/

































































































































































































































Текст
ИЗДАТЕЛЬСТВО
«M И Р»
Sigmar Spauszus
STREIFZUGE DURCH DIE
ORGANISCHE CHEMIE
URANIA —VERLAG
LEIPZIG • JENA • BERLIN • 1964
3. Шпаусцус
Путешествие
в мир
органической
химии
Предисловие д-ра хим. наук проф. А. Н. Коста
ИЗДАТЕЛЬСТВО «МИР»
МОСКВА 1967
Перевод с немецкого
И. М. ЧАПЛИНОЙ
Редактор
д-р хим. наук В. Р. СКВАРЧЕНКО
Редакция научно-фантастической и научно-популярной литературы
Предисловие
Органическая химия как наука сформировалась
сравнительно недавно. Тысячелетиями человечество осваивало
тайны химических превращений соединений углерода. Сначала
это были самые простые процессы окисления — горение
древесины, нефти, угля, полностью разрушающие органические
вещества. Затем были найдены способы выделения некоторых
природных химических соединений. Это были прежде всего
красители и лекарственные вещества, которые находили
практическое применение. Почти не располагая данными о
их составе и правильными представлениями о строении
атома и молекулы, первооткрыватели называли такие вещества,
руководствуясь их цветом, вкусом или источником
получения. И в наше время мы пользуемся этими названиями,
такими, например, как «борнеол» — маслообразное вещество,
которое содержится в эфирных маслах деревьев, растущих
на острове Борнео, — или «лимонная кислота». Зачастую и
сейчас исследователи присваивают новым выделенным
веществам названия, указывающие на источник их получения.
Так, грибок пенициллиум определил название пенициллина.
Постепенно накапливался опыт химических и
биохимических превращений органических веществ. На первых
ступенях это, как правило, была деструкция — превращение
сложных веществ в более простые. Уже давно из сахара
научились получать этиловый спирт и уксус. Окислением
ископаемой смолы — янтаря была получена янтарная кислота.
Были сделаны первые шаги и в области синтеза — Дюма
открыл галогенирование, а Пириа получил ацетон сухой
перегонкой кальциевых солей карбоновых кислот.
Однако дальнейшее развитие органической химии
преграждали два барьера.
Во-первых, в науке господствовали идеалистические
представления — считалось, что органическое содержит
некую «жизненную силу» и поэтому не может быть
искусственно получено из простых неорганических веществ. Этот барьер
был взорван открытием Велера, который синтезировал
мочевину — безусловно органическое соединение — из
неорганических исходных веществ.
Во-вторых, химики долгое время не знали основ строения
органического вещества, не понимали особенностей химии
углеродов, не представляли себе механизма процессов,
протекавших в их колбах, и не могли ими сознательно управлять.
Лишь в начале второй половины XIX века английский химик
Купер, немецкий ученый Кекуле с его тончайшей интуицией
и русский ученый Бутлеров — блестящий
химик-экспериментатор и в то же время мыслитель-материалист — независимо
друг от друга создали основы современных представлений о
строении молекул органических веществ. Александр
Михайлович Бутлеров не только интуитивно принял четырехва-
лентность атома углерода и направленность его связей в
пространстве, но и создал общую стройную теорию, объяснил
ранее непонятные явления изомерии, сумел предсказать
познаваемость строения молекулы и указал пути к этому.
Создание теории химического строения органических
соединений привело к быстрому развитию органического
синтеза и вызвало к жизни современную химию. Теперь
исследователь уже сознательно комбинировал фрагменты молекул,
открывая новые пути синтеза и новые перспективы
использования синтезированных соединений. Началась эпоха
органического синтеза. И сегодня человек может получать
структуры такой сложности, о которых полстолетия назад
трудно было даже мечтать.
Бурно развиваются новейшие физические и химические
методы выделения природных соединений и устанавливается
их строение. Ученые подбираются к синтезу тончайших и
сложнейших структур белковых тел и носителей
наследственности — нуклеиновых кислот. Отталкиваясь от созданного
природой, химики сделали то, что ей самой без
вмешательства человека оказалось не под силу. Так, начав с получения
соединений, подобных натуральному каучуку, пришли к
синтетическим полимерам, по своим свойствам намного
превосходящим природные соединения, В поисках путей синтеза
алкалоида хинина ученые давно решили задачу борьбы с
малярией, обнаружив гораздо более эффективные препараты.
На границе неорганической и органической химии выросла
химия элементорганических соединений. Для фтор- и крем-
нийорганических соединений давно нашли широкое
промышленное применение, совершив тем самым переворот в
отдельных областях технологии.
Эти колоссальные успехи опираются на непрерывно
развивающиеся теоретические основы органической химии. За
последние годы сделан новый скачок — выяснены механизмы
многих химических реакций, особенности пространственного
их протекания.
Наш современник, в какой бы отрасли народного
хозяйства ему не пришлось работать, должен иметь хотя бы
некоторое представление об органической химии, так как мы
встречаемся с "нею на каждом шагу. Осваиваются новые
волокна и красители в текстильной промышленности, широко
применяются гербициды в сельском хозяйстве, меняются
состав и тип топлива на различных видах транспорта,
повсеместно внедряются новые полимерные материалы, моющие
средства и т. п.
Предлагаемая вниманию читателя книга 3. Шпаусцуса
«Путешествие в мир органической химии», выпущенная в се-
рии «Основы знания» издательством научно-популярной
литературы ГДР, преследует именно эту цель. Она написана
необычно — в ней нет и следа сухости и академичности
изложения, читается она легко и увлекательно. Это цепь
рассказов о том, как шло формирование органической химии и
какие практические успехи ею достигнуты.
«Дремучий тропический лес, полный удивительных
вещей, куда не отважишься войти», — так охарактеризовал
органическую химию 125 лет назад Фридрих Велер. С рассказа
о Велере, который впервые синтезировал мочевину и
опроверг тем самым виталистическое учение о «жизненной силе»,
и начинается эта книга. Простейшие органические
соединения — метан, ряд предельных углеводородов, изомеры, тетра-
эдрическая структура, кратная связь, октет электронов —
так, шаг за шагом, в форме непринужденной беседы автор
вводит читателя в суть основных представлений
органической химии, на опушку «дремучего леса».
Также в форме занимательного рассказа излагается
богатая интересными событиями история открытия, применения
и промышленной добычи нефти, подробности о современных
методах ее переработки, крекинге, реформинге, получении
высокооктанового топлива, а также искусственном жидком
топливе.
С увлекательного повествования об открытии немецким
химиком Кекуле формулы бензола начинается знакомство с
соединениями ароматического ряда. Описав историю
открытия анилина и получения первых синтетических красителей,
автор рассматривает физические основы теории цветности,
давая читателю нить к пониманию явления (уже название
раздела интригует — «Почему помидор красный?»).
Так же интересно построены рассказы о природном и
искусственном каучуке, моющих средствах, пластмассах и
искусственном волокне.
Удачно дополняют книгу многочисленные шутливые
иллюстрации, а также упрощенные шаржированные схемы
лабораторных приборов и технологических процессов.
Вполне естественно, что при небольшом объеме и обилии
рассматриваемых вопросов нельзя требовать от автора
абсолютной точности и строгости в доказательствах тех или иных
положений. 3. Шпаусцус не претендует также на полноту
охвата всех проблем органической химии, и книга отнюдь не
предназначена заменить учебник.
Поскольку автор рассчитывал на читателя, незнакомого
с органической химией, он счел необходимым ввести в книгу
основные понятия, без которых трудно было бы изложить
материал. К сожалению, это сделано не всегда
последовательно — кое-где встречаются названия и термины, требующие
дополнительного разъяснения. Но это касается только
отдельных деталей изложения. В целом же книга будет
понятна школьникам старших классов и особенно интересна для
занимающихся самообразованием.
Следует отметить, что автор много внимания уделяет той
роли, которую сыграли его соотечественники, немецкие уче-
ные, в создании органической химии. Порою это приводит к
тому, что рассматриваются лишь те области органической
химии, где вклад немецких ученых и «инженеров наиболее
значителен (нефтехимия, красители, моющие вещества,
полимерные материалы). В то же время важные открытия,
сделанные учеными других стран, в том числе русскими
учеными, освещены слабее или совсем не упоминаются. Не нашли
отражения вопросы современной теории — и в этом, пожалуй,
основной недостаток книги.
Однако несмотря на отдельные недостатки, «Путешествие
в мир органической химии», безусловно, вызовет у читателя
интерес к предмету и проблемам этой удивительной области
науки.
Л. Я. К ост
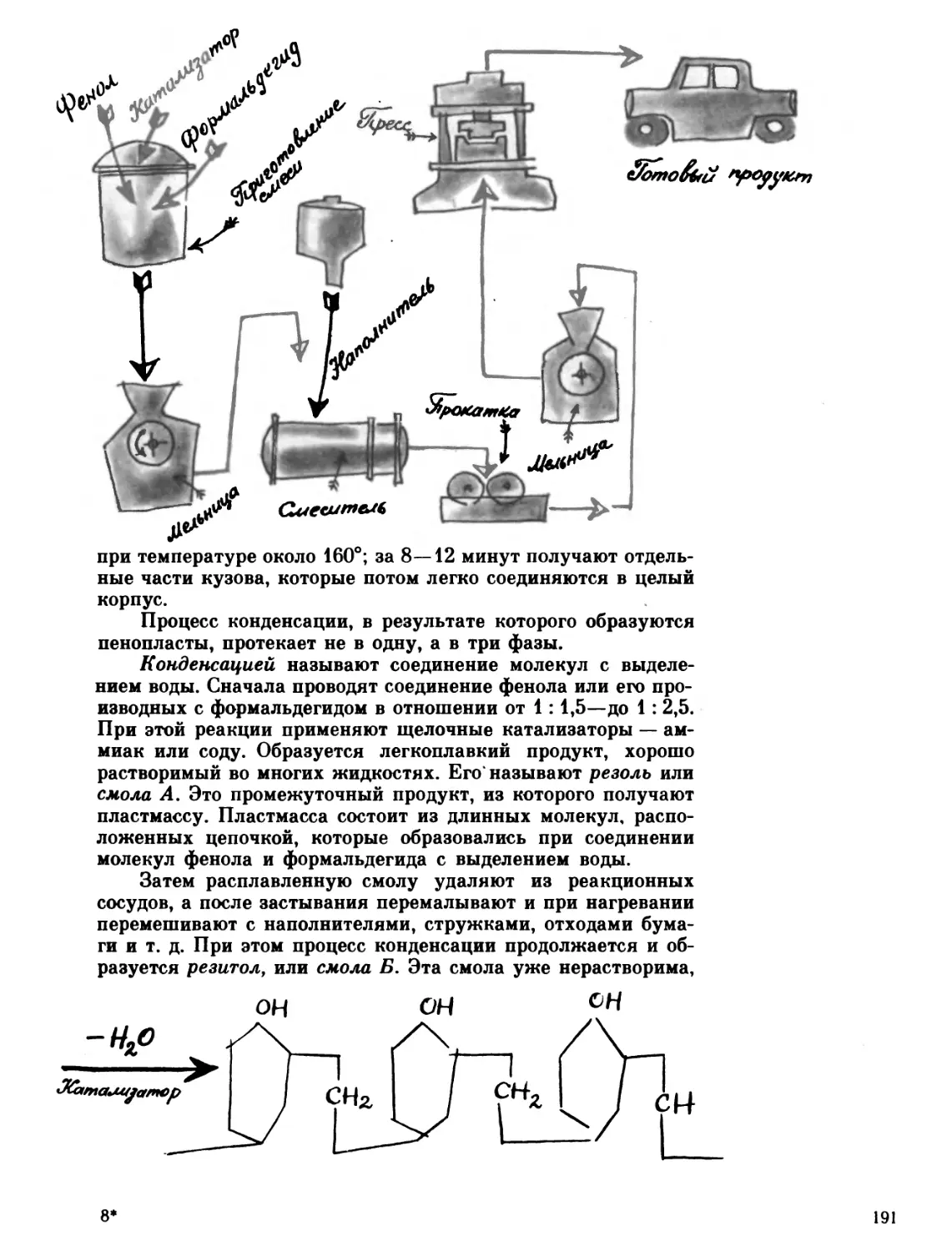
Первые шаги
22 февраля 1828 года молодой химик, преподаватель
Берлинского политехнического училища Фридрих Велер
написал письмо своему учителю и другу, знаменитому
шведскому ученому Якобу Берцелиусу. Велер не был новичком в
химии: несколько месяцев назад ему впервые удалось
получить из соединений металлический алюминий. И на этот раз
у него была важная новость.
«Я не в силах больше молчать и должен сообщить Вам,
что могу получать мочевину без помощи почек, без помощи
собаки, человека и вообще без участия какого-либо живого
существа...» — писал он. Подробно рассказывая о своих
опытах, Велер сообщал, что при получении мочевины он
использовал два широко известных неорганических соединения —
аммиак и циановую кислоту и вместо ожидаемого цианата
аммония получил белую соль, которая вступала в реакции,
совершенно не свойственные этому соединению. После
многочисленных сравнений и анализов Велер пришел в выводу,
что имеет дело с мочевиной — веществом, которое
вырабатывается живым организмом и выделяется вместе с мочой.
Синтез мочевины — это было нечто новое и неслыханное.
Именно он вошел в историю химии как... «первый пример
искусственного получения органического, «живого» вещества
из неорганических веществ». Чтобы лучше понять значение
этого события, начнем издалека.
Химию можно назвать наукой о веществах и их
превращениях. Такое определение сразу охватывает огромный круг
предметов и явлений. Ведь все, что нас окружает,
вещественно. Оконное стекло, сахар в свекле, воздух вокруг нас,
булыжник на мостовой, древесина состоят из различных
веществ. Естественно, эти вещества не находятся в состоянии
покоя, они подвергаются непрерывным изменениям, иногда
очень быстрым, а иногда настолько медленным, что мы их
совсем не замечаем. Так, превращаясь в другие вещества, сгорает
или сгнивает древесина. Под влиянием воздуха, воды и
углекислоты выветриваются даже прочные, нестирающиеся
камни мостовой. Воздух играет исключительную роль в
жизнедеятельности человека, животных и растений: он дает толчок
обмену веществ, а его компоненты в процессе этого обмена
превращаются в другие соединения. Примеры изменчивости
веществ можно приводить бесконечно.
В середине XVII века Юнгиус и Бойль сформулировали
понятие элемента. Элементами они назвали основные
вещества, которые не разлагаются на более простые. Из
отдельных элементов образуются соединения. Элементов
сравнительно немного — на сегодня их известно чуть более ста, и
это число в будущем вряд ли существенно увеличится.
Химических соединений существует несравненно больше — около
миллиона, и количество их ежедневно пополняется вновь
открытыми в природе или синтезированными в лаборатории.
Миллион соединений — прекрасно! Но может ли
человеческий разум охватить такое многообразие?
Вопрос этот вполне закономерен, и, если бы мы
рассматривали каждое вещество изолированно, без всякой связи с
другими веществами, ответить на него пришлось бы
отрицательно.
Однако ученые создали систему. Все соединения
расположены в ней по очень разумной схеме, благодаря которой
можно легко определить, к какой групйе следует отнести
каждое из них.
Итак, различают элементы и соединения. Соединения
делят на два больших класса: органические и неорганические.
Каждый класс распадается на множество мелких групп.
Необходимость, такого деления возникла в середине
XVIII века, когда ученым удалось открыть большое число
соединений растительного и животного происхождения. От
веществ «мертвой» природы они резко отличались по
составу и свойствам, поэтому вполне естественным было выделить
их в особую группу. В 1806 году Берцелиус впервые
употребил термин «органическая химия».
Считалось, что все органические вещества обладают
одной очень важной особенностью. Думали, что они образуются
только под действием особой «жизненной силы», которая
будто бы существует лишь в живой природе: в организме жи-
10
вотных и в растениях. «Грубые и низменные неорганические
силы» якобы не в состоянии создавать органические
соединения.
Эти представления преобладали до начала XIX века.
Берцелиус — один из главных сторонников теории жизненной
силы — писал в 1815 году:
«Когда мы рассматриваем наш организм как машину, то,
какими бы знаниями о его строении мы ни обладали, как бы
глубоко ни понимали взаимодействие веществ друг с другом,
причина большинства явлений в живом организме остается
так глубоко скрытой от нас, что мы, наверняка, никогда не
сможем обнаружить ее. Эту скрытую причину мы называем
жизненной силой». Еще определеннее высказался другой
химик: «То, что из основных веществ создает живой организм,
ни один химик никогда не сможет получить в колбе или
тигле».
Попытки синтезировать в лаборатории органические
вещества, такие, как жиры, спирты, красители, считались
бесплодными. Все эти вещества химики умели разлагать на
составляющие их элементы, но вновь получить исходное
вещество без таинственной жизненной силы считалось
невозможным. Господствовало мнение, что жизненная сила
существует вне неорганических элементов и не определяет их
первоначальных свойств, таких, как вес, плотность, поляризуемость
и т. п. Что же такое жизненная сила, как она возникает и
как исчезает, ученые не понимали.
Однако и не зная ничего о жизненной силе, человек
издавна умел получать различные «живые» вещества,
например спирт при брожении сахара. Конечно, это не был еще
настоящий синтез. Исходными веществами в этом процессе
оставались органические соединения, и происходило лишь
превращение одного органического вещества в другое. Хотя
человек уже умел вызвать процессы брожения и управлять
ими, превращение сахара в спирт полностью относили за
счет обмена веществ в дрожжах. Считалось принципиально
невозможным — приведем тот же пример — получить спирт
из составляющих его элементов водорода, углерода,
кислорода или из каких-либо других неорганических веществ.
Подобные воззрения были широко распространены и
полностью соответствовали господствовавшему тогда
идеалистическому представлению о мире. Время еще не опровергло
теорию жизненной силы. Напротив, к попыткам подобного
рода относились враждебно, скептически, считая их
богохульством. Именно поэтому особенно велико научное
значение открытия Велера. Ниспровержение догмы о какой-то
особой жизненной силе не только способствовало быстрому
развитию молодой органической химии, но и оказало сильное
влияние на философию и мировоззрение современников
Велера. По-видимому, Велер сам чувствовал и понимал
революционность своего открытия, потому что статью «Об
искусственном получении мочевины» он закончил такими осторожными
словами: «Я воздерживаюсь от каких-либо выводов, которые,
естественно, напрашиваются на основании этих фактов».
Работа Велера вызвала большой интерес и получила
широкое признание специалистов, хотя необходимые выводы
сумели сделать далеко не все. Даже Ббрцелиус, который
высоко ценил научные работы Велера, до конца жизни (он умер
в 1848 году) оставался сторонником теории жизненной силы.
И все же в 1831 году известный немецкий химик Либих
писал, что он рассматривает работу Берцелиуса о виноградной
кислоте и синтез мочевины Велером как поворотный пункт в
органической химии, после которого она действительно стала
наукой. После открытия Велера за короткий срок удалось
синтезировать из неорганических веществ много новых
органических соединений, и это еще сильнее пошатнуло теорию
жизненной силы. А спустя несколько лет в совместной
работе о мочевой кислоте Вел ер и Либих писали: «Философия
химии сделает из этой работы вывод, что получение всех
органических веществ вне живого организма в лаборатории
следует рассматривать не как вероятное, а как нечто безусловно
осуществимое. Правда, нам еще не известен точный путь,
так как мы не знаем исходных соединений, из которых будут
получены эти вещества, но в будущем этот путь будет, без
сомнения, найден».
Насколько точно оправдались эти предсказания ученых,
сейчас хорошо известно.
Шло время, смысл понятий «органическое» и
«неорганическое» вещество менялся, а названия оставались прежними.
Традиции часто бывают сильнее разумных доводов, но в
данном случае это было не так уж плохо. Сегодня никому не
приходит в голову, что органической химией правит
таинственная «жизненная сила». Для современного химика
органические соединения — вещества, содержащие связанный
углерод. Это относится ко всем без исключения органическим
соединениям и является настолько типичным, что
органическую химию справедливо называют химией соединений
углерода.
Не следует, однако, быть слишком педантичным, проводя
границу между органической и неорганической химией.
Между этими науками существует не пограничная линия, а,
скорее, пограничная область, куда входят многие соединения,
например, такие, как угольная кислота и ее соли. Их всегда,
и не без основания, причисляли к неорганическим веществам,
так как во многом они напоминают другие неорганические
кислоты и соли. В то же время некоторые производные
угольной кислоты, например мочевину, можно
рассматривать только как органические соединения. К органическим
соединениям можно причислить и угольную кислоту. К
счастью, подобных случаев не так уж много, и обычно провести
разграничение легче.
Итак, в состав органических соединений обязательно
входит углерод. Кроме того, для их получения нужны
водород, азот, кислород, сера, галогены, а в исключительных
случаях и некоторые другие элементы. Из ста с лишним
известных науке элементов для образования множества
органических соединений необходимы лишь 10.
Как это объяснить? Разгадку следует в первую очередь JtftlO*M */<?J€poya
искать в том, что именно атомы углерода обладают
исключительной способностью соединяться друг с другом. У атома
углерода всего 4 валентности. Если бы по этим валентностям
присоединялись только атомы других элементов, то
возможности образования соединений исчерпались бы очень быстро.
На самом деле часть валентностей замещается другими угле-"
родными атомами. Они объединяются в цепи и кольца,
иногда содержащие 50 и более атомов углерода. Благодаря этой
способности углерода возможность образования новых
веществ безгранично расширяется. Именно поэтому
органических соединений несравненно больше, чем неорганических.
Важно не только абсолютное количество атомов в молекуле,
\
л
г
^*щУ
I ^s^
/
/
но и то, как, каким образом эти атомы связаны друг с другом.
Например, спирт и эфир имеют одну и ту же суммарную
формулу СгНбО, тем не менее это два разных вещества, так как
расположение атомов и структура молекулы у них
совершенно различны.
Может возникнуть вопрос, целесообразно ли вообще де- >.
лить вещества на органические и неорганические? Конечно,
если разделить множество веществ на более мелкие группы,
их проще исследовать. Но вправе ли мы выделять именно
углеродные соединения?
У органических и неорганических веществ много общих
свойств, но есть и существенные различия. Большинство
органических соединений горит, разлагается или обугливается
при довольно низкой температуре. Вы, наверное, не раз
убеждались на собственном опыте, что суп может пригореть,
мясо — обуглиться, спирт, бензин или дерево — сгореть, а
неприятный запах нагретого жира свидетельствует о том, что
он разлагается. Как правило, неорганические вещества при
нагревании ведут себя иначе. Невозможно зажечь
поваренную соль или воду. Даже при сильном нагревании мел, гипс
и глина не меняют своего агрегатного состояния, не
разлагаются. Поскольку органические и неорганические вещества
ведут себя при нагревании неодинаково, то и приемы для
определения их класса применяют различные.
г
\
13
Мы уже знаем, что органические соединения построены
из сравнительно небольшого количества элементов. Это
обстоятельство не может не сказаться на" анализе таких
веществ. Однако достаточно ли знать, что какое-то соединение
состоит из углерода, водорода и кислорода? Ведь
существуют десятки тысяч самых разнообразных веществ,
достроенных из этих трех элементов. Химику-органику требуется
выяснить, в каком соотношении находятся эти элементы в
соединении, как велико абсолютное количество каждого из них,
из скольких атомов углерода, водорода и кислорода состоит
данная молекула.
Особенно кропотливой и напряженной работы требует
установление структуры молекул органических соединений.
При анализе неорганических соединений это тоже важно, но
в меньшей степени.
Тип связи атомов в молекулах органических и
неорганических веществ различен. Согласно современным
представлениям, любой атом состоит из ядра, в котором сосредоточена
большая часть массы атома, и электронов, вращающихся
вокруг ядра на разных расстояниях и расположенных слоями
или оболочками.
Ядро состоит из положительно заряженных протонов и
нейтральных, не имеющих электрического заряда нейтронов.
Таким образом, ядро атома заряжено положительно, и
величина его заряда зависит от числа протонов. Положительный
заряд ядра компенсируется отрицательным зарядом
электронов, число которых равно числу протонов. Следовательно,
атом в целом нейтрален.
Число электронов, окружающих атомное ядро, не
остается неизменным. При определенных условиях атом может
присоединять или отдавать их. Если атом принимает электроны,
то образуется избыточный отрицательный заряд и атом пре-
fbfjomoH ^' вращается в отрицательный ион, который называется анио-
~ ' ном. Если атом отдает электроны, то преобладает положи-
-HpJjmnoH * тельный заряд ядра и возникает положительно заряженный
- &и р ион _ катион
ч
Поясним это на примере. При столкновении атомов
лития и фтора атом лития отдает свой единственный на
внешней оболочке электрон во внешний электронный слой атома
фтора. Атом фтора получает, таким образом, дополнительный
электрон. В результате внешние электронные слои атомов
лития и фтора приобретают устойчивую конфигурацию, какую
1/ои cprnopQ
/
/
/
ч
ч
(С
ч v..
К.''
** "**
ф
X
^ -*
•ч
• \
N
I
/
/
—;^-*
имеют атомы благородных газов; после принятия одного элек-* /
трона внешняя оболочка атома фтора оказывается заполнен-^ j \ к \
ной восьмью электронами, а у лития, отдавшего один элект- \ / | I \
рон, на внешнем слое остаются два электрона (как у благо- I / / а"^
родного газа гелия). ф / " / '
Очевидно, что обмениваться могут только электроны * ! ' J
внешней атомной оболочки. После того как литий отдал свой v \ j .
внешний электрон фтору, трем положительно заряженным v \ 0 '
протонам его ядра противостоят только два отрицательно v \ / '
__ ______ •» _•* _ _ _ »» V _ . #"
заряженных электрона внешней оболочки: вместо нейтраль- < ч
ного атома лития возникает положительно заряженный ион
лития.
То же самое происходит со фтором. В этом случае
девяти протонам ядра противостоят десять электронов оболочки.
Естественно, это приводит к образованию отрицательно
заряженного иона фтора. Как известно, разноименные
заряды притягиваются друг к другу. Следовательно,
положительный ион лития и отрицательный фтора притягиваются —
образуется молекула фтористого лития. В воде эта молекула
диссоциирует, т. е. распадается на свободные ионы, но ионы
существуют и в твердом состоянии. Они располагаются в
определенном порядке в кристаллической структуре. Так как эт*.
связь осуществляется притяжением противоположно
заряженных ионов, то ее называют гетерополярной или ионной
связью.
15
В органических соединениях атомы в молекуле
соединяются в основном гомеополярной или атомной связью 1. В этом
случае ионы не образуются и нейтральные атомы
сохраняются в молекуле как. таковые. Рассмотрим, например,
углерод. В его ядре находятся 6 протонов. На близкой к ядру
/Т-оболочке вращаются 2 электрона, а на наружной L-обо-
лочке — 4. Таким образом, наружная оболочка заполнена
лишь наполовину. Достичь желаемой устойчивой оболочки
^ ---~0^*^
* ^ \ ^*.
/Ш J \
><,-« У^°Г
благородного газа можно в данном случае двумя способами:
атом углерода может отдать 4 электрона с L-оболочки, тогда
останется заполненной /^-оболочка, либо атом углерода может
дополнительно принять 4 электрона и таким образом
заполнить L-оболочку. И в том и в другом случае образовались бы
ионы углерода и нельзя было бы говорить об атомной
связи.
На самом деле процесс образования связей в углеродных
соединениях протекает иначе. Особенностью углерода
является то, что он находится в середине первого периода
периодической системы, следовательно, его наружная оболочка
заполнена лишь наполовину. Не так просто принять или
отдать 4 электрона. Предположим, что атом углерода отдал
сначала один электрон. Действие шести положительных зарядов
ядра на оставшиеся пять электронов становится сильнее, и
атому отдать второй электрон уже труднее. А если его и
удастся отдать, шесть протонов будут удерживать
оставшиеся 4 электрона еще сильнее. Избыточный заряд ядра
настолько увеличивается, притяжение между протонами и
электронами так возрастает, что отрыв других электронов
становится невозможным.
Такие же трудности возникают, если атом принимает
4 электрона. Чем больше электронов будет находиться на
L-оболочке, тем больше будет избыточный отрицательный
заряд. Одноименные заряды отталкиваются, поэтому
присоединение новых электронов будет все более и более затрудне*
но. Поскольку в нашем случае отдать или принять пришлось
бы довольно много электронов, то ионы углерода вообще не
образуются.
Как же объяснить образование ионов у атомов других
элементов, например свинца? На наружной оболочке атома
свинца тоже только 4 электрона, однако свинец образует
довольно устойчивые ионы.
Дело в том, что в данном случае действуют иные
энергетические соотношения, чем в атоме углерода. Очень важно,
сколько электронов находится на внешней оболочке и как
удалены они от ядра. Рассмотрим сначала атом углерода.
Между наружной оболочкой и ядром атома находится лишь
одна ЛГ-оболочка с двумя электронами. Из-за малого
удаления наружной оболочки от ядра его притягивающая и
отталкивающая силы особенно сказываются. В атоме свинца
расстояние между внешней оболочкой и ядром очень велико.
Оно заполнено многими внутренними электронными слоямц,
поэтому притяжение ядра ослаблено, электронам легче
покинуть наружную оболочку и образовать положительно
заряженные ионы. Атомы таких металлов, как хром или
молибден, могут отдавать со своей наружной оболочки шесть или
даже семь электронов. Это возможно только потому, что ядро
от внешней оболочки отделяют многочисленные электронные
слои.
Как же представить себе атомную связь, характерную
для большинства органических соединений? Рассмотрим
простейшее органическое соединение — метан. Его формула СН4.
Каждая молекула метана состоит из одною атома углерода
и четырех атомов водорода. Но как же в этом случае образу- /
c/ttnout
т сбинцй
ОС & Ж *V О Р
/
Ф
ется заполненная устойчивая оболочка, если обмен
электронов между атомами невозможен? Причины этого «запрета*
нам уже известны. Для построения устойчивого слоя один
электрон атома водорода и один из электронов атома углерода
соединяются в общую электронную пару, принадлежащую
сразу обоим атомам. В молекуле метана могут образоваться
четыре такие электронные пары.
4 0iu Таким образом, во внешнем слое атома углерода враща-
of о ются восемь электронов (как у благородного газа неона), на
орбите атома водорода — два (как у благородного газа гелия).
Благодаря образованию таких электронных пар слои
заполняются без присоединения электронов других атомов. На
самом деле, конечно, каждому атому принадлежит только
половина образовавшейся электронной пары. На языке обычных
химических символов каждая такая пара обозначается
черточкой валентной связи. Итак, молекулы органических
соединений построены из нейтральных атомов, а большинство
неорганических — из разноименных ионов.
Этим и определяются различные свойства этих классов
соединений. Примером может служить растворимость. Вода
является идеальным растворителем для большинства
неорганических веществ. Это происходит оттого, что в ее молекулах
преобладает ионная связь. Особое пространственное
положение разноименных зарядов создает в молекуле диполь. В
целом молекула нейтральна, поскольку положительные и
отрицательные заряды компенсируют друг друга. Когда диполь-
ные молекулы воды подходят к молекулам растворяемого
соединения ионного характера, они поворачиваются таким
образом, чтобы к ионам твердого вещества был обращен
заряд противоположного знака. Между молекулами твердого
вещества и молекулами воды возникает притяжение.
Молекулы воды энергично проникают в кристаллическую решет-
18
ку твердого вещества, разрушают ее и расталкивают ионы в
разные стороны. В этом случае говорят, что вещество
растворяется. При растворении его молекула диссоциирует на
составные части — ионы. В растворе каждый ион окружен
оболочкой ориентированных диполей молекул воды, что очень
затрудняет соединение ионов в молекулы.
У органических веществ это происходит иначе.
Поскольку их атомы нейтральны, электростатические силы диполей
воды проявиться не могут. Молекулы воды отскакивают от
молекул растворяемого вещества, как горох от стальной
пластины. Вещество с атомной связью в растворителе
размельчается только на молекулы, ионы же образоваться не могут.
При растворении органических веществ вода не проявляет
своих дипольных свойств, она ведет себя так же, как любой
другой растворитель. В ряду таких растворителей, как спирт,
эфир, бензол, диоксан, четыреххлористый углерод, вода
занимает далеко не первое место. Подтверждается старое
химическое правило: подобное растворяется в подобном. Конечно,
есть и органические соединения, более или менее сильно
диссоциирующие в воде на ионы. Так, многие органические
кислоты, как и всякие кислоты, диссоциируют с образованием
ионов водорода. Подвергаются диссоциации не только
кислоты, но и основания, соли и другие группы веществ.
Образование ионов в таких случаях происходит обычно не у
углеродного, а у кислородного или азотного атома. Но такие
соединения составляют лишь небольшую часть огромного мира
органических веществ.
Как правило, скорость реакции для многих процессов в
органической химии гораздо меньше, чем в неорганической,
и это также объясняется особым типом связи органических
соединений. При определенных условиях реакционная
способность ионов очень велика. Добавим к раствору хлористого
бария, содержащему ионы бария и хлора, серную кислоту.
Ионы бария соединяются с сульфат-ионами серной кислоты,
<s>
19
и образуется нерастворимый осадок сернокислого бария.
Время этой реакции очень мало.
Ba"+ZcC+ZH4So;- >Ы$(У+ + ZGT+ZH+
Обратимся к реакции нейтрализации. Смешаем раствор
едкого натра с соляной кислотой. Мгновенно образуются вода
и хлористый натрий, диссоциированный на свободные ионы:
Типичные органические вещества ионов не образуют, и
их реакции протекают поэтому очень медленно, в течение
нескольких часов, а иногда и дней. Так, значительно
медленнее протекает реакция между спиртами и кислотами с
образованием эфиров и воды, которая внешне очень похожа
на реакцию нейтрализации кислот и оснований.
сн5сюон + cz*5@\^p;CH5C00CzH's + hz<d
Определение веса воды, полученной при реакции между
известными количествами водорода и кислорода, —
простая стехиометрическая задача. Оба газа реагируют
полностью.
Однако конечный выход продуктов реакции между
органическими веществами в большинстве случаев заранее
определить трудно.
Правда, такой расчет можно сделать, предположив, что
исходные вещества полностью превращаются, но
вычисленные результаты редко совпадают с действительными. Обычно
истинный выход продуктов реакции бывает намного ниже
теоретического. Это происходит потому, что реакция
протекает в течение долгого времени, что в свою очередь
благоприятствует возникновению многих побочных реакций и
сдвигает равновесие основной реакции.
Мы успели заметить, что в органической химии имеются
свои особые закономерности. Но это не значит, что она
сложнее неорганической или ее изучение связано с большими
трудностями. Она просто иная. Пожалуй, органическая
химия по своему построению и применяемой классификации
даже проще неорганической. И что особенно удобно — это
почти полное постоянство валентностей в органических
соединениях. Выше уже говорилось, что в неорганических
соединениях одни и те же элементы могут проявлять различную
валентность, а в органических соединениях углерод всегда
четырехвалентен, кислород — двухвалентен, водород —
одновалентен. Азот и сера в органических соединениях также
проявляют значительно меньше валентных состояний, чем в
неорганических. Как из кубиков можно соорудить
разнообразные постройки, так, соединяя различные группы атомов,
можно получить сложнейшие органические соединения, во
всяком случае на бумаге. Так ли гладко это происходит на
самом деле — другой вопрос.
Простейшие органические вещества
«Органическая химия может свести с ума. Она
представляется мне девственным тропическим лесом, полным
неожиданностей, непроходимой чащей без конца и края, куда
страшно войти», — писал Велер Берцелиусу в 1838 году. Поскольку
Велер синтезом мочевины заложил основы органической
химии, можно отнестись к его словам с полным доверием.
А ведь во времена Велера число известных органических
соединений по сравнению с сегодняшним днем было невелико.
Такой химик, как Велер, должен был бы разбираться во всем
их разнообразии. А можно ли в наши дни вообще понять
органическую химию, если уже тогда один из ведущих
химиков видел в ней непроходимую чащу? И не была ли эта
попытка бесполезной с самого начала?
Начиная с 1838 года ученые не только открывали новые
соединения, но одновременно глубже и основательнее
исследовали ранее известные. Химики научились анализировать
вещества, объединять их в группы с родственной структурой
и сходными свойствами, устанавливали скрытые прежде
связи и закономерности. Предыдущие 125 лет органическая
химия развивалась так же, как и многие другие науки —
ученых интересовала количественная сторона, открытие все
новых и новых соединений, а теоретическому изучению их
уделялось мало внимания. И лишь позже на основе
накопленного обширного материала стало возможным вскрыть
взаимосвязь отдельных фактов, задать вопросы «как» и
«почему», овладеть всем многообразием известных данных. Все
это, конечно, не означает, что химики перестали или
перестанут когда-либо открывать и анализировать новые
вещества. Но в наше время такие исследования имеют прочную
научную основу, ясную цель, они углубляют и расширяют
уже известное ранее.
Представим себе, что мы расположились на поляне
посреди густого девственного леса. Слова Велера как нельзя
более справедливы: повсюду вокруг нас глушь,
непроходимая чаща. Но если расчистить этот лес, проложить в нем
дорожки, то темная непроходимая прежде чаща превратится
в благоустроенный парк, пройти по которому легко и просто.
И хотя кое-где еще встречаются заросли, непроходимый лес
остался только вдали, но и о нем мы уже сумели собрать
ценные сведения и знаем, что нас ожидает впереди и как
взяться за дело.
Так обстоит с любой наукой, в том числе и с
органической химией. Изученные области представляются нам
логичными и ясными, многие зависимости более простыми и
понятными, чем в неорганической химии. И все же в
органической химии скрыто еще много проблем, ожидающих
исследования и решения.
Поищем путь в мир органических веществ. Отправимся
сначала по наиболее прямой и широкой дороге.
Задумаемся, как построено простейшее органическое со-
единение? Мы уже знаем, что в его молекуле должен быть
по крайней мере один атом углерода. Кроме углерода, в
молекулу входит водород, четыре атома которого присоединяются
Наверное, таким же образом метан образуется и в
природных условиях, заполняя пустоты и трещины в
каменноугольных пластах. Как и многие органические соединения,
метан горюч, а в смеси с воздухом взрывается. Шахтеры
боятся «рудничного газа»: знакомство с ним стоило многим их
товарищам жизни. Взрывы на шахтах происходят так. Когда
шахтерский отбойный молоток, врубаясь в угольный пласт,
вскрывает трещину, заполненную метаном, газ смешивается
с воздухом, и достаточно искры, чтобы произошел взрыв.
Очень много метана содержится также в природных
газах. В некоторых районах они выделяются из земли в таких
количествах, что их используют в городах как дешевое
топливо. Расширяется применение природного газа как
основного сырья для химической промышленности. Содержащиеся
в нем компоненты перерабатываются во множество важных
продуктов. Природный газ часто сопутствует
месторождениям нефти.
Обнаружить метан можно и в любом болоте или пруду.
Целлюлоза погибших растений и другие вещества под
воздействием особых бактерий без доступа воздуха
превращаются в метан. Тронешь ил, и подымутся пузырьки болотного
газа, который также в основном состоит из метана.
В живом организме подобным образом перерабатывается
целлюлоза, съеденная вместе с пищей. Такой процесс
особенно интенсивно происходит у травоядных, например в
желудке жвачных животных. Небольшие количества метана
образуются и в кишечнике человека.
Метан получают и путем разложения фекалий и
отбросов с помощью соответствующих бактерий. Его легко можно
собрать и использовать. Уже существуют биогазовые
установки, которые могли бы обеспечить городской транспорт
топливом для двигателей внутреннего сгорания. Такие
установки были построены на сельскохозяйственных
предприятиях. Они, во-первых, помогают очистить хлев от навоза, а во-
вторых, снабжают предприятие энергией. Однако, несмотря
ЛЛ/
на успешную работу, биогазовые установки не получили
широкого распространения из-за высокой стоимости.
Известны и другие методы получения метана. Например,
с этой целью можно использовать карбид алюминия. Это
соединение образуется при пропускании двуокиси углерода
над раскаленным порошком алюминия. При взаимодействии
с водой карбид алюминия разлагается с выделением метана
и гидрата окиси алюминия:
&г
МА С5 +
П Hza
5 Ш4 + 4Ж(офд
Метан можно получить и путем прямого соединения
углерода с водородом при 1200° или, что еще лучше,
взаимодействием окиси углерода с водородом в присутствии
катализатора, тогда достаточна температура от 250 до 400°:
В этой реакции катализатором служат тонко
измельченные металлические никель или кобальт. Действуя как
ускоритель реакции, катализатор после окончания процесса
остается неизменным и, следовательно, практически не
расходуется. О катализаторах мы еще не раз будем говорить. Они
имеют большое значение, особенно для химической
промышленности. Лишь с их помощью вообще становится
возможным проведение многих реакций в техническом масштабе.
Катализаторы придают некоторым производственным
процессам экономическую целесообразность. От успешных поисков
подходящего катализатора очень часто зависит внедрение
того или иного химического метода в
промышленность.
Конечно, если использовать метан как горючий газ, то
такими способами его получать невыгодно, поскольку на
получение газа пришлось бы затратить гораздо больше энергии,
чем можно извлечь в дальнейшем при его сжигании. В
промышленности метан обычно получают из природного газа или
нефти.
Перейдем к другим углеводородам. В молекуле метана
содержится один атом углерода, четыре валентности которого
насыщены четырьмя водородными атомами. Возможностей
для присоединения других атомов нет. Но к молекуле метана
можно присоединить новые атомы углерода. Получается
обычная для органической химии связь углерод — углерод.
У нее такой же механизм, как и в связи углерод —
водород. Только второй электрон общей пары поставляет на
этот раз не водород, а вновь присоединившийся атом
углерода.
Итак, если мы заменим один атом водорода в метане
атомом углерода, обладающим тремя свободными
валентностями, по которым в свою очередь могут присоединиться
атомы водорода или других элементов, то получим этан с сум-
24
марной формулой СэНв. Более наглядна и понятна его
структурная формула
bf—е—с—и
I i
н и
Эту формулу можно написать еще и так: СНз—СНз.
И наконец, следует обозначить электронные пары, чтобы
показать образование конфигурации, подобной оболочке
благородного газа:
Н: с : с •. н
• • • •
н н
Две точки на схеме обозначают общие электронные пары.
Этан представляет собой газ, во многом похожий на
метан. Правда, он сжижается при более высокой
температуре — его точка кипения —89°, а точка кипения метана —162°.
В больших количествах этан встречается чаще всего в
природном гаае как спутник метана. В лабораторных условиях
этан легко может быть получен в чистом виде.
Ограничимся этими сведениями об этане и перейдем к
другому соединению. Если в молекуле содержатся три атома
углерода, то должно получиться соединение с формулой СзНв.
Как показывает структурная формула, средний атом
углерода удерживает два других углеродных атома
а оставшиеся свободные валентности связывают еще два
атома водорода. Здесь также следует обозначить электроны:
ИНН
Н'-С: с :'с: н
н н н
Этот газ называется пропаном. Кто из нас не видел
баллонов с такой надписью. Газ в них хранится под давлением.
На пропане могут работать автомашины, таким машинам не
нужен карбюратор. Тазовые баллоны не очень удобны, они
громоздки и сейчас в городах и особенно на автомашинах
встречаются редко. Зато в сельской местности и в кемпингах
баллоны с пропаном находят широкое применение.
Теперь нетрудно продлить этот ряд углеводородов.
Присоединив к молекуле пропана еще один атом углерода,
получим соединение с формулой С4Н10, или СН3—СНг—СНг—СНз.
Это бутан. Бутан стоит на границе между газом и жидкостью,
его точка кипения —0,5°. За бутаном следуют пентан С5Н12,
или СНз—СН2—СН2—СН2—СНз, гексан С6Н,4, гептан С7Н,6,
октан CeHie и т. д. Нетрудно заметить, что это однотипные
соединения. Однако физические свойства соединений этого
ряда, как видно из приведенной ниже таблицы, изменяются
в широких пределах.
Легко заметить, что атомы водорода и углерода в
соединениях этого ряда находятся в строго определенных
соотношениях. Если в молекуле п атомов углерода, то число атомов
водорода составит 2л + 2. Такая закономерность выводится
при систематизации структур этого ряда. А общая формула
членов гомологического ряда СпНгл+2. Следовательно,
формула гептаконтана, в молекуле которого содержатся 70
атомов углерода, С70Н142.
»■ . 1
Название
1 Метан
[ Этан
1 Пропан
I н-Бутан ,
I н-Пентан
1 н-Гексан
1 н-Гептан
I н-Октан
1 н-Нонан 1
I н-Декан
К н-Октадекан
1 н-Нона декан
1 и т. д
^^^•^и———
[Формула
СН4
С.Не
| с3нв
С4Ню
СбНи
с6н14
C?Hie 1
GgHie
С9Н10
С10Н22
Gi8H38
I C19H4o 1
г *
г
| Точка
1 плавления,
1 С°
—182,6
-172,0
-187,1
-135,0
-129,7
— 94,0
- 90,4
— 56,8
— 53,7
- 29,7
+ 28,0
+ 32,0
, 1
Г Точка
1 кипения,
-161,7
— 88,6
— 42,2
— 0,5
+ 36,1
+ 68,7
+ 98,4
+125,6
+150,7
+174,0
+308,0
+330,0
1
Плотность 1
0,424
0,546
0,582
. 0,579
0,626
| 0,659 1
0,684
0,703
0,718
0,736
0,777
0,778
|__ . __
Если все валентности углерода замещены водородом и
отсутствуют двойные связи, говорят о насыщенных или
предельных углеводородах. Соединения этого ряда отличаются
низкой реакционной способностью. Раньше считали, что
реакционная способность зависит от степени «химического
сродства», и назвали такие углеводороды парафинами — от
латинского parum affinis, т. е. мало сродства. Название это
сохранилось до сегодняшнего дня, хотя теперь известно, что
при определенных условиях парафины не так инертны, как
считали когда-то. Точное название парафинов в соответствии
с правилами Женевской номенклатуры — алканы.
Рассмотрим еще раз первые члены ряда парафинов,
обратив особое внимание на пространственное расположение
атомов углерода и водорода. Для метана возможна
единственная структура, о которой уже неоднократно говорилось.
Напишем ли мы формулу в виде структуры а или б,
естественно, никакого значения не имеет:
н
н
У с—н Л н-с-н
/ \ I
Химическая структурная формула отражает лишь
возможное расположение атомов в молекуле. Так ли они
размещены на самом деле, узнать из этой формулы нельзя. Вторая
структура удовлетворяет нашим представлениям о
симметрии и поэтому употребляется чаще. Следует подчеркнуть, что
двумерное изображение ни в коем случае не соответствует
действительности. Но сейчас это обстоятельство для нас
несущественно.
U 4пго; и другое — Mfonan
Так же как и формула метана, структурные формулы
этана и пропана очень просты. Напишем ли формулу
пропана в виде прямой или ломаной линии, значения не имеет.
Важно одно, каким образом соединены друг с другом атомы
углерода и водорода.
Иначе обстоит с бутаном. Раньше мы приводили для
него формулу
Ч V Н *
Н—с — с* '
н и и А
но ее можно представить и так:
Н (г)
л*
pa
Н**4
Эти формулы существенно отличаются друг от друга
распределением валентных связей. Атом углерода,
отмеченный звездочкой в формуле (1), связан с двумя атомами
углерода и с двумя атомами водорода, а в формуле (2) — с тремя
атомами углерода и одним атомом водорода. Такое различие
в структурах оказывает сильное влияние на свойства обоих
соединений. Фактически это два различных вещества, хотя
оба они имеют одну и ту же суммарную формулу и
называются бутаном.
В зависимости от того, соединен ли атом углерода в
молекуле с одним, двумя, тремя или даже четырьмя другими
атомами углерода, принято различать первичный, вторичный,
третичный или четвертичный углеродные атомы. В формуле
(1) имеется вторичный атом углерода, в формуле (2)
—третичный. Атом углерода в молекуле метана занимает особое
положение, так как все его четыре валентности насыщены
атомами водорода.
Для бутана возможны две структуры. Каждой из них
соответствуют различные вещества, отличающиеся друг от
друга по своим физическим свойствам. У бутана с неразветв-
ленной цепью углеродных атомов точка кипения —0,5°, а с
разветвленной —12°.
Явление, при котором вещества с различной структурой
имеют одну и ту же суммарную формулу, Берцелиус в
1830 году назвал изомерией. Это греческое слово, которое
обозначает «равные части». Понятие изомерии очень
многообразно, в данном случае говорят о структурной изомерии, или,
что точнее, об изомерии цепи. Бутан со структурой (1)
называют нормальным, или н-бутаном, в отличие от изобутана.
Этими обозначениями мы уже пользовались в таблице
парафинов.
А какова изомерия пентана? У н-пентана — прямая не-
разветвленная углеродная цепь
СИ,
•сНр—сн
•CHz—cH,
Кроме того, можно представить изомерное соединение
СНь СЦ снг—СНз
ОД3
и, наконец, структуру с четвертичным атомом углерода
СНъ
Во всех случаях суммарная формула одна и та же —
С5Н12. Но как различить по названию эти изомерные
соединения, ведь все они изопентаны?
Прежде чем решить этот вопрос, введем понятие
радикала. Так называются группы атомов, получающиеся, если от
молекулы отнимают один атом водорода. Если отнимают атом
водорода от метана с формулой СН4, то получается радикал
метил —СНз. Одна черточка указывает, что метильная
группа имеет одну валентность, или, говоря иначе, один
свободный электрон. Конечно, такая свободная валентность сразу
насыщается. Поэтому свободные радикалы не бывают
устойчивыми, они тут же вступают в реакцию с образованием
какого-нибудь соединения.
Назовем радикалы, соответствующие углеводородам
парафинового ряда: этил — С2Н5, пропил — С3Н7, бутил —С4Н9
и т. д. Теперь снова вернемся к изопентану и пронумеруем
углеродные атомы в его цепи справа или слева (это значения
не имеет)
л
д си, -cWhi - &> S) <Ч— с!
I
СН3 СНЪ СНз
В первом случае можно говорить о бутане, ко второму
атому углерода которого вместо атома водорода
присоединена метильная группа. Назовем это соединение 2-метил-бута-
ном, помня при этом, «то его следует отнести к ряду пентанов.
Если метильную группу присоединить к третьему атому
углерода, то строение молекулы не изменится и новое соединение
не образуется. И в том и в другом случае метильный радикал
соединен с атомом углерода, связанным с группой СНз и
группой СНз—СНг. Какую группу мы пишем справа ot атома
углерода, а какую слева, значения не имеет. Для простоты
пронумеруем атомы углерода так, чтобы замещение водорода
другими атомами и атомными группами приходилось на атом
углерода с меньшим номером. Предпочтительно писать 2-ме-
тил-бутан, а не 3-метил-бутан, хотя оба названия
обозначают одно и то же соединение.
А как назвать соединение б? Поскольку цепь состоит
из трех атомов углерода, речь идет о пропане, к среднему
атому углерода которого присоединены две метильные группы.
В соответствии с описанными выше правилами
номенклатуры это соединение следует назвать 2,2-диметил-пропаном.
Говорить надо не «два, запятая, два», а просто «два, два». Это
означает, что ко второму атому углерода присоединены две
метильные группы.
Число изомеров у более высокомолекулярных членов
этого ряда возрастает до бесконечности. Для гексана возможны
пять структур.
Прежде всего н-гексан:
сН3— c4z—chz~chz-chz—снъ
Затем 2-метил-пентан:
1 • 2. 3 4 5
сн,-рн-снг-снг-с4
В третьих, 3-метил-пецтан
1 2, 3 4 S
снъ — cnL—cti—Ctfo—ctj
Внимательно глядя на формулы, легко заметить, чем
отличается 2-метил-пентан от 3-метил-пентана. В первом
соединении второй углеродный атом соседствует с двумя
группами СНз и одной группой СНг, а во втором — третий атом
углерода связан с двумя группами СНг и одной группой СНз.
Четвертый изомер — 2,2-диметил-бутан:
са—
30
И наконец, 2,3-диметил-бутан:
%—СН СИ СН3
СН5 СН3
Точки кипения углеводородов этого ряда
соответственно 68, 60,3, 63,3, 49,7 и 58,0°. Как видно из этих данных,
изомерные формы вещества имеют более низкие точки кипения,
чем н-соединения. Это правило справедливо для любых
изомерных соединений.
Чему равно число изомеров для более
высокомолекулярных парафинов? У нонана СдНго их уже 35, и при желании
нетрудно вывести все изомерные структуры. У пентадекана
С15Н32 число возможных изомеров 4347, у триконтана СзоНвг
их уже 4111846763 и у тетраконтана — невероятное число —
62491178805831! Если для тетрадекана СиНзо еще можно
начертить на бумаге каждый из его «немногочисленных»
1858 изомеров, то для остальных соединений удается лишь
рассчитать их количество.
Итак, очевидно, что благодаря изомерии, во-первых,
безмерно возрастает число органических веществ, а во-вторых,
при их анализе недостаточно устанавливать только
суммарную формулу. Большая часть изомерных алканов
представляет лишь теоретический интерес, многие из них вообще
существуют только на бумаге.
Итак, группа алканов составляет гомологический ряд,
члены которого мало отличаются друг от друга по
химическим свойствам. В следующей главе мы рассмотрим
соединения другого ряда.
Вдвое —не всегда прочнее
Мы привыкли к тому, что если что-нибудь скрепить
двойной связью, то соединение» будет вдвое прочнее, чем
если бы связь была одинарной. Тройная связь крепче двойной.
