/




























































































































































































































































































































































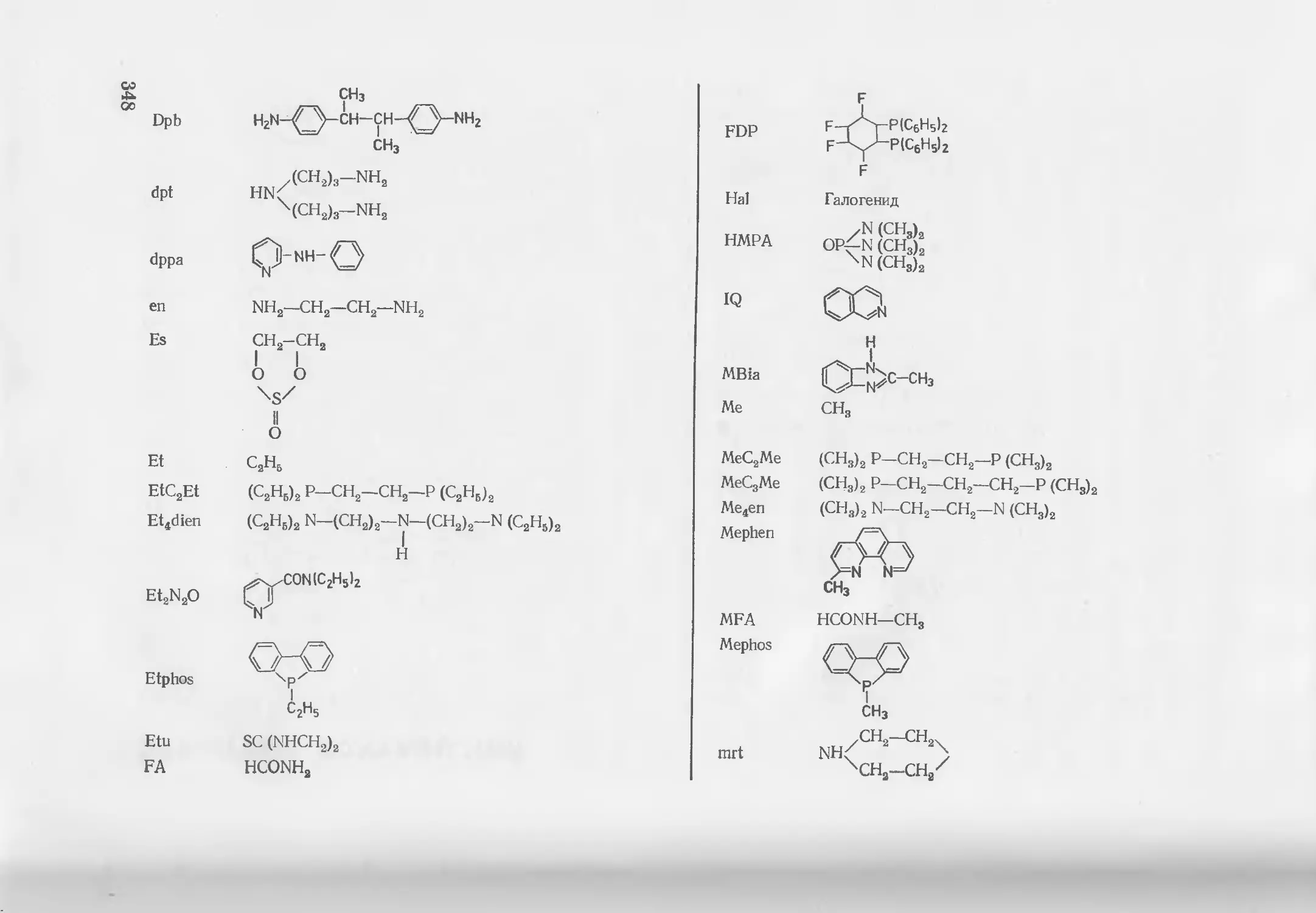
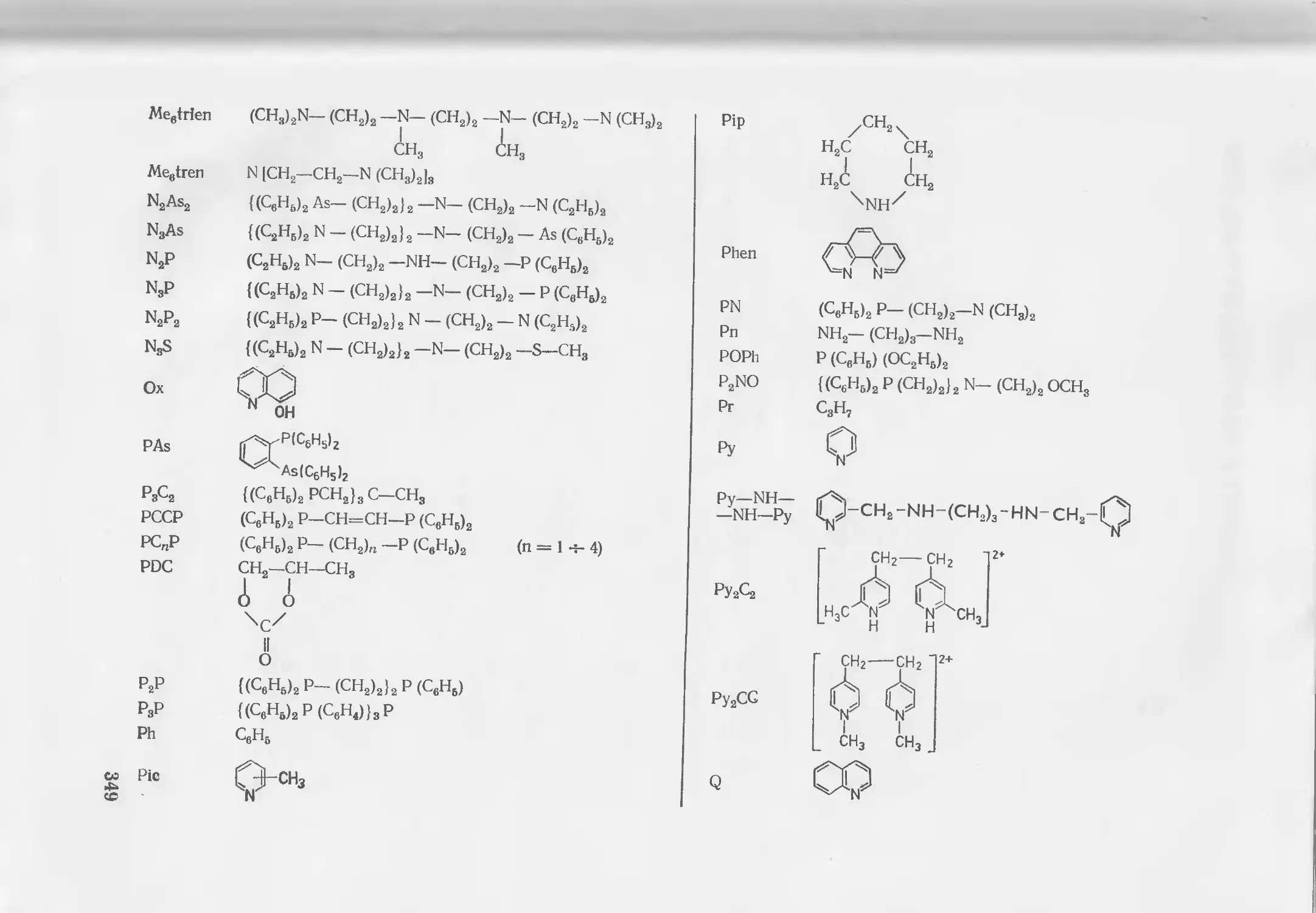
















Текст
АШИ
псевдвгвпвгенидов
Ймия
псевдогалогенидов
Под редакцией
А.М. Голуба,
• X. Кёлера,
В. В. Скопенко
Перевод с немецкого
КИЕВ
ИЗДАТЕЛЬСТВО ПРИ КИЕВСКОМ
ГОСУДАРСТВЕННОМ УНИВЕРСИТЕТЕ
ИЗДАТЕЛЬСКОГО ОБЪЕДИНЕНИЯ «ВИЩА ШКОЛА»
1981
УДК 546 4- 541.49
Химия псевдогалогенидов. Под ред. А. М. Голуба, X. Кёлера, В. В. Ско-
пенко. Киев, издательское объединение «Вища школа», 1981,360 с.
Монография посвящена одному из важных разделов неорганической хи-
мии — химии псевдогалогенидных соединений. Подробно рассмотрены соеди-
нения с линейными и нелинейными псевдогалогенидными группами. Всесто-
ронне обсуждены синтез псевдогалогенидных соединений, их устойчивость в
растворах, способы координации и др. Приведена обширная библиография.
Книга является совместным трудом ученых СССР и ГДР.
Для научных работников, преподавателей и студентов старших курсов
соответствующих специальностей. Табл. 81. Ил. 37. Списки лит.: 2856 назв.
Авторский коллектив: А. М. Голуб, X. Кёлер, В. В. Скопенко, X. Боланд.
Т. П. Лишко, В. М. Самойленко, Г. В. Цинцадзе
Рецензент акад. АН УССР А. В. Городыский
Редакция естественной литературы
Зав. редакцией Б. Н. Фляшников
X ----20502 . 081- 128а—81. 1 802000000
М224(04)—84
Перевод с издания:
Chemie der Pseudohalogenide.
VEB Deutscher Verlag der Wissen-
schaften. Berlin, 1979
©VEB Deutscher Verlag der
Wissenschaften, 1979
©Перевод на русский язык,
издательское объединение «В: ща
школа», 1981
ПРЕДИСЛОВИЕ К РУССКОМУ ИЗДАНИЮ
Предлагаемая читателю монография «Химия псевдогалогенидов» по-
священа рассмотрению интересного и важного класса простых и координа-
ционных соединений, содержащих азид-, цианид-, цианат-, тиоцианат-,
селеноцианат- и другие подобные группы. В научной литературе эти соеди-
нения обычно называют псевдогалогенидами. Написанная совместно со-
ветскими учеными и учеными ГДР, эта монография вышла в 1979 г. в ГДР
и в том же году переиздана в ФРГ. Книга нашла широкий отклик за рубе-
жом и в СССР.
В ней обобщен широкий круг теоретических вопросов химии псевдога-
логенидных соединений, рассмотрены перспективы дальнейшего исследова-
ния псевдогалогенидов, их практического применения в аналитической хи-
мии, цветной металлургии и гальванотехнике, техническом разделении
металлов, производстве новых синтетических материалов.
При подготовке настоящего издания разделы 3 «Цианиды» и 5 «Тиоциа-
натные соединения» дополнены новыми литературными источниками.
Разделы 1 «Введение» и 9 «Сравнительная характеристика псевдогалоге-
нидов» написаны проф. А. М. Голубом, раздел 2 «Неорганические азидные
соединения» — проф. X. Боландом, раздел 3 «Цианиды» — канд. хим. на-
ук Т. П. Лишко, проф. А. М. Голубом, раздел 4 «Цианаты и фульмина-
ты» — проф. Г. В. Цинцадзе, раздел 5 «Тиоцианатные соединения» — доц.
В. М. Самойленко, раздел 6 «Селеноцианатные соединения» — проф.
В. В. Скопенко, разделы 7 «Трицианметаниды» и 8 «Дицианамиды» — проф,
X. Кёлером.
Мы надеемся, что эта первая написанная совместно с учеными ГДР
монография будет полезна специалистам, работающим в области координа-
ционной химии, преподавателям, аспирантам и студентам вузов.
Авторы с благодарностью примут все замечания, пожелания и предло-
жения, которые просят направлять по адресу: 252017, Киев-17, Владимир-
ская, 64, Киевский государственный университет им. Т. Г. Шевченко, ка-
федра неорганической химии.
АВТОРЫ
ПРЕДИСЛОВИЕ К НЕМЕЦКОМУ ИЗДАНИЮ
Настоящая монография «Химия псевдогалогенидов» ставит перед собой
•цель дать обзор одного из актуальных разделов неорганической химии.
В работе проведено сравнительное и систематизированное сопоставление важ-
нейших псевдогалогенидов и их неорганических производных, обширная
библиография облегчает доступ к литературным источникам.
Псевдогалогениды образуют группу одновалентных анионов, которые
по своим химическим и физическим свойствам приближаются к галогенидам.
Этим аналогичным отношениям к галогенидам противостоят четко выражен-
ные особенности псевдогалогенидов. Они основаны на общих структурных
признаках ионов. Псевдогалогениды — это, как правило, многоатомные ли-
нейные или планарные анионы.
Большинство амбивалентных псевдогалогенидов по сравнению с гало-
генидами характеризуются особой реакционной способностью, разнообра-
зием образования соединений и многообразием их структурных типов, что
•составляет особую привлекательность химии псевдогалогенидов.
Прежде всего за последние два десятилетия ощутимо возрос интерес
ученых к псевдогалогенным соединениям. Этому способствовали не только
новые интересные результаты в области фундаментальных исследований,
"но и открытие многочисленных возможностей применения псевдогалогени-
дов. В связи с этим необходимо назвать применение тиоцианатов в экстрак-
ционной технологии разделения соединений урана, в металлургии редких
металлов и ту важную роль, которую играют цианидные комплексы в гальва-
нопластике и в металлургии благородных металлов.
Всевозрастающий в последнее время интерес к псевдогалогенидам и их
производным находит свое выражение в возрастающем количестве обзор-
ных статей по химии отдельных ионов, а также в актуальных монографиях
по отдельным разделам химии псевдогалогенов (А. А. Ньюмен. Химия и био-
химия тиоцианатной кислоты и ее производные. Нью-Йорк, 1975; А. Ц. Шарп.
Химия цианокомплексов переходных металлов. Нью-Йорк, 1976).
Отдельным аспектам химии псевдогалогенидов посвящены обзорные
статьи В. Бека и В. П. Фельхамера (Металлокомплексы с азотносодержащи-
ми псевдогалогенид-лигандами), А. Н. Норбери (Координационная химия
цианатных, тиоцианатных и селеноцианатных ионов), а также монография
Г. В. Цинцадзе (Смешанные псевдогалогенидоаминные соединения).
Названные обзоры по отдельным аспектам псевдогалогенидов убедитель-
но подчеркивают необходимость обобщения всего материала в целом. Это
становится очевидным, если учесть также, что последняя обобщающая, но не
всеохватывающая монография по химии некоторых обсуждаемых здесь клас-
сов соединений (X. Е. Вильямс. Соединения дициана) вышла еще в 1948 г.
Если же учесть всевозрастающее в последние годы количество оригиналь-
4
ных работ по химии цианид-, тиоцианат- и ацид-ионов (эти ионы стали во
многих лабораториях предметом интенсивных исследований) и принять во
внимание такие актуальные, но исчерпывающе не исследованные области
химии цианат-, фульминат-, селеноцианат-, дицианамид- или трицианмета-
нид-ионов, то станет очевидной потребность в обширном обобщении.
Нам представлялось необходимым сопоставить полученные ранее резуль-
таты с многочисленными новыми экспериментальными и теоретическими дан-
ными, причем основное внимание при этом было сосредоточено на химии не-
органических соединений псевдо галогенидов. Подробное рассмотрение мно-
гочисленных органических производных псевдогалогенов в этой работе
не предусматривалось.
В разработке монографии приняли участие авторы из СССР и ГДР. Так,
раздел 2 «Неорганические ацидные соединения» написан проф. X. Боландом
(Мюльхаузен); раздел 3 «Цианиды» — канд. хим. наук Т. П. Лишко и проф.
А. М. Голубом (Киев); раздел 4 «Цианаты и фульминаты» — проф. Г. В. Цин-
цадзе (Тбилиси); раздел 5 «Тиоцианатные соединения» — доц. В. М. Самой-
ленко (Киев); раздел 6 — «Селеноцианатные соединения» — проф. В. В. Ско-
пенко (Киев), разделы 7 и 8 —«Трицианметаниды» и «Дицианамиды»— проф.
X. Кёлером (Галле). Введение и заключительный раздел были разработаны
проф. А. М. Голубом, который одновременно являлся редактором первона-
чального русского варианта рукописи. Обработку и редактирование немец-
кого варианта рукописи выполнил проф. X. Кёлер.
При написании настоящей монографии авторы ставили перед собой цель
учесть прежде всего все наиболее важные публикации по химии псевдогало-
генидов. Эта цель могла быть в полной мере достигнута при рассмотрении
таких классов соединений, как селеноцианаты, цианаты и фульминаты, а
также дицианамиды и трицианметаниды. В области химии цианида, тио-
цианата и ацида в связи с изобилием опубликованных работ мы вынуждены
были пойти на определенное ограничение материала, сконцентрировав ос-
новное внимание на псевдогалогенидах металлов и их комплексах.
Настоящая монография рассчитана на химиков — научных работников и
практиков, особенно на неоргаников и химиков-комплексников, а также на
учителей химии и студентов-химиков.
Предпринятая авторами попытка представить в коллективной работе
область неорганической химии псевдогалогенов не претендует на полноту
изложения. Поэтому авторы просят коллег высказать свои замечания, со-
веты и пожелания.
Авторы выражают признательность и сердечную благодарность В. Ре-
шетиловскому за его тщательный перевод на немецкий язык глав книги, на-
писанных на русском языке.
Профессор Андрей Матвеевич Голуб вскоре после написания книги вес-
ной 1977 г. скончался. До последних дней своей жизни он уделял постоянное
и неустанное внимание завершению настоящей работы, инициатором кото-
рой он был. Мы сохраним благодарную память о нем.
Киев. Галле. 1977
АВТОРЫ
1. ВВЕДЕНИЕ
1.1. ПОНЯТИЕ «ПСЕВДОГАЛОГЕН».
ПСЕВДОГАЛОГЕНИДЫ. ПСЕВДОГАЛОГЕНЫ
В настоящее время понятие «псевдогалоген» стало более точным, и число
охватываемых им соединений значительно возросло. Это не удивительно,
так как наши представления об этом классе соединений постоянно расширя-
ются вследствие открытия многих новых фактов. Поэтому жизнь требует
дальнейшего уточнения этого понятия и более точной систематизации ве-
ществ, относящихся к классу псевдогалогенов.
Центральное место среди псевдогалогенид-ионов занимают: цианид CN-,
фульминат CNO~, цианат OCN~, тиоцианат SCN-, селеноцианат SeCN~,
азид Nr, дицианамид N (CN)F и трицианметанид С (CN)T- Общим призна-
ком всех этих ионов является их четко выраженное сходство с галоген ид-
ионами, аналогия некоторых производных галогенов и псевдогалогенов [1]:
— Как и галогениды, псевдогалогениды, Y~, образуют плохо раствори-
мые соли серебра, ртути (I) и свинца (II) — AgY, Hg2Y2 и PbY2.
— Подобно галогеноводородам существуют псевдогалогеноводороды HY,
водные растворы которых являются кислотами различной силы.
— По аналогии с различными типами галогенидов металлов, а также ко-
валентных галогенидов неметаллов, существуют различные типы псевдога-
логенидов металлов и ковалентных псевдогалогенидов неметаллов.
— Подобно галогенид-ионам псевдогалогениды образуют с металлами
псевдогалогенидные комплексы, например MY4--, MY^F- или MY2L2- и
MY2L4- типа.
— Псевдогалогенид-ионы Y~ воздействием соответствующих окислите-
лей превращаются в псевдо гало гены
2Y~-^Y2 + 2e~. (1.1)
Псевдогалогены представляют собой неустойчивые ковалентные соеди-
нения, обладающие способностью окислять металлы с образованием псевдо-
галогенидов металлов. Помимо псевдогалогенов, существуют ковалентные
галогенпсевдогалогениды (например, C1N3, BrCN, C1NO2, BrSCN) или же
межпсевдогалогенидные соединения Y — Y' (например, NCC (CN)3).
Характерной для галогенпсевдогалогенидов является их реакция с ко-
валентными галогенидами металлов [2]
МС1Л +ClY->MCln_iY + С12. (1-2)
Приведенный признак псевдогалогенидов — их способность окисляться
до псевдогалогенов — еще не реализован для всех представителей этой груп-
пы соединений. Так, еще не удалось получить такие молекулы, как (N3)2
или (NC)2 N — N (CN)2. Диоксициан (OCN)2 отличается крайне низкой ус-
тойчивостью.
6
Из псевдогалогенидных соединений первыми были получены цианиды.
Уже в 1704 г. Дисбах открыл берлинскую лазурь [3], а в 1782 г. Шееле по-
лучил цианиды калия, ртути и циановодород [3, 4]. В 1815 г. Гей-Люссак
термическим разложением цианида ртути получил дициан и установил его
аналогию с галогенами [51.
Либих, Гей-Люссак и Велер в 1824 г. доказали, что фульминат и цианат
серебра имеют одинаковый состав и плохо растворяются в воде [6а, 66].
А еще раньше Берцелиус [7] установил незначительную растворимость
AgSCN.
Цианаты [6а, 66, 81, тиоцианаты и селеноцианаты [7] щелочных металлов
получены в первой половине, а азиды в конце XIX в. (Куртиус, 1890, Вис-
лиценус, 1892 [9]).
В работах [10—19] проведено сравнение химических свойств CN~, NT,
XCN~ (X = О, S, Se) и галогенид-ионов между собой. Позднее к группе
псевдогалогенид-ионов были причислены трицианметенид- С (СЫ)Г [20] и
дицианамид-ионы N (CN)7 [21]. Подобие трицианметанид-ионов с галогенид-
ионами впервые установлено Биркенбахом [22]. Позже было замечено, что
трицианметанид и дицианамид как лиганды тесно примыкают к цианат- и
тиоцианат-ионам [31]. Кроме того, бромтрицианметанид, например, может
взаимодействовать с галогенидами металлов, что характерно для галоген-
псевдогалогенидов [23],
МВг4 + ВгС (CN)S -> МВг3С (CN)3 + Вг2. fl .3)
Таким образом, понятие «псевдогалогенид»-ион (или группа) распростра-
няется теперь как на CN~-, Nr-, NCO~-, CNO~-, SCN—-, SeCN-, так и на
N (CN)?-, С (СЫ)Г-ионы (группы), а в некоторой степени и на ЫО~-ионы
124, 25].
1.2. НОМЕНКЛАТУРА ПСЕВДОГАЛОГЕНИДНЫХ СОЕДИНЕНИЙ
Термин «псевдогалоген» впервые введен Биркенбахом в 1925 г. [13] и
подробно обоснован рядом дальнейших работ (см., например, [14, 15, 22]).
Этот термин относили и относят к молекулам (CN)2, (NCS)2, (NCO)2 и (NCSe)2.
К этому ряду иногда причисляют и соединения (SCSN3)2 [1] (см. 9.5).
Позже параллельно с термином «псевдогалоген» получили распростра-
нение и родственные ему названия «псевдогалогенид-ион» и «псевдогалоге-
нидная группа». Последние названия следует признать более общими, ибо
они позволяют причислить к соединениям псевдогалогенидов не только
свободные псевдогалогены, но и различные многочисленные их производные
как ионного, так и ковалентного характера.
Иногда термины «псевдогалогены», «псевдогалогенидные группы» и
«псевдогалогенид-ион» употребляют как синонимы. Однако более точно псев-
догалогенами следует называть лишь такие молекулярные соединения У2,
как дициан, диоксициан, дитиоциан и др. Термины «псевдогалогеногруппа»
или «псевдогалогенид-радикал», в отличие от псевдогалоген ид-ион а У-,
следует применять к соответствующим группам атомов, ковалентно связан-
ным с другими атомами и группами атомов.
Отсюда и вытекают названия производных псевдогалогенов, например
псевдогалогенидов металлов: NaN3 — азид натрия, Ba (CN)2 — цианид ба-
рия, La (NCO)3 — цианат лантана. Соответствующие водородные сседине-
ния следует назвать азоимид (HN3), оксициановодород (HNCO), цианово-
7
дород (HCN), селеноциановодород (HSeCN), так как все псевдогалогеново-
дороды являются ковалентными соединениями.
Если псевдогалогеноводороды растворять в полярных растворителях,
таких, как вода, то образуются псевдогалогенид-ионы Y-, например: CN~~,
NCX-, NO7, С (CN)r. Поэтому водные растворы этих соединений на-
зываются псевдогалогеноводородными кислотами. Так, циановодороду от-
вечает циановодородная или синильная кислота, а азоимиду — азидная (азо-
тистоводородная) кислота. Кислоту HCNO обычно называют гремучей.
Для производных псевдогалогенов различных металлов и неметаллов при-
нято общее название «псевдогалогениды» без учета возможных различий в
характере связи: ионной или ковалентной. Объясняется это тем, что иногда
трудно установить, какой тип связи преобладает. Это иллюстрируется на
примере псевдогалогенидов металлов, таких, как селеноцианат ртути (II)
Hg (SeCN)2, изоцианат свинца Pb (NCO)2, фульминат серебра AgCNO и циа-
нид арсена (III) As (CN)3.
Псевдогалогенидные комплексы, например К3 [Со (CN)6], [С5Н5Мо (SCN) •
• (СО)3], [Pd (NO2)2 (NH3)2] и [Rh (SCN) (NH3)5] SO4, называют в соответствии
с общими номенклатурными правилами названия координационных соеди-
нений: гексацианокобальтат (III) калия, тиоцианатотрикарбонил-л-цикло-
пентадиенил-молибден (I), динитродиаминпалладий, тиоцианато-пентамин-
родий (Ш)-сульфат.
Соединения, включающие М — NCX- или М — XCN-группировки (X =
= О, S, Se), называют изоцианаты (изотио-, изоселеноцианаты) или цианаты
(тио-, селеноцианаты).
1.3. ХАРАКТЕРИСТИКА ПСЕВДОГАЛОГЕНИДОВ
Псевдогалогениды представляют собой важный класс неорганических
соединений. Производные псевдогалогенов получены почти для всех хими-
ческих элементов, за исключением благородных газов. Их исследование ве-
дется широким фронтом.
Хотя псевдогалогениды сходны с галогенидами, они во многих отноше-
ниях отличаются от последних. Известно больше псевдогалогенидов, и,
кроме того, они имеют более сложную структуру. Подобие в химическом и
физическом поведении отдельных псевдогалогенидов основывается на об-
щих структурных признаках.
Все псевдогалогениды представляют собой отрицательно одновалентные
мезомерноустойчивые анионы. Большинство из них (CN—, Nr, CNO~, NCO~ ,
NCS~, NCSe-) имеют линейную структуру, в то время как N (CN)r, С (CN)r
и NO7 представляют собой плоские анионы. Среди псевдогалогенидов самым
простым по структуре является двухатомный цианид-ион, самым сложным
анионом — семиатомный трицианметанид-ион. Многоатомность псевдогало-
генидов обусловливает их больший (по сравнению с галогенидами) ионный
радиус и разнообразие строения.
Сложная структура псевдогалогенидов и наличие в них разных донорных
атомов приводят к интересным и разнообразным способам присоединения псе-
вдогалогенидных групп, что чаще всего проявляется в псевдогалогенидных
комплексах металлов [26].
Прежде всего псевдогалогениды могут выступать в качестве моноден-
татных лигандов. При этом в большинстве случаев преобладает координация
через один из донорных атомов, например углерод (для цианида и фульми-
8
ната) или азот (для азида и цианатов NCX“ (X = О, S, Se), дицианамида и
трицианметанида). Амбидентатный характер псевдогалогенидов может при-
вести также к образованию координационных соединений со связями XCN-
групп (X = О, S, Se) через кислород, серу или селен; С (СМ)3-группы могут
образовывать связи через атом углерода.
Примечательна для всех псевдогалогенидов их четко выраженная склон-
ность к образованию лигандных мостиков [26]. Так, известны многочисленные
цианокомплексы с цианидными мостиками типа М—С—N—М. Цианат и
азид (каждый в отдельности) образуют аналогичные мостики типа М—N—С—
X—М (X = О, S) и М—NNN—М, хотя для цианатных и для азидных комп-
лексов типичной является связь
М
M\nnn
Аналогичный мостиковый тип известен и для фульминатных соединений
M\cNO
Способность к мостикообразованию характерна для дицианамид- и
трицианметанид-ионов. Оба иона выступают преимущественно как биден-
татные мостиковые лиганды [25]
М—NCN
М'
Описаны также координационные соединения, в которых одна тиоциа-
нат- [26], дицианамид- и трицианметанид- [25] группа образует связи с тре-
мя атомами металла,
М М
Такие псевдогалогениды, как азид-, цианат-, тиоцианат-, дицианамид
и трицианметанид, в гомологических диметилталлийпсевдогалогенидах ко-
ординационно связаны с четырьмя атомами таллия (опять-таки образуя свое-
образный мостик) [39]
9
Мч /М
>NCX<
Mz ХМ
Итак, подобное поведение псевдогалогенидных лигандов, амбидентат-
ный характер большинства из них и тенденция к образованию мостиков —
все это можно считать характерными признаками псевдогалогенидов.
По сравнению с рассматриваемыми здесь группами нитрит-ион менее изу-
чен [24]. Можно предположить, что в ряду псевдогалогенидов МОГ занимает
особое место и является, как и СЮГ, связующим звеном между типичными
псевдогалогенидами и широким классом оксоанионов.
Подобно тиоцианату нитрит является амбидентатным лигандом, который
может образовывать координационные связи или через азот (нитрокомплек-
сы), или через кислород (нитритокомплексы). В противоположность другим
псевдогалогенидам нитрит-ион обладает свойством образовывать хелато-
комплексы, но в то же время подобен классическим псевдогалогенидам в их
способности выступать в качестве мостикового лиганда
/М
/СГ /О мх О
М—О—NZ М—N< >O-NZ
Nd—м
Сходство МОГ с галогенидами выражается в образовании малораство-
римой в воде соли AgNO2, которая реагирует с избытком МОГ, образуя
соединения типа М [Ag (NO2)2L Псевдогалогенидный характер N2O4 прояв-
ляется в реакциях этого соединения с металлами, в результате которых
образуются нитриты металлов.
Принадлежность нитрит-иона к группе псевдогалогенидов все еще не
очевидна. Поэтому химия нитрит-иона подробно не рассматривается, что
вызвано также ограниченным объемом монографии.
1.4 КЛАССИФИКАЦИЯ ПСЕВДОГАЛОГЕНИДНЫХ СОЕДИНЕНИИ
Относительно классификации псевдогалогенидных соединений не су-
ществует единого мнения. Очевидно, его и не может быть, ибо в зависимости
от признака, который положен в основу классификации, на передний план
могут выступать различные аспекты. Мы рассматривали два основных под-
хода к систематике псевдогалогенидных соединений: классификация по ти-
пам соединений; классификация по структурным признакам псевдогалоге-
нид-ионов (линейная или нелинейная структура).
Классификация по типам соединений. Этот принцип классификации вы-
10
двигает на передний план рассмотрение характера связи псевдогалогенид-
ной группы. Исходя из этого принципа, псевдогалогенидные соединения мож-
но ориентировочно разделить на следующие классы:
а) псевдогалогены ((CN)2, (NCS)2 и т. д.);
б) псевдогалогеноводороды и псевдогалогеноводородные кислоты;
в) ионные псевдогалогениды s-металлов и комплексных катионов типа
NHt, [Р (C6H5)J+ и т. п.;
г) простые и координационные псевдогалогениды р- и ё10-металлов;
д) простые и координационные псевдогалогениды d-переходных ме-
таллов;
е) псевдогалогениды f-металлов.
Соединения s-металлов и комплексных катионов представляют собой
преимущественно ионные солеобразные вещества (NaN3, NH4NO2 и т. д.).
Преобладающее большинство псевдогалогенидов p-элементов (ON3, Р (NCS)3,
Ga (NCO)3 и т. п.) ковалентны. Псевдогалогениды бго-элементов имеют, как
правило, координационную природу.
Производные переходных металлов являются координационными сое-
динениями. Комплексы богатых электронами центральных атомов (под-
группа меди, платиноиды и некоторые другие) имеют более высокую степень
ковалентности, чем комплексы бедных электронами металлов (подгруппы
ванадия, скандия и титана). В цианатах, нитритах, тиоцианатах, дициана-
мидах и трицианметанидах переходных металлов с низкой d-электронной
плотностью сравнительно высока ионная составляющая связи, тогда как в циа-
нидах и фульминатах переходных металлов преобладает ковалентная связь.
Поскольку большинство известных псевдогалогенидных соединений
принадлежит к группам в, д и е, их следует классифицировать, основываясь
на природе псевдогалогенид-ионов. Образование псевдогалогенид-ионов мож-
но рассматривать как следствие диссоциации соответствующих псевдога-
логеноводородных кислот, HN3, HCN, НС (CN)3. Эти кислоты в свою очередь
формально выводят из би- нарных водородных сое- динений неметаллов Н4С, H3N, Н2О, H2S, H2Se, за- мещая один или несколь- ко атомов водорода CN-, NO- или N^N-группой. Таблица 1.1. Расчет л-электронной плотности и поряд- ков л-связей в некоторых псевдогалогенид-ионах [37]
Ион Эффективные л-заряды атомов Порядок л-связей
Qa Qb Qc AB вс
При замещении в молекуле Н2О атома водорода CN- СЬГ —0,2501 —0,7499 — 1,9848 —
группой получим HOCN, N, —0,8060 +0,6121 —0,8060 1,3874 1,3874
а при замещении NO-rpyn- NCO~ —0,7712 +0,0442 —0,1846 1,5503 1,2629
пой — HONO. Замещение в NCS“ —0,4826 +0,1934 —0,7108 1,8243 0,7964
H3N двух атомов водорода NCSe" —0,3941 +0,2345 —0,8404 1,8943 0,5973
N=N-rpynnod приводит к образованию HN3, а двумя NCTe~ —0,4919 +0,1859 —0,6940 1,8156 0,8179
CN-группами — к образо- ванию HN (CN)2. Анало- CNO —1,1663 +0,6516 —0,4852 1,7402 0,9221
гичным образом из метана получают НС (CN)3.
По данным рентгеноструктурных исследований, псевдогалоген ид-ионы
типа NCX (X = О, S, Se), Г<з~ и CNO~ очень сходны между собой [26].
Вагнер [37] произвел расчет распределения л-электронной плотности
в некоторых псевдогалогенид-ионах по методу МО ЛКГО (линейная ком-
бинация групповых орбиталей). Он предположил, что крайние атомы АВС-
11
ионов образуют шесть «асимметричных групповых орбиталей», из которых
четыре относятся к о-, а две — к л-типу. Первые две из этих групповых
орбиталей сочетаются соответственно с s- и р-орбиталями атома В и образу-
ют ст-МО-систему. Другие две орбитали остаются несвязывающими и содер-
жат неподеленные пары электронов на двух крайних атомах. Пятая групповая
орбиталь сочетается с рх- и р^-атомными орбиталями атома В, образуя вы-
рожденные л-МО. Шестая групповая орбиталь остается несвязывающей
МО-л-типа. В соответствии с этим электронную структуру ABC-ионов мож-
но описать следующим образом: (оАВ)2 (оВС)2 (оА)2 (оС)2 (оул)4 (г/л)4, где ш
и и характеризуют обе л-МО, о характеризует орбитали концевых атомов,
имеющих неподеленные электронные пары.
В табл. 1.1 приведены результаты расчета распределения л-зарядов,
а также порядок л-связей в некоторых псевдогалогенид-ионах [37]. Так как
автор работы [37] для оценки диагональных матричных элементов исполь-
зовал представление об орбитальных электроотринательностях, расчет не-
достаточно точен. Однако он убедительно иллюстрирует неравномерное рас-
пределение заряда на атомах псевдогалогенидов типа АВС-.
Квантово-механический расчет по методу МО ЛКГО, где в отличие от
оценки кулоновских интегралов использовались потенциалы ионизации,
показывает, что в NCX--ионах эффективный отрицательный заряд на ато-
мах азота, как правило, наиболее высокий. Так, в NCS-группах заряд на
N равен —0,51, а на S — 0,48 [27].
Согласно работе [281, эффективные заряды на атомах NCO равны: —0,871
(N), +0,430 (С) и —0,648 (О). Селеноцианат-ион ведет себя подобно тиоциа-
нату. Что же касается азид-иона, то электронная плотность локализована
на концевых N-атомах.
Характер связи в NCS- можно проиллюстрировать, сопоставляя сило-
вые постоянные С—S- и N—С-связей. Силовая постоянная С—S-связн в
CH3SH равна 3,18 Н/см, а в CS2 6,88 Н/см. Для NCS- она равна 5,18 Н/см
и занимает промежуточное значение между величинами, характерными для
одинарной и двойной С—S-связей. Подобным образом силовая постоянная
С—N-связи для NCS- 15,95 Н/см имеет промежуточное значение между
17,8 Н/см (в CH3CN) и 13,2 Н/см (в HNCS). Аналогичные характеристики по-
лучены и для селеноцианат-иона [19].
Распределение заряда на атомах XCN можно схематически изобразить
следующим образом:
—(0,5 + 6) + 26^ — (0,5 + 6)
X — C = N
Из распределения заряда следует, что XCN-ионы могут образовывать
связи через N или X. В тио- и селеноцианатных соединениях преобладает
координация лиганда через N; через S или Se образуется связь прежде всего
в комплексах «мягких» металлов.
Фульминат-ион заметно отличается от псевдогалогенид-лигандов, ко-
торые координируются через N, своей четко выраженной тенденцией к об-
разованию связи через углерод, и в этом отношении он близок к цианиду.
Согласно работе [30], отрицательный заряд в фульминат-ионе локали-
зован на атоме углерода, однако расчеты [39] показывают, что распределе-
ние зарядов на концевых атомах CNO-, а также NCO-сравнительно равно-
мерное.
Исключительная координация фульмината через углерод (С-координа-
ция наблюдается и для мостиковых фульминатов, таких, как AgCNO)
12
и доминирующая координация NCO~ через азот обусловлены по [26] пре-
жде всего различием в расположении связывающих орбиталей линейных
лигандов.
Следовательно, псевдогалогениды по их преобладающей тенденции к
координации грубо можно разделить на три группы:
—N-донорные лиганды (N7, NCO~, NCS~, NCSe~, N (CN)C, C (CN).D;
—С-донорные лиганды (CN~, CNO~);
—О-донорные лиганды (NOT).
Амбидентатный характер некоторых псевдогалогенидов (наряду с NCS~
и NCSe~ прежде всего NO?, NCO”, а также С (CN)C) 1361 позволяет более
детально дифференцировать эти соединения.
Классификация по структурным признакам. Рассмотренную выше сис-
тематику псевдогалогенидных соединений и псевдогалогенидных ионов
нельзя считать исчерпывающей. С одной стороны, многие представители
того или иного класса веществ по своим свойствам являются переходными
звеньями между различными группами. С другой стороны, в основу класси-
фикации можно положить и другие отличительные признаки, например про-
странственное расположение и структуру псевдогалогенид-ионов. На по-
следнем критерии следует остановиться подробнее.
Азид (ИГ) и NCX (X = О, S, 5е)-группы имеют линейное или близкое
к линейному строение. Так, в газообразном диметилкремнийизоцианате най-
дены такие параметры NCO-группы: расстояния С—N 1,20 А, С—О 1,18 А;
Z.NCO = 180° [32]. Рентгеноструктурные исследования орторомбической
модификации фульмината серебра показали следующие результаты: длины
связей С—N 1,09 ± 0,03 A, N—О 1,25 + 0,03, Ag—С2,23 ± 0,02A; ZLCNO =
= 180° [32]. Для тригональной, модификации AgCNO найдены межатомные
расстояния Ag—С 2,16 ± 0,03 А и 2,19 ± 0,03 A, Z.CNO = 172° [33].
В структурном отношении тиоцианат и селеноцианат очень похожи на
цианат-ион. По данным работы [341, образование координационных связей
тиоцианатного лиганда через серу может привести к уменьшению угла связи
NCS до 160°.
Ввиду указанных выше структурных особенностей Nr, NCO~, CNO~,
NCS~ и NCSe~ называют линейными псевдогалогенид-ионами. К этой груп-
пе, конечно, относится и цианид-ион.
Второй структурный тип псевдогалогенид-ионов составляют дицианамид
N (CN)7, трицианметанид С (CN)3“ и нитрит NO7. Этот тип отличается от
первого нелинейной структурой. Из рентгеноструктурных данных известно,
что С (CN)r [35а] и N (CN)7 [356, 381 имеют плоское строение. Для С (СЫ)Г
угол связи ССС равен почти 120°. Подобно этому и в дицианамид-ионе угол
CNC равен ~120°. Строение плоских ионов С (CN)7, N (CN)7 и N07 можно
представить структурными формулами (лишь для одной из мезомерных гра-
ничных формул)
С
I
С
II!
N
О|в
К-
О|
13
Приведенные существенные различия в структурах обоих типов псевдо-
галогенидов приводят к интересным вариациям и в структурах однотипных
комплексов с линейными и нелинейными псевдогалогенид-лигандами. На-
пример, комплексы типа MY2L, (L = сс-производное Ру; М = Со, Ni) для
Y = NCO, NCS имеют мономерно-тетраэдрическое или мономерно-квадрат-
ное строение, в то же время вследствие сильно выраженной склонности не-
линейных псевдогалогенидов к образованию мостиков при Y = С (CN)3
или N (CN)o рассматриваемые соединения имеюг полимерно-октаэдрическую
структуру [25].
СПИСОК ЛИТЕРАТУРЫ
1. Коттон Ф., Уилкинсон Дж. Современная неорганическая химия. М., Мир, 1969,
т. 2. 420 с.
2. Dehnicke К.—J Inorg. Nucl. Chem., 1965, 27, р. 809.
3. Блох М. А. Хронология важнейших событий в области химии. М., 1940, с. 41, 69.
4. Джуа М. История химии. М., Мир, 1966, с. 155.
5. Gay-Lussac J. L.— Ann. Chim., 1815, 95, р. 156.
6а. Gay-Lussac J. L., Liebig J.— Ann. Chim. phys., 1824, 25, p. 285.
66. Wohler F.— Ann. Physik u. Chemie (Poggendorf), 1824, 1, S. 87, 117; 1825, 5, S. 385.
7. Berzelius J. J.— Jahresber. Fortschr. phys. Wiss., 1822, 1, S. 49.
8. Wohler F.— Gilberts Ann. Phys., 1822, 71, p. 95.
9. Curtius T.— Ber., 1891, 24, S. 3342; Wislicenus W.— Ber., 1892, 25, S. 2084.
10. Soderback E.— Liebigs Ann. Chem., 1919, 419, S. 217.
11. Soderback E.— Acta chem. Scand., 1957, 11, p. 1622.
12. Голуб A. M.— Журнал неорган. химии, 1956, 1, с. 2517.
13. Birckenbach L., Kellermann К-— Ber., 1925, 58, S. 786.
14. Birckenbach L., Hutter K-— Z. anorg. allg. Chem., 1930, 190, S. 1.
15. Birckenbach L., Linhard M.— Ber., 1930, 63, S. 2544, 2588.
16. Williams H. E. Cyanogen Compounds. 2nd ed. London, 1948.
17. Голуб A. M.— Журнал общей химии, 1956, 26 (88), с. 1837.
18. Lodzinska А.— Roezn. Chem., 1967, 41 (6), p. 1007.
19. Голуб A. M., Скопенко В. В.— Успехи химии, 1965, 34, с. 2098.
20. Schmidtmann Н.— Вег.. 1896, 29, S. 1172.
21. Madelung W., Kern F.— Ann. Chem., 1922, 427, p. 1.
22. Birckenbach L., Kellermann K-— Ber., 1925, 58, S. 2377.
23. Kohler H., Miilmann E.— Z. anorg. allg. Chem., 1970, 373, S. 222.
24. Голуб A. M., Акмырадов P.— Журнал неорган. химии, 1966, 11, с. 2347.
25. Kohler H.— Z. Chem., 1973, 13, S. 401.
26. Beck W., Fehlhammer W. P.— MTP Internat. Rev. of Science. Inorg. Chem., 1972,
Serie 1, Vol. 2, p. 253.
27. Di Sipio L., Oleari L., Michelis G. D.— Coord. Chem. Rev., 1966, 1, p. 7.
28. Yonezawa T., Kato H., Konishi H.— Bull. Chem. Soc. Japan, 1967, 40, p. 1071.
29. Харитонов Ю. Д., Икрамов X. У., Бабаева А. В.— Докл. АН СССР, 1964, 158,
с. 1412.
30. Bonaccorsi R., Petrongolo С.. Scrocco Е., Е. Tomasi J.— J. chem. Phys., 1968, 48,
р. 1500.
31. Кёлер X.— Координационная химия, 1977, 3, с. 139.
32. Kimura К-, Katada К., Bauer S. Н.— J. Am. Chem. Soc., 1966, 88 (3), p. 416.
33. Britton DDunitz J. D.— Acta Crystallogr., 1965, 19, p. 662.
34. Scoulondi H.— Acta Crystallogr., 1953, 6, p. 651.
35a . Andersen PKlewe B., Thom E.— Acta chem. Scand., 1967, 21, p. 1530.
356. Klewe B. Privatmitteilung.
36. Kuhn M., Mecke R.— Ber., 1961, 94, S. 3016.
37. Wagner E. L.— J. Chem. Phys., 1965, 43, p. 2728.
38. Chow Y. M., Britton D.— Acta Crystallogr., 1975, B31, p. 1922, 1934.
39. Holsboer F. J., Beck W.— Chem. Commun., 1970, p. 262.
2. НЕОРГАНИЧЕСКИЕ
АЗИДНЫЕ
СОЕДИНЕНИЯ
2.1. ВОДОРОДНЫЕ СОЕДИНЕНИЯ АЗОТА
Ковалентные водородные соединения азота можно представить следую-
щими гомологическими рядами: NnH2«+m (А, т= 1 или 2); NnH„ (В); N„Hn_2 •
• (С) и NnHn_4 (D). Представители этих рядов в качестве химических соеди-
нений известны лишь для определенных значений п, а также в форме раз-
личных органических производных. Примером соединений типа А являются
аммиак, аммоний (гг — 1), гидразин или соли гидразония (и = 2). К соеди-
нениям ряда В относятся имин-радикал NH, диимид N2H2, триазин N'3H3,
а также азид аммония (NH4N3 = N4H4) и гидразония (N5H5 = N2H4 •
. HN3 — N2H5N3). Важным соединением ряда С является азоимид (газо-
образный или безводный HN3) или азидная кислота (растворы HN3). Из ря-
да D определенный интерес представляет лишь состав N8H4 (м = 8), для ко-
торого известны различные органические производные [1]. »
2.2. АЗИДОВОДОРОД И АЗИДНАЯ КИСЛОТА
2.2.1. Газообразный и жидкий HN3 (азоимид)
Исходными веществами для получения водного или безводного HN3
служат ионные азиды или диазосоединения и другие органические производ-
ные [1—41. Газообразный HN3 образуется при взаимодействии сухого NaN3
с сиропообразной Н3РО4 15, 61 или расплавленной стеариновой кисло-
той [7].
Не содержащий растворителя азоимид HN3 представляет собой бес-
цветную жидкость (плотность 1,126 г/см3), которая кипит при 35,7° С и за-
мерзает при —80° С 11, 41 Это очень эндотермичное соединение, которое при
контакте с нагретыми предметами или ударе разлагается со взрывом
2HN3 (г) -> 3N2 + Н2, ЬН = — 600,51 кДж. (2.1)
Энтальпия образования равна 300,25 кДж/моль для газообразно-
го и 269,71 кДж/моль для жидкого HN3 17—101- Другие термодинамиче-
ские константы для HN3 (г) 19, 101: S0 = 239,16 Дж/(моль • град); =
43,74 Дж/(моль • град); (Н — Но)/Т = 36,54 Дж/(моль • град); —(F —>
— Fq)/T — 202,67 Дж/(моль - град).
Молекула азидной кислоты (как и молекула DN3) имеет угол связи Н—сс-
N—P-N 114°; расстояния cc-N—(3-N и (3-N—y-N линейной N—N—N-группы
в противоположность азид-иону и азидному радикалу неодинаковы. Такая
структура наблюдается и в других ковалентных азидах типа RN3 (R = СН3
15
галоген), где наблюдаются различия в расстоянии R—a-N и в величине угла
связи 111—15].
Н
Такая схема хорошо согласуется с данными колебательных спектров
112, 161. Так, из шести валентных колебаний (пять колебаний в плоскости
(А') и одно вне плоскости (А") для HN3 и DN3 были определены силовые
константы валентного взаимодействия, равные 10,1 Н/см (KjHN—N2) и
16,3 Н/см (K2HN2—N) (11, 14, 171. Были сняты ИК-спектры поглощения га-
зообразного (10, 18], твердого (91 HN3, а также водных (191 и неводных (9]
растворов азидной кислоты. Изучены спектры комбинационного рассеяния
газообразного HN3 (201.
Частота v (Н—N) = v1 (А') для газообразного HN3 найдена при 3336 см-1
(2480 см-1), vac (N—N—N) = v2 (А') при 2140 см-1 (2141 см-1) и vc (N—
N—N) = v3 (А') при 1274 см-1 (1183 см-1). Соответствующие деформа-
ционные колебания характеризуются следующими частотами: 6(Н—N—N) =
= v4 (А') 1150 см-1 (955 см-1); 6ас (N—N—N) = v5 (А') 522 см-1 (498 см-1)
и 6С (N—N—N) = v6 (А") 672 см—1 (638 см-1). В скобках приведены часто-
ты, соответствующие дейтерированному соединению DN3 (91.
Распад молекулы HN3 следует рассматривать как реакцию первого по-
рядка, причем часто термическое и фотохимическое разложение без взрыва
сопровождается образованием одних и тех же промежуточных продуктов
и однотипными реакциями (1, 4, 111. При этом особую роль должно играть
образование иминного радикала NH. Возникновение NH в возбужденном
состоянии (наряду с N2) из молекулы HN3 требует затраты энергии А// —
= 410,62 кДж/моль (1, 11, 12, 21—26]. Она соответствует разрешенному
переходу по уравнению
HN3 (W) NH RA) + N2 (xS+). (2.2)
При облучении (1, 22, 241 протекают реакции
HN3 + hv-+ NH 4- N2; (2.3)
NH + NH3 -> H2 + 2N2; (2.4)
HN3 + NH -> N2H2 + N2; (2.5)
N2H2 + HN3 -> NH3 + 2N2; (2 6)
3NH->NH3 + N2; (2.7)
NH3 + HN3 NH4N3. (2.8)
Существование имида NH в качестве промежуточного продукта доказано
появлением новых полос поглощения в УФ-спектрах 126] при 336 и 337 нм
(переход 3П-> 3S) [28] в области 253 нм (переход 1П->12+) (431 и 324 нм
(переход 41 ХА) (29].
Разложение молекул HN3co взрывом приводит к образованию водорода
и азота (7, 301, а в присутствии водорода — к образованию NH4N3 и N2H5N3
16
2.2.2. Азидная кислота
Химические свойства водных растворов HN3 предсказал Д. Менделеев
[32], который полагал, что N3-rpynna должна быть подобна атому хлора,
а соли азидной кислоты вследствие этого аналогичны галогенидам. Позд-
нее к такому же выводу пришел Биркенбах [33] при сопоставлении величин
напряжения разложения KN3 и галогенидов щелочных металлов.
Амид натрия реагирует при нагревании с N2O, нитрат калия или ам-
мония — с амидом калия в жидком аммиаке при нагревании в заплавленной
трубочке
2NaNH2 + N2O NaN3 + NaOH + NH3; (2.9)
KNO3 + 3KNH2 KN3 + 3KON + NH3. (2.10)
Азидную кислоту получают различными способами [1, 4], например при
взаимодействии N2H4 с HNO2, HNO3 и другими окислителями (Н2О2, Сг2О? »
МпОГ), по реакции
N,H4 + HNO2 ->HN3 + 2Н2О; (2.11)
N2H4 + NH2OH + 20 HN3 + 3H2O; (2.12)
При перегонке водных растворов HN3 получают фракцию с высоким со-
держанием HN3, а вначале даже газообразный HN3, но в интервале 90—100° С
отгоняется фракция, содержащая 27% HN3 [1]. Полученная многократной
дистилляцией 91%-ная HN3 имеет точку кипения 45° С [1]. Разбавленные
растворы HN3 (~3%) устойчивы, а большей концентрации — могут детони-
ровать [1, 4, 11]. Степень диссоциации HN3 (0,1 н, 20° С) составляет лишь
около 1% [1, 4, 11], а константа диссоциации равна 1,2 • 10~° (20° С). Для
DN3 в D2O при той же температуре она равна 0,98 • 10~° [1,4, 34].
Энтальпия образования водных растворов HN3 составляет 259,49 кДж/
моль (для недиссоциированной формы) и 274,57 кДж/моль (для диссоцииро-
ванной формы) [7—10]. Отсюда следует
HN3 (Н2О)Х Н+(H2O)V + Nr (H2O)Z; А/Дэй = 15,08 кДж/моль. (2.13)
Теплота диссоциации HN3, полученная из данных зависимости элек-
тропроводности HN3 от ее концентрации в интервале 10—15° С, равна
24,72 кДж/моль Ш. Теплоты нейтрализации HN3 растворами Ва (ОН)2
и NH4OH составляют соответственно 41,9 и 34,78 кДж/моль. Напряжение
разложения 1 н. HN3 равно 1,29 В [11. При электролизе 10%-ной кислоты
выделяются Н2 (на катоде) и N2 (на аноде) в соотношении 1 : 3 [1].
Спектры поглощения водных растворов HN3 отличаются от спектров
поглощения ионных азидов и указывают на ковалентный характер HN3
11]. УФ-облучение в присутствии H2SO, вызывает распад HN3
HN, (Н2О)Х Л». NH.2OH + (х - 1) Н2О + N2. (2.14)
Наряду с NH2OH в незначительных количествах образуется и NH3 [1].
В отсутствие H2SO4 при облучении образуются NH3 (~3%) и незначитель-
ные количества N2H4. При таком распаде возможно образование промежуточ-
ного продукта, NH-радикала, который ответствен за механизм и стехио-
метрию этой реакции [1].
Гидратированный азид-ион имеет слабые нуклеофильные свойства по
отношению к протону и ряду ионов тяжелых металлов. Водные растворы ази-
дной кислоты выступают в качестве окислителя ряда металлов. В этом
17
случае, кроме азида металла, образуются N2. NH3 и частично N2H4 [44]
М + 3HN3^M(N3)2 + NH3 + N2 (М = Zn, Мп, Fe, Ni). (2.15)
Смесь соляной кислоты с водным раствором азидной кислоты ведет себя
аналогично «царской водке» и растворяет платину [45]
Pt + 2HN3 + 4НС1 PtCl4 ± 2NH3 ± 2N2. (2.16>
В смеси НО—HN3, как и в смеси NaN3—HI, растворяется золото [44].
По отношению к КМпО4 азидная кислота выступает восстановителем, при-
чем в зависимости от концентрационных соотношений образуются соедине-
ния Мп (IV) или Мп (II) [11.
В практике широкое применение нашли эфирные [1, 351 и бензольные
[1] растворы HN3. Вероятно, в препаративной химии азидов можно исполь-
зовать растворы HN3 и в других растворителях, однако они изучены недо-
статочно.
Эфирные растворы HN3 получают действием смеси концентрированной
серной кислоты и диэтилового эфира на порошок NaN3. После многократ-
ного взбалтывания смеси нерастворимый сульфат натрия отфильтровывают
[1, 351.
Бензольные растворы HN3 получают многократной экстракцией бен-
золом из водных растворов, содержащих NaN3 и избыток 50%-ной серной
кислоты. После отделения бензольного слоя последний высушивается без-
водным сульфатом натрия [1].
2.3. АНАЛИТИЧЕСКИЕ И СТРУКТУРНЫЕ СВОЙСТВА
АЗИДНОЙ ГРУППЫ
2.3.1. Порядок связей и структура
Применяя правило электронейтральности и правило пограничных за-
рядов Полинга [46], для 16-электронной системы азидного иона можно
предложить две резонансные структуры I и II (рис. 2.1), каждая из которых
должна иметь длину связи 1,17 А. В ионных кристаллах KN3 и NaN3 азид-ион
линейный и симметричный (точечная симметрия DTCh)o-
Расстояния a-N—[3-N и [3-N—y-N равны 1,15 Д 0,02 А
147], в NH4N3 расстояние N—N равно 1,16 А [48], в
азиде стронция, напротив,— 1,12 А [49], т. е. в этих
случаях в мезомерии участвуют резонансные струк-
туры III и IV (ожидаемаоя длина связи 1,15 ±0,02 А).
Длина связи N—N 1,17 А несколько меньше, чем ожи-
даемая для N =N с зр2-гибридизированным атомом
азота. Длина связи зависит и от гибридизации атом-
ных орбиталей, участвующих в о-связях. На этом
основании связь N—N в азид-ионе рассматривается
как двойная ([3-N—sp-гибридизирован).
Расчет по методу МО [41, 50] дает для азидного иона, кроме двух о-ор-
битальных связей (между a-N—[3-N или [3-N—y-N), две связующие л-орби-
тали, которые образуют делокализованные л-связи (рис. 2.2). Каждый из
атомов азота имеет n-электронную пару, другие две электронные пары при-
сутствуют на несвязующих л-орбиталях.
Формальный заряд на крайних атомах азота составляет —0,8, а на сред-
//
е- ® ©
IN=N=M
Q © — О
IN=N=NI
ее- ©
IN—H=NI
© _©е
in=n-ni
2.1. Граничные
Рис.
формулы азид-иона
IV
18
нем атоме -4-0,6. Это указывает на существование обеих мезомерных граничных
форм
[|:N=N-N:| ----* |:N-^N=N:|J
При этом связующие л-электроны изображены различно в зависимости
от положения л-электронов в плоскости [131 (одна над другой стоящие чер-
точки обозначают положение л-
электронов в плоскости бумаги;
смещенные линии — положение
л-электронов пер пенди ку л яр но
к плоскости бумаги; свободная
электронная пара показана од-
ной рисочкой; л-электроны, не
образующие связей, изображе-
а б
Рис. 2.2. Связующие л-орбитали:
а — азид-ион; 6 — ковалентный азид
ны двоеточием.
Азидная группа при ковалентном связывании имеет расположение ато
мов, показанное на примере метилазида,
СН3
1,10А
Аналогичные расстояния для N—N-связей в азидной группе найдены
для триазидоциануровой кислоты 1511 C3N3 (N3)3 (1,26 и 1,11 А), а также для
азидной кислоты (см. 2.2). С учетом длин связей ковалентные азидные сое-
_ © е
I R —
© _е
©_ ©
В R—N—N=NI
динения могут рассматриваться как гибридные
молекулы граничных структур I и II (рис. 2.3).
Граничная структура IV в этих случаях не
участвует в резонансе, если принять во внима-
ние правило пограничных зарядов [461.
При одновременном присутствии граничных
структур I и III (рис. 2.3) в структуре молекул
® © _© ©
# R—N—N—NI
Рис. 2.3. Граничные струк-
туры ковалентных азидов
ковалентных азидов рассчитанные длины связи
N—N оказались равными 1,25 А для a-N—|3-N
и 1,12 А для p-N—y-N. Азидная группа асим-
метрична и редко нелинейна.
При ковалентном связывании в азидной груп-
пе присутствуют две различные связующие л-орбитали, причем одна де-
локализована между всеми атомами азота, а другая — между [3-N- и y-N-
атомами (рис. 2.2). Согласно расчетам [41], ковалентные азиды имеют раз-
личные формальные заряды на атомах азота: a-N —0,7; [3-N +0,9 и y-N
—0,2, что приводит к следующим граничным мезомерным структурам:
© © © ©
/:N-№NI
Rz
ZN_-N=N:|
Rz
Гибридизация концевых атомов азота подвержена влиянию соседних
атомов и может быть найдена из значений углов связи. Речь идет о sp-гиб-
ридизации, причем несвязующая электронная пара может занять одну sp-
орбиталь. В ионе N3 оба концевых атома азота не гибридизированы. Это
значит, что n-электроны находятся на s-орбитали, в то время как одна
р-орбиталь участвует в о-связи, а две р-орбитали — в л-связи. Энергия связи
19
Is атома азота в азидных комплексах металлов сравнима с энергией связи в
нитридных и динитридных комплексах переходных металлов [811.
Наблюдаемый угол связи у a-N атома (105—120°) позволяет сделать вы-
вод о 5р2-гибридизании (исключая влияние отталкивания п-электронов (521).
В соответствии с исследованием кристаллической и молекулярной структуры
триметилплатинаазида [531 впервые обнаружен пример зр3-гибридизации a-N-
атома; последний соединяется с тремя атомами металла
pt\
Pt-^N—N=N |.
Известны соединения и с тетрадентатными Ng-группами [262].
В комплексных соединениях N3 -группа, судя по значениям [3 для
[Со (N3)J2- [71], [О (N3)613- [421, [Rh (N3)6I3~ и [Pt (N3)6]2~ 142], прояв-
ляет наибольший нефелоксети-
ческий эффект по сравнению с гало-
генидами, что позволяет говорить
о высокой степени ковалентности
связи металл — азид,
F~ < NCO~ < NCS~ < СГ
« CNO~ CN~ < Br~ < Г^ Nr.
В соответствии с положением N3
в нефелоксетическом ряду в ИК-
Рис. 2.4. Структурная модель тетразидо- спектрах максимум поглощения
комплекса v (М—N) наблюдается в области
=400—450 см-1142]. Как следует из
положения в спектрохимическом ряду, азид-ион создает по сравнению с дру-
гими лигандами, связанными с металлом через азот, более слабое поле [42]
Г < Вг~ < СГ ~ SCN~ < (С2Н5О)2 Р (S) S~ < N~ < F~ <
< NCCT < NCS~ < NO? CNO“ < CN.
В модельной схеме для азидов переходных металлов (рис. 2.4) следует
принять, что азид-ион выступает в качестве донора л-электронов. Это зна-
чит, что происходит перекрывание занятых л-орбиталей азидного лиганда
d-орбиталью переходного металла.
В ИК-спектрах поглощения азидных комплексов металлов наблюдается
пять характеристических полос, которые лежат в области около 2050, 1300,
600 см \ от 300 до 400 см 1 и около 200 см-1. Эти полосы можно отнести к
валентным антисимметричным (vac (N3)), симметричным (vc (N3)), дефор-
мационным (6 (N3)) колебаниям No-группы, а также к валентным (v (М—N))
и деформационным (6 (М—N—N)) колебаниям [42, 263, 264].
Для гексазидных комплексов в случае линейной структуры металла-
зидной группы и Oh симметрии в ИК-спектре должно проявляться по одно-
му vac (N3), vc (N3), v (M—N), а также 6 (M—N—N) и 6 (N3) колебанию. He
исключается возможность появления и других полос, однако их отнесение
носит предположительный характер [42].
В диамагнитных тетразидных комплексах металл планарно окружен че-
тырьмя атомами азота [42]. Такая структура с точечной группой С4ь требует
по правилам отбора по одному vac (N3) — (Eu), vc (N3) — (feu) и v (M—N) —
(Eu) колебанию и по два 6 (N3) и 6 (М—N—N) (Eu + Au) колебания. Однако
20
экспериментально находят и другие частоты; это, вероятно, обусловлено тем,
что не все атомы находятся в одной плоскости. Для объяснения в этих слу-
чаях можно привлечь модель точечной группы Сь согласно которой выро-
жденное колебание Еи расщепляется на два активных в ИК-спектре коле-
бания (2Аи).
Комплексы с мостиковой азидной группой между двумя атомами металла
имеют полосу vac (N3) при 2060—2090 см-1 [42, 232], в то время как для азид-
ного мостика с «N-диазониевой структурой» значения vac (N3) значительно
выше [42, 173].
Азид-лиганд может проявлять би-, три- и даже тетрадентатный характер
[42, 262]. Его электроотрицательность, найденная из положения полосы
переноса заряда от лиганда к металлу, оценивается величиной 2,7—
2,8 [42].
2.3.2. Структура азидного радикала
и нитренной группировки
Сведения о структуре и свойствах линейного • Ng-радикала (основное со-
стояние 2Гу можно получить из сравнения его с такими изоэлектронными
группировками, как -NCO (цианат-радикал), СО2+ и N2O+. При этом
для азид-иона можно ожидать незначительных отклонений длин связей,
положения частот в ПК-спектрах поглощения (максимумы при 600, 1250,
1700, 2000 и 2140—2150 см-1) [54, 55] и силовых констант [12]. Как псевдо-
галоген N3 по величине энергии сродства к электрону должен располагаться
между хлором и бромом [33]. Эта энергия по данным измерения потенциа-
лов [56] и спектральным данным оценивается в 293,9 ± 21 кДж/моль
[8,34].
В видимой области спектра растворов Fe3+—NF отмечается полоса
при 460 нм, которую можно использовать для расчета энергии сродства к
электрону Ng-группы [8, 34]. Энтальпия образования газообразного азид-
ного радикала найдена равной 439,95 ± 12.6 кДж/моль [561 (рассчитан-
ная—473,47 кДж/моль [57]), энергия ионизации составляет 1185,77 ±
± 25,1 кДж/моль [56], электроотрицательность по Полингу равна 2,7 [46] и
находится между значениями для бромида (2,8) и иодида (2.5) [331.
Азидные радикалы образуются при фотолизе, пульсирующем радио-
лизе ковалентных азидных соединений в газовой фазе [36, 55, 82], а также
при термолизе твердых азидов металлов [12, 36], причем в начальный момент
реакция происходит по уравнению
NF—(ND* -> N3 4- ё~. (2.17)
В соответствии с исследованием электролиза водных растворов азидов
щелочных металлов, меченых изотопами в определенных местах азидной
группы (например, 14N15NUN), было установлено существование шести N-
содержащих промежуточных веществ [571. ЭПР-исследования облученных
азидов щелочных металлов указали на существование неустойчивых проме-
жуточных продуктов: радикал-ионов NT, N? или N2- [12, 58, 80]. Исследо-
вана кинетика их образования и проведен расчет электронной структуры ND
Ng, N^ [84].
Термическое и фотохимическое разложение ковалентных азидов на пер-
вой стадии реакции приводит к образованию нитренных соединений (биради-
калы структуры RN:) RN—N2->RN: +N2, которые способны к дальнейшим
21
превращениям (например, димеризации с образованием азосоединений
при R = фенил; для R = Н см. 2.2). Нитрен-бирадикалы склонны к много-
численным реакциям с участием простых С—Н- или С—С-связей, как, на-
пример, при реакциях алкалоидного синтеза [591.
2.3.3. Аналитическая химия азидных соединений
Типичные реакции осаждения азидного иона из водных растворов под-
тверждают его свойства как псевдогалогенидного иона. Растворимость
азидов аналогична хлоридам. Так, азиды Ag (I), Hg (I), Т1 (I) и Pb (II) плохо
растворяются в воде. В отличие от галогенидов все азиды этих металлов взры-
воопасны. Азид Ag (Г) устойчив на свету [11. Определение азота в азидах
сжиганием их с СнО (или с другими контактными материалами) возможно
для щелочных и щелочноземельных металлов, однако для азидов тяжелых
металлов этот метод неприменим. Гравиметрические и титриметрические ме-
тоды определения часто основаны на выделении свободной азидной кислоты;
газообъемные или йодометрические методы базируются на реакциях с рас-
твором 12 в присутствии тиосульфата или солей церия (IV).
Колориметрическое и фотометрическое определение азида основано на
использовании окрашенного в интенсивный красный цвет соединения же-
теза (III) [8, 341 в сочетании с другими методами (экстракция [681, микро-
анализ [69], комбинация с метиленовым голубым [1, 681). Разложение азид-
ной кислоты или ковалентных азидов концентрированной HI может быть
использовано для их количественного анализа. При этом определяются N2,
12 или NH3, образующиеся по реакции
HN3 + 3HI->NH4I N2 4-12. (2.18)
Иод-азидная реакция включает каталитическое разложение смеси азида
натрия с иодом в присутствии серы
12-г S2—-> 2I~ 4-S; (2.19)
S ф- 2N3~ -> S2~ 4- 3N2. (2.20)
Эти реакции применяют для обнаружения сульфидов, тиосульфатов,
тиоцианатов и органических серосодержащих соединений. Иод-азидные
реакции совершенствуются и используются для специальных систем [60,
611 Потенциометрические [621, кондуктометрические [631, [64] и амперо-
метрические [64, 65], осциллографические [851, а также электрохромато-
графические [66] методы используют преимущественно для определения и
отделения азидных соединений или комплексов [64, 65]. Титримегрический
анализ азидов окислением с HNO2 или нитрит-ионом в кислой среде [11 при-
годен также для комплексных соединений [67]. Обзор новых методов опре-
деления азидов приведен в работе [70], а в [861 описаны микротитриметри-
ческие методы. Тонкослойная хроматография [871 и масс-спектрометрия [881,
а также ЯМР-спектроскопия [89, 901 на ядрах 14N особенно пригодны для
исследования ковалентных азидов металлов или металлоорганических ази-
дов [90, 264]. Для линейного азидного иона (в водных растворах) наблюда-
ются резонансные сигналы 14N [911, а для ковалентно связанной азидной
группы, напротив, три сигнала 14N [92]. Это доказывает неэквивалентность
атомов азота. Появление трех сигналов 14N в ЯМР-спектрах ряда азидных
комплексов [891 указывает на ковалентный характер связи металл — азид
(М = Sn, Pb, Au, Pd, Pt). Для интерпретации спектров ЯМР азидов на яд-
рах 14N и электронных спектров использованы расчетные методы [931. Для
22
характеристики азидов металлов и сопоставления с другими данными часто
применяют ИК- и КР-спектры [42, 97, 263, 264], мессбауэровские спектры
[95, 96] и другие методы исследования [94, 95].
2.4. КЛАССИФИКАЦИЯ АЗИДОВ
К настоящему времени не существует общепринятой классификации азид-
ных соединений. На основании внутримолекулярных структур различают:
а) ионные азиды (содержащие симметрические азидные ионы); б) ковалентные
азиды (содержащие асимметрические азидные группы).
В качестве критерия для отнесения азидных соединений к одному из
названных типов можно принять длины и углы связей, а также ЯМР-спек-
тры. Ионный и ван-дер-ваальсовский радиусы атома азота азида находятся
в пределах г, = 1,55 А и rs = 1,65 А, в то время как ковалентный радиус
составляет гс = 0,70А [46, 47]. Химическая связь преимущественно ионного
характера осуществляется тогда, когда для всех длин связей атом—N —
атом азидной группы соблюдается соотношение d = ri гк (где d — длина
связи, г, — ионный или ван-дер-ваальсовский радиус; rk — радиус катио-
на). Ковалентное сочетание в азидной группе происходит в тех молекулах,
которые по крайней мере имеют одинаковые расстояния в связях, удовлет-
воряющие соотношению d — гс + га (гс — ковалентный радиус атома
азидной группы, га — ковалентный радиус атома, который непосредственно
связан с азидной группой). Отнесение азидных комплексов к одному из на-
званных типов азидных соединений возможно в том случае, если известны
соответствующие структурные данные. Однако в азидных комплексах мож-
но допустить координацию азидной группы многими атомами металлов,
что приводит к образованию координационной решетки олигомера или
полимера. Так, триметилплатинаазид [53] состоит из кубического каркаса
молекул [(CH3)3PtN3]4, так что каждый ct-N-атом азидной группы связан с
тремя атомами платины. Азидные комплексы металлов можно классифици-
ровать и по центральному атому металла. Для первого ряда переходных
металлов при ковалентной связи расстояние М—N составляет около 2 А
(ван-дер-ваальсовское расстояние ~3,6 А).
2.5. АЗИДЫ s-ЭЛЕМЕНТОВ
2.5.1. Азиды щелочных металлов
Азиды щелочных металлов являются исходными веществами для син-
теза других азидов. Сведения о получении этих соединений (включая NH4N3)
можно найти в учебниках препаративной химии [1241. Механизм образова-
ния азида натрия из амида натрия и гемиоксида азота исследован с помощью
азота 15N (O15NN), при этом установлено образование 22% Na15NNN и 78%
NaN15NN [122, 1251. Азид аммония выпадает в осадок при пропускании сухо-
го аммиака в эфирный раствор HN3 или же вследствие обменной реакции в
диметилформамиде между NaN3 и NH4C1 при 100° С с последующим охлаж-
дением (~60° С). Из этанольного раствора NH4N3 выделяется в виде бес-
цветных длинных игл. Азид аммония принадлежит к группе симметрии Ршап
(или D2h) и изоморфен дифториду аммония. Азид лития образуется в водно-
спиртовом растворе по уравнению
2NaN3 + Li2SO4 -> 2LiN3 ф- Na2SO4. (2.21)
23
Азиды щелочных металлов, как и азиды щелочноземельных металлов и
объемистых катионов (например, [R4N]+, [R4P]+, [R4As]+ и др.), принадле-
жат к ионным азидам. Они химически устойчивы, малочувствительны к
механическим воздействиям и разлагаются лишь при высокой температуре.
По аналогии с другими солями щелочных металлов азиды этих металлов ра-
створимы в полярных и малорастворимы в неполярных растворителях.
Большинство данных о кристаллической структуре ионных азидов ука-
зывают на их аналогию с хлоридами, а изменения симметрии в азидах вы-
зываются цилиндрическим азид-ионом. Замещение ионов хлора в струк-
Таблица 2.1. Некоторые структурные данные щелочных и щелочноземельных азидов
Соль Элементарная ячейка Угол p Пространст- венная груп- па 7. Литера- тура
a b c
LiN3 5,627 3,319 4,979 107,4° C2/m 2 [126]
LiN3-H2O 9,259 5,594 P63/mcm 6 [127)
a-NaN3 6,211 3,658 5,323 108,4° C2/m 2 [126]
P"NaN3 5,491 a = 38,7° P3m 1
nh4n3 8,930 8,642 3,800 Pm an 4 11281
KN3 6,113 7,094 I4/mcm 4 [129]
RbN3 6,310 7,519 14/mcm 4 »
CsN3 6,541 8,091 14/mcm 8 »
Ca (N3)2 11,32 11,07 5,95 Fddd 8 [130]
Ca (N3)2.3/2H2O 11,59 6,14 7,83 106,7° 4 »
Sr (N3)2 11,82 11,47 6,08 Fddd 8 Ц26]
Ba (N3)2 9,59 4,39 5,42 99,7° P2j/m 2 [131]
Ba (N3)2- H2O 7,29 10,84 6,96 104,7° C2/c 4 [132]
Ba (N3)2-3/2H2O 7,58 5,22 14,56 93,1° [133]
турном типе NaCl азид-ионами параллельно пространственным диагона-
лям элементарной ячейки NaCl приводит к удлинению последних и к об-
разованию гранецентрированной ромбоэдрической элементарной ячейки
(ромбоэдрический угол а = 38,7е, а' = 67,1°) для |3-NaN3. Данные струк-
турных исследований приведены в табл. 2.1. Ниже 18° С образуется моно-
клинный a-NaN3; в этой модификации отклонение угла связи от первона-
чальных пространственных диагоналей составляет 12° [126]. LiN3 имеет
аналогичную кристаллическую структуру [126], которая соответствует ше-
стикоординационному окружению катиона. Другие азиды щелочных метал-
лов (KN3, RbN3, CsN3) принадлежат к тетрагональной системе с октаэдри-
ческим окружением центрального атома. Они представляют собой слоистую
структуру, в которой слои расположены перпендикулярно к тетрагональ-
ной с-оси, причем каждый слой азидных ионов перекрывается следующим
слоем ионов азида. В этом случае восемь ближайших атомов азота так рас-
полагаются вокруг каждого иона металла, что наблюдается точечная сим-
метрия незначительно искаженной квадратной антипризмы. При нагрева-
нии RbN3 (точка превращения 315°С) и CsN3 (точка превращения 151°С)
образуют структуры, относящиеся к кубической модификации структурного
типа CsCl [134]. Азид аммония подобен азиду калия, он принадлежит к орто-
ромбической системе (искаженная CsCl-структура) с координацией 8 : 8,
24
в которой четыре атома азота находятся вблизи катиона, вследствие обра-
зования водородных мостиков (тетраэдрическое расположение). Из-за су-
ществования мостиковых водородных связей в азиде аммония он относится
к переходному типу.
2.5.2. Азиды щелочноземельных металлов
Be (N3)2 и Mg (N3)2 получают при взаимодействии эфирного раствора азид-
ной кислоты с диметиллатом бериллия [136] или с диэтилатом магния [137],
а также при действии на галогениды бериллия или магния триметилкремний-
азида [103]
MR2 + 2HN3 -> М (N3)2 4- 2RH (М = Be, R = СН3; М = Mg, R - С2Н5);
(2.22)
2BeCI2 • 20 (СгН,^ + 2 (СН8)8 SiNs
[BeCIN3 • О (С2Н6)2]2 + 2 (СН3)8 SiCl; (2.23)
ВеС12 + 2 (CHS)3 SiN8-2b2’- Be (N3)2 + 2 (СН3)3 SiCl; (2.24)
MgX2 • 20 (C2H5)2 + (CH8)8 SiN3
-> MgXN3 • nO (C2H6)2 + (CH3)S SiX (2.25)
(X = Cl, n = 0,5; X = Br, I, n = 1).
Реакция дигалогенида бериллия c Me3SiN3 в мольном соотношении 1 : 2
приводит к образованию в эфире димерного эфирата бериллийхлордиазида,
а в метиленхлориде при соотношении 1:2к образованию бесцветного, рас-
творимого в воде взрывоопасного полимера [Be (N3)2]n.
MgBrN3 и MgIN3 образуются в реакциях Ph3SiN3 с магнийфенилгало-
генидом в эфире в запаянной стеклянной трубке [138], a Mg (N3)2 получают
из Mgl2 и Me3SiN3 в соотношении 1 : 2 при 100° С в эфире или при взаимо-
действии PliMgBr с Me3SiN3. Полученный по этому методу Mg (N3)2 содержит
также MgBrN3 в качестве примеси. Наиболее подходящим растворителем для
этой реакции является тетрагидрофуран, причем при этом образуются
Mg (N3)2 • 2Thf или MgIN3 2Thf.
Азиды стронция и бария получают реакцией их гидроксидов или оксидов
с HN3 или NH4N3 [265, 266]; Ba (N3)2 образуется также при взаимодейст-
вии Ва (С1О4)2 с KN3 или посредством реакции Ва (ОН)2 с этилнитритом и
гидразином [266]. Ra (N3)2 получают из Ra СО3 и HN3 [267]. В системе
Sr (N3)2— Н2О наряду со Sr (N3)2 также существуют гидраты Sr (N3)2 • nH2O
(и = 2,4 или 6) [2871.
Азиды кальция и стронция образуют орторомбическую элементарную
ячейку с восьми координационным окружением катиона, а Ва (N3)2 кристал-
лизуется в моноклинной системе [131, 135], в которой барий имеет коор-
динационное число 9. Каждый ион бария находится в центре тригональной
призмы с наслоением прямоугольных пирамид с кристаллографически дву-
мя типами азидных ионов.
2.53. Свойства азидов щелочноземельных
и щелочных металлов
Отдельные данные о максимумах в ИК-спектрах поглощения азидов ще-
лочных и щелочноземельных металлов, а также силовые постоянные азид-
иона содержатся в табл. 2.2. Из сравнения ИК-спектров азиду бериллия
25-
Таблица 2.2. Характеристические колебания азидов щелочных
и щелочноземельных металлов (см-1) и силовые постоянные
азид-иона [16, 103]
Соединение Vac (N3) VC (N3) * 6(N3) 26 (N3) * Силовые постоянные, Н/см
kt kit
LiN3 2092 1369 635 1277 13,94 1,73 0,56
NaN3 2128 1358 639 1267 13,92 1,30 0,57
KN3 2041 1344 645 1273 13,36 1,74 0,58
RbN3 2024 1339 642 1271 13,21 1,78 0,58
CsN3 2062 1329 635 1267 13,31 1,45 0.56
Ca (N3)2 2114 1381 638 1267 14,21 1,74 0,57
Sr (^3)2 2096 1373 635 1273 14,01 1,75 0,56
Ba (N3)2 2123 1354 650 1278 14,15 1,97 0,55
2083 637
Mg (N3)2 2204 653
[Be (N3)2j;** 2217 2128 1255 *
]BeClN3L] **** 2200 1275 **
(L = (C2H5)2O) nh4n3 2145 2041 1259 1345 661 650 13,36 1,74 0,59
* Спектры комбинационного рассеяния.
** Перекрывается.
*** Другие максимумы поглощения: 824 широкий (v (BeN)); 710, широкий (v(BeN)); 533, широкий.
**** Другие максимумы поглощения: 814 (v (BeN)); 685 (v (BeN)); 571 (v (BeCl)); 483, 395 и по-
лосы эфира.
приписана ковалентная структура, а для бериллия найдено координационное
число 4 [103]. В то же время азид магния, как и азиды щелочных и тяжелых
щелочноземельных металлов, является солеобразным продуктом. Граница
между ионогенными и ковалентными азидными соединениями элементов в
периодической системе часто проходит между Li и Be, а также между Mg
Рис. 2.5. Строение [BeClN3 • О(С2Н5)2]2 и [Be (N3)2]n
и Al, Са и Ga. «Простой» азид бериллия, как и [BeClN3 OEt2]2, принадле-
жит к координационным соединениям. Их строение [103], вытекающее из
данных ИК-спектроскопии, представлено на рис. 2.5. Валентные колебания
азида в азиде бериллия (табл. 2.2) наблюдаются в области колебаний p-N3-
лигандов. Основные полосы N3-rpynn в ИК-спектрах азидгалогенидов бора
представлены в табл. 2.3. Полимерный [Be (N3)2i„ имеет очень широкие ИК-
полосы поглощения (табл. 2.2).
Значения энергии кристаллических решеток щелочных и щелочноземель-
26
Таблица 2.3. Волновые числа азидных колебаний
в соединениях (BX2N3)3 [ 103], см-1
Отнесение (BF2Ns)3* (BC12N,)3** (BRr2N3)3**
(vac 4“ гс) N3 3410 (оч. сл.) 3400 (оч. сл.) 3380 (сл.)
3270 (сл.) — —
2vc 2440 (сл.) 2430 (сл.) 2400 (сл.)
— 2390 (сл.) 2370 (сл.)
vac (Ng) 2210 (оч. с.) 2210 (оч.с.) 2200 (оч. с.)
2160 (с.) 2160 (ср.) 2155 (сл.)
vc (Ng) 1290 (пл.) 1240 (пл.) 1232 (пл.)
1210 (с.) 1198 (с.) 1195 (оч. с.)
26 (N3) 1100 (сл.) — —
T(N3) 670 (пл.) 670 (пл.) —
6 (N3) 587 (ср.) 535 (ср.) 518 (ср.)
* Другие поглощения при: 1350 пл., 1320 оч. с. (vac (BF2)); 1050 пл., 1010 оч. с. (v (BN)); 930 ср.,
910 ср. (v (BN)) 825 ср. (Vc (BF2)); 633 оч. с. (v (BN?)); 570 пл., 544 оч. сл., 508 сл., 475 оч. сл.
** Полный спектр приведен в [100].
Относительная интенсивность: сл.— слабая, оч. сл.— очень слабая, с.— сильная, оч. с.— очень
сильная, ср.— средняя, пл.— плечо.
ных азидов определялись с применением приближений цикла Борна — Га-
бера [11, 146], расчетными методами [8, 140] и из измерений теплот гидра-
тации [8]. При этом последний метод дает надежные результаты при опре-
делении энергий решеток азидов щелочных металлов. Некоторые эксперимен-
тальные данные об энергии
решетки азидов металлов и
энтальпии реакций разложе-
ния азидов металлов [139]
приведены в табл. 2.4.
Рассматривая оптические
свойства азидов щелочных и
щелочноземельных металлов,
приходится, к сожалению,
ссылаться только на спектры
поглощения, так как их мож-
но использовать для сопо-
ставления со спектрами погло-
щения других азидных сое-
динений. Точно так же азиды
щелочных и щелочноземель-
ных металлов могут быть ис-
пользованы при рассмотрении
реакций разложения и опре-
делении энергетического уров-
ня электронов [12].
В спектре отражения NaN3
[141] и KN3 [142] найдены
Таблица 2.4. Энергия решеток и энтальпия
реакций разложения
некоторых азидов [8,139]
Соединение Энергия решетки, кДж/моль Энтальпия реакции разло- жения или превращения, кДж
металл нитрид ОКСИД
LiN3 NaN3 KN3 RbN3 CsNs nh4n3 812,86 733,25 657,83 636,88 611,74 733,25 10,9 78,77 311,31
Ca (N3)2 2167,19 46,09 190,23 682,55
Sr (N3)2 2069,02 7,12 187,43 597,91
Ba (N3)2 CuN3 AgN3 T1N3 cx-Pb (N3)2 1964,27 951,97 857,69 685,10 1817,20 —22,21 98,88 506,99
максимумы поглощения соответственно при
240 и 248 нм, однако после облучения УФ-лучами при —196° С появляют-
ся новые полосы [143, 145]. Наблюдаемые полосы поглощения NaN3 [1411,
KN3 [8] и Ba (N3)2[141] соответствуют переходам от валентной зоны к зоне
проводимости.
По данным уровней энергетического состояния [12], границе непрерыв-
ного спектра соответствует ряд линий поглощения, которые отвечают дис-
кретным линиям перехода Однако они наблюдаются только тогда, когда
27
резко уменьшаются помехи, вызываемые колебаниями решетки (спектры при
пониженной температуре).
Дискретные (характеристические) спектры известны, например, для ази-
дов натрия, калия, рубидия и цезия [144]. Для NaN3/Na были проведены
исследования абсорбционных и ЭПР-спектров [147].
Многочисленные исследования других физических свойств щелочных
и щелочноземельных азидов относятся к чистым и частично разложенным
азидам и охватывают электрические (фотопроводимость, электропровод-
ность, энергия разложения), термические изменения, а также фотохимиче-
ские и радиационно-химические свойства при облучении [11, 12, 148—152],
внутреннюю фотоэмиссию [153], процессы диффузии [154], а также спектро-
скопию электрона [155]. В связи с рентгеновскими исследованиями описан
синтез азидогидратов стронция [156]. Известны кристаллографические,
ИК-спектроскопические исследования гидразонийазида (N2H5N3) [157, 158].
Возможности применения азидов щелочных и щелочноземельных металлов
в химических реакциях обсуждаются в 2.8.
2.6. АЗИДЫ р-ЭЛЕМЕНТОВ
2.6.1. Азиды неметаллов
Азиды неметаллов относятся к группе ковалентных (молекулярных)
азидов. Азидная кислота рассмотрена в 2.2. Сказанное о структуре молеку-
лы HN3 можно распространить и на другие ковалентные азиды типа RN3.
В качестве примера на рис. 2.6 представлены структуры хлоразида [72],
триазидокарбония [С (N3)3]+ [73] и азидодиаминокарбония [N3C (NH2)2]+
[74]. Сведения об электронной структуре получены, как и для цианазида,
на основании микроволновых спектров [104].
Азиды карбония (рис. 2.6) содержат азидные группировки с углом связи
107 или 108° и 160, 162 или 167° (например, ион [С (N3)3]+). Кроме того, груп-
па N3 наклонена к плоскости л-электронной системы. Угол наклона состав-
ляет 2, 3 и 8° для [С (N3)3]+, а расстояния связи N—N существенно разли-
чаются между собой, так что их следует рассматривать в качестве одинарных
или тройных связей [13, 73]. Напротив, в триазиде циануровой кислоты [51]
азидные группы лежат в плоскости цианурового кольца. Расстояния свя-
зей N—N в [N3C (NH2)2]+ [74] соответствуют ожидаемым для ковалентных
азидов (см. 2.2). Структурными исследованиями кристаллического [С (N3)3] •
• SbCl6 установлен положительный заряд на [S-N-атоме.
Азид бора В (N3)3 получают при взаимодействии В2Н6 с эфирным раство-
ром азидной кислоты (в диэтиловом эфире или в тетрагидрофуране) [75].
Структура соединения (BC12N3)3 [76] подобна (SbCl4N3)2 [77] и (TaCl4N3)2
[78]. В этих соединениях существуют многочисленные циклические системы,
так как a-N-атомы азидных групп связаны с двумя другими атомами. Цик-
лический каркас (В—a-N)3 от (BC12N3)3 представляет собой конформацию
скошенной ванны.
Для получения смешанных азидных соединений можно использовать
хлоразид [39, 40]. Последний реагирует с ВС13 при 0° С [79] по реакции
ЗВС13 + 3C1N3 -> [BC12N3]3 + ЗС12. (2.26)
Пиридинат азида бора получают обменом галогена на азид в пиридине
[98]. Соединения типа BX2N3 (X = Cl, Вг) образуются из тригалогенида
бора и LiN3 [99] или галогеназида [100]. Известны также азидные соединения
28
бора типа ХВ (N3)2 и Y2BN3 (X = Н; Y = Н, R, R2N, RO; R = органиче-
ский остаток) [98] и координационные соединения состава М [В (N3)4] (М =
= катионы щелочных металлов) [101]. LiB (N3)4 образуется при взаимодейст-
вии LiBH4 с азидной кислотой при 0° С.
Галогениды бора реагируют в метиленхлориде с Me3SiN3 с образованием
тримерных бордигалогенидазидов [102, 103]
3BX3 + 3Me3SiN3-£^-(BX2N3)3 + 3Me3SiX (X = F, Cl, Br). (2.27)
При этом образуются бесцветный взрывчатый (BF2N3)3 или (BC12N3)3
и (BBr2N3)3, которые получают и по другим реакциям [99, 1001. Однако по
этому методу нельзя синтезировать ВХ (N3)2 и В (N3)3 [103] и боридоазиды.
Рис. 2.6. Строение некоторых ковалентных азидов:
а — триазидкарбоний-ион; б — азидодиаминокарбоний-ион; в — хлоразид
Напротив, этот метод пригоден для азидирования органических бордигало-
генидов, а также бис-органических боргалогенидов [98]. Циклическая струк-
тура соединений типа (BX2N3)3 доказана ИК-спектроскопически [76]
(табл. 2.3).
Из азидных соединений Si и Ge, кроме тетразидов, например Si (N3)4,
имеют значение кремнийорганические и германийорганические азиды типа
R3MN3 и R2M (N3)2 [101] (R = Me, Ph и др.; M = Si, Ge). Si (N3)4 образуется
из SiH4 и HN3 [75, 101], а соединения типа R3SiN3 получены из R3SiX и ази-
дов лития или натрия [105]. Последние пригодны в качестве азидирующих
агентов кислот Льюиса, таких, как галогениды Si (образование SiCl3N3)
[103], Sn [106], Sb [107], Те [108], I, Be, Mg, Al [103], а также для получения
фосфорных гетероциклов [112]-
S S S S
II /Sx || || S. || .
R_pZ \p__R + Me3SiN3->R-P< >P-R + -j- Srt + N2 (2.28)
\SZ \nZ n
I
SiMe3
(R = Me, Et).
При этом отщепление серы и азота происходит уже при комнатной тем-
пературе. Избыток (СН3)3 SiN3 не приводит к образованию других продуктов.
Известна структура SiH3N3 [109] и GeH3N3 [110], а также данные о роли
d- состояния связи Si—N [111]. Реакции фотолиза кремнийазидов приводят к
образованию промежуточных соединений типа R2Si=NR с Si—N (рл—ря)-
связями [272].
29
Сведения об азидных соединениях фосфора относятся к катиону [Р (N3)4]+
[113], аниону [Р (N3)6]~ 1114], фосфорфторазидам [115], исследованиям реак-
ционной способности некоторых фосфоразидных соединений [116, 118],
а также реакций органических азидов с ос-, [3-ненасыщенными соединениями
фосфора (III) [119]. На основании ИК-спектров обсуждена электронная стру-
ктура трехмерных фосфорнитрилпсевдогалогенидных соединений [120].
Для арсенорганических соединений следует отметить реакции триарилар-
синов с нитренами [121].
Галогеназиды играют роль в образовании элементазидных группировок.
Наиболее важными являются C1N3 [122] и FN3 [И], получен также мало-
устойчивый нитрозилазид NON3 (ON4) [123].
2.6.2. Азиды алюминия, галлия, индия и таллия
Азиды образуются при взаимодействии соответствующих гидридов ме-
таллов с HN3 в эфире [159]
МН3 + 3HN3^- М(N3)3 + ЗН2 (М = Al, Ga). (2.29)
Кроме того, азид алюминия получают азидированием А1С13 [103] или ор-
ганоалюмогалогенидов [160], например
А1С13 + nMe3SiN^§- А1С13_п (N3)n + nMe3SiCl. (2.30)
Триметилат алюминия реагирует с HN3
А12 (СН3)6 + 4HN3 2СН3А1 (N3)2 + 4СН4. (2.31)
Азид алюминия образуется также в реакции А1С13 с NaN3 в безводном бен-
золе. Азидхлориды алюминия — твердые термически неустойчивые соедине-
ния: [AlCl2N3]n взрывается уже при соприкосновении с водой, а [А1С1 (N3)2K —
при температуре выше 100° С. Из ИК-спектров [103] можно заключить,
что азидхлориды алюминия являются ковалентными соединениями и имеют
полимерную структуру. В противоположность трехмерным азиддигалоге-
нидам бора [A1C12N3]„ растворяются в СН2С12 с образованием неассоцииро-
ванного соединения. Реакцией гидрида алюминия с HN3 можно получить
также азидоалюминаты типа Al (N3)f— и Al (К3)Г [159, 161]; алюминийорга-
нические азиды, например К [(CH3)3A1N3A1 (СН3)3], исследованы структурно-
химически [162]. По данным ИК-спектров, в соединении [Al (C2H5)2N3]3
азидная группа выступает в качестве мостиковой с образованием N-диазо-
ниевой структуры [173]. Органо галлий- и органоиндийазид соответству-
ют типу [R2MN3]n (М = Ga, In; п — 3 или 2; R = алкилрадикал) [160,
161, 173].
T1N3 осаждается из водных растворов в виде желтых кристаллов. Азид
таллия (I) кристаллизуется, как и азиды К, Rb и Cs (табл. 2.1), в виде тет-
рагональной слоистой структуры с восьмикоординационным окружением
центрального атома [129]. В спектре поглощения T1N3 проявляется широкая
полоса с максимумами при 320 и 275 нм с возрастанием поглощения до 200 нм
[142]. При облучении электромагнитными лучами определенной длины вол-
ны T1N3 проявляет фотопроводимость [163]. По величине энергии решетки
азид таллия (I) располагается в ряду азидов щелочных металлов (табл. 2.4).
Исследованы фазовые переходы при низких температурах, ИК-и КР-спект-
ры и влияние добавок других металлов [164—166, 289]. Водные растворы
Т1 (III) в НСЮ4 образуют с азидными ионами комплексы с максимальным
числом азидных лигандов, равным 3 [171]; колориметрическими исследова-
30
ниями было доказано также образование комплексов [Т1 (N3)6]3~ и [Т1 (\т3)4 •
• С12]3~ [172]. Из растворов Т1Вг3 и NaN3 можно выделить в виде желтых игл
соединения [Ph4As] [TJBr2 (N3)2] [42]. Ph2TlN3 образуется при взаимодейст-
вии водных растворов Ph2TlF и NaN3 [42].
2.6.3. Азиды олова, свинца, сурьмы и висмута
Азиды Sn (N3)4 или М2 [Sn (N3)6] (М = Na, объемистый катион) [167, 168]
образуются при реакциях обмена SnCl4 и NaN3 в бензоле или тетрагидрофу-
ране [167], в то время как соединение [Ph4As]2 [Sn (N3)6] может быть выделе-
но из сернокислых водных растворов (NH4)2 SnCl6 и NaN3 [42].
Хлорид олова (IV) реагирует с Me3SiN3 в метиленхлориде по правилу
замещения Виберга [103] с образованием [SnCl2 (N3)2]m [106, 107]. Для изу-
чения оловоорганических азидов [275], а также комплексов [SnCl4 (N3)2]2~
и [SnCl4N3]2~ [292] применены спектры Мессбауэра. Исследования диметилоло-
во (1У)азидных комплексов выполнены в водных растворах [170]. Азид свинца
(II) выделяется из водных растворов. Pb (N3)2 имеет различные модифика-
ции, которые можно отнести к координационным азидным соединениям (см.
табл. 2.4). a-Pb (N3)2 содержит четыре различные линейные азидные группы,
из которых две симметричны, одна несколько асимметрична, а еще одна от-
личается различным расстоянием N—N (1,21 и 1,15 А) [169]. Каждый атом
свинца окружен восьмью атомами азота, образуя искаженную тетрагональ-
ную антипризму.
a-Pb (N3)2 кристаллизуется в орторомбической модификации, а р-Pb (N3)2—
в моноклинной [12, 13]. |3-Pb (N3)2 можно получить специальным диффу-
зионным способом из раствора, содержащего N^, Pb (NO3)2 и NaNO3 [11].
Однако при этом возможно разложение азида свинца со взрывом. Кристалли-
ческий Pb (N3)2 имеет максимум поглощения при 400 нм [174] и проявляет
слабую фотопроводимость [142]. Максимум в спектральном распределении
фототока лежит при 365 нм, однако первичная фотоэмиссия зависит от вре-
мени облучения [175]. Исследованы рост кристаллов Pb (N3)2 [176], его тер-
мическая устойчивость [177, 178, 179], фоторазложение и электронная струк-
тура [180], ИК-спектры поглощения [181, 289] и электрохимические свойства
[182, 183].
Pb (N3)4 и азидоплюмбаты (IV) относятся к взрывчатым соединениям
[184]. Поэтому гексазидоплюмбат (IV) может быть получен в виде темно-
красных игл лишь при введении катионов тетрафениларсония [42]. Окра-
шенные в полярных органических растворителях в темно-красный цвет рас-
творы [Ph4Asl2 [Pb (N3)6] на свету сразу обесцвечиваются. При этом выделя-
ется азот, a Pb(IV) восстанавливается до РЬ(П) [184, 42]. Известны также
смешанные ацетатазидные соединения состава Pb (ООССН3)4—п (N3)n [185].
Хлорид сурьмы (V) реагирует с R3SiN3 с образованием димера [SbCl4N3]2
[77, 106], в котором a-N-атомы азидных групп связаны с двумя атомами
сурьмы с образованием четырехчленного цикла [77]. Последний является
напряженным и объясняет значительную реакционную способность
[SbCl4N3]2 и легкую замещаемость p-N3-rpynn. Известны данные о получении
и спектрах поглощения триметил- и трифенилдиазидов сурьмы [186]. BiON3
образуется при взаимодействии водного раствора Bi (NO3)3 с избытком NaN3
[289].
