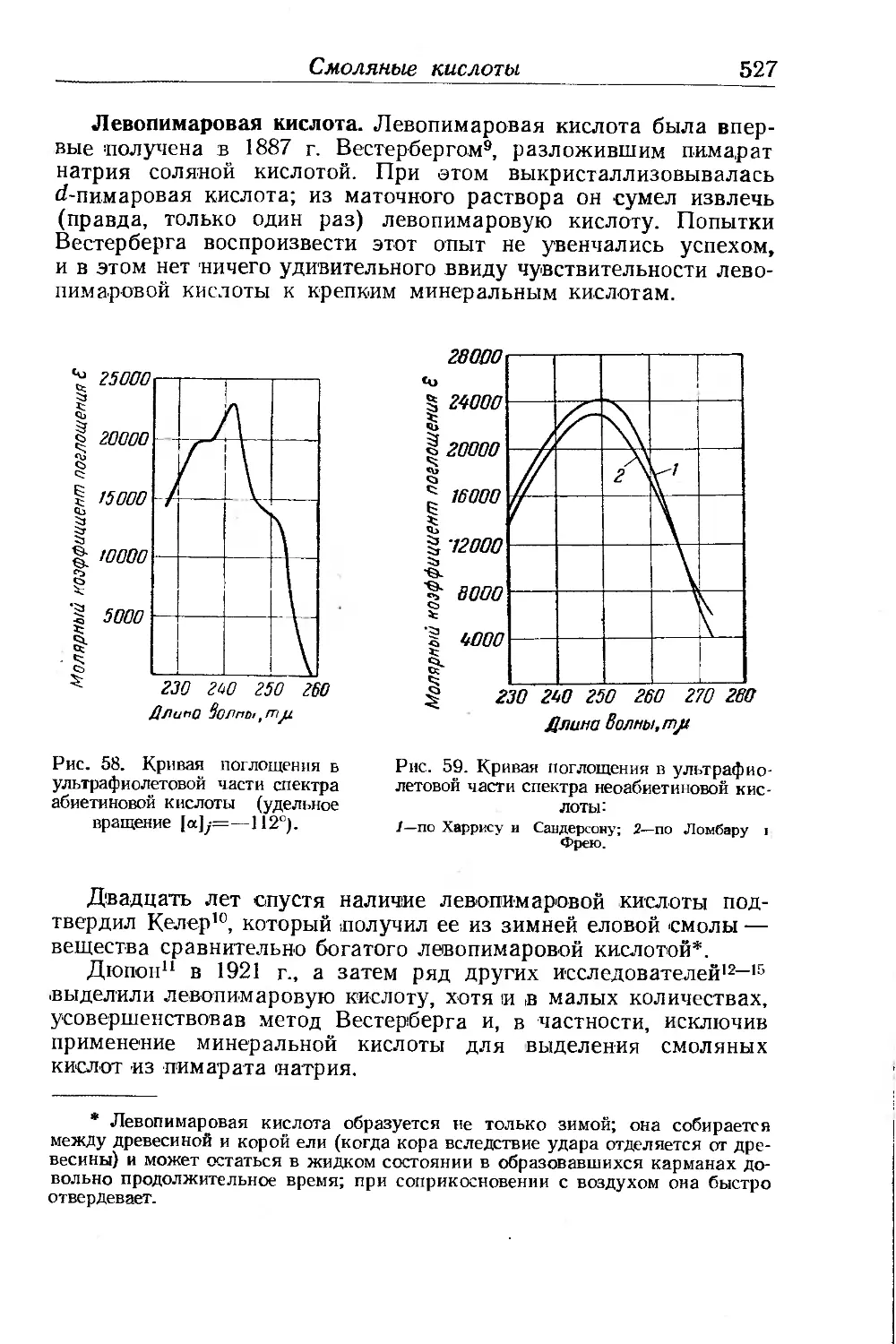
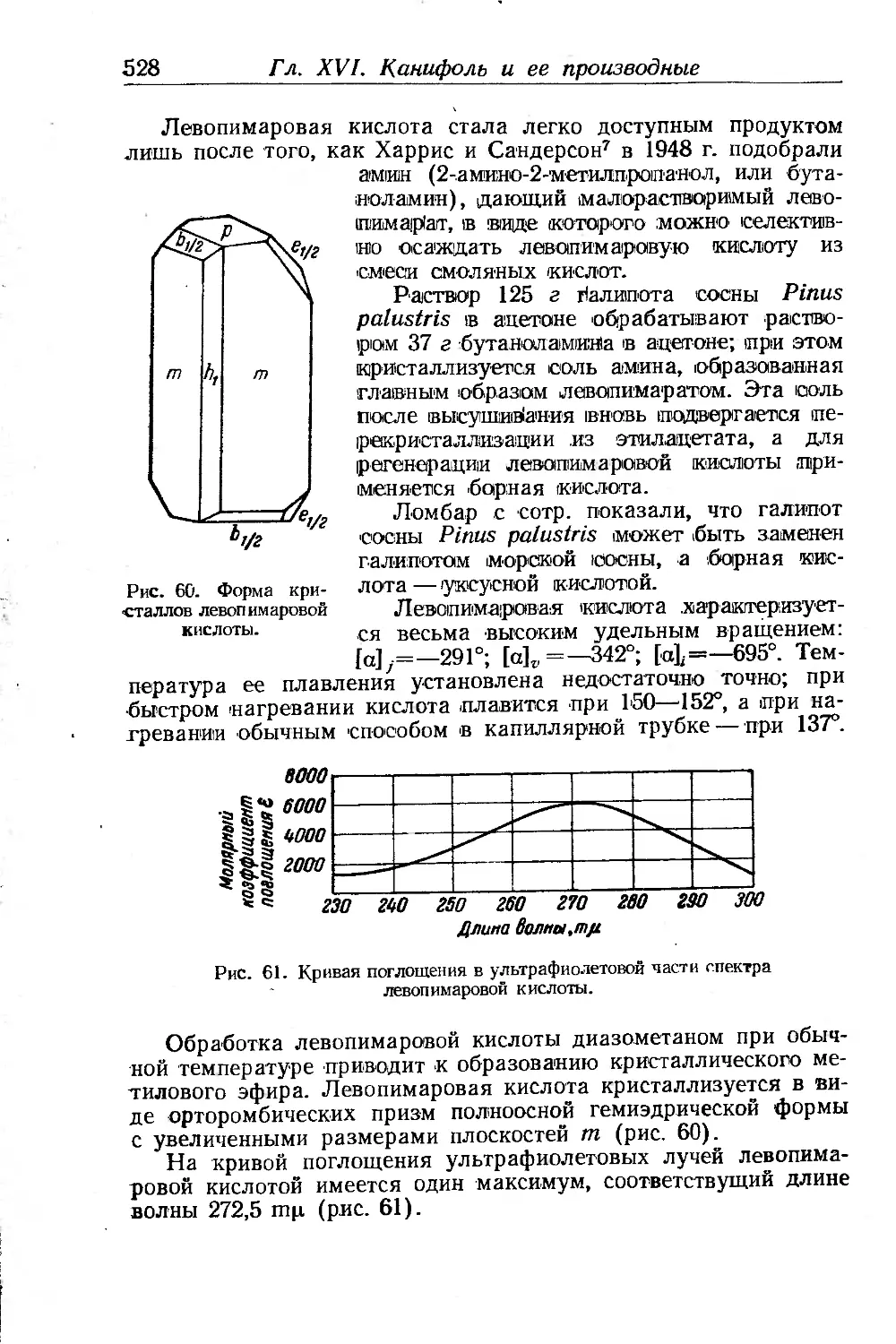
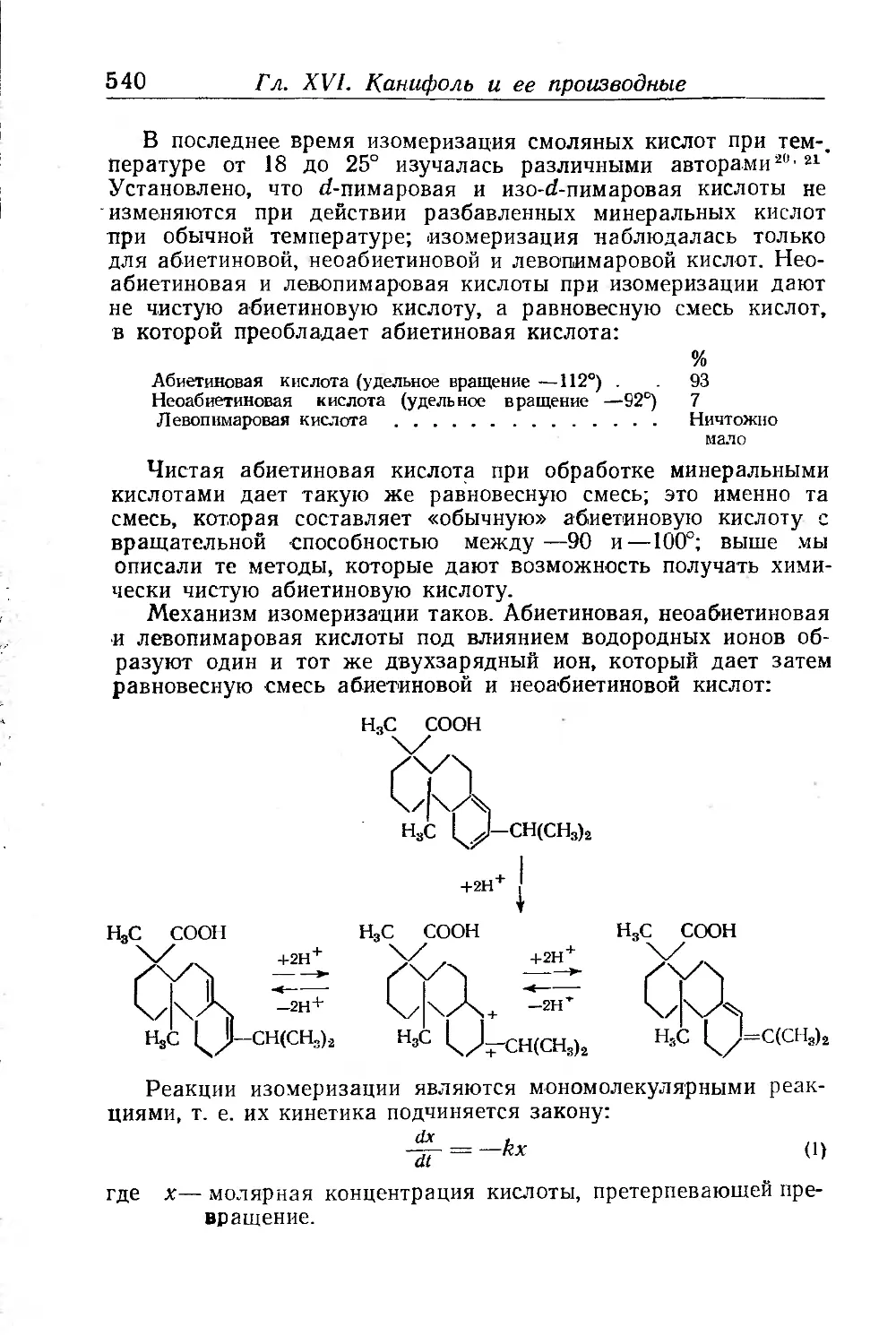
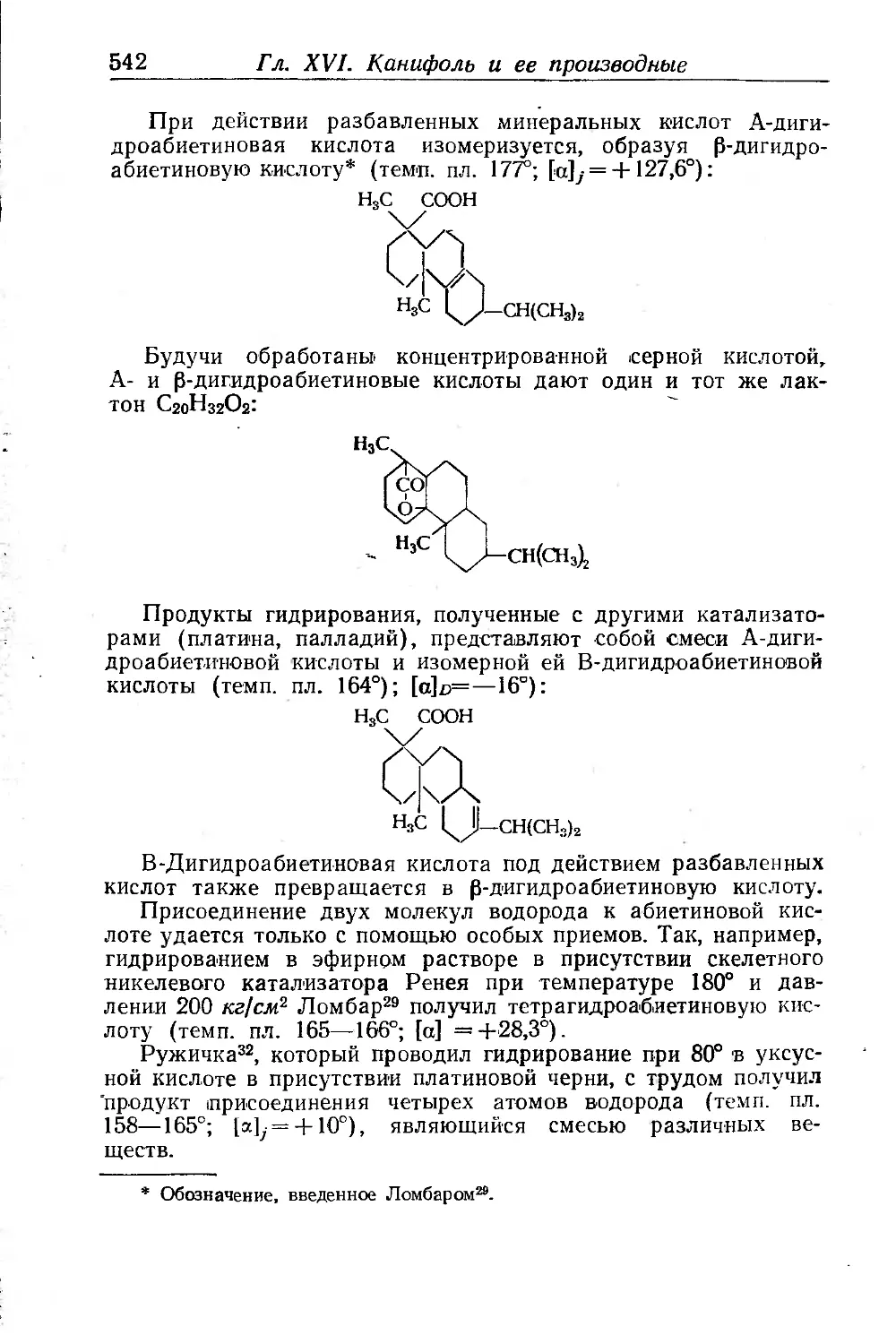
/





































































































































































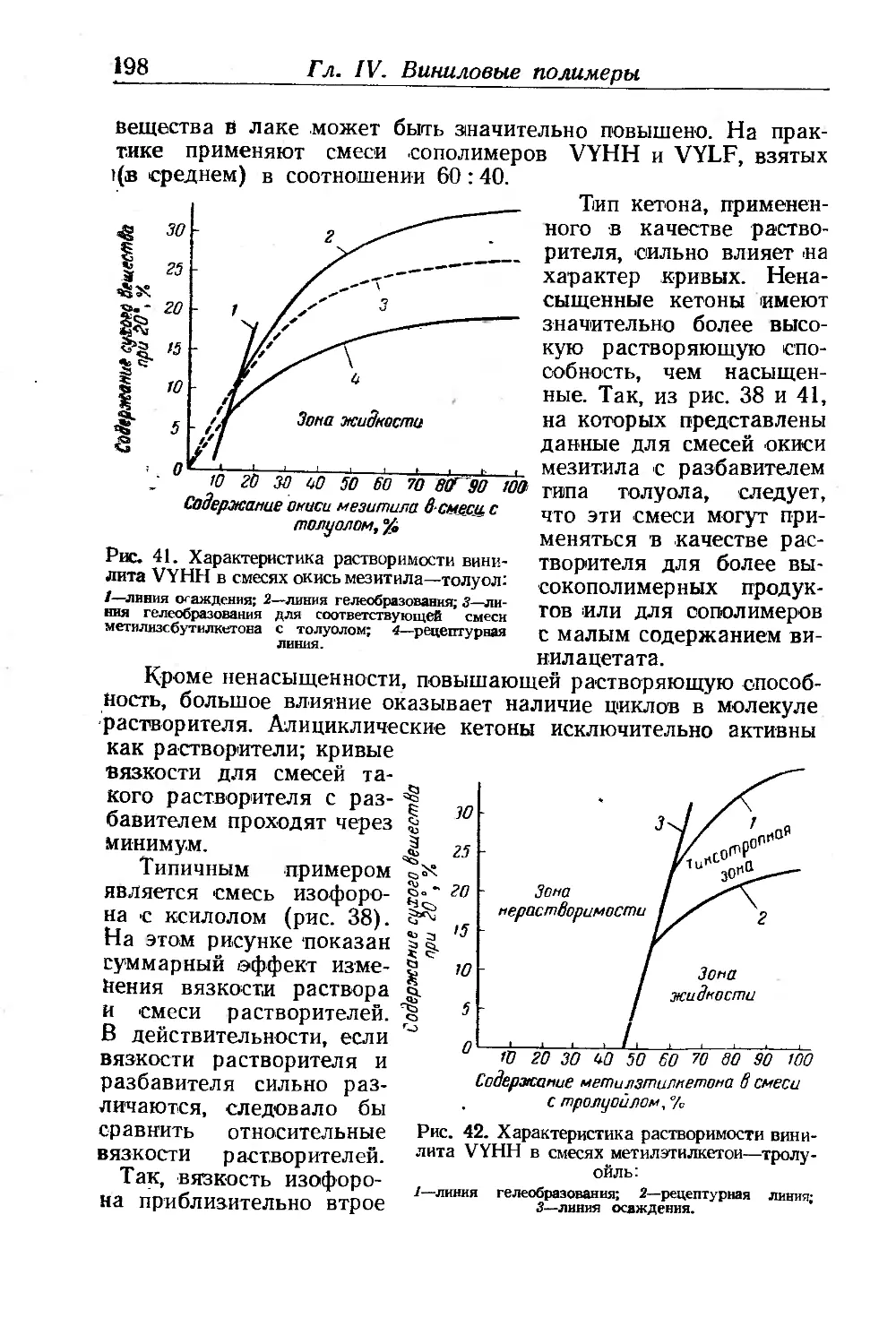
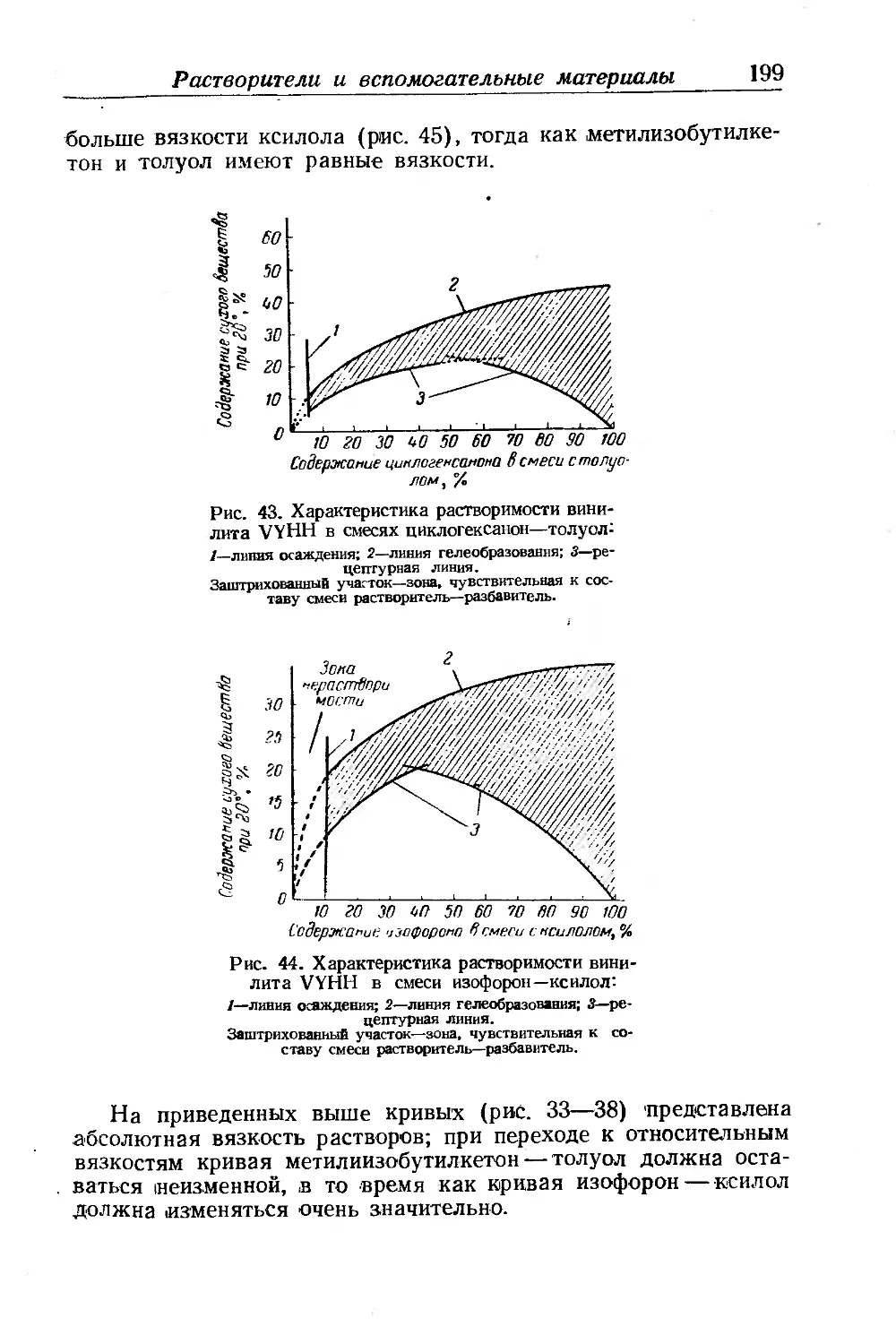
















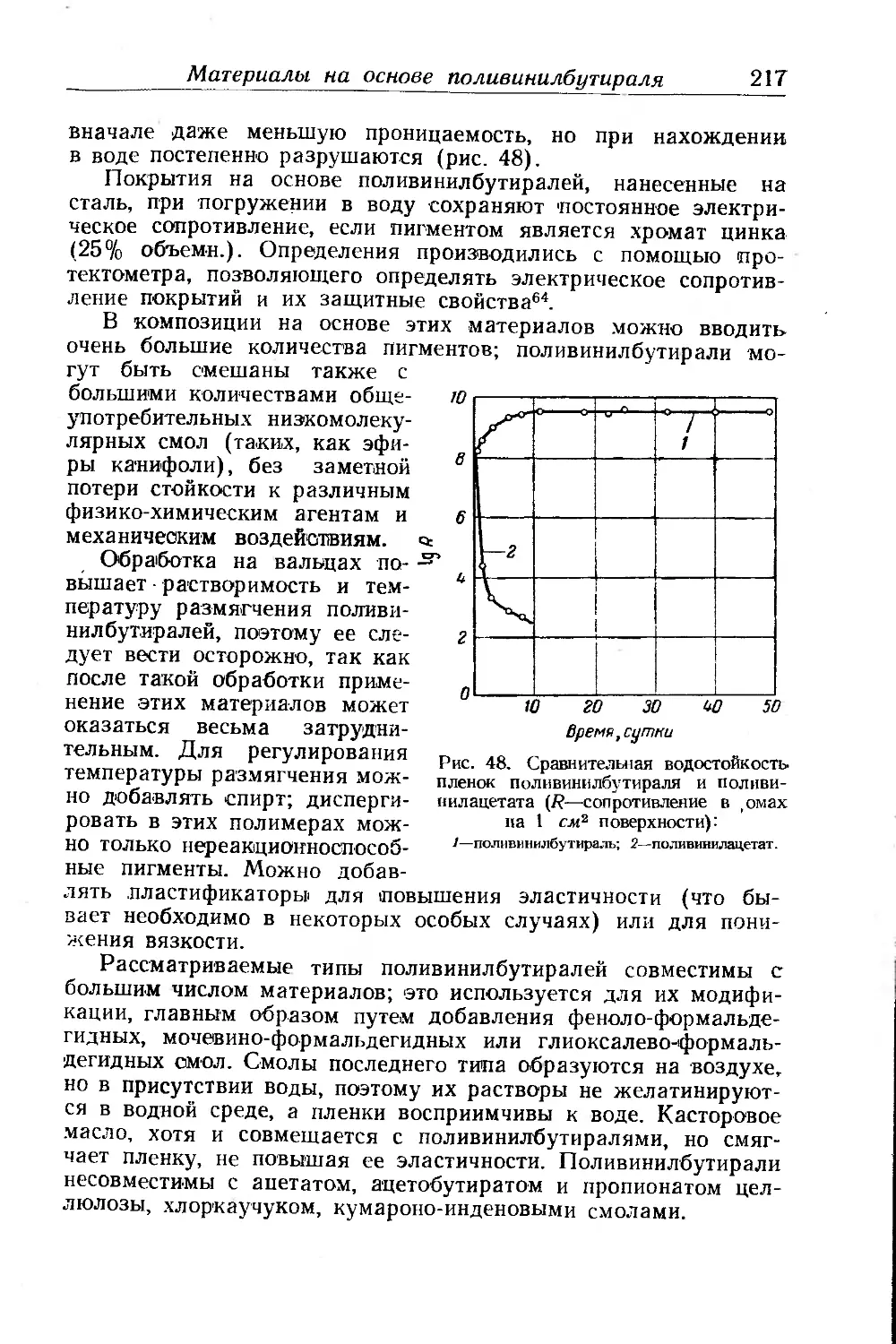


















































































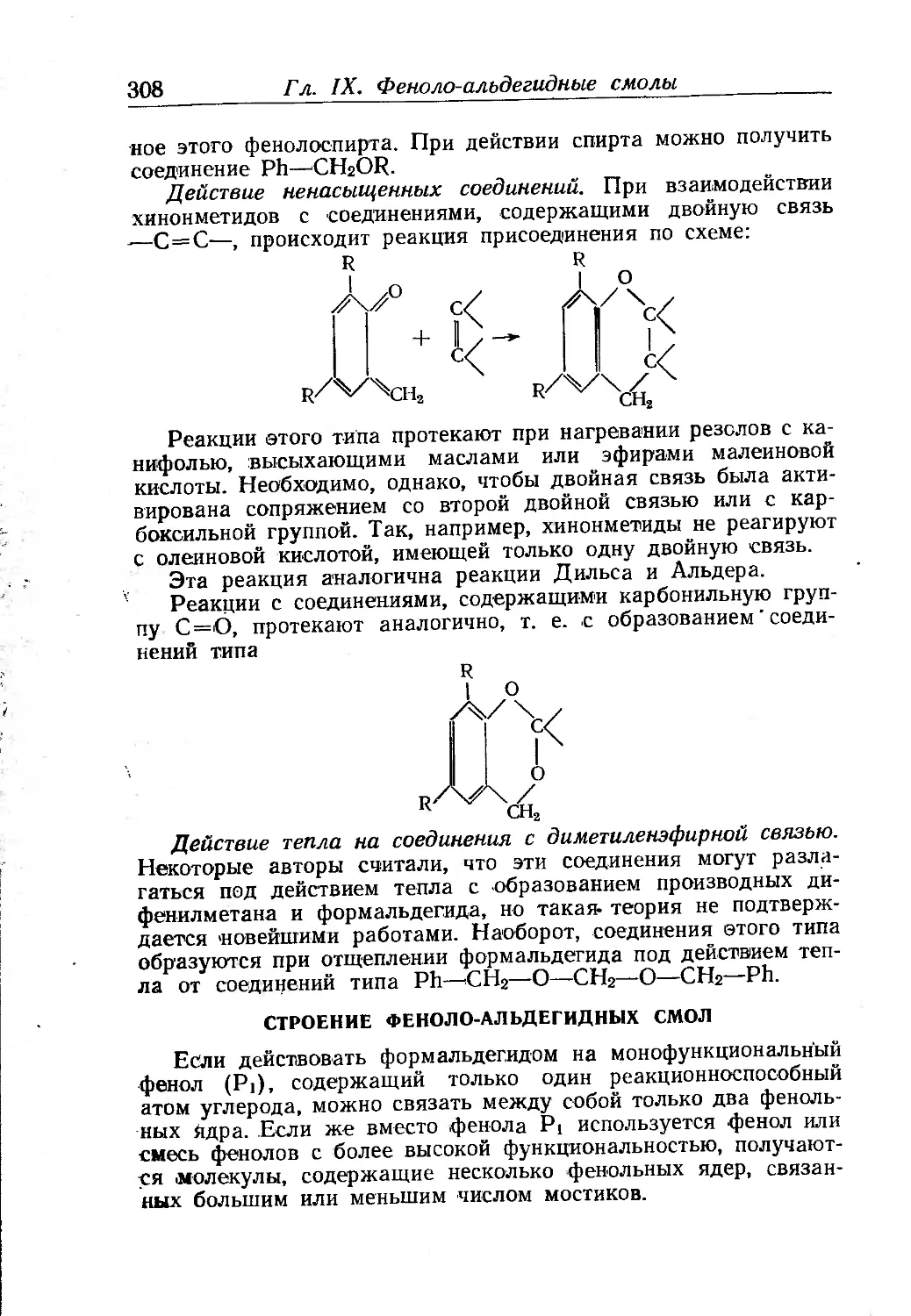


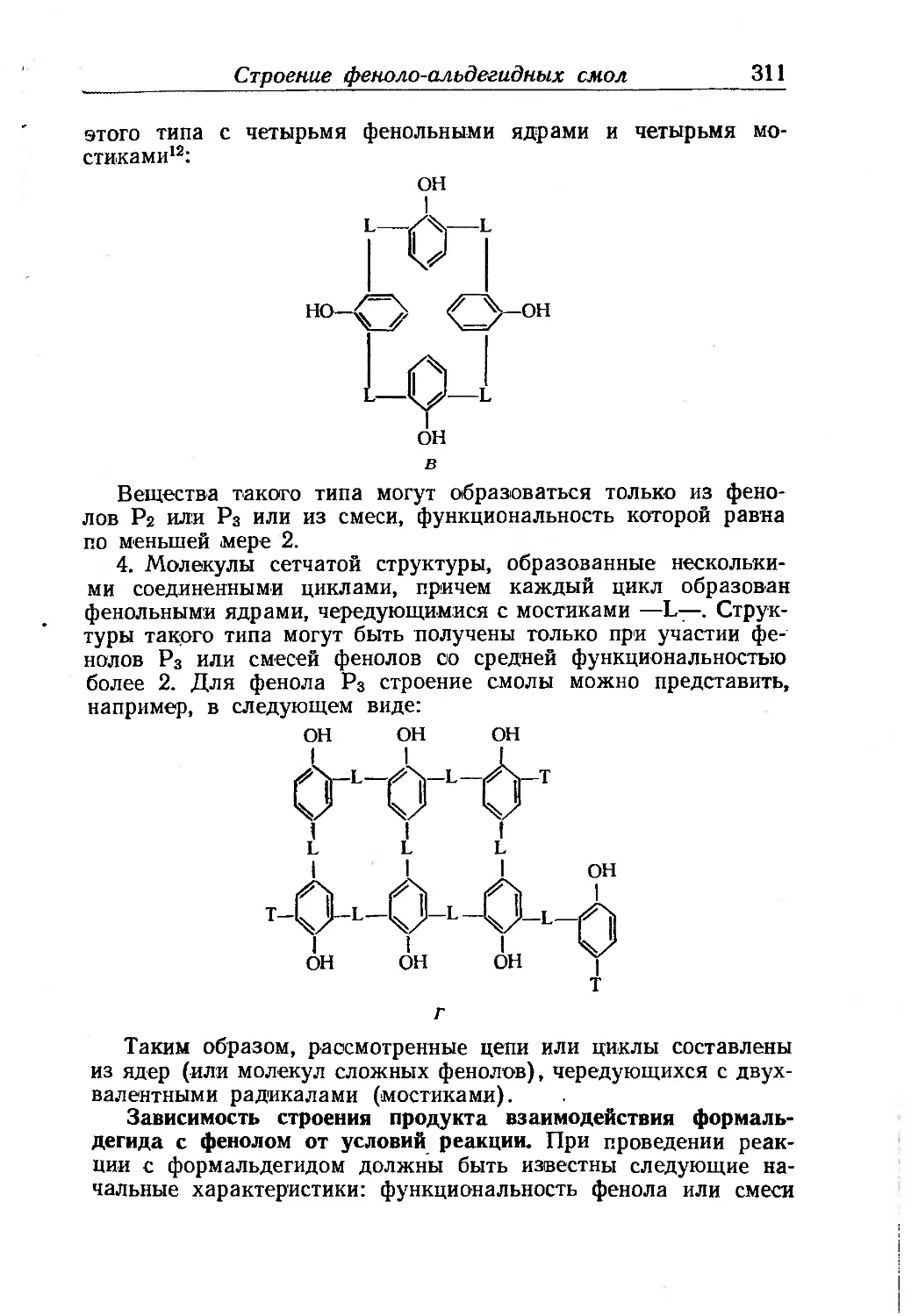












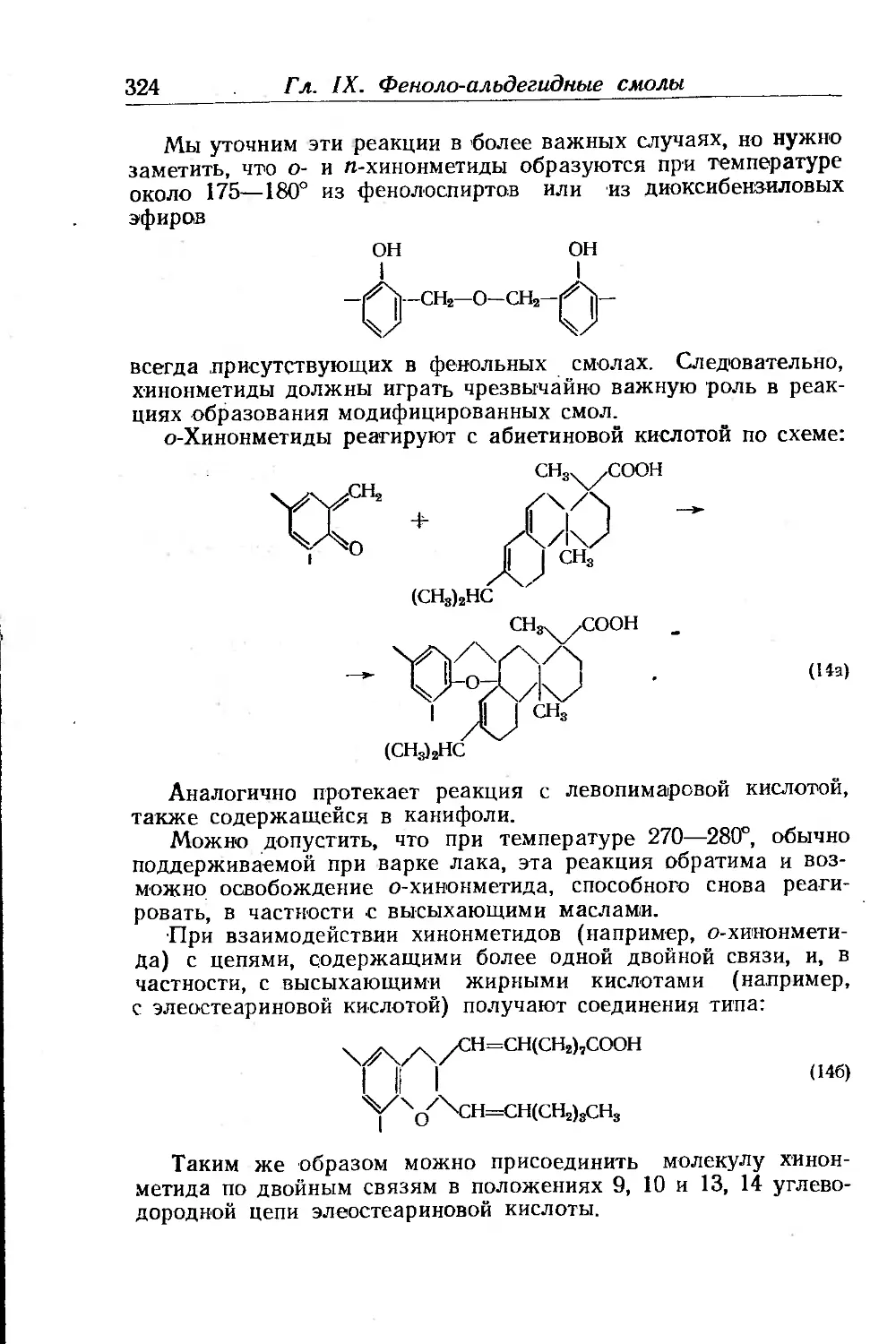






































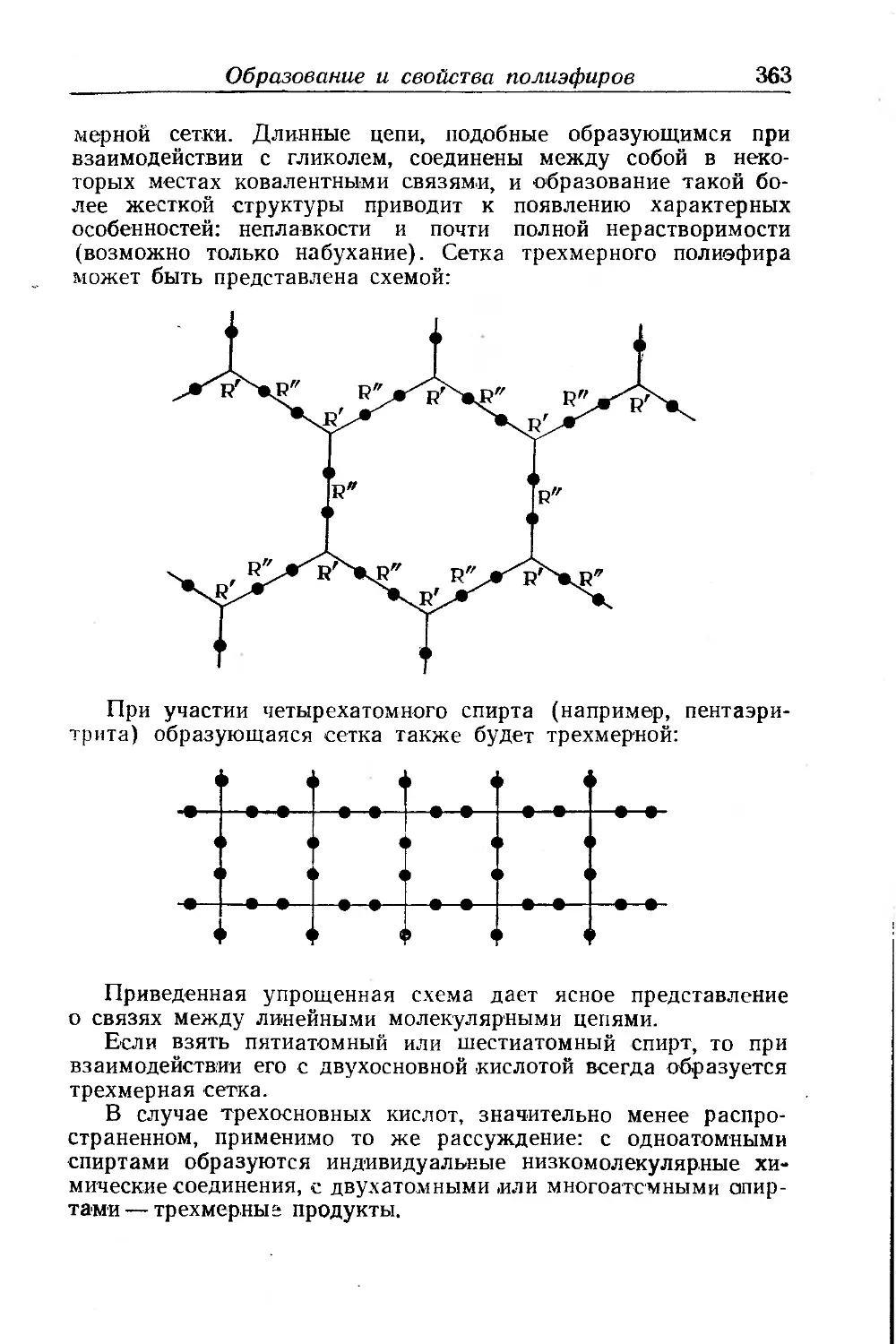





































































































































































































Текст
Г. ШАМПЕТЬЕ, Г. РАБАТЭ
!/ С Г‘,
hJ- /9*
й
i
'f4
ХИМИЯ ЛАКОВ, КРАСОК
И ПИГМЕНТОВ
&ЛСУб~
ТОМ I
ПЕРЕВОД С ФРАНЦУЗСКОГО
Н. П. Аграненко, Ю. Т. Беловицкой, Э. М. Левиной
и Ю. А. Холмогорцевой
Под редакцией А. А. Беловицкого
ГОСУДАРСТВЕННОЕ НАУЧНО-ТЕХНИЧЕСКОЕ ИЗДАТЕЛЬСТВО
ХИМИЧЕСКОЙ ЛИТЕРАТУРЫ
Москва • 1960
32—5—3
661-7
Ш-19
Книга представляет собой фундаментальную монографию
по лакам и краскам. В первом томе, посвященном пленкообра-
зующим веществам, содержатся сведения о многочисленных син-
тетических лаковых смолах (поливиниловых, полиакриловых,
полиэфирных, полиамидных, полиуретановых, эпоксидных, сили-
коновых и др.), а также об эфирах-целлюлозы, пленкообразую-
щих на основе каучука, высыхающих маслах.
Книга предназначена для работников различных отраслей
промышленности, изготовляющих и применяющих лаки и краски.
Она может быть полезна также для студентов вузов и техни-
кумов.
CHIMIE DES PEINTURES VERNIS ЕТ PIGMENTS
publiee sous la direction de
G. Champetier H. Rabate
Professeur a la Sorbonne, directeur des Fondateur—Redacteur et chef des'revues
etudes a 1‘ Ecole sup€rieure de Physique Peintures Pigments—Vernis et Travaux
et Chinlie de la Ville de Paris de Peintures
Secretaire general de la Redaction
J. L. Rabate
TOME I
Paris
Dunod
92, Rue Bonaparte (6е)
1956
СОДЕРЖАНИЕ
Предисловие........................................................ 8
Глава 1. Химия высокомолекулярных соединений и пленкообразующих
веществ. Г. Шампетье.............................................. 9
Синтез высокополимеров................................ 11
Пространственное строение макромолекул.................... 17
Свойства высокополимеров и способность к пленкообразованию 22
Глава II. Высыхающие масла. Ж. Пти , ............................. 25
Состав высыхающих масел....................................... 25
Важнейшие жирные кислоты и их производные ....... 26
Второстепенные компоненты высыхающих масел................ 33
Высыхание масел......................................... . 33
Действие кислорода иа двойные связи....................... 35
Возможные реакции пленкообразован и я . . 42
Физико-химические гипотезы плеикообразоваиия.............. 47
Старение пленок высыхающих масел ......... 48
Обработка высыхающих масел........... . . 52
Термическая полимеризация масел........................... 52
Окислительная полимеризация масел........................ 65
Дегидратация касторового масла............ . . , 66
Модификация масел стиролом................................ 69
Модификация масел малеиновым ангидридом . ... 73
Применение кислот высыхающих масел ....... . . 77
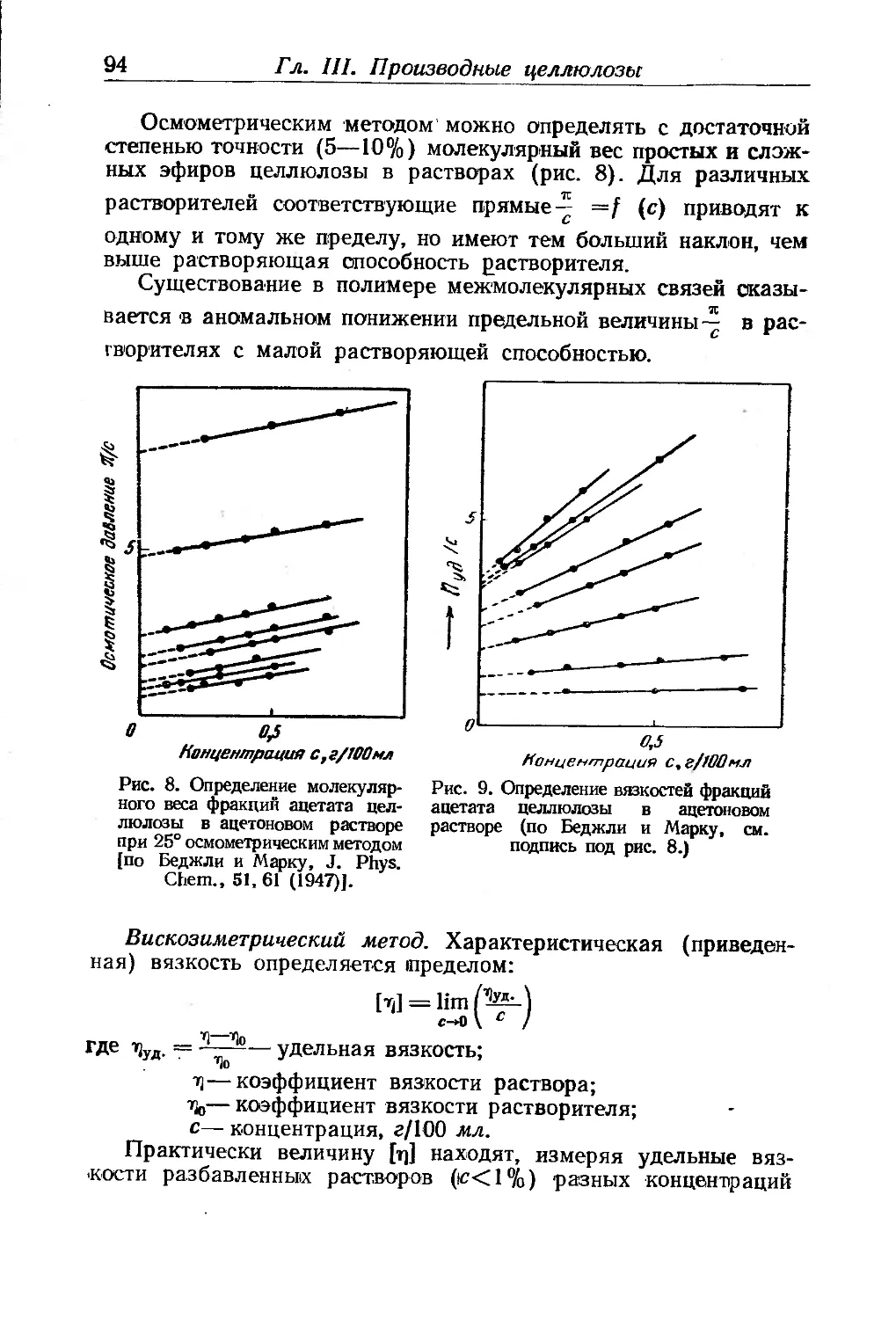
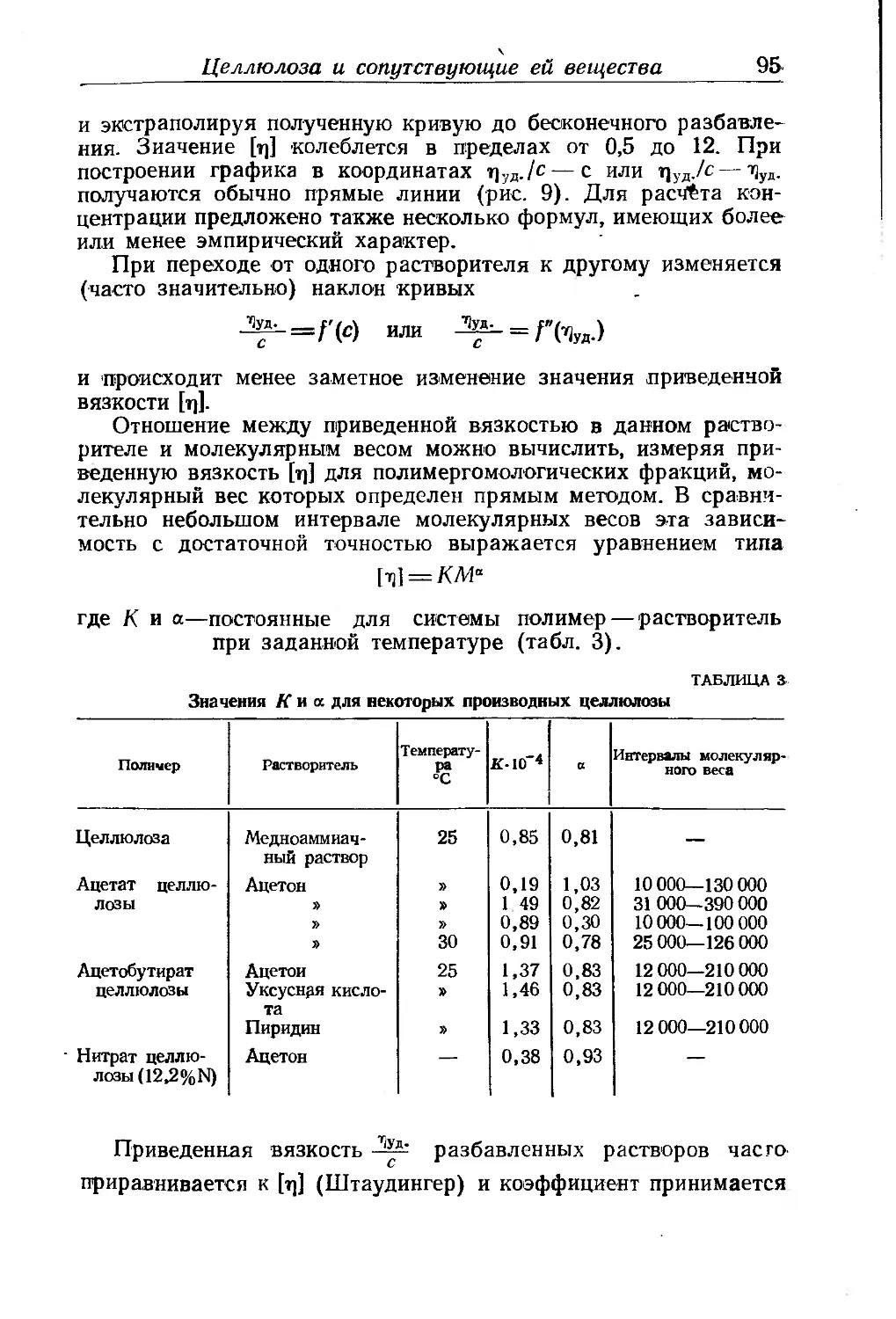
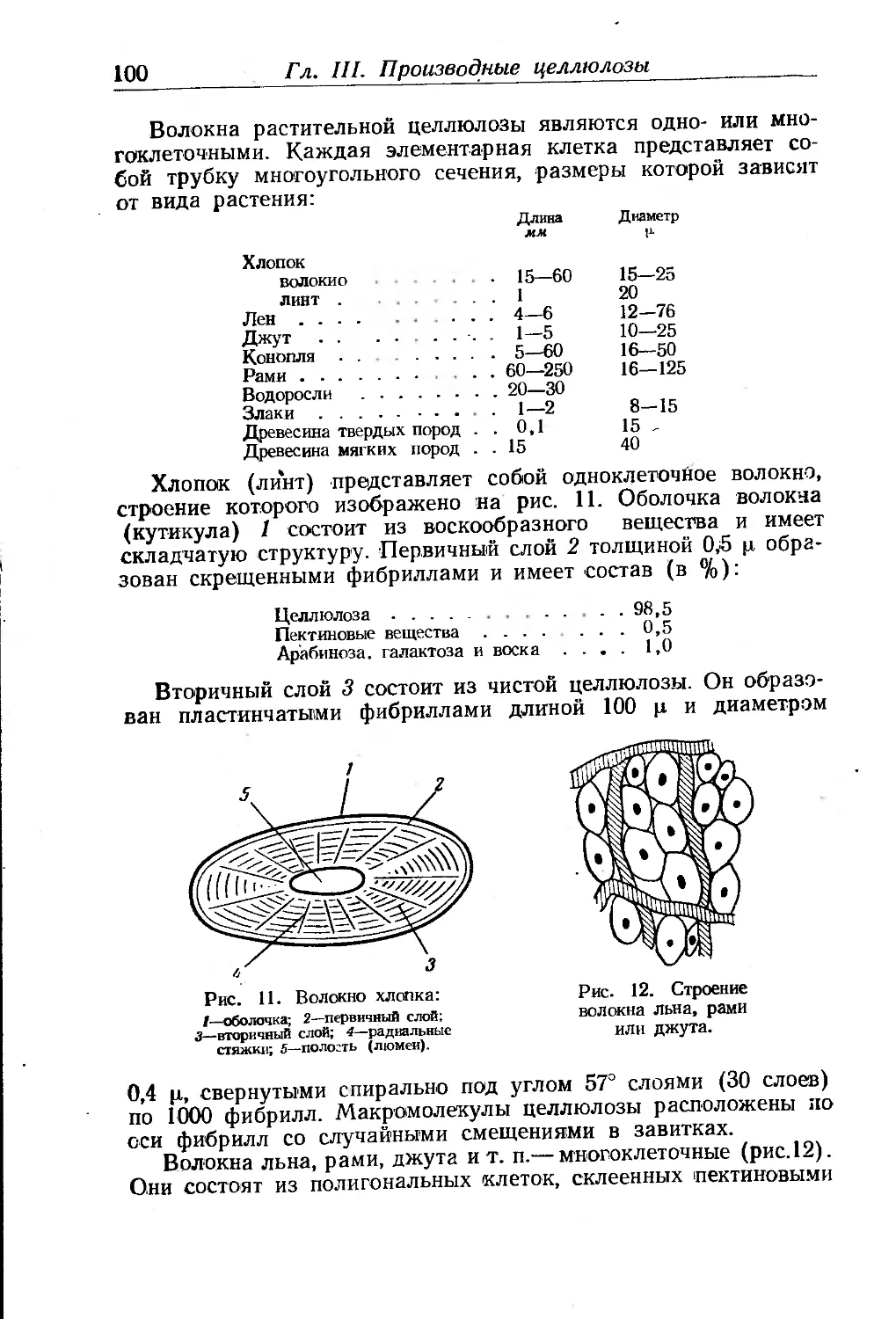
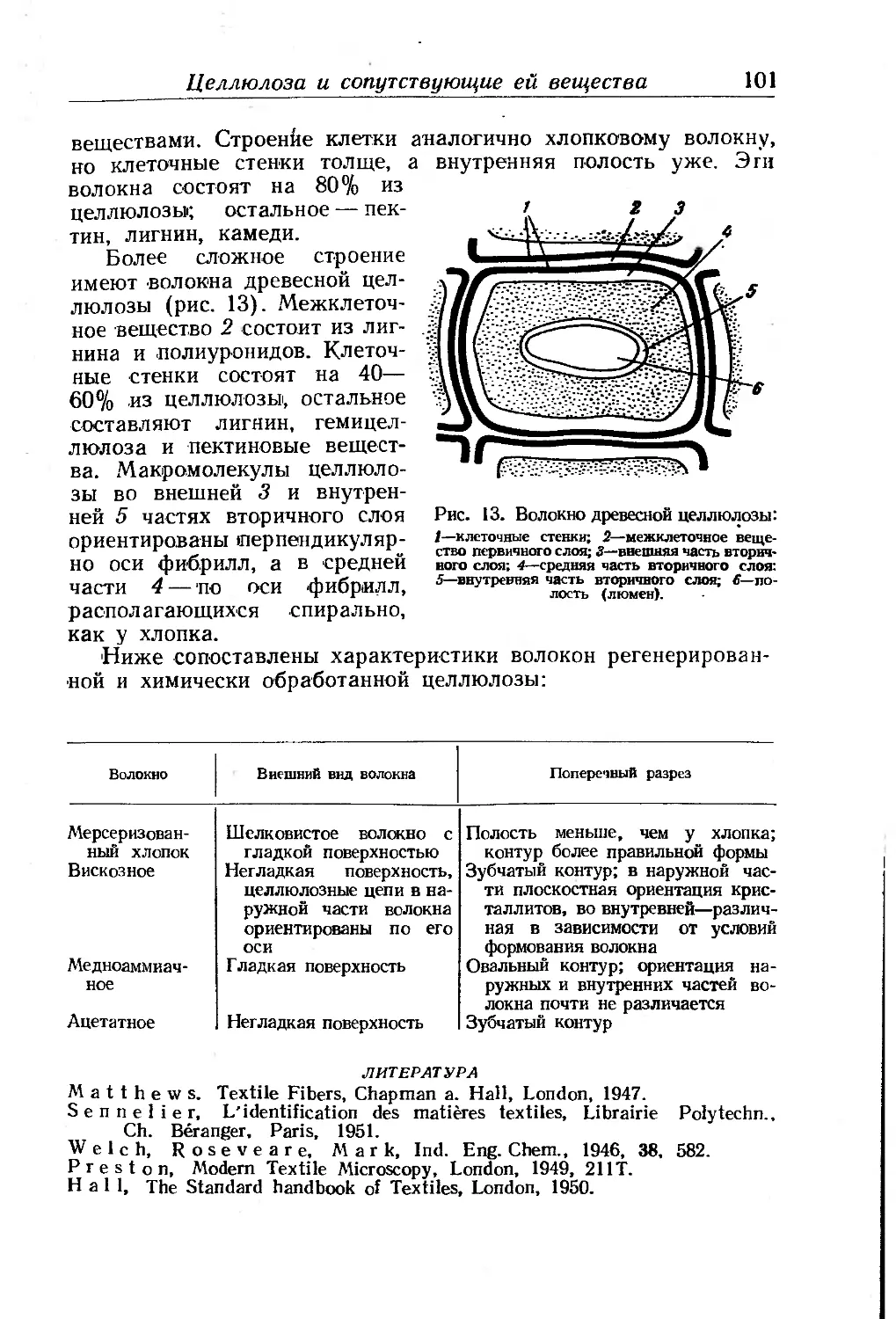
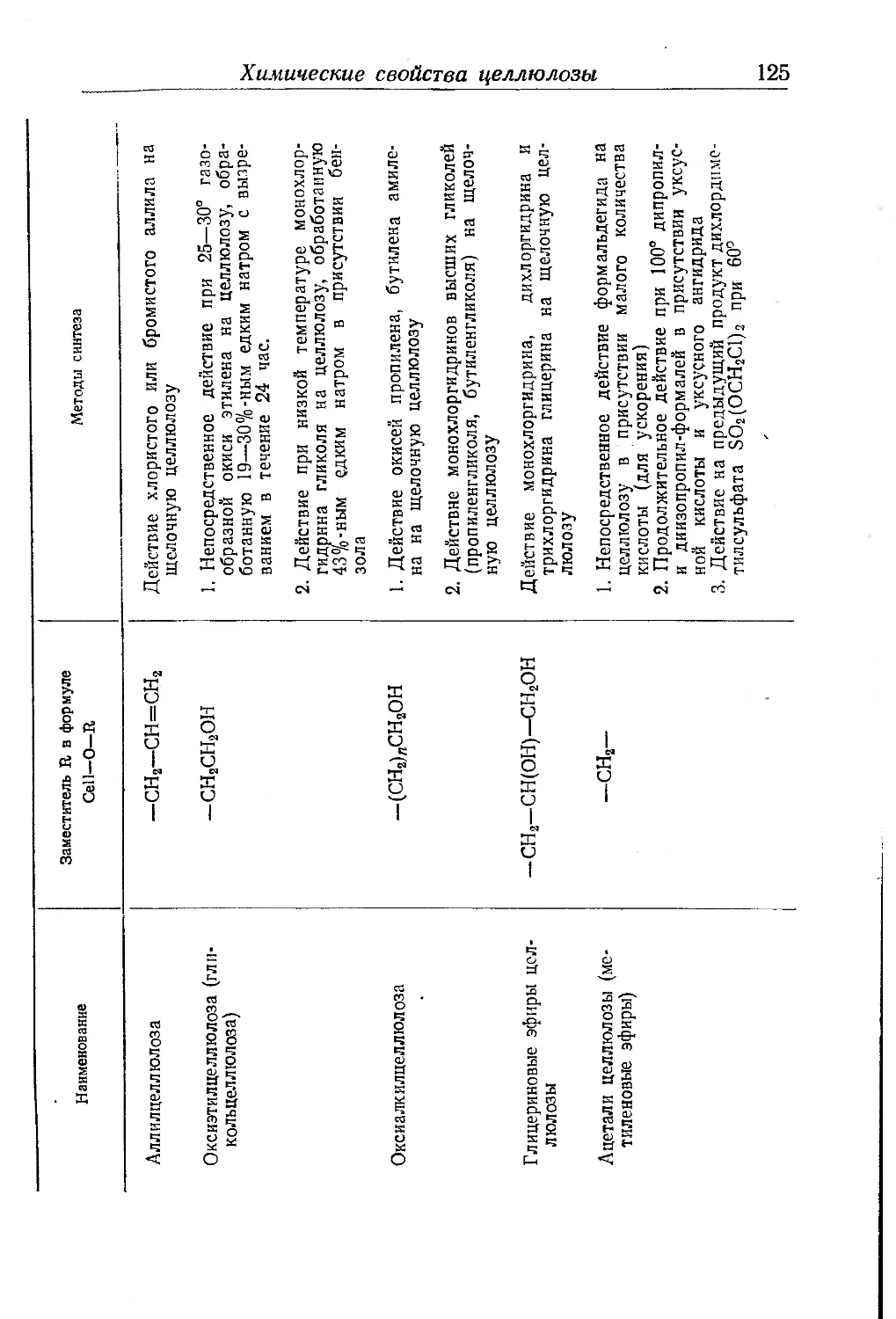
Глава III. Производные целлюлозы. П- Клеман ... . . 85
Целлюлоза и сопутствующие ей вещества............... .... 85
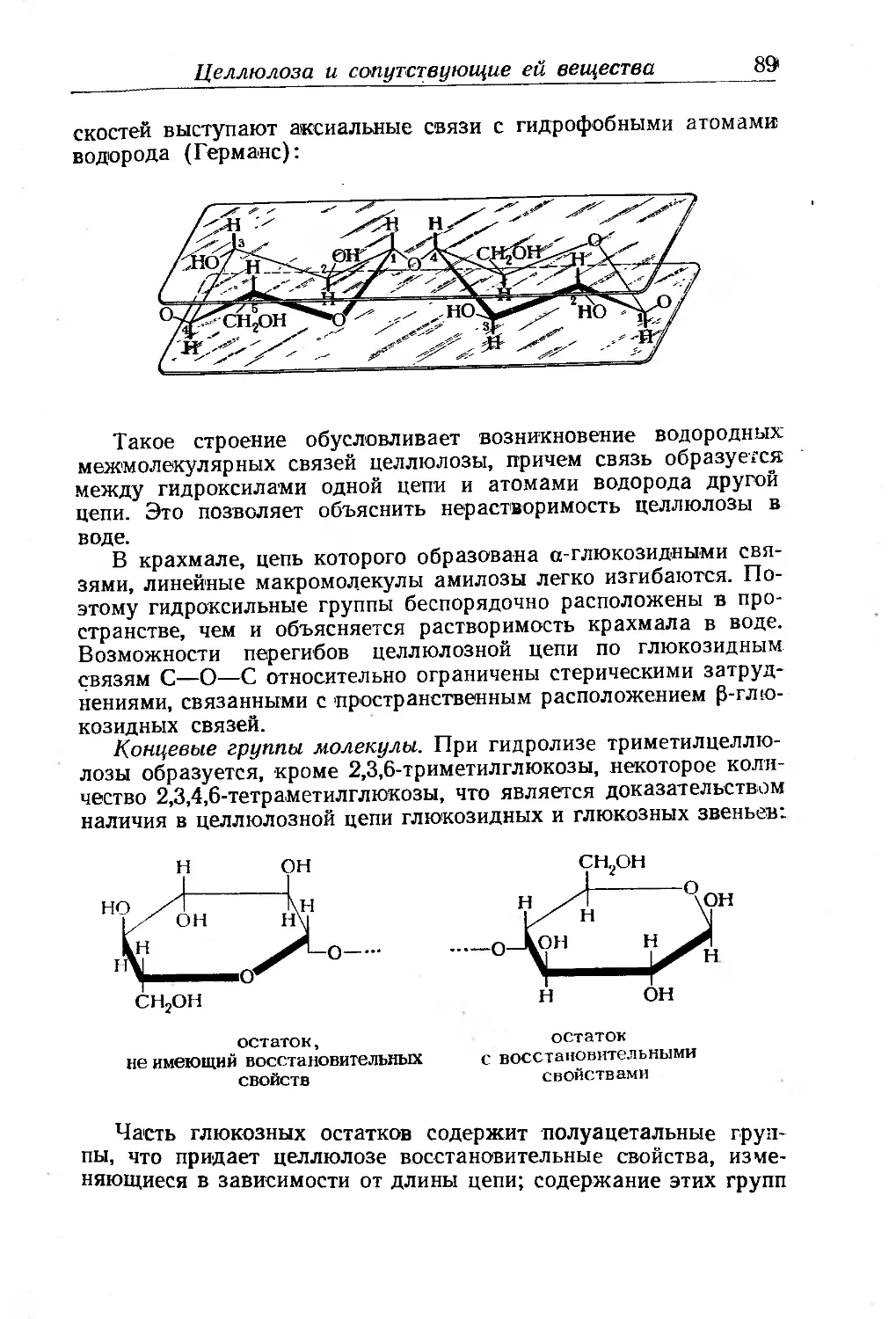
Химическое строение целлюлозы...................... 86
Молекулярный вес'целлюлозы......................... 92
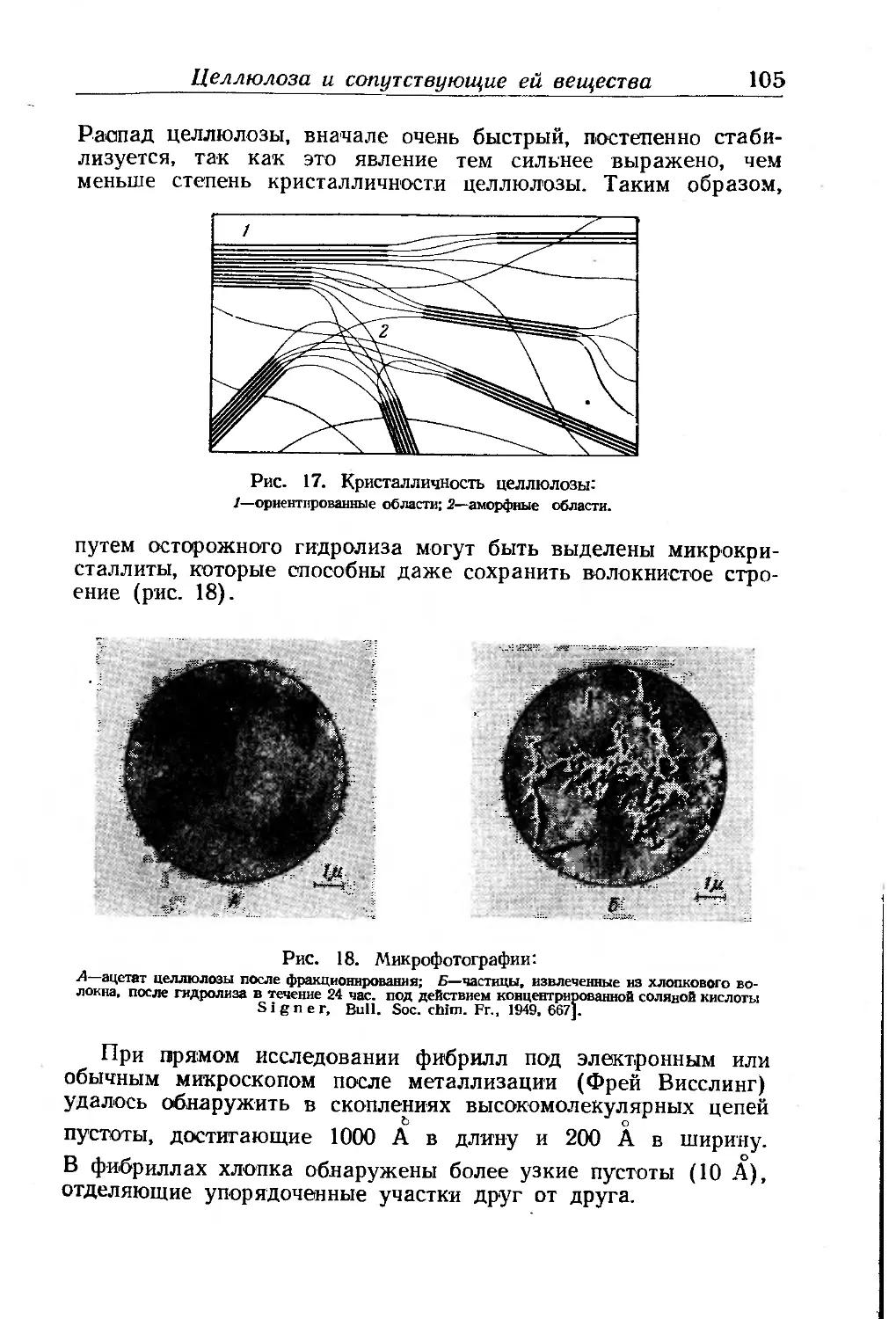
Полидисперсиость целлюлозы............................... 98
Микроскопическое исследование структуры целлюлозы ... 99
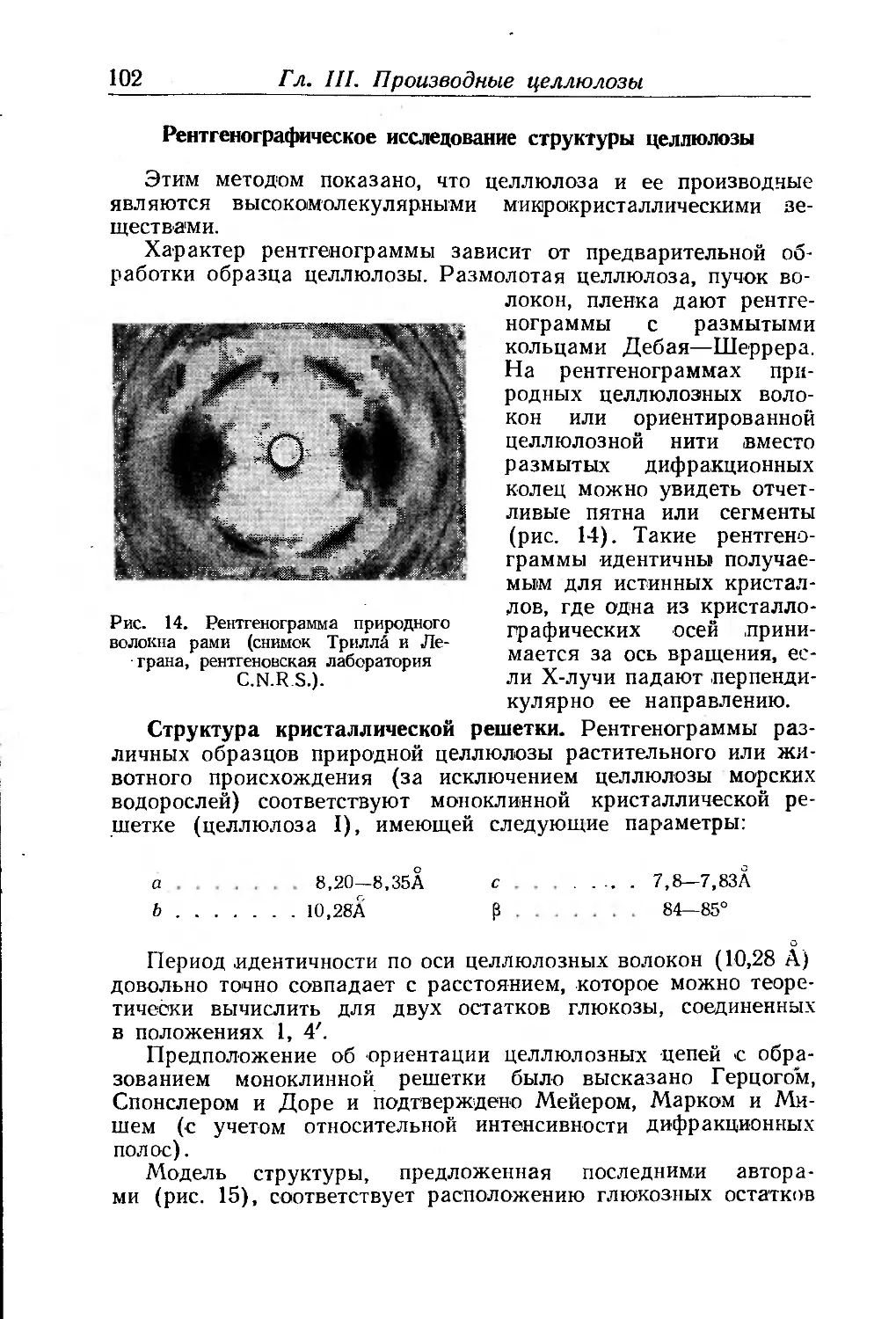
Рентгенографическое исследование структуры целлюлозы . . 102
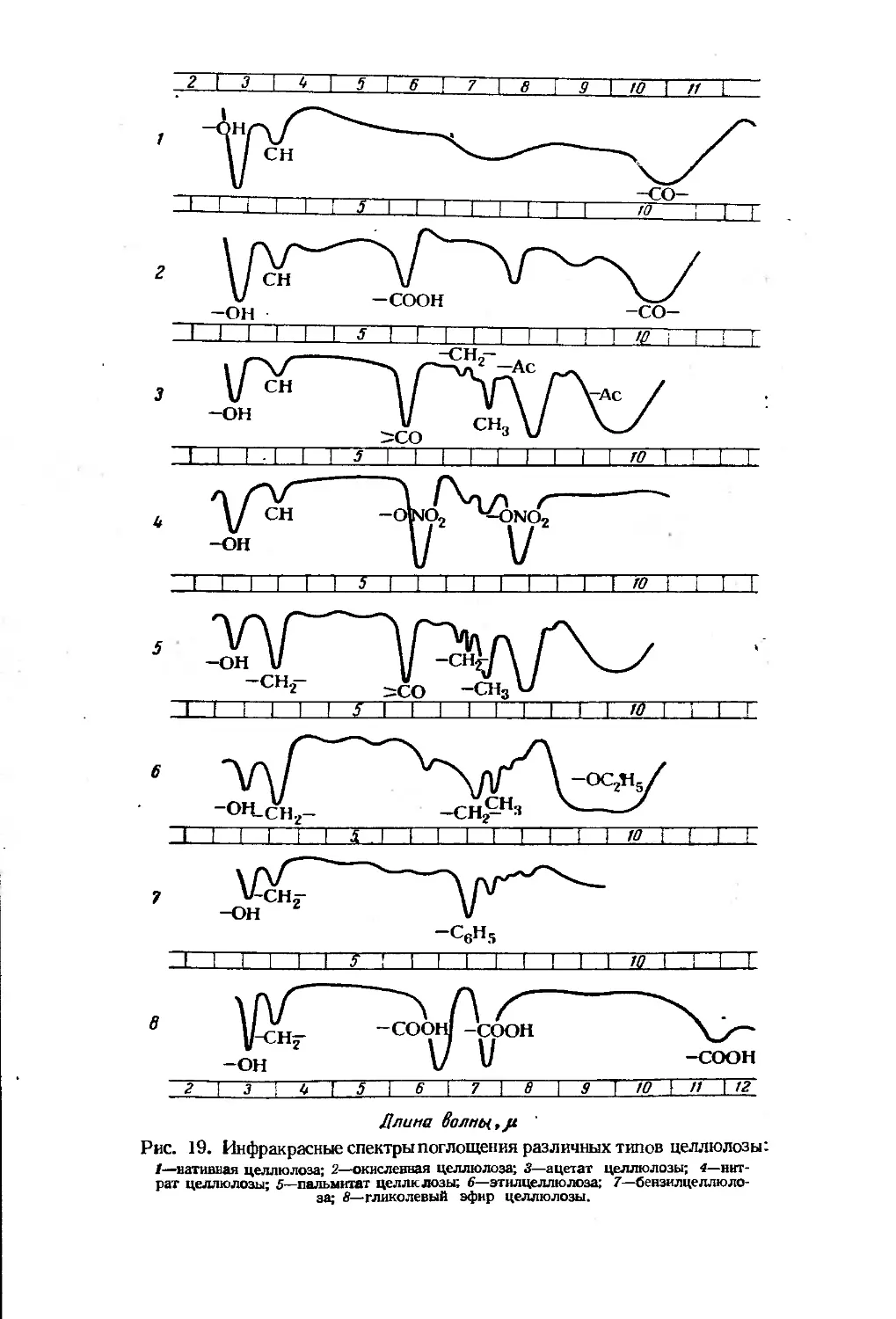
Исследование структуры целлюлозы методом спектроскопии в
инфракрасной области.................................... 10?
4
Содержание
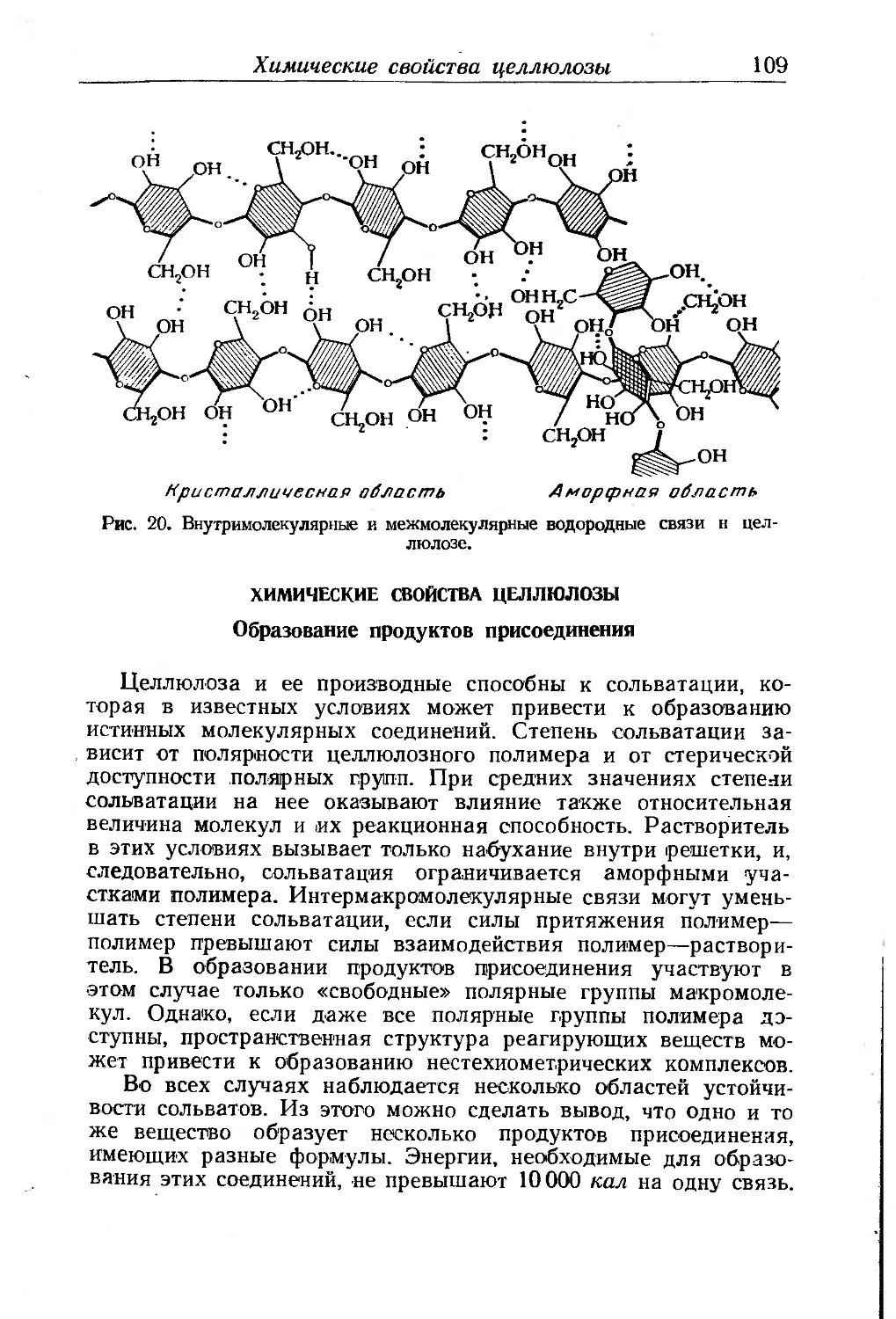
Химические свойства целлюлозы................................ 109
Образование продуктов присоединения . . ..........109
Реакции замещения..................................... . 112
Реакции окисления ............................... . . 130
Реакции деструкции.................................. ... 133
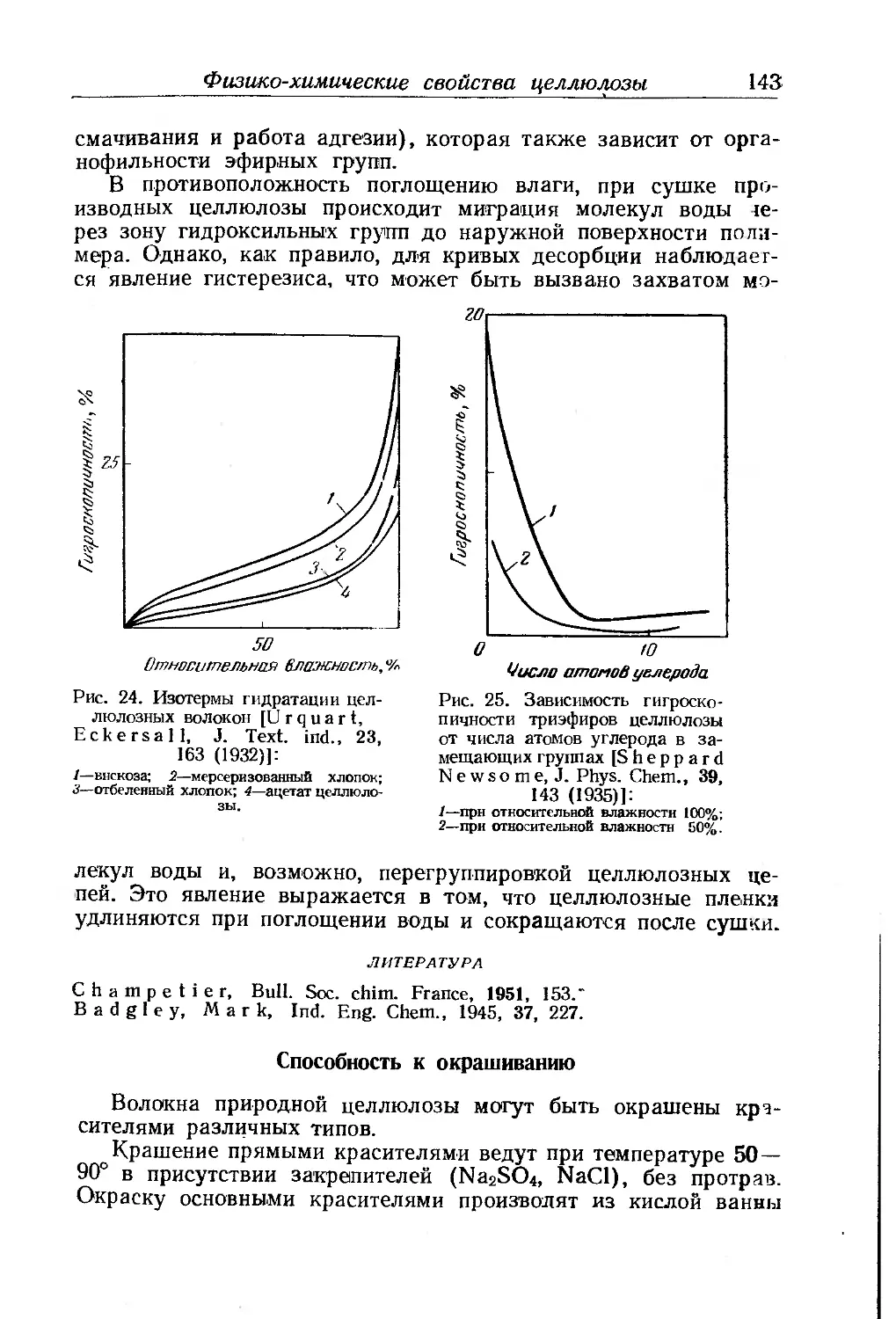
Физико-химические свойства целлюлозы . ... 135
Набухание и растворение ............ . . . 135
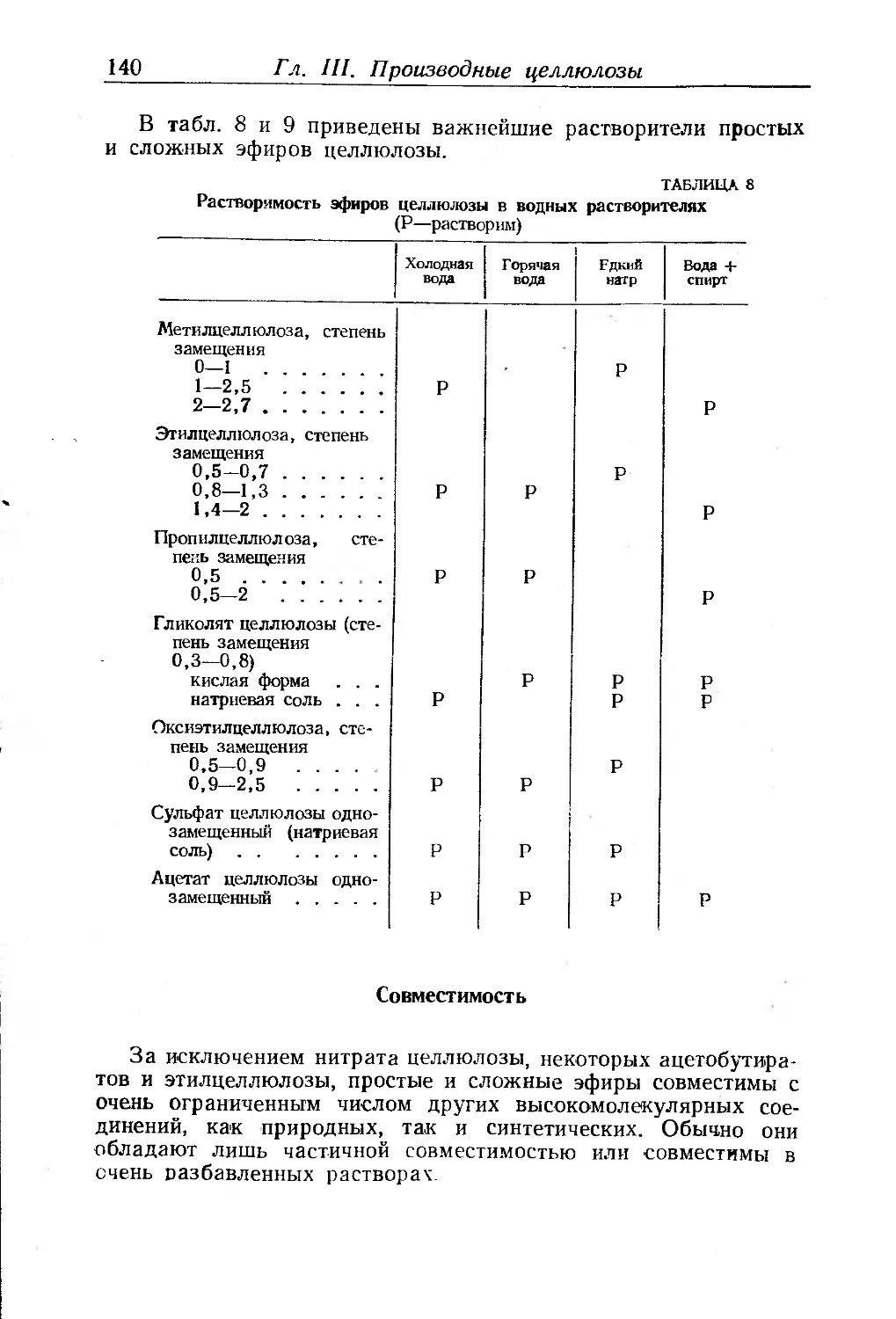
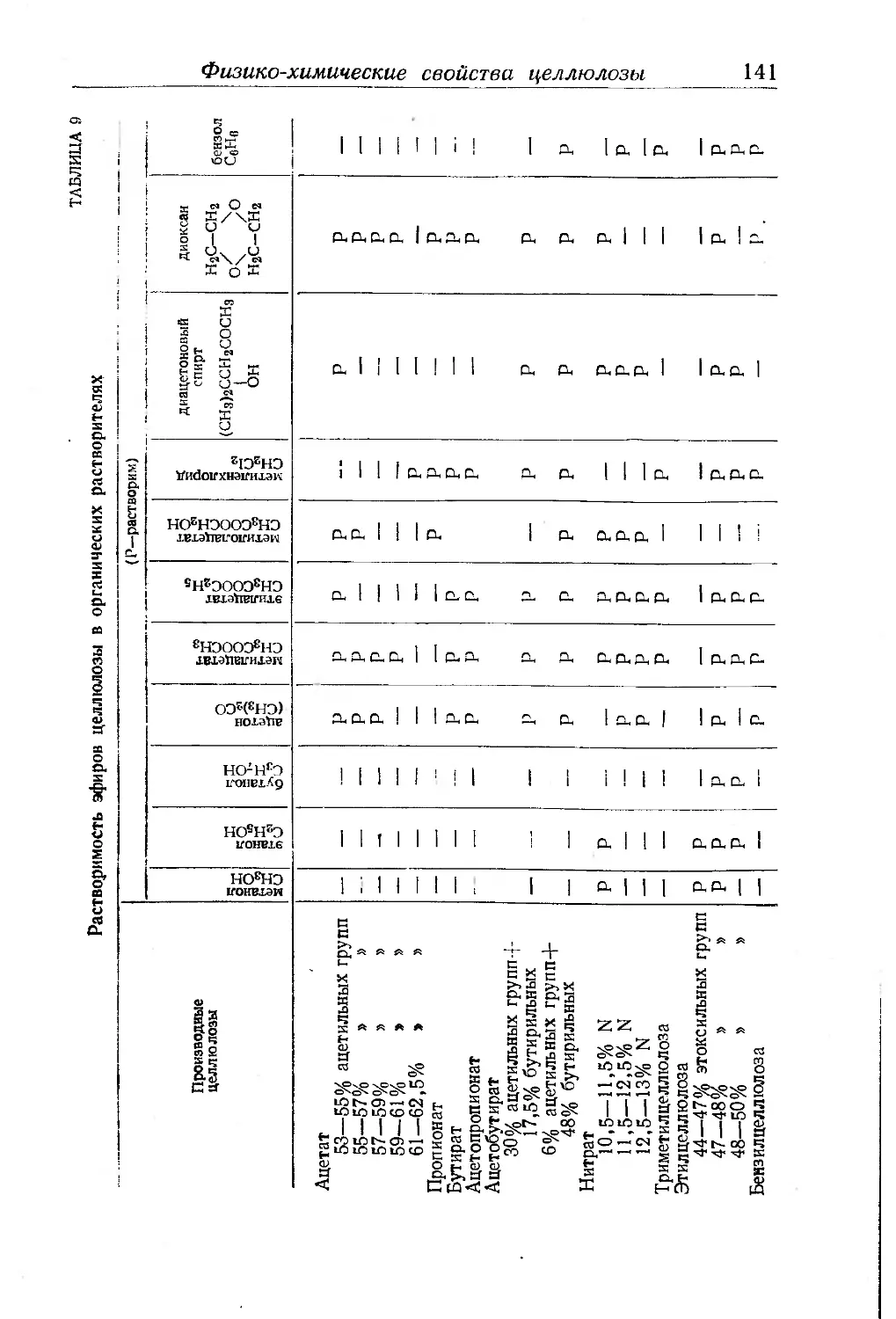
Совместимость............................ . . 140
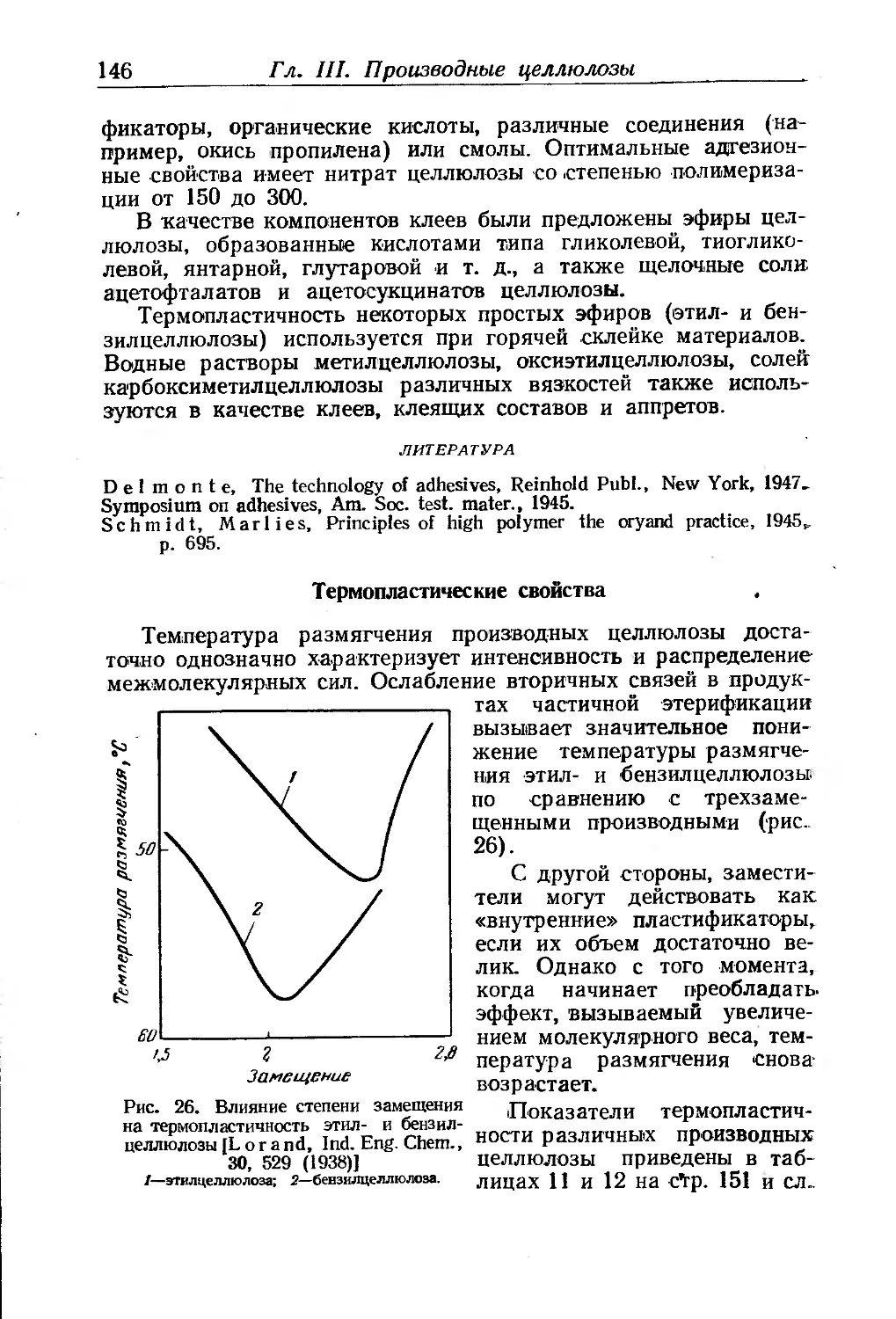
Гигроскопичность 142
Способность к окрашиванию . . ... 143
Адгезионные свойства ............ ............... . . 144
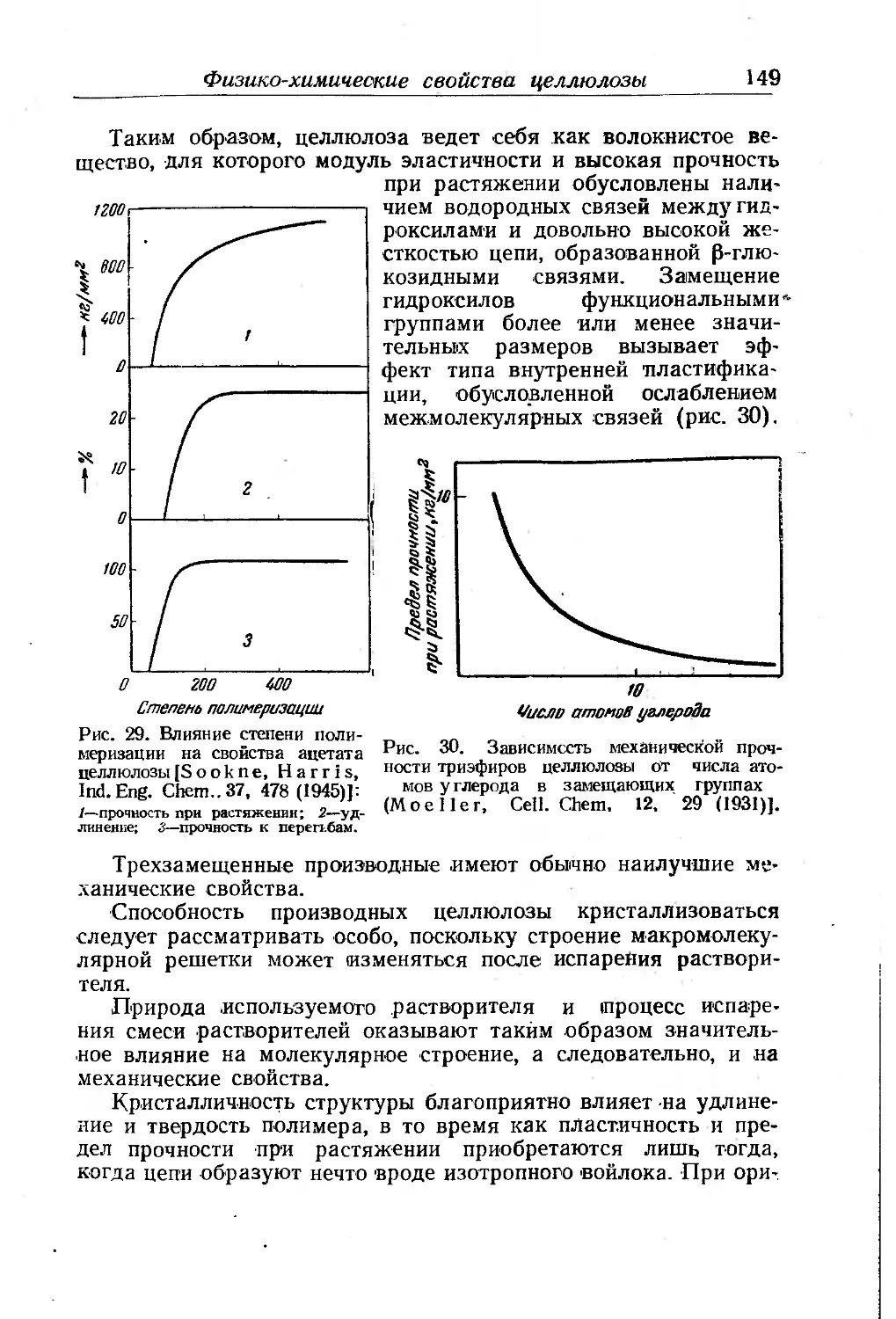
Термопластические свойства . . . . .............. ... 146
Электрические свойства.................................... 147
Оптические свойства .... 147
Механические свойства..................................... 148
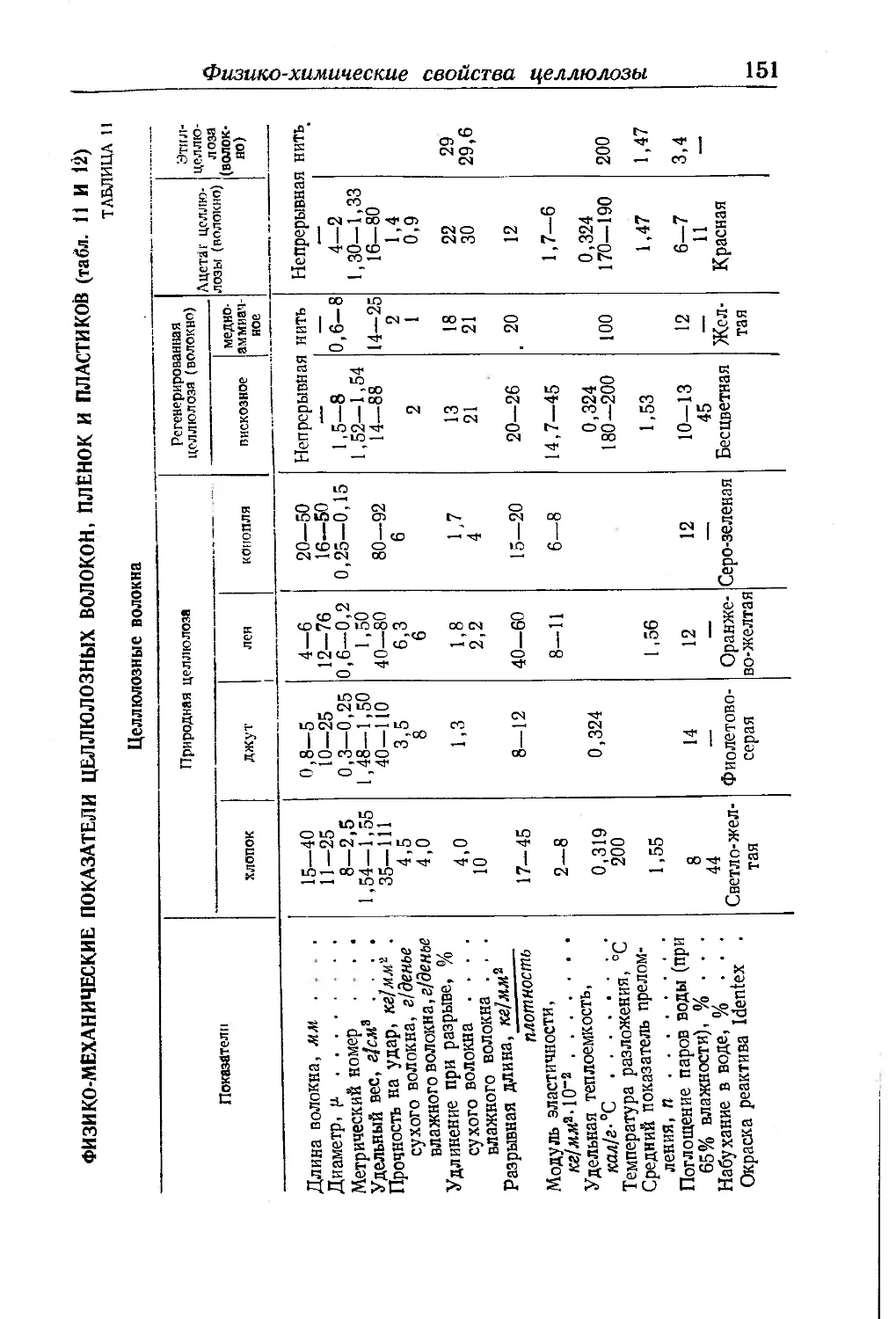
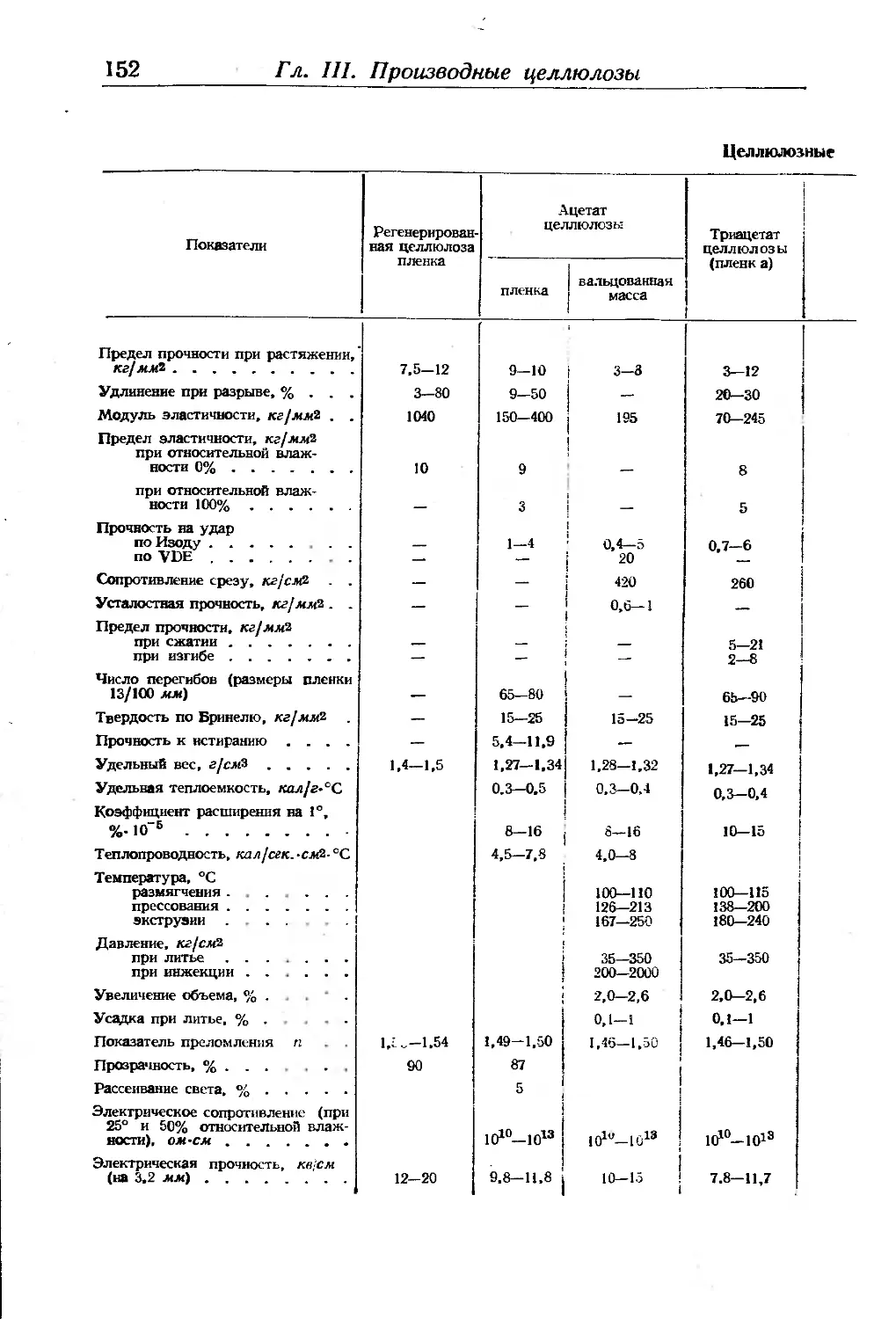
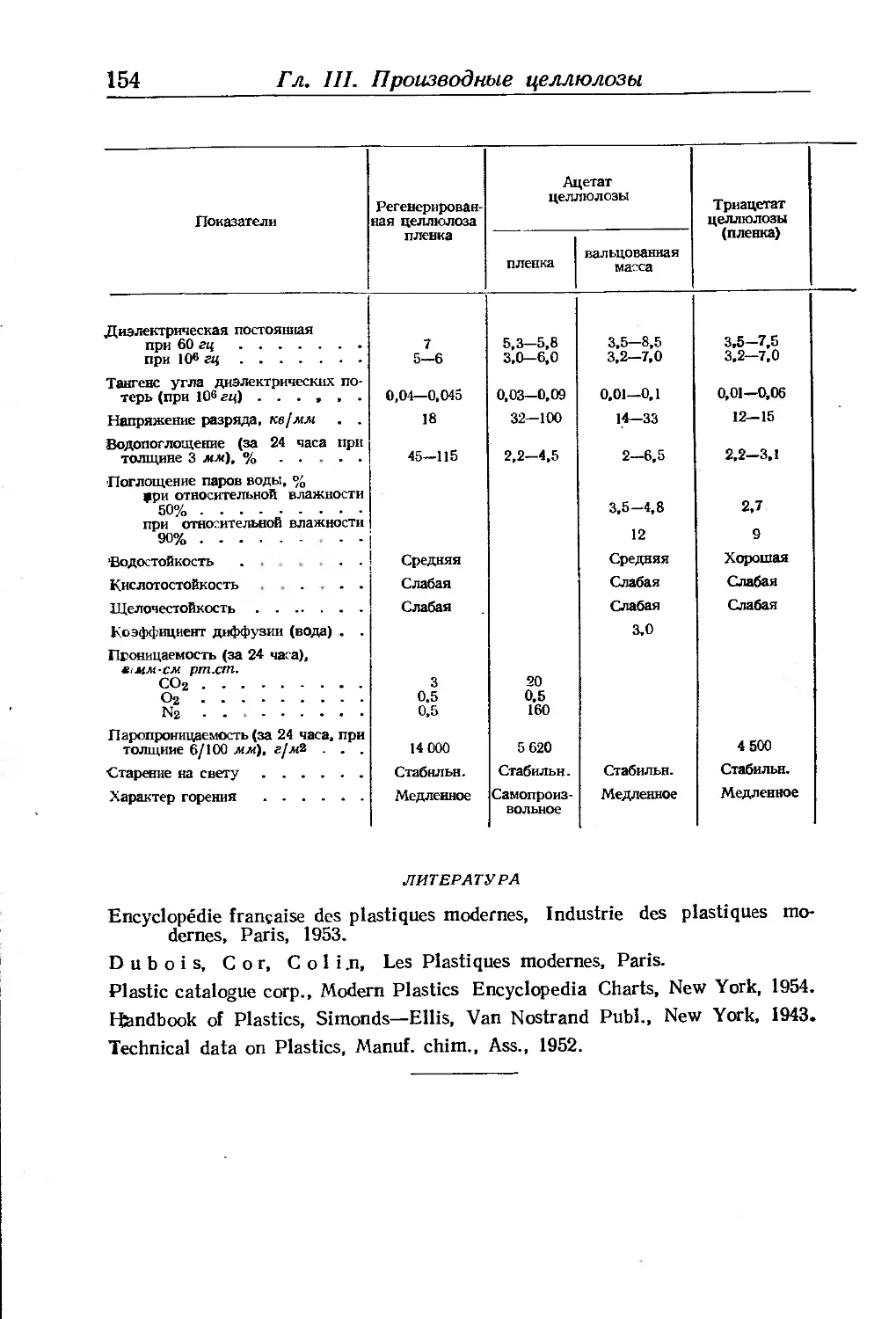
Физико-химические показатели целлюлозных волокон, пленок и плас-
тиков ...................................................... 151
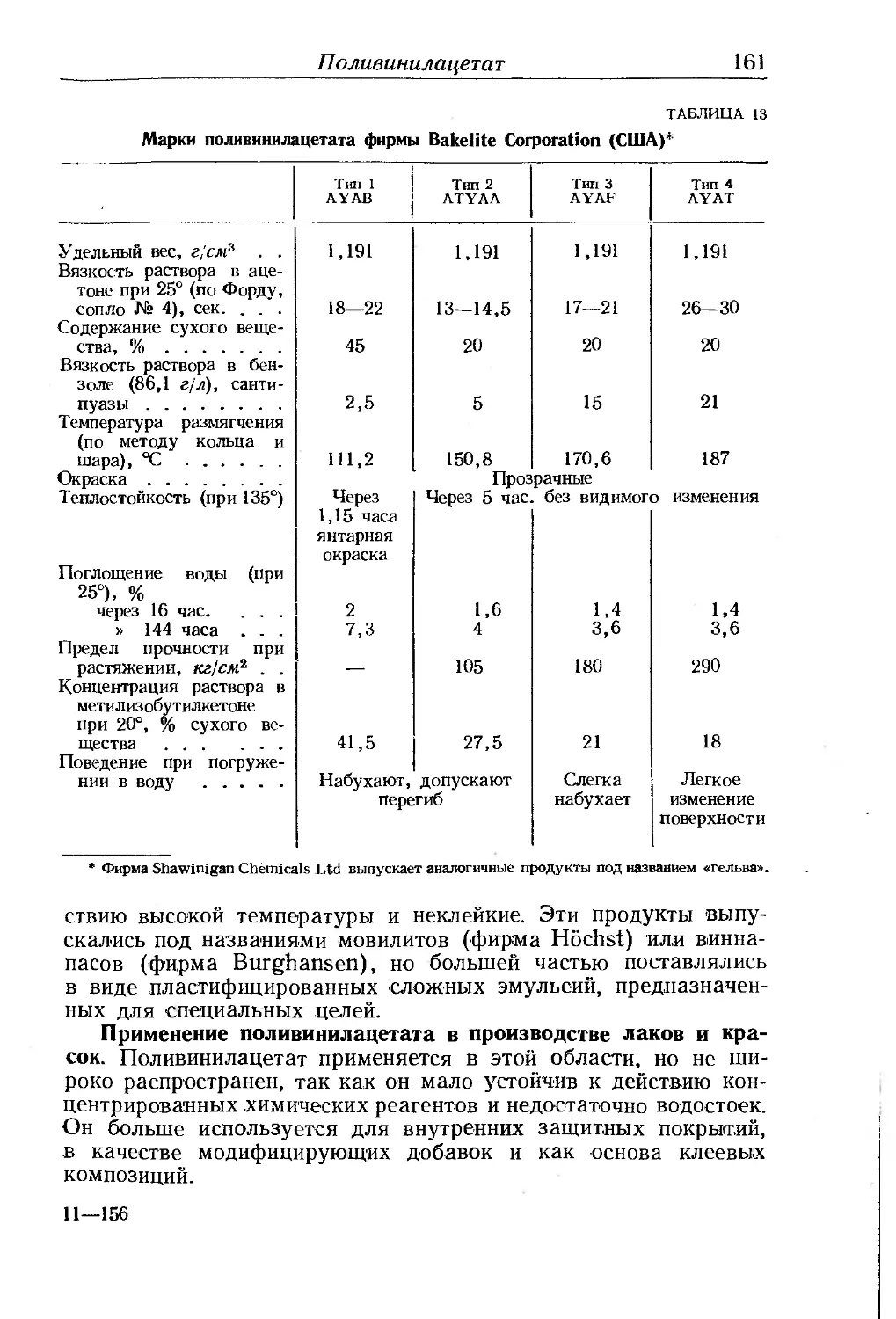
Глава IV. Виниловые полимеры. Ф. Шевассю......................... 156
Поливинилацетат..................................... - 158
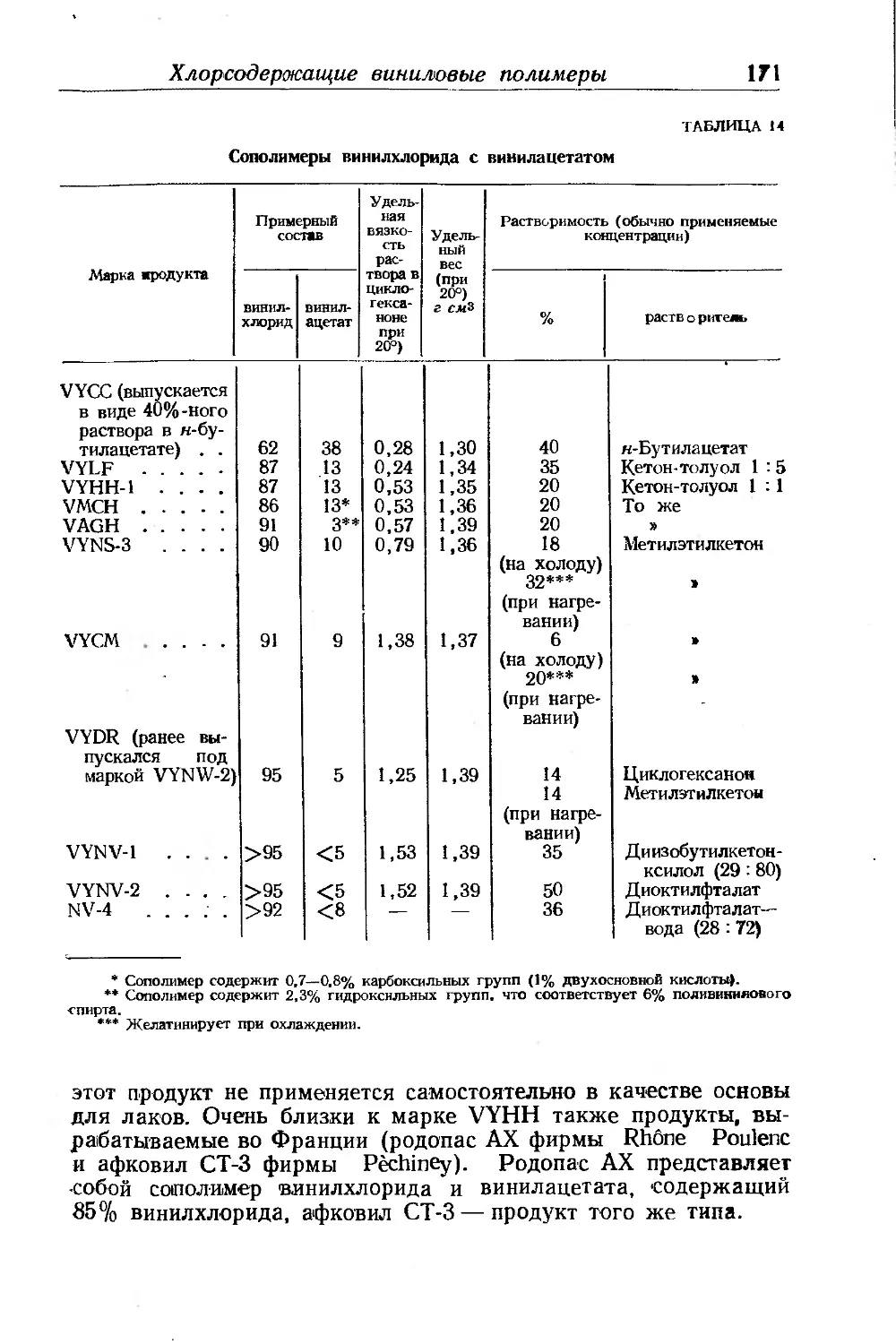
Хлорсодержащие виниловые полимеры............................ 165
Поливинилхлорид и его сополимеры с винилацетатом .... 165
Сополимеры винилхлорида с винилиденхлоридом .............. 173
Модифицированные виниловые сополимеры . ......... - 176
Винилит VMCH 176
Винилит VAGH . .......................................... 180
Сополимеры с высоким содержанием винилацетата............. 183
Растворители и вспомогательные материалы, применяемые совмест-
но с виниловыми полимерами.................................. 185
Товарные лакокрасочные материалы . . . 209
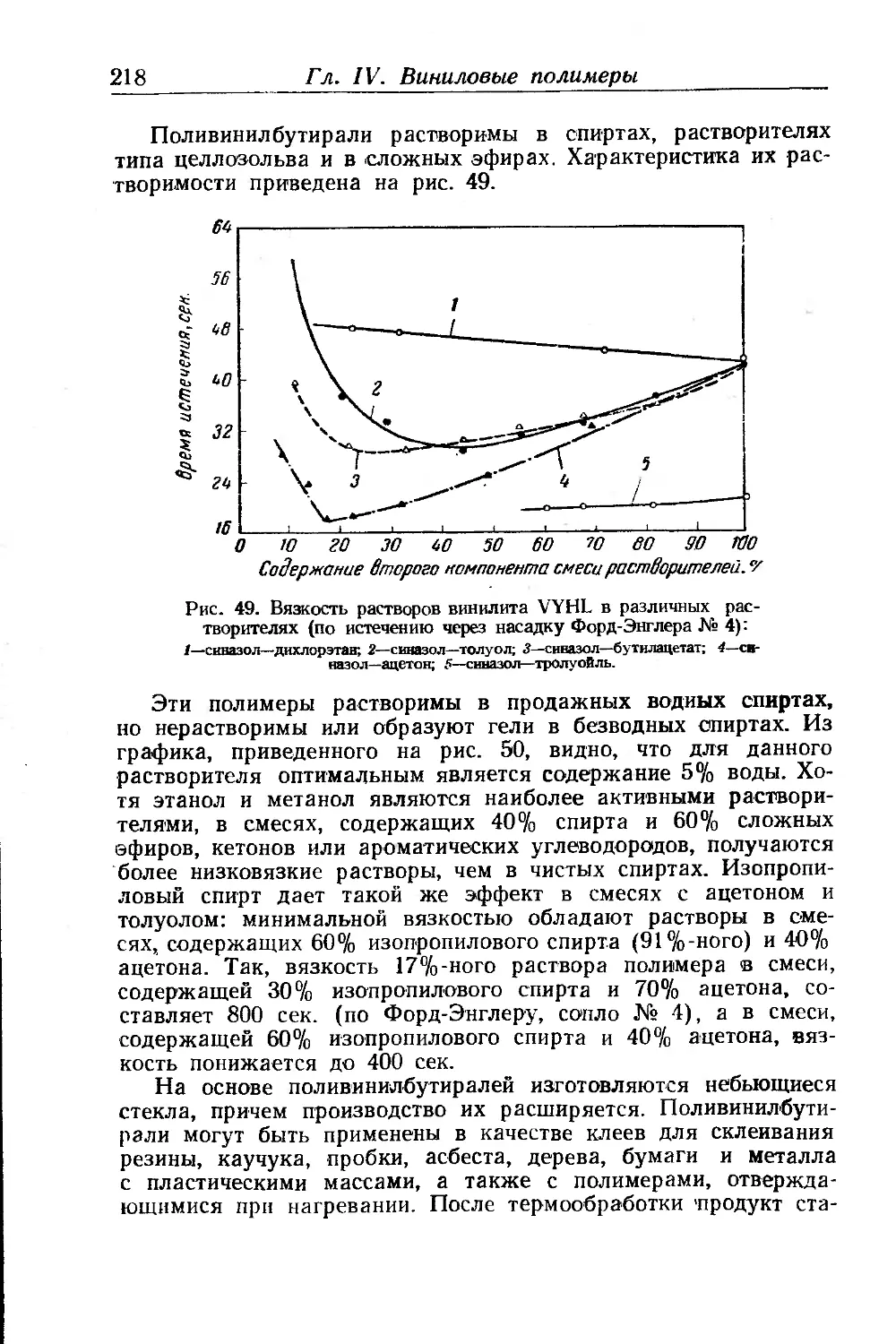
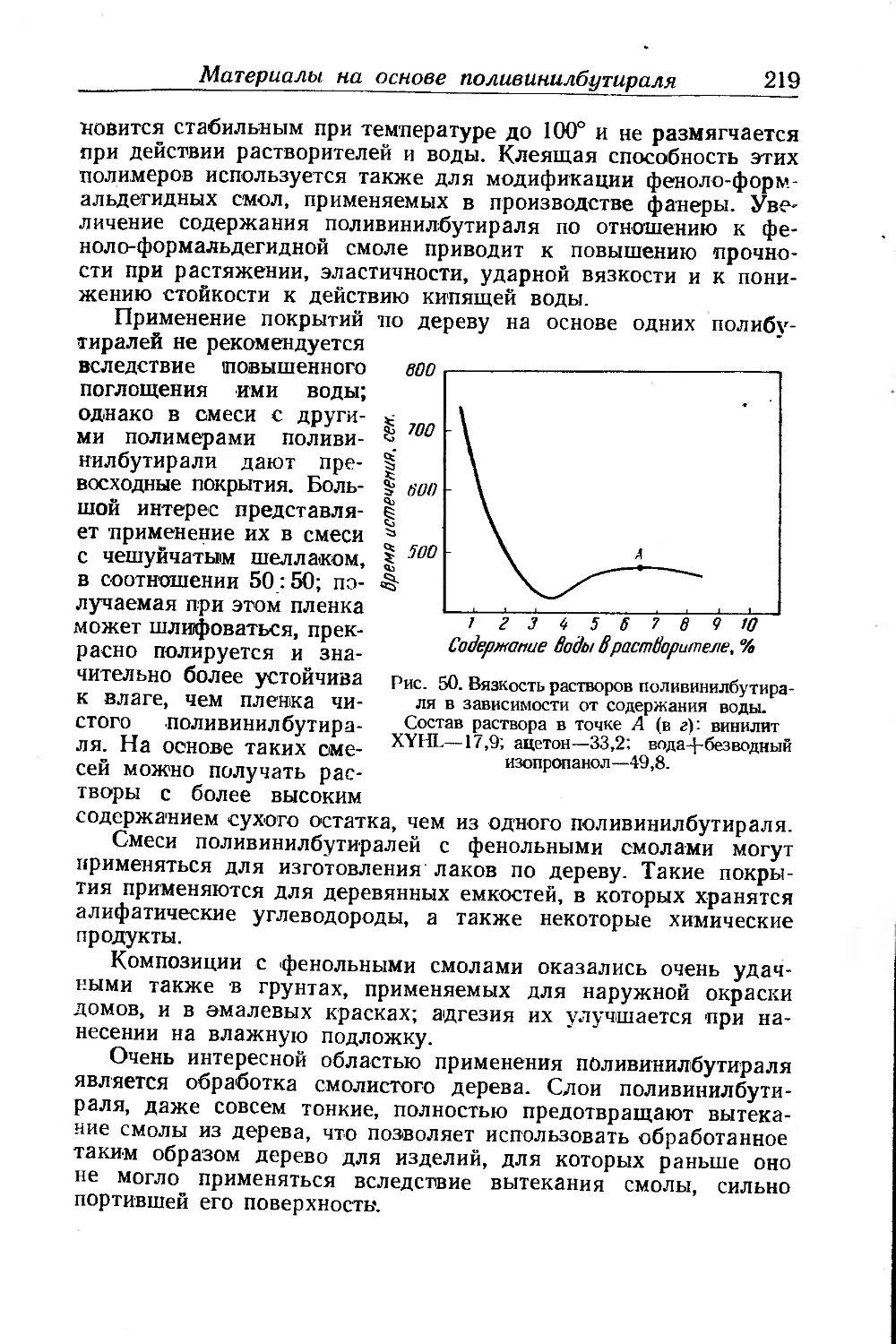
Материалы на основе поливинилбутираля ...................... 215
Фосфатирующие грунты Wash Primer......................... 220
Глава V. Полистирол. Е. Дорре, Л. Реморде , . 226
Стирол ..... ............................................ • 226
Полистирол................................................... 228
Пластифицированный полистирол....................... ... 230
Полистирол, модифицированный фенолами ..... . 230
Полистирол, модифицированный высыхающими маслами . . . 230
Полистирол, модифицированный непредельными кислотами . 231
Сополимеры стирола в виде водных эмульсий................. 232
Содержание
5
Глава VI. Полиакрилаты и полиметакрилаты. П. Тале.............. 235
Акриловые мономеры . . . .................................. 235
Акриловые полимеры............................. ... . 240
" Модифицированные полиакрилаты................. 247
Идентификация акриловых полимеров 247
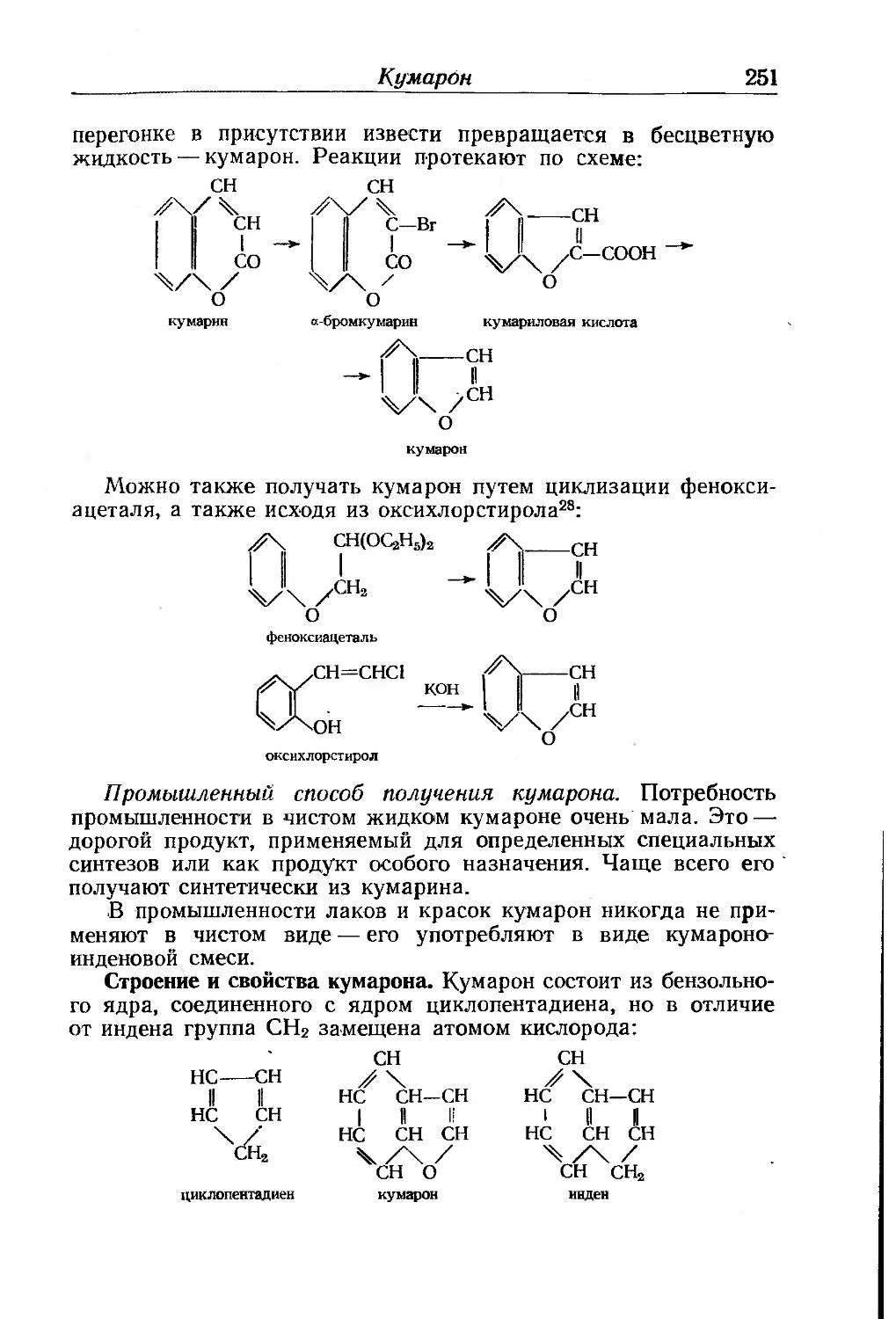
Глава VII. Кумароно-ииденовые смолы. А- Марти.................. 249
Кумарон..................................... . 250
Кумароно-инденовые смолы . 253
Лаки и краски на основе кумароновых смол .... 254
Другие области применения кумароновых смол................. 257
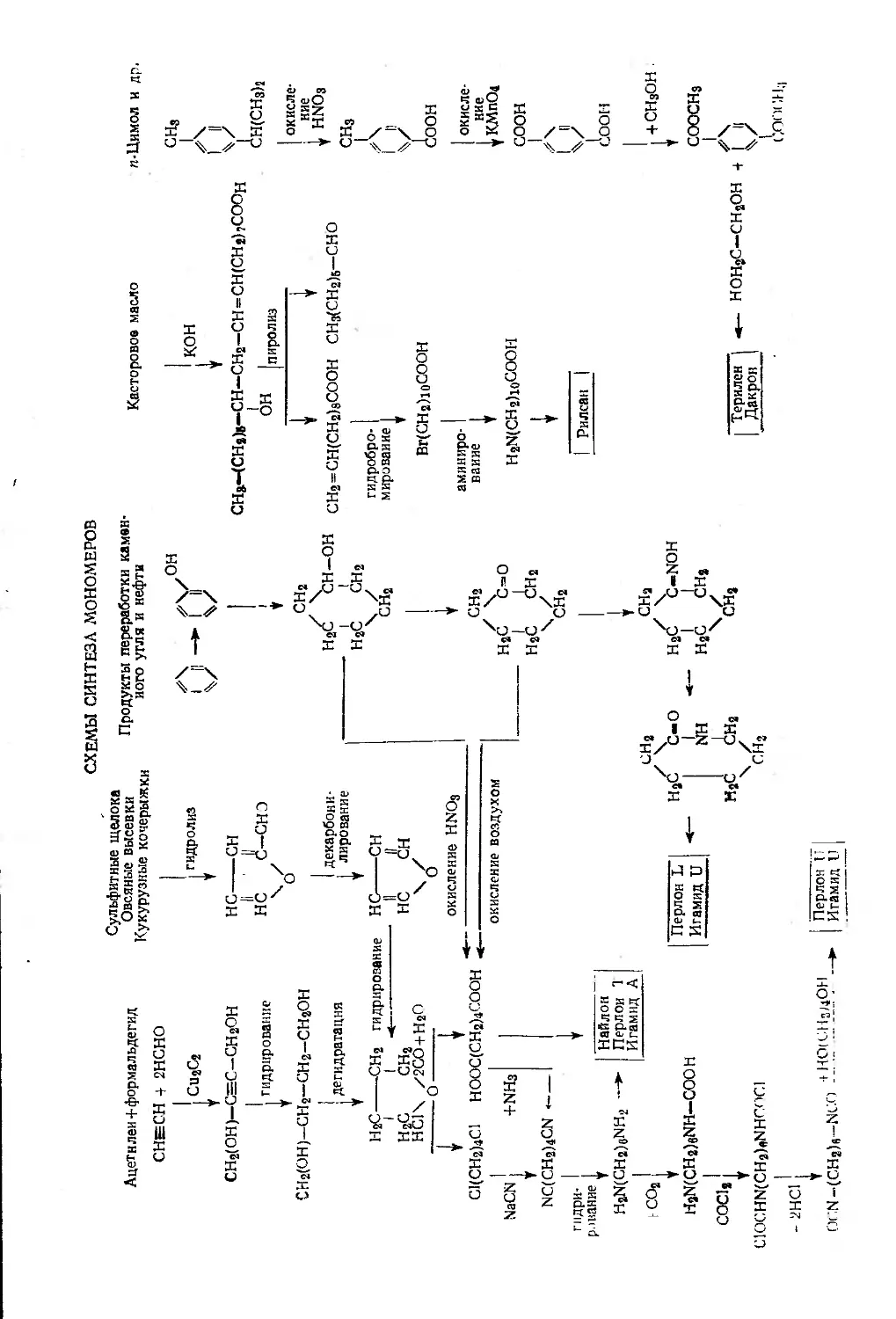
Глава VIII. Полиэфиры, полиамиды и полиуретаны. Р. Элион .... 261
Исходные материалы и методы синтеза мономеров . . 263
Синтез полимеров........................................ 265
Классификация полимеров . . . . . 267
Полиэфиры............................ .... . 267
Линейные ароматические полиэфиры........................ 268
Комплексные полиэфиры................................... 269
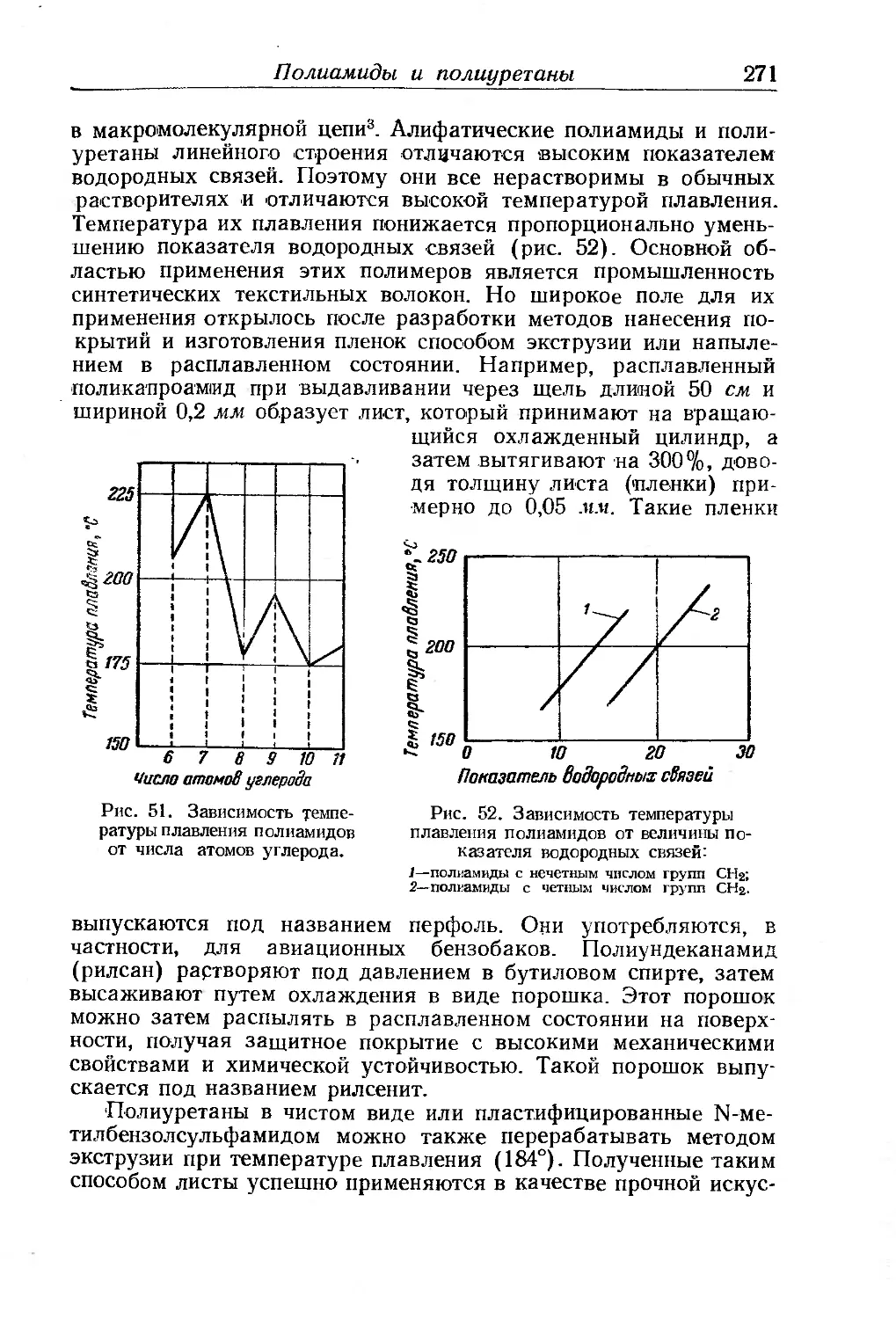
Полиамиды и полиуретаны.......... ... ......... 270
Линейные алифатические полиамиды и полиуретаны .... 270
Полиамиды с очень длинной линейной алифатической цепью . . 272
Продукты совместной поликонденсации и диспропорциониро-
вания ................................................. 272
Полимеры с разветвленной цепью ......... 274
Полимеры, содержащие гетероатомы в углеводородной цепи . . 275
Полимеры с ароматическими радикалами.................... 275
Полифункциональные полимеры................. . . 276
Модифицированные полиамиды ... . 277
Глава IX. Феноло-альдегидные смолы. М. Э. Мейер............. 279
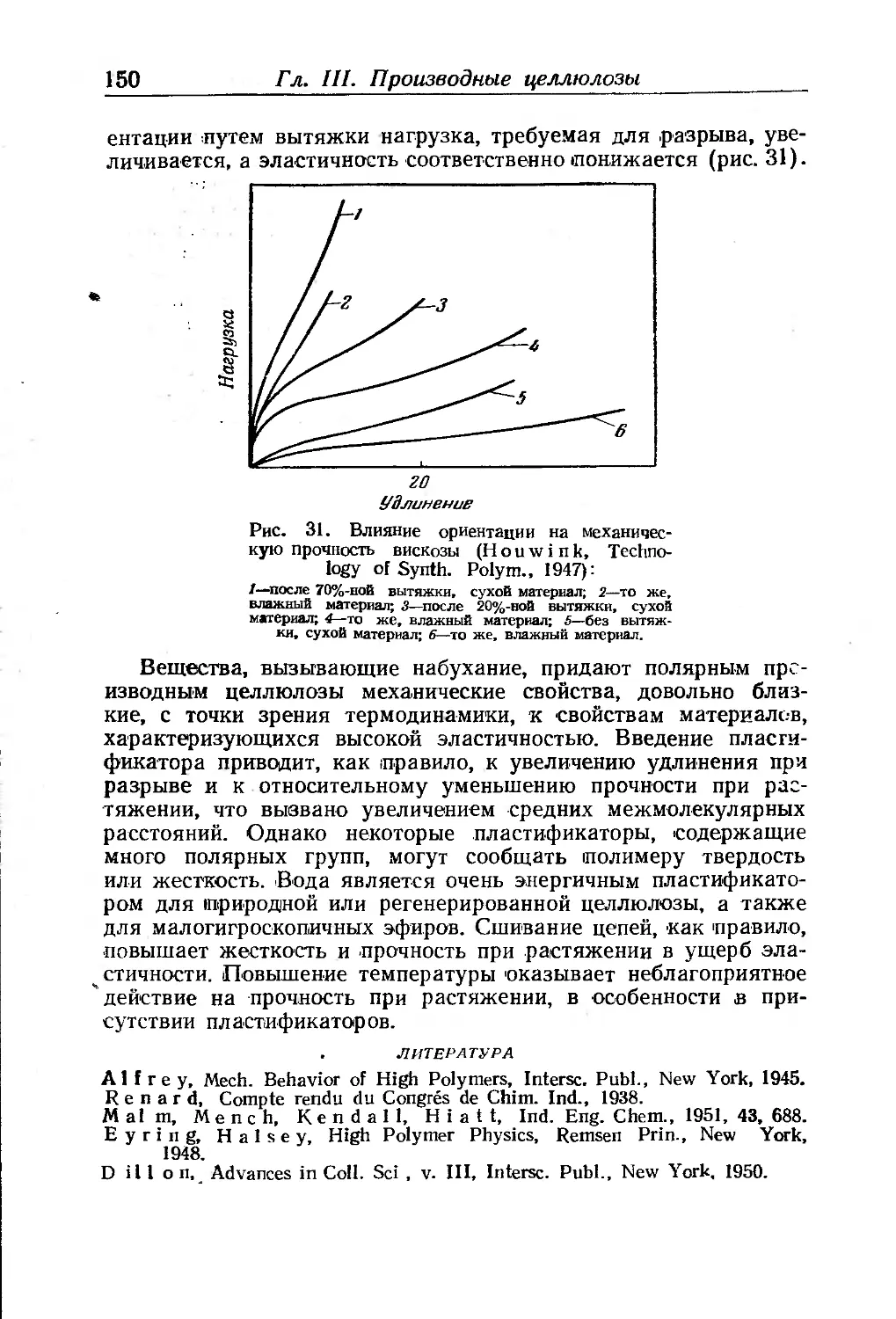
Основные сведения о синтезе феноло-альдегидных смол . 282
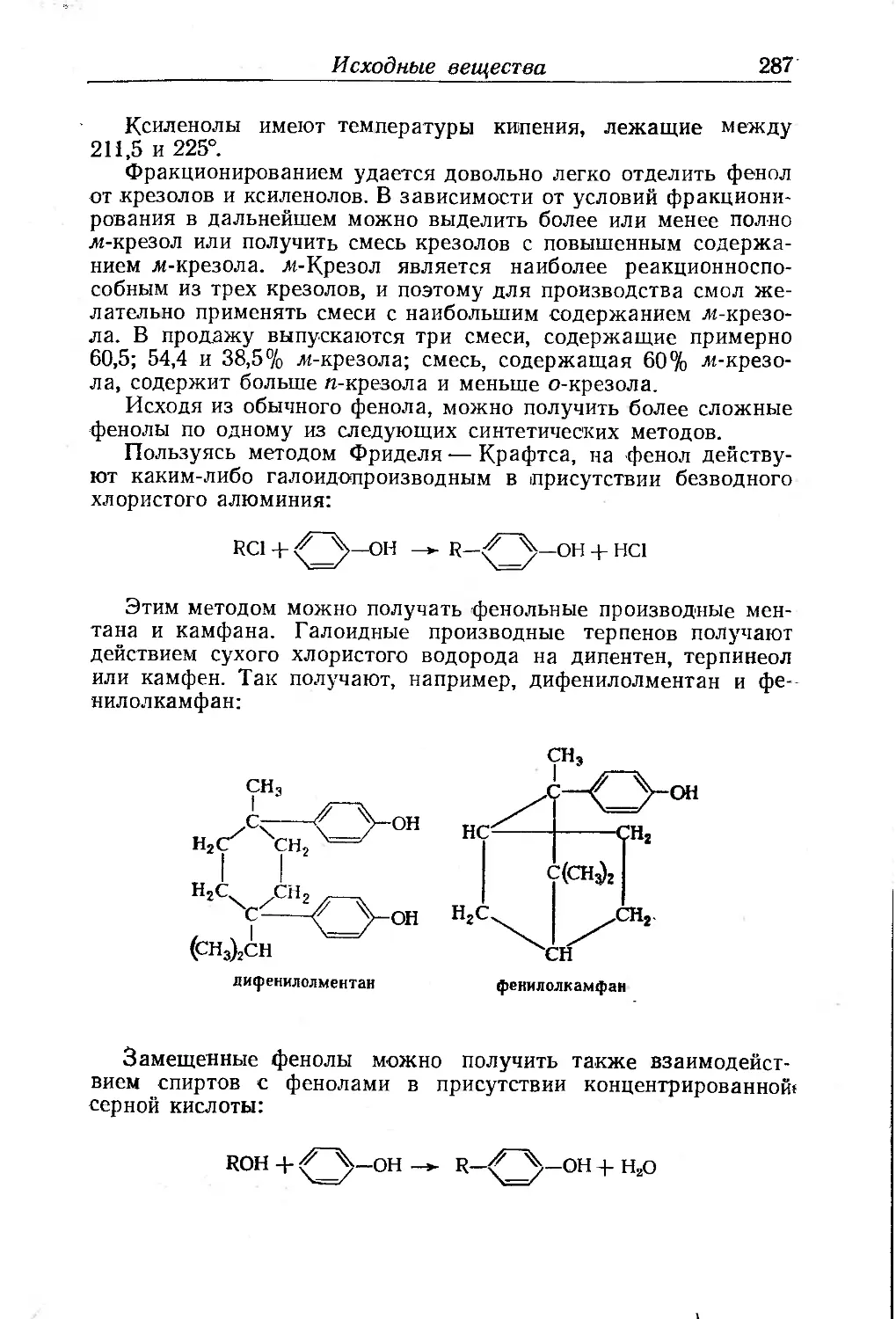
Исходные вещества .... .... 285
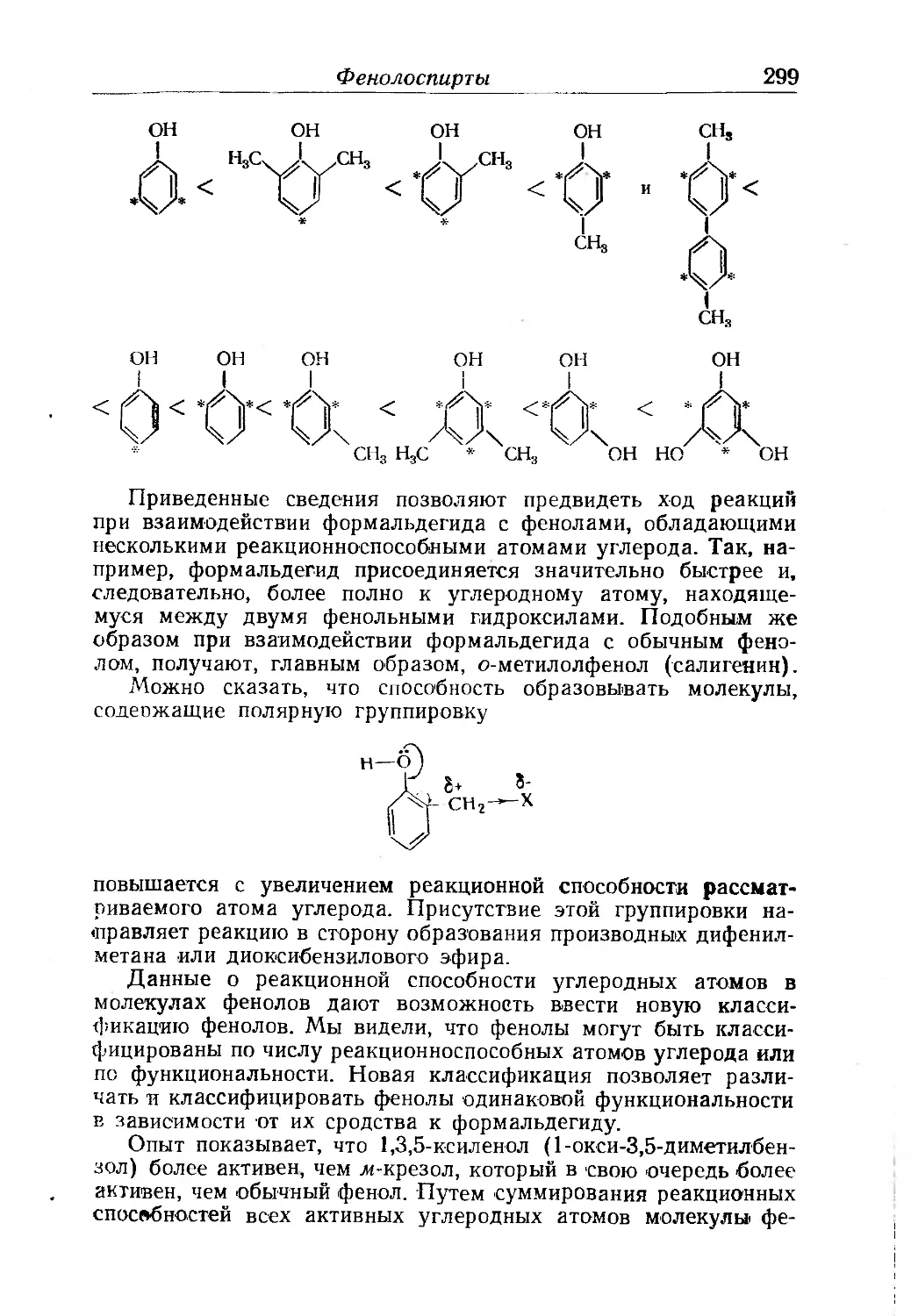
Фенолоспирты.......................................... . . 290
Условия образования фенолоспиртов....................... 291
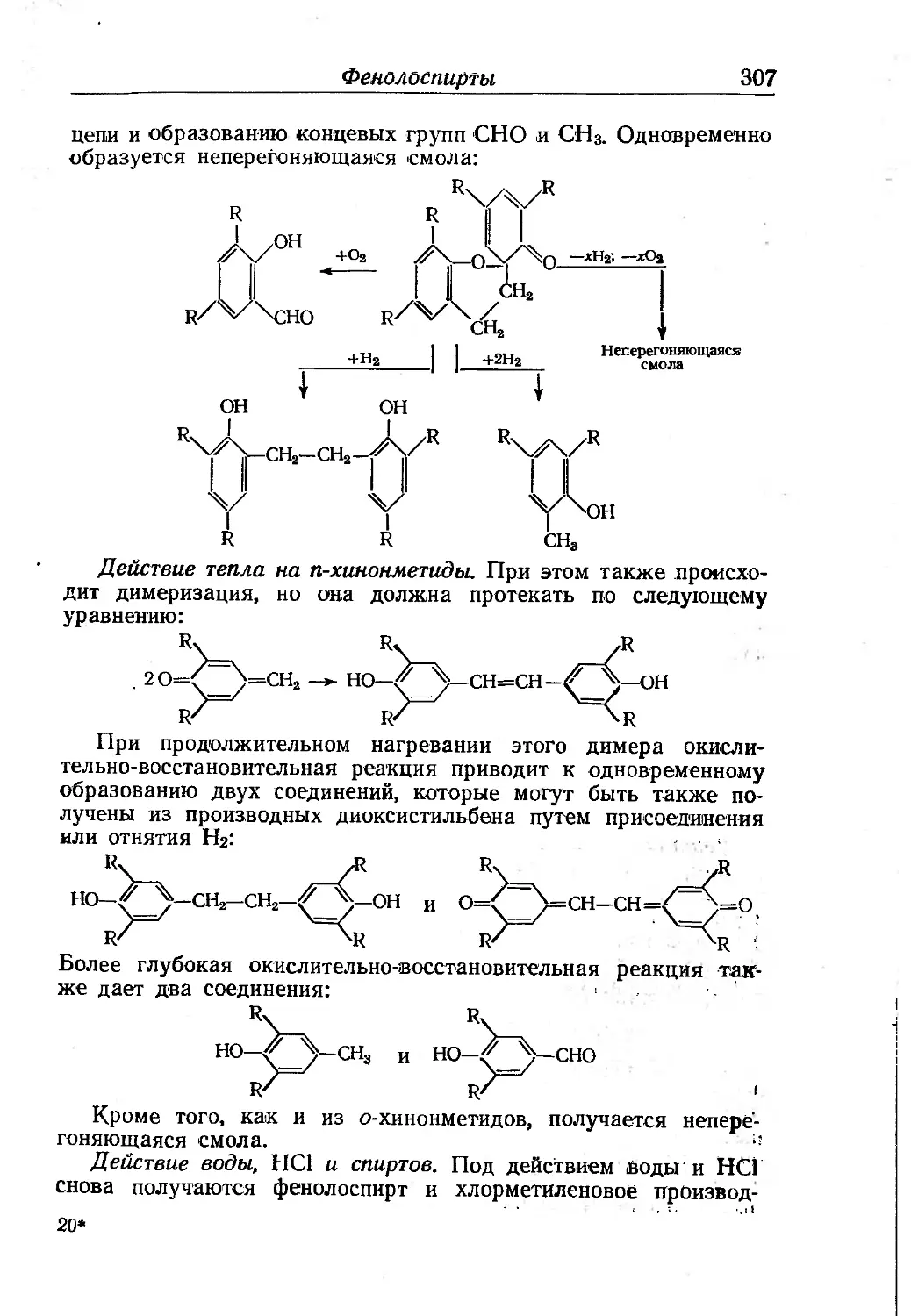
Химия фенолоспиртов............... 302
Строение феноло-альдегидных смол........................ 308
Модифицированные феноло-альдегидиые смолы и лаковые смолы . 319
Феноло-альдегидные смолы, растворимые в высыхающих мас-
лах .................................................. 321
Товарные феноло-формальдегидные смолы ..................... 326
6
Содержание
Глава X. Мочевиио-формальдегидные смолы и их аналоги. П. Тале . . 331
Мочевино-формальдегидные смолы.............................. 331
Мочевино-формальдегидные смолы для лаков и красок .... 335
Меламино-формальдегидные смолы............................ 340
Эмульсионные смолы............................. .... 347
Различные амино-альдегидные смолы.................... ..... 347
Глава XI. Алкидные смолы. И. Пети ........................... 356
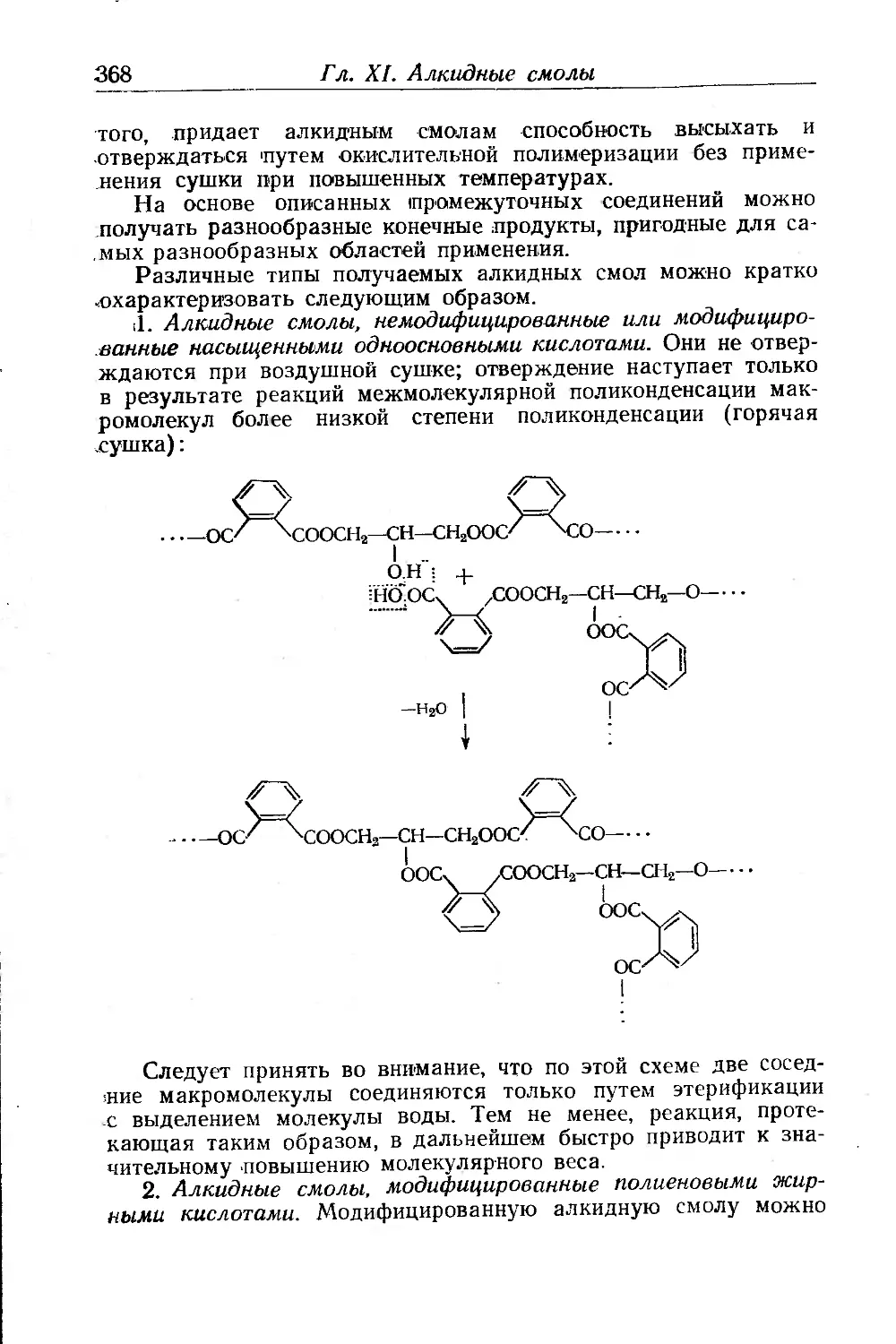
Исходные вещества для синтеза алкидных смол ..... 356
Образование и свойства полиэфиров .... . . 360
Модифицированные алкидные смолы............................. 367
Промышленное производство модифицированных алкидных
смол .'................................................. 370
Процесс образования моноглицеридов....................... 372
Взаимодействие фталевого ангидрида со смесью моноглицери-
дов .................................................... 378
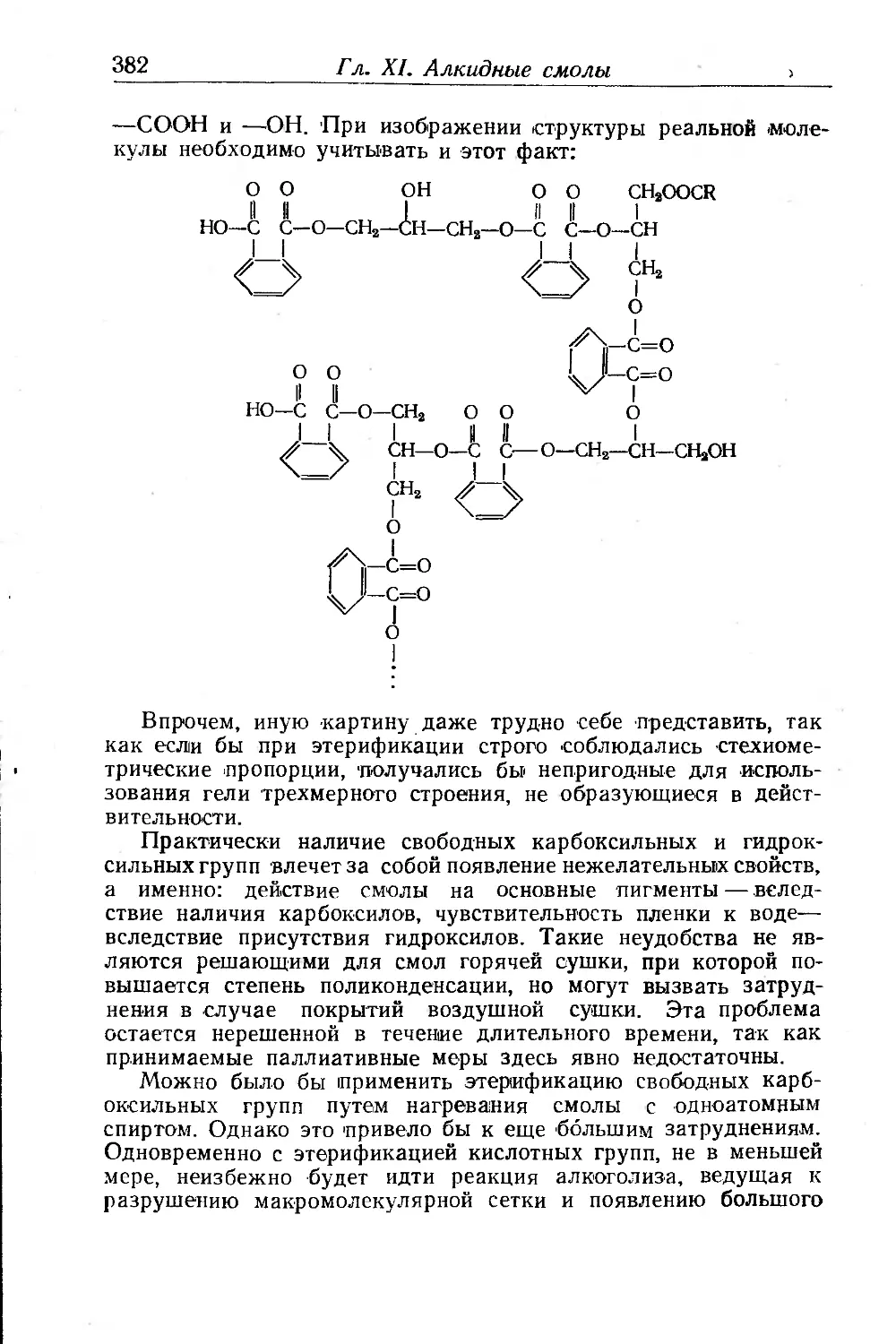
Глава XII. Малеиновые смолы. И. М. Буше .......... , . 385
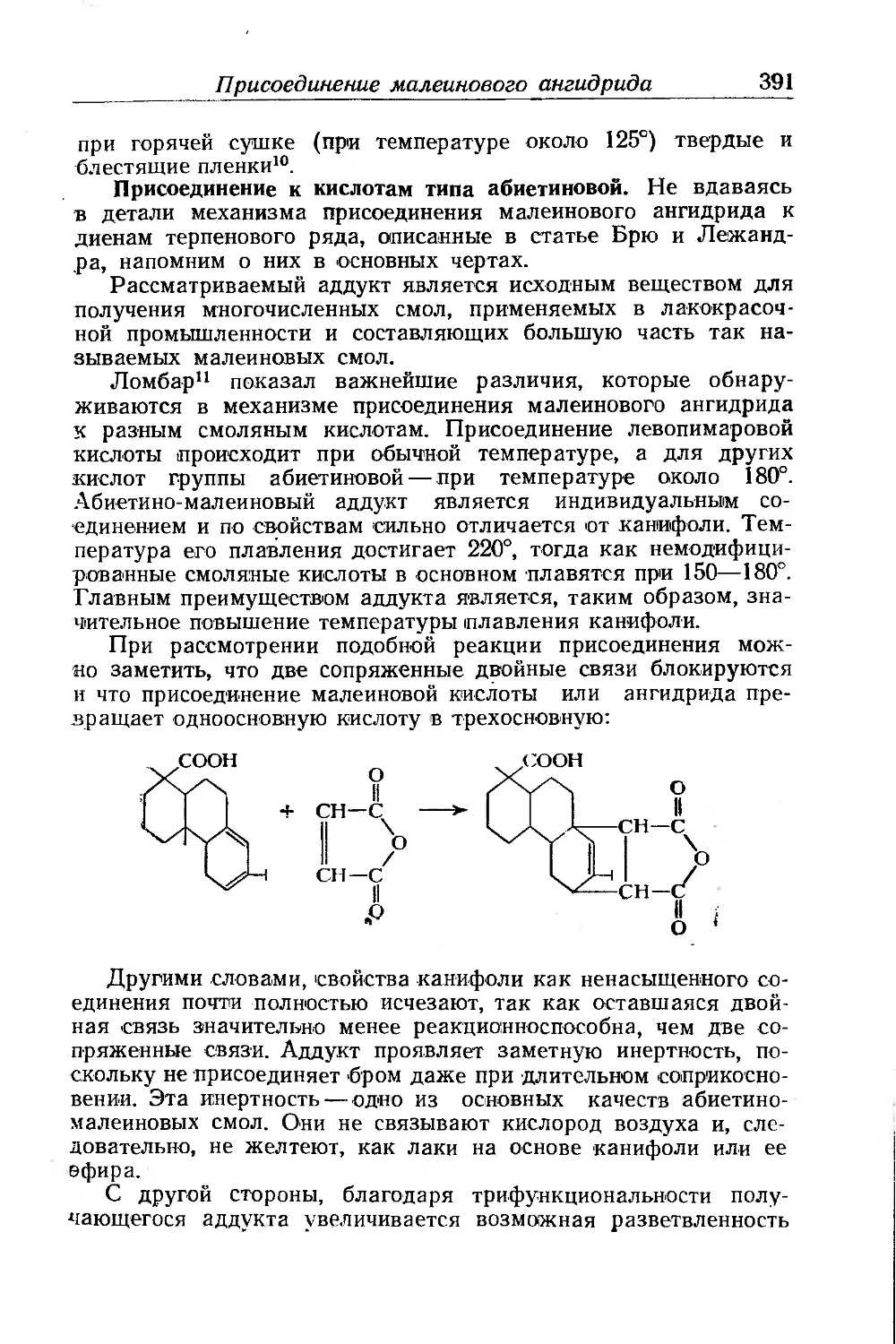
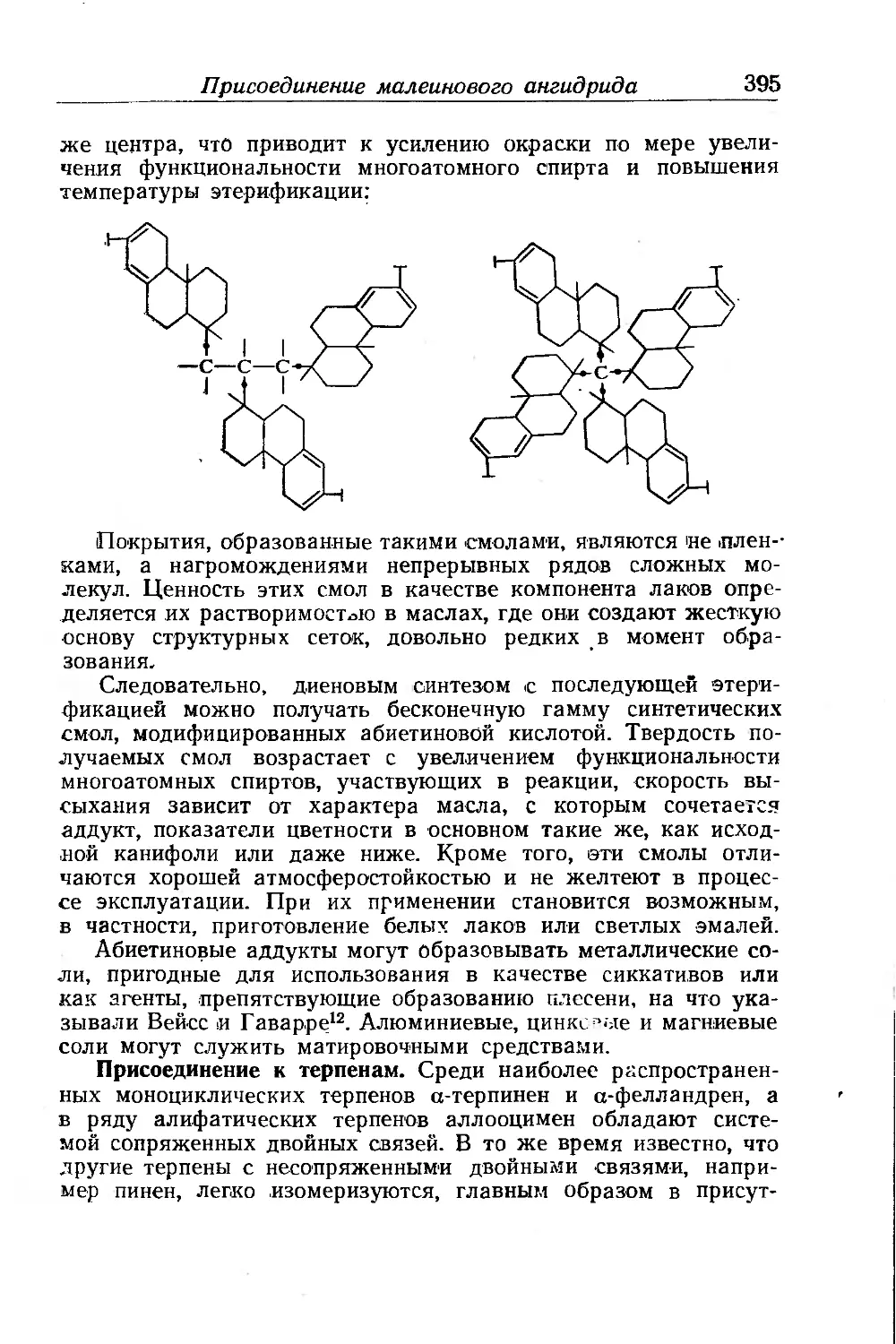
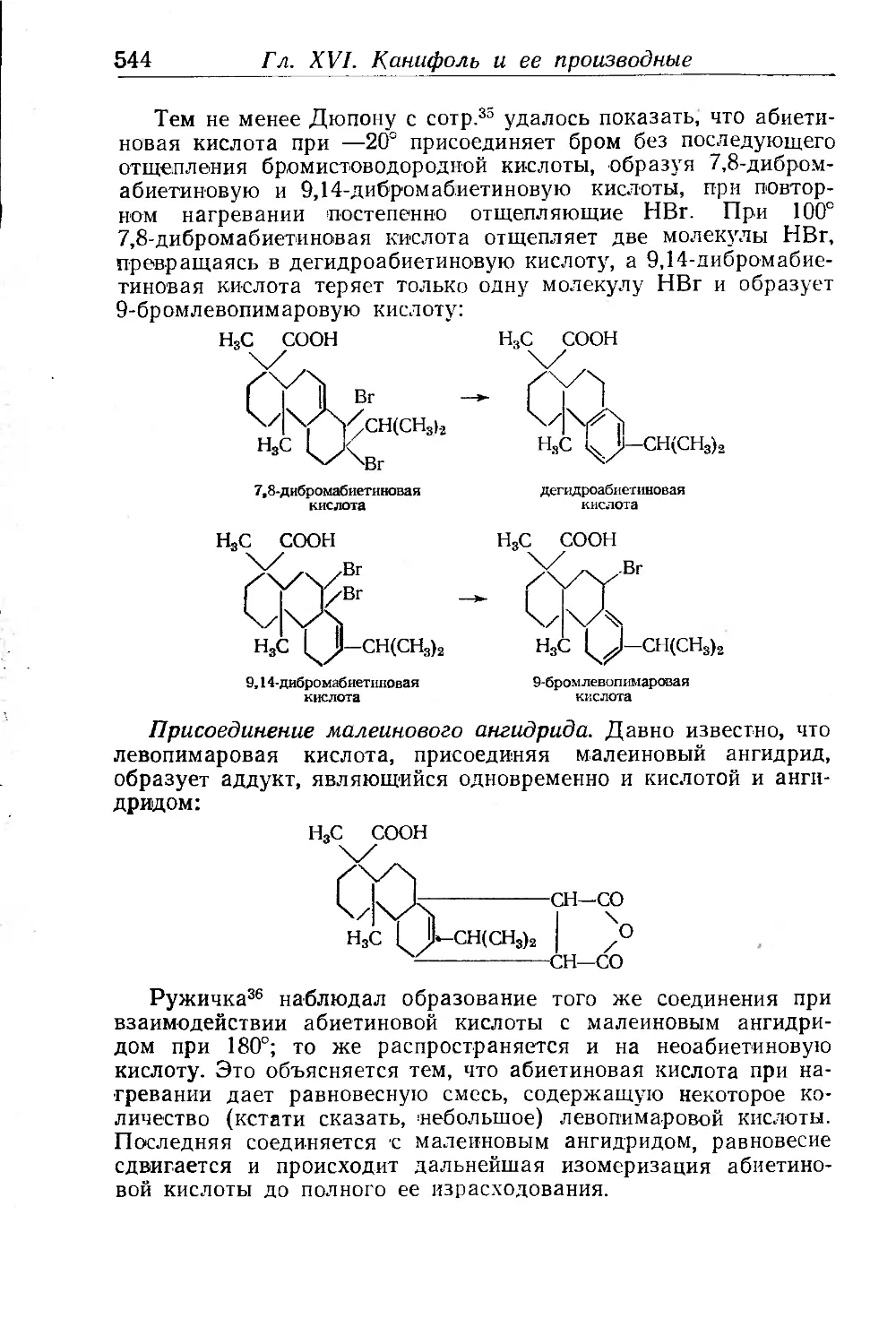
Присоединение малеинового ангидрида...................... 387
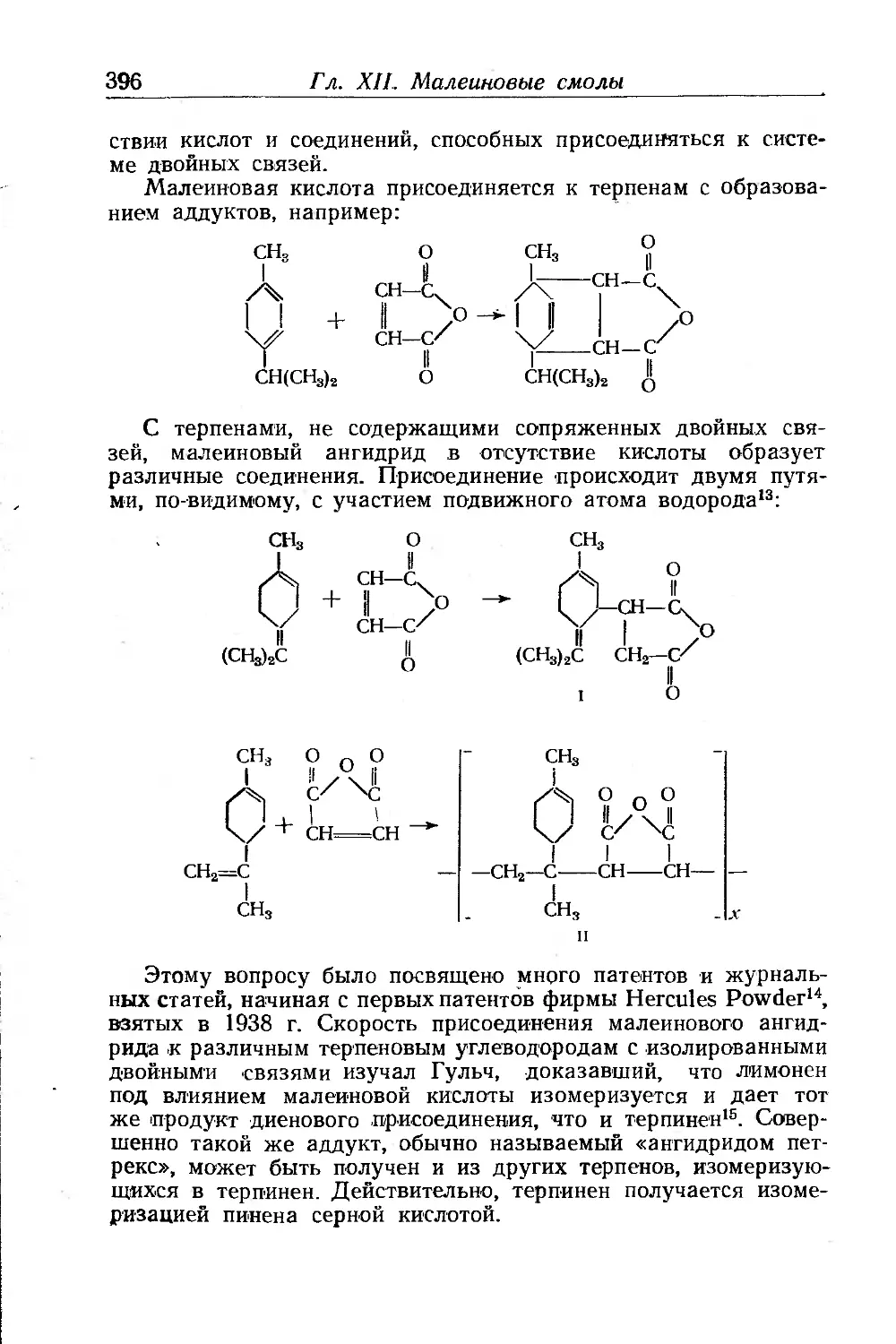
Линейная полиэтерификация.......................... .... 400
Поликонденсация и сополимеризация........................ 402
Реакции гомологов малеиновой кислоты . .............. 404
Анализ малеиновых смол.................................... 405 .
