава гумуса в дерново-
подзолистых почвах при внесении высоких доз органических
удобрений. — Зап. Ленингр. с.-х. ин-та, 1972, т. 165, вып. 2.
69. Волобуев В. Р. Система почв мира. Баку, «Элм», 1973.
70. Волощук 3. Ф., Жоробекова III., Бугу баев Я. Б, Некоторые данные
о строении гуминовых кислот. — Науч. докл. высш. школы. Биол.
науки, 1973, № 12.
71. Галле Р. Р., Николаев А. Г. К вопросу об определении конститутивных
групп в гуминовых кислотах. — Журн. прикл. хим., 1939, т. 12.
72. Ганжара Я. Ф. Фракционирование гумусовых веществ почв методом
гелевой фильтрации. — Докл. Тимиряз. с.-х. акад., 1969, вып. 149.
73. Ганжара Н. Ф., Кауричев Я. С, Рассохина В. В. Влияние поглощенных
катионов на процесс гумусообразования. — В кн.: Особенности
почвенных процессов дерново-подзолистых почв. М., 1977.
74. Гапон Е. Я. Адсорбция ионов и молекул коллоидной фракцией почвы
и строение почвенных коллоидов. — В кн.: Почвенно-поглощающий
комплекс и вопросы земледелия. М., 1937.
75. Гедройц К. К. Химический анализ почвы. М., Сельхозгиз, 1955, т. 2,
гл. VI.
76. Герасимов И. Я., Г лазовская М. А. Основы почвоведения и география
почв. М., Географгиз, 1960.
77. Герман Р. О химическом исследовании черноземных почв для
определения различных свойств их в наших южных губерниях. — Землед. журн.
Моск. о-ва сельск. хоз-ва, 1837, № 5.
78. Гиляров М. С. Роль почвенных животных в формировании гумусового
слоя почв. — Усп. соврем, биол., 1951, т. 31, №2.
79. Гиляров М. С. Зоологический метод диагностики почв. М., «Наука»,
1965.
80. Глазовская М. А. Почвы мира. М., Изд-во Моск. ун-та, 1972, т. I.
81. Глебко Л. Я., Максимов О. Б. О функциональном анализе гуминовых
кислот. — В кн.: Новые методы исследования гуминовых кислот.
Владивосток, 1972.
82. Глебко Л. Я., Максимов О. Б., Улькина Ж. Я., Кошелева Л. Я.,
Василевская Я. А. Новые методы определения функциональных групп гуминовых
кислот. — В кн.: Новые методы исследования гуминовых кислот.
Владивосток, 1972.
83. Глинка К. Д. Почвоведение. 4-е изд. М., Сельхозгиз, 1931.
84. Горбунов Я. Я. Минералогия и коллоидная химия почв. М., «Наука»,
1974.
85. Горбунов Я. Я., Орлов Д. С. Природа и прочность связи органических
веществ с минералами почвы. — Почвоведение, 1977, № 7.
86. Гринберг А. А. Введение в химию комплексных соединений. 4-е изд. М.,
«Химия», 1971.
87. Гришина Л. Л., Орлов Д. С. Система показателей гумусного состояния
почв. — Тез. докл. на V делегатском съезде Всесоюз. о-ва почвоведов.
Минск, 1977, т. 2.
88. Гукерт Я., Валла М., Жакен Ф. Адсорбция гуминовых кислот и
полисахаридов в почве на монтмориллоните. — Почвоведение, 1975, № 2.
265
89. Гумус и почвообразование. — Науч. тр. (Записки) Ленингр. с.-х. ин-та*
1972, т. 165, вып. 2; 1973, т. 206; 1974, т. 237; 1976, т. 296; 1977, т. 329;
1978, т. 354.
*90. Густавсон Г. (работа ст. Гаврилова). О составе гумусового вещества
чернозема. — Журн. Рус. физ.-хим. о-ва, 1883, т. 15, вып. 1.
91. Детерман Г. Гель-хроматография. М., «Мир», 1970.
92. Дмитриев Е. А. Математическая статистика в почвоведении. М., Изд-во
Моск. ун-та, 1972.
93. Докучаев В. В. Русский чернозем. Спб., 1883.
94. Долотов В. А. Специфика биологического круговорота элементов в
подзолистых почвах разных угодий. — В кн.: Биогеохимические процессы
в подзолистых почвах. Л., «Наука», 1972.
95. Дорфман Э. М. Природа соединений ионов железа и алюминия с
гумусовыми кислотами почв. Канд. дис. Моск. ун-т, 1968.
96. Драгунов С. С, Высоцкая П. Н. Химические исследования гумусовых
веществ некоторых почв. — Почвоведение, 1953, № 4.
97. Драгунов С. С, Желоховцева Н. Я., Стрелкова Е. Я. Сравнительное
исследование почвенных и торфяных гуминовых кислот.— Почвоведение,.
1948, № 7.
98. Драгунов С. С, Мурзаков Б. Г. Гетерогенность фульвокислот
обыкновенного чернозема. — Почвоведение, 1970, № 3.
99. Дубин В. Я. Применение сефадексов для фракционирования
гуминовых кислот. — Науч. докл. высш. школы. Биол. науки, 1968, №11.
100. Дубин В, Я., Филъков В. А. Фракционирование гуминовых кислот
некоторых почв Молдавии фильтрацией через сефадексы. —
Почвоведение, 1968, № 5.
101. Дьяконова К. В, Железогумусовые комплексы и их роль в питании
растений. — Почвоведение, 1962, № 7.
102. Дьяконова К. В. Природа гумусовых веществ почвенного раствора, их
динамика и методы изучения. — Почвоведение, 1964, № 4.
103. Дэвис Д., Джованелли Дж., Рис Т. Биохимия растений. Пер. с англ*
М., «Мир», 1966.
104. Дюшофур Ф. Основы почвоведения. Эволюция почв. Пер. с франц. М.,
«Прогресс», 1970.
105. Ершова А. Я. Некоторые данные по изучению разлагающего действия
гумусовых кислот на минералы. — В кн.: Кора выветривания, Л.,
1968, № 10.
106. Ефимов В. Я., Василъкова М. Г. К методике выделения гумусовых
веществ из торфяных почв. — Почвоведение, 1970, № 5.
107. Жигунов A. Я, Симаков В. Н. Состав и свойства гуминовых кислот,
выделенных из разлагающихся растительных остатков. —
Почвоведение, 1977, № 12. '
108. Запрометов М. Я. Фенольные соединения и методы их исследования. —
В кн.: Биохимические методы в физиологии растений. М., 1971.
109. Запрометова К. М., Мирчина Т. Г., Орлов Д. С, Юхнин А, А,
Характеристика черных пигментов темноокрашенных почвенных грибов. —
Почвоведение, 1971, № 7.
110. Зверева Т. С, Базилинская М. В. О взаимодействии воднорастворимого
вещества с бентонитом. — Почвоведение, 1976, №11.
111. Зонн С* В, О роли глинистых А1-, Fe-минералов в диагностике
современных процессов коро- и почвообразования. — Почвоведение, 1971, № 12.
112. Зонн С. В., Маунг Вин-Хин. О формах железа, методах их определения
и значении для диагностики тропических почв. — Почвоведение, 19711
№5.
ИЗ. Зырин Я. Г. Гумин органического вещества. — Вестн. Моск. ун-та,
1948, № 1.
113а. История агрикультуры почв. М., Изд-во АН СССР, 1940.
114. Каплин В. Г., Панченко С. ?., Шлычкова В. В. О сорбции многоатомных
фенолов и нафтолов ионообменными смолами. — Гидрохим. матер.,..
1967, т. 43.
266
115. Карамщук 3. П. Свободные аминокислоты в темно-каштановой почве
Северного Казахстана. — Почвоведение, 1975, № 11.
116. Карпухин А. И, Исследование взаимодействия фульвокислот и ионов
железа методом радиоактивных индикаторов. Автореф. канд. дис. М.,
1970.
117. Карпухин А. И., Фокин А. Д. Фракционный состав фульвокислот
некоторых типов почв. — Изв. Тимиряз. с.-х. акад., 1971, № 3.
118. Карпухин А. И.ч Фокин Л. Д. Включение продуктов разложения
растительных остатков (меченных 14С) в гумусовые вещества. —
Почвоведение, 1974, № 11.
119. Карпухин А. If., Фокин А. Д. Применение гелевой хроматографии для
изучения фульвокислот и железофульватных соединений. — В кн.:
Особенности почвенных процессов дерново-подзолистых почв. М., 1977.
120. Касаточкин В. И. О строении карбонизованных веществ. — Изв.
АН СССР, 1953, № 10.
121. Касаточкин В. И.., Жоробекова Ш. Ж., Волощук 3. Ф. Электрофоторе-
тические фракции и молекулярная структура гуминовых кислот
торфа. — В кн.: Тез. докл. к совещ. по физ. хим. торфа. Минск,
1977.
122. Касаточкин В. if., Кононова М. М.9 Ларина Н. К., Егорова О. И.
Спектральное и рентгеновское исследование химического строения
гумусовых веществ почвы. — В кн.: Физика, химия, биохимия и
минералогия почв СССР. М., «Наука», 1964.
123. Кауричев И. С. Особенности генезиса почв временного избыточного
увлажнения. Докт. дис. Тимиряз. с.-х. акад., 1965.
124. Кауричев И. С. Практикум по почвоведению. М., «Колос», 1973.
125. Кауричев И. С, Базилинская И, В, О миграции органического
вещества и железа в почвах солонцового комплекса лесостепи Западной
Сибири. — Изв. Тимиряз. с.-х. акад., 1965, № 4.
126. Кауричев И. С, Карпухин А. И., Степанова Л. П. Изучение
водорастворимых железоорганических соединений подзолистых и дерново-
подзолистых почв. — В кн.: Особенности почвенных процессов дерново-
подзолистых почв. М., 1977.
127. Кауричев И. С, Карпухин А. И., Степанова Л, П. Водорастворимые
железоорганические соединения почв таежной лесной зоны. — В кн.:
Проблемы почвоведения. М., «Наука», 1978.
128. Кауричев Л. С, Ноздрунова Е. М. О содержании низкомолекулярных
кислот в составе воднорастворимых органических веществ почв. —
Почвоведение, 1965, № 3.
129. Кауричев И. С, Ноздрунова Е. М., Цюрупа И. Г. Хелатные железо-
органические соединения в почвах. — В кн.: Докл. к VIII Междунар.
конгр. почвоведов. М., «Наука», 1964.
130. Кауричев И. С, Фролова Л. Н. Воднорастворимые органические вещества
индивидуальной природы в лесных подстилках. — Докл. Тимиряз.
с.-х. акад., 1965, т. 115, вып. 2.
131. Кауричев И. С, Яшин И. М., Кашинский А. Д. Применение метода
лизиметрических хроматографических колонок в почвенных
исследованиях. — В кн.: Методы стационарного изучения почв. М., «Наука»,
1977.
132. Качинский Н. А. Корневая система растений в почвах подзолистого
типа. — Тр. Моск. обл. с.-х. опытной станции, 1925, № 7.
133. Клейст X. Физические исследования гуминовых кислот и их
соединений с металлами. — Науч. докл. высш. школы. Виол, науки, 1960,
№5.