31
2.7. АЗИДЫ ЭЛЕМЕНТОВ ПОБОЧНЫХ ПОДГРУПП
Химия «простых» азидов элементов побочных подгрупп принципиально
относится к химии координационных соединений, ибо на основании струк-
турнохимического поведения и реакционной способности нельзя провести
резкого различия между ними. Поэтому можно объединить методы получе-
ния «простых» и анионных азидов металлов. Соединения с объемистыми ка-
тионами, как и ряд солей аммония, термически устойчивы и не чувствитель-
ны к перемешиванию (прикосновению). Однако в пламени они часто вспы-
хивают, а соли щелочных металлов взрывоопасны, и поэтому работа с ними
требует большой предосторожности (например, комплексы Си (II), Аи(Ш),
Pb(IV)). Повышенная устойчивость соединений с объемистыми катионами
[42] основывается на эффекте «разбавления» их большими катионами. Так,
эти соединения часто образуют кристаллы правильной геометрической струк-
туры, малочувствительные к воздействию света. Они преимущественно хо-
рошо растворимы в полярных органических растворителях (ацетон, нитро-
бензол, метиленхлорид и др.), и поэтому их легко очищать. Металл-азидная
группировка в соединениях переходных металлов, однако, довольно реак-
ционноспособна. На этом основании они используются в реакциях с различ-
ными веществами [38] (см. 2.8). Для получения азидных комплексов могут
быть использованы следующие методы:
1. Реакции обмена галогенидных комплексов с азидами щелочных ме-
таллов или азидом серебра (I) в водных и неводных средах [168]. Для полно-
го обмена галогенидных лигандов на азид в водных растворах часто необхо-
дим большой избыток азидных ионов. Введение органических растворите-
лей улучшает образование азидных комплексов [187—189].
2. Реакции соответствующих сульфатов и нитратов металлов (а также
других солей металлов) с азидом натрия в водном растворе. Из-за щелочно-
го характера растворов азида натрия при обменном взаимодействии для
ионов, образующих прочные гидроксокомплексы или оксигидраты, реакцию
необходимо проводить в кислом растворе.
3. В некоторых случаях образование азидных комплексов происходит
при внутримолекулярных реакциях окисления — восстановления азидных
комплексов, при этом выделяется азот и образуется азид металла более низ-
кой степени окисления (например, Au (N3)F [42]).
2. 7.1. Азиды элементов I и II побочных подгрупп
CuN3 кристаллизуется в тетрагональной структуре [190, 289], каждый
атом меди планарно окружен четырьмя N-атомами: двумя с расстоянием
2,23 А (транс-атомы) и двумя на расстоянии 2,30 А. Угол NCuN составляет
73 и 107° (табл. 2.5). Комплекс меди (I) [(Ph3P)2Cu (N3)]2 в твердом состоянии
образует восьмичленные кольца с азидными мостиками, но в растворе моно-
мерен [279]. Си (N3)2 образует бесконечные цепи, в которых каждые два атома
меди связаны двумя атомами азота с образованием четырехчленного цикла
(табл. 2.5, рис. 2.7). Каждая цепь смещена параллельно по отношению к
другой; половина a-N-атомов одной цепи связана координационно с медью
другой цепи [191, 192, 289], остальные азидные группы координированы
медью через y-N-атомы. Цифры возле атома азота на рис. 2.7 обозначают
расстояние, на котором эти атомы отстоят от плоскости цепи (кольца). В со-
единениях Си (NH3)2 (N3)2 и СиРу2 (N3)2 также присутствуют бесконечные
цепи с ковалентно связанными аммиаком или пиридином в транс-положен ш
[193].
32
Таблица 2.5. Структурные данные для координационных азидов
Соединение Элементарная ячейка. A Простран- ственная группа z Литература
CuNg a = 8,65, c = 5,59 I4j/a 8 [190]
Ou (N3)2 a = 13,45, b = 3,08 Pnma 4 [191, 192]
Си (N3)2 (NH3)2 a = 6,39, b = 7,45, c - 12,71 Pnma 4 [193]
Си (N3)2Py2 a = 13,88, b = 13,65, c = 6,41 Стс2г 4 »
AgN3 a = 5,62, b = 5,92, c = 6,01 Ibam 4 [194, 195]
Cd (N3)2 a = 7,82, b = 6,46, c - 10,04 Pbca 8 [196]
Cd (N3)2Py2 a = 15,79, c = 10,15 I4x/a 8 [197]
азиднои кислоты
Взаимодействие азида меди CuN3 с водным раствором
в присутствии катионов [Ph4P]+ приводит к образованию чувствительных
к воздуху диазидокупритов (I) [Ph4P] [Си (N3)2] [42]. Для азидных комплек-
сов меди (II) в зависимости от объема и заряда катиона получаются соеди-
нения типа [Cu(N3)3] , [Cu(N3)4]2~, [Си (N3)6]4~ или [Cu2(N3)5r [198].
При введении [РН4Р]+-катиона выделяется соединение состава [Ph4Pb
[Си (N3)3], которое в растворе аце-
тона имеет магнитный момент рЭф =
= 1,89 М. Б., соответствующий
d9-конфигурации ионов Си2+. Дан-
ные ИК-спектроскопии азидных со-
единений меди, а также других
азидных соединений металлов при-
ведены в табл. 2.6 [42]. Известны
исследования растворимости ази-
дов меди (II) [199], констант обра-
зования ионов [Си (N3)4]2~ и [Си •
• (N3)312~ [200], магнитные исследо-
Си
N-----N------N'
Си
N-------N------N
Си ,
Си Z54 N_U3_NJ2LN 'NJ2LN_ULN
-027 -0,16 +0,43
Си
Рис. 2.7. Фрагмент цепи в структуре Cu(N3)2
вания комплексов Cu-p-N3 и Ni-p-N3 [201], а также другие структурные
данные [13]. Магнитные свойства димера [Cu2 (dien)2 (N3)2] [BPh4]2 изуче-
ны в работе [284].
Азид серебра (I) кристаллизуется подобно азиду калия (табл. 2.1 и 2.5).
Элементарная ячейка имеет искаженную орторомбическую структуру с
координационным числом центрального атома 8, однако при этом четыре ато-
ма азота находятся в ближайшем окружении атомов серебра с тетрагональной
структурой и расстоянием Ag — N, равным 2,56 А. Азидные группы симме-
тричны и линейны [194, 195,289]. Ряд исследований относится к разложению
и взрыву AgN3 [202—204]. Азидные соединения золота соответствуют типам
[Au (N3)4]—, [Au (N3)2]_, [42, 187, 205] и [Me2AuN3]2 [209]. Взрывчатый
[Me2AuN3]2 выпадает из водного раствора по реакции
2 [Me2Au en] N3 + 2НС1 [Me2AuN3]2 + 2en НС1. (2.32)
Для Cd (N3)2 установлены параметры элементарной ячейки [196] (табл. 2.5),
а для комплекса CdPy2 (N3)2 — точная структура [197], причем это соедине-
ние рассматривается как высокополимерное. В комплексах цинка Zn (NH3)2-
• (N3)2 и ZnPy2 (N3)2 [206] атом цинка координирован тетраэдрически, точно
так же, как и в [Zn (N3)4]2~ [42, 168, 207, 296]. Соединения типа [RZnN3]n
относятся к полимерным с П3-мостиками [278]. Последние содержатся и в
2 2473
33
Таблица 2.6. Максимумы полос поглощения в ИК-спектрах азидных комплексов металлов
Соединение vac <N3) vc (N3)
(Ph4As]2 [Sn (N3)6J 2073, 2055 (оч. с., шир.) 1336 (ср.), 1276 (с.)
(Ph4AsJ2 [Pb (N3)6J 2045 (оч. с.) 1262 (сл.), 1253 (сл.)
Na[Me4N[2[Fe (N3)6[ 2064 (оч. с.) 1350 (с.)
[Ph4As]2 [Pd2 (N3)6J 2060, 2033, 2000 (оч. с.) 1283, 1271 (ср.)
[Ph4As]2 [Pt (N3)4] 2075 (сл,), 2024 (с.) 1276 (ср.)
[Et4N[2 [Mn (N3)4J 2046 (оч. с.) 1344 (ср.), 1287 (сл.)
[Ph4As]2 [VO (N3)4[ 2088 (ср.), 2051 (оч. с.) 1340 (ср.)
[Ph4As]2 [UO2 (N3)4] 2035 (оч. с.) 1321 (ср.), 1210 (сл.)
[Ph4Asj [Au (N3)4[ 2030 (оч. с.) 1261, 1251 (ср.)
[Ph4P]2[Cd2 (N3)6[ 2110 (оч. с.), 2085 (оч. с.), 2056 (пл.), 2046 (пл.), 2032 (оч. с.) 1333 (сл.), 1301 (ср.), 1279 (сл.)
[Ph4P] Hg (N3)3[ 2053 (оч. с.), 2020 (оч. с.) 1316 (ср.), 1272 (ср.)
[Ph4P] [Cu (N3)3J 2049 (оч. с.), 2018 (пл.) 1289 (сл.), 1278 (сл.)
Ph2TlN3 2012 (оч. с.) 1263 (сл.)
[Ph4As[ [TlBr2 (N3)2[ 2036 (оч. с.) 1265 (ср.)
* М = металл.
** Другие полосы при 969 (c.) = v(VO) и 125 (шир.).
*** бас (UO2): 890 (с.); vc (UO2):512 (оч. сл.); 6 (UO2): 295 (с.).
Относительная интенсивность: сл.— слабая; оч. сл.— очень слабая; с.— сильная; оч. с.— очень
CdPy2 (N3)2, в котором кадмий имеет гексакоординационное (октаэдрическое)
окружение [197]. Описаны устойчивые азидные комплексы цинка с радикаль-
ными лигандами, изучены их ЭПР-спектры [285]. Определены параметры
моноклинной решетки Zn (N3)2 - ЗН2О [291]. Для кадмия и ртути описаны
комплексы: [Cd2 (N3)5]— [42] (рассматриваемый как аддукт состава Cd(N3)2-
• [Cd (N,)sn. [Cd (N3)„l2-" (n = 1 4- 5) [208]; [Hg (N8)J2-, [Hg (NS)F,
[Hg (CNO)2N3] , а также [Hg (N3)2 • 2Ph3P] [42]. Азиды ртути (I) и (II) оса-
ждаются из водных растворов (см. 2.3.3), а взрывчатый p-Hg (N3)2 получает-
ся аналогично P-Pb (N3)2. Известны кристаллические структуры a-Hg (N3)2
и CH3HgN3 [210, 213], координационное число ртути в которых необычно —
равно 7. Ртуть-органические азиды синтезированы по реакциям обмена с га-
логеназидами [42]. Описаны смешанные полиядерные азидомеркураты (II)
[211] и их оптические свойства [212].
2. 7.2. Азиды элементов III — VII побочных подгрупп
Реакции ионов Се4+ с азидом представляют аналитический интерес (см.
2.3.3) [1]. Из водных растворов нельзя получить азиды Y, La, Се, UO2+ дей-
ствием на их соли раствором NaN3, так как при этом образуются основные
азиды [1 ]. Последние образуются и для легких лантаноидов [270] по реакции
М3+ + NY + ЗН2О М (ОН)2 N3 (Н2О) + 2Н+ (М - Рг, Nd, Sm) (2.33)
34
(нуйол и КВг) [42], см 1
6(N3) v (М— N) * 6 (М—N—N) б (N—М—N)
654,594 (ср.) 392 , 376 (с.) 231, 221. 207 (с.) 118 (сл.)
640 (сл., шир.), 597 (ср.) 327, 313 (с.) 185, 179 (с.) 117 (сл.)
643 (ср.), 609 (сл.) 330 (шир.) 231 (ср., с.) 137 (шир.)
583 (сл.), 563 (сл.) 417 (с.), 403 (с.) 227 (с.), 150 (ср.)
582 (сл.) 394 (сл.) 230 (ср.), 208 (ср.) 118 (сл.)
651 (сл.), 633 (сл.) 315 (с.), 272 (пл.)
652 (сл.) 442,405 (ср.) 276, 220 (ср.) **
629 (сл.) 410 (оч. сл.) 156, 154 (сл.)
578 (ср.) 432 (ср.) 236 (ср.) 118 (сл.)
621 (сл.), 599 (сл.) 446 (сл.), 226 147 (ср.)
287, 270 (ср.)
сильная; ср.— средняя; пл.— плечо.
Исследована кинетика восстановления N3 ионами Еи2+ в присутствии
VO2+ [214].
В системе UO2+ — NjE — Н2О обнаружены комплексы [UO2(N3)n]2—n
(п = 1 4- 3), а соединение [Ph4As]2 [UO2 (N3)4] изолировано [42, 215]. UC16
реагирует с C1N3 с образованием UC15N3, UC14 (N3)2, а в присутствии СС14
образуется [С (N3)31 UC16 [218]. Реакция TiCl4 с C1N3 приводит к образованию
TiCl3N3
TiCl4 + ClN3-> TiCl3N3 + Cl2.
(2.34)
Это — желтое взрывчатое соединение, которое в СС14 отщепляет хлор и
азот и превращается в TiNCl. В воде гидролизует до ТЮ2 (Н2О)Г [39, 40,
277]. В твердом состоянии TiCl3N3 ассоциирован посредством азидных мос-
тиков [277].
В неводных растворителях VO2+ образует с Ns" комплексы: [VON3]+,
VO (N3)2, [VO (N3)4]2~ и [VO (N3)5]3~ [42, 188]. Выделены соединения состава
M2[VO(N3)4] [42], M3[V (N3)el [222] и VOC12N3 [277]. [TaCl4N3]2 относится к
ковалентным азидам. Азидные группы находятся в углах плоскости пла-
нарного четырехчленного цикла [13, 216], так что в структурном отношении
[TaCl4N3]2 напоминает уже рассмотренные соединения состава [BC13N3]3
и [SbCl4N3]2. СгО2С12, МоС15 или WC16 реагируют с C1N3 с образованием взрыв-
чатых хлоридазидов металлов состава CrO2ClN3, MoCl4N3 и WC15N3, а из
последних двух соединений образуются MoNCl3 и WNC13 [39, 248]. Наряду
с другими псевдогалогенидными соединениями состава Cp2WO2X2 (X = NCS,
2:
35
SCN, NCO, OCN, CN, NC) получено и Cp2WO2(N3)2 [269]. Комплекс Cr (N3)|“
образуется в водном растворе [42]. Образование мостиковых азидных комп-
лексов Сг (III) может быть использовано для исследования механизма ре-
докс-процессов, что имеет значение для оценки устойчивости мостиковых
связей металл — азид [161]. Кинетические исследования основываются на
восстановлении комплексов (Н2О)5СгГ<+ цис-(Н2О)4Сг(Ка)^и (H2O)8Cr (N,),
солями ванадия (II) [286]. Светочувствительный комплекс [(С 2H5)4N]2 [Мп •
•(N3)4] вспыхивает в пламени [42]. Мп(СО)5Вг, а также Re (СО)5С1 реагируют
с NaN3, LiN3 или [E^N .]N3 в подходящем растворителе с образованием ди-
мерных анионов [М2 (СО)6 (N3)3]~ (М2 (СО)6 (N3)x (NCO)3_X]~ или [Re2 (СО)6-
-(NCO)2 N3)2[2~ (М — Мп, Re) [38, 217]. Реакции замещения в карбонилах
металлов с участием азидных ионов охватывают такие элементы, как Сг,
Mo, W, Мп, Со, Fe [219 — 221].
2. 7.3 Азиды элементов VIII побочной подгруппы
Азидные соединения железа (III) используются в анализе [8, 34], а вы-
деленные соединения соответствуют комплексам типа Fe (N3)i~ [421, Fe (N3)i~
[42, 223]. Пентаазидоферрат (III) образуется по реакции
[Et4N]2 FeBr4 + 5AgN3 [Et4N]2 [Fe (N3)5] + 4AgBr + Ag (2.35)
и, судя по данным ИК-спектров [42], содержит, вероятно, тетраэдрический
ион [Fe (N3)4]~, структура которого обсуждается в работе [206]. В соединении
[Ph4Asl2 [Fe (N3)5] атом железа окружен пятью cc-N-атомами азидогрупп,
образуя тригональную бипирамиду [226]. Аммонийные соли [Fe (N3)J3~-
иона принадлежат к высокоспиновым комплексам (рЭф = 5,85 М. Б.) [2231.
Известны разнолигандные азидные комплексы железа [224, 2251, а также
азидоферрат (III) тетрафенилфосфония, который может превращаться в ди-
мер с мостиковыми Ng-группами [273].
Однородные комплексы кобальта и никеля имеют состав [Со (N3)4]2~
[168, 189, 2271, [Ni (N3)6]4~ [228, 294, 295], [Ni2 (N3)51“ [42] или [Ni (N3)4]2~
[188, 229].
Октаэдрическая конфигурация кобальта (III) обнаружена в аминоа-
зидах [13, 231]. В качестве примера можно привести соединение [Со (NH3)5 •
•N3] (N3)2 [230]. Октаэдрическое окружение имеют оба атома кобальта в
[Со (NH3)4 (N3)21 [Со (NH3)2 (N3)J [281], причем в катионе все молекулы NH3
расположены в экваториальной плоскости, а азидные группы занимают
транс-положение. В анионе этого комплекса транс-положение занимают
NH3-rpynnbi. Соединение относится к триклинной сингонии (пространствен-
ная группа Р1) [281]. Катионы I(diphos) Ni (N3)2 Ni (diphos)]2^, содержащие
мостиковые Ng-группы, образуются из мономерных азидных комплексов
Ni (N3)2 • diphos и нитрозил- или триэтилоксониевых солей [232] (diphos =
= 1,2-бис (дифенилфосфано) этан). Механизм реакций обмена и кинетики,
а также другие физико-химические измерения комплексных соединений ко-
бальта, включающих азидные лиганды, изучены в работах [233—237, 271,
274, 282, 288].
Азидоплатинаты щелочных металлов являются взрывчатыми, точно
так же, как азиды других платиновых металлов. «Простые» азиды платино-
вых металлов в сухом состоянии чрезвычайно чувствительны к соприкосно-
вению, малодоступны исследованию, они также взрываются при встряхивании
с водой. В качестве примера может быть рассмотрен Pd (N3)2, который оса-
36
ждается в виде нерастворимого коричневого осадка из растворов солей пал-
ладия при добавлении к ним водного раствора NaN3 [187]. Он легко раство-
ряется в избытке азида натрия с образованием азидопалладатов, состав ко-
торых зависит от условий проведения реакции. Так, в водных растворах
получены [Pd (N3)2C12]2~ и [Pd (N3)4]2~ (солянокислый раствор), а также ком-
плекс с соотношением 1 : 3 (нитратные растворы) [238]. В водно-спиртовом
растворе образуются [Pd (N3)5]3~ и [Pd (N3)6]4~ [187]. Использование объе-
мистых катионов не только уменьшает опасность образования взрывоопас-
ных соединений, но оказывает влияние на состав осаждаемых соединений.
С катионами [Ph4As]+ получаются соединения типа [Ph4Asl2 [Pd (N3)J и
Ph4As]2 [Pd2 (N3)6] [42]. Азидные комплексы Pd (II) и Pt (II) реагируют с
Ph3P, Phen, diphos с замещением азидных групп и образованием термически
устойчивых неполярных соединений М (N3)2 • 2L (L = Ph3P, V2 Phen,
2 diphos) [42, 240, 242]. Изучены геометрия и способ координации азидных
групп в разнолигандных азидных комплексах палладия (II) [276]. Анион
tPd2 (N3)612— образует четырехчленный цикл, причем атомы палладия свя-
заны между собой двумя мостиковыми М3-группами [239]. Данные о струк-
туре соединения (CH3)3PtN3 приведены в работах [53, 245] (см. 2.3.1). Дру-
гие платиновые металлы образуют комплексы [М (N3)4]2~ и [М' (N3)6]3~ или
[М" (N3)6]2~ (М = Pt [42]; М' = Ru [2221, Rh [42, 222], Os [222, 243]; Ir [222,
224]; M" = Pt [42, 222]).
Известны катионы с мостиковыми азидными группами состава [(Ph3P)2
• (CO)MN3M(CO) (PPh3)2]+(M = Rh, Ir) [232], комплексы типа L3MN3 (M =
=Rh(I), Au (I)), [Ph3P]4Rh2 (N3)2, [Ph3Pl2 Ir (CO)N3 [240], [(Ph3P)2M (N3)2M •
•PPh3)2l2+ (M = Pt, Pd) 1241]. Комплексы палладия (II) и платины (II) с
двумя мостиковыми М3-группами образуются при реакции М (N3)2- 2Ph3P
с солями оксония и нитрозила [241]. Реакция с транс-[Р1 (NH3)4C12]2+-
катионом приводит к образованию транс-fPt (NH3)4C1N3]2+; исследована
также кинетика этого процесса [280].
Азидные соединения платиновых металлов склонны к многочисленным
превращениям и поэтому пригодны для синтеза ряда соединений, при этом
образуются промежуточные продукты нитренов металлов, которые отме-
чены для комплексов Ir, Rh и Ru [246, 247, 283].
2.8. ПРИМЕНЕНИЕ АЗИДОВ
Возможности практического использования азидов зависят от типа свя-
зи и структурно-химического поведения соответствующих соединений, а
также от возможности освоения путей синтеза и получения новых типов сое-
динений.
Особое значение в препаративной химии имеют алкил-, арил- и ацил-
азиды, а также металлоазидные соединения, которые способны к различным
превращениям (например, образование дифениламина, азобензола, анилина
или 1-фенилтриазола из фенилазида; реакции координационного фиксирова-
ния азидной группировки [38], синтез металлотиоцианатных комплексов
И Др.).
Из имеющихся в литературе данных, включая и обзорные работы [1—4,
И—13, 36—42] по неорганическим азидам, большая их часть посвящена ази-
дам щелочных и щелочноземельных металлов. Сведения об однородных ком-
плексных азидах тяжелых металлов пока еще малочисленны, хотя в послед-
37
нее время о них сообщается все чаще. Применение объемистых катионов типа
[Ph4P]+, [Ph4As]+, N [(PPh3)]^ и [R4N]+ должно существенно ускорить
синтез новых азидов тяжелых металлов [42]. В этой области еще не исполь-
зованы многочисленные возможности.
«Классическая» область применения азидов тяжелых металлов (напри-
мер, азида свинца) включает использование их в качестве детонаторов огне-
стрельных и взрывоопасных веществ, термическое и фотохимическое разло-
жение, вспышку, взрыв и детонацию азидов [11, 12, 36, 249]. При детона-
ции различают две фазы нарастания реакции [11]. Для азидов Tl+, Ag+,
Cu+ начальная стадия разложения соответствует фотохимическому взаимо-
действию, а вторичный процесс (прохождение реакции до взрыва) — тер-
мическому взаимодействию. В первичной стадии протекания реакции об-
/Н Нч@.
)N=N®
Н " ЬГ
Рис. 2.8. Изомеры диазена:
а — транс-диазен; б — изодиазен
разуются бис-азидные радикалы, затем молекулы азота в экзотермической
реакции, которые определяют скорость нарастания реакции [142]. Сказанное,
включая суммарное разложение, можно представить реакциями
NF~Ns + e~ (2.36)
2N3->-3Ns (ДН = —879,9 кДж); (2.37)
2MN3 -> 2М + 3N2 (М-тяжелый металл). (2.38)
Проведенные до настоящего времени исследования показывают, что чув-
ствительность к свету (начальная стадия) возрастает с увеличением потен-
циала ионизации металла. Однако вопрос о разложении азидов окончатель-
но еще не решен.
В зависимости от состава и обработки азиды могут использоваться в ка-
честве фоточувствительных материалов [250], фоторезисторов [251], а также
фототермографических материалов [252]. Предпринято математическое мо-
делирование фотолиза азидов тяжелых металлов [253]. При контролируемом
разложении азидов их применяют в качестве газовых генераторов азота [254],
горючего [255] для транспорта. Азиды участвуют в получении синтетических
полимеров со специфическими свойствами (диэлектрическое поведение, чув-
ствительность к облучению) [256], а также в различных реакциях катализа.
При определенных условиях их можно употреблять для получения чистых
металлов.
Азиды широко используются в биохимических процессах [258], специ-
альные азиды — в качестве акаризидов, инсектицидов и нематизидов [259,
293], а также денитрификаторов минеральных удобрений [2601.
Главной областью применения азидов является синтез ряда соединений
металлов, которые могут быть получены лишь с участием азидов [38]. Зна-
ние химии азидов важно при изучении комплексов переходных металлов с
молекулярным азотом [261]. Известные до настоящего времени реакции азид-
ных комплексов металлов [38, 242] охватывают 1,3-биполярные реакции ци-
клообразования координированной азидной группы; фотохимические или
термические, внутримолекулярные реакции окисления — восстановления.
Термолизом некоторых азидных соединений можно получить изомерные
диазены состава N2H2 [2681, строение которых показано на рис. 2.8. Изуче-
ны ИК-спектры поглощения твердого N2H2 и его дейтеропроизводных [2901.
38
СПИСОК ЛИТЕРАТУРЫ
1. Gmelins Handbuch der Anorgan. Chemie, 8. Auflage. Stickstoff-Syst.-Nr. 4. Berlin,
Verlag Chemie, 1936, S. 285 ff.
2. Grundmann C. Stickstoffverbindungen.— Houben-Weyl: Methoden der organ. Chemie,
Bd 10/3. Stuttgart, G. Thieme Verlag, 1965, S. 777 ff.
3. Andreev R. R.— Physik. Z. UdSSR, 193.4, 6, S. 121.
4. Audrieth L. F.— Chem. Rev., 1934, 15, p. 169.
5. Guenebaut H., GaydonA. G.— C. r. Acad. Sci., 1955, 240, p. 958.
6. Pannetier G.— C. r. Acad. Sci., 1951, 232, p. 817.
7. Gunther P., Mayer R., Miller-Skjold F.— Z. physik. Chem., 1935, 175, S. 154.
8. Gray P., Waddington T. C.— Proc. Royal Soc., 1956, A235, p. 106, 235, 481.
9. DowsD. A., Pimentel G. C.— J. Chem. Phys., 1955, 23, p. 1258.
10. Eyster E. H., Gillette R. H.— J. Chem. Phys., 1940, 8, p. 369.
11. Evans В. L., Yoffe A. D., Gray P.— Chem. Rev., 1959,59, p. 515.
12. Gray P.— Quart. Rev., 1963, 17, p. 441.
13. Muller U.— Struct. Bonding (Berlin), 1973, 14, S. 141.
14. Amble E., Dailey В. P.— J. Chem. Phys., 1950, 18, p. 1437.
15. Winnewisser M.,Cook R. L.— J. Chem. Phys., 1964, 41, p. 999.
16. Gray P., Waddington T. C.— Trans. Faraday Soc., 1957, 53, p. 901.
17. Eyster E. H.— J. Chem. Phys., 1940, 8, p. 135.
18. Davies M. M.— Trans. Faraday Soc., 1939, 35, p. 1184.
19. Buswell A. M., McMillan G. W., Rodebush W. H., Wall F. T.— J. Am. Chem. Soc.,
1939, 61, p. 2809.
20. Engler W., Rohlrausch R. W. F.— Z. physik. Chem., 1936, B34, S. 214.
21. Pannetier G., Gaydon A. G.— J.Chim. Phys., 1951,48, p. 221; Bull. Soc. chim. France,
1954, p. 1068; С. r. Acad. Sci., 1955, 240, p. 958.
22. Rice F. 0., Freamo M.— J. Am. Chem. Soc., 1951, 73, p. 5529.
23. Mador, Williams.— J. Chem. Phys., 1954, 21, p. 1627.
24. WhittleD. A., DowsD. A., Pimentel G. C.— J. Chem. Phys., 1954, 21, p. 1943; 1955,
22, p. 1606.
25. Meyer R., Schumacher H. J.— Z. physik. Chem., 1934, A170, S. 33.
26. Leermakers J. A.— J. Am. Chem. Soc., 1933, 55, p. 2719.
27. Baratt S.— Proc. Royal. Soc., 1921, A98, p. 44.
28. Mulliken R. S.— Phys. Rev. 2, 1927, 29, p. 646.
29. Meeks R., Guiliery M.— Phys. Z-, 1927, 28, S. 523.
30. Gray P., Waddington T. C.— Nature, 1957, 179, p. 576.
31. Alexejew D.— Z. physik. Chem. (UdSSR), 1931, 2, S. 535.
32. Mendelejeff DBer., 1890, 25, S. 3470.
33. Birckenbach L., Rellermann R.— Ber., 1925, 58, S. 787, 2377.
34. BunnD., Dainton F. S., Duckwarth S.— Trans. Faraday Soc. 1961, 57, p. 1131.
35. Booth H. S. Inorganic Synthesis, vol. 1. New York, Me Graw-Hill, 1939.
36. Захаров Ю. A.— Изв. Томск, политехи, ин-та, 1969, 199, с. 3.
37. Yoffe A. D.— Develop. Inorg. Nitrogen Chem., 1966, 1, p. 72.
38. Dori Z., Ziolo R. F.— Chem. Rev., 1973, 73, p. 247.
39. Dehnicke R.— Angew. Chem., 1967, 79, S. 253.
40. Dehnicke R.— Chimia (Aarau, Schweiz), 1973, 27, p. 309.
41. Patai S. The Chemistry of the azido group. London, Int. Publ., 1971.
42. Beck W., Pehlhammer W. P., Pollmann P., Schuierer B., Feldl R.— Chem. Ber., 1967,
100, 2335; Nachr. Chem. Techn., 1968, 16, Nr. 5, S. 80; Beck Z.— Jahrbuch d. Akad. d. Wiss.
Gottingen, 1967, S. 13.
43. Lunt R. W., Pearse R. W. B., Smith E. C. W.— Nature, 1935, 135, p. 508; 1935, 136,
p. 32.
44. Franklin E. C.— J. Am. Chem. Soc., 1934, 56, p. 569.
45. Turrentine J. W.— J. Am. Chem. Soc., 1912, 34, p. 385.
46. Pauling L.— Procnat. Acad. Sci. USA, 1932, 18, p. 498.
47- Hendricks S. B., Pauling L.— J. Am. Chem. Soc., 1925, 47, p. 2904; Frevel L. R.—
J. Am. Chem. Soc., 1936, 58, p. 779.
48. Frevel L. R.— Z. Kristallogr., 1936, 94, S. 197.
49. Llewellyn F. L., Whitmore F. E.— J. Chem. Soc., 1947, p. 881.
50. Wyatt J. F., Hillier I. H., Saunders U. R. a. o.— J. Chem. Phys., 1971, 54, p. 5311.
51. Hughes E. W.— J. Chem. Phys., 1935, 3, p. 1; Rnaggs I. E.— Proc. Royal Soc. (Lon-
don), 1935, A150, p. 576.
52. Gillespie R. J.— Angew. Chem., 1967, 79, S. 885.
53. Atam M., Muller U.— J. Organometal. Chem., 1974, 71, p. 435.
54. Milligan D. E., Brown H. W., Pimentel G. C.— J. Chem. Phys., 1956, 25, p. 1080;
1957, 26, p. 145.
39
55. Thrush G.— Proc. Royal Soc., 1956, A235, p. 143.
56. Franklin J. L., Dibeler V. H., Reese R. M., Krauss M.— J. Am. Chem. Soc.. 1958,
80, p. 298.
57. Clusius K., Schumacher H.— Helv. chim. Acta, 1958, 41, S. 972.
58. Neilson G. W., Symons M. C. R.— J. Chem. Soc. Faraday Trans., 1972, p. 1772; Clax-
ton T. A., Overill R. E., Symons M. C. R.— Mol. Phys., 1973, 26, p. 75.
59. Barton D. H. R., Morgan L. R.— J. Chem. Soc., 1962, p. 622.
60. W torkowska-Zaremba A.— Chem. Anal., 1969, 14, p. 847.
61. Сегеда A. C.— Укр. хим. журн. 1970, 36, с. 213; Utsumi S., Okutani T.— J. chem.
Soc. Japan, 1973, p. 75.
62. Botre C., Macini M., Bencivenga B., Pallotti G. F.— Ed. Prat., 1973 , 28, p. 218; CA
1973, 78, p. 157919.
63. Berneira L. T.— Rev. Fac. Farm. Bioquim., Univ. Fed. Santa Maria, 1970, 16, p. 75;
CA, 1971, 75, p. 126052.
64. Rao A. L. J., Puri В. K-— Z. analyt. Chem., 1969, 248, S. 33;1972, 258, S. 286.
65. Dziegiec J., Ignaczak M. — Soc. Sci. Lodz, Acta Chim., 1971, 16, p. 69.
66. MajumdarA., Mitra В. K-— Mikrochim. Acta, 1970, 3, S. 596.
67. BiggsW. R., Gaver R.— Anal. Chem., 1972, 44, p. 1870.
68. Kuroda R., Yoshikuni N. K- Y.— Anal. Chim. Acta, 1972, 60, p. 71.
69. Utsumi S., Okutani T.— Nippon Kagaku Kaishi, 1973, 1, p. 75.
70. JoharG. S.— Taianta, 1972, 19, p. 1461.
71. Cotton F. A., Goodgame D. M. L., Goodgame M.— J. Am. Chem. Soc., 1961, 83,
p. 4690.
72. Cook R. L.,Gerry M. C. L.— J. Chem. Phys., 1970, 53, p. 2525.
73. Milller U., Barnighausen H.— ActaCryst., 1970, B26, p. 1671.
74. Henke H., Barnighausen H.— ActaCryst., 1972, B28, p. 1100.
75. Wiberg E., Michaud H.— Z. Naturforsch., 1954, 9b, S. 495.
76. Milller U.— Z. anorg. allg. Chem., 1971, 382, S. 110.
77. Milller U.— Z. anorg. allg. Chem., 1972, 388, S. 207.
78. Strahle J.— Z. anorg. allg. Chern., 1974, 405, S. 141.
79. Paetzold P. J., Gayoso M., Dehnicke K-— Chem. Ber., 1965, 98, S. 1173.
80. Guha C., Bogan L. D., Gilliam 0. R.— Phys. Rev., 1973, 7, (9) p. 4097; Claxton T. A.,
Overill R. E., Symons M. C. R.— Mol. Phys., 1973, 26, p. 75.
81. FinnP., Jolly W. L.— Inorg. Chem., 1972, 11, p. 1434.
82. Dreyer J. W.,PernerJ.— Ber. Bunsenges. Phys. Chem., 1973, 77, S. 442.
83. Hayon E., Simic M.— J. Am. Chem. Soc., 1970, 92, p. 7486.
84. Archibald T. W., Sabin I. R.— J. Chem. Phys., 1971, 35, p. 389.
85. Blum A., Zagorski Z., Broszkiewicz P., Roman K-—Chem. Anal., 1973, 18, p. 57;
Zagorski Z. P., Blum A.— J. Electroanal. Chem., Interfacial Electrochem., 1973, 41, p. 447.
86. Selig W.— Microchim. Acta, 1971, p. 46.
87. Thielemann H.— Z. Chem., 1972, 12, S. 343.
88. Campell M. M., Dunn A. D.— Org. Mass Spectrom., 1972, 6, p. 599; Ribando Ch,,
Semel S., Blosser E. R.— US Clearinghouse Fed. Sci. Techn. Inform., AD 1970, No. 707934,
p. 43.
89. Beck W.,Becker W., Chew K- F. a. o.— J. Chem. Soc. Dalton, 1972, p. 245.
90. Mueller J.— J. Organometal. Chem., 1973, 51, p. 119.
91. Forman R. A.— J. Chem. Phys., 1963, 39, p. 2393; Kanda T., Saito Y., Kawatnu-
ra K-—Bull. Chem. Soc. Japan, 1962, 35, p. 172.
92. Witanowski M.— J. Am. Chem. Soc., 1968, 90, p. 5683.
93. HendricksonD., Kuznesov P. M.— Theor. Chim. Acta, 1969, 15, p. 57.
94. Fritzer H. P., ClarkD. T., Woolsey J. S.— Inorg. Nucl. Chem. Lett., 1974, 10, p. 247.
95. Holsboer F., Beck W.— Z. Naturforsch., 1972, 27b, S. 884.
96. Bartunik H. D., Potzel W., Mofibauer R. L., Kuindl G.— Z. Physik, 1970, 240, S. 1.
97. Oelker C. J., Beck W.— Spectrochim. Acta, 1973, A29, p. 1975.
98. Paetzold P.— Fortschr. chem. Forsch., 1967, 8, S. 437.
99. Paetzold P. J.— Z. anorg. allg. Chem., 1963, 326, S. 47.
100. Paetzold P. J., Gayoso M., Dehnicke K.— Chem. Ber., 1965, 98, S. 1173.
101. Lappert M. F., Pyszora H. — Adv. Inorg. Chem. and Radiochem., 1966, 9, p. 133.
102. Wiberg NSchmid К- H., Joo W. C.— Angew. Chem., 1965, 77, S. 1042.
103. Wiberg N., Joo W. C., Schmid К- H.— Z. anorg. allg. Chem., 1972, 394, S. 197.
104. Costain С. C., Kroto H. W.— Can. J. Phys., 1972, 50, p. 1453; Bolton K-,
Brown R. D.—~ Chem. Phys. Lett., 1972, 15, p. 79.
105. Wiberg N„ Neruda B.— Chem. Ber., 1966, 99, S. 740.
106. Wiberg N., Schmid К. H — Chem. Ber., 1967, 100, S. 748.
107. Wiberg N., Schmid K. H.— Chem. Ber., 1967, 100, S. 741.
108. Wiberg N., Schwenk G., Schmid К. H.— Chem. Ber., 1972, 105, S. 1209.
40
109. Anderson D. W. W., Rankin D. W. H., Robertson A.— J. Molec. Struc., 1972, 14,
p. 385.
110. Murdoch J. D., RankinD. W. H.— Chem., Commun., 1972, p. 748.
111. Baybutt P., Guest M. F., Hiller J. H.— Proc. Royal Soc. (London), 1973, A, p. 225.
112. Roesky H. W., Dietl M. — Angew. Chem., 1973, 85, S. 454.
113. Schmidt A.— Chem. Ber., 1970, 103, S. 3923.
114. Roesky H. W.— Angew. Chem. Int. Ed. Engl., 1967, 6, S. 363.
115. O'Neill S. R., Schreeve J. M.— Inorg. Chem., 1972, 11, p. 1629.
116. Buder W., Schmidt A.— Chem. Ber., 1973, 106, S. 3812.
117. Regitz M., Anschiltz W.— Chem. Ber., 1969, 102, S. 2216.
118. Пудовик A. H., Хусаинова H. Г., Бердников E. А., Насыбуллина 3. A.— Журнал
общей химии, 1974, 44, с. 222.
119. Галишев В. А., Чистоклетов В. Н., Петров А. А., Тамм Л. А.— Журнал общей
химии, 1973, 43, с. 1470.
120. Pulay Р., Lakatos В., Toth G.t Hesz A., Vetessy Z.— Acta Chim. (ВНР), 1969, 60,
p 333.
121. Cadogan J. J.G., Gosney J.— J. Chem Soc. Perkin Trans., 1974, p. 460.
122. Clusius K-, Knopf H. — Helv. Chim. Acta, 1956, 39, p. 681.
123. Lucien H. W.— J. Am. Chem. Soc., 1958, 80, p. 4458.
124. Booth H. S. (Editor) Inorg. Syn. Vol. 1. New York, McGraw-Hill, 1939; Inorg. Syn.
Vol. 2. New York, McGraw-Hill, 1946; Brauer G. Handbuch der Praparativen Anorg. Chemie.
Stuttgart, 1960.
125. Clusius K-> Effenberger E.— Helv. Chim. Acta, 1955, 38, p. 1834; Clusius K.,
Knopf H.— Helv. Chim. Acta, 1956, 39, p. 681.
126. Pringle G. E., Noakes D. E.— Acta Cryst., 1968, B24, p. 262.
127. Griffin J. F., Coppens P.— Chem. Commun., 1971, p. 502.
128. Frevel L. K-— Z. Krist., 1936, 94, S. 197.
129. Muller U.— Z. anorg. allg. Chem., 1972, 392, S. 159.
130. Krischner H.— Monatsh. Chem., 1968, 99, S. 2134.
131. Choi C. S.— Acta Cryst., 1969, B25, p. 2638.
132. Walitzi E. M., Krischner H.— Z. Krist., 1970, 131, S. 25.
133. Torkar K-, Krischner H., Radi H.— Monatsh. Chem., 1965, 96, S. 932.
134. Muller H. J., Joebstl J. A.— Z. Krist., 1963, 121, S. 385.
135. Walitzi E. M., Krischner H.— Z. Krist., 1970, 132, S. 19.
136. Wiberg E., Michaud H.— Z. Naturforsch., 1954, 9b, S. 502.
137. Wiberg E., Michaud H.— Z. Naturforsch., 1954, 9b, S. 501.
138. Wiberg N., Joo W. C.— J. Organometal. Chem., 1970, 22, p. 333.
139. Yoffe A. D.— Proc. Royal Soc., 1951, A208, p. 188.
140. Kapustinsky A. F.— Z. physik. Chem., 1933, B22, S. 257.
141. Jacobs P. W. M., Tompkins F. C.— Proc. Royal Soc., 1952, A215, p. 254.
142. Evans B. L., Yoffe A. D.— Proc. Royal Soc., 1959, A250, p. 346.
143. Tompkins F. C., YoungD. A.— Proc. Royal Soc., 1956, A236, p. 10.
144. Deb S. K-—J. Chem. Phys., 1961, 35, p. 2122.
145. BoganL.D., Bartram R. H., Owens F.— J. Phys. Rev., 1972, B618, p. 3090.
146. Danemar A., Welch D. 0., Royce В. S. M.— Phys, status solidi, 1973, B55, p. 201.
147. Smithard M. A.— Solid State Commun., 1974, 14, p. 407, 411.
148. Беломестник В. И., Балки А. А., Левочко Д. С.— Физика твердого тела, 1973, 15,
с. 3097.
149. Prout Е. G., Liddiard V. С.— J. Inorg. Nucl. Chem., 1973, 35, р. 2183, 3419.
150. Prout Е. G., Sears W. G.— Inorg. Nucl. Chem. Lett., 1972, 8, p. 937, 943; 1973, 9,
p. 31; J. Inorg. Nucl. Chem., 1973, 35, p. 2163.
151. Захаров Ю. A.— Изв. Томск, политехи, ин-та, 1969, 199, с. 3.
152. IgbalZ. — J. Chem. Phys., 1973, 59, p. 1769.
153. De Panafieu A., Royve B. S. H.— Phys. Lett., 1972, A39, p. 77.
154. Sharma I., Laskar A. L.— J. Phys. Chem. Solids, 1973, 34, p. 989.
155. Sharma I., Gora T., Rimstidt J.D., Staley R.— Chem. Phys. Lett., 1972, 15, p. 232.
156. Krischner H., Roth H. E.— Z. Kristallographie, Kristallgeometrie, Kristallphysik,
Kristallchemie, 1973, 137, S. 311.
157. Pannetier G., Margineanu F., Dereigne A., Bonnaire R.— Bull. Soc. chim. France,
1972, p. 2623, 3725.
158. КирпичевЕ. П., Алексеев А. П. Рубцов Ю. И., Манелис Г. В.— Журнал физ. химии,
1973, 47, с. 2492.
159. Wiberg Е., Michaud Н.— Z. Naturforsch., 1954, 9b. S. 496.
160. Wiberg E., Joo W. C., Hemke H. — Inorg. Nucl. Chem. Lett., 1967, 3, p. 267.
161. Beck WFehlhammer W. P.— MTP Int. Rev. Sci. Inorg. Chem. Serie 1, 1973, p. 253.
162. Atwood J. L., Nemberry W. R.— J. Organometal. Chem., 1972, 42, p.C77.
41
163. Evans В. L., Yoffe A. D.— Nature, 1959, 183, p. 4670.
164. Nauer F. A., Hubbard C. R., Hahu T. A.— J. Chem. Phys., 1973, 59, p. 3770.
165. IgbalZ., Malhotra M. L.— J. Chem. Phys., 1972, 57, p. 2637.
166. Owens F. J.— J. Chem. Phys., 1972, 57, p. 2349.
167. Wiberg E., Michaud H.— Z. Naturforsch., 1954, 9b, S. 500.
168. Forster D., Horrecks W. D.— Inorg. Chem., 1966, 5, p. 1510.
169. Choi C. S., Boutin H. P.— Acta Cryst., 1969, B25, p. 982.
170. Pellerito L.— Atti Accad. Sci. (Palermo), 1972, 31, p. 133.
171. Vogt G.— Ber. Bunsenges. phys. Chemie, 1965, 69, S. 648.
172. Srivastava T. N., Singh N.— Z. anorg. allg. Chem., 1966, 343, S. 211.
173. Dehnicke K., Strahle J., Seybold D., Milller J.— J. Organometal. Chem., 1966, 6,
p. 298; 1967, 7, p. 1; 1968, 12, p. 37.
174. McLaren A. C. Dissertation. Cambridge, 1957.
175. McLaren A. C., RogersG. T.— Proc. Royal Soc., 1957, A240, p. 484.
176. Garrett W. L.— Mater. Res. Bull., 1972, 7, p. 949.
177. Harris J.— U. S. Nat. Techn. Inform. Serv., AD Rep., 1972, No 743191; Habermann
JCastorina T. — U. S. Nat. Techn. Inform. Serv., AD Rep., 1972, No 743472.
178. Hutchinson R. W„ Stein F. P.— J. Phys. Chem., 1974, 78, p. 478; 1973, 77, p. 870.
179. Шечков Г. T.— Изв. вузов СССР. Химия и хим. технология, 1973, 16, с. 1794.
180. Hall R. В., Williams F.— J. Chem. Phys., 1973, 58, р. 1036.
181. VarmaS. Р., Williams F.— J. Chem. Phys., 1973, 59, p. 912.
182. СтенгахВ. В.— Журнал прикл. механики и техн, физики, 1972, 5, с. 128.
183. Сукусин. Ю. Н., Захаров Ю. А., Иванов Ф. И.— Химия высоких энергий, 1973, 7,
с. 261.
184. Muller Н.— Z. anorg. allg. Chem., 1949, 260, S. 249.
185. Stuetz A., Zbiral E.— Ann., 1972, 765, p. 34.
186. Joel R. G., Ridley D. R.— Inorg. Chem., 1974, 13, p. 1252.
187. Clem R. G., Huffman E. H.— J. Inorg. Nucl.Chem., 1965, 27, p. 365: Analyt. Chem.,
1965, 37, p. 86, 1155.
188. Gutmann V., Leitmann 0.— Monatsh. Chem., 1966, 97, S. 926.
189. Senise P.— J. Am. Chem. Soc., 1959, 81, p. 4196.
190. Wilsdorf H.— Acta Cryst., 1948, 1, p. 115.
191. Agrell J., Lannevik S. — Acta Chem. Scand., 1968, 22, p. 2038.
192. Soderquist R.— Acta Cryst., 1968, B24, p. 450.
193. Agrell J.— Acta Chem. Scand., 1966, 20, p. 1281; 1969, 23, p. 1667.
194. Bassiere M.— C. r. Acad. Sci, 1935, 201, p. 735.
195. West C.D — Z. Krist., 1936, 95, S. 42.
196. Bassiere M.— C. r. Acad. Sci., 1937, 204, p. 1573.
197. Agrell J.— Acta Chem. Scand., 1970, 24, p. 3575.
198. Straumanis M., CirulisA.— Z. anorg. allg. Chem., 1944, 252, S. 9, 121; Ber., 1943,
76, S. 825.
199. Neves E. F. A., Senise P.— J. Inorg. Nucl. Chem., 1972, 34, p. 1915.
200. Senise P., Neves E. F. A.— J. Inorg. Nucl. Chem., 1972, 34, p. 1923.
201. DugganD. M., HendricksonD. N.— Inorg. Chem., 1973, 12, p. 2422.
202. ГасьмаевВ. К., ЗахаровЮ. A. — Журнал физ. химии, 1972, 46, с. 2967.
203. Chandhri М. N.— Nature Phys. Sci., 1973, 242, р. ПО.
204. Рябых С. М., Мешков В. А.— Журнал физ. химии, 1973, 47, с. 740.
205. Шамовская Г. И., Пещевицкий Б. И.— Изв. Сиб. отд. АН СССР. Сер. хим. наук,
1972, № 6, с. 53.
206. Agrell J..VannerbergN. G.— Acta Chem. Scand., 1970, 24, p. 1247; 1971, 25, p. 1630.
207. Krischner H., Fritzer H. P.— Z. anorg. allg. Chem., 1970, 376, S. 162; Z. Kristallogr.,
Kristallogeom., Kristallphys., Kristallchem., 1972, 135, S. 459.
208. Senise P., Neves E.D. A.— J. Am. Chem. Soc., 1961,83, p. 4146.
209. Beck W., Fehlhammer W. P., Pollmann P., Tobias R. S.— Inorg. Chim. Acta, 1968,
2, p. 467.
210. Muller U.— Z. anorg. allg. Chem., 1973, 399, S. 183; Seibold D., Dehnicke K.—
Z. anorg. allg. Chem., 1968, 361, S. 277.
211. Stan M., Zalaru F.— An. Univ. Bucuresti. Chim., 1972, 21, p. 47.
212. Deb S. K.— J. Phys. Chem. Solids. 1973, 34, p. 2265.
213. Milller U.— Z. Naturforsch., 1973, 28b, S. 426.
214. Espenson J. H., Christensen R. J.— J. Am. Chem. Soc., 1969, 91, p. 7311.
215. Beck W., Schuierer F., Pollmann P., Fehlhammer W. P.— Z. Naturforsch., 1966, 21b,
S. 811.
216. Strahle J.— Z. anorg. allg. Chem., 1974, 405, S. 139.
217. MasonR., RusholmeG. A., Beck W. a. o.— Chem. Commun., 1971, p. 496.
218. Kolitsch W., Milller U.— Z. anorg. allg. Chem., 1974, 410, S. 24.
42
219. Werner И.— Angew. Chem., Int. Ed. Engl., 1968, 7, S. 930.
220. Beck W., Werner H., Englemann H., Smedal H.— Chem. Ber., 1968, 101, S. 2143;
Brower R. R-, Chen Toa-shing.— Inorg. Chem., 1973, 12, p. 2198.
221. Werner H., Beck W., Englemann H.— Inorg. Chim. Acta, 1969, 3, p. 331.
222. Schmidtke H. H., Garthoff D.— J. Am. Chem. Soc., 1967, 89, p. 1317; Z. Naturforsch.,
1969, 24b, S. 126.
223. Rrohnke F. Sander B.— Z. anorg. allg. Chem., 1964, 334, S. 66.
224. Angelici R. J.. Busetto L.— J. Am. Chem. Soc., 1969, 91, p. 3197.
225. GuttermanD. F., Gray H. B.— Inorg. Chem., 1972, 11, p. 1727.
226. Drummond J., Wood J. S.— Chem. Commun., 1969, p. 1373.
227. Abu-Eittah R., ArafaG.— Z. anorg. allg. Chem., 1973, 399, S. 244.
228. Fritzer H. P., Torkar R.— Monatsh. Chem., 1968, 99, S. 2333.
229. Abu-Eittah R., Elmakabaly S.— Bull. Chem. Soc. Japan, 1973, 46, p. 3427.
230. PalenikG. J.— ActaCryst., 1964, 17, p. 360.
231. Druding L. F., Wang H., Cohen R. E., Sancilio F. D.— J. Coord. Chem., 1973, 3,
p. 105.
232. Werner R. V., Beck W.— Chem. Ber., 1972, 105, S. 3209.
233. Ferrandi F., Endicatt J. F.— Inorg. Chem., 1973, 12, p. 2389; Chem. Commun.,
1973, p. 674.
234. ForsterD.— Inorg. Chem., 1973, 12, p. 4.
235. Yamasaki R., Hidake JShimura Yo.— Chem. Abstr., 1973, 78, p. 35834.
236. Swaddee T. W., Guastalla G.— Inorg. Chem., 1969, 8, p. 1604
237. Rawajuchi H., Rawajuchi S.— Bull. Chem. Soc. Japan, 1973, 46, p. 3453.
238. Sherif F. G., Michael R. F.— J. Inorg. Nucl. Chem., 1963, 25, p. 999; Sherif F. G.,
Awad A. M.— J. Inorg. Nucl. Chem., 1961, 19, p. 94; 1962, 24, p. 179.
239. Fehlhammer W. PDahl L. F.— J. Am. Chem. Soc., 1972, 94, p. 3377.
240. Beck W., Fehlhammer W. P., Pbllmann PSchacht H.— Chem. Ber., 1969, 102,
S. 1976.
241. Beck W., Rreutzer P., Werner R.— Chem. Ber., 1971, 104, S. 528.
242. Rreutzer B., Rreutzer P., Beck W.— Z. Naturforsch., 1972, 27b, S. 461.
243. Bell B., ChattJDilworth J. R., Leigh G. J.— Inorg. Chim. Acta, 1972, 6.
244. Bauer R. A., Basolo F.— Inorg. Chem., 1969, 8, p. 2237.
245. Dahlen R. H., Lorberth J. —J. organometal. Chem., 1974, 65, p. 275.
246. Gafney H.D., Reed J. L., Basolo F. — J. Am.Chem. Soc., 1973, 95, p. 7998.
247. Reed J. D., Gafney H. D., Basolo F.— J. Am. Chem. Soc., 1974, 96, p. 1363.
248. Dehnicke R., Strdhle J.— Z. anorg. allg. Chem., 1965, 339, S. 171.
249. Cowley B. R.,Oughton J. F.— Chem. Ind.,1973, p. 444; Cranston В. H.— US Patent,
1973, No 3 739 614; Baer M., Severini J. H.— US Patent, 1973, No 3718513; Chandh-
ri M. M.— Combust. Flame, 1972, 19, p. 419; Bartels.— Explosivstoffe, 1972, 20, p. 143;
Fox P. G.— J. Solid. State Chem., 1970, 2, p. 491.
250. Robillard J. J.— J. Photogr. Sci., 1971, 19, p. 25; Wiegand 0. A.— US Patent, 1973,
No 3778269; Ohtani H. a. o.— Japan Kokai, 1973, 73, p. 18, 320; Гаврищенко Ю. В., Са-
вельев Г. Г., Захаров Ю.— Изв. Томск, политехи, ин-та, 1970, № 185, с. 69; Testa J. F.„
Robinson D. W.— J. Chem. Phys., 1971, 55, p. 3056.
251. Clecak N. J., Cox R. J.— Ger. Offen, 1974, 2314 868; France Demande, 1974,
2 160 820; Yoichi Ohba.— Japan Kokai, 1974, 397602; Noguchi Mitsushije.— Japan Kokai,
1974, 7398904.
252. Reynolds G. A.— Ger. Offen., 1973, 2258452.
253. Гаврищенко IO. В., Савельев Г. Г.— Изв. Томск, политехи, ин-та, 1970, № 251,
с. 255.
254. Beck W., Schorpp R.— Angew. Chem. Int. Ed. Engl., 1970, 9, S. 735; Boyars C.,
Zovko С. T.— Ger. Offen., 1974, 2327741; US Patent, 1974, No 3775199; 1974, No 3773;
Taylor G. W. G.—Brit., 1974, 1336561; Brennan R. L. Laue G. A.— France Demande, 1974,
2160411; Marshall M. D— US Patent, 1973, No 3755182.
255. Passauer H., Huebel R. — Ger. Offen. 1974, 2236175; Price R. M., Reed R. — US
Patent, 1974, No 3779823; Rains W. A., Stang P. L.— US Patent, 1973, No 3727407.
256. Вавилов A. Г. и др.— Журнал науч, и прикл. фотографии и кинематографии, 1973,
18, с. 412; Труды Ленингр. ин-та киноис., 1972, № 19, с. 134; Nishikubo Т., Toyama Yo.,
Maki R., Imamura Y.— Japan Patent, 1973, No 7303219; Sayih A. A. R., Stuber F. A.,
Ulrich H.— Ger. Offen, 1972, 2222193.
257. Ralra D. S., Helmig J. H.— Hargana Agr. Univ. J., 1973, 1 (4), p. 77; Soiminen
R., Elifolk N.— Acta Chem. Scand., 1973, 27, p. 35; Samejima T., Rita M.— Biochem. Biop-
hys. Acta, 1969, 175, p. 24; Makino R., Jamazaki J.— Arch. Biochem. Biophys., 1973, 157,
p. 356.
258. Banerjee R., Henry Y., Cassoly R.— Eur. J. Biochem., 1973, 32, p. 173; Yonetani
T. et al.— J. Biol. Chem., 1972, 247, p. 863; Behlke J., Scheier W.— Stud. Biophys., 1970
43
24/25, р. 61; Nourbakhsh М., Baumgarten А.—Experientia, 1972, 28, р. 1419; Cornoin J.—
Stud. Cercet. Biochim., 1972, 15, p. 21.
259. Ennuel L., Staehler G.— Ger. Offen., 1974, 2227683.
260. Ichikawa T., Seto S., Goto K-— Japan Patent, 1973, No 7129765.
261. Nefedov V. J., Lenenko V. S., Shur V. B. a. o.— Inorg. Chim. Acta, 1973, 7, p. 499.
262. Chow J. M., Britton D.— Acta Cryst., 1975, B31, p. 1922.
263. Nakamoto K- IR-Srectra of Inorganic and Coordination Compounds. New Vork,
1963.
264. Thayer J. S.— Organometal. Chem. Rev., 1966, 1, p. 157.
265. Gmelins Handbuch der Anorganischen Chemie. 8. Aufl. Strontium, Syst. Nr. 29.
Berlin, Verlag Chemie, 1932.
266. Gmelins Handbuch der Anorganischen Chemie. 8. Aufl. Barium, Syst. Nr. 30. Ber-
lin, Verlag Chemie, 1932.
267. Gmelins Handbuch der Anorganischen Chemie. 8. Aufl. Radium, Syst. Nr. 31. Ber-
lin, Verlag Chemie, 1928.
268. Wiberg N., Fischer G., Вachhuber H.— Angew- Chem., 1976,88, S. 386.
269. Rastogi M. K-, Multani R. K-— J. Inorg. Nucl. Chem., 1975, 37, p. 1995.
270. El Ezaby M. S., Abdel-Aziz I. E.— J. Inorg. Nucl. Chem., 1975, 37, p. 2013.
271. Ferraudi G. J., Guillermo J., Barber Endicott J. R.— J. Am. Chem. Soc., 1975, 97,
p. 6406.
272. Parker D. R., Sommer L. H.— J. Am. Chem. Soc., 1976, 98, p. 618.
273. Summerville D. A., Cohen J. A.— J. Am. Chem. Soc., 1976, 98, p. 1747.
274. Miskowski V. M., Robbins J. L., Hammond G. S., Gray H. B.— J. Am. Chem. Soc.,
1976, 98, p. 2477.
275. Bancroft G. M., Das V. G. K-, Sham T. K-, Clark M. G.— J. Chem. Soc. Dalton,
1976, p. 643.
276. Bresciani Pahor N., Calligaris M., Randaccio L.— J. Chem. Soc. Dalton, 1976,
p. 725.
277. Dehnicke K-— J. Inorg. Nucl. Chem., 1965, 27, p. 809.
278. Muller H., Dehnicke K.— J. Organometal. Chem., 1967, 10, p. 1.
279. Ziolo R. F., Gaughan A. P., Dori Z. a. o.— J. Am. Chem. Soc., 1970, 92, p. 738.
280. Alden Bryan H., Pantaleo N. S., Dickinson W. L., Johnson R. C.— Inorg. Chem.,
1975, 14, p. 1336.
281. Druding L. F., Sancilio F. DLukaszewski D. M.— Inorg. Chem., 1975, 14, p. 1365.
282. Birk J. P.— Inorg. Chem., 1975, 14, p. 1724.
283. Brown G. M., Callahan R. W., Meyer T. J.— Inorg. Chem., 1975, 14, p. 1915.
284. HallG. R., Duggan D. M., Hendrickson D. N.— Inorg. Chem., 1975, 14, p. 1956.
285. Richter S., Daul C., Zelewsky A.— Inorg. Chem., 1976, 15, p. 943.
286. Thompson R. C.— Inorg. Chem., 1976, 15, p. 1080.
287. Krischner H., Torkar K-, Roth H. E.— Z. anorg. allg. Chem., 1974, 405, p. 119.
288. Seel F., Meyer D.— Z. anorg. allg. Chem., 1974, 408, S. 283.
289. Dehnicke K-— Z. anorg. allg. Chem., 1974, 409, S. 311.
290. Minkwitz R.— Z. anorg. allg. Chem., 1975, 411, S. 1.
291. Krischner H., Winkler H.— Z. anorg. allg. Chem., 1975, 413, S. 94.
292. Busch B.,Pebler J., Dehnicke K- — Z.anorg. allg. Chem., 1975, 416, S. 203.
293. Schroder H. F., Muller J.— Z. anorg. allg. Chem., 1975, 418, S. 247.
294. Krischner H., Fritzer H. P., TechtG.— Z. anorg. allg. Chem., 1976, 424, S. 190.
295. Krischner H., Dobramysl W., Fritzer H. P.— Z. anorg. allg. Chem., 1976, 423, S. 255.
296. Krischner H., Fritzer H.— Z. anorg. allg. Chem., 1970, 376, S. 162.
297. Linke К- H., Turley R.— Z. anorg. allg. Chem., 1970, 377, S. 139.
3. ЦИАНИДЫ
3.1. ПОЛУЧЕНИЕ И СВОЙСТВА ДИЦИАНА
Дициан (CN)2— легковоспламеняющийся бесцветный газ, обладающий
высокой токсичностью. Образование его из элементов связано с поглощени-
ем тепла и частично происходит при горении электрической дуги в атмосфе-
ре азота
2С + N2 (CN)2, AH = 309,39 кДж. (3.1)
Несмотря на высокую теплоту образования, (CN)2 довольно устойчив
и образуется при пиролизе азотсодержащих органических соединений [1].
(CN)2 получают при нагревании цианидов ртути или серебра, смесей
Zn (CN)2 с CuC12 при 160—170° Си К4 [(Fe (CN)6] с HgCl2, сухой перегонкой
оксалата аммония в присутствии Р2О5, пропусканием сухого циановодорода
над нагретым до 250° С МпО2 11, 2]. Дициан может быть получен также при
окислении газообразного HCN воздухом над серебряным катализатором,
хлором в присутствии активированного угля или кварца и NO2 в присутст-
вии кальциевых стекол [21. Из водного раствора дициан можно получить
при действии на цианиды s-металлов солей меди (II)
Cu2+ + 2CN~ = CuCN + V2 (CN)2 (3.2)
или подкисленного раствора пер оксо дисульфата калия K2S2O8.
Наилучшим методом получения сухого (CN)2 является нагревание
Hg (CN)2 или К4 [Fe (CN)61 с эквивалентным количеством HgCl2. Наряду с
(CN)2 при этом образуется также небольшое количество парациана (CN)X.
В чистом виде дициан устойчив, а при наличии различных примесей при
300—500° С полимеризуется с образованием твердого темноокрашенного
полимера (CN)X. Парациан можно превратить в дициан при нагревании до
800—850° С в токе азота [3].
По данным ИК-спектроскопии [4], парациан имеет следующую струк-
туру:
Он нерастворим в воде, спирте, жидком циановодороде, но растворяется
в холодной концентрированной H2SO4.
Известен и другой полимер — гексациан (бесцветные кристаллы с tnJ1 =
= 119° С), который в присутствии сильно нагретой платины разлагается с
образованием обычного дициана.
45
(CN)2 растворим в спирте, бензоле, уксусной кислоте, медленно гидро-
лизуется водой
(CN)2 + 4Н2О (NH4)2 С2О4, (3.3)
(CN)2 + 2Н2О -> HCN + NH3 + СО2. (3.4)
Структурные исследования показали, что молекула дициана симметрич-
на и линейна,о причем расстояния С—С и С—N равны 1,37 и 1,16 А 15] или
1,387 и 1,155 А [6], и его строение можно представить формулой | N=C—С ==
=N|.
Нагревание дициана выше 1000° С ведет к его диссоциации
C2N2<>2CN. (3.5)
Моноциан термически очень устойчив, поэтому его удалось обнаружить
даже в атмосфере Солнца.
При участии свободных электронных пар у атомов азота или л-электро-
нов связи C=N дициан может образовывать координационные соединения,
например (CN)2 • 2О1С1 [7], Na [Си (CN)2NCCN1 • 4Н2О [2061.
Дициан имеет сходство с галогенами и по своим свойствам располагается
между бромом и иодом. Аналогия с галогенами проявляется в способности
циана соединяться с металлами, растворяться в щелочах
(CN)2 + 2NaOH -> NaCN 4- NaNCO 4- H2O, (3.6)
а также реагировать со многими органическими соединениями [2]. Например,
с реактивом Гриньяра (CN)2 образует нитрилы
(CN)2 4- RMgHal RCN 4- Mg (CN) Hal. (3.7)
3.2. ЦИАНОВОДОРОД И ЕГО КИСЛОТНЫЕ СВОЙСТВА
3.2.1. Общие свойства HCN
Циановодород HCN представляет собой бесцветную и легколетучую
жидкость (/Кип = 25,7° С при 760 мм рт. ст.) [8] с запахом горького миндаля,
смешивающуюся с водой и многими органическими растворителями в любых
отношениях.
Так как циановодород проявляет кислотные свойства, его иначе называ-
ют синильной кислотой. HCN имеет высокую диэлектрическую проницае-
мость (114,9 при 20° С) [81, которая обусловлена, как и в случае воды, ассо-
циацией полярных молекул (р = 2,930) [9]. Степень ассоциации жидкого
HCN около двух; даже в парообразном состоянии при температуре кипения
циановодород ассоциирован на 3%.
Жидкий HCN неустойчив; при хранении в отсутствие стабилизаторов
(НО, СаС12 и др.) склонен к полимеризации. Как растворитель циановодород
обладает сильной ионизирующей способностью В водном растворе HCN
ведет себя как очень слабая кислота; его термодинамическая константа дис-
социации при 25° С очень мала (рК = 9,21) [101.
Цианидная кислота реагирует с оксидами и гидроксидами щелочных
и щелочноземельных металлов, но не взаимодействует с их карбонатами.
Однако при введении в такой раствор суспензии карбонатов d-металлов (Ni,
Со, Fe) происходит реакция с образованием комплексных цианидов и выде-
лением СО2.
HCN вытесняется из солей не только действием сильных кислот, но и
фенолом, боратной кислотой и СО2.
46
В силу слабовыраженной кислотной функции HCN цианид-ион легко ги-
дролизует
CN" + Н2О <> HCN + ОН“
(3.8)
3.2.2. Получение циановодорода и его строение
Известно много способов получения HCN, основанных на различных
источниках азота (NH3, N2, азотистые органические соединения) и углерода
(древесный уголь, СО, СН4) [1, 11]. В промышленности HCN получают ката-
литическим окислением смеси NH3 и СН4 воздухом [4, с. 142]
2СН4 + ЗО2 + 2NH3 KaggacTop-> 2HCN + 6Н2О, (3.9)
Д// = —946,9 кДж.
В лабораторных условиях его выделяют из доступных солей (NaCN, KCN
и К4 [Fe (CN)6]) действием разбавленных кислот при нагревании.
Циановодород состоит из линейных молекул с длинами связей CN и СИ
1,15 и 1,06 А, а его строение соответствует нитрильной форме Н—C=N |.
В то время как обсуждается [1, 11, 12, 794] вопрос о таутомерном равно-
весии
Н—C^N | <> | C=N—Н, (3.10)
из исследования спектров КР следует, что жидкий и газообразный циано-
водород не содержит изонитрильной формы [795].
Теоретический расчет обеих форм циановодорода показал, что внутри-
молекулярная перегруппировка с образованием HNC ограничена относитель-
но высоким энергетическим барьером [796].
В противоположность HNC алкил- и арилизоцианиды выделены и ин-
тенсивно исследуются [797].
3.2.3. Химические свойства HCN
Электронное строение циановодорода
Н—С-N | <-> Н—С=N | Н+ [| C=N | Г
обусловливает возможность реакций двух типов: присоединения по тройной
связи углерод — азот (аналогично ацетилену) и с предварительной диссо-
циацией на протон и цианид-ион. К первому типу реакций относятся: гид-
ролиз с образованием формиата аммония HCOONH4; гидрирование с прев-
ращением HCN в метиламин CH3NH2; взаимодействие с аминами
HCN + RNH2 Н—С—NHR.
II
NH
По другому типу взаимодействий, кроме реакций HCN с основаниями,
протекают также его полимеризация
HC№F->NC—CH=N-±si-NC—CH=NH и т. д.,
присоединение к ненасыщенным соединениям, например к ацетилену с
образованием СН2=СН—CN, к альдегидам и кетонам, взаимодействие с
47
галогенами и галогеносодержащими соединениями типа
HCN + RC1 -> RCN + НС1. (3.11)
Большая реакционная способность циановодорода привела к широкому
использованию его в промышленности органического синтеза. Благодаря
наличию л-связи CsN и неподеленной пары электронов на атоме азота ни-
трильной группы HCN (L) образует ряд координационных соединений:
А1С13 2L, SbCl5 • 3L, FeCl3 • 2L, SnCl4 • 2L, ВС13 • 2L, TiCl4 • 2L [11,
TiX4 • 2L (X = F, Cl, Вг, I) 1131, Lm - MCln (M = B, Ti, Sb, tn = 1 или 2)
[151, VML (M = Cr, Mo) [161, (Co)5Cl4 • 2L [14].
Методом ИК-спектроскопии установлено образование координационной
связи М—NCH в соединениях МХ4 - 2L (М = Ti, V), осуществленной через
атом азота, а не тройную связь C=N [13, 14]. Наличие такой же связи под-
тверждено исследованиями методом ЯМР-спектроскопии взаимодействия
между HCN и некоторыми цианидфосфиновыми комплексами никеля (II)
типа Ni2 (CN)4 (РС3Р)3 и Ni (CN)2 (PPr3)2 [17].
3.3. ЦИАНИДЫ АММОНИЯ, ЩЕЛОЧНЫХ МЕТАЛЛОВ
И МЕТАЛЛОВ ПОДГРУППЫ БЕРИЛЛИЯ
3.3.1. Получение солей
При нагревании протекают реакции [1]
Na4Fe (CN)e 4NaCN + Fe + C2N2, (3.12)
K4Fe (CN)6 + K2CO3 5KCN + KCNO + Fe + CO2, (3.13)
Rb4Fe(CN)6 + 2Rb->6RbCN + Fe, (3.14)
K2CO3 + 4C 4- N2 -> 2KCN + 3CO, (3.15)
NaNH2 + C NaCN 4- H2, (3.16)
CaCN2 + C -> Ca (CN)2, (3.17)
Ba (NCS)2-> Ba (CN)2 4- 2S. (3.18)
Значительно легче образуются цианиды щелочноземельных металлов при
их непосредственном соединении с цианом [1].
Пропусканием аммиака и CS2 над нагретыми до красного каления ме-
дью или железом получают цианид аммония [ 1 ]
2NH3 + CS2 4- 4Cu-> 2Cu2S 4-NH4CN 4- H2. (3.19)
Некоторые цианиды удобно получать посредством обменных реакций
Ba (CN)2 4- Li2SO4 -> 2LiCN 4- BaSO4, (3.20)
2CsCN 4- Mg (C1O4)2 -> 2CsC1O4 4- Mg (CN)2. (3.21)
В лабораторных условиях с целью синтеза цианидов чаще всего циано-
водород, образующийся при взаимодействии гексацианоферрата (II) щелоч-
ного металла с разбавленной серной кислотой, поглощают спиртовым ра-
створом щелочи.
3.3.2. Общие свойства ионных цианидов
Цианиды аммония, щелочных и щелочноземельных металлов хорошо
растворяются в воде, a NaCN и NH4CN — ив спирте. Из водных растворов
в чистом виде можно выделить лишь цианиды натрия и калия и с большим
48
трудом — лития, стронция и бария. Продолжительное кипячение ведет к их
полному разложению
KCN + 2Н2О -> КСНО2 + NH3. (3.22)
Цианиды щелочных металлов превращаются в тиоцианаты при сплавле-
нии или кипячении с серой или полисульфидами, a NH4CN и при выдержива-
нии его водного раствора со свежеосажденной серой.
Цианид щелочного металла легко переходит в цианат при нагревании на
воздухе или с легко восстанавливающимся оксидом, например МпО2. При
сплавлении NaCN с оксидами Pb, Sn легко получаются гранулы восстанов-
ленного металла.
При взаимодействии хлора, брома и иода с раствором цианида образу-
ются галогеноцианы
NaCN + С12 -> C1CN + NaCl. (3.23)
Действием SO2 на холоду на KCN получен цианосульфит калия KSO2CN,
раствор которого восстанавливает соли серебра и золота.
Цианиды щелочных металлов не изменяются при прокаливании без
доступа воздуха, а цианиды щелочноземельных, особенно Са (CN)2, частич-
но превращаются в цианамиды.
Вследствие образования комплексных соединений водные растворы
цианидов щелочных и щелочноземельных металлов хорошо растворяют
гидроксиды Zn, Cd, Си, Ni, Со, Fe (II) и нерастворимые цианиды Zn, Cd, Hg
и d-металлов.
3.3.3. Свойства отдельных соединений
элементов I и II главных подгрупп
NH4CN — бесцветное кристаллическое вещество тетрагональной струк-
туры, очень неустойчивое на воздухе, разлагается на NH3 и HCN при 36° С.
LiCN по свойствам близок к цианидам натрия и калия. Абсолютно без-
водную соль можно выделить из тетрагидрофурана при взаимодействии Li
с AgCN в присутствии нафталина [18]. При нагревании до высоких темпера-
тур LiCN устойчив, а в присутствии железа разлагается
2LiCN -> С + Li2CN2. (3.24)
NaCN в зависимости от температуры кристаллизуется из воды в виде
моно- или дигидрата. В его водных растворах в присутствии О2 растворяют-
ся многие мелкодисперсные металлы, кроме РЬ и Pt. В воде NaCN взаимо-
действует с тетратионатом натрия по реакции
3NaCN + Na2S4O6 + Н2О -> NaNCS + Na2S2O3 + Na2SO4 + 2HCN. (3.25)
Помимо спирта, NaCN растворим в DMFA (18,76 г на 100 г растворителя
при 25° С), претерпевая сольволиз в кипящем DMFA с образованием диме-
тиламида NaN (СН3)2 [191. Для цианида натрия выделено координационное
соединение типа Na (Dpb)3CN [201 и рентгенографически установлено, что
атом натрия окружен шестью атомами азота с расстоянием Na—N 2,608 А
[211.
KCN кристаллизуется из воды в безводном состоянии, плавится при
622° Сив отсутствие кислорода и влаги остается неизменным даже при
белом калении. Его водные растворы сохраняются относительно хорошо
в отсутствие воздуха, но постепенно KCN частично гидролизует с образова-
нием NH3 и формиата калия. При сплавлении цианида калия с K2SO4 обра-
зуются цианат и сульфид калия.
49
RbCN и CsCN хорошо растворимы в воде, но не растворимы в спирте или
эфире. Цианиды рубидия и цезия с цианидами переходных металлов легко
образуют соединения типа Kat4 [М (CN)61 и Kat3 [М (CN)6] (Kat = Rb, Cs).
Так, из водных растворов получены Kat4 [Fe (CN)61 (Kat = Rb, Cs) [22],
смешанные соли Rb и Cs c Mg и Ca, например Rb2Ca [Fe (CN)6] [23], c Ni —
CsNi [Fe (CN)6] [24], с редкоземельными элементами — RbGd [Fe (CN)6]
[25, 26].
Цианиды щелочных металлов имеют при комнатной температуре различ-
ную кристаллическую решетку: LiCN — орторомбическую, NaCN, KCN,
RbCN — кубическую типа NaCl, a NH4CN и CsCN — типа CsCl [27, 28].
Be (CN)2 может быть получен в виде белого осадка при добавлении эфир-
ного раствора диметилбериллия к избытку HCN в бензоле. Соединение при-
сутствует в продукте реакции сухого Ве12 с дицианом при 500° С, изученной
в 1898 г. [1]. Be (CN)2 нерастворим в органических растворителях, кроме
тех, которые вызывают сольволиз [29].
Выделены также металлоорганические цианиды бериллия (MeBeCN)n
и [(Me3N) MeBeCNl, содержащие, как показывают ИК-спектры, мостиковые
CN-группы [291.
Mg (CN)2 и Ca (CN)2 очень неустойчивы в растворе; их получают пропус-
канием HCN в раствор соответствующего гидроксида, a Mg (CN)2 — раство-
рением металла в 10—15%-ном растворе циановодорода. Водные растворы
солей нестабильны даже при хранении в присутствии HCN.
Известны аммиакаты М (CN)2 • 2NH3 (М = Mg, Са) и соль Na2Ca (CN)4
[1]. Цианиды магния и кальция более устойчивы в виде комплексных со-
единений с цианидами других металлов, например Mg [Ag (CN)2]2 • 4Н2О,
Ca [М (CN)4] (М = Zn, Ni) [11, Mg2 [Fe (CN)6] nH2O [26].
При кристаллизации растворов, полученных обработкой кристаллических
гидроксидов стронция и бария сухим HCN, выделены Sr (CN)2 • 4Н2О и
Ba (CN)2 2Н2О, а из петролейного эфира — безводный Ba (CN)2 [1]. Послед-
ний плавится при 600° С и заметно летуч уже при этой температуре. При
дальнейшем нагревании соль разлагается
Ba (CN)2 BaCN2 + С. (3.26)
Выделен ряд комплексных соединений Sr (CN)2 и Ba (CN)2 с цианидами
d-металлов, например Ba [Ni (CN)4] [1], Ва2 [Fe (CN6)]- 6НО, Sr2 [Fe (CN)6] •
- nH2O [26].
3.4. ЦИАНИД-ИОН И ЕГО
КОМПЛЕКСООБРАЗУЮЩАЯ СПОСОБНОСТЬ
3.4.1. Электронное строение
и донорная способность
В основном невозбужденном энергетическом состоянии цианид-ион опи-
сывается электронной конфигурацией (os)2 (о*)2 (лх;/)4 (ог)2, что отвечает
формальному образованию одной о- и двух л-связей [1421. Низшая, наибо-
лее стабильная занятая о-орбиталь, образованная преимущественно s-ор-
биталями атомов углерода и азота, отвечает обычной о-связи углерод — азот.
Остальные две занятые о-орбитали соответствуют двум парам электронов,
одна из которых (с наибольшим запасом энергии) локализована в основном
у атома углерода, другая (промежуточная) — около атома азота. Первая из
указанных орбиталей образована перекрыванием р-орбиталей атомов угле-
рода и азота, а промежуточная — за счет s- и р-орбиталей этих атомов [1431.
50
Это — две так называемые «свободные» неподеленные пары электронов циа-
нид-иона. Дважды вырожденная зх-орбиталь соответствует образованию двух
я-связей.
По расчетным данным [798—8001 заряд СЫ~-иона симметрично распре-
делен на обоих атомах. Благодаря наличию в цианид-ионе заполненных элек-
тронами о- и зх-орбиталей, в частности двух неподеленных пар электронов,
он обладает активными донорными свойствами.
Донорно-акцепторная о-связь М ч- CN в цианидных комплексах может
осуществляться за счет взаимодействия всех трех заполненных орбиталей
С№-иона (os, Os, сгг) и пустых (п — 1) do-, ns- и пр0-орбиталей атома-ком-
плексообразователя. Однако, поскольку потенциал ионизации азота (14,
53 эВ) выше, чем углерода (11, 26 эВ), а у последнего преимущественно
локализована наименее стабильная ог-орбиталь, координация CN-группы че-
рез атом углерода более предпочтительна, чем через атом азота. О таком спо-
собе координации свидетельствуют нейтронографические данные по кри-
сталлическим структурам К3 (Со (CN)6] [144], Hg (CN)2 [34], К2 [Zn (CN)4]
[145] и результаты исследования ИК-спектров комплексных цианидов пла-
тины и железа [146, 152].
В ряду изоэлектронных лигандов NO+, СО и CN~ наилучшим о-донором
является цианид-ион [142], что подтверждают полуэмпирические расчеты
по методу самосогласованного поля [147]. Это, возможно, обусловлено на-
личием отрицательного заряда у иона CN~, который способствует, при про-
чих равных условиях, повышению электронодонорных свойств лиганда.
В цианидных комплексах может возникнуть и зх-донорно-акцепторное
взаимодействие М -2- CN за счет комбинации занятых зх-орбиталей CN~-ио-
на и пустых прл- и (и — 1)бл-орбиталей (преимущественно последних)
атома-комплексообразователя. Очевидно, зх-связь тем более эффективна,
чем меньше дя-электронов имеет атом металла-комплексообразователя [148].
Следовательно, она играет более важную роль в комплексах ионов с элек-
тронной конфигурацией d1 — d3, чем с d5 или d6.
3.4.2. Акцепторная способность
В координационной связи М—CN имеет значение и зх-дативная компо-
нента. Связь М -> CN образуется в результате взаимодействия заполненных
(и — 1^я-орбиталей металла с вакантными зх*-орбиталями С№-иона и
наиболее эффективна в случае, когда последние более стабильны, чем р-ор-
битали центрального атома.
зх-Дативное взаимодействие понижает разрыхляющий характер рас-
сматриваемых орбиталей, в какой-то мере компенсируя уменьшение от-
рицательного заряда на лиганде, вызванное донорно-акцепторным взаимо-
действием М CN. Доля вклада зх-дативной составляющей уменьшается с
о
увеличением степени окисления металла.
зх-Дативная и о-донорно-акцепторная компоненты связи М—CN взаимо-
связаны. Увеличение отрицательного заряда на атоме металла в результате
о-взаимодействия компенсируется переносом части этой зарядовой плотно-
сти с металла на лиганд за счет зх-дативного взаимодействия. Не исключено,
что при прочих равных условиях усиление о-донорно-акцепторной состав-
ляющей связи М—CN способствует росту зх-дативной составляющей и нао-
борот.
51
Благодаря наличию л-дативных связей CN~ наравне с такими л-акцепто-
рами, как NO, СО, проявляет способность стабилизировать низшие степени
окисления центральных атомов. Так, известны комплексы со степенью окис-
ления металла 0 или +1 тетраэдрического [М (CN4)]4~ (М = Ni, Pd, Pt)
и октаэдрического строения [М (CN)6]5~ (М = Мп, Re), [М (CN)6]6~ (М =
= Сг, Мп) [142]. Полуэмпирические расчеты молекулярных орбиталей по-
казали, что л-акцепторная способность изоэлектронных лигандов (NO+,
СО, CN~) изменяется в ряду NO+ СО > CN~ [801, 839, 840].
Представления о возникновении л-дативных связей в цианокомплексах
согласуются с результатами изучения колебательных спектров. В частно-
сти, во многих случаях вследствие л-дативного взаимодействия силовые кон-
станты связей CN в циано комплексах меньше, чем у цианид-иона [26]. Ус-
тановлено, что для комплексов железа и платины, при переходе от их со-
единений с низшими степенями окисления к высшим, частоты v(CN) и силовые
константы связей CN возрастают [26, 146]. Это обусловлено понижением спо-
собности металла к л-дативному взаимодействию и возрастанием роли o'-
связывания.
На проявление л-дативного взаимодействия в комплексных цианидах
указывают и результаты исследования спектров Мессбауэра ионов
[Fe (CN)3L]"“ [ 149].
По величине изомерного сдвига 6 можно судить о л-связывающей способ-
ности лигандов L. По степени увеличения л-связывающей силы лиганды
располагаются в ряд NH3 < NO? < PPh3 < SO5~ < CN~ < CO < NO+.
Величины ЯМР (на F1S) параметров экранирования для фторофенильных
комплексов платины транс-IPt (FC6H4) (PEt3)2L] (L = CN, NCS, NCO, Cl,
Br, I и др.) указывают, что CN-группа проявляет сильные л-акцепторные
свойства [150].
Как известно, цианнд-ион обладает сильным трансвлиянием, что в свете
современных воззрений приписывается его л-связывающим свойствам [151].
Резюмируя сказанное выше, можно представить принципиальную схему
связи металл — цианид в виде
л
mZIccn
о
причем доля каждой составляющей компоненты во многом зависит от ин-
дивидуальных свойств и особенностей центрального атома.
3.4.3. Амбидентатность цианид-иона
Приведенные выше свойства цианид-иона относились к одноядерным ком-
плексам, в которых CN~ является монодентатным лигандом и связан через
атом углерода только с одним атомом металла. Однако ему свойствен и дру-
гой характер связывания. Возникновение о-донорно-акцепторной связи
через атом углерода приводит к дестабилизации о*-орбитали, локализован-
ной в основном на атоме азота и занятой второй неподеленной парой элек-
тронов. В результате этого CN~ может участвовать в донорно-акцепторном
взаимодействии М' <- NC [143]. Таким образом возникают мостиковые CN-
группы. При осуществлении структуры М—CN—М' связь CN, как и в слу-
чае образования о-донорно-акцепторной связи Мч-С, упрочняется [152,153],
ибо электроны, участвующие в образовании донорно-акцепторной связи
N->M', находятся на более разрыхляющей, чем в ионеС№, о-орбитали циа-
ногруппы [143].
52
Вследствие перераспределения электронной плотности образование мо-
стиковых связей сопровождается понижением отрицательного заряда на
CN-группах, что способствует усилению роли л-дативной компоненты
М -> CN и ослаблению донорно-акцепторного взаимодействия M-X-CN.
Часто в мостиковых CN-группах прочность связи М'—NC может прибли-
жаться к прочности связи М—CN [154].
Линейные мостики М—CN—М' имеют большое значение в структуре
многих цианидов. Так, кристаллы AuCN, Zn (CN)2, Cd (CN)2 являются поли-
мерами с бесконечной цепью.
3.5. ЦИАНИДЫ р- И d10-ЭЛЕМЕНТОВ
3.5.1. Простейшие цианиды и координационные
соединения несолевого типа
Наиболее изученными цианидными соединениями p-элементов являются
цианиды металлов подгруппы цинка. Эти металлы в указанных соединениях
проявляют степень окисления, равную 2. Соли одновалентной ртути с циа-
нид-ионами диспропорционируют, образуя цианид ртути (II).
Цианиды подгруппы цинка бесцветны и диамагнитны; их легко получить
при введении CN~-hohob в раствор соли соответствующего металла. Наиме-
нее растворим в воде и этаноле цианид цинка. Цианид ртути хорошо раство-
рим в воде, этаноле, метаноле, жидком аммиаке и этилендиамине [30].
Zn (CN)2 и Cd (CN)2 изоструктурны, имеют структуру антикуприта, ко-
гда каждый атом металла связан с четырьмя тетраэдрически расположенны-
ми CN-группами [31, 32].
Наиболее устойчивый из рассматриваемых цианидов Hg (CN)2 образует
в твердомосостоянии тетрагональную ячейку с параметрами а = 9,643, с —
= 8,880 А [33]. Молекулы Hg (CN)2 почти линейны в твердом состоянии
(AHgCN и ZLCHgC соответственно равны 177,7 и 175,0° [33, 34]), а также в
водных и метанольных растворах [35].
Цианид ртути мало диссоциирован в воде
к = [Hg2+] (CN—__ ш-35
4 [Hg(CN)2] ’
и только введением сульфид-ионов можно перевести ртуть в осадок.
Цианиды подгруппы цинка образуют многочисленные координационные
соединения с нейтральными лигандами: М (CN)2 • 2N2H4 (М = Zn, Cd),
Hg (CN)2 • NH3, Cd (CN)2 (C16H18N3S)2 [1], M (CN)2 • 2tu • rcH9O (n = 2(Zn),
4 (Cd), О (Hg) [36, 37]) и Hg (CN)2 • tu [381, M (CN)2 • 2RNH2 (R = Me,
Et, Pr, Bu) [39—41].
Для цианида ртути из воды или спирта выделены аддукты Hg (CN)2 • L
(L = mrf [42], en [43], 2-MePy, 2,4-диметилпиридин, 2,6-диметилпиридин
[44], Dipy, Phen [45], 3-цианопиридин, Me4en, DMSO [46], Diox [47], MeOH
[48]); Hg (CN)2 • 2L (L = Py, 3-MePy, 4-MePy [44], Ph3P [45]), а также
4Hg (CN)2 • Urt, 2Hg (CN)2 • Atp [42], Hg (CN)2 • 0,8Thf [49] и 5Hg (CN)2 •
• 4Thf [50].
Согласно ИК-спектрам поглощения полимерную октаэдрическую струк-
туру с мостиковыми CN-группами имеют аддукты с пиридинами [44] и со-
единения цианидов подгруппы цинка с алифатическими аминами [40, 41].
В соединениях же Hg (CN)2 • Diox [47], Hg (CN)2 • PC2P, Hg (CN)2 • AsAs
[51] атомы ртути имеют координационное число (к. ч.)4, а в аддуктах
53
Hg (CN)2 • L (L = Q и замещенные Q [52]) — к. ч. 3, причем в последних
CN-группы немостиковые.
Представляют интерес соединения TiCl4 • Hg (CN), и МХ4 • 2Hg (CN)2
(М — Ti (X = Br), Sn (X = Cl, Br) [53]), имеющие полимерную структуру
за счет мостиковых CN-групп, причем во втором случае одна мостиковая
CN-группа связывает атомы Hg и Ti (или Sn), а другая — атомы ртути.
На основе цианидов металлов получен ряд клатратных соединений.
Среди них Cd (NH3)2 Hg (CN)4 • 2С6Н6 имеет трехмерную структуру за счет
мостиковой функции CN-rpynn [54].
О получении В (CN)3 сообщалось в 1931 г. Однако в дальнейшем по реак-
ции между галогенидами бора и HCN или AgCN получили лишь неустойчи-
вые продукты неопределенного состава [55]. Взаимодействие LiBH4 или
LiAlH4 с HCN в эфирном растворе приводит к выделению комплексных циа-
нидов бора и алюминия [56, 57]
LiMH4 + 4HCN -> LiM (CN)4 + 4Н2 (М = В, А1). (3.27)
Как показали ЯМР-исследования, бор образует соединения B2H5CN • L
+ —
(L = Thf, Ру, NMe3), имеющие структуру L ВН2 N^C-ВН3. Они
получены при взаимодействии ВН3 • Thf в тетрагидрофуране с HCN и по-
следующем введении пиридина или триметиламина [58]. Для бора известен
устойчивый в водном растворе Li [BH3CN] [56].
Цианид алюминия осаждается при взаимодействии гидрида алюминия и
HCN в эфире при —20° С [59]
5А1Н3 + 5HCN -> 2A12H6CN • О(С2Н5)2 ф- Al (CN)3 ф- 5Н2. (3.28)
Растворимый в этих условиях гидридцианид алюминия, по данным ана-
лиза и ИК-спектров .поглощения, вероятно, имеет строение
Выделено также соединение А1Н (CN)2 при взаимодействии А1Н3 с
Hg (CN)2 в эфире при —20° С.
Образование газообразных AICN [60] и BCN [61] установлено масс-спек-
троскопически.
Для галлия известно лишь соединение [Me2GaCN]4, полученное при вза-
имодействии Me3Ga и HCN в бензоле [29]. Аналогичные соединения с линей-
ными MCNM-группами выделены для А1 и In. Таллий образует Me2TlCN,
диссоциирующий в водном растворе на [Ме2Т1]+ и CN~ [29]. Наличие ионной
структуры предполагается и для соединений R2T1CN (R = С5Н5, С9Н7) [62].
Для таллия (III) известны координационные соединения К [PhTl (CN)3]
[55] и Ме4 N [Me3TlCN] [63].
In (CN)3 можно получить при взаимодействии металлического индия со
смесью Н2 и HCN (1 : 1) при 350° С [64]; действии (CN)2 на InOI, Hg (CN)2
на In в жидком аммиаке, циановодорода на In (ОН)3 [65]. Побочным продук-
том синтеза [64] является соединение In (CN)2, имеющее, по данным ИК-
спектров, строение In+In (CN)r-
Ge (CN)4 образуется в бензоле по реакции [66]
Gel4 ф- 4AgCN -> Ge (CN)4 ф- 4AgI (3.29)
и при взаимодействии GeCl4 с Me3SiCN в ксилоле в токе N2 [67].
54
Аналогично по реакции
3Me3SiCN + МС13 -> М (CN)3 + 3Me3SiCl (3.30)
синтезированы цианиды трехвалентных фосфора, мышьяка и сурьмы. Для
висмута получается, вероятно, BiCi (CN)2 или смесь Bi (CN)3 и BiCl3 [67].
Для германия и кремния известны многочисленные цианидные произ-
водные типа MH3CN и MR3CN (М = Ge, Si; R = Me, Et, Ph, ц-МеС6Н4)
[55, 67—69].
По данным ИК-спектров, гермил- и силилцианиды имеют структуру нор-
мальных цианидов, а в соединениях MMe3CN присутствует около 10% изо-
цианидов MMe3NC [68].
Для олова выделены лишь соединения Sn (OH)3CN [1], R3SnCN и
R2Sn (CN)2 (R = Me, Et, Pr, Bu, Ph) [55].
По данным Я MP (на 119Sn) и мессбауэровских спектров, R3SnCN (R =
= Me, Et, Bu) имеют тригонально-бипирамидальную структуру с мостико-
выми бидентатными CN-группами [70]. Рентгенографически подтверждена
такая же структура для (CH3)3SnCN, причем связь Sn—CN (2,48 А) почти
ионная [71].
Цианид свинца Pb (CN)2 можно получить из водного раствора лишь в
присутствии HCN, иначе выделяются основные соли переменного состава,
например Pb (CN)2 • 2РЬО [1].
Известны цианиды четырехвалентного свинца R3PbCN (R = Et, Pr,
Ph), R2Pb (CN)2 (R = Me, Ph) и Ph2Pb (OH)CN [55]. Me2M (CN)2 (M = Si,
Ge, Sn, Pb) по структурным и ИК-спектральным данным являются циани-
дами, хотя не исключена и примесь изоцианидов [72].
Для неметаллов V группы выделены Р (CN)3 и As (CN)3 при взаимодей-
ствии AgCN с ЕС13 (Е = Р, As), а также Р2 (CN)4 по реакции между Р214
и AgCN в дихлорэтане [1].
Из сероуглерода при взаимодействии AgCN и SC12 получены ромбические
кристаллы S (CN)2 [74, 75], который является первым представителем так
называемых цианосульфанов NC—Sn—CN (п = 1 -4- 8) [76]. Цианиду серы
изоструктурен Se (CN)2 [77, 78], а Те (CN)2 имеет другое строение [79]. По-
следний образуется при взаимодействии TeBr4 с AgCN в бензоле в атмосфере
N2 [1]
TeBr4 + 3AgCN 3AgBr + BrCN + Те (CN)2. (3.31)
Детально изучены галогенцианы XCN (X = F, CI, Br, I) [80]. C1CN и
BrCN получают обработкой водных растворов цианидов хлором и бромом,
a ICN — при взаимодействии сухого цианида, обычно Hg (CN)2, с иодом.
FCN был получен путем крекинга фторцианура (FCN)3 [81] и при взаимодей-
ствии тетрацианметана с избытком CsF по реакции [82]
С (CN)4 + CsF CsC (CN)3 + FCN. (3.32)
В промышленных масштабах галогенцианы можно получить электро-
лизом водных растворов HCN и NH4X [83].
Фтор- и хлорциан — бесцветные газы, а остальные галогенцианы — лету-
чие кристаллические вещества. Их растворимость в воде в ряду С1, Вг,
I заметно уменьшается.
Молекулы галогенцианов линейны и имеют следующую электронную
структуру:
|X—C=N|<->[X =C==N| (X = F, Cl, Br, I),
причем последняя форма приобретает все большее значение с возрастанием
электроположительного характера галогена [4], на что указывают данные
спектров Мессбауэра [84].
55
Галогенцианы склонны к полимеризации с образованием галогенангид-
ридов циануровой кислоты и к образованию координационных соединений:
C1CN • МС15 (М = Nb, Та) [85], C1CN • МО2С12 (М = Mo, U) [86], 2XCN •
• TiCl4, XCN • AICI3, XCN • SbCl5, XCN -А1Вг3 (X = Cl, Br) [87], в кото-
рых ИК-спектрально установлено наличие связи металл — азот CN-группы
[85, 87]. При взаимодействии C1CN и AgCNO образуется межпсевдогалоге-
нидное соединение — изоцианатоциан [88]. Это полимер (NCNCO)n, который
выше 140° С деполимеризуется с образованием быстро полимеризующегося
мономера.
3.5.2. Анионные комплексы р- и б10-элементов
р- и <110-металлы образуют разнообразные цианидные ацидокомплексы
однородного и смешанного типа.
Для цианидов подгруппы цинка получены координационные соединения
с общей формулой Kat2 [М (CN)41 (Kat = NH^, щелочные металлы, Ag+,
Cu+, Tl+), Kat [M (CN)41 (Kat = Pb2+, Ni2+, Co2+, Mn2+) [1, 89] и
Kat [M (CN)31 (Kat = Cs+, Rb+; M = Cd2+, Hg2+) [90].
В литературе имеются противоречивые сведения о составе комплексов
цинка, кадмия, ртути в водных растворах и их устойчивости.
Потенциометрически, калориметрически и полярографически в системе
Zn2+ — CN~ — Н2О установлены комплексы Zn (CN)2, [Zn (CN)3] и
[Zn (CN)4]2~, энтальпии образования которых соответственно равны: —45,25;
—80,45 и —116,48 кДж/моль [91]. Образование ZnCN+ установлено при
потенциометрическом исследовании указанной системы с использованием
цинкамальгамного и стеклянного электродов [92].
Комплексы [М (CN)5]3~ и [М (CN)6]4~ (М = Zn, Cd), выявленные поляро-
графически [93, 94], не установлены ИК-спектральными исследованиями
[95, 961.
Хотя по данным электронных и спектров КР в системе Cd (CN)2 — KCN —
Н2О существуют только [Cd (CN)4]2~, методами растворимости и ИК-
спектроскопни установлено также образование [Cd (CN)3]~ [30]. Кадмий
образует с цианид-ионом и простейшие комплексы [CdCN]+ и Cd (CN)2,
устойчивость которых оценена потенциометрически [92, 971 (табл. 3.1). Для
ртути потенциометрически [98, 99] и полярографически [100, 101] установ-
лено существование и рассчитаны константы образования комплексов, со-
держащих 1, 2, 3 и 4 CN-лиганда.
В ПК- и спектрах КР водных растворов цианидных комплексов ртути
выявлены Hg (CN)2, Hg (CN)7, Hg (CN)2- [95, 102, 1031. По данным ИК-спек-
тров, при низких концентрациях CN~-ионов в системе существуют следую-
щие комплексы ртути: [Hg2CN]3+, [Hg3 (CN)2]4+ и [Hg (OH)CN] [102]. При
большом же избытке CN~-hohob (до 24-кратного) некоторые исследователи
установили комплексы [Hg (CN)5]3~, [Hg (CN)6]4~ и даже [Hg (CN)7]5~ по-
тенциометрическим методом [104, 105] и методом спектроскопии КР [106].
Однако большая разница в константах устойчивости |35 и |36 [104,105] и полу-
количественный характер исследования спектров КР [106] вызывают сом-
нение в существовании таких комплексов.
Полярографически установлено, что в растворах с соотношением
CN~: Hg2+ = 500 присутствуют только ионы [Hg (CN)4]2~ и CN~ [100].
Как видно из табл. 3.1, среди [М (CN)4]2— для Zn, Cd, Hg наибольшей
56
Таблица 3.1. Логарифмы констант образования [M(CN)n]2 п
Ион металла Метод исследования Температу- ра, °C Ионная сила 1g Pi 1g ₽2 1g ₽3 1g ₽4 1g Р8 1g ₽е 1g ₽7 Литература
Zn2+ Потенциометрия 25 0 11,067 16,05 19,62 [91]
» 25 3,0 5,34 11,02 16,68 21,57 [92]
Cd2+ » 25 3,0 5,54 10,60 15,30 18,85 [97]
» 25 3,0 5,62 10,84 15,72 19,20 [92]
Hg2+ » 25 0,1 18,00 34,71 38,54 41,52 [98]
» 25 0 17,00 32,75 36,31 38,97 [99]
» 40 0 16,26 31,28 34,65 37,07 »
Полярография 30 2,0 — 33,9 38,1 40,6 [ЮО]
» 25 0,2 — 35,25 38,96 41,51 ![101]
Потенциометрия 25 .— 36,6 40,6 43,5 44,2 45,2 — 4104]
» 30 3,0 — — — 40,53 42,57 38,24 4105]
прочностью обладает комплекс ртути. Этот вывод подтверждают и величины
силовых констант связей М—С, равных 1,30; 1,28 и 1,53 Н/см [30].