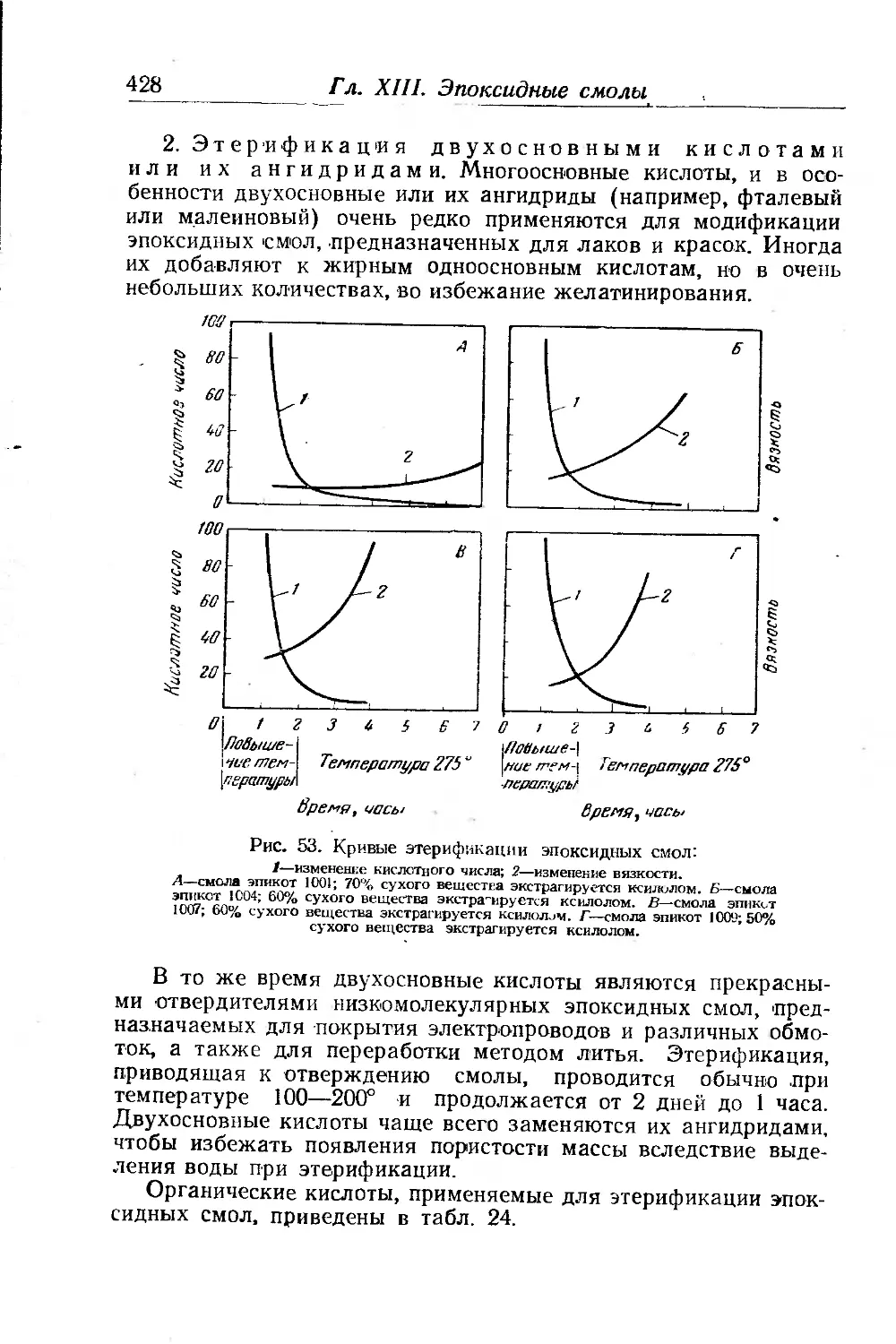
Г ла’ва XIII. Эпоксидные смолы. Р. Мартин, Ф. Саррю 408
Исходные вещества для синтеза эпоксидных смол............ 411
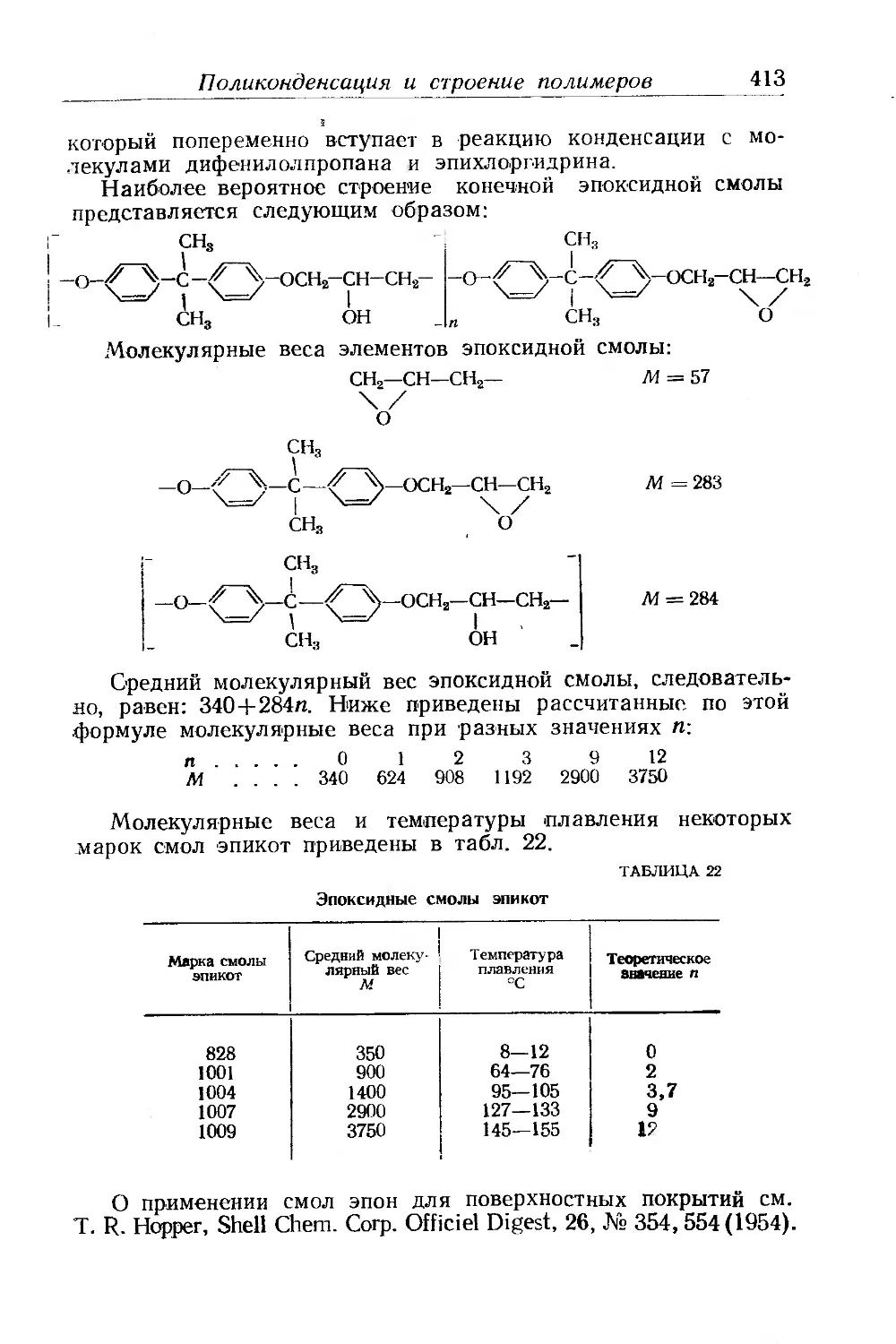
Поликонденсация и строение полимеров..................... 412
Физические свойства эпоксидных смол..................... 414
Химические свойства эпоксидных смол..................... 423
Анализ и идентификация эпоксидных смол................... 432
Применение эпоксидных смол............................... 435
Глава XIV. Каучук и продукты его превращений. Ш. П. Пинацци, Р. Ше-
рита......................................................... 437
Натуральный каучук.......................................... 437
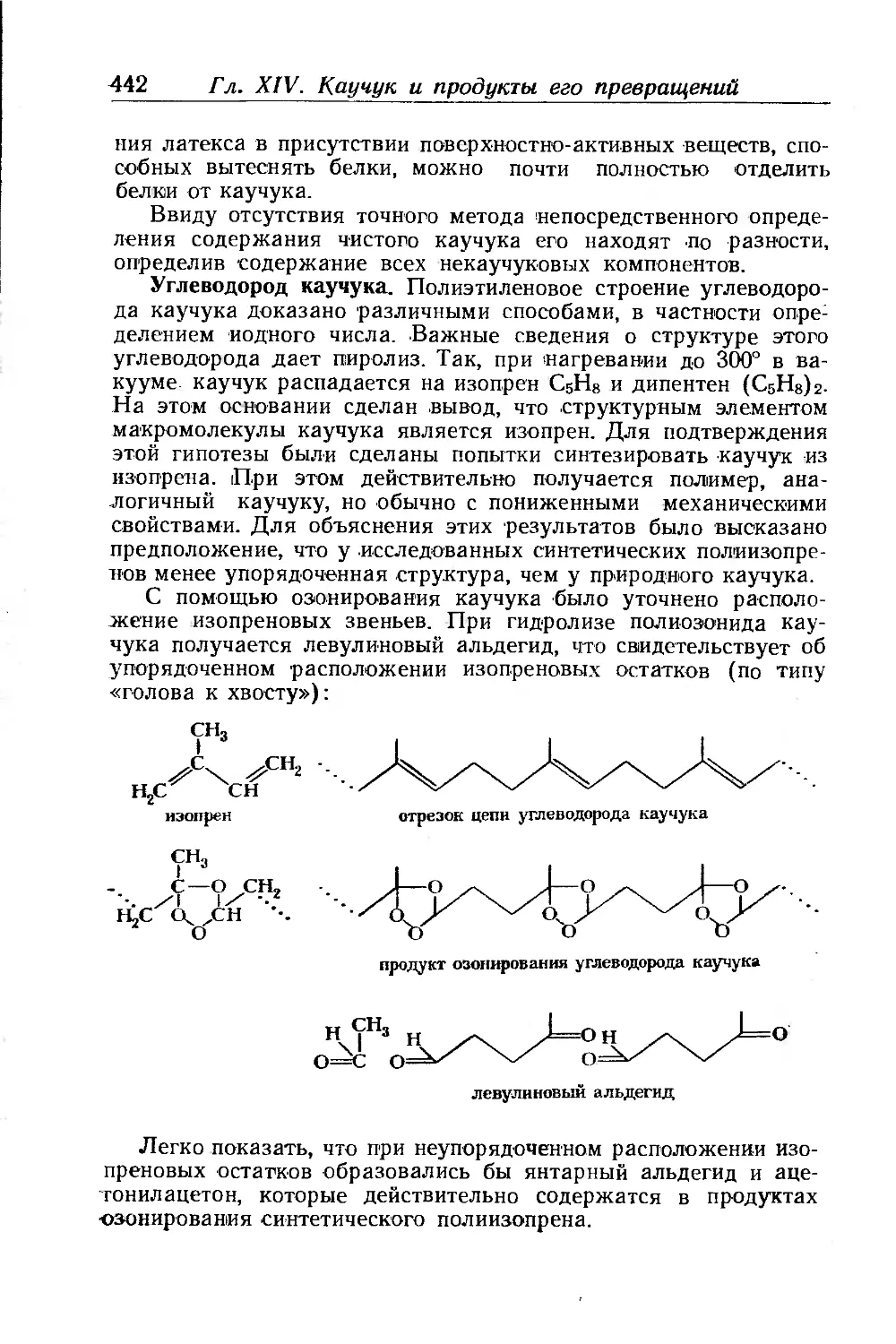
Состав сырого каучука.................................... 440
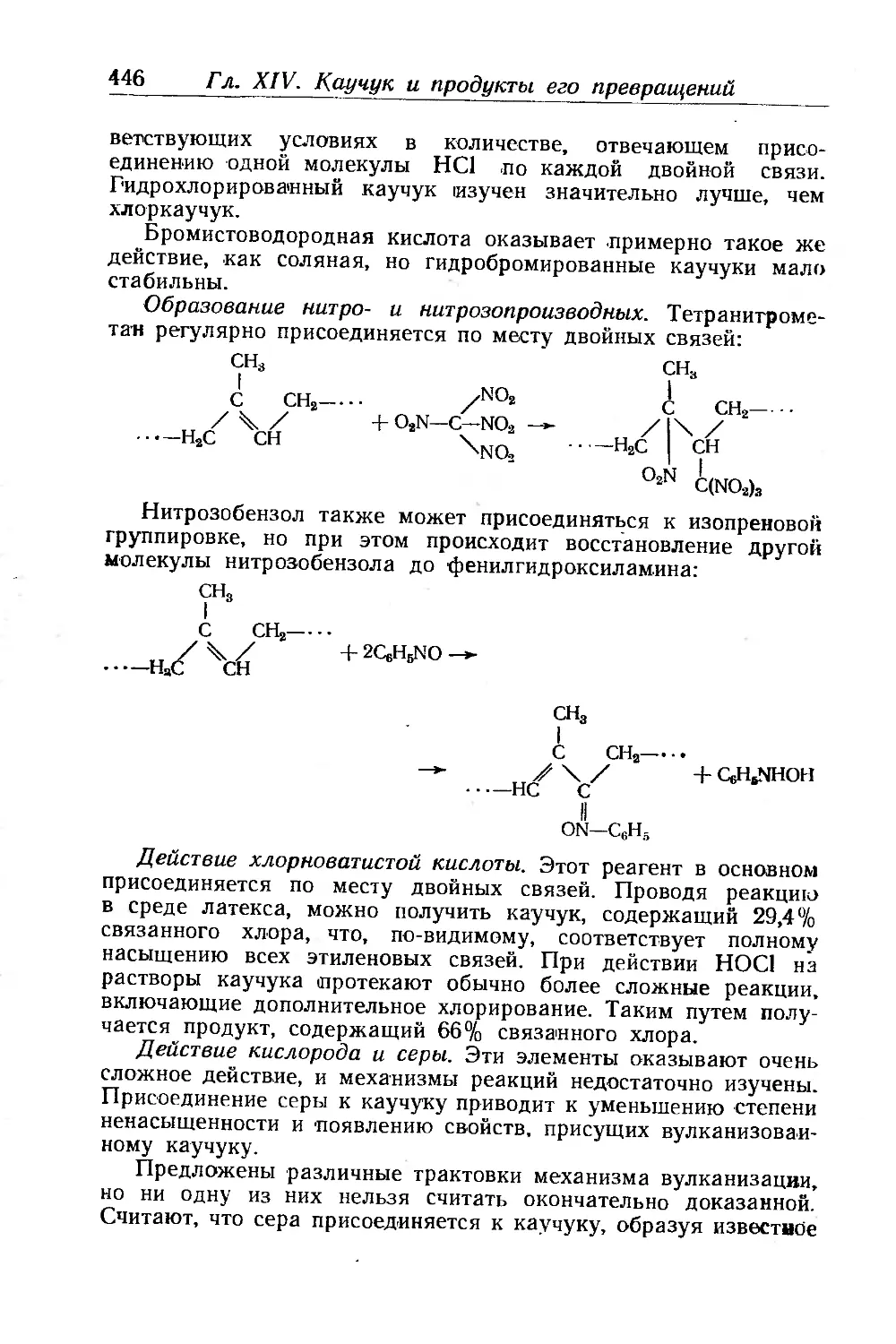
Химические свойства каучука.............................. 444
Применение сырого каучука................................ 448
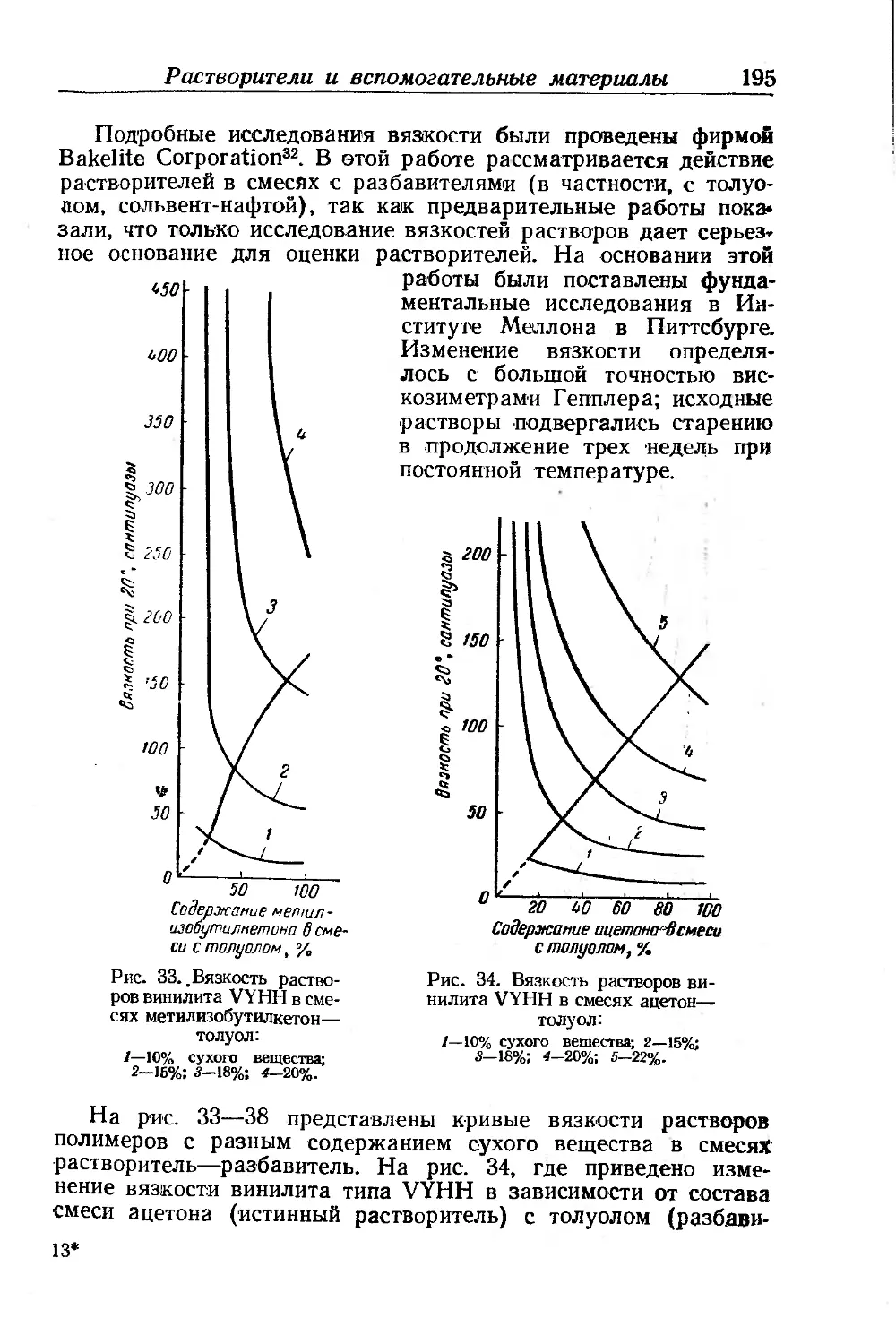
Гуттаперча и балата........................................ 449
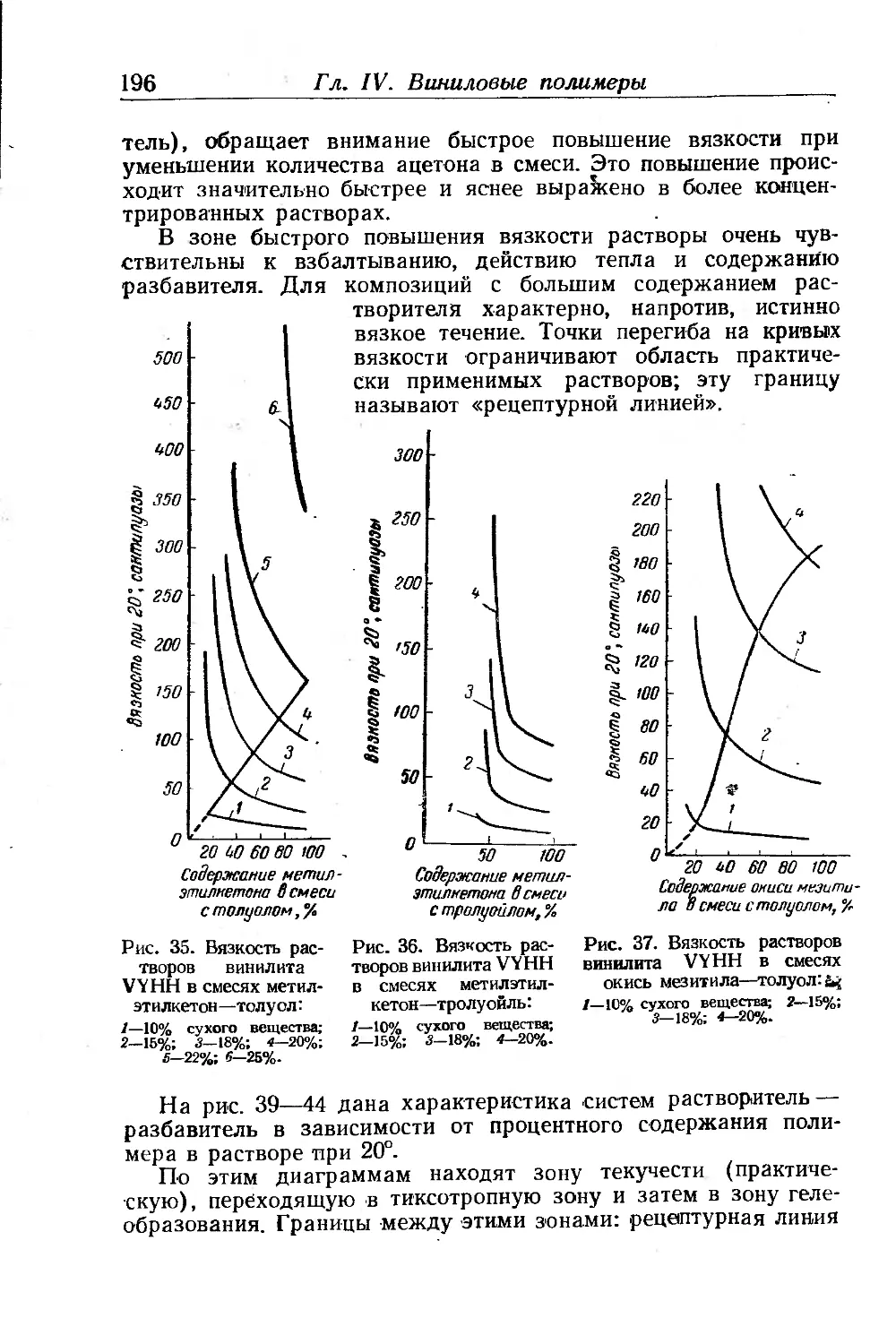
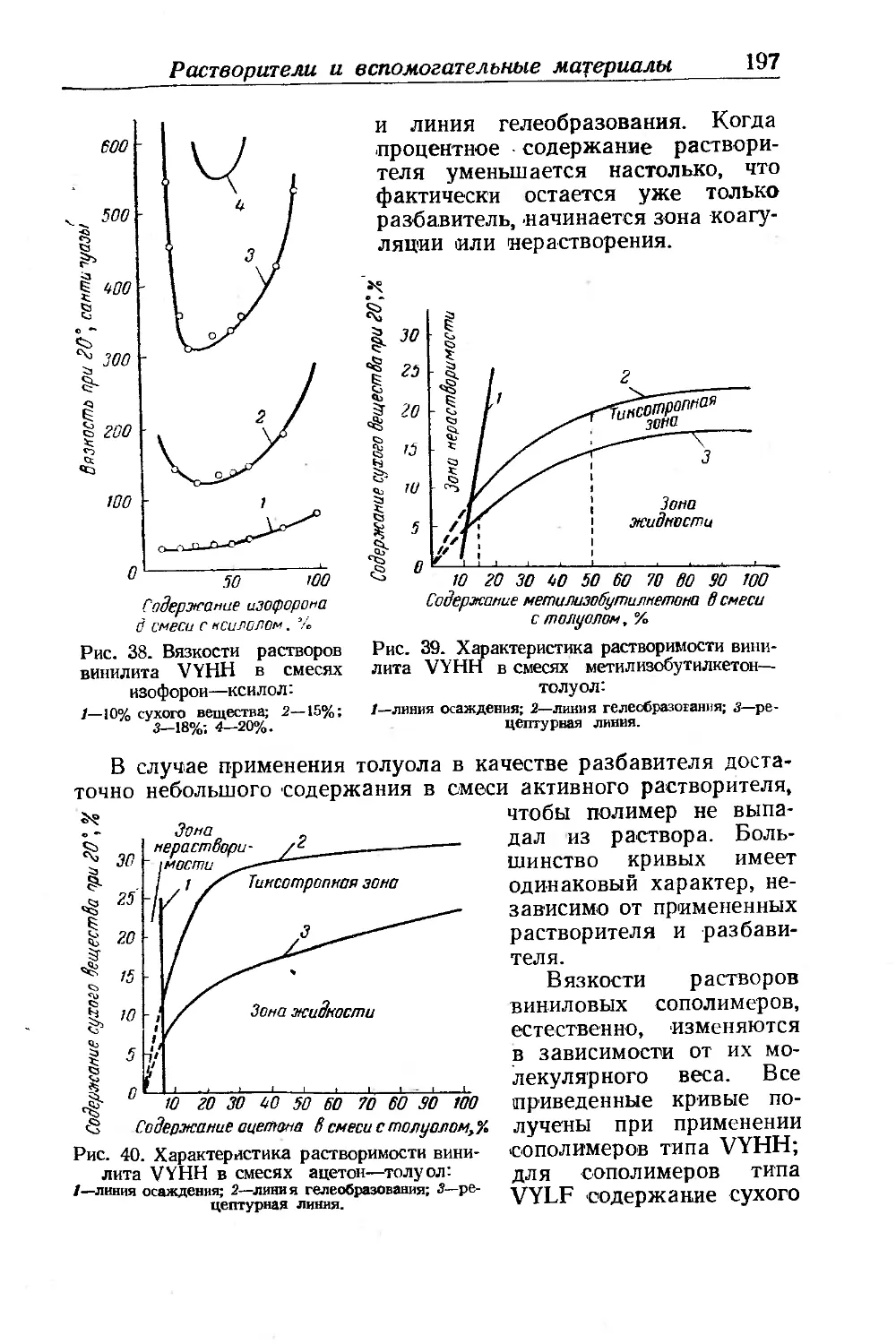
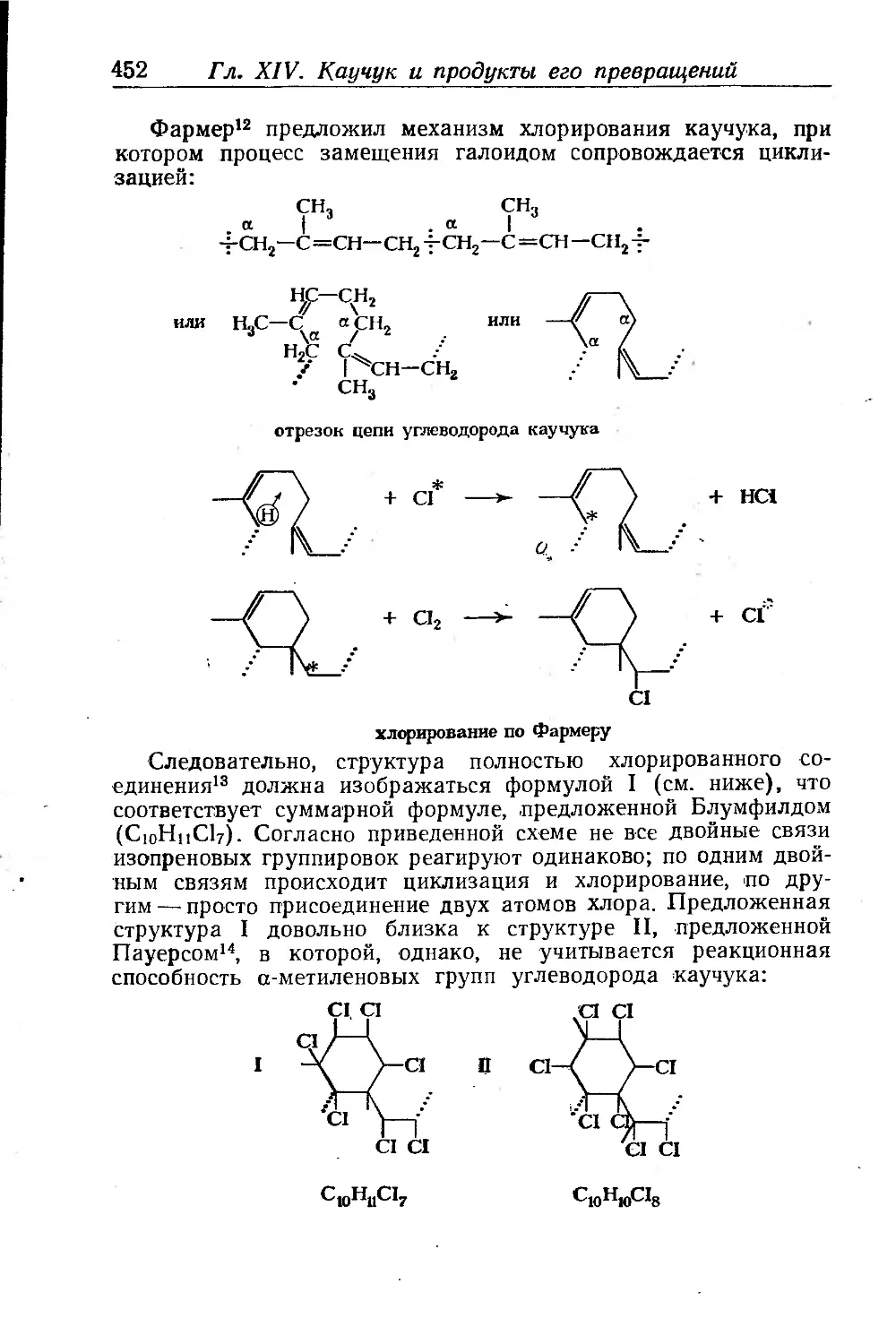
Хлоркаучуки.......................................... .... 451
Получение хлоркаучуков.................................. 453
Свойства хлоркаучуков.................................... 456
Применениехлоркаучуков................................... 460
Содержание
7
Гидрохлорированный каучук..................................... 463
Получение гидрохлорированного каучука .................. 464
Свойства гидрохлорированного каучука...................... 465
Применение гидрохлорированного каучука.................. 467
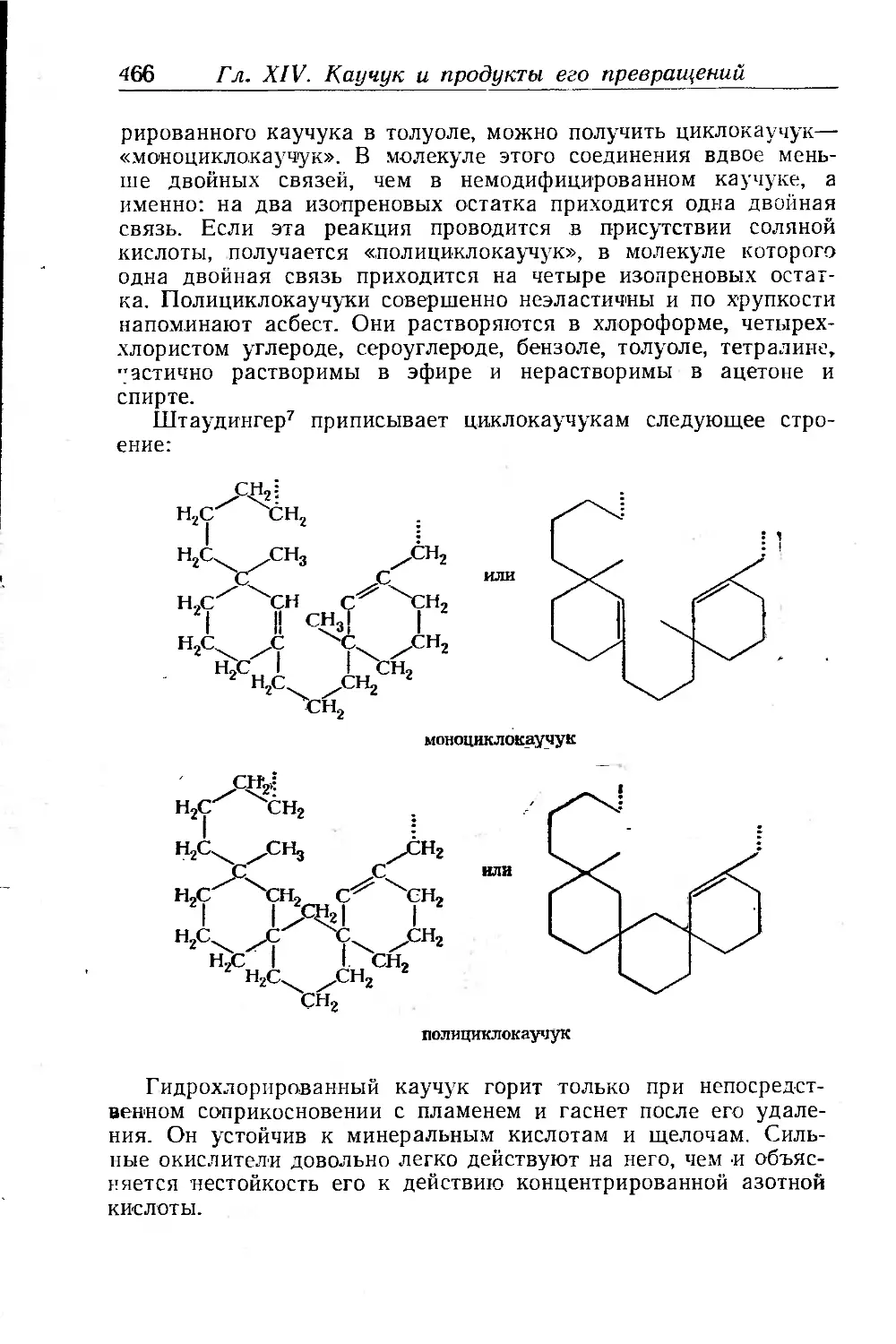
Продукты циклизации каучука (циклокаучуки).................. 468
Получение циклокаучуков...............................- • 468
Строение циклокаучуков . . ............................... 470
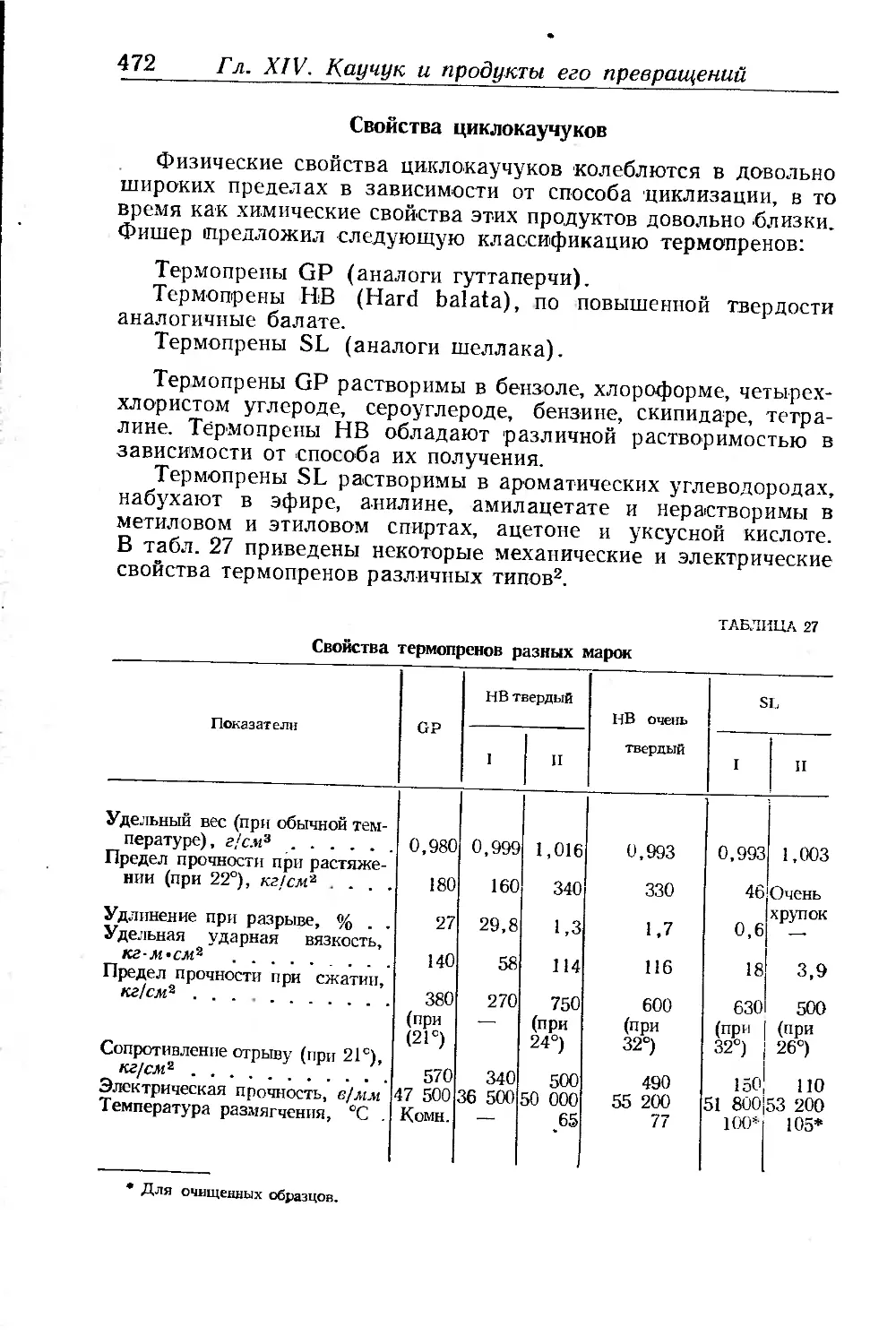
Свойства циклокаучуков.................................... 472
Применение циклокаучуков.................................. 475
Продукты окисления каучука . '................................ 478
Реакции окислении каучука ................................ 478
Получение окисленных каучуков............................. 481
Свойства окисленных каучуков...............................483
Применение раббонов и других окисленных каучуков .... 485
Синтетические каучукоподобные эластомеры...................... 489
Вулкапрены.............................................. 489
Блек-аут.................................................. 490
Синтетические каучуки..................................... 491
Торговые марки каучуков и продуктов их превращений............ 494
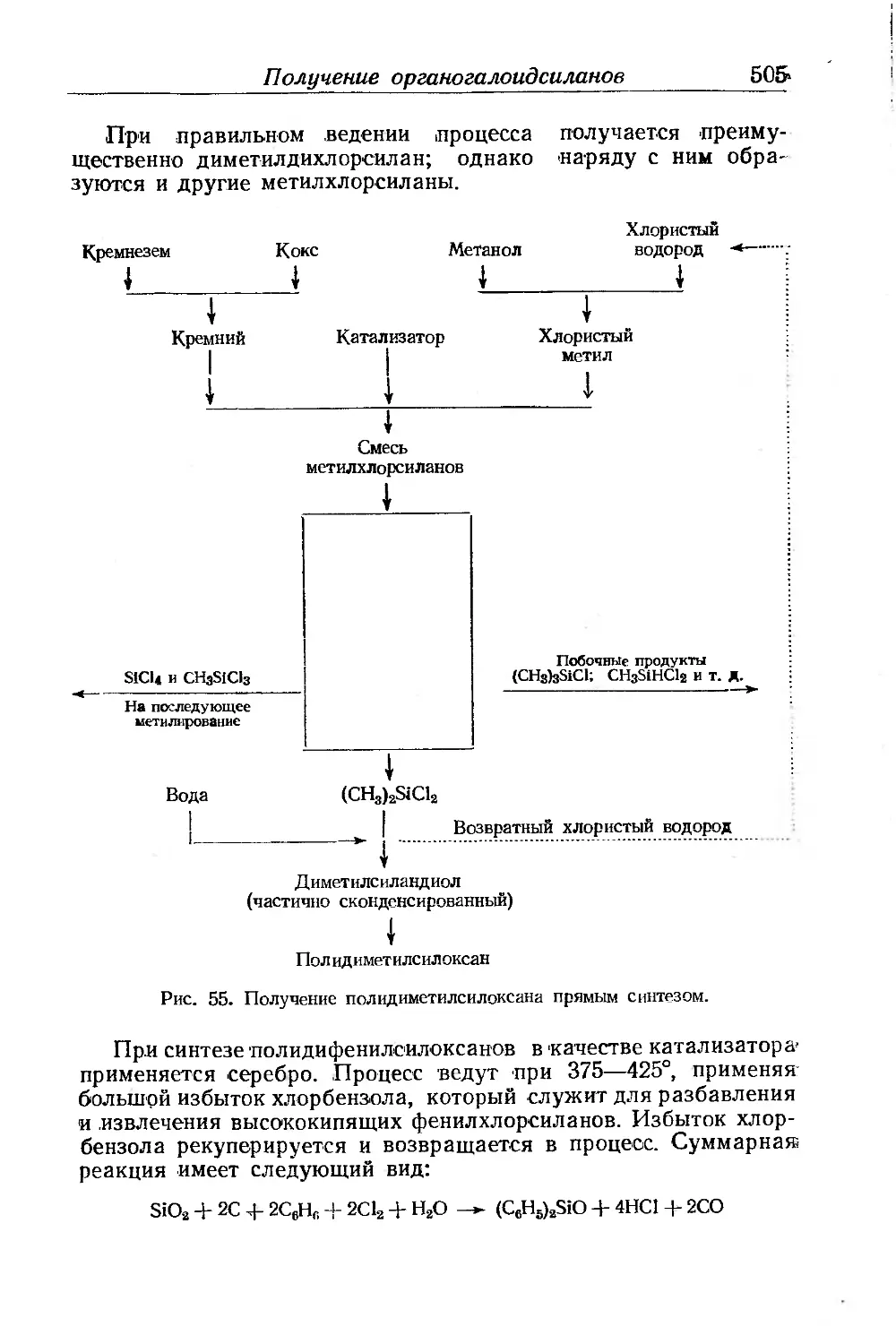
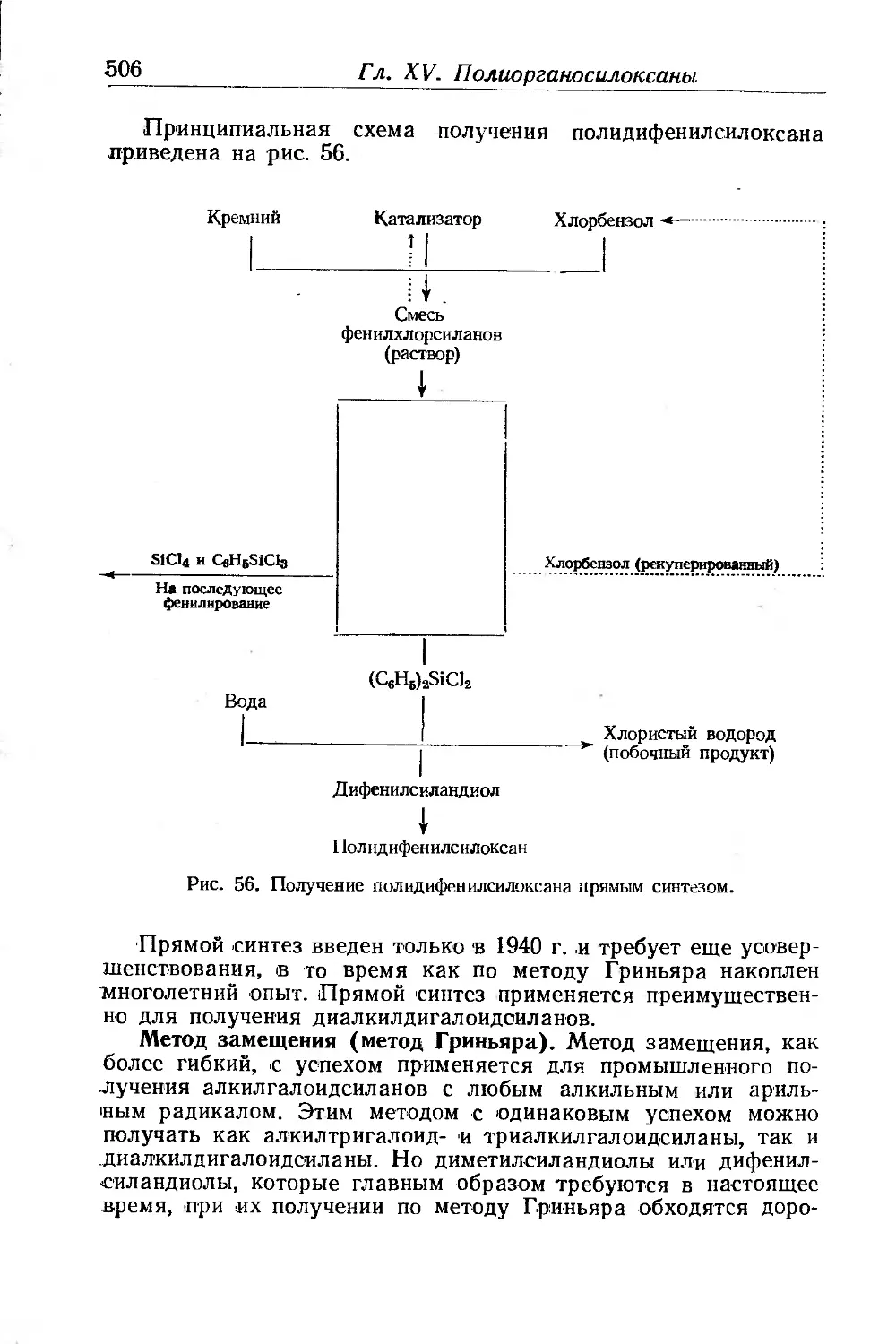
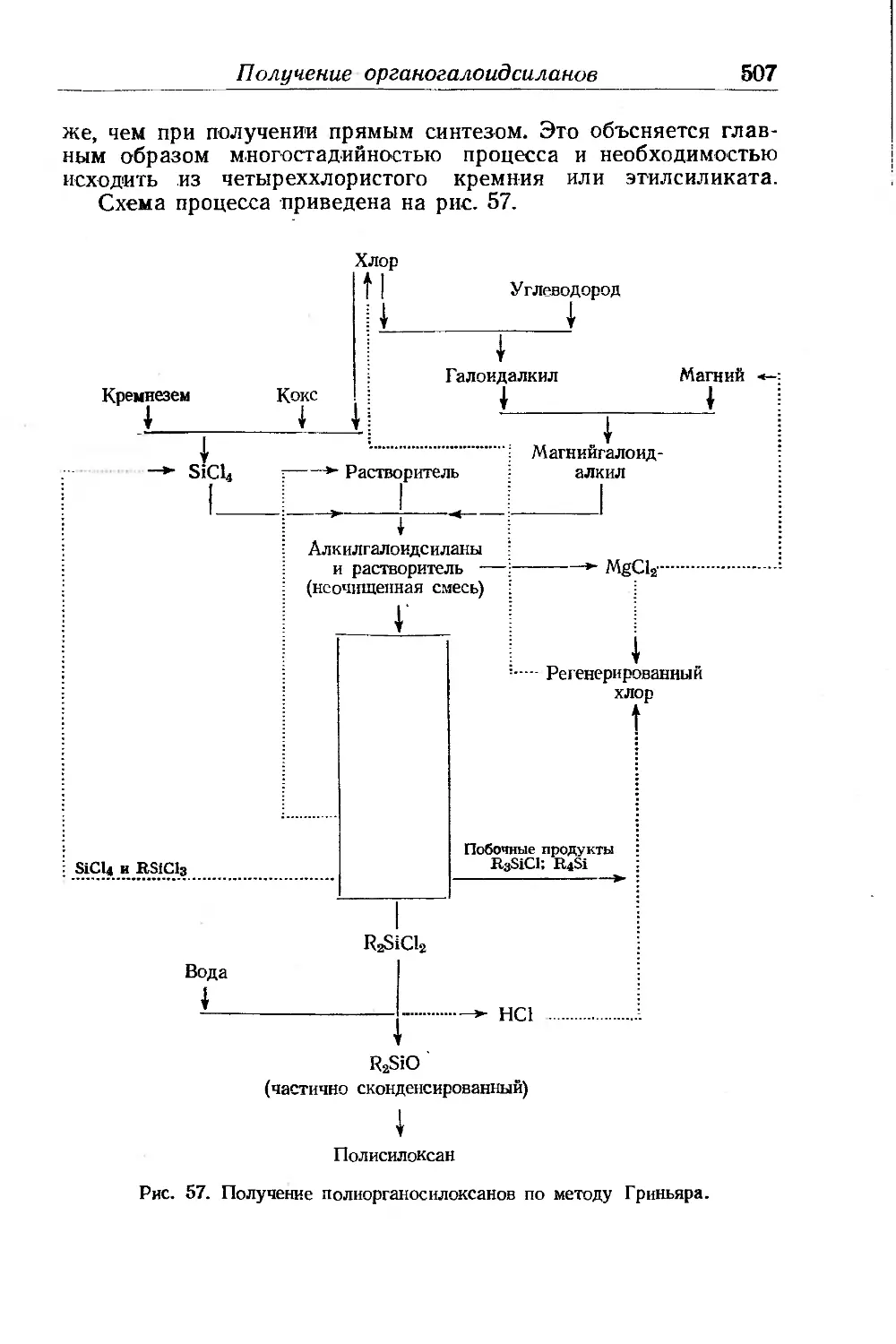
Глава XV. Полиоргаиосилоксаны. Ф. Аппель ......................... 499
Получение органогалоидсиланов............................. 501
Получение алкилсиланолов.................................. 504
Типы полиорганосилоксанов и их свойства .................. 508
Применение полиорганосилоксанов для красок................ 513
Совместимость полиорганосилоксанов с другими полимерами . 515
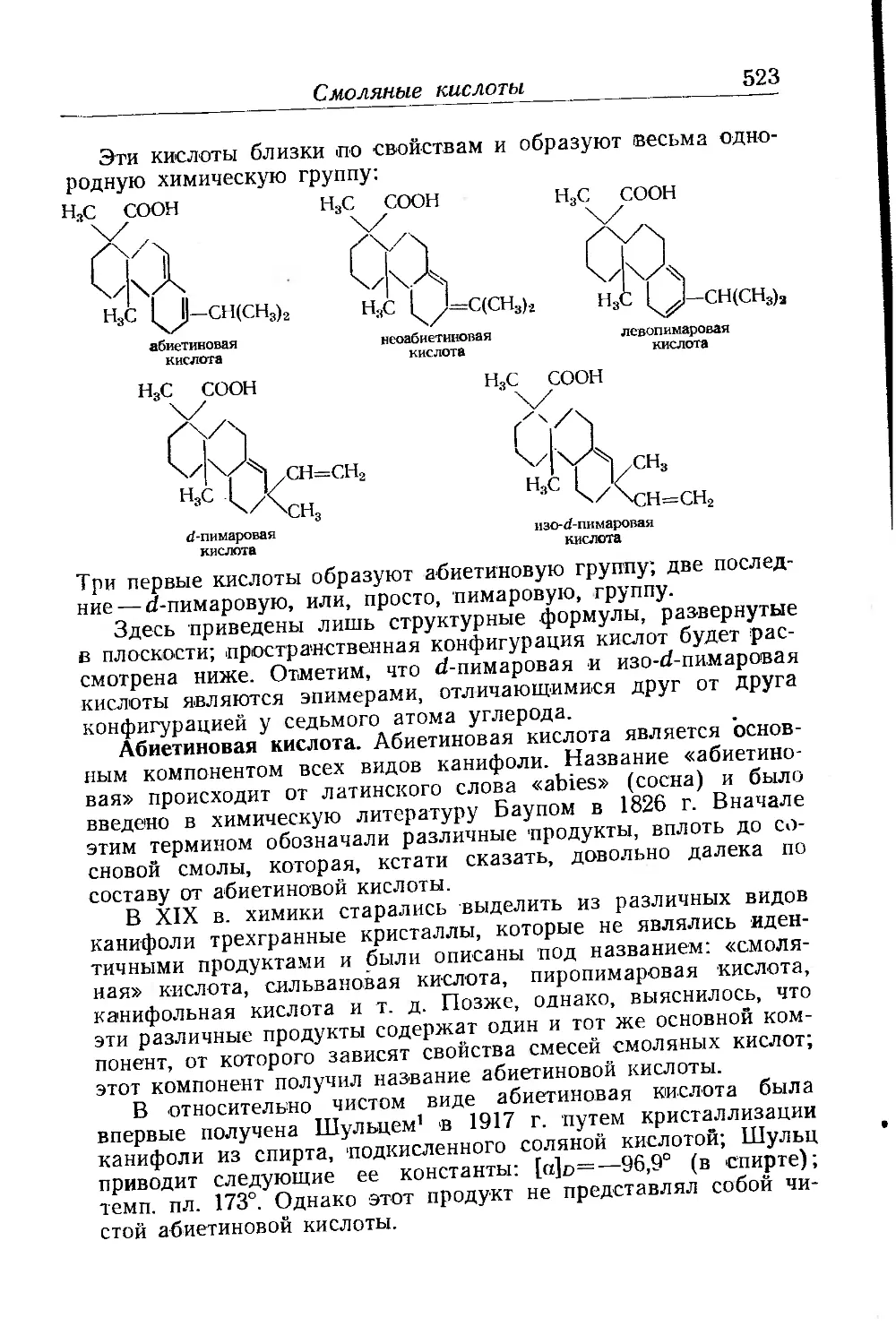
Глава XVI. Канифоль и ее производные. Р. Ломбар................... 518
Состав канифоли........................................... 519
Смоляные кислоты.............................................. 522
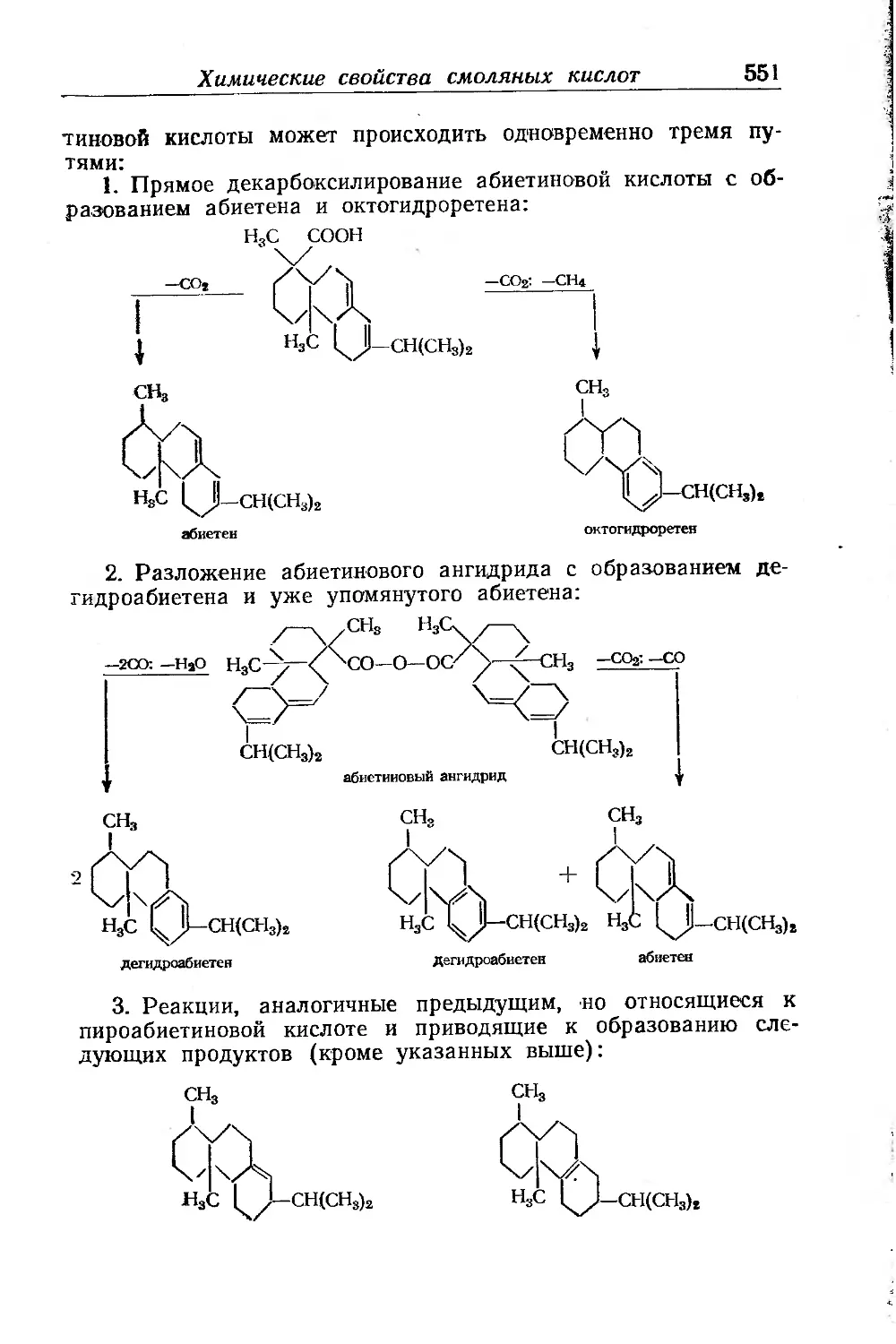
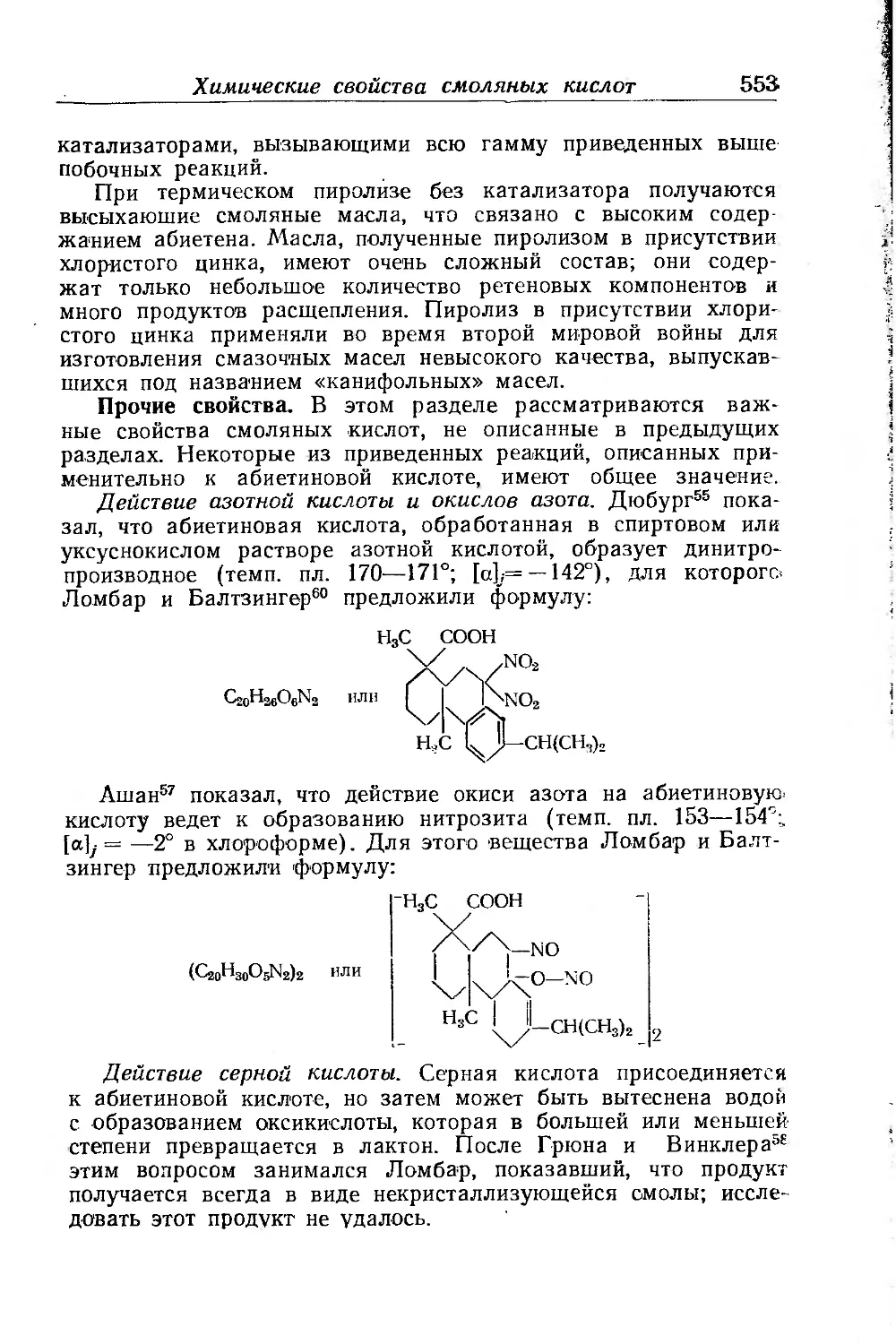
Химические свойства смоляных кислот и их производных . . 531
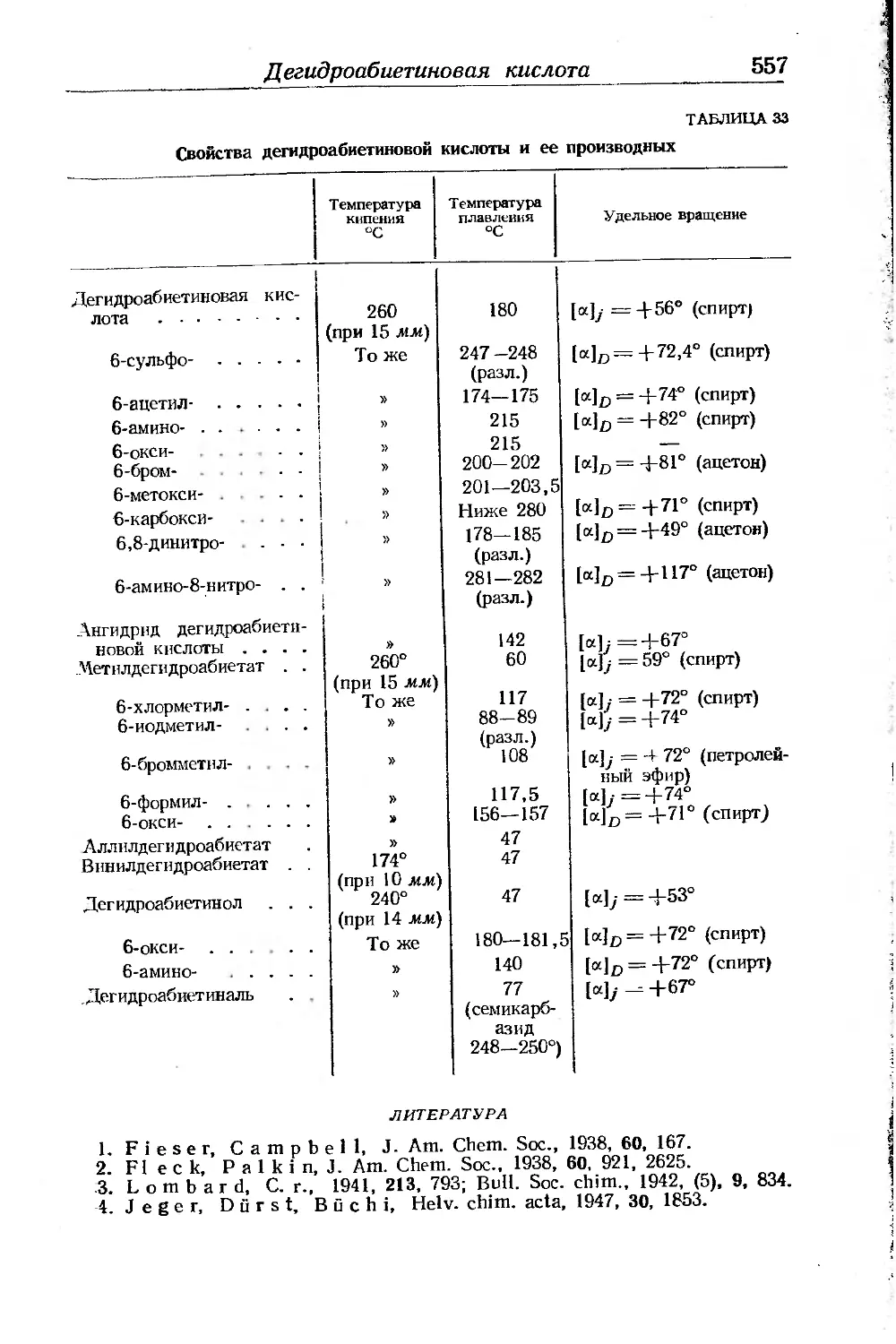
Дегидроабиетиновая кислота ............................... 556
Пространственное строение смоляных кислот................. 558
Химия канифоли и ее производных............................... 559
Предметный указатель............................................. 577
ПРЕДИСЛОВИЕ
Быстрое развитие лакокрасочной промышленности пред-
усматривает все более широкое использование новых видов
сырья и в частности многочисленных и разнообразных синте-
тических пленкообразующих веществ. Отсюда естественно выте-
кает необходимость изучения химии синтетических полимеров,
методов их получения, структуры, свойств и возможностей при-
менения в лакокрасочном производстве. В этом отношении пе-
ревод книги Г. Шампетье и Г. Рабата «Химия лаков, красок и
пигментов» представляет несомненный интерес, так как ,в книге
собран обширный материал о крайне разнообразном ассорти-
менте лакокрасочного сырья, включая все новые типы лаковых
смол.
Книга представляет собой двухтомную монографию, состоя-
щую из отдельных статей, написанных инженерами и учеными-
специалистами в различных узких областях, под общей редак-
цией Г. Шампетье и Г. Рабатэ. Первый том посвящен химии
синтетических пленкообразующих веществ, высыхающих масел
и канифоли (глава о тропических природных смолах при пере-
воде опущена). В этом же томе излагаются основы
химии полимеров, теория пленкообразования, теория старения
полимеров (лаковых пленок) и зависимость свойств лаковых
пленок от характера пленкообразователей. Во втором томе опи-
саны битумы и пеки, крахмал, казеин, а также сиккативы, рас-
творители и пластификаторы, минеральные и органические пиг-
менты, наполнители и вспомогательные материалы (дисперга-
торы, эмульгаторы, антиоксиданты и др.).
Недостатком книги является то, что авторы отдельных ста-
тей не придерживались какого-либо единого, общего для всей
книги принципа, и поэтому статьи довольно резко отличаются
друг от друга по построению, характеру изложения, объему и
глубине освещения материала. Кроме того, в книге часто недо-
стает практических указаний о возможности использования тех
или иных материалов для производства конкретных видов лако-
красочной продукции.
Несмотря на эти недостатки, книга, по нашему мнению,
должна служить полезным пособием для инженерно-техниче-
ских и научных работников лакокрасочной и смежных с нею
отраслей промышленности.
А. А. Беловицкий
ГЛАВА I
ХИМИЯ ВЫСОКОМОЛЕКУЛЯРНЫХ СОЕДИНЕНИЙ
И ПЛЕНКООБРАЗУЮЩИХ ВЕЩЕСТВ
Г. Шампетье
Окраска или лакировка поверхности это — покрытие ее тон-
ким слоем хорошо прилипающего вещества для защиты от кор-
розии или придания красивого внешнего вида. Если образовав-
шаяся на поверхности защитная пленка прозрачна, процесс на-
зывается лакированием. Если же пленка вследствие наличия
в ней пигментов непрозрачна, процесс носит собирательное на-
звание «окраска» (в это понятие входит нанесение на поверх-
ность различных покрытий, аппретов, порозаполнителей, мастик
и т. п.).
Большинство пигментов и наполнителей выполняет только
декоративные функции; благодаря своей непрозрачности они за-
крывают недостатки окрашиваемой поверхности или придают
ей красивый вид. Некоторые же из них выполняют непосредст-
венно защитные функции, например в антикоррозионных и не-
обрастающих красках. Как в защитных, так и в декоративных
покрытиях пигменты удерживаются на поверхности с помощью
обволакивающего их связующего вещества, так называемого
пленкообразующего. Пленкообразующие — это вещества, спо-
собные образовывать сплошную пленку, плотно прилегающую
к поверхности и облегающую все ее неровности без отслаива-
ния и растрескивания.
Таким образом, главной задачей в производстве лаков и
красок является подбор таких связующих веществ, пленки ко-
торых обладали бы адгезионными свойствами, эластичностью,
непроницаемостью, антикоррозионной стойкостью и твердостью.
Связующие вещества должны обладать различными свойствами
в зависимости от назначения изготовляемых на их основе по-
крытий: в строительстве, автомобильной или авиационной про-
мышленности, мебельной промышленности, для защиты метал-
лических изделий, конструкций или сооружений, в электропро-
мышленности, в производстве консервных банок, в живописи
или в других областях.
Применение красок восходит к глубокой древности. В каче?
стве первых связующих использовались природные вещества:
10 Гл. I. Химия высокомолекулярных соединений
крахмал, яичный белок, казеин, диспергированные в .воде. За-
тем стали применять «высыхающие» растительные масла, спо-
собные отверждаться, окисляясь в тонком слое под действием
кислорода воздуха, и в качестве растворителя — скипидар, а
также камедь, растворенную в спирте. В дальнейшем широкое
применение получили синтетические пленкообразующие и про-
дукты химической обработки природных веществ (нитроцеллю-
лозы, феноло-формальдегидные смолы и др.). В настоящее
время техника располагает огромным количеством разнообраз-
ных синтетических связующих веществ, в том числе аминопла-
стов, глифталевых смол, поливиниловых производных, полиами-
дов, силиконов и др.
Мало-помалу были разработаны теории, на основе которых
оказалось возможным более уверенно осваивать новые виды
связующих и подбирать их в соответствии с областями приме-
нения. Было установлено также, что все связующие вещества,
применяемые в лакокрасочной промышленности, являются высо-
комолекулярными соединениями, в изучении которых за послед-
ние двадцать лет достигнуты большие успехи.
Лакокрасочная промышленность в настоящее время стала
одной из самых значительных отраслей прикладной химии вы-
сокомолекулярных веществ. Поэтому в начале этой книги целе-
сообразно изложить основы этой новой .химической науки.
В течение более полутора веков органическая химия зани-
малась изучением природных и синтетических соединений, мо-
лекулы которых состоят лишь из нескольких десятков атомов.
Только к началу этого столетия было установлено существова-
ние молекул-гигантов, состоящих иногда из нескольких десят-
ков тысяч атомов. К таким веществам относятся белки — глав- ,
ная составная часть живого вещества, целлюлоза — опорное
вещество растительной клетки, крахмал — запасное вещество
растений, а также каучук. Возникла новая область химии, пред-
метом которой является изучение природных высокомолекуляр-
ных соединений, синтез высокополимеров и выяснение зависи-
мости их свойств от строения.
Химия высокомолекулярных соединений еще не достигла
успеха в синтезе таких природных веществ, как белки или цел-
люлоза, но уже освоен синтез веществ, близких по составу и
обладающих аналогичными свойствами, иной раз даже превос-
ходящими свойства природных веществ. Такие синтетические
макромолекулы получены путем соединения друг с другом боль-
шого количества одинаковых мономеров, состоящих из неболь-
шого числа атомов. Поэтому их следует рассматривать как
полимеры данного мономера и, поскольку степень их полиме-
ризации очень высока, их обычно называют «высокополиме-
рами».
Синтез высокополимеров
11
Синтез . высокополимеров
Полимеризация. Примером процесса полимеризации являет-
ся образование поливиниловых производных:
пСНа=СН -----сн2—сн—сн2—сн—сн2—сн—сн2—сн----
I 1111
R R R R R
В такие реакции вступают непредельные соединения, содер-
жащие двойные связи в определенных положениях. Образование
высокополимерных соединений состоит в непосредственном со-
единении мономерных звеньев, и конечный полимер имеет такой
же элементарный состав, как основной мономер.
Реакции полимеризации происходят иногда самопроизвольно
при обычной температуре (так полимеризуется, например, сти-
рол С6Н5—СН = СН2), но они ускоряются под влиянием тепла и
света, особенно ультрафиолетовых лучей, а также в присутст-
вии различных инициаторов и катализаторов: минеральных и
органических перекисей, некоторых галоидных соединений
(хлористый алюминий, фтористый бор, хлористое олово, пяти-
хлористая сурьма и др.), восстановительно-окислительных (ре-
докс-) систем и т. д. Реакции полимеризации это — цепные ре-
акции, для осуществления которых необходимо наличие активи-
рованных молекул мономера. Последние возникают в резуль-
тате различных процессов, приводящих к образованию свобод-
ных радикалов, к которым могут далее присоединяться моле-
кулы мономера (фаза инициирования).
Образование активных свободных радикалов под влиянием
света или в результате молекулярных столкновений состоит в
раскрытии двойных связей некоторого количества молекул мо-
номера:
сн2=сн • сн2—сн.
I I
R R
Перекиси, применяемые в качестве инициаторов реакции, яв-
ляются источниками свободных радикалов. Примером может
служить перекись бензоила, которая, распадаясь, образует сво-
бодные фенильные радикалы
С6Н5— СО—О-О—СО—С6Не — 2CeHs . + 2СОа
с которыми начинают соединяться молекулы мономера
CeHs.+ СН2=СН СвНБ—СН2—СН.
I I
R R
образуя в дальнейшем макромолекулярные цепи.
Активация галоидными солями металлов (типа катализато-
ров Фриделя-Крафтса) протекает по механизму типа образо-
1
Гл. I. Химия высокомолекулярных соединений
вания карбо пневых ионов. Так, хлористый алюминий в форме
комплексной кислоты Н + (А1С14)~ активирует мономер по схеме:
Н+(А1С14Г + СН2=СН — СН3—СН + (А1СЦ-
I I
R R
Карбониевый ион
СН3—СН
I
R
как и свободный радикал, может присоединять новые молеку-
лы мономеров.
В случае применения восстановительно-окислительной систе-
мы, например системы перекись водорода—соль двухвалент-
ного железа, инициирование реакции происходит в результате
образования свободных радикалов ОН», к которым могут при-
соединяться молекулы .мономеров, вызывая дальнейший рост
макромолекулярных цепей:
Fe2+ + Н2О2 Fe3+ 4- ОН" + ОН»
НО» + СН2=СН НО—СН2—СН»
I I
R R
За стадией инициирования следует стадия очень быстрого
роста цепи, в течение которой молекулярные цепи путем после-
довательного присоединения молекул мономера удлиняются и
образуют макрорадикалы:
СвН6—СН2—СН» +СН2=СН С6Н5—СН2—СН—СНа-СН»
I I II
R R R R
СвНь-СН2—СН—СН2—СН » + СН2=СН -►
I I I
R R R
-> СвН6—СН2—СН—СН2—СН—СН2—СН» и т. д.
I I I
R R R
Стадия роста цепи заканчивается обрывом цепи, когда рост
макрорадикалов прекращается. Полимеризация может прекра-
щаться в результате различных процессов. Двумя самыми важ-
ными из них являются дезактивация путем димеризации —со-
единения двух растущих макромолекул
С6НЬ—(СН2—СН)Я—СН2—СН » + » СН—СН2—(СН—СН2)р—с6н5 ->
I III
R R R R
-* СвН6—(СН2-СН)„—СН,—СН—СН-СНг-(СН—СН2)„—СвН5
I 1 I I
R R R R
Синтез высокополимеров
13
и дезактивация путем миграции атомов водорода, когда макро-
молекулы теряют характер свободных радикалов вследствие
перемещения атома водорода от одного радикала к другому:
С6Н5— (СН2—СН)„—СН2—сн. + .CH—СН2—(CH—СН2)О-С6Н5 —-
I II I
R R R R
-> С6Н5—(СН2—СН)„—СН2—СН2 + СН=СН—(CH—СН2)„—С6Н5
I II'
R R R R
Миграция водорода от одной активной макромолекулы к
другой приводит к тому, что рост обеих макромолекул оказы-
вается прерванным. Но возможна также миграция водородного
атома к активной макромолекуле,
рост которой приостанавливается,
от неактивной' молекулы (мономер
или растворитель), которая, превра-
щаясь в свободный радикал, может
вызвать рост новых макромолеку-
лярных цепей (дезактивация «путем
передачи цепи).
При полимеризации никогда не
получаются макромолекулы одина-
ковой степени полимеризации, а
«происходит образование целой гам-
Рис. 1. Кривая распределения
макромолекул по степени поли-
меризации.
мы продуктов с различными степе-
нями полимеризации, располагаю-
щимися на кривой вероятности,
имеющей максимум, соответствую-
щий преобладающей степени полимеризации. В зависимости от
условий реакции максимум может быть более или менее ярко
выражен, что соответствует большей или меньшей однородности
образовавшихся макромолекул по степени полимеризации (рис. 1).
Обычно при высокой температуре или в присутствии значи-
тельного количества катализатора процессы полимеризации
протекают с большей скоростью, но достигаемые при этом сте-
пени полимеризации в среднем ниже, чем при более низкой тем-
пературе и меньшем количестве катализатора.
При соединении отдельных молекул мономера в процессе по-
лимеризации могут образоваться макромолекулы, характеризу-
ющиеся некоторыми неравномерностями структуры. Прежде
всего присоединение мономеров друг к другу не всегда проис-
ходит однотипно, так, чтобы «голова» одной молекулы соединя-
лась с «хвостом» другой, создавая правильную структуру:
---СН2—СН—СН>—СН—СН.>—CH—СН2—СН-----
I I I
R R R R
14
Гл. I. Химия высокомолекулярных соединений
Некоторые молекулы соединяются также «голова к голове»
----сн2- сн—сн—сн.----
I I
R R
или «хвост к хвосту»:
----СН—CHj—сн2—сн-----
I I
R R
Таким образом, расположение боковых групп в образующей-
ся макромолекуле может быть нерегулярным и определяться
лишь законами вероятности; иногда может преобладать какое-
нибудь определенное расположение в соответствии с характером
исходных мономеров, например:
---СН2—СН—СН2—СН—СН—СН2—СН—СН2—СН2—СН—СН2—СН----
Illi i I
R R R R R R
Кроме того, боковые группы могут или регулярно распола-
гаться все с одной стороны макромолекулярной цепи, или же в
произвольном порядке размещаться по обе ее стороны.
R R R
• • —СН2—СН—СН2—СН—СН2—СН—СН2—СН—СН2—СН—СН2—СН-----
I I I
R R R
Поскольку возможно сочетание обоих типов структуры, в
определенных условиях полимеризации могут получаться мак-
ромолекулы весьма неправильного строения, что иногда очень
существенно сказывается на технических свойствах продукта.
Наконец, при активации молекул мономера возможно иногда
образование двух свободных валентностей у одного и того же
атома углерода:
I
—СН2—с—
I
R
В этом случае рост цепи происходит в двух различных на-
правлениях. Образовавшаяся при этом макромолекула имеет
более или менее значительные ответвления (боковые цепи), ко-
торые могут иногда соединять («сшивать») две макромолеку-
лярные цепи с образованием макромолекулярной сетчатой
структуры. Эти разветвления и мостики весьма легко обра-
зуются при полимеризации диенов в производстве синтетиче-
ских эластомеров.
% Синтез высокополимеров 15
• Из вышесказанного видно, что реакции полимеризации, про-
Й водимые без необходимых предосторожностей, могут привести
£ к образованию макромолекул очень сложного строения. Даже
| небольшие изменения условий производства могут вызвать
Л, существенное различие физических и механических свойств
| получаемых продуктов.
f Количество соединений, способных к полимеризации, очень
Й велико, причем технические возможности получения разнооб-
к разных полимеров еще более расширяются благодаря совмест-
| ной полимеризации двух или нескольких мономеров, входящих
Я в состав одних и тех же макромолекул. Так, хлористый винил,
полимеризованный отдельно, без других мономеров, дает по-
£ ливинилхлорид:
4 ---СН2—СН—СН3—СН—СН2—СН—СН2—СН-
1111
£ С1 С1 С1 С1
а винилацетат — поливинилацетат:
! ---СН2—СН—СН2—СН—СН2—СН—СН2—СН-
1111
О О О О
1111
СОСН3 СОСН3 СОСН3 СОСНз
При совместной же полимеризации хлористого винила и
винилацетата получаются сополимеры хлористого винила с ви-
нилацетатом, макромолекулы которых содержат одновременно
и атомы хлора и ацетильные группы, в произвольном порядке
; расположенные в макромолекулярных цепях:
---СН2—СН—СН2—СН—СН2—СН—СН2—СН—СН2—СН-СН2—СН------
I I I I I I
С1 О С1 О О С1
I II
СОСНз СОСНз СОСНз
Свойства таких сополимеров зависят от соотношения вхо-
дящих в их состав мономеров и отличаются от свойств полиме-
• ров, которые были бы получены при раздельной полимериза-
ции каждого из двух мономеров.
Методом совместной полимеризации можно получать широ-
t кий ассортимент продуктов, пригодных для самого различного
i применения в лакокрасочной промышленности.
К Поликонденсация. Одним из примеров поликонденсации яв-
ляется взаимодействие двухосновной кислоты с двухатомным
спиртом, приводящее к образованию полиэфира:
? норе—(сн2)„—соГон + ню—(сн2)р—о;н +’нб:ос—(сн2)„—со—loti -►
----ОС—(СН2)„—СО—О—(СНа)р—О—ОС—(СН2)„-со-
16 Гл. I. Химия высокомолекулярных соединений
В этом случае небольшие молекулы соединяются друг с дру-
гом, отщепляя какое-либо соединение, чаще всего воду, как
это показано в приведенном примере.
Реакции поликонденсации соответствуют обычным реак-
циям классической химии; по мере их течения происходит по-
степенное увеличение размеров макромолекул. Теоретически
реакции поликонденсации чистых оксикислот или строго эквимо-
лекулярных смесей чистой двухосновной кислоты и гликоля мо-
гут продолжаться до тех пор, пока все малые молекулы не сое-
динятся в одну гигантскую молекулу. Практически же рост
макромолекул ограничен присутствием монофункциональных
примесей или избытком одного из бифункциональных реаген-
тов (кислоты или алкоголя). В первом случае монофункци-
ональный реагент, присоединившись к макромолекуле, образует
нереакционноспособную концевую группу, прекращающую
рост цепи. При избытке одного из реагентов в конечном итоге
•образуются макромолекулы с одинаковыми концевыми функци-
ональными группами, не способными соединяться друг с дру-
гом. В промышленной технике эти два приема часто исполь^
зуют для ограничения роста макромолекул до определенной
величины, соответствующей требованиям, вытекающим из усло-
вий применения продукта.
Полимеризация с раскрытием циклов. Примеры таких про-
цессов пока еще немногочисленны. Наиболее характерным при-
мером является полимеризация окиси этилена в присутствии
воды или спирта как инициаторов реакции. В результате при-
соединения молекулы спирта к окиси этилена получается окси-
соединение
Н2С СН2 + ROH ro-ch2—сн2он
способное присоединить следующую молекулу окиси этилена:
Н2С--СН2 + RO—СН2—СН2—ОН —RO—СН2—СН2—О-СН2—СН2ОН
''о'/
и т. д. до очень высоких степеней полимеризации:
RO—СН2—СН2 - (О—СН2—СН2) О—СН2—СН2ОН
Высокомолекулярные пленкообразующие вещества могут
быть получены также путем введения в природные высокомоле-
кулярные соединения определенных функциональных групп,
придающих им требуемые свойства. Таким способом получают
растворимые производные целлюлозы, превращая ее при по-
мощи этерификации в нитрат, ацетат или простые эфиры (ме-
Пространственное строение макромолекул 17
тил-, этил- или бензилцеллюлозу), широко применяемые в про-
изводстве лаков и красок. Совершенно очевидно, что условия
проведения этих обменных реакций должны подбираться так,
чтобы не разрушалась структура макромолекул или, по край-
ней мере, не происходила деструкция, приводящая к изменению
основных технических свойств вещества. Однако некоторая де-
струкция иногда желательна, например, когда требуется облег-
чить растворение высокополимеров или уменьшить вязкость по-
К лучаемых растворов.
г* Пространственное строение макромолекул
'2 Соединение отдельных мономерных звеньев в высокополи-
к мере может происходить таким образом, что в результате обра-
зуются макромолекулы различного пространственного строения.
Мономеры могут соединяться друг с другом подобно
звеньям длинной цепи, образуя так называемые линейные, или
; * цепные, макромолекулы. К такому типу полимеров (если не
I Принимать во внимание возможные разветвления цепи) можно
< \~2>тнести полистирол:
---СН2—СН—СН2—СН—СН2—СН—СН2—СН----
Ох 1111
с6н5 с„н5 С6Н5 С6Н5
( Соединение мономерных звеньев может также происходить
двух пространственных направлениях, причем образуются
хоетчатые или двухмерные макромолекулы; примеры подобных
веществ немногочисленны. Возможно, что некоторые фено-
пласты или аминопласты на промежуточных стадиях поликон-
денсации (перед окончательным отверждением) приближаются
к такому типу строения макромолекул.
Наконец, некоторые мономеры могут образовывать связи
в трех пространственных направлениях, давая трехмерные ма-
кромолекулы. В качестве примера таких полимеров можно
назвать отвержденные фенопласты, аминопласты, полиэфиры.
Путем соответствующего подбора мономеров, участвующих
в реакциях полимеризации или поликонденсации, можно полу-
чать по желанию или линейные макромолекулы, или же трех-
!( мерные, характеризующиеся наличием более или менее плотной
I макромолекулярной сетки. Структура макромолекулярной сетки,
в конечном итоге, будет зависеть от функциональности моно-
мера или смеси молекул мономеров, вступающих в реакцию.
Две монофункциональные молекулы, например однооснов-
ная кислота и одноатомный спирт, могут соединяться, образуя
только молекулу эфира
RCOOH + HOCH2R ?
2—156 I ^аш^овиар*-1--'
| Уфимский химзавод
библии ‘
18 Гл. I. Химия высокомолекулярных соединений
имеющую совершенно определенные размеры; молекулярный
вес этого эфира равен сумме молекулярных весов кислоты и
спирта за вычетом молекулярного веса воды (рис. 2).
Рис. 2. Конденсация двух монофункциональных
соединений.
В противоположность этому, в результате соединения би-
функциональных молекул, например молекул оксикислоты
НООС-(СН2)„ ЮН’+НООС—(СН2)„— ОН фН ООС -(СН2)„— ОН —
-*----ООС—(СН2)„—О-ОС— (СН2)„—О-ОС— (СН2)Л-О-
или молекул двухатомного спирта с молекулами двухосновной
кислоты, могут образоваться линейные макромолекулы, длина
которых может быть сколь угодно велика (рис. 3).
Рнс. 3. Линейная поликонденсация двух различных
бифункциональных соединений (А) или гомополнкон-
денсация одного бифункционального соединения (Б).
Винильные производные, вследствие разрыва двойной связи
СН,=СН -> — СН2—СН—
I I
R R
ведут себя как бифункциональные молекулы и в результате
полимеризации также образуют линейные макромолекулы.
Для образования макромолекул трехмерного строения функ-
циональность реагирующих молекул должна быть равна по
меньшей мере трем (рис. 4).
Так, например, в молекулах триглицеридов жирных кислот
высыхающих масел каждый из трех радикалов жирной кислоты
может соединяться с радикалом соседнего триглицерида в про-
цессе окислительной или термической полимеризации.
Трехмерные макромолекулы (рис. 5) могут образовываться
также при поликонденсации какого-либо бифункционального
компонента с другим компонентом, по меньшей мере трифунк-
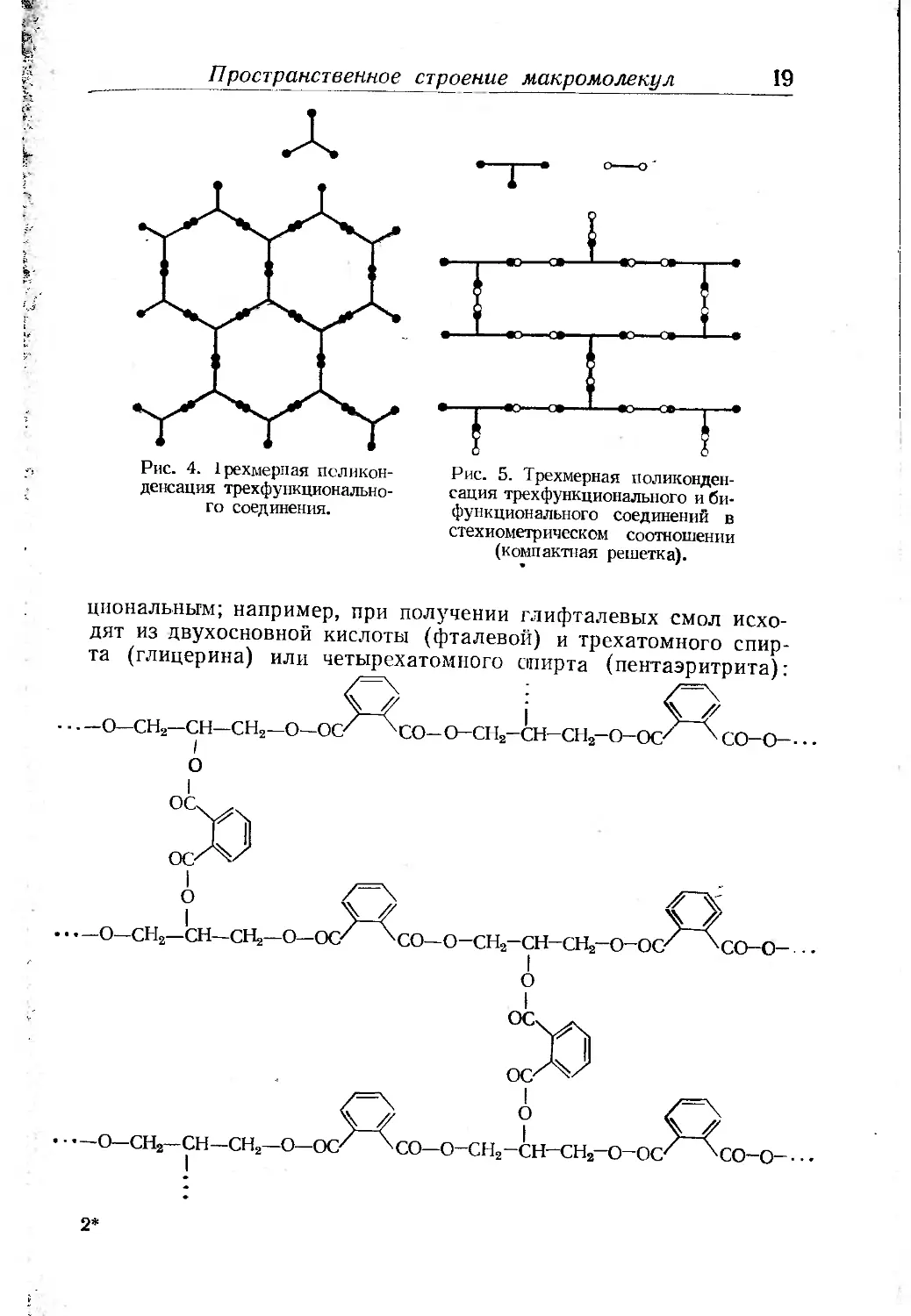
Пространственное строение макромолекул 19
Рис. 4. Срехмерная поликон-
денсация трехфункционально-
го соединения.
Рис. 5. Трехмерная поликонден-
сация трехфункциопального и би-
функционального соединений в
стехиометрическом соотношении
(компактная решетка).
дневальным; например, при получении глифталевых смол исхо-
дят из двухосновной кислоты (фталевой) и трехатомного спир-
та (глицерина) или четырехатомного спирта (пентаэритрита):
—О—СН2—СН—СН2—О—ОС
СО-О-СН,-СН
О
со— О-СН2-СН-СН2-О-О(/ \CO-O-
I
о
со—о-сн2-сн-сн2-о-ос
2*
20
Гл. I. Химия высокомолекулярных соединений
При частичной замене двухосновной кислоты на однооснов-
ную или трех-(четырех)-атомного спирта на двухатомный неко-
торые мостиковые связи не образуются, в результате чего полу-
чаются макромолекулы, сохраняющие трехмерное строение, но
содержащие ослабленные участки (рис. 6). Такое строение
имеют алкидные смолы, при синтезе которых часть фталевого
ангидрида заменена одноосновной жирной кислотой.
Компактные трехмерные структуры могут образовывать фе-
нопласты. Обычный фенол присоединяет формальдегид в трех
•положениях (два орто-положения и паращоложение), бла-
годаря чему образуются весьма плотные трехмерные струк-
туры.
» I' • 0—0
Рис. 6. Трехмерная поликонденсация трехфункцио-
нального, бифункционального и монофункциональ-
ного соединений в эквимолекулярных соотношениях
(решетка с ослабленными участками).
Таковы нерастворимые и неплавкие материалы типа бакелита.
Аналогично ведет себя л«-крезол, имеющий также подвижные
атомы водорода в трех положениях. В противоположность
этому, n-крезол, являющийся бифункциональным соединением,
вступая в реакцию с формальдегидом, образует линейные ма-
кромолекулы.
ОН
ОН
о-крезол
(f-2)
ОН
СН,
л-крезот
(1^)
фенол л-крезол
0=3) (f=3)
Пространственное строение макромолекул 21
Смеси м- и n-крезолов, имеющие среднюю функциональность,
промежуточную между 2 и 3, образуют макромолекулы доволь-
но плотной структуры. Некоторые из подобных продуктов рас-
творимы и пригодны для изготовления лаков и красок.
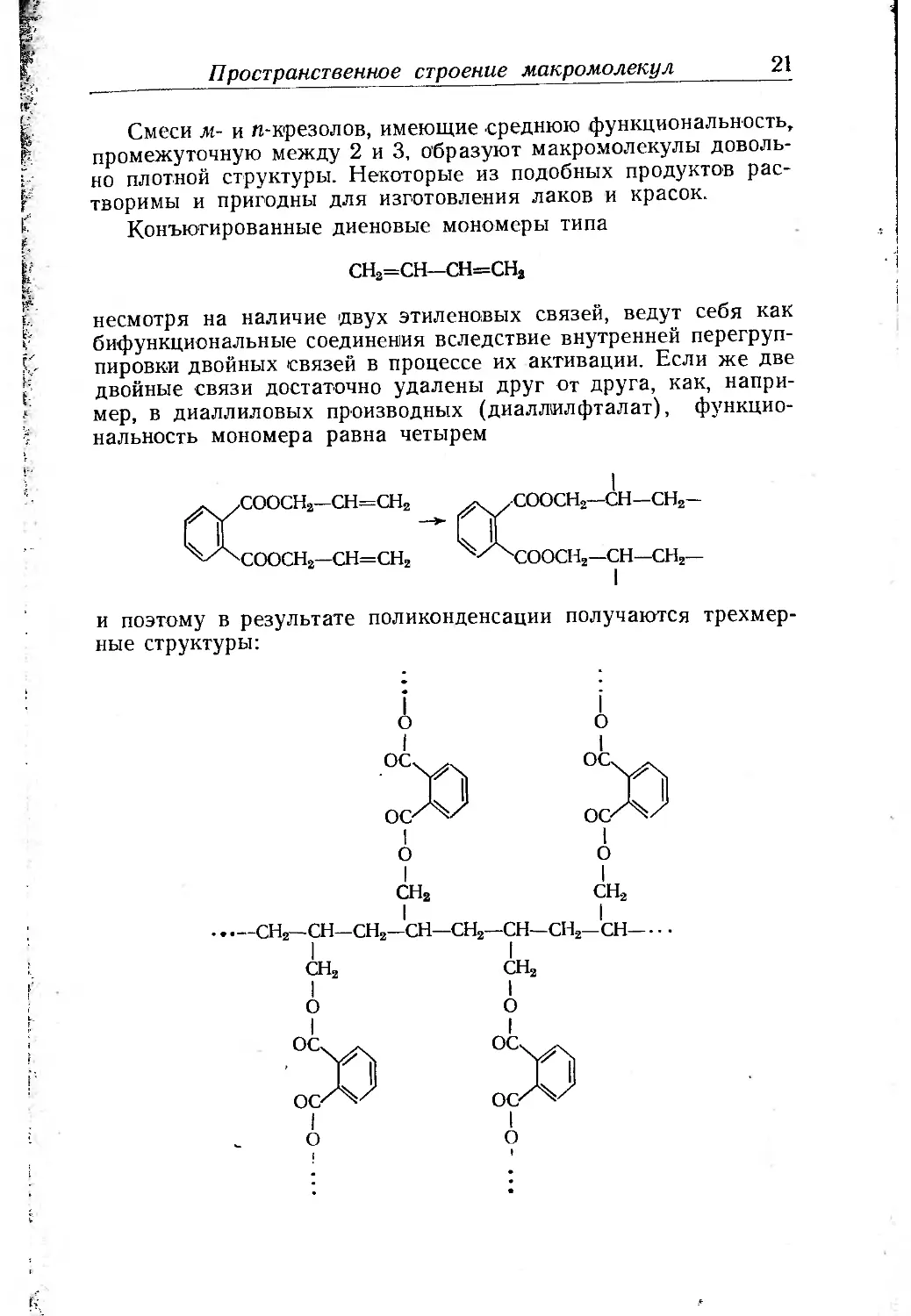
Конъюгированные диеновые мономеры типа
СН2=СН—СН=СН2
несмотря на наличие двух этиленовых связей, ведут себя как
бифункциональные соединения вследствие внутренней перегруп-
пировки двойных связей в процессе их активации. Если же две
двойные связи достаточно удалены друг от друга, как, напри-
мер, в диаллиловых производных (диаллилфталат), функцио-
нальность мономера равна четырем
СООСН2-СН=СН2
СООСН2-СН=СН2
соосн»—сн—сн2—
СООСН2—СН—СН2—
и поэтому в результате поликонденсации получаются трехмер-
ные структуры:
22 Гл. I. Химия высокомолекулярных соединений
Свойства высокополимеров и способность к пленкообразованию
Использование высокомолекулярных соединений в лакокра-
сочной промышленности определяется не только их химически-
ми, но и физическими и механическими свойствами. Химическое
строение полимера оказывает прямое влияние на его раствори-
мость и стойкость по отношению к воде и различным реагентам
при их воздействии на сухую пленку; эти свойства зависят
главным образом от строения мономерных звеньев и особенно
от характера заместителей. Физические свойства, в частности
растворимость и вязкость, также зависят от природы мономе-
ров, но, кроме того, в значительной степени от формы и раз-
мера макромолекул. В полимергомологическом ряду линейных
высокополимеров по мере возрастания степени полимеризации
растворимость уменьшается, а вязкость растворов при одина-
ковой их концентрации увеличивается. Для удобства использо-
вания красок и лаков следует применять при их изготовлении
высокомолекулярные 'соединения такой степени полимеризации,
чтобы растворимость их была достаточно большой, вязкость не
слишком высокой, но чтобы эти соединения в то же время об-
ладали требуемыми, механическими свойствами. Удовлетворить
всем этим противоречивым требованиям иногда бывает нелегко.
Следует также иметь в виду, что в то время как линейные
полимеры при соответствующем подборе растворителя можно
легко перевести в раствор, для трехмерных высокомолекуляр-
ных соединений в конечных стадиях их образования характерна
полная нерастворимость. Поэтому для окраски поверхностей мо-
гут быть применены только исходные мономеры или раствори-
мые начальные продукты поликонденсации. Последующее обра-
зование трехмерной структуры достигается (или завершается)
за счет протекания окислительных процессов (как в случае три-
глицеридов высыхающих масел) или путем дальнейшей поли-
меризации или поликонденсации под действием тепла или ката-
лизаторов (как в случае фенопластов). Преимущества способа
окраски с образованием в конце процесса трехмерных макро-
молекул, создающих сухую защитную пленку, основаны именно
на нерастворимости молекул-гигантов. Слой краски или лака
после высыхания перестает быть растворимым в растворителях,
которые применялись для нанесения на подложку.
Пленкообразуюшие вещества с линейными макромолекула-
ми сохраняют свою растворимость и после высыхания. Образо-
вание пленки происходит вследствие простого улетучивания
растворителя. Хотя при этом в результате взаимного переплете-
ния линейных макромолекул и образуется структура, напоми-
нающая трехмерную, но такая сетка не обладает должным
сцеплением. В то время как в трехмерных макромолекулах все
Свойства высокополимеров и пленкообразовшше
23
атомы соединены между собой ковалентными связями, которые
не разрываются под действием органических растворителей,
атомы в переплетенных линейных макромолекулах связаны
лишь относительно малыми силами межмолекулярного взаимо-
действия, уступающими влиянию применяемых растворителей.
Механические свойства высокомолекулярных соединений
также находятся в тесной зависимости от формы, величины й
химического состава макромолекул. Полимеры трехмерной
структуры вследствие наличия ковалентных связей между всеми
атомами образуют жесткие 'пленки, обладающие высокой ме-
ханической прочностью. При слишком большой уплотненности
трехмерной структуры, как, например, в продуктах поликонден-
сации обычного фенола с формальдегидом, получаются очень
твердые неэластичные пленки. В подобных случаях необходимо
уменьшить плотность структуры, например путем частичной за-
мены обычного трехфункционального фенола пара-замещен-
ным бифункциональным фенолом или (в глифталевых смолах)
путем частичной замены бифункционального фталевого ангид-
рида монофункциональными жирными кислотами.
Линейные макромолекулы образуют пленки, в которых мо-
лекулярные цепи, образованные ковалентными связями, скреп-
лены только за счет весьма слабых сил взаимодействия (силы
ван-дер-Ваальса, полярные взаимодействия) и при деформациях
подложки возможно некоторое скольжение цепей друг относи-
тельно друга. Благодаря этому пленки обладают эластичностью,
которая может быть еще увеличена путем введения особых до-
бавок, так называемых пластификаторов или мягчителей.
Молекулы пластификатора, внедряясь между макромолекуляр-
ными цепями, раздвигают их и несколько ослабляют их взаим-
ную связь. Чрезмерное ослабление межмолекулярных связей не-
желательно, так как полезна только возможность обратимых,
квази-эластических деформаций пленки ,и совершенно недопусти-
ма способность пленки к необратимым, т. е. истинно пластиче-
ским деформациям. Покрытия на основе линейных полимеров
иногда обладают недостаточной твердостью, которая, однако,
может быть повышена путем добавления различных природных
или синтетических смол. На механическую прочность лакокра-
сочных пленок влияют также пигменты и наполнители, входя-
щие в состав покрытий.
Благодаря исследованиям в области химии высокомолеку-
лярных -соединений оказалось возможным понять механизм
образования пленочных покрытий, объяснить их свойства и,
наконец, возникла возможность подбора пленкообразующих
связующих веществ или улучшения их свойств в соответствии с
назначением покрытий. Химия больших молекул может пред-
ложить в настоящее время лакокрасочной промышленности
24
Гл. I. Химия высокомолекулярных соединений
большой ассортимент продукции, удовлетворяющей самым раз-
нообразным техническим требованиям; при этом разнообразие
этой продукции продолжает расти. Как будет видно из 'сле-
дующих глав этой книги, некоторые синтетические материалы
в отдельных областях уже совершенно вытеснили применяв-
шиеся ранее связующие вещества, содержащие жирные кис-
лоты; другие, как, например, силиконы, только начинают нахо-
дить широкое применение, но все они при правильном исполь-
зовании могут открыть для лакокрасочной промышленности
огромные новые технические и экономические возможности.
ЛИТЕРАТУРА
G. Champetier, Les molecules geantes et leurs applications, Albin Mi-
chel, Paris, 1948.
J. D u с 1 a u x. Macromolecules et matieres plastiques, Presses Universitaires
de France, Paris, 1949.
P P i g a n i о 1, Macromolecules, t. I, Dunod, Paris, 1947.
A. V. Blom, Organic coatings in theory and practice, Elsevier Publishing Cy
Inc., Amsterdam, 1949.
J. J. M a 11 i e 1 1 o, Protective and decorative coatings, Chapman a. Hall,
London, 1946. •
К. H. Meyer, Natural and synthetic high polymers, Interscience publishers
Inc., New- York, 1950.
Comptes rendus du ler Congres International de 1’Industrie des Peintures et des
Industries associees, Paris, 1947.
ГЛАВА II
ВЫСЫХАЮЩИЕ МАСЛА
Ж. Пти
СОСТАВ ВЫСЫХАЮЩИХ МАСЕЛ
Природные растительные и животные высыхающие масла
являются триэфирами глицерина, трехатомного спирта, пропан-
триола строения СН2ОН—СИОН—СН^ОН.
Первичные спиртовые функциональные группы глицерина
обычно обозначаются буквой а, а средняя вторичная — бук-
вой р. Этот способ обозначения, хотя и не соответствует стан-
дартной химической номенклатуре, но часто употребляется на
практике.
В маслах три гидроксила глицерина этерифицированы жир-
ными кислотами, имеющими линейную углеродную цепь с чет-
ным числом атомов углерода. Некоторые масла составляют
исключение из этого правила, но они встречаются редко и не
относятся к группе высыхающих масел. Продукты этерифика-
ции глицерина жирными кислотами называются глицеридами.
Число атомов углерода в жирных кислотах колеблется для
большинства масел .между 14 и 22 (чаще всего 18), но строение
цепи может быть самым разнообразным (наличие одной или
нескольких двойных связей, тройных связей, гидроксильных
групп, кетонных групп). Поэтому существует довольно много
различных кислот и еще больше разнообразных глицеридов,
так как глицерин в маслах большей частью этерифицирован.
тремя различными кислотами:
СН2—OOCR
I
СН—OOCR'
I
СН2—OOCR"
где R, R', R" — радикалы, различающиеся между собой или
числом атомов углерода, или структурой углеродной цепи.
Такие смешанные глицериды весьма разнообразны; при ана-
лизе масел часто удается обнаружить присутствие одновремен-
но четырех, шести и даже более различных кислот.
26
Гл. II. Высыхающие масла
Важнейшие жирные кислоты и их производные
Предельные кислоты. Пальмитиновая кислота С15Н31СООН
встречается во многих маслах и жирах. Она представляет со-
бой воскообразное белое твердое вещество, плавящееся при
62,5—63°. Температура кипения:
Давление, мм pm. cm....... 1 2 4 8 15 20
Температура кипения, °C . . . 167,4 179 192,2 206,1 215 219
Коэффициент преломления Пд= 1,4385. Нерастворима в
воде; легко растворяется в горячем спирте и в хлороформе.
При нагревании 'растворяется в большом числе растворителей.
Метилшальмитат: темп. пл. 29°; темп. кип. 163° при
4 мм рт. ст., 196° при 15 мм рт. ст.
Этилиальмитат: темп. пл. 33,5°; темп. кип. 182° при
2 мм рт. ст., 213—215° при 15 мм рт. ст.
Амид пальмитиновой кислоты: темп. пл. 107°.
Анилид пальмитиновой кислоты: темп. ил. 90,2°.
Пальмитат свинца чрезвычайно плохо растворяется в эфире.
Стеариновая кислота С17Н35СООН очень часто входит в со-
став жиров. Она представляет собой белое твердое воскооб-
разное вещество; мол. в. 284,4; темп. пл. 69,5—71°. Температура
кипения:
Давление, мм рт. ст........ 1 2 4 8 15
Температура кипения, °C .... 183,6 195,9 209,2 224,1 232
Коэффициент преломления «0=1,4413. Кислотное число
197,2. Нерастворима в воде, растворима в горячем спирте. По
другим характеристикам растворимости аналогична пальмити-
новой кислоте.
Метилстеарат: темп. пл. 38—39°; темп. кип. 184° при
4 мм рт. ст., 214° при 15 мм рт. ст.
Этилстеарат: темп. пл. 33—34°; темп. кип. 182° при
2 мм рт. ст., 215° при 15 мм рт. ст.
Амид стеариновой кислоты: темп. пл. 109°
Анилид стеариновой кислоты: темп. пл. 95°.
Стеарат свинца при нормальной температуре практически
нерастворим в эфире и в спирте.
Арахиновая, или эйкозановая, кислота С19Н39СООН распро-
странена также довольно широко, но все же значительно мень-
ше, чем стеариновая кислота. Это белое твердое вещество;
мол. в. 312,5; темп. пл. 76—77°; темп. кип. 205° (при 1 ммрт.ст.).
Кислотное число 179,5. Нерастворима в воде; в различных дру-
гих растворителях растворима менее, чем стеариновая кислота.
Состав высыхающих масел 27
Метиларахинат: темп. пл. 46,5—47°; темп. кип. 216° при
10 мм рт. ст.
Этиларахинат: темп. пл. 41,5°; темп. кип. 295° при
100 мм рт. ст.
Амид арахиновой кислоты: темп. пл. 109°.
Анилид арахиновой кислоты: темп. пл. 96°.
Непредельные кислоты. Олеиновая кислота. Одноосновная
кислота этиленового ряда с 18-ю атомами углерода:
СН3(СН2)7—СН=СН—(СН2)7—СООН
Она весьма распространена в природе и содержится в боль-
шом количестве в некоторых маслах (оливковое, миндальное
и т. д.); в различных количествах присутствует во всех высы-
хающих маслах.
Природная олеиновая кислота является чис-изомером:
CHS—(СН2),Х /(СН2)7—СООН
/С=Сч
н/ Nd
Она представляет собой маслянистую жидкость, в чистом
виде бесцветную, но чаще всего имеющую бледно-желтый цвет.
Мол. в. 282,4; темп. пл. 13° (для кристаллической формы 16°);
темп. кип. 153° при 1 лои рт. ст. Кислотное число 198,6. Нерас-
творима в воде и легко растворима во многих органических
растворителях. Поверхностное натяжение при 20°—от 29 до
32 дин/см (в зависимости от чистоты кислоты), на поверхности
раздела с водой 15,6 дин/см.
Метйлолеат: темп. кип. 150° при 3 мм рт. ст.
Амид олеиновой кислоты: темп. пл. 75—76°.
Гидрогенизация олеиновой кислоты ведет к образованию
стеариновой кислоты.
Укажем некоторые особые свойства олеиновой кислоты. Под
действием азотистой кислоты, селена, серы и т. д. олеиновая
кислота изомеризуется в элаидиновую кислоту (транс-изомер)
СН8—(СН2)?Х /Н
/С=С.
н/ \сн2)7—СООН
твердую при обычной температуре (темп. пл. 44°).
Вследствие наличия двойной связи иодное число олеиновой
кислоты равно 89,9. Присоединением брома 1получается дибром-
стеариновая кислота (темп. пл. 29°), растворимая в петролей-
ном эфире. При окислении олеиновой кислоты перекисью водо-
рода образуется диоксистеариновая кислота (темп. пл. 95°),
при окислении щелочным раствором перманганата на холоду —
изомерная диоксистеариновая кислота (темп. пл. 132°). Окисле-
ние перманганатом в уксуснокислой среде приводит к разрыву
28
Гл. П. Высыхающие масла
двойной связи с образованием смеси азелаиновой и пеларгоно-
вой кислот. Озонирование ведет к сходному результату: обра-
зованию альдегида пеларгоновой и полуальдегида азелаиновой
кислоты.
В противоположность предельным кислотам, олеиновая кис-
лота образует свинцовую соль, хорошо растворимую в эфире,
спирте и петролейном эфире. Свинцовая соль элаидиновой кис-
лоты чрезвычайно мало растворима в этих растворителях.
Линолевая кислота. Непредельная кислота с двумя двой-
ными связями и 18-ю атомами углерода:
СН3(СН2)4-СН=СН-СН2—СН=СН(СН2)7—СООН
Она также чрезвычайно распространена в природе и содер-
жится во многих растительных маслах. Природная линолевая
кислота в чистом виде представляет собой бесцветную жид-
кость, которой обычно приписывают строение tiwc-tfwc-изомера.
Мол. в. 280,4; темп. пл. — 6° (не совсем точно); темп. кип. 186е
при 3 мм рт. ст. и 228° при 14 мм рт. ст.-, коэффициент прелом-
ления и^= 1,4698; уд. в. с^°=0,9025. Кислотное число 200,0. Не-
растворима в воде, растворима во многих органических рас-
творителях.
При полной гидрогенизации линолевая кислота превращает-
ся в стеариновую кислоту. Иодное число 181. Присоединяя
бром на холоду в эфирном растворе, линолевая кислота обра-
зует две тетрабромстеариновые кислоты. Одна из них, назван-
ная а-кислотой, плавится при 114—‘115°, другая — 0-кислота
при нормальной температуре является жидкостью’ (хотя не-
которые авторы указывают, что температура плавления 0-кис-
лоты равна 58°). Тетрабромпроизводное может быть очищено
путем перекристаллизации и использовано для получения чи-
стого метиллинолеата путем отщепления брома в присутствии
цинка и метилового спирта. Однако нет уверенности в том, что
кислота, полученная по такому методу, идентична природной.
Осторожным окислением марганцевокислым калием полу-
чают смесь тетраоксистеариновых кислот (сативиновые кис-
лоты), из которых одна плавится при 174° (а-кислота), а дру-
гая при 163,5° (0-кислота).
Линолевая кислота и ее соли весьма чувствительны к дей-
ствию атмосферного кислорода. Свинцовая соль ее легко рас-
творима в спирте или в эфире.
Изомеризованные линолевые кислоты. Под влиянием раз-
личных воздействий (азотистая кислота, повышенная темпера-
тура, сильные основания и т. д.) линолевая кислота может
подвергаться двум видам изомеризации: цис-транс-мзомериза-
ция с образованием элаидолинолевой кислоты и изменение
взаимного расположения двойных связей (образование кис-
Состав высыхающих масел
29
лоты с сопряженными двойными связями). По-видимому, эти
два превращения происходят одновременно, что усложняет
картину. При дегидратации рицинэлапдиновой кислоты удалось
выделить линолевую кислоту с сопряженными двойными свя-
зями в положениях 9, 11 (темп. пл. 53°). В других случаях вы-
воды делаются на основании данных по спектрам поглощения
в ультрафиолетовой области; кислоты с сопряженными двой-
ными связями дают характерные полосы. Эти данные являются
доказательством сопряжения, но до сего времени ни одна кис-
лота с сопряженными двойными связями не была выделена в
индивидуальном виде.
Линоленовая кислота — непредельная кислота с тремя двой-
ными связями и 18-ю атомами углерода:
GH3—СН2—СН=СН—СН2—СН=СН—СЙ2-СН=СН-(СН2)7—СООН
Линоленовая кислота очень распространена в растительных
маслах. Некоторые из них особенно богаты ею, например:
льняное, перилловое, ореховое и другие масла. Чистая линоле-
новая кислота представляет собой бесцветную жидкость, кото-
рой обычно приписывают строение цис-цис-цис-изомера.
Мол. в. 278,4; темп. пл. 15° (неточно); темп. кип. 231° при
17 мм рт. ст.; коэффициент преломления = 1,4678; уд. в. d2^—
=0,9046. Кислотное число 201,5. Нерастворима в воде, раство-
рима во многих органических растворителях.
Полная гидрогенизация линоленовой кислотьи приводит к
образованию стеариновой кислоты. Иодное число 273,5. В эфир-
ном растворе присоединяет бром на холоду, образуя изомерные
гексабромстеариновые кислоты, из которых одна нераствори-
ма в эфире (а-кислота, темп. пл. 180°). Из гексабромида мож-
но приготовить чистую линоленовую кислоту (учитывая те же
замечания, что и для линолевой кислоты).
При окислении перманганатом в водной среде образуется
смесь гексаоксистеариновых кислот (линузиновые кислоты), из
которых одна плавится при 203° (р-линузиновая), а другая —
при 173° (а-линузиновая, или изолинузиновая, кислота).
Линоленовая кислота и ее соли более чувствительны к кис-
лороду, чем линолевая кислота. Свинцовая соль линоленовой
кислоты хорошо растворяется в эфире или спирте.
Изомеризованные линоленовые кислоты. В тех же условиях,
что и линолевая кислота, линоленовые кислоты претерпевают мо-
лекулярную перегруппировку, в результате которой образуются
изомерные кислоты с сопряженными двойными связями. Для
триенов с сопряженными двойными связями характерна абсорб-
ция в ультрафиолетовой части спектра при 2700 А.
30
Гл. II. Высыхающие масла
Элеостеариновая кислота — одноосновная кислота с тремя со-
пряженными двойными связями и 18-ю атомами углерода
СН3—(СН2)з -СН=СН—СН=СН—СН=СН- (СН2)7—СООН
является изомером линоленовой кислоты и встречается в боль-
шом количестве в масле тунгового дерева (Aleurites). Она
представляет собой белое твердое вещество. Мол. в. 278,4;
темп. пл. 235° при 12 мм рт. ст.; коэффициент преломления
«£>=1,5112 с сильным рассеиванием; уд. в. d50=0,9028. Кис-
лотное число 201,5. Нерастворима в воде, растворима во мно-
гих растворителях.
Существует три изомерных кислоты:
а-Элеостеариновая кислота (основной компонент тунгового
масла); темп. пл. 48—49°.
0-Элеостеариновая кислота; темп. пл. 71°,
у-Элеостеариновая, или пунициновая, кислота: темп. пл.
43,5—44°.
Под влиянием ультрафиолетовых лучей или следов иода или
серы а- и у-изомеры превращаются в 0-изомер. Структура этих
изомеров еще не полностью изучена, однако есть предположе-
ние, что а-изомер является изомером цис-9, цис-11, транс-13,
в то время как 0-изомер должен соответствовать изомеру
транс-9, цис-11, цис-13.
В результате полной гидрогенизации элеостеариновой кис-
лоты образуется стеариновая кислота. Вследствие наличия со-
пряженных двойных связей наблюдаемое иодное число элео-
стеариновой кислоты существенно отличается от нормы. Тео-
ретически иодное число должно точно соответствовать иодному
числу линоленовой кислоты (278,4); наблюдаемые же величи-
ны иодного числа лежат между 160 и 180, что соответствует,
по-видимому, присоединению двух молекул иода вместо трех
(теоретическое иодное число для двух двойных связей 182,3).
При бромировании в обычных условиях получается лишь тетра-
бромид (темп. пл. 115°). Гексабромид образуется только под
действием ультрафиолетовых лучей. Вследствие этих особен-
ностей элеостеариновую кислоту долгое время рассматривали
как изомер линолевой кислоты (две двойные связи) и поэтому
называли ее элеомаргариновой кислотой. Действительный со-
став этой кислоты был установлен только к 1925 г. на основа-
нии определения молекулярной рефракции. Элеостеариновая
кислота присоединяет малеиновый ангидрид: при этом «-кис-
лота образует производное, плавящееся при 66,5°, а 0-кислота—
производное с темп. пл. 77°. Элеостеариновая кислота имеет
большую склонность к окислению и полимеризации. Свинцовая
соль ее трудно растворяется в спирте и эфире.
Состав высыхающих масел
31
Кислоты, имеющие более трех двойных связей. Эти кислоты
составляют особый ряд* и встречаются главным образом в ры-
бьем жире различного происхождения. Выделение индивидуаль-
ных кислот этого ряда затруднительно вследствие их большой
склонности к окислению. Самое важное свойство, используемое
для их распознавания, заключается в том, что, присоединяя
бром, они образуют окто- и декабромиды, практически нерас-
творимые в бензине, в то время как бромиды менее непредель-
ных кислот растворяются в этом растворителе. Полная же
гидрогенизация полиеновых кислот дает соответствующую на-
сыщенную кислоту. При осторожном окислении марганцовокис-
лым калием они образуют полиоксикислоты, легко раствори-
мые в воде и почти нерастворимые в эфире. Полиеновые кис-
лоты в тех же условиях, что и другие непредельные кислоты,
могут подвергаться изомеризации с образованием кислот с со-
пряженными двойными связями.
Изановая кислота содержится в большом количестве в масле
изано и обладает рядом свойств, отличающих ее от других
жирных кислот. Это октодецен-1-диин-7,9-овая-17-кислота с
18-ю атомами углерода:
СН2=СН—(СН2)4—С=С—С=С— (СН2)7—соон
Ее основные константы: мол. в. 274; темп. пл. 39,5°; коэф-
фициент преломления «о = 1,4865; плотность 0,9085 при 78°;
иодное число 274 (три двойные связи), диеновое число равно
нулю; оптически недеятельна.
Изановая кислота очень чувствительна к действию тепла
(вследствие этого практически не может подвергаться пере-
гонке), кислорода и света. Присоединяя 5 молекул водорода,
она переходит в стеариновую кислоту. При хранении образует
продукт красного цвета, за что и получила название эритроге-
новой кислоты.
Рицинолевая кислота
СН3—(СН2)В—СНОН—СН2—СН=СН—(СН2)7-СООН
не встречается в высыхающих маслах, но, являясь основным
компонентом касторового масла, представляет интерес вследст-
вие своей способности к многочисленным химическим превра-
щениям.
Основные константы: вязкая жидкость; мол. в. 298,4; темп,
пл. 4—5° (легко подвергается переохлаждению); темп. кип. 230е
при 9 мм рт. ст. (легкое разложение); показатель преломления
«0 = 1,4706; уд. в. d15=0,9509. Способна вращать плоскость по-
ляризации [а]о = 6,67° (в ацетоне). Нерастворима в воде и
* Автор называет эти кислоты «одоновыми»;. более принято называть их по-
лиеновыми—Прим. ред.
32
Гл. II. Высыхающие масла
петролейном эфире, но растворима в спирте и эфире. Свинцо-
вая соль ее растворяется в эфире. Кислотное число 188,3; аце-
тильное число 165.
Рицинолевая кислота присоединяет молекулу водорода,
превращаясь в 12-оксистеарпновую кислоту. Иодное число 85,1.
Образует дибромид. При окислении марганцевокислым калием
дает смесь триоксистеариновых кислот (темп. пл. 111° и 140°).
Под действием азотной кислоты превращается в рицинэлаиди-
новую кислоту (темп. пл. 52—54°).
При нагревании рицинолевая кислота частично превращает-
ся в гептиловый (энантовый) альдегид и ундециленовую кис-
лоту, в то время как другая большая часть ее дегидратируется
с образованием двух изомеров линолевой кислоты. Этот вопрос
будет детально рассмотрен при описании дегидратированного
касторового .масла.
Ликановая кислота (октадекатриен-5,7,9-он-15-овая-17-кис-
лота) с тремя сопряженными двойными связями и кето-
группой
СН3—(СН2)3— СН=СН—СН=СН—СН=СН —(СН2)4—СО—(СН2)2 - СООН
встречается в большом количестве в ойтисиковом масле, в кото-
ром она является основным компонентом. Ликановая кислота—
белое кристаллическое вещество, мол. в. 292,4; число нейтрали-
зации 192.
Состав ликановой кислоты был установлен около 1935 г.
Из общего числа возможных изомеров этой кислоты в настоя-
щее время установлены два: а-ликановая кислота (темп. пл.
74—75°) и р-ликановая кислота (темп. пл. 99,5°).
а-Кислота является природной; под влиянием ультрафиоле-
тового света, иода, серы и т. д. она легко превращается в р-кис-
лоту. Семикарбазон а-кислоты плавится при 110—111°, семи-
карбазон р-кислоты — при 138°. Продукты присоединения ма-
леинового ангидрида к этим двум кислотам плавятся при 79°
(а-) и при 97° (р-).
Ликановая кислота весьма легко присоединяет две моле-
кулы брома; при каталитическом гидрировании она присоеди-
няет три .молекулы водорода, образуя кетостеариновую кислоту
(темп. пл. 96,5°); семикарбазон кетостеариновой кислоты имеет
темп. пл. 119°. Кик и элеостеариновая кислота, ликановая кис-
лота обладает большой склонностью к окислению и полимери-
зации.
Метилликанат имеет следующие основные константы: темп,
пл. 242° при 2 мм рт. ст.; уд. в. ^44=0,952; коэффициент пре-
ломления Пв=1,5127.
Высыхание масел
33
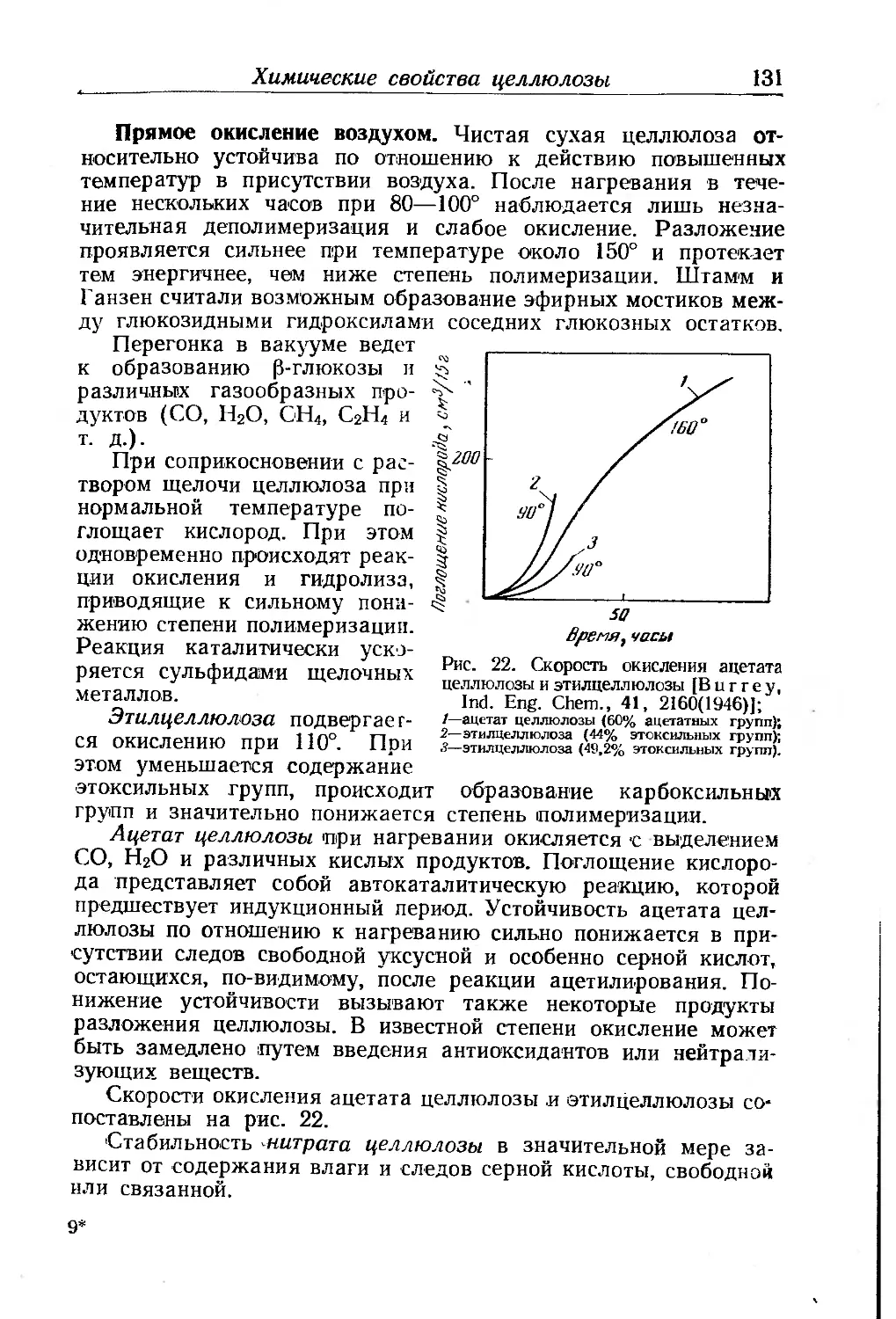
Второстепенные компоненты высыхающих масел
Кроме глицеридов, содержание которых в высыхающих
маслах составляет примерно 99%, в этих маслах содержатся
также так называемые «малые» компоненты, среди которых
следует упомянуть: стерины, хлорофиллы, каротиноиды, некото-
рые углеводороды, некоторые витамины (A, D, Е), фосфоами-
нолипиды (лецитины и кефалины), инозитофосфаты, расти-
тельные слизи, воска, диастазы и следы минеральных солей.
Рафинирование масел производится с целью удаления боль-
шей части этих «малых» компонентов, присутствие которых в
масле в некоторых случаях может быть вредным.
ВЫСЫХАНИЕ МАСЕЛ
Практическое применение лаков и красок целиком основано
на способности пленок, нанесенных в жидком виде, постепенно
переходить в твердое состояние — высыхать. Под термином вы-
сыхание (высыхаемость) подразумевается способность масел,
нанесенных тонким слоем, образовывать в результате химиче-
ских превращений прилипающую к подложке твердую пленку,
полностью утратившую свойства исходного масла.
Масла (триглицериды), нанесенные тонким слоем на стек-
лянную пластинку, могут вести себя по-разному. Масла одного
типа могут оставаться неопределенное время жидкими, но под
действием кислорода претерпевать различные изменения (про-
горкание, окисление и т. д.). Так ведут себя, например, олив-
ковое, миндальное, касторовое и другие масла. Такие масла,
как льняное, тунговое, ореховое, маковое, напротив, весьма бы-
стро образуют твердую пленку, которая не имеет ни одного
физического свойства, идентичного исходному маслу. Масла
этого типа называются высыхающими в противоположность
первым, называемым невысыхающими. Некоторые масла обла-
дают промежуточными свойствами и носят название полувы-
сыхающих, однако этот термин все же не совсем точен, так как
нет метода испытания, точно определяющего способность к вы-
сыханию.
При более детальном рассмотрении явления высыхания
обнаруживается, что в первом приближении способность масла
высыхать зависит от величины иодного числа данного
масла.
Ранее в научных трудах именно по этому признаку и клас-
сифицировали масла. Такая классификация позволяла пола-
гать, что масло тем легче образует сухую пленку, чем выше
его иодное число.
Но опыт показывает, что это не так; масла высокой непре-
дельности, как, например, рыбий жир, поглощают гораздо боль-
3—156
34
Гл. II. Высыхающие масла
ше кислорода, чем другие, но образуют пленки довольно плохо-
го качества — неводостойкие, склонные к пожелтению и быстро
стареющие.
Непредельность оказывает, несомненно, весьма большое
влияние на высыхание природных масел, но только непре-
дельностью нельзя объяснить явление высыхания. Установлено,
например, что жирные кислоты высыхающих масел, которые по
иодному числу всегда превосходят исходное масло, не обла-
дают способностью к пленкообразованию в условиях, в кото-
рых рассматриваемое масло образует пленку. То Ясе относится
к сложным эфирам непредельных жирных кислот и одноатом-
ных спиртов. Соответствующие эфиры двухатомных спиртов
(гликолей) могут образовывать пленки, но продолжительность
их образования очень велика, а полученные пленки сохраняют
растворимость. Реальная способность к пленкообразованию
проявляется только, начиная с эфиров трехатомных спиртов
(глицерина); пленка образуется достаточно скоро и обладает
ярко выраженной нерастворимостью.
Синтетические масла, полученные этерификацией много-
атомных спиртов (пентаэритрита, сорбита, маннита и т. д.)
полиеновыми жирными кислотами, обладают повышенной спо-
собностью к высыханию по сравнению с природными маслами.
Подобный эфир, полученный на основе неразделенных кислот
соевого масла, ведет себя в отношении высыхания так же, как
и соответствующий эфир льняного масла, в то время как в ис-
ходном состоянии эти масла обладают различной способностью
к высыханию.
Таким образом, природа спирта, этерифицированного непре-
дельными жирными кислотами, играет не менее важную роль,
чем иодное число. Однако значение природы спирта не следует
преувеличивать: нужно, чтобы среди этерифицирующих жир-
ных кислот в достаточном количестве были кислоты, по край-
ней мере, с двумя двойными связями. Так, если оливковое
масло само не высыхает, то эфиры пентаэритрита, образо-
ванные кислотами этого масла, также не способны к высы-
ханию.
Итак, в отношении эфиров полиеновых кислот можно сфор-
мулировать следующее правило: высыхание наблюдается в том
случае, если спирт, по меньшей мере трехатомный, этерифици-
рован достаточным количеством полиеновых кислот.
Эта формулировка, может быть, недостаточно полна, так
как не учитывает так называемых инвертированных масел, ко-
торые также хорошо высыхают. Под инвертированными подра-
зумеваются масла, представляющие собой эфиры трикарбал-
лиловой (пропан-1,2,3-трикарбоновой) кислоты и высших непре-
дельных алифатических спиртов (линолевый спирт и др.).
Высыхание масел
35
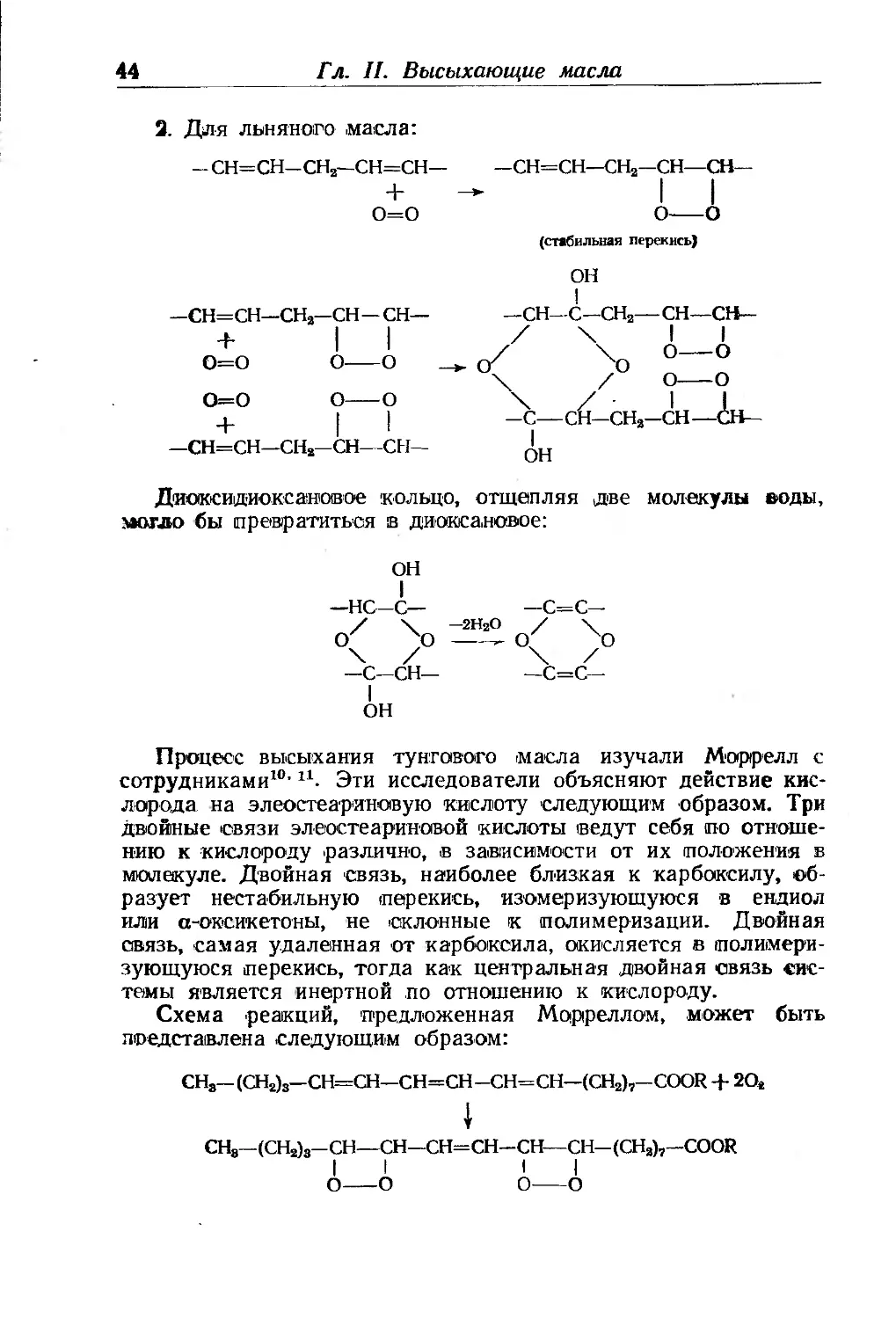
Действие кислорода на двойные связи
До сих пор мы довольствовались лишь общей констатацией
совокупности явлений, происходящих в то время, когда масло,
нанесенное тонким слоем, подвергается действию атмосферы.
Внешне действие кислорода проявляется весьма различно, по-
этому, чтобы лучше понять его механизм, нужно детально рас-
смотреть реакции, которые могут протекать с участием кисло-
рода.
Независимо от конечных результатов действия кислорода,
реакция всегда начинается с образования перекисей. Эти пе-
рекиси представляют собой продукты присоединения молеку-
лярного кислорода; стабильность
их бывает различна в зависимо-
сти от природы исходного веще-
ства.
Если мы в общем виде обо-
значим органическое соединение
через А, то общая формула пере-
киси будет АОг- При простом
•присоединении молекулы кисло-
рода к молекуле органического
соединения реакция может быть
изображена следующим образом:
А + О2 АОг
Время
Рис. 7. Изменение перекисного чис-
ла масла в процессе окисления.
Органические перекиси отличаются высокой реакционной
способностью, например выделяют иод из йодистого калия (ре-
акция, дающая возможность количественно определять содер-
жание перекисей). Перекисное число масла (число Леа) выра-
жается в миллимолях перекиси или в миллиэквивалентах актив-
ного кислорода на 100 г масла.
Изображая графически изменение числа Леа в зависимости
от времени соприкосновения масла с кислородом, получим
S-образную кривую, характерную для автокаталитических ре-
акций: поглощение кислорода сначала протекает медленно, за-
тем быстро возрастает и через некоторое время содержание кис-
лорода становится постоянным (рис. 7).
Эта кривая представляет, конечно, идеальный случай, так
как почти всегда происходят вторичные явления, которые нала-
гаются на основную реакцию. Такое окисление стали называть
автоокислением (аутооксидацией), в отличие от классических
реакций окисления, обычно встречающихся в органической
химии.
Аутооксидация, представляющая собой весьма распростра-
ненное явление в химии ненасыщенных соединений, была изуче-
3*
36
Гл. II. Высыхающие масла
на примерно в 1920 г. Мурэ и Дюфрессом. Эти авторы описы-
вали аутэоксидацию следующим образом: «Это такие реакции
окисления свободным кислородом, которые приводят к образо-
ванию перекисей, т. е. когда можно обнаружить наличие так
называемого «активного кислорода».
Для первичной перекиси Мурэ и Дюфресс предлагают фор-
мулу А (О) 2.
Молекула А, предварительно активированная, соединяясь с
молекулой активного кислорода (Ог), образует с поглощением
энергии молекулу перекиси А (О) 2. Эта молекула обладает
весьма большой реакционной способностью и не остается не-
изменной. Она может, например, диссоциировать
А(О2) —*- А -|- О2
образуя вновь первичные продукты. Тогда реакция дальше не
идет и сохраняется исходное состояние.
Возможен также переход перекиси в стабильную форму
А(О2) АО2
в которой она может быть практически выделена.
Активная перекись может реагировать с избытком веще-
ства А, образуя продукты окисления, уже не являющиеся пере-
кисными соединениями:
А(О2) + А 2А(О) -» 2АО
Типичным примером такой реакции является аутооксидация
бензойного альдегида в бензойную кислоту:
C6HSCHO + О2 С6Н5СНО(О2)
С6Н5СНО +- С6Н5СНО(О2) -*• 2СвН5СООН
Если наряду с веществом А присутствует другое вещество В,
способное окисляться под действием перекиси, может произойти
образование двух стабильных окисей (сопряженное автоокис-
ление)
А(Оа) + В -*- А(О) + В(О) -э- АО + ВО
или окиси только одного вещества В по схемам
А(О2) + В — А + В(О2) А + ВО2
А(О2) + 2В А + 2В(О) А + 2ВО
В двух последних случаях реакция может развиваться далее
различными путями, представляющими значительный интерес.
Так, по реакции
А(О3) + В - А(О) + В(О)
получается активная окись В(О), которая может реагировать
С веществом А с образованием окиси А (О) и восстановлением
вещества В:
В(О) + А -- А(О) у в
Высыхание масел
37
В целом реакция выражается следующим образом:
А(О2) В —► А(О) + В(О) + А —► 2А(О) + В —2АО + В
Продукт В остается неизменным и, следовательно, играет в
реакции автоокисления роль положительного катализатора.
В некоторых случаях активные окиси А(О) и В (О) не могут
существовать одновременно и взаимно разрушаются, регене-
рируя кислород и продукты А и В:
А(О) + В(О) А + В + О2
А(О2) + В —А(О) + В(О) А + В + О2
Тогда реакция автоокисления не может далее продолжаться
и процесс идет так, как будто автоокисление вовсе не происхо-
дило. В данном случае вещество В является отрицательным
катализатором автоокисления и называется антиоксидантом
(антиокислителем).
Известно, например, что следы гидрохинона полностью
предотвращают полимеризацию акролеина или стирола.
Антиоксидант играет очень важную роль. Согласно Мурэ и
Дюфрессу антиоксидантами могут быть только вещества, спо-
собные окисляться; очень часто эти функции выполняют фе-
нолы. Антиоксиданты имеют большое значение в резиновой про-
мышленности, а также при хранении пищевых жиров (для пред-
упреждения прогоркания). Они могут находиться (случайно
или естественным образом) в составе высыхающих масел и ока-
зывают на продолжительность их высыхания значительное
влияние, которое иногда недооценивается. Из веществ, обла-
дающих антиокислительной активностью, можно назвать не-
которые фенолы и амины, кефалины, токоферолы и др.
Следует также отметить, что активность антиоксидантов мо-
жет значительно усиливаться в присутствии некоторых веществ,
которые сами по себе не обладают заметными антиокислитель-
ными свойствами. Такие вещества носят название синэргиче-
ских. Этим свойством могут обладать очень немногие вещества,
чаще всего минеральные и органические кислоты (фосфорная,
лимонная, щавелевая, аскорбиновая и др.). Поэтому неудиви-
тельно, что глифталевые смолы, обладающие всегда заметной
свободной кислотностью, проявляют при сушке различные ано-
малии, если взятое для их изготовления масло содержало
следы антиоксиданта. Факты, подобные вышеприведенным, ча-
сто встречаются в органической химии.
Строение перекисей. Вопросу о строении перекисей посвя-
щено много исследований, однако большим препятствием к изу-
чению этих соединений является их нестойкость.
В настоящее время можно отметить наличие двух основных
теорий, существенно различающихся между собой.
38
Гл. 11. Высыхающие масла
Согласно теории Энглера1, перекиси олефинов имеют цикли-
ческое строение, возникающее в результате присоединения мо-
лекулы кислорода по двойной связи:
—СН=СН— -СН-СН—
+ | |
0=0 о—-о
Теория Энглера была единственной признаваемой в течение
40 лет, хотя подтвердить ее экспериментально не представля-
лось возможным. Она дает объяснение уменьшению иодного
числа, наблюдаемому при окислении льняного и других высы-
хающих масел: в начальной стадии окисления иодное число .по
мере образования перекисных групп уменьшается так, что одна
молекула иода соответствует одной молекуле присоединившего-
ся активного кислорода. Таким образом, на каждую образовав-
шуюся перекисную группу должна исчезнуть одна двойная
связь. Это соотношение справедливо только в узком промежут-
ке времени в самом начале процесса окисления, так как экспе-
риментально найдено, что иодное число монотонно уменьшает-
ся, тогда как перекисное число сначала стабилизируется, а за-
тем тоже начинает уменьшаться. Эти экспериментальные дан-
ные не подтверждают, таким образом, предложенной теории.
Согласно теории Криге2, олефины образуют не циклические
перекиси, а гидроперекиси за счет метиленовой группы, нахо-
дящейся в а-положении к двойной связи:
—сн2—сн=сн— + о2 —сн—сн=сн—
ООН
Эта теория во многих случаях находит серьезное экспери-
ментальное подтверждение3. Так, при окислении метилолеата
при обычной температуре иод действием ультрафиолетовых лу-
чей или же при 60°, Свифту, Доллеару и О’Коннору4 удалось
выделить и охарактеризовать гидроперекись метилолеата. Эту
гидроперекись выделяли из общей смеси продуктов реакции
посредством многократной перекристаллизации в ацетоне при
80°, поскольку при перегонке была опасность разрушения мо-
лекулы гидроперекиси. Из 500 г окисленного метилолеата
авторы после двух перекристаллизаций получили 15 г гидро-
перекиси метилолеата 90%-ной чистоты. Полученное соединение
является смесью двух изомеров:
СН3(СН2)6-СН—СН=СН—(СН,)7—СООСН,
ООН
а
СН3(СН,)7—СН=СН—СН—(СН2)6—СООСНз
ООН
Высыхание масел
39
Другие исследователи5, изучавшие процесс окисления чи-
стого этиллинолеата при 35°, пришли к выводу, что первично
образуется моногидроперекись строения
СН3—(СН2)4—СН=СН— СН— СН=СН—(СН2)7—СООС2Н2
ООН
которая быстро изомеризуется в более стойкие гидроперекиси с
сопряженными двойными связями:
СН3(СН2)4—СН=СН-СН=СН—СН—(СН2)7—СООС2Н6
I
ООН
СН3(СН2)4—СН—СН=СН— СН=СН—(СН2)7—СООС2НВ
ООН
Таким образом, выявился весьма важный факт: образование
гидроперекисей олефинов с изолированными двойными связями
сопровождается молекулярной перегруппировкой, в результате
которой двойные связи располагаются в сопряженном поло-
жении.
Это явление позднее было экспериментально установлено
многими исследователями и в настоящее время не подвергает-
ся сомнению. Между прочим, оно дает возможность объяснить
аномалии, которые были обнаружены для величин иодных чи-
сел, не связывая присоединение иода с исчезновением двойных
связей, так как в сопряженных системах значительно изменяет-
ся процесс присоединения иода.
Теория гидроперекисей, опирающаяся на серьезные экспери-
ментальные доказательства, в течение последних десяти лет
сплотила вокруг себя большое число исследователей и оказа-
лась чрезвычайно плодотворной.
Пако6 «показал на примере многих веществ, что чаще
всего перекиси образуются по обеим рассмотренным схемам,
и предложил две формулы для изображения структуры пере-
кисей.
Ниже (приведены ранее предложенные (формулы и формулы
Пако:
—СН—СН—
по Энглеру I |
О—О
—СН—СН=СН—
по Криге I
ООН
по Пако
—НС—СН—
Vo
1 —СН—СН=СН—
I
О: ОН
40
Гл. II. Высыхающие масла
В формулах Пако особо показан активный кислород пере-
киси, который вызывает выделение иода при определениях чис-
ла Леа.
Образование перекисей в начале процесса высыхания масла
неоспоримо доказано; перекиси могут существовать в той или
иной форме, ио одного факта их образования недостаточно для
объяснения механизма образования пленки.
Превращения первичных перекисей. Перекиси представляют
собой весьма нестабильные вещества и в силу своей большой
реакционной способности стремятся перейти в стабильные
окисные формы.
В зависимости от начального строения перекисей могут по-
лучаться различные конечные продукты.
Приняв формулу Энглера, можно ожидать следующих реак-
ций:
1. Превращение в эпоксиды
—СН—СН— —НС—сн—
I I -*• \/
о—о о
2. Превращение в а-кетолы
Н
_сн—СН— —с—с—
I I -II
о—о о он
которые при известных условиях могут существовать в изомер-
ной форме—в виде непредельных двухатомных спиртов:
Н
I
—С-С— —с=с—
II I — I I
о он он он
3. Дальнейшее окисление эпоксидов и а-кетолов кислоро-
дом (воздуха с образованием а-дикетонов
Н
I 1
—С—С-----1" о O2
II I 2
о он
4. Превращение перекисей под действием паров воды в
а-диолы
Н н
I I
—СН—СН—+ 2Н2О ——с—с—+ н2о2
II II
О о он он
Высыхание масел
41
5. Длительное действие кислорода может вызвать разрыв
углерод-углеродной связи с образованием двух альдегидных
или (кислотных групп:
. —С=О О=С —
—СН—СН— + 4 °2 I + | + Н2О
I I 2 н н
он он
-СН-СН- + 4 02 -* -С< + >с- + Н2О
I I 2 хон ней
он он
Рассмотренные реакции представляют собой все возможные
варианты окисления по двойной связи, начиная от простого при-
соединения молекулярного кислорода, вплоть до разрыва угле-
водородной цепи с образованием двух карбоксильных групп.
Приняв формулу Крше, можно ожидать след юн их реакций:
1. Взаимодействие гидроперекиси с двойной связью; в этом
случае образуется эпоксидный цикл, а гидроперекись превра-
щается в спирт
—СН—сн=сн— + —СН=СН----> — СН-СН=СН— 4- —СН—СН—
I I \ /
ООН он о
2. Превращение гидроперекиси в кетон при повышении тем-
пературы или под влиянием соответствующего катализатора
—СН—сн=сн— —с-сн=сн—
| —' + Н2О
ООН о
3. Превращение гидроперекиси в спирт в присутствии воды
или щелочных агентов
—СН - СН=СН— — СН—сн=сн—
I + Н2о I + Н2О2
ООН он
4. Превращение гидроперекиси в акролеин. Реакции этого
типа недавно установлены экспериментально7 и пока трудно
объяснимы:
-СН=СН-СН-
| —СН=СН—СНО + ?
ООН
Из рассмотрения всех приведенных выше возможных пре-
вращений совершенно ясно, что ни одно из них не может дать
исчерпывающего объяснения явлению высыхания масел. '
Основной вывод, который можно сделать, состоит в том,
что рассмотренные реакции представляют собой простые реак-
ции окисления, вызывающие изменения свойств исходных ве-
ществ. Однако, как бы значительны ни были эти изменения, они
не меняют химической сущности триглицеридов. Хорошо из-
вестны глицериды, содержащие гидроксильные или кетонные
42 Гл. II. Высыхающие масла
группы (касторовое масло, ойтисиковое масло), но они все
равно остаются маслами или жирами, не имеющими ничего об-
щего с сухими пленками высыхающих масел. Даже реакции
разрыва углеродных цепей жирных кислот не изменяют глице-
ридной структуры масла. Уменьшением молекулярного веса
триглицерида и изменением 'строения входящих в молекулу
радикалов нельзя объяснить образование пленки, т. е. переход
в нерастворимые и неплавкие соединения.
Реакции, рассмотренные выше, не являются единственными
реакциями образования перекисей, даже для простейших оле-
финов. Исследователи, изучавшие этот вопрос, наряду с обыч-
ными продуктами окисления наблюдали различные количества
не поддающихся перегонке маслянистых или смолообразнык
соединений, которые они называли полимерами и считали их
нежелательными. Пако6 отметил это .в своем труде и считал,
например, что при автсокислении углеводорода гексена-2
СН3—СН = СН—СН2—СН2—СН3 образуется множество диме-
ров и полимеров, составляющих 19% от количества исходного
углеводорода и 41% от количества окисленного углеводорода.
Таким образом, при дальнейших превращениях первичных пере-
кисей, наряду с простыми продуктами окисления и распада,
образуются полимеры, о строении которых точно ничего неиз-
вестно. Поэтому для того чтобы объяснить образование пленки,
необходимо рассмотреть все эти результаты с точки зрения хи-
мии высыхающих масел.
Возможные реакции пленкообразования
В процессе термической полимеризации масел происходит
постепенное образование трехмерной структуры, приводящее
в конце к желатинизации: масло сразу превращается в нерас-
творимую массу, непригодную для использования. При изго-
товлении линоксина путем продувания воздуха через льняное
масло, в конце концов также происходит желатинизация мас-
ла, которое становится нерастворимым. Отверждение пленки
высьихающего масла на подложке представляет собой анало-
гичное явление: здесь также происходит переход в нераствори-
мое состояние, что можно объяснить только образованием трех-
мерной высокомолекулярной структуры.
Способность масел к высыханию различна и зависит от их
химического строения. Масла, содержащие кислоты с сопря-
женными двойными связями, обычно высыхают быстрее, чем
масла иного 'Строения. Большинство исследователей различает
масла именно по этому признаку.
Маркуссон8 и Кауфманн считают возможным 'взаимодей-
ствие молекул первичной перекиси с двойной связью другой
Высыхание масел
43
цепи, е образованием диоксановопо кольца:
О
II / \
О—СН СН —НС СН—
Протекание такой реакции 'между двумя молекулами гли-
церидов привело бы к образованию связи между ними, а сле-
довательно, возникла бы возможность образования полимер-
ной цепи.
Диоксановое кольцо может образоваться также при взаимо-
действии двух эпоксидных соединений:
Трейбс9, являющийся сторонником образования диоксано-
вого кольца, пытается объяснить различие между- маслами,
содержащими и не содержащими системы двойных связей.
Перекиси элеостеаратов должны немедленно димеризоваться;
соединения же с изолированными двойными связями должны
сначала образовать стабильную моноперекись, которая впо-
следствии оказывает активирующее действие на перекиси, об-
разующиеся на месте других двойных связей. При этом полу-
чалось бы не диоксановое, а диоксидиоксановое кольцо. Эти
последовательные реакции могут быть представлены следую-
щими схемами:
1. Для тунгового масла:
-сн=сн—сн=сн—
+
0=0
0=0
+
—сн=сн—сн=сн—
- СН=СН—СН—СН—
—СН=СН—СН—СН—
(нестабильная перекись)
I
он
—сн=сн-с—СН—
°\ /°
—сн=сн—сн—с—
I
он
44
Гл. II. Высыхающие масла
2. Для льняного масла:
- СН=СН-СН2—СН=СН— —СН=СН—СН2—СН—СН—
+ —► II
0=0 о—о
—СН=СН—СН2—СН—СН—
+ I I
0=0 о—о
0=0 о—о
+ I I
—СН=СН—СН2—СН—СН—
(стабильная перекись)
Диоксидиоксаиовое кольцо, отщепляя две молекулы воды,
могло бы превратиться в диоисановое:
ОН
I
—НС-С— —с=с—
—С—СН— —с=с—
I
он
Процесс высыхания тунгового масла изучали Моррелл с
сотрудниками10, п. Эти исследователи объясняют действие кис-
лорода на элеостеариновую кислоту 'следующим образом. Три
двойные связи элеостеариновой кислоты ведут себя по отноше-
нию к кислороду различно, в зависимости от их положения в
молекуле. Двойная связь, наиболее близкая к карбоксилу, об-
разует нестабильную перекись, изомеризующуюся в ендиол
или а-оксикетоны, не 'Склонные к полимеризации. Двойная
связь, самая удаленная от карбоксила, окисляется в полимери-
зующуюся перекись, тогда как центральная двойная связь сис-
темы является инертной по отношению к кислороду.
Схема реакций, предложенная Морреллом, может быть
представлена следующим образом:
СНа— (СН2)3—СН=СН—СН=СН-СН=СН—(СН2),—COOR + 2О4
I
СНа—(СН2)а—СН—СН—СН=СН—СН—СН—(СН2),—COOR
II II
о—о о—о
Высыхание масел
45
СН,—(СН ),-СН-СН—СН=СН—СН-СН-(СН2)7- COOR
II I I
О—О I о—о
Y
СН,—(СН2)3—СН—СН—СН=СН—С—СН—(СН2)7—COOR
I I । 8 1
о—о I О ОН
О------------------------—
I
СН,—(СН2)3— СН—СН—СН=СН—С—СН—(СН2)7— COOR
II I
----------о о он
Такая ।полимеризация аналогична полимеризации синиль-
ных соединений, ио связи между молекулами осуществляются
с помощью кислородных мостиков —О—О— вместо обычных
углерод-углеродных связей —С—С—.
ЭльмII 12 и Штаудингер13 для объяснения полимеризации пе-
рекисей пользуются схемой, предложенной Морреллом.
Брэдлей14 предложил совсем другое объяснение связи меж-
ду двумя молекулами глицеридов. Он предполагает, что про-
межуточной стадией реакции является образование эфира. По
предложенной им схеме, молекула перекиси распадается, обра-
зуя легко летучий альдегид и альдегидную группу на конце
оставшейся цепи, все еще связанной с глицерином:
II /Н /Н
R—С—С—R' R-с; + R'-C
II ^О^О
0-0
Здесь R — углеводородный радикал, состоящий из неболь-
шого числа атомов, a R' — вся остальная часть молекулы гли-
церида. Кислород .воздуха, воздействуя на концевую альдегид-
ную группу, превращает ее в карбоксильную:
R'—ОНО + у 02 R'—СООН
При этом образуется вещество следующего строения:
СН2—ОСО—(СН2)Л—СООН
I
CH-OCOR
СН2—OCOR'
Такие кислоты могли бы вступать в реакцию с другими мо-
лекулами глицеридов, содержащими эпоксидные группы:
R—СН—CH-R' + R'COOH R— СН—СН-R'
ОН О—СО—R'
46
Гл. II. Высыхающие масла
В более (развернутой форме образующиеся соединения
имеют вид:
R—СО О—СН2
R'—СО-О-СН
R"—СН—СН—(СН2)„—СО—О—СНг
СН2—ОСО—(СН2)„—со—о он
СН— OCOR'"
СН2—OCOR”"
Подобные реакции между двумя молекулами глицеридов
должны привести, очевидно, к образованию трехмерной сетки.
Образование трехмерной сетки пытались объяснить15 также
и полимеризацией винилового типа. В этом случае перекисные
группы должны активировать двойные связи и вызывать диме-
ризацию по схеме:
2R—СН=СН—R
R-CH2—CH-R
R—C=CH—R
Представляется, однако, маловероятным, чтобы образова-
ние сетки осуществлялось путем димеризации, так как в этом
случае пленки масла были бы гораздо прочнее, чем это наблю-
дается. Результаты химических исследований также опровер-
гают это предположение.
Реакция Дильса — Альдера16 была также предложена
прежде (всего для объяснения образования енольных форм ке-
тонов, которые мо,гут (возникать в процессе окисления масел.
Некоторые исследователи полагают, что образование трех-
мерной сетки может происходить также частично за счет сил
побочных валентностей (водородные связи). Эти силы взаимо-
действия могли бы возникнуть между перекисными, гидроксиль-
ными и кислотными функциональными группами:
СН—о.-НС—О---
II I I
СН—О - -НС-О--.
-4С— Of 'O-Cv
;О- • -НО\
\он---о^
Однако, если даже допустить существование таких сил, то
они должны играть весьма ограниченную роль. Одни они не
смогли бы обеспечить необходимое сцепление внутри сетки.
Высыхание масел
47
Эти силы взаимодействия значительны только для высокополи-
меров с симметричным строением (целлюлоза, полиамиды) ,
обладающих явной кристаллической структурой, легко обна-
руживаемой рентгенографически. Пленки высыхающих масел
весьма далеки от такой кристаллической 'Структуры.
Физико-химические гипотезы пленкообразования
Некоторые .исследователи считают, что при образовании
пленки не происходит никакой химической реакции или, если
она и протекает, то играет весьма незначительную роль. Так,
например, Сланский17 полагал, что процесс высыхания масла
складывается из двух основных стадий:
1. Кислород действует .на масла с образованием окислен-
ных глицеридов.
2. Окисленные глицериды образуют с еще неокисленным
маслом коллоидную систему. При увеличении концентрации
окисленных глицеридов до определенной величины происходит
коагуляция системы и получается линоксин.
Образование линоксина, по Сланскому, осуществляется так-
же в два последовательных этапа: а) коагуляция дисперсной
фазы; б) превращение коагулировавшей дисперсной фазы в
однородный гель.
Теория, называемая «газовой коагуляцией», была предло-
жена Ауэром18, который совершенно пренебрегает химической
стороной явления. Ауэр исходит из следующих наблюдений.
Можно получить сухую пленку в присутствии гораздо мень-
шего количества кислорода, чем имеется обычно. Следователь-
но, поглощение маслом большого количества кислорода необя-
зательно. С другой стороны, масла даже перед окислением
должны представлять собой коллоидные системы. В качестве
доказательства этого Ауэр приводит факт разделения масла
на две фазы при добавлении муравьиной кислоты. Согласно
Ауэру, в этом случае нерастворимая фаза соответствует диспер-
гированной фазе, которая обусловливает процесс высыхания,
протекающий в результате коагуляции системы вследствие на-
личия в ней следов ионизированных газов. Поглощение более
значительных количеств кислорода может привести только к
протеканию вторичных реакций окисления, ухудшающих ка-
чество пленки.
Из рассмотрения приведенных выше различных реакций вид-
но, что для объяснения механизма образования пленки высы-
хающего масла предложено большое число гипотез. Такое
чрезвычайное разнообразие мнений уже само по себе свиде-
тельствует о том, что явление это еще мало изучено, в осо-
бенности, в части, касающейся процесса полимеризации.
48
Гл. II. Высыхающие масла
Превращение перекисей в стабильные кислородсодержащие
соединения или продукты деструкции изучено значительно луч-
ше. Эти два процесса, во всяком случае, протекают одновремен-
но и в совокупности приводят к образованию сухой пленки,
свойства которой могут изменяться в положительную или от-
рицательную сторону под влиянием каждого из этих процессов.
Старение пленок высыхающих масел
Пленка практически .начинает стареть еще до ее образова-
ния: молекулы глицеридов масла под влиянием кислорода под-
вергаются глубоким изменениям, ведущим к деструкции с
образованием отдельных осколков, среди которых легко обна-
ружить альдегиды. Другие реакции, .протекающие без разрыва
молекул, ведут к образованию спиртовых групп, понижающих
•водостойкость пленок. Кроме того, в сухой пленке остается не-
которое количество двойных связей: действие на них кислоро-
да продолжается, хотя и несколько медленее, и рано или позд-
но приводит к 'исчезновению двойных связей. Вследствие этого
трехмерная высокомолекулярная сетка, обусловливающая
основные свойства пленки, оказывается разорванной во многих
точках. В результате происходит превращение трехмерных мак-
ромолекул в более мелкие молекулы, отличающиеся по строе-
нию от исходных глицеридов, но сохраняющие характер моле-
кул масел. Пленка «осаливается» и в ней даже по запаху
можно обнаружить присутствие низших жирных кислот. На
этом заканчивается, в основном, образование собственно мас-
ляной пленки, не содержащей пигментов. В окрашенной пленке
могут протекать еще дополнительные реакции. Так, основные
пигменты (свинцовый сурик, окись цинка) будут нейтрализо-
вать получающиеся кислоты по мере их образования. Действи-
тельно, известно что такие пленки содержат некоторое (иногда
довольно значительное) количество металлических мыл.
Разрыв двойной связи с образованием двух карбоксильных
групп необязательно должен привести к разрыву макромоле-
кулярной сетки. Вместо двойной связи может образоваться
достаточно прочная связь, характерная для солей:
R—СН=СН—R' — R—СООН + НООС—R' — R—COO—Pb-OOC-R'
Именно этой реакцией, а не простым образованием мыл
объясняется общеизвестная высокая прочность красок на осно-
ве свинцового сурика, свинцовых белил, окиси цинка и других
основных пигментов. Эта реакция не может протекать в случае
применения кислых или инертных пигментов, которые не реаги-
руют с органическими кислотами, чем и вызвано резкое отли-
чие в свойствах получаемых пленок.
Высыхание масел
49
Водостойкость масляный пленок находится в непосред-
ственной зависимости от присутствия в них гидрофильных
групп (главным образом, гидроксильных, а также кислотных).
Различия между маслами, содержащими сопряженные двойные
связи, и другими маслами в отношении водостойкости их пле-
нок, связанные с присутствием гидроксильных групп, известны
и давно используются. Последние исследования (показали, что
масла с сопряженными двойными связями присоединяют гораз-
до меньше кислорода с образованием перекисей, чем масла, не
содержащие сопряженных систем19. Следовательно, последую-
щие превращения перекисей будут протекать в меньшей сте-
пени, конечное содержание гидроксильных групп будет меньше
и вследствие этого получится более водостойкая пленка.
Термическая полимеризация масел, не содержащих сопря-
женных двойных связей, также улучшает их водостойкость, так
как этот процесс приводит к исчезновению значительного коли-
чества двойных связей, по месту которых возможно образова-
ние перекисей.
Рассмотрение явления высыхания во всем его объеме пред-
ставляет собой очень 'сложную задачу. Количество продуктов,
образующихся в результате автоокисления даже простого оле-
фина, превосходит количество продуктов, получаемых при
классической реакции окисления. В еще большей степени эго
относится к полифункциональным соединениям таким, как
природные ненасыщенные глицериды.
Пожелтение высыхающих масел. Одновременно с отвержде-
нием пленки происходит ее пожелтение, являющееся с точки
зрения практики крайне нежелательным. Степень пожелтения
различна в зависимости от природы взятого масла; удовлетво-
рительного объяснения это явление, как и явление высыхания,
до сих пор не получило.
Можно отметить лишь основные фактические данные, кото-
рые сводятся к следующему.
1. Покрытие, изготовленное на белом или светлом масле,
всегда имеет тенденцию к пожелтению.
2. Пожелтение покрытий всегда сильнее в местах, защищен-
ных от света (за мебелью, картинами и т. д.).
3. Степень пожелтения обычно больше, если исходное масло
имеет повышенное иодное число.
4. Масла с сопряженными двойными связями при той же
степени непредельное™ образуют пленки с меньшей склон-
ностью к пожелтению, чем масла, не содержащие сопряжен-
ных систем.
5. Пленки на основе полимеризованного масла имеют за-
метно меньшую склонность к пожелтению, чем пленки, получен-
ные па основе соответствующего сырого масла.
4—156
50
Гл. II. Высыхающие масла
Пожелтение пленок не 'следует связывать с образованием
металлических мыл. Причина этого явления кроется исключи-
тельно в самом масле. Так, некоторые из многочисленных вто-
ричных реакций превращения перекисей могут приводить к
образованию хромофорных групп, в первую очередь кетонных.
Обычно присутствие одной этой группы недостаточно для того,
чтобы вызвать появление зоны поглощения в видимой части
спектра; окраска возникает, если вблизи кетонной группы
имеется другая хромофорная группа, и чаще всего, если двой-
ная связь карбонильной группы находится в сопряжении с дру-
гой двойной связью (а-дикетоны, а-кетоны с двумя этилено-
выми связями). Хорошо известны следующие примеры:
Ацетон СН3—С—СН3....................... Бесцветен
II
О
Диацетил СН3—С—С—СН3 .................Желтая окраска
' II II '
О О
Форон (СН3)2С=СН—С—СН=С(СН3)2............. То же
Дибензальацетон С6Н5—СН=СН—С—СН=СН—CgH5 То же
Вполне возможно, что в пленках окисленных масел встре-
чаются соединения подобного строения.
Другие группы, образующиеся при окислении, например
кетольные —С—СН—, ендиольные —С = С— иди спиртовые
II I II
О ОН он он
—СН—, сами по себе не вызывают окраски. Очень трудно пред-
ОН
/°
ставить себе действие альдегидной группы —Сх , не вызы-
\Н
вающей окраски самостоятельно, но способной вызвать пожел-
тение при сопряжении ее с другим хромофором. Во всяком
случае подобная сопряженная система не может существовать
без какой-либо перегруппировки, так как она слишком подвер-
жена окислению и быстро превращается в кислоту. Высказыва-
лось предположение, что пожелтение вызывается образованием
Высыхание масел
51
хинонов (типа ксилохинонов), которые могут получаться путем
конденсации двух молекул дикетонов:
СО
R—СО ^CHs-R'
+
R'—СН2 СО—R
\о
о
II
с
R— СХ ^С-R'
R'-C^/C-R + 2Н*°
С
о
Однако такое объяснение появления окраски не имеет
большого смысла, так как предполагает предварительное обра-
зование веществ, уже имеющих окраску.
Некоторые авторы полагали, что окрашивание может быть
вызвано находящимися в масле загрязнениями, которые могут
окислиться с образованием окрашенных веществ, например,
хроман-хинонных производных токоферолов.
Во всяком случае весьма возможно, что пожелтение масел
связано с наличием кетонных групп. Это позволяет очень легко
объяснить более интенсивное пожелтение неосвещенных участ-
ков покрытий и исчезновение окраски под действием света.
Действительно, как показали уже сорок лет назад Вертело
и Годешон20, ацетон под влиянием ультрафиолетовых лучей
быстро распадается на этан и окись углерода:
сн3—со—сн3 сн3—сн3 + со
Более или менее сильному фотолизу под действием ультра-
фиолетовых лучей подвергаются, как правило, все кетоны, что
иногда даже затрудняет определение их спектра поглощения.
Если пожелтение связано с наличием кетонных групп, то со-
вершенно естественно, что в темноте происходит более сильное
пожелтение, частично исчезающее ,на свету. Отчасти это под-
тверждается тем, что в продуктах распада масляной пленки
всегда присутствует окись углерода.
ЛИТЕРАТУРА
1. Engler, Wild, Вег., 1897, 30, 1669.
2. Criegee, Р i 1 z, F 1 у g а г е, Вег., 1939, 72, 1799
3. Farmer, Sundralingam, J. Chem. Soc., 1942. 121.
4. S w i f t, D о I 1 e a г, O'C о n n о r. Oil a. Soap, 1946, 23, 297.
5. Bolland, Koch. J. Chem. Soc., 1945, 445.
6. P a q u о t, Диссертация, Paris, 1944, Masson Ed.
7. Swif t и corp., J. Am. Oil Chem. Soc., 1949, 26, 297.
8. Mar cusson, Z. Angew. Chem., 1925, 38, 148.
9. T r e i b s, Серия статей в Вег., 1942, 75.
Ю. М о г г е 1 1, Marks, J. Soc. Chem. Ind.. 1931, 50, 27T.
4*
Ь2
Гл. II. Высыхающие масла
11. Morrell, Davis, J. Soc. Chem. Ind., 1936, 55, 237T, 261T.
12. E 1 m, Ind. Eng. Chem., 1931, 23, 881.
13. S t a u d i n g e r, Ber., 1925, 58, 1075.
14. Bradley, Paint Technol., 1941, 6, 134.
15. Powers, Overholt, Elm, Ind. Eng. Chem., 1941. 33, 1257.
16. Scheiber. Farbe u. Lack, 1929, 477.
17. S 1 a n s к y, Z. Angew. Chem., 1921, 34, 533; 1922, 35, 389.
18. Auer, Paint Manuf., 1948, 18, 41.
19. Allen, Jackson, К u m m e г г о v, J. Am. Oil Chem. Soc., 1949, 26, 395
20. В e r t h e 1 о t. G a u d e c h о n, C. r., 1910, 151, 478; 1912, 155, 209
обработка высыхающих масел
Многие высыхающие масла могут непосредственно приме-
няться для производства красок и лаков. В этом случае пленки,
полученные после высыхания сырого масла, уже представляют.
собой полноценный защитный и декоративный материал. Однако
некоторые сырые масла, содержащие сопряженные системы
двойных связей и применяемые сравнительно недавно (тунговое,
ойтисиковое и особенно дегидратированное касторовое масло),
часто образуют тусклые, белесые, хрупкие и сморщенные плен-
ки, не обладающие требуемыми защитными свойствами. Качест-
во пленок из сырого масла может быть заметно улучшено с по-
мощью различных несложных способов обработки, придающих
маслу ускоренную высыхаемость. а полученным пленкам —
блеск и водостойкость. Это позволяет использовать масла, содер-
жащие сопряженные системы двойных связей, для получения
пленок высшего качества.
Из способов подобной обработки масел можно назвать тер-
мическую полимеризацию (уплотнение), оксидацию и изомери-
зацию. Масла перед обработкой, разумеется, рафинируют.
Термическая полимеризация* масел
Уплотнение высыхающих масел («варка») является ^весьма '
простой и давно известной операцией. По одним данным, ее
впервые начали применять братья Ван-Эйк в XIV веке, по дру-
гим — еще раньше.
Вначале для улучшения качества высыхающих масел их под-
вергали длительному хранению, затем их стали подвергать «вар-
ке» при невысокой температуре, не принимая никаких мер для
предотвращения контакта масла с окружающим воздухом. В
настоящее время полимеризация масел проводится в атмосфере
* Автор пользуется для обозначения этого процесса термином standolisation,
поясняя, что слово standolie голландского происхождения и означает масло, ко-
торому дали отстояться. То же значение имеют английский термин standolie и
немецкий slandoel. В советской технической литературе терминами «стандойль»
или «штандойль» пользуются весьма редко.—Прим. ред.
Обработка высыхающих масел
53
инертного газа (СО2, N2 и др.). Температура обработки устанав-
ливается в зависимости от природы масла (обычно процесс
проводят при температуре около 300°). Продолжительность опе-
рации зависит от того, какой продукт требуется получить.
Воздействие температуры порядка 300° оказывает на разные
масла различное влияние. Невысыхающие масла заметно не из-
меняются (точнее изменение их свойств происходит только при
очень большой длительности обработки). Свойства высыхающих
масел при тех же условиях существенно изменяются уже через
несколько часов, причем сильнее всего изменяется вязкость. Вы
сыхающие масла с сопряженными двойными связями (тунговое
и ойтисиковое масла) уже через несколько минут превращаются
в нерастворимый гель, полностью теряя физические свойства,
присущие исходному маслу. Наконец, масло изано разлагается
быстро, со взрывной скоростью, что весьма опасно, если опыт
ведут со сколько-нибудь значительным количеством масла.
В процессе полимеризации главную роль играет природа
кислотных радикалов, связанных с глицерином. Поэтому можно
классифицировать масла по скорости полимеризации следующим
образом:
масла с несопряженными полиеновыми кислотами (полимери-
зация протекает нормально);
масла с моноэтиленовыми кислотами (полимеризация прак-
тически невозможна);
масла,с сопряженными полиеновыми кислотами (полимери-
зация протекает очень быстро);
масла с ацетиленовыми кислотами (реакция протекает со
взрывной скоростью).
При термической обработке сильнее всего изменяется вяз-
кость масел. Поэтому полимеризованные масла представляют
собой чрезвычайно вязкие жидкости, вплоть до консистенции,
предшествующей образованию геля. Цвет масла также меняет-
ся в результате полимеризации. При нагревании масла до 270°
прежде всего наблюдается резкое посветление; одновременно
происходит коагуляция слизистых веществ, если они имелись
в масле. Это явление, называемое осаждением (la «casse» de
1’huile), вызывается термическим разложением 'неглицеридных
окрашенных веществ (каротиноидов), содержащихся в масле.
Оно наблюдается даже у рафинированных масел, не содержа-
щих растительных слизей. При дальнейшем нагревании масли
темнеет.
Особенно сильно темнеет нерафинированное масло, так как
образуется много продуктов разложения, обугливающихся и
оседающих в виде черного налета на стенках аппарата. Хоро-
шие результаты дает проведение полимеризации в вакууме,
вследствие возможности непрерывного удаления легколетучих
54
Гл. II. Высыхающие масла
нестабильных веществ; цвет масла после полимеризации не
темнее цвета исходного масла. Современная техника дает воз-
можность получить путем полимеризации продукты высокого
качества, весьма слабо окрашенные.
В результате термической обработки увеличивается также
плотность масла (например, для льняного масла от 0,930 до
0,980 г[смГ) и поверхностное натяжение (для льняного масла
от 32,2 дин!см до 36 дин/см после 5 час. полимеризации при
295°). Именно изменением поверхностного натяжения можно
объснить лучшую поверхность пленок полимеризованного масла.
Однако трудно установить, вызвано ли это улучшение разруше-
нием поверхностно-активных примесей или изменением строения
молекул самого масла.
Коэффициент преломления масел при полимеризации может
изменяться по-разному в зависимости от химического строения
содержащихся в нем кислот. Для глицеридов кислот с несопря-
женными двойными связями (льняное масло) коэффициент пре-
ломления увеличивается, а в случае глицеридов кислот с сопря-
женными двойными связями (тунговое масло) — уменьшается.
Увеличением коэффициента преломления можно объяснить тот
общеизвестный факт, что полимеризованные масла дают более
блестящие пленки. Для масел с сопряженными двойными связя-
ми полимеризация целесообразна, несмотря на уменьшение
коэффициента преломления, так как сырые масла этого типа
образуют пленки, как бы покрытые изморозью.
Кислотные числа масел значительно изменяются в результа-
те полимеризации, обычно в сторону увеличения. Меньше изме-
няются числа омыления, уменьшающиеся лишь на несколько
процентов. Иодное число, напротив, уменьшается очень заметно.
Например, для льняного масла —от 185 до 100 или даже до бо-
лее низкой величины. Гексабромное число уменьшается очень
быстро — за короткое время становится равным нулю. Это осо-
бенно существенно, если сырое масло имеет высокое гексабром-
ное число.
Таким образом полимеризованное масло обладает совсем
иными свойствами, чем исходное. Чрезвычайно большим измене-
ниям подвергается при полимеризации химическое строение
масел.
Современные представления о строении полимеризованных
масел. После полимеризации масло по-прежнему имеет строение
глицерида. Любое полимеризованное масло, даже доведенное
до желатинизации, можно омылить точно так же, как исход-
ное масло, получив мыло и глицерин. Числа омыления полиме-
ризованного и сырого масла весьма близки. Непредельность же
полимеризованных масел, несомненно, значительно ниже, чем
исходных, о чем свидетельствует тюнижение иодного числа.
Обработка высыхающих масел
55
Кислоты полимеризованного масла существенно отличаются
от кислот, содержавшихся в исходном масле. Если гидролизо-
вать или омылить сырое масло, то образовавшиеся кислоты
при соблюдении соответствующих условий можно перегнать в
вакууме без остатка. Кислоты полимеризованных масел не
удается полностью отгонять в вакууме. Правда, при этом полу-
чается фракция, по свойствам не отличающаяся от кислот сыро-
го масла, но обязательно остается вязкий неперегоняющиися
остаток в количестве, иногда достигающем 65% от количества
кислот в полимеризованном масле. Этот осадок состоит также
из кислот. Поэтому совершенно естественно предполагать, что
именно неперегоняющиеся кислоты в конечном счете определяют
свойства полимеризованных масел.
Строение этих кислот было впервые приемлемым образом
объяснено Каппельмейером1. Убедившись, что тунговое масло
полимеризуется гораздо быстрее, чем другие масла, и что это
масло в большей своей части состоит из элеостеариновой кис-
лоты, содержащей систему сопряженных двойных связей, он
приписал реакции полимеризации тунгового масла механизм ди-
енового синтеза2.
Согласно этому механизму радикалы элеостеариновой кисло-
ты могут легко соединяться, образуя соответствующие димер-
ные кислоты:
СН3—(СН2)3—СН=СН—СН=СН- СН=СН—(СН2)7—соон
+
НООС—(СН2)7—СН=СН—СН СН—(СН2)3—сн3
''сн—сн
I
СН3—(СН2)3—СН—СН—СН—СИ—СН=СН—(СН2)7—соон
НООС—(СН2)7 —СН=СН—СН ХСН—(СН2)3—сн3
СН=СН
При диеновом синтезе могут образовываться многочислен-
ные изомеры, что чрезвычайно затрудняет изучение продуктов
реакции.
Эта теория представляется чрезвычайно убедительной и
вполне согласуется с экспериментальными данными, полученны-
ми при полимеризации масел, содержащих сопряженные диено-
вые системы (тунговое, ойтисиковое, дегидратированное касто-
ровое масло). Однако эту теорию труднее применить к маслам,
не содержащим сопряженных двойных связей.
56
Гл. II. Высыхающие масла
Шампетье и Пети3 экспериментально доказали, что масла,
не содержащие сопряженных двойных связей, полимеризуются
аналогично, так как в конечных продуктах содержатся димер-
ные кислоты с шестичленными углеродными циклами. Это
можно объяснить только тем, что под влиянием температуры
происходит предварительная изомеризация кислот, приводящая
к образованию сопряженных двойных связей.
Таким образом, механизм полимеризации одинаков для всех
масел. Одной из стадий этого процесса является образование
глицеридов, в которых гидроксилы глицерина этерифицированы
мономерными и димерными кислотами. Следовательно, конечные
продукты полимеризации должны быть сходны по строению с
полиэфирными смолами, модифицированными жирными кисло-
тами. Полимеризованное масло представляет собой смесь высо-
комолекулярных продуктов «полимеризации молекул различной
функциональности, содержавшихся в высыхающих маслах.
Представление о полимеризованных маслах как о высокопо-
лимерах позволяет обяснить значительно большее число фактов,
чем все ранее предложенные гипотезы. Так, с этих позиций очень
легко объяснить различие в поведении льняного и тунгового
масел. В тунговом масле содержится преимущественно тригли-
цериды элеостеариновой кислоты, которые могут рассматривать-
ся как трехфункциональные. Поэтому образование трехмерной
сетки должно происходить очень быстро, с обычными для подоб-
ных систем быстрым увеличением вязкости и образованием не-
растворимого конечного продукта. В противоположность этому,
льняное масло, содержащее гораздо большее количество раз-
личных кислот, нельзя рассматривать как трехфункциональное
соединение. Наличие многочисленных неполимеризующихся
радикалов олеиновой и насыщенных кислот ограничивает воз-
можность увеличения молекулярного веса и препятствует об-
разованию трехмерной сетки. Поэтому льняное масло желатини-
руется весьма трудно.
Полимеризованное масло имеет строение полиэфира, но спо-
соб его образования коренным образом отличается от образо-
вания таких классических полиэфиров, как алкидные смолы, по-
лисебацинаты гликолей и т. д. Классические полиэфиры полу-
чаются путем реакции поликонденсации многоосновных кислот
с многоатомными спиртами, протекающей с выделением воды.
В случае высыхающих масел, наоборот, природный эфир подвер-
гается полимеризации, возможной вследствие непредельное™
его углеводородных цепей. Характерным примером полиэфира
трехмерной структуры является полимер диаллилфталата, кото-
рый может быть получен как полимеризацией диаллилового
эфира фталевой кислоты, так и поликонденсацией диаллилгли-
коля с фталевой кислотой.
Обработка высыхающих масел
57
Процесс полимеризации высыхающих масел не является про-
цессом поликонденсации, его следует рассматривать как процесс
полимеризационный. Однако здесь невозможно выделить какую-
то единицу, которая, многократно повторяясь, образует полимер,
как наблюдается, например, для полистирола или полибута-
диена; в данном случае увеличение молекулярного веса происхо-
дит за счет непосредственного сращивания молекул глицеридов,
без отщепления какого-либо побочного продукта реакции. Вви-
ду того что в большинстве масел кислоты, этерифицирующие
глицерин, распределены нерегулярно, случайно, содержащиеся
в масле триглицериды могут быть весьма разнообразны. Но при
рассмотрении этих триглицеридов только с точки зрения их спо-
собности к реакциям полимеризации, их можно свести к срав-
нительно небольшому числу типов, а именно: нереакционноспо-
собные триглицериды, триглицериды с одной, двумя и тремя
реакционноспособными цепями.
К нереакционноспособным триглицеридам относятся глице-
риды, у которых все три кислотных радикала образованы пре-
дельными или моноэтиленовы1ми кислотами, не способными
вступать в реакцию полимеризации в обычных условиях. Эти
глицериды совершенно инертны и, если они не подвергнутся
переэтерификации, их можно обнаружить в продуктах реакции.
В молекулах триглицеридов с одной реакционноспособной цепью
один из трех кислотных радикалов способен вступать в реакцию
с другим подобным радикалом. Если этот второй радикал вхо-
дит в состав глицерида такого же типа, то реакция не пойдет
дальше образования димера. В молекулах триглицеридов с дву-
мя реакционноспособными цепями к димеризации способны уже
два радикала из трех. Такие глицериды, взаимодействуя между
собой, образуют линейные полимеры, т. е. ведут себя в отноше-
нии полимеризации аналогично стиролу, хотя характер связей
в цепи совершенно иной. Наконец, взаимодействие триглицери-
дов с тремя реакционноспособными цепями неизбежно приводит
к образованию трехмерной сетки. В отношении полимеризации
такие глицериды приближаются к дивинилбензолу.
Приведенными четырьмя типами глицеридов ограничивается
весь возможный набор молекул, входящих в состав всех масел
и природных жиров. От наличия и количественного соотношения
глицеридов этих типов зависит, относится ли масло к высыхаю-
щим или к невысыхающим, а также поведение масла в процес-
се термической полимеризации. Содержание реакционноспособ-
ных цепей может быть охарактеризовано величиной иодного
числа, но при одном и том же иодном числе распределение
молекул по указанным группам может быть различным и соот-
ветственно возможны большие различия в высыхаемости и спо-
собности к полимеризации.
о8
Гл. II. Высыхающие масла
Рассмотренные типы молекул могут быть условно изображе-
ны следующим образом:
Двойными линиями обозначены углеводородные цепи, способ-
ные вступать в реакции присоединения с другими аналогичными
цепями. При рассмотрении различных возможностей комбиниро-
вания этих молекул между собой, можно придти к следующим
выводам:
молекулы типа А не вступают в реакции полимеризации;
молекулы типа Б при соединении друг с другом образуют
только димеры, а при соединении с молекулами типов В и Г
могут далее расти с образованием полимерных молекул;
молекулы типа В, соединяясь друг с другом, образуют ли-
нейные макромолекулы, а соединяясь с молекулами типа Г —
трехмерные макромолекулы;
молекулы типа Г, соединяясь между собой, образуют трех-
мерные макромолекулы, обладающие самой высокой частотой
решетки.
В приведенной ниже таблице сопоставлены все возможности
попарного сочетания молекул типов А, Б, В и Г.
Типы исход- ных моле? кул Типы присоединяющихся молекул
А Б В г
А Никаких со- четаний Никаких сочетаний Никаких сочетаний Никаких сочетаний
Б То же Молекула димера Молекула димера, далее реагирую- щая по типу В Молекула димера, далее реагирую- щая по типу Г
В То же Молекула димера, далее реагирую- щая по типу В Линейные макромо- лекулы Трехмерные макро- молекулы
Г То же Молекула димера, реагирующая по типу I Линейные макромо- лекулы Трехмерные моле- кулы с более час- той решгткой, чем в случае В—Г
Обработка высыхающих масел
59
В таблице сделана попытка разграничить реакции, которые
могут происходить между отдельными мономерами, но в дей-
ствительности реакция не ограничивается взаимодействием двух
мономеров; она продолжается далее, в соответствии с возмож-
ностями взаимодействия образовавшихся димеров. В результа-
те возникает чрезвычайно сложная цепь реакций, приводящих к
образованию молекул все более и более значительных размеров,
и, если не будут приняты соответствующие меры, то получится
трехмерная сетка и вся масса быстро превратится в нераство-
римый полимер. Такой полимер не может быть обратно превра-
щен в исходные мономеры вследствие высокой прочности обра-
зовавшихся углерод-углеродных связей.
Получаемые полимеры представляют собой полиэфиры, а
следовательно, для них характерны следующие свойства:
1) способность омыляться так же легко, как и сырое масло,
с выделением глицерина и смеси кислот, в которой легко об-
наружить димерные кислоты;
2) легкость алкоголиза одноатомным спиртом, приводящего
к полному превращению желатинированного полимеризованного
масла в растворимые эфиры, отличающиеся по свойствам от на-
чального Полимера;
3) легкость ацидолиза и переэтерификации.
Особый интерес представляет возможность переэтерифика-
ции, так как для этого могут быть использованы те же моно-
меры, т. е. сырое масло, взятое для полимеризации. Методом
переэтерификации можно воспользоваться, если реакция зашла
слишком далеко.
Во всех этих случаях речь идет о разрыве эфирных связей
в трехмерной макромолекуле полимеризованного масла, приво-
дящем к весьма заметному уменьшению молекулярного веса
(в предельном случае получаются многоатомный спирт и смесь
кислот) и коренному изменению свойств. Однако трехмерную
сетку легко восстановить путем поликонденсации димерных двух-
основных кислот с глицерином, протекающей при простом на-
гревании.
Приведенные выше соображения достаточно убедительны
для доказательства того, что полимеризованное высыхающее
масло имеет строение полиэфира. С позиций химии высокомоле-
кулярных соединений может быть объяснен ряд специфических
свойств полимеризованных масел, например повышенная ско-
рость их высыхания по сравнению с сырым маслом. Как уже
указывалось ранее, иодное число полимеризованного масла все-
гда заметно ниже иодного числа исходного масла, но несмотря
на это высы.хаемость в результате полимеризации обычно улуч-
шается. Этот казалось бы парадоксальный факт вполне соот-
60 Гл. II. Высыхающие масла
ветствует нашим представлениям о химическом строении поли-
меризованных масел.
Для высыхания сырого масла с образованием твердой плен-
ки требуется присоединить относительно большое количество
кислорода, достаточное для связывания между собой множест-
ва разнообразных изолированных молекул глицеридов (эти свя-
зи могут образовываться в результате различных процессов).
В полимеризованном же масле уже имеется большое число проч-
ных химических связей между различными молекулами и роль
кислорода в этом случае будет ограничена образованием свя-
зей между молекулами довольно значительной величины. Таким
образом, при гораздо меньшем расходе кислорода трехмерная
макромолекулярная сетка образуется в полимеризованном мас-
ле значительно быстрее, чем в соответствующем сыром масле.
Легко объяснить также меньшую склонность пленок полиме-
ризованного масла к пожелтению, которое наиболее сильно
проявляется при наличии в масле триеновых и еще более нен^
сыщенных кислот. Поскольку при полимеризации в первую оче-
редь вступают в реакции димеризации именно эти кислоты и
степень ненасыщенности кислотных радикалов уменьшается,
естественно, что полимеризованные масла менее склонны к по-
желтению.
Различия между пленками полимеризованных льняного и
тунгового масла также связаны с высокомолекулярной структу-
рой этих соединений.
Известно, что пленки полимеризованного тунгового масла
высыхают быстрее, чем соответствующие пленки льняного масла,
и дают более прочные покрытия.
Тунговое масло можно рассматривать в первом приближе-
нии как триглицерид элеостеариновой кислоты. Ввиду наличия
в этой кислоте трех сопряженных двойных связей при терми-
ческой полимеризации и пленкообразовании получаются трех-
мерные структуры с большой частотой решетки (многочислен-
ные новые связи образуются как за счет полимеризации, так
и за счет окисления). Пленки же полимеризованного льняного
масла имеют малую частоту трехмерной решетки, так как в
состав этого масла входят насыщенные радикалы и остатки
олеиновой кислоты, инертные в отношении полимеризации и
окисления.
Частота трехмерной решетки, которую можно выразить чис-
лом поперечных связей на 100 ковалентных связей линейной
цепи, является одной из характеристик физических свойств вы-
сокомолекулярного вещества. Если этот показатель слишком
высок, то полимер твердый, нерастворимый, слабо набухает;
если он равен нулю, то высокополимер термопластичен. При
малой частоте решетки полимер нерастворим, но не обладает
Обработка высыхающих масел
61
достаточной твердостью и подвергается значительному набуха-
нию под действием растворителей. Последний случай характе-
рен для пленок высыхающих масел, как сырых, так и полиме-
ризованных.
Сетку полимеризованного льняного масла следует рассмат-
ривать как макромолекулу, обладающую внутренней пластич-
ностью (ограниченная величина «показателя частоты сетки»),
в то время как сетка полимеризованного тунгового масла не
обладает этой особенностью. Полимеризованные масла, как и
все высокомолекулярные соединения, полидисперсны, т. е. со-
стоят из макромолекул различного молекулярного веса. Таким
образом, свойства полимеризованного масла являются усред-
ненными по отношению к частным свойствам всех видов содер-
жащихся в нем макромолекул.
Известно, например, что сырое льняное масло при обычной
температуре полностью растворяется в ацетоне или бутиловом
спирте, между тем как после полимеризации оно растворяется в
этих растворителях только частично. Предпринятые попытки
улучшить в этом отношении свойства полимеризованных масел
(Текао!) привели лишь к повышению высыхаемости нераствори-
мой фракции.
Реакции, протекающие в процессе термической полимеризации.
Термическая полимеризация масел основана на способности ра-
дикалов полиеновых кислот к димеризации. Если бы эта реак-
ция была единственной, то процесс протекал бы одинаково при
всех условиях и мог бы быть описан определенными математи-
ческими уравнениями.
Однако при тщательном рассмотрении явления оказывается,
что процесс протекает гораздо сложнее, о чем свидетельствуют
следующие факты.
1. В процессе термической полимеризации кислотное число
увеличивается даже в том случае, если .исходное масло было
почти совершенно нейтрально.
2. Полимеризация всегда ведет к выделению более или менее
значительных количеств летучих продуктов, неправильно назы-
ваемых «парами масла».
3. При проведении полимеризации льняного масла в вакууме
происходит его желатинирование (структурирование), в то вре-
мя как при той же температуре, но при атмосферном давлении
это практически не происходит. Такое поведение масла в слу-
чае простой реакции полимеризации противоречило бы элемен-
тарным законам термодинамики.
Указанные факты, как и некоторые другие, были бы невоз-
можны, если бы при термической обработке протекала чистая
реакция полимеризации. Следовательно, основная реакция ос-
ложнена какими-то побочными процессами.
62
Гл. И. Высыхающие масла
Увеличение кислотного числа свидетельствует о появлении
в реакционной среде свободных карбоксильных групп, отсутст-
вовавших в исходном масле. Эти группы могли образоваться
только вследствие разрыва молекул по эфирным связям. Моно-
эфиры одноатомных спиртов под влиянием температуры обычно
расщепляются на кислоту и углеводород этиленового ряда:
RCOO - СН2—СН2—R' -* RCOOHСН3=СН—R'
В маслах содержатся не моноэфиры, а триглицериды, рас-
щепление которых при выделении первой молекулы кислоты
может протекать по реакции:
СН2—OCOR СН2
I «
CH-^OCOR' С—OCOR' + RCOOH
I I
CH2—OCOR" CH2—OCOR"
Но реакция на этой стадии не останавливается. О далеко
идущей реакции распада свидетельствует то, что в продуктах
омыления полимеризованных масел никогда не обнаруживали
нропандиола и в то же время в процессе полимеризации всегда
выделяется акролеин. Акролеин может образоваться при дегид-
ратации глицерина, но реакцию разложения триглицерида с об-
разованием только кислот и акролеина представить себе невоз-
можно. Однако, хотя точный механизм этой реакции еще не
установлен, бесспорно, что она происходит и играет существен-
ную роль в образовании трехмерных структур.