Это настолько очевидно, что не требует объяснений. А вот
если утверждать противоположное, то на первый взгляд это
будет выглядеть по меньшей мере странным, и тут каждый
вправе потребовать доказательств.
Прежде всего условимся о главном — наши обычные
представления о «технике крепления» неприменимы к
химическим связям. Существование двойной или тройной
валентной свяэи в химическом соединении вовсе не означает, что
оно стало более прочным. Наоборот, двойная связь разрыва-
I
•&—*o
С
/
ется гораздо легче, чем одинарная. Видимое противоречие
между общим правилом и многократно проверенным
химическим экспериментом настолько бросается в глаза, что
целесообразно рассмотреть двойные связи подробнее.
Начнем с того, существуют ли двойные связи на самом
деле.
Всегда следует помнить, что невозможно увидеть ни
простых, ни двойных связей, мы только предполагаем их
существование для объяснения тех или иных экспериментальных
фактов.
сцгсН-*Г
Само собой разумеется, что между атомами в молекуле
стабильного соединения имеется какая-то связь. А как же
доказать, что в некоторых случаях эта связь двойная или
даже тройная?
Приведем пример. Количественный анализ газа показал,
что в молекуле два атома углерода связаны с четырьмя
атомами водорода. Это экспериментально проверенный
факт. Попробуем представить себе структурную формулу
этого соединения. Вот возможные структуры, в которых
углерод остается четырехвалентным, а водород —
одновалентным:
а)
с ~ь
И
I
с
I
I
■с-
I
н
5)
н
V
-С-
I
Н
с—н
В каждом из трех приведенных случаев структурная
формула соответствует суммарной С2Н4, однако даже с
первого взгляда ясно, что схемы а и б невозможны и от них надо
отказаться. Такие радикалы весьма недолговечны, они могут
существовать только очень короткое время в качестве
промежуточных соединений. Вещество, о котором идет речь,
стабильно и, являясь составной частью природного газа,
может не изменяться миллионы лет. Итак, осталась только одна
структура, в которой ненасыщенные валентности атомов
углерода объединились и образовали двойную связь. Таким
образом, мы убедились, что если углерод всегда остается четы-
32
рехвалентным, то между углеродными атомами должна
осуществляться двойная связь. Схема в изображает структуру
газа этилена, который, как это видно по его названию, можно
получить из этана.
Вообще говоря, в существовании двойной связи нет
ничего необыкновенного. Мы уже знаем, что каждая черточка
в структурной формуле обозначает общую пару электронов.
В случае одинарной связи каждый углеродный атом
участвует в образовании только одной электронной пары, а если
связь двойная, он отдает сразу два электрона, и образуются
две электронные пары. У такого ненасыщенного соединения,
как этилен, это выглядит так:
И
С :
• •
н
И
: с
Н
Кажется странным, что двойная связь не приводит к
упрочению структуры, а, наоборот, является ахиллесовой
пятой каждого соединения. Дело в том, что двойная связь
легко разрывается, и к освободившимся валентностям
присоединяются другие атомы или атомные группы. Это
становится более понятным, если вспомнить, что дополнительная
связь возникла только потому, что в момент образования
молекулы не было никакой иной возможности насыщения
валентностей углерода.
Образование такого «ненасыщенного соединения» с
двойными и тройными связями было, так сказать, «выходом из
положения». И как только появляется возможность
насыщения углеродных валентностей, двойная связь тут же
переходит в одинарную. Теперь легко понять различия в
химических свойствах веществ с насыщенными и ненасыщенными
связями. Как известно, насыщенные парафины, например
этан, с трудом взаимодействуют с другими веществами.
Присоединить к молекуле насыщенного соединения другие
атомные группы или атомы можно только путем замещения
атомов водорода. Вот как протекает реакция между этаном и
элементарным бромом:
СНд сн5 + ЪЧ > снд CHZ
У этилена — представителя ненасыщенных соединений—
это происходит иначе. Если в сосуд с парами брома ввести
этилен, то красно-коричневый цвет этих паров очень быстро
исчезает — бром в чистом виде перестанет существовать.
Реакция протекает непрерывно и до конца. Никаких побочных
веществ не образуется. Происходит только присоединение
брома, двойная связь переходит в простую, и получается
2 3. ДЛпаусцус
дибромэтан. В подобных случаях мы говорим о реакциях
присоединения.
Ok=<%f Br,—> <Црг—CH,fir
Эта реакция лишний раз подтверждает, что двойная
связь нестабильна и при первой возможности стремится
перейти в устойчивую одинарную связь.
Это основная особенность двойных связей. А чем она
вызвана?
Созданная в 1885 году Адольфом Байером теория
напряжения наглядно объясняет причины этого явления. Чтобы
разобраться в ней, следует сказать несколько слов о
пространственной структуре органических соединений.
До сих пор наше изображение молекул походило на
почтовую марку. Мы строили молекулярную структуру в
плоскости бумаги, и, естественно, рисунок был двухмерным.
Такое изображение очень удобно и наглядно, но оно не
соответствует действительности, ибо молекула, как и всякое тело,
трехмерна.
Рассмотрим наиболее простой случай — структуру
метана. В пространстве вокруг углерода каким-то образом
располагаются четыре атома водорода. Их положение не
произвольно, а строго обусловлено энергетическими
соотношениями в молекуле. Ученые вычислили, что наиболее устойчивая
структура образуется, если углеродный атом расположен в
центре тетраэдра 2, а четыре атома водорода — по его углам.
На рисунке валентные связи между центральным атомом
углерода и атомами водорода обозначены сплошными
линиями, а пунктиром очерчен воображаемый тетраэдр.
Молекулу этана можно представить состоящей из двух
незавершенных углеродных тетраэдров, в каждом из которых
не хватает по одному атому водорода
меман ute*m(*t J/enwu Эм<*н
В этих местах незаконченные тетраэдры соединяются в
один двойной.
Мы сделали пока только первый шаг к пониманию
строения простейшего ненасыщенного соединения — этилена.
Если у этана отнять два атома водорода, то незамещенные ва-
34
лентности, за неимением другого партнера, насыщают друг
друга, возникает двойная связь углерод-углерод и образуется
этилен.
В действительности это так гладко не происходит. Как
видно из рисунка, чтобы соединиться, обе валентности
должны существенно отклониться от своего нормального
положения. На это затрачивается энергия, которая и накапливается
затем в двойной связи. Нечто подобное мы знаем по
собственному опыту. Чтобы растянуть стальную пружину,
необходимо мускульное усилие, затрата энергии. Энергия,
конечно, не пропадает — это противоречило бы законам физики,
происходит лишь преобразование ее в иной вид: пружина
переходит в другое состояние с большим запасом энергии. Если
ослабить усилие, пружина немедленно возвращается в
первоначальное состояние и запасенная энергия
освобождается.
Согласно байеровской теории напряжения, тот же
процесс происходит при возникновении двойной связи. В момент
образования этилена направления валентностей сильно
отклоняются от их нормального положения. Как и растянутая
стальная пружина, образовавшаяся связь запасает
добавочную энергию, становится неустойчивой. В момент
приближения других атомов или атомных групп двойная связь
разрывается, валентности принимают свое первоначальное
положение и насыщаются новыми атомами.
Теория напряжения наглядна, ясна и легко объясняет
образование двойных связей, однако не соответствует
новейшим представлениям о строении атома и сейчас имеет лишь
историческую ценность. Так, в байеровской схеме строения
молекулы не показано расположение электронов.
Те же недостатки имеет теория парциальных
валентностей, сформулированная в 1899 году Тиле. На первый взгляд
она кажется довольно странной.
Тиле считал, что у этилена и других ненасыщенных
соединений настоящих двойных связей вообще не существует.
По Тиле, одна из четырех валентностей углерода
расщепляется на парциальные или остаточные валентности. Таких
валентностей у двух атомов образуется четыре, две из них
2*
взаимно насыщаются с образованием слабой двойной связи,
а две остаются частично свободными, чем и объясняется у
ненасыщенных соединений способность к присоединению
других атомов. Более наглядно это представлено на схеме.
НоС—СН* + tir0
НоС-СИо
I /
Jhr J3r
В реакциях присоединения частично свободные
парциальные валентности и образуют новую связь.
Эта теория наглядно объясняет, почему реакции
присоединения протекают именно по двойной связи, однако она
обладает теми же недостатками, что и теория напряжения.
В настоящее время о природе двойной связи существуют
иные представления.
Как известно, черточка валентности на рисунке означает
общую электронную пару. Следовательно, двойная связь
соответствует двум общим электронным парам. Это понятно
без объяснений. Удивительно другое — эти общие
электронные пары неравноценны, а поэтому и связи, определяемые
ими, естественно, должны быть различны.
Суть атомной связи в органических соединениях в том,
что для образования общей электронной пары каждый
атом-партнер поставляет по одному электрону со своей
наружной оболочки. Общее число электронов остается, таким
образом, неизмененным, но у каждого атома возникает
конфигурация электронов, аналогичная оболочке
благородного газа. Мы уже поясняли это раньше на некоторых
примерах.
Такие электронные пары есть всюду, где существует
простая связь, например между атомами углерода и водорода
в метане или между двумя атомами углерода в этане.
Электроны таких пар назвали о-электронами. Они принадлежат
в равной степени обоим атомам и образуют прочную простую
связь.
Иначе ведет себя вторая электронная пара,
осуществляющая двойную связь. Оба электрона этой пары называют
я-электронами. Они более подвижны и не всегда в равной
степени принадлежат обоим атомам. Когда же это
происходит, электрический заряд молекулы полностью выравнивается
и в ней отсутствуют центры повышенной электронной
плотности. Но если я-электроны смещаются в сторону одного или
другого атома, возникают условия, отличные от тех, которые
наблюдались в основном состоянии. Возникают активные
предельные структуры, в молекуле появляются центры
повышенной электронной плотности. Это хорошо видно на рисун-
36
ке: а-электроны обозначены точками, а я-электроны —
крестиками.
С+) со с-) (+)
н н н н н н
c^fiftdmypct
структура, Z
В предельной структуре 1 оба л-электрона смещены к
правому-атому углерода, поэтому у него появляется излишек
электронов и, следовательно, отрицательный заряд.
Одновременно левый атом углерода теряет свой л-электрон и
приобретает положительный заряд. В предельной структуре 2
происходит обратный процесс. Молекула в основном состоянии
неактивна. В предельном состоянии в ней возникают центры
различной электронной плотности, и, к своему удивлению,
мы обнаруживаем картину, знакомую нам по ионной связи.
Сразу же отметим существенное отличие. Рассматривая
типично ионное соединение, например хлористый водород,
можно с уверенностью сказать, что водород в нем — катион,
следовательно, несет положительный заряд, а хлор — анион и
всегда заряжен отрицательно. А для молекулы этилена
характерны всевозможные переходы структур 1 и 2 друг в друга,
причем все эти переходы пребывают в состоянии подвижного
равновесия. Поэтому нельзя сказать точно, как заряжен в
данный момент каждый углеродный атом — положительно
или отрицательно и насколько силен в молекуле этот сдвиг
заряда. И наконец, молекула может находиться в основном
состоянии, т. е. иметь выравненный электрический заряд.
В отличие от ионной связи в данном случае речь идет о таком
виде ионов, которые могут быть заряжены как
положительно, так и отрицательно, и в зависимости от этого изменяется
положение центров различной электронной плотности в
молекуле.
Под влиянием внешних условий положение центров
повышенной электронной плотности изменяется, то есть
изменяется положение л-электронов, что приводит к образованию
предельных структур, которые показаны на рисунке.
Теперь нам должна быть ясна склонность такого типа
соединений к реакциям присоединения. Например, если
смещать этилен и бромистый водород, произойдет быстрая
реакция с образованием монобромэтана:
chz=chz+ щ
CHjflr~-CH-9
Как известно, бромистый водород — типичное ионное
соединение, диссоциирующее в водном растворе на
положительные ионы водорода и отрицательные, ионы брома. Когда
ион водорода или брома или молекула бромистого водорода
с четко выраженными центрами различной электронной
плотности приближаются к молекуле этилена, то под влиянием
внешних зарядов n-электроны этилена смещаются и
образуется активная предельная структура. Ион брома отдает
свой избыточный электрон и присоединяется к положительно
заряженному углеродному атому, а ион водорода — к
отрицательно заряженному, как показано на следующей
схеме:
С-) О)
Н+Вг"
Н Б/
Механизм реакции присоединения к этилену чистого
Вг2, связь в молекуле которого гомеополярна, совсем иной.
Ограничимся этими сведениями о двойных связях и
рассмотрим некоторые ненасыщенные соединения.
По аналогии с гомологическим рядом метана
ненасыщенные углеводороды также можно расположить в определенный
ряд. Если ввести в каждый член метанового ряда двойную
связь, то при этом число атомов водорода в молекуле
уменьшится на два и мы получим соответствующие соединения
ряда этилена. Тесная связь между углеводородами ряда
этилена и метана видна в названиях соединений. Названия
предельных углеводородов оканчиваются на «ан» (метан,
бутан и т. д.). Чтобы получить название ненасыщенных
углеводородов, к тому же корню просто прибавляют другое
окончание — «ен» (этен, бутен и т. п.). Эти названия
установлены Женевской номенклатурой. Раньше для
ненасыщенных соединений было принято окончание «илен» (этилен,
бутилен), по привычке и до сих пор часто пользуются
старыми названиями.
В молекуле метана только один атом углерода, и,
разумеется, в нее нельзя ввести двойную связь. Поэтому первый
член ряда ненасыщенных углеводородов образуют от этана
и приходят таким образом к этену, или этилену, о которых
мы уже не раз упоминали. При взаимодействии этилена с
галогенами образуются маслянистые жидкости. Это издавна
было хорошо известно французским химикам, которые
называли этилен gaz olefiant, что значило газ, образующий масло.
Все представители этого гомологического ряда получили
поэтому название олефины, сохранившееся до наших дней. Их
название алкены происходит от названия членов метанового
ряда «алканов», окончание «ен» указывает на наличие двои-
ной связи. Общая формула алканов СпНгп+г, а для алкенов
Cntbn, поскольку из-за двойной связи у них на два атома
водорода меньше.
В последнее время значение этилена все возрастает. В
промышленном масштабе его получают чаще всего из нефти
или ацетилена. В определенных условиях этилен полимери-
аувтся, т. е. его молекулы соединяются между собой, и
образуется полиэтилен. О механизме полимеризации мы
подробно расскажем в дальнейшем, а пока ограничимся лишь
некоторыми необходимыми сведениями. Полиэтилен— очень
ценная пластмасса, которую используют для самых
разнообразных целей.
Если ввести двойную связь в пропан СНз—СНа—СНз,
то получается второй представитель олефинового ряда —
пропен СНа —СН—СНз. Соответственно из н-бутана
образуется бутен-1 СНавСН—СНа—СНз или изомерный бутен-2
СНз—СН«*СН—СНз. Для бутена небезразлично, где
расположена в молекуле двойная связь. Если двойная связь
находится между первым и вторым атомами углерода, такое
соединение называют бутен-1, а если она расположена в
середине молекулы, между вторым и третьим атомами углерода,
то вещество называют бутен-2. Исходя из изобутана
СН3—°Н СН,
получают изобутилен.
CHj-C-CHj,
СНз
В изобутилене положение двойной связи роли не
играет. Но для следующих членов гомологического ряда с
разветвленной цепью атомов углерода это уже небезразлично,
для них существует сравнительно большое количество
изомеров.
Если отнять у парафинов один атом водорода, то
получаются неустойчивые радикалы, от которых, однако,
образуются названия всех устойчивых производных парафиновых
углеводородов. Так, от метана CHU образуется радикал метил
—СНз, от бутана С4Н10 — бутил —С4Н9 и т. п. Отнимая один
атом водорода у алкенов, мы также приходим к радикалам,
но их названия нельзя образовать от названия исходного
соединения.
Приведем несколько примеров: СНа—СНа —это этилен,
но получающийся из него радикал— СН—СНа называется
винилом. Поэтому соединение CHCl — СНа называют винил-
хлоридом. Запомним слово «винил». Мы еще не раз встретимся
с этим радикалом, потому что он участвует в образовании
многих важных веществ.
Если отнять один водородный атом от пропилена
СНз—СН = СН2, получаются радикалы пропенил цли аллил
в зависимости от того, где расположена свободная
валентность. СНз—СН = СН дто пропенил, а формула аллила
—СНг—СН—СНг. Число изомеров в последующих
соединениях этого ряда увеличивается.
УкрепиеныЬе н« оун&и оси otnfi^ujue ttfo*
$Ьуно (paufatomcjf £ lAscfytc сторону
Познакомимся еще с одним случаем изомерии в
ненасыщенных соединениях. Представим себе, что у нас есть ось
с двумя свободно вращающимися стрелками на концах. Такое
устройство легко изготовить из деревянного стержня и двух
картонных стрелок, которые можно вращать в разные
стороны независимо друг от друга. Установить точно, когда
острия стрелок направлены друг к другу, невозможно. Из-за
быстрого вращения положение стрелок постоянно
изменяется и его нельзя строго зафиксировать.
неп*су£ил€Ыб/,лосг
Сделаем в нашей модели еще одну ось - стержень,
параллельный первому. Картина совершенно меняется. Теперь
стрелки не смогут больше вращаться независимо друг от
друга и займут такое положение, какое было у них в момент
присоединения второго стержня. Этот второй стержень
делает всю систему неподвижной и прочной. Можно представить
себе два возможных случая — либо стрелки закреплены в
одинаковом положении, либо острия их расположены по диа-
40
гонали друг против друга, т. е. стрелки находятся в цис- или
rpawc-положении относительно оси.
В молекулах атомные группы, находящиеся по концам
простой связи, также могут свободно вращаться вокруг «оси».
В случае 1,2-дихлорэтана каждая группа имеет один атом
хлора, и неважно, как написать структурную формулу — в
виде а или в виде б, поскольку из-за свободного вращения
llfoutepov" не oSpajt/e+rtcj} — о£е uttuse —
hj/uu со&ершенно едина tic fac
нельзя установить точное расположение атомов в
пространстве. Обе записи относятся к одному и тому же соединению.
Совершенно иное положение в молекуле этилена, где
существует двойная связь, которая мешает свободному
вращению и делает структуру жесткой. Так, известны два
изомерных дихлорэтилена. Структура а соответствует цис-1,2-
дихлорэтилену, а структура б — гране-форме.
В этом случае оба соединения также имеют одну и ту же
суммарную формулу, но обладают различными структурами.
Л - о том fojopoycL
; - antcit хиора
у nujl радное cjnpce+tue,
Это типичный случай так называемой цис-транс- или
пространственной изомерии.
Перейдем к тройным связям. В этом случае между двумя
атомами углерода имеются не две, а три общие электронные
пары, состоящие из малоподвижных а-электронов и
подвижных я-электронов. Соединения с двойной связью легко всту-
41
пагот в реакции присоединения, тем более это свойственно
соединениям с тройной связью.
Простейший представитель этого" класса — ацетилен
С2Н2. Он общеизвестен, хотя бы по названию. Соединения
гомологического ряда ацетилена называют ацетиленовыми
углеводородами, а по Женевской номенклатуре — алкинами.
Названия соединений этого ряда оканчиваются на «ин»: этин
С2Н2, бутин СчНв и т. д. Ацетилен никогда не называют эти-
ном, и мы тоже не будем этого делать.
Познакомимся поближе с первым членом этого ряда —
ацетиленом. Это — бесцветный газ с неприятным, как
принято считать, запахом. Такое утверждение не совсем верно.
В чистом виде ацетилен почти не пахнет, но при его
получении образуются газообразные примеси, придающие
ацетилену типичный «карбидный запах».
Ацетилен можно получать из карбида, точнее, из
карбида кальция — соединения кальция с углеродом. Карбид
кальция в свою очередь получается при высокотемпературной
реакции углерода с жженой известью.
C&0-JT ЗС ► CatC^Ca
Под воздействием воды из карбида кальция образуются
газообразный ацетилен и применяемая в строительстве
гашеная известь:
CeC^TMfi >czHz + <ЪЩ
Ацетилен находит разнообразное применение. Кое-кто
еще помнит карбидные лампы на старых велосипедах.
Сделаны они были весьма просто: в сосуд с карбидом кальция
равномерно капала вода, а образовавшийся при этом
ацетилен поступал через сопло в лампу, где сгорал ярким
пламенем.
Для полного окисления ацетилена, как видно из
уравнения, требуется много кислорода. При неполном окислении в
лампе образовывались тонко измельченные частички
свободного углерода, которые, раскаляясь в пламени, давали
сильный, яркий свет. В горелке Даниеля смешивают ацетилен с
кислородом, при этим происходит полное сгорание.
Получается неяркое, но очень горячее пламя, которое применяют
для резки и сварки металлов:
Конечно, каждый раз получать ацетилен на месте
сварки очень неудобно. Установки для получения ацетилена
остались сейчас только в стационарных сварочных мастерских.
Чаще пользуются ацетиленом из стальных баллонов, где его
хранят под давлением. Заполнять пустые баллоны газом и
транспортировать их очень опасно. При давлении 45 ат
ацетилен становится жидким. В этом состоянии он может
взрываться без всякой видимой причины. Работать с жидким
ацетиленом в стальных баллонах сложно. Для безопасности
ацетилен под небольшим давлением нагнетают в стальной
баллон, заполненный ацетоном. При давлении приблизительно
12 ат ацетон поглощает 300-кратное количество газа. Если
открыть вентиль, ацетилен освобождается подобно тому,
как, вспенивая жидкость, выходит наружу избыточный
углекислый газ из только что откупоренной бутылки лимонада.
Карбид кальция производится в больших количествах
для получения ацетилена, однако в последнее время для этой
цели все больше используют нефть и природный газ. Лишь
незначительная часть производимого ацетилена находит
применение в сварочной технике. Гораздо бблыпую роль он
играет как исходный материал в химическом синтезе: поскольку
его молекула имеет тройную связь, ацетилен легко вступает
в реакции с разнообразными веществами. Из него получают
ацетальдегид, спирт, уксусную кислоту, ацетон. Эти
соединения в свою очередь перерабатывают во множество других
веществ. Самое главное, что при некоторых условиях
молекулы ацетилена могут объединяться в молекулы-великаны. Мы
уже упоминали о таком процессе и назвали его
полимеризацией. Путем полимеризации ацетилена получают
искусственный каучук и целое семейство пластмасс. Поэтому
государственный план развития химии в ГДР предусматривает
увеличить выпуск карбида кальция. Завод искусственного
каучука в Шкопау станет крупнейшим производителем карбида в
мире. Без карбида кальция, а следовательно, без ацетилена
был бы невозможен нынешний уровень развития химии.
Сегодня можно с полным правом говорить о специальной
химии ацетилена.
Следующий член ацетиленового ряда — пропин, или алли-
лвн, СНз—С = СН с суммарной формулой С3Н4. Это
вещество, как и следующие за ним бутин-1 СНз—СН2—С^СН и бу-
тин-2 СНз—С=С—СНз (суммарная формула в обоих случаях
С4Н6), не имеет такого значения, как ацетилен. Мы привели
формулы этих соединений только для того, чтобы
вывести общую формулу всех членов ряда алкинов —
СпН2п-2.
Конец венчает дело
Успех любого дела зависит прежде всего от того, какой
метод, какую систему избирают для его осуществления.
Такое утверждение неново. Оно относится к химии вообще и к
органической химии в частности. Сейчас известно более
миллиона органических соединений и ежедневно в лабораториях
открывают или синтезируют все новые и новые вещества. Без
определенной системы в таком многообразии не разобраться.
Если бы мы решили изучить органическую химию, просто
вызубрив все соединения в алфавитном порядке, то,
разумеется, потерпели бы неудачу. Подавленные обилием
разрозненных фактов, мы потеряли бы вскоре всякую ориентацию.
В нашей голове накопились бы отрывочные знания, мертвые
и бесполезные.
Гораздо плодотворнее отыскать у различных веществ
общие свойства и, выделив из всего многообразия типичное,
объединить их в группы и подгруппы. Тогда станет ясно, что
многие соединения по своим свойствам мало отличаются друг
от друга, вступают в одинаковые реакции, а изучив одно из
веществ той или иной группы, можно многое сказать о
других.
Систематизация выявляет взаимосвязи и очень облегчает
работу. В сложных формулах и длинных названиях
отражается хорошо построенная и строго продуманная система.
Название 2,4-диамино-5-дихлорацетиламино-6-оксипирими-
дин — это не плод разгоряченной фантазии или
отрывок химической скороговорки, а научное название,
которое многое говорит химику о структуре и свойствах
соединения.
В неорганической химии, насчитывающей 60000
различных соединений, существует своя классификация, работать
без которой было бы невозможно. Согласно этой
классификации различают элементы и соединения. Элементов
немногим более ста, все остальное — соединения. Так как
соединений очень много, их разделяют на группы кислот, оснований
и солей. Эти три группы охватывают почти все
неорганические соединения. Отдельные представители этих групп
иногда сильно отличаются друг от друга, но в принципе имеют
много общего.
Так, все кислоты в большей или меньшей степени
диссоциируют на положительно заряженные ионы водорода и от-
рицательно заряженные кислотные остатки. Кислота
окрашивает лакмусовую бумагу в красный цвет и имеет типичный
кислый вкус. Основания состоят из характерных для них
гидроксильных ионов и ионов металлов. В отличие от кислот
они окрашивают лакмусовую бумагу в синий цвет и имеют
щелочный вкус. При нейтрализации кислот и оснований
получаются соли, содержащие отрицательно заряженный
кислотный остаток и положительно заряженный ион металла.
Классификация хорошо видна из такой небольшой таблички.
с=
Общая формула
Диссоциируют на
Реакция
I
Кислоты
HR
Н+ и R"
Кислая
Основания
МеОН
Ме+ и ОН-
Щелочная
4-
Соли
MeR
Ме+ и R"
Нейтральнйя
Заменяя у соли кислотный остаток, получают различные
подгруппы соединений: сульфаты, нитраты, хлориды, иодаты
и т. д., а заменяя ион металла — другие подгруппы: соли
натрия, соли железа и т. п.
И наконец, вспомним, что элементы сгруппированы в
периодическую систему. Поскольку органическая и
неорганическая химия отличаются друг от друга в своей основе, эту
систематизацию, конечно, нельзя перенести на органические
соединения. Как уже говорилось, у органических соединений
преобладает гомеополярная атомная связь, а у
неорганических — ионная, допускающая диссоциацию веществ в водном
растворе. Кроме того, органические вещества состоят в
основном только из четырех элементов: углерода, водорода,
кислорода и азота, а неорганические — более чем из ста.
Несмотря на то что органические соединения строятся всего
из четырех элементов, количество их огромно. Этим
многообразием природа обязана способности углеродных
атомов соединяться друг с другом в прямые и разветвленные
цепи.
Открытую цепную структуру имеют молекулы
алифатических соединений. Иногда углеродные цепочки
замыкаются, образуя кольцо. Вещества, молекулы которых имеют фор-
VH*
му кольца, называются циклическими соединениями. Особую
роль играет кольцо бензола, состоящее из шести атомов
углерода и шести атомов водорода. Подробнее мы расскажем о
бензоле несколько ниже. От бензольного кольца берет начало
целый ряд веществ — ароматических соединений. Свое
название они получили потому, что некоторые из них имеют
приятный, душистый запах.
Познакомившись с алифатическими соединениями, мы
узнали о существовании гомологических рядов метана,
этилена и ацетилена, представители каждого из которых
отличаются друг от друга на одну и ту же группу — СНг. Мы
установили, что физические свойства членов таких рядов
изменяются по определенному закону, например температура
плавления и кипения соединений увеличивается с
возрастанием молекулярного веса, в то время как их химические
свойства в основном не меняются. Как мы уже знаем, алканы
отличаются очень низкой реакционной способностью, потому
что в их молекулах все валентности насыщены атомами
водорода или углерода.
Для алканов возможны только реакции замещения. В
отличие от них алкены и алкины со своими ненасыщенными
двойными и тройными связями всегда готовы вступить в
реакцию. Двойные и тройные связи легко превращаются в
простые, а к свободным валентностям присоединяются другие
атомы или атомные группы. Повторим еще раз: алкены и
алкины отличаются от алканов более сильной способностью к
реакциям.
Если в молекулах углеводородов любой группы
заместить один или несколько водородных атомов на другие атомы
или атомные группы, образуются новые классы соединений.
При замещении на гидроксильную группу ОН получают
спирты. Бели обозначать углеводородные остатки через R, то
формула любого спирта будет ROH. Спирты можно получить,
воздействуя влажной окисью серебра на галоидозамещенные
углеводороды (влажную окись серебра можно рассматривать
как гидроокись):
Лс£ -/- o4<jOH ■—>&оц +J3c€
Примером может служить реакция между производным
метана — хлористым метилом и гидроокисью серебра,
приводящая к образованию метилового спирта (метанола),
ZCH56H ~\-Z№ >Z%OM -f- Hz
До сих пор мы рассматривали спирты как производные
парафиновых углеводородов, в формуле которых буквой R
обозначен углеводородный остаток. Если R заменить
водородным атомом, получится НОН, или НгО, т. е* обычная
вода.
Следовательно, мы могли бы рассматривать спирты как
производные воды, где один атом водорода замещен
углеводородным остатком. Как ни поразительно это утверждение на
первый взгляд, оно совершенно справедливо. У первых членов
ряда спиртов — метанола и этанола, действительно,
наблюдается известное сходство с водой. Так, их точки кипения
мало отличаются от точки кипения воды. Это сходство
подтверждается и старым правилом — подобное растворяется в
подобном. Метиловый, этиловый и пропиловый спирты
смешиваются с водой в любых соотношениях. Чем тяжелее
углеводородный остаток, тем сходство с водой меньше,
и, наконец, такой спирт, как гексиловый, с формулой
СН3СН2СН2СН2СН2СН2ОН в воде почти нерастворим. Из
формулы этого спирта видно, что углеводородный остаток в
его молекуле преобладает, чем и определяются ее
свойства.
Легко заметить, что формулы спиртов похожи на
формулы оснований, известных из неорганической химии, в
которых тоже содержится группа ОН, но соединенная с ионом
металла. Может возникнуть предположение, что спирты —
это органические гидроокиси, основания. Однако это не так.
Сравнивая основания и спирты, нетрудно установить, что
сходство между ними чисто формальное. Уже в механизме
связи обнаруживаются существенные различия. Для
оснований — типичных неорганических веществ — характерна ярко
выраженная ионная связь, и значит, в воде они подвергаются
электролитической диссоциации. А у спиртов преобладает
гомеополярная, атомная связь, и никакой диссоциации в воде
не наблюдается.
Различие отчетливо проявляется и в характерных
реакциях. При реакции спирта с металлическим натрием
образуется алкоголят и выделяется водород:
сн^ + ^ой- > снъо\) \-J<jc£
Главное свойство гидроокисей — их основной характер.
Поэтому они антиподы кислот. Гидроокиси окрашивают
красную лакмусовую бумажку в синий цвет и имеют щелочной
вкус. Мы хорошо знаем из собственного опыта, что у спиртов
вкус совсем иной. Они могут опьянять, как этиловый спирт,
или бывают сладкими, как глицерин, но щелочного вкуса
никогда не имеют.
Вполне очевидно, что гидроокиси и спирты надо
рассматривать как две различные группы веществ.
Гидроксильная группа в спиртах в значительной мере
определяет все их свойства и реакции. Такие группы,
которые характеризуют весь класс соединений в целом, принято
называть функциональными группами. Наличие таких групп
определяет общие черты у различных соединений одного
класса. Например, поведение всех спиртов по отношению к
щелочным металлам и кислотам одинаково. Конечно, на
свойства спиртов влияют и алкильные остатки. Метиловый и эти-
ловый спирты резко отличаются друг от друга, если не по
физическим свойствам, то по вкусу и воздействию на
организм. Однако оба спирта синтезируются Щ) одному и тому же
принципу и вступают в одинаковые реакции с натрием и
кислотами. Функциональные группы обычно пишут на конце
формулы, но это, разумеется, не всегда соответствует
истинному положению радикалов в молекуле.
Функциональные группы определяют типичные свойства
и реакции соответствующих классов веществ.
Объясним это на примере.
Паровоз, тепловоз, электровоз. Слыша «воз» в конце
слова, мы понимаем, что речь идет о чем-то движущемся, скорее
всего по рельсам. Неважно, что в одном случае движущая
сила — пар, а в другом — электричество и они отличаются
друг от друга. Главное у всех этих средств передвижения —
общие функции, на что и указывает одинаковое окончание
«воз».
Спирты — большой класс органических соединений,
который в свою очередь можно подразделить на более мелкие
группы. По числу гидроксильных групп в молекуле
различают одноатомные и многоатомные спирты. Самые известные
одноатомные спирты метанол СНзОН и этанол С2Н5ОН.
Метиловый спирт относится к тем химическим соединениям,
которые известны человечеству с давних пор. Этиловый спирт
для пищевой промышленности получают при брожении
картофеля, зерна и фруктов. Кроме того, его можно
синтезировать искусственным путем.
Из трехатомных спиртов наиболее известен глицерин. Он
образуется при замещении в пропане СН3СН2СН3 трех атомов
водорода гидроксильными группами по одной у каждого
атома углерода. Получается соединение с суммарной формулой
СзН5(ОН)3 и структурной
н
14—с-—оН
И с—еИ
А
В связанном виде глицерин является составной частью
всех жиров. В чистом виде — это густая гигроскопичная
сладковатая жидкость. Глицерин употребляется в косметике
и в больших количествах идет для изготовления
нитроглицерина.
К классу спиртов близки альдегиды. Под воздействием
на спирт какого-либо окислителя протекает реакция
дегидрирования (отнятия водорода) и образуется альдегид. 3jot
способ получения определил название всего класса веществ.
Слово альдегид происходит от alkohol dehydrogenatus, что
значит спирт, у которого отнят водород. Для метилового
спирта эта реакция протекает по уравнению
Hi—с—он + iPi- ►**—с<
+
it
чн
Получается формальдегид. Его название происходит от
названия муравьиной кислоты acidium formicum, образующейся
при дальнейшем окислении формальдегида. В больших
количествах формальдегид получают пропусканием смеси метана
и воздуха через раскаленные сетки из платины, меди или /
серебра, которые в этом процессе играют роль катализаторов. vvv
Катализаторы приходится применять потому, что
кислород воздуха недостаточно сильный окислитель для реакции
дегидрирования. Эта реакция протекает с отдачей тепла — она
экзотермична. Поэтому катализатор остается раскаленным
за счет тепла реакции и его не требуется разогревать
дополнительно. Если горячую медную спираль, покрытую тонким
слоем окисла, предварительно раскалить на воздухе и
опустить в метиловый спирт, то кислород окиси меди окислит
спирт, и образующийся формальдегид легко можно
обнаружить по специфическому острому запаху. В то же время
темная проволока становится красной, так как окисел
восстанавливается до меди:
енът+Си0 —> нсно+нген-сч,
В этом процессе медь является переносчиком кислорода.
При комнатной температуре формальдегид — газ, легко
растворяющийся в воде. Под названием формалин продается
35%-ный водный раствор формальдегида, который нашел
широкое применение как дезинфицирующее средство. В
дыме, образующемся при неполном сгорании дерева, угля,
содержатся небольшие количества формальдегида.
Консервирующее действие дыма при копчении мяса и колбасных
изделий, по крайней мере частично, следует приписать
формальдегиду. В наши дни формальдегид производят в
промышленном масштабе и используют главным образом как исходное
сырье для изготовления пластмасс и других полимеров.
Другой альдегид, также имеющий важное значение,
получают путем окисления этанола. Отняв от этанола два
атома кислорода, получают ацетальдегид:
сцъсня&н-\-о -^снъс -н tizo
И
6
Acidum acetum — латинское название уксусной кислоты,
образующейся при его дальнейшем окислении. Вот откуда
происходит название ацетальдегид. В промышленном
производстве обычно исходным продуктом является ацетилен. В
присутствии катализатора к молекуле ацетилена присоединяется
молекула воды и образуется ацетальдегид3.
WOssrCH + Н-оН
Для всех альдегидов характерна карбонильная группа
—с
Любое соединение с такой группой можно смело отнести к
альдегидам. Как видно из формулы, карбонильная группа
содержит двойную связь и, следовательно, реакционноспо-
собна. Действительно, альдегиды легко вступают в реакции с
другими веществами и играют поэтому важную роль в
химическом синтезе. Формальдегид и ацетальдегид реагируют
между собой с разрывом двойных связей и образованием
полимерных молекул. Альдегиды — очень важный класс
веществ.
К спиртам и альдегидам близки органические карбоно-
вые кислоты. Наиболее распространенный способ их
получения — окисление спиртов, причем как промежуточный
продукт образуется сначала соответствующий альдегид.
Разумеется, альдегид можно окислить далее до кислоты:
__ Фкисилние, а Окиси&нме* ~
(^штгку — —>- 'Лшуегиу ^t/fapfo/tofysi, кцсиогксс
Если, например, исходить из этанола, схема реакции
такова:
Для всех карбоновых кислот характерна карбоксильная
группа п\
— С^
которую обычно записывают проще — СООН.
50
У карбоновых кислот в основном такие же свойства, как
у известных нам неорганических минеральных кислот. В
водном растворе они окрашивают лакмус в красный цвет,
обладают кислым вкусом и нейтрализуются щелочами с
образованием солей. Так что ничего принципиально нового по
сравнению с минеральными кислотами у карбоновых нет. Для
них также характерна более или менее ярко выраженная
диссоциация на кислотный остаток и ион водорода. В
конечном счете диссоциация и обусловливает кислотные свойства
соединения. Однако если разбавленные минеральные
кислоты распадаются на ионы практически на 100%, то степень
диссоциации карбоновых кислот значительно меньше.
Равновесие между диссоциированной и недиссоциированной карбо-
новой кислотой значительно смещено влево, поэтому такие
кислоты являются слабыми.
При окислении простейшего спирта — метанола
образуется простейшая кислота — муравьиная:
Ctf,©H -—*Н-СГ
Кроме карбоксильной группы, в ней содержится только
один атом водорода, соединенный с углеродом. Это придает
муравьиной кислоте особые свойства, выделяющие ее среди
других кислот. Муравьиную кислоту можно рассматривать
как альдегид, у которого группа ^
-<
связана с гидроксильным остатком. И в самом деле,
муравьиная кислота вступает в некоторые реакции, характерные
только для альдегидов.
А есть ли у муравьиной кислоты что-либо общее с
муравьями?
Да, эта кислота — главное оружие рыжих лесных
муравьев (Formica rufa). Им они парализуют добычу и убивают
своих врагов. Каждый, кто когда-нибудь разорял
муравейник, помнит жгучую боль, которую причиняет муравьиная
кислота.
Более важна уксусная кислота СНзСООН — основная
составляющая часть различных сортов уксуса. Уксус известен
человечеству очень давно, его легко получить окислением
вина и других алкогольных напитков. Если оставить
открытой бутылку вина, то через некоторое время оно станет
кислее, чем было раньше. Спирт превратился в уксусную
кислоту, пить его теперь нельзя, он годится только на заправку
салатов. Мы получили винный уксус. Конечно, такая реакция
не всегда удается. Из портвейна, в котором много спирта,
уксус не образуется. Можно спокойно оставлять открытыми
ликер, водку и другие крепкие напитки: они могут
испариться, но не прокиснуть.
Объяснить это нетрудно. В этом случае происходит
реакция окисления кислородом воздуха, и уксусная кислота
образуется в большем количестве, чем ацетальдегид, но скорость
этой реакции при комнатной температуре так мала, что ее
течение практически незаметно. В подобных случаях
повышению скорости реакции, а иногда и вообще ее
осуществлению способствует, как известно, присутствие
соответствующего катализатора. Для превращения вина или, точнее
говоря, спирта в вине в уксусную кислоту без катализатора тоже
не обойтись. В этом случае такие катализаторы выделяют
живые организмы. В процессе жизнедеятельности некоторых
бактерий выделяются биокатализаторы, или ферменты,
способствующие окислению спирта в уксусную кислоту.
Разумеется, бактерии делают это не по злому умыслу и
не для того, чтобы заставить нас сразу выпить только что
откупоренную бутылку. Дело в том, что освобождающаяся
при таком окислении энергия обеспечивает их
жизнедеятельность. Для бактерий превращение спирта в уксусную
кислоту — необходимое условие их существования.
Поскольку такие бактерии обитают практически повсюду, то на
воздухе в жидкостях, содержащих спирт, всегда происходит
окисление спирта в уксусную кислоту при условии, что
количество спирта не слишком велико. Если концентрация спирта
повышается, бактерии погибают, и всякое выделение
ферментов прекращается. Концентрированные растворы спирта
могут служить отличным консервирующим средством.
Раньше получали уксус только ферментативным
окислением спирта и для ускорения этого процесса применялись
особые приемы. Например, спирт пропускали через буковые
стружки, богатые уксуснокислыми бактериями; при этом
также увеличивалась поверхность жидкости, кислород
получал более свободный доступ к ней, и реакция заканчивалась
очень скоро.
Позднее научились получать древесный уксус. При
нагревании без доступа воздуха из-за недостатка кислорода
дерево не сгорает, а разлагается. Наряду с газом и смолой
отгоняется жидкость, окрашивающая синюю лакмусовую
бумагу в красный цвет. Простое исследование показывает,
что это уксус.
В наши дни известны более рациональные методы
получения уксусной кислоты, которая в больших количествах
используется в промышленности. К ацетилену в присутствии
катализатора присоединяется вода, получается ацетальдегид,
легко окисляющийся в уксусную кислоту. Поскольку
ацетилен получают из карбида кальция, в этом случае говорят о
карбидном уксусе. По своим химическим свойствам он не
отличается от уксуса, полученного из естественного сырья.
Жирные кислоты — производные более
высокомолекулярных углеводородов — имеют большое значение: они
являются составной частью животных и растительных жиров.
До сих пор мы рассматривали кислоты только с одной
карбоксильной группой в молекуле. Помимо таких монокар-
боновых кислот, существуют дикарбоновые — с двумя
карбоксильными группами. Известны также трикарбоновые и тетра-
карбоновые кислоты.
Простейшая дикарбоновая кислота — щавелевая
(СООН)2. Ее структурная формула показывает, что она
состоит из двух карбоксильных групп, связанных между собой.
Название она получила по своей калийной соли,
содержащейся в щавеле.
Если в углеводородных соединениях заместить один или
несколько атомов водорода на галогены, то получается
многочисленная и важная группа галоидных производных. Такое
замещение иногда можно осуществить прямым действием
галогенов на углеводороды. Другой метод их получения
заключается во взаимодействии спиртов с галогеноводородами,
например
ct+3 он + Нес < > СНЪ£С + #гО
Гидроксильная группа спиртов замещается на атом
хлора хлористого водорода, и образуется вода и хлористый метил
(метилхлорид).
Мы уже говорили, что гидроксильная группа спирта
существенно отличается от гидроксильной группы щелочи.
Однако только что описанная реакция настолько похожа на
реакцию нейтрализации, что может возникнуть сомнение, не
происходит ли в этом случае нейтрализация кислоты
основанием.
Рассмотрим еще раз типичную реакцию
нейтрализации — взаимодействие едкого натра с соляной кислотой
При этой реакции образуется соль—хлористый натрий—
и вода. Поскольку едкий натр и соляная кислота
диссоциированы почти на 100%, речь может идти только об ионной
реакции. Поэтому превращение протекает мгновенно и до конца
в направлении, указанном стрелкой:
Л5г+ + 0Н~+ И1" + СС~ »1кЧ<кг+ #2р
При образовании хлористого метила происходит совсем
иное. Для молекулы органических соединений — а метанол,
естественно, относится к ним — характерна атомная связь,
никакой диссоциации и, следовательно, образования ионов
ОН произойти не может. У спирта нельзя обнаружить
никаких основных свойств, ведь эти свойства как раз
обусловливаются присутствием гидроксильных ионов. Взаимодействие
метанола с кислотой протекает медленно и не до конца,
поскольку получающийся хлористый метил и вода реагируют
между собой с образованием исходных соединений. Чем
больше получается хлористого метила и воды, тем заметнее
протекает обратная реакция. Наконец, наступает такой
момент, когда в единицу времени распадается столько же
молекул хлористого метила, сколько образуется вновь.
Наступает состояние равновесия, определяющее конечный
выход реакции.
Возможно ли сместить это равновесие так, чтобы
повысился выход хлористого метила? Решить эту задачу можно
двумя способами. Во-первых, экспериментальным путем, но
для этого пришлось бы провести большое число опытов,
каждый раз изменяя условия реакции, и получить результат
только после долгой и терпеливой работы. Проще применить
к этому частному случаю общие законы, например закон
действующих масс.
Возьмем за основу уже известное нам уравнение реакции
сН3оН t Htf <=^снвс£+ НьО
и рассмотрим отдельно каждую его часть. Во-первых,
образование хлористого метила
си5ои + №-**сн5се+ Hzo (1)
а во-вторых, обратную реакцию — разложение хлористого
метила
Ш3с{ + цр -^СН3оН + НсС (г)
Соответствующие скорости реакции обозначим и\ ш V2,
понимая под этим выход вещества в единицу времени.
Если метиловый спирт и соляная кислота реагируют
друг с другом согласно уравнению (1), то одна молекула
спирта сталкивается с одной молекулой хлористого водорода.
Происходит ли при этом между ними взаимодействие —
другой вопрос, во всяком случае предпосылка для него
существует. Чем чаще случаются такие столкновения в единицу
времени, тем вероятнее, что реакция произойдет, и тем выше
ее скорость. Число столкновений зависит в основном от
количества молекул в единице объема. Если в темной комнате
ходят два человека, то они будут сталкиваться («вступать в
реакцию» друг с другом) реже, чем если бы в том же
помещении быстро двигались сто человек. Следовательно,
повышение концентрации увеличивает скорость образования
хлористого метила
где Сен .он и Chq — концентрация метанола и хлористого
водорода. Вместо С обычно пишут химическую формулу
соединения в квадратных скобках. Такое написание обозначает
то же самое, но проще и нагляднее:
v, = £, • \сн3е>н] • [на]
здесь к\ — коэффициент, который для нашей эЬдачи
значения не имеет. Этот коэффициент учитывает температуру,
давление, присутствие катализатора и некоторые другие
факторы, влияющие на скорость реакции. Так как мы не
предполагаем никаких внешних влияний, т. е. считаем все эти
факторы неизменными, к\ в нашем случае есть величина
постоянная.
То же самое относится и к скорости обратной реакции
разложения хлористого метила, выраженной уравнением (2).
Взаимодействие хлористого метила и воды происходит только
при столкновении друг с другом молекул обоих соединений.
Разумеется, частота таких столкновений зависит от
концентрации обоих веществ. Для v% это можно записать так:
где &2 — постоянная величина, к ней относится все,
сказанное о Ль
Рассмотрим еще раз подробнее ход реакции. В качестве
исходных веществ мы берем смесь метанол — хлористый
водород. Вначале налицо лишь молекулы, реагирующие друг с
другом со скоростью реакции, равной v\. Естественно, что в
ходе реакции концентрация метанола и хлористого водорода
снижается, тогда как концентрация конечных продуктов
реакции постоянно возрастает. Они могут вступить во
взаимодействие друг с другом согласно уравнению (2). Вначале скорость
этой реакции (Рг) невелика, так как молекул хлористого
метила и воды еще мало. Но постепенно в ходе реакции (1)
концентрация конечных продуктов возрастает и соответственно
увеличивается i>2. В то же время концентрация исходных
веществ падает, а значит, уменьшается v\. И наконец,
наступает момент, когда скорости обеих реакций выравниваются и
i>i = i>2. Наступает состояние равновесия, когда соотношение
между исходными и конечными веществами не меняется,
несмотря на то что эти вещества продолжают
взаимодействовать между собой. Такое состояние равновесия типично для
многих химических реакций, его называют подвижным
равновесием.
ПОСКОЛЬКУ У1 — У2, ТО
Если перенести константы в одну сторону, получится
где &2/&1, конечно, останется постоянной величиной.
Обозначим ее К. Мы установили таким образом для нашего
конкретного случая закон действующих масс.
В состоянии равновесия устанавливается совершенно
определенное соотношение между концентрациями исходных
и конечных продуктов реакции. Естественно, это
соотношение справедливо только при том условии, что другие
факторы, такие, как давление, температура и т. п., не изменяются.
Как применяют закон действующих масс?
Обычно мы стремимся к тому, чтобы выход конечного
продукта реакции был как можно больше. В нашем случае
желательно получить наиболее высокую концентрацию
хлористого метила. Если ее величина возрастет, изменятся и
концентрации других веществ, так как величина всей дроби
должна оставаться постоянной, равной К. Если хотят
повысить содержание СНзС1, то либо добавляют спирт, либо
каким-то образом уменьшают количество НгО. На практике
пользуются обоими способами. В смесь добавляют вещество,
связывающее воду, тем самым концентрация образующейся
воды все время уменьшается, а равновесие сдвигается.
Можно добавлять концентрированную серную кислоту или
хлористый цинк. Оба вещества очень гигроскопичны, т. е.
хорошо поглощают воду. Для получения большого количества
конечного продукта обычно повышают концентрацию наибо-
56
лее дешевого из двух исходных веществ. В нашем случае
стоимость метанола и хлористого водорода примерно
одинакова, но если бы пришлось проводить реакцию между
дорогой кислотой и дешевым спиртом, разумеется, нужно было
бы в избытке употребить спирт.
Итак, закон действующих масс позволяет проводить
реакцию количественно, сознательно направляя ее ход. Вот
почему этот закон так важен для химика.
Реакцию между спиртом и кислотой называют реакцией
этерификации, а образовавшийся наряду с водой продукт
реакции — сложным эфиром. В нашем случае мы
использовали неорганическую кислоту и получили солянокислый эфир
метилового спирта. Но часто для реакции этерификации
применяют и органические кислоты.
Сложные эфиры играют большую роль в органической
химии. Все растительные и животные жиры — это сложные
эфиры жирных кислот и трехатомного спирта — глицерина.
В дальнейшем мы еще вернемся к ним. Большое значение в
технике имеет ряд галоидопроизводных, в том числе и
сложные эфиры галогеноводородных кислот. Об этом мы также
расскажем подробнее в следующих главах.
Невозможно сразу рассмотреть все другие группы
веществ со сходными структурами, свойствами и реакциями.
Мы еще успеем познакомиться с ними дальше. А сейчас
хочется подчеркнуть еще раз, что огромное многообразие
органических веществ укладывается в строгую и точную систему.
Конечно, число соединений от этого не становится меньше,
но появляется возможность обоснованно предсказывать
свойства, способы получения и реакции еще не известных
соединений.
Путь к нефти
В 1849 году немного было мест более глухих, чем городок
Торентум, раскинувшийся у подножия Аллеганских гор
недалеко от Питтсбурга. Мало кто оседал здесь, да и
путешественники показывались тут нечасто. Один из жителей этого
городка — мистер Кайер владел в окрестностях несколькими
соляными источниками. Большого богатства они ему не
сулили — источники были плохие: из-под земли редко бил
чистый солевой раствор. Обычно он был загрязнен бурой
неприятно пахнущей жидкостью.
Кайер знал, что это нефть. И когда однажды вместо
солевого раствора забила чистая струя нефти, он решил
обратить несчастье в удачу.
В то время нефть употребляли в качестве смазочного
материала и в лечебных целях. Надобность в ней была
невелика, и, конечно, большого дохода Кайер получить не мог. Вот
если бы из нефти можно было получать масло для
светильников или использовать ее в качестве горючего для ламп, то
можно было бы поправить пошатнувшиеся дела, — думал
неудачливый предприниматель.
По своим собственным чертежам Кайер построил
небольшой куб для перегонки неочищенной нефти. В 1850 году
ему впервые удалось получить керосин, пригодный для
использования в лампах. С каждым днем спрос на него
возрастал, дело процветало, и через пять лет Кайер, уже ничем не
рискуя, построил куб для перегонки емкостью 600 литров.
Теперь он мог получать сколько угодно ценного горючего, но
сами источники были бедны нефтью, и производство Кайера
имело только местное значение.
Шло время, интерес к нефти возрастал. Жители городка
Титусвилла в штате Пенсильвания знали, что вокруг
имеется множество природных нефтяных источников. Эти
источники вполне покрывали потребности в нефти как в
лекарстве и смазочном материале, но никто не знал, куда девать ее
излишки. Тут и вспомнили о перегонном кубе Кайера.
Выбросив на рынок большое количество керосина, можно было
бы наверняка сделать хороший бизнес. Эта мысль пришла
в голову двум дельцам Джорджу Бисселю и Джонатану Эве-
лету, и они за довольно крупную сумму приобрели
земельные участки вокруг Титусвилла с находящимися на них
нефтяными источниками. Вместе с банкиром Таунсендом они
основали компанию «Пенсильвания Рок ойл». Однако
природные источники давали мало нефти, и рассчитывать
только на них было невыгодно. Природе надо было помочь,
пробурив в недра земли скважины.
Таунсенд, который к тому времени стал единственным
владельцем фирмы, нашел человека, взявшегося
осуществить зту задачу. Его звали Эдвин Дрейк.
Дрейк начал действовать обдуманно и планомерно,
используя опыт, полученный при эксплуатации соляных
источников. Он привлек к делу опытных работников, купил
необходимый инструмент и машины и приказал выстроить
десятиметровую буровую вышку.
К концу весны 1859 года все было готово, и можно было
начинать бурение. Однако вскоре после начала работ
возникла трудность, казавшаяся на первый взгляд непреодолимой.
Нефтяную скважину все время засыпало породой, и всю
работу приходилось начинать сначала.
Дрейк был близок к отчаянию, но тут ему пришла в
голову блестящая идея — по мере проходки скважины
опускать в нее толстую стальную трубу, предотвращая тем
самым обрушение стенок скважины.
Работы продолжались, и, наконец, 27 августа 1859 года
на глубине двадцати одного метра была обнаружена нефть.