Из водных растворов выделены изоструктурные соединения К2 (М (CN)4]
(М — Zn, Cd, Hg). К2 (Hg (CN)4] получен также из метанола и жидкого HCN
[102]. Соединение KHg (CN)3, выделенное ранее из жидкого HCN [107], ока-
залось смесью Hg (CN)2 и К2 [Hg (CN)4] [102].
Как установлено методом ЭПР [108], под действием рентгеновского и
7-излучения в солях К2 [М (CN)4], имеющих структуру шпинелей, образуют-
ся комплексы симметрии СзУ парамагнитных одновалентных ионов Zn,
Cd, Hg.
Спектрофотометрически и потенциометрически доказано, что при взаимо-
действии Hg (CN)2 с Кп [Fe (CN)6] (и = 3 и 4), К4 [Mo (CN)8] и К4 [Ru (CN)J •
• ЗН2О в водном растворе протекает ступенчатое образование полиядерных
комплексов типа M(CN)6 • nHg (CN)^~ (М = Fe (II), Mo (IV), Ru (II), n =
= 14-3) [109].
Для металлов подгруппы цинка, особенно для ртути, характерно об-
разование смешанных координационных соединений. Выделены соединения
К2 [М (CN)2 (SCN)2] • пН2О, в которых цианидные группы координированы
с металлом через углерод, а NCS-лиганды — через азот (для цинка) и через
серу (для кадмия и ртути) [ПО].
Методом ИК-спектроскопии установлено [102] образование цианид-
галогенидных комплексов [М (CN)2CI]” и [М (CN)3C1]2~ для кадмия и
ртути, а по данным ультрафиолетовых и спектров КР-комплексов Hg (CN)I,
[Hg (CN)I3]2—, [Hg (CN)2In]n~ [111]. Кроме того, для ртути выделены
Kat [Hg (CN)2X] (X = Cl, Br, I; Kat = щелочной металл), соединения
Hg (CN)2 с сульфатами и нитратами Ni и Co, с хроматами и бихроматами
щелочных металлов, с тиоцианатами К, Са, Ва, Cd, Со, Ni [1].
В водном растворе полярографически при 20° С и ионной силе 2 выявле-
ны смешанные комплексы [Hg (CN)2X]~ и [Hg (CN)3X]2— (X = Cl, Br, NCS),
устойчивость которых (1g |3) соответственно равна 36,19 и 39,43 (Cl); 35,99
и 39,10 (Br); 36,30 и 38,74 (NCS) [112].
При потенциометрическом исследовании системы Zn2+ — гистидин —
CN~ [113] установлено образование комплексов Zn (Hist) CN, [Zn (Hist)
57
(CN)2] , [Zn (Hist) (CN)3]2 , характеризующихся следующей устойчивостью
(1g 0) при 25° С и р з= 0,1: 1g 0J = 11,76; 1g 02 = 16,71; 1g 03 = 20,48.
Цианид-ион в координационной сфере ацидокомплексов цинка и кадмия
может быть замещен на нейтральный лиганд. Интересно соединение К [Cd -
•(CN)3tu] (tu)H2O, в котором, по данным ИК-спектров поглощения, наряду
с координированной тиомочевиной, имеется и внешнесферная молекула тио-
мочевины [36].
Смешанные цианидные комплексы известны для Р (V), Sb (V) и Bi (III).
Выделены тетрахлородицианофосфаты (Bu4N) [РС14 (CN)2] и (Pr4N) [PC14(CN)2]
при взаимодействии РС15, KCN и R4NC1 (R = Pr, Bu) в ацетонитриле
[114] и дицианодитиофосфат (Bu4N) [PS2 (CN)2] по реакции между P4S4O6,
KCN и Bu4NC1 [115].
При взаимодействии SbCl5 с KCN в среде жидкой SO2 образуется
К [SbCl5CN] [116].
Для висмута получены гидролитически неустойчивые бромид- и иодид-
цианидные соединения Kat [BiX (CN)31 (Kat = Ag+, K+, Cu+, Hg+ и X =
= Br, I) по реакции между BiCl3 и цианидом металла в сухом ксилоле [1].
Среди элементов третьей группы наиболее устойчивый цианид образует
таллий (I). T1CN, имеющий структуру типа CsCI, можно получить из водно-
го раствора. Соединение по многим свойствам напоминает цианиды щелоч-
ных металлов, в частности при образовании координационных соединений
Tl2 [Hg (CN)J, TI [Ag (CN)2], Tl2 [Zn (CN)4] [1]. Кристаллооптическое, рент-
генографическое и ИК-спектральное исследование системы KCN — T1CN —
Н2О при 25° С [117] не подтвердило образование комплекса KTI (CN)2, на
что указывалось ранее [73].
На основании влияния С№-иона на скорость обмена Т13+ — Т1+ в вод-
ном растворе было сделано предположение о существовании комплексов
T1CN, [TI (CN)2]~, [TI (CN)3]2~, образующихся при высоких соотношениях
CN~ : Т1+, и комплексов [TI (CN)]2+, [TI (CN)2]+, TI (CN)3, [TI (CN)4]~ 1118].
Константы образования [TI (CN)rt]3~n (n = 1; 2) при 8—32° С и p = 0,5,
рассчитанные по катионообменному методу, составляют < 1011 [119].
При выпаривании под вакуумом раствора Т12О3 в цианидной кислоте вы-
делен цианид TI (CN) TI (CN)3, который подобно смешанным галогенидам,
по-видимому, состоит из ионов Т1+ и [TI (CN)4]~ [73].
По данным полярографического исследования, в системах М — CN~ —
Н2О (М = Pb2H, Ge4+) образуются комплексы [Pb (CN)41 ~ и [Ge (CN)6]2~
[73].
3.6. ПРОСТЫЕ И КООРДИНАЦИОННЫЕ
ЦИАНИДЫ d- И f-МЕТАЛЛОВ
Не все d- и f-металлы, для которых известны многочисленные коорди-
национные соединения на основе цианидов, образуют простые цианиды.
Комплексные же цианиды получены в настоящее время почти для всех пере-
ходных металлов, кроме Hf, Та, Ac, Th и трансурановых элементов.
Для многих переходных металлов оценена устойчивость цианидных
комплексов в водных растворах. Однако эти данные в большинстве случаев
не приведены к стандартным условиям, а потому и трудно сопоставимы.
Я. Бьеррум [1551 на основе расчета энергии Гиббса реакций комплексо-
58
образования показал, что в большинстве случаев цианидным комплексам
металлов отвечает большее сродство металл — лиганд по сравнению с акво-,
амино- и галогенокомплексами.
3.6.1. Цианиды металлов I побочной подгруппы
3.6.1.1. Простые цианиды
При введении С№-ионов в водный раствор соли Си2+ протекает реакция
[11
Cu2+ + CN~ Си (CN)2 -> 2CuCN • Си (CN)2 -а-> CuCN. (3.33)
Стабильным цианидом является нерастворимый в воде CuCN, который
можно получить в водном растворе при взаимодействии NaHSO3, KCN и
CuSO4 [30].
AgCN осаждается в виде белого осадка при взаимодействии ионов Ag+
и CN~ и в отличие от AgCI разлагается горячей HNO3 [1]. В твердом состоя-
нии AgCN состоит из линейных цепей — Ag—Ch=N—Ag—C=N с расстоянием
Ag—Ag5,26 A [30]. Частота валентных колебаний v(CN) ковалентного AgCN
2178 см-1, а ионного NaCN 2080 см-1.
Для золота известны два цианида AuCN и Ati (CN)3 [73]. Желтый кристал-
лический AuCN можно получить нагреванием при 50° С раствора KAu (CN)2
с соляной кислотой. Соединение нерастворимо в воде и при сильном нагрева-
нии разлагается с выделением дициана и золота.
Структуры AuCN и AgCN подобны, однако соединения не изоструктурны
[120], а частота v (CN) (2261 см-1) значительно выше, чем у AgCN [30].
При действии H2SiFfi на раствор KAu (CN)4 выделен бесцветный, раство-
римый в воде гидратированный Au(CN)3, который, по-видимому, является
комплексной кислотой Н3О+ [Au (CN)4]— • пН2О (и = 1 или 2) [1, 30, 73].
3.6.1.2. Однородные комплексные цианиды Си, Ag, Аи
Для металлов подгруппы меди характерно образование устойчивых
соединений, содержащих от двух до четырех CN-лигандов. Сильной тенден-
цией к комплексообразованию с CN-лигандом объясняется растворимость
металлической меди в концентрированных растворах щелочных цианидов
[156]
Си + 2CN~ + Н2О -> [Си (CN)2r + ОН~ + (3.34)
Серебро и золото также легко растворяются в этих растворах, однако
лишь при доступе кислорода воздуха
2Au + Ч2О2 4- Н2О + 4CN~-> 2 [Au (CN)2]~ + 2ОН~. (3.35)
Помимо указанных комплексных ионов [М (CN)2]~, известны также
[M(CN)312-, [М (CN)4]3~ [30, 73, 157—179].
Изучение ИК-, УФ- и спектров КР, потенциометрическое и калориметри-
ческое исследование водных растворов цианидных комплексов Си (I), Ag
(I), Au (I) [157—179] позволило количественно оценить их устойчивость
(табл. 3.2). Так, величины констант устойчивости дицианидных комплексов
характеризуются следующей последовательностью: Au > Си Ag.
Повышение устойчивости комплексов от [Ag (CN)2]~ к [Au (CN)2]~ кор-
релирует с ростом частот валентных колебаний и силовых констант связи
59
g Таблица 3.2. Константы устойчивости цианидных комплексов меди, серебра, золота
Ион ме- талла Метод исследования t, °C Ионная си- ла ц 1g Pi 1g ₽» lgKs 1g 3г 1g < ig₽4 Литера- тура
Си2+ ИКС 25 0 4,59 28,59 1,70 30,30 [157]
Потенциометрия 25 0 24,00 [158]
Спектрофотометрия 25 0,001 4,10 [159]
» 25 0 4,59 1,72 »
Калориметрия 25 ^0,6 5,0 2,64 [160]
Потенциометрия 25 0,025 21,7 26,8 27,9 [164]
Спектрофотометрия 25 5,20 [165]
» 25 2,62 [166]
Спектрофотометрия, рН-потенциометрия 25 0,01 16,26 5,39 [167, 802]
Потенциометрия, спектрофото- метрия 20 Переменная 21,7 4,6 2,3 [168]
Потенциометрия 18 0,05 20,77 [169]
» 25 0 [170]
Ионный обмен 25 0,0001 4,60; 4,85 1,85; 2,03 27,56 [171]
рН-Потенциометрия 25 0 5,30 1,5
Калориметрия 25 0 ❖ ** [172]
Спектрофотометрия 25 0,01 5,34 1,74 [173]
Ag+ Растворимость 25 0 19,85 [97]
икс 25 0 0,70 21,8 — 1,13 20,68 [161]
» 25 0,1—1,0 0,95 —0,50 »
Потенциометрия 25 0,005—1 20,44 21,98 [162]
» 20,5 0 <9,60 20,85 0,95 21,80 [174]
» 30 1 20,30 20,79 [175]
» 25 1 20,37 [176]
» 21 4,25 20,23 [177]
Калориметрия 25 0 *** [172]
1Тотенциометрия 25 0,25 20,9 21,8 [164]
Спектрофотометрия, рН-Потен- циометрия 25 1 20,14 [178]
Калориметрия 25,3 2 0,78—0,85 —0,26— (—0,19) [179]
Au+ 11отенциометрия Переменная >29,4 [97]
Термодинамический расчет 25 0 38,3 »
Потенциометрия 25 0,025 36,6 [164]
* \Н° — —46,4 кДж/моль; Д£° = —56,1 Дж/(моль-К).
** &Н° = — 46,8 кДж/моль; AS0 = —129,7 Дж/(моль-К)-
\/i° = —134,4 кДж/моль; AS0 =-|-67 ДжДмоль •К).
**** Д№ =—2,5 кДж/моль; Д£° = 20,9 Дж/(моль-К).
М—С в этих комплексах (390, 427 см-1 и 1,8; 2,8 Н/см). Это связано с боль-
шей величиной л-дативной компоненты связи М—С в [Au (CN)2]~ [30].
Методом ЯМР (на 63Си и 6бСи) в водных растворах CuCN в присутствии
избытка цианид-ионов выявлены как [Си (CN)4]3~, [Си (CN)3]2~, так и ком-
плексы [Си2 (CN)5]3~, причем преобладающим является первый из них [180].
Из систем MCN — KCN — Н2О (М = Си, Ag, Аи) выделены следующие
координационные соединения: К [M(CN)2], К [М2 (CN)3]-H2O, К3 [Ag (CN)4] •
• Н2О (или безводный), К3 [Си (CN)41 и моногидрат последнего [30]. В ра-
боте [1] сообщалось о выделении M2Cu3(CN)5 (М = Rb, Cs), Na2Cu (CN)3 •
• 3H2O, однако строение этих соединений не рассматривалось.
Известно большое количество соединений М [Ag (CN)2]3 • иН2О (М =
= La, Се, Pr, Nd, Sm, Tb, Но, Dy, Er, Y, Gd; n = 3 или 4) [122, 181].
Дицианокомплексы Ag и Au имеют линейную структуру как в растворе,
так и в твердом состоянии [182, 183].
В отличие от серебра и золота достоверные данные о существовании ком-
плекса [Си (CN)2]~ отсутствуют, и по ИК-спектру поглощения водного рас-
твора К [Си (CNy [157] сделано лишь предположение [187] о линейной струк-
туре [Си (CN)2]~. Согласно же рентгеноструктурным данным [184, 185],
в К [Си (CN)21 медь связана через углерод с двумя CN-лигандами и через
азот — с третьим. Координационное число 3 для меди наблюдается и в со-
единении К [Cu2 (CN)3] • Н2О [186].
Из металлов подгруппы меди лишь для золота выделена устойчивая ком-
плексная кислота HAu (CN)2, содержащая симметричные N—Н—N водо-
родные связи [188].
Комплексные цианиды, в которых степень окисления металла выше 1,
известны для Си (II) и Au (III).
При потенциометрическом и спектрофотометрическом исследовании сис-
темы Си2+ — С№ в метанольно-водных растворах (60; 80% СН3ОН) при
t = —45°, —70° С установлено существование [Си (CN)4]2— и оценена ус-
тойчивость комплекса (табл. 3.5) [189, 190]. Наличие этого комплекса пока-
зали также ЭПР- и электронные спектры водных, диметилформамидных и
ацетонитрильных растворов при температуре от —30 до —60° С [191].
Из водного раствора при взаимодействии К3 [Си (CN)4] с CuSO4 выделено
соединение Cu3 (CN)4 • 5Н2О, которое, по данным магнитных и ИК-спект-
ральных исследований, является смешанным цианидом меди (I) и (II) —
[Си11 (Н2О)4] [Си1 (CN)2]2 • Н2О [192].
Для золота (III) получено соединение К [Au (CN)4] • Н2О, которое при
нагревании выше 200° С превращается в К [Au (CN)2] [30, 73].
Данные ИК- и КР-спектров К [Au (CN)4] указывают на сильное о- и сла-
бое л-взаимодействие для связи металл — углерод, как и в Hg (CN)2 [193].
Ионообменным методом получена Н [Au (CN)4] • 2Н2О, содержащая ион
гидроксония [194].
3.6.1.3. Смешанные соединения металлов
подгруппы меди
Действием хлора, брома или иода, растворенных в метаноле, на водный
раствор К [Au (CN)2] выделены смешанные соединения К [Au (CN)2X2] (X =
= Cl, Br, I) [194]. Синтезированы также К [Au (CN)3C1] и К [Au (CN)3Brl,
в которых комплексные анионы, по данным И К- и спектров КР, имеют пло-
скую конфигурацию симметрии Сг¥ [194].
61
Из соединения К [Au (CN)2 С121 путем замещения хлора на азид-ионы
получен дицианодиазидоаурат (III) калия К lAu (CN)2(N3)2] [195].
Смешанные цианидные комплексы ацидотипа для меди долгое время не
были известны, хотя растворение CuCN в сильных галогенидных кислотах
связывали с образованием [Си (CN) Х3]3~ [73].
Спектры КР систем CuCN — NaCN — NaX — Н2О, CuCN — NaX —
— H2O, CuCN — HX — H2O показали существование лишь однород-
ных комплексов [Си (CN)2]~ и [Си (CN)3]2— [196], однако методом ЯМР уста-
новлено образование в небольших концентрациях ионов [Си (CN)3L](2+n)~
(L = СГ, Br~, Г, SCN~, и, tu, NH3) [180].
Таблица 3.3. Константы образования смешанных цианидных комплексов серебра
Комплексный ион Метод исследования /°, с Ионная сила 1g ₽ Литера- тура
[Ag(S2O3)2CN]4~ Потенциометрия 25 1 15,68 [176]
[Ag (S2O3)2 (CN)2J5— » » » 18,15 »
[Ag (S2O3) (CN)3]4— » » 21,28 »
[Ag en2CN] » » » 10,57 [201]
[Ag en2 (CN)2]~ » » » 14,14 »
[Agtu3CN] » » » 15,53 [202]
[Ag tv2(CN)2r » » » 18,83 »
[Ag (SO3)2 CN]4~ » » » 10,79 [200]
[Ag (SO3)2 (CN)2]5- » » » 13,96 »
[Ag (CN) OH]- Амперометрия 25 0 13,22 [97]
[Ag (CN) OH]- Потенциометрия 21 4,25 13,23 [177]
[Ag (CN) OH]” Спектрофотометрия, рН-потенциометрия 25 1 12,80 [178]
Наконец, было выделено соединение К3 [Си (CN)3SCN] • 1/2Н2О, в кото-
ром SCN-группа координирована атомом меди через серу [197]. Такая же
координация наблюдается в К [Ag (CN) (SCN)] и К2 [Ag (CN) (SCN)2] [198].
Потенциометрически установлено образование смешанного комплекса
[Au (SCN) (CN)]- в системе [Au (SCN)2]~ — CN~ — H2O [199].
В водных растворах Ag образует ряд смешанных цианидных комплексов,
устойчивость которых оценена потенциометрически (табл. 3.3).
Гидроксид серебра лучше растворим в водном растворе К [Ag (CN)2],
чем в воде, вследствие образования комплексного иона [Ag (CN) ОН]~ [203].
В то время как комплекс [Си (CN)4]2~ малоустойчив, из водных растворов
выделены соединения меди (II) состава (CuL)2CN (С1О4)3 и CuL' (CN)C1O4 •
• Н2О (L и L' = тетрадентатные циклические основания Шиффа)
[204], а также [CuPhen2CN] [CuPhen (CN)21 • 6Н2О, [CuPhen (CN)2] и
[CuPhen2(CN)]X-иН2О (X = СГ, ВГ, Г, NOT, п =1; СЮГ, п = 0,5) [250].
В водном растворе спектрофотометрически установлено образование ком-
плексного катиона [CuL'CN]+ с 1g р = 2,42 4- 2,95 при 22° С и р =0 [205].
В системе Си2+ — CN~ — Н2О спектрофотометрически обнаружен ком-
плекс [Си (CN)2NCCN]~. Присутствие в нем координированного через азот
62
дициана подтверждается ИК-спектральным и термографическим изучением
соли Na [Си (CN)2NCCN] - 4Н2О [206].
Помимо описанных выше, для металлов подгруппы меди известны разно-
образные смешанные цианидные соединения: MCN • L (М = Си, Ag, a L =
— NH3, N2H4, PrNH2, BuNH2) [1, 30, 207]. По рентгеноструктурным данным
[208, 209] CuCN • L состоят из полимерных слоев (CuCN)co с молекулами
NH3 и N2H4 между ними.
AgCN хорошо растворим в водном растворе NH3 с образованием
[Ag (NH3)21 [Ag (CN)23 [30], а из безводного этилендиамина можно получить
аналогичного типа соединение меди, только катионом при этом является
комплекс двухвалентной меди: [Cuen3] [Си (CN)3] [2101.
Обычно устойчивые к окислителям цианидные комплексы меди (I) в при-
сутствии NH3 или еп при мольном соотношении CN~ : Cu+ < 3 легко оки-
сляются кислородом воздуха с образованием Cu3 (NH3)4 (CN)4 и Cu3 (en)2 •
•(CN)4 [211]. Соединения Cu3 (NH3)n (CN)4 (n =2 ч-4) выделены также при
различном молярном соотношении CuSO4, NH3 и KCN в водном растворе
[213—215].
Изучение ИК-, электронных, спектров КР и рентгеноструктурные исследо-
вания показали, что образуются [Си (NH3)4] [Си2 (CN)4] и [Cuen2l [Cu2 (CN)4],
в которых разновалентные атомы меди связаны через азот мостиковой
CN-группы [211, 212, 216, 217].
По данным ЭПР и ИК-спектров мостиковая функция CN- и SeCN-лиган-
дов наблюдается и в соединениях Cu3en2 (SeCN)n (CN)4_П-Н2О (n =1; 2),
построенных из квадратно-пирамидальных катионов [Cuen2 • Н2О]2+ и по-
лимерных анионов [Cu2 (SeCN)„ (CN)4_ ,712^ [803].
Образование бесцветной соли К2 [Cu2 (CN)4 (NH3)2] наблюдалось при изу-
чении взаимодействия в системе К4 [W (ОН)4 (CN)4] — CuCl2 — NH3 —Н2О
[218]. Легче образуются смешанные соединения Си (I) с к. ч. 4типа CuL (CN)
(L = Dipy, Phen) и CuL2CN (L = PPh3, AsPh3), имеющие полимерную струк-
туру за счет мостиковой функции CN-лиганда [219].
К. ч. 4 проявляет и золото в соединениях К [Au (CN)2L] (L = Dipy,
Phen) 30].
С тиомочевиной выделены соединения [Cu2 (CN)2tul • 0,5 H2O [2201,
Agtu2CN [221], причем в последнем более прочная связь Ag—S, чем Си—S.
Большую группу координационных цианидных соединений Си, Ag,
Аи составляют смешанные фосфиноцианидные соединения [222]. Кроме
упомянутого выше [CuCN (PPh3)2] [219], для меди выделены соединения с
триалкилфосфитами Р (OR)3 типа 1 : 1 и 1 : 2 (R = Me, Et, Pr, i-Pr, Bu).
Для AgCN выделены из толуола комплексные соединения [AgCNL2]
и [AgCNL3] (L = Р (МеС6Н4)3), которые, по данным ЯМР (на 31Р), имеют
тригональную и тетраэдрическую структуру [223].
Соединение [Au (CN) PPh3] в этаноле при взаимодействии с избытком
фосфина переходит в [Au (PPh3)3] [Au (CN)2] [2221. Имеются сведения о по-
лучении [Au (CN) РМе3], [Au (CN)P (С1С6Н4)3] и кластерного соединения
Au3 (CN) (PPh3)2 [2221
3.6.2. Цианиды металлов III побочной подгруппы
3.6.2.1. Цианиды лантаноидов
Простые цианиды лантаноидов состава М (CN)3 (М = Се, Pr, Sm, Eu,
Но, Yb) и М (CN)2 (М = Sm, Eu, Yb) можно получить при взаимодействии
указанных металлов в жидком аммиаке при t = —63,5° С и ниже с Hg (CN)2
или с NH4CN [123, 124].
ИК- и спектры Мессбауэра цианидов М (CN)3 и М (CN)2 указали на зна-
чительную долю ковалентности связи М—CN, соответственно равную 37
или 26% для Eu (CN)2 и Eu (CN)3 1123], и, вероятно, мостиковый характер
CN-rpynn.
Из тетрагидрофурана по реакции между МВг3 • nThf (и = 3; 3,5 и 4)
и LiCN • Thf на холоду или при слабом нагревании выделены легко гидро-
лизующие соединения М (CN)3 • nThf (М = Y, Nd, Sm, Eu, Gd, Tb, Dy,
Ho, Er, Tu, Yb, Lu, n = 2), n = 2,5 (Pr), n = 3,25 (La) и 6,4 (Ce) [121, 122].
В водном растворе при pH == 7 4- 11 и р = 0,5 экстракционным методом
показано существование следующих комплексов гадолиния: [GdOHCN]+
и [Gd (OH)2CN] (при pH > 8), [Gd (ОН) (HCN)]2+ или [GdCNl2+ (pH около
8) и [Gd (HCN)]3+. устойчивость которых (1g К) соответственно равна 1,9;
—5,7; —13,9; —22,9 [224].
3.6.2.2. Цианиды актиноидов
Из актиноидов комплексные цианиды известны лишь для урана. Еще
в начале нашего столетия действием KCN на ацетат уранила получено рас-
творимое в воде соединение К2IUO2 (CN)4] и определен ионный вес комплекса
[225], однако более поздние исследования подвергли сомнению его существо-
вание [30, 226].
При взаимодействии UX4 (X = Cl, Br, I) c NaCN в жидком аммиаке
выделены соединения UX3 (CN) • 4NH3. Из UC13 (CN) • 4NH3 затем полу-
чен UC13CN. В жидком циановодороде образуется координационное соедине-
ние вероятного состава [Et4N]2 [UC14 (CN)2] [2271.
Имеются сведения о получении ThCN при обработке порошкообразного
карбида тория азотом при 1800—1850° С [125]. Исходя из рентгенограмм и
нейтронограмм порошка, авторы считают это соединение моноцианидом
тория, однако его можно принять и за твердый раствор карбида и нитрида
тория.
3.6.3. Цианидные комплексы металлов
IV побочной подгруппы
Простые цианиды металлов подгруппы титана неизвестны. Из жидкого
аммиака по реакции TiBr3 с избытком KCN получен темно-зеленый
К5 [Ti (CN)8] (р — 1,74 М. Б.) вероятного строения К3 [Ti (CN)6] • 2KCN
[228], что вызвало дискуссию [229]. По новым данным предложена формула
К4 [Ti (CN)J • KCN [231].
Описанный ранее синтез К3 [Ti (CN)6] из водных растворов [230] не был
воспроизведен [231].
При взаимодействии TiBr31 KCN и К в жидком аммиаке выделено также
64
соединение К4 [Ti (CN)J [231]. Для циркония в жидком аммиаке получены
комплексы M5Zr (CN)5 (М = К, Rb) [838]. Цианидные соединения четырех-
валентных Ti, Zr, Hf пока еще не известны.
3.6.4. Цианиды металлов V побочной подгруппы
В этой подгруппе простой цианид и наиболее многочисленные комплексы
с цианид-лигандом образует ванадий.
Методами потенциометрического и калориметрического титрования
установлено образование VO (CN)2 в системе VOSO4 — KCN — Н2О [126].
Соединение К4 [V (CN)6] • ЗН2О, легко окисляющееся на воздухе до
К3 [VO (CN)5], выделено из водных растворов [232, 233], например, по реак-
ции (pH = 9,5)
V (ОН)2 + 4KCN + 2HCN + Н2О К4 [V (CN)6] • ЗН2О. (3.36)
Теплота образования комплекса [V (CN)6]4~ в водном растворе по кало-
риметрическим данным составляет —196,9 кДж/моль [238].
Для безводного К4 [V (CN)6] [232] установлена кристаллическая струк-
тура [234] и определен магнитный момент 3,78 М. Б. [235] и 3,5 М. Б. [236,
237].
Среди цианованадатов наиболее полно исследован гептацианованадат
(III) калия К4 [V (CN)7] • 2Н2О. Вначале это соединение, полученное еще в
1898 г., было охарактеризовано как К3 [V (CN)6] [1,239,240], и лишь в 1966 г.
Чедвик и Шарп [30] установили его правильный состав.
К4 [V (CN)7] • 2Н2О получают действием H2S на щелочной раствор
NH4VO3 и KCN [259] или по реакции V2 (SO4)3 с KCN [241]. Наряду с моно-
гидратом [235] описан и безводный K4[V(CN)7], выделенный из метанола
[236]. Из жидкого аммиака по реакции ацетата ванадия (III) с NaCN полу-
чен Na3 [V (CN)6] [236].
Рентгеноструктурное [242, 243] и ИК-спектральное [236, 243, 246] ис-
следования К4 IV (CN)7] • 2Н2О в твердом состоянии и в растворе указали
на пентагонально-бипирамидальную структуру комплекса (симметрия Dsh).
На основании этого полосы 20 600, 22 800 и 28 600 см—1 в электронных спек-
трах отражения отнесены к соответствующим d — d-переходам [244, 245].
Как показали ЭПР-спектры, комплекс [V (CN)7I4~ в разбавленных рас-
творах диспропорционирует на [V (CN)6]4- и [VO (CN)5]3~ [246, 804].
На воздухе К4 [V (CN)7] • 2Н2О окисляется с образованием соли
К3 [VO (CN)5], которую можно получить по обменной реакции между вана-
дилацетатом и KCN в водном растворе и жидком NH3 [235]. Были исследова-
ны ИК- и электронные спектры солей Kat3 [VO (CN)5] (Kat = Cs+, Me4N+,
ЕЦИ4-) [247]. Обсуждается вопрос о соотношении о- и л-компонент в связи
металл — углерод для комплексов [V (CN)6]4~, [V (CN)7]4~ и [VO (CN)5]3~~
[248, 249].
По данным ИК- и электронных спектров, анион [VO (CN)5]3~ имеет сим-
метрию Ок,, которая подтверждена рентгеноструктурными исследования-
ми [747].
Известны цианиды ванадия и с более низкими степенями окисления.
Восстановлением К4 [V (CN)7] раствором калия в жидком NH3 получены
К5 [V (CN)eJ (р = 2,85 М. Б.) [236] и К7 [V (CN)7] (р = 3,2 М. Б.) [235].
Последнее соединение, возможно, имеет состав К6 (V (CN)6] • KCN [235]
3 2473
65
и значительно отличается по свойствам от диамагнитного К5 [V (CN)5], полу-
ченного при восстановлении К3 [V (CN)5NO] водородом при 200° С [258].
Из щелочного раствора в результате восстановления KVO3 гидроксил-
амином при избытке KCN выделен К5 [V (CN)5NO1 • Н2О [251], однако после
тщательного препаративного и структурного анализа показано, что состав
пентацианонитрозилванадата отвечает формуле К3 [V (CN)5NO1 • 2Н2О
[252, 254—2561.
Хотя [V (CN)5NO]3~ можно формально рассматривать как комплекс ва-
надия (I) с лигандом NO н, следует учитывать и увеличение электронной
плотности на NO-лиганде, вызванное — ря*-переходом с d-орбиталей ме-
талла на антисвязывающие л-орбитали NO. Подобный перенос заряда ха-
рактерен для других членов гомологического ряда [М (CN)5NO]"~ (М = Сг,
Мп, Fe, Со) [257, 805].
Действием гидроксиламина и H2S на щелочной раствор NH4VO3 и KCN мо-
жно получить цианонитрозилванадат (I) калия состава К4 [V (CN)6NO] -Н2О,
диамагнетизм которого связывают с возможным повышением к. ч. ванадия
до 7 или 8 [259].
Сообщалось [235] о получении соединения Кз.гУ (CN)s,5 (ОН)0>5, в кото-
ром, вероятно, ванадий содержится в различных степенях окисления
[K4V (CN)6] [K3VO (CN)5] • 0,25H9O. Соединение такого типа известно и для
марганца 1,2 [K4Mn (CN)6J • 0,7^ [K3Mn (CN)61 • 0,25Н2О [2601. Подобные
соединения иногда называют веществами «второго класса» [261].
Для ниобия (V) известны лишь смешанные координационные цианиды.
При взаимодействии NbCl5 с HCN в эфире образуется соединение
NbCl4CN • Et2O [127].
Выделены также кислоты Н [NbCl5CNl, Н [NbBr5CN] и соль
[Et3NH] [NbCl5CN] [127,806]. При действии KCN на продукт электроли-
тического восстановления NbCl5 в метаноле получены устойчивая соль
К4 [Nb (CN)81 • 2Н2О, изоморфная с К4 [Mo (CN)8] • 2Н2О [262], а также
Kat4 [Nb (CN)]8- /гН2О (Kat = №+, п = 4; Cs+, п = 2; Tl+, Pr2NHf) [807].
При восстановлении калиевой соли амальгамой калия выделена соль
К5 [Nb (CN)g] [262, 807].
ЭПР-спектры указывают на додекаэдрическую структуру (D2ci) аниона
[Nb (CN)814~ в твердом состоянии и антипризматическую (D4d) в водном рас-
творе [262, 8081.
Из NbCl4 и KCN в ацетонитриле синтезировано октаэдрическое соедине-
ние [NbCl3 (CN) (CH3CN)2] [263].
3.6.5. Цианиды металлов V! побочной подгруппа
Среди многочисленных координационных соединений на основе циани-
дов Сг, Mo, W наиболее стабильны производные Сг (III), Mo (IV), W (IV) и
W(V).
Один из методов получения Кг [Сг (CN)6] сводится к действию избытка
KCN на ацетат хрома (III) в водном растворе с последующим высаливанием
его этанолом [1, 73]. Октаэдрический анион [Сг (CN)6]3~ довольно устойчив
в водных растворах [239, 266, 2691, и лишь нагревание со щелочами, кипя-
чение, с разбавленными сильными кислотами, хлорной и бромной водой вы-
зывают его разложение [30]. Под действием света и в кислых растворах на-
блюдается замещение CN-лигандов в [Сг (CN)6]3~~ молекулами Н2О [265,
268—270].
Известны многочисленные гексацианохроматы (III) щелочных металлов,
66
NHf, Tl+, Ag+, Bu4N+, Ph4N+, Ph4As+, Zn2+, Cd2+, Pb2+, Mn2+, Fe2+, Co2+.
Ni2+ [1, 273], а также соли с комплексным катионом [Сг L6]3+ (L = NH3,
и, atp), [CrL3]3+ (L = en, Pn) [1, 274], [CrPy2 (H2O)4]3+ и др. [1]. K3 [Cr (CN)61
имеет орторомбическую структуру [275], а соли Kat3 [Сг (CN)6]2- 6Н2О
(Kat = Mn2+, Fe2+, Со2+, Ni2+, Cu2+, Zn2+, Cd2+) [276] образуют кубиче-
ские решетки и изоструктурны с соответствующими гексацианосоединениями
мангана, железа, кобальта, родия и церия (III) [2771.
В водном растворе получена кислота Н3 [Cr (CN)61 [73].
Для трехвалентного молибдена известны координационные соединения
Kat4 [Mo (CN)7] • nH2O (Kat = К+, Cs+), легко окисляющиеся в соедине-
ния Mo (IV) [30]. Установлено, что в комплексе [Mo (CN)7]4~ как в твердом
состоянии, так и в растворе к. ч. молибдена равно 7 [279], хотя ранее пред-
полагалось существование в растворе додекаэдрического [Mo (CN)7H2O14~
[142, 278].
При взаимодействии расплавленного К4 [Mo (CN)8] с KCN получена соль
К2 [Mo (CN)5] [280], которая в неводных средах частично окисляется с обра-
зованием К3 [Мо2 (CN)10] [281].
Имеются данные о получении Кз IM (CN)6] (М = Mo, W) при обработке
метанолом солей К4 IM (CN)6] [282] и К2 IW (CN)5H2O] [281].
Для трехвалентных металлов рассматриваемой подгруппы получены и
смешанные координационные соединения: цис- и транс-изомеры Kat3 [Сг -
• (CN)„ (NCS)6_п] [283] и Kat3 [Сг (CN)„ (NCSe)6_„] [284] (Kat = К+, Bu4N+,
п = 1 ~ 5), К [Сг (CN) (NCS)3 en], [Cr (CN) (NCS) en2] SCN [264],
K2 [Cr (CN)4L], K2 [Cr (CN)2L2] (L == глицин, гистидин, аспарагиновая кис-
лота) [271], К3 [MoS (CN)4] • 2Н2О [30, 285] и К6 [Мо2О2 (NO)2 (CN)l0] 1254].
В отличие от молибдена и вольфрама четырехвалентное состояние для
хрома не характерно. Можно отметить лишь соединение К3 [Cr (О2)2 (CN)3i
со структурой искаженной пентагональной бипирамиды [286] и выделенные
еще в начале века К5 [Cr2 (О2)4 (CN)5] • 5Н2О и К2 [Cr (О2)2 (CN)2NH3] [2541.
Из цианосоединений молибдена (IV) весьма устойчива диамагнитная соль
К [Mo (CN)5] [287], но наиболее характерны для молибдена и вольфрама изо-
структурные диамагнитные соли К4 LM (CN)8] • 2Н2О, которые можно по-
лучить из соединений Mo (III), Mo (IV), Mo (V), а также из Na2WO4,
Кз [W2C19], (NH4)2 [WOC15] [30, 288—291]. Выделено много солей типа
Kat4 [М (CN)8] для щелочных металлов, Ag, TI; Kat2 [М (CN)81 для Mg, Sr,
Zn, Cd, Мп [1, 290]; Kat2 [Mo (CN)8] (Kat = [Cu (NH3)4]2+, [Cuen2]2+) и
Kat2 [W (CN)8] (Kat = [Cd (NH3)2]2+, [Zn (NH3)2]2+) [1]. В водном растворе
и в твердом состоянии получены и кислоты Н4 [М (CN)8] (М = Mo, W)
[73, 292].
Октацианомолибдат-ион обычно образует с ионами переходных металлов
нерастворимые в воде соединения. Однако в ряде случаев образуются рас-
творимые комплексы [293—297, 299, 304].
Спектрофотометрически в растворе выявлены комплексы [CrMo (CN)8]~
[293, 294]; [FeMo (CN)8]~ [295]; [UO2Mo (CN)8]2~ [296], устойчивость которо-
го при 30° С характеризуется 1g ₽ = 2,97 [299], а амперометрически, кондук-
тометрически и потенциометрически — комплексы [ММо (CN)8]2~ (М = Си,
Со, Ni) [297]. Из растворов выделено соединение K2Cr [Mo (CN)81, содержа-
щее, по-видимому, Сг2+-ионы [298], и соль (UO2)2Mo (CN)8 • пН2О (и = 6 -г-
8). Для последней предполагается полимерная структура [UO2 (Н2О)в]и •
• IUO2 (Н2О)2 Mo (CN)8ln с мостиковой связью Мо—C=N—UO2 [300].
В водных растворах кондуктометрически установлено образование
3* 67
соединений M2Fe [Mo (CN)8](M=Li+, Na+, K+, Rb+, Cs+, NH^), из которых
соли K+, Rb+, Cs+, NH^ нерастворимы и выделены в виде дигидратов (для
калия — тригидрат) [301]. Получены также соли MFe [Mo (CN)8] • лН2О
(M=NH^, Rb+, Cs+ (и =2), К+ (и =4) [3021), которые имеют кубиче-
скую структуру [303] и, по данным ПК-спектров, как и соединения Fe (II)
[301], содержат мастиковую связь Мо—CN->Fe [302]. Гетерополиядерные
комплексы образуются в водных растворах и по реакции Hg (CN)2 с
[Mo (CN),!4-, (Mo (CN),]3-, [Fe (CN)el3~, [М (CN)el4- (М = Fe, Ru) [3041.
Согласно теории поля лигандов, [М (CN)814~ должен иметь структуру анти-
призмы (симметрия D4d) или додекаэдра (D2d) [305, 306]. Рентгенографически
для кристаллического К4 [Mo (CN)8] • 2Н2О [307], которому изоморфен
К4 IW (CN)8] • 2Н2О [308], установлена додекаэдрическая структура, а для
Н4 [W (CN)8] • 6Н2О — конфигурация тетрагональной антипризмы (сим-
метрия D4d) [309].
Додекаэдрическая структура [М (CN)8]4~ подтверждена ИК-, спектрами
КР и Мессбауэра соответствующих солей в кристаллическом виде [310—
313, 317, 318], в то время как об их структуре в водных растворах отсутству-
ет единое мнение. По данным ПК- и спектров КР, антипризматическая сим-
метрия [М (CN)8] ’ более вероятна, чем додекаэдрическая [312—317], однако
ЯМР-исследования [Mo (CN)/" [319] и электронные спектры поглощения
солей типа К4 [М (CN)8] (М = Mo, W) [278, 322] не дают однозначного ответа
на этот вопрос [320, 3211.
Результаты изучения спектров КР, ИК- [323] и спектров магнитного
кругового дихроизма [324], полученные в последнее время для К4 [М (CN)8] -
• 2НО (М=Мо, W), показывают, что додекаэдрическая структура сохра-
няется и в водных растворах.
Ионы [Mo (CN)8]4 и [W (CN)8]4~ в водных растворах фотохимически ак-
тивны. Длительное воздействие излучения с X = 370 нм приводит к частич-
ному выходу CN-лигандов из координационной сферы и к образованию ком-
плексов [Мо (ОН) (CN)4]3~ [325—329].
Ранее полагали [330—332], что конечными продуктами гидролиза яв-
ляются комплексы красные [М (ОН)4 (CN)4]4~ и синие [М (ОН)3Н2О (CN)4]3~.
Частичное рентгеноструктурное исследование К4 IM (ОН)4 (CN)J и ИК-спек-
тральное исследование К3 [М (ОН)3 (CN)4 (Н2О)] показало, что эти соедине-
ния имеют состав К4 [МоО2 (CN)4] • 6Н2О и К3 [МоО (ОН) (CN)4] • 6Н2О
[333, 334], т. е. содержат шестикоординированный Мо (IV). Правиль-
ность этого вывода подтверждена полным рентгеноструктурным исследова-
нием соли NaK3 [МоО2 (CN)J • 6Н2О [335].
Такое же строение имеют и соединения Kat4 [МО2 (CN)4] • nH2O (Kat =
= Na+, К+, п =0 4- 12), К3 [МоО (ОН) (CN)41 [338, 339], выделенные по
реакции между МО (ОН)3 и KCN в щелочном растворе с последующим вве-
дением твердого КОН. Полученные ранее по этой схеме соединения рассмат-
ривали как соли с анионами [М (ОН)4 (CN)4]4~ (М = Мо, W) [340—342].
Из облученных растворов выделены и другие кристаллогидраты:
К3 [МоО (ОН) (CN)4] • 2Н2О и К4 [МоО2 (CN)4] • 6Н2О [336, 337].
Наличие восьмикоординационной конфигурации Мо (IV) предполагается
в соединениях Kat [Мо (CN)4 (ОН)3Н2О] (Kat = [KUO213+, IKVO]3+ [343,
344], Сг3+, Fe3+, [KMn]3+ [345]) и солях калия, серебра и меди (II) с анио-
ном [Мо (CN)2 (ОН)6]4~ [346]. Однако без полного рентгеноструктурного ис-
следования такое утверждение нельзя считать достоверным.
G8
Известны и другие оксо- и гидроксоцианиды Мо и W, например
К2 [Mo (ОН)2 (CN)41, Мо (ОН)2 (CN)2 • Н2О [338], W (ОН)2 (CN)2 • Н2О [342],
и К6 [Мо3 (CN)8O6] • 2Н2О [338].
Относительно природы красных промежуточных продуктов фотолиза
отсутствует единое мнение. Ряд авторов полагают, что образуются десяти-
координационные комплексы [M(CN)8(H2O)2]4~, тем более, что из растворов в
аммиаке или гидразине удалось выделить смешанные соединения
Kat2 [М (CN)8L21 • мН2О (Kat = Cd2+, Mn2+; М = Мо, W; L = NH3, N2H4)
[326—328]. Другие [337, 347—349] предлагают восьмикоординационную
структуру образующихся комплексов [М (CN)7 (Н2О)13~ или [М (CN)7 (ОН)]4~,
для которых выделены в твердом виде соли К4 [Мо (CN)7OH] [337] и
Ag3[Mo (CN)7H2O1 [349].
При фотолизе (NH4)4 [Мо (CN)8] в жидком аммиаке образуется ряд про-
межуточных комплексов [Мо (CN)8—п (NH3)n]"~ , имеющих различные спек-
тральные характеристики. Выделен продукт фотолиза [Мо (CN)4 (NH3)21
[350].
Известны также смешанные серосодержащие цианиды Мо (IV): К5 [MoS-
• (CN)7] • Н2О [3511, кислота Н3 [MoS (ОН) (CN)4 (Н2О)2], при разложении
которой образуется MoS (CN)2 [352], ее соли с катионами: К+ [285], Сн2+,
Ag+, Ag2K3+, Zn2+, Cd2+, Hg2+, Pb2+, Mn2+, Fe2+, Co2+, Ni2+ [353], а также
соединение Кб LMo2S (CN)12] • 4H2O [354].
Действием алкилгалогенидов на Ag4 [M (CN)81 выделены диамагнитные
изоцианидные комплексы цианидов Мо (IV) и W (IV) состава [М (CN)4 •
• (RNC)J (R Me, Et, Pr, i-Рг, ABu, Ph — CH2) [355, 356].
Сильные окислители (МпОГ, Ce4+) превращают ион [М (CN)8]4— в
[М (CN)8]3- (М — Мо, W) [30], причем вольфрам окисляется легче, чем мо-
либден. Известны соли Kat5 [М (CN)8] (Kat = щелочные и щелочноземельные
металлы, Ag+ [11, Bu4N+ [357]) и соответствующие кислоты Н3 [М (CN)8]
• пН2О (п = 3 (Мо), 6 (W) [73, 2921).
Действие излучения (X = 370 нм) на цианидные соединения Мо (V) и
W (V) в твердом состоянии и в растворе приводит к восстановлению металла
до степени окисления 4 [325].
Парамагнитные ионы [М (CN)8]3— могут иметь антипризматическую или
додекаэдрическую структуру в растворе и в твердом состоянии. ЭПР-спектры
К3 [Мо (CN)8] и К3 (W (CN)8] указывают на конфигурацию антипризмы для
ионов [М (CN)8]3- в растворе [358, 359]. Такое же строение имеют эти комп-
лексы в кристаллических изоструктурных солях натрия [360]. Однако
рентгеноструктурное исследование [Bu4N]3 [Мо (CN)8] и ЭПР-спектры этого
соединения в твердом состоянии и в растворе свидетельствуют о додекаэд-
рической структуре аниона [357]. Его геометрия, как показали ЭПР-спектры
Kat3 [W (CN)8] • иН2О (Kat = К+, Na+, Ag+, Bu4N+), в твердом состоянии
или в растворе (Н2О, МеОН, BuOH, MeCN) зависит от природы катиона и рас-
творителя [324].
Для металлов подгруппы хрома выделены цианидные комплексы одно-
родного и смешанного типа со степенью окисления металла 4-2, 4~1,0. Так,
ацетат хрома (II) с KCN в инертной атмосфере образует К4 [Сг (CN)6] - ЗН2О
(р= 3,2 М. Б.) [30], а ацетат молибдена (II)—диамагнитную соль К5 [Мо •
• (CN)7], легко окисляющуюся в К4 iMo (CN)7] [253].
Восстановление водородом при 200—400° С цианидных соединений Сг (III),
Мо (IV), W (IV) приводит к образованию К3 (Сг (CN)4] [258], К4 IM (CN)6]
(М '= Мо, W) [361], К4 [Мо (CN)J и К4 [МоО (CN)J [272, 362], а восстанов-
69
ление К3 [Сг (CN)6I калием в жидком аммиаке — К61Сг (CN)6], который обра-
зует с СО соединение К3 [Сг (СО)3 (CN)31 [363].
Известны и другие карбонилцианиды хрома, молибдена и вольфрама:
[Сг (CN) (СО)6] [364], Na2 [М (СО)4 (CN)21 (М = Сг [365], Mo, W [3661),
К3 [Мо (СО)3 (CN)3], К4 [М (CN)4 (СО)2] (М = Сг [367], W, Мо [3681),
Na [М (CO)5CN1 • иН2О (М = Сг [365], W; п = 1 [366]), [М2 (СО)10 (CN)]“
(Сг, W) [369], [LM (CO)3ICN] (М = Мо, W; L = Dipy, Phen) [370]. Все со-
единения бесцветны и диамагнитны.
Для Сг, Мо, W получены цианонитрозильные соединения. При вос-
становлении СгО3 гидроксиламином в присутствии KCN образуется
К3 [Cr (CN)5NO1 • Н2О (р = 1,9 М. Б. и v (NO+) = 1645 см-1) [371—373],
который в кислых водных растворах гидролизует с образованием комплексов
[Сг (NO) (CN)n (Н2О)5_п]2~п [254]. Электролитическим восстановлением
Кз [Сг (CN)5NO1 получен К4 [Сг (CN)5NO] -2Н2О (v (NO)+ = 1515 см-1) [374].
Аналогичному цианонитрозилмолибдату долгое время приписывали раз-
личный состав [371, 375, 376], и только рентгеноструктурное исследование
подтвердило правильность формулы К4 [Мо (CN)5NO1 [377].
Хотя NO-группа в исследованных соединениях рассматривается как
носитель положительного заряда, (1Я — ря*-взаимодействие М -> NO при-
водит к появлению на ней эффективного отрицательного заряда, соответст-
венно, —0,24 эВ и —0,34 эВ в соединениях К3 [Сг (CN)5NO] Н2О и
К4 [Мо (CN)5NO] [257, 805].
3.6.6. Цианиды металлов VII побочной подгруппы
Простой цианид марганца не выделен. В жидком аммиаке получен про-
дукт Hg (CN)2Mn (CN)2 • 2...3NH3 [30], а в водных растворах — осадок ве-
роятного состава KMn (CN)3 или K2Mn [Мп (CN)J [73].
Для Мп, Тс, Re, как и для подгруппы хрома, наблюдается рост устой-
чивости цианидных комплексов металлов в высоких степенях окисления с
увеличением атомного номера.
По реакции между МпСО3 или Мп (СН3СОО)2 с избытком KCN в инертной
атмосфере получен гексацианоманганат (II) калия К4 [Мп (CN)6] • ЗН2О
[30] (для которого известна соответствующая кислота [73]), а при недостатке
KCN — соль KMn (CN)3 (р = 4,53 М. Б., v (CN) = 2062 см-1) [30, 3781.
Для последнего соединения предполагают структуру типа берлинской ла-
зури К2Мп [Мп (CN)6] [379].
По данным ЭПР-спектров, в водных и смешанных (МеОН—Н2О, CH3CN—
Н2О) растворах К4 [Мп (CN)6] образуются комплексы [(CN)5Mn—C==N—
Мп (CN)5]7~ с мостиковой CN-группой [380]. Такая же функция CN-лиганда
обнаруживается в соединениях Мп [М (CN)6] 8Н2О (М = Fe, Ru, Os) и
Мп3 [М (CN)612 пН2О (М = Сг, Со) [254].
Исследованы колебательные (ИК-, КР-), электронные и ПМР-спектры
К4 [Мп (CN)eJ в твердом состоянии и в растворе [254].
Для рения (II) известны комплексы [Re (CN)5H2O]3~ и [Re (CN)G14 \
Восстановлением перрената натрия амальгамой Na при избытке NaCN по-
лучена соль Na3 [Re (CN)5H2O], а из нее соединения Katn [Re (CN)5H2O]
(Kat = H+, Ag+, Cu+, (T12H)3+, Co2+, [Co (NH3)6]3+) и K3 [Re (CN)5] [381,
382]. Кипячение Kat3 [Re (CN)5H2O] с избытком KCN приводит к образова-
нию Kat4 [Re (CN)6] • nH2O (Kat = K+ (n = 3), Na+ (n = 5)), из которых
получены соли Со2+ и Си2+ [383]. В водном растворе гексацианоренаты (II)
70
Рис. 3.1. Структура аниона комплексной соли
К7 [Мп2О (CN)10] CN [395]
калия и натрия гидролизуют с образованием [Re (CN)5H2O]3 . Из последне-
го получен ряд цианидных комплексов смешанного типа Nan [Re (CN)5L]
(L = NH3, O2~ [384], SOi~, CO, NO, NO? [254]), Katn [Re (CN)5SCN] (Kat =
= K+, Ag+, Cu2+) [385], Kat„ [Re (CN)4L] (Kat= Na+, L = en [386], Phen,
Dipy [254]; Kat = K+, L = HTart (H2Tart — винная кислота) [387]).
В результате взаимодействия Ag4[Re (CN)J с избытком Mel в метаноле или
эфире получено соединение [Re (CN)2 (MeNC)J в виде двух изомеров [388].
Для марганца (II) также известны цианиды смешанного типа: парамаг-
нитный [Мп Phen2 (CN)21 (р = 5,95 М. Б.) [389] и пентацианонитрозилман-
ганат К2 [Мп (CN)5NO], полученный окислением Кз [Мп (CN)5NO] азотной
кислотой или хлором и ис-
следованный спектроско-
пическими методами [254].
Окисление К4 [Мп (CN)6]
кислородом воздуха, вза-
имодействие МпРО4 с KCN
илиМпСОо с Н2О2 и KCN
[254] приводит к образова-
нию Кз [Мп (CN)6], изо-
морфного с Кз [Fe (CN)6]
[390, 391].
Сопоставление резуль-
татов исследования элек-
тронных и ИК-спектров с
параметрами кристалличе-
ской решетки для соедине-
ний Кз [М (CN)61 указало
на падение прочности свя-
зи М—С в ряду Fe >• Со >
>Мп>Сг [254].
Октаэдрический [Мп (CN)6]3~ (р = 3,50 М. Б.) относится к числу низко-
спиновых комплексов [392].
В противоположность более ранним данным [393, 394] рентгеноструктур-
ное исследование показало, что конечным продуктом взаимодействия КМпО4
и KCN в воде является соединение К7 f(NC)5MnOMn (CN)51 CN [395] (рис. 3.1).
К3 [Мп (CN)6] медленно гидролизует в водных растворах с образованием
Кз [Мп (CN)5OH1 (р= 2,90 М. Б.) [396], а затем и комплекса [Мп (CN)2(OH) •
• (Н2О)3] [254].
Использование NOC1 как окислителя позволило получить из неводных
сред соединения KMn/,z [Ain" (CN)6], К2 [Мп (CN)6] [281] и К2 [Мп (CN)6] •
• 3DMFA [398].
Для Re (III) известны комплексные цианиды как однородного, так и
смешанного типа. При действии KCN на K2ReCl6 в расплавленном KSeCN
при 250° С получен К4 [Re (CN)7] • 2Н2О [253], а в водном растворе —
Кз [Re (CN)61 • ЗН2О, образующий кубическую решетку с а = 12,19 А
[397]. Такое же октаэдрическое окружение имеет Re (III) и в соединениях
[Со (NH3)6] [Re (CN)6] [399] и К3 [Re (CN)3 (ОН)3] [400].
Из неводных растворов выделены кластеры типа [Et4N]3 [Re3Cl9 (CN)31
и [Et4N]3 [Re3Cl3 (CN)J [401].
Окисление K3 [Re (CN)5H2O1 бромом или H2O2 привело к образованию
ионов [Re (CN)5H2O]2“. Из таких растворов получено соединение типа нит-
ропруссида Na2 [Re (CN)5NO1 [73].
71
Выделены также гептацианонитрозилренаты состава Ag3 [Re (CN)7NO]
[3991 и К3 [Re (CN)7NO] • 4Н2О [809].
Цианидные производные Мп, Тс, Re со степенью окисления металла +4
мало характерны. Выше упоминалось соединение Мп (IV) — К2 [Мп (CN)6]
[281]; для Re (IV) и Тс (IV) известны лишь гидроксо- и оксоцианиды типа
К3 [ReO (CN)4OHI, К4 [ReO2 (CN)J, Т13 [Тс (ОН)3 (CN)J (или Т13 [ТсО (ОН)
• (CN)J) [30]. Соединение К4 [ReO2 (CN)4], по-видимому, правильнее счи-
тать производным рения (III) и (V), а именно К6 [Re2O3 (CN)8] • 4Н2О [254].
Наиболее распространены цианидные производные рения (V). Из K2[ReI6]
[30] и К2 [ReCl6] [402, 403] можно получить К3 [Re (CN)81, для которого, как
и для продукта гидролиза К3 [ReO2 (CN)4], предполагалась додекаэдрическая
симметрия аниона в водном растворе [404, 405].
Позднее в К3 [ReO2 (CN)4] установлено октаэдрическое окружение Re
(V) как в твердом состоянии [406], так и в водном растворе [407]. Такое же
координационное число имеет Re и в соединениях Kat [Re (CN)6] • nH2O
(Kat = Ph4As+ (ti = 1) и Bu4N+ (n = 0) [408]).
При подкислении растворов [ReO2 (CN)4]3~ превращается в [ReO (ОН) •
• (CN)4]2M для которого известны соли с катионами Ва2+, Ph4As+ и
[DipyH2]2+ [4041, а затем в ион [Re2O3 (CN)8]4 с мостиковым атомом кисло-
рода между мономерными частицами [ReO (CN)4]“ [409, 410]. Исследование
структуры и ИК-спектров соли [Pt (NH3)4]2 [Re2O3 (CN)8] показало присутствие
линейного звена О = Re — О — Re = О [254]. Действием разбавленной
НС1 на К3 [ReO2 (CN)4] выделено кристаллическое вещество состава ReO-
- (CN)3 • 2Н2О, водный раствор которого является равновесной смесью двух
кислот Н [ReO (ОН) (Н2О) (CN)3] и Н2 [ReO (ОН)2 (CN)3L Для обеих кислот
получены соли Na, К, Rb [411]. Известны и другие оксопроизводные:
[ReO2 (CN)6Fe]3“, [Re2O3 (CN)4], H2 [(H2O) О (NC)3ReORe (CN)3O (Н2О)1
[254], Kat [ReOF (H2O) (CN)3ln (Kat = H+, Na+, K+, NHt, Tl+, Ni2+,
[Co (NH3)6]3+, [Coen3]3+; n = 1 4- 3) [412].
Цианонитридоренаты составляют другой тип смешанных соединений ре-
ния (V): К2 [ReN (CN)4]-H2O, К3 [ReN (CN)61 [404, 413, 414], [FeReN (CN)4 -
• Н2О]. Согласно ИК-спектрам, в последнем имеются CN-мостики между Re
и Fe [414], а в К2 [ReN (CN)4] • Н2О показано существование октаэдрическо-
го аниона транс-tReN (CN)4H2O12~ в водном растворе и бесконечных цепей
—N—Re—N—Re— в твердом состоянии [413]. По рентгенографическим дан-
ным [415] в К2 [ReN (CN)J • Н2О CN-группы координированы атомом рения
через азот, что вызывает сомнение [414].
При окислении кислых растворов К3 [Re (CN)8] кислородом воздуха об-
разуются цианиды рения (VI) состава Katm [Re (CN)8]n (Kat ~ [Co (NH3)6]3+,
Ph4As+) [30, 4041.
Для металлов рассматриваемой подгруппы выделены цианиды со степе-
нью окисления металла +1: изоструктурные К5 (М (CN)6] (М = Мп, Тс,
Re) [254], смешанные соединения состава К2 [М (СО)3 (CN)3], К3 [М (СО)2
• (CN)4] (Мп, Re) [254, 422, 423], Na3 [Re (NO)(CN)5J, К [Re (CN)2(CO)4],
К [Re (CN)2 (CO)3 NH3], [Re (CN) (CO)3 (NH3)21, H [Re (CN)2 Dipy2],
Na3 [Re (CN)4 Phen] • H2O [254], Mn (CN) (CO)6 [424] и Na [M (CO)4 (CN)X1
(M =Mn, Re; X = Cl, Br, I) [425].
Моноклинный K3 [Mn (CN)5 (NO)] • 2H2O, полученный действием гидро-
ксиламина на К4 [Мп (CN)6] или К3 [Мп (CN)6] [416, 417], формально считает-
ся комплексом Мп (I) с ПО+-лигандом, на что указывают диамагнетизм сое-
динения, положение v (NO) (1730 см-1) и приблизительная линейность фраг-
72
мента М — NO [256]. Сравнение экспериментальных данных: длина связи
NO (1,21 A), v (NO) (1700 см-1) и энергия N1S(NO) (401 эВ) для закомплексо-
ванного NO с соответствующими данными для свободного NO+-лиганда ука-
зывают на ярковыраженный перенос заряда металл -> NO, и благодаря зт-ак-
цепторному характеру NO-группы точное определение степени окисления
металла и NO-группы в К3 iMn (CN)5NO1 • 2Н2О и в других цианонитрозиль-
ных соединениях вряд ли допустимо [30, 73, 142, 254, 257, 418—421,805, 810].
Из жидкого аммиака выделены цианиды марганца со степенью окисления
меньше нуля: цис- и транс-К4 [Мп (NO)2 (CN)2]2 и К3 [Мп (CN)2 (NO)21 [422].
3.6.7. Цианиды семейства железа
3.6.7.1. Простые цианиды
Рассматриваемые здесь бинарные соединения М (CN)n (М = Fe, Со, Ni)
часто содержат неэквивалентные атомы металла и имеют комплексную при-
роду, являясь координационными полимерами.
Нерастворимый Fe (CN)2 получен при взаимодействии FeCl2 с Са (CN)2
в водном растворе [П или при нагревании (NH4)4 [Fe (CN)6] под вакуумом
[73]. О его строении чока нет определенных данных.
Некоторые соединения, имеющие суммарную формулу Fe (CN)3, пред-
ставляют собой коричневый дигидрат Fe [Fe (CN)61 [30].
Безводный Со (CN)2 (р =3,30 М. Б.) [129] образуется при нагревании
СоС12 с безводным NaCN в присутствии порошка Fe как катализатора или
при нагревании гидратов Со (CN)2 • пН2О в токе N2 при 250° С. Соединение
имеет структуру, подобную солям М3 [М (CN)6]2 (Со, Fe), и является коорди-
национным полимером [130].
Из водных растворов выделен ряд кристаллогидратов Со (CN)2 • пН2О
(и = 1,2; 2; 2,5; 3) [30, 73, 129, 131, 1321. Сравнение ИК-спектральных дан-
ных (v (CN) при 2130 и 2160 см-1) и параметров кубической решетки для
гидратов Со (CN)2 и гексацианокобальтата (III) кобальта (II) (а = 10,12 4-
4- 10,25 А) показывает, что Со (CN)2 • пН2О приближается по составу и
строению к Со3 [Со (CN)6]2 • пН2О [132].
Предположено [301 существование Со (CN)3 в виде темно-синего порошка,
устойчивого в отсутствие влаги (р = 2,79 М. Б.).
При нагревании гидратов кислот НСо (CN)4 и Н2Со (CN)5 получен красно-
коричневый Со (CN)3 • 2Н2О (р = 3,09 М. Б.) [30].
Цианид никеля (I), образующийся в виде оранжевого осадка при под-
кислении раствора K4Ni2 (CN)6, способен поглощать СО с образованием про-
дукта состава (NiCNCO)M [731. Парамагнитные соединения Ni (CN)2 • nH2O
(п — 1,5; 2; 3; 4; 7) (р = 2,2—2,3 М. Б.) можно получить из водных раство-
ров различными способами [1, 30, 129, 133—136].
Произведение растворимости Ni (CN)2 • пН2О в 0,1 м. NaCl при 25° С
(1,7 • 10“ ) не зависит от способа получения соединения: по реакции между
Ni2+ и CN или между Ni2+ и [Ni (CN)412“. Это привело к допущению [137]
что Ni (CN)2 • пН2О является координационным соединением [Ni (Н2О)П]2+-
•[Ni (CN)4]2“.
Существование атомов Ni разного сорта, а именно квадратно-плоскост-
ных групп Ni (CN)4 и псевдооктаэдрических NiN4 (Н2О)2 (соотношение 1 : 1),
связанных мостиками Ni — CN — Ni, установлено рентгенографическими,
73
спектральными и магнитными исследованиями Ni (CN)2 • пН2О (п =1,5
и 2) [133, 1341. Такая же картина наблюдается и в других гидратах [1361.
Для Ni (CN)2 характерно образование разнообразных клатратов общей
формулы Ni (CN)2 • NH3 • L (L = бензол, тиофен, анилин, фенол, пиррол,
фуран) [30].
3.6.7.2. Комплексные цианиды
Исследованию цианидов металлов семейства железа посвящено много
работ, в том числе и обзоров [1, 30, 73, 142]. Поэтому имеет смысл подробно
рассмотреть наиболее поздние работы.
Железо, кобальт, никель образуют устойчивые в водных растворах ком-
плексы [Fe(CN)6]4~, [Fe (CN)613~, [Со (CN)5]3~, [Со (CN)6]3“ и [Ni (CN)412-
[426—440], причем константы образования для первых двух можно опреде-
лить лишь непрямыми методами (табл. 3.4). Для Со (II) и Ni показано на-
личие ступенчатого комплексообразования [427, 428].
Энтальпии образования октаэдрического комплекса [Fe (CN)6]4~ [4291,
квадратно-пирамидального [Со (CN)5]3— [430] и квадратно-плоскостного
[Ni (CN)4]2— [431] составляют —359,1; —257,4 и —180,8 кДж/моль, что в
пересчете на одну CN-связь дает величины —59,8; —51,5 и —45,2 кДж.
Уменьшение этих значений в ряду Fe < Со < Ni можно связать [430] с ла-
билизирующим действием на связь металл — цианид последовательного
увеличения электронной плотности на ей-орбитали [432].
Хотя ион [Со (CN)5]3~ является объектом многих исследований [30, 432—
435, 487, 818, 819], структура его в водном растворе окончательно не уста-
новлена. Большинство исследователей на основании данных оптических
и ЭПР-спектров считают, что в растворе доминируют мономерные парамаг-
нитные комплексы [Со (CN)5]3~ со структурой квадратной пирамиды [433—
435, 817]. Однако одни авторы полагают, что [Co(CN)5]3~ в действительности
является октаэдрическим комплексом [Со (CN)5H2O]3— [432], другие допу-
скают существование димеров [Со2 (CN)JO16_ [818].
Водные растворы [Со (CN)5]3— изменяются со временем благодаря взаимо-
действию комплекса с водой [819]
2 [Со (CN)5]3~ + Н2О -> [Со (CN)5 И]3' + [Со (CN)5 ОН]3-. (3.37)
Эта реакция не протекает в метаноле и этаноле [488].
В твердом виде из системы Со2+ — CN~ — Н2О выделяются диамагнит-
ные соединения Katm [Со2 (CN)1O1 • пН2О (Kat = Na+, п = 4; К+, п = 6
30]; Ва2+, п = 13 [724, 725]). Мономерные соединения Kat3 [Со (CN)51
(Kat = Li+ [820], Et4N+, Bu4N+ [445], [NEt2 (CPr)2]+ [814]) удалось синте-
зировать из неводных сред (ЕЮН, DMFA, НМРА).
Пентацианидные комплексы в водных растворах известны также для
никеля [436—439] и железа (II), причем ионы [Fe (CN)5H2O13~ образуются
при действии света [325], в присутствии кислот и при кипячении растворов
[Fe (CN)6]4— [30].
Образование комплексов [Со (CN)eI4~ и [Ni (CN)6I4- в растворе представ-
ляется маловероятным [427, 432, 438]. Комплекс же [Ni (CN)5]3~ существует
не только в растворе [437—439], но и в соединениях К3 [Ni (CN)rJ • 2Н2О
[4411, [CrLj [Ni (CN)5] • nH,0 (L = NH3 (m = 6, n =2), en (tn = 3,
n = 1,5) 1442, 4431 и Pn (m = 3, n = 2) [444]).
74
Таблица 3.4. Константы устойчивости цианидных комплексов металлов VIII группы
Ион ме- талла Метод исследования 1, °C Ионная сила И 1g 1g 1g ₽3 1g 04 1g к8 lg 05 lg 1% Лите- ратура
Fe2+ Термодинамический расчет 25 0 35 1177]
Калориметрия, 25 » [430]
Термодинамический расчет 35,4
Fe3+ Калориметрия, 25 » »
Термодинамический расчет 43,6
Термодинамический расчет 25 » 42 [177]
Со2+ Калориметрия 25 0 sjs*# IQ OQ [430] IQ71
Полярография 20 5 (У/J
Со3+ Потенциометрия 30 Переменная 17,7 [177]
Ni2+ Спектрофотометрия, 25 0 30,3 [431]
рН-Потенциометрия
Спектрофотометрия, 25 р -» 0 31,0 [97]
рН-Потенциометрия
Спектрофотометрия 25 0,1 30,5 [440]
Спектрофотометрия, ИКС 25 4 0,03 [438]
Потенциометрия 25 3 7,03 <13,95 <22 31,06 [427]
Спектрофотометрия, ИКС 25,2 1,34 —0,72 [439]
Спектрофотометрия 23 2,5 —0,69 [437]
Pd2+ Потенциометрия 25 Переменная 51,6 [575]
Калориметрия 25 0 ❖ [576]
Потенциометрия » » 42,4 2,9 »
Растворимость 20 0,1 10,5 [577]
Спектрофотометрия 25 63,0 [811]
Pt2+ Потенциометрия 18 1 41,0 [578]
* ДН = —359,38 кДж/моль.
** Д/7 =—293,9 кДж/моль.
ДН =—257,7 кДж/моль.
ДН =—386,4 кДж/моль.
qJ Д Д >= —0,8 кДж/моль.
Многочисленную группу соединений составляют соли гексацианоферрат-
ной (II) кислоты Н4 [Fe (CN)J, известной в растворе и в твердом состоянии
[30]. Эти соли получены почти для всех металлов [26]; мало изученными яв-
ляются лишь соединения с V (II), Cr (II), Ti (II) и (III), TI (III), Мо (IV),
Та (V), Nb (V), Re (V).
Известны также соли с комплексным катионом, например [Со (NH3)6]4-
•[Fe (CN)6]3 • 10Н2О[446], и с катионами, содержащими три различных ме-
талла,— (NH4) KNa2[Fe (CN)6J • nH2O, К2 (NH4)8Ca5 [Fe (CN)6]5-2H2O [26].
Растворимые в воде соли щелочных и щелочноземельных металлов яв-
ляются ионными соединениями. Если же анион [Fe (CN)6]4~ связан с переход-
ным металлом, то благодаря существованию мостиковых групп Fe — CN-^-M
образуются координационные полимерные структуры [448, 4491.
Наблюдение эффекта Мессбауэра показало, что формулы берлинских
лазурей правильнее записывать не в виде К [Fe"'Fe" (CN)6] и Fe"' [FewFe" •
• (CN)6]3, а по типу «зонального» строения К {Few [Fe"(CN)6]} и Fe"'IFe"' •
• [Fe"(CN)6]}3 [450].
Образование донорно-акцепторных связей между атомом азота CN-rpynn
и «внешнесферными» катионами является, по-видимому, главной причиной
термолиза гексацианоферратов (II) по схеме [451]
М4 [Fe (CN)e]„ 4М (CN)n + nFe (CN)2. (3.38)
Термолиз солей с комплексным катионом протекает с образованием про-
межуточных полиядерных цианидов. Так, [Со (NH3)6]4 [Fe (CN)6]3 • ЮН2О
при 215—320° С разлагается с образованием аниона [(CN)5Co"NCFe" (CN)517~,
для которого выделены соли Na, Fe (III), Ni, Си (II), Мп (II), Со (II) [452].
Замена одного из CN-лигандов в октаэдрическом комплексе [Fe (CN)6]4~
на ацидогруппу или нейтральный лиганд (Н2О, NH3, NO, СО) приводит к об-
разованию смешанных соединений.
Взаимосвязь между некоторыми из них видна из схемы [30]
Все пентацианиды Fe (II) диамагнитны, а образующиеся в результате их
окисления производные Fe (III) парамагнитны.
Первый пентацианид железа (II), нитропруссид натрия Na2 [Fe (CN)5NO1 •
• 2Н2О был выделен в 1850 г. [1] по реакции
[Fe (CN)J4~ + 4Н+ + NO? -> [Fe (CN)5NO[’~ + CO2 + NH+. (3.39)
К настоящему времени выделены и исследованы электронные и ПК-спек-
тры поглощения нитропруссидов щелочных металлов, Ag+, Си'1', Си2+, Мп2+,
76
Fe2+, Co2+, Ni2+, Zn2+, Cd2+, Ti4+, [Bu4N]+ [447, 453—455] и R4P+ [821];
получена и соответствующая кислота Н2 [Fe (CN)5NO1 [73].
Поданным ИК- [456] и рентгеноэлектронных спектров [257],
[Fe (CN)5NO12~ содержит NO+-rpynny с эффективным зарядом + 0,35 эВ,
хотя иногда считают NO в нитропруссиде нейтральной молекулой [457, 458].
Замещение NO-группы другими лигандами, как показывают электрон-
ные, ИК- и мессбауэровские спектры, приводит к тетрагональному искаже-
нию октаэдрической симметрии комплексов. По силе действия эти лиганды
можно расположить в ряд: NO+ > Н2О > RNH2 > NH3 > SOf“ >
> NOF > CO > CN~ (R = Me, Et, Pr, Bu) [454, 459].
Согласно ИК-спектрам, в комплексе [Fe (CN)5NO2]4~ имеется связь
Fe—NO2 [460]. Тем не менее этот комплекс в водном растворе превращается
в пентацианоаквоферрат (П)-ион (АТ/ обратимой реакции = 51,5 кДж/моль)
[461]. В щелочной среде протекает внутрисферный обмен NO и Н2О в комплек-
сах [Fe (CN)5LJn на NCS-ион с образованием [Fe (CN)5NCS]4~, постепенно
окисляющегося в комплекс Fe (III) [Fe (CN)5NCS13“, причем промежуточ-
ными комплексами являются семикоординационные [Fe (CN)5NCS (ОН)]4'
и [Fe (CN)5 (NCS) Н2О13~ [462, 463]. Из раствора в присутствии щелочи вы-
делена соль Na2K2 [Fe (CN)5NCS], а при pH 7,6 из водно-ацетонового рас-
твора — соединение Na2K [Fe (CN)5NO NCS] [464]. Интермедиат такого
же ассоциативного типа получен и с тиомочевиной Na2 [Fe (CN)5NO - tu]
[465, 822].
Помимо рассмотренных выше пентацианидов, установлено существова-
ние комплексов [Fe (CN)5Lln~ (L = S2O2~, AsOC [73], CNO~ [466], PBu3,
PPh3 [222], tu [465, 822], нитрозобензол C6H5NO [467, 468], DMSO [469]').
Из этанол-метанольных растворов при взаимодействии нитропруссида
натрия с гидразином выделено соединение Na4 [Fe (CN)5N3] [470], с гидро-
ксиламином Na3 [Fe (CN)5N2O] и с аммиаком Na3 [Fe (CN)5N21 [471]. Послед-
ний продукт, по данным ИК- и рентгеноэлектронных спектров, содержит
координированный молекулярный азот, причем фрагмент Fe — N — N
имеет почти линейную структуру [471]. Кроме упомянутых выше продук-
тов, при взаимодействии Na2 [Fe (CN)5NO] • 2Н2О с гидразином в водно-
спиртовом растворе при pH = 9 синтезирована соль Na3 [Fe (CN)5N2H4] •
• Н2О [4721.
Из нитропруссида натрия получены также соединения [FeNO (CN)3L21
(L = Ру, y-Pic) и [FeNO (CN)3Dipy] [473—475].
По данным ИК- и мессбауэровских спектров, промежуточным продуктом
реакции [Fe (CN)J3' с NO2 в водном растворе является биядерный комплекс
IFe2 (CN), (NO)2)3“ ’, содержащий линейную мостиковую группу Fe" — NO —
— Fe" и цис-расположенную концевую NO+-rpynny. Выделена диамагнит-
ная соль серебра с этим анионом [476].
Кроме реакций замещения, нитропруссид натрия может вступать в реак-
ции присоединения. Цветной реакцией на SH-группу долгое время служи-
ло взаимодействие [Fe (CN)5NO12~ с SH~, исследование кинетики которого
Г г
указало на существование продукта A Fe (CN)5N\^^^
под действием щелочи в продукт В Fe (CN)5N\
, превращающегося
[477]. Ярко-красное
окрашивание растворов появляется при взаимодействии [Fe (CN)5NO12~’
77
c SO3 (реакция Бодекера). Равновесие
[Fe (CN5 NO]2- + SOf~ <> [Fe (CN)5 NOSO3]4~ (3.40)
исследовано в водных и неводных растворах [478—480]. На примере выде-
ленной соли цезия ЙК-спектрально показано, что в анионе [Fe (CN)5NOSO3]4~
SO3-группа связана с NO через кислород [479]. Ион [Fe (CN)5NO]2~ взаимо-
действует также с кетонами и альдегидами [481].
Восстановление нитропруссид-иона водородом при 230° С приводит к
образованию NO, HCN и K2[Fe(CN)4], который содержит мостиковые
CN-группы [258].
Кроме рассмотренных выше смешанных соединений, координационное
число 6 характерно для Fe (II) в комплексных цианидах типа [FeL2 (CN)21 •
- nH2O (L = Dipy, Phen), К2 [FePhen (CN)4] -4H2O [482—484], [Fe (CH3CN)4 •
• (CN)2] [73,485], [Fe (P3P) (CN)2] [222] и в аддуктах состава [FePhen2 (CN)2] •
• nMX (M = Si, Ge, Sn; X = F, Cl; n = 1 4- 3), в которых наблюдается
мостиковая связь Fe—CN—М [486].
Интересным свойством комплекса [Со (CN)5]3~ является его каталити-
ческая активность в реакциях гидрирования, полимеризации, изотопного
обмена. Это связано с взаимодействием [Со (CN)5]3— с молекулярным водо-
родом, протекающим как в водных [487], так и в водно-этанольных растворах
[488],
2 [Со (CN)5]3~ 4- Н2 2 [Со (CN)5 Н]3~ (3.41)
Из водных растворов можно выделить соль Cs2Na [Со (CN)5H], причем
наряду с этим предполагается образование комплексов [Со (CN)4H2]3~ и
[Со (CN)6F~ [490]. Некоторые авторы считают комплекс [Со (CN)5H]3~ про-
изводным Со (I) [491].
В водном растворе [Со (CN)5]3~ поглощает СО2, превращаясь в комплекс
[Со2 (CN)10CO2]6~, который выделяет водород из воды [492]. При взаимодей-
ствии с различными реагентами ион [Со (CN)5]3“ образует ряд биядерных
комплексов: [(NC)5Co—L—Со (CN)516- (L = Cd (II), Hg (II) [823], TI (I)
[824], SnCl2, SO2 [493], C2F4 [548], S [825]), [Co2L2 (CN)10l~6 (L = S, Se) [826],
[Co2en (CN)8]4—. Последний легко окисляется водой с образованием соли
К [Coen (CN)4J - Н2О [494].
Для Со (II) известны и смешанные моноядерные соединения: [Со (CN)otu2],
К2 [Со (CN)2 (NCS)2] [465, 827], [Со (CN)2L3] и [Со (CN)2 (PR3)2CO1
(L = вторичные и третичные фосфины HPPh2, PEt2Ph, PEtPh2, PMe2Ph,
PMePh2), [Co (CN)2 (PC2P)21, [Co (CN)2L1>5]2 (L = PC3P, PC4P) [222, 495].
Соединения с фосфинами получены и для Ni (II): [Ni (CN)2 (PR3)2] (R =
= Me, Et, Pr, Bu, Ph), [Ni (CN)2L3] (L = PMe3, PMePh2, PEt2Ph, PBu3,
HPEtPh [222, 496], MeP (OMe)2, Me2P (OMe) [828] и некоторые серосодержа-
щие фосфины [829]). Дифосфины в комплексах с Ni (CN)2 ведут себя как хе-
латообразующие, мостиковые или монодентатные лиганды. Первый тип ком-
плексов — четырехкоординационные соединения [Ni (CN)2L] (L = EtC2Et,
PQP, МеСзМе), второй — четырехкоординационные [Ni (CN)2 (PC4P)]2 и
пятикоординационные [Ni (CN)2Li>512 (L = MeC3Me, PC3P, PC4P). Моноден-
татным лигандом является одна молекула РС2Р в комплексе [Ni (CN)2 (РС2Р)21
с координационным числом никеля 5.
С трех- и четырехдентатными лигандами получены пятикоординацион-
ные соединения [Ni (CN)2L1 (L = Р2Р, Р3С2, As2P), [Ni (CN) Ll X (X= BPhT,
78
СЮ4 ; L = As3P) и четырехкоординационный комплекс [Ni (CN) (N3P)]+
[222].
Цианид никеля образует координационные соединения с тиомочевиной
[Ni (CN)2tu2 - 2Ас], для которого с учетом магнитных свойств предполагают
октаэдрическое строение [497], с шиффовыми тетрадентатными основаниями
[NiL (CN)1C1O4 (координационное число никеля 5 в растворе и 6 в твердом
состоянии за счет мостиковой CN-группы) и шестикоординационный
[NiL (CN)2] с концевыми CN-группами [204, 205]; с Phen типа
[Ni (CN)2Phen2] • 2Н2О [498].
Ni (CN)2 сравнительно легко присоединяет аммиак с образованием крис-
таллических соединений Ni (CN)2NH3 • пН2О (и = 0; 0,5; 0,25) [499—501],
в которых 50% атомов Ni имеет к. ч. 6, остальные — 4 [448, 499]. Внедрение
молекул органических веществ в эту сформированную полимерную струк-
туру приводит к образованию соединений включения типа Ni (CN)2 • NH3< L
(L == пирол, фуран, тиофен, пиридин, фенол, анилин, бензол) [30, 502].
Подобные клатраты образуют и тетрацианоникелаты с комплексным катио-
ном. Выделены соединения общей формулы М (NH3)2Ni (CN)4 • 2L (М = Мп,
Fe, Со, Ni, Си, Zn, Cd; L - С6Н6, PhNH2, C4H4NH, C4H4S) [503], Cd (en) •
• Ni (CN)4 • 2L (L = C6H6, C4H4S), причем для последнего типа клатратов
ИК-спектрально и рентгенографически установлено существование мостиков
Cd—en—Cd [504, 505].
Спектрофотометрически показано образование в водном растворе сме-
шанных цианоацидоникелатов|[№ (CN)X]3—, устойчивость которых изменяет-
ся в ряду SCN~ > 1~ > Вг~ [506]. В твердом виде выделены соединения
К3 [Ni (CN)3S] • Н2О [5071 и К2 [Ni (CN)2 (NCS)21 [508].
Цианиды со степенью окисления металла +3 известны для железа и ко-
бальта. Октаэдрический парамагнитный ион [Fe (CN)6J3-~, будучи более реак-
ционноспособным, чем диамагнитный [Fe (CN)6]4' , ведет себя как сильный
окислитель, особенно в щелочной среде. К примеру, молибден и вольфрам
легко растворяются в присутствии К3 [Fe (CN)6] [509]
М + 6К3 [Fe (CN)61 + 8КОН -> К2МО4 + 6К4 [Fe (CN)6] + 4Н2О. (3.42)
Гексацианоферраты (III) получены для большинства металлов [26, 693]
и катионов типа Bu4N+, Ph4P+, Ph4As+ [510], R4P [511, 821]. Они построены
в основном подобно цианидам железа (II), т. е. образуют ионные структуры
или полимерные сетки. Однако при переходе от иона [Fe (CN)6]4~ к [Fe (CN)6]3~
ослабляется роль л-дативной компоненты связи Fe Д CN и увеличивается
роль о-донорно-акцепторной компоненты Fe Д CN, что находит отражение
в ПК- и мессбауэровских спектрах [451].
Долгое время предметом дискуссии была структура берлинской лазури,
растворимой KFe"' [Fe" (CN)6] и нерастворимой Fef [Fe" (CN)6]3 форм, и
турнбуллиевой сини Fe3 [Fez,/ (CN)6]2. Результаты исследования спектров
Мессбауэра и ЯМР-спектров показали, что все соединения являются гекса-
цианоферратами (II) железа (III) [513]. Гексацианоферрат (III) железа (II)
можно получить в восстановительной среде, например при взаимодействии
FeCl3 и К4 [Fe (CN)6] в присутствии KI или сахарозы [514].
Диамагнитный комплекс [Со (CN)J3~ характеризуется большей устой-
чивостью в растворе, чем [М (CN)6]3~ (М = Cr, Мп, Fe) [30, 73]. Получены
многочисленные гексацианокобальтаты с катионом — атомом металла [1,
838], комплексным катионом типа [Сг (en)3]3+, [Сг (NH3)6]3+, [IrDipy2Cl2]+,
[Cru6]3+, [RuDipy3]3+ [515] и с катионом R4P (R = Bu, Ph и др.) [512]. Для
79
Fe (III) и Co (III) в свободном состоянии выделены изоструктурные кислоты
Н3 [M(CN)6], причем кристаллы гексацианоферратной (III) кислоты облада-
ют пьезоэлектрическим эффектом [489]. Известно много смешанных, и в част-
ности пентацианидных, комплексов [М (CN)5L]"— (М = Fe3+, Со3+; L =
- Н2О, NH3, NO, RNH2 (R -Me, Et, Pr, Bu, PhCH2), OH~, СГ, Br~, Г,
SCN“, NF, NOF, SO2-, S2O2~, HCOO~) [30, 731.
Соединения [Fe (CN)5L]'l— могут быть получены окислением соответству-
ющих пентацианоферратов (II) [30], a [Fe (CN)5H2O]2~-hoh образуется так-
же при действии света, кислоты или при нагревании [Fe (CN)613~ [30].
Предполагают существование в водных растворах и димерных форм
пентацианидных ионов Fe (II) и Fe (III), соответственно [Fe2 (CN)10]6~ и
[Fe2 (CN)1o]4~ [516,5171.
Введение SCN~-Hona в раствор [Fe (CN)5H2O12~ приводит к образованию
комплекса [Fe (CN)5SCN13 , для которого наблюдается изомерия связи
Fe—SCN^FFe—NCS [463]. Из Na2 [Fe (CN)5NH3] были синтезированы соли
[Bu4N]3 [Fe (CN)5L1 (L- NF, NCS~ [518, 519], NCSe-[519]) и Kat3 [Fe(CN)5 •
• NCS] (Kat- Et4N+, Ph4As+) [519], в которых, по данным ИК- и спектров
КР, осуществляется связь Fe—NCX (X — S, Se) [519]. Известно соединение
Кз [Fe (CN)4 (CNS)2], содержащее тиофульминатный лиганд [520].
Величина производимого тетрагонального искажения октаэдрической
симметрии при замене лиганда С№ в ионе [Fe (CN)6J3- другими лигандами
более ощутима для комплексов Fe (III), чем для Fe (II) [459].
Смешанные координационные соединения с анионом [Со (CN)5L]n— так-
же многочисленны. Так, еще в 1937 г. проведена реакция [30]
[Со (NH3)5 S2O3]+ —- lCo <CN)s S2°^4’ *Co <CN)s H2°l2~• (3-43)
Последний комплекс является также продуктом фотоактивации [Со (CN)613~
при 300—700 нм [325, 521]. В зависимости от условий фотохимической реак-
ции (pH, температура, время облучения) установлено образование и других
комплексов [Со (CN)5OH]3~ [522], [Со (CN)4 (Н2О)2Г, [Со (CN)3 (Н2О)31,
[Со (CN)4 (ОН)213~, [Со (CN)3 (ОН)3]3~ [523].
В результате замещения молекулы воды в [Со (CN)5H2O1 ~ получены ком-
плексы [Со (CN)5X]3—, устойчивость которых изменяется в ряду Вг~>
> СГ [524], и [Со (CN)5L12~ (L - NH3, N2H4, Ру) [525]. Галогеноцианид-
ные комплексы, а также соединения [Со (CN)5Lln- (L — NF, NOF, SCN~,
OH , SO2-) образуются при взаимодействии соответствующих ионов
[Co(NH3)5L]“+cCN~'-ионом [526]. Такие же комплексы получены и из солей
[Со (NH3)4 (CN)LlL (L = Г, NF, NOF, SCN”), [Co (NH3)4 (CN)L] (L =
= SO2~, S2O2~) [527]. В свою очередь пентацианиды легко превращаются в
[Со (CN)6]3~ в присутствии CN~-hohob и [Со (CN)5]3~ как катализатора [528].
Замещение Н2О в [Со (CN)5H2O]2~ HaSCN~-HOH приводит к образованию
N-связанного изомера, выделенного в виде калиевой соли [529], в то время
как по реакции
[Со (NH,)S NCSj2+ + [Со (CN),]3- — [Со (CN)6 SCN]3- + [Со (NHS), НаО]2+
(3.44)
образуется тиоцианатный изомер [5301. В водном растворе превращение изо-
тиоцианата в тиоцианат протекает очень медленно, а при нагревании до 128°С
80
OJ • nL
Рис. 3.2. Структура аниона [Co(CN)5O2J3
(пентагидрат тетраэтиламмонийной соли
[5351)
соль K3lCo (CN)5NCS] изомеризуется в К3 (Со (CN)5SCN] [531]. Такие же изо-
меры выделены и для тетрабутиламмония [531], причем соединение [Bu4Nb
- [Со (CN)5NCS] более устойчиво [532]. Для NCSe-лиганда известны лишь изо-
селеноцианаты Kat3 [Со (CN)5NCSe] (Kat = К-1- [533], Bu4N+ [532]) и
[Со (NH3)4 (CN) (NCSe)lCl [533].
Из соединения [Со (NH3)5NO]C12 был получен диамагнитный К3 [Со (CN)5 •
• NO] - 2Н2О [534], содержащий, по данным ИК-спектров и кинетики изо-
топного обмена, NO~-rpynny [30, 421, 465].
В результате взаимодействия солей Kat3 [Со (CN)5] с кислородом воздуха
в DMFA и НМРА образуются соед
= Et4N+, Et3BzN+,Me2Bz2N+; L =
= Н2О (и = 1,5; 3; 5), DMFA (п =
= 0,5, 1)) [445, 536].
По рентгеноструктурным данным
[Et4N]3 (Со (CN)5O2] • 5Н2О и
[NBz2Me2l3 • [Со (CN)5O2] содержат
в себе октаэдрический комплекс-
ный анион с оксогенидным (ОГ) ли-
гандом (рис. 3.2) [535, 536]. Извес-
тен и биядерный комплекс [Со2 •
• (CN)4 (PMe2Ph)5O2], в котором
молекула кислорода координиро-
вана лишь с одним атомом кобаль-
та, являясь бидентатным лигандом,
а атомы кобальта связаны мости-
ковой CN-группой [5371.
Процесс оксигенации [Co(CN)5l3~
протекает в водном растворе с об-
разованием диамагнитного перок-
социанокомплекса [(NC)5CoO2Co •
• (CN)5]6~, который при дальней-
шем окислении Вг2 переходит в парамагнитный комплекс I(NC)5Co02Co«
• (CN)5]5~ [538, 830].
Исследование ЭПР-спектра первого соединения [539] подтверждает осу-
ществление связи молекулы О2 с двумя атомами кобальта с помощью четырех-
центровой МО, построенной из d^-AO кобальта и луМО кислорода. Выделена
соль с этим анионом состава К8 [Со2О2 (CN)3O1 (NO3)2 • 4Н2О и исследована
ее кристаллическая структура [540].
Окисление [Со (CN)513~ гексацианоферрат (Ш)-ионом приводит к обра-
зованию биядерного комплекса [(CN)5CowNCFe* (CN)5J€~ [538], состав ко-
торого подтвержден полярографически [5411. Мостиковая функция CN-груп-
пы осуществляется и в соединении кобальта (III) состава [(NH3)5CoNCCo •
• (CN)5] 1542].
Существование биядерных комплексов в водном растворе установлено
также для железа (III) и (II) состава l(CN)5Fe (CN)Fe (CN)5]7~, [(CN)4Fe •
• (CN)2Fe (CN)4]4~, f(CN)5Fe (CN)Fe (CN)5]5~ [543].
Помимо описанных выше комплексов октаэдрическая конфигурация
Со (III) наблюдается в смешанных соединениях Na [Co(CN)4 (Н2О)2] [731,
К4 [Со (CN)3S2 (Н2О)] - 4Н2О [544], К4 [Со (CN)5SO3b 4Н2О и солях
LF1-, К+, Ag+, TI, Sr2+, Ва2+ с анионом [Со (CN)4 (SO^jjf— [5451, К2 [Со •
• (CN)5PPh3] • ЗН2О [2221; Kat [Со (CN)4L2b мН2О (Kat = Na, К; L =
81
= PPh3, AsPh3 [222, 546], en [547] и Kat = Ba, L = Pn [547]); K3[Co(CN)5 •
• (LH)] (L = C2F4) [548]; Na [Co(CN)2L] (H2L = основание Шиффа)
[831]; Kat [Co (CN)2 (DH)2p ziH2O (Kat = H+, K+, Ag+ [549], Ph2I+, PyH+,
PicH+ [550]); Kat [Co (OH) (CN) (DH)21 (Kat = K+, [Co (DH)2Py2l+,
[Co (DH)2 (NH3)2]+ [549]). Соединение К [Co (CN)2 (DH)2I • 1,5H2O имеет
конфигурацию и ведет себя в растворе подобно витамину В12 [551].
Известны смешаные цианидные комплексы кобальта (III) катионного
типа: цис- и транс-изомеры [Со (CN)2L2I+ (L = en, Dipy, Phen [552—558],
Pn [5471, PC2P [222]); [Co (CN)2 (NH3)J+ [554, 558]; цис- [Co (CN)2en (NH3)2]+
[554]; цис- и транс- [Co (CN)en2NH3]2+ [559]. Получены и нейтральные комп-
лексы: [Со (CN)3en (NH3)] • Н2О [554], [Со (CN)3dien] [558], цис- [Со (CN)3 •
• (NH3)3] [560], [Со (CN) (DH)2PPh3] [222].
Цианидные координационные соединения, содержащие металл в необыч-
ных степенях окисления, выделены для железа, кобальта, никеля.
Полярографическое восстановление нитропруссида натрия в DMFA
ведет к образованию Na3 [Fe (CN)5NO1, в котором предполагается наличие
железа (I) [561]. Однако по другим сведениям [454, 562] эта соль является
производным Fe (II) с нейтральной NO-группой, как и соединения
Na3 [Fe (CN)5NO1 2NH3, [Et4Nl3 [Fe (CN)5NO1 и Kat2 [Fe (CN)5NOH]
(Kat == Et4N+, Ph4P+, Ph4As+), выделенные из жидкого NH3 [5641.
Методом ЭПР установлено существование ионов железа (I) [Fe (CN)4NO12~,
образующихся при термолизе Kat [Fe (CN)5NO] пН2О (Kat = Zn, Cd)
[565].
Пентацианокобальт (1)-ион [Co (CN)5]4— образуется при радиолизе вод-
ных растворов [Со (CN)5]3— [566] и при химическом и электрохимическом
окислении [Со (CN)5H13— [567]. Более устойчивы смешанные цианидные пя-
тикоординационные комплексы кобальта (I) [Со (CN)2 (CO)2L]~, [Со (CN)2 •
. (CO)L2]~, [Со (CN) (CO)2L2] (L = PPr3, PEt3, PEt2Ph, PPh3) [567, 568],
[Co (CN)3 (CO)2]2~ [569] и соли Kat2 [Co (CN)3 (CO)2] (Kat = PPhf, AsPht,
[Fe Phen3]2+) [570].
Широко известно производное никеля (I), например соль Белуцци,
К4 [Ni2 (CN)6], анион которой, по спектральным и рентгенографическим дан-
ным, содержит связь Ni—Ni [30, 571]. Взаимодействие этой соли с СО при-
водит к образованию смеси К2 [Ni (CN)41 и К2 [Ni (CN)2 (СО)2] [572].
Помимо Kat2 [Ni (CN)2 (СО)21 (Kat = К+, Ph4P+) [572], к производным
никеля (О) относятся тетраэдрические соединения К4 [Ni (CN)41 и
К2 [Ni (CN)3NO1 [30].
Интересны четырехкоординационные цианидные соединения кобальта,
железа, никеля с отрицательной степенью окисления металла. Из карбонил-
нитрозилов Со (NO) (СО)3 и Fe (NO)2 (СО)2 в жидком NH3 получены соли
К3 [CoNO (CN)3], К2 [CoNO (СО) (CN)21, К2 [Fe (NO)2 (CN)2] [573], а из нитро-
зилбромидов [М (NO)9Br]2 — соединения Na [М (NO)2 (CN)2J, [М (NO)2 •
• (CN)L], [Ni (NO) (CN)L21, Na [Ni (NO) (CN)2L1, Na2 [Ni (NO) (CN)31
(M = Fe, Co; L = PPh3, AsPh3, SPPh3, SePPh3) [574].
3.6.8. Цианиды платиновых металлов
3.6.8.1. Простые цианиды
Простые цианиды платиновых металлов получены для рутения, родия,
иридия, палладия и платины. При действии H2SO4 на раствор К4 (Ru (CN)61
выделяется эквимолярная смесь Ru (CN)2 и Ru (CN)3. Если раствор
К4 [Ru (CN)e] предварительно обработать хлором, можно получить Ru (CN)3 •
•5Н2О, растворимый в аммиаке с образованием Ru (CN)3 • Н2О • 4NH3
[1]. При нагревании в вакууме до 200° С Ru (CN)3 • 5Н2О переходит в моно-
гидрат.
Аналогично цианиду рутения (III), только без хлорирования, получен
желто-коричневый 2Rh (CN)3 • 7Н2О, который, по-видимому, имеет поли-
мерный характер [30, 73].
При взаимодействии RhCl3 с KCN (1:5) получен Rh (CN)3 • ЗН2О, со-
держащий как мостиковые, так и концевые CN-группы [139]. Полимерное
строение имеют и М (CN)3 (М = Rh, 1г) [140], полученные термическим раз-
ложением (NH4)3 [М (CN)6], а также Pd (CN)2 и Pt (CN)2 [30].
Диамагнитный светло-желтый Pd (CN)2 осаждается при введении стехио-
метрического количества CN~-hohob в раствор нитрата или сульфата палла-
дия или при подкислении нагретого раствора К2 [Pd (CN)J • ЗН2О. Pt (CN)2
выделяется при действии кислот на К2 [Pt (CN)J, при взаимодействии
Hg (CN)2 и К2 [PtCl4] в водном растворе, а также при нагревании (NH4)2 •
• [Pt (CN)4L Соединение содержит мостиковые CN-группы [141].
3.6.8.2. Комплексные цианиды
Комплексные цианиды известны для всех платиновых металлов. Они
характеризуются большей устойчивостью по отношению к реакциям гидро-
лиза, окислительно-восстановительным, а также к реакциям замещения CN-
группы, чем соединения железа и кобальта.
В водных растворах установлено образование [М (CN)6]4— (М = Ru, Os),
[М (CN)6]3— (М - Ru, Os, Ir), [M (CN)4]2~ (M = Pd, Pt), [Pd (CN)5]3~ и
[Pt (CN)6]2~ [30, 575—578]. Что касается констант их образования, то име-
ются лишь данные о тетрацианидных комплексах платины и палладия, а
также о [Pd (CN)5]3~, которые более устойчивы, чем аналогичные соеди-
нения никеля (II) (табл. 3.4).
К. К- Клаус более ста лет назад синтезировал соли К4 [М (CN)6] • ЗН2О
(М = Ru, Os), изоструктурные с К4 [Fe (CN)6], путем сплавления хлорсо-
держащих соединений этих металлов с KCN [1]. В настоящее время выделе-
ны соли Katm [Ru (CN)6] • nH2O (Kat - NF# Li+, Na+, Rb+, Cs+ [579],
Ag+, Sr2+, Ba2+, Cu2+ [1], Al3+, Fe3+ [580, 581], Pr3+ [582]), KM [Ru (CN)6] *
• nH2O (M == Y3+, Ln3+) [583, 584, 832]. В свободном состоянии существуют
кислоты Н4 [М (CN)6] (М = Ru, Os) [73], в которых, как и в Н4 [Fe (CN)6],
по данным ИК-спектров, все четыре атома водорода образуют водородную
связь типа N—H...N [585].