Разрыв эфирных связей в процессе полимеризации, приво-
дящий к уменьшению величины макромолекул, является неже-
лательным, так как затрудняет желатинирование масла.
Поскольку термическая полимеризация проводится при темпе-
ратуре около 300° и в течение довольно длительного времени,
часть макромолекул, естественно, подвергается более или менее
глубокому крекингу. Реакции процесса крекинга, широко ис-
пользуемого в нефтяной промышленности, пока еще не при-
влекли внимания .исследователей процесса полимеризации ма-
сел, вероятно, ввиду сложности и 'недостаточной изученности их
механизма.
Однако эти реакции, безусловно, протекают и могут вносить
некоторые осложнения в течение основного процесса полимери-
зации. Так, при нагревании льняного масла в течение 55 мин.
при остаточном давлении 20 мм рт. ст. (почти желатинирован-
ное масло) выделяется до 18% летучих веществ, конденсирую-
щихся при охлаждении водой. Сконденсировавшиеся вещества
имеют характерный для лаковарочных цехов запах, не похожий
ни на запах сырого масла, ни на запах акролеина (который к
Обработка высыхающих масел
63
тому же в этих условиях не конденсируется), ни на запах жир-
ных кислот. Эти вещества имеют кислую реакцию и обычными
техническими приемами могут быть разделены на две фракции:
Нейтральная фракция ... 21%
Кислая фракция ... 78%
Нейтральная фракция представляет собой подвижную желто-
ватую жидкость и, как установлено анализом, состоит из угле-
водородов этиленового ряда, характерным запахом которых она
и обладает.
Идентифицировать отдельные продукты не удается ввиду
очень широких пределов кипения, например:
Температура кипения
Фракции °C
1 2 3 4 5 170 (при 760 мм) 170—210 (при 760 мм) 100—150 (при 20 мм) 150—210 (при 20 мм) 210—260 (при 20 мм)
Образование широкой гаммы непредельных углеводородов
не является неожиданным, поскольку речь идет о продуктах
крекинга.
Кислоты, входящие в состав кислой фракции, также очень
отличаются от кислот, входивших в состав исходного масла.
Так, при разгонке под давлением 20 мм рт. ст. получаются
следующие фракции:
Фракции
1
2
3
Остаток
Температура кипения
°C
70—150
155-175
175—215
Кислотное
число
424
329
250
200
Кислотные числа выделенных фракций показывают, что в
них содержатся продукты распада («осколки») исходных кис-
лот. Полученные кислоты отличаются ярко выраженной непре-
дельностью, причем первые фракции имеют характерный запах
низших гомологов соответствующего ряда.
На основании полученных данных можно утверждать, что
летучие продукты, выделяющиеся в процессе полимеризации,
отличаются от исходных компонентов масла и что они образуют-
ся в результате крекинга углеродных цепей кислотных радика-
лов и разрыва эфирных связей. В процессе крекинга происхо-
дит шромежуточное образование свободных радикалов, чрезвы-
чайно реакционноспособных и способных рекомбинироваться по
законам вероятности с образованием разнородных соединений.
64 Гл. II. Высыхающие масла
Самой простой реакцией, протекающей при крекинге, яв-
ляется разрыв цепи с образованием двойной связи с разных
сторон от места расщепления:
<-► R—СН=СН2 + СН3—СН2—R'
R—СН2 СН2—СН2—СН,—R' —
I-»- R--CH.,-CHS + СН2=СН—R'
Эта реакция дает возможность объяснить различные явле-
ния, наблюдаемые при термической полимеризации масел. Так,
запах «вареного масла» является в действительности запахом
олефиновых углеводородов, образующихся в результате кре-
кинга углеродных цепей. Сильный запах рыбы, который ощу-
щается при варке некоторых масел, обусловлен аминами, обра-
зующимися в результате пиролиза фосфоаминолипидов, содер-
жащихся в масле. Хорошо рафинированные масла вообще не
имеют этого запаха.
Термическая полимеризация при атмосферном давлении и
нормальной продолжительности обычно не вызывает желатини-
рования масел с несопряженными двойными связями, так как
разрыв эфирных групп ограничивает размеры макромолекул, а
крекинг углеродных цепей вызывает образование сравнительно
низкомолекулярных углеводородов, которые только частично
выделяются из масла, большей частью оставаясь в сухой плен-
ке и играя роль пластификаторов. По этой причине маслам, при-
меняемым в производстве клеенки и обычно подвергаемым дли-
тельной варке при высоких температурах, нужно стараться
придать большую эластичность, а не стремиться только к повы-
шению степени их полимеризации. Наиболее высокополимери-
зованное масло является полностью структурированным и по
эластичности совершенно не отвечает предъявляемым требованиям.
При проведении термической полимеризации в вакууме, про-
дукты крекинга удаляются по мере их образования. Благодаря
этому полимеризованное масло сохраняет строение глицеридов
и, кроме того, уменьшается возможность вторичных реакций
между образовавшимися углеводородами и непредельными кис-
лотными радикалами глицеридов. Возможность таких реакций
полностью не исключена; если они происходят, то уменьшается
степень полифункциональности триглицеридов вследствие бло-
кирования некоторого количества двойных связей. В то же время
вследствие крекинга углеродных цепей образуются более реак-
ционноспособные концевые двойные связи в кислотных радика-
лах глицеридов, что увеличивает их полифункциональность.
Совокупностью всех перечисленных особенностей протекания
процесса в вакууме объясняется желатинирование масла, прак-
тически не наблюдающееся при атмосферном давлении. Теми
же причинами вызывается желатинирование в вакууме полу-
высыхающих и даже невысыхающих масел.
Обработка высыхающих масел
65
Хотя термическая полимеризация 'высыхающих масел долж-
на рассматриваться прежде всего как полимеризационный про-
цесс, не следует упускать из виду побочных реакций, (прида-
ющих этому процессу более сложный характер.
ЛИТЕРАТУРА
1. Kappeimeier, Farben Ztg., 1933, 38, 1018.
2. D i е 1 s, Alder, Die Methoden der Dien Synthese, Handbuch der bio-
logischen Arbeitsmethoden, Abderhalden, Berlin, 1933.
3. J. Petit, Peint. Pigm. Vern., 1946, 23, 3, 41, 73, 118; G. C h a m p e-
t i e r, J. P e t i t, Bull. Soc. Chim., 1945. 12, 680, 689; C. r., 1945,
220, 748; J. Petit, C. R„ 1945, 220,829.
Окислительная полимеризация масел
Этим термином принято обозначать процесс, состоящий в
пропускании потока воздуха или кислорода через всю массу
масла при нормальной или повышенной температуре, практиче-
ски не превышающей 150°. Такая обработка, особенно если она
проводится при низкой температуре, вызывает изменения хими-
ческой структуры, аналогичные изменениям, происходящим в
процессе высыхания масла (см. стр. 33 и сл.).
Предельным результатом окислительной полимеризации
льняного масла является его загустевание; получаемый продукт
носит название линоксина. Такая обработка применяется в про-
изводстве линолеума, где линоксин является основным сырьем.
Линоксин начали изготовлять начиная примерно с 1850 г.
Первоначально линоксин получали путем орошения вертикально
подвешенных кусков ткани льняным маслом, содержавшим свин-
цовый сиккатив. После отверждения образовавшейся пленки
операцию повторяли и заканчивали процесс, когда пленка до-
стигала толщины около 2 см. Таким образом, техника производ-
ства линоксина базировалась на использовании простого явле-
ния высыхания масел.
Вследствие длительности этой процедуры, она была заме-
нена окислением всей массы масла путем интенсивного проду-
вания воздуха или пропускания потока воздуха при постепен-
ном повышении температуры для ускорения процесса поглоще-
ния кислорода.
Все химические реакции, происходящие в ходе этих опера-
ций, уже были рассмотрены выше.
При окислительной полимеризации масел происходят значи-
тельные изменения их свойств. Приведенные ниже данные
(табл. 1), заимствованные у М. Каррьера, хорошо иллюстри-
руют эти изменения.
5—156
66
Гл. И. Высыхающие масла
ТАБЛИЦА 1
Изменение свойств льняного масла при продувании воздуха при 125°
Показатели Продолжительность продувания воздуха, часы
0 1 3 6 12 18 24
Иодное число 183,5 180 170 159 141,2 123 112,5
Родановое число .... 118,5 114,2 106,5 100 87,5 77 69,3
Число омыления . .... 192,5 194 196 198 205,5 214,5 218
Диеновое число 11,8 — 4,75 2,76 1,5 1,16 —
Гексабромное число 54,2 51 45 37 26,3 18,8 14,9
Количество кислородсодержа-
щих кислот, % 0,02 1,56 4,55 9,2 21,5 35 40
Кислотность 0,75 0,9 1,10 1,14 1,65 1,71 1,74
Гидроксильное число . . . 0,8 4 8,3 18,1 39 57 —
Перекисное число 0 27,5 28,5 40,8 21,6 13,4 —
Вязкость (при 61,5°) 0,130 0.150 0,199 0,319 1,066 3,80 47,15
Молекулярный вес .... 640 690 785 878 962 1250 —
Удельный вес (при 0е), е/с.и3 0,9315 0,9320 0,9416 0,9517 0,9727 0,9913 1.0009
Коэффициент преломления 1,4818 1,4821 1,4833 1,4847 1,4881 1,4893 1,4908
Удельная рефракция .... 0,3059 0,3059 0,3034 0,3009 0,2961 0,2912 0,2892
Молекулярная рефракция . . 195,7 211 238,1 264,2 284,8 364
Из этой таблицы видно, что при окислительной полимериза-
ции масел большинство показателей подвергается изменению.
Особенно заметно увеличиваются удельный вес, вязкость и
количество кислородсодержащих кислот. Содержание переки-
сей, как и при высыхании, сначала увеличивается, проходит
через максимум, а затем начинает уменьшаться.
Полученные путем окислительной полимеризации высоко-
вязкие масла находят различное применение. Так, благодаря
наличию гидроксильных групп эти масла растворимы в спирте
и совместимы с нитроцеллюлозными лаками, что позволяет
использовать их в качестве пластификаторов. В большом коли-
честве такие масла расходуются в производстве линолеума; под-
вергнутый окислительной полимеризации рыбий жир исполь-
зуется в производстве искусственных жиров (ворвани) и для
получения искусственной кожи. Для малярных красок подобные
масла применяются меньше, чем масла, обработанные различ-
ными другими способами.
Дегидратация касторового масла
Касторовое масло, извлекаемое из семян Ricinus communis L,
само по себе не принадлежит к числу высыхающих масел.
Основным компонентом касторового масла, содержащимся в
Обработка высыхающих масел 67
количестве 80—90%, является триглицерид рицинолевой кис-
лоты:
СН3(СН2)5—СНОН— СН2—СН=СН- (СН2)7-СООН
В касторовом масле содержатся также в небольших коли-
чествах диоксистеариновая и олеиновая кислоты. Все эти ком-
поненты не могут, естественно, придать касторовому маслу
способности к высыханию.
Касторовое масло используют в производстве эфироцеллю-
лозных лаков как пластификатор, но наибольшее применение
оно нашло в качестве высыхающего масла после соответству-
ющей обработки.
При известных условиях обработки рицинолевой кислоты про-
исходит отщепление молекул воды и на месте вторичной спир-
товой группы образуется двойная связь. При этом атом водо-
рода может отщепляться как от метиленовой группы, менее
удаленной от карбоксильной группы
СН3—(СН2)8—СН—СН—СН=СН—(СН2)7—СООН
он Н |_н2О
I
СН3—(СН2)5—СН=СН—СН=СН—(СН2)7—СООН
так и от метиленовой группы, более удаленной от карбоксиль-
ной группы:
СН3—(СН2)4—СН—СН—СН»—СН=СН—(СНо)7—СООН
I I
н он
СН,-(СН2)4-СН=СН—СН2— СН=СН— (СН2)7—СООН
Поэтому в результате дегидратации получается смесь двух
изомеров: линолевой кислоты 9,11, имеющей сопряженные двой-
ные связи, и линолевой кислоты 9,12, представляющей собой
собственно линолевую кислоту. Многочисленные примеры реак-
ции дегидратации, известные в органической химии, свидетель-
ствуют о бессмысленности попыток получить в подобном случае
преимущественно какой-либо один из изомеров.
Основные условия, необходимые для проведения направлен-
ной дегидратации, не осуществимы в отношении молекулы ри-
цинолевой кислоты. Кроме того, под действием тепла может
произойти пиролиз, приводящий к расщеплению молекулы ри-
цинолевой кислоты на молекулу гептилового альдегида (энан-
тола) и молекулу ундециленовой кислоты:
CH3—(CH2)S—СНОН—СН2—СН=СН—(СН2)7—СООН
СН3—(СН2)5—СНО + СН2=СН—(СН2)8—СООН
—Н2О
5*
68
Гл. II. Высыхающие масла
Эта реакция, полезная в других случаях, в данном случае
совершенно нежелательна.
Реакция дегидратации при соответствующих условиях про-
текает очень легко даже в отсутствие катализатора. Поэтому
неудивительно, что почти всякое вещество, введенное в масло,
рассматривали как катализатор дегидратации. Однако можно
считать безусловно установленным каталитическое действие,
оказываемое на эту реакцию кислыми веществами и прежде
всего серной кислотой (даже очень разбавленной). Можно пред-
полагать, что сначала происходит этерификация спиртовой
группы
СН3—(СН2)5—СНОН -СН2 - СН=СН—(СН2)7—СООН + H2SO4 -*
->- СН3—(CH2)S—СН—СН2—СН=СН—(СН>)7—СООН + Н2О
I
OSO3H
но образовавшийся эфир тотчас же расщепляется с выделением
вновь серной кислоты и возникновением двойной связи:
СН3-(СН,)6—СН—СН2— СН=СН—(СН2)7—СООН ->-
I
OSO3H
СН3— (СН2)6— СН=СН—СН=СН—(СН2)7—СООН 4- H2SO4
На первых установках (1923 г.), где дегидратация касторо-
вого масла, основанная на аналогичной реакции, была осуще-
ствлена как длительный и многостадийный процесс, технологи-
ческой схемой предусматривались следующие операции:
1) омыление касторового масла для получения рицинолевой
кислоты;
2) ацетилирование спиртовой группы (получение ацетилри-
цинолевой кислоты);
3) термическое разложение ацетилрицинолевой кислоты с
получением смеси двух линолевых кислот;
4) этерификация глицерина смесью двух полученных кислот.
В дальнейшем две операции удалось сначала свести к одной,
производя ацетилирование непосредственно самого глицерида,
а затем было обнаружено, что реакция дегидратации может
протекать и без ацетилирования.
Практически процесс дегидратации сводится к нагреванию
масла до относительно умеренной температуры (200—250°) в
присутствии катализатора и постоянному контролю процесса
с целью своевременного удаления выделяющейся воды.
Однако, если выходы до 90% достигаются сравнительно лег-
ко, то, напротив, приблизиться к теоретическому выходу очень
трудно, так как удаление последних следов воды связано с
особенно большими трудностями. Увеличение же продолжи-
Обработка высыхающих масел
69
тельности операции приводит к тому, что высыхающее масло
все больше полимеризуется; поэтому нередко приходится пре-
кращать процесс на стадии, когда масло еще обладает замет-
ным гидроксильным числом.
Дегидратированное касторовое масло по технологическим
свойствам занимает промежуточное положение между льняным
и тунговым маслами. Вследствие отсутствия радикалов с тремя
двойными связями оно имеет то достоинство, что получаемые
пленки относительно мало склонны к пожелтению.
Модификация масел стиролом
Модификацию масел стиролом начали применять сравни-
тельно недавно (начиная с 1944 г.), хотй возможность введения
в высыхающие масла винильных соединений была известна еще
с 1930 г. Модификация высыхающих масел стиролом может
производиться в различных условиях. Согласно патентным дан-
ным, реакция может проводиться непосредственно в массе (про-
стое смешение чистого масла со стиролом-мономером) или в
присутствии растворителя (обычно ксилола) с введением за-
медлителей полимеризации (а-метилстирола, дипентена, фено-
лов, серы и т. д.) или без них. Содержание стирола можно изме-
нять в зависимости от требуемых свойств конечного продукта,
но обычно оно не превышает 45%.
Стиролом модифицируют различные масла: льняное, дегид-
ратированное касторовое, тунговое, как сырые, так и частично
полимеризованные или окисленные. Реакционная смесь состоит
из 55% льняного масла и 45% смеси стирола с а-метилстиролом.
Для инициирования полимеризации мономеров добавляется 3%
вес. перекиси бензоила. Ниже приводится режим процесса мо-
дификации льняного масла смесью стирола (70%) и а-метил-
стирола (30%):
Температура °C Продолжитель- ность операций часы
160 6 Нагревание чистого
масла
160—250 5 Введение стирола и
перекиси бензоила
250 6
250—300 3
300 1
Полученные таким образом чрезвычайно вязкие масла име-
ют консистенцию мягкой смолы, иногда мутны, обладают ха-
рактерным запахом стирола. Они хорошо растворимы в арома-
тических растворителях, а также во многих алифатических рас-
творителях.
70
Г л. II. Высыхающие масла
Масла, модифицированные стиролом, могут иметь различное
строение. Их можно рассматривать как сополимеры, если они
являются продуктам'и соединения многих молекул стирола и
.многих молекул глицеридов в единую макромолекулу. Если же
стирол полимеризовался отдельно и цепи полистирола не свя-
заны ковалентными связями с углеродными цепями глицеридов,
такие модифицированные масла следует рассматривать как
смесь полимеров. В то же время, если две молекулы А и В ка-
ким-либо (путем образуют соединение АВ и реакция оста-
навливается на этой стадии, нет никаких оснований считать это
полимеризацией. Для возможных соединений полистирола с ра-
дикалами непредельных жирных кислот были предложены два
основных типа структурных формул.
Если радикалы масла имеют несопряженные двойные связи,
то полистирольная цепь должна присоединяться в а-положении
по отношению к двойной связи:
СН3-(СН2)4—СН=СН—СН2—СН=СН-СН— (СН2)в—СООН
Если же в кислотных радикалах масла имеются сопряжен-
ные двойные связи, т. е. они напоминают по строению молеку-
лу бутадиена, то строение модифицированного масла будет
сходным со строением бутадиен-стирольных сополимеров.
Кислотный радикал рассматривается здесь как замещенный
бутадиен, сополимеризованный со стиролом:
R
Обработка высыхающих масел
71
Однако, если бы реакция протекала по последнему меха-
низму, происходило бы быстрое желатинирование реакционной
массы, в действительности наблюдаемое лишь в весьма слабой
степени. Следует иметь в виду, что среди молекул масла мно-
гие содержат две и более цепей, сходных с замещенным бута-
диеном, причем значительное количество молекул содержит по
три таких цепи. Поэтому если принять указанный выше меха-
низм реакции, имеются все условия для образования трехмер-
ной решетки. Напротив, при механизме реакции, предложенном
для масел с несопряженными двойными связями, не должно
происходить желатинирование масла, так как полистирольные
цепи не образуют мостиков между различными молекулами
глицеридов.
В маслах, модифицированных стиролом, можно очень легко
определить, полностью ли присоединился стирол к глицеридам.
Известно, что наиболее вероятным местом разрыва связей в
макромолекулах масла являются легко омыляемые эфирные
г руппы.
Получаемые в результате омыления калийные соли раство-
ряются в водно-спиртовой смеси, в то время как полистирол, не
связанный с глицеридами, нерастворим в этой смеси. Таким
образом, теоретически их можно разделить. Если же опыт по-
кажет, что количество несвязанного полистирола значительно
превышает ожидаемое, следует воспользоваться весьма распро-
страненным в химии высокомолекулярных соединений методом
фракционированного осаждения и отделить этим методом по-
листирол от более низкомолекулярных жирных кислот. Для
этого к растворителю, общему для обоих типов веществ, посте-
пенно добавляют осадитель, не растворяющий полистирол, но
растворяющий жирные кислоты. Прекрасный эффект дает осаж-
дение метиловым спиртом из эфира, но для получения точных
результатов совершенно обязательно многократное фракциони-
рование, позволяющее избежать взаимного захвата разделяе-
мых веществ. Путем фракционированного осаждения можно
получить чистый полистирол и кислоты с примесью полистиро-
ла. Количество последнего в кислотной фазе относительно мало,
поэтому его можно полностью удалить по первому методу, рас-
смотренному выше.
В результате такого фракционирования можно обнаружить,
пто в процессе модификации стиролом льняного масла, его жир-
ных кислот и жирных кислот тунгового масла значительное
количество стирола (85—90% от взятого мономера) не всту-
пает в реакцию с маслом. Этот стирол содержится в модифи-
цированном масле в виде (полимера, что видно по очень малой
величине кислотного числа (близкого к единице) и подтвер-
ждается данными элементарного анализа.
72
Гл. II. Высыхающие масла
По-видимому, большая часть стирола полимеризуется неза-
висимо от масла и находится, следовательно, в виде полисти-
рола, растворенного в масле. Однако небольшое количество
стирола вступает в соединение с жирными кислотами. Эти про-
дукты в среднем имеют кислотное число около 160 и путем
экстрагирования из них можно выделить только те кислоты, ко-
торые перегонкой в вакууме могут быть разделены на две фрак-
ции. Одна из этих фракций соответствует обычным кислотам
исходных масел, а другая по составу точно соответствует про-
дукту присоединения одной молекулы стирола к одной моле-
куле кислоты.
Таким образом путем фракционирования продуктов омыле-
ния масла, модифицированного стиролом, удается выделить
следующие три составные части:
1) полистиролы различной степени полимеризации;
2) обычные кислоты масла;
3) кислоты, присоединившие молекулу стирола.
Такой состав продуктов омыления заставляет полностью
отбросить предположение о возможности сополимеризации в
процессе получения модифицированных масел. Модифицирован-
ное стиролом масло следует рассматривать как лак в класси-
ческом понимании этого слова (раствор смолы в высыхающем
масле), имея при этом в виду, что только небольшое количе-
ство стирола весьма ограниченно реагирует с некоторыми це-
пями непредельных жирных кислот.
Что же касается продукта присоединения стирола к кислоте,
то можно предположить следующий механизм его образования:
1) изомеризация кислоты, не содержащей сопряженных двой-
ных связей (в случае масел, содержащих подобные кислоты);
2) реакция Дильса—Альдера между стиролом и кислотой с
системой сопряженных двойных связей:
CH3-(CH2)S—СН=СН-СН=СН—(СН2)7—СООН
+
сн=сн2
сн=сн
СН3 - (СН2)5-НС ^СН—(СН£)7-СООЯ
^СН—СН»
Обработка высыхающих масел
73-
Эта реакция может с первого взгляда .показаться аномаль-
ной для стирола, который обычно полимеризуется только по
типу винильных соединений. Однако в данном случае нет ника-
ких препятствий для протекания реакций Дильса—Альдера при
действии стирола на систему сопряженных двойных связей. К то-
му же одновременно протекает обычная реакция полимериза-
ции, в которую вступает от 80 до 85% стирола.
Модифицированные масла не являются сополимерами, так
как этому противоречат следующие факты:
1) большая часть стирола полимеризуется сама по себе,
не вступая в реакцию с маслом, которое в данном случае ведет
себя, как инертный растворитель;
2) небольшое количество стирола, вступающее в реакцию,
не образует длинных полистирольных цепей, а дает эквимо-
лекулярные продукты присоединения в соответствии с представ-
лениями классической химии.
Не будучи сополимерами, модифицированные масла имеют
определенные достоинства. Путем модификации удается прак-
тически осуществить «in situ» введение в масло высококаче-
ственного полимера полистирола, который сам по себе трудно
растворяется в высыхающих маслах вследствие весьма высокого
молекулярного веса. Благодаря высокому содержанию поли-
стирола первая фаза образования покрытия — высыхание — яв-
ляется скорее физическим, чем химическим процессом, и поэтому
протекает очень быстро. Получаемая пленка благодаря повы-
шенному коэффициенту преломления полистирола имеет пре-
восходный блеск. Однако, наряду с этими ценными свойствами,
модифицированные стиролом масла имеют и некоторые недо-
статки, из которых основным является чувствительность пленки
к растворителям. Кроме того, возможно ухудшение совмести-
мости с маслами при наличии в композиции каких-либо дру-
гих пленкообразующих веществ. Краски, изготовляемые на ос-
нове масел, модифицированных стиролом, как и любые другие-
краски, не универсальны. Для некоторых назначений они могут
оказаться незаменимыми, а для других непригодными.
Масла, модифицированные стиролом, являющиеся сравни-
тельно новыми пленкообразующими веществами, безусловно,
имеют перспективы широкого применения в будущем.
Модификация масел малеиновым ангидридом
Мысль о введении малеинового ангидрида в молекулы вы-
сыхающих масел возникла приблизительно в 1935 г. Первые
опыты модификации масел с несопряженными двойными свя-
зями были поставлены с целью приближения технологических
свойств этих масел к свойствам масел типа тунгового.
Г л. 11. Высыхающие масла
Малеиновый ангидрид является чрезвычайно реакционноспо-
собным веществом, могущим вступать в реакции двух типов.
Он может реагировать как двухосновная кислота и в этом
отношении сходен с фталевым ангидридом. Кроме того, малеи-
новый ангидрид может вступать в реакции, характерные для
соединений с двойными связями. При раскрытии двойной связи
возникают свободные валентности:
ОС—сн
\ I
ос-сн
ОС—сн
°\ I
ос—сн
В таком виде малеиновый ангидрид может присоединяться
к большому числу непредельных соединений. Иногда эта реак-
ция имеет характер цепной полимеризации типа винильных
соединений и приводит к образованию сополимеров:
СН=СН+СН=СН СН—СН—СН—СН—~
11 II —*• 1111
R R' СО СО R R' СО СО
Хо _ _
Другой характерной реакцией является диеновый синтез, со-
стоящий в присоединении одной молекулы малеинового ангид-
рида на каждую систему сопряженных двойных связей:
Реакции второго типа, гораздо более распространенные, по-
зволяют осуществлять синтез многочисленных циклических со-
единений.
Наконец, между малеиновым ангидридом и непредельными
углеводородами могут протекать реакции еще одного типа. Ма-
леиновый ангидрид за счет раскрытия двойной связи присоеди-
няется к углеродному атому непредельного углеводорода с обра-
зованием замещенного янтарного ангидрида с непредельной
Обработка высыхающих масел
75
боковой цепью. Так протекает реакция с пропиленом, приводя-
щая к образованию аллилянтарного ангидрида:
СН2=СН—СН3 + нс=сн
I I
ОС со
о
СН2=СН—СН2—НС —сн2
I I
ОС со
В некоторых случаях эта реакция может протекать с пере-
мещением двойной связи в замещающей цепи. Это характерно
главным образом для аллиловых производных, которые превра-
щаются в пропениловые:
С6Н5-СН2—СН=СН2 + НС=СН С6Н5—СН=СН—СН,—НС—СН2
II— II
ОС со ОС со
Хо/ Хо/
Как видно из приведенных примеров, малеиновый ангидрид
является весьма реакционноспособным соединением, легко об-
разующим различные производные.
Для выяснения механизма присоединения малеинового ан-
гидрида к высыхающим маслам были предложены различные
реакции. В случае масел с сопряженными двойными связями
наиболее вероятной является реакция типа Дильса-Альдера:
СН3—(СН2)3—СН=СН—СН=СН—СН=СН—(СН2)7—СООН
НС=СН
I I _
ОС со
V
нс=сн
— СН3—(СН2)3—НС^ СН—СН=СН—(СН2)7—СООН
НС—сн
I I
ОС со
Если в кислотном радикале имеется не одна пара сопря-
женных двойных связей, реакция может протекать с любой из
них. При наличии одной системы сопряженных двойных связей
остается лишь одна возможность присоединения.
Для присоединения к маслам с изолированными двойными
связями можно допустить различные механизмы реакций: без
перемещения и с перемещением двойных связей. Если реакция
протекает без перемещения двойных связей, т. е. путем ‘простого
76
Гл. II. Высыхающие масла
присоединения в a-положении по отношению к двойной связи,
получаются различные производные янтарного ангидрида:
СН3—(СН2)4—СН=СН—СН2—СН=СН—СН2—(СН2)в—соон
+
нс=сн
I I
ОС со
СН3— (СН2)4— СН=СН—СН2— СН=СН—СН—(СНа)в—соон
н2с—сн
I I
ОС со
''с/
Если же взаимодействию с малеиновым ангидридом пред-
шествует перемещение двойных связей, то механизм реакции
аналогичен описанному для масел с сопряженными двойными
связями. Возможен и более сложный случай, когда перемеще-
ние двойной связи происходит в результате взаимодействия с
молекулой малеинового ангидрида:
СН3— (СН2)4—СН=СН—СН2—СН=СН-(СН2)7—соон
+
нс=сн
I
ОС СО "*
-> СН3—(СН2)4—СН=СН—СН=СН—СН—(СН2)7—соон
НС—сн2
I I
ос со
чо
Образующееся соединение может присоединить еще одну
молекулу малеинового ангидрида по реакции Дильса—Альдера,
в результате чего получится следующий конечный продукт:
нс=сн
СН3-(СН2)4—НС7 ^СН-СН—(СН2)7—СООН
НС —СН НС—сн2
'll II
ос со ОС со
V
Применение кислен высыхающих масел
77
Молекула линолевой кислоты может присоединить три мо-
лекулы малеинового ангидрида — две путем присоединения к
и-метиленовой группе и одну — по реакции Дильса—Альдера.
На основании имеющихся в настоящее время данных еще
трудно сделать точное заключение о достоверности той или
иной гипотезы. Возможно, что каждая из них содержит долю
истины, так как доказано, что малеиновый ангидрид способен
вступать в реакцию как с маслами, так и с непредельными жир-
ными кислотами и их эфирами. Во всех образующихся соеди-
нениях ангидридная группа чрезвычайно устойчива по отноше-
нию к омылению, что характерно для малеиновых аддуктов.
Масла, модифицированные малеиновым ангидридом, имеют
ряд преимуществ по сравнению с обычными маслами. Они лег-
че подвергаются термической полимеризации, при их высыха-
нии достигается более глубокое отверждение пленки, облада-
ющей лучшей атмосферостойкостью. Модификация малеиновым
ангидридом препятствует также желатинированию готовых ла-
кокрасочных материалов. Окончательное суждение о качестве
масел, модифицированных малеиновым ангидридом, затрудняет-
ся тем, что их изготовление и применение обусловлено в боль-
шей степени экономической конъюнктурой, чем техническими
соображениями.
ПРИМЕНЕНИЕ КИСЛОТ ВЫСЫХАЮЩИХ МАСЕЛ
Кислоты высыхающих масел представляют для производства
лаков и красок все возрастающий интерес. Находясь обычно
в связанном состоянии в виде триглицеридов, они не могут
участвовать в обычных для кислотной группы реакциях, приво-
дящих к созданию различных новых веществ с гораздо более
разнообразными свойствами, чем свойства самих масел.
Здесь не рассматриваются способы выделения кислот, кото-
рые весьма разнообразны. Из областей применения кислот, вы-
деленных тем или иным способом из высыхающих масел, самой
важной является производство модифицированных алкидных
смол (см. главу XI).
Сами кислоты и образуемые ими эфиры одноатомных спир-
тов не обладают способностью к высыханию, так как не могут
образовать трехмерных структур. Этой способностью обладают
эфиры многоатомных спиртов, в частности трехатомных (таковы
природные высыхающие масла). Если кислоты этерифициро-
ваны высшими полиолами (пентаэритритом, сорбитом, манни-
том и т. д.), способность к высыханию выражена еще лучше.
Из пентаэритрита и жирных кислот соевого масла можно
получить эфиры, у которых способность к высыханию такая же,
как и у льняного масла. Но не следует забывать, что высыха-
78
Гл. II. Высыхающие масла
ние возможно только в том случае, если сама кислота способна
к димеризации. Поэтому бесполезно пытаться этим методом при-
дать способность к высыханию кислотам, выделенным из масел,
которые сами не обладают этим свойством.
Полиамиды. Амиды жирных кислот сами по себе не пред-
ставляют интереса как вещества, способные к пленкообразова-
нию, но им можно придать это свойство, используя основные
принципы химии высокомолекулярных соединений.
Амиды мономерных кислот обычно являются кристалличе-
скими веществами, не обладающими способностью к пленкооб-
разованию. Иначе ведут себя амиды, полученные на основе ди-
аминов и димерных кислот. Так, например, продукты поликон-
денсации димерных кислот (подобных полученным путем тер-
мической полимеризации высыхающих масел) и алифатических
диаминов (например, этилендиамина или гексаметилендиамина)
дают пленкообразующие вещества типа термопластов, облада-
ющие высокой эластичностью. Такие пленкообразующие веще-
ства были впервые применены в США (норлак). Они относятся
к классу полиамидов, представителем которого является, на-
пример, найлон, но ввиду различия в строении исходной кисло-
ты отличаются по свойствам от найлона, в частности, раство-
римы в гораздо большем числе растворителей. Полиамиды в от-
личие от полиэфиров весьма устойчивы к омылению.
Полиамиды могут быть получены двумя методами: путем
поликонденсации или полимеризации. При получении полиами-
дов методом поликонденсации жирные кислоты сначала под-
вергают полимеризации или в свободном состоянии или в виде
триглицеридов, после чего отделяют недимеризовавшиеся кис-
лоты путем перегонки в вакууме. Димеризовавшиеся кислоты
конденсируют затем с диамином:
2R—СООН »- НООС—R—R—СООН
HOOC-R-R—СООН + H2N—(СН2)2—NH3 + НООС— R-R—СООН —
—>---ОС- R—R—СО—HN—(СН2)2—NH—ОС—R—R—СО------
Первой стадией процесса получения полиамидов методом
полимеризации является образование диамидов взаимодействи-
ем мономерных жирных кислот с диаминами:
2RCOOH + H2N—(СН2)2—NH2 R—СО—HN—(СН2)2—NH—ОС—R + 2Н2О
Образовавшиеся диамиды полимеризуются в условиях, ана-
логичных условиям термической полимеризации масел. При
этом происходит димеризация радикалов R и образуются ли-
Применение кислот высыхающих масел
79
нейные макромолекулы, по строению сходные с макромолеку-
лами, полученными методами поликонденсации:
.. —ОС—R—R—СО—HN—(СН2)2—NH—OC-R-R— CO-HN—(CH2)-NH— .
Поскольку не все кислотные радикалы способны димеризо-
ваться, в смеси остается некоторое количество незаполимери-
зовавшихся диамидов, которые необходимо отделить путем ва-
куумной перегонки.
Практически операции полимеризации и отделения не во-
шедших в реакцию мономеров могут быть осуществлены в од-
ном и том же аппарате, что является преимуществом данного
метода по сравнению с предыдущим.
Димеризованные кислоты высыхающих масел можно при-
менять как обычные двухосновные кислоты, но необходимо по-
мнить, что их молекулярный вес довольно велик и полиамиды,
получаемые на их основе, могут существенно отличаться по
свойствам от продуктов, полученных на основе обычных срав-
нительно низкомолекулярных кислот.
Продукты сложноэфирной конденсации. Алифатические слож-
ные эфиры одноатомных спиртов могут конденсироваться в при-
сутствии соответствующих конденсирующих агентов с образо-
ванием эфиров р-кетонокислот:
R—СН2—COOC2HS R—СН—COOC2HS
+ -► | + С2Н£ОН
R—СН2—СО—ОС2Н5 R—СН2—СО
В американском патенте (USP *2361027) указываются на
возможность сложноэфирной конденсации метиловых эфиров
кислот, полученных из различных высыхающих масел (льняно-
го, соевого, рыбьего жира и т. д.). Согласно этому патенту, та-
кие продукты представляют большой интерес вследствие боль-
шой скорости высыхания; будучи добавлены к высыхающим
маслам, они весьма существенно сокращают время их сушки.
Само собой разумеется, что такие пленкообразующие вещества
заслуживают внимания.
Совершенно очевидно, что для получения продукта с хоро-
шими свойствами в отношении сушки радикалы R должны иметь
непредельный характер.
Конденсирующим агентом является гидрид натрия, который
для достижения лучшего выхода должен быть чистым. Присут-
ствие в нем металлического натрия, едкого натра, карбоната
натрия вызывает нежелательные вторичные реакции.
Молекулы p-кетоэфиров могут претерпевать в зависимости
от условий вторичные превращения, характерные для этого
класса соединений: кислотное и кетонное расщепление. Реакция
80 Гл. II. Высыхающие масла
кислотного расщепления особого интереса не представляет, так
как при этом легко и просто выделяется обратно первоначаль-
ный эфир (или кислота):
R—СН2—СО +н2о R—СН2—СООН
R—сН~1 СО°С2Н5 r-ch2—соос2н5
Реакция кетонного расщепления, происходящая в щелочной
среде, ведет к образованию симметричного кетона:
R—СН2—СО +2кон
| ----* R—СН2—СО—СН2—R + С2Н5ОН Ч- К2СО3
R-CH— COOC2HS
Практически в процессе производства р-кетоэфиров очень
трудно избежать этих вторичных реакций, но, как указывается
в упомянутом патенте, образование подобных продуктов не
тшляется в данном случае помехой, так как образующиеся не-
предельные кетоны также улучшают высыхаемость масел.
Пока имеются только данные о возможности приготовления
Р-кетоэфиров, но нет опыта их практического применения. По-
этому в настоящее время нельзя даже примерно оценить, ка-
кое значение они могут иметь в дальнейшем в производстве
защитных покрытий.
Показатели химических свойств масел. Химия жиров вклю-
чает определение множества различных «показателей», не яв-
ляющихся константами, как показатели индивидуальных ве-
ществ, так как масла представляют собой смеси различных
соединений. Необходимо, однако, иметь возможность различать
между собой типы масел, чтобы быстро и безошибочно иден-
тифицировать льняное масло, подсолнечное масло, рыбий жир
и т. д. Эту задачу не так просто разрешить, так как большое
количество природных масел, даже абсолютно чистых, обла-
дает химическими и физическими свойствами, весьма близкими
к свойствам других чистых масел. С другой стороны, различ-
ные образцы масла, полученного из каких-либо определенных
семян (например, образцы льняного масла), могут иметь раз-
ные показатели, хотя все это образцы льняного масла.
«Показателям» масел продолжают и в настоящее время
придавать большое значение, пытаясь объяснить чрезвычайно
сложные явления ненадежными данными об изменении раз-
личных показателей.
Однако нельзя не признать, что эти показатели, разумно
примененные в отношении чистых продуктов, дают ценные све-
дения о них и в большинстве случаев остаются средством опре-
деления и достаточного контроля этих продуктов в лаборато-
риях, не имеющих специального оборудования.
Применение кислот высыхающих масел
81
Кислотное число (/а). Этот показатель выражается количе-
ством едкого кали (в миллиграммах), необходимым для ней-
трализации на холоду одного грамма масла. Нейтрализация
протекает по реакции:
RCOOH + КОН ->- RCOOK + Н2О
На основании кислотного числа можно с уверенностью уста-
новить лишь процентное содержание карбоксильных групп в
смеси и только в исключительных случаях удается узнать при-
роду радикала R. Кислотное число имеет большое значение
при анализе смеси кислот.
Число омыления (Is) выражается количеством едкого кали
(в миллиграммах), необходимым для расщепления одного грам-
ма триглицерида (или другого эфира, не являющегося произ-
водным глицерина). Реакция омыления имеет следующий вид:
RCOOR' + КОН RCOOK + R'OH
Число омыления глицеридов дает некоторое представление
о длине кислотных радикалов: чем больше число омыления, тем
короче кислотные радикалы. Если этот показатель слишком
низок по сравнению с обычным его значением для данного
масла, то можно подозревать, что исследуемое масло содержит
примесь какого-либо неомыляемого вещества.
Иодное число (/j) выражается количеством иода (в санти-
граммах), которое может присоединить один грамм масла в оп-
ределенных условиях опыта. Чистый иод с трудом присоеди-
няется по месту двойных связей, поэтому применяют уксусно-
кислые растворы галоидоиодидов (хлористый или бромистый
иод) и проводят реакцию в присутствии солей ртути (хлори-
стая или уксуснокислая ртуть), действующих каталитически.
Чтобы избежать возможных реакций замещения, реакционный
сосуд рекомендуется оставлять в темноте. Эта реакция являет-
ся фактически присоединением галоидоиодидов, а потому для
замены атомов других галоидов атомами иода титрование не-
обходимо проводить в присутствии избытка йодистого калия.
Избыток иода затем оттитровывается тиосульфатом калия. Ос-
новные реакции будут следующие:
... -СН=СН-----f-JCl ----СНС1—CHJ----
----СНС1—CHJ---|-KJ —КС1 4------CHJ—CHJ-----
Следует иметь в виду, что иодное число триглицерида дает
представление только об общей его непредельности. Было бы
слишком смелым на основании иодного числа делать опреде-
ленные выводы о природе самих кислотных радикалов. Напри-
мер, значение иодного числа льняного масла позволяет пред-
6—156
82
Гл. II. Высыхающие масла
полагать, что это масло представляет собой триглицериды кис-
лот с двумя двойными связями, тогда как в действительности
в нем присутствуют гораздо более разнообразные кислоты.
Определение иодного числа не имеет никакого смысла для
масел с системой сопряженных двойных связей. Следует пом-
нить, какие неправильные выводы относительно структуры элео-
стеариновой кислоты были сделаны вследствие пренебрежения
этим основным фактом. Присоединение иода в данном случае
приводило лишь к частичному раскрытию двойных связей, что
обесценило сделанные выводы. Присоединение иода, не приво-
дящее к насыщению всех двойных связей, наблюдается также
для терпеновых производных.
Содержание неомыляемых веществ. Этот показатель харак-
теризует процентное содержание веществ, не поддающихся омы-
лению (стерины, углеводороды и т. д.) и составляет обычно
1% для всех масел за исключением некоторых сортов рыбьего
жира. При определении неомыляемых веществ не удается обна-
ружить фосфатиды, которые оказывают заметное влияние на
свойства масел.
Гексабромное число. Определение этого числа проводят с
целью установить наличие линоленовой кислоты в высыхающих
маслах и количественно оценить ее содержание. Гексабромное
число выражает процентное отношение кислот, образующих не-
растворимые в эфире гексабромиды, к общему количеству кис-
лот, содержащихся в масле. Метод определения гексабромного
числа основан на следующей реакции:
СН3—СН2-СН=СН—СН2—СН=СН—СН2—СН=СН—(СН^—COOH4-3Brg —
— СН3—СН2—СНВг—СНВг—СН2—СНВг—СНВг—СН2—СНВг—СНВг—(СН2)7—СООН
Родановое число представляет интерес при определении раз-
личных компонентов смеси жирных кислот.
Родан (CNS)2, присоединяясь к олеиновой и линолевой кис-
лотам, образует продукты присоединения, состоящие из одной
молекулы родана и одной молекулы кислоты. Молекула лино-
леновой кислоты способна присоединить две молекулы родана.
Однако все эти соотношения верны лишь в первом приближении.
Техника определения роданового числа сложна и заклю-
чается в выделении родана из тиоцианата свинца во время
реакции. Подробно рассматривать здесь это число не имеет
смысла, поскольку оно является малоупотребительным.
Ацетильное число выражает количество (в миллиграммах)
едкого кали, необходимое для нейтрализации уксусной кислоты,
выделившейся при омылении одного грамма ацетилированного
масла. Этот показатель определяют только для масел, содер-
Применение кислот высыхающих масел 83
жащих гидроксильные группы, например для касторового масла.
Реакция ацетилирования протекает следующим образом:
R-OH4. (СН3СО)3О CH3COOR 4-СН3СООН
В процессе ацетилирования происходит выделение уксусной
кислоты, что может привести к искажению истинного значения
ацетильного числа, так как под действием свободной кислоты
может происходить ацидолиз глицеридов, т. е. замещение ради-
кала жирной кислоты ацетильным радикалом. Вследствие аци-
долиза изменяются результаты определения ацетильного числа.
Перекисное число (число Леа). Этот показатель, сравнитель-
но редко применяемый для характеристики высыхающих масел,
выражает содержание активного кислорода (в миллиэквива-
лентах) на 100 г масла.
Метод определения основан на том, что при взаимодействии
окисленного масла с раствором йодистого калия в уксусной
кислоте выделяется иод, который оттитровывают раствором
тиосульфата калия. Реакция протекает по схеме:
R—О—ОН— 4- 2KJ ♦ 2СН3СООН ROH -|- J2 + 2СН3СООК + Н2О
Совершенно очевидно, что перекисное число не представляет
интереса для характеристики высыхающих масел, которые все
склонны к окислению, всегда происходящему при высыхании
масляной пленки. Этот показатель обычно используется при
анализе пищевых жиров.
Другие показатели, рассматриваемые в книгах по химии жи-
ров, совершенно (или почти совершенно) не представляют ин-
тереса при изучении высыхающих масел.
Обычные физические константы: плотность, коэффициент
преломления, температура начала отверждения, способность
вращать плоскость поляризации и т. д. — при изучении масла!
так же необходимы, как и описанные выше показатели хими-
ческих свойств.
Масла не являются индивидуальными веществами и по не-
большому изменению аналитических показателей нельзя судить
о их качестве. Правда, можно омылить масло и, выделив со-
держащиеся в нем жирные кислоты, подвергнуть их всем необ-
ходимым операциям, принятым в органической химии для иден-
тификации той или иной кислоты (см. выше). Но в общем слу-
чае показатели химических и физических свойств масел дают
лишь качественные характеристики. Так, масло, имеющее опре-
деленное гексабромное число, ббразует пленку, имеющую тен-
денцию к пожелтению; масло с иодным числом 145 относится
к высыхающим маслам, но неизвестно, будет ли масло с иод-
ным числом 150 высыхать быстрее. Данные, получаемые при
6*
84 Гл. II. Высыхающие масла
определении показателей, следует истолковывать именно та-
ким образом, т. е. как относительные, а отнюдь не абсолютные
величины.
В табл. 2 приведены приближенные значения показателей
главнейших высыхающих масел.
ТАБЛИЦА 2
Свойства главнейших высыхающих масел
Название масла Показатели
Удельный вес г/см? Показатель преломления Число омыления Иодное число Содержание неомыляе- мых веществ % Гексабром- ное число
Льняное" 0.927—0,931 (при 20°) 1,4786— —1,4815 (при 25°) 189—196 170—204 0,5—1,6 42—52
Перилловое’ 0,930-0,937 (при 15°) 1,4800— —1,4820 (при 25°) 180—197 185—208 0,6—1,3 45—54
Сафроловое 0,925—0,928 (при 15°) 1,4679— —1,4693 (при 40°) 188—194 140—150 0,5—1,3 0,4—1,6
Маковое 0,924—0,927 (при 15°) 1,4750— —1,4753 (при 20°) 189—196 132—140 0,5—1,0 0
Соевое 0,922—0,925 (при 15°) 1,4722— —1,4750 (при 25°) 190—194 124-136 0,5—1,8 —
Подсолнеч- ное 0,922—0,926 (при 25°) 1,4734— — 1,4740 (при 20*) 189—194 120-136 0,7—1,2 —
Тунговое 0,934—0,937 (при 20°) 1.5163— —1,5213 (при 20°) 189—195 157—172 0,4-0,8 —
Ойтисиковое 0,963—0,969 (при 25°) 1,5130— —1,5158 (при 25°) 186—195 140—152 0,4—0,9 —
ГЛАВА Hl
ПРОИЗВОДНЫЕ ЦЕЛЛЮЛОЗЫ
П. Клеман
ЦЕЛЛЮЛОЗА И СОПУТСТВУЮЩИЕ ЕЙ ВЕЩЕСТВА
Целлюлоза является основным химическим веществом кле-
точных оболочек высших растений (явнобрачных). Она присут-
ствует также в некоторых низших растениях и, в виде исклю-
чения, в тканях некоторых животных.
Независимо от происхождения целлюлозы ей сопутствуют в
относительно постоянных количествах некоторые другие химиче-
ские вещества как сходного с ней строения, так и сильно от
нее отличающиеся.
Лигнин. Полимер (молекулярный вес от 10 000 до 20 000),-
структурной единицей которого является бензольное ядро со
сложной боковой цепью. Лигнин, представляющий собой важ-
ный побочный продукт в производстве бумаги и гидролизного
спирта, находит применение в производстве пластических масс.
Гемицеллюлозы могут быть извлечены из целлюлозы экс-
трагированием 17%-ным спиртовым раствором щелочи на хо-
лоду.
К этим веществам относятся как целлюлозы с низким моле-
кулярным весом (а- и р-целлюлозы), так и высокомолекуляр-
ные полисахариды, отличающиеся от целлюлозы по своему
строению:
Гексозаны (маннаны, галактаны);
Пентозаны (ксиланы, арабаны):
н н он
ксилан
Полиурон иды.
86
Гл. III. Производные целлюлозы
Пектиновые вещества. 1) Пектин: цепь галактуронидных
остатков, соединенных в положении 1,4' глюкозидными связями;
2) Пектиновая кислота: цепь остатков галактуроновой кислоты,
содержащая несколько карбоксильных групп, этерифицирован-
ных метиловым спиртом
3) Протопектин, или пектоза.
Кроме того, присутствуют и некоторые другие вещества,
содержание которых изменяется в зависимости от природы рас-
тительного материала; к ним относятся азотсодержащие про-
дукты, жиры, красители, воска, смолы, масла, таннины, мине-
ральные вещества.
Наличие сопутствующих веществ вызывает затруднения при
выделении целлюлозы из растительного сырья и ее очистке, так
как необходимо, чтобы операции очистки оказывали как можно
меньшее влияние на химические свойства и степень полимери-
зации целлюлозы.
ЛИТЕРАТУРА
Целлюлоза:
Ward, Cellulose and cellulose derivatives, E. Ott, Ed Intersc. Pub., New
York, 1943.
Л и г и и н:
Wacek, Kratel, J. Polym. Sci., 1948, 3, 539.
В a u n s, Cellulose and cellulose derivatives (см. выше).
Traynard, Assoc. Techn. de 1’Ind. Papetiere, 1951, 2, 49.
Пектин:
Palmer, High Polymer Physics (A. Robinson), Remsen Press Div., 1948.
Извлечение целлюлозы'-
Koon и corp., Cellulose and cellulose derivatives (см. выше).
Химическое строение целлюлозы
Установление химической формулы. Химический анализ очи-
щенной целлюлозы приводит к основной формуле (СбН10О5)я,
отвечающей ангидриду гексозы. Определение молекулярного
веса и физико-химических свойств целлюлозы показывает, что
она является высокополимерным веществом.
Целлюлоза и сопутствующие ей вещества
87
Реакции гидроксильных групп. Действием минеральных или
органических кислот можно произвести этерификацию всех гидр-
оксильных групп целлюлозы:
ныоз
(СвН10О6)„--* [C6H,O2(ONO2)3]„
трииитрат целлюлозы
(СН3СО)2О
(С6Н10О6)„ ~ * [С6Н7О2(ОСОСН3)3]„
триацетат целлюлозы
Действуя метилирующими агентами в определенных усло-
виях, можно получить триметильные производные:
(CH3)2SO4
(С6Н10О5)„ * [СеН7О2(ОСН3)3]„
При осторожном окислении целлюлоза превращается в цел-
луроновую кислоту, содержащую одну карбоксильную группу
в каждом остатке СеНщОй-
Эти характерные реакции указывают на присутствие в каж-
дом остатке СбНюОб трех спиртовых групп, из которых одна
является первичной. Строение это подтверждается спектрогра-
фическим анализом в инфракрасных лучах.
Реакции полного или частичного гидролиза. Анализ про-
дуктов кислотного гидролиза целлюлозы дает возможность до-
полнить сведения о ее строении.
Концентрированные кислоты и ферменты количественно пе-
реводят целлюлозу в глюкозу:
HSO
(С6Н10О5)„ *" nCsHjaOg
Гидролиз в мягких условиях приводит к образованию гомо-
логов глюкозы, а также некристаллизующихся целлодекстри-
нов (олигосахаридов). Примеры:
Целлобиоза:
' н он сн2он
Ь VT у
сн2он н он
Целлотриоза (СеНщСМзОг
Целлодекстрины или амилоиды Н — (CfiH10Os)n— ОН
(где5<п<10)
Под воздействием разбавленных кислот целлюлоза превра-
щается в гидратцеллюлозу со степенью полимеризации поряд-
ка 100.
88 Гл. III. Производные целл юлозы
На основании этих данных можно считать, что целлюлоза
представляет собой высокомолекулярное соединение линейного
строения
' —С6Н10О5 —С6Н10О5— • •
Линейная цепь при гидролизе разрывается на сегменты различ-
ной длины, вплоть до глюкозы.
Реакции гидролиза целлюлозы и ее метильных иЛи ацетиль-
ных производных, кроме того, показывают, что элементарным
звеном макромолекулы целлюлозы является целлобиоза, пред-
ставляющая собой два остатка глюкопиранозы, соединенные
в положении 1,4':
целлобиоза
Пространственное строение целлюлозы. Считается доказан-
ным, что глюкозные остатки в целлюлозе связаны между со-
бой в положениях 1,4' р-глюкозидными связями, причем кольцо
менее напряжено, если оно имеет форму кресла:
Спиртовые группы располагаются между двумя параллель-
ными плоскостями, на которых располагаются соответственно
четные и нечетные атомы колец глюкозы. За пределы этих пло-
Целлюлоза и сопутствующие ей вещества
89
скостей выступают аксиальные связи с гидрофобными атомами
водорода (Германе):
Такое строение обусловливает возникновение водородных
межмолекулярных связей целлюлозы, причем связь образуется
между гидроксилами одной цепи и атомами водорода другой
цепи. Это позволяет объяснить нерастворимость целлюлозы в
воде.
В крахмале, цепь которого образована а-глюкозидными свя-
зями, линейные макромолекулы амилозы легко изгибаются. По-
этому гидроксильные группы беспорядочно расположены в про-
странстве, чем и объясняется растворимость крахмала в воде.
Возможности перегибов целлюлозной цепи по глюкозидным
связям С—О—С относительно ограничены стерическими затруд-
нениями, связанными с пространственным расположением [3-глю-
козидных связей.
Концевые группы молекулы. При гидролизе триметилцеллю-
лозы образуется, кроме 2,3,6-триметилглюкозы, некоторое коли-
чество 2,3,4,6-тетраметилглюкозы, что является доказательством
наличия в целлюлозной цепи глюкозидных и глюкозных звеньев:.
остаток,
не имеющий восстановительных
СВОЙСТВ
остаток
с восстановительными
свойствами
Часть глюкозных остатков содержит полуацетальные груп-
пы, что придает целлюлозе восстановительные свойства, изме-
няющиеся в зависимости от длины цепи; содержание этих групп
'90
Гл. III. Производные целлюлозы
может быть определено химическими методами (медное число,
реакция с фенилгидразином, метилгидроксиламином или этил-
меркаптаном).
Остатки глюкозы, сохранившие глюкозидный гидроксил,
очень чувствительны к действию окислителей и в щелочной сре-
де обычно окисляются с образованием глюконовой или сахар-
ной кислот.
Глюкозные остатки второго типа (не имеющие глюкозид-
ного гидроксила) содержат по четыре гидроксильные группы,
что позволяет определить химическими методами степень поли-
меризации целлюлозы.
При изучении механизма деполимеризации целлюлозы и ее
производных было обнаружено, что гидролиз высокомолекуляр-
ных цепей не подчиняется статистическому закону. Это явле-
ние получило разные объяснения, основанные на двух предпо-
ложениях.
Согласно одной гипотезе, в макроцепи целлюлозы имеются,
кроме остатков глюкозы и глюкофуранозы, также окисленные
кольца:
Монотонное чередование этих групп приводит при гидролизе
к образованию молекул меньших размеров, но более однород-
ных по величине, например, содержащих по 500 остатков глю-
козы (по Веберу и Шульцу) или по 150 остатков глюкозы (по
Кинсингеру и Гуку).
Другая гипотеза предполагает наличие в макромолекуле
целлюлозы также цепей других типов. Так, Паксу предложил
для целлюлозы формулу, в которой элементарные звенья
Целлюлоза и сопутствующие ей вещества
91
(660 А) соединяются полуацетальными связями с остатками
глюкозы в открытой форме, образующими мостики:
I
О
Хэуорз с сотр. выдвинули предположение, что целлюлозные
цепи могут полностью состоять из циклических глюкозных
остатков, но содержать поперечные связи, легче подвергающие-
ся гидролизу:
Аналогичное строение предполагают Гесс и Штейрер. Обе
эти формулы требуют подтверждения.
92
Гл. III. Производные целлюлозы
ЛИТЕРАТУРА
Р. Н. Hermans, Physics and Chemistry of cellulose fibres, Elsevier Pub!..
New York, 1949, p. 6—13.
R о u d i e r, Ass. Techn. de 1'Ind. Papetiere, 1951, 2, 56.
H a 1 s a 1 и сотр., Nature, 1947, 159, 97.
Re eve,s, Mazzeno, Hoffpauir, Text. J. Res., 1951, 21, 78.
S i p p e 1, Koll. Z., 1950, 119, 42.
Молекулярный вес целлюлозы
Точный молекулярный вес нативной целлюлозы неизвестен.
При выделении и очистке целлюлозы молекулярный вес ее зна-
чительно понижается и одновременно нарушается однородность
по величине частиц. '
Тем не менее можно предполагать, что степень толимериза-
ции природной целлюлозы не ниже 3000, причем частицы отно-
сительно однородны по молекулярному весу (Гуземан и
Шульц).
Определение молекулярного веса химическими методами.
Молекулярный вес целлюлозы можно вычислить по содержа-
нию концевых групп. Найденная величина отвечает среднечис-
ловому значению молекулярных весов отдельных цепей.
Определение альдегидных групп. Содержание альдегидных
групп определяют, используя их восстановительные свойства.
Так, восстановлением фелинговой жидкости с последующим оп-
ределением закиси меди СигО по перманганату калия находят
медное число.
Если р вес (в граммах) закиси меди, образовавшейся при
восстановлении 100 г целлюлозы, то степень полимеризации
составляет:
Величина р обычно находится в пределах от 0,01 до 3.
По иодному числу, выражаемому объемом (в миллилитрах)
0,1 н. раствора иода, поглощенным одним граммом целлюлозы,
степень полимеризации вычисляют по формуле:
где I — иодное число.
Эти два метода основаны на предположении, что восстанав-
ливающие группы не подверглись окислению при обработке и
очистке целлюлозы и что последняя не содержит других вос-
станавливающих групп.
Целлюлоза и сопутствующие ей вещества
93
Серное число определяется по реакции альдегидных групп с
этилмеркаптаном:
Н Н
/ I zSQIi,
Целлюлоза—С 2C2H5SH —Целлюлоза—С< 4-Н»О
xsc2H5
о
Если S количество присоединенной серы в процентах, то
степень полимеризации выразится уравнением:
Образование фенилгидразона при взаимодействии с фенил-
гидразином также может служить методом определения степени
полимеризации (Шампетье). Можно также определять количе-
ство концевых альдегидных групп, пользуясь реакцией с метил-
гидроксиламином (Мезук и Пурвес).
Определение концевых тетраоксиглюкозных остатков (см.
стр. 89). Их содержание определяется косвенно после выделе-
ния тетраметилглюкозы (Хэуорз и Мэгемер).
Определение карбоксильных групп. Концевые альдегидные
группы целлюлозы можно перевести в карбоксильные группы,
не окисляя остальных гидроксилов макроцепи. Эти карбоксиль-
ные группы можно определять прямым ацидиметрическим тит-
рованием.
Определение молекулярного веса физическими методами.
Осмометрический метод состоит в определении осмотического
давления л (в сантиметрах водяного столба) разбавленных
растворов различных концентраций, выражаемых в а/100 мл.
Функция
для очень разбавленных растворов имеет обычно линейный ха-
рактер, а в пределе (при бесконечном разбавлении) величина
у связана со среднечисловым молекулярным весом отноше-
нием:
Мв='^
11 It
_ с _с—0
где тг— осмотическое давление;
с—< концентрация;
R— газовая постоянная;
Т — абсолютная температура.
94
Гл. III. Производные целлюлозы
Осмометрическим методом можно определять с достаточной
степенью точности (5—10%) молекулярный вес простых и слож-
ных эфиров целлюлозы в растворах (рис. 8). Для различных
растворителей соответствующие прямые— —f (с) приводят к
одному и тому же пределу, но имеют тем больший наклон, чем
выше растворяющая способность растворителя.
Существование в полимере межмолекулярных связей сказы-
вается в аномальном понижении предельной величины-^ в рас-
творителях с малой растворяющей способностью.
Рис. 8. Определение молекуляр-
ного веса фракций ацетата цел-
люлозы в ацетоновом растворе
при 25° осмометрическим методом
[по Беджли и Марку, J. Phys.
Chem., 51, 61 (1947)}.
Рис. 9. Определение вязкостей фракций
ацетата целлюлозы в ацетоновом
растворе (по Беджли и Марку, см.
подпись под рис. 8.)
Вискозиметрический метод. Характеристическая (приведен-
ная) вязкость определяется пределом:
[т(] = 1пп
с-»0 \ с /
Ч—То
где чуд. — — удельная вязкость;
Ч—коэффициент вязкости раствора;
Чо— коэффициент вязкости растворителя;
с— концентрация, е/100 мл.
Практически величину [р] находят, измеряя удельные вяз-
кости разбавленных растворов (!С<1%) разных концентраций
Целлюлоза и сопутствующие ей вещества 95
и экстраполируя полученную кривую до бесконечного разбавле-
ния. Значение [rj] колеблется в пределах от 0,5 до 12. При
построении графика в координатах т]уд./с— с или Чуд./С— 'Чул-
получаются обычно прямые линии (рис. 9). Для расчета кон-
центрации предложено также несколько формул, имеющих более
или менее эмпирический характер.
При переходе от одного растворителя к другому изменяется
(часто значительно) наклон кривых
^=Г(С) или ^ = Г(Ъ)
и происходит менее заметное изменение значения приведенной
вязкости [-Г)].
Отношение между приведенной вязкостью в данном раство-
рителе и молекулярным весом можно вычислить, измеряя при-
веденную вязкость [т]] для полимергомологических фракций, мо-
лекулярный вес которых определен прямым методом. В сравни-
тельно небольшом интервале молекулярных весов эта зависи-
мость с достаточной точностью выражается уравнением типа
[ т) ] = КМа
где К и а—постоянные для системы полимер — растворитель
при заданной температуре (табл. 3).
ТАБЛИЦА 3
Значения К и а. для некоторых производных целлюлозы
Полимер Растворитель Температу- ра °C К-10 4 а Интервалы молекуляр- ного веса
Целлюлоза Медноаммиач- ный раствор 25 0,85 0,81 —
Ацетат целлю- Ацетон » 0,19 1,03 10000—130000
лозы » » 1 49 0,82 31 000—390 000
0,89 0,30 10000—100 000
» 30 0,91 0,78 25 000—126 000
Ацетобутират Ацетон 25 1,37 0,83 12 000—210000
целлюлозы Уксусндя кисло- та » 1,46 0,83 12 000—210 000
Пиридин » 1,33 0,83 12 000—210000
Нитрат целлю- лозы (12,2%Ы) Ацетон — 0,38 0,93 —
Приведенная вязкость разбавленных растворов часто
приравнивается к [rj] (Штаудингер) и коэффициент принимается
96
Гл. Ш. Производные целлюлозы
близким к единице, что упрощает уравнение, приводя его к ви-
ду Пул. = КМ*Л4. Значения постоянной Ки обычно находятся в
пределах от 4* 104 до 20-104.
Рассеяние света. Способ состоит в измерении коэффициента
рассеяния света (т) разбавленным раствором полимера. Моле-
Н-с
кулярныи вес вычисляют, экстраполируя выражение —— до
бесконечного разбавления:
j____г II-с 1
м ~ L z L-o
где т — мутность (рассеяние при 90°);
с— концентрация;
Н— постоянная, зависящая от длины волны падающего
света и показателя преломления раствора.
Рассеяние света было использовано, в частности, для изме-
рения молекулярных весов фракций ацетилцеллюлозы, размеры
молекул которых оказалось возможным определить путем иссле-
дования дисимметрии распределения и деполяризации рассеян-
ного света (Штейн и Доти).
Ультрацентрифугирование. Этим методом оказалось воз-
можным установить связь между средним молекулярным весом
целлюлозы и ее производных и их приведенной удельной вяз-
костью (Грален, Кремер, Зигнер). Преимуществом этого метода
является возможность определения абсолютного значения
молекулярного веса и непосредственного контроля полидисперс-
ности.
Метод порога осаждения. Относительное количество (у) оса-
дителя, требуемое для осаждения сильно разбавленного рас-
твора, может быть связано со средним молекулярным весом
эмпирической формулой (Шульц):
Коэффициенты а и С можно вычислить, выражая как
функцию у и градуируя кривую по фракциям с известным зна-
чением молекулярного веса. Этот простой способ, являющийся
развитием старого метода определения «точек помутнения» по
Мардлю, был применен, в частности, при изучении молекуляр-
ных характеристик нитроцеллюлозы (От). Турбидиметрическое
определение может быть автоматизировано при помощи фото-
электрических методов (Морей и Тамблин) и дает кривую,
очень близкую к интегральной кривой распределения.
Целлюлоза и сопутствующие ей вещества 97
Поверхностные пленки. Изотермическое сжатие пленки по-
лимера, полученной (из сильно разбавленного раствора) в вице
мономолекулярного слоя на поверхности воды, позволяет вы-
числить величину Мп:
Мп =
Г Кс I
L Р JC--0
где с — поверхностная концентрация,
р —поверхностное натяжение, 10—3 дин]см.-,
/(=2,43-10-6 (при 20°С).
Получаемая кривая позволяет также определять простран-
ственную структуру молекулы ацетата целлюлозы в мономоле-
кулярном слое.
В табл. 4 .приведены примерные молекулярные веса цел-
люлозы и ее производных.
ТАБЛИЦА 4
Примерные молекулярные веса целлюлозы и ее производных
Производные целлюлозы Молекулярный вес Производные целлюлозы Молекулярный вес
Природный хлопок От 500000 до не- скольких миллио- нов 1 Вискоза (волокна и пленки) .... Медноаммиачное 30 000—70 000
Отбеленный линт Отбеленная хлопча- тобумажная пря- жа Древесная целлю- лоза сульфитная . . сульфатная . . Щелочная целлюло- за 500000—700000 150 000—200000 700000 600 000 200 000 волокно .... Нитрат целлюлозы лаковый .... для пластмасс и пленок . . . для порохов . . Ацетат целлюлозы (пленки и искус- ственное волокно) Ацетобутират цел- 70000—100 000 30 000—80000 50 000—120000 400000—800000 40 000— 80 000
З-Целлюлоза . . . у-Целлюлоза . . . Ксантогенат целлю- 2 000—10 000 2000 люлозы .... Триацетат целлю- лозы (пленка) 60 000—90 000 50000—120000
лозы 80000-120000 Метилцеллюлоза . Этилцеллюлоза . . Бензилцеллюлоза . 40 000—80 000 30000—150 000 10000—60000
Из приведенных данных видно, что значения среднего моле-
кулярного веса производных весьма различны и, как правило,
указывают на более пли менее сильную деполимеризацию по
сравнению с исходной целлюлозой. Деструкция вызвана боль-
шей частью химической обработкой (выделение и очистка цел-
люлозы, реакции замещения). Вторичным проявлением этого
гидролитического распада целлюлозных цепей является усиле-
7—156
98
Гл. III. Производные целлюлозы
ние или появление некоторой неоднородности в распределении
по молекулярным весам (полидисперсность).
Полвдисперсность целлюлозы
Общее представление о степени однородности по молеку-
лярному весу можно получить при лабораторном контроле,
определяя растворимости в специально подобранном раство-
рителе или, наоборот, числа коагуляции при добавлении нерас-
творителя. Достоинствами этих методов являются быстрота и
простота, что позволяет пользоваться ими при решении техни-
ческих задач в производстве. Более точные сведения о полидис-
персности могут быть получены путем сопоставления средних
молекулярных весов, найденных разными методами (для моно-
дисперсных полимеров эти средние значения должны быть оди-
наковы); но чаще применяется
способ фракционирования, по-
зволяющий непосредственно
построить кривую распределе-
ния частиц полимера по моле-
кулярным весам. Для относи-
тельно однородных систем
фракционирование может быть
заменено отделением наиболее
высокомолекулярных и наибо-
лее низкомолекулярных фрак-
ций и простым определением
среднего молекулярного веса
остальной части полимера.
Полидисперсность производного целлюлозы можно, наконец,
оценить непосредственно нефелометрически (получая кривую,
близкую к интегральной кривой распределения) или косвенно—
путем измерения двойного лучепреломления потока. Характер
кривой растворимости также дает представление о распределё-
нии по молекулярному весу.
В опубликованных работах имеется значительное количе-
Рис. 10. Примерная кривая распреде-
ления ацетата целлюлозы по моле-
кулярному весу
ство кривых распределения для целлюлозы или ее производных
(рис. 10), но в их форме нет никаких общих признаков (раз-
мытость, дисимметрия), по которым можно было бы характе-
ризовать отдельные образцы. Кривые распределения неотбе-
ленной древесной целлюлозы часто имеют два максимума, из
которых один указывает на присутствие примесей низкомолеку-
лярных частиц. Неотбеленный хлопковый линт, напротив, пред-
ставляется более гомогенным, хотя, согласно Сведбергу, кри-
вая распределения имеет несколько максимумов, отвечающих
полимерам, содержащим примерно ГСЮ, 200 и 300 остатков
глюкозы. В результате отбелки и превращения в вискозу в
Целлюлоза и сопутствующие ей вещества
99
обоих случаях уменьшается средняя степень полимеризации и
материал становится более однородным по молекулярному ве-
су, особенно целлюлоза, регенерированная из линта.
Кривые распределения по молекулярным весам сложных
эфиров целлюлозы редко бывают симметричны и обычно имеют
несколько максимумов.
ЛИТЕРАТУРА
Концевые группы:
Heuser, Cellulose Chemistry, Wiley a. Johnson, New York, 1946.
Unruh, Kenyon, J. Am. Chem. Soc., 1942, 64, 127.
Вискозиметрия:
Newman, J. Coll. Sci., 1950, 54, 964.
Осмометрия:
Philipp, Bjork, J. Polymer Sci., 1951, 6, 383, 549.
Рассеяние света:
Stein, Doty, J. Am. Chem. Soc., 1946, 68, 159.
Ультрацентрифугирование:
S ved berg, Cellulosechemie, 1943, 21, 57.
Пороги осаждения:
О t h, Bull. Soc. chem. Beiges, 1949, 58, 285.
Morey, T a m b 1 у n, J. Phys. Coll. Chem., 1947, 51, 721.
Поверхностные пленки:
Clement, Pouradier, J. chim. Phys., 1949, 46, 620.
Фракционирование производных целлюлозы:
Tamblyn, Morey, Wagner, Ind. Eng. Chem., 1945, 37, 573.
J u 1 1 a n d e r, J. Polymer Sci., 1947, 1, 2.
Herren t, Govaerts, J. Polymer Sci., 1949, 4, 289.
Beauvalet, Clement, Bull. Soc. chim., 1949, D. p. 1, Comm. 22-e,
Congres de Chinlie Ind., Barcelone, 1949.
Cocpick, Battista, Lytton, Ind. Eng. Chem., 1950, 42, 2533.
Billmeyer, Stockmayer, J. Polymer Sci., 1950, 5, 121.
Schultz, J. Makromol. Chem., 1950, 83.
Mitchell и сОтр., Ind. Eng. Chem., 1953, 45, 2482.
Микроскопическое исследование структуры целлюлозы
Структура целлюлозных волокон обусловливает как физи-
ческие свойства природного волокна, так и его поведение по
отношению к некоторым химическим реагентам. Изучение струк-
туры макромолекул может быть углублено посредством микро-
скопических исследований в естественном или поляризованном
свете нативных волокон, а также набухших и окрашенных.
Более полные данные можно получить при использовании ми-
кроскопа с фазоконтрастным устройством, электронного микро-
скопа или рентгенографических методов.
7*
100
Гл. III. Производные целлюлозы
Волокна растительной целлюлозы являются одно- или мно-
гоклеточными. Каждая элементарная клетка представляет со-
бой трубку многоугольного сечения, размеры которой зависят
от вида растения:
Длина
мм
Хлопок
волокно . 15—60
линт . . . 1
Лен.....................4—6
Джут..................... . 1—5
Конопля . . .......5—60
Рами................. . . 60—250
Водоросли ............20—30
Злаки.................. . 1—2
Древесина твердых пород . . 0,1
Древесина мягких пород . . 15
Диаметр
р-
15-25
20
12-76
10—25
16—50
16—125
8-15
15 -
40
Хлопок (линт) представляет собой одноклеточное волокно,
строение которого изображено на рис. 11. Оболочка волокна
(кутикула) 1 состоит из воскообразного вещества и имеет
складчатую структуру. Первичный слой 2 толщиной 0,5 р обра-
зован скрещенными фибриллами и имеет состав (в %):
Целлюлоза ................ . . . . 98,5
Пектиновые вещества................0,5
Арабиноза, галактоза и воска .... 1,0
Вторичный слой 3 состоит из чистой целлюлозы. Он образо-
ван пластинчатыми фибриллами длиной 100 р и диаметром
Рис. 11. Волокно хлопка:
/—оболочка; 2—первичный слой;
3—вторичный слой; 4—радиальные
стяжки; 5—пологть (люмен).
Рис. 12. Строение
волокна льна, рами
или джута.
0,4 р,, свернутыми спирально под углом 57° слоями (30 слоев)
по 1000 фибрилл. Макромолекулы целлюлозы расположены по
оси фибрилл со случайными смещениями в завитках.
Волокна льна, рами, джута и т. п.— многоклеточные (рис. 12).
Они состоят из полигональных клеток, склеенных пектиновыми
Целлюлоза и сопутствующие ей вещества
101
веществами. Строенйе клетки аналогично хлопковому волокну,
но клеточные стенки толще, а внутренняя полость уже. Эги
волокна состоят на 80% из
целлюлозы; остальное — пек-
тин, лигнин, камеди.
Более сложное строение
имеют волокна древесной цел-
люлозы (рис. 13). Межклеточ-
ное вещество 2 состоит из лиг-
нина и полиуронидов. Клеточ-
ные стенки состоят на 40—
60% из целлюлозьи, остальное
составляют лигнин, гемицел-
люлоза и пектиновые вещест-
ва. Макромолекулы целлюло-
зы во внешней 3 и внутрен-
ней 5 частях вторичного слоя
ориентированы •перпендикуляр-
но оси фибрилл, а в средней
части 4 — по оси фибрилл,
располагающихся спирально,
Рис. 13. Волокно древесной целлюлозы:
/—клеточные стенки; 2—межклеточное веще-
ство первичного слоя; 3—внешняя часть вторич-
ного слоя; 4—средняя часть вторичного слоя:
5—внутренняя часть вторичного слоя; 6— по-
лость (люмен).
как у хлопка.
Ниже сопоставлены характеристики волокон регенерирован-
ной и химически обработанной целлюлозы:
Волокно Внешний вид волокна Поперечный разрез
Мерсеризован- ный хлопок Вискозное Медноаммиач- ное Ацетатное Шелковистое волокно с гладкой поверхностью Негладкая поверхность, целлюлозные цепи в на- ружной части волокна ориентированы по его оси Гладкая поверхность Негладкая поверхность Полость меньше, чем у хлопка; контур более правильной формы Зубчатый контур; в наружной час- ти плоскостная ориентация крис- таллитов, во внутренней—различ- ная в зависимости от условий формования волокна Овальный контур; ориентация на- ружных и внутренних частей во- локна почти не различается Зубчатый контур
ЛИТЕРАТУРА
Matthews. Textile Fibers, Chapman a. Hall, London, 1947.
S e n n e 1 i e r, L’identification des matieres textiles, Librairie Polytechn.
Ch. Beranger, Paris, 1951.
Welch, Roseveare, Mark, Ind. Eng. Chem., 1946, 38, 582.
Preston, Modern Textile Microscopy, London, 1949, 21 IT.
Hall, The Standard handbook of Textiles, London, 1950.
102
Гл. III. Производные целлюлозы
Рентгенографическое исследование структуры целлюлозы
Этим методом показано, что целлюлоза и ее производные
являются высокомолекулярными микрокристаллическими ве-
ществами.
Характер рентгенограммы зависит от предварительной об-
работки образца целлюлозы. Размолотая целлюлоза, пучок во-
Рис. 14. Рентгенограмма природного
волокна рами (снимок Трилла и Ле-
грана, рентгеновская лаборатория
C.N.R.S.).
локон, пленка дают рентге-
нограммы с размытыми
кольцами Дебая—Шеррера.
На рентгенограммах при-
родных целлюлозных воло-
кон или ориентированной
целлюлозной нити вместо
размытых дифракционных
колец можно увидеть отчет-
ливые пятна или сегменты
(рис. 14). Такие рентгено-
граммы идентичны получае-
мым для истинных кристал-
лов, где одна из кристалло-
графических осей .прини-
мается за ось вращения, ес-
ли Х-лучи падают перпенди-
кулярно ее направлению.
Структура кристаллической решетки. Рентгенограммы раз-
личных образцов природной целлюлозы растительного или жи-
вотного происхождения (за исключением целлюлозы морских
водорослей) соответствуют моноклинной кристаллической ре-
шетке (целлюлоза I), имеющей следующие параметры:
а 8,20—8,35А
b ............ 10.28А
с ...........7,8—7.83А
Р . ......... 84—85°
Период идентичности по оси целлюлозных волокон (10,28 А)
довольно точно совпадает с расстоянием, которое можно теоре-
тически вычислить для двух остатков глюкозы, соединенных
в положениях 1, 4'.
Предположение об ориентации целлюлозных цепей с обра-
зованием моноклинной решетки было высказано Герцогом,
Спонслером и Доре и подтверждено Мейером, Марком и Ми-
шем (с учетом относительной интенсивности дифракционных
полос).
Модель структуры, предложенная последними автора-
ми (рис. 15), соответствует расположению глюкозных остатков
Целлюлоза и сопутствующие ей вещества
ЮЗ
в одной плоскости ab. Расстояние 2,6 А, разделяющее гидро-
ксильные остатки двух соседних цепей, благоприятствует об-
разованию водородных связей, которые частично обусловливают
кристалличность целлюлозы.
Ввиду относительно не-
большого количества дифрак-
ционных полос и пятен и воз-
можности взаимного наложе-
ния некоторых других полос
нет оснований утверждать, что
эта модель является единст-
венной, правильно отража-
ющей пространственное стро-
ение целлюлозы. Хэуорз, в ча-
стности, предложил модель,
основанную на предположении рис ]5_ Структура целлюлозы по
О сгибаемости целлюлозных це- Мейеру, Марку и Мишу.
пей под действием вторичных
интер- или интрамолекулярных связей между гидроксилами.
Пьерсе предложил другую конфигурацию, получившую крити-
ческую оценку Мейера. Все это свидетельствует о том, что окон-
чательно строение целлюлозьи еще не установлено.
При замене гидроксилов алкоксильными или ацильными
группами различных размеров происходит значительное изме-
нение кристаллической решетки (Трилла). Период идентич-
ности 10,3 А в общем сохраняется (хотя возможны и величины,
некратные 5,15 А), но только при условии замещения всех спир-
товых групп.
Кристаллический характер всех производных целлюлозы вы-
ражен тем яснее, чем больше заместителей одинакового раз-
мера содержат высокомолекулярные цепи; это условие выпол-
нимо лишь в том случае, если все гидроксилы заменены на
идентичные группы (Матье).
Полиморфизм. Указанные выше размеры кристаллической
решетки целлюлозы относятся к нативной целлюлозе, или цел-
люлозе I. Иногда наблюдаются другие виды решеток, соответ-
ствующие различным полиморфным разностям,’ достаточно хо-
рошо исследованным. Одна из этих полиморфных форм — цел-
люлоза II, или гидратцеллюлоза, получается при осаждении
целлюлозы из раствора.
Обычным примером гидратцеллюлозы является мерсеризо-
ванная целлюлоза, которая после обработки достаточно кон-
центрированной щелочью отмыта от щелочного раствора. По-
лиморфизм выражается в перемещении и незначительном (на
104
Гл. III. Производные целлюлозы
27°) повороте плоскостей, в которых расположены остатки цел-
лобиозы (рис. 16). В литературе описаны также другие поли-
морфные разности.
Для различных простых и сложных эфиров целлюлозы так-
же, по-видимому, возможно явление полиморфизма типа цел-
Рис. 16. Структура нативной
(А) й мерсеризованной (5)
целлюлозы.
люлоза I—целлюлоза II. Форма зави-
сит от того, получено ли производное
в гетерогенной среде (тогда четкая
рентгенограмма свидетельствует о со-
хранении волокнистого строения ис-
ходной целлюлозы) или в гомогенной
среде, в которой макромолекулы пол-
ностью диспергированы.
Полиморфное превращение целлю-
лозы может совершаться также при
ее набухании. Набухание должно про-
исходить в этом случае интрамолеку-
лярно (в отличие от интермолекуляр-
ного набухания, когда реагенты, вы-
зывающие набухание, не могут про-
никнуть внутрь кристаллической ре-
шетки полимера).
Кристалличность целлюлозы. Сте-
пень кристалличности различна для
различных типов целлюлозы и может изменяться при физиче-
ской и химической обработке.
В опровержение старых гипотез относительно мицеллярно-
го строения целлюлозы (Нагели) в настоящее время можно
считать установленным, что целлюлоза не состоит из изолиро-
ванных кристаллитов, сцементированных «аморфной» целлю-
лозой. Надо считать, что псевдокристаллы в действительности
представляют собой отдельные участки ориентированных цел-
люлозных цепей. Каждая цепь может участвовать в образо-
вании ориентированных областей 1, определяющих кристалли-
ческий характер, и одновременно в образовании аморфных
областей 2, где беспорядочно переплетаются высокомолеку-
лярные цепи (рис. 17).
У хлопка размер ориентированных участков достигает в
° • о
среднем 50 А в ширину и от 600 до 1000 А в длину (общая
длина молекулы хлопковой целлюлозы 10000 — 20000 А). Эти
псевдокристаллиты могут быть выделены путем осторожного
гидролиза целлюлозных цепей. Действительно, опыт показы-
вает, что гидролитическое расщепление целлюлозных цепей
идет быстрее в той части полимера, где макромолекулы рас-
положены наиболее беспорядочно (Левель и Гольдшмидт).
Целлюлоза и сопутствующие ей вещества
105
Распад целлюлозы, вначале очень быстрый, постепенно стаби-
лизуется, так как это явление тем сильнее выражено, чем
меньше степень кристалличности целлюлозы. Таким образом,
Рис. 17. Кристалличность целлюлозы:
1—ориентированные области; 2—аморфные области.
путем осторожного гидролиза могут быть выделены микрокри-
сталлиты, которые способны даже сохранить волокнистое стро-
ение (рис. 18).
Рис. 18. Микрофотографии:
А—ацетат целлюлозы после фракционирования; Б—частицы, извлеченные из хлопкового во-
локна, после гидролиза в течение 24 час. под действием концентрированной соляной кислоты
Signer, Bull. Soc. chim. Fr., 1949, 667].
При прямом исследовании фибрилл под электронным или
обычным микроскопом после металлизации (Фрей Висслинг)
удалось обнаружить в скоплениях высокомолекулярных цепей
пустоты, достигающие 1000 А в длину и 200 А в ширину.
В фибриллах хлопка обнаружены более узкие пустоты (10 А),
отделяющие упорядоченные участки друг от друга.
106 Гл. III. Производные целлюлозы
Соотношение кристаллических и аморфных участков из-
меняется в зависимости от природы исходной целлюлозы. Сте-
пень кристалличности может составлять от 70% для природ-
ных волокон до 10% для аморфной целлюлозы, полученной
при сухом измельчении вискозного волокна. Для регенериро-
ванной целлюлозы или омыленных ацетатов целлюлозы она
составляет около 40%. Под действием различных химических
и физических факторов (например, температуры) степень кри-
сталличности может изменяться.
Рентгенограммы волокон природной целлюлозы показывают,
что кристаллиты имеют преимущественную ориентацию вдоль
оси волокна. В .хлопке они перекрещиваются под углом 57°
в соответствии со спиральной структурой, устанавливаемой пу-
тем оптических наблюдений.
В волокнах регенерированной целлюлозы или ее сложных
эфиров не обнаруживается ориентации кристаллитов, если они
в процессе формования специально не подвергались вытяжке.
Этим способом изменения структуры пользуются, кстати, для
улучшения некоторых механических свойств волокон, например
при получении искусственного шелка типа фортизан (волок-
но G, состоящее из регенерированного триацетата, омыляемо-
го под натяжением).
Ацетатные или нитроцеллюлозные пленки, получаемые пу-
тем испарения растворителя без вытяжки, слабо ориентиро-
ваны. Ориентация, возникающая при истечении из отливоч-
ного приспособления, может быть усилена путем вытяжки или
сушки под натяжением.
Взаимное переплетение волокон в целлюлозных пастах
можно исследовать при помощи пропитки волокон иодом или
сернокислым свинцом, что делает их непрозрачными для мяг-
ких рентгеновых лучей (Пелгромс).
Механические испытания волокон рами показали, что макро-
молекулы целлюлозы располагаются не параллельно оси во-
локна, а по спирали с большим шагом (Шевенар и Шампетье).
Растягивающее усилие приводит к выпрямлению спирали за
счет изменения валентных углов оксидных связей. Остаточная
деформация, сопровождающая эластические деформации, вызы-
вается продольным скольжением макромолекул относительно
друг друга.
О степени ориентации целлюлозных цепей производных цел-
люлозы можно судить также по характеру кривых удлинения
и по величинам модулей эластичности, определяемым по этим
кривым. Модуль эластичности изменяется с удлинением, и мо-
мент, когда предел эластичности превзойден, соответствует
необратимой ориентации макромолекул в направлении растя-
гивающего усилия.
Целлюлоза и сопутствующие ей вещества
107
ЛИТЕРАТУРА
Hermans, Physics and'Chemistry of Cellulose fibres, Elsevier, 1949.
Meyer, van der Wy'k, J. chim. phys., 1951, 48, 33.
Legrand, C. R., 1948, 226, 1938.
Mathieu, La gelatinisation des nitrocelluloses, Hermann Co., Paris, 1936.
Hermans, Wei di nger,' J. Polymer Sci., 1951, 6, 533.
Frey Wy ssHng, Makromol. Chem., 1951, 6, 7.
Mukherjee, Sikorski, Woods, Nature, 1951, 167, 821.
С a 1 v e t, Hermans, J. Polymer Sci., 1951, 6, 33.
Segal, Nelson, Conrad, J. Phys. Coll. Chem., 1951’, 55, 324.
Chevenard, Champetier, Bull. Soc. chim., 1946, 13, 464.
Исследование структуры целлюлозы методом спектроскопии
в инфракрасной области
Наличие характерных полос разных функциональных групп
и интенсивность этих полос в спектрах поглощения, снятых
при длине волны более 1 р. для различных производных целлю-
лозы (рис. 19), подтверждают их химическое строение, уста-
новленное другими методами. Спектроскопия в инфракрасных
лучах показывает кроме того, что большая часть гидроксиль-
ных групп участвует в образовании водородных связей. Такое
взаимодействие между гидроксилами соседних цепей может
привести к возникновению межмолекулярных связей в кри-
сталлических областях полимера (рис. 20). Высокополимерные
цепи при этом образуют двухмерные сетки в плоскостях 001.
Весьма вероятно, однако, что эти явления происходят также
в аморфных областях целлюлозы вследствие способности цепей
изгибаться при возникновении межмолекулярных связей. Проч-
ность этих связей может сильно колебаться, если целлюлоза
гидратирована или мерсеризована.
Незамещенные гидроксилы производных целлюлозы также,
по-видимому, участвуют в образовании водородных мостикэв.
В производных целлюлозы интер- или интрамолекулярные свя-
зи могут возникать, кроме того, за счет электростатического
взаимодействия между простыми и сложными эфирными груп-
пами, обладающими наведенным или постоянным дипольным
моментом. Энергия подобных связей невелика, но пренебрегать
ими нельзя. Ее значения, вычисленные Марком на основании
изучения молекулярной когезии различных органических групп,
достигают 6000 кал для целлюлозы и 4800 кал для ацетата
целлюлозы (средние значения для элементарного звена в 5 А,
окруженного четырьмя прилегающими цепями).
ЛИТЕРАТУРА
Cubbing, Sinton, J. Opt. Soc., 1950, 40, 537.
В 1 о u t, В i r d, J. Opt. Soc., 1950, 40, 304.
Clement, Ind. Plast. Mod., 1951, 3, 5.
Длина волны,ft. '
Рис. 19. Инфракрасные спектры поглощения различных типов целлюлозы'-
/—нативная целлюлоза; 2—окисленная целлюлоза; 3—ацетат целлюлозы; 4—нит-
рат целлюлозы; 5—пальмитат целлюлозы; 6—этилцеллюлоза; 7—бензилцеллюло-
аа; 8—гликолевый эфир целлюлозы.
Химические свойства целлюлозы
109
ХИМИЧЕСКИЕ СВОЙСТВА ЦЕЛЛЮЛОЗЫ
Образование продуктов присоединения
Целлюлоза и ее производные способны к сольватации, ко-
торая в известных условиях может привести к образованию
истинных молекулярных соединений. Степень сольватации за-
висит от полярности целлюлозного полимера и от стерической
доступности полярных групп. При средних значениях степени
сольватации на нее оказывают влияние также относительная
величина молекул и их реакционная способность. Растворитель
в этих условиях вызывает только набухание внутри решетки, и,
следовательно, сольватация ограничивается аморфными уча-
стками полимера. Интермакромолекулярные связи могут умень-
шать степени сольватации, если силы притяжения полимер—
полимер превышают силы взаимодействия полимер—раствори-
тель. В образовании продуктов присоединения участвуют в
этом случае только «свободные» полярные группы макромоле-
кул. Однако, если даже все полярные группы полимера до-
ступны, пространственная структура реагирующих веществ мо-
жет привести к образованию нестехиометрических комплексов.
Во всех случаях наблюдается несколько областей устойчи-
вости сольватов. Из этого можно сделать вывод, что одно и то
же вещество образует несколько продуктов присоединения,
имеющих разные формулы. Энергии, необходимые для образо-
вания этих соединений, не превышают 10000 кал на одну связь.
по
Гл. III. Производные целлюлозы
Реакции присоединения частично объясняют поведение поли-
меров целлюлозы по отношению к химическим реактивам, уча-
ствующим в образовании кислотно-целлюлозных комплексов в
первой фазе этерификации (см. ниже), к веществам, вызываю-
щим набухание (например, при образовании комплексов типа
щелочной целлюлозы), к желатинирующим веществам, раство-
рителям и некоторым пластификаторам.
Сведения об образовании и свойствах различных комплек-
сов приведены в табл. 5. ,
ТАБЛИЦА 5
Продукты присоединения, образуемые целлюлозой и ее производными
Производные целлюлозы Среда Состав комплекса (в молях на I остаток Се) Способ идентификации комплекса
Нативная целлю- лоза (целлюло- за 1) Вода 1/2 Избирательное поглоще- ние в присутствии соли
Мерсеризованная целлюлоза (цел- люлоза П) Вода 4/5 То же
Регенерированная целлюлоза (цел- люлоза II) Вода 1 »
Целлюлоза I Азотная кислота 1/2, 1 Избирательное поглоще- ние из водного раство- ра
Соляная кислота 1/2 Рентгенограмма
Едкий натр 1/2, 2/3, 3/4, 1 Избирательное поглоще- ние из водного раство- ра
Куприэтилен- диамминовый раствор 1 Химическое определение поглощенной меди
Ацетат целлюлозы Вода до 3* Избирательное поглоще- ние в присутствии соли
Хлорированные углеводороды [** Избирательное поглоще- ние из раствора; спектры поглощения в инфра- красной области
* Зависит от степени ацетилирования.
** В молях на одну ацетатную группу.
Химические свойства целлюлозы
111
Продолжение табл. 5
Производные целлюлозы Среда Состав комплекса (в молях на 1 остаток С6) Способ идентификации комплекса
Вторичный ацетат целлюлозы Ацетон 3/2 Избирательное поглоще- ние в присутствии соли
Ацетон + мета- нол 3-6 Изменение диэлектричес- ких свойств
Триацетат и вторич- ный ацетат цел- люлозы Ацетон 2 Тепловой эффект при по- глощении в жидкой фазе
Нитрат целлюлозы Вода 1* Избирательное поглоще- ние в присутствии соли
Ацетон Измерение вязкости; ад- сорбция в паровой фазе; рентгенограмма; тепло- вой эффект при поглоще- нии в жидкой фазе; ди- намометрические свой- ства при поглоще- нии в паровой фазе; избирательное поглоще- ние в жидкой фазе
6 Тепловой эффект при по- глощении в жидкой фазе
Камфора Мочевина 1 Поглощение в жидкой фазе; скорость высыхания пла- стифицированной плен- ки; рентгенограмма; оп- тическая анизотропия при вытяжке
Трибутилфосфат 2/5 Избирательное поглоще- ние из раствора
Трикрезилфос- фат + спирт 1/4 То же
Метилцеллюлоза Вода Избирательное поглоще- ние в присутствии соли
* Н молях на одну незамещенную гидроксильную группу.
** В молях на одну нитратную группу.
***^В молях на одну метоксигруппу.
ЛИТЕРАТУРА
1. Ch ampe tier, Bull. Soc. chim. France, 1951, 3, 153.
2. Clement, Ann. chim., 1947, 12, 420.
3. Farrar Neale, Williamson, Nature, 1951, 167, 525.
4. Lauer, Roll. Z., 1951, 121, 33.
5. Moore, J. Polymer Sci., 1950, 5, 91.
112 . Гл. III. Производные целлюлозы
Реакции замещения
Спиртовые группы каждого глюкозного остатка целлюлозы
могут замещаться функциональными группами с образованием
сложных и простых эфиров и т. п. При рассмотрении этих ре-
акций должны учитываться следующие факторы:
1. Реакционная способность спиртовых групп, которая мо-
жет быть оценена по степени метилирования, достигаемой при
непосредственном воздействии на целлюлозу диазометана или
йодистого метила в присутствии этилата таллия, а также опре-
делена оптическими методами (рентгеноскопические исследова-
ния частично этерифицированных волокон).
2. Свойства целлюлозы как высокомолекулярного соедине-
ния, обусловливающие статистическое распределение замещен-
ных гидроксилов по длине целлюлозной цепи.
3. Зависимость реакционной способности спиртовых групп
от их характера и положения (первичная спиртовая группа в
(положении 6, вторичные спиртовые группы в положениях 2
и 3).
На кинетику реакций замещения сильно влияют сила и
взаимное расположение связей между гидроксилами. Замещаю-
щий реагент, как правило, быстрее взаимодействует с наибо-
лее доступными гидроксилами, расположенными главным обра-
зом в аморфной области (в случае целлюлозных волокон).
Исключение представляют реакции нитрования, которые про-
текают при непосредственном воздействии нитрующей смеси
на волокна и при которых, по-видимому, происходит одновре-
менное замещение всех гидроксилов в макромолекулах (пер-
мутоидная реакция). Замещение гидроксилов протекает рав-
номерно во времени, если в реакцию вступает целлюлозное
волокно, набухшее (например, в щелочном растворе) или
растворенное (например, в пиридине, четвертичном амине и др.),
а также, если в процессе реакции происходит растворение в
самой реакционной среде при достижении определенной
степени замещения (ацетилирование гомогенным способом
в присутствии уксусной кислоты, растворение щелочной
целлюлозы в смеси NaOH+CSz). Если структура волокна
сохраняется вследствие добавления разбавителя (ацетилиро-
вание в гетерогенной фазе), считают, что на поверхно-
сти волокна образуется непосредственно трехзамещенное произ-
водное.
Эффект предварительной обработки целлюлозных волокон
кислотой или щелочью перед ацетилированием и «созревания»
перед ксантогенированием объясняется, таким образом, увели-
чением числа свободных гидроксилов, реакционная способность
которых возрастает вследствие частичной деполимеризации
Химические свойства целлюлозы
ИЗ
целлюлозы и полиморфных превращений ее первичной кристал-
лической решетки.
При определении химического строения производных целлю-
лозы возникают затруднения, связанные с тем, что не все мак-
ромолекулы имеют одинаковую степень полимеризации, не все
глюкозные остатки одинаково замещены и, кроме того, цепь
может содержать разные замещающие группы (например, смесь
простых и сложных эфирных групп). Только трехзамещенные
производные отвечают более или менее определенной химиче-
ской формуле (рис. 21).
б
в
Рис. 21. Изменение строения целлюлозы при замещении (по Отту):
А—нативная целлюлозы; Б—частично замещенное производное целлюлозы (степень замеще-
ния 2,33/3); В—трехзамещенное производное.
Изменение рентгенограмм при реакциях замещения свиде-
тельствует о топохимическом характере большинства из этих
реакций.
Статистическое распределение замещенных групп под-
тверждается к тому же фракционированием или определе-
нием степени замещения, которая не совпадает со стехиомет-
рическими соотношениями. Шпарлин вычислил таким образом,
что для цепи со степенью полимеризации СП = 100 имеется 1089
возможностей расположения 1,5 (в среднем) простых или слож-
ных эфирных групп по отдельным звеньям р-глюкозы. Топохи-
8—156
114 Гл. HI. Производные целлюлозы
мический характер наиболее вероятен для смешанных произ-
водных целлюлозы, которые могут рассматриваться как сопо-
лимеры с той только разницей, что распределение боковых групп
более или менее беспорядочно.
Реакционная способность трех гидроксилов неодинакова.
Изучение этерификации целлюлозы в гомогенной среде с по-
мощью rt-толуолсульфокислоты показывает, что вторичные
спиртовые группы в положении 3 наименее реакционноспособ-
ны, в то время как реакционная способность первичной спир-
товой группы вдвое больше. Галоидирование реакционноспособ-
ных групп в положении 6 позволяет к тому же определить
количество первичных функциональных групп в частично за-
мещенном эфире (Пурвес, Мальм), в то время как расположе-
ние остальных заместителей может быть определено анализом
полностью деполимеризованного производного. Реакция этери-
фикации трифенилкарбинолом наиболее энергично протекает
в положении 6 (Сакурада). Напротив, скорость этерификации
окисью этилена больше для вторичных спиртовых групп (Коэн).
Возможно, наконец, что на скорость реакции влияет
соседство уже замещенной группы в том же элементарном
звене.
Сложные и простые эфиры целлюлозы. Описано большое
число сложных, простых и смешанных эфиров целлюлозы. Прин-
ципы их получения сопоставлены в табл. 6, в которой
различные замещенные производные классифицированы то
функциональным группам. Методы синтеза, свойства и приме-
нение главных замещенных производных целлюлозы явля-
ются предметом отдельной книги и здесь не рассматри-
ваются.
ЛИТЕРАТУРА
Champetier, Derives cellulosiques, Dunod, Paris, 1947.
Barsha, Malm, Fordyce, Cellulose et celluloses derivatives, E. Ott,
Inters. Publ., New York, 1943.
Malm, Mench, Kendall, Hiatt, Ind. Eng. Chem., 1951, 43.
685.
Kenyon, Ind. Eng. Chem., 1951, 43, 820.
D о r e e, The Methods of Cellulose Chemistry, Chapman a. Hall, London,
1947.
Heuser, The Chemistry of Cellulose, Wiley a. Sons, New York,
1946.
S t a n n e t. Acetate Plastics, Temple Press Ltd., London, 1950.
В u t t r e y, Cellulose Plastics, Cleaver Hume Press, London, 1947.
Fa b e 1, Nitrocellulose, Ferdinand Enke, Stuttgard, 1950.
Brown, Crawford, A Survey of nitrocellulose lacques, Reinhold Publ.
Corp., New York.
0© * ТАБЛИЦА 6 Принципы синтеза замещенных производных целлюлозы
Наименование Заместитель R в формуле Cell—О—С—R II О Степень замеще- ния (число заме- стителей в глю- козном остатке) Методы синтеза
I. Сложные эфиры органических кислот
Эфиры незамещенных жирных кислот: Формиат Н =#=1 1—3 • Действие муравьиной кислоты на гидратцеллю- лозу. Катализаторы: НС1 газообразный, H2SO4, Н3РО4, PCI5, монохлоруксусный ангидрид, перхлораты Повторная этерификация при —5°
Ацетат -сн3 7^3 1. Гомогенный способ: действие уксусного ан- гидрида на целлюлозу в кислой среде Катализаторы: H2SO4, Н3РО4, SO2C12, НС1О4, HNO3, ZnCl's и т. д. Растворитель: уксусная кислота, растворяющая ацетат целлюлозы по мере его образования 2. Гетерогенный способ: ацетилирование уксус- ным ангидридом в присутствии разбавителя (бензол и его гомологи, метиленхлорид, четы- реххлористый углерод, пиридин и т. д.) с целью сохранения волокнистой структуры цел- люлозы 3. Ацетилирование хлористым ацетилом в при- сутствии растворителей (этилацетат, хлоро- форм, нитробензол) Катализаторы: ацетаты натрия, циика, магния 4. Действие кетена СН2 = С = О на целлюлозу в присутствии разбавителя (эфир, бензол и его гомологи) и уксусной кислоты Катализаторы: ZnCl2, H2SC>4
СП
Химические свойства целлюлозы
Наименование Заместитель К в формуле Cell—О—С—R II О Степень замеще- ния (число заме- стителей в глю- козном остатке)
Ацетат -сна <3 <1
Пропионат -СН2СН3 ¥=2
Бутират — (СН2)2СН3 *3 ¥=3 .
От валерата до миристата Эфиры замещенных жирных кислот: —(СН2)ЛСН3 (п=3—12) 1-2 #=3
Хлорацетаты -СН„С1 -СНС12 -СС13 ¥=3
Продолжение табл. 6
Методы синтеза
5. Приготовление «вторичных ацетатов» путем
гидролиза в ацетилирующей ваине первичных
ацетатов, полученных гомогенным способом
6. Кислотный гидролиз вторичного ацетата в сре-
де органического растворителя
1. Действие пропионовой кислоты на гидратцел-
люлозу в присутствии разбавителя (толуол) и
катализатора (хлоруксусный ангидрид)
2. Действие пропионового или масляного ангид-
рида на целлюлозу в присутствии катализа-
тора (Нг5О4, ZnCls)
3. Действие хлорангидрида соответствующей кис-
лоты на целлюлозу в присутствии пиридина и
катализатора (хлоруксусная кислота или хлор-
уксусный ангидрид)
1. Действие кислоты на гидратцеллюлозу в при-
сутствии хлоруксусного ангидрида и сериой
кислоты
2. Действие хлорангидрида иа щелочную цел-
люлозу или гидратцеллюлозу в присутствии
пиридина и толуола
3. Повторная этерификация при избытке хлор-
ангидрида кислоты
1. Действие моно-, ди- или трихлоруксусного
ангидрида на целлюлозу, предварительно обра-
ботанную хлоруксусной кислотой или хлоро-
формом
Катализаторы: H2SO4, ZnCl2, SO2CI2
2. Действие PCls на триацетат целлюлозы
Гл. III. Производные целлюлозы
Гликолят —СНаОН
Лактат Гидракрилат -СН(ОН)СН3 -СНаСН2ОН
Ацетилацетат —СН2СОСН3
Карбонат (метилкарбо- нат) Эфиры ненасыщенных жирных кислот: -ОСНа
Акрилат Кротонат -СН=СН2 —СН=СН-СН3
Ундециленат -(СНа)8-СН=СН2
Олеат, элаидат, линолв'
ат, линоленат
Нафтенаты —СЯН2И_!
1. Действие гликолевого ангидрида на целлюло-
зу в присутствии уксусной кислоты и ката-
лизатора
2. Действие гликолевого ангидрида в присутствии
четвертичного органического основания
3. Гидролиз монохлоруксусных эфиров
Действие соответствующей кислоты на целлюлозу
в присутствии SO2CI2
Действие дикетена
СН2=С—СН2
I I
О-СО
на гидролизованный ацетат целлюлозы в при-
сутствии пиридина
*2
Действие метилхлорформиата на гидратцеллю-
лозу в щелочной среде
Действие соответствующей кислоты на гидрат-
целлюлозу в присутствии разбавителя. Катали-
заторы: хлоруксусный ангидрид, перхлорат
магния, .метилсерная кислота
Действие хлорангидрида ундециленовой кислоты
на гидратцеллюлозу в присутствии пиридина
и толуола
Действие хлорангидрида соответствующей кисло-
ты на целлюлозу в присутствии пиридина или
диметиланилина
2—3
Химические свойства целлюлозы
Действие хлорангидридов нафтеновых кислот на
целлюлозу в присутствии хлорбензола
Наименование Заместитель R в формуле Cell—0—С—R 11 О Степень замеще- ния (число заме- стителей в глю- козном остатке)
Эфиры ароматических кислот: Бензоат -свн6 У=2
Фенилацетат Эфиры многоосновных кислот: Алкилоксалаты (метил-, этил-, изоамил-, аллил-, циклогексил-, цетил- и т. д.) Сукцинат и глутарат о-Фталат —СН3С6Н6 -СООА1К -(СН2)„-СООН (п=от 2 до 3) тМ
-уУ СООН 3
Продомюение табл, 6
Об
Методы синтеза
Действие бензоилхлорида иа щелочную целлю-
лозу в присутствии бензола или пиридина
1. Действие хлорангидрида фенилуксусной кисло-
ты на целлюлозу в присутствии пиридина
2. Действие фенилуксусного ангидрида на цел-
люлозу в присутствии метилсерной кислоты
Хлорангидрид щавелевой кислоты в присутствии
пиридина не реагирует с целлюлозой. Этерифи-
кация идет только с алкилзамещенными окса-
лилхлоридами
Действие ангидрида соответствующей двухоснов-
ной кислоты на целлюлозу в присутствии пири-
дина
I. Действие фталоилхлорида на целлюлозу в при-
сутствии нитробензола и хинолина при 120°
2. Действие фталевого ангидрида на целлюлозу
или гидратцеллюлозу в среде пиридина
Гл. III. Производные целлюлозы
Алкил-о-фталаты
Эфиры сульфобензойных
кислот
Ацетоформиат
Ацетопропионат и ацето-
бутират
Ацетокротонат
Прямая этерификация кислого трифталата цел-
люлозы
Действие ангидрида о-сульфобензойной кислоты
на целлюлозу в присутствии пиридина
II. Сметанные эфиры органических кислот
(См. соответствующие
эфиры каждой кис-
лоты)
То же
(См. соответствующие
эфиры каждой кис-
лоты)
I. Действие смеси муравьиной и серной кислот
на целлюлозу с последующим ацетилирова-
нием в сернокислой среде
2. Растворение первичного ацетата целлюлозы в
концентрированной муравьиной кислоте с до-
бавлением или без добавления алкилформиата
1. Действие пропионовой или масляной кислот
на частично омыленный ацетат целлюлозы.
Катализатор: монохлоруксусный ангидрид
2. Действие пропионилхлорида или бутирилхло-
рида на ацетат целлюлозы в среде пиридина
3. Действие смеси уксусного и пропионового
(или масляного) ангидридов на целлюлозу в
присутствии хлоруксусной кислоты и перхло-
рата магния
4. Действие смеси уксусной и пропионовой (или
масляной) кислот и уксусного ангидрида на
целлюлозу
1. Действие кротоновой кислоты и хлорангид-
рида уксусной кислоты на ацетат целлюлозы
в присутствии перхлората магния
2. Действие ацетнлхлорнда н кротонилхлорида
на целлюлозу
Химические свойства целлюлозы
о
Продолжение табл. 6
Наименование Заместитель R в формуле Cell—О—С—R 6 Степень замеще- ния (число заме- стителей в глю- козном остатке) Методы синтеза
Аиетопропионатобутират Ацетофталат То же Действие смеси кислот на ацетат целлюлозы Действие фталевого ангидрида иа ацетат цел- люлозы в присутствии пиридина
III. Смешанные эфиры органических и минеральных кислот
Ацетонитрат
(См. соответствующие
эфиры каждой кис-
лоты)
Сульфоацетат То же
Нитросульфоацетат
Ацетосиликат
Нитросиликат
То же
1. Ацетилирование нитрата целлюлозы уксусной
кислотой, 'ее ангидридом или хлорангидридом
Катализатор: серная кислота
2. Нитрование ацетата целлюлозы
3. Одновременное нитрование и ацетилирование
целлюлозы смесью азотной кислоты с уксус-
ным ангидридом
1. Действие уксусного ангидрида на целлюлозу
в присутствии серной кислоты
2. Действие хлорсульфоновой кислоты на аце-
тат целлюлозы, растворенный в диоксане, в
присутствии пиридина
Гл. III. Производные целлюлозы
Действие смеси уксусной и серной
нитрат целлюлозы
Действие четыреххлористого кремния
но омыленный ацетат или нитрат
кислот на
иа частич-
целлюлозы
Наименование
Заместитель Ас в формуле
Cell—О—Ас
Степень замеще-
ния (число заме-
сителей в глю-
козном остатке)
Методы синтеза
Сульфат
Сульфит
Фосфат
IV. Эфиры минеральных кислот
-SO9H
у=з
—SO2N а
-РО3Н2
1. Действие концентрированной серной кислоты
на целлюлозу в присутствии уксусной кислоты
или без нее
2. Действие раствора серного ангидрида в пи-
ридине
3. Действие хлорсульфоновой кислоты на целлю-
лозу или щелочную целлюлозу в присутствии
пиридина
4. Действие газообразного серного ангидрида
на сухую целлюлозу в присутствии пиридина
или действие раствора серного ангидрида в се-
роуглероде
5. Действие хлористого сульфурила на щелоч-
ную целлюлозу в присутствии бензола
6. Действие пиросерной кислоты на целлюлозу в
присутствии пиридина
7. Действие смеси серной кислоты и сульфата
аммония на целлюлозу в присутствии спиртов
от С3 до С»
Действие жидкого сернистого ангидрида на ще-
лочную целлюлозу
1. Действие смеси фосфорной кислоты и хлор-
окиси фосфора на щелочную целлюлозу в при-
сутствии разбавителя (бензол-пиридин)
2. Действие смеси фосфорной кислоты с моче-
виной или гуанидином иа целлюлозу
3. Действие треххлористого фосфора и эфира
фосфорной кислоты (монофенилфосфата) на
производные целлюлозь;
Химические свойства целлюлозы
Наименование Заместитель Ле в формуле Cel 1-0-Ас Степень замеще- ния (число заме- стителей в глю- козном остатке)
Перхлорат Нитрит —С103 —NO
Нитрат -no2 1 1,4 2,4 от 0 до 2,8 /3 /3 =/=2,8 от 0 до 2,8
от 0 до 2,6 от 0 до 2,9 /3 от 2 до 2,5
Продолжение табл, 6
ND
ND
Методы синтеза
Прямое действие хлорной кислоты на целлюлозу
Действие окислов азота на гидратцеллюлозу
(механизм не установлен)
1. Действие на целлюлозу
75%-ной азотной кислоты
87%-ной азотной кислоты
безводной азотной кислоты
2. Действие смеси азотной и серной кислот на
целлюлозу
3. Дополнительное нитрование полного нитрата
целлюлозы 87%-ной азотной кислотой
4, Действие безводной азотной кислоты в при-
сутствии 10%-ного нитрата калия или мета-
фосфата натрия
5, Действие азотного ангидрида, газообразного
или растворенного в азотной кислоте
6. Действие двуокиси азота, растворенной в азот-
ной кислоте
7. Действие смеси двуокиси азота и серной
кислоты
8. Нитрование целлюлозы насыщенными парами
азотной кислоты
9. Нитрование смесью азотной и уксусной кислот
в присутствии или без уксусного ангидрида
10. Нитрование смесью азотной и фосфорной кис-
лот
11. Нитрование целлюлозы, первичные спиртовые
группы которой предварительно окислены
/п лс е л/
Гл. III. Производные целлюлозы
Кремний орган ические производные SiRg Действие алкилхлорсиланов на дегидратирован- ную целлюлозу
Заместитель R в формуле Степень замеще- ния (число заме- Методы синтеза
Наименование •Cell—О—R стнтелей в глю- козном остатке)
Простые эфиры целлюлозы
Метилцеллюлоза -СН3 0—3 1. Действие диметилсульфата при 20° на цел- люлозу, предварительно обработанную 15— 30%-ным едким натром. Триметилцеллюлоза получается путем многократной этерификации 2. Действие йодистого или хлористого метила на целлюлозу, предварительно обработанную 55— 75%-ным едким натром при 80° и 5 ат 3. Действие метилсульфата на целлюлозу в го- могенной фазе (раствор в триметилбензил- аммонии) 4. Действие метилгалогенида на целлюлозу, об- работанную едким натром в присутствии ам- миака 5. Действие йодистого метила на целлюлозу, об- работанную этилатом таллия (поверхностная этерификация) 6. Действие диазометана в растворе серного эфи- ра на нативную или мерсеризованную целлю- лозу (поверхностная этерификация)
Этилцеллюлоза —СП»СН3 э^2 3 1. Действие днэтилсульфата на целлюлозу, об- работанную 44%-ным едким натром при тем- пературе 80—200°. Среда: бензол+эмульгатор Получение триэтилцеллюлозы путем последова- тельной этерификации 2. Действие хлористого этила на целлюлозу, об- работанную едким натром
Химические свойства целлюлозы
КЗ
w
Наименование Заместитель R в формуле Cell—0—R Степень замеще- ния (число заме- стителей в глю- козном остатке)
Пропилцеллюлоза -(СН2)аСН3
Бутилцеллюлоза Амилцеллюлоза Гексилцеллюлоза —(СН2)„СН3 (п=от 3 до 6)
Бензилцеллюлоза -CH2CeHs М
¥=2
Монотрифенилметилцел- люлоза (тритилцеллю- лоза) —С(СвН6)8
Винилцеллюлоза -сн=сн2
Продолжение табл. 6
to
Методы синтеза
Действие хлористого или бромистого пропила на
щелочную целлюлозу при 100—130°
Действие соответствующего хлорпроизводного
на щелочную целлюлозу при 120—125° в ав-
токлаве
Действие соответствующего хлоропроизводного
целлюлозу при температуре 60—100°
1. В присутствии избытка порошкообразного ед-
кого иатра
2. Реакция проводится сначала в разбавленном
растворе щелочи (15—20%) с небольшим коли-
чеством хлористого бензила, затем в 50%-ном
растворе едкого натра в присутствии избытка
хлористого бензила
Товарный продукт, полученный после нескольких
операций этерификации
Действие хлористого трифенилметила при 100°
на гидратцеллюлозу
Действие ацетилена на щелочную целлюлозу в
присутствии различных катализаторов
Температура 120—150°; продолжительность 4—
48 час.
Гл. III. Производные целлюлозы
Наименование Заместитель R, в формуле Cell—О—R
Аллилцеллюлоза -сн2—сн=сн2
Оксиэтилцеллюлоза (глп- кольцеллюлоза) -СНаСН2ОН
Оксиалкилцеллюлоза -(СН2)„СН2ОН
Глицериновые эфиры цел- люлозы -СН2-СН(ОН)-СН2ОН
Ацетали целлюлозы (ме- тиленовые эфиры) -сн2-
Методы синтеза
Действие хлористого или бромистого аллила на
щелочную целлюлозу
1. Непосредственное действие при 25—30° газо-
образной окиси этилена на целлюлозу, обра-
ботанную 19—30%-ным едким натром с вызре-
ванием в течение 24 час.
2. Действие при низкой температуре монохлор-
гидрнна гликоля на целлюлозу, обработанную
43%-ным едким натром в присутствии бен-
зола
I. Действие окисей пропилена, бутилена амиле-
на на щелочную целлюлозу
2. Действие монохлоргидринов высших гликолей
(пропиленгликоля, бутиленгликоля) на щелоч-
ную целлюлозу
Действие монохлоргидрина, дихлоргидрина и
трихлоргидрина глицерина на щелочную цел-
люлозу
1. Непосредственное действие формальдегида на
целлюлозу в присутствии малого количества
кислоты (для ускорения)
2. Продолжительное действие при 100° дипропил-
и диизопропил-формалей в присутствии уксус-
ной кислоты и уксусного ангидрида
3. Действие на предыдущий продукт дихлордпме-
тилсульфата S02(OCH2C1)2 при 60°
Химические свойства целлюлозы
Наименование
Заместитель R в формуле
Cell—О—R
Этиленовый эфир целлю-
лозы
Карбоксиметил цел л юл оз а
(целлюлоз or ликолевая
кислота)
^>сн-сн<^
-сн2соон
Этилпропилцеллюлоза,
этилбензилцеллюлоза
и т. д.
(См. соответствующие
простые эфиры)
Метилэтилпропилцеллю-
лоза и т. д.
Нитраты диметил- и ди-
этилцеллюлозы, фосфат
этилцеллюлозы, ацета-
ты метил- и бензнлцел-
люлозы
Фталат этилцеллюлозы
То же
(См. соответствующие
простые и сложные
эфиры)
Продолжение табл. 6
Методы снжтеза
Непосредственное действие глиоксаля на цел-
люлозные волокна
1. Действие монохлоруксусной кислоты на ще-
лочную целлюлозу (40%-ный раствор едкого
натра)
2. Действие монохлорацетата натрия на щелоч-
ную целлюлозу (20%-ный едкий натр)
1. Действие смеси соответствующих алкилхлори-
дов на щелочную целлюлозу (более низкомо-
лекулярный алкил вступает в реакцию в боль-
шем количестве)
2. Этерификация в две стадии
Одновременное или последовательное действие
алкилхлоридов на щелочную целлюлозу
Этерификация обычными методами
Гл. III. Производные целлюлозы
Действие фталевого ангидрида на этилцеллюло-
зу в присутствии пиридина или бензола
Заместитель В в формуле
Cell—В
Наименование
Методы синтеза
Галоидпроизводные
Ам инопроизводные
Амиды
Уретаны
Гетеропроизводныс
—CH2J
—OCORC1
-nh2
—ch2nh2
—och2ch2nh2
-0-C2H4-N(C2H4OH)2
-O-C-NHCgH4N (CH3)2
—0—CH2CH2NH—C—NHR
0
—О—C—RNHCOCHg
II
о
-O-C-NHR
о
—О—C—NHRNH—-С—O--
целлюлозы
Действие NaJ или хлористого пиридиния на
n-толуолсульфонат или нитрат целлюлозы
Действие галоидокислот более чем с 5 атомами
углерода на целлюлозу или пятихлористого
фосфора на ацетат целлюлозы
Действие амида натрия на нитрат целлюлозы,
аммиака на w-толуолсульфонат или галоидо-
производные целлюлозы
Присоединение к целлюлозе этиленимина
Действие диэтаноламина на п-толуолсульфоно-
вый эфир оксиэтилцеллюлозы
Действие аминоизоцианатов на целлюлозу
Присоединение продуктов конденсации этилен-
имина с изоцианатом
Действие N-ацетил-а-аминомасляной кислоты на
ацетат целлюлозы в присутствии хлоруксус-
ного ангидрида
Присоединение изоцианата или диизоцианата к
целлюлозе
Химические свойства целлюлозы
Наименованиа Заместитель R в формуле Cell- R
Уретаны -0 -С—CHuS—С—NHR i А
Тиоуретаны -C-C-NHR
Нитрилы —OCH2CH2CN
Изотиоцианаты —СН2—N=C=S
Эфиры, содержащие серу и фосфор —О—С—CH2S2O3Na А _о—С—CH2SCH2—С— II II 0 0 /ОС2нв -О-С-СН2-Р< 1) 0 0R >ОС2НВ -0-Р< II \ОС2Н6 - 0
Продолжение табл. 6
Методы синтеза
Присоединение эфира или соли тиокарбаминоаой
кислоты к хлоруксусному эфиру целлюлозы
Присоединение тиоизоцианата к целлюлозе или
действие амина на целлюлозоксантогенуксусную
кислоту
Действие акрилонитрила на щелочную целлюлозу
Действие роданидов щелочных металлов на
n-толуолсульфонат целлюлозы
Действие тиосульфата на хлорированный эфир
целлюлозы
Окисление предыдущего продукта
Действие триэтилфосфата на хлорацетат цел'
люлозы
Гл. III. Производные целлюлозы
Действие днэтилхлорфосфата на эфир целлюлозы
Химические свойства целлюлозы
129
Ксантогенаты целлюлозы. Щелочная целлюлоза (иногда на-
зываемая алкалицеллюлозой) при действии сероуглерода об-
разует ксантогенат, или ксантат (Кросс и Бивен), по реакции:
Cell—ОН-р NaOH+ CS2 Cell—ОС—SNa + Н2О
1
Полученный ксантогенат целлюлозы может быть дисперги-
рован в щелочной среде с образованием раствора (вискозы).
Триксантогенаты могут быть получены только в присутствии
четвертичных аммониевых оснований или из целлюлозы, об-
работанной предварительно металлическим натрием в среде
жидкого аммиака.
Приготовление ксантогената целлюлозы включает следую-
щие стадии:
1. Получение щелочной целлюлозы путем обработки целлю-
лозы 18%-ным раствором едкого натра, не содержащим следов
металлов, иногда в присутствии смачивающих веществ, в те-
чение 1—2 час. при 30°. После отжима жидкости до примерно
утроенного по сравнению с первоначальным веса продукта
щелочную целлюлозу измельчают и затем подвергают «созре-
ванию» в течение 3 суток при 25—28°. Во время «созревания»
целлюлоза поглощает кислород и деполимеризуется. Продолжи-
тельность и температура созревания сильно влияют на вязкость
растворов ксантогената целлюлозы.
2. Ксантогенирование путем обработки сероуглеродом во
вращающихся барабанах, в которых поддерживается темпера-
тура 22—27°. После двухчасового воздействия сероуглерода
ксантогенат растворяют при перемешивании в едком натре при
низкой температуре с тем, чтобы получить раствор вискозы,
содержащий 6,4% NaOH и 7,5% целлюлозы.
3. «Созревание». Раствор фильтруют и выдерживают при
температуре около 15° в течение 3—4 суток, чтобы сделать его
пригодным для прядения. «Созревание» контролируют по по-
нижению вязкости, пробами на коагуляцию (солевая точка),
осаждением или по иодному числу.
При обработке сероуглеродом и при созревании происходит
сложный комплекс накладывающихся друг на друга реакций,
приводящих к установлению равновесия собственно процесса
ксантогенирования (Геррент и Инофф). После созревания со-
держание связанной серы в продукте соответствует довольно
сильно омыленному ксантогенату состава:
/(ОН),
ХОС—SNa
9—156
130 Гл. HI. Производные целлюлозы
Созревание сопровождается глубокими изменениями колло-
идной структуры раствора, вызванными изменением агрегации
диспергированных макромолекул целлюлозы или продолжаю-
щимся процессом ксантогенирования в кристаллических обла-
стях щелочной целлюлозы. Раствор вискозы, окрашенный в
красно-оранжевый цвет, становится более вязким и способен
коагулировать при действии раствора соли или кислоты. Такая
вискоза пригодна для прядения, т. е. для регенерации целлю-
лозы путем коагуляции в форме волокна или пленок.
Созревание может быть замедлено действием сульфита или
цианистого натрия, алкилксантогенатов, фенолов, галловой кис-
лоты и др. или ускорено путем повышения температуры, до-
бавления электролитов, перекиси водорода, глицерина и поли-
сульфидов.
Производные ксантогенатов. Под действием этерифицирую-
щих агентов тиокарбоновые группы могут замещаться по ре-
акции:
Cell—О—С—SNa + RX --*• Cell—О—С—SR + NaX
И II
s s
где RX — иодистый метил, диэтилсульфат, дихлоргидрин глико-
ля, монохлоруксусная кислота.
Натриевая соль ксантогенуксусной кислоты реагирует с ами-
ном, образуя тиоуретан типа
Cell—О—C-NHR
II
S
который можно получать прямым действием ксантогената на
амин.
Диазометан CH2N2, наоборот, превращает тиокарбоновую
группу в метоксил. Реакция не идет при непосредственном воз-
действии на целлюлозу.
Производные диазония также дают продукты замещения.
ЛИТЕРАТУРА
Kline, Cellulose and Cellulose derivatives, E. Ott., Ed. Intercs. Publ., New
York, 1943.
Herren t, Jnoff, J. Polymer Sci., 1950, 5, 727.
Sanford, A. Mossjr., Colloid Chemistry, Alexander, v. VI, Reinhold
Publ., New York, 1946.
Inskeep van Horn, Ind. Eng. Chem., 1952, 44, 2511.
Реакции окисления
Эти реакции обычно рассматривают при изучении реакций
разложения. Однако в некоторых случаях окисление затраги-
вает только глюкозидные гидроксилы, а высокомолекулярная
цепь остается неизменной.
Химические свойства целлюлозы
131
Рис. 22. Скорость окисления ацетата
целлюлозы и этилцеллюлозы [В и г г е у,
Ind. Eng. Chem., 41, 2160(1946)];
1—ацетат целлюлозы (60% ацетатных групп);
2—этилцеллюлоза (44% этоксильных групп);
3—этилцеллюлоза (49,2% этоксильных групп).
Прямое окисление воздухом. Чистая сухая целлюлоза от-
носительно устойчива по отношению к действию повышенных
температур в присутствии воздуха. После нагревания в тече-
ние нескольких часов при 80—100° наблюдается лишь незна-
чительная деполимеризация и слабое окисление. Разложение
проявляется сильнее при температуре около 150° и протекает
тем энергичнее, чем ниже степень полимеризации. Штамм и
Ганзен считали возможным образование эфирных мостиков меж-
ду глюкозидными гидроксилами соседних глюкозных остатков.
Перегонка в вакууме ведет
к образованию р-глюкозы и $
различных газообразных про-
дуктов (СО, Н2О, СН4, С2Н4 и
т. Д.).
При соприкосновении с рас-
твором щелочи целлюлоза при
нормальной температуре по-
глощает кислород. При этом
одновременно происходят реак-
ции окисления и гидролиза,
приводящие к сильному пони-
жению степени полимеризации.
Реакция каталитически уско-
ряется сульфидами щелочных
металлов.
Этилцеллюлоза подвергает-
ся окислению при 110°. При
этом уменьшается содержание
этоксильных групп, происходит образование карбоксильных
групп и значительно понижается степень полимеризации.
Ацетат целлюлозы при нагревании окисляется -с выделением
СО, Н2О и различных кислых продуктов. Поглощение кислоро-
да представляет собой автокаталитическую реакцию, которой
предшествует индукционный период. Устойчивость ацетата цел-
люлозы по отношению к нагреванию сильно понижается в при-
сутствии следов свободной уксусной и особенно серной кислот,
остающихся, по-видимому, после реакции ацетилирования. По-
нижение устойчивости вызывают также некоторые продукты
разложения целлюлозы. В известной степени окисление может
быть замедлено путем введения антиоксидантов или нейтрали-
зующих веществ.
Скорости окисления ацетата целлюлозы и этилцеллюлозы со-
поставлены на рис. 22.
Стабильность нитрата целлюлозы в значительной мере за-
висит от содержания влаги и следов серной кислоты, свободной
или связанной.
9*
132
Гл. III. Производные целлюлозы
Очень сложный процесс окислительной деструкции нитрата
целлюлозы может быть разделен на две стадии:
1. Период индукции с малой скоростью разложения, резко
увеличивающейся при повышении температуры.
2. Разложение со скоростью взрыва, связанное с нестабиль-
ностью эфирных групп —ONO2. Эта стадия разложения пред-
ставляет собой автокаталитический процесс, вызываемый дей-
ствием на эфирные группы окислов азота и азотной кислоты,
образующихся в первой стадии окисления. Скорость разложе-
ния сильно уменьшается, если эти. вещества по мере их обра-
зования удалять или нейтрализовать путем добавления соот-
ветствующих стабилизаторов.
Конечными продуктами разложения нитрогрупп и пирано-
вых циклов являются NO2, N2, СО2, СО, Н2 и СН4.
Окисление с помощью окисляющих агентов. Гидроксильные
группы в глюкопирановьих кольцах под действием окислителей
могут превращаться в альдегидные или карбоксильные группы.
Окислители могут таким же образом действовать на кислород-
ные мостики, изменяя структуру концевых групп молекулы.
Полученные таким путем окисленные целлюлозы могут быть
разделены на две категории:
1. Окисленные целлюлозы, содержащие восстанавливающие
группы. Они характеризуются высоким медным числом, неста-
бильностью к щелочам, восстанавливающей способностью по
отношению к фенил гидр азину.
2. Окисленные целлюлозы, содержащие кислотные группы.
Для них характерны низкое медное число, сильное поглощение
метиленовой сини, довольно слабая чувствительность к щело-
чам.
Химическое строение и степень полимеризации окисленной
целлюлозы зависят в основном от природы окислителя и усло-
вий окисления.
Иодная кислота вызывает раскрытие ядра с превращением
двух спиртовых групп в альдегидные (Джаксон—Резерфорд).
Конечные продукты гидролиза окисленной целлюлозы — гли-
оксаль и d-эритроза.
Действие хромовой кислоты ограничивается, по-видимому,
только аморфными участками целлюлозы (Давидсон) и вы-
ражается в превращении глюкозидных мостиков в эфирные
связи, легко омыляемые в щелочной среде (Штаудингер). Хро-
мовая кислота, с другой стороны, может окислять спиртовую
группу в положении 6 с образованием альдегида, способного
превращаться в кислоту при последующей обработке гипохло-
ритом натрия (Пинт и Роша).
Двуокись азота производит более мягкое окисление, при
котором сохраняется волокнистая структура целлюлозы. Пер-
Химические свойства целлюлозы 133
вичные спиртовые группы превращаются в карбоксильные без
существенного гидролитического расщепления (Кеньон с сотр.)
и без изменения исходной кристаллической решетки (Невелл).
Содержание образовавшихся карбоксильных групп зависит от
условий окисления, но может достигнуть теоретического зна-
чения (25,56% СООН).
Озон в присутствии влаги окисляет, по-видимому, первичные
спиртовые группы до альдегидных, а вторичные — до кетон-
ных групп (Доре).
Отбеливающие агентьи (гипохлориты) вызывают одновремен-
но образование карбоксильных групп и деполимеризацию, ко-
торая достигает максимальной величины при рН~6 (Вебер и
Гуземан). Окисление ведут в присутствии следов тяжелых ме-
таллов (Си, Мп, Fe), действующих каталитически. Перекись
водорода (60%-ная) растворяет целлюлозу. Для эфиров целлю-
лозы, содержащих группы СООН, обнаруживается повышение
вязкости растворов после обработки поливалентными солями
(например, солями Са + +). Это явление вызывается, по-видимо-
му, агрегацией за счет интермолекулярных связей; аналогич-
но ведут себя эфиры целлюлозы, содержащие свободные суль-
фогруппы (Кемпбелл, Мальм). >
ЛИТЕРАТУРА
Heuser, Cellulose Chem., Wiley, New York, p. 477.
Davidson, J. Text. Inst., 1951, 42, 141.
Pinte, Rochas, Bull. Inst. Text, de Fr., 1950, 21, 9.
G 1 e g g, Text. Res. J„ 1951, 21, 143.
Kenyon и сотр., Ind. Eng. Chem., 1949, 41, 2.
Neve 11, J. Text. Inst., 1951, 42, 130.
Kleiner t, Wincor, Svensk Papperstidn, 1950, 53, 638.
R о we n, P 1 у 1 e r, J. Res. Nat. Bur. St., 1951, 46, 288.
Campbell, Johnson, J. Polymer Sci., 1948, 3, 735.
Malm, Tanghe, Smith, Ind. Eng. Chem., 1950, 42, 730.
Реакции деструкции
Гидролиз под действием кислот. Кислородные мостики в по-
ложениях 1,4' при действии кислот разрываются, в результате
чего образуются концевые полуацетальные группы. Для депо-
лимеризованной таким образом целлюлозы (гидрат целлюлозы)
характерны низкая вязкость, ухудшение механических свойств,
повышенная восстановительная способность, растворимость в
щелочных растворах, преимущественно при низкой температуре,
и сильное сродство к основным красителям.
Глюкозидные связи, расположенные у концов цепей, по-ви-
димому, наиболее чувствительны к гидролизу, но некоторые ав-
торы полагают, что разрыв цепей облегчается благодаря регу-
лярному распределению наиболее реакционноспособных групп.
134 Гл. III. Производные целлюлозы
Скорость деполимеризации производных целлюлозы при том
же pH меньше, чем у самой целлюлозы, в связи с меньшей
доступностью эфирных кислородных мостиков.
Реакции кислотного гидролиза играют значительную роль
в так называемых процессах предварительной обработки (ла-
пример, при ацетилировании) и в реакциях собственно замещен
ния. Протеканием реакций гидролиза объясняется химическая
неустойчивость сложных эфиров целлюлозы по отношению к
остаточным кислотам (серной, азотной) или к кислотам, обра-
зующимся вследствие омыления самих эфирных групп. Стаби-
лизация .нитратов и ацетатов целлюлозы состоит поэтому в пол-
ном удалении этих примесей, во введении в полимеры солей,
способных нейтрализовать их по мере образования, или в ней-
трализации кислотных заместителей (—OSO3H) с превраще-
нием их в стабильные негидролизующиеся продукты.
Действие ультрафиолетовых лучей. Деполимеризация про-
изводных целлюлозы под влиянием света имеет большое зна-
чение в текстильной и швейной промышленности.
Быстрая деструкция производных целлюлозы, происходящая
под действием ультрафиолетовых лучей, объясняется главным
образом окислением, так как сами ультрафиолетовые лучи в
отсутствие кислорода оказывают лишь относительно слабое дей-
ствие. Под действием дальнего ультрафиолета происходит об-
разование карбоксильных групп (с последующим декарбокси-
лированием) и альдегидных и кетонных групп, распадающихся
с образованием СО, СО2 и Н?О, а также понижение степе-
ни полимеризации. В ближнем ультрафиолете разложение про-
исходит лишь при условии фотосенсибилизации различными
красителями, пигментами, наполнителями и т. д. В присутствии
влаги эти сенсибилизаторы могут образовывать перекись во-
дорода, каталитически ускоряющую окисление. Другие веще-
ства (антрахиноновые и азокрасители) вызывают противопо-
ложный эффект.
В нитрате целлюлозы одновременно происходит разложение
нитрогрупп и деполимеризация, усиливающаяся в присутствии
некоторых пластификаторов. Напротив, нитрат целлюлозы отно-
сительно стабилизуется в присутствии веществ основного ха-
рактера (двузамещенные мочевины, амины), которые могут
•поглощать выделяющиеся окислы азота. Ацетат целлюлозы,
ацетобутират и этилцеллюлоза также способны деполимеризо-
ваться, в то время как бензилцеллюлоза и оксиэтилцеллюлоза
менее подвержены деструкции.
В настоящее время в качестве замедлителей деструкции
целлюлозных пластиков под действием ультрафиолетовых лу-
чей предложено много различных веществ: фенилсалицилат,
резорциндисалицилат, п-метоксифенил-п-анизол и др.
Физико-химические свойства целлюлозы
135
Действие микроорганизмов. Некоторые микроорганизмы пол-
ностью разлагают целлюлозу до СОг и НгО. Промежуточные
стадии процесса разложения представляют собой сложные про-
цессы, включающие образование алифатических кислот или
спиртов и газообразных углеводородов. Деструкция вызывает-
ся аэробными и анаэробными, мезофильными или термофиль-
ными бактериями, а также грибками и некоторыми простейши-
ми одноклеточными организмами.
Механические воздействия. Продолжительное измельчение
целлюлозных волокон сопровождается понижением степени по-
лимеризации, которое приписывают разрыву ковалентных свя-
зей полимерных цепей (Гесс). Эффект, вызываемый действием
на целлюлозное волокно ультразвука, зависит от частоты и
состоит главным образом в дезагрегации фибрилл и измене-
нии кристаллической структуры. Химические процессы! типа
гидролиза не наблюдались.
ЛИТЕРАТУРА
Г идролиз:
Mark, Harris, Cellulose Chemistry, E. Ott. Ed. Intersc. Publ., 1943,
New York.
Действие ультрафиолетового света:
E g e r t о n, Photochemistry in relation to textiles. Symposium, Harrogat,
1949.
Heuser, Chamberlin, J. Am. Chem. Soc., 1946, 68, 79.
Evans, M с В u r n e y, Ind. Eng. Chem., 1949, 41, 1236.
T i c h e n о r, J. Polymer Sci., 1946, 9, 217.
Clark, Techn. Ass. of Lithog. Ind., 1950.
Winogradoff, Nature, 1950, 165, 72.
Действие микроорганизмов:
Hajny, Gardner, Ritter, Ind. Eng. Chem., 1951, 43, 1384.
Механическое воздействие
и действие ультразвука:
Fromm и сотр., Koll. Z., 1942, 98, 148.
Simpson, Mason, Pulp. Paper Mag. Canada, 1950, 51.
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА ЦЕЛЛЮЛОЗЫ
Набухание и растворение
Переход высокомолекулярного вещества из твердого состоя-
ния в растворенное состоит в постепенном превращении из ас-
социированной формы (например, волокна, пленки, осадки) в
неассоциированную, при которой макромолекулы стремятся к
максимальной дезагрегации, соответствующей в пределе мо-
лекулярному раствору.
136
Гл. III. Производные целлюлозы
Процесс может проходить через промежуточные стадии ог-
раниченного набухания, при котором кристаллическая струк-
тура полностью не нарушается, затем желатинирования и,
наконец, разрушения коллоидной системы, когда подвижность
целлюлозных цепей приближается к их подвижности в истин-
ных растворах. Постепенные переходы из стадии в стадию мож-
но наблюдать при обработке полимера растворителем с воз-
растающей растворяющей способностью. Обратный переход к
ассоциированной форме происходит при добавлении к раство-
ру нерастворителя; тогда вязкость возрастает вплоть до осаж-
дения, а в общем случае — до образования пленки.
Энергия ассоциации типа полимер—полимер максимальна
у целлюлозы (водородные мостики между гидроксилами). Она
уменьшается у простых и сложных эфиров по мере замещения
гидроксилов и снова увеличивается, когда все гидроксилы за-
мещаются полярными группами.
Диспергирование целлюлозы в растворителе, по-видимому,
обусловливается в значительной мере сродством полимера к
растворителю, которое можно заранее предугадать, исходя из
положения, что в результате сольватации полимерные цепи
становятся неполярными. Тот же механизм имеет химическая
пластификация; после удаления растворителя некоторые пла-
стификаторы, образующие сольватьи, понижают полярность цел-
люлозных цепей, придавая им таким образом большую под-
вижность.
Соотношением сил ассоциации полимер—полимер и поли-
мер—растворитель частично объясняется изменение раствори-
мости производных целлюлозы в зависимости от степени заме-
щения. Целлюлоза нерастворима в воде вследствие сильного
межмолекулярного взаимодействия гидроксильных .групп, ко-
торое ослабляется только при сольватации в присутствии силь-
но ионизированных веществ. Гибкостью макромолекул частично
объясняются отличия растворимости целлюлозы, цепи которой
относительно мало подвижны', и полимеров типа поливинило-
вого спирта.
При частичной или полной этерификации нарушаются свя-
зи ОН ОН и появляется растворимость в веществах, имею-
щих сродство к свободным гидроксилам. Например, одноза-
мещенные ацетаты и простые эфиры целлюлозы (этилцеллю-
лоза и метилцеллюлоза) растворяются в воде и щелочах. При
замене первичных спиртовых групп целлюлозы (СН2ОН) океи-
этильными группами (СН2СН2ОН) гидроксилы становятся бо-
лее доступными и появляется растворимость в водных раство-
рах. Если замещены все гидроксилы, растворимость образо-
вавшегося трехзамещенного производного зависит от силы вза-
имодействия типа полимер—полимер и определяется полярным
Физико-химические свойства целлюлозы 137
характером новых заместителей. Это правило хорошо оправ-
дывается, например, для этилцеллюлозы, которая в зависимо-
сти от степени этерификации растворима либо в гидрофильных,
либо в неполярных растворителях. Из трехзамещенных эфиров
алифатических кислот в неполярных растворителях растворимы
только высшие эфиры (валерат, стеарат). Растворимость вто-
ричных ацетатов и нитрата целлюлозы в ацетоне может быть
объяснена полярным взаимодействием типа кетон—эфир или
водородными связями между свободными гидроксилами и мо-
лекулами кетона.
По-видимому, можно считать доказанным, что повышен-
ная способность хлорированных углеводородов к желатиниро-
ванию ацетата целлюлозы обусловлена сольватацией ацетиль-
ных групп. Поэтому четыреххлористый углерод желатиниро-
вания не вызывает.
Присутствие различных полярных групп в одной и той же
цепи позволяет объяснить механизм растворения в бинарных
смесях растворителей двойной сольватацией производного цел-
люлозы. Так, важную роль при растворении целлюлозных про-
изводных играет спирт. Присоединение его к свободным гидро-
ксилам способствует проникновению внутрь решетки «непря-
мого» растворителя или пластификатора, размеры молекул ко-
торого столь велики, что он сам без второго компонента не
может быть растворителем.
Производные целлюлозы, содержащие много групп —СООН
или —SO3H (оксицеллюлоза, гликолят, фталат, сульфат цел-
люлозы и др.), растворимы в щелочных растворах; напротив,
нейтрализация этих кислых групп ионами поливалентных ме-
таллов приводит к нерастворимости, по-видимому, вследствие
образования солевых межмолекулярных мостиков. Полная не-
растворимость может быть вызвана также наличием химиче-
ских мостиков, соединяющих целлюлозные цепи; в этом случае
полимер имеет трехмерную структуру и способен лишь к не-
значительному набуханию.
Явление внедрения. Пластификация. Явление внедрения со-
стоит в проникании слабо полярной молекулы между цепями
целлюлозы, предварительно диспергированной в растворителе
и вновь агрегированной после его испарения. Это явление очень
четко выражено при образовании покрытия из раствора, содер-
жащего неполярный разбавитель. Оно происходит, по-видимо-
му, также при пластификации целлюлозных производных не-
желатинирующими пластификаторами (рис. 23).
Пластификаторы играют важную роль в отношении меха-
нических свойств и гигроскопичности, так как они заполняют
пустоты между цепями, особенно значительные в аморфной
области.
1,38
Гл. III. Производные целлюлозы
А
б
Захваченные молекулы
Q — Моле нулы растворителя
Рис. 23. Пластификация целлюлозы:
А—ра.'твор в смешанном растворителе; Б—макромолекулярная решетка после испарения раство-
рителя.
Физико-химические свойства целлюлозы
139
Пластификаторы целлюлозы и ее производных
ТАБЛИЦА 7
Производные целлюлозы Область применения Пластификаторы
Целлюлоза Пленки Глицерин, пентаэритрит
Ацетат Пластические массы Диметил- и диэтилфталаты, метилол-
Ацетобутират Пленки Покрытия Пластические массы фталат, триацетин—три-(хлорэтил)- фосфат, n-толуолсульфамид, три- фен илфосфат Трифенчл- и трикрезилфталаты Алкилфосфаты и алкилфталаты, бутил- или амилтартрат Дибутилсебацинат + диметил- или
Нитрат Покрытия Пластические массы Пленки Покрытия диэтилфталаты Алкилфталаты Камфора Камфора г трифенилфосфат Дибутилфталат 4- трикрезилфосфат, касторовое масло + метилциклогек- силадипат + алкилрицинолеаты
В табл. 7 «риведены важнейшие пластификаторы целлюлозы
и ее производных,
Ниже приведены наиболее распространенные растворители
целлюлозы:
Концентрированные растворы галогенидов и роданидов (тио-
цианатов) щелочных или щелочноземельных металлов.
Растворы галогенидов тяжелых металлов, например хлористо-
го цинка*.
Раствор куприаммингидрата в аммиаке [Си(МНз)4](ОН)2
(медно-аммиачный раствор, или реактив Швейцера**) и рас-
твор куприэтилендиаммингидрата***.
Четвертичные аммониевые основания: гидроокиси тетраме-
тил- или диметилдибензиламмония (тритон F) .
Растворы минеральных солей в органических растворителях.
Для производных целлюлозы имеется значительно более
широкая гамма растворителей, особенно среди органических
веществ. Растворимость производных целлюлозы зависит от
характера замещающих групп, степени замещения, темпера-
туры и степени полимеризации.
* Желатинирование целлюлозного волокна в концентрированном 75%-ном
растворе хлористого цинка положено в основу производства пластических масс
типа фибры.
** Растворимость целлюлозы в реактиве Швейцера использовалась в процес-
се производства медноаммиачного шелка (искусственное волокно Бемберга и
Глаицштофа).
*** Куприэтилендиаммингидрат используется главным образом при определе-
нии вязкости целлюлозы.
140
Гл. III. Производные целлюлозы
В табл. 8 и 9 приведены важнейшие растворители простых
и сложных эфиров целлюлозы.
ТАБЛИЦА 8
Растворимость эфиров целлюлозы в водных растворителях
(Р—растворим)
Холодная вода Горячая вода Едкий натр Вода + спирт
Метилцеллюлоза, степень замещения 0—1 1—2,5 2—2,7 р р р
Этилцеллюлоза, степень замещения 0,5-0,7 0,8—1,3 1,4—2 р р р р
Пропилцеллюлоза, сте- пень замещения 0,5 0,5-2 р р р
Гликолят целлюлозы (сте- пень замещения 0,3—0,8) кислая форма . . . натриевая соль . . . р р р р р р
Оксиэтилцеллюлоза, сте- пень замещения 0,5—0,9 .... 0,9—2,5 р р р
Сульфат целлюлозы одно- замещенный (натриевая соль) р р р
Ацетат целлюлозы одно- замещенный р р р р
Совместимость
За исключением нитрата целлюлозы, некоторых ацетобутира-
тов и этилцеллюлозы, простые и сложные эфиры совместимы с
очень ограниченным числом других высокомолекулярных сое-
динений, как природных, так и синтетических. Обычно они
обладают лишь частичной совместимостью или совместимы в
очень цазбавленных растворах.
Растворимость эфиров целлюлозы в органических растворителях
ТАБЛИЦА Я
(Р—растворим)
1Л « о 5 диацетоновый диоксан
Производные ..5, §• спирт Н2С—сн2 °\ /° н2с-сн2
целлюлозы метанол СНзОН этанол С2Н5ОН бутанол С3Н7ОН ацетон (СНз)зСО “О й su этилацета! СН3СООС< 40 20 so метиленхл СН2С12 (СН3)2ССН2СОСН3 ОН бензол СвНв
Ацетат
53—55% ацетильных групп — — — р р р р — р р —
55-57% » » - - — — р р — р — — р —
57—59% » » — —• — р р — — — — р —
59-61% > » — — — — р —. —— —- —— р —
61—62,5% > » — — — — — — — р — — —
Пропионат — — ... — — — р р — р —
Бутират — — — р р р р — р —
Ацетопропионат Ацетобутират — — р р р р р
30% ацетильных групп-1- 17,5% бутирильных 6% ацетильных групп-)- — — р р р — р р р —
48% бутирильных — — — р р р р р р р р
Нитрат р
10,5-11,5% N р р — — р р — р р —
11,5-12,5% N —. — — р р р р — р — р
12,5—13% N — — — р р р р — р — —
Триметилцеллюлоза Этилцеллюлоза — — — — р р — р — — р
44—47% этоксильных групп р р — — — — — — — — —
47—48% » » р р р р р р — р р р р
48-50% » » — р р — р р — р р — р р
Бензилцеллюлоза — — — р р р р — р .
Физико-химические свойства целлюлозы
142
Гл. III. Производные целлюлозы
Ниже приведены данные о совместимости некоторых важ-
нейших производных целлюлозы с другими высоко1полимерными
веществами:
Ацетат целлюлозы
Ацетобутираты целлюлозы
с высоким содержанием
бутирильных групп
с высоким содержанием
ацетильных групп
Этилцеллюлоза
Нитрат целлюлозы
Метилцеллюлоза
Оксизтилцеллюлоза
Полиэфирные (алкидные) смолы чистые или
модифицированные; акриловые полимеры
Многочисленные синтетические и природные
смолы
Виниловые и акриловые полимеры; феноло-
формальдегидные, ку мароно- инденовые
смолы
Природные смолы (канифоль и т. д.); фено-
ло-формальдегидные и полиэфирные смолы;
нитрат целлюлозы
Природные смолы; виниловые полимеры
(поливинилхлорид и поливинилиденхлорид,
поливиниловый спирт, поливинилацетат,
полистирол); феноло-формальдегидные и
мочевино-формальдегидные смолы; поли-
эфиры; этилцеллюлоза
Оксизтилцеллюлоза
Желатин, метилцеллюлоза, казеин, вискоза
ЛИТЕРАТУРА
С о 1 b or n е, Elastomers and Plastomers, v. I Houwink, Ed. Elsevier
Publ., 1949.
D u r r a n s, Solvents, Chapman a. Hall, London.
В u 11 r e y, Plasticizers, Cleaver-Hume press, Ltd, London, 1947.
Gu i n о t, Solvants et Plastifiants, Dunod, Paris, 1948.
Honnelaitre, La solubilite des hauts polymeres, Conference du Centre de
Perfectionnement Technique, n° 415.
Howlett, Urquhart, Chem. Ind., 1951, 5, 82.
Fordyce, Simonsen, Ind. Eng. Chem., 1949, 41, 104.
Dobry, Bove r-K a w e n о k i, Ind. Plast. Mod., 1948, 4, 553.
Г игроскопичность
Кривая поглощения паров воды в зависимости от величины
относительной влажности воздуха имеет S-образную форму
(рис. 24). Такой характер кривой объясняется одновременным
протеканием двух процессов: капиллярной конденсации воды
в виде жидкости в порах производных целлюлозы и молеку-
лярной абсорбции воды. Можно считать, что при относитель-
ной влажности до 6% вся абсорбированная вода связывается
с полимером химически. Эта реакция протекает одновременно
с поглощением воды в виде жидкости. Самый механизм гид-
ратации пока мало выяснен.
Гигроскопичность довольно быстро понижается по мере
этерификации целлюлозы и особенно при переходе к сложным
эфирам с длинной углеводородной цепью (рис. 25). Эта за-
кономерность справедлива и для смачиваемости (краевой угол
Физико-химические свойства целлюлозы 143
смачивания и работа адгезии), которая также зависит от орга-
нофильности эфирных групп.
В противоположность поглощению влаги, при сушке про-
изводных целлюлозы происходит миграция молекул воды ie-
рез зону гидроксильных групп до наружной поверхности поли-
мера. Однако, как правило, для кривых десорбции наблюдает-
ся явление гистерезиса, что может быть вызвано захватом мо-
Относительная влажность, %
Рис. 24. Изотермы гидратации цел-
люлозных волокон [Urquart,
Eckersall, J. Text, ind., 23,
163 (1932)]:
1—вискоза; 2—мерсеризованный хлопок;
3—отбеленный хлопок; 4—ацетат целлюло-
зы.
Рис. 25. Зависимость гигроско-
пичности триэфиров целлюлозы
от числа атомов углерода в за-
мещающих группах [Sheppard
Newsome, J. Phys. Chem., 39,
143 (1935)]:
1—при относительной влажности 100%;
2—при относительной влажности 50%.
лекул воды и, возможно, перегруппировкой целлюлозных це-
пей. Это явление выражается в том, что целлюлозные пленки
удлиняются при поглощении воды и сокращаются после сушки.
ЛИТЕРАТУРА
Champetier, Bull. Soc. chim. France, 1951, 153.'
В ad gley, Mark, Ind. Eng. Chem., 1945, 37, 227.
Способность к окрашиванию
Волокна природной целлюлозы могут быть окрашены кра-
сителями различных типов.
Крашение прямыми красителями ведут при температуре 50—
90° в присутствии закрепителей (Na2SO4, NaCl), без протрав.
Окраску основными красителями производят из кислой ванны
144 Гл. III. Производные целлюлозы
с нанесением на волокно протравы (таннина) и последующим
закреплением. Из числа других красителей для крашения цел-
люлозы пригодны сернистые красители, протравные красители
(красный ализариновый), азокрасители (красный п-нитроани-
линовый), нафтоловые и нафтазоловые, черный анилин, кубовые
красители, индантреновые, индигозоли, а также хромирующиеся
красители (хром желтый и оранжевый).
Те же красители применяются и для окраски регенерирован-
ных целлюлоз (вискозы).
Для крашения ацетатного волокна субстантивные красители
непригодны. Для этой цели могут быть использованы основные
красители с протравой и без нее, кислотные красители и диазо-
красители, проявляемые путем азосочетания на волокне.
Окрашивание лаков и пластических масс на целлюлозной
основе обычно производится нерастворимыми в воде органиче-
скими или минеральными пигментами. Среди нерастворимых
красителей, список которых более ограничен, можно назвать
некоторые основные красители, азокрасители в виде комплексов
с металлами (лаков) и кислотные азиновые красители.
Скорость поглощения красителя зависит от степени ориен-
тации полимера и расстояния между макроцепями.
ЛИТЕРАТУРА
Battegay, Deniveil е, La cellulose, Hermann Co., Paris, 1934.
D i s e r e n s, Progres realises dans 1’application des matieres colorantes, I?d.
Teintex, Paris, 1950.
Capron, Technologie chimico—textile, Les Editions Text, et Techn., Paris,
1948.
Robinet, Precis de teinture des fibres textiles, Ed. Teintex, 1951.
Адгезионные свойства
Адгезия производных целлюлозы к различным подложкам
имеет большое значение в технике нанесения покрытий и клеев.
Физико-химически механизм адгезии мало изучен. Известно,
однако, что адгезия зависит от химического строения и поляр-
ности подложки и целлюлозного полимера, от молекулярного
веса полимера и его поверхностной активности по отношению
к подложке (т. е. от смачивающей способности), а также от
механических свойств сформированной пленки.
Связь между подложкой и полимером возникает, по-видимо-
му, за счет электростатических сил взаимодействия полярных
веществ (Мак-Ларен).
Некоторую качественную характеристику адгезии производ-
ных целлюлозы к различным подложкам дает табл. 10.
Физико-химические свойства целлюлозы
145
Адгезионные свойства производных целлюлозы
ТАБЛИЦА 10
Подложка Этилцел- люлоза Нитрат целлюлозы Ацетат- целлюлозы Ацетобутн- рат целлю- лозы Целлюлоза
Ацетат целлюлозы Хор. Хор. — Хор. Средн.
Нитрат целлюлозы Хор. — Хор. Хор. Средн.
Ацетобутират целлюлозы Средн. Средн. Средн. Хор. Средн.
Этилцеллюлоза — Хор. Средн- Хор. Хор.
Поливииилацетат Хор. Хор. Хор. Хор. Средн.
Сополимер винилхлорида Удовл. Удовл. Удовл. Удовл. Удовл.
и винилацетата
Поливинилбутираль Средн. Средн. Средн. Средн. Хор.
Полиметилметакрилат Средн. Средн. Удовл. Удовл. Средн.
Поливинилхлорид Средн. — — —• Средн.
Полистирол Удовл. — —. — Удовл.
Каучук Средн. Средн. Средн. Средн. Средн.
Бутилкаучук Удовл. Удовл. Удовл. Удовл. Удовл.
Неопрен Удовл. — — — Средн.
Буна N Хор. Хор. Хор. Хор. Хор.
Политетрафторэтилен Удовл. Удовл. Удовл. Удовл. Удовл.
Хлорированный каучук Средн. — — Средн.
Фенопласты Хор. Удовл. Удовл. Удовл. Средн.
Аминопласты Удовл. Удовл. Удовл. Удовл. Хор.
Силиконы Удовл. Удовл. Удовл. Удовл. Удовл.
Алкидные смолы Средн. Средн. Средн. Средн. Хор.
Кумароно- инденовые
смолы Средн. — — •—— Средн.
Шеллак Удовл. Удовл. Удовл. Удовл. Средн.
В отношении адгезии к металлическим подложкам пока не
удалось установить какой-либо закономерности. Известно лишь,
что адгезия сильно зависит от физического состояния поверх-
ности и может быть улучшена путем специальной обработки,
например путем травления в кислой среде.
Адгезию производных целлюлозы по отношению к заданной
подложке можно изменять путем выбора соответствующей сте-
пени замещения или подбора замещающего радикала. Так, в
ряду эфиров жирных кислот с содержанием от 3 до 18 ато-
мов углерода оптимальными адгезионными свойствами обла-
дают, по-видимому, эфиры капроновой кислоты (Мальм и
Хиат) Ацетопропионат прилипает к стеклу или к производным
целлюлозы при нанесении в виде раствора в ацетоне. Адгезион-
ные свойства ацетата целлюлозы повышаются при частичном
окислении (Фордайс). Нитрат целлюлозы является основой мно-
гочисленных клеев, в состав которых обычно входят пласти-
10—156
146
Гл. III. Производные целлюлозы
фикаторы, органические кислоты, различные соединения (на-
пример, окись пропилена) или смолы. Оптимальные адгезион-
ные свойства имеет нитрат целлюлозы со .степенью полимериза-
ции от 150 до 300.
В качестве компонентов клеев были предложены эфиры цел-
люлозы, образованные кислотами типа гликолевой, тиоглико-
левой, янтарной, глутаровой и т. д., а также щелочные соли
ацетофталатов и ацетосукцинатов целлюлозы.
Термопластичность некоторых простых эфиров (этил- и бен-
зилцеллюлозы) используется при горячей склейке материалов.
Водные растворы метилцеллюлозы, оксиэтилцеллюлозы, солей
ка'рбоксиметилцеллюлозы различных вязкостей также исполь-
зуются в качестве клеев, клеящих составов и аппретов.
ЛИТЕРАТУРА
Del monte, The technology of adhesives, Reinhold Publ., New York, 1947„
Symposium on adhesives, Am. Soc. test, mater., 1945.
Schmidt, Marlies, Principles of high polymer the oryand practice, 1945,.
p. 695.
Термопластические свойства
Температура размягчения производных целлюлозы доста-
точно однозначно характеризует интенсивность и распределение
межмолекулярных сил. Ослабление вторичных связей в продук-
тах частичной этерификации
вызывает значительное пони-
Рис. 26. Влияние степени замещения
на термопластичность этил- и бензил-
целлюлозы [Lorand, Ind. Eng. Chem.,
30, 529 (1938)]
1—этилцеллюлоза; 2—бензилцеллюлоза.
жение температуры размягче-
ния этил- и бензилцеллюлозьп
по сравнению с трехзаме-
щенными
(рис..
производными
26).
С другой стороны, замести-
тели могут действовать как
«внутренние» пластификаторы,
если их объем достаточно ве-
лик. Однако с того момента,
когда начинает преобладать,
эффект, вызываемый увеличе-
нием молекулярного веса, тем-
пература размягчения снова-
возрастает.
.Показатели термопластич-
ности различных производных
целлюлозы приведены в таб-
лицах 11 и 12 на cYp. 151 и сл..
Физико-химические свойства целлюлозы
147
ЛИТЕРАТУРА
Т е е р 1 е, Elastomers and Plastomers, v. Ш, Houwink, Ed. Elsevier Publ.,
New York, 1945.
Extrusion, Reinhold Publ. corp., 1952.
Электрические свойства
Показатели электроизоляционных свойств (удельное объем-
ное или поверхностное сопротивление) целлюлозных пластиков
довольно высоки, вследствие чего эти материалы применяются
в качестве компонентов лаков для изоляции электрических про-
водов. Однако следует иметь в виду, что электрические харак-
теристики производных целлюлозы зависят от их гигроскопич-
ности. Диэлектрические свойства производных целлюлозы связа-
ны также с их полярностью. При трении производные целлюло-
зы приобретают электрический заряд—обычно отрицательный.
ЛИТЕРАТУРА
Granier, Gran i е г, Les proprietes electriques des resines synthetiques,
Dunod, Paris, 1948.
Fuoss, The Chemistry of large molecules, Intersc. Publ., New York, 1943.
J ou a us t, Dielectriques solides,- Ed. de la Revue d’optique, 1949.
Laurent, Les materiaux electrotechn. modernes, 1945.
Seve, Диссертация, Paris, 1951.
Оптические свойства
Спектры поглощения света с длиной волны от 2400 до
3400 А сплошные, за исключением ацетата целлюлозы, который
дает слабо выраженную полосу поглощения около 2680А. Про-
зрачность сильно зависит от пластификации. Кривые поглоще-
ния целлюлозы и ее производных приведены па рис. 27.
Производные целлюлозы
обладают слабой флуорес-
ценцией. (ацетат — голубой,
нитрат — темно-желтой).
Высокомолекулярный ха-
рактер производных целлю-
лозы способствует явлениям
двойного преломления, на-
блюдаемым у натуральных
волокон. Эти явления могут
Рис. 27. Кривые поглощения пле-
нок (толщина 0,09 хя) в ультрафио-
летовой области [Е s р i п a s s е, Ре-
int, pigm., vemis, 19, 42 (1943)]:
1—регенерированная целлюлоза; 2—нит-
рат целлюлозы; 3—ацетат целлюлозы;
4—стекло.
10*
148 Гл. III. Производные целлюлозы
также вызываться плоскостной ориентацией (при образовании
пленки на подложке) или чаще одноосной ориентацией (волок-
но) или двухосной ориентацией (пленки). Растворы производ-
ных целлюлозы также обладают двойным лучепреломлением
при нетурбулентном истечении. Угол двойного лучепреломления
изменяется в зависимости от природы заместителей.
Растворы целлюлозы и ее производных вращают плоскость
поляризации, причем знак вращения, видимо, зависит от при-
роды растворителя, вызывающего некоторые изменения конфи-
гурации макромолекул.
ЛИТЕРАТУРА
McNally, Sheppard, J. Phys. Chem., 1930, 34, 165.
West, M a k a s, J. Opt. Soc., 1949, 39, 791.
Connor, Donnely, Ind. Eng. Chem., 1951, 43, 1136.
Malm. Mench, Kendall, Hiatt, Ind. Eng. Chem., 1951, 43, 688.
Механические свойства
Производные целлюлозы начинают проявлять заметную ме-
ханическую прочность лишь при степени полимеризации выше
100. Показатели механических свойств возрастают затем по ме-
Степень полимеризации
Рис. 28. Зависимость разрывной
прочности целлюлозного волокна от
степени полимеризации [Р 1 о t z е,
Person, Z. Phys. Chem., 45, 193
(1940)]:
/—рами; 2—хлопок.
механические свойства.
ре увеличения молекулярного
веса, причем для целлюлозы
это возрастание значительно
замедляется по достижении
СП = 1500 (рис. 28), а для ее
эфиров уже при СП=200
(рис. 29).
Как правило, для производ-
ного заданного типа повыше-
ние степени полимеризации вы-
зывает повышение прочности
при растяжении, модуля эла-
стичности и твердости. Откло-
нения от этого правила стано-
вятся весьма значительными
при высокой полидисперсности
материала, если макромоле-
кулы с низким молекулярным
весом оказывают неблагопри-
ятное .влияние на некоторые
свойства производных
Механические
целлюлозы сильно зависят также от химического строения по-
лимера, определяющего энергию межмолекулярных связей.
Физико-химические свойства целлюлозы 149
Таким образом, целлюлоза ведет себя как волокнистое ве-
щество, для которого модуль эластичности и высокая прочность
при растяжении обусловлены нали-
Степень полимеризации
Рис. 29. Влияние степени поли-
меризации на свойства ацетата
целлюлозы[Sookne, Harris,
Ind. Eng. Chem.. 37, 478 (1945)]:
/—прочность при растяжении; 2—уд-
линение; 3—прочность к перегибам.
чием водородных связей между гид-
роксилами и довольно высокой же-
сткостью цепи, образованной р-глю-
козидными связями. Замещение
гидроксилов функциональными'-
группами более или менее значи-
тельных размеров вызывает эф-
фект типа внутренней пластифика-
ции, обусловленной ослаблением
межмолекулярных связей (рис. 30).
Число атомов углерода
Рис. 30. Зависимость механической проч-
ности триэфиров целлюлозы от числа ато-
мов углерода в замещающих группах
(Moeller, Cell. Chem, 12, 29 (1931)].
Трехзамещенные производные имеют обычно наилучшие ме-
ханические свойства.
Способность производных целлюлозы кристаллизоваться
следует рассматривать особо, поскольку строение макромолеку-
лярной решетки может изменяться после испарения раствори-
теля.
Природа используемого растворителя и процесс испаре-
ния смеси растворителей оказывают таким образом значитель-
ное влияние на молекулярное строение, а следовательно, и на
механические свойства.
Кристалличность структуры благоприятно влияет на удлине-
ние и твердость полимера, в то время как пластичность и пре-
дел прочности при растяжении приобретаются лишь тогда,
когда цепи образуют нечто вроде изотропного войлока. При ори-.
150
Гл. III. Производные целлюлозы
ентации путем вытяжки нагрузка, требуемая для разрыва, уве-
личивается, а эластичность соответственно понижается (рис. 31).
Рис. 31. Влияние ориентации на механичес-
кую прочность вискозы (Houwink, Techno-
logy of Synth. Polym., 1947):
/—после 70%-ной вытяжки, сухой материал; 2—то же,
влажный материал; 3—после 20%-ной вытяжки, сухой
материал; 4—то же, влажный материал; 5—без вытяж-
ки, сухой материал; 6—то же, влажный материал.
Вещества, вызывающие набухание, придают полярным про-
изводным целлюлозы механические свойства, довольно близ-
кие, с точки зрения термодинамики, к свойствам материалов,
характеризующихся высокой эластичностью. Введение пласти-
фикатора приводит, как травило, к увеличению удлинения при
разрыве и к относительному уменьшению прочности при рас-
тяжении, что вызвано увеличением средних межмолекулярных
расстояний. Однако некоторые пластификаторы, содержащие
много полярных групп, могут сообщать полимеру твердость
или жесткость. Вода является очень энергичным пластификато-
ром для природной или регенерированной целлюлозы, а также
для малогигроскопичных эфиров. Сшивание цепей, как правило,
повышает жесткость и прочность при растяжении в ущерб эла-
стичности. Повышение температуры оказывает неблагоприятное
действие на прочность при растяжении, в особенности в при-
сутствии пластификаторов.
. ЛИТЕРАТУРА
Al f ге у, Meeh. Behavior of High Polymers, Intersc. Publ., New York, 1945.
Renard, Compte rendu du Congres de Chim. Ind., 1938.
Malm, Mench, Kendall, Hiatt, Ind. Eng. Chem., 1951, 43, 688.
E у ring, Halsey, High Polymer Physics, Remsen Prin., New York,
1948.
D ill on. Advances in Coll. Sci , v. Ill, Intersc. Publ., New York, 1950.
ФИЗИКО-МЕХАНИЧЕСКИЕ ПОКАЗАТЕЛИ ЦЕЛЛЮЛОЗНЫХ ВОЛОКОН, ПЛЕНОК И ПЛАСТИКОВ (табл. И И 12)
ТАБЛИЦА И
Целлюлозные волокна
Показатели Природная целлюлоза КОНОПЛЯ Регенерированная целлюлоза (волокно) Ацстаг целлю- лозы (волокно) Этил- целлю- лозя (волок- но)
ХЛОПОК джут ЛСН вискозное медно- аммиач- ное
Длина волокна, мм . Диаметр, р Метрический номер ... Удельный вес, г/см3 : • Прочность на удар, кг/мм . сухого волокна, г/денье влажного волокна, г/денье 15-40 11-25 8-2,5 1,54-1,55 35-111 4,5 4,0 0,8-5 10-25 0,3—0,25 1 48—1,50 4-6 12-76 0,6-0,2 1,50 20—50 16—50 0,25—0,15 Непрерывная 1,5-8 1,52-1,54 НИТЬ 0,6—8 Непрерывна) 4-2 1,30-1,33 1 нить
40-110 3,5 8 40—80 6,3 6 80—92 6 14—88 2 14-25 2 1 16—80 1,4 0,9
Удлинение при разрыве, /о 4,0 10 17-45 1,3 1 8 1,7 13 18 22 29
сухого волокна • • • • влажного волокна . . • Разрывная длина, кг/мм _ плотность 8-12 2,2 40—60 4 15-20 21 20—26 21 20 30 12 29,6
Модуль эластичности, 9 Я 8-11 6—8 14,7-45 1,7-6
Удельная теплоемкость, 0,319 200 0,324 0,324 100 0,324 200
180—200 170—190
Температура разложения, с Средний показатель прелом- 1,56 1,53 1,47 1,47
1,55
ления, п 3,4
Поглощение паров воды (при 14 12 12 10-13 12 6-7
65% влажности), % . • • Набухание в воде, % • • Окраска реактива Identex . 44 Светло-жел- 45 — 11 —-
Фиолетово- Оранже- Серо-зеленая Бесцветная Жел- Красная
тая серая во-желтая тая
Физико-химические свойства целлюлозы
152
Гл. III. Производные целлюлозы
Целлюлозные
Показатели Регенерирован- ная целлюлоза пленка Ацетат целлюлозы Триацетат целлюлозы (пленк а)
пленка вальцованная масса
Предел прочности при растяжении, кг/мм2 7.5-12 9—10 3—3 3—12
Удлинение при разрыве, % . . . 3—80 9—50 — 20—30
Модуль эластичности, кг/мм2 . . 1040 150—400 195 70—245
Предел эластичности, кгf мм2 при относительной влаж- ности 0% 10 9 __ 8
при относительной влаж- ности 100% — 3 — 5
Прочность на удар по Изоду по VDE 1-4 0,4—5 20 0,7—6
Сопротивление срезу, ка/слЗ . . — — 420 260
Усталостная прочность, кг] мм2 . . — — 0.6-1 —
Предел прочности, кг/мм2 при сжатии при изгибе — — 5—21
Число перегибов (размеры пленки 13/100 мм) — 65—80 — 65—90
Твердость по Бринелю, кг/мм2 — 15—25 15-25 15-25
Прочность к истиранию .... — 5,4—11,9 — —
Удельный вес, г]см2 1,4—1,5 1,27—1,34 1,28—1,32 1,27—1,34
Урцльвая теплоемкость, кал]г*сС 0.3—0.5 0.3—0,4 0,3—0,4
Коэффициент расширения на 1°, %. 10“5 8—16 8—16 10—15
Теплопроводность, кал/сек. • сл&- °C 4,5—7,8 4,0—8
Температура, °C размягчения прессования экструзии . . . . 100-110 126—213 167—250 100—115 138—200 180—240
Давление, кг (см2 при литье при инжекции 35—350 200—2000 35—350
Увеличение объема, % . 2.0-2.6 2,0—2,6
Усадка при литье, % 0,1-1 0,1—1
Показатель преломления п . . l.i„—1.54 1,49-1,50 1.46—1.50 1,46-1,50
Прозрачность, % . . . . ... 90 87
Рассеивание света, % 5
Электрическое сопротивление (при 25° и 50% относительной влаж- ности), ож-слс 10*°—1013 IO1"—ю18 Ю10—1013
Электрическая прочность, кв-см (на 3,2 мм) 12—20 9,8—11,8 10—15 7,8—11,7
Физико-химические свойства целлюлозы
153-
пленки и пластики
ТАБЛИЦА 12
Нитрат целлюлозы Пропионат целлюлозы Ацетобутират целлюлозы Этил- целлюлоза Бензил- целлюлоза (непластифици - рованная)
непластифи- цированиый пластифици- рованный - альцованная масса
7—13 2,8—4,4 4-6 3-6
34 20—42 — — 8-14 19-55
215 140—280 — 140 200—300 200
— 10 — — 5 4
— 8 — — 3 3 •
— 2—8 — 1—9 0,6-11
— 6 — — — —
— — — — — —
— — — — — —
17 5-15 8-14
— 6 — 4,5—7 6-8,5 —
— 95—100 — — — —
— 15—25 15—25 15-25 15—25 —
— 10 4,5 — 4,3-6,3 —
1,35—1,4 1,35—1,4 1,16—1,21 1,15—1,25 1,09-1,17 1,17—1,22
0,3-0,4 1.49-1,51 Ю10- Ю11 6—16 3,0—5,1 60 85—120 140-350 88 5 1О10— 10й 10-24 0,4 13-16 60-105 120—190 175—260 35—350 100-2450 1,7—2 0,4—0,6 1,49—1,48 Ю12—1O1S 0,3—0,4 11—17 4,8 60—105 130—200 165—240 35-350 2,0—2,4 0,1—0,9 1,47—1,46 89 8 10*°— 10м 8-16 0,3—0,75 10—20 50-130 120-245 240—280 35—360 150—1500 1.8—2,4 0,2—0,9 1,47 89 5—12 1012-10*5 10-18 120—155 1,47—1,60
154
Гл. III. Производные целлюлозы
Показатели Регенерирован- ная целлюлоза Ацетат целлюлозы Триацетат целлюлозы
пленка пленка вальцованная масса (пленка)
Диэлектрическая постоянная при 60 гц при 106 гц 7 5—6 5,3—5,8 3.0—6,0 3,5—8,5 3,2—7,0 3,5—7,5 3,2—7,0
Тангенс угла диэлектрических по- терь (при 106 гц) ...... 0,04—0,045 0,03—0,09 0,01—0,1 0,01—0,06
Напряжение разряда, кв/мм . . 18 32—100 14—33 12—15
Водопоглощение (за 24 часа при толщине 3 мм), % 45—115 2,2—4,5 2—6,5 2,2—3,1
Поглощение паров воды, % ури относительной влажности 50% при относительной влажности 90% 3,5-4,8 12 2,7 9
'Водостойкость . . . Средняя Средняя Хорошая
Кислотостойкость .... Слабая Слабая Слабая
Щелочестойкость Слабая Слабая Слабая
Коэффициент диффузии (вода) . . 3.0
Проницаемость (за 24 часа), внмм-см рт.ст, СО2 О2 N2 - - 3 0.5 0,5 20 0,5 160
Паропроницаемость (за 24 часа, при толщине 6/100 aijw), г/м% . . . 14 000 5 620 4 500
Старение на свету Стабнльн. Стабильн. Стабильн. Стабильн.
Характер горения Медленное Самопроиз- вольное Медленное Медленное
ЛИТЕРАТУРА
Encyclopedic fransaise des plastiques modernes, Industrie des plastiques mo-
dernes, Paris, 1953.
Dubois, Cor, С о 1 i .n, Les Plastiques modernes, Paris.
Plastic catalogue corp., Modern Plastics Encyclopedia Charts, New York, 1954.
Handbook of Plastics, Simonds—Ellis, Van Nostrand Publ., New York, 1943.
Technical data on Plastics, Manuf. chim., Ass., 1952.
Физико-химические свойства целлюлозы
155
Продолжение табл. 12
Нитрат целлюлозы Пропионат целлюлозы Ацетобутират целлюлозы Этил- целлюлоза Бензил- целлюлоза ( иеп ластифици- рованная)
непластифи- цированный пластифици- рованный вальцованная масса
6,7—7,3 6,0—6,2 3.0—0,5 3,3-3,55 3.5—6,4 3,2—6.2 2,5—4 2,0-4
0,07—0.10 0.01—0,04 0,005—0.04
24—40 10 10—16 16-60
1—2 1,2—1,7 1,1—2,2 0,8—1,8 60
0,6—1.7
2-6 1,3—1,5
Хорошая Хорошая Хорошая Хорошая Хорошая
Слабая Слабая Слабая Плохая Плохая
Слабая Слабая Слабая Хорошая Хорошая
1480 5 120 1460
Нестабильн. Нестабильн. Стабильн. Стабильн. Стабильн.
Очень быстрое Очень быстрое Медленное Медленное Медленное Медленное
ГЛАВА IV
ВИНИЛОВЫЕ ПОЛИМЕРЫ
Ф. Шевассю
Класс виниловых производных известен с 183,8 г., когда
Реньо осуществил синтез хлористого винила, действуя дихлор-
этаном на спиртовый раствор едкого кали. Он же открыл вини-
лиденхлорид. Бауман4 в 1872 г. путем полимеризации винил-
галогенидов получил пластическую массу, устойчивую к дейст-
вию большинства кислот. Наибольшее значение, однако, имели
работы Остромысленского5, который, проводя исследования в
области синтеза каучука, детально изучил полимеризацию хло-
ристого и бромистого винила и их производных.
Материалы на основе виниловых полимеров получили ши-
рокое распространение в период второй мировой войны, когда
возникла необходимость изыскания синтетических заменителей.
Однако в промышленности лаков и красок, в связи с получе-
нием важных результатов, работы в этом направлении прово-
дились не с целью получения заменителей, а имели самостоя-
тельное значение. Полученные продукты оказались не только
удовлетворительными, но отличались целым рядом весьма важ-
ных свойств, что привело к широкому развитию производства
•виниловых полимеров в последующие годы. Большая заслуга
в этом отношении принадлежит фирме Carbide and Carbon
Chemicals Corporation, которая уже с 1926 г. специализировалась
в области производства виниловых полимеров. Фирма
И. Г. Фарбениндустри не заинтересовалась промышленным раз-
витием этих продуктов; немецкие специалисты считали, что
хлорированный каучук вполне может заменить поливиниловые
производные.
Поливинилацетаты применяются для нанесения покрытий
с 1920 г. (.метод их производства запатентован Клаттом2
в 1912 г.). Сополимеризация винилацетата с другими мономе-
рами, а также его полимеризация в присутствии катализаторов
была открыта в 1917 г. Клаттом и Роллетом3. В 1926—1933 гг.
было освоено производство сополимеров винилхлорида и винил-
ацетата для лакокрасочной промышленности, но только во
время второй мировой войны (1939—1944 гг.) выпуск этих про-
дуктов начал быстро возрастать.
Виниловые полимеры
157
В 'настоящее время фирма Bakelite Division (дочерняя фир-
ма Union Carbide) производит 1300 т виниловых смол в месяц,
покрывая 90% потребности США.
Типы виниловых полимеров. Термин «виниловые полимеры»
применяется для определения класса синтетических продуктов
(в большинстве случаев производных виниловых эфиров), по-
лучаемых путем полимеризации соответствующих мономеров.
Полимеризация поддается регулированию и приводит к получе-
нию продуктов с определенными свойствами. Молекулы вини-
ловых полимеров представляют собой высокомолекулярные ли-
нейные цепи, в которые объединяются мономеры.
Свойства виниловых полимеров находятся в прямой зави-
симости от молекулярного веса, соотношения мономеров в со-
ставе сополимера, а также от природы сополимеров. Некоторые
свойства этих полимеров мало изменяются в зависимости от
молекулярного веса; растворимость же их в органических рас-
творителях изменяется обратно пропорционально молекуляр-
ному весу. Хотя такие пластики, как «толиакрилаты! или поли-
стирол, также могут быть отнесены к виниловым производным,
поскольку мономеры содержат винильную группу —СН = СНг,
в области лаков и красок термином «виниловые полимеры» при-
нято обозначать только поливинилацетат, поливинилхлорид,
поливинилиденхлорид, сополимеры винилхлорида с винилаце-
татом или винилиденхлоридом, поливиниловый спирт и поли-
винилацетали. Эти продукты составляют ’целую серию поли-
мерных производных. Только некоторые из них применяются
в производстве лаков и красок.
Ниже приведены наиболее важные виниловые полимеры и
'Мономеры, из которых они получаются:
Мономеры
Сложные виниловые эфиры
ch2=choocr
Винилхлорид
СН2=СНС1
Простые виниловые эфиры
СН2=СН—OR
Виниловые тиоэфиры
СН2=СН—SR
Винил амины
СН2=СН—NR2
Полимеры
---СН2—СН—СН2—СН---
I I
RCOO RCOO
----сн2—СН—сн2—СН---
I I
С1 С1
----сн2—сн—сн2—сн---
' I
OR OR
---сн2—СН—сн2—СН---
I I
SR SR
----СН2—СН—сн2—СН---
I I
nr2 nr2
158
Гл. IV. Виниловые полимеры
Мономеры
Винилцианид (акрилонитрил)
СН2=СН—CN
В иннлиденхлорид
СН2=СС12
Винилацетат
СН2=СН
I
ОСОСН3
То же
Полим еры
----СН2—СН—сн2- сн----
I I
CN CN
С1 С1 С1
' I I
----сн2—с—сн2—с—сн2—С------
I 1 I
С1 С1 С1
»
»
----сн2-сн—сн2—сн-----
I I
ОСОСНз ОСОСНз
Поливиниловый спирт
----СН2—СН—СН2—СН----
I I
ОН он
Поливин илформаль
• • ♦—СН2—СН—СН2—СН---
I I
О—сн2—о
Поливинилацеталь
----СН2—СН=СН2—СН----
I I
О—СН—О
I
СНз
Поливинилбутираль
----СН2—СН—СН2—СН-----
I I
О—сн—о
I
С3Нг
ПОЛИВИНИЛА ЦЕТАТ
Поливинилацетат получают полимеризацией винилацетата,
образующегося при взаимодействии ацетилена с уксусной кис-
лотой в присутствии солей ртути как катализатора:
HCsCH -|г СНзСООН СН,=СН—ОСОСНз
Получаемый таким путем винилацетат представляет собой
продукт очень высокой степени чистоты (99,5%-ный); темп,
кип. 72—73°; темп, замерз. — 7°; уд. в. 0,984 г/см?.
Поливинилацетат
159
При полимеризации винилацетата в растворе можно по-
лучить одну из следующих структур:
---сн2-сн—СН2—СН—СН2—сн—сн2- сн-----
I I I
ОАс ОАс ОАс ОАс
или
----СН2—СН—СН—СН2—СНг—СН—СН—сн2-----
II II
ОАс ОАс ОАс ОАс
Сочетание обеих структур со случайным их чередованием
для виниловых полимеров встречается чрезвычайно редко.
Поливинилацетат не может быть получен также в кристалли-
ческой форме; рентгенограммы свидетельствуют о полной
аморфности структуры. Наибольший достижимый молекуляр-
ный вес составляет около 80000, что соответствует макромо-
лекуле из 900 остатков мономера (Штаудингер и Швальбах6).
Растворители, используемые при реакции полимеризации,
различны по своему ингибирующему действию. Соответствен-
но различен и молекулярный вес получаемого полимера. По-
лимеры винилацетата, содержащие от 150 до 250 винильных
остатков в макромолекуле, эластичны. Продукты, имеющие
от 30 до 70 остатков, при тех же условиях неэластичны, а при
степени полимеризации 300 и более размягчению предшествует
деполимеризация, достаточная для проявления эластичности.
Физические свойства полимеров зависят, следовательно, от
степени полимеризации, характеризуемой на практике различ-
ной вязкостью получаемых растворов.
Свойства поливинилацетата. Поливинилацетат растворим
в низкокипящих спиртах, сложных эфирах, альдегидах, кето-
нах, алифатических кислотах, ароматических углеводородах и
в некоторых хлорированных углеводородах. Он нерастворим в
углеводородах жирного ряда, растительных и животных жи-
рах, а также в жирных кислотах. Поливинилацетат нераство-
рим в воде, хотя имеет склонность (при продолжительном воз-
действии) набухать в ней и размягчаться.
Полимеры винилацетата частично или полностью совмести-
мы с некоторыми типами нитроцеллюлозы, с феноло-формаль-
дегидными смолами, .хлорированным каучуком, терпеновыми
смолами, а также с небольшим количеством кумароно-индено-
вых смол. Они имеют ограниченную совместимость с природ-
ными смолами (даммарой, каури, копалами и т. д.) и совмести-
мы с большинством пластификаторов типа сложных эфиров,
таких, как дибутилфталат, диоктилфталат, трикрезил- и триок-
тилфосфаты, запатентованные фирмой Monsanto сантицайзеры
160
Гл. IV. Виниловые полимеры
М-8 (смесь о- и п-метилтолуолсульфамидов), М-17 (этиленгли-
колевый эфир метилфталевой кислоты) и В-16 (бутиленглико-
левый эфир бутилфталевой кислоты), запатентованные фирмой
Union Carbide флексолы 3GO (ди-2-этилгексоат триэтилен-
гликоля) и 3GH (ди-2-этилбутират триэтиленгликоля).
Поливинилацетатные смолы свето- и теплостойки, не подвер-
жены старению и устойчивы к разбавленным кислотам, рас-
творам щелочей и солей. Они могут быть пластифицированы
для получения желаемой степени эластичности.
Торговые марки поливинилацетата. Поливинилацетат произ-
водится во Франции (фирма Societe Rhone—Poulenc), ,в США
(фирма Bakelite Corporation), в Канаде (фирма Shawinigan),
в Англии (фирма I.C.I), в ГДР и ФРГ (бывшие заводы I. G.
Farben Industrie). В табл. 13 приведены показатели некоторых
торговых марок поливинилацетатов.
Французская фирма Rhone Poulenc поставляет серию, про-
дуктов под общим названием родопас, включающую следую-
щие 6 марок: В — низковязкий, М — средней вязкости, Н — вы-
соковязкий, НН, HV1, HV2—продукты с очень высокой вяз-
костью, возрастающей от НН к HV2. Эти марки отвечают возра-
стающим степеням полимеризации и различаются между собой
лишь некоторыми свойствами, в частности вязкостью раство-
ров (возрастающей с увеличением степени полимеризации), а
также теплостойкостью и эластичностью.
Поливинилацетат марки родопас В плавится при темпера-
туре —80°. Растворы его имеют низкую вязкость, после испа-
рения растворителя остается пленка невысокой твердости, но
с очень хорошей адгезией ко всякого рода подложкам. Родо-
пас В пригоден, в частности, для приготовления клеев и лаков,
для грунтов, которые должны обладать высокой адгезией.
К тому же он хорошо совмещается с нитроцеллюлозными ма-
стиками, повышая их светостойкость и адгезию. Родопас М
плавится при более высокой температуре, чем родопас В
{—100°), и дает более вязкие растворы. Эти растворы после
испарения образуют эластичную твердую пленку с хорошей
адгезией. Родопас М пригоден для производства всевозмож-
ных лаков, в частности, в тех случаях, когда требуются про-
зрачные тепло- и светостойкие покрытия.
Родопасы Н и НН трудноплавки. Их растворы очень вязки
и образуют после испарения растворителя эластичную пленку.
Эти марки рекомендуются для пропитки фанеры, тканей и т. д.,
-а также для некоторых специальных лаков, от которых тре-
буется высокая эластичность.
Родопасы HV1 и HV2 дают еще более вязкие растворы, чем
родопас НН. Растворы их после испарения растворителя об-
разуют очень эластичные пленки, мало чувствительные к дей-
Поливинилацета т
161
ТАБЛИЦА 13
Марки поливинилацетата фирмы Bakelite Corporation (США)*
Тип I AYAB Тип 2 ATYAA Тип 3 AYAF Тип 4 AYAT
Удельный вес, г;см3 . . Вязкость раствора в аце- тоне при 25° (по Форду, 1,191 1,191 1,191 1,191
сопло № 4), сек. . . . Содержание сухого веще- 18—22 13—14,5 17—21 26—30
ства, % Вязкость раствора в бен- золе (86,1 г/л), санти- 45 20 20 20
пуазы Температура размягчения (по методу кольца и 2,5 5 15 21
шара), °C Окраска 111,2 150,8 Проз 170,6 рачные 187
Теплостойкость (при 135°) Поглощение воды (при 25°), % Через 1,15 часа янтарная окраска Через 5 час без видимог о изменения
через 16 час. . . . 2 1,6 1,4 1,4
» 144 часа . . . Предел прочности при 7,3 4 3,6 3,6
растяжении, кг/см? . . Концентрация раствора в метилизобутилкетоне при 20°, % сухого ве- 105 180 290
щества Поведение при погруже- 41,5 27,5 21 18
нии в воду Набухают, допускают перегиб Слегка набухает Легкое изменение поверхности
* Фирма Shawinigan Chemicals Ltd выпускает аналогичные продукты под названием «гельва».