134. Ковалев В. А., Генералова В. А. О взаимодействии гуминовых
и фульвокислот торфяных почв с железом. — Почвоведение, 1967,
№ 9.
135. Ковда В. А. Основы учения о почвах. М., «Наука», 1973, т. 1.
136. Козловский С, О растворимости перегноя. — Матер, по изучению рус.
почв, 1893, вып. 8.
267
137. Колесников Af. Я. Молекулярно-весовое распределение гуминовых
кислот по данным гельхроматографии на сефадексах. — Почвоведение,
1978, № 4.
438. Комиссаров И. Д., Логинов Л. Ф. Структурная схема и моделирование
макромолекул гуминовых кислот. -— Науч. тр. Тюменск. с.-х. ин-та,
1971, т. XIV.
139. Комиссаров И. Д., Стрельцова И. Я. Влияние способа извлечения
гуминовых кислот из сырья на химический состав получаемых
препаратов. — Науч. тр. Тюменск. с.-х. ин-та, 1971, т. XIV.
140. Кононова М. М. К изучению процесса новообразования гумусовых
веществ. — Почвоведение, 1944, № 10.
141. Кононова М. М. О биохимизме процесса образования гуминовых
кислот. — Микробиология, 1949, т. 18, вып. 2.
142. Кононова М. М. Проблема почвенного гумуса и современные задачи его
изучения. М., Изд-во АН СССР, 1951.
143. Кононова М. М. Органическое вещество почвы. М., Изд-во АН СССР,
1963.
143а. (Кононова М. Af.) Kononowa Af. M. Gedanken zur Nomenklatur der im
Boden vorhandenen organischen Stoffe. — Im: Trans. 8th Intern, congr.
soil sci. Bucharest, 1964, vol. II.
144. Кононова Af. Af. Процессы превращения органического вещества и их
связь с плодородием почвы. — Почвоведение, 1968, № 8.
145. Кононова М. М. Проблема органического вещества почвы на
современном этапе. — В кн.: Органическое вещество целинных и освоенных почв.
М., «Наука», 1972.
146. Кононова М. М., Александрова Я. В. Применение метода
распределительной хроматографии на бумаге при изучении форм азота гумусовых
веществ. — Почвоведение, 1956, № 5.
147. Кононова М. Af., Александрова Я. В. Процессы гумусообразования как
звено круговорота углерода в почве. — В кн.: Тр. X Междунар. конгр.
почвоведов. М., «Наука», 1974, т. II.
148. Кононова М. Af., Александрова И. В., Белъчикова Я. Я. Биосинтез
гумусовых веществ и их превращение в процессе почвообразования. —
В кн.: Докл. сов. почвоведов к VII Междунар. конгр. почвоведов
в США. М., Изд-во АН СССР, 1960.
149. Кононова М. Af., Александрова И. В., Ларина Я. К. Включение
органического азота в гуминовые кислоты в процессе их формирования. —
В кн.: Проблемы почвоведения. М., «Наука», 1978.
150. Кононова М. Af., Александрова Я. В., Титова Я. А. Разложение
силикатов органическими веществами почвы. — Почвоведение, 1964, № 10.
151. Кононова М. Af., Белъчикова Я. Я. Ускоренные методы определения
состава гумуса минеральных почв. — Почвоведение, 1961, № 10.
152. Кононова М. Af., Белъчикова Я. Я. Применение Na пирофосфата для
выделения и характеристики железо- и алюмоорганических соединений
почвы. — Почвоведение, 1970, № 6.
153. Кононова М. Af., Титова Я. А, Применение электрофореза на бумаге
для фракционирования гумусовых веществ почвы и изучения их
комплексных соединений с железом. — Почвоведение, 1961, № 11.
154. Короткое А. А. Процессы накопления и выноса веществ в дерново-
подзолистых пахотных и луговых почвах. Докт. дис. Ленингр. с.-х.
ин-т, 1970.
155. Короткое А. А., Ипполитова Л. Ф. О круговороте элементов питания
в дерново-подзолистых пахотных песчаных почвах. — В кн.: Науч.
тр. Северо-Западного науч.-исслед. ин-та сельск. хоз-ва. Л., 1977.
156. Корюшкина С. Р. Природа и свойства фульвокислот основных типов
почв. Канд. дис. Ленингр. с.-х. ин-т, 1976.
157. Корюшкина С. Р., Садовникова Л. К. Об участии углеводов в составе
фульвокислот. — Зап. Ленингр. с.-х. ин-та, 1975, т. 269.
158. Костычев Я. А. Образование и свойства перегноя. — Тр. Спб. о-ва есте-
ствоиспыт., 1889, т. 20, отд. бот.
268
159. Костцчев П. А, О некоторых свойствах и составе перегноя. — Сельск.
хоз-во и лесоводство, 1890, № 10.
160. Костычев Я. А. Почвы черноземной области России, их происхождение,
состав и свойства. Спб., 1885, ч. 1.
161. Красильников Я. А. Микроорганизмы почвы и высшие растения. М.,
Изд-во АН СССР, 1958.
162. Кревелен Ван, Графостатический метод изучения структуры и
процессов образования углей. — В кн.: Химия твердого топлива. М., 1961,
т. П.
163. Кретович В, Л. Основы биохимии растений. М., «Высш. школа», 1964.
164. Крупский Л. #., Черников В. А., Кончиц В. А. Элементный состав и ИК
спектры фульвокислот, фракционированных методом гельхроматогра-
фии. — Изв. Тимиряз. с.-х. акад., 1976, № 4.
165. Крупина Г, Я. Изменение содержания некоторых воднорастворимых
органических соединений в процессе разложения растительных остат^
ков. — Науч. тр. Ленингр. с.-х. ин-та, 1978, т. 354.
166. Кудрина О. С. Влияние гуминовых кислот на некоторые группы
почвенных микроорганизмов и их значение для этих организмов как
источник питательных веществ. — Тр. Почв, ин-та АН СССР, 1951, т. 33.
167. Кулаковская Т. Н. Современные данные о роли органического вещества
в плодородии почв. — В кн.: Проблемы накопления и использования
органических удобрений. Минск, 1976.
168. Купревич В, Ф.у Ивашкевич Т. Г., Щербакова Т. Я. Свободные
аминокислоты в почве. — ДАН БССР, 1962, т. 6, № 8.
169. Купревич В. Ф., Щербакова Т. Я. Почвенная энзимоЛогия. Минск,
1966.
170. Кухаренко Т, А. Реакции гуминовых кислот с нейтральными солями. —
Хим. твердого топлива, 1937, т. 8.
171. Кухаренко Т. Л., Беликова В, Я., Мотовилова Л, В. Исследование
структуры гуминовых кислот окислением перманганатом калия в
щелочной среде. '— Почвоведение, 1969, № 2.
172. Кухаренко Т. А., Екатеринина Л. Я. Гиматомелановые кислоты
ископаемых углей. — Почвоведение, 1960, № 12.
173. Кухаренко Т. Л., Екатеринина Л. Я. Окисление гиматомелановых и
гумусовых кислот углей перманганатом калия в щелочной среде. —
Почвоведение, 1965, № 11.
174. Кухаренко Т. А., Екатеринина Л. Я. Метод определения хиноидных
групп в гуминовых кислотах. — Почвоведение, 1967, № 7,
175. Ландсберг Г. Я. Изменение состава органических веществ торфяных
почв Северо-Запада СССР при их сельскохозяйственном использовании.
Канд. дис. Ленингр. с.-х. ин-т, 1973.
176. Ларина Я. Я., Касаточкин Я. Я. Ионный обмен и строение гуминовых
кислот. — Почвоведение, 1957, № 9.
177. Ларина Я. #., Касаточкин В, Я. Спектральные методы исследования
гуминовых веществ почвы. — В кн.: Физико-химические методы
исследования почв. М., «Наука», 1966.
178. Левашкевич Г. А. Взаимодействие гумусовых кислот с гидроокисями
железа и алюминия. — Почвоведение, 1966, № 4.
179. Левин Ф. Я. Окультуривание подзолистых почв. М., «Колос», 1972.
180. Левина В. Я., Конасова А, А., Белокурова А. Я., Серова Л. В.
Биологическая продуктивность сельскохозяйственных культур и круговорот
элементов питания в севооборотах на дерново-подзолистых почвах
разной окультуренности. — В кн.: Науч. тр. Северо-Западного науч.-ис-
след, ин-та сельск. хоз-ва. Л., 1977.
181. Лейн 3. Я. Обменная способность различных веществ гумуса. — Тр*
Почв, ин-та АН СССР, 1940, т. 23. . .
182. Ленинджер А. Биохимия. Пер. с англ. М., «Мир», 1976.
183. Лесневский С, Сравнительное определение растворимости перегноя
различных почв в воде и несколько данных о составе гуминовой кислоты. —
Зап. Ново-Александр, ин-та, 1897, т. X, вып. 2,
269
/
!
184. Липовский Я. Критический разбор мнений ученых об условиях
плодородия земли. Спб., 1846.
185. Лиштван Я. Я., Круглицкий Я. Я., Третинник В. Ю.
Физико-химическая механика гуминовых веществ. Минск, «Наука и техника», ;
1976.
186. Лукошко Е. С, Раковский В. Е. Механизм образования гуминовых
веществ в процессе торфообразования. — В кн.: Химия и генезис торфа
и сапропелей. Минск, Изд-во АН БССР, 1962.
187. Лыков А. М. Органические вещества и плодородие дерново-подзолистых
почв в условиях интенсивного земледелия. Автореф. докт. дис. М., 1976.
188. Люжин М. Ф. Минерализация и гумификация растительных остатков
в почве. Канд. дис. Ленингр. с.-х. ин-т, 1968.
189. Люжин М. Ф. Минерализация и гумификация растительных остатков
в почве. — Зап. Ленингр. с.-х. ин-та, 1968, т. 117, вып. 1.
190. Люжин М. Ф. Влияние условий разложения на процессы
минерализации растительных остатков и изменение их химического состава. — Зап.
Ленингр. с.-х. ин-та, 1970, т. 137, вып. 4.
191. Мазур Т. Я., Смирнова Я. Я. Применение метода бумажной
хроматографии для изучения превращения свободных аминокислот в почве. —
Докл. Тимиряз. с.-х. акад., 1963, вып. 84.
192. Максимов О. Я., Ребачук Я. М., Гонтарь Я. Использование диметил-
сульфоксида при экстракции гуминовых кислот из природных
объектов. — Почвоведение, 1974, № 2.
193. Малянов А. Я. Физические свойства почв и корневые системы растений
Башкирии в пределах юго-западного Предуралья. — Учен. зап. Моск.
ун-та, 1937, вып. 12.
194. Мамченко О. А. Свободные аминокислоты в некоторых почвах
Украины. — Почвоведение, 1970, № 2.
195. Шанская СМ., Дроздова Т. В. Геохимия органического вещества. М.,
«Наука», 1964.
196. Манская С М., Дроздова Т. В., Емельянова М. Я. Связывание меди
различными формами природных органических соединений. —
Почвоведение, 1958, № 6.
197. Матвеева В. И., Зенюк, Е. В., Переднее Я. Я., Пшонка Л. Я. Влияние
минеральных и различных видов органических удобрений на гумусона-
копление в почве. — В кн.: Проблемы накопления и использования
органических удобрений. Минск, 1976.