Диамагнитные ионы [Pd (CN)4]2— и [Pt (CN)4]2— имеют плоскоквадратное
строение; они входят в состав солей KatOT [М (CN)4] (Kat = щелочные ме-
таллы, NF#, Ag+, Ва2+, Cu2+, Hg2+ и др.) [1, 30].
83
Для рассмотренных платиновых металлов известны многочисленные
цианидные координационные соединения смешанного типа.
Под действием света, кислоты, брома и при нагревании К4 [Ru (CN)61
в водном растворе переходит в К3 [Ru (CN)5H2O] [586, 587]. Из раствора этой
соли в концентрированном аммиаке выделен ряд амминоцианидов типа Kat
[Ru2 (CN)5 (NH3)4lm • nH2O (Kat = NHt (m = 1); Ni2+, Cu2+ (m = 2) [1]).
Рутений и осмий подобно железу образуют нитропруссиды Kat2 [М (CN)5 •
• NO] -2Н2О (Kat = Na+, K+) [1, 588], в которых степень окисления металла
четко не установлена. По данным ИК- и спектров Мессбауэра, одни авторы
[588, 589] предполагают наличие ИО+-группы и степень окисления металла
+2, другие считают, что вследствие координации молекулы NO как электро-
нейтралыюго или ацидолиганда степень окисления Os и Ru +3 и выше
[465, 590, 591]. Из нитрозилцианидов Ru и Os получены промежуточные
продукты К2М (CN)5NO-tu, K2Os (CN)5NO • KNCS, а также соли K3 IM (CN)5 •
• tu] -nH2O (n = 1, 2), K2 [Os (CN)5SCN1 -3H2O [590, 591], K4 [Ru (CN)5NO2] -
• nH2O (n = 0 и 2). Последние содержат нитрогруппу (связь Ru—N)
[592].
Выделены и другие шестикоординационные соединения: К2 [Ru (CN)4 •
• (N2H4)2] • 2Н2О [5931, [Ru (CN)2L21 (L = Dipy [594], MeC2Me [222]),
[Ru (CN)2P3P], [Ru (CN)HL2] (L = MeC2Me, EtC2Et) [222].
Из смешанных комплексов Pd (II) и Pt (II) следует указать соединения
транс- [Pd (CN)2 (NH3)2] и [Pd (CN)2 (NH3)1, причем в последнем содержатся
концевые и мостиковые CN-группы, образующие тетрамерную структуру,
[Pd (CN)2en] и [Pd4 (CN)8en2] [595]. При взаимодействии соединений [PtX2 •
• (NH3)2] с KCN получен транс- [Pt (CN)2 (NH3)2] [5961, а соединение, ранее
принятое за цис-изомер, представляет собой iPt (NH3)4] [Pt (CN)4] [5971.
В то же время, согласно [141, 598], Pt (CN)2 образует цис-соединения
[Pt (CN)2enl и [Pt (CN)2L2] (L = NH3, Py, tu).
В этом ряду следует рассмотреть также координационные соединения
[Pt (CN)2U * ftH2O (L = Dipy, Phen) [599], [M (CN)2L2] (L = PMe3, PEt3,
PPh3; M = Pt, Pd), [Pd (CN)2L1 (L = EtC2Et, PC2P, PC3P), [Pt (CN)XLJ
(X = Cl, Br, I; L = PEt3), [Pt (CN) HL2] (L = PEt3, PPh3) [222]. Инте-
ресно, что последние образуют аддукты типа [Pt (CN)H (PEt3)2l • L (L —
= A1C13, AlMe3, ZnCl2, NiCl2, СоС12 и т. п.) [600].
Для Pd и Pt получен ряд клатратов общей формулы М' (NH3)2M (CN)4 •
• 2L (М' = Mn, Fe, Co, Ni, Cu, Zn, Cd, L = C6H6, PhNH2, C4H4S, C4H4O,
C4H4NH) [503, 833], структура которых подобна клатрату Гоффмана
Ni (NH3)2Ni (CN)4 • 2C6H6.
Представляют интерес яркоокрашенные цианокомплексы платины, об-
разующиеся при частичном окислении К2 [Pt (CN)4] хлором или бромом со-
става К2 IPt (CN)4]Xo,3 • nH2O (п = 2,5 [601] и 3 [602]). По данным анализа,
соединения имеют состав К2 [Pt“84 Ptoje (CN)4]C1O,32 • 2,6 Н2О. Рентгено-
графическое исследование показало, что избыточный заряд платины (фор-
мально +2,3) не локализован и компенсируется нестехиометрическим коли-
чеством ионов С1~ или Вг~, статистически заполняющих пустоты структуры.
Ее характерная особенность — образование колонок из плоских квадрат-
ных групп [Pt (CN)41 и цепочки из атомов Pt с расстоянием Pt—Pt 2,88—
2,887 А, близким к длине металлической связи [601, 602]. Подобное наблю-
дается и в других соединениях Pt (II), например солях Kat [Pt (CN)J •
- пН2О (Kat = Mg2+, Ca2+, Sr2+, Ba2+) [603], Mg [Pt (CN)4]C1O28 - 7H2O
[604].
Цианидные соединения co степенью окисления металла +3 известны для
84
Ru, Rh, Ir. В водном растворе K4 [Ru (CN)61, в отличие от К4 [Os (CN)61,
электролитически и под действием сильных окислителей (Н2О2, Се4+, NaBiO3)
окисляется до [Ru (CN)613~ [605]. Однако из таких растворов выделяются
соединения Ru (II), как, например, соль [Со (NH3)5H2O]4 [Ru (CN)6]3 • 5Н2О
[6061. А для Os (III) выделены соли Kat3 [Os (CN)6] (Kat = К4" [607], Bu4N+
[2391).
Изоструктурные соли К3 [М (CN)6] (М = Rh, 1г) можно получить при
взаимодействии хлоридных соединений Rh и Ir с KCN [30]. Выделены также
соединения Katw [М (CN)6]„ • пН2О (М = Rh, Ir; Kat = NH^-, Ag+, Cu2+,
Ni2+) [1, 1401 и Kat3 [Ir (CN)6]2 • nH2O (Kat = Zn2+, Mn2+, Fe2+, Co2+) [6081.
Соли переходных металлов с указанными анионами изоструктурны соот-
ветствующим солям [Со (CN)6]3~ и благодаря образованию связей М—C=N—
Kat относятся к классу координационных полимеров [608, 834]. Гекса-
цианородиаты (III) Na, Mg, Са, Ва получены в виде соединений с уротропи-
ном, например КВа [Rh (CN)6]2 • 2Urt • 4Н2О [1].
Под действием УФ-облучения в кислых растворах комплексы [М (CN)613~
(М = Ir, Rh) превращаются в пентацианоаквокомплексы [М (CN)5H2O12~,
из которых получены смешанные комплексы [М (CN)5L]3~ (L = СГ, Вг—,
Г, ОН~ [609] и N7 [835]). Выделены кристаллические соли Kat [М (CN)5L1
(Kat = К+, [Со (NH3)6]3+; L = СГ, ВГ, Г) [609] и К3 [М (CN)5N3] • Н2О
[8351.
Для Ir (III) и Rh (III), как и для Со (III), известны гидридные цианоком-
плексы. Действием KCN на водно-метанольный раствор [Rh (СО)2С1]2 вы-
делено соединение К2 IRhH (CN)4 (Н2О)], содержащее, по данным ИК- и
ЯМР-спектров, связь Rh—Н. Из последнего при действии О2, NO2 и C2F4
получены, соответственно, К2 IRh (О2Н) (CN)4 (Н2О)] [6101, К2 [Rh (CN)4 •
NO2 (Н2О)] и Кз [Rh (CN)5C2F4H1 [139].
Оказалось, что в абсолютно безводных условиях образуются гидридные
комплексы типа К3 [МН (CN)5] (М = 1г [6111 и Rh [612]). Для аниона [RhH •
• (CN5]3~ выделены соли щелочных металлов (кроме Li) [613], причем авторы
полагают, что ранее описанный гидрид К2 [RhH (CN)4 (Н2О)] [139] представ-
ляет собой соль Кз IRhH (CN)5J • nH2O.
Ранее комплекс [RhH (CN)5]3~ был обнаружен по ЯМР-спектрам водных
растворов смеси RhCl3, NaBH4 и KCN, однако отнесен к производным родия
(I) [493].
Из других смешанных соединений иридия (III) и родия (III) можно при-
вести цис-tRh (CN)9L2]C1 • гсН2О (L = Dipy (п = 4) и Phen (п = 2,5) [614]);
[Rh (CN)3 (NH3)3] [140]; [RhX (CN)2 (PPh3)2 (NCR)] (X = Cl, Br; R = Me,
Ph, CH3—CH =CH); flrCl (CN) (NCS) (CO)L2] (L = PPh3, PMePh2) [222,
6151, [Rh Py2 (CN)2CH • 2H2O [595].
Гексацианидные комплексы платины (IV) не удается получить при вза-
имодействии соединений Pt (IV) с KCN и при окислении [Pt (CN)J2~ даже
сильными окислителями [616]. К2 [Pt (CN)6] синтезирован по схеме [617]
К2 [Pt (CN)J — ~ К2 [Pt (CN), I] К2 [Pt (CN)6[. (3.45)
Выделены в чистом виде сильная кислота Н2 [Pt (CN)6] [6181 и ряд солей
Kat [Pt (CN)61 • пН2О (Kat = Mg2+, Ca2+, Cr2+, Mn2+, Fe2+, Co2+, Ni2+,
Cu2+, Zn2+, Cd2+ [619] и Bu4N+ [624]).
Окисление K2 [Pt (CN)41 галогенами (Cl2, Вг2, I2) приводит к образованию
K2 [Pt (CN)4X2], K2 [Pt (CN)4ClBr] [620—623], в которых, по данным ИК-
и спектров КР, связь Pt—С имеет сильный о- и слабый л-характер [622].
85
Пентацианидные соединения типа К2 fPt (CN)5X] (X = Вг—, I-) можно
получить при окислении К2 [Pt (CN)J бром- или иодцианом [617, 625, 626]
либо действием С12 или Вг2 на К2 [Pt (CN)5H [627]. По обменной реакции
получена соль с комплексным катионом и анионом [Pt(NH3)2(H2O)2] [Pt (CN)5 •
•I] [6281.
Большая заслуга в исследовании смешанных цианидных соединений пла-
тины (IV) принадлежит школе И. И. Черняева. Выделены и изучены разно-
образные шестикоординационные производные типа [Pt (CN)4 (NH3)2] [620],
[Pt(CN)2(NH3)2L2] (L = C1- Вг~, I“, OH- NO?), [Pt (CN)3 (NH3)2X[ (X =
= C1-, Br~, Г) [629], [Pt (CN)3 XL2] (X = СГ, Br“ I~; L = MeNH2) [630],
[Pt (CN)2 X2en] (X = СГ, Br- 1“ NOF, OH~) [631], [Pt(CN) X3L2] (L =
— MeNH2, X — Cl , Br , I ) [632] и другие [633].
Гексацианопалладат (IV) калия можно получить при окислении соот-
ветствующего хлоридного соединения пероксодисульфатом калия K2S2O8
по схеме [6341
К2 [PdCl6] + 6KCN -^-8- К2 [Pd (CN)6] + 6КС1. (3.46)
Из цианидов с высшей степенью окисления металла известна соль
К2 [OsO2 (CN)J [11, которая при 260—400° С переходит в К4 [Os (CN)6] [635].
Для платиноидов с аномально низкими степенями окисления получены
соли родия (I): [М (en)3l [Rh (CN)41 (М = Сг, Со) [636], полимерные продук-
ты [Rh (CO)2CN]n и [(Ph3P)2 RhCNl2 с мостиковыми CN-группами [139].
По данным ЯМР-, электронных и колебательных спектров, в метанольных
растворах [Rh (СО)2С112 и NaCN предполагается существование промежуточ-
ных комплексов родия (I) [Rh (COR (CN)ClJ-, [Rh (CO)2 (CN)2]~, [RhCO •
• (CN)312~ [6131.
Мономерное соединение [RhCN (PPh3)3l реагирует с CO с образованием
четырех- и пятикоординационных соединений [Rh (CN) (СО) (PPh3)2l и
[Rh (CN) (CO) (PPh3)3] (аналогичное соединение известно и для иридия
[5631), обратимо присоединяет водород и кислород, образуя, соответственно,
[Rh (CN) Н2 (PPh3)2] и [Rh (CN)O2 (PPh3)2l. Получены и другие соединения
Rh (I): четырехкоординационные [RhCN (HPPh2)3], [RhCN (СО)РС2Р1 и пяти-
координационный [Rh (CN) (РС2Р)2] [2221.
Восстановлением К2 [Pt (CN)41 калием в жидком NH3 получены соедине-
ния платины со степенью окисления 0, а именно: Kat2 [Pt (CN)2I (Kat = K+,
Ph„P+), [Ph4Pl2 [Pt (CN), (NH,),I, [Ph4PI2 [Pt (CN), (CO),1, K2 [Pt (CN),I •
♦ 5Phen. Структура последнего соединения по криоскопическим и кондукто-
метрическим данным отвечает формуле [KPhen2]2 [Pt (CN)2Phen] [637].
При восстановлении К2 [Pd (CN)41 калием в жидком аммиаке выделены
К2 [Pd (CN)21 • nNH3, Kat [Pd (CN)CO] (Kat = Ph4P+, Me Ph3P+, Ba [Pd •
• (CN) COR - 0,25NH3 [6381, Ba [Pd (CN)21, [R3R'P12 [Pd (CN)21, [R3R'PR .
- [Pd (CN)3Phl и [R4Asl2 [Pd (CN)3Phl (R = Ph, R' = Me, Ph) [8361.
3.7. ЦИАНИДНЫЕ КОМПЛЕКСЫ В НЕВОДНЫХ СРЕДАХ
При исследовании цианидных комплексов в неводных средах нашли
применение не только органические растворители, жидкие HF, HCN, амми-
ак, но и расплавленные соли (тиоцианат калия, хлориды, нитраты щелочных
металлов и их эвтектические смеси). Однако комплексы далеко не всех ме-
таллов одинаково успешно исследуются в неводных средах.
86
3.7.1. Цианокомплексы материалов I и II
побочных подгрупп
В 1938 г. Германн кондуктометрически показал образование цианидных
комплексов [Си (CN)3]~ в метиловом и [Си (CN)4]2— в этиловом спиртах
[6391. Эти комплексы крайне неустойчивы и быстро распадаются с образова-
нием дициана и комплекса меди (I) [Си (CN)312- в водных, водно-метапольных
и метанольных растворах [189, 190, 640]. Однако введение метанола в водный
раствор [Си (CN)4]2~ значительно уменьшает скорость этой реакции, что
связано с сольватационными процессами [6401.
Цианиды Ag, Си (I) нерастворимы в DMSO, но растворяются в нем в при-
сутствии малорастворимого KCN. По калориметрическим данным [6411,
предполагается существование в DMSO комплексов [Ag (CN)2]~, [Си (CN)21~,
[Си (CN)41^, а потенциометрически показано образование [Ag2CN]+ (lg [3 =
= 15,0), AgCN и [Ag (CN)2]~ [642].
В безводном этилендиамине [97] и нитрометане [631] существует комплекс
[Ag (CN)2]~, устойчивость которого оценена потенциометрически
(табл. 3.5).
Для золота экстракцией циклогексаноном и н-амиловым спиртом в бен-
золе из водных растворов установлено образование Н [Au (CN)2] • nL, при-
чем степень сольватации комплексной кислоты зависит от концентрации
экстрагента в бензоле [644].
Цианид серебра при растворении в жидком HF диссоциирует по схеме
2AgCN + 2HF 2Ag (HCN)+ + 2F~ Ag (HCN)^ + Ag+ + 2F~. (3.47)
На образование таких комплексов указывают данные ПМР- и ИК-спек-
тров растворов AgCN—HF и AgCN—HCN—HF, причем в первой си-
стеме присутствуют 19% Ag (HCN)+ и 40,5% Ag (HCN)^" (данные ИК-спектров)
[6451.
Цианид ртути также хорошо растворим в безводном HF с образованием
токопроводящих растворов. Ранее [646] полагали, что проводимость обус-
ловлена процессом протонизации
Hg(CN)2 + 2HF<>Hg(CN)2H+ + HFr. (3.48)
Однако, как установлено кондуктометрическими, криоскопическими,
ИК-спектральными и ЯМР (19Р)-исследованиями [647], при t > —70° С
протекает процесс ионизации по схеме
2Hg (CN)2 + 2HF Hg2 (CN)3+ + HCN + HF?. (3.49)
Катион Hg2 (CN)+ имеет, вероятно, линейную мостиковую структуру
[N=C—Hg—C=N—Hg—C=N]+.
Возможна и дальнейшая ионизация
Hg2(CN)3+ + 2HF«>2HgCN+ + HCN + HFr, • (3.50)
ускоряемая при введении BF3 и AsF3, которые образуют нерастворимые соли
HgCNBF4 и HgCNAsF6.
В отличие от цианидов натрия, калия, никеля, цинка, кобальта,
Hg (CN)2 и Cd (CN)2 растворяются в DMSO [641].
В метанольных растворах Hg (CN)2, К2 [Hg (CN)4], Hg (CN)2 (избыток
твердого вещества) и KCN по ИК-спектральным данным существует комплекс
[Hg (CN)3]~. При удалении метанола выделяется смесь Hg (CN)2 и К2 [Hg •
• (CN)J. Такая же смесь образуется при взаимодействии Hg (CN)2 и KCN
в жидком HCN, хотя ИК-спектрально обнаруживается [Hg (CN)3]~ [102].
87
0° Таблица 3.5. Константы образования цианидных комплексов некоторых металлов в неводных и смешанных растворах
Ион ме- талла Растворитель Метод исследования t, °C ц 1К ₽, 1g К2 1g ₽2 Ig К3 1g Рз 1g К4 1g ₽4 Литера- тура
Ag+ Нитрометан DMSO Потенциометрия » 25 25 25 0 0,1 34 23,4 13,3 [643] [642] [97]
en NaI-3,3NHs » 20 4,9 [653]
NH4NO8- 1,3NH3 Вольт-амперометрия, потенциометрия 20 6,45 11,9 26,70 [654]
Cu2+ H20 — 60 вес. % MeOH Потенциометрия —45 0,05—0,1 [189]
Cd2+ en Полярография » 25 25 0,25 1 2,56 2,31 4,87 3,8 2,00 6,87 1,7 * 8,87 [97] [650]
H2O —0,66 об. % en » 25 0,5 4,7 8,3 [649]
H2O —6,25 об. % en » 25 » 3,4 5,8 »
H2O — 14,3 об. % en » 25 » 3,1 5,0
H2O —25 об. % en DMFA » Растворимость 25 25 » 0,1 4,7 4,4 2,8 9,1 3,08 12,18 4,5 [651]
Hg2+ en Полярография 25 1 13,7 [650]
H2O— 10 об. % en » 25 0,5 13,2 [649]
H2O — 30% en » 25 » 12,2
H2O — 50% en » 25 » 11,6
H2O — 70% en » 25 » 13,5
HoO— Юоб. % L » 25 0,5 16
H2O — 30% L » 25 » 13,4 13 »
H2O — 50% L » 25 »
H2O — 70% L » 25 » 13,8
H,O — 99,9% L » 25 » 16
H2O — 10 об. % Py Потенциометрия, 0,5 26,1 26,5 27,3 28,9 28,4 28,4 28,8 18,6 [648] » » » » [653]
H2O — 20% Py H,O-40% Py h;o—60% py HoO — 80% Py NH4NO3- 1,3NH3 полярография » » » » Потенциометрия 25 20 7,3 22,9 22,9 26,5 28,6 12,5
• 1g ₽</Р2 = 3.2,
L •—этаноламин
Существование комплекса [Hg (CN)3]~ наряду с Hg (CN)2 и [Hg (CN)J2-
установлено электрохимическими методами и в водно-пиридиновых раство-
рах [648] (табл. 3.5).
В этаноламине, этилендиамине и в смесях этих растворителей с водой на-
блюдается образование лишь Hg (CN)2 (649, 650]; кадмий же в указанных
растворах (кроме этаноламина и его смесей с водой) образует комплексы
[CdCN]+, Cd (CN)2, [Cd (CN)3]~, [Cd (CN)J~2 [97, 649, 650], устойчивость
которых падает с увеличением содержания неводной добавки (табл. 3.5).
В DMFA методом растворимости выявлены лишь первые три комплекса
кадмия. Рост растворимости Cd (CN)2 в DMFA с увеличением ионной силы
раствора указывает на частичную диссоциацию Cd (CN)2 на [CdCN]+ и
[Cd (CN)3]“ [651].
Представляют определенный интерес как растворители растворы некото-
рых солей щелочных металлов и аммония, например NH4I, NH4NCS, NH4NO3,
LiNO3, Nal в аммиаке [652]. Так, в жидкости состава NH4NO3 • 1,3NH3
установлено образование комплексов [HgCN]+, Hg (CN)2, [Hg (CN)4]2~
[653] и AgCN, [Ag (CN)2]~ [654], а в Nal • 3,3NH3 — [Ag (CN)2]“ [653].
Образование смешанных цианид-галогенидных комплексов [Hg (CN)X]
(X = СГ, Br-, I~) спектрофотометрически изучено в диоксане. Величины
констант равновесия реакций
Hgx2 + Hg (CN)2<> Hg (CN) X (3.51)
составляют 0,2 (I-); 1,74 (Br“); 12,0 (Cl~), что указывает на возрастание ус-
тойчивости комплексов в ряду I- < Вг~ < С1— [655].
В системах Н2О—L (L = Act, ацетонитрил, изопропанол, DMFA) ис-
следовано влияние растворителей на протекание равновесной реакции
[М14]2~ + CN-[MI3CN]2“ + Г (М = Hg2+, Cd2+). (3.52)
Изменение содержания неводных растворителей заметно не влияет на
константы равновесия этих реакций [656].
3.7.2. Цианокомплексы металлов семейства железа
Большое число работ [443, 657—662] посвящено изучению образования
цианидных комплексов Fe, Со, Ni в нитрометане, PDC, ES, DMSO, DMAA,
НМРА. Эти исследования носят чисто качественный характер, так как в них
отсутствуют данные об устойчивости комплексов.
Кондуктометрически в системе Fe (С1О4)3 — [Et4N]CN — DMSO установ-
лено существование комплексов Fe (CN)jt, Fe (CN)3 и Fe (CN)E, а по данным
спектрофотометрии предполагается образование Fe (CN)E и, возможно,
[Fe (CN)613—. Подобие спектров FeLE (L == NE, CN") в DMSO и спектров
[FeL6l3- (L = CN“, NCS~) указывает на октаэдрическое окружение Fe3+
за счет присоединения двух молекул DMSO к тетрацианоферрат (Ш)-иону
[6571.
Для никеля спектрофотометрическое, кондуктометрическое и потенцио-
метрическое исследования показали образование малорастворимого
Ni (CN)2 и квадратно-плоскостного комплекса [Ni (CN)412~ в DMSO, DMAA,
PDC [6581.
При введении CN~ в систему Со (С1О4)2 — нитрометан (L) выделяется
Со (CN)2 • 2L, который растворяется в избытке цианид-ионов с образованием
89
тетраэдрического комплекса [Со (CN)J2 , а при большем избытке CN и
комплекса [Со (CN)5L]3— [6591.
По спектрофотометрическим, потенциометрическим и кондуктометри-
ческим данным в пропандиолкарбонате (L) присутствуют комплексы
[Со (CN)5L]3~ (тригонально-бипнрамидальный) и [Со (CN)2L2] (тетраэдриче-
ский) [6601.
Напротив, в этиленсульфите ион [Со [CN)5ES]3- имеет псевдооктаэдри-
ческую структуру [661]. Такой же комплекс образуется и в НМРА, наряду
с тетраэдрическими [Со (НМРД)2 (CN)21 и [Со (НМРА) (CN)3]- [662].
В ацетонитриле (L) установлено образование комплексов [Со (CN)2L21,
[Со (CN)3L]~ и [Со (CN)5]3~. Два последних комплекса электрохимически
обратимо восстанавливаются до [Со (CN)3L]2“ [445]. Из цианидных комплек-
сов металлов других групп следует упомянуть [Сг (CN)5 (DMFA)]2-, обра-
зующийся при фотолизе [Bu4Nl3 [Сг (CN)61 в DMFA [663].
3.7.3. Исследование цианидов неметаллов
Наиболее полно изучен трицианид фосфора, который согласно данным
ИК-спектроскопии, масс-спектрометрии и эбулиоскопии существует в виде
ассоциатов [Р (CN)3]W (п = 2 в диоксане, ацетонитриле, бензоле и п = 4 в
нитрометане) [664].
Как показало изучение НК- и ПМР-спектров [665], Р (CN)3 в ацетонитри-
ле взаимодействует с пиридин-И-оксидом, трнфенилфосфиноксидом и DMSO
с образованием Оксицианида фосфора РО (CN)3.
Акцепторная способность Р (CN)3 подтверждается данными изучения
ПМР-спектров систем Р (CN)3 — Ру — ацетонитрил, Р (CN)3 — L — бен-
зол (L = DMFA, DMAA). В последней системе образуются сольватокомплек-
сы состава 1 : 6. Подобные комплексы образуются в бензоле между PhP-
-(CN)2 и DMFA состава 1 : 4 и ICN и DMFA типа 1 : 2. Ранее было выделено
соединение ICN * Ру [666].
Цианиды S (CN)2, Se (CN)2 и Те (CN)2 в ацетонитриле реагируют с CN-
с образованием соответствующего иона NCX (X = S, Se, Те) [667].
Теллуроцианаты образуются также при взаимодействии KatCN (Kat =
= Et4N+, Me4N+, Ph4As+) с теллуром в DMFA [6681 и ацетонитриле [669].
3.7.4. Исследование цианокомплексов
в солевых расплавах
Цианидные комплексы образуются и в расплавленных солях щелочных
металлов.
В расплавленном KNCS показано существование цианидных комплексов
Сг3+ [6701, Cu+, Ag+, Au+, Hg2+, Zn2+, Cd2+ [6711, Pb2+ [672] (табл. 3.6).
Для Co2+ имеет место взаимодействие
ЗСо2+ + NCS~ + 1 ICN- -> CoS + 2 [Co(CN)6]3~, (3.53)
а при наличии избытка CN- CoS растворяется
2CoS + 11CN~ + NCS~-> 2 [Co (CN)6]3~ + 2S2-. (3.54)
Fe2+ с цианид-ионом образует осадки K4 [Fe (CN)6] и K2 Fe [Fe (CN)6]
[67Й. В этом же расплаве при 190° С спектрофотометрически выявлены
90
Таблица 3.6. Логарифмы констант образования цианидных комплексов в расплавленных средах
Ион металла Метод Исследования Состав расплава t, °C 1g Pi 1g P2 1g Рз lg P4 Литера- тура
Си+ Полярография KNCS 195 5,70 1671]
*Си+ Потенциометрия КС1 —59 мол.% LiCl 370 2,38 [680]
Ag+ Полярография KNCS 195 3,20 1671]
*** Ag+ Потенциометрия KNCS 178—204 3,23 [672]
*** Ag+ » KNCS —25 мол. % NaNCS 140 3,15 »
Cu+ Спектрофотометрия KNCS 190 7,25 1674]
** Ag+ Потенциометрия NaNO3 — KNO3 (1:1) 250 10,32 [675]
** Ag+ » » 246 4,04 7,89 [676|
** Ag+ » » 248 4,04 8,04 [677]
Ag+ » KNO3 — 43 мол.% LiNO3 150 13,5 [6781
* Ag+ » KC1 — 59 мол.% LiCl 370 1,16 (1,H) 1,79 (1,85) [679]
Au+ Полярография KNCS 195 11,23 1671]
Zn2+ » KNCS 195 5,1 6,6 8,65 »
Cd2+ » KNCS 195 1,8 3,9 5,15 »
Hg2+ » KNCS 195 6,50 »
*** рь2+ Потенциомет рия KNCS 180—185 2,36 [6721
* Pb2+ » KC1 —NaCl (1:1) 706 1,99 3,39 4,72 1681]
* Co2+ » KC1 —59 мол.% LiCl 450 1,38 2,11 [680]
Ni2+ Спектрофотометрия KNCS 190 19,5 [673]
Ni2+ » » 190 16,45 [674]
* Sn2+ Потенциометрия KC1 — NaCl (1 : 1) 700 1,68 [812]
* Константы образования в моляльных единицах.
** То же 6 молярных единицах.
Концентраций в ионных долях.
комплексы Fe [Fe (CN)6F , [Fe (CN)6]4 , [Co (CN)6]3 , [Ni (CN)J2 , [Cu •
• (CN)21—[673, 674].
В эквимолярной смеси NaNO3 — KNO3 установлены комплексы Ag2CN+,
AgCN, [Ag (CN)2]- [675—677], а в эвтектике KNO3 — 43 мол.% LiNO3 —
только [Ag (CN)2]_ [678] (табл. 3.6). Такой же комплекс, имеющий, вероят-
но, искаженную тетраэдрическую структуру, существует в расплаве КО —
59 мол. % LiCl при 370° С [679]. В этой системе по потенциометрическим дан-
ным предполагается образование CuCN, Pd (CN)2, [Ni (CN)412-’, [Co (CN)4]2-,
причем для последних двух вероятен переход от тетраэдрической к квад-
ратно-плоскостной структуре [6801 (табл. 3.6).
В хлоридных расплавах не исключено существование смешанных комп-
лексов, например [PbCNCl3J2~, [Pb (CN)2C12]2~, [Pb (CN)3C1]2~, образование
которых предполагается в эквимолярном расплаве КО—NaCl [681]. По
потенциометрическим данным рассчитаны константы образования цианидных
комплексов PbCN+, Pb (CN)2, Pb (CNK [681] и SnCN+ [812] (табл. 3.6).
3.8. СТРУКТУРА ЦИАНИДОВ
Строение цианидных соединений исследуется с помощью таких методов,
как электронная, колебательная, радио- и у-спектроскопия, измерение маг-
нитной восприимчивости и рентгеноструктурный анализ [254].
3.8.1. Применение колебательных спектров
для выяснения строения цианидных комплексов
Частоты v (CN) преимущественно валентных колебаний связей азот —
углерод в спектрах водных растворов цианидов щелочных металлов близки
к значению 2080 см-1 [161, 682], a v (CN) линейной молекулы дициана со-
ставляет 2068 см-1 [683]. Для кристаллических ионных цианидов эта частота
в зависимости от природы катиона может несколько смещаться в ту или иную
сторону.
Координированная CN-группа имеет три системы полос в ПК-спектрах
поглощения и спектрах КР: одну в области 1900—2250 см-1 и две в области
от ~100 до ~600см~1 [146, 682]. Полосы в области 1900—2250 см-1 обычно
относят к валентным колебаниям связей CN, а низкочастотные полосы — к
валентным колебаниям связей М—С и деформационным колебаниям М—C=N
и С—М—С.
Частоты v (CN) в цианидных комплексах имеют как повышенные, так и
пониженные (чаще всего повышенные) значения по сравнению с частотой
С№"-иона.
Образование сильной М—С о-связи приводит к упрочнению связи CsN
(поэтому v (CN) растет), в то время как проявление — рл-взаимодействия
металл — лиганд ослабляет ее, увеличивая вклад резонансной структуры II
(v (CN) уменьшается),
М—(=NoM=C=NC
I II
Следовгтельно, наблюдаемое положение частоты v (CN) в комплексных
цианидах является результатом комбинации о- и л-связывания [142, 254].
92
Мерой л-связывания металл — углерод в гексацианидных комплексах
может служить интегральная интенсивность одной из полос валентных ко-
лебаний CN-группы, активной в ИК-области (Iv6) [2671. Ее значение растет
параллельно увеличению частоты валентных колебаний металл — углерод
v (МС) в ряду Сг < Мп < Fe < Со для соединений Cs2Li [М (CN)6]
(табл. 3.7) [254].
Положение частот валентных колебаний v (CN) в ИК-спектрах циано-
комплексов зависит от: 1) электроотрицательности; 2) степени окисления и
3) координационного числа металла. Влияние электроотрицательности вид-
но из повышения v (CN) в последовательности [Ni (CN)4l2-< [Pd (CN)412~ <
< [Pt (CN)4]2~, где электроотрицательность Ni (II) наименьшая [684], и в
ряду [Pd (CN)2 (PEt3)2] < [Pt (CN)2 (PEt3)2] [222]. С ростом степени окисле-
ния металла увеличивается v (CN), например, для комплексов [V (CN)61"—
(п = 2 - 3) [685], [Мп (CN)6]"~ (п = 3 5) [254], [Fe (CN)6F~ (п = 3, 4),
цианидных соединений Pt (II, IV) [146]. Для [Со (CN) (РС2Р)2] и [СО (CN) •
• (РС2Р)2]+ v (CN) соответственно равна 2045 и 2080 см-1 [222]. Повышение
координационного числа металла вызывает уменьшение v (CN), как, напри-
мер, в рядах: [М (CN)4]3~ < [М (CN)3]2~ < [М (CN)2]~ (М = Ag, Си)
[157, 16П; [Hg (CN)4]2~ < [Hg (CN)31“ < Hg (CN)2 [102]; [Ni (CN)2 (PEt2
• Ph)3] < [Ni (CN)2 (PEt2Ph)2l [2221.
Такие изменения связаны с перераспределением электронной плотности
внутри CN-группы (структура I <-> структура II), т. е. с уменьшением или
увеличением роли дативной л-компоненты связи М—CN. В частности, уве-
личением роли связи М-2- CN в цианокомплексах с ростом числа d-электро-
нов атома металла и уменьшением степени его окисления можно объяснить
изменения колебательных частот у многих гексацианокомплексов металлов
(табл. 3.7 и 3.8).
Как показал теоретический анализ колебаний координированных CN-
групп [153], повышение частоты v (CN) при комплексообразовании мо-
жет быть обусловлено не только увеличением силовой постоянной CN-свя-
зи, но также изменением механики колебаний системы. В этих случаях вклад
эт-связи в связь металл — углерод невелик.
По характеру полос v (CN) при наличии в комплексе двух CN-групп мож-
но различить цис- и транс-расположение этих лигандов. Синглетный ха-
рактер полос указывает на транс-, а дублетный — на цис-структуру. По это-
му признаку приписывают транс-структуру [Pd (CN)2 (NH3)2] и цис-строение
[Pd (CN)2en] [5951, [Pt (CN)2L1 (L = en, 2NH3, 2tu, 2Py) [1411; различают
также цис- и транс-изомеры [Pt (CN)2 (NH3)21 и транс-соединения [Pt (CN)2X2
(NH3)21 (X = Cl~, Br“, I~, OH") [6861.
Тем не менее еще нет единого взгляда на природу низкочастотных полос,
обнаруживаемых в цианидных соединениях. Одни авторы [146, 465, 682,
687] считают, что в области 600—400, 400—250 и около 100 см-1 в ИК-спек-
трах поглощения соответственно проявляются валентное колебание М—С
и деформационные колебания М—C^N и С-—М—С. На соединениях плати-
ны замечено, что изменение ее степени окисления и переход от одного типа
цианокомплексов к другим (в случае немостиковых цианогрупп) влияет в
основном на положение частот ~450— 550 см-1. Это подтверждает правиль-
ность отнесения этих величин к v (МС) [146].
Другие исследователи [254, 267, 685, 688], напротив, относят большую
частоту к деформационным колебаниям 6 (MCN) (табл. 3.8). Методом мече-
ных атомов 13С и 15N была показана правильность такого отнесения на
93
Таблица 3.7. Колебательные частоты v (МС) и v (CN) некоторых цианидных
комплексов d-металлов, см-*
Комплекс Число d-электро- нов на t2g МО Vi (CN) * v2 (МС) * v6 (CN) v8 (MC) Jve
[Сг (CN)6]3- 3 2142 380 2138 362 2100
[Mn (CN)6J3— 4 2138 395 2125 384 8200
[Fe (CN)6]3~ 5 2140 411 2128 407 12 300
[Со (CN)6]3~ 6 2161 432 2140 428 18 300
* Спектры КР.
Таблица 3.8. ИК-спектры поглощения некоторых немостиковых цианидов
в твердом состоянии *, см—1
Соединение v (CN) v (MC) 6 (MCN) Литература
К [An (CN)2 Cl2] 2181 430 456 [688]
K[Au(CN)2 I2] 2166 430 448 »
K3 [V(CN)e] 2105, 2087, 2062 367, 340 478 [685]
К* [V (CN)6] 2083, 2066 349 448 »
K4 [Mo(CN)8] 2130, 2125, 2097 420, 373, 361 [317]
Ka[W(CN)8] 2130, 2125, 2097 486, 405 »
K5 [Mn (CN)6] 1934 452, 380 ** 605 [254]
K5 [Tc(CN)6] 1949 463, 416** 568 »
K5 [Re (CN)e] 1932 436** 570 »
K4 [Mn (CN)6[ 2060 386 526 »
K3 [Mn (CN)J *** 2112 361 483 »
Ca2 [Fe (CN)6] 2060 595 447 [146]
Cas [Fe (CN)6]2 2134 506 410 »
K4 [Ru (CN)6[ 2048 546 374 [465]
K2 [Pt(CN)6] 2190 484 416 [146, 153]
K2 [Fe (CN)5 NO] 2156, 2144 424, 417 [465]
K2 [Ru (CN)5 NO] 2215, 2165 •415 »
K2 [Os (CN)5 NO] 2200, 2160 449
K2 [Pt (CN)J 2150 [684]
инс-jPt (NH3)2(CN)2] 2138, 2123 452, 441 397 [686]
транс-[Pt (NH3)2 (CN)2] 2123 458 407 »
транс-fPt (NH3)2(CN)2C1J 2174 474 41'2 »
♦ Степень гидратации солей не указана.
•* Спектры КР.
*** Водные растворы.
Таблица 3.9. Некоторые цианидные соединения с мостиковой CN-группой
Соединение v (CN), cm —1 Тип связи Литера- тура
Cu3 (NH3)4 (CN)4 2130, 2100 Cu—CN—Cu [2H]
Си (Ph3As)2 CN 2125 Cu—CN—Cu [219]
CuCN-PrNH2 2128, 2098 Cu—CN—Cu, Cu—CN [207]
AuCN 2239 Au—CN—Au [157]
Zn (PrNH2)2 (CN)2 2200, 2155, 2100 Zn—CN—Zn [41]
Cd (BuNH2)2 (CN)2 2180 Cd—CN—Cd [40]
Hg (CN)2 2193 Hg-CN-Hg [157]
HgPy2 (CN)2 2205, 2195, 2185 Hg-CN-Hg [44]
K2Fe [Mo (CN)8] 2135, 2122, 2100 Mo—CN—Fe [301]
KNi [Cr (CN)6[ 2172 Cr—CN—Ni [G92]
Cu2 [Fe (CN)e] 2103 Fe—CN—Cu [154]
Al4[Fe(CN)6]3 2128, 2078 Fe—CN—Al »
Ba3 [FeCo (CN)n]- 16H2O 2130, 2090, 2055 Co—CN, Fe—CN, Fe—CN— [691]
Co
Ag3 [Co (CN)6[ 2187, 2130 Co—CN, Co—CN—Ag »
KMn [Co (CN)6] 2168 Co—CN—Mn [692]
(NH3)5 CoNCCo (CN)5 2180, 2120 Co—CN—Co, Co—CN [542]
Pd (CN)2.NH3 2210, 2145, 2135 Pd—CN, Pd—CN—Pd [595]
Pt (CN), 2218 Pt—CN—Pt [141]
Cu [Pt (CN)J 2207, 2177 Pt—CN—Cu [695]
Rh (CN)2Cl-2Py 2201, 2150 Rh—CN, Rh—CN—Rh [595]
примере комплексов [Au (CN)2X2]“ (X = С1_, Вг“, Г) [688]. Эти частоты,
как и v (CN), сильно зависят от природы центрального атома металла (степе-
ни окисления, координационного числа, числа d-электронов), природы внеш-
несферных ионов, а для тетрацианокомплексов платины (II) — еще и от
степени гидратации комплексов. Обезвоживание последних ведет к исчезно-
вению высокочастотных полос v (CN) [689].
Характеристические частоты CN-групп изменяются при замещении од-
ной из них другим лигандом [690]. Так, введение более сильного л-акцептора
NO приводит к сильному увеличению v (CN) и уменьшению v (МС) соответ-
ственно на 150 и 130 см-1 (табл. 3.8). Замечено, что для соединений
К2 [М (CN)5NO1 • 2Н2О (М == Fe, Ru, Os) наиболее сильная л-связь М—NO
наблюдается в случае рутения [465].
Амбидентатность CN--иона (см. 3.4.3) проявляется в существовании мо-
стиковых цианидных соединений, как однородных (связь М—CN—М), так
и смешанных (М—CN—М') (табл. 3.9). Первый тип таких структур наблю-
дается у простых цианидов Ag, Au (I), Си (I), Pd, Ni, Hg [6911, в координа-
ционных соединениях [M (CN)2L1 (М = Zn, Cd, Hg; L = MeNH2, PrNH^
BuNH2, Py) [40, 41, 44], CuCN • 2L (L = NH3, PrNH2, BuNH2) [207] и др.
К другому типу относятся гексацианоферраты (II) р- и d-металлов (кро-
ме Cd) [154], гексацианоферраты (III) Си, Ni, Со, Zn, Cd [6921 и РЗЭ [6931,
гексацианокобальтаты (III) и хроматы (III) Мп, Fe, Со, Ni, Си, Zn [694]
и РЗЭ [733], гексацианоиридиаты (III) Мп, Fe, Со, Ni, Си, Zn [610] и тетра-
95
цианоплатинаты (II) Си, Ag, Zn, Cd, Hg, Ni [695] и частично Mg, Ca, Sr,
Ba [696].
Образование мостиковых CN-групп обычно сопровождается повышением
v (CN), тем большим, чем прочнее связь N->M' [146, 691, 697]. Частоты
v (CN) одноядерных комплексов, имеющих одинаковое отношение (п) коорди-
национного числа и формальной степени окисления центрального атома,
попадают в относительно узкий частотный интервал. Например, для п = 2
v (CN) близки к 2130 см-1, а для мостиковых структур они повышаются на
-—30—70 см-1 [153, 691, 695, 697]. Так, частотный интервал для немостико-
вых цианидов Pt (II) и Pt (IV) составляет соответственно —2120—2140 и
—2160—2190 см-1, а для мостиковых —2150—2210 и 2200—2240 см~* [697].
Образование дополнительных связей атома металла с азотом упрочня-
ет связь CN. Помимо изменения силового поля внутри CN-группы, что
является основным фактором, изменение механики колебаний также вызы-
вает рост v (CN) [153].
Чем прочнее связь N ->М', тем менее растворимым должно быть соеди-
нение. Такая зависимость обнаружена для мостиковых гексацианоферратов
(II) [154] и тетрацианоплатинатов (II) р- и d-металлов [695].
Для мостиковых CN-групп наблюдается также повышение частот 6 (MCN)
по сравнению с концевыми CN-группами. Например, частотный интервал
6 (MCN) для солей типа Na2 [Pt (CN)4] составляет 400—420 см-1, а для Kat •
• [Pt (CN)4] (Kat = Cu, Zn, Ni) — 475—500 см-1 [695].
В некоторых мостиковых цианидах, например Fe3 [Мп (CN)6]2, Kat3 [Сг •
• (CN)6]2 (Kat = Со2+, Fe2+), KCr [Fe (CN)6], наблюдается изомерия свя-
зи цианомостиков (перестройка типа М—C=N—М' -> М—\^С—М')
[692, 698].
Весьма вероятно, что цианидные мостики образуются в редокс-реакциях,
которые проходят с участием цианокомплексов, причем связь осуществляет-
ся цианосоединением М—CN и окислителем или восстановителем М'. После-
довательное расщепление этих мостиков может приводить к образованию
малоустойчивого N-связанного (изоцианидного) комплекса. Так, комплекс
[Сг (H2O)5NC12+ с периодом полураспада 1,78 мин при 15° С обнаружен в
процессе восстановления Сг (II) соединений [Со (NH3)5CN)]2+, транс-[Со (CN) •
• (NH3)4 (НоО)]2+, транс-[Соеп2 (H2O)CN]2+ в кислых перхлоратных раство-
рах [698].
ИК-спектрально показано присутствие изоцианидов Me3MNC (М = Si,
Ge) как примеси (10%) к нормальным цианидам [68] и NaBH3NC в смеси
(1 : 4) с NaBH3CN [699].
Хотя изоцианидные соединения имеют пониженную устойчивость, по-
средством электронных и ИК-спектров показано, что цис-а-[Со (trien) (NC)2]
и цис-|3-[Со (trien) (NC)2], где а и [3 относятся, к различным типам координа-
ции тетрадентатного лиганда trien, весьма стабильны [700]. Другие авторы
подвергли сомнению существование таких необычных структур, полагая,
что они являются на самом деле транс-цианосоединениями [588].
Исследование ИК- и спектров КР цианидных соединений может быть так-
же полезным в установлении стереохимической конфигурации комплексно-
го аниона, например додекаэдрической структуры [М (CN)8]4~ (М = Мо, W)
в твердом состоянии [310—313, 317, 323], пентагонально-бипирамидальной —
иона [Мо (CN)7]4~ в растворе и тригонально-призматической в кристалли-
ческом состоянии [279], квадратно-пирамидальной структуры [М (CN)513~
(М = Со, Ni) в водном растворе [7011.
Однако из колебательных спектров не всегда можно сделать однозначный
96
вывод о структуре цианидных соединений. Это, в частности, касается дис-
куссионного вопроса о строении ионов [Мо (CN)8]4~ и [W (CN)8]4— в водном
растворе (симметрия D2d или D4d) [312, 317, 3231. Более достоверные сведе-
ния дает рентгеноструктурное исследование цианидов.
3.8.2. Изучение кристаллической структуры
цианидных соединений
нейтронографически для
Долгое время по данным рентгеновского исследования не могли одно-
значно установить способ координации CN-группы.Лишь в 30—40-х годах
нашего столетия для соединений К [Ag (CN)2], К4 [Мо (CN)81 и К4 [Fe (CN)6]
удалось показать, что атом углерода наиболее близок к атому металла [73].
Такой способ координации был установлен и
Hg(CN)2[34], К2 [Zn (CN)J
[145], К3 [Co(CN)6] [144].
Для кристаллохимии ко-
ординационных цианидов ха-
рактерно большое многооб-
разие кристаллических струк-
тур с координационными чис-
лами атома металла от 2 до 8.
Линейная структура с
к. ч. 2 наблюдается в простых
цианидах серебра и золота
(I), имеющих полимерный ха-
рактер в кристаллическом со-
стоянии, и в комплексных со-
единениях К [М (CN)2] (М =
= Ag, Au) [27, 182, 183], со-
единениях AgCN • 2AgNO3,
О (HgCN)2, Hg (CN)2 [27],
Hg (CN)2 • AgNO3 • 2H2O
[702]. Структуры линейных
комплексов [M (CN)2]~ (М =
= Ag, Au) отличаются лишь
упаковкой катионов и анио-
нов, поэтому К [Au (CN)2]
имеет ромбоэдрическую ячейку [183], а К'[Ag (CN)2] — гексагональную
[182].
Структура Hg (CN)2 • AgNO3 • 2Н2О состоит из почти линейных цепей
—Ag—NC—Hg—CN—Ag— [702].
В отличие от серебра и золота, медь (I) проявляет тригональную коор-
динацию в солях К [Си (CN)2] [184, 185] и К [Cu2 (CN)3] • Н2О [186].
В К [Си (CN)2] медь связана через углерод с двумя CN-группами (Z_CCuC =
= 134,2°) и через азот с третьим CN-лигандом. Анион имеет вид спиралевид-
ной цепи, состоящей из атомов Си и CN-rpynn [184] (рис. 3.3). К- ч. 3 имеет
медь и в соединении К [Cu2 (CN)3] • Н2О, содержащем плоские полимерные
анионы [Cu2 (CN)3]oo [186].
В соли Cs [Hg (CN)3] анионы также практически плоские; отклонение
атома ртути от плоскости ЗС составляет лишь 0,169 А [703]. А в соединениях
Kat [Cd (CN)3] (Kat = Rb, Cs) образуются зубчатые цепи тетраэдров Cd (CN)4,
имеющих общие вершины [90].
4 2473
97
Таблица 3.10. Основные структурные и кристаллографические данные некоторых цианид
Соединение К. 4. Координационный полиэдр
CuCN-N2H4 4 Искаженный тетраэдр С, N, N2H4
Снзеп2 (CN)4-H2O 4 Тетраэдр 4С
Cuatren2 (CN)2 (BPh4)2 5 Тригональная бипирамида С, 4N (tren)
K5 {[Au (CN)2]4 [Au (CN)J21) -2Ho0 1—2 Гантель 2С
I II II—4 Квадрат 2С, 21
H |Au (CN)4] • 2H2O 4 Квадрат 4С
Cs2 [Zn (CN)4] 4 Тетраэдр 4С
Cs[Hg(CN)3] 3 Треугольник ЗС
Hg (CN)2-2tu 6 Искаженный октаэдр 2С, 4S
Hg (CN)2. AgNO3-2H2O 2 Гантель 2С
Cd (NH3)2-Hg (CN)4-2CeHe 4 (Hg) Тетраэдр 4CN
6 (Cd) Октаэдр 2NH3, 4NC
K4 [V (CN)7[-2H2O 7 Пентагональная бипирамида 5С, 2С
[Co en3] [Cr (CN)5 NO]-2H2O 6 Искаженный октаэдр 4С, С, NO
Кз [Cr (O2)2 (CN)3J 7 Пентагональная бипирамида 40, С, 2С
K4 [MoO2 (CN)4].6H2O 6 Октаэдр 4С, 20
(Bu4N)3 [Mo (CN)8] 8 Додекаэдр 8С
Na3 [W (CN)8]-4H2O 8 Антипризма 8С
K3 [ReO2 (CN)4] 6 Октаэдр 4С, 20
Fe4[Fe (CN)6]3.14H2O 6 Октаэдр 6С
Ba3 [Co2(CN)10]-13H2O* 5 Квадратная пирамида 5С
[Co dien2] [Co (CN)6] • 2H2O 6 Октаэдр 6С
[(NHS)5 CoNCCo (CN)5]-H2O 6 Октаэдр 5С, С
[Cr (NH3)6][Ni (CN)5]-2H2O 5 Искаженная квадратная пирамида 4С, С
[C14H18N3[ [Ni (CN)4J-3H2O 4 Квадрат 4С
Rb4 [Ni2 (CN)6] 4 Искаженный квадрат
fNi (Etphos)3 (CN)2] 5 Тригональная бипирамида 2С, ЗР
Mn2 [Ru (CN)6]-8HaO 6 Октаэдр 6С
Cd [Pd (CN)6] 6 Октаэдр 6С
[Hi en2] [Pd (CN)4[ 4 Квадрат 4С
[Et2NH2]2 [Pt (CN)J 4 Квадрат 4С
Kat [Pt (CN)6[ (Kat = Mg, Ca, Mn, Fe, Co, Ni, Zn, 6 Октаэдр 6С
Cd)
* Указанный полиэдр относится к труппам ' [Со (CN)b]3~, соединенным связью Со-= Со <2,794 А).
98
ных соединении
а ь С а V Z Простран- ственная группа Литера- тура
4,684 9,172 7,830 4 Pbcm [209]
14,774 7,749 14,272 112,65 4 Сс [212]
13,792 10,338 20,316 94,27 2 P2i/c [726]
11,592 7,309 9,825 111,27 99,91 92,46 1 Pl [704]
7,881 10,129 6,798 123,3 2 P2i/c [708]
14,469 9,198 8,575 109,69 4 С2/с [90]
8,247 16,880 9,969 8 Pbca [703]
30,18 19,32 8,00 16 С222 [727]
16,743 7,251 10,622 90,05 6 Cg—12 [702]
8,534 8,537 14,619 90,5 89,2 89,3 2 Pl [54]
9,229 9,097 9,341 90,02 92,49 90,00 2 II [242, 243]
13,472 11,591 13,392 100,5 4 Р2г/с [745]
11,526 7,628 12,1:8 111,12 4 P2/a [286]
7,245 13,548 9,434 113,7 2 P2t/c [748]
17,009 22,784 4 P4/ncc [357]
6,126 16,13 17,42 94,75 4 P2i/c [360]
0,766 0,745 0,629 109,6 105,9 115,4 1 pF [406]
10,179 1 Pm3m [738]
9,411 20,822 15,13 93,58 4 C2/c [724]
13,942 32,370 9,418 8 P22t2 [735]
17,359 12,187 13,936 8 Pbca [751]
23,102 11,529 11,735 8 Pcca [722]
7,02 19,31 15,53 4 Pnnm [714]
12,26 16,80 7,65 90,0 100,6 110,0 4 pT [720]
11,67 26,60 15,02 125,51 4 P2,/b [723]
9,488 12,494 7,606 98,8 2 P2i/n [744]
10,911 4 Fm3m [742]
7,174 10,740 10,135 115,0 2 P2i/n [717]
15,85 9,06 6,35 83,18 93,3 94,1 2 СГ [716]
10,60 4 Fm3m [619]
4*
99
Заслуживает внимания структура соли К5 [Au5 (CN)10I2] 2Н2О, в ко-
торой золото находится в степенях окисления +1 и 4-3. В этом соедине-
нии имеются слои линейных [Au (CN)2]~ и квадратно-плоских комплексов
[Au (CN)2I2]~ в отношении 4:1, которые поочередно связаны со слоями
ионов К+ и молекул воды [704] (табл. 3.10).
Для цианидов Си (I) и металлов подгруппы цинка характерно тетраэд-
рическое расположение лигандов, а для соединений Ni, Pt (II), Pd — квад-
ратно-плоскостное .
Наличие тетраэдрической конфигурации лигандов установлено в
К3 ICu (CN)J [705], простых цианидах цинка и кадмия [27], солях К2 IM (CN)4]
(М = Zn, Cd, Hg [271, Cs2 [Zn (CN)4], изоструктурных Tl2 [M (CN)4] (M =
= Zn, Cd, Hg) [90], соединениях типа Me2M (CN)2 (M = Si, Ge) [706], а квад-
ратно-плоскостной — в Kat [Au (CN)J • /гН2О (Kat = H+ [708] и K+ [709]),
а также в [M (CN)4]2~ (M = Ni, Pd, Pt) [603, 710, 7171. Соли Na9 [M (CN)41 •
• 3H2O 1710], Ca IM (CN)4] - 5H2O [711] и [Py2CCl IM (CN)4] [712] изострук-
турны для M = Ni, Pd, Pt, a Rb2 [M(CN)4] • H2O — для M = Pd, Pt [713].
Исследована кристаллическая структура цианидов с объемными органиче-
ским и комплексным катионами: [Ру2С9] [Ni (CN)41 [714], (Et2NH9)2 [Pd (CN)J
[715], Kat2 [Pt (CN)4] (Kat = EtNH3, Et2NH2, Et3NH) [7161 и [Nien2] [Pd -
• (CN)41. В последнем соединении два транс-атома азота квадрата [Pd (CN)4]2~
связаны с никелем в виде бесконечных цепей —Pd—С—N—Ni—N—С
[717].
Приблизительно квадратная координация атома ртути существует в соль-
вате Hg (CN)2 • МеОН [48].
Интересно, что структуры квадратно-плоскостных солей Kat [Pt (CN)4]
(Kat = Mg2+, Ca2+, Sr2+, Ba2+) содержат непрерывные Pt-цепи с различным
расстоянием Pt — Pt, наименьшим (3,09 А) для соли Sr [Pt (CN)4] • ЗН2О
[603]. Существование таких цепей отмечается и в структурах соединений
К2 lPt(CN)4] Хо.з • ЗН2О (X = О, Вг) [602, 718], причем нейтронографиче-
ски показано некоторое отклонение конфигурации [Pt (CN)4]2~ от плоской
[718].
Искаженный тетраэдр реализуется в соединениях CuCN • L (L = NH3,
N2H4). В первом соединении он образован двумя атомами углерода соседних
CN-групп, молекулой NH3 и атомом азота CN-группы [2081, а во втором —
двумя группами NH2 —, одним атомом N и одним атомом углерода CN-rpynn
[209]. В соединении CuCN • NH3 наблюдается изогнутый CN-мостик с углом
CuCCu 69,9° и расстояниями Си—С 2,13 А, С—N 1,13 А и Си—Си 2,418 А
следующего вида:
Си
in
/Сч
— C=N —СиК \Cu-N=C-
XCZ
• II!
N
Си
I
100
Полимерные цепи в обеих структурах соединены молекулами NH3 или
N2H4.
В цианидах меди, содержащих разновалентные атомы металла, существу-
ет различная координация для Cu (I) и Си (II).
Структура Cu3en2 (CN)4 • Н2О построена из трехмерного каркаса полимер-
ных анионов [Cu2 (CN)4J^~, содержащих атомы Cu(I) тетраэдрической коор-
динации, которые объединены при помощи мостиковых CN-групп. Коорди-
нация Cu (II) в катионе квадратно-пирамидальная с двумя бидентатными мо-
лекулами еп и молекулой Н2О в вершине [212]. Другие авторы [7071 полага-
ют, что в рассматриваемом соединении, как и в Cu3 (NH3)3 (CN)4, Cu3 (NH3)4-
• (CN)4, координационный по-
лиэдр для Cu+ — деформирован-
ный тетраэдр, а ион Си2+ имеет
октаэдрическое окружение.
Ранее указывалось на су-
ществование искаженного тет-
рагонально - бипирамидального
окружения Cu (II) в соедине-
нии Cu3 (NH3)3 (CN)4 [2171. Од-
нако последние рентгенострук-
турные данные показали нали-
чие искаженной тригонально-
плоскостной координации CN- Рис. 3.4. Структура аниона lNi2 (CN)6]4~ (калие-
группами атомов Cu (I) и иска- вая соль [719])
жен но-октаэдр ической — атомов
Cu (II) (три молекулы NH3, распределенные по четырем положениям, и
два атома азота CN-групп). Интересно, что четыре атома Cu (I) и атом Си
(II) соединены мостиковыми CN-группами с образованием почти плоского
пятиугольника [2161
Тщательное рентгеноструктурное исследование солей Kat4 [Ni2 (CN)6
(Kat = K+ 17191, Rb+ 17201) однозначно решило спорный вопрос о структуре
димерного аниона в пользу осуществления сильной связи Ni—Ni длиной
2,29—2,32 А (рис. 3.4).
Большой интерес представляет структура цианидных клатратов, напри-
мер М (NH3)2M' (CN)4 • L (М = Мп, Fe, Со, Ni, Cu, Zn, Cd; M' = Ni, Pd,
Pt; L = C6H6, C6H5NH2, C4H5N) [541, Cd (en)Ni (CN)4 • 2L (L = C6H6 [7211,
C4H5N [813]) и Cd (NH3)2 Hg (CN)4 • 2C6H6 [54]. Рентгенографически пока-
зано, что в соединениях Ni, Pd, Pt осуществляется квадратно-плоскостная
координация атома М' и октаэдрическая — атома М, а в Cd (NH3)2Hg (CN)4 •
- 2С6Н6 атом Hg имеет тетраэдрическое и атом Cd — октаэдрическое окру-
жение (табл. 3.10). За счет мостиковой функции CN-групп структура клат-
ратов носит трехмерный сетчатый характер с вкрапленными в сетку молеку-
лами L.
Рентгеноструктурно установлено, что в пентацианоникелатах и кобаль-
татах осуществляется квадратно-, тетрагонально-пирамидальная и триго-
нально-бипирамидальная координация центрального атома. Искаженное
квадратно-пирамидальное окружение имеет Ni в соединениях [CrL3] [Ni •
• (CN)51 • 2Н2О (L = Pn, 2NH3). Все CN-группы участвуют в образовании
водородных связей с молекулами Н2О [7221. Однако в соединении [Сгеп3] •
• [Ni (CN)51 • 1,5 Н2О существуют две стереохимические формы никеля:
квадратно-пирамидальная и искаженная тригонально-бипирамидальная
[4431, которая по данным ИК- и спектров КР превращается в первую ппи
растворении в воде или дегидратации соединения [4441.
10-1
Наличие двух стереохимических форм (тригонально-бипирамидальной
и тетрагонально-пирамидальной) наблюдается и в соединении [Ni (Mephos)3 •
• (CN)2], в то время как однотипный [Ni (Etphos)3 (CN)2] имеет лишь триго-
нально-бипирамидальную структуру [7231. В соли Ва3 [Со2 (CN)1O1 • 13Н2О
каждая группа [Со (CN)5]3~ димерного аниона (расстояние Со—Со 2,79 А)
имеет квадратно-пирамидальную конфигурацию [724, 725]. Такую же кон-
фигурацию имеет атом Со (II) и в соли [NEt2 (i -Рг)2]3 [Со (CN)J [814]. В би-
ядерном соединении меди (II) [Cu2 trien2 (CN)2] [BPh4]2 димерная молекула,
находящаяся в центре симметрии, образована за счет двух Н-связей длиной
3,05 А, в которых участвуют атом азота CN-лиганда и группа NH тетраден-
татного trien Си—CN ... Н—N—Си. Координация атома Си (II) — триго-
нально-бипирамидальная [726].
Октаэдрическое расположение лигандов характерно для большинства
цианидов переходных металлов. Для p-металлов октаэдр реализуется в ад-
дуктах цианида ртути: Hg (CN)2 • 2tu [727] и 5Hg (CN)2 • 7Thf [501/Zn (NO3)2-
• 2Hg (CN)2 • 7H2O. Последнее соединение по структурным данным можно
представить как (Zn (Н2О)4 [Hg (CN)212[ (NO3)2 - ЗН2О [728, 815].
Гексацианидные комплексы d-металлов типа [М (CN)6]4~, [М (CN)6]3~
и [М (CN)612- имеют правильную или искаженную октаэдрическую конфигу-
рацию [737]. Это подтверждено рентгеноструктурными исследованиями изо-
структурных соединений Na4 [М (CN)61 • ЮН2О (М = Mn2+, Fe2+) [729];
Kat2 [Fe (CN)6J (Kat = Cu2+, Fe2+, Co2+, Ni2+) [26]; Cs2M [Fe (CN)61 (M =
= Zn2+, Cd2+, Co2+, Ni2+) [730]; Kat4 [Ru (CN)6]3 • 10H2O (Kat = Fe3+,
AF+) и KAI [Ru (CN)6] • 5H2O [731]; K3 [M (CN)6] (M = Mn3+, Fe3*, Co3+)
[7321; Kat [M (CN)61 • цН2О (M = Fe3+, Co3+; Kat = Ln3+, n = 4 [693, 733],
[Coen3]3+, n = 2 [734]); [Co (dien)2] (Co (CN)6] • 2H„O [735]; Kat [Pt (CN)61 •
- nH2O (Kat = Mg2+, Ca2+, Cr2+, Mn2+, Fe2+, Co2+, Ni2+, Cu2+, Zn2+, Cd2+),
причем для солей Сг2+ и Cu2+ наблюдается тетрагональное искажение окта-
эдра [619]; соединений Pd (IV) Katn [Pd (CN)6] (Kat = щелочной металл,
Ag+, NHf, Mn2+, Zn2+ [736], Cd2+ [742]).
Если катионом соли служит металл, способный к донорно-акцепторному
взаимодействию с азотом CN-группы, то такие гексацианиды переходных
металлов имеют структуру координационных полимеров. Примером такого
типа структур является кубическая гранецентрированная решетка берлин-
ской лазури Fe4 [Fe (CN)6]3 с константой около 10 А [738]. В этом соединении
CN-группы, расположенные на ребрах куба, осуществляют мостиковую
связь между высокоспиновыми (Fe3+) и низкоспиновыми (Fe2+) ионами желе-
за. Первые окружены по октаэдру шестью атомами азота, а вторые — шестью
атомами углерода [30]. Образуется трехмерная полимерная сетка.
Такой тип кубической структуры имеют полиядерные цианиды простого
или смешанного типа Katm [М (CN)6]n • ftEEO (М = Pd4+, Cr3^-, Mn3+,
Fe3+, Co3+, Rh3+, Ir3+, Fe2+, Ru2+, Os2+; Kat" = Mn2+, Fe2+, Co2+, Ni2+,
Cu2+, Zn2+, Cd2+, Cr3+, Fe3+, Ln3+) [26, 737, 739—743, 610, 730, 816].
Иной структурный тип представляет соль Mn2 [Ru (CN)6] • 8Н2О. Трех-
мерный каркас моноклинной решетки составлен последовательностью ато-
мов Ru, Mn, Мп, Ru, а не Мп, Со, Мп, Со, как в кубическом Мп3 [Со (CN)6]2 •
• /гН2О [741]. Характерно наличие искаженных октаэдров RuC6 и MnN3 •
• (Н2О)3 [744].
Рентгеноструктурно установлено, что искаженное (тетрагональное или
тригональное) октаэдрическое окружение центрального атома наблюдается,
например, в [Соеп3] [Сг (CN)5 NO] • 2Н2О [745]; Na? [Fe (CN)5NH3] • 2H2O
[7461; Km [M (CN)5NO] • nH2O (M = V [256], Мп “[416] (т = 3, п = 2),
102
Mo (m = 4)) [3771; K3 [VO (CN)J [747]; K3 [ReO2 (CN)4J [748,406]; Kat [MoO2 ♦
• (CN)41 • 6H2O (Kat = NaK3 [335] и K4 [333, 7491); Na3 [MoO (OH) (CN)4] •
• 4H2O [750]; [(NH3)5CoCNCo (CN)5] • H2O [751] и его цианоизомере [(NH3)5 •
• CoNCCo (CN)5] • H2O [752]; K7 [Mn2O (CN)1O1 CN [395] (рис. 3.1), кислород-
содержащих соединениях K5 [(CN)5CoO2Co (CN)5] • H2O [753] и [Со2 (CN)4 •
• (PMe2Ph)5O2l [537], причем в первом осуществляется мостик Со — О —
О — Со, а в последнем Со — CN — Со.
Исследование структуры солей К4 [Мо02 (CN)4] • 6Н2О и NaK3 [МоО2 •
• (CN)4] • 6Н2О [333, 335, 748] имело особо важное значение, так как позво-
лило установить, что соединения содержат октаэдрический анион [МоО2 •
• (CN)4]4~ с транс-расположением атомов кислорода, а не восьмикоордина-
ционный ион [Мо (ОН)4 (CN)4]4—, как предполагалось ранее [330—332L
Группировка МоО2, как и ReO2 в диамагнитнойосоли К3 [ReO2 (CN)4]o[748],
имеет линейное строение с длиной М—О 1,83 А (Мо) [335] и 1,78 A (Re)
[748]. Такая же линейная группировка, но с мостиковым атомом кислорода
типа О = Re — О — Re = О имеется в соединении [Pt (NH3)4]2 [Re2O3 (CN)8]
с октаэдрическим окружением каждого атома Re [753].
Рентгеноструктурное исследование нитрозилцианида молибдена [377]
окончательно решило вопрос о его составе как К4 [Мо (CN)5NOJ [376], а не
К4 [Мо (ОН)2 (CN)6NO1 [371].
Для семикоординационных цианидных комплексов К4 [V (CN)7] • 2Н2О
[242, 243] и К3 [Сг (О2)2 (CN)3] [286] установлена конфигурация анионов в ви-
де пентагональной бипирамиды. Во втором из приведенных соединений би-
пирамида образована из атомов О и группы CN в основании и двух других
групп CN в вершинах с расстояниями Сг — О = 2,090 А, О — О = 1,446 А,
Сг — С = 2,090 А и £ ОСгО = 45°.
Дополнительным фактором, стабилизирующим семикоординационный
анион [V (CN),]4-, является образование «кольцевой» экваториальной л-ор-
битали за счет большего перекрывания л-орбиталей экваториальных CN-rpynn
по сравнению с октаэдрическими и квадратными комплексами [243].
Из цианидов других d-металлов следует привести комплексы [Мо (CN)8]4—
и [W (CN)8]4~, которые, как и [М (CN)8]3~ (Мо, W), имеют в кристаллическом
состоянии структуру додекаэдра или антипризмы.
Соединения [Bu4N]3 [Мо (CN)8] [357] и К4 [М (CN)8] • 2Н2О (М = Мо,
W) [305—308] имеют додекаэдрическую структуру аниона (симметрия D2d),
в то время как в соли Na3 [W (CN)8] • 4Н2О, изоморфной с Na3 [Мо (CN)8] •
• 4Н2О, наблюдается антипризматическая конфигурация (D4d) [360]. Подоб-
ное явление пытаются связать с различной упаковкой молекул воды вокруг
катиона [360]. По данным ЭПР-спектров Kat3 [W (CN)8] • пН2О на геомет-
рию аниона влияет природа катиона (К+, Na+, Ag+, Bu4N+) [324].
В соединении К4 [Мо (CN)8] • 2Н2О длины связей Мо—С примерно оди-
наковы (2,16 А), хотя додекаэдрическая координация требует большей ве-
личины (Мо — С)д, чем (Мо — С)в- На основании этого полагают, что в до-
декаэдре, в противоположность антипризме, отсутствует существенная до-
полнительная л-дативная стабилизация [305].
Антипризматическая конфигурация установлена для анионов соедине-
ний Н4 [М (CN)81 • 6Н2О (М = Мо, W) [309] и Н4 [W (CN)81 • 4НС1 12Н2вО
[755]. В первом соединении имеются водородные связи О—Н ...N (2,58 А)
со всеми атомами азота аниона и О—Н...О (2,89 А) [3091, а во втором — шесть
атомов N входят в цепи W—C=N...H—C1...N=C—W, которые стабили-
зируют каркас структуры (N...C1 2,38 А). По-видимому, четыре протона об-
разуют линейные цепи C^N...H+ ...Cl [7551.
103
По предварительным рентгеноструктурным данным наблюдается анти-
призматическое окружение молибдена (IV) и в соединении Cd2 [Мо (CN)R] •
• 2N2H4 - 4Н2О, которое ранее считали десятикоординационным [328].
Молекулы гидразина координированы вокруг атомов Cd и образуют водо-
родные связи с азотом CN-групп, а вода в структуре имеет цеолитный
характер [756].
Додекаэдрическую структуру имеют смешанные цианидные соединения
[Мо (CN)4 (CNMe)4] по данным рентгеновского исследования [757] и по дан-
ным спектров Мессбауэра [758]. Последний метод подтвердил антипризмати-
Таблица 3.11. Длина связей и валентные углы в некоторых цианидных комплексах
(экв. — экваториальное положение, акс. — аксиальное направление)
Соединение Длина связи, A ^MCN Литера- тура
M—C C=N
KCu2 (CN)3 1,89 1,15 174° [186J
K3Cu (CN)4 1,99—2,01 1,13 174—180° [705]
К [Ag (CN)2] 2,13 1,15 180° [183]
K2 [Zn (CN)4] 2,024 1,157 180° [145]
K2 [Ni (CN)4J 1,86 1,13 180° [758]
[Ni (Etphos)3 (CN)2] Ba3 [Co2(CN)10]-13H2O 1,82—1,85 1,885 экв. 1,15 176° 173,9—176,8° [723] [724]
Na4 [Mn (CN)6]- 10H2O 1,946 акс. 1,95 1,16 178,4° [729]
K3 [Mo (NO) (CN)5[ 2,135 1,17 (5) 175° [377]
K4[V(CN)7]2H2O 2,15 1,14 178° [243]
[Bu4N]3 [Mo (CN)8] 2,11 1,16 176° [357]
H[W (CN)8]-6H2O 2,18 1,12 (7) 177° [309]
ческую структуру аниона в соединениях Katm [W (CN)8] • nH2O (Kat =
= Н , Na+, Cd +) и додекаэдрическую — в солях К4 [W (CN)J • 2Н,О и
Li4 [W (CN)8] • пН2О [758].
Согласно рентгеноструктурным данным расстояние CN в координирован-
ных цианидах составляет около 1,15 А, что мало отличается от d (CN) в ионе
(1,16 А), дицианес(1,172 А) и HCN (1,156 А) [27] (табл. 3.11). Наименьшая
величина — 1,11 А в Cd [Pd (CN)6] [742], а наибольшая — 1,175 А в К3 [Мо •
• (NO) (CN)5] [377].
Расстояние М—С находится в пределах 1,89—2,2 А, незначительно из-
меняясь с изменением степени окисления металла (например, в [Мо (CN)8]3~
d = 2,14 А, а в [Мо (CN)J4-d — 2,16 А) или положения рассматриваемого
металла в подгруппе (так, d (Сг — С) в [Cr (CN)6]3~ только на 0,1 А короче,
чем d (Мо — С) в К4 [Мо (CN)8] • 2Н2О [254]). Однако в ряду Сг, Мп, Fe,
Со, 1г для соединений Cs2Li [М (CN)6] длина связи М—С уменьшается до
Со (2,046; 1,976; 1,926 и 1,886 А), а затем резко увеличивается для 1г
(2,003 А). Это свидетельствует об усилении dл — ря-взаимодействия от Сг
к Со [759].
В большинстве случаев отклонение от линейности для М—С—N-rpynn
составляет 2—6°. Однако для соединения К4 [V (CN)7] • 2Н2О углы VCN и
104
CVC в аксиальном направлении составляют 172,5 и 171°, что вызвано взаимо-
действием лиганд — катион [243].
Значительное отклонение от линейности может наблюдаться в мостиковых
цианидах, например в биядерных изомерах [(CN^Co1 —С—N—Со2 (NH3)5] •
• Н2О и [(CN)5Cox —N—С—Со2 (NH3)5] • Н2О. В первом соединении
y^biCoxCN hCNCo2 равны 172,4 и 159,8°, а во втором углы CoxNC и NCCo2
составляют 166,5 и 165,6° [751, 752]. Эти отклонения приписывают внутри-
молекулярному отталкиванию лигандов [751].
Некоторые угловые и линейные характеристики CN-групп в цианидных
комплексах металлов приведены в табл. 3.11.
3.9. ПРИМЕНЕНИЕ ЦИАНИДОВ
3.9.1. Цианиды в химическом анализе
Цианиды натрия и калия находят применение в комплексометрии при
определении серебра, никеля, ртути (II) [7611, спектрофотометрическом оп-
ределении Ni2+ в виде [Ni (CN)4]2" [762], микроопределении меди (II) по-
тенциометрическим [763], спектрофотометрическим методами [764], а также
по хемилюминесценции люминола при окислении его смесью Си2+ и CN~
[765]. Серебро и ртуть (II) гасят свечение, на чем основан чувствительный
метод определения этих металлов [766].
Раствор NaCN в изопропиловом спирте применяют для быстрого опре-
деления серы в рудах и промышленных водах [767].
Цианид калия иногда используют для маскирования Со, Ni, Си, Zn, Cd,
Hg при определении свинца, марганца, щелочноземельных металлов, а так-
же для связывания Fe3+ при титровании цинка ЭДТА. Большое преимущест-
во KCN состоит в возможности выделения маскируемых примесей из их циа-
нидных комплексов (после титрования) при действии формальдегида или
хлоральгидрата [768].
В объемном анализе как окислитель успешно применяется BrCN для оп-
ределения Sn2+, 82Оз—, SO2' , S2~, аскорбиновой кислоты [769] и различных
алкил- и арилдитиокарбаматов (наряду с использованием ICN) [770].
Находят применение и комплексные цианиды. Так, [Ni (CN)4]2— исполь-
зуется для косвенного определения Ag, Au, Pd, Pt при анализе их трудно-
растворимых соединений [768]; [Fe (CN)6]3— — в качестве окислителя при
определении гидроксиламина [761], анилина [771]; [Мо (CN)g]3— — для спек-
трофотометрического определения AsO? [772].
На способности [Fe (CN)6]4~ образовывать малорастворимые осадки с не-
которыми органическими основаниями (антипирин, красители и др.) основа-
но его применение в органическом и фармацевтическом анализах, а также
для обнаружения ионов щелочных и щелочноземельных металлов, Ag+,
Cu2+, Zn2+, Cd2+, Hg2+, Fe3+, UO2+ и гравиметрического определения Cd2+
в виде Cd2 [Fe (CN)6], a Ce3+ — в виде KCe[Fe(CN)6] • 4H2O [26].
Известно много титриметрических методик определения металла при
помощи К4 [Fe (CN)e]. Точка эквивалентности устанавливается как с помощью
индикаторов, например определение Zn2+, Cu2+, Ni2+, Cd2+, Pb2+, Fe+2,
Mn2+, Ga3+, In3+, Bi3+, Th4+ в присутствии З^'-диметилнафтидина, так и
кулонометрическими, кондуктометрическими, амперометрическими, потен-
циометрическими и фотометрическими методами ]26].
105
Кондуктометрическое титрование с помощью К4 [Fe (CN)61 предложено
для определения Cd2+, Pb2+, Tl+, La3+ и других катионов [26], а потенцио-
метрическое — для Се4+ [773], Аи3+ [774], VOf [775] и анионов С1~, Вг~,
С1СГ, ВгСГ, сюг, ВгОГ [776], Сг2О2~ [773], МпОГ [775].
3.9.2. Цианидные соединения
в цветной металлургии и гальванотехнике
Давно известно использование разбавленных растворов цианидов ще-
лочных металлов для выделения золота из руд (метод «цианидного выщела-
чивания»). Иногда такой же метод применяют для серебряных руд [156].
Для получения металлических покрытий электролитически осаждают
Ag, Au, Си, Zn, Cd, Hg и некоторые другие металлы, а также их двойные
сплавы из цианидных ванн, содержащих комплексы [М (CN)2]~ (М = Ag,
Au), [Си (CN)3]2-\ [Zn (CN)4]2~, [Cd (CN)3r, [Cd (CN)4]2~, [Hg (CN)4]2“ и
следы [Hg (CN)3] [777]. Для электроосаждения благородных металлов ис-
пользуют также растворы ферроцианидов, так как они гораздо менее ядови-
ты, чем чисто цианидные ванны [26].
Запатентован и неэлектролитический способ покрытия серебром и зо-
лотом металлических поверхностей из ванн, содержащих амины и цианид-
ные комплексы этих металлов [778].
Для нанесения покрытий из платиновых металлов проводят электролиз
расплавов, содержащих цианиды щелочных металлов, при температурах
350—600° С [779, 780].
Обработка металлических поверхностей, например титана или его спла-
вов, цианидами при 800° С улучшает их механические и антикоррозионные
свойства благодаря образованию нитридов или карбидов [781].
Антикоррозионные покрытия металлических поверхностей, находящих-
ся в контакте с водой, получают из ванн, содержащих водные растворы циа-
нидов Kat [М (CN)J (Kat = NH^ или щелочной металл, М = Си+, Со2+,
Ni2+, Ag+, Fe2+) [782]. Ингибиторами коррозии являются ионы [Fe (CN)e]4~
вследствие образования плотной пленки малорастворимого ферроцианида
на поверхности металла [26].
3.9.3. Каталитические процессы
Важной областью использования цианидных соединений, в основном
координационных, является катализ ряда химических процессов. В качестве
катализатора гомогенной гидрогенизации, в частности олефинов и сопря-
женных диолефинов (бутадиен, стирол), применяется комплекс [Со (CN)5]3~.
Последний обратимо присоединяет водород с образованием [НСо (CN)J3~,
который взаимодействует по схеме [487]
[НСо (CN)г>Г - + С4Н6 -> С4Н,Со (CN)!-,
(о.оо)
С4Н7Со (CN)s + [НСо (CN)S]3~ -> С4Н8 + 2 [Со (CN)5[3-.
В состав катализаторов полимеризации виниловых соединений (стирол,
метилметакрилат, бутадиен) входят К4 [М (CN)6] (М = Мп, Fe) и К, [М (CN)J
(М = Ni, Pd) [783].
106
Активными катализаторами гидрирования (Pt, Pd), оксосинтеза (Со),
димеризации диенов (Ni) являются соединения [М (CN)2L21, где L = моно-
или полифункциональные лиганды, содержащие N, Р, As, Sb [784].
Многие гексацианоферраты (II) А1 и некоторых d-металлов (Мо, V, Сг,
Os, Ru) смешанного типа являются исходными соединениями для приготов-
ления катализаторов синтеза аммиака [26, 249, 785, 786].
3.9.4. Другие области применения цианидов
Соединения LHCo3 [Fe (CN)6]2 и L2Co3 [Fe (CN)6]2 (L = различные амины)
[787], Zr[Fe (CN)6] [788], Sn [Fe (CN)6] [789] и другие [26] иногда применяются
в качестве неорганических ионообменников. Характерно окрашенные фер-
роцианиды используются в производстве пигментов, малярных и типограф-
RCN
RR‘С (ОН) С ООН
ор’'^'
Рис. 3.5. Циановодород в органическом синтезе
ских красок (например, всем известные краски берлинская лазурь, «мос-
ковская красная» (NH4)2 CuFe (CN)6 [790]).
В электротехнике находят применение некоторые гексацианоферраты
(II) как низкотемпературные ферромагнетики, а соединения MCN • 4AgI
(М = К, Rb, Cs) — как ионные проводники в батарейках с твердым электро-
литом [791].
Проводятся исследования по использованию XCN (X — Br, Cl), (CN)2
и HCN в качестве компонентов газовых химических лазеров [792, 837].
Широкое применение в органическом синтезе имеет и сам циановодород
[11, 793], что видно из рис. 3.5.
СПИСОК ЛИТЕРАТУРЫ
1. Williams Н. Е. Cyanogen Compounds. 2nd ed. London, E. Arnold and Co, 1948.
2. Brotherton T. K., Lynn J. W.— Chem. Reviews, 1959, 59, p. 841.
3. Cullis C. F., Gates J. G.— J. Chem. Soc., 1964, p. 2833.
4. Коттон Ф., Уилкинсон Дж. Современная неорганическая химия, т. 2. М., Мир,
1969.
5. Pauling L., Springall N. D., Palmer К. G.— J. Am. Chem. Soc., 1939, 61, p. 927.
107
6. Morino Y., Kuchitsu K., Hori Y„ Tanimoto M. — Bull. Chem. Soc. Japan, 1968, 41,
p. 2349.
7. Смирнов С. K-, Струхов О. Г., Дубов С. С. и dp.—Журнал неорган. химии, 1968,
13, с. 2993.
8. Coates G. E., Coates J. E.— J. Chem. Soc., 1944, p. 77.
9. Iguchi K-— J. Chem. Phys., 1956, 25, p. 217.
10. Izatt R. M., Christensen J. J., Pack R. T., Bench R.— Inorg. Chem., 1962, 1, p. 828.
11. Бобков С. C-, Смирнов С. К. Синильная кислота. M., Химия, 1970.
12. NelsonD. F., Kirk P. L.— J. Chromat., 1963, 12, p. 167.
13. Constant G., Daran J.-C., Jeannin Y. — C. r. Acad. Sci., 1969, C269, p. 975.
14. ConstantG., Jeannin Y., Morancho R. — C. r. Acad. Sci., 1970, C271, p. 361.
15. Kawai K-. Kanesaka J.— Spectrochim. Acta, 1969, 25A, p. 1265.
16. Guttenberger J.-F.— Chem. Ber., 1968, 101, S. 403.
17. Corain B.— Coord. Chem. Rev., 1972, 8, p. 159.
18. Rossmanith K.— Monatsh. Chem., 1965, 96, p. 1690.
19. Paul R. C., Sreenathan B. R.— Indian J. Chem., 1966, 4, p. 382.
20. Marullo N. P., Allen J. F., Cochran G. T., Lloyd R. A.— Inorg. Chem., 1974, 13,
p. 115.
21. Duvall L. A., Miller D. P.— Inorg. Chem., 1974, 13, p. 120.
22. Сейфер Г. Б.— Журнал неорган. химии, 1962, 7, с. 1242.
23. Тананаев И. В., Сейфер Г. Б.— Журнал неорган. химии, 1957, 2, с. 600.
24. Тананаев И. В., Дарчиашвили Т. В.— Журнал неорган. химии, 1960, 5, с. 80.
25. Глушкова М. А., Петушкова С. М.— Журнал неорган. химии, 1961, 6, с. 349.
26. Тананаев И. В., Сейфер Г. Б., Харитонов Ю. Я. и др. Химия ферроцианидов. М.,
Наука, 1971.
27. Britton D. Perspectives in Structural Chemistry. New York — London — Sydney, J.
Wiley and Sons, 1967, 1, p. 109.
28. Ladd M. F. C. — Trans. Faraday Soc., 1969. 65, p. 2712.
29. Coates G. E., Mukherjee R. N.— J. Chem. Soc., 1963, p. 229.
30. Chadwick В. M., Sharpe A. G.— Advan. Inorg. Chem. Radiochem., 1966, 8, p. 84.
31. Жданов Г.С — Докл. АН СССР, 1941, 31, с. 350.
32. Шугам Е. А., Жданов Г. С. — Журнал физ. химии, 1945, 19, с. 515.
33. Seccombe R. С.. Kennard С. Н. L.— J. Organometal. Chem., 1969, 18, р. 243.
34. Hvoslef J.— Acta Chem. Scand., 1958, 12, p. 1568.
35. Woodward L. A., Owen H. F.— J. Chem. Soc., 1959, p. 1055.
36. Сергеева A. H., КиселеваЛ. A.— Укр. хим. журн., 1970, 36, с. 435.
37. Сергеева А. Н., КиселеваЛ. А., Галицкая С. М.— Журнал неорган. химии, 1971,
х с. 1782.
38. Perez Rodriguez М., Cubero М.. Lopez-Castro A., Moreno-Bajo С.— Nature, 1965,
206, № 4982, р. 392.
39. Михалевич К. Н.. Галицкая С. М., Скопенко В. В.— Укр. хим. журн., 1971, 37,
с. 289.
40. Михалевич К. Н., Галицкая С. М.— Укр. хим. журн. 1972, 38, с. 713.
41. Mbckel К-, Muller-Litz W.— Z. anorg. allg. Chem., 1972, 393, S. 81.
42. Галицкая С. M., Михалевич К- Н — Вкник Льв1в. гкмптех. in-ту. 1971, № 60, с. 28.
43. Ahuja I. S., Singh R.— Inorg. Nucl. Chem. Lett., 1973, 9, p. 289.
44. Ahuja I. S., Carg A.— J. Inorg. Nucl. Chem., 1972, 34, p. 2681.
45. Jain S. C., Rivest R.— Inorg. Chim. Acta Revs., 1970, 4, p. 291.
46. Jain S. C., Rivest R. — Inorg. Chim. Acta Revs., 1969, 3, p. 552.
47. Ahuja 1. S., Carg A.— J. Inorg. Nucl. Chem., 1971, 33, p. 1515.
48. Ledesert M., Frey M., Nakajima S., Monier J. C.— Bull. Soc. Fr. Mineral. Cristal-
logr., 1969, 92, p. 342.
49. Ledesert M., Frey M., Monier J. C.— Bull. Soc. Fr. Mineral. Cristallogr., 1967, 90, p. 36.
50. Frey M., Ledesert M.— Acta Crystallogr., 1971, B27, p. 2119.
51. Jain S. C.— J. Inorg. Nucl. Chem., 1973, 35, p. 413.
52. Ahuja 1. S., Sambastva Rao K-—J- Inorg. Nucl. Chem., 1975, 37, p. 586.
53. Jain S. C., Rivest R. — Canadian J. Chem., 1969, 47, p. 2209.
54. Kuroda R.— Inorg. Nucl. Chem. Lett., 1973, 9, p. 13.
55. Lappert M. F., Pyszora H.— Advan. Inorg. Chem. Radiochem., 1966, 9, p. 133.
56. Wittig G., Raff P.—Z. Naturforsch., 1951, 6b, S. 225.
57. WittigG.,Bille H.— Z. Naturforsch., 1951, 6b, S. 226.
58. Лаврентьева E. А., Лагодзинская Г. В., Хидекелъ М. Л. и др.— Журнал общей
химии, 1973, 43, с. 295.
59. Кораблев Л. Г., Лаврентьев И. П., Лаврентьева Е. А.. Хидекель М.Л.— Изв.
АН СССР. Сер. хим., 1968, № 12, с. 2826.
60. Gingerich К. А.— Naturwissenschaften, 1967, 54, S. 646.
108
61. Gingerich К. A.— Chem. Commun., 1969, 13, p. 764.
62. Kumar N., Kalsotra B. L., Multani R. K-— J- Inorg. Nucl. Chem., 1974, 36, p. 1157.
63. Ehemann F., Dehnicke K. — J. Organometal. Chem., 1974, 71, p. 191.
64. Goggin P. L., McColm I. J.— J. Less-Common Metals, 1966, 11, p. 292.
65. Goggin P. L., McColm I. J., Shore R.— J. Chem. Soc., 1966, A, p. 1314.
66. Menzer W.— Angew. Chem., 1958, 70, S. 656.
67. Bither T. A., Knoth W. H., Lindsey R. V., Jr., Sharkey W. H.— J. Am. Chem. Soc.,
1958, 80, p. 4151.
68. Goldfarb T. D.— J. Chem. Phys., 1962, 37, p. 642.
69. Srivastava T. N., Tandon S. K.—J- Inorg. Nucl. Chem., 1968, 30, 1399.
70. Gassenheimer B., Herber R. H.— Inorg. Chem., 1969, 8, p. 1120.
71. Schlemper E. 0., Britton D.— Inorg. Chem., 1966, 5, p. 507.
72. Konnert J., BrittonD., Chow I. M.— Acta Crystallogr., 1972, B28, p. 180.
73. Ford-Smith M. H. The Chemistry of Complex Cyanides. London, 1964.
74. Linke К- H., Lemmer F.— Z. anorg. allg. Chem., 1966, 345, S. 203.
75. Pierce L., Nelson R., Thomas C. — J. Chem. Phys., 1965, 43, p. 3423.
76. Feher F., Linke K.-H.— Z. anorg. allg. Chem., 1964, 327, S. 151.
77. Linke К.-H., Lemmer F.— Z. anorg. allg. Chem., 1966, 345, S. 211.
78. Linke К.-H., Lemmer F.— Z. Naturforsch., 1966, 21b, S. 192.
79. Linke K.-H.— Z. Naturforsch., 1966, 21b, S. 1108.
80. Paul R. C., Chauhan R. K-, Parkash R.— J. Sci. and Ind. Res., 1974, 33, p. 31.
81. Fawcett F. S., Lipscomb R. D.— J. Am. Chem. Soc., 1964, 86, p. 2576.
82. Mayer E. — Angew. Chem., 1969, 81, p. 627.
83. Foremann R.W., Veatch F. — Chem. Abstr., 1966, 64, P 13767b.
84. Pasternak M., Sonnino I.— J. Chem. Phys., 1968, 48, p. 2009.
85. McCordick J.— C. r. Acad. Sci., 1968, 266C, p. 1296.
86. McCordick J., Kaufmann G.,Rohmer R.— Rev. Chim. Miner., 1968, 5, p. 629.
87. KawaiK., Kanesaka I.— Spectrochim. Acta, 1969, 25A, p. 263.
88. Mayer E. — Monatsch. Chem., 1970, 101, p. 834.
89. Bok L. D. C., Leipoldt J. G.— Z. anorg. allg. Chem., 1966, 344, S. 86,
90. Bauer R. Dissertation. Erlangen — Niirnberg, 1973; РЖ Химия, 1974, 95436Д.
91. Izatt R. M., Christensen J. J., Hansen J. W., Watt G. D.— Inorg. Chem., 1965, 4,
p. 718.
92. Perrson H. — Acta Chem. Scand., 1971, 25, p. 543.
93. 0sterud T., Prytz M.— Acta Chem. Scand., 1950, 4, p. 1250.
94. Prytz M., 0sterud T.— Acta Chem. Scand., 1952, 6, p. 1534.
95. Chantry G. N.,Plane R. A.— J. Chem. Phys., I960, 33, p. 736.
96. Ashurst K. G., Finkelstein N. P., Goold L. A.— J. Chem. Soc., 1971, A, p. 1899.
97. Sillen L. G., Martell A. E. Stability Constants of Metal Ion Complexes. London, The
Chem. Soc., 1964.
98. Anderegg G.— Helv. Chim. Acta, 1957, 50, p. 1022.
99. Christensen J. J., Izatt R. M., Eatough D. — Inorg. Chem., 1965, 4, p. 1278.
100. Newman L., de 0. Cabral J., Hume D. N.— J. Am. Chem. Soc., 1958, 80, p. 1814.
101. Tanaka N., Murayama T.— Z. Physik. Chem. N. F., 1957, 1.1, S. 366.
102. Penneman R. A., Jones L. H.— J. Inorg. Nucl. Chem., 1961, 20, p. 19.
103. Woodward L. A., Owen H. F.— J. Chem. Soc., 1959, p. 1055.
104. Azzam A. M., Shimi I. A. W.— Z. anorg. allg. Chem., 1964, 327, p. 89.
105. Ammar I. A., Darwish S., Rizk K.— Electrochim. Acta, 1968, 13, p. 797.
106. Saraf J. R., Aggarwal R. C., Prasad J. — J. Inorg. Nucl. Chem., 1969, 31, p. 3839.
107. JanderG., Grilttner B.— Chem. Ber., 1948, 81, p. 114.
108. Isoya J., Fujiwara S. — Bull. Chem. Soc. Japan, 1972, 45, p. 2182.
109. Beck M. T., Porzsolt E. Cs.— J. Coord. Chem., 1971, 1, p. 57.
НО. Сергеева A. H., Киселева А. Л., Галицкая С. M.— Журнал неорган. химии,
1971, 16, с. 2057.
111. Coleman J. S., Penneman R. A., Jones L. H., Kressin T. K.— Inorg. Chem., 1968,
7, p. 1174.
112. Agrawal A. K-, Vishnu, Mehrotra R. C.— Proc. Indian Acad. Sci., 1968, A68,
p. 123.
113. Martin R.-P., BlancM.— Bull. Soc. chim. France, 1969, №6, p. 1866.
114. Roesky H. W.— Angew. Chem., 1967, 79, p. 316.
115. Roesky H. W.— Chem. Ber., 1967, 100, p. 2138.
116. Muller U.— Z. anorg. allg. Chem., 1973, 396, S. 187.
117. Penneman R. A., Staritzky E. —J. Inorg. Nucl. Chem., 1958, 6, p. 112.
118. Penna-Franca E., Dodson R. W.— J. Am. Chem. Soc., 1955, 77, p. 2651.
119. Horne R. A.— J. Inorg. Nucl., Chem., 1958, 6, p. 338.
120. Жданов Г. С., Шугам Е. А.— Журнал физ. химии, 1943, 19, р. 519.
109
121. Rossmanith К.— Monatsh. Chem., 1965, 96, S. 1407.
122. Rossmanith K.— Monatsh. Chem., 1966, 97, S. 1698.
123. McColm I. J., Thompson S.— J. Inorg. Nucl. Chem., 1972, 34, p. 3801.
124. Colquhoun I., Greenwood N. N., McColm 1. J., Turner G. E.— J. Chem. Soc., Dal-
ton Trans., 1972, p. 1337.
125. Benz R., Arnold G. P., Zachariasen IT. H.— Acta Crystallogr., 1972, 28B, p. 1724.
126. Rivenq F.— Bull. Soc. chim. France, 1947, № 11/12, p. 971.
127. Brauer G., Walz H.— Z. anorg. allg. Chem., 1963, 319, S. 236.
128. Ripan R., Ceteanu J.— Chimia Metalelor. Vol. 2. Bucuresti, Editura didactics si
pedagogica, 1969.
129. Ray P., Sahu H.— J. Indian Chem. Soc., 1946, 23, p. 161.
130. Weiss A., Rothenstein W.— Angew. Chem., 1963, 75, S. 575.
131. Poskozim P. S.— Inorg. Nucl. Chem. Lett., 1969, 5, p. 933.
132. Poskozim P. S., Shute R., Taylor R., Wysocki J.—J. Inorg. Nucl. Chem., 1973, 35,
p. 687.
133. LudiA., Hiigi R. — Helv. Chim. Acta, 1967, 50, p. 1283.
134. Ltidi A., Hugi R.— Helv. Chim. Acta, 1968, 51, p. 349.
135. Walker G. F., Hawthorne D. G.— Trans. Faraday Soc., 1966, 63, p. 166.
136. Mathey Y., Mazieres C. — Canadian J. Chem., 1974, 52, p. 3637.
137. HumeD. N., Kolthoff J. M.— J. Am. Chem. Soc., 1950, 72, p. 4423.
138. DowsD. A., Haim A., Wilmarth W. K-—J. Inorg. Nucl. Chem., 1961,21, p. 33.
139. LawsonD. N., Mays M. JWilkison G.— J. Chem. Soc., 1966, A, p. 52.
140. Барановский M. Б.— Журнал неорган. химии, 1969, 14, с. 222.
141. Барановский И. Б., Харитонов Ю. Я.— Докл. АН СССР, 1966, 169, с. 1335.
142. Griffith W. Р.— Quart. Rev., 1962, 16, р. 188.
143. Purcell К. F.— J. Am. Chem. Soc., 1967, 89, p. 247.
144. Curry N. A., Runeiman W. A.— Acta Crystallogr., 1959, 12, p. 674.
145. Sequeira A., Chidambaram R. — Acta Crystallogr., 1966, 20, p. 910.
146. Харитонов Ю. Я-— В кн.: Колебательные спектры в неорганической химии. М.,
Наука, 1971, с. 139—181.
147. Purcell К. F.— J. Am. Chem. Soc., 1969, 91, р. 3487.
148. Gray Н. В., Beach N. А.— J. Am. Chem. Soc., 1963, 85, р. 2922.
149. Burger К.— Inorg. Chim. Acta, 1972, 6, p. 31.
150. Parshall G. W.— J. Am. Chem. Soc., 1966, 88, p. 704.
151. Basolo F., Pearson R. G. Mechanisms of Inorganic Reactions. New York, John Wiley
and Sons Inc., 1967.
152. Харитонов Ю. Я*> Евстафьева О. Н., Барановский И. Б. и др.— Журнал неорган.
химии, 1966, 11, с. 1733.
153. Харитонов Ю. Я-> Евстафьева О. Н.— Журнал неорган. химии, 1969, 14, с. 770.
154. Харитонов Ю. Я-, Сейфер Г. Б.— Изв. АН СССР. Неорган. материалы, 1966,
2»Гс. 124.
155. Bferrum J.— Chem. Rev., 1950, 46, р. 381.
156. Remy Н. Lehrbuch der Anorganischen Chemie. Bd. 2. Leipzig, Akademische
Verl.-Ges. Geest and Portig K.-G., 1961.
157. Penneman R. A., Jones L. H.— J. Chem. Phys., 1956, 24, p. 293.
158. Владимирова M. Г., Каковский И. A.— Журнал прикл. химии, 1950, 23, с. 580.
159. Simpson Е. A., WaindG. М.— J.Chem. Soc., 1958, р. 1746.
160. Brenner А.— J. Electrochem. Soc., 1965, 112, р. 611.
161. Jones L. H., Penneman R. A.— J. Chem. Phys., 1954, 22, p. 965.
162. Azzam A. M., Shimi I. A. W.— Z. anorg. allg. Chem., 1963, 321, S. 284.
163. Jones L. H.— J. Chem. Phys., 1958, 29, p. 463.
164. Hancock R.D., Finkelstein N. P., Evers A..— J. Inorg. nuci. Chem., 1972, 34, p. 3747.
165. Pierrad J.-C., Kappenstein C., Hugel R. — Rev. chim. miner., 1971, 8, p. 11.
166. Kappenstein C., Hugel R.— Rev. chim. miner., 1969, 6, p. 1107.
167. Kappenstein C., Hugel R.— J. Inorg. Nucl. Chem., 1974, 36, p. 1821.
168. Rothbaum H. P.— J. Electrochem. Soc., 1957, 104, p. 682.
169. Стабровский A. И.— Журнал физ. химии, 1952, 26, с. 949.