ствию высокой температуры и неклейкие. Эти продукты выпу-
скались под названиями мовилитов (фирма Hochst) или винна-
пасов (фирма Burghansen), но большей частью поставлялись
в виде пластифицированных сложных эмульсий, предназначен-
ных для специальных целей.
Применение поливинилацетата в производстве лаков и кра-
сок. Поливинилацетат применяется в этой области, но не ши-
роко распространен, так как он мало устойчив к действию кон-
центрированных химических реагентов и недостаточно водостоек.
Он больше используется для внутренних защитных покрытий,
в качестве модифицирующих добавок и как основа клеевых
композиций.
11—156
162 Гл. IV. Виниловые полимеры
Первые опыты применения поливинилацетата в качестве
основы антикоррозионных красок8 относятся к 1928 г.
Наилучшей адгезии для эмалевых красок удалось добиться
при смешении полимеров различной вязкости (поливинил-
ацетата или шоливинилбутирата высокой и низкой вязкости9).
Если поливинилацетат употребляется в качестве грунта под
нитроцеллюлозные эмали, необходимо наносить очень тонкие
пленки, чтобы предотвратить растрескивание поверхностного
слоя. Можно также придать непроницаемость этой пленке, до-
бавляя к полимеру парафин и пластификаторы10.
Введение поливинилацетата в нитроцеллюлозные эмали с
целью повышения 'блеска и адгезии покрытий изучалось Гех-
нелем и Германом11 в 1928 г.; применение этих продуктов для
поверхностной обработки кожи запатентовано в 1930 г. Дор-
ном, и Гофманом12.
Поливинилацетат, обычно несовместимый с ацетатом целлю-
лозы, приобретает способность совмещаться с ним при введе-
нии третьего полимера, смешивающегося с двумя первыми.
В качестве таких добавок можно использовать смолы типа
сульфонамидо-формальдегидной, фурфурольной, фенол-фурфу-
рольной, ацетон-фурфурольной, анилино-фурфурольной.
Области применения поливинилацетата могут быть значи-
тельно расширены, так как его водо- и теплостойкость могут
быть повышены.
Растворение поливинилацетата может производиться в обыч-
ных аппаратах. В качестве растворителей пригодны многочис-
ленные вещества.
Спирты. Поливинилацетат не растворим в безводных спир-
тах, за исключением метилового спирта, но растворим во всех
низших спиртах, содержащих определенное количество .воды.
Так, 95%-ный этиловый спирт является хорошим растворите-
лем; получаемые растворы мутнеют на холоду, но осветляются
при добавлении 10% бензола или этил ацетата.
Ароматические углеводороды. Интересно отметить, что неко-
торые растворители, содержащие 90% ароматических соедине-
ний и 3—5% бутилового спирта, дают растворы (для полимеров
со средним молекулярным весом) такой же вязкости, как рас-
творы в чистом толуоле, хотя ни одна из двух жидкостей не
является истинным растворителем. Бензол и толуол являются
хорошими растворителями при обычной температуре: помутне-
ния растворов при стоянии можно избежать, добавляя 10%
этилацетата или спирта.
Кетоны. Кетоны пригодны только в смесях, так как вследст-
вие высокой скорости их испарения происходит побеление плен-
ки за счет конденсации воды; растворы получаются низко-
вязкие.
Поливинилацетат
163
Сложные эфиры. Этил- и бутил ацетат являются превосход-
ными растворителями. Они дают растворы, не мутнеющие при
охлаждении.
Хлорсодержащие растворители. Дихлорэтан и трихлорэтан
как негорючие растворители применяются при покрытии кар-
тона. Тетрахлорэтилен и четыреххлористый углерод не являют-
ся растворителями, но применяются как разбавители, пони-
жающие воспламеняемость.
Наполнители, пигменты и другие компоненты. Для поливи-
нил ацетатных композиций применяются компоненты тех же ти-
пов, что и для композиций на основе поливинилхлорида (см.
далее).
При введении пигментов (после тщательного их пере-
тирания) в количестве до 100% от веса полимера получаются
блестящие пленки. При более высоком содержании пигмента
получаются матовые однородные пленки, нехрупкие и обладаю-
щие кроющей способностью благодаря сильной непрозрачности.
Нейтральность поливинилацетата позволяет применять его в ка-
честве основы лаков для металлов.
Лаки и краски на основе поливинилацетата применяются
для покрытия многих материалов (во всех случаях наблюдает-
ся прекрасная адгезия). При нанесении этих лаков и красок по
цементу или камню следует принимать во внимание недоста-
точную водостойкость покрытий. Наибольшее распространение
эти материалы получили для отделки кож и бумаги, а также
в производстве печатных красок. Следует упомянуть о внедре-
нии их в производство клеев.
При отделке кож применяются в основном продукты с вы-
соким молекулярным весом. Они наносятся в виде эмульсий
методом окунания или лучше пульверизатором. Пластифика-
торы, применяемые с целью придания высокой эластичности
пленкам, вводятся в большом количестве (25—30% от веса по-
лимера). Выбор пластификатора имеет большое значение, так
как для предотвращения быстрого старения пленки нужно,
чтобы пластификатор не мигрировал в кожу.
Лаки на основе поливинилацетата благодаря их бесцветно-
сти, отсутствию запаха, хорошей светостойкости, высокой адге-
зии и прочности пленок нашли широкое применение для покры-
тия бумаги. Между прочим, бумага, покрытая таким способом,
может легко склеиваться при горячем проглаживании или при
действии растворителей. Температура размягчения полимеров,
применяемых для этих целей, может быть повышена путем
введения в них нитрата или ацетопропионата целлюлозы, коли-
чество которых доводят до 10—20%.
Для получения непроницаемых покрытий употребляются в
основном эмульсии поливинилацетата.
11*
164
Гл. IV. Виниловые полимеры
Получение эмульсий. Растворы полимеров, содержащие
жирные кислоты, эмульгируются за счет образования мыл.
Чаще всего вводят водный раствор олеата аммония, содержа-
щий избыток аммиака; хорошие результаты дает олеат триизо-
пропанол амина, а также лаурил сульфат натрия, введенные в
водную среду. Количество эмульгатора колеблется от 0,5 до
5% от веса полимера.
Водостойкость покрытий на основе этих эмульсий невелика,
.потому что всегда возможно эмульгирование. Особенно значи-
тельна эта опасность при применении олеата аммония, так как
аммиак испаряется при сушке. Для эмульгаторов типа морфо-
лина этот недостаток смягчается.
Стабильность эмульсий при хранении достигается при до-
бавлении от 0,2 до 0,5% (по отношению к полимеру) бенто-
нита, предварительно диспергированного в шаровой мельнице
и добавленного к готовой эмульсии.
Типовой состав эмульсии (вес. ч.):
Лаковая фаза
поливинилацетат среднего молекулярного веса (AYAA
или родопас М).................................... ... 50
флексол 3 GO ......................................5,5
толуол........................................... 43
олеиновая кислота ................................ 1,5
Водная фаза
дистиллированная вода..............................92
аммиак (28%-ный) ..............................8
Такие эмульсии широко применяются. Их преимуществами
являются ограниченное проникание в пористые поверхности,
пониженная огнеопасность и легкость нанесения материалов
с высоким содержанием сухого остатка.
ЛИТЕРАТУРА
1. R e g n a u I t, Ann. phys. chim., 1838, 65, 157.
2. KI at te, герм. пат. 281687, 1912 г.
3. Klatt e, Rollett, пат. США, 1241738.
4. Baumann, Ann. phys. chim., 1872, 163708.
5. Ост p ом ы с ленский, Chem. Ztg., 1912, 36, 199, <Le caoutchouc
et ses homologues»; ЖРФХО, 1916, 48, 114; пат. США, 1721034, 1929 г.
6. H. Staudinger, A. Schwalbach, Ann. Physique, 1931, 488, 8.
7. Герм. пат. 291299, 1915 г., дополненный пат. 290544; J. Soc. Chem. Ind.,
1916, 35, 698.
8. Англ. пат. 314499, 1928 г., Consort. Elektrochemie I. G., С. A., 1930, 24,
1527.
9. Фр. пат. 712590, 1931 г., Consort. Elektrochemie ind. G. m. b. H.; англ,
пат. 361768, 1931 г.; British С. А., В. 1932, 235; пат. США 1984678, 18/XII,
1934 г., Chemische Forschungs G.; см. также W. Rep ре, W. Stark,
A. Voss. герм. пат. 593399. 1934 г., I. G. Farben W. S.; C. A., 1934,
28, 3255; E. W. Reid, англ. пат. 406338, 1932 г., Carbide and Carbon
Chemical Coro.
Хлорсодержащие виниловые полимеры
165
10 W. Н. С h а г с h, англ. пат. 380483, 1931, Du Pont Cellophane Со., Bri-
tish С. А., В. 1932, 1041.
11. W. Hachnel, W. О. Hermann, герм. пат. 559402, 1928 г., Con-
sort. Elektrochemie I. G. m. b. H.
12. O. Dorr, M. Hoffmann, фр. пат. 705219, 1930 г.; англ. пат.
369260, 1930 г.; British С. А., В. 1932, 475.
ХЛОРСОДЕРЖАЩИЕ ВИНИЛОВЫЕ ПОЛИМЕРЫ
Поливинилхлорид и его сополимеры с винилацетатом
Поливинилхлорид получается при полимеризации винилхло-
рида по схеме:
Н CI С1 С1 С1
Il III
С=С —-----сн2-сн—сн2—сн—сн2—сн---
I
н н
При увеличении степени полимеризации получается нерас-
творимый продукт1,2.
Изучая полимеризацию винилхлорида, Штаудингер, Брун-
нер и Файсет3 получили полимеры, нерастворимые в мономере
(исключительный случай для виниловых полимеров), что по-
зволяет предположить очень высокую степень разветвленности
полимеров. Вместо сиропообразной жидкости получалось твер-
дое вещество, состоящее из смеси высоко- и низкомолекуляр-
ных продуктов. Средняя степень полимеризации составляла
около 50. Низкомолекулярные полимеры относительно лучше
растворимы в растворителях, но зато более чувствительны к
действию света и тепла. Более высокомолекулярные поливинил-
галогениды, содержащие до 100 остатков мономера, исключи-
тельно плохо растворимы и соответствуют полимерам, описан-
ным Остромысленским. Другие типы (X, В, D) вовсе нераство-
римы в ароматических углеводородах.
Однако Саусон и Вейнц4 получили путем специальной обра-
ботки продукт с более низким молекулярным весом, чем тип X.
В то время Как 30%-ный раствор полимера X в 'этилацетате
при нормальной температуре представляет собой гель, из поли-
мера нового типа получался низковязкий раствор, несовмести-
мый с высыхающими маслами (продукт типа X совмещался
с ними). Из этого полимера, полученного в среде углеводоро-
дов бензольного ряда, была приготовлена краска по следующей
рецептуре (в %):
Полимер....................16
Дибутилфталат............ 2
Сольвент-нафта ..........36
Толуол ..................27
Двуокись титана............19
166
Гл. IV. Виниловые полимеры
Низкая концентрация полимера в получаемых растворах
(вследствие пространственного строения макромолекул, со-
стоящих из очень длинных сшитых цепей) сильно затрудняет
пользование этими растворами. Введением модифицирующих
групп можно предотвратить сближение длинных цепей. Проч-
ность получаемых продуктов при этом уменьшается, а пластич-
ность повышается.
Такие группы могут быть введены химическим путем при
совместной полимеризации, например при получении сополиме-
ров винилацетата с хлористым винилом. Ацетатная группа
играет в сополимере роль внутреннего пластификатора. Неко-
торое количество пластификатора все равно необходимо добав-
лять, но для сополимера требуется значительно меньше пласти-
фикатора, чем для чистого поливинилхлорида. Облегчается так-
же растворение полимера.
Интересно проследить, насколько глубокое влияние оказы-
вает введение заместителей на пластические свойства полимера.
Хотя речь идет о пространственных полимерах, здесь мы рас-
сматриваем только идеальные линейные цепи.
Цепи поливинилхлорида расположены очень близко одна
к другой. Между .ними вследствие полярности атомов хлора
•действуют значительные силы когезии (силы ван-дер-Ваальса).
Эти силы достаточно велики, чтобы помешать раздвижению це-
пей, . а следовательно, проявлению текучести при достижении
известной температуры. По этой же причине затрудняется дей-
ствие пластификаторов и растворителей.
В сополимере, содержащем ацетатные группы, картина ме-
няется: цепи раздвинуты и силы когезии ослаблены. Поэтому
температура размягчения понижается, для пластификации тре-
буется меньше пластификатора, растворы при более низкой
вязкости содержат больше сухого вещества.
В схеме, приведенной ниже, для наглядности цепи раздви-
нуты до предела, что в действительности не происходит, но
сущность явления показана правильно:
---сна—сн—сн—сна—сн—сн2—сн—сн---
С1 о С1 С1 о
1 СОСНз • СОСНз
СОСНз 1 СОСНз
С1 1 о С1 С1 о
II I I I
---сн2—СН—СН—СН2—СН—СН2—СН—сн--
сополимер винилхлорида и винилацетата
Хлорсодержащие виниловые полимеры. 167
---СН—СН—СН—СН—СН—СН-------
I I I I I I
Н С1 Н С1 Н С1
С1 Н С1 Н С1 н
I I I I I I
---СН—СН—СН—СН—СН—СН-------
поливинилхлорид
Сополимеры винилхлорида и винилацетата имеют низкие
молекулярные веса (12000-—25000) и, следовательно, повы-
шенную растворимость. Образующийся сополимер не обяза-
тельно имеет тот же состав, что исходная смесь мономеров6. Так
как хлористый винил полимеризуется быстрее, чем винилаце-
тат, образующийся сополимер вначале содержит больше
звеньев хлористого винила. Чтобы получать продукты одина-
кового состава, необходимо соблюдать большую осторожность;
недостаточно точное воспроизведение условий ведения процесса
приводит к получению неоднородных продуктов.
Еще в 1933 г. Рейд7 указывал на пластифицирующее влия-
ние ацетатных групп в сополимере; при сополимеризации по-
лучался внутренне пластифицированный продукт, обладавший
наряду с прочностью и эластичностью также и превосходной
агдезией. Фракционирование путем экстракции, позволяющее
отделять растворимые фракции от нерастворимых, стало с того
времени предметом патентных заявок .и исследований фирмы
Carbide and Carbon Chemicals Corporation8.
Сополимеры, содержащие 30 вес. ч. хлористого винила и
70 вес. ч. винилацетата, с 1927 г. испытывались в качестве
основы лаков и красок9. Такие лаки имели большую светостой-
кость, повышенную устойчивость к растворам поваренной соли,
щелочей и кислот. Эти данные подтвердились при испытаниях,
проведенных американской фирмой Stonite в 1932 г. Испыты-
вались лаки на основе сополимеров фирмы Carbide and Car-
bon Chemicals Corporation, нанесенные на стены силосных ба-
шен10.
Покрытия на основе сополимеров обладают следующими
общими свойствами. Получаемые пленки термопластичны, ней-
тральны, не подвержены окислительной деструкции (при усло-
вии надлежащей стабилизации) и не изменяются под дейст-
вием высоких температур (при условии стабилизации). Для
них характерно: полное отсутствие запаха, вкуса, токсичности;
большая химическая инертность, нерастворимость в спирте, мас-
лах, жирах, алифатических углеводородах; высокая водостой-
кость; твердость и эластичность; невоспламеняемость; прозрач-
ность, очень светлая окраска и высокий коэффициент прелом-
ления 1,53; легкость 'нанесения; большая стабильность эмуль-
сий; термопластичность, позволяющая получать в определен-
168
Гл. IV. Виниловые полимеры
ных условиях очень гладкую поверхность; очень высокая стой-
кость к старению, а также очень большая атмосферостойкость
(при правильном изготовлении).
Промышленные типы сополимеров винилхлорида с винил-
ацетатом. На основе работ фирмы Carbide and Carbon Chemi-
cals Corporation, начатых в 1926 г., в 1928 г. был построен опыт-
ный завод. В результате улучшения качества продукции и от-
крытия более широких областей применения были созданы но-
вые типы смол, и производство начало постепенно увеличивать-
ся. В 1936 г. была введена в строй промышленная установка,
которая должна была полностью удовлетворить спрос амери-
канской клиентуры. Этот завод продолжал далее расширяться
и в настоящее время фирма производит 92% всего количества
виниловых полимеров, применяемых в промышленности, вла-
деет большей частью патентов.
Первоначально предполагалось изготовлять лишь сополи-
меры винилхлорида с винилацетатом* ввиду исключительно хо-
рошего качества этих продуктов. Путем регулирования степени
полимеризации удавалось получать продукты различного моле-
кулярного веса, что приводило к большому разнообразию поли-
меров. Вначале довольствовались полимером, содержавшим
87% винилхлорида и 13% винилацетата и выпускаемым в на-
стоящее время под маркой «винилит VYHH». В условиях испы-
тания, которые будут описаны ниже, этот сополимер имеет
относительную вязкость 0,53 и удачно сочетает химическую
устойчивость по отношению к различным реагентам с раство-
римостью и хорошими свойствами пленки.
Сополимер указанного состава производился в 1932 г.,-
когда единственным доступным растворителем типа кетонов
был ацетон. В дальнейшем были освоены другие более слож-
ные кетоны, что позволило развивать производство и других
разновидностей сополимера.
Торгующие организации очень скоро обратили внимание
на то, что в большинстве областей применения выгодно пожерт-
вовать твердостью, химической стойкостью и другими свойст-
вами для повышения содержания сухого остатка. В соответст-
вии с этим было организовано производство винилита VYLF,
обладавшего низким молекулярным весом, удельной вязкостью
0,24 и почти полной растворимостью в ароматических углево-
дородах. При нормальной температуре (20°) можно получить
35%-ный раствор этого сополимера в смеси, состоящей из 107о
кетонов и 90% толуола. Добавление 60—100 вес. ч. сополимера
VYLF к раствору 100 вес. ч. винилита VYHH приводило лишь
* Пути совершенствования подобных полимеров изучал Паттон в лаборато-
риях фирмы Bakelite Corporation11.
Хлорсодержащие виниловые полимеры
169
к незначительному повышению вязкости раствора. В результате
получались покрытия с лучшим блеском, облегчалось нанесе-
ние лака, а при обработке бумаги достигалась более низкая
температура склеивания. Благодаря сходству в химическом
строении сополимеров сохранялась устойчивость покрытия к
химическим реагентам.
К сожалению, даже сополимеры этого типа имели плохую
адгезию к металлам и полированным поверхностям. Очень
ограниченная совместимость с другими полимерами уменьшала
возможность их применения совместно с другими продуктами,,
имеющими хорошую адгезию.
Некоторые успехи были достигнуты при применении фено-
ло-формальдегидных смол, диспергированных в тунговом мас-
ле (бакелит В К 3692). Эта смола по сравнению с другими су-
ществующими типами смол значительно более устойчива к рас-
творителям и может применяться для грунтовок (другие смолы,
пригодные для этой цели, растворяются при перекрытии лака-
ми на основе виниловых полимеров). Дальнейшее улучшение
было получено при смешении с акриловыми или метакриловы-
ми полимерами. Было найдено, что при добавлении в грунтовку
на основе винилового полимера некоторого количества пигмен-
та (примерно 100% от количества полимера) адгезия улучша-
лась, особенно, если вводили также небольшое количество сла-
бо совместимых смол, таких как глицериновый эфир канифоли,
модифицированный малеиновой кислотой. История этих усовер-
шенствований изложена в работе Дулиттла и Пауэла12.
При изучении этой проблемы установлено, что адгезия пле-
нок к металлическим поверхностям может быть значительно
улучшена при введении в молекулу некоторых функциональных
групп (остаток малеиновой кислоты), в то время как простое
смешение с веществами, содержащими полярные группы, не
дает хорошего результата. Сополимер винилхлорида с винил-
ацетатом, модифицированный малеиновой кислотой17, выпу-
скается под названием винилит VMCH.
Вскоре было найдено, что замена части винилацетатных
групп на гидроксильные приводит к получению продуктов,
имеющих значительно лучшую совместимость, в частности, с
парафином. Такой сополимер18 был выпущен под названием
винилит VAGH. Это позволило совмещать виниловые полимеры
с масляно-смоляными лаками или с алкидными смолами. При
уменьшении содержания винилхлорида в сополимере был по-
лучен продукт, совместимый с нитратом целлюлозы: винилит
VYGC с содержанием 65% винилхлорида и 35% винилацетата20.
Сополимеры с повышенным содержанием винилхлорида
(90% винилхлорида и 10% винилацетата) и более высоким мо-
лекулярным весом (относительная вязкость от 0,79 до 1,53>
170 Гл. IV. Виниловые полимеры
обладают лучшими физическими и химическими свойствами.
Это полимерьи VYNS, или дисперсионные полимеры21 VYNV-1,
VYNV-2, VYNV-3. Температура, при которой эти полимеры,
нормально пластифицированные и нанесенные на ткань, при-
обретают способность склеиваться под умеренным давлением,
равна приблизительно 70°.
Увеличивая содержание винилхлорида в сополимере до 95%,
можно повысить температуру склеивания до 105°, что приводит
также к повышению прочности толстых пленок на изгиб (от
300 тыс. до 3 млн. циклов для полимера VYDR)21. Понижая со-
держание винилхлорида до 91% и повышая молекулярный вес,
получают полимер типа винилит VYCM, имеющий примерно
те же физические свойства, что и полимер VYDR с 95% хлори-
стого винила, но с растворимостью, очень близкой к полимеру
VYNS, содержащему 90% хлористого винила.
За последние годы оптимальные свойства в отношении рас-
творимости были достигнуты при применении дисперсий поли-
меров в пластификаторах; эти дисперсии можно наносить прак-
тически без применения растворителей. Продукты, содержащие
только полимеры и другие компоненты, диспергированные в
пластификаторе, называются пластизолями, а смеси, содержа-
щие дополнительно летучие растворители,—органозолями.
Основная часть летучих компонентов, содержащихся в органо-
золях, испаряется при быстром нагревании; дальнейшее повы-
шение температуры приводит к растворению полимера в пласти-
фикаторах, что дает возможность получать сплошное непорис-
тое покрытие.
Производство этих полимеров начало развиваться в Герма-
нии (Игелит PCU тип Р), а затем в США (жеон 121 фирмы
В. F. Goodrich Chemical Со, винилиты VYNV-1 и VYNV-2 фирмы
Bakelite Corporation и т. д.). Английские фирмы производят
различные типы полимеров: вельвик (Imperial Chemical Indu-
stry) и разновидности жеона (Britisch Geon Ltd), поступаю-
щие в продажу под названием бреон. Во Франции фирма Kuhl-
mann изготовляет экавиль, а фирма Rhone-Poulenc —диспер-
сии под названием «пасты X».
В результате работ, проведенных фирмой Bakelite Division,
была создана целая гамма полимеров, приведенных в табл. 14.
Свойства сополимеров винилхлорида с винилацетатом. При-
водимые ниже данные относятся главным образом к винилиту
VYHH (Bakelite Corporation), поскольку другие полимеры
очень близки к нему по своим общим свойствам. Сополимер
VYLF (Bakelite Corporation) со значительно более низким мо-
лекулярным весом растворяется в очень многих растворите-
лях и дает более низковязкие растворы при том же сухом
остатке. Не будучи таким устойчивым, как винилит VYHH,
Хлорсодержащие виниловые полимеры
1Л
Сополимеры винилхлорида с винилацетатом
ТАБЛИЦА 14
Марка яродукта Примерный состав Удель- ная вязко- сть рас- твора в цикло- гекса- ноне при 20°) Удель- ный вес (при 20°) г см3 Растворимость (обычно применяемые концентрации)
винил- хлорид винил- ацетат о/ /о раств о ригель
VYCC (выпускается в виде 40%-ного раствора в н-бу- тилацетате) . . 62 38 0,28
VYLF 87 13 0,24
VYHH-1 .... 87 13 0,53
VMCH 86 13* 0,53
VAGH 91 3** 0,57
VYNS-3 .... 90 10 0,79
VYCM ..... 91 9 1,38
VYDR (ранее вы- пускался под маркой VYNW-2) 95 5 1,25
VYNV-1 .... >95 <5 1,53
VYNV-2 .... >95 <5 1,52
NV-4 ..... >92 <8 —
1,30 40 н-Бутилацетат
1,34 35 Кетон-толуол 1 :5
1,35 20 Кетон-толуол 1 :1
1,36 20 То же
1,39 20 »
1,36 18 (на холоду) Метилэтилкетон
32*** (при нагре- вании) >
1,37 6 (на холоду) »
20*** »
(при нагре- вании)
1,39 14 Циклогексанои
14 (при нагре- вании) Метилэтилкетон
1,39 35 Диизобутилкетон- ксилол (29 80)
1,39 50 Диоктилфталат
— 36 Диоктилфталат— вода (28 : 72)
* Сополимер содержит 0,7—0,8% карбоксильных групп (1% двухосновной кислоты).
** Сополимер содержит 2,3% гидроксильных групп, что соответствует 6% поливинилового
спирта.
*** Желатинирует при охлаждении.
этот продукт не применяется самостоятельно в качестве основы
для лаков. Очень близки к марке VYHH также продукты, вы-
рабатываемые во Франции (родопас АХ фирмы Rhone Poulenc
и афковил СТ-3 фирмы Pechiney). Родопас АХ представляет
•собой сополимер винилхлорида и винилацетата, содержащий
85% винилхлорида, афковил СТ-3 — продукт того же типа.
172
Гл. IV. Виниловые полимеры
Адгезия пленок.. На большинстве поверхностей лучшая адге-
зия пленок из обычных сополимеров винилхлорида и винилаце-
тата достигается путем горячей сушки при температурах, до-
статочных для удаления остатков растворителя и для расплав-
ления полимера. При этой операции используются преимущества
термопластичное™ смол.
Для полированных поверхностей (металлы, стекло, слоистые
покрытия) горячая сушка обязательна. На большинстве по-
ристых или шероховатых поверхностей удовлетворительную
адгезию пленок можно получить при воздушной сушке. Это
особенно относится к тканям, бумаге и поверхностям строитель-
ных материалов.
Очень большое значение имеет степень пигментирования
грунтов и покрывных слоев: на некоторых типах грунтов можно
добиться хорошей адгезии, нанося сильно пигментированный
промежуточный слой. Тем не менее, в большинстве случаев,
результаты, достигаемые при горячей сушке, всегда превос-
ходят получаемые при воздушной сушке. Важно нагреть плен-
ку до 160°, чтобы полимер расплавился. Продолжительность
нагревания зависит от размеров и материала изделия.
До указанной температуры должна обязательно нагреться
сама пленка. Более длительное нагревание при более низкой
температуре, как это обычно практикуется для других типов
покрытий, в данном случае не дает эффекта. Для достижения
хорошей адгезии к мягкой обыкновенной стали, олову, меди,
бронзе, к поверхностям, металлизированным гальваническим
способом, достаточно 30—60 мин. нагревания при 170°. Те же
условия применимы также и к стеклу. Для алюминия требуется
температура не ниже 185°, но в этом случае приходится счи-
таться со стабильностью смолы (вопросы стабилизации будут
рассмотрены в специальной главе). Нестабилизованный продукт
не может выдержать такую температуру, в особенности, нахо-
дясь в соприкосновении с металлом, ускоряющим процесс раз-
ложения.
Очень тонкие пленки (0,02 мм или менее), нанесенные на
олово или сталь, не изменяют цвета, но при нанесении более
толстых пленок на химически активные поверхности нужно
принимать тщательные меры предосторожности. Эти предосто-
рожности могут быть разными;
1) добавление стабилизаторов с целью замедлить процесс
разложения полимера;
2) нанесение полимеризованного масла или грунта на
основе синтетической смолы для изоляции поверхности от плен-
ки сополимера винилхлорида и винилацетата;
3) обработка металла с целью пассивирования его поверх-
ности.
Хлорсодержащие виниловые полимеры
173
Когда на предварительно прогретый в печи первый слой по-
лимера наносится второй слой, достаточно нагревания до тем-
пературы, требуемой для полного испарения растворителей.
Если в состав покрытия входят низкомолекулярные полимеры,
такие как винилит VYLF, верхний слой перед горячей сушкой
можно шлифовать. Благодаря присутствию низкомолекуляр-
ного полимера пленка при нагревании приобретает текучесть,
следы шлифовки во время сушки затекают и достигается очень
высокий блеск покрытия.
Кроме описанных выше основных продуктов, за последние
годы 1ПОЯ'Вились другие сополимеры, которые мы рассмотрим до
перехода к модифицированным полимерам специального назна-
чения.
Сополимеры винилхлорида с винилиденхлоридом
Растворимость этих продуктов была повышена путем увели-
чения количества винилиденхлорида по отношению к винилхло-
риду. Эти продукты выпускаются фирмой В. F. Goodrich Chemi-
cal под названием жеон 200 X 20. Они отличаются от виниловых
полимеров, имеющихся в настоящее время в продаже, способ-
ностью непосредственно растворяться в толуоле и ксилоле,
сольвент-нафте и других углеводородах. При медленном введе-
нии полимера в растворитель при обычной температуре и интен-
сивном механическом перемешивании можно получать рас-
творы с содержанием сухого остатка до 50%.
Интересно отметить, что растворы, даже с высоким содер-
жанием сухого вещества, не желатинируются при охлаждении
до 0° и восстанавливают свою первоначальную вязкость при
последующем нагревании до комнатной температуры.
Сополимер винилхлорида • и винилиденхлорида с высоким
содержанием последнего (марка жеон 200x20) выпускается в
виде тонкого белого порошка со следующими показателями:
Прохождение через сито № 100 (США), %............ 80
Относительная вязкость (0,4%-ный раствор в нитро-
бензоле) ......................................0,17
Вязкость раствора в толуоле при 25°, сантипуазы
20% сухого вещества............................ . 41
30% » » . . . 285
40 % » » ...................... 2000
Содержание нерастворимого остатка в смеси толуол-
метилэтилкетон 90: 10, %, не более .............0,3
Удельный вес пленки, г/см3.......................0,54
Обычными способами нанесения растворов являются пуль-
веризация, нанесение кистью, накатка валиком, а при высоких
174
Гл. IV. Виниловые полимеры
вязкостях — намазывание штапелем или раклей. Растворам по-
лимера жеон 200X20 с содержанием до 30% сухого остатка
легко могут быть приданы свойства, требуемые при том или
ином способе нанесения: .большая скорость высыхания или
лучшая растекаемость. Растворы в ароматических углеводоро-
дах можно разбавлять в любом соотношении кетонами, в не-
сколько меньшем количестве — сложными эфирами и ограни-
ченно — алифатическими углеводородами. Спирты также в из-
вестных пределах могут применяться как разбавители.
На рис. 32 приведена вязкость сополимеров жеон 200 X 20,
растворенных в некоторых товарных растворителях, как функ-
ция от процентного содержания сухого остатка.
Приведем некоторые основные характеристики пленки жеон
200 X 20 (толщина 0,1 мм)-.
Предел прочности при растяжении, кг/см2 . . . .........316—386
Удлинение при разрыве, %................................. . 0—10
Сопротивление раздиру (по Гревсу), кг!см .............. 125—178
Истирание по Таберу (потеря веса после 500 оборотов), г 0,006
Огнестойкость ...............................................Не горит
Пластификаторы применяются те же, что для сополимеров
с винилацетатом, но благодаря заутренней пластификации,
происходящей при наличии винилиденхлорида. количество пла-
стификатора может не г.оевынщ;и 13%. Стабилизаторы должны
применяться в больших количествах, чем для сополимеров ви-
нилхлорида и винил ацетата, так как винилиденхлорид пони-
жает стабильность сополимера. Учитывая значительную спо-
ТАБЛИЦА 15
Механические свойства пластифицированных пленок сополимера винилхлорида
с вниилиденхлоридом
Примененный пластификатор Предел прочности при растяжении кг/см^ Удлинение при разрыве % Сопротивление раздиру (по Гревсу) К2;СМ
Трибутилацетоцитрат 74 500 27
Дутрекс 141 275 98
Флексол TOF (триоктилфосфат) 148 325 67
Герколин 167 200 70
Санти цайзер В-16 . . 167 215 63
Адиполь ВСА ... 167 350 85
Моноплекс DOS (диоктилсебацинат) 171 200 130
Параплекс G-25 176 200 70
Параплекс G-40 183 150 63
Сантицайзер 141 204 85 178
Бальцер Р4С . . 211 275 76
Флексол 4GO 218 275 125
Хлорсодержащие виниловые сополимеры
175
собность сополимеров винилхлорида с винилиденхлоридом к
поглощению пластификаторов, не рекомендуется применять их
в качестве верхнего покрытия по пластифицированным вини-
ловым полимерам, так как при этом происходит миграция пла-
стификатора из нижнего слоя в верхний.
Состав смеси растворителей, %
Рис. 32. Вязкость растворов сополимеров жеон 200x20:
1—толуол-ксн лол; 2-толуол-ацетон; 3—-толуол-метилизобутилке-
тон; 4—толуол-метилэтилкетон.
В табл. 15 приведены механические свойства пленок, содер-
жащих 95% сополимера и 5% различных пластификаторов и
полученных из 35%-ных растворов в толуоле. Испытания про-
водились после сушки в течение 4 час. на воздухе и 1 час при
100°.
Хорошая адгезия пленок жеон 200 X 20, как и сополимеров
винилхлорида и винилацетата, достигается путем горячей суш-
176
Гл. IV. Виниловые полимеры
ки окрашенного изделия при температуре 150—190° в продол-
жение 3—5 мин.
Сополимер совместим во всех отношениях с синтетическими
каучуками типа GRA (паракрил-хайкар) и лишь ограниченно
совместим с некоторыми алкидными и карбамидными смолами,
.с касторовым маслом.
МОДИФИЦИРОВАННЫЕ ВИНИЛОВЫЕ СОПОЛИМЕРЫ
Винилит VMCH
Основной недостаток полимера VYHH (жеона 200X20) за-
ключается в необходимости высокотемпературного нагрева для
достижения удовлетворительной адгезии к металлическим по-
верхностям. Поэтому уже давно ощущалась потребность в свя-
зующем, которое, будучи нанесено непосредственно на металл,
давало бы пленки с достаточной адгезией при воздушной
сушке. Для получения такого связующего пытались смешивать
сополимер описанного выше типа с феноло-формальдегидными
.смолами, диспергированными в маслах, например со смолой
ВК 3692 фирмы Bakelite Corporation (эта смола значительно
менее подвержена действию растворителей, чем другие подоб-
ные смолы).
Адгезию удалось улучшить путем смешения сополимеров с
метакрилатами или дезакрилатами, содержание которых дости-
гало 100% от веса винилового сополимера. Улучшают адге-
зию также небольшие количества частично совместимых про-
дуктов, таких, как эфиры канифоли, модифицированные ма-
.леиновой кислотой, но в этом направлении 'проведены лишь
первые опыты, и полученные результаты еще далеко не удовле-
творительны.
При исследовании сополимеров винилхлорида с винилацета-
том было найдено, что адгезию к металлическим поверхностям
пленок, высушиваемых на воздухе, можно улучшить путем вве-
дения химических групп, непосредствено реагирующих с поверх-
ностью металла; однако введение в сополимер высокополярных
групп, обладающих этим свойством, вызвало затруднения.
Вследствие большой длины цепи сополимера происходило
ухудшение растворимости и понижение способности к ориен-
тации; усадка пленки при сушке повышалась настолько, что
местами пленка отслаивалась. Поэтому скоро пришли к заклю-
чению, что полярные группы следует вводить непосредственно
в смолу.
Дальнейшие исследования привели к получению сополимера
с очень малым количеством карбоксильных групп, имевшего
после сушки на воздухе значительно лучшую адгезию к метал-
Модифицированные виниловые сополимеры
V71
лическим поверхностям (работа Дулиттла и Пауэла12, амери-
канский патент Кэмпбелла13).
Был намечен полупроизводственный выпуск сополимера, со-
держащего 82% винилхлорида и 0,3% малеиновой кислоты,
остальное — винилацетат. Молекулярный вес этого продукта14,
определенный по методу Штаудингера, составлял около 10 000.
Возможность полимеризации малеиновой кислоты с винило-
выми производными не явилась новостью, так как подобный
процесс был запатентован Фоссом и Дикхаузером15, но вопрос
о влиянии малых количеств малеиновой кислоты на адгезию
сополимера к металлическим поверхностям ранее не подвергал-
ся достаточно глубокому исследованию.
В дальнейшем были предприняты тщательные исследования
явления адгезии, в частности Зельденом и Пруттоном16. Эти
авторы показали, что карбоксильные группы благодаря явле-
нию адсорбции увеличивают адгезию, но при введении их в
слишком большом количестве адгезия ухудшается вследствие
химического взаимодействия этих групп с поверхностью метал-
ла. Улучшение адгезии у модифицированных смол безусловно
вызвано присутствием в полимере активных карбоксильных
групп; доказательством служит полная потеря адгезии при
нейтрализации этих групп.
Диэфиры малеиновой кислоты обусловливают очень слабую
адгезию, в то время как эфиры, подобные мономалеиновым, по
эффективности почти не уступают самой кислоте.
Тот же эффект был получен при сополимеризации с очень
малыми количествами других ненасыщенных кислот; однако
во’ всех случаях карбоксильные группы должны оставаться
свободными.
Исследования целой гаммы образцов показали, что для
обеспечения хорошей адгезии к стали при воздушной сушке
требуется не менее 0,25—0,30% малеиновой кислоты. Явное
улучшение адгезии отмечается уже при 0,1%, а при высоком
содержании малеиновой кислоты (5—<10%) появляются ослож-
нения (коррозия тары, слабая тепло-, свето- и водостойкость).
В результате большого количества опытов, проводившихся в
течение нескольких лет, было установлено, что оптимальным
вариантом является продукт сополимеризации с 1% малеино-
вой кислоты, который к тому же совместим с немодифицирован-
ны.ми смолами.
Такой модифицированный сополимер выпускается фирмой
Bakelite Corporation под названием винилит VMCH. Его состав
(в %):
Винилхлорид ...............................86,00
Винилацетат................................13,00
Малеиновая кислота ........................ 1,00
12—156
178
Гл. IV. Виниловые полимеры
Наиболее важным свойством этого 'сополимера является
адгезия к металлическим поверхностям после очень кратковре-
меной сушки; адгезия становится еще лучше при несколько по-
вышенной температуре, которая ускоряет ’испарение раствори-
телей и повышает твердость поверхностной пленки. Другие пока-
затели этого продукта — вязкость растворов, растворимость,
устойчивость пленок по отношению к кислотам, щелочам и воз-
действию воздуха те же, что у винилитов типа VYHH. Однако
пленки из винилита VMCH менее устойчивы к свету и теплу,
чем пленки, полученные из материала VYHH.
Способность растворов этих сополимеров вызывать корро-
зию стальных или луженых резервуаров была предметом мно-
гих исследований. Сама коррозия фактически ничтожна, но тем
не менее она очень заметна для растворов в метилэтилкетоне
или ацетоне. Поэтому нельзя пользоваться для этих растворов
тарой из черных металлов; тара из луженого железа очень мед-
ленно подвергается коррозии. К растворам подобных сополи-
меров, с целью избежать изменения их цвета и коррозии тары
из листовой стали, рекомендуется добавлять очень небольшие
количества окиси пропилена. Стеклянная тара, конечно, вполне
пригодна.
Вопрос о коррозии должен быть тщательно изучен, так как
адгезия пленок, полученных из растворов,, которые хранились
в таре, подвергавшейся коррозии, значительно ухудшается, а
иногда и вовсе теряется.
Вследствие присутствия свободных карбоксильных групп,
растворы этих полимеров имеют тенденцию вступать в реакцию
с. основными пигментами, что ведет к образованию необрати-
мых гелей. Поэтому не рекомендуется вводить в композиции
сильно основные пигменты; если же их все-таки приходится
употреблять, следует вводить специальные добавки: малеино-
вую кислоту, дифенилгуанид'ин и особенно монобутилфосфат.
Чтобы избежать всякой возможности реакции с пигментом,
лучше применять немодифицированные полимеры типа хлораце-
тата. Полученный таким образом раствор добавляется к рас-
твору смолы VMCH на месте применения.
Адгезионные свойства винилита VMCH. Адгезия этого по-
лимера к стали при воздушной сушке очень хорошая, в то
время как для материала VYHH и других полимеров этого
типа наблюдается слабая адгезия. После нагревания до 110°
адгезия винилита VMCH не изменяется, тогда как адгезия ма-
териалов типа VYHH остается все еще очень слабой. После
сушки при температуре 170° оба материала имеют примерно
одинаковые адгезионные свойства.
Превосходная адгезия достигается на всех металлах, содер-
жащих железо, на кадмии, олове, меди. На цинке, напротив,
Модифицированные виниловые сополимеры
179
адгезия со временем может сильно ослабляться вследствие бы-
строго взаимодействия модифицированного полимера с этим
металлом. В этом случае было предложено предварительно
обрабатывать металл водным раствором фосфорной кислоты.
Адгезия к дереву лишь немногим лучше, чем у обычного сопо-
лимера. Напротив адгезия к камню, бумаге, тканям значитель-
но лучше.
Условия применения. Для покрытий воздушной сушки реко-
мендуется применять смеси равных количеств винилита VMCH
и 1немодифицированного сополимера. Можно также применять
большее количество смолы VMCH, но при этом адгезия замет-
но не улучшается. Если сушка проводится при невысокой тем-
пературе, то при соотношении 1 вес. ч. винилита VMCH на
4—5 вес. ч. немодифицированного сополимера получаются
пленки с удовлетворительной адгезией к большинству подло-
жек.
Эти соображения относятся, конечно, к грунтам, так как для
промежуточных .и поверхностных слоев покрытия не требуется
применять модифицированный сополимер.
Устойчивость пленок к растворителям может быть значи-
тельно улучшена при смешивании полимера с желтым или
оранжевым кроном, с некоторыми солями металлов и с неко-
торыми смолами типа меламино-мочевино-альдегидных.
Модифицированный сополимер рекомендуют как грунт для
последующего нанесения обычных виниловых полимеров.
В противоположность применяемым в качестве грунтов сополи-
мерам винилхлорида с винилацетатом, в которые рекомендует-
ся вводить пигменты, адгезия смолы VMCH не улучшается при
таких добавках. Ни в коем случае не следует увеличивать ко-
личество пигмента свыше 20—25% объемн. по отношению к
сухой пленке.
Если в качестве антикоррозионных агентов в грунты на
основе винилита VMCH вводят хроматы цинка и стронция,
окиси цинка или железа, не рекомендуется повышать содержа-
ние этих пигментов сверх 50% вес. (10—15% объемн. по отно-
шению к сухой пленке). При введении пигментов не следует
забывать о возможности желатинирования, поэтому предпочти-
тельнее такие пигменты, как двуокись титана, ортофосфат свин-
ца, а также пигменты с нейтральной или слабокислой реакцией.
Потребители должны <по возможности использовать только
инертные пигменты; если же это невозможно, раствор немоди-
фицированного полимера, содержащий пигмент, надо смешивать
с раствором винилита VMCH непосредственно перед употреб-
лением.
Наиболее эффективными стабилизаторами, оказывающими
тем не менее лишь временное действие, являются монобутил-
12*
180 Гл. IV. Виниловые полимеры
фосфат и аммиак (особенно, если его применять в растворе раз-
бавителя) .
Для грунтов горячей сушки следует предпочесть малеино-
вую кислоту или дифенилгуанидин, так как в этом случае явно
достигается лучшая адгезия. Другой способ состоит в блоки-
ровании карбоксильных групп такими веществами, как основ-
ной сульфат свинца: реакция, протекающая при нагревании до
150—160°, .ведет к получению менее растворимых пленок. Удо-
влетворительный результат дают также соли других металлов
и смолы типа мочевино-формальдегидных. Добавка 10—20 вес. ч
такой типа смолы к 100 вес. ч. винилита VMGH дает после
нагревания удовлетворительные результаты. Здесь, по-види-
мому, происходит взаимодействие карбоксильных групп с моче-
винными смолами. Водостойкость таких продуктов, а также
продуктов, модифицированных меламино-формальдегидными
смолами, улучшается добавлением к ним небольших количеств
(0,2%) фосфорной кислоты, которая, по всей вероятности, мо-
жет играть роль катализатора в протекающих сложных реак-
циях.
Винилит VAGH.
Путем сополимеризации с мономерами, содержащими гидр-
оксильные группы, был получен продукт марки VAGH прибли-
зительного состава (в %):
Винилхлорид................................91
Винилацетат................................ 3
Виниловый спирт............................ 6
Этот модифицированный сополимер, содержащий около
2,3% гидроксильных групп, по большинству свойств сходен
с винилитом VYHH; введение гидроксильных групп обус-
ловливает лучшую совместимость, значительно лучшую адге-
зию, более высокую температуру размягчения, а также повы-
шенную реакционную способность.
Растворимость. Винилит VAGH растворяется в растворите-
лях того же типа, что и обычные сополимеры винилхлорида с
винилацетатом, но растворы его имеют несколько более высо-
кую вязкость. Благодаря присутствию гидроксильных групп
растворы полимера могут быть разбавлены спиртами, но они
более чувствительны к алифатическим углеводородам. Можно
получать растворы 10%-ной концентрации при 25° в смеси рас-
творителей: метилизобутилкетон (25%), безводный изопропи-
ловый спирт (25%), толуол (50%). Толуол может быть ча-
стично заменен алифатическими углеводородами, но в этом слу-
чае для получения 10%-ного раствора необходимо нагревание
до 50°.
Модифицированные виниловые сополимеры
181
В качестве примера приведем следующий состав смеси рас-
творителей (в %):
Метил изобут илкетон.................25
Изопропиловый спирт (бесцветный) . . 25
Толуол...............................25
«Тролуойль»..........................25
Растворимость гидроксил содержащих полимеров значитель-
но лучше в алифатических растворителях (несовместимых или
слабо совместимых с обычными виниловыми полимерами). По-
этому при смешивании растворов винилита VAGH с раство-
рами обычных полимеров необходимо соблюдать предосторож-
ности.
Совместимость. Полимер VAGH совместим с большим ко-
личеством продуктов, в частности с необработанными маслами
или сиккативированными олифами, с алкидными смолами. Тем
не менее совместимость его с некоторыми сортами льняного
масла не очень хороша, в то время как совместимость с сырым
или дегидратированным касторовым маслом превосходная.
Температура размягчения. Этот полимер имеет более высо-
кую температуру размягчения, чем полимер типа VYHH.
В обычных условиях температура размягчения непластифици-
рованного полимера VYHH равна «примерно 80°, а для VAGH
она составляет 100°. После пластификации путем введения 25%
диоктилфаталата температура размягчения понижается соот-
ветственно до 65° и 80°.
Пластификация. Пленки на основе VAGH требуют для до-
стижения одинаковой степени эластичности несколько больше
пластификатора, чем пленка на основе VYHH. Найдено, что
для получения пленок одинаковой эластичности на 100 вес. ч.
полимера VYHH требуется 25 вес. ч. диоктилфталата, тогда как
для полимера VAGH необходимо ввести 30 вес. ч. того же
пл а стифика тор а.
Водостойкость. Водостойкость пленок на основе VAGH лишь
ненамного ниже, чем пленок на основе VYHH, несмотря на
присутствие гидроксильных групп. Однако можно получать зна-
чительно менее водопроницаемые пленки путем добавления
восков, совместимых с полимером VAGH.
Пигментирование. Для этого типа полимеров могут приме-
няться те же пигменты, что и для винилита VYHH.
Адгезия. Адгезия к металлам покрытий на основе VAGH
значительно лучше адгезии обычных виниловых сополимеров, но
основное достоинство этого материала состоит в том, что он
придает превосходную адгезию фосфатирующим грунтам. Эти
грунты были разработаны во время второй мировой войны в
США, где выпускались под названием Wash Primers.
182
Гл. IV. Виниловые полимеры
Придание нерастворимости. Нерастворимость этим полиме-
рам можно придать путем связывания гидроксильных групп.
Совместимость их с термореактивными смолами дает возмож-
ность получать пленки с очень ценными свойствами. Наиболее
простой способ состоит в смешении модифицированного поли-
мера с термореактивными смолами (от 5% до 10% мочевияо-
форм альдегидной, феноло-формальдегидной, меламино-формаль-
дегидной и др.). Полезно применять в этих случаях раствори-
тели типа целлозольва.
Нерастворимость достигается также при действии аминов:
полиаминов, продуктов конденсации аминобензальдегида, уско-
рителей вулканизации каучука (например, 808—833, гептил-
анилин с тиурамом). К сожалению, получаемые продукты окра-
шены. В большинстве случаев для отверждения достаточно
обработки в течение одного часа при температуре 160°.
Интересные результаты, в частности, дает реакция с гекса-
метилендиизоцианатом, добавление которого в небольших ко-
личествах может привести к образованию почти нерастворимых
пленок при воздушной сушке. К сожалению, высокая реакцион-
ная способность изоцианатов затрудняет их практическое при-
менение.
Следует остановиться еше на реакции с производными гли-
цидиловых эфиров (прототипом является стабилизатор А-5 фир-
мы Carbide and Carbon Corporation). Гидроксилсодержащий
виниловый полимер, модифицированный эфирами этого типа
(5—40%) и таким же количеством смолы типа мочевино-фор-
1мальдегидной, после быстрого нагревания образует теплостой-
кие, очень твердые и неистирающиеся покрытия. Полученные
таким способом покрытия значительно более устойчивы к горя-
чей воде и пару, чем большинство виниловых покрытий.
На основе материала VAGH можно получать разнообразные
покрытия, так как эти полимеры совмещаются с алкидными
смолами и маслами. После воздушной сушки достигается хо-
рошая адгезия к грунтам на основе шеллака; она может быть
еще улучшена путем добавления некоторых смол. Улучшенной
полируемости можно добиться при добавлении эфиров кани-
фоли и малеиновых производных.
Несомненно, однако, что основное назначение полимера
VAGH — введение в фосфатирующие грунты (Wash Primers).
Такой грунт, обладающий исключительно высокой адгезией, и
пассивирующий металл, был получен при применении раство-
ров (полимеров типа поливинилбутираля (полимер XYHL) с
хроматом цинка (в нерастворимой форме) и фосфорной кис-
лотой.
Фосфатирующий грунт, нанесенный в виде чрезвычайно тон-
кого слоя (—1 yr), проявляет исключительную адгезию почти ко
Модифицированные виниловые сополимеры
183
всем металлам. Было установлено, что на таком слое грунта
пленки на основе винилита VAGH держатся очень хорошо. Это
позволило создать краску для подводной части судов, значитель-
но превосходящую по качеству все другие типы защитных по-
крытий, применяемых в настоящее время. В практике амери-
канской морской службы по слою фосфатирующего грунта нано-
сят промежуточный слой на основе винилита VAGH, а затем —
специальное необрастающее покрытие. Эти система имеет пре-
восходную адгезию, высокую твердость и длительный срок
службы. Суда, защищенные таким способом, не подвергаются
никаким изменениям в течение многих лет.
Покрытия для дерева. Полимер VAGH прекрасно прилипает
при нанесении на всевозможные подслои, за исключением нит-
роцеллюлозных.
Покрытия для бумаги. Очень высокая адгезия, полимеров
типа VAGH к печатным краскам представляет большой инте-
рес, так как дает возможность защищать нанесенные на бумагу
масляные печатные краски покрытиями на основе этих полиме-
ров. Совместимость полимера VAGH с винилацетатом позволяет
улучшить свойства покрытий для бумаги.
Следует отметить также явно выраженное улучшение свойств
алкидных смол при добавлении к ним небольших количеств
полимеров этого типа. При этом сокращается время сушки,
повышается твердость пленок, их эластичность, особенно в
процессе формования, улучшается водо- и щелочестойкость.
Однако нельзя забывать, что винилит VAGH особо чувстви-
телен к алифатическим растворителям, поэтому при составле-
нии композиций следует соблюдать предосторожности. Если
применен раствор алкидных смол в алифатических раствори-
телях, необходимо вводить растворитель, активный по отно-
шению к виниловым полимерам; этот растворитель должен со-
держать небольшое количество компонента с более высокой
точкой кипения с тем, чтобы на последних стадиях сушки в
пленке оставалось вещество, являющееся растворителем для
полимера VAGH.
Сополимеры с высоким содержанием винилацетата
Проведенные исследования показали, что поливинилацетат
в известных условиях совместим с нитроцеллюлозными лаками,
улучшая в отдельных случаях их общие свойства. Такие совме-
щенные продукты несколько чувствительны к воде и нестойки
к некоторым химическим реагентам, вследствие чего их при-
менение ограничено.
Фирмой Carbide and Carbon Corporation разработан сопо-
лимер, выпускаемый под маркой винилит VYCC, в виде
184 Гл. IV. Виниловые полимеры
40%-ного раствора в бутил ацетате. Это сополимер, содержа-
щий 62% винилхлорида и 38% винилацетата.
Была специально исследована возможность применения
этого материала в сочетании с нитроэмалями. Оказалось, что
эмали, «подкрепленные» таким способом, обладают хорошей
атмосферостойкостью при продолжительных испытаниях в тя-
желых условиях. Второй отличительной чертой эмалевых кра-
сок на основе винилита VYCC является повышенная стой-
кость к поту, к спиртам и коррозионноактивным агентам.
Важной областью применения винилита VYCC является ис-
пользование его в качестве основы красок для печатания на
хлорвиниловых пленках, а также для промышленных покрытий.
Общие свойства. Пленки, получаемые на основе винилита
VYCC, прозрачны и бесцветны. Их инертность по отношению к
химическим реагентам примерно такая же, как у остальных
рассмотренных полимеров. Температура размягчения немного
ниже, чем у винилита VYHH, .но тепло- и светостойкость почти
одинаковы.
Материал VYCC испытывался вначале с целью повышения
качества нитроцеллюлозных эмалей. Он имеет более низкий мо-
лекулярный вес (близкий к молекулярному весу полимеров ти-
па VYLF) и поэтому пленки, получаемые из одного винилита
VYCC, обладают некоторой хрупкостью. Этот полимер совме-
стим с продажными типами нитрата целлюлозы; так как в су-
хих пленках может появляться легкое помутнение, рекомендует-
ся вводить небольшое количество (5—10%) пластификатора ти-
па сложных эфиров. Поскольку в большинство эмалевых красок
в действительности приходится вводить более Значительные ко-
личества пластификатора, это не является существенным недо-
статком. Совместные растворы с нитратом целлюлозы совер-
шенно стабильны в течение очень продолжительного времени.
Винилит VYCC совместим с другими сополимерами, а также
с большинством .полимеров типа акриловых и метакриловых
эфиров, но несовместим с поливинилбутиралем и поливинилаце-
татами. Он несовместим или ограниченно совместим со слож-
ными и простыми эфирами целлюлозы, с алкидными смолами,
модифицированными высыхающими и невысыхающими мас-
лами, с мочевино-меламиновыми и большинством феноло-форм-
альдегидных смол, с глицериновыми эфирами канифоли и хло-
рированными каучуками. Можно с успехом применять для его
пластификации большинство известных пластификаторов, за
исключением касторового масла и его производных.
Растворы этого полимера можно разбавлять толуолом и
ксилолом до бесконечности, не опасаясь выпадения полимера.
Практически большая часть растворителей активна по отноше-
нию к этому материалу и может добавляться для разбавления.
Растворители и вспомогательные материалы
185
Применение. Основное назначение этого полимера — повы-
шение атмосферостойкости красок для внешних покрытий.
РАСТВОРИТЕЛИ И ВСПОМОГАТЕЛЬНЫЕ МАТЕРИАЛЫ,
ПРИМЕНЯЕМЫЕ СОВМЕСТНО С ВИНИЛОВЫМИ ПОЛИМЕРАМИ
Виниловые полимеры обычно применяются в виде компози-
ций, содержащих кроме полимера следующие вспомогательные
материалы: 1) стабилизаторы; 2) пластификаторы; 3) раство-
рители; 4) пигменты.
Стабилизаторы. При применении большей части рассмотрен-
ных виниловых полимеров необходимо вводить специальные
стабилизаторы, чтобы предотвратить разложение полимера под
действием света, тепла или соприкасающихся с пленками ме-
таллов. Вопросы стабилизации поливинилхлорида и виниловых
сополимеров имеют исключительно большое значение, потому
что стабильность определяет ценность торговых продуктов22—200
Разложение поливинилхлорида может быть представлено
схематически следующим образом (Фокс, Генрике и Ретти23).
Под действием света и тепла поливинилхлорид (I) подвергает-
ся разложению, приводящему к отщеплению соляной кислоты:
и возникновению двойных связей в цепях (II). Последующее
действие кислорода приводит к образованию продукта, имею-
щего строение III:
нннн нннн нннн
Здесь рассмотрен частный случай разложения поливинил-
хлорида, но другие виниловые сополимеры претерпевают ана-
логичные изменения. При их разложении могут образоваться
также многие другие вещества, но для простоты мы ограничи-
ваемся рассмотрением двух приведенных типов конечных про-
дуктов.
При действии только тепла около 70% конечного продукта
получаются в форме II и 30% в форме III, тогда как при дей-
ствии только света эти соотношения несколько изменяются.
Так как соединения типа II способны действовать каталитиче-
ски на процессы разложения или превращения, то изменения в-
системе происходят очень быстро.
Соединения типа III обычно мало окрашены, но они обра-
зуют разные модификации в зависимости от типа пластифика-
тора. Иногда продукт после старения склонен к размягче-
186
Г л. IV. Виниловые полимеры
нию, в других случаях, напротив, становится чрезвычайно хруп-
ким.
Стабилизация хлорсодержащих виниловых полимеров сла-
гается из стабилизации к действию тепла, света, предотвраще-
ния окисления и стабилизации диэлектрических свойств. Типо-
вой стабилизатор должен обладать способностью поглощать со-
ляную кислоту, придавать полимеру' непроницаемость для уль-.
трафиолетовых лучей, действовать в качестве антиокислителя,
легко диспергироваться.
Применяемые стабилизаторы в большинстве случаев пред-
ставляют собой слабые основания, соли слабых кислот или
такие соединения металлов, которые могут реагировать с соля-
ной кислотой. Выбор стабилизатора в значительной мере опре-
' деляется его ценой и цветом.
Следует отметить, что действие стабилизатора является из-
бирательным по отношению к пластификаторам, содержащим-
ся в смеси: стабилизатор может давать хорошие результаты
для одного типа пластификатора и оказаться непригодным при
другом типе пластификатора.
Пигментированный грунт, содержащий соли свинца (фосфо-
ристые, сернокислые или даже ортосиликат) в количестве 3%
по отношению к полимеру, приобретает способность выдержи-
вать действие температуры 180° в течение 1 часа и более без
всяких признаков разложения па любых металлических по-
верхностях.
Соли жирных, кислот. Большая часть стабилизаторов пред-
ставляет собой соли жирных кислот. Первостепенную роль в
отношении стабилизирующего действия, естественно, должен
играть катион, но природа аниона также сказывается на цве-
те, запахе, водостойкости и даже собственно на стабильности
полимера. Как правило, можно считать, что жирные кислоты
с низким молекулярным весом (масляная, пропионовая) по-
нижают водостойкость; соли нафтеновых кислот способствуют
получению прозрачных смесей.
По данным Салли и Ганзена (фирма Ferro Chemicals Corpo-
ration1), при сравнении разных солей одного и того же металла
оказывается, что на стабилизирующее действие влияет при-
сутствие гидроксильных групп и двойных связей в кислотном
остатке. Так, из кадмиевых солей олеиновой, стеариновой,
оксистеариновой, рпцинолевой кислот (все они содержат 18 уг-
леродных атомов) наилучшую стабильность при действии вы-
сокой температуры дает рицинолеат, который содержит гидр-
оксильную группу и двойную связь. Эти наблюдения были уч-
тены при разработке стабилизаторов на основе солей бария,
стронция, свинца, кальция, олова. Что касается солей олова,
то здесь имеются в виду образованные жирными кислотами
Растворители и вспомогательные материалы
187
соли диалкилолова, представителем которых является дилаурат
дибутилолова (в его молекуле две валентности четырехвалент-
ного атома олова насыщены двумя алкильными группами, а
две другие — остатками органических кислот). Эти стабилиза-
торы могут реагировать с соляной кислотой, образуя хлорид
диалкилолова и освобождая при этом органическую кислоту,
которая «вытесняется» на поверхность (это особенно заметно
при разложении хлорсодержащих полимеров под действием
тепла или света). Главный из стабилизаторов этого типа — ди-
лаурат дибутилолова—придает конечным продуктам запах
лауриновой кислоты; диацетат дибутилолова сообщает им от-
четливый запах уксусной кислоты и придает мутность.
При совместном применении в качестве стабилизаторов со-
лей двух или более разных металлов при повышенной темпера-
туре в большинстве случаев набюдается значительное синэрги-
ческое действие. Так, работы фирмы Ferro Chemicals показали,
в частности, что при введении в качестве стабилизатора 3% ри-
цинолеата бария появление заметной окраски образца, (подвер-
гнутого действию высокой .температуры (метод A. S.T. М.), на-
блюдалось через 400 мин. (то же время зафиксировано и для
рицинолеата кадмия). Однако, если взять */« соли бария и 3Д
соли кадмия, то время увеличивается до 900 мин.
В отношении светостойкости приводились весьма различ-
ные данные. Можно только указать, что стабильность систем,
стабилизованных производными кадмия, пропорциональна ко-
личеству взятого кадмия.
Соли минеральных кислот. Эти соли при использовании их
в качестве стабилизаторов ведут себя по-разному. Так, из сред-
них солей слабых кислот (карбонат или силикат кальция) со-
ляная кислота вытесняет кислоту, образуя хлористую соль
металла. В основных солях сильных кислот (основной суль-
фат свинца) соляная кислота нейтрализует свободные гидр-
оксильные группы с выделением воды и образованием комп-
лексного сульфохлорида свинца.
Главнейшими из стабилизаторов этого типа являются соли
свинца, из которых чаще всего употребляются наиболее актив-
ные основные соли (основные фосфаты, сульфаты, силикаты).
Так как они применяются в большом количестве (до 10% от
веса полимера при стабилизации материалов для электроизо-
ляционных целей), то хорошую светостойкость следует отне-
сти скорее за счет пигментирования, чем за счет химического
взаимодействия.
Силикаты, фосфаты, лактаты натрия придают полимерам
превосходную свето- и теплостойкость, но они неудобны вслед-
ствие их растворимости в воде. Пленки, полученные с их при-
менением, имеют тенденцию тускнеть во влажной атмосфере.
188
Гл. IV. Виниловые полимеры
Эпоксипроизводные. Эти соединения характеризуются нали-
чием кольца, состоящего из атомов углерода и одного атома
кислорода. В стабилизаторах этого типа эпоксидный цикл свя-
зан с длинной углеводородной цепью. Вещества с низким мо-
лекулярным весом, как правило, растворимы в воде и потому
непригодны. Эпоксигруппа может реагировать с соляной кис-
лотой, образуя очень стабильную группировку, содержащую
гидроксил и атом хлора у соседних атомов углерода:
О НО С1
—С^С — + НС1 ->- —с—с—
II II
Если молекула стабилизатора содержит две эпоксигруппы,
она может связывать между собой две молекулы полимера.
Такие стабилизаторы, придающие полимерам замечательную
теплостойкость, выпускаются фирмой Carbide Corporation под
названием «стабилизатор G-18». Они представляют собой све-
тло-желтые жидкие смолы и применяются для получения про-
зрачных или непрозрачных пленок, например для пищевой
промышленности.
.. Антиоксиданты. В качестве антиоксидантов могут быть ис-
пользованы фенолы, нафтолы. Одним из лучших, особенно для
эмалей, является фенилсалицилат (салол). Могут применяться
также амины, легко реагирующие с соляной кислотой, но вслед-
ствие их ясно выраженного основного характера при этом
ускоряется образование соляной кислоты, а следовательно, и
разложение полимера. К тому же, образующиеся продукты
при температуре выше 150° вновь выделяют соляную кислоту.
Выбор стабилизатора зависит от состава композиции (типа
полимеров и (пластификаторов), условий ее приготовления и
назначения конечного продукта. Испытания стабилизаторов
должны проводиться в соответствующих условиях, а нормы их
промышленного применения должны устанавливаться лишь пос-
ле неоднократных опытов, так как поспешное заключение мо-
жет привести к очень серьезным неполадкам.
Стабилизаторы для прозрачных пленок должны быть, само
собой разумеется, бесцветными и прозрачными (поэтому в ка-
честве стабилизаторов и были предложены эпоксисоединения).
В отдельных случаях эффективными стабилизаторами против
действия тепла являются кальциевые и свинцовые соли орга-
нических кислот, но они не могут применяться вследствие пло-
хой водостойкости или токсичности.
'При стабилизации прозрачных пленок против действия тепла
лучше всего вводить в виниловые сополимеры небольшие ко-
личества мочевино-формальдегидных и меламино-формальде-
Растворители и вспомогательные материалы
189
гидных смол (от 1 до 3 вес. ч. на 100 вес. ч. винилового сопо-
лимера в расчете на сухой вес). Замечено, что после продол-
жительной сушки пленки иногда приобретают с поверхности
некоторую липкость (отлип); этого можно избежать, добав-
ляя в композицию небольшие количества невысыхающих смол
(1 вес. ч. на 100 вес. ч. винилового полимера).
Хотя большая часть продажных мочевино- и меламино-
формальдегидных смол не совмещается с другими пленкооб-
разующими веществами, бакелит UREA XVU 15765, пластифи-
цированный раствором бакелита XJ 14424 или оксидированным
косторовым маслом, дал очень хорошие результаты29. В про-
зрачные или белые составы вводили также мочевино-формаль-
дегидные смолы с добавкой небольшого количества оксидиро-
ванного касторового масла, присутствие которого не влияло за-
метным образом на другие свойства покрытия, например на
адгезию.
Такие смолы, как бакелит BR 3360, BR 8900, BR 302, дюрез
»202, теглак 161, петрекс 1 и 5, некоторые алкидные или ви-
ниловые полимеры, будучи взяты в очень небольших количе-
ствах, проявляют хорошее стабилизирующее действие. Вероят-
но, оно вызвано некоторой незначительной несовместимостью
с сополимерами или виниловыми смолами, приводящей на прак-
тике к легкому «выпотеванию», предотвращающему непосред-
ственный контакт между виниловым полимером >и металлом. Тем
не менее в промышленной практике применение этих продуктов
не очень желательно, так как получаемые пленки сильно окра-
шены и менее устойчивы.
Грунты на основе невиниловых полимеров. Другим методом
улучшения термостабильности виниловых полимеров на актив-
ных поверхностях является применение лакового или смоляно-
го грунта, препятствующего прямому контакту винилового по-
лимера с активной поверхностью. Эти грунты могут готовить-
ся на основе смолы типа бакелит BV 1600, оксидированного
тунгового масла, полимеризованного льняного масла. После
нанесения по грунту винилового полимера покрытие можно
подвергать горячей сушке. Было также найдено, что с некото-
рыми из этих грунтов совместим поли-н-бутилметакрилат, кото-
рый при введении его в грунт в небольшом количестве (около
10%) придает покрытию лучшую адгезию при более низких
температурах сушки. Большую часть этих грунтов можно на-
носить на тонкие подложки и высушивать при температуре
180—190° в течение 15—30 мин.; для слабо высыхающих грунтов
требуются часто особые сиккативы.
Виниловые сополимеры, нанесенные на подобные грунты,
обнаруживают хорошую адгезию после горячей сушки при тем-
пературе 170°, но если в состав грунта ввести поли-н-бутилмет-
190
Г л. IV. Виниловые полимеры
акрилат, то для получения хороших результатов горячую суш-
ку достаточно вести при температуре 130—145°.
Пассивирование металлов. Третий метод ослабления воз-
действия активных поверхностей на пленку виниловых сопо-
лимеров при нагревании состоит в химической обработке по-
верхности металла перед нанесением покрытия. При такой об-
работке улучшаются свойства конечного покрытия, так как
достигается высокая чистота поверхности, не всегда достигае-
мая при других способах обработки. Адгезия улучшается не
только благодаря чистоте поверхности, но и механически бла-
годаря ее шероховатости. Наконец, такая обработка значитель-
но уменьшает необходимость термической стабилизации вини-
ловых полимеров и удлиняет продолжительность службы по-
крытий вследствие ингибирования коррозии металлической под-
ложки.
Пассивирование металлов тем не менее не исключает необ-
ходимости термической стабилизации пленок, кроме особых слу-
чаев применения. Благодаря пассивированию металлов стано-
вится возможным применение красителей светлых оттенков и
достигается удовлетворительная адгезия при более низкой тем-
пературе сушки.
Известны следующие способы обработки черных металлов:
процессы типа бондеризации, процессы кромодин, гранодин
(Parker Rust Proof Со, Detroit), преп-райт (Neilson Chemical Co,
Detroit), для цинка — процесс Паркера и процессы фирмы
American Chemical Paint Со. Возможность пассивирования цин-
ковых сплавов в значительной степени зависит от содержа-
ния в них цинка, причем наилучшие результаты получаются при
применении сплавов, содержащих более 20% цинка. Для сплавов
магния фирма Dow Chemical Со, Midland предлагает разные типы
обработки с использованием хромовых солей, несколько способов
обработки сплавов алюминия описано фирмой Aluminium Со of
America (Pittsburg, Pennsylvania). Два последние типа сплавов
нейтральны по отношению к виниловым полимерам, но обработка
поверхности необходима для достижения лучших адгезионных
свойств.
Пластификаторы. При изготовлении пленок из виниловых
полимеров для улучшения их эластичности в большинстве слу-
чаев необходимо добавлять некоторое количество пластифика-
торов.
Количество пластификаторов, вводимое в композиции на
основе полимеров типа VYHH, примерно вдвое меньше требуе-
мого для нитролаков на основе нитрата целлюлозы с вязкостью
0,5 сек. В композиции, содержащие полимеры типа VYLF,
требуется вводить больше пластификаторов, чтобы компенсиро-
вать меньшую эластичность этих полимеров.
Растворители и вспомогательные материалы
191
Возможно применение большей части общеизвестных пла-
стификаторов, используемых в лаках, но многочисленные ра-
боты, проведенные в этом направлении, показали, что для до-
стижения заданных свойств необходимо вводить в каждом от-
дельном случае определенный пластификатор. В принципе при-
меняемый пластификатор должен быть совместим с виниловыми
полимерами, возможно медленнее испаряться, не обесцвечивать
композицию в процессе ее приготовления, нагревания или есте-
ственного старения, не понижать атмосферостойкость пленки.
Кроме того, он должен быть нетоксичным, не иметь вкуса,
обладать максимальной влагоустойчивостью и эластичностью
без отлипа, а также повышенной устойчивостью к кислотам,
щелочам, жирам и маслам.
В большинстве случаев вполне удовлетворительные резуль-
таты дают трикрезилфосфат и бутоксигликольфталат, обла-
дающие превосходной влаго- и атмосферостойкостью при ма-
лой скорости испарения. Пластические свойства получаемых
пленок удовлетворительны. Для некоторых областей примене-
ния рекомендуются другие продукты, такие, как ацетилрицино-
леат метоксигликоля (пластификатор КР-120 фирмы Ohio Apex
Corporation), главным образом в тех случаях, когда требуется
очень высокая пластичность и способность складываться без
слипания (например, для покрытия бумаги или тканей). Могут
применяться хлорпроизводные типа галовакса и арохлора, а
также хлорпарафины. Часто применяется диоктилфталат (ДОФ)
и некоторые другие пластификаторы, характеристики которых
приведены ниже.
Флексол 4GO (ди-2-этилгексоат полиэтиленгликоля) значи-
тельно дешевле диоктилфталата и применяется в виде смесей
с ним в отношении 1:1.
Флексол 3GO (ди-2-этилгексоат триэтиленгликоля) также
употребляется совместно с диоктилфталатом, но более дорог.
Флексол TOF (триоктилфосфат) придает пленкам гибкость,
при температуре до —50°, одновременно сообщая им лучшие
свойства, чем трикрезилфосфат.
Флексол 8-N-8 [ди-(2-этилгексоил)-2'-этилгексанимин] обла-
дает исключительно интересными свойствами: очень слабой ле-
тучестью (ниже, чем у трикрезилфосфата), высокой водостой-
костью, хорошей эластичностью при низких температурах. Он
применяется в США как основной пластификатор в производ-
стве лаков и пластических масс.
Флексол TWS (сульфофенолят, подобный немецкому меза-
молу) отличается очень слабой миграцией, очень слабо летуч—
потеря веса пленки толщиной 0,1 мм в течение 10 дней при 60е
составляет 0,3%, по сравнению с 0,5% для трикрезилфосфата.
Водостойкость этого пластификатора также очень высока: после
192 Г л. IV. Виниловые полимеры
пребывания пленки толщиной 0,1 мм в течение 10 дней в воде
при 25° набухания не наблюдается, в то время как набухание
пленок, пластифицированных трикрезилфосфатом, составляет
0,7%. Флексол TWS отличается к тому же очень большой ста-
бильностью. Он употребляется в производстве лаков и пласти-
ческих масс, но вследствие наличия в нем серы не рекомендует-
ся для покрытий по меди и по грунтам, стабилизованным со-
лями свинца (образуются черные сульфиды свинца).
Флексол 3GH (ди-2-этилбутират триэтиленгликоля) придает
пленкам высокую эластичность при низких температурах, а
также очень большую светостойкость. К сожалению, он отно-
сительно летуч, даже более, чем дибутилфталат.
Кроме перечисленных пластификаторов, можно назвать па-
раплексы типов R. G.6— R. G.6.0 фирмы Rhom et* Haas (по-
лиэфиры, устойчивые к углеводородам), сантицайзеры В. 16 и
М.17 (бутилфталилбутилгликолят и метилфталилэтилгликолят),
сантицайзер 141 (дифенилмонооктилфосфат) фирмы Monsanto
Chemical Со. Все эти продукты нетоксичны.
Большинство пластификаторов, употребляемых в обычных
композициях на основе поливинилхлорида, пригодны и для
красок. Не рекомендуется применять пластификаторы, вовсе не
растворяющие сополимеры; в некоторых случаях их можно
применять в небольших количествах, но действие таких пла-
стификаторов-разбавителей заключается в повышении термо-
стойкости, водостойкости и т. п., т. е. в улучшении свойств, не
связанных с пластифицирующим действием.
При применении пленкообразующих веществ их приходит-
ся в некоторых случаях нагревать до температуры около 150°.
Поэтому необходимо учитывать скорость испарения пластифи-
катора, так как при высокой температуре пленка может поте-
рять значительное количество пластификатора и приобрести
ломкость. Дибутил фтал ат, хотя и применяемый во Франции
как основной пластификатор для большого числа лаков, после
5 мин. при 175° полностью улетучивается из пленок на основе
сополимеров винилхлорида и винилацетата толщиной 0,2 мм.
Поз тому при определении возможности применения различных
пластификаторов чрезвычайно важно учитывать их относитель-
ную летучесть.
Следует отметить, что пластификатор влияет также на
улетучивание растворителя. Установлено, что в непластифици-
рованных пленках, (полученных из растворов, остается прибли-
зительно 4—5% растворителя, удерживающегося в пленке да-
же после очень длительной сушки на воздухе или в сушильной
камере. Это явление проявляется даже при нанесении пульве-
ризатором растворов полимера в низкокипящих растворителях,
так как в результате очень быстрого высыхания на поверхности
Растворители и вспомогательные материалы
193
пленки образуется твердая и сухая корка, препятствующая
удалению растворителя. Применение 15—25% пластификатора,
очевидно, замедляет образование сухой корки на поверхности
пленки и удаление растворителя облегчается. Это явление ска-
зывается сильнее, когда применяемый пластификатор принад-
лежит к типу истинных пластификаторов, т. е. желатинирует
пленкообразующее вещество. Таким образом, вопрос выбора
пластификатора является первостепенным. Этот вопрос подроб-
но рассмотрен в специальных работах30 , 33, зв-в\
Модификаторы. В большинстве случаев добавление других
пленкообразующих к сополимерам не дает удовлетворительных
результатов. Тем не менее отметим применение полиметилмет-
акрилата в некоторых материалах, используемых для наруж-
ных покрытий. Иногда можно улучшить водостойкость или по-
высить твердость покрытия путем добавления некоторых типов
смол или восков, но во всех случаях основные свойства пленки
будут несколько ухудшены.
Совместимость сополимеров детально рассмотрена в одной
из работ Bakelite Corporation31.
Растворители и свойства растворов. Сополимеры типа VYHH
или родопаса АХ легко растворяются при нормальной темпе-
ратуре в растворителях типа кетонов, в производных типа азот-
нокислых эфиров, в хлорированных углеводородах, образуя
прозрачные растворы и в некоторых случаях коллоидные дис-
персии. Альдегиды, простые и смешанные эфиры также могут
в известной степени действовать как растворители. Ароматиче-
ские углеводороды типа толуола, ксилола вызывают набухание
и в некоторых случаях растворяют виниловые полимеры, осо-
бенно при высоких температурах. Напротив, спирты и алифа-
тические углеводороды не являются растворителями, а дей-
ствуют как осадители. Алифатические соединения типа нафте-
нов занимают промежуточное место.
Растворимость виниловых сополимеров изменяется в зави-
симости от молекулярного веса и содержания винилацетата.
Так, продукты типа VYLF вследствие более низкого молеку-
лярного веса значительно лучше растворимы, чем продукты
типа VYHH. При определенной температуре винилит VYLF дает
«ежелатинирующиеся растворы со значительно более высоким
содержанием сухого остатка; он растворим также в большем
числе растворителей.
Здесь будет рассмотрена растворимость наиболее распростра-
ненного типа сополимеров: родопаса АХ, или винилита VYHH.
Лучшими растворителями для них являются кетоны. Они
дают наиболее концентрированные нежелатинирующиеся рас-
творы, имеющие к тому же при равных концентрациях наимень-
шую вязкость. Преимуществом их является также возможность
13—156
194
Гл. IV. Виниловые полимеры
введения больших количеств разбавителей. Большинство рас-
творителей этого класса стабильны и не вызывают коррозии.
Некоторые хлорпроизводные имеют те же свойства, но при-
менение их требует специальных условий (приточной или вы-
тяжной вентиляции).
Нитропроизводные (нитропарафины, нитроэтан, нитропро-
пан) также отличаются превосходной растворяющей способно-
стью, но вязкость получаемых растворов, как правило, выше,
чем при применении кетонов. В некоторых случаях в этих рас-
творителях удается получать нежелатинирующиеся растворы с
высоким содержанием сухого остатка.
Сложные эфиры отличаются более низкой растворяющей
способностью и образуют растворы с более высокой вязкостью.
Их иногда применяют в качестве добавок, например с целью
придания растворам более приятного запаха.
Оценка растворителей или смесей разбавителей с раство-
рителями может быть произведена по кривым вязкостей полу-
чаемых растворов. Для виниловых полимеров кривые вязкости
разделяются на три области.
Если увеличивать процентное содержание смолы в лаке,
вязкость раствора возрастает сначала равномерно, а выше
определенного содержания сухого вещества — значительно бы-
стрее, чем концентрация. Было найдено, что в этом интервале
концентраций явления вязкого течения проявляются слабее,
чем пластические свойства. Вязкость таких растворов чрезвы-
чайно чувствительна даже к небольшим изменениям темпера-
туры. Большинство растворов обнаруживает в этом интервале
концентраций сильно выраженную тиксотропию и обозначается
поэтому как тиксотропная фаза, а иногда как пластическая фаза.
При дальнейшем повышении концентрации получаются же-
латинированные растворы, т. е. раствор переходит в фазу геля.
Эта область концентраций не представляет большого интереса
для производства лаков и красок.
Вследствие низкой газопроницаемости поливинилхлоридных
пленок следует принимать определенные меры предосторожно-
сти при их сушке, так как в процессе удаления растворителя,
наряду с пластической, или тиксотропной, фазой в пленке может
возникнуть и гелеобразная фаза.
Изменение соотношения между разбавителями и растворите-
лями может привести к таким же изменениям вязкости, без из-
менения конечного содержания сухого вещества. Если содержа-
ние разбавителя будет очень велико, полимер в такой смеси не
будет растворяться.
Соотношение разбавителя и растворителя оказывает влияние
на вязкость растворов; исследование получаемых кривых вяз-
кости представляет большой интерес.
Растворители и вспомогательные материалы
195
Подробные исследования вязкости были проведены фирмой
Bakelite Corporation32. В этой работе рассматривается действие
растворителей в смесях с разбавителями (в частности, с толуо-
лом, сольвент-нафтой), так как предварительные работы пока*
зали, что только исследование вязкостей растворов дает серьез-
ное основание для оценки
Рис. 33. .Вязкость раство-
ров винилита VYHH в сме-
сях метилизобутилкетон—
толуол:
/—10% сухого вещества;
2—15%; 3—18%; 4-20%.
растворителей. На основании этой
работы были поставлены фунда-
ментальные исследования в Ин-
ституте Меллона в Питтсбурге.
Изменение вязкости определя-
лось с большой точностью вис-
козиметрами Гепплера; исходные
растворы подвергались старению
в продолжение трех недель при
постоянной температуре.
Рис. 34. Вязкость растворов ви-
нилита VYHH в смесях ацетон—
толуол:
7—10% сухого вещества; 2—15%;
3-18%; 4—20%; 5-22%.
На рис. 33—38 представлены кривые вязкости растворов
полимеров с разным содержанием сухого вещества в смесях
растворитель—разбавитель. На рис. 34, где приведено изме-
нение вязкости винилита типа VYHH в зависимости от состава
смеси ацетона (истинный растворитель) с толуолом (разбави-
13*
196
Гл. IV. Виниловые полимеры
тель), обращает внимание быстрое повышение вязкости при
уменьшении количества ацетона в смеси. Это повышение проис-
ходит значительно быстрее и яснее выражено в более концен-
трированных растворах.
В зоне быстрого повышения вязкости растворы очень чув-
ствительны к взбалтыванию, действию тепла и содержанию
разбавителя. Для композиций с большим содержанием рас-
Содержание метил-
этилнетона В смеси
с толуолом, %
Рис. 35. Вязкость рас-
творов винилита
VYHH в смесях метил-
ат илкетон—толуол:
1—10% сухого вещества;
2—15%; 3-18%; 4-20%;
5—22%; 5—25%.
творителя характерно, напротив, истинно
вязкое течение. Точки перегиба на кривых
вязкости ограничивают область практиче-
ски применимых растворов; эту границу
называют «рецептурной линией».
Рис. 36. Вязкость рас-
творов винилита VYHH
в смесях метилэтил-
кетон—тролуойль:
7—10% сухого вещества;
2—15%; 3-18%; 4—20%.
Рис. 37. Вязкость растворов
винилита VYHH в смесях
окись мезитила—толуол:^
/—10% сухого вещества; 2—15%;
3—18%; 4—20%.
На рис. 39—44 дана характеристика систем растворитель —
разбавитель в зависимости от процентного содержания поли-
мера в растворе при 20°.
По этим диаграммам находят зону текучести (практиче-
скую), переходящую в тиксотропную зону и затем в зону геле-
образования. Границы между этими зонами: рецептурная линия
Растворители и вспомогательные материалы
197
Рис. 38. Вязкости растворов
винилита VYHH в смесях
изофорон—ксилол".
1—10% сухого вещества; 2—15%;
3—18%; 4—20%.
и линия гелеобразования. Когда
процентное содержание раствори-
теля уменьшается настолько, что
фактически остается уже только
разбавитель, начинается зона коагу-
ляции или иерастворения.
Содержание метилизобутилнетона в смеси
с толуолом. %
Рис. 39. Характеристика растворимости вини-
лита VYHH в смесях метилизобутилкетон—
толуол:
/—линия осаждения; 2—линия гелеобразогания; з—ре-
цептурная линия.
В случае применения толуола в качестве разбавителя доста-
точно небольшого содержания в смеси активного растворителя,
й И ---------------------1-----1----1-----1----1..- 1 - I______L-
10 го 30 50 60 70 60 30 100
<S Содержание ацетона в смеси с толуолом, X
Рис. 40. Характеристика растворимости вини-
лита VYHH в смесях ацетон—толуол:
/—линия осаждения; 2—линия гелеобразования; 3—ре-
цептурная линия.
чтобы полимер не выпа-
дал из раствора. Боль-
шинство кривых имеет
одинаковый характер, не-
зависимо от примененных
растворителя и разбави-
теля.
Вязкости растворов
виниловых сополимеров,
естественно, изменяются
в зависимости от их мо-
лекулярного веса. Все
приведенные кривые по-
лучены при применении
сополимеров типа VYHH;
для сополимеров типа
VYLF содержание сухого
198
Гл. IV. Виниловые полимеры
Вещества в лаке может быть значительно повышено. На прак-
тике применяют смеси .сополимеров VYHH и VYLF, взятых
>(в среднем) в соотношении 60 : 40.
Рис. 41. Характеристика растворимости вини-
лита VYHH в смесях окись мезитила—толуол:
/—линия (каждения; 2—линия гелеобразования; 3—ли-
ния гелеобразования для соответствующей смеси
метилизсбутилкетона с толуолом; 4—рецептурная
линия.
Кроме ненасыщенности, повышающей растворяющую способ-
ность, большое влияние оказывает наличие циклов в молекуле
растворителя. Алициклические кетоны исключительно активны
как растворители; кривые
вязкости для смесей та-
кого растворителя с раз-
бавителем проходят через
минимум.
Типичным примером
является смесь изофоро-
на с ксилолом (рис. 38).
На этом рисунке показан
суммарный эффект изме-
нения вязкости раствора
и смеси растворителей.
В действительности, если
вязкости растворителя и
разбавителя сильно раз-
личаются, следовало бы
сравнить относительные
вязкости растворителей.
Так, вязкость изофоро-
на приблизительно втрое
Тип кетона, применен-
ного в качестве раство-
рителя, сильно влияет на
характер кривых. Нена-
сыщенные кетоны имеют
значительно более высо-
кую растворяющую спо-
собность, чем насыщен-
ные. Так, из рис. 38 и 41,
на которых представлены
данные для смесей окиси
мезитила с разбавителем
типа толуола, следует,
что эти смеси могут при-
меняться в качестве рас-
творителя для более вы-
сокополимерных продук-
тов или для сополимеров
с малым содержанием ви-
нилацетата.
IV 20 30 40 50 60 70 00 90 100
Содержание метилзтилкетопа в смеси
с гпролцойлом,
Рис. 42. Характеристика растворимости вини-
лита VYHH в смесях метилэтилкетон—тролу-
ойль:
1—линия гелеобразования; 2—рецептурная линия;
3—линия осаждения.
Растворители и вспомогательные материалы
199
больше вязкости ксилола (рис. 45), тогда как метилизобутилке-
тон и толуол имеют равные вязкости.
Рис. 43. Характеристика растворимости вини-
лита VYHH в смесях циклогексанон—толуол-
/—линия осаждения; 2—линия гелеобразования; 3—ре-
цептурная линия.
Заштрихованный участок—зона» чувствительная к сос-
таву смеси растворитель—разбавитель.
Рис. 44. Характеристика растворимости вини-
лита VYHH в смеси изофорон—ксилол:
1—линия осаждения; 2—линия гелеобразования; 3—ре-
цептурная линия.
Заштрихованный участок—зона, чувствительная к со-
ставу смеси растворитель—разбавитель.
На приведенных выше кривых (рис. 33—38) представлена
абсолютная вязкость растворов; при переходе к относительным
вязкостям кривая метилиизобутилкетон— толуол должна оста-
ваться неизменной, ,в то время как кривая изофорон — ксилол
должна изменяться очень значительно.
200
Гл. IV. Виниловые полимеры
На рис. 46 приведены кривые относительных вязкостей
(при 20°) растворов с разным содержанием полимера в равных
объемах растворителей.
Рис. 45. Вязкости смесей растворителей:
I—изофорон—ксилол; 2—метилизобутилкетон—
толуол.
пульверизации, могут
растворители.
На рис. 47 закономерности
изменения вязкости винилита
VYHH сопоставлены с поведе-
нием нитрата целлюлозы (вяз-
кость 0,5 сек.). Нитроцеллю-
лозные лаки, не обнаруживаю-
щие тиксотропных свойств, мо-
гут содержать значительно
больше сухого вещества. Зато
для 'приготовления лаков на
основе виниловых нтолимеров
при малых количествах рас-
творенного вещества, когда
возможно нанесение путём
применяться значительно менее активные
Содержание изофорапа в
смеси с ксилолом, °/„
Рис. 47. Сравнение рецептурных линий рас-
творов винилита VYHH и нитрата целлюлозы
(вязкость 0,5 сек) :
Рис. 46. Кривые относи-
тельной вязкости растворов
винилита VYHH в смеси
растворитель—разба-
витель:
Z—10 г полимера в 100 мл смеси;
2—15 а/100 лл; 3—18 а/100 мл-,
4—20 а/100 мл.
/—линия осаждения винилита VYHH; 2—линия осаж-
дения нитроцеллюлозы; 3—линия гелеобразования ни-
троцеллюлозы; 4—линия гелеобразования винилита
VYHH; 5—рецептурная линия винилита VYHH; 6—ре-
цептурная линия нитроцеллюлозы.
Д—зона жидкости VY’HH; В—тиксотропная зона вини-
лита VYHH; С—область пластического течения нитро-
целлюлозы; О—зона жидкости нитроцеллюлозы; Е— зо-
на осаждения винилита VYHH; F—зона осаждения
нитроцеллюлозы.
Растворители и вспомогательные материалы
201
Составление рецептур. При составлении рецептур лаков на
основе виниловых сополимеров, кроме растворяющей способ-
ности растворителей, следует учитывать также скорость испаре-
ния, токсичность, негорючесть, растворимость в воде, стабиль-
ность, запах и, наконец, цену. Трудности выбора растворителя
хорошо известны потребителям и здесь нет необходимости их
обсуждать.
Для лаков, наносимых путем пульверизации, в качестве
растворителей можно применять метилэтилкетон, метилизобу-
тилкетон или метиламилкетон с такими разбавителями, как:
толуол и ксилол. Метилэтилкетон, отличающийся высокой рас-
творяющей способностью и относительно дешевый, не следует
вводить в композиции в очень больших количествах, так как
возможно побеление пленок, в особенности, если нанесение ла-
ка производится во влажной атмосфере. Кроме того, метил-
этилкетон может вызывать образование пузырей при нанесении
Или во время сушки, если работа ведется при повышенных тем-
пературах. Метиламилкетон, напротив, замедляет сушку, улуч-
шает растекаемость лака и исключает побеление1. Можно ис-
пользовать также изофорон, но он значительно повышает склон-
ность пленки удерживать загрязнения.
Для покрытий по металлам, подлежащим обработке при вы-
сокой температуре, могут применяться многие растворители и1
разбавители. Для покрытий по тканям и бумаге могут упот-
ребляться сравнительно легко летучие растворители (например,
ацетон). Более высокая летучесть растворителей допустима
вследствие более низкой теплоемкости бумаги, и ткани, а так-
же потому, что благодаря большой скорости теплопередачи
этих материалов уменьшается возможность конденсации воды
Можно использовать смесь ацетона и толуола, так как потеря
растворяющей способности вследствие улетучивания большей
части активного растворителя компенсируется улучшением рас-
творяющей способности толуола при повышении температуры..
Для некоторых специальных целей могут применяться раствори-
тели с очень низкой летучестью.
Известны также негорючие смеси растворителей. Среди них
в литературе часто упоминается хлоразол, состоящий из трех
объемов дихлорэтилена и одного объема четыреххлористого
углерода. Эта негорючая смесь отличается хорошей растворяю-
щей способностью по отношению к виниловым сополимерам.
С этой же целью применяется метиленхлорид и трихлорэтилен
(чаще всего в смеси с дихлорэтаном). Хлорированные раство-
рители более токсичны, чем кетоны и сложные эфиры. Поэтому
при работе с ними нужно принимать меры предосторожности.
Применение более высокомолекулярных полимеров (напри- '
мер, винилитов VYNS и VYNW фирмы Bakelite Corporation) „
202
Гл. IV. Виниловые полимеры
используемых для особых целей, когда требуется эластичность,
твердость, химическая стойкость, значительно осложняется
вследствие высокой вязкости их растворов. Для этих полиме-
ров почти всегда ограничиваются 'применением активных рас-
творителей без разбавителей; в частности, для винилита VYNS
применяют смеси метилэтилкетона или метилизобутилкетона с
диоксаном, окись мезитила, циклогексанон и изофорон. Уста-
новлено, что некоторое понижение вязкости растворов высоко-
молекулярных полимеров или сополимеров с низким содержа-
нием ацетатных групп достигается при предварительном сма-
чивании полимера более активным растворителем.
Несколько композиций растворителей и разбавителей при-
ведено в табл. 16.
ТАБЛИЦА 16
Некоторые композиции растворителей для виниловых полимеров
Растворители Композиции (состав в %)
А в в г д Е
Метилэтилкетон ....... 10 35 5
Метилизобутилкетон 40 50 40 35 — —
Метиламилкетон . . — — 10 — ..— 10
Изофорон ... — — — — 20
Ксилол . . — — 10 — — —•
Толуол 49,8 49,8 39,8 29,8 95 —
Сольвессо № 3 (ароматический рас-
творитель) — — — —_ — 39,8
Тетралин — — — — —- 30
Окись пропилена 0,2 0,2 0,2 0,2 — 0,2
Примечание. Смеси А, Б и В предназначены для эмалевых красок, наносимых пульве-
ризатором; качество их улучшается от А к В. Смесь Г применяется при нанесении краски вали-
ком на бумагу или ткань. Смесь Е наиболее изучена для покрытий по металлу. Смесь Д—
основная композиция, применяемая для разбавления с целью уменьшения вязкости.
Пигменты. При составлении рецептуры и изготовлении ла-
кокрасочных материалов на основе хлорсодержащих виниловых
полимеров, кроме выбора пленкообразующего вещества, тре-
буется выбрать также соответствующие пигменты. Пигменты
не должны содержать солей металлов (главным образом желе-
за и цинка), способных оказывать влияние на теплостойкость
полимера, так как это приводило бы к быстрому обесцвечива-
нию пленок на металлической поверхности (особенно на по-
верхности цинка) при горячей сушке. Как общее правило, сле-
дует избегать железных цинковых пигментов. Тем не менее для
некоторых материалов, подвергаемых сушке при нормальной
температуре или при температурах ниже 150°, можно иногда
применять железоокисные пигменты и окись цинка. Большинство
других пигментов можно успешно применять во всех случаях.
Растворители и вспомогательные материалы
203
Свинцовые пигменты дают очень хорошие результаты, так
как они оказывают стабилизирующее действие на виниловые
полимеры. Эти пигменты применяются очень широко (за исклю-
чением немногих областей, где недопустима их токсичность
или возможно соприкосновение с сернистыми соединениями).
Они пригодны также для применения в присутствии солей же-
леза и цинка. Вполне пригодны также многие другие пигменты,
в частности производные сурьмы, кадмия и титана. Хорошие
результаты дают органические пигменты, например индантро-
новые и фталоцианиновые красители.
Газовую сажу можно применять в качестве пигмента, но
для ее хорошего диспергирования необходимо употреблять ам-
миачные растворы.
Диспергирование пигментов. Пигменты следует вводить в
высокодисперсном состоянии. Наилучшие результаты дают так
называемые «чипсы», т. е. пигментные пасты, получаемые ме-
тодом вальцевания. Диспергирование можно облегчить путем
введения специальных диспергаторов, которые нужно выбирать
очень тщательно, так как они имеют склонность понижать во-
достойкость покрытий. Смачиватели типа аэрозолей, выпускае-
мые фирмой American Cyanamid and Chemical Corporation, ока-
зались очень полезными при диспергировании двуокиси титана.
Недавно предложенный новый метод диспергирования пиг-
ментов состоит в смачивании пигмента растворителями (в при-
сутствии специального смачивателя или без него) в шаровой
<мельнице, куда затем добавляется полимер (сухой или в виде
концентрированного раствора) до получения густой пасты, ко-
торая в дальнейшем разбавляется. В композициях с высоким
содержанием пластификаторов пигменты могут быть сначала
диспергированы в пластификаторе, а затем уже смешаны с
раствором полимера.
Некоторые рецептуры, применяемые при таком методе дис-
пергирования пигментов, приведены в табл. 17.
Процесс диспергирования пигментов на вальцах очень прост.
Пигменты, полимер и пластификаторы предварительно очень
тщательно смешивают. Вальцы нагревают примерно до 70° и
смесь вальцуют до получения ровного полотна. К этому вре-
мени вальцы охлаждают и смесь обрабатывают на холодных
вальцах в продолжение 5 мин., все время подрезая массу для
полной гомогенизации. При последнем пропускании массы че-
рез вальцы зазор уменьшают до минимума, чтобы создать высо-
кое давление, требуемое для полного диспергирования. После
вальцевания полученные тонкие листы снимают с вальцов и
охлаждают. В таком виде пигментная смесь пригодна для даль-
нейшего употребления. При вальцевании необходимо очень тща-
тельно контролировать температуру, так как при ее повышении
204
Гл. IV. Виниловые полимеры
сверх 100° свойства конечного продукта сильно изменяются.
Кроме того, чем .ниже температура, тем больше внутреннее тре-
ние смеси, способствующее лучшему ее диспергированию.
ТАБЛИЦА 17
Рецептуры пигментированных смесей
Компоненты Композиция (состав в %)
1 11 III IV 1 V
Пигмеи ты:
Двуокись титана А-168 LO 40,0 20,5 — — —
Аэрозоль ОТ (смачиватель) 0,4 0,2 — 0,3 —
Кронизол (дибутилцеллозольвфталат) . . . 5,0 2,0 2,9 4,6 2,4
Масло Бэкер №4 0,2 — — — — 0,4
Смесь растворителей (смеси А, Б или В,
см. табл. 16) 8,9 7,0 0,7 6,7 3,2
Основной сульфат свинца — — 48,5 — —
Цинковый крои (8S3 Империэл) — — — 30,7 -—
Свинцовый оранжевый крои (12011-SW) . . — — — — 20,1
Циклогексиламинолеат — — — — 0,2
Полимер:
Родопас АХ или винилит VYHH-1 .... 13,0 20,0 12,1 16,2 20,1
Смесь растворителей и разбавителей (А, Б) . 32,5 50,3 35,8 41,5 53,6
100,0 I 100,0 100,0 100,0 100,0
П р и м е ч а в и е. Полимер и растворители добавляются после перетира остальных компо-
нентов в течение не менее 12 час.
Рецептуры пигментных паст. При составлении рецептур пиг-
ментных паст (чипсов) рекомендуется брать максимальное ко-
личество пигментов по отношению к пленкообразующему. При
этом улучшается диспергирование пигментов, облегчается валь-
цевание и в дальнейшем получаются пленки с более высоким
блеском.
Количество вводимых пластификаторов должно быть тща-
тельно рассчитано: при слишком большом количестве пласти-
фикатора смесь прилипает к вальцам и ухудшается дисперги-
рование, в то время как недостаток пластификатора приводит
к затруднениям при переработке готовой смеси. Желатиниру-
ющие пластификаторы должны применяться в меньшем коли-
честве, чем нежелатинирующие, так как они придают клейкость
смеси. Для уменьшения клейкости композиций к желатиниру-
ющим пластификаторам добавляют оксидированное касторовое
масло, которое оказывает к тому же некоторое стабилизирующее
действие на виниловые полимеры. Количество касторового мас-
ла не должно превышать 1—2% по отношению к основному
полимеру. При более высоком соотношении дисперсия слишком
сильно разбавляется и адгезия пленок понижается.
----------------------------------------------------------------
Растворители и вспомогательные материалы 205
Для переработки методом вальцевания практически при-
меняют винилит VYHH или родопас АХ. Можно перерабаты-
вать на вальцах и винилит VYLF, а также сополимеры с более
высоким молекулярным весом (VYNS, VYNW). Для этих плен-
кообразующих требуется значительно более высокое содержа-
ние пластификаторов и вальцевание должно проводиться при
более высокой температуре (120°). При соблюдении этих усло-
вий достигается прекрасное диспергирование пигментов.
Полуфабрикатные пигментные пасты. После вальцевания
масса, снимаемая с вальцов в виде листов, дробится на мелкие
кусочки (чипсы или стружки) и либо хранится в таком виде,
либо смешивается с растворителями. При этом получаются так
называемые полуфабрикатные пигментные пастьи, легко смеши-
вающиеся с растворами пленкообразующих и с другими ингре-
диентами при изготовлении конечного лакокрасочного материала.
Процесс приготовления пасты-полуфабриката состоит в до-
бавлении к диспергированным пигментам небольшого количе-
ства растворителей и перемешивании массы после ее набуха-
ния в двухлопастном смесителе типа «Вернер» до достижения
гомогенности. Нагревание в конце операции улучшает гомоге-
генизацию и приводит к повышению блеска покрытия. Тот же
результат достигается при добавлении в конце операции не-
больших количеств смеси растворителей и разбавителей до
получения требуемой консистенции.
Паста-полуфабрикат может быть разбавлена раствором
пленкообразующего или смесью растворителей. Соотношение
между растворителями и разбавителями зависит от содержа-
ния полимера в смеси, типа применяемого растворителя и спо-
собности данного пигмента повышать вязкость; паста должна
быть достаточно вязкой, чтобы не происходило осаждение пиг-
мента, но в то же время достаточно текучей, чтобы в даль-
нейшем ее легко было смешать с другими ингредиентами, вхо-
дящими в состав готового материала. Если растворитель (ме-
тилизобутилкетон) и разбавитель (толуол) взяты в соотноше-
нии 55:45, хорошие результаты получаются при содержании
в пасте 20—23% винилита VYHH.
Приготовление паст и «чипсов» на основе газовой сажи.
Диспергирование газовой сажи с низким показателем черноты
очень затруднено вследствие наличия чрезвычайно мелких ча-
стиц и их склонности к поглощению жидкостей и газов. Поэтому
газовую сажу рекомендуется предварительно обрабатывать ам-
миаком или смачивать водным или спиртовым раствором ам-
миака. Спирт во время работы быстро испаряется, тогда как
вода удаляется труднее.
При введении слишком большого количества аммиака па-
ста-полуфабрикат приобретает явную тенденцию к желатини-
206
Гл. IV. Виниловые полимеры
рованию при хранении; кроме того, количество аммиака долж-
но быть минимальным для достижения высокого блеска плен-
ки (|так как блеск зависит от содержания газовой сажи, дать
какие-либо точные цифры затруднительно). Тем це менее для
сажи с показателем черноты, близким к 60, можно использо-
вать приблизительно 0,3%-ные (по отношению к весу сажи)
растворы аммиака в метиловом спирте. Пасты газовой сажи
можно также обрабатывать аммиаком, растворяя аммиак в том
растворителе, который применяется для растворения чипса, но
этот метод приводит к частичному желатинированию. Углево-
дороды ухудшают качество пасты газовой сажи; ароматиче-
ские углеводороды сильно уменьшают блеск пленки. Поэтому
чипсы растворяют обычно в истинных растворителях, а затем
добавляют ароматические углеводороды.
Пигментная смесь на основе газовой сажи имеет следующий
примерный состав (в %):
Обработанная газовая сажа . . .... 20
Винилит VYHH ......................... .... 60
Бутоксигликольфталат ........14
Паста-полуфабрикат содержит 75% пигментной смеси и 25%
метилэтилкетона.
Пасты на основе красных пигментов. Большинство красных
кадмиевых пигментов пригодно для паст. Для исходной пиг-
ментной смеси принимается следующий примерный состав
(в %):
Красный кадмиевый ... . ..........80
Винилит VYHH....................... ..... 15
Бутоксигликольфталат................... . 4
Касторовое масло оксидированное ... . 1
Паста-полуфабрикат содержит 70% пигментной смеси и
30% смеси растворителей и разбавителей. Применяемая в этом
случае смесь «Т» имеет состав (в %):
Метилизобутилкетон ....................... 40
Метилэтилкетон . . . 10
Толуол . 40
Ксилол .... 10
Толуидиновые красные пигменты также пригодны для паст
и позволяют получать блестящие покрытия, имеющие, однако,
склонность тускнеть с течением времени. Эти покрытия устой-
чивы даже при высокой влажности. Основная пигментная смесь
может иметь состав (в %):
Толуидиновый красный .................... .35
Винилит VYHH.............................. 55
Бутоксигликольфталат ................. . . 8
Касторовое масло оксидированное............ 2
Растворители и вспомогательные материалы
207
Паста-полуфабрикат содержит 35% этой смеси и 65% смеси
летучих компонентов «Т» (см. стр. 206).
Индантреновые красные пигменты дают очень устойчивые
блестящие покрытия, пригодные для наружных окрасок. Эти
пигменты не мигрируют (хотя некоторые из них имеют склон-
ность к миграции) при следующем составе (в %) исходной
пигментной смеси:
Индантреновый красный................. 15
Винилит VYHH . . ......... 75
Бутоксигликольфталат ....... 8
Касторовое масло оксидированное .... 2
Полуфабрикат содержит 25% исходной смеси в 75% сме-
си «Т» (см. стр. 206).
Пасты на основе желтых пигментов. В качестве желтых
пигментов могут применяться пигменты типа «Ганза», кото-
рые благодаря непрозрачности имеют хорошую укрывистость,
но склонны к миграции. Желтые кроны, рекомендуемые для
наружных окрасок, имеют очень хорошую укрывистость, хоро-
шо диспергируются и оказывают стабилизирующее действие на
виниловые полимеры.
Исходная пигментная смесь имеет следующий примерный
состав (в %):
Желтый крон . . ..........80
Винилит VYHH . . . . 15
Бутоксигликольфталат . .... 5
Представляют интерес также кадмиевые желтые пигменты,
которые могут применяться по той же рецептуре.
Могут применяться и другие органические красители (на-
пример, типа литозола); они склонны к миграции, хотя во
всем остальном дают хорошие результаты. Применяют также
стронциевые кроны, особенно для антикоррозионных грунтов.
Пасты на основе оранжевых пигментов. Оранжевые кроны
дают удовлетворительные результаты, они имеют хорошую
укрывистость, легко диспергируются и хорошо стабилизируют
виниловые полимеры при нагревании. Могут применяться тай-
же органические оранжевые пигменты, но их недостатком яв-
ляется склонность к миграции.
Пасты на основе синих пигментов. Синие индантреновые пиг-
менты яркого цвета склонны к миграции, имеют слабую укры-
вистость и к тому же трудно диспергируются. Состав исходной
пигментной смеси (в %):
Индантреновый синий ................. .... 15
Винилит VYHH ..............................75
Бутоксигликольфталат ......................10
208
Гл. IV. Виниловые полимеры
Паста-полуфабрикат содержит 25% исходной смеси и 75%
растворителей «Т» (см. стр. 206).
Синие фталоцианиновые пигменты дают прекрасные пасты
при том же составе.
Пасты на основе зеленых, пигментов. Окись хрома обнару-
живает очень большую светостойкость при хорошей укрыви-
стости. Ее применяют в смесях следующего состава (в %):
Окись хрома .... 79
Винилит VYHH .... 15
Бутоксигликольфталат .... 5
Касторовое масло оксидированное . 1
Полуфабрикат содержит 67% исходной смеси и 33% раство-
рителей «Т» (см. стр. 206).
Зеленые фталоцианиновые пигменты применяются в тех же
условиях, что и синие фталоцианиновые.
Пасты на основе белых пигментов. В качестве белого пиг-
мента применяется окись сурьмы в смеси с двуокисью титана
(на I вес. ч. окиси сурьмы берут 1 вес. ч. двуокиси титана).
Кроющая способность окиси сурьмы ниже, но она повышает
устойчивость к мелению, особенно в наружных окрасках, и ока-
зывает стабилизирующее действие на виниловые полимеры по
отношению к световому и тепловому старению.
Двуокись титана —очень хороший пигмент (в форме ру-
тила). Исходная пигментная смесь имеет следующий состав
(в %):
Двуокись титана (рутил) ... 68,5
Окись сурьмы ............... . . 7,5
Винилит VYHH ... ... .15
Бутоксигликольфталат............... 8
Касторовое масло оксидированное......... 1
Паста-полуфабрикат содержит 67% исходной смеси и 33%
растворителей «Т» (см. стр. 206).
Титанат свинца, несмотря на слегка желтоватый оттенок,
часто применяется для наружных окрасок, так как повышает
устойчивость к мелению. Он применяется или один, или в сме-
сях, содержащих 3—4 вес. ч. двуокиси титана на 1 вес. ч. ти-
таната свинца. Титанат свинца замедляет выцветение красок
вследствие своей способности поглощать ультрафиолетовые лу-
чи. Он также оказывает стабилизирующее действие на вини-
ловые полимеры при высоких температурах.
Окись цинка каталитически ускоряет термическое разложе-
ние поливинилхлорида. Поэтому ее не следует вводить в мате-
Товарные лакокрасочные материалы
209
риалы, которые могут подвергаться действию температуры вы-
ше 130°. Тем не менее она может применяться в смеси с дру-
гими пигментами, например с двуокисью титана. Смешение
должно производиться перед вальцеванием. Основное назначе-
ние окиси цинка — повышение устойчивости к мелению и твер-
дости пленки. Рекомендуется применять препараты окиси цин-
ка, содержащие некоторое количество гидроокиси.
Состав пигментной смеси (в %):
Двуокись титана........................ 34,5
Окись цинка .................... ... 35
Окись сурьмы . .7,5
Винилит VYHH ...... . . 15
Бутоксигликольфталат ... . . . 7
Касторовое масло оксидированное ......... 1
Пигменты, придающие термостабильность. В большинстве
случаев это свинцовые пигменты, содержащие основной суль-
фат свинца, основной фосфит (наиболее часто применяется),
силикат и основной карбонат свинца. Может применяться и
свинцовый глет, но он имеет склонность темнеть. Все эти про-
дукты можно вводить в количестве 3—6% по отношению к
полимеру.
Основная рабочая смесь для вальцевания имеет следующий
состав (в %):
Соли свинца . .80
Винилит VYHH ... 15
Бутоксигликольфталат 5
ТОВАРНЫЕ ЛАКОКРАСОЧНЫЕ МАТЕРИАЛЫ
Получение товарных продуктов несложно, если правильно
приготовлены пигментная паста и раствор полимера. Предвари-
тельно составленную пигментную пасту гомогенизируют путем
непродолжительного перемешивания, затем добавляют пласти-
фикаторы и другие ингредиенты. По окончании этой операции
продукт рекомендуется профильтровать.
Эмалевые краски для наружных покрытий. В краски для
наружных покрытий необходимо вводить достаточное количе-
ство пигмента для обеспечения хорошей укрывистости. Однако
избыток пигмента может ухудшать свойства конечного про-
дукта. Очень важна светостойкость пигмента, который должен
в течение длительного времени сохранять свой цвет и одновре-
менно оказывать стабилизирующее действие на полимер. Наи-
лучшие результаты дают пигменты, отличающиеся наиболее
высоким поглощением ультрафиолетовых лучей; с этой точки
14—156
210 Гл. IV. Виниловые полимеры
зрения лучшим является титанат свинца. Некоторые новые
сорта двуокиси титана дают те же результаты.
Устойчивость прозрачных материалов, в частности красок
на основе синих органических красителей, может быть улуч-
шена путем добавления продуктов типа фталата свинца (ди-
тал фирмы National Lead Company), обладающего слабой укры-
вистостью, но большой способностью к поглощению ультрафи-
олетовых лучей.
Для белых красок эта задача более трудна. Кроме разно-
видностей двуокиси титана, можно добавлять большие коли-
чества окиси сурьмы, уменьшающей явление меления. Ти-
танат свинца также сильно уменьшает меление; окись цинка,
хотя и несколько хуже, но тоже может применяться при усло-
вии смешения ее перед диспергированием с другим пигментом,
например с двуокисью титана. Кроме того, пленки, содержа-
щие окись цинка, не должны подвергаться действию темпера-
туры выше 130°. Для белых эмалей рекомендуется применять
пленкообразующие типа VYLF (2 вес.’ ч.) в смеси с 3 вес. ч.
полимера с более высоким молекулярным весом (это позво-
ляет увеличить содержание сухого остатка и облегчает обра-
ботку). Такие продукты имели очень хорошие показатели в
условиях естественного старения.
Материалы для внутренних покрытий. К наполнителям ма-
териалов, предназначенных для внутренних покрытий, где не
требуется атмосферостойкость, предъявляются менее серьезные
требования. Однако выбор пигмента для покрывных белых эма-
лей, наносимых в один слой, — весьма ответственная задача.
В этом случае рекомендуется применять двуокись титана с ма-
лой маслоемкостью. Введение оксидированного касторового
масла (или его производных) нецелесообразно, так как при
этом не достигается хорошего диспергирования при вальцева-
нии. Касторовое масло можно, правда, вводить после вальце-
вания, так как оно обусловливает лучшую светостойкость при
последующей термообработке, в особенности, если компози-
ция не содержит окиси сурьмы.
В матовые покрытия можно добавлять от 3 до 6% (по от-
ношению к полимеру) наполнителей типа силикатных диато-
митов.
Горячая сушка покрытий. Операцию горячей сушки (или
запекания в печи) следует тщательно контролировать; должна
поддерживаться минимальная температура, необходимая для
хорошей адгезии пленки к подложке. Кроме того, следует стре-
миться к равномерному распределению тепла, чтобы избежать
местных перегревов. Замена длительной сушки на холоду крат-
ковременной сушкой при высокой температуре практически воз-
можна только для покрытий, высыхающих за счет окисления или
Товарные лакокрасочные материалы 211
содержащих термореактивные, а не термопластичные полимеры.
Тогда при горячей сушке вначале происходит испарение раство-
рителей, а затем размягчение материала покрытия, который
как бы частично расплавляется на поверхности, что приводит
к улучшению адгезии и повышению блеска.
Горячую сушку лучше всего проводить в сушильных каме-
рах с циркуляцией воздуха и с хорошим распределением тепла.
Для сушки темных покрытий, нанесенных на тонкие металли-
ческие листы, на бумагу или ткань, применяются отражатель-
ные печи. В отражательных печах можно устанавливать лампы
инфракрасного света, но при этом покрытие необходимо
предварительно прогревать для испарения растворителя; в зоне
радиации также должна быть предусмотрена циркуляция
воздуха.
При сушке в инфракрасных лучах могут быть достигнуты
высокие температуры, вызывающие плавление пленкообразу-
ющего; облучение инфракрасными лучамй может быть исполь-
зовано дополнительно в установках, не позволяющих достигать
достаточно высоких температур.
Стадия расплавления полимера должна наступать после пол-
ного испарения смеси растворителей; если в состав этой смеси
входят тяжелые растворители, время их улетучивания может
быть продолжительным. При нанесении покрытий на бумагу,
напротив, лучше результаты достигаются, если тепловое воздей-
ствие начинается раньше, чем растворитель полностью испа-
рится. Однако сразу начинать сушку при высокой температуре
можно только при использовании тяжелых растворителей, так
как при наличии легко летучих растворителей непосредствен-
ное помещение в зону высокой температуры приведет к обра-
зованию пузырей. В этом случае нагревание должно произво-
диться постепенно, что облегчается при использовании длинной
печи. Такую печь разделяют на несколько зон: первая — низ-
котемпературная, в которой происходит удаление растворителя
при низкой температуре, и следующие высокотемпературные, в
которых завершается сушка.
Циркуляцию воздуха следует регулировать так, чтобы не
происходило скопление паров, могущее привести к взрыву. Надо
отметить, что при концентрации паров порядка 2% объемн.
самовоспламенение или взрыв могут произойти при темпера-
турах, начиная с 275°.'
Основные рецептуры. Ниже приводится несколько практи-
чески испытанных рецептур, которые могут быть приняты за
основу при промышленном производстве; эти рецептуры раз-
работаны фирмой Bakelite (отделение Union Carbide and Carbon
Chemicals).
14
212
Гл. IV. Виниловые полимеры
I. Подводные краски для пресной воды
Грунт — 1 слой, сушка на воздухе 1 час.
Покрытие — 2 слоя, сушка на воздухе.
Состав грунта (в %):
Двуокись титана................................................... 12
Винилит VYHH-1 или родопас АХ....................................... 8
Винилит VMCH ....................................................... 8
Трикрезилфосфат..................................................... 3
Растворители (смесь Б, см. табл. 16 на стр. 202) . ................ 69
Состав покрытия (в %):
Окись цинка (Horse Head ХХ50).........................................4,5
Двуокись титана .......................................................5,2
Окись сурьмы........................................................ 2,3
Винилит VYHH-1 или родопас АХ...................................... 16,0
Кронизол (дибутилцеллозольв-фталат)...................................1,3
Касторовое масло оксидированное ....................................... 0,3
Трикрезилфосфат........................................................ 2,3
Растворители (смесь Б, см. табл. 16 на стр. 202) . ......... . . 67,7
В эту рецептуру могут быть введены также диоктилфталат или пластицайзер Е-40
II. Подводные краски повышенного качества
Грунт—1 слой, сушка на воздухе 1 час.
Покрытие (состав см. выше) — 2 слоя, сушка на воздухе.
Применяемый в данных композициях грунт был разработан специально для
подводных окрасок. Он получается при смешении равных количеств композиций
а и б непосредственно перед употреблением.
Состав композиции а (в %):
Тетраоксихромат цинка ............................................. 7,5
Мультифлекс ММ (карбонат кальция)...................................2,5
Винилит VYHH-1 .....................................................9,9
Трикрезилфосфат.....................................................1,7
Растворители (смесь Б, см. табл. 16 на стр. 202).................. 78,4
Состав композиции б (в %):
Винилит VMCH . 10,0
Трикрезилфосфат . 2,4
Метилизобутилкетон . 22,4
Толуол.......................................................... 58,1
Бутилацетат (98%-ный) ......................................... 7,1.
III. Краски для холодильников (по листовой стали)
Грунт— 1 слой, сушка 30 мин. при 175°.
Покрытие — 1 слой, сушка 30 мин. при 150°.
Поквытие устойчиво при очень высокой влажности воздуха.
Товарные лакокрасочные материалы 213
Состав грунта (в %):
Основной сульфат свинца 15,3
Двуокись титана . ....... 6,7
Окись сурьмы......... ... . 0,7
Винилит VYHH-1 ..................... . . . 14,9
Кроннзол (дибутилцеллозольв-фталат) 4,8
Касторовое масло оксидированное........... . . . . 0,3
Растворители (смесь В, см. табл. 16 на стр. 202) . ... 57,3
Состав покрытия (в %):
Двуокись титана . ......... .......... 10,5
Окись сурьмы............... . . . . ... 1,2
Винилит VYHH-1 . . ........................... 17,5
Кронизол .................. . 1,2
Растворители (смесь В, см. табл. 16 на стр. 202) .... 69,4
Касторовое масло оксидированное............................. . 0,2
IV. Краски для медицинского оборудования
Покрытие—2 слоя, сушка 15 мин. при 165°.
Состав покрытия (в %):
Двуокись титана................................................ 12,2
Винилит VYHH-1 ............................................ ... 15.9
Сантицайзер В-16 (фирмы Monsanto)............................ . 4,3
Мочевино-формальдегидная смола................................ 0,4
Алкидная смола (немодифицированная) ............................ 0,2
Окись пропилена- ............................................... 0,5
Целлозольв.................................................... 2,7
Растворители (смесь В, см. табл. 16 на стр. 202)............... 63,8
V. Краски общего назначения для наружных работ
Грунт—1 слой, сушка 30 мин. при 180°.
[ Покрытие—1 слой, сушка 15 мин. при 145°.
Состав грунта (в %):
Металлический свинец (сублимированный) .................. ... 31,3
Винилит VYHH-1 ......................................... .... 13,0
Кронизол (дибутилцеллозольв-фталат) ............................... 3,4
Растворители (смесь В, см. табл. 16 на стр. 202) .... .... 52,3
Состав покрытия (в %):
Красный кадмиевый пигмент .......................................... 10,7
Винилит VYHH-1 ................................................... 9,0
Винилит VYLF....................................................... 6,0
Кронизол .......................................................... 2,5
Касторовое масло оксидированное..................................... 0,1
Растворители (смесь В, см. табл. 16 на стр. 202).................... 71,7
214
Гл. IV. Виниловые полимеры
VI. Покрывные краски для наружных работ
Грунт (см. Краски для холодильников)—! слой, сушка 30 мин. при 360°.
Покрытие—I слой, сушка 30 мин. при 290°.
Состав покрытия (в %):
Двуокись титана...................................................... 6,8
Окись цинка . ...................................................... 5,8
Окись сурьмы......................................................... 2,8
Винилит VYHH-1....................................................... 16,0
Трикрезилфосфат...................................................... 3,0
Кронизол (дибутилцеллозольв-фталат)................................. 1,4
Касторовое масло оксидированное ..................................... 0,2
Растворители (смесь В, см. табл. 16 на стр. 202)..................... 64,0
Для повышения светостойкости рекомендуется вводить стабилизатор—основ-
ной фосфит свинца (днфос).
VII. Химически стойкие покрытия
Грунт—1 слой, сушка на воздухе 1 час.
Покрытие—1 слой, сушка на воздухе.
Состав грунта (в %):
Хромат цинка ............ ........................................
Желтый крон ......................................................
Винилит VYHH-1 или родопас АХ............................... . . .
Винилит VYDR .....................................................
Винилит VMCH .....................................................
Трикрезилфосфат...................................................
Кронизол (дибутилцеллозольв-фталат)...............................
Циклогексанон.....................................................
Метилэтнлкетон ...................................................
Растворители (смесь В, см. табл. 16 на стр. 202)..................
3,0
3,0
1,2
5,5
7,0
23’,О
17,5
37,8
Состав покрытия (в %):
Газовая сажа ......................................................... 0,9
Винилит VYHH-1 или родопас АХ................................... 3,0
Винилит VYDR ......................................................... 8,4
Кронизол (дибутилцеллозольв-фталат) .................................. 0,6
Трикрезил фосфат.................................................... 1,5
Циклогексанон...................................... . 35,0
Метилэтнлкетон ..................................................... 26,7
Растворители (смесь В, см. табл. 16 на стр. 202) . . .......... 24,0
VIII. Покрытия для водных бассейнов
Грунт—1 слой, сушка 12 час.
Покрытие—2 слоя, сушка на воздухе.
Состав грунта (в %):
Двуокись титана . ......... . . . 6,9
Окись сурьмы . . ... ... 3,9
Винилит VYHH-1 . . ...... . . 9,7
Винилит VMCH ...................... 8,0
Трикрезилфосфат ... ......................................... 3,6
Растворители (смесь Б, см. табл. 16 на стр. 202)........ . . 68,2
Материалы на основе поливинилбутираля 215
Состав покрытия (в %):
Двуокись титана 4,57
Окись сурьмы . . (|,50
Винилит VYHH-1 , . 15,00
Кронизол (дибутилцеллозольв-фталат) 0,53
Касторовое масло оксидированное 0,07
Трикрезилфосфат.................................................... 2,50
Растворители (смесь Б, с.м. табл. 16 на стр. 202) ................ 76,83
IX. Покрытия для промышленных металлических конструкций
Грунт—1 слой, сушка 30 мин. на воздухе.
Промежуточный слой—1 слой, сушка на воздухе.
Покрытие—1 слой (состав см. Покрывные краски для наружных работ), сушка
на воздухе.
Такие трехслойные покрытия применяются для производственных помещений
и для наружных окрасок.
Состав грунта (в %):
Окись цинка . . . . ......................................18,7
Хромат цинка ....................................................... 7,5
Кремнезем .... ...................................... 4,2
Асбестин ........................................ ............. . - . 4,2
Бакелит ВК-3962 . ................................'.............. 27,0
Бакелит XJ-14424 .... .............. 11,4
Ксилол ......................................................... 27,0
Состав промежуточного слоя (в %):
Окись цинка (Кадоке).............................. . . . . 39,5
Винилит VYHH-1 . . .... .... 12,6
Бекацит 1120 . ............... .............. 2,0
Кронизол (дибутилцеллозольв-фталат)................................. 3,0
Растворители (смесь В, см. табл. 16 на стр. 202) .... .... 42,8
X. Покрытия для наружной окраски консервных банок
Покрытие—1 слой, сушка 15 мин. при 165е.
Состав покрытия (в "о):
Винилит VYHH-1 или родопас АХ.................................... 19,4
Мочевино-формальдегидная смола.................................... 0,2
Алкидная смола невысыхающая ................................. . . 0,1
Целлозольв . .1,4
Растворитель (смесь Б, см. табл. 16 на стр. 202)................. 78,9
Покрытие наносится на хромированные поверхности.
МАТЕРИАЛЫ НА ОСНОВЕ ПОЛИВИНИЛБУТИРАЛЯ
За последние годы особенно широкое распространение при-
обрели грунты на основе поливинилбутираля, содержащие хро-
маты цинка и свинца, фосфат хрома и фосфорную кислоту. Та-
кие грунты были разработаны фирмой Union Carbide and
Chemicals Corporation совместно с морским ведомством США.
216
Гл. IV. Виниловые полимеры
В .настоящее время проведены теоретические исследования этих
грунтов, позволяющие глубже познать механизм пассивирова-
ния металлов62. Рецептуры фосфатирующих грунтов были опу-
бликованы в журнале «Peintures, Pigments, Vernis»63. Здесь
будут указаны лишь главные компоненты этих грунтов и при-
ведены примерные рецептуры специальных лаков, в состав ко-
торых входят поливинилбутирали.
Применяемые поливинилбутирали должны быть близки по
свойствам к продуктам, выпускаемым фирмой Bakelite Corpo-
ration в США или по лицензиям в ФРГ. В частности, они долж-
ны содержать не более 2% ацетильных групп; сорта этих поли-
меров, выпускаемых в Европе с большим содержанием ацетиль-
ных групп (20%), непригодны для этих целей.
Показатели поливинилбутиралей американской фирмы
Bakelite:
Винилит Винилит
XYHL XYSG
Внешний вид Белый порошок То же
Относительная вязкость 0,81 1,16
Удельный вес, г/сл8.................... 1,12 1,12
Примерный состав, %:
винильные группы . 54,4 54,4
бутиральные группы . . . 38,3 38,3
ацетильные группы . . . 0,3 0,3
гидроксильные группы . . 7 7
Подобные продукты изготовляются также фирмой Monsanto
под названием бутвар. Продукт марки бутвар В 76 представ-
ляет собой низковязкий полимер с низким содержанием гидр-
оксильных групп (11,5—12,5% поливинилового спирта и менее
2,5% поливинилацетата); бутвар В 72-А, напротив, является
высоковязким полимером с ‘повышенным содержанием гидрок-
сильных групп (17,9—19,8% поливинилового спирта).
Покрытия на основе всех этих материалов характеризуются
высокой ударной вязкостью, исключительной адгезией к дереву,
металлам, коже и к большей части пластических «масс, способ-
ностью не изменять цвета под действием тепла и света, очень
большой твердостью и одновременно хорошей эластичностью.
Кроме того, одним из основных достоинств этих покрытий яв-
ляется их способность переходить со временем в необратимое
состояние; их поверхность приобретает при этом очень боль-
шую устойчивость к растворителям, повышается температура
размягчения покрытия и значительно улучшается водостой-
кость. При погружении в воду покрытие набухает лишь до
определенного максимума, выше которого поглощение воды
прекращается. Поэтому такие покрытия значительно устойчи-
вее дпугих лакокрасочных материалов, которые могут иметь
Материалы на основе поливинилбутираля
217
вначале даже меньшую проницаемость, но при нахождении
в воде постепенно разрушаются (рис. 48).
Покрытия на основе поливинилбутиралей, нанесенные на
сталь, при погружении в воду сохраняют 'постоянное электри-
ческое сопротивление, если пигментом является хромат цинка
(25% объемн.). Определения производились с помощью про-
тектометра, позволяющего определять электрическое сопротив-
ление покрытий и их защитные свойства64.
В композиции на основе этих материалов можно вводить
очень большие количества пигментов; поливинилбутирали мо-
гут быть смешаны также с
большими количествами обще-
употребительных низкомолеку-
лярных смол (таких, как эфи-
ры канифоли), без заметной
потери стойкости к различным
физико-химическим агентам и
механическим воздействиям.
Обработка на вальцах по-3
вышает растворимость и тем-
пературу размягчения поливи-
нилбутиралей, поэтому ее сле-
дует вести осторожно, так как
после такой обработки приме-
нение этих материалов может
оказаться весьма затрудни-
тельным. Для регулирования
температуры размягчения мож-
но добавлять спирт; дисперги-
ровать в этих полимерах мож-
но только нереакционноспособ-
ные пигменты. Можно добав-
Рис. 48. Сравнительная водостойкость
пленок поливинилбутнраля и полнви-
нилацетата (R-—сопротивление в ,омах
па 1 см2 поверхности):
/—поливинилбутираль; 2—поливинилацетат.
лять пластификаторы' для повышения эластичности (что бы-
вает необходимо в некоторых особых случаях) или для пони-
жения вязкости.
Рассматриваемые типы поливинилбутиралей совместимы с
большим числом материалов; это используется для их модифи-
кации, главным образом путем добавления феноло-формальде-
гидных, мочевино-формальдегидных или глиоксалево-формаль-
дегидных смол. Смолы последнего типа образуются на воздухе,
но в присутствии воды, поэтому их растворы не желатинируют-
ся в водной среде, а пленки восприимчивы к воде. Касторовое
масло, хотя и совмещается с поливинилбутиралями, но смяг-
чает пленку, не повышая ее эластичности. Поливинилбутирали
несовместимы с ацетатом, ацетобутиратом и пропионатом цел-
люлозы, хлоркаучуком, кумароно-инденовыми смолами.
218
Гл. IV. Виниловые полимеры
Поливинилбутирали растворимы в спиртах, растворителях
типа целлозольва и в сложных эфирах. Характеристика их рас-
творимости приведена на рис. 49.
Содержание второго компонента смеси растворителей.
Рис. 49. Вязкость растворов винилита VYHL в различных рас-
творителях (по истечению через насадку Форд-Энглера № 4):
/—синазол—дихлорэтан; 2—синазол—толуол; 3—сивазол—бутилацетат; 4—св-
назол—ацетон; 5—синазол—тролуойль.
Эти полимеры растворимы в продажных водных спиртах,
но нерастворимы или образуют гели в безводных спиртах. Из
графика, приведенного на рис. 50, видно, что для данного
растворителя оптимальным является содержание 5% воды. Хо-
тя этанол и метанол являются наиболее активными раствори-
телями, в смесях, содержащих 40% спирта и 60% сложных
эфиров, кетонов или ароматических углеводородов, получаются
более низковязкие растворы, чем в чистых спиртах. Изопропи-
ловый спирт дает такой же эффект в смесях с ацетоном и
толуолом: минимальной вязкостью обладают растворы в сме-
сях, содержащих 60% изопропилового спирта (91%-ного) и 40%
ацетона. Так, вязкость 17%-ного раствора полимера в смеси,
содержащей 30% изопропилового спирта и 70% ацетона, со-
ставляет 800 сек. (по Форд-Энглеру, сопло № 4), а в смеси,
содержащей 60% изопропилового спирта и 40% ацетона, вяз-
кость понижается до 400 сек.
На основе поливинилбутиралей изготовляются небьющиеся
стекла, причем производство их расширяется. Поливинилбути-
рали могут быть применены в качестве клеев для склеивания
резины, каучука, пробки, асбеста, дерева, бумаги и металла
с пластическими массами, а также с полимерами, отвержда-
ющимися при нагревании. После термообработки ‘продукт ста-
Материалы на основе поливинилбутираля
219
Содержание воды В растворителе, %
Рис. 50. Вязкость растворов поливинилбутира-
ля в зависимости от содержания воды.
Состав раствора в точке А (в г): винилит
XYHL—17,9; ацетон—33,2; водаЦ-безводный
изопропанол—49,8.
новится стабильным при температуре до 100° и не размягчается
при действии растворителей и воды. Клеящая способность этих
полимеров используется также для модификации феноло-форм-
альдегидных смол, применяемых в производстве фанеры. Уве'
личение содержания поливинилбутираля по отношению к фе-
ноло-формальдегидной смоле приводит к повышению прочно-
сти при растяжении, эластичности, ударной вязкости и к пони-
жению стойкости к действию кипящей воды.
Применение покрытий по дереву на основе одних полибу-
тиралей не рекомендуется
вследствие повышенного
поглощения ими воды;
однако в смеси с други-
ми полимерами поливи-
нилбутирали дают пре-
восходные покрытия. Боль-
шой интерес представля-
ет применение их в смеси
с чешуйчатым шеллаком,
в соотношении 50; 50; по-
лучаемая при этом пленка
может шлифоваться, прек-
расно полируется и зна-
чительно более устойчива
к влаге, чем пленка чи-
стого поливинилбутира-
ля. На основе таких сме-
сей можно получать рас-
творы с более высоким
содержанием сухого остатка, чем из одного поливинилбутираля.
Смеси поливинилбутиралей с фенольными смолами могут
применяться для изготовления лаков по дереву. Такие покры-
тия применяются для деревянных емкостей, в которых хранятся
алифатические углеводороды, а также некоторые химические
продукты.
Композиции с фенольными смолами оказались очень удач-
ными также в грунтах, применяемых для наружной окраски
домов, и в эмалевых красках; адгезия их улучшается при на-
несении на влажную подложку.
Очень интересной областью применения поливинилбутираля
является обработка смолистого дерева. Слои поливинилбути-
раля, даже совсем тонкие, полностью предотвращают вытека-
ние смолы из дерева, что позволяет использовать обработанное
таким образом дерево для изделий, для которых раньше оно
не могло применяться вследствие вытекания смолы, сильно
портившей его поверхность.
220
Гл. IV. Виниловые полимеры
Эластичность и ударная прочность покрытий на основе фе-
нольных смол также могут быть повышены путем добавления
поливинилбутираля; в частности, при обработке консервных
банок рекомендуется вводить не менее 20% поливинилбутираля
по отношению к фенольной смоле, что во много раз повышает
эластичность пленки.
Фосфатирующие грунты WASH PRIMER
Производство фосфатирующих грунтов было реализовано
во время второй мировой войны в США, но сведения о них не
публиковались до 1948 г.
Фосфатирующий грунт выполняет две основные функции:,
придает покрытию превосходную адгезию, не превзойденную ни-
каким из грунтов, и хорошие антикоррозионные свойства. Ра-
зумеется, фосфатирующий грунт не заменяет многослойных
антикоррозионных покрытий, но применяется как грунт для
первичного покрытия. Комплекс полимер—фосфат—хромат об-
разуется из смеси поливинилбутираля, хромата и фосфорной
кислоты в растворе. Этот комплекс создает превосходную адге-
зию к большинству металлических подложек. Применение рас-
творителя, состоящего в основном из спирта, позволяет нано-
сить грунт в большом диапазоне температур и в условиях влаж-
ной атмосферы; можно наносить пленки различной толщины,
причем они очень быстро высыхают на воздухе, образуя эффек-
тивное антикоррозионное покрытие. Большая часть красок и
покрытий имеют прекрасную адгезию к фосфатирующему
грунту.
Первоначально разработанный грунт составлялся из двух
компонентов: основы, содержавшей поливинилбутираль с хро-
матом цинка или хроматом свинца, и кислотного разбавителя,
содержавшего фосфорную кислоту; оба компонента смешива-
лись перед употреблением.
Состав фосфатирующих грунтов WP-1 и ХЕ-5220 приведен
в табл. 18.
Основа приготовляется путем перетира в шаровой мельнице
с фарфоровыми шарами или галькой; применения стальных
мельниц и стальных шаров следует избегать, чтобы не допу-
стить загрязнения железом. Компоненты грунта должны
выпускаться в инертной таре (либо стеклянной, либо по-
крытой феноло-формальдегидной смолой или виниловым по-
лимером) .
Нанесение фосфатирующего грунта может производиться
кистью, пульверизатором или методом окунания; при очень ма-
лой концентрации (в расчете на сухое вещество) он проникает
во впадины, а не покрывает их пленкой.
Материалы на основе поливинилбутираля
221
ТАБЛИЦА 18
Фосфатирующие фунты
Компоненты Состав, %
WP-I XE-5220
Основа
Винилит XYHL или бутвар В 76
фирмы Monsanto ..................
Основной хромат цинка (тетраокси-
хромат цинка ....................
Хромат свинца с малым содержа-
нием растворимых солей . . . .
Тальк (асбестин ЗХ)..............
Изопропиловый спирт (99%-ный) .
Толуол ..........................
Метилэтилкетон...................
7,2 9,0
6,9 —
8,6
1,1 1,4
50,4 53,0
14,4 —
— 13,0
80,0 85,0
2,9
9,2
2,9
Кислотный разбавитель
Фосфорная кислота (85%-ная) . .
Изопропиловый спирт (99%-ный) .
Вода...........................
3,4
13,2
3,4
20,0
15,0
Рекомендуется предварительная подготовка поверхности пу-
тем пескоструйной обработки или обезжиривания. Хотя доста-
точная адгезия фосфатирующего грунта достигается даже при
необработанной поверхности, однако, если поверхность не обез-
жирена, не получается хороших результатов.
Фосфатирующий грунт типа WP-1 благодаря своей хорошей
адгезии применяется для нанесения по большинству металлов
(сталь, цинк, кадмий, олово, алюминий, оцинкованное железо);
особенно он рекомендуется для стали. Его жизнеспособность
после смешения с раствором кислоты составляет 8 час.; далее
свойства пленки ухудшаются. Можно предположить, что слож-
ный процесс взаимодействия поливинилбутираля с хроматом
цинка и металлом, который должен происходить после нанесе-
ния грунта, начинается еще в смеси, теряющей вследствие этого
свои адгезионные свойства. Следует отметить, что этот про-
цесс не сопровождается никакими видимыми изменениями
раствора; поэтому необходимо точно соблюдать даваемые здесь
указания, чтобы не получить неожиданно плохих результатов.
В рецептуре WP-1 на 1 вес. ч. кислотного разбавителя при-
ходится 4 вес. ч. основы. Так как удельные веса основы и рас-
222 ’ Гл. IV. Виниловые полимеры
твора кислоты приблизительно равны, можно пользоваться как
весовыми, так и объемными соотношениями. Добавление раз-
бавителя следует производить при перемешивании, чтобы из-
бежать местной коагуляции.
Основа ХЕ-5220 значительно более стабильна, что позволяет
применять ее даже через несколько недель после смешения,
но она пригодна для нанесения только на сталь и ее адгезия
проявляется значительно медленнее. Лишь через несколько дней
адгезия начинает 'приближаться к адгезии основы WP-1.
Оба типа фосфатирующих грунтов можно покрывать слоем
антикоррозионной или любой другой краски после сушки в
течение 15 мин. при комнатной температуре или 1—2 мин. при
повышенной температуре.
В США были разработаны также другие типы стабильных
фосфатирующих грунтов, пригодных для нанесения на все ме-
таллы. К этим грунтам хорошо прилипают краски всех типов,
за исключением некоторых нитроцеллюлозных эмалей и эма-
левых красок, изготовленных на основе таких виниловых со-
полимеров, как VYHH, родопас АХ и VMCH. Напротив, покры-
тия на основе винилита VAGH, содержащего гидроксильные
группы, обнаруживают превосходную адгезию и составляют
основу подводных красок, превосходящих по свойствам все ти-
пы красок, применявшихся до сих пор. Эти покрытия не реко-
мендуется наносить без грунта, но они создают временную за-
щиту, длительность которой зависит от условий эксплуатации.
Считается, что защита может быть достаточной в продолжение
нескольких месяцев, если объекты (например, стальные) нахо-
дятся в закрытом помещении, но если металл подвергается
непосредственному воздействию атмосферы, защитные свойства
проявляются в течение не более 2—3 недель. Такого рода ма-
териалы приобрели чрезвычайно широкое распространение для
подводных окрасок. Они действительно обладают, по сравне-
нию с ранее применявшимися продуктами, значительно луч-
шими свойствами (твердостью, адгезией, химической стойко-
стью), быстро высыхают (средняя продолжительность сушки
30—40 мин.), нечувствительны к влаге: при содержании не ме-
нее 25% бутанола их можно наносить на поверхность, на кото-
рой имеется небольшое количество влаги. Грунты на основе
этих продуктов замедляют коррозию, так как они пассивируют
металл. При покрытии обыкновенными красками коррозия на-
чинает развиваться в местах повреждения окрашенной поверх-
ности; при применении фосфатирующего грунта поцарапанная
часть окрашенной поверхности может быть очищена и снова
обработана грунтом, а последний — слоем защитной краски.
Обычно при обработке больших поверхностей производят
пескоструйную обработку по частям и немедленно наносят ан-
Материалы на основе поливинилбутираля 223
тикоррозионный слой. После полного высыхания окрашенной
части приступают к пескоструйной обработке соседней части,
так как песчинки могут прилипнуть к невысохшей пленке. Для
фосфатирующего грунта это несущественно, так как продол-
жительность его сушки составляет всего несколько минут.
Кроме применения для подводных покрытий, фосфатиру-
ющие грунты в сочетании с поливиниловыми покрытиями по-
лучили значительное распространение в нефтяной промышлен-
ности (покрытие нефтепроводов, цистерн для бензопродуктов
и масел), в автомобильной и химической промышленности, при
защитной окраске аппаратуры установок для кондиционирова-
ния воздуха, в частности лопастей вентиляторов, края которых
подвергаются особенно сильной коррозии.
В этих случаях важно, что фосфатирующий грунт создает
очень хорошую адгезию, а виниловые полимеры образуют
пленки, очень устойчивые к истиранию.
При применении фосфатирующих грунтов можно наносить
обычные краски на алюминий; грунт предварительно наносят
на листовой материал, затем штампуют изделия и после этого
покрывают их краской. Такая грунтовка рекомендуется для
улучшения адгезии при маркировке алюминиевых листов. При
обработке металла фосфатирующими грунтами значительно
улучшается адгезия и повышаются антикоррозионные свойства
алкидных, меламиновых, фенольно-масляных лаков и высыха-
ющих масел. Тем не менее, если требуется повышенная хими-
ческая стойкость покрытия, рекомендуется наносить на фос-
фатирующий грунт промежуточный слой винилового полимера.
ЛИТЕРАТУРА
1. Фр. пат. 719032, 1931 г.; 1. G. Farbenindustrie A. G.
2. Фр. пат. 741657, 1932 г.; англ. пат. 406338, 1914 г.. Carbide a. Carbon Chemi-
cals Corp.
3. Staudinger, Brunner, Flisst, Helv. chim. acta, 1930, 13,
805; C. A., 1931, 25, 487.
4. Sawson, Weintz, пат. США 1775832, 30/VIII 1932 г., (Du Pont
de Nemours); фр. пат. 709562, 1931 г.
5. Англ. пат. 379292 (Е. I. du Pont de Nemours, 1931); фр. пат. 717887, 1931 г •
British С. А., В., 1932, 997.
6. Marvel, Horning, Gilman’s Organic Chemistry, v. 1, p. 701.
7. E. V. R e i d, пат. США 1935577, 14/11 1933 г.. Carbide a. Carbon Chemi-
cals Corp., См. также Frazier—Groff, англ. пат. 388309, 1933 г., Carbide
a. Carbon Chemicals Corp.; фр. пат. 740962, 1932 г.
8. С. О. Young, S. D. D ou gl a s, пат. США 1990685,12/11 1935 г.,
Carbide a. Carbon Chem. Corp.; канал, пат. 333391, 1933 г.
9. Фр. пат. 640364, 1927 г.; англ. пат. 285049, 1927 г.; герм. пат. 590858,
1934 г.; пат. США 1838368, 29/ХП 1931 г.. Carbide a. Carbon Chem
Corp.
10. G. F. S t e i ger wa I t, Agr. Eng., 1933, 14, 154.
11. C. W. Patton, Bakelite Corp. New York; Paint a. Varnish Production
Club, February 1948; Official Digest, May 1948.
224
Гл. IV. Виниловые полимеры
12. A. R- Doolittle, G. М. Powell, New Vinyl resins for air dry
and low bake coatings, Paint, Oil Chem. Rev., April, 6, 1944.
13. W. E. Campbell, пат. США 2329456.
14. H. S t a u d i n ge r, Die Hochmolekularen organischen Verbindungen,
Berlin, Springer, 1932.
15. A. Voss, E. D i с к a u s e г, пат. США 2047398.
16. G. Selden, C. F. P r u 11 о n, Federation of Paint a. Varnish Produc-
tion Clubs Convention Chicago, Official Digest, 1941.
17. Resine Vinylite VMCH, Bakelite Corporation.
18. Resine Vinylite VAGH, Bakelite Corporation.
19. Resine Vinylite XYHL, Bakelite Corporation.
20. Resine Vinylite VYCC.
21. Resines Vinylites VYNS, VYCM, VYDR.
22. F. Chevassus, Stabilisation des chlorures de vinyie et copolymeres
vinyliques. Rev. Gen. Caoutchouc, 1950. 26, 337, 341.
23. V. W. F о x-H e n d r i с к s, H. J. R a 11 i, Ind. Eng., 1949, 41, 1774.
24. Handbook on stabilizer for vinyl resins, National Lead Company, New York
25. Advance Solvents and chemicals Corporation, New York.
26. A. W. Johnson, G H. Young, пат. США 2208216.
27. V. Y n g u e, пат. США 2267777, 2267778. 2267779.
28. G. Y о u n g, пат. США 2169717.
29. Bakelite wea XVU 15 765—XJ 14 424, Bakelite Corp.
30. D. L. Kent, P. J. Weaver, Methods for evaluating plasticizer for
polyvinyl chloride, India Rubb. World, March, 1947, p. 813, 817.
31. Vinylite Resins for Surface Coatings, p. 31, 37, Bakelite Corporation.
32. R. Quarles, Ketones as solvents for vinyl resins. Ind. Eng. Chem., 1943,
35, 1033.
33. K’e nneth Graver, The mechanism of plasticization in plastics. Bull.
*ASTM, 1948, May, № 152.
34. New Vinyl Resins for air dry and low bake coatings, Paint, Oil Chem. Rev.,
6/IV 1944.
35. W. E., пат. США 2329456.
35. Пат. США 2047398.
37. Official Digest Federation of Paint a. Varnish, Chicago, 1941, p. 149.
38. Plasticizers, Carbide a. Carbon Chem. Corp.
39. M. C. Reed, Behavior of plasticizers in vinyl chloride, Acetate resins,
Ind. Eng. Chem., August, 1943, 35.
40. M. C. Reed, L. Connor, то же, Ind. Eng. Chem., August, 1948, 40.
41. M. C. Reed, James Harding, Ind. Eng. Chem., 1949, 41.
42. R e e d, Survey of plasticizers for vinyl resins, J. Polymer Sci., 1947, 2,
115, 120.
43. R. F. Clash, R. M. Berg, Vinyl elastomers, Ind. Eng. Chem., 1942,
34, 12.
44. С1 a s h, Berg, Stiffness and brittleness of non-rigid vinyl chloride acetate
resin compounds, Mod. Plast. mag., July, 1944.
45. С 1 a s h, R у n к i e w i c z, Thermal expansion properties of plastic mate-
rials, Ind. Eng. Chem., 1944, 36.
46. Meyers, Impact strength of plastic sheet, Mod. Plast., October, 1942.
47. F. W. D u g g a n, Abrasion resistance of flexible vinyl plastics, Prod.
Eng., January,' 1943.
48. H. B. McClure, M. C. Reed, Plasticizers, Plastics Catalog, 1944
p. 300.
49 A. K. Doolittle, Mechanism of Solvent Action, Ind. Eng. Chem., 1944,
36, 239.
50. H. Mark, Intermolecular Forces and Mechanical Behaviour of High
Polymers. Ind. Eng. Chem., 1942, 34, 1343.
Материалы на основе поливинилбутираля
225
51. Н. I- Cramer, Industrial Progress in Synthetic Rubberlike Polymeis,
Ind. Eng. Chem., 1942, 34, 243.
52. G. E. King, Bend—Brittle and Shatter Points of Rubberlike Materials,
Ind. Eng. Chem., 1943, 35, 949.
53. Vinylite Polyvinyl Acetate Resins, Carbide a. Carbon Chem. Corp., 1942.
54. H. A. L i e b h a f s к y, A. L. Marshall, F. G. V e r h о e k, Loss
of Plasticizers from Polyvinyl Chloride Plastics in Vacuum, Ind. Eng.
Chem., 1942, 34, 704.
55. H. L e a d e r m a n, Creep a. Creep. Recovery in Plasticized Polyvinyl
Chloride, Ind. Eng. Chem., 1943, 35, 374.
56. F. W. Duggan, К. K. Fligor, Fatigue Resistance.of Flexible
Plastic Sheetings, Ind. Eng. Chem., 1943, 35, 172.
57. M. C. Reed, Stress—Strain Characteristics of Vinyl Elastomers, Ind.
Eng. Chem., 1943, 35, 429.
58. D. E. S t r a i n, R. G. Ke njn e Г1 у, H. R. D i t t m a r. Meth-
acrylate Resins, Ind. Eng. Chem." 1939, 31, 382.
59. W. C. G о g g i n, R. D. Lowry, Vinylidene Chloride Polymers, Ind.
Eng. Chem., 1942, 34, 327.
60. R. C. Reinhardt, Vinylidene Chloride Polymers, Ind. Eng. Chem.,
1943 35 422.
61. J. W. Reynolds, M. R. Radcliffe, Mr. R. Vogel, Sol-
vents and Plasticizers for Chlorinated Rubber, Critical Data, Ind. Eng.
Chem., 1942, 34, 466.
62. H. Rosenbloom, Chemistry of Wash Primers, Ind. Eng. Chem.,
November, 1953, 2561, 2568..
63. F. Chevassus, Importance des Wash Primers dans la lutte contre la
corrosion, Peintures. Pigments, Vernis, aout, 1953. 630—635.
64. Bacon, Smith, Rugg, Electrolytic Resistence as a Means ‘of eva-
luating Protective Merit of Coating on Metals, Ind. Eng. Chem., January,
1948.
15—156
ГЛАВА V
ПОЛИСТИРОЛ
Е. Дорре, Л. Реморде
Полимеры стирола начали применяться в производстве ла-
ков и красок лет двадцать тому назад, причем их применение
расширялось по мере их удешевления. Распространение этих
полимеров объясняется их хорошей атмосферостойкостью, стой-
костью к химическим реагентам и высокими диэлектрическими
показателями.
Полистирол имеет линейное строение и состоит из групп
Жирно-ароматический характер полистирола обусловливает
его химическую инертность и диэлектрические свойства.
СТИРОЛ
Мономерный стирол имеет следующее строение:
СН=СНа
I
С
- /Ч
НС сн
II I
НС сн
\н
Физико-химические свойства стирола:
Молекулярный вес ................................. 104,15
Температура замерзания, °C........................ —30,5
Температура кипения (при 760 мм рт. ст.), °C . . 145,2
Показатель преломления Пр . . . . ................. 1,5437
Плотность с?20’ Лем*.............................. 0,9074
Вязкость (при 25°), сантипуазы ................... 0,72
Удельная теплоемкость (при 25°), кал/г-°С.......... 0,416^
Теплота испарения, кал[г ......................... 102,65
Стирол
227
Впервые стиролом начали заниматься французы Бонастр и
Дарсе. Бонастр1 в 1831 г., перегоняя 100 г копалмского баль-
зама (liquidambar styraciflua), получил 7 г бесцветного, име-
ющего неприятный кислый вкус легколетучего масла с удель-
ным весом выше, чем у спирта. Дарсе2 в 1837 г. описал то же
масло в своей работе по исследованию действия железа' на
камфару при повышенной температуре; по данным его анализа
это масло содержало 92,35% углерода и 7,65% водорода. По-
этому Дарсе счел его изомером бензола. В 1839 г. Симон3,
получив то же масло при перегонке styrax liquidus, предложил
назвать его стиролом. Анализируя это вещество, он нашел в нем
92,46% углерода и 7,54% водорода.
Стирол, способный превращаться при хранении в стеклян-
ной посуде в твердую прозрачную смолу, вызвал большой инте-
рес у химиков, но промышленное производство его начало раз-
виваться только после первой мировой войны, одновременно с
широким развитием производства пластических масс.
В 1925 году фирма Naugatuck стала изучать способ полу-
чения стирола из а-хлорэтилбензола. Позже (в 1928 г.) фирма
J. G. в Урдингене применила метод получения стирола из а-фе-
нилэтилового спирта; в 1929 г. фирмой J. G. в Людвигсгафене
был успешно осуществлен процесс получения стирола дегидри-
рованием этилбензола; метод в 1931 г. был положен в основу
промышленного производства стирола.
В США освоением производства стирола занималось не-
сколько фирм (Dow, Koppers, Monsanto). В промышленном
масштабе стирол начали получать в 1935 г.
Способы получения стирола. Исходным продуктом для полу-
чения стирола служит этилбензол, получаемый этилированием
бензола при 90° в присутствии хлористого алюминия:
СвН6 + СН2=СН2 С6Н5—сн2—сн3
Для получения стирола из этилбензола пользуются различ-
ными способами.
По способу фирмы Naugatuck этилбензол хлорированием
превращают в а-хлорэтилбензол, из которого при отщеплении
молекулы НС1 получают стирол:
CeH6—СНС1—СН3 -* С6Н5—СН=СН2 + НС1
В Урдингене этилбензол окисляют хроматом в ацетофенон:
СвН5— СН2-СН3 + О2 СвН8—СОСН3 + НгО
Ацетофенон гидрируют, превращая его в а-фенилэтиловый
спирт
С..Н5-СОСН3 + Н2 -> С6Н6—СН(ОН)--СН3
15*
228
Гл. V. Полистирол
от которого отнимают молекулу воды и получают стирол:
С6Н6—СН(ОН)-СН3 с6н6—сн=сн2 + Н2О
Фирме Carbide Со удалось получить а-фенилэтиловый спирт
непосредственным окислением этилбензола воздухом.
Самым простым является разработанный в Людвигсгафене
способ прямого дегидрирования этилбензола:
СвН6—СН2-СН3 с6н6-сн=сн2 + н2
Дегидрирование ведут под действием окислов алюминия,
цинка, кальция, хрома, калия в качестве катализаторов, при
температуре 580—610° в присутствии паров воды. Выход в рас-
чете на этилбензол достигает 88—92%.
ПОЛИСТИРОЛ
Полимеризация стирола. Классический метод полимериза-
ции стирола — это полимеризация в блоке. Ее осуществляют,
нагревая мономер в продолжение нескольких часов при тем-
пературе 70—80°, затем при 80—90° и к концу доводят темпе-
ратуру до 150—180°. Общая продолжительность полимеризации
50—70 час. Процесс можно вести непрерывно в аппаратуре
из алюминия или нержавеющей стали.
Полимеризация может проводиться также в среде раство-
рителя, путем нагревания растворов стирола в бензоле, толуоле,
хлорированных углеводородах под давлением.
Как при блочной полимеризации, так и при полимеризации
в растворе, в качестве катализатора применяют перекиси (на-
пример, перекись бензоила).
Другой способ состоит в полимеризации стирола в эмуль-
сий, содержащей 25—50% мономера стирола. Эмульсию гото-
вят, добавляя в воду эмульгаторы: сульфонаты, мыла. В каче-
стве инициаторов применяют персульфаты или гидроперекиси
или проводят реакцию в окислительно-восстановительной (Re-
dox) системе. Полимеризацию ведут при температуре 50—80*
в продолжение нескольких часов. По окончании процесса поли-
меризации полимер высаживают, отделяют от воды и промы-
вают. Полимер в виде отдельных крупинок (гранулированный,
бисерный) получают, если исходят -не из тонких эмульсий, а
из водных суспензий, образующихся при добавлении таких
эмульгаторов, как поливиниловый спирт.
Свойства полистирола. Чистый полистирол растворим в аро-
матических углеводородах, хлорированных и бронированных
углеводородах, кетонах (метилэтилкетоне, циклогексаноне), ди-
оксане. Он менее растворим в сложных эфирах и нафтеновых
углеводородах и нерастворим в углеводородах жирного ряда и
в спиртах. Растворимость полистирола и его сополимеров за-
Полистирол
229
висит в известной степени от их молекулярного веса и про-
странственной структуры.
Сополимеризация стирола. Кроме чистого полистирола, су-
ществует большое число сополимеров, т. е. продуктов, образу-
ющихся при полимеризации смесей стирола с другими полиме-
ризующимися веществами. Путем сополимеризации можно по-
лучать продукты с разнообразными свойствами, имеющие более
широкое применение, чем сам полистирол.
Чистый полимер отличается прекрасными диэлектрическими
и ’химическими свойствами и может перерабатываться мето-
дами шприцевания и литья. С увеличением молекулярного веса*
полимера возрастает твердость изделий. Чистый полистирол,
вследствие хрупкости и твердости, непригоден для производства
лаков и красок; для придания ему необходимых свойств потре-
бовалось проведение длительных исследований.
Примерно в 1932 г. фирме I. G. в Людвигсгафене удалось
получить полистирол (под маркой ронилла) с хорошими хими-
ческими и диэлектрическими свойствами, пригодный в качестве
связующего для лаков и красок. Этот продукт, получавшийся
путем полимеризации стирола в присутствии небольшого коли-
чества растворителя (толуола), имел меньший средний моле-
кулярный вес и меньшую твердость (благодаря присутствию
низкомолекулярных полимеров, оказывающих пластифицирую-
щее действие). Дополнительная пластификация может быть
достигнута путем введения пластификаторов (трикрезилфосфа"
та, фталатов, хлорированных полициклических углеводородов),
но пластификаторы ухудшают химические и диэлектрические
свойства полистирола.
В 1938 г. цена на стирол понизилась настолько, что его при-
менение в производстве лаков и красок могло быть расширено;
тогда начали прибегать к сополимерам, в основном к сополи-
мерам с винилацетатом, молекулярный вес которых колеблет-
ся от 5000 до 120 000. Кроме того, в качестве синтетических плен-
кообразующих для лаков начали выпускаться полимеры и со-
полимеры а-метилстирола с более низким молекулярным весом
(от 500 до 10 000). При таком молекулярном весе эти полимеры
практически представляют собой пластификаторы.
Широкое промышленное развитие лаковые смолы на основе
полистирола получили лишь после 1945 г. Эти смолы резко
отличались по химическому строению и молекулярному весу
от полистиролов, применяемых для пластических масс.
При получении полимеров стирола, предназначенных для
лаков, руководствуются двумя основными требованиями, предъ-
являемыми потребителями:
1. Хорошая растворимость в обычных органических раство-
рителях, достижимая -при сравнительно низком молекулярном
230
Гл. V. Полистирол
весе полимеров, из которых благодаря этому можно получать
растворы достаточно высокой концентрации.
2. Достаточная эластичность без значительного ухудшения
химических и диэлектрических свойств.
Ниже будут рассмотрены различные пути получения про-
дуктов, удовлетворяющих этим требованиям.
Пластифицированный полистирол
Как указывалось ранее, для пластификации полистирола
* применяют классические пластификаторы!4: эфиры фталевой,
фосфорной, лауриновой кислот, ароматических хлорорганиче-
ских кислот,, нафтенаты. Пленки, получаемые таким путем, или
хрупки или чрезмерно мягки и частично лишены свойств, при-
сущих чистому полистиролу.
Полистирол, модифицированный фенолами
Более ценные и более пригодные для лаков и красок поли-
меры получают при взаимодействии стирола с фенолом или
крезолом. Винильная группа стирола реагирует с фенолом, что
приводит к частичному его алкилированию. Одновременно сти-
рол полимеризуется, в результате чего фенольные ядра ока-
зываются связанными более или менее длинными цепочками
полистирола. Подобное химическое соединение фенола с поли-
стиролом придает полимерам способность совмещаться с вы-
сыхающими маслами и другими полимерами. Дальнейшее улуч-
шение свойств модифицированного полистирола (растворимости,
химической стойкости, механических и диэлектрических свойств)
может быть достигнуто действием на него альдегидом (напри-
мер, формалином). Такие полимеры и сейчас еще применяются
в некоторых случаях4. Их получают путем конденсации стирола
с фенолом при высоких температурах (200—450°) и под высо-
ким давлением. Взамен фенола применяют также его сложные
эфиры. Обычно в качестве катализаторов реакции конденсации
употребляют слабокислые соли металлов, чаще всего фосфат
или метафосфат церия. Изменяя условия конденсации, можно
в широких пределах изменять свойства полимеров, например
температуру размягчения, и повышать их совместимость с дру-
гими смолами, маслами, наполнителями, пигментами.
Полистирол, модифицированный высыхающими маслами
Установлено, что высыхающие масла благодаря наличию
двойных связей могут реагировать с мономерным стиролом и с
продуктами его конденсации с фенолом. Механизм этой реак-
Полистирол
231
ции точно не выяснен, но известно, что между цепями высыха-
ющего масла и молекулами стирола образуются истинные хи-
мические связи. Для получения таких модифицированных поли-
меров растворы стирола в толуоле или ксилоле обрабатывают
льняным или дегидратированным касторовым маслом при вы-
сокой температуре в продолжение 20—30 час. Этим способом
получают пленкообразующие, пригодные для производства вы-
сококачественных лаков и красок.
За последние годы поступает много предложений по замене
стирола метилстиролом, хлорстиролом и т. д. Это дает возмож-
ность расширить еще более диапазон свойств полимеров, на-
пример, от продуктов с очень высокой твердостью—-к мягким,
как воск, и одновременно изменить другие физические и хими-
ческие свойства. Однако до настоящего времени не предложено
способа, позволяющего получать полимеры, имеющие явное
превосходство над другими.
В общем все масляно-стирольные пленкообразующие очень
прозрачны и слабо окрашены. Благодаря способности хорошо
совмещаться с различными пигментами, стабилизаторами, на-
полнителями, они нашли широкое применение.
Полистирол, модифицированный непредельными кислотами
По окончании второй мировой войны получил развитие но-
вый класс пленкообразующих с чрезвычайно разнообразными
свойствами. Речь идет о полиэфирах, сшитых стиролом. Для
получения этих материалов сначала проводят поликонденсацию
гликолей и двухосновных кислот, часть которых является не-
насыщенными, как, например, малеиновая кислота. После по-
ликонденсации эти полиэфиры содержат в цепи двойные связи,
за счет которых может идти их сополимеризация со стиролом,
с образованием трехмерных структур. Такие полиэфиры пред-
ставляют собой жидкости, имеющие консистенцию меда, или
более .или менее вязкие.
После добавления мономера стирола и катализатора поли-
меризации непредельные полиэфиры отверждаются, образуя
продукты с высокой механической прочностью и химической
стойкостью. Полимеры этого типа приобрели уже широкое рас-
пространение как пластические массы и представляют также
большой интерес в качестве связующих для лакокрасочных ма-
териалов (лаков, красок и различных покрытий).
Недавно в США стали выпускать полиэфиры, сшитые сти-
ролом, хлорированным в ядре. При этом получаются продукты,
хорошо совмещающиеся с пластификаторами, растворителями,
различными смолами, а также малогорючие, химически стой-
кие или с хорошими диэлектрическими свойствами. Эти ком-
232
Гл. V. Полистирол
'плексные полимеры настолько широко развились, что получили
специальное название «стиролизованные полиэфиры».
Можно сказать, что в настоящее время стирол стал одним
из основных продуктов, применяемых при сополимеризации и
сополиконденсации для получения полимерных материалов, ис-
пользуемых в качестве лакокрасочного сырья.
Сополимеры стирола в виде водных эмульсий
В последние годы в США стали выпускать сополимеры в
виде водных эмульсий, которые успешно конкурируют с крас-
ками, содержащими в своем составе растворители. Это сополи-
меры стирола с относительно небольшими количествами бу-
тадиена или других виниловых или дивиниловых производных.
В Германии, начиная с 1935 г., и в США со времени второй
мировой войны, наряду с твердыми полимерами стирола, при-
меняемыми для прессования и литья, вырабатываются в про-
мышленном масштабе каучукоподобные сополимеры стирола с
мономерами, содержащими две коньюгированные винильные
группы. Путем сополимеризации стирола с бутадиеном полу-
чался синтетический каучук (Buna S или GR-S) с содержа-
нием 20—25% и максимально 30% стирола. Эти сополимеры
получали эмульсионным методом.
Когда в конце войны США пришлось резко сократить вывоз
синтетического каучука, возникла необходимость изыскания
новых областей применения этих продуктов. Тогда стали по-
лучать сополимеры с более высоким содержанием стирола —
от 50% До 60—70%. Из литературных данных известно, что
с 1946 г. многочисленные сополимеры такого типа с превосход-
ными диэлектрическими свойствами и хорошей химической
стойкостью применяются в виде водноэмульсионных красок (ла-
тексов). Выпуск этих красок в США быстро возрастает, что
видно из приведенных ниже данных (в тоннах латекса, содер-
жащего 15—20% сополимеров стирола):
Года
1948
1949
1950
1951
1952
350
7000
12 000
105 000
120 000
Такой быстрый рост применения эмульсий объясняется нали-
чием опыта использования эмульсионных красок на основе хло-
рированного, циклизованного и гидрохлорированного каучука.
Изучению свойств латексов такого типа и химических харак-
теристик пленок и покрытий, получаемых после испарения во-
Полистирол
233
ды, было посвящено много научных и технологических иссле-
дований. В литературе5 эти вопросы подробно разбираются.
При исследовании сополимеров приходится встречаться с
рядом неожиданных явлений. Например, при увеличении до
80% содержания стирола в его сополимерах с бутадиеном резко
повышаются такие показатели, как прочность к истиранию, отно-
сительное удлинение при разрыве и термостойкость, что пред-
ставляет большой интерес при использовании этих сополимеров,
для лаков и красок. Содержание стирола 80% оказывается
оптимальным в отношении свойств сополимера; при дальней-
шем увеличении содержания стирола в сополимере начинают
ухудшаться эластичность и адгезия. Эмульсионным краскам
на основе сополимеров стирола с бутадиеном посвящены мно-
гочисленные работы. Так, перед полимеризацией пробовали вво-
дить в смесь мономеров также другие компоненты: акрилонит-
рил, винилацетат, циклопентадиен, аллиловый спирт, полные или
кислые эфиры малеиновой и фумаровой кислот, высыхающие масла.
Кроме сополимеров, эмульсии содержат защитные коллои-
ды или загустители, наполнители, пигменты, смачивающие ве-
щества, вещества, предотвращающие пенообразование, ингиби-
торы коррозии6. Благодаря специальным работам в этой обла-
сти в последние годы была найдена возможность получать ра-
нее неизвестные водноэмульсионные краски, устойчивые к кис-
лотам и щелочам и не желтеющие на свету.
Единственной нерешенной задачей является придание вод-
ноэмульсионным краскам атмосфероустойчивости; сейчас в раз-
ных странах проводятся многочисленные исследования для.
разрешения этого вопроса. Во всяком случае краски и покрытия
на основе латексов, применяемые для внутренней окраски,
устойчивы к действию мыльной воды (это важно при окраске
кухонь, ванных комнат).
Из других специальных областей применения стирольно-бу-
тадиеновых эмульсий следует упомянуть покрытия для бумаги,,
и ткани, клеи, составы для пропитки волокна. Имеются ука-
зания на применение сополимеров с более высоким содержа-
нием бутадиена и, следовательно, более эластичных в качестве-
связующих для штукатурки. Из этого перечисления видно на-
сколько широка гамма свойств и возможных способов приме-
нения сополимеров стирола с бутадиеном. Разнообразие свойств-
сополимеров представляет большой интерес для производствен-
ников, так как (независимо от области применения) все эти
продукты можно получать в одной и той же аппаратуре, что
способствует их удешевлению.
Само собой разумеется, что из эмульсий после коагуляции:
и сушки можно получать продукты, пригодные для использо-
вания в виде растворов в органических растворителях.
234
Гл. V. Полистирол
В пределах этой монографии нет возможности шире рас-
смотреть эмульсионные краски на основе сополимеров стирола,
тем более, что по этому вопросу есть много литературы. На-
пример, одна из работ Фридленгера7 посвящена краскам на
основе стирольных полимеров; очень важное сообщение по ©тому
вопросу имеется в Industrial and Engineering Chemistry.8 Onyfr
ликаваны и другие работы9,10.
* *
*
За последние 20 лет развитие промышленности синтетиче-
ских полимеров привело ко все большему проникновению их
в область производства лаков и красок. Аналогично тому как
30—40 лет назад с развитием производства нитроцеллюлозы и
других эфиров целлюлозы появилось новое направление в раз-
витии лакокрасочной промышленности, так теперь освоение
промышленного производства относительно недорогих поливи-
ниловых полимеров, и особенно полистирола, вызвало важный
сдвиг в этой области.
Полистирол и особенно продукты его химической модифи-
кации путем конденсации и сополимеризации, образуют новый
класс лакокрасочных материалов, обладающих прекрасными
механическими и химическими свойствами. Несомненно, в бли-
жайшие годы появятся новые виды материалов с интересными
свойствами. Судя по тому, как идет развитие промышленности
синтетических полимеров, полистирол, на основе которого полу-
чаются материалы с самыми разнообразными свойствами, будет
все шире применяться для лаков и красок.
ЛИТЕРАТУРА
1. М. В о п a s t г е, J. pharm. chim., 1831, 17, 338.
2. Е. d’A г с е t, Ann. chim. phys., 1837, 66, ПО—111.
3. E. Simon, Ann., 1839, 31, 265—277.
4. R. M. Boundy, R. F. Boyer, (Dow Chemical), Styrene its polymers,
copolymers and derivatives, New York, 1952.
5. I. D. d'J a n n i, L. D. Hess, W. C. Mast, Ind. Eng. Chem., 1951,
319.
6. L. Berthelot Ind. Plast. Mod., Avril, 1953, 27.
7. I. H. F г у d 1 e n g e r, La Rev. Prod, chim., 1951, 54, 169.
8. Симпозиум по применению красителей в виде водных эмульсий, Ind. Eng.
Chem., 1953, 45, 709—754.
"9. Ch. Parker, Homopolymeres et copolymeres dans les peintures emul-
sions, Federation of Paint a. Varnish Production Clubs, Official Digest,
1952, № 332, p. 320—625.
30. W. L. Grave, Am. Paint. J., 1952, 37. № 14, 78—86; 1953, 37, № 16,
60—70.
ГЛАВА VI
ПОЛИАКРИЛАТЫ И ПОЛИМЕТАКРИЛАТЫ
П. Тале
Акриловая кислота была открыта в 1843 г., а ее полимеры
впервые исследованы в 1873 г. Каспари и Толленсом. Однако
только в 1927 г. после работ Рема была создана первая полу-
производственная установка для синтеза акриловых эфиров,
исходя из монохлоргидрина гликоля.
Из эфиров, замещенных в a-положении, промышленное зна-
чение получили только метакрилаты. Опытные работы по их
•синтезу были закончены в 1934 г., а вскоре после этого был
налажен (промышленный выпуск ©тих продуктов в значитель-
ных масштабах.
Для производства лаков и красок представляют наиболь-
ший интерес амиды, нитрилы и сополимеры акриловой и мет-
акриловой кислот.
Полиакрилаты являются высококачественными материалами
для лакокрасочной промышленности. Их широкому использо-
ванию мешает только относительно высокая цена. Новые ме-
тоды синтеза, при которых образуется меньше побочных про-
дуктов и возможно применение такого более доступного сырья,
как этилен, пропилен, ацетилен, синильная кислота, расширяют
возможность развития в ближайшем будущем производства раз-
нообразных акриловых полимеров.
АКРИЛОВЫЕ МАНОМЕРЫ
Ввиду ценности акриловых мономеров были предложены
многочисленные способы их получения, но только некоторые
из этих способов приобрели промышленное значение.
Акриловая кислота и ее производные. Этиленциангидрин,
получаемый из этилена через стадию образования монохлор-
гидрина гликоля или проще действием синильной кислоты на
окись этилена, при дегидратации дает акрилонитрил, а при
гидролизе — акриловую кислоту:
носн2—ch2-cn —СН2=СН—CN
НОСН2—СН2—CN + H2SO4 + Н2О -* СН2=СН—СООН + NHtHSO*
2сб Г л. VI. Полиакрилаты и полиметакрилаты_________
Эфиры могут быть получены из кислоты классическими ме-
тодами, причем можно проводить этерификацию в момент по-
лучения кислоты, без ее выделения. Второй способ получения
эфиров состоит в пиролизе ацетилированной молочной кислоты
в паровой фазе при 450—500°:
СН3—СН—COOR -* СН2=СН—COOR + СН3СООН
I
ОСОСНз
Успешно применяется для этой цели также непосредствен-
ное карбонилирование ацетилена, приводящее в присутствии
воды к акриловой кислоте:
сн=сн + со + Н2О СН2=СН—СООН
в присутствии спиртов — к эфирам
СН=СН + СО + ROH СН2=СН—COOR
а в присутствии аминов — к амидам:
СН=СН + СО + RNH2 СН2=СН—CONHR
Эфиры акриловой кислоты — это бесцветные жидкости, ки-
пящие при следующих температурах (в °C):
Метиловый...................... 77.4
Этиловый..................... 99,4
Бутиловый..................... 138
Метакриловая кислота и ее производные. При промышлен-
ном синтезе метакриловой кислоты и ее эфиров обычно исхо-
дят из ацетонциангидрина (процесс аналогичен процессу по-
лучения эфиров акриловой кислоты) :
ЮН
€Н3—С< + H2SO4 + Н2О -* СН2=С—СООН + NH4HSO,
I 4CN I
СН3 СН3
Прямая дегидратация ацетонциангидрина приводит к полу-
чению нитрила:
/ОН -н2о
СН3—С<--------» СН2=С—CN
I 4CN |
СН« CHS
Если реакцию гидролиза, при которой образуется метакри-
ловая кислота, остановить на промежуточной стадии метакрил-
амида, получается раствор этого амида в серной кислоте. После
разбавления этого раствора и нейтрализации его карбонатом
магния можно обычными способами проводить полимеризацию.
При добавлении метанола в реакционный раствор можно
получать метилметакрилат, выделяемый далее путем перегонки.
Акриловые мономеры
237
Новейший способ синтеза состоит в присоединении двух мо-
лекул ацетона к одной молекуле ацетилена с образованием ди-
метилгексиндиола, превращаемого в а-оксиизомасляную кислоту.
Дегидратацией эфира этой кислоты получают соответствующий
эфир метакриловой кислоты:
СН3 СН3
I I
НС=СН + 2СН3—СО—СН3 -> СН3-С—с=с-с—сн3
I I
он он
сн3 сн3 сн3
I I I
СН3—С—СООН -»- СН3—С—COOR -> СНа=С—COOR
I I
он он
Эфиры метакриловой кислоты имеют следующие темпера-
туры кипения (в °C):
Метиловый...........................................100
Этиловый................................117
н- Бутиловый............................163
Полимеризация акрилатов и метакрилатов. Двойная связь
в акриловых и метакриловых производных придает им способ-
ность чрезвычайно легко полимеризоваться под влиянием раз-
личного рода воздействий. Полимеризация может инициировать-
ся радикалами, ионами, действием света или тепла.
При полимеризации эфиров акриловой кислоты образуются
линейные цепи следующего строения:
----сн2—сн—сна---сн-сн2-----сн----
СООСН3 СООСНз COOCHs
Эфиры метакриловой кислоты ведут себя аналогично акри-
ловым. Однако при полимеризации проявляется заметная раз-
ница, вызванная тем, что понижение электронной плотности
двойной связи в молекуле метакрилатов менее значительно
вследствие электронодонорного действия группы СН3, которая
вызывает, кроме того, пространственные затруднения.
Практически полимеризацию чаще всего проводят по ради-
кальному механизму, используя в качестве инициаторов пере-
кись водорода, соли высших кислородных кислот, органические
перекиси и гидроперекиси. Кислород воздуха также способен
вызывать образование перекисей мономера и, следовательно,
инициировать полимеризацию при хранении продукта. Поэтому,
если хранение должно быть продолжительным или если тем-
пература окружающей среды относительно высока, следует до-
бавлять ингибиторы. Обычно для этого применяют гидрохинон
или его растворимые производные.
238
Гл. VI. Полиакрилаты и полиметакрилаты
Один из промышленных способов последних лет позволяет
получать высокомолекулярные полимеры, благодаря относи-
тельно низкой температуре реакции. По этому способу иници-
ирование производится с помощью окислительно-восстанови-
тельных систем, состоящих из гидроперекисей и восстановите-
лей, в присутствии активаторов.
Акрилаты и метакрилаты полимеризуются в промышлен-
ности в аналогичных условиях. Однако вследствие того, что
тепловой эффект полимеризации акрилатов выше, чем метакри-
латов, первые полимеризуются быстрее и более бурно.
Полимеризация в блоке требует больших предосторожно-
стей, так как реакция может легко привести к взрыву. Чтобы
избежать этой опасности, полимеризацию ведут при низкой
температуре, постепенно доводя массу до вязкого состояния и
медленно повышая температуру к концу реакции. Этот способ
полимеризации применяют только при изготовлении органиче-
ского стекла или при покрытии некоторых изделий.
Полимеризация в блоке зависит в основном от температуры,
концентрации катализатора и концентрации мономера, тогда
как при полимеризации в растворе необходимо считаться с
природой растворителя, которая сильно влияет на степень по-
лимеризации конечного продукта. Особенно понижают степень
полимеризации хлорсодержащие растворители вследствие воз-
можности реакций передачи цепи. Аналогичное действие ока-
зывают добавляемые в процессе полимеризации меркаптаны,
которые уменьшают полидисперсность полимера. В последнее
время получают даже полимеры таких низких степеней поли-
меризации, при которых полиакрилаты (этиловые или бутило-
вые эфиры) представляют собой жидкости, действующие как
превосходные пластификаторы. Активными растворителями в
этом случае являются производные изопропилбензолов1.
Твердые полимеры могут быть получены при проведении
полимеризации в среде жидкости, растворяющей мономер, но
не растворяющей полимер. Последний выпадает в осадок и
может быть легко выделен. Наиболее распространенным про-
мышленным способом является, по-видимому, полимеризация
в водной среде. В зависимости от того, требуется ли в дальней-
шем получить сухой полимер или использовать всю дисперсию,
как таковую, полимеризацию проводят либо суспензионным,
либо латексным способом. При суспензионной, или гранульной,
полимеризации вокруг частиц • цолимера образуется защитная
оболочка из поливинилового спирта, метилцеллюлозы, карбок-
симетилцеллюлозы, полиакриловой кислоты и т. д. При выборе
инициаторов отдают предпочтение тем, которые нерастворимы
в воде, но растворимы в мономере. При латексной полимери-
зации необходимо избегать осаждения, а частицы полимера
Акриловые мономеры
239
должны быть очень мелкими. Это достигается путем примене-
ния специально подобранных эмульгаторов и инициаторов,
обычно растворимых в воде. Полезно заметить, что акриловые
эфиры гидролизуются легче метакриловых; при латексной по-
лимеризации это может настолько снизить pH, что потребуется
добавлять щелочные агенты или соответствующим образом по-
добранные буферные смеси.
Полимеризация других акриловых производных. Акриловая
кислота очень легко полимеризуется, чаще всего в виде 50%-не-
го водного раствора, но в качестве загустителей водных кра-
сок больше всего применяются натриевые или аммонийные соли
полиакриловой кислоты. Они могут получаться путем нейтра-
лизации полимерной кислоты, но в промышленности их обычно
получают омылением полиметилакрилата. Полученный раствор
отделяют затем от выделившегося при этом метанола. Поли-
акриламид и полиметакриламид (полиакрилатьи-основания) яв-
ляются также ценными продуктами. Но в основном использу-
ются их сополимеры. Они получаются омылением сополимеров
акрилонитрила или метакрилонитрила с метиловыми эфирами
соответствующей кислоты.
Сополимеризация. Несмотря на достаточно широкую гамму
свойств полимеров акриловых и метакриловых производных со-
вокупность требуемых свойств в одном гомополимере часто
не достигается. Если же просто смешать растворы различных
полимерных акрилатов или метакрилатов, то получаемые плен-
ки после удаления из них растворителя могут оказаться него-
могенными. Напротив, при совместной полимеризации двух раз-
ных мономеров конечный раствор дает совершенно прозрачные
пленки. Возможна сополимеризация не только смеси акрилового
и метакрилового эфиров, но также акриловых мономеров с мо-
номерами других классов, например со стиролом, винилацета-
том, винилхлоридом и др. Состав сополимеров, получающихся
из этих смесей, обычно достаточно сложен, так как он почти
никогда не соответствует соотношению мономеров в исходной
смеси.
Кривые, позволяющие предвидеть строение сополимера
в зависимости от состава смеси мономеров, могут быть построе-
ны или по данным определения относительной реакционной спо-
собности исследуемой пары мономеров или путем применения
более общих констант Прайса (константы Q и е).
Большие перспективы в области получения смешанных по-
лимеров открываются в связи с развитием нового направления,
а именно синтеза привитых полимеров. Новые методы позво-
ляют присоединять к уже образованным полимерам цепочки
различных мономеров, подвергаемых дальнейшей полимериза-
ции. В этом процессе участвуют активные функциональные
.’240 Гл. VI. Полиакрилаты, и полиметакрилаты
группы, не прореагировавшие при первичной полимеризации.
Можно также путем реакции передачи цепи вызывать образо-
вание полимерных радикалов, способных инициировать новую
реакционную цепь, в которой участвуют молекулы присутству-
ющего второго мономера. Полиметилметакрилат оказался осо-
бенно способным к образованию макрорадикалов и, следова-
тельно, к образованию привитых полимеров, свойства которых
„довольно значительно отличаются от свойств обычных сополи-
.меров2.
АКРИЛОВЫЕ ПОЛИМЕРЫ
Акриловые эфиры. Свойства полимерных акриловых эфиров,
жак и свойства всех других полимеров, зависят в известной
мере от степени полимеризации и от условий реакции. Пре-
обладающее влияние имеет длина основной цепи полимера, а
также степень ее разветвленности.
Температура стеклования, являющаяся весьма важной ве-
личиной, соответствует температуре, при которой образец раз-
рушается от удара. Для эфиров алифатических спиртов нор-
мального строения температура стеклования вначале понижает-
ся по мере увеличения молекулярного веса спирта, а начиная
с полиоктилакрилата — повышается3.
Морозостойкость понижается, если спиртовая группа имеет
изостроение. Например, температура стеклования 2-этилгексил-
полиакрилата равна —55° и превышает на 10° температуру
стеклования н-октилполиакрилата.
Полиметилакрилат эластичен и относительно мягок. Пленки
очень устойчивы к изгибу, их относительное удлинение при
разрыве достигает 1000%. Они не липкие или обладают лишь
весьма слабой липкостью при нормальной температуре. Поли-
этилакрилат значительно мягче полиметилакрилата, имеет еще
большее относительное удлинение и меньшую прочность при
растяжении. Пленки н-бутилцолиакрилата липки уже при нор-
мальной температуре. При удлинении цепи алифатического
спирта свойства полиакрилатов сначала изменяются так же,
как было указано выше, а начиная со спиртов с восемью угле-
родными атомами, наблюдается переход к воскообразным про-
дуктам и повышение температуры стеклования.
Удельный вес полимера при 20° значительно выше, чем мо-
номера: 1,223 г/смй против 0,956 г/смй для мономера. Та же зако-
номерность наблюдается и для показателя преломления, ко-
торый изменяется от и о = 1,4117 до и о= 1,4720, что в практи-
ческих условиях позволяет использовать этот показатель для
контроля процесса полимеризации (путем применения эталон-
ной кцивой).
Акриловые полимеры
241
Полиакрилаты бесцветны и не изменяются под действием
света. Атмосферостойкость метил- и этилакридатов значитель-
ная. После трехлетней экспозиции пленок не отмечено ни изме-
нений цвета, ни трещин; блеск сохраняется4.
Полиметилакрилат растворяется в большом числе органи-
ческих растворителей — в бензольных углеводородах, в дихлор-
этане, в метиленхлориде, ацетоне, этилацетате, этиленгликоль-
ацетате и т. д. Бутилацетат является плохим растворителем.
Спирты, а также алифатические углеводороды, как правило,
не растворяют полиакрилаты. Хорошими пластификаторами яв-
ляются ди бутилфтал ат, трикрезилфосфат, пентахлордифенил;
касторовое масло и алкидные смолы не совместимы с полиме-
тилакрилатом.
Полиметилакрилат совместим с поливинилацетатом, ацетил-
целлюлозой, нитроцеллюлозой (вязкость 0,5 сек.). Он не сов-
местим с даммарой (исключение составляют полимеры высокой
степени полимеризации, которые обнаруживают некоторую сов-
местимость), с кумароновыми смолами, с этил- и бензилцел-
люлозой, ацетобутиратом целлюлозы и феноло-формальдегид-
ными смолами.
При 300° полиакрилаты разрушаются, образуя смесь моно-
мера с ди- и тримерами. Сильные кислоты и основания при
нагревании омыляют полиметилакрилат.
Часто бывает необходимо придать нерастворимость пленке
после нанесения лака на подложку. Акрилаты легко поддаются
такой «вулканизации». Для этой цели было предложено много
вспомогательных средств, действие которых усиливается под
влиянием тепла. Например, при 160° оказывают действие гидро-
окись кальция, свинцовый глет, щелочные станнаты, ванадаты,
силикаты5. Установлено, что во время этой операции выделяет-
ся некоторое количество спирта. Другими «вулканизующими»
агентами являются двухатомные спирты, их сложные эфиры
и т. д.
Для получения нерастворимых покрытий обычно предпочи-
тают пользоваться сополимерами акриловых эфиров с мономе-
рами, содержащими хлор, например с хлорпропилакрилатом,
2-хлораллиловым спиртом, хлорэтилвиниловым эфиром и т. д.
Можно получать таким путем настоящие эластомеры, которые
вулканизуются под влиянием серы в присутствии некоторых
ускорителей6. Такие синтетические каучуки, известные под на-
званием лактопренов, превосходит GR-S по маслостойкости и
устойчивости к старению. Их способность к вулканизации обус-
ловливается так же, как у натурального каучука, наличием двой-
ных связей.
Полимеры с двойными связями легко получаются при поли-
меризации мономеров с функциональностью больше двух: аллил-
16—156
242
Гл. VI. Полиакрилаты и полиметакрилаты
акрилата или аллилметакрилата, акриловых эфиров много-
атомных спиртов, дивинилсульфида и т. д.
Метакриловые эфиры. В ряду метакриловых эфиров обнару-
живаются те же закономерности свойств, что и в ряду акрило-
вых эфиров, но с некоторым сдвигом. Полиметакриловые эфиры
одного и того же спирта более жестки и тверды, чем полиакри-
латы. Температуры стеклования полиметакрилатов выше, чем
соответствующих пол и акрилатов, вплоть до эфиров нормального
спирта, содержащего 10 атомов углерода. Введение радикалов
изостроения, как правило, приводит к повышению температуры
плавления: н-бутилтюлиметакрилат при обычной температуре
гибок и липок, в то время как трет-бутилметакрилат образует
полимер даже более твердый, чем полиметилакрилат.