198. Минеев В. Г.у Шевцова Л, К. Влияние длительного применения
удобрений на гумус почвы и урожай культур. — Агрохимия, 1978, № 7.
199. Мистерски В., Логинов В. Исследование некоторых физико-химических
свойств гуминовых кислот. — Почвоведение, 1959, № 2.
200. Мишустин Е. Я., Никитин Д. Я. Атакуемость гуминовых кислот
почвенной микрофлорой. — Микробиология, 1961, т. 30, вып. 5.
201. Надь М, О методах пептизации коллоидов почвы. — Почвоведение,
1957, № 9.
202. Назарова А. В. Некоторые основные константы гуминовых кислот
различного происхождения. — Зап. Ленингр. с.-х. ин-та, 1975,
т. 269.
203. Назарова А. В. Гетерогенность гуминовых кислот различного
происхождения-. — Науч. тр. Ленингр. с.-х. ин-та, 1976, т. 296.
204. Назарова А. В. Исследование гуминовых кислот различного
происхождения методом ИК спектроскопии. — Науч. тр. Ленингр. с.-х. ин-та, 1977,
т. 329.
205. Назарова А. Я. Сравнительная характеристика гуминовых кислот
различного происхождения. Канд. дис. Ленингр. с.-х. ин-т, 1977.
206. Найденова О. А. К вопросу о природе гуминов почвенного гумуса. —
Учен. зап. Ленингр. ун-та. Сер. биол., 1951, № 140, вып. 27.
207. Найденова О. А. Опыт применения высокочастотного титрования для
определения функциональных групп в гумусовых кислотах. — В кн.:
Химия, генезис и картография почв. М., «Наука», 1968.
270
208. Найденова О, Л. Использование неводных растворителей и
высокочастотного титрования при определении функциональных групп в
гумусовых кислотах. — Зап. Ленингр. с.-х. ин-та, 1968, т. 117, вып. 1.
209. Найденова О. Л. К методике получения чистых препаратов фульвокис-
лот. — Зап. Ленингр. с.-х. ин-та, 1973, т. 206.
210. Найденова О. А. О путях использования УФ спектроскопии для
определения природы фенолгидроксильных групп в фульвокислотах. —
Зап. Ленингр. с.-х. ин-та, 1974, т. 237*
211. Найденова О. Л., Андроничева Л. Е. Сравнительная характеристика
фульвокислот основных типов почв СССР методом гельфильтрацшь —
Зап. Ленингр. с.-х. ин-та, 1972, т. 165, вып. 2;
*!l2. Найденова О. А., Васильева Т. M.t Капранова В. Б, К вопросу о кислых
группах фульвокислот. — Зап. Ленингр. с.-х. ин-та, 1973, т. 206;
213* Найденова О. А., Корюшкина С. Р. Изучение природы
фенолгидроксильных групп фульвокислот методом УФ спектроскопии в сочетании с бор-
гидридным методом. — Зап. Ленингр. с.-х. ин-та, 1974, т. 237.
214. Найденова О. А., Корюшкина С. Р. Сравнительная характеристика
фульвокислот дерново-подзолистой почвы и чернозема. — Зап. Ленингр,
с.-х. ин-та, 1975, т. 289.
215. Найденова О. Л., Корюшкина С. Р. К характеристике фульвокислот
серозема. — Науч. тр. Ленингр. с.-х. ин-та, 1976, т. 296.
216. Наканиси К. Инфракрасные спектры и строение органических
соединений. М., «Мир», 1965.
217. Некрасов Б. В. Основы общей химии. 3-е изд. М., «Химия», 1974, т. I, II»
218. Неунылов Б. А., Хавкина Я. В» Изучение скорости разложения и
процессов превращения в почве органического вещества, меченного 14С. —
Почвоведение, 1968, № 2.
219. Никитин Д. И. Разложение почвенных гуминовых кислот
микроорганизмами. — Изв. АН СССР. Сер. биол., 1960, № 4.
220. Никитин Б. Л. Почвы Горьковской области. Горький, 1978.
221. Новицкий М> В. Превращение растительных остатков и вынос
органических веществ в дерново-подзолистых суглинистых почвах. — Канд.
дис. Ленингр. с.-х. ин-т, 1971.
222. Новицкий М. В. К методике изучения состава низкомолекулярных
органических веществ в лизиметрических водах. — Зап. Ленингр. с.-х.
ин-та, 1973, т. 206.
223. Новицкий М. В., Крупина Г. Я. О методах изучения состава воднорас-
творимых органических веществ, образующихся при разложении
растительных остатков. — Науч. тр. Ленингр. с.-х. ин-та, 1978, т. 354.
224. Овчаренко Ф. Д., Вдовенко И. В. Адсорбция аминокислот
монтмориллонитом. —¦ Укр. хим. журн., 1969, т. 35, № 2.
225. Овчинникова М. Ф. Органические соединения азота в гумусовых
веществах почвы. Автореф. канд. дис. М., 1965.
226. Овчинникова М. Ф., Орлов'Д. С. Распределение азота по фракциям
органических веществ почвы. — Вестн. Моск. ун-та. Сер. биол., 1964, № 3.
227. Одинцов Б, К вопросу о растворимых в воде органических продуктах
разложения растительных остатков. — Матер, по изучению рус. почв,
1912, вып. 22.
228. Окороков В, В., Черников В. А. Исследование кислотных свойств
фракций фульвокислот многонатриевого солонца. — В кн.: Вопросы
генезиса, мелиорации и охраны почв Северного Казахстана. Алма-Ата,
1972.
229. Орлов Д. С. Особенности спектров поглощения и распространение
гуминовых кислот Р-типа в почвах СССР. — Почвоведение, 1968, № 10.
230. Орлов Д. С, Гумусовые кислоты почв. М., Изд-во Моск. ун-та, 1974.
231. Орлов Д, С. Вопросы идентификации и номенклатура гумусовых
веществ. — Почвоведение, 1975, № 2.
232. Орлов Д. С. Кинетическая теория гумификации и схема вероятного
строения гуминовых кислот. — Науч. докл. высш. школы. Биол»
науки, 1977, № 9.
271
233. Орлов Д. С, Аммосова Я. М., Глебова Г. Я., Горшкова Е. И., Ильин П. Я.,
Колесников Т.П. Молекулярные веса, размеры и конфигурация частиц
гумусовых кислот. — Почвоведение, 1971, № 11.
234. Орлов Д. С, Аранбаев М. Я., Осипова П. Я. Молекулярно-спектрофото-
метрические методы исследования почв и гумусовых соединений. —
В кн.: Методы изучения минералогического состава неорганического
вещества почв. Ашхабад, «Ылым», 1975.
235. Орлов Д. С, Гришина Л. А., Ерошичева Л. Я. Практикум по биохимии
гумуса. М., Изд-во Моск. ун-та, 1969.
236. Орлов Д. С, Денисова М. Ф. Об ароматической природе ядра гуминовых
кислот из чернозема и дерново-подзолистой почвы. — Науч. докл.
высш. школы. Биол. науки, 1962, № 3.
237. Орлов Д, С, Пивоварова И. Я., Горбунов Я. И. Взаимодействие
гумусовых веществ с минералами и природа их связи. — Агрохимия, 1979,
№9.
238. Орлов Д. С, Розанова О. Я., Матюхина С. Г. Инфракрасные спектры
поглощения гуминовых кислот. — Почвоведение, 1962, № 1.
239. Орлов Д. С, Садовникова Л. К., Садовников Ю. Я. Углеводы в почвах. —
Агрохимия, 1976, № 3.
240. Орлов Д. С, Шулъман Ю. А., Юхнин А. А. Инфракрасные спектры
гумусовых кислот. — В кн.: Органическое вещество целинных и
освоенных почв. М., «Наука», 1972.
241. Пестряков Я. К. Окультуривание почв Северо-Запада. Л., «Колос»,
1977.
242. Пивоварова И. А. Количественные закономерности поглощения гумусовых
кислот почвы минералами и природа их взаимодействия. Автореф. канд.
дис. М., 1975.
243. Пихо А. Я. Влияние органических удобрений и плодородие почв
Эстонской ССР. — В кн.: Проблема накопления и использования
органических удобрений. Минск, 1976.
244. Полинг Л. Общая химия. Пер. с англ. М., «Мир», 1974.
245. Пономарева В. Я. О методах выделения и химической природе фульво-
кислот. — Почвоведение, 1947, № 12.
246. Пономарева В. В. О реакциях взаимодействия группы креновой и апо-
креновой кислот с гидроокисями оснований. — Почвоведение, 1949,
№ И.
247. Пономарева В, В. О роли гумусовых веществ в процессах
почвообразования. — В кн.: Проблемы почвоведения. М., Изд-во АН СССР, 1962.
248. Пономарева В. В. Теория подзолообразовательного процесса. М.; Л.,
«Наука», 1964.
249. Пономарева В. В., Николаева Т. А. Методы изучения органического
вещества в торфяно-болотных почвах. — Почвоведение, 1961, № 5.
250. Пономарева В. В., Плотникова Т. А. Методика и некоторые
результаты фракционирования гумуса черноземов. — Почвоведение, 1968,
№11.
251. Пономарева В. Я., Плотникова Т. А. Сравнительное изучение принятых
в СССР схем определения группового и фракционного состава гумуса. —
Почвоведение, 1972, № 7.
252. Пономарева Я. Я., Плотникова Т. А. Определение группового и
фракционного состава по схеме И. В. Тюрина в модификации В. В.
Пономаревой и Т. А. Плотниковой. — В кн.: Агрохимические методы
исследования почв! М., «Наука», 1975.
253. Пономарева Я. Я., Плотникова Т. А. Миграционная и седиментацион-
ная способность черных и бурых гуминовых кислот и их соединений
с кальцием. — В кн.: Проблемы почвоведения. М., «Наука», 1978.
254. Пономарева Я. Я., Рагим-заде А. Я. Сравнительное изучение фульво-
кислот и гуминовых кислот как агентов разложения силикатных ми*
нералов. — Почвоведение, 1969, № 3.
255. Пономарева С. И. Влияние жизнедеятельности дождевых червей на
минерализацию растительных остатков. — Почвоведение, 1950, № 8.
272
256. Потоп Ж,, де Варжак Г. Почвенная микробиология. Пер. с франц.
М., ИЛ, 1960.
257. Прох И. Состав и свойства воднорастворимых гумусовых веществ.
Канд. дис. Ленингр. с.-х. ин-т, 1960.
258. Пупков А. М. Влияние органических удобрений на содержание гумуса
в дерново-подзолистых почвах. — Науч. тр. Ленингр. с.-х. ин-та, 1977,
т. 329.
259. Пупков Л. М., Колодка В. Я. Влияние навоза и ТМАУ на содержание
гумуса и его групповой состав в дерново-подзолистых песчаных и
легкосуглинистых почвах. — Науч. тр. Ленингр. с.-х. ин-та, 1978, т. 354.
260. Раковский В. Е. Химическая сущность процессов диагенеза торфа. —
В кн.: Органическое вещество современных и ископаемых осадков. М.,
«Наука», 1970.
261. Рассохина В. В. Влияние обменных катионов на процессы гумификации
растительных остатков и поглощение водорастворимых продуктов
гумификации минеральной частью почв. Автореф. канд. дис. М., 1977.