170. Suzuki S. — Sci. Reports Res. Inst., 1953, A5, p. 311; Chem. Abstr., 48, 7491h.
171. Coleman J. S., George R., Allaman L., Jones L. H.— J. Phys. Chem., 1968, 72,
p. 2605.
172. Izatt R. M., Johnston H.D., WattG. D., Christensen J. J.— Inorg. Chem., 1967, 6,
p. 132.
173. Baxendale J. H., WestcottD. T.— J. Chem. Soc., 1959, p. 2347.
174. Zsako J., Petri E.— Rev. Roumanine Chim., 1965, 10, p. 571.
175. Ammar I. A., Darwish S., Rizk K.— Electrochim. Acta, 1967, 12, p. 647.
176. Боос Г. А., Попель A. A.— Журнал неорган. химии, 1967, 12, с. 2086.
ПО
177. Sillen L. G., Martell A. E. Stability Constants of Metal Ion Complexes. London, The
Chem. Soc., 1971.
178. Gl'ibeli A. 0., Cote P. A.— Canadian J. Chem., 1972, 50, p. 1144.
179. Cudey G.— Bull. Soc. chim. France, 1968, 8, p. 3177.
180. Jamamoto T., Haraguchi H., FujiwaraS.— J. Phys. Chem., 1970, 74, p. 4369.
181. Чупахина P. А., Индукаев Ю. В., Серебренников В. В.— Журнал неорган. химии
1961, 6, с. 2713.
182. Hoard J. L.— Z. Kristallogr., 1933, 84, S. 231.
183. Rosenzweig A., Cromer D. T.— Acta Crystallogr., 1959, 12, S. 709.
184. Graybeal J. D., McNownG. L.— Inorg. Chem., 1966, 5, p. 1909.
185. Cromer D. T.— J. Phys. Chem., 1957, 61, p. 1388.
186. Cromer D. T., Larson A. C.— Acta Crystallogr., 1962, 15, S. 397.
187. Mason W. R.— J. Am. Chem. Soc., 1973, 95, p. 3573.
188. EvansD. F., Jones D., Wilkinson G. — J. Chem. Soc., 1964, p. 3164.
189. Paterson R., Bjerrum J.— Acta Chem. Scand., 1965, 19, p. 729.
190. M&nsted 0., Bjerrum J.— Acta Chem. Scand., 1967, 21, p. 1116.
191. Longo A., Buch. T.— Inorg. Chem., 1967, 6 (3), p. 556.
192. Kappenstein C., Hagel R.— C. r. Acad. Sci., 1972, C274, p. 362.
193. Jones L. H., Smith J. M.— J. Chem. Phys., 1964, 41, p. 2507.
194. Smith J. M., Jones L. H., Kressin J. Д.. Penneman R. A.— Inorg. Chem., 1965, 4,
p. 369.
195. Negoiu D., Baloiu L. M.— Z. anorg. allg. Chem., 1970, 374, S. 105.
196. Cooper D., Plane R. A.— Inorg. Chem., 1966, 5, p. 16.
197. Сергеева A. H., Павленко JI. И.— Укр. хим. журн., 1972, 38, с. 418.
198. Сергеева А. Н., Семенишин Д. И., Мазепа А. В.— Журнал, неорган. химии, 1973,
18, с. 2956.
199. Poradier J., Cugnac-Pailliotet A., Pontoreau V.— С. г. Acad. Sci., 1971, С272,
р. 865.
200. Попель А. А., Боос Г. А.— В кн.: Исследования по электрохимии, магнетохимии и
электрохимии методом анализа. Казань, 1969, вып. 2, с. 23.
201. Попель А. А., Боос Г. А.— Там же, с. 237.
202. Боос Г. А., Попель А. А.— Там же, с. 243.
203. Kolthoff I. М., Stock J. Т.— J. Am. Chem. Soc., 1956, 78, р. 2081.
204. Curtis Y. М., Curtis N. F.— Austral. J. Chem., 1966, 19, p. 609.
205. CurtisY. M., Curtis N. F.— Austral. J. Chem., 1965, 18, p. 1933.
206. Tobia S. Д., El-Shahat M. F.— J. Chem. Soc. A., 1968, p. 2444.
207. Muller-Litz IF. — Z. Chem., 1968, 8, S. 389, 432.
208. Cromer D. T., Larson A. C., Roof R. B., Jr.— Acta Crystallogr., 1965, 19, p. 192.
209. Cromer D. T., Larson A. C., Roof R. B., Jr.— Acta Crystallogr., 1966, 20, p. 279.
210. Nicholas M., Wolford T. — Inorg. Nucl. Chem. Lett., 1975, 11, p. 157.
211. Cooper D., Plane R. A.— Inorg. Chem., 1966, 5, p. 1677.
212. Williams R. J., Larson A. C., Cromer D. T.— Acta Crystallogr., 1972, B28, p. 858.
213. Сергеева A. H., Павленко JI. И.— Журнал неорган. химии, 1967, 12, с. 2063.
214. Сергеева А. Н., Павленко Л. И.— Журнал неорган. химии, 1967, 12, с. 3295.
215. Павленко Л. И., Сергеева А. Н.— Журнал неорган. химии, 1969, 14, с. 680.
216. Williams R. J., Cromer D. Т., Larson А. С.— Acta Crystallogr., 1971, В27, р. 1701.
217. Dunaj-Jurco М., Poraj-KosicМ. А.— Chem. Zvesti, 1967, 21, s. 241.
218. Михалевич Д. И., Литвинчук В. М.— Журнал неорган. химии, 1964, 9, с. 2391.
219. Cooper D., Plane R. А.— Inorg. Chem., 1966, 5, p. 2209.
220. Сергеева A. H., Павленко Л. И.— Журнал неорган. химии, 1970, 15, с. 2085.
221. Сергеева А. Н., Семенишин Д. И.— Журнал неорган. химии, 1973, 18, с. 2081.
222. Rigo Р., Turco А. —Coord. Chem. Rev., 1974, 13, р. 133.
223. Muetterties Е. L., Alegranti C. W.— J. Am. Chem. Soc., 1972, 94, p. 6386.
224. Dautet H., Guillaumont R.— Radiochem. Radioanal. Lett., 1971, 8, p. 175.
225. Britzinger H., Jahn F.— Z. anorg. allg. Chem., 1938, 235, S. 244.
226. Комплексные соединения урана. M., Наука, 1964, с. 246.
227. Bagnall Д. W ..Baptista J. L.— J. Inorg. Nucl. Chem., 1970, 32, p. 2283.
228. Schlafer H. L., Gotz R.— Z. anorg. allg. Chem., 1961, 309, S. 105.
229. Bose S. Д., Basu S. — J. chim. Phys, et phys.-chim. biol., 1969, 66, p. 1985.
230. Heintz E. A.—Nature, 1963, 197, 690.
231. NichollsD., Ryan T. A., Seddon Д. R.— Chem. Commun., 1974, p. 635.
232. Peterson E.— Z. anorgan. Chem., 1904, 38, S. 342.
233. Довгей В- В., Михалевич Д. Н., Сергеева А. Н.— Журнал неорган. химии, 1974,
19, с. 943.
234. Jagner S.— Acta Chem. Scand., 1975, A29, p. 255.
235. BennettB.G., NichollsD —J. Chem. Soc., 1971, A, p. 1204.
Ill
236. Nast R., Rehder D —Chem. Ber., 1971, 104, S. 1709.
237. Behrens H., Lutz K. — Z. anorg. allg. Chem., 1967, 354, S. 184.
238. Guzzetta F. H., Hadley W. B.— Inorg. Chem., 1964, 3, p. 259.
239. Alexander J. JGray H. B.— J. Am. Chem. Soc., 1968, 90, p. 4260.
240. Nicholls D.— Coord. Chem. Rev., 1966, 1, p. 379.
241. Довгей В. В., Сергеева А. Н., Михалевич К. И.— Журнал неорган. химии, 1974,
19, с. 1527.
242. TownsR. L. R., Levenson R. А.— J. Am. Chem. Soc., 1972, 94, p. 4345.
243. Levenson R. A., TownsR. L. R.— Inorg. Chem., 1974, 13, p. 105.
244. Levenson R. A., Dominguez R. J. G.— Inorg. Chem., 1973, 12, p. 2342.
245. Levenson R. A.— Chem. Phys. Lett., 1973, 22, p. 293.
246. Levenson R. A., Dominguez R. J. G., Willis M. A., Young F. R.— Inorg. Chem.,
1974, 13, p. 2761.
247. Selbin J., HolmesL. H., Jr.— J. Inorg. Nucl. Chem., 1962, 24, p. 1111.
248. Wasson J. R.— J. Inorg. Nucl. Chem., 1968, 30, p. 171.
249. Доегей В. В. Координационные цианиды ванадия и их каталитические свойства.
Автореф. канд. дис. Львов, 1975.
250. Wicholas М., Wolford Т. — Inorg. Chem., 1974, 13, р. 316.
251. Griffith W. Р., Lewis J., Wilkinson G. — J. Chem. Soc., 1959, p. 1632.
252. Masek J., Pribil R. — Inorg. Chim. Acta, 1971, 5, p. 499.
253. Kiernan P. M., Griffith W. P.— Inorg. Nucl. Chem. Lett., 1976, 12, p. 377.
254. Griffith W. P.— Coord. Chem. Rev., 1975, 17, p. 177.
255. Jagner S., Vannerberg N.-G.— Acta Chem. Scand., 1968, 22, p. 3330.
256. Jagner S., Vannerberg N.-G.— Acta Chem. Scand., 1970, 24, p. 1988.
257. Folkesson B.— Acta Chem. Scand., 1974, A28, p. 491.
258. BanksD. F.,Kleinberg J.— Inorg. Chem., 1967, 6, p. 1849.
259. Milller A., Werle P., Diemann E., Aymonino P. J.— Chem. Ber., 1972, 105, S. 2419.
260. McCarthy A. E.— J. Chem. Soc., A, 1970, p. 1379.
261. Robin M. B., Day P.— Advan. Inorg. Chem. Radiochem., 1967, 10, p. 247.
262. Kiernan P. M., Gibson J. F., Griffith W. P.— Chem. Commun., 1973, p. 816.
263. MachinD. J., Sullivan J. F.— J. Less-Common Metals, 1969, 19, p. 413.
264. Botar A., Blasius E., Augustin H.— Z. anorg. allg. Chem., 1976, 422, S. 54.
265. Moggi L., Bolletta F., Balzani V., Scandola F. —J. Inorg. Nucl. Chem., 1966, 28,
p. 2589.
266. Jensen P. W.— J. MoL Struct., 1973, 17, p. 377.
267. JonesL. H.— Inorg. Chem., 1963, 2, p. 777.
268. Birk J. P., Espenson J. H.— J. Am. Chem. Soc., 1968, 90, p. 2266.
269. Birk J. P., Espenson J. H.— J. Am. Chem. Soc., 1968, 90, p. 1153.
270. Espenson J. H., Birk J. P.— J. Am. Chem. Soc., 1965, 87, p. 3280.
271. Malik W. U., Ramesh B., Teotia M. P., Goyal R. N.— J. Indian Chem. Soc., 1975,
52, p. 605.
272. Врецена H. Б., Лишвинчук В. M., Черняк Б. И.— Координационная химия, 1976,
2, с. 1559.
273. Schlafer Н. L., Wagener Н., Wasgestian F. и. а.— Ber. Bunsenges. Phys. Chem.,
1971, 75, S. 878.
274. Schlafer H. L., Gausmann H., Mobius С. H.— Inorg. Chem., 1969, 8, p. 1137.
275. Kohn J. A., Townes W.D.— Acta Crystallogr., 1961, 14, p. 617.
276. Ferrari A., Tani M. E., Morisi E.— Gazz. Chim. I tai., 1961, 91, p. 1142.
277. Ferrari A., Tani M. E., Morisi E.— Acta Crystallogr., 1962, 15, p. 90.
278. Basu M., Basu S.— J. Inorg. Nucl. Chem., 1968, 30, p. 467.
279- RossmanG. R., Tsay F.-D., Gray H. B.— Inorg. Chem., 1973, 12, p. 824.
280. Magnusson W. L., Griswold E., Kleinberg J.— Inorg. Chem., 1964, 3, p. 88.
281. Fowler J. R., Kleinberg J.— Inorg. Chem., 1970, 9, p. 1005.
282. Yoo J. S., Griswold E., Kleinberg J.— Inorg. Chem., 1965, 4, p. 365.
283. Blasius E., Augustin H.— Z. anorg. allg. Chem., 1975, 417, S.-47, 55.
284. BotarA., Blasius E., Augustin H.— Z. anorg. allg. Chem., 1975, 417, S. 93.
285. Михалевич К- H., Сергеева А. Н.— Научные зап. Львов, политехи, ин-та. Сер.
химико-технол., 1956, вып. 22, с. 11.
286. Stomberg R. — Arkiv Kemi, 1965, 23, S. 404.
287. Steele H. C.— Austral. J. Chem., 1957, 10, p. 404.
288. Pribush R. A., Archer R. D — Inorg. Chem., 1974, 13, p. 2556.
289. Leipoldt. J. G., Bok L. D. C., Cilliers P. J.— Z. anorg. allg. Chem., 1974, 409, S. 343.
290. Leipoldt J. G., Bok L. D. C., Cilliers P. J.— Z. anorg. allg. Chem., 1974, 407, S. 350.
291. Литеинчук В. M., Михалевич К. Н., Зубрицкая Д. И,—Журнал неорган. химии,
1972, 17, с. 1357.
292. Samotus A., Kosowicz-Czajkowska В*—Rocz. Chem., 1971, 45, S. 1623.
112
293. Malik W. U., Ali S. I.— Naturwissenschaften, 1959, 46, S. 579.
294. Malik W. U., Ali S. I.— J. Inorg. Nucl. Chem., 1961, 20, p. 155.
295. Malik W. U., Ali S. I.— Taianta, 1961, 8. p. 737.
296. KhwajaN., BegM. A.— J. Prakt.Chem., 1969,311, p. 102.
297. Malik W. UAli S. I.— Bull. Chem. Soc. Japan, 1961, 34, p. 1306.
298. Kabir-Ud-Din, BegM. A. —J. Indian Chem. Soc., 1970, 47, p. 719.
299. JoshiD. P., Sharma K- N.— Z. phys. Chem., 1971, 246, S. 281.
300. Allen M., Lippard S. J.— Inorg. Chem., 1970, 9, 991.
301. Зубрицкая Д. И., Михалевич К. Н., Литвинчук В. М., Любченко Ю. А.— Журнал
неорган. химии, 1974, 19, с. 706.
302. Михалевич К. Н., Зубрицкая Д. И., Литвинчук В. М., Любченко Ю. А.— Журнал
неорган. химии, 1973, 18, с. 418.
303. Зубрицкая Д. И., Семенишин Д. И., Любченко Ю. А., Михалевич К. Н.— Журнал
неорган. химии, 1973, 18, с. 2289.
304. Beck М. Т., Porzsolt Е. Сг.— Magyar kern, folyoirat, 1971, 77, old. 543.
305. Hoard J. L., Silverton J. V.— Inorg. Chem., 1963, 2, p. 235.
306. Golebiewski A., Nalewajski R.— Z. Naturforsch., 1972, 27a, S. 1672.
307. Hoard J. L., Hamor T. A., Glick M. D.— J. Am. Chem. Soc., 1968, 90, p. 3177.
308. Baadsgaard H., Treadwell W. D.— Helv. Chim. Acta, 1955, 38, p. 1669.
309. Basson S. S., Bok L. D. C., Leipoldt J. G.— Acta Crystallogr., 1970, B26, p. 1209.
310. Kettle S. F. A., Parish R. V.— Spectrochim. Acta, 1965, 21, 1087.
311. Parish R. V.— Spectrochim. Acta, 1966, 22, 1191.
312. Parish R. V., Simms P. G., Wells M. A., Woodward L. A.— J. Chem. Soc., 1968, A,
p. 2882.
313. Hartman K. 0., Miller F. A.— Spectrochim. Acta, 1968, 24A, 669.
314. Stammreich H., Sala 0.— Z. Elektrochem., 1960, 64, S. 741.
315. Stammreich H., Sala 0.— Z. Elektrochem., 1961, 65, S. 149.
316. Hidalgo A., Matthieu J.-P.— C. r. Acad. Sci., 1959, 249, p. 233.
317. Konig E.— Z. Naturforsch., 1968, 23a, S. 853.
318. Clark M. G., Cancedo J. R., Maddock A. G., Williams A. F.— J. Chem. Soc. Dalton
Trans., 1975, p. 120.
319. Muetterties E. L.— Inorg. Chem., 1965, 4, p. 769.
320. Konig E.— Theoret. Chim. Acta, 1962, 1, 23.
321. Perumareddi J. R., Liehr A. D., Adamson A. W.— J. Am. Chem. Soc., 1963, 85,
p. 249.
322. Basu M., Basu S.— J. Inorg. Nucl. Chem., 1969, 31, p. 2989.
323. LongT. V., Vernon G. A.— J. Am. Chem. Soc., 1971, 93, p. 1919.
324. Pribush R. A., Archer R. D.— Inorg. Chem., 1974, 13, p. 2556.
325. Adamson A. W., Waltz W. L., Zinato E., Watts D. W. a. o.— Chem. Rev., 1968, 68,
p. 541.
326. Jakob W., Samotus A., Stasicka Z., Golebiewski A.— Z. Naturforsch., 1966, 21b,
S. 819.
327. StasiskaZ., Samotus A., Jakob W.— Roczn. Chem., 1966, 40, S. 967.
328. Jakob W., Jakob Z.— Roczn. Chem., 1962, 36, S. 601.
329. Perumareddi J. R.— Z. Naturforsch., 1966, 21b, S. 22.
330. Bucknall W. R., Wardlaw W.— J. Chem. Soc., 1927, p. 2981.
331. Bertoluzza A., Carassiti V., Marinangeli A. M.— Ann. Chim. (Rome), 1960, 50,
p. 806.
332. Михалевич К- H., Литвинчук В. М.— Докл. Львов, политехи, ин-та. Химия
и хим. технология, 1963, 5, № 1—2, с. 127.
333. Lippard S. J., Russ В. J.— Inorg. Chem., 1967, 6, р. 1943.
334. Lippard S. J., Nozaki H., RussB. J.— Chem. Commun., 1967, p. 118.
335. Day V. W., Hoard J. L.— J. Am. Chem. Soc., 1968, 90, p. 3374.
336. Marks R. L., Popielski S. E.— Inorg. Nucl. Chem. Lett., 1974, 10, p. 885.
337. Mitra R. P., Mohan H.— J. Inorg. Nucl. Chem., 1974, 36, p. 3739.
338. Van de Poel J., Neumann H. M.— Inorg. Chem., 1968, 7, p. 2086.
339. Samotus A., Dudek M., Kanas A.— J. Inorg. Nucl. Chem., 1975, 37, p. 943.
340. Jakob Z.— Roczn. Chem., 1957, 31, S. 681.
341. Михалевич К. H., Литвинчук В. М.— Журнал неорган. химии, 1959, 4, с. 1775.
342. Литвинчук В. М., Михалевич К- Н.— Укр. хим. жури., 1959, 25, с. 503.
343. Kabir-Ud-Din, Beg М. А.— J. Indian Chem. Soc., 1969, 46, р. 503.
344. Kabir-Ud-Din, Beg М. A.— J. Indian Chem. Soc., 1968, 45, p. 455.
345. Kabir-Ud-Din, Khan A. A., Beg M. A.— J. Indian Chem. Soc., 1968, 45, p. 841.
346. Saha H. K., Saha В. C., Mondal S. S.— Z. anorg. allg. Chem., 1975, 412, S. 179.
347. Adamson A. W., Perumareddi J. R.— Inorg. Chem., 1965, 4, p. 247.
348. Balzani V., Manfrin M.-F., Moggi L.— Inorg. Chem., 1969, 8, p. 4749.
113
349. Mitra R. P., Sharma В. K., Mohan H.— Canadian J. Chem., 1969, 47, p. 2317.
350. Archer R-D..DrutnD. A.— J. Inorg. Nucl. Chem., 1974,36, p. 1979.
351. Sergeyeva A. N., Mikhalevich C. N.— Proceed. 7th ICCC. Sweden, (Stockholm, Upp-
sala), 1962, p. 311.
352. Сергеева A. H., Михалевич К. H.— Научные зап. Львов, политехи, ин-та. Сер.
химико-технол., 1958, вып. 50, №3, с. 28.
353. Сергеева А. Н.— Научные зап. Львов, политехи, ин-та. Сер. химико-технол.,
1958, вып. 50, № 3, с. 22.
354. Mueller A., Christophliemk Р.— Angew. Chemie Intern. Ed., 1969, 8, S. 753.
355. Latka H.— Z. anorg. allg. Chem., 1967, 353, S. 243.
356. Novotny M., Lewis D. F., Lippard S. J.— J. Am. Chem. Soc., 1972, 94, p. 6961.
357. Corden B. J., Cunningham J. A., Eisenberg R.— Inorg. Chem., 1970, 9, p. 356.
358. McGarvey B. R.— Inorg. Chem., 1966, 5, p. 476.
359. Hayes R. G.— J. Chem. Phys., 1966, 44, p. 2210.
360. Bok L. D. C., Leipoldt J. G., Basson S. S.— Acta Crystallogr., 1970, B26, p. 684.
361. Yoo G. S., Grisfold E-, Kleinberg J.— Inorg. Chem., 1965, 4, p. 365.
362. Литвинчук В. M., Врецена Н. Б., Михалевич К. Н.— Координационная химия,
1975, 1, с. 1328.
363. Heintz Е. А.— J. Inorg. Nucl. Chem., 1961, 21, р. 262.
364. Behrens Н., HermannD.— A. Naturforsch., 1966, 21В, S. 1236.
365. Behrens H., Kohler J. —Z. anorg. allg. Chem., 1960, 306, S. 94.
366. Behrens H., Vogl J.— Chem. Ber., 1963, 96, S. 2220.
367. Behrens H., Midler A.— Z. anorg. allg. Chem., 1965, 341, S. 124.
368. Behrens H., Lindner E., Rosenfelder J.— Chem. Ber., 1966, 99, S. 2745.
369. Ruff J. K-— Inorg. Chem., 1969, 8, p. 86.
370. MemeringM. N., DobsonG. R.— J. Inorg. Nucl. Chem., 1973, 35, p. 665.
371. Griffith W. P., Lewis JWilkinson G.— J. Chem. Soc., 1959, p. 872.
372. Kuska H. A. Rogers M. T.— J. Chem. Phys., 1965, 42, p. 3034.
373. Vannerberg N.-G.— Acta Chem. Scand., 1966, 20, p. 1571.
374. Griffith W. P.— J. Chem. Soc., 1963, p. 3286.
375. Nast R., Gehring G.— Z. anorg. Chem., 1948, 256, S. 169.
376. Riley R. F., Ho L.— J. Inorg. Nucl. Chem., 1962, 24, p. 1121.
377. SwedungD. H., Vannerberg N.-G.— Acta Chem. Scand., 1968, 22, p. 1551.
378. Qureshi A. M., Sharpe A. G.— J. Inorg. Nucl. Chem., 1968, 30, p. 2269.
379. Robin M. B.— Inorg. Chem., 1962, 1, p. 337.
380. Rotlevi E., EatonD. R.— Canadian J. Chem., 1970, 48, p. 1073.
381. Sen S.— Z. anorg. allg. Chem., 1962, 315, S. 315.
382. SenS.— Z. anorg. allg. Chem., 1964, 333, S. 160.
383. Sen S.— Z. anorg. allg. Chem., 1965, 340, S. 82.
384. Sen S.— Z. anorg. allg. Chem., 1965, 335, S. 222.
385. Sarkar S.— J. Indian Chem. Soc., 1969, 46, p. 871.
386. Sarkar S.— J. Indian Chem. Soc., 1968, 45, p. 1186.
387. Sarkar S.— Indian J. Chem., 1969, 7, p. 940.
388. Sarkar S., Bandyopadhyay P., Sarkar P. B.— J. Indian Chem. Soc., 1968, 45,
p. 922.
389. Morcom R. E., Bell C. F.— J. Inorg. Nucl. Chem., 1973, 35, p. 1865.
390. Gupta M. P., Milledge H. J., McCarthy A. E. — Acta Crystallogr., 1974, B30, 656.
391. Vannerberg N. G.— Acta Chem. Scand., 1970, 24, p. 2335.
392. Levason IF., McAuliffe C. A.— Coord. Chem. Rev., 1972, 7, p. 353.
393. Yakimach A.— C. r. Acad. Sci., 1930, 190, p. 681.
394. Bailey R. A., Balko E. N.— J. Inorg. Nucl. Chem., 1972, 34, p. 2668.
395. Ziolo R. F., Staford R. H., Rossman G. R., Gray H. B.— J. Am. Chem. Soc., 1974,
96, p. 7910.
396. Chawlal.D., Frank M. J.— J. Inorg. Nucl. Chem., 1970,32, p. 555.
397. Сколоздра О. E., Сергеева A. H., Михалевич К. H.— Журнал неорган. химии,
1971, 16, с. 1628.
398. TrageserG., Eysel Н. Н.— Z. anorg. allg. Chem., 1976, 420, S. 273.
399. Colton R., Peacock R.D., WilkinsonG.— J. Chem. Soc., 1960, p. 1374.
400. Walter P. H. L., Kleinberg J., Griswold E.— Inorg. Chem., 1962, 1, p. 10.
401. Gutmann V., Paulsen G.— Monatsch. Chem., 1969, 100, S. 358.
402. Сколоздра О. E., Михалевич К- H., Сергеева A. Н.— BicHiiK Льв1в. полИехн. iH-ту,
1971, 60, с. 23.
403. Сколоздра О. Е., Сергеева А. Н.— Журнал неорган. химии, 1973, 18, с. 552.
404. Lock С. J. L., Wilkinson G.— J. Chem. Soc., 1964, р. 2281.
405. Basu М., Basu S.— J. Inorg. Nucl. Chem., 1969, 31, p. 3669.
406. Fenn R.H., Graham A. J., Johnson N. P.— J. Chem. Soc., 1971, A, p. 2880.
114
407. Johnson N. P.— J. Inorg. Nucl. Chem., 1972, 34, p. 2875.
408. Bailey R. A., Elguindy M., Kozak S. L.— J. Inorg. Nucl. Chem., 1969, 31, p. 2275.
409. ToppenD. L., Murmann R. K-— Inorg. Nucl. Chem. Lett., 1970, 6, p. 139.
410. Nadziefa L., Stasicka Z.— Rocz. Chem., 1974, 48, S. 1195.
411. Chakravorti M. C.— J. Inorg. Nucl. Chem., 1972, 34, p. 893.
412. Chakravorti M. C., Chaudhuri M. K-—J. Inorg. Nucl. Chem., 1972, 34, p. 3479.
413. Johnson N. P.— J. Chem. Soc., 1969, A. p. 1843.
414. Сколоздра О. E. Синтез и исследование комплексных цианидов рения. Автореф.
канд. дис. Львов, 1973.
415. Davies W. D., Johnson N. Р., Johnson Р., Graham A. J.— Chem. Commun., 1969,
.Ms 13, p. 736.
416. Tullberg A., Vannerberg N .-G.— Acta Chem. Scand., 1966, 20, p. 1179.
417. Tullberg A., Vannerberg N.-G.— Acta Chem. Scand., 1967, 21, p. 1462.
418. Monoharan P. T., Gray H. B.— Inorg. Chem., 1966, 5, p. 823.
419. Poletti A., Santucci A., Paliani G.— Spectrochim. Acta., 1971, 27A, p. 2061.
420. Jakob W., Senkowski T.— Rocz. Chem., 1966, 40, S. 1601.
421. Jezowska-Trzebiatowska В., Ziolkowski J.— Chem. Zvesti, 1965, 19, S. 177.
422. Behrens H., Lindner E., Schlindler H.— Z. anorg. allg. Chem., 1969, 365, S. 119.
423. Behrens H., Ruyter E., Lindner E.— Z. anorg. allg. Chem., 1967, 349, S. 251.
424. Chiswell B., Venanzi L. M.— J. Chem. Soc., 1966, A, p. 417.
425. Behrens H., Ranly H.-J., Lindner E.— Z. anorg. allg. Chem., 1974, 409, S. 299.
426. Suzuki T., Kwan T.— Chem. Abstr., 1966, 65, p. 13190d.
427. Persson H.— Acta Chem. Scand., 1974, A28, p. 885.
428. Shibata NFujinaga T., Yoshinara K., Kamchiku Y.— Radiochim. Acta, 1966,
5, 238.
429. Watt G. D., Christensen J. J., Izatt R. M.— Inorg. Chem., 1965, 4, p. 220.
430. Izatt R. M., Watt G.D., Bartholomew С. H., Christensen J. J.— Inorg. Chem., 1968,
7, p. 2236.
431. Christensen J. J., Izatt R. M., Hale J. D. a. o.— Inorg. Chem., 1963, 2, p. 337.
432. Pratt J. M., Williams R. J. P.— J. Chem. Soc., 1967, A, p. 1291.
433. Tsay F.-D., Gray H. B-, Danon J.— J. Chem. Phys., 1971, 54, p. 3760.
434. Alexander J. J., Gray H. B.— J. Am. Chem. Soc., 1967, 89, p. 3356.
435. Griffith W. P., Lane J. R.— J. Chem. Soc. Dalton Trans., 1972, p. 158.
436. Penneman R. A., Bain R., Gilbert G. a. o.— J. Chem. Soc., 1963, p. 2266.
437. Van Geet A. L., HumeD. N.— Inorg. Chem., 1964, 3, p. 523.
438. Coleman J. S., Petersen H. Jr., Penneman R. A.— Inorg. Chem., 1965, 4, p. 135.
439. McCullough R. L., Jones L. H., Penneman R. A.— J. Inorg. Nucl. Chem., 1960,
13, p. 286.
440. Kolski G. B.,MargerumD. W.— Inorg. Chem., 1968, 7, p. 2239.
441. Anderson W. C., Harris R. P.— Inorg. Nucl. Chem. Lett., 1966, 2, p. 315.
442. Raymond K- NBasolo F.— Inorg. Chem., 1966, 5, p. 949.
443. Raymond K. N., Corfield P. W. R., Ibers J. A.— Inorg. Chem., 1968, 7, p. 1362.
444. Terzis A., Raymond K. N. ,Spiro T. G.— Inorg. Chem., 1970, 9, p. 2415.
445. White D. A., Solodar A. J., Baizer M. M.— Inorg. Chem., 1972. 11, p. 2160.
446. Верендякина H. А., Сейфер Г. Б., Харитонов Ю. Я., Борщаговский Б. В.— Жур-
нал неорган. химии, 1971, 16, с. 2714.
447. BrownD. В.— Inorg. Chem., 1975, 14, р. 2582.
448. Хайдук И.— Успехи химии, 1961, 30, с. 1144.
449. Тананаев И. В., Сейфер Г. Б., Яковлева Р. Т.— Докл. АН СССР, 1974, 219, с. 1389.
450. Борщаговский Б. В., Гольданский В. И., Сейфер Г. Б., Стукан Р. А.— Изв.
АН СССР. Сер. хим., 1968, 87.
451. Харитонов Ю. Я-, Гольданский В. И., Сейфер Г. Б. и др.— Изв. АН СССР. Сер.
хим., 1970, с. 271.
452. Верендякина Н. А., Сейфер Г. Б., ХаритоновЮ. Я-, Борщаговский Б. В.— Жур-
нал неорган. химии, 1973, 18, с. 753.
453. Ayers J. В.— J. Inorg. Nucl. Chem., 1969, 31, р. 2045.
454. Manoharan Р. Т., Gray Н. В.— J. Am. Chem. Soc., 1965, 87, р. 3340.
455. Tosi L.— С. г. Acad. Sci., 1972, С275, р. 439.
456. Gans Р., Sobatini A., Sacconi L.— Inorg. Chem., 1966, 5, p. 1877.
457. Jezowska-Trzebiatowska B., Ziolkowski J.— Bull. Acad. Pol. Sci., Ser. Chim., 1964,
12, s. 503.
458. Сейфер Г. Б., Тарасова 3. А.— Журнал неорган. химии, 1972, 17, с. 1361.
459. Успехи химии координационных соединений. Киев, Наукова думка, 1975, с. 84.
460. WestD. X.— J. Inorg. NucL,Chem., 1967, 29, с. 1163.
461. Swinehart J. H., Rock P. A.— Inorg. Chem., 1966, 5. c. 573.
462. Andrade C., Swinehart J. H.— Inorg. Chim. Acta, 1971, 5, p. 207.
115
463. Espenson J. H., Wolenuk S. G., Jr.— Inorg. Chem., 1972, 11, p. 2034.
464. Сергеева A. H., Семенишин Д. И., Мазепа Л. В.— Журнал неорган. химии, 1975,
20, с. 396.
465. Sergeyeva A. N., Pavlenko L. I., Kohut L. К., Tkachenko J. I.— J. Mol. Struct.,
1973, 19, p. 513.
466. Beck W.— Z. anorg. allg. Chem., 1964, 333, S. 115.
467. Malik M. U., От H.— J. Electroanalyt. Chem., 1966, 11, p. 467.
468. Dezsi I., Molnar B., Szalay T., Jaszberenyi I.— Chem. Phys. Lett., 1973, 18, p. 598.
469. Toma H. E., Malin J. M., Giesbrecht E. — Inorg. Chem., 1973, 12, p. 2084.
470. Жилинская В. В., Назаренко Ю. П., Братушко Ю. И., Дцимирский К- Б.— Жур-
нал неорган. химии, 1974, 1S, с. 2186.
471. Дцимирский К. Б., Немошкаленко В. В., Назаренко Ю. П. и др.— Докл АН СССР
1974, 217, с. 1374.
472. Сергеева А. Н., Зубрицкая Д. И., Семенишин Д. И.— Журнал неорган. химии.
1975, 20, с. 1925.
473. Sarkar S., Mishra R., Singh R.— Indian J. Chem., 1974, 12, p. 523.
474. Sarkar S., Singh R., Mishra R.— Curr. Sci (India), 1974, 43, p. 475.
475. Sarkar S.,Singh R., Mishra R.— J. Inorg. Nucl. Chem., 1975, 37, p. 578.
476. Bates J. C., Davies К- M., Stedman G.— J. Chem. Soc. Dalton Trans., 1974, p. 2182.
477. Rock P. A.,Swinehart J. H.— Inorg. Chem., 1966, 5, p. 1078.
478. Moser W., Chalmers R. A., Fogg A. G.— J. Inorg. Nucl. Chem., 1965, 27, p. 831.
479. Fogg A. G., Norbury A. H., Moser W.— J. Inorg. Nucl. Chem., 1966, 28, p. 2753.
480. Fogg A. G., Jones A. D., Moser W.— J. Inorg. Nucl. Chem., 1966, 28, p. 2427.
481. Swinehart J. H., Schmidt W. G.— Inorg. Chem., 1967, 6, p. 232.
482. Schilt A. A.— Inorg. Syn., 1970, 12, p. 247; Chem. Abstr., 1971, 74, p. 60360e.
483. Archer R. D., Suydam L. J., DollbergD. D.— J. Am. Chem. Soc., 1971, 93, p. 6837.
484. Schilt A. A., Cresswell A. M.— Taianta, 1966, 13, 911.
485. Condorelli G., Giallongo L., Giuffrida A., Romeo G.— Inorg. chim. Acta, 1973,
7, p. 7.
486. Rupp J. J., Shriver D. F.— Inorg. Chem., 1967, 6, p. 755.
487. Kwiatek J.— Catal. Rev-, 1967, 1, p. 37.
488. Гуцыков В. В., Ермаков В. И., Сахаровский Ю. А.— Труды Моск, хим.-технол.
ин-та, 1974, вып. 81, с. 39.
489. Haser R., Broin С.-Е., Pierrot М.— С. г. Acad. Sci., 1971, С272, р. 1308.
490. Banks R. G. S., Pratt J. M.— J. Chem. Soc., 1968, A, p. 854.
491. Griffith W. P., Wilkinson G.— J. Chem. Soc., 1959, p. 2757.
492. Suzuki T., Kwan T.— Chem. Abstr., 1966, 65, p. 14487e.
493. Vlcek A. A., Basolo F. Inorg. Chem., 1966, 5, p. 156.
494. RipanR., FdrcasA., PiringerO.— Z. anorg. allg. Chem., 1966, 346, S. 211.
495. Bresson M., Rigo P.— Inorg. Chem., 1975, 14, p. 38.
496. Dartiguenave M., Dartiguenave Y., Zinoune M.— J. Inorg. Nucl. Chem., 1974, 36,
p. 2253.
497. Сергеева A. H., Когут JI. H.— Журнал неорган. химии, 1973, 18, с. 3245.
498. Morgenthaler L. P., MargerumD. W.— J. Am. Chem. Soc., 1962, 84, p. 710.
499. Rayner J. H., Powell H. M.— J. Chem. Soc., 1958, p. 3412.
500. Bhatnagar V. M.— Z. anorg. allg. Chem., 1967, 350, p. 214.
501. Bhatnagar V. M.— Журнал структурной химии, 1967, 8, с. 698.
502. Rayner J. H., Powell H. M.— J. Chem. Soc., 1952, p. 319.
503. Iwamoto T., Nakano T., Morita M., Miyoshi T. a. o.— Inorg. Chim. Acta, 1968, 2,
p. 313.
504. Iwamoto T.— Inorg. Chim. Acta, 1968, 2, p. 269.
505. Iwamoto T., Ohtsu Y.— Chem. Lett., 1972, p. 463.
506. Beck M. T., Bjerrum J.— Acta Chem. Scand., 1962, 16, p. 2050.
507. Сергеева A. H., Михалевич К. H.— Журнал неорган. химии, 1962, 7, с. 686.
508. Сергеева А. Н., КогутЛ. Н.— Журнал неорган. химии, 1973, 18, с. 2281.
509. Kiss А.— Acta Chim. Acad. Sci. Hung., 1965, 45, p. 267.
510. Schmidtke H.-H., Eyring G.— Z. phys. Chem. (BRD), 1974, 92, S. 211.
511. Papp S., Dombi A.— Monatsh. Chem., 1973, 104, S. 885.
512. Papp S., Kovacs S. — J. Inorg. Nucl. Chem., 1976, 38, p. 231.
513. Ito A., Suenaga M.. Ono K.— J- Chem. Phys., 1968, 48, p. 3597.
514. Robinette R., Collins R. L.— J. Coord. Chem., 1974, 3, p. 333.
515. Cervone E., Conti C., Sartori G. — Gazz. chim. ital., 1973, 103, p. 923.
516. Emschwiller G.— C. r. Acad. Sci., 1967, 265C, p. 281.
517. Emschwiller G., jQrgensen С. K.— Chem. Phys. Lett., 1970, 5, p. 561.
518. JaselskisB.— J. Am. Chem. Soc., 1961, 83, p. 1082.
519. GuttermanD. F., Gray H. B.— Inorg. Chem., 1972, 11, p. 1727.
116
520. Cole S. В., Kleinberg J.— Inorg. Chim. Acta, 1974, 10, p. 157.
521. Wrighton M., Hammond G. S., Gray H. B.— J. Am. Chem. Soc., 1971, 93, p. 5254.
522. WHghton M., BredesenD.— Inorg. Chem., 1973, 12, p. 1707.
523. Viaene L., D’Olieslager J., De Jaegere S.— J. Inorg. Nucl. Chem., 1975, 37, p. 2435.
524. Grossi R., Haim A., Wilmarth W. K-— Inorg. Chem., 1967, 6, p. 237.
525. Barca R., Ellis J., Tsao М.-S., Wilmarth Z. K.— Inorg. Chem., 1967. 6, p. 243.
526. Candlin J. P., Halpern J., Nakamura S.— J. Am. Chem. Soc., 1963, 85, p. 2517.
527. Maki N.— inorg. Chem., 1974, 13, p. 2180.
528. Birk J. P., Halpern J.— J. Am. Chem. Soc., 1968, 90, p. 305.
529. Stotz I., Wilmarth W. K., Haim A.— Inorg. Chem., 1968, 7, p. 1250.
530. Burmeister J. L.— Inorg. Chem., 1964, 3, p. 919.
531. Gutterman D. F., Gray H. B.— J. Am. Chem. Soc., 1969, 91, p. 3105.
532. Gutterman D. F., Gray H. B.— J. Am. Chem. Soc., 1971, 93, p. 3364.
533. Burmeister J. L., Al-Janabi M. Y.— Inorg. Chem., 1965, 4, p. 962.
534. Nast R., Rohmer M.— Z. anorg. allg. Chem., 1956, 285, S. 271.
535. Brown L. D., Raymond K. N.— Chem. Commun., 1974, p. 470.
536. Brown L. D., Raymond K. N.— Inorg. Chem., 1975, 14, p. 2595.
537. Halpern J., Goodall B. L., Khare G. P. a. o.— J. Am. Chem. Soc., 1975, 97, p. 230L
538. Haim A., Wilmarth W. K-— J. Am. Chem. Soc., 1961, 83, p. 509.
539. Mori M., Well J. A., Kinnaird J. K-— J. Phys. Chem., 1967, 71, p. 103.
540. Frortczek F. R., Schaefer W. P.— Inorg. Chim. Acta, 1974, 9, p. 143.
541. Hanzlik J,,Vicek A A.— Collect. Czech. Chem. Commun., 1973, 38, p. 3019.
542. Castello R. A., Mac-Coll С. P., Egen N. B., Haim A.— Inorg. Chem., 1969, 8,
p. 699.
543. Emschwiller G.— C. r. Acad. Sci. 1969, 268C, p. 692.
544. Сергеева A. H., Михалевич К. H.— Докл. АН СССР, 1960, 131, с. 327.
545. Siebert Н., Siebert С.— Z. anorg. allg. Chem., 1971, 383, S. 165.
546. Maki N., Ohshima K.— Bull. Chem. Soc. Japan, 1970, 43, p. 3970.
547. Ohkawa K-, Fujita J., Shimura Y.— Bull. Chem. Soc. Japan., 1965, 38, p. 66.
548. Mays M. J., WilkinsonG.— J. Chem. Soc., 1965, p. 6629.
549. Аблое А. В., Сырцова Г. П.— Журнал неорган. химии, 1965, 10, с. 1980.
550. Egen N. В., Krause R. А.— Inorg. Chem., 1972, 11, р. 1327.
551. Maki N.— Bull. Chem. Soc. Japan, 1965, 38, p. 2013.
552. Maki N., Hamazaki T., Sakuraba S.— Bull. Chem. Soc. Japan, 1968, 41, p. 1735.
553. Muto M., Baba T., Yoneda H.— Bull. Chem. Soc. Japan, 1968, 41, p. 2918.
554. Maki N., Mochizuki M., Sakuraba S.— Bull. Chem. Soc. Japan, 1968, 41, p. 1971.
555. Maki N.— Bull. Chem. Soc. Japan, 1969, 42, p. 2275.
556. Maki N., Sakuraba S.— Bull. Chem. Soc. Japan, 1969, 42, p. 1908.
557. Паладе Д. M., Борейко M. K-, Волох T. H.— Изв. вузов СССР. Химия и хим. тех-
нология, 1971. 14, с. 824.
558. Konya К., Nishikawa Н., Shibata М.— Inorg. Chem., 1968, 7, р. 1165.
559. Norris A. R., Wilson J. W. L.— Canadian J. Chem., 1973, 51, p. 4152.
560. Siebert H., Siebert C., Wieghardt K-— Z. anorg. allg. Chem., 1971, 380, S- 30.
561. McNeil D. A. C., Raynor J. B., Symons M. C. R.— J. Chem. Soc., 1965, p. 410.
562. Gray H. B-, Manoharan P. T., Pearlman J., Riley R. F.— Chem. Commun., 1965,
p. 62.
563. ClarkG. R., Reed C. A., Roper W. R. a. o.— Chem. Commun., 1971, p. 758.
564. Nast R., Schmidt J.— Angew. Chem., 1969, 81, S. 399.
565. Brennan R. L., Symons M. C. R., West D. X.— Inorg. Nucl. Chem. Lett., 1975, 11,
p. 61.
566. Venerable II,G.D., Halpern J.— J. Am. Chem. Soc., 1971,93, p. 2176.
567. Halpern J., GuastallaG., Bercaw J.— Coord. Chem. Rev., 1972, 8, p. 168.
568. Bressan M., Corain B., Rigo P., Turco A.— Inorg. Chem., 1970, 9, p. 1733.
569. Halpern J., Pribanic M.— J. Am. Chem. Soc., 1971, 93, p. 96.
570. Farkas A., Lupu D.— J. Inorg. Nucl. Chem., 1975, 37, p. 837.
571. Jarchow 0.— Z. Kristallogr., 1972, 138, S. 122.
572. Nast R., Schulz H., Moeler Д.-D.—Chem. Ber., 1970, 103, S. 777.
573. Behrens H., Lindner E., Schindler H.— Chem. Ber., 1966, 99, S. 2399.
574. Hieber W., Fiihrling H.— Z. anorg. allg. Chem., 1970, 373, S. 48.
575. Фасман А. Б., Кутюков Г. Г., Сокольский Д. В.—Журнал неорган. химии, 1965,
10, с. 1338.
576. Izatt R. М., Watt G. D., Eatough D., Christensen J. J.— J. Chem. Soc., 1967, A,
p. 1304.
577. Гринберг А. А., Гельфман M. И., Киселева H. B.— Журнал неорган. химии,
1967, 12, с. 1171.
578. Гринберг А. А., Гельфман М. И.— Докл. АН СССР, 1960, 133, с. 1081.
117
579. Павленко Л. И., Окорская А. П., Сергеева А. Н.— Координационная химия, 1975,
1, с. 510.
580. Павленко Л. И., Гундорина Л. А., Сергеева А. Н.— Журнал неорган. химия, 1975,
20, с. 1026.
581. Семенишин Д. И., Павленко Л. И., Врублевская Т. И.— Журнал неорган. химии,
1975, 20, с. 532.
582. Kralik F., Meindl J., Tauer Z., MaleticD.— Collect. Czech. Chem. Commun., 1973
38, p. 1466.
583. Krdlik F.— Collect. Czech. Chem. Commn., 1969, 34, p. 1327.
584. Krdlik F.— Collect. Czech. Chem. Commun., 1970, 35, p. 1916.
585. Ginsberg A. P., Koulek E.— Inorg. Chem., 1965, 4, p. 1186.
586. Emschwiller G.— C. r. Acad. Sci., 1959, 248, p. 959.
587. Legros J.— C. r. Acad. Sci., 1959, 248, p. 1339.
588. Baran E. J., Muller A.— Z. anorg. allg. Chem., 1969, 370, S. 283.
589. Good M. L., Clausen C. A., Prados R. A.— J. Amer. Chem. Soc., 1970, 92, p. 7482.
590. Сергеева A. H., Окорская А. П., Ткаченко Ж. И., Семенишин Д. И.— Журнал
неорган. химии, 1975, 20, с. 1031.
591. Сергеева А. П., Ткаченко Ж. И., Семенишин Д. И.— Журнал неорган. химии,
1975, 20, с. 1314.
592. Baran Е. JMilller А.— Chem. Вег., 1969, 102, S. 3915.
593. Павленко Л. И., Окорская А. П., Сергеева А. Н.— Журнал неорган. химии, 1975,
20, с. 820.
594. Demas J. N., Turner Т. F., Crosby G. А.— Inorg. Chem., 1969, 8, p. 674.
595. Gillard R. D.— J. Inorg. Nucl. Chem., 1965, 27, p. 1321.
596. Черняев И. И., Желиговская Н. Н., Бабков А. В.— Журнал неорган. химии, 1961
6, с. 2627.
597. Евстафьева О. Н., Барановский И. Б., Бабаева А. В.— Журнал неорган. химии,
1965, 10, с. 27.
598. Черняев И. И., Бабков А. В., Желиговская Н. Н.— Журнал неорган. химии, 1963,
8, с. 1355.
599. Bielly Е., Gidney Р. М., Gillard R. D., Heaton В. Т.— J. Chem. Soc. Dalton Trans.,
1974, p. 2133.
600. Manzer L. E., Parshall G. W.— Chem. Commun., 1975, p. 227.
601. Krogmann K-, Hausen H.-D.— Z. anorg. allg. Chem., 1968, 358, S. 67.
602. Deiseroth H. J., Schulz H.— Phys. Rev. Lett., 1974, 33, p. 963.
603. Krogmann K., Stephan D.— Z. anorg. allg. Chem., 1968, 362, S. 290.
604. Krogmann K., Ringwald G.— Z. Naturforsch., 1968, 236, S. 1112.
605. Deford D. D., Davidson A. W.— J. Am. Chem. Soc., 1951, 73, p. 1469.
606. Vogler A., Kunkely H.— Ber. Bunsenges. Phys. Chem., 1975, 79, S. 83.
607. Mikhalevich К. H., Sergeeva A. N., Dzyana D. I., Okorskaya A. P.— Chem. Abstr.,
1972, 77, p. 50901e.
608. Inoue H., Wada M., Yanagisawa S.— Inorg. Chim. Acta, 1973, 7, p. 129.
609. Geoffroy G. L., Wrighton M. S., Hammond G. S., Gray H. B.— Inorg. Chem., 1974,
13, p. 430.
610. Roberts H. L., Symes W. R.— J. Chem. Soc., 1968, A, p. 1450.
611. Krogmann K-, Binder W.— Amgew. Chem., 1967, 79, S. 902.
612. Krogmann K., Binder W.— J. Organometallic Chem., 1968, 11, № 2, p. P27.
613. Jewsbury R. A., Maher J. P.— J. Chem. Soc., 1971, A, p. 2847.
614. Gidney P. M., Gillard R. D., Heaton В. T.— J. Chem. Soc. Dalton Trans., 1972,
p. 2621.
615. Ibers J. A., HamiltonD. S., Baddley W. H.— Inorg. Chem., 1973, 12, p. 229.
616. Птицын Б. В., Земсков С. В., Николаев А. В.— Докл. АН СССР, 1966, 167, с. 112.
617. Бабков А. В.—Докл. АН СССР, 1967, 117, с. 337.
618. Черняев И. И., Бабков А. В.— Журнал неорган. химии, 1964, 9, с. 2253.
619. Siebert Н., Weise М.— Z. Naturforsch., 1975, 30b, S. 33.
620. Черняев И. И., Бабков А. В., Желиговская Н. Н.— Журнал неорган. химии, 1963,
8, с. 2441.
621. Mason W. R.— Inorg. Chem., 1971, 10, р. 1914.
622. Jones L. H., Smith J. M.— Inorg. Chem., 1965, 4, p. 1677.
623. Mason W. R.— Inorg. Chem., 1970, 9, p. 1528.
624. SwihartD. L., Mason W. R.— Inorg. Chem., 1970, 9, p. 1749.
625. Бусев А. И., Бабков А. В., Бусурманова Б. T.— Кинетика и катализ, 1973, 14,
с. 1154.
626. Бусев А. И., Бабков А. В., Бусурманова Б. Т.— Кинетика и катализ, 1975, 16,
с. 1945.
627. Барановский И. Б., Бабаева А. В.— Журнал неорган. химии, 1966, 11, с. 1732,
118
628. Евстафьева О. Н., Барановский И. Б., Бабаева А. В.— Журнал неорган химии,
1966, 11, с. 1333.
629. Черняев И. И., Бабков А. В.— Журнал неорган. химии, 1965, 10, с. 802.
630. Черняев И. И., Бабков А. В.— Журнал неорган. химии, 1964, 9, с. 2307.
631. Черняев И. И., Бабков А. В., Желиговская Н. Н.— Журнал неорган. химии, 1964,
9, с. 576.
632. Черняев И. И., Леонова Т. Н.— Журнал неорган. химии, 1965, 10, с. 1935.
633. Черняев И. И. Комплексные соединения платины. М., Наука, 1973.
634. Siebert Н., Siebert А.— Angew. Chem., 1969, 81, S. 575.
635. Михалевич К- Н., Сергеева А. Н., Ткаченко Ж. И., Окорская А. П.— Изв.
СО АН СССР. Сер. хим. наук, 1970, вып. 4, с. 47.
636. Halpern J., Cozens R.— Coord. Chem. Rev., 1975, 16, p. 141.
637. NastR., Moerler H.-D.—Chem. Ber., 1969, 102, S. 2050.
638. Nast R., Buick J.— Z. anorg. allg. Chem., 1975, 416, S. 285.
639. German IF. L.—J. Chem. Soc., 1938, p. 1027.
640. NordG., Matthes H.— Acta Chem. Scand., 1974, 28A, p. 13.
641. Dodd R. E., Gasser R. P. H.— Proc. Chem. Soc., 1964, p. 415.
642. LeDemezet M., MadecC., L’Her M.— Bull. Soc. chim. France, 1970, p. 365.
643. Barden J.-C.— J. Electroanal. Chem., 1970, 28, p. 157.
644. Звягинцев О. E., Захаров-Нарциссов О. И.— Журнал неорган. химии, 1960, 5,
с. 124.
645. Dove М. F. A., Hallett J. G.— Chem. Commun., 1967, р. 571.
646. Klatt W.— Z. phys. Chem., 1939, 185A, S. 306.
647. Gillespie R. J., Hulme R., HumphreysD. A.— J. Chem. Soc., 1971, A, p. 3574.
648. Payen-Baldy N., Machtinger M.— Bull. Soc. chim. France, 1968, p. 2221.
649. Touller J. C., Grail M., Bigois M., Tremillon B.— Bull. Soc. chim. France, 1965,
p. 1853.
650. Bigois M., Touller J.-C., Tremillon B.— Bull. Soc. chim. France, 1965, p. 1847.
651. Jagtiani I. A., Siddigi I. IF., Tyrrell H. J. V.—J. Chem. Soc. Faraday Trans. I, 1972,
68, p. 2090.
652. Sellers D. E., Leonard G. W., Jr.— Analyt. Chem., 1962, 34, p. 1457.
653. Thiebault A.— Bull. Soc. chim. France, 1968, p. 3429.
' 654. Thiebault A., Herlem M., Badoz-Lambling J.— Rev. chim. miner., 1966, 3, p. 1005.
655. Beck M., Gaizer F.— Magyar kem. folyoirat, 1963, 69, old. 560.
656. Gellin N., Marcus Y.— J. Inorg. Nucl. Chem., 1974, 36, p. 1325.
657. Csiszar B., Gutmann V., Wychera E.— Monatsh. Chem., 1967, 98, S. 12.
658. Gutmann V., Bardy H.— Z. anorg. allg. Chem., 1968, 361, S. 213.
659. Gutmann V., Wegleitner К- H.— Monatsh. Chem., 1968, 99, S. 368.
660. Gutmann V., Bohunovsky 0.— Monatsh. Chem., 1968, 99, S. 751.
661. GutmannV., Scherhaufer A.— Inorg. Chim. Acta, 1968, 2, p.325.
662. Weisz A., Gutmann V.— Monatsh. Chem., 1970, 101, S. 19.
663. Wasgestian H. F.— J. phys. Chem., 1972, 76, p. 1947.
664. Kirk P. G., Smith T. D.— J. Inorg. Nucl. Chem., 1968, 30, p. 892.
665. Kirk P. G., Smith T. D.— J. Chem. Soc., 1969, A, p. 2190.
666. Dahl T., Hassel 0., Sky K-— Acta Chem. Scand., 1967, 21, 592.
667. Austad T., Esperas S.— Acta Chem. Scand., 1974, 28A, 892.
668. Downs A. W.— Chem. Commun., 1968, p. 1290.
669. Austad T., Songstad J., Ase K.— Acta Chem. Scand.. 1971, 25, 331.
670. Harrington G.,Sundheim B. R.— Ann. N. Y. Acad. Sci., 1960, 79, p. 950.
671. Eluard A., Tremillon B.— J. Electroanalyt. Chim., 1967, 13, p. 208.
672. Bennett W. E., Jensen W. P.— J. Inorg. Nucl. Chem., 1966, 28, p. 1829.
673. Hennion J., Nicole J., Tridot G.— C. r. Acad. Sci., 1968, 267C, p. 831.
674. Hennion J., Nicole J., Tridot G.— Analysis, 1972, 1, p. 48.
675. Flengas S. NRideal E.— Proc. Roy. Soc. (London), 1956, 233A, p. 443.
676. Manning D. L., Blander M.— Inorg Chem., 1962, 1, p. 594.
677. Ti Tien H. — J. Phys. Chem., 1965, 69, p. 3763.
678. Bombi G. G., Fiorani M., Mazzocchin G.-A.— J. Electroanalyt. Chem., 1965, 9,
p. 457.
679. White S. H., InmanD., Jones B.— Trans. Faraday Soc., 1968, 64, p. 2841.
680. InmanD., JonesB., WhiteS. H.— J. Inorg. Nucl. Chem., 1970, 32, p. 927.
681. ДелимарскийЮ. K-, Грищенко В. Ф., ЗарибицкаяЛ. И.— Журнал неорган. химии,
1975, 20, с. 921.
682. Накамото К- Инфракрасные спектры неорганических и координационных соеди-
нений. М., Мир, 1966.
683. Poletto G., Rigutti М.— Nuovo cimento,1965, 39, р. 519.
684. El-Sayed М. F., Sheline R. К.— J- Inorg. Nucl. Chem., 1958, 6, p. 187.
119
685. Griffith W. P., Turner G. T.— J. Chem. Soc., 1970, A, p. 858.
686. Харитонов Ю. pl., Евстафьева О. H., Барановский И. Б., Мазо Г. pl.— Журнал
неорган. химии, 1969, 14, с. 485.
687. Nakagawa I., Shimanouchi Т.— Spectrochim. Acta, 1960, 16, 424.
688. Jones L. H.— Inorg. Chem., 1965, 4, p. 1473.
689. Харитонов Ю. p{., Евстафьева О. H., Барановский И. Б., Мазо Г. pl.— Журнал
неорган. химии, 1968, 13, с. 827.
690. Schleinitz К- D— Z. Chem., 1971, 11, S. 189.
691. DowsD- A., Haim A., Wilmarth W. К.— J- Inorg. Nucl. Chem., 1961, 21, p. 33.
692. Shriver D. F., Shriver S. A., Anderson S. E.— Inorg. Chem., 1965, 4, p. 725.
693. Bonnet M. C., Paris R. A.— Bull. Soc. chim. France, 1975, part 1, № 5—6, p. 1062.
694. Ghosh S. N.— J. Inorg. Nucl.Chem., 1974, 36, p. 2465.
695. Харитонов Ю. Я-, Евстафьева О. H., Барановский И. Б., Мазо Г. Ж—Журнал
неорган. химии. 1969, 14, с. 2111.
696. Харитонов Ю. Я., Евстафьева О. Н., Барановский И. Б., Мазо Г. Я-— Журнал
неорган. химии, 1969, 14, с. 1016.
697. Харитонов Ю. Я-, Евстафьева О. И., Барановский И. Б. и др.— Журнал неорган.
химии, 1966, 11, с. 1733.
698. Beck W., Tehlhammer W. Р.— М. Т. Р. Int. Rev. Sci. Inorg. Chem., 1972, Serie 1,
7, p. 253.
699. Wade R. C.,Sullivan E. A.,Berschied J. R., Jr., Purcell K. F.— Inorg. Chem., 1970,
9, p. 2146.
700. Kuroda K-, Gentile P. S.— Inorg. Nucl. Chem. Lett., 1967, 3, p. 151.
701. Griffith W. P., Lane J. R.— J. Chem. Soc. Dalton Trans., 1972, p. 158.
702. Mahon C., BrittonD.— Inorg. Chem., 1971, 10, p. 586.
703. Thiele G., Bauer R., Messer D.— Naturwissenschaften, 1974, 61, S. 215.
704. Bertinotti C., Bertinotti A.— Acta Crystallogr., 1972, 28B, 2635.
705. Roof R. B., Larson A. C., Cromer D. T.— Acta Crystallogr., 1968, 24B, 269.
706. Konnert J., BrittonD., Chow Y. M.— Acta Crystallogr., 1972, 28B, 180.
707. Dunaj-Jurco M., Caraj J., Gazo J.— Chem Abstr., 1972, 76, 384905.
708. Penneman R. A., Ryan R. R.— Acta Crystallogr., 1972, 28B, 1629.
709. Bertinotti C., Bertinotti A.— Acta Crystallogr., 1970, 26B, 422.
710. Ledent J.— Bull. Soc. roy. sci. Liege, 1972, 41, p. 537.
711. Holt E. M., Watson K- J.— Acta Chem. Scand., 1969, 23, p. 14.
712. Leipoldt J. G., Basson S. S., Bok L. D. C.— Acta Crystallogr., 1970, 26B, 361.
713. Dupont L.— Bull. Soc. Roy. Sci. Liege, 1967, 36, p. 471.
714. Bok L. D. C., Basson S. S., Leipoldt J. G., Wessels G. F. S.— Acta Crystallogr., 1971,
27B, 1233.
715. Jerome-Lerutte S.— Acta Crystallogr., 1971, 27B, p. 1624.
716. Jerome-Lerutte S.— Bull. Soc. Roy. Sci. Liege, 1967, 36, p. 49.
717. Ruegg M., Ludi A.— Theor. Chim. Acta, 1971, 20, p. 147.
718. Williams J. M., Petersen J. L., Gerdes H. M., Peterson S. W.— Phys. Rev. Lett.,
1974, 33, p. 1079.
719. Jarchow 0., Schulz H., Nast R.— Angew. Chem., 1970, 82, S.43.
720. Jarchow 0.— Z. anorg. allg. Chem., 1971, 383, S. 40.
721. Miyoshi T., Iwamoto T., Sasaki Y.— Inorg. Chim. Acta, 1972, 6, 59.
722. Jurnak F. A., Raymond K-N.— Inorg. Chem., 1974, 13, p. 2387.
723. Powell H. M., WatkinD. J., Wilford J. B.— J. Chem. Soc., 1971, A, p. 1803.
724. Brown L. D., Raymond K- N., Goldberg S. Z.— J. Am. Chem. Soc., 1972, 94, p. 7664.
725. Simon G. L., Adamson A. W., Dahl L. F.— J. Am. Chem. Soc., 1972, 94, p. 7654.
726. Duggan D. M., Jungst R. G., Mann K. R- a. o.— J. Am. Chem. Soc., 1974, 96,
p. 3443.
727. Moreno E., Lopez C. A.— An. fis. Real soc. esp. fis. у quim., 1971, 67, p. 371; Chem.
Abstr., 1972, 77, 10736y.
728. Mahon C.— Inorg. Chem., 1971, 10, p. 1813.
729. Tullberg A., Vannerberg N.-G.— Acta Chem. Scand., 1974, A28, p. 551.
730. Кузнецов В. Г., Максимова С. И.— Журнал структ. химии, 1973, 14, с. 849.
731. Семенишин Д. И., Павленко Л. И., Врублевская Т. И.— Журнал неорган. химии,
1975, 20, с. 532.
732. Gupta М. Р., Milledge Н. J., McCarthy А. Е — Acta Crystallogr., 1974, ВЗО, 656.
733. Bonnet М. С., Paris R. А.— Bull. Soc. chim. France, 1975, part 1, № 5—6, p. 1067.
734. Bok L. D. C., Leipoldt J. G., Basson S. S.— Z. anorg. allg. Chem., 1972, 389, S. 307.
735. Konno M., Marumo F., Saito Y.— Acta Crystallogr., 1973, 29B, 739.
736. Siebert H., Siebert A.— Z. anorg. allg. Chem., 1970, 378, S. 160.
737. Wyckoff R. W. G.— Crystal Structures. Vol. 3. 2nd ed. New York, Interscience, 1965,
p. 382.
120
738. Buser H.-J., Ludi A., Petter W., SchwarzenbachD.— Chem. Commun., 1972, p. 1299.
739. Giidel H.— Acta Chem. Scand., 1972, 26, p. 2169.
740. Giidel H., Stucki H., Ludi A.— Inorg. Chim. Acta, 1973, 7, 121.
741. Ludi A., Giidel H., Ruegg M.— Inorg. Chem., 1970, 9, p. 2224.
742. Buser H.-JRon G., Ludi A., Engel P.— J. Chem. Soc. Dalton Trans., 1974, p. 2473.
743. Ludi A., Giidel H. U.— Structure and Bonding, 1973, 14, p. 1.
744. RileggM., Ludi A., Rieder R.— Inorg. Chem., 1971, 10, p. 1773.
745. Enemark J. FL, Quinly M. S-, Reed L. L. a. o.— Inorg. Chem., 1970, 9, p. 2397.
746. Tullberg A., Vannerberg N.-G.— Acta Chem. Scand., 1974, 28A, p. 340.
747. Jagner S., Vannerberg N.-G.— Acta Chem. Scand., 1973, 27, p. 3482.
748. Murmann R. R., Schlemper E. 0.— Inorg. Chem., 1971, 10, p. 2352.
749. Schlupp R., Le Carpentier J. M., Weiss R.— Rev. Chim. Miner, 1970, 7, p. 63.
750. Stadnicka R.— Rocz. Chem., 1973, 47, S. 2021.
751. Fronczek F. R., Schaefer W. P.— Inorg. Chem., 1974, 13, p. 727.
752. Wang В.-C., Schaefer IF. P., Marsh R. E.— Inorg. Chem., 1971, 10, p. 1492.
753. Fronczek F. R., Schaefer W. P., Marsh R. E.— Inorg. Chem., 1975, 14, p. 611.
754. Shandies R., Schlemper E. 0., Murmann R. R.— Inorg. Chem., 1971, 10, p. 2785.
755. Bok L. D. C., Leipoldt J. G , Basson S. S.— Z. anorg. allg. Chem., 1972, 392, S. 303.
756. Chojnacki J., Crochowski J., Lebioda L. i i.— Rocz. Chem., 1969, 43, S. 273.
757. Novotny M., LewisD. F., Lippard S. J.— J. Am. Chem. Soc., 1972, 94, p. 6961.
758. Clark M. G., Cancedo J. R., Maddock A. G., Williams A. F.— J. Chem. Soc. Dalton
Trans., 1975, p. 120.
759. Ryan R. R., Swanson В. I.— Inorg. Chem., 1974, 13, p. 1681.
760. Vannerberg N.-G.— Acta Chem. Scand., 1964, 18, p. 2385.
761. Шарло Г. Методы аналитической химии. Количественный анализ неорганических
соединений. М.—Л., Химия, 1965. 448 с.
762. Scoggins М. W.— Anal. Chem., 1970, 42, р. 301.
763. Parkash R., Zyka J.— Microchem. J., 1972, 17, p. 309.
764. Glasner A., SarigS., WeissD., ZidonM.— Taianta, 1972, 19, 51.
765. Пилипенко A. T., Ангелова Г. В., Налиниченко И. В.— Докл. Болг. АН, 1973,
26, с. 1359.
766. Пилипенко А. Т., Ангелова Г. В., Налиниченко И. В.— Укр. хим. журн., 1974,
40, с. 1302.
767. Пантелеева Л. Занько А. А., Дегтяренко Л. А.— В1сник Льв1в. пол1техн.
1н-ту, 1966, № 10, с. 130.
768. Umland F., Janssen A., Thierig D., Wilnch G.— Theorie und praktische Anwendung
von Komplexbildnern. Frankfurt-am-Main, Akademische Verlagsgesellschaft, 1971.
769. Paul R. C., Sharma N. C., Chauran R. R., Parkash R.— Indian J. Chem., 1971, 9,
p. 879.
770. Paul R. C., Sharma N. C., Chauran R. R., Parkash R.— Indian J. Chem., 1972,
10, p. 227.
771. RoporriKoea В. И., Носков В. В.— Журнал аналит. химии, 1972, 27, с. 580.
772. Duque-Macias F., Lucena-Conde F.—An. Quim., 1970, 66, p. 79; Chem. Abstr., 1970,
73, 52055y.
773. Eross R., Buzas J., Erdey L.— Fresenius’ Z. anal. Chem., 1971, 255, S. 273.
774. Ольхович П. Ф., Пилипенко A. T.— Укр. хим. журн., 1970, 36, с. 388.
775. Eross R., Buzas I., Erdey L.— Fresenius’ Z. anal. Chem., 1971, 255, S. 271.
776. Eross R., Buzas L, Erdey L.— Fresenius’ Z. anal. Chem., 1971, 255, S. 275.
777. Бейлар Дж. Химия координационных соединений. М., Изд-во иностр, лит., 1960.
526 с.
778. Schneble F. W., McCormack J. F., Rau T. A.— Chem. Abstr., 1972, 77, 51802k.
779. Appleby A. J.— J. Electrochem. Soc., 1970, 117, p. 1610.
780. Smith G. R., Renahan С. B., Andrews R. L., SchlainD.— Plating, 1969, 56, p. 805.
781. Finnern B.— Chem. Abstr., 1971, 75, 90684b; 1972, 77, 104715t.
782. Chem. Abstr., 1966, 64, 3144h.
783. Takahashi M., limura R.— Chem. Abstr., 1969, 70, 115690n.
784. Rick E. A., McReon J. E., Fitton P., Argento B. J. Пат. США, № 3678085 от
18.02.72. РЖхим, 1973, 104197 П.
785. Зубрицкая Д. ТЛюбченко Ю. А., Михалевич R. Н. и др.— Журнал неорган. хи-
мии, 1973, 46, с. 329.
786. Любченко Ю.А., ТкаченкоЮ. И., Павленко Л. И. и др.— РЖхим, 1975, 17Л204П.
787. Hahn R. В., Rlein Н. С.— Anal. Chem., 1968, 40, р. 1135.
788. Rourim V.— Chem. Abstr., 1966, 64, 15023h.
789. Qureshi M., Varshney R. G., Israili A. H.— J. Chromatogr., 1971, 59, p. 141.
790. Нозлов A. C.— Изв. АН СССР. Неорганические материалы, 1965, 1, с. 730.
791. Mellors G. IF.—Chem. Abstr., 1971, 74, 36188j.
121
792. Shortridge R. G., Lin M. C.— J. Phys. Chem., 1974, 78, p. 1451.
793. Okada T.— CEER Chem. Econ. and Eng. Rev., 1973, 5, № 7, c. 21.
794. Мюллер E. Новые воззрения в органической химии. М., Изд-во иностр, лит.,
1969, 580 с.
795. HerzbergG.— J. Chem. Phys., 1940, 8, р. 847.
796. Loew G. H., Chang S.— Tetrahedron, 1971, 27, p. 3069.
797. Yamamoto Y., Yamazaki H.— Coord. Chem. Rev., 1972, 8, p. 225.
798. Doggett G., McKendrick A.— J. Chem. Soc., 1970, A, p. 825.
799. Dixon M., Doggett G.— J. chem. Soc. Faraday Trans. II, 1973, 69, p. 298.
800. Bonaccorsi R., Petrongolo C., Scrocco E., Tomasi J.— J. Chem. Phys. 1968 48,
p. 1500.
801. Beach N. A., GrayH. B.— J. Am. chem. Soc., 1968, 90, p. 5713.
802. Hancock R.D., Finkelstein N. P., Evers A.— J. Inorg. Nucl. Chem., 1976, 38, p. 343.
803. Vrebel V., Kello E., Dunaj-Jurco M. u. a.— Z. anorg. allg. Chem., 1975, 417, S. 154.
804. Маров И. H., Беляева В. К., Довгей В. В., Сергеева А. Н.— Журнал неорган. хи-
мии, 1976, 21, с. 3238.
805. Vannerberg H.-G., Jagner S.— Chem. Scr., 1974, 6, p. 19.
806. Chavant C., Constant G., Jeannin Y., Morancho R. — Acta Crystallogr., 1975, B31,
№ 7, 1823.
807. Kiernan P. M., Griffith W. P.— J. Chem. Soc. Dalton Trans., 1975, № 23, p. 2489.
808. Kiernan P. M.— Inorg. Chim. Acta, 1976, 20, p. 89.
809. Сергеева A. H., Мазепа А. В., Мазурок В. С.— Координационная химия, 1975,
1, с. 1681.
810. Нефедов В. И., Синицын Н. М., Салынь Д. В., Байер Л.— Координационная хи-
мия, 1975, 1, с. 1618.
811. Hancock R. D., Evers А.— Inorg. Chem., 1976, 15, р. 995.
812. Делимарский Ю. К-, Зарубицкая Л. И., Грищенко В. Ф.— Журнал неорган. хи-
мии, 1976, 21, с. 413.
813. Iwamoto Т., Kiyoki М.— Bull. Chem. Soc. Japan, 1975, 48, p. 2414.
814. Brown L. D., Raymond K- N.— Inorg. Chem., 1975, 14, p. 2590.
815. Power L. F., King J. A., Moore F. H.— J. Chem. Soc. Dalton Trans., 1975, № 20,
p. 2072.
816. Bonnet M. C., Paris R. A.— Bull. Soc. chim. France, 1975, part 1, № 5—6, p. 1067.
817. Raynor J. B., Nye R. L.— J. Chem. Soc. Dalton Trans., 1976, p. 504.
818. Halpern J., Pribanic M.— Inorg. Chem., 1970, 9, p. 2616.
819. Sokol C. S., Brubaker С. H., Jr.— J. Inorg. Nucl. Chem., 1968, 30, p. 3267.
, 820.Pregaglia G., Morelli D., Conti F. a. o.— Discuss. Faraday Soc., 1968, p.110.
821. PappS., KovacsS., LisziJ.— J. Inorg. Nucl. Chem., 1972,34, p. 3111.
822. Сергеева A. H., Семенишин Д. И., Мазепа А. В.— Укр. хим. журн., 1975, 41,
с. 1048.
823. Lim Н. S., Anson F. С.— Inorg. Chem., 1971, 10, р. 103.
824. Crouch Е. С. С., Pratt J. 7И.— Chem. Commun., 1969, р. 1243.
825. Poskozim Р. S.— J. Inorg. Nucl. Chem., 1975, 37, p. 2342.
826. Siebert H., Thym S.— Z. anorg. allg. Chem., 1973, 399, S. 107.
827. Кривлюк A. H., Семенишин Д. И., Сергеева A. H.— Координационная химия,
1976, 2, с. 176.
828. Lukosius Е. JCoskran К- J— Inorg. Chem., 1975, 14, р. 1922.
829. Rigo Р., Bressan М.— Inorg. Chem., 1975, 14, р. 1491.
830. Strekas Т. С., Spiro Т. G.— Inorg. Chem., 1975, 14, р. 1421.
831. Ebina F., Куипо Е., Uehara A., Tsuchiya R.— Bull. Chem. Soc. Japan, 1975, 48,
p. 3120.
832. Павленко Л. И., Семенишин Д. И., Сергеева А. Н., Любченко Ю. Д.— Журнал
неорган. химии, 1976, 21, с. 1961.
833. Skorsepa JChotnic J., Matejcikova E.— Monatsh. Chem., 1976, 107, S. 297.
834. Inoue EL, FluckE., YanagisawaS.— Z. Naturforsch., 1976, 31b, S. 167.
835. Miskowski V. M., NobingerG. L., Hammond G. S.— Inorg. Chem., 1976, 15, p. 2904.
836. Nast R., Biilck J., Kramolowsky R.— Chem. Ber., 1975, 108, S. 3461.
837. Lourtioz J. M., Adde R.— Rev. Phys. AppL, 1976, 11, p. 533.
838. NichollsD., Ryan T. A.— Inorg. Chim. Acta, 1977, 21, № 1, p. L17—L18.
839. Manaharan P. T., Gray H. B.— Inorg. Chem., 1966, 5, p. 823.
840. Gray H. B., Alexander J. J.— Coord. Chem. Rev., 1967, 2, p. 29.
4. ЦИАНАТЫ И ФУЛЬМИНАТЫ
Цианат (NCO-)- и фульминат (CNO-)-ионы являются изомерами. Как
известно, понятие изомерии впервые было введено Берцелиусом [1], после то-
го как Либих и Велер независимо друг от друга установили различие в свой-
ствах фульмината и цианата серебра [2, 3]. Кроме того, как показал Бек [4],
возможна изомеризация, т. е. переход фульминат -+ цианат.
Различие между цианат- и фульминат-ионами проявляется как в стабиль-
ности образованных ими соединений, так и в способах их координации.
В противоположность взрывчатым фульминатам тяжелых металлов их циа-
наты довольно устойчивы. Помимо этого цианат-ион, подобно тиоцианат-
и селеноцианат-ионам, координируется преимущественно атомом азота,
а фульминат-ион присоединяется к металлу через углерод. В этом отноше-
нии фульминат-ион напоминает ион цианида.
4.1. ДИОКСИЦИАН
Факт существования (OCN)2 до настоящего времени продолжает оставать-
ся спорным. Первые попытки получения диоксициана анодным окислением
цианатов относятся к 1925 г. [5]. Сообщается об образовании (OCN)2 путем
реакции обмена между AgNCO и 12 [61. Согласно [6], (OCN)2 — бесцветное,
устойчивое лишь при низких температурах (до —12° С) соединение. По-
следние данные об анодном окислении цианатов в ацетонитриле ставят под
сомнение существование (OCN)2. Конечным продуктом анодного окисления
цианатов является (CN)2 [7].
По формальной аналогии с галогенами (OCN)2 может реагировать с не-
которыми неметаллами, так как ковалентные изоцианаты обычно получают
обменными реакциями галогенидов неметаллов с цианатами металлов
(см. 4.4).
4.2. ЦИАНАТНАЯ КИСЛОТА. СТРУКТУРА И СВОЙСТВА
В свободном виде цианатная кислота (оксициановодород) получена Ве-
лером и Либихом [8, 9]. Это — бесцветная жидкость (т. пл. —86° С), устой-
чивая при низких температурах. Выше —20° С она быстро разлагается, и
образуется смесь циановой кислоты, циана и циамелида [11]. Ниже —100° С
образуется другая модификация HNCO [12]. Цианатную кислоту получают
путем термолиза циануровой кислоты (HNCO)3 с последующей конденсацией
123
паров HNCO и очисткой ее вакуумной дистилляцией при низких температу-
рах [10]
(HNCO)3 3HNCO. (4.1)
Исследование структуры кристаллической и газообразной циановой кис-
лоты показывает, что она представляет собой изосоединение HNCO [13—
15], причем параметры молекул кристаллической и газообразной HNCO
совпадают. HNCO имеет ромбическую структуру (а — 10,82; b = 5,52;
с = 3,57 A; Z = 4; пространственная группа, вероятно, Рата). При темпе-
ратуре < —100° С существует вторая модификация HNCO, для которой рас-
стояния N—С и С—О составляют 1,183 и 1,184 А, а ее молекулы связаны
водородными мостиками N—H...N в зигзагообразные цепи [12]. Согласно
[13, 1о4], для газообразной HNCO найдено: длины связей N—С 1,19 или
1,20 A; N—О 1,19 или 1,17 A; N—Н 1,01 или 0,987 A; Z_ HNC = 125 или
128,5°; Z. NCO — 180°. Силовые константы связей: н — 6,752; kc-н =
= 13,35; kc—o — 15,34 Н/см. Порядок связей цианатной кислоты описывает-
ся граничными мезомерными формулами
Н—N=C=O| <-> Н—N—С ~ О | Н—N = С—О|
Вследствие значительной летучести HNCO имеет резкий запах, напоми-
нающий запах уксусной кислоты. Это слабая кислота (k = 2 • 10-4). Ее вод-
ные растворы быстро гидролизуют
HNCO + Н2О НОС< -> СО2 + NH3. (4 2)
xnh2
В спиртовом растворе HNCO превращается в эфир карбаминовой кисло-
ты, а в эфирных растворах устойчива
/OR
.OR О=С<
HNCO + ROH->О=С< -> >NH (4.3)
\NH« О=С<
xnh2
Так как HNCO образуется при нагревании мочевины с последующей
тримеризацией в циануровую кислоту, то эту реакцию можно применить для
синтеза циануровой кислоты [15]
ЗОС (NH2)2 3HNCO -> (HNCO)3. (4.4)
Циануровая кислота кристаллизуется в виде бесцветных игл и существу-
ет в кристаллической форме как изоциануровая кислота
101
HNI INH
I I
о=с. х=о
I
н
Для планарного кольца (HNCO)3 найдены расстояния 1,34—1,36 А
(С—N) и 1,21 А (С—О). Циануровая кислота образует слоистую структуру
124
с сильными N—H...O-водородными связями [16]. Выше —20° С, кроме циа-
нуровой кислоты, из HNCO образуется полимер циамелида со структурой
[17]
... о—С—О—С—О—С—О—С...
II II II II
NH NH NH NH
4.3. ПОЛУЧЕНИЕ И СВОЙСТВА ЦИАНАТОВ АММОНИЯ,
ЩЕЛОЧНЫХ И ЩЕЛОЧНОЗЕМЕЛЬНЫХ МЕТАЛЛОВ
Хотя первые цианатные соединения были получены в начале XIX в.,
простые цианаты многих металлов до сих пор неизвестны [18]. Это объясня-
ется слабостью и неустойчивостью цианатной кислоты в водном растворе,
а также заметной склонностью цианат-ионов к образованию малораствори-
мых двойных цианатов. Сравнительно хорошо изучены цианаты щелочных
металлов.
Цианат натрия (т. пл. 550° С) — белые пластинчатые кристаллы. Для
синтеза NaNCO разработано несколько методов. Его можно получить сплав-
лением натрия с мочевиной или взаимодействием тех же компонентов, дис-
пергированных в бензине или толуоле. Выход NaNCO составляет —90%
[19]. Ход реакции описывается уравнениями
2ОС (NH2)2 + 2Na 2NaNHCONH2 + Н2;
2NaNHCONH2 — - 2NaNCO + 2NH3. (4.5)
Соли MNCO (M = Na, К, Rb, Cs) образуются при сплавлении М2СО3
или МНСО3 с мочевиной [20—22], а также при взаимодействии мочевины с
М2СО3 (М = Li, Na, К, Rb, Cs) в полярных органических растворителях,
например в DMSO [21]. NaNCO можно синтезировать, нагревая эквимоляр-
ные количества уретана и натрия в абсолютном эфире [23]. Описан синтез
MNCO (М = Na, К) окислением MCN активированной Н2О2 в спиртово-
щелочной среде. Чистота солей составляет 95—98% [24].
KNCO получают окислением К4 [Fe (CN)6] бихроматом калия [25]. В от-
сутствие влаги KNCO хорошо сохраняется, во влажном воздухе расплы-
вается и подвергается гидролизу. Водные растворы KNCO неустойчивы
и гидролизуют [26]
KNCO + 2Н2О -> КНСО3 + NH3. (4.6)
LiNCO получают взаимодействием металлического лития с AgNCO в Thf
в присутствии нафталина в атмосфере аргона [27] или нагреванием до 550° С
смеси LiOH и (HNCO)3, взятых в мольном соотношении 3 : 1 [28].
NH4NCO, а также соли типа [R4N] NCO весьма сходны с цианатами ще-
лочных металлов. NH4NCO можно получить взаимодействием газообразных
NH3 с HNCO или их эфирных растворов [29, 30]. Его также получают об-
меном NH4C1 с AgNCO. Цианат аммония при нагревании или растворении
в водном растворе быстро превращается в мочевину.
Открытие в 1828 г. Велером [31] равновесия
NH4NCO <> NH2CONH2 (4.7)
сыграло огромную роль в развитии синтетической органической химии.
Соль [Me4N] NCO получают из метанольного раствора взаимодействием
эквимолярных количеств [Me4N] Br и AgNCO [32, 33].
125
Цианаты щелочноземельных металлов подобно цианатам щелочных ме-
таллов получают по [21]. Са (NCO)2 можно также получить нагреванием до
200—300° С известково-мочевинной смеси [34, 35], a Ba (NCO)2 — реакцией
обмена метанольных растворов Ва (С1О4)2 или Ba (SCN)2 с водным раствором
NaNCO [36].
4.4. ИЗОЦИАНАТЫ ЭЛЕМЕНТОВ III—VII ГЛАВНЫХ ПОДГРУПП
Синтезу, свойствам и структурным особенностям элементоорганических
изоцианатов неметаллов III—V групп посвящены обширные обзорные статьи
[37, 38]. Интересна обзорная работа по химии изоцианатов фосфора [39].
В то же время сведений о простых цианатах указанных элементов значитель-
но меньше.
Цианат бора получен реакцией обмена ВВг3 с AgNCO в бензоле [40].
Это — полимерное соединение состава [В (NCO)3]X. В газообразных продук-
тах реакции ВС13 или ВВг3 с AgNCO удалось доказать существование неус-
тойчивых соединений BC12NCO и BBr2NCO [41]. Более устойчивы органо-
боризоцианаты R2BNCO [37]. В качестве интересного «неорганического»
изоцианата бора следует назвать соединение [NHB (NCO)]3 [42].
Прозрачные кристаллы Ga (NCO)3 • Thf получают в реакции между эк-
вивалентными количествами Ga (NO3)3 • 8Н2О и KNCO в Thf. Аддукт
Ga (NCO)3 • Thf гидролизует в воде, растворяется в DMFA и этаноле [32,
33]. Аналогично из 1пС13 и AgNCO в ацетоне получают In (NCO)3. Он очень
гигроскопичен, гидролизует в воде, хорошо растворяется в DMSO, DMFA,
ацетоне, этаноле [32, 33]. Полный обмен 1~ на NCO~ происходит в реакции
Inl3 с AgNCO в этаноле. Однако вторичные реакции с растворителем при-
водят к тому, что чистый In (NCO)3 получить нельзя [43]. Довольно устой-
чив цианат таллия (I). Эту соль получают реакцией T12SO4 с цианатом ще-
лочного металла в воде с последующим добавлением этанола [44].
Цианат углерода не получен, однако известны алкил- и арил-цианаты,
которые представляют практический интерес [45—47, 127, 253].
Реакцией C1CN с AgNCO получен изоцианат циана NCNCO [48]. Удоб-
ным методом получения его является термолиз AgNCO при ~335° С [49]
По данным ИК-спектроскопии молекула NCNCO линейна [48]. Соединение
легко подвергается гидролизу или сольволизу спиртами
/.О
- NCN—NH2CN + С02
b он
NCNCO _ (4.8)
ncn-c(:
Получены соединения состава X—СО—NCO (X = F, С1, Вг). Фторкарбо-
нилизоцианат (т. кип. 26—28° С) получают реакцией обмена между Si (NCO)4
или Me3SiNCO с COF2 [50], а аналогичные соединения хлора и брома — фото-
лизом соответствующих галоген-изоцианатов [94, 96].
Si (NCO)4 (т. пл. 26° С, т. кип. 186° С) впервые получен обменной реак-
цией SiCl4 с AgNCO в бензоле [40]. Его также получают взаимодействием
SiCl4 с KNCO в расплаве LiCl—КО [51] или SiCl4 с HNCO в эфире в присут-
ствии триметиламина при охлаждении [52]. Наряду-с Si (NCO)4 авторы [401
126
предполагали также образование Si (OCN)4, но, как показали новые данные
ИК-спектроскопии [53], это вещество является смесью Si (NCO)4 и Si2O (NCO)6
[53]. Последнее соединение можно получить прямой реакцией Si2OCl6 с
AgNCO или частичным гидролизом Si (NCO)4. Аналогично получено соеди-
нение Si3O2 (NCO)8 [54]. Хлоризоцианаты кремния SiCl4—я (NCdj„ (п = 1
3) образуются при взаимодействии SiCl4 с Si (NCO)4 [55], а аналогичные
фторизоцианаты — из Si (NCO)4 и SbF3 [56]. Из данных И К- и КР-спектров
следует, что молекулы SiCl3NCO и SiCl (NCO)3 имеют С3у-симметрию, а груп-
па Si—N—С—О линейна [137].
Ge (NCO)4 (т. пл. 8° С, т. кип. 204° С) получен аналогично Si (NCO)4
[40]. Если для триклинно кристаллизующегося Si (NCO)4 (пространственная
группа Р1 или Р1) предполагается тетраэдрическое строение [571, то для
Ge (NCO)4 наблюдаются значительные отклонения от Та-симметрии [571. Про-
ведены структурные исследования SiH3NCO и GeH3NCO [282]. Из микровол-
нового спектра для SiH3NCO следует линейное расположение связей Si—N—
С—О и найдены следующие длины связей [58]: Si—N 1,699; N—С 1,150 и
С—О 1,179 А. Для GeH3NCO, полученного из GeH3I и AgNCO [59], по дан-
ным ИК- [60] и микроволнового [61] спектров, установлена нелинейность
GeNC-остова, для которого Z GeNC = 154 или 143° [61]. Измерения ди-
фракции электронов Me3SiNCO дают: угол связои SiNC 150°; расстояния Si—N,
N—С и N—О соответственно 1,76; 1,20; 1,18 А [62]. Описаны соединения ти-
па R3SnNCO и R2Sn (NCO)2 [37, 63].
Цианат олова (II) Sn (NCO)2 • Diox образуется при реакции SnCl2 с
Pb (NCO)2 в диоксане [32, 33].
В литературе имеются сведения о цианатах Pb (II) и Pb (IV) [43, 64, 65].
Pb (NCO)2 получен сплавлением РЬО с мочевиной при 400—450° С [65],
a Pb (NCO)4 образуется при взаимодействии РЬ (СН3СОО)4 и HNCO [431.
Описаны органоцианаты свинца (IV) состава R3PbNCO [37, 38].
Р (NCO)3 (т. кип. 169,3° С) получен реакцией РС13 с AgNCO в кипящем бен-
золе [40, 661. Аналогично получают изоцианаты мышьяка (III) и сурьмы
ЕС13 4- 3AgNCO Е (NCO)3 + 3AgCl (Е = Р, As, Sb). (4.9)
Предложен модифицированный метод синтеза Р (NCO)3 в жидком SO2
или бензоле с применением NaNCO вместо AgNCO [67]. Если реакцию обме-
на РС13 с AgNCO проводить в присутствии избытка первого, то образуется
РС12 (NCO) и PCI (NCO)2 [68, 69]. Частичное фторирование Р (NCO)3 при
помощи SbF3 приводит к образованию РЕ2 (NCO) и PF (NCO)2[70]. Соедине-
ния фосфора (V) РО (NCO)3 получают реакцией РОС13 с AgNCO в бензоле
[71] или окислением Р (NCO)3 триоксидом серы, a PS (NCO)3 — окислением
Р (NCO)3 серой. Выделены РОС12 (NCO) [74] и POF2 (NCO) [50, 75, 76]
РС15 + H2N—СО—ОС2Н5->РС13 = N—СО—ос2н5->-
POC12NCO + QHCl + НС1; (4.10)
4POF3 + Si (NCO)4 -> 4POF2 (NCO) + SiF4; (4.11)
KNCO + F2P (O) -OP (O) F2-> POF2 (NCO) + К [PO2F2]. (4.12)
Соединения РХз-« (NCO),, (X = F, Cl) и POCI3—n (NCO)n изучены ме-
тодом ЯМР. По величине химического сдвига аналогичных соединений уста-
новлено, что электроотрицательность лигандов изменяется в ряду С1 >
> NCO > NCS > Вг [77].
Описаны соединения N3PBrn (NCO)6-/? [72] и N3P3C14 (NH2) (NCO) [73],
которые получены при взаимодействии (NPBr2)3 с AgNCO в нитрометане или
же реакцией фосгенирования N3P3C14 (NH2)2 в хлорбензоле.
127
Прекрасный обзор по синтезу и реакционной способности фосфороргани-
ческих изоцианатов приведен в работе Деркача [39]. Получены важные ал-
килфосфорцианаты (X = О, S) [78, 80, 246]
~Х /X
(RO)2 Р< 4- СОС12 (RO)2 P<f + 2НС1 + 2СО;
\nh2 xNCO
//X
(4.13)
(4.14)
ROH + (RO)2
NH—COOR XNCO
Впервые в соединении (PhO)2 Р (О) OCN обнаружена связь Р—OCN.
Для получения белых игл As (NCO)3 проводят реакцию между AsCl3
и AgNCO, Pb (NCO)2 или NaNCO в бензоле или ацетонитриле [81]. Реакция-
ми обмена галогенидных групп на NCO получают органоарсен-, -сурьма-
и -висмутцианаты [37, 38, 63, 82, 83]; Ph3As (NCO)2 образуется также в ре-
зультате прямого взаимодействия Ph3AsO с мочевиной [63]. Реакция SbCl5
с KNCO в жидком SO2 приводит через К [SbCl5NCO] к димеру [SbCl^NCOL
с мостиковыми NCO-группами, который выше —10° С превращается в три-
мер [SbCl4NCO]3 [269, 270].
Попытка получить S2 (NCO)2 предпринята в работе [64]. При взаимодей-
ствии S2C12 и AgNCO в бензоле образуется желтоватый продукт, представ-
ляющий, очевидно, полимерное вещество (SNCO)X.
Описаны изоцианатные соединения серы SO2 (NCO)2 и S?O5 (NCO)2 [85,
86], синтезирован SO2F (NCO) [87]. Дисульфурилизоцианат S2O5 (NCO)2
образуется также при взаимодействии KNCO с жидким SO3 [88] или (наряду
с другими продуктами) при реакции SO3 с KCN [89].
Интересны галоген-изоцианаты XNCO (Х = С1, Br, I), первые попытки
синтеза которых были предприняты Биркенбахом [90]. В то время как иод
взаимодействует при низкой температуре в эфире
I2 + AgNCO INCO + Agl, (4.15)
в случае хлора и брома в этих условиях образуются X2N—СО—NCO
(X = Cl, Вг) [91, 92]. Соединения C1NCO можно получить термолизом в ва-
кууме трихлорциануровой кислоты. Это—желто-зеленая устойчивая лишь
при низких температурах жидкость (т. пл.—98,5° С) [93]. BrNCO (жидкость,
т. пл.— 48° С) (разлагается) получают аналогично из трибромциануровой
кислоты [94] или из AgNCO и Вг2 при повышенной температуре (150° С и
р = 1 атм) с последующей конденсацией при •—195° С [96]. При относитель-
но низких температурах XNCO (X = Br, I) склонны к димеризации и об-
разуют N-дигалогенкарбомоилизоцианат X2NCO—NCO [90, 94], a C1NCO
при нагревании до комнатной температуры образует 1,3-дихлорэтидиндон
[96, 97]
О
II
С\
Cl—N<f >N—Cl
XCZ
li
О
Это соединение при нагревании в вакууме до 300° С превращается в
хлоризоцианат [97].
Изучены электронные [98] и ИК-спектры [99] C1NCO; из данных мик-
128
роволновой спектроскопии следует, что расстояние С1—N равно 1,688 А,
a Z.C1NC = 123° [1001.
Хлоризоциан реагирует со щелочами, как типичный галогенопсевдо-
галогенид [931,
C1NCO + 2ОБГ-^С1О’ + NCCT + Н2О. (4.16)
При фотолизе XNCO наряду с другими продуктами образуется XCONCO,
а обменные реакции элементоорганических галогенидов (R3SiCl, RHgCl)
с C1NCO ведут к образованию элементоорганических изоцианатов
(R3SiNCO, RHgNCO) [101].
4.5. ЦИАНАТЫ d- И f-МЕТАЛЛОВ
Простые цианаты d- и f-металлов до сих пор малоизучены. Один из
важнейших цианатов AgNCO, описанный еще Велером [31, выпадает из вод-
ных растворов при взаимодействии Ag+ и NCO~. Хорошо образованные
кристаллы AgNCO можно получить при длительном отстаивании водного
раствора, содержащего, кроме AgNCO, мочевину [2571. Устойчивая при ком-
натной температуре модификация AgNCO (см. 4.11.1, рис. 4.4) превращает-
ся при 120° С в высокотемпературную модификацию, которая выше 335° С
разлагается с образованием N2, СО, СО2, (CN)2 и NCNCO [49]. Малораствори-
мый в воде AgNCO легко растворяется в NH4OH и HNO3, находит широкое
применение в синтезе цианатных соединений.
Светло-зеленый микрокристаллический порошок Си (NCO)2 можно по-
лучить из CuBr2 и Hg (NCO)2 в ацетоне [36] или разложением при нагрева-
нии Си (NCO)2 • 6Ру [103]. Восстановлением водных растворов, содержащих
Си2+ и NCO-, получают бесцветную соль CuNCO, которая по химическим
свойствам напоминает CuCl, однако в отличие от последнего относительно
хорошо растворима в воде [36].
Реакциями обмена CdSO4 с Ba (NCO)2 в воде или Cdl2 с AgNCO в ацетоне
получают цианат кадмия. Из воды кристаллизуется Cd (NCO)2 • 2,5 Н2О,
который загрязнен основной солью. Относительно низкую растворимость
этой соли в метаноле можно объяснить ауто комплексообразованием [36].
Для получения Hg (NCO)2 используют следующие методы: обмен HgCl2
с AgNCO [36, 104], реакцию эфирного раствора HNCO и HgO или диспропор-
ционирование Hg2 (NCO)2 [36]. Hg (NCO)2 — бесцветные призматические
кристаллы, темнеющие под действием света. Цианат ртути (II) напоминает
HgCl2. Как и хлорид, цианат ртути хорошо растворяется в воде и полярных
органических растворителях, являясь в растворе неэлектролитом. Бесцвет-
ные растворимые в бензоле кристаллы CH3HgNCO получают по уравнению
[101]
CH3HgCl + C1NCO CH3HgNCO + Cl2. (4.17)
Бледно-голубой Со (NCO)2 и бледно-зеленый Ni (NCO)2 получают тер-
молизом соответствующих пиридиновых комплексов М (NCO)2 • 6Ру [103].
Для Со (NCO)2 и Ni (NCO)2 • Н2О определены магнитные моменты, изучены
электронные и ИК-спектры [136]. Реакцией обмена UO2 (NO3)2 с KNCO в
этаноле получен золотисто-желтый UO2 (NCO)2 [105].
5 2473
4.6. ГРЕМУЧАЯ КИСЛОТА
История исследования гремучей (фульминатной кислоты) связана с име-
нами таких выдающихся химиков, как Берцелиус, Бертло, Дэви, Дюма,
Гей-Люссак, Либих, Кекулеи Тенар. Виланд объяснил образование фульми-
натной кислоты из спирта и азотной кислоты [106], после того как еще рань-
ше Неф [107] установил ее мономерную формулу CNOH. Долгое время гре-
мучая кислота рассматривалась как оксим оксида углерода. Однако Бек
[108], исследуя ИК-спектр газообразной гремучей кислоты, однозначно оха-
рактеризовал ее как формонитрил-оксид HC=N—О. Линейная структура
гремучей кислоты подтверждена микроволновой спектроскопией. Найдены
расстояния [109]: Н—С 1,027 А; С—N 1,168 А и N-—О 1,200 А. Фульминатную
кислоту можно представить двумя мезомерными формами, причем пред-
почтение отдается структуре (I) [110],
+ + _ + _
Н—C=N—О| (I) Н—C=N=О | (II).
Газообразную HCNO получают взаимодействием водного раствора
NaCNO и H2SO4 при охлаждении льдом с последующей отгонкой ее в токе
N2 или реакцией HICNOH с NaOH [ПО]
CNO- — -HCNO; (4.18)
HICNOH -+°»~ ->HCNO. (4.19)
-i--h2o v '
Так как HCNO в воде быстро полимеризуется, ее необходимо быстро уда-
лить из раствора и высушить. Более целесообразно высушивать HCNO над
Р2О5 или Mg (СЮ4)2, а затем конденсировать охлаждением жидким азотом.
Очистку проводят сублимацией в высоком вакууме [ПО]. HCNO имеет
т. пл. —10° С. Ниже этой темературы она представляет собой бесцветные
кристаллы. При комнатной температуре быстро полимеризуется. По вели-
чине р/( она примерно в пять раз сильнее HN3 [1101.