Как и в случае акрилатов, присутствие в спиртовых остатках
метакриловых эфиров циклических или гетероциклических ядер
приводит к образованию более твердых и менее пластичных
полимеров, чем при этерификации метакриловой кислоты али-
фатическими спиртами нормального строения с тем же числом
углеродных атомов. Например, полифенилметакрилат имеет тем-
пературу размягчения 120°, в то время как полигексил метакри-
лат мягок при обычной температуре.
Введение хлора в спиртовый радикал мало влияет на темпе-
ратуру размягчения, в то время как этоксигруппы придают по-
лимеру гибкость и эластичность. Напротив, если заменить ме-
тильную группу метакриловой кислоты на атом хлора, темпе-
ратура размягчения повышается, а также возрастают твердость
и механическая прочность7. Так как недостатком полиметилмет-
акрилата являются пониженная теплостойкость и малая устой-
чивость к истиранию, было предложено заменить его полимера-
ми метил-а-хлоракрилата, превосходящими полиметилметакри-
латы, кроме того, по водостойкости и химической стойкости.
К. сожалению, как и большинство хлорпроизводных, эти шласти-
ки склонны под атмосферным влиянием или под действием от-
носительно высокой температуры отщеплять хлористый водород.
Для того чтобы преодолеть этот недостаток, предпринимались
различные меры8.
Удельный вес полиметакрилата составляет 1,213 г!см3, в то
время как удельный вес мономера 0,944 г/см3. Таким образом,
при полимеризации происходит сокращение первоначального
объема на 22%, которое в некоторых случаях необходимо учи-
тывать. Это сокращение объема можно уменьшить путем пред-
варительной полимеризации. Показатель преломления мономера
п— 1,414; у полимера он достигает 1,488.
Огранические стекла устойчивы к воздействию атмосферы и
не желтеют на свету; поглощение света составляет всего 8—9%.
Разложение их при нагревании начинается около 200°; полное
Акриловые полимеры
243
разложение вплоть до 'мономера происходит при 370—400°. При
температуре выше 125° полимер становится мягким и легко фор-
муется.
Некоторые свойства полиметакрилатов приведены ниже:
Предел прочности (при 30°), кг!смг
при растяжении . . . 400—500
при изгибе ............... 1000—1100
Модуль упругости (при 30°), кг]мм2 . . ......... 210—350
Диэлектрическая постоянная (при частоте 60 гц)......... 3—3,6
Тангенс угла диэлектрических потерь (при частоте 60 гц) . 0,05—0,06
Удельное электрическое сопротивление, ом-см 1015
Электрическая прочность, кв/см ......... 20
Растворимость полимеров изменяется в зависимости от сте-
пени полимеризации и способа получения. Как правило, хоро-
шими растворителями являются сложные ефиры, кетоны, арома-
тические углеводороды, хлорорганические растворители, безвод-
ные органические кислоты. Одноатомные алифатические спирты,
простые эфиры и масла не растворяют полиметакрилаты. На них
не действуют 70%-ная фосфорная кислота, соляная кислота при
30°, 60%-ная серная кислота и 30%-ные растворы едкого натра
и аммиака. Водные растворы гипохлорита и бихромата натрия
не оказывают заметного влияния.
Для повышения эластичности пленок пригодно большое
число пластификаторов. Наиболее широко применяются эфиры
фталевой, винной, адипиновой или себациновой кислот, триэти-
ленгликолевые эфиры 2-этилмасляной или 2-этилкапроновой кис-
лоты, триацетин, метилабиетат, трифенилфосфат. Трикрезилфос-
фат лишь частично совместим и употребляется только в смеси
с одним или несколькими из ранее названных пластификаторов.
Для некоторых областей применения целесообразно комбини-
ровать полиметилметакрилат с другими пластиками. До извест-
ной степени с ним совместимы: Хлорированный каучук, кумаро-
новые смолы, эфиры канифоли, канифоль, даммара, пропионат
целлюлозы, глифталевые смолы, а также некоторые типы фено-
ло-формальдегидных смол. Нитроцеллюлоза совместима с поли-
метилметакрилатом в любых соотношениях, в то время как
этилцеллюлоза, бензилцеллюлоза и высыхающие масла несов-
местимы.
Применение акриловых полимеров. Акрилаты оказались вы-
сококачественными пленкообразователями для лакокрасочной
промышленности. Благодаря им удается избежать многих часто
встречающихся затруднений: пожелтения пленок на свету', пло-
хой водостойкости, текучести при умеренной температуре, по-
вреждения плесенью и т. д. Их ценность подтверждается широ-
чайшей гаммой эластических свойств и растворимости, удовле-
творительной адгезией к многочисленным подложкам.
16*
244
Гл. VI. Полиакрилаты и полиметакрилаты
Эмульсии полиакрилатов. Водные эмульсии успешно (приме-
няются в производстве красок, так как они позволяют обходить-
ся без органических растворителей и избавляют от связанных
с этим неудобств. Эмульсии полиметилакрилата, его сополиме-
ров с метилметакрилатом и вообще всех сополимеров с высо-
ким содержанием акрилатов высоко дисперсны. Диаметр их
частиц колеблется от 0,05 до 0,3 р, что обусловливает ничтож-
ную седиментацию, даже при продолжительном хранении.
Кислотность эмульсий выражается относительно низким pH
(4—5), часто повышающимся до 8—9. Эмульсии стабильны' в
интервале pH от 3 до 9 в зависимости от эмульгатора.
Если при применении возникает необходимость повысить
pH, к эмульсии осторожно добавляют какое-либо • относительно
слабое основание (аммиак, бикарбонат натрия, триэтаноламин,
морфолин и т. д.). Если нужно понизить pH, следует применять
слабую кислоту, например уксусную. Температура при хранении
не должна быть ниже 0°, но слишком высокая температура тоже
не рекомендуется, так как возможна коагуляция.
Акрилатные эмульсии находят применение в лаках и красках
как порозаполнители, промежуточные слои, отделочные лаки и
краски. Они пригодны в качестве порозаполнителей для кожи,
штукатурки, цемента, бетона, кирпича и дерева. Для кожи со-
став эмульсии подбирают так, чтобы пленки получались эла-
стичными (это достигается путем введения пластификаторов
или применения соответствующих сополимеров). Для пористых
кож следует применять высококонцентрированные эмульсии
(40%-ные), для плотных кож желательно их разбавлять. Кроме
образования эластичных пленок, придающих коже приятную
мягкость, акрилатные порозаполнители облегчают получение
зернистых покрытий. Прочно удерживая пластификаторы,, поли-
акрилаты задерживают миграцию пластификатора в кожу из
покровного слоя.
Количество пигмента в эмульсиях может быть доведено до
70—75% сухого вещества без ущерба для механических свойств
пленки. Можно применять большое число пигментов: окись же-
леза, двуокись титана, цинковые белила и т. д. При перетире с
основными пигментами величину pH полезно доводить до 8—9.
Диспергирование цинковых белил проходит без осложнений,
если окись цинка и пластификатор предварительно перетереть
t 5—20% (в расчете на пигмент) 12%-ного раствора полиакри-
лата натрия в присутствии достаточного количества воды. Гото-
вая дисперсия вводится в эмульсию и гомогенизацию заканчи-
вают в шаровой мельнице, заполненной шарами, цилиндрами
или галькой.
Преимуществом полиакрилатных эмульсий является возмож-
ность их нанесения на влажные поверхности. Если их применять
Акриловые полимеры
245
в качестве грунта, для отделочных слоев можно использовать
казеиновые, нитроцеллюлозные, масляные, глифталевые краски,
материалы на основе сополимеров винилхлорида с винилацета-
том и, конечно, сами полиакрилаты. Если требуется повышен-
ная устойчивость покрытия к истиранию, полезно ввести неболь-
шое количество анионного смачивателя и повысить pH до 8.
Акрилатные эмульсии пригодны для лакировки целлофана,
бумаги или картона. В последнем случае достигается исключи-
тельная стойкость к воде и маслам, еще более повышающаяся
после горячей сушки при 100—110°. Эти покрытия пригодны так-
же для предохранения резины от действия масла,, керосина и
бензина. Возможна модификация акрилатных эмульсий путем
смешивания их с каучуковым латексом, поливинилхлоридными
эмульсиями, растворами крахмала, декстрина, карбоксиметил-,
целлюлозы, метилцеллюлозы и щелочных солей полиакрилатов.
Продукты этого типа выпускаются во Франции под марками:
нобельакрилаты 24019, 24022, 24028, 24029, 24030; в Германии
выпускаются акроналы и плекстолы; в Англии — вина мулы;
в США — роплексы и полико и т. д.
Растворы полиакрилатов. Растворы полиакрилатов приме-
няются главным образом для покрытий по металлам: меди,
латуни, алюминию, магнию, цинку, олову, стали, хромирован-
ным металлам. Полиметакрилаты дают хорошие результаты на
серебре.
Адгезия тем лучше, чем ниже температура размягчения по-
лимера. Поэтому полиэтил- и полибутилакрилаты обнаружива-
ют лучшую адгезию к алюминию, чем полиметилакрилаты. Хо-
рошая адгезия ко многим подложкам позволяет применять акри-
латные лаки как грунты для повышения адгезии красок, напри-
мер масляных или на основе хлорированного каучука, к алюми-
нию. Метакрилатные лаки были, кроме того, предложены для
защиты поверхности эфироцеллюлозны.х и глифталевых эмале-
вых покрытий. Ценным качеством акрилатных лаков, является
их безвредность, позволяющая применять эти лаки для нужд
пищевой и фармацевтической промышленности.
Полиакрилаты, как правило, выпускаются в виде растворов
в этилацетате или толуоле, в то время как метакрилаты посту-
пают в продажу в виде сухих полимеров. При составлении
рецептур надо учитывать строение акриловых эфиров^ а также
других полимеров, входящих в состав композиции. Хотя раство-
рители для одного гомологического ряда эфиров близки между
собою, все же полезно напомнить, что полимеры этилового и бу-
тилового эфиров легче растворяются в неполярных раствори-
телях, чем полимеры метилового эфира, и что во всех случаях
более активные растворители труднее удаляются из шщнкщ
На растворимость влияет также степень полимеризации, поэто-j
246
Гл. VI. Полиакрилаты и полиметакрилаты
му необходимо применять относительно низкомолекулярные по-
лимеры, чтобы чрезмерно высокая вязкость растворов не созда-
вала неудобств при их нанесении.
Как и при использовании эмульсий, свойства пленок зависят
от строения полимера или сополимера. Для сополимеров акри-
латов с метакрилатами установлено, что акрилаты сообщают
пленкам эластичность, а метакрилаты повышают их твердость
и устойчивость к химическим реагентам.
Во Франции фирма Societe Alsthom освоила в настоящее вре-
мя выпуск метакрилового полимера Р 725 с относительно низ-
ким молекулярным весом (около 100 000). Для растворения
этого полимера можно применять хлорорганические раствори-
тели, например дихлорэтан, а также бензол, толуол и др. В со-
четании с хлорированным каучуком на основе полимера Р 725
•получаются хорошие лаки для консервных банок.
Лак нобельакрил 21874 представляет собой раствор полиме-
тилакрилата.
Акриловые полимеры, растворимые в воде. Акриловые про-
изводные дают много различных ценных водорастворимых про-
дуктов, которые применяются в производстве красок и аппретов.
Полиакриловая и полиметакриловая кислоты выпускаются
в виде 20 и 30%-ных растворов и применяются как загустители
при добавлении оснований. Действительно, соли этих кислот
дают значительно более вязкие растворы, чем сами кислоты.
Благодаря этому свойству некоторые эмульсии, содержащие не-
большое количество акриловой кислоты, входящей в состав сопо-
лимера, заметно загустевают при добавке аммиака.
Некоторым полимерам можно придать адгезию к алюминию
путем проведения полимеризации в присутствии 2—5% поли-
акриловой или полимета криловой кислоты9.
Широко применяются натриевые и аммониевые соли поли-
акриловой и полиметакриловой кислот. Натриевые соли явля-
ются очень ценными добавками при диспергировании пигментов
или .нерастворимых красителей в воде. Они применяются также
как загустители эмульсий (в этом случае., величина pH должна
быть возможно ближе к 6 или выше). В сочетании с казеином
эти соли дают возможность эмульгировать такие полимеры, как
нитроцеллюлоза, бензилцеллюлоза, хлорированный каучук, с
образованием эмульсий типа «масло в воде»10.
Аммониевые соли полиакриловых кислот при том же моле-
кулярном весе образуют более вязкие растворы, чем соли на-
трия, и без специальной обработки дают более водостойкие
пленки.
Полиакрилаты щелочных металлов смешиваются с метано-
лом, этанолом и ацетоном, пластифицируются гликолем и гли-
церином. Они эмульгируют гидрофобные растворители.
Акриловые полимеры
2А7
Выпускаемые во Франции нобельакрилы 24031 и 24047 пред-
ставляют собой полиакрилаты щелочных металлов. В других
странах эти продукты известны под названиями: латеколи,
плейкслеймы, акризоли и т. д.
Полиакрил- и полиметакриламиды, получаемые полимериза-
цией мономерных амидов или омылением полимерных нитрилов,
являются хорошими загустителями. При их нагревании выде-
ляется аммиак и образуются мостиковые связи, придающие
материалу нерастворимость в воде, кислотах и щелочах даже
при кипячении.
Модифицированные полиакрилаты
Акриловые эфиры могут давать сополимеры .не только с мет-
акриловыми эфирами, но также со многими другими мономера-
ми, способными к полимеризации.
Сополимер винилацетата с бутил акрил атом дает эластичные
лаки с небольшим отлипом; стирол повышает твердость и улуч-
шает эластические свойства полиакрилатов; винилизобутиловый
эфир придает эластичность, адгезию и очень большую клейкость;
винилбензоат повышает температуру размягчения и улучшает
механические свойства. Тройные сополимеры имеют прекрасный
блеск, очень высокую твердость и водостойкость. В эти ком-
позиции могут входить: винилхлорид, акрилонитрил, а также
вышеперечисленные мономеры.
Путем полимеризации винилхлорида в присутствии метил-
акрилата получаются полимеры, весьма рекомендуемые для тек-
стильной промышленности и производства антикоррозионных
красок. С каолином эти полимеры дают эмульгирующие порош-
ки11. Винилиденхлорид ведет себя, как винилхлорид; получаемые
сополимеры образуют эмульсии, которые применяются для при-
дания непроницаемости целлофану12. Наконец, водные дисперсии
сополимеров акрилатов с бутадиеном находят применение в ко-
жевенной, бумажной и текстильной промышленности; получа-
емые покрытия отличаются прекрасной эластичностью в сочета-
нии с хорошей масло- и водостойкостью.
< Идентификация акриловых полимеров
Часто бывает полезно простыми способами установить со-
став краски, лака или пленки. Для идентификации акрилатов
имеется несколько качественных реакций: _
1) Флуоресценция в свете, проходящем через фильтр, про-
пускающий только ультрафиолетовые лучи:
Полиметилакрилат — бело-лиловая.
Полиметилметакрилат — не флуоресцирует.
248
Гл. VI. Полиакрилаты и полиметакрилаты
Полиакрилонитрил — интенсивная светло-желтая.
Полиакриловая кислота — синяя с ярко-розовым.
2) Пиролиз:
Полиметилакрилат — деполимеризация при 280—300°; ха-
рактерный запах мономера.
Полиметилметакрилат: то же.
3) Число омыления:
Полиметилакрилат — найденное число омыления очень близ-
ко к теоретическому (651).
Полиметилметакрилат — найденное число омыления значи-
тельно ниже теоретического.
ЛИТЕРАТУРА
1. С. Е. R е h b е г g, Ind. Eng. Chem., December, 1952, p. 2864.
2. G. S m e t s, M. С 1 a e s e n, J. Polymer. Sci., March, 1952, p. 289.
3. S. E. Reh berg, С. H. Fisher, Ind. Eng. Chem., August, 1948,
p. 1429.
4. L. Klein, W. T. Pearce, Ind. Eng. Chem., June, 1936, p. 635.
5. Goodrich B. F. Co., фр. пат. 971643, 1947 г.
6. W. C. Mast, С. H. Fisher, Ind. Eng. Chem., January, 1948, p. 107.
7. M. C. Slone, J. J. Lamb, Mod. Plast., June, 1952, p. 109.
8. General Aniline a. Film Corp., фр. пат. 939190, 1946 г.: Kodak—Pathe,
фр. пат. 946236, 1947 г.
9. Lonza Elektrizitatswerke, фр. пат. 1008440, 1950 г.
10. Rohm a. Haas G. m. b. H., фр. пат. 904760, 1944 г.
11. Badische Anilin u. Soda-Fabrik, фр. пат. 1020170, 1950 г.
12. E. I.jiu Pont de Nemours, фр. пат. 1020354, 1950 г.
ГЛАВА VII
КУМАРОНО-ИНДЕНОВЫЕ СМОЛЫ
А. Марти
Кумарон (или бензофуран) был открыт Кремером и Шпиль-
кером1 в 1890 г. во фракции легкого масла каменноугольной
смолы, перегоняемой в пределах 168—175°. Примерно в то же
время Фиттих и Эберт2 также изучали строение производных
этого ряда.
Вплоть до 1930 г. этим соединениям посвящались многочис-
ленные патенты, книги и научные работы, авторы которых оспа-
ривали друг у друга открытие и разработку способов получения
кумароновых смол. Надо особо отметить работы Уэндринера3,
Боттлера4 и Игтфага5 в Германии, Шпеера6, Даррина6, Андер-
сона7 и Ирвина8 в США, Глазера9, Маркуссона10, Кесслера11,
фирмы Баррет12.
Некоторые авторы подробно описывали области применения
кумароно-инденовых смол13-15. После 1930 г. также было опуб-
ликовано несколько печатных работ16-19, но появившиеся син-
тетические полимеры затмили на многие годы кумароно-индено-
вые смолы и оставили их в тени. Истинной причиной такого
пренебрежительного отношения к этим смолам явилось недо-
статочно продуманное их использование в областях, где они
легко могли быть заменены другими вновь открытыми мате-
риалами.
Однако с 1939 г. во Франции, Англии и Германии вследствие
недостатка сырья вновь пробудился интерес к кумарону и инде-
ну, причем их начали применять более разумно, чем прежде.
Даже в США, несмотря на обилие синтетических продуктов, в
ряде случаев признавалась необходимость применения этих
смол, длительное пренебрежение которыми было совершенно
не оправдано 20-26 .
В .настоящее время наши знания в области кумароно-инде-
новых смол сильно обогатились. Теперь полимеризацию можно
регулировать в зависимости от назначения материала и во мно-
гих случаях эти смолы даже не могут быть заменены никакими
другими.
Многочисленные отрасли промышленности нуждаются в
большом количестве кумароновых смол. Для производства этих
250
Гл. VII. Кумароно-инденовые смолы
смол используют богатые кумароном и инденом фракции камен-
ноугольной смолы.
Фракция каменноугольной смолы, кипящая в интервале
160—200°, содержит наряду с инденом и кумароном также
метил- и этилкумароны. При попытках экстрагирования этих
производных они полимеризуются, и получается смола, свойства
которой зависят в основном от условий экстрагирования. В ка-
честве катализаторов полимеризации можно использовать хлор-
ное железо, хлористый алюминий, хлорокись фосфора и др.31,32,
но чаще всего полимеризация проводится в присутствии серной
кислоты •• 91247. В зависимости от концентрации кислоты,
температуры экстрагирования и, вероятно, чистоты выделяемой
фракции, которую называют «кумароновым маслом», получают-
ся более или менее интенсивно окрашенные смолы с различной
температурой плавления и разной растворимостью в ароматиче-
ских углеводородах. При низкой концентрации кислоты получа-
ются легкоплавкие смолы, высокая концентрация содействует
образованию твердых смол с высокой температурой плавления,
а также повышению выходов. Высокая температура экстраги-
рования приводит, как правило, к сильному потемнению поли-
меров; то же явление наблюдается при очень высоких концен-
трациях кислоты. Количество применяемого катализатора, как
правило, не превышает 5% от веса обрабатываемого кумароно-
вого масла.
Обычный промышленный процесс состоит в обработке на
холоду 80—98%-ной серной кислотой фракции каменноугольной
смолы, перегоняющейся в пределах 160—180°. Перегонкой с
водяным паром образовавшуюся кумароно-инденовую смолу
отделяют от углеводородов, сопровождающих смолистые продук-
ты в исходной фракции. Затем следует перегонка в вакууме,
после чего готовая смола остается в котле.
Светлые смолы с высокой температурой плавления, которые
требуются для приготовления бесцветных лаков, получаются
обработкой фракции, перегоняющейся в интервале 160—200°,
небольшим количеством концентрированной серной кислоты при
очень низкой температуре.
Кумарон
Лабораторные способы получения кумарона. Кумарон полу-
чается из кумарина27, 28. Кумарин — вещество с запахом лан-
дыша или свежескошенного сена, применяемое в качестве отдуш-
ки в производстве многочисленных парфюмерных изделий. Он
представляет собой лактон о-оксикоричной кислоты. Бромиро-
ванием этого лактона с последующим омылением образовавше-
гося продукта получается кумариловая кислота, которая при
Кумарон
251
перегонке в присутствии извести превращается в бесцветную
жидкость — кумарон. Реакции протекают по схеме:
кумарон
Можно также получать кумарон путем циклизации фенокси-
ацеталя, а также исходя из оксихлорстирола28:
феноксиацеталь
оксихлорстирол
Промышленный способ получения кумарона. Потребность
промышленности в чистом жидком кумароне очень мала. Это —
дорогой продукт, применяемый для определенных специальных
синтезов или как продукт особого назначения. Чаще всего его
получают синтетически из кумарина.
В промышленности лаков и красок кумарон никогда не при-
меняют в чистом виде — его употребляют в виде кумароно-
инденовой смеси.
Строение и свойства кумарона. Кумарон состоит из бензольно-
го ядра, соединенного с ядром циклопентадиена, но в отличие
от индена группа СН2 замещена атомом кислорода:
СН СН
НС---СН \
II II НС СН—СН НС СН—сн
нс СН | II II I || II
\ / нс СН СН НС сн сн
сн2 v /\ / 'Ч /\ /
сн о сн сн2
циклопентадиен кумарон инден
252
Гл. VII. Кумароно-инденовые смолы
Кумарон представляет собой бесцветную жидкость, запах
которой нельзя назвать неприятным. Темп. кип. 173° при
760 мм рт. ст., уд. в. 1,077 г/см3 при 15°. Кумарон нерастворим в
воде; неосмолившийся кумарон может перегоняться с водяным
паром. Кумарон совместим с ароматическими соединениями,
мало или совершенно несовместим с соединениями парафино-
вого ряда.
Химические свойства27~3(3. Благодаря наличию двойной свя-
зи кумарон способен к реакциям присоединения. Бромирование
кумарона, кроме того, представляет своего рода исторический
интерес, так как впервые он был получен именно в виде бром-
производного1. Присоединение хлора, брома и иода приводит
чаще всего к образованию дизамещенных продуктов. Действием
окиси азота NO получаются нитрозопроизводные, при гидриро-
вании легко образуется дигидрокумарон, хлорноватистая кисло-
та образует с кумароном монохлоргидрин, превращающийся в
диоксикумарон.
Под действием катализаторов или даже просто при нагрева-
нии разрываются двойные связи в молекуле кумарона и про-
исходит полимеризация, приводящая к образованию смолы.
Смола после перегонки регенерирует кумарон, но основная
часть ее карбонизуется, причем образуются продукты деструк-
ции. В определенных условиях при перегонке может получаться
фенол.
По Штаудингеру33, процесс полимеризации кумарона и инде-
на идет следующим образом. Эти вещества содержат в цикло-
пентадиеновом или фурановом ядре двойную связь, которая
под влиянием катализаторов (как это уже было сказано) может
раскрываться с образованием цепных молекул:
НС—СН
нс-сн
нс-сн-
полиннден
поликумарон
По мнению того же автора, при взаимодействии двойных
связей концевых молекул возможно образование циклического
полимера. Однако большая часть проведенных работ свиде-
тельствует о протекании цепной полимеризации, при которой
Кумароно-инденовые смолы
253
сохраняется двойная связь в концевых группах полимера. Это
подтверждается реакцией бромирования полииндена.
Реакция полимеризации экзотермична, причем для кумарона
тепловой эффект меньше, чем для индена. Смолы, получаемые
полимеризацией индена, к тому же менее стабильны.
Продажные продукты представляют собой сополимеры, в
которых группы кумарона и индена распределены беспорядочно:
Н2С
Н2С
---НС-СН—нс-сн—нс-сн—нс-сн—нс-сн—
Полимеризованный кумарон, как и мономерный, не подвер-
жен действию ни кислот, ни щелочей, даже очень концентриро-
ванных, при высокой температуре. Однако в известных условиях
спиртовая калийная щелочь вызывает деструкцию. Окислители,
напротив, бурно реагируют со смолой; однако с озоном реакция
идет сравнительно медленно.
Кумароно-инденовые смолы
Кумароно-инденовыс смолы не имеют запаха. Они стабильны
при высокой температуре, но некоторые из них медленно депо-
лимеризуются при продолжительном нагревании. Кумароновые
смолы трудно воспламеняются, горят коптящим пламенем, рас-
плавляясь. Температуры плавления и размягчения смолы зави-
сят главным образом от способа ее получения.
Кенни26 в 1930 г. предложил следующую классификацию
смол по температуре плавления (способ определения темпера-
туры плавления автором не указан):
Температура
плавления
°C
Мягкая . . 26,7—37,8
Лаковая 126—132
Твердая 148,9
Цвет смол также зависит от способа получения,, но совершен-
но не связан со степенью полимеризации; цветность можно опре-
делять путем сравнения в колориметре бензольного раствора
смолы с эталонным раствором бихромата калия в серной кис-
лоте или хлорного железа в воде.
Твердость смолы является важным свойством, имеющим
решающее значение при применении смол в лакокрасочной про-
мышленности. Можно измерять твердость либо пенетрометром,
применяемым для измерения твердости битумных материалов,
254
Гл. VII. Кумароно-инденовые смолы
либо оценивать ее по растворимости в спирто-эфирной смеси,
так как количество растворившейся смолы обратно пропорцио-
нально ее твердости.
Растворимость смолы имеет очень большое значение. Кума-
роно-инденовые смолы, как правило, легко растворимы в арома-
тических углеводородах, хлорорганических растворителях, ски-
пидаре, труднее в уайт-спирите и нефтепродуктах. Они совмес-
тимы с обычно применяемыми для лаков и красок пластифика-
торами (с фталатами, трикрезилфосфатом и т. д.) и могут сме-
шиваться в любых соотношениях с растительными маслами и
большинством природных и синтетических полимеров.
Кумароно-инденовые смолы имеют высокую диэлектрическую
постоянную (3,5) и по диэлектрическим свойствам близки к
каучуку или шеллаку (диэлектрическая постоянная шелла-
ка 3,1). Они светостойки, но под действием света желтеют при
старении. Вязкость растворов кумароно-инденовых смол при
равных концентрациях выше, чем других смол, например кани-
фоли или ее эфиров. Хрупкость смолы, по Штаудингеру, зависит
от длины цепи; чем выше степень 1полимеризации, тем меньше
хрупкость.
Лаки и краски на основе кумароновых смол
«Кумароновой смолой» мы будем называть смолу, получен-
ную при полимеризации самого кумарона, этил- и метилкумаро-
на и индена, которые находятся в промышленных фракциях
каменноугольного дегтя, отгоняющихся при 160—200°.
Самым важным потребителем кумароновой смолы является
лакокрасочная промышленность, потребляющая в настоящее
время большую часть производимых смол. Довольно большое
количество этих смол используется в резиновой промышленно-
сти, где кумароновая смола является необходимым компонен-
том некоторых резиновых смесей.
В производстве красок используется вся гамма продуктов
разной степени полимеризации. При производстве масляных ла-
ков отдают предпочтение очень твердым смолам с высокой тем-
пературой плавления. Вязкие или пастообразные смолы, тем-
пература размягчения которых не превышает 30°, находят при-
менение для теплостойких покрытий или в качестве пластифи-
каторов других полимеров13.
Приготовление жирных лаков, содержащих кумароновую
смолу, не представляет особых затруднений при варке. Смола,
даже высокой степени полимеризации, растворима в обычных
растворителях, совместима с растительными маслами и с боль-
шинством природных и синтетических полимеров. Легко полу-
чаются очень твердые быстросохнущие лаки для внутренних
Лаки и краски на основе кумароновых смол
255
покрытий или для наружных грунтов, называемые 4-часовыми
лаками, или несколько медленнее высыхающие 12-часовые ла-
ки (для их отверждения требуется по меньшей мере 24 часа),
а также более жирные лаки для наружных покрытий.
В качестве примера можно привести рецептуру (в %), пред-
ложенную Кенни 26-34 в 1930 году:
Кумароновая смола ... 16
Льняное масло .... 4
Тунговое масло . . 25
Растворитель .......... 55
По этой рецептуре получается 12-часовый лак. Тунговое мас-
ло нагревают до 245°, добавляют постепенно % смолы при пе-
ремешивании, после чего повышают температуру до 280—290°
и поддерживают ее в течение 15—30 мин. Окончание варки опре-
деляют по нанесенной на стекло капле, которая должна застыть
в твердый шарик. Тогда добавляют остальную смолу и льняное
масло, охлаждают и вводят растворитель и 'примерно 1% сик-
катива (линолеата кобальта). Чтобы получить светлый лак,
надо избегать длительного нагревания при 280°, вызывающего
потемнение смолы; лучше поддерживать температуру около 220°,
увеличив продолжительность процесса. Получаемые таким спо-
собом светлые пленки, однако, более проницаемы. Поэтому для
наружных покрытий (за исключением грунтов) не рекомендует-
ся добиваться получения светлого продукта.
Ценным качеством лаков, получаемых указанным способом,
является их нейтральность, замедляющая желатинирование тун-
гового масла, что позволяет удлинить время варки. Однако при
использовании некоторых сиккативов, чтобы предотвратить их
выпадение в осадок, требуется небольшая кислотность; поэтому
приходится добавлять небольшое количество канифоли, имеющей
кислую реакцию.
Кумароновая смола не реагирует с основными пигментами,
например с цинковыми белилами, и поэтому удается избежать
явления «загустевания», наблюдаемого при применении других
смол. Для красок с металлическими пигментами нейтральность
смолы необходима для того, чтобы металл сохранил свою отра-
жающую способность. В случае алюминиевой пудры было за-
мечено, что присутствие кумарона содействует лучшему рас-
слаиванию блестящих чешуек и придает пленке более яркий
металлический блеск.
Кумароновая смола не омыляется и благодаря этому на ее
основе можно получать щелочестойкие лаки; кислоты также не
действуют на пленки с большим содержанием кумароновой смо-
лы. Такие агрессивные агенты, как солевой туман или морская
256
Гл. VII. Кумароно-инденовые смолы
вода только очень медленно разрушают защитные покрытия на
основе кумароновых смол.
Кумароновые лаки или краски рекомендуется применять
как в условиях приморского климата, так и в условиях произ-
водственных помещений химических заводов.
Будучи мало чувствительной к действию высокой темпера-
туры, кумароновая смола, входящая в состав лаков и красок,
придает им устойчивость к высоким температурам — до 300°.
Благодаря непроницаемости, адгезионным свойствам и ней-
тральности кумароновые смолы успешно используются для по-
крытия цементов и бетонов. Смесь этой смолы с каолином и кау-
чуком представляет собой превосходную замазку для заделки
трещин, образующихся в силосных ямах.
В резиновой промышленности добавление кумароновой смо-
лы придает резине лучшую устойчивость к истиранию; в произ-
водстве красок это свойство также очень полезно, в частности
для красок, предназначенных для дорожных знаков.
Диэлектрические свойства кумароновых смол допускают их
использование в изоляционных лаках для кабелей; по изоля-
ционным свойствам такие лаки равноценны шеллаку. В смеси
с другими полимерами, например с виниловыми, кумароно-
вые смолы могут быть использованы как изоляционные мате-
риалы.
Как уже указывалось, в 1939 г., ввиду крайнего недостатка
лакокрасочного сырья, начало развиваться производство лако-
красочных материалов на основе каменноугольной смолы21.
Однако пленки, получавшиеся по ранее разработанным рецеп-
турам, часто были хрупкими и очень чувствительными к изме-
нениям температуры. К этому времени благодаря усилиям авто-
ров данной главы краски на основе каменноугольных пеков
и битума были значительно улучшены путем введения мягких,
очень клейких и пластичных кумароновых смол. Эти смолы хо-
рошо совместимы с пеком, так как они родственны ему по хими-
ческой структуре; они придают пеку пластичность и позволяют
получать более однородный материал. Улучшилась также адге-
зия пленки к подложке. В битумных красках, пигментирован-
ных алюминиевой пудрой, благодаря присутствию кумароновой
смолы значительно улучшается также «всплывание» алюминия.
Пластифицирующие свойства кумароновых смол облегчают
применение некоторых очень ценных синтетических смол36, пе-
реработка которых вызывает затруднения. Кумароновые смолы
добавляются к этим полимерам в одних случаях в очень ма-
лых количествах, а в других — до 30—40%.
Виниловые полимеры, например полистиролы, отличающие-
ся большим электрическим сопротивлением даже при высокой
частоте, очень хрупки (Флакс и Монтериоль) и мало эластичны.
Лаки и краски на основе кумароновых смол 257
На их основе нельзя изготовлять изоляционные прокладки для
конденсаторов и лаки для изоляции кабелей без добавки пласти-
фикаторов; последние же часто ухудшают диэлектрические свой-
ства полимера. Введение кумароновых смол позволяет приме-
нять эти полимеры без ухудшения их диэлектрических свойств.
Добавление кумароновых смол к поливинилхлориду практи-
куется с 1940 г. ввиду недостатка обычных пластификаторов
(органических фосфатов)14. Вначале это вызвало ухудшение
качества пластиков, но впоследствии ухудшения удалось избе-
жать. В настоящее время кумароновая смола добавляется к по-
ливинилхориду при пропитке тканей, устойчивых к химическим
реагентам. Добавление этого пластификатора даже в большом
количестве не вызывает ухудшения качества готового продукта;
такой пластификатор обладает хорошей адгезией к волокнам
и устойчив к коррозии.
Благодаря введению кумароновой смолы становится возмож-
ным использование поливинилового спирта в качестве связую-
щего, а также приготовление атмосферостойких лаков на осно-
ве метакриловых полимеров. В сочетании с модифицированными
глифталевыми смолами кумароновые смолы могут использо-
ваться в производстве эмалевых красок для мебели. Совместно
г феноло-формальдегидными смолами они применяются в основ-
ном для приготовления изоляционных лаков или изоляционных
пленок, а также для изготовления многочисленных типов анти-
коррозионных лаков.
Добавка 5% кумароновой смолы к нитрату целлюлозы, кото-
рый сам по себе дает хрупкие пленки35, дозволяет получать
более эластичные покрытия; улучшается адгезия пленок к под-
ложке, повышается атмосферостойкость, пленка приобретает
очень хороший блеск.
Другие области применения кумароновых смол
Второе место по потреблению кумароновых смол занимает
резиновая промышленность, где эти смолы применяются в сме-
сях с каучуком. Вначале кумароновую смолу использовали в
резиновой промышленности для пластификации регенерирован-
ных каучуков. Это — прекрасная адгезионная добавка. Она
смягчает каучук в процессе смешения его с остальными ингре-
диентами и препятствует выцветанию и окислению резиновых
смесей. Кроме того, даже небольшая добавка смолы (5% или
менее) облегчает процессы вальцевания и каландрования. Ку-
мароновую смолу добавляют в резиновые смеси в виде порошка
или расплавленную. При этом повышаются также адгезионные
свойства, особенно для тиоколовых, пербунановых и неопрено-
вых каучуков.
17—156
258
Гл. VII. Кумароно-инденовые смолы
Обработка на каландрах при добавлении кумароновой смо-
лы производится легче, чем при добавлении канифоли. В отли-
чие от канифоли, кумароновую смолу можно применять и для
хлорированных каучуков.
Для латекса кумароновая смола является прекрасным пла-
стификатором, играющим, кроме того, роль антиоксиданта и
придающим изделиям более высокую адгезию. Однако при вул-
канизации необходимо добавлять несколько большее количест-
во серы.
Кумароновая смола улучшает клеящую способность проре-
зиненных тканей29. Она употребляется как добавка к вальцо-
ванным смесям при изготовлении пневматических шин, ремней
и обуви, так как повышает устойчивость резины к истиранию.
В полиграфии при изготовлении пастообразных или полу-
жидких красок вводят некоторое количество кумароновой смолы,
растворенной в скипидаре. Из смесей кумароновых смол с кар-
наубским воском и канифолью получаются высококачественные
восковки. В смеси с этил- и бензилцеллюлозой кумароновая
смола применяется для изготовления литографских красок.
В 1937 г. вся выпускавшаяся в Германии кумароновая смола
использовалась для нужд полиграфической промышленности46.
Кумароновая смола используется для производства искус-
ственных изоляционных восков. Так, в 1942 г. в США38 карнауб-
ский воск заменялся смесью кумароновой смолы с температурой
плавления выше 100° и стеарина или пальмитина с температурой
плавления выше 20°. Эта смесь применялась также как электро-
изоляционный материал, так как ее диэлектрическая постоян-
ная (3,5) была очень близка к постоянной шеллака и каучука
(3,1).
В чистом виде или в смеси с другими смолами кумароновые
смолы применяются также в производстве непроницаемых тка-
ней, бумаги, текстильного волокна. Для придания непроницае-
мости коже, материалу для верха автомобилей и т. д. картон
окунают в раствор кумароновой смолы в бензоле (Эльдис39)
или в смесь смолы с парафином, гуттаперчей и резиной (Шаф-
фер40) . Для проклейки бумаги смолу сплавляют с жирными кис-
лотами и бентонитом в виде геля12.
Сульфированная кумароновая смола в виде солей железа,
хрома или алюминия является хорошим дубителем, который
закрепляется на растительных и животных волокнах в виде
окрашенных пленок37. Как указывали Дендер и Кох41 еще в
1912 г., а позже и другие авторы44, 4S, кумароновые смолы —
хорошее связующее для производства линолеума, клеенки и то-
му подобных тканей; смеси кумароновой смолы, окисленного
льняного масла и древесного масла с добавкой пигмента,, проб-
кового порошка и древесной муки дают превосходные результа-
Лаки и краски на основе кумароновых смол
259
ты. Смешивая кумароновые смолы с асбестовым волокном, крем-
неземом и другими минеральными пигментами, можно получать
пластичный материал, перерабатываемый в листы для покры-
тия полов. Этот материал представляет интерес, так как он
обладает очень высокой устойчивостью к истиранию и стоимость
его невысока.
Следует упомянуть здесь еще о некоторых, несколько не-
обычных областях применения кумароновой смолы. Она упо-
требляется при изготовлении паст для отпечатков в зубовра-
чебной технике, а специально обработанная гидрированная или
дегидрированная кумароновая смола42"43 является основным
элементом жевательной резины.
Приведенный здесь очень сжатый обзор областей примене-
ния и свойств кумароно-инденовых смол подтверждает их боль-
шое значение. Получаемые из фракций каменноугольной смолы,
они являются важным видом полупродуктов для промышлен-
ности лаков и красок. Возможности этих смол еще далеко не
исчерпаны и работы в этом направлении должны продолжаться,
ЛИТЕРАТУРА
1. Kraemer, S р i I к е г. Вег.. 1890, 23, 3276; 1900, 33, 2262; J. Chem.
Soc., 1900, 78, 656.
2. Malatesta, Le goudron et ses derives, Dunod, Paris, 1918, p. 225.
3. Wendriner, герм. пат. 270993, 20/VIII 1912 г.; герм. пат. 281432,
8/XI 1913 г., дополнит.
4. Bottler, Kunststoffe, 1915, 5, 227.
5. I g i f a g, англ. пат. 310816, 1928 г. и 332963, 1929 г.; фр. пат. 677512,
1929 г.
6. S р е е г—D а г г i п, пат. США 1263813. 23/IV 1917 г.; англ. пат. 123806,
1918 г.; Chim. Ind., 1920, 2, 380.
7. Anderson, пат. США 2047245.
8. Irvin, пат. США 1684868, 1928 г.
9. Glaser, Brennstoff-Chem., 1921, 2, 99, ИЗ.
10. Marcusson, Chem. Ztg., 1919, 43, 93, 109, 122.
11. Kessler, фр. пат. 51411035, 28/Ш 1922 г.
12. Barret Company, англ. пат. 149982, 1920 г.; Procede de fabrication de resi-
nes, фр. пат. 524238, 1921 г.; фр. пат. 520850, 1921 г.; англ. пат. 225216,
1923 г.; пат. США 2024568; Chim. et Ind., 1936, 2, 585; пат. США 2217119,
13. Е. du Pont de Nemours, пат. США 1334049, 8/VIII 1917 г.
14. G i b е 1 I о, Resines vinyliques, Dunod, 1947, 181.
15. Gerard, J. Pharmac. Chim., 1927, 6, 110.
16. Morgan, Harrisson, англ. пат. 319444, 23/VII 1938 г,
17. Rev. Gen. mat. plast., 1936, 12, 361.
18. Svensson, англ. пат. 468374; Chim. et Ind., 1938, 1, 1172.
19. P у т м а н, Кокс н химия, 1937, 7, 19; Chim. et Ind., 1938, 2, 554.
20. C h о m a r d Emile, Rev. Gen. mat. plast., 1939, 15 et 16, 200.
21. Marty, J. U. G., 1939, 56, 420; Gaz de France, Sauret, Monaco, 1948,
22. W e i n d e 1, герм. пат. 698855, 1940 г.
23. Darmi er, Paquin, T г о и. герм. пат. 721197, 1942 г.
24. Powers, пат. США 2287535, 1943 г.
25. Cline, пат. США 2345962, 1944 г.; пат. США 2344676, 1944 г.
17*
260
Гл. VII. Кумароно-инденовые смолы
26. Кеппу, Paint, Oil Chem. Rev., 1941, 20 a. 22, 32; Drugs, Oil a. Paint,
1930, 45, 261; Am. Paint J., 14, 20, 1930; Paint, Oil Chem. Rev,, 1930,
89, 12.
27. Lamirand, Pariselle, Cours de chimie organique, Masson, Paris,
1947, p. 621.
28. Richter, Traite de chimie, Librairie Polyt. Beranger, Paris, 1918. 2,
821. v .
29. T о u г n a i r e, Benzene, Goudrons et derives, Nouvelle Societe d'Edi-
tion, Paris, 1946.
30. Champet ier, Les hauts polymeres, Cent, de Doc. Tournier et Con-
stsns Paris.
31. Miller, пат. США 1360665, 1921 -.
32. H i г а п о, J. Chem. Ind. Japan, 1922. 25, 827.
33. Staudinger, Helvetia chem. acta, 1922, 5, 787; 1929, 12, 934; Ber.,
1920, 53, 1073; J. Am. Chem. Soc., 1928. 50, 1162; Ind. Eng. Chem., 1934,
26, 663.
34. King, Bayard, Rhodes, Ind. Eng. Chem., 1920, 12, 549; J. Chem.
Ind., 1921, 1, 328.
35. Brit. Ind. Fin., 1931, 2, 36; Rev. Gen. mat. plast., 1937. 13, 334.
36. D u b о i s, Cor, Collin, Manuel des Plastiques, Presses doc., Paris,
246.
37. Gert und Farbstoffwerke—Renner et Co., англ. пат. 173757.
38. С о г к e г у, пат. США 2255242.
39. Е 11 i s, пат. США 1451092; пат. США 1412014; пат. США 1570584; Chem.
Ind., 1926, 1, 95Э.
40. Schaffer, пат. США 183191.
41. Lender, Koch, пат. США 1019666.
42. М a t h е г е 1 1, пат. США 2070047.
43, Carmody, Sherhan, Kelly, Ind. Eng. Chem., 1938, 30, 245.
44. E. du Pont de Nemours, пат. США 1334049.
45. The Solden Co., пат. США 1892769.
46. Rev. Gen. mat. plast., 1937, 13, 108.
47. Masson. Ind. Plast., 1948, 4, 82.
ГЛАВА VI11
ПОЛИЭФИРЫ, ПОЛИАМИДЫ И ПОЛИУРЕТАНЫ
Р. Элион
Изучение синтеза полимеров путем поликонденсации, нача-
тое 25 лет тому назад Карозерсом, привело к освоению в про-
мышленном масштабе производства многочисленных типов по-
лимеров, из которых важнейшими являются полиэфиры и поли-
амиды. Карозерс1, установивший на основе глубоких теорети-
ческих исследований закономерности поведения исходных моно-
меров, использовал эти данные прежде всего для синтеза али-
фатических полиэфиров из (о-оксикислот. Стремясь получить
полимеры, пригодные для получения синтетических волокон,
Карозерс был разочарован физическими свойствами полиэфиров,
в частности их низкой температурой плавления и неустойчи-
востью по отношению к обычным растворителям, что препятство-
вало применению полиэфиров в качестве текстильного сырья.
Тогда Карозерс решил получить полимеры, по химическому
строению сходные с шерстью и шелком, вводя в цепь амидные
группы. Он синтезировал линейные полиамиды, промышленное
производство которых, так же как и производство синтетическо-
го волокна, было осуществлено на заводах фирмы Дюпон.
Широкие изыскания, предпринятые затем в США и в Германии,
привели к развитию производства полиамидных пленок, красок,
обмазок и клеев.
То же произошло с ароматическими полиэфирами, которые,
возбудив сначала очень большой интерес в качестве возмож-
ного сырья для синтетического волокна, нашли широкое рас-
пространение в пленочной промышленности и в производстве
тонких прозрачных листов, выпускаемых под названием милара.
Усиленные изыскания немецких химиков, главным образом кон-
церна И. Г. Фарбениндустри, сосредоточились вначале на син-
тезе полиамидов, а затем на полиуретанах — полимерах, по хи-
мической природе близких к полиамидам. Успехи в этом на-
правлении оказались настолько значительными, что полиуретаны
стали применяться не только для синтетических волокон, но и в
качестве пленкообразующих в лакокрасочной промышленности.
Полиамиды и полиэфиры являются классическим примером
высокомолекулярных соединений, получаемых поликонденса-
цией.
262 Гл. VIII. Полиэфиры, полиамиды и полиуретаны
Полиэфиры синтезируют, исходя либо из оксикислот
«НО—R—СООН -> Н-[—О—R—СО—J„—ОН + (п — 1)Н2О
либо из двухосновных кислот и двухатомных спиртов:
nHO—R—ОН + пНООС—R'—СООН ->-
-* Н—[—О—R—ООС—R'—СО—ОН + (2л — 1)Н2О
Для получения полиамидов поликонденсацию можно прово-
дить, исходя из аминокислот
nH2N—R—СООН — Н—[—HN—R—СО—]л -ОН + (п — 1)Н2О
или из двухосновных кислот и диаминов:
nHjN—R-NH2 + «НООС—R'—СООН
Н— [—HN—R—NHCO—R'—СО—ОН + (2п— 1)Н2О
Во всех приведенных реакциях R и R' являются двухвалент-
ными радикалами.
Полиуретаны, близкие к полиамидам по химическому строе-
нию, могут тем не менее получаться еще одним путем, так как
способ их синтеза является промежуточным между поликонден-
сацией и полимеризацией. Как при поликонденсации, мономеры
бифункциональны и образуют низкомолекулярные промежуточ-
ные продукты реакции, которые могут быть выделены, но в то
же время, как при полимеризации, в процессе роста цепи не
происходит выделения простых молекул, в частности молекул
воды. Этот синтез основан на реакции между спиртовыми и изо-
цианатными группами по схеме:
R—N=C=O4-H—O-R' -* R—NH—С^°
Для цепи полиуретанов характерно присутствие уретановой
группы
—О—С—NH—
II
О
Полиуретаны образуются при взаимодействии двухатомных
спиртов с диизоцианатами:
nHO—R—ОН + nO=C=N—R'—N=C=O -*
---[—О—R—O-CONH—R'—NHOC—]„----
Здесь также R и R' являются двухвалентными радикалами.
Все эти типы полимеров различаются по химическим и физи-
ческим свойствам, сообщаемым эфирными, амидными и урета-
новыми группами, входящими в основную цепь.
Исходные материалы и методы синтеза мономеров 263
Исходные материалы и методы синтеза мономеров
Ко времени промышленного освоения синтеза полиэфиров,
полиамидов и полиуретанов доступным сырьем являлись только
двухатомные спирты и, в меньшей степени, двухосновные кис-
лоты. Синтез аминокислот, диаминов и диизоцианатов осущест-
влялся в масштабах, не превышавших лабораторные. Труд-
ности промышленного освоения синтеза подобных мономеров
были быстро преодолены в нескольких отдельных случаях, но
без общего подхода к проблеме сырья. Поэтому промышленные
способы синтеза мономеров до сих пор основаны на применении
ограниченного числа исходных материалов: фенола или бензола,
циклогексана, ацетилена и формальдегида, фурфурола,, полу-
чаемого из растительного сырья, и, наконец, касторового масла.
На стр. 264 приведены схемы современных промышленных син-
тезов.
Первым синтезом, получившим промышленное значение, был
осуществленный фирмой Дюпон процесс получения адипиновой
кислоты. Адипиновая кислота (двухосновная кислота для про-
изводства найлона) получается по этому методу, исходя из фе-
нола, каталитическим его гидрированием с последующим окис-
лением. По немецкому методу исходным сырьем для синтеза
адипиновой кислоты служит тетрагидрофуран, получаемый либо
из ацетилена и формальдегида, либо кислотным гидролизом
овсяных высевков, кукурузных кочерыжек или сульфитных ще-
локов (отходов производства целлюлозы). Адипиновая кислота
получается из тетрагидрофурана при непосредственном присое-
динении окиси углерода и воды под давлением.
Синтез е-капролактама, освоенный фирмой И. Г. Фарбенин-
дустри, основан на взаимодействии циклогексанона с водным
раствором гидроксиламина. Получаемый при этом оксим под
действием концентрированной серной кислоты претерпевает бек-
мановскую перегруппировку и образует е-капролактам, исполь-
зуемый в качестве мономера при синтезе перлона*, или игамида L.
Гексаметилендиамин получается гидрированием адипонитри-
ла, полученного из дихлорбутана и цианида натрия, или взаимо-
действием аммиака и паров адипиновой кислоты в присутствии
борфосфатного катализатора. Гексаметилендиаминадипат, кото-
рый является основным мономером при получении найлона,
образуется при смешении спиртовых растворов адипиновой кис-
лоты и гексаметилендиамина при нагревании.
Гексаметилендиизоцианат (один из мономеров полиуретано-
вого волокна перлона U) (получается из гексаметилендиамина
* В СССР получаемый таким путем поликапроамид выпускается под назва-
нием «капрон». Прим. ред.
СХЕМЫ СИНТЕЗА МОНОМЕРОВ
Ацетилен -f-формальдегид
CHECH + 2НСНО
|cu2C2
СН2(ОН)-С=С-СН2ОН
^гидрирование
СНа(ОН)-СН2-СН2-СН2ОН
I дегидратация
Сульфитные щелока
Овсяные высевки
Кукурузные кочерыжки
।гидролиз
НС----СН
НС С:—СНЭ
V
I декарбон и-
I лирование
Продукты переработки камеи- v____________
иого угля и нефти Касторовое масло
н-Цимол и др.
Н2С СН2
НС1\ /2СО+НаО
Н2С-----СН2 гидрирование
окисление HNO3
С1(СН2)4С1 НООС(СН2)4СООН
NaCN^ +NH3
NC(CH2)4CN ---
гпдри- у
рииание I
Т Найлон
H2N(CH2)eNH2 —Перлой 1
। Игамид А
fCOgj
H2N(CH2)eNH-COOH
cocij I
C10CHN(CH2)«NHCOCI
окисление воздухом
Перлон L
Игамид U
- 2НС1 j
or.N-(CH2)fl-NC.O |и&1|
СН2
Н^ \-0
I NH
Н2С (!н2
\н2
СН2
н2<^ чсн-он
I I
н2с сн2
Хсн^
I
сн2
НзС С=*О
н2с ёна
\н2
сн2
H2(f XC-NOH
I I
H2C CHj
\н2
кон
Т
СН»-(СН2)в-СН-СН2-СН=СН(СН2)7СООн
I
он пиролиз
I I
СН2=СН(СН2)аСООН СН3(СН2)6-СНО
гидробро-1
мироваиие |
Вг(СН2)юСООН
аминиро- I
ваиие |
H2N(CH2)i0COOH
I
| Рилсан |
I Терилен I •*— НОН2С—СН2ОН
I Дакрон |
СНз
ч/
^Н(СНз)!
окисле-
ние
| HNO3
СНз
II
Ч/
J.OOH
| окисле-
I ние
jKMnO4
СООН
I
СООН
+CH3OH
СООСНз
(';оо( :н4
Синтез полимеров
265
через стадию образования монокарбамата гексаметилендиами-
на, на который действуют фосгеном при температуре —5°.
Для полиундеканамида, выпускаемого под названием рилса-
на, исходным сырьем служит касторовое масло, основной со-
ставной частью которого является триглицерид рицинолевой
кислоты. После омыления и пиролиза эта кислота распадает-
ся на ундециленовую кислоту и энантол. Мономер для по-
лучения рилсана (ю-аминоундекановая кислота) получается
в результате присоединения бромистоводородной кислоты по
месту двойной связи ундециленовой кислоты с последующим
замещением брома на аминогруппу.
Для производства терилена (полиэтилентерефталата) исход-
ным сырьем служит углеводород бензольного ряда с боковыми
цепями в пара-положении. Углеводород окисляется, образуя
терефталевую кислоту. Терефталевая кислота этерифицируется
метанолом в присутствии серной кислоты при нагревании. Диме-
тилтерефталат (темп. пл. 142°) выделяют вымораживанием с
последующей перегонкой в вакууме.
Синтез полимеров
При синтезе полиэфиров и полиамидов рост полимерной
цепи осуществляется путем последовательных реакций этери-
фикации и амидирования. Поэтому, приостановив в определен-
ный момент реакцию поликонденсации, можно выделить поли-
меры заданного молекулярного веса. Рост цепи, как правило,
можно проследить, отбирая пробы по ходу реакции и исследуя
изменение относительной вязкости растворов или вязкости рас-
плавленной массы. ДАолекулярный вес продажных полиэфиров
и полиамидов колеблется от 10 000 до 30 000, в зависимости
от их назначения.
Полиуретаны, получаемые путем ступенчатой полимериза-
ции, имеют более низкие молекулярные веса (10 000— 12 000),
так как синтез их проводится в среде растворителя.
Рассматриваемые полимеры часто синтезируют в темпера-
турных условиях, при которых реакция поликонденсации разви-
вается с очень большой скоростью. Это приводит к тому, что
выделяющиеся пары воды остаются в виде пузырей в массе по-
лимера, свойства которого перестают поддаваться контролю.
Поэтому возникает необходимость регулировать скорость реак-
ции. Замедление реакции поликонденсации достигается либо
путем понижения реакционной способности концевых групп вы-
сокомолекулярных цепей, либо путем введения небольшого ко-
личества моно- или бифункционального реагента, а в некоторых,
случаях — путем создания небольшого избытка одного из моно-
меров. Химическая природа стабилизатора зависит от назначе-
ния полимера.
266 Гл. УШ. Полиэфиры, полиамиды и полиуретаны
При синтезе полиэфиров и полиамидов стабилизаторами мо-
лекулярного веса служат обычно уксусная или адипиновая кис-
лота, гексаметилендиамин и т. д. Реакции образования поли-
уретанов также замедляют путем введения небольшого коли-
чества монофункционального реагента или небольшого избытка
мономерного диизоцианата.
Промышленные способы получения полимеров. При синтезе
поликонденсационных линейных полимеров должны быть реше-
ны три основные технические задачи:
1) возможно более полное удаление воды из сферы реакции;
2) выбор химически инертного металла для конструирования
реакционного аппарата;
3) создание инертной атмосферы и постоянной температуры.
Обычно достаточная скорость реакции достигается при тем-
пературе кипения мономеров. Следовательно, удаление воды,
образующейся при поликонденсации, должно проводиться при
высокой температуре. При поликонденсации одного мономера,
стабильного при температуре кипения, реакция легко осуществ-
ляется без катализатора. Если же в поликонденсации участвуют
два мономера с различной температурой кипения или способ-
ных возгоняться, необходимо сначала получить ониевую соль
(например, гексаметилендиаминадипат) и затем дегидратиро-
вать эту соль. Ниже приведено несколько примеров поликонден-
сации в 1производственных условиях.
Полиамиды. В автоклав с двойными стенками, между кото-
рыми циркулирует высокотемпературный теплоноситель (дау-
терм), загружают мономер (аминокислоту или ониевую соль)
в виде пасты или водного раствора в присутствии неболь-
шого количества (от 1 до 10%) стабилизатора. Затем постепен-
но повышают температуру до температуры кипения мономера,
в результате чего в автоклаве за счет выделяющейся воды уста-
навливается высокое давление. По мере протекания реакции
давление постепенно понижают, а когда оно снизится до атмо-
сферного, путем циркуляции азота создают инертную атмосферу
над жидкой реакционной массой. Поликонденсацию, продол-
жающуюся много часов, заканчивают в вакууме, чтобы получить
максимальный выход. Готовый полимер под небольшим давле-
нием а'зота выдавливают из реактора через щель в нижней части
аппарата. Полученную ленту полимера быстро отверждают,
охлаждая струей воды во избежание окисления, а затем для
удобства применения превращают в стружку.
Ароматические полиэфиры. Алифатические полиэфиры бла-
годаря низкой температуре плавления двухосновных алифатиче-
ских кислот образуются сравнительно легко. Труднее получить
ароматические полиэфиры, так как двухосновные ароматиче-
ские кислоты имеют очень высокую температуру плавления и
Полиэфиры
267
большую склонность к возгонке. Поэтому приходится пользо-
ваться эфирами этих кислот, обычно диметиловыми. При этом
вместо сложноэфирной конденсации происходит переэтерифика-
ция, т. е. реакция обмена между метиловым спиртом и мономер-
ным двухатомным спиртом; эта реакция протекает труднее, чем
отщепление воды, и поэтому для ее осуществления требуется
катализатор.
При синтезе полиэтилентерефталата реакция диметилтере-
фталата с избытком гликоля проводится в присутствии маг-
ниевой ленты, служащей катализатором. В первой стадии про-
цесса (при температуре кипения эфира) протекает переэтерифи-
кация диметилтерефталата гликолем и образующийся метиловый
спирт удаляют в токе азота. Во второй стадии (при более высо-
кой температуре) удаляется избыток гликоля; реакция закан-
чивается в вакууме.
Полиуретаны. Реакция поликонденсации диизоцианата с
двухатомным спиртом проводится обычно в прйсугствии рас-
творителя. Синтез перлона U, или игамида U, из гексаметилен-
диизоцианата и 1,4-бутандиола 1проводится в среде раствори-
теля, состоящего из 90% хлорбензола и 10% дихлорбензола.
Регулятором реакции служит небольшой избыток диизоцианата.
Следует отметить, что диизоцианаты токсичны и при их пере-
работке должны соблюдаться определенные условия.
Классификация полимеров
Полиэфиры, полиамиды и полиуретаны удобно классифици-
ровать по характеру связей между макромолекулами. Разли-
чают три типа межмолекулярных связей:
1) связи за счет сил ван-дер-Ваальса (энергия связи
1,5—5 ккал/моль, радиус действия 3—5 А);
2) связи за счет сил электростатического взаимодействия по-
лярных групп или групп, способствующих поляризации молеку-
лы (энергия связи 5—10 ккал/моль, радиус действия 2—3 А);
3) ковалентные связи в трехмерных полимерах (энергия
связи 50—200 ккал/моль, радиус действия 1—2 А).
В зависимости от типа межмолекулярных связей полимеры
имеют различные физические и механические свойства.
ПОЛИЭФИРЫ
Полиэфиры относятся к полимерам, для которых характер-
ны очень слабые межмолекулярные связи типа электростати-
ческого взаимодействия. Эфирные группы очень слабо шолярны,
й цепи полимера сохраняют большую гибкость. Этим объяс-
няется сравнительно низкая температура плавления полиэфиров:
268 Гл. VIII. Полиэфиры; полиамиды и полиуретаны
она даже ниже, чем у полиэтилена, имеющего чисто парафино-
вую цепь. Такая структура полимеров обусловливает их высо-
кую растворимость не только в обычных органических раство-
рителях, но и в воде. Физические свойства лишь незначительно
изменяются с увеличением числа метиленовых групп в цепи,
приближаясь к свойствам полиэтилена.
Ниже приводятся температуры плавления полимеров с раз-
личной длиной парафиновой цепи:
Температура
плавления
°C
Адипиновая кислота + этиленгликоль...................... 50
Адипиновая кислота + гексаметиленгликоль .... 56
Адипиновая кислота + декаметиленгликоль ... 77
Себацнновая кислота 4- декаметиленгликоль .... 74
Указанные свойства алифатических полиэфиров препятствуют
их применению в производстве синтетических волокон или пла-
стических масс. Эти полиэфиры применяются в производстве
пластификаторов (так как алифатические полиэфиры совмести-
мы со многими другими полимерами, например с меламино-аль-
дегидными смолами, смолами на основе эфиров диметилол-
мочевины и т. д.), клеящих составов и лакокрасочных мате-
риалов.
Полиэфиры с длинной парафиновой цепью выпускаются под
торговой маркой параплекс. Алифатические полиэфиры приме-
няются также в виде сополимеров с полиамидами, обладающих
повышенной растворимостью.
Линейные ароматические полиэфиры
Физические свойства полиэфиров можно значительно изме-
нять, понижая чрезмерную гибкость высокомолекулярных
цепей.
Различные исследователи2, главным образом из фирмы
Calico Printers, пытались синтезировать такие ароматические
полиэфиры, в которых присутствие циклических ядер уменьшает
возможность свободного вращения цепей. В результате был по-
лучен полиэтилентерефталат, который вначале использовался
только для синтетического волокна, выпускаемого в Англии под
названием терилена и в США под названием дакрона. В настоя-
щее время фирма Дюпон изготовляет на основе этого полимера
путем экструзии пленки и тонкие прозрачные листы с замеча-
тельными механическими свойствами. Этими свойствами они
обязаны сильной ориентации макромолекул путем вытяжки
пленки в двух взаимноперпендикулярных направлениях. Этот
Полиамиды и полиуретаны
269
листовой материал, выпускаемый под названием милара, отли-
чается большой стабильностью размеров в двух направлениях
и химической инертностью.
Ниже изображено строение ориентированного полиэтилен-
терефталата:
О О
с—о—сн2-сн2—о—с—с—о—сн2—сн2-------
Л о h
i О 10.86А О
Комплексные полиэфиры
К комплексным полиэфирам относятся линейные полиэфиры
с очень длинной парафиновой цепью и трифункциональные
полиэфиры. Соединения этого класса являются одними из важ-
нейших пленкообразующих в производстве красок.
В главе XI, посвященной алкидным смолам, рассматривают-
ся полиэфиры, получаемые путем поликонденсации фталевого
ангидрида с многоатомными спиртами, например с глицерином
и дипентаэритритом. Здесь мы ограничимся разбором синтеза
полиэфиров из малеинового ангидрида. При взаимодействии
двухатомного спирта с этим ангидридом в первой стадии полу-
чаются линейные полиэфиры, содержащие конъюгированные
двойные связи типа —С = С—С=О:
СН=СН
nHO—R—ОН 4- .1С
О
HO-
CH =сн
I I
—R—О—С С-О--------Н 4- (п — 1)Н2О
II II
О О
Благодаря наличию этих двойных связей, при действии воз-
духа, по тому же механизму, который приводит к высыханию
масел, образуются межмолекулярные мостиковые связи и полу-
чаются трехмерные полимеры, выпускаемые в виде пленок под
названиями селектрон, талид, вибрин и т. д.
Наконец, многочисленные полиэфиры, содержащие больше
гидроксильных групп, чем карбоксильных, были синтезированы
в Германии как исходные продукты для получения специальных
полиуретанов путем реакции с изоцианатами. Эти полиэфиры
выпускаются под торговым названием десмофен.
270 Гл. VIII. Полиэфиры, полиамиды и полиуретаны
ПОЛИАМИДЫ И ПОЛИУРЕТАНЫ
Амидные и уретановые функциональные группы весьма по*
лярны. Поэтому при высокой степени симметрии макромолекул
таких шолимеров силы межмолекулярного взаимодействия
должны быть очень значительными и следует ожидать высо-
ких механических свойств, сочетающихся с инертностью по отно-
шению к растворителям и химическим реагентам. Эти межмоле-
кулярные связи могут быть в большей или меньшей степени
нарушены, и таким образом может быть получена целая гамма
полимеров, пригодных для таких разнообразных областей, как
производство волокон, пластических масс, красок, лаков, клеев
и клеящих веществ. Изменяя длину и химическую природу ради-
калов R и R', расположенных между функциональными группа-
ми, а также варьируя функциональность .мономеров или реак-
ционную способность амидных или уретановых групп, получают
различные полимеры.
Линейные алифатические полиамиды и полиуретаны
Для этих полимеров характерно наличие межмолекуляриых
сил электростатического притяжения, которые усиливаются во-
дородными связями между группами СО одной цепи и группа-
ми NH соседней цепи:
Температура плавления полимеров этого типа понижается по
мере удлинения цепи, причем полимеры с четным числом ато-
мов углерода в цепи плавятся при более высоких температурах,
чем полимеры с нечетным числом углеродных атомов (рис. 51).
Межмолекулярные связи, действующие вдоль цепей, харак-
теризуются специальным показателем (показатель водородных
связей). Он определяется числом водородных связей между
группами СО и NH, приходящимся на 100 ковалентных связей
Полиамиды и полиуретаны
271
в макромолекулярной цепи3. Алифатические полиамиды и поли-
уретаны линейного строения отличаются -высоким показателем
водородных связей. Поэтому они все нерастворимы в обычных
растворителях и отличаются высокой температурой плавления.
Температура их плавления понижается пропорционально умень-
шению показателя водородных связей (рис. 52). Основной об-
ластью применения этих полимеров является промышленность
синтетических текстильных волокон. Но широкое поле для их
применения открылось после разработки методов нанесения по-
крытий и изготовления пленок способом экструзии или напыле-
нием в расплавленном состоянии. Например, расплавленный
поликапроамид при выдавливании через щель длиной 50 см и
шириной 0,2 мм образует лист, который принимают на вращаю-
Рис. 51. Зависимость темпе-
ратуры плавления полиамидов
от числа атомов углерода.
щиися охлажденный цилиндр, а
затем вытягивают на 300%, дово-
дя толщину листа (пленки) при-
мерно до 0,05 -ii.w. Такие пленки
Показатель Водородных связей
Рис. 52. Зависимость температуры
плавления полиамидов от величины по-
казателя водородных связей:
/—полиамиды с нечетным числом групп СН2;
2— полиамиды с четным числом групп СНг-
выпускаются под названием перфоль. Они употребляются, в
частности, для авиационных бензобаков. Полиундеканамид
(рилсан) рартворяют под давлением в бутиловом спирте, затем
высаживают путем охлаждения в виде порошка. Этот порошок
можно затем распылять в расплавленном состоянии на поверх-
ности, получая защитное покрытие с высокими механическими
свойствами и химической устойчивостью. Такой порошок выпу-
скается под названием рилсенит.
Полиуретаны в чистом виде или пластифицированные N-ме-
тилбензолсульфамидом можно также перерабатывать методом
экструзии при температуре плавления (184°). Полученные таким
способом листы успешно применяются в качестве прочной искус-
272 Гл. VIII. Полиэфиры, полиамиды и полиуретаны
ственной кожи, пленок или оболочек. Эти полимеры не нашли
применения в лакокрасочном, производстве 'по причине нерас-
творимости в обычных растворителях. Растворимость была до-
стигнута при создании новых типов полимеров со значительно
более низким показателем водородных связей. Ниже приведены
основные методы их получения и области промышленного при-
менения.
Полиамиды с очень длинной линейной алифатической цепью
Самый простой способ понижения показателя водородных
связей — это разделение амидных групп длинными парафино-
выми цепями. Для достижения этой цели были применены двух-
основные кислоты, содержащие 16 и более атомов углерода.
Однако при поликонденсации таких кислот с диаминами не
удалось получить полимеры с достаточно высоким молекуляр-
ным весом. Такие полимеры, плавящиеся при низкой темпера-
туре, не обладают тем не менее удовлетворительной раствори-
мостью. Другой, более интересный способ состоит в непосред-
ственном использовании одноосновных кислот, полученных омы-
лением высыхающих масел (льняного, тунгового)4. При вза-
имодействии с диамином, например с этилендиамином, получает
соответствующий диамид, содержащий в цепи сопряженные
двойные связи и, следовательно, реакционноспособный. При
термической полимеризации этого полиамида получается линей-
ный полимер с длинной цепью, по строению аналогичный про-
дуктам термической полимеризации триглицеридов. Подобные
полиамды растворимы при нагревании в растворителях высы-
хающих масел. В результате окисления кислородом воздуха
полиамиды образуют трехмерную решетку вследствие «сшива-
ния» цепей кислородными мостиками.
Полиамиды, полученные таким путем, были использованы
в промышленности для получения пленок более высокого каче-
ства, чем пленки льняного масла, образующиеся в тех же усло-
виях. Эти полимеры продаются под названием норлак.
Продукты совместной поликонденсации и диспропорционирования
Показатель водородных связей может быть сильно понижен
путем применения смесей мономеров с разной длиной алифати-
ческой цепи. Реакция совместной поликонденсации двух или
нескольких аминокислот, или нескольких двухосновных кислот
с диаминами, или, наконец, смеси аминокислоты, двухосновной
кислоты и диамина приводит к образованию высокомолекуляр-
ных цепей, в которых мономеры располагаются случайно и рас-
стояния между функциональными группами в цепи различны.
Вследствие этого очень ограниченное количество групп окажет-
Полиамиды и полиуретаны
273
ся на расстоянии, достаточном для образования водородных
связей, и показатель водородных связей будет, следовательно,
невысоким. Минимальное значение этого показателя достигает-
ся при эквимолекулярном соотношении мономеров, так как в
этом случае достигается максимальная нерегулярность располо-
жения амидных группировок.
Продукты такого типа могут быть получены также путем
смешения двух разных расплавленных полимеров. В результате
диспропорционирования по амидным (группам быстро дости-
гается нерегулярное распределение отрезков алифатических це-
пей. Здесь также при равных количествах полимеров создается
максимальная нерегулярность строения.
Эти наиболее простые способы понижения показателя водо-
родных связей получили широкое промышленное применение.
Пользуясь ими, можно получать продукты' совместной поликон-
денсации или диспропорционирования с повышенной раствори-
мостью, сохраняющие удовлетворительные механические свой-
ства и приемлемые температуры плавления. Разнйца в длине
отрезков алифатических цепей может быть небольшой. Фирма
И. Г. Фарбениндустри применяет, например, продукты совмест-
ной поликонденсации гексаметиленадипамида и капролактама
следующего состава:
Игамид 6А
Игамнд’БА
Игамид 1С
{60% гексаметиленадипамида
40% капролактама
(50% гексаметиленадипамида
[ 50% капролактама
Тройная смесь гексаметиленадипамида,
капролактама и циклогексилметан-
адипамида
Добавлением к игамиду 1С полиамида, полученного из ади-
пиновой кислоты, и п,п'-диаминодициклогексилметана
удается получить прозрачный полимер.
Фирма Rhodiaceta выпускает смешанные полиамиды под
торговыми марками В60, В50, МВ1 и МВ2, английская фирма
I.C.I выпускает растворимые полиамиды под маркой «найлон
растворимый типа СА».
Смешанные полиамиды отличаются от индивидуальных поли-
амидов более низкой и менее четкой температурой плавления.
Зона пластических деформаций у них значительно больше.
Растворением в смесях спирт—вода и спирт—хлорированный
углеводород можно получать вязкие растворы с содержанием
до 30% полимера. Растворение осуществляется обычно при на-
гревании, и в зависимости от значения показателя водородных
18-156
274 Гл. VIII. Полиэфиры, полиамиды и полиуретаны
связей смешанного полиамида раствор остается стабильным
или желатинируется при охлаждении. Ниже приведено несколь-
ко примеров применения таких «полимеров.
Смешанный полиамид растворяют в автоклаве при темпе-
ратуре 70—75° в водном 71%-ном спирте. На 300 л раствори-
теля берут примерно 100 кг полиамида. Для повышения адге-
зии и эластичности пленок можно добавлять пластификаторы:
изододецилфенол, деллатол (смесь 75% толуолметилсульфами-
да и 25% толуолбутилсульфамида), резорцинфосфат, диоксиди-
фенилсульфон, бензилсалицилат. Добавление некоторых высо-
кокипящйх растворителей, таких, как бензиловый спирт, позво-
ляет замедлить испарение раствора и сушку пленки.
Путем налива раствора на бесконечную ленту, обдуваемую
горячим воздухом, можно получать пленку с производитель-
ностью от 160 до 200 м/час. Растворитель регенерируют, пленку
нарезают и затем кондиционируют в помещении, где происхо-
дит досушка в мягких условиях. Толщина пленки может коле-
баться от 0,2 до 0,002 мм. Такие пленки выпускаются под на-'
званием лиафоль.
Во Франции смешанные полиамиды применяются в самых
разнообразных областях: для лакокрасочных покрытий, пропит-
ки, лакировки и даже для нанесения покрытий путем окунания.
Это возможно благодаря совместимости полиамидов с мочеви-
но-формальдегидными смолами и шеллаком.
Полимеры с разветвленной цепью
Совместная поликонденсация позволяет снизить показатель
водородных связей, внося некоторый беспорядок в расположе-
ние амидных групп в цепи высокополимера. Наличие боковых
цепей ослабляет межмолекулярное Взаимодействие макромоле-
кул полиамидов и полиуретанов, увеличивая расстояние между
цепями. Так как расстояние между атомами, соединенными во-
дородной связью, составляет приблизительно 2,8 А, то доста-
точно заместить один атом водорода в алифатической цепи
(или даже у атома азота) на группу, представляющую доста-
точное пространственное препятствие, чтобы равномерно осла-
бить силы когезии по всей длине цепи.
Аналогичным способом частичного ослабления водородных
связей между цепями является совместная поликонденсация
N-ациламидов с амидами кислот нормального строения. При
изменении степени замещения водородных атомов амидных
групп на ацильный радикал можно получить целый ряд полиме-
ров’ свойства которых являются промежуточными между свой-
ствами нормальных полиамидов и полиамидов, полностью за-
мещенных при азоте6.
Полиамиды и полиуретаны
275
Полимеры, содержащие гетероатомы в углеводородной цепи
Все рассмотренные способы синтеза полимеров, пригодных
для лакокрасочной промышленности, приводят к повышению
растворимости при одновременном ухудшении механических
свойств. Чтобы устранить этот недостаток, у некоторых авторов
появилась идея не изменять структуру полимеров для уменьше-
ния вторичных сил взаимодействия, а добиться растворимости,
вводя в цепь атомы элементов, обладающих повышенным срод-
ством к органическим растворителям. Например, введение в
алифатическую цепь атомов кислорода или серы придает поли-
мерам растворимость без ослабления сил межмолекулярного
взаимодействия6.
Полимеры с ароматическими радикалами
Гибкость, или свободное вращение, высокомолекулярной
цепи оказывает решающее влияние как на растворимость поли-
мера, так и на температуру плавления. Например, применяя
для конденсации с этиленгликолем терефталевую кислоту вме-
сто адипиновой, можно получить полиэфир, плавящийся при
температуре 256°. При синтезе полиамидов поликонденсация
той же двухосновной кислоты с гексаметилендиамином приво-
дила к получению полимера, плавящегося при 80°, т. е. при
более высокой температуре, чем полигексаметиленадипамид.
Полимерам этого типа можно придать хорошую растворимость
путем введения в ароматическое ядро функциональных групп,,
имеющих сродство к растворителям. Поскольку синтез аромати-
ческих полиамидов затруднен, главное внимание было уделено
полиуретанам. Действительно, ароматические диизоцианаты
сравнительно просто синтезируются и благодаря невысокому
давлению паров не оказывают токсического действия. Большой
число диизоцианатов выпускается в продажу под названием
десмодуров. Например, десмодур Т является смесью 60%,
2,4-толуилендиизоцианата и 40% 2,6-толуилендиизоцианата:
2, 6-толуилендиизоцманат
2,4-толу илендиизоцианат
Смесь готовят, действуя фосгеном на соответствующие аро-
матические диамины, причем нет необходимости блокировать
18*
276 Гл. VIII. Полиэфиры, полиамиды и полиуретаны
одну из аминогрупп перед реакцией и получать сначала моно-
карбамат. Реакции протекают по схеме:
Взаимодействием
спиртом получаются
получаемых диизоцианатов с двухатомным
разветвленные растворимые полиуретаны.
Полифункциональные полимеры
Когда силы электростатического притяжения между высо-
комолекулярными цепями заменяются силами ковалентных свя-
зей, расстояния между цепями значительно сокращаются и
образуется трехмерная решетка. Для синтеза таких полимеров
требуются мономеры, содержащие более двух функциональных
групп. Две реакционноспособные группы обусловливают рост
линейной цепи, остальные могут либо оставаться несвязанными
и сообщать линейным полимерам растворимость,, либо непо-
средственно или при определенных условиях реагировать с об-
разованием мостиковых связей, вызывающих нерастворимость
и неплавкость полимера. Оба типа полимеров широко применя-
ются в производстве лакокрасочных материалов.
Из полиамидов для этих целей применяются только поли-
меры, «полученные из двухосновных кислот и полиамино- или
диаминокислот. Например, можно указать на продукты поли-
конденсации диаминокислот7
H2N—(СН2)2—NH—(СН2)10—СООН
или амидов двухосновных кислот с диэтилентриамином и три-
этилентетрамином. Однако эти термореактивные продукты мало
интересны, так как они очень легко окисляются. Более важный
класс этих полимеров составляют полиамиды и полиуретаны с
избытком гидроксильных групп.
В зависимости от реакционной способности гидроксильной
группы получаются трехмерные, нерастворимые термореактив-
ные полиамиды или, напротив, линейные и до некоторой сте-
пени растворимые полимеры. Трехмерные полиамиды полу-
чаются из мономеров, содержащих гидроксильные группы в
«-положении к реагирующей функциональной группе, напри-
мер при поликонденсации с диамином а.а'-диоксиадипиновой
кислоты8, N-этанол амино-11-ундекановой кислоты9, 11-амино-
10-оксиундекановой кислоты, или при совместной поликонден-
сации этих компонентов с компонентами нормальных полиами-
Полиамиды и полиуретаны
277
дов. Линейные полимеры получаются из мономеров, содержа-
щих нереакционноспособные гидроксильные группы, располо-
женные, по меньшей мере, у шестого атома углерода от карб-
оксильной группы и, по крайней мере, у четвертого атома угле-
рода от аминогруппы10. Могут также применяться продукты
совместной поликонденсации, содержащие в цепи полиэфирные
и полиамидные звенья. Для получения полиуретанов линейного
строения диизоцианаты полимеризуют с полифункциональны-
ми спиртами, получая растворимые нетоксичные продукты, ко-
торые далее могут образовывать трехмерную решетку. Такие по-
лиуретаны применяются главным образом в производстве лаков.
Ниже приведено несколько примеров торговых «продуктов.
Десмодур НН — полимер, полученный конденсацией трех
молекул 1,6-гексаметилендиизоцианата и одной молекулы три-
метилолпропана. Десмодур TH, получаемый взаимодействием
пяти молекул десмодура Т (смесь 2,4- и 2,6-толуилендиизоциа-
натов) с одной молекулой триметилолпропана и одной молеку-
лой 1,4-гександиола. Получаемый полимер растворим в этил-
ацетате (концентрация раствора может достигать 75%); его
можно превратить в трехмерный полимер путем добавления
полифункционального спирта в достаточном количестве. Могут
применяться также полифункциональные изоцианаты, такие, как
десмодур DR (1дифенил-4,6,4'-триизоцианат) и десмодур R [три-
(4-изоцианофенил) -метан]..
Десмодуры выпускаются в виде смесей с полиэфирами (дес-
мофены), содержащими в избытке гидроксильные группы. Дес-
модуры и десмофены, растворимые в обычных органических рас-
творителях, являются важными полупродуктами для лаков.
После нанесения раствора и сушки с помощью горячего воздуха
образуется нерастворимая пленка трехмерного полимера. Такие
пленки нашли применение для покрытия кожи, шелка, бумаги,
резины и дерева, которым они сообщают большую устойчивость
к воде, органическим растворителям и химическим реагентам.
Композиции десмодура с десмофеном могут быть пластифици-
рованы или смешаны с пигментами. На их основе получаются
пленки с отличными физико-механическими свойствами.
Модифицированные полиамиды
Формальдегид в смеси со спиртом может взаимодействовать
с амидными группами полиамидов с образованием N-алкокси-
метилполиамидов. В первой стадии формальдегид присоединяет-
ся к азоту амидной группы по схеме:
ДЭ лР
—С< + НСНО >- —С<
XNH xNCH2OH
278 Гл. УШ. Полиэфиры, полиамиды и полиуретаны
N-Метилолполиамид является разветвленным амидом и со-
держит активную гидроксильную группу. Реакционная способ-
ность метилольной группы используется' для синтеза раствори-
мых полиамидов при действии спиртов или меркаптанов. Взаи-
модействием со спиртами, например с бензиловым спиртом, по-
лучаются алкоксиметилполиамиды типа
4NCH2OR
которые растворяются в спирте при нагревании.
Растворы N-метоксиметилполиамидов применяются для свое-
го рода накрахмаливания тканей из синтетического волокна.
Например, воротничкам рубашек из найлона придают жест-
кость путем обработки препаратами N-метоксиметилполиами-
дов.
*
* *
Рассмотренные в данном кратком обзоре полимеры, содер-
жащие эфирные, амидные и уретановые группы, включают тер-
мопластичные и термореактивные материалы, техническое зна-
чение которых было быстро оценено. Термопластичные продук-
ты образуют покрытия или пленки, области применения кото-
рых, несмотря на сравнительную дороговизну, еще далеко не
исчерпаны. Эти покрытия отличаются очень высокими механи-
ческими показателями и большой химической устойчивостью.
ЛИТЕРАТУРА
I. Mark, Whitby, Collected papers of W. H. Carothers on high polyme-
ric substances, Interscience Publishers, New York, 1940.
2. Whinfield, Dickson, англ. пат. 578079; J. Hardy, Soc. Chem.
Ind., 1948, 67, 426.
3. Champetier, Aelion, Bull. Soc. chim., 1948, 15, 683.
4. Champet ier, Fournier, Bull. Soc. chim., 1950, 17, 881.
5. Baker, Fuller, J. Am. Chem. Soc., 1943, 65, 1120.
6. Du Pont de Nemours, англ. пат. 487734.
7. S. Z. Szewczyk, Ann. chim., 1951, 6, 53.
8. Beauvalet, Ann. chim., 1950, 5, 313.
9. Aelion, Champetier, Bull. Soc. chim., 1949, 16, 529.
10. Du Pont de Nemours, фр. пат. 867502.
ГЛАВА IX
ФЕНОЛО-АЛЬДЕГИТНЫЕ СМОЛЫ*
М. Э. Мейер
Феноло-альдегидными смолами принято называть смеси вы-
сокомолекулярных продуктов, полученных взаимодействием сое-
динений, содержащих альдегидные группы, с различными фено-
лами. Этим названием обозначают также продукты взаимодей-
ствия ацетона и ацетилена с фенолами и в более общем смыс-
ле— смолообразные продукты, в состав которых входят в доста-
точно большом количестве фенолы. Наибольший технический
интерес представляют продукты, получаемые взаимодействием
формальдегида с фенолами. Этим продуктам и посвящена в
основном данная глава.
Некоторые феноло-альдегидные смолы обладают способно-
стью формоваться (литьевые смолы) и представляют собой ос-
новные связующие вещества для формующихся пластмасс,
обычно называемых бакелитами. Другие смолы этого типа ис-
пользуются для приготовления лаков и красок. Они могут при-
меняться для этой цели или в чистом виде или в сочетании с
другими пленкообразующими веществами, например с высыхаю-
щими маслами, природными (канифоль) или синтетическими
смолами (полиэфирные и эфироцеллюлозпые).
Химия литьевых смол не отличается от химии лаковых смол;
при их образовании протекают примерно одинаковые реакции.
Различие состоит в том, что смолы второго типа должны быть
растворимыми в растворителях или в высыхающих маслах.
Первые работы, приведшие к получению феноло-альдегид-
ных смол, приписываются Жерару1, получившему в 1853 г. смо-
листый продукт путем взаимодействия хлорокиси фосфора с
салицилатом натрия. Байер2 в 1872 г. шире распространил эту
реакцию, изучая последовательно действие бензойного альдеги-
да на пирогалловую кислоту и на резорцин, а затем действие
ацетальдегида на фенол в присутствии серной кислоты. Михаэль
и Комей3 изучали в 1883—1884 гг. действие ацетальдегида на
обычный фенол в присутствии соляной кислоты. Действие форм-
альдегида не было изучено ввиду трудности его приготовле-
* Широко применяемые автором данной главы условные обозначения, не
являющиеся общепринятыми и затрудняющие понимание текста, при переводе
частично опущены, а частично заменены обычными структурными химическими
формулами.—Прим. ред.
280
Гл. IX. Феноло-альдегидные смолы
ния в то время; этот продукт начали производить в промыш-
ленном масштабе в 1880 г., после чего он стал легко доступным
материалом. Клееберг4, по совету Эмиля Фишера, изучал взаи-
модействие формальдегида с фенолом, резорцином и галловой
кислотой.
В результате всех перечисленных работ получались только
нерастворимые смолы, непригодные для производства лаков.
Первые феноло-формальдегидные смолы с низким молекуляр-
ным весом, растворимые в спирте, были приготовлены в 1894 г.
Манассе и в 1895 г. Ледерером5,6, которым удалось получить
о-метилолфенол в форме кристаллов; под влиянием тепла и ми-
неральных кислот этот продукт конденсируется, образуя фе-
нольную смолу. В то же время были начаты поиски замените-
лей шеллака, и в 1902 г. Блюмер7 запатентовал смолу, получае-
мую взаимодействием формальдегида с фенолом в присутствии
виннокаменной кислоты и способную при растворении в спирте
давать лак (отсюда происходит название «новолак»), Блюмер
рекомендовал также заменять для этой цели фенол a-наф-
толом. ,
Манассе и Ледерер работали с концентрированными щелоч-
ными растворами, стремясь превратить в фенолят весь фенол,
находящийся в реакционной среде. Бакеланд (именем которого
был назван бакелит) сделал решительный шаг вперед, рекомен-
довав применять небольшие количества щелочных катализато-
ров8. Его основной целью было получение формующихся смол,
но в то же время в 1910 г. он указывал10, что резолы, получен-
ные этим способом, растворимы в спирте или ацетоне и могут
быть использованы для приготовления лаков.
Следовательно, здесь имеется два первых примера изготов-
ления спиртовых лаков путем растворения смолы, приготовлен-
ной в кислой среде (новолак) и в щелочной среде (резол). Эти
смолы не всегда растворимы в высыхающих маслах, и лаки„
приготовленные из них, имеют только ограниченное примене-
ние: они непригодны для окрашивания ни с помощью кисти, ни
пульверизатором. Поэтому целью последующих исследований
было получение смол, растворимых в маслах, т. е. аналогичных
распространенным в то время копалам. Первые достижения в
этой области принадлежат Курту Альберту и Фонроберу
(1912 г.), которые получили путем сочетания феноло-альдегид-
ных смол с природными смолами, главным образом cjcaHH-
фолью, продукты, непосредственно растворимые в высыхаюпГих
мЯсЛах. Результатом этого сочетания явилось «облагоражива-
ние» канифоли, давшее возможность применять ее вместо твер-
дых копалов. При этом преимуществом являлось то, что кани-
фоль, в отличие от копалов, для придания ей растворимости в:
высыхающих маслах не нужно было подвергать трудно кон-
Основные сведения
281
тролируемой операции предварительной плавки. Однако хотя
полученные лаки были безусловно лучше лаков на основе обыч-
ной канифоли, они все же уступали по качеству лучшим лакам
на основе твердых копалов. С другой стороны, преимуществом
лаков на основе фенольных смол, модифицированных кани-
фолью, является возможность обходиться без требуемого для
копаловых лаков длительного периода созревания.
Эти смолы, названные альбертолями, были выпущены на
рынок в 1912 г. и начали широко применяться лишь после пер-
вой мировой войны10. Увеличение производства бутилового
спирта привело к значительному развитию производства и по-
требления эфироцеллюлозных лаков. Эти лаки начали серьезно
конкурировать с лаками на основе копалов, по сравнению с ко-
торыми они имели двойное преимущество: быстрое высыхание
и отсутствие длительного (до нескольких месяцев) периода
созревания. Масляные лаки на основе альбертолей оказались по
этим признакам конкурентоспособными и по отношению к эфи-
роцеллюлозным лакам.
В 1928 г. Морган приготовил первые феноло-формальдегид-
ные смолы, модифицированные высыхающими маслами, после-
довательно вводя во взаимодействие с маслами сначала фенол,
затем формальдегид. Это производство, однако, в большей сте-
пени относится к химической промышленности, а не к промыш-
ленности лаков и красок.
Затем на основе оксибифенила были получены феноло-альде-
гидные смолы, непосредственно растворимые в маслах и обра-
зующие лаки, стойкие к атмосферным влияниям. Недостатками
этих смол являются способность темнеть в процессе старения и
необходимость применения ароматических растворителей. Боль-
шой успех был достигнут Хонелем и фирмой Бек Коллер
(1929 г.), которые, использовав п-алкилфенолы (замещенные в
пара-положении пропил-, бутил- и амилфенолы), приготовили
чистые маслорастворимые смолы, не темнеющие со временем и
растворимые в алифатических углеводородах. В результате
других работ были получены смешанные смолы — водостой-
кость полиэфирных (алкидных) смол была улучшена путем со-
вмещения их с феноло-формальдегидными смолами; этот важный
прием позволил получать большое число различных ценных ла-
ков для защиты металлов от коррозии (люфены, бекозоли).
Была изучена также возможность применения модифицирован-
ных феноло-альдегидных смол вместо мягких копалов для
улучшения адгезии и блеска нитроцеллюлозных лаков.
Наконец, 'Следует перечислить ряд новых смол: феноло-аль-
дегидные смолы, модифицированные спиртами, используемые
для приготовления лаков горячей сушки; смолы, полученные-
взаимодействием фенолоспирт.ов с эпихлоргидрином, совмести-
282
Гл. IX. Феноло-альдегидные смолы
мые с высыхающими маслами (эпикоты, смолы Шелла); смолы
на основе спиртов ряда терпенов; феноло-альдегидные смолы,
модифицированные мочевинными смолами; смолы, полученные
взаимодействием с фенолом ацетилена, кетонов или смеси форм-
альдегида с кетонами. Из этого перечня видно, что область
феноло-альдегидных лаковых смол весьма обширна. В первом
приближении может быть принята следующая их классифи-
кация:
1) нерастворимые в маслах и растворимые в кислородсодер-
жащих растворителях;
а- 2) модифицированные канифолью, растворимые в маслах
(заменившие твердые копалы);
3) чистые феноло-альдегидные, растворимые в маслах;
4) модифицированные высыхающими маслами;
5) модифицированные полиэфирными (алкидными) смола-
ми, аминопластами и другими синтетическими смолами;
6) модифицированные высшими спиртами;
7) прочие смолы (резоло-эпихлоргидриновые, феноло-кето-
новые, феноло-ацетиленовые, смолы на основе сложных терпе-
новых спиртов, не подвергнутые действию карбонилсодержащих
соединений, и т. д.).
Изучению феноло-альдегидных смол вообще и лаковых в
частности посвящено огромное количество работ. Большая часть
этих работ, как правило неопубликованных, проводилась с
целью приготовления смол, пригодных для технического приме-
нения. Значительно меньшее число теоретических работ посвя-
щено установлению строения макромолекул, входящих в состав
смол. Подобные работы могут быть отнесены к двум периодам.
До 1935 г. были созданы различные гипотезы, более или менее
доступные для проверки. Истолкование полученных результатов
•было затруднено тем, что для реакции применяли фенолы (и в
особенности обыкновенный фенол CqHsOH), легко дававшие
•сложные смеси продуктов с высоким молекулярным весом,
трудно разделяемые и с трудом характеризуемые. После 1935 г.
работы проводились с замещенными фенолами, дающими при
взаимодействии с формальдегидом низкомолекулярные продук-
ты, легко поддающиеся очистке и определению строения мето-
дами классической химии. Этот метод работы будет описан и
уточнен несколько позже, но предварительно необходимо опи-
сать приготовление чистой феноло-формальдегидной смолы и
дать несколько определений.
Основные сведения о синтезе феноло-альдегидных смол
При синтезе феноло-формальдегидной смолы формальдегид
применяют обычно в самой доступной и дешевой форме — в ви-
де водного раствора 30%-ного по весу, т. е. содержащего—40 г
Основные сведения
283
СН2О в 100 мл воды. Реакция протекает в водной среде, а сле-
довательно, величина pH должна иметь важное значение.
Смешивают F молей формальдегида (в растворе) с Р моля-
ми фенола и прибавляют основание, соль или кислоту, предна-
значенные, с одной стороны, для поддержания постоянного зна-
чения pH и, с другой стороны, играющие роль катализатора.
Смесь с обычным фенолом, растворимым в воде, в начале опе-
рации представляет собой однофазную систему; с малораство-
римым фенолом образуется двухфазная смесь. Эту смесь выдер-
живают при постоянной температуре более или менее длитель-
ное время (температуру выбирают в интервале между комнат-
ной и температурой кипения, близкой к 100°; во втором случае
приемник снабжают обратным холодильником). При этом про-
текают реакции двух типов: 1) реакции присоединения форм-
альдегида к фенолам; 2) реакции конденсации продуктов при-
соединения.
В результате реакции присоединения -получаются только
низкомолекулярные вещества. Реакции конденсации могут при-
вести к образованию высокомолекулярных продуктов, строение
которых зависит от относительной скорости протекания реак-
ций присоединения и конденсации в данных условиях (катали-
затор, температура, продолжительность соприкосновения), а
также от начальных параметров — молярного соотношения форм-
альдегида и фенолов (F/Р) и функциональности взятого фе-
нола (и). О протекании реакции присоединения можно судить
по ослаблению запаха формальдегида; в зависимости от условий
этот запах может исчезнуть полностью, что свидетельствует об
окончании реакции, или сохраняется вплоть до того времени,
когда скорость реакции присоединения практически станет рав-
ной нулю.
Если скорость реакции конденсации больше скорости реак-
ции присоединения (это наблюдается при pH < 7, когда F/P
практически меньше единицы), то выделить продукты присое-
динения не удается. Конечные .продукты реакции называются
новолаками (предполагается, что речь идет об одноатомных
фенолах; с резорцином результаты будут другими).
При щелочном характере реакционной смеси скорость реак-
ций конденсации значительно меньше и можно выделить про-
дукты присоединения. В этом случае F/Р может иметь значе-
ние, превышающее единицу, и полученные продукты реакции
(неконденсированные или слабо конденсированные продукты
присоединения) называются резолами.
Новолаки являются термопластическими материалами, а
резолы обычно термореактивны. Эти свойства зависят от значе-
ний F/Р и п. Резолы имеют высокую реакционную способность,
что соответствует их термореактивности.
284
Гл. IX. Феноло-альдегидные смолы
Продукты реакции могут содержать не весь взятый для
реакции фенол или формальдегид, так как эти вещества могут
частично испариться или перейти в водную фазу, не вступая в
реакцию. Поэтому для смолы вводят характеристику, обозна-
чаемую отношением f/p, т. е. молекулярное соотношение форм-
альдегида и фенола (или фенолов, если взята смесь фенолов),
непосредственно участвующих в образовании смолы. Величина
f/p может отличаться от величины F/P.
Теперь определим характеристику п, которую мы назвали
функциональностью фенола. Как будет видно из дальнейшего,
максимальное число молекул формальдегида, способных при-
соединиться к фенольному ядру, равно числу углеродных ато-
мов, находящихся в орто- и пара-положениях по отношению к
гидроксильной группе фенола й не имеющих заместителей (т. е.
связанных лишь с водородом). Это число равно трем для обыч-
ного фенола СбН5ОН; оно может быть равно двум, единице или
даже нулю для замещенных фенолов и может быть более трех
для сложных фенолов с несколькими бензольными ядрами.
Условимся обозначать Pi, Р2, Р3, ..., Рл фенолы с функцио-
нальностью 1, 2, ..., п.
Знание функциональности фенола очень полезно для опреде-
ления наиболее вероятного строения феноло-альдегидной смолы.
Для приготовления фенольной смолы часто применяют смесь
фенолов с различной функциональностью, например смеси кре-
золов или ксиленолов. Понятие функциональности, как мы ее
определили, не имеет в этом случае физического смысла. Функ-
циональность такой смеси можно определить иначе. А именно,
если смесь фенолов содержит Ni молей фенолов Pi, N2 молей
фенолов Рг, ..., Nn молей фенолов Рл, то функциональность
смеси определяется величиной отношения:
А/i + 2#2 +• • •+ nN„
+ ^2 +’ • ’
Очевидно, что эта величина не обязательно выражается це-
лым числом. Она соответствует максимальному числу молекул
формальдегида, могущих присоединиться к одной молекуле фе-
нола в смеси, т. е. представляет собой среднюю величину.
Так, например, если функциональности о-нрезола, м-крезола
и n-крезола равны 2, 3 и 2, то функциональности продажных
смесей, содержащих 60,5%, 54,4% и 38,5% лг-крезола, будут
равны соответственно 2,60, 2,54 и 2,38.
Это означает, что на каждую фенольную группу ОН, содер-
жащуюся в смеси, может присоединиться максимально 2,60,
2,54 и 2,38 моля формальдегида. На практике этот максимум не
Исходные вещества 285
всегда достижим, но знание ©той величины позволяет определить,
какой продукт может получиться при взаимодействии данной
смеси фенолов (или данного фенола) с формальдегидом: макро-
молекулы сетчатой структуры, линейной структуры или же мо-
лекулы не смолообразных веществ.
Значение величины pH будет подробно разобрано далее, но
следует указать, что с достаточной точностью бывает известно
только начальное значение pH. В ходе реакции эта величина
может меняться, в особенности вследствие превращения форм-
альдегида в муравьиную кислоту и метиловый спирт (реакция
Каниццаро, см. стр. 289).
Однако изучение реакции между формальдегидом и фенолом
предполагает знание следующих величин: отношение числа мо-
лекул формальдегида к фенолу F/Р, (первоначальное значе-
ние pH смеси и условия реакции (продолжительность и темпе-
ратура).
Свойства полученной смолы будут определяться природой
исходного фенола или смеси фенолов, функциональностью п
этого фенола (или смеси), молярным отношением f/p формаль-
дегида и фенола, вошедших в состав смолы.
Литературные указания
В историческом обзоре развития синтезов фенольных смол был приведен
ряд библиографических ссылок и указано, что систематические работы по хи-
мии этих смол, выполненные главным образом после 1935 г., представляют
объект обширной библиографии. Однако представляется целесообразным ука-
зать четыре основных труда, широко использованные в данной главе:
1) критический библиографический обзор Лиллея11; 2) статья Цинке и
Циглера12, 3) работа Гульча13, озаглавленная «Химия фенольных смолг, очень
полная и содержащая обширную библиографию; 4) работа Эллиса10, содержа-
щая также обширную блиблографию.
Кроме того, были использованы данные, изложенные в учебном курсе
Высшей военной школы14.
Исходные вещества
Фенолы. Фенол — это химическое соединение, полученное
путем замещения одного водородного атома в бензоле на гидр-
оксильную группу. Свойства фенола являются промежуточ-
ными между свойствами спиртов и свойствами органических
кислот. В результате повторных операций замещения в одном
и том же ядре получается многоатомный фенол (полифенол).
Моноциклические замещенные фенолы получаются из обычного
в результате замещения одного или нескольких водородных ато-
мов алкильными или арильными радикалами.
286
Гл. IX. Феноло-альдегидные смолы
В соответствии с этим имеются три крезола, которые могут
быть представлены схематически следующим образом:
ОН
о-крезол
ОН
л-крезол
Точно так же имеется шесть ксиленолов, в которых металь-
ные радикалы находятся соответственно в положениях: 2,3-;
2,4-; 2,5-; 2,6-; 3,4- и 3,5- по отношению к фенольному гидрок-
силу. Из фенолов более сложного строения можно указать п-ок-
сибифенил, побочный продукт производства фенола из бензола
(см. далее), многоядерные фенолы, например дифенилолпро-
пан, и продукты взаимодействия фенола с галоидными произ-
водными терпенов, примеры которых приведены на стр. 287.
Обычный фенол является главным исходным продуктом для
получения замещенных фенолов (за исключением крезолов И
ксиленолов). Это — кристаллическое вещество, плавящееся '
при 42,3°. Для облегчения работы с фенолом в лабораторных
условиях полезно иногда прибавить к нему 8% вес. воды, чтобы
получить смесь, жидкую при 20°. В большинстве случаев фено-
ло-альдегидные смолы получают в водной фазе; наличие такого
небольшого количества воды не препятствует изготовлению смол
в условиях, предусмотренных режимом их производства.
Фенол, крезолы, и ксиленолы выделяют из каменноугольной
смолы, а также получают из бензола по одному из следующих
методов*: 1) прямое каталитическое окисление; 2) хлорирова-
ние и последующий гидролиз хлоропроизводных (в качестве
побочного продукта получаются оксибифенилы); 3) сульфи-
рование и щелочная плавка сульфокислот.
Растворимые в щелочах фракции дистиллятов каменноуголь-
ной смолы содержат, наряду с фенолом, крезолы и ксиленолы.
Температуры кипения этих продуктов следующие (в °C):
Фенол . 182,1
о-Крезол...................190,9
.и-Крезол................. 202,2
я-Крезол.................. 202,1
* В 1939 г. в СССР П. Г. Сергеевым, Р. Ю. Удрисом, Б. Д. Кружаловым
и др. разработан метод получения фенола совместно с ацетоном через гидропе-
рекись изопропилбензола. Этот изящный и весьма экономичный метод получил
широкое промышленное применение в СССР, а позднее н в других странах.—
Прим. ред.
Исходные вещества
287
Ксиленолы имеют температуры кипения, лежащие между
211,5 и 225°.
Фракционированием удается довольно легко отделить фенол
от крезолов и ксиленолов. В зависимости от условий фракциони-
рования в дальнейшем можно выделить более или менее полно
/«-крезол или получить смесь крезолов с повышенным содержа-
нием /«-крезола. /«-Крезол является наиболее реакционноспо-
собным из трех крезолов, и поэтому для производства смол же-
лательно применять смеси с наибольшим содержанием jw-крезо-
ла. В продажу выпускаются три смеси, содержащие примерно
60,5; 54,4 и 38,5% /«-крезола; смесь, содержащая 60% /«-крезо-
ла, содержит больше n-крезола и меньше о-крезола.
Исходя из обычного фенола, можно получить более сложные
фенолы по одному из следующих синтетических методов.
Пользуясь методом Фриделя — Крафтса, на фенол действу-
ют каким-либо галоидопроизводным в присутствии безводного
хлористого алюминия:
Rci+О-°н ”“* R—он+НС1
Этим методом можно получать фенольные производные мен-
тана и камфана. Галоидные производные терпенов получают
действием сухого хлористого водорода на дипентен, терпинеол
или камфен. Так получают, например, дифенилолментан и фе-
нилолкамфан:
дифенилолментан
Замещенные фенолы можно получить также взаимодейст-
вием спиртов с фенолами в присутствии концентрированной»
серной кислоты:
ROH + ОН -> R—<Л-ОН + Н2О
288
Гл. IX. Феноло-альдегидные смолы
Аналогичным путем может быть получен дифенилолпропан
действием ацетона на фенол в присутствии серной кислоты:
СН3 СН3
НО—+ со + он —► НО—ОН +Н2о
I \=/ , \=/
сн3 сн3
Можно также конденсировать ненасыщенные углеводороды
с фенолами; некоторые из этих углеводородов, полупродукты
нефтяной промышленности, производятся в большом количестве
в США. /По этому способу готовят п-изобутилфенол алкилиро-
ванием фенола изобутиленом:
С4НВ—Z~\-OH
Для изготовления феноло-формальдегидных смол применя-
ют также иногда многоатомные фенолы, в которых несколько
гидроксильных групп находятся в одном и том же ядре в мета-
положении друг к другу. Чаще всего используют резорцин и
флороглюцин:
ОН
ОН
резорцин । флороглюцин
Резорцин применяется или один или в смеси с другими фе-
нолами; получаемая смесь приобретает повышенную реакцион-
ную способность. На основе резорцина получают лаки, отверж-
дающиеся на холоду (см. ниже). Для получения люфена L берут
смесь, состоящую из 99 вес. ч. фенола и 1 вес. ч. резорцина.
Флороглюцин применяется в смеси с другими фенолами.
Наконец, некоторое применение имеют нафтолы. Один из
первых чистых фенольных лаков, предложенных Блюмером7,
был изготовлен на основе а-нафтола:
ОН
00
Не все фенолы представляют одинаковый интерес для про-
изводства феноло-альдегидных смол вообще и лаковых в част-
ности. При выборе фенола учитывают цену, реакционную спо-
собность по отношению к формальдегиду, совместимость с вы-
Исходные вещества
289
сыхающими маслами и с алифатическими растворителями (на-
пример, с уайт-спиритом), а также качество получаемого лака.
Наиболее употребительными являются следующие фенолы:
фенол, крезолы, ксиленолы;
замещенные в пара-положении изобутил- и изоамилфенолы;
п-фенилфенол;
дифенилолпропан и диоксибифенил (бис-фенол);
сложные фенолы терпенового ряда, полученные действием
фенола на полупродукты синтеза камфоры. *
В США применяют также природные фенолы, извлеченные
из орехов «кажу».
Формальдегид. Это — простейший альдегид строения
/°
Н—<С. . В промышленных масштабах его получают катали-
ЧТ
тическим окислением метилового спирта кислородом воздуха в
присутствии восстановленного серебра. При обычной темпера-
туре формальдегид представляет собой газ с острым запахом;
темп. кип. —19,2°.
Формальдегид обладает очень высокой реакционной способ-
ностью; он быстро полимеризуется, образуя параформаль-
дегид— белый порошок, состоящий из полимеров разного сос-
тава, плавящийся с обратным переходом в мономер. Мономер
хорошо растворим в воде.
Формальдегид выпускается или в форме параформальдеги-
да (параформа), или в виде водного раствора 40%-ного (по
объему) и 37%-ного или 30%-ногох(по весу); 30%-ный раствор
содержит 30 г формальдегида на 100 г раствора, так называе-
мый 40% раствор —40 г формальдегида на 100 см3 раствора.
Водные растворы формальдегида содержат не свободный
альдегид, а либо одноводный гидрат СН2(ОН)2, либо полимер-
ный продукт состава НОН2СО(СН2О)ИСН2ОН. Параформаль-
дегид имеет аналогичное строение, причем п равно от 6 до 50.
Концентрированные растворы формальдегида нестабильны;
концентрация их уменьшается за счет осаждения нераствори-
мого твердого полимера. Осаждение сильно замедляется в при-
сутствии метилового спирта, а потому в продажные растворы
формальдегида для повышения устойчивости всегда вводят 8—
10% СНзОН.
Продажные растворы подвергаются также изменениям
вследствие протекания реакции Канниццаро с образованием му-
равьиной кислоты и метилового спирта:
2НСНО + Н2О -► СНзОН + нсоон
По этой причине в растворах содержится 0,2—0,3% мура-
вьиной кислоты. Реакция Канниццаро протекает быстро и пол-
19—156
290
Гл. IX. Феноло-альдегидные смолы
но при кипячении в присутствии какого-либо основания; мура-
вьиная кислота переходит в соответствующий формиат.
Поскольку растворы формальдегида обычно хранятся в же-
лезной таре, они содержат следы железа в виде формиата же-
леза. Присутствие железа в формальдегиде вызывает окраши-
вание феноло-формальдегидных смол, изготовляемых с приме-
нением такого продукта.
Формальдегид легко конденсируется с аммиаком и с амина-
!йи с выделением воды; с аммиаком образуется гексаметилен-
тетрамин C6Hi2N4, используемый в производстве формовочных
феноло-альдегидных смол для достижения определенной степе-
ни отверждения материала, предназначенного для формования.
При взаимодействии формальдегида с другими веществами
интенсивный запах его исчезает только тогда, когда формаль-
дегид прореагирует полностью; этим признаком пользуются при
изготовлении феноло-формальдегидных смол.
В водной среде, кислой или щелочной, формальдегид реаги-
рует как диалкоголь:
zONa ГСН2—О~1
СН2ОNaOH —Н2С< или I1 Na+
ЧЭН [он J
СН2О 4- НС1
или
СН2 cj-
ОН .
Реакции протекают в
формальдегида
соответствии с полярной структурой
Д-
Н2С=О
Эти сведения будут использованы в следующем разделе, где
будет рассматриваться взаимодействие формальдегида с фено-
лом в щелочной и кислой средах.
ФЕНОЛОСПИРТЫ
При взаимодействии фенола РИН с альдегидами или кето-
нами общей формулы R—СО—R' может быть получено боль-
шое число различных продуктов. Но можно предположить, что
первой стадией реакции является реакция присоединения, в ре-
зультате которой образуется фенолоспирт:
R
I
PhH+ R—СО—R' —» Ph—с—ОН (1)
I
R'
Фенолоспирты
291
В некоторых случаях в действительности не удается обнару-
жить присутствие фенолоспирта, а находят производное дифе-
нилметана, образующееся согласно реакции:
R
2PhH + R—СО—R' -- Ph—С—Ph-f- Н2О (2)
I
R*
Однако даже в этом случае можно предполагать, что реак-
ция (/) действительно протекает, но сопровождается реак-
цией (3)
R R
I I
Ph—С—ОН + PhH -> Ph—с—Ph + Н2О (3)
I I
R' R'
причем скорость реакции (3) достаточно велика,, чтобы >присуг-
ствие продуктов присоединения не могло быть обнаружено.
Более новые работы подтверждают эту гипотезу, и можно
предполагать, что во всех подобных случаях сначала обра-
зуются фенолоспирты, далее превращающиеся в более слож-
ные продукты.
Реальный технический интерес представляют продукты, по-
лученные действием формальдегида на фенолы, данная глава
посвящена поэтому почти исключительно изучению этих про-
дуктов. Смолы, получаемые действием других кетонов и альде-
гидов, кроме формальдегида, будут кратко рассмотрены в раз-
деле, посвященном модифицированным феноло-альдегидным
смолам и лаковым смолам.
Здесь же приводятся условия, необходимые для того, чтобьи
задержать реакцию формальдегида с фенолом на стадии обра-
зования фенолоспиртов, и современная интерпретация реакции
образования этих спиртов; подвижность водородных атомов
в фенольном ядре, т. е. возможность присоединения формальде-
гида в различных положениях по отношению к фенольному
гидроксилу .в зависимости от природы и положения других за-
местителей и, наконец, реакционная способность различных фе-
нолов, с учетом числа подвижных водородных атомов, характера
и положения заместителей в фенольном ядре.
Условия образования фенолоспиртов
Первой стадией взаимодействия формальдегида с фенолом
является образование фенолоспиртов, которые нелегко выде-
лить или воспрепятствовать их взаимодействию между собой
или с фенолами. Скорости этих вторичных реакций зависят
19*
292
Гл. IX. Феноло-альдегидные смолы
от вида фенола, температуры и, главным образом, от значения
pH реакционной среды.
Когда pH меньше 7 (реакционная среда кислая), скорости
вторичных реакций и в особенности реакций, приводящих к
образованию производных дифенилметана, очень велики. Если
реакция протекает в присутствии соляной кислоты НС1, то это
ускорение обусловлено образованием весьма активных хлор-
метиленовых производных РЬСН2С1, реагирующих мгновенно
с фенолами и фенолоспиртами.
Чтобы выделить фенолоопирты, необходимо создать щелоч-
ную среду. При pH больше 7 возможны два случая:
1. Реакция протекает в присутствии незначительного коли-
чества щелочного продукта и большая часть фенола находится
в свободном состоянии; в данном случае основание играет толь-
ко роль катализатора.
2. Количество основания достаточно для превращения всего
фенола в фенолят; основание и в этом случае играет роль ка-
тализатора, но, кроме того, содействует растворению фенола и
фенолоспирта в виде их натриевых производных; поэтому ре-
акция протекает в гомогенной среде.
Установлено, что во втором случае скорости реакций пре-
вращений фенолоспиртов весьма малы или равны нулю, и такой
способ проведения реакции является единственным, позволяю-
щим выделять эти продукты.
Следует отметить, что некоторые фенолы всегда проявляют
большую склонность к образованию дифенилметановых про-
изводных, независимо от значения pH реакционной среды, кис-
лой и щелочной. Такими фенолами являются 1-окси-2,6-диме-
тилбензол, 1-окси-2,4,5-триметилбензол и р-нафтол:
ОН
В молекулах первых двух фенолов имеется только по од-
ному подвижному атому водорода, и следовательно, они не
способны давать смолы и поэтому не представляют практиче-
ского интереса.
Для максимального замедления реакций конденсации целе-
сообразно работать при низкой температуре.
Таким образом, фенолоспирты следует изготовлять путем
прибавления к фенолам эквимолекулярных количеств основа-
Фенолоспирты
293
ния для превращения их в феноляты; реакция должна прово-
диться при возможно более низкой температуре (обычно при
комнатной температуре). В этих условиях реакция может
продолжаться несколько дней (конец реакции может быть
иногда установлен по исчезновению запаха формальдегида).
Растворы фенолоспиртов следует нейтрализовать слабыми
кислотами, например уксусной кислотой; сильные кислоты
как, например, соляная кислота, являются катализаторами ре-
акции конденсации.
Реакция присоединения формальдегида к фенолу. Попы-
таемся определить причины стойкости фенолоспиртов в щелоч-
ной и в нейтральной средах. Эти реакции происходят в водной
фазе, и, поскольку их течение изменяется в зависимости от
pH, они являются реакциями ионного типа.
Рассмотрим сначала случай, когда pH выше 7. Предполо-
жим, что реакция проводится в присутствии сильного основа-
ния, например едкого натра. Можно допустить, что формальде-
гид при взаимодействии с едким натром образует неполный
алкоголят следующего строения
Н2С—ONa
1
ОН
т. е. реагирует в соответствии с полярностью его молекулы
Д-
Н2С=О
Известно также, что в молекуле фенола* взаимодействие
свободной электронной пары атома кислорода с системой со-
пряженных связей бензольного кольца приводит к повышению
электронной плотности в орто- и пара-положениях:
* Приведенные ниже общепринятые объяснения реакционной способности
фенолов составлены редактором. Устаревшие трактовки, данные автором на
основании работ11,16 при переводе опущены.—Прим. ред.
294
Гл. IX. Феноло-альдегидные смолы
Присоединение полярной молекулы формальдегида в этих
положениях облегчается:
Стабильность о-метилолфенола можно объяснить образова-
нием водородной связи между водородным атомом фенольного
гидроксила и кислородным атомом гидроксила метйлольной
группы. Для п-метилолфенола можно допустить наличие меж-
молекулярных водородных связей:
n-Фенолоспирты, впрочем, несколько менее стабильны, чем
о-феполоспирты.
При проведении реакции в присутствии кислоты водородные
связи не могут образоваться. Этим и объясняются различия,
обнаруженные между реакциями, протекающими в щелоч-
ной и кислой средах.
Действительно, в присутствии кислоты, например, соляной,
формальдегид присоединяет НС1
СН2С1-
он
и реагирует с фенолом с образованием соединения, в котором
вследствие индукционного смещения электронной плотности к
атому хлора углеродный атом метиленовой группы приобре-
тает положительный заряд:
ОН
Ввиду подвижности водородного атома, присоединенного к
атому углерода молекулы фенола, находящемуся в орто-по-
ложении по отношению к фенольному гидроксилу, указанное
Фенолоспирты
295
соединение может вступать в реакцию с фенолом с отщепле-
нием НС1, образуя производное дифенилметана:
n-Фенолоспирт в присутствии минеральной кислоты превра-
щается в то же самое соединение:
он
Следовательно, соляная кислота, действуя на фенолоспирт
или на фенол в момент его взаимодействия с формальдегидом,
препятствует образованию водородных связей, обусловливаю-
щих стабильность фенолоспирта. Образующиеся соединения,
содержащие группировку
мгновенно реагируют с фенолом с образованием дифенилмета-
нового производного.
Можно предположить, однако, что когда количество НС1
очень мало (pH близко к 7), водородные связи ослаблены и
поляризованные метилольные группы в орто-положениях фе-
нольных ядер могут реагировать с образованием диоксибен-
зилового эфира:
+ н2О
296
Гл. IX. Феноло-альдегидные смолы
Влияние температуры на стабильность фенолоспиртов мож-
но объяснить ослаблением водородных связей под влиянием
тепла. Тогда молекула оксибензилового спирта, как и в присут-
ствии НС1, реагирует с молекулой фенола с отщеплением воды
и образованием дифенилметанового производного:
Вследствие ослабления водородной связи метилолфенолы
могут взаимодействовать также с образованием диоксибензи-
ловых эфиров.
Рассмотрим теперь продукт присоединения, полученный дей-
ствием формальдегида, например, на м-диоксифенол (резор-
цин). Можно предположить, что в молекуле резорцина наиболее
подвижным является атом водорода у углеродного атома, нахо-
дящегося между двумя атомами углерода, несущими фенольные
гидроксилы. Следовательно, сначала получится монометилол-
резорцин
он
который не может быть полностью стабилизован образованием
внутримолекулярных водородных связей. Как и в предыдущих
случаях, метилольная группа в этом соединении поляризована
и реакция дальнейшей конденсации протекает с образованием
дифенилметановых производных.
Таким образом, полярная группировка
Z^-ch2—x
может образовываться в трех следующих случаях:
1) в присутствии сильной кислоты;
2) под влиянием тепла;
3) при применении ж-диоксисоединений.
Фенолоспирты
297
Эта весьма активная группировка реагирует непосредствен-
но с фенолами и фенолоспиртами с образованием мостиков
— СН2— или — СН2—О—СН2—. Поэтому реакция между форм-
альдегидом и фенолом не может быть прервана на стадии
образования фенолоспирта, если соблюдается, по крайней мере,,
одно из указанных трех условий; такой вывод подтверждается
практикой.
Заметим, что группа —СН3 может играть ту же роль, что
группа ОН, но в значительно меньшей степени. Например, в
молекуле м-крезола сильно повышена подвижность атома во-
дорода, находящегося в положении 2:
Зная функциональность фенола (или смеси фенолов), мож-
но определить строение соединений, получаемых при его взаи-
модействии с формальдегидом. Однако в отдельных случаях,
не все атомы углерода в ядре фенола реагируют с формальде-
гидом с одинаковой скоростью; скорость реакции может изме-
няться в зависимости от присутствия в фенольном ядре заме-
стителей.
Рассмотрим сначала незамещенный фенол СбН5ОН. Спо-
собность к электрофильному замещению максимальна у ато-
мов углерода, находящихся в о-положении по отношению к
фенольному гидроксилу, немного меньше — у атома углерода,,
находящегося в «-положении, и совершенно отсутствует у угле-
родного атома, находящегося в ^-положении. Можно считать
даже, что влияние фенольного гидроксила в последнем случае
является отрицательным, например, если рассматривать пове-
дение гидрохинона по отношению к формальдегиду18. На при-
мере пирокатехина, который может образовывать только диме-
тилолпроизводные следующего строения19
ОН
НОСН2Ч1/ОН
XX
^/\СН2ОН
можно видеть, что к атомам углерода, находящимся в «-поло-
жении «о отношению к одному гидроксилу и в M-положении-
по отношению к другому гидроксилу, — формальдегид не при-
соединяется.
298
Гл. IX. Феноло-альдегидные смолы
Углеводороды бензольного ряда не реагируют непосредст-
венно с формальдегидом, но на примере реакции крезола с
формальдегидом можно видеть, что присутствие метильного
радикала в ядре изменяет поведение атомов углерода. Под
влиянием метильной группы происходит увеличение или умень-
шение реакционной способности атома углерода в соответствии
с тем, в каком положении он находится по отношению к заме-
щающей водород метильной группе, в орто-, пара- или мета-
положении:
1. Подвижность атома водорода в о-положении превосхо-
дит его подвижность в «-положении по отношению к феноль-
ному гидроксилу. Это правило сохраняет силу при электро-
фильном замещении и для других заместителей первого рода,
которые могут присутствовать в ядре.
2. Углеводородные радикалы также усиливают подвижность
водородных атомов в о- и «-положениях; ото активирование
постепенно уменьшается с переходом от арильного радикала
к метильному, а затем к алкильному радикалу с несколькими
атомами углерода.
3. Активирование может уменьшаться или увеличиваться;
когда реакционная способность незначительна, то при повыше-
нии температуры реакция присоединения формальдегида к фе-
нолу может стать обратимой.
В качестве примера можно указать, что продукт конденса-
ции 1,2,6-ксиленола (1-окси-2,6-диметилбензола) с формальде-
гидом при температуре 140° частично разлагается20, выделяя
формальдегид:
С другой стороны, из 1,2,6-ксиленола можно получить про-
изводное дифенилметана, хотя реакционная способность атома
углерода, к которому присоединяется формальдегид, вследствие
наличия двух метильных групп в мета-положении, весьма не-
значительна.
Ниже приведены формулы различных фенолов в порядке
возрастания реакционной способности отмеченных звездочка-
ми углеродных атомов:
Фенолоспирты
299
Приведенные сведения позволяют предвидеть ход реакций
при взаимодействии формальдегида с фенолами, обладающими
несколькими реакционноспособными атомами углерода. Так, на-
пример, формальдегид присоединяется значительно быстрее и,
следовательно, более полно к углеродному атому, находяще-
муся между двумя фенольными гидроксилами. Подобным же
образом при взаимодействии формальдегида с обычным фено-
лом, получают, главным образом, о-метилолфенол (салигенин).
Можно сказать, что способность образовывать молекулы,
содеожащие полярную группировку
повышается с увеличением реакционной способности рассмат-
риваемого атома углерода. Присутствие этой группировки на-
правляет реакцию в сторону образования производных дифенил-
метана или диоксибензилового эфира.
Данные о реакционной способности углеродных атомов в
молекулах фенолов дают возможность ввести новую класси-
фикацию фенолов. Мы видели, что фенолы могут быть класси-
фицированы по числу реакционноспособных атомов углерода или
по функциональности. Новая классификация позволяет разли-
чать и классифицировать фенолы одинаковой функциональности
в зависимости от их сродства к формальдегиду.
Опыт показывает, что 1,3,5-ксиленол (1-окси-3,5-диметилбен-
зол) более активен, чем лг-крсзол, который в свою очередь более
активен, чем обычный фенол. Путем суммирования реакционных
способностей всех активных углеродных атомов молекулы фе-
300
Гл. IX. Феноло-альдегидные смолы
Итак, установлено, что фенолы, очень активные по отно-
шению к формальдегиду, дают с ним продукты присоединения,
также весьма активные и легко реагирующие между собой.
Приведенные данные позволяют, в конечном счете, определить
относительную способность различных фенолов образовывать
смолы при взаимодействии с формальдегидом. Эксперименталь-
ные данные, описываемые в следующем разделе, находятся в
согласии с теоретическими выводами.
Взаимодействие формальдегида с различными фенолами
(экспериментальные данные). Для получения фенолоспиртов
фенол должен быть превращен в фенолят (при pH от 10 до 12)
и реакция должна проводиться при возможно более низкой
температуре.
При применении монофункционального фенола (Pi) полу-
чается только один фенолоспирт. Если же применяют двух-,
трех- или полифункциональные фенолы (Ро, Рз или Ря), полу-
чают всегда сложные смеси, состав которых зависит от про-
должительности соприкосновения реагирующих веществ, вели-
чины отношения F/Р и реакционной способности примененных
фенолов, т. е. от числа молекул формальдегида, приходящихся
на одну молекулу фенола.
Фенолоспирты
301
Для примера ниже будут приведены результаты, получен-
ные Гренджером21 при взаимодействии формальдегида с обыч-
ным фенолом, крезолами и ксиленолами. Следует, однако, за-
метить, что самые сложные смеси получаются при использо-
вании обычного фенола, ж-крезола и 1-окси-3,6-диметилбензола,
которые обладают наибольшим числом подвижных атомов во-
дорода.
Действием, формальдегида на фенол СбН5ОН можно получить
пять различных веществ: два монометилолфенола, два диме-
гилолфенола и один триметилолфенол.
1. F/P=l; комнатная температура. Смесь вносят в 10%-ный
раствор едкого натра; после 24 час. выдержки запах формаль-
дегида исчезает. Смесь нейтрализуют уксусной кислотой и из-
влекают эфиром. На один моль израсходованного фенола по-
лучают 7з моля фенола и 1/з моля монометилолфенола в эфир-
ной фазе; водная фаза содержит ’/з моля фенола в форме ди-
и триметилолфенолов. Установлено, что по мере разбавления
содержание монометилолфенола повышается.
2. f/P=2; комнатная температура. Запах формальдегида
исчезает только через несколько дней; образуется большое ко-
личество монометилолфенола и остается свободный фенол.
3. F/P=3; комнатная температура. Запах формальдегида
остается неопределенное время; получается очень мало сво-
бодного фенола и монометилолфенола, но в то же время полу-
чается смесь ди- и триметилолфенолов.
Действие формальдегида на о-крезол. F/P=2-, комнатная
температура. Получаемые продукты растворимы в 20%-ном
растворе едкого натра; запах формальдегида исчезает через
два дня. Подкисляя смесь уксусной кислотой, получают кри-
сталлический диметилолкрезол с хорошим выходом. При по-
пытке приготовить монометилолфенол при F/P=l получается
смесь моно- и диметилолфенолов.
Действие формальдегида на м-крезол. Можно выделить три-
метилолпроизводное, но оно очень быстро осмоляется и сохра-
нить его в чистом виде не удается.
Действие формальдегида на ксиленолы. Монофункциональ-
ные ксиленолы (Р]) довольно легко образуют соответствующие
метилолпроизводные, за исключением 1,2,6-ксиленола (1-окси-
2,6-диметилбензола), который дает производные дифенилметана.
Бифункциональные ксиленолы (Р2) легко присоединяют одну
молекулу формальдегида, с большим трудом — вторую.
1,3,5-Ксиленол легко присоединяет две молекулы формаль-
дегида; при попытке присоединить третью — происходит осмо-
ление.
Эти факты находятся в соответствии с ранее сделанными вы-
водами.
302
Гл. IX. Феноло-альдегидные смолы
Таким образом, при взаимодействии с формальдегидом фе-
нолов, имеющих в ядре более одного подвижного атома водо-
рода, могут наблюдаться два случая. Либо реакция останавли-
вается на стадии образования фенолоспиртов, но получаются
сложные смеси, которые трудно или невозможно разделить,
либо очень быстро происходит осмоление. Поэтому подробное
изучение феноло-альдегидных смол весьма затруднительно;
строение этих смол было открыто только недавно, когда от-
казались от исследования продуктов, получаемых из обычного
фенола, и перешли к работе с фенолами Pi и Рг, дающими легче
выделяемые соединения.
На практике при работе с крезолами или ксиленолами при-
меняют довольно сложную смесь, поведение которой изменяется
в зависимости от ее состава: реакционная способность повы-
шается с увеличением количества лг-крезола или 1,3,5-ксиле-
нола. Иногда к обычному фенолу прибавляют небольшое коли-
чество многоатомных фенолов, таких как резорцин или фло-
роглюцин, чтобы получить смесь с повышенной реакционной
способностью (см., например, люфен L, стр. 288).
Химия фенолоспиртов
Зная условия образования фенолоспиртов из чистых фе-
нолов или их смесей, можно приступить к изучению химии этих
соединений. При этом следует выяснить характер мостиков,
образующихся между фенольными ядрами в процессе отверж-
дения смол, т. е. исследовать процессы образования макромо-
лекул. Эти вопросы изучались в течение 15 лет четырьмя
школами, во главе которых стояли Цинке, Ханус, Хульч и
Эйлер. В отличие от более ранних работ, в этих исследованиях
использовались не фенолы типов Р3 и Рг, а фенолы типа Pi,
дающие с формальдегидом соединения ограниченного молеку-
лярного веса, которые можно было очищать и точно определять
их строение классическими методами органической химии. Ре-
зультаты, полученные в этих простых случаях, могут быть
распространены на более сложные фенолы, содержащие любое
количество реакционноспособных атомов углерода.
Продукты взаимодействия формальдегида с фенолами де-
лятся на два типа в зависимости от того, получены ли они в
щелочной или нейтральной, или же в кислой среде. Смолы,
полученные в кислой среде, имеют более простое строение, но
во всех случаях начальным продуктов конденсации является
фенолоспирт. Из дальнейшего будет видно, что новолаки и
смолы, полученные из фенолоспиртов, не отличаются друг ог
друга.
Фенолоспирты
303-
Рассмотрим свойства метилольной группы СН2ОН, связан-
ной с фенольным ядром, и изменения, которые она претерпе-
вает при повышенной температуре или под действием различ-
ных химических реагентов.
При температуре ниже 175° происходит прежде всего обра-
зование мостиков —СН2— или —СН2—О—СН2—. Выше 175е
происходят глубокие изменения, которые некоторыми исследова-
телями приписываются реакциям хинонметидов.
Реакции, протекающие в процессе отверждения феноло-форм-
альдегидных смол, могут быть также разделены на четыре
типа:
1) реакции образования термостойких мостиков;
2) реакции, ведущие к образованию реакционноспособных
концевых групп, реагирующих в дальнейшем с другими про-
дуктами реакции;
3) реакции, приводящие к образованию нереакционноспособ-
ных концевых групп;
4) реакции, вызывающие разрыв нетермостойких мостиков,
и приводящие к протеканию одного из трех типов реакций,
перечисленных выше.
В этом разделе мы будем рассматривать монофункциональ-
ные фенолы (Pi), содержащие один активный углерод в орто-
или пара-положении, х^ринимая, что два других активных атома
углерода блокированы нереакционноспособными радикалами R.
Реакции метилольной группы. Действие тепла. Установлено,
что-при нагревании до 110—135° происходит отщепление воды
от фенолоспирта (примерно 1 моль воды на две метилольных
группы) по следующей реакции:
Ph—СН2ОН + НОСН2—Ph Ph—СН2— О—СН.2—Ph Н2О (1)
При дальнейшем нагревании и повышении температуры вы-
деляется формальдегид. Это приписывалось протеканию реак-
ции:
Ph—СН3— О—СН2—Ph — Ph—СН2—Ph + НСНО (2)
Однако, согласно последней, еще не опубликованной работе
М. Жюиллара, мостик —СН2—О—СН2— весьма устойчив. От-
щепление формальдегида в действительности вызвано разло-
жением мостика —СН2—О—СН2—О—СН2—, превращающегося
в—СН2—О—СН2—, или же разложением фенолоспирта на фе-
нол и формальдегид, что особенно легко осуществляется, когда
метилольная группа присоединена к 1малореакционноспособному
углеродному атому. Эта теория основывается на наблюдении,
что эфирные мостики, образованные оксибензиловыми спирта-
ми без участия формальдегида, никогда не разлагаются с от-
щеплением формальдегида.
304
Гл. IX. Феноло-альдегидные смолы
Метиленовая группа эфирного мостика присоединена к без-
вольному ядру очень прочной углерод-углеродной связью; эфир-
»:ная связь, образующаяся при реакции (1), значительно менее
прочна и может быть разорвана действием бромистоводородной
кислоты:
Ph—СН2—О—СНг—Ph + 2НВг —► 2Ph—СН2Вг + Н2О
Реакция (2), наблюдаемая при температуре 175—180°, со-
'провождается одновременным выделением воды, что является
основанием для предположения об образовании соединения но-
вого типа — хинонметида, получаемого также из фенолоспирта
в результате потери одной молекулы воды:
ОН ОН О
Дегидратацией п-метилолфенола
-чается п-хинонметид:
аналогичным путем полу-
Хинонметиды могут также образоваться непосредственно из
-соответствующих фенолоспиртов путем отнятия одной моле-
кулы воды:
Таким образом, при отсутствии всех других влияний, в усло-
виях нейтральной среды под действием тепла происходит сна-
чала превращение фенолоспиртов в смесь диоксибензиловых
эфиров, производных дифенилметана и хинонметидов. Хинонме-
тиды могут, кроме того, полимеризоваться с образованием ди-
меров или тримеров.
Действие фенола, формальдегида и соляной кислоты. При
промышленном изготовлении смол обходятся без предваритель-
..ного выделения фенолоспиртов; таким образом, последние на-
Фенолоспирты
305
ходится в присутствии свободных фенола, формальдегида и
хлористого водорода или сильной кислоты, если реакция про-
водится в кислой среде (новолаки).
Фенол реагирует с фенолоспиртом, образуя молекулу воды
и производное дифенилметана:
Ph-CH2OH + PhH Ph—СН2—Ph + Н2О (5)
В кислой среде эта реакция является преобладающей.
Формальдегид с метилольным гидроксилом дает ацеталь
или полуацеталь (эти реакции протекают преимущественно в
кислой среде):
Ph—СН2ОН+НСНО Ph—СН2—О-СН2ОН (6)
2Ph—СН2ОН 4- НСНО —> Ph—СН2—О—СН2—О—СН2—Ph + Н2О (7)
Наконец, с соляной кислотой образуется хлорметильное про-
изводное согласно следующей обратимой реакции:
Ph—СН.2ОН + НС1 Ph—СН2С1 + Н2О (8)
Действие спиртов и жирных кислот. Эти реакции использу-
ются при образовании модифицированных феноло-формальде-
гидных смол. Метилольный гидроксил блокируется вследствие
образования простого или сложного эфира и поэтому теряет
свою реакционную способность:
Ph—СН2ОН + ROH -* Ph—CH2OR + Н.гО (9)
Ph—СН2ОН 4- RCOOH Ph-CH2OCOR 4 Н2О (10)
Действие веществ, содержащих азот. С аммиаком протекает
следующая реакция:
Ph—СН2ОН + NH3 — Ph—CH2NH2 + Н2О (11)
Полученные соединения могут реагировать со второй моле-
кулой фенолоспирта, образуя вещества типа
Ph—СН,—NH—СН2—Ph
Вещества этого типа получаются, в частности, при примене-
нии .аммиака в качестве катализатора реакции образования
фенолоспиртов. Полученные таким путем смолы содержат мо-
стик —СНг—iNH—СНг—. Азот может быть обнаружен в них
только при весьма энергичном нагревании: при температуре
240°, вызывающей «пиролитическое разложение.
Амины образуют аналогичные продукты общей формулы
Ph—СНг—NHR; с мочевиной получаются продукты
Ph—СН2—NH—СО—NH2 и Ph—СН2—NH—СО—NH—СН2—Ph
находящиеся в смешанных смолах на основе фенола и мо-
чевины.
20—156
06
Гл. IX. Феноло-альдегидные смолы
Реакции хлорметильной группы. Хлорметильные произ-
водные фенолов, содержащие группу CH2CI, при взаимодей-
ствии с водой вновь образуют фенолоспирты. С фенолами и
фенолоспиртами они реагируют с образованием веществ, по-
добных получаемым по реакциям (1) и (2):
Ph—СН2С1 + НОСН2—Ph -»- Ph—СН2—О—СН2—Ph -р НС1 (12)
Ph— CH2Cl-f-PhH -* Ph—СН2—Ph+HCl (13)
Выделяющаяся в обоих случаях соляная кислота может
вновь вступать в реакцию с фенолоспиртом, давая снова хлор-
метиленовые производные. Этим и объясняется каталитическое
действие НС1 в процессе образования новолаков.
В присутствии оснований хлорметиленовое производное те-
ряет НС1 с образованием хинонметидов— реакция, обратная
реакции (8), где происходит замещение гидроксила хлором.
Реакции фенольного гидроксила. Известно, что фенольный
гидроксил с альдегидами образует ацетали, но полученные
соединения при нагревании в присутствии катализаторов легко
превращаются в фенолоспирты. Основная' реакция может, на-
пример, быть изображена следующим образом:
ОН О—СН2ОН он
A A AzH,°"
Q+HCHO —у — Qr
В старых гипотезах о строении фенолочформальдегидных
смол этой реакции придавалось большое значение. Предпола-
галось, что фенольный гидроксил участвует, таким образом, в
построении мостиков, возникающих между фенольными ядрами.
В настоящее время такое толкование представляется непра-
вильным.
Взаимодействием фенолов со спиртами можно получать про-
стые эфиры с блокировкой фенольного гидроксила; таким же
образом при получении модифицированных фенольных смол
кислоты образуют сложные эфиры фенолов.
Фенольный гидроксил участвует также в реакции образова-
ния хинонметидов и в реакциях этих соединений, которые бу-
дут рассмотрены ниже.
Свойства хинонметидов. Хинонметиды, образовавшиеся из
о- ,или и-метилолфенолов, ведут себя по-разному.
Действие тепла на о-хинонметиды. Образовавшиеся о-хи-
нонметиды сразу обнаруживают склонность полимеризоваться,
превращаясь в тримеры или димеры.
При нагревании димера до температуры около 200° происхо-
дят окислительно-восстановительные реакции, ведущие или к
образованию этиленового мостика —СН2СН2—, или к разрыву
Фенолоспирты
307
цепи и образованию концевых групп СНО и СНз. Одновременно
образуется неперегоняющаяся смола:
Действие тепла на п-хинонметиды. При этом также происхо-
дит димеризация, но она должна протекать по следующему
уравнению:
Rx Rx zR
. 2О=/~/=СН2 НО—СН=СН-/~^—ОН
r/^ r/ ^<R
При продолжительном нагревании этого димера окисли-
тельно-восстановительная реакция приводит к одновременному
образованию двух соединений, которые могут быть также по-
лучены из производных диоксистильбена путем присоединения
или отнятия Нг:
Более глубокая окислительно-восстановительная реакция так-
же дает два соединения:
Rx Rx
НО—<2V-CHg и НО—СНО
R' RS I
Кроме того, как и из о-хинонметидов, получается неперё-
гоняющаяся смола.
Действие воды, НС1 и спиртов. Под действием Воды и НСГ
снова получаются фенолоспирт и хлорметиленовое производ-
20*
308
Г л. IX. Феноло-альдегидные смолы
ное этого фенолоспирта. При действии спирта можно получить
соединение Ph—CH2OR.
Действие ненасыщенных соединений. При взаимодействии
хинонметидов с соединениями, содержащими двойную связь
—С=С—, происходит реакция присоединения по схеме:
Реакции этого типа протекают при нагревании резолов с ка-
нифолью, высыхающими маслами или эфирами малеиновой
кислоты. Необходимо, однако, чтобы двойная связь была акти-
вирована сопряжением со второй двойной связью или с кар-
боксильной группой. Так, например, хинонметиды не реагируют
с олеиновой кислотой, имеющей только одну двойную связь.
Эта реакция аналогична реакции Дильса и Альдера.
Реакции с соединениями, содержащими карбонильную груп-
пу С=О, протекают аналогично, т. е. с образованием' соеди-
нений типа
Действие тепла на соединения с диметиленэфирной связью.
Некоторые авторы считали, что эти соединения могут разла-
гаться под действием тепла с образованием производных ди-
фенилметана и формальдегида, но такая- теория не подтверж-
дается ‘новейшими работами. Наоборот, соединения этого типа
образуются при отщеплении формальдегида под действием теп-
ла от соединений типа Ph—^СН2—О—СН2—О—СН2—Ph.
СТРОЕНИЕ ФЕНОЛО-АЛЬДЕГИДНЫХ СМОЛ
Если действовать формальдегидом на монофункциональный
фенол (Pi), содержащий только один реакционноспособный
атом углерода, можно связать между собой только два феноль-
ных ядра. Если же вместо фенола Pi используется фенол или
смесь фенолов с более высокой функциональностью, получают-
ся молекулы, содержащие несколько фенольных ядер, связан-
ных большим или меньшим числом мостиков.
Строение феноло-альдегидных смол
309
Условимся обозначать символом —L— двухвалентные ра-
дикалы, образующие связи или мостики между двумя феноль-
ными ядрами. В соединениях, получающихся при действии форм-
альдегида на фенолы, реакционноспособные атомы углерода,
т. е. атомы углерода, находящиеся в орто- и пара-положениях
по отношению к фенольному гидроксилу и не блокированные
углеводородными радикалами, связаны или с атомами водоро-
да, или с мостиками —L— или же, наконец, с концевой груп-
пой, которую обозначим символом Т.
Концевые группы Т — это радикалы —СН2ОН и = СН2; если
реакция происходит в присутствии НС1, то нужно прибавить
еще группу —СН2С1. Все эти концевые группы реакционно-
способны, т. е. способны реагировать или с атомом водорода,
связанным с активным атомом углерода, или между собой
с образованем нового мостика —L—.
Группа —СН3 образуется наряду с группой —СНО при
окислительно-восстановительной реакции группы (мостика)
—СН=СН—. Группа —СН3 нереакционноспособна, а группа
—СНО может реагировать с двумя фенольными ядрами, име-
ющими подвижные водородные атомы, по схеме:
,РЬ
—СНО + 2РЬН -> —СН< 4- Н2О
xPh
Таким образом, можно считать, что в среднем каждая кон-
цевая группа может принимать участие в реакции образова-
ния мостика, так как неактивность группы —СН3 компенси-
руется способностью группы —СНО реагировать с образова-
нием двух мостиков.
Приводим перечень мостиков —L— с указанием реакций ;
их образования:
I -СН2—
II —СН2—О-СНо—
III — сн2— сн2—
IV —СН=СН—
V =сн—сн=
VII — СН2—О—СН2—О—СН2—
VIII —СН2—NH—СН2—
Реакции (2) и (3)
Реакция (1)
См. стр. 306—307
См. стр. 307
См. стр. 307
Связь VI образована одновременно двух-
валентным радикалом и кислородным
мостиком
сн2
сн2
Конденсация в присутствии параформа
Конденсация в присутствии аммиака
310
Гл. IX. Феноло-альдегидные смолы
Мостики могут включать одну, две или три молекулы форм-
альдегида, т. е. содержать один, два или три атома углерода.
Мы будем различать эти три типа мостиков, обозначая их сим-
волами —Ц—, —L2— и —L3—.
Некоторые из перечисленных мостиков способны изменяться
при нагревании до более или менее высокой температуры:
мостики I и III являются термостойкими; мостик II, считав-
шийся ранее нестойким и способным при повышении темпе-
ратуры превращаться с выделением НСНО в мостик — СН2—,
по новым данным, следует считать также термостойким. Мо-
стик VII при повышении температуры укорачивается за счет
выделения формальдегида. Наконец, очень прочный мостик VIII
при температурах выше 240° способен превращаться в мостик I
с выделением аммиака:
Ph—СН2—NH—CH2-Ph + 2PhH 2Ph—СН2—Ph + NH3
По возрастающей сложности можно установить следующие
четыре типа молекул феноло-формальдегидных смол.
1. Молекулы, содержащие два фенольных ядра и мостик;
ОН ОН
А
Орто- и пара-положения в фенольных ядрах могут быть
незамещены или содержать концевые группы или же нереак-
ционноспособные углеводородные радикалы. Молекулы этого
типа могут быть получены с фенолами типов Рь Р2, Рз-
2. Молекула содержит несколько фенольных ядер, располо-
женных в виде прямой или разветвленной цепи:
он он он он
Б
Молекулы этого типа могут быть получены из фенолов ти-
пов Р2, Р3, но не фенолов Рь Если для синтеза взята смесь
фенолов, средняя функциональность ее должна быть больше
единицы.
3. Молекулы представляют собой циклы, образованные фе-
нольными ядрами, связанными мостиками. Цепи могут быть
разветвленными или неразветвленными. Известны молекулы
Строение феноло-альдегидных смол
311
этого типа с четырьмя фенольными ядрами и четырьмя мо-
стиками12:
он
в
Вещества такого типа могут образоваться только из фено-
лов Рг или Р3 или из смеси, функциональность которой равна
по меньшей мере 2.
4. Молекулы сетчатой структуры, образованные нескольки-
ми соединенными циклами, причем каждый цикл образован
фенольными ядрами, чередующимися с мостиками —L—. Струк-
туры такого типа могут быть получены только при участии фе-
нолов Р3 или смесей фенолов со средней функциональностью
более 2. Для фенола Рз строение смолы можно представить,
например, в следующем виде:
Таким образом, рассмотренные цепи или циклы составлены
из ядер (или молекул сложных фенолов), чередующихся с двух-
валентными радикалами (мостиками).
Зависимость строения продукта взаимодействия формаль-
дегида с фенолом от условий реакции. При проведении реак-
ции с формальдегидом должны быть известны следующие на-
чальные характеристики: функциональность фенола или смеси
312 Гл. IX. Феноло-альдегидные смолы
фенолов (и); pH реакционной смеси; величины исходного мо-
лекулярного отношения формальдегида к фенолу F/Р; реак-
ционная способность различных атомов углерода и функция
7’=/(0), представляющая зависимость температуры реакцион-
ной смеси от времени 0.
В результате реакции получают продукт большей или мень-
шей степени полимеризации, характеризуемый величиной моле-
кулярного отношения f/p, где f и р — количества формальде-
гида и фенола, участвовавших в образовании этого продукта
(значение f/p может быть отличным от F/Р и может быть экс-
периментально определено). Попытаемся определить влияние пе-
ременных величин: п, pH, F/Р и режима нагревания на вели-
чину f/p, а также на величины р (число фенольных остатков)
и t (число реакционноспособных концевых групп Т).
Таким путем мы можем предвидеть структуры продуктов,
получаемых при различных условиях реакции. Дальнейшее
рассмотрение, однако, может относиться лишь к средней мо-
лекуле и позволяет только статистически определить характер
явления.
Влияние нагревания и катализаторов. Реакция взаимодей-
ствия формальдегида с фенолом протекает в две стадии: сна-
чала образуются продукты присоединения, а затем происходит
конденсация этих продуктов. Однако реакции конденсации мо-
гут происходить и одновременно с начальной реакцией, а по-
тому образовавшаяся смесь является уже частично сконден-
сированной. Конденсации может быть подвергнута также вы-
деленная смесь начальных продуктов реакции.
Конденсация (отверждение) происходит либо при повыше-
нии температуры, либо при изменении pH реакционной среды.
Она может осуществляться двумя различными способами:
1. Внутренняя конденсация с увеличением числа мостиков
—L— за счет уменьшения числа реакционноспособных углерод-
ных атомов в фенольном ядре и концевых групп Т. При этом
молекулярный вес продукта не меняется, но внутренняя струк-
тура его усложняется.
2. Конденсация уже образовавшихся начальных продуктов
либо путем взаимодействия двух концевых групп Т, либо путем
взаимодействия концевой группы Т одной молекулы с реакцион-
носпособным углеродным атомом фенольного ядра другой мо-
лекулы. При этом молекулярный вес повышается без уплотне-
ния сетчатой структуры.
Конденсация с возрастанием молекулярного веса протекает
до тех пор, пока не получится молекула, совершенно не содер-
жащая концевых реакционноспособных групп или содержащая
лишь несколько таких групп, не могущих реагировать друг с
другом вследствие их относительной отдаленности и жесткости
Строение феноло-альдегидных смол 313-
молекулы, препятствующей их относительному смещению и сра-
щиванию.
Влияние pH. В зависимости от величины pH реакции кон-
денсации могут протекать более или менее быстро. Для одно-
атомных фенолов эти реакции протекают с особенно большой
скоростью при рН<7; при pH, близком к 4, первичный про-
дукт присоединения выделить не удается, так как сразу обра-
зуется конечная структура. Этот случай, в частности, соответ-
ствует процессу изготовления новолаков.
При .использовании многоатомных фенолов, например ре-
зорцина, скорость конденсации максимальна в среде, близкой
к нейтральной.
Если конденсацию проводят при величине pH, не соответ-
ствующей высокой скорости реакции, возможно прекращение
реакции, причем получаются несконденсированные или частич-
но сконденсированные продукты присоединения.
Влияние отношения f/p. Молекулы формальдегида, участвую-
щие в построении молекулы (их число мы обозначаем через /),
используются для образования различных типов мостиков, со-
держащих один, два или три атома углерода, или же для обра-
зования концевых групп. Если обозначить через / число молекул
формальдегида, участвующих в образовании мостиков, то мож-
но сделать следующие выводы. Величина отношения lip всегда
меньше величины f/p; отношение Z/p тем больше, чем большее
число мостиков из двух или трех атомов углерода содержится в-
молекуле, и тем меньше, чем большее число реакционноспособ-
ных концевых групп Т осталось несвязанным. Если процесс про-
водится в таких условиях, когда реакции последующей конден-
сации оказываются задержанными, то можно получать реакци-
онноспособные молекулы простой структуры (следовательно,
мягкие продукты).
Если после завершения процессов конденсации, когда реак-
ционноспособные концевые группы все израсходованы, отноше-
ние f/p<l, то отношение Цр<\ и получаемые продукты термо-
пластичны (линейные цепные молекулы).
Влияние функциональности фенола п. Пока процесс конден-
сации не закончился, в реакционной смеси могут содержаться
молекулы, для которых величина Z/p возрастает от ее минималь-
ного значения !/г до предельного значения п/2 при любом зна-
чении величины Z/p.
В этих (пределах следует рассмотреть несколько возможных
случаев:
1. Для монофункциональных фенолов (п=1) величина 1/р-
никогда не превышает V2.
2. При /<п<2 в начале конденсации отношение l/р равно V2,
а затем повышается, всегда оставаясь менее 1. Соответствую-
314
Гл. IX. Феноло-альдегидные смолы
щие молекулы могут иметь, следовательно, лишь линейную
структуру, каково бы ни было значение f/p.
3. При п>2 отношение l/р увеличивается от У2 до величины
п/2; при достаточно большой величине f/p можно получать мо-
лекулы, имеющие любую из структур А, Б, В, Г (см. схемы на
стр. 310—311).
Итак, исходя из допущений, сделанных в начале этого раз-
дела, можно с достаточным приближением предвидеть форму
молекул, получаемых в зависимости от условий реакции. Остает-
ся, однако, некоторая неопределенность, обусловленная тем,
что распределение мостиков, состоящих из одного, двух и трех
атомов углерода, нельзя установить заранее.
Влияние реакционной способности различных углеродных
атомов фенольного ядра. Знание реакционной способности раз-
личных углеродных атомов позволяет установить, какие из них
реагируют с формальдегидом быстро (а следовательно, и более
полно), и. получить указания о строении молекулы, в частности,
когда F/P<n. Например, при получении новолака из обычного
фенола формальдегид связывается главным образом с атомами
углерода в о-положении, которые по реакционной способности
значительно превосходят атомы углерода в и-положении.
Все приведенные сведения будут использованы для изучения
структуры смол, представляющих технический интерес.
Предварительно необходимо сделать краткий обзор развития
гипотез о строении смол.
Старые теории строения феноло-формальдегидных смол.
Раньше в схемах строения смол всегда исходили из обычного
фенола; допускалось существование мостиков —СН2— и
—СН2—О—СН2—, а также мостиков, полученных взаимодей-
ствием двух фенольных гидроксилов или одного фенольного
гидроксила и метилольной группы. Существование мостиков
двух последних типов в настоящее время отрицается.
Бакеланд8 высказал в 1909 г. предположение, что первая
стадия реакции формальдегида с фенолом состоит в образова-
нии о-оксибензилового спирта (салигенина) и что две молекулы
фенолоспиртов соединяются путем этерификации одной феноль-
ной группы спиртовой группой соседней молекулы. Кроме того,
Бакеланд считал, что образуются молекулы циклического строе-
ния, которые могут быть изображены следующей схемой:
сн2
Строение феноло-альдегидных смол
315
Рашиг22 в 1912 г. допускал существование трех видов мо-
стиков: мостиков —СН2—, —СН2—О—СН2— и кислородных
мостиков, образующихся при взаимной этерификации двух фе-
нольных гидроксилов; он также предполагал, что молекулы
имеют циклическую структуру, выражаемую схемами:
Блюмфельд23 (1921 и 1929 гг.) сначала допускал наличие
мостиков —СН2—, но в дальнейшем предположил, что углерод-
углеродные связи образуются с выделением Н2О за счет от-
дачи атома водорода группы —<СН2— одной молекулой и фе-
нольного гидроксила другой молекулой. По Блюмфельду,
строение резольной смолы выражается схемой:
Для новолаков Блюмфельд предложил новые схемы, в ко-
торых допускалось существование метиленовых мостиков с
316
Гл. IX. Феноло-альдегидные смолы
взаимной этерификацией фенольных гидроксилов или без нее:
ш
Наконец, Морган и Мегсон24 в 1933 г. .предполагали суще-
ствование только метиленовых мостиков, что дает следующую
схему:
Современные представления о строении феноло-формальде-
гидных смол. Различают два типа смол: смолы, полученные при
рН>7 и при рН<7.
Смолы, полученные в кислой среде (новолаки). В кислой
среде фенолоспирт с момента своего образования реагирует с
кислотным катализатором. Например, с НС1 образуется весьма
реакционноспособное хлорметиленовое производное, реагирую-
Строение феноло-альдегидных смол
317
щее с фенолоспиртами или с фенолами по следующим схемам
(см. также стр. 306):
Ph-CHaCl ф НОСН2 -Ph Ph—СНг—О—СН2—Ph ф НС1 (12)
Ph—СН2С1 ф PhH —► Ph—СН2 -Ph ф НС1 (13)
Освобождающаяся соляная кислота снова реагирует с ме-
тилольными группами, образуя радикал —СН2С1, и цикл возоб-
новляется до полного исчезновения свободных метилольных
групп. Скорость реакций (112) и (13) очень велика, и поэтому
никогда не удается выделить промежуточный продукт присое-
динения; непосредственно получаются только продукты конден-
сации.
Согласно старым теориям, для новолаков допускалось су-
ществование только мостиков —СН2—; реакция (112) пока-
зывает, что следует ожидать также образования мостиков строе-
ния —СН2—О—СН2—. Кроме того, имеются концевые группы
—СН2—О—СН2—ОН и образующиеся в результате их цикли-
зации бензо-1,3-диоксановые группы:
СН2
о хо
I сн2
Веглер25 доказал образование мостиков —СН2—О—СН2—,
действуя формальдегидом на пара-алкилфенолы в кислой среде.
Поскольку в большинстве случаев смолы получают при F/P,
равном 0,8—0,9 (вследствие полного использования реагентов
для образования смолы F/Р=f/p), можно допустить, что мости-
ки —СН2— преобладают и число мостиков, состоящих из двух
и трех углеродных атомов^ пренебрежимо мало. Тогда
F/Р — f/p=1/р и, следовательно, l/р также равно 0,8—0,9. При
этом получаются молекулы линейного или разветвленного строе-
ния, а длина цепи оказывается тем больше, чем ближе к еди-
нице отношение l/р. Ниже, в качестве примера, приводится ха-
рактеристика новолаков, полученных с обычным фенолом при
возрастающем процентном содержании формальдегида26:
f/p
0,5
0,75
1
Температура
плавления
°C
52—57
76—86
100—110
Свойства полученной смолы
Клейкая и растворимая в С2Н8ОН
Неклейкая и растворимая в С2Н8ОН
Твердая и труднорастворимая в С2Н8ОН
318
Гл. IX. Феноло-альдегидные смолы
Из этих данных видно, что по мере удлинения цепи темпера-
тура плавления продуктов повышается, а растворимость их
уменьшается. Формулы новолаков соответствуют схеме Б на
стр. 310.
Смолы, полученные в щелочной среде (резолы). Если про-
цесс ведут в щелочной среде, то реакции конденсации проте-
кают тем медленнее, чем выше pH. Чтобы получить несконден-
сированные фенолоспирты, необходимо проводить реакцию в
присутствии некоторого количества основания, достаточного
для превращения фенолов в феноляты. Фенолоспирты можно
затем выделить, нейтрализовав раствор органической кислотой,
например уксусной. Нейтрализация соляной кислотой ускоряет
процесс конденсации. Раствор можно затем декантировать и
экстрагировать эфиром или кислородсодержащим растворите-
лем, не смешивающимся с водой.
Полученные продукты могут быть в дальнейшем отверж-
дены каталитически или путем повышения температуры. От-
верждение происходит, как описано на стр. 312.
Продукты конденсации могут содержать все типы мостиков,
перечисленных на стр. 309. Однако нужно различать два слу-
чая в зависимости от температуры, при которой смола была
получена. Если эта температура не превышала 110°, средний
молекулярный вес смолы незначителен и образовывались толь-
ко мостики типов I, II и VII (см. стр. 309). Мостики типа I,
содержащие один атом углерода, преобладают при малой ве-
личине f/p (от ’/2 до 1). Мостики типа II (или VII) образуют-
ся при большом количестве формальдегида, в особенности ког-
да F/Р приближается к п или больше п. При температуре выше
110° структура смол весьма сходна со структурой новолаков,
с преобладанием эфирных мостиков, если f/p, как обычно бы-
вает, больше 1.
Если температура образования смолы близка к 150°, то не-
которое количество мостиков —СН2—О—СН2— превращается
в мостики типа I, а мостики типа VII превращаются в мостики
типа II согласно реакции:
—СН2—О—СНг—о—СНа------ —СН2-О-СН2—Ч-НСНО
Выделение значительного количества формальдегида наблю-
дается тогда, когда в реакционной смеси не остается фенолов со
свободными реакционноспособными атомами углеродов. В про-
тивном случае формальдегид присоединяется к одному из этих
атомов углерода, образуя дополнительную концевую группу,
способную реагировать с образованием в дальнейшем новых
мостиков.
При еще более высокой температуре, достигающей 175°,
происходит образование хинонметидов и мостиков типа IV и
Модифицированные феноло-альдегидные смолы
313
VI; если же температура достигнет 200°, вследствие протека-
ния окислительно-восстановительных реакций образуются мо-
стики типа III и V, появляются инертные группы —СНЭ и реак-
ционноспособные концевые группы —СНО, образующие мости-
ки типа
Ph—СН—Ph
I
Ph
Поскольку метилольная группа занимает преимущественно
о-положение, вероятность образования мостиков, связанных с
атомами углерода в о-положении,, больше, чем мостиков, свя-
занных с углеродными атомами в n-положении. Это дает осно-
вания для вывода различных теоретических схем макромоле-
кул с учетом значений р, п, f/p и температурных пределов, пр»
которых происходит образование смолы.
Для смол, приготовленных в присутствии аммиака или гек-
саметилентетрамина, нужно учитывать, кроме того, возмож-
ное наличие мостиков типа —СН2—NH—СН2— и мостиков, в
которых один атом азота связан с тремя фенольными ядрами12:.
—СН2—N—СН2—
I
сн2
I
Если температуру повысить до 240°, эти мостики обрывают-
ся, одновременно взаимодействуя с атомами водорода, связан-
ными с реакционноспособными углеродными атомами. При
этом образуются мостики —СН2— и выделяется аммиак.
Наблюдения, описанные в начале этой главы, дают возмож-
ность установить последовательность различных типов резолов
с учетом вероятного значения l/р, значения п и максимальной
температуры, при которой происходит образование этих резо-
лов. Когда F/Р меньше п, устанавливая реакционную способ-
ность каждого атома углерода, можно определить, какие атомы
углерода реагируют быстрее (и, следовательно, более полно)
с формальдегидом.
МОДИФИЦИРОВАННЫЕ ФЕНОЛО-АЛЬДЕГИДНЫЕ СМОЛЫ
И ЛАКОВЫЕ СМОЛЫ
Если феноло-альдегидные смолы (новолаки и резолы) изго-
товлены на основе обычного фенола, то они растворимы только
в сложных эфирах, кетонах и вообще в кислородсодержащих
растворителях, при условии малых размеров молекул смолы и
линейности их структуры. Эти смолы нерастворимы в аромати-
ческих или алифатических углеводородах и высыхающих мас-
лах.
320
Гл. IX. Феноло-альдегидные смолы
Спиртовые растворы этих смол применялись вначале как
заменители шеллачных лаков, но в дальнейшем поиски были
направлены на приготовление производных этих смол, раствори-
мых в углеводородах и совместимых с высыхающими масла-
ми или даже растворимых в высыхающих маслах. Рассмотрим
причины такого направления работ и общие характеристики мо-
дифицированных смол, растворимых в маслах.
Можно дать следующее определение лаку: это раствор или
коллоидная дисперсия смеси А больших молекул органического
вещества, которая, будучи нанесена тем или иным способом на
поверхность, после испарения растворителей, сопровождающе-
гося иногда химическим превращением смеси А, образует
сплошную, тонкую, плотно прилипающую и прочную пленку.
Превращение растворимой смеси А в нерастворимую смесь
Б достигается путем образования соединительных мостиков
между молекулами и увеличения среднего молекулярного веса.
При этом 1протекает один (или несколько) из следующих про-
цессов.
1. Когда смесь А содержит непредельные соединения, на-
пример высыхающие масла, химическое превращение смеси А
в нерастворимое соединение является результатом окисления
масел кислородом воздуха. Этот процесс может быть ускорен
повышением температуры или действием сиккативов (органи-
ческих солей металлов переменной валентности). В данном
случае происходит главным образом образование кислородных
мостиков —О—, возникающих вследствие образования проме-
жуточных гидроперекисей. Превращение происходит довольно
медленно, без доступа воздуха оно вовсе не происходит.
2. Смесь А содержит молекулы, способные реагировать
между собой и полимеризоваться или конденсироваться без
участия кислорода воздуха. Процесс конденсации или отверж-
дения чистых фенольных смол (стр. 312) требует или примене-
ния катализаторов, изменяющих pH среды, или повышения тем-
пературы. В первом случае ускорение конденсации или отверж-
дение достигается сразу после добавления катализатора (от-
вердителя); во втором случае требуется сушильная камера до-
статочного размера, чтобы вместить покрытый данным лаком
предмет.
Таким образом, можно различить четыре процесса сушки ла-
ковых пленок:
1) испарение растворителя без химического превращения;
2) испарение растворителя с последующим окислением;
3) испарение и .последующая каталитическая конденсация
или полимеризация;
4) испарение и последующая конденсация или полимериза-
ция при повышенной температуре.
Модифицированные феноло-альдегидные смолы 321
Для лаков, процесс сушки которых 'протекает по 'первому
типу, затруднительно нанесение нескольких последовательных
слоев: при нанесении следующего слоя лака сухая пленка рас-
творится.
Недостатком лаков третьего типа является их способность
изменяться во времени и ограниченность срока их хранения на
складе или срока применения. Лаки четвертого типа имеют
лишь ограниченное применение, поскольку для отверждения ла-
ковых пленок требуется сушильная камера. Лаки второго типа,
высыхающие при окислении, не представляют никаких не-
удобств при применении и могут использоваться во всех слу-
чаях, особенно если для их сушки требуется достаточно ко-
роткое время.
По указанным причинам поиски были направлены в сторону
изыскания смол, растворимых в высыхающих маслах или содер-
жащих ненасыщенные радикалы (обычно цепи жирных кис-
лот выедающих .масел) и. отверждающихся при окислении.
Феноло-альдегидные смолы, растворимые в высыхающих маслах
Для того чтобы какая-либо природная или искусственная
смола могла растворяться в том или ином растворителе, необ-
ходимо, чтобы были выполнены следующие два условия:
1) смола должна состоять йз молекул малых размеров; если
она высокомолекулярна, то макромолекулы должны иметь ли-
нейную структуру;
2) должно существовать определенное сходство в строении
смолы и растворителя.
Резолы и новолаки, растворимые в спиртах, удовлетворяют
этим условиям.
Для придания феноло-формальдегидным смолам раствори-
мости в углеводородах и маслах необходимо ограничить раз-
меры их молекул и ввести в их состав углеводородные цепи.
Ограничение размера молекул может быть достигнуто, если
l/р не превышает единицы; это достигается путем применения
бифункциональных фенолов (п=2) или блокирования реак-
ционноспособных концевых групп инертными радикалами, чтобы
ограничить величину I, или же, когда п>2, путем уменьшения
величины F/P.
Углеводородные цепи могут быть присоединены или к фе-
нольным ядрам, или к концевым активным группам.
Как мы увидим далее, все растворимые феноло-альдегид-
ные смолы удовлетворяют одному или нескольким из указан-
ных условий. Следует, однако, заметить, что «растворение» фе-
нол о-альдегидных смол в высыхающих маслах всегда произво-
дится при повышенной температуре и при проведении этой опе-
21—156
322
Гл. IX. Феноло-альдегидные смолы
рации всегда происходят сложные химические реакции, в ре-
зультате которых молекулы смолы соединяются с молекулами
масла. Получаемый продукт является не раствором смолы в
масле, а сложным соединением, более или менее полно сохра-
няющим способность масла к высыханию. Если вначале и обра-
зуется истинный раствор, то он представляет интерес главным
образом с точки зрения получения гомогенной смеси, в которой
легче могут протекать дальнейшие химические реакции.
Перейдем теперь к изучению сложных веществ, содержащих
в молекуле фенольные ядра и длинные ненасыщенные цепи.
Такие молекулы частично обладают свойствами феноло-альде-
гидных смол и частично также свойствами высыхающих масел.
Феноло-альдегидные смолы в чистом виде имеют следующие
свойства:
Твердость .................................. Хорошая
Сопротивление удару ...................... Слабое
Стойкость к действию воды и кислот .... Очень хорошая
Стойкость к действию щелочей .............. Хорошая
Лаки на основе только одних высыхающих масел имеют
следующие характеристики:
Твердость...................................Плохая
Сопротивление удару.........................Очень хорошее
Стойкость к действию воды и кислот .... Плохая
Модифицированные смеси имеют хорошую твердость и хоро-
шее сопротивление удару; стойкость к воде, кислотам и щело-
чам улучшается по сравнению с соответствующими свойствами
лаков на основе только одних высыхающих масел.
Было бы естественным попытаться приготовить феноло-аль-
догидные смолы, модифицированные высыхающими маслами,
так, чтобы модификация происходила без варки лака. Такие
смолы существуют и имеются в продаже.
Наконец, для получения лаков горячей сушки можно также
совместить в одной и той же молекуле элементы и, следова-
тельно, свойства феноло-альдегидных смол, алкидных смол и
высыхающих масел или феноло-альдегидных и алкидных смол.
Ниже приводится перечень чистых и модифицированных фе-
нольных смол, практически используемых в технике изготов-
ления лаков:
Феноло-альдегидные смолы неотверждающиеся (ново-
лаки).
Феноло-альдегидные смолы, отверждающиеся при [нагре-
вании или с помощью катализатора.
Феноло-альдегидные смолы, модифицированные высшими
спиртами.
Модифицированные феноло-альдегидные смолы
323
Феноло-альдегидные смолы, модифицированные природ-
ными смолами и, в частности, канифолью.
Феноло-альдегидные смолы чистые, растворимые в мас-
лах.
Феноло-альдегидные смолы, модифицированные высы-
хающими маслами.
Феноло-альдегидные смолы, модифицированные алкид-
ными смолами, высыхающими и невысыхающими маслами.
Различные смолы, содержащие фенолы.
В дальнейшем характеристики каждого из этих типов смол
будут уточнены, но перед этим рассмотрим исходные вещества(
применяемые для получения смол.
Компоненты чистых феноло-альдегидных смол (их получе-
ние из природных или синтетических продуктов было описано
на стр. 285—290):
Фенол обычный, крезолы и ксиленолы.
Фенолы, замещенные в пара-положении каким-либо али-
фатическим радикалом.
н-Оксибифенил.
S бис- Фенолы.
Сложные терпеновые фенолы, содержащие обычно не-
сколько фенольных ядер.
Продукты, используемые для модификации феноло-альде-
гидных смол:
Природные смолы на основе смоляных кислот (например,
абиетиновой кислоты).
Высыхающие и невысыхающие масла и входящие в их
состав насыщенные и ненасыщенные жирные кислоты (сте-
ариновая, олеиновая, линолевая, линоленовая, дегидратиро-
ванная рицинолевая).
Спирты алифатические (бутиловый, амиловый, октило-
вый) и многоатомные спирты (гликоль, глицерин, пента-
эритрит).
Дикарбоновые кислоты, компоненты алкидных смол (ади-
пиновая, малеиновая, фталевая).
Реакции, позволяющие изготовлять модифицированные
смолы:
Образование простые и сложных эфиров с участием фе-
нольного гидроксила.
Образование простых и сложных эфиров с участием ме-
тилольной группы.
Реакции присоединения о-хинощиетида к спиртам или
жирным кислотам, а также к соединениям, содержащим со-
пряженные двойные связи.
21
324
Гл. IX. Феноло-альдегидные смолы
Мы уточним эти реакции в более важных случаях, но нужно
заметить, что о- и n-хинонметиды образуются при температуре
около 175—180° из фенолоспиртов или из диоксибензиловых
эфиров
всегда .присутствующих в фенольных смолах. Следовательно,
хинонметиды должны играть чрезвычайно важную роль в реак-
циях образования модифицированных смол.
о-Хинонметиды реагируют с абиетиновой кислотой по схеме:
(11а)
Аналогично протекает реакция с левопимаровой кислотой,
также содержащейся в канифоли.
Можно допустить, что при температуре 270—280°, обычно
поддерживаемой при варке лака, эта реакция обратима и воз-
можно освобождение о-хинонметида, способного снова реаги-
ровать, в частности с высыхающими маслами.
При взаимодействии хинонметидов (например, о-хинонмети-
Да) с цепями, содержащими более одной двойной связи, и, в
частности, с высыхающими жирными кислотами (например,
с элеостеариновой кислотой) получают соединения типа:
/СН=СН(СН2),СООН
I || I (14б)
YXO/XcH=CH(CH»)sCHs
Таким же образом можно присоединить молекулу хинон-
метида по двойным связям в положениях 9, 10 и 13, 14 углево-
дородной цепи элеостеариновой кислоты.
Модифицированные феноло-альдегидные смолы
325
Аналогичные результаты получаются, когда применяют не
жирные кислоты, а непосредственно высыхающие масла. По-
скольку эта реакция обусловливает исчезновение двойной связи
в молекуле масла, способность высыхать на воздухе, или, дру-
гими словами, способность этой молекулы присоединяться к со-
седней молекуле путем окисления, уменьшается. Следовательно,
пленка лака будет образовываться не путем присоединения друг
к другу молекул масла, а путем присоединения значительно
более крупных молекул, быстрее переходящих при окислении
в нерастворимое состояние. Вызываемое этим уменьшение ско-
рости высыхания не является, однако, особо существенным.
С малеиновой кислотой и эфирами малеиновой кислоты
о-хинонметиды реагируют по механизму, аналогичному реакции
Дильса—Альдера:
СН—СООН
+ II
СН—СООН
(Ив)
о-Хинонметиды способны реагировать со спиртами и жирны-
ми кислотами с образованием продуктов, аналогичных получае-
мым по реакциям (9) и (10) (см. стр. 305):
(15)
(16)
При достаточно высоких температурах эти реакции, вероят-
но, обратимы.
Реакции (15) и (16) показывают, что фенольные смолы
путем переэтерификации могут быть превращены в невысыхаю-
щие алкидные смолы.
Все описанные реакции вызывают блокирование метилоль-
ной группы; это или ограничивает увеличение молекулярного
веса, или же способствует его увеличению, в зависимости от
того, используются ли для этого блокирования монофункцио-
нальные или полифункциональные соединения. Например, реак-
ция с одноосновной кислотой ограничивает дальнейший рост
молекулы феноло-формальдегидной смолы; реакция с кислотной
группой, входящей в молекулу алкидной смолы, способствует
этому росту.
326
Гл. IX. Феноло-альдегидные смолы
ТОВАРНЫЕ ФЕНОЛО-ФОРМАЛЬДЕГИДНЫЕ СМОЛЫ
В заключение рассмотрим разные типы немодифицирован-
ных и модифицированных феноло-альдегидных смол и попы-
таемся установить их химическое строение и механизм их
отверждения или сушки. Составление перечня торговых смол с
их качественной оценкой оказалось невозможным, поскольку от-
сутствуют подробные сведения о производстве этих продуктов.
Поэтому ниже будет приведено по нескольку примеров каждого
типа смолы, причем авторы заранее просят извинения за воз-
можные ошибки.
Неотверждающиеся смолы; полученные в кислой среде и
растворимые в спирте. Такие смолы могут быть приготовлены
на основе любого фенола, но чаще всего для их изготовления
применяется обычный фенол, причем F/Р находится в пределах
между 0,8 и 0,9. Эти смолы до сих пор еще иногда применяют-
ся как заменители шеллака, но основной областью их приме-
нения являются термореактивные прессовочные порошки, полу-
чаемые посредством смешения смолы с гексаметилентетрамином
и наполнителями. После отверждения смолы отношение ftp боль-
ше единицы.
В новолачных смолах содержатся почти исключительно мо-
стики —СН2—, но возможно также наличие мостиков типа
СН2—О—СН2— и —'СН2—О—СН2—О—СН2 —. Реакционно-
способные атомы углерода фенольных ядер, не участвующие в
образовании мостиков, связаны с водородом. Молекулы смол
имеют линейное строение и состоят из 4—8 соединенных фе-
нольных ядер или молекул фенола.
Немодифицированные резолы, применяемые для лаков. Для
изготовления резолов применяют фенолы с функциональностью
выше 2. В результате получаются термореактивные смолы, рас-
творимые в спиртах, кетонах, простых и сложных эфирах.
Отверждение лаковой пленки может быть достигнуто или нагре-
ванием, или соответствующим .подбором значения pH.
Резолы на основе одноатомных, фенолов. Отверждение их
Достигается или повышением температуры, или при рН^4, т. е.
обязательно в кислой среде, если желают обойтись без нагре-
вания лакируемого изделия. Отверждение на холоду возможно
только при лакировании неметаллических изделий; эти смолы
очень удобны для лаков по дереву. В качестве катализаторов
отверждения применяют соляную или фосфорную кислоты25.
Резолы на основе многоатомных фенолов^6. Отверждение
достигается повышением температуры или путем доведения pH
до 7. Лаки на основе, например, резорцина до применения не-
обходимо хранить при рН = 13—14, но даже в этих условиях
скорость конденсации нельзя считать ничтожно малой. Доведе-
Товарные феноло-формальдегидные смолы
327
ние же pH до 7 непосредственно перед применением лака пред-
ставляет собой весьма тонкую операцию. Поэтому предпочи-
тают применять следующий метод использования лаков этого
типа. Готовят водно-спиртовой раствор смолы с pH, близким к
7, и с F/Р значительно меньше единицы (0,5—0,6), следователь-
но, неотверждающийся. В момент применения лака прибавляют
формальдегид в количестве, достаточном для доведения F/Р до
величины более единицы, например до 1,5. Приготовленный та-
ким образом лак быстро отверждается, а поэтому должен быть
использован в довольно короткое время26.
Линейные смолы после отверждения приобретают сетчатую
структуру. Характер мостиков и концевых групп изменяется в
зависимости от температуры, при которой готовится смола (это
было подробно рассмотрено в предыдущем разделе). Получае-
мые лаки обладают хорошей стойкостью к действию химических
реагентов; они дают твердую пленку, прочно прилипающую к
стали, но недостаточно эластичную и сильно окрашенную.
В качестве примера можно указать бакелитовые лаки 260 и
265А, приготовленные соответственно на основе крезолов и
обычного фенола.
Резолы, модифицированные спиртами. Они могут быть ис-
пользованы для приготовления лаков путем простого растворе-
ния, без введения каких-либо других компонентов. Модифика-
ция производится путем обработки резолов высшими спиртами
(бутиловым, амиловым, октиловым), причем происходит частич-
ная этерификация фенольных и метилольных гидроксилов.
Вследствие блокирования концевых групп реакционная спо-
собность смолы уменьшается. В то же время уменьшается рас-
творимость в спиртах и увеличивается растворимость в углево-
дородах. Сначала получаются продукты, растворимые в арома-
тических углеводородах. Если обработка продолжается доволь-
но долго, то образующиеся смолы становятся нерастворимыми
в спиртах и растворимыми в алифатических углеводородах.
Полученные лаки отверждаются при нагревании или при дове-
дении до соответствующего pH. Твердые пленки имеют сетчатую
Структуру с меньшей частотой сетки, чем у соответствующего
резола. При- этом они более эластичны и значительно светлее.
Феноло-альдегидные смолы, модифицированные природными
смолами. Такие смолы получаются взаимодействием резолов с
некоторыми природными смолами, например с деполимеризо-
ванным копалом Конго, и главным образом с канифолью. По-
лученные продукты затем этерифицируются глицерином или
другим многоатомным спиртом, например пентаэритритом27.
Описан также способ изготовления модифицированной смолы,
состоящий во взаимодействии смеси фенола и формальдегида с
канифолью28 в кислой среде.
328
Г л. IX. Феноло-альдегидные смолы
Количество вводимой феноло-альдегидной смолы состав-
ляет от 10 до 20%.
Модифицированная смола имеет температуру размягчения
примерно на 50° .выше, чем этерифицированная природная
смола (глицериновый эфир канифоли). С повышением содер-
жания феноло-альдегидной смолы температура размягчения
повышается, а растворимость в растворителях и в маслах
уменьшается. Применяемые феноло-альдегидные смолы изго-
товляются на основе обычного фенола, бис-фенолов и п-алкил-
фенолов.
Выше было показано, что реакция 'протекает с обра-
зованием производных хромана за счет раскрытия одной из
двойных связей абиетиновой или левопимаровой кислоты.
Если взять абиетиновую кислоту (М=378) и фенолоспирт
в виде производного бчс-фенола (Л1—290), то можно видеть,
что в практически применяемых рецептурах одна молекула фе-
нолоспирта приходится на 4—8 молекул абиетиновой кислоты.
Так как к одной молекуле фенолоспирта можно присоединить
от одной до двух молекул абиетиновой кислоты, то, очевидно,
что имеется избыток свободной абиетиновой кислоты, в кото-
рой должно быть растворено соединение абиетиновой кислоты
с фенолоспиртом. Предельная величина молекулярного веса
такого продукта присоединения близка к 1 000.
Смолы, модифицированные канифолью, растворимы в высы-
хающих маслах. Не исключено также, что при высокой темпе-
ратуре выделяется фенолоспирт, который вступает в реакцию
с жирными кислотами. Растворение в маслах сопровождается,
по всей вероятности, реакциями, глубоко модифицирующими
структуру .продукта, входящего в дальнейшем в состав лака.
Из модифицированных смол этого типа можно назвать гли-
кофены, апьбертоли (111L, 209L, 116Q, 118S), бекациты 1100
и 1102.
Немодифицированные феноло-альдегидные смолы, раство-
римые в маслах. Приготовление их описано Шривом и Гол-
дингом29. Это резолы или .новолаки, полученные из фенолов, За-
мещенных в пара-положении (следовательно, с функциональ-
ностью 2); используют также бпс-фенол (п=4), на основе ко-
торого готовят только новолаки.^Растворимость в маслах .до-
стигается ограничением размера молекул (причем lip всегда
должно быть меньше /) и присоединением углеводородных це-
пей к фенольным ядрам. Структура этих смол всегда линейная.
Растворение .в маслах, вероятно, сопровождается (как и в
случае смол, модифицированных канифолью) разрывом мости-
ков —СН2—О—СН2— с промежуточным образованием хинон-
метидов и соединением их с ненасыщенными жирными кисло-
тами масел.
Товарные феноло-формальдегидные смолы
329
Эти смолы разделяются на реакционноспособные (вероятно,
относящиеся к смолам типа резола) и нереакционноспособные
(относящиеся к типу новолаков). Первые при растворении в
маслах образуют обильную пену. Метод исследования этих смол
описывается в статье Чатфилда30.
В качестве смол такого типа можно упомянуть супербека-
циты 1001 и 1002, бакелиты BR 3360 и BR 14634.
Феноло-альдегидные смолы, модифицированные высыхаю-
щими маслами. Они получаются взаимодействием высыхающих
масел с резолами или с фенолами, а затем с формальдегидом.
На стр. 308 была рассмотрена реакция присоединения к фено-
лоспирту углеводородных цепей, содержащих более одной двой-
ной связи.
Из смол этого типа можно указать бакелит 2175 и дурофен
306 V.
Феноло-альдегидные смолы, модифицированные алкидными
смолами. Высыхающие алкидные смолы. При взаимодействии
резолов с алкидными смолами, образованными ненасыщенны-
ми жирными кислотами, происходит реакция присоединения,
аналогичная протекающей при применении высыхающих масел.
В качестве смол этого типа можно упомянуть бекозолы 1А, 40,
1316 и 1326.
Невысыхающие алкидные смолы. Смолу, в которой мости-
ками служат простые или сложные эфиры, можно получать пу-
тем соединения чистых фенольных смол с жирными кислотами,
многоосновными кислотами, жирными спиртами и многоатом-
ными спиртами.
Примерами подобных смол являются люфены АТ и В на
основе фенолов, формальдегида, адипиновой кислоты, бутано-
ла и триметилолпропана. Они отверждаются путем поликонден-
сации и путем повышения температуры.
Феноло-альдегидные смолы, не содержащие формальдегида,
и прочие смолы. Смолы первого типа приготовляются обычно из
сложных фенолов терпенового ряда (например, супербекацит -
2000).
Из прочих смол следует упомянуть продукты конденсации
фенола с 'циклогексаноном (смола AW2).
Недавно появившиеся эпоксидные смолы эпон получаются
взаимодействием эпихлоргидрина с бпс-фенолами (см. стр. 412 $
и сл.).
Эти смолы совместимы с маслами и, по-видимому, обла-
дают прекрасными свойствами.
Заканчивая это перечисление, необходимо указать, что оно
не является полным. Можно предвидеть, что на основе феноль-
ных смол будут приготовлены другие смолы для лаков, причем
эти новые продукты будут иметь следующие улучшенные каче-
330
Гл. IX. Феноло-альдегидные смолы
ственные показатели: лучшая совместимость с высыхающими
маслами и растворимость в уайт-спирите; повышенная темпера-
тура плавления; большая легкость обработки; возможность по-
лучения быстросохнущих лаков; высокая стойкость к химиче-
ским реагентам и газам.
ЛИТЕРАТУРА
1. Gerhardt, Annalen, 1853, 87, 159.
2. Baeyer, Вег., 1872, 5, 25, 280, 1094.
3. Michael, Come у, Am. Chem. J., 1883—1884, 5, 347.
4. К 1 e e b e r g, Annalen, 1891, 263, 283.
5. M a n a s s e, Ber., 1894, 27, 2408.
6. L. Lederer,!. Soc. Chem. Ind., 1895, 14, 297.
7. В I u m e r, J. Soc. Chem. Ind., 1903, 22, 706; 1904, 23, 448.
8, Baekeland, Ind. Eng. Chem., 1909, 1, 151.
9. В a e k e 1 a n d, C. A., 1910, 4, 1689.
10. Ellis, Chemistry of Synthetic Resins, Rheinhold, New York, 1935, v. 1,
p. 412.
11. Lilley, J. Oil Col. Chem. Ass., May 1943, 26.
12. Z i n k e, Ziegler, Fette u. Seifen, 1950, № 10.
13. Hultzsch, Chemie der Phenolharze, Springer, 1950.
14. Meyer, Introduction a I'etude des Matieres Plastiques, Dunod, 1948.
15. Cm. A u w e r s, K. Ann., 1907, 356, 124.
16. Hultzsch, Angew. Chem., 1948, A60, 179.
17. Price, Ind. Eng. Chem., 1948, 40, 992.
18. Cm. Euler, Adler и др., Ark., 1941, 14B, № 9.
19. К у r n i n g, Ark., 1941, 15B, № 11,
20. Adler, cm. Euler, Cedwall, Ark., 1941, 15A, № /,
21. F. S. Granger, Ind. Eng. Chem., 1932, 24, 442; 1937, 29, 860. 1125,
1305.
22. R a s c h i g, Angew. Chem., 1912, 25, 1945.
23. Bl umf el d, Chem. Ztg., 1929, 53, 493; 1931, 55, 551.
24. Morgan, Megson, J. Soc. Chem. Ind., 1933, 52, 420T.
25. F о n t г о b e r t, Fab. Zeit., 1943, 38. № 6, 26—29.
26. L i t 11 e, Tepper, Sci. Techn. Memorandum Ministry of Supply, July
1947, № 13—47.
27. P. O. Powers, Ind. Eng. Chem., 1951, 43, 8, 1770.
28. Champetier, Laporte, Recherches et Inventions, 1934, p. 230,
252 291
29. R. N’ Shreve, B. Golding, Ind Eng. Chem., 1951, 43, № 1, 134.
30. H. W.- C h a t t f i e 1 d. Paint Manufacture, 1951, 21, 1.
ГЛАВА X
МОЧЕВИНО-ФОРМАЛЬДЕГИДНЫЕ СМОЛЫ И ИХ АНАЛОГИ
77. Тале
МОЧЕВИ НО ФОРМАЛЬДЕГИДНЫЕ СМОЛЫ
Основные этапы процесса получения смолы. В этой главе
рассмотрено образование мочеви но-форм альдегидных смол;
общая схема процесса не изменяется, если вместо мочевины
используются тиомочевина, меламин и т. д.
Исключительная реакционная способность формальдегида
позволяет ему вступать в реакцию со многими органическими
соединениями, имеющими подвижные атомы водорода, в част-
ности с аминами и амидами. Присоединением .молекулы альде-
гида к амиду в первой стадии получаются производные, содер-
жащие радикал —СН2ОН; в следующей стадии выделяется мо-
лекула воды за счет гидроксила и атома водорода, освобож-
даемого, например, аминогруппой при образовании метилено-
вой связи.
Процесс конденсации с фенолом обычно принято подраз-
делять на три последовательные стадии: А, В и С. Аналогич-
ным образом можно различать три стадии при превращении
смеси мочевины и формальдегида в смолу.
Стадия А. Если мочевину растворять на холоду в растворе
формальдегида, то состав образующихся продуктов будет раз-
личным в зависимости от молекулярного отношения мочевины
к формальдегиду (U/F) и pH реакционной среды. Начало реак-
ции определяется по повышению температуры.
U]F=\. В нейтральной и слабощелочной среде получается
раствор монометилол мочевины:
H2N— CO-NH,pCH2O -»- H2N—СО—NH—СН2ОН
В кислой среде выделяется одна молекула воды и образует-
ся монометиленмочевина, что часто приводит к получению не-
растворимых продуктов.
Были идентифицированы также соединения, образующиеся
при взаимодействии молекулы метилолмочевины с молекулой
метиленмочевины:
H2N—СО—NH—СН2—NH -СО—N=CH2
332 Гл. X. Мочевино-формальдегидные смолы и их аналоги
UlF—'lz- В нейтральной или слабощелочной среде получает-
ся раствор диметилолмочевины:
HjN—СО—NH2-f-2CH2O НОСН2—NH—СО—NH—СН2ОН
В кислой среде одна молекула формальдегида, присоеди-
няясь к одному атому азота, образует метилольный радикал,
а вторая молекула реагирует с выделением воды. В результате
образуется метилолмегиленмочевина:
H2N—СО—NH2 + 2CH2O —>- НОСН2—NH—СО—N=CH2
UjF^li. В нейтральной или щелочной среде возможно при-
соединение четырех молекул формальдегида с образованием
тетраметилолмочевины. Это вещество не было выделено; в дей-
ствительности происходит дегидратация с образованием следую-
щего соединения:
СО
НОН2С—N \[-СН2ОН
I I
н2с сн2
Диметиловый эфир этого соединения был выделен.