Каждый день скважина давала 1500 литров нефти —
огромное количество по тем временам. Добыча быстро покрыла
все расходы.
Слух об этом событии разнесся повсюду, в Америке
началась нефтяная лихорадка. Одна за другой сооружались
нефтяные вышки, и уже через десять лет в Пенсильвании
добывалось около 300 миллионов литров нефти в год. Нефть
начинали применять во все новых и новых областях. После
изобретения дизельных двигателей ее стали использовать в
качестве горючего и смазочного материала. Постепенно нефть
как горючее вытесняла уголь. В последние десятилетия она
служит ценнейшим сырьем для химической
промышленности.
В наши дни нефть наряду с углем является важнейшим
химическим сырьем и энергетическим источником. В 1962
году ее мировая добыча составила 1,1 миллиарда тонн.
Каждый год открывают все новые нефтяные месторождения.
И все-таки нельзя вести историю добычи нефти с
1859 года. Действительно, с этого времени стала развиваться
нефтяная промышленность, именно тогда были начаты в
крупных масштабах буровые работы, но впервые человек
начал использовать нефть еще в глубокой древности.
Точно не известно, когда человек впервые применил
летучие нефтяные фракции. При исследовании этого вопроса
мы вынуждены довольствоваться рисунками, реже
надписями. Однако чем древнее эпоха, в которую мы стараемся
проникнуть, тем их становится меньше и понять их труднее.
Во всяком случае, достоверно установлено, что уже
более 6000 лет назад шумерам, населявшим до вавилонян и
ассирийцев территорию между Тигром и Евфратом, был
известен вязкий смолообразный нефтяной битум, образующийся из
выступившей на поверхность нефти, после того как из нее
испарились наиболее летучие части. Шумеры использовали
его как вяжущее и уплотняющее вещество. Там, где жили
шумеры, было мало природных строительных материалов —
дерева и камня. Поэтому шумеры возводили свои постройки
из кирпича, который приготовляли из смеси песка, глины или
гравия и битума (около 35%). Этот кирпич обладал
поразительной прочностью.
■UpnuZs
Шумеры, разумеется, видали и жидкую нефть и знали,
что если она останется на воздухе, то превратится в битум.
Они называли ее esir, что означает светящаяся вода,
ff*^ Применяли нефть и для лекарственных целей. Битумны-
■fc ми мазями лечили чесотку и нарывы, а длительными «ванна-
/*« ми» в нефтяных лужах пытались избавиться от боли в
суставах. При болезнях желудка жевали пилюли из нефтяного
битума. Теперь кажется сомнительным, чтобы эти лекарства
помогали.
Огромные сооружения городского типа, обнаруженные
при раскопках вавилонских и ассирийских поселений,
удалось воздвигнуть только благодаря широкому применению
природных битумов.
Битум применялся для постройки зданий, мостовых —
прообраза наших асфальтовых дорог, каналов, для
укрепления берегов рек и искусственных водоемов. Жидкую нефть,
как показывают археологические раскопки, уже тогда
использовали как горючее в светильниках.
Еще в XVIII веке до нашей эры знали нефть и в
Китае. Для ее добычи там строились специальные нефтяные
колодцы. Китайцы употребляли нефть для освещения, как
лекарство и в военных целях. Китайские воины из «огненных
повозок» бросали горшки с горящей нефтью в ряды врагов.
Во время своих походов грекам и римлянам стали
известны многие месторождения нефти в Малой Азии. Римляне
называли нефть oleum petrae — каменное масло. Это
название перешло в другие языки. Отсюда произошло и русское
«петролеум», а один из продуктов перегонки нефти
называется петролейным эфиром.
Римляне привозили нефть и битум к себе на родину и
использовали их так же, как и покоренные ими народы.
У греков и римлян нефть, кроме того, применялась для
военных целей.
уЛссурд,
Уже давно были известны огненные стрелы, острия
которых обматывались паклей, пропитанной нефтью, а в VII веке
нашей эры византийцы создали так называемый «греческий
огонь». «Возьми чистую серу, нефть, винный камень, смолу,
поваренную соль, деревянное масло; хорошенько провари все
вместе, пропитай этим составом паклю и подожги. Такой
огонь можно погасить только песком или винным уксусом» —
так написано в одном из многочисленных рецептов, которые
греки хранили в глубочайшей тайне.
Применялся «греческий огонь» так.
Горючую смесь из насосов направляли на вражеские
корабли или просто выпускали в воду, а потом поджигали ее
огненными стрелами. Часто к ней примешивали негашеную
известь, которая реагирует с водой, выделяя большое количе
ство тепла, и тогда нефть загоралась. Огонь нефтяного
пламени, загасить которое было почти невозможно, полностью
уничтожал деревянные суда противника.
«У греков есть огонь, подобный небесной молнии. Они
направляли его на нас и сжигали дотла все попадавшееся на
пути, поэтому победить их невозможно», — так рассказывал
один из оставшихся в живых о морском сражении с греками.
Позднее арабы, познакомившиеся с достижениями
соседних народов, тоже узнали о нефти и внесли значительный
вклад в развитие методов ее переработки. Около 950 года
арабы построили первые установки для перегонки нефти. Они
делали перегонные кубы из обожженной глины или свинца.
В Германии нефть открыли в средние века. Известно, что
примерно к 1450 году в Тегернзее уже добывали тягучий
нефтяной продукт, который местные монахи — люди
предприимчивые — успешно продавали как средство против
подагры, боли в ушах и мозолей. Многие крестьяне
использовали его для смазки телег, положив начало нынешнему
применению нефти в качестве смазочного материала.
61
,j№
9>*%**f»'
vuateMHutc
Vo&torojapeA*
^«$й.
f*e«.
Уже в 1550 году около Витце и Ольхайма, недалеко от
Ганновера, была обнаружена выступающая на поверхность
смола. Около Витце в 1857 году под руководством профессора
Гунойса бурили скважины в поисках бурого угля, так как
тогда считали, что появление смолы и есть признак его
залегания.
За два года с большим трудом была пробурена скважина
глубиной 36 метров. 29 мая 1859 года на этой глубине
обнаружили нефть. Ежедневно из скважины добывали около
15 литров нефти — примерно ведро. Даже по тем временам
это не считалось большой удачей.
Систематическая разработка месторождений нефти
началась здесь намного позднее, чем в Америке. Эти участки
остаются нефтеносными и сейчас: они продолжают давать в
год свыше одного миллиона тонн нефти.
Еще задолго до того, как нефть приобрела такое значение
в мировом хозяйстве, какое она имеет сейчас, ученые многих
стран начали заниматься разгадкой тайны происхождения
нефти и возникновения ее огромных подземных
месторождений. Ответ на этот вопрос имеет не только чисто научное
значение, но и позволяет вести поиски нефти более
целенаправленно и успешно.
Как мы вскоре узнаем, нефть представляет собой смесь
углеводородов, имеющих различную структуру и длину цепи.
Эти соединения, входящие в состав нефти, уже давно были
известны химикам, и они умеют получать их в лаборатории.
При реакциях между карбидами — соединениями металлов с
углеродом — и водой образуются углеводороды. Мы уже
знаем, например, что при реакции карбида кальция с водой
получается газообразный ацетилен, а карбида алюминия —
метан. Карбид урана при реакции с водой также дает в
основном метан, но, кроме того, образуются в незначительных
количествах жидкие и даже твердые углеводороды, т. е.
соединения с относительно большими молекулами.
Может быть, много тысячелетий назад, когда земная
кора была намного тоньше, жидкая расплавленная магма
гораздо чаще, чем теперь, вырывалась на ее поверхность, где
вступала в реакцию с углеродом органических соединений, и
62
образовывались карбиды. Карбиды рано или поздно вступали
в реакцию с водой, и возникали углеводороды. Иногда этот
процесс мог проходить под землей. Высокие температура и
давление совместно с каталитическим воздействием каких-то
веществ превратили в течение длительного времени
первичные продукты в то, что сегодня мы добываем в виде нефти4.
На первый взгляд теория неорганического
происхождения нефти кажется весьма разумной и ясной. Не
удивительно, что у нее было много приверженцев. Однако через
некоторое время против этой теории появились веские
возражения, показавшие ее несостоятельность. В наши дни трудно
найти ее сторонников.
Прежде всего установили, что нефть никогда не находят
в формациях вулканического происхождения. А по
карбидной теории этого как раз и следовало бы ожидать, потому что
именно здесь, в местах вторжения расплавленной магмы в
земную кору, должны были создаваться наиболее
благоприятные условия для образования карбидов.
Кроме того, в нефти было обнаружено множество
сложных органических соединений, которые никак не могли
возникнуть в результате чисто неорганических процессов.
Например, такие соединения, как холестерин, хлорофилл,
половые гормоны, образуются только в живом организме.
Все это укрепляло предположение о том, что нефть,
подобно каменному углю и торфу, возникла на органической
основе. Более детальное изучение вопроса о происхождении
нефти заставило отказаться от предположения, что нефть
образовалась из отмирающих частей растений.
В настоящее время наиболее вероятным считают, что
нефть в течение тысячелетий образовывалась из тел
мельчайших живых организмов, обитавших в морской воде. В пользу
этого свидетельствует расположение нефтяных
месторождений.
Итак, наши сегодняшние представления о возникновении
нефти сводятся к следующему.
В спокойных прибрежных водах, отделенных от морей
песчаными отмелями, только в верхнем слое воды в
достаточном количестве содержался кислород. С глубиной
содержание кислорода в воде уменьшалось и возрастала
концентрация ядовитого сероводорода. Нечто подобное наблюдается
сейчас в Черном море, которое, как известно, соединяется со
Средиземным только узким проливом.
w*
j«**
<UCope~
о<з
/°Ь° Сушей
ov**-
Обитатели моря, попадая в эти прибрежные воды, уже на
незначительных глубинах встречали здесь такие условия, в
которых они не могли существовать, погибали и падали на
дно. Вымиранию организмов способствовало также и высокое
содержание соли. Концентрация солей повышалась из-за
сильного испарения мелких прибрежных вод, особенно
интенсивного в субтропическом и тропическом климате.
Так как на дне заливов концентрация сероводорода
была очень высокой, погибшие организмы не разлагались и все
более толстым слоем накапливались в иле. Колебания и
различные смещения земной коры загоняли ил в более глубокие
слои земли, где под влиянием давления, умеренных
температур и катализаторов из жиров и белков этих организмов
образовывались природный газ и нефть.
Только в редких случаях нефть оставалась там же, где
образовывалась — в пластах глины. Давление горных пород
выжимало ее в пустоты, трещины и расселины, а также в
пласты песка, между зернами которого много пустот. Пески,
как губка, впитывали нефть. В их слоях она и накапливалась.
Можно ли поднять на поверхность нефть, залегающую в
недрах земли?
&С&афиН<ит
64
машина
Уже очень давно на Кавказе5, где в некоторых местах
нефть просачивалась на поверхность, рыли колодцы
глубиной до 35 метров. Эти колодцы постепенно заполнялись
нефтью, и ее ковшами вычерпывали наверх.
Пока нефти требовалось немного — для приготовления
лекарств или для наполнения ламп, этот способ добычи был
пригоден. Но когда потребности в нефти сильно возросли,
пришлось искать другие способы.
Уже в XVIII веке при поисках подземных источников
воды использовалось так называемое ударное бурение.
Применение этого метода позволило осуществить и
рациональную добычу нефти.
Около вырытой вручную ямы ставили высокий хорошо
укрепленный столб. На верхнем конце его устанавливали
журавль, подобно тому как это сейчас можно увидеть в
деревенских колодцах. К нему подвешивали на цепи тяжелую
ударную штангу, на конце которой укрепляли стальную
головку. Если журавль с другого конца опускать и поднимать
вручную или с помощью машины, то ударная штанга,
поднимаясь, а затем падая, с силой ударяется в дно ямы. Для
очистки ямы от разрушенной породы через определенные
промежутки времени к цепи вместо штанги подвешивали
грейфер. Позднее этот метод усовершенствовали. Вместо
столбов стали строить высокие буровые вышки, которые
облегчили подъем и опускание штанги. Постепенно процесс
бурения механизировали. Но потребовалось еще очень много
терпения и изобретательности, пока научились бурить
глубокие скважины. Чтобы стенки скважины не
обрушивались, в нее опускали длинные железные трубы. За год
этим способом можно было пробурить скважину глубиной не
более 200 м. Методом ударного бурения, конечно, нельзя
было достичь больших глубин. Современные скважины глубиной
от 2000 до 7000 м стали возможны лишь после изобретения
вращательного бурения, которое явилось большим шагом
вперед в развитии техники бурения.
Буровая штанга, на нижнем конце которой укреплена
буровая коронка, опускается в скважину. С поверхности ей
придают вращательное движение, и коронка разрушает поро-
#
//
*
\
3 3. Шпаусцус
65
ду. В зависимости от свойств пород применяют разные
буровые коронки. В настоящее время за год удается пробурить
скважину глубиной до 4000 ле.
Методы бурения совершенствовались. Научились
вымывать из забоя частицы разрушенной при бурении породы —
теперь не нужно было, как раньше, через короткие
промежутки времени вынимать и разбирать став буровых штанг,
длина которого нередко достигала нескольких километров.
Работа эта очень трудоемкая и дорогая. Промывка забоя
суспензией иэ глины или цемента позволяла извлекать на
поверхность тяжелые частицы и одновременно укреплять и
уплотнять стенки скважины. Наконец, советские инженеры
применили для вращения бура турбины. Теперь уже во
вращение приводится не весь длинный и тяжелый буровой став,
а лишь буровая коронка, находящаяся в забое скважины.
Турбина приводится в движение потоком глинистого
раствора, нагнетаемого в скважину. Иногда буровая коронка
приводится в движение электродвигателем, также находящимся
в скважине. Буровой став при этих методах остается
неподвижным.
Говоря о нефтедобыче, следует помнить, что большей
частью нефть залегает глубоко в недрах земли под огромным
давлением. Требуется высокое искусство и остроумные
технические приспособления, чтобы не допустить возникновения
могучего, высотой с многоэтажный дом, нефтяного фонтана,
разрушающего буровую вышку и другие сооружения. При
этом теряется много нефти и может возникнуть пожар.
Погасить такой огромный нефтяной факел — труднейшая и
опаснейшая задача. Современная техника позволяет нефтяникам
110чти во всех случаях взять нефтяной поток под контроль
и без потерь доставить нефть в резервуары или к местам
переработки.
Многоликое вещество
Нефть, которую обнаружили когда-то в природных
источниках, а теперь все в больших масштабах добывают с
помощью бурения, называют сырой нефтью. Описать сырую нефть
в нескольких словах невозможно, потому что она изменчива,
как хамелеон.
В зависимости от происхождения цвет нефти меняется от
светло-коричневого до черного, в ярком свете на поверхности
любой нефти видны зеленоватые блики. Сорта нефти
отличаются и по запаху. Запах некоторых нефтей очень приятен,
другие пахнут чесноком. У различных нефтей различная
вязкость. Нефти Суматры или Пенсильвании негустые,
маловязкие. Литр такой нефти весит не более 700 г. Нефть из Баку
или западноамериканских месторождений гораздо более
вязкая и тяжелая. Литр ее весит около 900 г, а иногда и больше.
Все это указывает на то, что нефть неоднородна по
своему химическому составу, т. е. не является индивидуальным
веществом, как, например, вода или бензол. В ней
содержится множество различных соединений, дающих в
совокупности нефть. В зависимости от изменения состава меняются и
ее свойства.
Некоторые сорта нефти состоят в основном из
насыщенных углеводородов — парафинов с прямыми и
разветвленными цепями. Представители ряда парафинов до бутана С4Н10
газообразны и сопровождают нефть в виде природных газов.
Следующие члены ряда парафинов, содержащие в прямых
цепях до 16 углеродных атомов, жидкие и входят в состав
нефти. Чем выше точка кипения парафинового углеводорода,
тем длиннее его цепь, тем больше у него изомеров с
разветвленной цепью. Нефти, основным компонентом которых
являются парафиновые углеводороды, называют парафиновыми.
Некоторые насыщенные углеводороды способны также
образовывать замкнутые цепи атомов углерода. Назовем для
примера циклогексан СвН^. Нефти, содержащие такие
углеводороды, называются нафтеновыми. Иногда, правда довольно
редко, в нефти содержатся бензол и его производные.
Такие смеси, как нефть, отдельные компоненты которых
обладают различными точками кипения, сравнительно легко
разделить путем перегонки на составные части. В практике
органической химии перегонку используют очень часто,
поэтому стоит поговорить о ней подробнее.
Мы знаем, что почти все вещества могут существовать
в трех агрегатных состояниях. Рассмотрим, например, воду.
При температурах ниже 0° вода превращается в твердый лед.
В интервале от 0 до 100° — это обычная жидкая вода, выше
100° она переходит в газообразное состояние. При переходе
от льда к жидкости, а затем к пару никаких химических
превращений не происходит. При изменении агрегатных
состояний мы имеем дело только с физическими процессами.
:\*
100
5о°\
ffia
,*■
ар
04
*~~<*J8o&z
„Sew?" „n
Вода состоит из молекул. Каждая из этих бесчисленных
молекул обладает определенным запасом кинетической
энергии, и, следовательно, молекулы находятся в постоянном
движении. Если к этой системе подвести тепло извне — нагреть
ее — и тем самым увеличить запас кинетической энергии, то
движение молекул станет более быстрым. В
противоположном случае, т. е. при охлаждении системы, энергия
уменьшается и движение молекул замедляется.
Кроме энергии движения, приводящей к беспорядочному
распределению молекул в газообразном состоянии, на
частицы действуют еще силы притяжения. Если силы притяжения
по сравнению с кинетическими силами очень велики, как это
наблюдается у твердых веществ, молекулы занимают
определенное место, и это приводит к созданию прочной
малоподвижной системы. Правда, и в этом случае сохраняется
некоторое движение частиц, но колебания их незначительны.
Менее прочно связаны молекулы на поверхности вещества,
так как связывающие их силы действуют только с одной
стороны.
И наконец, следует иметь в виду, что общее количество
энергии при данной температуре постоянно, но энергия,
приходящаяся на каждую молекулу, распределяется
статистически, т. е. существуют частицы, энергия которых больше или
м&ыпе средней.
/
9<->0<~>Ф<^>&^
■ 9
68
Статистическое распределение энергии можно сравнить,
скажем, с такой картиной. Город в определенное время,
предположим в 8 часов вечера, расходует постоянное количество
электроэнергии. Если разделить всю энергию на число семей,
то на долю каждой придется примерно по 120 ватт, и в
большинстве случаев это соответствует действительности. Но
может случиться, что в то время как Майеры ушли из дому и
погасили свет, у Шульцев — гости и все комнаты ярко
освещены. В этот вечер Шульцы расходуют больше, а Майеры
меньше электроэнергии, чем обычно.
Если температура повышается, колебания молекул
усиливаются. Наконец, средняя кинетическая энергия достигает
такой большой величины, что жесткая связь между
молекулами ослабляется и молекулы переходят в менее
упорядоченное состояние. Они еще подчиняются силам притяжения, о
которых мы говорили, но становятся гораздо подвижнее.
s
I
/
9 *
\
В этом случае мы говорим о плавлении твердого вещества,
переходе его в жидкость. В жидкости молекулярные связи
гораздо слабее, и с ее поверхности некоторые молекулы могут
отрываться, переходя в газовую фазу. При этом переходе
молекулы должны преодолевать давление воздуха. Часть
«воздушных молекул» возвращается в жидкость, однако
некоторые из них отрываются с поверхности — жидкость
испаряется, и ее объем медленно уменьшается.
При дальнейшем повышении температуры
соответственно увеличивается кинетическая энергия молекул жидкости,
наконец, силы притяжения и давление воздуха на
поверхность уравновешиваются, и жидкость начинает кипеть —
происходит бурный переход в газообразное агрегатное состояние.
При этом переходе молекулы приобретают энергию.
Увеличение энергии молекул приводит к тому, что они
располагаются в беспорядке. Вся энергия, которую система получает
извне, тратится на испарение, поэтому дальнейшего
повышения температуры при кипении жидкости не происходит.
Кипящая вода не становится горячее от того, что мы ее сильнее
нагреем, она только быстрее испарится.
69
Л З&лсперапура
1оо° f-
Хиление
—jS>-
Sobpfoyuusoc ме/гио
Итак, если равномерно нагревать жидкость, ее
температура постоянно повышается до тех пор, пока не будет
достигнута точка кипения. После этого, несмотря на дальнейшее
нагревание жидкости, температура ее остается постоянной, так
как вся энергия расходуется на переход из жидкого
состояния в газообразное.
Разумеется, этот процесс можно проследить и в другом
направлении. Если охлаждать газообразную воду, т. е.
отнимать у нее энергию, то она снова конденсируется в жидкость,
а при более сильном охлаждении застынет в твердый лед.
Мы уже знаем, что давление атмосферного воздуха на
поверхность воды препятствует отрыву молекул с этой
поверхности, поэтому оно влияет на процесс кипения. Если
уменьшить давление атмосферного воздуха, то кипение
облегчается и становится возможным при более низких
температурах, так как при более низких давлениях молекулам
легче преодолеть силы притяжения. Это хорошо известно
альпинистам, которым приходится долго возиться, когда после
тяжелого подъема они хотят сварить себе пищу. На
Монблане, например, высота которого 4810 ж, а давление 420 мм
ртутного столба, температура кипения воды всего 84°,
следовательно, и яйца приходится варить там гораздо дольше, чем
обычно. Если на самолете подняться еще выше, наступит
такой момент, когда вода закипит при 65°, т. е. она будет
кипеть при температуре ниже температуры свертывания белка.
В этом случае сварить яйцо совсем невозможно.
Если повысить давление на поверхность жидкости,
молекулам, отрывающимся с ее поверхности, при переходе из
жидкого в газообразное состояние придется преодолевать
большее сопротивление. Для кипения потребуется больше
энергии — температура его возрастет. Это явление
используют в быту. В кастрюлях для быстрой варки пищи делают
плотно прилегающую крышку. При нагревании жидкости в
такой кастрюле давление пара превышает нормальное, и
точка кипения жидкости в ней повышается. Например, при
повышении давления на одну атмосферу вода кипит при 120°.
Конечно, в такой кастрюле пища варится быстрее, чем в
кастрюле, прикрытой обычной крышкой. В ней можно
сварить еду и на высокой горе, и в самолете и, если хотите, даже
на Луне, где давление атмосферы вообще отсутствует и вода
в открытом сосуде должна была бы закипеть при 0°.
При работе с органическими веществами также часто
пользуются уменьшением давления, чтобы снизить темпера-
70
туру их кипения. Многие органические смеси можно было бы
путем перегонки разделить на компоненты или очистить от
примесей, если бы они не разлагались при температуре
кипения. Перегонка таких веществ возможна в системе, из
которой вакуумным насосом откачан воздух, потому что в этих
условиях температура кипения понижается. Это можно
продемонстрировать на таком опыте. Вода в перегонном кубе
нагрета до 50° и, следовательно, при обычных атмосферных
условиях не кипит. Если мы начнем откачивать из куба воздух,
то при давлении 92 мм рт. ст. (примерно Ув обычного
атмосферного давления) вода закипит.
Теперь, зная, как протекает процесс кипения и как
можно воздействовать на него, мы легко поймем процессы
перегонки и фракционирования нефти.
Фракционировать означает разделять. При
фракционировании нефть разделяется на отдельные компоненты.
Конечно, разделить нефть на все компоненты, входящие в нее,
невозможно, так как их несколько сотен. Разделяют только
группы веществ, резко отличающихся друг от друга по
температурам кипений.
С 1859 года, когда началось бурение нефтяных скважин,
нефть еще длительное время употребляли в основном как
горючее для ламп. В наши дни для этой цели используют
керосин — компоненты нефти, кипящие от 150 до 300°. В нефти,
которую раньше применяли для заправки ламп,
присутствовали бензиновые фракции с более низкой температурой
кипения. Применять такую нефть было довольно опасно, так
Соа/у J*UL SitcJrtpou
барки
TCokfentamop
CkpaJL
НСфтЬ
перегони
'190'-
Z50
VM хорошо, если взрыв оказывался не слишком сильным.
Поставщикам пришлось в конце концов найти способы удаления
из нефти низкокипящих компонентов. Для этого нефть
нагревали в больших чугунных котлах-кубах до 150°. При
этом бензин отгонялся из нефти, затем его конденсировали и
собирали в специальные приемники. Оставшуюся в кубе
жидкость можно было спокойно использовать как горючее для
ламп. А вот что делать с отогнанным бензином, долгое время
не знали. Его либо сжигали, либо спускали в реки.
Уничтожение бензина прекратилось только после изобретения
двигателей внутреннего сгорания. Его стали употреблять как
горючее. В наши дни бензин считается одной из самых
ценных фракций, получающихся при перегонке нефти.
Техника развивалась, наступил момент, когда
потребовалось разделить фракцию, кипящую выше 150°, на керосин
и газойль, смазочные масла и мазут. Процесс перегонки в
кубах не был непрерывным и вскоре перестал удовлетворять
инженеров. Сначала его усовершенствовали, соединив
несколько кубов в батарею и снабдив каждый куб специальной
насадкой. С помощью этих насадок осуществлялось
фракционирование — отделение газообразных продуктов перегонки от
жидких компонентов с более высокой температурой кипения.
В наши дни применяют не батареи перегонных кубов, а
очень экономичные колонны для фракционной перегонки.
Предварительно нагретая и освобожденная от легкоплавких
составных частей сырая нефть поступает в трубчатые печи,
где ее постепенно нагревают до 300° и выше, т. е. до темпера-
туры кипения высококипящих компонентов. Эта
температура, конечно, должна быть ниже температуры разложения
углеводородов.
При сильном нагревании сырой нефти в трубчатой печи
сначала испаряются компоненты, кипящие при более низкой
температуре. Постепенно давление в системе повышается до
6—7 ату в результате чего температура кипения нефти
также повышается и высококипящие компоненты остаются
жидкими даже при 350°. Из печи нефть поступает в испаритель.
Здесь давление уменьшается, и компоненты, кипящие ниже
350°, сначала испаряются, а затем частично конденсируются
на тарелках колонны для фракционной перегонки и стекают
вниз. Остаток, состоящий из тяжелых масел, смолы и
асфальта, поступает в другую систему, где подвергается перегонке
под уменьшенным давлением во избежание разложения.
Легкокипящие фракции поступают в верхнюю часть
колонны, где проходят через большое число колпачковых
тарелок, Колпачковые насадочные тарелки — это
горизонтально расположенные перегородки, имеющие многочисленные
отверстия с крышками в форме колпачков. Каждая из
тарелок связана с двумя соседними специальными отводными
трубками, одна из которых направлена вверх, а другая вниз.
Снизу вверх колонны температура понижается, и на каждой
тарелке конденсируются фракции, кипящие при
определенной температуре.
Поднимающиеся вверх пары проходят через жидкость на
тарелке, захватывая из конденсата наиболее летучие
компоненты. Для перемешивания паров с конденсатом на нижнем
крае колпачков имеются зубцы. Происходит быстрое
разделение компонентов, испаряющихся при разных температурах.
При этом дистиллят из верхней тарелки постоянно поступает
в нижнюю тарелку, а уже с нее стекает вниз. В некоторых
местах колонны для отбора отдельных фракций сделаны
съемные тарелки, из которых жидкость может поступать в
специальные баки.
Летучие фракции из верхней части колонны поступают
в конденсатор, в котором еще раз происходит разделение на
жидкую и газообразную фракции. Жидкий конденсат частим*
но отбирают, а частично он вновь поступает в верхнюю часть
колонны и стекает навстречу поднимающимся парам. Такой
противоток позволяет добиться более полного
фракционирования и регулирует температуру в колонне. Чем больше
обратный поток жидкости, направляющийся в колонну, где он
снова частично испаряется, тем сильнее понижается
температура в колонне, так как на испарение уходит много тепла.
Даже при изменении температуры снаружи колонны
температура внутри системы может оставаться постоянной. Такие
колонны работают непрерывно, и производительность их
может быть огромна.
Теперь поговорим об очистке нефти. Под очисткой
понимают удаление ненужных, вредных веществ, присутствие
которых затрудняет или препятствует применению нефти в
специальных целях. Такими вредными примесями могут быть
красящие, неприятно пахнущие вещества или соединения,
легко осмоляющиеся и снижающие качеотво смазок. Чаще
всего приходится удалять содержащие серу вещества,
присутствие которых ускоряет коррозию металла и способствует
тем самым его разрушению.
Уже известный нам Сэмюэль М. Кайер первым произвел
очистку нефти, обработав ее концентрированной серной
кислотой. После этого нефть становилась светлей, утрачивала
свой резкий запах и ее можно было употреблять для
освещения жилых помещений.
Способ очистки нефти и продуктов ее перегонки
концентрированной серной кислотой долгое время был
основным. Серная кислота поглощала часть связанной серы.
Остатки кислоты сразу же отмывали разбавленной щелочью.
В конце XIX века нефть начали обрабатывать
отбеливающими глинами, которые хорошо поглощали пахнущие и
красящие компоненты. Отбеливающие глины применяются и
сейчас.
Методы очистки нефти продолжали совершенствоваться.
Появлялись все новые и новые способы. Так, серу
связывают и удаляют, добавляя соединения тяжелых металлов.
Кроме того, соединения, содержащие серу, можно окислять.
Первый опыт по их окислению был проведен в 1875 году. В
качестве окислителя применяли хлор. После обработки хлором в
нефти всегда оставались нежелательные продукты
хлорирования, поэтому позже при окислении стали добавлять гипо-
хлорит, что дало хорошие результаты.
Исключительное значение имеют экстракционные
методы, впервые разработанные румынским ученым Эдельману.
В 1906 году он применил для экстракции примесей жидкий
сернистый ангидрид; сейчас с этой целью используют
множество веществ, большинство из которых избирательно удаляет
тот или иной ненужный компонент. Экстракция позволяет
полностью удалить определенную примесь, которая особенно
мешает при дальнейшем использовании нефти. Удаление
одного определенного вредного компонента снижает и стоимость
очистки.
И наконец, следует упомянуть о методах каталитической
очистки нефти, значение которых сильно возросло в
последнее время.
Нефть — почти идеальное горючее. Она обладает очень
высокой теплотворной способностью — от 10 000 до
11000 ккал/кг. Это значит, что при сгорании одного
килограмма нефти можно, если не учитывать потерь
выделяющегося тепла, нагреть 100 литров воды от 0 до 100°. По своей
теплотворной способности нефть превосходит все другие
виды технического горючего. Есть у нее и другие преимущества.
Нефть удобна для перевозки и хранения. Производство
нефтяного горючего очень рациональное, простое и чистое.
Поэтому понятно, что в наше время нефть и продукты ее
перегонки покрывают значительную часть мировой потребности в
горючем.
Сказанное выше не исчерпывает все области применения
нефти. В течение последних 20 лет она все шире
используется как химическое сырье. Весьма вероятно, что в этом ее
будущее. Многие химики сейчас жалеют каждую каплю нефти,
использованную в качестве горючего. Молодую отрасль
химии, которая занимается производством полупродуктов и
продуктов из нефти, называют нефтехимией. Программа
развития химии в ГДР предусматривает ускоренное развитие
нефтяной химии. Из-за бедности собственных месторождений
необходимая для этой цели нефть будет ввозиться
преимущественно из СССР.
Что представляет собой нефть как химическое сырье?
Известно, что она в основном состоит из алифатических и
циклических насыщенных углеводородов. Прямым
окислением газообразного пропана и бутанов можно получать спирты,
альдегиды и кетоны. Эти вещества либо поступают в
продажу, либо подвергаются дальнейшей переработке.
Хлорированием низкокипящих углеводородов получают также ценные
полупродукты, из которых изготовляют множество важных
веществ.
Но гораздо больше, чем насыщенные углеводороды с их
довольно слабой реакционной способностью, химиков интере-
75
суют олефины с их двойными связями, а также
ароматические углеводороды.
Однако ароматические соединения типа бензола и его
производных встречаются в нефти очейь редко и в
незначительных количествах, а олефины в сырой нефти практически
вообще не встречаются. Необходимо поэтому превратить
насыщенные углеводороды нефти в ненасыщенные или в
ароматические углеводороды, обладающие высокой реакционной
способностью.
Давно было установлено, что при некоторых процессах
переработки нефти образуются ненасыщенные соединения.
Это происходит, например, при процессе риформинга, с
помощью которого получают высокооктановые бензины. В
термическом варианте этого процесса бензин при повышенном
давлении подвергают кратковременному воздействию
высоких температур. При этом образуются ароматические
соединения: бензол и его производные — толуол, ксилол
и т. д.
Кроме того, образуются олефины и разветвленные
насыщенные углеводороды, улучшающие качество бензина. Из
бензина физическими и химическими методами выделяют
ароматические соединения и олефины.
При каталитических процессах риформинга выход
ароматических углеводородов увеличивается, но олефиновых
уменьшается. Между тем потребность в этилене и
пропилене настолько возросла, что пришлось изыскивать
специальные методы получения их в больших количествах из низко-
кипящих компонентов нефти. Наиболее важным из этих
методов является пиролиз, в процессе которого бензиновые
углеводороды расщепляются, и благодаря частичному
дегидрированию в молекуле возникают двойные связи. Так, из
низкосортного бензина можно получать этилен, пропилен,
бензол, толуол, ксилолы.
Этилен перерабатывается главным образом в широко
известный пластический материал — полиэтилен, а пропилен
служит для изготовления полипропилена и используется в
промышленном методе получения фенола из кумола.
Коксование угля в настоящее время не удовлетворяет все
возрастающую потребность в феноле, поэтому фенол стали
получать синтетическим путем. Бензол при взаимодействии
с пропиленом превращается в изопропилбензол, или, как его
называют иначе, кумол, из которого в результате дальнейшей
обработки получают фенол и ацетон. Фенол служит сырьем
для получения не только фенольных смол, но и капролакта-
ма, из которого изготовляют капроновое волокно
полиамидного типа, известное в продаже под названием капрон или
дедерон.
Можно без преувеличения утверждать, что во всем мире
большая часть продуктов органического синтеза
изготовляется из этилена, пропилена, бензола и ксилола, сырьем для
получения которых служат нефть или природный газ. К
важнейшим продуктам органического синтеза принадлежит ряд
ценных пластмасс, синтетических волокон, лекарственных
препаратов, синтетический каучук, моющие вещества,
растворители и красители.
Тетраэтил-
свинец
(антидетонационная
добавка)
Этиленгликоль|
(антифриз)
i
моющие
средства
Этилен,
Этиловый
спирт
I
ацетальдегид
уксусная
кислота
растворители
i
бутадиен
К синтетический
каучук)
Поливинил-
хлорид
(ПВХ)
I
полистирол
тетрафтор-
этилен
I
полиэтилен
Синтетические
масла
Теперь нам ясно, какую важную роль играет нефть в
производстве химических продуктов. Понятно, что
химическая промышленность мира все больше ориентируется на
нефть. Чтобы идти в ногу со временем, химическая
промышленность ГДР также будет перестроена на использование
нефтяного сырья.
gjQ4>M30Us
Фенол
I
Пластмассы
i
(Синтетические
волокна
(дедерон)
Толуол
I
Пластмассы!
i
Взрывчатые]
вещества
Ксилолы
i
Красители
i
Смягчители
I
Пластмассы
i
Синтетические волокна
(ланон)
Красители
I
Фармацевтические
препараты
i
Растворители
I
Горючее
Синтетические
моющие средства
i
Средства борьбы
с сорняками
и вредителями
Строительство трубопровода протяженностью 2800 км, по
которому нефть из Советской Татарии будет
транспортироваться в резервуары и перегонные установки сооруженного
недавно нефтеперерабатывающего завода в Шведте,
обеспечит непрерывное снабжение нефтехимической
промышленности ГДР этим ценным сырьем.
Октановое число 80
Может показаться, что в наше время не стоит подробно
говорить о бензине. Каждому ребенку известна эта жидкость,
о которой он знает, что ею наполняют баки автомашин, чтобы
привести в движение двигатель или, проще говоря, чтобы
машина поехала. Однако бывает и так, что о спутнике или
атомной электростанции знают подчас гораздо больше, чем о
такой обычной, простой и будничной вещи, как бензин.
Конечно, о том, сколько он стоит и как его применить, известно
каждому водителю, но на этом его познания обычно и
кончаются.
Наш рассказ лучше начать с общих, но очень нужных
'сведений. Бензин служит главным образом горючим для
двигателей внутреннего сгорания. Горючая жидкость
смешивается с воздухом в карбюраторе, а затем распыляется,
образуя легко воспламеняющуюся смесь. \ f
с&2 -'
Эта смесь всасывается в цилиндр двигателя и при
движении поршня сжимается до 1/6 или 1/8 первоначального
объема, а затем воспламеняется от электрической искры,
которую дает свеча зажигания. Смесь сгорает. Получающиеся
при сгорании и внезапном повышении температуры (До
1500—2200°) газы быстро расширяются и давят на
окружающие их стенки цилиндра и поршень с силой в 30—40 ат.
Подвижный поршень опускается вниз, а энергия через шатун,
коленчатый вал, приводы и дифференциал передается на
колеса, которые вращаются и двигают машину вперед.
Не всякое вещество может служить горючим. Для этого
необходим целый ряд условий, о которых мы сейчас коротко
расскажем.
Прежде всего, горючее должно быть удобным для
транспортировки, хранения, дозировки. Этому требованию лучше
всего отвечает жидкость. И хотя применение пропана в ка-
78
честве горючего делает ненужным карбюратор, но пропан —
газ, и пользоваться им неудобно.
Эта жидкость, естественно, должна гореть. Необходимо,
чтобы при сгорании выделялось возможно больше энергии,
то есть чтобы соотношение между расходом горючего и
расстоянием, проделанным автомашиной, было выгодным.
Кроме того, необходимо, чтобы продукты сгорания были
газообразными при той температуре, которая возникает в
цилиндрах, и не разрушали бы их металлические части. Жидкости,
которые в большом количестве содержат серу в связанном
виде, не могут служить горючим: при их сгорании образуется
двуокись серы, которая интенсивно разрушает стенки
цилиндров и поверхность поршней.
Температура кипения горючего также должна
удовлетворять известным требованиям. Точка кипения не должна
быть слишком высокой. Важно, чтобы в любое время года,
даже зимой, жидкость в карбюраторе распределялась
равномерно. Но точка кипения не должна быть ниже определенного
предела. Горючее, кипящее при 25°, имело бы прекрасную
стартовую готовность. Но в этом случае образовывалось бы
так много легковзрывающихся газов, что курение в машине
или даже рядом с ней могло бы стоить человеку жизни. К
тому же летом чересчур возрастали бы потери на испарение.
И наконец, легко летучее горючее образует в системе газовые
пробки, которые мешают равномерному притоку топлива из
бака в карбюратор.
Короче говоря, далеко не всякая горючая жидкость
может служить в качестве топлива для двигателей. В то же
время можно назвать многие жидкости, которые обладают всеми
указанными выше качествами. И если в основном
применяется только бензин, это происходит оттого, что его легче
всего получать в больших количествах и, следовательно, он
довольно дешев. Жидкость, литр которой стоит 20 марок
(около 15 рублей), не подходит для горючего, даже если она
наилучшим образом удовлетворяет всем остальным требованиям.
Такое горючее можно употреблять лишь для специальных
целей, но его ни в коем случае не станут предлагать на
заправочных станциях водителям автомашин.
То, что мы называем бензином, химически не
представляет собой однородного вещества. На самом деле он состоит
из нескольких десятков жидких углеводородов, содержащих в
молекуле от 5 до 8 атомов углерода. Речь идет о пентанах,
гексанах, гептанах и октанах. Содержание пентана в бензине
очень незначительно и составляет не более пяти процентов.
Поскольку бензин — смесь, то и определенной точки кипения
он не имеет. Кипение происходит в интервале от 40 до 200°.
Жидкости, кипящие при более низкой температуре, создают
нежелательные газовые пробки в трубах, по которым
поступает горючее. Жидкости, кипящие при температуре выше
200°, также не должны содержаться в горючем в большом
количестве. Такие жидкости сгорали бы в цилиндре
неполностью и, кроме того, смывали бы защитную масляную пленку
на стенках цилиндра.
Нужную смесь углеводородов — бензин можно получить
при перегонке нефти. Такой бензин прямой гонки
применяют для двигателей внутреннего сгорания, но весьма
ограниченно. Хотя его точка кипения и удовлетворяет всем
требованиям, любой шофер назвал бы его неодобрительно — «вода
со звоном» и, конечно, добавил бы в его адрес много других
нелестных слов. Октановое число такого бензина от 40 до 65.
Для наших нынешних двигателей оно в любом случае
слишком низко, и при сгорании такого топлива в цилиндре
возникает четкое постукивание.
Антидетонационные свойства и октановое число — эти
два понятия решающим образом определяют ценность
горючего. Поговорить более подробно о них интересно не только
для шоферов.
Как мы уже знаем, смесь бензин — воздух сжимается в
цилиндре при обратном движении поршня. Когда поршень
достигает своего наивысшего положения, в свече
проскальзывает искра, которая и вызывает вспышку. Процесс сгорания
протекает не просто. Сначала напомним, что сгорание
происходит не мгновенно, оно требует известного, хотя и очень
незначительного отрезка времени — 1/300 секунды. За это
время пламя распространяется от свечи до противоположного
конца цилиндра. В то время как фронт пламени
продвигается через смесь, в ее сгоревшей части сильно повышаются
давление и температура. Давление и тепло распространяются
по смеси быстрее, чем пламя. При этом происходит, вероятно,
частичное окисление еще не сгоревших углеводородов —
процесс, в ходе которого температура самовоспламенения
бензина понижается.
При достаточном сжатии в цилиндре остаток смеси
загорается преждевременно и мгновенно сгорает со взрывом.
Вызванное таким сгоранием стремительное повышение
давления приводит к коротким и сильным толчкам в отдельных
участках цилиндра. Кажется, что кто-то молотком стучит по
стенкам камеры сгорания и слышится звон. Процесс, из-за
которого возникают толчки, пока не исследован во всех
деталях. Однако эти звуки являются серьезными признаками
того, что процесс сгорания протекает неблагоприятно,
цилиндр и приводы получают большую нагрузку, чем нужно.
Поршень не может справиться со слишком быстро
освобождающейся энергией, и возникают значительные потери
мощности. И наконец, энергия сгорания должна двигать только
поршень, а не трясти стенки цилиндра. Слишком высокая
температура тоже может повредить цилиндр и поршень.
Кроме того, сильно возрастает расход масла, и оно часто
застывает в кольцах поршня. Все эти явления нежелательны.
Избавиться от такого стука можно, во-первых,
целесообразным устройством цилиндра внутреннего сгорания.
Например, можно поместить свечу зажигания в самое горячее
место, рядом с наружным вентилем. Очень важно также создать
хорошую систему охлаждения. Во-вторых, необходимо
повысить антидетонационные свойства горючего. Чем они выше,
тем большей может быть степень его сжатия, а при этом дви-
гатель, расходуя то же количество горючего, развивает ббль-
шую мощность.
В 1910 году степень сжатия у двигателей грузовиков не
превышала в общем 4:1, т. е. была довольно низкой, поэтому
и антидетонационные свойства у бензина того времени могли
быть невысокими. По этой причине литровая мощность этих
двигателей была невелика, а рабочий объем цилиндров не мог
быть меньше 2000 см3. Соответственно бблыпим был и расход
бензина.
тоМй* "t
7:1
В современных моделях машин двигатели работают со
степенью сжатия 7:1 и более, поэтому их мощность в
лошадиных силах при меньших объемах цилиндров и расходе
горючего гораздо выше. Зато требуются такие виды горючего,
которые по антидетонационным свойствам резко отличаются
от бензина прямой гонки.
Для того чтобы определить антидетонационные свойства
горючего, нельзя, конечно, руководствоваться чутьем. Эти
свойства по-разному будут проявляться в разных двигателях.
Форма камеры сгорания, температура зажигания, степень
сжатия и другие факторы, например такой субъективный,
как манера езды водителя, определяют, будет ли двигатель
стучать при использовании этого горючего. Следовательно,
нужно выработать определенные условия, при которых
оценка горючего будет наиболее надежной. Для этого, во-первых,
необходимо иметь четкую систему измерения. В данном
случае мерой является октановое число, дающее однозначную
характеристику антидетонационных свойств топлива.
Во-вторых, необходимы стандартные условия испытания, при
которых могут быть получены ясные и воспроизводимые
результаты. С этой целью сконструирован испытательный двигатель,
в котором горючее сгорает при совершенно
определенных точно установленных условиях. Испытательный
двигатель имеет цилиндр определенного типа и размеров, причем
степень сжатия в нем можно изменять, укорачивая или
удлиняя поршень. Начало стука в цилиндре устанавливают,
конечно, не на слух, а регистрируют надежными
измерительными приборами. Бензин, который хотят испытать, сравнива-
81
ют с горючим, антидетонационные свойства которого заранее
известны.
А нтидетонационные свойства выражают октановым
числом, сокращенно — 04. Но так как речь идет не об
абсолютной величине, нужно найти какие-то отправные точки для ее
определения. Подобным образом поступали во многих
случаях, например при установлении температурной шкалы.
Итак, среди известных углеводородов выбрали один
н-гептан, который обладал ярко выраженной склонностью к
детонации, и произвольно стали считать его октановое число
равным 0. Также произвольно выбрали изооктан, или,
точнее, 2, 2, 4-триметилпентан СНз-С(СН3)2-СН2-СН(СНз)-
—СНз, и приняли его антидетонационные свойства за
наиболее высокие. Его октановое число приняли за 100. Смешивая
оба эти углеводорода, получили промежуточные октановые
числа. Бензин с 04 70 обладает такими
антидетонационными свойствами, как смесь из 70% изооктана и 30% н-гептана.
Чем выше октановое число, тем более высокими
антидетонационными свойствами обладает горючее.
Из гептана и изооктана можно получить смеси с
октановым числом от 0 до 100. Если сжигать такие смеси в
испытательном двигателе, изменяя степень сжатия, то чем больше
доля изооктана в смеси, следовательно, чем выше октановое
число соответствующего горючего, тем при более высокой
степени сжатия будет зарегистрирован стук. Каждому
октановому числу соответствует определенная степень сжатия, и
наоборот. При определении октанового числа неизвестного
горючего в испытательном двигателе степень сжатия
определяют в тот момент, когда стук слышен наиболее отчетливо.
На сегодня октановое число установлено практически у всех
жидкостей, которые могут быть компонентами горючего.
В отдельных случаях это число выше, чем у вещества,
взятого для верхней точки шкалы, и, следовательно, больше 100.
В связи с этим особенно интересно, существует ли связь
между антидетонационными свойствами соединения и
структурой его молекул. В настоящее время установлено, что такая
связь существует. Теперь с помощью определенных методов
и добавок горючее с плохими антидетонационными
свойствами можно превратить в горючее с высоким ОЧ.
Если посмотреть на эту таблицу, легко увидеть
определенную связь между структурой соединения и октановым
числом. Так, соединения первой группы показывают, что у
насыщенных углеводородов с прямой цепью атомов углерода
октановое число уменьшается с увеличением длины цепи.
Для соединений с разветвленными углеродными цепями
октановое число увеличивается. Это особенно ясно видно на
примере гексана и его изомеров. На антидетонационные
свойства углеводородов также влияет наличие двойных
связей. Так, у олефинов антидетонационные свойства гораздо
выше, чем у соответствующих насыщенных соединений,
причем существенное влияние оказывает положение двойной
связи в молекуле. Чем ближе двойная связь к середине
молекулы, тем выше октановое число (группа III). Замыкание
Соединение
I Пропан
н-Бутан
н-Пентан
н-Гексан
н-Гептан
Формула
СНз"~СН|—СНз
СНз~~~СНз—С Hj—СНз
СНз™"GHj—CHf—CHf—'СНз
СНз"~GHj—GH|—СНз-~СН|—СНз
СНз—СН|—СН|—СНз—СНз—СНз—СНз
О.Ч.
) 102
| 92
62
26
0
II н-Гексан
2-Метилпентан
2,2-Диметидбутан
III н-Гептан
Гептен-1
Гептен-2
Гептен-3
IV н-Гексан
Циклогексан
СНз—СН|—СНз—СНз—СНз—СНз
СИ,—СН(СНз)—СНз—СНз—СН8
СНз—С(СН8)з—СИ.—СН8
СНз-"СНз"~СНз—СН3-—СНз-~"СНз"~~СНз
СН8—СИ.—СН,—СНз—СНз—СН=СНз
СН8—СНз—СИ.—СНз—СН=СН-СН,
СН8—СНз—СНз-СН=СН—СНз—СН8
СНз—СНз—СН3—"СНз—СН3—СНз
н н н н
н' \с о/ ^н
н н н н
26
73
92
О
54
70
84
26
78
102
105
углеводородных цепей в кольца повышает антидетонационные
свойства соединения. Это видно из сравнения н-гексана с
циклогексаном. И наконец, следует упомянуть, что бензол и
его производные обладают высокими октановыми числами
(группа IV).
Обычный бензин прямой гонки состоит из смеси
насыщенных углеводородов с прямой цепью и поэтому имеет
такое низкое октановое число, что вообще не может служить
горючим для двигателей современных автомашин. Для
повышения октанового числа можно избрать два пути. Во-первых,
в горючее можно ввести добавки, которые либо регулируют
процесс сгорания, либо обладают высоким октановым числом
83
и увеличивают октановое число смеси. Во-вторых, качество
бензина прямой гонки можно улучшить, подвергнув его
различным химическим воздействиям. В этом случае
компоненты с плохими антидетонационными свЪйствами превращаются
в компоненты о высоким октановым числом. Это происходит
обычно благодаря циклизации алифатических углеводородов.
Рассмотрим сначала добавки.
Очень действенным оказался тетраэтилсвинец —
бесцветное жидкое соединение свинца с четырьмя этильными
остатками, т. е. РЬ(СгНб)4. Всего 0,05% тетраэтилсвинца,
добавленного к горючему, способствуют значительному
повышению октанового числа. При реакции, происходящей в
цилиндре, из тетраэтилсвинца выделяется металлический
свинец, который регулирует процесс сгорания.
Антидетонационное действие тетраэтилсвинца, по-видимому, состоит в
том, что на большой поверхности тонко измельченного
металлического свинца, возникающего при распаде, адсорбируются
многие активные атомы и радикалы еще не сгоревшей смеси
и их активность понижается. Процесс сгорания становится
спокойнее, не происходит самовозгорания несгоревшего
остатка смеси до тех пор, пока фронт пламени не подойдет
вплотную к горючему.
Этот способ повышения октанового числа, вообще
говоря, удобен, однако у него есть опасные недостатки.
Тетраэтилсвинец очень ядовитое соединение. Длительное вдыхание
бензина, «отравленного» тетраэтилсвинцом, приводит к
понижению кровяного давления и нервным заболеваниям.
В быту при использовании такого горючего необходимы
известные меры предосторожности: бензин не должен попадать
на кожу, им ни в коем случае нельзя чистить вымазанные
маслом руки, вещи или двигатель. Этим горючим нельзя
наполнять керосинки, примусы, керогазы. Во избежание
опасных недоразумений существует правило — горючие с
добавками соединений свинца окрашивают различными
красителями.
Следует обратить внимание и вот на что. Возникающий
при распаде тетраэтилсвинца тонкоизмельченный свинец
существует в металлической форме лишь одно мгновение и тут
же сгорает в окись свинца — твердое нелетучее соединение,
остающееся частично в камере сгорания. Со временем это
может привести к нежелательным очень вредным отложениям
на стенках камеры. Поэтому в горючее добавляют еще и ди-
бромэтан. Тогда окись свинца превращается в более летучий
бромистый свинец, легко удаляющийся при выхлопе.
К заметному увеличению октанового числа приводят
самые незначительные добавки тетраэтилсвинца, но даже и они
небезопасны для здоровья. Химики приложили немало
усилий, чтобы найти другие, менее ядрвитые антидетонационные
вещества. Их попытки бывали удачны, но найденные
вещества либо не были столь эффективны, как тетраэтилсвинец,
либо также образовывали в камере сгорания нелетучие окиси
металлов. Все сказанное в равной мере относится и к тетра-
этилжелезу и к тетраэтилникелю.
Чтобы избавиться от тетраэтилсвинца и все же
увеличить октановое число смеси, следовало бы добавлять в бензин
такие антидетонационные жидкости как изооктан, бензол или
толуол. Это, конечно, привело бы к желаемой цели, но все эти
вещества относительно дороги, а так как они должны были
бы составлять от 30 до 50% горючего, то стоимость
улучшенного бензина была бы очень высока.
В качестве антидетонационной добавки использовали
бензол, который иногда содержится в горючих, хотя, конечно,
не в тех количествах, которые необходимы для достижения
нужного октанового числа. Гораздо успешнее оказались
попытки превратить некоторые компоненты бензина в
соединение с более высокими антидетонационными свойствами и тем
самым улучшить качество горючего. В настоящее время
существует много таких методов.
При перегонке нефти выход бензина в общем невелик.
В среднем получают не более 15—20% бензина прямой
гонки, который имеет низкое октановое число и не может быть
использован как горючее. Бензин прямой гонки ни по
качеству, ни по количеству не удовлетворяет современным
требованиям. К тому же при перегонке остается много тяжелых
масел. Как изменить это соотношение в пользу бензиновой
фракции?
Уже в 1825 году английский естествоиспытатель Фара-
дей нашел в конденсате пара, полученного при нагревании
свежих масел, незначительные количества бензола. Это
открытие не было сенсацией, но все же оно ясно показывало,
что нагревание масел приводит к химическому превращению,
так как в конечном продукте были найдены новые вещества.
Во второй половине XIX века ученые получили
подтверждение этим наблюдениям. Бертло показал, что
термическим разложением углеводородов можно получать
ароматические соединения, например бензол. В это же время Юнг,
нагревая тяжелые масла под давлением, получил из них
легкие.