262. Роде А. А. К вопросу о степени подзолистости. — Тр. Почв, ин-та
АН СССР, 1936, т. 13.
263. Роде А. А. Подзолообразовательный процесс. М., Изд-во АН СССР, 1937.
264. Роде А. А. Система методов исследования в почвоведении. Новосибирск,
«Наука», 1971.
265. Родин Л. Е., Базилевич Н. И, Динамика органического вещества
и биологический круговорот в основных типах растительности. М.; Л.,
«Наука», 1965.
266. Рубилин Е. В., Козырева М, Г., Зубков А. И. О возрасте гумуса
черноземной европейской части СССР. — В кн.: Тр. X Междунар. конгр.
почвоведов. М., «Наука», 1974, т. VI (II).
267. Румянцева Э. А. О скорости минерализации гумусовых веществ. — Зап.
Ленингр. с.-х. ин-та, 1970, т. 137, вып. 4.
268. Румянцева Э. А. Процессы превращения гумусовых веществ. Канд. дис.
Ленингр. с.-х. ин-т, 1971.
269. Саввинов Н. if., Панкова Н, А, Корневая система растительности
целинных участков степей Заволжья и новый метод ее изучения. — В кн.:
Сб. памяти акад. В. Р. Вильямса. М., Изд-во АН СССР, 1942.
270. Семенов А. Д. Методы исследования органического вещества
природных вод. — Гидрохим. матер., 1967, т. 45.
271. Семенов А. Д., Ивлева И. #., Дацко В, Г. Определение аминокислот
в природных водах. — Гидрохим. матер., 1961, т. 33.
272. Семенов А. Д., Ивлева И. #., Дацко В. Г. Спектрофотометрическое
определение редуцирующих Сахаров в пресных водах. — В кн.:
Современные методы анализа природных вод. М., 1962.
273. Семенов А. Д., Немцева Л, И,, Бражникова Л. В., Демченко А, С, О
составе органических веществ в паводковых водах, формирующихся
на малых водосборах засушливой зоны. — Гидрохим. матер., 1966,
т. 42.
274. Семенов А. Д., Немцева Л. И., Кишкинова Т. С. О составе органических
веществ в водах водохранилищ и озер. — Гидрохим. матер., 1968, т. 46.
275. Семенов А. Д., Залетов В. Г., Сойер В. Г. Спектры электронного
парамагнитного резонанса фульвокислот и гуминовых кислот природных
вод. — Гидрохим. матер., 1966, т. 41.
276. Симаков В. Я., Алябина Г. А. Изучение фракционного состава
гуминовых кислот некоторых типов почв методом гельфильтрации. —
Почвоведение, 1972, № 7.
277. Скоропанов С, Г. Современные проблемы органических удобрений. —
В кн.: Проблемы накопления и использования органических удобрений.
Минск, 1976.
278. Соколов А. А. Значение дождевых червей в почвообразовании. Алма-Ата,
Изд-во АН КазССР, 1956.
279. Сотникова Г. Ф. Влияние температурного фактора на разложение
минералов гумусовыми кислотами. — Тр. ВАСХНИЛ, 1972, вып. V.
18 Л. Н. Александрова 273
280. Сотникова Г. Ф. Особенности разложения силикатных минералов
гумусовыми кислотами при разных температурах и условиях увлажнения.
Автореф. канд. дис. Л., 1977.
281. Ставников Г. Л. Химия торфа. М.; Л., Госхимиздат, 1932*
282. Ставников Г. Л. Химия угля. М.; Л*, ГОНТИ, 1932.
283. Ставников Г. Л., Сысков К. И., Ушакова А * А. О гуминовых кислотах. —i
Хим. твердого топлива, 1934, т. 5.
284. Станчев Л. Б. Върху характера на свързване на микроэлементе с ху-
миновите киселини. — Почвознан. и агрохим.* 1971 * т> 6, № 2*
285. Степаненко Л. С, Максимов О. Б. Исследование гуминовых кислот
гельхроматографией на сефадексах в ДМСО. — Почвоведение * 1976*
№ 10.
286. Степанов В. В. Взаимодействие гуминовых кислот с некоторыми азот^
содержащими соединениями. — Почвоведение, 1969, ч№ 3.
287. Степанов И. С. Органо-минеральные вещества почвы. — В кн.: Органо-
минеральные вещества почв Нечерноземной зоны. М., 1977.
288. Суворов А. К. О методах выделения почвенных коллоидов. — Зап. Ле-
нингр. с.-х. ин-та, 1974, т. 234.
289. Таусон В. О, Основные положения растительной биоэнергетики. М.,
Изд-во АН СССР, 1950.
290. Титова Н. А. Железо гумусовые комплексы некоторых почв. —
Почвоведение, 1962, №12.
291. Тищенко В. В., Рыдалевская М. Д. Опыт химического исследования
гуминовых кислот различных почвенных типов. — ДАН СССР, 1936, т. 4,
№3.
292. Тоуэрс Г. X, Метаболизм фенолов в высших растениях и
микроорганизмах. — В кн.: Биохимия фенольных соединений. Пер. с англ. М,,
«Мир», 1968.
293. Трусов А. Г. Материалы к изучению почвенного гумуса. Процессы
образования «гуминовой кислоты». Пг., 1917, ч. 1.
294. Туренков Н. Я., Вавуло Ф. Я. Роль органического вещества в процессах
почвообразования. — В кн.: Проблемы накопления и использования
органических удобрений. Минск, 1976.
295. Тюкавкина Н. А., Литвиненко В, Я., Шостаковский'М. Ф.
Хроматография на полиамидных сорбентах в органической химии. Новосибирск,
«Наука», 1973.
296. Тюлин А. Ф. О формах связи гуминовых веществ с минеральной частью
почвенных коллоидов. — Почвоведение, 1938, № 7—8.
297. Тюрин Я. В. Органическое вещество почв. М.; Л., Сельхозгиз, 1937.
298. Тюрин И. В, К вопросу о природе фульвокислот почвенного гумуса. —
Тр. Почв, ин-та АН СССР, 1940, т. 23.
299. Тюрин И. В. О количественном участии живого вещества в составе
органической части почв. — Почвоведение, 1946, № 1.
300. Тюрин И. В. О формах связи органического вещества в почве. Докл.
на учен. сов. Почв, ин-та АН СССР. М., Изд-во АН СССР, 1948.
301. Тюрин Я. В. Географические закономерности гумусообразования. —
В кн.: Тр. юбил. сессии, посвящ. 100-летию со дня рождения В. В.
Докучаева. М., Изд-во АН СССР, 1949.
302. Тюрин И. В. К методам анализа для сравнительного изучения состава
почвенного перегноя или гумуса. — Тр. Почв, ин-та АН СССР, 1951,
т. 38.
303. Тюрин И. В., Гуткина Е. Л. Материалы по изучению природы гуминов
чернозема. — Тр. Почв, ин-та АН СССР, 1940, т. 23.
304. Улановская О. Я., Розинер Я. М., Харитонов Г. В. Органические
вещества в воде Воронежского водохранилища в первый год его
становления. —- Гидрохим. матер., 1975, т. 64.
305* Умаров М. М., Асеева Я. В. Свободные аминокислоты в некоторых
почвах СССР. — Почвоведение, 1971, № 10.
306. Фидер Л., Физер М. Органическая химия. Пер, с англ* М., «Химия»,
1969, т. I; 1970, т. II.
274
307. Филатов М. П. Степень однородности гумусового горизонта,
формирующегося при окультуривании дерново-подзолистой
тяжелосуглинистой почвы. — Зап. Ленингр. с.-х. ин-та, 1966, т. 105, вып. 1.
308. Филиппов П. М. Фотометрическое определение галактуроновой
кислоты в смеси с нейтральными моносахаридами. — Аналит. хим., 1970,
т. 25, вып. 12.
309. Фильков В. А., Пилипепко А. Д. Элементный состав гуминовых и фуль-
вокислот почв Молдавии. — Почвоведение, 1972, №11.
310. Фильков В. А., Пилипепко А. Д. Некоторые термические показатели
гумусовых кислот почв Молдавии. — Почвоведение, 1977, № 1.
311. Фокин Л. Д. Исследование процессов трансформации, взаимодействия
и переноса органических веществ, железа и фосфора в подзолистой почве.
Автореф. докт. дис. М., 1975.
312. Фомин Ю. И. Адсорбция воднорастворимых органических веществ
глинистыми минералами. — Зап. Ленингр. с.-х. ин-та, 1972, т. 165, вып. 2.
313. Фонквн Г., Джонсон Р. Микробиологическое окисление. Пер. с англ.
М., «Мир», 1976.
314. Фотиев А. В. К изучению гумуса болотных почв. — Почвоведение, 1964,
№ 12.
315. Фридлянд Е. В. Липидная (спиртобензольная) фракция органического
вещества различных типов почв. — Почвоведение, 1976, № 10.
316. Хан Д. В, К вопросу о методике выделения из подзолистых почв
нерастворимой фракции (гумин). — Докл. ВАСХНИЛ, 1945, вып. 7—8.
317. Хан Д. В, Органо-минеральные соединения и структура почв. М.,
«Наука», 1969.
318. Царева Р. И. Химизм торфяной почвы и рост растений. Минск, 1976.
319. ЦибаН. #., Столярова А. В. Органическое вещество и микрофлора
грунтовых вод Волгоградской области. — Гидрохим. матер., 1968, т. 47.
320. Чебаевский А. #., Туев Н. А. Изотермическая деструкция гуминовых
кислот. — Почвоведение, 1976, № 1.
321. Чичибабин А. Е. Основные начала органической химии. 4-е изд., М.; Л.,
Госхимиздат, 1932, вып. 1, 2.
322. Шаймухамедов М. Ш., Воронина К. Л. Методика фракционирования
органо-глинных комплексов почв с помощью лабораторных
центрифуг. — Почвоведение, 1972, № 8.
323. Шалыт М. С, Калмыкова А. А. Корневая система в основных
почвенных типах Украины. — Бот. журн. СССР, 1945, т. 20.
324. Шилова Е. И. Метод изучения почвенного раствора в природных
условиях. — Почвоведение, 1955, №11.
325. Шлегель Г. Общая микробиология. Пер. с нем. М., «Мир», 1972.
326. Шуберт В. Д. Биохимия лигнина. Пер. с англ. М., «Лесная пром-сть»,
1968.
327. Шурухина С. И., Чебаевский А. И. О природе спирторастворимой
фракции гуминовых кислот. — Почвоведение, 1973, № 2.
328. Шурыгина Е. А., Ларина Н. /Г., Чубарова М. Л., Кононова М. М.
Дифференциально термический и термовесовой анализы гумусовых веществ
почвы. — Почвоведение, 1971, № 6.
329. Юрлова О. В. Сравнительная оценка условий экстракции гумусовых
веществ в некоторых типах почв. -— В кн.: Химия, генезис и
картография почв. М., «Наука», 1968.
330. Юхнин А. А., Орлов Д. С. Фракционирование фульвокислот на угле. —
Науч. докл. высш. школы. Биол. науки, 1972, № 5.
331. Ярошевич М. И. Растительные остатки полевых культур — важный
источник органических веществ пахотных почв. — В кн.: Проблемы
накопления и использования органических удобрений. Минск, 1976.