Для газообразной HCNO найдены следующие ИК-частоты (см-1): 3336
(v (СН)), 2198 (vac (CNO)), 1256 (vc (CNO)), 538 (6 (CNO)), 2500 (2vac (CNO)).
Силовые постоянные связей: ken = 5,92; ken = 15,5 и &no = 12,0 Н/см
[ПО, 2731.
4.7. ФУЛЬМИНАТЫ МЕТАЛЛОВ
Фульминаты металлов — обычно нестойкие и очень ядовитые соедине-
ния, поэтому работа с ними требует чрезвычайной осторожности. Вследствие
склонности фульминатов к детонации их часто называют гремучими солями.
Ионную структуру имеют фульминаты щелочных металлов и таллия (I).
Фульминаты тяжелых металлов, например AgCNO, Hg (CNO)2,— преиму-
щественно ковалентные соединения со связью М—С.
Ключевым соединением, дающим представление о свойствах большинства
фульминатов металлов, является фульминат ртути (II) (гремучая ртуть)
Hg (CNO)2 [111—114]. Способ ее получения описан в работах [112—114]
и сводится к прямому взаимодействию ртути, азотной кислоты и этанола.
Hg (CNO)2 — бесцветные кристаллы, взрывающиеся при ударе, толчке
или трении. При нагревании они разлагаются по схеме [113]
Hg (CNO)2 -> Hg + 2СО + N2. (4.20)
130
Фульминат ртути был использован Нобелем как детонатор, но позже был
заменен более удобным для хранения Pb (N3)2 [115]. Имеется обширная ли-
тература по свойствам и практическому применению Hg (CNO)2 [115].
AgCNO — светочувствительная и очень взрывоопасная соль — стала
широкоизвестной благодаря знаменитой юношеской работе Либаха [116].
Ее можно получить реакцией Ag+ с CNO- или прямым взаимодействием
Ag, HNO3 и С2Н5ОН [107]. AgCNO существует в двух модификациях
(см. 4.11.1, рис. 4.4).
Чрезвычайно взрывоопасны и фульминаты щелочных металлов. NaCNO
кристаллизуется изоморфно с NaN3 и NaNCO [117]. Для получения безвод-
ных MCNO (М = Na, К, Rb, Cs) действуют при соблюдении мер предосто-
рожности избытком амальгамы щелочного металла на раствор фульмината
ртути в абсолютном этаноле [118—120].
На примере KCNO Бек показал, что при осторожном нагревании ионных
и ковалентных соединений возможна реакция изомеризации фульминатов
в цианаты [121, 134].
Фульминаты М (CNO)2 • СН3ОН (М = Са, Sr, Ва) получены при взаи-
модействии Hg (CNO)2 с соответствующей амальгамой в метаноле [119].
Подобрав растворитель, этим же методом можно получить Cd (CNO)2,
T1CNO и CuCNO [122].
Синтезирован ряд металлоорганических фульминатов типа Ph3MCNO
(М = Si, Ge, Sn, Pb); C3H7PbCNO [123], R2T1CNO (R - C6Hr„ CH3) и
RHgCNO (R = арил) [124]
Ph3MX + AgCNO -> Ph3MCNO + AgX (X = Cl, M = Si; X = Br, M = Ge).
(4.21)
Соединения Ph3MCNO подобно Ph2TlCNO относительно устойчивы, в пла-
мени спокойно сгорают.
Алкилпроизводные C3H7PbCNO и Me2TlCNO — чрезвычайно взрывчатые
вещества. Ph2TlCNO при нагревании приблизительно до 200° С превра-
щается в Ph2TlNCO. Арилфульминаты ртути RHgCNO термически несколько
более устойчивы, чем Hg (NCO)2, растворимы в бензоле и имеют, вероятно,
линейное строение. Получают их по схеме
Hg (CNO)2 + HgR2 -> 2RHgCNO. (4.22)
Представляет интерес прямое взаимодействие PhHgOH (или бис(метил-
ртуть) оксида) с нитрометаном [125]
PhHgOH + CH3NO2 -> PhHgCNO + 2Н2О. (4.23)
4.8. СТРУКТУРА И СПОСОБ КООРДИНАЦИИ
ЦИАНАТ- И ФУЛЬМИНАТ-ИОНОВ
Отношение связей в цианат- и фульминат-ионах позволяет представить
их в виде мезомерных граничных форм
0 q ф
|N=C-O| |N = C=O| |N-C=O|
I II III
© e -S. 2q ф
|C=N —O|<-> |C=N = O|
IV V
5*
131
При этом для цианата [126] и фульмината [121, 127] из предложенных ме-
зомерных форм отдается предпочтение формам (I) и (1\). Межатомные рас-
стояния равны для цианата: N—С 1,17 и С—О 1,23 А; для фульмината:
С—N 1,17 и N—О 1,23 А [1281.
Для цианат-иона рассчитаны энергии молекулярных орбиталей (эВ):
1 о — 549,06; 2о — 413,33; Зо —301,17; 4о —27,51; 5о — 22,77; 6о—
7,63; 7о — 4,25; 1зт — 6,56; 2л — 0,43 [129]. МО-расчет при условии при-
менения групповых орбиталей указывает на неравномерное л-распределе-
ние зарядов на атомах трехатомных псевдогалогенидов. Для NCO" и CNO"
вычислена относительная высокая л-плотность зарядов на N- и С-атомах
[1281 (табл. 1.1); для NCX [129] (X = О, S, Se) методом 14М-ЯМР-спектро-
скопии определены значения химических сдвигов (относительно МОГ,
м. д.): +288 (NCO~), +165 (NCS~), +135 (NCSe~), которые хорошо кор-
релируют с уменьшением л-плотности зарядов N-атомов в этих анионах
[130]. Для С NO" значение 6-сдвига равно +176 м. д. [138].
С помощью ЭСХА измерений оценена энергия N-ls-связи для NCO"
в зависимости от природы катиона: KNCO (398,3 эВ), [Ph4As] NCO (397,0 эВ).
Аналогично рассчитана энергия N-ls-связи для NCSe" (KNCSe 398,5 эВ)
и NCSe" ([Ph4As]NCSe 397,0 эВ) [131, 132]. Следовательно, имеющиеся дан-
ные ЭСХА указывают на сравнимые суммарные заряды (о + л) на N-ато-
мах гомологов цианатов.
В противоположность данным [128] о различном распределении зарядов
на атомах азота псевдогалогенидов, для цианата расчет [133] показал при-
мерно равномерное распределение зарядов на атомах: Qn = —0,781, Qc =
= +0,430 и Qo = —0,649. Относительно равномерное распределение за-
рядов на N и О (цианат), С и О (фульминат) следует и из работы [134]:
NCO" — Qn = —0,637, Qc = +0,291; Qo - = —0,654; CNO" — Qc =
= —0,578; Qn = +0,223; Qo - —0,645.
Из имеющихся данных следует, что CNO" координируются исключитель-
но через углерод, a NCO" — преимущественно через азот. Однако приве-
денные выше данные по распределению зарядов не допускают явного пред-
почтения этих лигандов образовывать связи через С или N. В то же время ряд
других факторов создает условия предпочтительной М — С-координации
для фульминатов и М — N-координации для цианатов [135].
4.9. ЦИАНАТНЫЕ И ИЗОЦИАНАТНЫЕ КОМПЛЕКСЫ
4.9.1. Анионные соединения
Однородные анионные изоцианатные комплексы металлов главных под-
групп получены лишь для галлия, индия, олова и свинца. Такие коорди-
национные соединения, как [Me4N]3 [Ga (NCO)6] [36], [Me4N][In (NCO)J
[33] и Kn [Pb (NCO)2+J (n = 1, 2, 4), получают совместной кристаллиза-
цией цианата металла и цианата тетраметиламмония или калия в полярных
органических растворителях в виде бесцветных хорошо растворимых
в высоко до норных растворителях кристаллов. Соль [Me4N]2 [Sn (NCO)6J
образуется в результате реакции обмена между [Me4NL[SnCl6] и AgNCO
[1391.
Аналогично однородным изоцианатным соединениям получают разно-
лигандные псевдогалогенидные координационные соединения рассматрива-
132
емых металлов. Совместной кристаллизацией KNCS и соответствующих циа-
натов металлов в растворителях средней донорной силы получены
К3 [Ga (NCO)3(NCS)3] • Thf, К [In (NCO)3NCS] • Act, К, [Sn (NCO)2 •
• (NCS)2] [32, 33] и Kn [Pb (NCO)2 (NCS)J (n = 1, 2, 4) [36]. Все они хо-
рошо растворимы в DMSO, DMFA и, за исключением комплексов свинца,
в этаноле, плохо или нерастворимы в ацетоне и эфире.
Изоцианатная координация доминирует в однородных комплексах эле-
ментов побочных подгрупп. Примерами важнейших типов комплексов
могут быть
[M(NCO)3r (М = Ag, Au); [M(NCO)3]“ (M = Cd, Си);
[M(NCO)4]2“ (M = Zn, Cd, Hg, Mn, Fe, Co, Ni, Pd, Pt, Си);
[Fe(NCO)4]~ и [M(NCO)6]3~ (M = Cr, Ln).
К этим комплексам примыкают цианаты [Мо (OCN)6]3—; [Re (OCN)6]2—
и [Re (OCN)6]“.
Довольно хорошо изучен тетраизоцианатокупрат (II), который можно
получить, например, из ацетона [140],
[R4N]2 [CuBr4] + 4AgNCO -> [R4N]2 [Си (NCO)4] + 4AgBr. (4.24)
Этот прием можно использовать и для получения анионных изоцианат-
ных комплексов ряда других d-металлов. Анион [Си (NCO)4]2~ имеет иска-
женную тетраэдрическую структуру [140, 141]. Из водного раствора реак-
цией Си2+ с KNCO выделен К [Си (NCO)3], содержащий как концевые, так
Мк
и мостиковые NCO-группы (тип ^NCO) [142]. Информация о структуре
ряда цианокупритов (I) К [Си (NCO)2], МСи3 (NCO)4 (М = К, РЬ) и NaCu4 •
• (NCO)5 отсутствует [36].
Характерный для серебра тип комплекса [Ag (NCO)2]~ [144], судя по
структурному исследованию [Bu4N][Ag (NCO)2], имеет линейное строение
(см. также 4.11). Соль серебра этого комплексного аниона Ag [Ag (NCO)2],
существование которой вначале было доказано изучением электропроводнос-
ти в растворе [148], можно получить в кристаллической форме из AgNCO
и KNCO в DMFA [32, 147]. Из этого же растворителя взаимодействием
AgNCO и KNCS выделено соединение К [Ag (NCO) (SCN)].
Соединение золота (I) [Ph4As] [Au (NCO)2] получают реакцией [Ph4As] •
• [Au (N3)2] c CO (cm. 4.9.2 и 2.8).
Тетраэдрические изоцианаты цинка и кадмия [М (NCO)4]2— подобно
[Си (NCO)4]2— образуются при реакции обмена галогенидных групп на
псевдогалогенид в ацетоне или нитрометане [140, 141], в то время как соеди-
нения типа К [М (NCO)3] (М = Cd, Hg) можно получить реакцией Cd2+ с
KNCO в воде [145, 146] или из Hg (NCO)2 и KNCO в водно-ацетоновом рас-
творе [142]. В ацетоне взаимодействие Hg (NCO)2 с KNCS ведет к образованию
К„ [Hg (NCO)2 (SCN)J (и = 1, 2).
В соединении К [Cd (NCO)3] обнаружены мостиковые NCO-группы типа
Cd\
cd^>NCO [142, 147] (см. 4.11.1).
Кроме упомянутых выше тетраизоцианатометаллов (II), по уравнению
(4.24) получен ряд других соединений состава [Me4N]2 [М (NCO)4] (М - Мп,
Fe, Со, Ni, Pd, Pt). Из этого ряда прежде всего следует выделить соединение
133
кобальта; образование Со (NCO)4 более ста лет назад наблюдал Бломст-
ранд при взаимодействии Со2+ с NCO- в воде [150], а позже получены из
воды К2 [Со (NCO)J и [BuPh3P]2 [Со (NCO)J, изучены УФ-спектры и маг-
нитные свойства этих солей [1511. Рентгенограммы соединений [Et4N]2
[М (NCO)J (М = Мп, Fe, Со, Ni, Zn) указывают на их изоморфность.
ИК- и УФ-спектры темно-синего [Et4Nl2 [Ni (NCO)4] (рЭф = 3,70 М. Б.)
[140] и К2 [Со (NCO)4] (рЭф = 4,32 М. Б.) [151] свидетельствуют о тетраэд-
рической структуре этих соединений [140, 141, 151—153].
Для железа (III) описан тетраэдрический изоцианатокомплекс [Ph4As] •
[Fe (NCO)J (оранжевые кристаллы, полученные из [Ph4Asl [FeClJ и
AgNCO в ацетоне) [154].
Тетраизоцианатные комплексы [Me4N]2 [Pd (NCO)41 и Kat2 [Pt (NCO)4]
(Kat = Et4N, Ph4As) представляют собой квадратно-плоскостные соедине-
ния [126, 139].
Для хрома (III) из СгС13, [Ph4As] Cl и AgNCO в ацетоне получен [PhAsl3-
• [Сг (NCO)6I, октаэдрическая структура которого подтверждается спект-
ральными данными [157]. Для металлов подгрупп титана и ванадия анион-
ные цианаты не получены. Описан ряд изоцианатов лантаноидов и урана:
[Bu4N13 [Er (NCO)6] [158], [Et4N]3 [Ln (NCO)6] (Ln = Nd, Er, Yb) [159],
Kat2 [UO2 (NCO)41 (Kat = К [105], Et4N (157]) и K3 [UO2 (NCO)5] [105].
Реакцией K2 [ReCl6] и K3 [MoClJ c KNCO в диметилсульфоне с после-
дующим обменом с [Ph4As] Cl в этаноле выделены гексацианаты состава
[Ph4As]2 [Re (OCN)6] и [Ph4As]3 [Мо (OCN)6L Для рения (V) из ReCl5 и
KNCO в присутствии Cs+ получен Cs [Re (OCN)6L Эти соединения сравни-
тельно малоустойчивы, связь М—OCN установлена по данным их ИК-спект-
ров [160].
4.9.2. Координационные соединения цианатов
с другими лигандами
Для получения координационных соединений изоцианатов металлов глав-
ных подгрупп прежде всего привлекались реакции изоцианатов металлов
с соответствующими донорами, а также реакции обмена согласно уравнению
MXnLm -F nAgCO -> М (NCO)nLm -ф nAgX (X=галоген). (4.25)
Так, соединения типа Ga (NCO)3 • 3L (L = Dipy, Phen) получают дей-
ствием диаминов на тетрагидрофурановый раствор Ga (NCO)3 [32, 33]. Вза-
имодействие In (NCO)3 с этими же диаминами в ацетоне ведет к образованию
In (NCO)3 • Dipy, In (NCO)3 • Phen • Act, в то время как в нитрометане
Inl3 • Dipy или 1пВг3 • Phen в присутствии избытка диамина реагируют
с AgNCO, образуя In (NCO)3 • 1,5L (L = Dipy, Phen) [162]. Если цианатом
серебра действовать на растворы In 13 в DMSO или пиридине, то выделяются
комплексы состава In (NCO)3 • 3L (L = DMSO, Py) [43]; в ацетоне In (NCO)3
в присутствии пиридина образует соединение In (NCO)3- 2Ру • Act. Все
приведенные изоцианатные соединения индия хорошо растворимы в рас-
творителях средней и большой донорной силы.
Sn (NCO)2 образует с Dipy и Phen соединения типа Sn (NCO)2 • L2.
При нагревании они хорошо растворимы в DMEA, нерастворимы в эфире,
ацетоне, этаноле, воде [32, 33, 163]. Описан аналогичный цианат свинца
Pb (NCO)2 - nL (n = 1, 2) [33].
Для перечисленных выше цианатов металлов принята М—N-координа-
ция. Этот тип связи преобладает в разнолигандных комплексах d-металлов,
134
однако могут присутствовать и мостиковые группы (в основном типа
Мк
\NCO), а в некоторых соединениях М—О-связи.
М/
Довольно детально исследованы изоцианаты двухвалентных Зд-металлов
с гетероциклическими аминами, например пиридиновые комплексы состава
М (NCO)2 • 6Ру (М == Мп, Fe, Ni, Си, Cd), М (NCO)2 - 4Ру (М = Fe, Мп,
Со, Ni), Fe (NCO)2 - ЗРу • Н2О, М (NCO)2 - 2Ру (М = Си, Zn, Cd),
AgNCO ♦ Ру, полученные добавлением пиридина к водным растворам циа-
натов [161].
Так как соединения М (NCO)2Ln (L=гетероциклический аммин), как пра-
вило, плохо растворимы в воде, синтезировать их сравнительно просто
М2+ + 2NCO~ -Ь nL М (NCO)2L„. (4.26)
Поскольку соединения М (NCO)2 • 2L не всегда можно выделить непо-
средственно из водного раствора, их получают или термическим разложением
тетраминов в вакууме, или обработкой тетраминов инертным растворителем
[172]. Иногда при синтезе аминоцианатных соединений применяют органи-
ческие или смешанные водноорганические растворители [162, 185]. Напри-
мер, соединения М (NCO)2L2 (L = Dipy, Phen) получены из водно-спирто-
вых растворов [148, 165, 175].
ИК- и УФ-спектральные структурные исследования, магнитные измере-
ния цианатов переходных металлов позволяют достаточно четко говорить
о строении многочисленных аминоизоцианатов металлов. При этом, кроме
мономерных комплексов с монодентатными NCO-лигандами, существует ряд
соединений с мостиковой NCO-группой (табл. 4.5). Рассмотрим, например,
комплексы с пиридином и некоторыми его производными. Гексапиридинаты
[М (NCO)2 • Ру4] - 2Ру содержат, наряду с координированными, внешне-
сферный пиридин (М = Fe [164], Ni [166]). Соединения [М (NCO)2 Ру41
(М = Fe, Со, Ni) имеют октаэдрическое строение [169, 170, 173, 174, 187,
162]. Мономерно-тетраэдрическую конфигурацию имеет Zn (NCO)2L2 (L =
= 3-, 4-цианопиридин), в то время как М (NCO)2L2 (L = тот же ли-
ганд; М = Mn, Fe, Со, Ni) имеют полимерно-октаэдрическую структуру
[172].
Наряду с уже перечисленными соединениями Sd-металлов описаны мно-
гочисленные изоцианаты металлов и с другими аминами, например соеди-
нения марганца (II): [Мп (NCO)2L2] (L = Dipy, Phen) [165, 175, 232];
[Мп (NCO)2Urt (Но0)2] [177]; мономерно-октаэдрические комплексы желе-
за (II): [Fe (NCO)2L4] (L = y-Pic, IQ, 3-CNPy) [172, 165]; [Fe (NCO)2 (Phen)2],
который получают реакцией [Fe (NCO)2 PyJ c Phen в пиридине [188], а так-
же соединения c y-Pic ([Fe (NCO)2 (y-Pic)4] • y-Pic [164] и [Fe (NCO)2Urt •
• (H2O)2] [1771). Аминокомплексы цианата кобальта [II] имеют преимущест-
венно октаэдрическую структуру, например [Со (NCO)2L4] (L = y-Pic
[1691, IQ [185], Р-Pic, y-EtPy [1861, Н,Н-диэтилникотинамид [230]),
[Co (NCO)2 Urt (H2O)2] [177] или тетраэдрическую [Co (NCO)2L2J (L = a-,
Р-Pic, Q, IQ алкил-Ру [169, 178, 185, 186], а также аминопиридины [148,
175]). Тригонально-бипирамидальное строение имеет [Со (NCO)2 (Et4dien)]
[179].
Описанные до сих пор разнолигандные аминоцианатные комплексы ни-
келя имеют, за исключением планарного [Ni (NCO) (Et4 dien)] BPh4 [179],
октаэдрическую структуру. Мономерно-октаэдрическими являются соеди-
нения [Ni (NCO)oL4] (L = р-, y-Pic, IQ) [146, 171], [Ni (NCO)2L2] (L =
= Dipy, Phen) [146, 148, 165, 175, 232] и [Ni (NCO)2Urt (H2O)2] [177]. Ha-
135
против, комплексы типа [Ni (NCO)2L2] (L = 4-карбометоксипир идин,
V2 Dipy, х/2 Phen) являются полимерно-октаэдрическими [146, 172]. Мостико-
вый тип связи Ni—NCO—Ni доказан для [Ni2 (tren)2 (NCO)2] (BPh4)2 [180].
Для меди (II) известны многочисленные комплексы типа [Си (NCO)2L2].
Эти соединения, где L — IQ, 3-, 4-метилизохинолин, 6-ПО2-хинолин, про-
изводные пиридина [146], a-Pic, 2,4- и 2,5-лутинид, 2,4,6-коллидин [181],
анилин, n-X-анилин (X = Cl, Br, I), м-, п-толуидин [182, 183], являются
планарными с незначительными отклонениями в аксиальном направлении,
а для L == р-, y-Pic [181], хинолин [146, 181], о-толуидин [182] имеют псев-
дооктаэдр ическую структуру с мостиковыми NCO-группами [181].
В ацетоне и метаноле соединения типа Си (NCO)2L2 (L = п-галогенани-
лин, м- и n-толуидин) присоединяют две молекулы растворителя и образуют
искаженно-октаэдрическую структуру [182]. Аддукты состава Си (NCO)2 • L
(L = a-Pic, 2,4-лутидин, Q, 3-метилизохинолин) имеют относительно высо-
кий магнитный момент (рЭф > 2,0) и содержат мостиковые NCO-группы
[181а, 1836]. Диамины образуют соединения типа [Си (NCO)2L21 (L = Dipy,
Phen) [165, 1751 Координационное число 4 имеет медь в [Си NCO) (Et4 •
• dien)] BPh4 [179]. С уротропином получены [Си (NCO)2UrtL2] (L = Н2О,
СН3ОН, С2Н5ОН) [155, 176, 177].
Цинк, кроме уже упомянутых тетраэдрических соединений типа
Zn (NCO)2L2 (L = Ру, CN-Ру) [172], амино-Ру [148, 179], образует октаэд-
рические комплексы состава Zn (NCO)2L4 (L = 3- и 4-CN-Py) [172], 1/2 Di-
py, Phen [148, 165, 175].
Известен также ряд соединений кадмия и ртути. Это, например,
[Cd (NCO)2Py2] [161, 162, 165] и [Cd (NCO)2L2] (L = Dipy, Phen) [165, 175],
[Cd (NCO)2Urt (H2O)21 [177], [Hg (NCO)2L21 (L = Py, a-Pic) и [Hg (NCO)2 •
Dipy] [165, 184]. Описано соединение AgNCO • L (L = Py, a-Pic),
AgNCO • 2Py и AgNCO • 1,5 a-Pic [165, 184].
Для ряда аминоизоцианатов d-металлов доказано образование мостико-
вых связей, образуемых нейтральными лигандами, например [М (NCO)2L21
(L = N,N-диэтил никотинамид; М = Мп, Со [230]; L = никотинамид, М =
= Со [231]); [М (NCO)2L] (L = гидразид изоникотиновой кислоты; М =
= Мп, Со, Cd, Zn) [233]; [Си (NCO)2 (С1-пиридинальдоксим)2] [146].
В соединениях [М (NCO)2 (НМРТ)2] (М = Со, Zn) реализуется тетраэд-
рическая структура [187].
Плоские изоцианаты Pd (II) и Pt (II) [М (NCO)2L21 (М = Pd, L == NH3,
Ру, а- и y-Pic, х/2 Dipy, V2 Phen, Ph3E [126, 156]; М = Pt, L = Ph3E,
1/2 Dipy [1561); [Pd (NCO)LI BPh4 (L = dien, Et4dien) [179], а также транс-
[PtH (NCO) L2] (L = Ph3P, Ph2EtP, Et3P, Bu3P) [189] и транс-iPt (CN) •
• (NCO) (Ph3P)2] [195] получены в большинстве случаев в ходе реакции меж-
ду соответствующими хлорокомплексами и цианатом. Соединение
[Pd (NCO)2 (Ph3P)2] также можно получить реакцией [Pd (NCO)2Py2] или
[Pd (NCO)4]2~ с Ph3P [156, 251].
Подобно комплексам Зd-мeталлoв, в которых обнаружена лишь М—N-
координация, в названных соединениях благородных металлов также имеет
место исключительно N-координация. Никакого влияния координированных
нейтральных лигандов на тип связи металл — цианат (в противоположность
тиоцианатным комплексам) в цианатных комплексах не наблюдается. Так,
ни лиганды, образующие прочные л-связи [156], ни пространственные фак-
торы не могут вызвать изомерию (М—N или М—О) связи [179]. Однако высо-
ковалентные металлы, по-видимому, способны образовывать О-цианатные
соединения. Так, Голуб и Лишко получили цианатокомплексы [М (OCN)4L21
(М = Zr, Hf; L. = Phen, Dipy) и [M (OCN)4LJ (L = Py, DMSO) по реакции
136
между МС14 и KNCO или Pb (NCO)2 в Thf в присутствии соответствующих
донорных лигандов [190].
г Авторы работ [191, 192] независимо друг от друга получили цианаты
состава Ср2 М (OCN)2 (М = Ti, Zr), которым Бурмейстер [193, 194] припи-
сал, как и аналогичным соединениям гафния, О-цианатную связь. Однако
рентгеноструктурный анализ соединения титана (IV) указывает на N-коор-
динацию (Cp2Ti (NCO)2) [196].
N-координация имеет место в комплексах Ti (IV) типа [Ti (NCO)nLm
(i-OC3H7)4—л] (L = CH3CN, CH3COOC3H7; tn = 1, 2) [197], а также в
Cp./FiNCO [193, 229], CpCr (NCO) (NO)2 [200], CpMo (NCO) (CO) (Ph3P)2
[201] и CpW (NCO)2 [202].
Особого внимания заслуживают цианатные комплексы богатых электро-
нами ЗсЬметаллов с мягкими лигандами, как R3P, СО или NO. В принципе
соединения этого типа могут быть получены путем замещения галогенидных
групп на цианатные подобно упоминавшемуся выше синтезу комплексов пал-
ладия и платины ([М (NCO)2 (R3P)21) [156]. Так, [М (NCO) (СО) (Ph3P)2]
синтезирован из [MX (СО) (Ph3P)2l и MNCO [217], а комплекс [Сг (CO)5NCO]
образуется из Сг (СО)51— и KNCO [203]. Для получения [М (CO)5NCO]~
(М = Cr, Мо, W) используют реакцию [204]
М (СО)6 + NCO“ -> [М (СО)5 NCO]” + СО. (4.27)
Интересно, что изоцианатокарбонилметаллаты в виде солей тетраэтил-
аммония (для вольфрама с катионом [Ph4As]+) образуются при действии ази-
дов на карбонилы [205, 204]
М (СО)6 + NV [М (СО)5 NCO]- + N2. (4.28)
Азидные комплексы с концевыми или мостиковыми Ng-группами могут
превращаться под действием СО в соответствующие изоцианатные соедине-
ния [149, 206]
М (N3)2 (R3P)2 + 2СО + М (NCO)2 (R3P)2 + 2N2 (М = Pd, Pt). (4.29)
Наряду с азид-ионом для превращения координированного СО-лиганда
в NCO-группу используются и другие соединения азота, например N2H4
[207], NH2OH или NH2C1 [208],
[CpFe (СО)3]+ + N2H4 CpFe (СО)2 CONHNH2 + NH2NH^. (4.30)
CpFe(CO)2 NCO + NH3
Кинетические исследования показывают, что азотсодержащие основания
NT, NH2X (X = NH2, ОН, Cl) нуклеофильно взаимодействуют с СО с обра-
зованием промежуточных продуктов MCON3 или MCONH2X+, которые затем
превращаются в устойчивые М—NCO-группировки [135, 204, 205, 207].
При УФ-облучении CpMo (NO) (СО)2 и Ph3P образуется соединение
CpMo (NCO) (СО) (Ph3P)2 [2011.
Превращением азидных комплексов в соответствующие изоцианатные
получены CpMo (NCO) (СО) (Ph3P)2 и CpMo (NCO) (СО)2 (Ph3P) [2011,
[Мп (NCO) (СО)3 (Ph3P)2] [211], CpFe (NCO) (СО)2 [207], [Pd (NCO)2 (R3P)21
[149], [Rh (NCO) (CO) (Ph3P)2l, [Ir (NCO) (CO) (Ph3P)2] [149, 209], [Rh (NCO)«
. (CO)2]2 [216], [Au (NCO) (Ph3P)] [149].
[Re (CO)61+ реагирует c NJ', образуя Re (CO)5NCO [215] , тогда как
Mn (CO)5Br и Re (СО)5С1 с азидом дают многоядерные комплексы [Мп , (СО)6 •
• (N3)x (NCO)s—*1“ и [Re2 (СО)4 (NCO)2(N3)2]2~, которые, присоединяя
Ph3P, превращаются в моноядерные [М (NCO) (CO)3Ph3P] [2111.
137
При взаимодействии карбонилокомплексов металлов с гидразином об-
разуются изоцианатные соединения: [Мп (NCO) (СО), (N2H4) (R3P)21,
[Мп (NCO) (CO)3(R3P)2], [Мп (NCO) (СО)2 (R3P)3] [212], [Re (NCO) (CO), •
• (N2H4) (R3P)], [Re (NCO) (CO)2 (N2H4) (R3P)21, [Re (NCO) (CO)2 (R3P)31,
[Re (NCO) (CO)3 (R3P)2] [2131, [Re (NCO) (CO)4 (NH2CH3)] [214], [Re (NCO) •
- (CO)3L] (L -Phen, Dipy) [215], CpFe (NCO) (CO)2 [207, 215]. Заслуживают
также внимания многоядерные N-цианаты марганца и рения: [(СО)3Мп •
• (NCO)3 Мп (СО)3Г [211] и [(CO)4Re (NCO)2 Re (СО)4Г [220].
К группе цианатов металлов с «мягкими» лигандами относят также соеди-
нения [Rh (NCO) (Ph3P)3] (желтый) и [Rh (OCN) (Ph3P)3] (оранжевый),
которые являются первым примером изомерии связи с участием NCO-
групп; синтез их из RhCl (Ph3P)3 и [Ph4As] NCO дает возможность просле-
дить за влиянием растворителя на способ координации NCO-группы [218].
4.93. Реакции изоцианатных комплексов
Представляют интерес некоторые вопросы реакционной способности изо-
цианатных комплексов, например окислительные реакции присоединения
галогенов к таким соединениям, как Au (NCO) L (L = Ph3P, Ph3As,
V2 PC2P) и Ir (NCO) (CO) (Ph3P)2, тетрацианоэтилена к Ir (NCO) (CO) •
• Ph3P)2, а также NO+ к M (NCO) (CO) (Ph3P)2 (M — Rh, Ir), которые при-
водят к образованию изоцианатных комплексов AuBr2 (NCO) L [217],
Ir (NCO) Х2 (СО) (Ph3P)2 [221], [Ir (NCO) (СО) (Ph3P)2TCNE [222],
[М (NCO) (СО) (NO) (Ph3P)2]+ [2231.
Необычное поведение координированных NCO-групп наблюдается при
замене 2,4-лутидина в [Си (NCO)2 (2,4-лутидин)2] 3,5-диметилпиразолом
(Dmpz). Образующийся [Си (NCO)2 (Dmpz)2l существует в двух формах.
Структурное исследование a-формы указывает на транс-планарное строение
комплекса с новым лигандом, а образовавшаяся через атом углерода (NCO-
группы) связь с некоординированным азотом пиразола приводит к нару-
шению линейности NCO-остатка.
В кислой среде протекает протонирование N-цианата в комплексах
[М (NH3)5NCO12+ (М = Со [225, 226], Rh, Ru [2271), присоединение воды и
образование промежуточного комплекса карбаминовой кислоты, который
затем отщепляет СО2 и превращается в [М (NH3)613+. Как промежуточный
продукт карбаматокомплекс образуется при реакции [Со (NH3)5H2O12+ с
NCCT [228].
Изоцианатокомплексы М (NCO) (СО) (PPh3)2 (М = Ir, Rh) превращаются
7 /NH2\
в этаноле в присутствии HBF4 в М t °C^q ) (СО) (PPh3)2 • BF4 [2341.
Соединения палладия и платины [М (NCO)2 (Ph3P)2l реагируют с этано-
лом и СО, образуя (алкоксикарбонил)-изоцианатокомплексы [М (NCO) -
• CO2R) (Ph3P)2]. Движущей силой этой реакции является образование
легко отщепляемой NH2CO2Et-rpynnbi [234]
—М—NCO + EtOH -> —MNHCO2Et
-> [—M—CO]+ ОЕГ -> —MCO2Et. (4.31)
138
Взаимодействие тех же соединений с SO2 или CS2 и СН3ОН при-
водит к образованию метоксисульфонил- или дитиокарбонатокомплексоз
[М (SO2OCH3)2 (R2P)21 или [М (S2COCH3)2 (Ph3P)2] [235].
Частичным алкилированием псевдогалогенид-лигандов, например при
взаимодействии [М (NCO)2 (Ph3P)2] (М = Pd, Pt) с Et3O BF^, можно по-
лучить биядерные соединения типа
О
С
,N\
[(Ph3P)2M< >M(Ph3P)2](BF4)2
С
О
[236], которые также образуются при действии М (NCO)2 (Ph3P)2 на HBF4
и EtOH [234] или при реакции Pd (NCO)2 (Ph3P)2 с NOBF4.
4.10. ФУЛЬМИНАТНЫЕ КОМПЛЕКСЫ
Первый фульминатный комплекс К [Ag (CNO)2] описан Либихом еще
в 1823 г. [237]. За ним последовал синтез желтых игл Na4 [Fe (CNO)6] •
18 Н2О [238]. Хотя Неф [238], а за ним Велер, которому удалось получить
ряд других фульминатных комплексов [239], указывали на значительную
аналогию между цианидными и фульминатными комплексами, до 1960 г.
почти отсутствовали сведения о структуре комплексных фульминатов. Лишь
в начале 60-х годов Беку удалось доказать координацию фульминат-лиган-
да исключительно через атом углерода [240—245, 251]. Следовательно, по
способу координации фульминаты напоминают цианиды. Так, комплексы
[Fe (CNO)6]~4, [Со (CNO)6]3~ и [Ni (CNO)4]2~, каки аналогичные цианидные
комплексы этих металлов, являются диамагнитными [240, 241]; CNO-ли-
ганд, создает сильное поле, близкое по силе к таковому CN-лиганда [242].
Подобно цианиду, фульминат проявляет сильный транс-эффект, что следует
из сопоставления данных ПМР комплексов типа транс-fPtXH (Et3P)2l
[243]. Изучение колебательных спектров [М (CNO)4]2~ указывает на симмет-
рию D4h этих анионов для М = Ni, Pd, Pt и симметрию Td для М = Zn,
Cd, Hg с линейной М—CNO-группой [244]. Наконец, восстановление
[Fe (CNO)6]4~ при помощи Fe (ОН)2 и [Ni (CNO)4]2~ амальгамой натрия при-
водит к цианидным комплексам [Fe (CN)6]4~ и через [Ni (CN)4]2~ к
[Ni2 (CN)6]4~ [245]. Твердые тетрафульминатоплатинаты (II) щелочных и
щелочноземельных металлов, как и аналогичные цианидные соединения,
проявляют дихроизм, рентгенофлуоресценцию, имеют металлический блеск,
изменяют цвет при обезвоживании, например Na2Pt (CNO)4 • 5Н2О (бес-
цветный), Na2Pt (CNO)4 (темно-красный) [239]. Вероятно, в этих соедине-
ниях, как и в подобных цианидных, содержатся линейные цепочки Pt—Pt с
четким обменным взаимодействием металл — металл [273].
Координация М—CNO доказана рентгеноструктурными исследованиями
координационно-полимерного AgCNO [102] (см. 4.11), как было установлено
раньше для Ag (CNO)F [290].
Синтез анионных фульминатных комплексов осуществляется действием
фульминатов щелочных металлов на соли металлов в воде или органиче-
ских растворителях [119, 237—239]. Так, из метанольно-эфирного раствора
139
получены NH4 [Ag (CNO)2] и Mg [Ag (CNO)2]2 [119], а из воды — Na2 [Ni •
• (CNO)4] • 5H2O (светло-коричневые иглы), Na2 [M (CNO)4j • 5H2O (M =
= Pd, Pt, бесцветные иглы), Na2 [Au (CNO)2] • 3H2O (бесцветные пластин-
ки), Na [Cu (CNO)2] и Na2 [Cu (CNO)3] - 3H2O. Реакцией обмена щелочного
иона на М2+ (М2+ = Са2+, Sr2+, Ва2+) получены фульминаты щелочноземель-
ных металлов М [Pt (CNO)J • иН2О, М [Cu (CNO)2]2 - иН2О и М [Си •
• (CNO)3] • пН2О.
Подобным способом синтезировано соединение Си [Си (CNO)2]2 (светло-
зеленые пластинки) [239]. Упомянутый комплекс Au (I) можно получить,
подобно фульминату ртути, в результате прямого взаимодействия АиС13,
HNO3 и С2Н5ОН в присутствии соли натрия [239].
Фульминат Со (III), описанный Нефом как соль К5 [Со2 (CNO)UJ,
получен позже в виде желтой соли [Сг en3] [Со (CNO)6] • ЗН2О при окисле-
нии кислородом воздуха раствора Со2+ и CNO- [240]. В то время как боль-
шинство комплексных фульминатов щелочных металлов взрывоопасны
(особенно в безводном состоянии), замена последних большими объемными
катионами резко повышает стабильность фульминатов. Так, получены до-
вольно устойчивые [NiL3] [Ni (CNO)J иН2О (L = Dipy, Phen) и [CoL3] •
• [Co (CNO)6] • nH2O [241]. Бесцветный [Ph4As]2 [Hg (CNO)J выделен из
воды при взаимодействии Hg (CNO)2 с KCNO в присутствии [Ph4As] Cl
[247]. Ряд других комплексных фульминатов, которые по строению похожи
на цианиды соответствующих металлов, также можно стабилизировать че-
рез объемные катионы, такие, как [Ph4As]+, [R4N]+ (R = Et, н-Pr). В про-
тивоположность взрывчатым Ва2 [Fe (CNO)61 • 4Н2О и К4 [Ru (CNO)6] •
• пН2О, соединения [Ni (Phen)3]2 [Ru (CNO)6] • nH2O, [Ph4As]2 [OsO2 •
• (CNO)2] (светло-коричневый), [Ph4As]3 [Co (CNO)6] - 3H3O, [Ph4As]3 -
• [Rh (CNO)6] (светло-желтый), [Ph4As]3 [Ir (CNO)6] • 6H2O (желтый)
[(H-Pr)4N]2 [Ni (CNO)4], а также (последующие соединения бесцветны)
[(h-Pt)4N]2 [М (CNO)J (М = Pd, Pt), [Et4N] [Ag (CNO)21, [Ph4Asl [Au (CNO)2],
Kat2 [M (CNO)4J (Kat — [(H-Pr)4N]+, [Ph4P]+; M — Zn, Cd) в большинстве
своем относительно устойчивы [248].
Однородные анионные фульминаты известны для элементов VIII группы
и I—II побочных подгрупп.
Известны некоторые разнолигандные анионные комплексы, которые по-
лучают реакциями Hg (CNO)2 или AgCNO с солями щелочных металлов:
Na [Hg (CNO)2X] (X = Cl, Br), Na2S2O3 2Hg (CNO)2 [239] и К [Ag (CNO) •
• Cl] [249]. Соединение Na4 [Fe (CN)5 CNO] • nH2O (диамагнитное, желтые
иглы) получают, действуя на [Fe (CNO)5L13- (L = Н2О, NH3) CNO-; при
помощи Fe (ОН)2 оно, как и [Fe (CNO)6]4’ , превращается в [Fe (CN)6]3—
[250]. Разнолигандные фульминатогалогенидные комплексы [Ph4As]
• [Au (CNO)2X2] (X = Br, I), а также комплексы типа [M (CNO)2X2L2]
(М == Pt; X = Br, L = Et3P; M = Pd; X = Br, I, L == (C6HU)3P; M = Pd,
Pt; X — Br, L = V2 (PC2P)) получают окислительным присоединением гало-
гена к соответствующему комплексу Au (I), Pd (II) или Pt (II) [248].
При одновременном воздействии CNO- и УФ-облучения на М (CO)5Thf
образуются карбонилфульминатные комплексы металлов [248]:
М (СО)5 Thf + CNO- -> [М (СО)5 CNO]- + Thf; (4.32)
[М (СО)5 CNO]- + CNO-—[М (СО)4 (CNO)2]2- + СО
(М — Сг, Мо, W),
140
из которых [Cr (СО)5 CNO1- подтвержден ИК-спектроскопией. Выделены
соединения типа [Ph4As]2 [М (СО)4 (CNO)2] [248].
Аминофульминатокомплексы [М (CNO)2 Ру2] (М = Zn, Cd, Hg) и
[М (CNO)2 (NH3)2] (М = Мп, Zn) описаны Велером [119, 239], a AgCNO •
• 2NH2Ph — Виландом [249].
Диамагнитные [Fe (CNO)2L2] • //I I2O (L = Dipy, Phen) являются не-
электролитами и образуются по схеме
FeL2+ + 2CNO~ [Fe (CNO)2 L2] + L, (4.33)
в то время как с Mn2+, CNO~ и L дают электролитный комплекс [Мп (CNO)2 •
• L2], а [Со (Phen)3]2+ с CNO~ — соединение Со (Phen)2 (CNO)2 • Н2О
[241]. BHg(CNO)2- Phen предполагается наличие непрочной связи амина
[247].
Интересные результаты получены для комплексов фульминатов металлов
с фосфинами. Бесцветные невзрывчатые соединения MCNO • 2R3P (М = Си,
Ag) и Hg (CNO)2 (R3P)2 (R=C6H5, C6Hu) получают взаимодействием фуль-
мината металла с R3P в неводном растворителе [247]. При синтезе соедине-
ний типа транс-[М (CNO)2 (RSP)2J используют также реакции тетрафульми-
натомета ллов (И) с фосфинами в смеси СНС13 — С2Н/)Н (для Ni) в воде
или водно-спиртовой смеси (для Pd, Pt) [251]
[М (CNO)4]2~ + 2R3P -> [М (CNO)2 (R3P)2] + 2CNO ’ (4.34)
(M = Ni, Pd, Pt).
В комплексах [M (CNO)2 (PC2P)] (M = Pd, Pt) CNO-лиганды находятся
в цис-положении [251].
Реакциями обмена хлора на фульминат при взаимодействии [М (CNO) •
• C1L2] с [Ph4As] CNO в хлороформе выделены соединения [М (CNO)2L21
(М = Pd, Pt; L = BuPh2P, Bu2PhP, Ph3P, Bu3P, Ph3As, Ph3Sb); в аналогич-
ных условиях из [МХС1 (Ph3P)2] можно получить [М (CNO)XL2] (X = Н,
CH3CN, NCO, М = Pt; X = СО, М - Rh) [252].
Интересен синтез [Pt (CNO)2 (Ph3P)2] путем окислительного присоедине-
ния нитрометана к Pt (Ph3P)4 в смешанных растворителях (вода — эта-
нол — бензол — нитрометан) [154] по предполагаемому механизму
Pt (Ph3P)4 Pt (Ph3P)2 + 2Ph3P;
PPh3 PPh3
I /CH2 x |
Pt (Ph3P)2 + CH3NO2 -> H—Pt< >NO -h-c^n°2-> ONC—Pt—CNO.
..................... I'O / H= I
PtPh3 PPh3
(4.35)
Детально исследована реакционная способность фульминат-лигандов
в относительно устойчивых соединениях транс-fPt (CNO)2 (Ph3P)2] и транс-
fPt (CNO) (CN) (Ph3P)2] [254]. При нагревании транс-fPt (CNO)2 (Ph3P)2]
превращается в цис-fPt (NCO)2 (Ph3P)2]
PPh3 PPh3
| | Ph3P. .NCO
ONC—Pt—CNO -> ONC—Pt—NCO -> >Pt<
| ) Ph3Pz XNCO
PPh3 PPh3
(4.36)
В присутствии кетонов изомеризация CNO NCO протекает в более
мягких условиях. Эти комплексы также легко реагируют с тиокетонами или
141
CS2, образуя цис-IPt (NCS)2 (Ph3P)2] или транс-fPt (NCS) (CN) (Ph3P2)l,
в то время как фосфины восстанавливают фульминат до цианида, при этом
оба исходных соединения превращаются в [Pt (CN)2 (Ph3P)2] [253, 254].
4.11. СТРУКТУРА ЦИАНАТНЫХ
И ФУЛЬМИНАТНЫХ СОЕДИНЕНИЙ
4.11.1. Рентгеноструктурные исследования
До настоящего времени исследовано сравнительно мало структур цианат-
ных и фульминатных соединений. Выше (см. 4.2, 4.4 и 4.6) уже приводились
структурные данные для некоторых ковалентных изоцианатов гремучей
кислоты, полученные на основании рентгеновских исследований, микровол-
новых спектров или дифракционных измерений. Ниже приводятся резуль-
таты рентгеноструктурных исследований изоцианатов и фульминатов
металлов.
В табл. 4.1 приведены параметры решетки некоторых солеобразных циа-
натов. Цианаты натрия и калия изоморфны с азидами натрия и калия.
Цианаты рубидия, цезия, таллия, как и цианат калия, имеют объемно-
Таблица 4.1. Кристаллографические
характеристики цианатов
некоторых металлов
Соедине- ние Простран- ственная группа Параметры решетки, A z Лите- ратура
a c
NaNCO R3m 5,44 — 1 [255]
KNCO 14/mcm 6,07 7,03 4 [256]
RbNCO 14/mcm 6,35 7,38 4 [20]
CsNCO 14/mcm 6,71 8,04 4 »
nh4nco — 3,64 5,57 1 »
T1NCO 14/mcm 6,23 7,32 4 »
Таблица 4.2. Некоторые
кристаллографические параметры
изоструктурных комплексов типа
[M(NCO)2 (Urt) <Н2О)2]
Металл Пераметры решетки, A ₽ П ро- стр яв- ствен- ная группа
a b c
Mn 8,50 12,75 11,10 93° B2/b
Co 8,45 12,62 11,20 93° В2/Ь
Ni 8,50 12,75 11,10 93° В2/Ь
Cd 8,50 12,8 11,20 93° В2/Ь
центрированную тетрагональную кристаллическую решетку. Для NHdNCO
можно предположить наличие водородной связи между NH^ и OCN~ [20].
Полные структурные исследования выполнены для некоторых изоцианат-
ных комплексов металлов, а также для цианата и фульмината серебра. Ис-
следованные до настоящего времени цианаты металлов [Со (NCO)2 Urt •
• (Н2О)2] [155], [Bu4N] [Ag (NCO)2J [144], [M (NCO)2 (Phen)2] [232],
CpCr (NCO) (NO)2 [200] и CpMo (NCO) (CO) (Ph3P)2 [201] содержат изоциа-
Mx
натные группы. Мостиковые цианатные группы >NCO содержатся, на-
Мх
пример, в AgNCO [257], М—NCO—М в Си (NCO)2Py2 [258], [Ni2 (NCO)2 •
Мх /М
• tren2] (BPh4)2 [180]; mJ>NCO<^ — в (СН3)2 T1NCO [259]. Общим
признаком изоцианатов является линейность NCO-rpynn.
142
Для [Ag (NCO)2] найдены следующие величины углов и длин связей:
Z.NAgN = 177,2°, Z_AgNC = 170,0°, Z.NCO = 178,2 и 179,8°; расстояния
Ag—N 2,068 и 2,015 A; N—С 1,111 и 1,1076 А; С—О 1,129 и 1,200 А [144],
Рис. 4.1. Фрагмент структуры ICo(NCO)., Urt - (Н2О)21 вдоль оси b
Соединения [М (NCO)2 (Urt) (Н2О)2] (М = Со, Мп, Ni, Cd) изоструктур-
ны [155] (табл. 4.2). На рис. 4.1 представлен фрагмент структуры [Со (NCO)2 •
• (Urt) (Н2О)2], а в табл. 4.3 — результаты рентгенографического иссле-
дования. Уротропин выступает в роли мостика, атом кобальта имеет окта-
эдрическое окружение: 2Nnco,
20н2о> 2Nurt [155].
Различные координиро-
ванные цианатогруппы со-
держатся в К [М (NCO)3]
(М = Cd, Hg). В К [Cd
•(NCO)3] кадмий имеет иска-
женное октаэдрическое окру-
жение атомами азота с рас-
стояниями Cd—N между 2,24
и 2,46 А. Три различно ко-
ординированные NCO-группы
имеют следующие параметры:
Z_NCO— 175,2; 176,9; 178,1°,
расстояния N—С и N—О 1,119
и 1,198;о1,170 и 1,198; 1,186
и 1,226 А. Аналогичный комп-
Таблица 4.3. Структура комплекса
[Co(NCO)2 (Urt) (Н2О)21
Расстояние, А
Комплекс
Со—^nco ~ 1,93 -+- 0,02
Со—NUrt = 2,26 ± 0,02
Со~°н2о = 2,17 ± 0,01
NCO-epynna
С—N = 1,10 ±0,03
С—О = 1,26 ± 0,02
Углы
N ncoC°Oh2o = 83,9°
NUrtCoNNCO = 78,5°
OH2oCoNUrt- 83,9°
NCO = 171,7°
CoNC = 168°
леке ртути следует рассматривать как двойную соль KOCN • Hg (NCO)2.
Расстояния N—С и N—0о свободных и ковалентно связанных цианатных
групп равны 1,23 и 1,19 А или же 1,15 и 1,17 А.
Соединения типа [М (NCO)2 (Phen)2] (М = Мп, Ni) являются орторомби-
ческими, пространственная группа РЬсп. NCO-группа практически линейна,
а расстояния N—С и С—О для цис-октаэдрических комплексов равны:
1,11 и 1,26 А (М = Ni) или 1,20 и 1,22 А (М = Мп) [232].
143
Концевыми являются изоцианатные группы в соединениях CpCr (NCO) •
1 (NO)2 [200] и СрМо (NCO) (СО) (Ph3P)2 [201]. Оба соединения моноклин-
йы и относятся к пространственной группе Р21ш. Для первого найдено:
а = 6,18; b = 9,00; с = 7,52 А; |3 = 94° 54'; Z = 2; для второго: а =
= 16,65; b = 13,21; с - 18,14 А; |3 = .
= 90° 12'; Z = 4. Углы CrNC и (NCO)
для обоих соединений равны соответст- чг
*1 ф
Рис. 4.2. Структура цианата серебра [257]
Рис. 4.3. Строение комплекса
[CuPy2(NCO)2] [258]
венно 180 и 178,6°. Расстояния в соли хрома равны: Сг—N 1,982, N—С
1,126, С —01,179 A. ZMoNC = 178,3°; ANCO = 179,1°. Расстояния в
соединении молибдена: Мо—N 2,127; N—С 1,118, С—О 1,238 А.
Цианат серебра AgNCO является координационно-полимерным соедине-
нием с мостиком & /NCO. Он кристаллизуется моноклинно и относится
Н пространственной группе P2jm; а = 3,473; b = 6,372; с = 3,416 А; |3 =
= 91,5°; Z = 2 [257] (согласно [20] AgNCO орторомбичен, пространственная
группа Ibam; а = 6,37; b = 6,52; с = 5,48 A; Z— 4). Рис. 4.2 дает представ-
ление о структуре AgNCO, для которой характерны угловые цепи Ag—N—
Таблица 4.4. Кристаллографические параметры некоторых фульминатов
Соединение Пространствен- ная группа Параметры решетки, А • Z Литерату- ра
а ь С
NaCNO R32 или R3m 4,95 — — 1 [264]
KCNO 14/mcm 5,81 — 7,16 4
T1CNO 6,39 6,41 4 »
AgCNO Cmcm 3,86 10,42 5,85 4 [Ю2]
R3 9,11 — — 6 »
Hg(CNO)2 — 7,71 5,48 10,83 4 [261]
144
<5
©N ©0 ©Лу
Рис. 4.4. Структура фульмината се-
ребра на плоскости (001) и (100):
а — орторомбическая модификация;
б — тригональная модификация
Ag—N с углами связи NAgN 180° и AgNAg 97,7°. Z_AgNC = 128,7°, а
zCNCO = 178,2° [257].
Связь типа М—NCO—М установлена в Си (NCO)2Py2 [2581 (см. рис. 4.3).
Мостики Ni
/NCO^
Xqcn/
Ni содержатся в соединении [Ni2 (NCO)2 (tren)2J •
• (BPh4)2 [180]. Из ацетонитрила или воды выделена моноклинная модифи-
кация этого соединения с параметрами: а =
= 3,236; b = 10,65; с = 19,319 А; [3 =
==119,18°; Z = 4. Z.NiNC = 156°; zlNiOC =
119°. Z.NCO = 172° [180].
Мостики
М\ /М
>NCO<
М/ \М
обнаружены в обе-
их модификациях (СН3)2 T1NCO (ортором-
бическая, пространственная группа Реша;
а = 11,09; b = 4,09; с = 12,281 A; Z = 4;
тригональная, пространственная группа R3m;
а = 18,28; с = 9,474; Z = 18). Для ромби-
ческой модификации расстояния Т1—О и Т1—
N равны 2,66 и 2,85 А, а расстояния N—С и
С—О 1,16 и 1,18 A. Z.NCO = 179°. Для три-
гональной модификации расстояния Т1—О
2,85; TI—N 2,58; N—С 1,23; С—О 1,19;
Z.NCO - 180° [259].
Определены также параметры решеток для
некоторых фульминатов металлов (табл. 4.4).
Установлено, что AgCNO существует в двух
фНд QCO-CN0
Рис. 4.5. Структура фульмина-
та ртути
145
полиморфных модификациях: орторомбической (пространственная группа
Cmcm) и тригональной [102]. Это дало возможность внести поправки в дан-
ные, полученные в работе [262]. Обе модификации содержат мостики типа
CNO. На рис. 4.4, а представлена орторомбическая модификация
м/
AgCNO, которая отличается от тригональной модификации тем, что атом
углерода в вершине мостика заменен атомом азота. Межатомные расстояния
для этой формы следующие: С—N 1,09, N—О 1,25, Ag—С 2,23; Ag ... О
2,77; Ag...Ag 2,93 A; Z.CNO = 180°, Z_AgCAg = 82°, Z_CAgC = 180°.
Ромбоэдрическая форма (рис. 4.4, б) содержит циклические гексамеры с
расстоянием Ag —С 2,16 и 2,19, N—О 1,20, С — N 1,12, Ag —О 2,45; Ag...Ag
2,83 A; Z_CNO = 172°, Z-AgCAg = 81°, Z_CAgC = 163°. Сравнивая рас-
стояния Ag...Ag в этих двух модификациях (2,93 А и 2,83А), можно увидеть,
что в орторомбической модификации оно больше, чем в ромбической.
В последней форме оно немного короче, чем в металлическом серебре (2,89 А)
[102].
Фульминат ртути рентгеноструктурно исследовали неоднократно [261,
263, 164]. Кристаллы этого соединения относятся к ромбической сингонии.
На рис. 4.5 представлена структура Hg (CNO)2 по данным работы [261].
4.11.2. ИК-спектральные исследования
Немногочисленность рентгеноструктурных данных по цианатам и фуль-
минатным соединениям делает ИК-спектры поглощения ценным материалом
для изучения природы этих соединений. При этом в первую очередь рас-
сматривается вопрос о типе связи и способе координации цианатной и фульми-
натной групп. Цианат- и фульминат-ионы, как и другие трехатомные псев-
догалогениды, имеют четыре колебательные степени свободы и три ИК-ак-
тивные частоты (A), v2 (Е) и v3 (А). В цианат-ионе этим частотам соответ-
ствуют v (CN) или vac (NCO), 6 (NCO) и v (СО) или vc (NCO). Так как ва-
лентные колебания касаются главным образом связей CN и СО, то в дальней-
шем частоты и v3 будем обозначать как v (CN) и v (СО) [196, 268]. Основ-
ные частоты фульминат-иона следующие: = v (NO) или vc (CNO), v2 =
= 6 (CNO) и v3 = v (CN) или vac (CNO) [121].
Экспериментально для цианат-иона (KCNO в матрице KI) наблюдаются
такие основные частоты: v (CN) = 2165, 6 (NCO) = 628, 637, а также дуб-
лет при 1207 и 1301 см-1, отнесенный к v (СО) и 26 (NCO) [274]. Расчет по-
казывает, что частота v (СО) должна быть при 1254 см~’, но из-за резонан-
са Ферми между v (СО) и 26 (NCO) появляется указанный дублет средней
интенсивности [265].
Основные частоты для фульминат-иона (KCNO в нуйоле) следующие:
2042 (v (CN)), 1057 и 1064 (у (NO)), а также 473 и 486 см-1 (6 (CNO)). Допол-
нительно проявляется интенсивное колебание 2v (NO) при 2126 см-’ [121].
Силовые постоянные связей цианат-иона равны: &nc — 15,879, kco —
= 11,003 Н/см [2651; для фульминат-иона: £cn — 15,6, Ano = 6,81 Н/см [121].
146
4.11.2.1. ИК-спектральные исследования цианатов
Изоцианаты элементов главных подгрупп, структуры которых частично
установлены из микроволновых спектров или дифракционных измерений
[58, 61, 62], в большинстве характеризуются значительными сдвигами час-
тот v (CN) и v (СО) к более высоким волновым числам (табл. 4.5).
Органоцианаты и (PhO)2P (О) OCN являются единственными представи-
телями О-циаиатов элементов главных подгрупп. Частоты v (CN) арилциа-
натов и арилизоцианатов смещены по сравнению со свободными NCO~ в
высоковолновую область и лежат в области 2200—2300 см-1. К тому же
полосы валентных колебаний v (CN) часто расщеплены на две-три компоненты,
Таблица 4.5. Характеристические частоты в ИК-спектрах* некоторых
изоцианатов элементов главных подгрупп, см—|
Соединение v (CN) v (CO) (26 (NCO)) 6 (NCO) Литература
Тип связи: М—NCO
CH3NCO 2231 1412 [268]
(СН3)3 CNCO 2270 603, 627 [137]
(СН3)3 SiNCO 2282 1435 (наложение) [53]
Cl3SiNCO 2296 1474 714 [137]
Si (NCO)4 2284 1482 608, 542 [57]
2260 1475, 1218 704 [53]
(ONC)3 SiOSi (NCO)3 2268 1475, 1215 704 »
Ge (NCO)4 2247 1426 608, 528 [57]
H3GeNCO 2272 1420 [60]
P (NCO)3 2239 1421, 1206 603, 577 ** [279]
OP (NCO)3 2265 1429, 1283 600, 620 ** »
C1NCO 2215 1429, 1309 708 [99]
Тип связи: /NCO
[Cl4SbNCO]2 2200 1303 728 [269, 270]
* Данные для твердых веществ или их растворов в CCL.
** 6 (MNCO).
а полосы валентных колебаний v (СО) лежат в диапазоне 1150—1230 см-1.
Алкилцианаты характеризуются частотами v (CN) и v (СО) в интерва-
ле 2240—2280 и 1060—1300 см-1 (например, v (CN) для PhOCN — 2235;
2261 и 2282, для MeOCN — 2248, для EtOCN — 2248 и 2282 см-1 [267]).
Так как для органоцианатов и органоизоцианатов в одинаковой мере наблю-
дается повышение v (CN), то для определения структурного различия меж-
ду ними можно привлечь интегральные интенсивности (А) м (СИ)-полос [253].
Примером цианата металла главной подгруппы с мостиковой NCO-rpyn-
пой является fCl4Sb (NCO)]2. Как и в цианатах d-металлов с /NCO-мос-
м/
тиком, в противоположность изоцианатам с концевой NCO-группой, для этого
соединения характерно относительно незначительное смещение v (CN) и
v (СО) в высоковолновую область (табл. 4.5) [269, 270].
Особенно обстоятельные исследования имеются по изоцианатам d-ме-
таллов. Подобно ковалентным цианатам в изоцианатах наблюдается
147
повышение частот валентных колебаний v (CN) и v (СО) (характеристические
области 2170—2250 и 1320—1350 см-1). Типичный для NCO-ионов резонанс
Ферми между v (СО) и 26 (NCO) в изоцианатных комплексах не наблюдает-
ся; полосы 6 (NCO) понижаются 585—630 см-1) (табл. 4.6) [196, 266].
Полосы -V (MN) для изоцианатных комплексов наблюдаются в области 300—
400 см-1 (в табл. 4.6—4.8 приведены частоты, полученные при снятии ИК-
спектров веществ в нуйолеили в КВг). Для ряда комплексов палладия мож-
но наблюдать хорошую корреляцию между понижением частот v (CN) и по-
вышением v (MN) соответственно для мостиковой связи металл — лиганд
[280].
Характерным для цианатных комплексов (М—OCN) является пониже-
ние V (СО) и 26 (NCO) до ~1070—1300 см-1 (резонанс Ферми при этом оста-
ется). Полосы v (CN) в ИК-спектрах цианатов, как и в спектрах изоциана-
тов, повышены и наблюдаются в области ~2200—2240 см-1, а 6 (NCO) на-
ходятся в области 590—630 см-1 [196, 266] (табл. 4.6).
Для различия связи p-NCO и p-OCN можно воспользоваться величиной
интегральной интенсивности полосы v (CN) (А). Эта величина, как правило,
для изоцианатов выше, чем для цианатов [196, 266]. Например, для изоме-
ров [Rh (NCO) (Ph3P)3] и [Rh (OCN) (Ph3P)3] А соответственно равна 12,7 •
• 104 и 9,0 • 104 см2/моль [218].
В
ИК-спектрах соединений с мостиковыми
М^
М/'
NCO-группами
положе-
ние v (CN) и v (СО) в общем мало отличается от такового для изоцианатов
(полоса v (CN) лежит в области 2150—2210, a v (СО) — 1300—1340 см-1)
[196, 266]. Для установления способа координации NCO-групп можно ис-
пользовать значения 6 (NCO). В ИК-спектрах изоцианатов типа М—NCO
часто наблюдается расщепление 6 (NCO) на две близко лежащих полосы,
Мх
в то время как в соединениях с мостиковой ^J^NCO-связью это расщепле-
ние заметно больше (полоса наблюдается в области 610—660 см ') и со-
ставляет 30—50 см-1. Это хорошо видно из табл. 4.6 при сравнении положе-
ния 6 (NCO) для соединений [М (NCO)2L4] (NCO — концевые) и [М (NCO)2 •
• L2] (NCO — мостиковые) [172].
Для координированных цианатов типа М—NCO и М—OCN проведен ин-
тересный модельный расчет, из которого, допуская линейность М—NCO-
и MOCN-связей и варьируя массой атома металла и силовых постоянных,
можно сделать следующие теоретические предсказания [271]:
Соединения типа М—NCO (изоцианатные комп-
лексы)
— Частоты v (CN) и v (СО) увеличиваются в сторону больших волновых
чисел при неизменных значениях силовой постоянной &mn.
— Частоты v (CN) и v (СО) не зависят от массы металла М, в то время
как частота v (MN) линейно возрастает с увеличением &мы-
— v (MN) возрастает с увеличением массы М и является линейной функ-
цией силовой постоянной &MN.
— v (CN)-, v (СО)- и v (MN)-4acTOTbi не соответствуют свободным CN-,
СО- и MN-полосам валентных колебаний, а представляют собой наложение
различных колебаний.
— Для типа координации М—NCO экспериментально найденные сме-
щения частот v (CN) и v (СО) в длинноволновую область нельзя объяснить
148
Таблица 4.6. Характеристические частоты в ИК-спектрах некоторых
изоцианатор М—NCO *, см 1
Соединение v (CN) v (СО) 6 (NCO) v (MN) Лите- ратур а
[Et4N]2 [Cu (NCO)J 2247 (пл.), 2183 2251, 2179 (пл.) 1328 619, 617, 612 338 [141]
[Ph4As] [Au (NCO),] 1355 623 1149]
[Et4N]2 [Zn (NCO)4] 2174 1335 321 |141|
[Bu4N]3[Er (NCO)e] 2155 1322 608 [158]
[Et4N]2 [Mn (NCO)J 2222 (пл.), 2174 2182 1335 623 325 [141]
[Et4N]2 [Fe (NCO) J 1337 619 325 »
[Ph4P] [Fe (NCO)4] 2208 (пл.), 2171 1370 626, 619 410 »
[Et4N]2 [Co (NCO)4] 2217 (пл.), 2179 1335 629, 617 (пл.) 345, 213 »
[Et4N]2 [Ni (NCO)J 2237 (пл.), 2186 1330 619 (пл.), 617 341 »
[Me4N]2 [Pd (NCO)J 2190—2200 1319 613, 604, 594 408 (пл.), 384, 350,274 [139]
[Et4N] [Cr (CO)5 NCO] 2224 1288 595 280 [204]
[Et4N] [Mo (CO. NCO] 2220 1302 592 »
[Et4N] [W (CO)5 NCO] 2230 1320 292 »
[Mn (NCO)2 (3-CNPy)4J 2205 1334 628, 620 [172]
[Fe(NCO)2 (3-CNPy)4] 2216 1350 620, 613 »
[Co (NCO)2 (3-CNPv)4] 2222 1334 620, 613 »
[Ni (NCO)2 (3-CNPy)4] 2222 1330 620, 612 »
[Zn (NCO)., (3-CNPy)4] 2230 1337 627, 614 »
[Pd(NCO)2Py2] 2180—2210 1332 586 406 [156, 280] [280]
[Pd (NCO), (y-Pic)2] 2195—2215 1332 586 416
[Pd (NCO)2Dipy] 2195—2215 1340 587 389, 374 1156,
(пл.) 280]
[Pd (NCO)2 (Ph3P2] 2235 1350 615, 610, 690 350 То же
[Pd (NCO)2 (Ph3As)2] 2230 1325— 1340 585 360 » »
Pd (NCO)2 [Ph3Sb)2] 2170, 2210 (пл.) 1330 600 367, 339 [156]
Pt (NCO)2 (Ph3P)2] 2230 (пл.), 2200 1355, 1312 590 [126]
[Ir (NCO) (CO) (Ph3P)2] 2240 1353 [149]
[Rh(NCO)(CO) (Ph3P)2] 2239 1340 592 »
[Rh (NCO) (Ph3P)3] 2230 [218]
* ИК-спектры веществ сняты в нуйоле или КВг.
Пл.— плечо.
Таблица 4.7. Характеристические частоты в ИК-спектрах некоторых
цианатов М—OCN *, см-1
Соединение v (CN) v (CO), 26 (NCO) 6 (NCO) Литература
[Ph4As]2 [Re (OCN)eJ 2224 1306,1138 595 [160]
[Ph4As] [Re (OCN)6] 2220 »
[Ph4As]3 [Mo (OCN)6] 2205 1296,1140 595 »
[Cp2Zr (OCN)2] 2233,2200 1257, 1070 631, 607 [193]
[Zr (OCN)4 Phen] 2190 1210,1070 640, 625 [190]
[Zr (OCN)4 (DMSO)2] 2220 1280, 1150 630 »
[CpHl (OCN)2] 2246, 2211 1257, 1071 632, 606 [193]
[Hf (OCN)4 Phen] 2195 1210, 1070 640, 625 [190]
[Hf (OCN)4 (DMSO)2] 2225 1280, 1160 625 »
[Rh (OCN) (Ph3P)3] 2215 1318 607, 590 [218]
* ИК-спектры веществ сняты в нуйоле или КВг.
149
Таблица 4.8. Характеристические частоты в ИК-спектрах некоторых
цианатов с мостиковыми ^^NCO-связями *, см-1
Соединение v (CN) v (СО) б (NCO) Лите- ратура
AgNCO 2170 1350 657, 598 [172[
I(CH8)2AuNCO]2 2192 1323 711, 723 [219[
[Mn (NCO), (3-CNPy)2l 2185 1300 658, 629 [172]
[Fe (NCO)2 (3-CNPy)2j 2180 1305 661, 622 »
[Co (NCO)2 (3-CNPy)2] 2185 1303 667, 623 »
[Ni (NCO)2 (3-CNPy)2[ 2180 1300 671, 621 »
[Ni (NCO)2 (4-CNPy).] 2210 1303 670, 622 »
[Cu (NCO)2 (о-толуидии)2[ 2258, 2207 1367 683, 634, [1821 [1491
[Pd (NCO)2PPh3]2 2231, 2173 1352, 1334 613 (пл.), 604
[(Ph3P)2 Pd (NCO)2 Pd (PPh3)2l 2172 [2361
(BF4)2 [(Ph3P)2Pt (NCO)2 Pt (PPh3)2[ 2152
2192 »
(BF4)2 2182
[(CO)3 Mn (NCO)3 Mn (CO)3J~ 2183 [211]
* ИК-спектры веществ сняты в нуйоле или кВг. Пл.— плечо.
на основании расчета модели М—N-связи, так как это потребовало бы вы-
соких значений силовых постоянных. Главным фактором при повышении
частот v (CN) и v (СО) является изменение механики колебаний и незначи-
тельное возрастание силовых постоянных &cn и &no-
Соединения типа М—OCN (цианатные комплек-
с ы)
— При неизменных силовых постоянных &cn и /гСо частота v (СО) воз-
растает в большей степени, чем для N-координации.
— Частота v (CN) не зависит, а частота v (СО) практически не зависит
от массы атома М.
— Рост /гмо не влияет на частоту v (CN), повышает частоту v (СО)
и заметно увеличивает частоту v (МО).
— Существенные различия между экспериментальными (повышение
v (CN) и понижение v (СО)) и теоретически предсказанными данными (v (CN)
не должна изменяться, a v (СО) должна возрастать) свидетельствуют о том,
что в отличие от структуры М—NCO в случае М—OCN-координации про-
исходит существенное изменение силовых постоянных связей СО и CN
(повышение /ген и понижение /гео) вследствие перераспределения электрон-
ной плотности.
4.11.2.2. ИК-спектральные исследования фульминатов
ИК-спектры фульминатов металлов весьма характерны и поэтому ока-
зались ценным средством в исследовании и описании этого класса соедине-
ний [121, 123, 124, 240, 241, 244, 248, 264, 272, 273]. Частоты валентных ко-
лебаний v (CN) и v (NO) лежат в области ^2100—2200 и ^1050—1200 см-1
и четко повышены по сравнению с частотами фульминат-иона. Характерной
для ИК-спектров фульминатов является также полоса 2v (NO), лежащая в
области ^2200—2300 см-1. Вследствие резонанса Ферми интенсивность
2v (NO) достаточно высока и даже может превосходить интенсивность полосы
V (CN) [244]. Деформационные колебания 6 (CNO) для большинства метал-
150
лов проявляются в области ^480—500 см"1 и незначительно смещены в
в высоковолновую область по сравнению с фульминат-ионом.
В табл. 4.9 приведены характеристические частоты ИК-спектров ряда
фульминатов металлов. Если сопоставить положение полос v (CN) и v (NO)
различных фульминатов металлов с частотами нитрилоксида RCNO, то
можно заметить, что повышение частот v (CN) и v (NO) наблюдается по мере
возрастания ковалентности связи М—CNO: ионные фульминаты < фульми-
натные комплексы металлов < ковалентные фульминаты тяжелых металлов
< арилметаллофульминаты < (RCNO, HCNO). Аналогичные смещения
v (CN) и v (С—С) наблюдаются в ряду циан- и алкильных соединений в за-
висимости от увеличения доли ковалентности [273]. Повышение частот
v (CN) и v (NO) наблюдается для комплексов [М (CNO)4]2~ в последователь-
ности Ni < Pd < Pt, причем увеличение доли ковалентности связи
одновременно ведет к возрастанию их термической устойчивости [253].
Таблица 4.9. Характеристические частоты ИК-спектров фульминатов
и комплексных фульминатов металлов *, см~*
Соединение 2v (NO) v(CN) v (NO) | 6 (CNO) v (MC) Литера- тура
NaCNO 2188 2098 1112 471 [121, 264]
KCNO 2126 2042 1064, 1057 473, 468 .— »
CuCNO 2175 2089 1104 458 355 [273]
AgCNO 2244 2102 1135 493, 461 — [272, 273[
2256 2178 1155 495, 461 338 [264]
Hg(CNO)2 2410 2200 1120 488, 481 348 [124, 264, 272, 273]
PhHgCNO 2382 2194 1200 484 314 [124]
Ph3SiCNO 2594 2200 1302 526 562 [123]
Ph3GeCNO 2540 2164 1276 — »
Ph3SnCNO 2315 2156 1165 484 499 »
Ph3PbCNO 2294 2123 1149 483 492
NH4 [Ag (CNO)2| 2233 2150 1135, 1121 477, 485 328 [244, 273]
[Ph4As] [Au (CNO)J 2368 2173 1180 — 278 [248]
[Pr4N]2 [Zn (CNO)4J 2307 2118 1154 475 282 [244, 248]
[Pr4N[2 [Cd (CNO) J 2283 2119 1162 478 244 »
[Ph4Asj2 [Hg (CNO),] 2272 2120 1141 462 360 [244]
[Pr4N]2 [Ni (CNO)J 2293, 2255 2156 1127 483 423 [244, 248}
[Pr4N]2 [Pd (CNO)J 2279 2164 1131 480 366 »
[Pr4N]2 [Pt (CNO)4J 2273 2177 1127 470 (пл.) 347 [244]
[Ph4As]3 [Co (CNO)e]-3H2O 2244 2167 1114 — [248]
[Ph4AsJ3 [Ir (CNO)6]-6H2O 2286 2126 1146 — »
Ba2 [Fe (CNO)g] - 4H2O 2041 2187 1040 514, 466 445, 396 »
[Hg(CNO)2 Dipy] 2407 2194, 2146 1213, 1203 489, 480 [247]
[Mn (CNO)2 (Phen)2] 2275 2077 1141 497, 492 [241]
[Fe (CNO)2 (Phen)2J.H2O 2200 2119 1100, 1085 — »
[Ni (CNO)2 (Et3P)2] 2314 2164 1150 480 [251]
[Pd (CNO)2 (Et3P)2j 2321 2160 1153 483 »
[Pt (CNO)2 (Et8P)2] 2323 2189 1155 480
[Pt (CNO) (H) (Et3P)2] 2268 2110 1136 483 [243]
[Ph4As] [Cr (CO)5 CNO] 2225 2128 1112 — [248]
[Ph4As]2 [Cr (CO)4 (CNO)2] 2173 2090 1082 —
[Ph4Au]2 [Mo (CO)4 (CNO)2] 2187 2094 1088 — »
[Ph4As]2 [W (CO)4 (CNO)2] 2194 2101 1094 — »
к КВг или нуйол.
151
В соответствии с линейностью связи М—CNO в ИК-спектрах октаэдри-
ческих, тетраэдрических, плоскостных и линейных фульминатных компле-
ксов наблюдаются лишь полосы v (CN) и v (NO). Расщепление этих полос
в спектрах твердого вещества ограничено [244, 273].
Колебания v (МС) проявляются в фульминатных комплексах в области
ниже 400 см-1 (см. табл. 4.9). Как в цианидных соединениях, в фульмина-
тах часто проявляется значительный вклад л-дативной связи. Так, для фос-
финовых комплексов типа транс-[М (CNO)2 (R3P)2] (М = Pd, Pt) наблюда-
ется монотонное понижение частот v (CN) и v (NO) при повышении основно-
сти и уменьшении л-акцепторной способности фосфинов. Это можно объяснить
увеличением прочности л-дативной связи за счет заполненных d-ор-
биталей металла и л*-орбиталей фульминат-лигандов [253]. В разнолиганд-
ных комплексах [Pt (CNO) X (Ph3P)2] полосы v (CN) (при X = CN или
CNO) по сравнению с [Pt (CNO)2 (PPh3)2] не смещены. Напротив, введение
о-доноров (X = Н или СН3) понижает v (CN) (фульмината), что обусловлено
увеличением вклада л-дативной связи Pt—CNO [253].
Особенно заметны смещения частот валентных колебаний фульминатной
группы в соединениях металлов с низкими степенями окисления, например
[M(CO)4(CNO)J2~ (М = Сг, Мо, W) [248].
М\
Примером связи /CNO являются обе модификации AgCNO (см.
4.11.1). Они различаются по положению частот v (CN), v (NO) и 6 (CNO),
которые, однако, находятся в тех же интервалах, что и соответствующие час-
тоты концевых фульминат-лигандов [264, 272, 273]. Это затрудняет исполь-
зование ИК-спектроскопии для исследования подобных фульминатов.
Приближенный теоретический (полуэмпирический) анализ колебания
координированных MCNO-групп показал, что при неизменном силовом поле
фульминат-иона координирование CNO-групп через атомы углерода увели-
чивает частоту v (CN). Частота v (NO) также повышается по сравнению с
фульминат-ионом, обе частоты не зависят от массы атома М, а частота v (СО)
заметно понижается с ростом массы атома М.
Сравнение расчетов фульминато- и изоцианатокомплексов, с одной сто-
роны, а также изотио- и изоселеноцианатокомплексов — с другой [275]
показывает, что при изотио- и изоселеноцианатной координации незначи-
тельное изменение силовых постоянных £nc и kcx приводит к повышению
v (СХ) и v (CN), напротив, для изоцианатов и фульминатов только заметное
увеличение силовых постоянных /?nc и &cn, а также kco и /eno может рассмат-
риваться как фактор повышения v (CN), v (СО) и v (NO) [271].
4.11.3. ,4М-ЯМР-исследования цианатов и фульминатов
Для псевдогалогенид-ионов NCO~ и CNO- измерены химические сдвиги
сигналов 14N (относительно 6 NO3), которые равны +300 [2761, +288 [129]
и +176 м. д. [277].
Изоцианатные комплексы характеризуются сдвигом 14N -ЯМР-сигнала
в область высоких полей. Так, для комплексов [Et4N] [Ag (NCO)2], [Pt •
• (NCO)2 (Ph3P)2] и [Me4N]2 [Hg (NCO)4] определены химические сдвиги
в +344 [276], +340 [273] и +312 м. д. [276]. Сдвиг в область высоких полей
наблюдается и для EtNCO (+346 м. д.) [277, 278]. Напротив, для ROCN
типичным является сдвиг в сторону низкого поля (EtOCN +222 [276];
PhOCN +208 м. д. [273]).
152
^N-сигналы комплексных фульминатов по отношению к таковым фуль-
минат-иона сдвинуты в сторону низких полей и появляются в относительно
узком интервале (20 м. д.): [Et4N] [Ag (CN0)9] +164 [277], [Pt (CNO)2 •
• (Ph3P)2l + 148 [273] и Na2 [Ni (CNO)4] +163 м. д. [277].
4.12. ПРИМЕНЕНИЕ ЦИАНАТОВ И ФУЛЬМИНАТОВ
Цианаты щелочных металлов находят ограниченное применение в ана-
литической химии. Они могут использоваться для осаждения металлов в
форме их гидроксидов. Так, KNCO предложен для осаждения А1 (ОН)3
[283], для отделения трехвалентных Сг, Al, Fe от двухвалентных элементов
группы (NH4)2S [283—285], для отделения индия от цинка и никеля [286],
а также для количественного определения олова и сурьмы [287]. KNCO
можно применять для обнаружения кобальта в виде темно-синей соли Блом-
странда К2 [Со (NCO)4L Чувствительность реакции возрастает в водно-
ацетоновом растворе [161, 288, 289].
Псевдогалогенокомплексы типа [МХ2 (4-алкил Ру)41 образуют клатра-
ты с ароматическими соединениями и могут применяться для разделения
изомеров. Так, при помощи [Mn (NCO)2 (4-EtPy)4] можно разделить о-
и и-ксилол [290, 291].
Среди фульминатов лишь Hg (CNO)2 имеет пректическое применение.
Под названием «гремучая ртуть» это соединение используется в качестве
детонирующего вещества, но в настоящее время его заменяют более удобным
для хранения Pb (N3)2. Hg (CNO)2 имеет скорость детонации 4250 м/с, теп-
лоту сгорания 3,93 кДж/г, теплоту взрыва 1,79 кДж/г [115]. Детонирующая
смесь состоит из трех компонентов: инициатора (Hg (CNO)2), окислителя
(КС1О3) и горючего состава (Sb2O3) [292].
В противоположность цианатам металлов, которые пока не нашли ши-
рокого применения, органоизоцианаты в последнее десятилетие имеют важ-
ное значение в технике. Их мировое производство в 1974 г. составляло бо-
лее 1 млн. т в год [293]. Алкил- и арилмоноизоцианаты являются исходными
продуктами для производства замещенных мочевины, карбаматов и уре-
танов, гербицидов и средств борьбы с вредителями. Ряд органодиизоциана-
тов, например толуолдиизоцианат, гексаметилендиизоцианат и др., исполь-
зуются в производстве полиуретанов. Ввиду большого практического значе-
ния органоизоцианатов число публикаций по этим соединениям с каждым
годом увеличивается. Укажем лишь на работы обобщающего характера
[294—296].
Изучение органоизоцианатов начато Вюрцем, который в 1848—1849 гг.
получил эти соединения реакциями цианатов металлов с алкил- и арилхло-
ридами или с арилсульфатами [297]
RC1 + NaNCO -> RNCO + NaCl; (4.37)
(RO)2 SO2 + 2KNCO -> 2RNCO + K2SO4. (4.38)
В настоящее время для промышленного получения разнообразных али-
фатических и ароматических моно- и диизоцианатов используют метод фос-
генизации первичных аминов [298]
RNH2 RNHCOC1 лзЕЕЦ RNCO. (4.39)
Предложен весьма интересный метод получения арилизоцианатов из
нитросоединений и СО в присутствии благородных металлов как катализа-
153
торов [296, 299, 300], который, возможно, найдет техническое применение
+ 6 со
+ 4 С02
(4.40)
Разностороннее применение изоцианатов объясняется в первую очередь
их исключительной реакционной способностью по отношению к веществам
с подвижным атомом водорода. Например, спирты или вторичные амины
образуют с изоцианатами уретаны или замещенные мочевины
О
II (4.41)
RNCO + НХ R—NH—С—X (X = OR или NR2).
Реакции органоизоцианатов с полимерами, которые имеют подвижные
атомы водорода, можно использовать при модификации полимеров для полу-
чения новых синтетических материалов, среди которых разностороннее
использование (производство искусственной кожи, пластмасс, пенопластов,
лаков и др.) находят полиуретаны [301, 302],
г о о
nOCNRNCO + nHOR'OH ->
—С—NH—R—NH—С—О—R'—О
п-
(4.42)
Наконец следует указать на производство и использование органо-
цианатов ROCN, открытых лишь в начале 60-х годов. Это — высокореак-
ционные вещества, которые можно получать, например, при взаимодействии
NaN3 с PhOC (S) Cl с последующим термолизом образовавшегося тиотриа-
зена 145]
S
II
С6Н5О—С—Cl + NaN3 С6Н5О-С-----S C6H5OCN. (4.43)
II I "
N N
\n^
В промышленных масштабах цианаты получают в мягких условиях
(низкая температура, добавка Et3N для связывания НС1) [461
ArOH + ClCN-z^g^ci- ArOCN. (4.44)
Органоцианаты при нагревании превращаются в органоизоцианаты.
Практическое применение находят реакции арилцианатов с соединениями,
содержащими подвижные атомы водорода. В результате этих реакций обра-
зуются имино- или тиоиминоэфиры угольной кислоты [471
NH
II
ArOCN + НХ Аг—О—С—X (X = OR, SR, NRO) (4.45)
СПИСОК ЛИТЕРАТУРЫ
1. Berzelius J.— Ann. Physik u. Chemie (Poggendorff), 1830, 19, S. 326.
2. Gay-Lussac J. L., Liebig J.— Ann. Chim. Phys., 1824, 2, 25, p. 285.
3. Wohler F.— Ann. Chim. Phys., 1824, 2, 27, p. 199.
4. Beck W., Schorpp K-, Oetker Ch.— Chem. Ber., 1974, 107, S. 1380.
5. Birckenbach L., Kellermann K-— Ber., 1925, 58, S. 787.
6. Hundt H.— J. Am. Chem. Soc., 1932, 54, p. 907.
154
7. Cauqtiis G., Pierre G.— Bull. Soc. chim. France, 1975, p. 997.
8. Wohler F.— Ann. Phys. Chem., 1829, 15, p. 622.
9. Liebig J., Wohler F.— Ann. Phys. Chem., 1830, 20, p. 373.
10. Linhard M.— Z. anorg. allg. Chem., 1938, 236, S. 200.
11. Werner E. A., Fearon W. R.— J. Chem. Soc., 1920, 117, p. 1356.
12. Bohlen W. C., Carpenter С. B.— Acta Crystallogr., 1955, 8, p. 649.
13. Eyester E. H., Gillette R. H., Brockway L. 0 — J. Am. Chem. Soc., 1940, 62, p.3236.
14a . Jones L. H., Shoolery J. H., Shulman R. G., YostD. M.— J. Chem. Phys., 1950, 18,
p. 900.
14b . ReidC.— J. Chem. Phys., 1950, 18, p. 1544; Hersberg E., Reid C.— Discuss. Faraday
Soc., 1950, 9, p. 92.
15. Walter R.-— J. prakt. Chem., 1909, 2, S. 128.
16. Wiebenga E. H.— J. Am. Chem. Soc., 1952, 74, p. 6156.
17. Beyer H.— Lehrbuch der Organ. Chem., 15/16. Auflage. Leipzig, S. Hirzel Verlag,
1968, S. 291.
18. Некрасов Б. В. Основы общей химии. М., Химия, 1967, т. 2, с. 37.
19. De Pree D. О., Oldenburg Е. В., Burns J. A., Jr.— J. Inorg. Nucl. Chem., 1960, 15,
p. 287.
20. Waddington T. C.— J. Chem. Soc., 1959, p. 2499.
21. Williams W.— US Patent, 1960, №316783.
22. Scattergood A., McAdams D. R., McReynolds J. P.— Inorg,. Synth., 1946, 2, p. 88.
23. Leuchs FI., Ceserick A.— Ber., 1908, 41, S. 4171.
24. Flachbaur E.-—Monatsh. Chem., 1969, 100, S. 1998. ;
25. Палаузов В. А. Химические реактивы и их свойства, получение, методы исследова-
ния и применение. Харьков — Киев, 1936.
26. Vanderzee С. Е., Myers R. А.— J. Phys. Chem., 1961, 65, р. 153.
27. Rossmanith R.— Monatsh. Che., 1967, 98, S. 501.
28. Nachbaur E., Schober W.— Monatsh. Chem., 1973, 104, S. 538.
29. Walker J., Wood J. R.— J. Chem. Soc., 1900, 77, p. 21.
30. Liebig J., Wohler F.— Ann. Phys. Chem., 1830, 20, p. 393.
31. Wohler F.— Ann. Phys. Chem., 1828, 12, p. 253.
32. Цинцадзе Г. В., Голуб А. М., Махатадзе Ц. Л., Цивадзе А. Ю.— Труды Груз, по
литехн. ин-та, 1972, 5 (153), с. 25.
33. Махатадзе Ц. Л. Автореф. канд. дис. Тбилиси, Политехи, ин-т, 1973.
34. Baskin Н.— US Patent, 1965, № 3222126.
35. Baskin Н.— US Patent, 1964, № 3131023.
36. SoderbackE.— Acta Chem. Scand., 1957, 11, p. 1622.
37. Lap pert M. F., Pyszora H.— Adv. Inorg. Chem. and Radiochem., 1966, 9, p. 133.
38. Thayer J. S., West R.— Adv. Organometal. Chem., 1967, 5, p. 169.
39. Derkac G. I.— Angew. Chem., 1969, 81, S. 407.
40. Forbes C. S., Anderson H. H.— J. Am. Chem. Soc., 1940, 62, p. 761.
41. Mengeot H., Dasord J., Atcheksat H. R., Carrd J.— C. r. Acad. Sci., 1975, C280,
p. 1109.
42. Lappert M. F., Pyszora H.— J. Chem. Soc., 1963, p. 1744.
43. Patel S. J., TuckD. G.— J. Chem. Soc., 1968, A, p. 1870.
44. Huttner R., Rnappe S.— Z. anorg. allg. Chem., 1930, 190, S. 34.
45. Martin D.— Angew. Chem., 1964, 76, S. 303.
46. Grigat E., Putter R.— Chem. Ber., 1964, 97, S. 3012.
47. Grigat E., Putter R.—Angew. Chem., 1967, 79, S. 219; 1972, 84, S.1008.
48. Mayer E.— Monatsh. Chem., 1970, 101, S. 834.
49. Cottardi 117.— Monatsh. Chem., 1971, 102, S. 264.
50. Glemser 0 , Biermann H., Fild M.— Chem. Ber., 1967, 100, S. 1082.
51. Sundermeyer IF.— Z. anorg. allg. Chem., 1961, 313, S. 290.
52. Steyermark P P.— J. Org. Chem., 1963, 28, p. 586.
53. Coubeau J., Heubach R., Paulin D., Widmaier I.— Z. anorg. allg. Chem., 1959, 300.
S. 194.
54. Forbes G. S., Anderson FI. H.— J. Am. Chem. Soc., 1947, 69, p. 3048.
55. Anderson H. H.— J. Am. Chem. Soc., 1944, 66, p. 934.
56. ForbesG. S., Anderson H. H.— J. Am. Chem. Soc., 1947, 69, p. 1241.
57. Miller F. A., Carlson G. L.— Spectrochim. Acta, 1961, 17, p. 977.
58. Gerry M. C. L., Thompson J. C., Sugden T.M.— Nature, 1966, 211, p. 846.
59. Srivastava T. E., Griffiths J. H., Onyszchuk M.— Can. J. Chem., 1962, 40, p. 739.
60. Griffiths J. E.— J. Chem. Phys., 1928, 48, p. 278.
61. Ramaprasad R. R., Varma R., Nelson R.— J. Am. Chem. Soc., 1968, 90, p. 6247.
62. Rimura R., Ratada R., Bauer S. H.— J. Am. Chem. Soc., 1965, 88, p. 693.
63. Stamm W.— J. Org. Chem., 1965, 30, p. 693.
155
64. Nelson S. M., Gruner E.— US Patent, 1963, № 311610.
65. Ganner N.— US Patent, 1963, № 311607.
66. Anderson H. H.— J. Am. Chem. Soc., 1942, 64, p. 1757.
67. JenkinsL. H.,SearsD. S.— US Patent, 1959, №2873171.
68. Anderson H. H.— J. Am. Chem. Soc., 1945, 67, p. 223.
69. Anderson H. H.— J. Am. Chem. Soc., 1945, 67, p. 2176.
70. Anderson H. H.— J. Am. Chem. Soc., 1947, 69, p. 2495.
71. Anderson H.H.— J. Am. Chem. Soc., 1942,64, p. 1757.
72. Steger E., Bachmann G.— Z. Chem., 1970, 10, S. 306.
73. Tesi G., Zimmer-Galler R.— Chem. and Ind., 1964, p. 1916.
74. Кирсанов А. Б.— Журнал общ. химии, 1954, 24, с. 1033; 1959, 29, с. 2256.
75. Kuhn S. J., Olah G. A.— Can. J. Chem., 1962, 40, p. 1951.
76. Roesky H. W.— Angew. Chem., 1967, 79, S. 61.
77. Fluck V. E.— Z. Naturforsch., 1964, 19b, S. 869.
78. Самарай Л. И., Деркач Г. И.— Журнал общ. химии, 1965, 35, с. 755.
79. Самарай Л. И., Деркач Г. И.— Журнал общ. химии, 1966, 36, с. 1433.
80. Hofmann A. W.— Вег., 1870, 3, S. 693.
81. Fielding Н. С., Polleck I. М.— Brit. Patent № 941240; Chem. Abstr., 1963, 59, 8384a.
82. Challenger F., Wilson V. K.— J. Chem. Soc., 1927, p. 209.
83. Beaumont R. E., Goel R. G.— J. Chem. Soc. Dalton, 1973, p. 1394.
84. Forbes G. S., Anderson H. H.— J. Am. Chem. Soc., 1943, 65, p. 2272.
85. Graf B.— Chem. Ber., 1956, 89, S. 1071.
86. Appel R., Gerber H.— Chem. Ber., 1958, 91, S. 1200.
87. Appel R., Ritterebacher H.— Chem. Ber., 1964, 97, S. 849.
88. Appel R., Gerber H.— Angew. Chem., 1958, 70, S. 271.
89. Lehmann H. A., Riesel L., Flohne K., Maier E.— Z. anorg. allg. Chem., 1961, 310,
S. 298.
90. Birckenbach L., Linhard H.— Ber., 1930, 63, S. 2944.
91. Birchenbach L., Linhard H.— Ber., 1929, 62, S. 2261.
92. Birchenbach L., Linhard H.— Ber., 1930, 63, S. 2928.
93. Hachbaur E., Cottardi W.— Monatsh. Chem., 1966, 97, S. 115.
94. Cottardi W.— Monatsh. Chem., 1972, 103, S. 1150.
95. Cottardi W.— Angew. Chem., 1971, 83, S. 445.
96. Cottardi W., HennD.— Monatsh. Chem., 1969, 100, S. 1860; 1970, 101, S.l 1.
97. Cottardi W., HennD.— Monatsh. Chem., 1970, 101, S. 264.
98. Oberhammer H. E.— Naturforsch., 1971, 26a, S. 280.
99. Eysel H. H., Nachbauer E.— Z. anorg. allg. Chem., 1971, 381, S. 71.
100. Hacking W. H., Gerry M. C. L.— Chem. Commun., 1970, p. 448.
101. Leimeister H., Dehnicke K-— J. Organometal. Chem., 1971, 31, p. C3.
102. Britton D., Dunitz J. D.— Acta Crystallogr., 1965, 19, p. 662.
103. Davis T. L., Logan A. V.— J. Am. Chem. Soc., 1935, 54, p. 2155.
104. Birckenbach L., Kolb H.— Ber., 1935, 68, S. 895.
105. Pascal P.— C. r. Acad. Sci., 1913, 157, p. 932.
106. Wieland H.— Ann., 1925, 444, p. 7.
107. Nef I. U.— Ann., 1894, 280, p. 303.
108. Beck W., Peldi K.— Angew. Chem., 1966, 78, S. 746.
109. Wiennewieser M., Bodenach H.— Z. Naturforsch., 1967, 22a, S. 1724; 1973, 24a,
S. 1966.
110. Beck W., Swoboda F., Feldi K., Tobias R. St.— Chem. Ber., 1971, 104, S. 533.
111. Liebig J.— Ann. Chem. Pharm., 95, p. 284.
112. Beckmann E.— Ber., 1886, 19, S. 993.
113. Gmelin’s Handbuch der anorganischen Chemie. Verlag Chemie, 1968, Bd 34, B3,
S. 1229.
114. Wohler L., Berthmann A.— Angew. Chem., 1930, 43, S. 59.
115. Ulimann’s Enzyklopadie der techn. Chemie. Munchen, Verlag Urbana Schwarzenberg,
1965, 3, Au flage 16, p. 95.
116. Gay-Lussac, Liebig J.— Ann. chim. phys., 1849 (2), 25, p. 295.
117. Bassiere M.— Mem. Serve Chim. Etat, 1942, 30, p. 30.
118. WohlerL.— Ber., 1905,38, S. 1351.
119. Wohler L., Weber A.— Ber., 1929, 62, S. 2742.
120. Hackspill, Schumacher.— C. R. Acad. Sci., 1936, 202, p. 70.
121. Beck W.— Chem. Ber., 1962, 95, S. 341.
122. Wohler L., Martin F.— Ber., 1917, 50, S. 586.
123. Beck W., Schairer E.— Chem. Ber., 1964, 97, S. 3517.
124. Beck W., Schairer E.— J. Organometal. Chem., 1965, 3, p. 59.
125. Kasutina M. B., Oklobystin 0. Yu.— J. Organometal. Chem., 1967, 9,.p, 5.
156
126. Nor bur у A. H., Sinha A. J. P.— J. Chem., Soc., 1968, A, p. 1598.
127. Stroh R., Gerber H.— Angew. Chem., 1960, 72, S. 1000.
128. Wagner E. L.— J. Chem. Phys., 1965, 43, p. 2723.
129. Bonaccorsi R., Petrongolo C., Scrocco E., Tomasi J.— J. Chem. Phys., 1968, 48,
p. 1500.
130. Kent J. E., Wagner E. L.— J. Chem. Phys., 1966, 44, p. 3590.
131. Hendrickson D. H., Hollander J. M., Jolly W. L.— Inorg. Chem., 1969, 8, p. 2642.
132. Norbury A. H., Thompson M., Sengatad J.— Inorg. Nucl. Chem. Lett., 1973, 9,
p. 347.
133. Yonezawa T., Kato H., Konishi H.— Bull. Chem. Soc. Japan, 1967, 40, p. 1071.
134. Holeboer F. J., Beck W.— Chem. Commun., 1970, p. 262.
135. Beck W., Fehlhammer W. P.— MTP Internat. Rev. Sci., Inorg. Chem., 1972, Ser.
1, 2, p. 253.
136. Micu-Semenine R., Macarovici G. G., Lupu D.— Rev. Roum. Chim., 1972, 17(6),
p. 993; CA, 1972, 77, p. 121613a.
137. Koster D. F.— Spectrochim. Acta, 1968, 24A, p. 395.
138. Becker W., Beck W.— Z. Naturforsch., 1970, 25b, S. 101.
139. Forster D., Goodgame D. M. L.— J. Chem. Soc., 1965, p. 1286.
140. Forster D., Goodgame D. M. L.— J. Chem. Soc., 1964, p. 2790.
141. Forster D., Goodgame D. M. L.— J. Chem. Soc., 1965, p. 262.
142. Цивадзе А. Ю., Цинцадзе Г. В., Харитонов Ю. Я- и др.— Журнал неорган хи-
мии, 1970, 15, с. 1818.
143. Norbury А. Н., Sinha А. I. Р.— J. Inorg. Nucl. Chem., 1971, 33, р. 2683.
144. Aarflot К-, Ase К-— Acta Chem. Scand., 1974, A28, p. 137.
145. Ripan R.— Bull. soc. StiinteCluj, 1929, 4, p. 499.
146. Burmeister J. L., O'Sullivan T. F.— Inorg. Chim. Acta, 1969, 3, p. 479.
147. Голуб A. M., Цинцадзе Г. В., Махатадзе Ц. Л.— Труды Груз, политехи, ин-та,
1970, № 4 (139), с. 23.
148. Мамулашвили А. М. Автореф. канд. дис. Тбилиси, Груз, политехи, ин-т,
1971.
149. Beck W., Fehlhammer W. Р., Pollmann Р., Schdchl Н.— Chem. Ber., 1969, 102,
S. 1976.
150. Blomstrand C. W.— J. prakt. Chem., 1871, 2, S. 221.
151. Cotton F. A., Goodgame M.— J. Am. Chem. Soc., 1961, 83, p. 1777.
152. Sabatini A., Bertini I.— Inorg. Chem., 1965, 4. p. 959.
153. Fackler J. P., Dolbear G. E.„ Coucouvanis D.— J. Inorg. Nucl. Chem., 1964, 26,
p. 2035.
154. Forster D., Goodgame D. M. L.— J. Chem. Soc., 1965, p. 454.
155. Цинцадзе Г. В. Смешанные псевдогалогенидо-аминные соединения некоторых ме-
таллов. Тбилиси, 1974.
156. Nor bury А. Н., Sinha A. I. F.— Inorg. Nucl. Chem. Lett., 1967, 3, p. 355.
157. Bailey R. A., Michelsen T. W.— Inorg. Nucl. Chem. Lett., 1972, 34, p. 2935.
158. Burmeister J. L., Patterson S. D., Deardorff E. A.— Inorg. chim. Acta, 1969, 3,
p. 105.