Итак, применяя тепло и давление, можно расщеплять
большие молекулы тяжелых масел на более малые и таким
образом получать легкие масла. Такое расщепление с
помощью давления и нагревания назвали крекингом. Это слово
английского происхождения; оно означает расщепление.
В 1865 году большое значение приобрело получение
керосина из тяжелых масел. В то время преобладали
керосиновые лампы, и потребность в горючем для них была велика.
Результаты Юнга были восприняты радостно, и тут же
началось строительство примитивных установок для крекинга.
Конечно, при крекинге получали не только керосин, но и
низкокипящие компоненты, бензины, которые в то время
использовать не умели и, как уже говорилось, просто сжигали.
Позднее появились светильный газ и электричество, и
керосиновые лампы стали исчезать. Потребность в горючем
для ламп сокращалась. Больше не были заинтересованы в
крекинге тяжелых масел, и установки одна за другой
исчезали. Но когда появились автомобили, потребность в бензине
стала из года в год возрастать. Снова вспомнили о низкоки-
пящих компонентах, которые прежде получали при крекинге.
Сооруженные вновь установки для крекинга на этот раз
служили уже для получения бензина. Выяснили, что такое
горючее лучше, чем бензин, полученный прямой гонкой, и
крекинг-процесс утвердился окончательно. Сегодня бблыпую
часть бензина получают на крекинг-установках. Выход
бензина из сырой нефти возрос благодаря крекингу от 15—20 до
40-60%.
При нагревании углеводородов до 300—600° под
давлением от 5 до 80 ат происходит частичное расщепление
больших молекул. Этот процесс происходит в основном между
двумя атомами углерода, и при этом возникают две молекулы
углеводорода с меньшим числом углеродных атомов в
каждой. Например, молекула декана СюН22 может расщепиться
по следующему уравнению:
СфНк ^СбИм + С^Ию +С
Из уравнения видно, что из молекулы декана не могут
получиться две молекулы пентана с пятью атомами углерода
каждая, так как в этом случае число атомов водорода было
бы 12X2, то есть 24. А в декане всего 22 атома водорода.
Поэтому происходит отщепление элементарного углерода,
который осаждается на стенках крекинг-установок и должен быть
немедленно удален. Это требует не только лишних затрат
труда, но и остановки на некоторое время всего процесса,
следовательно, уменьшает выход продукции.
Конечно, возможно такое расщепление длинных
углеводородных цепей, в ходе которого образуются ненасыщенные
соединения. Для декана этот процесс можно представить так:
C10hfzz ^С5ъ°
Поскольку в этом случае образуется пентен, то общее
число атомов водорода не превышает 22 и свободный углерод
не выделяется. Но на практике крекинг нефти целиком не
протекает ни по первой, ни по второй схеме. Образуются
одновременно и нефтяной кокс, и ненасыщенные углеводороды,
причем удается лишь влиять на относительное количество
того и другого.
Как мы видим, нефтяной кокс и ненасыщенные
соединения образуются при крекинге из-за недостатка водорода.
Если в камеры, где происходит реакция, ввести водород, то
образование углерода и олефинов почти прекратится. В этом
случае говорят о деструктивной гидрогенизации.
Вспомним еще раз таблицу, которая показывала связь
между октановым числом и структурой молекул. Из этой
таблицы видно, что у ненасыщенных углеводородов октановое
число больше, чем у соответствующих алканов. Естественно
было бы предположить, что образование большого количества
олефинов весьма желательно, так как такой бензин должен
обладать высокими антидетонационными свойствами. Но
олефины склонны образовывать смолы, что крайне
неблагоприятно для горючего, так как трубки карбюратора имеют
очень маленькие отверстия, которые легко забиваются.
Поэтому олефины либо удаляют из бензина, либо путем
дальнейшей обработки превращают в другие соединения.
Ненасыщенные соединения также нежелательны в
горючем, но они необходимы химикам, потому что из них легко
можно получать многие ценные продукты. Как известно,
олефины отличаются высокой реакционной способностью. В
некоторых случаях специально создают такие условия
крекинга, чтобы образовалось особенно много олефинов типа
пропилена и этилена. До сих пор мы говорили о термическом
крекинге — расщеплении углеводородов при нагревании под
давлением. Именно такой крекинг был впервые осуществлен
в промышленном масштабе. В 1913 году в так называемом
процессе Бюртона газонефтяную фракцию подвергали
медленной перегонке в реакционных камерах при 370—400° и
давлении 7 ат. В этом случае выделялся в значительном
количестве чистый углерод, который нужно было время от
времени (чтобы избежать прогорания стенок) удалять, от чего
установка работала с перерывами. Немного позже стали
применять более рациональные установки, имеющие более
высокую производительность. При этом температуру повысили
до 500—600°, а давление до 20—70 ат.
В принципе этот процесс протекает следующим образом:
нефть накачивается в трубчатый нагреватель, где при
температуре 500° и давлении 70 ат протекает частичный крекинг.
Далее в колонне происходит разделение смеси. Пары бензина
конденсируются в холодильнике в верхней части колонны и
лишь небольшая часть их используется для обратного
орошения. Тяжелые масла, содержащие углерод, собираются в
нижней части колонны, их можно употреблять, например,
как топливо. Промежуточные фракции многократно проходят
повторный цикл крекинга.
При этом процессе в трубчатой печи образуется кокс,
поэтому через определенное время нужно останавливать
установку и удалять его из труб — работа не из легких.
В наши дни печь и реакционная камера уже не
составляют одно целое. Нагретую смесь направляют в одну из двух
коксовых камер. Здесь в течение определенного времени и
происходит собственно крекинг. Пока одна камера работает,
другую очищают. Такая установка может работать
непрерывно. Разделение продуктов крекинга на отдельные фракции и
в этом случае происходит в колонне. На таких установках
остатки тяжелых масел также можно расщеплять на легкие
масла и бензин, что в хозяйственном отношении очень
выгодно.
Сейчас преимущественно ведут каталитический крекинг,
предложенный Гудри в 1934 году. В большинстве случаев
катализатором служит смесь соединений алюминия, магния
Но гъоусг/?<>&
и кремния с добавкой незначительного количества окислов
тяжелых металлов. Каталитический крекинг протекает
преимущественно при 500° и давлении всего 2 ат. Времени,
необходимого для осуществления процесса каталитического
крекинга, требуется гораздо меньше. Сокращается и количество
газообразных продуктов реакции и олефинов, потому что
под влиянием катализатора они либо превращаются в
изомеры, либо полимеризуются. Поэтому повышается октановое
число бензиновой фракции. Катализатор может быть
закреплен неподвижно. Через короткое время он покрывается
слоем кокса, который сильно снижает его эффективность,
поэтому катализатор регулярно обжигают для регенерации, а
приблизительно по истечении года его заменяют. Более
современный метод состоит в том, что катализатор в тонко
измельченном виде непрерывно подают в реакционную камеру,
а затем выводят для регенерации. В этом случае говорят о
движущемся слое катализатора и такой процесс называют
процессом с ожиженным или вихревым катализатором6. Как
видно из схемы, активный катализатор вместе с нагретой
исходной смесью, поступающей из печи, попадает в
реакционный сосуд, где и происходит собственно крекинг.
Использованный катализатор оседает вниз, и воздушный поток несет
его в установку для регенерации, где происходит обжиг.
При обжиге он снова становится активным.
Регенерированный катализатор вновь поступает вместе с исходной смесью
в реакционные камеры.
Применение каталитического крекинга позволяет
повысить октановое число бензина приблизительно на 10 единиц
по сравнению с термическим крекингом. Таким образом, без
дополнительных затрат или дополнительной обработки сразу
получают высококачественное горючее.
88
В то время как крекинг-бензины имеют
удовлетворительное октановое число, соответствующие фракции прямой
гонки нельзя применять как горючее для двигателей мк^ы^
очень низкого октанового числа. Для таких бензинов нашли
способ повысить их антидетонационные свойства с помощью
риформинг-процесса.
Риформинг похож на процесс крекинга, он
осуществляется в паровой фазе под давлением в присутствии
катализаторов. Расщепления и без того уже довольно маленьких
молекул углеводородов при этом не происходит. Молекулы с
нормальной цепью атомов углерода превращаются в
разветвленные, т. е. имеет место изомеризация их, либо образуются
ароматические соединения7.
В любом случае происходит повышение октанового
числа горючего. Конечно, в незначительной степени протекают
и процессы расщепления. Образующиеся при этом газы
отделяются от риформинг-бензина ректификацией на
колонке.
Различают также чисто термический вариант
риформинг-процесса, осуществляемый под давлением 50 ат при
температуре около 500°, и каталитический, который
происходит в присутствии соединений молибдена или платины.
Для каждого из этих двух вариантов существуют различные
методы, которые отличаются друг от друга незначительными
деталями; при любом варианте качество горючего
существенно улучшается. В большинстве случаев удается удвоить
октановое число. Если исходный бензин имеет октановое число
от 40 до 45, то из него получают горючее с октановым чис-
89
$Г*'«5Ч*
а Исходное
л
крекинга
kr Acouowe
<^ *%#*
лом 85—88, удовлетворяющее большинству современных
требований.
Обобщая сказанное выше, можно сделать вывод, что
смесь углеводородов, называемую бензином, нельзя получить
только прямой перегонкой сырой нефти. Кроме перегонки,
необходима дальнейшая переработка, требующая большого
технического мастерства и более сложных установок.
Только таким путем можно получить горючее, пригодное для
современных двигателей и имеющее максимальную
теплотворную способность.
Горючее иа угля
Выделение бензина иа нефти, без сомнения, самый
простой путь получения горючего. Бензин содержится в нефти
как готовый компонент и его можно выделить просто
перегонкой. Кроме этого, правда, необходимы некоторые
преобразования — часть углеводородных молекул следует
подвергнуть циклизации или изомеризации, но по существу это
ничего не меняет. Рассказ о горючем, однако, был бы неполным,
если бы мы не упомянули такой важный для Германии
метод, как метод каталитического гидрирования угля. Этот
метод связан с работами Бергиуса и Пира, а также Фишера и
Тропша.
90
Из сравнения химического состава угля, нефти и
бензина видно, что нефть и бензин содержат значительно больше
связанного водорода, чем уголь. Зато в угле имеются
связанный кислород, сера и углекислый газ, которые представляют
собой нежелательные «примеси». Их особенно много в буром
угле. Если нужно превратить такое сырье в бензин,
дизельное топливо и т. д., от этих примесей избавляются путем
гидрирования под давлением. При этом примеси
восстанавливаются, переходят в летучие водородные соединения и
отгоняются. Для такого процесса необходимо значительное
количество водорода, который вместе с оставшимся углеродом
образует углеводородные смеси, подобные нефтяным фракциям.
Этот процесс очень легко представить на бумаге, но
осуществить его практически гораздо сложнее. Прежде чем из
угля удалось получить первые капли бензина, ученые
преодолели множество препятствий, проделав громадную работу, и
проявили выдержку и настойчивость.
Химика Фридриха Бергиуса особенно интересовали
возможности применения высоких давлений в химии, и после
окончания высшей школы в 1910 году он решил посвятить
себя разработке этой проблемы.
В маленькой частной лаборатории вместе с несколькими
сотрудниками Бергиус занялся исследованием угля.
Как возник уголь? Нет сомнения в том, что уголь —
вещество растительного происхождения, образовавшееся из
деревьев, т. е. в конечном счете — из целлюлозы. В течение
тысячелетий при отсутствии воздуха происходили процессы
обугливания, благодаря которым и возник, наконец,
каменный уголь. Бергиус надеялся с помощью высоких давлений
и температуры получить уголь из целлюлозы в лаборатории
и, разумеется, за более короткое время, чем понадобилось
природе. Опыты завершились успешно. Бергиус рредполо-
жил, что, если искусственный уголь и дальше подвергать
воздействию сильного давления и высокой температуры,
отдельные молекулы его расщепятся. Если все это будет происхо-
ТСаменнЫи уеоиб *—
ьЬурый уг<мб »■
3&<рт(> *
I [ Уиерч □ *Ь»*/>у □ *Wf
дить в атмосфере водорода, то, возможно, в активных местах
разрыва произойдет присоединение атомов водорода. Это
привело бы к образованию углеводородов с гораздо меньшим
молекулярным весом.
1 оССоив*
ЦиЯрНЬи fee I
1 Каменный уголь Около 5000 |
1 Бурый уголь 1
1 Полукоксовая смола :
1 Нефть 1
1 Мазут 1
1 Дизельное масло \
1 Бензин 1
> 5000
> 250
> 400
> 400
> 200
> 100
Одновременно примеси, содержащиеся в угле, —
кислород, сера и углекислый газ — были бы восстановлены до
летучих водородных соединений, и удалить их из угля было
бы просто. Такие опыты были проведены Бергиусом летом
1913 года. Он получил весьма обнадеживающие результаты:
твердый уголь на 80% был превращен в газообразные и
жидкие углеводороды. Те же результаты были получены и для
природного угля. Предположения Бергиуса подтвердились.
В том же году под номером 301231 был зарегистрирован
патент на гидрирование угля под давлением.
Эти результаты не вызвали никакой сенсации. Во
Франции их приняли к сведению, но технического применения они
не находили, во-первых, потому что было достаточно
углеводородов, получаемых из нефти, причем они обходились
гораздо дешевле, а во-вторых, инженерам казалось
невозможным создать в техническом масштабе установки, работающие
под давлением выше 200 ат при 400°. Единственный отклик
современников Бергиуса заключался в совете воздержаться
от дальнейших опытов.
О работах Бергиуса вспомнили только во время первой
мировой войны, когда в Германии стала ощущаться
нехватка нефтяных продуктов, тем более что новые опыты по
гидрированию малолетучих нефтяных остатков проходили
очень успешно. В Маннгейм-Райнау была построена
опытная установка. Но война окончилась, и интерес к установке,
а вместе с ним и финансовая поддержка иссякли.
Шли годы. Бергиус, уверенный в плодотворности своей
идеи, неустанно продолжал работу над совершенствованием
процесса гидрирования угля под давлением. Тысячами
опытов он сумел показать, что даже низкокачественный уголь, а
значит и бурый, могут быть выгодно использованы для
гидрирования под давлением. Ему удалось провести
производственный эксперимент на опытной установке, и когда, наконец, он
изложил свои результаты в докладе, ими заинтересовались
не только в Германии, но и в других бедных нефтью, но
обладающих достаточными запасами угля странах. Различные
промышленные группы стали бороться за овладение патен-
том. И наконец, тогдашней И. Г. Фарбен удалось выиграть
эту борьбу и получить от Бергиуса все права на
использование результатов экспериментов, касающихся гидрирования
угля под высоким давлением. Процессы, дотоле
проводившиеся только на опытных установках, стали внедрять в
производство.
Эти работы проводились под руководством Пира на ани-
лино-содовой фабрике в Бадене. Внедрение в производство
потребовало серьезной доработки процесса. К этому времени
уже был собран ценный опыт в области промышленного
синтеза аммиака при высоком давлении. Потребовались новые,
особопрочные сорта стали.
Продолжительными были поиски катализатора для
гидрирования. Инженеры знали, что все катализаторы под
действием серы быстро теряют свою активность. А как раз серы
в угле было довольно много. Наиболее подходящими
катализаторами, устойчивыми по отношению к сере, оказались
соединения вольфрама и молибдена. Одновременно выяснили, что
более целесообразно проводить весь процесс не на одной
установке, а разделить его на жидкофазную и газофазную
ступени.
На заводе Лейна в Мерзебурге в 1927 году была
сооружена первая большая установка для гидрирования бурого
угля под давлением. Позднее производительность этого
предприятия достигла 600000 т бензина в год. Здесь исходный
бурый уголь почти полностью превращали в жидкие
углеводороды. С учетом угля, необходимого для производства
энергии, и огромных количеств водорода из 3,5—4 т сухого
бурого угля добывали 1 т бензина.
После прихода к власти Гитлера основным
направлением в политике и экономике Германии стало стремление к
так называемой автаркии, т. е. независимости от подвоза
сырья извне. Так как бензин являлся одним из основных
видов стратегического сырья, стали расширять уже имеющиеся
установки для гидрирования и построили ряд новых. В 1943
году гидрированием угля при высоких давлениях уже
получали 3 409 000 т горючего, а еще 425000 т давал синтез по
методу Фишера—Тропша.
Технологический процесс каталитического гидрирования
под давлением с течением времени претерпел некоторые
изменения. В отдельных случаях повышали давление до 700 ат
и в некоторых пределах изменяли температуру реакции. Но
принцип метода сохранялся.
Уголь непрерывно поступает из бункеров, измельчается,
смешивается с катализатором и высушивается в
специальных сушилках, теряя большую часть воды. В смесителе этот
порошок перемешивают с тяжелым маслом. Полученную
густую массу с помощью насосов по трубам транспортируют
дальше. В жидкофазной ступени масса прессуется в
атмосфере водорода под высоким давлением, нагревается, а затем
поступает в реакционные печи. Здесь она претерпевает
значительную часть превращений. В отделителе, примыкающем к
печам, большая часть непревращенных тяжелых масел с со-
держащимися в них твердыми контактными веществами
отделяется от среднего масла. После очистки от зольных
примесей масло вместе с ценными контактными веществами
снова направляется в реакционные печи. В перегонной установке
выделяют среднее масло (А) вместе с образовавшимся
бензином, а также остатки тяжелого масла, которые
возвращаются в цикл, где снова смешиваются с угольным порошком.
Далее среднее масло поступает на газофазную ступень,
где оно подвергается предварительному гидрированию. Его
снова сжимают до 200, а иногда и до 700 ат и в атмосфере
водорода доводят до необходимой температуры реакции.
В бензиновой печи среднее масло каталитически
расщепляют. Катализаторами служат сульфиды никеля и вольфрама,
которые неподвижно закреплены в реакционной печи. Здесь
происходит также восстановление примесей — кислород, сера
и азот превращаются соответственно в воду, сернистый
водород и аммиак. Вот почему этот процесс называют
рафинирующим предварительным гидрированием.
94
В перегонной установке исходные продукты разделяют,
выделяя непревращенные средние масла (А), которые снова
поступают в цикл, и средние масла (Б), которые находят
применение как дизельное топливо или используются в
последующем процессе бензинирования. В этом процессе
среднее масло снова подвергают воздействию высокого давления
и температуры, каталитически расщепляют и гидрируют в
реакционной печи.
Сразу после этого проводится перегонка. В верхней
части колонны конденсируется высококачественный бензин, а
непревращенное среднее масло снова масляным насосом
накачивают в реакционные камеры. Образовавшиеся
газообразные углеводороды служат для отопления или используются в
качестве сырья в органических синтезах.
Как мы видели, непревращенную исходную смесь
стремятся вновь и вновь использовать в реакции и по
возможности без потерь. В результате достигают практически
стопроцентного использования исходного сырья.
Примерно в то же время, когда Бергиус проводил свои
опыты по гидрированию угля под высоким давлением, в
институте угля в Мюльгейме (Рур) над проблемой получения
жидких углеводородов из угля работали директор этого
института профессор Ф. Фишер вместе с химиком X. Тропшем.
Они избрали совершенно иной путь, использовав результаты
опытов Сабатье и Сендеренса. Этим ученым в 1902 году
удалось получить метан из газовой смеси окиси углерода и
водорода на никелевом катализаторе при 250° и нормальном
давлении. Они решили попытаться таким же путем получить
более высокомолекулярные углеводороды.
В 1925 году им удалось, применяя катализаторы из
кобальта и железа, впервые синтетически получить жидкие
углеводороды из окиси углерода и водорода. Но прошло еще
много лет напряженной работы, прежде чем в 1932 году этот
метод получил промышленное оформление. Через четыре
года на одном из предприятий началось производство жидких
горючих по этому принципу.
В основе синтеза горючего по Фишеру — Тропшу лежит
следующее суммарное уравнение реакции:
Zc + o +#zo->2cc+hz
Исходные продукты — окись углерода и водород —
получают из угля. Для этого над раскаленным углем пропускают
воздух и водяной пар:
При этой реакции в небольшом количестве образуется
также двуокись углерода, и таким образом в результате
получается смесь газов, состоящая из окиси углерода, водорода
и двуокиси углерода. Последняя составляет очень небольшую
часть и удаляется промывкой. Остающаяся газовая смесь
подвергается основательной очистке, в ходе которой
соединения серы полностью удаляются, иначе они за короткое
время отравили бы катализатор. И только теперь смесь
поступает в контактную печь. При давлении, которое в
зависимости от того, какой конечный продукт хотят получить,
колеблется между 1 и 20 ат, и температуре примерно 250° смесь
превращается в углеводороды. Реакция протекает экзотер-
мично, и дополнительное нагревание не требуется. Наоборот,
реакционные камеры охлаждают проточной водой, чтобы не
превышалась оптимальная температура 250°.
В общем первичным продуктом синтеза является смесь
насыщенных углеводородов и олефинов с различной длиной
цепи атомов углерода. При разгонке получают газы типа
пропана и бутана, а также бензин, дизельное масло, смазочное
масло и высокомолекулярные парафиновые масла. Из-за
довольно низкого октанового числа и присутствия примесей
олефинов, превращающихся в смолу, полученный таким спо
$o<jcl
ПГ^п
\
SSenjcuc
собом бензин нужно подвергнуть риформингу или другой
дополнительной обработке.
На первый взгляд может показаться поразительным, что
в наше время установки для гидрирования имеются повсюду,
и даже такие богатые нефтью страны, как Советский Союз
или Соединенные Штаты Америки, применяют гидрирование
под давлением. Но сейчас в качестве сырья используют не
уголь, а дистиллятные тяжелые масла или отходы крекинг-
процесса.
В ГДР также заменили уголь нефтью.
Такая замена была вызвана рядом причин. Как мы уже
говорили, при гидрировании бурого угля нужно гораздо
больше водорода, чем при гидрировании нефти. Для изготовления
1 г бензина из бурого угля требуется 2500 м3 водорода, а из
нефти — вполне достаточно 500 мг. Для получения водорода
нужны дополнительная энергия и уголь.
Гидрирование нефтяных продуктов позволяет
значительно сократить материальные затраты, экономить мощности и
рабочие руки.
4 3. Шпаусцус
Замена сырья на заводах ГДР стала возможна только
благодаря возросшему импорту советской нефти. Если к тому
же принять во внимание, что Гайзельтальские
месторождения бурого угля будут исчерпаны в недалеком будущем, а
транспортировка его из отдаленных шахт делает этот метод
совершенно неэкономичным, значение проведенных
мероприятий еще больше возрастает. Наша социалистическая
экономика, которой чуждо стремление к автаркии, построена на
основе тесного и планомерного сотрудничества партнеров-
единомышленников, что позволяет достигать огромной
производительности при очень высоком качестве продукции. Все
это дало возможность положить конец эпохе нерентабельного
использования угля при превращении его в жидкие
углеводороды.
Бензольное кольцо
В 1828 году Велер, получив мочевину в лаборатории,
нанес удар витализму и проложил путь к синтезу органических
веществ. Все больше становилось соединений, полученных в
реакционных сосудах химиков.
В 1831 году Либих значительно усовершенствовал и
упростил элементарный анализ, что очень облегчило
исследование состава органических соединений. С этого времени
было открыто, синтезировано и подвергнуто анализу огромное
число соединений. Причем выяснили, что есть вещества,
имеющие одинаковый химический состав, но различные
свойства. Другими словами, обнаружилось, что «равные
количества одних и тех же элементов могут быть связаны друг с
другом неодинаково». Все более настоятельно вставал вопрос
о структуре, о расположении атомов в молекуле. Поиски
ответа привлекли внимайие профессора Гентского
университета Августа Кекуле.
Существовали различные точки зрения. Была выдвинута
теория радикалов, много спорили о так называемом учении
о типах. Но ни то, ни другое толкование не в состоянии было
удовлетворительно объяснить все экспериментальные факты.
Противоречия между экспериментальными данными и
теорией требовали неотложного решения.
Для Кекуле было ясно, что дальнейшая работа над
теорией радикалов непременно приведет в тупик, что она
бессмысленна, бесцельна. Ему казалось гораздо более разумным
исходить из самих элементов и искать ответ на вопрос, по
каким законам атомы этих элементов соединяются в
молекулы.
Кекуле тщательно познакомился со всей имевшейся
литературой по этому вопросу и нашел кое-что,
соответствовавшее его представлениям. Однако со многими утверждениями
он не мог согласиться. В 1854 году он закончил свою первую
большую работу, посвященную валентности.
Как же обстояло дело с «атомностью» 8 углерода,
содержащегося во всех органических соединениях? Со сколькими
эквивалентами других элементов он мог соединяться?
Подобными вопросами Кекуле обстоятельно занимался. На
примере гремучей ртути, соединения, которое так успешно
исследовал его учитель Либих, он провел серию экспериментов
и показал, что углерод четырехвалентен. «Рассмотрим
простейшие соединения углерода. Бросается в глаза, что масса
углерода, которую химики признали наименьшей, равной
одному атому, всегда присоединяет четыре атома
«одноатомного» или два атома «двухатомного элемента». Таким
образом, общая сумма химических единиц элементов,
связанных с одним углеродом, равна 4. Эти данные приводят к
заключению, что углерод четырехатомен», — писал
Кекуле.
Практически в молекулах почти всех соединений
содержится по нескольку углеродных атомов.
Как можно представить себе образование цепей из них
и какие выводы следуют из этого для теории валентности?
В конце концов помог случай. Кекуле вспоминал
впоследствии: «Во время своего пребывания в Лондоне я долгое время
жил в Клепхем-Роуд, а вечера часто проводил у моего друга
Хьюго Мюллера в Айлингтоне, на другом конце этого
огромного города... Однажды прекрасным летним вечером я ехал
последним омнибусом по пустынным в это время улицам
города. Я погрузился в мечты. Перед моими глазами двигались
атомы. Я всегда видел их в движении, эти маленькие
существа, но мне никогда не удавалось распознать характер их
движения. А в тот день я увидел, как два маленьких
существа соединялись в пары, большие обнимали двух меньших,
еще большие удерживали три и даже четыре маленьких, и все
это вертелось в круговом вихре. Я видел, как большие
образовывали ряды и только на концах цепи тащили самых
маленьких. Возглас кондуктора: — «Клепхем-Роуд!», —
заставил меня очнуться, но дома я провел полночи, изображая на
бумаге эскизы этих фантастических картин. Так возникла
структурная теория!»
Так выражал Кекуле само собой разумеющееся в наши
дни понятие о том, что «атомы углерода соединяются между
собой, затрачивая при этом часть химического сродства друг
на друга. Из восьми единиц сродства (два раза по четыре)
двух атомов углерода две единицы используются для
соединения этих атомов между собой и остаются еще шесть, по
которым могут быть присоединены атомы других элементов.
Если таким образом соединяются больше чем два атома
углерода, то с каждым вновь присоединенным атомом
основность углеродной группы повышается на две единицы».
Эти положения имели огромное значение. Только теперь
можно было разрешить многие противоречия, только теперь
стало понятным, как соединяются атомы углерода в
линейные молекулы. Теперь можно было по-новому взглянуть на
4*
множество известных соединений и объяснить ход их
реакций. Теперь структуру органических молекул выводили из
числа и валентности атомов. Это подтвердили многие
реакции обмена.
Кекуле был, конечно, далек от того, чтобы рассматривать
символы элементов, связанные в определенной
последовательности в цепи, как реальное отражение строения
молекулы. Он был совершенно уверен в пространственном
расположении атомов. «Само собой понятно, что положение атомов
в пространстве, даже если бы оно было установлено, нельзя
изобразить в плоскости бумаги. Для этого по крайней мере
необходим рисунок, сделанный с учетом перспективы, или
модель». Кекуле привлекали те представления, которые в
1874 году высказали независимо друг от друга Вант-Гофф
и Ле-Бель, и которые впоследствии были развиты в тетра-
эдрическую модель атома углерода, принятую и в наши
дни.
Гораздо больше пришлось поломать голову над
строением молекул ароматических соединений. Оказалось, что
учение о валентности не применимо к некоторым органическим
соединениям, что строение многих из них не укладывается в
схему, о которой мы рассказывали. Такими соединениями
являлись ароматические соединения. Их называли так
потому, что некоторые из них обладали приятным запахом.
Именно тем, что их нельзя было уложить в схему, эти
соединения заинтересовали Кекуле. Он установил, что во всех
ароматических соединениях содержится группировка из
шести атомов углерода, которая сохраняется в некоторых
продуктах превращения этих веществ. Эта группировка Се была
и у бензола СбНб — простейшего представителя
ароматических соединений. Если бы удалось выяснить структуру
бензола, то, наверняка, это явилось бы ключом к пониманию
строения всех ароматических соединений.
Принимая углерод четырехвалентным, а водород
одновалентным, в чем Кекуле не сомневался, было невозможно
образовать молекулярную цепь из шести углеродных и шести
водородных атомов, а экспериментальные факты
свидетельствовали именно об этом. Особенно трудно было объяснить
строение продуктов замещения. Казалось, что теория и
практика противоречат друг другу. \
Мысли о формуле бензола не покидали Кекуле. В его
мозгу проносились пляшущие атомы и длинные их цепи,
крепко связанные между собой. Все это в непрерывном-дви-
жении вертелось и извивалось подобно змеям. Но что это?
Одна змея схватила свой собственный хвост. Это может быть
решением! Никакой цепи нет. Кольцо из атомов углерода^ДЧ
такова формула бензола.
Однако прежде чем написать формулу бензола, удовлет- /
воряющую всем известным экспериментальным фактам,
следовало еще очень многое осмыслить. Как происходит
объединение шести атомов углерода в кольцо, все еще было не
совсем понятно. В полученной таким образом молекуле углерод
должен был быть трехвалентным. Но это противоречило
теории об обязательной четырехвалентное™ углерода,
признанной Кекуле. Тогда он пришел к выводу, что остающиеся
шесть валентностей шести атомов углерода объединяются в
три двойные связи, которые в кольце чередуются с
одинарной связью.
Кекуле принялся исследовать группу ароматических
соединений. Уже давно было установлено, что некоторые
свойства членов этой группы сильно отличаются от свойств
других соединений. Принципы строения формулы бензола
Кекуле распространил на всю эту группу. Полученные сведения
он изложил в своих «Исследованиях об ароматических
соединениях».
«Когда несколько атомов углерода соединяются друг с
другом, это может, во-первых, произойти таким образом, что
на каждую единицу сродства одного атома приходится одна
единица сродства соседнего атома. Далее можно
предположить, что многие атомы расположены так, что они всегда
соединяются по двум единицам сродства. Затем можно
предположить, что связь осуществляется поочередно: то через
одну, то через две единицы сродства... Если шесть атомов
углерода соединены по этому закону симметрии, то
получилась бы группа, которая, если представить ее в виде
открытой цепи, содержала бы еще восемь ненасыщенных единиц
сродства.
— с = С
/ /
/
I /
\
ъ>
Если теперь предположить, что два атома углерода,
замыкающие цепь, связаны через одну единицу сродства, мы
получим замкнутую цепь (симметричное кольцо),
содержащую еще 6 свободных единиц сродства. Если эти 6 единиц
сродства насыщены водородом, получается бензол. Водород
в бензоле можно полностью или частично заменить хлором,
бромом или иодом».
Далее Кекуле доказывал, что в любом ароматическом
соединении, известном до сих пор, содержится по крайней мере
одно бензольное кольцо, представляющее собой характерную
составную часть, основной конструктивный элемент
ароматических соединений.
Предположив, что 6 углеродных атомов бензольного
кольца образуют симметричный шестиугольник, легко было
прийти к выводу, что все шесть атомов водорода
равноценны. Например, не играет роли, у какого атома углерода —
первого, второго, третьего или пятого — произошло
замещение атома водорода на атом хлора, поскольку все атомы
водорода одинаково расположены в пространстве и имеют
одинаковые связи. Тот факт, что у однозамещенных производных
бензола отсутствует изомерия, вполне может служить
доказательством регулярного расположения атомов углерода в
шестиугольнике бензола.
Эту хорошо обоснованную теорию о строении
бензольного кольца Кекуле представил на обсуждение специалистов.
Практика должна была решить, насколько она верна.
Формула бензола, сконструированная Кекуле, была
принята подавляющим большинством химиков. Оставалось еще
много неясного, но работать органикам стало гораздо
проще. Теперь можно было рассматривать всю химию
ароматических соединений под единым углом зрения, можно было
уД а объяснить строение многих соединений, вскрыть аналогии,
.L^A*5 истолковать различия и, таким образом, создать основу для
л Iri синтеза природных веществ. О чем бы ни шла речь — о
каменноугольных красителях, о лекарственных препаратах,
пластмассах или природных соединениях, выясняется, что в
основе их структуры всегда лежит формула бензола.
Несмотря на славу, Кекуле оставался скромным
человеком и никогда не преувеличивал своих личных заслуг. Он
чувствовал себя звеном в общей цепи исследователей, всегда
рассматривал свою работу в связи с работами своих совре-
h менников и предшественников. Он знал, что в России химик
Бутлеров в тяжелых условиях получил ценные сведения о
химической структуре органических соединений9. Он ценил
работы Франкланда и Купера. Позже Кекуле узнал, что уже
в 1861 году физик Лошмидт в своей работе «Структурные
формулы в органической химии в графическом изображении»
3 П рассматривал бензол как кольцо и предположил существова-
£) 5 / ~*-—/-/ ние «шестивалентного бензольного ядра». Но представления
\\ *^S^' Лошмидта оставались несовершенными, и он был
недостаче точно твердо убежден в их справедливости, чтобы развивать
дальше. Кекуле утверждал: «Мы все стоим на плечах наших
предшественников, и поэтому не удивительно, что видим
н
■Н-
102
дальше их. Никогда и никто не придумывал ничего абсолютно
нового, тем более в химии. Каждый, кто подобно мне с
юношеских лет с любовью изучает историю развития своей
науки, а позднее, как и подобает возрасту, углубляется в новое,
более глубокое изучение классиков, может с уверенностью
заявить, что ни одна наука не развивалась так непрерывно,
как химия».
Кекуле умер в 1896 году в Бонне в возрасте 67 лет.
Мы рассказали далеко не все о формуле бензола. Ряд
особенностей делает целесообразным дальнейшее ее
рассмотрение.
Расшифровывая структуру молекулы бензола, Кекуле
расположил атомы углерода в правильный плоский
шестиугольник. Каждый атом углерода в нем соединен двумя
валентностями с соседними атомами углерода и одной
валентностью с атомом водорода (а). Такой способ, несомненно,
наиболее прост. Однако кольцо, построенное таким образом, не
удовлетворяло бы существующему представлению о четырех-
валентности углерода. Поэтому Кекуле считал единственно
возможным оставшиеся 6 валентностей шести углеродных
атомов соединить в двойные связи, расположенные
равномерно в кольце (б).
Qtofutyuaf
Tenjoucf
ho <Лоши/идти
а)
Н —С
«
Таким образом, получилась структурная формула,
которая принята и в наши дни. Такая формула бензола выдвигает
новые проблемы. С появлением в молекуле двойной связи
структура бензола приобретает ненасыщенный характер. Но
согласуется ли это с реальным положением вещей?
Действительно ли бензол обнаруживает свойства и реакции
ненасыщенного соединения?
Вспомним свойства алкенов или алкинов, содержащих
в молекуле двойные и тройные связи. Мы уже знаем, что
именно по месту двойной и тройной связи проявляется
реакционная способность молекулы, причем кратная связь стре-
103
мится превратиться в простую. Например, алкен — пропен
уже при комнатной температуре присоединяет бром,
превращаясь в 1,2-дибромпропан:
СЦ =CHZ +#гг > СН3 CHJir СЦг£>г
Искать подобные реакции для бензола бесполезно.
Обычно бром вообще не реагирует с бензолом. Только в
присутствии катализаторов происходит реакция, заключающаяся в
замещении атома водорода атомом брома:
1
л А
Это, по-видимому, указывает на то, что двойные связи
в молекуле бензола особенные. Только в специфических
условиях, когда отсутствуют катализатор и кислород, а смеси
чистого бензола и брома подвергаются ультрафиолетовому
облучению, двойные связи разрываются и происходит
присоединение 6 атомов брома к молекуле бензола.
4f tor
К X /«
+з^—**j _
" \
Jbr
При определенных условиях к бензолу можно
присоединить и водород. В этом случае двойные связи также
разрываются и бензол превращается в циклогексан.
.с— н
ч
и
V,/
~н
• с
I
н
Н Нх/Н
1
Можно приводить много примеров, но уже ясно, что
предположение о существовании двойных связей в молекуле
бензола вполне оправданно. Конечно, эти связи весьма
отличаются от тех двойных и тройных связей, которые знакомы
нам по алифатическим углеводородам. К этому вопросу мы
еще вернемся. Но сначала рассмотрим возможности изомерии
замещенных производных бензола.
Чтобы не писать каждый раз подробно довольно
сложную молекулу бензола, химики позволяют себе некоторые
упрощения. В дальнейшем мы будем изображать бензол в
виде шестиугольника, в котором обозначены только двойные
связи. В вершинах шестиугольника мы подразумеваем по
одному атому углерода, связанному с одним атомом
водорода. Если вместо атома водорода в молекуле находится какой-
нибудь другой атом или радикал, такой заместитель
обязательно обозначается, чтобы показать, что теперь мы имеем
дело уже не с бензолом, а с одним из его производных.
Чтобы однозначно определить положение этого заместителя,
атомы углерода принято нумеровать определенным образом.
Верхний атом углерода обозначается цифрой 1, остальные по
часовой стрелке нумеруются до шести.
Начнем с однозамещенных производных. Если ввести в
кольцо вместо атома водорода атом брома, можно заместить
любой водород, скажем водород в положении 1. Можно
представить себе, что такое замещение произойдет также в
положении 2 или 5. В зависимости от места замещения мы
получили 1-бромбензол, 2-бромбензол или 5-бромбензол.
/?\**
Как мы уже установили, не играет роли, у какого
именно атома углерода в кольце происходит замещение.
Поскольку все атомы водорода в бензольном кольце равноценны, то
равноценность, естественно, сохраняется и для заместителей.
Итак, если мы имеем дело с леоно-бромбензолом, то
безразлично, на каком месте мы напишем в формуле заместитель.
hi- Лг
•О" *СК "О
(1,2- gufyojuiinsouj (1уз -yvfyoutfcrtjo^J (l,4- guV/>o*u$<HjouJ
Совсем иначе обстоит дело, если в бензольном кольце
два атома водорода замещены на другие атомы или
радикалы. В этом случае возможны три расположения
заместителей, реальное существование которых подтверждается нали
чием трех различных изомерных соединений. При структуре
(а) оба атома брома соседствуют друг с другом (орго-поло-
жение), при структуре (б) они отделены друг от друга одним
атомом углерода (леега-положение) и при структуре (в)
расположены друг против друга (шгра-положение). Конечно,
совершенно ясно, что 1,3-положение идентично 1,5-; 2,4-;
2,6-; 3,5- и 4,6-положениям. В каждом случае речь идет об
одном и том же соединении, и только способ написания
формулы различен. Мы будем оперировать самыми меньшими
цифрами и в случае орго-бромбензола обозначать 1,2-положе-
ние, а в случае леега-бромбензола — 1,3-положение.
А* ' Яг
1j5- Z,4- Z,6- $5- 4,6-
Для трибромзамещенного бензола существуют 3
изомерных соединения:
1,Z,d-tofHfSjxutFeMJo* IjZ/i-byufptbuSeHjcuL ЩбГ-тирЛръаЯуои
При структуре (а) атомы брома соседствуют друг с
другом, при (б) — асимметричны и при (в) расположены
симметрично. Конечно, все эти рассуждения относятся только к
тому случаю, когда 3 заместителя одинаковы. Когда
заместители разные, количество возможных изомерных соединений
увеличивается.
Такое 'же число изомеров существует и для четырехза-
мещенных бензолов.
J5i
Лг
•Лг
Лг
fir
,Лг
'Лг
12,3,4-memfiJ^cutitHjcu. Wfifi-nienifa^fcu&WJ 1J2,4J6-mMpaf/><w(u#jAl
Сказанное об изомерии бензола не является чем-то
поразительным. Все эти случаи изомерии следовало предвидеть.
Однако если мы более тщательно займемся дибромбензолом,
то убедимся, что положение гораздо более запутанно, чем
кажется сначала.
Мы уже зарегистрировали три изомера: орто-, мета- и
ядра-соединения. Рассмотрим повнимательнее орго-форму.
Проанализировав тип связи атомов углерода в кольце, мы
придем к выводу, что должны существовать еще и другие
изомерные соединения. Замещение атомами брома у атомов
углерода может происходить либо по простой связи (а), либо
по двойной связи (б). В каждом случае в молекуле должны
возникать разные энергетические соотношения, которые
должны были бы проявляться в различии свойств полученных
соединений. Для различных заместителей в леега-форме
легко можно представить себе существование двух изомерных
соединений (а) и (б). Предположим, что бензольное кольцо
имеет двойные связи, а опираясь на предыдущие
рассуждения, мы имеем на это полное право, тогда в случае орто- и
леега-форм наличие изомеров явится обязательным
следствием.
И тут мы сталкиваемся с поразительным фактом — такие
изомерные соединения никогда не были найдены. Итак, хотя
существование в бензоле «неких» двойных связей мы
считали доказанным и единственно правильным, изомеры,
образованные на основании такой структуры, отсутствуют — факт
более чем загадочный!
С этим противоречием пришлось столкнуться уже Кеку-
ле и его современникам. Л. Майер считал, что эти изомеры
существуют, но так мало отличаются друг от друга, что
разделить их нельзя. Кекуле не сомневался в существовании
двойных связей в кольце, но не рассматривал их как
неподвижные и неизменные. Он считал, что их место в молекуле
должно меняться. «Нужно принять, что атомы в системах,
Jbr
Jbr
d)
«О
Лг
Jr Jbr
Я)
Ч/
ъ
м
107
которые мы называем молекулами, находятся в постоянном
движении... Движение должно быть такого типа, при котором
все атомы системы удерживаются в одной и том же
относительном порядке, т. е. всегда возвращаются к среднему
положению равновесия... Отдельные атомы системы могут в
процессе своего основного прямолинейного движения
натолкнуться друг на друга и затем, как упругие тела, оттолкнуться.
Тому, что в химии называют валентностью, можно дать с
этой точки зрения механическое истолкование. Валентность—
это относительное число толчков, которые один атом
получает от других атомов за определенный промежуток
времени». Такое утверждение недостаточно убедительно, и сложная
форма изложения не может скрыть известной неуверенности
автора. Утверждение, что «молекула в зависимости от
желания химика, экспериментирующего с ней, может изменять
свое строение и располагаться наиудобнейшим образом»,
давало лишь повод к шуткам.
Уже во времена Кекуле было множество попыток
разрешить возникшее противоречие, придав другое
расположение атомам в молекуле бензола. Из этих многообразных
моделей следует выделить модель Тиле. Мы упоминали о Тиле,
когда говорили о теории парциальных валентностей в
образовании двойных связей в алифатических соединениях.
Можно представить себе такие же валентности и у бензола. Если
каждую из трех двойных связей, обозначенных на рисунке
пунктиром, разложить на парциальные валентности, которые
взаимно насыщают друг друга, то все связи в молекуле,
естественно, становятся равноценными. Тогда уже никакой роли
не играет, произойдет ли в случае opro-бромбензола
замещение в 1,2-положении или в 2,3-положении. Случаи
изомерии, которые неизбежно возникали на основе структуры
молекулы с двойными связями, теперь невозможны.
Несмотря на наглядность теории парциальных
валентностей, мы для алифатических соединений заменили ее
новейшей электронной теорией и установили, что только она
удовлетворительно объясняет экспериментальные факты.
Поэтому, безусловно, стоит применить электронную теорию и для
108
объяснения свойств молекулы бензола. Это означает, что
реальным содержанием черточки-связи является общая
электронная пара. Одинарные связи образуются а-электронными
парами, обеспечивающими прочную связь. У ненасыщенных
молекул существуют л-электронные пары, которые, как
известно, гораздо более подвижны. То же самое мы наблюдаем и
в молекуле бензола, где 3 пары образованы шестью я-элект-
ронами.
4-
•С
:С
С:
XX
•' с Ъ
с-
С;
Для алифатических ненасыщенных соединений
подвижность я-электронов распространяется только на два атома
углерода, а у бензольного кольца я-электроны могут
занимать любое положение в молекуле. Поэтому нельзя говорить
об определенном месте двойной связи в молекуле бензола.
Для бензола характерно явление, называемое мезомерией.
При мезомерии равноценные пограничные формы,
показанные на рисунке, различаются между собой лишь
распределением я-электронов. Подлинная структура бензола является
совокупностью пограничных форм.
Благодаря мезомерии, которая существует, по всей
вероятности, из-за подвижности я-электронов, двойные связи
бензола резко отличаются от двойных связей алифатических
соединений. Они не локализованы, подвижны, и поэтому у ор-
го-бромбензола мы не можем найти двух изомеров.
Одновременно становится понятным более насыщенный характер
молекулы бензола и, следовательно, его склонность к реакциям
замещения.
бен]
родЬ
Эти соображения позволяют упростить запись формулы
бензола. Поскольку положение двойных связей в молекуле
казать точно нельзя, в будущем мы вообще не будем их пи-
. Рисунок простого шестиугольника обозначает молекулу
ла, состоящую из 6 атомов углерода и 6 атомов водо-
10
Благодаря своей электронной конфигурации это кольцо
обнаруживает ненасыщенный характер в незначительной
степени, и положение двойных связей, место я-электронов,
укавать точно нельзя.
Первые ароматические соединения
В предыдущей главе мы подробно рассказали о бензоле.
Это было необходимо потому, что только знание структуры
молекулы бензола дает возможность полностью понять его
свойства и открывает огромное царство ароматических
соединений. Но нельзя забывать, что, рассказывая о бензоле, мы
говорили лишь об одном соединении, в то время как класс
ароматических соединений охватывает почти неисчерпаемое
количество различных веществ. Как метан можно считать
простейшим соединением и первым членом ряда
алифатических углеводородов, так и бензол можно рассматривать как
родоначальника всех ароматических соединений. Любое
соединение, в молекуле которого имеется хотя бы одно бензольное
кольцо, следует относить к группе ароматических
соединений. Подчеркнем еще раз, что ученые пришли к такому
выводу не на основе формальных умозаключений. Наличие
бензольного кольца в молекуле обязательно приводит к
свойствам, которые делают целесообразной такую
классификацию.
Замещением одного или нескольких водородных атомов
бензольного кольца на другие атомы или радикалы
теоретически можно образовать бесконечное множество производных.
Конечно, группы, присоединенные к бензольным ядрам,
тоже в значительной мере определяют характер образующихся
соединений, поэтому возникает необходимость разделить
производные бензола на группы и подгруппы. Рассмотрим
подробнее хотя бы наиболее важные из них.
Суммарная формула бензола СбНб. Если отнять один
ато|к водорода, получается фенильная группа — С$Нъ —
одновалентный радикал. Соединение CeHsBr можно назвать или
но-бромбензолом или фенилбромидом. Отняв один атом
водорода от любого гомолога бензола, получают радикал,
который обычно называют арилом. Для алифатических
углеводородов в подобном случае говорят об алкиле.
О галоидопроизводных бензола мы уже упоминали, когда
знакомились с возможностями изомерии замещенных
бензола, и не будем на них останавливаться подробнее.
Более интересны фенолы. Все они образуются от
фенола — простейшего члена этой группы веществ. Фенол полу-
110
чается при замещении одного атома водорода бензольного
кольца на гидроксильную группу. Судя по названию, речь
должна была бы идти о спирте. Как раз на это указывает
окончание «олж Вспомним о метаноле, этаноле или бутаноле.
Об этом же говорит и ОН-группа, связанная с фенильным
остатком СвН.5.
А каковы на самом деле свойства фенола?
Одной из характерных особенностей спиртов является
способность к образованию сложных эфиров. Кроме того, они
вступают в реакции обмена с кислотами и сложными эфира-
ми. Эти реакции свойственны также фенолу и его
производным. Поразительно то, что в водном растворе фенолы
обнаруживают отчетливую кислую реакцию, тогда как для
спиртов такой реакции установить нельзя. Итак, фенолы
обладают кислотным характером. Поэтому фенол иногда называют
карболовой кислотой. Степень кислотности в фенольных
растворах по сравнению с известными минеральными
кислотами довольно низка. Она значительно повышается при
введении в молекулу нитрогруппы NO2. Известное взрывчатое
вещество пикриновая кислота — СеНг^ОгЬОН, 1-окси-2,4,6-
тринитробензол, уже настолько сильная кислота, что вполне
может конкурировать с настоящими карбоновыми кислотами.
Пикриновая кислота сильнее уксусной. Но мы не будем
просто сравнивать фенолы и кислоты, а рассмотрим фенолы как
отдельную группу веществ, стоящую по своим свойствам
между спиртами и кислотами.
Кислотный характер фенолов подчеркивает их реакция
с гидроокисями, при которой наряду с водой образуются соли,
так называемые феноляты. Происходит реакция, которую
вполне можно считать реакцией нейтрализации.
Сб ti5OH +/A ОН —* С6 н5 ОМ + UzO
Мы с удивлением отмечаем, что группа ОН в различных
соединениях может проявлять различную реакционную
способность. Так, у гидроокиси натрия, молекула которой
содержит типичную гетерополярную связь и легко диссоциирует
на отрицательно заряженный гидроксильный ион и
положительный ион натрия, гидроксильная группа обусловливает
основные свойства вещества. У спиртов диссоциация
отсутствует, и они имеют нейтральную реакцию. Тем более
поразительно, что диссоциация в значительной мере свойственна
фенолам, в которых диссоциирует только атом водорода гид-
роксильной группы в виде положительно заряженного
иона — вид диссоциации, характерный для кислоты.
Мы теперь уже представляем, что тип связи и
распределение подвижных электронов внутри молекулы чрезвычайно
важны и определяют поведение ОН-группы. У фенолов из-за
влияния соседней фенильной группы атом кислорода в ОН-
группе прочно связан с ядром. Общая электронная пара
между кислородом и водородом сдвинута к кислороду, так что
атом водорода довольно подвижен и может легко
диссоциировать в виде положительного иона.
Фенол — сильное дезинфицирующее средство, т. е. он
убивает бактерии. О его ярко выраженном бактерицидном
действии знали уже в 1867 году, и с тех пор фенол все шире
используют для дезинфекции. Разбавленный водный раствор
фенола известен под названием карболовой кислоты.
Феноловый, или карболовый, запах — типичный запах больниц и
госпиталей. К сожалению, фенол убивает не только бактерии,
но в больших концентрациях поражает и здоровые ткани.
Поэтому в наше время его заменили более эффективными и
безопасными препаратами.
В то же время фенол остается незаменимым исходным
сырьем для промышленного органического синтеза. Он
необходим для изготовления целого ряда экономически важных
пластмасс, лекарственных препаратов, красителей, ионно-об-
менных смол, дубильных веществ. Почти 50% фенола,
вырабатываемого в ГДР, используют для производства капролак-
тама, из которого затем получают дедероновое волокно.
Если в бензольное кольцо ввести 2 или 3 гидроксильные
группы, то можно образовать следующий ряд изомерных
соединений:
ОН М
£l,2-<JfUo*W&'0ou) (ly3-guo*tufo*jOJi) (1,4-yuoXcufeHjouJ
S/[upobcU4*e*A СриорогиюциК, Оха/гиурохиноК
(1}2,3 -mpuoKci/$uijA4} (1,3,f-nrpuoKutfcHjou} (tj2,4-HipidoKUtScHjoa)
Многоатомные фенолы, такие, как флороглюцин и окси-
гидрохинон, имеют большое хозяйственное значение: их
применяют как лекарственные препараты, они являются
компонентами красителей и составной частью фотопроявителей.
112
Особенности фенолов обусловлены тем, что группа ОН в
них связана непосредственно с бензольным кольцом. Это,
например, вызывает диссоциацию иона водорода. Если гидрок-
сильная группа находится в боковой цепи, то она не
испытывает влияния бензольного кольца, и соединение дает
реакции, свойственнее спиртам. Соединение CeHs—CH2OH — ашз
матический бензиловый спирт. Ароматические спиртылщя
нас не представляют особого интереса, и мы о них подробно
говорить не будем. [
При окислении ароматических спиртов образуются
[ароматические альдегиды. Подобная реакция известна нам по
алифатическим соединениям. Из бензилового
спирта—(первого представителя группы ароматических спиртов —
образуется бензальдегид, простейший ароматический альдегид.
CUZ<DH
+ 0
->
8>e#jaj6fee(/o
Бензальдегид — бесцветная маслянистая жидкость с
типичным запахом горького миндаля. В связанном виде
бензальдегид содержится в горьком миндале и в косточках
многих фруктов. На воздухе такие соединения разлагаются, и
некоторое количество бензальдегида освобождается в чистом
виде. Этот альдегид и придает специфический запах,
например, горькому миндалю. В технике бензальдегид применяют
как промежуточный продукт в синтезе красителей и пахучих
веществ. Для этой цели его получают не окислением
бензилового спирта, а совершенно иным путем.
Ароматические альдегиды в общем обнаруживают
реакции, знакомые нам по свойствам соответствующих
алифатических соединений. Так, при дальнейшем окислении они
переходят в кислоты. В случае бензальдегида окисление
протекает настолько легко, что он на воздухе превращается в
бензойную кислоту.
снгсм
У
&ен}аиуешу
tOeHjouHty Киеиома
В таких случаях говорят о самопроизвольном окислении.
При этом сложные реакции проходят несколько ступеней.
Они довольно точно исследованы, и сейчас известно, что
малейшие примеси, которые, как правило, присутствуют в бен-
зальдегиде, определяют ход этого самопроизвольного
окисления. У совершенно чистого альдегида процесс
самоокисления не происходит. Бензойную кислоту можно получать
окислением такого альдегида только при высоких
температурах. Окисление — наиболее простой путь получения
ароматических кислот. Существуют и другие промышленные
методы получения бензойной кислоты, в которых исходят не из
бензальдегида. Об этом важно упомянуть, так как бензойная
кислота широко используется в промышленности для
синтеза красителей. Кроме того, бензойная кислота убивает
бактерии и употребляется как консервирующее средство.