332. Acle de Careres J. A., Sanchez A., Camazano. Estudio termico differencial
у termogravimetrico de sustincias humicas. —- Ann. edafol. agrobiol.,
1963, vol. 22, N 11-12.
275
18*
333. Adams W. A., Perry D. R. The effect of pH on the incorporation of amino
acids into humic acid extracted from soil. — J. Soil Sci., 1973, vol. 24,
N1.
334. Adhikari M.t Chakravorty G., Hazra G. C. Fulvic acid metal complexes. —
J. Indian Soc. Soil Sci., 1972, vol. 20, N 4.
335. Adler E. Neuere Erkenntnisse auf dem Gebiet der Lignin Chemie.— An-
gew. Chem., 1956, Bd 68, N 2.
336. Akio Inoko, Makoto Tama. Ноге* гидзюцу кэнкюсё. — Bull. Nat. Inst.
Agr. Sci., 1976, B, N 28.
337. Allison F. E., Roller E. M. Fixation and release of ammonium ions by
clay minerals. — Soil Sci., 1955, vol. 80, N 6.
338. Alvasaker E.t Michelesen K. Carbohydrates in a cold water extract of a pine
forest soil. — Acta chem. scand., 1957, vol. 11.
339. Anderson G. Identification of derivatives of deoxyribonucleic acid in
humic acid. — Soil Sci.» 1958, vol. 86, N 4.
340. Anderson G. Estimation of purines and pyrimidines in soil humic acid. —
Soil Sci., 1961, vol. 91, N 3.
341. Anderson G. Investigations on the analysis of inositol hexaphosphate in
soils. — In: Trans. 8th Intern, congr. soil sci. Bucharest, 1964, vol. IV.
342. Anderson G. The isolation of nucleoside diphosphates from alkaline
extracts of soil. — J. Soil Sci., 1970, vol. 21, N 1.
343. Anderson H. A., Fraser A. R., Hepburn A., Russell J. D. Chemical and
infrared spectroscopic studies of fulvic acid fraction from a podsol. — J. Soil
Sci., 1977, vol. 28, N 4.
344. Anderson H. A., Hepburn A. Fractionation of humic acid by gel permeation
chromatography. — J. Soil Sci., 1977, vol. 28, N4.
345. Anstett A. Sur une metode rapide de determination du rapport G : N dans
les sols, les amendemants organiques et les vegeraux, — In: VI congr.
Intern, sci. sol. Paris, 1956, vol. B.
346. Basu A. JV., Mukherjee D. C, Murherjee S. K, Interaction between humic
acid fraction of soil and trace element cations. — J. Indian Soc. Soil
Sci., 1964, vol. 12, N4.
347. Batistic L. Les acides aromatique et volatile dans les sols. — Plant a. Soil,
1974, t. 41, N 1.
348. Batistic L., Mayaudon J. Stabilisation biologique dans le sol de l'acide
ferulique C14, de l'acide vanillique C14 et de l'acide p-coumarique C14. —
Ann. Inst. Pasteur, 1970, t. 118.
349. Bernier B. Characterization of polysaccharides isolated from forest soils. —
Biochem. J., 1958, vol. 70.
350. BerzeliusJ. J. Lehrbuch der Chemie. Dresden; Leipzig, 1839, Bd VII, VIII.
351. Braids O. C., Miller R. H. Fats, Wates and Resins in Soil. In: Soil
components. Berlin et al., Springer Verlag, 1975, vol. 1.
352. Bremner /. M. Studies on soil humic acids. I. The chemical nature of
humic nitrogen. — J. Agr. Sci., 1955, vol. 46, N 2.
353. Bremner /. M. Nitrogeneous compounds. — In: Soil biochemistry,
Madison, Wisconsin, 1966.
354. Breng R. Proteins in humic acids. — Proc. 5th Intern. Peat Congr. Poz-
nan, 1976, vol. 24.
355. Broadbent F. E. Modification in chemical properties of straw during
decomposition. — Soil Sci. Ann. Proc, 1954, vol. 18, N 2.
356. Broadbent F. E. Basic problems in organic matter transformations. — Soil
Sci., 1955, vol. 79, N2.
357. Broadbent F. E. Soil organic-metal complexes. I. Factors affecting
retention of various cations. — Soil Sci., 1957, vol. 83, N 6.
358. Broadbent F. E. Soil organic matter-metal complexes. II. Cation exchange
chromatography of copper and calcium complexes. — Soil Sci., 1957,
vol. 84, N 2.
359. Bruckert S., Jaquin F., MetcheM. Contribution a l'etude des acids phenol
presents dans les sols. — Bull. Ecole nat. super, agr. Nancy, 1967, t. IX,
N11.
276
360. Surges R. The nature and distribution of humic acid. — Sci. Proc. Roy.
Dublin Soc. Ser. A, 1960, vol. 1.
361. Burgess N. A., Hurst H. M., Walkden B. The phenolic constituents of
humic acid and their relation to the lignin of the plant cover. — Geochim.
cosmochim. acta, 1964, vol. 28.
362. Butler J. #., Ladd J. N. Effect of extraction and molecular size on the
optical and chemical properties of soil humic acids. — Austral. J. Soil Sci.,
1969, vol. 7, N 3.
363. Cameron R. S., Thornton B. K., Swift R. S., Posner A. M. Molecular weight
and shape of humic acid from sedimentation and diffusion measurements on
fractionated extracts. — J. Soil Sci., 1972, vol. 23, N 4.
364. Cheam V., Gamble D. S. Metalfulvic acid chelation equilibrium in aqueous
NaN03 solution Hg(II), Cd(II) and Gu(II) fulvate complexes. — Canad.
J. Soil Sci., 1974, vol. 54, N4.
365. Chen J., Senasi N., Schnitzer M. Information provided on humic
substances by EjEe ratios. — Soil Sci. Soc. Amer. J., 1977, vol. 41, N 2.
366. Cheshire M. V., Anderson G. Soil polysaccharides and carbohydrate
phosphates. — Soil Sci., 1975, vol. 119, N 5.
367. Cheshire M. V., Berrow M. L., Goodman B. A., Mundie С. M. Metal
distribution and nature of some Gu, Mn, and V complexes in humic and ful-
vic acid fraction of soil organic matter. — Geochim. cosmochim. acta,
1977, vol. 41, N 8.
368. Coulson С. В.у Davies R. /., Khan E. Humic acid investigation. Studies
on the chemical properties of certain humic acid preparations. — Soil
Sci., 1959, vol. 88, N4.
369. Coulson С. В., Davies R. I., Lewis D. A. Polyphenols in plant, humus and
soil. — J. Soil Sci., 1960, vol. 11, N 2.
370. Danneberg 0. 15N-Untersuchungen als Beitrag zur Kenntnis der Humi-
fizierungsprozesse. I. Die Synthese von Huminstoffen — ein Teilproze3
des Bodenstickstoffkreiselaufs. — Bodenkultur, 1971, Bd 22,. N 1.
371. Danneberg O. 15N-Untersuchungen als Beitrag zur Kenntnis der Humi-
fizierungsprozesse. II. Huminstoffaufbau und Stickstofumbau wahrend
der Rotte von Maisstroh. — Bodenkultur, 1971, Bd 22, N 3.
372. Dawson I. E., Nair С. K. The copper amalgam electrode and its
applications. IV. The chemical nature of the copper complexes in peat soils and
plants. — In: Sympos. on copper metabolism. Baltimore, 1950.
373. DelVAgnola G., Ferrari G. Molecular-sizes and functional groups of humic
substances extracted by pyrophosphate from soil aggregates of different
stability. — J. Soil Sci., 1971, vol. 22, N3.
374. Dijk H. Electrometric titration of humic acids.— Sci. Proc. Roy. Dublin
Soc, 1960, vol. 1, N4.
375. Dijk H. Cation binding of humic acids. — Geoderma, 1971, vol. 5, N 1.
376. Dormaar J. F. Polysaccharides in chernozemic soils of Southern Alberte. —
Soil Sci., 1967, vol. 103, N 6.
377. Dormaar J. F. Molecular-sieve chromatography of humic substances
extracted with chelating resin from chernozemic Ah horizons. — Plant
a. Soil, 1974, vol. 41, N 1.
378. Duchaufour Ph. Humification et ecologie. — Gah. ORSTOM. Pedol..
Paris, 1970, t. 8, N 4.
379. Duchaufour Ph. Action des cations sur les processes d'humification. — Sci.
Sol, 1973, N 3.
380. Duchaufour Ph., Jacquin F. Recherche d'une methode d'extraction et de
fractionnement des composes humiques controlee par 1'electrophorese. —
Ann. Agr., 1963, t. 14.
381. Duchaufour Ph., Jacquin F. Resultats des recherches recentes sur
revolution de la matiere organique dans les sols. — С. г. Acad, agric. France,
1964, t. 50, N 4.
382. Duchaufour Ph., Mangenot F. Recherches sur revolution experimental de
certains humus. I, II. Humification biologique et abiologique. — Ann.
Agr., 1956, t. 2; 1957, t. 4.
277
383. Duff R. В. The occurence of methylated carbohydrates and rhamnose as
components of soil polycaccharides. — J. Sci. Food. Agr., 1952, N 3.
384. Evans L. Г., Russell E. W. The adsorption of humic acids and fulvic acids
by clays. — J. Soil Sci., 1959, vol. 10, N 1.
385. Felbeck G. T. Structural chemistry of soil humic substances. — Rhode
Jsland Agric. Exper. Stat. Adv. Agr., 1965, vol. 17.
386. Felbeck G. T. Normal alkanes in muck soil organic matter hydrogenoly-
sis products. — In: Trans. Gomm. Ha. IV Intern, soc. soil sci. Aberdeen,
1967.
B87. Felbeck G. T. Structural hypotheses of soil humic acids. -— Soil Sci., 1971,
vol. Ill, N 1.
388. Fiedler H. /., Reissig H. Lehrbuch der Bodenkunde. Jena, Fischer Verlag,
1964.
389. Filip Z. Prispevek к spektrometrickemu studiu humosojiloveho komp-
lexu. — Rostl. vyroba, 1968, t. 14, N 7.
390. Filip G., Tatar L. Nehany hazaitalaj oldhato huminsavkomponensenek
frakcionalasa gelsziiressec. — Agrocem. Talajtan, 1974, t. 23, N 1—2.
391. Finkle J. Soil humic acid as a hydroxypolystyrene: a biochemical
hypothesis. — Nature, 1965, vol. 207, N 4997.
392. Flaig W. Zur Grundlagenforschung auf dem Gebiet des Humus und der
Bodenfruchtbarkeit. Koln; Opland, 1956.
393. Flaig W. Zur Umwandlung von Lignin in Humusstoffe. — Freiberger For-
schungen., 1962, A, N 254.
394. Flaig W. Gedanken zur Nomenklatur der im Boden vorhandenen organi-
schen Stoffe. — In: Trans. 8th Intern, congr. soil sci. Bucharest, 1964,
vol. III.
395. Flaig W. Ghemische Untersuchungen an Humusstoffen. — Z. Chem.,
1964, Bd 4, N 7.
396. Flaig W. Veranderungen am Lignin und Anlagerung von Stickstoffhalti-
gen Verbindungen zu Beginn der Bildung und bei der Nutzung von Torf. —
Landbauforschung Volkenrode, 1967, Bd 17, H. 1.