Отношение U/F превышает единицу только при получении
мочевино-формальдегидных смол.
Известно, что амины и амиды ведут себя в присутствии
формальдегида довольно различно. Марвел допускает, что мо-
чевина ведет себя одновременно и как амин, и как амид. Ре-
агируя сначала как амин, мочевина дает метиленмочевину, ко-
торая, как первичные амины, циклизуется с образованием три-
мера, сходного с меламином:
СН2
H2NOC—Ы \—CONH,
I I
Н2С сн2
^N—CONH2
Боковые группы —CONH2 ведут себя в этом случае как
амидные радикалы, образуя следующее соединение:
СН2
НОСН2—HNOC—N CONH—СН2ОН
I I
Н2С сн2
^N— CONH—СН2ОН
Мочевино-формальдегидные смолы
333
Однако это вещество вследствие его весьма большой реак-
ционной способности выделено не было.
В стадии А в реакционной смеси присутствуют описанные
вещества или начальные продукты их конденсации в виде истин-
ного или коллоидного раствора.
Стадия В. Стадия В соответствует положению, когда тредва-
рительная реакция конденсации прошла достаточно глубоко, и
молекулы состоят из 10—20 структурных единиц мочевины. Эти
вещества относительно легкоплавки, чувствительны к действию
воды, иногда даже растворимы.
Следует указать, что наши знания в отношении стадии В ме-
нее надежны, чем в отношении предыдущей стадии. Если рас-
твор А вначале содержит монометилолмочевину, то образование
линейных молекул с метиленовыми мостиками может осуще-
ствляться с выделением воды по схеме:
H2N—СО—NH—СН2;ОН + ffc NH—СО—NH—СН2: ОН + H:NH-CO~NH-CH2OH
Весьма вероятно, что одновременно возникают эфирные свя-
зи согласно схеме
H2N—СО—NH—CHjfOH+'HlOCHa—NH—СО—NH2
Наконец, .метилениминные группы метиленмочевины могут
также образовывать метиленовые мостики, присоединяя по-
движный атом водорода:
—N=CH2 4-HN—СО----*- —XH-CHj—N—СО—
1 1
Стадия С. Для получения неплавких смол, нерастворимых и
главным образом мало чувствительных к действию воды, необ-
ходимо, чтобы среда была кислой. По мере протекания конден-
сации увеличивается количество групп —СНгОН. Необходимым
условием максимального смолообразования является то, чтобы
отношение U/F было меньше единицы, так как монометилолмо-
чевина образует не смолы, а только аморфные массы в виде
нерастворимых хлопьев. На холоду диметиленэфирные связи,
образующиеся по реакции
—NH—СН2:ОН"+ HOCHs-NH----— NH—СН2—О—СН2—NH-
не нарушаются, и обнаруживается только выделение воды. При
повышенной температуре или в сильнокислой среде эти связи
разрушаются, как это наблюдалось при конденсации с фенолом.
Формальдегид освобождается в результате следующего превра-
щения:
R—СН2—О—СН2—R' ->- R-CH2-R' 4- СН2О
334 Гл. X. Мочевино-формальдегидные смолы и их аналоги
Сетка становится более частой и вследствие этого увеличи-
вается плотность смолы, ее твердость и сопротивление действию
различных химических реагентов.
Строение мочевино-формальдегидных смол. При взаимодей-
ствии мочевины с формальдегидом образуется смола. В резуль-
тате последовательных превращений начального продукта — ме-
тилолмочевины — происходит прогрессирующее уменьшение рас-
творимости по мере потери воды и альдегида. Что же известно
о структуре конечных продуктов?
Первые авторы, которые упоминают о мочевино-формальде-
гидных смолах, получали их случайно как нежелательные про-
дукты реакции. В настоящее время структура и точный меха-
низм образования этих смол еще далеко не ясны.
Можно допустить, что в предельной идеальной структуре ос-
новным звеном является остаток диметиленмочевины:
I
----N—СО—N—СН2—N—СО—N— СН2----
СН2 СН2 СН2
I I I
N—СО—N—СН2— N—СО—N-----
I I
СН2 СН,
I I
Конечно, логично предположить, как это сделал Хувинк, что
внутри сетки остается небольшое количество метилольных ра-
дикалов. Некоторые из них остались свободными, не оказав-
шись ни вблизи подвижного атома водорода, ни вблизи второй
свободной метилольной группы. Такие «дефекты» структуры ха-
рактерны не только для мочевино-формальдегидных смол —они
имеются в большей части материалов, образованных из простей-
ших структурных звеньев, соединившихся между собой в ре-
зультате конденсации или полимеризации. «Дефекты» струк-
туры влекут за собой значительное ослабление механических
свойств по сравнению с величинами, рассчитанными исходя из
прочности первичных или вторичных связей. Они оказывают
влияние не только на механические свойства, но и на стойкость
к воде и химическим реагентам, гак как в дефектных участках
находятся гидрофильные и химически активные группы.
Интересно выяснить, в какой мере возможно проведение ре-
акции таким образом, чтобы оставалось меньшее число нежела-
тельных остатков. Медленное проведение реакции, когда сна-
чала образуются изолированные цепи, которые затем постелен-
Мочевино-формальдегидные смолы
335
но соединяются между собой, конечно, наиболее благоприят-
но. При этом необходимо задерживать образование сетки, чтобы
в реакции могли участвовать все реакционноспособные группы.
Мочевино-формальдегидные смолы для лаков и красок
Водорастворимые мочевино-формальдегидные смолы. Водные
растворы метилолмочевины или продуктов низкой степени мх
конденсации, после того как была установлена их относитель-
ная стабильность, были предложены в качестве лаков.
Как связующие вещества для красок водорастворимые мо-
чевино-формальдегидные смолы не получили того широкого
применения, которого можно было ожидать, учитывая их твер-
дость, прозрачность, бесцветность, негорючесть, стойкость к хи-
мическим воздействиям и т. д. Достижения последних лет в
этой области, по нашему мнению, должны снова вызвать внима-
ние к этим продуктам.
Мочевино-формальдегидные смолы, растворимые в органи-
ческих растворителях. Несмотря на определенные достоинства
водорастворимых мочевино-формальдегидных смол, применяв-
шихся в лакокрасочной промышленности, они плохо совмеща-
лись со сложной гаммой продуктов, используемых этой отраслью
промышленности. Общеизвестно, что свойства пленкообразую-
щего вещества могут быть улучшены путем введения в него
других природных или синтетических смол, которые в конце
концов придают пленке комплекс свойств, требуемых для тех-
нических целей. 'Поскольку большая часть промышленных свя-
зующих веществ применяется в виде растворов в органических
растворителях, полезно добиться растворимости в них и моче-
вино-формальдегидных смол, чтобы можно было вводить эти
смолы в эфироцеллюлозные, глифталевые, масляные и другие
лаки.
Высокая реакционная способность метилолмочевины позво-
ляет преобразовывать ее молекулу путем взаимодействия груп-
пы —СН2ОН с различными функциональными группами, ис-
пользуя, например, следующие реакции:
Дегидратация при взаимодействии со второй метилольной
группой:
—NH—СН2;ОН + Н:ОСН2—NH-->- —NH—СН2—О—СН2—NH— + Н2О
Этерификация спиртом:
—NH—СН2 ОН + HpR >- —NH—СН2—OR + Н2О
Этерификация кислотой:
-NH—СН2'ОН + HJOCOR - >- —NH—СН2—OCOR + Н2О
336 Гл. X. Мочевино-формальдегидные смолы и их аналоги
Взаимодействие с аминами, сопровождающееся выделением
NH3:
—NH—CH2O:H4-H2N;—R —- —NH—CH2—OR-f-NH2
Взаимодействие с амидами с выделением NH3:
—NH—CH2OB+H2N:COR >- —NH—CH2—OCOR + NH.
Присоединение ацетилена:
—NH—CH2 ОН + HteCH -» — NH—CH2—C==CH > H.O
Эфиры моно- и диметилолмочевины. Для придания рассмат-
риваемым смолам растворимости в органических растворителях
чаще всего пользуются .реакциями этерификации. Связывание
гидроксильных групп обеспечивает смолам стабильность, кото-
рой часто недоставало .продуктам конденсации в водной среде;
при этом сохраняется возможность проведения последующей
конденсации до стадии С. В распоряжении химика вместо двух
основных исходных веществ оказывается большое количество
разнообразных спиртов, фенолов и различных, оксипроизвод-
ных, с помощью которых можно получать самые разнообразные
комбинации для придания конечным продуктам желаемых
свойств.
Моно- и диметилолмочевины являются спиртами, образую-
щими эфиры следующих типов:
zNH2 zNH-CH2OH znh—ch2or
ос/ ос/ oc<
w-сад \nh-ch2or xNH-CH2OR
(R— алкильный, арильный или аралкильный остаток).
Все эти эфиры при нагревании и при действии кислот обра-
зуют смолистые продукты. Нашример, эфиры диметилолмоче-
вины с выделением (п—1) ROH образуют цепь:
RO- CH2-N—СН» 1 СО —N—СН2- 1 СО —N— - СО —II
1 NH 1 NH NH
1 ch2or ch2or 1 ch2or_ п
Такие линейные молекулы образуются в относительно мяг-
ких условиях конденсации. При более высокой температуре по-
лучаются неплавкие и нерастворимые продукты, образование
Мочевино-формальдегидные смолы
337
которых вновь сопровождается выделением спирта. Реакция
будет происходить за счет алкоксигрупп, реагирующих со вто-
ричными аминогруппами:
i I
HN-CH2OR n—ch2or
+ -* |
----HN—CH2OR------------------------HN-CHg
I I
При этом будут образовываться также поперечные связи,
что необходимо для придания твердости и химической инерт-
ности. От числа реакционноспособных групп и от степени за-
вершенности реакций внутренней этерификации зависит число
образующихся связей, а следовательно, и твердость получае-
мой пленки.
В условиях промышленного производства метилольные про-
изводные вообще не выделяют, а сразу после образования эте-
рифицируют, причем операции присоединения формальдегида
и этерификации метилольных групп часто выполняются одно-
временно. Для этерификации чаще всего применяют бутиловый
спирт.
При конденсации берут примерно 2 моля формальдегида на
1 моль мочевины. Процесс большей частью проводят в щелоч-
ной среде, прибавляя бутиловый спирт в конце первой фазы
реакции или вводя его с самого начала. Во второй фазе реак-
ции доводят pH реакционной смеси до значений, соответствую-
щих кислой среде, отгоняют воду и отделяют бутиловый спирт
от дистиллята путем декантации. По мере удаления воды эте-
рификация становится все более и более полной, но в то же
время происходят реакции конденсации между реакционноспо-
собными группами и, вероятно, группы —NH—GH2OH обра-
зуют метиленовые мостики —NH—<СНг—NH—. При этом оказы-
вается, что по мере развития процесса смолообразования за-
трудняется реакция этерификации. бутиловым спиртом. Слиш-
ком быстрое смолообразование приводит к присоединению мень-
шего количества бутилового спирта, а следовательно, к умень-
шению совместимости смолы с углеводородами. Иногда проис-
ходит даже желатинирование массы.
После полной отгонки воды получается раствор бутаноли-
з'ированной смолы в избытке бутанола.
Были испытаны также спирты, растворимые в воде (мети-
ловый, этиловый и .пропиловый), но оказалось, что для улуч-
шения растворимости смол в углеводородах и повышения их во-
достойкости целесообразнее вводить возможно более длинные
22—156
338 Гл. X. Мочевино-формальдегидные смолы и их аналоги
парафиновые цепи, чтобы уменьшить относительное .влияние ди-
польного момента молекул мочевины. Было установлено, что
желательное удлинение алифатической цепи может быть достиг-
нуто путем полной или частичной замены бутанола высшими
жирными спиртами с числом атомов углерода не менее 8. Таки-
ми спиртами являются природные цетиловый или миристиновый
спирты, а также спирты, полученные гидрогенизацией насыщен-
ных или из ненасыщенных жирных кислот, выделенных из ма-
сел или животных или растительных жиров. Соответствующие
спирты образуются также при гидрировании кислот, получен-
ных окислением парафинов.
Вместо алифатических спиртов могут быть использованы
также алициклические спирты, а именно: циклогексанол, мен-
тол, абиетинол и др.
Из сказанного видно, что число соединений, пригодных для
получения ценных пленкообразователей на основе метилолмоче-
вин, весьма велико,, и трудно определить границы синтеза подоб-
ных продуктов в будущем. По мере развития химической про-
мышленности становятся доступными в качестве исходного
сырья продукты, выпускавшиеся долгое время лишь в лабора-
торных масштабах. Это позволяет осуществлять значительно
более разнообразные промышленные синтезы и расширять об-
ласти применения получаемых продуктов.
Мочевино-формальдегидные смолы, модифицированные по-
лиэфирами. Этерификация метилолмочевины может быть с успе-
хом осуществлена путем взаимодействия со спиртовыми гидр-
окислами, оставшимися свободными при конденсации много-
атомных спиртов с многоосновными кислотами. Для этого часто
применяются глифталевые, смолы, а также многочисленные
аналогичные смолы такого типа.
Диметилолмочевина, вступая в реакцию с продуктами кон-
денсации фталевого ангидрида с гликолями, может образовать
модифицированную смолу, строение которой схематически изо-
бражается следующей примерной формулой:
---N—СН2----
СО
---N-CH2-O-CH2-CH2—
---СН2—СН2—ООС—L I
Ходжинс и Ховей1, специально исследовавшие такие смолы,
предполагают, что одновременно с образованием простых 'Эфи-
ров метилольные остатки этерифицируются также оставшимися
свободными карбоксильными группами молекулы полиэфира.
Процесс ведут путем смешения бутанолизированной мочевино-
Мочевино-формальдегидные смолы
339
формальдегидной смолы с глифталевой смолой, имеющей высо-
кое кислотное число, или же путем конденсации мочевины с
формальдегидом в присутствии полиэфира.
Было испытано большое число сочетаний многоатомных
спиртов и многоосновных кислот, выпускаемых в настоящее
время в промышленном масштабе. Оказалось, что эластичность,
тепло- и водостойкость, стойкость к атмосферным воздействиям
и твердость получаемых продуктов зависят как от природы
исходных веществ, так и от способа конденсации. Из спиртов,
пригодных для применения, укажем пентаэритрит, гексантриол,
триметилолэтан, триметилолпропан, бутиленгликоль. Соответ-
ствующие двухосновные кислоты обычно принадлежат к пара-
финовому или циклопарафиновому ряду и должны содержать
по меньшей мере пять атомов углерода. Наиболее употреби-
тельными кислотами являются глутаровая, адипиновая,' себаци-
новая, пимелиновая, кетопимелиновая и циклогексанондиуксус-
ная. Избыток кислотности часто оказывается вредным, .поэтому
рекомендуется вести реакцию конденсации при кислотном
числе 30—40.
Обычно принято смешивать мочевино-формальдегидную смо-
лу с глифталевой смолой, модифицированной в той или иной
степени жирными кислотами высыхающих или невысыхающих
масел. В этом случае реакция между двумя компонентами,
по-видимому, осуществляется в конце процесса варки лака —
при высокой температуре. Но более высокая гомогенность лака
достигается при проведении предварительной конденсации двух
смол.
Мочевино-формальдегидные смолы, модифицированные фе-
ноло-альдегидными смолами, и прочие модифицированные
смолы. Известно, что формальдегид реагирует с фенолами по
реакциям, аналогичным реакциям мочевины. В щелочной среде
подвижный атом водорода принимает участие в образовании
радикала — СН2ОН и получается моно- .или диметилолфенол.
Эти фенолоспирты в присутствии диметилолмочевины или ее
бутилового эфира вступают в реакции внутримолекулярной кон-
денсации, приводящие в конце концов к образованию комплекс-
ных смол. Твердость таких смол обусловливается участием мо-
чевины, а водостойкость — участием фенолов.
Смолы такого типа, полученные конденсацией бутанолизиро-
ванной мочевино-формальдегидной смолы с диметилолкрезолом
в циклогексан оловом растворе, применяются для эмалирования
электрических проводов.
Наибольшая эластичность пленки достигается при совмеще-
нии феноло-мочевино-формальдегидного комплекса, этерифици-
рованного бутанолом, с продуктами конденсации типа алкид-
ных смол с кислотным числом, очень близким к 50. Этот же
22*
340 Гл. X. Мочевино-формальдегидные смолы и их аналоги
принцип используется при изготовлении лаков, отверждаю-
щихся на холоду в присутствии кислоты.
В заключение отметим, что арилсульфамиды, большое чис-
ло дикарбоновых кислот, ацетилен, некоторые хлорзамещен-
ные альдегиды или кетоны, аминокислоты, эфиры глицерина,
сероуглерод, казеин, виниловые или акриловые производные
образуют с мочевино-формальдегидной смолой модифицирован-
ные смолы, не нашедшие еще применения в массовом масштабе
в лакокрасочной промышленности.
МЕЛАМИНО ФОРМАЛЬДЕГИДНЫЕ СМОЛЫ
Меламин ведет себя по отношению к формальдегиду почти
так же,, как мочевина. Следовательно, данные, полученные для
мочевино-формальдегидных смол, применимы часто к мелами-
но-формальдегидным смолам. Однако необходимо учесть, что
меламин представляет собой циклическое соединение, содержа-
щее три симметричные аминогруппы, что увеличивает склон-
ность к быстрому смолообразованию с образованием трехмер-
ной сетчатой структуры.
В присутствии воды в нейтральной или щелочной среде ме-
ламин легко присоединяет три молекулы формальдегида с обра-
зованием симметричной молекулы:
NH—СН2ОН
I
С
ьЛ \
II I
НОСН2—HN—С С—NH—СН2ОН
Введением в реакцию большого избытка формальдегида
может быть достигнуто замещение шести атомов водорода с
получением гексаметилолмеламина.
Метилольные производные меламина представляют собой
кристаллические вещества, мало растворимые в воде при обыч-
ной температуре, но легко растворимые в кипящей воде. Они
более устойчивы, чем соответствующие производные мочевины.
Температура плавления гексаметилолмеламина не установ-
лена, так как этот продукт разлагается ниже температуры его
плавления. Начиная от 90°, происходит отщепление формальде-
гида. Путем быстрого нагревания можно определить только
примерный интервал температуры плавления, который находит-
ся между 140 и 170°.
Установлено, что реакция образования метилольных произ-
водных при 60—80° в нейтральной или слегка щелочной среде
Меламино-формальдегидные смолы
341
является реакцией первого порядка. Реакция образования ме-
тиленмеламинов в нейтральной или предпочтительно кислой
среде при 60—90° протекает как реакция второго порядка.
Скорость реакции в нейтральной или слабощелочной среде
невелика; .реакция весьма заметно ускоряется даже в слабокис-
лой среде. Прибавление избытка щелочи также увеличивает
скорость реакции.
Продолжительность конденсации и температура влияют на
строение смол, изолированных после отделения воды. Так, при
быстром охлаждении растворов, содержащих меламин и форм-
альдегид, получается большое количество кристаллизующихся
продуктов, представляющих собой метилолмеламины. После
более продолжительной конденсации образуется гидрофобная
смола, выпадающая из раствора. Пока глубина полимеризации
относительно мала,, эти гидрофобные смолы способны перехо-
дить в раствор в присутствии кислот. Прибавление едкого натра
или едкого кали вызывает их осаждение. Если продолжать на-
гревание, смолистые осадки становятся твердыми, неплавкими
и нерастворимыми.
Таким образом, можно различить несколько стадий про-
цесса. Сначала происходит образование метилолмеламинов, за-
тем быстро наступает стадия предварительной конденсации, в
процессе которой .происходит повышение вязкости при сохране-
нии гомогенности массы и растворимости ее в воде; при быст-
ром охлаждении образуется нерастворимый осадок, впоследст-
вии отделяющийся. На следующей стадии масса уже не может
быть разбавлена водой без образования коллоидной мути. При
дальнейшем нагревании появляются две совершенно отчетливые
фазы. Наконец, масса желатинируется.
Как и при получении мочевино-формальдегидных смол,
смолообразование осуществляется путем взаимодействия мети-
лольных групп или раскрытия метилениминовых двойных свя-
зей. Метилольные группы сначала постепенно дегидратируются
с образованием эфирных мостиков —СН2—О—СН2—. В зави-
симости от температуры и pH эти мостики превращаются бо-
лее или менее полно в метиленовые мостики —СН2— с выделе-
нием формальдегида:
—NH—СН2—О—СН2—NH------>- —NH—СН2—NH— + СН2О
Метиленовый мостик может образоваться и непосредствен-
но, если радикал — СН2ОН реагирует с подвижным атомом
водорода .свободной аминогруппы.
В кислой среде нельзя исключить возможность образования
связей за счет азометинрвых групп —N = CH2 меламинмети-
дов, которые не были выделены в мономерном состоянии. При
взаимодействии двойной азометиновой связи с подвижным ато-
342 Гл. X. Мочевино-формальдегидные смолы и их аналоги
мом водорода должен образоваться метиленовый мостик путем
миграции атома водорода согласно схеме:
..—О—СН2—NH + CH2=N ••—О—СН2—N—СН2—NH
II II
• • •
Были изучены смолы, полученные не из заранее выделен-
ного триметилолмеламина, а из смесей меламина и формальде-
гида2, взятых в молярном ‘отношении 1 :3. Реакционную смесь
при pH=7,2 нагревают в течение нескольких часов с обратным
холодильником. После перегонки в вакууме (получается про-
зрачная смола, растворимая в 0,5—1,0 вес. ч. воды с образова-
нием ‘бесцветного вязкогб раствора. Пробы этой смолы нагре-
вали при 100° в течение различного времени и определяли по-
терю веса, которая увеличивалась со временем при одновремен-
ном уменьшении растворимости в пиридине. В процессе отверж-
дения (продукта производились определения свободного форм-
альдегида. После 5 час. нагревания при 100° никаких следов
формальдегида обнаружено не было, а при 130° выделившиеся
количества были незначительны. Из этого можно заключить,
что мостики —СНг—О—СН2— в указанных условиях не были
разрушены.
Готовые смолы содержат свободные метилольные группы в
количестве, зависящем от степени конденсации, достигнутой до
отверждения смолы. Содержание метилольных групп тем мень-
ше, чем глубже степень предварительной конденсации.
Полученные результаты3 приведены ниже:
Продолжительность конденса-
ции (при 100° и pH=5), часы 0
Число метилольных групп на
молекулу меламина после
отверждения............... 2,03
Смешиваемость с водой . . . —
1,80
1,43
1 объем рас-
твора сме-
шивается
с 5 объ-
емами воды
1,16
После охла-
ждения
до 78° об-
разуются
2 слоя
1
Следует также отметить, что чем меньше отношение количе-
ства меламина к формальдегиду, тем ‘больше метилольных
групп содержится в готовой смоле.
Водорастворимые меламино-формальдегидные смолы. Эти
смолы быстро нашли применение в лакокрасочной промышлен-
ности вследствие наличия опыта (работы с аналогичными моче-
вино-формальдегидными смолами.
Как мы видели выше, водорастворимые смолы получаются
путем конденсации при нагревании с обратным холодильником
в слабокислой .или слабощелочной среде.
Меламино-формальдегидные смелы
343
Обычно эти смолы сохраняют растворимость значительно
дольше, чем мочевино-формальдегидные, особенно, если они
получены в щелочной среде. Их рекомендуется хранить при тем-
пературе, не превышающей 15°. Растворение их непосредственно
перед применением облегчается в присутствии кислот; в присут-
ствии едкого натра происходит осаждение смолы. Прибавление
кислоты к раствору ускоряет отверждение лаковой пленки, но,
начиная с некоторого количества сильной кислоты, процесс
отверждения пленки больше не ускоряется, а при определенном
избытке кислоты — даже замедляется. Относительно слабые
кислоты или вещества со слабокислой реакцией вызывают уско-
рение отверждения, быстро увеличивающееся .по мере прибав-
ления отвердителя, а затем уменьшающееся; далее скорость
отверждения перестает зависеть от количества введенной кис-
лоты. В случае слабых кислот отверждение можно регулиро-
вать, изменяя отношение меламин—формальдегид; при увеличен-
ном количестве формальдегида слабая кислота ведет себя так
же, как сильная. Повышение температуры также сближает кри-
вые для слабых и сильных кислот.
Для получения краски смолу растирают с такими пигмен-
тами, как литопон или каолин. Получаются матовые покрытия,
устойчивые к действию воды и к трению. Если поверхность
окрашиваемого предмета имеет щелочную реакцию, полезно
сначала нанести слой разбавленной кислоты или раствора ам-
монийной соли сильной кислоты.
Следует упомянуть также о катионных коллоидных раство-
рах мел амино-форм альдегидных смол, хотя эти растворы мало
применяются в лакокрасочной промышленности. Если 15%-ный
водный раствор метилолмеламина, содержащего более 2,5 молей
формальдегида на 1 моль меламина, подкислить до pH=0,5—
3,5, то происходит полимеризация — частицы увеличиваются и
достигают через некоторое время коллоидных размеров. Ча-
стицы смолы оказываются положительно заряженными и, если
суспензию подвергнуть электрофорезу, направляются к катоду.
Это дает возможность наносить смолу в виде тонкой пленки
на металлическую подложку4.
Такие дисперсии находят применение в качестве подслоя
для нанесения лаков на целлофан. Перед внесением пленки из
регенерированной целлюлозы в дисперсию смолы к этой диспер-
сии прибавляют глицерин в количестве, достаточном для при-
дания лаковой пленке эластичности. Сушка производится в
течение нескольких минут.
Этерифицированные меламино-формальдегидные смолы. Ме-
тилольные группы, присоединенные к меламиновому циклу,
легко этерифицируются спиртами, если вести (реакцию при на-
гревании и в кислой среде. В результате получаются продукты
344 Гл. X. Мочевино-формальдегидные смолы и их аналоги
конденсации, растворимые в органических растворителях. Это —
прекрасные связующие вещества, производство которых родст-
венно производству аналогичных мочевино-формальдегидных
смол. Путем подбора спирта и степени алкоксилирования мож-
но придать смоле хорошую растворимость в органических рас-
творителях и высокую водостойкость при вполне достаточной
способности к отверждению.
Совместимость с углеводородами и эластичность пленки мо-
гут быть улучшены путем применения спиртов с довольно вы-
соким молекулярным весом.'Но эти спирты реагируют медлен-
но, что приводит к недостаточной степени этерификации в мо-
мент, когда приходится прекратить реакцию во избежание
желатинирования. Эти две реакции — смолообразование и эте-
рификация могут быть до некоторой степени разделены, если
создать для этого соответствующие условия. Действительно, при
температуре ниже 50° и в присутствии относительно больших
количеств сильной кислоты этерификация происходит энергич-
но, в то время как смолообразование почти не протекает. Путем
более или менее полной нейтрализации с последующим повы-
шением температуры до 90° можно превратить образовавшиеся
эфиры в смолы. Для осуществления этого способа в качестве
высокомолекулярного спирта должен применяться моно- или
диглицерид жирной кислоты в смеси со спиртом, содержащим
от одного до пяти атомов углерода. Такой спирт участвует в
реакции образования смешанного эфира и играет в то же
время роль разбавителя. Получаемые лаки можно разбавлять
алифатическими углеводородами. После горячей сушки они
дают эластичные пленки, мало чувствительные к действию
воды.
Если исходные глицериды получены из высыхающих жир-
ных кислот, то лаки высыхают на воздухе при обычной тем-
пературе после прибавления сиккатива5.
Описанная технология применима также к спиртам с мень-
шим молекулярным весом; в этом случае, при условии дове-
дения предыдущей стадии до образования гексаметилолмел-
амина, достигается более высокая степень алкоксилирования.
Применение большого избытка формальдегида в присутствии
небольших количеств кислоты приводит к высокой степени
алкоксилирования одноатомными спиртами Ci—С4.
-Модифицированные меламино-формальдегидные смолы. Мел-
амино-формальдегидные смолы, этерифицированные спиртами,
содержащими от одного до четырех атомов углерода, образуют
твердые, водостойкие, но неэластичные лаковые пленки. Уста-
новлено, что продукты конденсации многоатомных спиртов с
МНОГООСНОВНЫМ1И кислотами являются прекрасными пластифи-
каторами для этих смол.
Меламино-формальдегидные смолы 345-
Модификация меламино-формальдегидных смол полиэфи-
рами легко осуществима, если сначала частично этерифициро-
вать многоатомные спирты многоосновными карбоновыми кисло-
тами, а затем этерифицировать метилолмеламины, используя сво-
бодные гидроксильные группы полиэфира. Получение сначала
простых эфиров действием многоатомных спиртов с последую-
щим образованием сложных эфиров дает худшие результаты.
Промышленные алкидные смолы, полученные этерифика-
цией многоатомных спиртов многоосновными и одноосновными
кислотами, оказывая на меламино-формальдегидные смолы
пластифицирующее действие и улучшая их водостойкость,,
одновременно сообщают им способность к высыханию.
Ввиду того, что алкоксигруппы этерифицированных мелами-
но-формальдегидных смол могут вступать в обменные реакции
со спиртовыми остатками полиэфира, заменяя .в этом отноше-
нии метилольные группы, чаще всего исходят из продуктов, бо-
лее или менее полно этерифицированных спиртами Ci—С4.
Лишь немногие многоатомные спирты, многоосновные и
одноосновные кислоты с длинными цепями не были упомянуты
в многочисленных патентах, посвященных этому вопросу; выбор
реагирующих веществ и способа проведения реакций зависит
от заданных свойств лака. Из экономических соображений
обычно используют в качестве исходных спиртов, в зависимости
от соотношения цен, глицерин, этиленгликоль или пентаэритрит.
В качестве многоосновной кислоты основным продуктом остает-
ся фталевый ангидрид, хотя промышленность органической хи-
мии все больше и больше осваивает производство других при-
годных для данной цели соединений с несколькими карбоксиль-
ными группами.
Получаемые модифицированные смолы растворяются в
обычных органических растворителях и выдерживают разбавле-
ние самыми разнообразными разбавителями. Они совместимы
также с другими пленкообразующими веществами, которым
сообщают соответствующие свойства; упомянем, например, нит-
рат и ацетат целлюлозы, некоторые виниловые производные
и т. п.
Обычно желательно, чтобы .покрытие отверждалось как мож-
но скорее. Активные ускорители для рассмотренных выше лаков
были найдены. Наряду с кислотами, являющимися классически-
ми ускорителями, рекомендуются хлорированные триазины.
Хлорангидрид циануровой кислоты, растворенный в диоксане,
очень активен даже в концентрации 1 % по отношению к смо-
лам6.
Улучшение свойств меламино-формальдегидных смол, со-
вмещенных с полиэфирными, достигается при взаимодействии,
стирола с алифатическими цепями кислот, имеющих три со-
-346 Г л. X. Мочевино-формальдегидные смолы и их аналоги
•пряженные двойные связи, как, например, в тунговом или ойти-
сиковом масле. Сополимеры стирола с продуктами совместной
конденсации многоатомных спиртов с многоосновными кислота-
ми и жирной кислотой с тремя двойными связями реагируют
при нагревании с меламиновыми смолами. Получаемые плен-
кообразующие вещества после сушки дают пленки с хорошей
адгезией, механической прочностью, эластичностью и твер-
достью7.
Кроме модифицированных смол, рассмотренных выше в разде-
ле о 'мочевине (в этих смолах мочевину можно легко заменить
меламиноьм), упомянем также несколько аналогичных продуктов.
Способность метилольной группы, как спиртовой, к образо-
ванию простых эфиров, дает основания полагать, что возможно
также образование сложных эфиров. Такие эфиры, действитель-
но, легко образуются с кислотами, имеющими высокий молеку-
лярный вес. Однако при непосредственной этерификации за-
метная склонность метилолмочевины к гомополиконденсации
при повышенной температуре быстро приводит к желатиниро-
ванию. Чтобы избежать коагуляции реакционной массы, прихо-
дится прекращать нагревание, когда реакция образования
сложного эфира еще далеко не закончена. Если же исходить из
метиловых или бутиловых эфиров, более стойких, чем незаме-
щенные метилолмеламины, становится возможным .получение
при 100—200° сложных эфиров различных кислот8. При этом
протекает следующая реакция:
R—СООН +JCH3OCH2-NH— — R—СООСН2—NH— + СНо0Н
В результате образуется смола, твердая или вязкая, окра-
шенная или неокрашенная, в зависимости от взятой кислоты.
Так, с линолевой кислотой получается прозрачная масса с кон-
систенцией бальзама, растворимая в обычных растворителях.
Пленка высыхает в течение 15—20 час. при обычной темпера-
туре, образуя эластичное и прочное покрытие. При этерифика-
ции олеиновой кислотой образуется маслообразный .продукт с
более интенсивной коричневой окраской. С бензойной кислотой
получается хрупкая прозрачная смола, слегка желтеющая в
конце этерификации.
Меламино-формальдегидные смолы применяются также в
сочетании с продуктами конденсации замещенных фенолов с
формальдегидом. Особенно рекомендуются для этой цели про-
стые эфиры метилольных производных фенола и триазина. Реак-
ция осуществляется при 100—150° с отгонкой растворителя. Для
придания полученной смоле хорошей растворимости в высыхаю-
щих маслах следует брать относительно небольшое количество
меламиновой смолы при высокой степени этерификации как ме-
•тилолмеламина, так и метилолфенола, и проводить реакцию
зпри возможно более низкой температуре9.
Эмульсионные смолы
347
Процесс получения маслорастворимых смол еше более упро-
щается, если вводить в реакцию гексаметилол мел амин с фено-
лом, замещенным преимущественно в пара-положении. Процесс
проводят при нагревании в углеводородной среде — в толуоле
или в ксилоле. Кислые конденсирующие агенты ускоряют реак-
цию, но не являются необходимыми; катализаторами реакции
могут служить также карбонаты щелочных металлов10.
ЭМУЛЬСИОННЫЕ смолы
В некоторых случаях требуется приготовление пленкообра-
зующих веществ в виде эмульсий. В текстильной промышлен-
ности, например, нанесение эмульсии на ткань методом набив-
ки или тиснения часто дает более эластичную пленку, чем обыч-
ный пластифицированный лак. Эмульсия покрывает волокна
каждое в отдельности, не связывая их жестко между собой.
Кроме того, и некоторые красители легче всего использовать в
водной среде. Наконец, при применении эмульсий не расходуют-
ся органические растворители.
Вопросами эмульгирования карбамидных смол и использо-
вания эмульсий для печатания на тканях начали заниматься
сравнительно давно. Исходную мочевино-формальдегидную смо-
лу этерифицировали и растворяли в растворителе, не смеши-
вающемся во всех отношениях с водой. Возможен следующий
метод изготовления эмульсии. К вязкому циклогексановому
раствору мочевино-формальдегидной смолы прибавляют этил-
целлюлозу и смесь выдерживают при 80° до полного растворе-
ния или по крайней мере до получения однородной смеси. При-
бавляют полиэфир и продолжают нагревание до полной гомо-
генизации смеси. В энергично размешиваемую массу затем
постепенно вводят аммиачный раствор Казеина. Полученную
при этом эмульсию стабилизуют, пропуская через коллоидную
мельницу. Введение пигментов легко осуществляется на крас-
котерке11. Для закрепления печати на тканях (т. е. для прида-
ния смоле нерастворимости) материал выдерживают в течение
5 мин. при 150°. Рисунок получается замечательно четким и
прекрасно выдерживает стирку.
Бутанолизированные меламино-формальдегидные смолы,
обработанные аналогичным способом, также дают эмульсии,
пригодные для печати и крашения текстильных материалов.
РАЗЛИЧНЫЕ АМИНО-АЛ ЬДЕГИДНЫЕ СМОЛЫ
Описав мочевино-формальдегидные и меламино-формальде-
гидные смолы, мы охватили в значительной мере самые важные
амино-альдегидные смолы, используемые в настоящее время
для лаков .и красок. В то же время большое значение имеют и
348 Гл. X. Мочевино-формальдегидные смолы и их аналоги
азотсодержащие материалы на основе других исходных ве-
ществ, например анилина, дициандиамида, п-толуолсульфами-
да. Из более сложных .продуктов были изучены те, которые об-
разуют смолы с формальдегидом или другими альдегидами.
Быстрое расширение областей применения пластических
масс требует все большего и большего разнообразия смол, при-
годных в различных условиях применения; при этом трудно
предполагать, что новые продукты долго сохранят то довольно
незначительное место, какое они занимают теперь. Поэтому
представляется полезным хотя бы кратко рассмотреть их в этой
главе.
Производные анилина. Анилин при конденсации с ацеталь-
дегидом или формальдегидом образует довольно ценные смолы,
давно уже применяемые в электропромышленности.
Производные п-толуолсульфамида. Многие сульфамиды спо-
собны при конденсации с формальдегидом образовывать смолы;
из них чаще всего применяется п-толуолсульфамид — продукт,
производимый промышленным способом в больших количест-
вах.
Молекула п-толуолсульфамида монофункциональна и не мо-
жет поэтому образовывать с формальдегидом твердые и неплав-
кие смолы. Молекулярные цепи получаются в результате кон-
денсации сначала в щелочной среде с образованием метилоль-
ных производных, а затем в кислой среде, где путем последо-
вательных дегидратаций получается линейный полимер:
Получаемые смолы термопластичны, прозрачны, имеют
полужидкую консистенцию и невысокую температуру плавле-
ния, бесцветны или слегка окрашены в желтый цвет. Они не
растворяются ни в воде, ни в разбавленных кислотах, но в ще-
лочах растворяются, несколько разлагаясь. Растворяются они во
многих органических растворителях: в спиртах, кетонах, про-
стых эфирах, ароматических углеводородах, но нерастворимы в
алифатических углеводородах и высыхающих маслах. Их рас-
творяют также глицерин и уксусная кислота.
Наряду с линейными полимерами не исключена возможность
образования при конденсации п-толуолсульфамида с форм-
альдегидом неполимеризующихся циклических производных
Различные амино-альдегидные смолы
349
меламина; такое предположение было высказано Мак-Мастером
,и Хьюгом. Циклизация при pH>7, вероятно, происходит при
взаимодействии трех молекул промежуточно образующегося
метиленсульфамида, в результате чего получается следующее
•соединение:
СН2
НзС—/~\-SO2—N ^N—SO2—S—< СН3
। | \==/
* Н2С сн2
I
so2
9
СН3
п-Толуолсульфамидо-формальдегидные смолы совместимы с
нитратом и ацетатом целлюлозы, а также с этилцеллюлозой.
Производные дициандиамида. Дициандиамид, или цианогу-
анидин, получается из цианамида путем соответствующего пре-
вращения в водном растворе; максимальная скорость .превра-
щения достигается при pH =9,6. Реакция может быть выражена
схемой:
/NH2
2H2N—CN — HN=C<_
'Ч'Н-CN
Это бесцветные моноклинные кристаллы, плавящиеся при
207°. Дициандиамид — малостойкое вещество, разлагающееся
в водном растворе, начиная с температуры 80°. В кислой среде
он превращается в весьма гигроскопичный дициандиамидин;
конденсируется также предпочтительно в кислой среде при
рН = 8—10 с образованием гидрофобных смол, нашедших глав-
ное применение для отделки текстильных материалов. Его спо-
собность отверждаться в щелочной среде может быть полезной
при нанесении на грунты основного характера. Дициандиамид
часто используют для совместной конденсации с мочевиной,
меламином или фенолом.
Ацетилендимочевина. Газообразный ацетилен под давлением
реагирует с диметилолмочевиной как кислота; в качестве ката-
лизатора применяется хлористая медь. В среде метанола этим
путем были получены смолы. Можно предположить для соот-
ветствующего мономера следующее строение:
НОСН2—NH—СО—NH—СН2—С==СН
350 Гл. X. Мочевино-формальдегидные смолы и их аналоги
Но при взаимодействии мочевины с глиоксалем получаете®
ацетилендимочевина, или глиоксальдиуреид, строения
NH—СН—NH
ОС\ | /СО
NH—СН—NH
Наличие четырех атомов водорода, присоединенных к ато-
мам азота, приводит к образованию стабильного тртрамети-
лольного соединения, растворимого в воде, в то время как аце-
тилендимочевина мало растворима. При конденсации в щелоч-
ной среде получаются водные растворы, которые в тонких слоях
под действием температуры 100° отверждаются, образуя про-
зрачные пленки, нерастворимые в воде и растворителях.
Аналогичные смолы получаются при конденсации в слабокис-
лой среде. Такие продукты выпускаются под названием каурита
AF. Они легко зтерифицируются спиртами в присутствии кислот,
причем получаются смолы, совместимые с эфирами целлюлозы12.
Замещенные мочевины. Замещение в мочевине водородных-
атомов аминогруппы алифатическими радикалами препятст-
вует образованию смолы, так как при этом уменьшается число-
подвижных атомов водорода. Однако некоторые замещенные
мочевины оказались пригодными для производства смол. В част-
ности, при конденсации диэтилдифенил мочевины с формальде-
гидом в сильнокислой сернокислотной среде получается смола,
пригодная для производства лаков13.
Уреилметиламины. Монометилолмочевина реагирует на хо-
лоду с аммиаком или с аминами, давая при этом вещества, спо-
собные, как и мочевина, реагировать с формальдегидом.
Аммиак или амин (первичный или вторичный) медленно добав-
ляют к концентрированному раствору монометилолмочевины.
Температура поддерживается между —20 и +9°. При соотно-
шении 1 моль аммиака на 3 моля монометилолмочевины полу-
чается триуреилметиламин:
zCH2—NH—CONH2
N—СН2—NH—CONH2
\СН2—NH—CONH2
Такие производные мочевины реагируют с формальдегидом,
образуя метилольные соединения, способные непосредственно
•превращаться в смолы; можно также предварительно этерифи-
цировать их спиртами в известных условиях. По этому способу
получаются твердые, прозрачные, бесцветные лаки, совмеща-
ющиеся с многочисленными синтетическими и природными, смо-
лями14.
Различные амино-альдегидные смолы
351
Триазиновые производные. Смолы, сочетающие свойства мо-
чевинных и меламиновых смол, получаются на основе продукта
взаимодействия дициандиамида с моногидразиндиаминотри-
азином15.
В молекуле меламина одна или несколько аминогрупп могут
быть замещены уреиловыми радикалами. Реакция состоит во
взаимодействии полигалоидного производного 1,3,5-триазина-
с мочевиной, замещенной щелочным металлом. Остальные
галоиды затем замещаются аминогруппами путем взаимодей-
ствия с аммиаком. Эти продукты при конденсации с формаль-
дегидом дают смолы, применяемые для лаков16.
Некоторое промышленное значение имеет 2,6-диамино-4-фе-
нил-1,3,5-триазин
который получается из меламина путем замещения одной'
аминогруппы фенильной группой.
Этот продукт получается при действии бензонитрила на
дициандиамид. Конденсацией его с формальдегидом в присут-
ствии бутанола получаются лаки, блеск и стабильность которых
выше, чем лаков на основе меламина (Мапреналь).
Уретаны. Уретаны—сложные эфиры карбаминовой кислоты,,
отвечают общей формуле NH2—COOR. Благодаря наличию
аминогруппы они способны реагировать с альдегидами с обра-
зованием метилольных производных в щелочной среде и мети-
леновых производных — в кислой среде. Эти промежуточные
продукты превращаются в смолы, свойства которых меняются
в зависимости от строения радикала R.
Молекула формальдегида реагирует с двумя молекулами
уретана по схеме:
О
ROOC—NH2 4- || 4- H2N— COOR -►
СН2
-► ROOC—NH—СН2—NH—COOR 4- H2O
Образующиеся по этому механизму линейные полимеры
получаются в результате взаимодействия формальдегида с:
диуретанами. Так, из 1,4-бутилендиуретана
H2N—СОО-(СН2)4—ООС—nh2
352 Гл. X. Мочевино-формальдегидные смолы и их аналоги
получается полимер
---СН2—N Н—СОО(СН2)4ООС—N Н—СН2—NH—СОО(СН2)4ООС—NH---
Водородные атомы метиленовых групп, заключенных между
иминными группами, способны присоединять альдегиды .по
схеме
—NH—СН2—NH— — NH—С—NH—
+ | + Н2О
сн=о сн
I I
R R
что позволяет получать структуры, довольно сильно различаю-
щиеся в зависимости от характера присоединенного альдегида17.
Продукты конденсации мочевины с альдегидами, кетонами и
другими продуктами. Ацетальдегид. Ацетальдегид реагирует с
мочевиной в присутствии кислого катализатора с образованием
смол. В качестве катализатора были предложены уксусная кис-
лота, соляная кислота, аммонийные соли. При ведении процесса
нельзя допускать полимеризации ацетальдегида в паральдегид.
Вполне возможно, что в качестве промежуточного продукта об-
разуется ацетилиденмочевина:
h2n—со—nh2 + О=СН—СН3 -* H2N—СО—N=CH—СН3+Н2О
Получаемые смолы растворимы в спирте. Для облегчения
смолообразования в конце конденсации часто прибавляют не-
которое количество формальдегида. Тогда получаются водорас-
творимые смолы18.
Акролеин и ненасыщенные альдегиды. Акролеин реагирует с
мочевиной легче, чем ацетальдегид, вследствие чего этому про-
цессу посвящено много работ. Саме и Видмер установили, что
при конденсации под давлением 1 моля акролеина и 2 молей
мочевины образуются сиропообразные продукты, с 1,5 моля
акролеина получается гель, при большем содержании альдеги-
да — твердые смолы. Конденсацию необязательно проводить
под давлением, она осуществляется в щелочной или кислой сре-
де. В щелочной среде кристаллический промежуточный продукт
не выделяют, как и при синтезе мочевино-формальдегидной
смолы.
Получаемые смолы сильно окрашены. Прибавление к кисло-
му катализатору небольшого количества тиомочевины заметно
увеличивает скорость реакции, позволяет регулировать смоло-
образование и дает возможность получать слабоокрашенные
смолы19.
Различные амино-альдегидные смолы
353
Пакэн выделил следующее кристаллическое соединение:
сн,—сн2—сн—nh—со—nh2
I I
NH—СО—NH
. Оно получается при взаимодействии 1 моля акролеина с 2
молями мочевины; компоненты растворяют в смеси равных ве-
совых количеств спирта и воды, содержащей 6% концентриро-
ванной соляной кислоты. Предложенная формула представ-
ляется вероятной, так как выделенные кристаллические соедине-
ния имеют насыщенный характер, что .позволяет отказаться от
строения оснований Шиффа.
Реакция аналогично протекает с кротоновым альдегидом,
причем получаются кристаллические продукты следующего
строения:
СН3—СН—СН2—CI I—N Н—СО—NH2
I I
NH—СО—NH
Они разлагаются20 при 250°.
Указанные циклические соединения вследствие подвижности
атомов водорода, связанных с азотом, могут давать относитель-
но стойкие метилольные производные, в кислом растворе реаги-
рующие со спиртами, образуя простые эфиры. При повышенной
температуре эти эфиры конденсируются до состояния восков или
смол, по-видимому, представляющих технический интерес.
Фурфурол. При конденсации фурфурола с мочевиной полу-
чаются сильно окрашенные неплавкие смолы, обладающие хо-
рошей стойкостью к щелочам и разбавленным кислотам. Реак-
ция протекает по схеме:
НС СН НС СН
II II II II
h2n—со—nh2 + О=СН—С СН —H2N—СО—N=CH—С СН
Х'о/ ^о^
Образующееся соединение является мономером, из- которого
далее получается смола. Реакция протекает легко и может быть
осуществлена в щелочной или кислой среде и даже в отсутст-
вие катализатора.
Фурфурол не реагирует с меламином, но вступает во взаимо-
действие с продуктами конденсации меламина и формальде-
гида. Полимеризация три 150° приводит к получению светлой
смолы с хорошей механической прочностью и теплостойкостью21.
Фенилмеламины, в противоположность меламину, реагируют
непосредственно с фурфуролом, образуя смолы, остающиеся
термопластичными в щелочной среде и приобретающие термо-
реактивность в кислой среде22.
23—156
354 Гл. X. Мочевино-формальдегидные смолы и их аналоги
Кетоны. Кетон реагирует с одной или двумя аминогруппа-
ми мочевины с выделением одной молекулы воды. В солянокис-
лой среде при 50—60° три молекулы ацетона образуют с одной
молекулой мочевины кристаллический продукт следующего
строения:
CH^^NH—СО—N=C(CH8)2
енХ 'Чн -СО—N=C(CH8)2
Ацетофенон и пропиофенон дают аналогичные соединения.
Эти соединения содержат двойные связи и, следовательно, спо-
собны образовывать полимеры, имеющие в зависимости от усло-
вий их образования характер масел или смол. Если в реакции
конденсации участвуют спирты или фенолы, получаются либо
маслообразные продукты, пригодные в качестве пластификато-
ров, либо смолы, аналогичные шеллаку. Продукты конденсации
мочевины с циклогексаноном, дополнительно обработанные
формальдегидом, даже превосходят в некоторых отношениях
шеллак23:
Окись этилена. Окись этилена, введенная в расплавленную
мочевину, реагирует с ней при 135° под давлением или без него
по схеме:
NH—СН2СН2ОН
ос/
ТМН—СН2СН2ОН
NH—СН2СН2ОН H2N— СО—NHa nh—СН2СН2—oconh2
ос/ + -* ос/
^NH—сн2сн2он h2n—со—nh2 ^nh—сн2сн2—oconh2
Образовавшееся соединение теряет две молекулы двуокиси
углерода:
NH—СН2СНа—NH«
ос/
.N'H—СН2СН2—NH2
Это соединение снова присоединяет две молекулы окиси эти-
лена и реакция продолжается до наступления смолообразова-
ния. Концевые гидроксильные группы сообщают смоле раство-
римость в воде, которая может быть устранена путем этерифи-
кации с образованием простых и сложных эфиров.
Если к первичному продукту конденсации прибавить форм-
альдегид, нерастворимость может быть достигнута путем на-
гревания при 125—135°.
Различные амино-альдегидные смолы
355
ЛИТЕРАТУРА
1. Т. S. Н о d g i n s, A. G. Hovey, Ind. Eng. Chem., 1938, 30, 1021.
2. R. Kohler, Koll. Z., 1943, 103, 2.
3. A. Grams, G. Widmer, W. Fisch, Brit. Plast., 1943, 15, 808.
4. Am. Cyanamid Co., фр. пат. 917895, 1945 г.
5. Soc. pour 1 Ind. Chim. a Bale, фр. пат. 867109, 1940 г.
6. Am. Cyanamid Co., фр. пат. 967251, 1948 г.
7. Lewis Berger a. Sons Ltd., фр. пат. 959024, 1947 г.
8. Soc. pour Find. Chim. a Bale, фр. пат. 890760, 1943 г.
9. Imperial Chemical Ind. Ltd., фр. пат. 933009 и 933010, 1946 г.
10. Imperial Chemical Ind. Ltd., фр. пат. 953962, 1947 г.
11. Am. Cyanamid Co., фр. пат. 862945, 1940 г.
12. I. G. Farben A. F„ фр. пат. 839875, 1938 г.; 858223, 1939 г.; 885927,
1942 г.
13. T.i, W a a g, фр. пат. 967347, 1948 г.
14. Comp, franc. Thomson—Houston, фр. пат. 953893, 958656, 1947 г.; 964832,
1948 г.
15. Comp. franc- Thomson—Houston, фр. пат. 954108, 1947 г.
16. Comp, franc- Thomson—Houston, фр. пат. 962875, 1948 г.
17. А. М. Paquin, Kunststoffe, 1947. 37, 25.
18. A. S. Bata, фр. пат. 898915, 1943 г.
19. R о h m a. Haas, фр. пат. 884091, 1942 г.
20. А. М. Р a q u i п, Kunststoffe, 1947, 37, 165.
21. Am. Cyanamid Co., фр. пат. 967063, 1948 г.
22. Am. Cyanamid Co., фр. пат. 967064, 1948 г.
23. А. М. Р a q u i n, Kunststoffe, 1947, 37, 25.
24. А. М. Р a q и i n, Kunststoffe, 1947, 37, 165.
Дополнительная справочная литература
Р. Т а 1 е t, Les aminoplastes, Dunod, Paris, 1951.
M. Fournier, Industries des Plastiques, 1947 . 3, 403; 1947, 3, 464; 1948,
4, 25.
G. H. Ott, Premier congres international de 1’Industrie des peintures, Paris,
1947, p. 530.
ГЛАВА XI
АЛКИДНЫЕ СМОЛЫ
И. Пети
Термином «алкидная смола» принято обозначать совокуп-
ность соединений, образующихся при взаимодействии много-
* атомных спиртов и многоосновных кислот*. Следовательно, ал-
кидные смолы относятся к классу полиэфиров. Алкидные смолы
могут отличаться друг от друга, поскольку они образуются
обычно в результате реакции трехатомного или многоатомного
спирта с кислотой, содержащей более двух карбоксильных
групп.
Более точным является определение алкидных смол как
полиэфиров трехмерной структуры.
Алкидные смолы принято подразделять на немодифициро-
ванные и модифицированные. Немодифицированные алкидные
смолы — это продукты .поликонденсации многоатомных спиртов
и многоосновных кислот; модифицированные алкидные смолы
содержат, кроме того, радикалы одноосновных кислот (обычно
ненасыщенных жирных кислот), вводимые для изменения фи-
зических свойств получаемых продуктов.
Практически в лакокрасочной промышленности используют-
ся только модифицированные алкидные смолы, но при теоре-
тических исследованиях ограничиваются обычно рассмотрением
только реакций образования немодифицированных смол.
Исходные вещества для синтеза алкидных смол
Исходными веществами для получения алкидных смол слу-
жат многоосновные карбоновые кислоты, многоатомные спирты
и некоторые одноосновные карбоновые кислоты, применяемые
* Определение алкидных смол как продуктов поликонденсации много-
атомных спиртов и многоосновных кислот охватывает не только синтетические
алкидные смолы, но и некоторые другие исходные материалы лакокрасочной
промышленности. Например, полимеризованные масла точно соответствуют
определению модифицированных алкидных смол: они представляют собой про-
дукты этерификации глицерина двухосновными и одноосновными карбоновыми
кислотами. Копалы Конго и шеллак являются продуктами поликонденсации
природных спиртов и кислот. При исследовании их методами, используемыми
в химии модифицированных алкидных смол, получаются вполне сопоставимые
результаты.
Исходные вещества для синтеза
357
для изготовления модифицированных смол. Модификация мо-
жет быть с таким же успехом осуществлена с помощью одно-
атомных спиртов, но в технике эта возможность пока не. исполь-
зуется.
Ввиду невозможности и нецелесообразности приведения пол-
ного перечня всех веществ, пригодных для получения алкидных
смол, здесь будут указаны только основные исходные про-
дукты, вошедшие в современную промышленную практику.
Двухосновные кислоты. Важнейшей из этих кислот является
фталевая, применяемая в виде фталевого ангидрида
В зависимости от условий изготовления фталевый ангидрид
получается в форме белых игл или пластинок, образующихся
вследствие агломерации большого числа мелких кристаллов.
Запах промышленного продукта обусловлен наличием в нем
следов альдегидных соединений. При медленном нагревании
фталевый ангидрид возгоняется, выделяя очень едкие пары.
При быстром нагревании фталевый ангидрид плавится, пере-
ходя в бесцветную жидкость, перегоняющуюся без разложения
при соблюдении элементарных мер предосторожности. Фтале-
вый ангидрид плавится при 128°; растворим в эфире, бензоле,
хуже — в спирте. В водных растворах он превращается в кис-
лоту, но эта реакция все же протекает довольно медленно.
Иногда считают, что некоторые затруднения в производстве
алкидных смол вызваны недостаточно высоким качеством ис-
пользуемого фталевого ангидрида; это часто несправедливо,
так как промышленный продукт получается всегда высокой сте-
пени чистоты. В сомнительных случаях качество фталевого
ангидрида можно проверить самым простым способом — путем
определения обычным методом числа омыления. Теоретическая
величина числа омыления чистого продукта составляет 756, что
соответствует реакции:
аС\ ^\/СООК
О 4- 2КОН f Y + Н2О
/ ^/\СООК
Малеиновый ангидрид используется в производстве алкид-
ных смол вследствие легкости образования эфиров при взаимо-
358
Гл. XI. Алкидные смолы
действии его со спиртами. Но малеиновый ангидрид способен
реагировать и другим способом: это замечательная филодиено-
вая компонента синтезов Дильса—Альдера, образующая про-
дукты присоединения с аминами, фенолами и некоторыми аро-
матическими углеводородами.
Подобно фталевому ангидриду, малеиновый ангидрид при
нагревании выделяет едкие пары, но сублимируется значительно
труднее. Несмотря на наличие в молекуле двойной связи, его
иодное число равно нулю. Эту двойную связь можно количе-
ственно определить путем бромирования в определенных усло-
виях или гидрирования. Число омыления малеинового ангид-
рида равно 1142.
Адипиновая кислота НООС—(СН2)4—СООН в очень боль-
ших масштабах применяется в .производстве суперполиамидов,
но мож,ет найти применение также и для получения алкидных
смол. Она плавится при 150° и кипит при 205° при 10 мм рт. ст.
Три перечисленные двухосновные кислоты наиболее распро-
странены (чаще всего применяется фталевый ангидрид). Могут
применяться,, однако, также и многие другие кислоты — прак-
тически, все известные двухосновные кислоты, достаточно стой-
кие к нагреванию. Но одних только технологических сообра-
жений не всегда достаточно: первостепенное значение имеет
возможность организации производства соответствующей двух-
основной кислоты простым и дешевым способом.
Для иллюстрации приведем перечень некоторых насыщен-
ных двухосновных кислот:
Температура
плавления
°C
Малоновая НООС—СН2—СООН .... 35,6
Янтарная НООС—(СН2)2—СООН .... 185
Глутаровая НООС—(СН2)3—СООН ... 97,5
Адипиновая НООС—(СГУд—СООН ... 1 51
Пнмелиновая НООС—(СН2)5—СООН . . 105,5
Пробковая НООС—(СН2)6—СООН ... 144
Азелаиновая НООС—(СН2)7—СООН . . 107
Себациновая НООС—(СН2)8—СООН 134
Было предложено применять также непредельные кислоты.
Нельзя не упомянуть здесь бесчисленные возможности, предо-
ставляемые диеновыми производными малеинового ангидрида,
замещенными производными фталевого ангидрида, и т. д. Трех-
основные кислоты, теоретически пригодные для изготовления
алкидных смол, более труднодоступны.
Спирты. Из многоатомных спиртов наиболее широко при-
меняется глицерин (пропантриол) СН2ОН—СНОН—СН2ОН.
До последних лет единственным источником глицерина были
триглицериды, получаемые путем омыления жировых веществ
Исходные вещества для синтеза
359
щелочами или водой (гидролиз) в присутствии катализаторов
или без них. В период недостатка жировых веществ делались
попытки получать глицерин путем брожения. Однако широкое
использование этого способа в настоящее время мало вероятно.
Со времени последней войны появился новый промышленный
способ, позволяющий получать глицерин синтетически из угле-
водородов крекинга нефти, в особенности из пропилена.
В чистом виде глицерин представляет собой бесцветную,
вязкую жидкость, со сладким и несколько жгучим вкусом. Эта
жидкость ниже 18° существует практически в состоянии пере-
охлаждения и при сильном охлаждении можно нередко наблю-
дать кристаллизацию всего глицерина в сосуде. Температура
кипения глицерина 290° при 760 мм рт. ст. и 180° при 12 лш рт. ст.
Глицерин растворим в воде, с которой он смешивается во всех
отношениях; он весьма гигроскопичен и при хранении должен
быть тщательно закупорен. Следует также указать, что гли-
церин растворяет в значительных количествах большое число
различных минеральных солей.
Пентаэритрит. Пентаэритрит (тетраметилолметан, или 2,2-ди-
метилол пропандиол-1,3) С(СН2ОН)4 представляет собой четы-
рехатомный спирт, получаемый синтетически и широко приме-
няемый в производстве алкидных смол.
Пентаэритрит образует кристаллы, хорошо растворимые в
горячей воде. В чистом виде он плавится при 260,5°, но часто
оказывается загрязненным примесями, главным образом про-
стым эфиром, неправильно называемым дипентаэритритом:
СН2ОН СН2ОН
I I
HOH2C—С—СН2—О—СН2—С—СН2ОН
I I
' СН2ОН СН2ОН
Этот эфир в чистом виде плавится при 221° и образует с
пентаэритритом эвтектическую смесь, плавящуюся при 190° и
не разделяемую путем кристаллизации.
Высшие многоатомные спирты (полиолы). Из более слож-
ных многоатомных спиртов было предложено применять, на-
пример. сорбит и маннит:
Н Н ОН Н
1111
НОСН2—С—С—С—С—СН2ОН
1111
ОН ОН Н ОН
Н Н ОН ОН
носн2—с—с—i—с—сн2он
он он н н
сорбит маннит
Эти спирты обладают шестью спиртовыми группами; при
повышении температуры происходит внутренняя этерификация,
ведущая к уменьшению числа спиртовых групп. В результате
360
Гл. XI. Алкидные смолы
получаются соединения (сорбитан и маннитан), реагирующие
в форме тетролов. Кроме того, они часто претерпевают весьма
нежелательную «карамелизацию».
Было предложено также применять диолы (гликоли), но
они могут дать хорошие результаты только в сочетании с
трехосновными кислотами. Наконец, в патентной литературе
есть указания на возможность применения таких спиртов, как
поливиниловый, и даже целлюлозы. К оценке результатов при-
менения подобных веществ следует подходить чрезвычайно
осторожно.
Что касается двухосновных кислот, к ним применимы те
же соображения, что и для многоатомных спиртов.
Одноосновные кислоты. В производстве модифицированных
алкидных смол находит широкое применение вся группа одно-
основных жирных кислот (см. главы о высыхающих маслах).
Эти жирные кислоты весьма разнообразны, так как .в зави-
симости от назначения они могут быть получены из различ-
ных высыхающих или невысыхающих масел. Обычно приме-
няют не чистые жирные кислоты, сравнительно трудно выде-
ляемые, а смеси кислот, получаемые при омылении или гид-
ролизе различных масел.
Технологические свойства смол улучшаются при примене-
нии перегнанных жирных кислот и более постоянны, если свой-
ства различных партий смесей кислот одинаковы. Так, напри-
мер, в США выпускаются кислоты высыхающих масел с по-
стоянным иодным числом.
Абиетиновая кислота (см. главу, посвященную канифоли)
также находит применение в производстве некоторых алкидных
смол.
Таким образом, имеется исключительно широкая гамма ис-
ходных веществ для изготовления покрытий на основе алкид-
ных смол.
Образование и свойства полиэфиров
Основные понятия химии высокополимеров дают возможность
непосредственно уяснить условия, необходимые для образо-
вания алкидных смол, и в то же время объясняют их свой-
ства. Первым условием образования смолы является получе-
ние вещества с высоким молекулярным весом.
При получении сложных эфиров высокомолекулярные цепи
могут быть образованы только при участии некоторых опре-
деленных групп кислот и спиртов. При взаимодействии одно-
основной кислоты с одноатомным спиртом может образоваться
лишь низкомолекулярный сложный эфир, например этилацетат
и все эфиры этого типа.
Образование и свойства полиэфиров
361
Соединение одноосновной кислоты и одноатомного спирта
может быть представлено схематически следующим образом:.
В F R'
*
где R— ацил;
R' — алкоксигруппа;
• —сложноэфирная связь.
Принимая те же обозначения, реакцию одноосновной кис-
лоты с двухатомным спиртом можно представить в виде:
R т R' у R
С трехатомными спиртами одноосновные кислоты образуют
эфиры следующего строения (например, тристеарин):
Четьврехатомные спирты, такие как пентаэритрит, образуюг
сложные эфиры, которые символически можно изобразить сле-
дующим образом:
Следовательно, какова бы ни была функциональность спир-
та, сложный эфир, полученный путем его взаимодействия с
одноосновной кислотой, не будет высокомолекулярным соеди-
362
Гл. XI. Алкидные смолы
пением. Не имеет также значения, этерифицированы ли раз-
личные спиртовые группы одной и той же или различными
одноосновными кислотами и даже все ли спиртовые группы
замещены.
Иные продукты получаются при использовании двухоснов-
ных кислот. Так, если эфир двухосновной кислоты и одноатом-
ного спирта (например, диэтилсукцинат или диэтилфталат) еще
представляет собой определенное соединение, которое можно
представить схемой
р" R'
-------•----
где R" — радикал двухосновной кислоты, то с двухатомным
спиртом реакция может протекать по-разному. Кроме особых
случаев, очень трудно осуществить этерификацию двух карб-
оксилов одной и той же молекулы кислоты двумя гидрокси-
лами одной и той же молекулы двухатомного спирта, получая
циклический диэфир, хотя эта возможность не должна быть
отброшена. Реакция этерификации будет протекать главным
образом между карбоксилами'и гидроксилами, принадлежащи-
ми к разным молекулам:
НО(СН2)аОН + НООС(СН2)2СООН + НО(СН2)2ОН + НООС(СН2)2СООН4-►
----О—(СН2)2ООС(СН2)аСОО(СН2)2ООС(СН2)2СОО--h хН2О
При этом получается не какой-то один определенный эфир,
-а смесь линейных полиэфиров следующего строения:
R' R" R' R" R' R" Rz R"
----•------•------•------•------•------Я------♦----
Полученные таким образом сравнительно высокомолекуляр-
ные продукты могут сильно различаться в зависимости от усло-
вий проведения реакции. Величина молекулярного веса опре-
деляет и физические свойства этих полиэфиров, но вследствие
линейной структуры макромолекулы они всегда сохраняют спо-
собность плавиться и заметную растворимость.
Если двухосновная кислота реагирует с трехатомным спир-
том (например, с глицерином), то реакция может развиваться
во всех пространственных направлениях с образованием трех-
Образование и свойства полиэфиров
363
мерной сетки. Длинные цепи, подобные образующимся при
взаимодействии с гликолем, соединены между собой в неко-
торых местах ковалентными связями, и образование такой бо-
лее жесткой структуры приводит к появлению характерных
особенностей: неплавкости и почти полной нерастворимости
(возможно только набухание). Сетка трехмерного полиэфира
может быть представлена схемой:
При участии четырехатомного спирта (например, пентаэри-
трита) образующаяся сетка также будет трехмерной:
Приведенная упрощенная схема дает ясное представление
о связях между линейными молекулярными цепями.
Если взять пятиатомный или шестиатомный спирт, то при
взаимодействии его с двухосновной кислотой всегда образуется
трехмерная сетка.
В случае трехосновных кислот, значительно менее распро-
страненном, применимо то же рассуждение: с одноатомными
спиртами образуются индивидуальные низкомолекулярные хи-
мические соединения, с двухатомными или многоатомными апир-
тами — трехмерные продукты.
364
Гл. XI. Алкидные смолы
Рассмотренные закономерности сведены в таблицу:
Кислоты Спирты
одноатомные двухатомные трехатомные высшие
Однооснов- Низкомолеку- Низкомолеку- Низкомолеку- Низкомолеку-
ные лярные ин- дивидуаль- ные соедине- ния лярные ин- дивидуаль- ные соедине- ния лярные ин- дивидуаль- ные соедине- ния лярные ин- дивидуаль- ные соедине- ния
Двухоснов- ные То же Термопластич- ные линей- ные полиме- ры Трехмерные полимеры Трехмерные полимеры
Трехоснов- ные То же Трехмерные полимеры То же То же
Высшие То же То же То же То же
Эта таблица в сжатом виде суммирует представления Кинле
о функциональности исходных веществ. Функциональными груп-
пами в данном случае являются карбоксильные группы кислот
и гидроксильные группы спиртов.
Чтобы избежать возможных ошибок, необходимо постоянно
иметь в виду приводимые ниже основные положения:
1. Высокомолекулярное соединение не может быть получено
взаимодействием монофункционального соединения с другим
моно- или полифункциональным, даже если функциональность
второго соединения очень велика.
2. Реакция между двумя бифункциональными соединениями
приводит к образованию термопластичных и растворимых по-
лимеров линейного строения.
3. Во всех остальных случаях образуются полиэфиры трех-
мерного строения.
Эти положения соблюдаются, когда одна кислота реагирует
с одним определенным спиртом, т. е. когда карбоксильные
группы кислоты могут реагировать только с гидроксильными
группами данного спирта. Совершенно очевидно, что для смеси
многоатомных спиртов, или многоосновных кислот, или в при-
сутствии соединений, способных полимеризоваться, могут по-
лучаться иные результаты.
Если бы в технике использовались только рассмотренные
выше идеальные системы, полиэфиры были бы в большинстве
случаев неприменимыми; помимо некоторых трудностей син-
теза, полимеры трехмерного строения было бы трудно исполь-
Образование и свойства полиэфиров
365
зовать вследствие отсутствия растворителей, позволяющих при-
менять их в тонком слое. В то же время линейные немоди-
фицированные полиэфиры мало используются в лакокрасочной
промышленности (применяются 'немногие из них, например
гликольмалеаты) и лишь некоторые подобные полиэфиры пред-
ставляют интерес для других отраслей промышленности.
Следовательно, задача специалистов-лакокрасочников состоит
в использовании полимеров с трехмерной сеткой путем
•придания им формы, удобной для применения в данной обла-
сти.
Самое простое решение этого вопроса состоит в- получении
полимеров относительно низкой степени поликонденсации.
Тогда смола сохраняет еще заметную растворимость и может
быть использована в виде раствора. После удаления раство-
рителя проводится дальнейшая поликонденсация под действием
тепла, приводящая к образованию твердой и нерастворимой
лЛенки. Однако такой путь довольно мало применим, так как
получаемые пленки крайне хрупки.
Кроме указанных выше главнейших свойств трехмерных
полимеров, имеют значение также и другие факторы, в осо-
бенности степень полимеризации или поликонденсации. Осо-
бенно существенно это в отношении линейных полимеров.
Для иллюстрации в табл. 19 показаны изменения свойств
полимеров окиси этилена в зависимости от степени полимери-
зации.
ТАБЛИЦА 19
Свойства полимеров окиси этилена в зависимости от степени полимеризации
Степень полимери- зации Молекуляр- ный вес Температура плавления °C Состояние полимера Растворимость в воде Растворимость в эфире
10 440 Жидкость Вязкая жидкость Растворим Растворим на холоду
30 1 300 40 Мягкий воск » Растворим при нагревании
100 4 400 55 То же » Нерастворим
300 13000 70 Твердый воск Сильно на- бухает »
•2 500 100 000 175 (с разложе- нием) Твердый волокни- стый Слабо на- бухает
366
Гл. XI. Алкидные смолы
В случае .трехмерных полимеров существенную роль играет
другой фактор: показатель частоты сетки. Этот показатель мож-
но определить как число ковалентных межмолекулярных свя-
зей, соединяющих две линейные цепи, на 100 ковалентных
связей в линейной цепи.
При высоком показателе частоты сетки продукты характе-
ризуются твердостью, неплавкостью, нерастворимостью и стой-
костью к набуханию. Такими свойствами обладают, например,
^модифицированные феноло-альдегидные смолы, полученные
из обычного фенола, или ^модифицированные глифталевые
смолы. С уменьшением показателя частоты сетки эти свойства
ослабляются и образуются менее жесткие макромолекулы,
сильнее набухающие.
Величину показателя частоты сетки можно изменять, ис-
пользуя мономеры более высокого молекулярного веса (при за-
мене фталевого ангидрида себациновой кислотой в алкидных
смолах получаются более эластичные покрытия) или уменьшая
среднюю функциональность мономеров (например, применяя
смеси многоатомного спирта с одноатомным или же двухоснов-
ной кислоты с одноосновной).
Последний способ, широко применяемый при синтезе совре-
менных модифицированных алкидных смол, нуждается в не-
которых объяснениях.
При поликонденсации двухосновной кислоты с трехатомным
спиртом реакция быстро развивается в направлении образова-
ния трехмерных макромолекул. Если бы вместо двухосновной
кислоты была применена одноосновная, то получалось бы толь-
ко низкомолекулярное соединение определенного состава.
Однако .возможен промежуточный случай, когда одни гидр-
оксилы трехатомного спирта этерифицированы двухосновными
кислотами, а другие—-одноосновными. Тогда получаются весь-
ма разнообразные молекулы. Наличие двухосновной кислоты
благоприятствует образованию полимера, тогда как присоеди-
нение молекулы одноосновной кислоты может в любой момент
ограничить рост макромолекул, блокируя некоторое число гидр-
оксилов, которые после этерификации уже не участвуют в даль-
нейшем росте цепи. Такой процесс является совместной поли-
конденсацией и свойства получаемых продуктов будут в ос-
новном зависеть от относительного содержания двухосновных
и одноосновных кислот. Само собой разумеется, что общее чис-
ло наличных карбоксилов всегда должно быть равно общему
числу гидроксилов.
Схематическое изображение подобных молекул затрудни-
тельно, так как реакции этерификации протекают по законам
случая и приводят к большому разнообразию размеров полу-
чаемых молекул.
Модифицированные алкидные смолы
367
Увеличивая содержание одноосновных кислот, можно достиг-
нуть некоторого уменьшения показателя частоты сетки, а также
уменьшения размеров макромолекул. При более высоких кон-
центрациях одноосновных кислот получаемые продукты нельзя
даже относить к высокомолекулярным веществам.
Аналогичным образом можно регулировать свойства поли-
эфиров путем применения смесей двухатомного и одноатомного
спиртов.
Существует еще один крайне простой способ ограничения
размера макромолекул полиэфиров: он состоит в применении
исходных реагентов не в эквифункциональных количествах, а
при избытке одного из них. Благодаря этому некоторое число
функциональных групп, например карбоксильных, не прореаги-
рует с гидроксильными функциональными группами вследствие
их недостатка и рост макромолекул во многих точках будет
ограничен.
В заключение следует подчеркнуть, что молекулярный вес
полиэфира, как и любого другого полимера, не может рассмат-
риваться как молекулярный вес соединения определенного со-
става. В действительности имеется смесь молекул всех разме-
ров, а физические и химические свойства получаемых продуктов
зависят в основном от тех молекул, которые преобладают в дан-
ной смеси. Можно говорить только о среднем молекулярнрм
весе, точное определение которого весьма затруднительно я
возможно лишь при использовании классических кривых распре-
деления.
МОДИФИЦИРОВАННЫЕ АЛКИДНЫЕ СМОЛЫ
В лакокрасочной промышленности за исключением некото-
рых особых случаев используются только модифицированные
алкидные смолы. Нерастворимость немодифицированных алкид-
ных смол исключает возможность их применения в качестве
пленкообразующих веществ.
Исходная макромолекулярная сетка может быть модифи-
цирована с помощью спирта или кислоты. Практически к фта-
левой кислоте добавляют другие кислоты, что обусловливает
изменение структуры. Применение любой одноосновной кислоты,
заменяющей эквифункциональное количество фталевого ангид-
рида, позволяет уменьшить частоту сетки и размеры макро-
молекул, а также значительно расширить гамму растворимости
получаемых продуктов по сравнению с немодифицированными
смолами. При последующем нагревании поликонденсация за-
вершается и продукты приобретают требуемую твердость и
нерастворимость (эмали горячей сушки).
Введение полиеновых одноосновных кислот, полученных из
высыхающих масел, приводит к тем же результатам, но, кроме
368
Гл. XI. Алкидные смолы
того, придает алкидным смолам способность высыхать и
отверждаться ‘путем окислительной полимеризации без приме-
нения сушки при повышенных температурах.
На основе описанных промежуточных соединений можно
получать разнообразные конечные продукты, пригодные для са-
мых разнообразных областей применения.
Различные типы получаемых алкидных смол можно кратко
«охарактеризовать следующим образом.
J. Алкидные смолы, не модифицированные или модифициро-
ванные насыщенными одноосновными кислотами. Они не отвер-
ждаются при воздушной сушке; отверждение наступает только
в результате реакций межмолекулярной поликонденсации мак-
ромолекул более низкой степени поликонденсации (горячая
сушка):
Следует принять во внимание, что по этой схеме две сосед-
ние макромолекулы соединяются только путем этерификации
с выделением молекулы воды. Тем не менее, реакция, проте-
кающая таким образом, в дальнейшем быстро приводит к зна-
чительному повышению молекулярного веса.
2. Алкидные смолы, модифицированные полиеновыми жир-
ными кислотами. Модифицированную алкидную смолу можно
Модифицированные алкидные смолы
369
рассматривать как сложную цепь, образованную из различных
соединенных между собой элементарных звеньев, например сле-
дующих:
—ООС^^СО( )СН2 СН—СН2—о—
I
OOCR
—оос^~^соосн2—сн—о—
I
ch2oocr
соосн2—сн—ch2oocr
OOCR
Здесь R обозначает радикал ненасыщенной кислоты,
пример линолевой:
на-
—(сн2)7—СН=СН—СН2—СН=СН—(СН2)4—сн3
Следовательно, молекулу алкидной смолы, содержащую та-
кие радикалы, можно рассматривать как молекулу высыхаю-
щего масла. Ненасыщенная кислота этерифицирует фактически
многоатомный спирт высокой функциональности, состоящий из
остатков глицерина, частично этерифицированных и связанных
между собой радикалами ортофталевой кислоты. Поэтому ал-
кидные смолы этого типа должны иметь повышенную способ-
ность к высыханию, что и наблюдается в действительности (см.
главу «Высыхающие масла»).
Отверждение подобной смолы, т. е. соединение различных
макромолекул, о величине которых мы практически не имеем
сведений, происходит в результате обычных реакций, проте-
кающих при высыхании масел; явления поликонденсации не
играют в этом процессе никакой роли. Такие смолы после вве-
дения сиккатива образуют пленку на холоду и притом значи-
тельно быстрее, чем обычные высыхающие масла.
Однако нельзя упускать из виду, что модифицированные
алкидные смолы, фактически представляющие собой высыхаю-
щие масла с повышенной функциональностью, содержат, как
и немодифицированные алкидные смолы, свободные карбок-
сильные и гидроксильные группы. Благодаря этому можно еще
больше повысить показатель частоты сетки в уже готовой плен-
ке путем последующего повышения температуры, 'приводящего
к образованию новых мостиков между макромолекулами. Та-
24—156
370
Гл. XI. Алкидные смолы
ким путем получаются высококачественные эмали горячей
сушки.
Все модифицированные алкидные смолы относятся к описан-
ным выше типам. Алкидные смолы, модифицированные пре-
дельными кислотами, иногда называют «невысыхающими ал-
кидными смолами», так как они образуют пленку только путем
поликонденсации.
Изменения свойств алкидных смол в результате модифици-
рования. Свойства алкидных смол могут весьма различаться
в зависимости от соотношения взятых для реакции однооснов-
ной и двухосновной кислот. Кроме того, их свойства могут
изменяться в широких пределах при переходе от чистых алкид-
ных смол (т. е. продуктов конденсации двухосновной кислоты
с многоатомным спиртом) до чистых высыхающих масел, пред-
ставляющих собой сочетание непредельных жирных однооснов-
ных кислот с глицерином.
Изменяя содержание одноосновных кислот, можно получать
неограниченное количество веществ, свойства которых будут
переходными между свойствами чистого высыхающего масла и
свойствами немодифицированной алкидной смолы. Эти вари-
ации схематически представлены в таблице:
Содержа- ние немо- дифициро- ванной алкидной смолы % Скорость Возможность сушки Растворимость Эластич- ность пленки
высыхания отвержде- ния воздуш- ной горячей в алифати- ческих углеводо- родах в спиртах
100 0 t 1 t 1 1 t 1 1 1 1
Промышленное производство модифицированных
алкидных смол
Для производства модифицированных алкидных смол мо-
гут быть использованы различные способы. Самый простой со-
стоит во введении в реакционный аппарат смеси различных
компонентов: фталевого ангидрида, глицерина и соответствую-
щих жирных кислот в заранее рассчитанных количествах. Эту
смесь нагревают до температуры, достаточной для протекания
процессов этерификации, приводящих к образованию смолы.
При слишком продолжительном нагревании возможно даль-
нейшее течение реакции, приводящее иногда к нежелательным
результатам: повышению вязкости, уменьшению кислотного чис-
ла, образованию нерастворимой массы (гелеобразование).
Модифицированные алкидные смолы
371
Указанный способ осуществим при условии применения
жирных кислот высыхающих масел. Преимуществом его яв-
ляется возможность получения конечных продуктов практи-
чески 1постоян‘ного качества при условии соблюдения одинако-
вого режима проведения операции и одинакового состава смеси
жирных кислот.
В ходе производства некоторое, иногда довольно значи-
тельное, количество фталевого ангидрида возгоняется и, чтобы
сократить продолжительность нагревания в присутствии фта-
левого ангидрида, можно разделить процесс на две стадии.
Сначала все количество глицерина, необходимое для образо-
вания смолы, частично этерйфицировать всем количеством жир-
ных кислот, а затем полученную смесь, богатую неполными
эфирами (моноглицеридами), ввести в реакцию с фталевым
ангидридом. Этот способ в американской литературе называется
«кислый моноглицеридный процесс» и также предусматривает
применение жирных кислот.
При других способах вместо жирных кислот используют
непосредственно высыхающие масла. Эти так называемые «мо-
ноглицеридные способы» широко известны вследствие своей
очевидной простоты и относительной дешевизны. Во всех вари-
антах процесс осуществляется в две стадии:
1. Приготовление моноглицеридов, т. е. алкоголиз, или гли-
церолиз, масел. Высыхающее масло нагревают в присутствии
глицерина, взятого в количестве, соответствующем схеме:
СН2—OOCR СН2ОН СН2ОН
I I I
СН—OOCR + 2 СНОН -* 3 СНОН
I I I
СН2—OOCR сн2он ch2oocr
2. Поликонденсация фталевого ангидрида с моноглицерида-
ми. При нагревании вводят фталевый ангидрид для этерифика-
ции свободных гидроксильных групп моноглицеридов.
Этот способ известен в американской литературе под на-
званием «масляный моноглицеридный процесс». Применяя раз-
личные количества масла, этот способ можно осуществить в
двух вариантах. Можно подвергнуть глицеролизу все количе-
ство масла, вводя все требуемое количество глицерина. Но
можно также проводить глицеролиз масла всегда одним и тем
же количеством глицерина (например, взятым в стехиометри-
ческом соотношении), а остальное количество многоатомного.'
спирта вводить в дальнейшем одновременно с фталевым ан-
гидридом. Полученные таким образом модифицированные ал-
кидные смолы имеют менее стандартные свойства, чем про-
дукты, полученные из жирных кислот, но этот способ все же
применяется весьма широко.
24*
372 Гл. XI. Алкидные смолы
Состав модифицированных алкидных смол. В зависимости
от способа изготовления различают смолы, модифицированные
жирными кислотами или высыхающими маслами. В действитель-
ности модификация связана только с введением жирных кислот,
и такое деление сохраняется исключительно по привычке. Кро-
ме того, принято указывать, сколько в модифицированной ал-
кидной смоле содержится масла по отношению к немодифици-
рованной смоле, что также не имеет химического смысла, так
как при модификации протекает химическая реакция, а не
растворение алкидной смолы в масле. Тем не менее, различают
просто жирные глифталевые лаки, вдвойне жирные, сильно
масляные, средне масляные, тощие масляные и т. д. Эти на-
звания, разумеется, не могут служить основой технической но-
менклатуры. То же относится к весьма широко применяемым
цифровым обозначениям. Эти обозначения образуются из двух
цифр, из которых первая выражает процентное содержание
чистой алкидной смолы, а вторая — содержание масла. Сле-
довательно, «глифталевая алкидная смола 60/40» состоит из
60% чистой алкидной смолы и 40% масла. Немодифицирован-
ная алкидная смола получается в действительности конден-
сацией 29,3% глицерина с 70,7% фталевого ангидрида, в то
время как масло можно считать соединением 90,2% жирной
кислоты и 9,8% глицерина. Таким образом, модифицированная
алкидная смола 60/40 в действительности получена при сле-
дующем соотношении компонентов (вес. ч.):
Фталевый ангидрид.................. 424
Жирная кислота С18 .................. 360,8
Глицерин .......................... 215,2
Можно было бы рассмотреть все вероятные комбинации в
пределах 0/100 и 100/0, но в действительности состав смол,
применяемых в лакокрасочной технике, лежит в интервале меж-
ду 30/70 и 70/30. Композиции, содержащие более 45% алкидной
смолы, применимы только для эмалей горячей сушки.
Процесс образования моноглицеридов
Приготовление моноглицеридов методом -алкоголиза масел
является главным процессом при получении модифицирован-
ных алкидных смол. Теоретически глицеролиз триглицерида
представляет собой весьма простую реакцию:
СН„—OOCR сн2—ОН СН2—ОН
I I I
СН—OOCR + 2 СН—ОН -> 3 СН—ОН
I 1 I
СН»—OOCR СН2—ОН СН2—OOCR
Модифицированные алкидные смолы
373
В патентной литературе обычно отсутствуют точные ука-
зания о технике проведения этой операции, о температуре и
применяемых катализаторах. Однако знание таких произволь-
ных параметров, зависящих от случайных местных условий,
не так важно для производственника, как понимание сущности
процесса.
Реакция образования моноглицеридов является обратной
реакции этерификации, которая, как известно, обратима, т. е.
степень превращения ограничивается реакцией алкоголиза. По-
этому количественный выход моноглицерида принципиально
недостижим.
Если для упрощения обозначить радикал —СНг—СН—'СНг—
буквой G, а радикал жирной кислоты RCOO — буквой А, то
различные реакции, протекающие при нагревании глицерина
с маслом, можно представить следующими схемами.
1. Образование моноглицерида:
/А /ОН /ОН
G—А + 2G—ОН 3G—ОН
\А \он \А
2. Образование диглицерида:
/А /ОН /ОН
2G—А + G—ОН 5*: 3G—А
\д чон \а
3. Превращение диглицерида в моноглицерид:
/ОН /ОН /ОН
G—А + G—ОН 2G—ОН
\д "Ч)Н \д
Ни один продукт этих реакций не был выделен, так как в
действительности образуется смесь различных компонентов, т. е.
неизмененного триглицерида, диглицерида, моноглицерида и не
вступившего в реакцию глицерина. Концентрации этих веществ
при данной температуре и начальной концентрации будут
определяться различными константами равновесия указанных
реакций. Если бы можно было определить константы равно-
весия этих реакций, то проблема оказалась бы совсем простой;
применение закона действия масс дает:
[G(A)3]-[G(OH)3p
[GA(OH)S]S
(1)
[G(A)3]2[G(OH)3J
[G(A)2OH]3
= К" ± <Э2
(2)
374
Г л. XI. Алкидные смолы
[G(A)3OH]-[G(OH)3]
[GA(OH)2J2 v ~
Величины, заключенные в квадратные скобки, означают мо-
лярные концентрации реагентов, приводящие к константам
равновесия К', К", К'" при температуре t. Каждая реакция
сопровождается положительным или отрицательным тепловым
эффектом Qi, Q2, <2з-
Зная эти теплоты реакции, можно определить влияние тем-
пературы на константы равновесия, применив формулу
dlnK _ Q
dt ~~ RT2
где /? — газовая постоянная, а Т—абсолютная температура.
После этого остается узнать для данной температуры зна-
чение констант равновесия путем химического анализа смеси.
К сожалению, этот анализ оказывается крайне сложным и
требующим ряда предосторожностей, и, кроме того, он может
дать только кажущееся значение К, которое определяется со-
отношением констант К', К" и К'". То же относится и к тепло-
вому эффекту Q. Поэтому все теоретические изыскания в этой
области являются иллюзорными и в лучшем случае представ-
ляют собой лишь более или менее обоснованные приближения.
Термическая стойкость чистого моноглицерида, как пред-
ставляется на первый взгляд, должна определяться легче, чем
для обычных сложных смесей, но в действительности трудность
определения не уменьшается. Поскольку моноглицерид являет-
ся продуктом неполной этерификации глицерина, находящимся
в состоянии подвижного равновесия, всякое термическое воздей-
ствие приводит неизбежно к нарушению этого неустойчивого
состояния.
Все изложенное касается термодинамики процесса, но суще-
ствует также не менее важный кинетический аспект. Действи-
тельно, можно создать прекрасные термодинамические условия,
сильно смещающие равновесие в сторону преимущественного
образования моноэфира, но это оказывается излишним, если
для установления такого равновесия требуется значительное
время. В этом случае выход не имеет практически никакого
значения; выгоднее работать в худших термодинамических ус-
ловиях, ускоряя реакцию при помощи катализаторов. При этом
нельзя забывать, что никакой катализатор не способен изме-
нить конечное равновесие реакции. Он позволяет лишь значи-
тельно быстрее подойти к состоянию равновесия.
Кажущееся противоречие может возникнуть, если возмож-
но протекание нескольких реакций, развивающихся с различ-
ными скоростями. Вторичная реакция может ускоряться в зна-
Модифицированные алкидные смолы 375
чительно большей мере, чем основная реакция. Наблюдаемые
результаты будут тогда обусловлены вторичной реакцией, что
и создает впечатление противоречия, в действительности не
существующего.
В случае приготовления моноглицеридов катализаторы, при-
меняемые в количестве тысячных долей от веса масла, доволь-
но различны, но все они представляют собой металлические
мыла. К маслам прибавляют окислы, карбонаты и т. п„ ко-
торые, взаимодействуя с маслами, быстро превращаются в мы-
ла, участвующие в дальнейшей реакции.
Вторичные реакции. Приведенные выше реакции являются
лишь теоретическими, но они, однако, охватывают большую
часть веществ, образование которых возможно в этих условиях.
Помимо этих реакций могут протекать побочные реакции, спо-
собные изменять более или менее глубоко природу полученных
веществ, а следовательно, и свойства алкидных смол, полу-
чаемых при последующем взаимодействии с фталевым ангид-
ридом. В побочных реакциях могут участвовать как исходные
реагенты (масло и глицерин), так и вещества, образующиеся
из них (диэфиры и моноэфиры).
Реакции, протекающие с участием масла. Учитывая относи-
тельно высокие температуры проведения процесса (200—250°),
можно было бы опасаться возможности частичной термической
полимеризации масла. В этом случае изменялась бы структура
масла и появившиеся радикалы двухосновных кислот при вза-
имодействии с глицерином образовали бы соединения, отли-
чающиеся от продуктов реакций с одноосновными кислотами.
Практика, однако, показала, что этого не происходит.
Реакции, протекающие с участием глицерина. Глицерин,
как трехатомный спирт, склонен к дегидратации при темпера-
туре, близкой к его температуре кипения, а при продолжитель-
ном воздействии даже при более низкой температуре.
Дегидратация глицерина может осуществляться различны-
ми путями.
1. Образование акролеина:
СН2ОН—СИОН—СН2ОН СН2=СН—СНО + 2Н2О
При получении моноглицеридов эта реакция приводит к по-
тере эквивалентного количества глицерина. Дегидратация с об-
разованием акролеина протекает очень часто, но с пренебре-
жимо малой скоростью.
Возможны также побочные реакции, при которых из одной
молекулы глицерина выделяется только одна молекула воды.
2. Образование ацетола:
СН2ОН—СНОН—СН2ОН СН3—СО—СН2ОН + Н2О
376
Гл. XI. Алкидные смолы
3. Образование эпоксидов:
СН2—ОН СН2—ОН
I I
сн—он сн х + Н2О
। । ;о
сн2—он сн/
Две последние реакции специально не изучались, так же
как реакции дегидратации, протекающие с отщеплением воды
от двух или нескольких молекул глицерина с образованием
диглицеринов и триглицеринов:
сн2—он но—сн2 сн2—О—сн2
I I II
но—СН + сн—он -* НО—СН СН—ОН + Н2О
I I II
НО—СН2 СН2—ОН НО—сн2 сн2—ОН
В этой реакции могут участвовать гидроксилы в а- или
р-положениях, что приводит к образованию различных изоме-
ров, содержащих 4 гидроксильные группы в случае диглицери-
на и 5—в случае триглицерина. Действительно, можно показать
наличие полиглицериновых спиртов в процессе приготовления
моноглицеридов. В противоположность предыдущим случаям,
в результате реакции не теряется эквивалентное количество
глицерина, поскольку функциональность вновь образующихся
соединений выше, чем у глицерина. Однако в случае неправиль-
ного дозирования фталевого ангидрида эти реакции влияют
на качество конечной смолы.
Выделение воды в ходе реакции очень легко доказать, при-
меняя абсолютно сухой реакционный прибор и обезвоженные
реактивы. Имеется также возможность собрать воду и взвесить
ее. Учитывая, что выделение 1 моля воды (18 г) сопряжено со
связыванием двух спиртовых групп, количество фталевого ан-
гидрида должно быть уменьшено (на 1 моль воды следует
уменьшить количество фталевого ангидрида на 1 моль, т. е. на
148 г). Другими словами, образование каждого кубического
сантиметра воды, выделившейся в процессе приготовления мо-
ноглицерида, приводит к уменьшению на 8,22 г количества
требуемого ангидрида, избыток которого нежелателен, так как
приводит к нежелательному повышению кислотности.
Впрочем, это имеет значение только в том случае, если
исходные продукты не содержат воды, т. е. в условиях, реаль-
но неосуществимых при промышленном производстве.
Аналогичные реакции протекают также с пентаэритритом,
склонность которого к этерификации очень велика.
Реакции диглицеридов. Диэфиры глицерина (диглицериды)
являются вторичными продуктами, образующимися из исход-
ных веществ. Они ведут себя как одноатомные спирты и огра-
Модифицированные алкидные смолы
3>7Т
ничивают реакцию поликонденсации с фталевым ангидридом.
Однако диглицериды 'принимают участие в пленкообразовании
при воздушной сушке вследствие присутствия в них радикалов
ненасыщенных кислот.
Диглицериды могут также взаимно этерифицироваться с
образованием тетраэфиров диглицерина, неспособных к поли-
конденсации:
СН2—СН—СН2ОН НОСН2—СН—СН2 -Н2О
I I + II --------------*
RCOO OOCR RCOO OOCR
СН2—СН—СН2—о—сн2—сн—сн2
— II II
RCOO OOCR RCOO OOCR
Реакции моноглицеридов. Главным компонентом смеси ос-
тается все же моноглицерид. Благодаря .наличию двух спирто-
вых функций он играет главную роль в построении молекулы
смолы, но при нагревании может изменяться вследствие вторич-
ных реакций этерификации. Эти вторичные реакции могут про-
текать по следующим схемам.
1. Внутримолекулярная этерификация:
CH2—OOCR СН2—OOCR
I —Н2О I
СН—он-------► СН х
I I >О
сн2—он сн/
Образующийся эпоксид с трудом вступает в реакции поли-
конденсации.
2. Межмолекулярная этерификация:
СН2-СН—СН2ОН НОСН2— СН—СН, —н2о
II + II-------------------------*
RCOO ОН ОН OOCR
сн,—сн—сн2—о—сн2—сн—сн2
I I II
RCOO ОН ОН OOCR
В этом случае образуется диэфир диглицерина, который мо-
жет в дальнейшем принимать участие в построении макромо-
лекулы, так как он имеет две спиртовые группы.
Межмолекулярная этерификация может протекать также с
выделением двух молекул воды:
сн2—сн—сн2 сн2—сн—сн,
I I ! Ill'
RCOO ОН ОН _2Н2О RCOO О О OOCR
НО ОН OOCR СН2—СН—СН2
I I I
сн2—СН—сн2
Подобные вещества не способны к поликонденсации.
378
Г л. XI. Алкидные смолы
Таким образом, кроме глицеролиза — реакции, протекаю-
щей без выделения воды, могут протекать также многочислен-
ные нежелательные побочные реакции. Тот факт, что образо-
вание воды наблюдается довольно часто, является безусловным
.доказательством наличия побочных реакций.
Среди приведенных побочных реакций некоторые представ-
ляют лишь теоретический интерес, но другие, главным образом
реакции образования полиглицеринов, подтверждены совершен-
но бесспорными опытными фактами. Следовательно, .суммарный
процесс в действительности значительно сложнее, чем простое
образование моноглицерида.
Контроль процесса образования моноглицеридов. Довольно
трудно точно установить момент, когда к реакционной смеси
следует прибавлять фталевый ангидрид. Обычно критерием
служит проба на растворимость в спирте: моноглицериды, со-
держащие гидроксильные группы, растворимы, в то время как
масла нерастворимы. Обычно считают, что когда достигается
полная растворимость, реакция закончена. Такая упрощенная
проба не дает, однако, точных показаний относительно содер-
жания моноглицерида; кроме того, растворимость колеблется
в широких пределах в зависимости от концентрации спирта. Не
исключена возможность контроля процесса по изменению пока-
зателя преломления после отделения свободного глицерина, но
эти изменения слишком малы и трудно улавливаются.
Более наглядный, хотя может быть и менее удобный ме-
тод состоит в удалении свободного глицерина из смеси промыв-
кой эфирного раствора водой. После испарения эфира доста-
точно определить число омыления полученного продукта. Так,
число омыления моноглицерида линолевой кислоты равно
157,3; путем простого вычисления можно легко определить со-
держание моноглицерида в смеси, если исходить из предполо-
жения, что он является единственным продуктом реакции.
Взаимодействие фталевого ангидрида со смесью моноглицерндов
С полученным на первой стадии сложным промежуточ-
ным продуктом фталевый ангидрид реагирует с образованием
модифицированных алкидных смол. Хотя различные реакции
этерификации осуществляются по законам случая, необходимо
для ясности рассмотреть их отдельно.
Масла. При любом способе ведения процесса в реакционной
смеси всегда остается более или менее значительное количество
триглицеридов, не подвергшихся глицеролизу. Вследствие от-
сутствия в их молекулах спиртовых групп фталевый ангидрид
.не может вступить с ними во взаимодействие и полученная
«мола будет содержать неизмененное масло.
Модифицированные алкидные смолы
379
Глицерин. В исходной смеси глицерин находится всегда в
избытке (при стехиометрическом количестве по отношению к
маслу перед реакцией с фталевым ангидридом необходимо до-
бавлять глицерин) и энергично реагирует с фталевым ангидри-
дом. При этом быстро образуется трехмерная сетчатая струк-
тура. При образовании немодифицированной глифталевой смо-
лы реакция протекает по схеме:
Диглицериды. Фталевый ангидрид реагирует с присутствую-
щими в смеси диглицеридами с образованием соединений, мо-
лекулярные веса которых не могут более увеличиваться путем
поликонденсации:
ОН OOCR
I I
сн2—сн—ch2oocr
сн2—сн—ch2oocr
I I
ОН OOCR
О OOCR
II 1
.с—осн2—сн—ch2oocr
•с—осн2—сн—ch2oocr
II I
О OOCR
380
Гл. XI. Алкидные смолы
Полученный таким образом дифталат имеет относительно
низкий молекулярный вес — около 1370. Однако он может уско-
рять высыхание пленки.
Моноглицериды. Этот компонент наиболее интересен, так
как он образуется при этерификации двухатомного спирта
двухосновной кислотой. В результате взаимодействия фталевого
ангидрида с моноглицеридами образуется линейный полиэфир:
С—ОСН2—СН—СН2О—С С—ОСН2—СН—СН2О—- •
•—О-С
II II I II II I
О О OOCR О О OOCR
Этот полиэфир можно рассматривать как идеальный пример
модифицированной алкидной смолы воздушной сушки.
Полиглицерины. Неэтерифицированные полиглицерины ре-
агируют с фталевым ангидридом таким же образом, как и гли-
церин. Вследствие их более высокой функциональности трех-
мерная сетка образуется с их участием еще быстрее, чем в
случае глицерина.
Полиглицерины, частично этерифицированные жирными кис-
лотами, ведут себя как глицерин, если остаются неэтерифици-
рованными три гидроксильные группы, как моноглицериды,
если сохранились лишь два гидроксила, и как диглицериды,
если в молекуле имеется лишь одна спиртовая группа.
Если бы указанные реакции протекали при взаимодействии
фталевого ангидрида с каждым из перечисленных индивидуаль-
ных соединений, химия алкидных смол оказалась бы значитель-
но упрощенной. Но в действительности картина существенно
отличается от описанной в каждом отдельном идеальном слу-
чае. В самом деле, различные рассмотренные выше соедине-
ния находятся в виде смеси, и было бы нарушением элемен-
тарных принципов термодинамики, если бы фталевый ангид-
рид выборочно реагировал только с некоторыми из имеющихся
гидроксилов. В действительности реакции этерификации про-
исходят лишь по законам случая и предусмотреть размеры и
форму образующихся макромолекул совершенно невозможно.
Могут быть указаны лишь следующие основные принципы:
1. Участие в реакции глицерина обусловливает образова-
ние сетки.
2. Участие моноглицеридов снижает показатель частоты
сетки.
3. Наличие диглицеридов ограничивает рост макромолекул.
В соответствии с относительным содержанием этих трех
типов компонентов, вступающих в реакцию с фталевым ангид-
Модифицированные алкидные смолы
381
рядом, .полученные модифицированные алкидные смолы будут
приближаться по свойствам либо к полимерам трехмерного
строения, либо к термопластичным смолам, либо, наконец, к
исходному маслу. Переходы от одного типа к другому происходят
постепенно, по мере изменений в соотношениях компонентов.
Мы считаем неправильным приводить здесь схемы упорядо-
ченных структур, встречающиеся во всех опубликованных сооб-
щениях. Можно лишь привести в качестве примера следующую
схему строения части макромолекулы модифицированной ал-
кидной смолы:
Ч
/CHgOOCR
о=с с—о—сн
| 'xch2oocr
о о о о о ch2oocr
I) II I II II I
---ОС С—О—СН2—СН—СН2—О—С С—о-сн
I I
СН2 О О СН2
I II II I
О О СН—О—С С—О—СН
I
CHaOOCR
Нерешенным остается также важный вопрос, каким обра-
зом заканчиваются молекулы. В приведенной схеме этот во-
зрос умышленно обойден. Можно предполагать, что молекула
растет до бесконечности в соответствии с наличием свобод-
ных валентностей, но возможно также, что диглицерид этери-
фицирует кислотную группу молекулы фталевой кислоты, огра-
ничивая в этом месте рост макромолекулы.
Опыт показывает, что все алкидные смолы имеют весьма
значительное кислотное число, т. е. содержат свободные карбок-
сильные группы, а следовательно, также и свободные гидрокси-
лы. В макромолекулах модифицированных алкидных смол
должны иметься такие как бы торчащие, свободные группы
382
Гл. XL Алкидные смолы
—СООН и —ОН. При изображении структуры реальной моле-
кулы необходимо учитывать и этот факт:
О О ОН О О CHjOOCR
II II I II II I
НО—С с—о—сн2—сн—сн2—о—с с—о—сн
но—с с—о—сн2 о о о
Впрочем, иную картину даже трудно себе представить, так
как если бы при этерификации строго 'Соблюдались стехиоме-
трические пропорции, получались бы непригодные для исполь-
зования гели трехмерного строения, не образующиеся в дейст-
вительности.
Практически наличие свободных карбоксильных и гидрок-
сильных групп влечет за собой появление нежелательных свойств,
а именно: действие смолы на основные пигменты — вслед-
ствие наличия карбоксилов, чувствительность пленки к воде—
вследствие присутствия гидроксилов. Такие неудобства не яв-
ляются решающими для смол горячей сушки, при которой по-
вышается степень поликонденсации, но могут вызвать затруд-
нения в случае покрытий воздушной сушки. Эта проблема
остается нерешенной в течение длительного времени, так как
принимаемые паллиативные меры здесь явно недостаточны.
Можно было бы применить этерификацию свободных карб-
оксильных групп путем нагревания смолы с одноатомным
спиртом. Однако это привело бы к еще большим затруднениям.
Одновременно с этерификацией кислотных групп, не в меньшей
мере, неизбежно будет идти реакция алкоголиза, ведущая к
разрушению макромолекулярной сетки и появлению большого
Модифицированные алкидные смолы 383
числа дополнительных свободных гидроксилов. Качество смол
при этом сильно ухудшается.
Можно также пытаться приготовить смолу только алкого-
лизом, например обработкой диметил фталата моноглицеридом:
СОО—CHs + НО—R —>• COO—R СН3—ОН
^1-СОО—СН3 ООО—сн3
Образовавшийся метиловый спирт легко удаляется при на-
гревании. В этом случае кислотные группы всегда будут ос-
таваться этерифицированными или моноглицеридом или мети-
ловым спиртом. Практика показывает, что этим методом мож-
но приготовить модифицированные алкидные смолы, но, к со-
жалению, кислотное число постепенно повышается в ходе опе-
рации до исходного значения, соответствующего смеси моно-
глицерида и фталевого ангидрида.
Напомним, что существуют методы, включающие нейтрали-
зацию кислотных групп с образованием металлических солей.
Но в отношении гидроксильных групп проблема остается нере-
шенной, так как возникают те же препятствия: повышение
кислотного числа и опасность ацидолиза макромолекул.
Теоретически данная проблема легко разрешима: достаточ-
но обработать смолу на холоду диазометаном для метилирова-
ния кислотных групп, а затем фенилизоцианатом для блокиро-
вания гидроксильных групп до образования фенилуретана.
Можно было бы применить для этой цели также кетен. Однако
помимо того, что эти операции являются весьма тонкими, на-
званные реактивы при нынешнем уровне техники мало доступ-
ны для применения в промышленных масштабах.
Внимание специалистов-лакокрасочников привлекают кис-
лотные числа смол как константы, удобные для практического
пользования; определение содержания гидроксилов в алкид-
ных смолах осложняется необъяснимыми явлениями. Это про-
исходит потому, что в химии модифицированных алкидных смол
возможно протекание разнообразных побочных реакций, ко-
торые в большинстве случаев практически не удается разгра-
ничить, даже производя многочисленные анализы и измерения.
Эти трудности, несомненно, являются причиной скудости на-
учной литературы по этому вопросу. Техническая же литера-
тура изобилует подобными данными; не объясняя явление,
она удовлетворяется в большинстве случаев тем, что излагав!
полезные результаты различных опытов, бесконечные варианты
изменения многочисленных параметров.
Чтобы устранить некоторые недостатки алкидных смол,
прибегают к их модифицированию мочевино-формальдегидными,
384
Гл. XI. Алкидные смолы
меламино-формальдегидными, феноло-формальдегидными смо-
лами, абиетиновой кислотой, диизоцианатами, виниловыми по-
лимерами и т. д. Практические результаты иногда бывают
.интересны, но реакции, приводящие к образованию сложных
высокомолекулярных структур, остаются все еще гипотетиче-
скими.
ЛИТЕРАТУРА
"G. Champetier, Les molecules geantes et leurs applications, Albin Michel,
Paris, 1948.
H. R. Flee k, The theory of polymerisation, The English Universities Press
Ltd. London, 1946.
J. i. M a t t i e 1 1 o, Protective and decorative coatings, Wiley a. Sons, New
York.
.J.; В о u r r y, Resines alkydes Polyesters, Dunod, Paris, 1952.
'Von, Fischer, Paint and varnish technology, Reinhold, New York, 1948.
ГЛАВА XII
МАЛЕИНОВЫЕ СМОЛЫ
И. М. Буше
Первые работы, посвященные малеиновому ангидриду (Фор-
лендер), появились еще в 1894 г. Однако основные успехи в
этой области были достигнуты главным образом благодаря ра-
ботам Дильса и Альдера. Их исследования позволили выяснить
механизм диенового синтеза и показали ценность малеинового
ангидрида как реагента для количественного определения со-
пряженных двойных связей и как исходного продукта для син-
тезов.
В 1907 г. Альбрехт описал 'продукты реакции между цикло-
пент адиеном и некоторыми хинонами, а в 1920 г. Эйлер ука-
зал на возможность получения соединений на основе изопре-
на и бензохинона. В 1928 г. Дильс и Альдер1 опубликовали
результаты работы, проведенной ими с целью идентификации
некоторых ранее описанных соединений. Они показали возмож-
ность взаимодействия двух ненасыщенных соединений, одно
из которых является диеном (т. е. содержит в молекуле две со-
пряженные двойные связи С = С в положении 1,3), а другое
соединение—филодиеновая компонента — содержит одну эти-
леновую связь, активированную соседними функциональными
группами. В качестве одного из таких соединений они указали
на малеиновый ангидрид:
В дальнейшем им удалось обобщить явление присоединения,
которое может быть представлено следующей схемой:
25—156
386
Гл. XII. Малеиновые смолы
Для объяснения процесса присоединения филодиена к дие-
ну и отличительных особенностей, которыми должен обладать
филодиен, ими были высказаны два положения.
Первое состоит в том, что соединения, обладающие систе-
мой сопряженных двойных связей, присоединяют к концам этой
конъюгированной системы соединения, содержащие одну акти-
вированную двойную или тройную связь, образуя цикл из шести
атомов углерода. Сопряженные двойные связи раскрываются и
насыщают активированную этиленовую связь, в результате чего
в цикле остается одна двойная связь. Активирование двойных
или тройных связей осуществляется сопряжением с другой
двойной связью (углерод-углерод или углерод-гетероатом), на-
пример, со следующими группами:
Таким образом, сами диены тоже являются филодиенами
и способны соединяться между собой, образуя полимеры. Дей-
ствительно, хорошо известны полимеры бутадиена и изопрена.
В некоторых случаях, например для пентадиена, полимеризация
ограничивается димеризацией. Возможно и соединение друг
с другом различных диенов, как это .показал, например, Дюпон
для изопрена и диметилбутадиена.
Второе положение заключается в сходстве производных, по-
лученных с участием малеиновой кислоты, со структурой фта-
левого ангидрида и фталевой кислоты. Это сходство особенно
заметно в случае реакции бутадиена с малеиновым ангидридом,
протекающей с образованием тетрагидрофталевого ангидрида:
I
СН
СН2
СН2
Делаби2 дает весьма полный обзор различных механизмов
диенового синтеза и работ Дильса и Альдера.
С помощью малеинового ангидрида можно переходить от
полимеризации свободно-радикального типа к поликонденсации.
Сочетая эти два способа образования полимеров, можно полу-
чать разнообразнейшие конечные продукты сетчатой структуры.
Образование полимеров происходит в две стадии: присоедине-
ние диена и поликонденсация путем этерификации. В этой гла-
ве будет рассмотрена только стадия присоединения диенов и
основные свойства продуктов присоединения малеинового ан-
Присоединение малеинового ангидрида 387
гидрида к простым и сложным молекулам. Механизм поли-
конденсации в данном случае идентичен механизму образования
алкидных смол, на который автор в дальнейшем будет часто
ссылаться.
Присоединение малеинового ангидрида
Присоединение к простым молекулам. Малеиновый ан-
гидрид (или малеиновая кислота) присоединяется к си-
стеме сопряженных двойных связей с образованием цик-
лического ангидрида (или двухосновной кислоты), способ-
ного к этерификации. Филодиены представляют большой ин-
терес для синтеза полимеров, если образуемые ими комплексы
сохраняют функциональность, равную двум или более. Дей-
ствительно, совершенно очевидно, что для последующих реак-
ций образования цепей требуется полифункциональность ис-
ходных материалов. Однако при соединении двух веществ опре-
деленных функциональностей используется часть имеющихся
валентностей (четырех валентностей в случае присоединения ма-
леинового ангидрида), т. е. происходит уменьшение функци-
ональности. В качестве примера достаточно привести реакцию
присоединения акролеина к бутадиену.
СН2 СН2
СН2 / \
СН Ц НС сн2
I 4- СН || I
СН \ НС СН—СНО
'Ч сно \ /
СН2 CHS
Здесь бутадиен тетрафункционален, акролеин — трифункци-
онален, а получаемый тетрагидробензойный альдегид — также
' трифункционален.
Следовательно, нужно учитывать функциональности диена
и филодиена. Продукты присоединения малеинового ангидрида
имеют функциональность, по меньшей мере равную четырем,
причем две кислотные группы способны к этерификации. Во
всех этих .случаях получающийся аддукт может образовывать
линейные макромолекулы при этерификации двухатомными
спиртами и трехмерные макромолекулы при этерификации спир-
тами, содержащими более двух гидроксильных групп. Трехмер-
ные макромолекулы образуются также при этерификации двух-
атомными спиртами, если в аддукте сохранились другие функ-
циональные группы, имевшиеся в молекуле диена.
Присоединение малеинового ангидрида к простым молеку-
лам с образованием ангидридов двухосновных кислот, весьмр
близких, к фталевому ангидриду, позволяет затем путем поли-
25*
388
Гл. XII. Малеиновые смолы
конденсации получать полимеры, по свойствам близкие к фта-
левым полиэфирам. Однако присоединение филодиена вносит
в свойства аддуктов определенные особенности.
Если исходный диен тетрафункционален, то получающиеся ад-
дукты лишь теоретически обладают бифункциональностью как
производные этиленового ряда. В действительности, оставшаяся
двойная связь в положении 2,3 теряет свою реакционную способ-
ность, в особенности если исходный диен является замещенным.
Пространственные препятствия и электростатическое влияние
соседних групп препятствуют раскрытию этиленовой связи.
Поскольку присоединение малеинового ангидрида позво-
ляет синтезировать полимеры с особыми свойствами, таким пу-
тем можно получать интересные продукты. Так, можно по-
лучать смолы или пластификаторы из диолефинов, содержа-
щихся в погонах каменноугольной смолы и в крекинг-дистил-
ляте нефти4. Двухстадийный способ синтеза был описан фир-
мой British Celanese. Сначала путем пропускания сернистого
ангидрида при нагревании через смесь углеводородов получают
сульфон, после чего смесь нагревают с эфирами незамещенной
или замещенной малеиновой кислоты. С бутилмалеатом обра-
зуется дибутилтетрагидрофталат, пластификатор для целлю-
лозных или поливиниловых производных5:
/ CH-COOCJL
I + II
СИ—СООС4Н0
< СООС^Нд
^—СООС4Н9
Этерифицированные продукты присоединения малеинового
ангидрида к циклопентадиену — более или менее твердые смо-
лы. Неэтерифицированный аддукт представляет собой эндоме-
тилентетрагидрофталевый ангидрид (так называемый «ангид-
рид карбик»):
Гликолевые моноэфиры эндометилентетрагидрофталевой
кислоты можно использовать в качестве пластификаторов или
модификаторов восков. Они пригодны также как растворители и
пластификаторы для виниловых полимеров6.
Ацетилен в присутствии медных катализаторов образует
продукты полимеризации, которые могут присоединять малеи-
новый ангидрид. Так, полимеры дивинилацетилена присоеди-
Присоединение малеинового ангидрида
389
няют малеиновый ангидрид в количестве, соответствующем
числу имеющихся сопряженных связей.
Новый диен, имеющий систему сопряженных двойных свя-
зей, соединенных с насыщенным циклом, был получен Бейли7-
Это диметиленциклогексан
СН2 СН2
\
H2(Z /СН2
н2с-сн2
который можно рассматривать как бутадиен-1,3, для которого
невозможно свободное вращение вокруг С—С-связи, так как
она входит в насыщенный шестичленный цикл. Из этого соеди-
нения путем полимеризации получают твердые кристалличе-
ские полимеры. Можно полагать, что реакция с малеиновым
ангидридом протекает следующим образом:
Попвидимому, увеличение числа сопряженных двойных свя-
зей или их особое расположение может привести к новым про-
дуктам диенового синтеза. Некоторые подобные соединения уже
синтезированы и после освоения их промышленного производ-
ства, несомненно, будут играть важную роль. В качестве при-
мера можно привести циклооктатетраен, недавно полученный
Реппе с сотрудниками путем полимеризации -ацетилена под
умеренным давлением в присутствии особых катализаторов8.
Строение циклооктатетраена позволяет предвидеть очень боль-
шое число изомеров, еще не разделенных до настоящего време-
ни. Присутствие четырех сопряженных двойных связей обуслов-
ливает его высокую реакционную способность. Изучены
продукты присоединения малеинового ангидрида к двум изо-
мерным формам циклооктатетраена, содержащим сопряженные
двойные связи:
00=1
Малеиновый ангидрид может также присоединяться к раз-
личным видам виниловых соединений без образования циклов,
подобно тому как присоединяется заместитель к стиролу или
стильбену. В данном случае малеиновый ангидрид присоеди-
390
Гл. XII. Малеиновые смолы
няется к мономерному углеводороду только одной связью, при-
чем полимеры могут быть получены по двум механизмам: путем
присоединения и полиэтерификацией. Характерно, что эквимоле-
кулярное присоединение происходит при любых начальных соот-
ношениях исходных веществ. Избыточный малеиновый ангидрид
в процессе аддитивной полимеризации остается неизмененным и
при последующей этерификации ведет себя как обычная двух-
основная кислота.
Полимеризация путем присоединения приводит к образова-
нию только линейных полимеров, растворимых в ацетоне и вод-
ных щелочах и пригодных в качестве аппретов для текстиль-
ных материалов. Поликонденсация всегда сопровождается поли-
меризацией путем присоединения; поэтому с увеличением
молекулярного веса .полимера уменьшается возможность полу-
чения совершенно нейтральных смол.
Замещенные стиролы также дают эквимолекулярные .продук-
ты, присоединения (реакция протекает в присутствии перекисей):
СН=СН8
НС==СН
4- ОС
В патентах английской фирмы Beckacite указывается воз-
можность получения продуктов конденсации кетонов или аль-
дегидов с малеиновым ангидридом или его гомологами. Обра-
зовавшиеся аддукты при этерификации многоатомными спир-
тами образуют твердые полимеры, совместимые с нитроцеллю-
лозой и растворимые в кислородсодержащих полярных раство-
рителях (ацетон, спирты), а также в ксилоле. Эги полимеры
могут быть модифицированы маслами; получаемые таким об-
разом продукты образуют после горячей сушки твердые и
гибкие пленки. Возможна также модификация таких полиме-
ров с целью получения пленок, высыхающих на воздухе. Кроме
того, не исключена возможность совмещения их с фенольными
смолами и полистиролом. В зависимости от метода получения
продукты имеют различную кислотность и вязкостьВ 9.
(Получаемые полимеры приближаются к полимерам, образую-
щимся путем присоединения диенов к р-нафтолу. >В этом случае,
как показал Зальфельд, получается «изоаддукт». Диены могут
присоединяться только к одному углеродному атому р-нафтола,
а именно — в орто-положении ,по отношению к гидроксилу.
Этерификация полученного продукта одновалентными спиртами
или моноэфирами гликоля приводит к пластификаторам или к
прозрачным твердым смолам с низким кислотным числом. Такие
смолы растворимы в ароматических растворителях и образуют
Присоединение малеинового ангидрида
391
при горячей сушке (при температуре около 125°) твердые и
блестящие пленки10.
Присоединение к кислотам типа абиетиновой. Не вдаваясь
в детали механизма присоединения малеинового ангидрида к
диенам терпенового ряда, описанные в статье Брю и Лежанд-
ра, напомним о них в основных чертах.
Рассматриваемый аддукт является исходным веществом для
получения многочисленных смол, применяемых в лакокрасоч-
ной промышленности и составляющих большую часть так на-
зываемых малеиновых смол.
Ломбар11 показал важнейшие различия, которые обнару-
живаются в механизме присоединения малеинового ангидрида
к разным смоляным кислотам. Присоединение левопимаровой
кислоты происходит при обычной температуре, а для других
кислот группы абиетиновой — при температуре около 180°.
Абиетино-малеиновый аддукт является индивидуальным со-
единением и по свойствам сильно отличается от канифоли. Тем-
пература его плавления достигает 220°, тогда как немодифици-
рованные смоляные кислоты в основном плавятся при 150—180°.
Главным преимуществом аддукта является, таким образом, зна-
чительное повышение температуры плавления канифоли.
При рассмотрении подобной реакции присоединения мож-
но заметить, что две сопряженные двойные связи блокируются
и что присоединение малеиновой кислоты или ангидрида пре-
вращает одноосновную кислоту в трехосновную:
Другими словами, свойства канифоли как ненасыщенного со-
единения почти полностью исчезают, так как оставшаяся двой-
ная связь значительно менее реакционноспособна, чем две со-
пряженные связи. Аддукт проявляет заметную инертность, по-
скольку не присоединяет бром даже при длительном соприкосно-
вении. Эта инертность — одно из основных качеств абиетино-
малеиновых смол. Они не связывают кислород воздуха и, сле-
довательно, не желтеют, как лаки на основе канифоли или ее
эфира.
С другой стороны, благодаря трифункциональности полу-
чающегося аддукта увеличивается возможная разветвленность
392
Гл. XII. Малеиновые смолы
продуктов взаимодействия с многоатомными спиртами. Темпе-
ратура плавления значительно повышается, если для повыше-
ния водостойкости смолы этерификация проведена настолько
глубоко, что прореагировали все кислотные группы и не оста-
лось свободных гидроксилов. Температура плавления повы-
шается с увеличением степени этерификации.
Температуры плавления сложных эфиров абиетиновой кис-
лоты в зависимости от молекулярного веса приведены в табл. 20.
Такие же закономерности наблюдаются для абиетино-малеи-
новых эфиров, но полнота этерификации достигается здесь
очень быстро.
' Сложные эфиры абиетиновой КИСЛОТЫ ТАБЛИЦА 20
Эфиры Молекуляр- ный вес Температура кипения .°C Температура плавления °C
Метиловый . Этиловый Этиленгликолевый Глицериновый Пентаэритритовый Эфир канифоли (триабиетат глице- рина) Пенталин (тетраабиетат пентаэрит- рита) Льюисол (абиетат-малеат глице- рина) 316 330 632 944 1272 360—365 205 (при 4 мм рт. ст.) Жидкость То же 45—60 150—162 115—160 90—110 116—160 140—160
Пр именами е. Последние три эфира являются торговыми продуктами.
На основе трифункциональных продуктов присоединения ма-
леинового ангидрида могут быть получены всеми обычными
методами алкидные смолы. При этом можно получать смолы
из слабо этерифицированных аддуктов, содержащие кислотные
группы и растворимые в спирте, твердые смолы, нераствори-
мые в спиртах, но даже при высоком молекулярном весе рас-
творимые в ароматических углеводородах и маслах, а также не-
растворимые смолы, способные изменяться под действием тепла
и пригодные для прессовочных порошков. Смолы последнего
типа мало применяются из-за слишком высокой стоимости.
Смолы, содержащие кислотные группы и растворимые в
спирте, изготовляются путем нагревания до 200° канифоли с
малеиновым ангидридом с последующей частичной этерифика-
Присоединение малеинового ангидрида
393
цией многоатомными спиртами. Операция проводится в токе
инертного газа, причем температура не должна превышать 200°,
чтобы структурирование было минимальным и сохранялась
достаточная растворимость смолы в спирте и стабильность рас-
творов. Конечное кислотное число должно составлять от 80 до
110, а окраска получающегося продукта должна быть по край-
ней мере такой же бледной, как у исходной канифоли. Полу-
ченные смолы вследствие наличия свободных кислотных групп
растворимы в разбавленных щелочах и в водных растворах
солей щелочных металлов, например в растворах буры или би-
карбоната натрия.
Растворимые в маслах канифольно-малеиновые смолы с низ-
ким кислотным числом могут быть изготовлены таким же об-
разом, но при более высоких температурах (225—260°). Соот-
ношения канифоли, малеинового ангидрида и многоатомного
спирта подбирают из расчета полной этерификации кислотных
групп аддукта. Практически для повышения водостойкости
смолы количество многоатомного спирта должно быть несколько
меньше стехиометрического. В данном случае приходится делать
выбор между двумя возможностями: или ввести в реакцию
больше многоатомного спирта, чтобы сместить равновесие в
сторону более полной этерификации, или же допустить, чтобы
продукт имел определенное кислотное число во избежание не-
удобств, обусловливаемых присутствием свободных гидроксиль-
ных групп. Выпускаемые в продажу смолы имеют кислотные
числа от 15 до 40 и в то же время превосходную атмосферо-
стойкость. Высокая атмосферостойкость достигается удалением
в конце операции фракций, содержащих продукты декарбок-
силирования канифоли, могущие образоваться в процессе кон-
денсации при высокой температуре и весьма чувствительные
к действию света. Удаление этих фракций достигается путем
выдерживания смолы в течение некоторого времени под ва-
куумом.
Для получения абиетинового аддукта требуется около 35%
малеинового ангидрида по отношению к канифоли (98 и
302 вес. ч.), но промышленные малеиновые смолы обычно не
являются продуктами конденсации глицерина с полным абие-
тиновым аддуктом, так как их стоимость была бы слишком
высокой. Кроме того, при этерификации трехосновной кислоты
спиртом с функциональностью выше двух, получаются очень
сложные соединения, быстро переходящие в стадии нераство-
римости и неплавкости. Поэтому пришлось уменьшить коли-
чество малеиновой кислоты и подобрать количества аддукта,
достаточные для сообщения смоле высокой стойкости. В со-
став конечного продукта абиетиновая кислота входит в избыт-
ке. ограничивая, таким образом, степень его структурирования..
394
Гл. XII. Малеиновые смолы
Одной из лучших смол, выпускаемых в настоящее время, яв-
ляется продукт конденсации восьми молей абиетиновой кислоты
и четырех молей фталевого ангидрида, этерифицированный
пятью молями глицерина. Свойства смол существенно зави-
сят от количества взятого фталевого ангидрида (табл. 21).
ТАБЛИЦА 21
Смолы на основе абиетино-малеинового аддукта и глицерина
Содержа-
ние
малеино-
вого ангид-
рида
%
Темпера-
тура
плавления
°C
Растворимость
Совместимость
с маслами
7
8
9,5
11—12
14
82—90
87—96
93—102
101—114 1
108—119 |
Легко растворяют-
ся в смеси
этилацетат-бен-
. зол-уайт-спирит
Растворяются в
отношении 1 : 1
в уайт-спирите
Дают прозрачный рас-
твор в льняном мас-
ле средней степени
полимеризации
Необходимо нагрева-
ние при 250°
При нагревании малеиновых смол с маслами кривая вяз-
кости повышается круче, чем для глифталевых смол, модифи-
цированных тем же количеством масла. По эластичности, твер-
дости, совместимости и стойкости малеиновая смола, модифи-
цированная на 60% маслом, приближалась к глифталевой смо-
ле, модифицированной на 50%. Продолжительность варки мас-
ляного лака, даже при достаточно низком кислотном числе при-
меняемой смолы, ограничена и зависит от частоты молеку-
лярной сетки. Поскольку малеиновые смолы получаются из
трифункциональных мономеров, в то время как фталевый ангид-
рид бифункционален, структурирование их проходит быстрее
и они содержат значительно больше жирных радикалов в том
же объеме сетки.
Твердые абиетино-малеиновые смолы совместимы с нитро-
целлюлозой и сообщают лаком высокий блеск. Вследствие боль-
шой твердости смол в лаки на их основе приходится вводить
больше пластификатора. Существенным преимуществом этих
смол является то, что они не удерживают растворителей.
Необходимо отметить, что главный эффект присоединения
фталевого ангидрида к канифоли состоит в превращении веще-
ства, не являющегося пленкообразующим, в трехмерное соеди-
нение. Применяемые в лакокрасочной технике глицериновые
или пентаэритритовые эфиры абиетиновой кислоты концентри-
руют двойные связи природной кислоты вокруг одного и того
Присоединение малеинового ангидрида
395
же центра, что приводит к усилению окраски по мере увели-
чения функциональности многоатомного спирта и повышения
температуры этерификации:
Покрытия, образованные такими смолами, являются не плен-
ками, а нагромождениями непрерывных рядов сложных мо-
лекул. Ценность этих смол в качестве компонента лаков опре-
деляется их растворимостью в маслах, где они создают жесткую
основу структурных сеток, довольно редких в момент обра-
зования.
Следовательно, диеновым синтезом с последующей этери-
фикацией можно получать бесконечную гамму синтетических
смол, модифицированных абиетиновой кислотой. Твердость по-
лучаемых смол возрастает с увеличением функциональности
многоатомных спиртов, участвующих в реакции, скорость вы-
сыхания зависит от характера масла, с которым сочетается
аддукт, показатели цветности в основном такие же, как исход-
ной канифоли или даже ниже. Кроме того, эти смолы отли-
чаются хорошей атмосферостойкостью и не желтеют в процес-
се эксплуатации. При их применении становится возможным,
в частности, приготовление белых лаков или светлых эмалей.
Абиетиновые аддукты могут образовывать металлические со-
ли, пригодные для использования в качестве сиккативов или
как агенты, препятствующие образованию плесени, на что ука-
зывали Вейсс и Гаварре12. Алюминиевые, цинке г,ые и магниевые
соли могут служить матировочными средствами.
Присоединение к терпенам. Среди наиболее распространен-
ных моноциклических терпенов а-терпинен и а-фелландрен, а
в ряду алифатических терпенов аллооцимен обладают систе-
мой сопряженных двойных связей. В то же время известно, что
другие терпены с несопряженными двойными связями, напри-
мер пинен, легко изомеризуются, главным образом в присут-
396
Гл. XII. Малеиновые смолы
ствии кислот и соединений, способных присоединяться к систе-
ме двойных связей.
Малеиновая кислота присоединяется к терпенам с образова-
нием аддуктов, например:
С терпенами, не содержащими сопряженных двойных свя-
зей, малеиновый ангидрид в отсутствие кислоты образует
различные соединения. Присоединение происходит двумя путя-
ми, по-видимому, с участием подвижного атома водорода13:
Этому вопросу было посвящено много патентов и журналь-
ных статей, начиная с первых патентов фирмы Hercules Powder14,
взятых в 1938 г. Скорость присоединения малеинового ангид-
рида к различным терпеновым углеводородам с изолированными
двойными связями изучал Гульч, доказавший, что лимонен
под влиянием малеиновой кислоты изомеризуется и дает тот
же продукт диенового присоединения, что и терпинен15. Совер-
шенно такой же аддукт, обычно называемый «ангидридом пет-
рекс», может быть получен и из других терпенов, изомеризую-
щихся в терпинен. Действительно, терпинен получается изоме-
ризацией пинена серной кислотой.
Присоединение малеинового ангидрида
397
Ангидрид петрекс является смесью соединения типа аддукта
Дильса и Альдера и сополимера продуктов присоединения, от-
вечающих формулам 1 и II (см. стр. 396). Продукт диенового
синтеза образует с двухатомными спиртами линейные полиэфи-
ры, в то время как сополимер продуктов присоединения дает
трехмерное соединение. Линейный полимер играет в смеси роль
пластификатора трехмерного полимера. Изменяя число атомов
углерода, разделяющих две гидроксильные группы бифунк-
ционального спирта, можно получать продукты различной сте-
пени твердости.
Продукты присоединения описанного типа получаются про-
стым нагреванием компонентов при температуре кипения угле-
водорода. Для пинена, как показал Дюпон16, внутренний мо-
стик по реакционной способности близок к двойной связи и
образует сопряженную систему с двойной связью шестичлен-
ного кольца. Смесь пинена и малеиновой кислоты нагревают
с обратным холодильником, повышая температуру от 156° до
180° в течение 2—3 час. По мере повышения температуры пи-
нен изомеризуется в терпинен и лимонен, температура кипения
которых равна 178°. Изомеризацию можно рассматривать как
обратимую реакцию, равновесие которой непрерывно смещается
в сторону образования изомеров, поскольку они непрерывно
удаляются из системы, связываясь с малеиновой кислотой и
образуя соответствующий аддукт, относительно очень стойкий.
Этот аддукт, являющийся ангидридом двухосновной кислоты,
может быть этерифицирован таким же образом, как фталевый
ангидрид, с образованием светопрочных, очень светлых смол,
модифицируемых маслами или природными смолами. При эте-
рификации глицерином получаются твердые и хрупкие смолы,
растворимые в спирте и применяемые в качестве заменителей
шеллака.
Можно использовать метод, немного отличающийся от обыч-
ного метода получения немодифицированных смол, вводя во
взаимодействие, как в случае абиетино-малеиновых смол, не
малеиновую кислоту, а уже полимеризованный эфир, получен-
ный нагреванием малеинового ангидрида с многоатомным спир-
том в течение получаса при 150°. Таким путем быстрее полу-
чаются смолы с высоким молекулярным весом. Эти немодифи-
цированные смолы растворимы в ароматических углеводородах,
сложных эфирах и кетонах и применяются для производства
безопасных стекол.
Этерификацией двухатомными спиртами или простыми эфи-
рами, имеющими два свободных гидроксила, можно получать
более или менее твердые смолы. В частности, при этерификации
этиленгликолем получаются твердые светлые смолы с кислот-
ным числом 40, плавящиеся при температуре около .100°. Этери-
398
Гл. XII. Малеиновые смолы
фикация одноатомными спиртами приводит к образованию-
жидкостей -с высокой температурой кипения, которые могут
служить пластификаторами или высококипящими раствори-
телями.
Васильев17 указывал на аномалии, встречающиеся при опре-
делении химических констант продуктов присоединения. О»
предложил для терпеновых аддуктов своеобразную структуру,
предполагающую наличие одной свободной кислотной группы:.
.О
СН—с'
II /О
сн—
хо
Ангидрид петрекс и ангидрид карбик имеют достаточное
сходство с фталевым ангидридом, чтобы применяться вместо
него в производстве алкидных смол и пластификаторов.
При этерификации ангидрида петрекс аллиловым или мет-
аллиловым спиртом получается мягкая смола, способная по-
лимеризоваться .в присутствии перекиси бензоила. Этерификация
проводится в присутствии таких катализаторов, как п-толуол-
сульфокислота или ее натриевая соль. Продукт реакции может
служить интересным образцом композиций, применяемых в каче-
стве покрытий, без растворителей. Тщательно подбирая заме-
стители, можно получать продукты довольно низкой вязкости.
Образование сетки в таких системах обусловливается наличием
аллильной группы.
Следует подчеркнуть возможность использования диеново-
го синтеза для непосредственного присоединения малеинового
ангидрида или кислоты к природным терпеновым кислотам,
содержащимся в виде раствора в выделениях деревьев хвойных
пород. При этом нужно подбирать соотношение углеводорода
и природной кислоты и температуру присоединения, которая
должна повышаться лишь постепенно, по мере превращения
терпена.
Образование хроманового ядра. Механизм диенового син-
теза может быть различным. Известно присоединение малеино-
вого ангидрида к кетонам этиленового ряда и к нафтолам, не
замещенным в орто-положении. Возможно также присоединение
этиленовых соединений к о-метилольным производным фенола.
Присоединение малеинового ангидрида
399
Реакция, протекает, по-видимому, с потерей молекулы воды и
промежуточным образованием о-хинонметида:
L СН2:ОН; | |
................. V^ch,
сн—с<
+ I
'^/Хсн2 сн—с<
о-Метилольные производные фенолов являются первичным
продуктом процесса образования феноло-формальдегидныхсмол.
Гульч18, а затем Генель изучили этот процесс конденсации и
показали переход к хромановому кольцу. Этим путем можно
значительно умножить число сочетаний алкидных смол с фе-
нольными, которые в свою очередь могут присоединять диены,
образующиеся при нагревании масел.
Присоединение к маслам. Малеиновый ангидрид присоеди-
няется к природным маслам, главным образом к тем, в кото-
рых имеется система сопряженных двойных связей, как, на-
пример, в тунговом масле, изученном Морреллем. Фармер по-
казал возможность присоединения малеинового ангидрида к
льняному маслу, имеющему только одну двойную связь, путем
замещения подвижного водородного атома активированной двой-
ной связью метиленовой группы с образованием ангидрида за-
мещенной янтарной кислоты:
R—СН2—СН=СН—R—СООН R—СН—СН=СН—R—СООН
+ |
СН—С=О СН—С=О
I > I >
СН—С=О СН2—С=О
Каппельмейер и ван Гоор19 показали, что при температурах,,
при которых невозможна термическая полимеризация (200°),
наблюдается присоединение малеинового ангидрида к системе
сопряженных двойных связей. При этом значительно умень-
шается иодное число и повышается показатель преломления.
Эта реакция широко используется для отверждения масел,
преимущественно в США. Малеиновый ангидрид берут обычно
в количествах порядка 5—10%. Можно также присоединить
малеиновый ангидрид к изомеризованным льняному и соевому
маслам и к дегидратированному касторовому маслу, содержа-
щим сопряженные двойные связи.
400
Гл. XII. Малеиновые смолы
Присоединение к сложным молекулам. Ранее была рассмот-
рена возможность переходить с помощью малеинового ангид-
рида от механизма полимеризации путем присоединения к меха-
низму 1поликонденсации. Эта возможность сохраняется также для
полимеров, содержащих сопряженные двойные связи. В сетке
трехмерного полимера в процессе ее образования или в уже
образовавшейся возможно возникновение новых внутримолеку-
лярных связей, число которых может быть увеличено путем эте-
рификации многоатомным спиртом. Такие реакции протекают в
макромолекулах большинства продуктов полимеризации или по-
ликонденсации, получаемых из рассмотренных выше мономеров.
Благодаря этому путем обработки каучука одновременно
малеиновой кислотой и фенолом могут быть получены смолы,
пригодные для литья. Присоединением малеинового ангидрида
к димеризованной канифоли и к политерпеновым смолам до-
стигается повышение их вязкости или температуры плавления.
Малеиновый ангидрид присоединяется также к димеризованным
жирным кислотам, образующимся при термической полимери-
зации тунгового или ойтисикового масла. Аналогичным обра-
зом он способствует значительному повышению вязкости алкид-
ных смол, модифицированных высыхающими маслами. Введе-
нием малеиновой или фумаровой кислот в алкидные смолы
достигается более быстрое повышение вязкости, становится
светлее их окраска и улучшается водостойкость вследствие об-
разования более плотной трехмерной структуры.
Линейная полиэтерификация
Если подвергать этерификации эквимолекулярные количества
малеиновой кислоты и двухатомного спирта, то первая стадия
реакции будет состоять в образовании бифункциональной окси-
кислоты
НОСН2—СН2ООС—СН=СН—СООН
При последующей этерификации образуются линейные по-
лимеры, молекулярный вес которых будет постепенно увеличи-
ваться по мере протекания процесса. Конечный продукт, при
условии отсутствия побочных реакций, можно представить сле-
дующим образом:
НО—I—СН2—СН2ООС—СН=СН—СОО—]„—н
Степень полимеризации может быть вычислена путем опре-
деления концевых активных групп. Она будет выражаться отно-
шением числа исходных молекул к числу непрореагировавших
молекул:
Линейная по.шэтерификация
401
Если принять, что в реакцию вступило 95% функциональ-
ных групп, то степень полимеризации будет равна:
пр —_____!__— 20
" 1—0,95
Таким образом, каждая молекула конечного полиэфира бу-
дет состоять в среднем из двадцати звеньев первичного слож-
ного эфира.
В случае малеиновой кислоты и гликоля трудно довести
этерификацию до полной нейтрализации. При этом могут про-
текать побочные реакции, ограничивающие основную реакцию,
тем более что выделение воды превышает теоретическое и про-
дукт имеет склонность к очень быстрому желатинированию. Ос-
новная реакция также не протекает до конца прежде всего по-
тому, что в результате присоединения малеиновой кислоты
бифункциональный эфир превращается в тетрафункциональнэе
соединение (Брадлей, Джонстон и Кропа20):
I
СН—СООСН,—СН2ОН
СН—СООСН2—СН2ОН
I
Кроме того, малеиновая кислота сама способна полимери-
зоваться, образуя полиангидрид
сн=сн сн сн сн=сн | 1 сн=сн i I
1 1 п СО СО —► со 1 ОС СО ОС 1 । со ОС
1 1 / X
он он —о о' о О—
чем объясняется образование воды в количестве, превышающем
стехиометрическое.
Гликоль также может образовывать линейные полиэфиры:
----О—СН2—СН,—О—СНг—СН2—О—СН,—СН2—О-------
Как показали Кинле и Петке21, средний молекулярный вес
малеиновых полиэфиров, имеющих сложное строение, состав-
ляет от 1000 до 2000, что соответствует степени этерификации
(степени превращения) от 66 до 80%-
Под влиянием кислорода в присутствии катализаторов окис-
ления мягкие смолы на основе малеиновых полиэфиров могут
переходить в стадию неплавкости и нерастворимости. Этот про-
цесс, по-видимому, протекает параллельно с образованием плен-
ки— по механизму, аналогичному процессу высыхания масел.
Винцент22, как и Брадлей, считал, что строение активной
26—156
402
Гл. XII. Малеиновые смолы
группы гликольмалеата аналогично строению активной группы
тунгового масла:
—СН=СН—С=О и —сн=сн—сн=сн—
!
Однако Френч23 подчеркивал, что только под действием кис-
лорода невозможна полимеризация смол на основе ненасыщен-
ных полиэфиров. Но кислород, реагируя с некоторыми восста-
новителями, может образовать активный катализатор, пригод-
ный в качестве агента полимеризации. Меркаптаны, в частно-
сти, вызывают структурирование, очень быстро приводящее к
желатинированию. Превращение ненасыщенных полиэфиров в
смолы ускоряется перекисями, триэтилацетатом свинца, кобаль-
товыми солями органических кислот, а также облучением уль-
трафиолетовым светом.
Если малеиновую кислоту етерифицировать ненасыщенными
спиртами, то получаются полиэфиры, сходные с высыхающими
маслами. При сушке на воздухе они даже без применения сик-
кативов быстро образуют прозрачные, блестящие и эластичные
пленки, обладающие высокой адгезией24.
Смолы на основе гликольмалеата применяются главным
образом для модификации полимеризационных смол. Бутилен-
гликольмалеаты используются в качестве пластификаторов по-
ливинилацетата, придавая ему морозостойкость и повышая
адгезию. Их можно с тем же успехом использовать для пла-
стификации акриловых полиэфиров.
Поликонденсация и сополимеризация
Неоднократно отмечалась способность ангидрида малеиновой
кислоты и ее эфиров к реакциям присоединения с образованием
продуктов, имеющих строение замещенных производных янтар-
ной кислоты, а также к полимеризации с образованием линей-
ных молекул, содержащих повторяющийся остаток янтарной
кислоты. При присоединении к стиролу протекают обе эти реак-
ции. То же самое происходит, когда малеиновый ангидрид
взаимодействует с соединениями, содержащими виниловые, ак-
риловые или метакриловые радикалы:
I
—СН=СН + СН=СН — СН—СН— СН—СН— - —
I II— III
ОС со ОС с о
\ _ 'Чо'/ _ х
Такое многообразие реакций позволяет, разработать методы,
в которых способность различных веществ к совместной поли-
Поликонденсация и сополимеризация 403
меризации используется для получения многочисленных и раз-
нообразных продуктов. Соотношения компонентов играют при
этом весьма важную роль. Если в смеси стирола с малеиновым
ангидридом содержится более 20% мол. ангидрида, получается
полимер с постоянным молярным содержанием ангидрида, рав-
ным 49%, что соответствует образованию эквимолекулярного
продукта присоединения. Получаемый продукт является гете-
росополимером, так как малеиновый ангидрид не способен
полимеризоваться самостоятельно. Однако он может давать по
аналогичному механизму гетеросополимеры с виниловыми или
акриловыми эфирами. Эфиры малеиновой кислоты, образован-
ные одноатомными спиртами, имеют очень малую склонность
давать сополимеры, в то время как эфиры многоатомных спир-
тов легко полимеризуются совместно со стиролом, акриловы-
ми или виниловыми производными. Гликольмалеаты, раство-
ренные в стироле-мономере, используются в качестве связую-
щего при изготовлении слоистых пластиков. Малеаты, нераство-
римые в углеводородах, часто используются в форме терпено-
вых аддуктов25.
Можно провести сравнение между способностью к образо-
ванию сополимеров со стиролом у эфиров малеиновой и фума-
ровой кислот. Фумаровые эфиры лучше полимеризуются и лег-
че дают сополимеры. Например, они полимеризуются совместно
с изобутиленом, тогда как соответствующие малеиновые эфи-
ры к этому не способны. Прайсе26 объяснил эти различия в
поведении разной степенью поляризации двойных связей, а так-
же пространственными затруднениями, возникающими у ма-
леиновых эфиров и отсутствующими у фумаровых эфиров и
малеинового ангидрида.
Путем совместной полимеризации удается вводить в моле-
кулу полимера новые функциональные группы и получать про-
дукты, свойства которых обусловлены этими группами. Раз-
личия свойств могут быть вызваны также изменением относи-
тельного расположения молекулярных цепей вследствие сосед-
ства полярных групп. Так, в соединениях, полученных путем
совместной полимеризации винилхлорида и малеинового ангид-
рида, атомы хлора более подвижны, чем в поливинилхлориде.
Сополимеризация гликольмалеинатов с виниловыми производ-
ными приводит к образованию смол, твердость и эластичность
которых зависят от примененного катализатора, температуры
процесса и соотношения компонентов. Таким путем могут быть
получены продукты, устойчивые в водных растворах. Можно
также изменить растворимость поливинилхлорида сополимери-
зацией с малеиновыми эфирами насыщенных спиртов. При со-
полимеризации винилацетата с малеиновым ангидридом полу-
чаются твердые, слабоокрашенные смолы, растворимые в аце-
26*
404
Гл. XII. Малеиновые смолы
тоне и пригодные для шлихтовки и в качестве текстильных ап-
претов. Такие полимерные сложные эфиры могут быть омылены
или переэтерифицированы многоатомными спиртами.
Реакции гомологов малеиновой кислоты
Наряду с отмеченным сходством реакций малеиновой и фу-
маровой кислот, а также их эфиров, между ними имеются суще-
ственные различия, вызванные разным расположением карбок-
сильных групп по отношению к двойной связи. Фумаровая кис-
лота легче дает линейные полимеры, а образуемые ею аддукты
устойчивы только в присутствии ингибиторов полимеризации.
Благодаря высокой реакционной способности фумаровая кис-
лота может давать также продукты, аналогичные получаемым
из ее изомера. Фумаровые алкидные смолы не изменяют окрас-
ки при кипячении и весьма быстро приготовляются.
Аллилфумараты представляют собой жидкости, способные
полимеризоваться под влиянием света, при нагревании и под
влиянием перекисных катализаторов. Они могут полимеризо-
ваться совместно со стиролом, но эта реакция требует весьма
тщательного проведения и контроля ее течения. Получающиеся
пленкообразующие вещества совмещаются с большинством смол
и полимеризованных масел и дают пленки, очень стойкие к
свету, теплу, воде и щелочам. Одним из больших преимуществ
фумаровой кислоты является отсутствие токсичности.
Итаконовая кислота содержит винильную двойную связь:
СН2=С—СООН
I
СН2—СООН
Она представляет собой продукт разложения лимонной кис-
лоты и может быть получена путем брожения. Образование
аддуктов возможно только в редких случаях, вследствие асим-
метрической активации двойной связи. Например, итаконовая
кислота легко присоединяется к маслам, но только с трудом
образует абиетиновые аддукты. Алкидные смолы на основе
итаконовой кислоты имеют низкую вязкость, но медленно поли-
меризуются. При сравнительной легкости изготовления они об-
ладают глубокой окраской и получаются с плохими выходами.
Линейные итаконовые полиэфиры типа гликолевых легко по-
лимеризуются под влиянием перекисей и образуют твердые смо-
лы, тогда как соответствующие эфиры фумаровой и малеино-
вой кислот пластичны. Сополимеризацией итаконовых эфиров
со стиролом или акриловыми эфирами могут быть получены
ценные продукты, представляющие интерес для лакокрасочной
Анализ малеиновых смол
405
промышленности. Пока итаконовая кислота производится толь-
ко методом брожения, она может применяться в производстве
алкидных смол в качестве 15%-ной добавки к фталевому ан-
гидриду.
Мезаконовая и цитраконовая кислоты являются изомерами
итаконовой кислоты и соответствуют метилфумаровой и метил-
малеиновой кислотам:
СН—СООН
I
ноос—с—сн8
мезаконовая кислота
СН—СООН
II
СН3—СН—СООН
цитраконовая кислота
Анализ малеиновых смол
Изодиеновый показатель. Малеиновый ангидрид, применя-
емый, как известно, для определения количества сопряженных
двойных связей, кроме того, дает возможность определять спо-
собность соединения реагировать три высокой температуре с
изолированными двойными связями. Реакция протекает по ме-
ханизму изомеризации, замещения или же путем активации со-
седней метиленовой группы. Дюрр и Вельдинг определяли таким
образом изодиеновый показатель27.
Идентификация двухосновных кислот. Пфейффер и Беттлер
предложили метод качественной идентификации малеинового
ангидрида, основанный на сходстве его структуры со структу-
рой бензохинона:
Действительно, малеиновый ангидрид дает с диметил’анили-
ном продукт присоединения, имеющий интенсивную красно-
оранжевую окраску. Чувствительность реакции28 позволяет об-
наружить количества ангидрида порядка 0,1%.
Хансон показал возможность возгонки малеинового ангид-
рида путем нагревания его соединений в сухом виде. Раствор
сублимата в хлороформе дает оранжевое окрашивание с три-
фенилфосфином29.
С другой стороны, можно определять двухосновные кислоты
методом, основанным на образовании анилиновых производных.
Эти кислоты могут реагировать с анилином двумя способами.
Каппельмейер приводит пример фталевой кислоты, образующей
406
Гл. XII. Малеиновые'смолы
симметрический фталанил с выделением воды или производное
фталиминовой кислоты:
О
В ацетоновой среде и при известных условиях ведения опе-
раций получают количественно анилиновые производные, ко-
торые могут быть затем оттитрованы спиртовым раствором ед-
кого кали в присутствии фенолфталеина30.
Кроме того, фталевую кислоту можно осадить из водного
раствора, даже очень разбавленного, бензилизотиокарбамидом.
Другие двухосновные кислоты осаждаются только по истече-
нии некоторого времени31. Как отмечает Садолин, малеиновая
кислота может быть осаждена в виде малеата свинца32.
Кислотные числа малеиновых смол. Малеиновые смолы очень
легко омыляются и для определения их кислотного числа нужно
осторожно подходить к выбору реактива. Действительно, при
измерении кислотного числа классическим методом с помощью
спиртового раствора едкого кали получаются завышенные зна-
чения. Большинство изготовителей смол указывают кислотные
числа, найденные по этому методу, но для некоторых смол при-
водятся числа, составляющие немного менее половины зна-
чений, получаемых титрованием спиртовым раствором едкого
кали. Эти низкие значения получаются при титровании водным
раствором КОН, но конец реакции довольно трудно определить.
Вблизи точки перехода титрование необходимо проводить очень
медленно и значение кислотного числа следует указывать, как
наименьшее из найденных величин, так как зона перехода сме-
щается очень быстро.
ЛИТЕРАТУРА
1.0. Diels, К. Alder, Ann., 1928, 460, 98.
2. R. D е 1 а b у, Bull. Soc. chim., 1937, 4, 765.
4. G. T i о u t i о u n n i k о v, Chim. Ind., 1939, 42, 398D.
5. Англ. пат. 596463.
6. Carbide a. Carb. Chem. Co., пат. США 2311260.
Анализ малеиновых смол
407
7. R. J. Bailey, Conf. Am. Chem. Soc., Detroit, April, 1950.
8. W. R e p p e, Ann., 1948, 560, 1.
9. E. A. Bevan, R. S. R о b i n s о n-Beck, Koller Co., англ. пат. 600408,
600618, 607031, 608023, 608027.
10. J. Cr. S a 1 f e 1 d, Ber., 1940, 73, 376; см. также Johnson Co., англ. пат.
572455.
11. R. Lombard. Produits Resineux в серии «Materiaux de synthese»,
Dunod, Paris, 1946.
12. A. Weiss, J. G a v a r r e t, Congres Intern. Ind Peint., Paris.. 1947.
13. E. R. L i t t m a n n, Ind. Eng. Chem., 1936, 28, 1150.
14. Пат. США 1882298, 1978598, 1993025—1993037.
15. К. Hultzsch, Ber., 1939, 72, 1173.
16. G. Dupont, Bull. Soc. chim., 1931, 49, 478.
17. G. A. Vassilie-v, Congres Intern. Ind. Peint., Paris, 1947.
18. K. Hultzsch, J. prakt. Chem., 1941, 158, 275.
19. С. P. А. К a p p e 1 m e i e r, Congres Ind. Peint., Paris, 1947.
20. R. F. Bradley, W. B. Johnston, E. L. К r op a, Ind. Eng.
Chem., 1937, 29, 1270.
21. R. H. Kienle, F. E. Petke, J. Am. Chem. Soc., 1940,62,1053.
22. H. L. Vincent, Ind. Eng. Chem., 1937, 29, 1267.
23. D. M. French, Paint Oil Chem. Rev., 1949, 112, 15.
24. Rohm et Haas, фр. пат. 860842 1939 г.; I. G. F., фр. пат. 898257»
1943 г.
25. E. L. Kropa, T. F. Bradley, Ind. Eng. Chem., 1939, 31, 1512.
26. С. C. Price, Mecanisme des reactions de la double liaison Carbone—
Carbone, Dunod, Paris, 1951.
27. A. Durr, R. Wendling, Peint. Pigm. Ver., 1948, 24, 106.
28. P. Pfeiffer, T. Bottler, Ber., 1918, 51, 1820.
29. N. W. H a n s о n, J. Oil Col. Chem. Ass., 1949, 32, 137.
30. С. P. А. К a p p e 1 m e i e r, W. R. Van G о о r, Anal. Chem. Acta,
1948, 2. 146.
31. G. La Parola, Ann. chim. appl., 1940, 30, 275.
32. E. S a d о 1 i n, Ind. Eng. Chem., Anal. Ed., 1939, 11, 608.
ГЛАВА XIII
ЭПОКСИДНЫЕ СМОЛЫ
Р. Мартин, Ф. Саррю
Эпоксидные смолы, иногда называемые также эпихлоргидри-
новыми или этоксилиновыми, являются полимерами продуктов
конденсации эпихлоргидрина глицерина с многоатомными спир-
тами или бис-фенолами, чаще всего с дифенилолпропаном.
Эпоксидные смолы представляют собой новый тип синтети-
ческих смол и лишь сравнительно недавно появились на миро-
вом рынке. Количество эпоксидных смол, производимых в на-
стоящее время в ОША и в Европе, еще относительно невелико
по сравнению с общим производством синтетических смол, при-
меняемых в лакокрасочной промышленности и в производстве
пластических масс. Однако оно непрерывно возрастает благо-
даря ряду исключительных свойств, которыми обладают эти по-
лимеры.
Эпоксидные смолы можно рассматривать как линейные мно-
гоатомные спирты с концевыми эпоксидными группами. Высо-
кая реакционная способность концевых групп создает возмож-
ность сочетания эпоксидных смол с различными модификато-
рами, щелочными или кислотными отвердителями и термореак-
тивными смолами в широком интервале температур. Получен-
ные продукты после отверждения или полной полимеризации
имеют трехмерную структуру и обладают комплексом ценных
свойств, редко достижимым для других полимеров в столь ярко
выраженном виде. Из этих свойств следует особо отметить вы-
сокую адгезию к большинству обычно применяемых материа-
лов, прекрасную твердость и большую эластичность пленок, а
также очень хорошую стойкость к действию растворителей и
многочисленных химических агентов. Благодаря этим свойствам
эпоксидные смолы используются для различных защитных по-
крытий, как клеящие вещества, как компоненты печатных паст
для текстильной промышленности, как связующие для слоистых
пластиков, а также как электроизоляционные материалы, при-
меняемые для покрытия или пропитки обмоток и проводов.
В патентной литературе имеется большое число указаний на
различные области применения этих смол и приводятся много-
численные отвердители и модифицирующие агенты, вводимые
пои получении полупродуктов и готовых материалов.
Общие сведения
409'
Из эпоксидных смол наиболее известны смолы эпон фирмы
Shell, распространенные во Франции под названием эпикот, н
смолы аральдит фирмы Ciba.
Смолы эпон (эпикот) являются эпоксидными смолами мас-
сового производства и в основном применяются в качестве ис-
ходных веществ для производства лаков, красок и защитных по-
крытий воздушной и горячей сушки. Они пригодны также для
производства клеев, слоистых пластиков и покрывных составов.
Смолы аральдит, запатентованные в Европе фирмой-Ciba, пред-
ставляют собой полупродукты для лаков, в некоторых случаях
пигментированные; они используются также как клеи или
литьевые смолы. Чаще всего смолы аральдит выпускаются с
соответствующими кислыми или же щелочными отвердителями,,
состав которых не сообщается.
В США эпоксидные смолы и их производные выпускаются фир-
мой Shell Chemical Corporation, а также фирмами Devoe et Ray-
nolds. Bakelite, R. С. I. Beckacite, Ciba. В Европе производителя-
ми этих смол являются фирма Ciba a Bale, фирма В. Р. М.
(Shell), имеющая завод в Перни (Голландия), фирма Shell Petroleum
в Англии (завод Stanlow), английские фирмы Leicester Lovell, Bri-
tish Resin Products, Aero Research, а также фирма Chemische Wer-
ke Albert. Создание производственных установок для выпуска эпок-
сидных смол намечено и в других европейских странах, в частности
во Франции.
Эпоксидные смолы появились, настолько недавно, что пока
нет возможности охватить все области их применения и опи-
сать с достаточной полнотой их многочисленные модификации,
тем более, что состав некоторых из них, а также состав соот-
ветствующих отвердителей еще не опубликован. Здесь будет
сделана попытка дать лишь некоторые сведения о производстве
эпоксидных смол и сырья для их получения, о структуре, физи-
ческих и химических свойствах этих смол, а также об их про-
мышленном применении.
Мы надеемся, что данный краткий очерк возбудит у читате-
лей интерес к этим новым конденсационным смолам, удачно
дополняющим гамму синтетических смол, применение кото-
рых непрерывно возрастает в различных отраслях промышлен-
ности.
Возможность взаимодействия эпихлоргидрина глицерина с
полиоксисоединениями типа многоатомных спиртов или поли-
фенолов и получения таким образом смолообразных продуктов
поликонденсации была известна еще пятнадцать лет назад. Од-
нако развитие промышленного производства этого класса син-
тетических смол было невозможно до организации крупнотон-
нажного производства эпихлоргидрина из пропилена по спо-
собу английской фирмы Shell.
410
Гл. XIII. Эпоксидные смолы
Братья де-Трей в Швейцарии (франц, пат. 859061, 1938 г.;
франц, пат. 907172, 1943 г.) впервые установили, что термопла-
стичные смолы, полученные конденсацией эпихлоргидрина с
дифенилолпропаном, могут быть отверждены с переводом в не-
плавкое состояние путем взаимодействия с ангидридами кислот
или аминами. Они .показали также исключительную адгезию
этих смол ко многим материалам, например к дереву, стеклу,
некоторым пластическим массам, а также к металлам, в осо-
бенности к алюминию и легким сплавам. Через некоторое вре-
мя в'США Гринли с сотр. (фирма Devoe and Raynolds) осуще-
ствили ‘синтез, аналогичный синтезу братьев де-Трей, и пока-
зали, что для получения смол можно использовать большое
число эпоксисоединений и ароматических полиоксисоединений;
кроме того, они' предложили новую гамму лаковых смол, полу-
чаемых этерификацией вторичноспиртовых групп эпоксидных
смол жирными кислотами.
Эпоксидные смолы могут быть также конденсированы с дру-
гими термореактивными смолами типа мочевино-формальде-
гидных, мел амино-форм альдегидных или феноло-формальдегид-
ных с образованием пленкообразующих веществ, представляю-
щих большой интерес вследствие высокой адгезионной способ-
ности, эластичности и химической стойкости.
Интересно отметить, что производство эпоксидных смол,
впервые появившихся на рынке в 1940 г., в течение последних
лет увеличилось почти в 10 раз —от 3000 т в 1952 г. почти до
30 000 т в 1955 г.
До настоящего времени основная часть выпускаемых эпо-
ксидных смол используется в промышленности лаков и красок.
•Однако на основании работ, проведенных в других областях
промышленности, можно предвидеть их широкое применение в
машиностроительной, электротехнической, химической промыш-
ленности, в авиа- и автомобилестроении и т. п. в виде плит,
фитингов, труб, конструкций, соединяемых путем склейки, слои-
стых (пластиков, стеклопластиков, аппаратуры, моделей и т. п.
Для иллюстрации возможностей сбыта эпоксидных смол
можно указать, что только для производства шлангов, предна-
значенных для транспортировки кислых нефтепродуктов, может
потребоваться около 20006 т эпоксидных смол. При этом вы-
годы от их применения компенсируют значительно более высо-
кую стоимость эпоксидных смол по сравнению с применявши-
мися до сих пор для этой цели поливинилхлоридными пласти-
ками или синтетическими каучуками.
Эпоксидные смолы получаются конденсацией эпихлоргидрина
СН2—СН—СН2—С1
Исходные вещества для синтеза 411
с многоатомным спиртом или бис-фенолом в присутствии едкого
натра при температуре около 10(Г. Обычно применяемым бис-
фенолом является 2,2-(4,4'-диоксидифенил)-пропан, называе-
мый иногда дифенилолпропаном или бис-фенолом А.
В условиях, при которых осуществляется эта реакция, и
эпихлоргидрин и дифенилолпропан бифункциональны вследст-
вие наличия в молекуле эпихлоргидрина эпоксидной группы и
атома хлора, а в молекуле дифенилолпропана — двух феноль-
ных гидроксилов в пара-положениях. Следовательно, теорети-
чески при ^конденсации должно получаться соединение линей-
ной структуры.
В действительности установлено, что эпоксидные смолы этого
типа термопластичны.
Изменяя условия реакции конденсации и соотношения ис-
ходных компонентов, можно получить серию смол, средний
молекулярный вес которых изменяется в довольно широких пре-
делах, обычно между 350—400 и 3500—4000. Эти смолы пред-
ставляют собой более или менее густые жидкости или твердые
вещества янтарного цвета; температура размягчения их по мере
повышения степени конденсации повышается, обычно ступен-
чато, от 10 до 150°.
Исходные вещества для синтеза эпоксидных смол
Дифенилолпропан. Этот продукт используется для производ-
ства некоторых типов феноло-формальдегидных смол. Однако
к дифенилолпропану, предназначенному для производства эпо-
ксидных смол, необязательно должны предъявляться те же
требования, что при синтезе феноло-формальдегидных смол.
Только на основе данных о синтезе эпоксидных смол в лабора-
торных или полупроизводственных условиях, согласованных с
данными эксплуатации полученных полимеров, можно устано-
вить пригодность дифенилолпропана.
Лабораторный метод синтеза дифенилолпропана с выходом
50—60% состоит в конденсации ацетона с фенолом в присут-
ствии серной кислоты*.
По другому методу ацетон с фенолом нагревают в присут-
ствии уксусной и соляной кислот**.
Эпихлоргидрин получают присоединением хлорноватистой
кислоты к хлористому аллилу, с последующим выделением со-
ляной кислоты. Для получения хлористого аллила пропилен
хлорируют при нагревании по способу, разработанному и запа-
тентованному фирмой Shell.
*Schmidler, Lang, Вег., 43, 2814 (1910).
** A. Di ami п, Ber., 25, 334 (1892).
412
Гл. XIII. Эпоксидные смолы
Реакции, приводящие к образованию эпихлоргидрина, можно
представить следующим образом:
С12
СН3-СН=СН2-------► СН2—СН=СН2
I
С1
СНа—СН=СН2
<L
нею
—►СН,—СН—СН2
I I \
С1 С1 он
-*сн2—сн—сн2
I I I
Ci ОН С1
-> СН2—СН— СНа H IC1
\ / I
О Ci
Наряду с эпихлоргидрином в небольших количествах обра-
зуются также дихлоргидрин глицерина и трихлорпропан.
Поликонденсация и строение полимеров
Конденсация эпихлоргидрина с дифенилолпропаном приво-
дит к образованию термопластичных 'продуктов, сходных по
строению с многоатомными спиртами с длинными цепями, в ко-
торых гидроксильные группы .находятся на большом расстоянии
одна от другой. Синтез осуществляется таким образом, что на
конце каждой такой цепи сохраняется эпоксидная группа, от-
куда и 'произошло название этого класса смол.
В процессе конденсации может последовательно протекать
ряд реакций. Первой стадией является взаимодействие эпокси-
групп с фенольными гидроксилами:
СН2—I
—ОСН2—СН—СНа
I I
ОН С1
он
СН3
Образующийся хлоргидрин неустойчив м в присутствии
едкого натра немедленно превращается в диэпоксид
СН3
СНз—СН—СН2О-<^^>—С—ОСН»—СН—СНа
О> СН3 'о'/
Поликонденсация и строение полимеров
413
который попеременно вступает в реакцию конденсации с мо-
лекулами дифенилолпропана и эпихлоргидрина.
Наиболее вероятное строение конечной эпоксидной смолы
представляется следующим образом:
СН3 - СН3
-О-<<^ГЛ-С-/~Ч“ОСН2-СН-СН2-----о-^А-с-/^-осн2-сн—сн2
\=/ ( \=/ I \=/ I \=/
сн3 он _ „ сн3 о
Молекулярные веса элементов эпоксидной смолы:
М = 57
сн2—сн—сн2—
сн3
_ о— С —осн2—сн—сн2—
\=/ , \=/ I
сн3 он
М = 283
/И = 284
Средний молекулярный вес эпоксидной смолы, следователь-
но, равен: 340+284/г. Ниже приведены рассчитанные по этой
формуле молекулярные веса при разных значениях п:
п....... О 1 2 3 9 12
М .... 340 624 908 1192 2900 3750
Молекулярные веса и температуры плавления некоторых
марок смол эпикот приведены в табл. 22.
ТАБЛИЦА 22
Эпоксидные смолы эпикот
Марка смолы эпикот Средний молеку- лярный вес М Температура плавления °C Теоретическое значение п
828 350 8—12 0
1001 900 64—76 2
1004 1400 95—105 3,7
1007 2900 127—133 9
1009 3750 145—155 12
О применении смол эпон для поверхностных покрытий см.
Т. R. Hopper, Shell Chem. Corp. Officiel Digest, 26, № 354, 554(1954).
414
Гл. XIII. Эпоксидные смолы
. Физические свойства эпоксидных смол
Чистые эпоксидные смолы, т. е. состоящие исключительно
из .продуктов конденсации эпихлоргидрина и бнс-фенола или
двухатомного спирта, представляют собой более или менее
вязкие жидкости (эпикот 562, 828, 834) или твердые продукты
в виде кусков или зерен (эпикот 864, 1001, 1004, 1007, 1009) от
янтарного до буро-красного цвета; температура размягчения
тем выше, чем больше степень конденсации и средний моле-
кулярный вес смолы.
Эпоксидные смолы в чистом виде не обладают особо инте-
ресными свойствами. Только после добавления к ним соответ-
ствующих модификаторов и отвердителей они приобретают ха-
рактерный комплекс ценных свойств и, в частности, исключи-
тельную адгезию к большинству материалов. В зависимости
от природы модифицирующих веществ отверждение и поли-
меризацию слоя проводят при обычной температуре или при на-
гревании.
Модифицированные эпоксидные смолы выпускаются в виде
жидкотекучих масс (аральдиты D, F и М) или хлопьев (араль-
дит В), эпоксидных клеев, порошкообразных или в палочках
(аральдит 1), жидкостей, не содержащих растворителей (араль-
диты 101 и 103) или растворов (аральдиты 15 и 102), пигмен-
тированных густых паст без растворителя (аральдит 121),
лаковых смол кусковых (аральдит 961А) или в виде растворов
(аральдиты 985 Е и 985 F). Соответствующие отвердители иног-
да поставляются фирмами вместе со смолами. Они представ-
ляют собой белые порошки (отвердители 901, 902, 903) или бо-
лее или менее вязкие жидкости различной окраски (отверди-
тели 930, 951, 952).
Выпускаются также стабилизаторы (для входящих в состав
эпоксидных смол хлорсодержащих соединений), комбинирован-
ные с металлическими мылами и пластификаторами; они
представляют собой более или менее вязкие жидкости или
пасты.
Наконец, укажем продукты (в. сущности они уже не могут
рассматриваться как истинные эпоксидные смолы), представ-
ляющие собой продукты модификации эпоксидных смол жир-
ными кислотами высыхающих масел (эфиры эпоксидных смол)
или термореактивными смолами (начальные продукты конден-
сации). В зависимости от условий приготовления эти модифи-
цированные смолы представляют собой растворы или твердые
продукты.
Эфиры эпоксидных смол чаще всего выпускаются в виде рас-
творов с переменным содержанием сухого остатка (обычно от
50 до 80%), причем растворителями в зависимости от длины
Физические свойства эпоксидных смол
415-
цепи этерифицирующей жирной кислоты служат ароматиче-
ские или алифатические углеводороды. Из них можно назвать
следующие марки: эфиры 40RD и 40L, бекозол Р786, араль-
диты 801L и 802R, врезинол 9001 и др., синолак E100W, креста-
пон 600, сепокс и др.; предконденсаты — аральдит 961А, син-
резен W604 и т. д.
Совместимость. Эпоксидные смолы практически никогда не-
применяются одни, а чаще всего в сочетании с отвердителями
или модификаторами, вызывающими полимеризацию; к ним
иногда прибавляют также термопластичные смолы и различ-
ные ингредиенты, назначением которых является облегчение
процесса (переработки эпоксидного соединения или модифика-
ция некоторых свойств применительно к условиям эксплуа-
тации.
Для разработки рецептур необходимы сведения о совмести-
мости эпоксидных смол с другими термореактивными или тер-
мопластичными синтетическими смолами и пленкообразовате-
лями. Однако невозможно дать точные указания по этому воп-
росу и только лабораторные испытания позволят установить,
какие из продажных синтетических смол обладают частичной
пли полной совместимостью с эпоксидными смолами. Обычно
эпоксидные смолы, как и другие синтетические смолы, совме-
стимы с данной смолой тем в большей мере, чем меньше их
средний молекулярный вес.
Модификация эпоксидных смол, естественно, влияет на их
совместимость. Так, например, эпоксидные смолы, этерифици-
рованные жирными кислотами, становятся совместимыми, если
не полностью, то, по меньшей мере, частично, с сырым или по-
лимеризованным льняным маслом .и с различными алкидными
смолами, а также с феноло-форм альдегидными смолами, моди-
фицированными тунговым маслом или кетоновыми смолами,
(смола AW2 фирмы В. A. S. F.).
Уксуснокислые эфиры низкомолекулярных эпоксидных смол
хорошо совмещаются с нитроцеллюлозой, тогда как исходные
эпоксидные смольи совместимы с ней лишь в очень малой
степени.
Следует отметить, что во многих случаях совместимость эпо-
ксидной смолы и смолы-модификатора, так же как и легкость
переработки полученных продуктов, может быть улучшена
предварительным нагреванием смеси двух соответствующих
смол в чистом виде или в растворах.
Часто нет необходимости в том, чтобы эпоксидная смола
была совместима с полимером или смолой-модификатором во
всех отношениях. Такие условия создаются, когда эпоксидная
смола является вспомогательным агентом, например, вводится
для стабилизации хлорсодержащих полимеров, в частности
416
Гл. XIII. Эпоксидные смолы
поливинилхлорида, или входит в состав тройных систем: эпоксид-
ная смола — алкидная смола, модифицированная кокосовым
маслом — карбамидная смола. Содержание эпоксидной смолы
по отношению к смоляной фазе в этих случаях не превышает
обычно 25%.
Нет необходимости в полной совместимости и тогда, когда
эпоксидная смола является основным компонентом и в нее вво-
дят кроме отвердителя или смолы-модификатора небольшие ко-
личества вспомогательной смолы, например поливинилацеталя,
этилцеллюлозы, винилового сополимера (винилит VAGH), си-
ликонового масла, полисульфида и т. д.
Ниже приводятся некоторые указания о совместимости эпо-
ксидных смол (в частности, смол эпикот) с различными продаж-
ными термопластичными и термореактивными смолами.
Совместимость с термореактивными смолами. Эпоксидные
смолы обычно совместимы во всех отношениях с мочевино-
формальдегидными смолами, модифицированными бутиловым
спиртом. Из числа таких смол можно назвать:
Бекамины Р 138 и 354 (R. С. I. Beckacite)
Уропал FB 46 (R. V. А.)
Аминолак ND (Kuhlmann)
Гедамин (Н. G. D.)
Битл 610 и 640 (British Industrial Plastics)
Паралак 6001 (I. С. I.)
U900/60 British Resin Products Ztd
Скадонур M-6 (Scado-Zwolle, Голландия)
Нобель 24604
Перамин 1260 (Svanska Atti Fabrik, А. В., Швеция)
'Соамины 833 и 834 (Soab)
Сакопал B/3 (Bayer, Leverkusen)
Виафен 301 (Vianova, Австрия)
Врезилак 385 и 2694 (Resinous Chemicals Ztd, Англия)
Совместимость с меламино-формальдегидными смолами, мо-
дифицированными бутиловым спиртом, обычно ограниченная, в
особенности для эпоксидных смол с высоким молекулярным
весом. Некоторые меламино-формальдегидные смолы, напри-
мер меламин М86 (Ciba), заметно лучше совместимы. Наконец,
эпоксидные смолы совместимы с некоторыми сульфонамидными
смолами.
Из феноло-формальдегидных смол чаще всего совмещаются
с эпоксидными смолами низкомолекулярные резолы на основе
фенола, крезола или ксиленола, иногда бутанолизированные, а
также маслорастворимые, так называемые 100%-ные смолы на
основе замещенных фенолов. Обычно несовместимы с эпоксид-
Физические свойства эпоксидных смол
417
ними смолами феноло-формальдегидные смолы, модифициро-
ванные жирными кислотами или смолами типа альбертолей. Из
феноло-формальдегидных смол, совместимых с эпоксидными
смолами, можно указать следующие:
Плиофен 5030 и 5344
Супербекацит 1001 и 1002 (R. С. I. Beckacite, Франция)
Бакелит R 17620, R 5468/1, R 5173, R 5363, В 85, А 77,
R 534 (Bakelite)
АР 26,750 и 717 (R. V. А., Франция)
Скадоформы L 5 и L 9 (Scado, Голландия)
R 108 (General Electric Со., США)
Z 400, врезинилы 265, 2602 (James Ferguson G. В,, Resinous
Chemicals G. В.)
Резинокс P 97 (Monsanto)
Синрезен U 363 и 440 W (Synres, Голландия)
Дурез 15956 (Durez Plastics and Chemicals, США)
Варкум 2896 (Varcum Chemical Corp., США).
Совместимость с термопластичными смолами. С эпоксидны-
ми смолами совместимы в определенных соотношениях сле-
дующие поливинилацетали:
Бутвары В-72-А, В-73, В-76 (Monsanto)
Винилиты ХУНЬ, XYSG (Bakelite Division)
Ревилы F-25.702, F-25.005, В-25.235, В-25.705, В-25.114,
В-25.704, В-25.708, 25.004, 25.045 (Nobel fran^aise)
Ровиналы А, В, FM, FH, F (Rhone-Poulenc)
Некоторые марки низкомолекулярных эпоксидных смол со-
вместимы с поливинилацетохлоридами, особенно с винилитом
VAGH. Эпоксидные смолы совместимы также в некоторых со-
отношениях с поливинилхлоридами, что позволяет применять
их для стабилизации этих хлор содержащих полимеров по отно-
шению к действию тепла.
Наконец, эпоксидные смолы совместимы в ограниченных пре-
делах с некоторыми полигликолями.
Большинство глифталевых смол, немодифицированных или
модифицированных маслами (за исключением некоторых невы-
сыхающих глифталевых смол, модифицированных, например,
кокосовым маслом), несовместимы с эпоксидными смолами.
Однако низкомолекулярные эпоксидные смолы обладают все же
ограниченной совместимостью с глифталевыми смолами, моди-
фицированными высокомолекулярными жирными кислотами
или стиролом.
Благодаря совмещению невысыхающей глифталевОй смолы
с эпоксидной смолой возникает возможность получения эмалей
27-Г-156
418
Гл. XIII. Эпоксидные смолы
горячей сушки, причем для придания пленке эластичности при-
бавляют меламино-формальдегидную или бутанолизированную»
мочевино-формальдегидную смолу.
К нитроцеллюлозе можно прибавлять только крайне неболь-
шие количества низкомолекулярных эпоксидных смол, совме-
стимость которых может быть, однако, улучшена внесением-
пластификаторов и особенно дибутилфталата.
В заключение укажем еще на совместимость эпоксидных
смол с жидкими полисульфидами (тиокол LP-3), которые мо-
гут быть введены для придания пленке большей эластичности
или гибкости при низких температурах.
Растворимость. Эпоксидные смолы являются сильно поляр-
ными полимерами и поэтому для их растворения обычно при-
меняют растворители с резко выраженной полярностью: кисло-
родсодержащие растворители типа спиртов, кетоноспиртов, ке-
тонов, простых и -сложных эфиров, а также некоторые хлорсо-
держащие растворители и различные фенолы. Это относится к.
немодифицированным эпоксидным смолам, так как смолы, моди-
фицированные жирными кислотами (т. е. представляющие со-
бой сложные эфиры), обычно растворимы в углеводородных рас-
творителях.
Ароматические углеводороды обычно^ не растворяют эпоксид-
ных смол, за исключением жидких -смол с очень низким моле-
кулярным весом. Они, однако, часто используются в качестве
разбавителей в смеси с кислородсодержащими растворителями
для удешевления композиций и улучшения некоторых свойств
конечных продуктов.
Следует отметить, что некоторые спирты, не являющиеся рас-
творителями эпоксидных смол, в смесях с ароматическими
разбавителями приобретают растворяющую способность. Такие
смешанные растворители иногда применяются для растворе-
ния низкомолекулярных эпоксидных смол и, в частности, для
приготовления лредконденсатов или композиций эпоксидных
смол с полиаминами или аминоамидами.
Эпоксидные смолы, как и многие другие высокополимеры.
могут смешиваться с некоторыми растворителями лишь в огра-
ниченных пределах. Так, например, из эпоксидной смолы со
средним молекулярным весом от 1000 до 2000 можно пригото-
вить концентрированные растворы (с содержанием сухого ве-
щества больше 50%) в ацетоне, этил- или бутил ацетате, метил-
изобутилкетоне, но в тех же растворителях нельзя приготовить
растворы с малым содержанием сухого вещества (например.
20%-ные).
Далее приведены некоторые из растворителей и разбавите-
лей, наиболее часто применяемых для растворения эпоксидных
смол.
Физические свойства эпоксидных смол
419
У глеводороды
Бензол
Толуол
Ксилол
Спирты (компоненты
Метиловый спирт
Этиловый спирт
н-Пропиловый спирт
Изопропиловый спирт
н-Бутиловый спирт
(разбавители)
Тетралин
Сольвент-нафта
растворяющих смесей)
emop-Бутиловый спирт
Бензиловый спирт
Циклогексанол
Метил из обут илкарбинол
Терпинеол
Фенолы (растворители)
Крезолы
Ксиленолы
Кетоны (растворители)
Ацетон
*Метилзтилкетои
Метилизобутилкетон
‘Диацетоновый спирт
Циклогексанод
’Метилциклогексанон
Простые эфиры (растворители)
Монометиловый эфир этиленгликоля (метилцеллозольв)
*Моноэтиловый эфир этиленгликоля (целлозольв)
*Монобутиловый эфир этиленгликоля (бутилцеллозольв)
Сложные эфиры (растворители)
Метилацетат
Этилацетат
Бутилацетат
Метилцеллозольв-ацетат
* Целлозольв -ацетат
Этиллактат
Хлорированные углеводороды (растворители)
Монохлорбензол
о-Дихлорбензол
Указанные растворители (практически всегда применяются в
виде .смесей, состав которых варьируется в зависимости от сред-
него молекулярного веса эпоксидной смолы, природы модифи-
катора или отвердителя, а также в соответствии с намечаемым
способом применения (окрашивание кистью, распылителем, ла-
кирование и т. д.) красок и лаков. Смеси растворителей вклю-
чают чаще всего растворитель, компонент, не являющийся рас-
творителем, и ароматический разбавитель. В табл. 23 приведены
некоторые смеси растворителей, применяемые в рецептурах ком
позиций на основе эпоксидных смол.
Рецептуры, содержащие растворители типа фенола, приме-
няются в основном для производства лаков, предназначенных
для эмалирования электропроводов.
* Звездочкой отмечены вещества, растворяющие смолы эпикот 1C0I и 1007 с образованием
растворов любой концентрации.
27*
420
Гл. XIII. Эпоксидные смолы
Растворители эпоксидных смол
ТАБЛИЦА 23
1 Q.3 Компоненты Относительные Раство ряемые смолы
количества
Бинарные смеси
1 Днацетоновый спирт Толуол или ксилол 1 1 Эпикот 1007 Аральдит 961А
2 Этилечгликольацетат 1 Эпикот 1007
Толуол или ксилол 1
3 «-Бутиловый или втор-бутиловый
спирт 1 ‘ Эпикот 1001
Толуол ... 5
4 Метилизобутилкетон 1 То же
Толуол ‘ I
5 «-Бутиловый спирт 1 Эпикот 1007
Толуол . . . . 2
6 emop-Бутиловый спирт 1 То же
Толуол 2
7 Этиленгликоль 1 Аральдит 961А
Толуол 1
8 Метилэтилкетоп 1 То же
Толуол 1
9 «-Бутиловый спирт 1 То же
Ксилол 1
10 н-Проп иловый спирт 1 То же
Этиленгликоль 1
11 «-Бутиловый спирт 1 То же
Этиленгликоль —•
12 Этиловый эфир этиленгликольаце-
тэта (целлозольв-ацетат) . . . . 1 Эпикот 1007
Толуол 1
Трехкомпонентные смеси
1 ‘Диацетоновый спирт 1 Аральдит 961А
Бутиловый спирт Толуол . . . . 1 1
2 Метилэтилкетоп . . . . > Этиленгликоль 1 1 То же
Кснлол 1
3 Метилизобутилкетон 1 То же
Этиленгликоль 1
Ксилол 1
Физические свойства эпоксидных смол
421
Продолжение табл. 23
№ рецеп-
туры
Компоненты
Относительные
количества
Растворяемые смолы
4 Бутилацетат .... . . Этиленгликоль ... Ксилол 3 4 3 Аральдит 961А
5 Диацетоновый спирт . 3 Аральдит 985F
Бутилацетат . . . 3
Толуол или ксилол 4
6 Диацетоновый спирт . ... 455 Аральдит 985Е
Толуол или ксилол 130
Метил- или этилацетат . 80
7 Диацетоновый спирт 3 То же
Толуол или ксилол . . . 17
Метил- или этилацетат 30
8 Диацетоновый спирт 80 То же
Толуол 35
Метил- или этилацетат 360
9 Диацетоновый спирт 70 То же
Ксилол 115
Тетралин 115
10 Метилизобутилкетон 45 Эпикот 1001
Моно-бутиловый эфир этнленгли-
коля (бутилцеллозольв) 5
Толуол 50
11 Метилизобутилкетон 25 То же
Циклогексанол 2b
Толуол 50
Диац&тоновый спирт . 20 То же
12 я-Бутиловый спирт 10
Толуол 70
Метилизобутилкетон 20 То же
13 Моноэтиловый эфир этиленгликоля
(целлозольв) 5
Толуол 25
Метилизобутилкетон - 20 То же
н-Бутиловый или втор-бутиловый
спирт 5
Толуол 25
15 Метилэтилкетон ... 20 То же
Моиоэтиловый эфир этиленгликоля
(целлозольв) 5
Толуол 25
16 Метилэтилкетон 33 То же
Метилизобутилкетон 33
Толуол 34
422
Гл. XIII. Эпоксидные смолы
Продолжение табл. 23
№ рщсч туры Компоненты Относительные количества Растворяемые смолы
17 Диацетоновый спирт Метилэтилкетон Толуол . . ... 20 10 70 Эпикот 1001
Четырех к ом поне1 т н ы е смеси
1 Этиловый или изопропиловый спирт Метил=тилкетон Сосновое масло или бутилцелло- зольв . . . . Толуол 25 15 10 50 Эпикот 1007
2 Метилэтилкетон Метилизобутилкетон Монобутиловый эфир этиленглико- ля (бутилцеллозольв) Толуол 10 10 5 25 Эпикот 1001
3 Метилизобутилкетон Моноэтиловый эфир этиленгликоля (целлозольв) Монобутиловый эфир этиленглико- ля (бутилцеллозольв) Изопропиловый спирт 20 5 5 20 То же
4 Метилэтилкетон . . Метилизобутилкетон . . . Моноэтилэвый эфир этиленгликоля (целлозольв) Толуол . . . . 20 20 10 50 То же
5 Диац 'тоновый спирт Ксиленол Монохлорбензол Сольвент нафта . . 240 360 280 120 Аральдит 970В
6 Днацет-.'.новый спирт Ксилол Мо.нохлорбснзол Этилен!ликоль . . . 300 100 300 300 То же
• Диацетоновый спирт Ксиленол . . Монохлэрбспзол . . . . . Ксилол 200 .360 280 160 То же
8 Крезол . . ... Бутиловый спирт . . . . Ксилол Ацетон 400 200 200 200 Аральдит 985F
Химические свойства эпоксидных смол
423
Продолжение табл. 23
Компоненты
Относительные
количества
Растворяемые смолы
10
Диацетоновый спирт...........
Бхтилацетат ... ....
н-Бутиловый спирт ...
Этилацетат . . ..............
Циклогексанол ...................
Мсноэтиловый эфир этиленгликоля i
(целлозольв) ................I
Бутил ацетат............. .....[
Толуол или ксилол . . . . . . .1
Диацетоновый спирт.............।
Этиловый спирт (95%-ный) ...
Бутилацетат
Метилэтнлкетон ......... i
Диацетоновый спирт.............I
МеТИЛИЗСмуТИЖСТоп . ........
Моноэтиловый эфир этиленгликоля i
(целлозольв) ...................
Толуол ... ...................
Диацстоновый спирт............. .
Моноэтпловын эфир этиленгликоля 1
(целлозольв) ...............'
Бут лацетат ...................;
Циклогексан
Д-гацетановый спирт ...........!
Метилэтнлкетон . . ....
Бутилацетат .....................
Толуол . .... . . .
1
1
1
1
20
25
25
30
1
1
1
1
15
25
40
2
2
3
3
1
1
1
1
Аральдит 985F
То же
Аральдит 961А
То же
Аральдит 985F
То же
9
Химические свойства эпоксидных смол
Химические свойства эпоксидных смол определяются строе-
нием их молекул и, в особенности, зависят от наличия вторич-
ных спиртовых групп в углеродной цепи полимеров, а также от
присутствия концевых эпоксидных групп.
В молекулах эпоксидных смол имеются только углерод-
углеродные или эфирные связи, чем объясняется повышенная
химическая стойкость этих полимеров, в частности по отноше-
нию к щелочам. Особенно устойчивы смолы, модифицированные
некоторыми термореактивными смолами типа мочевино-форм-
альдегидных, меламино-формальдегидных или феноло-формаль-
дегидных.
Знание химических свойств эпоксидных смол дает возмож-
ность выбирать способы их переработки и позволяет понять
424
Гл. XIII. Эпоксидные смолы
механизм их взаимодействия с различными отвердителями или
модифицирующими агентами, приводящего к образованию
трехмерной структуры,. После такой обработки эпоксидные смолы
приобретают прекрасную стойкость к растворителям и к много-
численным химическим агентам, органическим или минеральным.