Бензойная кислота — это белое кристаллическое
вещество, малорастворимое в воде. В воде она, как всякая кислота,
диссоциирует на ионы водорода и кислотный остаток.
Бензойной кислоте свойственны реакции, характерные для любой
карбоновой кислоты. Карбоксильная группа — СООН — вот
что определяет ее основные свойства и реакции. Нам они
известны по алифатическим карбоновым кислотам. Так,
бензойная кислота образует соли — бензоаты и сложные эфиры.
Если в бензольное кольцо ввести две карбоксильные
группы, получаются двуосновные фталевые кислоты,
существующие в трех изомерных формах:
СООН- СООН СООН
СООН
cootj соон
CP/rtofUC^a-f Уиыапа t/jccprna^^fctJf имсиема даерефмаиЖц Кислом*
Особенно важны для различных отраслей
промышленного органического синтеза фталевая и терефталевая
кислоты.
Фталевая кислота применяется в синтезе многих важных
красителей. Назовем, например, известные каждому химику
фенолфталеин и индиго. При соединении фталевых кислот
с трехатомным спиртом — глицерином образуются так
называемые глифтали, сложные эфиры с более или менее
длинными разветвленными цепями. Они широко применяются
как кислые искусственные смолы и в качестве основы для
лаков. При синтезах глифталей применяют не фталевую
кислоту, а ее ангидрид — вещество, в которое превращается
кислота, отдавая при нагревании воду. Так как образование фта-
114
левой кислоты происходит при высоких температурах, то
всегда сначала образуется ангидрид.
ФтаигАг^ Зелота ^HUffpuj (ртме&ш л!и<иат6(
В заключение еще несколько слов о терефталевой
кислоте. С двухвалентным спиртом — гликолем она образует
сложные эфиры, которые могут полимеризоваться в длинные
цепи. Эти полиэфирные волокна перерабатываются в
текстильные товары, очень стойкие к химическим воздействиям
и отличающиеся высокой прочностью на разрыв. Кроме того,
изделия из этих волокон очень мало мнутся.
Если в бензольное ядро вводят аминогруппу ГШг, то
образуется фениламин, или анилин, простейший ароматический
амин. Его формула C6H5NH2. Этот амин можно рассматривать
как аммиак, в котором один атом водорода замещен на фе-
нильную группу. Анилин, который встречается в
каменноугольной смоле, а также может быть получен синтетически,—
основа химии каменноугольных красителей. О нем речь
пойдет ниже.
До сих пор в рассуждениях мы исходили только из одной
молекулы бензола, в которой заменяли один или несколько
атомов водорода на различные радикалы. Многообразие
соединений, образованных от бензола, значительно возрастает,
если учесть, что в одной молекуле могут быть два и более
бензольных кольца.
В простейшем случае каждая из двух молекул бензола
теряет по одному атому водорода и обе образующиеся фе-
нильные группы объединяются в новое соединение — ди-
фенил.
0>н+н<3—> ОО+Ъ,
Связь между бензольными ядрами может
осуществляться и с помощью алифатических цепей. Это еще более
расширяет круг возможных ароматических соединений. Рассмот-
рим, например, структурную формулу диаминодифенил-
метана
W-
-си,
■ш
Существуют и другие возможности для связи между
бензольными ядрами. Очень часто встречается случай, когда два
или более бензольных кольца соединены друг с другом
настолько прочно, что практически возникает новое ядро. В этом
случае атомы углерода одного бензольного кольца
принадлежат одновременно и другому. Тогда говорят о
конденсированных циклических системах. Простейший представитель этих
соединений — нафталин CioHg хорошо известен как
вещество, предохраняющее вещи от моли. Нафталин — одна из
важнейших составных частей каменноугольной смолы. В чистом
виде он представляет собой белое твердое вещество с
характерным запахом, применяющееся в химической
промышленности для многих важных синтезов.
н—с
с
С
II
\
н
\
Ре*
'at/,
»*f>&/
с С
I
Н Н дъшпрацен
Из структурной формулы видно, что кольца обладают
двумя общими атомами углерода. Эти оба атома насыщают
свои четыре валентности только за счет соседних атомов
углерода. Отсюда суммарная формула нафталина СюНв.
Простейшая конденсированная система, состоящая из
трех бензольных колец, является антраценом СмНю. Он
содержится в каменноугольной смоле. Сейчас без антрацена
невозможно представить себе химию красителей. Если
кольца расположены так, что между ними образуется угол,
получается изомер антрацена — фенантрен. Другие бензольные
ядра могут присоединяться к антрацену либо линейно, как,
например, в случае пентацена, имеющего синий цвет, либо
образовывать более сложные системы, например 3,4-бензпи-
рен. Бензпирен интересен тем, что встречается в
каменноугольной смоле. Это соединение способно 'вызывать
116
рак. Ег апример, нанести бензпирен на кожу мышей, то
на этих местах образуются раковые опухоли.
Э;4- fenj парен
Ябенпау&с
Оваиек,
Следует, наконец, указать еще на одно соединение —
овален, молекула которого образуется из 10 симметрично
расположенных бензольных колец.
Производные конденсированных ароматических
углеводородов встречаются в красителях и других природных
соединениях буквально на каждом шагу. Например, в красителе
ализариновом прямом синем А легко найти циклическую
систему антрацена.
° JWZ
I
В заключение нужно сказать, что для ароматических
соединений, так же как и для алифатических, существует ряд
строгих правил и законов. Группы, замещающие атомы
водорода в бензольном кольце, как и в линейных углеводородах,
в значительной степени определяют свойства и реакции
новых соединений. Наличие той или иной группы позволяет
причислить соединение к определенному классу веществ.
Совершенно новые типы соединений образуются при
конденсации бензольных ядер. Это делает многообразие
органических соединений неисчерпаемым.
117
Краска из угля
В 1862 году в Лондоне состоялась международная
выставка. В выставочных павильонах толпилось множество
посетителей, они с интересом, иногда с удивлением, рассматривали
экспонаты из многих стран. Тут было что посмотреть.
Это было время бурного развития естественных наук и
техники. Ни один год не проходил без открытия или
изобретения, которое не заставляло бы говорить о себе. Фотография
перестала быть курьезом, заслуживающим лишь насмешки,
электричество начало свой победный марш, и первые
лампочки накаливания загорались по вечерам рядом с привычными
газовыми светильниками. Телеграф и телефон позволяли
людям общаться на больших расстояниях. Все больше и больше
рельсовых путей покрывало землю. По этим стальным
магистралям железнодорожные поезда быстро доставляли к цели
людей и грузы. На дорогах появились первые велосипеды с
педалями. Продувка в конвертерах позволила Бессемеру
наладить производство стали в соответствии с возросшими
требованиями.
Выставка проходила под знаком новых открытий. Не
удивительно, что многие из посетителей не всегда знали, на
какие экспонаты стоит обратить особое внимание.
Тем более удивительно, что поток посетителей
останавливался не только перед многочисленными новинками
техники, но и перед некоторыми незаметными витринами, где
экспонировались химические процессы и продукты. В этих
витринах были выставлены окрашенные ткани, перья и меха.
Само по себе это не было удивительно, но поражало
многообразие красок, их яркость, глубина и блеск, невиданные до
той поры.
Это демонстрировались красители. Они находились в
стеклянных пробирках рядом с окрашенным предметом и
представляли собой «кристаллы, переливающиеся зеленым
металлическим блеском, как крылья жука». Однако
посетители замечали и еще кое-что. Совсем рядом с прекрасно
окрашенными тканями, мерцающими, блестящими красителями,
резко контрастируя с ними, находилось странное неприятное
вещество. Черное, липкое и тягучее, с неприятным запахом,
оно, казалось, не имело никакого отношения к выставленным
рядом красителям. Может быть, его показывали только за
тем, чтобы еще больше оттенить чистоту и яркость красок?
И почти каждый посетитель обращался к путеводителю
по выставке, чтобы узнать, что же написал об этом веществе
известный немецкий ученый профессор Гофман, который в то
время жил в Англии и преподавал в королевском химическом
колледже. В путеводителе было сказано, что это неприятное
вещество — побочный продукт при производстве светильного
газа — газовая смола. Эта смола образовывалась в больших
количествах и долгое время считалась просто
обременительным побочным продуктом, устранение которого требовало
лишних затрат денег и труда. С удивлением и недоверием
посетители выставки прочитали, что «все эти радующие взор
краски получены путем еще более удивительных
превращений из одного и того же исходного материала, из
отвратительной смолы».
Гофман не ограничивался простым описанием
существующих фактов. Он предсказывал именно такой путь развития
производства красителей, который осуществился лишь
несколько десятилетий спустя. «Вместо веществ животного и
растительного происхождения, употребляемых для
изготовления красок, в любом искусстве и ремесле будет
применяться минеральное вещество, преобразованное искусственным
путем с помощью химических процессов. Как ни велико уже
сейчас количество красителей, добываемых из
каменноугольной смолы, этот новый источник еще только открыт и
предварительные результаты позволяют надеяться, что скоро
мы сумеем получать тончайшие оттенки красок, источником
которых до сих пор было дорогостоящее животное или
растительное сырье — насекомые, кора, цветы и корни».
Крашение — одно из древнейших ремесел. Даже в очень
далекие времена были народы, которые умели окрашивать
ткани и различные предметы.
Человека всегда радовало в природе разнообразие
красок, и он пытался перенести это красочное разнообразие на
окружающие его предметы. Конечно, люди довольствовались
такими красителями, которые уже существовали в природе,
и нужно было лишь выделить и очистить их. Пользовались и
минеральными красками, но главным образом употребляли
красители животного или растительного происхождения. До
второй половины прошлого века в основном ничего не
изменилось. Методы добычи красок совершенствовались, люди
научились повышать качество добываемых красителей,
разводили и возделывали соответствующие растения на
плантациях, но в конце концов всегда приходилось довольствоваться
тем, что давала природа.
Важнейшим красителем XIX века было, без сомнения,
индиго — «король красителей». Это — один из старейших
красителей. Ленты, окрашенные индиго, находят на
египетских мумиях, погребенных более 4000 лет назад. В Европе
индиго знали уже в IX веке. Этот красивый темно-синий
краситель добывали из вайды. Это растение в метр высотой
содержит индиго в небольших количествах. Добыча красителя
из вайды была очень сложной, требовала много времени.
Искусство «синего крашения» было мало распространено.
Не удивительно, что красящие вещества, содержащие индиго,
были очень дороги и пользоваться ими могли только
богатые.
Сначала ничего не изменилось и тогда, когда европейцы
узнали об индийском индиго. В XVI веке в Европу привезли
первое индийское индиго. Оно было лучше европейского,
добывать его было легче, и ввоз его стал непрерывно
увеличиваться. Это свело на нет значение местной вайды.
Благополучие земледельцев, разводивших ее, оказалось под угрозой.
Тогда тех, кто ввозил индийское индиго, стали облагать
большими штрафами. Но даже введение смертной казни не смогло
бы теперь приостановить импорт индиго. Его привозили в
Европу во все возрастающих количествах, и, наконец,
пришлось снять всякие ограничения. Теперь крашение индиго
стало гораздо более распространенным и дешевым.
В Бенгалии начали разводить индиго на огромных
плантациях. В лице местных жителей британские плантаторы
получили неограниченное количество дешевой рабочей силы,
которой можно было распоряжаться по своему усмотрению.
В Америке и на Яве также были заложены большие
плантации индиго. Здесь белые господа столь же
бесчеловечно эксплуатировали местное население.
В 1880 году пятую часть возделываемого в мире индиго
доставляли в Германию.
Так же было и с ярко-красным красителем ализарином,
который добывался из корня марены. Это растение уже
тысячелетия назад было известно в странах Малой Азии и на
Кипре. В древности египтяне, персы, индусы, греки и римляне
красили ализарином. В средние века марену привезли в
Европу, и она хорошо прижилась. Карл V распорядился
разводить марену в Эльзасе. Это было очень выгодно: из 60
миллионов марок, которые в 1869 году выручили за краситель из
марены, 25 миллионов притекло в Эльзас. К этому времени
самым значительным производителем маренового красителя
была Франция.
Ализарин и индиго были наиболее выгодными
растительными красителями. Им значительно уступали красители из
красного и синего деревьев, растущих в Америке. Из их
древесины экстрагировали красители, которые также служили
для крашения тканей и производства типографских красок.
Некоторые животные тоже вырабатывают красители.
С давних пор из высушенных тел самок насекомого
кошениль, живущего в Америке, добывали кошенильный
краситель.
Красящий компонент кошениля — карминовая кислота.
Этот краситель привозили в Европу в больших количествах
для крашения шерсти и шелка и для изготовления красных
чернил.
И наконец, следует упомянуть пурпур, краску, которой
окрашивали одеяния королей и князей. Крашение пурпуром
считалось искусством, мало кто владел им, тонкости его
долгое время хранились в тайне. Этот краситель был очень
дорог и его ценили на вес золота. Пурпур добывали из
пурпурной улитки, живущей в Средиземном море. В ее железах
содержится чрезвычайно малое количество красителя. Нужно
было поймать громадное число улиток, очень тщательно
обработать их, чтобы получить крохи пурпурного красителя,
который еще предстояло очистить.
В первой половине XIX века несколько химиков
проводили экспериментальную работу с этими природными
красителями, стремясь открыть секрет их строения. Но об их
искусственном изготовлении, о синтезе их даже не думали.
Ведь в то время ничего не было известно ни о их составе, ни
о их строении. Эти вещества смешивали с различными
другими соединениями, подвергали воздействию кислот и
оснований, нагревали, перегоняли, стремясь установить, как они
будут вести себя при этих реакциях. Химиков привлекало
новое, неизвестное.
В 1826 году в Берлине химик Унфердорбен проводил
эксперименты с индиго. Ему нравилось это красивое
вещество, работа с ним доставляла эстетическое удовольствие. Он
проводил самые разнообразные реакции. Например,
смешивал индиго с известью и подвергал эту смесь сухой перегонке.
Так он получил новое, неизвестное вещество, которое назвал
кристаллином из-за его способности к образованию
кристаллов. Его открытие не вызвало большого интереса у химиков,
скоро об этих опытах забыли, а может быть, их не заметили
вообще.
Восемь лет спустя химик Рунге занимался в Берлине
исследованием каменноугольной смолы, большие количества
которой скапливались на заводах при получении светильного
газа. Использование этого ненужного продукта было бы очень
выгодно любой химической фабрике. Рунге удалось выделить
из смолы соединение, которое при реакции с хлорной
известью давало прекрасную фиолетовую краску. Он назвал это
вещество кианолом.
Несколько лет спустя в Петербурге Ю. Ф. Фрицше, как и
Унфердорбен, проводил опыты с индиго, воздействуя на него
едким кали. При последующей перегонке он тоже получил
новое вещество — анилин. Это благозвучное слово он
произвел от испанского названия индиго (anil — синий). В том же
году русский профессор Н. Н. Зинин восстановлением
нитробензола получил вещество, которое он назвал бензидамом п.
Но как ни интересны были эти работы, они
представляли собой чисто научные достижения, знали о них очень
немногие. Никто в то время не подозревал, какую огромную
роль вскоре станет играть анилин и как развитие химической
промышленности во второй половине XIX века будет
зависеть от этого незаметного вещества.
Большой вклад в развитие химической промышленности
в прошлом столетии внес немецкий химик Август Вильгельм
Гофман.
Гофман родился в 1815 году в Гисене. Там же поступил в
университет, собираясь посвятить себя изучению права.
В это время в Гисене жил и преподавал химию
профессор, который, как никто другой, умел пробуждать интерес и
любовь к своей науке у студенческой молодежи. Студенты из
разных стран приезжали послушать его лекции, теснились в
лабораториях, где он вел практикум. Его лекции
приковывали внимание слушателей, его методы обучения были
совершенно новыми, революционными. Молодой профессор, а это
был 33-летний Либих, не только учил химии, но ежедневно
восхищал своих учеников этой юной и в то же время древней
наукой о веществах и их превращениях. Гофман тоже
посетил несколько лекций Либиха и был захвачен этой наукой.
Оставив изучение права, он посвятил себя химии.
Гофман работал без устали, он был талантливым
экспериментатором, страсть и вдохновение учителя передались
ему. Вскоре Либих обратил на него внимание, и после
окончания учебы Гофман стал его приват-ассистентом. В качестве
первого самостоятельного задания ему было поручено
исследование органических оснований каменноугольной смолы.
Один из бывших учеников Либиха после окончания
университета построил в Оффенбахе установку для перегонки смолы
и прислал Либиху пробу полученного им вещества. Гофман
нашел в нем в небольшом количестве основание, но для
успешного продолжения исследований требовалось гораздо
больше исходного материала. Он отправился в Оффенбах,
провел у перегонной установки неделю и за это время
экстрагировал основание более чем из 600 т основной смеси.
Гофман установил, что выделенное им вещество идентично крис-
таллину Унфердорбена, кианолу Рунге, анилину Фрицше и
бензидаму Н. Н. Зинина. Он выбрал для этого вещества
название — анилин.
С этого момента и до конца жизни Гофман занимался
анилином. Сначала нужно было определить состав и харак-
терные реакции нового химического соединения. В 1845 году
Гофман нашел более простой и дешевый способ получения
анилина из бензола. Еще раньше Гофман установил, что
бензол является главной составной частью низкокипящих
фракций каменноугольной смолы. При воздействии на бензол
крепкой азотной кислотой получается нитробензол, который
атомарным, т. е. особенно реакционноспособным, водородом
восстанавливается в анилин — фениламин:
с^+нщ-—>с6н5хо2 н- н2о
C6HbtfOz+6H >C6h5SThz +3-Н*0
фенил, сслсин-
В этом же году Гофману предложили должность
профессора химии в Боннском университете, и почти
одновременно он получил приглашение в Англию. Согласившись поехать
в Лондон лишь на два года, он проработал там 20 лет.
И в Англии Гофман занимался главным образом
исследованиями соединений, входящих в состав каменноугольной
смолы. Благодаря его работам химики нашли источник
получения анилина в больших количествах. Правда, анилин в то
время интересовал только специалистов-химиков. Однако
вскоре судьба его резко изменилась.
В 1856 году в институте, где работал Гофман, получили
первый анилиновый краситель, а через некоторое время его
уже изготовляли промышленным способом. Был осуществлен
синтез и других важных анилиновых красителей. Анилин и
красители стали единым понятием.
Хинин представляет собой белый легкий порошок
горького вкуса. Он плохо растворяется в воде и несколько лучше
в спирте.
Хинин содержится в коре хинного дерева, растущего в
Перу. Издавна высушенную кору этого дерева размельчали
и обрабатывали кипящей водой. Полученная настойка была
действенным средством против малярии, воспаления легких
и других заболеваний типа лихорадки. В начале XIX века из
настойки выделили белый хинин, который и стали
употреблять как лекарство. Хинин вывозили из Перу. Монополия на
этот продукт позволяла устанавливать высокие цены.
Гофман также заинтересовался хинином. Подвергнув его
анализу, он пришел к выводу, что хинин должен быть близок
к основаниям, выделенным из каменноугольной смолы, а
значит к анилину. Он установил также, что если нагревать
хинин с едкой щелочью, то при разложении образуется
немного анилина. Дальнейшие исследования хинина Гофман
поручил своему ассистенту Вильяму Генри Перкину. Восем-
надцатилетний химик с жаром принялся за порученное ему
дело.
Нельзя ли не из хинина получать анилин, а, наоборот,
синтезировать хинин из оснований каменноугольной смолы?
Это было бы не только большим научным достижением, но и
открыло бы заманчивые экономические возможности.
Весной 1856 года Перкин не уехал на пасхальные
каникулы. В маленькой домашней лаборатории он работал над
осуществлением своих замыслов. Одно за другим брал
Перкин основания, содержащиеся в каменноугольной смоле,
обрабатывал их различными веществами, нагревал, исследовал,
но каждый раз его ожидало разочарование. Не было и следа
белых кристаллов хинина, которые он так жаждал получить.
Однажды Перкин добавил к анилину серную кислоту,
несколько кристаллов бихромата калия и нагрел смесь.
Полупилась какая-то темная масса. Значит, опять напрасный
опыт. Огорченный, он хотел вымыть реакционный сосуд и
налил туда воды. Перкин с трудом подавил волнение. Что
это? Вещество или по крайней мере часть его растворилась
в воде, придав ей прекрасный ярко-фиолетовый цвет.
Перкин забыл о хинине. Его теперь гораздо больше
интересовал новый краситель. Он надеялся, что сможет получать
его в больших количествах и продавать. Но сначала нужно
было проверить, надолго ли сохраняет это вещество свою
красящую способность, насколько оно водоустойчиво и
светопрочно. Для этого ему требовалось получить гораздо большие
количества красящего вещества. Упорно работая, Перкин
получил около 100 г мовеина, как он назвал новое вещество (от
французского слова mauve, которым французы обозначают
цвет красной мальвы). Его надежды оправдались. Краситель
был светопрочным и водостойким, хорошо ложился на ткань.
26 августа 1856 года Перкин взял патент на первый
анилиновый краситель. Это событие открыло новую эпоху в
развитии химической промышленности.
Перкин решительно избрал свой жизненный путь.
Гофман был поражен, когда его самый способный, подающий
блестящие надежды ассистент попросил об увольнении и
основал фирму, чтобы осуществить открытый им синтез в
промышленном масштабе.
Трудности, возникшие при промышленном изготовлении
красителя, были столь велики, что кого-нибудь другого могли
бы и поколебать. Но Перкин проявил настойчивость.
Промышленная химия только здрождалась. Почти не
было опыта фабричного изготовления веществ, полученных
предварительно в лабораторных условиях, Все приходилось
придумывать заново. Анилина — исходного вещества для
синтеза — в то время в продаже не было. Приходилось
получать его на своей же фабрике из бензола. Но и производство
бензола первое время наталкивалось на серьезные трудности.
Однако уже через 6 месяцев после основания фабрики
на рынке появился первый краситель. Почти невероятное
достижение. Руководителю и совладельцу фабрики в это
время только что исполнилось 19 лет.
Вскоре возникли совершенно неожиданные трудности.
Мовеин почти не покупали. Новому красителю необходимо
было завоевать доверие консервативных английских
текстильных фабрикантов и потребителей. Для каждого сорта
ткани Перкин сам разработал наилучшие методы крашения.
Для своих клиентов, которые очень недоверчиво и неохотно
покупали краситель, он организовал специальные
консультации. Сначала казалось, что его инициатива разобьется о
равнодушие и косность.
Толчок для решительного поворота пришел из-за
границы, из Франции, где более благосклонно отнеслись к мовеину.
Ткани, окрашенные им, стали пользоваться большим спросом
и в Англии. Это заставило английских фабрикантов
применить новый краситель, иначе они не смогли бы выдержать
конкуренции с Францией. Правда, сначала цена мовеина
почти равнялась цене платины, и им окрашивали только
отдельные шелковые нити, которые потом вплетали в
дорогие ткани. Но вскоре краситель подешевел, вошел в моду и
пользовался исключительной популярностью. Дошел анекдот
того времени, в котором даже полицейский вместо «move on»
(вперед), командует «mauve on».
Перкин положил начало промышленности анилиновых
красителей. Он синтезировал первый из них и изготовил его
фабричным способом. Это был огромный успех, который
привлек всеобщее внимание к анилину.
В 1862 году на международной выставке в Лондоне
всеобщее внимание и восхищение вызвали уже различные
анилиновые красители. А через пять лет общая стоимость
изготовленных к тому времени красок из каменноугольной смолы
составляла 1 250 000 фунтов стерлингов. Большую их часть
выпустила английская промышленность.
Центром бурных поисков анилиновых красителей был
институт Гофмана в Лондоне. Гофман сам участвовал в
совершенствовании их палитры. Так он открыл способ
изготовления прекрасного красителя — фуксина. Получив
розанилин, он открыл основное вещество для синтеза других
красителей из каменноугольной смолы. Гофман нашел чудесный
«гофмановский фиолетовый», который много лет не выходил
из моды, красители йодистый зеленый и метиловый зеленый.
Все они созданы на основе анилина.
В 1865 году Гофман уехал из Лондона. После
двадцатилетнего отсутствия он принял предложение Берлинского
университета, где в его распоряжение предоставили новый
химический институт. Это событие означало нечто большее,
чем просто перемена кафедры. Спустя почти сорок лет
английский ученый Вильям Рамзай сказал: «Мы все знаем,
какую потерю понесла Англия с отъездом Гофмана в
Германию. Я, наверное, выражу чувства всех английских химиков,
сказав, что если бы мы удержали его в Лондоне, то быстрое
развитие химической промышленности в Германии может
быть и не прекратилось бы, но было бы надолго
приостановлено, и Англия заняла бы то передовое место, которое
принадлежит теперь Германии».
К 1892 году — времени смерти Гофмана — развитие
химической промышленности в Германии продвинулось далеко
вперед. Многие фабрики, на которых раньше было занято
лишь несколько рабочих, превратились в огромные
предприятия. Промышленное производство красителей не только
достигло уровня Англии, но и превзошло его. Предпосылками
этого явились, с одной стороны, научные исследования и
отличная подготовка специалистов в институтах, а с другой —
бурное развитие промышленности и концентрация
производства.
Полученный Перкином мовеин был первым анилиновым
красителем и первым синтетическим красителем вообще. Он
был создан в химической реторте, это было соединение, для
которого, как и для многочисленных красителей, полученных
из анилина в последующие годы, не существовало образца в
природе. Но все же основными красителями оставались два
важнейших природных вещества — ализарин, полученный из
корня марены, и индиго.
Между тем практический опыт и теоретические работы
уже открывали широкие возможности для объяснения
структуры ароматических соединений.
Для получения какого-либо вещества прежде всего
необходимо знать его состав и структуру. Определить суммарную
формулу ализарина не составляло труда. Его молекула
построена из 14 углеродных, 8 водородных и 4 кислородных
атомов, следовательно, суммарная формула СмНвО^ Но этого
мало. Гораздо важнее было выяснить структуру молекулы,
ответить на вопрос, как связаны друг с другом углеродные,
кислородные и водородные атомы.
Это сделал 27-летний ассистент Берлинского института
Карл Грабе, который с 1868 года вместе со своим коллегой
Карлом Либерманом работал над выяснением структуры
ализарина. Ализарин, красный краситель корня марены,
оказался 1,2-диокси-антрахиноном: /^
Итак, ученые с удивлением установили, что речь шла о
соединении с определенной и относительно простой
структурой. После выяснения структуры синтез этого вещества не
представлял трудностей и был лишь вопросом времени.
За эту задачу снова взялся Грабе. Разлагая мареновый
краситель, он получил антрацен. Французские химики Дюма
и Лорен еще в 1832 году выделили это вещество из
каменноугольной смолы, где оно содержится в количестве от 0,5 до
5%. Суммарная формула антрацена была хорошо известна —
СмНю. Его структура ясно указывала на тесное родство с
ализарином — 1,2-диокси-антрахиноном. Чтобы получить из
каменноугольной смолы антрацен, нужно было достаточно
сильно ее нагреть. Только при температуре около 300°
антрацен начинает отгоняться и собирается в приемнике в
виде бесцветных или бледно-желтых пластинчатых
кристаллов.
При перегонке с цинковой пылью, т. е. при реакции
восстановления, Грабе сумел отнять от ализарина кислород и
перевести его в антрацен. Если бы удалось, наоборот,
окислить антрацен, то ализарин был бы одним из продуктов его
окисления. Эта реакция была осуществлена гораздо скорее,
чем ожидали ученые.
Уже 11 января 1869 года на заседании химического
общества Грабе сообщил о синтезе ализарина и
продемонстрировал несколько образцов окрашенных им тканей.
Требовался еще один решительный шаг. Синтез
ализарина нужно было сделать экономически выгодным, вывести
за пределы лаборатории. Необходимо было разработать
такой промышленный процесс, который позволил бы
производить ализарин, способный конкурировать по цене с
натуральным.
Грабе понимал, что в его маленькой, плохо
оборудованной лаборатории невозможно разработать синтез в
промышленном масштабе. Он принял предложение мощной анили-
но-содовой фабрики, обладавшей большими капиталами,
наладить в Людвигсгафене технический синтез красителей.
Грабе положил в основу своего метода окисление щелочного
плава антрахинонсульфокислоты. Анилино-содовая фабрика
немедленно взяла на него патент во всех странах. 25
июня 1869 года был получен английский патент. Это было
очень своевременно, так как уже 26 июня Перкин подал
заявку на изготовление ализарина из антрацена. Он опоздал
лишь на сутки.
В 1870 году один килограмм синтетического ализарина
стоил еще 60 марок, а через 30 лет цена на него упала до
1 марки за килограмм. К началу XX века разведение марены
Вт Европе прекратилось. Но природное индиго все еще
занимало господствующее положение* в промышленности.
Химики не могли пока заменить его соответствующим
синтетическим красителем, но уже начали заниматься
выяснением его структуры. Проникнуть в тайну строения индиго
было куда труднее, чем установить структуру молекулы
ализарина. Последователю Либиха преподавателю химического
института в Мюнхене Адольфу Байеру пришлось потратить
18 лет, прежде чем, наконец, в 1883 году он окончательно
выяснил структурную формулу индиго, определив место
каждого атома в молекуле этого красителя.
—С = 0 С —
Uhuuso
Структурная формула, которую вывел Байер,
свидетельствовала о том, что молекула индиго довольно сложна.
Байер вскоре сумел осуществить и синтез индиго, но это
представляло лишь научную ценность. Для промышленности
метод был не пригоден, поскольку Байер использовал
исходные вещества, которых не было в достаточном для
.широкого производства количестве. Синтетическое индиго,
изготовленное таким способом, было бы слишком дорого и не
смогло бы конкурировать с более дешевым натуральным
красителем.
И снова анилино-содовая фабрика приобрела права на
открытия Байера, хотя было хорошо известно, какие
трудности ждут впереди.
В 1880 году в Людвигсгафене начались первые работы,
но и через десять лет они еще не дали существенных
результатов, хотя уже стоили миллионы. В это время из Цюриха
пришли одновременно ошеломляющие и обнадеживающие
известия. Профессор химии Карл Гейман разработал метод,
по которому исходя из анилина и хлоруксусной кислоты
многоступенчатым синтезом было получено индиго. Анилина, как
и хлоруксусной кислоты, всюду было достаточно.
Но и этот метод не давал окончательного решения
проблемы. На одной из промежуточных ступеней синтеза
требовалось щелочное плавление при температуре около 300°. Такое
нагревание приводило к разложению значительного
количества конечного продукта, что снижало его выход и повышало
цену полученного индиго.
В 1893 году Гейман использовал для синтеза индиго
нафталин, который можно было добывать из каменноугольной
смолы в любом количестве. Из нафталина получали антрани-
ловую кислоту, а из нее — индиго. В 1897 году на фабрике
было получено наконец первое синтетическое индиго. С
начала работ прошло 17 лет, опыты стоили более 18 миллионов
марок. Если в этом же году килограмм синтетического индиго
еще продавали за 17 марок, а такое же количество
натурального красителя стоило от 18 до 20 марок, то в 1913 году цена
химического продукта упала до 8 марок за килограмм, а в
1927 году она составляла всего лишь 2,7 марки. Фирмы по
производству натурального индиго некоторое время пытались
еще задержать это падение цен безжалостной эксплуатацией
128
своих рабочих, но долго выдерживать конкуренцию они уже
не могли и вынуждены были сложить оружие. Плантации
индиго сокращались, и доля природного красителя в общем
количестве производимого продукта стала ничтожной.
На основе индиго была открыта целая группа
красителей. Фридлендер с удивлением установил, что античный
пурпур — очень близкий родственник голубого индиго. Только
вместо водорода в молекуле пурпура находятся 2 атома брома:
Замещая NH-группы индиго на атомы серы, Фридлендер
сумел получить новый класс индигоподобных красителей,'
обладающих не только синим, но и желтым, красным,
зеленым цветами.
Следует упомянуть еще один важный этап на пути
химии красителей, который привел к созданию чрезвычайно
ценного их класса.
В 1901 году химик Рене Бон — специалист по крашению
на анилино-содовой фабрике—попытался получить индиго из
антрацена — исходного вещества при синтезе ализарина. Он,
действительно, выделил голубой краситель, который
отличался хорошими красящими свойствами, не линял при стирке и
был чрезвычайно светопрочен. Бон назвал его индантреном,
образовав это слово от индиго и антрацен. Новый краситель
сразу нашел промышленное применение. Оказалось, что с
помощью незначительных изменений исходных веществ и
метода их обработки можно получить палитру этих
красителей, обладающих чрезвычайно высокой стойкостью.
Количество синтетических красителей, известных в
настоящее время, необозримо. Химики открывают все новые и
новые красящие соединения, но лишь немногие из них
находят промышленное применение, так как требуется, чтобы
краситель не только хорошо ложился на волокно, но и имел
высокую свето- и водостойкость, а кроме того, был бы еще
дешевым. Эти требования не всегда легко удовлетворить.
Натуральные красители, которые применялись сто лет назад,
сегодня уже не играют никакой роли. Химическая
промышленность производит гораздо более разнообразные и дешевые
красители, чем создает природа,
ЛЬ 3. Шпаусцус
Краски и крашение
Обычно под словом краска понимают и охру, и индиго,
и ализарин. Но это не совсем верно. Мы постараемся быть
более точными и различать краски и красители.
Красками, основными красками или пигментами
называют твердые частицы неорганических веществ,
нерастворимые в обычных растворителях. Для крашения краску тонко
измельчают, смешивают с каким-либо связующим веществом
и в таком виде наносят на нужный предмет. После того как
связующее вещество застывает или испаряется, равномерно
распределенные в нем пигменты закрепляются на
поверхности. Так происходит, когда мы рисуем акварельными
красками, белим стены или покрываем что-либо разнообразными
цветными лаками.
Иное дело — красители. Среди многих других к ним
относится большое число каменноугольных красителей и
ализарин, о котором мы уже говорили.
Красители — это органические соединения естественного
или синтетического происхождения, растворимые в воде и
других жидкостях. В процессе крашения волокно
воспринимает их в растворенном виде. Практическое значение имеют
только красители, растворимые в воде.
Красить — это не единственное качество, которым
должно обладать вещество, чтобы можно было говорить о его
практическом применении. Сегодня к краскам и красителям
предъявляется много других требований. Если голубая
краска наружной побелки дома смывается первым дождем, то,
разумеется, от нее мало толку. Если краситель, который
придает вашей косынке красный цвет, после стирки остается в
щелочном растворе, то, конечно, его нельзя применять для
крашения тканей, каким бы красивым и ярким он ни был.
Красители пестрых тканей летнего платья тоже не должны
линять при стирке и, кроме того, не должны блекнуть от
ярких солнечных лучей, во всяком случае до тех пор, пока
платье носится. Такой краситель или краску, в котором бы
все эти качества проявлялись одновременно и в полной мере,
найти невозможно. Но всегда легко установить разницу в
поведении краски или красителя в каждом данном случае и
выбрать подходящую технику окраски и крашения.
Рассмотрим, например, такое качество красок, как свето-
прочность. Мы стоим перед полотнами старых мастеров и
поражаемся свежести красок, сохранившихся в течение
нескольких столетий. Если некоторые картины и потемнели
немного, то в этом виноваты не краски, а лак, который за эти
долгие годы изменился. Нам известны также и светопрочные
красители, но все же у них это качество выражено гораздо
слабее. Это не удивительно. Большая часть пигментов —
естественного происхождения. В виде неорганических соединений
они существуют на земле с незапамятных времен и
выдержали своеобразный «естественный» отбор. Поэтому сейчас
встречаются только такие природные краски, которые оказа-
лись особенно прочными. Понятно, что эти пигменты должны
отличаться полной нерастворимостью в воде. Если бы они
были даже мало растворимы, то в течение тысячелетий
дожди смыли бы их и унесли в землю или моря.
Подобно всем неорганическим соединениям основные
краски гораздо менее чувствительны к высоким
температурам. Они часто способны выдерживать температуру свыше
1000°, в то время как красители, будучи органическими
соединениями, полностью разлагаются при температуре менее
100°.
Конечно, в некоторых случаях основные краски хуже,
чем красители. Они обычно красят слабее, и чтобы придать
цвету большой блеск и глубину, приходится добавлять к ним
осветляющие примеси и красители. Красители и пигменты
наносят на предмет по-разному. Пигменты приходится
наносить, как правило, гораздо более толстым слоем, чем
красители. Это менее удобно. К тому же пигменты нужно
смешивать с более или менее густым связующим веществом.
Поэтому ясно, что для крашения тканей употребляются
красители, так как только они оставляют неизменными качество и
структуру ткани.
Из сказанного можно сделать вывод, что на практике
пигменты и красители не заменяют, а взаимно дополняют
друг друга. Выбор красителя или пигмента зависит от того,
что мы хотим окрасить и какую цель преследуем. Но
подумайте, какое огромное количество предметов нужно окрасить
и какой широкой и разнообразной должна быть палитра
пигментов и красителей, чтобы удовлетворить все требования.
А теперь поговорим о пигментах. Их значение обычно
недооценивают. Краски и красители в мировом производстве
химических товаров вместе с лаками и растворителями
составляют 10%. Из них девять десятых приходится на
пигменты. Производство каменноугольных красителей, бурно
возросшее с 1860 года, сильно влияет на развитие
химической промышленности, но доля их и сейчас еще относительно
мала. Правда, это не умаляет их значения. Следует помнить,
что красители применяются преимущественно при крашении
тканей, а область применения пигментов значительно шире.
И еще, красители, как и пигменты, нужны всюду. Но
производство красителей требует высокого развития науки и
техники и концентрируется поэтому в странах с современным
уровнем промышленности, а минеральные краски можно
производить со значительно меньшими затратами. Часто весь
процесс их приготовления сводится к измельчению и очистке
естественных минералов. В Германии процент
каменноугольных красителей по отношению ко всей продукции
химической промышленности наиболее высокий в мире. Иная
картина в странах с менее развитой промышленностью, особенно
если они владеют богатыми запасами сырья, так как добыча
естественных и искусственных минеральных красок типа
охры или цинковых белил не требует больших затрат.
Мы уже говорили, что для окраски недостаточно одних
пигментов. Рыхлый мелкозернистый порошок пигментов не
5 3. Шпаусцус
закрепляется и отпадает. Этому препятствуют связующие
вещества, которые переводят нерастворимые пигменты в
гомогенную, более или менее вязкую форму. В таком виде они
обычно наносятся кистью и прочно закрепляются на
поверхности окрашиваемого предмета. Связующие вещества можно
рассматривать как разновидность клея. И не случайно для
этой цели применяется ряд обычных клеев.
При окрашивании клеевой краской употребляют
растворимые в воде связующие вещества. Естественно, они не могут
быть водоустойчивыми. Требование к такой краске —
хорошая кроющая способность, экономичность, привлекательный
вид и прочность на истирание. Ведь никому не нравится,
случайно прислонившись к стене, испачкать свой костюм.
Клеевыми красками покрывают преимущественно
внутренние стены помещений.
Значительно прочнее, но и гораздо дороже клеевых
масляные краски, которые получают, смешивая пигменты с
маслом. Однако не всякое масло подходит для этой цели. Масло
должно быстро застывать и, как говорят, «не давать отлипа».
Есть целый ряд масел, которые сохраняют свое вязко-
текучее состояние даже через несколько месяцев после
окраски. Их нельзя употреблять как связующее средство. В основу
масляной краски входят такие масла, которые сразу после
нанесения застывают в виде мягкой и эластичной пленки,
т. е. быстро сохнут.
Высушивание масел происходит на воздухе, причем
существенную роль в этом процессе играет кислород. Чтобы
ускорить окисление, в краску добавляют осушающие вещества,
так называемые сиккативы. Смешанные с маслом в
количестве от одного до пяти процентов, они действуют как
катализатор и ускоряют сушку. Точный химизм процесса
неизвестен, но в конечном итоге он сводится к окислению
ненасыщенных жиров, входящих в состав масел. Количество
известных сиккативов огромно. К ним относятся такие соединения,
как пиролюзит, свинцовые белила, окись свинца, т. е.
вещества, которые содержат много кислорода и сравнительно
легко его отдают. В последнее время все большее применение
в качестве сиккативов находят соли тяжелых металлов
пальмитиновой, олеиновой и линолевой кислот.
Всем известная льняная олифа состоит из льняного
масла, к которому примешано от 1 до 5% сиккатива. Не позднее
чем через 24 часа после покраски она застывает в виде неот-
липающей твердой массы.
В настоящее время получают органические красители
самых различных цветовых оттенков. Они отличаются особой
интенсивностью цветов. К сожалению, использовать их
вместо пигментов нельзя, так как они легко растворяются в
различных жидкостях и не обладают такой кроющей
способностью, как более грубые пигменты. Чтобы придать
красителям свойства пигментов, их превращают в сравнительно
большие нерастворимые частицы. Для этого различные
конденсаты и поверхностноактивные бесцветные пигменты
обрабатывают раствором красителя. Частицы глинозема, каолина
и ряда других веществ обладают способностью присоединять,
поглощать и прочно удерживать мелкие крупицы красителя.
Цвет дают красители, а крупные твердые и белые частицы
служат носителем. Такие краски называют цветными лаками
или фарблаками. Применяют их так же, как и чистые
неорганические пигменты.
\
• 1 • • • * • * ' *
1 • . ч * % '
7Г 0 •*.»• •**•.•• *
* * t
i» * «
От фарблаков следует отличать так называемые
эмалевые краски. Их значение сейчас особенно велико, так как они
решают сразу две задачи. Окрашенные эмалевой краской
корабли, машины, оконные рамы и т. д. имеют
привлекательный вид, и, кроме того, прочная водо- и
воздухонепроницаемая пленка краски защищает их поверхность от коррозии.
Основу эмалевых красок составляют смолы или
смолоподобные вещества, которые после окраски застывают в виде
прочной и блестящей защитной пленки. Смолу растворяют
специальными растворителями для того, чтобы ее можно
было нанести кистью или распылить пульверизатором. После
нанесения растворитель, представляющий собой
органическую жидкость, испаряется, а лаковое покрытие закрепляется.
До сих пор мы говорили о бесцветных лаках. Их
используют, если с помощью защитной лакировки хотят сохранить
собственный цвет предмета или его рисунок. Но чаще
употребляют цветной лак. Для его получения к бесцветному лаку
добавляют соответствующий красящий порошок, который
после тщательного размешивания равномерно распределяется
в нем. Выбирая соответствующие пигменты, можно получить
все известные нам оттенки цветов.
Итак, мы видим, что получать окрашивающие материалы
гораздо труднее, чем обычно себе представляют. Вид и
прочность окраски зависят часто от правильного выбора
пигмента, связующего вещества, других примесей, а кроме того, от
предварительной обработки поверхности.
Процесс крашения еще более сложен, так как в этом
случае должно произойти взаимодействие между красителем и
5*
133
волокном. Трудность заключается в том, что, с одной
стороны, существует очень много различных по структуре
волокон, а с другой стороны, очень велико число красителей. Для
каждого волокна нужно найти такой краситель, который
полностью воспринимается тканью и хорошо закрепляется на
ней. Изменить состав волокна и красителя, как правило,
нельзя, это величины постоянные. И натуральные, и
синтетические волокна приходится брать в том виде, в каком их
производят. Никакие химические и структурные изменения
вообще невозможны. Добиться нужного цвета ткани можно,
лишь заменив краситель, способ предварительной обработки
или технологию крашения.
Уже сотни лет назад человек научился красить ткани.
Искусство крашения во все времена пользовалось большим
уважением. Но еще недавно приемы крашения были чисто
эмпирическими, они основывались на результатах
многочисленных практических опытов и лишь постепенно
совершенствовались. Чтобы как можно скорее создать технологию
крашения новых тканей и красители для них, очень важно
было разработать теорию и понять все происходящие при
крашении процессы.
И сегодня исследовано далеко не все. В представлениях
о процессе крашения, о силах, которые удерживают
краситель на волокне, имеется много пробелов. Однако некоторые
ценные выводы можно уже сделать.
При крашении одинаково важны свойства и поведение
как красителя, так и волокна, поскольку они оба вступают
во взаимодействие. Чтобы лучше понять процесс
крашения, расположим и волокна и красители по группам в
соответствии с их химическим составом и поведением при
крашении.
Различают следующие группы волокон,
1. Целлюлозные волокна — к ним относятся натуральные
хлопок и лен, а также искусственное штапельное волокно и
ацетатный шелк. Штапельное волокно и ацетатный шелк
состоят из целлюлозы, хотя она и подвергалась химической
обработке в процессе производства.
2. Белковые волокна — это прежде всего шерсть и шелк.
3. Химические волокна получают только синтетическим
путем. На сегодняшний день их количество огромно — деде-
рон, ланон, прелан, капрон и др. В будущем их будет еще
больше. Состав таких волокон может быть самым
различным, в некоторых случаях они напоминают волокна первой
и второй групп.
В отличие от волокон для красителей важен не столько
химический состав, сколько их поведение при крашении.
Рассмотрим так называемые субстантивные красители. Это
вещества, окрашивающие волокна целлюлозы непосредственно,
т. е. они переходят из красильной ванны прямо на волокно.
Это не настолько само собой разумеется, как может
показаться на первый взгляд.
Другая группа — кубовые красители. Из них самым
известным является индиго. Для крашения кубовые красители
сначала переводят в растворимую форму, которая, как
правило, другого цвета, и только на волокне восстанавливается
первоначальный состав и цвет красителя.
Совершенно иначе ведут себя проявляющиеся красители.
Чем менее растворим краситель на волокне, тем прочнее
будет окраска. Но нерастворимый краситель не может
перейти из красильной ванны на волокно. Поэтому его разлагают
на растворимые в воде компоненты. Эти компоненты один за
другим наносят на волокно, где они превращаются в
нерастворимый и нелиняющий краситель.
Основные красители содержат группы основного
характера, например — Ntb, которые соединяются в соли с
отрицательно заряженными кислотными остатками. Эти соли и
окрашивают волокно. Совершенно аналогично ведут себя
кислотные красители, только здесь соотношение обратное. И
наконец, рассмотрим протравные красители. Соединяясь с
различными солями металлов, такие красители дают трудно- или
совсем нерастворимые лаки, которые прочно закрепляются на
волокне. Протравные красители окрашивают особенно прочно.
Вернемся к механизму крашения. Сначала рассмотрим
белковые волокна, из которых состоят шелк и шерсть. Как и
во всех.белковых веществах со сложным строением, в их
молекулах содержатся одновременно кислотные группы,
например карбоксильные — СООН, и основные, например — РШг.
В так называемой изоэлектрической точке, которая возникает
при рН = 4,6, обе группы диссоциированы, следовательно, они
ось.
Щ* Jti&
#>*
рН4,6
° "К cod *»; со0'
Жг,о
Ы%о
существуют как заряженные частицы, имеющие характер
ионов. При этом карбоксильная группа несет отрицательный
заряд ( —СОО~), а аминогруппа — положительный ( —NH*
ион). Причем число положительно и отрицательно
заряженных групп одинаково. Если повысить концентрацию
свободных ионов водорода и тем самым снизить значение рН, то
снова образуется группа — СООН и в растворе будут
преобладать положительно заряженные радикалы — NHg •
Волокно тоже заряжается положительно. Его можно
рассматривать как катион. Если в окружающем растворе содержатся
анионы, т. е. отрицательно заряженные частицы красителя,
это приводит к образованию соли и, следовательно, к
окрашиванию. В этом случае процесс крашения нужно
рассматривать как химическую реакцию. При низкой кислотности, т. е.
при значении рН = 4,6-7,0, на белковом волокне
преобладают отрицательно заряженные анионы кислоты. Волокно
имеет отрицательный заряд, и положительно заряженные
катионы основных красителей связываются и фиксируются на
волокне.
Целлюлозные же волокна химически относительно
индифферентны, так что в процессе крашения ни при каких
условиях не происходит химических реакций между
веществом волокна и молекулой красителя, как у шерсти или шелка.
В этом случае, по-видимому, осуществляется
коллоидно-химический процесс адсорбции, при котором краситель
присоединяется к волокну за счет остаточных свободных связей.
Хлопчатобумажные волокна состоят из вытянутых в длину
молекул целлюлозы. Молекулы не содержат никаких
химически активных групп, по крайней мере таких, которые легко
диссоциируют. Поэтому окраска с помощью солеобразования
исключается. Конечно, молекула целлюлозы содержит
многочисленные ОН-группы, но они не обладают основными
свойствами. Однако существует группа красителей, имеющих
радикалы, которые могут свободно соединяться с этими ОН-
группами с помощью так называемых водородных связей.
При этом важную роль играет форма молекулы красителя.
У «конго красного» образование водородных связей происхо-
Цеи*Л4ои<унаЛ Кипб
дит за счет NH2-rpynn. Молекула красителя вытянута в
длину, причем расстояние —NH2—NH2 в молекуле красителя
соответствует расстоянию —ОН—ОН в молекуле волокна.
Процесс заканчивается адсорбцией молекулы красителя на
поверхности волокна с образованием водородной связи. А
закрепившиеся на волокне молекулы красителя медленно
диффундируют внутрь волокна и внедряются между молекулами
целлюлозы. При этом происходит более сильное поглощение
красителя веществом волокна. При добавлении солей
адсорбция усиливается.
В нашей жизни все большую роль играют сейчас
синтетические волокна, которые продаются в магазинах под
названиями дедерон, нейлон, ланон, капрон и др. Окраска этих
волокон очень сложна, так как они имеют плотную
структуру, которую вода не разрыхляет,, отчего молекулы красителя
не могут проникнуть в волокно. Если к тому же на
поверхности волокна отсутствуют активные группы, то это сильно
затрудняет процесс крашения. В таких случаях приходится
покрывать волокно носителем, который и связывает
краситель.
Некоторые синтетические волокна типа дедерона и
нейлона ведут себя подобно шерсти, поэтому для крашения
применяются те же методы, что и для белковых волокон.
Мы видим, как трудно красить даже однородные
волокна. Каждое волокно, каждая ткань требует индивидуальной
технологии крашения. Представляете, как трудно окрасить
смешанные ткани, которые состоят, например, из шерсти и
хлопка, ведь оба вида волокна требуют разной обработки.
В таких случаях необходимы наиболее рациональные методы,
которые дают возможность красить ткань равномерно и
прочно.
В заключение нужно сказать еще несколько слов о
кубовом крашении, которое практикуется в Германии со средних
веков. Самый известный кубовый краситель индиго
практически нерастворим в воде. Очень удобно, когда краситель
находится уже на волокне и не смывается ни при стирке, ни
от дождя и не выцветает. Но как нанести его на волокно, если
он нерастворим? Ведь только в растворенном виде он может
вступить в реакцию с волокном. Вот какая особенность
кубовых красителей приходит на помощь в этом случае:
кубовые красители восстанавливаются в так называемые лейко-
соединения. Лейкоформа индиго хорошо растворяется в
слабой щелочи, но при этом ее цвет меняется. Раствор лей-
коформы индиго или совсем бесцветен или окрашен в бледно-
желтый цвет, в то время как натуральный цвет индиго —
ярко-синий. Лейкосоединения хорошо наносятся на
волокно. При окислении на воздухе снова образуется
первоначальный краситель, который теперь уже находится на волокне в
нерастворимом виде.
Почему помидор красный?
Согласитесь, что вопрос этот необычен и в первый
момент хочется отделаться от него пожатием плеч, отнестись
как к шутке. Если кто-нибудь и ответит на него, то скорее
всего так: помидор красен, когда он созрел, так же как слива
синяя, а тыква желтая. Это хорошо всем известно, так было
всегда. Зачем зря ломать себе голову?
Верно, но почему он именно красный, почему не голубой
или фиолетовый? Мы можем совершенно правильно ответить
на это, что красная окраска зависит, по-видимому, от
присутствия какого-то красителя, от красного соединения,
вырабатываемого растением.
Однако это опять не ответ — мы лишь перенесли вопрос
с помидора на краситель. И наконец, перед нами возникла
необходимость решить, рассматриваем ли мы цвет всех
красителей как случайность или существует определенная связь
между строением молекулы красителя и его цветом. Если
верно последнее положение, то ответ на поставленный вопрос
должен одновременно вскрыть общие связи между
химическим строением и свойствами вещества. Химик мог бы,
синтезируя новое вещество, знать заранее, какую окраску оно
будет иметь.
Соединение, придающее помидору желто-красную
окраску, сейчас хорошо известно химикам — это ликопин, кароти-
ноидный краситель, состав которого можно считать точно
установленным. Ликопин имеет на первый взгляд
устрашающую по сложности формулу
сн,
/
i
СН,
'1
сн5 , сн5
Г3 ' I ! I " I
(W^C=CHCH£-curc=cH-+-CH=CH-c=CH-hcH=cH-c=cH-rCM<
UXJ
копии
На самом деле структура его молекулы довольно проста.
Она состоит из 40 углеродных и 56 водородных атомов,
следовательно, является ненасыщенным углеводородом с
двойными связями, отделенными друг от друга простыми
связями. Такие двойные связи называют сопряженными, и мы уже
знаем, что они отличаются высокой реакционной
способностью. Вернемся еще раз к формуле ликопина. Легко заметить,
что определенные группировки атомов повторяются. Это
относится к группировке
-сИ—сн—с=сн —
138
которую надо рассматривать как радикал изопрена
СНл
chz=ch-c^chz
Позднее, говоря о каучуке и резине, мы расскажем об
изопрене подробнее. Сейчас он интересует нас лишь как
составная часть молекулы ликопина.
Но пока ответ на первоначальный вопрос не стал яснее,
и мы по-прежнему не знаем, почему ликопин с его похожей
на червя формулой придает помидору красный цвет.