397. Flaig W. Organic compounds in soil. — Soil Sci., 1971, vol. Ill, N 1.
398. Flaig W., Beutelspacher H. Physikalische Chemie der Huminsauren/—
Landbowkund Tijdschr., 1954, Bd 66, H. 5/6.
399. Flaig W., Beutelspacher H., Riets B. Chemical composition and physical
properties of humic substances. — In: Soil components. Berlin et al.,
Springer Verlag, 1975.
400. Flaig W., Breyhan Th. Uber das Vorkommen von Indolverbindungen in
Schwarzerdehuminsauren. — Z. Pflanz. Dung. Bodenk., 1956, Bd 75, H. 2.
401. Flaig W., Haider K. Uber die Beteiligung von Phenolen am Aufbau von
Huminsauren. — In: Trans. 9th Intern, congr. soil sci. Adelaide, 1968,
vol. III. .
402. Flaig W., Salfeld /. C, Haider K. Zwischenstufen bei der Bildung von
naturlichen Huminsauren und synthetischen Vergleichsubstanzen. —-
Landwirtsch. Forsch., 1963, Bd 16, H. 2.
403. Flaig W., Schobinger #., Deuel H. Umwaldung von Lignin in
Huminsauren bei der Verrottung von Weizenstroh. — Chem. Berichte, 1959, Bd 92,
H. 8.
404. Freudenberg K. Entwurf eines Konstitutionsschemas fur das Lignin der
Fichte. — Holzforschung, 1964, Bd 18.
405. Freytag H. E., Horst I. Mineralisirung und Humifizierung von applizier-
ter Glukose and Ruckwirkung auf die Zersetzung der organischen Boden-
substanz in der Proben einiger Parzellen der Dauerversuche «Halle». —
Albrecht-Thaer-Arch., 1968, Bd 12, H. 4.
406. Frissel G., Bolt G. Interection between certaine ionizable organic compounds
(herbicides) and clay minerals. — Soil Sci., 1962, vol. 94, N 5.
407. Gonzales A., Hubert G. Proprietes chimiques et physiques de la matiere
organique nondialysable extraite de quatre podzols du Quebec. — Canad.
J. Soil Sci., 1972, vol. 52, N 1.
278
408. Qoh к. М., Reid M. &. Molecular weight distribution of soil organic matter
as affected by acid prctrcatment and fractionation into humic and fulvic
acids.— J. Soil Sci., 1975, vol. 26, N3.
409. Goh K, M., Stevenson F. J. Comparison of infrared spectre of synthetic and
natural humic and fulvic acids. — Soil Sci., 1971, vol. 112, N 6.
410. Greenland D. J. The adsorption of sugars by montmorillonite. — J. Soil
Sci., 1956, vol. 7, N 3.
411. GreenlandD. J. Interaction between clays and organic compounds in soil. —
Soil a. Fertilizers, 1968, vol. 28, N 5—6.
412. Greenland D. /. Interactions between humic and fulvic acids and clay. —
Soil Sci., 1971, vol. Ill, N 1.
413. Greenland D. /., Laby i?., Quirk /. Adsorption of amino acids and peptides
by montmorillonite and illite. II. Physical adsorption. — Trans. Faraday
Soc, 1965, vol. 61, N 9.
414. Greenland D. /., Oades J. M. Saccharides. — In: Soil components. Berlin
et al., Springer Verlag, 1975, vol. 1.
415. Griffith S. M.y Schnitzer M. Oxidative degradation of humic and fulvic
acids extracted from tropical volcanic soils. — Ganad. J. Soil Sci., 1975,
vol. 55, N 3.
416. Groy A, Carbohydrates in cold water extracts of a pine forest soil. — Acta
chem. scand., 1963, vol. 17, N 8.
417. Guardino F., Bazan E., Averna V. Techica di separatione di una miscela
di acidi di nature fenolica par T. L. C. Successive. — Agrochimica, 1975,
t. 19, N 6.
418. Guenzi W. />., McCalla Т. М. Phytotopic substances exracted from soil. —
Soil Sci. Soc. Amer. Proc, 1966, vol. 30.
419. Guidi G., Petruzzelli G. Carboidrati negli acidi fulvici e loro frazion. —
Agr. ital., 1974, t. 74, N 1.
420. Guidi G., Petruzzelli G., Sequi P. Characterization of aminacid and
carbohydrate components in fulvic acid. — Canad. J. Soil Sci., 1976, vol. 56,
N 3.
421. Gupta U. С Soil biochemistry. New York; London, 1967, vol. 1.
422. Gupta U. C, Sowden F. I, Accurence of free sugars in soil organic matter. —
Soil Sci., 1963, vol. 96, N 3.
423. Hansen R. #., Schnitzer M. The alkaline permanganate oxidation of Danish
illuvial organic matter. — Soil Sci. Soc. Amer. Proc, 1966, vol. 30.
424. Hansen R. #., Schnitzer M. Zn-dust distillation and fusion of soil humic
and fulvic acid. — Soil Sci. Soc. Amer. Proc, 1969, vol. 33, N 1.
425. Haworth R. D. The chemical nature of humic acid. — Soil Sci., 1971,
vol. Ill, N 1.
426. Hayashi Т., Nagai T. On the components of soil humic acids. VIII. The
oxidative decomposition by alkaline permanganate and nitric acid. — Soil
a. Plant Food, 1961, vol. 6, N 4.
427. Hayashi Т., Nagai T. On the components of soil humic acids. IX. The
carbohydrate composition of different components. — Soil Sci. a. Plant Nutr.,
1962, vol. 8, N 4. *"
428. Hayes M. H., Stacey'M., Standley J. Studies on the humification of plant
tissue. — In: Trans. 9th Intern, congr. soil sci. Adelaide, 1968, vol. Ill,
429. Hoppe-Seyler F. Uber Huminsubstanzen, ihre Entstehung and ihre Eigen-
schaften. — Z. physiol. Chem., 1889, Bd 13.
430. JacquinF., Guckert A., Gallali T. Evolution des polysaccharides et des Ami-
nopolysaccharides au cours de Г humification. — Тр. Х Междунар. контр,
почвовед. М., 1974, т. 2.
431* Jenkinson D. S. Decomposition of labelled plant material in soil. Experi"
mental pedology. London, Butterworths, 1965.
432. Jnoue Т., Wada K. Adsorption of humified clover extracts by various
clays. — In: Trans. 9th Intern, congr. soil sci. Adelaide, 1968, vol. III.
433. Kawaguchi К., Куата К. On the complex formation between soil humus
and polyvalent cations. — Soil a. Plant Food, 1959, vol. 5> N 2.
279
434. Kevan t). К. Soil fauna and humus formation.— In.: Trans. 9th Intern,
congr. soil sci. Adelaide, 1968, vol. II.
435. Khan S. ?/., FriesenD. Gel-filtration of humic acid extracted from the black
solonetzic and black chernozemic soils of Alberta. — Soil Sci., 1972,
vol. 114, N 1.
436. KhanS. U., Schnitzer M. Sephadex gel-filtration of fulvic acid: the
identification of major components in two low-molecular weight fractions. —
Soil Sci., 1971, vol. 112, N 4.
437. Khan S. U., Schnitzer M. Permanganate oxidation of humic acids, fulvic
acids and humins extracted from Ah horizons of black chernozemic,
a black solod and black solonetz soil. — Canad. J. Soil Sci., 1972, vol. 52,
N 1.
438. Khan S. ?/., Sowden F. J. Distribution of nitrogen in fulvic acid fraction
extracted from the black solonetzic and black chernozemic soils
of Alberta. — Canad. J. Soil Sci., 1972, vol. 52, N 1.
439. Kleinhempel D. Ein Beitrag zur Theorie des Huminstoffzustandes. —
Albrecht-Thaer-Arch., 1970, Bd 14, H. 1.
440. Kleist #., Miicke D. Die Stalle freie Radikale in Huminsauren. — Expe-
rientia, 1966, N 22.
441. Kobo К: Л., Tatsukava R. On the colored material of fulvic acid. —
Z. Pflanz. Dung. Bodenk., 1959, Bd 84, N 1—3.
442. Kodama #., Schnitzer M. Kinetics and mechanism of the thermal
decomposition of fulvic acid. — Soil Sci., 1970, vol. 109, N 5.
443. Kodama H., Schnitzer M. Evidence for interlamellar adsorption of organic
matter by clay in a podzol soil. — J. Soil Sci., 1971, vol. 51, N 3.
444. Kosaka /., Honda C, Izeki A. Fractionation of humic acid by organic
solvents. — Soil Sci. a. Plant Nutr., 1961, vol. 7, N 2.
445. Kumada K. Several properties of humic acids. — Soil a. Plant Food, 1956,
vol. 2.
446. Kumada K., Miyara E. Sephadex gel fractionation of humic acids. —
Soil Sci. a. Plant Nutr., 1973, vol. 19, N 4.
447. Kumada K., Matsui J. Studies on the composition of aromatic nuclei
of humus. I. Detection of some condenced aromatic nuclei of humic acid. —
Soil Sci. a. Plant Nutr., 1970, vol. 16, N 6.
448. Kunal Chosh, Mukherjee S. K. The infrared spectra of natural and synthetic
hymatomelanic acids. — Indian Agr., 1971, vol. 15, N 1—2.
449. Kiister B. Untersuchungen uber die Bildung und Zersetzung von Humus-
stoffen durch Microorganismen. — Arch. Mikrobiol., 1960, Bd 15, N 1.
450. Ladd J. N. The extinction coefficients of soil humic acids fractionated
by sephadex gel filtration. — Soil Sci., 1969, vol. 107, N 4.
451. Leon Serve M. Etude par chromatographic sur gels de dextran des acides
humiques de quelques sols siliceux de haute montagne. — C. r. Acad, sci.,
1975, t. 280, N 20.
452. Leonowicz A., Trojanowski J. Exoenzgmes in fungi degrading lignin.
I. Pholiota mutabilis. — Acta microbiol. polon., 1965, vol. 14.
453. MacCarty P., O'Cinneide S. Fulvic acid. I. Partial fractionation. II.
Interaction with metal ions. — J. Soil Sci., 1974, vol. 25, N 4.
454. Macnamara Y., Tuth S. Adsorption of linuron and malathion by soils
and clay minerals. — Soil Sci., 1970, vol. 109, N 4.
455. Martin Martinez F. Estudio de los acides humicos у fulvicos extraidos
de un podsol. I. Garacteristicas analiticas. — Ann. edafol. у agrobiol.,
1975, t. 34, N 9-10.
456. Martin F., Dubach iV., Mehta N. C, Deuel H. Bestimmung der funktionel-
len Gruppen von Huminstoffen. — Z. Pflanz. Dung. Bodenk., 1963,
Bd 103, H. 1.
457. Martin /., Haider K. Microbial activity in relation to soil humus
formation. — Soil Sci., 1971, vol. Ill, N 1.
458. Martin M., Petre A. Presencia de comprestos fenolicos libres en tipos de sue-
los distintos. — Ann. edafol. у agrobiol., 1965, t. 24, N 5—6.
280
459. Martin F., Saiz-Jimenez C, Gonzalez F. I. Humic matter from vertsols.
II. Fulvic acids. — Ann. edafol. у agrobiol., 1974, t. 33, N 5—6.
460. Matsuda K., Schnitzer M. The permanganate oxidation of humic acids
extracted from acid soils. — Soil Sci., 1972, vol. 114, N 3.