Хотя в некоторых случаях эпоксидные группы ведут себя
подобно гидроксилам, однако для упрощения мы разделим
реакции эпоксидных смол на две категории в зависимости от
того, какие группы участвуют в этих реакциях, эпоксидные или
гидроксильные.
Реакции, характерные для эпоксидных групп. Эпоксидные
группы крайне реакционноспособны и склонны к многочислен-
ным реакциям, из которых многие протекают при обычной тем-
пературе. Часть этих реакций в известной мере аналогична ре-
акциям альдегидов, являющихся изомерами а-эпоксидов:
R—СН—СН2 R—СН2—С—Н
\/ II
О . о
Из важнейших реакций, протекающих благодаря наличию
эпоксидной группы, необходимо отметить следующие.
Реакции присоединения первичных и вторичных аминов:
R—СН—СН2 -|- R'—NH2 — R-CH—СН2—NHR*
ОН
R—СН—СН2—NHR' 4- СН2—СН—R —- R—СН—СН.,— NR'—СН2—СН—R
I \/ I I
ОН о он он
С некоторыми первичными аминами или полиаминами (>в ча-
стности, с этилендиамином или диэтилентриамином) реакции
присоединения протекают при обычной температуре. Установ-
лено, что при наличии в молекуле такого амина четырех или
пяти подвижных атомов водорода продукты реакции имеют
трехмерную структуру, если в молекуле исходной эпоксид-
ной смолы содержалось по меньшей мере две эпоксидные
группы.
Варьируя соотношение амина и эпоксидной смолы, можно
получать смолообразные продукты со свободными эпоксидны-
ми группами (например, предконденсат, полученный при избыт-
ке эпоксидной смолы) или со свободными аминогруппами
(например, аддукты, полученные при избытке амина). Эти смо-
лообразные производные, приближающиеся в первом случае
к эпоксидным смолам, а во втором к аминам, способны в свою
очередь реагировать соответственно с аминами или эпоксидны-
Химические свойства эпоксидных смол
425
ми смолами. Можно также комбинировать продукты присоеди-
нения двух указанных типов, получая трехмерные полимеры.
Аналогично аминам или аддуктам, содержащим аминогруп-
пы, ведут себя низкомолекулярные полиамиды, получаемые,
например, взаимодействием триаминов с димерами или гриме-
рами ненасыщенных жирных кислот. Они вызывают отвержде-
ние эпоксидных смол иногда уже при обычной температуре.
Некоторые третичные амины также могут .вызывать полиме-
ризацию эпоксидных смол. Однако механизм отверждения от-
личен от предыдущего и аналогичен ионной полимеризации оки-
си этилена; третичный амин является в этом случае катализа-
тором полимеризации, протекающей при участии 'эпоксидных
групп. Это объяснение подтверждается тем фактом, что про-
дукты реакции сохраняют некоторую термопластичность, а, на-
пример, в случае фенилглицидного эфира также тем, что содер-
жание азота в них ничтожно мало.
Реакции присоединения галоидоводородных кислот. Эпоксид-
ные смолы могут присоединять галоидоводородные кислоты,
особенно соляную кислоту:
R—СН—СН2 + НС1 R—СН—СН2С1
\ / I
О он
Эта реакция позволяет объяснить стабилизирующее действие,
которое оказывают эпоксидные смолы при нагревании хлорсо-
держащих соединений и, в частности, поливинилхлорида, поли-
хлоропрена и различных сополимеров винилхлорида. Метод ко-
личественного определения эпоксидных групп в эпоксидных
смолах основан также на этой реакции.
Реакции присоединения фенолов:
R—СН—СН2 + НО—Ar R—СН—СН2—О—Аг
ОН
В начальной стадии получения эпоксидных смол эти реак-
ции приводят к образованию нестойкого дихлоргидрина в ре-
зультате присоединения двух молекул эпихлоргидрина к мо-
лекуле дифенилолпропана, а в конечной стадии —к присоеди-
нению других молекул дифенилолпропана к диэпоксиду, обра-
зующемуся при разложении дихлоргидрина в присутствии
едкого натра. На этой же реакции основан процесс предвари-
тельной конденсации, позволяющий получать смолообразные
комплексы присоединением эпоксидных смол к феноло-форм-
альдегидным смолам. В зависимости от соотношения эпоксид-
ной и феноло-формальдегидной смол можно получать либо пред-
конденсаты, не содержащие эпоксидных групп и отверждаю-
426
Гл. XIII. Эпоксидные смолы
щиеся в результате взаимодействия метилольных групп феноло-
формальдегидной смолы, либо предконденсаты разветвленного
строения, содержащие концевые эпоксидные группы, способные
отверждаться полиаминами (см., выше).
Реакции, характерные для гидроксильных групп. Отверж-
дение или модификация эпоксидных смол жирными кислотами
или термореактивными смолами может осуществляться при
участии гидроксильных групп. При этом протекают либо реак-
ции образования сложных эфиров (взаимодействие с жирными
кислотами), либо реакции образования простых эфиров (кон-
денсация щд'ермореактивными смолами).
Кроме того, вторичные гидроксильные группы и эпоксидные
группы могут реагировать с диизоцианатами, в частности с то-
луилендиизоцианатом и гексаметилендиизоцианатом с образо-
ванием полиуретанов трехмерного строения.
Реакции сложноэфирной конденсации. Эпоксидные смолы
подобны полиспиртам линейного строения и поэтому могут быть
этерифицированы различными кислотами или их ангидридами:
(OOCR')„,
R(OH)„ -J- n'R'—СООН — R Z + n'H2O
X(OH)n_n,
Чаще всего для этой цели применяются жирные кислоты высы-
хающих, полувысыхающих или невысыхающих растительных
масел и некоторые органические двухосновные кислоты или их
ангидриды. В случае жирных кислот величина п' обычно лежит
между 0,3(п+4) и 0,9(и4-4).
Принято считать, что эпоксидные группы (каждая из них
в процессе этерификации равноценна двум гидроксильным
группам) вступают в реакцию первыми, так как они более ре-
акционноспособны, чем вторичные гидроксильные группы.
1. Этерификация одноосновными кислотами.
Модификация эпоксидных смол органическими одноосновными
кислотами обычно используется для получения связующих ве-
ществ воздушной или горячей сушки (в зависимости от стро-
ения кислот), предназначенных для изготовления лаков и кра-
сок, печатных красок по металлу и паст для набивки текстиля.
Этерификация эпоксидных смол осуществляется при доволь-
но высокой температуре (около 250°) в обычных аппаратах для
производства глифталевых смол. Пропуская инертный газ (азот
или двуокись углерода) в продолжение всей операции, проводи-
мой в закрытом котле, можно получать менее окрашенные про-
дукты.
Введение в реактор небольшого количества азеотропного
растворителя (ксилола или ароматических высококипящих
углеводородов) дает возможность уменьшить длительность на-
Химические свойства эпоксидных смол
427
гревания, требуемую для достижения кислотного числа, обеспе-
чивающего слабую окраску этерифицированной смолы.
Многообразие этерифицированных эпоксидных смол прак-
тически неограниченно, так как свойства эпоксидных смол мо-
гут изменяться в зависимости от жирной кислоты, примененной
для модификации, и, кроме того, этерификацию можно произ-
водить смесями кислот, изменяя в довольно широких пределах
соотношения эпоксидной смолы и жирной кислоты. Это соотно-
шение может колебаться в пределах 30—60 вес. ч. жирной кис-
лоты на 70—40 вес. ч. эпоксидной смолы в зависимости от тре-
бований, предъявляемых к конечному продукту.
При выборе жирной кислоты и установлении количества ее,
требуемого для этерификации эпоксидной смолы, руководст-
вуются теми же соображениями, что и для алкидных смол; уве-
личение количества жирной кислоты при равном содержании
сухого остатка повышает растворимость смолы в алифатиче-
ских растворителях и уменьшает ее твердость. При более вы-
соком молекулярном весе эпоксидной смолы при прочих рав-
ных условиях твердость этерифицпрованного продукта повы-
шается, а растворимость его ухудшается. На практике для
этерификации применяют эпоксидные смолы с относительно
малым средним молекулярным весом (от 1000 до 2000 'макси-
мально) и жирные кислоты сильно высыхающих масел, напри-
мер дегидратированного касторового масла. Так поступают для
того, чтобы, с одной стороны, получить эфиры, растворы ко-
торых имели бы допустимую вязкость, и, с другой стороны,
чтобы избежать риска желатинирования, возможного при при-
менении эпоксидной смолы с более высоким молекулярным
весом и, следовательно, с большей функциональностью. По
тем же причинам и, кроме того, для улучшения адгезии, сте-
пень этерификации, т. с. отношение числа гидроксильных групп
(или соответственно эпоксидных групп), вступивших в реакцию
с жирными кислотами, к общему числу реакционноспособных
групп, способных к этерификации, должна составлять 30—50%
для эфиров с малой жирностью и 50—80% для эфиров со сред-
ней или большой жирностью.
Контроль протекания реакции этерификации состоит в пе-
риодическом определении кислотного числа эфиров, которое для
массовой продукции находится в пределах от 1 до 10, в. зави-
симости от жирности эфиров и природы жирных кислот, входя-
щих в их состав. Кислотное число системы эпоксидная смола-—
жирная кислота обычно тем меньше, чем меньше количество
жирной кислоты,.
Изменение кислотного числа и вязкости растворов этерифи-
цированных эпоксидных смол в зависимости от продолжитель-
ности варки показано на рис. 53.
428
Гл. XIII. Эпоксидные смолы
2. Этерификация двухосновными кислотами
или их ангидридами. Многоосновные кислоты, и в осо-
бенности двухосновные или их ангидриды (например, фталевый
или малеиновый) очень редко применяются для модификации
эпоксидных смол, предназначенных для лаков и красок. Иногда
их добавляют к жирным одноосновным кислотам, но в очень
небольших количествах, во избежание желатинирования.
Время, uacbi время, чаем
Рис. 53. Кривые этерификации эпоксидных смол:
1—изменение кислотного числа; 2—изменение вязкости.
А—смола эпикот 1001; 70% сухого вещества экстрагируется ксилилом. Б—смола
эпикот 1004; 60% сухого вещества экстрагируется ксилолом. В—смола эпикит
1007; 60% сухого вещества экстрагируется ксилолам. Г—смола эпикот 1009; 50%
сухого вещества экстрагируется ксилолом.
В то же время двухосновные кислоты являются прекрасны-
ми отвердителями низкомолекулярных эпоксидных смол, 'пред-
назначаемых для покрытия электропроводов и различных обмо-
ток, а также для переработки методом литья. Этерификация,
приводящая к отверждению смолы, проводится обычно при
температуре 100—200° и продолжается от 2 дней до 1 часа.
Двухосновные кислоты чаще всего заменяются их ангидридами,
чтобы избежать появления пористости массы вследствие выде-
ления воды при этерификации.
Органические кислоты, применяемые для этерификации эпок-
сидных смол, приведены в табл. 24.
Химические свойства эпоксидных смол
429
ТАБЛИЦА 24
Кислоты, применяемые для этерификации эпоксидных смол марки эпикот
Кислоты Эквивалентный вес Свойства кислоты (или эфира)
Невы с ы х а ю щ ие кислоты
Уксусная кислота 60 Кислота невысыхающая. Получаемые эфиры имеют вид твердой смолы светлого желто-янтарного цвета; со- вместимы с нитроцеллюлозой
Кислоты кокосового масла 215 Кислоты невысыхающие. Образуют светлые продукты, не желтеющие в сушильных камерах; применяются с добавкой меламино- или феноло-форм- альдегидных смол для эмалевых красок типа глянцевых эмалей
Кислоты хлопкового масла 280 Получаемые эфиры образуют светлые высокоэластичные пленки. Примене- ние то же, что и жирных кислот ко- косового масла
Лауриновая кислота 202 Вводится для улучшения качества жир- ных кислот кокосового масла
Ангидрид малеиновой кислоты 49 Применяется в небольших количест- вах в сочетании с канифолью для повышения температуры размягчения и улучшения цвета эфиров. Может применяться также совместно с тал- ловым маслом для улучшения стой- кости окраски, повышения вязкости эфиров и получения более твердой пленки
п-шре/п-Бутилбензойная кислота 178 Образует светлые и стойкие эфиры, дающие твердые, блестящие, тепло- стойкие, быстро высыхающие пленки
Канифоль (димеризован- ная на 85 %) 398 Образует быстро отверждающиеся эфи- ры, но несколько окрашенные. Свя- зующее высокой вязкости; должно вво- диться в небольших количествах во избежание желатинирования
Канифоль (экстракцион- ная или подсочная) 348 Скорость этерификации невелика. Эфи- ры хорошо растворяются в алифати- ческих углеводородах (уайт-спирите, особых сортах бензина и у. д.), быст- ро затвердевают При сушке на воз- духе. Пленки хрупкие, несколько окрашенные
430
Гл. XIII. Эпоксидные смолы
Продолжение табл. 24
Кислоты Эквивалентный вес Свойства кислоты (илн эфира)
Канифоль гидрогенизиро- ванная 344 Свойства, аналогичные свойствам про- дуктов на основе дегидрогенизиро- ванной канифоли, но выше стойкость и светлее окраска
Кислоты п о л у в ы сыхающих масел
Жирные кислоты соевого масла 275—284 Стоимость несколько повышенная. Вы- сыхание эфиров медленное до сред- него; достаточная стойкость, светлая окраска, хорошая растворимость в алифатических углеводородах. Плен- ки рыхлые, но эластичные и с хо- рошей поверхностной твердостью
Жирные кислоты талло- вого масла 300—350 Смеси полувысыхающих кислот; стои- мость несколько повышенная. Со смолами эпикот образует связующее высокого качества, в особенности если талловое масло предварительно перегнано
Кислот ы в ы с ы хающих масел
Димеры 305 Применимы для повышения твердости, ускорения сушки, улучшения высы- хания внутри пленки и повышения вязкости
Жирные кислоты льняно- го масла 280- 284 Высыхание быстрое, химическая стой- кость хорошая, окраска темная, стой- кость посредственная. Хорсшая по- верхностная твердость пленки жир- ных эфиров при надлежащей пиг- ментации
Жирные кислоты ойтиси- кового масла 301 Высыхание очень быстрое, химическая стойкость хорошая; образуют твер- дые пленки. Вводятся в небольших количествах во избежание желатини- рования эфиров
Жирные кислоты дегид- ратированного касторо- вого масла 284—288 Позволяют получать быстросохнущие продукты горячей сушки с хорошей окраской и хорошей химической стойкостью
Жирные кислоты тунго- вого масла 284 Высыхание очень быстрое, окраска по- средственная, трудно полимеризуют- ся; вводятся в небольших количест- вах во избежание желатинизации. Высокая твердость пленки, высокая стойкость к истиранию и к действию химических агентов
Химические свойства эпоксидных смол
431
Образование простых эфиров:
R—ОН + R'—ОН * R-O-R' + Н2О
lev—zoo
Эпоксидные смолы обладают способностью реагировать при
нагревании с термореактивными смолами типа карбамидных 4}
или феноло-формальдегидных. Получаемые продукты имеют
трехмерную структуру; предполагается, что эта структура воз-
никает в результате образования простых эфирных связей
между молекулами двух различных смол. Это предположение
подтверждается тем, что исчезновение эпоксидных групп в ре-
зультате их гидратации (образование гликоля) или полной
гидрогенизации не изменяет характера отверждения комплекса
эпоксидная смола — термореактивная смола и не влияет замет-
но на адгезию, эластичность или химическую стойкость полу-
ченных продуктов.
Кроме того, если в производстве эпоксидных смол дифени- ?
лолпропан заменить бис-фенолом аналогичной структуры, в ко-
тором все водородные атомы бензольных ядер замещены ме-
тильными группами, получаются смолы, также способные-
отверждаться термореактивнымн смолами указанных типов. Та-
кие смолы по свойствам очень сходны^ со смолами, получающи-
мися при поликонденсации эпихлоргидрина и дифенилолпропана.
Из этого следует, что в реакции с метилольными группами
смолы-модификатора участвуют не фенольные гидроксилы эп-
оксидной смолы, а эпоксидные или гидроксильные группы по-
следующей схеме:
R'—СН2—ОН + СН2—СН—R —R'—СН2—О—СН2—СН—R
\) ОН
Реакция с диизоцианатами (образование полиуретанов). Хо-
рошо известно, что реакции с некоторыми диизоцианатами (на-
пример, с толуилендиизоцианатом или с гексаметилендиизоци-
анатом) могут быть использованы для изменения молекулярного
веса и структуры большого числа органических соедине-
ний и полимеров, содержащих подвижные атомы водорода и в
особенности такие реакционноспособные группы, как гидр-
оксильные, карбоксильные, аминогруппы и т. д.
Макромолекулы эпоксидной смолы способны соединяться с
диизоцианатами с образованием полимеров трехмерной струк-
туры, причем в реакции участвуют спиртовые гидроксилы или
эпоксидные группы.
Эпоксидные смолы, модифицированные жирными кислотами,
в молекулах которых всегда имеются неэтерифицированные
гидроксилы, также могут соединяться с диизоцианатами. Эти
432
Гл. XIII. Эпоксидные смолы
реакции (протекают или при обычной температуре, или же в
случае применения так называемых скрытых (блокированных)
изоцианатов—три нагревании. Реакция с толуилендиизоциа-
яатом протекает по схеме:
СН3
I
R—ОН -J- O=C=N—N=C=O 4- НО—R -►
толуилендиизоцганат
сн3
-*• RO—С—N11—NH—С—OR
Анализ и идентификация эпоксидных смол
Эпоксидные смолы характеризуются наличием концевых
эпоксидных групп; кроме этих групп, в их молекулах имеются
лишь связи углерод-углерод и простые эфирные связи. Способ-
ность эпоксидных смол к различным реакциям присоединения
позволяет производить их количественное определение.
Следует заметить, что для немодифицированных эпоксидных
смол число омыления теоретически должно быть равно нулю.
Для смол, модифицированных жирными кислотами, это число
значительно «иже, чем для смол, модифицированных высыхаю-
щими маслами (например, льняным маслом), и для глифтале-
вых смол, модифицированных маслами, жирными кислотами или
стиролом*. Хотя методика определения числа омыления не по-
зволяет с уверенностью различать модифицированные и немоди-
фицированные эпоксидные смолы, она дает сведения о природе
связующего, дополняя или подтверждая результаты, получен-
ные другими, более сложными методами.
Установлено, что с помощью специфических методов каче-
ственного анализа эпоксидных смол действительно можно
определять, содержит ли неотвержденный полупродукт (напри-
мер, лак или краска) эпоксидную смолу, модифицированную
жирными кислотами или начальными продуктами конденсации
феноло-формальдегидных смол.
Укажем некоторые методы качественного или количествен-
ного анализа, к которым можно прибегать при исследовании
эпоксидных смол.
* Скоростные качественные пробы для определения компонентов лаков
и синтетических смол—см. CD1C Club, Official digest, 731—735 (1952).
Анализ и идентификация эпоксидных смол
433
Методы качественного анализа. Некоторые методы качест-
венного анализа, позволяющие обнаружить присутствие моди-
фицированных или немодифицированных эпоксидных смол в
неотвержденных продуктах, были предложены Фукри*. Из этих
методов два дают положительные результаты для эпоксидов и
отрицательные — для феноло-формальдегидных, мочевино-форм-
альдегидных и глифталевых смол.
При анализе смолы или продуктов, не содержащих ни рас-
творителя, ни наполнителя, ни пигмента, навеску около
1 г после измельчения (в случае твердого продукта) растворяют
в 100 мл 98%-ной серной кислоты при охлаждении и переме-
шивании. При исследовании продуктов, содержащих раствори-
тели (например, красок или лаков), берут навеску около 3 г
лака (после центрифугирования для отделения пигментов в
случае краски) и нагревают ее в шкафу при 100—105° в тече-
ни 3 час. Остаток измельчают и 1 а полученного порошка пере-
водят в раствор, как указано далее.
Затем в пробирку вносят 1 мл сернокислотного раствора
экстракта смоляной основы лака и определяют возможное при-
сутствие эпоксидной смолы путем одной из следующих проб:
а) Прибавляют 1 мл 63%-ной азотной кислоты, хорошо пе-
ремешивают и оставляют стоять 5 мин. Содержимое (пробирки
выливают в 100 мл 'нормального раствора едкого натра
и перемешивают. В присутствии эпоксидной смолы появ-
ляется оранжево-красное окрашивание, в присутствии феноль-
ной — желтое.
б) Прибавляют 5 мл реактйва Дениже, хорошо перемеши-
вают и оставляют стоять. В случае эпоксидной смолы на дне
пробирки оседает оранжевый осадок, при фенольной смоле из
желтого раствора выделяется белый осадок.
Методы количественного анализа. Для определения содер-
жания эпоксидных или гидроксильных групп, а также эквива-
лента этерификации** эпоксидной смолы были разработаны
различные методы, основанные на простых химических реак-
циях и требующие большой тщательности выполнения.
Полное описание этих методов выходит из рамок данного
труда; поэтому здесь будут указаны только принципы, положен-
ные в их основу.
Определение числа эпоксидных групп. Определение числа
эпоксидных групп основывается на способности каждой
эпоксидной группы связывать одну молекулу соляной кислоты.
* М. F о и с г у, Peint. Pigm. Ver., nowembre 1954, 925.
** Эквивалент этерификации выражается весом смолы в граммах, необ-
ходимым для полной этерификации одной грамм-молекулы одноосновной кис-
лоты (например, для этерификации 280 г жирных кислот С18 или 60 г уксусной
кислоты).
28-156
434
Гл. XIII. Эпоксидные смолы
После растворения пробы исследуемой смолы в подходящем
растворителе (диоксан) и прибавления смеси соляной кислоты
с диоксаном определяют по разности количество связанной кис-
лоты, что позволяет установить число эпоксидных групп, содер-
жащихся в определенном весовом количестве смолы. При рас-
чете, разумеется, учитывают свободную кислоту или свободное
основание, могущие присутствовать в пробе.
Навеска, требуемая для определения, тем больше, чем мень-
ше содержание эпоксидных групп в исследуемой смоле:
Число эпоксидных
групп в 1 г смолы
0,002
0,001
0,0005
Навеска
г
0,9—1,5
2—3
3,5—4,5
Определение числа гидроксильных групп. Это определение
состоит в измерении объема водорода, выделившегося при
взаимодействии органических соединений, содержащих подвиж-
ные атомы водорода, с литийалюминийгидридом в эфирном
растворе. Данный метод может быть использован для анализа
спиртов, органических кислот, аминов, меркаптанов и некоторых
фенолов.
Реакции с гидроксильными и эпоксидными группами
эпоксидной смолы протекают следующим образом:
4ROH 4- LiAlH4 LiAl(OR)4 -j- 4Нг
4R—СН—СНг + LiAlH4 LiAlfO—СН—СН3’
\ zz I
О R ]4
Поскольку при взаимодействии с эпоксидными группами про-
исходит выделение водорода, этим .путем можно определить
содержание гидроксильных групп в эпоксидной смоле.
Следует заметить, что присутствие эпоксидных групп пре-
пятствует применению классических методов определения гидр-
оксильных групп (метод с уксусным ангидридом и пиридином, с
хлористым ацетилом и т. д.), осложненных, кроме того, труд-
ностью подбора растворителя.
Определение эквивалента этерификации. Эта характеристика
особенно существенна при получении эпоксидных смол, моди-
фицированных жирными кислотами.
Определив эквивалент этерификации с помощью реакции
эпоксидной смолы с жирными кислотами и внеся соответствую-
щую поправку на присутствие эпоксидных групп (см. выше
определение числа эпоксидных групп), можно найти содержа-
ние в смоле гидроксильных групп, которое может быть также
Применение эпоксидных смол
435
установлено непосредственно (см. выше определение числа
гидроксильных групп). При расчете принимают, что каждая
эпоксидная группа связывает две молекулы жирной кислоты.
Предварительное определение эквивалента этерификации
эпоксидной смолы дает ценные сведения, используемые при про-
изводстве эфира, который в дальнейшем должен служить свя-
зующим в лаке или краске. Это может быть показано на следую-
щем примере.
Пример. Эпоксидную смолу с эквивалентом этерификации 200 требуется
этерифииировать на 75% жирными кислотами льняного масла со средним мо-
лекулярным весом 280. Для этого на 200 г эпоксидной смолы нужно взять
следующее количество кислот льняного масла:
Количество выделившейся веды (теоретические потери в ходе этерификации
при этом составит:
18-75
ТОСТ = 13’5 г
Расчет сделан в предположении, что кислотное число полученного эфира
будет равно нулю, что на практике никогда не достигается.
Применение эпоксидных смол
Способы применения и отверждения эпоксидных смол в про-
изводстве лаков, красок, клеев, слоистых пластиков и материа-
лов, предназначенных для облицовки поверхностей или изгото-
вления изделий методом литья, определяются физическими и
химическими свойствами этих смол. Важнейшими из этих
свойств являются растворимость и совместимость с другими
смолами или отвердителями, а также свойства, приобретаемые
при взаимодействии с веществами, содержащими подвижные
атомы водорода, например с жирными кислотами, некоторыми
полиаминами и полиамидами.
При выборе эпоксидной смолы для определенной области
применения руководствуются, с одной стороны, требованиями,
предъявляемыми к свойствам готового изделия, и, с другой сто-
роны, возможностью подвергать изделие горячей сушке или по-
лимеризации, а также существующими способами производства.
Обычно низкомолекулярные эпоксидные смолы вследствие
их малой вязкости и большой реакционной способности при
обычной температуре используются в сочетании с отвердителя-
ми типа полиаминов или многоосновных кислот, в основном для
покрытий или для изготовления литьевых масс. Эпоксидные
смолы со средними молекулярными весами чаще всего модифи-
цируются жирными кислотами высыхающих масел для получе-
28*
436
Гл. XIII. Эпоксидные смолы
ния связующих воздушной или горячей сушки, применяемых
для изготовления лаков или эмалевых красок. Высокомолеку-
лярные эпоксидные смолы .переводят в раствор и добавляют
вязкий раствор (сироп) мочевино- или меламино-формальдегид-
ной смолы в бутиловом спирте или раствор феноло-формальде-
гидного резола. При этом получаются связующие вещества,
служащие основой для лаков или эмалей горячей сушки высо-
кого качества.
ЛИТЕРАТУРА
История вопроса:
Вег., 1891, 24, 2145.
Братья De Trey, Ър. пат. 859061; англ. пат. 518057, 1949 г.
I. G. Farbenindustrie, англ. пат. 477843, 1938 г.
S. О. Greenlee, пат. США 2456408, 1948 г.; 2503726, 1950 г.; 2493486,
1950 г.
Du Pont, пат. США 2060715, 1946 г.
Физические и химические свойства;
W. Rubin, J. Oil Col. Chem. Ass., 1952, 418.
P. Bruin, Kunststoffe, 1955, 8, 335.
Verity, Smith, British Plastics, August 1954, 307.
T. F. Bradley, Official Digest, 1950, № 310.
J. W. McNabb, H. F. Payne, Ind. Eng. Chem., 1952, 2394.
R. W. Tess, C. A. Ma y. Official Digest, December 1950.
R. N. Wheeler, J. Oil Col. Chem. Ass., June 1953, 305.
T. G. Harris и др., Modern Plastics, February 1954, 146.
Анализ:
D. Swer n, T. W. Findley, G. M. Biller, J. T. Scanlan,
Anal. Chem.. 1947, 19, 414.
G. King, Nature, 1949, 164, 706
J. L. Jungnicker, A. P о 1 g a n, F. T. Weiss, E. D. Peter,
Methods for determination of alpha-epoxides, Shell Development Report,
S-5 101, 1951.
ГЛ A BA XIV
КАУЧУК и ПРОДУКТЫ ЕГО ПРЕВРАЩЕНИЙ
Ш. П. Пинацци, Р. Шерита
НАТУРАЛЬНЫЙ КАУЧУК
При изучении натурального каучука и его производных как
возможных пленкообразующих веществ для производства не-
которых красок необходимо знать основные свойства этого ве-
щества и продуктов его превращений. Несмотря на то, что сам
каучук сравнительно мало используется в производстве лаков
и красок (за исключением материалов для окраски резины),
мы сочли полезным уделить ему значительное место, с
тем чтобы сделать более понятным описание важнейших произ-
водных каучука, рекомендуемых в качестве пленкообразующих
веществ.
Плантационные каучуки. В странах тропического пояса, где
имеются плантации гевеи, сосредоточено 95% мировой добычи
натурального каучука.
Каучук содержится в растении в виде водной дисперсии (ла-
текса), локализованной в млечных сосудах. Латекс вытекает из
регулярно возобновляемых надрезов при подсочке гевеи. Его
собирают в большие чаши, гомогенизируют, процеживают и
разбавляют, после чего переносят в резервуары, где коагули-
руют, добавляя разбавленную уксусную кислоту. Концентрации
латекса и кислоты подбирают с таким расчетом, чтобы коагуля-
ция не происходила мгновенно и коагулят постепенно оседал
на дно. Таким путем получают пропитанные серумом гели в
форме довольно широких лент. Эти губчатые листы пропускают
через вальцы для удаления серума, промывают водой, валь-
цуют и, наконец, сушат в коптильных камерах, получая таким
образом так называемые «копченые листы», известные под тор-
говым названием смокед-шитс.
Для получения «крепа», а не смокед-шитса, каучук, извле-
ченный из коагуляционных резервуаров, отжимают и пропу-
скают через вальцы с рифлеными валками, вращающимися с
разной скоростью. Во время промывки на вальцах (более тща-
тельной, чем в предыдущем случае) листы разрываются «а ча-
сти. Полученный креп высушивают в сушильных камерах горя-
чим воздухом.
438 Г л. XIV. Каучук и продукты, его превращений
Латекс. Латекс, являющийся биологической жидкостью,
представляет собой многокомпонентную смесь различного со-
става, зависящего от климатических условий, почвы и вида кау-
чуконосного растения. Латекс изменяется при хранении и яв-
ляется благоприятной средой для развития некоторых видов
микроорганизмов.
Свежий латекс состоит главным образом из водной фазы и
углеводорода каучука. В водной фазе содержатся минеральные
соли, углеводы и белковые вещества. Частицы каучука имеют
форму глобул диаметром от 0,1 до 2 ц. Эти частицы окружены
слоем белков — защитных коллоидов, сообщающих глобулам
каучука отрицательный заряд. Обычно латекс мало устойчив и
вскоре начинает коагулировать. Для предупреждения коагуля-
ции в латекс вводят консервирующее .вещество, препятствующее
развитию микроорганизмов и повышающее pH среды до 10—11.
Большей частью для этой цели применяют аммиак, который вво-
дят в количестве от 0,5 до 1%, но используются и другие кон-
сервирующие вещества, например пентахлорфенолят натрия.
Обычно в сыром латексе содержится 38—42% углеводорода
каучука, но при неблагоприятных условиях сбора каучука со-
держание углеводорода может снизиться до 30% и менее. Та-
кие латексы вполне пригодны для производства смокед-шитса
и крепов на месте, но латекс, предназначенный для дальней
транспортировки, должен быть сконцентрирован до содержания
каучука приблизительно 60%. Концентрирование производится
путем центрифугирования, отстаивания, выпаривания или элек-
тродекантации. В латексах, сконцентрированных выпариванием,
может содержаться до 70% каучука; они консервируются силь-
ной щелочью (едким натром или едким кали).
'Сконцентрированный и консервированный латекс в процессе
хранения претерпевает изменения, связанные, вероятно, с био-
химическими изменениями защитных коллоидов. Происходящие
изменения можно установить, определяя устойчивость латекса
при нагревании в присутствии окиси цинка, измеряемую време-
нем, требуемым для желатинирования латекса при заданной
температуре (обычно при 70°). Об изменениях, происходящих в
латексе, можно судить также по кислотному числу и по так на-
зываемой «механической» стабильности, т. е. скорости коагуля-
ции латекса при определенном режиме перемешивания (в ста-
билометре).
Разрушение коллоидной структуры латекса происходит путем
коагуляции, желатинирования или флокуляции. Эти явления
вызываются действием тепла, кислот, минеральных солей двух-
или трехвалентных металлов (Zn, Mg, Ba, Са, Al), протеолити-
ческих энзимов, поверхностно-активных катионных агентов и
некоторых коллоидов (раствор фторсиликата натрия).
Натуральный каучук
439
Товарные каучуки. Исходный каучук принято называть сы-
рым каучуком в отличие от вулканизованного каучука, или ре-
зины. Пользуются также названиями сухой или твердый каучук
в отличие от каучука в виде латекса. Наконец, добавляют при-
лагательное натуральный, чтобы отличить природный продукт
растительного происхождения от синтетических эластомеров,
получаемых полимеризацией или сополимеризацией соответст-
вующих мономеров.
Имеется .много различных видов товарных сырых каучуков:
лесные каучуки, или паракаучуки, туземные и плантационные
каучуки. Лесные каучуки имеют только историческое значение.
Однако в периоды острого дефицита в каучуке сбор его в лесах
возобновляется. Туземные каучуки, поступающие только на рын-
ки стран тропического .пояса, собираются туземцами и продают-
ся в нестандартном виде. Их скупают на месте промышленники,
перерабатывающие этот каучук и .выпускающие его в виде
«караваев».
В табл. 25 приведена классификация выпускаемых стандарт-
ных плантационных каучуков.
ТАБЛИЦА 25
Плантационные каучуки
Наим енование Сорт (в нисходящем порядке от 1 до 5) и характеристика
Обычные каучуки
Смокед-шитс рифленый 1 X
Крепы 1 X
Вторичные (темные) крепы 1 X,
2 X, 3 X
Крепы повторной переработки
Рекуперированные каучуки
1—2—3—4 и 5 (обрезки)
1—2
1—2—3—4 и 5
Специальные каучуки
Сернанби белый и черный
Скреп (оскребки с подсочечных чаш)
Каучук с земли
Каучук с коры (смесь каучука с расти-
тельными остатками)
Улучшенные каучуки, особо приготов-
ленные для придания им специальных
свойств, но без добавки химических
продуктов (марки TRS, USF и др.)
Каучуки, модифицированные химически-
ми продуктами для изменения некото-
рых свойств (искусственно пластициро-
ванные каучуки: пласторуб)
Порошкообразные каучуки, сырые или
вулканизованные (распыленный каучук,
илиамилоруб)
440 Гл. XIV. Каучук и продукты его превращений
Смокед-шитс, составляющий большую часть сырых каучуков
промышленного назначения, выпускается в кипах спрессован-
ных листов (вес кипы немного более 100 кг), обмазанных
суспензией, содержащей тальк и мел. При высыхании суспензии
на поверхности кипы остается нелипкая белая пленка, предот-
вращающая слипание кип во время их транспортировки. Кипы
маркируются: указывается происхождение и качество каучука.
В результате серьезных работ, проводившихся на планта-
циях стран тропического пояса, начиная с 1951 г., оказалось
возможным поставлять каучуки, сортированные не по внешнему
виду листов, а по их технологическим свойствам. Такие сорти-
рованные каучуки удовлетворяют определенным требованиям
в отношении скорости их вулканизации.
Состав сырого каучука
Вследствие растительного происхождения каучука следует с
известной осторожностью подходить к понятию его химического
состава. Естественно, что в крепе или смокед-шитсе содержат-
ся известные количества органических или минеральных
веществ, сопутствующих углеводороду каучука в латексе гевеи.
Следовательно, необходимо отличать каучук как химическое
вещество от сырого каучука, т. е. материала, содержащего все
эти примеси. Пожалуй, было бы правильнее называть сырой
каучук соответственно крепом или смокед-шитоом и сохранить
наименование каучук для очищенного продукта.
Из сказанного выше можно видеть, что в сыром каучуке
содержится большое количество различных «некаучуковых ком-
понентов».
Некаучуковые компоненты. Эти компоненты можно разде-
лить на две группы: минеральные компоненты (вода, соли) и
органические компоненты, относящиеся к разным классам
соединений: белки и продукты их распада (полипептиды, амино-
кислоты, амины), жироподобные вещества (лецитины, стерои-
ды, жирные кислоты), стероидные гликозиды или родственные
им соединения (например, квебрахит).
В каучуке обычно содержится от 0,3 до 1,2% воды. Ее содер-
жание явно зависит от содержания гигроскопичных белковых
веществ. При чрезмерной влажности каучука облегчается раз-
витие микроорганизмов, что приводит к его разрушению.
Минеральные компоненты чаще всего представлены соедине-
ниями, содержащими натрий, калий, кальций, магний, железо,
фосфор, серу и хлор. Кроме того, часто можно обнаружить
следы еще некоторых элементов, например меди и марганца,
способных играть роль катализаторов окислительной деструк-
ции сырого и вулканизованного каучука. Эти примеси вредны,
если их содержание превышает 10~4—10-3%.
Натуральный каучук
441
Органические компоненты, сопутствующие каучуку, можно
разбить на две группы по растворимости в ацетоне.
Ацетоновый экстракт. Продукты, извлекаемые ацетоном и»
сырого каучука, были на основании их консистенции неправиль-
но названы смолами. В действительности фракция ацетоновой
вытяжки, стоящая на первом месте в весовом отношении,
состоит в основном из жирных кислот Си. Сложные жироподоб-
ные .вещества должны были бы быть представлены лецитинами,
но лецитины мало растворимы в ацетоне и мало устойчивы. Все
же в ацетоновом экстракте должны присутствовать некоторые
продукты их распада: жирные кислоты, холин.
Ацетоновый экстракт содержит стероиды и, в частности,,
некоторые стерины. Удалось выделить два стерина (С20Н30О и
С27Н42О3), являющиеся ингибиторами окисления каучука; в кре-
пе содержится около 0,1% таких стеринов-антиоксидантов.
Приходится допустить, что довольно значительная часть вы-
сокомолекулярных веществ нерастворима в ацетоне. Эта часть,
состоящая преимущественно из восков, может составлять до
1% сырого каучука. Глюкозидная фракция некаучуковых ком-
понентов состоит преимущественно из стероидных глюкозидов;
и многоатомного спирта квебрахита (метил-1-инозита), который
можно обнаружить также в водной вытяжке сырого каучука.
Белковая фракция представлена аминокислотами, в том числе
d-валином; полипептиды в ацетоне нерастворимы.
Как установлено, количество веществ, извлекаемых ацетоном;
из одного и того же образца сырого каучука, колеблется в за-
висимости от степени его окисления. В ацетоновой вытяжке-
окисленного каучука можно обнаружить продукты окисления
углеводорода каучука, которые еще легче выделить путем
экстрагирования спиртовым раствором едкого -кали.
Наконец, в ацетоновом экстракте содержатся красящие ве-
щества, относящиеся к политерпенам. Эти вещества содержатся
в неомыляемой части экстракта.
Белковые вещества. Содержание белков в латексе сильно
колеблется в зависимости от биологических и -климатических
факторов. При переработке латекса в смокед-шитс или в креп
значительная, но непостоянная часть белков теряется; поэтому
точное их содержание трудно установить. Считают, что в боль-
шинстве случаев содержание белков составляет 2,5—3,5% от
веса каучука. Часть органически связанного азота входит в со-
став аминокислот и аминов жирного ряда — конечных продук-
тов распада белков. Аминокислотный состав этих белков мало-
изучен.
Можно отметить, что белки, содержащиеся в латексе,
играют роль защитных коллоидов и придают стабильность дис-
персии глобул каучука. Путем многократного центрифугирова-
442 Гл. XIV. Каучук и продукты его превращений
ния латекса в присутствии поверхностно-активных веществ, спо-
собных вытеснять белки, можно почти полностью отделить
белки от каучука.
Ввиду отсутствия точного метода непосредственного опреде-
ления содержания чистого каучука его находят по разности,
определив содержание всех некаучуковых компонентов.
Углеводород каучука. Полиэтиленовое строение углеводоро-
да каучука доказано различными способами, в частности опре-
делением иодного числа. Важные сведения о структуре этого
углеводорода дает пиролиз. Так, при нагревании до 300° в ва-
кууме. каучук распадается на изопрен С5Н8 и дипентен (С5Н8)2.
На этом основании сделан вывод, что структурным элементом
макромолекулы каучука является изопрен. Для подтверждения
этой гипотезы были сделаны попытки синтезировать каучук из
изопрена. При этом действительно получается полимер, ана-
логичный каучуку, но обычно с пониженными механическими
свойствами. Для объяснения этих результатов было высказано
предположение, что у исследованных синтетических полиизопре-
нов менее упорядоченная структура, чем у природного каучука.
С помощью озонирования каучука было уточнено располо-
жение изопреновых звеньев. При гидролизе полиозонида кау-
чука получается левулиновый альдегид, что свидетельствует об
упорядоченном расположении изопреновых остатков (по типу
«голова к хвосту»):
СНз
с ^сн2
н2с сн
изопрен
СН.,
I J
С—О^_СН2
н!сЛх .сн
о
отрезок цепи углеводорода каучука
продукт озонирования углеводорода каучука
левулиновый альдегид
Легко показать, что при неупорядоченном расположении изо-
преновых остатков образовались бы янтарный альдегид и аце-
тонилацетон, которые действительно содержатся в продуктах
озонирования синтетического полиизопрена.
Натуральный каучук.
443
Структура, предложенная для углеводорода каучука, до-
пускает возможность чпс-транс-стереоизомерии:
Н3С
=сн сн2 сн2 с=сн
СН2 С=СН Х'сн2 ^СНл—
цис-конфигурация
Н3С сн2—
Н3С СН2ЧС=СН
н3с сн2 с=сн ^сн,
ХС=СН ^СНг
—сн2
/принт-конфигурация
zjtzc-Конфигурация соответствует каучуку, транс-конфигура-
ция — углеводородам балаты и гуттаперчи. Разной пространст-
венной структурой объясняются различия механических свойств
этих двух групп продуктов.
Строение молекулы каучука нельзя считать полностью вы-
ясненным, так как на основе предложенной модели не удается
объяснить всех свойств сырого каучука.
При действии растворителей, например серного эфира, кау-
чук разделяется на две фракции: сравнительно растворимую
фракцию — золь-каучук и .нерастворимую фракцию — гель-кау-
чук. Поскольку это различие трудно объяснить разным разме-
ром линейных молекул, приходится допустить наличие межмо-
лекулярных связей, образующих сетчатую структуру. Простран-
ственным структурированием можно объяснить малую раство-
римость части сырого каучука и его механические свойства, не-
сколько отличающиеся от свойств, которыми должен был бы
обладать полимер строго линейного строения.
Если фракционирование проводится в присутствии кисло-
рода (даже очень небольших его количеств), количество золь-
фракции непрерывно увеличивается за счет геля. Если же тща-
тельно изолировать полимер от доступа .кислорода, количество
растворимой фракции будет зависеть от степени окисления ве-
щества перед фракционированием. Следовательно, золь-каучук
состоит из продуктов деструкции гель-каучука. В некоторых
случаях возможен обратный переход золь-каучука в гель-кау-
чук путем образования межмолекулярных мостиков (анало-
гично процессу вулканизации).
444 Гл. XIV. Каучук, и продукты его превращений
Молекулярный вес каучука обычно определяют осмометри-
чески или вискозиметрически, причем надежность вискозимет-
рического метода вызывает сомнение, так как он применим,
по-видимому, только к макромолекулам линейного строения.
Расхождения между данными вискозиметрических и осмомег-
рических определений привели Штаудингера к заключению,
что не все молекулы каучука имеют линейное строение.
Осмометрическим методом установлено, что молекулярный
вес наиболее медленно диффундирующих фракций лежит в
пределах от 200 000 до 300 000. При фракционировании без
доступа кислорода молекулярный вес наиболее растворимых
фракций в среднем равен 50 000.
Не следует забывать, что каучук 'неустойчив по отношению
к кислороду, особенно в присутствии растворителя. Поэтому
никогда нельзя быть уверенным, что измерен истинный моле-
кулярный вес каучука, а не молекулярный вес частично окис-
ленных молекул.
Пластикация сырого каучука сопровождается интенсивным
окислением,, приводящим к уменьшению полидисперсности, и
молекулярный вес приближается к средней величине около
30 000. При очень длительной пластикации на холоду молеку-
лярный вес каучука может быть снижен до 10 000 и менее.
Химические свойства каучука
Углеводород каучука обладает свойствами полиенового
соединения, но конфигурация изопренового остатка, высокий
молекулярный вес и наличие некаучуковых компонентов вносят
ряд особенностей в его химические свойства. Как правило, про-
дукты реакций не удается выделить в чистом виде и о проис-
ходящих превращениях чаще всего приходится судить по изме-
нению технологических свойств. Физические константы непри-
годны для характеристики каучука, поскольку он представляет
собой смесь различных веществ.
Подобно обычным олефиновым углеводородам, каучук спо-
собен к реакциям присоединения, деструкции и полимеризации.
Кроме того, он может подвергаться изомеризации, циклизации
и даже способен к реакциям замещения. Классифицировать
эти реакции довольно трудно, так как многие реагенты всту-
пают с каучуком в реакции нескольких типов.
Почти во всех превращениях каучука принимает участие
кислород; ввиду многообразия реакций с кислородом моле-
кула каучука еще более усложняется.
Наиболее распространенным агентом вулканизации каучука
является сера. Вулканизация представляет собой совокупность
не вполне выясненных химических процессов, включающих ре-
акции присоединения, замещения, возможно, даже циклизации.
Натуральный каучук
445
приводящие к увеличению молекулярного веса и сшиванию
макромолекулярных цепей. Эти изменения структуры вызы-
вают изменение технологических свойств каучука, у которого в
сыром виде пластичность преобладает над эластичностью, а в
вулканизованном состоянии преобладает эластичность.
Реакции присоединения. Углеводород каучука может присо-
единять по месту двойных связей водород, галоиды, галоидо-
водородные кислоты, некоторые нитро- и нитрозопроизводные,
хлорноватистую кислоту, кислород и озон, а также серу.
Все эти реакции присоединения обычно сопровождаются дру-
гими превращениями, например вызываемыми повышенной
реакционной способностью а-метиленовых группировок, легко
отщепляющих атом водорода:
СН3
I
С СН2
сн2 сн
а
В то же время следует всегда учитывать, что некоторые двой-
ные связи ,не затрагиваются обычной реакцией присоединения.
Гидрирование. Водород присоединяется по двойным связям
в 'присутствии катализатора и под высоким давлением. В этих
условиях нельзя избежать деструкции, приводящей к такому
снижению молекулярного веса гидрированного продукта, что
образуются маслообразные вещества.
Галоидирование. Все галоиды способны вступать в реакцию
с каучуком, но каждый из них оказывает несколько отличное
действие. Действие фтора на'каучук пока мало изучено, но,
учитывая интерес, проявляемый в последнее время к фториро-
ванным полимерам, можно ожидать развития исследований в
этой области. Реакции взаимодействия хлора с каучуком ши-
роко изучены. Теоретически каучук должен был бы присоеди-
нять одну молекулу Ch 'По каждой двойной связи с образова-
нием соединения, содержащего 51% хлора. Однако практически
трудно остановить хлорирование на 51%, и хлорированные кау-
чуки содержат от 65 до 70% связанного хлора, что примерно
соответствует четырем атомам хлора на один изопреновый оста-
ток (см. стр. 451 и сл.). При действии брома 1получается продукт
более определенного состава, соответствующий присоединению
одной молекулы брома по каждой двойной связи. Наиболее
интересным свойством бромированного каучука является его
способность вступать в реакции Фриделя — Крафтса.
Действие галоидоводородных кислот. Фтористый водород
вступает с каучуком в сложные реакции и благоприятствует
его циклизации. Хлористый водород присоединяется в соот-
446
Г л. XIV. Каучук и продукты его превращений
ветствующих условиях в количестве, отвечающем присо-
единению одной молекулы НС1 ,по каждой двойной связи.
Гидрохлорированный каучук изучен значительно лучше, чем
хлоркаучук.
Бромистоводородная кислота оказывает примерно такое же
действие, как соляная, но гидробромированные каучуки мало
стабильны.
Образование нитро- и нитрозопроизводных. Тетранитроме-
тан регулярно присоединяется по месту двойных связей:
СН3 сн3
С СН2------ /NO« с СН2-----
/ Ч / +OsN-C-NO2 — /1\ /
---Н2С СН \Na ---------н2с | сн
°2N C(NO2)3
Нитрозобензол также может присоединяться к изопреновой
группировке, но при этом происходит восстановление другой
молекулы нитрозобензола до фенилгидроксиламина:
СН,
I
С СН2-------
/ \ / + 2CeH5NO
----НаС СН
СН3
I
С СН2—
У \ / + CsHjNHOH
----НС с
ON—С6Н5
Действие хлорноватистой кислоты. Этот реагент в основном
присоединяется по месту двойных связей. Проводя реакцию
в среде латекса, можно получить каучук, содержащий 29,4%
связанного хлора, что, по-видимому, соответствует полному
насыщению всех этиленовых связей. При действии НОС1 на
растворы каучука протекают обычно более сложные реакции,
включающие дополнительное хлорирование. Таким путем полу-
чается продукт, содержащий 66% связанного хлора.
Действие кислорода и серы. Эти элементы оказывают очень
сложное действие, и механизмы реакций недостаточно изучены.
Присоединение серы к каучуку приводит к уменьшению степени
ненасыщенности и появлению свойств, присущих вулканизован-
ному каучуку.
Предложены различные трактовки механизма вулканизации,
но ни одну из них нельзя считать окончательно доказанной.
Считают, что сера присоединяется к каучуку, образуя известное
Натуральный каучук
447
число межмолекулярных мостиков, определяющих свойства вул-
канизата. Однако эти мостики не должны быть слишком ча-
стыми; при введении более 30% серы получается твердый н
жесткий материал — эбонит.
Сера является не единственным реактивом, способным вы-
зывать вулканизацию; аналогичное действие могут оказывать-
различные аминопроизводные, нитропроизводные и перекиси.
Под влиянием некоторых соединений действие серы на кау-
чук ускоряется. Такими ускорителями вулканизации служат
меркаптобензотиазолы, дитиокарбаматы, тиурамы, ксантогенаты
и дифенилгуанидин. Для повышения эффективности действия
ускорителей добавляют активаторы, например окись или стеарат
цинка.
Кислород и окислители вызывают значительные изменения
каучука. Кислород действует как на сырой, так и на вулкани-
зованный каучук и служит непосредственной причиной его
«старения». Подобно сере кислород может вступать в различ-
ные реакции с каучуком: он может присоединяться, вызывать
разрыв цепей, циклизацию, образование межмолекулярных мо-
стиков и т. д. Механизм этих реакций недостаточно выяснен и
уточнен.
Кислород воздуха постепенно присоединяется к каучуку, но-
не установлено, в какой форме содержится связанный кисло-
род. Поскольку в каучуке обычно содержатся антиокислители,
счищенный каучук окисляется гораздо быстрее, чем сырой.
Действие окислителей. Озон реагирует с каучуком, образуя
полиозониды, которые дают при гидролизе левулиновый аль-
дегид и левулиновую кислоту. Слабо озонированный воздух
вызывает особенно сильное разрушение вулканизованного кау-
чука; появляются глубокие параллельные трещины, перпенди-
кулярные к направлению приложенных напряжений.
При действии надбензойной кислоты по каждой двойной
связи присоединяется по одному атому кислорода. Другие окис-
лители, например, перекись бензоила, нитробензол, хлоранил
и т. д., оказывают вулканизующее действие.
Реакции расщепления и деструкции. При некоторых воз-
действиях растворимость каучука в растворителях увеличивает-
ся, а вязкость растворов уменьшается, что свидетельствует о
снижении молекулярного веса. Трудно‘установить, вызывается
ли это реакциями деполимеризации или реакциями окислитель-
ной деструкции; поэтому мы будем применять термин «де-
струкция», который не связан с представлением об определен-
ном механизме реакции.
Под действием высокой температуры каучук превращается
в вязкую массу; при пиролизе без доступа воздуха образуются
изопрен и терпеновые углеводороды.
448 Гл. XIV. Каучук и продукты его превращений
Кислород вызывает пластикацию сырого каучука и старение
?зулканизованного каучука. Вообще не существует химических
.реакций или превращений каучука, в которых не участвовал
<бы кислород.
Деструкция каучука может ускоряться ‘при помощи катали-
заторов окисления (солей меди, кобальта, марганца или же-
леза), перекисей или действием света и нагревания. Замедле-
ние деструкции вызывают антиокислители, например фенил-
Р-нафтиламин, альдоль-а-нафтиламин, динафтил-п-фенилен-
.диамин.
Реакции изомеризации и циклизации. Это сложные реакции,
гпри которых происходит перемещение двойных связей или цик-
.лизация после раскрытия этиленовых связей. Реакции цикли-
зации каучука будут подробно рассмотрены в разделе, посвя
щенном циклизованным каучукам.
Применение сырого каучука
Сырой каучук, подвергнутый частичной деструкции путем
пластикации, может вводиться в масляные краски для прида-
ния им некоторых свойств1. Он легко смешивается с высыха-
ющими маслами и облегчает образование прочной пленки.
Краска, содержащая равные количества каучука и масла, хо-
рошо наносится на поверхность. Пигменты, содержащиеся в
таких красках, не склонны оседать на дно тары при хранении
красок. Этот вопрос подробно рассматривается в разделе «Про-
дукты окисления каучука».
Раствор пластицированного каучука в соответствующем рас-
творителе может быть использован также как основа красок,
предназначенных для резиновых изделий2. В такие краски
можно вводить любые твердые компоненты, за исключением
графита и сульфата бария.
В качестве примера приведем рецептуру смеси (вес. чд,
которая после растворения в соответствующем растворителе
(предпочтительно в хлорированном углеводороде жирного ряда)
дает обычно хорошие результаты:
Светлый креп ..........100
Стеариновая кислота . . 0,6
Окись цинка............0,5—1
Сера ..................1,5
Ускоритель ............ 1
Цветные пигменты .... X
Стеариновая кислота должна вводиться в малых количествах
во избежание выпотевания, ухудшающего адгезию пленки. Ре-
комендуется применять только так называемые активные сорта
окиси цинка. Выбор ускорителей зависит от условий вулкани-
зации. Хорошие результаты дает применение в качестве уско-
рителя тетраметилтиурамдисульфмда, активированного неболь-
Гуттаперча и балата
449
шим количеством диэтилдитиокарбамата цинка. Ускорители (или
смесь ускорителей) не должны обладать активностью при обыч-
ной температуре, чтобы во время хранения краски не происхо-
дила вулканизация. Сушка таких красок при 100—110° должна
продолжаться 20—30 мин. Количество вводимого цветного пиг-
мента колеблется в зависимости от требуемого цвета и может
достигать 25%.
ГУТТАПЕРЧА И БАЛАТА
Эти вещества естественного происхождения содержатся в
млечниках некоторых деревьев семейства Sapotacea", млечники
этих деревьев нечленистые, но сегментированные удлиненные.
Латекс очень сходен с латексом гевеи, но значительно более
вязкий. Гуттаперча извлекается из латекса видов Palaquiutn,
в частности Inosandra percha. Балата получается коагуляцией
латекса Mimosaps globosa.
Гуттаперча и балата — вещества, довольно твердые при
обычной температуре, но в то же время термопластичные.
Гуттаперча малоэластична при 15°; при нагревании до 50° она
размягчается и при этой температуре поддается вальцеванию
и растяжению. При 70° гуттаперча легко формуется, при 100°
становится текучей, при 110° плавится и при 180° разлагается.
Если температура нагревания не превышает 120°, то при по-
следующем охлаждении гуттаперча снова приобретает исход-
ную консистенцию и жесткость. Балата несколько отличается
по свойствам от гуттаперчи. Она мягче при обычной темпера-
туре и сохраняет большую эластичность при низких темпера-
турах. Основной составной частью гуттаперчи и балаты яв-
ляется углеводород полиизопрен (транс-изомер углеводорода
каучука). Этот углеводород связан со смолами, состоящими
в основном из сложных эфиров: спиртов фитостеринового ря-
да, этерифицированных различными жирными кислотами,
главным образом уксусной, капроновой, каприловой и янтар-
ной (эфиры янтарной кислоты содержатся только в смолах
гуттаперчи). Экстрагированием спиртом удалось выделить два
смолистых вещества: альбан и флюавиль. Альбан — белая смо-
ла, плавящаяся при температуре 160—175°, растворимая в ки-
пящем этиловом спирте и нерастворимая в холодном; флю-
авиль— желтая смола, плавящаяся при 100—110° и раство-
римая в холодном этиловом спирте. Смолы гуттаперчи состоят
из двух частей альбана и одной части флюавиля. Этим раз-
личием в составе отчасти объясняется разный внешний вид
товарных продуктов.
Углеводород гуттаперчи и балаты является твердым веще-
ством, несколько напоминающим рог; в чистом виде при обыч-
29—156
450 Гл. XIV. Каучук и продукты его превращений
ной температуре бесцветен. Он термопластичен и приобретаег
небольшую липкость при 100°; при этой температуре легко
деформируется при сжимании пальцами. По химическим свой-
ствам этот углеводород близок к углеводороду каучука, но
более инертен. Он растворяется в ароматических углеводоро-
дах, но нерастворим на холоду в углеводородах жирного ряда;
растворим в хлороформе, четыреххлористом углероде и серо-
углероде, плохо растворим в сложных эфирах и вовсе не рас-
творяется в спиртах и кетонах. Балата частично растворяется
в скипидаре, оливковом масле, сланцевом масле и обладает
незначительной гигроскопичностью.
Гуттаперча и балата устойчивы по отношению к разбав-
ленным растворам щелочей и кислот, к растворам солей и к
плавиковой кислоте. Они быстро разрушаются концентрирован-
ными кислотами, серной и азотной; после обработки концен-
трированными растворами щелочей становятся очень чувстви-
тельными к окислению.
Гуттаперча очень легко окисляется. При длительном воз-
действии воздуха и света она окисляется с поверхности, теряя
вязкость и эластичность, и приобретает растворимость в без-
водном спирте. Балата хотя и склонна к окислению, но все
же устойчивее гуттаперчи. Антиоксиданты, применяемые в ре-
зиновой промышленности, достаточно хорошо предохраняют от
окисления гуттаперчу и балату. Гуттаперча и балата могут
вулканизоваться серой; при этом они становятся менее плав-
кими и лучше противостоят старению. Из них можно получать
особый вид эбонита, поддающийся обработке, как слоновая
кость и рог, и хорошо полирующийся.
Применение гуттаперчи и балаты постепенно сокращается,
но во многих областях они могут с успехом использоваться.
Благодаря хорошим диэлектрическим свойствам они представ-
ляют большую ценность в качестве изоляции электрических
проводов и кабелей, а также для изготовления мастики Чаг-
тертона.
Вследствие высокой прочности при растяжении и малой
эластичности под действием растягивающего усилия балата
используется для изготовления транспортерных лент и для
покрытия грунтовых дорог. Эти смолы входят в состав неко-
торых мастик для литья и для заливки стыков и швов. Бла-
годаря термопластичности они используются для получения
оттисков и изготовления форм. Для нанесения покрытий при-
меняют бензольные растворы гуттаперчи или балаты. Под-
лежащее покрытию изделие обливают или окунают в эти до-
вольно вязкие растворы.
Гуттаперча и балата мало используются в качестве пленко-
образующих веществ. Это объясняется их быстрым старением,.
Хлоркаучуки
451
при котором они приобретают липкость, а затем затвердевают
и растрескиваются. Однако их термопластичность и клеящая
способность могут представить в некоторых случаях большую
ценность.
ХЛОРКАУЧУКИ
В настоящее время хлоркаучуки выпускаются в промыш-
ленных масштабах и вследствие своей коррозионной стойко-
сти являются ценными связующими для некоторых лакокра-
сочных материалов.
Действие хлора на каучук. Механизму хлорирования на-
турального каучука было посвящено довольно много работ1-9.
Однако сравнительно точное представление о протекании этой
реакции было получено только в 1943 г. в результате работ
Блумфилда10.
Действуя на раствор каучука в четыреххлористом углероде
хлором в количестве одного, двух и трех молей на каждую
изопреновую группу, Блумфилд показал, что только первые
два атома хлора замещают атомы водорода. Следующие два
атома входят в молекулу углеводорода как заместители и ча-
стично присоединяются по двойным связям. Третья молекула
хлора реагирует только по реакции замещения, но хлор всту-
пает в реакцию уже значительно труднее. Схематически реак-
ции элементарного звена, состоящего из двух изопреновых
групп, могут быть изображены следующим образом:
(C10Hlc)„ J- 2пС12 —*• (С10Н14С12)„ + 2пНС1 (замещение)
(CioHidClaL + 2пС12 —* (Ci0H13Cl6)n + пНС1 (замещение и присоединение)
(СюН13С16)„ + 2пС12 —> (CioHuCl7)n + 2пНС1 (замещение)
Хлорпроизводное, соответствующее первой из приведенных
реакций, можно получить также, применяя в качестве реагента
фенилиодидхлорид CeHgJCh или хлористый сульфурил в при-
сутствии перекиси бензоила11. Следовательно, при этом не полу-
чается тетрахлорпроизводное суммарного состава (СщНщС^и,
как предполагали Кирхгоф7 и Нильсон8.
Блумфилд определял одновременно степень ненасыщенно-
сти, количество связанного хлора и количество выделяющегося
в процессе хлорирования хлористого водорода. Он обнаружил
расхождение между содержанием связанного хлора, количе-
ством выделившегося хлористого водорода и степенью нена-
сыщенности и приписал его реакции циклизации. Однако не-
достаточная точность определения степени ненасыщенности
не позволяет точно установить истинную структуру хлоркау-
чука.
29*
452 Гл. XIV. Каучук и продукты его превращений
Фармер12 предложил механизм хлорирования каучука, при
котором процесс замещения галоидом сопровождается цикли-
зацией:
СН, сн,
. « I
-т-СН2—с=сн— сн2 ч-сн2—с=сн —сн2 4-
или
или
отрезок цепи углеводорода каучука
хлорирование по Фармеру
Следовательно, структура полностью хлорированного со-
единения13 должна изображаться формулой I (см. ниже), что
соответствует суммарной формуле, .предложенной Блумфилдом
(СюНпСЬ). Согласно приведенной схеме не все двойные связи
изопреновых группировок реагируют одинаково; по одним двой-
ным связям происходит циклизация и хлорирование, по дру-
гим— просто присоединение двух атомов хлора. Предложенная
структура I довольно близка к структуре II, предложенной
Пауерсом14, в которой, однако, не учитывается реакционная
способность а-метиленовых групп углеводорода каучука:
СюНиС17
Хлоркаучуки
453.
Штаудингер15 установил, что молекулярный вес хлоркаучу-
ков колеблется от 100 000 до 400 000. По его мнению, более
низкая вязкость растворов хлоркаучуков по сравнению с ис-
ходными каучуками объясняется циклизацией, захватывающей
большое число звеньев.
Таким образом, структура хлоркаучуков точно не установ-
лена. По-видимому, в зависимости от количества присоеди-
ненного хлора и характера реакции они имеют различное строе-
ние. Кроме того, нет твердой уверенности в том, что одно
и то же звено повторяется на протяжении всей цепи.
Получение хлоркаучуков
Хлорирование каучука в растворе. Хлоркаучук, предна-
значенный для производства лакокрасочных материалов, не дол-
жен обладать слишком большой вязкостью. Поэтому перед
хлорированием сырой каучук подвергают предварительной де-
струкции путем механической обработки или непосредственного
окисления (кислородом, озоном, органическими перекисями
или в присутствии катализаторов деструкции). Для растворе-
ния каучука применяются растворители, устойчивые к хлору,
чаще всего четыреххлористый углерод, реже — хлорбензол, гек-
сахлорэтан и смеси гексахлорэтана с четыреххлористым угле-
родом.
Чтобы получить при хлорировании однородный продукт,
нужно создать условия для наиболее тесного соприкосновения
каучука с хлором. Для этого процесс ведут в разбавленном
маловязком растворе, ограничивая концентрацию каучука пре-
делами 4—7%. Для улучшения контакта с хлором пользуются
либо механическими способами (непрерывное перемешивание
или пропускание раствора через вертикальные колонны, запол-
ненные стеклянными кольцами при введении хлора снизу),
либо распыляют раствор смесью хлора с воздухом, подава-
емой под давлением. Реакция, вначале экзотермическая, ста-
новится затем эндотермической и требует подвода тепла.
Предпочтительно сначала применять умеренный подогрев,
а затем нагревать реакционный раствор до более высокой тем-
пературы. При кипении растворителя происходит дополнитель-
ное перемешивание и облегчается удаление хлористого водо-
рода.
Некоторые авторы рекомендуют применять для ускорения
реакции хлорирования иод16, окислители17 и галогениды железа,
цинка и алюминия18.
При слишком высокой концентрации раствора в процессе
реакции может произойти полное и необратимое желатини-
рование. По данным Швейцера19, такое гелеобразование можно
454 Гл. XIV. Каучук и продукты его превращений
предотвратить, добавляя некоторые вещества. Растворитель
удаляют перегонкой или отгонкой с водяным паром.
При перегонке необходимо принимать соответствующие ме-
ры предосторожности, чтобы избежать местных 'перегревов, ко-
торые могут вызвать разложение продукта.
Хлористый водород при перегонке удаляется не полностью.
При отгонке с водяным паром конечный продукт приходится
подвергать дополнительной сушке (этот способ принят фирмой
Societe d’ Electrochimie d’ Ugine20).
Хлорирование набухшего и сухого каучука. Некоторые ав-
торы 211 zz, стремившиеся упростить получение хлоркаучуков,
пытались непосредственно действовать хлором на тонкие ли-
сты каучука, пропитанные четыреххлористым углеродом23, а
также проводить хлорирование тонких листов .каучука под
давлением. Эти попытки не увенчались успехом, так как при
хлорировании сухого каучука трудно обеспечить тесный кон-
такт хлора с каучуком.
Хлорирование латекса. Для получения хлоркаучука можно
использовать латекс, но при этом следует иметь в виду необ-
ходимость предотвращения нежелательной коагуляции, вызы-
ваемой хлором или образующимся хлористым .водородом.
С этой целью были испытаны различные способы, в частности
процесс И. Г. Фарбениндустри24, заключающийся в окислении
латекса, стабилизованного анионными поверхностно-активными
веществами, и последующем его хлорировании. Получающиеся
таким путем продукты содержат 40—60% хлора.
Мартин, Дэви и Бэкер25 рекомендуют стабилизовать латекс
сапонином и подкислять его точно определенным количеством
соляной кислоты. Реакция протекает под действием хлора, вы-
деляющегося при окислении хлоратами соляной кислоты. Полу-
ченные хлорпроизводные содержат менее 60% хлора.
Мак-Гавак26 осуществил хлорирование вулканизованного ла-
текса, представляющего собой значительно более стойкую дис-
персию. Анализ очищенного продукта реакции показал отсут-
ствие в нем серы, ранее связанной с каучуком. По-видимому,
сера отщепляется в процессе хлорирования.
Промышленное производство хлоркаучуков. В промышлен-
ных условиях хлорирование каучука проводится в растворе.
Схема производства изображена на рис. 54.
Каучук, предварительно обработанный на вальцах 1, рас-
творяют в емкости 2, получая 4—5%-ный раствор. Хлорирова-
ние проводят в реакторе 3, куда под давлением подается хлор.
По завершении реакции раствор сливают в сборник 5 и сушат
в сушилке 6. Рекуперация растворителя производится в агре-
гате 7. Выделяющийся при хлорировании хлористый водород
в смеси с парами растворителя (четыреххлористого углерода)
Хлоркаучуки
455
поступает в конденсатор 4, откуда направляется на фракцио-
нирование и очистку в аппарат 8.
По завершении процесса из раствора удаляют растворен-
ную в нем соляную кислоту путем промывки щелочным рас-
твором.
При получении хлоркаучуков можно регулировать их вяз-
кость. На вязкость влияют предварительная деструкция каучу-
ка, условия хлорирования и метод удаления растворителя.
Рис. 54. Схема установки для хлорирования
каучука:
/—вальцы; 2—емкость; 3—реактор; 4—конденсатор;
5—сборник; 6—сушилка; 7—рекуператор; 8—устрой-
ство для выделения и очистки НС1.
Для получения очищенного хлоркаучука из него удаляют
белки путем обработки раствора активированным углем или
кипячением со щелочью с последующей промывкой и сушкой.
Стабилизация хлоркаучуков. Полностью хлорированные кау-
чуки довольно стабильны, в то время как хлоркаучуки с ма-
лым содержанием хлора не отличаются стойкостью. Нестой-
кость каучуков вызывается не только наличием следов кис-
лоты, оставшейся после хлорирования; небольшие количества
хлористого водорода отщепляются под действием света и тепла.
Для стабилизации хлоркаучуков и нейтрализации выделя-
ющихся кислот рекомендуются некоторые реагенты, представ-
ляющие собой вещества основного характера27,28 (окиси цинка,
бария и магния, анилин, толуидин, мочевина). Однако обра-
зующиеся при связывании кислот хлориды или хлоргидраты
гидрофильны, вследствие чего они ухудшают прозрачность рас-
творов и водостойкость пленок. Кроме того, после полной ней-
456
Гл. XIV. Каучук и продукты его превращений
трализации образующиеся хлоргидраты вызывают каталити-
ческое разложение хлоркаучуков29.
Для связывания кислоты можно с 'прекрасными результа-
тами использовать окись этилена и ее производные, но их
защитное действие носит лишь временный характер, так как
эти реагенты в конечном итоге насыщаются. Поэтому хлоркау-
чук рекомендуется применять в условиях, в которых он до-
статочно устойчив и без добавления специальных стабилиза-
торов. Входящий в состав лакокрасочных композиций дибутил-
фталат, при содержании -.в нем небольших количеств перекиси
бензоила, ограничивает фотохимическое разложение хлоркау-
чука30.
Товарные хлоркаучуки. Из товарных хлоркаучуков первым
«появился на рынке дюропрен (1921 г.), затем торнезит (1930 г.),
пергут (1933 г.), тогофан (1934 г.). В настоящее время раз-
ными фирмами выпускаются следующие хлоркаучуки:
Пергут (I. G. Farbenindustrie)
Тогофан (Togofan A. G.)
Дартекс (Dartex A. G.)
Аллопрен—вместо дюропрена (Imperial Chemical Industries)
Парлон (Hercules Powder Company)
Детел (Detel Product)
Паравар (Naugatuck Chemical Corporation)
Реолин (Raolin Corporation)
Электрогам (Societe d’ Electrochimie d’ Ugine)
Пешини (Compagnie Alais, Froges' et Camargues)
Хлортекс и протеке (Societe Elettrica et Electrochimica del Caffaro)
Выпускаются хлоркаучуки различной вязкости: 5, 10, 20, 125
и 1000 сантипуаз. Хлоркаучуки с вязкостью 10 и 20 санти-
пуаз пригодны для лаков, с вязкостью 125 сантипуаз — для
цементных красок.
Свойства хлоркаучуков
Товарные хлоркаучуки содержат 65—68% хлора. Они имеют
вид белых порошков без запаха и вкуса. Их действительный
удельный вес колеблется от 1,63 до 1,66 кажущийся
удельный вес очень мал.
Хлоркаучуки растворимы в ароматических углеводородах,
во многих галоидопроизводных углеводородов, в сложных эфи-
рах жирного ряда, циклогексаноне, диоксане, нитробензоле,
пиридине и в высыхающих маслах. Не растворяются они в
воде, диэтиловом эфире, спиртах, минеральных маслах, бен-
зине и керосине. Как и сырой каучук, хлоркаучуки сначала
набухают. Ацетон и метилэтилкетон, не являющиеся раство-
Хлоркаучуки
457
рителями хлоркаучука, улучшают растворяющую способность
истинных растворителей.
Теплостойкость хлоркаучуков зависит от степени их хлориро-
вания. Она максимальна и примерно .постоянна при 65—
70 %-ном содержании хлора31. По данным Хекстра32, хлоркау-
чук, содержащий 65—70% хлора, выделяет 0,2% хлора после
нагревания в течение 700 час. при 100°, после 7 час. при 130°
и после 40 мищ при 150°.
При длительном выдерживании на свету выделяется хлори-
стый водород, вызывающий легкое побурение хлоркаучуков.
Сильнее всего поглощаются лучи с длиной волны менее 3000.
Устойчивость к фотохимическому разложению можно улучшить
путем применения пластификаторов с явно выраженной склон-
ностью к поглощению лучей с длиной волны менее 3000 А.
Таким пластификатором является дибутилфталат.
Коэффициент преломления хлоркаучука по Бакстеру9
nD—1,596.
Хлоркаучуки обладают довольно низкой теплопроводностью
(1,20-10“4 для порошкообразных продуктов и 1,51 • 10 4 для
хлоркаучуков в виде пористой массы), прекрасными электро-
изоляционными свойствами. Ниже приведены некоторые элек-
трические характеристики хлоркаучуков:
Диэлектрическая постоянная.............4
Удельное объемное сопротивление (при
25°), ом -см!см2.......................7-1 (Г16
Удельное поверхностное сопротивление
(при 75% относительной влажности и
25°), ом/см2...........................5-10-п
Электрическая прочность33, в/мм .... 5000—7000
Химические свойства. Хлоркаучуки прекрасно противостоят
действию химических реагентов. При соприкосновении с пла-
менем они обугливаются, выделяя хлористый водород, но при
удалении пламени горение прекращается. Под действием как
холодной, так и горячей воды хлоркаучуки очень слабо и очень
медленно гидролизуются.
Хлоркаучуки реагируют с некоторыми органическими осно-
ваниями. Реакции протекают в растворе и приводят к геле-
образованию. Моно-, ди-, триметиламин, бутиламин, пипери-
дин, триметилендиамин придают хлоркаучуку нераствори-
мость34, 35. Для повышения устойчивости хлоркаучука к воде,
кислотам и другим реагентам рекомендуется36 вводить небольшие
количества соединений, содержащих группы >C = S, >C = NH
или >C = NR—. При этом твердость и плотность хлоркаучука
увеличиваются, термопластичность уменьшается и адгезия пле-
нок улучшается. Мочевина, тиомочевина, анилиды, гуанидин,
458 Гл. XIV. Каучук и продукты, его превращений
тиурамсульфиды, ксантогенаты, дитмокарбаматы, меркаптоти-
азолы и продукты взаимодействия альдегидов с аминами так-
же улучшают устойчивость пленок хлоркаучука.
Изучая действие пиперидина на хлоркаучук, Жаригон37
обнаружил постепенное отщепление хлористого водорода и из-
менение структуры. Можно .предполагать13, что первая ста-
дия дегидрохлорирования хлоркаучука протекает с образованием
трехмерной решетки вследствие отщепления НС! от разных мо-
лекул:
Штаудингер15 установил, что хлор прочно связан в мо-
лекуле хлоркаучука. По аналогии со структурой сильно хло-
рированных циклических терпенов он ‘пришел к выводу, что
хлоркаучук имеет циклическое строение.
Данные о химической стойкости хлоркаучуков38 приведены
в табл. 26.
ТАБЛИЦА 26
Химическая стойкость хлоркаучуков
Среда Потеря веса (через 7 дней), % Изменение цвета пленка
при обыч- ной темпе- ратуре ! приП00°, прн обычной температуре при 100°
Серная кислота (97%-ная) 0,5 1,2 Без измене- ний Слегка буреет
Соляная кислота (37%-ная) 0,4 0,8 То же Слегка мутнеет
Азотная кислота (65%-ная) 0,4 0,5 » То же
Царская водка 4-0,4 4 1,2 Слегка мут- неет Становится не- прозрачной
Едкое кали (50 %-ное) . . . Бихромат калия и серная 0,5 1,0 Без измене- ний Слегка буреет
кислота Перманганат калия и серная 0,5 1,3 .— —
кислота ... 0,5 —- Слегка бу- реет —
Перекись водорода .... 0,6 — Без измене- ний —
Дистиллированная вода . . 0,5 1,2 — Становится не- п розрачной
Хлоркаучуки
459
Технологические свойства. Хлоркаучуки хорошо растворя-
ются в большинстве обычных растворителей. Несмотря на то,
что растворы почти всегда опалесцируют, они образуют про-
зрачные пленки. Некоторые товарные хлоркаучуки образуют
желтые или светло-коричневые растворы, что обычно вызывает-
ся наличием белков или других некаучуковых компонентов, со-
путствующих углеводороду каучука. После осаждения этих при-
месей метанолом получаются бесцветные растворы.
Пластификаторы повышают эластичность и уменьшают хруп-
кость пленки хлоркаучуков, что весьма важно при использо-
вании хлоркаучука в лаках и красках. Наиболее употреби-
тельными пластификаторами являются: трифенил- или трикре-
зилфосфаты, бутил- и амилстеараты, диэтил- и дибутилфтала-
ты, эфиры лимонной кислоты, о-дихлорбензол, хлорированные
дифенилы, хлорированные парафины, пропил- и бутилнафтали-
ны, высыхающие масла, хлорированные высыхающие масла,
пальмовое масло.
Непластифицированная пленка хлоркаучука толщиной
0,02 мм обладает прочностью при растяжении 450 кг/см2, но
она хрупкая; введение 25% дибутилфталата понижает проч-
ность при растяжении до 230 кг!см2. Относительное удлинение
непластифицированной пленки мало, а при введении трикре-
зилфосфата или бутилстеарата достигает 150—250%- При над-
лежащей пластификации хлоркаучуковые пленки имеют пре-
красный внешний вид.
Для улучшения адгезии, придания блеска и твердости в
краски на основе хлоркаучука часто вводят смолы. Копалы и
смола даммара совмещаются с хлоркаучуком, но копалы ка-
ури, манилла, а также шеллак могут вводиться только после
соответствующего нагревания их с маслами. Прекрасные ре-
зультаты дают кумароновые, глифталевые смолы и низковяз-
кий сополимер винилхлорида с винилацетатом; производные
целлюлозы для этой цели не пригодны.
Можно вводить в хлоркаучуки и некоторые воска: карна-
убский воск, монтан-воск. Шевассю39 собрал много практиче-
ских данных по использованию этих продуктов.
Для красок на основе хлоркаучука пригодны различные
минеральные пигменты: окиси титана, железа, цинка, хрома,
свинцовые белила, киноварь, алюминиевая пудра, бронзиро-
вочные порошки, а также наполнители: асбест, кварц, слюда
и некоторые растворимые органические пигменты40.
На основе хлоркаучука в смеси с пластификаторами и рас-
творителями или некоторыми синтетическими смолами можно
получать пластические массы. Совмещение хлоркаучука со
смолами может производиться, например, путем нагревания
раствора хлоркаучука с фенолом и формальдегидом 41,4г; мож-
460
Гл. XIV. Каучук и продукты его превращений
но также синтезировать глифталевые смолы в водной эмуль-
сии, содержащей глицерин и раствор хлоркаучука42. При этом
получаются термопластичные порошки. Следует упомянуть так-
же композиции хлоркаучуков с мочевино-формальдегидными43
и меламино-формальдегидными44 смолами, с полиакрилатами и
полиметакрилатами45, с нитроцеллюлозой46. Фирма Marbon-
Согр.47 получала пластические массы, нагревая хлоркаучук при
110—125° с сульфидами и полисульфидами металлов.
Применение хлоркаучуков
Благодаря высокой коррозионной стойкости хлоркаучук наи-
более широко применяется для производства лаков и кра-
сок. При правильном подборе композиции краски на основе
хлоркаучука дают покрытия, очень стойкие к действию влаги
и особенно устойчивые к неорганическим и многим органиче-
ским агрессивным агентам. Кроме того, эти пленки негорючи
и обладают хорошими электроизоляционными свойствами. Ни-
же приводится несколько рецептур красок (вес. ч.), собран-
ных Ле-Бра и Делаландом13.
Защитная краска для легких
металлов
Хлоркаучук средней вязкости ............ 50
Хлоркаучук высоковязкий.............. 50
Ксилол ......................... .... 130
Пластификатор (хлорированный бифенил) ... 30
Алкидаль Т ......................... - . 30
Альбертоль 209 L..................... 30
Бутилацетат ............................. 20
Скипидар ................................200
Твердые компоненты (титановые белила и ши-
ферная пудра) ...........................175
Антикоррозионная краска
Хлоркаучук низковязкий ...................10
Полимеризованное льняное масло............ 6
Слабо полимеризованное тунговое масло ... 6
Скипидар .................................30
Толуол ...................................30
Жидкий сиккатив .......................... 1
Твердые компоненты .......................15
Краска, стойкая к действию
морской воды
Хлоркаучук низковязкий ....................14
Бекозол 1 (гл и фталевая смола)............14
Полимеризованное тунговое масло ..........3,5
Толуол ...................................57
Сиккатив .................................0,5
Титановые белила........................11,9
Сажа .....................................0,2
Хлоркаучуки
461
Щелочеустойчивые и кислото-
стойкие краски
Хлоркаучук низковязкий.......................18
Полимеризованное тунговое масло . . 9
Толуол ......................................43
Скипидар .................................... 9
Тетралин..................................... 4
Амилацетат................................... 1
Красная окись железа .......................17
Краска для цемента
Хлоркаучук низковязкий . . . .'..........10
Альбертоль 209 L ........................10
Полимеризованное льняное масло...........Ю
Сырое тунговое масло.....................20
Толуол ..................................30
Дипентен ................................20
Свинцово-марганцевый сиккатив............0,3
Краска высокой атмосферостой-
ко с т и
Хлоркаучук высоковязкий...................100
Слабо полимеризованное льняное масло .... 50
Альбертоль 111 L..........................15
Ксилол ...................................220
Скипидар .... 180
Бутилацетат...............................Ю
Пластификатор (хлорированный бифенил) ... 10
Свинцово-кобальтовый сиккатив..............0,5
Твердые компоненты .......................210
Краска для дерева
Грунтовка Лак
Хлоркаучук низковязкий ................10 13
Полимеризованное льняное масло .... 10 13
Скипидар...............................10 17
Толуол................................ 38 40
Тетралин . . . .........................— 3
Красная окись железа...................32 14
Подлежащие покрытию поверхности должны быть тща-
тельно очищены, обезжирены и высушены. Для достижения
максимальной долговечности пленки первый слой краски или
грунтовка не должны содержать высыхающих масел.
Лаки и краски на основе хлоркаучука пригодны для нане-
сения на металлы и бетон. Краска хорошо держится на де-
реве и пробке; в тропиках хлоркаучук, по-видимому, не разру-
шается термитами. При достаточной пластификации эти краски
можно наносить на бумагу и на ткани, а соответствующие лаки
пригодны для.отделки кожи.
462
Гл. XIV. Каучук и продукты его превращений
Краски на основе хлоркаучука могут применяться в судо-
строении. Опыты погружения стальных пластинок в воду на.
четыре года показали превосходную стойкость этих красок
к воде48.
Растворы хлоркаучука могут быть использованы также для
пропитки тканей, разных волокон, бумаги, кожи с целью пред-
охранения их от огня, влаги, бактерий и улучшения изолиру-
ющих свойств этих материалов. Делались попытки использо-
вать листовой хлоркаучук в качестве упаковки; однако гидро-
хлорированный каучук и целлофан превосходят хлоркаучук по
механическим свойствам.
Было предложено использовать хлоркаучук в виде волокон,
хлопьев или пористых плит для внутренней облицовки резер-
вуаров, труб и различных сборников. Такое невоспламеняющее-
ся покрытие обладает одновременно электро-, тепло- и зву-
коизоляционными свойствами. Введение хлоркаучука в чер-
нила придает им стойкость; небольшая добавка хлоркаучука
в битумные краски значительно снижает их склонность к рас-
трескиванию.
Таким образом, хлоркаучуки представляют весьма боль-
шой интерес для лакокрасочной промышленности. Они
позволяют разрешить некоторые проблемы, кажущиеся нераз-
решимыми при использовании других пленкообразуюших ве-
ществ.
ЛИТЕРАТУРА
1. Н. J. Gladstone, W. Hibbert, J. Chem. Soc., 1888, 53, 682.
2. W. A. C a s p a r i, J. Soc. Chem. Ind., 1905, 24, 1275.
3. R. D i t m a r, Gummi-Ztg., 1906, 20, 844.
4. F. W. Hinrichsen, H. Quensell, E. Kin dscher, Ber..
1913, 46, 1283.
5. S. J. Peachey, англ. пат. 1894, 1915 г.
6. J. M a c G a v a c k, Ind. Eng. Chem., 1923, 15. 961.
7. F. К i r c k h о f, Gummi-Ztg., 1932, 46, 497.
8. A. Nielsen, Kautschuck, 1933, 9, 167.
9. J. P. Baxter, J. Soc. Chem. Ind., 1936, 55, 407.
10. G. F. Bloomfield, J. Chem. Soc., 1943, 289.
11. G. F. Bloomfield, J. Chem. Soc., 1944, 114.
12. E. H. F a r m e r, Advances in Colloid Science, New York, 1946, p. 304.
13. J. L e Bras, A. D e 1 a 1 a n d e, Les derives chimiques du caoutchouc
naturel, Dunod, Paris, 1950.
14. P. O. Power s, Chemical Formular of plastics, Plastics catalogue,
1945, p. 116—117.
15. H. S t a u d i n g e r, Hg. Staudinger, J. prakt. Chem., 1941, 157,
19, 158.
16. A. N i e 1 s e n, Chlorkautschuk, Leipzig, 1937, S. 19.
17. D. F. Twiss, J. A. Wilson, Dunlop Rubber Co., англ. пат. 414862,
1934 г.
18. I. G. Farbenindustrie A. G., пат. США 2040460, 1936 г.
19. О. Schweitzer, Congres international du caoutchouc, Paris, 1937,
p. 210.
Гидрохлорированный каучук.
463
20. Societc d'Electrochimie d’Ugine, фр. пат. 800624, 1936 г.
21. T. А. Е d i s о п, пат. США 1495580, 1924 г.
22. С. Е 1 1 i s, пат. США 1544529, 1925 г.
23. С. Е 1 1 i s, пат. США 1544531, 1925 г.
24. I. G. Farbenindustrie A. G.. англ. пат. 390097, 1933 г.: фр. пат. 744565,
1933 г.; пат. США 2005320, 1935 г.
25. G. Martin, W. S. Dave у, Н. С. Baker, Rubber Produ-
cers’ Research Association, англ. пат. 476743.
26. J. MacGa vac k, International Latex Processes Ltd., пат. США
2021318, 1935 г.
27. С. Е 1 1 i s, пат. США 1695641. 1928 г.
28. R. Yamamoto, J. Soc. Rub. Ind., Japan, 1934, 7, 711.
29. W. Becker, Proc. Rubb. Techn. Confer., London, 1938, p. 342.
30. Imperial Chemical Industries Ltd., англ. пат. 432905, 1935 г.
31. J. P. Baxter, J. G. Moor e, J. Soc. Chem. Ind., 1938, 57, 327.
32. J. Hoekstra, Rubber Chem. Techn., 1940, 13, 582.
33. R. H a s к o, J. Soc. Rub. Ind., Japan, 1934, 7, 504.
34. Chemische Fabrik Buckan, фр. пат 749553, 1935 г.; англ. пат. 461631, 1937 г.
35. Т. Goldschmidt A. G., герм. пат. 702172. 1941 г.; 725848, 1942 г.
36. Raolin Corporation, англ. пат. 491075, 491141, 1938 г.; пат. США
2148831, 1939 г.
37. A. J а г г i g о n. F. L е р е t i t, Rev. Gen. Caout., 1945, 22, 49.
38. J. G F о 1, А. В. В i j 1, Chem. Weekblad, 1932, 29, 126.
39. F. Chevassus, Caout. et Gutta-percha, 1939, 36, 86, 117.
40. F. von Artus, Korrosion u. Metallschutz, 1941, 17, 361.
41. J. Mac G a vac к, пат. США 1640363, 1927 г.
42. D. F. Twiss, J. A. Wilson, Dunlop Rubber Co., англ. пат. 417273,
1934 г.
43. H. Р 1 a u s а, герм. пат. 560260, 1931 г.
44. R. С. Swain, Р. Adams, American Суanamid Со., пат. США 2290132,
1942 г.
45. J. W. R а у п ol ds, М. R. R a d с 1 i f f е, Raolin Corporation, пат. США
2364589, 1944 г.
46. Н. Michaelis, jr., англ. пат. 495967, 495968, 1938 г.
47. Marbon Corporation, пат. США 2115053, 1938 г.
48. Hercules Powder Со., фр. пат. 750013, 1933 г.
ГИДРОХЛОРИРОВАННЫЙ КАУЧУК
Из гидрогалоидированных каучуков промышленное приме-
нение получил только гидрохлорированный каучук.
Хлористый водород присоединяется к молекуле каучука по
двойным связям:
+ НС1 +HCI ci CI
По данным Буйна и Гарнера1, присоединение HCI происхо-
дит по правилу Марковникова, т. е. хлор присоединяется к
наименее гидрогенизированному атому углерода. Как показал
Ван Феерзен2, на скорость гидрохлорирования оказывает боль-
шое влияние растворитель. Так, при добавлении к толуолу
464
Гл. XIV. Каучук и продукты его превращений
диоксана или этилацетата скорость гидрохлорирования очень
заметно увеличивается, вероятно вследствие промежуточного
образования комплексных соединений оксониевого типа. При-
соединение НС1 протекает с выходом гидрохлорированного кау-
чука около 90%.
Получение гидрохлорированного каучука
Каучук можно гидрохлорировать в растворенном или на-
бухшем состоянии, в сухом виде, а также в виде латекса.
Гидрохлорирование каучука в растворе. Для получения про-
изводных, пригодных для лаков, необходимо предварительно
провести деструкцию каучука путем длительной его пластика-
ции. Обычно безводный хлористый водород пропускают через
раствор каучука в бензоле, толуоле, хлороформе, дихлорэтане
или тетрахлорэтане. По мере протекания реакции гидрохлори-
рованный каучук можно осаждать гептаном, изопропиловым
эфиром, смесью 70% бензина с 30% этилацетата или диэтило-
вого эфира.
Гидрохлорированием окисленных производных каучука, на-
пример так называемых раббонов, можно получить связующие
вещества для красок, обладающие большой устойчивостью к
щелочам3. В этом случае хлористый водород пропускают в
охлажденный раствор раббона в ксилоле.
Гидрохлорирование набухшего и сухого каучука. Тонкие
пленки каучука, набухшие в растворителе, подвергают дей-
ствию хлористого водорода. Применяя диоксан, можно за один
цикл провести гидрохлорирование на 5—15%. Для гидрохло-
рирования сухого каучука применяется ожиженный хлористый
водород и реакцию проводят при низкой температуре. Таким
путем получается продукт, содержащий 28% и более хлора.
Гидрохлорирование латекса. В присутствии поверхностно-
активных неионогенных или катионных реагентов в латекс
пропускают хлористый водород, который растворяется в серуме.
После насыщения серума хлористый водород начинает реаги-
ровать с каучуком.
Ван Феерзен2 изучил влияние температуры и давления на
скорость гидрохлорирования латекса.
Обработка гидрохлорированного каучука. После удаления
избыточного хлористого водорода гидрохлорированный каучук
пластифицируют и стабилизуют. Пластификаторами служат:
парафин, бутилстеарат, трикрезилфосфат, различные фталаты,
себацинаты и т. д. В качестве стабилизаторов применяют фе-
ноло-анилино-формальдегидные или дифениланилин-формальде-
гидно-дициклогексиламинные смолы, а также гексаметилентетр-
амин.
Гидрохлорированный каучук
465
Свойства гидрохлорированного каучука
Теоретически в гидрохлорированном каучуке должно содер-
жаться 33,9% хлора. Товарные продукты содержат меньше хло-
ра; наилучшие свойства достигаются при 28—30% хлора. Про-
дукты, содержащие свыше 30% хлора, — ломкие, содержащие
менее 28% хлора — смолистые.
Исследования рентгенограмм гидрохлорированного каучука
показали, что в нерастянутом состоянии он обладает кристал-
лической структурой4. При температуре выше 90° он стано-
вится очень эластичным и его можно растянуть и затем охла-
дить в растянутом состоянии. Такая обработка придает гидро-
хлорированному каучуку волокнистую форму.
Гидрохлорированный каучук растворим в хлорированных
углеводородах, набухает на холоду в ароматических углеводо-
родах и растворяется в них при нагревании. Он набухает при
нагревании в сложных эфирах, нерастворим в воде, спирте, дч-
этиловом эфире, ацетоне и бензине. Совмещается с хлоркау-
чуком, но не совмещается с сырым каучуком, нефтяными мас-
лами и жирами. Его плотность ниже плотности 'производных
целлюлозы. Коэффициент преломления по Нильсену5 равен
1,533.
Пленки гидрохлорированного каучука мало проницаемы
для воды. При 39° они в 17 раз менее водопроницаемы, чем
ацетилцеллюлоза6. Их прочность при растяжении вдвое боль-
ше, чем для листовой целлюлозы той же толщины; они не
сминаются.
Гидрохлорированный каучук значительно более термопла-
стичен, чем хлоркаучук, но легко разлагается при нагревании.
Он чувствителен к температурам около 130°, однако кипящая
вода оказывает на него слабое действие.
Нагревая гидрохлорированный каучук с органическими ос-
нованиями, например с анилином, 'пиридином или пипериди-
ном, можно вызвать отщепление части хлористого водорода.
Нагреванием с безводным пиридином при 125—145° можно
полностью отщепить хлористый водород. В таких условиях
получается «изокаучук» — ненасыщенное соединение, более
пластичное и менее эластичное, чем невулканизованный каучук.
Изокаучук, по-видимому, построен из структур трех ти-
пов:
Цинковая пыль восстанавливает гидрохлорированный кау-
чук. Действуя цинковой пылью на кипящий раствор гидрохло-
30—156
466 Гл. XIV. Каучук и продукты его превращений
рированного каучука в толуоле, можно получить циклокаучук—
«мо'ноциклокаучук». В молекуле этого соединения вдвое мень-
ше двойных связей, чем в немодифицированном каучуке, а
именно: на два изопреновых остатка приходится одна двойная
связь. Если эта реакция проводится в присутствии соляной
кислоты, получается «полициклокаучук», в молекуле которого
одна двойная связь приходится на четыре изопреновых остат-
ка. Полициклокаучуки совершенно неэластичны и по хрупкости
напоминают асбест. Они растворяются в хлороформе, четырех-
хлористом углероде, сероуглероде, бензоле, толуоле, тетралине,
"астично растворимы в эфире и нерастворимы в ацетоне и
спирте.
Штаудингер7 приписывает циклокаучукам следующее стро-
ение:
полициклокаучук
Гидрохлорпрованный каучук горит только при непосредст-
венном соприкосновении с пламенем и гаснет после его удале-
ния. Он устойчив к минеральным кислотам и щелочам. Силь-
ные окислители довольно легко действуют на него, чем и объяс-
няется нестойкость его к действию концентрированной азотной
кислоты.
Гидрохлорированный каучук 467
Потеря веса пленки гидрохлорированного каучука после
нахождения в 2 н. соляной кислоте, 4 н. серной кислоте,
20%-ном растворе едкого натра или 2 н. растворе едкого кали
составляет от 0,5 до 1,2%. В концентрированной серной кислоте
потеря веса составляет 3,4%.
Гидрохлорированный каучук устойчив по отношению к озону
и кислороду; с течением времени незначительно ухудшаются
только его механические свойства. По водостойкости пленки
гидрохлорированного каучука значительно превосходят целлю-
лозные пленки.
Применение гидрохлорированного каучука
Гидрохлорированный каучук в виде растворов, содержащих
также пластификаторы и стабилизаторы, используется для про-
питки, нанесения покрытий, изготовления пленок.
В производстве лаков применяются бензольные растворы8
или водные эмульсии, содержащие гидрохинон9. В смеси с ка-
менноугольной смолой или каменноугольными маслами он при-
меняется для дорожных покрытий10. Для придания водонепро-
ницаемости оберточной бумаге ее пропитывают бензольным
раствором, содержащим гидрохлорированный каучук и сополи-
мер винилхлорида и винилиденхлорида11.
Особенно широко гидрохлорированный каучук применяется
в виде пленок. Листовой материал, получаемый испарением
растворов, содержащих пластификаторы и стабилизаторы, вы-
пускается под названием плиофильм. Этот материал применяет-
ся главным образом для упаковки пищевых продуктов. Плио-
фильм имеет значительные преимущества перед целлюлозными
пленками ввиду чрезвычайной устойчивости к влаге и жирам;
его прочность при растяжении вдвое выше, а эластичность
также весьма высока. Листы плиофильма можно склеивать пу-
тем простого нагревания. Он прозрачен, не имеет ни запаха, ни
вкуса, равномерно окрашивается и может быть использован для
печати.
Выпускаются четыре типа плиофильма: обычный плио-
фильм— марка N; специально пластифицированный — марка Р;
для упаковки замороженных пищевых продуктов — марка FF и
для упаковки механических деталей (авиационных моторов)—
марка аэро. После букв N и Р ставят цифры, указывающие
вид и количество введенного пластификатора.
Вытяжкой нагретого листа гидрохлорированного каучука и
охлаждением его в растянутом состоянии удается получить более
тонкие пленки (0,002 ж), чем плиофильм, выпускаемые под*на-
званием тенсолит, и очень прочные нити.
Плиофильм и тенсолит производятся фирмой Goodyear Tire
and Rubber Co.
30*
468 Гл. XIV. Каучук и продукты его превращений
ЛИТЕРАТУРА
I. С. W. Bunn, Е. V. Garner, J. Chem. Soc., 1942, 654.
2. G. J. Van V e e r s e n, Proc. Rubber Technol. Conf., London, 1948,
№ 6,
3. H. P. Stevens, F. J. W. Pop h am, англ. пат. 499550, 1939 г..
4. S. D. G e h m a n, J. E. Field, R. P. Dinsmore, Proc. Rubb.
Technol. Conf., London, 1938, p. 961.
5. A. Nielsen, Chlorkautschuk, p. 87.
6. C. R. О s w i n, Chem. a. Ind., 1943, 62, 114.
7. H. S t a u d i n g e r, W. Widmer, Helv. Ch. Acta, 1929, 468,
12, 48.
8. W. P. Bradley, W. A. Gibbons, пат. США 1627725, 1927 г.
9. Goodyear Tire and Rubber Co., фр. пат. 753027, 1934 г.
10. D. D. Pratt, R. Handley, R. G. A. В u 1 1, 1936,18, 391.
11. F. H. Manchester, Wingfoot Corporation, пат. США 2386700, 1945 г.
ПРОДУКТЫ ЦИКЛИЗАЦИИ КАУЧУКА (ЦИКЛОКАУЧУКИ)
Получение циклокаучуков
Молекулы каучука склонны к изомеризации и циклизации с
уменьшением степени ненасыщенности и возникновением вну-
тримолекулярных связей. Как было указано ранее, циклиза-
ция часто сопутствует другим превращениям каучука.
Циклизацию можно осуществить длительным нагреванием,
действием электрического разряда или химических агентов. То-
варные циклокаучуки получаются действием химических аген-
тов. Они представляют ценность для ряда областей применения,
в частности как связующее для лаков и красок. Можно назвать
два типа применяемых для этой цели товарных продуктов:
термопрены и группа смол плиоформ и плиолит NR.
Термопрены образуются при действии на каучук неоргани-
ческих или органических соединений общей формулы R—SO2X,
где R—органический радикал или гидроксил, X —хлор или
также гидроксил. Смолы плиоформ и плиолит NR получаются
действием на каучук комплексных оловогалоидоводородных
кислот.
Циклокаучуки могут получаться также под действием фено-
лов в кислой среде12, галоидуксусных кислот13, уксусного и фта-
левого ангидридов14, трехфтористого бора15. Как правило, хи-
мические реагенты, вызывающие циклизацию, представляют со-
бой соединения, обладающие кислой реакцией или способные
выделять сильные кислоты.
..Циклизация под действием реагентов типа R—SO2X. При
действии этих реагентов на каучук получаются термопрены.
Процесс можно проводить путем непосредственного смешения
каучука с реагентом (реакция продолжается при последующем
Продукты циклизации каучука (циклокаучуки)
469
нагревании), а также в растворе и даже в латексе. Было испы-
тано много реагентов указанного типа.
При обработке тонко измельченного каучука серной кисло-
той получается темноокрашенная твердая и хрупкая масса.
Фишер23, проводивший циклизацию путем непосредственного
смешения каучука с серной кислотой, нашел, что реакция про-
текает слишком быстро, и предпочел перейти к изучению дей-
ствия менее энергичных реагентов—сульфокислот.
Серная кислота реагирует с каучуком также в растворе.
Кирхгоф1 действовал H2SO4 на бензольный раствор каучука
(крепа), температура которого поддерживалась в течение не-
скольких часов на уровне 60—100°. После очистки получался
белый порошок, набухающий в уксусном ангидриде и плавя-
щийся при температуре около 180°.
С помощью серной кислоты можно проводить циклизацию
каучука в латексе4. Ван Феерзен5 разработал метод получения
циклокаучука из латекса, стабилизованного неионогенным по-
верхностно-активным веществом. Реакция с серной кислотой,
концентрация которой в серуме достигает 75%, продолжается
4 часа при 70—90°. Латекс циклокаучука подвергают диализу
через коллодионные мембраны или осаждают в виде хлопьев
теплой водой, фильтруют и промывают для удаления кислоты.
Получаемый продукт представляет собой порошок кремового
цвета, обладающий термопластическими свойствами. Латекс
циклокаучука, очищенный диализом, можно смешивать с обыч-
ным латексом. Непосредственным формованием из него можно
получать изделия повышенной твердости с повышенным моду-
лем эластичности.
Хлорсульфоновая кислота HSO3CI реагирует'с сухим каучу-
ком еще быстрее, чем серная кислота2. Медленнее действуют
сульфохлориды R—SO2C1 благодаря наличию органического
радикала R, замедляющего течение реакции3. Хлорангидрид
п-толуолсульфокислоты СНз—С6Н4-—SO2C1 можно вводить при
пластикации; последующее нагревание смеси приводит к обра-
зованию термопластичной массы. Удобно проводить циклиза-
цию с помощью хлорангидридов р-нафталинсульфокислоты и
м-нитробензолсульфокислоты.
Хлорангидрид бензолсульфокислоты, жидкий при комнатной
температуре, применяется в тех случаях, когда реакцию ведут
в растворе.
Термопластичные массы можно получать также из вулкани-
зованного каучука; его измельчают совместно с хлорангидри-
дом zz-толуолсульфокислоты, а затем нагревают в течение
8 час. при 140е; в результате получается циклокаучук.
Циклизация с помощью галогенидов металлов. Безводное
хлорное олово реагирует с растворенным каучуком в инертной
470 Гл. XIV. Каучук и продукты его превращений
среде при обычной температуре. Образующееся соединение
предположительно имеет состав (С5Н8) ioSnCl4. Оно разлагается
спиртом или ацетоном с образованием белого аморфного порош-
ка состава (Сг,И8)х. Аналогично реагируют с каучуком четырех-
хлористый титан, хлорное железо и пятихлористая сурьма6.
Реакции циклизации могут протекать под действием хлористой
сурьмы, хлористого алюминия и ряда других галогенидов ме-
таллов7, 8.
С использованием комплексных кислот, образуемых четырех-
хлористым оловом, а также хлороловянных и хлороловянистых
кислот производятся в промышленном масштабе циклокаучуки9,
известные под названиями плиоформ и плиолит NR. Гидратиро-
ванная хлороловянная или хлороловянистая кислота вводится
при пластикации; затем смесь нагревают при температуре
около 150°. Получаются растворимые в бензоле вещества, плас-
тичные при низкой температуре, легко окисляющиеся. Их можно
предохранить от окисления, покрывая тонкой пленкой мыла,
воска или жирной кислоты. Циклизация каучука действием хлор-
оловянных или хлороловянистых кислот может проводиться в
растворе; по завершении реакции раствор эмульгируют водой,
удаляют растворитель и осаждают циклокаучук10,11.
Для циклизации каучука можно применять также галоидо-
производные фосфора 1&~18 и хлорокись селена19.
Строение циклокаучуков
Строение продуктов циклизации каучука точно не установ-
лено. Штаудингер и Гейгер20 считают, что в процессе циклиза-
ции степень непредельности уменьшается в 4 раза. Кирхгоф21 и
Брюзон6 предполагают, что образуются четырех- или восьми-
членные углеродные циклы. Против этой гипотезы имеются
возражения, важнейшее из которых базируется на правиле
Байера — Сакса. С другой стороны, при проведении циклизации
в растворе образование мостиков должно было бы вызвать
гелеобразование, в действительности не наблюдающееся.
По Пауерсу22 повторяющееся звено в молекуле циклокаучу-
ка имеет строение (I), а по Янни23 — строение (II):
Продукты циклизации каучука (циклокаучуки) 471
Структура (II) согласуется с гипотезой Тиманна24 о цикли-
зации гераниола и с гипотезой Блумфилда25 о циклизации
дигидромирцена. Краузе26 предлагает следующий механизм
циклизации каучука:
По Ван Феерзену27 циклизация каучука может быть пред-
ставлена схемой:
В действительности в последней формуле должна быть одна
двойная связь, положение которой неизвестно. Приняв эту фор-
мулу, можно объяснить результаты, полученные Ван Феерзеном
при определении ненасыщенности циклокаучука. Многие вопросы
еще остаются невыясненными.
Таким образом, продукты циклизации каучука содержат
метилциклогексильные группировки, которые могут чередовать-
ся с незатронутыми отрезками полиизопреновых цепей.
472 Гл. XIV. Каучук и продукты его превращений
Свойства циклокаучуков
Физические свойства циклокаучуков колеблются в довольно
широких пределах в зависимости от способа циклизации, в то
время как химические свойства этих продуктов довольно близки.
Фишер предложил следующую классификацию термопренов:
Термопрены GP (аналоги гуттаперчи).
Термопрены НВ (Hard balata), по повышенной твердости
аналогичные балате.
Термопрены SL (аналоги шеллака).
Термопрены GP растворимы в бензоле, хлороформе, четырех-
хлористом углероде, сероуглероде, бензине, скипидаре, тетра-
лине. Термопрены НВ обладают различной растворимостью в
зависимости от способа их получения.
Термопрены SL растворимы в ароматических углеводородах,
набухают в эфире, анилине, амилацетате и нерастворимы в
метиловом и этиловом спиртах, ацетоне и уксусной кислоте.
В табл. 27 приведены некоторые механические и электрические
свойства термопренов различных типов2.
Свойства термопренов разных марок
ТАБЛИЦА 27
Показатели GP НВ твердый НВ очень твердый SL
I II I II
Удельный вес (при обычной тем-
пературе), г/см3 Предел прочности при растяже- 0,980 0,999 1,016 0,993 0,993 1,003
нпи (при 22°), кг1см3 . . . 180 160 340 330 46 Очень
Удлинение при разрыве, % . . Удельная ударная вязкость, 27 29,8 1,3 1,7 0,6 хрупок
кг-м-см3 Предел прочности при сжатии, 140 58 114 116 18 3,9
кг/см3 380 270 750 600 630 500
Сопротивление отрыву (при 21°), (при (21°) •— (при 24°) (при 32°) (при 32°) (при 26°)
кг/см3 570i 340 500 490 150 НО
Электрическая прочность, в/мм 47 500 36 500 50 000 55 200 51 800 53 200
Температура размягчения, °C . Комн. — 65 77 100s- 105*
Для очищенных образцов.
Продукты циклизации каучука (циклокаучуки)
473
Растворимость каучуков, циклизованных галогенидами
металлов, зависит от степени полимеризации исходного каучу-
ка. Если каучук был подвергнут предварительной пластикации,
то продукты его циклизации полностью растворяются в бензоле.
При проведении циклизации без предварительной деструкции
получаются нерастворимые циклокаучуки.
Продукты, полученные из каучуков, предварительно подверг-
нутых деструкции, представляют собой белые порошки, раство-
римые в обычных растворителях каучука, мало растворимые в
эфире, нерастворимые в этиловом спирте и ацетоне, смешиваю-
щиеся с парафинами. Они быстро окисляются. Растворы их до
50%-ной концентрации низковязки и после испарения раствори-
теля образуют неэластичные, ломкие, бесцветные и прозрачные
пленки. Такие растворимые циклокаучуки размягчаются при
185—220°, а при 280° разлагаются, причем расплавление может
начаться при 250°.
Продукты, получаемые при обработке пластицированного
каучука хлороловянной или хлороловянистой кислотами, извест-
ные под названиями плиоформ и плиолит NR, мало изучены.
Некоторые из них имеют сходство с балатой, другие — с эбони-
том. Все они растворимы в углеводородах жирного и аромати-
ческого рядов, нерастворимы в этиловом спирте и ацетоне. Это
термопластичные вещества с хорошими электроизоляционными
свойствами.
Товарные марки плиоформ № 40 и № 20 отличаются друг от
друга по термопластичности. Ниже приведены некоторые свой-
ства продуктов типа плиоформ:
Удельный вес, г!см3 ..............................1,06
Температура размягчения, СС
плиоформ Xs 20 ........................105
» № 40 ............................. 80—90
Пластическая деформация (под давлением 140 кг!см2
при 50е), см на 1 см длины........................ 0,00035
Температура прессования, СС
плиоформ №20 ..........................155
« № 40 . . . ..............125
Коэффициент термического расширения . . ... 0,00008
Усадка линейная, % ........ ..................0,35
Поверхностное электрическое сопротивление, ом/см .
при 90% относительной влажности................4-1010
при 75% » » .............4<10и
Предел прочности, кг] см?
при растяжении ................................... 280—350
при сжатии ................................... 600—800
при изгибе . . ............... 500—650
Водопоглощение (за 24 часа), %....................0,03
Смолы плиолит отличаются от смол плиоформ только темпе-
ратурой размягчения. Температура размягчения смол плиолит
474
Гл. XIV. Каучук и продукты его превращений
колеблется от 55 до 65°; .смолы некоторых марок размягчаются
даже начиная с 45°.
Химические свойства. Изучены реакции термопренов, осо-
бенно термопренов SL. Степень ненасыщенности очищенных
термопренов SL на 55—60% меньше, чем у исходного каучука28.
Присоединяя серу, термопрены теряют термопластичность и
превращаются в твердые нерастворимые вулканизаты. При
выдерживании смеси 10 вес. ч. циклокаучука с 2 вес. ч. серы в
течение 15 час. при 141° получается .материал, аналогичный эбо-
ниту. Если сера вводится при пластикации одновременно с
циклизующим агентом, то вулканизация не происходит, а обра-
зуется растворимое и термопластичное производное. При
взаимодействии избытка хлористой серы с термопренами, рас-
творенными в бензоле или четыреххлористом углероде, обра-
зуются продукты присоединения. Это порошкообразные вещест-
ва, нерастворимые в ацетоне, не термопластичные и содержащие
от 16 до 17,5% серы и от 10,6 до 12,1% хлора.
Термопрены SL связывают НС1 и НВг, а также бром. При
этом происходят реакции присоединения и замещения. Очищен-
ные термопрены SL при нагревании до 130° на воздухе присо-
единяют 5,63% кислорода. При взаимодействии с озоном обра-
зуется нерастворимое производное желтого цвета. Под дейст-
вием .надбензойной кислоты термопрены образуют растворимый
в ацетоне белый порошок, состав которого должен отвечать
формуле (СбН8)Л. Азотная кислота дает с очищенными термо-
пренами SL довольно стойкий желтый порошок, растворимый
в ацетоне и плавящийся при 260°.
Продукты циклизации, образующиеся при действии галоид-
ных соединений мышьяка, олова, сурьмы, титана, алюминия или
железа, менее реакционноспособны, чем каучук. Они образуют
гели с хлористой серой и окрашенные соединения с галогени-
дами металлов.
Смолы плиоформ и плиолит NR по химическим свойствам
довольно близки к производным, образующимся при действии
галогенидов перечисленных металлов. Они весьма устойчивы к
основаниям и к влаге. Погружение в 43%-ную азотную кислоту
на 24 часа не вызывает никакого разрушения плиоформа; по-
гружение в 65%-ную азотную кислоту вызывает только изме-
нение цвета.
Технологические свойства. Циклокаучуки за немногими ис-
ключениями растворяются в жирных и ароматических углево-
дородах и мало растворимы в кислородсодержащих раствори-
телях. Термопрены могут пластицироваться путем вальцевания.
Для этой цели пригодны обычные вальцы с обогреваемыми вал-
ками. Смолы типа плиоформ также можно вальцевать в горячем
состоянии, но расход энергии при этом слишком велик.
Продукты циклизации каучука (циклокаучуки) 475
При использовании циклокаучуков для многих целей их при-
ходится очищать, удаляя гигроскопические .вещества. Для очист-
ки термопрена SL Фишер растворял его в четыреххлористом
углероде, перемешивал полученный раствор с инфузорной землей
и осаждал циклокаучук, повторяя все эти операции несколько
раз. Гир29 удалял некаучуковые компоненты из каучука до про-
ведения циклизации, исключая таким путем образование гигро-
скопичных соединений.
К циклокаучукам применимы обычные пластификаторы кау-
чука. Для пластификации термопренов рекомендуются30 дре-
весная смола, парафины, воска, растительные масла, жирные
кислоты, кумароновые смолы, трикрезил- и трифенилфосфаты,
а также эфиры типа диамилфталата. Могут применяться для
этой цели также фенолы, анизол, альдоль, фурфурол, бензаль-
дегид, фталевый ангидрид, толуол, ксилол, n-цимол, трихлор-
этилен. Циклокаучуки, полученные действием галоидных соеди-
нений олова, пластифицируются растительными или животными
восками, хлорнафтал'ином или алкоксинафталином, хлорзаме-
щснным бифенилом и всеми обычными пластификаторами
каучука.
Продукты циклизации, полученные действием на каучук
кислот, очень чувствительны к кислороду и требуют стабилиза-
ции. Стабилизация достигается введением соединений щелочных
или щелочноземельных металлов (окисей, гидроокисей, стеара-
тов, олеатов, пальмитатов, тартратов) или азотсодержащих ве-
ществ (казеина, желатина, альбумина, мочевины, анилидов,
дитиокарбаматов цинка) 3,-34.
Применение циклокаучуков
Почти все виды циклокаучуков были предложены в качестве
связующих для лакокрасочных покрытий, так как их коррозион-
ная стойкость очень высока. Грей35 показал, что краски на осно-
ве термопрена превосходят обычные краски как по водонепро-
ницаемости пленок, так и по адгезии, эластичности и устойчи-
вости к температурным колебаниям. Краски асидсил, изготовляе-
мые на основе смол типа термопрена. применяются в качестве
антикоррозионных покрытий и прекрасно предохраняют внут-
реннюю поверхность резервуаров и трубопроводов от действия
щелочных веществ или растворов едких щелочей (в производ-
стве красителей, вискозном производстве и т. д.). Они приме-
няются также для окраски стен в лабораториях и лабораторных
установок.
Смолы типа плиолит также представляют большую ценность
для защитных покрытий. Андр36 запатентовал 32 рецептуры ла-
ков и красок на основе каучука, циклизованного хлороловян-
476 Гл.. XIV. Каучук и продукты его превращений
ной кислотой. Состав лаков и красок на основе таких циклокау-
чуков может быть весьма многообразен.
Фирма Goodyear Tire and Rubber Co выпускает смолы типа
плиолит или в виде твердых веществ, которые надо смешивать
с наполнителями и пигментами, входящими в рецептуру красок,
или в виде основы (Pliolite Base), уже содержащей наполните-
ли, которую достаточно только растворить. В качестве раство-
рителей и разбавителей могут применяться обычные бензины.
Лаки на основе смол типа плиолит могут наноситься путем
пульверизации, а также методом окунания. Пленки легко высы-
хают, причем при высыхании они окисляются и затвердевают.
Эти пленки обладают хорошей адгезией к любой правильно
подготовленной поверхности, сохраняют свой цвет и не желтеют.
Покрытия на основе плиолита противостоят действию воды,
морского воздуха, а также щелочных очищающих составов. Они
не разрушаются бензином, керосином, минеральными маслами
и жировыми веществами, особенно если применяется очищенный
циклокаучук. Пленки устойчивы к кислотам и щелочам. Обра-
зец не изменяется после трехнедельного воздействия на него
50%-ного раствора едкого натра при 70°. Серная, соляная и
азотная кислоты ниже 50%-ной концентрации не действуют на
плиолитовые пленки, но при воздействии SO2 и С1г эти пленки
разрушаются.
Пленки плиолита устойчивы к трению, а потому эта смола
пригодна для введения в лаки и краски для окраски полов.
Одной из наиболее важных- областей применения смол типа
плиолит являются покрытия по цементу. Цементные поверхности
обычно плохо поддаются окраске вследствие наличия пор, обра-
зующихся при испарении воды в процессе схватывания цемента
и особенно вследствие наличия свободной щелочи. Циклокаучук
прекрасно противостоит этой щелочности, а также заполняет
поры цемента.
Кроме лаков и красок продукты циклизации каучука ис-
пользуются для крепления резины к металлу в качестве формо-
вочных порошков, а также пропиточных и электроизоляционных
материалов.
Для крепления резины к металлам применяются преимущест-
венно термопрены. Клей вулкалок (фирмы В. F. Goodrich Со)
приготовляется на основе продукта циклизации, получаемого
действием на каучук «-фенолсульфокислоты по Фишеру. Этот
клей служит связующим между металлом и резиной; поэтому
с его помощью можно гуммировать стальные емкости и трубо-
проводы, предназначенные для агрессивных сред.
Циклокаучуки, получаемые действием галогенидов металлов,
также являются прекрасными клеями для крепления резины к
металлам 371 38.
Продукты циклизации каучука (циклокаучуки)
477
Пропиточные составы на основе термопренов и смолы типа
плиолита применяются для придания водонепроницаемости,
волокнистым материалам, например тканям и коже.
Из электроизоляционных материалов следует назвать мар-
бон В (фирмы Marbon Corporation), получаемый циклизацией
каучука под действием фенола в кислой среде с последующей
тщательной очисткой для удаления гигроскопичных примесей.
Марбон В используется преимущественно в рецептурах изоля-
ционных лаков, в которые можно вводить небольшие количества
бутил- и амилацетата, метилэтилкетона, ацетона, амилового и
бутилового спирта и др., не вызывая выделения осадка. К лаку
следует добавлять антиокислитель, чтобы предотвратить геле-
образование, которое может быть вызвано окислением.
литература
1. F. Kirch hof, Koll. Z„ 1920, 27, 311.
2. Н. L. Fisher, Ind. Eng. Chem., 1927, 19, 1325—1328.
3. H. L. Fisher, B. F. Goodrich Co., пат. США 1605180, 1926 г.
4. F. К i r c h h о f, Kautschuk, 1926, 2, 1.
5. G. J. van V i e r s e n, Rev. gen. Caout., 1950, 27, 473.
6. H. А. В г u s о n, L. B. Se b u i i, W. С. С a 1 о u t, Ind. Eng. Chem.,
1927.
7. H. Gray, B. F. Goodrich Co., пат. США 1719930, 1929 г.
8. W. N, J ones, H. A. Winkelmann, B. F. Goodrich Co.,
пат. США 1751817, 1930 г.
9. H. А. В г u s о n, Goodyear Tire a. Rubber Со., пат. США 1797188, 1931 г.
10. S. S. Kurt z, Wingfoot Corporation, пат. США 2052411, 1936 г.
11. L. В. S е b г е 1 1, Wingfoot Corporation, пат. США 2052423, 1936 г.
12. Н. L. Fisher, В. F. Goodrich Со., канад. пат. 282778, 1928 г.
13. И. L. F i s h е г, В. F. Goodrich Со., пат. США 1642018, 1927 г.
14. I. G. Farbenindustrie A. G., фр. пат. 723838, 1932 г.
15. Н. А. В г u s о n, Goodvear Tire and Rubber Co., пат. CHIA 1846247,
1932 r.
16. H. A. В r u s о n, Goodvear Tire and Rubber Co., пат. США 1853334.
1932 г,
17. E. H. Farmer, J. W. Ro w e, H. P. Stevens, Rubber Produ-
cers Research Association, англ. пат. 467531, 1937 г.
18. R. L. S i b 1 e y, Monsanto Chemical Co., пат. США 2364394, 1943.
19. С. E. F г i c h, J. Am. Chem. Soc., 1923, 45, 1800.
20. H. Staudinger, E. Geiger. Helv. chim. acta, 1926, 9, 549.
21. F. К i r c h h о f, Kautschuk, 1928, 4, 143.
22. P. O. Powers, Synthetic Resins and Rubbers, Weley, New York, 1943,
p. 266.
23. J. D. D'l a n n i, F. J. Naples, J. W. Marsh. J. L. Z a г n e y,
Ind. Eng. Chem., Ind. Ed.. 1946, 38, 1174.
24. F. T i e m a n n, Ber., 1900, 33, 3710.
25. G. F. В 1 о о m f i e 1 d. J. Chem. Soc., 1944, 114.
26. A. H. Krause, H. E. Scott, A. G. Susie, Rubb. Age, New
York, 1948, 64, 189.
27. G. J. Van V i e r s e n, Rev. gen. Caout., 1951, 28, 34.
28- H. L. Fisher, E. M. McColm, Ind. Eng. Chem., 1927, 19, 1328—
1333.
29. W. C.’ Geer, B. F. Goodrich Co., пат. США 1731485, 1731849, 1929 г.
478 Гл. XIV. Каучук и продукты его прев ращений
30. J. L е Bras, A. De 1 а 1 a n de, Derives chimiques du caoutchouc na-
turel, Dunod, Paris, 1950, p. 362.
31. W. C. Gee г, пат. США 2035062, 1936 г.
32. H. J. Osterhof, Wingfoot Corporation, пат. США 2381180, 1945 r.
33. С. M. Carso n, Wingfoot Corporation, пат. США 2371736, 2371737,
1945 г.
34. С. М. Carson, Wingfoot Corporation, пат. США 2414018, 1947 г.
35. Н. Gray, Ind. Eng. Chem., 1928, 20, 156.
36. H. A. Ender s. Wingfoot Corporation, пат. США 2052391, 1936 г.
37. L. В. S e b r e I 1, Goodyear Tire and Rubber Co., пат. США 1781649,
1930 г.
38. T. C. Morri s, Wingfoot Corporation, пат. США 2084042, 1937 г.
ПРОДУКТЫ ОКИСЛЕНИЯ КАУЧУКА
Склонность каучука к окислению установлена очень- давно.
Ван Россем1 показал, что с уменьшением молекулярного веса в
результате окислительной деструкции пластичность каучука и
его растворимость увеличиваются. По имеющимся данным, су-
ществуют многочисленные «окисленные каучуки», строение ко-
торых точно не установлено. Свойства продуктов окисления кау-
чука зависят от применяемого реагента и условий проведения
реакции, в первую очередь от катализаторов окисления.
Реакции окисления каучука
Реакции окисления каучука можно разбить на две группы:
прямое окисление и каталитическое окисление. Практический
интерес представляет только каталитическое окисление, приво-
дящее к образованию пленкообразующих веществ.
Некаталитическое окисление каучука. Окисление кислородом.
Наиболее простое предположение о присоединении кислорода
по каждой двойной связи углеводорода каучука не подтверж-
дается, так как, кроме того, происходит окислительная деструк-
ция и в то же время очень часто сохраняется ненасыщенность
Молекулы, свидетельствующая о том, что не все двойные связи
раскрылись.
Заслуживают упоминания работы Пичи2,3, предложившего
«приближенную» формулу СюНтеО^ для продукта прямого окис-
ления каучука кислородом, а также работы Пумерера4 щ Босу-
элла5, выделивших ацетонорастворимые смолы, легко превра-
щающиеся в твердое вещество состава Ci0Hi6O и смолу соста-
ва С25Н40О9. Кемп6 показал, что при прямом окислении каучука
образуются даже летучие продукты реакции, а в осколках мо-
лекул, появляющихся при окислительной деструкции, имеются
карбонильные, лактонные и гидроксильные группы. Наконец,
новые сведения по этому вопросу получены в результате работ
Исследовательской ассоциации Британских производителей
каучука.
Продукты окисления каучука
479
Окисление неорганическими и органическими окислителями.
Босуэлл5 обрабатывал концентрированным водным раствором
перманганата раствор каучука в четыреххлористом углероде
без доступа воздуха. При этом получался продукт состава
'C2SH40O, нерастворимый в этиловом спирте и способный допол-
нительно окисляться на воздухе с образованием соединений
C2SH40O2. Из продуктов окисления каучука, выделенных Роберт-
соном и Мейром7, некоторые обладали эластичностью, в то
время как другие представляли собой хрупкие смолы. В общем
продукты окисления каучука перманганатом, по-видимому,,
довольно мало отличаются от продуктов прямого окисления
кислородом. Возможно, что при окислении перманганатом цепь
разрывается в еще большем числе мест, чем при действии кис-
лорода. Примечательно, что почти во всех продуктах деструк-
ции содержатся незатронутые двойные связи. Опыты различных
исследователей по окислению каучука перекисью водорода не
дали определенных результатов, тем более что окислитель боль-
шей частью разбавляли уксусной кислотой. Блумфилд и Фар-
мер8 полагают, что этот окислитель действует аналогично над-
уксусной кислоте, которая, вероятно, образуется в смеси
Н2О2 + СН3СООН. Как показали Робертсон и Мейр7, при дейст-
вии надуксусной кислоты в момент ее образования на каучук,
растворенный в хлороформе, получается продукт состава
С59НЮ2О16. Это белый порошкообразный продукт со значитель-
ной степенью ненасыщенности, растворимый в этиловом спирте,
ацетоне и уксусной кислоте.
Блумфилд и Фармер8 предлагают окислять перекисью водо-
рода и уксусной кислотой тонко измельченный каучук. Таким
путем получается белый порошкообразный продукт, для которого
характерно наличие карбонильных и гидроксильных групп, от-
сутствие двойных связей и более высокий молекулярный вес,
чем у ранее полученных продуктов.
По-видимому, по химической структуре эти вещества не
являются ни окисями, ни эпоксидами, а скорее продуктами окис-
лительного расщепления с большим числом гидроксильных
групп, способных далее этерифицироваться уксусной кислотой.
При действии перекиси бензоила при нагревании в отсутст-
вие растворителя и кислорода каучук вулканизуется. Это озна-
чает, что молекулярный вес каучука не только не уменьшает-
ся, а наоборот, увеличивается вследствие образования меж-
молекулярных мостиков. Действие перекиси бензоила на рас-
твор каучука без доступа воздуха вызывает гелеобразование,
что также свидетельствует об образовании сетчатой структуры.
Зато в присутствии кислорода перекись бензоила является ка-
тализатором окисления каучука. Компаньон и Делаланд9, на-
гревая с обратным холодильником в присутствии воздуха рас-
480 Г л. XIV. Каучук и продукты его превращений
творы каучука с перекисью бензоила, получали полужидкие
вещества с довольно большой степенью ненасыщенности.
Окисление каучука в присутствии катализаторов. Вследст-
вие устойчивости каучука против действия некоторых химиче-
ских агентов его неоднократно пытались вводить в состав кра-
сок, но этому препятствовала высокая вязкость растворов кау-
чука. Частично окисленные каучуки имеют значительно мень-
шую вязкость и благодаря этому могут использоваться в ка-
честве пленкообразующих веществ. Особенно энергичное уско-
ряющее действие на процесс окисления каучука оказывают
«доноры кислорода». Для быстрого 'И глубокого изменения как
сырого, так и вулканизованного каучука достаточно их нали-
чия в количестве 10—3%. Такими катализаторами являются в
первую очередь соли меди, марганца, кобальта и железа, при-
чем активность их возрастает с увеличением растворимости в
углеводороде каучука. Наиболее активны линолеаты и нафте-
наты меди и марганца. После введения линолеата марганца,
растворенного в минимальной количестве растворителя, в сы-
рой пластицированный каучук через короткое время проис-
ходит сильная деструкция углеводорода.
Пропускание в течение двух суток тока кислорода в бензоль-
ный раствор, содержащий 1% крепа и 0,025% линолеата кобаль-
та, приводит к образованию сильно окисленных продуктов,
именуемых раббонами. По степени окисления раббоны делятся
на три типа:
Раббон А — растворим в ароматических углеводородах и
уайт-спирите, но нерастворим в ацетоне и этиловом спирте.
Раббон В —растворим в ароматических углеводородах,
уайт-спирите и ацетоне, но нерастворим в этиловом спирте.
Раббон С (наиболее окисленный продукт) — растворим в
ароматических углеводородах, ацетоне и этиловом спирте, но
почти нерастворим в уайт-спирите.
Механизм окисления сырого каучука. Предполагалось, что
в большинстве реакций каучука участвуют этиленовые связи.
Однако Фармер показал, что активными центрами молекулы
являются также метиленовые группы, находящиеся в «-поло-
жении к двойной связи. Эти метиленовые группы могут отщеп-
лять атом водорода, в результате чего образуется свободный
радикал, способный вступать в цепную реакцию:
а
На стадии инициирования могут образовываться также пе-
рекисные радикалы, причем присоединение кислорода к
гоуппе —СН— в «-положении приводит к образованию ради-
Продукты окисления каучука
481
кала типа (I), а присоединение его с раскрытием двойной
связи — к образованию бирадикала типа (II):
Отщепление атомов водорода в положении а обеспечивает
протекание реакции передачи цепи. Перекисный свободный ра-
дикал присоединяет атом водорода, отщепившийся из а-положе-
ния другого изопренового звена:
При этом перекись превращается в а-гидроперекись, а изо-
преновая группировка, потерявшая атом водорода, — в ра-
дикал, продолжающий цепную реакцию.
Фармер уточнил некоторые детали механизма окисления и,
в частности, предположил, что некоторые перекиси имеют ци-
клическую структуру. Эти гипотезы удалось построить на
основе изучения полиизопреновых соединений более простой
структуры, чем у каучука10. Поэтому 'Следует пользоваться
этими положениями с большой осторожностью.
Образование перекисей, как оно было представлено выше,
не влияет на величину молекулы каучука. Но вследствие не-
стойкости перекисей происходят реакции их изомеризации и
расщепления, приводящие к быстрому изменению некоторых
свойств каучука (понижению вязкости и обычно увеличению
растворимости). По этому поводу были высказаны различные
гипотезы; общее представление об этом вопросе дают исследо-
вания Ле-Бра11.
Получение окисленных каучуков
В патенте Стсвенса и Ропгема12 описан удобный способ
получения раббона действием воздуха под давлением «а
50%-ный раствор каучука. По другому патенту13 растворитель
не применяют, а увеличивают поверхность соприкосновения
31—156
482
Гл. XIV. Каучук и продукты его превращений
каучука с кислородом путем введения наполнителя — древес-
ной муки. Следует отметить также способ, заключающийся
в вальцевании измельченного каучука или даже регенерата в
присутствии нафтената кобальта совместно с древесной пуль-
пой с высоким содержанием а-целлюлозы.
Было изучено влияние, оказываемое наполнителем, напри-
мер древесной мукой, на окисление. При этом установлено,
что целлюлозные материалы (бумага, древесная пульпа или
древесная мука, хлопок и т. д.) оказывают на реакцию до-
полнительное весьма значительное каталитическое действие.
Если не принимать специальных мер предосторожности, окис-
ление протекает так быстро, что может произойти обуглива-
ние смеси. Впрочем, разные виды целлюлозы обладают раз-
личной активностью. Так, если принять коэффициент актив-
ности древесной муки за единицу, то коэффициент активности
целлюлозы будет равен 2,5, а хлопка — 3,25.
Минеральные наполнители (асбест, шифер, кизельгур),
по-видимому, не влияют на скорость окисления каучука.
Фрейндлих и Талалай14 исследовали каталитическое окис-
ление в латексе с применением в качестве катализаторов рас-
творимых солей меди, марганца, кобальта и трехвалентного
железа. Они установили, что существует оптимальная доза
катализатора, превышение которой ослабляет каталитическое
действие и меняет направление реакции. Стивенс15 рекомендует
применять линолеат свинца с вольфраматом цинка для полу-
чения слабоокисленного каучука в латексе. Для глубокой де-
струкции латекса он предлагает применять линолеаты или
ацетаты марганца, меди, кобальта, свинца и цинка, причем эти
соли должны вводиться в виде хорошо стабилизованных вод-
ных дисперсий. Например, к 1 л латекса, содержащего 40%
каучука, можно добавить 240 г эмульсии линолеата марганца
(что соответствует 0,15% марганца от веса сухого каучука).
Через такой латекс, нагретый до 70°, пропускают примерно в
течение суток ток воздуха. После коагуляции латекса уксусной
кислотой и промывки коагулята получается окисленный каучук,
пригодный для производства красок.
Помимо солей перечисленных металлов, в некоторых 'слу-
чаях можно, по-видимому, использовать в качестве катализа-
торов окисления соли магния16.
Окисление может 'инициироваться также световыми лучами.
Фармер и Сандралингем10 облучали очень разбавленный бен-
зольный раствор каучука ультрафиолетовыми лучами в присут-
ствии кислорода. При соответствующей длительности облуче-
ния они получали каучуки, содержащие до 28,5% кислорода.
Такие каучуки растворимы в этиловом спирте; они имеют кис-
лую реакцию; молекулярный вес их порядка 1300, в то время
Продукты окисления каучука
483
как средний молекулярный вес недеструктированного сырого
каучука равен 350 000.
Процесс производства окисленных каучуков (раббонов)
заключается в вальцевании каучука с линолеатом кобальта
и затем с древесной мукой12,13. Продукты окисления извле-
кают растворителем.
На практике предварительно понижают вязкость каучука
путем пластикации его в закрытом смесителе (типа Вернера)
при температуре до 80° в присутствии 2,5% линолеата кобаль-
та. Примерно через 20 мин. медленно загружают в смеситель
целлюлозную массу. О протекании реакции окисления свиде-
тельствует выделение кислых паров и увеличение расхода
анергии на перемешивание. Как только начнут появляться
пары, необходимо прекратить нагрев смесителя, так как сильно
экзотермическая реакция окисления начинает развиваться
слишком быстро, что может привести к пирогенетическому раз-
ложению загрузки. Иногда бывает полезным энергичное
охлаждение аппарата. К концу процесса получают белый, 'слегка
липкий порошок, который после охлаждения обрабатывают
растворителем.
Образовавшийся раббон переходит в раствор, а нераствори-
мый остаток отделяют на .фильтрпрессе.
Окисленный каучук после удаления растворителя может
быть расфракционирован на три вида раббонов. Экстрагиро-
ванием этиловым спиртом выделяют раббон С, затем ацето-
ном — раббон В, а оставшийся раббон А очищают растворе-
нием в бензоле.
Линолеат кобальта почти нацело адсорбируется целлюлозой
и поэтому полностью отсутствует в раббоне. Регенерировать
катализатор трудно; во всяком случае целлюлозная масса, со-
держащая соль кобальта, не может быть повторно использо-.
вана.
Свойства окисленных каучуков
Окисленные каучуки представляют собой светло-желтые
вещества с удельным весом около единицы (для раббона типа В
^27=0,966 г/слс3), 'безвкусные, со слабым запахом. Они очень
устойчивы к влаге, малогигроскопичны (поглощают 0,25%
воды за сутки, 0,75% за неделю), не меняют цвета и не белеют,
хорошо совмещаются с маслами, смолами и пластификаторами.
При переходе от раббона типа А к типу С вязкость снижается,
но сильно возрастает при переходе от типа С к типу D.
Раббоны обладают хорошими диэлектрическими свойствами:
пробивное напряжение 57—76 кв на 4 мм, объемное сопротив-
ление 2,2-Ю9 ом-см2/см, после трехчасового нагревания при
31*
Гл. XIV. Каучук и продукты его превращений
110° и последующего охлаждения до 20° повышается до
2,8 • 10й ом • см2/см.
Качество раббонов хорошо характеризуется тремя показате-
лями, значения которых для раббона В приведены «иже:
Кислотное число .... ..............5
Число омыления ..........................60
Иодное число..............................305
Раббоны весьма термостойки; длительное нагревание при
350° вызывает потерю веса около 10% за счет удаления воды,
дипентена и маслянистых веществ26.
По химическим свойствам окисленные каучуки довольно
сильно отличаются от исходного каучука; в частности, различие
в остаточной ненасыщенности более или менее значительно в
зависимости от вида раббона. Они могут вступать в реакции
гидрохлорирования, галоидирования, вулканизации, хорошо из-
вестные в химии каучука.
В молекулах окисленных каучуков можно обнаружить кисло-
родсодержащие функциональные группы: гидроксильные17, окис-
ные и перекисные18, карбоксильные19.
Робертсону и Мейру20 удалось получить дихлоргидрат при
пропускании хлористого водорода через раствор окисленного
каучука в эфире или уксусной кислоте. Присоединение брома
к окисленному каучуку дает неясную картину. Между тем при-
соединение иода легко устанавливается путем определения
иодного числа, для раббона В равного 290 (по данным Хил-
тона), а для раббона С равного 260 (по данным Нейлора19).
Из продуктов обработки раббонов хлором и хлористым во-
дородом удается получить пленкообразующие вещества, изу-
ченные Стивенсом и Попгемом21. Эти вещества, пригодные для
красок, термопластичны и с этой точки зрения представляют
известный интерес. Пленки из гидрохлорированного раббона
обладают замечательной устойчивостью к щелочам. Хлориро-
ванные раббоны (сильно отличающиеся от хлоркаучуков) пред-
ставляют собой липкие смолы, которые после растворения и
добавления пигментов дают краски, образующие пленки с вы-
соким блеском.
Окисленный каучук ввиду наличия в его молекулах двой-
ных связей можно вулканизировать. При нагревании раб-
бона В в течение 2 час. при 100° в присутствии 10% серы полу-
чаются твердые, но термопластичные продукты. Кроме того,
установлено, что раббоны медленно затвердевают на воздухе,
причем нагревание содействует затвердеванию. Это явление,
по-видимому, связано с увеличением молекулярного веса и,
несомненно, с такими изменениями структуры, как циклизация,
межмолекулярная конденсация и т. д., точная природа кото-
Продукты окисления каучука
485
рых неизвестна. Такой продукт превращения раббона С являет-
ся очень термопластичным нерастворимым веществом. Он
именуется раббоном D.
Применение раббонов и других окисленных каучуков
Раббоны А, В и С — это каучуки разной глубины окисления,
отличающиеся по растворимости в некоторых растворителях;
раббон D является продуктом полимеризации раббона С, для
которого характерна очень малая растворимость. Помимо этих
продуктов фирма Scott Bader and Со., производящая раб-
боны, выпустила два новых раббона SH и SL, рекомендуемые
для отделки дерева и дающие лаки с высоким блеском.
Необходимо отметить, что «раствор В», применяемый в
Англии для производства красок, не является раствором раб-
бона В. Раббон В, по-видимому, наиболее распространенный
из окисленных каучуков, содержит около 10% •связанного кис-
лорода и совершенно лишен каучукоподобных свойств вслед-
ствие малого молекулярного веса (около 3000); он растворим
в ацетоне и уайт-спирите, причем вязкость его 50%-ного рас-
твора в уайт-спирите равна 3 пуазам. Между тем раствор В
получается из каучука, содержащего около 1% связанного
кислорода. Этот продукт нерастворим в ацетоне; вязкость
50%-ного раствора в уайт-спирите 3000 пуазов; молекулярный
вес порядка 100000.
Возможность использования раббонов в производстве лако-
красочных материалов, печатных красок и клеев несомненно
представляет интерес.
Наибольшее значение имеет возможность применения раб-
бонов в производстве изоляционных лаков горячей сушки.
Такие лаки обладают превосходными электроизоляционными
свойствами. Кроме того, они очень хорошо пропитывают бу-
магу и асбест, прекрасно противостоят действию влаги и
масел22. а
Фирма Scott Bader and Со. опубликовала следующую при-
мерную рецептуру лака23 (в вес. ч.):
Раббон В ...................................100
Полимеризованное льняное масло (25 пуаз при
25°)........................................200
Уайт-спирит ................................250
Нафтенат кобальта............................ 6
Для получения лака по этой рецептуре раббон В раство-
ряют в масле, нагревают в течение часа при 260°, затем охлаж-
дают до 100° и разбавляют уайт-спиритом, толуолом или кси-
лолом. После испарения растворителя раббон отверждают
486
Гл. XIV. Каучук и продукты его превращений
(окисляют) путем кратковременного (около 30 мин.) нагрева-
ния при 120°. Полученная таким способом лаковая пленка
очень устойчива к нагреванию и способна в течение довольно
длительного времени выдерживать температуру 200° (обычные
лаки начинают разрушаться уже при 150°). Лак на основе раб-
бона весьма хорош для защиты жести; он противостоит дейст-
вию кислых растворов, но разрушается при соприкосновении с
крахмалистыми веществами.
Лак, полученный по приведенной рецептуре, может приме-
няться без сиккатива; в этом случае он требует горячей сушки
в течение часа при 150°; пленка очень устойчива к воде, кисло-
там, растворителям и обладает высокой термостойкостью. При
замене льняного масла тунговым применяется рецептура сле-
дующего типа (в вес. ч.) :
Раббон В.....................................100
Крестин SB 208 (или другая 100%-ная феноль-
ная смола)................................... б
Сырое тунговое масло.........................200
Уайт-спирит .................................300
Нафтенат кобальта (50%-ный)................... 4
Твердость и эластичность получаемых лаковых пленок за-
висят от взятых масел. Эти пленки нерастворимы в обычных
растворителях, не разрушаются ни кислотами, ни щелочами,
ни растворами солей. Свойства их мало изменяются при на-
гревании и при резких колебаниях температуры.
Смеси раббонов с льняным маслом обладают высокой ад-
гезией к асбесту, в то время как обычные лаки часто дают мало
удовлетворительные покрытия по асбесту. Раббоны можно при-
менять также для первого покрытия по стали.
Раббон В прекрасно совмещается с нитроцеллюлозными ла-
ками22, 23, которым он придает прекрасный блеск; кроме того,
он улучшает адгезию покрытия, его устойчивость к старению
и к.действию солнечного света. Раббон совмещается с пласти-
фикаторами, например с трикрезилфосфатом, хлорированными
бифенилами, ланолинами, кумароновыми смолами. Получаю-
щиеся лаки быстро высыхают и рекомендуются для покрытия
легких сплавов, применяемых в авиастроении. Благодаря устой-
чивости к химическим агентам они могут успешно применяться
для защиты различных аппаратов в химической промышленно-
сти.
Следует указать, что раббоны смешиваются с хлоркаучу-
ками, но не в любых соотношениях. Лучше всего совмещаются
с раббонами низковязкие хлоркаучуки. Смеси раббон — хлор-
каучук рекомендуется применять для повышения прочности ас-
бестового шнура при растяжении и для пропитки древесины.
Продукты окисления каучука.
487
Раббоны применяются также для электроизоляции, в частно-
сти для покрытия медных проводов или, предпочтительно,
проводов из луженой меди, так как раббоны обладают прекрас-
ной адгезией к олову. Лаки на основе раббонов, совмещенных
•с 'полимеризованными маслами, отличаются эластичностью пле-
нок, теплостойкостью, превосходными диэлектрическими свой-
ствами и поэтому представляют ценность для покрытия обмо-
ток и пропитки электроизоляционной бумаги.
Делались попытки использовать раббоны в производстве
печатных красок. Оказалось, что они являются подходящей
средой для диспергирования металлических пигментов, напри-
мер алюминиевой пудры.
Раббоны преимущественно используются для изготовления
лаков горячей сушки и мало применяются для лаков холодной
сушки. Изучение соединений, менее глубоко окисленных, чем
раббоны, показало их полную пригодность для изготовления
обычных красок. Первые опыты в этом направлении были про-
ведены Стивенсом и Хитоном24.
Для получения таких материалов каучук пластицируюч в
закрытом смесителе с 2,5% линолеата кобальта и после дис-
пергирования катализатора всю массу растворяют при переме-
шивании в равном по весу количестве уайт-спирита. Это рас-
твор В, изготовляемый в Англии (см. стр. 485). Раствор Б
совмещается с высыхающими маслами и в него можно вводить
обычные пигменты. Ниже приводится одна из предложенных
рецептур (в вес. ч.):
Литопон................................... 80
Цинковые белила........................ . . 20
Слабополимеризованное масло.................30
Раствор В ..................................15
Жидкий сиккатив ............................15
Уайт-спирит.................................20
Никакой трудности не представляет применение и других
пигментов. Перетир пигментов и наполнителей '.можно про-
изводить также в смеси с маслом и окисленным каучуком.
Преимуществом таких красок по сравнению с краска-
ми близкого состава, но не содержащими каучука, является
большая легкость нанесения и отсутствие следов кисти на
пленке.
Частично окисленные каучуки рекомендуется применять для
матовых красок и для антикоррозионных покрытий, в которые
обычно вводят большое количество 'пигментов с высоким удель-
ным весом (например, сурика), склонных оседать и образовы-
вать осадки, с трудом поддающиеся повторному диспергиро-
ванию. Краски такого типа трудно наносить, и они не всегда
488 Гл. XIV. Каучук и продукты его превращений
дают достаточно равномерное покрытие. Применение в этих
красках частично окисленного каучука обеспечивает равномер-
ность пленки. Повторное суспендирование пигментов и напол-
нителей даже после многомесячной седиментации значительно
облегчается.
Ниже приводится 'рецептура матовой краски, содержащей
довольно большое количество твердых компонентов25 (в вес. ч.):
Частично окисленный каучук (50%-ный раствор)
Полимеризованное льняное масло ............
Жидкий сиккатив ... ...........
Уайт-спирит................................
Двуокись титана . . ...
Литопон .... . . ............
Асбест............... .....................
20
10
1
50
65
65
15
Окисленные каучуки не вызывают затруднений при сушке-
и дают твердую и прочную пленку.
Матовые быстровысыхающие краски можно получать, заме-
нив льняное масло эфирами канифоли или их смесью с уплот-
ненным тунговым маслом; в такой рецептуре окисленный кау-
чук является основным пленкообразующим веществом. Следует
подчеркнуть, что частично окисленные каучуки хорошо совме-
щаются с высыхающими маслами, но не совмещаются с
природными .смолами, введения которых в смесь следует
избегать.
ЛИТЕРАТУРА
1. А. V. Van Rossem, Caout. et Gutta-percha, 1913, 107, 6961.
2. J. J. Peachey, J. Soc. Chem. Ind., 1912, 31, 1103.
3. J. J. Peachey, M. Leon, J. Soc. Chem. Ind., 1918, 37, 55T.
4. R. Pummerer, A. Burkard, Ber., 1922, 55, 3462.
5 M. C. Boswell и corp., Cour. Chem. a. Metal!., 1922, 6, 237; Indian
Rubb. J„ 1922, 64, 981.
6. A. R. Kemp н сотр., Ind. Eng. Chem. Ind., 1931, 23, 1444.
7. J. M. Robertson, J. M a i r, J. Soc. Chem. Ind., 1927, 46, 41T.
8. G. F. Bloomfield, E. H. Farmer, J. Soc. Chem. Ind., 1934,.
53. 121T.
9. P. Comp agnon, A. Del a 1 a n de, Rev. gen. Caout., 1943, 20,
133; 1947, 24, 4.
10. E. H. Farmer, A. Sundral i ngam, J. Chem. Soc., 1942, 121;
E. H. Farmer и сотр., Trans. Farad. Soc., 1942, 38, 348; E. H. F a r-
m e r, D. A. Sutton, J. Chem. Soc., 1942, 139; E. H. Farmer,
A. Sundralingam, J. Chem. Soc., 1943, 356; E. H. Farmer,.
H. P. Koch, D. A. Sutton, J. Chem. Soc., 1943, 541; J. L. В о 1-
l a n d, India Rubb. J., 1946, 111, 213.
11. J. Le Bras, Rev. gen. Caout., 1944, 21, 243.
12. H. P. Stevens, F. J. W. P о p h a tn, Rubber Producers’ Research.
Association, англ. пат. 462613, 1937 г.
13. H. Р. Stevens, F. J. W. P о p h a m, Rubber Producers’ Research
Association, англ. пат. 462627, 1937 г.
14. H. F г е u n d 1 i с h, N. T a 1 a 1 a y, Kautchuk, 1933, 9, 34.
Синтетические каучукоподобные эластомеры 489'
15. Н. Р. Stevens, Rubber Producers' Research Association, англ. пат.
442136, 1936 г.
16. A. Davies, англ. пат. 373228, 1932 г.
17. G. F. Bloomfield, Е. Н. Farmer, J. Soc. Chem. Ind., 1934,
53, 121T; J. L. В о 11 a n d, Trans. Inst. Rubb. Ind., 1941, 16, 267.
18. H. Lund, Ber., 1937, 70, 1520.
19. F. Hilton, Trans. Inst. Rubb. Ind., 1942, 17, 319.
20. J. M. Robertson, J. A. Mair, J. Soc. Chem. Ind., 1927, 46, 41T.
21. H. P. Stevens, F. J. W. P о p h a m, Rubb. techn. confer., London,
1938, p. 336.
22. F. J. W. P о p h a m, Rev. gen. Caout., 1944, 21, 255.
23. Scott Bader a. Co. Ltd., Rubbone, London, 1939.
24. H. P. Ste vens, N. Heaton, Bull. R. G. A., 1933, 15, 600.
25. J. Le Bras, A. D e 1 a 1 a n d e. Les derives chimiques du caoutchouc?
naturel, Paris, 1950.
СИНТЕТИЧЕСКИЕ КАУЧУКОПОДОБНЫЕ ЭЛАСТОМЕРЫ
На основе некоторых синтетических эластомеров, например-
вулкапренов и блек-аута, можно получать эластичные окра-
шенные пленки, способные выдерживать деформацию покры-
ваемого объекта.
Материалы, получаемые растворением эластомера в раство-
рителе с введением необходимых ингредиентов и пигментов,,
приобретают в настоящее время распространение для опреде-
ленных областей применения.
Вулкапрены
Вулкапрены (фирмы Imperial Chemical Industries) являют-
ся эластомерами сложного строения1. Они получаются поли-
конденсацией двухосновной кислоты, двухатомного спирта и
диамина. Однако в результате этой реакции можно получить,
вещества с молекулярным весом не более 5 000. Для получения,
же эластичного продукта необходимо увеличить молекулярный
вес, что может быть достигнуто действием диизоцианатов на
концевые группы —СООН, —ОН или —NH2 продукта поликон-
денсации. При этом группа —СООН образует амид, группа
—ОН —уретан, а группа —ЫН2 —замещенную мочевину. Мо-
лекулярный вес увеличивается, и образуются линейные цепи,,
придающие продукту каучукоподобные свойства.
Вулкапрены можно вулканизовать, но не с помощью серы,,
а с помощью формальдегидных производных (вулкафор VHM),.
реагирующих с амидными группировками. Ускорителями вулка-
низации служат галоидопроизводные, известные под названием
вулкафор VDC. В некоторых случаях, когда необходимо вести;
вулканизацию при комнатной температуре, применяются по-
лиизоцианаты (вулкафор VCC).
490_____Гл. XIV. Каучук и продукты его превращений_________
Получаемые насыщенные полимеры прекрасно противостоят
действию кислорода и озона, но чувствительны к химическим
реагентам и нагреванию.
Выпускается шесть товарных сортов вулкапренов: марки А,
АС 50, АС 90, АС 160, АС 230 и PN.
Вулканизованные вулкапрены А устойчивы к маслам и бен-
зинам. Они применяются преимущественно в качестве специ-
альных каучуков для непосредственного формования.
Вулкапрен PN представляет собой смесь вулкапрена А и
продуктов гидролиза кожи. Путем нанесения его на ткань на
каландрах можно получать клеенку.
Вулкапрены АС растворимы в ацетоне (вязкость раствора
обозначается цифрами в их марках); вулкапрен АС 230 об-
разует очень вязкие растворы, а вулкапрен АС 50 — низко-
вязкие.
Специально изучалась возможность применения вулкапре-
нов АС для лаков. Оказалось, что они дают очень ценные
лаки, так как образующаяся пленка обладает такой же эла-
стичностью, как сам каучук. Эти лаки могут применяться для
защиты натурального каучука от вредного воздействия кисло-
рода воздуха, приобретающего особую активность под влия-
нием солнечных лучей.
Вулкапрены А и АС выпускаются в виде плотных пластин
кремового цвета, напоминающих креп. Перед растворением в
смеси растворителей их необходимо подвергнуть кратковремен-
ному вальцеванию. Эту операцию используют для введения
цветных пигментов. Растворы вулкапрена мало изменяются при
хранении (ускоритель вулкафор VCC вводится только
непосредственно перед использованием). Краски на основе
вулкапрена могут наноситься кистью и пульверизатором.
Лаковые пленки вулкапренов очень стойки к истиранию, причем
эту стойкость можно еще более повысить введением ацетил-
целлюлозы.
Нитроцеллюлозы тоже совмещаются с вулкапренами. Такие
лаки сохраняют известную эластичность и весьма пригодны для
покрытия кожи.
Блек-аут
Блек-аут (фирмы Vanderbilt Со.), строение которого точно
не установлено, представляет собой модифицированный синте-
тический эластомер, выпускаемый в виде раствора в летучих
органических растворителях. Получаемые на его основе плен-
ки обладают эластичностью.
Блек-аут может наноситься на резину, металлы, дерево,
пластические массы, бумагу, кожу и другие подложки. Его
Синтетические каучукоподобные эластомеры
491
совместимость с некоторыми цветными пигментами позволяет
получать широкую гамму красок4.
Пленки блек-аута защищают грунт от разрушения, вызывае-
мого солнечным светом, ультрафиолетовыми лучами, атмосфер-
ным воздействием. Они устойчивы по отношению к маслам,
основаниям и кислотам; сопротивление истиранию удовлетвори-
тельное.
Лаки на основе блек-аута можно наносить методами окуна-
ния и пульверизации.
Блек-аут термопластичен и обладает высокой когезией. То-
варный продукт выпускается черного, белого цвета или бес-
цветный, что позволяет путем смешения с окрашенными дис-
персиями получать желаемые оттенки.
Синтетические каучуки
Применение синтетических каучуков в производстве лаков
и красок быстро расширяется, так как их свойства пред-
ставляют большую ценность для некоторых специальных
целей.
Бутадиен-акрилонитрильные сополимеры, полихлоропрены
и органические .полисульфиды после вулканизации приобрета-
ют более высокую устойчивость к растительным маслам и рас-
творителям. Полихлоропрены негорючи, устойчивы к окисли-
телям и предохраняют грунт, на который они нанесены, от
разрушающего действия кислорода. Изобутилен-изопреновые
сополимеры, содержащие около 2% изопрена, отличаются осо-
бой газонепроницаемостью. Они хорошо противостоят окисле-
нию и чрезвычайно .устойчивы к действию тепла, света и мед-
ных солей. Органические полисульфиды обладают неприят-
ным запахом и дают вулканизаты с большим остаточным
удлинением, но весьма устойчивы к органическим раствори-
телям.
Сополимеры изобутилена менее эластичны, чем натуральный
каучук, и имеют очень большое остаточное удлинение. Они до-
вольно чувствительны к растворителям.
Покрытия на основе синтетического каучука могут нано-
ситься путем наклеивания каландрюванных листов на пред-
охраняемую поверхность. Такой метод требует применения раз-
личных клеев на основе хлоркаучука, циклокаучука, полихло-
ропрена (неопрена), бутадиен-стирольных полимеров (клеи
плиогрип фирмы Goodyear) и т. д.
В производстве красок применяются различные синтетиче-
ские латексы, в частности полихлоропреновые и бутадиен-сти-
рольных полимеров с различным содержанием стирола. Из
них следует назвать латекс хемигама 101 (фирмы Goodyear)5,
492 Гл. XIV. Каучук и продукты его превращений
неопреновый латекс 842 А (фирмы Du Pont de Nemours) и ла-
тексы хемигама 200 и 245 (фирмы Goodyear), представляющие
собой, по-видимому, бутадиен-акрилонитрильные сополи-
меры.
Ниже приводятся две рецептуры красок на основе синтети-
ческого латекса.
Рецептура для покрытия резиновых изделий включает сле-
дующие компоненты (в вес. ч.):
Неопрен (латекс 842 А) .... .... 100
Цинковые белила............................ 15
Антиокислитель.............................. 2
Силикат натрия.............................0,25
Т иокарбанилид.............................. 1
Сера........................................ 1
Стабилизатор (акварекс D фирмы Du Pont de
Nemours)..................................0,25
Этот состав дает пленки с хорошей адгезией при нанесении
на чистую поверхность; при желании в него могут быть введе-
ны цветные пигменты, диспергирующиеся в латексе. Для полу-
чения «пастельных» тонов можно добавлять 5—10 вес. ч.
двуокиси титана.
Введение серы и ускорителя не обязательно, но вулканизация
повышает прочность пленки.
Рецептура композиции для внутренних покрытий стен5 со-
стоит из трех частей: А представляет собой дисперсию пиг-
ментов, Б — раствор а-протеина, В — синтетический латекс.
Компоненты смешивают в алфавитной последовательности:
А растворяют в Б, а полученную смесь — в латексе хемига-
ма 101.
На 100 вес. ч. латекса хемигам 101 (55% сухого вещеетва)
берут компоненты А и Б в следующих количествах (в вев. ч.):
А. Литопон ...............................273
Слюда '................................ 88
Титановые белила ...................... 65
Вода ...................................145
Смачиватель (дарван 1)................1,1
Трифосфат натрия .......................2,2
Б. а-Протеин..............................1,5
Аммиак (уд. в. 1,22 г/см3)..............2,2
Пентахлорфенолят натрия (10%-ный раствор) 2,2
Вода ...................................160
Такая краска выдерживает длительное хранение, если не
допускать присутствия растворимых солей двух- и трехвалент-
ных металлов, которые вызывают коагуляцию латекса. Слюду
Синтетические каучукоподобные эластомеры
493
можно заменять каолином или аморфным кремнеземом (диа-
томитом) .
а-Протеин можно заменять казеином, метилцеллюлюзой или
карбоксиметилцеллюлозой.
Подобно натуральному каучуку синтетические каучуки в ви-
де растворов могут применяться для изготовления антикорро-
зионных лаков и красок. Выпускаются бутадиен-стирольные со-
полимеры, специально предназначенные для этой цели; из них
очень ценными являются плиолиты S (фирмы Goodyear), в
частности плиолиты S5, S4, S3, растворимые в хлорорганическик
растворителях.
Для изготовления лаков и красок применяются также поли-
хлоропрены (неопрен), растворенные в хлорорганическом рас-
творителе.
Ниже приводится рецептура такого лака (в вес. ч.):
Неопрен W...................................100
Обожженная магнезия (экстра легкая) . . 4
Цинковые белила................ ... 5
Пигменты .................... ... X
Растворитель ' По выбору
Пигменты подбирают в соответствии с требуемым оттенком;
для пастельных красок можно на 100 вес. ч. неопрена вво-
дить 15 вес. ч. двуокиси титана. В качестве растворителя 'мо-
жет применяться трихлорэтилен или ароматический углеводо-
род.
Для удешевления таких смесей в них можно вводить в каче-
стве наполнителя карбонат кальция.
В настояшее время изучается возможность применения на-
ряду с самими синтетическими каучуками их производных, в
частности хлоркаучуков и циклокаучуков.
Можно назвать несколько работ, посвященных этому во-
просу ®’718.
ЛИТЕРАТУРА
1. A. J а г г i j о n, Premier Congres Intern. Industr. Peintures, Bruxelles,
1—5 octobre, 1948.
2. J. M. F. D e 1 1 о n e, фр. пат. 965077.
3. Le manuel des Vulcaprenes, Notices, Imperial Chemical Industries Ltd.
4. S. S. Rogers, The Vanderbilt News, January, February, 1950, 16, 8.
5. Anonyme, La chimie des peintures, Bruxelles, 1951, 14, 75.
6. H. A. E n d г e s, Rubber Age, N. Y., 1944, 55, 351—366.
7. Borrow, Jordan, Kingcome, Robinson, Waters, BIOS,
final report, № 727, London, 1947.
,8. Anonyme, Prod. Finishing, avril 1948, 12, 86.
Торговые марки каучуков и продуктов их превращений
Торговое название Характеристика Фирма
Аллопрен (АПоргёпе) В, С, D Аморит (Amorite) Айцен (Aizen) Асидсил (Acidseal) Е,МА,РА Балата Блек-аут (Black-out) Буна 85 115 S N Бутапрен NAA, NF, NL, NMX н NI S Бутил 15, 17, 18, 50, R-2 Бутилкаучук CCq , СС4, ссв Вистанекс (Vestanex) Вулкабонд (Vulcabond) Вулкалок (Vulcalock) Вулкапрен (Vulcaprene) Вулколлан^ (Vulcollan) Хлорированный натуральный каучук Клей на основе циклизованного натурального каучука Хлорированный натуральный каучук Краски на основе циклизованного натурального кау- чука Клен на тойже основе /праяс-Изомер натурального каучука Модифицированный сополимер Полибутадиен Продукт полимеризации бутадиена ,в присутствии металлического натрия Сополимеры бутадиена со стиролом Сополимеры бутадиена с акрилонитрилом Сополимеры бутадиена с акрилонитрилом Сополимер бутадиена со стиролом Сополимеры изобутилена с изопреном Сополимеры изобутилена с изопреном Хлорированный натуральный каучук Полимеры изобутилена Клеи на основе циклизованного натурального кау- чука для крепления резины к металлам; вулка- бонд S может применяться только в виде 10%-ного раствора, вулкабонд R— в виде растворов до 50% -ных Клей на основе циклизованного каучука для креп- ления резины к металлам Смешанный полиэфир-полиамид-полиуретан Полиэфир-изоцианат Imperial Chemical Industries (Англия), I. С. I. (Франция) В. F. Goodrich Со. (США) Hadogaya Soda КК (Япония) В. F. Goodrich Со. (США) Kleber-Colombes (Франция) Vanderbilt Со. (США) 1. G. Farbenindustrie То же » » Firestone Tire and Rubber Co. (США) «Esso» Standard Standard Oil Co. (США) Pechiney (Франция) Standard Oil Co. (США) I. С. I. (Англия) В. F. Goodrich Co. (США) (Kl£ber-Colombes, Франция) I. С. I. (Англия) Farben Fabrik Leverkusen
Гл. XIV. Каучук и продукты его превращений
Продолжение
Торговое название Характеристика Фирма
Гуттаперча Дартекс (Dartex) и дартекс АС Детель (Detel) Дуропрен* (Duroprene) Изолак (Isolac) Крилле (Grille) Марбон В V и X S и Sj 8000 Метилкаучуки Н и W Неопреновые латексы 571, 572, 601,601 А, 700, 735, 842, 842 А Неопрены AC, CG, Е, FR, GN, GNA, KNR, RT, S, W Ниппон (Nippon) Новоплас A (Novoplas А) Оппанолы В 100 и В 200 Паравар (Paravar)} Паракрил 26 и 35 (Paracril) Парлон (Parlon) Пепрен (Рёргеп) Пербунан Пергут (Pergut) Пердурены G и М (Perduren) /праяс-Изомер натурального каучука Хлорированный натуральный каучук То же » Циклизованный натуральный каучук типа термо- прена Гидрохлорированный.натуральный каучук, подверг- нутый растяжению в набухшем состоянии Очищенный циклизованный натуральный каучук Продукты гидрохлорирования очищенного циклизо- ванного натурального каучука Сополимеры бутадиена со стиролом; высокое содер- жание стирола Сополимеры бутадиена со стиролом Продукты полимеризации диметилбутадиена Латексы полихлоропрена Полихлоропрены Хлорированный натуральный каучук Органические полисульфиды Полимеры изобутилена Хлорированный натуральный каучук Сополимеры бутадиена с акрилонитрилом Хлорированный натуральный каучук Циклизованный натуральный каучук Сополимеры бутадиена и акрилонитрила Хлорированный натуральный каучук Органические полисульфиды Metallgesellschaft A. G. Detel Products (США) I. С. I. (Англия) Du Pont de Nemours (США) Goodyear Tire and Rubber Co. (США) Marbon Corporation (США) To же » » » Du Pont de Nemours (США) (Alcan, Франция) To же Nippon Soda KK (Япония) I. С. I. (Англия) Naugatuck Chemical Corp. (США) «Esso» Standard Hercules Powder Co. (США) I. G. Farbenindustrie To же » »
в настоящее время выпускается под маркой аллопрен.
Торговые марки каучуков 495
Продолжение
Торговое название Характеристика Фирма
Плиоваксы (Pl iowaxes) Смесь циклизованного натурального каучука со смолами или восками; применяется для придания водонепроницаемости бумаге или целлофану Goodyear Tire arid Rubber Co. (США)
-Плиолестик (Pliolastic) Плиолит (Pliolite) Гидрохлорированный натуральный каучук, вытяну- тый в набухшем состоянии в нити для эластич- ных тканей To же
NR Натуральный каучук, циклизованный действием галоидоловянных кислот »
Сополимер нерасшифрованного строения »
Ss, Se, S, Сополимеры бутадиена со стиролом с различным содержанием стирола »
Плиолит латекс 160 FR Сополимеры бутадиена со стиролом; стойкий латекс »
Плиофильм (Pliofilm) Пленки гидрохлорированного натурального каучука »
Плиоформ (Plioform) Полисар Натуральный каучук, циклизованный галоидными солями олова; № 20 и № 40 различаются по термопластичности »
N и NP Сополимеры бутадиена с акрилонитрилом Polymer Согрог. (Канада)
S 50, S 65, SX 371, SS 250 Сополимеры бутадиена со стиролом To же
Полисар-бутил (Polysar-Butyi) Сополимеры изобутилена с изопреном »
Полисар-крилен (Polysar-Krylene) Сополимеры бутадиена со стиролом; низкотемпера- турная полимеризация »
Протеке (Protex) Раббоны Хлорированный натуральный каучук Ste Elettrica ed Elettrochimica del Caffaro (Италия)
А, В, C, D Окисленный науральный каучук различной раство- римости Scott Bader Co. Ltd (Англия)
SH и SL Окисленный натуральный каучук, для обработки дерева (SH—высоковязкий; SL-низковязкий) To же
496 Гл. XIV. Каучук и продукты его превращений
Продолжение
32—156
Торговое название Характеристика Фирма
Раствор В Раствор окисленного натурального каучука Англия
Резинит (R£sinite) Органические полисульфиды СССР
Реолин (Raolin) Хлорированный натуральный каучук Raolin Corpor. Продается фир- мой Binney and Smith (США)
Санипрен (Sanipr£ne) Футеровка гибким полуэбонитом В. F. Goodrich Со. (США) Kleber-Colombes (Франция)
СК Полибутадиен СССР
Спектрафильм (Spectrafilm) Пленки гидрохлорированного каучука Goodyear Tire and Rubber Co. (США)
Суперфлексайт регулярный и А (Superflexite) Тай-плай (Ту-Р1у) Футеровка гибким эбонитом Клеи на основе гидрохлорированного каучука: В. F. Goodrich Co. (США) Kleber-Colombes (Франция)
R для приклеивания натурального каучука к черным металлам R. T. Vanderbilt Co. (США)
q улучшенный R To Же
S для синтетических каучуков »
qA и SA для широкого применения »
Тегофан (Tegofan) Хлорированный натуральный каучук T egofan-Gesel Ischaft
Тенейлон (Tensylon) Нити, полученные круткой тенсолайта, одного или вместе с хлопчатобумажными нитями Goodyear Tire and Rubber Co. (США)
Тенсолайт (Tensolite) Гидрохлорированный натуральный каучук, растяну- тый при нагревании и охлажденный в растянутом состоянии To же
Термопрены GP, НВ, SL Циклизованный натуральный каучук В. F. Goodrich Co. (США)
Тиокол А Органические полисульфиды Thiokol Gesellschaft
A, FA, ST, PR-1 То же Thiokol Corpor. (США)
Торнезит (Tornesit) Хлорированный натуральный каучук, замененный в США парлоном Hercules Powder Co.
Три флекс К (Triflex) Клей на основе циклизованного натурального кау- чука для приклеивания к металлам B. F. Goodrich Co. (США) Kleber-Colombes (Франция)
Торговые марки каучуков 497
Продолжение
Торговое название Характеристика Фирма
Фенолак (Phenolac) Хайкар (Hycar) Клей на основе натурального каучука, циклизован- ного фенолсульфокислотой; применяется для гум- мирования металлических поверхностей Kleber-Colombes (Франция)
OS Сополимеры бутадиена со стиролом; хайкар OS-10 обладает хорошими диэлектрическими свойствами В. F. Goodrich Со. (США)
OR-15 н OR-25 Сополимеры бутадиена с акрилонитрилом То же
РА Сополимеры акрилатов и галоидсодержащих моно- »
Хемигам (Chemigum) N3-NS, Goodyear Tire and Rubber Co.
N4-NS-3O, N4-NS-50 Хемнгам латекс Сополимеры бутадиена с акрилонитрилом (США)
101А Сополимер бутадиена со стиролом To же
200 Сополимер бутадиена с акрилонитрилом »
Хлортекс (Clortex) Хлорированный натуральный каучук Sa Elettrica ed Elettrochimica del Caffaro (Италия) Ugine (Франция)
Электрогам (Electrogum) То же
QR-A Сополимеры бутадиена с акрилонитрилом Государственные заводы США
GR-I Сополимеры изобутилена с изопреном То же
GR-S стандартный Сополимеры бутадиена со стиролом, содержащие 1,5% антиокислителя (фенил-₽-нафтиламина) »
QR-S низкотемпературный получаются при низких температурах »
GR-S 5U GR-S 38 содержат в качестве антиокислителя сталайт получаются непрерывным методом »
GR-S 10 содержат абиетиновую кислоту вместо жирной »
GR-S 20 • пластичность выше, чем стандартного »
GR-S 65 пластичность ниже, чем стандартного; меньше содержание сажи, лучшие электрические »
GR-S 85 пластичность значительно ниже, чем стандартного »
GR-S AC 12AC, 20AC коагуляция квасцами »
GR-S блэк 1 на 100 в. ч. GR-S до 50 в. ч, усиливающей сажи
Гл. XIV. Каучук и продукты его превращений
ГЛАВА XV
ПОЛИОРГАНОСИЛОКСАНЫ *
Ф. Аппель
До последнего времени кремнезем и силикаты применялись
в промышлености в качестве строительных материалов, гидрав-
лических ^кидкостей и сырья для керамического производства.
Органические производные кремния не вырабатывались в
промышленном масштабе и синтезировались лишь в лабо-
раториях. Промышленное производство эфиров кремневых кис-
лот, а затем полиорганосилоксанов было начато совсем не-
давно.
В 1899 г. английский ученый Киппинг24 приступил к серьез-
ным исследованиям органических производных кремния, про-
должавшимся более сорока лет. Он заложил основы этой
области химии, которая, судя по последним работам, по-види-
мому, получит такое же широкое развитие, как органическая
химия. Подобно всем химикам начала этого столетия, Киппинг
стремился выделить индивидуальные химические вещества. Он
охарактеризовал мономеры и низшие полимеры, но не интере-
совался многочисленными образующимися смолами. Работы,
проводившиеся начиная с 1939 г., показали, что в этих смолах
содержатся высокополимерные кремнийорганические соеди-
нения, являющиеся переходным звеном от высокополимерных
неорганических соединений типа силикатов к органическим
высокополимерам.
В институте Меллона (фирмы Corning Glass Works) были
обнаружены замечательные свойства высокополимерных крем-
нийорганических соединений (неправильно названных силико-
нами), а фирма Dow Chemical Со., впоследствие объединившаяся
с Coming Glass Works в фирму Dow Corning Corporation, органи-
зовала их промышленное производство. Производством и реа-
лизацией этих своеобразных продуктов занялась также фирма
General Electric Со. и ее филиалы. Каждая из этих двух фирм
разработала свой производственный метод получения кремний-
органических полимеров.
* Глава была написана в 1951 г. и не обновлялась ввиду смерти ее автора.
32*
5С0
Гл. XV. Полиорганосилоксаны
Силиконы (полиорганосилоксаны, полисилоксаны) представ-
ляют собой полимеры, цепи которых построены из последова-
тельно соединенных атомов кислорода и кремния, а остальные
валентности атомов кремния насыщены органическими радика-
лами.
Существуют полисилоксаны различный физических со-
стояний: начиная от жидкостей и кончая твердыми смолами и
каучуками. Они отличаются превосходной теплостойкостью,
морозостойкостью, стойкостью к химическим агентам, гидрофоб-
ностью и ценными диэлектрическими свойствами. Благодаря
всем этим свойствам полисилоксаны с успехом используются
для придания различным материалам водонепроницаемости
в производстве красок, в электротехнике и радиотехнике. Ши-
рокому распространению пол и сил окса нов препятствует их вы-
сокая стоимость (примерно 6000 франков за килограмм во
Франции в 1951 г.). .Поэтому они до сих пор применяются лишь
в таких областях, где стоимость их не имеет большого значения,
например в авиастроении.
Мы ограничимся здесь рассмотрением полимерных органо-
силоксанов, не затрагивая эфиров ортокремневой кислоты
Si (ОН) ч, в которых, в отличие от силоксанов, углеводородные
радикалы связаны с атомом кремния не непосредственно, а
через кислород.
По кремнийорганическим соединениям имеется очень бога-
тая литература. Так, в книге Поста3, вышедшей в 1949 г., при-
водится 700 литературных ссылок. В работе Рохова33, вышед-
шей в 1946 г., сознательно приведена ограниченная библиогра-
фия, но в 1947 г. Рохов совместно с другими авторами опубли-
ковал обзор9, в котором приведена 371 литературная ссылка*.
Обзорные статьи по кремнийорганическим соединениям по-
явились в США9,12, Англии21, Франции3,8> 10’17 ’18 ’ 19, Бельгии8,
ФРГ25.
Имеется несколько сот патентов на синтез и применение
полисилоксанов. Мы сознательно ограничили приводимые би-
блиографические ссылки перечислением трех десятков новей-
ших книг и статей, представляющих наибольший интерес.
Полиорганосилоксаны (силиконы) могут быть линейными
или трехмерными (обладать пространственной структурой).
Они получаются поликонденсацией продуктов гидролиза орга-
ногалоидсиланов. Например, из диалкилдихлорсилана R2SiCI2
получается диалкилсиланднол R2Si(OH)2, поликонденсация
которого происходит более или менее самопроизвольно.
* Более полный обзор состояния химии кремнийорганических соединений
дан в монографии К. А. Андрианова «Кремнийорганические соединения»,
Госхимиздат, 1955 и в последующих обзорных статьях того же автора. Прим. ред.
Получение органогалоидсиланов 501
Получение органогалоидсиланов
Для получения органогалоидсиланов в лабораторных усло-
виях можно использовать различные методы органического
синтеза, которые Рохов33 делит на 4 группы:
1) прямой синтез, состоящий во взаимодействии галоидного
алкила (арила) с кремнием;
2) метод замещения, к которому относятся классические
методы алкилирования галогенидов кремния металлорганиче-
окими соединениями, в частности реактивом Гриньяра (истори-
чески это первый из примененных методов);
3) присоединение силана к ненасыщенному органическому
соединению;
4) миграционный метод, при котором под действием ката-
лизатора происходит перемещение атомов в молекуле кремний-
органического соединения.
В принципе все эти методы представляют одинаковую цен-
ность и один метод дополняет другой. В этой бурно развиваю-
щейся области химии непрерывно предлагаются все новые ме-
тоды. Несмотря на это, в 1951 г. в промышленности примени
лись только два метода: прямой метод, по которому работала
фирма General Electric Со. и ее филиалы, и метод замещения
с помощью реактива Гриньяра, применявшийся фирмой Dow
Corning Corporation и ее филиалами.
Рассмотрим механизм этих двух реакций и их использо-
вание в лабораторных и промышленных условиях.
Прямой синтез. Этот метод разработан Роховым. Галоид-
ные алкилы или арилы в жидкой или газообразной форме ре-
агируют с металлическим кремнием в присутствии катализатора
или без него; образуется смесь галоидалкил (арил)силанов
R3SiX; R2SiX2 и RSiX3. Состав этой смеси зависит от темпера-
туры реакционной массы, типа применяемого катализатора,
способа смешения его с кремнием и, вероятно, от длительно-
сти контакта.
Реакцию можно направить на преимущественное образова-
ние диалкилдигалоидсиланов R2SiX2 (где X — обычно хлор
или бром); для этого следует, в частности, поддерживать воз-
можно более низкую температуру реакции (от 275 до 375°).
Из диалкилдигалоидсиланов R2SiX2 можно в случае необ-
ходимости получить по методу Гриньяра соединения R3SiX
или R<Si.
На практике порошкообразный кремний, смешанный с ката-
лизатором, нагревают в реакционной трубке, через которую
пропускают пары галоидалкилов. Получающиеся продукты
реакции конденсируют и разделяют кремнийорганические
галоидпроизводные перегонкой.
502
Гл. XV. Полиорганосилоксаны
Выбор катализатора зависит от радикала R. Для метиль-
ного радикала очень .подходит медь, для фенильного — се-
ребро.
Синтез метилхлорсил анов по этому методу был изучен
Гарди21 в теддингтонсиой лаборатории (Англия).
Получаемые органохлорсиланы представляют собой бес-
цветные- подвижные низкокипящие жидкости, очень чувстви-
тельные к влаге; они оказывают разрушительное действие на
прокладки и уплотнения аппаратуры; применимы наждачные
прокладки, пропитанные вазелином.
Смесь полученных продуктов разделяют перегонкой. Раз-
делить органохлорсиланы очень трудно, так как они кипят при
очень близких/температурах (см. табл. 28). Например, после
трехкратной перегонки метилтрихлорсилана получается про-
дукт не более 92 %-ной чистоты.
ТАБЛИЦА 28
Физические константы некоторых органогалоидсиланов
Название Формула Темпера- тура плавления °C Температура кипения °C
Триметилхлорсилан ..... (CH3)3SiCl —40 57,6
Трифенилхлорсилан (C6H5)3SiCl 111 365
Трибензилхлорсилап (C6HsCH2)3SiCl 141 300—360 (при 100 мм)
Триметилбромсилан . (CH3)3SiBr — 80
Трифенилбромсилан . (CeH5)3SiBr 120 —
Диметилдихлорсилан (CH3)2SiCl2 —86 70
Диэтилдихлорсилан (C2H5)2SiCl2 — 129
Дивинилдихлорсилан .... (CH2=CH)2SiCl2 — 118
Дифен илдихлорсилан (C6H5)2SIC12 —22 303
Дифенилдибромсилан (СбН5)251Вг2 — 180 (при 12 мм)
Дибензилдихлорсилан (C6H5CH2)2Si Cl2 51 243 (при 100 мм)
Метилтрихлорсилан ...... CH3SiCl3 — 66
Этилтрихлорсилан QHeSiCl., — 100
Изоамилтрихлорсилан z-CsH^SiClg — 56 (при 17 мм)
Фепилтрихлорсилан CeHgSIClg •—- 199
Получение органогалоидсиланов
503
Метилхлорсиланы легче разделить после перевода их в ме-
тилалкоксисиланы, сильнее различающиеся по температурам
кипения, лучше поддающиеся переработке и легче транспор-
тируемые.
Метилалкоксисиланы получают взаимодействием метилхлор-
силанов со спиртом по реакции:
2ROH + (CH3)2SiCl2 (CH3)2Si(OR)2 + 2HCi
Реакцию трудно довести до конца, так как НС1 реагирует со
спиртом с образованием воды, которая вызывает гидролиз
алкилхлорсиланов. Следовательно, необходимо связывать хло-
ристый водород по мере его образования, например, диметил-
анилином. При обычной температуре в присутствии инертного
растворителя, такого, как, например, петролейный эфир (темп,
кип. 40°), можно осуществить конверсию хлорпроизводпых на
90%. Чистые метилтриэтокси- и диметилдиэтоксисиланы полу-
чаются этим путем довольно легко.
Органоалкоксисиланы можно получать также непосредствен-
но взаимодействием ортокремневых эфиров с алкилпроизводным
цинка или реактивом Гриньяра в отсутствие эфира2.
Метод замещения. Первые органогалоидсиланы были полу-
чены с помощью органических соединений цинка, ртути или
алюминия или по реакции Вюрца с натрием. Киппинг применил
для этой цели только что появившийся в то время реактив
Гриньяра, оказавшийся наиболее удобным. Одна из реакций
этого классического синтеза такова:
SiCl4 -с 2RMgBr -► R2SiCl2 + 2MgBrCl
По этой реакции Киппинг получил в 1912 г. фенилхлорси-
ланы24.
Вместо алкилмагнийбромидов можно применять значитель-
но более дешевые хлорпроизводные, которые, однако, полу-
чаются со значительно большим трудом и менее реакционно-
способны10. При получении фейилмагнийгалогенидов использо-
вание хлорбензола вместо бромбензола создает дополнитель-
ные преимущества, а именно: позволяет вести реакцию в отсут-
ствие огнеопасного диэтилового эфира, так как фенилмагний-
хлорид растворяется в избытке хлорбензола.
Разделение и гидролиз органогалоидсиланов. Каким бы спо-
собом ни были получены органогалоидсиланы, при их перера-
ботке возникают одни и те же проблемы, связаннные с пере-
гонкой, гидролизом и конденсацией продуктов гидролиза.
Переработка и перегонка органогалоидсиланов является одной
из самых сложных проблем. Работу необходимо вести в совер-
шенно безводной среде, чтобы избежать выделения хлористого
504
Гл. XV. Полиорганосилоксаны
водорода; только при этом условии можно применять аппара-
туру из обычной малоуглеродистой стали.
Часто вместо органогалоидсиланов предпочитают приме-
нять органоалкоксисиланы, получаемые действием спирта на
органогалоидсиланы и легче поддающиеся переработке, разде-
лению и транспортированию.
Гидролиз этил- и изобутилэтоксисиланов детально изучал
К. А. Андрианов2.
Для последующей переработки кремнийорганических поли-
меров применяется обычная аппаратура промышленности пла-
стических масс; возможные затруднения связаны с необходи-
мостью вести процесс при более высоких температурах. В част-
ности, широкому распространению эмалей на основе полиоргано-
силоксанов, обладающих несомненными достоинствами, препят-
ствует высокая температура их сушки, часто недостижимая
в сушильных камерах, применяемых для сушки обычных эма-
лей.
Получение алкилсиланолов33
В настоящее время из существующих методов получения
алкил сил андиолов в промышленности применяются два. Фирма
Dow Corning Corporation применяет метод Гриньяра, а фирма
General Electric Со. и ее филиалы (в частности, Rhone Pou-
lenc — единственный производитель полисилоксанов в Европе)
в 1951 г. применяла прямой синтез. В будущем можно ожи-
дать внедрения в промышленность новых методов, например
метода присоединения к олефинам.
Прямой синтез. Режим проведения прямого синтеза должен
быть изучен применительно к каждому радикалу в отдельности;
протекающие реакции во всех случаях одни и те же, но приме-
няемые катализаторы и условия проведения реакции различны.
Для получения диметилсиландиолов газообразный хло-
ристый метил примерно при 250° пропускают над брикетиро-
ванной смесью кремния (90%) и меди (10%). Хлористый ме-
тил получают из метана или метилового спирта, кремний —
восстановлением кремнезема. Суммарная реакция может
быть изображена следующим образом:
SiO2 + 2С + 2СН3ОН (CH3)2Si(OH)2 + 2СО
На рис. 55 приведена принципиальная схема получения
полидиметилсилоксанов.
Этот метод проще метода Гриньяра. Для хлорирования
кремния не требуется свободный хлор, а хлористый водород,
образующийся при гидролизе хлорсиланов, полностью расхо-
дуется на реакцию с метиловым спиртом.
Получение органогалоидсиланов
50&
При правильном ведении процесса
щественно диметилдихлорсилан; однако
зуются и другие метилхлорсиланы.
получается преиму-
наряду с ним обра-
Кремнезем
Кокс
Метанол
Хлористый
водород
Кремний
Катализатор
Хлористый
метил
Смесь
метилхлорсиланов
S1C14 И CH3SiCl3
На последующее
метилирование
Побочные продукты
(CH3)3SiCl; CH3SU ICI2 и т. д.
Вода
(CH3)2SiCl2
Возвратный хлористый водород
Диметилсиландиол
(частично сконденсированный)
Пол идиметилсилоксан
Рис. 55. Получение полиднметилсилоксана прямым синтезом.
Пр.и синтезе полидифенилсилоксанов в качестве катализатора-
применяется серебро. Процесс ведут при 375—425°, применяя
большой избыток хлорбензола, который служит для разбавления
и извлечения высококипящих фенилхлорсиланов. Избыток хлор-
бензола рекуперируется и возвращается в процесс. Суммарная
реакция имеет следующий вид:
SiO2 + 2С 4- 2С6Н6 + 2С12 + Н2О —- (CeHs)2SiO + 4НС1 + 2СО
506
Гл, XV. Полиорганосилоксаны
Принципиальная схема получения полидифенилсилоксана
приведена на рис. 56.
Кремний
Хлорбензол
Катализатор
На последующее
фенилирование
S1C14 и CeHBSiCl3
Смесь
фенилхлорсиланов
(раствор)
(C6H6)2SiCl2
Хлорбензол (рекуперированный)
Вода
Хлористый водород
(побочный продукт)
Дифенилсиландиол
I
Полидифенилсилоксан
Рис. 56. Получение полидифенилсилоксана прямым синтезом.
Прямой синтез введен только в 1940 г. и требует еще усовер-
шенствования, в то время как по методу Гриньяра накоплен
многолетний опыт. Прямой синтез применяется преимуществен-
но для получения диалкилдигалоидсиланов.
Метод замещения (метод Гриньяра). Метод замещения, как
более гибкий, с успехом применяется для промышленного по-
лучения алкилгалоидсиланов с любым алкильным или ариль-
ным радикалом. Этим методом с одинаковым успехом можно
получать как алкилтригалоид- и триалкилгалоидсиланы, так и
диалкилдигалоидсиланы. Но диметилсиландиолы или дифенил-
силандиолы, которые главным образом требуются в настоящее
время, при их получении по методу Гриньяра обходятся доро-
Получение органогалоидсиланов
507
же, чем при получении прямым синтезом. Это объсняется глав-
ным образом многостадийностью процесса и необходимостью
исходить из четыреххлористого кремния или этилсиликата.
Схема процесса приведена на рис. 57.
Хлор
Углеводород
Кремнезем
Кокс
Галоидалкил
Магний
-► SiCU
Растворитель
Магнийгалоид-
алкил
I
Алкилгалоидсиланы
и растворитель -
(неочищенная смесь)
MgCl*
Ре генерировании й
хлор
SiCl4 и RSiCl3
Побочные продукты
R3SICI; H4S1
R^iClg
Вода
НС1
R2SiO
(частично сконденсированный)
Полисилоксан
Рис. 57. Получение полиорганосилоксанов по методу Гриньяра.
508
Г л. XV. Полиорганосилоксаны
Типы полиорганосилоксанов и их свойства
Независимо от степени 'полимеризации <и физического со-
стояния полиорганосилоксаны' обладают рядом замечательных
свойств: термостойкостью, морозостойкостью, устойчивостью к
действию химических реагентов и окислению, гидрофобностью
и ценными диэлектрическими свойствами.
Строение линейных полисилоксанов выражается общей фор-
мулой R3SiO(R2SiO)xSiR3, а циклических полисилоксанов —
более простой формулой (R2SiO)x.
Линейные полисилоксаны образуются обычно в результате
межмолекулярной конденсации диалкилсиландиолов, получае-
мых гидролизом диалкилдигалоидсиланов:
R R
2R2Si(OH)t -> НО—Si—О—Si—ОН Н2О
I I
R R
R R R
। 1 1
3R2Si(OH)2 -»• НО—Si—О—Si—О—Si—ОН + 2Н2О
I I I
R R R
И т. д.