Вскоре мы убедимся, что этому, действительно, есть
объяснение. Но придется начать издалека и прежде всего
познакомиться с понятиями света и цвета и с тем, как эти понятия
связаны между собой.
Начнем с понятия свет. Все на Земле было бы мертво,
если бы бесчисленные источники, из которых самым важным
для нас и самым мощным является Солнце, не излучали свет
и энергию. Человечество всегда понимало, какое значение
имеет для жизни свет. Его рассматривали как атрибут
божественного, пока, наконец, трезвое естественнонаучное
исследование не определило свет как электромагнитные колебания
с определенной длиной волны. Источники света,
следовательно, можно рассматривать как датчики этих волн — место их
возникновения и испускания.
Лучи света, естественно, несут определенную энергию.
Чтобы тело начало излучать, оно должно затратить какую-то
энергию, которая может переходить в световую. На Солнце в
Г -'-I _Л ГЦ
, Г / 3 » •
:СЯ^сн=с-сН^СН1-сн=0-сН=сН-гСН=С-сн—сНРСИ
результате ядерных реакций освобождается тепловая и
световая энергия, доходящая до Земли. Включенная
электрическая лампа потребляет электрическую энергию. Спичка,
сгорая, дает свет, потому что благодаря химическим
преобразованиям во время горения освобождается энергия.
Займемся более подробно электромагнитными
колебаниями, выясним сущность возникновения волн.
Нам хорошо известны волны на воде. Если бросить
камень на гладкую и спокойную поверхность озера, частицы
воды начинают колебаться в вертикальном направлении. От
того места, где камень коснулся поверхности воды и отдал
часть энергии движения, распространяются концентрические
волны, состоящие из гребня и впадины. Гребень и впадина
/§
сменяют друг друга. Число таких изменений в секунду
называют частотой и обозначают греческой буквой v. Частоту
измеряют в герцах, 1 герц (гц) — это одно колебание в
секунду.
Другая величина, характерная для колебательной формы
движения — длина волны X. Под длиной волны понимают
расстояние от ее гребня до впадины. Легкий порыв ветра
вызывает на гладкой поверхности воды волны небольшой
длины, а ветер на море может создать волны длиной во много
метров.
И наконец, интересна скорость распространения волны
С, измеряемая в метрах в секунду. Чем больше С, тем
быстрее распространяется колебание. Для волн на воде, которые
вызваны брошенным камнем, С можно определить с помощью
секундомера и рулетки. Скорость распространения в этом
случае составляет всего несколько метров в секунду, а часто
бывает еще меньше. А свет распространяется с огромной
быстротой. Его скорость так велика, что ее уже нельзя
измерить обычными методами. Эту скорость определяли
различными способами и установили, что она составляет
300000 км/сек.
Между этими основными величинами существует
соотношение .
с = Л- v
(1)
140
т. е. скорость распространения волны равна произведению
длины волны на число колебаний. Для электромагнитных
колебаний, а значит и для света, С постоянно.
Так как величина С постоянна, то X и v зависят друг от
друга — при уменьшении длины волны частота
увеличивается и наоборот. Это можно пояснить на рисунке. Если длина
волны Д.2 составляет Vs от Xi, то число колебаний v2 должно
быть в 5 раз больше, чем vi, если эти колебания
распространяются с равной скоростью.
Нам уже известно, что свет не является единственным
видом электромагнитных колебаний, что спектр этих
колебаний гораздо шире. Он начинается радиоволнами, которые
разделяются на длинные, средние и короткие, а с некоторого
времени и на ультракороткие. Длинные волны с частотой
примерно в 300 кгц, т. е. колеблющиеся 300 000 раз в секунду,
имеют длину волны
300 000 000 : 300 000 = 1000 м.
Мы преобразовали уравнение (1) в c/v = X и приняли
для С значение 300 000 000 метров в секунду.
Уменьшая длину волны, мы получим сначала важные
для телевидения ультракороткие радиоволны, а затем
инфракрасные, или тепловые волны длиной от 1 до 0,001 мм. Эти
волны невидимы, но мы можем воспринимать их как тепло в
отличие от радиоволн, которые мы вообще не воспримем.
Колебания с еще более короткой длиной волны от
10 000 до Тообб" мм мы °ЩУЩаем как свет. В
соответствии с уравнением \ = с/Х можно вычислить частоты —
они будут колебаться от 8«1014 до 4 • 1014, т. е. от 800
триллионов до 400 триллионов колебаний в секунду. Подобные
числа ничего не говорят нашему воображению. Для
измерения таких малых длин волн пришлось ввести специальную
единицу; их измеряют в миллимикронах (ммк). 1 ммк —
это миллионная доля миллиметра, т. е. Ю^6 мм или 10~9 м.
Иногда употребляют другую единицу — ангстрем. 1А —
десятимиллионная доля миллиметра, т. е. 10~7 мм. Длины волн
видимого света лежат между 400 и 800 ммк или 4000 и
8000А.
Однако спектр электромагнитных колебаний охватывает
еще более короткие волны. При длинах волн менее 400 ммк
говорят об ультрафиолетовом излучении, которого мы не
видим, но ощущаем по его воздействию на наш организм.
Ультрафиолетовые лучи составляют часть солнечного
излучения. Именно они вызывают ожоги, если слишком
интенсивно принимать солнечные ванны. Из этого факта мы можем
сделать еще и такой вывод: энергия излучения возрастает с
увеличением частоты. Это естественно, так как чем чаще
колебания волны, тем система должна быть богаче энергией.
За ультрафиолетовыми лучами следуют рентгеновские лучи,
а за ними, наконец, гамма-лучи, возникающие при ядерных
реакциях. Их длина измеряется десятками ангстрем.
Итак, мы повсюду встречаем электромагнитные
колебания с длиной волны от многих километров до десятимиллион-
/амнЫ
k- Umqgg?"*
}%щи«*е и eft,»"* «*
-Я^ГвменН**
motO
ных долей миллиметра. И только очень незначительную часть
этих волн человек ощущает как видимый свет.
Мы уже знаем, что электромагнитные волны несут
энергию. В 1900 году немецкий физик Макс Планк сумел
доказать, что эта энергия делится на мельчайшие порции, так
называемые кванты. Кванты для энергии то же самое, что
атомы для вещества. Энергия кванта выражается уравнением
E = h-v
д
и, следовательно, зависит только от числа колебаний v, так
как h — постоянная. Чем больше v, или, что то же самое, чем
меньше X, тем больше будет энергия кванта излучения. Так
как излучение следует рассматривать как сумму
соответствующих квантов, то увеличение кванта энергии означает
увеличение энергии излучения.
Так, коротковолновые рентгеновские лучи обладают
большей энергией, чем, например, длинноволновые
инфракрасные лучи. Свет с длиной волны 800 ммк несет меньше
энергии, чем свет с Х = 400 ммк.
После этих предварительных замечаний мы остановимся
на излучении с длинами волн между 800 и 400 ммк —
видимом свете. Видимый свет представляет собой сумму волн
различной длины — от 400 до 800 мМк. Разложить этот свет по
длинам волн очень легко, и каждый может проделать опыт,
для которого нужно иметь лишь стеклянную призму. На
призму под определенным углом направляют луч света. Сте-
142
пень его отклонения зависит от длины волны, и поэтому все
цвета, составляющие белый свет, преломляются по-разному.
Если перед призмой поместить однородный экран, то
вместо ожидаемого светлого светового пятна мы получим
цветную спектральную полосу, состоящую из семи цветов
радуги: красного, оранжевого, желтого, зеленого, голубого, синего
и фиолетового. Это основные цвета, между которыми
существует много переходных тонов. Всего в солнечном спектре
человеческий глаз различает примерно 160 различных тонов.
Наибольшая длина волны 800 ммк соответствует
красному цвету — границе видимого спектра. Из уравнений c = Xv
и E = hv следует, что в случае длинных волн число
колебаний относительно мало. Значит, и энергия этих световых
квантов должна быть незначительна, по крайней мере
меньше, чем для фиолетового излучения, у которого длины волн
составляют 400 ммк.
Таким образом, количество энергии светового излучения
возрастает от красного к фиолетовому. Это хорошо видно,
если наблюдать за проволокой, которую раскаляют,
пропуская через нее электрический ток. Если ток мал, проволока
накаляется слабо и становится красной. По мере увеличения
тока увеличивается и энергия возбуждения. Окраска света,
излучаемого раскаленной проволокой, переходит из красной
сначала в желтую, а затем в белую. В то время как при
красном калении проволока испускает только лучи с длиной
волны от 700 до 800 ммк, при большем разогреве возрастает
количество лучей с меньшей длиной волны. В сумме все эти
лучи дают белый свет такого же состава, как солнечный.
Источники цветного света испускают лучи лишь одной
длины волны или принадлежащие к ограниченной области
спектра. Например, источник, дающий зеленый свет,
испускает лучи только с длинами волн между 500 и 550 ммк.
Однако только очень немногие из окружающих нас
предметов являются прямыми источниками света, образующими
и испускающими лучи. Большинство предметов лишь
отражает падающий на них свет и поэтому становится видимым.
Но почему же у нас на ногах черные ботинки, на столе
белый лист бумаги, а чашки в шкафу синие? Как объяснить,
почему окраска этих предметов разная — ведь во всех
случаях состав падающего света одинаков. Это понять нетрудно.
Предмет кажется белым тогда, когда отражает весь
падающий на него свет. В этом случае меняется только путь,
направление луча, а его состав остается неизменным.
Если тело воспринимает весь свет, полностью поглощает
всю падающую энергию и, следовательно, ничего не
отражает, то от тела не исходят световые лучи, которые могли бы
попасть в наш глаз; в этом случае мы воспринимаем предмет
черным. Для синих чашек в шкафу, о которых мы
упоминали, существуют две возможности. Во-первых, из световых
волн могут быть поглощены все, за исключением тех, которые
вызывают у нас ощущение синего.
Возможен и другой случай, наиболее часто
встречающийся в действительности. Поглощаются только волны длиной в
590 ммк. Мы воспринимаем эти волны как оранжевый свет,
а остаток отраженного излучения в целом вызывает
ощущение синего. Оранжевый и синий — две краски, вместе даю-
144
щие белый цвет. Цвета, образующие такие пары, называют
дополнительными цветами. Известно несколько таких пар.
Они представлены в таблице, и их нужно знать, чтобы понять
дальнейшие рассуждения.
Поглощаемый свет
1 Длина волны в ммк
400
425
450
490
510
530
550
590
640
730
__———— ч
Соответствующий цн<
Фиолетовый
Индиго-синий
Синий
Сине-зеленый
Зеленый
Желто-зеленый
Желтый
Оранжевый
Красный
Пурпурный
Наблюдаемый цвет
т
Зеленовато-желтый
Желтый
Оранжевый 1
Красный
Пурпурный
Фиолетовый
Индиго-синий 1
Синий 1
Сине-зеленый
Зеленый
Следовательно, если какой-либо предмет поглощает из
падающего белого света лучи с длиной волны в 490 ммк, то
сумма волн отраженного излучения дает красный цвет.
Вернувшись к исходной точке наших рассуждений, можем
сказать, что именно так это происходит и у красных помидоров.
Мы уже знаем, что их цвет вызван присутствием красителя
ликопина. Но нас интересуют более глубокие причины.
Как известно, молекула состоит из атомов, а атомы в
свою очередь состоят из ядра и электронной оболочки. У
большинства органических соединений, а нас интересуют сейчас
только они, связь между атомами осуществляется общими
электронными парами. Простая связь — это одна общая
электронная пара, двойная связь — две, а тройная — три. Именно
эти электронные пары в состоянии принимать энергию
излучения, поглощать ее. При этом они переходят в состояние с
более высокой энергией, возбуждаются, как говорят физики.
Однако такая энергия не накапливается подобно воде в
большом водохранилище. Возбужденное состояние электрона
очень кратковременно, а затем поглощенная энергия
отдается, конечно, уже в другой форме, и электроны возвращаются
в первоначальное состояние, которое сохраняется до
столкновения со следующим квантом излучения.
Эта игра повторяется снова и снова. Похоже на автомат,
в который бросают 1 марку, а он выбрасывает 2 монеты по
50 пфенигов. Опустили 1 марку, а автомат отдал то же
количество денег, хотя и в других монетах. При этом общее
количество денег в нем остается неизменным. Лишь на короткий
промежуток времени, между опусканием монеты и выдачей,
автомат имеет на одну марку больше, чем обычно. В этот
момент он находится в «возбужденном состоянии».
Здесь мы имеем дело с динамическим состоянием
равновесия электронов в молекулах соответствующих соединений.
Пары электронов, участвующие в образовании простой
связи, довольно прочны и неподвижны. Это проявляется в
стабильности насыщенных соединений. Такие электроны
трудновозбудимы, и энергии световых квантов в диапазоне
видимого света для их возбуждения не хватает. Достаточна
лишь энергия квантов коротковолнового ультрафиолетового
излучения. Ультрафиолетовые лучи для нас невидимы, в то
же время видимая часть излучения отражается полностью.
Соединения кажутся бесцветными или белыми, хотя они и
поглощают лучи, но только в ультрафиолетовой области.
Таковы бензол или парафин и многие другие белые и
бесцветные соединения. С физической точки зрения они цветные, но
человек этого не ощущает. Живые существа, которые
обладали бы органом для восприятия волн подобной длины, смогли
бы зарегистрировать в этом случае «различия в цвете» и,
следовательно, рассматривали бы бензол или эфир как
«цветные» вещества.
Электроны двойных связей более подвижны. Поэтому
они возбуждаются уже при облучении длинноволновыми
лучами. Чем больше в молекуле таких двойных связей, тем
дальше отодвигается граница поглощения в видимую область
спектра, и вещества начинают восприниматься нами как
цветные. Сказанное особенно справедливо для сопряженных
связей. Из таблицы видно, как наблюдаемый цвет меняется
в соответствии с длиной Болны поглощенного света от
зеленовато-желтого через красный и голубой в зеленый.
Представим эти соотношения более наглядно. Бензол
имеет три двойных связи, но мы уже знаем, что они
стабильны и их нельзя сравнивать с обычными двойными связями.
Возбуждать их может только излучение, достаточно
коротковолновое, с высокой энергией, и поглощают они только
ультрафиолетовый свет с длиной волны 270 лелея.
Видимый свет в диапазоне между 400 и 800 лелея
полностью отражается. Поэтому бензол подобно воде кажется нам
бесцветной и прозрачной жидкостью.
Два бензольных радикала, связанных друг с другом,
образуют дифенил СвНб—СвНб. Удвоение числа двойных связей
в молекуле проявляется поглощением лучей с длиной волны
315 лелея. И в данном случае это поглощение находим еще в
ультрафиолетовой области спектра, дифенил — это
бесцветное соединение. Если в молекуле содержится больше
ненасыщенных связей, то граница поглощения отодвигается все
дальше в область излучения с большей длиной волны.
Наконец, начинается поглощение видимого света, и соединения
становятся окрашенными, что видно из таблицы на стр. 147.
Рассмотрим теперь еще раз формулу красного красителя
в помидорах — ликопина. Несмотря на различие, она имеет
и много общего с последним соединением в этой таблице,
однако у ликопина больше сопряженных двойных связей.
Поэтому спектр поглощения его еще сильнее сдвинут в
длинноволновую область; поглощение происходит в сине-зеленой
области видимого спектра (490 лелея). Соответственно
отраженная часть света кажется нам красной.
CgHe • .
бесцветный
свн5 — свн5
бесцветный
СфН}—СН=СН—С(|Н5 . . . .
бесцветный
СвН5— СН=СН—СН=СН—СвН6
бесцветный
СвН5— СН=СН—СН=СН— СН=
желто-зеленый
CeH5—CH=CH—GH=CH—CH:
желтый
СвН5— СН=СН—СН=СН—СН=
=СН—СвНб
оранжевый
СвН5—СН=СН—СН=СН—СН=
=СН—СН=СН—СвН5 . . .
красно-оранжевый
Поглощение волн длиной:
270 ммк
(ультрафиолетовая область
315 ммк
(ультрафиолетовая область)
340 ммк
(ультрафиолетовая область)
360 ммк
(ультрафиолетовая область)
GH—СвН6 400 ммк
(фиолетовая область
видимого спектра)
=СН—СН-СН—СвН6 . 425 ммк
(темно-синяя область
видимого спектра)
СН—СН =СН— СН=
450 ммк
(синяя область видимого
(спектра)
СН— СН=СН— СН=
480 ммк
(сине-зеленая область
видимого спектра)
Легко заметить, что при увеличении числа сопряженных
двойных связей произойдет дальнейшее смещение
поглощаемого света в длинноволновую область, причем цвет
соединения соответственно изменится.
Вернемся к нашему примеру: соединение CeHs— (CH =
= СН)ц—СвНб обладает максимумом поглощения при
А, = 530 ммк, следовательно, цвет его может меняться от
фиолетового до черного. Прекрасным подтверждением
сказанного являются ароматические соединения с конденсированными
кольцами, т. е. соединения с несколькими бензольными
кольцами в молекуле. Бензол СвНб
нафталин СюНв
и антрацен СмН
ю
нам уже известны. Они поглощают излучение только с
высокой энергией, а это значит, что для нашего иИ&а ор&^а^^тсяА,
бесцветными или белыми. Нафтацен CieHij . ^f ^>f \
Ш-V
оранжевого цвета, пентацен
— голубого, а гексацен СгвН^
зеленого. Из этих примеров становится ясно, Что с
увеличением количества бензольных ядер в молекуле, а значит, с
147
увеличением числа двойных связей соединение поглощает
излучение, менее богатое энергией, и цвет его углубляется.
Мы уже вскрыли существенные связи между строением
молекулы и цветом соединения. Но тут же мы сталкиваемся
с другими особенностями.
Во второй половине XIX века пытались установить связь
между строением и цветом всех известных тогда красителей.
Химия красителей в то время была молодой отраслью науки.
Только в 1856 году Перкин синтезировал мовеин — первый
промышленный синтетический краситель. В последующие
годы были синтезированы многие новые красители — и такие,
которые прежде находили в природе, и такие, для которых
природных образцов не было. Стало очевидным, что открыта
новая обширная область, скрывающая множество чудес.
Чтобы дальнейшие исследования не были случайными, ученые
искали связь между химическим строением и цветом.
Представим себе местность со множеством озер,
расположенных на большом расстоянии друг от друга. Мы намерены
побывать у каждого из них. Если бродить без карты, наугад,
можно как-то случайно наткнуться на озеро, но можно и не
заметить его. Если же у нас будет план местности, то свою
задачу мы выполним быстрее и успешнее. Чем точнее план,
чем надежнее карта, тем нам будет легче.
Так и с красителями. Если бы удалось найти
взаимосвязь между строением молекулы и цветом, то дальнейшая
работа не зависела бы только от случая. Больше того, можно
было бы вести направленную работу, сознательно изменяя
состав молекул, чтобы получить краситель желаемого цвета.
Однако такими сведениями в полной мере химия в то
время не располагала, но уже было получено много новых
данных о красках и обобщены ранее известные
закономерности.
В 1876 году Витт, который работал в области химии
красителей, установил, что в любом окрашенном органическом
соединении содержатся определенные группы молекул. Он
назвал эти группы хромофорами, носителями цвета. Уже
Витт отнес к хромофорам приведенные ниже радикалы.
Присутствие каждого из них придает соединению окраску,
которая углубляется в ряду
_сн=сн- ^с=о -сн^~ -Я-Сп
В каждой хромофорной группе есть по крайней мере одна
двойная связь.
Значение хромофоров можно показать на следующем
наглядном примере. Мы уже знаем, что соединение СбНб—СН =
= СН—СвНб бесцветно — оно поглощает ультрафиолетовый
свет. Если —СН = СН— заменить другими хромофорными
группами, то поглощаться будет свет с другой длиной волны.
Так, бензилиденанилин CeHs—CH = N—СбНб поглощает
свет с длиной волны 330 ммк, т. е. это соединение все еще бес-
цветно. Азобензол CeHs—N = N—CeHs кажется оранжевым
потому, что его максимум поглощения равен 450 ммк. У тио-
бензофенона CeHs—CS—CeHs основное поглощение в области
более длинных волн, а именно при А, = 620 ммк. Цвет
соединения синий.
Иногда достаточно ввести одну хромофорную группу,
чтобы сдвинуть поглощение в видимую область спектра.
В большинстве случаев, как мы уже видели на предыдущих
примерах, необходимо несколько таких групп. У бесцветного
ацетона СНз—СО—СНз только одна хромофорная группа,
диацетил СН3—СО—СО—СН3 окрашен в желтый цвет, а
трикетопентан СНз—СО—СО—СО—СНз, содержащий три
хромофора, — оранжевого цвета. Максимум поглощения
сместился из ультрафиолетовой области в синюю.
Витт установил, что некоторые другие радикалы также
влияют на цвет соединения. Эти группы сами по себе не
придают окраску бесцветной молекуле, значит хромофорами их
считать нельзя, но они усиливают окраску, обусловленную
уже имеющимися хромофорными группами. Витт назвал
такие группы ауксохромами, т. е. усилителями цвета. Ауксо-
хромный характер имеют —О—СНз, —ОН, —NH2, —NHR
и — NR2, где R — любой радикал, например —СН3.
Красящее действие этих групп возрастает в такой
последовательности:
02N—CeH4—N=N—С„Н4—ОН
Максимум поглощения при 425 ммк
02N—CeH4—N=N—CeH4—NH2
Максимум поглощения при 450 ммк
02N-GeH4-N=N-CeH4-N(CH3)2
Максимум поглощения при 490 ммк
Темно-желтый цвет
Желто-красный цвет
Темно-красный цвет
За исключением конечных заместителей формула всех
трех соединений совершенно одинакова. Если все-таки цвет
переходит от желтого к желто-красному и к красному, то это
связано только с заменой ауксохромных групп от —ОН через
—NH2 к — М(СНзЬ, так как именно в такой
последовательности цветоусиливающее действие этих групп возрастает.
Роль двойных связей в хромофорных группах можно
продемонстрировать на кубовых красителях. Возьмем снова
индиго. Из предыдущей главы мы уже знаем, что синий
краситель при восстановлении переходит в желтоватую или
белую лейкоформу. Тогда нас не интересовали причины
изменения цвета. Рассмотрим теперь уравнение реакции
м
ItHoueofouHuii)
A,—C-0H 0U-C—/ |
t/teurto-индиго
(SeuQCiJ
Мы видим, что в ходе реакции происходит разрыв трех
двойных связей в молекуле индиго. Максимум поглощения в
лейкосоединениях сдвигается в ультрафиолетовую область
спектра, соединение переходит в бесцветное состояние.
Несмотря на то что взгляды на причины возникновения
цвета расширялись и углублялись, классические воззрения
Витта сохранили свое значение и до нашего времени и лежат
в основе учения о цвете.
В 1924 году было установлено, что полярный характер
молекулы, возникающей, например, при солеобразовании,
существенным образом влияет на углубление цвета
соединений. Введение ауксохромов способствует также
возникновению центров различной электронной плотности, т. е.
обусловливает полярность молекулы.
Приведем конкретный пример. Растворяющемуся в воде
и спирте ярко-красному красителю парафуксину
приписывают структурную формулу
Смещение электронов в молекуле, а именно у правого
бензольного кольца и аминогруппы — NH2, приводит к
образованию хиноидной структуры, углубляющей цвет, а также
к возникновению положительно и отрицательно заряженных
центров. Все это, а также наличие ауксохромных аминогрупп
в молекуле создает условия для поглощения в видимой
области спектра, что и определяет цвет парафуксина.
Хиноновая структура
Как уже точно установлено, более сильное ауксохромное
действие оказывают — NR2-rpynnbi, например — 1Ч(СНз)г.
Если мы в парафуксине заменим аминогруппы на диметил-
аминные группы — ЩСНзЬ, то получим кристаллический
фиолетовый краситель с формулой
Как показывает название, это краситель яркого
фиолетового цвета, что обусловлено смещением максимума
поглощения в область более длинных волн.
В сильно кислом растворе кристаллический фиолетовый
поляризован еще сильнее. Это видно по формуле
Молекула становится все более похожей на молекулу
соли, максимум поглощения отодвигается в область более
длинных волн, примерно до 640 ммк, и цвет раствора
переходит в сине-зеленый.
Во многих случаях молекулы красителей обнаруживают
характер амфотерного иона. Положительно и отрицательно
заряженные центры распределены внутри такой недиссоции-
руемой молекулы равномерно. Благодаря этому происходит
углубление цвета.
Хотя в исследовании связей между структурой и цветом
органических соединений достигнуты довольно
значительные успехи, нельзя умолчать о том, что наши знания в
этой области все еще очень несовершенны. Особенно это
касается соединений сложного строения. Известны случаи, в
которых трудно согласовать теорию с практикой.
Итак, хотя нет безупречной и окончательной теории о
зависимости между строением и цветом, мы уже сейчас в
состоянии понять некоторые закономерности и в известной
степени заранее делать определенные заключения.
Глава о чистоте
Чистоту любили уже древние римляне. Правда, им не
было известно мыло. При мытье или стирке они пользовались
чистой водой, золой растений и ароматическими маслами.
При такой стирке масло и зола растений, состоящая, как
известно, в основном из карбоната калия, являлись
исходными веществами, образующими мыло. Где и когда впервые
смешали жир с золой растений, сварили эту смесь и таким
путем получили мыло — не известно. Римский историк
Плиний, еще в I веке нашей эры, сообщил о мыле из Галлии, а сто
лет спустя римский врач Гален упоминал галльское и
германское мыло, способы применения и преимущества
которого он подробно описывал.
Мыло изобрели, но прошли еще столетия, прежде чем
его научились изготовлять в промышленном масштабе.
Начало мыловарения относится к IX веку. В это время
мыло уже варили в Германии и Франции. Позднее стало
известно об английском мыле, а в 1400-х годах
исключительной славой пользовалось мыло из Венеции. Но мыло в то
время было предметом роскоши, и лишь очень немногие
могли позволить себе покупать его. В 1850 году Англия
повысила налог на мыло. Поэтому мылись в основном без него,
употребляя дешевую древесную золу.
В XIX веке произошел решительный поворот в
производстве мыла. Научное исследование шло рука об руку с
техническим прогрессом, взаимно обогащая друг друга. Благодаря
французскому химику Шеврелю, исследовавшему состав
жиров и механизм мылообразования, столетняя эмпирика
начала уступать место точному познанию. Важно отметить, что
уже тогда умели в нужном количестве производить дешевую
соду и каустик. Не было недостатка также в жирах и
маслах. Их поставляли из заморских колоний. Известную роль
сыграли общественные изменения: наступило время, когда
маленькие мануфактуры были поглощены огромными
промышленными предприятиями, которые могли производить
больше дешевых товаров. В Англии это преобразование
совершилось раньше. В результате в период с 1810 по 1880 год
производство мыла увеличилось там в 10 раз, и цены на него
значительно упали.
Мыло превратилось из предмета роскоши в предмет
широкого потребления. Люди уже регулярно стирали белье,
ежедневно мыли тело, а это положительно сказалось на
здоровье народа.
В наши дни мыло и моющие средства, которые
объединяются под названием моющих веществ, являются настолько
привычными и само собой разумеющимися, что о них вряд
ли кто-нибудь задумывается. Это не справедливо, так как
не только состав и способ изготовления этих веществ, но и
причины их моющего действия очень интересны.
Чтобы понять механизм мылообразования, необходимо
прежде всего выяснить состав исходных веществ. Для соды и
гидрата окиси натрия это сделать просто. С химической
точки зрения сода — это карбонат натрия Na2CC>3, а формула
гидрата окиси натрия NaOH.
Труднее с жирами и маслами, необходимыми для
изготовления мыла. Это продукты растительного и животного
происхождения, которые содержатся в тканях организма и
могут составлять значительную долю их общего веса (как,
например, у откормленной утки или у жирной свиньи). Зерна
мака содержат около 43% масла, мякоть плодов оливы —
до 60%. Сегодня известно более 1300 различных жиров и
масел, встречающихся в природе.
У жиров и масел, несмотря на различие, есть нечто
общее. Их можно рассматривать как нейтральные сложные
эфиры, в которых спиртовым компонентом всегда является
трехатомный спирт — глицерин, а остаток кислоты жирного
ряда содержит от 8 до 18 атомов углерода. Чаще всего
встречаются жиры, являющиеся эфирами пальмитиновой
С15Н31СООН, стеариновой С17Н35СООН или ненасыщенной
олеиновой С17Н33СООН кислот.
Конечно, жирные кислоты не исчерпываются
перечисленными выше. Глицерин как трехатомный спирт содержит
в молекуле три ОН-группы, которые можно этерифицировать.
Так как жирные кислоты имеют только одну карбоксильную
группу — СООН, принимающую участие в образовании
сложных эфиров, то при этой реакции к одной молекуле
глицерина присоединяются три молекулы жирной кислоты. Прежде
считали, что к одной молекуле глицерина могут
присоединяться только одинаковые кислоты, напшшео
ООС— С 1у И
СН,0Ц *НОос-с,7Нз!>
I z
СНОН + Ноос-с17%
CHZ ~^~ ^17^35-
>CH-<Ooc-Cl7Ub5 -idHjfi
CH2-Ooc-c1?tf35
Унристеарсип
9СиСи&тсь
Сейчас известно, что так бывает не всегда. Преобладают
не «симметричные сложные эфиры», у которых с глицерином
связаны три одинаковые молекулы жирной кислоты, а
«смешанные сложные эфиры», в которых все три молекулы
кислоты жирного ряда различны. В природе едва Ли можно
встретить тристеарат, легче найти такой эфир, который
образовался этерификацией глицерина олеиновой, пальмитиновой
и стеариновой кислотами:
СИ, ОН i НООС— CiiH'ii
15 Н3 7
снон +воос-с
I
сц2сн+ ноос— спи
chz— ooc-cnft33
с
Этот процесс может протекать также и в обратном
направлении — справа налево. Существуют различные
технические способы подобного расщепления жиров. В процессе
такого расщепления образуются глицерин и свободные кислоты
жирного ряда, существовавшие в жире или масле в связанном
состоянии.
Если подействовать на жир или масло едким кали или
едким натром, также происходит расщепление жира, однако
при этом свободные кислоты жирного ряда не образуются.
Эти кислоты вступают в реакцию с избытком щелочи, и
образуются калиевые и натриевые соли кислот жирного ряда.
Именно эти соли мы и называем мылами, а весь процесс
омылением жиров.
В принципе изготовление мыла — очень простой процесс,
и человечество давно владеет им. Преимущества нынешних
заводов по сравнению с прежними мыловарнями — в богатом
техническом оснащении, лучшем сырье и применении
добавок. Все это вместе с более глубоким знанием химических
процессов позволяет изготовлять продукцию хорошего
качества. Но и сегодня, как и в давние времена, также нагревают,
перемешивая, жир и щелочь в огромных котлах, где
происходит процесс омыления. Через некоторое время получают
вязкую массу, мыльный клей, состоящий из собственно мыла,
глицерина, воды и избыточной щелочи.
Эту массу, содержащую еще некоторые примеси, можно
отправить прямо в продажу как клеевое мыло. Но такое
мыло содержит очень много воды и глицерина и слишком быстро
расходуется при стирке, поэтому его чаще всего
перерабатывают в ядровое мыло. С этой целью в мыльный клей
прибавляют поваренную соль. В насыщенном растворе соли,
диссоциирующей на ионы, получаются натриевые соли жирных
кислот. Они-то, собственно, и являются мылом. Такое мыло
гораздо менее растворимо в воде и отделяется в виде
полужидкого верхнего слоя смеси. Остается «подмыльный щелок»,
который состоит из глицерина, воды, соли и щелочи, не
вступившей в реакцию. Ядровое мыло отделяется от подмыльного
щелока. В него добавляют немного воды, чтобы масса не
крошилась, различные разбавители, ароматические вещества,
красители, и мыло, которое мы потом покупаем, готово. Если
при омылении применить едкий натр, получаются твердые
мыла. Если заменить его едким кали, то образуются
калийные соли жирных кислот. Эти соли не затвердевают и
образуют жидкое мыло.
В зависимости от чистоты жиров, от качества и
количества примесей, а также от способа их прибавления в
мыльную массу получаются различные мыла, от грубого ядрового
для стирки до ароматного и мягкого туалетного мыла.
Все мыла служат для растворения и удаления грязи из
тканей или кожи. С водой мыла образуют более или менее
сильную пену. Моющая способность мыльного раствора больше,
чем у чистой воды. Рассмотрим подробнее процесс мытья.
Займемся прежде всего водой. Вот кран, из которого
капает вода. Почему она капает? Почему бы ей не течь более
или менее тонкой струйкой?
Если мы присмотримся внимательнее, то заметим, что
вода, вытекая из крана, как бы наполняет тончайшую
оболочку, которая постепенно растягивается. Когда капля
становится большой и тяжелой, она срывается с крана и падает вниз.
Если добавить в воду немного маленьких твердых частиц, то
можно отчетливо наблюдать движение жидкости под
пленкой. Именно на поверхности водяной капли действуют силы,
играющие важную роль и существующие только там. Эти
силы называют поверхностным натяжением, или, лучше,
силами на поверхности раздела, так как именно на поверхности
капли существует раздел между водой и другой средой,
обычно воздухом. Силы поверхностного натяжения сжимают кап-
\
if
лю в шар, то есть делают ее поверхность наименьшей для
данного объема жидкости.
Припомните, если стакан осторожно наполнять водой, то
уровень жидкости может подняться выше его края, образуя
выпуклость, горку. И в этом случае избыток воды
удерживается особой водяной пленкой, не позволяющей жидкости ^> ,,, , о
стечь вниз е%* «*£>**
Как объяснить особое поведение молекул воды на по- 4 >
верхности раздела? Рассмотрим сначала молекулу в большой
массе воды, например в середине полной ванны. Физик знает,
что все тела воздействуют друг на друга, чаще всего они
взаимно притягиваются. Это справедливо для молекул воды так
же, как и для Солнца и Земли. И наша молекула также
воздействует на соприкасающиеся с ней другие молекулы и в
свою очередь испытывает их воздействие. Так как эти
воздействия одинаковы со всех сторон, все силы, действующие на
молекулы, взаимно уравновешиваются. Примерно то же
самое происходит с человеком, которого тянут со всех сторон с
равной силой. Он остается на том же месте, так как силы
взаимно компенсируются. Картина резко изменится, если его
тянуть с трех сторон. В этом случае действие сил будет
преобладать в одном направлении. То же самое происходит с
молекулами воды на границе раздела, на поверхности
жидкости. Силы действуют тут только с одной стороны, молекула
притягивается к воде — происходит образование так
называемой поверхностной пленки.
Есть целый ряд веществ, которые уменьшают силы на
поверхности раздела. К ним относится мыло. Это доказывает
простой опыт. Пусть из небольшого отверстия бюретки
капает вода — 1 миллилитр ее распадается примерно на 20 капель.
Если мы заполним ту же бюретку слабым мыльным
раствором, то капли станут меньше и будут падать быстрее — на 1
миллилитр приходится уже около 40 капель. Это
происходит потому, что силы на поверхности раздела стали меньше,
водяная оболочка уже не так прочна и капли раньше
отрываются от бюретки. Это можно продемонстрировать также на
другом опыте, который легко провести самому. Очень
осторожно положим швейную иглу, лучше всего с помощью двух
ниток, на спокойную поверхность воды. Если и не сразу, то
после нескольких попыток нам определенно удастся
заставить ее плавать.
155
Стальная игла с большим удельным весом благодаря
поверхностному натяжению удерживается на водяной пленке и
не тонет. Если осторожно добавить к этой воде мыльный
раствор, игла сразу упадет на дно и больше уже невозможно
будет заставить ее плавать. Происходит это потому, что силы
на поверхности раздела с добавлением мыла уменьшаются.
Поверхностнее натяжение чрезвычайно затрудняет
процесс мытья, так как оно препятствует быстрому и полному
смачиванию текстильных волокон или кожи тела, обычно
покрытых очень тонкой пленкой жира. А интенсивное
смачивание является первой предпосылкой того, что моющая
жидкость и частицы грязи вступят в контакт и будут унесены
в процессе мытья. Рассмотрим под микроскопом текстильное
волокно или кожу нашего пальца. Мы не увидим ровной
поверхности, а обнаружим неровное поле с бесчисленными
трещинами, щелями и порами, в которых оседает грязь и откуда
ее трудно удалить. Эту большую поверхность полностью
смочить чистой водой нельзя.
Если кусок материи положить на чистую воду,
понадобится довольно много времени, прежде чем она пропитается
ею и утонет. Это произойдет гораздо быстрее, если в воде
содержится немного стирального порошка или мыльного
раствора, так как теперь она имеет более сильную способность
к смачиванию.
То же явление произойдет и при таком опыте. Утка, как
известно, хорошо плавает, и представить тонущую утку
довольно трудно. Ее перья покрыты водоотталкивающим слоем
жира, благодаря которому вода не может вытеснить
заключенный между крыльями воздух, и удельный вес всей птицы
остается меньше удельного веса воды. Если добавить в воду
поверхностноактивное вещество, смачивающая способность
ее возрастет, перья птицы вскоре пропитаются водой, она
опустится на дно и утонет. Это показывает, что увеличение
смачивающей способности жидкости связано с
соответствующим уменьшением сил поверхностного натяжения. Как это
объяснить?
156
Каждому известно, что водяной мыльный раствор
обладает слабой основной реакцией. Если красную лакмусовую
бумажку погрузить в такой раствор, она окрашивается в
синий цвет. Причина устанавливается просто. Как известно,
мыла являются солями щелочных металлов очень слабых
кислот жирного ряда. Соли, состоящие из сильного
основания, т. е. из NaOH или КОН, и слабой кислоты, вступают с
водой в реакцию, которую называют гидролизом:
Jlcoojta, + няо >Лсоон + XdOH
При этом образуются исходные жирные кислоты и
гидрат окиси натрия. Кислоты жирного ряда слабые и
практически вообще не диссоциируют, а гидрат окиси натрия,
напротив, диссоциирует очень сильно. Возникают свободные
гидроксильные ионы, которые и обусловливают щелочную реакцию
раствора.
За уменьшение поверхностного натяжения ответственны,
однако, не гидроксильные ионы, а как раз молекулы жирных
кислот и особенно мыла. Эти молекулы построены из двух
полярных компонентов, обладающих противоположными
свойствами. Многоатомная алифатическая часть,
представляющая собой типично гомеополярное органическое
соединение, имеет малое сродство к воде, она обладает как бы
«водобоязнью». Мы назовем эту часть «враждебной воде» —
гидрофобной. Другой компонент — карбоксильная группа СООН
построена полярно и может диссоциировать, она
«дружественна воде» — гидрофильна. На поверхности воды будут
накапливаться как молекулы кислоты жирного ряда, так и
<%о&$
молекулы мыла, причем их гидрофильная составная часть —
СООН или COONa — погружается в воду, а гидрофобная
часть выталкивается из воды. Если можно было бы сильно
увеличить эту картину, то мы увидели бы своеобразного
ежика, длинные иглы которого были бы гидрофобными
алифатическими частями молекулы. Благодаря такой ориентации
молекул поверхностное натяжение воды уменьшается, так
157
- Jfc
coofa
v&jf.
как слой, покрывающий поверхность воды, обладает теперь
более низкой энергией. Такой раствор будет смачивать
лучше.
Мыльные растворы и растворы моющих средств
обнаруживают гораздо большую способность к диспергированию и
эмульгированию, чем чистая вода. Это легко показать на
примере. Нальем в бутылку воду и масло. Два слоя жидкости
отчетливо разделяются, так как они не смешиваются и не
растворяются друг в друге. Если мы сильно встряхнем
бутылку, а затем подождем, масляный слой разобьется на
капли, но капельки масла будут относительно большими и вскоре
снова сольются друг с другом. Через некоторое время обе
жидкости опять будут разделены на два слоя.
Добавим в бутылку немного мыльного раствора и снова
взболтаем смесь. Масло будет разбито на более мелкие капли,
мы легко заметим это по молочному цвету смеси. Мелкие
капельки масла уже не сольются так быстро в большие, и
полученная эмульсия довольно долго сохраняется. Это явление
как раз и вызывается полярным характером молекул мыла.
«Подобное растворяется в подобном». На поверхности
раздела масла и воды особенно отчетливо проявляется полярность
молекул мыла. Гидрофобные алифатические остатки молекул
мыла втягиваются в масло, а гидрофильная карбоксильная
группа остается в воде. Все больше молекул воды
располагается таким образом, что масляная капля впитывает
гидрофобную часть молекул, а гидрофильный карбоксильный
остаток окружает масляную каплю тонким слоем.
Карбоксильный остаток частично диссоциирует и тем самым заряжает
всю каплю отрицательно. Одноименно заряженные капельки
отталкиваются друг от друга и лишь с большим трудом
снова соединяются в большие капли. Группы COO, COONa или
СООН гидрофильны, они притягивают молекулы воды и
окружают капельку масла прочной гидратной оболочкой.
Результат в этом случае тот же: каплям трудно соединяться
друг с другом.
При исследовании моющих процессов нельзя не сказать
об образовании электрических зарядов. Часть молекул мыла
диссоциирует по уравнению
JLCGcjfz —> Jtcco~+Na +
Почти все текстильные волокна и частицы грязи в воде
заряжены слабо отрицательно. Полагают, что это связано с
небольшой адсорбцией свободных гидроксильных ионов,
всегда имеющихся в воде.
В мыльном растворе вследствие адсорбции
отрицательных анионов жирных кислот и гидроксильных ионов,
возникающих при гидролизе, этот отрицательный заряд
увеличивается. Одинаковые заряды, т. е. текстильные волокна и
грязь, отталкиваются друг от друга, что благоприятствует
моющему действию раствора.
Не следует переоценивать моющего действия пены.
Часто способность образовывать пену приравнивают к моющей
158
силе раствора. Конечно, мыло, которое образует хорошую
пену, очень приятно. Правда, чаще это зависит не от мыла,
а от того, насколько мягка, т. е. лишена кальциевых солей,
вода. Пена увеличивает поверхность моющей жидкости,
впитывает в себя свободные частички грязи и уносит их. Но
существует много хороших моющих средств, употребляемых
при стирке в стиральных машинах, которые не образуют
пены. Другие вещества, например сапонин, образуют в воде
обильную пену, но совершенно не обладают моющей
способностью.
Итак, мы видим, что процесс мытья сложен. Он связан
с рядом условий и реакций, которые и определяют
различное действие тех или других моющих средств. Только
хорошая смачивающая способность раствора не определяет еще
его хорошую моющую способность.
Обратное утверждение тоже справедливо. Знание
процессов, происходящих при мытье, позволяет более
целеустремленно осуществлять дальнейшее развитие производства мыла
и моющих средств, позволяет искать наилучшие моющие
вещества для различных волокон. Ниже мы расскажем об этом
подробнее. Но сначала сообщим еще некоторые сведения о
воде и мыле.
Каждый знает, что одно и то же мыло в разных условиях
обладает различной моющей способностью. Например, в Иене
оно дает умеренную пену и очень плохо удаляет грязь, а в
Карл-Маркс-Штадте, наоборот, показывает себя наилучшим
образом. Это, конечно, не зависит ни от города, ни от людей,
живущих в нем, а только от воды, которая имеет различную
жесткость. Чистой воды в природе нет. Она всегда содержит
соли или по крайней мере газы, как, например, дождевая
вода. Это очень хорошо, потому что совершенно чистая вода
безвкусна и, кроме tojto, неполезна.
Просачиваясь через различные слои земли, любая
природная вода растворяет соли, а поскольку она содержит
углекислый газ, то известняк тоже растворяется. При этом
образуется кислый карбонат кальция, который в отличие от
нерастворимого карбоната кальция СаСОз слабо растворим в воде:
Са С03 + -^Q+ C0Z JZT* C4 tHCo3Jz
Наряду с кислым карбонатом кальция в воде часто
встречается в растворенном виде бикарбонат магния,
образующийся подобным же образом:
Мисод + -н^с + сог-< У Лу СНсо3)г
Если в породах, по которым течет река, достаточно
железа, то в воде растворяется бикарбонат железа. Кроме
бикарбонатов, в воде часто находятся сульфаты кальция и магния.
Сульфат кальция CaSC>4, правда, растворим в воде очень
мало, но все же достаточно, чтобы его можно было
обнаружить.
Все эти растворенные соли придают воде жесткость.
Следует различать бикарбонатую, или временную,
жесткость и сульфатную, или постоянную, жесткость. Бикарбо-
натную жесткость можно относительно легко устранить. Если
воду нагреть, бикарбонаты разлагаются с выделением
двуокиси углерода и нерастворимых карбонатов, которые
осаждаются на стенках сосуда в виде накипи. Вода становится
мягче и тем самым пригоднее для стирки. Сульфатная
жесткость зависит от растворенных сульфатов, и кипячением ее
не удалишь.
Вообще-то мы должны радоваться, что в воде растворено
столько солей. Они делают воду вкусной, и, кроме того,
содержащиеся в ней соли необходимы организму. Но наличие в
воде солей имеет и свою оборотную сторону. Во-первых, это
влечет за собой образование накипи, а накипь делает
непригодными кастрюли, забивает трубы котлов и препятствует
их равномерному прогреву. Во-вторых, жесткость воды
снижает моющее действие мыла. Из-за образования
нерастворимых соединений кальция и магния мыло практически
уничтожается. Потери мыла при употреблении жесткой воды
часто очень значительны. Во многих местностях большие
количества мыла расходуются зря, не выполняя своего
назначения.
Если для одного человека эти потери малозаметны, то
для такой отрасли коммунального хозяйства, как, например,
прачечная, которая потребляет много мыла, они весьма
значительны. Многие другие отрасли промышленности также
вынуждены пользоваться мягкой водой. В городах с
развитой текстильной промышленностью, как, например, Карл-
Маркс-Штадт, Плауен, Коттбус, вода мягкая, иногда даже
слишком мягкая. Это не случайно, ведь раньше приходилось
пользоваться такой водой, какая была в природе, поэтому
текстильные фабрики строили лишь там, где имелась
мягкая вода.
Если мыло попадает в жесткую воду, вместо ионов
щелочных металлов образуются ионы кальция и магния:
ZAsCGOMsl + Cz
CAcoq)ca-+Z№+
160
Образуются кальциевые или магниевые мыла, которые в
воде нерастворимы и выпадают в виде беловатых хлопьев.
Это мыло не обладает моющим действием, а, наоборот, делает
ткань жесткой, ее волокна покрываются серым неприятным
налетом. И лишь когда все ионы щелочноземельных
металлов, имеющиеся в воде, выпадут в осадок, мыло начнет
пениться и проявлять моющие свойства.
Мыльный раствор известной концентрации служит
обычно для определения степени жесткости воды. Для этого к
заранее отмеренному количеству воды прибавляют
небольшими порциями мыльный раствор и каждый раз смесь
взбалтывают. Момент, когда впервые образуется пена, означает, что
реакция обмена мыла с ионами щелочноземельных металлов
закончена. Зная, сколько мыльного раствора израсходовано,
можно вычислить количество этих ионов. Степень жесткости
воды обозначают 1, если в 100 л содержится 1 г СаО.
В наше время можно уменьшить или вообще устранить
жесткость воды. Для этой цели особенно часто применяется
сода, которая вступает в реакцию обмена с бикарбонатом.
Образуется карбонат, который выпадает в осадок:
К жесткой воде нечувствительны так называемые
синтетические моющие средства, известные под торговыми
названиями «Фева», «Фай», «Мильвок» и т. д.12 Кроме того, они
обладают и другими преимуществами, поэтому мы
рассмотрим их внимательнее.
Мыло — главное моющее средство для кожи, и здесь его
ничем нельзя заменить. Но для прачечных оно имеет много
недостатков. Как уже говорилось, в жесткой воде оно
образует нерастворимое мажущееся кальциевое мыло, которое
совсем не моет ткань и делает ее жесткой. Кроме того, шерсть
и синтетические волокна не переносят обработки щелочным
раствором, образующимся при гидролизе мыла водой.
Поэтому уже давно стремились заменить мыло другими моющими
веществами. Нужно было заменить чувствительную к
жесткости воды и склонную к гидролизу COONa-группу на другую
или по меньшей мере ослабить ее влияние. При этом
следовало сохранить полярный характер молекулы мыла.
Жиры или масла, содержащие в молекуле двойные
связи или свободные ОН-группы, при обработке
концентрированной серной кислотой присоединяют ее по месту двойной связи
и образуют сложные эфиры. Таким путем с 1875 года
получали в больших количествах так называемое красное турецкое
масло — натриевую соль сернокислого эфира и рицинолевой
кислоты. Название этой коричнево-черной жидкости связано
с ее применением. Бе употребляли при крашении турецким
красным ализарином. Сначала действуют концентрированной
серной кислотой при постоянном помешивании на рициноле-
вую кислоту и этерифицируют таким образом имеющуюся
в ней гидроксильную группу:
он
o-so3M
(Тернии э<рир pu<ji</io*<e etuis /ьис~сот£<
Затем образовавшийся эфир нейтрализуют едким натром и
получают
O-SO3W
cHf-CcHjgr-cH-cH£-cH=cH-($H2)fCQOMa+2^0
Серная кислота может присоединяться также по
двойным связям, и тогда образуются еще более сульфированные
соединения.
Натриевые соли этих соединений обладают свойствами
мыла. Они хорошо растворимы в воде, и на них меньше
сказывается ее жесткость, поскольку кальциевые соли эфиров
серной кислоты растворимы, обладают хорошим моющим
действием и хорошо смачивают. Но все же они хуже
мыла.
Кроме того, у этих соединений имеется еще COONa-rpyn-
па, чувствительная к кальциевым солям. Поэтому химикам
следует работать над тем, чтобы совершенно исключить из
соединения эту карбоксильную группу, не понижая
моющего действия вещества.
162
Простейший путь состоит в блокировании карбоксильной
группы низкомолекулярными спиртами, которые
присоединяются к молекуле с образованием сложного эфира по схеме
Х-СсоН + СмНд0Н *Л-СоО-сАНэ + Нг°
ОН ОН
Жирном) /cacuona SbyrnuAoSbu Zbj/Htu+tofifat* Эфир
Спирт э/шрно# KucucntU
Далее присутствующая в кислоте гидроксильная группа
этерифицируется серной кислотой, а затем после
нейтрализации едким натром получается вещество
Л~ сое — сцНэ
Моющие средства, которые получают по такой схеме,
отличаются очень хорошей смачивающей способностью, не
боятся жесткой воды, но их моющее действие не очень
велико. Однако его можно усилить, если блокировать
карбоксильную группу такими соединениями, как, например, амины;
вещества, образующиеся в этом случае, лучше растворяются
в воде.
Можно получить еще лучшие результаты, если избрать
совершенно иной путь и вовсе отказаться от жирных кислот
как исходных соединений. Для этого обычно исходят из
высокомолекулярных жирных спиртов. Под действием
концентрированной серной кислоты при низких температурах
гидроксильная группа в них этерифицируется по уравнению
Л-ОН Чг Hz Щ >A-0-So5H+HlO
Затем реакцией обмена с едким натром получают
соответствующую натриевую соль R—О—SC^Na, которая
отличается прекрасной моющей способностью и нечувствительна
к кальциевым солям и кислотам. Водный раствор этого
вещества имеет нейтральную реакцию, а это значит, что в нем не
происходит гидролиза, как это наблюдалось в растворе мыла.
Подобные сульфонаты жирных спиртов — правильнее,
сульфаты спиртов жирного ряда — играют сейчас очень большую
роль. Так, моющее средство «Фева», изготовляющееся на этой
основе, обладает в растворе нейтральной реакцией и поэтому
безвредно для тканей из шерсти, дедерона или других
волокон, чувствительных к щелочи.
Спирты жирного ряда сейчас в большинстве случаев
получают из парафинов, которые образуются как побочные
продукты реакции при сухой перегонке бурого угля и поэто-
му имеются в больших количествах. Сначала парафины
окисляют воздухом при температуре от 80 до 170° в присутствии
катализатора и получают кислоты жирного ряда, содержащие
в молекуле от 12 до 18 углеродных атомов. Затем эти кислоты
гидрируют также в присутствии катализаторов при высоком
давлении и получают соответствующие спирты жирного ряда.
Если сульфогруппа SO3H присоединена непосредственно
к углероду, а не через атом кислорода, как это было в
предыдущем случае, то получают сульфокислоты жирного ряда или
их соли с общей формулой R—SC^Na. Соль можно получить
и непосредственно из углеводорода, вводя в него группу
SO3H, а затем воздействуя едким натром. Этим способом
получают очень хорошие моющие средства, обладающие такими
же высокими свойствами, как «Фева».
Сульфогруппа может находиться в конце молекулы
углеводорода или у одного из средних углеродных атомов.
о?,—сн—л
\ -
SOzM
Это немаловажно. Гидрофильные группы, находящиеся
в середине молекулы, улучшают смачивающую способность,
а стоящие на концах — моющее действие. Поэтому при
получении моющего вещества можно придать ему то или иное
необходимое качество.
Преимущества, которыми обладают современные
моющие средства, лучше всего известны домашним хозяйкам. Но
еще более они важны в промышленности. С помощью ста
килограммов природной кислоты жирного ряда, переработанной
в мыло, можно выстирать 2800 кг овечьей шерсти. Если из
такого же количества кислоты жирного ряда изготовить
моющие средства, которым не страшна жесткая вода, можно
выстирать овечьей шерсти в 10 раз больше.
Такое совершенствование современных моющих средств
стало возможно только после того, как была достигнута
полная ясность в теории моющих процессов и когда научились
получать вещества в соответствии с заданными требованиями.
164
Каучук натуральный и
синтетический
Римская мифология рассказывает о двуликом Янусе —
боге домашнего очага, который мог смотреть одновременно
вперед и назад, потому что у него была голова с двумя
лицами. О Янусе невольно вспоминаешь, знакомясь с историей
каучука — одного из важнейших промышленных продуктов
нашего времени. У этой истории также две стороны, два лица.