461. Mayaudon /., Batistic L. Degradation biologique de la lignine Gu dans
le sol. — Ann. Inst. Pasteur, 1970, t. 118.
462. Mehta N. C, Dubach P., Deuel H. Untersuchungen uber die Molekular-
gewichtsverteilung von Huminstoffen durch gelfiltration on Sephadex. —
Z. Pflanz. Dung. Bodenk., 1963, Bd 102, H. 2.
463. Morrison R. I. Products of the alkaline nitrobenzene oxidation of soil
organic matter. — J. Soil Sci., 1963, vol. 14.
464. Mulder G. J. Die Chemie der Ackerkrume. Berlin, 1861—1862, Bd 1.
465. Mundie С. М. The identification und determination of glucuronic and
galacturonic acids in Scottish soils and soil fractions using ion-exchange
and gas-liquid chromatography. — J. Soil Sci., 1976, vol. 27, N 3.
466. Murphy O., Moore A. W. A possible structural basis of natural humic
acid. — Sci. Proc. Roy. Dublin Soc. Ser. A, 1960, vol. 1.
467. Neyroud J. A., Schnitzer M. The exhaustive alkaline cupric oxide oxidation
of humic acid and fulvic acid. — Soil Sci. Amer. Proc, 1974, vol. 38, N 6.
468. Neyroud J. A., Schnitzer M. The alkaline hydrolysis of humic substances. —
Geoderma, 1975, vol. 13, N 3.
469. Neyroud J. A., Schnitzer M. Structure chimique des acides humiques
et fulviques du sol. — In: Soil organ, matter stud. Vienne, 1977, vol. 2.
470. Nguyen Kha, Bruckert S. The effect of pH in the extraction of ironorganic
matter complexes by sodium hydroxide and sodium phyrophosphate. —
С. г. Hobdomadairon Senneas Acad, sci., 1972, 274D (5).
471. Oades J. M. Studies on soil polysaccharides. III. Composition of
polysaccharides in some Australien soils. — Austral. J. Soil Res., 1972, vol. 10, N 1.
472. Oden S. Die Huminsauren. — Kolloidchem. Beihefte, 1919, Bd 11.
473. Ogner G. Oxidation of nonhydrolyzable humic residue and its relation
to lignin. — Soil Sci., 1973, vol. 116, N 2.
474. Ogner G., Gronneberg T. Permanganate oxidation of methylated fulvic
and humic acids in chloroform. — Geoderma, 1977, vol. 19, N 3.
475. Parson J. W., Tinsly J. Nitrogeneous substances. — In: Soil components.
Berlin et al., Springer Verlag, 1975, vol. 1.
476. Payne Т. ЛГ., Rouatt J. W., Katznelson H. Determination of free amino-
acids in soil. — Soil Sci., 1956, vol. 82, N 6.
477. Perraud A., Nguyen Kha, Jacquin F. Essai de caracterisation des formes
de Thumine dans plusieurs types de sols. — C. r. Acad, sci., 1971, D272,
N 12.
478. Piper T. /., Posner A. M. Sodium amalgam reduction of humic acid.
I. Evaluation of the method. — Soil Biol. a. Biochem., 1972, vol. 4, N 4.
479. Posner A. M., Creeth J. M. A study of humic acid by equilibrium ultra-
centrifugation. — J. Soil Sci., 1972, vol. 23, N 3.
480. Pospisil F. Gel chromatography of humic acids. — Rostl. vyroba, 1974,
vol. 20, N 8.
481. Roulet JV., Mehta N. C, Dubach P., Deuel H. Abtrennung von Kohlehydra-
ten und Stickstoffverbindungen aus Huminstoffen durch Gelfiltration
and Ionenaustausch-Chromatographie. — Z. Pflanz. Dung. Bodenk., 1963,
Bd 103, H. 1.
482. Saeki #., Azuma J. Studies on the accumulation and the quality of humus
["derived from various organic materials unter different conditions. —
Soil a. Plant Food, 1956, vol. 2, N 1.
'483. Samuelson O. Ion exchanges in analytical chemistry. New York; Stockholm
1958.
484. Sato O., Kumada K. The chemical nature of the green fraction of P-type
humic acid. — Soil Sci. a. Plant Nutr., 1967, vol. 13, N 4.
485. Satoth Tsutomu. Выделение органо-минеральных компонентов из
почдеы. — Пэдородсисуто, 1974, т. 18, № 2.
281
486. Scharpenseel H. W. Aufbau und Bindugsform der Ton-Huminsaurekom-
plex. IV. Radiometric mit Aminosaure-belegtem Ton. — Z. Pflanz. Dung.
Bodenk., 1970, Bd 125, N 2.
487. Scharpenseel H. W. Naturliche Radiokohlenstoffmessungen als Mittel
zur Untersuchung von Bodenprozessen und deren Dynamik. — Тр. Х Меж-
дунар. конгр. почвовед., М., 1974, т. VI (II).
488. Scharpenseel H. W., Albersmeyer W. Infrarotspektroskopische Untersuchun-
gen an Huminsauren, Huminsaureaufschlussen und phenolisch-chinoi-
den Vergleichssubstanzen. — Z. Pflanz. Dung. Bodenk., 1960, Bd 88,
N 3.
489. Scharpenseel H. W., Kruse E. Aminoacids in clayhumic acid complex
formation. — In: Isotopes and radiation in soil plant relationships include
Forestry. Vienna, 1972.
490. Scheele W. Beitrage zur Charakterisierung naturlicher Humusstoffe. —
Kolioidchem. Beihefte, 1937, Bd 46.
491. Scheffer F., Schachtschabel P. Lehrbuch der Agrikulturchemie und Boden-
kunde. Stuttgart, Enke. Verlag, 1960, T. 1.
492. Scheffer F., Schliiter H. Uber Aufbau und eigenschaften der Braun-Grauhu^
minsauren. — Z. Pflanz. Dung. Bodenk., 1959, Bd 84, H. 1—3.
493. Scheffer F., Ziechmann W. Huminstoffe als Radikale. — Z. Pflanz. Dung.
Bodenk., 1967, Bd 116, H. 2.
494. Schlichting E. Zur Kenntnis des Heidenhumus. II. Die Fulvosaurefrak-
tion. — Z. Pflanz. Dung. Bodenk., 1953, Bd 61, H. 1—2.
495. Schnitzer M. Characteristics of organic matter extracted from podzol В ho-
rizone. — Canad. J. Soil Sci., 1970, vol. 50, N 2.
496. Schnitzer M. Alkaline cupric oxide oxidation of a methylated fulvic
acid. — Soil Biol. a. Biochem., 1974, vol. 6, N 1.
497. Schnitzer M. Investigations on the chemical structure of humic substances
by gas chromatography-mass spectrometry. — Tp. X Междунар. конгр.
почвовед. М., «Наука», 1974, Т. 2.
498. Schnitzer M. Recent findings on the characterization of humic substances
extracted from soils from widely differing climatic zones. — In* Soili
organ, matter stud. Vienna, 1977, vol. 2.
499. Schnitzer 71/., Desjardins /. G. Alkaline permanganate oxidation of
methylated and unmethylated fulvic acid. — Soil Sci. Soc. Amer. Proc, 197<0t,
vol. 34, N 1.
500. Schnitzer M., Khan S. U. Humic substances in the environment. New York,
Marcel Dekker, 1972.
501. Schnitzer M., Kodama H. An electron microscopic examination of fulvic
acid. — Geoderma, 1975, vol. 13, N 4.
502. Schnitzer M., Neyroud J. Alkanes and fatty acids in humic substances. —
Fuel, 1975, vol. 54, N 1.
503. Schnitzer M., Skinner S. /. Organo-mettallic interactions in soils. I.
Reactions between a number of metal ions and the organic matter of a podsol
Bh horizon. — Soil Sci., 1963, vol. 96, N 2.
504. Schnitzer M., Skinner S. I. Organo-metallic interactions in soils. II.
Reactions between different forms of iron and aluminium and the organic
matter. — Soil Sci., 1963, vol. 96, N 3.
505. Schnitzer Af., Skinner S. I. Organo-metallic interactions in soils. HI. Pro-
pertyes of Fe-Al-organic matter prepared in the laboratory and extracted
from a soil. — Soil Sci., 1964, vol. 98, N 3.
506. Schnitzer M., Skinner S. I. Organo-metallic interactions in soils. IV. Car-
boxyl and hydroxyl groups in organic matter and metal retention. — Soil
Sci., 1965, vol. 99, N 4.
507. Schnitzer M., Skinner S. I. Organo-metallic interaction in soils. VI.
Stability constante of Pb2+, Ni2+, Mn2+, Co2+, Ca2+, Mg2+-fulvic acid
complexes. — Soil Sci., 1967, vol. 103, N 4.
508. Schnitzer M., Sowden F. J., Ivarson К. С Humic acid reactions with апф
noacids. — Soil Biol. a. Biochem., 1974, vol. 6, N 6.
282
509. Schnitzer Л/., Wright J. R. Studies on the oxidation of the organic matter
of the A0 and Bh horizons of a podzol. — In: Trans. 7th Intern, congr.
soil sci. Madison, 1960, vol. II.
610. Schonwalder H. I. Uber die Verwertung von Huminsauren als Nahrstoff-
quelle durch Mikroorganismen. — Arch. Mikrobiol., 1958, Bd 30, H. 2.
611. Shah R. K., Chokshi M. R., Joshi В. С Development studies on soil organic
matter: humus. — Viscwakarma, 1975, vol. 16, N 1.
512. ShahR. K., Chokshi M. #., Joshi В. С Development studies on soil organic
matter: humin. — Viscwakarma, 1975, vol. 16, N 3.
513. Shan R. K., Chokshi M. R., Joshi В. С Development studies on soil organic
matter: hymatomelanic acid — a review. — Viscwakarma, 1975, vol. 16,
N 5.
514. Shigemitsu A., Kumada K. An interpretation of the conductometric
titration curve of humic acid. — Geoderma, 1977, vol. 19, N 1.
515. Shiroya i?., Kumada K. Combination reaction between humic acid and
calcium ions. — Soil Sci. a. Plant Nutr., 1976, vol. 22, N 3.
516. Simonart P., Batistic L., Mayaudon J. Isolation of protein from humic
acid extracted from soil. — Plant a. Soil, 1967, vol. 27, N 2.
517. Sinha M. K. Organic matter transformation in soils. I. Humification
of 14C tagged oat roots. — Plant a. Soil, 1972, vol. 36, N 2.
518. Sinha M. K. Organic matter transformations in soils. HI. Nature of amino-
acids in soils incubated with 14G tagged oat roots under aerobic and
anaerobic conditions. — Plant a. Soil, 1972, vol. 37, N 2.
519. Sinha M. K. Organic matter transformation in soils. IV. Aromatic
compounds in fulvic acid of soil incubated with 14G tagged oat roots under
aerobic and anaerobic conditions. — Plant a. Soil, 1972, vol. 37, N 2.
520. Sklodowski P. Characteristics of absorption spectre in infrared of humic
acids isolated from blak earth. — Rocz. glebozn., 1974, vol. 25.