159. Harris M. B., Thompson L. C.— J. Inorg. Nucl. Chem., 1974, 36, p. 212.
160. Bailey R. A., Kozak S. L.— J. Inorg. Nucl. Chem., 1969, 31, p. 689.
161. Ripan R.— Bull. Soc. Stiinte Cluj., 1927, 3, p. 176; 1928, 4, p. 144; Ch. Z., 1927,
98, S. 2388; Ch. Z„ 1929, 100, S. 2905.
162. Голуб A. M., Цинцадзе Г. В., Мамулашвили А. М.— Укр. хим. журн., 1970, 36,
с. 1207.
163. Голуб А. М., Цинцадзе Г. В., Махатадзе Ц. Л.— Сообщ. АН Груз. ССР, 1971,
61, с. 1, 57.
164. Burbridge С. D., Goodgame D. М. L.— Inorg. chim. Acta, 1970, 4, р. 231.
165. Голуб А. М., Цинцадзе Г. В., Мамулашвили А. М.— Журнал неорган. химии,
1969, 14, с. 3013.
166. Norbury А. Н., Ryder Е. A., Williams R. F.— J. Chem. Soc., 1967, А, р. 1439.
167. Цинцадзе Г. В.— Сообщ. АН Груз. ССР, 1970, 57, с. 57.
168. Ford Р. С.— Inorg. Chem., 1971, 10, р. 2153.
169. King Н. С. A., Koros Е., Nelson S. М.— J. Chem. Soc., 1969, р. 5449.
170. Nelson S. М.— Proc. chem. Soc., 1961, p. 372.
171. Nelson S. M., Sheppard T. M.— Inorg. Chem., 1965, 4, p. 813.
172. Nelson J., Nelson S. M.— J. Chem. Soc., 1969, p. 1597.
173. King H. C. A., Koros E., Nelson S. M.— Nature (London), 1962, 196, p. 572.
174. Schmidtke H.-HCarthoff D.— Helv. chim. Acta, 1967, 50, p. 1631.
175. Цинцадзе Г. В. Автореф. докт. дис. Тбилиси, 1973.
176. Ripan R.— Bull. Soc. Stiinte Cluj., 1928, 4, p. 29; Ch. Z., 1928, 99, S. 2938.
157
177. Цинцадзе Г. В., Мамулашвили А. М., Демченко П. Л.— Журнал неорган. химии,
1970, 15, с. 276.
178. Lever А. В. Р., Nelson S. М.— J. Chem. Soc., 1966, А, р. 859.
179. Burmeister J. L., O’Sullivan Т. Р., Johnson К. А.— Inorg. Chem., 1971, 10,
р. 1803.
180. DugganD. М., Hendrickson D. N.— Chem. Commun., 1973, p. 411.
181. Kohout J., Quastlerovd-Hvastijovd M., Kohutova M., Gaso J.— Monatsh. Chem., 1971,
102, S. 350; Monatsh. Chem., 1973, 104, S. 779.
182. Quastrelovd-Hvas M., Kohout J., Gaso J.— Z. anorg. allg. Chem., 1973, 396.
S. 341.
183. Kohout J., Quastlerovd-Hvastijovd. M., Kohutova M.— Z. Naturforsch. 1971 265,
S. 1366; 1969, 24b, S. 134.
184. Chauhan S. S., Sinha P. C.— J. Indian Chem. Soc., 1963, 40, p. 769.
185. King H. C. A., Koros E., Nelson S. M.— J. Chem., Soc., 1964, A, p. 4832.
186. Cabral J. deO., King H.C.A., Nelson S. M. a. o.— J. Chem. Soc., 1966 A,
p. 1348.
187. Schafer Sr., M., Curran C.— Inorg. Chem., 1965, 4, p. 623.
188. Madeja K., Wilke W., Schmidt S.— Z. anorg. allg. Chem., 1966, 346, S. 306.
189. Adlard M. W., Socrates G.— J. Chem. Soc., 1972, p, 797.
190. Голуб A. M., Лишка T. П.— Журнал неорган. химии, 1974, 19, с. 1509.
191. Samuel Е.— Bull. Soc. chim. France, 1966, p. 3548.
192. Coutts R. S. P., Wailes P. C.— Aust. J. Chem., 1966, 19, p. 2069.
193. Burmeister J. L., Deardorff E. A., Jenson A., Christiansen V. H.— Inorg. Chem.,
1970, 9, p. 58.
194. Burmeister J. L., Deardorff E. A., Van Dyke С. E.— Inorg. Chem., 1969, 8,
p. 170.
195. Beck W. , Schorpp K.— Chem. Ber., 1975, 108, S. 3317.
196. Nor bury A. H.— Adv. in Inorg. Chem. and Radiochem., 1975, 17, p. 382.
197. Gorsi B. L., Kapoor 0. N., Mehrotra R. C.— Indian J. Chem., 1975, 13(H),
p. 1200.
198. Coutts R. S. P., Wailes P. C.— Inorg. Nucl. Chem. Lett., 1967, 3, p. 1.
199. Doyle C., Tobias R. St.— Inorg. Chem., 1968, 7, p. 2479.
200. Bush M. A., Sim G. A.— J. Chem. Soc., 1970, A, p. 605.
201. McPhail A. T., Knox G. R., Robertson C. G., Sim G. A.— J. Chem. Soc., 1971, A,
p. 205.
202. Green M. L. H., Lindsell W. E.— J. Chem. Soc., 1969, p. 2150.
203. Behrens H., Herrmann D.— Z. anorg. allg. Chem., 1967, 351, S. 225.
204. Beck W., Werner H., Engelmann H., Smedal H. S.— Chem. Ber., 1968, 101,
S 2143.
205. Beck WSmedal H. S.— Angew. Chem., 1966, 78, S. 264.
206. Beck W., Fehlhammer W. F.— Angew. Chem., 1967, 79, S. 146.
207. Angelici R. J., BusettoL.— J. Am. Chem. Soc., 1969, 91, p. 3197.
208. Beck W., Lindenberg B.— Angew. Chem., 1970, 82, S. 701.
209. Colimann J. P., Kubota M., Hosking J. W.— J. Am. Chem. Soc., 1967, 89,
p. 4809.
210. VaskaL., Peone J.— Chem. Commun., 1971, p. 419.
211. Mason R., Rusholme G. A., Beck W., Englemann H. a. o.— Chem. Commun., 1971,
p. 496.
212. Moelwyn-Hughes J. T., Garner A. W. B., Howard A. S.— J. Chem. Soc., 1971, A,
p. 2370.
213. Moelwyn-Hughes J. T., Garner A. W. B., Howard A. S.— J. Chem. Soc., 1971, A,
p. 2361.
214. Angelici R. J., Kruse A. E.— J. Organometal. Chem., 1970, 22, p. 461.
215. Angelici R. I., Faber G. C.— Inorg. Chem., 1971, 10, p. 514.
216. Busetto L., Palazzi A., Ros R.— Inorg. Chem., 1970, 9, p. 2792.
217. Burmeister J. L., De Stefano N. J.— Inorg. Chem., 1971, 10, p. 998.
218. Anderson S. J., Norbury A. HSongetad J. — Chem. Commun., 1974, p. 37.
219. Stocco F., Stocco G. C., Scovell W. W., Tobias R. S.— Inorg. Chem., 1971, 10,
p. 2639.
220. Saillant R. E.,— J. Organometal. Chem., 1972, 39, p. C 71.
221. GachD. M., Harris R. 0.— Can. J. Chem., 1971, 49, p. 867.
222. Baddley W. H.— J. Am. Chem. Soc., 1968, 90, p. 3705.
223. Beck W., Werner K.— Chern. Ber., 1973, 106, S. 868.
224. Kohout I., Valach F., Hvastijova M. a. o.— Chem. Commun., 1976, p. 903.
225. Balahura R. J., Jordan R. B.— Inorg. Chem., 1970, 9, p. 1567.
226. Balahura R. J., Jordan R. B.— Inorg. Chem., 1971, 10, p. 198.
558
227. Ford P. C.— Inorg. Chem., 1971, 10, p. 2153.
228. Sargeson A. M., Taube H.— Inorg. Chem., 1966, 5, p. 1094.
229. Courts R. S. P., Wailes P. C.— Inorg. Nucl. Chem. Lett., 1967, 3, p. 1.
230. Цивадзе А. Ю., Цинцадзе Г. В., Гонгадзе Н. П., Харитонов Ю. Я-— Координа-
ционная химия, 1975, 1, с. 1084.
231. Цивадзе А. Ю., Цинцадзе Г. В., Гонгадзе Н. П., Харитонов Ю. Я.— Координа-
ционная химия, 1975, 1, с. 1221.
232. Цинцадзе Г. В., Цивадзе Т. Л., Орбеладзе В. В.— Журнал структурной химии,
1975, 16, с. 320.
233. Цивадзе А. Ю., Харитонов Ю. Я-, Цинцадзе Г. В., Петриашвили Ж. Д.— Коорди-
национная химия, 1975, 1, с. 525.
234. Werner К.., Beck W.— Chem. Ber., 1972, 105, S. 3947.
235. Werner К., Beck W„ BohnerH.— Chem. Ber., 1974, 107, S. 2434.
236. Beck W., Werner K.— Chem. Ber., 1971, 104, S. 2901.
237. Liebig J.— Ann. chim. phis., 1823, p. 315.
238. Nef J. U.— Ann., 1894, 280, p. 334.
239. Wohler L.„ Berthmann A.— Ber., 1929, 62, S. 2748.
240. Beck W.— Z. Naturforsch., 1962, 17b, S. 130.
241. Beck W., Schuierer R.— Chem. Ber., 1962, 95, S. 3048.
242. Beck W., Feldl K.— Z. anorg. allg. Chem., 1965, 341, S. 113.
243. Beck W., Feldl K.— Z. Naturforsch., 1966, 21b, S. 588.
244. Beck WOetker C., Swoboda P.— Z. Naturforsch., 1973, 28b, S. 229.
245. Beck W., Lux F.— Chem. Ber., 1962, 95, S. 1683.
246. Кирсанов А. Б., Жмурова T.— Журнал общей химии, 1966, 36, с. 1433.
247. Beck W., Schuierer E.— Z. anorg. allg. Chem., 1966, 347, S. 304.
248. Beck W., Swoboda F., Feldl K-, Schuierer E.— Chem. Ber., 1970, 103, S. 3591.
249. Wieland H.— Ber., 1909, 42, S. 820.
250. Beck W.— Z. anorg. allg. Chem., 1964, 333, S. 115.
251. Beck W., Schuierer E.— Chem. Ber., 1965, 98, S. 298.
252. Beck W., Schorpp K-. Cetker C.— Chem. Ber., 1974, 107, S. 1371.
253. Hoyer H.~ Chem. Ber., 1961, 94, S. 1042.
254. Beck W., Schorpp K-— Chem. Ber., 1975, 108, S. 3317.
255. Bassiere.— C. r. Acad. Sci., 1938, 206, p. 1309.
256. Hendricks S. B., Pauling L.— J. Am. Chem. Soc., 1926, 47, p. 2904.
257. BrittonD., Dunitz J. D.— Acta Cristallogr., 1965, 18, p. 424.
258. Walach G., Dunai-Jurco M., Goraj J., Nemcok P.— IVth Conf. Coord. Chem., Il
Seminar on Crystallochem. Coord, and Metallogr. Compounds. Smolenice — Bratislave, 1973.
259. Chow Y. M., BrittonD.— Acta Cristallogr., 1975, B31, p. 1922.
260. Yonesawa T., Kato H., Konishi H.— Bull. Chem. Soc. Japan., 1967, 40, p. 1071.
261. Suzuki A.— J. Industr. Explosives Soc. Japan, 1953, 14, p. 142.
262. Singh K-— Acta Cristallogr., 1959, 12, p. 1053.
263. Pandey S. N.— Indian J. Phys., 1962, 26, p. 657.
264. Iqbal Z., Joffe A. D.— Proc. Royal Soc., 1967, 1, p. 302, 3549.
265. Maki A., Decius J. C.— J. Chem. Phys., 1959, 31, p. 772; 1958, 28, p. 1003.
266. Bailey R. A., Kozak S. L., Michelsen T. W., Mills W. N.— Coord. Chem. Rev.,
1971, 6, p. 407.
267. Reich P., Martin D.— Chem. Ber., 1965, 98, S. 2063.
268. Eyeter E. H., Gilette R. H.— J. Chem. Phys., 1940, 8, S. 369.
269. Muller V.— Z. anorg. allg. Chem., 1973, 396, S. 187.
270. Milller V.— Z. anorg. allg. Chem., 1976, 422, S. 134.
271. Цивадзе А. Ю., Харитонов Ю. Я-> Цинцадзе Г. В.— Журнал неорган. химии,
1972, 27, с. 2706.
272. Singh К., Banerji N. N.— J. Chem. Soc., 1965, р. 1513.
273. Beck W.— Organometal. Chem. Rev., 1971, A7, p. 159.
274. Decine I. G., Gordon D. I.— J. Chem. Phys., 1967, 47, p. 1286.
275. Харитонов Ю. Я., Цинцадзе Г. В., Порай-Кошиц М. А,— Докл. АН СССР, 1965,
160, с. 1351.
276. Chew К. F., Derbyshire W., Logan N. а. о.— J. Chem. Soc. Dalton, 1970,
р. 1970.
277. Becker W., Beck W.— Z. Naturforsch., 1970, 25b, S. 101.
278. Chew K. F., Derbyshire W., Logan N. a. o.— Chem. Commun., 1970, p. 1708.
279. Miller F. A., Baer W. K.— Spectrochim. Acta, 1962, 18, p. 1311.
280. Norbury A. H., Sinha A. I. P.— J. Inorg. Nucl. Chem., 1973, 35, p. 1211.
281. Beck W., Feldl K.— Z. Naturforsch., 1965, 20b, S. 272.
282. Ebsworth E. A. V., Mays M. J.— J. Chem. Soc., 1962, p. 4844.
283. Ripan R.~ Bull. Soc. Stiinte Cluj, 1927, 3, p. 311. Цит. Ch. Z., 1927, 98, S. 2389.
159
284. Ripan R.~ Bull. Soc. Stiinte Cluj, 1928, 4, p. 57. Цит. Ch. Z., 1928, 99, S. 2973.
285. Ripan R.— Bull. Soc. Stiinte Cluj, 1928, 4, p. 104. Цит. Ch. Z., 1928, 99.
S. 2974.
286. Moser L., Siegmann F.— Monatsh. Chem., 1930, 55, S. 18.
287. Dalietos J.— Z. anorg. allg. Chem., 1934, 217, S. 381.
288. Dorrington В. 1. F., Ward A. M.— Analyst, 1929, 54, p. 327.
289. Аналитическая химия элементов. Кобальт. М., Наука, 1966.
290. Schaeffer W.D., Dorsey W. С., Skinner D. A.— J. Am. Chem. Soc., 1957, 7y.
p. 5870.
291. Gamalek G. Einechlufiverbindungen. Berlin, Deutscher Verlag der Wissensch., 196.
292. Бубнов Д. Ф., Сухов И. П. Средства инициирования. М., Оборонгиз, 1945.
293. Twitchett Н. J.— Chem. Soc. Rev., 1974, 3, р. 209.
294. Saunders J. H., Slocombe R. J.— Chem. Rev., 1948, 43, p. 203.
295. Arnold R. G., Nelson J. A., Verbang J. J.— Chem. Rev., 1957, 57, p. 47.
296. Ozaki S.— Chem. Rev., 1972, 72, p. 457.
297. Wurtz A.— C. r. Acad. Sci., 1848, 27, p. 241; Ann., 1849, 71, p. 326.
298. Hentschel W.— Ber., 1884, 17, S. 1284.
299. Hardy W. B., Bennet R. P.— Tetrahedron Letters, 1967, p. 961.
300. Sherwin M. S., Spitz R. H.— Chem. Eng. News, 1971, p. 61.
301. Bayer 0.— Angew. Chem., 1947, 59, p. 257.
302. Благонравова А. А., Левкович Г. A.— Успехи химии, 1955, 24, с. 93.
5. ТИОЦИАНАТНЫЕ СОЕДИНЕНИЯ
Хотя тиоцианатам посвящено много работ, в том числе и обзорных [1—•
4,941], еще нет достаточной четкости в определении комплексообразующих
свойств SCN~ [5, 61, а также перспективных путей исследования и примене-
ния тиоцианатных соединений [7—9].
5.1. ПОЛУЧЕНИЕ И СВОЙСТВА ДИТИОЦИАНА
Свободный дитиоциан был получен при действии иода на тиоцианат серебг
ра в диэтиловом эфире [10]
I2 + 2AgSCN 2AgI + (SCN)2. (5.1)
Однако эта реакция не протекает до конца, и эфирный раствор тиоциа-
на содержит также иод.
Лучшие результаты были получены при замене иода на бром [11]
2AgSCN 4- Br2 2AgBr + (SCN)2; (5.2)
М (SCN)2 + Br2->MBr2 + (SCN)2 (M = Pb, Hg).
Дитиоциан можно получить также при химическом (МпО2, РЬ (СН3 •
• СОО)4 и др.) [14] или электрохимическом [12, 13] окислении тиоцианат-?
нона
Pb (СН3СОО)4 + 4HNCS -> Pb (SCN)2 + (SCN)2 + 4СН3СООН; (5.3)
2SCN~ (SCN)2 + 2ё. (5.4)
Так как дитиоциан неустойчив и легко полимеризуется, образуя твердый
оранжевый продукт (SCN)X [13, 15], для получения небольших его коли-
честв предпочтителен первый метод. Этим методом пользуются в настоящее
время для получения тиоциана. Раствор же тиоциана может быть использо-
ван для синтеза новых тиоцианатов [16].
Охлаждением раствора дитиоциана с CS2 можно получить кристаллы с
температурой плавления —3° С [10]. Водные растворы дитиоциана разлага-
ются по схеме [19]
3 (SCN)2 + 4Н2О -> 5HNCS + H2SO4 + HCN. (5.5)
С химической точки зрения дитиоциан во многом напоминает галогены.
Подобно иоду он с тиосульфатом натрия образует тетратионат
(SCN)2 + 2Na2S2O3 2NaNCS + Na2S4Oe. (5.6)
161
6 2473
Дитиоциан легко окисляет сероводород [18]
(SCN)2 4- H2S -> S + 2HNCS. (5.7а)
При этом, однако, могут образовываться и другие продукты [181
2 (SCN)2 + 2H2S S (SCN)2 + 2HNCS. (5.76)
Соединения S2 (SCN)2, Se2 (SCN)2, S (SCN)2 и Se (SCN)2 напоминают соот-
ветствующие соединения серы и селена с хлором. Их получают реакцией об-
мена в органических растворителях между S2C12, Se2Cl2, SC12 или SeCl2 и тио-
цианатами щелочных металлов или ртути [19—21]; S (SCN)2 и Se (SCN),
можно получить также по реакциям [17, 18]
4AgCN + 2S2C12 -> 4AgCl + S (SCN)2 + CN (SCN); (5.8)
3H2SeO3 + 7HNCS 3Se (SCN)2 + HCN + H2SO4 + 5H2O. (5.9)
По-видимому, перечисленные соединения имеют полимерный характер,
как, например, красное твердое вещество [S2 (SCN)2]X [21].
Полимерные соединения [SO (SCN)2]X [211 и [SO2 (SCN)2] [21, 22] анало-
гичны SOC12, SOBr2 и SO2C12. Однако они устойчивы только в растворе.
По данным Яндера [22], получившего тионилтиоцианат по реакции
NH4NCS + SOC12 NH4C1 + SO (SCN)2, (5.10)
Из концентрированных растворов выделяется аморфный полимерный про-
дукт. Эти же соединения были получены и в бензольных растворах [24].
С металлоорганическими соединениями дитиоциан реагирует с образова-
нием в большинстве случаев металлоорганических тиоцианатов
R„+iM + (SCN)2 RnMSCN + RSCN. (5.11)
По этой схеме можно получить, например, мономерные металлооргани-
ческие тиоцианаты Me3SnNCS, Ph3SnNCS [797], Ph2BNCS [798], димеры,
например [PhHgSCNl2 [799], тримерные [Et2MNCS]3 (М = Ga, In) [518],
а также полимерные металлоорганические тиоцианаты [RZnNCS]* [800],
[RCdSCNR [7981, [R2BiSCN]x [798] (R = Me, Et, Ph).
Образующиеся в результате взаимодействия дитиоциана с Rn_|_iM сое-
динения RSCN можно получить и термолизом алкоксикарбонилтиоцианатов
и изотиоцианатов RCOC (О) SCN. При этом одновременно образуются
как изо-, так и тиоцианаты. Например, при термолизе изотиоцианата
СН3СОС (О) NCS образуется 81% CH3SCN и 4% CH3NCS [8011.
5.2. ТИОЦИАНАТОВОДОРОД И ТИОЦИАНАТНАЯ КИСЛОТА
Тиоцианатоводород, или родановодород, получают при взаимодействии
сероводорода с тиоцианатами ртути, свинца. При этом получают бесцветное
вещество 99,6—99,7% чистоты и температурой плавления 4-5° С [25].
HNCS с выходом продукта около 100% образуется также при взаимодейст-
вии сухих KHSO4 и KNCS [26, 27].
Тиоцианатоводород устойчив лишь при низких температурах; выше
4-10° С полимеризуется с образованием окрашенных продуктов, которые
разлагаются при дальнейшем повышении температуры. Криоскопические
измерения в бензоле, нитробензоле и ледяной уксусной кислоте указывают
на частичную димеризацию HNCS [25].
162
Родановодород неоднократно подвергался структурному исследованию
[28—32]. Из возможных резонансных структур
H-N = C = s| о Н —N=C —F| Н—N —C=S|
наиболее приемлемой является первая; межатомные расстояния в ней
Н—N 0,9887, N—С 1,2164 и С—S 1,5605 А. Угол HNC равен 134° 59' [31].
Получение водных и неводных растворов HNCS не представляет трудно^
стей. Водные растворы кислоты могут быть получены реакцией обмена, на-
пример
Ba (NCS)2 + H2SO4—>BaSO4 + 2HNCS; (5.12)
Pb (SCN)2 + H2S PbS + 2HNCS, (5.13)
а также ионообменным методом [33—35] с использованием катионитов:
вофатита KS [33], амберлита IR-120 [34] и КУ-2 [35]. При этом получаются
растворы значительной концентрации и высокой степени чистоты.
Достаточно чистые растворы тиоцианатной кислоты умеренной концент-
рации можно получить экстракцией HNCS в 100%-ный трибутилфосфат и в
другие фосфорорганические соединения, а также в диэтиловый эфир из
подкисленных серной кислотой растворов тиоцианата аммония с последую-
щей реэкстракцией в бидистиллат [36, 37]. Однако при экстракции с увели-
чением концентрации SCN-ионов может происходить снижение степени из-
влечения HNCS в органическую фазу вследствие димеризации [38]
2SCN" (SCN)|~. (5.14)
Водные растворы HNCS устойчивы лишь при концентрациях не выше
5%. Они отличаются повышенными кислотными свойствами (ЛдИС =0,11)
[39]. Концентрирование растворов тиоцианатной кислоты приводит к ее
разложению по схемам [23, 40]
HNCS + Н2О + Н+ ->NH,k + COS;
3HNCS HCN + H2C2N2S3; (5.15)
HNCS + H2O -> COS + NH3.
Тиоцианатная кислота легко окисляется сильными окислителями
(КМпО4, Н2О2, Вг2), образуя при этом H2SO4, HCN, BrCN; мягкое окисле-
ние приводит к образованию тиоциана [12]
2HNCS + О -> (SCN)2 + Н2О. (5.16)
В отличие от цианистоводородной кислоты и ее солей тиоцианатная кис-
лота и ее соли не токсичны. HNCS присутствует в слюне человека и в луке.
5.3. ТИОЦИАНАТЫ s-МЕТАЛЛОВ И АММОНИЯ
Тиоцианаты щелочных металлов и аммония хорошо изучены; они служат
исходными продуктами для получения многочисленных простых и комплек-
сных тиоцианатов других элементов. Получены монокристаллы NaNCS
и KNCS [802].
За исключением соли лития, тиоцианаты щелочных металлов можно легко
получить в безводном состоянии. Так, безводный тиоцианат натрия образу-
ется при нагревании кристаллогидрата NaNCS • 2Н2О при 100° С. RbNCS
и CsNCS в лабораторной практике синтезируют обменной реакцией между
6
163
сульфатом щелочного металла и тиоцианатом бария. Безводный LiNCS
трудно получить из его дигидрата даже при длительном нагревании в вакуу-
ме. Его получают сплавлением смеси NH4NCS и LiOH с последующей экс-
тракцией диэтиловым эфиром. После отгонки эфира остается сильно гигро-
скопичный продукт, содержащий 98—99% основного вещества [41].
С донорным растворителем тиоцианаты щелочных металлов могут обра-
зовывать сольваты, например KNCS • 2DMFA [42], LiNCS - НМРА и
LiNCS • 2НМРА, NaNCS • НМРА и NaNCS - 2НМРА [43].
Тиоцианаты щелочных металлов термически неустойчивы. Термическое
разложение KNCS можно выразить схемами [44]
2KNCS K2S 4- (CN)a; K2S + KNCS KCN + K2S2 (желтый);
350° (5.17)
K2S2 + 2KNCSz=Jt K2s4 + 2KCN; K2S4<>2K++ 2S? (синий).
Способы получения и свойства тиоцианата аммония описаны в работе
[1]. Для синтеза тиоцианатных соединений металлов часто используют тио-
цианаты тетралкиламмония [R4N] NCS. Последние получают по обменной
реакции между неводными растворами соответствующего хлорида или бро-
мида тетралкиламмония и тиоцианата калия [68, 84] или же методом ионного
обмена с использованием анионитов в NCS-форме [84]. Тиоцианаты тетрал-
киламмония можно также получить при взаимодействии растворов HNCS
и [R4N] ОН, причем раствор последнего готовят по схеме [184]
AgOH + [R4N| X AgX + [R4N] ОН. (5.18)
Кристаллогидраты магния Mg (NCS)2 • 4Н2О и щелочноземельных ме-
таллов Са (NCS)2 - ЗН2О, Sr (NCS)2 • ЗН2О и Ва (NCS)2 2Н2О получают
при взаимодействии М (ОН)2 и MN4NCS [1] или растворением соответствую-
щих карбонатов в тиоцианатной кислоте [45].
Как и тиоцианаты щелочных металлов, эти соединения обычно применяют
Для получения двойных тиоцианатов или для синтеза тиоцианатов других
металлов, в том числе и щелочных. За исключением тиоцианата магния, со-
держащего мостиковые тиоцианат-группы, рассматриваемые соединения
других металлов имеют ионную природу [46]. Известны аддукты тиоциана-
тов магния и щелочноземельных металлов с азот- и кислородсодержащими
лигандами. Так, Mg (NCS)2 с N-окисью 2,6-лутидина образует соединение
Mg (NCS)2 • 2L, в котором координационное число магния равно 5. Органи-
ческий лиганд служит мостиком между атомами магния, а NCS-группы яв-
ляются концевыми (Mg—NCS) [803]. С Dipy и Phen из ацетоновых раство-
ров выделены М (Dipy)2 (NCS)2 (М — Mg, Са, Sr, Ва), М (Phen)2 (NCS)2
(М = Mg, Са, Ва) и [М (Phen)3] (SCN)2 (М = Sr или Ва). В соединениях ти-
па ML2 (NCS)2 SCN-группы связаны с центральным атомом через азот, а в
[М (Phen)3] (SCN)2 — имеют ионную природу [804]. Для тиоцианатов каль-
ция и бария получены криста л лосольваты с диметилформамидом Са (NCS)2 •
• 4DMFA, Ва (NCS)2 • 3DMFA [805], а для тиоцианатов магния и щелочно-
земельных металлов с уротропином: М (NCS)2 • 4Н90 • 2Urt (М = Mg,
Са), Sr (NCS)2 6Н2О 2Urt и Ва (NCS)2 • 2Urt [842]/
До сих пор попытки получить чистый Be (NCS)2 не увенчались успехом.
Аддукт тиоцианата бериллия Be (NCS)2 • 2Et2O был получен при взаимо-
действии металлического бериллия с дитиоцианом в диэтиловом эфире,
из которого выделены соединения с диоксаном Be (NCS)2 • 2Diox и пириди-
ном Be (NCS)2 • 2Ру [16]. При нагревании Be (NCS)9 • 2CH3CN можно полу-
чить [Be (NCS)2 • 0,5 CH3CN]X и [Be (NCS)2 • CH3CNR [47].
Двойные тиоцианаты бериллия типа М [Be (NCS)3 CH3CN](n — 1) •
164
• CH3CN (M = Rb, Cs) и M2 [Be (NCS)J • n CH3CN (M = K, Rb, Cs, NH4)
были получены из ацетонитрильных растворов по реакции ВеС12 с MNCS.
В этих соединениях бериллий проявляет типичное для него координацион-
ное число 4. Соединения NH4Be2 (NCS)5 • 4CH3CN и CS3Be2 (NCS)7 •
• 5CH3CN оказались соответственно смесью NH4Be (NCS)3 • 2CH3CN и
Be (NCS)2 • 2CH3CN, а также CsBe (NCS)3 • mCH3CN и Cs [Be (NCS)4] •
• (5 — n) CH3CN [48].
5.4. ПОЛУЧЕНИЕ И СВОЙСТВА ТИОЦИАНАТНЫХ
СОЕДИНЕНИИ р- И б10-ЭЛЕМЕНТОВ
5.4.1. Тиоцианаты металлов
II побочной подгруппы
Простые тиоцианаты металлов этой подгруппы могут быть получены
как обменными реакциями
ZnSO4 + Ba (NCS)2 BaSO41 + Zn (NCS)2; (5.19)
Cd (С1О4)2 + 2KNCS 2КС1О41 + Cd (SCN)2; (5.20)
Hg (NO3)2 + 2NaNCS 2NaNO3 + Hg (SCN)2 |, (5.21)
так и при взаимодействии тиоцианатной кислоты с оксидом (гидроксидом)
соответствующего металла.
Zn (NCS)2, Cd (SCN)2, Hg (SCN)2 — бесцветные кристаллические вещест-
ва, растворимость которых в воде заметно снижается от цинка к ртути. Они
имеют сравнительно низкие температуры разложения. Так, Hg (SCN)2 на-
чинает распадаться уже при 150° С. Разложение протекает в несколько ста-
дий с образованием Hg (CN)2 в качестве промежуточного продукта и выде-
лением CS2, (CN)2 и N2 [812].
В то время как тиоцианат ртути имеет концевые SCN-группы, тиоциана-
ты Zn и Cd содержат мостиковые SCN-группы [46, 49]. В случае присоедине-
ния к тиоцианату цинка двух монодентатных органических лигандов происхо-
дит разрыв мостиков с образованием соединений, имеющих тетраэдрическую
структуру с двумя координированными молекулами L и координированны-
ми через атом азота NCS-группами [50—55]: Zn (NCS)2 • 2L (L = Ру и
его производные [51, 54]), DMSO [53], RNH2 (R = Me, Et, Pr, Bu) [501,
HMPA [55]. Однако в тетраэдрическом соединении Zn (NCS)2 • L (L =
= Et2N • CH2 • CH2 • NH2) предполагается Zn—SCN-связь, что необычно
для тиоцианатов цинка [56]. С пропиоамидом тиоцианат цинка образует
Zn (NCS)2 • 2L (часть NCS-групп — мостиковые) и Zn (NCS)2 • 4L (конце-
вые NCS-группы) [806]. Концевые NCS-группы содержат комплекс тиоциана-
та цинка с 4-винилпиридином Zn (NCS)2 • 4L [849] и с y-Pic: Zn (NCS)2 •
• 4y-Pic [558].
В аддуктах тиоцианата цинка с полидентатными лигандами в зависимо-
сти от их числа и химической природы NCS-группа может быть концевой,
мостиковой или внешнесферной. Так, в Zn (NCS)2 • 2L (L = нитробензгид-
разиды, Phen, амид никотиновой кислоты) NCS-группы являются концевы-
ми [807, 808, 816], ас тиоморфолином и тиоазолидином предполагается ион-
ная природа SCN [809]. Концевые изотиоцианатные группы (Zn—NCS-коор-
динация) имеет тиоцианат цинка и в соединениях с двумя диаминами [810].
С полиаминами, например с trien, возможно одновременное наличие как
165
координированной концевой, так и внешнесферной SCN-групп: [М (trien)
• NCS] SCN (М = Zn, Cd, Hg) [811].
В некоторых случаях из-за сложности ИК-спектра затруднительно оце-
нить функцию SCN-групп в аддуктах тиоцианатов металлов с органическими
лигандами, например в соединениях М (NCS)2 • 2L (М = Zn, Cd; L = ди-
этилникотинамид [815]), Cd (NCS)2 • 2N (L = пара-нитробензоилгидразин)
[816]. В Cd (NCS)2 • 2L (L = орто- и пара-метоксибензоилгидразин), по-
видимому, NCS-группы расположены в цис-положении друг к другу, а кад-
мий имеет октаэдрическое окружение [335].
В аддуктах Cd (SCN)2 • 2L полимерная структура с мостиковыми SCN-
группами обычно сохраняется [51, 53, 57, 349, 809], но в имеющем тетраэд-
рическое строение Cd (NCS)2 • 2НМРА NCS-группы функционируют как
концевые [55]. Концевыми (Cd—NCS-связь) являются тиоцианатные группы
в комплексах Cd (NCS)2 • 2L с диаминами [810].
Мостиковые SCN-группы обнаруживаются и в некоторых соединениях
ртути, например в 2Hg (SCN)2 • L, Hg (SCN)2 • L (L = Py, Ph3P, Ph3As
и др.). Приведенные соединения являются димерами [51, 58, 59, 807]. Тет-
раэдрические димеры образуют также соединения типа М (SCN)2 •
L : Cd (SCN)2 • L (L = изоксазол) [60], Hg (SCN)2 • 3-CNPy [52] и, возмож-
но, Hg (SCN)2 • L (L - нитробензоилгидразины) [816]. С бидентатными ли-
гандами N,N'-диоксидом 2,2'-дипиридила тиоцианаты кадмия и ртути об-
разуют мономерные тетракоординационные комплексы с М—SCN-связями
[61]. Тетракоординационную мономерную структуру с псевдотетр аэдриче-
ским окружением атома ртути (Hg—SCN-связь) образует тиоцианат ртути
с N-оксидом лутидина [803], пиперидином [813], этилендиамином [811].
Но в соединениях с trien, Ме6 trien одна SCN-группа является ионной [811].
Некоторые аддукты цинка и кадмия М (NCS)2 • 2L, например с изохи-
нолином, термически устойчивы и могут быть использованы для гравиметри-
ческого определения этих металлов [62].
При взаимодействии простых тиоцианатов металлов подгруппы цинка
с тиоцианатами щелочных, щелочноземельных металлов и аммония образуют-
ся координационные соединения с комплексными анионами [М (NCS)3]“,
[М (NCS)4]2— и даже [М (NCS)614~. Пентатиоцианатные комплексы для этих
металлов не характерны. Однако пентакоординационные соединения, содер-
жащие SCN-группы, известны. К примеру, с тетрадентатными азотсодержа-
щими лигандами tren и Ме6 tren получены соответственно [Hg (tren) SCN]
- SCN и [Hg (Me6 tren) SCN] BF4 [63].
Тритиоцианатные соединения цинка и кадмия исследовались методом
ИК-спектроскопии [49, 64]. При этом установлено наличие мостиковых тио-
цианатных групп в соединениях обоих металлов. Для цинка предположи-
тельно тетраэдрическое окружение; структуру тритиоцианатов кадмия пред-
сказать затруднительно. По-видимому, в них проявляется октаэдрическая
конфигурация по аналогии со структурно исследованными соединениями.
Даже в комплексах кадмия типа Cd (SCN)2 • 2L центральный атом имеет
октаэдрическое окружение (табл. 5.12).
Тритиоцианаты ртути так»е имеют тетраэдрическое строение. Данные о
структуре MHg (SCN)3 (М = К, Cs, NH, Me4N) приведены в табл. 5.12.
Соли калия, рубидия и аммония изоморфны и имеют характер двойной со-
ли [65].
Наибольшее число работ посвящено тетратиоцианатам цинка, кадмия
и ртути. Все соединения этс;с типа можно ориентировочно разделить на
две группы. Первую групп) ((ставляют комплексы, в которых в качестве
внешнесферных катионов выступают ионы щелочных и щелочноземельных
166
металлов аммония и им подобных. Вторая группа соединений содержит в
своем составе ионы переходных металлов.
В комплексах первой группы ионы цинка и ртути, как правило, имеют
характерное для них тетраэдрическое окружение, что подтверждается ре-
зультатами рентгеноструктурных исследований. Тиоцианатная группа коор-
динируется с атомом ртути посредством серы (табл. 5.12), в комплексах цин-
ка — через азот [66, 67, 549, 851].
Более сложными являются тетратиоцианатные соединения второй груп-
пы, которые представлены комплексами ртути и частично кадмия. В них
в качестве ионов-комплексообразователей выступают два иона: ртути (или
кадмия) и переходного металла. В этом случае можно ожидать, что тиоци-
анатная группа будет играть роль бидентатного лиганда, образуя мостики
между ионами. Большая группа таких соединений исследована в работах
[66, 68, ПО]. Было показано, что в соединениях MHg (SCN)4 (М = Си,
Zn, Mn, Fe, Со, Ni) и CoCd (SCN)4 тиоцианатная группа действительно имеет
бидентатный характер. Определены параметры решетки и установлен изо-
морфизм для пар соединений MnHg (SCN)4, FeHg (SCN)4 и ZnHg (SCN)4,
CoHg (SCN)4 [110].
Интересные данные получены для аддуктов CoHg (SCN)4 с основаниями
Льюиса (Ру, Diox, Ph3P, Dipy, Phen, en и др.) [69]. Присоединение органи-
ческих лигандов к этому комплексу существенно изменяет характер струк-
туры последнего, состоящей из тетраэдрических узлов CoN4 и HgS4
(табл. 5.12). В аддуктах MHg (SCN)4 • 2Thf и MHg (SCN)4 • 2Py (M = Cu, Zn,
Cd, Mn, Fe, Co, Ni) ион так называемого внешнесферного металла имеет ок-
таэдрическое, а ион ртути тетраэдрическое окружение. Но в аналогичных
по составу аддуктах, содержащих трифенилфосфин — MHg (SCN)4 • 2Ph3P
(М = Zn, Cd, Co), ионы металлов M2+ и Hg2+ имеют четырехкоординацион-
ное, а в NiHg (SCN)4 • 4Ph3P ртуть имеет шестикоординационное окруже-
ние [71]. Тетраэдрическое окружение имеет атом ртути в мономерных комп-
лексах CuHg (SCN)4 • 2L (L = никотинамид, 2-аминопиримидин, Ph3P,
Ph3As) и FeHg (SCN)4 • 2Ph3P с мостиковыми и концевыми SCN-группами.
При этом лиганды Ph3P и Ph3As координируются с атомом ртути [814].
Для тетратиоцианатов кадмия и цинка MZn (NCS)4 и MCd (SCN)4 (М =
= Со или Ni) также получены аддукты с основаниями Льюиса: Thf, Ру,
Phen, Dipy и др. [72]. Согласно ПК-спектрам и магнитным измерениям,
предложены структуры этих соединений, например [СоРу6] [М (CNS)4],
[Со (Dipy)3l [М (CNS)4] или [Со (Phen)3] [М (Phen) (CNS)4] [72].
Для некоторых из координационных соединений второй группы опреде-
лен тип решетки. Соединение ZnHg (SCN)4 имеет тетрагональную (а = 11,06;
с = 4,43 A), CuHg (SCN)4 — орторомбическую (а = 7,65; b — 9,02; с =
= 15,17 А) [68], a CoHg (SCN)4 • 2Ру — моноклинную (а — 11,46; Ь =
= 13,24; с = 14,50 А; [3 = 105,5°) [69] решетки.
Гексатиоцианатоцинкаты и гексатиоцианатокадматы исследовались с
помощью ПК- и ЯМР-спектроскопии [67, 73, 74]. В солях цинка тиоцианат-
ная группа координируется через азот [74], а в гексатиокадматах, за ис-
ключением Cs4 [Cd (NCS)61, посредством серы [73].
5.4.2. Тиоцианаты элементов III главной подгруппы
Первая попытка получить тиоцианат бора была предпринята в 1908 г.
[751, однако эти данные не подтвердились [76]. Позднее синтез В (NCS)3
был осуществлен по реакции [77]
ВС13 + ЗК (Na) SCN ЗК (Na) Cl + В (NCS)s. (5.22)
В (NCS)3 — бесцветная жидкость, кипящая при 92° С (0,1 мм рт. ст.).
Выделены твердые аддукты В (NCS)3 • Ру, В (NCS)3 • CH3CN, В (NCS)3 •
• PhCN, В (NCS)3 • Et3N и В (NCS)3 • PhCH2CN [77].
Для алюминия посредством обменной реакции
Д12 (SO4)3 + ЗВа (NCS)2 -> 3BaSO4 | + Al (NCS)3 (5.23)
получены кристаллогидраты: Al (NCS)3 - ЗН2О (7разл > 100° С) и
Al (NCS)3 • 6Н2О [78]. Во избежание гидролиза [781 кристаллизация тиоциа-
ната алюминия проводилась в вакууме.
Синтезированы также аддукты Al (NCS)3 Phen и Al (NCS)3 • 2cc-Pic,
являющиеся электролитами типа 1 : 1 в нитробензоле, и розовый (из-за сле-
дов железа) Al (NCS)3- 3DMSO [78]. Розенгеймом и Коном [79, 342] получены
соли М3 [Al (NCS)4] • 4Н2О (М = Na, К, NH4). Соль К3 [Al (NCS)6] • CH3CN
получена из ацетонитрильного раствора по обменной реакции между А1С13
и KNCS [80].
Более полно в литературе представлены тиоцианаты галлия и индия.
Так, реакцией обмена между GaBr3 и KNCS в ацетонитриле был получен
К [Ga (NCS)4] [81]. Более сложный продукт К3 [Ga (NCS)6] • 3CH3CN, по
данным рентгенофазового анализа, оказался смесью К [Ga (NCS)4] CH3CN
и KNCS. Из других соединений галлия на основе его тиоцианата известны
соединения щелочных М [Ga (NCS)J [82] и щелочноземельных металлов
М [Ga (NCS)J2 • «Н2О [45], [РуН] [Ga (NCS)J [83], [R4N] [Ga (NCS)J (R =
= Me, Et, Bu) [84—86] и M3 [Ga (NCS)J (M = K, Rb, Cs [82], Me4N, Et4N,
Bu4N [84—86] и PyH [83]).
Получены также аддукты Ca (NCS)3 • 3H2O [87], Ga (NCS)3 • 3L (L =
= Py, DMSO, V2 Dipy, V2 Phen), Ga (NCS)3 - 2Diox, Ga (NCS)3 2,5 •
• CH3OH и др. [87,88,820]. Индивидуальность соединений подтверждена рент-
генографически [88]. Соединения тиоцианата галлия с 2,2'-дипиридилом или
1,10-фенантролином другого состава приведены в работе [89].
Все указанные соединения галлия можно получать как из воды, так и из
неводных растворов; они представляют собой белые кристаллические вещест-
ва. По данным физико-химических исследований, в этих соединениях
галлий проявляет координационное число 4 или 6. Координация NCS-груп-
пы с металлом, как правило, осуществляется через азот. В случае гексг тио-
цианатов, по данным ИК-спектров и дериватографического анализа, пред-
полагается, что по крайней мере две NCS-группы в [Ga (NCS)6]4~ менее проч-
но связаны с металлом [84].
Термическая устойчивость всех синтезированных соединений невысока.
Для тетра- и гексатиоцианатогаллатов тетраалкиламмония характер разло-
жения не зависит от природы внешнесферного катиона. Температуры плав-
ления (или разложения) находятся в пределах 80—230° С, причем более
устойчивыми являются соли тетраметил аммония [84].
Что касается тиоцианатных соединений индия, то о них первые сведения
приведены в работе [90]. Из метанольных растворов реакцией обмена между
перхлоратом индия и тиоцианатом калия были выделены In (NCS)3 • ЗРу,
Kin (NCS)4 • 2Ру, К3 [In (NCS)el • СН3ОН и К3 [In (NCS)6] • 4 Diox. Од-
168
нако детальному исследованию эти соединения не подвергались. Позднее
In (NCS)3 был получен из спиртовых растворов смешиванием 1пС13 и NaNCS
с последующей экстракцией продукта диэтиловым эфиром (выход 90%),
а также посредством реакций [91]
3Hg (SCN)2 + 2 In 2In (NCS)3 + 3Hg; (5.24a)
In (CN)3 + 3S In (NCS)3. (5.246)
Из водных растворов это соединение можно получить в результате реак-
ции Ba (NCS)2 с In2 (SO4)3 [92]. Прочность связи In — NCS довольно высо-
кая, поэтому в водном растворе In (NCS)3 практически не подвергается гид-
ролизу. Белый кристаллический In (NCS)3 имеет полимерное строение с мос-
тиковыми NCS-группами; решетка кубическая с а = 12,00 А [91].
На основе тиоцианата индия были получены многочисленные координа-
ционные соединения с азот-, кислородсодержащими и другими лигандами:
In (NCS)3 • 3L (L = Ру, y-Pic, DMSO, DMA A, Ph3PO, Ph3P, tu, V2 Dipy,
x/2 Phen) [92] и азолами [820]. Все соединения представляют собой белые
(или с розовым оттенком — следы железа) кристаллические вещества, не
проводят ток, что свидетельствует о координационном числе индия в них,
равном 6.
Сведения о тетратиоцианатных соединениях индия в литературе практи-
чески отсутствуют, кроме М [In (NCS)4 (CH3CN)2I (М = К, Na, NH4) [821].
Значительно более полно представлены гексатиоцианатные комплексы с
большими катионами, легко выделяемые из водных растворов. Получены
соли катионов: Me4N ', Et4N+, Bu4N+, Et3NH+ и Me3PhN+ [93—961, а так-
же [Ph4As]2 [In (NCS)5] и [Ph3PCH2Ph]2 [In (NCS)5] [94]. По данным KP-
спектров, предполагается, что ион [In (NCS)5]2~ имеет моноядерную струк-
туру (Dsh- или С^-симметрию) с координационным числом 5. Об этом гово-
рит частота колебания связи In—N, равная для пентакоординационных
комплексов 270—285 см-1, в то время как для октаэдрических гексатиоциа-
натов индия (Oh-симметрия) v (InN) 250 см“' [94].
Следует отметить, что и галлий может образовывать соединения, содер-
жащие пять NCS-групп, например [РуН] [Ga (NCS)51 [83], но никаких
данных о структурных особенностях таких соединений в литературе не име-
ется.
Крайне малочисленны сведения о тиоцианатных соединениях TI (I) и
Т] (III). Предполагается, что в тиоцианате таллия (I) преобладающей явля-
ется ионная связь, причем тиоцианатная группа обращена к иону таллия ато-
мом азота, т. е. Т1—NCS [96]. Это предположение находится в согласии с ре-
зультатами работы [97], где показано, что в кристаллическом T1NCS доля
ионности составляет 37%. Именно поэтому двойные тиоцианаты таллия в
кристаллическом виде получить довольно трудно. Известны только некото-
рые соединения щелочных металлов, например КТ1 (NCS)2 [1], CsTl4 (NCS)5
981 и другие.
Тиоцианат таллия (III) не получен, вероятно, из-за большого редокс-
потенциала пары Т13+/Т1 . При добавлении KNCS к раствору соли таллия
(III) в результате окислительно-восстановительной реакции образуется
T1NCS 199]
ЗТ1С13 + 4KNCS + 4Н2О -> T1NCS + 4КС1 + 5НС1 + H2SO4 + HCN.
(5.25)
169
Тем не менее известны соединения R2T1NCS (R = Me, Et, Ph) [100.
1011, PhTl (SCN)2 [102, 103] и Cp2TlNCS [104], TI (SCN) X2 - 3H2O (X -
= Br, I) [971.
5.4.3. Тиоцианаты элементов iV главной подгруппы
Для углерода тиоцианат пока не известен. Для кремния же получен
Si (NCS)4 в виде бесцветных призм [105] по обменной реакции SiCl4 с
Hg (SCN)2 или AgSCN в неполярном растворителе. Его можно также полу-
чить из ацетонитрильного раствора при взаимодействии SiCl4 и NH4NCS
[106, 1071. Из ацетонитрильных растворов получены CH3Si (NCS)3, (СН3)2 Si •
• (NCS)2 и (СН3)3 SiNCS [1071. Метильные производные являются жидкос-
тями, а тетратиоцианат кремния — твердое вещество.
Германий образует твердое соединение Ge (NCS)4 [107] и ряд соединений
типа R„Ge (NCS)4_rt [108, 109, 111, 112]. Из ацетонового раствора получен
ряд аддуктов Ge (NCS)4 • 4L (L = Dipy, Phen и др.), Ge (NCS)4 2L
(L = y-Dipy). Соединения первого типа можно рассматривать как [GeL3l •
• (SCN)4 • L [ИЗ]. С y-Dipy предполагается образование димера [Ge2L4 •
• (NCS)4) (SCN)4 [113]. Получены соединения тиоцианата германия с основа-
ниями Шиффа: Ge (NCS)4 • 4L (L = шиффово основание), в которых содер-
жатся как координированные (Ge—NCS), так и свободные SCN-груп-
пы [823].
Для олова (II) известны как простой Sn (NCS)2, так и координационные
тиоцианаты. Sn (NCS)2 можно получить по реакции между SnSO4 и Pb (SCN)3
в воде [1] или SnSO4 и NaNCS [114]. Соединение имеет полимерную структуру
с мостиковыми NCS-группами и относится к триклинной сингонии с парамет-
рами а = 5,68; b = 4,97 и с = 10,30 А; а = 74,0°; В = 103,5° и у = 73,5°
[114].
Синтезированы аддукты Sn (NCS)2 • L (L = Py, Ph3PO) и Sn (NCS)2 • 2L
(L = Py, DMSO, Ph3PO), [115. 1161, из которых более стабильны соединения
состава Sn (NCS)2 • L, так как второй лиганд связан с атомом олова слабее.
Известны также три- и тетратиоцианатостаниты (И): MSn (NCS)3 (М = Cs,
Me4N, Et4N и РуН) [114, 1161 и K2Sn (NCS)4 • СН3ОН [1171.
Тиоцианат олова (IV) можно получить окислением олова тиоцианом,
растворенным в сероуглероде или диэтиловом эфире,
Sn ф- 2 (SCN)2 -> Sn (NCS)4. (5.26)
Из эфирного раствора выделены Sn (NCS)4 • 2Et2O [101 и Sn (NCS)4
[1071.
Из бензольного или этанольного растворов при взаимодействии Ph2SnCl2
с AgSCN или NaNCS получены Ph2Sn (NCS)2 и аддукты Ph2Sn (NCS)2 •
• Dipy (или Phen) и Ph2Sn (NCS)2 • 2DMSO, в которых олово имеет коорди-
национное число 6 [1191. Аналогичного типа аддукты с Phen, Dipy, Ру,
Ph3PO, Ph3AsO и другими лигандами описаны в работах [118, 121, 122].
Все соединения олова рассмотренного типа являются изотиоцианатами.
Bu2Sn (NCS)2 и Ph2Sn (NCS)2 в твердом состоянии являются полимерами,
а бутильные и фенильные группы находятся в транс-положении. Транс-
положение этих групп сохраняется и в Bu2Sn (NCS)2 • L (L = Dipy, Phen).
В аналогичных по составу фенильных производных Ph-группы находятся
в цис-положении. Влияние типа радикала сказывается и на структуре соеди-
нений с оксихинолином: для Bu2Sn (NCS) (Ох) предполагается мостиковый
димер; в фенильном производном олово имеет координационное число 5
170
[122] . Установлено образование аддуктов R3SnNCS (R = Me, Et, Pr, Bu,
Ph). В присутствии третичных аминов и фосфинов R3SnLCS в большинстве
случаев диспропорционирует с образованием R4Sn [8241.
Белые кристаллы К2 [Sn (NCS)6] получены обменной реакцией между
SnCl4 и KNCS в ацетонитриле [825]. В этой же работе сообщается о получе-
нии координационных соединений Со [Sn (NCS)6] и аддуктов на его основе —
Со [Sn (NCS)61 • 4L (L =Ру, никотинамид, этилникотинат) и Со [Sn (NCS)J •
• 6L (L - никотинамид, CNPy, гидразид изоникотиновой кислоты, г/2 еп).
В К2 [Sn (NCS)6] реализуется изотиоцианатная структура, а Со [Sn (NCS)61
имеет как мостиковые (Со—SCN—Sn), так и концевые (Sn—NCS) SCN-
группы. При присоединении органического лиганда происходит разрыв
мостиков с образованием тетраэдрического или октаэдрического ионов
[CoLJ2+ и октаэдрического [Sn (NCS)6]4 [825].
Из тиоцианатных соединений свинца наиболее детально изучен Pb (SCN)2
и его аддукты с 2,2'- и 4,4'-дипиридилом Pb (SCN)2. Dipy или 2Pb (SCN)2 •
• y-Dipy, в которых тиоцианатная группа координируется с ионом свинца
через серу [123]. Структурные особенности Pb (SCN)2 рассмотрены ниже
(см. табл. 5.12). В кристаллическом состоянии выделен Ке [Pb (CNS)8] •
• 2Н2О [98].
5.4.4. Тиоцианаты элементов V—VII главных подгрупп
N (NCS)3 не получен, а тиоцианатные соединения трехвалентного и пяти-
валентного фосфора Р (NCS)3, РО (NCS)3, PS (NCS)3, PhPO (NCS)2 и PhPS •
• (NCS)2 описаны в работах [106, 107]. Значительный интерес представля-
ют также три- и тетрамер [PN (NCS)2]„, которые получены реакцией обмена
из ацетоновых растворов
[PNC12U 4- 2nKNCS -> [PN (NCS)2]n + 2nKCl (5.27)
и имеют циклическое строение [124, 125].
As (NCS)3 можно получить либо взаимодействием мышьяка с тиоцианом
в сероуглероде [10], либо реакцией обмена между AsCl3 и NH4NCS в жидкой
SO2 при —25° С [106]. При этом образуется белое кристаллическое вещество;
выход чистого продукта 69%. Получен R3As (NCS)2 (R = Me, Ph) [798].
Попытка получить Sb (NCS)3 путем взаимодействия стибия и тиоциана
в диэтиловом эфире не привела к успеху [10]. Для пятивалентной сурьмы,
как и для таллия (III), удалось получить лишь соединения R3 Sb (NCS)2
(R = Me, Ph) [130, 131, 798]; в них тиоцианато-группа связана с металлом
посредством азота.
Из соединений этой группы наиболее изучены тиоцианаты висмута.
Получены соединения, содержащие от трех до шести SCN-групп. Кроме
Bi (SCN)3 и его гидрата [126, 127], описаны [Bi (DMFA)3 (SCN)3],
[Bi (HMPA)3 (NCS)3] и [BiL3l (SCN)3 (L = Dipy, Phen) [128, 178], в кото-
рых проявляется характерное для висмута координационное число 6. По-
следнее также реализуется в пентатиоцианатовисмутитах, содержащих коор-
динируемую молекулу монодентатного органического лиганда, например
в солях типа М [Bitu (SCN)5] (М = Mg, Са, Sr или Zn) [129].
Систематическое исследование тетра- и гексатиоцианатовисмутитов про-
ведено в работах [132—137]. Установлено, что М [Bi (SCN)4] для Rb и Cs
изоструктурны; они относятся к пространственной группе Р24242 и имеют
следующие параметры кристаллической решетки: Rb [Bi (SCN)41: а == 7,65;
b = 11,24; с = 6,52 A; Cs [Bi (SCN)J: a = 7,88; b = 11,33; c = 6,57 A [1381.
171
Из гексатиоцианатовисмутитов получены: М3 [Bi (SCN)6] (М — Li, Na, К,
NH4, Me4N, Et4N, Bu4N) [126, 132, 1391, M3 [Bi (SCN)6]2 • nH2O (M = Mg,
Ca, Sr, Ba, Mn) [1351, а также соли с двумя одновалентными внешнесферны-
ми катионами типа М2М' [Bi (SCN)6] [133, 137] и М'М" [Bi (SCN)6] • nH2O
[136]. Для солей последних типов исследована зависимость степени гид-
ролиза и растворимости от природы внешнесферного катиона. Устойчивость
соединений с двумя катионами щелочных металлов зависит от разности их
ионных радиусов [137]. Замечено, что легко получить соединения, содержа-
щие один катион с малым и два с большим радиусом, например Cs2Na [Bi •
• (SCN)6], но не удалось получить соли с обратным соотношением катио-
нов [133]. Сведения о более ранних работах по синтезу тиоцианатовисмути-
тов представлены в монографии Вильямса [1].
Что касается тиоцианатных соединений халькогенов, то о них упомина-
лось выше. Из галогенидных соединений тиоциана в твердом виде выделены
(SCN) С1 и (SCN) С13, которые гидролизуют с образованием HNCS, H2SO4
и других продуктов [1401. В водных растворах тиоцианатов щелочных ме-
таллов могут образовываться комплексы I (SCN)7* [141] и I2 (SCN)“ [1421.
Согласно [142], продуктом взаимодействия 12 и SCN~ является комплекс
с переносом заряда I2 (SCN)-, а не 1 (SCN)y. Константа устойчивости
I2 (SCN)- при 25° С равна 53,3 [142].
5.5. ТИОЦИАНАТЫ ПЕРЕХОДНЫХ МЕТАЛЛОВ
5.5.1. Тиоцианаты металлов i побочной подгруппы
Тиоцианаты меди (1) обычно получают восстановлением солей меди (II)
в присутствии тиоцианата щелочного металла
2CuSO4 + 2NaNCS + SO2 + 2Н2О -> 2CuSCN + Na2SO4 ф- 2H2SO4.
(5.28)
CuSCN растворяется в растворах тиоцианатов щелочных и щелочнозе-
мельных металлов с образованием координационных соединений, например
Cs [Си (SCN)2], Na3 [Си (SCN)41 • 4Н2О и др. [98].
CuSCN образует координационные соединения с азот-, фосфор-, серосо-
держащими и другими лигандами. Эти соединения типа CuSCN nL [1431
имеют, по-видимому, полимерный характер за счет связывания атомов меди
мостиковыми SCN-группами. Мостиковая функция SCN-группы, вероятно,
проявляется и в соединениях тиоцианата меди (1) с тиосемикарбазонами:
в комплексах CuSCN • L и CuSCN • R • Н2О (L — бензальдегидтиосемикар-
базон, R == диметиламинобензальдегидтиосемикарбазон) [144]. Первое из
них является диамагнитным, второе — парамагнитным. Тригональная струк-
тура с S- и N-координированными мостиковыми SCN-группами реализует-
ся в аддукте CuSCN • L (L = метил изоникотинат) [940].
Представляет интерес метод получения тиоцианата меди (I) из ее азид-
ных производных [146]. Реакции протекают по схемам
Си (Ph3P)2N3 + CS2 Си (Ph3P)2 NCS + S + N2 (5.29)
ИЛИ
2Cu (Ph3P)2 N3 + CS2 CuSCN + Си (Ph3P)2 NCS + N2 + PSPh3.
(5.30)
172
Этим методом можно получить и тиоцианат серебра. В работах [145,
146] описаны также CuSCN 2L (L = MePh2P, Ph2 (МеО) Р, Ph2 (EtO) Р,
Ph2 (С6Н4СН3О) Р). Для соединений CuSCN • 2L предполагается димерное
строение с двумя мостиковыми SCN-группами. Димерное строение имеет
соединение (CuSCN)2 - 3 (Ph2PC = CPPh2), но мостиками служат три моле-,
кулы органического лиганда [529].
Тиоцианаты меди (II) в литературе представлены более полно, чем сое-
динения меди (I). Простой тиоцианат меди неустойчив, но на его основе полу-,
чено много координационных соединений, в которых медь проявляет коор-
динационные числа 4, 5 и 6. При этом реализуются структуры квадрата, как,
например, в комплексах Cu (NCS)2 • 2L (L = хинолин, производные пири-
дина [147, 849], имидазол и его производные [147, 148]), тригональной бипи-
рамиды или квадратной пирамиды. Тетраэдрическую конфигурацию в этих
соединениях обнаружить не удалось. В то же время искаженно-тетраэдри-
ческое окружение иона меди возможно, например, в [CuxZni—х] [Hg (SCN)J,
что подтверждается при сопоставлении спектра отражения этого соедине-
ния со спектром тетратиоцианатокупритов (II), имеющих тетрагонально-
октаэдрическое строение [68].
В табл. 5.12 приведены структурные данные соединений меди (II) с пи-
ридином, аммиаком, этилендиамином и другими аналогичными лигандами.
Некоторые из них заслуживают детального рассмотрения. Так, соединения
[Cu (NH3)2 (NCS)2] и [СиРу2 (NCS)21 существуют в ос- и ^-модификациях
[149, 150], различающихся некоторыми свойствами. Например, магнитные
моменты а- и |3-[Cu (NH3)2 (NCS)2] равны соответственно 1,86 и 1,82 М. Б.
при 21° С и подчиняются закону Кюри — Вейса. Сравнение электронных и
ЭПР-спектров указывает на большую степень искажения тетрагональной
бипирамиды, особенно в экваториальной плоскости, в случае Р-изоме-
ра [149].
Интерес представляют комплексы [Cu (dppa) (CNS)2] и [Cu (tripyam) •
• (CNS)2] [151]. Каждый из них имеет потри модификации. Модификациям
[Cu (tripyam) (CNS)2] присуща темно-зеленая, желто-зеленая и коричневая
окраска. Подобное явление характерно и для комплекса с dppa. Наличие
нескольких модификаций у рассматриваемых комплексов связано с различ-
ным способом присоединения SCN-группы к меди. Взаимные превращения
этих изомеров зависят от температуры и природы растворителя.
Заслуживает внимания также соединение [Cu2 (Dben)4 (SCN) (C1O4)J •
Cu
• C1O4, в котором предполагается N=C=S<^ -мостик [152]. В соедине*
Си
ниях Си (Dben)2 (NCS)A (А = NO3 или Br) нитрат-ион входит во внутреннюю
сферу комплекса, представляющего собой искаженный октаэдр, а бромид-
ион — во внешнюю, и катион [Cu (Dben)2 NCS]+ имеет квадратно-пирами-
дальную конфигурацию. Такую же конфигурацию имеет катион [CuPn2 •
- (NCS)]+ комплекса CuPn2 (NCS)2 в DMFA [153, 157]. Координационное
число 5 (квадратная пирамида) медь имеет в катионных комплексах, которые
образуются при диссоциации соединений [Cu (trien) SCN] SCN или
[Cu (Ме6trien) NCS] C1O4 в метаноле и нитрометане [153, 157], а также в ди-
ядерном соединении Cu2L (SCN)2C1O4 (L = тетрадентатный азотсодержащий
лиганд) [154].
Типичным представителем пятикоординационных тиоцианатов меди (II)
является [Cu (tren) NCS] X (Х= SCN, BPh4) [155, 8271, катион которого име-
ет форму тригональной бипирамиды. Структурные данные [Cu (tren) NCS] •
173
• SCN [155] приведены в табл. 5.12. Атом меди в димере [Cu2 (tren)2 (NCS)21 •
• (BPh4)2 также имеет структуру тригональной бипирамиды, поскольку
димеризация обусловлена образованием связей NCS...HN, а не за счет
мостиковых SCN-групп [826]. Аналогичное окружение атома меди (II)
наблюдается также в комплексе [Си (NH3)2 Ag (SCN)3], который имеет почти
одинаковый электронный спектр отражения и значение магнитного момента
с таковыми для структурно изученного [Си (tren) NCS] SCN [156]. Триго-
нально-бипирамидальное окружение для атома меди предполагается в
Си (Dipy)2 (NCS)2 [817].
Координационное число 5 для меди (II) предполагается также в соеди-
нениях [Си (dpt) (NCS)2] [157] и [Си (Et2en)2 NCS] С1О4 [158]. Если послед-
ним двум соединениям предположительно приписывается форма квадратной
пирамиды, то комплексу [Си (Me6trien) NCS]+ — тригонально-бипирами-
дальная конфигурация. Однако известно, что [Си (trien) NCS]+ имеет форму
квадратной пирамиды (табл. 5.12).
Координационное число 6 для атома меди реализуется в координацион-
ных соединениях тиоцианата меди (II) с Phen, Dipy и аминами: CuDipy •
• (NCS)2, Си Phen (NCS), [817, 818], Си (Phen), (NCS)2 [808], Си (RNH2)2 •
• (NCS)2 (R = Me, Pr, Bu) [8171; октаэдрическое окружение с мостиковыми
SCN-группами имеет атом меди в димерном соединении [CuL2 (NCS)2]2
(L — N-оксид 2,6-лутидина) [803]. Из диметилформамида выделено соедине-
ние тиоцианата меди (II) с производными меламина CuL (NCS)2 [819].
Для серебра известны многочисленные координационные соединения,
например одно- и полиядерные тиоцианатные комплексы Cs [Ag (SCN)2],
Cs2 [Ag (SCN)3] 2H2O [98], Cu [Ag (SCN)21 и [Cu (NH3)4] [Ag (SCN)3J [164],
Ba [Ag2 (SCN)4] • 2H2O [98] и AgSCN nL [159—161], где n = 1 или 2. Судя
по результатам исследования структур AgSCN и других соединений, ион
серебра в них чаще всего проявляет координационное число 4 и имеет тетра-
эдрическое окружение (табл. 5.12). В некоторых соединениях серебро имеет
координационное число 2. Вероятно, такие комплексы имеют линейное строе-
ние, так как при их образовании используются sp-гибридные орбитали,
например в AgSCN • L (L = хинолин, изохинолин и 2-метилхинолин)
[159]. В качестве L могут выступать также Ме3Р, Ph3P, Ph3As, Ph3Sb и др.
[146, 160—162], причем в зависимости от числа присоединенных органиче-
ских лигандов SCN-группа может быть либо концевой (AgSCN • 3Ph3P и
AgSCN • 3Ph3As [160]), либо чаще мостиковой (для комплексов с двумя или
одним нейтральным лигандом [146, 162]).
Аддукты [AgSCN • zzL] можно получить при взаимодействии солей сереб-
ра с органическими лигандами в присутствии SCN-ионов или же, как было
показано на примере соединений меди (I), путем фотолиза соответствующего
азида серебра и сероуглерода [146].
Для золота известны тиоцианаты как одновалентного металла К [Au •
• (SCN)2], NH4 [Au (SCN)2], растворы которых неустойчивы и диспропор-
ционируют с выделением Аи, так и трехвалентного золота в виде солей
М [Au (SCN)4] (М = К, NH4, Me4N, Et4N) [1,66], М [Au (SCN)4]2 (M = Zn,
Co) II]. Для одновалентного золота получены линейные аддукты AuSCN •
• L (L = Ме3Р, Ph3P, Ph3As и до.) [146, 1631 и кластерные соединения со*
става [AunL7 (SCN)3], [Au9L8] (SCN)3 и [Au6L6] (SCN)2 (L = Ph3P, (CH3C6H4)3P
и др.) [785].
С бидентатными лигандами, которые служат мостиками, Au(I) образует
(Au SCN)2 • L (L = Ph2PC s= CPPh2 [529], PC2P 1559]) или (AuSCN)2 •
• 3L [529], в которых координационное число Au (I) равно 2 или 4.
В М [Au (SCN)4] тиоцианатные группы являются концевыми и связаны
174
с атомом золота посредством серы 166]. Соединение [Me2Au SCN]2 [165] яв-
ляется димером с двумя мостиковыми SCN-группами. Поэтому координаци-
онное число в этом соединении равно 4. Присоединение к нему двух молекул
тиомочевины приводит к разрыву мостиков и образованию соединения
Me2Au (tu)2SCN.
5.5.2. Тиоцианаты редкоземельных элементов
Первые тиоцианаты металлов подгруппы скандия были получены Клеве
[1], который выделил R (NCS)s • 7Н2О и некоторые двойные соли. Боль-
шинство работ по синтезу и исследованию тиоцианатов этих металлов выпол-
нено лишь в последние годы.
Тиоцианаты РЗЭ, т. е. Sc, Y и лантаноидов, можно получить при раство-
рении их гидроксидов в тиоцианатной кислоте и последующей кристаллиза-
ции растворов, а также обменными реакциями между R2 (SO4)3 и Ba (NCS)2.
Будучи хорошо растворимыми в воде, они обычно кристаллизуются в виде
гидратов R (NCS)3 • 7Н2О (для Sc, Y, La, Се, Pr, Nd, Gd), R (NCS)3 • 6H2O
(Sm, Dy, Er).
Кроме кристаллогидратов, тиоцианаты РЗЭ образуют многочисленные
сольваты и продукты присоединения органических лигандов: R (NCS)3 •
• nL (L = EtOH, Diox, DMFA, DMSO, Ph3PO, Ph3AsO, Py, Dipy, Phen,
пиколинамид, тетраметилалонамид и др.; п = 1, 2, 3 и реже 4 или 5 [166—
178, 180, 181,829]). Большая часть указанных работ посвящена тиоцианатам
скандия [167—172, 178]. По данным физико-химических исследований,
скандий в большинстве из этих соединений должен проявлять координа-
ционное число 6. Например, в комплексах Sc (NCS)3 - 3L (L — монодентат-
ный лиганд) все три тиоцианатные группы связаны с металлом, что под-
тверждается данными ИК-спектроскопии и молярной электропроводности
[167, 168, 178]. В соединениях с бидентатными лигандами, как Dipy, Phen,
тиоцианатные группы занимают внешнесферное положение, например
[RL3I (SCN)3 [167/ 172—174, 8281 и [RL2 (NCS)2J SCN. Для последних воз-
можны цис- и транс-изомеры [167, 171]. Однако предполагают, что в соеди-
нениях R (NCS)3 ntmu (n = 4 или 5) координационные числа равны соот-
ветственно 7 и 8 [176]. Координационное число 8 имеют атомы лантаноидов
в соединениях состава [Dipy Н] [R (DipyR (NCS)4J (R = Y, La, Sm, Yb)
[828].
Тетратиоцианатоскандиаты щелочных и щелочноземельных металлов
кристаллизуются из водных растворов в виде кристаллогидратов. Число
молекул воды в ряду комплексов щелочных металлов изменяется от 11
(для лития) до 15 (для рубидия) [179]. Соответственно из этанольных рас-
творов выделены KR (NCS)4 • 4ЕЮН и NaR (NCS)4 • ЗЕЮН [175] (R —
= La, Рг, Nd). Для скандия в указанных соединениях, за исключением
Rb [Sc (NCS)4] • 1,5 Н2О, координационное число равно 6, а для лантана,
вероятно, 7.
Из гексатиоцианатных соединений в литературе наиболее полно представ-
лены комплексы скандия [179, 182]. В безводном состоянии кристаллизу-
ются гексатиоцианатоскандиаты рубидия и цезия и соли с большими орга-
ническими катионами типа [R4N]+, [PhenH]1”, [DipyH]+, [Me3PhNl+ и
др. [183—185].
В силу специфики электронного строения ионы лантаноидов La3+, Се3+,
Nd3+ и т. д. в отличие от Sc3+ и Y3+ в тиоцианатных соединениях могут иметь
175
координационные числа больше 6. Тем не менее тиоцианаты лантаноидов с
количеством тиоцианатных групп больше шести пока не известны. Как вид-
но из табл. 5.12, в некоторых из этих соединений координационное число мо-
жет достигать 9, и соль Dy (NCS)3 • 7Н2О можно интерпретировать как
[Dy (NCS)3 (HSO>„) • Н2О.
Как семикоординационные комплексы можно рассматривать сольвати-
рованные тетрагидрофураном тиоцианаты состава R (NCS)3 • 4Thf, которые
при нагревании в интервале температур 120—150° С в вакууме отщепляют
координированный растворитель и превращаются в безводные соли [186].
Из неводных растворов для празеодима, неодима и самария получены
R (NCS)3 • 4L (L = Ру, Diox) 1175, 187], а для самария также Sm (NCS)3 •
• 7Diox [188]. Обработкой R (NCS)3 • nH2O избытком tmu получены кри-
сталлические R (NCS)3 • 5tmu (R = Се, Pr, Nd), R (NCS)3 • 4tmu (R =
= Sm, Eu, Tb, Dy, Ho, Er) и R (NCS)3 • 3tmu (R = Tm, Yb, Lu) [176].
Соединения аналогичного состава образуют тиоцианаты РЗЭ с Ph3PO и
Ph3AsO: R (NCS)3 • 3L (L = Ph3PO; R = Y, Sm, Eu, Gd, Dy, Ho, Er,
Tm, Lu; L = Ph3AsO; R = Nd, Sm) и R (NCS)3 • 4L (L = Ph3PO; R = La,
Ce, Pr, Nd, Sm) [189].
Соединения R (NCS)3 • 3L в кристаллическом виде имеют полимерный
характер с Ph3PO в качестве мостика [189]. Семикоординационные комплек-
сы с тетраметиленсульфоксидом образуют тиоцианаты почти всех РЗЭ
[190]. Повышение числа координированных лигандов в приведенных выше
соединениях подтверждается данными кондуктометрических измерений в
нитрометане и нитробензоле, а также изучением ИК-спектров [176, 190].
Так как с л-лигандами редкоземельные металлы не склонны образовы-
вать комплексы, их тиоцианаты с бидентатными лигандами образуют шести-
координационные соединения типа [RLS] (SCN)3 [172, 174, 177] или [RL2 •
• (NCS)al SCN [172].
Наблюдаются некоторые закономерности в термической устойчивости
координационных соединений R (NCS)3 с Dipy и Phen. Оказалось, что сое-
динения с Dip}' более устойчивы, чем с Phen. Термическая устойчивость сое-
динений цериевой группы с последним лигандом значительно выше, чем
иттриевой; она резко падает на границе Gd (260°) и ТЬ (170° С) [172].
Для празеодима, гадолиния и самария из этанольных растворов получе-
ны соединения KR (NCS)4 • 4ЕЮН (R = Pr, Nd, Gd) [175, 1871 и MSm •
• (NCS)4 - nEtOH (M = K, Rb, Cs, n = 4, 5 и 6). Получить гексатиоциа-
натные соединения самария не удалось даже при некотором избытке тио-
цианата щелочного металла [188]. Этот факт может служить косвенным до-
казательством того, что в спиртовом растворе прочность тиоцианатных ком-
плексов самария незначительна.
Гексатиоцианаты состава [Bu4N]3 [R (NCS)6] (R = Pr, Nd, Sm, Eu,
Ho, Er, Tm, Yb) получают реакцией обмена между RC13 • /гН2О и [Bu4N] •
NCS в этанольном растворе [191]. Для некоторых лантаноидов известны
гексатиоцианаты с катионом тетраметиламмония [192], а для неодима —
(NH4)3Nd (NCS)6 • 4ЕЮН 1175].
Вероятно, все гексатиоцианатные комплексы имеют октаэдрическое стро-
ение, что подтверждается результатами рентгеноструктурного исследова-
ния и магнитных измерений соединения эрбия [Bu4N]3 [Er (NCS)61 [191].
5.5.3. Тиоцианаты актиноидов
В отличие от лантаноидов, для которых, за исключением прометия, тио-
цианатные соединения известны, сведения о синтезе тиоцианатов актинои-
дов ограничены лишь соединениями тория и урана. Для тория и урана были
получены октатиоцианаты щелочных металлов М4 [Th (NCS)8] • нН2О (М =
= К, Rb, Cs, NH4) [193, 194, 1961, а для урана (IV) М4 [U (NCS)8] (М =
= Rb, Cs, NH4, Et4N, DipyH) [195—197]. Комплексные октатиоцианатные
анионы тория и урана имеют додекаэдрическую конфигурацию [196]. Cs4 •
• [Th (NCS8)] • 2Н2О имеет моноклинную сингонию, пространственная
группа Р2т/п, параметры кристаллической решетки: а = 13,5; b = 14,0;
с = 16,0 A; Z = 4 [193]. Более точные значения параметров решетки этого
соединения приведены в [204]: а — 13,520; b = 13,693 и с = 16,226 А.
Расшифрована и структура соли [Et4NI4 [U (NCS)8] [197], a Cs [U (NCS)8] •
- 2H2O имеет следующие кристаллографические характеристики: а =
= 13,15; b = 13,20; с = 15,75А; Z — 4. Пространственная группа РпаЬ
[204].
В работах [193, 194] были предприняты попытки получить все коорди-
национные соединения генетического ряда от [Th (NCS)4 (Н2О)4] до
М4 (Th (NCS8)]. Из представителей этого ряда были синтезированы [Th •
• (NCS)4 (Н2О)4], Rb (Th (NCS)5 (H2O)3] и (NH4)s [Th (NCS)7H2OJ • nH2O.
Гексатиоцианатотораты из водных растворов получить не удалось. Тем не
менее имеются сведения о существовании соединений К? [Th (NCS)6] и
K4ThO (NCS)6 [198].
Значительно полнее представлены в литературе тиоцианатные соедине-
ния урана (VI). Описаны как кристаллогидраты простого тиоцианата урани-
ла [1,199], так и большое количество координационных соединений [1,199—
201]. Безводный тиоцианат уранила можно получить при нагревании UO2 •
• (NCS)2 • ЗН2О до 125° С [199]. Аналогично получены безводные три- и
тетратиоцианатоуранаты щелочных металлов и аммония из соответствующих
кристаллогидратов. Например, отщепление двух молекул воды соответ-
ственно при 27 и 125° С от соли KUO2 (NCS)3 • 2Н3О [200] приводит к обра-
зованию KUO2 (NCS)3.
Пентатиоцианатоуранаты щелочных металлов кристаллизуются также
в виде кристаллогидратов [1], а с большими катионами (QH+, DipyH+,
Et3NH+) в виде безводных солей [199, 202].
Получена также [Соеп3]2 [UO2 (NCS)6] (NO3)2 • 5Н2О [201], структура
которой нуждается в уточнении. Предполагается, что анион [UO2 (NCS)6]4~
устойчив и в растворе [203].
Свойства и структура тиоцианатов уранила исследованы недостаточно.
Имеются лишь отрывочные сведения о том, что тиоцианатная группа связа-
на с атомом урана посредством азота, например в Cs3UO2 (NCS)5 [204]. Од-
нако в аддуктах UO2 (SCN)2 • 3L, где в качестве L выступают N-оксид пири-
дина и его производные, по данным ИК-спектров, осуществляется связь
U—SCN [205], в то время как UO2 (NCS)2 • 2L (L = N-оксид хинолина)
имеет U—NCS-координацию [206]. Аналогичны по составу соединения тио-
цианата уранила с оксидами трифенилфосфина и трифениларсина [207].
С гексаметилфосфортриамидом тиоцианат уранила образует как UO2 •
• (NCS)2 • 2НМРА, так и UO2 (NCS)2 - ЗНМРА [830].
177
5.5.4. Тиоцианаты металлов IV побочной подгруппы
Из этой подгруппы только титан образует тиоцианаты, в которых он
имеет степень окисления +3. Так, были синтезированы NH4 [Ti (NCS)4
NH4 [Ti (NCS)4 (H2O)2] [208]. Соли K3 [Ti (NCS)6] - 6NH3 [209] и [Bu4N]3 •
• [Ti (NCS)6] [210] были синтезированы из жидкого аммиака и ацетонитрила
взаимодействием TiCl3 и KNCS. [Bu4N]3 [Ti (NCS)6] имеет кубическую сингс-
нию и относится к пространственной группе РаЗ [210].
Простые тиоцианаты металлов подгруппы титана не получены. Обра-
боткой NH4NCS тетрагидрофуранового или ацетонитрильного растворов
TiCl4 получены [TiCl4—п (Thf), (NCS)J (п = 1 ~ 3) и [TiCl4-« (CH3CN)2 •
• (NCS)2] (п = 14-3) [211]. Известны соединения типа МО (NCS)O • 2ELO
(М = Ti, Zr) [212—214], Ср2М (NCS)2 (М = Ti, Zr, Hf) [215—218], а для цир-
кония и гафния также М (NCS)4 5Diox [219] и [М (DMFA)4 (NCS)4] [220].
С Dipy и Phen тиоцианаты обоих металлов присоединяют по две молекулы
лиганда и также образуют соединения [ML2 (NCS)4] [221 j. Во всех приведен-
ных соединениях на основе тетратиоцианатов циркония и гафния металл
имеет координационное число 8. Титан же в аналогичных соединениях,
например в [TiL2 (NCS)2] (SCN)2 [2221 (L = Dipy, Phen), имеет координа-
ционное число 6. Для циркония и гафния получены также соединения
М [ZrO (NCS)3 (Н2О)2] (М = NH4, К, Rb, Cs) [214] и Cs [HfO (NCS)3 (H2O)21
[223], которые в кристаллическом состоянии, вероятно, содержат циркониль-
ную и гафнильную группы [224—226]. Титанильную группу содержат со-
единения M2TiO (NCS)4 (М = К, NH4, РуН, QH) [212].
Из спиртовых растворов выделен ряд соединений типа [R4N]O [TiOR4 •
• (NCS)5], [Ti (OR4)2L (NCS)2] (R = Me или Et; Rx = Me, Et“ Pr, Bu;
L = Dipy, Phen) и [Ti (ORJgLg (NCS)2] (Rx = Me или Et; L = Ph3PO или
Ph3AsO) [227, 831]. Соединения [Et4N]2 [Ti (OEt) (NCS)51 и [T (OEt) -
• (Ph3PO)2 (NCS)3] кристаллизуются в ромбической сингонии и относятся
к пространственным группам Рпа2, и РЬса [227].
Из алкоксопроизводных тиоцианатов титана (IV) получен ряд оксоком-
плексов: [Ti3O4L3 (NCS)J (L = Phen или Dipy), [Et4N]2 [TiO (NCS)4|,
[Ti2O (Acac)4 (NCS)2] и [Ti2O R4 (NCS)2] • 1,2 Act. Для последних трех сое-
динений получены монокристаллы и определены параметры их кристал-
лических решеток [831].
Гексатиоцианатные координационные соединения элементов подгруппы
титана получены для всех металлов. В качестве катионов выступают ионы
щелочных металлов [220, 222, 228—230, 686], производные аммония R4N+
[222, 227, 231], а также пиридиния [214, 223] и [DiantH]+ [232]. Соединения
титана, циркония и гафния с катионами щелочных металлов были получе-
ны из ацетонитрильных растворов. Интересно отметить, что соединения
[Et4N]2 [Zr (NCS)61 и [Et4N]2 [Hf (NCS)6] были выделены из водных раство-
ров без признаков гидролитического разложения [231].
Из этанольного раствора получены Со [Ti (NCS)6I, аддукты CoTi (NCS)6 •
• 4L (L = Py, Et-никотинат, никотинамид) и CoTi (NCS)6 • 6L (L =
никотинамид, CNPy, 1/2en, гидразид никотиновой кислоты). В Co [Ti (NCS)61
присутствуют как мостиковые (Со—SCN—Ti), так и концевые NCS-группы,
а его аддукты состоят из катионов [CoL„]+2 и анионов [Ti (NCS)6]2~ О- -сим-
метрии [8251.
278
5.5.5. Тиоцианаты металлов V побочной подгруппы
Комплексные тиоцианаты ванадия изучены достаточно полно [1], од-
нако простые тиоцианаты, а также их аддукты получены сравнительно
недавно. Черно-фиолетовый V (NCS)3 выделен при нагревании в вакууме
V (NCS)3 • 3Et2O [2331. Кроме соединений с диэтиловым эфиром, тиоцианат
ванадия (III) образует также аддукты с тетрагидрофураном, пиридином
и ацетонитрилом: V (NCS)3 • 3L [234]. Для V (NCS)3 • 3L (L = CH3CN и
Thf) измерены магнитные моменты, соответственно равные 2,46 и 2,56 М. Б.
(25° С), что несколько меньше теоретической величины (2,7 М. Б.) для пра-
вильных октаэдрических комплексов. Получены гексатиоцианатованадиты
(III) М3 [V (NCS)6]-2L (L = CH3CN, М = Na, К, NH4, РуН, Ph4N) [234],
а также [Bu4N13 [V (NCS)61 и [Ph4Asl3 [V (NCS)6] [93]. Ввиду заниженных
значений рЭф для солей пиридиния и калия [234] эти соединения, как и упо-
мянутые выше аддукты, имеют искаженно-октаэдрическую конфигурацию.
Для четырехвалентного ванадия известен только Cp2V (NCS)2, образую-
щийся по обменной реакции между Cp2VCI2 и KNCS в водном растворе [235],
а тиоцианат ванадила легко образует как продукты присоединения с Diox,
НМРА, DMSO, Dipy, Phen и др. [236—238, 8521, так и комплексные соли
состава М3 [VO (NCS)5J [93, 236, 239]. В соединениях М2 VO (NCS)4 • пН2О
ванадий имеет координационное число 6 за счет вхождения во внутреннюю
сферу комплекса одной молекулы воды (табл. 5.12). Из ацетонитрила выделе-
на соль К2 [V (NCS)6] [832].
Тиоцианаты ниобия и тантала выделены из апротонных растворителей,
в частности из ацетонитрила; исходными веществами являются пентахло-
риды ниобия или тантала и соответствующего тиоцианата одно- или двух-
валентного катиона. Попытки получить тиоцианаты из метанола или этано-
ла приводят к образованию продуктов частичного замещения хлора на RO-
группу. Например, из метанола выделен KNb (ОСН3)2 (NCS)4 • СН3СН
[2401, из метанола и этанола [М (OR)3 Dipy (NCS)2]2 (М = Nb, Та) [241].
Выделены также соединения [NbPy2 (NCS)4], [Nb (Dipy)2 (NCS)4] [242],
К., [Nb (NCS)6] [244], M [TaX (NCS)J • nCH3CN (X = O, S; M = Na, K,
NH4), TaOCh-n (NCS)n • 2CH3CN [943], M [Ta (NCS)61 • n CH3CN (M = Li,
Na, K, NH4) и Sr [Ta (NCS)6]2 • 4CH3CN [944].
Несольватированные димеры [M (NCS)5]2 с мостиковыми NCS-группами
получены десольватацией ацетонитрильных аддуктов М (NCS)5 • 3CH3CN
[245], для которых в ацетонитриле характерна реакция аутокомплексооб-
разования
2Nb (NCS)5 + 2CH3CN [Nb (CH3CN)2 (NCS)4]+ + [Nb (NCS)6]~. (5.31a)
Гексатиоцианатониобаты и танталаты щелочных и щелочноземельных
металлов кристаллизуются из ацетонитрила, присоединяя молекулы раство-
рителя. Последние при нагревании в вакууме отщепляются, и образуются
АГ [М (NCS)6] или М" [М (NCS)6]2 [246]. С большими катионами [Ph4N]+
и [Ph4As]+ и другими выделяются несольватированные комплексы [243,
244, 8331. Данные электропроводности этих солей в ацетонитриле указывают
на значительную устойчивость анионов. Для ниобия получены соединения
состава М2 [NbO (NCS)51 (М = Et4N, Ph4N или Ph4As) [833].
179
5.5.6. Тиоцианаты металлов VI побочной подгруппы
Синтез и некоторые свойства тиоцианатных соединений хрома и молиб-
дена подробно описаны еще Вернером, Рейнеке, Розенгеймом и др. [1]. Рас-
смотрим лишь некоторые соединения металлов этой группы.
Соль Рейнеке NH4 [Сг (NH3)2 (NCS)J и ее аналоги были объектом иссле-
дования многих авторов [247—251]. В работах [247, 248] рассмотрены соли
двух- и трехвалентных металлов (Си, Hg, Со, Mn, Ni (II) и Fe (III)) с анио-
ном соли Рейнеке. Получены соли катионов [Mtu2]+ (М = Си, Ag, Аи) и
[Mtu4P+ (М"+ - Pd2+, Pt2+, Bi3+) с анионом [Cr (NH3)2 (NCS)4]~ За исклю-
чением [Pdtu4] [Сг (NH3)2 (NCS)4], остальные комплексные соли не содержат
мостиковых SCN-rpynn [834].
Было показано, что природа внешнесферного катиона заметно влияет
на параметры элементарной ячейки и магнитные свойства комплексов. Если
ион хрома в соли Рейнеке имеет октаэдрическое окружение (табл. 5.12),
то в солях Mn (II), Ni (II), Fe (III) оно ближе к тетрагональному [248].
Замена двух молекул аммиака в анионе [Сг (NH3)2 (NCS)4]~ на два других
монодентатных лиганда не изменяет, за некоторыми исключениями, транс-
октаэдрическую структуру этого аниона [249—254]. Это подтверждается
величинами магнитных моментов, например для комплексов с бензимидазо-
лом рЭф » 3,70 ~ 3,90 М. Б., значения которых характерны для октаэд-
рических комплексов хрома (III) [250, 252, 253]. Целый ряд новых соедине-
ний с комплексными катионами кобальта (III) и анионом [CrL2 (NCS)4]~
(L = анилин и его производные) получены путем внедрения амина во внутрен-
нюю координационную сферу К3 [Сг (NCS)6] в расплавленном состоянии по
схеме [835]
[Cr (NCS)6]3- + 2Am -> [CrAm2 (NCS)J- + 2NCS-. (5.316)
Октаэдрическое строение имеют и аддукты тиоцианата хрома с DMFA,
DMSO, Ph3P, Ру, y-Pic и другими азотсодержащими лигандами. Незначи-
тельная электропроводность аддуктов с монодентатными лигандами в аце-
тонитриле и ацетоне подтверждает шестикоординационное строение этих
комплексов [255, 872].
Из гексатиоцианатохромитов (III) интерес представляют соли серебра,
таллия, свинца, кадмия и др. Некоторые из этих соединений описаны в ра-
боте [1], другие получены и исследованы позднее [256, 836]. Соли тяжелых
металлов, за исключением таллия, содержат мостиковую SCN-rpynny,
которая координирует с ионами этих металлов через серу. Окружение иона
хрома в этих солях близко к октаэдрическому [256]. Вероятно, такое окру-
жение свойственно всем гексатиоцианатохромитам (III). К сожалению, де-
тальному структурному исследованию подвергалось лишь соединение
[Но (C5H4NCOOH)3 (Н2О)2] [Сг (NCS)6] • 2Н2О (табл. 5.12). Имеются сведе-
ния о существовании К4 [Сг (NCS)6] [837].
Гексатиоцианатные соединения Мо (III) типа М3 [Мо (NCS)6] • пН2О
получены для калия, аммония и пиридиния [257]; известен также комплекс
[Me4N]3 [Мо (NCS)6] [258]. Согласно данным [259], значение магнитного мо-
мента для аммонийных и калийных солей колеблется в пределах 3,70—3,80
М. Б. (18° С), что удовлетворительно согласуется с теоретической величи-
ной 3,88. Рентгеноструктурное исследование соли К3 LMo (NCS)61 • Н2О •
• СН3СООН подтверждает октаэдрическое строение [Мо (NCS)6]3~ (табл.
5.12). Октаэдрическое окружение, по данным магнитных измерений, имеет
атом молибдена также в соединениях [MoL3] (SCN)3 (L = диэтиленамин,
180
орто-фенилендиамин и другие бидентатные лиганды) и [Мо (HL)2 (NCS)2] -
• SCN (L = дигуанид [260] или его производные).
Соединения К2 [Мо (NCS)6], [Bu4N]2 [Мо (NCS)6] и [Ph4As]2 [W (NCS)6]
были получены из ацетонитрильных растворов. По величинам магнитных
моментов анионы этих солей можно отнести к октаэдрическим комплексам
[261].
Октаэдрическое строение имеют анионы также в солях К IW (NCS)6]
и [Bu4N] [W (NCS)61 [261].
Попытки выделить однородные тиоцианаты молибдена (V) из бутанола
не увенчались успехом. Была получена соль К2 [Мо (OBu) (NCS)6] • 2ВиОН
[262]. При взаимодействии МоС15 с KNCS в ацетоне и метилэтилкетоне были
выделены MoCl (NCS)4 • 2L и Мо (NCS)5 • L (L = диоксан, ацетон, метилбу-
тилкетон) [263]. При получении этих комплексов следует учитывать возмож-
ность восстановления в присутствии тиоцианата калия [261]. Свойства оксо-
тиоцианатных соединений молибдена приведены в обзорах Митчелла [264],
Вильямса [1] и в других работах [265—268, 840]. В частности, оксотиоциана-
ты молибдена (V) с Dipy, Phen, пирамидоном, антипирином описаны в ра-
ботах [838, 839].
Оксотетратиоцианаты вольфрама состава WO (NCS)4 • 2L и гексатиоциа-
наты (W (NCS)6 • 2L получены при взаимодействии WOC14 или WC16 с
KNCS в неводном растворителе [263, 269]. Синтезирован М2 [WO (NCS)5]
[2701. Из тетрагидрофурана выделены изомеры CpWO (NCS)» и Ср WO (SCN)»
[841].
Отдельно можно выделить группу карбонилтиоцианатных комплексов:
[Сг (CO)5NCS] [276], М' [М (CO)5NCS] (М' = К, Me4N; М = Сг, Мо, W)
[271, 273, 277] и [СрМ (CO)3NCS] для молибдена и вольфрама [272], диядер-
ные однородные [Cr2 (CO)10SCN] [275], [М2 (CO)10SCN]rt- [273, 276] и неодно-
родные [Me4N] (ОС)5М—NCS—М (СО)5] [274] и некоторые другие комп-
лексы, для большинства из которых предложены оригинальные методы
получения. Например, соединение [Сг (СО)5 NCS] [276] получают при
взаимодействии Сг (CO)5I с ISCN, а СрМо (CO)3CNS — по схеме
СрМо(СО)3 Н + (SCN)2 СрМо (СО)3 CNS + HNCS. (5.32)
Это соединение интересно тем, что может существовать в виде двух изо-
меров, в которых SCN-группа координирована с молибденом или через азот
либо посредством серы [272].
5.5.7. Тиоцианаты металлов подгруппы марганца
Простой тиоцианат марганца получают реакцией обмена между его суль-
фатом и тиоцианатом бария; кристаллизуется из водных растворов в виде
Mn (NCS)» • 4Н2О [1]. В присутствии донорного растворителя тиоцианат
марганца также образует сольваты с четырьмя молекулами органического
лиганда Mn (NCS)2 • 4L (L = Ру, FA, DMFA, DMSO, 4-винилпиридин и
пара-толуидин) [53, 278, 279, 843, 849]. С бидентатными лигандами Dipy
и Phen [280], никотингидразидом [844], диэтиламидом никотиновой кислоты
[845], производными бензоилгидразина [335] и бензимидазола [846] получены
Mn (NCS)2 • 2L. Можно предположить, что перечисленные выше соединения
имеют октаэдрическую структуру. Действительно, значения магнитных мо-
ментов при 13° С для комплексов с Dipy и Phen равны соответственно 5,92
и 5,90 М. Б., что подтверждает октаэдрическое окружение иона марганца в
181
высокоспиновых комплексах [281]. В комплексе с 2 (2-пиридил)-бензимида-
золом ион марганца также находится в октаэдрическом окружении (цЭф =
= 5,99 М. Б.) [846]. Правда, и тетраэдрические комплексы имеют значения
магнитного момента примерно 5,8—5,9 М. Б. [282], но октаэдрические комп-
лексы обычно имеют розовую окоаску, в то время как тетраэдрические -
желто-зеленую. Координационное число 4 марганец, по-видимому, имеет
в комплексах с бидентатными гидразидом изоникотиновой кислоты (коорди-
нация по атому азота гетероцикла отсутствует) [847] и изомерами анизидина
(координация гетероцикла через азот) [848], причем для Mn (NCS)2 L
(L = орто-анизидин) предполагается димерное строение с мостиковыми
SCN-группами).
Тетратиоцианатоманганиты щелочных металлов выделяются в виде
кристаллогидратов М2 [Mn (NCS)4] • пН2О [1], а с большими катионами
обычно получаются безводные соли, например [BuPh3P]2 [Mn (NCS)J [282].
Судя по электронным спектрам, тетратиоцианатоманганиты имеют те-
траэдрическое строение [ПО, 282]. Но в комплексе [UrtH]2 ]Mn (NCS)4]
марганец имеет октаэдрическую конфигурацию за счет связей Мп—[UrtH],
находящихся по отношению друг к другу в транс-положении [283].
Гексатиоцианатоманганиты, как и тетратиоцианатоманганиты, можно
получить либо в форме кристаллогидратов М4 [Mn (NCS)6] пН2О [1], либо
безводными [R4N]2 [Мп (NCS)6] [93], [QH]4 [Мп (NCS)6] [279]. В этих соеди-
нениях ион Мп (II) должен иметь октаэдрическое окружение [281], посколь-
ку даже в расплавленных тиоцианатах щелочных металлов NCS-группы
октаэдрически расположены относительно Mn (II) [284].
Для марганца (III) получены пента- и гексакоординационные изотиоци-
анаты состава Mn (Асас)2 NCS (цЭф = 4,90 М. Б. при 24° С) и Мп (Асас)2 •
• (NCS) Ру [850].
В отличие от марганца соединения технеция и рения, в частности четы-
рех- и пятивалентных металлов, были получены сравнительно недавно.
При этом исходили из пертехнецатов и перренатов; для синтеза соединений
рения используют также ReCl5 и K2ReCl6 [285—288].
Для технеция получены соединения [Me4N]2 [Тс (NCS)61 [286], Т1[Тс •
• (NCS)fi] и [Me4N] [Тс (NCS)6] [285, 286], которые в ацетонитриле ведут
себя как электролиты типа 2 : 1 и 1 : 1. Тиоцианат технеция (V) кристалли-
зуется в ромбоэдрической системе и имеет конфигурацию тригонально растя-
нутого октаэдра [286].
Для рения также известны тиоцианаты Re (IV) и Re (V). К2 [Re (NCS)6]
получают сплавлением K2ReClfi с KNCS при 250° С с последующей обработ-
кой сплава этанолом. Из калийной соли были синтезированы соответствую-
щие соединения цезия, таллия и серебра [287]; получены [Ph4AsL [Re (CNS)6]
[289, 290] и [Bu4N12 [Re (NCS)J [289].
Значения магнитных моментов приведенных соединений 3,4—3,9 М. Б.
показывают, что степень окисления рения в них равна 4 [288, 289, 291].
Это удовлетворительно согласуется с цЭф для октаэдрического комплекса
с конфигурацией d3. Для соединений Tl2 [Re (CNS)6], [Ph4As]2 [Re (NCS)6]
и [Bu4N]2 [Re (NCS)6] величина рЭф равная 3,4—3,5 M. Б. [287, 289], не-
сколько меньше ожидаемой для правильного октаэдрического комплекса.
Тиоцианаты рения (V) можно получить взаимодействием ReCl5 с расплав-
ленным KNCS. Обменной реакцией спиртового раствора К [Re (NCS)6]
с CsCl выделен Cs [Re (NCS)6L Эффективный магнитный момент этого соеди-
нения отвечает степени окисления рения, равной 5 [288].
При восстановлении перренатов хлоридом олова (II) в солянокислой сре-
де в присутствии NCS- возможно образование следующих продуктов [288,
182
289]:
2 экв. Sn2 1
ReO4 -
2—4 экв. Sn2+
---------------
4 экв. Sn2-b
Re (CNS)r
Re(CNSK + Re (CNS)I~
Re (CNS)i~
(5.33)
Re? -> ReO (NCS)1“ -> Re2O2 (NCS)|~ Re (NCS)I~.