В корне отличные друг от друга, они характерны для истории
многих природных продуктов.
С одной стороны, мы видим неустанный труд химиков
и физиков, техников и инженеров, их стремление отвоевать
у природы секрет каучука и заменить его новым
синтетическим веществом. Ученые добились успеха, и сегодня более
одной трети резины, производимой в мире, изготовляется из
синтетического каучука. Каучук и резина внесли огромный
вклад в технический прогресс последнего столетия. Вспомним
хотя бы о шинах и разнообразных изоляционных материалах,
и нам станет ясна роль каучука в важнейших отраслях
хозяйства. Каучук делает нашу жизнь удобнее.
Но вряд ли найдется другое природное сырье, добыча
которого так была бы связана с кровью, произволом и
безграничной колониальной эксплуатацией. Сотни тысяч негров
и индейцев погибли от болезней и непосильного труда на
плантациях белых колонизаторов. Их насмерть забивали
бесчеловечные надсмотрщики — Европа и Америка все
настоятельнее требовали каучука, и бесправные рабы-туземцы
вынуждены были добывать его.
Когда испанские конкистадоры в XVI веке высадились в
Южной Америке, их внимание привлекли мячи, которыми
индейцы пользовались в спортивных играх. Эти мячи были
сделаны из неизвестного в Испании упругого и пластичного
вещества, получаемого индейцами из сока каких-то деревьев.
Индейцы находили ему и другое применение. Изготовляли из
него водонепроницаемую обувь или обрабатывали им ткань,
чтобы сделать ее непромокаемой.
Слухи о странном веществе достигли Испании. Вначале
это показалось интересным, но вскоре о диковинных ИГруШ-
^У 3. Шпаусцус
ках просто забыли. И суда отправляли в опасные
путешествия не за ними, а за золотом. Когда гораздо позже
французский ученый Шарль де ля Кондамин напомнил об этом
веществе, его сообщение восприняли как занятный курьез.
Однако этим деревом, которое росло в огромных
девственных лесах Амазонки и получило у биологов название
Hevea brasiliensis, в последующие годы продолжали
интересоваться и наблюдали, как местные жители добывали его
сок — каучу. При этом ножом делали косой надрез на коре,
укрепляли под ним маленький сосуд, и в него капля за
каплей вытекал сок. Примерно через час, когда надрез
затягивался тонкой пленкой, снова делали надрезы, на этот раз более
глубокие. Из них стекал уже млечный сок — латекс. Этот
процесс повторялся несколько раз, пока дерево совсем не
переставало давать сок.
Напоминающая молоко белая жидкость, полученная
таким образом, еще не была каучуком. Она состояла на 65—
75% из воды, в которой в коллоидном состоянии были
диспергированы 25—35% каучуковых частиц. Кроме того, в
растворе содержалось 1,5—2% белка, 1,5—2% смолы и 0,5—1%
минеральных солей. Чтобы получить каучук, местные
жители поступали так. Они укрепляли на двух вилках из сучьев
обтесанный ствол дерева и разводили под ним костер,
который не давал большого пламени, но зато сильно дымил.
Бревно все время поворачивали и одновременно капали на него
латекс. Вода испарялась, а каучук оставался в твердом виде.
Правда, дым окрашивал такой каучук в коричневый цвет,
отчего он становился похожим на копченое сало. Каучук,
добытый таким образом, не был чистым, так как содержал
минеральные соли, а также пыль и золу.
Ввозимый в Европу и Америку в небольших количествах
каучук употребляли для пропитывания тканей. Научились
изготавливать непромокаемую одежду, но каучук имел
неприятное свойство — на холоде он становился жестким и
ломким, а в тепле — вязким и пачкающим. Конечно,
покупатели по достоинству оценили эти малоприятные качества.
Кому же может понравиться, если, вставая со стула, он
обнаружит, что пальто прилипло. Поэтому дело, так много
сулившее вначале, отнюдь не процветало. И хотя скоро узнали еще
об одном свойстве каучука — способности стирать с бумаги
карандашные надписи, производство каучука в Бразилии
существенно не увеличилось.
Но вскоре все изменилось. В 1839 году два американца
Гудьир и Хейворд после многолетних и упорных опытов
обнаружили, что при обработке сырого каучука серой
происходит его вулканизация. После такой обработки каучук
теряет вязкость, становится гораздо эластичнее и сохраняет
эту эластичность в широком температурном интервале.
Теперь открылись новые области применения каучука. Резину,
полученную из него, начали применять в качестве
амортизаторов на автомашинах и мотоциклах. Позднее такие
амортизаторы превратились в современные шины и камеры.
Бурное развитие электротехники сделало резину
необходимым изоляционным материалом для электрических
проводов и кабелей. Каучук очень подходил для этой цели, так
как не проводил тока, а его эластичность делала провода с
изоляцией гибкими. Уже в 1840 году было переработано
400 т каучука, и в последующие десятилетия потребление
его непрерывно возрастало. В 1890 году оно составило уже
свыше 29 000 г.
Все надежды тогда возлагали на росшую в необозримых
лесах Бразилии гевею. Жители многочисленных поселений,
разбросанных по побережью океана, вдоль рек и в глубине
страны, собирали сок, чтобы добывать из него желанный
каучук. Уже давно перестали получать каучук испарением воды
из латекса. Было установлено, что при подкислении каучук
свертывается, как молоко, и осаждается.
В огромных сосудах сок гевеи разбавляли так, чтобы
концентрация твердых частиц составляла от 15 до 20%, и
затем осторожно добавляли уксусную или муравьиную
кислоту. Каучук отделялся в виде губчатой массы. Сгустки
каучука вынимали и пропускали через рифленые вальцы, после
чего он превращался в тонкую пленку. В течение всего
процесса каучук постоянно промывали водой, освобождая его
таким образом от избыточной кислоты, минеральных солей и
других примесей. После сушки каучук прессовали в кипы,
которые из бразильских портов отправлялись в Европу для
переработки в резину.
Существовал и другой способ добычи каучука. В латекс
быстро добавляли избыток кислоты, и каучук тотчас же осаж-
I
дался. После вальцовки под струей воды следовала сушка и
консервирование его в специальной коптильной камере.
Необходимо было добывать очень много латекса, и
тысячи «mangeveroe» — искателей каучука с привязанными за
спиной сосудами потянулись в леса в поисках драгоценного
сока. Неосмотрительное выкачивание сока из деревьев
приводило к тому, что они переставали давать сок, а потребность
в каучуке росла из года в год.
Поиски гевеи уводили людей все глубже в тропический
лес. Все труднее, все опаснее становились поиски каучука,
белые не могли переносить эту работу. Наконец, и индейцы,
которые сначала за ничтожную плату добровольно
участвовали в поисках, отказались продолжать их. Стали применять
силу. Индейцев гнали в чащу леса, и горе было тому, кто не
набирал определенного количества сока. Бесчеловечные побои
были еще не самым страшным наказанием, которое
применяли белые надсмотрщики. В бассейне реки Путамайо, где
были найдены большие заросли гевеи, насчитывалось около
50 тысяч индейцев. В течение нескольких лет хорошо
организованной эксплуатации иностранным компаниям удалось
отгрузить отсюда свыше 4000 т каучукового сырья, зато
число живущих там индейцев уменьшилось до 8000. Оставшиеся
в живых, избитые и измученные индейцы скрылись в лесах.
Это не был единичный случай. Тысячи туземцев оплатили
своей жизнью и здоровьем огромные прибыли компаний. На
каждой кипе каучука, которая отправлялась из Южной
Америки в далекую Европу или Соединенные Штаты, была кровь
замученного индейца.
В глубине Африки, в бассейне реки Конго, также были
открыты обширные леса каучуконосных деревьев. Король
Бельгии Леопольд II основал «Свободное государство Конго».
В этой колонии нещадно разорялись огромные богатства.
Здесь происходило то же, что и в Бразилии. Жестокие белые
надсмотрщики, применяя варварские методы, принуждали
местных жителей добывать каучук. В то время как в Европе
говорили о культуре, свободе и гуманизме, со всех амвонов
проповедовали любовь и смирение и посылали миссионеров к
бедным «дикарям», в Конго за несколько лет более 30 000
негров поплатились жизнью за каучук.
Из-за хищнических приемов добычи дикорастущих
каучуконосных деревьев становилось все меньше. В Бразилии
стали закладывать плантации каучуконосов. Под страхом
смертной казни было запрещено перевозить через границу
семена или молодые растения, так как Бразилия желала
сохранить монополию в этой области и устанавливать цены на
каучук по своему усмотрению.
Англичане давно стремились принять участие в
каучуковом бизнесе. Для этого нужно было разводить гевею в
собственных колониях.
В 1870 году англичанину Генри Викхему после
тщательной подготовки удалось контрабандой вывезти из Бразилии
70 000 семян гевеи и доставить их в Англию. Семена тотчас
же высадили в Лондоне — в крупнейшем ботаническом саду
я
Англии Нью-Гардене. Но из семян выросло всего 3000 MOJiSfe^
дых растений, и лишь 1100 деревцев гевеи сумели высадить
на уже подготовленных плантациях на Цейлоне. Это была
основа для создания больших плантаций в захваченных
англичанами государствах Малайи. Вскоре монополия Бразилии
была сломлена, и Англия стала главным производителем
каучука. Из 869 000 т каучукового сырья, которые добывались
в 1936 году, только 2% шли из Бразилии.
.К началу XX века было уже достаточно плантаций,
чтобы добыть любое количество каучука, Однако в некоторых
странах каучук продолжал доставлять много забот.
Деревья, дающие латекс, росли только в экваториальном поясе,
и некоторые индустриальные государства в любой момент
могли лишиться его привоза. С другой стороны, они были
вынуждены платить за каучук любую запрашиваемую. цену.
Особенно тревожным это положение оказалось для Германии.
Тогда вспомнили о химии. Может быть, она сумеет
сломить монополию колониальных стран. Началась новая эпоха
в истории каучука.
Чтобы изготовить машину, нужно знать количество,
форму и назначение отдельных деталей, а затем по
определенному плану собрать их в общий агрегат. То же самое
необходимо и при синтезе химических соединений. Прежде чем
думать об искусственном изготовлении каучука, нужно было
установить его состав и структуру. Анализ был первой
ступенью к синтезу.
Когда химики во второй половине XIX века начали
заниматься природным каучуком, они и не подозревали, какая
это тяжелая работа. Каучук оказался веществом со слабой
реакционной способностью. Тогда часто применяли сухую
перегонку. Поэтому методу исследовали многие вещества,
например древесину. При нагревании вещество начинало
разлагаться, а продукты разложения можно было собрать и
исследовать. Нагревая каучук, англичанин Вильяме в 1860
году получил масло, которое назвал изопреном. Он сумел
определить его состав: CsHe. Только 22 года спустя Тильдену
удалось установить структурную формулу изопрена
сН£=с—сц
6 3. Шпаусцус
Изопрен оказался ненасыщенным соединением с двумя
двойными связями в молекуле; это жидкость с низкой
температурой кипения 34,3°.
В 1879 году французскому химику Бушарду действием
соляной кислоты на изопрен, полученный им при сухой
перегонке каучука, удалось получить массу, которая вела
себя, как первоначальный каучук, т. е. ...«обладала
эластичностью и другими качествами природного каучука. Она не
растворялась в спирте, набухала в эфире и сероуглероде и
растворялась в них так же, как природный каучук», — с
радостью сообщал он. Если продукт, полученный таким образом,
fyjcnpe*
?Hi
Н,с=
снова подвергали сухой перегонке, то вновь получали
изопрен. Сделали вывод, что каучук и изопрен тесно связаны
между собой и что молекула каучука, возможно, образована
из нескольких молекул изопрена. При нагревании без доступа
воздуха молекула каучука снова расщеплялась на составные
части и отгонялся изопрен.
Этот процесс соединения многих одинаковых молекул в
одну большую был известен и раньше на других веществах и
его называли полимеризацией. Значит, каучук можно
рассматривать как продукт полимеризации мономерного,
простого изопрена.
Что же представляет собой процесс полимеризации?
Молекула изопрена, конечно, насыщена, не располагает
свободными связями, и, следовательно, ее нельзя рассматривать
как свободный радикал, который в процессе полимеризации
вступает в химическую реакцию с другими соединениями. Но
молекула изопрена обладает двумя двойными связями. А нам
известно, что они неустойчивы, лабильны и в реакции в
первую очередь подвергаются атаке. Двойные связи у изопрена
расположены между первым и вторым и межДу^ третьим и
четвертым углеродными атомами, т. е. занимают полонЧение-1,3.
—сн
з
СН;
4
ас
сн,
i:
-Си
Z
СН
з
снп
Двойные связи, которые, как у изопрена, разделены
одной простой связью, называют сопряженными двойными
связями^ Благодаря этим связям у первого и четвертого
углеродных атомов может возникнуть по одной свободной валентно-
170
сти. Молекула таким образом переходит в более активное
состояние и может теперь вступать в реакцию с другой такой
же активной молекулой. При таком взаимодействии
происходит насыщение свободных двойных связей. Большое
количество молекул изопрена соединяется в одну длинную цепь —
в молекулу каучука, точный молекулярный вес которой
неизвестен, но достаточно велик.
сн3
I 3
си* |
I СНз
*ис^С = СН СНг
СН,
I
Hzc^C—ch^=ch9 ^н-с—с^си—сн
\
\
\
св5
Н±с—с -сН^СНг
сн
\ I
^Н^С—С^СН-СНъ
I
Формулу каучука можно записать так:
[-снг— с—с н —сн—.D»
где п — степень полимеризации, число молекул изопрена в
молекуле каучука. Долгое время считали, что п не
превышает 5, но более поздние исследования показали, что степень
полимеризации равна примерно 3000. Так как молекулярный
вес изопрена в соответствии с формулой CsHg составляет 68,
jro для этой молекулы-великана он равен примерно 200000.
Нитеобразные макромолекулы каучука не располагаются
в строгом порядке, а образуют спутанный клубок. Этим
объясняется прочность полимерного продукта в отличие от
жидкого изопрена, маленькие молекулы которого находятся друг
около друга и их легко разделить.
Дальнейшее изучение каучука привело к выводу, что
отдельные цепи молекул в нормальном состоянии
располагаются в виде клубков. Благодаря непрерывному движению
конфигурация цепей постоянно изменяется, все же в конце
6*
171
концов образуется, как говорят специалисты, «свободный
клубок».
При воздействии внешней силы молекула
растягивается, причем часто ее длина становится во много раз больше
первоначальной. Такое растянутое напряженное состояние
нельзя считать естественным, оно существует лишь
постольку, поскольку действуют внешние силы. Если действие
внешней силы прекращается, то молекула принимает
нормальную форму и снова свертывается в клубок. Вспомните о
щетке из проволоки для чистки кастрюль. Здесь мы тоже видим
сплетение стальной проволоки в форме свободного клубка.
Если потянуть за конец, т. е. приложить силу, все
сооружение вытягивается, а как только действие этой силы
прекращается, тотчас же проволока переходит в первоначальное
состояние. При таком воздействии не вытягивается отдельная
проволочка, отдельная «ниточка молекулы», а благодаря
взаимодействию многих нитей возникает эластичная деформация
всего клубка. Так объясняется эластичность щеток из
стальной проволоки или некоторых пластмасс, а также
эластичность каучука.
Итак, связь между изопреном и каучуком была
установлена. Далее выяснили, что изопрен может полимеризоваться
в каучук. Потом узнали, что натрий является веществом,
которое значительно ускоряет полимеризацию. Но все эти
сообщения не слишком огорчали владельцев каучуковых
плантаций по ту сторону экватора. Ведь пока не приходилось и
думать о химическом синтезе каучука в промышленном
масштабе. Однажды, правда, был получен продукт, но он по
качеству не выдерживал никакого сравнения с натуральным
каучуком. И наконец, изопрен умели получать только из
натурального каучука путем его сухой перегонки. Нужно
была быть безумцем, чтобы добывать из природного
каучука изопрен, а из него полимеризацией снова получать
каучук.
Для химиков достигнутые в этом направлении успехи
были только началом. Чтобы серьезно приступить к синтезу
каучука, нужно было сначала из легкодоступных исходных
дешевых материалов получить изопрен или другие
мономерные соединения, которые можно было бы полимеризовать в
продукты, аналогичные каучуку.
В начале XX века с изопреном экспериментировал
молодой химик Фриц Гофман. Эта своеобразная жидкость
полностью овладела его воображением. Он знал ее состав, ее
свойства и реакции, умел, наконец, полимеризовать ее в каучук.
Но еще больше его интересовал вопрос о синтетическом
получении изопрена.
Гофман попробовал извлечь его из каменноугольной
смолы, которая уже дала химикам так много. Ему это удалось.
Правда, метод был дорог и слишком сложен, чтобы его
можно было превратить в промышленный и нанести какой-либо
ущерб йроизводству натурального каучука. Снова и снова
исследовал Гофман изопрен, получал разнообразные его
производные и подвергал их полимеризации. В этих работах он
натолкнулся на 2,3-диметилбутадиен, молекула которого
содержит в отличие от изопрена еще одну метильную группу.
снз <Ч
сНг=с с - CHZ
Действуя на 2,3-диметилбутадиен едким кали, при
нагревании он получил каучукоподобную массу. Но гораздо
важнее был тот факт, что сам диметилбутадиен сравнительно
просто можно было получить из ацетона. Ацетон в свою
очередь можно было получить синтетически, и стоил он не
слишком дорого. В присутствии алюминия и ртути ацетон
восстанавливался водородом и через промежуточную стадию
мог быть превращен в желаемый диметилбутадиен.
Но и каучук из диметилбутадиена не мог конкурировать
с натуральным. Стоил он дороже, а по качеству был хуже.
Он имел лишь одно преимущество — его можно было
получать вдали от тропиков и плантаций.
Когда в 1914 году началась война и Германия не смогла
больше импортировать каучук, вспомнили об этом метилкау-
чуке. Тогда-то его и стали изготовлять в значительном
количестве. Конечно, качество этого каучука было очень низким,
и его ни в какой мере нельзя было рассматривать как
заменитель природного каучука. Шины, изготовленные из него,
редко выдерживали более 3000 километров, а плащи вообще
нельзя было делать. Но путем вулканизации большим
количеством серы из этого каучука удавалось получать более или
менее пригодный эбонит.
После войны, когда снова стал доступен природный
каучук, производство метилкаучука было прекращено как
нерентабельное. Однако работы по синтезу искусственного каучука
не были прерваны. Эти работы стимулировались
предыдущими обнадеживающими результатами, монополией на
природный каучук, а также сильно колеблющимися ценами на него.
Вскоре были сделаны новые важные открытия. До сих
пор пытались полимеризовать исходные мономерные
соединения сплошной массой, при этом для реакции
требовалось довольно длительное время, теперь вспомнили, что в
природе твердый каучук встречается в виде маленьких
шариков, диспергированных в жидкости. Диметилбутадиен
размельчили в воде и добились тем самым значительного
ускорения процесса полимеризации. Там, где прежде требовались
дни, теперь достаточно было нескольких часов.
В то же время интенсивно изучали структуру каучука,
процессы, происходящие при полимеризации и
вулканизации. Однако решающие успехи были достигнуты, когда
отказались от изопрена как исходного вещества и стали
осуществлять полимеризацию других мономеров.
В Советском Союзе и США исходным мономером являлся
хлоропрен СН2 = СНС1—СН = СНг. Хлоропрен — это изопрен,
у которого вместо метильной группы стоит один атом хлора.
Хлоропрен можно получать сравнительно дешево из
ацетилена и быстро полимеризовать в каучук, который по многим
качествам превосходит натуральный. В 1931 году этот
синтетический каучук появился на американском рынке под
названием неопрен и в Советском Союзе — под названием совпрен,
В Германии дальнейшие работы тоже шли успешно.
Исходным веществом в немецких работах был бутадиен
СНг = СН—СН = СНг. Это соединение — близкий родственник
изопрена. Вскоре бутадиен стал в Германии важнейшим
исходным веществом для синтеза каучука.
Бутадиен чаще всего получают из ацетилена СН=СН,
который уже известен рам как первый и важнейший член
гомологического ряда алкинов. Мы уже знаем, что ацетилен
образуется из карбида кальция и воды. Итак, необходимо
было большое количество карбида кальция. Каучуковый завод
в Шкопау стал поэтому крупнейшим в мире производителем
карбида.
Ацетилен превращают в ацетальдегид присоединением
одной молекулы воды ,3.
сЦ==сН + НгР : >СН—С^
Эту реакцию очень просто представить на бумаге, но
технически осуществить ее было очень сложно. Прежде всего
нужно было найти подходящий катализатор. Таким
катализатором стала контактная кислота, представляющая собой
разбавленную серную кислоту, в которой растворены
сульфат ртути и соли железа. Недостаток этого катализатора
состоит в том, что во время процесса выделяется
металлическая ртуть, которая особенно опасна из-за своей летучести и
ядовитости. Необходимо было принимать специальные меры
для удаления ртути из продуктов реакции и реакционных
сосудов. Оказалось более выгодным и удобным сделать всю
аппаратуру открытой и, чтобы не отравлять парами
окружающую атмосферу, снабдить установку только защитной
крышей.
В трубы генератора при 980° снизу накачивают
ацетилен и контактную кислоту. Сверху собирают получающийся
ацетальдегид, а также непрореагировавший ацетилен,
который снова направляют в генератор.
Если к ацетальдегиду прибавить очень слабый раствор
щелочи, то происходит своеобразная реакция. Две молекулы
ацетальдегида реагируют друг с другом таким образом, что
один водородный атом молекулы II присоединяется к
кислороду карбоксильной группы молекулы I, образуется углерод-
углеродная связь и получается альдегидоспирт, альдоль.
0^ * & И f H О
Ц гЧ / II / I / II
с :н\-с—6 > н—с—с—с—с
I '■ II I I / I
Мы можем представить себе ацетальдегид как
производное метана, у которого один водородный атом замещен на
альдегидную группу:
^0
н
В то время как у метана реакционная способность
водородных атомов относительно слаба, в ацетальдегиде они
вступают в реакцию гораздо легче. Альдегидная группа
активирует водородные атомы у соседнего с ней углеродного атома,
делает их более реакционноспособными. Приведенный
только что пример — не единственный случай. Выяснилось, что
у более высокомолекулярных альдегидов только СНг-группа,
находящаяся по соседству с карбонильной группой, может
отдавать свой водород второй альдегидной молекуле.
Полученный таким образом неустойчивый альдоль поступает на
следующую ступень. Его сжимают примерно до 300 ат и при
температуре от 100 до 120° в присутствии катализатора,
состоящего из тонкоизмельченной меди, никеля и хрома,
нанесенных на силикагель, проводят реакцию восстановления
тщательно очищенным водородом. Водород присоединяется
к альдолю — говорят, что происходит гидрирование альдо-
ля, — и образуется бутиленгликоль-1,3. На четвертой ступени
после отщепления воды получают бутадиен. И здесь не
обходится без катализатора. Применяют фосфат натрия на коксе
или графите. После прохождения реагирующих веществ
через главную контактную печь их очищают и получают
бутадиен 99%-ной чистоты.
Ниже приведены еще раз отдельные стадии получения
бутадиена из ацетилена.
1) Образование ацетальдегида
% сн== сн +z uzo —> ш5—с^
1илен W
Лцетс
2) Образование альдоля
4 он
3) Образование бутиленгликоля-1,3
/^ !Ka*nciUHjamop
-н-
СНЪ- ы~с"г\ + Ъ -—> сн3—сн
юн он
4) Получение бутадиена
cH5-ch—chz-chz ^Wz-=eH-cH=cHz-hZHzo
он он
Следует упомянуть, что это не единственно возможный
способ получения бутадиена. Наиболее современен метод, при
котором исходными соединениями являются ацетилен и
формальдегид. Сначала получают бутин-2-диол-1,4, который
далее после отщепления воды превращается в бутадиен. Этот
процесс отражает следующая схема:
НОСНъ—CHZ—CHl-CHjLoH--+CMz ?Hi+*k°
СН.-СНг
хсц chz »•<?«—сн-сн«снг-|-й*о
о
В СССР уже в тридцатые годы исходным веществом для
получения бутадиена служил спирт, а позднее с этой целью
применяли смесь этилового спирта и ацетальдегида 14.
Бутадиен получают также каталитическим дегидрированием газов
крекинга.
В Германии в 1935 году началось производство
синтетического каучука в больших количествах. Во вращающиеся
горизонтально расположенные автоклавы накачивают бутадиен
и при охлаждении прибавляют регулятор полимеризации —
диоксан и ускоритель — металлический натрий. От слов
бутадиен и натрий образовано название буна. Так назвали
продукт полимеризации бутадиена, который, выгрузив из
автоклавов, промывают, вальцуют, высушивают и скатывают
в большие кипы, после чего он пригоден для дальнейшей
обработки. В зависимости от степени полимеризации получают
буна-85 или буна-115. Цифры означают приблизительный
176
молекулярный вес полимера в тысячах, т. е. 85 000 или
115000. Если этот буна-каучук с высоким молекулярным
весом подвергнуть вулканизации, получается резина, которая
имеет высокую прочность на истирание, теплостойка и не
стареет, однако обладает низкой эластичностью и невысокой
прочностью на разрыв и растяжение. Поэтому такой каучук
только условно можно было рассматривать как заменитель
натурального. И лишь твердая резина, изготовленная из
буна-85, в некоторой степени удовлетворяла необходимым
требованиям.
Каучук лучшего качества удалось получить с
применением смешанной полимеризации, которая теперь главным
образом и осуществляется в промышленных масштабах. Как
правило, полимер состоит из одинаковых структурных
элементов. Если, например, отдельные составные части
обозначить через А, то весь процесс полимеризации схематично
можно представить так: А + А + А + А + ...-*А'—А'_а'_
-А'-А'-...
При смешанной полимеризации в большинстве случаев в
реакцию вступают молекулы двух соединений, которые
связываются друг с другом примерно по такой схеме: А + В +
+ А + В...->А'—В'—А7—В'... Так, например, полимеризуют
не один бутадиен, а добавляют к нему стирол CeHs—CH =
= СНг или нитрил акриловой кислоты СНг = СН—CN. В обоих
соединениях имеется по одной двойной связи, благодаря
чему они могут участвовать в полимеризации. Так как их
молекулы нитевидны, реже образуются поперечные связи,
снижающие эластичность каучука. Было установлено, что в
натуральном каучуке отдельные молекулы изопрена соединены
в длинные, не связанные между собой цепи.
У высокомолекулярного буна-каучука при
полимеризации хотя и возникают длинные нитеобразные молекулы, но
они во многих местах соединены с соседними молекулами.
Это происходит потому, что после расщепления обеих
двойных связей в молекуле бутадиена образуется новая
непрочная связь, которая, даже в мягких условиях полимеризации,
может быть нарушена. При этом возникают свободные
валентности, по которым нитевидные молекулы связываются
друг с другом. Поэтому буна-85 или -115 плотнее и прочнее
на истирание, чем натуральный каучук, но зато менее
эластичен и труднее поддается обработке.
,JCeS<fU/6af^ajupo^aHH6tu ьрцреднЬи, Щ/Щ^
э*^Г
Введение в цепь полимера молекул стирола или нитрила
акриловой кислоты уменьшает количество перекрестных
связей, и полученные таким образом сорта искусственного
каучука по некоторым важным качествам приближаются к
натуральному. Вместо молекулы бутадиена в продукте
полимеризации находится стирол, у которого после внедрения в
макромолекулу полимера обнаруживаются только простые
связи, как видно из следующей схемы.
-сн2-сн=сн-сн;>,
Sbyfnayu&t
сн^-сн
Стирои
С, И
б^х
е%у/каус/е/€. Смцрои.
Таким образом, в цепи уменьшается количество молекул
бутадиена, способных к образованию перекрестной связи, а
следовательно, и тенденция к переплетению нитевидных
молекул. То же самое происходит при внедрении в молекулу
полимера нитрила акриловой кислоты.
—ИЗ —Шк~
CUxxdo в^ибанияиро^анНЫи синтенчигесКоси Kayty/d
Продукт полимеризации бутадиена и стирола называется
буна-S. Содержание стирола может составлять от 10 до 45%.
Чем процент стирола выше, тем эластичнее каучук.
Самый важный синтетический каучук — буна-ЭЗ. Во
всем мире он находит применение для изготовления шин,
ленточных транспортеров, прокладок и т. д. По прочности на
178
истирание, по устойчивости к старению и термостойкости
искусственный каучук во многих случаях превосходит
натуральный, но он менее эластичен и хуже обрабатывается.
Дальнейшее развитие промышленности синтетического
каучука (СК) привело к изготовлению буна-Э4 и буна-в4Т,
которые легко поддаются обработке. Эти каучуки часто не
нуждаются в предварительной термической пластификации,
поэтому в будущем буна-ЭЗ будет, по-видимому, вытеснен
этими сортами синтетического каучука. Особенно это
относится к буна-Э4Т, температура полимеризации которого
всего 5° вместо обычных 50°.
Полимеризационная цепь пербунана — продукта
полимеризации бутадиена и нитрила акриловой кислоты — похожа
на цепи каучуков, описанных выше.
^сн2-сн=сн-сн, снг-сн —си
СНо СН
* I
Бутадиен Акрилонитрил Бутадиен
Прибавив к бутадиену 27% нитрила акриловой кислоты,
получают пербунан или буна-N. Пербунан экстра или бу-
на-NN содержит 35% нитрила акриловой кислоты. Оба сорта
легко обрабатываются и обладают высокой устойчивостью по
отношению к маслам и бензину.
Последним достижением является буна-NW, главное
преимущество которого в том, что он обрабатывается проще
всех иных сортов, поэтому потребление его все время
возрастает.
Производство синтетического каучука продолжает
развиваться, и, может быть, нас ждут в этой области большие
сюрпризы.
Давно уже не стремятся получить продукт, подобный
природному каучуку. В наши дни из новых исходных веществ
изготовляют совершенно новые материалы со свойствами,
которых нет у натурального каучука. Так, при полимеризации
силиконов, органических соединений кремния, с
соответствующими наполнителями получают силиконовый каучук,
который, как все силиконы, отличается особенно высокой
жаростойкостью. Он переносит довольно высокую для резины
температуру 200° и в то же время очень устойчив к низким
температурам.
Не меньший интерес представляют собой
полисульфидные каучуки, относящиеся к серусодержащим пластическим
материалам. Их будущее значение часто оспаривается, и мы
не будем останавливаться на них подробнее.
Сырой каучук, будь он естественного происхождения или
получен на химическом заводе, не может прямо
перерабатываться в готовые изделия. Он должен сначала пройти соот-
снг си
Акрилонитрил
ветствующую подготовку и обработку, которые в зависимости
от его происхождения и последующего употребления могут
быть различны. При этом часто его первоначальные свойства
значительно изменяются.
Большие каучуковые кипы, прибывающие с заморских
плантаций или с химических заводов, сначала измельчают с
помощью больших гидравлических ножей, которые
разрезают кипы на мелкие куски. После измельчения каучук-сырец
путем мастификации переводят в пластическое состояние
(мастификация — нечто вроде разжевывания). Конечно, это
«разжевывание» проводят с помощью машин. Кусочки
каучука помещают между двумя катками, которые вращаются с
различной скоростью в противоположных направлениях,
причем расстояние между ними все время изменяется. Кроме
того, масса интенсивно размешивается и прокатывается.
Расстояние между нитевидными молекулами увеличивается,
и они начинают свободнее двигаться относительно друг
друга, каучук переходит в пластическое, удобное для
деформации состояние, которое облегчает его дальнейшую обработку.
Конечно, этот процесс проводят лишь до определенного
момента, после которого масса может стать слишком рыхлой
и липкой.
Мастификация природного каучука осуществляется
просто, но различные сорта синтетического каучука необходимо
дополнительно обрабатывать. Мы уже знаем, что цепи
молекул буна-каучука в большей или меньшей степени связаны
перекрестно, в то время как у натурального каучука нити
макромолекул между собой не связаны. В результате такой
сетчатой молекулярной структуры буна-каучук прочнее, он
плотнее, но обладает гораздо меньшей пластичностью.
При мастификации не происходит разрыва перекрестных
связей, поэтому искусственный каучук нельзя сделать
пластичным только с помощью механической обработки.
Предварительно необходимо провести, как говорят специалисты,
окислительное расщепление, при котором происходит разрыв
перекрестных связей и молекулярных цепей, отчего число
сетчатых молекул и длина молекулярных цепей
уменьшаются.
При окислительном расщеплении куски буна-каучука
помещают на подвижные решетки в куб. Под действием
кислорода воздуха при определенном давлении и температуре
происходит расщепление молекул. После этого масса может
быть мастифицирована, и ее пластичность становится такой
же, как у натурального каучука. Нужно заметить, однако, что
вскоре вновь образуется некоторое количество ранее
разорванных перекрестных связей. Буна-каучук «отдыхает»,
восстанавливается, причем пластичность его частично
утрачивается. Поэтому вся дальнейшая обработка должна следовать
непосредственно после окислительного расщепления.
Открытие Гудьира и Хейворда, которые в 1840 году
обнаружили, что каучук-сырец, смешанный при нагревании с
серой, превращается в эластичную массу, создало основу для
широкого применения каучука. Ведь только при вулканиза-
ции каучук-сырец теряет свою клейкость, прибретает
прочность и эластичность — становится резиной с ее ценными
качествами. В зависимости от содержания серы и состава
наполнителей, добавляемых при вулканизации, получают
различные сорта резины, отвечающие любым требованиям.
Какую роль в вулканизации играет сера?
Распределяется ли она в каучуке механически, образуя с ним лишь смесь,
или вступает в химическую реакцию? Сейчас нам известно,
что происходит именно последнее. Доказывают это
следующие факты. Во-первых, после вулканизации серу невозможно
вымыть каким-нибудь растворителем, обычно растворяющим
элементарную серу. А если бы сера и каучук образовывали
смесь, то сера должна была бы раствориться. Во-вторых,
оказывается, что с увеличением количества серы число двойных
связей в продукте полимеризации пропорционально
уменьшается. Это позволяет сделать предположения о
возникновении связи сера — каучук. Поскольку при вулканизации под
действием серы часть ненасыщенных связей исчезает, можно
предположить, что введенная сера присоединяется по
двойной связи и при этом две цепочные молекулы соединяются
друг с другом. У натурального каучука, который, как
известно, образуется благодаря полимеризации изопрена,
возможна такая схема:
?-
СН3
СИ,
~CHZ-C -СИ-Щь—CHi-C =СН —СН£-СН£-С~СН-СН£-
S S
/ /
-(%-с—с\\-снгсц£-с=-сн-снг-снх-с —сн-снг
м
I
СИ
I
СИ,
L
®
©
arrouU/ ctfito ~и&НеМ сера pajopfaua финке е&цо
oShaioSauot ,t ucoanutfu "
Небольшое количество серы при вулканизации
превращает пластический каучук в эластичную резину.. Уже при
введении 0,15% серы каучук меняет свои свойства. Вообще
же количество вводимой при вулканизации серы колеблется
от 2 до 5%. Повышенное содержание серы способствует
образованию сетчатых молекулярных структур, отчего
снижается эластичность, но одновременно увеличивается прочность
материала. Примерно при 30% -ном содержании серы
получится эбонит, свойства которого хорошо известны.
Мы уже знаем, что для получения высококачественной
резины, которую можно переработать в различные изделия, в
каучук необходимо добавить ряд примесей.
Большую роль среди них играют так называемые
ускорители вулканизации — органические соединения,
содержащие серу или азот. Они значительно сокращают время и
снижают температуру процесса, а иногда позволяют проводить
его вообще без нагревания (холодная вулканизация).
Благодаря этим добавкам можно уменьшить количество
вводимой серы.
Очень важны также противостарители, которые
уменьшают влияние кислорода воздуха на резину. С течением
времени кислород присоединяется к оставшимся в молекулах
резины двойным связям и усиливает тем самым образование
сетчатых молекул, при этом резина теряет свои характерные
качества и становится твердой и ломкой. Противостарители —
это антиокислители.
Еще на заре применения каучука-сырца, когда он был
довольно дорог, предприимчивые фабриканты нашли
дешевый способ увеличивать его количество. В каучук-сырец
стали добавлять наполнители — сажу, мел, окись цинка и т. п.
Каково же было всеобщее удивление, когда оказалось, что
обработанный таким образом каучук не только увеличивался в
весе, но и в ряде случаев приобретал лучшие свойства —
увеличивались сопротивление разрыву и растяжению, твердость.
Вскоре стали различать две группы наполнителей:
1) активные наполнители, которые улучшают качество
каучука. К ним среди прочих относятся активная газовая
сажа, окись цинка и каолин;
2) инертные наполнители, которые лишь увеличивают
вес продукта, например сажа, мел и тяжелый шпат.
Наиболее активным наполнителем оказалась поверхност-
ноактивная газовая сажа, которая может быть получена
сжиганием газа при недостатке кислорода. Сегодня нет ли
одного сорта резины, который не содержал бы различные
примесей и наполнителей. Правильный выбор и
соответствующее соотношение количества этих примесей определяют
качество резины. Б этой области, несомненно, предстоят еще
интересные и важные открытия.
Примеси и наполнители смогут составлять значительную
часть общего веса, а нередко вообще превышают вес самого
каучука. Как многообразны и слонЩы могут быть примеси,
добавляемые в каучук-сырец, видно на щшмере резины для
автомобильных шин.
ByHa-S3
Пластификаторы
Канифоль
Стеариновая кислота
Церезин
П ротивоста рители
Цинковые белила
Газовая сажа
Ускоритель вулканизации
Сера
Готовый продукт
Более ста лет прошло с тех пор, как химики занялись
изучением каучука, привезенного из бразильских
девственных лесов. За это время они не только подробно исследовали
его состав и структуру, но и открыли пути, которые
позволили получать синтетический каучук на химических заводах.
Однако не потерял своего значения и натуральный каучук.
Две трети мировой продукции каучука-сырца по-прежнему
поступает с плантаций. А вот монопольное положение
натурального каучука исчезло навсегда,.
Наконец, были созданы полимеры, которые по некоторым
свойствам превосходят натуральный каучук. В наши дни
синтетический и натуральный каучук не конкурируют
между собой, а дополняют друг друга, тем самым значительно
расширяются возможности их применения. Поэтому
синтетический каучук производится и в тех государствах, которые
располагают достаточным количеством натурального каучука.
Пластмассы в наступлении
В 1920 году в мире было изготовлено около 20000 т
пластических масс. Исходными материалами для них служили
преимущественно натуральные высокополимерные
соединения типа целлюлозы или казеина. 30 лет спустя производство
пластмасс составляло уже 1,5 млн. г, а в 1961 году около^
8,6 млн. т. Для их изготовления теперь применяются в
основном простейшие органические соединения, получаемые из
нефти или угля. Сейчас эти цифры еще выше, и конца
бурному развитию промышленности пластмасс не предвидится.
В шипи дни пластмасс производится больше, чем всех
цветных и благородных металлов. Это соотношение из года в год
увеличивается ft пользу тигасТм^сг Можно смело утверждать,
что мы идем навстречу веку пластмасс. Химики открывают
все новые и новые пластмассы с исключительно
разнообразными свойствами, которым Доводят применение в самых раз-
100,0 вес.
7,0 вес.
1,5 вес.
1,0 вес.
1,0 вес.
0,7 вес.
5,0 вес.
45,0 вес.
1,5 вес.
1,8 вес.
ч.
ч.
ч.
ч.
ч.
ч.
ч.
ч.
ч.
ч.
164,5 вес. ч.
личных областях техники и в быту. Это лишь начало пути, и,
конечно, нам еще придется столкнуться с некоторыми
сюрпризами.
Каждый из нас постоянно пользуется предметами,
сделанными из пластмасс. Они все глубже проникают в нашу
жизнь. Футляр для зубной щетки, различные виды линкрус-
тов или посуда — это лишь некоторые предметы из
пластмасс, с которыми мы ежедневно имеем дело.
Преимущества пластмасс по сравнению с обычными
материалами чрезвычайно велики. Малый вес, гладкая и легко-
моющаяся поверхность, простота обработки, абсолютная
устойчивость к коррозии, а часто и к толчкам и ударам,
разнообразная окраска объясняют их широкую популярность. Но
пластмассы обладают и существенными недостатками, в
первую очередь низкой жаропрочностью. Многие из них
размягчаются уже при 80°, а при соприкосновении с кипящей водой
теряют форму.
Конечно, пластмассы в быту необходимы далеко не
всегда. Бокал или ключ могут быть изготовлены также и из
металла, стекла или керамики. Вряд ли столовый сервиз для
праздничных случаев следует делать из пластмасс. Пол
можно покрыть деревом или ковром. Совершенно иное дело —
посуда для туриста.
А вот в технике пластмасса удобна и часто просто
незаменима. Именно в технике пластмассы в будущем найдут
основное применение. Развитие электротехники немыслимо без
пластмасс, являющихся отличным изоляционным
материалом. В области фотографии пластмассы позволили перейти от
стеклянной пластинки к катушечной пленке, благодаря чему
стала развиваться кинотехника. В машиностроении и
строительстве, на транспорте и в химической промышленности
требуется все больше пластмасс. Разнообразие свойств
синтетических материалов делает их незаменимыми во всех областях
народного хозяйства.
Во многих случаях пластмассы заменили другие
материалы. Это очень важно, так как дерево и металл далеко не
всегда есть в неограниченном количестве и их следует
использовать экономно и применять только там, где это целесообразно
и они незаменимы. Конечно, можно делать водосточные
трубы из поливинилхлорида и таким образом экономить цинк,
но газету нельзя выпустить без дерева, а кабель изготовить
без металла. Однако назвать пластмассы просто веществами-
заменителями было бы ошибкой, хотя часто недостаток
обычных материалов вынуждал применять их там, где они не
удовлетворяли или лишь частично удовлетворяли
необходимым требованиям.
Появляются новые области применения пластмасс, без
которых и в наши дни уже невозможно представить ^себсг
жизнь. Пластмассы — это следует подчеркнуть ещехи&—
обладают своеобразными свойствами. Их примеденяв^как и
любых других веществ, должно соответстравяггь этим свойствам.
Вряд ли кому-нибудь придет в гомр*$ сделать окно из жести,
несмотря на то, что она прочна а для окна прочность необ-
ходима. Для этой цели все же берут легко бьющееся стекло,
потому что в данном случае решающим фактором является
прозрачность материала. С другой стороны, нельзя
представить себе, чтобы кто-нибудь использовал стекло для
изготовления печных колосников — в данном случае' важны прежде
всего такие свойства материала, как жаростойкость и
прочность. Принцип использования пластмасс — не исключение.
£/***•*
Баллон для химикалиев из полиэтилена, который не бьется и
практически не разрушается ни кислотами, ни щелочами, ни
большинством других реактивов, является почти идеальным
примером применения пластмасс. Но из этого материала не
следует изготовлять трубы для горячей воды или пард,
поскольку полиэтилен, как и другие пластмассы, уже при
довольно низких температурах размягчается и теряет форму и
прочность.
Пластмассы получают химическим путем, это
высокомолекулярные соединения. В пластическом состоянии они
поддаются формовке. Хотя бы один раз в процессе изготовления
или обработки эти вещества бывают пластичными, такое
состояние является их характерным признаком. В этом случае
говорят о пластмассах в отличие от других
высокомолекулярных соединений, таких, как белковые вещества, целлюлоза
и т. д.
У пластмасс при всем различии их видов есть еще один
общий признак. Все они состоят из макромолекул, из
молекул-великанов, которые, как правило, содержат более
2000 атомов. Все высокомолекулярные соединения
подчиняются особым законам и правилам, которые отличают их от
низкомолекулярных соединений. Сейчас существует
специальная наука — химия высокомолекулярных соединений,
которая занимается вопросами изготовления и исследования
этих веществ.
В то время как для низкомолекулярных соединений —
вспомним мыло или этиловый спирт—можно указать их
точную формулу и свойства, для пластмасс этого сделать нельзя.
При работе с ними всегда имеются лишь приблизительные
данные. Все их свойства и реакции зависят от степени
полимеризации, т. е. от числа отдельных элементов структуры,
Ыьеыи^
185
составляющих молекулу-великан, и от того, сколько
образуется сетчатых молекул в молекулярных цепях. Но ни степень
полимеризации, ни возможности образования сетчатых
молекул нельзя знать точно. Однако это не всегда является
недостатком.
Возвратимся еще раз к спирту. При любом синтезе мы
всегда получаем одно и то же соединение С2Н5ОН. В данном
случае мы вообще не можем варьировать свойства в каких-то
границах и изменять их так, как нам хотелось бы. Совсем
иначе обстоит дело с пластмассами. Управляя процессом
полимеризации и влияя на степень образования сетчатых
молекул, можно получить различные качества у одной и той же
пластмассы так же, как это было с синтетическим каучуком.
Это позволяет синтезировать пластмассы с оптимальными
свойствами для той или иной цели. Уже эти примеры
показывают, как целесообразно выделить макромолекулярные
соединения в специальный класс.
В природе существует огромное число макромолекуляр-
ных соединений. С ними связано все живое, вспомним хотя
бы о сложных белковых соединениях или об углеводах.
Казалось само собой разумеющимся, что следует использовать эти
уже готовые полимерные вещества как исходные и с помощью
химических или физических методов превратить их в новые
вещества со свойствами, которых не было у природных
полимеров. Так и поступали до тех пор, пока не были точно
известны состав и структура этих макромолекул, а также
элементы структуры и способы построения из них больших
молекул.
Уже с 1869 года из природного сырья получали в
промышленном масштабе целлулоид, который до сих пор
производят в значительном количестве. Исходным веществом для
его изготовления служит природная целлюлоза — главная
составная часть клетчатки растений, которая является самым
распространенным на Земле органическим веществом.
Правда, целлюлоза растений в той или иной мере загрязнена
различными другими веществами. Довольно чистую целлюлозу
представляет собой только хлопок. Однако целлюлозу чаще
добывают из дерева, а примеси удаляют химическими
методами. Вата или бумага для фильтров — это также почти
совершенно чистая целлюлоза.
Натуральная целлюлоза состоит из огромных вытянутых
молекул. Ее молекулярный вес определялся различными
методами и оказался неслыханно огромным — более 200 000.
Несмотря на это, структуру молекулы целлюлозы удалось
выяснить очень точно, и строение этой молекулы сейчас
известно лучше, чем строение молекул некоторых других
веществ с гораздо меньшими молекулярными весами.
Из этого можно сделать вывод, что величина молекулы
не обязательно влечет за собой дополнительные трудности
при ее изучении. У целлюлозы и некоторых других
высокомолекулярных веществ молекула состоит из простых, часто
даже очень простых, элементарных частиц, связанных друг с
другом по одному и тому же принципу. В известной степени
это можно сравнить с пуловером, состоящим из многих тысяч
петель, которые расположены по определенной периодически
повторяющейся системе. Чтобы описать пуловер,
необязательно перечислять петлю за петлей, достаточно назвать
лишь повторяющуюся группу петель. То же самое у
целлюлозы. Известный элементхтруктуры и способ образования цепи
позволяют видеть всю цепь целиком и понять ее строение,
даже если молекула состоит из многих тысяч таких элементов.
chzoh
CHrpH
CHzoH
Целлюлоза построена из углевода, состоящего из
молекул глюкозы, т. е. молекул сахара. Эта целлюлоза (вспомним,
например, о вате) не обладает, конечно, никакими
пластическими свойствами. Благодаря своей волокнистой структуре
она может быть переработана в бумагу и нитки. Такая
целлюлоза практически нерастворима в воде и во всех других
жидкостях.
Свойства целлюлозы можно изменить только химической
обработкой. Зная ее состав, нетрудно осуществить с ней
различные химические превращения. Формула целлюлозы
показывает, что на каждые шесть углеродных атомов приходятся
три свободные гидроксильные группы, следовательно, из нее
можно получить сложные эфиры. В 1846 году профессор Шен-
байн, действуя на целлюлозу азотной и серной кислотами,
впервые получил эфир азотной кислоты—нитрат целлюлозы:
Шенбайн не без удивления обнаружил, что эта
«нитроцеллюлоза», как не совсем точно называют это вещество,
оказалась очень опасной. Она чрезвычайно легко
воспламеняется и быстро сгорает, иногда со взрывом. Поэтому
нитроцеллюлоза сразу нашла широкое применение для взрывных работ.
Ее быстрое сгорание объясняется содержанием большого
количества атомов кислорода, который освобождается при
нагревании и окисляет остальную молекулу. Сгорание
происходит не только на поверхности нитроцеллюлозы, где она
соприкасается с кислородом воздуха, но и изнутри.
187
Понятно, что нитроцеллюлоза слишком опасна для
изготовления каких-либо предметов. Если бы мы употребляли
изделия из этого вещества, то постоянно подвергали бы себя
смертельной опасности. Однако если проводить нитрование
не очень интенсивно и превратить в эфирные группы лишь
около двух третей свободных гидроксильных групп, то
получается продукт, который менее опасен и по отношению к
растворителям ведет себя совершенно иначе. Нитроцеллюлоза
растворяется в ацетоне, этилацетате и некоторых других
органических жидкостях, а продукт частичного нитрования
растворяется в спирте или в смеси спирта и эфира. Такие
растворы называют коллодием. Из него можно делать
искусственные волокна, которые играли в прошлом большую роль.
Если частично этерифицированную целлюлозу
растворить в насыщенном спиртовом растворе камфоры, тщательно
перемешать и испарить спирт, то получается бесцветный
продукт реакции, твердый при нормальной температуре. При 80°
он размягчается и в таком состоянии легко поддается
прессованию, прокатке или другой обработке. Это вещество — всем
известный целлулоид. Добавляя различные красители, ему
можно придать любую окраску. Из целлулоида делают мячи,
куклы, гребни, посуду и пленки.
Применение целлулоида ограничено тем, что он легко
воспламеняется и быстро сгорает. Это его большой
недостаток. Например, раньше немало горя причиняла целлулоидная
кинопленка. В целлулоидную массу можно добавлять
снижающие горючесть вещества, такие, как гипс, но тогда она
становится непрозрачной, и из нее нельзя делать кинопленку.
Свойства, подобные целлулоиду, имеет ацетат целлюлозы, но
он загорается гораздо труднее, и поэтому его применение
более универсально. Если зажечь ацетат целлюлозы, он сгорит
зеленым пламенем и будет чувствоваться запах уксуса, так
как уксусная кислота — необходимый компонент этого
вещества. Для его изготовления целлюлозу обрабатывают
ангидридом уксусной кислоты и серной кислотой, которая служит
катализатором. Полученный ацетат растворяют в
соответствующей жидкости. Конечный продукт — пластмасса целлон,
которая внешне почти не отличается от целлулоида.
Целлон хорошо поддается обработке, особенно
прессованием и выдавливанием. В наши дни из этой пластмассы
изготовляют много видов изделий. В автомобильной
промышленности США из ацетата целлюлозы делают более 200
деталей автомобиля. Целлон нашел также широкое применение
в самолетостроении.
Очень важным продуктом переработки целлюлозы
является целлофан, из которого делается пленка, применяемая
как прозрачный гигиенический и пыленепроницаемый
упаковочный материал. В 1958 году мировое производство
целлофана составило около 300 000 т.
Целлофан тоже получают из целлюлозы, но его
изготовление протекает по совершенно иной технологии. Целлофан
получают либо вискозным, либо медно-аммиачным методом,
которые применяют при изготовлении искусственных шелка и
шерсти.
При вискозном методе целлюлозу сначала обрабатывают
едким натром, отчего образуется так называемая натронная
целлюлоза. Ее на некоторое время оставляют для созревания,
а затем обрабатывают сероуглеродом. Таким образом
получают растворимый в воде и щелочи ксантогенат целлюлозы.
После созревания ксантогенат целлюлозы превращается в
вискозу. Эта вязкая масса поступает через фильеру в осади-
тельную ванну, заполненную серной кислотой с добавками
солей. В ванне выделяется гидрат целлюлозы, который после
промывки и просушки представляет собой прозрачную, как
стекло, фольгу. Схематически этот процесс можно
записать так:
целлюлоза -> натронная целлюлоза ->
-* ксантогенат целлюлозы -*- вискоза ->
-> гидрат целлюлозы (целлофан).
Аналогичен процесс изготовления целлофана медно-ам-
миачным методом, только в этом случае растворителем
целлюлозы служит аммиачный раствор солей меди.
Совершенно иначе получают искусственный рог —
галалит, который известен с 1897 года. Сейчас он играет меньшую
роль, чем другие пластмассы. Сырьем для его изготовления
служит молоко, точнее, казеин молока — белковое вещество,
содержание которого в молоке составляет около 3%. Если
добавить в молоко немного кислоты, оно скисает, и казеин
выпадает в виде белой рыхлой массы. То же самое происходит
при добавлении в молоко сычужного фермента, выделенного
из телячьего желудка. Этот фермент может окислить в
400 000 раз больше молока, чем кислота.
Для изготовления пластмассы в казеин добавляют
смягчители типа глицерина, а также красители. Смесь
механически перемешивают, и полученное вещество прессуют в
бруски, пластины или трубы. Изделия, пока они еще очень мягкие
и бесформенные, обрабатывают водным раствором
формальдегида, который вызывает отверждение массы. Правда,
отверждение толстых пластин или блоков может длиться очень
долго — до 6 месяцев. Понятно, что метод, требующий так
много времени, дорог. После просушки получается галалит,
или искусственный рог. Он находит большое применение при
8 3. Шпаусцус
изготовлении пуговиц и художественных изделии, так как
похож на рог и легко поддается обработке.
В свое время производство пластмасс из натурального
сырья было чрезвычайно прогрессивным, потому что
позволяло изготовлять вещества с новыми свойствами, однако
остановиться на этом было нельзя. Во-первых, природные
источники сырья были ограничены и не позволяли достаточно
расширять производство. Во-вторых, с самого начала было ясно,
что на природные макромолекулы с их специфическими
свойствами можно повлиять, можно изменить их в известной
степени, но совершенно новых свойств получить нельзя. Перед
учеными встала задача создать путем полимеризации
высокомолекулярные соединения из простых структурных
элементов, т. е. полностью синтетические вещества.