521. Snyder H. Composition of humus. — J. Amer. Chem. Soc, 1897, vol. 19.
522. Sowden F. J. Action of proteolytic enzynes on soil organic matter. —
Canad. J. Soil Sci., 1970, vol. 50, N 2.
523. Sowden F. /., Parker D. I. Amino nitrogen of soil and of certain fractions
isolated from them. — Soil Sci., 1959, vol. 76, N 2.
524. Sposito G., Holtzclaw К. М. Titration studies on the polynuclear, polyaci-
die nature of fulvic acid extracted from sewage sludge-soil mixtures. —
Soil Sci. Soc. Amer. J., 1977, vol. 41, N 2.
525. 'Springer U. Zur Kenntnis der Bindungsformen der Humusstoffe, besonders
in Waldboden. — Z. Pflanz. Dung. Bodenk., 1936, Bd 45, H. 3.
526. Steelink C, Tollin G. Stable free radicals in soil humic acid. — Biochim.
biophys. acta, 1962, vol. 59, N 1.
527. Stevenson F. /. Lipids in soil. — J. Amer. Oil Chem. Soc, 1966, vol. 43,
N 4.
528. Stevenson F. J. Stability constants of Cu2+, Pb2+ and Cd2+ complexes with
humic acids. — Soil Sci. Soc. Amer. J., 1976, vol. 40, N 5.
529. Stevenson F, /., Goh K. Infrared spectre of humic and fulvic acids and their
methylated derivatives: evidence for nonspecificity of analytical methods
for oxygen-containing functional groups. — Soil Sci., 1972, vol. 113, N 5.
530. Stevenson F. /., Mendez /. Reductive clearage products of soil humic
acids. — Soil Sci., 1967, vol. 103, N 6.
531. Swaby R. J. The influence of humus on soil aggregation. — J. Soil Sci.,
1949, vol. 1, N 2.
532. Swaby R. /., Ladd J. N. Chemical nature, microbial resistance and origin
of soil humus. — In: Intern, soil conf. Wellington, 1962,
533. Swift Л. S., Posner A. M. Gel chromatography of humic acid. — J. Soil
Sci., 1971, vol. 22, N 2.
534. Swift R. S., Posner A. M. Nitrogen, phosphores and sulphur contents
of humic acid fractionated with respect to molecular weight. — J. Soil
Sci., 1972, vol. 23, N 1.
535. Swift R. «S., Posner A. M, Humification of plant materials. Properties
of humic acid extracts. — In: Soil org. matter stud. Vienne, 1977, vol. 1.
283
536. Swift R. S., Thornton B. if., Posner A. M. Spectral characteristics of a hu-
mic acid fractionated with respect to molecular weight using an agar gel. —
Soil Sci., 1970, vol. 110, N 2.
537. Tahoun S., Mortland M. Complexes of montmorillonite with primary,
secundary and tetriaryamides. I. Protonation of amides on the surface
of montmorillonite. — Soil Sci., 1966, vol. 102, N 4.
538. Tan К. H. Infrared absorption similarities between hymatomelanic acid
and methylated humic acid. — Soil Sci. Soc. Amer. Proc, 1975, vol. 39,
N 1.
539. Tan К. Н. The catalytic decomposition of clay minerals by complex
reaction with humic and fulvic acid. — Soil Sci., 1975, vol. 120, N 3.
540. Tan К. H. Infrared spectroscopy of pyrolyzates of soil humic and fulvic
acides. — Soil Sci., 1976, vol. 122, N 1.
541. Tan K. #., Clark F. E. Polysacharide constituents in fulvic and humic
acids extracted from soil. — Geoderma, 1970, vol. 4, N 2.
542. Tan K. #., Giddens J.N. Molecular weights and spectral characteristics
of humic and fulvic acids. — Geoderma, 1972, vol. 8, N 2.
543. Tate K. R., Goh К. М. Reductive degradation of humic acids from three
New Zealand soils. — N. Z. J. Sci., 1973, vol. 16, N 1.
544. Theng В. К. G. The chemistry of clay-organic reactions. New York, Marcel
Dekker, 1974.
545. Theng B. K. G. Note on the bonding of humic acid to montmorillonite. —
N. Z. J. Sci., 1976, vol. 19, N 1.
546. Theng В. К., Posner A. M. Nature of the carbonyl groups in soil humic
acid. — Soil Sci., 1967, vol. 104, N 3.
547. Theng В. К., Wake J. R.> Posner A. M. The humic acid extracted by various
reagents from a soil. — J. Soil Sci., 1967, vol. 18, N 2.
548. Thiele #., Kettner H. Uber Huninsauren. — Kolloid. Z., 1953, Bd 130.
549. Thomas R. ?., Mortenson I, L., Himes F. L. Fractionation and
characterization of a soil polysaccharide extract. — Soil Sci. Soc. Amer. Proc, 1967,
vol. 31, N 4.
550. Touati В., Guckert A., Valla M. Etude^au moyen du 46Ca des liaisons acides
humiques-montmorillonite. — Bull. Ecole nat. super, agr. ind. alim.,
1977, vol. 19, N 1—2.
551. Trojanovski J. Zastosowanie chromatografii do rozdzielania substranjii
pochniczych. — Ann. Univ. M. Curie-Sklodowska, 1952, vol. 6; 1957,
vol. 10, 12.
552. Trojanovski /., Leonowicz A.t Hampel B. Exoenzymes in fungi degrading
lignin. II. Demethoxylation of lignin and vanillic acid. — Acta microbiol.
polon., 1966, t. 15.
553. Turchenek L.W., Oades J. M. Size and density fractionation of naturally
occuring organo-mineral complexes. — Тр. Х Междунар. контр,
почвовед. M., 1974, т. 2.
554. Turner R. С, Schnitzer M. Thermogravimetry of the organic matter
of a podzol. — Soil Sci., 1962, vol. 93, N 4.
555. Valla M., Guckert A., Jacquin F. Formation de complexes entre acides
humiques et montmorillonite. I et II. — Bull. Ecole nat. super, agr. Nancy,
1972, t. XIV.
556. Valla M., Guckert A., Jacquin F.y Pavel L. Molecular weight of soil humic
acids from their diffusivity in the agar gel. — Rostl. vyroba, 1974, t. 20,
N 5.
557. Viser S. A. A physico-chemical study of the properties of humic acids
and their changes .during humification. — J. Soil Sci., 1964, vol. 15, N 1.
558. Whitehead D. C. Identification of p-hydroxybenzoic, vanillic, p-cumaric
and ferulic acids in soils. — Nature, 1964, vol. 202.
559. Witthauer /., Klocking R. Bindungsarten des Stickstoffs in Huminsau-
ren. — Arch. Acker- u. Pflanzenbau u. Bodenk., 1971, Bd 15, N 8.
560. Wojcik-Wojtkowiak D. The transformation of nitrogen and carbon in the soil
during humification of straw labelled with ^N. — Plant a. Soil, 1972,
vol. 36, N 2.
561. Wottny Ё. Die Zersetzung der organischen Stoffe und die Humusbildung.
Heidelberg, 1897.
562. Wright J, i?., Schnitzer M> Oxygen contaning functional groups in the
organic matter of the A0 and Bh horizons of a podsol. — In: Trans. 7th Intern,
congr. soil sci. Madison, 1960, vol. II.
563. Zakomiec I. Aminokwasy i gropy funkcyjne w czasteczkach fulwokwa-
sow. — Rocz. nauk rol., 1976, A, t. 101, N 3.
564. Ziechmann W. Zwischenmolekulare Krafte und die Struktur von Humin-
stoffen. — Z. Pflanz. Dung. Bodenk., 1977, Bd 140, H. 2.
565. Ziechman W. Molekiilkomplexe bei Huminstoffen durch e-Donator- und
e-Acceptor-Structuren. — Z. Pflanz. Dung. Bodenk., 1977, Bd 140, H. 2.
огЛаёлёнйе
Предисловие 3
Глава I. Источники органической части почвы 5
Биомасса растительного, микробного и животного происхождения,
поступающая в почву 5
Химический состав органических остатков 10
Органические удобрения 27
Глава II. Состав гумуса, природа и свойства главнейших
компонентов 31
Общие представления о гумусе и вопросы номенклатуры 32
Гумусовые кислоты как специфический класс органических
соединений 37
Номенклатура гумусовых кислот 38
Гуминовые кислоты 42
«Гиматомелановые» кислоты 80
Ульминовые кислоты 82
Гумины 82
Фульвокислоты 85
Промежуточные продукты разложения 97
Глава III. Процессы трансформации органических веществ в почве 106
Общая схема процессов трансформации 107
Процессы разложения—минерализации органических остатков в почве 108
Скорость разложения—минерализации органических остатков ... 118
Деятельность почвенной фауны 122
Процессы гумификации органических остатков в почве и вероятные
компоненты ее 122
Факторы гумификации 151
Глава IV. Органо-минеральные производные гумусовых кислот
и их состав 158
Общие представления и номенклатура 159
Гетерополярные соли 163
Комплексно-гетерополярные соли 166
Адсорбционные комплексы 174
Природа органо-минеральных коллоидов и их состояние 184
Глава V. Гумус пахотных дерново-подзолистых почв и влияние
органических удобрений на его содержание и состав 188
Гумусовый профиль и гумусовый режим, их основные параметры 188
Особенности формирования гумусового горизонта в пахотных
дерново-подзолистых почвах 192
286
Трансформация органических удобрений в пахотном слое дерново-
подзолистых почв и пути оптимизации гумусового режима в нем .... 198
Глава VI. Методы изучения органического вещества почвы ... 212
Методы определения общего содержания углерода и состава
органических веществ в минеральных почвах 212
Методы определения общего содержания углерода, азота и состава
органических веществ в органогенных материалах (торфяных почвах,
органических удобрениях, гумифицирующейся растительной массе) 218
Выделение и изучение состава органо-минеральных производных
гумусовых кислот 223
Выделение чистых препаратов гуминовых кислот и фульвокислот и
определение некоторых основных констант 228
Методы моделирования процессов гумификации органических
остатков 239
Методы исследования процессов миграции органических и
органо-минеральных соединений 243
Методы изучения состава индивидуальных (неспецифических)
органических веществ в почве 245
Заключение " 256
Литература
262
Людмила Николаевна Александрова
ОРГАНИЧЕСКОЕ ВЕЩЕСТВО ПОЧВЫ
И ПРОЦЕССЫ ЕГО ТРАНСФОРМАЦИИ
Утверждено к печати
Всесоюзным обществом почвоведов
и Институтом агрохимии и почвоведения
Академии наук СССР
Редактор издательства Г. И. Киселева
Художник Д. С. Данилов
Технический редактор Г. А. Бессонова
Корректоры Г. А. Александрова и Ж. Д. Андронова
ИБ № 9072
Сдано в набор 20 11.79. Подписано к печати 29 02 80. М-20768.
Формат 60x90V,e. Бумага типографская № 2. Гарнитура
обыкновенная. Печать высокая. Печ л 18 Уел печ л. 18
Уч.-изд л. 20.44. Тираж 1650. Изд. № 7457. Тип. зак. № 904.
Цена 3 р. 40 к.
Ленинградское отделение издательства «Наука»
199164, Ленинград, В-164, Менделеевская линия, 1
Ордена Трудового Красного Знамени
Первая типография издательства «Наука»
199034, Ленинград, В-34, 9 линия, 12