Циклические полисилоксаны (R2SiO) v также образуются в
результате конденсации.22 Выделены тример, тетрамер, пента-
мер, гексамер и т. д. вплоть до циклов, состоящих из 18 и даже
большего числа звеньев.
тример
тетрамер
Силоксаны сетчатой (пространственной) структуры полу-
чают введением наряду с бифункциональными также трех-
функциональных алкилсиланолов. Такие полимеры построены
Типы полиорганосилоксанов и их свойства
509
из цепей или циклов, связанных кислородными мостиками,
например:
; ' R R R R R R
---Si—о—Ь—о—Si—О—Si—О—ii—О—Si—О—Si—О-----
В подобных полимерах среднее отношение числа органиче-
ских радикалов к числу атомов кремния (R : Si) уже не равно
двум, как в линейных полисилоксанах, а всегда меньше двух.
Трехфункциональные кремнийорганические соединения
можно получать двумя способами:
1. В соединениях, для которых R : Si=2, удаляют окислением
или гидролизом группы R и замещают их атомами кислорода.
2. Подвергают гидролизу смесь диалкилдигалондсиланов и
моноалкилтригалоидсиланов (или соответствующих алкокси-
производных), взятых в требуемом соотношении:
R2SiCl2-f-2II2O R2Si(OH)24-2HCl
RSiClg + ЗН2О RSi(OH)s 4- 3HCI
При дальнейшей конденсации протекают следующие реакции: .
2xRtSi(OH)s 4- xRSi(OH)3
- R R R
I I 1
—Si—О—Si—О—Si—
I I I
R О R
I
4- -g- HtO
Из этого краткого обзора видно, что химия полиорганосил-
оксанов, которая только начинает развиваться, может дать
такое же многообразие соединений, как химия чисто органи-
ческих высокополимеров. Радикал R в этих соединениях может
представлять собой любую алкильную или арильную группу, «
510
Гл. XV. Полиорганосилоксаны
в лабораторном масштабе уже получены соединения с различ-
ными радикалами. Промышленные силиконы содержат почти
исключительно метильные или фенильные радикалы или их
сочетание. Поэтому исследователи до последнего времени за-
нимались преимущественно химией метил- и фенилсилоксанов.
Алкилсилоксаны. Лучше всего изучен класс метилсилокса-
нов, включающий самые простые соединения, наиболее близ-
кие к силикатам, так как в эти соединения входят самые корот-
кие органические радикалы и в них нет связей С—С. Линейные
метилсилоксаны даже при наибольшей возможной длине це-
пи представляют собой масла. Метилсилоксаны пространствен-
ного строения, и молекулы которых входят линейные и цикли-
ческие цепи, являются смолами. В отдельную группу можно
выделить эластомеры (силастики), имеющие значительно более
высокий молекулярный вес и, по-видимому, линейное строение.
Отметим, наконец, «пружинящую шпаклевку» (bouncing
putty) со своеобразными свойствами: пластичную при медлен-
ном увеличении напряжения и ломкую при действии сильных
ударов.
Метилсилоксановые масла. Как уже указывалось, при гид-
ролизе диметилдихлорсилана или диметилдиэтоксисилана обра-
зуются цепи, состоящие из структурных звеньев
СН3
I
—Si—О—
I
СН3
с реакционноспособными гидроксильными группами на концах
цепи. Чтобы получить цепи определенной однородной длины,
необходимо блокировать их концы монофункциональными звень-
ями, например триметилсилоксановыми (продукт гидролиза
триметилхлорсилана):
СН3
I
СН,—Si—О—
I
СН3
Метилсилоксановые масла общей формулы23
(CH3)3Si О
сн3
I
-Si—О—
I
. СН3
Si(CH3)3
п
получаются при гидролизе смеси диметилдихлорсилана с три-
метилхлорсиланом или диметилдиэтоксисилана с триметилэток-
сисиланом. При частичном гидролизе диметилдиэтоксисилана
Типы полиорганосилоксанов и их свойства
511
могут образоваться цепи с концевыми этоксильными радика-
лами2, “.
В настоящее время обычно отдают предпочтение так назы-
ваемому методу «каталитического диспропорционирования»28.
Он заключается в смешении продукта гидролиза диметилди-
хлорсмлана с соответствующим количеством гексаметилдисил-
оксана, образовавшегося при гидролизе триметилхлорсилана.
Добавляют серную кислоту, которая вызывает разрыв связей
и беспорядочное образование новых связей до тех пор, пока
не будет достигнуто равновесное распределение длин цепей.
После этого кислоту нейтрализуют. Этим методом удается
получать масла, состоящие исключительно из линейных поли-
меров.
Свойства метилсилоксановых масел детально изучены теоре-
тически и практически5’13Л4,1в’40.Подобно всем полисилоксанам,
силоксановые масла обладают исключительной термостой-
костью и стойкостью к окислению. Но самым характерным их
свойством является малая зависимость вязкости от температу-,
ры. Например, если сравнить полисилоксановое масло с мине-
ральным маслом той же вязкости (100 сантистоксов при +38°),
то окажется, что при охлаждении до —37° вязкость полисил-
оксанового масла возрастает в 7 раз, а вязкость минерального
масла в 1800 раз.
Метилсилоксановые смолы33. Эти термореактивные смолы
получаются взаимодействием диметилсилоксановых, мономе-
тилсилоксановых и, в известных случаях, четырехфункциональ-
ных мономеров. Существует три метода получения метилсилок-
сановых смол:
1. Гидролизуют диметилдихлорсилан (или его алкоксиль-
ное производное) и продукты гидролиза окисляют воздухом в
присутствии катализатора до достижения заданного отноше-
ния СНз/Si.
2. Гидролизуют смесь диметилдихлорсилана, метилтрихлор-
силана и четыреххлористого кремния, взятых в соответствую-
щих отношениях.
3. Проводят реакцию между четыреххлористым кремнием и
магнийхлорметилом до достижения требуемого отношения
СНз/Si и гидролизуют продукт реакции.
Преимуществом первого метода является возможность не-
посредственного регулирования конечной вязкости. Этот метод
может применяться в сочетании с третьим методом.
По второму методу гидролиз необходимо проводить в рас-
творителе, способном растворять и хлорсиланы и воду, чтобы
смола оставалась в растворе, так как в противном случае в
присутствии соляной кислоты может произойти гелеобразова-
ние. Преимуществом метода является возможность проведения
512
Гл. XV. Полиорганосилоксаны
реакции при более высокой температуре. Этот метод выгодно
применять для получения смол с малым отношением CH3/Si,
когда во избежание гелеобразования процесс приходится вести
в разбавленном растворе и на холоду. При его применении со-
храняется больше метильных групп, чем при первом способе,
но требуется предварительное разделение хлорсиланов, свя-
занное с большими трудностями.
Третий метод самый простой, но состав смеси трудно регу-
лировать.
Свойства метилсилоксановых смол определяются величиной
отношения СНз/Si, которое всегда меньше двух. При
СНз/Si <1,2 эти смолы представляют собой клейкие, вязкие
жидкости, которые при обычной температуре могут быть пре-
вращены в стекловидные твердые и хрупкие массы. При
СНз/Si < 1,5 смолы, полученные по второму методу, это—бес-
цветные жидкости с удельным весом от 1,20 до 1,06 г/см3. Для
-отверждения эти жидкости нагревают до 100°. Маслянистые
летучие продукты, для которых CH3/Si > 1,5 (в частности,
СНз/Si=7), желатинируются при 20° в течение времени от не-
•скольких дней до нескольких недель.
Метилсилоксановые смолы медленно окисляются на возду-
хе только при температуре выше 300°. Они обладают превос-
ходными диэлектрическими свойствами: диэлектрическая по-
стоянная равна, например, 3,7 при 26° и 2,6 при 56°, тангенс
угла диэлектрических потерь (при 60 гц) изменяется от 0,008
при 26° до 0,0045 при 56°.
Метилсилоксановые эластомеры. Они построены, по-видимо-
му, из очень длинных цепей. Получаются эти полимеры гидро-
лизом диметилдихлорсилана, причем их приготовление требует
'большой тщательности. Чтобы получить вещества, которые
действительно являются каучуками, необходимо подвергать
диметилдихлорсилан весьма тонкой очистке.
Прочие алкил сил оксаны 1,а, кроме этилсил оксанов, как пра-
вило, представляют собой медленнее отверждающиеся, более
мягкие и леУче окисляющиеся смолы.
Арилсилоксаны. Арилсилоксаны резко отличаются от алкил-
силоксанов. В то время как при гидролизе диметилдихлорси-
лана получается смесь полимеров, из которой невозможно вы-
делить мономерный диметилсиландиол, гидролиз дифенилди-
хлорсилана дает дифенилсиландиол (C6H5)2Si(OH)2 почти с
теоретическим выходом. Дифенилсиландиол можно перекрис-
таллизовать, после чего он плавится33 при 148°. Конденсирует-
ся он только при температуре выше комнатной, причем сначала
образуются циклы, а при температуре выше 200° получаются
некристаллизующиеся смолы. Эти смолы готовят из дифенил-
“силандиола, фенилсилантриола или их смесей.
Применение полиорганосилоксанов для красок
513
Алкиларилсилоксаны. Алкилсилоксаны с большим значе-
нием отношения R/Si являются маслянистыми жидкостями или
гелями33. В обычных метилсилоксановых смолах отношение
R/Si составляет примерно 1,5, в бутил- и бензилсилоксановых —
около 1,0. В то же время арилсилоксаны с высоким отноше-
нием R/Si представляют собой плавкие и растворимые стекло-
видные вещества, слишком хрупкие для использования, напри-
мер, в качестве покрытий.
Свойства веществ этих двух классов можно сочетать, но
это не достигается при простом смешении, так как они зачастую
несовместимы. Зато путем совместной поликонденсации можно
получить полисилоксаны с одним алкильным и одним арильным
радикалом при одном и том же атоме кремния 26127. Преиму-
ществом совместной конденсации является возможность осу-
ществить большое число комбинаций и, следовательно, достичь
большого разнообразия свойств (разной эластичности, термо-
пластичности и механической прочности). Таким путем были
Получены метилфенилсилоксаны 26 27. Наилучшие результаты
были достигнуты при почти эквивалентном соотношении обеих
групп и среднем общем отношении R/Si около 1,8. Чтобы пред-
отвратить хрупкость, вводят в избытке метильные группы и
таким путем получают метилфенилсилоксановые смолы, пре-
восходящие по диэлектрическим свойствам метилсилоксановые
смолы.
Исследование процессов окисления при повышенных темпе-
ратурах показало, что полиметилфенилсилоксаны более устой-
чивы, чем полиметилсилоксаны. |Полиметилфенилсилоксаны
при 225° не претерпевают заметного изменения; при 250° их
вязкость увеличивается и выделяется заметное количество ле-
тучих продуктов окисления. Инфракрасные спектры поглоще-
ния позволяют одновременно определить структуру силоксанов
и изменения, происшедшие в процессе их окисления. Эти спект-
ры показывают, что количество метильных групп постепенно
уменьшается, в то время как количество фенильных групп
остается почти постоянным.
Применение полиорганосилоксанов для красок6,7,10~12,20,29,32
В качестве связующего для красок применимы, по-видимо-
му, полимерные алкиларилсилоксаны, например фенилметил-
силоксаны. Несомненно, это первоклассное сырье для лакокра-
сочной промышленности, точнее для красок, от которых тре-
буется тепло- и морозостойкость, а также для обычных декора-
тивных красок. Их применение ограничивается двумя факто-
рами: с одной стороны, сравнительно высокой стоимостью
(около 6000 франков в 1951 г. и около 2500 франков в 1955 г.),
33—156
514 Гл. XV. Полиорганосилоксаны
с другой стороны, необходимостью либо модернизировать су-
ществующее сушильное оборудование, на котором нельзя до-
стигнуть высоких температур, либо разработать новые методы,
позволяющие понизить температуру сушки.
Краски на основе силоксановых смол можно перетирать на
обычных мельницах. Связующее обычно представляет собой
сочетание твердого полисилоксана с пластифицирующей поли-
силоксановой смолой. В эти краски можно вводить обычные пиг-
менты; для термостойких красок должны применяться пигменты,
стойкие к высоким температурам, например: титановые белила,
литопон, сажа, красная окись железа, красный кадмий и т. д.
Силиконовые краски имеют чрезвычайно чистые оттенки:
применяя их, можно с успехом использовать дешевые белые
пигменты.
В 1939 г. К- А. Андрианов2 сделал сообщение о полисилок-
санах, дающих пленки, неплавкие и нерастворимые при обыч-
ной температуре. Большинство полисилоксановых красок все
же приходится подвергать горячей сушке при очень высоких
температурах (300°). Для ускорения высыхания к ним можно
добавлять некоторые катализаторы, например соли железа,
кобальта или цинка.
Полисилоксановые краски обладают рядом свойств, недо-
стижимых для других красок. Они прекрасно противостоят
действию высоких температур (например, устойчивы при 530°
в течение нескольких часов или при 260° в течение многих сотен
часов), морозостойки, стойки к атмосферным влияниям, хими-
ческим агентам и пищевым продуктам, на них не налипает
пыль12. Эти краски не только противостоят действию высоких
температур, но даже не обугливаются при превышении темпе-
ратуры разложения, а разлагаются с образованием кремния.
Поэтому, нагревая полисилоксановые краски, пигментирован-
ные алюминиевым порошком, при температуре выше 370°,
можно получить прочно связанные с основой термостойкие и
совершенно не разрушающиеся покрытия сетчатой структуры,
построенной из атомов кремния и алюминия7. Таким путем
можно разрешить проблему, считавшуюся до сих пор нераз-
решимой: заменить сталь легкими сплавами. Полисилоксанами
можно «эмалировать» алюминий, что представляет большой
интерес в авиастроении, где для облегчения веса самолетов
применяется листовой алюминий, а также в производстве бы-
товых нагревательных приборов.
Белые эмалевые краски на основе полисилоксанов могли
бы с успехом заменить стекловидные эмали, но на существую-
щих установках нельзя проводить сушку при достаточно высо-
ких температурах6. Между тем таким путем можно было бы
уменьшить толщину, а следовательно, и вес применяемого
Совместимость полиорганосилоксанов
515
листового металла, наносить только один слой «эмали» вместо
трех и таким образом сократить время, требуемое для нанесения
эмалей. Таким образом, полное «эмалирование» можно было
бы провести за 1 час при 300°.
Полисилоксаны могли бы служить заменителями фарфора,
но они не обладают для этого достаточным сопротивлением к
.истиранию.
Ввиду высокой стоимости полисилоксанов краски на их
основе применяются в США преимущественно для специальных
целей, когда существенную роль играет ,их термостойкость,
морозостойкость и стойкость к химическим реагентам: в авиа-
строении, для защиты химической аппаратуры, покрытия кухон-
ных плит, дымовых труб, кипятильников, моторов. Но при уме-
ренной стоимости полисилоксаны могли бы с успехом приме-
няться для окраски автомобилей, холодильников и кухонной
посуды, 'больничного оборудования, бензонасосов, сельскохо-
зяйственных машин и т. д.
Совместимость полиорганосилоксанов с другими полимерами
Недостатком первых полисилоксановых красок являлась их
небольшая стойкость к некоторым растворителям; она была
повышена путем применения некоторых добавок. Для улучше-
ния устойчивости к растворителям, понижения температуры
сушки и удешевления красок было предложено совмещать поли-
силоксаны с обычными смолами. Некоторые полисилоксаны со-
вмещаются с алкидными, феноло-формальдегидными, мочевино-
или меламино-альдегидными смолами6, и-29, но не совмещаются
с хлоркаучуком, нитроцеллюлозой и виниловыми .полимерами.
Силоксановые полимеры применяются также в качестве
вспомогательных материалов в технологии красок и покрытий.
Так, в производстве фенольных смол можно использовать сил-
оксановые .масла в качестве пеногасителей. Они препятствуют
также образованию пузырей при нанесении красок кистью. С
другой стороны, введение некоторых силоксановых масел в
минимальных количествах (порядка 1 : 10 000) предотвращает
седиментацию красок, готовых к употреблению, и образование
так называемой «апельсиновой корки». Поразительные резуль-
таты были достигнуты при окраске методом пульверизации
конторской мебели или железнодорожных вагонов полисилок-
сановыми красками — в этих красках не наблюдается мигра-
ции пигментов. Наконец, нанося на наружные стены домов
1%-ный раствор полисилоксановой смолы в низкокипящем рас-
творителе, можно предохранить их от разрушения, в частности
•от выветривания. После такой обработки стены легко поддают-
ся окрашиванию обычным способом.
33*
516
Гл. XV. Полиорганосилоксаны.
ЛИТЕРАТУРА
(по май 1951)
Обзорные статьи отмечены звездочкой
1. Т. А 1 f г е у, F. J. Honn, Н. Mark, J. Polymer Sci., 1946, 1, 102.
2 К. Андрианов, ЖОХ, 1938, 8, 552, 557; С. А., 1938, 32, 7892; ЖОХ,
1938, 8, 558, 562; ЖОХ, 1938, 8, 969; ЖОХ, 1938, 8, 1255; ЖОХ, 1946,
16, 633; ЖОХ, 1947, 17, 1522; ПОХ, 1939, 6, 203; ДАН СССР, 1940, 28,
нов. сер., 8, 66.
3. *F. Appell, Rev. Gen. electr., 1946, 55, 99; P. P. V., 1946, 22, 145,
190; Assoc, anciens eleves E. P. С. I., 1948, 238.
4. E. A. Bassett, H. G. Emblem, M. Frankel, D. Ridge,
J. Soc. Chem. Ind., 1948, 67, 177.
5. О. K. Bates, Ind. Eng. Chem., 1949, 41, 1966.
6. Chicago Club, Off. Dig., 1948, 286, 876.
7. E. G. Bromstead, M. A. Glaser, Org. Finishing, 1948, 9, Xs 3, 21.
8. *M. Der i b ёгё, M. de Buccar, Ed. par E. L. P. I., Bruxelles, 1949
Сборник статей из Chimie des Peintures, 1948, 11, 38, 70, 134, 183, 284,
326, 358; 1949, 12, 118, 300, 353, 364, 455; Ind. Plastiques, 1946, 2, 8, 49;
Rev. Gen. Electr., 1948, 57, 93; Брошюра, Paris, Elzevir, 1947. •
9. *G. A. Burkhard, E. G. R oc h ow, H. S. В о о t h, J. H a r 11,
Chem. Rev., 1947, 41, 97.
10. *G. Champetier, Mem. Soc. Ing. Civils Fr., 1947, 100, 432; La Na-
ture, 1949, 38; Bull. Soc. Encour, Industr. Nationale, 1950, 138.
11. T. Chapman, J. Oil Col. Chem. Ass., 1948, 31, 205.
12 *W. R. Collings (Dow Corning Corp.), Paint Oil Chem. Rev., 1949, 32,
121; Off. Dig., 1949, 296, 625.
13 С. C. Currie, B. F. Smith (Dow Corning Corp.), Ind. Eng. Chem.,
1950, 42, 2457.
14 С. C. Currie, M. С. H о m m e 1 (Dow Corning Corp.), Ind. Eng.
Chem., 1950, 42, 2452.
15. H. J. Fletcher, M. J. Hunter, J. Am. Chem. Soc., 1949, 71,
Xs 8, 2918.
16. H. W. Fox, P. W. Taylor, W. A. Zisman, Ind. Eng. Chem.,
1947, 39, 1401.
17. *R. Gay, Ind. Plast. Mod., 1951, 3, 8, 13, 15.
18. *G. Genin, Electricite Fr., 1946, 30, 199.
19. *A. G i b e 1 1 o, Ind. Plastiques, 1946, 2, 358; 1947, 3, 6.
20. M. A. Glaser, Product. Eng., 1950, 21, Xs 2, 109.
21. *D. V. N. Hardy, Paint Technol., 1949, 14, 114; J. Oil Col. Chem.
Ass., 1949, 32. 202.
22. M. J. Hunter, J. F. Hyde, E. L. Warrick, H. J. Flet-
cher (Dow Corning Corp.), J. Am. Chem. Soc., 1946, 68, 667.
23. M. J. Hunter, E. L. Warrick, J. F. Hyde, С. C. Currie
(Dow Corning Corp.), J. Am. Chem. Soc., 1946, 68, 2284.
24. F. S. Kipping и сотр., Proc. Chem. Soc., 1899, 15, 174; J. Chem.
Soc., 1901, 79, 449; Proc. Chem. Soc., 1904, 20, 15; J. Chem. Soc., 1907,
91, 209, 717; 1908, 93, 198, 439, 457, 2004, 2090; 1909, 95, 69, 302, 488;
Proc. Chem. Soc., 1910, 26, 65; J. Chem. Soc., 1910, 97, 142, 755; 1911,
99, 138; Proc. Chem. Soc., 1912, 28, 243; J. Chem. Soc., 1912, 101, 2106,
2108, 2123, 2141, 2156, 2353; 1914, 105, 40, 484, 679; 1915, 107, 459; 1921,
119, 647, 830, 848; 1923, 123, 2190, 2598, 2830; 1924, 125, 2291, 2616; 1927,
104, 2719, 2728,2734; 1928, 1427, 1431; 1929, 357, 360, 1176; 1180- 2545;
1930, 1020 1029; 1931, 1290, 2774, 2840, 1932, 2200, 2205; 1933, 1040;
1935. 1085. 1088; Proc. Roy. Soc., 1937, A139; J. Chem. Soc., 1944, 81.
Совместимость полиорганосилоксанов
517
25. Н. W. Kohlschutter, Fortschr. Chem. Forsch. Dtsch., 1949, 1, Ks 1, 2.
26. R. N. Lewi s, (Gen. Electr. Co), J. Am. Chem. Soc., 1948, 70, 1115.
27. С. M. Murphy, С. E. Saunders, D. C. Smith (Naval Research
Lab. Washington), Ind. Eng. Chem., 1950, 42, 2462.
28. W. P a t п о d e, D. F. W i 1 с о c k, J. Am. Chem. Soc., 1946, 68, 358.
29. J. R. Patterson, Ind. Eng. Chem., 1947, 39, 1376.
30. M. P e у г о t, Ind. Plastiques, 1947, 3, 201, 246.
31. *H. W. Post, Silicones and other organic silicon compounds, Reinhold
Publich Corp., New York, 1949.
32. J. W. Reynolds (Gen. Electr. Co.), Off. Dig., 1950, 310, 896.
33. *E. G. R о c h о w (Gen. Electr. Co.), An Introduction to the Chemistry
of Silicones, New York, Wiley, 1946.
34. T. G. a. E. G. Rochow, Science, 1950, 111, 271.
35. W. L. Roth, J. Am. Chem. Soc., 1947, 69, 474.
36. L. H. Sommer, E. W. P i e t r u s z a, F. C. Whitmore,
J. Am. Chem., Soc., 1948, 70, 484.
37. L. W. Spooner, Prod. Eng. U. S. A., 1950, 21, № 1, 90.
38. Union Internationale de Chimie Pure et Appliquee, Amsterdam, 5—10 sep-
tembre 1949, Comptes rendus de la XV-e Conference, p. 27.
39. F. S. Whitmore, L. H. Sommer, J. Gold, J. Am. Chem.
Soc., 1947, 69, 1976.
40. D. F. W i 1 с о c k, Meeh. Eng., 1944, 66, 736; 1945, 67, 202; Gen. Electr.
Rev., 1946, 49, 14; J. Am. Chem. Soc., 1946, 68, 691; 1947, 69, 477.
ГЛАВА XVI
КАНИФОЛЬ И ЕЕ ПРОИЗВОДНЫЕ
Р. Ломбар
Канифоль является самым важным из так называемых «смо-
ляных» продуктов, в число которых входят: сосновая смола,
скипидар, смоляные кислоты, сосновый деготь, талловое мас-
ло и т. д. Смоляные продукты получаются главным образом из
сосны (а также из других хвойных деревьев).
Основными способами получения смоляных продуктов яв-
ляются: подсочка деревьев, экстрагирование смолистой древе-
сины растворителями, приготовление бумажной массы из смо-
листой древесины (при этом в качестве побочных продуктов
получают талловое масло, сульфитную и сульфатную живи-
цу), сухая перегонка смолистой древесины, ведущая к образо-
ванию смолы.
Подсочка. Если сделать надрез на растущей сосне, из него
вытекает смолисто-маслянбе вещество (живица), сохраняющее
консистенцию жидкости или частично кристаллизующееся.
Кристаллизующиеся вещества называются сосновой смолой (га-
липот), а некристаллизующаяся прозрачная жидкость — ски-
пидаром или бальзамом (венецианский скипидар, канадский
бальзам). Остающийся после дистилляции живицы нелетучий
осадок и является канифолью.
Совокупность операций, заключающихся в регулярном про-
ведении надреза деревьев и в сборе живицы, называют под-
сочкой.
Основные сорта сосны, которые подвергаются подсочке:
1. Сосна морская (Pinus maritima); широко используется во
Франции (произрастает в гасконских лесах).
2. Сосна (Pinus halepensis)', произрастает на всем сре-
диземноморском побережье, во Франции, Греции, Испании
и т. д.
3. Сосна черная австрийская (Pinus laricio austriaca)',
произрастает в Австрии.
4. Сосна итальянская (Pinus piuea); используется в не-
большом количестве в Италии и Португалии.
5. Сосна Pinus silvestris произрастает в Германии;
используется в небольшом количестве и дает невысокий выход
продукции.
Состав канифоли
519
6. Сосна болотная (Pinus palustrisY, произрастает в США.
7. Сосна караибская (Pinus caribaea, Pinus heterophylla)',
произрастает также в США.
8. Сосна (Pinus longifolia); произрастает на южном склоне
Гималаев.
Различные сорта канифоли, полученные из разных сортов
сосны, с точки зрения их применения очень сходны. Канифоль
обычно сливают прямо в деревянные бочки емкостью 400 кг
чистого веса.
Экстрагирование смолистой древесины. Когда сосна сруб-
лена или свалена бурей, пень и дерево, лежащее на земле,
полностью не сгнивают; остающиеся части такого дерева со-
ставляют так называемое смолье, или осмол. В южных лесах
США осмол содержит до 25% смолы; путем экстрагирования
растворителями из него извлекают скипидар (скипидар из ос-
мола) и канифоль (экстракционная канифоль). В лесах Евро-
пы смолистые пни не так богаты смолой и их экстракция не-
рентабельна.
Экстракционная канифоль не очень сильно отличается от
смолы, полученной из живицы.
ЛИТЕРАТУРА
I. V ё z е s, Dupont, Resines, -terebenthines, Bailliere ed., Paris, 1924’
2. Tschirch, Stock, Die Harze, Gebriider Borntrager ed., Berlin, 1933,
3. Lombard, Produits resineux: gemmes, colophanes, derives, Dunod ed.,
Paris, 1946.
4. Par de, Les coniferes, La Maison rustique ed., Paris, 1937.
5. Dupont, Les essences de terebenthine, Masson ed., Paris, 1926.
Состав канифоли
Для большей части областей применения канифоли все сор-
та сосны, из которых она добывается, равноценны; поэтому,
говоря о канифоли, обычно не уточняют ее происхождение.
Канифоль состоит на 90% и более из кислот и менее чем
на 10% из нейтрального вещества, составляющего неомыляе-
мую часть канифоли. Кислотное число стандартных сортов ка-
нифоли близко к 170; нестандартные продукты имеют более
низкое кислотное число.
Неомыляемые компоненты канифоли представляют собой
очень вязкую жидкость соломенно-желтого цвета, неустанов-
ленного состава; они составляют наиболее высококипящую
фракцию скипидара. Кислотные компоненты относятся к смо-
ляным кислотам, образующим большую группу соединений,
представители которой содержатся в различных природных
смолах.
520 Гл. XVI. Канифоль и ее производные
Главными компонентами различных сортов канифоли яв-
ляются три смоляные кислоты: абиетиновая, неоабиетиновая
и d-пимаровая. Большей частью в канифоли содержится также
четвертая кислота — изо-й-пимаровая. Кроме этих кислот, ка-
нифоль содержит продукты их окисления, а также в различных
соотношениях некристаллизующиеся кислоты не вполне уста-
новленного строения; их немного в канифоли, слитой в бочки,
но много в порошкообразной канифоли и в канифоли, нахо-
дившейся на воздухе.
Непосредственное исследование кислот, входящих в состав
канифоли, было связано со множеством трудностей, и химики
смогли установить состав канифоли лишь после того, как было
выяснено строение этих кислот, получивших название «смоля-
ных кислот»*. В процессе перегонки живицы смоляные кислоты
претерпевают различные изменения; изучение превращений,
которым они могут подвергаться при нагревании, позволяет
внести ясность в вопрос о составе канифоли.
Растворы канифоли довольно легко кристаллизуются, обра-
зуя смесь кристаллов, в которой преобладают треугольные
пластинки, характерные для абиетиновой кислоты (такая кри-
сталлическая форма не является, однако, доказательством чи-
стоты, так как возможна совместная кристаллизация). Кани-
фоль кристаллизуется при выдерживании ее расплава в тече-
ние продолжительного времени при температуре несколько ни-
же температуры ее плавления, а также при возгонке; полу-
ченные кристаллические смеси не удается разделить. Путем не-
посредственной кристаллизации канифоли нельзя выделить из
нее индивидуальные компоненты.
Вопрос о составе канифоли был несколько запутан различ-
ными авторами, утверждавшими, что канифоль состоит из ан-
гидридов l,2,s. Изучение смоляных кислот оказалось более пло-
дотворным для выяснения истинной природы канифоли.
Лоран в 1839 г. назвал пимаровой кислотой продукт, полу-
ченный при кристаллизации нз спирта4 смолы морской сосны.
Дювернуа (1868 г.) установил, что этот продукт образует в
водной среде пимарат натрия, кристаллизующийся в виде пер-
ламутровых чешуек, что дало возможность его очистить5.
Кальо (1874 г.) выделил из пимаровой кислоты правовращаю-
щую кислоту, которую он назвал декстропимаровой (теперь
ее называют rZ-пимаровой) кислотой6, а Вестерберг (1885 г.) вы-
делил из нее второй компонент, который он назвал левопимаро-
вой кислотой7. Это было подтверждено двадцать лет спустя
Колером8. Наконец, в 1921 г. Дюпон показал, что так называе-
мая «пимаровая» часть канифоли содержит только два упомя-
* Эти кислоты называют также «первичными кислотами» (acides primaires).
Состав канифоли
521
нутых выше компонента — d-пимаровую и левопимаровую кис-
лоты8. Таким путем эти компоненты оказались идентифициро-
ванными.
Идентификация других компонентов канифоли, не кристал-
лизующихся в виде натриевых солей из водной среды, оказа-
лась затруднительной. Только в 1948 г. Харрису и Сандерсону
удалось выделить новый компонент — неоабиетиновую кислоту10;
несколько позднее Ломбар и Фрей показали, что неоабиетиновая
кислота является постоянным и важным компонентом кани-
фоли11. Наконец, Харрис и Сандерсон выделили пятый компо-
нент, который они назвали изо-й-пимаровой кислотой12; послед-
няя, будучи выделена вначале из смолы Pinus palustris, была
впоследствии обнаружена в смолах других видов сосны.
В итоге из галипотов были выделены следующие пять чи-
стых смоляных кислот: левопимаровая, декстропимаровая, изо-
декстропимаровая, абиетиновая и неоабиетиновая. Ломбар (не-
опубликованная работа) количественно определил эти кислоты
поляриметрическим и спектрографическим методами и показал,,
что ими почти исчерпывается состав галипота. Состав галипо-
тов и канифоли, выделенных из некоторых видов сосны, при-
веден в табл. 29.
ТАБЛИЦА 29'
Состав (в %) галипотов и канифоли разных пород сосны
Кислоты Pinus mariiitna Pinus halepensis Pinus laricio austriaca Pinus silvestris
галипот канифоль галипот канифоль галипот галипот
Левопимаровая 35. 0 24 0 38 36
d-Пимаровая и изо-й-пимаровая 27 27 0 0 19 25
Абиетиновая . . 17 52 42 66 19 17 «
Неоабиетиновая . 21 21 34 34 24 22
К приведенным в таблице данным о составе галипотов необ-
ходимо добавить следующие данные:
1. Галипот не вполне идентичен совокупности найденных
кислот, но в первом приближении близок к ней, хотя соотно-
шения различных компонентов могут колебаться.
2. Приведенные в таблице данные являются усредненными;
в зависимости от времени года и породы сосны соотношения мо-
гут меняться, но компоненты те же.
3. Некоторые галипоты отличаются по содержащимся в них
компонентам; например, галипот сосны Pinus halepensis не
содержит d-пимаровой кислоты.
522
Гл. XVI. Канифоль и ее производные
Зная состав галипота, можно определить состав канифоли.
В процессе перегонки компоненты галипота изменяются следу-
ющим образом: левопимаровая кислота превращается в абие-
тиновую (см. табл. 29); d-пимаровая кислота, изю-й-пимаровая,
неоабиетиновая кислота и сама абиетиновая кислота изменяют-
ся лишь очень немного. Если принять во внимание, что неоабие-
тиновая кислота .по химическим свойствам обычно не отличается
от кислоты абиетиновой, то канифоль сосны Pinus halepensis
можно рассматривать как чистую абиетиновую кислоту.
Следует иметь в виду, что в приведенных данных не учи-
тывается содержание неомыляемых веществ, поэтому их сле-
дует уменьшить примерно на 10%; далее, они относятся только
к составу галипота, а не к составу общей смеси кислот. Нако-
нец, левопимаровая кислота претерпевает при нагревании более
сложные изменения, чем было указано; это требует внесения
дополнительных поправок в приведенные данные.
ЛИТЕРАТУРА
1. Maly, Ann. 1864, 132, 249.
2. F 1 u с k i g е г, J. prakt. Chem., 1867, (1), 101, 235.
3. Knecht, Hibbert, Mon. Sci., 1921, (5), 11, 107.
4. Laurent, Ann. phys. et chim., 1839, (2), 72, 383.
5. Duvernoy, Ann., 1868, 148, 143.
6. C a 11 i о t, Bull. Soc. chim., 1874 (2), 31, 387.
7. Vesterberg, Ber., 1885, 18, 3331; 1886, 19, 2167; 1887, 20, 3248.
8. Kohler, J. prakt. Chem., 1911, (2), 85, 534.
9. Dupont, Bull. Soc. chim., 1921, (4), 29, 718.
10. Harris, Sanderson, J. Am. Chem. Soc., 1948, 70, 334.
11. L о m b a г d, F г e у, C. R., 1950, 231, 445; Bull. Soc. chim., 1954, M.,
35.
12. Harris, Sanderson, J. Am. Chem. Soc.. 1948, 70. 2079, 2081.
СМОЛЯНЫЕ кислоты
Левопимаровая, d-пимаровая, изо-г/-пимаровая, абиетиновая
и неоабиетиновая кислоты являются практически единствен-
ными существенными компонентами кислотной фракции сосновой
смолы. Эти пять кислот являются изомерами состава C2oH30Ov,
причем их структурные формулы довольно близки между собой,
так как все они имеют скелет фенантрена:
Смоляные кислоты
523
Эти кислоты близки .по свойствам и образуют весьма одно-
родную химическую группу.
СООН
Н3С
кислота
^-СН(СН3)2
абиетиновая
кислота
Три первые кислоты образуют абиетиновую группу; две послед-
ние— d-пимаровую, или, просто, пимаровую, группу.
Здесь приведены лишь структурные формулы, развернутые
в плоскости; .пространственная конфигурация кислот будет рас-
смотрена ниже. Отметим, что d-пимаровая и изо-с^-пимаровая
кислоты являются эпимерами, отличающимися друг от друга
конфигурацией у седьмого атома углерода.
Абиетиновая кислота. Абиетиновая кислота является основ-
ным компонентом всех видов канифоли. Название «абиетино-
вая» происходит от латинского слова «abies» (сосна) и было
введено в химическую литературу Баупом в 1826 г. Вначале
этим термином обозначали различные продукты, вплоть до со-
сновой смолы, которая, кстати сказать, довольно далека по
составу от абиетиновой кислоты.
В XIX в. химики старались выделить из различных видов
канифоли трехгранные кристаллы, которые не являлись иден-
тичными продуктами и были описаны под названием: «смоля-
ная» кислота, сильвановая кислота, пиропимаровая кислота,
канифольная кислота и т. д. Позже, однако, выяснилось, что
эти различные продукты содержат один и тот же основной ком-
понент, от которого зависят свойства смесей смоляных кислот;
этот компонент получил название абиетиновой кислоты.
В относительно чистом виде абиетиновая кислота была
впервые получена Шульцем1 в 1917 г. путем кристаллизации
канифоли из спирта, подкисленного соляной кислотой; Шульц
приводит следующие ее константы: [а]о=—96,9° (в спирте);
темп. пл. 173°. Однако этот продукт не представлял собой чи-
стой абиетиновой кислоты.
524
Гл. XVI. Канифоль и ее производные
Химическое строение абиетиновой кислоты было установ-
лено в 1948 г. Ломбаром и Фреем2, которые выделяли абиети-
новую кислоту из ее кристаллических соединений: солей, эфи-
ров и т. д. Эти авторы заметили, что смеси смоляных кислот
часто кристаллизуются, не изменяя своего состава, даже из
различных растворителей, т. е. представляют собой особые сме-
си постоянного состава (теоретически образование таких сме-
сей пока не обосновано). Поэтому неизменность свойств про-
дукта после повторных кристаллизаций не служит критерием
чистоты. Такие смеси постоянного состава особенно часто обра-
зуются при работе со свободными кислотами и не обнаружива-
ются для кристаллических производных, в которых кислотные
группы замещены, например для солей этих кислот, их эфиров
и т. д. Эти производные можно очистить путем многократной
кристаллизации и выделить из них соответствующие кислоты
в чистом виде.
Обычный способ выделения абиетиновой кислоты состоит в
кристаллизации канифоли из спиртового раствора, подкислен-
ного соляной кислотой; этот раствор следует в течение несколь-
ких минут выдержать при температуре кипения, чтобы полностью
превратить неоабиетиновую кислоту в абиетиновую. Для при-
мера опишем способ, рекомендованный Дюпоном4.
Канифоль растворяют в спирте (приблизительно 1,5 л спирта на 1 кг ка-
нифоли), доводя раствор до кипения; после растворения добавляют 30 мл
продажного водного раствор а соляной кислоты, который желательно разбавить
спиртом". Кипячение продолжают еще в течение 15 мин., добавляют к раствору
гидрохинон (чтобы предотвратить окисление абиетиновой кислоты), после чего
раствор охлаждают при перемешивании во избежание образования на поверх-
ности кристаллической корки. Получаемую кристаллическую массу подвер-
гают сушке. Самый большой выход (75%) был получен из канифоли сосны Pinus
halepensis.
Последующая очистка абиетиновой кислоты производилась
в течение длительного времени по Дюпону5 — через кристалли-
ческий кислый абиетат натрия C20H2SO2Nа ЗС2оНзо02. Удельное
вращение плоскости поляризации выделенной кислоты едва до-
стигает —103°. Ломбар и Фрей2 разработали более эффектив-
ный способ очистки, который заключается в переводе кислоты
в кристаллический абиетат пиперидина; впрочем, и другие соли
абиетиновой кислоты дают аналогичные результаты.
Физические константы чистой абиетиновой кислоты*:
[“]/=—И2° (в спирте); т. пл. =170°; вращательная дисперсия
— = 1,151.
* В. Н. Крестинский и И. И. Бардышев3 приводят для абиетиновой кис-
лоты несколько отличающиеся константы: [а]р=—115,6°, темп. пл. 174—175°,
но эти константы не были подтверждены другими авторами.
Смоляные кислоты
525
Удельное вращение абиетиновой кислоты сильно изменяется
в зависимости от растворителей6, как это видно из приведен-
ных ниже данных автора:
Растворитель Удельное вращение абиетиновой кислоты Растворитель Удельное вращение абиетиновой кислоты
Спирт (95%-ный) —112° Петролейный эфир . —66,3°
Диэтиловый эфир —117,4° Бензол —21,3°
Ацетон .... —101,9° Ксилол —16,1°
Уксусная кислота _ 90,4° Сероуглерод . . . + 9,9°
Хлороформ . . —81,7° Расплавленная абиети-
Четыреххлористый новая кислота . . —49,7°
углерод . . . — 74,4°
Изменение способности вращать плоскость поляризации за-
висит, по-видимому, от сольватации абиетиновой кислоты в
различных растворителях. В пентане, петролейном эфире, че-
тыреххлористом углероде угол вращения не зависит от концен-
трации и равен углу вращения расплавленной кислоты; следо-
вательно, в этих трех растворителях измерялся угол вращения
чистой абиетиновой кислоты, а в других случаях — угол вра-
щения различных сольватов.
Кроме того, удельное вращение изменяется в зависимости
от концентрации (например, при увеличении концентрации обыч-
но несколько уменьшается его абсолютное значение), а также
в зависимости от температуры. Отсюда очевидна необходимость
уточнять условия измерения угла вращения (растворитель, кон-
центрация, температура); обычно, за исключением особо огово-
ренных случаев, указывают угол вращения 2%-него спиртового
раствора при комнатной температуре.
Абиетиновая кислота кристаллизуется, образуя характерные
треугольные пластинки, которые при рассмотрении в поляри-
зационный микроскоп дают очень яркую окраску; эти кристал-
лы принадлежат к моноклинной системе, >но являются квази-
орторомбическими. Кристаллографические константы абиетино-
вой кислоты еще не определены с достаточной точностью; дан-
ные, которые встречаются в литературе, относятся к недоста-
точно чистым продуктам и нуждаются в пересмотре.
Отметим наряду с треугольной формой кристаллов призмати-
ческую, а также копьевидную. Очевидно, из спирта чистая абие-
тиновая кислота кристаллизуется всегда в виде треугольников,
а другие кристаллические формы появляются только при нали-
чии загрязнений. Авторам удалось получить крупные кристаллы
призматической формы при кристаллизации из ацетона.
Ультрафиолетовый спектр поглощения абиетиновой кисло-
ты* характеризуется очень резким максимумом при длине вол-
* Указанные длины волн соответствуют определениям в спиртовом рас-
творе; они не очень сильно изменяются, если растворителем является гексаи.
526 Гл. XVI. Канифоль и ее производные
ны 241,5 ту. и двумя перегибами при 235 ту и 250 ту (рис. 58).
Выпуклость при 235 ту является, по мнению некоторых иссле-
дователей, второстепенным максимумом; наличие выпуклости
при 250 ту, совпадающей с максимумом абсорбции неоабиети-
новой кислоты, оспаривалось, но последние определения авторов
подтвердили ее существование.
Неоабиетиновая кислота. Эта кислота была получена впер-
вые в 1948 г. Харрисом и Сандерсоном7; ее можно выделить
обычным путем из галипота или из канифоли методом фрак-
ционной кристаллизации солей кислоты с аминами при много-
кратной замене аминов на последовательных этапах кристал-
лизации. Упомянутые авторы указали следующий способ полу-
чения неоабиетиновой кислоты.
Ацетоновый раствор 125 г галипота из сосны Pinus palustris обрабаты-
вают 37 г бутаноламина (2-амино-2-метилпропанола), также растворенного
в ацетоне. При этом выпадает кристаллическая соль бутаноламина, состоя-
щая в основном из левопимарата бутаноламина. Маточный раствор концен-
трируют в .несколько приемов: сначала получают левовращающую фракцию
(—60°), затем правовращающую фракцию (+12° и +15°). Для получения
неоабиетиновой кислоты последние фракции сначала превращают в соли ди-
этиламина, кристаллизацией которых доводят угол вращения до +70°, а
затем вновь переходят к солям бутаноламина, что дает возможность повы-
сить угол вращения до +il02°. Регенерированная из солей кислота имеет
уже угол вращения +159°.
Ломбар и Фрей8 показали, что неоабиетиновая кислота
вместе с абиетиновой составляют так называемую «сосновую»
часть галипота. Они предложили также другие сочетания ами-
нов, дающие возможность выделить неоабиетиновую кислоту.
Однако выделение неоабиетиновой кислоты представляет со-
бой чрезвычайно трудную операцию, несмотря на то, что этот,
компонент содержится в большом количестве и* в канифолп и
в галипоте.
Физические константы неоабиетиновой кислоты: [а]7-=+168°
(в спирте); темп. пл. 167—169°. Кристаллографические констан-
ты неоабиетиновой кислоты до сих пор не установлены.
Ультрафиолетовый спектр поглощения неоабиетиновой кис-
лоты (рис. 59) характеризуется единственным максимумом* при
длине волны 250 тр.
В качестве характерных кристаллических производных нео-
абиетиновой кислоты можно назвать метиловый эфир**, кото-
рый плавится при температуре 61,5—-62°.
* Спектры поглощения определялись в спиртовом растворе; длина вол-
ны, соответствующая максимуму поглощения, практически не изменяется,
если определение производят в стандартном гексане.
** Этот эфир получают обработкой неоабиетиновой кислоты диазометаном
при обычной температуре, чтобы избежать полимеризации кислоты.
Смоляные кислоты
527
Левопимаровая кислота. Левопимаровая кислота была впер-
вые получена в 1887 г. Вестербергом9, разложившим пимарат
натрия соляной кислотой. При этом выкристаллизовывалась
rf-пимаровая кислота; из маточного раствора он сумел извлечь
(правда, только один раз) левопимаровую кислоту. Попытки
Вестерберга воспроизвести этот опыт не увенчались успехом,
и в этом нет ничего удивительного ввиду чувствительности лево-
пимаровой кислоты к крепким минеральным кислотам.
Рис. 58. Кривая поглощения ь
ультрафиолетовой части спектра
абиетиновой кислоты (удельное
вращение [<х]у=—112°).
Рис. 59. Кривая поглощения в ультрафио-
летовой части спектра неоабиетиновой кис-
лоты:
1—по Харрису и Сандерсону; 2—по Ломбару i
Фрею.
Двадцать лет спустя наличие левопимаровой кислоты под-
твердил К^лер10, который получил ее из зимней еловой смолы —
вещества сравнительно богатого левопимаровой кислотой*.
Дюпон11 в 1921 г., а затем ряд других исследователей12-15
.выделили левопимаровую кислоту, хотя и в малых количествах,
усовершенствовав метод Вестерберга и, в частности, исключив
применение минеральной кислоты для выделения смоляных
кислот из пимарата натрия.
* Левопимаровая кислота образуется не только зимой; она собирается
между древесиной и корой ели (когда кора вследствие удара отделяется от дре-
весины) и может остаться в жидком состоянии в образовавшихся карманах до-
вольно продолжительное время; при соприкосновении с воздухом она быстро
отвердевает.
528
Гл. XVI. Канифоль и ее производные
Левопимаровая кислота стала легко доступным продуктом
лишь после того, как Харрис и Сандерсон7 в 1948 г. подобрали
амин (2-.амино-2-метилпрс)паН'0Л, или бута-
Рис. 60. Форма кри-
сталлов левопимаровой
кислоты.
ноламин), дающий 1малорастворимый лево-
1пима|р1ат, ib 'виде которого можно селектив-
но осаждать левой им аровую кислоту из
смеси смоляных кислот.
Раствор 125 г Галипота сосны Pinus
palustris ib ацетоне обрабатывают раство-
ром 37 г бутаиаламин1а в ацетоне; при этом
кристаллизуется соль амина, образованная
главным образом левопимаратом. Эта соль
после 1высуш;ив1ания вновь подвергается пе-
рекристаллизации из этилацетата, а для
регенерации левопимаровой кислоты /при-
меняется борная кислота.
Ломбар с сотр. показали, что галипот
сосны Pinus palustris может быть заменен
галипотом морской сосны, а ,борная кис-
лота — уксусной кислотой.
Левопимаровая кислота .характеризует-
ся весьма высоким удельным вращением:
[«],-=—291°; [а]г, =—342°; Н=—695°. Тем-
пература ее плавления установлена недостаточно точно; при
быстром нагревании кислота плавится при 150—152°, а при на-
гревании обычным способом в капиллярной трубке — при 137°.
Рис. 61. Кривая поглощения в ультрафиолетовой части спектра
левопимаровой кислоты.
Обработка левопимаровой кислоты диазометаном при обыч-
ной температуре приводит к образованию кристаллического ме-
тилового эфира. Левопимаровая кислота кристаллизуется в ви-
де орторомбических призм полноосной гемиэдрической формы
с увеличенными размерами плоскостей т (рис. 60).
На кривой поглощения ультрафиолетовых лучей левопима-
ровой кислотой имеется один максимум, соответствущий длине
волны 272,5 пщ (рис. 61).
Смоляные кислоты
529
d-Пимаровая кислота. Это первая из смоляных кислот, кото-
рая была выделена в чистом виде (она была получена Калли-
стом16 в 1874 г.). Метод ее получения был затем усовершен-
ствован различными авторами 20- 22, причем обычно проме-
жуточной стадией являлось образование пимарата натрия, кри-
сталлизующегося при обработке галипота содой. После много-
кратной перекристаллизации из воды пимарат натрия получает-
ся в виде красивых перламутровых листочков. Его разлагают
какой-либо кислотой и выделенную d-пимаровую кислоту вновь
подвергают кристаллизации. Заключительной операцией очи-
стки является сплавление кислоты с малеиновым ангидридом
для окончательного и полного удаления примесей, содержа-
щих в молекуле сопряженные двойные связи. Ломбар и его
сотрудники значительно упростили этот метод, исходя непо-
средственно из канифоли, перекристаллизованной из ацетона.
Харрис и Сандерсон23 предложили метод выделения d-пима-
ровой кислоты, основанный на другом принципе. гДПимаровая
кислота — самая летучая из всех смоляных кислот и поэтому
содержится в наибольшем количестве в головном продукте
дистилляции канифоли, из которого и может быть извлечена
описанным выше способом в виде соли бутаноламина. Однако
этот метод не имеет преимуществ по сравнению с предыдущими.
d-Пимаровая кислота кристаллизуется в орторомбической си-
стеме в виде плоских прямоугольных пластин. На рис. 62 пока-
заны кристаллы, полученные Дюпоном путем кристаллизации
из эфира; аналогичные результаты получаются и с другими
растворителями.
Рис. 62. Форма кристаллов d-пимаровой
кислоты.
Удельное вращение d-пимаровой кислоты [ct]y=+77°; темп,
пл. 218—219°. Она образует кристаллический метиловый эфир
с темп. пл. 69° и характерное дигидропроизводное19, *1, 24~26,
легко получаемое гидрированием на скелетном никелевом ка-
тализаторе при обычной температуре (в этих условиях происхо-
дит селективное гидрирование виниловой двойной связи). Ди-
гидро-й-пимаровая кислота очень мало растворима в большин-
34—156
530
Гл. XVI. Канифоль и ее производные
стве растворителей и плавится при 249—250°. Поскольку d-пи-
маровая кислота не содержит сопряженных двойных связей,
она не дает характерного спектра поглощения ультрафиолето-
вых лучей.
Изо-с/-пимаровая кислота. Изо-б/-пимаровая кислота — спут-
ник (Лпимаровой кислоты, являющийся ее эпимером, отлича-
ющимся стереохимической конфигурацией при седьмом атоме
углерода.
Изо-й-пимаровая кислота оставалась длительное время не-
замеченной, так как ее натриевая соль очень хорошо раствори-
ма и остается в маточной жидкости при классическом методе
выделения d-пимаровой кислоты в виде лимарата.
Харрис и Сандерсон23 впервые получили изо-б/-пимаровую
кислоту, обрабатывая смесь кислот* смолы Pinus palustris ма-
леиновым ангидридом в бензоле при кипении в присутствии
соляной кислоты. Кислоты группы абиетиновой кислоты при-
соединяют малеиновый ангидрид и могут быть отделены мето-
дом фракционного осаждения от группы d-пимаровых кислот;
последние разделяются при помощи селективных аминов. Этим
методом авторы выделили изо-й-пимаровую кислоту, но полу-
ченный продукт был далеко не чистым. Изо-й-пимаровую кис-
лоту выделили также Бросси и Егер26.
Продукты, описанные под названием изо-б/-пимаровой кис-
лоты, не являются чистыми и их константы настолько недосто-
верны, что указывать их было бы нецелесообразным.
ЛИТЕРАТУРА
1. Schultz, Chem. Z., 1917, 41, 666; Mon. Sei., 1920 (5), 10, 102.
2. Lombard, Frey, Bull. Soc. chim., 1948, M., 1194
3. Крестинский, Бардышев, ЖОХ, 1940, 10, 1894; ЖОХ, 1941,
11. 996.
4. Dupont, Bull. Soc. chim.. 1924, (4), 35, 1234.
5. Dupont, Desalbres, Bernet t e, Bull. Soc. Chim., 1926, (4),
39, 491.
6. Dupont, R ou i n, Dubourg, Bull. Inst. Pin, 1927, 157; Lom-
bard и сотр., не опубликовано.
7. Harris, Sanderson, J. Am. Chem. Soc., 1948, 70, 334, 339.
8. Lombard, Frev, C. r., 1950, 231, 445.
9. Vesterberg, Ber., 1887, 20, 3248.
10. Kohler, J. prakt. Chem., 1912, (2), 85, 534.
11- Dupont, Bull. Soc. chim., 1921, (4), 29, 729.
12. R u z i с k а и сотр., Helv. chim. acta, 1937, 20, 1547.
13. P al k i n, Harris, J. Am. Chem. Soc., 1933, 55, 3677.
* Как уже указывалось, галипот не является суммарной кислотной фрак-
цией смолы. «Суммарные кислоты» получаются путем осаждения всех смоля-
ных кислот циклогексиламином (образующим с ними со всеми нерастворимые
соли) или, еще лучше, аммиаком; после промывания осадка выделяют свобод-
ные кислоты. Таким образом, суммарные кислоты и галипот состоят из одних
и тех же компонентов, но в разных соотношениях.
Химические свойства смоляных кислот
531
14. Kraft, Ann., 1935, 520, 136.
15. Lombard, Bull. Soc. chim., 1948, M., 1149.
16. Call i о t, Bull. Soc. chim., 1874, (4), 31, 387.
17. Vest er berg, Ber., 1885, 18,3331; Ber., 1886, 19,2167; Ber., 1887, 20, 3248.
18. Dupont, Bull. Soc. chim., 1921, (4), 29, 718, 727.
19. P a 1 к i n, Harris, J. Am. Chem. Soc., 1933, 55, 3677.
20. F 1 e c k, Palkin, J. Am. Chem. Soc., 1940, 62, 2044.
21. Lombard, Bull. Soc. chim., 1946, M., 109.
22. R u z i с к а и сотр., Helv. chim. acta, 1923, 6, 677; 1924, 7, 875; 1931,
14, 233; 1932, 15, 915, 1289, 1300; 1940, 23, 124.
23. H a r r i s, Sanderson, J. Am. Chem. Soc., 1948, 70, 2079, 2081.
24. Чугаев, T e e a p у, Ber., 1913, 46, 1769.
25. Johansson, Mon. Sci., 1921 (5), 11, 73.
26. Br ossi, Jeger, Helv. chim. acta, 1950, 33, 722.
Химические свойства смоляных кислот и их производных
Химические .свойства смоляных кислот достаточно близки
между собой, чтобы их можно было рассматривать параллель-
но, группируя по следующему принципу:
1. Свойства, связанные с наличием карбоксильной группы,—
образование солей, эфиров, ангидридов и т. д.
2. Свойства, связанные с непредельностью, — ультрафиоле-
товые спектры, изомеризация минеральными кислотами, гидри-
рование, присоединение бромистоводородной кислоты, галоге-
нов, малеинового ангидрида и т. д.;
3. Дегидрирование и окисление—превращение в дегидро-
абиетиновую кислоту, ретен или пимантрен, различные процес-
сы окисления, аутооксидация и т. д.;
4. Действие тепла — образование пироабиетиновых и пиро-
смоляных кислот, смоляных и канифольных масел.
5. Прочие свойства.
Свойства смоляных кислот, связанные с наличием карб-
оксильной группы. Образование солей. Соли смоляных кислот,
образованные щелочными металлами, обычно кристалличны и
имеют хорошо выраженное строение, тогда как соли тяжелых
металлов аморфны и не имеют определенного строения.
Из многочисленных солей щелочных металлов рассмотрим
кратко (натриевые соли смоляных кислот, кристаллизующиеся из
водных растворов. Нейтральный кристаллический d-пимарат
иатрия нерастворим в воде при обычной температуре; нейтраль-
ный кристаллический левопимарат натрия очень плохо раство-
рим в воде при обычной температуре; нейтральный абиетат на-
трия1, образующий игольчатые кристаллы, довольно хорошо
растворим в воде (32 г/л при обычной температуре).
Натриевые соли смоляных кислот во многом аналогичны
мылам жирных кислот: в водных растворах они сильно гидро-
34*
532
Гл. XVI. Канифоль и ее производные
лизованы и с трудом выделяются в виде хорошо выраженных
кристаллов вследствие склонности к образованию мезоморфных
фаз, содержащих большое количество воды. Аммонийные соли
смоляных кислот обладают способностью к гелеобразованию.
Кроме того, известны многочисленные кристаллические соли
щелочных металлов, нейтральные или кислые, выделяемые из
спиртовых растворов, и множество кристаллических солей ами-
гнов, выделяемых из растворов в спирте, ацетоне, углеводоро-
дах и т. д. Соли, образуемые смоляными кислотами с аминами,
обычно растворимы как в воде, так и в углеводородах, и их
можно в случае надобности использовать для приготовления
водных растворов углеводородов.
О резинатах тяжелых металлов имеются недостаточные све-
дения. В 1914 г. Эллингсон2 изучал соли абиетиновой кислоты
с тяжелыми металлами. Он получал их обменным разложением
в водном растворе щелочного абиетата и соли заданного ме-
талла; осажденный абиетат тяжелого металла растворяли в бен-
золе, фильтровали для отделения могущей образоваться гидро-
окиси металла и, наконец, подвергали сушке. Продукты, полу-
ченные Эллингсоном, аморфны; анализ их приводит к формулам
типа 6NiA2 -7AH (АН обозначает абиетиновую кислоту). Обыч-
но это кислые соли, реже основные, и только в виде исключения
«нейтральные. В табл. 30 показана растворимость абиетатов в
различных растворителях, согласно Эллингсону.
ТАБЛИЦА зо
Растворимость абиетатов металлов в различных растворителях
(P—хорошая растворимость; С—средняя растворимость; Н—не растворяется)
Растворитель Абиетат
Ва Са Sr Си Cd Z11 Сг А1 Со N1 Мп Ag Fe Na]
Пиридин .... р Р р р р р р р р р р р р р
Анилин р Р р р р р р р р р р р р н
Метиловый спирт. с С с с р с с н с с с н н р
Этиловый спирт . с р с с р с с н с с с н С р
Амиловый спирт . р р р р р р р р р р р р р р
Бензол р р р р р р р р р р р р р н
Скипидар .... р р р р р р р р р р р р р н
Гептан н р р с р р с р с с р н н н
Керосин .... н с с р р р с р р р р с н н
Сероуглерод . . р р р р р р р р р р р р р н
Диэтиловый эфир. с р р р р р р р р р р с с с
Ацетон .... н с н р р р р н р р р с с н
Четыреххлористый
углерод . . . р р р р р р р р р р р р с н
Хлороформ . . . р р р р р р р р р р р р р н
Этила цетат . . . р р р р р р р р р р р р с н
Вода н н н н н н н н н н н н н р
Химические свойства смоляных кислот
533
В 1926 г. Дюпон3 также изучал абиетаты тяжелых метал-
лов, которые он получал в спиртовом растворе; в противопо-
ложность данным Эллингсона, абиетаты Дюпона часто ней-
тральны и, во всяком случае, имеют более определенное строе-
ние. Ломбар4 тоже получал абиетаты различных металлов, пред-
почитая спиртовый раствор водному, чтобы ограничить гидро-
лиз; рентгенограммы всех испытанных абиетатов (абиетаты
кальция, никеля, магния) не показали кристаллической струк-
туры.
Резинаты тяжелых металлов обычно растворимы в органи-
ческих растворителях, особенно хорошо в расплавленной кани-
фоли; благодаря этому способность канифоли растворять ме-
таллические окислы широко используется в технике (см.
стр. 560).
Образование сложных эфиров. Эфиры смоляных кислот
представляют известную ценность в качестве пленкообразу-
ющих для лаков и в качестве пластификаторов. Они трудно
омыляются.
Из эфиров смоляных кислот изучались только эфиры абие-
тиновой кислоты5; полученные результаты могут быть отнесены
к большинству смоляных эфиров.
Методы этерификации смоляных кислот можно разде-
лить на три группы: прямые, каталитические и косвенные
методы.
По методу прямой этерификации смоляную, кислоту нагре-
вают с избытком спирта при температуре свыше 250° в авто-
клаве из нержавеющей стали*. Если на одну молекулу абие-
тиновой кислоты приходится четыре молекулы этилового спир-
та, через 10 час. при температуре 300° происходит полная эте-
рификация; при этом давление повышается до 300 ат. Подоб-
ный же результат получается при этерификации другими спир-
тами, причем требуемое давление изменяется в зависимости
от взятого спирта.
Нужно отметить, что при проведении реакции в автоклаве
нельзя своевременно удалять из реакционной среды воду, и по-
этому для достижения полной этерификации требуется большой
молекулярный избыток спирта. При этерификации нелетучими
спиртами, например цетиловым спиртом, процесс можно вести
при обычном давлении в открытом перегонном кубе; в этом
случае образующаяся при реакции вода может быть легко и
своевременно удалена; кроме того, реакцию можно проводить
при стехиометрическом соотношении смоляной кислоты и
спирта.
* Применение нержавеющей стали необходимо для получения прозрач-
ных продуктов.
534 Гл. XVI. Канифоль и ее производные
Этерификация смоляных кислот многоатомными спиртами
протекает гораздо легче, чем одноатомными спиртами; при эте-
рификации канифоли глицерином реакцию ведут при 260—290°.
Практически прямая этерификация была осуществлена в
США; во Франции ввиду трудности изготовления из нержавею-
щей стали автоклавов большого размера, рассчитанных на
высокое давление, не удалось применить методы прямой этери-
фикации летучими спиртами. Но отсутствие автоклавов не яв-
ляется непреодолимым препятствием. Для тех же целей можно
использовать различные аппараты, работающие под давлением,
например колонны, в которых пары спирта барботируют через’
канифоль, нагретую до 300°, а сконденсировавшиеся пары ка-
нифоли возвращаются обратно в цикл. Можно также в ко-
лонну, заполненную спиртом, нагретым до 300°, вбрызгивать
сверху расплавленную канифоль, опускающуюся навстречу под-
нимающимся парам спирта. Хорошие результаты дает также
введение в канифоль активной глины, которая играет роль ка-
тализатора и легко отделяется от образовавшегося эфира про-
стой декантацией.
При каталитической этерификации можно заметно понизить
температуру реакции, а следовательно, и давление в автоклаве.
Самым активным катализатором является серная кислота; в
качестве катализаторов могут применяться также борная и
фосфорная кислоты, металлическая пудра, хлориды и окислы
металлов, активные глины, но эти катализаторы не так ак-
тивны.
В присутствии минеральных кислот скорость этерификации
при 100° еще не велика, что позволяет получать абиетиновую
кислоту путем изомеризации канифоли в спиртовом растворе,
не опасаясь этерификации продукта. При 160—170° этерифи-
кация канифоли этиловым спиртом в присутствии серной кис-
лоты проходит полностью в течение 15 час.; с фосфорной кис-
лотой и хлористым цинком для полной этерификации требуется
40 час. при 190—200°.
Применение серной кислоты в качестве катализатора пред-
ставляет особый интерес, так,как для этерификации этиловым
спиртом при 160° требуется давление 12 ат и материал авто-
клава не обязательно должен обладать особой стойкостью.
Неудобство метода заключается в том, что для удаления
серной кислоты требуется последующая промывка продукта
водой.
Другим интересным катализатором является трехфтористый
бор: он дает возможность этерифицировать абиетиновую кис-
лоту фенолами при обычной температуре.
Методы, при которых кислота или спирт принимают участие
не как таковые, а в виде более активных производных, носят
Химические свойства смоляных кислот
535
название методов косвенной этерификации. Главнейшими из них
являются следующие:
1. Этерификация диазометаном на холоду (для получения
метиловых эфиров).
2. Действие хлоралкилов и алкилсульфатов на абиетат ка-
лия или натрия.
3. Действие спирта 'на ангидрид абиетиновой кислоты.
4. Действие алкоголятов и фенолятов натрия на хлорангид-
рид абиетиновой кислоты.
5. Действие на абиетиновую кислоту эфиров летучих кислот,
например фталатов, ацетатов и т. д.; абиетиновая кислота
вытесняет летучие кислоты из их эфиров.
Из этих методов заслуживают внимание только два первых.
Смоляные кислоты (как и большинство других органиче-
ских кислот) при смешении с диазометаном в эфирном растворе
при обычной температуре образуют метиловые эфиры по реак-
ции:
CH2N2 -р R—СООН -► R—СО—ОСН3 -f- N2
Метод применяется для быстрой этерификации малых коли-
честв смоляных кислот или для этерификации нестойких смо-
ляных кислот, таких, как, например, левопимаровая кислота.
Классическим методом получения эфиров абиетиновой кис-
лоты является действие алкилхлоридов или алкилсульфатов на
щелочные абиетаты. Реакция легко протекает при действии
диметилсульфата на абиетат натрия или калия в аппарате
с обратным холодильником в спиртовой или бензольной среде;
с первичными алкилхлоридами этот метод всегда дает количе-
ственные выходы; с вторичными алкилхлоридами выход полу-
чается плохой и, наконец, для третичных алкилхлоридов этот
метод вообще непригоден.
Некоторые смоляные кислоты (неоабиетиновая кислота, ле-
вопимаровая, J-пимаровая) образуют кристаллические метило-
вые эфиры, в то время как метилабиетат нельзя получить в
кристаллической форме. Кристаллические метиловые эфиры
имеют важное значение для лабораторных исследований, так
как они обычно могут быть получены в чистом виде путем
многократной перекристаллизации и часто служат для иден-
тификации кислот.
Большая часть эфиров абиетиновой кислоты представляет
собой высоковязкие масла. Кристаллические абиетаты состав-
ляют исключение; 'они могут быть получены только из спиртов,
которые сами способны кристаллизоваться. Следует отметить,
что известен кристаллический абиетат глицерина, но промыш-
ленные эфиры глицерина не кристаллизуются.
536 Гл. XVI. Канифоль и ее производные
В табл. 31 приведены физические свойства важнейших абие-
тиновых эфиров, в большинстве случаев по данным Кеслера,
Лови и Фарагера6.
ТАБЛИЦА 3J
Физические свойства эфиров абиетиновых кислот
Углеводородный радикал Температура кипения °C Температура плавления °C Удельный вес (при 15°) г/см*
Метил 210 (при 4 мм) и 1,050
225—226 (при 16 мм)
Этил 204—207 (при 4 мм) — 1.032
н-Пропил 237—240 (при 4 мм) — 1,015
Изопропил 214—217 (при 4 мм) — 1.010
н-Бутил 247—250 (при 3 jhjm) — 1,014
Изобутил 222—225 (при 4 мм) — 1,008
Изоамил 254—257 (при 4 мм) — 1,001
Аллил 282—285 (при 5 мм) — 1,024
Фенил 330—333 (при 4 мм) — 1,056
Гексил 299—302 (при 4 мм) — 1,061
ж-Толуил 310—313 (при 6 мм) — 1,039
а-Нафтил 290 (при 2 мм) — 1,116
Терпенил 310 (при 2 мм) — 1,082
Бензил . 294—297 (при 4 мм) — 1.036
Холестерил — 122—125 —
Ментил — 77—83 —
Борнил — 75—80 —
Цетил — 42 —
Тритил* — 77 —
Глицерин (триабиетат)** — 150—162 —
* По Ломбару.
♦* По Фонроберу.
По химическим свойствам, не связанным с эфирной группой
(действие водорода в присутствии катализаторов, действие ма-
леинового ангидрида и т. д.), эфиры смоляных кислот подобны
соответствующим кислотам. Здесь будут рассмотрены только
свойства, специфические для эфиров.
Эфирные группы гораздо более устойчивы к действию высо-
ких температур, чем карбоксильные группы; эфиры без раз-
ложения перегоняются при пониженном давлении, в то время
как перегонка кислот даже при низком давлении всегда сопро-
вождается некоторым декарбоксилированием.
Смоляные эфиры устойчивы также и к действию омыляющих
веществ, так как карбоксильная группа связана с третичным
атомом углерода. Поэтому абиетиновая кислота, которая с
трудом этерифицируется, дает особенно стойкие эфиры. Такая
стойкость затрудняет определение коэффициента омыления
Химические свойства смоляных кислот
537
смоляных эфиров; классический метод определения, применя-
емый для эфиров жирных кислот, не всегда пригоден для эфиров
смоляных кислот.
Для полного омыления 0,1 и. раствором поташа в этило-
вом спирте требуется примерно 8 час.; если поташ применять
в виде раствора в бутаноле, в бензиловом спирте или в глико-
лях, продолжительность нагревания сокращается приблизи-
тельно до 2 час. При омылении необходимо принимать различ-
ные меры предосторожности, например работать в атмосфере
инертного газа, применять щелочестойкую стеклянную посуду
и т. д.
Всегда следует находить предельную величину коэффи-
циента омыления, проводя одновременно несколько проб и по-
степенно увеличивая время испытания.
Восстановление эфирной группы. Эфиры смоляных кислот
могут быть восстановлены в смоляные спирты двумя известны-
ми классическими методами: каталитически или по Буво и
Блану.
Для каталитического гидрирования эфиров в присутствии
хромата -меди не требуется повышенной температуры и высо-
кого давления. Это весьма тонкая операция, которая, однако,
была использована в промышленных условиях фирмой Hercules
Powder7 для получения дигидроабиетинола и тетрагидроабиети-
нола.
Метод гидрирования по Буво и Блану был недавно усовер-
шенствован Дюпоном с сотр.8; они диспергировали натрий в
углеводороде при повышенной температуре (160°) и постепенно
вводили в полученную суспензию смесь испытуемого эфира и
втор-бутилового спирта. Благодаря проведению процесса в кол-
бе с обратным холодильником создается возможность все время
регулировать температуру.
При восстановлении небольших количеств эфира можно с
успехом применять литийалюминийгидрид.
Образование ангидридов смоляных кислот и других их про-
изводных. В принципе каждой смоляной кислоте должен соот-
ветствовать ангидрид, но известен только ангидрид абиетиновой
кислоты.
Вопрос о существовании абиетинового ангидрида долгое
время был весьма спорным. Мали9 в 1864 г, считал, что кани-
фоль представляет собой абиетиновый ангидрид; Флукигер10,
Бишоф и Настфогель11, Эстерфилд и Баглей12, Кнехт и Гиб-
берт13 придерживались противоположного мнения, но фактически
никто из них не работал с абиетиновым ангидридом. Впервые
этот ангидрид был получен в 1926 г. Фонробером и Палло13.
Предложенный ими метод его получения, усовершенствован-
ный Ломбаром и Фреем14, состоит в следующем.
538
Гл. XVI. Канифоль и ее производные
В колбе с обратным холодильником кипятят в течение 4 час. концентриро-
'ванный раствор абиетиновой кислоты в уксусном ангидриде (475 г уксусного
•ангидрида на 1 г-мол абиетиновой кислоты). Абиетиновый ангидрид постепенно
кристаллизуется при охлаждении, но отделение кристаллов от маточного рас-
твора весьма затруднительно. Кроме того, его кристаллизации препятствует
уксусный ангидрид, который удаляют дистилляцией в вакууме, после чего абие-
тиновый ангидрид перекристаллизовывают из ацетона.
Абиетиновый ангидрид плавится при 130—131°; он плохо
.растворяется в ацетоне и спирте при обычной температуре;
омыляется с трудом, но легче, чем эфиры.
Метод Фоиробера и Палло можно распространить и на дру-
гие смоляные кислоты (за исключением таких нестойких кислот,
как левопимаровая).
Из других производных смоляных кислот известны, главным
образом, производные абиетиновой кислоты.
Хлор ангидрид абиетиновой кислоты можно получить, сме-
шивая в стехиометрических соотношениях сухую абиетиновую
кислоту с пятихлористым фосфором: смесь постепенно раство-
ряется с одновременным выделением соляной кислоты. В ре-
зультате получается вязкая масса, состоящая из хлорангиц-
рида абиетиновой кислоты и небольшого количества Хлорокиси
фосфора, которую можно удалить путем дистилляции в ваку-
уме. В промышленных масштабах хлорангидрид абиетиновой
кислоты получался действием фосгена на абиетиновую кислоту.
Хлорангидрид, реагируя с аммиаком, образует амид, с ани-
лином— анилид, со спиртами—соответствующие эфиры. Ни
.хлорангидрид, ни его производные не кристаллизуются.
Свойства смоляных кислот, связанные с их непредельностью.
Ультрафиолетовые спектры. Для кислот группы абиетиновой
кислоты, т. е. для смоляных кислот, содержащих сопряженные
двойные связи
СООН
абиетиновая
кислота
СООН
неоабиетиновая
кислота
СООН
левопимаровая
кислота
характерны ультрафиолетовые спектры с интенсивной полосой
поглощения в области 230—280 mg. Абиетиновая и неоабиети-
новая кислоты близки по интенсивности поглощения; интенсив-
ность поглощения левопимаровой кислоты примерно в четыре
раза слабее. Поэтому спектрофотометрическим методом можно
количественно определять абиетиновую и неоабиетиновую кис-
лоты, но не левопимаровую кислоту.
Химические свойства смоляных кислот
539
Спектр поглощения дегидроабиетиновой кислоты, содержа-
щей ароматическое ядро
СООН
—СН(СН3)2
характеризуется двумя максимумами (при 268 и 276 mpt). Со-
ответствующие интенсивности поглощения света примерно в
30 раз меньше, чем для абиетиновой кислоты.
Смоляные кислоты группы rf-пимаровой кислоты имеют по-
лосы поглощения, сдвинутые в сторону более коротких волн,
в область, где эти полосы уже не являются характерными, что
обусловлено отсутствием сопряженных двойных связей в мо-
лекулах этих кислот.
Основные спектральные характеристики кислот группы аби-
етиновой кислоты (включая дегидроабиетиновую кислоту) при-
ведены в таил. 32 по литературным данным 16-19 и данным ав-
тора.
ТАБЛИЦА 32
Ультрафиолетовые спектры поглощения смоляных кислот
группы абиетиновой кислоты
Кислота
Положение
максимума
поглощения
Ш|Х
Молярный
коэффициент
поглощения
Абиетиновая . . .
Неоабиетиновая . .
Левопимаровая . .
Дегидроабиетиновая
241,5
250
272,5
268 и 276
23 000
24 300
5 800
690 и 760
Связь между структурой диенов и их ультрафиолетовыми
спектрами в настоящее время установлена: правила Вудварда
дают возможность вычислить положение максимума поглоще-
ния, соответствующего данной структуре. Структуры, принятые
для смоляных кислот группы абиетиновой кислоты, согласуются
с этими правилами.
Изомеризация минеральными кислотами. Давно известно,
что минеральные кислоты способствуют превращению в абиети-
новую кислоту некоторых кислот, содержащихся в галипоте или
в канифоли. На этом основан способ приготовления абиетино-
вой кислоты, описанный на стр. 524.
540 Гл. XVI. Канифоль и ее производные
В последнее время изомеризация смоляных кислот при тем*,
пературе от 18 до 25° изучалась различными авторами20,21
Установлено, что rf-пимаровая и изо-й-пимаровая кислоты не
изменяются при действии разбавленных минеральных кислот
при обычной температуре; изомеризация наблюдалась только
для абиетиновой, неоабиетиновон и левопимаровой кислот. Нео-
абиетиновая и левопимаровая кислоты при изомеризации дают
не чистую абиетиновую кислоту, а равновесную смесь кислот,
в которой преобладает абиетиновая кислота:
%
Абиетиновая кислота (удельное вращение —112°) . 93
Неоабиетиновая кислота (удельное вращение —92°) 7
Левопимаровая кислота ......................... Ничтожно
мало
Чистая абиетиновая кислота при обработке минеральными
кислотами дает такую же равновесную смесь; это именно та
смесь, которая составляет «обычную» абиетиновую кислоту с
вращательной способностью между—90 и—100°; выше мы
описали те методы, которые дают возможность получать хими-
чески чистую абиетиновую кислоту.
Механизм изомеризации таков. Абиетиновая, неоабиетиновая
и левопимаровая кислоты под влиянием водородных ионов об-
разуют один и тот же двухзарядный ион, который дает затем
равновесную смесь абиетиновой и неоабиетиновон кислот:
Реакции изомеризации являются мономолекулярными реак-
циями, т. е. их кинетика подчиняется закону:
где х— молярная концентрация кислоты, претерпевающей пре-
вращение.
Химические свойства смоляных кислот
541
Характерная для данной кислоты константа скорости изо-
меризации не зависит от исходной концентрации смоляной кис-
лоты, но зависит от характера изомеризующего агента и его
концентрации, от температуры и т. д. По величине'константы
можно идентифицировать левопимаровую и неоабиетиновую
кислоты.
Гидрирование. Гидрирование канифоли как промышленная
операция будет рассмотрено позднее. Здесь будут описаны толь-
ко реакции гидрирования различных смоляных кислот, недавно
изученные Ломбаром и Эбеленом27 и Беллю61. Все эти кислоты
легко присоединяют одну молекулу водорода, но с трудом при-
соединяют две молекулы, требуемые для превращения в насы-
щенные продукты.
При гидрировании расплавленной d-пимаровой кислоты или
ее натриевой соли, независимо от природы растворителя, тем-
пературы и катализатора, всегда получается одна и та же
дигидро-сГпимаровая кислота22-26 , имеющая темп. пл. 242° и
удельное вращение [а]у = +24°. Эта кислота мало растворима
в большинстве растворителей и с трудом поддается перекри-
сталлизации (ее удается перекристаллизовать только из этил-
ацетата) .
Абиетиновую кислоту можно гидрировать в растворе при
обычной температуре в присутствии платинового или палла-
диевого катализаторов, а также при более высокой темпера-
туре в присутствии скелетного никелевого катализатора. Рас-
плавленную абиетиновую кислоту можно гидрировать без рас-
творителя, но в этом случае применение никеля требует осто-
рожности, так как никель является катализатором декарб-
оксилирования.
Возможно, наконец, гидрирование абиетата натрия в водном
растворе.
Продукт наиболее определенного состава получается на ске-
летном никелевом катализаторе Ренея; он состоит главным об-
разом из А-дигидроабиетиновой кислоты* (темп. пл. 145°;
[а]у = +43,9°):
HSC СООН
Обозначение, введенное Брюсом28.
542
Гл. XVI. Канифоль и ее производные
При действии разбавленных минеральных кислот А-диги-
дроабиетиновая кислота изомеризуется, образуя р-дигидро-
абиетиновую кислоту* (теми. пл. 177°; [«],- = +127,6°):
Н3С СООН
Н3С
<-СН(СН3)2
Будучи обработаны! концентрированной серной кислотой,
А- и p-дигидроабиетиновые кислоты дают один и тот же лак-
тон С20Н32О2:
сн(сн3)2
Продукты гидрирования, полученные с другими катализато-
рами (платина, палладий), представляют собой смеси А-диги-
дроабиетиновой кислоты и изомерной ей В-дигидроабиетиновой
кислоты (темп. пл. 164°); [а]о=—16°):
Н3С СООН
нзс l^JJ—СН(СН,)2
В-Дигидроабиетиновая кислота под действием разбавленных
кислот также превращается в p-дигидроабиетиновую кислоту.
Присоединение двух молекул водорода к абиетиновой кис-
лоте удается только с помощью особых приемов. Так, например,
гидрированием в эфирном растворе в присутствии скелетного
никелевого катализатора Ренея при температуре 180° и дав-
лении 200 кг/см2 Ломбар29 получил тетрагидроабиетиновую кис-
лоту (темп. пл. 165—166°; [а] =4-28,3°).
Ружичка32, который проводил гидрирование при 80° в уксус-
ной кислоте в присутствии платиновой черни, с трудом получил
продукт (присоединения четырех атомов водорода (темп. пл.
158—165°; [а]? = 4-10°), являющийся смесью различных ве-
ществ.
Обозначение, введенное Ломбаром29.
Химические свойства смоляных кислот
5431
В отношении гидрогенизации других смоляных кислот ука-
жем лишь, что гидрирование левопимаровой и неоабиетиновой
кислот приводит к А-дигидроабиетиновой кислоте (см. выше).
Присоединение галоидоводородов. Эта реакция еще недо-
статочно изучена применительно к смоляным кислотам, но мож-
но ожидать, что все кислоты группы абиетиновой кислоты дают
продукты присоединения, способные к изомеризации с обра-
зованием того же конечного продукта, что и абиетиновая кис-
лота.
Особо изучалась реакция присоединения к абиетиновой кис-
лоте НВг, которую следует рассмотреть подробнее. Установ-
лено, что абиетиновая кислота легко присоединяет две моле-
кулы бромистого водорода33, но получаемую дибромдигидро-
абиетиновую кислоту трудно очистить кристаллизацией. Гидр-
оксилсодержащие растворители, в которых дигидробромид до-
вольно хорошо растворяется, разлагают его; инертные раство-
рители, хлороформ, четыреххлористый углерод, сероуглерод,
этилацетат и т. д. или плохо растворяют его или не раство-
ряют совсем; дигидробромид легко разлагается даже при уме-
ренном нагревании. В качестве растворителей могут быть ис-
пользованы только диоксан и тетрагидрофуран.
Наиболее чистый дигидробромид, который был получен,,
плавился при 186—488°; его удельное вращение было равно
—21,7° (в тетрагидрофуране). При нагревании и обработке
спиртами, щелочами, растворами аминов и т. д. этот продукт
теряет две молекулы бромистоводородной кислоты, образуя
смесь, которую Сандерман34 назвал «изоабиетиновой кислотой».
Несмотря на многочисленные попытки, компоненты этой смеси
не были достаточно точно определены, и поэтому приводить
характеристики изоабиетиновой кислоты нецелесообразно. Ниже-
приведено схематическое (плоскостное) изображение структуры,
дигидробромида абиетиновой кислоты:
Н3С СООН
Присоединение галоидов. Смоляные кислоты вследствие-
своей ненасыщенности легко присоединяют галоиды, однако,
получить какие-либо определенные продукты присоединения
весьма трудно и даже почти невозможно, так как они самопро-
извольно разлагаются с выделением бромистоводородной кис«-
лоты.
544
Гл. XVI. Канифоль и ее производные
Тем не менее Дюпону с сотр.35 удалось показать, что абиети-
новая кислота при —20° присоединяет бром без последующего
отщепления бромистоводородной кислоты, образуя 7,8-дибром-
абиетиновую и 9,14-дибромабиетиновую кислоты, при повтор-
ном нагревании постепенно отщепляющие НВт. При 100°
7,8-дибромабиетиновая кислота отщепляет две молекулы НВт,
превращаясь в дегидроабиетиновую кислоту, а 9,14-дибромабие-
тиновая кислота теряет только одну молекулу НВт и образует
9-бромлевопимаровую кислоту:
7,8-дибромабиетиновая
кислота
дегидроабиетиновая
кислота
Н3С СООН
9,14-дибромабиетиновая
кислота
Н3С СООН
Вг
-СН(СН3)2
9-бром левопимаровая
кислота
Присоединение малеинового ангидрида. Давно известно, что
левопимаровая кислота, присоединяя малеиновый ангидрид,
образует аддукт, являющийся одновременно и кислотой и анги-
дридом:
Н3С СООН
CU - -
Н3с I У«-СН(СН3)2
----------------------сн—со
СН—СО
О
Ружичка36 наблюдал образование того же соединения при
взаимодействии абиетиновой кислоты с малеиновым ангидри-
дом при 180°; то же распространяется и на неоабиетиновую
кислоту. Это объясняется тем, что абиетиновая кислота при на-
гревании дает равновесную смесь, содержащую некоторое ко-
личество (кстати сказать, небольшое) левопимаровой кислоты.
Последняя соединяется с малеиновым ангидридом, равновесие
сдвигается и происходит дальнейшая изомеризация абиетино-
вой кислоты до полного ее израсходования.
Химические свойства смоляных кислот
545
По Сандерману34, изоабиетиновая кислота ведет себя так
же, как и абиетиновая, но это трудно объяснить, так как состав
изоабиетиновой кислоты неизвестен.
Продукт присоединения малеинового ангидрида к абиети-
новой кислоте может быть получен также в кипящем бензоле
в присутствии хлористого водорода, наличие которого, как от-
метили Харрис и Сандерсон37, обязательно.
Малеиновый аддукт левопимаровой кислоты может быть
получен непосредственно из канифоли. Для этого к расплав-
ленной канифоли, нагретой до 180°, добавляют теоретическое
количество малеинового ангидрида; кристаллизацией из уксус
ной кислоты получают твердый продукт. Уксусная кислота
обязательна в качестве растворителя лишь при первой кри-
сталлизации; последующие кристаллизации могут проводиться
в любом другом растворителе.
Малеиновый аддукт левопимаровой кислоты имеет темп. пл.
232°, [а],=—23,6°, [а]г, =—27°. Он содержит одну малореак-
ционноспособную двойную связь, в частности не присоединя-
ющую брома. Малеиновый аддукт левопимаровой кислоты яв-
ляется основным компонентом канифольно-малеиновых смол, ко-
торые будут рассмотрены далее.
Присоединение различных филодиенов. Левопимаровая кис-
лота представляет собой весьма активный диен, способный при-
соединять большинство классических филодиенов, хиноны, на-
фтохинон и т. д. Получаемые аддукты, по-видимому, не имеют
промышленного значения.
Дегидрирование и окисление. Превращение в дегидроабие-
тиновую кислоту, ретен или пимантрен. Обработка смоляных
кислот группы абиетиновой кислоты дегидрирующими агентами
(сера, селен, палладированный уголь) при высокой температуре
приводит к образованию ретена, а кислоты группы d-пимаровой
кислоты дают в тех же условиях пимантрен:
ретен пимантрен
Более энергичная обработка теми же дегидрирующими аген-
тами кислот группы абиетиновой кислоты дает частично арома-
тическую дегидроабиетиновую кислоту (см. стр 556). Флек
и Палкин38 получили дегидроабиетиновую кислоту дегидриро-
ванием на палладированном угле. Ломбар39 получил эту кислоту
путем обработки абиетиновой кислоты серой при 200°, а Дю-
35—156
546
Гл. XVI. Канифоль и ее производные
пои40 показал, что очень хорошим катализатором дегидриро-
вания является формиат никеля*. С помощью этого катализа-
тора оказывается возможным получать дегидро абиетиновую
кислоту в промышленном масштабе из канифоли сосны Pinus
halepensis, содержащей кислоты исключительно группы абиети-
новой кислоты.
К образованию дегидроабиетиновой кислоты приводят так-
же некоторые окислители, являющиеся дегидрирующими аген-
тами, например селеновый ангидрид, с помощью которого Фи-
зер41 впервые получил дегидроабиетиновую кислоту. Возможно,
что все окислители, способные вызывать окисление в а-поло-
жении, ведут себя по отношению к соединениям с двойными
связями совершенно так же.
Аутооксидация. Известно, что канифоль очень чувствительна
к действию кислорода, причем эта чувствительность возрастает
со степенью измельчения. Поэтому лучше хранить канифоль
в больших кусках, чем в порошке; хранение канифоли в боч-
ках, в которых она образует единый слиток, вполне гарантирует
ее сохранность.
Отметим прежде всего, что из всех смоляных кислот наибо-
лее склонна к аутооксидации абиетиновая кислота. Если вы-
держивать на воздухе образцы чистых смоляных кислот, то
быстрее всех желтеет абиетиновая кислота; еще быстрее про-
исходит аутооксидация абиетата натрия. d-Пимаровая кислота,
напротив, мало чувствительна к кислороду, что объясняется ее
строением — отсутствием сопряженных двойных связей. Нако-
нец, кислоты, содержащие только одну двойную связь (гидри-
рованные кислоты, присоединившие два атома водорода), прак-
тически уже не способны к аутооксидации. Поэтому в случаях,
когда имеется опасность самоокисления, представляет интерес
гидрированная канифоль.
Аутооксидация чистых смоляных кислот систематически не
изучалась; единственные точные данные, которые имеются в
отношении абиетиновой кислоты, приведены ниже.
Дюпон и Дюбур42 выдерживали на воздухе при рассеянном
свете образец чистой абиетиновой кислоты в течение пяти лет;
за это время удельное вращение изменялось с —100 до —15°,
в то время как кислотное число почти не изменялось (с 185 до
184,8). Неизмененная абиетиновая кислота сохраняет раство-
римость в петролейном эфире, но продукты окисления в нем
не растворяются, что дает возможность разделять их. Окислен-
ная фракция представляет собой некристаллизующийся аморф-
ный порошок.
* Следует отметить, что Дюпон работал с метилабиетатом, а не со свобод-
ной абиетиновой кислотой.
Химические свойства смоляных кислот
547
Следует отметить, что кристаллическая абиетиновая кис-
лота окисляется только с поверхности, тогда как в толще она
не изменяется.
Аморфный порошок, состоящий из смеси продуктов окисле-
ния, при нагревании до 130° легко теряет 6,5% воды; его кис-
лотное число равно 177; коэффициент омыления равен 344,
т. е. вдвое более кислотного числа. Таким образом, этот про-
дукт содержит как кислотные,
При омылении он расщепляет-
ся на оксикислоту и смесь ле-
тучих кислот, содержащую,
как было установлено, уксус-
ную кислоту. Это согласуется
с тем фактом, что при шере-
гонке канифоли (всегда более
или менее окисленной) полу-
чаются кислые воды, содер-
жащие уксусную, изомасляную
и валериановую кислоты, а
также с тем, что окисление
абиетиновой кислоты перман-
ганатом дает как раз перечис-
ленные кислоты. Окисленный
продукт больше не обладает
фенантреновым скелетом, так
как содержание в нем ретена
так и сложноэфирные группы.
время, часы
Рис. 63. Кинетика абсорбции кислоро-
да абиетиновой кислоты в ксилольном
растворе в присутствии абиетата ко-
бальта.
ничтожно —одно из трех ядер, несомненно, раскрывается в са-
мом начале окисления. Предполагают, что продукт аутооксида-
ции абиетиновой кислоты отвечает общей формуле.
Cj(;H2(;O4 или
/СООН
(С14Н21)—О—СО—СН3
4 ОН
Ясно, что, поскольку продукт не был очищен, это предпо-
ложение является гипотетическим.
Скорость присоединения кислорода к абиетиновой кислоте,
растворенной в ксилоле в присутствии абиетата кобальта, ил-
люстрируется кривой43 на рис. 63. На рисунке отчетливо видны
три стадии окисления: период индукции, стадия быстрого окис-
ления и стадия, когда окисление замедляется и прекращается.
Такой ход кинетической кривой объясняется тем, что основным
процессом является образование перекисей в результате цеп-
ных реакций. Перекиси разлагаются различными путями: с
образованием конечных продуктов реакции или свободных ра-
дикалов, инициирующих новые цепи. Первая стадия (индукци-
35*
548 Гл. XVI. Канифоль и ее производные
онный период) соответствует возникновению свободных радика-
лов и инициированию реакционных цепей; вторая стадия (харак-
теризующаяся возрастающей скоростью) соответствует развитию
цепного процесса; в третьей стадии новые реакционные цепи
уже не возникают, а имеющиеся цепи одна за другой обрывают-
ся. Это — стадия замедления и прекращения реакции.
Действие тепла. Пироабиетиновые и пиросмоляные кислоты,
смоляные и канифольные масла. При нагревании смоляные
кислоты претерпевают различные очень сложные превращения.
При температуре не выше 200° карбоксильная группа кисло-
ты остается неизменной, но структура кислоты изменяется, что
видно по уменьшению удельного вращения. Когда температу-
ра превышает 200°, происходят другие изменения, сопровож-
дающиеся выделением газообразных веществ и понижением
кислотного числа; удельное вращение, естественно, продолжает
уменьшается. Скорость, с которой происходят все эти измене-
ния, быстро возрастает с повышением температуры; ее можно
также увеличить с помощью различных катализаторов,
которые, кроме того, влияют на характер образующихся
веществ.
Для исследования продуктов, полученных в результате
нагревания смоляных кислот, нужно прежде всего разделить
их на кислую и нейтральную фракции. Для этого смолу рас-
творяют в эфире и ее кислые компоненты извлекают раство-
ром соды, разлагают 'натриевые соли уксусной кислотой и рас-
творяют выделенные смоляные кислоты в спирте, из которого
они кристаллизуются. Таким довольно обычным путем дости-
гается только грубое разделение на нейтральную и кислую фрак-,
ции, так как водные растворы резинатов натрия способны эмуль-
гировать масла. Чтобы избежать эмульгирования, нужно при-
менять в процессе разделения достаточно разбавленные щелоч-
ные растворы.
Отметим, что разделение может быть значительно улучшено
путем осаждения смоляных кислот циклогексиламином, с кото-
рым они все образуют нерастворимые соли.
Кислая фракция представляет собой сложную смесь, со-
держащую некоторое количество исходной кислоты и различ-
ные кислоты, образовавшиеся в результате ее превращения.
Эти кислоты называют «пиросмоляными кислотами», а часто
также и «пироабиетиновыми кислотами» (это название сле-
довало бы сохранить только за продуктами превращения са-
мой абиетиновой кислоты). Кстати, пироабиетиновая кислота
(полученная в результате пиролиза абиетиновой кислоты) яв-
ляется единственной хорошо известной из пиросмоляных кис-
лот; кроме того, кислые продукты пиролиза канифоли состоят
главным образом из пироабиетиновой кислоты.
Химические свойства смоляных кислот
549
Кислые продукты пиролиза канифоли были исследованы
уже давно. В 1890 г. Бишоф и Настфогель44, перегоняя кани-
фоль неустановленного происхождения, получили правовращаю-
щий продукт ([а]о=+63°), который они назвали «изосильви-
новым ангидридом», хотя он, конечно, не являлся ангидридом.
В 1906 г. Классон и Колер45, перегоняя под вакуумом смолу
стокгольмской сосны, также получили правовращающий про-
дукт ([а]о=+28°), который сени разделили на а-канифольную
левовращающую кислоту и 0-канифольную правовращающую
кислоту. Продукты, аналогичные промышленным смоляным
маслам, были выделены рядом авторов (Кельбе46, 1890 г.; Чирх
и Вольф47, 1907 г.; Шульц48, 1917 г.). Шульц получил право-
вращающую кислоту из смоляного масла и назвал ее «олео-
сильвиновой» кислотой. Позднее, в 1932 г. Ружичка49, перегоняя
предварительно подвергнутую пиролизу канифоль, наблюдал из-
менение ее удельного вращения вправо.
Кристаллические продукты, полученные при различных ви-
дах термической обработки канифоли разного происхождения,
безусловно, родственны между собой, но это родство требует
уточнения.
В 1926 г. Руэн50 показал, что из абиетиновой кислоты после
длительного нагревания при 250° и последующей перегонки
.при давлении 20 мм образуется правовращающая кислота; наи-
большим углом правого вращения ( + 58°) обладал продукт,
практически не содержавший более абиетиновой кислоты. Этот
продукт получил название пироабиетиновой кислоты. Другие
авторы51 показали идентичность этого продукта с правовра-
щающей кислотой, выделенной из канифоли сосны Pinus ha-
lepensis.
Истинную природу пироабиетиновой кислоты, которую дол-
гое время считали химически чистым продуктом, установили
в 1938—1939 гг. Флек и Палкин38. Эти авторы прежде всего
показали, что кислота, обладающая всеми свойствами пиро-
абиетиновой кислоты и, в частности, таким же удельным вра-
щением, получается при нагревании в течение пяти часов абие-
тиновой кислоты в присутствии палладия, нанесенного на уголь.
При проведении этого процесса они сделали следующие на-
блюдения. При 225° реакция идет без заметного выделения га-
зов с образованием- продукта состава С20Н30О2; при 275° вы-
деляется одна грамм-молекула водорода и получается про-
дукт состава С2оН2802, т. е. дегидроабиетиновая кислота (см.
стр. 556). Продукт состава С20Н3о02, получаемый при более
низкой температуре без выделения водорода, является смесью
дегидроабиетиновой, дигидроабиетиновой и тетрагидроабиети-
новой кислот, образующейся вследствие реакций изомеризации
абиетиновой кислоты. Те же авторы далее показали, что пиро-
550 Гл. XVI. Канифоль и ее производные
абиетиновая кислота Руэна является смесью дегидроабиетиновой
и дигидроабиетиновой кислот, так как при нагревании проте-
кает реакция диспропорционирования:
ЗСаоНзцОг —► С20Н28О2 - С20Н32О2
Состав пироабиетиновой кислоты можно, таким образом,
считать установленным. Состав же нейтральной фракции до
сих пор окончательно не установлен, несмотря на большое
число исследований, проводившихся главным образом с целью
выяснения состава смоляных масел, являющихся продуктами
термического разложения канифоли. Заметим кстати, что про-
мышленные смоляные масла, рафинированные различными ме-
тодами (например, перегонкой с известью, продуванием воз-
духом, обработкой серной кислотой и т. д.), в значительной
степени отличаются от нейтральных продуктов, образующихся
при пиролизе смоляных кислот.
Нейтральные продукты, образующиеся при термическом раз-
ложении собственно абиетиновой кислоты, были предметом
лишь немногих исследований. Основные работы, проводившие-
ся с практическими целями, касаются пиролиза канифоли,
что в первом приближении сводится к пиролизу абиетиновой
кислоты. Поэтому часто приходится совместно излагать резуль-
таты исследований пиролиза абиетиновой кислоты и канифоли.
При длительном нагревании абиетиновой кислоты при
240—270° получается примерно 70% пироабиетиновой кислоты;
остальное приходится на долю нейтральных продуктов. Таким
образом, в этом интервале температур можно, в частности,
получать пироабиетиновую кислоту. При температуре выше
300° все смоляные кислоты, являющиеся компонентами пиро-
абиетиновой кислоты, теряют кислотный характер за время от
24 до 48 час. При этом происходят одновременно декарбок-
силирование (с образованием главным образом углеводородов)
и дегидратация, приводящая к образованию абиетинового ан-
гидрида. Долгое время принимали во внимание только процесс
декарбоксилирования, сопровождающийся выделением дву-
окиси и окиси углерода. Образование абиетинового ангидрида
было обнаружено лишь недавно благодаря работам Василь-
ева52. Этот исследователь установил, что абиетиновая кислота,
будучи подвергнута пиролизу при относительно высокой тем-
пературе, теряет воду, вследствие чего ее кислотное число
понижается, а коэффициент омыления остается неизменным.
Отсюда логически следует вывод о возможности образования
в данном случае абиетинового ангидрида. При более высокой
температуре разлагается также и абиетиновый ангидрид. По-
этому можно предположить, что декарбоксилирование абие-
Химические свойства смоляных кислот 551
типовой кислоты может происходить одновременно тремя пу-
тями:
1. Прямое декарбоксилирование абиетиновой кислоты с об-
разованием абиетена и октогидроретена:
абиетен октогидроретен
2. Разложение абиетинового ангидрида с образованием де-
гидроабиетена и уже упомянутого абиетена:
3. Реакции, аналогичные предыдущим, но относящиеся к
пироабиетиновой кислоте и приводящие к образованию сле-
дующих продуктов (кроме указанных выше):
552 Гл. XVI. Канифоль и ее производные
Различные авторы пытались определить, какие из приве-
денных выше реакций преобладают. Пока можно лишь ука-
зать, что абиетиновый ангидрид, по-видимому, относительно
устойчивое соединение, разлагающееся только при высокой
температуре, дегидроабиетиновая кислота с трудом декарбок-
силируется при температуре выше 320°, а дигидроабиетиновая
кислота декарбоксилируется гораздо легче53.
Чтобы иметь более полное представление о компонентах
исследуемой нейтральной фракции, нужно учесть реакции де-
гидрогенизации, изомеризации, полимеризации и т. д., которые
могут накладываться одна на другую. Получаемая смесь на-
столько сложна, что невозможно перечислить все возможные
ее компоненты. При ректификации этой смеси получается (с
выходом примерно 60%) смесь углеводородов состава
С18_!9 Н24—32 , имеющих скелет ретена. Это — углеводороды, яв
ляющиеся основными компонентами смоляных масел.
Рассмотренные процессы термического разложения абиети-
новой кислоты, протекающие очень медленно (по крайней мере
при температуре не выше 300°), могут быть ускорены с помо-
щью многочисленных катализаторов, из которых достаточно
указать54 фосфорную кислоту, хлористый цинк, порошкообраз-
ные железо и никель и, наконец, хлористый алюминий. Эти
катализаторы, с одной стороны, ускоряют декарбоксилирова-
ние, а с другой,—-вызывают много изменений в самой струк-
туре продуктов декарбоксилирования. Процесс каталитическо-
го декарбоксилирования, по-видимому, отличается от обычного
термического процесса только тем, что он протекает гораздо
быстрее. Последующие изменения продуктов декарбоксилиро-
вания состоят главным образом в следующем:
1) перемещение двойных связей;
2) обрыв боковых цепей, связанных с ядром;
3) перемещение боковых цепей;
4) раскрытие циклов;
5) отщепление водорода и ароматизация гидроароматиче-
ских ядер;
6) гидрирование некоторых молекул водородом, выделяю-
щимся в процессе реакции;
7) реакции конденсации и полимеризации;
8) реакции расщепления и т. д.
Фосфорная кислота не способствует последующим превра-
щениям, и при пиролизе в ее присутствии образуются масла,
сходные с продуктами термической обработки без катализа-
тора. Железо и никель благоприятствуют, главным образом,
последующему дегидрированию, и получаемое в их присут-
ствии масло обогащено дегидроабиетеном. Хлористый цинк и
особенно хлористый алюминий являются весьма активными
Химические свойства смоляных кислот
553
катализаторами, вызывающими всю гамму приведенных выше
побочных реакций.
При термическом пиролизе без катализатора получаются
высыхающие смоляные масла, что связано с высоким содер-
жанием абиетена. Масла, полученные пиролизом в присутствии
хлористого цинка, имеют очень сложный состав; они содер-
жат только небольшое количество ретеновых компонентов и
много продуктов расщепления. Пиролиз в присутствии хлори-
стого цинка применяли во время второй мировой войны для
изготовления смазочных масел невысокого качества, выпускав-
шихся под названием «канифольных» масел.
Прочие свойства. В этом разделе рассматриваются важ-
ные свойства смоляных кислот, не описанные в предыдущих
разделах. Некоторые из приведенных реакций, описанных при-
менительно к абиетиновой кислоте, имеют общее значение.
Действие азотной кислоты и окислов азота. Дюбург55 пока-
зал, что абиетиновая кислота, обработанная в спиртовом или
уксуснокислом растворе азотной кислотой, образует динитро-
производное (темп. пл. 170—171°; [сф= —142°), для которого
Ломбар и Балтзингер60 предложили формулу:
Н3С СООН
Ашан57 показал, что действие окиси азота на абиетиновую-
кислоту ведет к образованию нитрозита (темп. пл. 153—154°:.
[а]у = —2° в хлороформе). Для этого вещества Ломбар и Балт-
зингер предложили формулу:
(C^I i.i0O5N2)2 или
Действие серной кислоты. Серная кислота присоединяется
к абиетиновой кислоте, но затем может быть вытеснена водой
с образованием оксикислоты, которая в большей или меньшей
степени превращается в лактон. После Грюна и Винклера56
этим вопросом занимался Ломбар, показавший, что продукт
получается всегда в виде некристаллизующейся смолы; иссле-
довать этот продукт не удалось.
554
Гл. XVI. Канифоль и ее производные
Действие хлорноватистой кислоты и гипохлоритов. Хлорно-
ватистая кислота (в виде гипохлоритов щелочных металлов),
присоединяясь к абиетиновой кислоте, образует дихлордиокси-
абиетиновую кислоту С2оНзо02С12(ОН)2 (темп. пл. 95°). Путем
ее омыления можно получить тетраоксиабиетиновую кислоту59.
Полимеризация. Полимеризация канифоли проводилась
фирмой Hercules Powder. Процесс проводится в присутствии
катализатора, например H2SO4, SnCl4, Z11CI2, BF3, в растворе
(растворителем может служить бензол, толуол, бензин, четы-
реххлористый углерод и т. д.) при температуре от —10 до
+ 65°. Таким путем получают смолу полипал, основным компо-
нентом которой является димер абиетиновой кислоты (гипоте-
тическая формула):
НООС СООН
Конденсация. Канифоль вступает в реакции конденсации
со многими соединениями: с альдегидами, кетонами, фенолами,
аминами. Эти реакции представляют интерес потому, что та-
ким путем может быть достигнуто заметное повышение твер-
дости канифоли. Реакции конденсации до сих пор никогда не
изучались теоретически; практические же сведения об этих ре-
акциях будут приведены далее, при рассмотрении химических
свойств канифоли.
литература
1. Lombard, Bull. Soc. chim., 1945, (5), 12, 8.
2. E 1 1 i n g s о n, J. Am. Chem. Soc., 1914, 36, 325.
3. Dupont, Desalbres, Bull. Soc. chim., 1926, (4), 39, 492.
4. Lombard не опубликовано.
5. Lombard, Premier Congres International de 1’Industrie des Peintures,
Paris, 1947, p. 393.
6. Kesler, Lowy, Faragher, J. Am. Chem. Soc., 1927, 49, 2902.
7. Hercules Powder, пат. США 2146897, 1934 г.
8. Devillers, Dupont, Dulou, Congres des Matieres Plastiq les,
1949, p. 57.
9. M a 1 v, Ann., 1864, 132, 249.
10. F 1 u c k i g e r, J. prakt. Chem., 1867, (1), 101, 235.
11. Bischoff, Nastvogel, Ber., 1890, 23, 1921.
12. Easterfield, Bagley, J. Chem. Soc., London, 1904, 85, 1243.
13. Knecht, Hibbert, Mon. Sci., 1921, (5), 11, 107.
14. Fonrobert, Pallauf, C., 1926, II, 199; Fonrobert, C., 1930,
1,828.
15. Lombard, Frey, Bull. Soc. chim., 1948, M., 1196.
Химические свойства смоляных кислот
555
16. Kraft, Ann., 1935, 520, 133.
17, Fie se г, Campbell, J. Am. Chem. Soc., 1938, 60, 159.
18. S a n d e r m a n n, Ber., 1941, 74, 154.
19. Harris, Sanderson, J. Am. Chem. Soc., 1948, 70, 334.
20. Lombard, Bull. Soc. chim., 1948, M., 1186.
21. R i t c h i e, Mac В ur ney, J. Am. Chem. Soc., 1949, 71, 3736.
22. Чугаев, Teeapy, Ber., 1913, 46, 1769.
23. Johansson, Mon. Sci., 1921, (5), 11, 73.
24. R u z i с к а и сотр., Helv. chim. acta, 1923, 6, 677; 1924, 7, 875, 1931, 14,
233; 1932, 15, 915, 1289, 1300; 1940, 23, 124.
25. P a 1 к i n, Harris, J. Am. Chem. Soc., 1933, 55, 3677.
26. Lombard, Bull. Soc. chim., 1946, M., 109.
27. Lombard, Ebelin, Bull. Soc. chim., 1953, M., 930.
28. Bru s, Bull. Soc. chim., 1939, (15), 6, 780; 1947, M„ 955.
29. Lombard, Bull. Soc. chim., 1944, (5), 11, 98, 526.
30. R u z i с к а и сотр., Helv. chim acta, 1933, 16, 178.
31. F 1 e c k, Palkin, J. Am. Chem. Soc., 1939. 61, 1230, 3197.
32. R u z i с к а и сотр., Helv. chim. acta, 1922, 5, 324.
33. Levy, Ber., 1907, 40, 3658; Ray, Simonsen, Indian Forest
Records, 1924, 11, 211; Ruzicka, Meyer, Helv. chim. acta, 1922,
5, 315; A s c h a n, Ann., 1930, 483, 124.
34. Hassel st rom, MacPherson, J. Am. Chem. Soc., 1939, 61, 2247;
Sanderman n, Ber., 1943, 76, 1257, 1261; Г a p у ш а, ЖОХ, 1938,
8, 1042.
35. Dupont, Dulou, Leon, Bull. Soc. chim., 1951, M., 61.
36. Ruzicka и сотр., Helv. chim. acta, 1932, 15, 1289; 1933, 16. 176; 1937,
20, 1546.
37. Harris, Sanderson, J. Am. Chem. Soc., 1948, 70, 2079.
38. Fl e c k, Palkin, J. Am. Chem. Soc., 1938, 60, 921, 2625.
39. Lombard, C. r., 1941, 213, 793; Bull. Soc. chim., 1942, (5), 9, 834.
40. Dupont, Dulou, Leon. Bull. Soc. chim., 1951, M., 239.
41. Fieser, Campbell, J. Am. Chem. Soc., 1938, 60, 167.
42. D u p о n t, D u b о u r g, Bull. Inst. Pin., 1928, 205.
43. D u p о n t, Levy, Allard, Bull. Soc. chim., 1930, (4), 47, 60, 147,
942, 1216.
44. Bischoff, Nastvogel, Ber., 1890, 23, 1921.
45. К 1 a s о n, Kohler, J. prakt. Chem., 1906, (2), 73, 353.
46. К e 1 b e, Ber., 1880, 13, 888.
47. Tschirch, Wolff, Arch, der Pharm., 1907, 245, 1.
48. Schultz, Mon. Sci., 1920, (5), 10, 103.
49. Ruzicka и сотр., Helv. chim. acta, 1922, 5, 337; 1923, 6, 833; 1925, 8,
632.
50. R о u i n, Bull. Inst. Pin, 1926, 479.
51. D u p о n t, D u b о u r g, Bull. Inst. Pin, 1928, 181.
52. Vassiliev, Bull. Soc. chim., 1947, M., 1080; 1948, M„ 178.
53. D e s a 1 b r e s, Chim. et Ind., 1946, 56, 363.
54. Vassiliev, Chim. et Ind., 1942, 47, 543; Easterfield, Bagley,
J. Chem. Soc. (London), 1904, 85, 1246: Brodschi, Bull. Inst. Pin,
1938, 89, 118, 159, 203.
55. D u b о u r g, Bull. Inst. Pin, 1929, 138.
56. Goldblatt, Lowy, Burnett, J. Am. Chem. Soc., 1930, 52, 2132.
57. A s c h a n, Ber., 1922, 55, 2944.
58. Gr u n, Winkler, Mon. Sci., 1921, (5), 11, 159.
59. M. P. R. L a m p r e c h t, Institute Forestal de Investigaciones у Expe-
riencias, Madrid, 1949, XX, 43, 93.
60. L о m b a r d, В a 1 t z i n g e r, C. r., 1953, 236, 1794.
61. Veil uz и сотр., Bull. Soc. chim., 1954, M., 401.
556
Гл. XVI. Канифоль и ее производные
Дегидроабиетиновая кислота
Дегидроабиетиновая кислота может быть получена дегидри-
рованием абиетиновой кислоты многими методами1,21 ®' *'5-
дегидроаэиетиновая кислот
Физические константы дегидроабиетиновой кислоты: темп,
пл. 180°; [а]у=+62°; [a]^=+70°. Она образует кристаллический
метиловый эфир с температурой плавления 60°, который может
быть получен в очень чистом виде: путем омыления этого эфи-
ра получается чистая дегидроабиетиновая кислота. Метиловый
эфир можно получить непосредственно дегидрированием мс-
тилабйетата6.
Дегидроабиетиновая кислота кристаллизуется в виде тре-
угольных или трапецеидальных пластинок; ее спектр погло-
щения в ультрафиолетовой области характеризуется дву-
мя близкими по интенсивности максимумами7 при 268 и
270 Ш|1.
Благодаря наличию ароматического ядра дегидроабиетино-
вая кислота способна давагь очень большое число производных,
замещенных в ядре (обычно в положении 6, в исключительных
случаях — в положении 8). Известно несколько двузамещен-
ных производных в положениях 6 и 8.
В табл. 33 приведены важнейшие из производных дегидро-
абиетиновой кислоты и указаны их константы8-12.
Самым доступным производным является замещенная де-
гидроабиетиновая кислота, содержащая сульфогруппу .в поло-
жении 6; она представляет интерес как поверхностно-активное
вещество. При щелочной плавке эта сульфокислота разлагает-
ся, не образуя ожидаемого оксипроизводного, а при обработке
раствором брома в бромиде калия дает бромопроизводное.
6,8-Динитродегидроабиетиновая кислота легко получается пу-
тем нитрования дегидроабиетиновой кислоты концентрирован-
ной азотной кислотой.
Дегидроабиетинол получают восстановлением метилдегидро-
абиетата по Буво и Блану. Следует указать, что 6-оксидегидро-
абиетинол обладает эстрогенными свойствами и по активности
близок к эстрону13.
Дегидроабиетиновая кислота
557
ТАБЛИЦА 33 Свойства дегидроабиетиновой кислоты и ее производных
Температура Температура
кипения °C плавления °C Удельное вращение
Дсгидроабиетиновая кис- 260 (при 15 мм) 180 [а]у =-|-56о (спирт)
лота
6-сульфо- То же 247 -248 (разл.) [a]D = 4-72,4° (спирт)
6-ацетил- » 174—175 Мд = 4-74° (спирт)
6-амино- в 215 [Дд = 4 82° (спирт)
6-окси- . . в 215 —
6-бром- - - в 200-202 Ыд=4-8Г (ацетон)
6-метокси- . - . в 201—203,5
6-карбокси- в Ниже 280 (а]о=+71° (спирт)
6,8-динитро- . в 178-185 (разл.) [а]о = +49° (ацетон)
6-амино-8-нитро- . . в 281—282 [а]о =4"И7О (ацетон)
(разл.)
Ангидрид дегидроабиети- 142
новой кислоты .... » [а], =4-67°
Метилдегидроабиетат . . 260° (при 15 мм) 60 [а]j = 59° (спирт)
6-хлорметил- .... То же 117 [“1; = +72° (спирт)
6-иодметил- ... В 88—89 (разл.) [а]< = 4-74°
6-бромметил- . . . в 108 [а]у = -+ 72° (петролей- ный эфир)
6-формил- ..... в 117,5 {а]у-=+74°
6-окси- в 156—157 Мд =+71 (спирт)
Аллилдегидроабиетат в 47
Винилдегидроабиетат . . 174° (при 10 мм) 47 [а],-=4*53°
Дегидроабиетинол . . . 240° (при 14 мм) 47
6-окси- То же 180—181,5 1“]д = +72° (спирт)
6-амино- В 140 (а1д = 4*72° (спирт)
Де.гидроабиетиналь в 77 (семикарб- азид 248—250°) [а], =4*67°
ЛИТЕРАТУРА
1. Fieser, Campbell, J- Am. Chem. Soc., 1938, 60, 167.
2. Fl e c k, Palkin, J. Am. Chem. Soc., 1938, 60, 921, 2625.
.3. Lombard, C. r., 1941, 213, 793; Bull. Soc. chim., 1942, (5), 9, 834.
4. J e g e r, Durst, В u c h i, Helv. chim. acta, 1947, 30, 1853.
558
Г л. XVI. Канифоль и ее производные
5. D и р о п t, Dulou, D е v i 1 1 е г s, Bull. Soc. chim., 1949, M., 315.
6. D u p о n t, Dulou, Leon, Bull. Soc. chim., 1951, M., 239.
7. Fieser, Campbell, J. Am. Chem. Soc., 1938, 60, 159.
8. Fieser, Campbell, J. Am. Chem. Soc., 1938, 60, 159, 2633; 1939
61, 2528.
9. Campbell, Morgana, J. Am. Chem. Soc., 1941, 63, 1838.
10. C a m p b e 1 1, Todd, J. Am. Chem. Soc., 1940, 62, 1287; 1942, 64, 932.
11. Dupont, Chem. Zeit., 1951, 75, 207.
12. В r u s и сотр., Bull. Soc. chim., 1950, M., 1035.
13. Zeiss, J. Am. Chem. Soc., 1948, 70, 858.
Пространственное строение смоляных кислот
Смоляные кислоты принадлежат к двум группам — абиети-
новой (абиетановой) и пимаровой (.пимарановой) в зависи-
мости от того, имеет ли молекула скелет ретена или пиман-
трена:
СН3 сн3
ретен пммантрен
Кислоты этих двух групп являются производными углево-
дородов абиетана и пимарана:
Абиетан, согласно терминологии, принятой для стероидов,
имеет транс-антн-транс-конфигурацию.
К абиетановой группе относятся абиетиновая, неоабиети-
новая и левопимаровая кислоты, к пимарановой группе d-пи-
маровая и изо-б/-пимаровая кислоты.
Ниже приведена стереохимическая формула, принятая для
абиетиновой кислоты:
6
Химия канифоли и ее производных
559
ЛИТЕРАТУРА
1- Vesterberg, Вег., 1903, 36. 4200.
2. R uzi с к а и сотр., Helv. chim. acta, 1933, 16, 842.
3. Ruzicka и сотр., Helv. chim. acta, 1923, 6, 677; 1924, 7, 875; 1931, 14,
233.
4. Lombard, Диссертация, Paris, 1943, p. 39.
5. Levy, Ber., 1907, 40, 3658; 1909, 42, 4305; Z. anorg. Chem., 1913, 81,
149.
6. Ruzicka и сотр., Helv. chim. acta, 1925, 8, 645; 1931, 14, 565; 1938,
21, 573.
7. Levy, Ber., 1929, 62, 2497.
8. Lombard, Bull. Soc. chim., 1942, (5), 9, 834; Paris, 1943, p. 40.
9. Vocke, Ann., 1932, 497, 247.
10. R u z i с к а и сотр., Helv. chim. acta, 1933, 16, 169.
11. R о u i n, Bull. Inst. Pin, 1928, 203; Ruzicka и сотр., Helv. chim. acta.
1925, 8, 637; Fieser, Campbell, J. Am. Chem. Soc., 1938, 60,
168.
12. R uz i с к а и сотр., Helv. chim. acta, 1940, 23, 355.
13. Wood war d, J. Am. Chem. Soc., 1942, 64, 75; Harris, Sander-
son, J. Am. Chem. Soc., 1948, 70, 335
14. Арбузов, C., 1942, II, 892—893.
15. H a r r i s, Sanderson, J. Am. Chem. Soc., 1948, 70, 339; Frey,
Диссертация, Strasbourg, 1951, p. 53.
16. Lombard, Bull. Soc. chim., 1942, (5), 9, 834.
17. Ruzicka и сотр., Helv. chim. acta, 1932, 15, 915.
18. R u z i с к а и сотр., Helv. chim. acta, 1940, 23, 124.
19. Fl e c k, Palkin, J. Am. Chem. Soc., 1940, 62, 2044.
20. Harris, Sanderson, J. Am. Chem. Soc., 1948, 70, 2079, 2081.
21. Lombard, Bull. Soc. chim., 1946, M., 430.
22. C a m p b e 1 I, Todd, J. Am. Chem. Soc., 1942, 64, 928.
23. В a r t о n, J. Chem. Soc. (London), 1948, 1197; 1949 (дополнит.) 232.
24. Haworth, Barker, J. Chem. Soc. (London), 1939, 1299.
25. В a c h m a n, J. Am. Chem. Soc., 1950, 72, 2000.
26. К1 у n e, J. Chem. Soc. (London), 1953, 3073.
ХИМИЯ КАНИФОЛИ И ЕЕ ПРОИЗВОДНЫХ
Канифоль представляет собой стекловидное, ломкое, твер-
дое, прилипающее к рукам вещество; уд. в. 1,070—1,085 г/см3-,
размягчается между 70 и 80°, превращается в жидкость при 120°.
Как уже указывалось, канифоль в основном является сме-
сью абиетиновой, неоабиетиновой и d-пимаровой кислот, в ко-
торой сильно преобладает абиетиновая кислота (количествен-
ные соотношения были указаны (на стр. 521). Кроме того, ка-
нифоль содержит также некоторые второстепенные компоненты:
неомыляемые вещества, смоляные оксикислоты, смоляные ан-
гидриды и, наконец, несколько смоляных кислот вполне опре-
деленного строения. Из них можно назвать левопимаровую
кислоту, которая содержится в очень ограниченном количестве,
изо-4/-пимаровую кислоту, являющуюся малоизученным спут-
ником d-пимаровой кислоты, количество которого недостаточно,,,
точно определено, и, наконец, пироабиетиновую кислоту, об-
разующуюся в перегретых частях перегонного аппарата.
.560 Гл. XVI. Канифоль и ее производные
Хорошая канифоль имеет кислотное число, близкое к 170,
достигающее иногда 175 (кислотное число чистой кислоты
С20Н30О2 равно 185); 5фирное число очень низкое (2 или 3);
содержание неомыляемых веществ обычно около 5%, но может
достигать и 10%.
Некоторые сорта канифоли при поверочном анализе дают
иногда совсем не те результаты, которые ранее были указаны
в паспортах. Перегретая канифоль может содержать значи-
тельное количество ангидридов, окислившаяся канифоль со-
держит эфиры, образующиеся ib результате взаимной этери-
фикации гидроксильных и карбоксильных групп. В обоих слу-
чаях коэффициент омыления превышает кислотное число.
Раньше, когда канифоль вырабатывалась кустарными спо-
собами, часто получались продукты со значительным эфирным
числом; при современных методах непрерывной перегонки с
водяным паром получается канифоль с весьма незначитель-
ным эфирным числом. Можно сделать аналогичные выводы в
отношении канифоли, полученной при сухой перегонке смоли-
стой древесины; благодаря современным методам очистки та-
кая канифоль стала практически идентичной хорошей канифоли,
полученной из живицы.
Поэтому в настоящее время вполне правомерно рассматри-
вать канифоль, независимо от ее происхождения, йак смесь
смоляных кислот состава С2оН3002, сопровождаемую 5—10%
неомыляемых веществ. Однако в действительности это соблю-
дается только для канифоли из смолы, собранной с деревьев
путем подсочки поздней осенью.
До сих пор рассматривались лишь чистые смоляные кисло-
ты; так как при необходимости технического применения смо-
ляных кислот используют канифоль, целесообразно рассмот-
реть ее свойства, ограничившись реакциями, имеющими тех-
ническое значение.
-Согласно принятой ранее схеме, распределим свойства ка-
нифоли следующим образом:
1. Свойства, связанные с наличием карбоксильной группы.
2. Свойства, связанные с непредельностью.
3. Дегидрирование и окисление.
4. Действие тепла.
5. Прочие свойства.
Свойства канифоли, связанные с наличием карбоксильной
группы. Основными промышленными производными канифоли, в
образовании которых участвует карбоксильная группа, являют-
ся резинаты металлов, натриевые мыла, эфиры и ангидрид
канифоли, смоляные спирты.
Резинаты металлов. Резинаты представляют собой продук-
ты полного или частичного замещения металлами кислотного
Химия канифоли и ее производных
561
водородного атома канифоли. Резинаты щелочных металлов
гидрофильны, растворимы в воде и гидроксилсодержащих
растворителях и нерастворимы в углеводородах; резинаты
остальных металлов, напротив, гидрофобны и растворяются в
неполярных органических растворителях, особенно в углево-
дородах.
Резинаты нещелочных металлов, в зависимости от способа
получения, делят на плавленые и осажденные. Плавленые ре-
зинаты обычно изготовляются путем введения тонкораздроб-
ленных окислов металлов при перемешивании в расплавлен-
ную канифоль при 200—270°; если окись металла мало реак-
ционноспособна, ее заменяют солями, легко обменивающими
металл, обычно солями уксусной кислоты. Осажденные рези-
наты получают путем обработки в растворе натриевого кани-
фольного мыла солью соответствующего металла; в лабора-
торных условиях лучше всего в качестве растворителя брать
спирт, в котором мыло мало гидролизуется, в промышленных
условиях обычно берут воду.
Резинаты металлов используются как связующие для лаков
и как сиккативы.
В качестве связующих для лаков используются резинаты
кальция, цинка и алюминия, в которых примерно половина
кислотных групп канифоли остается незамещенной. При вве-
дении в плавленую канифоль большего количества окиси ме-
талла не удается получить прозрачные и однородные продукты.
Перечисленные резинаты представляют собой прозрачные, слег-
ка окрашенные смолы, растворимые в органических раствори-
телях и маслах, гораздо более твердые, чем канифоль: они
размягчаются только при 120°, в то время как канифоль раз-
мягчается при 70—80°. Эти резинаты выпускаются в твердом
виде или в виде 50%-ного раствора в соответствующем рас-
творителе, что очень удобно для потребителя.
В качестве сиккативов применяют главным образом рези-
наты магния и кобальта, плавленые или осажденные. Осаж-
денные резинаты обычно содержат 8% металла. Это количе-
ство вдвое превышает количество металла в плавленых рези-
натах, полученных с применением окислов металлов. Однако
плавленые резинаты могут содержать и больше металла, чем
осажденные, если изготовлять их с применением солей органи-
ческих кислот, а не окислов металлов. В этом случае получают-
ся смешанные соли с повышенным содержанием металла, со-
храняющие в то же время растворимость в органических рас-
творителях.
Все большие количества резинатов металлов выпускаются
в виде растворов, а не в виде порошков, что облегчает введение
их в краски и лаки и повышает их сохранность (порошкообраз-
36—156
562 Гл. XVI. Канифоль и ее производные
ные резинаты окисляются и в конце концов теряют раствори-
мость в органических растворителях).
Более подробные сведения о плавленых резинатах имеются
в статье Борглина, Мошера и Эллиота1.
Натриевые канифольные мыла. Как указывалось ранее,
многие смоляные кислоты при нейтрализации содой в водном
растворе образуют легко кристаллизующиеся натриевые соли.
Канифоль не дает кристаллических солей, так как в растворе
происходят сложные коллоидно-химические процессы.
'При обработке канифоли соответствующим количеством
5%-ного водного раствора соды при 100° получается прозрач-
ный раствор, который при охлаждении густеет и желатини-
руется. Это загустевание часто сопровождается помутнением,
что придает массе вид сливок. Однако в определенных преде-
лах концентраций раствор .может оставаться прозрачным без
гелеобразования. Если раствор слишком разбавлен, то вслед-
ствие гидролиза он мутнеет через несколько часов; кроме того,
в прозрачных растворах часто образуется гелеобразный осадок.
Эти явления сходны с явлениями, характерными для мыл
жирных кислот, с той разницей, что канифольные мыла никогда
не бывают твердыми.
Смоляные мыла хорошо растворяются и являются сильны-
ми эмульгаторами. Достаточно концентрированные водный рас-
творы смоляных мыл дают совершенно прозрачные коллоидные
растворы при смешении со всеми органическими растворителя-
ми и в первую очередь с углеводородами. Если органический
растворитель, не смешивающийся с водой, добавить к раствору
канифольного мыла, частично выпавшего в осадок, происходит
растворение этого осадка, и раствор становится совершенно
однородным. Наконец, канифольные мыла, при высокой кон-
центрации действующие как растворители, при низкой концент-
рации являются стабилизаторами эмульсий.
Для объяснения всех этих коллоидных явлений достаточно
точных данных не имеется.
Смоляные мыла являются если не единственными, то во
всяком случае важнейшими из клеев, применяемых в писче-
бумажном производстве. Целесообразно дать некоторые сведе-
ния об этих клеях, хотя они имеют только отдаленное отноше-
ние к лакокрасочному производству.
Клеи для писчебумажного производства всегда кислые; со-
держание в них свободной смолы колеблется в пределах от
15 до 85%. Обычно различают: мыла с низким содержанием
свободной смолы (15—25%), со средним' содержанием (35—
45%) и смоляные суспензии (65—85% свободной смолы).
Мыла с низким содержанием свободной смолы могут быть
получены холодным способом путем обработки канифоли со-
Химия канифоли и ее производных 563
ответствующим количеством едкого натра в аппаратах с ме-
шалкой в течение 6—7 час. Они могут быть также получены
при повышенной температуре (100—120°) в варочных котлах,
обогреваемых паром. В этом случае можно использовать в ка-
честве омыляющего агента не едкий натр, а карбонат натрия.
Мыла со средним содержанием свободной смолы также
могут изготовляться как на холоду, так и при нагревании; обыч-
но они, как и смоляные суспензии, требуют применения за-
щитных коллоидов.
Смоляные суспензии приготовляются исключительно при на-
гревании в варочных котлах; повышенное содержание свобод-
ной кислоты может быть достигнуто только путем применения
защитных коллоидов, таких, как казеин или желатин. На 100 кг
канифоли требуется примерно 5 кг казеина.
Во Франции применяются для писчебумажной промышлен-
ности три сорта клеев, выпускаемых фирмами Delthirna, Gillet
и Bewoid. По способу Delthirna канифоль обрабатывают
содой при обычной температуре и получают мыло с малым
содержанием свободной смолы. Клей фирмы ETewoid изготов-
ляется по методу Wieger’a и содержит до 92% свободной смолы.
Отличительная особенность способа Gillet заключается в том,
что получаемые мыла содержат значительное количество рези-
ната аммония.
При использовании смоляных мыл в качестве клеев на бу-
мажных фабриках их разбавляют, превращая в так называе-
мое «клеевое молоко». Это «молоко» является дисперсией с
размером частиц 0,05 р при малом содержании свободной смо-
лы в мылах и 0,5 р. для смоляных суспензий.
Канифольные мыла находят также некоторое применение
в мыловарении, где канифоль (в количестве не более 10%)
добавляют к жирам. Нужно отметить, что канифольные мыла
сами по себе обладают плохим моющим действием; они быстро
образуют обильную, но не стойкую пену; кроме того, они «лип-
нут»— покрывают предметы, подлежащие мойке, пленкой сво-
бодной смолы, образующейся в результате гидролиза. Все эти
недостатки не ощущаются в присутствии большого количества
(80—90%) мыла жирных кислот; в этих условиях смоляные
мыла оказывают полезное действие: они размягчают и застав-
ляют пениться твердые мыла, например мыла на основе твер-
дых жиров и пальмового масла.
Канифольное мыло, представлявшее собой соль канифоли,
подвергнутой диспропорционированию, т. е. соль пироабиетино-
вой кислоты, применялось в качестве эмульгатора2 в производ-
стве синтетического каучука GR-S 10.
Эфиры канифоли. Ранее были подробно рассмотрены про-
цессы образования и свойства эфиров смоляных кислот. Здесь
36*
564 Гл. XVI. Канифоль и ее производные
следует добавить, что в производстве используются по возмож-
ности прямые методы этерификации или, если это необходимо,
каталитические методы.
Эфиры канифоли получают следующим образом. К кани-
фоли, нагретой до 200°, добавляют глицерин в количестве от 11
до 15% от веса канифоли, т. е. в небольшом избытке; получен-
ную смесь нагревают до 270—290° и выдерживают при этой тем-
пературе в течение получаса. Реакцию можно ускорить путем
применения в качестве катализаторов солей цинка или алюми-
ния и особенно путем введения тунгового масла. Процесс реко-
мендуется проводить в алюминиевой аппаратуре.
Хотя некоторые смоляные кислоты образуют кристалличе-
ские эфиры, эфиры, полученные, исходя из канифоли, никогда
не кристаллизуются; они не омыляются и, следовательно, очень
стойки, что придает им большую ценность.
Одноатомные спирты образуют с канифолью очень вязкие
маслянистые эфиры, которые могут применяться в качестве пла-
стификаторов, в особенности для нитроцеллюлозы. Таковы ме-
тиловые, этиловые, бензиловые и другие подобные эфиры. Мно-
гоатомные спирты, например глицерин или пентаэритрит, об-
разуют эфиры, являющиеся очень твердыми, неклейкими смо-
лами, совместимыми с основными пигментами. Эти эфиры на-
зывают эфирами канифоли или эфирами смоляных кислот.
Канифоль можно этерифицировать свободными спиртовыми
группами глифталевых смол, получая модифицированные гли-
фталевые смолы.
Ангидрид канифоли. Ангидриды смоляных кислот могут быть
легко получены по методу Фонробера, который заключается
в обработке смоляных кислот уксусным ангидридом. Метод
этот применим и к канифоли. По патенту Витекса3, канифоль
растворяют в смеси толуола, ксилола и уксусного ангидрида и
нагревают до кипения, отгоняя уксусную кислоту по мере ее
образования в виде азеотропной смеси.
Ангидрид канифоли должен представлять известный инте-
рес как полупродукт для получения смоляных эфиров; однако
он пока не вырабатывается в промышленных масштабах.
Смоляные спирты. Восстановление метилабиетата в абие-
тинол по методу Буво и Блана впервые было осуществлено'
Ружичкой4 в 1922 г. Этот метод был усовершенствован Дюпо-
ном5, который проводил реакцию в среде инертного раствори-
теля со строго рассчитанным количеством соды и спирта; при
этом выход достигал 87% от теоретического.
Во время второй мировой войны фирмой Hercules Powder6
был осуществлен в промышленном масштабе процесс восста-
новления эфиров смоляных кислот водородом под давлением
в присутствии катализатора на основе хромата меди. Процесс,
Химия канифоли и ее производных______ 565
однако, не останавливается на стадии образования абиетинола;
происходит более или менее полное гидрирование двойных свя-
зей С=С абиетиновой кислоты и в соответствии с условиями
реакции образуется дигидроабиетинол, тетрагидроабиетинол или
смесь этих спиртов. В промышленной практике исходят, разу-
меется, непосредственно из метилового эфира канифоли и полу-
чают смесь гидрированных смоляных спиртов*. При гидриро-
вании хотя бы одной двойной связи продукт становится стой-
ким к окислению. Получаемые спирты устойчивы и бесцветны.
Аналогично протекает гидрирование канифоли (см. стр. 566).
Смоляные спирты представляют собой очень вязкие, слабо
окрашенные жидкости, не смешивающиеся с водой, но раство-
римые в большинстве органических растворителей. Это первич-
ные спирты, которые можно легко этерифицировать, окислять
в альдегиды, превращать в сульфокислоты и т. д. Они совме-
щаются с натуральным и синтетическими каучуками, с произ-
водными целлюлозы и с многочисленными синтетическими по-
лимерами, например с виниловыми.
С одноосновными кислотами эти спирты образуют жидкие
эфиры, с многоосновными кислотами — твердые эфиры. Со смо-
ляными кислотами они также образуют твердые эфиры, пред-
ставляющие интерес как связующие для лаков.
Все эти продукты имеют перспективы широкого применения
в промышленности.
Свойства канифоли, связанные с ее непредельностью. Не-
предельность канифоли обусловливает ее реакционную спо-
собность, которая в одних случаях нежелательна, а в других
является ценным свойством, на котором основаны многочис-
ленные промышленные синтезы.
Кислоты группы абиетиновой кислоты, обладающие сопря-
женными двойными связями, весьма склонны к аутооксидации,
что является причиной многих недостатков материалов, изго-
товленных с применением канифоли. Из этих недостатков можно
упомянуть пожелтение лаков, окрашивание мыл, ослабление
проклейки бумаги. Лучший способ устранения этих явлений —
гидрирование канифоли.
Из положительных свойств, связанных с непредельностью
канифоли, укажем способность присоединять малеиновый ангид-
рид с образованием малеиновых смол, реагировать с хлором,
с гипохлоритами, с серной кислотой, конденсироваться с форм-
альдегидом, с фенолами, с аминами, с феноло-формальдегид-
ными смолами, а также способность к полимеризации. Ниже
* Промышленный продукт содержит также некоторое количество дегидро-
абиетинола, наличие которого объясняется частичным диспропорционирова-
нием. Дегидроабиетинол, не чувствительный к действию кислорода, нисколько
не ухудшает качества продукта.
566 Гл. XVI. Канифоль и ее производные
будут рассмотрены реакции гидрирования канифоли, получения
малеиновых смол, реакции конденсации и полимеризации. Дей-
ствие хлора, гипохлорита натрия и серной кислоты удобнее
рассмотреть далее при описании реакций окисления и прочих
свойств канифоли.
Гидрирование канифоли. Смоляные кислоты легко присоеди-
няют два атома водорода при жидкофазном гидрировании в
присутствии катализатора; такое частичное гидрирование про-
исходит легко для группы d-пимаровой кислоты и труднее —
для группы абиетиновой кислоты. Тем не менее со скелетным
никелевым катализатором можно гидрировать абиетиновую
кислоту при обычной температуре и нормальном давлении.
Канифоль гидрируется, разумеется, труднее, чем смоляные
кислоты, но ее также можно гидрировать в растворе (обяза-
тельно при нагревании) или в расплаве. Фирма Hercules
Powder8 использует непрерывный способ гидрирования, при ко-
тором плавленая канифоль стекает на последовательно распо-
ложенные полки для катализатора (активированный сплав ни-
кель—алюминий), где (продувается током водорода. Катализа-
тор периодически заменяют, причем самый свежий катализа-
тор помещают в конце потока. Процесс ведут при давлении
10 кг/см2 и температуре от 70 до 300°. В результате такой обра-
ботки присоединяется в среднем по три атома водорода на
каждую молекулу содержащихся в канифоли смоляных кис-
лот. Получаемый продукт представляет собой смесь дигидро-
и тетрагидрокислот и выпускается под маркой стейбелит. Этот
продукт рекомендуется длц тех областей применения канифоли,
когда нежелательна ее аутооксидация; для проклейки бумаги, в
мыловарении, для изготовления сиккативов. Склонность стей-
белита к кристаллизации меньше, чем у исходной канифоли.
Образование малеиновых смол. Известно, что из всех смо-
ляных кислот левопимаровая кислота является самым актив-
ным диеновым соединением; поэтому она присоединяет мале-
иновый ангидрид при обычной температуре. Абиетиновая и нео-
абиетиновая кислоты, также содержащие сопряженные двой-
ные связи, присоединяют малеиновый ангидрид только при от-
носительно высокой температуре (180°), но образуют при этом
такой же аддукт, как и левопимаровая кислота.
При обработке расплавленной канифоли малеиновым ан-
гидридом последний присоединяется к кислотам абиетиновой
группы; в результате этого температура размягчения канифоли
повышается в соответствии с количеством присоединенного ма-
леинового ангидрида и может достигнуть 25°. Такие продукты не
используются, однако, непосредственно как смолы для лаков.
Промышленные абиетиново-малеиновые смолы получают
обычно путем этерификации глицерином канифольно-малеино-
Химия канифоли и ее производных
567
вого аддукта, представляющего собой внутренний ангидрид
трехосновной кислоты и образующего соответствующие полиэфи-
ры. Абиетинов'О-малеиновые смолы можно получать также, дей-
ствуя на глицериновый эфир канифоли малеиновым ангидри-
дом. Другие абиетиново-малеиновые смолы получаются путем
этерификации различными спиртами канифольно-малеиновых
аддуктов с последующей обработкой окислами металлов. Та-
ким образом получают широкую гамму ценных продуктов.
Реакции конденсации канифоли*. Канифоль конденсируется
со многими соединениями. Конденсация канифоли с фенола-
формальдегидными смолами, приводящая к образованию аль-
бертолей, имеет большое промышленное значение. Предметом
многочисленных заявок является конденсация канифоли с форм-
альдегидом; кроме того, канифоль конденсируется с фено-
лами, альдегидами и т. д. Эти промышленно важные реакции
конденсации еще очень плохо исследованы. Здесь мы ограни-
чимся рассмотрением процессов конденсации канифоли с фе-
ноло-формальдегидными смолами. Один из промышленных спо-
собов проведения этого процесса состоит в том, что продукт
конденсации фенола и формальдегида в стадии резола** нагре-
вают с канифолью при 290°. Можно также нагревать смесь фе-
нола, канифоли и триоксиметилена до температуры около 200°.
Не рассматривая подробно вопросы синтеза альбертолей,
ограничимся изложением гипотезы, которая, по-видимому, луч-
ше всего согласуется с экспериментальными данными.
Резолы представляют собой фенолоспирты
в которых п относительно невелико. По Гульчу9 эти феноло-
спирты реагируют в виде о-хинонметидов
* Чтобы не усложнять изложение, здесь объединены реакции конденсации
и реакции присоединения.
** Резолы—продукты конденсации фенолов с формальдегидом в щелоч-
ной среде. Степень поликонденсации резолов невелика; они растворяются в
органических расторителях.
568
Гл. XVI. Канифоль и ее производные
взаимодействующих с непредельными соединениями по меха-
низму, аналогичному реакции Дильса:
о °
СН—R ^\/ \
I + II - ! R
С112
Реакция салигенина с абиетиновой кислотой может быть
изображена схемой:
Подробнее эти вопросы освещены в литературе10.
В действительности резолы сложнее салигенина (они обла-
дают по меньшей мере двумя реакционноспособными мети-
лольными группами), а потому продукты присоединения имеют
гораздо более сложное строение. Имеются и другие сложности,
о которых мы не будем здесь упоминать.
В результате реакций получаются большие молекулы, в
свободном состоянии обладающие остаточными кислотными
группами канифоли — альбертолевые кислоты. Эти кислоты мо-
гут быть этерифицированы многоатомными спиртами, в част-
ности глицерином, причем в зависимости от условий реакции
и взятых реагентов можно получить обширный ряд продуктов
конденсации.
Полимеризация канифоли. Полимеризация канифоли была
предметом длительных исследований фирмы Hercules Powder,
зафиксированных в виде большого числа заявок11. В результате
критического изучения этих заявок можно выделить следующий
.практический способ. Раствор канифоли в ароматическом (бен-
зол, толуол) или алициклическом (газолин) растворителе вы-
держивают в присутствии катализатора при температуре от
О до 100° в течение нескольких часов. Лучшими катализаторами
являются: серная кислота, хлористое олово, хлористый цинк;
для диспергирования катализаторов необходимо хорошее пере-
мешивание. I
Реакция под действием серной кислоты была изучена Брю-
сом12 применительно к абиетиновой кислоте; она приводит к
образованию смеси, содержащей 5% дигидроабиетиновой кис-
лоты в форме лактона, 5% дегидроабиетиновой кислоты и 10%
Химия канифоли и ее производных
569
димера абиетиновой кислоты, плавящегося при 395° (вероятную
формулу его см. на стр. 554). Два первых компонента обра-
зуются в результате диспропорционирования водорода. Обра-
зование лактона сопровождается заметным уменьшением кис-
лотного числа продуктов.
Полимеризованная канифоль фирмы Hercules Powder, вы-
пускаемая под торговым названием полипал, имеет более вы-
сокую температуру размягчения, чем канифоль (90—100° вме-
сто 70—80°). Она содержит от 30 до 40% димерных кислот,
остальное приходится на долю мономерных кислот. Смола пэ-
липал не имеет склонности к кристаллизации; ее кислотное
число меньше, чем у канифоли (150 вместо 170), что объяс-
няется частичной лактонизацией.
Области применения полимеризованной канифоли в общем
те же, что и канифоли: получение эфиров, резинатов, модифи-
кация глифталевых и феноло-формальдегидных смол, получе-
ние малеиновых смол и т. д.
Дегидрирование и окисление канифоли. Окисление кани-
фоли и ее дегидрирование, являющееся частным случаем окис-
ления, имеют большое практическое значение. Аутооксидация
канифоли представляет собой обычно нежелательное явление.
На самопроизвольном окислении смоляных кислот в сосновой
древесине основано изготовление смол винсол (см. стр. 571).
Окисление канифоли различными агентами позволяет повы-
сить ее твердость, а дегидрогенизация канифоли дает возмож-
ность преодолеть ее чувствительность к кислороду. Все эти во-
просы будут рассмотрены ниже.
Дегидрирование. Дегидрирование канифоли в жестких усло-
виях, так же как и дегидрирование в этих условиях абиети-
новой кислоты, приводит р образованию ретена. Проводились
исследования с целью разработки промышленного процесса
получения ретена из канифоли, который, по-видимому, легко
осуществим.
При осторожном дегидрировании абиетиновой кислоты по-
лучается дегидроабиетиновая кислота, менее чувствительная к
действию кислорода и близкая в этом отношении к дигидро-
абиетиновой кислоте. Дегидроабиетинол, эфиры, твердые соли
и натриевые мыла дегидроабиетиновой кислоты обладают теми
же достоинствами, что и соответствующие производные стей-
белита, причем изготовление их экономически более выгодно.
Другие производные дегидроабиетиновой кислоты, например, ее
сульфопроизводные, также могли бы представлять интерес.
В промышленном производстве нельзя исходить из кристал-
лической абиетиновой кислоты, но в этом нет и необходимости,
так как при дегидрировании канифоли получается смесь дегидро-
абиетиновой и d-пимаровой кислот, не склонных к аутоокси-
37—156
570
Гл. XVI. Канифоль и ее производные
дации*. Более чистый продукт может быть получен дегидри-
рованием канифоли сосны Pinus halepensis, содержащей кис-
лоты только абиетиновой группы и дающей почти чистую де-
гидроабиетиновую кислоту.
Указанные промышленные методы дегидрирования приме-
нимы как к канифоли, так и к ее эфирам, и не требуют ни
особых материалов, ни сложного оборудования14.
Аутооксидация. Как и смоляные кислоты (в особенности
абиетиновая кислота), канифоль очень склонна к аутооксида-
ции, но нужно заметить, что этот процесс происходит быстро
лишь при условии, что канифоль тонко размолота.
Фарион14, выдерживая на воздухе 1 г порошкообразной ка-
нифоли, наблюдал следующее увеличение ее веса:
Время Увеличение Время Увеличение
сутки веса, мг сутки веса, мг
2 4 30 32,5
5 9 40 36
10 18 50 38,5
20 26 60 42,5
При аутооксидации канифоли происходят существенные из-
менения ее свойств, что видно из приведенных данных:
Исходный Через 14
продукт месяцев
Содержание веществ, нерастворимых в
петролейном эфире, % ............. 4,4 54,7
Кислотное число (определено ациди-
метрически при обычной температуре) 159 151,2
Коэффициент омыления................ 165,8 175,5
Элементарный состав, %
С................................. 76,66 72,23
Н.................................. 9,63 8,98
О................................. 12,71 17,79
Неокисленная канифоль почти полностью растворяется в
петролейном эфире, но по мере аутооксидации образуются в
возрастающем количестве продукты, нерастворимые в нем; при
плавлении эти продукты вновь приобретают растворимость.
Аутооксидация канифоли приводит к повышению коэффициента
омыления без заметного изменения кислотного числа, что объ-
ясняется взаимной этерификацией кислотных и спиртовых групп.
При аутооксидации канифоль не теряет растворимости в
большинстве органических растворителей (в спирте, ацетоне,
эфире, в сложных эфирах, уксусной кислоте, хлороформе и т.д.),
исключая алифатические углеводороды. Отметим еще, что
окисленная канифоль не содержит перекисей.
* d-Пимаровая кислота и остальные кислоты этой группы не обладаюг
склонностью к аутооксидации.
Химия канифоли и ее производных 571
Смола винсол. Следует привести некоторые сведения отно-
сительно смолы винсол, являющейся побочным продуктом при
получении экстракционной канифоли16.
Пни, богатые смолой (20—25%), раскалывают в щепу («чип-
сы») и экстрагируют эту щепу растворителем нефтяного про-
исхождения. При последующей дистилляции отгоняют сначала
растворитель, затем терпены и сосновое масло. Осадок вновь
экстрагируют растворителем нефтяного происхождения и рас-
твор обрабатывают фурфуролом, который не смешивается с
алифатическими углеводородами и образует слой, в котором
собираются все окрашенные (продукты. Этот слой отделяют и
отгоняют фурфурол. Остается осадок — смола винсол.
В какой же мере смола винсол является продуктом окисле-
ния смоляных кислот? Смола винсол безусловно содержит
фенольные смолы, которые, по-видимому, находятся наряду со
смоляными кислотами в большинстве выделений хвойных де-
ревьев16, и значительное количество фенолов. Кроме того, в
смоле винсол содержатся компоненты кислотного характера
(смола имеет кислотное число 100), являющиеся, по-видимому,
продуктами окисления смоляных кислот. Приводимые ниже ха-
рактеристики образца смолы винсол подтверждают наличие в
ней компонентов фенольного и кислотного характера:
Температура размягчения, °C ................116
Содержание веществ, нерастворимых в пстролей-
ном эфире, % .................98
Кислотное число (определено ацидиметрически
при обычной температуре) ........ 93
Коэффициент омыления ..........135
Содержание метоксильных групп, % . . . 5,18
Содержание фенольных гидроксильных групп, % 3—4
Содержание неомыляемых веществ, % .... 7,7
Смола винсол мало чувствительна к действию кислорода.
Повышение твердости канифоли путем окисления. Окисление
смоляных кислот обычно повышает температуру их плавления.
Так, например, 7,8,9,14-тетраокситетрагидроабиетиновая кислота
(по другой номенклатуре тетраоксиабиетиновая) плавится при
250°. Обрабатывая измельченную канифоль воздухом при тем-
пературе, близкой к температуре ее размягчения, можно повы-
сить температуру ее плавления. Аналогичный результат полу-
чается при окислении канифольного мыла гипохлоритом нат-
рия с последующим осаждением смолы17; как было показано
ранее, при этом можно выделить промежуточную дихлор-
диоксикислоту, которая плавится при 95°.
В некоторых патентах предлагается повышать твердость
канифоли, действуя на нее хлором18; часть хлора, присоединив-
шегося к канифоли, затем отщепляют путем дегидрохлориро-
572 Гл. XVI. Канифоль и ее производные
вания; получаемый продукт является смесью дегидроабиетино-
вой кислоты и хлорпроизводных неопределенного состава.
Действие тепла. При нагревании канифоли получаются глав-
ным образом газообразные продукты, кислоты (пиросмоляные
кислоты) и нейтральные продукты; кроме того, образуется
вода, которая, выделяясь, увлекает с собой легкие жирные кис-
лоты и легкие органические продукты, не смешивающиеся с
водой и составляющие так называемый пинолин. В первом при-
ближении можно принять, что пиролиз канифоли протекает
подобно описанному выше пиролизу абиетиновой кислоты: пи-
росмоляные кислоты практически представляют собой пироаби-
етиновую кислоту, а нейтральная часть образована главным
образом абиетиновым ангидридом и гаммой указанных выше
углеводородов — абиетена, дегидроабиетена, октогидроретена
и т. д. В присутствии катализаторов происходит, кроме того,
большое количество различных вторичных процессов, в част-
ности деструкция, полимеризация, перегруппировки и т. д.
Термическое разложение канифоли имеет известное про-
мышленное значение: оно применяется с целью получения смеси
углеводородов, которая называется смоляным маслом.
В промышленности пиролиз канифоли проводят с дистил-
ляцией образующихся продуктов;, реакцию ведут в больших
чугунных котлах, снабженных медным шлемом, соединенным
с медным холодильником, погруженным в бак, наполненный
холодной водой. Установка может работать периодически или
непрерывно; в последнем случае реактор сообщается с пла-
вильным котлом, в котором производится предварительная не-
прерывная плавка канифоли; уровень канифоли в котле авто-
матически поддерживается постоянным. Установка может ра-
ботать в течение месяца без перерыва; если в котле имеется
всегда достаточное количество канифоли, можно избежать пе-
регревов, и образование кокса будет незначительным. Получае-
мое масло имеет однородный состав и постоянные свойства.
При пиролизе канифоли получается продукция пяти видов:
газы, кислые воды, несмешивающаяся с водой легкая фракция
(пинолин), смоляные масла и, наконец, осадок, который мо-
жет быть мягким, как битум (если перегонка производи-
лась не слишком быстро), или, наоборот, полностью закоксо-
ванным.
В качестве иллюстрации приводим материальный баланс
промышленной установки (в % вес.):
Газы....................................9
Кислые воды ............................3,5
Масло (с большей или меньшей примесью пиро-
смоляных кислот) ......................77
Пек неперегоняющийся....................4
Пинолин.................................3,5
Химия канифоли и ее производных
573
Газы состоят из окиси и двуокиси углерода в равных соот-
ношениях. Кислые воды содержат около 3% жирных кислот,
уксусную кислоту и высшие кислоты.
Пинолин—это весьма сложная смесь, перегоняющаяся в
очень широком диапазоне температур; главными компонентами
этой смеси являются метилциклогексен и терпены.
Смоляное масло также является важным продуктом; оно
перегоняется при 290—350° и содержит в первых погонах не-
большое количество пинолина. При быстрой перегонке полу-
чается вязкое масло, богатое пиросмоляными кислотами (так
называемое «тяжелое масло»); если же перегонка проводится
медленно, то декарбоксилирование проходит гораздо полнее
и сразу получается почти нейтральное масло. Масло обычно
разделяют на несколько фракций. Общее количество получае-
мого масла (77% от веса продуктов пиролиза) распределяется
по фракциям, например, следующим образом (в %):
Светлое масло мутное (содержит некоторое ко-
личество воды) ............... 5
Светлое масло (основная фракция) . 56
Синее масло............... . 8
Зеленое масло ................................ 8
Самым ценным техническим продуктом наиболее постоян-
ного состава является основная фракция светлого масла, со-
стоящая главным образом из углеводородов (абиетен, дегидро-
абиетен, октогидроретен и т. д. в различных соотношениях).
Содержание значительного количества абиетена придает маслу
в некоторой степени способность к высыханию. Это масло вы-
пускается в качестве товарного продукта после сложной обра-
ботки: нейтрализации, обесцвечивания, дезодорации и удале-
ния следов воды, придающих маслу мутность. Если при этом
проводятся только такие сравнительно мягкие операции, как
нейтрализация содой, то химический состав масла в процессе
очистки не изменяется. Масла, полученные каталитическим де-
карбоксилированием или подвергнутые рафинированию в жест-
ких условиях (например, перегонкой над окислами металлов),
содержат компоненты, не имеющие ретенового скелета, и лишь
в весьма слабой степени обладают способностью к высыханию.
Такие изменения, по-видимому, обусловлены реакциями де-
струкции и изомеризации.
В период второй мировой войны под названием «канифоль-
ное масло» выпускали масла, полученные путем плавления ка-
нифоли в присутствии хлористого цинка; эти продукты, вовсе
не обладающие способностью к высыханию, могли применяться
в качестве смазочных масел. Исходя из реакций, которые пред-
положительно должны происходить при пиролизе, возможными
574 Гл. XVI. Канифоль и ее производные
компонентами смоляного масла являются: абиетен, дегидро-
абиетен, октогидроретен и т. д. Многие авторы пытались уточ-
нить эти данные. Ломбар19 предложил определять диеновое
число и выход ретена из смоляных масел.
Диеновым числом называют количество (в миллиграммах)
малеинового ангидрида, которое присоединяется к 1 г масла
при 190°. Предполагается, что это число пропорционально со-
держанию в масле абиетена.
Для определения диенового числа к 1 г масла, пометенному в пробирку,
добавляют 0,5 г малеинового ангидрида, пробирку встряхивают и нагревают
при 190° в течение 45 мин. Затем смесь растворяют в толуоле и обрабатывают
кипящей водой, чтобы гидролизовать и растворить не вошедший в реакцию
малеиновый ангидрид. Определив его количество, по разности находят коли-
чество присоединившегося к маслу малеинового ангидрида.
Выход ретена указывает, в какой степени в масле сохрани-
лись вещества со скелетом ретена.
Выход ретена определяют следующим способом. Навеску исследуемого
масла (5 г) смешивают с 7 г селена и нагревают при перемешивании в течение
8 час. в металлической бане, температура которой поддерживается в преде-
лах 335—345°. Охлажденную смесь экстрагируют бензолом, фильтруют и пе-
регоняют. Фракция, перегоняемая между 200 и 300° при давлении 15 мм, со-
держит ретен; ее охлаждают во льду, а выпавшие кристаллы освобождают от
масла, отжимая на фильтровальной бумаге, и взвешивают.
Масла, декарбоксилированные с применением фосфорной
кислоты, богаты веществами с ретеновой структурой и обла-
дают особенно высоким диеновым числом, соответствующим
содержанию 70% абиетена. Продажные смоляные масла обычно
богаты веществами с ретеновой структурой, но рафиниро-
ванные масла имеют низкое диеновое число, соответствующее
содержанию 10% абиетена; тяжелые масла имеют более высо-
кое диеновое число. Наконец, масла, полученные с участием
хлористого цинка, бедны веществами с ретеновой структурой,
а масла, полученные при помощи кислых земель, вовсе не со-
держат этих веществ.
Ван Той провел тщательную ректификацию основной фрак-
ции светлого масла20 и выделил в чистом виде 1,2,3,4-тетрагидро-
ретен (темп. пл. 92°):
СН3 Смоляные масла применяются в качест-
I ве высыхающих масел и как смазочные
масла; в (первом случае они конкурируют
с жирными высыхающими маслами, а во
[ Н riHCH втором — с нефтяными продуктами. В обо-
1 3)2 их .случаях смоляные масла представляют
собой продукты невысокого качества и их
надо рассматривать главным образом как заменители.
Следует отметить, что сульфированные смоляные масла, по-
видимому, представляют интерес как детергенты.
Химия канифоли и ее производных 575
Прочие свойства канифоли. В этом разделе рассматрива-
ются некоторые реакции, позволяющие получать на основе ка-
нифоли технически важные продукты неизвестного или не пол-
ностью установленного строения.
Действие формальдегида. Из большого количества име-
ющихся патентов следует рассмотреть патент Бурна21, в кото-
ром описано получение смол путем взаимодействия порошко-
образной канифоли, с добавкой извести, с 40%-ным водным
раствором формальдегида. В результате конденсации образует-
ся продукт, представляющий интерес как лаковая смола.
Действие фенолов. Конденсации канифоли с фенолами по-
священо много патентов. Согласно патенту I. G.22, канифоль об-
рабатывают фенолами в присутствии трехфтористого бора, со-
ляной кислоты, четыреххлористого титана, хлористого олова
и т. д. в каком-либо растворителе. Канифоль, обработанная
крезолом в присутствии трехфтористого бора в четыреххло-
ристом углероде в течение 4 час., при 10°, имеет кислотное чис-
ло 114, ацетильное число 64,8 и температуру размягчения 105°.
Действие серной кислоты. Как и большинство ненасыщен-
ных веществ, смоляные кислоты реагируют с серной кислотой.
В некоторых случаях при этом образуются совершенно опре-
деленные продукты: сульфированная дегидроабиетиновая кис-
лота, лактоны дигидроабиетиновой, d-пимаровой и дигидро-
zf-пимаровой кислот.
При взаимодействии серной кислоты с канифолью индиви-
дуальные продукты не получаются; кроме того, происходит
разложение серной кислоты с выделением сернистого ангид-
рида. Можно избежать восстановления серной кислоты, про-
водя реакцию при достаточно низкой температуре, но получить
при этом кристаллизующиеся продукты не удается.
Грюн и Винклер22 путем разложения водой продуктов взаи-
модействия канифоли с серной кислотой при —5° получили
оксипроизводные.
Согласно американскому патенту24, канифоль растворяют в
жидком сернистом ангидриде (при —10°) и добавляют дымя-
щуюся серную кислоту, предпочтительно в присутствии бор-
алюминиевого катализатора или хлористого цинка. Для рас-
творения предельных соединений, нерастворимых в серной кис-
лоте, добавляют органический растворитель. Кроме того, до-
бавляют лед и нейтрализуют массу водой; в результате полу-
чается диспергированный продукт.
Следует напомнить, что при действии серной кислоты на
канифоль, растворенную в каком-либо растворителе, полу-
чается так называемая полимеризованная канифоль, о которой
указывалось выше.
576 Гл. XVI. Канифоль и ее производные
Канифоль является очень важным и сравнительно дешевым
исходным сырьем для производства лаков и красок. Она пред-
ставляет собой вещество с 20 атомами углерода в молекуле,
образующими плотную структуру фенантрена. Молекула содер-
жит реакционноспособную карбонильную группу и систему со-
пряженных двойных связей.
Канифоль используется для изготовления сиккативов; ее
натриевые мыла образуют коллоидные растворы и эмульсии.
Основной интерес представляет использование канифоли для
получения лаковых смол. При непосредственном использовании
канифоли в производстве лаков не удается получать материалы
высокого качества, но на основе канифоли можно получать ши-
рокую гамму лаковых смол. Таким образом проложен уже
широкий путь и работы то синтезу лаковых смол на основе
канифоли получают все большее развитие.
ЛИТЕРАТУРА
1. В о г g 1 i n, Mosher, Elliot, Ind. Eng. Chem., 1944, 36, 752.
2. Amberg, Ind. Eng. Chem., 1948, 40. 487.
3. V i t e x. фр. пат. 971653, 1951 г.
4. Ruzicka и сотр., Helv. chim. acta, 1922, 5, 588.
5. Devillers, Dupont, Dulou, Congres des Matieres plastiques,
Paris, 1949, p. 57.
6. Hercules Powder, пат. США 214689/, 1934 г.
7. News, 1947, 3670 (December).
8. Hercules Powder, пат. США 2155036, 1939 г.; пат. США 2174651, 1940 г.
9. Hultzsch, Вег., 1941, 74, 898.
10. G г е t h, Angew. Chem., 1938. 51, 719; Fonrobert, Fette u. Seifen,
1943, 514. Hultzsch. J. prakt. Chem., 1941, (2), 158, 275; 1941, 159,
155, 180; Ber., 1941, 74, 898, 1533, 1539; 1942, 75, 106, 363; v a n Der
Meer, Rec. trav. chim. Pays-Bas, 1944, 63, 147—156.
11. Hercules Powder, пат. США 2108928, 1938 г.; 2124675, 1938 г.; 2154704,
1939 г.; 2302576, 1943 г.; 2302577, 1943 г.; 2302632, 1943 г.; 2306650,
. 1943; г. 2342382, 1944 г.; 2307641. 1943 г.; 2310374, 1943 г.; 2327165,
1944 г.; 2329515, 1944 г.; 2288659. 1943 г.; 2375618, 1945 г.
12, В г u s и сотр., Bull. Soc. chim., 1950, М., 1032.
13. D и р о п t, Dulou, Leon, Bull. Soc. chim., 1951, M., 239.
14. F a h r i о n. Mon. Sei., 1903, (4), 17, 350; 1907, (4), 21. 478.
15. S h e a г о n, Patton, S h i n g 1 e r, Ind. Eng. Chem., 1948. 40, 1695;
Schantz, Marvin, Ind Eng. Chem., 1939, 31, 585; Highto-
wer. Chem. Eng., 1947, 54, 119—121; Humphrey, Ind. Eng. Chem.,
1943, 35, 1062-1067.
16. Lombard, Produits resineux. Dunod, Paris, 1946, p. 248.
17. I. G., герм. пат. 514151, 1925 г.
18. Павловский, Дмитров, сов. пат. 8907, 1927 г.
19. Lombard, Produits resineux, Dunod, Paris, 1946, p. 132, 184, 191, 199.
20. Le van Thoi, E zp on da, Bull. Soc. chim., 1950, M., 1035.
21. В о о r n e, англ. пат. 247620, 1924 г.
22. I. G., фр. гат. 734390, 1932 г.
23. G г й n, Winckler, Mon. Sci., (5), 11, 159.
24. Пат. США 2348200, 1944 г.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Абиетан 558
Абиетен 551, 572, 573
Абиетиновая кислота 323, 360, 520,
523
аутооксидация 546
малеиновые аддукты 391, 544
эфиры 392, 536
Абиетиновый ангидрид 537, 550
Абиетииол 564
Абиетино-малеиновые смолы 566
Адипиновая кислота 263, 358
Адиполь. 174
Азелаиновая кислота 28
Айцен 494
Акризоли 247
Акрилат целлюлозы 117
Акриловые
мономеры 235
пластификаторы 238
полимеры 240 и сл.
сополимеры 404
Акролеин 375
конденсация с мочевиной 352
Акроналы 245
Алкидаль 460
Алкидные смолы 329, 356 и сл.
малеииизация 392, 400
модификация феиоло-формальде-
гидиых смол 281, 323
модифицированные 367
сочетание с меламиио-формальде-
гидиыми 344
— с мочевиио-формальдегидными
338
фумаровые 403, 404
Алкиларилсилоксаны 513
Алкилоксалаты целлюлозы 118
Алкилсилаиолы и силандио-
лы 504
Алкилсилоксаны 510
Алкоголиз 371, 372
Алкоксисиланы 503
Аллилфумараты 404
Аллилцеллюлоза 125
Аллопрен 456, 493, 494
Альбан 449
Альбертолевые кислоты 568
Альбертоли 281, 328, 460, 461, 567
Амидопроизводные целлюлозы 127
Амилцеллюлоза 124
Амино-альдегидные смолы 347 и сл .
Аминопроизводные целлюлозы 127
Аморит 493
«Ангидрид карбик» 388, 398
«Ангидрид петрекс» 396, 398
Аиилино-альдегидные смолы 348
Антиоксиданты 188, 448
Аральдит 409
Арахиновая кислота 26
Арилсилоксаны 512
Арохлор 191
Асидсил 475, 494
Аутооксидация
канифоли 570
масел 35
смоляных кислот 546
Афковил СТ-3 171
Ацетали целлюлозы 125
Ацетальдегид, конденсация с мочеви-
ной 352
Ацетаты
метил- и бензилцеллюлозы 126
целлюлозы 97, НО, 115, 131, 151
152—155
Ацетилендимочевина 350
Ацетилиденмочевина 352
Ацетил рицинолевая кислота 68
Ацетильное число 82
Ацетобутират целлюлозы 97, 119
153—155
Ацетокротоиат целлюлозы 119
Ацетол 375
Ацетон-мочевинные смолы 354
Ацетоиитрат, ацетопропиоиат, ацето-
пропионатобутират, ацетосили-
кат целлюлозы 119, 120
Ацетофенон 354
Ацетоформиат и ацетофталат целлюло-
зы 119, 120
Бакелитовые лаки 327
Бакелиты 169, 176, 189, 279, 329
Балата 449, 494
Бальцер 174
Бекациты 328
Бекозолы 281, 329, 415, 461
Бензилцеллюлоза 97, 124, 153—155
578
Предметный указатель
Бензоат целлюлозы 118
Битумно-кумароновые краски 256
Блек-аут 490, 493
Бондеризация 190
Бреон 170
Буиа 232, 493
Бутадиен-акрилонитрильиые сополи-
меры 491
Бутапрен 493
Бутвар 216, 221
Бутнл 493
Бутилкаучук 493
Бутилцеллюлоза 124
Бутират целлюлозы 116
Валерат целлюлозы 116
«Варка» масел 52
Вельвик 170
Вибрин 269
Винамулы 245
Винилиты
VAGH 169, 180, 222, 417
VMCH 169, 176, 212, 214, 222
VYCC 169, 183
VYCM 170
V.'DR 170, 214
VYHH 168, 170, 193, 195, 197 и
сл., 204, 213, 222
VYHL ' 216,- 221
VYLF 168, 171, 173, 193, 197,
205, 210, 213
VYMC 212
VYNS 170, 201, 205
VYNV 170
VYNW 201, 205
f XYSG 216
Виниловые полимеры 156 и сл.
Вииилцеллюлоза 124
Виннапасы 161
Винсол 569, 571
Вискоза 97, 151
Вистанекс 494
Водноэмульсионные краски 164, 232,
244, 437
Воски кумароновые 258
Врезинол 415, 417
Вулкабонд 494
Вулкалок 476, 493
Вулканизация 446
акриловых эластомеров 241
Вулкапрены 489 сл., 494
Вулкафоры 489
Вулколлан 494
Высыхание
масел 33, 59, 77
модифицированных алкидных смол
369
Высыхающие масла 25 сл., 230, 281,
320, 321, 360
смоляные 573, 574
Галипоты 518
Галовакс 191
Галоидпроизводиые целлюлозы 127
Гексабромное число 54, 82
Гексаметилендиамин 263, 264
Гексаметилеидиизоциаиат 263
Гексаоксистеариновая кислота 29
Гексилцеллюлоза 123
Гельва 161
Гемицеллюлозы 85
Герколин 174
Гидракрилат целлюлозы 117
Гидрохлорированные
каучуки 445, 463 и сл.
раббоны 484
Гликольмалеат 402, 403
Гликольцеллюлоза 125
Гликолят целлюлозы 117
Гликофены 328
Глиоксальдиуреид 350
Глифталевые смолы
367 и сл.
Глицериды 25, 372
Глицерин 358, 379
абиетаты 394
Глицериновые эфиры целлюлозы 125
Глицеролиз 371, 372
Глутарат целлюлозы 118
Гранодин процесс 190
Гриньяра метод получения алкилга-
лоидсиланов 506
Гуттаперча 449, 495
Дакрон 264, 268
Дартекс 456, 495
Дегидратированное касторовое масло
52, 66
Дегидроабиетен 551, 572, 573
Дегидроабиетиновая кислота 539, 544,
545, 556
Дегидроабиетинол 556
Декстропимаровая кислота 520
Деллатол 274
Десмодуры
изоцианаты 275
полиуретаны 277
Десмофены 269, 277
Деструкция
каучуков 447
целлюлозы 133
Детел 456, 495
«Дефекты» структуры 334
Джи-ар-эс 498
Предметный указатель
579
Диазометаи 383
Ди аллилфтал ат 56
Дибутилтетрагидрофталат 388
Дибутилцеллозольв-фталат 212, 213
Дигидроабиетииовая кислота 541
Дигидроабиетинол 537, 565
Дигидро-«1-пимаровая кислота 529,
541
Диглицериды 376, 379
Диеновое число 574
Диеновый синтез 55, 72, 74, 75, 308,
325, 385, 568
Диизоцианаты 431
Диметиленциклогексан 389
Диметилфталат 383
Диоксистеариновая кислота 27, 67
Дифенилолпропан 288, 41!
Дифос 214
Дициандиамид 349
Диэтилдифенилмочевина 350
Дурез 189, 417
Дуропрен 456, 494
Дурофен 306V, 329
Дутрекс 174
Жевательная резина 259
Жеоны 170, 173
Загустители 239, 246, 247
Игамиды 263, 264, 267, 273
Игелит PCU 170
Идентификация
акриловых полимеров 247
двухосновных кислот 405
эпоксидных смол 432
Изаио масло 53
Изановая кислота 31
Изоабиетиновая кислота 543
йИзоаддукт» 390
Изоамилоксалат целлюлозы 118
Изобутилен-изопреновые полимеры
491
/i-Изобутилфенол 288
Изодиеновый показатель 405
Изокаучук 465
Изолак 4S5
Изолинузиновая кислота 29
Изомеризация смоляных кислот 540
Изомеризованные линолевые кислоты
28
ИзочКпимаровая кислота 520, 523,
530
Изосил ьеиновый ангидрид 549
Изотиоцианатные производные целлю-
лозы 128
Изоцианаты 275 431
Инвертированные масла 34
Инфракрасные спектры поглощения
целлюлозы 107, 108
Иодное число 57, 81
Итаконовая кислота 404
Канифоль 518 и сл.
гидрирование 565, 566
модификация фенольных смол, 327
— эпоксидных смол 429
облагороженная 280
полимеризация 554, 568
эфиры 533, 563
Канифольная кислота 523
Канифольно-малеиновые смолы 391,
293, 566
Канифольные
масла 553, 573
мыла 562
Карбоксиметилцеллюлоза 126
Карбонат целлюлозы 117
Касторовое масло 31, 52, 66, 204
Каурит 350
Каучуки 257, 400, 437 и сл.
гидрохлорированные 463 и сл.
натуральный 437 и сл.
окисленные 478 и сл.
— частично 487
синтетические 232, 241, 489 и сл.
хлорированные 451 и сл.
циклизованные 468 и сл.
Кетостеариновая кислота 32
Кислотное число 81
Кислоты высыхающих масел 25, 77,
360
Клеи
поливинилбутиральные 218
резиновые 419
смоляные 562
термопреиовые (циклокаучуковые)
476
эфироцеллюлозные 146
Крезолы 286
Кремиийорганические
полимеры 499 и сл.
производные целлюлозы 123
Креп 437
Крестапон 415
Крестин 486
Крилле 495
Кромодин процесс 190
Кротонат целлюлозы 117
Кротоновый альдегид 353
Ксаитогеиат целлюлозы 97, 129
Ксиленолы 286, 287
Кумарон (беизофуран) 249, 250
Кумароно-индеиовые смолы 249 и сл.,
580
Предметный указатель
Лактат целлюлозы 117
Лактопрены 241
Латеколи 247
Латексные краски 232, 244
Латексы
гидрохлорирование 464 ]
натуральный 437, 438
неопреновые 495
гллиолитовые 495
синтетические 491
хемигама 498
хлорирование 454
циклизация 469
Леа число 40, 83
Левопимаровая кислота 520, 523, 527
малеиновый аддукт 544
Лиафоль 274
Лигнин 85
Ликановая кислота 32
Лнноксин 42, 47, 65
Линолеат и линоленат целлюлозы 117
Линолевая кислота 28, 29, 67, 77,
346, 369
Линоленовая кислота 29, 82
Линолеум 65
Линузиновая кислота 29
Льняное масло 44, 56
окислительная полимеризация 66
Льюисол 392
Люфены 281, 288, 302, 329
Малеиновые
аддукты 325, 387, 399, 544, 566
линейные полиэфиоы 400
смолы 385 и сл., 566
сополимеры 177, 339, 402
Малеиновый ангидрид (кислота) 385
в алкициых смолах 357
в винилитах 169
в полиэфирах, сшитых стиролом
231
модификация высыхающих масел 73
Маннит и маннитан 359
Марбон 477, 494
Масла
высыхающие 25 и сл.
малеиновые аддукты 399
метилсилоксановые 511
модифицикация полистирола 230
— фенольных смол 230, 329
Масляно-стирольные пленкообразую-
щие 230
Мезаконовая кислота 405
Мезамол 191
Меламинметиды 341
Меламино-формальдегидные смолы
340 и сл.
Метакриловые полимеры 237 и сл.,
257
Метиларахииат 27
Метиленмочевины 331
Метиленовые эфиры целлюлозы 125
Метил карбонат целлюлозы 117
Метил каучуки 494
Метилликанат 32
Метилоксалат целлюлозы 118
Метилолмеламины 340
Метилолмочевины 331, 349
Метилпальмитат 26
Метилсилоксановые масла, смолы,
эластомеры 510, 511, 512
Метилстеарат 26
а-Метилстирол 69, 229
Метилфенилсилоксаиы полимерные
513
Метилцеллюлоза 97, 111, 123
Метилэтилпропилцсллюлоза 126
N-Метоксиметилполиамиды 278
Милар 261, 268
Милоруб 439
Миристат целлюлозы 116
Мовилиты 161
Моноглицериды 372, 377, 378
Моноплекс 174
Монотрифенилметилцеллюлоза 124
Мочевино-глиоксалевые смолы 350
Мочевино-формальдегидные смолы
" 331 и сл., 416
Найлон СА растворимый 273
Нафтенаты целлюлозы 117
Нафтолы 288
Невысыхающие масла 33, 429
Неоабиетиновая кислота 520, 523, 526
Неопрены 491, 495
Ниппон 495
Нитраты
диметнл- и диэтилцеллюлозы 126
целлюлозы 97, 111, 122, 131, 155,
257
Нитрит целлюлозы 122
Нитросульфоацетат и нитросиликат
целлюлозы 120
Нитроцеллюлозные эмали 162, 183
Нобельакрилы 245, 246, 247
Новолаки 280, 283, 305, 316
Новоплас 494
Норлак 78, 272
«Одоновые» кислоты 31
Ойтисиковое масло 32, 52, 53
Окислительная полимеризация 65,
320
Предметный указатель
581
Окись этилена
конденсация с мочевиной 354
оксиэтилироваиие целлюлозы 124
полимеры 365
Оксиалкилцеллюлоза 125
Оксистеариновая кислота 32^
Оксиэтилцеллюлоза 125
Октогидроретен 551, 5/2, 573
Олеат целлюлозы 117
Олеиновая кислота 27, 60, 67, 346
Олеосильвиновая кислота 549
Омыления число 81
Оппанолы 495
Органогалоидсиланы 501
Органозоли 170
Осмол 519
Отвердители эпоксидных смол 414,
425 ,
Пальмитиновая кислота 26
Паравар 456, 4S5
Паракрил 176, 495
Параплексы 174, 192, 268
Параформальдегид (параформ) 289
Паркера процесс 190
Парлои 456, 495
Пассивирование металлов 190, 216
Пасты пигментные 203
Пасты X 170
Пекн каменноугольные 256
Пектиновые вещества 86
Пеларгоновая кислота 28
Пенталин 392
Пентаэритрит 359
абиетаты 394
Пепрен 495
Пербутан 495
Пергут 456, 495
Пердурены 495
Перекисное число масла 35, 83
Перлон 263, 264, 267
Перфоль 271
Петрекс 189
Печатные краски 183, 184, 487
Пешиии 456
Пигменты
для винилита VMCH 178
для хлорвиниловых полимеров 202
для хлоркаучуков 459
Пимантрен 545, 558
Пимаран 558
d-Пимаровая кислота 520, 523, 529
Пииолин 572, 573
Пироабиетиновые кислоты 548
Пиропимаровая кислота 523
Пиросмоляные кислоты 548, 572
Пластизоли 170
Пл астифи каторы
акриловых полимеров 241, 243
виниловых полимеров 159, 174,
190, 204
гидрохлоркаучуков 464
кумароио-инденовых смол 254
полиамидов 274
полистирола 229, 230
хлоркаучуков 459
целлюлозы и ее производных 137
циклокаучуков 475
Пласторуб 439
Плейкслеймы 247
Плекстолы 245
Пленкообразование 22, 42, 47
Плиоваксы 496
Плиогрип 491
Плиолестнк 496
Плиолиты 468, 470, 473, 474, 476,
493, 495
Плиофильм 467, 496
Плиоформы 468, 470, 473, 474, 496
Подводные краски 212, 222, 461
Подсочка 518
Пожелтение высыхающих масел 49
Показатель
водородных связей 270, 272
частоты сетки 60, 356, 369
Полиакриламид 239
Полиакрилаты 235 и сл.
Полиамиды 78, 261 и сл., 277
Полнангидрид малеиновый 401
Поливинилацетали 215, 417
Поливинилацетат 158 и сл.
Поливинилбутираль 215
Поливиниловый спирт 228, 257
Поливинилхлорид 165 и сл., 257
Полиглицерины 376, 378
Полиеновые кислоты 31, 360, 368
Полииндеи 252
Пол и ко 245
Поликонденсация 15, 261 и сл.
Поликумарои 252
Полимеризация П и сл.
акрилатов 237
канифоли 554
масел 52 и сл.
стирола 228
Полиметакриламид 239
Пол и метакрилаты 235 и сл.
Полиорганосилоксаны 499 и сл.
Поливал 554, 569
Полисар-бутил и полисар-крилен 496
Полистирол 72, 226 и сл., 256
модифицированный маслами 230
— непредельными кислотами 231
— фенолами 230
582
Предметный указатель
Полисульфиды 491
Полиундеканамид 265, 271
Полиуретаны 261 и сл., 269, 275, 431
Полихлоропрены 491
Полиэтилентерефталат 265, 267, 268
Полиэфиры 261, 267 и сл., 356 и сл.
в меламино- и мочевино-формаль-
дегидных смолах 338, 344
природные 56
«стиролизованные» 231
Полувысыхающие масла 33
Порозаполнители 244
Преп-райт процесс 190
Пропилцеллюлоза 124
Пропионат целлюлозы 116, 153—155
Пропиофенон 354
Пространственное строение макромоле-
кул 17
Протеке 456, 496
Протектометр 217
Пружинящая шпаклевка 510
Пунициновая кислота 30
Раббоны 464, 480, 481 и сл., 496
хлорированные 484
Растворители
акрилатов 241, 245
виниловых полимеров 162, 193
гидрохлоркаучуков 465
кумароно-инденовых смол 254
метакрилатов 243, 246
феноло-альдегидных смол 319, 320
целлюлозы и ее производных 135
139 и сл.
эпоксидных смол 418, 420—423
Рафинирование масел 33
Резинаты 560
Резинит 497
Резолы 280, 283, 318, 567
немодифицированные 326
модифицированные спиртами 327
Резорцин 288 _
Реолин 453, 497
Ретен 545, 558, 574
«Рецептурная линия» 196
Рилсан 264, 265, 271
Рилсенит 271
Рицинолевая кислота 31, 67
соли 183
Рицинэлаидиновая кислота 29, 32
Родановое число 82
Родопасы 160, 171, 193, 204, 212 и сл.
Ронилла 229
Роплекс 245
Рыбий жир 31, 66
Санипрен 497
Сантицайзеры 159, 174, 192
Сативиновые кислоты 28
Селектрон 269
Сепокс 415
Сернанби 439
Сиккативы резинатные 561
Силастики 510
Силиконы 500
Силоксановые краски 514
Сильваиовая кислота 523
Синолак 415
Синрезен 415
Синтетические каучуки 176, 232, 241,
489 и сл.
Синэргические вещества 37, 187
Скреп 439
Смокед-шитс 437
Смоляные
ангидриды 537, 550, 564
кислоты 323, 391, 520 и сл.
масла 553, 572
мыла 562
спирты 537, 564
Совместимость
акриловых полимеров 241
кумароно-индеиовых смол 254
метакриловых полимеров 243
полиорганосилоксанов 515
феноло-альдегидных смол 320
хлоркаучуков 460
эпоксидных смол 415.
эфиров целлюлозы 140
Сольвессо 202
Сополимеры
акриловых мономеров 239, 247"
виниловых мономеров 166, 173,
177, 180, 183
малеинового ангидрида 177, 389,
402
стирола 229, 232, 404
Сопряженное автоокисление 36
Сорбит и сорбитан 359, 360
Спектрофильм 497
Стабилизация 414, 425
виниловых полимеров 179, 185-
гидрохлорированного каучука 464
хлоркаучуков 455
циклокаучуков 475
«Стандойль» 52
Стеариновая кислота 26, 30
Стейбелит 566
Стирол 226
модификация масел 69
— меламино-формальдегндиых
смол 345
присоединение малеинового ангид-
рида 390, 403
хлорированный в ядре 231
Предметный указатель
583
Стирол изованные полиэфиры 232
Стирольно-бутадиеиовые эмульсии 232
Сукцинат целлюлозы 118
Сульфат, сульфит и сульфоацетат
целлюлозы 120, 121
Сульфобензойные эфиры целлюлозы
119
Супербекациты 329
Суперфлексайт 497
Гай-план 497
Талид 269
Теглак 189
Тенсилон 497
Тенсолит 467, 497
Терилен 264, 268
Термопрены 468, 472, 474, 497
Терпеновые фенолы 323, 329
Терпены
малеиновые аддукты 396, 398
фенольные производные 287
Тетрагидроабиетинол 537, 565
Тетрагидроретен 574
Тетраоксистеариновая кислота 28
Тиксотропия 194
Тиоколы 418, 497
Тиомочевина 352
Тиоурета новые производные целлю-
лозы 128
Тогофан 456, 497
л-Толуолсульфамидо-альдегидные
смолы 348
Торнезит 456, 497
Триазино-формальдегидные смолы 351
Триацетат целлюлозы 97, 111, 152—
154
Триоксистеариновая кислота 32
Тритилцеллюлоза 124
Триуреилметиламин 350
Трифлекс 497
Тролуойль 181
Тунговое масло 30, 43, 52, 53, 55, 56
Ундециленат целлюлозы 117
Ундециленовая кислота 32, 67
Уреилметиламины 350
Уретановые производные целлюлозы
127
Уретаны 351
I
Фенилацетат целлюлозы 118
Фенилизэцианат 383
Фенилмеламины 353
Фенилметилсилоксаны 513
Фенилуретан 383
Фенолак 498
Феноло-альдегидные смолы 257, 279
и сл., 416, 417
Фенолоспирты 290, 339, 346, 567
Фенолы 285, 323
реакционная способность 299
б/«-Фенолы 289, 323, 329, 411,
431
Фибра 139
Филодиеновая компонента 385
Флексолы 160, 174, 191
Флороглюцин 288
Флюавиль 449
Формальдегид 289
Формиат целлюлозы 115
Фосфатирующие грунты 181, 182,
216, 220 и сл.
Фосфорсодержащие производные цел-
люлозы 121, 128
Фталаты целлюлозы и этилцеллю-
лозы 118, 126
Фталевый ангидрид 357 , 378
Фумараты 403, 404
Фурфурольные смолы 162, 353
Хайкар 176, 498
Хемигам 491, 498
Хинонметиды 303, 304, 306, 323, 324,
399, 567
Хлоразол 201
Хлоракриловые сополимеры 241
Хлорацетаты целлюлозы 116
Хлоркаучуки 445, 451 и сл.
Хлорметакриловые полимеры 242
Хлорвиниловые полимеры 165 и сл.
Хлортекс 456, 498
Xроманы 398
Целлобиоза 87
Целлюлоза и ее производные 85'и сл.
Целлюлозогликолевая кислота 126
Цетилоксалат целлюлозы 118
Цпаногуанидин 349
Цианопроизводные целлюлозы 128
Циклогексаноновые смолы 329, 354
Циклогексилоксалат целлюлозы 118
Циклокаучуки 466, 468
Циклооктатетраен, малеиновые аддук-
ты 389
Цитраконовая кислота 405
«Чипсы» 203, 571
Число
ацетильное 82
гексабромиое 54, 82
диеновое 574
иодное 57, 81
кислотное 81
584
Предметный указатель
Число
Леа 40, 83
омыления 81
перекисное 35, 83
родановое 82
Шелла смолы 282
Шпаклевка пружинящая 510
«Штандойлы» 52
Эйкозановая кислота 26
Экавиль 170
Эквивалент этерификации 434
Элаидат целлюлозы 117
Элаидиновая кислота 27, 28
Эландолинолевая кислота 28}
Электрогам 456, 493
Элеомаргариновая кислота 30
Элеостеариновая кислота 30, 43, 44,
55, 60, 324
Эмульсии
гидрохлорированного каучука 467
полиакрилатные 244
поливинилацетатные 164
сополимеров стирола 232
Эмульсионные
краски 232, 244
Смолы мочевино- и меламино-фор-
мальдегидные 347
Эпикоты 282, 409, 413, 414
Эпихлоргидрин 329, 409, 411
Эпоксидные смолы 329, 376, 408 и сл.
модифицированные 423
этерифицированные 415, 426
Эпоксидные стабилизаторы 188
Эпон 329, 409, 414
Эритрогеновая кислота 31
Этерификация
алкидных смол 382
метилолмочевин 336
смоляных кислот 533
целлюлозы 112 и сл.
эпоксидных смол 426
Этилбензил- и этилпропилцеллюлоэа
126
Этилоксалат целлюлозы 118
Этилцеллюлоза 97, 123, 131, 151, 153—
155
Этоксилиновые смолы, см. Эпоксид-
ные смолы
Эфиры
канифоли 392
метилолмочевины 336
40 RD и 40 L 415
эпоксидных смол 414
Янтарный ангидрид, производные 75,
76
Г. ШАМПЕТЬЕ, Г. РАБАТЭ
ХИМИЯ ЛАКОВ, КРАСОК И ПИГМЕНТОВ
Редактор В. Э. Вассерберг Техн. ред. А. А. Скеранская
108468 Подписано к печати 25/VI1 1960 г. ,
Бумага 60x921/16 = 18.5 бум. л. —37 печ. листа. Уч.-издат. 37.5 л. \
Тираж 10000 экз. Цена 24 руб. С 1/1 1961 г. цена 2 р. 40 к. Заказ 156
Типография Госхимиздата. Москва. 88. Угрешская