Работая с органическими соединениями, химики давно
заметили, что часто легкоподвижные жидкости вследствие
неизвестных превращений застывают в более или менее
твердые смолообразные массы, которые в большинстве случаев
трудно удалить из реакционных сосудов. Чистить такие
сосуды было практически невозможно, и их приходилось
выбрасывать вместе с содержимым. Именно так и поступил
Рудольф фон Байер в 1872 году со смолообразной массой,
которую он получил, нагревая фенол с формальдегидом. Он не
обратил на эту реакцию никакого внимания.
Иначе поступил живший в Америке голландский
профессор Л. Н. Бакеланд, когда получил такие же
смолообразные продукты конденсацией фенола и формальдегида. Он не
выбросил массу, а стал очень тщательно исследовать ее,
одновременно изучая и варьируя условия реакции и добавляя
вещества-наполнители типа древесной муки или асбеста.
Таким путем ему удалось получить первую пластмассу, годную
к техническому применению, которая с 1908 года начала
победное шествие по всему миру под названием баяе./Ш7\ И
сегодня фенопласты составляют значительную долю
синтетических пластмасс. В ГДР их производят в больших
количествах. Мы встречаем бакелит на каждом шагу. Из него
изготовлены корпуса телефонов и радиоприемников, крышки,
абажуры для ламп и т. д. Из бакелита, спрессованного вместе
с другими материалами, получают очень ценные слоистые
пластики. Вспомним о кузове маленькой машины «Трабант»,
где впервые был применен и блестяще выдержал
испытание «Р70». Изготовляют этот пластик из многих слоев хлопка
и искусственной смолы прессованием под сильным давлением
ЯН
Н
И
+7 О
Л.Гн
^CH,+
L--
н
0'eCH +
^ .У' jt
*VZ*£ /*A
при температуре около 160°; за 8—12 минут получают
отдельные части кузова, которые потом легко соединяются в целый
корпус.
Процесс конденсации, в результате которого образуются
пенопласты, протекает не в одну, а в три фазы.
Конденсацией называют соединение молекул с
выделением воды. Сначала проводят соединение фенола или его
производных с формальдегидом в отношении от 1:1,5—до 1 : 2,5.
При этой реакции применяют щелочные катализаторы —
аммиак или соду. Образуется легкоплавкий продукт, хорошо
растворимый во многих жидкостях. Его'называют резоль или
смола А. Это промежуточный продукт, из которого получают
пластмассу. Пластмасса состоит из длинных молекул,
расположенных цепочкой, которые образовались при соединении
молекул фенола и формальдегида с выделением воды.
Затем расплавленную смолу удаляют из реакционных
сосудов, а после застывания перемалывают и при нагревании
перемешивают с наполнителями, стружками, отходами
бумаги и т. д. При этом процесс конденсации продолжается и
образуется резитол, или смола Б. Эта смола уже нерастворима,
8*
но при нагревании размягчается, становится пластичной и в
этом состоянии ее можно формовать. Смола Б поступает на
горячее прессование, где ей придают нужную форму.
• Наконец, конденсация может быть продолжена при 150°,
когда происходит образование сетчатых макромолекул,
вытянутых в длину. Образуется нерастворимый и не
поддающийся формовке резит, или смола С. При нагревании до высокой
температуры она разлагается, но не размягчается, сохраняя
приданную ей форму. Такие пластмассы называют
термореактивными. Перед резит-фазой их обрабатывают прессованием
или литьем. Поэтому корпус «Трабанта» и в жаркой пустыне
не становится мягким и бесформенным, каким стал бы поли-
винилхлорид, а остается твердым и прочным.
От термореактивных пластмасс отличаются
термопластические, или термопластики, которые при повышенной
температуре становятся мягкими, пластичными, а при охлаждении
снова застывают. К термопластикам относится полиэтилен,
или винидур, о котором мы еще будем говорить.
Если вместо фенола берут мочевину или амины и
проводят конденсацию их с формальдегидом, то получают
соответствующие фенопласты — аминопласты, которые могут быть
окрашены в белый цвет или другие нежные тона. Поэтому их
особенно охотно используют в быту, и в будущем
производство этих пластмасс сильно возрастет. Белая тарелка из ами-
нопласта весит около 260 г, а из глины 480 г. Это очень важно,
когда приходится подавать много тарелок. Каждый официант
будет доволен, если, подавая 5 тарелок, ему придется нести
на 1 кг меньше. Причем такая посуда не бьется. Вот почему
этот материал так быстро приобрел популярность.
Из меламина и формальдегида получают меламиновую
смолу, из которой делают разноцветные предметы домашнего
обихода, обивку для мебели, кузовы машин, детали для
электропромышленности и т. д.
Если через смолу из мочевины в состоянии резоля
продувать газ, то получается очень ценное вещество — пенистый
пластический материал — пенопласт. Он в 70 раз легче воды;
пенистая структура делает его плохим проводником тепла и
звука. Поэтому пенопласты находят широкое применение в
качестве теплоизоляционных прокладок в
вагонах-рефрижераторах и холодильниках. Так как пенопласт, поглощая звук,
на 50% снижает слышимость, его применяют для
звукоизоляции, особенно в концертных и театральных залах.
Следует упомянуть о полиэфирных смолах, которые
образуются при конденсации ненасыщенных дикарбоновых
кислот, например малеиновой кислоты \-\Q С00\-\
\\с—соен
и двухатомного спирта, например гликоля ni± (ПЦ
Cttz—OH
При этом получают ненасыщенные полиэфиры с
двойными связями в молекуле. К этой массе можно добавить также
ненасыщенные вещества, например стирол CeHs—СН = СНг.
В присутствии катализатора без давления за короткое время
двойные связи разрываются, образуя сетчатые молекулы, и
получается твердая пластмасса. Особое значение приобрели
сейчас полиэфирные смолы, армированные стекловолокном.
Такой материал пронизан множеством тончайших
стеклянных нитей. Они не затрудняют обработку, но придают
материалу такую прочность, что его называют «стеклянной
сталью». Конечно, он гораздо легче настоящей стали, не
поддается коррозии, пропускает свет и может быть любого цвета.
Лыжные палки, удочки, пропеллеры для самолетов, кузовы
автомобилей, несущие части машин, корпуса для лодок
изготовляют сейчас из армированных стеклом эфирных смол.
Этой пластмассе, без сомнения, можно предсказать большое
будущее.
Все рассмотренные до сих пор пластики получали
соединением атомов по меньшей мере двух различных веществ с
отщеплением воды. Такой процесс, который не обязательно
приводит к образованию макромолекул, называют
конденсацией. В нашем случае речь шла о поликонденсации. Но
пластмассы можно получать и полимеризацией только одного
соединения. При этом возникают многоатомные, часто слегка
переплетающиеся между собой молекулы. Этот процесс,
который схематически можно представить как
d+d+d+d+d+d > d'-d'-dLd'-d'-d
называют полимеризацией, а полученные таким образом
продукты — полимеризационными пластиками.
Нам понятно, что не может быть и речи о полимеризации
насыщенных соединений. Для метана, например, у которого
все валентности углеродного атома насыщены водородом,
можно представить себе только реакцию замещения.
Простейшее вещество с двойными связями в молекуле, годное для
полимеризации, это этилен СН2 = СН2. Его можно получать в
большом количестве из угля и извести или еще дешевле —
из нефти.
Нам пришлось бы долго ждать, пока при нормальных
условиях из газообразного этилена получилось бы твердое
высокополимерное соединение.
Молекулы могут вступать в реакцию друг с другом
только тогда, когда образуются центры полимеризации, которые
возникают только при разрыве двойной связи в молекуле.
Образовавшиеся при этом свободные валентности способствуют
разрыву двойной связи следующей молекулы, и обе
молекулы объединяются в новую — с удвоенным
молекулярным весом.
Итак, первой ступенью процесса полимеризации
является активирование, или образование центров полимеризации.
Для этого необходима энергия, например, в виде светового
или какого-нибудь иного излучения. Во многих случаях
193
(Зародышей] ГЮил**М€рК- . / X
I \\\
ofpyjofaHue целей
иии процесс />осща
бывает достаточно тепловой энергии. Очень часто
активирование осуществляется катализаторами. Они снижают
требуемую энергию активации до технически достижимой
величины. При активации у некоторых мономеров разрываются
двойные связи и образуются активные участки, на которых
начинается процесс полимеризации. Число таких центров
зависит от количества полученной мономером энергии и
величины энергии активации, необходимой для данного процесса.
Чем энергия активации ниже, тем больше образуется
центров полимеризации.
Двойная связь обладает более высокой энергией, чем
одинарная. Если двойные связи разрываются, освобождается
тепловая энергия, которая способствует активации других
молекул.
Освободившиеся валентности немедленно насыщаются, и
таким образом из мономеров образуются все более длинные
молекулы. Это вторая фаза процесса полимеризации —
образование цепей, или процесс роста. Конечно, образование
цепей не протекает бесконечно долго. В какой-то момент оно
прерывается, например если неподалеку от растущей
молекулы нет больше других активированных молекул или если
происходит насыщение свободных валентностей внутри
макромолекулы. Этой третьей фазой — обрывом цепи
заканчивается процесс полимеризации.
194
Произойдет ли вообще полимеризация, зависит от
первой фазы — от образования центров полимеризации.
Образование центров определяет и ход всего процесса. Если центров
мало, образуется относительно мало макромолекул, зато они
обладают очень высоким молекулярным весом. Если же
центров образуется много, и макромолекул получается много, но
на этот раз со значительно меньшим молекулярным весом.
Так как от длины цепи и степени образования сетчатых
молекул в значительной мере зависят свойства пластиков, то,
управляя ходом полимеризации, мы можем соответственно
«шить пластмассу по мерке». Как это делается, наглядно
показывает небольшая схема.
Малое количество ка- ч Мало центров и мед-
тализатора и (или) ленная полимериза- _►
малое подведение / ция
энергии
Большое количество \ Много центров и быст-
рсатализатора и (или) ^ рая полимеризация __►
большое
энергии
подведение
/
Вые окополимерное
соед инение (длинная
ните видная
молекула)
Низкополимерное
соединение (короткая
нитевидная
молекула)
Если исходным продуктом служит этилен, то ход
полимеризации будет выглядеть так
Jt/eptvJL
Jfontaut/jamcp
Сначала мономерные молекулы расположены рядом (А).
Под влиянием катализатора и энергии происходит разрыв
двойной связи в молекуле, образуются центры полимеризации
и свободные валентности (В). Благодаря освободившейся
энергии двойная связь у второй молекулы тоже разрывается,
и две валентности объединяются, образуя димерное
вещество, состоящее из двух молекул этилена (С). Этот процесс
может продолжаться до тех пор, пока не образуются
молекулы, содержащие более 1000 атомов углерода.
Однако осуществить на практике этот столь просто
описываемый процесс было несравненно труднее. В 1931 —
1932 годах английским химикам впервые удалось, применяя
сверхвысокое давление (2000 аг), получить из газообразного
этилена твердый продукт полимеризации — полиэтилен.
Такой процесс назвали синтезом при высоком давлении. Этилен
смешивают с 0,05% кислорода, играющего роль
катализатора, и при давлении от 1500 до 2000 ат направляют по трубам,
нагретым до 180—200°. За один цикл полимеризуется от 8 до
10% этилена. Готовый полиэтилен собирают в приемник, а
непревращенный газ вторично подвергают тому же процессу.
Полиэтилен — термопластическая прозрачная масса.
Величина его молекулы может быть очень различна, в нее
входят от 2000 до 4000 углеродных атомов. Свойства материала
зависят от молекулярного веса и строения полимера,
поэтому сырой полиэтилен, как правило, в горячем состоянии
хорошо перемешивают.
Полиэтилен находит разносторонние применения. Он
очень стоек, легко поддается обработке и его широко
применяют в химической промышленности и в лабораториях.
Полиэтилен употребляют для изготовления труб, сосудов,
электроизоляции, в качестве упаковочного материала. Очень
удобны пульверизаторы из полиэтилена. Достаточно просто
сжать стенки эластичного сосуда, и жидкость
разбрызгивается.
Создать давление свыше 1500 ат, необходимое для
получения полиэтилена, и работать при таком давлении, конечно,
непросто. В 1955 году Циглеру и его сотрудникам в
институте имени Макса Планка удалось полимеризовать этилен без
применения давления. Были найдены специальные
катализаторы, способствующие образованию центров полимеризации.
Циглер установил, что такими свойствами обладают
определенные металлоорганические соединения, например триэтил-
алюминий АЦСгНбЬ — вещество, вполне доступное для
промышленного применения. Вместе с тетрахлоридом титана,
растворенным в бензине или дизельном масле, он образует
чрезвычайно эффективный катализатор. Выход полиэтилена
достигает 100%.
Однако вскоре было установлено, что сорта полиэтилена,
полученные с помощью катализаторов при низком давлении,
в некоторых отношениях очень резко отличались от
полиэтилена, полученного под давлением. Этилен, полученный при
низком давлении, прочнее, эластичнее и имеет более
высокую точку размягчения, но его изоляционные свойства
оказались гораздо ниже. Такое повышение электропроводности
вызывают остатки катализатора. Поэтому в наши дни оба
способа получения полиэтилена сосуществуют.
Полиэтилен, открытый только в 1932 году, является
сейчас исключительно ценным пластическим материалом с
разнообразными свойствами. Не нужно быть пророком, чтобы
предсказать ему большое будущее.
Применяя в качестве исходного вещества пропилен,
аналогичным путем получают полипропилен — пластмассу,
производство которой сейчас только начинает развиваться. По
сравнению с полиэтиленом он обладает некоторыми более
ценными свойствами, поэтому в будущем его, вероятно, будут
изготовлять в большом количестве.
По-видимому, самая известная пластмасса поливинил-
хлорид. В ГДР он известен под торговыми названиями PVC,
винидур, игелит, миполам, деселит и т. д. Исходным
мономером для получения поливинилхлорида служит ненасыщенное
соединение — хлористый винил СНг = СНС1. Хлористый
винил можно получать каталитическим присоединением
хлористого водорода к ацетилену.
CHz=CH+НсС > cHz—CHcC
В 1938 году был найден способ полимеризации
хлористого винила, а с 1942 года полихлорвинил стали получать в
промышленных масштабах.
-сигсц - [-снг-сн-г-снх-сн-
I
В ГДР поливинилхлорид — пластмасса № 1. В 1956 году
его выпустили 46 000 г, а в 1965 году —около 120000 г. Во
всем мире наибольшим производство поливинилхлорида было
в 1960 году, когда оно составило 1500000 г.
Поливинилхлорид получают в виде белого или желтого
порошка, который размягчается при температуре немного
выше 60°. При температуре более 100° его формуют в
разнообразные изделия. Поливинилхлорид не разрушается
большинством химических реактивов, устойчив к коррозии и
благодаря высокому содержанию связанного хлора (50%)
негорюч. При температуре выше 200° он разлагается с выделением
хлористого водорода и обугливается.
<рери/а
'WHcttc
9[оршемб
3&*гре&
пЯ*
Поливинилхлорид относится к термопластикам, но при
низких температурах (около 0°) он очень хрупок. Может
быть, кто-нибудь из вас в послевоенные годы носил плащ из
поливинилхлорида. В солнечные дни такие плащи мягки,
носить их удобно и приятно, но в холодную погоду они
становятся жесткими и даже не падают, когда их просто ставят
на землю.
Для получения поливинилхлорида, который всегда
оставался бы мягким, необходимо тщательно перемешивать
пластмассу со смягчителями. Смягчители — это высококипящие
жидкости, которые благодаря своему строению обладают
способностью комплексно связываться с макромолекулами
пластмассы и придавать им пластичность.
Конечно, для изготовления предметов домашнего
обихода можно применять только такие смягчители, которые
совершенно безвредны для человека. Если поливинилхлорид
/ъ подвергнуть хлорированию, доведя количество хлора до 64%,
***Ииро*( получается полимер, устойчивый по отношению к кислотам
и щелочам, но растворимый во многих органических раство-
ЦС = СН^ рителях.
1 Поливинилхлорид применяют очень широко — для
изготовления мячей, фольги, пластика для покрытия пола,
предметов домашнего обихода и т. д.
Не следует забывать и о полистироле, производство
которого непрерывно возрастает. Полистирол прозрачен, как
стекло, и может быть окрашен в любой цвет. Из него делают
многие предметы — молнии, пластинки, корпуса, оправы
для очков, посуду. Так как полистирол очень хороший
диэлектрик, его применяют как электроизоляционный
материал.
Сырьем для производства полистирола также служит
этилен, который, как мы уже знаем, получают из ацетилена или
нефти. При реакции этилена с бензолом получают этилбензол,
который превращают в винилбензол — стирол. Винильная
группа содержит двойную связь, благодаря которой
происходит полимеризация стирола в полистирол
L — cooti
Метилированием и этерификацией ненасыщенной
акриловой кислоты получают сложный эфир метакриловой
кислоты, имеющий общую формулу
■соол,
сн3
(R — радикал, зависящий от того, какой спирт
употребляется при этерификации). Сложные эфиры метакриловой
кислоты — это прозрачные, как вода, низкокипящие и неприятно
пахнущие жидкости. В настоящее время их производят из
ацетона и синильной кислоты. Сначала получают амид
метакриловой кислоты, который путем обменной реакции со
спиртом, например метиловым, превращают в сложный эфир.
Благодаря двойной связи в молекуле эфиры метакриловой
198
кислоты легко полимеризуются в высокомолекулярные с©- Metnuuatfpuuctutuj
единения CH_C_CQA/"^
СИ-.
- С
Сооц
~СН,
сн.
сн.
c-ch,— c~chz-
COOfC
COCK,
сн.
В 1900 году молодой химик Рем изучил продукты
полимеризации эфиров метакриловой кислоты и получил
прозрачную как стекло, пластическую массу. Результаты работы
были так заманчивы, что решили начать промышленное
производство этой пластмассы. В 1928 году полученное им
вещество под названием плексиглас впервые поступило на рынок
и сразу стало пользоваться большим спросом во всем
мире.
Пиакрил — так называется пластик, выпускаемый в
ГДР,— также представляет большой интерес. Он вдвое легче
неорганического стекла и в 10 раз прозрачнее его. Следует
особенно подчеркнуть, что пиакрил не бьется и хорошо
поддается обработке, так что ему можно придать любую форму.
Из этого «органического стекла» делают кабины для
самолетов, защитные стекла и очки, линзы, демонстрационные
модели и многое другое. Но по сравнению с силикатным
стеклом пиакрил обладает одним недостатком. Он не так тверд,
и его легче раздавить, поэтому из него нельзя делать
прочные автомобильные стекла. Однако, если поместить между
двумя силикатными стеклами пиакриловую фольгу, можно
сделать небьющееся автомобильное стекло. Это «слоеное»
стекло при толчках и ударах никогда не разбивается в куски,
так как легко крошащееся силикатное стекло приклеено к
пластмассе.
Нужно упомянуть также и о эпоксидных смолах,
значение которых возросло в последние годы. Исходным
веществом при их получении служит эпихлоргидрин. При сополи-
меризации его с другими веществами образуются ценные
высокомолекулярные искусственные смолы. В
зависимости от исходного соединения и от процесса сополимериза-
ции можно получать различные покрытия и твердые
пластмассы.
Особенно важны клеи, созданные на основе эпоксидных
смол. Они чрезвычайно прочно склеивают металлы, керамику,
дерево и т. д. В ряде случаев такие клеи заменяют сварку и
клепку металлов. Очень ценными являются также твердые
эпоксидные смолы, которые находят особенно широкое
применение в электропромышленности.
При сополимеризации образуются также и
полиуретаны, на которых, однако, мы не будем останавливаться.
'U6rnOn?
У9«ръ
С€сн-сН-СИ7
\/ Z
о
199
Пластмассы, о которых шла речь выше, далеко не
исчерпывают палитру известных в наше время искусственных
веществ. Существует еще много других пластмасс, которые
также выпускаются в большом количестве. Число их
непрерывно возрастает. Сейчас главное заключается в том, чтобы
найти более совершенные способы переработки пластмасс и
новые области их применения, а также изменить и улучшить
свойства уже известных пластических материалов. Особенно
необходимо повысить температуру размягчения пластмасс,
так как именно это ограничивает их применение.
Благодаря развитию ядерной техники наука располагает
сейчас мощными источниками излучения. Когда ученые
подвергли пластмассы действию такого облучения, получаемого
в ядерных реакторах или от радиоактивных изотопов,
оказалось, что оно сильно влияет на структуру органических
макромолекул. В то время как в большинстве случаев
излучение приводит к разложению вещества, у пластмасс при
сильном облучении происходит образование сетчатых
нитевидных молекул.
Как мы уже знаем, такая важная пластмасса, как
полиэтилен, состоит • преимущественно из нитевидных молекул,
расположенных рядом друг с другом и связанных между
собой очень слабо.
Отсюда относительно легкая подвижность нитевидных
молекул по отношению друг к другу, что делает материал уже
при низких температурах довольно мягким, способным
терять форму и прочность. В результате образования большого
количества сетчатых вытянутых молекул возникает более
компактная структура, материал становится прочнее и
размягчается при более высоких температурах.
Облучение выбивает из молекул полиэтилена атомы
водорода. Такой атомарный водород обладает большой
реакционной способностью. Он может вырвать из соседней
молекулы еще один водородный атом и соединить с ним
молекулу, а затем медленно выделиться из вещества. В тех местах
молекулы, где прежде находились водородные атошл/
остаются свободные валентности, которые взаимно насыщаются и
тем самым образуют сетчатые молекулы. Схематически этот
процесс можно представить так:
н
и
-с-с— с—с
' I 1
Н Н Н- ц
ПН
-с-с-с—С—
и н н «
В то время как необлученный полиэтилен плавится
примерно при 110°, после сильного облучения он сохраняет
форму до 300° и его термопластические свойства исчезают. Таким
образом, можно необлученному и термопластическому
полиэтилену придавать любую форму, а затем, облучив его,
получать изделия, прочные при высоких температурах.
Конечно, под действием излучения большой энергии в
пластмассе происходят и другие реакции, до сих пор
окончательно не изученные. Широкому использованию этого метода
мешает в настоящее время слишком высокая стоимость
облучения. Работы в этой области сейчас только начинаются.
По-видимому, в будущем их значение существенно
возрастет.
Принципиально иной путь повысить температуру
размягчения в том, чтобы сразу получать новые пластмассы с
добавкой таких компонентов, которые придавали бы им
большую теплостойкость. Такими компонентами являются
неорганические вещества, которые выдерживают, как правило, без
изменения более высокие температуры, чем органические.
Поэтому можно предположить, что введение неорганических
компонентов в органические пластмассы повысит
теплостойкость последних, а такие положительные их свойства, как
малый вес, устойчивость к коррозии и легкость в обработке,
сохранятся.
В этом направлении уже достигнуты некоторые успехи,
которые вместе с новыми открытиями приведут к
решительным изменениям в производстве и применении
пластмасс.
Особый интерес представляют силиконы. Кипплинг
исследовал их еще в 1900 году, но лишь в последние годы их
стали производить в промышленном масштабе.
Как и углерод, атом кремния имеет четыре валентности.
Поэтому ученые давно уже предполагали, что на его основе
можно создать такое же многообразие соединений, какое уже
сейчас получено на основе углерода. Правда, связь кремний—
кремний далеко не так прочна, как связь между двумя
углеродными атомами. Однако если атомы кремния связаны
друг с другом не непосредственно, а через атомы кислорода
и свободные валентности насыщены алкильными остатками,
например — СНз, то получаются высокомолекулярные
силиконы — соединения, занимающие по своему строению и
свойствам среднее положение между органическими и
неорганическими веществами.
В промышленном масштабе силиконы получают
восстановлением двуокиси кремния (песка), в избытке
имеющегося в природе, прокаливая его вместе с углем:
Sioz+c->Si+coz
Элементарный кремний в присутствии хлористого
метила и меди при 300° превращается в дихлордиметилсилан:
доо
Z СИЛ + Ъь "
> (<***lSiCtz
Си,
Полученные таким образом силаны подвергаются
гидролизу, при этом образуются спирты, силиколы, которые сразу
же конденсируются в высокополимерные силиконы:
(CHS\ SiC4 + ZHv, О > (сН3\ Si (ОН J2+ Z UCC
Cuautiout,
ь I НО—St-OH]
Chi
-hzo
— St — O-l
L CH3
M
Так как при полимеризации с образованием
кислородных мостиков образуются не только линейные, но дву- и
трехмерные молекулы, можно получить любые продукты
полимеризации — от летучих до твердых, применение которых очень
разнообразно.
Силиконовые масла не смачиваются водой, кроме того,
при высоких температурах они являются хорошим смазочным
202
материалом. При введении атомов кремния в молекулу
значительно повышается теплостойкость полимеров. В
большинстве случаев эти материалы сохраняют устойчивость до 200°,
что открывает совершенно новые возможности для их
применения. Например, вместо эмалей во многих случаях можно
применять негорючие силиконовые лаки. Применение
силиконов как электроизоляционного материала позволяет
значительно повысить нагрузку агрегатов.
Следовательно, силиконы являются такими
искусственными веществами, которые во многих отношениях
совершенно отличны от известных до сих пор пластмасс.
Получение их еще очень сложно и дорого, а высокая стоимость
изделий препятствует широкому применению силиконов, но в
дальнейшем они, безусловно, будут играть значительную
роль.
Поликарбонаты — вид полимерных материалов, который
тоже еще только начинает применяться. Макромолекулы с
неорганическими карбонатными группами образуют
пластмассы с очень высокой теплостойкостью. Высокая
теплостойкость характерна также и для тех пластмасс, которые
получают полимеризацией тетрафторэтилена F2C = CF2 или три-
фторхлорэтилена F2C = CFC1.
Политетрафторэтилен и соответственно политрифтор-
хлорэтилен обнаруживают такие же ценные свойства, как
силиконы. Это объясняется большим количеством
«неорганических» атомов в молекуле. Эти полимеры не изменяются
до 300°, обладают исключительными изоляционными
свойствами и почти не поддаются воздействию химических
реактивов.
Следует подчеркнуть, что мы находимся лишь у истоков
развития химии пластмасс. Из числа пластмасс, уже
известных сейчас, а также из тех, которые будут получены, человек
отберет материалы, наилучшим образом отвечающие
требованиям техники. В будущем появится возможность
применять в каждом случае наиболее подходящую пластмассу.
Это, конечно, не означает, что перестанут пользоваться
такими традиционными материалами, как дерево, глина или
металл. Наоборот, пластмассы способствуют расширению
спроса на традиционные материалы и одновременно
открывают перед инженерами и конструкторами новые
возможности.
Развитие промышленности искусственных материалов
предусматривается во всех индустриальных странах, в том
числе и в ГДР. Пластмассы будут играть все большую' роль в
нашей жизни. Это яркий пример того, как человек, исследуя
природу и происходящие в ней процессы, все больше
освобождается от зависимости от нее.
Волокна из реторты
Адам и Ева не заботились об одежде, чтобы прикрыть
наготу, им хватало фигового листка. В странах с теплым
климатом и где это позволяют общественные отношения, еще в
наше время есть племена, одежда которых мало чем
отличается от библейского фигового листка. Но это исключение, не
касающееся всей остальной части человечества. Одежда
повсюду необходима, защищает ли она от капризов погоды,
является ли моральной необходимостью или атрибутом моды.
И вот человек отложил в сторону легендарный фиговый
листок и начал обрабатывать звериные шкуры. Потом он
догадался из подходящих растений добывать волокна, которые
можно было бы прясть и делать из них ткани. Как ни
велико было количество исходных материалов для волокон, как ни
разнообразна была техника их обработки, до последних
десятилетий все они были изготовлены только из сырья, которое
имелось в природе в более или менее готовом виде.
Для удовлетворения растущего спроса на волокна
человек выращивал специальные растения, разводил овец и
шелковичных червей. Но и этого все увеличивающемуся
населению земного шара становилось недостаточно. К тому же
производство естественных текстильных волокон было связано
с определенными климатическими условиями (например,
хлопок рос только на юге). Именно в высокоразвитых
странах возникло несоответствие между мощностью
перерабатывающей промышленности и собственными источниками
сырья. И наконец, была еще одна причина. Естественные
волокна имеют определенные свойства. Человек вынужден брать
их такими, какими их создала природа, и уже
соответственно создавать технику переработки, т. е. соизмерять свои
потребности с природой.
Как введение механических ткацких станков и других
машин произвело революцию в технике переработки
волокон, так и создание полусинтетических и полностью
синтетических волокон вызвало в текстильной промышленности
решительный переворот, последствия которого необозримы.
Сейчас мы владеем множеством совершенно новых волокон
с новыми, разнообразными качествами.
Одним из самых важных растительных волокон был и
остается хлопок. Уже тысячелетия назад его разводили в
Египте, Китае, Индии. Говоря о хлопке, имеют в виду нити
семян хлопка, состоящие почти из чистой целлюлозы
(подробнее о строении и свойствах целлюлозы говорилось в
предыдущей главе). Когда стали заниматься составом и
происхождением целлюлозы, установили, что она встречается з
природе очень часто, являясь существенной составной частью
всех растений. Конечно, лишь в исключительных случаях она
встречается в таком чистом виде, как в волокнах хлопка.
В большинстве случаев ей сопутствуют примеси — другие
высокомолекулярные вещества, к которым в особенности
следует отнести лигнин и так называемые гемицеллюлозы. Древе-
сина состоит преимущественно из целлюлозы с примесью 40%
лигнина и других веществ. Совершенно естественно возникла
мысль использовать это огромное количество целлюлозы, но
тут встал вопрос об отделении целлюлозы от примесей.
Дальнейшей переработкой целлюлозы в текстиль тогда
интересовались мало, целлюлоза была нужна главным образом для
изготовления бумаги.
В начале XIX века стали усиленно искать сырье,
необходимое для производства бумаги в больших количествах. Оно
должно было не только обладать подходящими качествами,
но быть также дешевым и легко доступным.
JtOQCueufuu'
-tf UMQCCQ
\*J\
Было очевидно, что легче всего изготовлять бумагу из
древесины. Стволы деревьев измельчались в щепу,
смешивались с водой, и полученная древесная масса
перерабатывалась в бумагу или картон. Поскольку такая масса содержала
не только целлюлозу, но и другие составные части древесины,
качество конечного продукта сильно снижалось. Нужно было
попытаться избавиться от этих примесей, отделить их от
целлюлозы.
Первые попытки разложить древесину химическим
путем относятся к началу прошлого века. Во второй его
половине были разработаны и внедрены в производство два
метода, которые в усовершенствованном виде и сегодня широко
применяются в промышленности.
Измельченную древесину обрабатывают в больших
чанах под давлением (3%-ным раствором едкого натра). При
этом лигнин и другие примеси растворяются в щелочи, а
целлюлоза остается. Применение вместо разбавленного едкого
натра сульфита натрия или сульфата натрия улучшило
процесс и повысило качество получаемой сульфатной целлюлозы.
Кроме щелочного метода превращения древесины в
массу, удобную для переработки, существует также кислый
метод, впервые разработанный еще американцем Тильгменом.
205
ч
Щеиои
Огистк*
Хлор
У
JcpeccotfaHue-
uOMoHtu
В 1863 году он запатентовал получение сульфитной
целлюлозы путем обработки древесины раствором бисульфита
кальция и разбавленной серной кислотой под давлением и при
высокой температуре. Первым, кто применил этот метод на
практике, был швед Экман. В Германии техническое
развитие этого метода осуществил Митчерлих в 1877 году.
Однако по какому бы методу ни изготовляли целлюлозу,
раньше ее перерабатывали только в бумагу или картон.
Поначалу из древесины выделяли целлюлозу, подобную той, что
встречается в хлопке, но она имела короткие волокна, и
поэтому из нее можно было вырабатывать не очень прочную
бумагу, но нельзя было прясть нити. Получить из такой
целлюлозы нить можно было, лишь подражая пауку или
шелковичному червю, которые выдавливают из своего тела
полужидкую массу, затвердевающую на воздухе в прочную нить.
Ъии/С
Следовательно, и целлюлозу сначала нужно ра^гво^ить, в
жидком виде переработать ее в нить и_зате»/дать еу затве
деть. Выше уже говорили о том, как были найдены пути
вращения неприступной на первый взгляд целлюлозы в в)
ство, растворимое в различных жидкостях. Шбнбайн д^б
ся успеха, используя нитроцеллюлозу и получив п
продукт переработки целлюлозы — целлуюид. I В
французский химик Шардоне, выдавливая да^рнкйх от
стий спиртово-эфирный раствор нитроцеллюлозы, получи,
блестящую нить, очень похожую на натуральный щелк.
Так был впервые получен искусственный шелк\Тка^и,
которые Шардоне создал в 1886 году, имели, правда, тят н
достаток, что состояли в некоторой степени из пироксилина.
Искры от сигареты, даже сильного удара было достаточн
чтобы обладатель такой одежды превратился в горящий фа
кел. Но Шардоне вскоре нашел выход. Он предложил
обработать нити растворами, которые удаляли опасные нитрогруп-
пы и превращали таким образом нитроцеллюлозу в чистую
целлюлозу. Свойства нити при этом не менялись.
Так был получен безопасный искусственный шелк,
который очень быстро получил признание во всем мире. В 1891
году на построенной Шардоне фабрике ежедневно
вырабатывали 50 кг искусственного шелка. Для того времени это было
очень много. В наши дни коллодиевый или искусственный
шелк Шардоне больше не изготовляют, так как в
последующие годы появились другие методы, менее опасные и более
простые и дешевые.
Еще в 1857 году химик Швейцер установил, что
целлюлоза растворяется в аммиачном растворе гидрата окиси меди.
В конце столетия это былр использовано для получения мед-
но-аммиачного искусственного шелка. Вязкий темно-синий
медно-аммиачный раствор целлюлозы через очень тонкие
отверстия выдавливался в жидкость и там застывал в тонкую
нить. Жидкость, в которой нить затвердевает, содержит
от 5 до 10% серной кислоты, благодаря чему из волокон
удаляются остатки медно-аммиачного раствора. После
промывки, ч;ушки и намотки нить подвергается дальнейшей
переработке.
Самым распространенным является вискозный метод,
который начали разрабатывать с 1892 года. По этому методу
размельченную целлюлозу обрабатывают разбавленным
раствором едкого натра. Затем в процессе созревания, который
продолжается несколько часов, происходит разделение
длинных цепей молекул — деполимеризация их, и образуется
белая хрупкая щелочная целлюлоза. Бе отжимают и
направляют в большие закрытые баки, где обрабатывают
сероуглеродом CS2 — ядовитой и чрезвычайно огнеопасной жидкостью
с отвратительным запахом.
При этом образуется желто-оранжевый ксантогенат
целлюлозы, превращающийся в вязкую вискозу, которую
выдерживают в особых сосудах несколько часов или даже несколько
дней.
207
14**
jjfi'
j&
и
ytQHHOL
1* л***»*
S^SSSit^
&SCM03U
CycuuuiHifu*
аппарат
После созревания вискоза становится годной для
прядения. Через насадки, каждая из которых имеет до 15 000
тончайших отверстий диаметром 0,08 мм, вискоза под давлением
тонкими струйками вытекает в ванну, наполненную
разбавленной серной кислотой и солями.
В этих ваннах молекулы едкого натра и сероуглерода
отщепляются, и образуются тончайшие целлюлозные нити.
После многократной промывки и сушки процесс получения
шелка для химика закончен, дальнейшей переработкой его
занимаются текстильщики.
При всех этих методах целлюлоза в процессе
переработки подвергается различным изменениям, но конечный
результат одинаков — та же чистая целлюлоза, только в виде
нитей.
208
Ацетатный искусственный шелк, который особенно
ценится потому, что по фактуре и блеску похож на
натуральный, является исключением, так как он состоит не из чистой,
а из ацетатной целлюлозы. В предыдущей главе мы уже
рассказывали об ацетате целлюлозы и поэтому ограничимся
здесь лишь краткими сведениями.
У искусственного и натурального шелка волокно
состоит из длинных нитей, которые и поступают на дальнейшую
переработку. Шерсть, так же как и хлопок, состоит из более
или менее коротких волокон, из которых сначала прядут
нити, а уже из них вырабатывают легкие, теплые, хорошо
впитывающие влагу ткани.
Нетрудно догадаться, что для получения искусственной
шерсти нужно получить целлюлозу с короткими волокнами.
Просто разрезая^, длинные нити на короткие, этого добиться
нельзя. Потребовалось провести большую работу, прежде чем
в 1920 году в Премнице, на нынешнем заводе искусственного
шелка им. Фридриха Энгельса, удалось получить волокно,
напоминающее шерсть. Оно стало широко известно под
названием вистра. Вначале к «шерсти из дерева»
относились настороженно, но вскоре она сумела проложить
себе широкую дорогу не только в Германии, но и во всем
мире.
В последующие годы возникло много фабрик,
вырабатывающих такую шерсть. В настоящее время во всем мире
искусственной шерсти производят больше, чем овечьей.
Особенно хорошие материалы получают, когда смешивают
искусственную шерсть с другими волокнами. Сегодня
искусственная шерсть — это не низкосортный заменитель, а
полноценное текстильное сырье.
peu/qpo/jo 4а кие
и
Транца
^Й°^ Щомъри
^"Уъ^ахъо^
>Н«*"
УСтноАс^
^QTCC7CC/<
^U^p^
Искусственные шелк и шерсть относятся к
полусинтетическим волокнам. Исходными материалами для них — будь
то хлопок, дерево, тростник или солома — является
природное сырье, из которого получают целлюлозу.
Но количество этого сырья в природе ограничено. Не
только в Германии поредели леса; деревья вообще растут
гораздо медленнее, чем потребность в древесине, поэтому
совершенно необходимо экономно и обдуманно использовать
это ценное сырье. И ученые стали работать над получением
полностью синтетических волокон.
Первое полностью синтетическое волокно было получено
из перхлорвинилхлорида.
Полимер из перхлорвинилхлорида растворяется в
ацетоне и его легко можно «прясть» через фильеры. Как и
пластмассы, это волокно отличается исключительной
устойчивостью к щелочам и кислотам, водостойко, устойчиво против
бактерий гниения, обладает водоотталкивающими
свойствами. Все это позволяет употреблять перхлорвиниловые ткани
для изготовления фильтров, спецодежды, палаток,
рыболовных сетей и т. д. Так как эти волокна легко заряжаются
электричеством, они служат основой для изготовления
хлорвинилового лечебного белья.
Гораздо большее значение имеют волокна, получаемые
полимеризацией аминов и известные во всем мире под
названиями нейлон, капрон, дедерон, перлон. В 1930 году в США
ученый Карозерс проводил важные исследования
полимерных веществ. В ходе этих исследований он получил и изучил
много новых веществ. Среди прочих был получен также
полиамид е-аминокапроновой кислоты. Карозерс убедился, что
210
для синтеза волокна это вещество не годится, и занялся
другими исследованиями, которые в 1935 году привели его к
получению полимерной макромолекулы гексаметилендиамина
и адипиновой кислоты.
Из этого полимера удалось выткать волокно с
исключительными свойствами. Уже в 1940 году на больших
предприятиях по этому методу производили 4000 тонн нейлона,
который вскоре стал известен во всем мире.
В Германии тоже занимались проблемой изготовления
полностью синтетических волокон. Уже в начале XX века
было установлено, что из е-капроновой кислоты можно
получить капролактам, который при полимеризации становится
вязким и плотным. Это отмечал еще Карозерс, считая, однако,
что из такой массы волокна получить нельзя.
Несмотря на это, в 1936 году в Берлине химик Пауль
Шлак еще раз занялся капролактамом. На первый взгляд
это было напрасной тратой времени, так как Карозерс уже
получил отрицательный результат. Но Шлак хотел сам
убедиться в правильности его выводов. Полимеризацией капро-
лактама Шлак получил поликапролактам, из которого ему
удалось сделать волокно, равноценное нейлону. С 1938 года
в Германии начали в промышленном масштабе изготовлять
полностью синтетическое волокно перлон, сейчас известное в
ГДР под названием дедерон. В 1943 году на заводах Лейна
было получено 1000 т капролактама, а в 1956 году его
производили уже 6200 т. Это количество сегодня давно уже
превышено и по программе развития химии будет
увеличиваться еще больше.
Исходным сырьем для производства дедерона в ГДР
является сейчас бурый уголь, при сухой перегонке которого
получают фенол, после очистки поступающий на капролакта-
мовую фабрику. Когда будет введен в строй
химический завод Лейна II, будут работать преимущественно на
синтетическом феноле. Его получают кумольным методом
из нефти, он чище и более пригоден для синтеза
капролактама.
Сначала фенол гидрируют и превращают в спирт — цик-
логексанол
Цихиоъейсанои.
211
Затем циклогексанол окисляют над цинк-железным
катализатором и получают кетон — циклогексанон
нг9 t °Н ^ | ' Г
\ ?
Hi Н*
Циклогексанон реагирует с гидроксиламином, который
по техническим причинам вводится в реакцию в виде
аммиачного раствора сернокислого гидроксиламина, и образуется
оксим циклогексанона
ъидрок&циаи&иы Чикио2е/сс<*ноня.
Тепло, выделяющееся во время реакции, повышает
температуру до 90°, и образующийся оксим выделяется в виде
мельчайших капелек. К расплавленному оксиму при
перемешивании прибавляют дымящуюся серную кислоту. Оксим
претерпевает своеобразное превращение, названное по имени
химика, открывшего его, «бекмановской перегруппировкой».
В этом процессе серная кислота является катализатором. Атом
азота переходит из боковой цепи в кольцо, и таким образом
образуется капролактам
Vt Wt ft
Олени* Xanfouamutui
Капролактам отправляют на дедероновые заводы. Там
его снова плавят, смешивают с катализаторами и получают
поликапролактам, который под давлением продавливается
через прядильную фильеру и на воздухе затвердевает в тонкую
нить. После вытяжки, а это процесс, при котором длина нити
увеличивается в 4—5 раз по сравнению с первоначальной,
благодаря упорядочению разрозненных прежде молекул
получают очень прочную нить, готовую для дальнейшей
переработки.
Прочность дедероновой нити вошла в поговорку. Деде-
рон можно получать в виде бесконечного волокна, сматывать,
а затем перерабатывать подобно натуральному шелку в
тончайшие чулки, легкие ткани или тонкое белье. Дедероновую
нить можно также разрезать, а пучки волокон смешивать с
хлопком, шерстью или штапельным волокном.
Дедерон принадлежит к числу наиболее
распространенных химических волокон. Нить из дедерона легче, чем такая
же нить из шерсти или хлопка. Изделия из дедерона не
боятся моли и, так как дедероновое волокно плохо поглощает
воду, быстро сохнут. Плохое водопоглощение может быть в
некоторых случаях недостатком. Тем, кто сильно потеет, не
рекомендуется носить дедероновое белье, так как оно не
впитывает пот. Однако недавно получили дедероновое
полотно, способное хорошо поглощать пот. Для этого
дедероновую нить покрывают тонким слоем гигроскопического
вещества.
Дедерон исключительно прочен. Дедероновым канатом
толщиной с обычную бельевую веревку можно тянуть боль-
шой автомобиль — даже тонкую дедероновую нить разорвать
очень трудно,
Синильная
Ацетилен кислота Акрилонитрил
НС=СН + Н-ех —> Нгс^снсх
%hlc^chcj>c —^сн^—сиА
V С/ГД
Износоустойчивость дедеронового волокна в 10—20 раз
выше, чем у хлопка или шерсти, а по прочности на
растяжение оно во много раз превосходит натуральные
волокна.
Другое чрезвычайно ценное волокно производят из поли-
акрилонитрила. Мономерный акрилонитрил получают из
ацетилена. Немецкая промышленность выпускает его с 1942
года. В ГДР на заводе Буна из ацетилена и синильной кислоты
производят акрилонитрил — ядовитую прозрачную жидкость
с резким запахом, которую сразу полимеризуют в белый
порошкообразный полиакрилонитрил.
Из этого вещества делают волокно, известное под
названием волприла. Прядение полиакрилонитрила доставило хек-
стильщикам немало хлопот. Дело в том, что он не плавится и
его нельзя выдавить через прядильные отверстия —
фильеры подобно дедерону. Так же безрезультатны были попытки
растворить его в каком-нибудь растворителе. Только после
многочисленных опытов нашли несколько растворителей, из
которых самым подходящим оказался диметилформамид.
Вязкий раствор полиакрилонитрила в диметилформамиде
через узкие отверстия поступает в осадительную ванну, где и
образуются волокна. Их соединяют в пучки, вытягивают,
моют, сушат. Дальнейшая переработка происходит на
текстильных предприятиях.
Полиакриловое волокно обладает рядом преимуществ,
которые обеспечивают ему многостороннее применение. Оно
отличается высокой свето- и теплостойкостью и химической
устойчивостью. Оно чрезвычайно водоустойчиво и почти не
мнется. Прочность на разрыв у полиакрилового волокна
выше, чем у шерсти, и при этом оно сохраняет мягкость и
легкость. Его можно соединять с другими волокнами,
изготовлять плащи, купальные костюмы, вязаные вещи, зонтики и
многое другое, не говоря уже о возможностях применения
этого волокна в технике.
В заключение нужно сказать о полиэфирных волокнах.
Из этиленгликоля и терефталевой кислоты получают
сложный эфир, при полимеризации которого образуется ланоно-
вое волокно. Это волокно не портится при кипячении и
совершенно не мнется. Юбку или платье из ланона можно мять
как угодно, но оно все равно будет выглядеть как новое. Это
214
волокно не играло до сих пор большой роли, так как из-за
недостатка исходного сырья его производилось мало. Но в
недалеком будущем производство полиэфирного волокна
возрастет.
Наша одежда проделала длинный путь от звериной
шкуры до синтетической ткани. Еще 100 лет назад никто не мог
бы предсказать, что в наше время именно химия сумеет
удовлетворить потребность людей в лучшем текстильном сырье.
И конечно, в этой области нас ждет еще много
неожиданностей.
Примечания редактора
1 Гомеонолярную связь часто называют также ковалентной.
2 О тетраэдрическом расположении четырех атомов в
пространстве вокруг атома углерода впервые независимо друг от друга
высказали предположение (в 1874 г.) французский ученый Ле-Бель и
голландский ученый Вант-Гофф.
3 Эта реакция была открыта в 1881—1884 годах Михаилом
Григорьевичем Кучеровым, профессором Земледельческого (ныне
Лесного) института в Петербурге.
4 Теорию неорганического происхождения нефти развивал
Д. И. Менделеев; наличие в нефти веществ растительного и животного
происхождения объяснялось тем, что эти вещества просто
оказывались растворенными в ней и не имели отношения к ее
первоначальному происхождению.
5 В России, в районе Баку, нефть добывали с X века. В начале
XVIII века в этом районе насчитывалось более 50 нефтяных колодцев
глубиной до 40 м. В 1844 году чиновник горного управления Ф. А.
Семенов предложил проводить «земляное бурение» для углубления
колодцев, а в 1848 году закончили бурение первой нефтяной скважины.
Промышленное использование таких скважин в России началось в
60-е годы прошлого века. Добыча нефти из них проводилась в
основном фонтанным и поршневым методами. В конце 80-х годов
несколько скважин было оборудовано глубинными насосами, однако из-за
специфических особенностей кавказских месторождений этот метод в
то время не получил достаточного развития. В начале XX века на
бакинских промыслах нашел практическое применение компрессорный
способ, предложенный русскими инженерами В. Г. Шуховым и
А. Б. Бари. Следует отметить, что в дореволюционной России
нефтяная промышленность была несколько более развита, чем другие
отрасли тяжелой промышленности.
8 Распространено также название «процесс в кипящем слое*,
поскольку пылевидный катализатор, взвешенный в парах
перерабатываемого нефтепродукта, по виду похож на кипящий.
7 В 1936—1937 годах советскими химиками Б. А. Казанским и
А. Ф. Платэ, Б. Л. Молдавским и Г. Д. Камушер, В. И. Каржевым,
216
М. Г. Саверьяновой, А. П. Сновой почти одновременно впервые было
показано, что предельные и непредельные алифатические
углеводороды при повышенной температуре под различными катализаторами
превращаются в ароматические углеводороды. Это превращение,
называемое ароматизацией, протекает через стадию промежуточного
образования циклических предельных углеводородов,
дегидрирующихся далее в ароматические.
Каталитическая дегидрогенизация углеводородов ряда циклогек-
сана была открыта в начале XX века Николаем Дмитриевичем
Зелинским (1861—1953) — выдающимся советским химиком-органиком.
Н. Д. Зелинскому принадлежат многие важные открытия в
области синтеза и каталитических превращений углеводородов
различных классов, а также работы по изучению состава нефти
советских месторождений, по получению синтетического жидкого
топлива и др.
8 Под «атомностью» следует подразумевать валентность.
9 Несомненно, что именно Кекуле внес большой вклад в науку,
установив строение бензола, однако основоположником теории
химического строения, автором общей теории строения органических
соединений является великий русский химик Александр Михайлович
Бутлеров (1828—1886). Созданная им теория (основные положения
которой были сформулированы в 1861 г.) явилась передовой
химической теорией прошлого столетия и, что особенно важно, определила
основное направление развития теоретической и экспериментальной
органической химии XX века.
А. М. Бутлеров показал, что в молекуле органического соединения
имеется определенная последовательность химической связи атомов,
которую можно установить химическим путем. Он экспериментально
подтвердил это и таким образом доказал, что по химическим
свойствам вещества можно установить строение молекулы и, наоборот, зная
строение, можно предсказать многие свойства соединения. Бутлеров
наиболее последовательно выступил против так называемой теории
типов, в то время как Кекуле, по существу, оставался сторонником
этой теории и не смог полностью освободиться от ее представлений.
По мнению Кекуле, химические формулы могут выражать лишь
реакционную способность веществ, но не их истинное строение. А. М.
Бутлеров считал, что для каждого вещества возможна только одна
рациональная формула, которая и определяет его химические свойства,
дает основу для понимания возможных химических превращений,
позволяет предсказывать новые пути синтеза.
А. М. Бутлеров первый сформулировал идею о пространственных
изомерах. Следует также отметить, что он впервые открыл и
объяснил явление таутомерии (динамической изомерии), которое
заключается в том, что два соединения с одинаковой общей формулой, но
различным строением могут в определенных условиях легко
переходить друг в друга.
10 Такое написание в химической литературе принимается не
для бензольного, а для циклогексанового кольца; бензольное кольцо с
делокализованными л-электронами изображается так: /\
Ю|
\/
11 Николай Николаевич Зинин (1812—1880) — профессор, затем
академик, а с 1868 года первый президент Русского химического
общества, любимый учитель выдающегося химика А. М. Бутлерова.
Синтетическим путем впервые получил анилин (назвав его бензидамом)
путем восстановления нитробензола сернистым аммонием, а также
впервые получил ряд других ароматических аминов, имеющих в
настоящее время важное значение. «Если бы Зинин не сделал ничего
более, кроме превращения нитробензола в анилин, то и тогда его
имя осталось бы записанным золотыми буквами в истории», — писал
о нем немецкий химик Гофман.
12 У нас также выпускается много стиральных порошков —
«Новость», «Астра», «Эра», «Лотос» и др., — обладающих высокими
моющими качествами.
13 Эта реакция каталитической гидратации ацетилена в ацеталь-
дегид была открыта М. Г. Кучеровым в 1881—1884 годах.
14 Промышленный метод получения синтетического каучука из
спирта впервые был осуществлен в СССР в 1932 году по способу
С. В. Лебедева. Метод получения бутадиена из ацетальдегида и
спирта разработал И. И. Остромысленский.
Содержание
ПРЕДИСЛОВИЕ 5
ПЕРВЫЕ ШАГИ 9
ПРОСТЕЙШИЕ ОРГАНИЧЕСКИЕ ВЕЩЕСТВА 21
ВДВОЕ — НЕ ВСЕГДА ПРОЧНЕЕ 31
КОНЕЦ ВЕНЧАЕТ ДЕЛО 44
ПУТЬ К НЕФТИ 57
*
МНОГОЛИКОЕ ВЕЩЕСТВО 67
ОКТАНОВОЕ ЧИСЛО 80 78
ГОРЮЧЕЕ ИЗ УГЛЯ 90
БЕНЗОЛЬНОЕ КОЛЬЦО 98
ПЕРВЫЕ АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ НО
КРАСКА ИЗ УГЛЯ 118
КРАСКИ И КРАШЕНИЕ 130
ПОЧЕМУ ПОМИДОР КРАСНЫЙ? 138
ГЛАВА О ЧИСТОТЕ 151
КАУЧУК НАТУРАЛЬНЫЙ И СИНТЕТИЧЕСКИЙ 165
ПЛАСТМАССЫ В НАСТУПЛЕНИИ 183
ВОЛОКНА ИЗ РЕТОРТЫ 204
3. Шпаусцус
ПУТЕШЕСТВИЕ
В НИР ОРГАНИЧЕСКОЙ ХИМИИ
Редактор А. Г. Белевцева
Художник В. Ф. Лактиопов
Художественный редактор Ю. А. Максимов
Технический редактор Е. С. Потапенкова
Корректор Л. Я. Шехтер
Сдано в производство 23/IX 1966 г.
Подписано к печати 20/1V 1967 г.
Бумага 70Xi081/u=6,88. бум. л.,
19,25 печ. л. Уч.-изд. л. 16,93.Изд. j* 12/3480
Цена 88 коп. Зак. J4 695.
Темплан изд-ва «Мир» 1966 г., пор. J4« 247
ИЗДАТЕЛЬСТВО «МИР»
Москва, 1-й Рижский пер., 2
Ярославский полиграф комбинат
Главполиграфпрома Комитета по печати
при Совете Министров СССР
Ярославль, ул. Свободы, 97
0jt Ж
vfi
of
c?YT Л *. •*
*cv->^
i*
_ 1 О