Ослабление интенсивной красно-оранжевой окраски комплекса
[Re2O2 (NCS)8]3— происходит за счет восстановления последнего до [Re -
• (NCS)6]2— [290]. Можно ожидать, что в присутствии больших катионов
из водных растворов будут кристаллизоваться соответствующие тиоциана-
ты рения по аналогии с соединениями технеция [286].
Возможные способы присоединения тиоцианатной группы к рению об-
суждались неоднократно [192, 287—289]. Однако интерпретация ИК-спект-
ров не дает однозначного вывода о порядке координации SCN-группы.
Достаточно указать на тот факт, что при практически одинаковых значениях
частот v (CN), v (CS) и 6 (SCN) авторы работ [290] и [292] приходят к проти-
воположным выводам о способе координации тиоцианатной группы рением.
Соединения [Bu4N12 [Re2(NCS)8], [Ph4As]2 [Re2 (NCS)8], [Ph3MeAs]2 •
• [Re2 (NCS)8] и [Bu4N]2 [Re2 (NCS)8 (Ph3P)2] получены посредством вза-
имодействия между [Bu4N]2 [Re2CI8] и NaNCS в тетрагидрофуране и в мета-
ноле [289]. Атомы рения в этих соединениях связаны между собой мости-
ковыми тиоцианатными группами.
Кроме однородных комплексов, получен ряд оксотиоцианатных соедине-
ний рения: [Pt (NH3)4]3 [ReO2 (CNS)4]2, [ReO2tu4]3 [ReO2 (CNS)4] [293],
M [ReO2 (SCN)2 (Ph3P)J [294], а также Cs3H [Re2O (SCN)10] и Cs2H2 [Re2O •
• (SCN)10L Последние имеют небольшую величину магнитного момента, что
свидетельствует о двухъядерном характере этих соединений. ИК-спектры
указывают, что связь между атомами рения осуществляется через кислород,
а тиоцианатные группы координируются посредством серы [292]. В растворе
между Cs2H [Re2O (SCN)10J и Cs2H2 [Re2O (SCN)10] существует равновесие:
зеленый цвет соответствует первому из них, а при подкислении переходит
в коричневый, характерный для второго [292]. Синтезированы также карбо-
нилтисцианатные соединения рения [Bu4N]3 [Re2 (NCS)8 (СО)2], [Ph,As]3 •
• [Re2 (NCS)10 (CO)2] [289] и [Et4N]2 [Re (NCS)3 (CO)3 ] [277].
5.5.8. Тиоционаты металлов семейства железа
Простые тиоцианаты Fe (II), Со (II), Ni (II) можно получить посредством
обменных реакций из перхлоратов соответствующих металлов и KNCS в
ацетоне. Из водных растворов при действии HNCS на гидроксиды металлов
получают обычно кристаллогидраты этих соединений.
На основе простых тиоцианатов железа, кобальта и никеля получены
многочисленные координационные соединения. Стереохимия их хорошо изу-
чена. Наиболее часто тиоцианатные комплексы имеют октаэдрическую,
тетраэдрическую, а никелевые — плоскую конфигурацию. Кроме того,
для пятикоординационных комплексов кобальта и никеля возможны конфи-
гурации тригональной бипирамиды или квадратной пирамиды. Вероятны
взаимные переходы от одной структуры к другой.
Из всех d-металлов тетраэдрическое окружение наиболее характерно для
кобальта. В качестве примеров тетраэдрических комплексов кобальта могут
служить соединения типа [R4Nl2 [Со (NCS)J (R = Me, Et, Bu [282, 295]),
183
[CoL2] [Co (NCS)J (L = 2-аминопиримидин [851]), а также аддукты
Co (NCS)2 • 2L (L = а-, р-пиколин [296, 317], Ph3P, Ph3PO [295], HMPA
[55], производные изоксазола [852, 853] и имидазола [854]). Тетраэдриче-
ское окружение кобальт, по-видимому, имеет и в аддуктах Со (NCS)2 • L,
где L — бидентатные аминоацетамиды, которые координируются атомом
кислорода или азота [855]. Значение магнитного момента в тетраэдрических
комплексах кобальта колеблется в пределах 4,3—4,7 М. Б. Так, комплексы
[Со (Ph3P)2 (NCS)2] и [Со (Ph3PO)2 (NCS)2J имеют почти одинаковые с
[Me4N]2 [Со (NCS)4] значения магнитных моментов (4,46 М. Б. при —79° С
[295]).
Тетраэдрическую конфигурацию имеют и некоторые комплексы железа
HI) и никеля (II): [R4N12 [Fe (NCS)J (R=Me, Et) [286], [Ph4As]2 [Ni (NCS)4]
[58]. Последняя соль может существовать в форме изомеров, имеющих жел-
тую и сине-зеленую окраску. Сине-зеленый изомер имеет магнитный момент
~3,6 М. Б. при 25° С, что подтверждает его тетраэдрическую конфигурацию.
Структура комплекса не изменяется при растворении его в ацетоне. В то же
время для желтой формы значение магнитного момента меньше, что харак-
терно для октаэдрических комплексов никеля. Вероятно, тетраэдрическое
окружение железо имеет в высокоспиновом комплексе Fe (NCS)2 • 2L
(L = 2-(2-пиридил)бензимидазол [856]).
Квадратно-плоскостные тиоцианатные комплексы встречаются для ни-
келя, например оранжевая соль [NiEt4dien (NCS)] BPh4 [297]. В то же время
[NiEt4dien (NCS)2] в твердом состоянии имеет октаэдрическую конфигурацию
с двумя мостиковыми NCS-группами. Плоско-квадратное строение имеют
комплексы [NiL2 (NCS)2] (L = гомопиперазин [298] и бис(дифенилфосфино)-
метан [299, 300]). С последним никель образует транс-планарный комплекс,
в котором органический лиганд проявляет монодентатный характер. Плос-
кие комплексы тиоцианат никеля образует также с трифенилфосфином и
триизопропилфосфином [301], пиперидином [302], а- и у-пиколином, 2,6-
лутидином [303, 304], бидентатным фосфорсодержащим лигандом FDP
[305], 1,8-нафталин-бис(диметиларсином) [857]. Получены комплексы нике-
ля квадратной конфигурации, содержащие одну NCS-группу: [NiL (С6С15) •
• NCS] (L = 1,2-бис(дифенилфосфин)этан) [858] и [Ni (Ph3P)2 (С6С15) NCS]
[8591.
Пятикоординационные комплексы чаще других металлов образует ко-
бальт (II). С фосфорсодержащими лигандами получены низкоспиновые со-
единения {Со [PhP (OEt)2]4 NCS] BPh4 и {Co [PhP (OEt)2]3 (NCS)2) [306].
Однако окончательный вывод об их строении сделать затруднительно. Из
других пятикоординационных комплексов кобальта (II), содержащих до-
норные атомы фосфора, можно назвать [Со (R3P)3 (NCS)2] [307], а также низ-
коспиновые комплексы {Со [Et2P (СН2)3 PEt2h,5 (NCS)2] и [Со (PC4P)i,s •
• (NCS)2], которые не содержат мостиковых тиоцианатных групп, а следо-
вательно, кобальт имеет координационное число 5 [308]. Пентакоординацион-
ные комплексы кобальт (II) образует с вторичными фосфинами [CoL3 (NCS)2]
(L = PH (C6Hn)2 или PH (C6Hn) Ph [860]). Известны комплексы co смешан-
ными N, P- и N, As-содержащими лигандами, например [Co (N2P) (NCS)2]
[309], [Co (N3P) (NCS)2], [Co (N3As) (NCS)21 и др. [310].
С азотсодержащими органическими лигандами тиоцианат кобальта (И)
образует тригонально-пирамидальные комплексы [Со (tren) NCS] SCN [311],
[Со (MeG tren) NCS] SCN • H2O [312] и высокоспиновый [Co (Et4dien) •
- (NCS)2] [297, 313]. Структура последнего не установлена. Возможно, что
этот комплекс, как и [Со (Et4dien) 12], в неполярных растворителях имеет
искаженно-тетраэдрическую конфигурацию, когда Et4dien выступает как
184
бидентатный лиганд [313]. Координационное число 5 предполагается и в
[Ni (Et4dien) (NCS)2] [297], а также в упомянутом выше комплексе с бис(ди-
фенилфосфино)метаном, димер которого [Ni2L2 (NCS)J имеет искаженную
квадратно-пирамидальную геометрию с двумя мостиковыми SCN-группами
[299, 300]. Квадратно-пирамидальная конфигурация реализуется в изотио-
цианатных комплексах Ni2L (NCS)4 и Ni2L3 (NCS)4 (L = бис(3-диметилар-
синопропил)фениларсин [861]), а также в соединениях с другими полиден-
татными лигандами [310, 314, 315], включая тетрадентатные, например в
[NiL (NCS)] SCN, [NiL (NCS)] BPh4 (L = трис(орто-диметиларсинофенил)-
стибий [862]) и др. [8031.
Тиоцианат железа (II) также может образовывать пентакоординационные
комплексы, например с тридентатными шиффовыми основаниями [316].
Значительно чаще приходится встречаться с шестикоординационными
комплексами. Мономерные октаэдрические комплексы тиоцианатов Fe
(II), Со (II) и Ni (II) образуются присоединением четырех монодентатных
или бидентатных, координирующихся через один атом лиганда, например
[МРу4 (NCS)2] [296, 317, 8631, IM (DMFA)4 (NCS)21 [322], [ML4 (NCS)2J (L =
= 1,2-диметилимидазол) [323], [NiL4 (NCS)2] (L = у-пиколин, изохинолин
[317], тиомочевина и ее производные [319], N-эти л имидазол [148], пиперидин
[302], 4-метилпиридин [324], 3,5-диметилизоксазол [853], аммиак [869]).
Для кобальта и никеля получены изотиоцианаты металлов состава [ML4 •
• (NCS)2] с конденсированными азолами [870], З-метил-5-фенилизоксазолом
[852], метоксигидроксиламином [871] и 1-винилимидазолом [854]. Для ад-
дуктов состава [ML4 (NCS)2] и [ML2 (NCS)2] (М = Со, Ni; L = 4-метилпири-
дин) определены параметры их решеток и тип сингонии [325]. Октаэдриче-
скую структуру, вероятно, имеют комплексы [М (FLO)4 (NCS)21 • 2Urt
(М = Со, Ni) [842].
Результаты ИК-спектральных и магнитных исследований подтверждают
октаэдрическое строение комплексов типа [ML2 (NCS)2] (L = монодентатный
лиганд). Так, соединения тиоцианатов Fe (II), СО (II) и Ni (II) с пиридином
[МРу2 (NCS)2] и [NiL2 (NCS)2] (L = [3-, у-пиколин, хинолин и др.) состоят
из полимерных октаэдрических комплексов [60, 296, 317, 349]. Полимерными
октаэдрическими комплексами являются соединения [NiL2 (NCS)2J (L =
= анилин, орто-, мета-, пара-толуидин, изохинолин и др.). Магнитный момент
этих соединений равен 3,0—3,1 М. Б., что характерно для октаэдрических
комплексов никеля [317, 318]. Из октаэдрических узлов состоят структуры
изоморфных [Mtu2 (NCS)2] (М = Fe, Со, Ni), в которых мостиковыми явля-
ются атомы серы тиомочевины и некоторых ее производных [319, 320] (см.
табл. 5.12), комплексы с уротропином [321], а для никеля — с пиридином,
(3-пиколином, хинолином [303].
Комплексы [ML2 (NCS)2] (L = бидентатный лиганд) имеют октаэдриче-
ское или тетрагонально-бипирамидальное строение. Для кобальта и никеля
эти комплексы с дибензилэтилендиамином (Dben) [М (Dben)2 (NCS)2] имеют
значения магнитных моментов 3,20 М. Б. (Ni) и 5,09 М. Б. (Со), характерные
для высокоспиновых октаэдрических комплексов. Соединения тех же метал-
лов с бензилэтилендиамином являются тетрагонально-искаженными [326].
Октаэдрическая конфигурация характерна и для комплексов с Dipy и его
производными, Phen в изотиоцианатах [ML2 (NCS)2] [61, 327—329, 808,
822], причем для [Fe (Dipy)2 (NCS)21 октаэдрическое окружение иона железа
сохраняется в высоко- и низкоспиновой модификациях [329]. В растворе
цис-октаэдрический [Со (Phen)2 (NCS)2] диспропорционирует по схеме
2 [Со (Phen)2 (NCS)2] [Со Phen (NCS)2] + [Со (Phen)3] (SCN)2 (5.33a)
185
с образованием псевдооктаэдрического и тетраэдрического комплексов [864].
Комплексы [М (Phen)2 (NCS)21 (М = Fe, Ni) изоструктурны [328]. Октаэд-
рическое окружение железо имеет в аддукте Fe (NCS)2 с бидентатным этил-
пиколинатом [340], в транс-[РеЬ2 (NCS)2] (L = AsAs [865]), кобальт — с ди-
этилфосфино-метилпиридин-2 [331], диэтиламидом никотиновой кислоты
состава CoL4 (NCS)2 и CoL2 (NCS)2 (в последнем соединении мостиковую
функцию выполняют молекулы L) [845], железо (II), кобальт (II) и никель
(II) с производными 1,3-пропандиамина [298], с гидразидами кислот (аце-
тил-, бензоилгидразином и др.) [330,332—335]. Предполагают, что в комплек-
сах с гидразидами NCS-группы находятся в транс- (для кобальта и никеля)
или цис-положении (для железа) друг к другу. С 2,2'-дипиридилом для тио-
цианата никеля получены два структурных изомера [Ni (Dipy)2 (NCS)21 и
INi2 (Dipy)4 (NCS)2] (SCN)2, которые имеют цис-строение [336]. В димере
[Ni2 (tren)2 (NCS)2] (BPh4)2 октаэдрическое окружение никеля достигается
за счет мостиковой Ni—NCS—Ni-связи [826].
Известны октаэдрические аддукты типа М (NCS)2 • L с четыредентатными
макроциклическими лигандами для тиоцианатов железа (II) [337, 866,
873] и никеля [338, 339, 348, 367, 368]. С тридентатными лигандами октаэд-
рическое окружение никель и кобальт образуют за счет мостиковых тиоци-
анатных групп [314, 315, 341].
Октаэдрическое строение имеют упоминавшаяся выше желтая модифика-
ция соли [Ph4Afi]2 [Ni (NCS)41 (цЭф = 3,33 М. Б., 25° С) [68] и соединение
[UrtH]2 [М (NCS)41 (М = Fe, Со, Ni) [283]. Эффективные магнитные моменты
этих соединений в ряду Fe, Со, Ni равны соответственно 5,3; 4,9 и 3,2 М. Б.,
что свидетельствует об их октаэдрической конфигурации, за исключением
[UrtH ]2 [Fe (NCS)4], в котором анион имеет значительное тетрагональное
искажение, и соединения кобальта, имеющего в нитрометане тетраэдрическое
строение. Соединения изоструктурны и относятся к моноклинной сингонии
(пространственная группа Р2х/с). Так, [UrtH2] [Fe (NCS)4I имеет следующие
параметры решетки: а = 6,42; b = 11,01; с = 17,43 А; р = 107° 25'. Близ-
кие к указанным параметры имеют решетки солей кобальта и никеля [283].
Пентатиоцианатные комплексы состава [Cr (DMSO)61 [М (NCS)6 Н2О] по-
лучены для кобальта и никеля, в которых анион и катион имеют октаэдри-
ческую симметрию [872].
Гексатиоцианатные комплексы известны лишь для Fe (II) и Ni (II) [68,
279, 342]. Они имеют октаэдрическое строение [68].
Из простых тиоцианатов трехвалентных металлов известны только
Fe (NCS)3 • ЗН2О и многочисленные его аддукты с основаниями Льюиса
[1]. С монодентатными лигандами обычно образуются соединения типа
[FeL3 (NCS)3] (L = Ру, N-оксид Ру, хинолин, N-оксид хинолина и др.)
[1,343], а с бидентатными — Fe (NCS)3 • 2L (L = Dipy, Phen) [344]. Значе-
ния магнитных моментов комплексов с монодентатными лигандами (рЭф —
= 5,84-6,0 М. Б.) указывают на их октаэдрическое строение [344]. Комплексы
с бидентатными лигандами можно представить как [FeL2 (NCS)2] SCN, по-
скольку в ацетонитриле они являются электролитами типа 1:1, например
[Fe (AsAs)2 (NCS)2] PF6 [865], [Fe (Dipy)3 (NCS)2I и [Fe (Phen)2 (NCS)2] SCN
[344]. Замена в последнем соединении двух NCS-групп на Phen приводит к
образованию [Fe (Phen)3]3+, который в кислой среде окисляет SCN-ион до
CN~ и SO1” [874].
Для кобальта (III) получены координационные соединения с одной,
двумя и тремя тиоцианатными группами. Однако соединения с тремя коор-
динированными группами в координационной сфере неизвестны. Вот не-
которые соединения этого типа: [Со (tren) (NCS)J С1О4 [345], [Со (Dipy)2 •
186
• (NCS)2] C1O4 • H2O, [Co (Dipy)2 (NCS)2] SCN • 5/2H2O, [Co (Phen)2 •
• (SCN)2j SCN [346, 875] и др. Выделен ряд соединений кобальта (III) типа
[Со (DH)2 X (CNS)] • nH2O (X = органический лиганд [876—879]), коор-
динационных соединений, содержащих два псевдогалогенидных лиганда во
внутренней сфере, например Kat [CoL2 (N3) NCS] (Kat = H, [Coen2Cl2],
[CoPy4] и др., HL = диацетилдиоксим) [880]. Комплексы имеют характер-
ное для кобальта (III) октаэдрическое строение даже с tren [345] и другими
тетрадентатными азотсодержащими макроциклическими лигандами, напри-
мер в [CoL (NCS)2] SCN • 2Н2О [881], [CoL (NCS)2]+ и [CoLNO2NCS]+ [882].
Для никеля методом ЭПР установлено образование в растворе комп-
лексов [Ni (DH)2L (NCS)] и [Ni (DH)2 (NCS)2]~ (ПН2=дипропилглиоксим,
L — метанол, ацетон [347]).
Гексатиоцианатные соединения известны только для железа (III). По-
лучен малорастворимый в воде (Сг (DMSO)6] [Fe (NCS)6] [872]. Синтез и
свойства М3 [Fe (NCS)6] (М = Na, К, NH4, Cs) детально описаны [1, 258,
282, 342]. Атом железа в этих соединениях имеет октаэдрическое окружение
[282]. Структурные данные группы тиоцианатных соединений кобальта и
никеля приведены в табл. 5.12.
Представляют интерес семикоординационные комплексы железа (III)
с пентадентатными азотсодержащими микроциклами. Соединения состава
[FeL (NCS)2] С1О4 [883] и [FeL (NCS)21 SCN [884] представляют собой не-
сколько искаженную пентагональную бипирамиду с пятью атомами азота
в экваториальной плоскости и двумя изотиоцианатными группами в акси-
альных положениях. Структура [FeL (NCS)2] С1О4 приведена (табл. 5.12).
В димерном комплексе [(SCN) LFe—О—FeL (NCS)] (С1О4)2 железо также
имеет координационное число 7 [884].
5.5.9. Тиоцианаты платиновых металлов
Известны многочисленные тиоцианатные соединения платиновых метал-
лов, например М2 [М (SCN)41 (М = Pd (II), Pt (II)), М2 [Pt (SCN)4],
Мз [Rh (SCN)6], где в качестве внешнесферного катиона М' выступает
ион щелочного металла или аммония, а также тиоцианаты Pt (IV) с катио-
нами переходных металлов [1]. Комплексные соли с большими катионами
получены для всех платиновых металлов: [R4N]2 [Al (SCNIJ (М = Pd, Pt),
[R4N]2 [Pt (SCN)J (R = Me, Et [66, 350]), [Bu4N]3 [M (CNS)6J (M = Ru,
Rh, Os, Ir) [258], [Cr (DMSO)61 [Rh (SCN)c] [872]. Для платины (II) выделена
соль [Pt (Ме3Р)2 en2] [Pt (SCN)4] [885]. При синтезе соединений рутения
и осмия обычно исходят из К2 [МС16] (М = Ru, Os), которые в присутствии
тиоцианат-ионов восстанавливаются до степени окисления 4-3. Имеются
сведения, что иридий в этих же условиях также восстанавливается до 1г
(II), a Ru (III) — частично даже до Ru (II) [351].
Структура всех гексатиоцианатных комплексов вероятнее всего октаэд-
рическая. Даже комплекс [Ir (Ph3P)2 (СО) Cl (CN) (NCS)] имеет конфигура-
цию искаженного октаэдра [352]. В тетратиоцианатах платины реализуется
квадратно-плоскостная структура. Для аналогичных соединений Pd (II)
квадрат [Pd (SCN)4]2~ может дополняться до искаженного октаэдра за счет
атомов серы двух других ионов [Pd (SCN)4]2— (табл. 5.12). Однако в раство-
ре возможно существование пятикоординационных частиц палладия (II)
и платины (II), содержащих тиоцианатные и органические лиганды [357]
187
Из ацетонитрильных растворов реакцией между кристаллогидратами
RuCl3 • 3,5 Н2О, RhCl3 • ЗН2О и KNCS получены безводные Ru (NCS)3
и Rh (NCS)3 [353], из этанольного раствора — полимерный продукт
[Rh (SCN)2]n родия (II) [886]. Имеются сведения и о безводном Pd (SCN)_
[1]. Простые тиоцианаты других металлов, по-видимому, не получены. Но
известны многочисленные аддукты различного состава, в частности [ML2 •
• (SCN)2], для двухвалентных платины и палладия (см. табл. 5.8—5.10).
Если L — бидентатный лиганд, то соединения Pd (II) и Pt (II) состава ML2 •
• (SCN)2 являются электролитами типа 1:2с двумя внешнесферными
SCN-группами [890]. Внешнесферными являются SCN-группы и в соедине-
ниях типа ML4 (SCN)., (L = однодентатный лиганд), например [PdL4] (SCN),
(L = (RO)3P [889]). Для этих же металлов синтезированы соединения с од-
ной SCN-группой, например [Pd (С6С15) (Ph3P)2 NCS] [859], цис- и транс-
IPt (SCF3) (Et3P)2 NCS] [887], транс-tPt (Ph3P)2 (CN) SCN] [888].
Значительное число соединений смешанного типа получено и для других
платиновых металлов. Это, например, [R4NL [Ru (СО)2 (NCS)4] и [R4N]2 •
[Ru2 (СО)4 (NCS)6] (R = Me, Et) [356], [RhNO (Ph3P)2 (SCN)21 [354] и
[Rh (Ph3Sb)2 C12NCS] [355], [M (Me2PhP)2Cl2NCS] (M = Ph, Ir) [891, 892]
и [Ir (Ph3P)2 (CO) (NO) Cl (NCS)] [893].
5.6. ОБРАЗОВАНИЕ ТИОЦИАНАТНЫХ КОМПЛЕКСОВ
В ВОДНОМ И НЕВОДНОМ РАСТВОРАХ
Сведения о составе и устойчивости тиоцианатных комплексов в литера-
туре представлены для большого числа металлов [358—3611. Тем не менее
далеко не все имеющиеся данные достаточно надежны. Например, при
исследовании тиоцианатных комплексов железа (III) в качестве фона исполь-
зовали 0,665 М раствор хлорида натрия, что привело к значительному зани-
жению константы устойчивости комплекса FeNCS2+ [362], а при полярогра-
фическом изучении тиоцианатов индия в качестве «индифферентного» элек-
тролита был использован раствор нитрата натрия [363] без учета того, что
ионы индия могут образовывать комплексы с нитрат-ионом [364]. Поэтому
найденные константы устойчивости тиоцианатных комплексов (|3n; п =
= 1-4-6), соответственно равные 9,3; 7,5; 13,3; 6,0; 3,1 • 102 и 8,2 • 103 [363],
не согласуются с результатами, полученными в тех же условиях и тем же
методом, но на фоне перхлората натрия (1,2 • 102; 1,6 • 103; 1,75 • 104;
1,7 • 104; 5,5 • 104; 6,9 • 104) [365].
В литературе долго дискутировался вопрос о возможности существования
в растворе комплекса Cd (SCN)-T. Лишь в 1969 г. тщательная статистическая
обработка результатов полярографического исследования тиоцианатных
комплексов кадмия в воде показала, что на фоне нитрата калия полученное
значение р3 0 возможно вследствие образования смешанных комплексов
Cd (SCN) NO3, Cd (SCN)2NO?, а также CdNOt [366].
Немаловажное препятствие в сопоставлении результатов разных иссле-
дователей составляет различие температур и величин ионной силы иссле-
дуемых систем. Кроме того, специфика каждого метода исследования и рас-
чета констант устойчивости накладывает свой отпечаток на конечный
результат. Поэтому ниже мы будем указывать не только значение той или
другой константы, но и условия, при которых она определялась.
188
5.6.1. Состав и устойчивость тиоцианатных
комплексов в водном растворе
Систематическое изучение состояния тиоцианатных комплексов в вод-
ных растворах проводилось многими исследователями. Однако в большинст-
ве своем полученные константы устойчивости комплексов являются кон-
центрационными. Систематические данные о термодинамических константах
устойчивости тиоцианатов отсутствуют. В некоторых случаях, когда реакции
комплексообразования исследовались при небольшой ионной силе раство-
ра и температуре 25° С, можно использовать для расчета коэффициентов ак-
тивности реагирующих ионов различные уравнения, в частности уравнение
Дэвиса [367, 368, 437] или уравнение Васильева [369] (для расчета (5°)
IgP^lgPg- +ЬЦ- (5-34)
1 + 1,6 У р.
Правда, при этом как минимум надо знать константы устойчивости комплек-
са при трех значениях ионной силы раствора.
В табл. 5.1 приведены данные о константах устойчивости и составе тио-
цианатных комплексов в водном растворе. В первую очередь представлены
значения констант, найденные при р, = 0 и температуре 25° С. По возмож-
ности отобраны результаты тех работ, условия эксперимента которых были
близкими.
Для сравнения прочности тиоцианатных комплексов различных метал-
лов наиболее правильно пользоваться первой ступенчатой константой
устойчивости. При образовании монотиоцианатных комплексов химическая
связь между ионами металла и лиганда не осложнена взаимным влиянием
последних, а также, вероятно, возможным конфигурационным равновеси-
ем, что может иметь место в случае присоединения нескольких лигандов.
Так, сравнение ступенчатых констант устойчивости комплексов кадмия
и цинка показывает, что на первых двух ступенях кадмий прочнее связы-
вает тиоцианат-ион, но третий и четвертый лиганды сильнее удерживаются
ионом цинка [379]. Аналогичная закономерность наблюдается для этих ме-
таллов и в цианидных комплексах [370].
Ориентировочная оценка устойчивости монотиоцианатных комплексов
для октаэдрических и тетраэдрических конфигураций на основании электро-
отрицательности SCN-иона показала, что для одного и того же иона металла
можно ожидать более почную связь М—SCN в октаэдрических комплексах
[371]. Еще более наглядно влияние конфигурационного равновесия прояв-
ляется на тиоцианатных комплексах ртути. Если присоединение к иону
ртути (II) двух SCN-ионов приводит к образованию линейного комплекса
с логарифмами ступенчатых констант устойчивости 9,08 и 7,78, то превраще-
ние линейной конфигурации комплекса в тетраэдрическую сопровождает-
ся значительно меньшими изменениями в третьей и четвертой ступенчатых
константах [327].
В соответствии с теорией кристаллического поля константы устойчивости
однотипных комплексов двухзарядных ионов с азотсодержащими лигандами
изменяются в такой последовательности: Mn < Fe <Z Со < Ni < Си > Zn,
которая называется рядом Ирвинга — Вильямса [373]. С учетом двухвалент-
ных ионов титана, ванадия и хрома этот ряд будет иметь вид: Ti < V <С
< Cr > Мп < Fe < Со < Ni < Си > Zn [359].
Анализ приведенных в табл. 5.1 констант устойчивости монотиоцианат-
ных комплексов двухвалентных металлов первого переходного ряда пока-
189
“ Таблица 5.1. Устойчивость тиоцианатных юмплексов в воде
© Ион ме- талла Метод исследования Температу- ра, сС Ионная сила
Ве2+ Ионный обмен Комнатная 1,0
Zn2+ Потенциометрия 20 3,3
» » 25 0
» » 25 1,0
Cd2+ » 25 0
» Ионный обмен Комнатная 0
» Потенциометрия 25 1,0
» Полярография 25 2,0
» Потенциометрия 25 3,0
Hg2+ » 25 1,0
Ga3+ Спектрофотометрия 30—32 0
» Полярография 20 0
In3+ Спектрофотометрия 30—32 0
» Поте нциометрия 20 2,0
» » 20 1,6
T1+ » 25 0
» Растворимость 25 0
» » 25 0
Sn2+ Потенциометрия 20 2,2
» » 20 1,6
Pb2+ Калориметрия 25 0
» Потенциометрия 25 4,0
» » 20 Переменная
» Растворимость 25 3,0
Bi3+ Потенциометрия 25 0
Cu2+ Спектрофотометрия 25 0
Ag+ Потенциометрия 25 0
» Растворимость 25 0
Логарифмы ступенчатых констант устойчивости Литера- тура
lg К, lg «2 lg Ks lg Kt lg Kb Ig Ke
0,13 —0,14 — - — — [378]
1,47 0,70 0,16 —0,32 — — [379]
1,33 0,58 0,09 —0,37 — —- [381]
0,708 0,33 0,13 —0,33 — — [380]
1,89 0,89 0,07 —0,59 — — [369]
1,74 0,66 — 1,0 1,54 — — [382]
1,32 0,67 0,04 —0,15 — — [383]
1,26 0,87 —0,20 0,33 — — [366]
1,39 0,20 0,01 — — — [384]
9,08 7,78 2,84 1,97 — — [372]
2,15 — — — — — [368]
2,32 — — — — — [385]
3,15 — — — — — [368]
2,58 0,42 1,63 — — — [386]
2,51 1,31 0,92 0,05 — — [90]
0,56 0,19 —0,68 — — — [387]
0,80 — — — — — [3881
0,85 — — — — — [389]
1,17 0,60 —0,05 “ — — [390]
1,02 0,52 —0,05 — — — [H7]
1,06 — — — — — [375]
1,08 0,40 1,10 — — — [392]
1,09 1,43 — — — — [391]
0,42 0,29 0,21 —0,34 0,40 — [393]
2,21 1,5 0,7 0,6 0,4 —0,4 [394]
2,30 1,35 — — — — [377, 395]
4,75 3,48 1,22 0,22 — — [396]
— 8,39 (₽2) 1,23 0,28 — — [397]
Au+ Потенциометрия 25 3,0 15,27
Sc3+ » 20 0,1 1,87
» Распределение 20 1,0 0,27
» Спектрофотометрия 20 0,6 0,20
y3+ » 20 0,6 —0,07
La3*- Распределение 27 1,0 0,06
» » 25 5,0 0,24
» » 30 1,0 0,12
Ce3+ » 25 5,0 0,58
Nd3*- » 25 1,0 0,81
Sm3 Спектрофотометрия 20 0,6 0,10
Eu3+ Распределение 25 1,0 0,70
» » 25 5,0 0,32
» » 30 1,0 0,13
» » 25 5,0 0,43
Gd3+ Спектрофотометрия 20 0,6 0,21
Tb3+ Распределение 30 1,0 0,23
Dy3+ Спектрофотометрия 20 0,6 0,12
Er3'** » 20 0,6 0,16
Lu3+ Распределение 25 5,0 0,45
» » 30 1,0 0,21
u4+ Потенциометрия 20 1,0 1,49
» Распределение 25 2,0 1,49
U0£+ Спектрофотометрия 20 1,0 0,73
» Калориметрия 25 1,0 0,75
Pu3+ Распределение 25 1,0 0,46
» » 30 1,0 0,34
Am3+ » 25 1,0 0,50
» » 25 1,0 0,36
» » 30 1,0 0,17
» » 25 5,0 0,48
— — [398]
_ _ _ — [399]
_ - — »
— _ _ [400]
— — - — »
— — [401]
— — - [402]
— — - [403]
— — - [404]
— — - - [405]
— — - - [400]
—0,26 - — [405] - [402] - [403]
— — — [404]
— — — »
— — - [403]
— — [400]
— — _ — »
— — _ — [402]
— — [403]
0,23 — — [406] - [407]
—0,16 — 1,46 — — [408] — [380]
— — _ — [405]
— — [403]
—0,19 — — [405] — [409] — [403]
— — _ — [404]
~ Продолжение табл. 5.1
ГФ Ион ме- талла Метод исследования Температу- ра, °C Ионная сила и
Ст3+ Распределение 25 1,0 с
» » 25 1,0 с
» » 30 1,0 с
» » 25 5,0 с
Вк3+ » 25 1,0 с
» » 25 5,0 с
Cf3+ » 25 1,0 с
» » 25 1,0 с
Es3+ » 25 1,0 с
v2+ Спектрофотометрия 25 1,34
» » 25 1,0 ]
v3+ » 25 1,0 2
» » 23 1,0 2
VO2+ Потенциометрия 25 0 2
Cr2+ Спектрофотометрия 25 0
Cr3+ » 30 0
» » 25,1 0
Mn2+ Спектрофотометрия 23—24 0
Fe2+ » 23—24 0 1
Fe3+ » 22—23 4 г
» » 22—23 0 .*
» Распределение 25 3,0 2
» Спектрофотометрия 25 3,0 2
Co2+ » 25 0
» » 25 0 1
Ni2+ » 25 0 1
» » 22—23 0 1
» Распределение 20 3,0 1
» Потенциометрия 25 0 1
Pd2+ Спектрофотометрия 25 1,0 5
Логарифмы ступенчатых констант устойчивости Литера- тура
1 Ki 1g кг 1g Кз 1g К4 1g к# 1g К,
1,43 0,42 — — — — [405]
1,45 —0,52 —0,01 — — — [409|
>.18 0,43 — — — — [403]
1,61 — — — — — [404]
>,49 — — — — — [409]
>,86 — — — — — [404]
>,49 — — — — — [405]
1,57 —1,00 0,88 — — — [4091
1,56 -1,96 1,87 — — — »
,43 — — — — — ИЮ]
,43 — — — — — [411]
1,06 — — — — — [412|
1,18 — — — — — [413]
1,32 — — — — — [414]
,09 —0,32 — — — [374]
1,04 — — — — — [415[
1,09 — — — — — [416|
,23 — — — — — [376]
,31 — — — — — »
5,1 1,32 0,43 0,00 —0,04 —0,11 [417]
1,1 2,2 0,9 — — — [418|
’,18 1,42 1,40 1,30 —0,07 —0,09 [419]
!,19 1,48 — — — — [420]
,72 . — — — — [395]
,77 — — — — — [421]
,76 — — — — — [395]
,67 — — — — — [376]
,19 0,49 -0,38 0,24 — — [422|
,89 — — — — — [437|
,65 4,81 3,97 3,13 —
зывает, что последовательность Ирвинга — Вильямса соблюдается. Сравне-
ние констант устойчивости комплексов при ионной силе, равной нулю, дает
ряд: Сг2+ < Мп2+ < Fe2+ <С Со2+ < Ni2+ < Cu2+ > Zn2+ и соответствен-
но lg pi 1,09 [3741; 1,23 [376]; 1,31 [376]; 1,72 [395]; 1,76 [395]; 2,30 [377];
1,33 [381].
При сопоставлении констант устойчивости тиоцианатных комплексов
с110-металлов в подгруппах, как, например, в ряду Zn, Cd, Hg, можно заме-
тить, что с увеличением радиуса центрального иона прочность комплексов
не уменьшается, а возрастает. Последнее объясняется увеличением способ-
ности иона кадмия и в особенности ртути к образованию прочных л-связей
за счет вакантных d-орбиталей атома серы тиоцианат-иона. С этих же пози-
ций можно объяснить ослабление прочности связи М — SCN в ряду ионов
Ag+, Cd2+, In3+, поскольку прочность дативных л-связей уменьшается с
увеличением заряда центрального атома. По той же причине высокозаряд-
ные и имеющие сравнительно небольшие радиусы ионы трехвалентных эле-
ментов подгруппы скандия образуют малопрочные тиоцианатные комплексы.
5.6.2. Состав и устойчивость тиоцианатных
комплексов в смешанных и неводных растворителях
Как правило, введение органического растворителя в водный раствор
обусловливает увеличение прочности комплексов. Были предприняты попыт-
ки показать, что реакции
М°+ + /Lm“ ±5 ML!n“*“)+
подчиняются закономерностям электростатического взаимодействия. Так
Турьян [423] предложил уравнение, которое связывает константу диссоциа-
ции комплекса с величиной диэлектрической проницаемости среды. В общем
виде это уравнение можно записать как
рК = const + (В/е). (5.35)
В этом уравнении величина В не зависит от растворителя, и, следователь-
но, зависимость
рК = /=(1/е) (5.36)
должна быть прямолинейной. Действительно, в смешанных водно-спирто-
вых растворителях устойчивость комплексов находится в прямой зависимо-
сти от обратной величины диэлектрической проницаемости растворителя.
В литературе имеется большое количество примеров, подтверждающих ли-
нейную зависимость между lg Рп комплекса и 1/е смешанного растворителя
при условии, что можно пренебречь изменениями диэлектрической проница-
емости раствора по сравнению с таковой чистого растворителя [423—425L
Ирвинг и Россотти [426] предложили уравнение, связывающее величины
констант диссоциации в данном растворителе и воде с мольной долей перво-
го в смешанном растворителе. Линейная зависимость между указанными ве-
личинами должна наблюдаться в тех случаях, когда отношение коэффициен-
тов активности находящихся в растворе частиц в смешанном растворителе
и в воде равно или близко к единице или же пропорционально мольной доле
неводного растворителя.
По всей вероятности, оба уравнения справедливы лишь в том случае,
если образующие смешанный раствор компоненты близки по химической
природе и по энергии сольватационных процессов.
7 2473
193
Повышение устойчивости комплексов в смешанном растворителе
сравнению с водным раствором отчасти обусловлено уменьшением диэлек-
трической проницаемости среды. Однако более существенной причиной явля-
ется изменение в сольватной оболочке металла. Уменьшение е растворител
повышает ионное взаимодействие между М"+ и отрицательно заряженньг
лигандом. Чем выше степень ионности связи, тем сильнее должно сказы-
ваться влияние диэлектрической проницаемости среды и тем больше В
в зависимости (5.35).
Между тем линейность зависимости (5.36) соблюдается для смесей раство-
рителей одинаковой или близкой химической природы (водно-спиртовые и
спиртовые растворы). Для других растворителей, имеющих разные функцио-
Таблица 5.2. Общие константы устойчивости тиоцианатных комплексов индия
в смешанных растворителях (20° С, р. = 1,6 для NaC104; содержание неводного
растворителя в объемных процентах)
Растворитель In NCS2+ In (NCS)+ In (NCS), In (NCS)~ Литера- тура
н20 3,2-102 6,7-IO3 5,5-104 6,2-105 [90]
Н2О—МеОН (25) .— — 1,6-106 2,8-106 »
Н2О— МеОН (50) — — 1,3-107 4,0-Ю7
Н2О —МеОН (70) — — — 1,0-108 »
Н2О—DMFA (25) 5,9-102 3,1-104 3,2-105 8,3- IO8 »
Н2О —DMFA (50) 6,3-102 4,3-104 9,1-106 4,0-106 »
Н2О—DMFA (70) 7,1 • 102 9,1 -104 4,3-106 3,6-107 »
Н2О —CH3CN (25) — — — 3,3-106 [431]
Н2О—CH3CN (50) 9,b 108 »
нальные группы, линейность не соблюдается [427, 428]. Поэтому было выска-
зано предположение, что изменение состава комплексов и повышение их
прочности в водноорганических растворителях при значительной мольной
доле органического компонента обусловлено понижением степени гидрата-
ции ионов металла за счет связывания воды молекулами донорно-активных
растворителей. Это повышает конкурирующую способность лиганда в ко-
ординационной сфере иона металла [429]. Как следует из табл. 5.2, устой-
чивость тиоцианатных комплексов индия в смешанном растворе выше, чем
в водном растворе.
С повышением концентрации неводного раствора, помимо ослабления
гидратации ионов, более или менее заметно проявляется и пересольватация
последних. Следствием этого в смешанных растворителях, содержащих
80—100 об.% неводного растворителя, наблюдается резкое увеличение проч-
ности комплексов [430], а часто и уменьшение количества ацидолигандов
в координационной сфере [415, 429, 434].
Обращает на себя внимание тот факт, что равные добавки разных по хи-
мической природе растворителей в неодинаковой степени повышают проч-
ность комплексов. Например, в растворителях, содержащих 50% неводно-
го компонента, устойчивость комплекса In (NCS)T увеличивается в ряду:
диметилформамид < метанол < ацетонитрил, что согласуется с уменьше-
нием их сольватирующей способности.
Согласно [432], влияние растворителя на устойчивость тиоцианатных
комплексов по сравнению с галогенидными должно быть менее заметным.
Однако, как показывает анализ литературных данных, это не всегда под-
тверждается. Так, устойчивость тиоцианатных и галогенидных комплексов
в протонных диполярных растворителях изменяется в такой же последова-
194
тельности, как и в воде. В метаноле, например, устойчивость тиоцианатов
ртути [433], свинца [425], олова [117, 895], кадмия [434], кобальта [897]
и никеля [898] значительно выше, чем в водном растворе. В то же время
для индия в FA (е = 109,5 при 25° С) устойчивость тиоцианатных и галоге-
нидных комплексов несколько ниже, чем в воде, хотя последовательность
лигандов при этом не изменяется [435].
В апротонных диполярных растворителях устойчивость тиоцианатных
комплексов изменяется по-разному. При этом, к сожалению, нельзя уста-
новить корреляцию между устойчивостью комплексов и донорным числом
растворителей [432, 436]. Это вид-
но уже при сопоставлении значе-
ний а = [NCS-] : [М2+], необходи-
мых для полного превращения в
Таблица 5.3. Значения а для полного
превращения Со2+ и Ni2+ в комплексы
Со (NCS)2- [438J и Ni (NCS)|~ [439]
анионные тиоцианатные комплексы
ионов кобальта и никеля в раство-
рителях с разным донорным чис-
лом (табл. 5.3). При почти равных
значениях донорных чисел DMSO
и DMFA для образования Co(NCS)?-
требуются 200- и 13-кратные из-
бытки лиганда, а в случае триме-
тилфосфата и пропиленкарбоната
наблюдается даже обратная зави-
симость: большему значению
DNsbciB соответствует меньшее со-
отношение [NCS] : [Со2+]. Послед-
Растворитель
Диметилсульфоксид
Диметил формамид
Т риметилфосфат
Пропиленкарбонат
Ацетонитрил
Нитрометан
29,8 200 500
27,8 13 250
23,0 5 250
15,1 7 70
14,1 4 250
2,7 4 —
нее, по-видимому, связано с большой относительностью известных DNsbci6.
В неводных растворах по своей комплексообразующей способности SCN
далеко не всегда сохраняет постоянное место в ряду галогенид-ионов. В аце-
тонитриле, например, монотиоцианатный комплекс железа (II) имеет 1g 0Х
такой же, как и FeBr-1- [440]. В то же время в DMFA, DMSO, НМРА различие
в устойчивости галогенидных и тиоцианатных комплексов столь заметно,
что в них изменяется даже общая для водных растворов последовательность
комплексообразующей способности лигандов [441—448, 895, 896]. В DMSO
и DMFA, например, устойчивость хлоридных комплексов значительно вы-
ше устойчивости иодидов и тиоцианатов свинца, кадмия и даже ртути.
Резкое изменение донорной активности SCN- по сравнению с галогенид-
ионами в апротонных диполярных растворителях лучше согласуется с пред-
ставлениями об их сольватации [449, 895, 899], чем с выводами о донорных
числах самих растворителей [432].
Вероятно, обращение ряда устойчивости галогенидных и тиоцианатных
комплексов в апротонных диполярных растворителях характерно не только
для высокодонорных растворителей типа DMFA и DMSO, но и для сред,
имеющих среднее и, возможно, малые значения донорных чисел. Это под-
тверждается результатами потенциометрического титрования перхлората
кадмия галогенид-ионами в ацетонитриле. Устойчивость три- и тетрагалоге-
нидных комплексов кадмия увеличивается в ряду I- < Вг— < С1— [4501.
При этом следует отметить, что стабильность тиоцианатных комплексов ме-
таллов в ацетонитриле довольно высока [431, 900].
195
5.7. КООРДИНАЦИОННЫЕ ТИОЦИАНАТОГАЛОГЕНИДНЫЕ
СОЕДИНЕНИЯ И ИХ УСТОЙЧИВОСТЬ В РАСТВОРЕ
Несмотря на то, что систематические исследования тиоцианатогалогенид-
ных соединений начались сравнительно недавно, получены интересные ре-
зультаты, позволяющие проследить закономерности их образования.
Установлено [451—455], что одни лиганды легко образуют смешанные
комплексы, а другие трудно. Образование тиоцианатогалогенидных комп-
лексов можно представить равновесием
М + nNCS + mHal М (NCS)„ Halm (заряды опущены) (5.37J
с константой
R _ [м (NCS)nHalTO]
Рпт [М] [NCSf [Hair ’
С другой стороны, смешанные комплексы можно представить как продук-
ты взаимодействия соответствующих однолигандных комплексов
М (NCS)„+m + MHal„+m 2М (NCS)„ Halm. (5.39)
Константа этого равновесия Kd характеризует устойчивость смешанного
комплекса по сравнению с однолигандными
к - tM (NCS)n Halw]2
' (5-40)
Если Kd > 1, то смешанный комплекс более прочен, а при Kd <Z 1 ме-
нее прочен, чем однолигандные. Для многих систем статистически можно
рассчитать как 7Q, так и ^пт [456]. Последняя, кстати, определяется как
1OR _ п te PnNCS + т lg PmHal , (п-{-/п)!
Р™ ----------—------------+ lg -, -j- (5.41)
При образовании двухкоординационных комплексов мерой совместимо-
сти NCS- и Hal- в комплексе [MHal NCS] может служить константа
Kncs =
[MHal (NCS)]
[MHal] [NCS]
(5.42)
или
к _ [M (NCS) Hal] - q
AHal— [MNCS][Hal] •
Однако для установления стабилизации или дестабилизации смешан-
ных комплексов константы, рассчитанные с помощью уравнений (5.38) —
(5.43), должны иметь высокую точность [457]. В противном случае сделанные
выводы могут быть ошибочными, как и прогнозы на основе представлений
Пирсона [458]. Так, устойчивость комплексов [CuIHal]- (Hal- = NCS-,
Br , СГ) возрастает в ряду NCS- <С Вг— < СГ [453]. Для комплексов
[Hg (SCN) Hal] прочность возрастает в ряду Г < Вг— < СГ [459].
В табл. 5.4 и 5.5 приведены константы устойчивости смешанных тиоциа-
натогалогенидных комплексов, из которых можно видеть проявление «ста-
билизации» и «дестабилизации» комплексов вследствие простого сочетания
лигандов, а также их взаимного влияния, которое выражается в расхожде-
нии статистических и экспериментальных значений этих констант. Напри-
мер, значения логарифмов статистических констант устойчивости комплек-
сов Hg (SCN) I и Hg (SCN) Вг, вычисленные по уравнению (5.41), равны
196
20,7 и 17,45 соответственно. Экспериментальные значения этих величин —
19,9 (Hg (SCN) I) и 17,3 (Hg (SCN) Br) (для расчета использованы константы
устойчивости однородных комплексов [454]). Полученные таким образом
константы устойчивости смешанных комплексов согласуются с результатами
спектрофотометрического исследования этих соединений [459].
Для разнолигандных соединений железа значения статистических конс-
тант значительно выше экспериментальных, что, возможно, обусловле-
но неточностью определения самих
констант [460, 461]. Практически
отсутствует взаимное влияние ли-
гандов в комплексе TI (NCS) С1~, о
чем свидетельствуют незначитель-
ные расхождения между статисти-
ческими и экспериментальными
константами устойчивости этого
комплекса [462].
Помимо результатов исследова-
ния смешанных тиоцианатогалоге-
нидных комплексов в растворе,
значительную информацию о вза-
имном влиянии лигандов в смешан-
ном соединении можно получить
при физико-химическом исследо-
вании тиоцианатогалогенидных ко-
ординационных соединений. Изуче-
ние термической устойчивости, ИК-
спектров ит. п. дополняют сведе-
ния об их устойчивости. В качест-
ве примера рассмотрим смешанные
тиоцианатогалогенидные соедине-
ния металлов подгруппы цинка,
которым посвящено много работ
(см. табл. 5.4 и 5.5). Так, для кад-
мия и ртути известны смешанные
тиоцианатогалогенидные соедине-
ния Mz [М" (SCN)2A] и М2 [М" •
Таблица 5.4. Константы образования
смешанных тиоцианатогалогенидных
комплексов кадмия в водном растворе
при 25° С (потенциометрический метод,
ионная сила 5,0 М) [470]
Комплекс Комплекс J bp
Cd (SCN) Cl 2,8 Cd (SCN)2 BrJ“ 3,65
Cd (SCN) Cl~ 3,1 Cd (SCN)3 Br2“ 3,36
Cd (SCN) Clf~ 3,07 Cd (SCN)3 Br|~ 3,62
Cd (SCN) Cl|~ 2,67 Cd (SCN)3 Br|~ 3,56
Cd (SCN)2 СГ 3,14 Cd (SCN)4 Br3- 3,15
Cd (SCN)2 Cl2- 3,36 Cd (SCN)4 Br4~ 3,28
Cd (SCN)2 Clf- 3,19 Cd (SCN)5 Bl4- 2,7
Cd (SCN)3 Cl|~ 3,27 Cd (SCN) 17 4,65
Cd (SCN)3C1|“ 3,18 Cd (SCN) l|- 5,4
Cd (SCN)4 Cl3- 2,95 Cd (SCN) I3- 5,08
Cd (SCN) Br Cd (SCN) Br~ Cd (SCN) Br2~ Cd (SCN) Br3~ Cd (SCN)2 Br- Cd (SCN)2 Br^- Cd (SCN)2 Br|- 3,23 3,59 3,74 3,66 3,33 3,74 3,81 Cd (SCN)2 I- Cd (SCN)2 Cd (SCN) 21|~ Cd (SCN)3 I2- Cd (SCN)3 I3- Cd (SCN)4 I3- 3,77 4,77 5,41 3,81 4,41 3,54
• (SCN)a А2] (М' = катион щелоч-
ного металла или аммония; М" =
= Cd, Hg; А = С1, Br или I) [463].
Для кадмия и ртути получены
также соединения [Kat] [М (SCN) 13] [464]. В роли [Kat] выступает [Меп2]2+
(М = Cu, Ni). Получены смешанные кислоты Н2 [Cd (SCN)2 А2] • 3Et2O
(А = Br, I), Н [Zn (NCS) А2] • 2Et2O [465]. Смешанные тиоцианатогалогени-
ды цинка имеют такой же состав, как и приведенные выше соединения кад-
мия и ртути [466]. Для ртути известны смешанные тиоцианатогалогениды с
катионами двухвалентных переходных металлов: М [Hg (SCN)2 А2] • пЕЮН
(М = Со, Ni; А = О или Вг) [467].
ИК-спектральное изучение соединений К2 [Zn(NCS)2A2] показывает, что
в случае хлоридного комплекса, по-видимому, структура Zn—NCS преоб-
ладает над мостиковой Zn—NCS—Zn, а в бромидном они проявляются в оди-
наковой степени. Предполагается, что в К2 [Zn (NCS)2 С12] дополняющие
до октаэдрической конфигурации связи Zn—SCN значительно слабее, чем
197
Таблица 5.5. Константы образования смешанных тиоцианатогалогенидных
комплексов некоторых металлов в водном растворе при 25° С
Комплекс 6 о g 5 H s о Д о Я < a cq Ионная сила £ са Литера- тура Комплекс Метод исследо- вания Ионная сила Рпт Литера- 1 урн
Hg (SCN) Cl Спектро- 0,35 18,80 [471] Ag (SCN)3 Br3- Потен- 5,0 10,78 [476]
фото- циомет-
метрия рия
Hg (SCN) Br 0,35 16,06 » Ag (SCN)3 Br4~ » 10,33 >
Hg(SCN)!2- 1,0 29,30 [472] Ag (SCN)4 Br4~ » » 10,60
Hg (SCN)2 i2- » 27,76 Ag (SCN) I- Раство- » 10,15 [477]
Bi (SCN) C1+ » 2,0 4,71 [473] Ag(SCN) I3' римость
Bi (SCN)2C1 » 5,67 Потен- » 13,96 [476]
Bi (SCN) Br+ 4,08 » рия
Bi (SCN)2 Br * Cu (SCN) Cl“ » Раство- 4,0 5,30 7,31 » [474] Ag (SCN) It- » » 13,80 »
римость Ag (SCN)2 if- » 13,50
* Cu (SCN) Br~ Cu (SCN) I~ Cu (SCN) I22~ » » » 2,0 5,0 » 7,72 11,45 10,40 » [475] » Ag (SCN)3 I3- Fe (SCN) F+ Fe (SCN) C1+ » » Спектро- » 0,5 1,2 13,10 3,14 2,42 » [460] [461]
Cu (SCN) » 10,45 » фото-
Cu (SCN)212- » » 10,78 » Fe (SCN) Br+ метрия
Cu (SCN)2 I3~ » » 10,95 » » » 1,32
Pd (SCN) Clf » 1,1 17,15 [478]
Cu (SCN)3 I3~ » 10,40
Ag (SCN)3C13- Потен- » 9,94 [476] Pd (SCN)2 Cl|“ » » 22,05 »
циомет- Pd (SCN)3 Cl2- » » 25,64 »
Ag (SCN) Br3“ рия » » 9,7 Pd (SCN) Br|~ » 18,15 [479]
Pd (SCN)2 Br2" » » 22,25 »
Ag (SCN)2 Br3- 5,0 10,60 »
Pd (SCN)3 Br2“ » 25,85 »
Ag (SCN)2 Br|- » » 10,20
* 20° С.
в случае аналогичного бромидного комплекса. Возможно, что это является
причиной различия рентгенограмм указанных соединений [468].
Рентгенографическое и ИК-спектральное исследование смешанных тио-
цианатогалогенидных соединений К2 [Cd (SCN)2 Cl2], К2 [Cd (SCN)2 Вг2] •
• ЗН2О и К2 [Cd (SCN)2 I21 • ЗН2О показало, что прочность связи Cd—SCN
уменьшается в ряду: Cl > SCN ж Вг > I [468]. Показатель константы сту-
пенчатой диссоциации [Cd (SCN)2 А2]2- [Cd (SCN) А2]~ + SCN- для хло-
ридного комплекса (рК = 0,36) больше бромидного (рК == 0,16) [452],
т. е. результаты исследования устойчивости смешанных комплексов кадмия
в кристаллическом состоянии и в растворе коррелируют между собой. Мож-
но предположить, что и для комплексов цинка в растворе связь Zn—NCS
окажется более прочной в комплексе [Zn (NCS)2 С12]2-, а в соответствующем
бромидном соединении она будет менее устойчивой.
Изучение ИК-спектров поглощения кристаллов упоминавшихся выше
смешанных координационных соединений М [Hg (SCN)2 Ag], где М — ион
198
переходного металла, и некоторых тиоцианатоселеноцианатных соединений
[467] показывает, что значение частоты v (CN) закономерно увеличивается
в ряду Мп <С Со < Ni <С Zn, соответствующем повышению устойчивости
комплексов в известном ряду Ирвинга — Вильямса. Как следует из резуль-
татов термографического исследования дитиоцианатодихлоро- и дитиоциана-
тодибромогидраргиратов марганца и никеля, в той же последовательности
изменяется и термическая устойчивость указанных соединений [469].
5.8. ХАРАКТЕРИСТИКА КОМПЛЕКСООБРАЗУЮЩЕЙ
СПОСОБНОСТИ ТИОЦИАНАТ-ИОНА
Многочисленные сопоставления и образование соответствующих смешан-
ных комплексов свидетельствуют о том, что как по устойчивости, так и по
структурным особенностям тиоцианатные комплексы аналогичны галоге-
нидным. Иногда считают, что тиоцианат-ион является более сильным донор-
ным лигандом, чем галогенид-ионы [432]. В таком случае с большинством ме-
таллов он должен образовывать более прочные комплексы, чем соответству-
ющие галогениды. В действительности же это свойство проявляется лишь
для кобальта, никеля, цинка, индия. Правда, в этом случае не соблюдается
предполагаемая зависимость, отражающая увеличение прочности в ряду га-
логенидных лигандов, ибо донорные свойства последних увеличиваются от
хлорид-иона к иодид-иону, а последний с упомянутыми металлами образует
самые неустойчивые комплексы.
Для другой группы хорошо изученных металлов, например ртути, кад-
мия, устойчивость тиоцианатных комплексов близка к бромидам.
Следовательно, донорную способность тиоцианатного лиганда нельзя
объяснить без учета свойств иона-комплексообразователя. Для ионов ме-
таллов, способных образовывать дативные л-связи, можно ожидать, что
прочность тиоцианатных комплексов при прочих равных условиях будет
уменьшаться с увеличением заряда иона-комплексообразователя, так как
склонные к образованию таких связей лиганды стабилизируют низшие сте-
пени окисления. Поэтому, например, прочность комплексных тиоцианатов
падает от меди (I) к галлию и от серебра к индию. Однако, как следует из
табл. 5.1, тиоцианатные комплексы цинка и кадмия менее устойчивы, чем
соответствующие соединения галлия и индия. Эти факты трудно объяснить,
ибо радиусы изоэлектронных ионов при переходе от М+ к М3+ закономерно
уменьшаются в рядах: Cu+, Zn ь, Ga3+ и Ag+, Cd2+, In3+, а значения потен-
циалов ионизации ионов Мя+ увеличиваются от Си1- к Ga3+ и от Ag+ к 1п3+,
что свидетельствует об уменьшении подвижности электронов d-орбиталей
ионов металлов в том же направлении. Следовательно, это должно приво-
дить к ослаблению прочности л-дативной связи металл — лиганд. Быть
может, по причине ослабления л-дативной связи цинк, галлий и индий коор-
динируют тиоцианат-ионы через атом азота, а не серы.
При переходе к ионам б10з2-металлов вследствие экранирования d-элек-
тронов s-электронами понижается возможность их участия в образовании да-
тивных л-связей, и поэтому, например, тиоцианатные комплексы изоэлек-
тронных Tl+, Pb2+, Bi3+, а также Sn2+ и, вероятно, Sb3 + менее устойчивы,
чем соответствующие галогенидные комплексы. В ряду Tl+, Pb2+, Bi3+ ус-
тойчивость тиоцианатных комплексов увеличивается, что можно объяснить
преобладающим электростатическим взаимодействием в комплексах этих
металлов.
199
Неспособностью к образованию дативных л-связей, по-видимому, объя^
няется и низкая устойчивость тиоцианатных комплексов металлов подгр>-
пы скандия. Тиоцианат-ион является типичным линейным псевдогалогенил
ионом амбидентатного типа. Поэтому механизм образования его комплеь-
сов для разных металлов может быть различным.
Строение и, в частности, распределение зарядов на составляющих NO-
атомах можно представить в виде нескольких резонансных форм [481]
е © _ ©_® m
|N=C—_S| |N = C=S| о ]N-C=S|
I
Как следует из табл. 5.6, отсутствует удовлетворительная сходимость дл
всех значений эффективных зарядов атомов, полученных разными авторами.
Сопоставление зарядов атомов N и X в NCS~ (X = О, S и Se) показывает,
что наименьшее различие в их величинах наблюдается для тиоцианат-иона
Таблица 5.6. Распределение заряда на атомах в ионе NCS- [482]. Поэтому результаты работ [483] и [484] следует считать бо- лее достоверными.
Заряд [482] [483] [484] * nd основании данных распре- [3j деления зарядов в NCS-иона мож- но считать естественными три спо-
Cs Сс Cn CTBOlv C1BOK- рез c< —0,71 +0,19 —0,48 атома a серы—к spy с ион —0,59 +0,1 —0,5 зота, яв; 4ЯГКИМ.Г ами —м —0,48 +0,08 —0,51 1яется ж [оэтому э ягкими к соба координации: -О,18 1) М — NCS 2) М — SCN +0,08 изотиоцианаты тиоцианаты -0,90 3) М —NCS —М мостиковые связи Согласно концепции [5, 61, SCN- группа, координируемая посред- естким основанием, а присоединяемая посред- тот лиганд легче должен координироваться че- ислотами, а через азот — с жесткими кислота-
ми. Наличие вакантных d-орбиталей серы в SCN делает ее значительно луч-
шим акцептором, чем атом азота. Несмотря на повышенную электронную плот-
ность на атоме азота, при взаимодействии с ионами Ag+, Hg2+ и Au3+ проис-
ходит дополнительная стабилизация М—SCN-связи. Однако, во-первых,
такое деление на «жесткие» и «мягкие» кислоты, как и классификация метал-
лов Арланда — Чатта — Девиса [485], носит качественный характер;
во-вторых, из такой классификации естественно вытекает необходимость
выделения промежуточного класса кислот (ионов металлов), которые могут
реагировать как с жесткими, так и с мягкими основаниями, например:
Fe (II), Со (II), Ni (II), Cu (II), Zn (II), Pb (II), Sn (II), Sb (III), Bi (III),
Rh (III) и др. [6].
Условность классификации Пирсона очевидна, если обратиться к кон-
кретным фактам. Так, Zn (II) относится, по Пирсону, к пограничной области
кислот, но известно, что он, как правило, координирует SCN-группу через
азот [46, 486], т. е. «жестким» концом, и его можно отнести к типичным жест-
ким кислотам. Кадмий отнесен Пирсоном к мягким кислотам, в то время
как даже в однородных тиоцианатных комплексах кадмия SCN-группа не
всегда присоединяется посредством серы [73, 486].
К недостаткам классификации кислот Пирсона относится и то, что при-
надлежность металла к тому или иному классу кислот основывалась на оцен-
ке констант равновесия в водном растворе, хотя, как отмечалось выше,
фактор среды часто является определяющим. Не удивительно, что в водном
200
растворе Na2Cd (SCN)4 координация SCN-иона осуществляется через серу,
а в метаноле — через азот [487]. Координация через азот в случае тиоциана-
тов кадмия наблюдается и в расплаве KNCS [488].
5.9. ИК-СПЕКТРАЛЬНОЕ, ЭЛЕКТРОНОГРАФИЧЕСКОЕ
И РЕНТГЕНОСТРУКТУРНОЕ ИССЛЕДОВАНИЯ
ТИОЦИАНАТНЫХ СОЕДИНЕНИЙ
Многочисленные исследования, начало которым было положено в рабо-
тах [489—492], показали, что в координационных соединениях тиоцианатные
группы могут занимать внешнесферное, внутрисферное положение или и то
и другое одновременно, как это имеет место в [Cu (trien) SCN] SCN [493],
а также играть роль мостиков между атомами металла.
5.9.1. ИК-спектральный критерий
способа координации SCN-группы
Систематические ИК-спектральные исследования тиоцианатов металлов
впервые выполнены в работах [46, 494].
Известно, что колебательный спектр трехатомной линейной группы имеет
в инфракрасном спектре три активные частоты [495]: (A), v2 (А) и v3 (Е),
из которых V! (А) и v3 (Е) — частоты валентных колебаний v (CN) и v (CS)
соответственно, v2 (А) — частота дважды вырожденных деформационных
колебаний 6 (CNS).
Для типичного ионного кристаллического соединения KNCS частоты
v (CN), v (CS) и 6 (NCS) будут равны 2049; 747; 484; 471 см-1 [46]. Согласно
другим данным [496], изолированный в матрице CsI ион NCS~ при 27° С
имеет значения 2066,3; 744; 468,1 см-1. При этом частота валентного колеба-
ния v (CN) менее чувствительна к изменению температуры, чем v (CS) [901].
Приближенный полуэмпирический анализ колебаний SCN-группы в KNCS
дает значения v (CN) 2046 см-1 и v (CS) 748 см“’ [497]. Частоты, близкие к
экспериментальным частотам тиоцианата калия, получены и для тиоциана-
тов рубидия, цезия, таллия [497], а также [Me4N] NCS [498].
Если учесть, что частоты v (CN) тройной связи C=N [499, 500] и двойной
C=N [500] лежат в области спектра ~2250 и 1600—1700 см-1, а частоты
v (CS) одинарной связи С—S [500] и двойной C=S [495, 501] — в области
600—700 и 960—990 см-1, то сопоставление этих частот с частотами валент-
ных колебаний v (CN) и v (CS) KNCS и других ионных тиоцианатов [46] по-
зволяет предполагать, что кратность связи в CN и CS тиоцианат-иона долж-
на быть меньше трех (но больше двух) в CN и больше единицы (но меньше
двух) в CS. С учетом данных по распределению заряда (табл. 5.6) тиоцианат-
ион схематически можно представить как
—(0.5+6) +26 _(0,5+б)
N = С — S .
К такому же выводу можно прийти при сопоставлении соответствующих
значений длин связей G^N, C=N, C=S и С—S [502] с найденными для
KNCS [481], а также при сравнении экспериментальных силовых констант
Knc и Kcs [481] с Kc==n, Кс—s и Kc=s [46].
201
Естественно, что способ координации тиоцианатной группы должен ска-
зываться на степени кратности связей CN и CS в координированной SCN-
группе, и, следовательно, это изменение должно найти отражение в смеще-
нии полос частот v (CN) и v (CS) по сравнению с соответствующими частота-
ми для SCN-иона. Было замечено, что повышение значения частоты v (CN)
тиоцианатных комплексов увеличивается в ряду: NCS" <Z М—NCS < М—
—SCN < М—NCS—М' [494], причем частота v (CN) мостиковой группы по-
вышается тем больше, чем выше электронегативность М'. Увеличение час-
тоты v (CN) в случае мостиковых SCN-групп по сравнению с таковой для
иона может достигать 70—120 см"1 [46],
Таблица 5.7. Значения основных частот
изотиоцианатов ниобия и тантала [242]
Соединение v (CN) v (CS) 6 (NCS)
[Та Dipy (NCS)4] SCN 1975, 1915 918 494
[Та Ру (NCS)5] 1975, 1920, 1820 922 499
[Nb (Dipy)2 (NCS)4] 2075, 2035, 2010 906 492
[Nb Py2 (NCS)4] 1970 892 508
что позволяет относительно легко
фиксировать существование мос-
тиков в тиоцианатных комплек-
сах при помощи ИК-спектраль-
ного метода.
Поскольку частоты валентных
колебаний v (CN) тиоцианатов
и изотиоцианатов часто доволь-
но близки, то тип координации
(М — SCN или М—NCS) обычно
определяют по положению час-
тоты v (CS). При координации
через серу реализуется резо-
нансная форма N^C—S, что со-
провождается понижением час-
одновременном повышении частоты
при
тоты v (CS) (630—730 см"1) _ __________
v (CN). Для изотиоцианатов кратность связи CS увеличивается, что приво-
дит к повышению частот v (CS) по сравнению с ионом до 780—850 см"1.
Анализ частоты v (CS) изотиоцианатных комплексов показывает, что для
тетраэдрических комплексов (например, кобальта, железа) частота v (CS)
значительно выше (840—850 см-1), чем для октаэдрических (~800 см"1)
[296]. Повышение частоты v (CS) наблюдается и для изотиоцианатов с вы-
сокой степенью окисления металла, например циркония (824—836 см~!)
[220, 225]. Но для соединений [М (DMFA)4 (NCS)J и К2 IM (NCS)6] (М = Zr,
Hf) значения v (CS) лежат в области 770—790 см-1 [220]. Для гексаизотио-
цианэтов ниобия (V) и тантала (V) частота v (CS) достигает ~ 890 см-1 и бо-
лее [242, 244]. В табл. 5.7 приведены значения v (CN), v (CS) и 6 (NCS) для
аддуктов пента- и тетратиоцианатов ниобия и тантала.
Частоты деформационных колебаний 6 (SCN) и 6 (NCS) также несколько
различаются по величине (410—460 и 470—490 см"1) [259, 350], и это может
служить дополнительной информацией при определении М—N- или М—S-
координации.
Ценную информацию также можно получить из ИК-спектров в дальней
области (200—400 см"1), поскольку тут фиксируются те колебания, которые
в наибольшей степени деформируют связи М—N и М—S. Однако в отличие от
валентного колебания изотиоцианатов и тиоцианатов, величина которого
не зависит или почти не зависит от массы металла [497], в дальней области
ИК-спектра частоты колебаний v (MN) и v (МС) в значительной степени за-
висят от природы металла и конформации комплекса. Так, для [Me4N]3 •
• [М (NCS)6] (М = Сг, Мо) найдено v (CrN) 364 см"1 и v (MoN) 303 см"1.
Для тетраэдрических [Et4N]2 [Со (NCS)J и [Et4N]2 [Zn (NCS),J соответ-
ственно 304 и 287 см"1 [66]. Проанализировав ПК-спектры нескольких десят-
202
ков соединений тетра- и гексатиоцианатов, Фостер и Гудгейм [66] пришли
к выводу, что вопрос о координации тиоциана т-группы для большинства
случаев можно решить однозначно, если сопоставить спектры тиоцианатов и
галогенидов одних и тех же металлов. Средней интенсивности полоса v (MS)
лежит в области между v (МС!) и v (МВг), а большей интенсивности полоса
v (MN) — несколько выше, чем v (MCI), т. е. v (MN) > v (MS). Например,
для аналогичных соединений палладия v (PdS) ~ 300 см a v (PdN) ~
~ 340 см-1 [504]. Для указанных частот в соединениях типа [PdL (CNS)2]
и [PdL2 (CNS)2] наблюдается обратная зависимость: v (PdS) > v (PdN)
[505]. При интерпретации частот в дальней ИК-области, в частности для
изотиоцианатов, необходимо учитывать геометрию комплекса, поскольку
-v (MN)TeTP > v (MN)Okt [296].
Для мостиковой SCN-группы, по-видимому, главным критерием является
значение частоты v (CN), которая превышает таковую для тиоцианат-ионов
на 70—120 см-1 и лежит в области 2130—2180 см-1 [46]. Частота v (CS)
в этом случае может повышаться и понижаться и не быть определяющей при
оценке мостикообразования, а доказать присутствие мостиков по частотам
v (MN) и v (MS) в дальней ИК-области спектра удается очень редко [192].
По характеру полос валентных колебаний v (CN) и v (CS) при наличии
в комплексе двух SCN-групп можно различать цис- и транс-расположение этих
лигандов в комплексе. Синглетный характер полос этих колебаний указы-
вает на транс-, а дублетный — на цис-структуру комплекса [333—335,
506]. По этому признаку предполагают цис-расположение изотиоцианатных
групп в соединениях тиоцианатов кадмия и железа (II) с бидентатными гид-
разидами кислот, а изотиоцианаты цинка, марганца (II), кобальта (II) и
никеля (II) с теми же лигандами имеют транс-строение [333—335].
В табл. 5.8—5.11 представлена часть известных соединений с М—NCS-,
М—SCN- и М—SCN—М (М')-координацией. В них не приведены значения
характерных частот колебаний v (CN), v (CS) и 6 (NCS), так как запись ИК-
спектров проводилась в неидентичных условиях.
В табл. 5.8 из-за обилия данных не представлены соединения элементов
семейства железа, в которых SCN-группа практически всегда координирует-
ся посредством азота [46, 66, 68, 93, 259, 296, 322, 494 и др.]. Немногочислен-
ные случаи инверсии связывания для некоторых соединений семейства же-
леза, например для кобальта (III) [346, 508, 509], будут рассмотрены ниже.
Из соединений платиновых металлов, в которых SCN-группа координиро-
вана через азот, здесь приведены лишь результаты работ [192, 258], соглас-
но которым М—N-координация для осмия, иридия и рутения в гексаизотио-
цианатах [Bu4N]3 [М (NCS)6] объясняется тем, что они проявляют невысо-
кую dn-активность. Координация через азот наблюдается в соединениях
К2 [RuNO (NCS)J [487] и (RuNO (NH3)4 NCS] (SCN)2 [587], в Ru (NCS)3
[353] и M (Ph3P)2 CO (NCS) (M = Ir, Rh) [559].
Наибольший интерес представляют соединения с мостиковой SCN-груп-
пой. Для металлов II группы мостиковая связь реализуется в соединениях
магния и элементов подгруппы цинка. Мостиковую структуру, по всей веро-
ятности, должен иметь и Be (NCS)2 (подобно ВеС12), однако в чистом виде
он не получен. Аддукты его с ацетонитрилом, диоксаном и диэтиловым эфи-
ром, по-видимому, являются изотиоцианатами с концевой NCS-группой,
а бериллий имеет координационное число 4 [16, 17]. Мостиковая структура
присуща диизотиоцианатам и триизотиоцианатам цинка [49]. Для последних
наблюдаются полосы как мостиковой (2165 и 770 см-1), так и концевой
(2110—2120 и 830 см-1) тиоцианато-групп, что предполагает существование
203
Таблица 5.8. Изотиоцианатные соединения
1. Соединения s-, р- и d10-элементов
Cs2Be (NCS)4- 2Н2О [48] (M = Ca, Sr, Ba)
М2Ве (NCS)4-nMeCN [48] [PyH]2 Ga (NCS)5 [83]
МВе (NCS)3-nMeCN (М = NH4, К, Rb, Cs) Zn(NCS)2-4L [48] |R4N]3 Ga (NCS)6 (R = Me, Et, Bu) [84]
[53, 280,552, [PyH]3Ga (NCS)6 [83]
(L = DMSO, DMFA, FA, 806] M3Ga (NCS)6 (M = NH4, K, Rb, Cs) [503]
пропиоамид) In(NCS)3-nL (n = 1 или 2; [820]
Zn (NCS)-2L [51, 807— = производные анилина или
(L = Py и производные Py, 809, 815, имидазола)
Phen, гидразиды кислот и др.) 816, 849] In (NCS)3-3L [92, 178]
ZnL (NCS)2 (L — диамины, триамин) [810, 811] (L = Py, y-Pic, tu, DMSO, DMFA, HMPA, Ph3PO, Ph3P,
M2Zn (NCS)4-nH2O [49] 1/2 Dipv, 1/2 Phen)
MM'Zn (NCS)4-nH2O (M, M' = Na, K, Cs, NH4 [49] Mln (NCS)4-2CH3CN (M = Na, K, NH4) [821]
и др.) [Ph4N]2 In (NCS), [94]
MZn (NCS)4-nH2O [548] [Ph3PCH2Ph]2 In (NCS). [94]
(M = Ca, Sr, Ba) [R4N]3 In (NCS)g [93—95]
[Me4N]2 Zn (NCS)4 [66] (R = Me, Et, Bu)
M4Zn (NCS)6-nH2O [74] Si (NCS)4 [106, 547]
M3M'Zn (NCS)6-mH2O [74] Ge (NCS)4 [Ю7]
M2MgZn (NCS)6-nH2O (M, M' = Na, K, NH4, V2Ba) [74] Ge(NCS)4-4L (L-Dipy, Phen, шиффово ос- нование и др.) [113, 823]
B(NCS)3-Et3N [77] R3GeNCS [H2]
В (NCS),-RCN (R = Me, PhCHo) [77] [R4N] Sn (NCS)3 (R = Me, Et) [H4]
Al (NCS3-3DMSO [78] MeSn (NCS)3-Phen [120]
Al (NCS)3-2Pic [78] MeSn(NCS)3-2Ph3PO [120]
Ga(NCS)3-nL (« = 1,2; L = =производные анилина и ими- дазола) [820] Me2Sn (NCS)2-2L (L = Py, Ph3PO, Ph3AsO, x/2 Dipy, x/2 Phen) [120]
Ga (NCS)3-3H2O [87] Me3SnNCS-L [120]
Ga (NCS)3-3L (L = DMSO, Py, V2 Dipy, [87, 480] (L = Dipy, Phen)
K2Sn (NCS)g [825]
i/2 Phen) P (NCS)3 [106]
[R4N] Ga (NCS)4 [84] PO (NCS)3 [Ю6]
(R = Me, Et, Bu) PS (NCS)3 [Ю6]
MGa (NCS)4 [503] As (NCS)3 [Ю6]
(M — NH4, K, Rb, Cs) M [Ga (NCS)4]2-nH.2O [45] Bi (NCS)3-3HMPA [178]
2. Соединения d- и [-элементов
(CuNCS)2-3 (Ph2PC=CPPh2) [529] M [Sc (NCS)4]2-nH2O [179]
Cu (NCS)2-4Py [296] (M = Mg, Ca, Sr, Ba)
Cu (NCS)2-L [550] Y (NCS)3-3HMPA [178]
(L = Dipy, Phen) [Bu4N]3 Y (NCS)g [191]
Cu (NCS) (BPhj)-tren [827] La (NCS)s.3L [178, 189,
[Et4N]2 Cu (NCS)4 [68] 1 829]
Sc (NCS)3-3L [168] (L = HMPA, Ph..PO, PhoAsO,
(L = DMSO, DMFA, Ph3PO, PhgA^O, PyO) Dipy NO) [Et4N]3 La (NCS)g И92]
Sc (NCS)3 • 2L [167, 171] Pr (NCS)3.7H9O* [553]
(L = Dipy, Phen) Dy (NCS)3-7H2O * [554]
Sc (NCS)2-nL [169, 170, 178] R(NCS)3-3L (R = Ce, Pr, Nd; L = Ph3PO, [189]
(L — Py, ROH, Diox, HMPA) Ph3AsO)
MSc (NCS)4./rH2O [179] R(NCS)3-4L [189]
(M = Li, Na, K, Rb, Cs, NH4) (R = Ce, Pr, Nd; L - Ph3PO)
204
Продолжение табл. 5.8
R(NCS)3-nL [176] MNb (NCS)g [242, 833]
(R — РЗЭ, кроме Pr; L = (M = nh4, K, (C2h2n2s3)2 H)
= Me4U) R2 [NbO (NCS)5J [833]
Yb (NCS)3-2Phen [172] (R = EtN, Ph4P, Ph4As)
KNd (NCS)4-4EtOH [187] Ta (NCS) 5-Dipy [242]
MSm (NCS)4-4EtOH [187] Cr (NCS)4-3L [255]
(M = K, Rb, Cs) (L = DMFA, DMSO, Ph3PO,
Na2Gd (NCS)5-5Diox [187] Py, V-Pic)
[Et4N]3 R (NCS)6 [192] MCr (NCS)4-2L [249, 259,
(R = Ce, Pr, Nd, Yb) [Bu4N]3 R (NCS)e [191, 510] (M = K, NH4, Et4N и др.; 494, 835]
(R = Pr, Nd, Sm, Eu, Dy, (L = NH3, Py, ароматические
Ho, Er, Tm, Yb) амины)
M4Th (NCS)8-2H2O [196] K3Cr (NCS)e [249]
(M — Rb, Cs) [196] M3Cr (NCS)6.4H2O [46, 259]
M4U (NCS)8.(0 —1)H2O (M = NH4, K, TI)
(M = NH4, Rb, Cs) [RL„] Cr (NCS)g [551]
[Et4N]4 U (NCS)g [197] (R = РЗЭ, Y; L = DMFA, Py, Et3N; n = 6-н8)
Cs3UO2 (NCS)* [204] M3Mo(NCS)6-4H2O (M = nh4, K) [259, 494]
[Et4N]2 TiO (NCS)4 [831] (NH4)3Mo (NCS)6-EtOH [259]
Ti2O (Acac)4 (NCS)2 [831] (NH4)3 Mo (NCS)6-HCbH2O [259]
Ti3O4 (NCS)4-3L [831] Cs3Mo (NCS)6 [192]
(L — Dipy, Phen) [216, 217] M2Mo (NCS)6 [258, 261]
Cp2Ti (NCS)2 (M = к, Bu4N)
Ti(NCS)4-2L (L = Dipy, Phen) M2Ti (NCS)6 [222] [222, 229, MoO (NCS)3-2L (L = Dipy, Phen) [838]
230, 686] M2W (NCS)6 [261]
(M = Na, K, Rb, Cs, Bu4N) (M = K, Ph4N)
Cp2Zr (NCS) о [216] MW (NCS)6 [261]
Zr (NCS)4-4DMFA [220] (M == K, Bu4N)
Zr (NCS)4-2Dipy [221] (Mn (NCS)2-2L [335, 843,
Zr (NCS)4-3Phen [221] 846—849]
MZrO (NCS)3-2H2O [225] (L = ароматические амины,
(M = NH4, K, Rb, Cs, PyH) [225] гидразиды кислот и др.)
[PyH], Zr (NCS)fi-2H2O Mn (NCS),-4L [53, 278,
K2Zr (NCS)6-4DMFA [220] 322, 849]
[Et4N]2Zr (NCS)e [231] (L = DMSO, DMFA, FA, про-
Hf (NCS)4-4DMFA [220] изводи ые Ру)
Hf (NCS)4-2Dipy Hf (NCS)4-3Phen CsHfO (NCS)3.2H2O [221] [221] [226] [BuPh3P]2 Mn (NCS)4 K2Mn (NCS)4-6H2O [282] [494]
[PyH]2 Hf (NCS)6 [226] [R4N]2 Mn (NCS)6 [93]
K2Hf (NCS)6-4DMFA [220] (R = Me, Et)
[Et4N]2 Hf (NCS)6 M3V (NCS)e [231] [93, 192, 259] [Me4N]2 Tc (NCS)6 [Me4N] Tc (NCS)6 [286] [286]
(M = K, NH4, Et4N, Ph4As) [Ph4As]2 Re (NCS)g [290]
Cp2V (NCS)2 [216, 235] [Bu4N]2 Re2 (NCS)8 [289]
[Et4N]2 VO (NCS)4 VO(NCS)2-3L [192] [236, 832] [Bu4N]3 [M(NCS)6] [192, 258]
(L = 1/2 Dipy, l/2 Phen, DMSO, (M = Ru, Os, Ir)
HMPA) Rh (Ph3Sb) C12NCS [355]
[R4N]3 VO (NCS)6 [93, 236, 239] Pd (C6C15) (Ph3P)2 NCS [859]
K2v (NCS)6 [832] Pd (NCS)2-2RPh2Sb [890]
Nb (NCS)4-2Py [242] M(NCS)2-R (Me2As)2 [857]
Nb (NCS)4-2Dipy [242] (M = Pd, Pt)
* Структурные исследования.
205
Таблица 5.9. Тиоцианатные соединения
Zn (SCN),-N,N — Et2en [56]
M2Cd (SCN)4-/iH2O * [511]
(M2 = K2, Cs2, KNH4, CsNH4, Na[C(NH2)3]) M4Cd (SCN)6-nH2O [73]
(M4 = Na4, Na2Ba, Na, (NH4)2, Na3K, Na2K2, K3NH4“ NaCs3, K3Cs, K2Cs2, Cs4) Hg(SCN)2 [46]
Hg (SCN)2-L [810, 811]
(L = en, trien, Me6trien и другие полиамины) Hg(SCN)2-2L [50, 809, 813]
(L — RNH2, тиоморфолип, пиперидин) M2Hg (SCN)4 [46, 66, 494,
(M = NH4, K, Cs, TI) PhTl (SCN)2 486,511,555] [Ю2]
K„Pb (SCN)H 2 (расплав) [488]
Bi (SCN)3-3DMFA [128]
K3Bi (SCN)6 [556]
[R4N]3Bi (SCN)6 [139]
(R = Me, Et, Bu) Cu(SCN)2-2L [560, 566]
(L = Pn, en) AgSCN - riL [159]
(L — Q, IQ, 2-mctiuiIQ) AgSCN -3L [160, 162]
(L = Ph3P, Ph3As, EtPh2P) MAg (SCN)2 [46, 498]
(M = K, NH4, Me4N)
RAuSCN-L (L = Ph3P, Ph3As) [559]
(AuSCN)2-PC2P [559]
Au (SCN)2 Cl-Ph3P MAu (SCN)4 (M = K, Me4N, Et4N) M,Re (SCN)6 [559] [66, 192, 350]
[192, 287]
(M = Cs, Ag, TI)
CsRe (SCN)6 [192, 288]
RhCO (Ph8P), X2SCN [H]
(X = Cl, Br) RhCO (Ph3P)2 X (SCN)2 [И]
(X = Cl, Br) M3Rh (SCN)6 (M = K, Me4N, Et4N) [258, 259, 494]
[Bu4N]3 Ir (SCN)e [258]
Pd (SCN)2-2L [557, 885, 912]
(L — Pr3As, Bu3As, Me3P и ДР-)
Pd (SCN),-Dipy [557]
M2Pd (SCN)4 [66, 192, 259, 350]
(M = K, NH4, Me4N, Et4N) Pt (SCN)2-2L [557]
(L = Pr3As, BugAs) [46, 66, 192,
M2Pt(SCN)4 259]
(M = K, NH,4 Me4N) M2Pt (SCN)6 [66, 192, 259, 350]
(M = K, Me4N, Et4N)
* В цезиевых солях предполагается как Cd — SCN, так и Cd — NCS.
Таблица 5.10. Соединения с мостиковыми SCN-группами
1. Однородные мостиковые соединения типа М—SCN—М
Mg (NCS)2-4H2O [46] Hg (SCN)2-
Zn (NCS)-L [576] • [Ph2As (CH2)2 AsPh2] [59]
(L = (ЕЮ)2 POCH-CO • In (NCS)3 [91, 92]
• CH2NR2; R = Me, Et) Zn (NCS)2 • 2H2O [49] In (NCS)3-2L (L = Diox, Et2O, морфолин) ‘ [561]'
MZn (NCS)3-nH2O [49] Sn (NCS) 2 [H4]
(M = Na, K, NH4, Cs, C(NH2)g, i/gBa) Cd (SCN)2-nH2O (/1 = 0, 1) [46, 491 Me2Sn (NCS)^ Me3Sn (NCS)* [562] [563]
Cd (SCN)2-2L [53, 278, 280, Pb (SCN)* [564, 565]
(L = DMSO, DMFA, FA, 552, 809] CuSCN [46]
CuSCN-2Ph3P [145]
метил-FA и др.) CuSCN-nL [143, 940]
MCd (SCN)g-nH2O [64] (L = Q, Py, a-, y-Pic, лутидип
(M = Na, K, NH4, C(NH2)3, И др.)
x/2Ba) Cu (NCS)2 [46,143]
M2Cd (SCN)4-/iH2O (M2 = Na2, K2, NaNH4, KNa) Hg(SCN)a-L [46, 492, 494, 511] [51, 807] Cu (NCS)2-L (L = en, Pn) [Et4N]2 Cu (NCS)4 AgSCN Me9AuSCN [566] [66] [494, 567] [65]
(L = PhgAs, PhgSb, Ру и его Nb\NCS)3 Cl [569]
производные) [M(NCS)5]2 [245]
206
Продолжение табл. 5.10
[516]
(М = Nb, Та) [Ni (NO) NCS]X [516]
[Мо (СО)4 (NCS)2]„ [570] [Ni (NO) P (C6Hji)3 NCS]2 [826]
Mn(NCS)2-L [46] [Ni (tren) (NCS)]2 (BPh4)a
(L = о-анизидин) [848] [R4N]2 Ru (CO)2 (NCS)4 [356]
[Bu4N]2 Re2 (Ph3P)2 (NCS]8 [289] [R4N]2 Ru (CO)4 (NCS)e [356]
[Fe (CO)2 (NCS)2]„ [570] (R = Me, Et)
[Fe (NO)2 NCS], [516] Rh (SCN)3 [353]
Co (NCS)2-2L (L — Py, y-Pic, Urt) [317, 321] [RhL (SCN)]2 (L = R3P) [573]
[Co (NO)2 ncsj2 [516] [PdL (SCN)];* [574]
Ni (NCS)2-2H2O [46, 571] (L = 2-метилаллил)
Ni (NCS)2-2Q-2H2O [572] [PdH (SCN)]2 [575]
MNi (NCS)3-nH2O [571] (L = p-пинен)
(M = Na, K, NH4, Cs, г/2Ва) Pd (SCN)2-L [9П]
[Ph4As]2 Ni (NCS)4 [68] (L = изоксазол)
2. Смешанные мостиковые соединения типа М—SCN—М'
Mg (NCS)4 Zn-4H2O [548] Tl2 (NCS)4 Hg [192]
M(NCS)4 Cd-nH2O (M = Mg, Ca, Sr, Ba) [548] L5Co (NCS) Hgw+ *** [578]
M (NCS)4 Cd [66, 296, 579] (L5 = (NH3)6, en2Cl, en2H2O,
(M = Co, Ni) M (NCS)4 Hg [46, 66, 577] Et2NO2) M (NCS)6 Co [825]
(M = Cu, Zn, Cd, Pb, Mn, (M = Sn, Ti)
Fe, Co, Ni) Cr (NCS)6 Ag3 [46, 256]
L2M (NCS)4 Hg [69, 70, 71, [Cr (NCS)6]2 m3 [46, 256]
814] (М = Cu, Cd, Hg, Pb)
(M = Zn, Cd, Co, Cu, Fe; [Cr(NH3)2 (NCS)4]2M-H2O [247, 248,
L = Thf, Diox, Py, Ph3P, PhNH2, Ph3As) (M = Hg, Mn, Co, Ni) 494|
[Me4N] (OC)5 Cr — NCS — [274] M (NCS)6 Pt [192, 579]
M (Co)6J (M = Cu, Cd, Hg, Pb, Fe, Co,
(M = MO, W) [Ni (Phen)3] [(OC)6 W—NCS— [274] Ni)
Mo (CO)&]2
* Структурные исследования.
** ПМР-метод.
*** Образуются как промежуточные (в растворе) при взаимодействии Hg2+ и L5Co (NCS)n+.
Таблица 5.11. Примеры инверсии SCN-группы в соединениях одного и того же
переходного металла
Cu (tripyam) (SCN)2 цис-Mn (CO)3 (Ph3As)Q SCN [580]
Cu (tripyam) (SCN) (NCS) [151] транс-Mn (CO)3 (Ph3As)2 NCS
Cu (tripyam) (NCS)2 цис-Mn (CO)3 (Ph3Sb)2 SCN [580]
транс-Mn (CO)3 (Ph3Sb)2 NCS
Cu (dppa) (SCN)2 CpFe (CO)2 SCN [581, 908]
Cu (dppa) (SCN) (NCS) [151] K3Co (CN)5 SCN [508, 525]
Cu (dppa) (NCS)2 K3C0 (CN)5 NCS
[Co (NH3)6J [Co (CN)6 SCN] [904]
CpMo (CO)3 SCN [272] [Co (NH3)6] [Co (CN)6 NCS]
CpMo (CO)3 NCS [Me4N]3 [Co (CN)6 SCN] [904]
Cp2WO (SCN)2 [8411 [Me4N]3 [Co (CN)6 NCS]
Cp2 WO(NCS)a Mn (CO)5 SCN [Co (NH3)5 SCN] Cl2-1,5 H2O [582]
Mn (CO)5 NCS [542] [Co (NH3)6 NCS] Cl2
207
Продолжение табл. 5.11
[Ph4As] Со (DH)2 (SCN)2
[Ph4As] Со (DH)2 (SCN) (NCS) [Ph4As] Co DH)2(NCS)2 [543]
CoL (DH)2 SCN) * [583, 905,
CoL (DH)2 NCS] (L = CNPy, Py, Bu3N, Ph3P и др.) 906]
Co (DH2) (DH) (SCN)2 Co (DH2) (DH) (NCS)a [910]
[Rh (NH3)5 SCN]2+) ** [Rh (NH)3)5 NCS]2+J [584, 585]
Rh (Me2PhP)2 C12NCS Rh (Me2PhP)2 C12SCN [892]
* ПМР-метод.
** Анион не указан. Вероятно, SCN.
*** Анионы PFg . SCN
CpFe (СО)2 NCS
[Ir (NH3)5 SCN]2+) **
[Ir(NH3)6 NCS]2+J
Pd (Ph3As)2 (SCN)0
Pd (Ph3As)2 (NCS);
Pd Dipy (SCN)2
Pd Dipy (NCS)2
[Pd (Et4dien) SCNj+l ***
[Pd (Et4dien) NCS]+j
транс-[Р( (SMe2)2 (SCN)2]
транс-[Pt (SMe2)2 (SCN) (NCS)]
транс-jpt (SMe2)2(NCS)2]
[584, 585]
[504, 505,507,
522, 908]
[507, 586]
[533]
[909]
в кристаллическом состоянии димерных ионов
NCS ^NCS
Zn
SCN^ \nCS
Аналогичное строение, вероятно, имеют и тритиоцианаты кадмия; для
последних мостиковая связь в растворе сохраняется [64]. Что же касается
тетратиоцианатов кадмия в комплексах К2 ICd (SCN)4] • 2Н2О и (NH4)2 •
• [Cd (SCN)4] • 2НоО, то мостиковая функция SCN-группы носит лишь
формальный характер, поскольку значение v (CN) в этих соединениях не пре-
вышает 2100 см-1 [46, 494, 511]. Поэтому результаты структурного исследо-
вания первого соединения [492], в котором предполагалось наличие мости-
ковой SCN-группы, вызвали сомнения в их достоверности [3]. В соединениях
NaNH4 [Cd (SCN)4], Na2 [Cd (SCN)4] и NaK [Cd (SCN)4] мостик упрочняется
и появляется высокочастотная компонента 2150 см-1 [511].
К числу соединений с мостиковыми SCN-группами относится [Ph4As]2 •
[Ni (NCS)4], которое существует в двух модификациях, имеющих олив-
ково-желтую и сине-зеленую окраску. Для модификации желтого цвета маг-
нитный момент равен 3,33 М. Б. (25° С) и близок к магнитному моменту
[Et4N]2 [Ni (NCS)4] (3,25 М. Б.). Данные ИК-спектров подтверждают окта-
эдрическую конфигурацию формы, имеющей желтый цвет, с мостиковой
группой [68]. По этим же данным предполагается октаэдрическая структура
с двумя мостиковыми SCN-группами и в комплексе [Et4N]2 [Cu (NCS)4J
[66]. аким образом, с помощью ИК-спектральных данных можно сделать
вывод о координационном числе центрального атома.
По сравнению с однометалльными мостиковыми соединениями (тип
М—SCN—М) более полно исследованы разнометалльные тиоцианаты (тип
М—SCN—М') и, в частности, комплексы, содержащие Hg или Pt, а также один
из d-металлов 4-го периода (Сг, Mn, Со, Ni) или Cd, Zn. Некоторые из них
изучены рентгеноструктурно. В соединениях типа MHg (SCN)4 (М = Си,
Cd, Mn, Fe, Со, Ni и др.) ион ртути обычно имеет тетраэдрическое окружение,
а ион другого металла может быть окружен четырьмя или шестью лигандами.
Например, в CoHg (SCN)4 [512] ион кобальта имеет тетраэдрическое окру»
208
Рис. 5.1. Предполагаемое строение
[(CH3)3PtSCN]4
жение, а ионы никеля и меди в аналогичных комплексах имеют координа-
ционное число 6. Медь в CuHg (SCN)4 окружена четырьмя атомами азота и
двумя атомами серы (квадратная бипирамида) [513]. Координационное чис-
ло 6 реализуется и в соединениях типа CoHg (SCN)4 • 2L [69] (L = Thf,
Diox, Py, PhNH2), в которых ион кобальта дополняет свое координацион-
ное число до 6 за счет донорных молекул органических лигандов. В этих
соединениях предполагается наличие четырех мостиковых SCN-групп.
В то же время соединение CoHg (SCN)4 •
• 2Ph3P имеет большое значение магнит-
ного момента, что свидетельствует о
существовании как мостиковых, так и
концевых SCN-групп у ртути и коорди-
нированных молекул трифенилфосфина
у кобальта, а оба иона металла имеют
тетраэдрическое окружение [69], кото-
рое сохраняется также в MHg (SCN)4 •
• 2Ph3P (М = Zn, Cd) [71].
Для гексатиоцианатохромитов Ag,
TI, Hg, Pb мостиковая структура фик-
сируется в ИК-спектре довольно четко
(у (CN) > 2100 см-1), а связь Сг—NCS
(концевая) легко определяется по дан-
ным частоты v (CS) (800—820 см-1) и
v (CrN) [256]. Если для Ag, Hg и Pb об-
разование мостиковых структур с ионом
хрома вполне закономерно, то образо-
вание мостика Т1—SCN—Сг является
несколько необычным фактом, посколь-
ку связь SCN-группы с ионом таллия (I)
на 37% ионная [96, 97].
В соединениях М [Сг (NH3)2 (NCS)4]2
(М — Mn, Со, Ni) затруднительно определить наличие мостиков меж-
ду хромом и металлом, хотя и предполагается образование структур
Сг — NCS — М [247, 248]. Затруднения в образовании мостиковых структур
Cd — SCN — М можно предполагать и для MCd (SCN)4 (М = Со, Ni) [256, 511].
Кроме мостиков М — SCN — М и М — SCN — М', которые легко обна-
ружить по значительному смещению полосы v (CN) относительно к таковой
тиоцианат-иона, существует еще одна разновидность координации металла,
в которой атом серы SCN-группы одновременно координирован двумя цент-
ральными атомами,
м м
А) М—NCS<^ или Б) NCS<^
ХМ ХМ
Координация типа А имеет место в соединении CoHg2 (SCN)6 • С6Н6.
При этом Со имеет октаэдрическое (6N), a Hg тетраэдрическое (4S) окруже-
ние, а все SCN-группы образуют мостики между Со и Hg, причем четыре из
/Hg
них — Со—NCS—Hg, а два — Со—NCS\ [514]. Подробнее о
\Hg
структуре этого соединения см. ниже.
209
Такой же тип координации SCN-групп проявляется и в соединении
Ag (Ph3P)oNCS, в котором два удлиненных контакта Ag...S длиной 2,83
и 2,88 А «сшивают» отдельные молекулы в цепи с двумя мостиковыми
SCN-группами [515]
zAg
Ag-NCSC
''Ag
/М
По-видимому, мостики М—NCSy образуются и в соединении
[Ni (NO) (NCS)JX [5161, строение которого можно представить в виде поли-
мерной сетки с тетраэдрическими уз-
лами [3]
ON—Ni—NCS
NCS ON—Ni—NCS/
1 / \
ON—Ni—NCS
Тип координации Б предполагает-
ся в тетрамере [Me3PtSCN |4, исследо-
ванном ИК-спектральным и ЯМР-ме-
тодами [517] (рис. 5.1), циклическом
тримере [Et2MSCN]3 (М = Al, Ga, In)
[518] и в соединении [Cu2 (Dben)4 •
• SCN (С1О4)21 С1О4 [152]. Как проме-
жуточный радикал NCSy может
Рис. 5.2. Фрагмент структуры комплекса
[Cuen2NCS]ClO4 [520] образовываться в растворе при об-
/Pt
разовании активированного комплекса NCS'
[519].
Что же касается возможности реализации мостиковой структуры за счет
/ >М\
атома азота I SCNy^ I, то о ней свидетельствуют лишь результаты рентге-
ноструктурного анализа, например, Cuen2 (NCS) С1О4 [520], для которого
.Си
предполагается существование мостика SCN:' (рис. 5.2). Однако ча-
‘••Си
стота у (CN) тиоцианатной группы этого соединения близка к таковой
Му ZM
NCS-иона. Мостики pNCS\ образуются в (CH3)2T1NCS [937].
ХМ
210
5.9.2. Инверсия связи SCN-группы
Как видно из табл. 5.8—5.10, одни металлы присоединяют SCN-группы
через атом азота, другие — посредством атома серы. А иногда один и тот же
металл присоединяется как к жесткому (N), так и к мягкому (S) концу тио-
цианатной группы. Можно предположить, что инверсия связи с SCN-груп-
пой в соединениях металлов промежуточной области (Pb (II), Fe (II),
Со (II), Ni (II), Bi (III), Со (III), Rh (III), Ir (III) и др.) должна проявлять-
ся довольно часто. И действительно, для большинства из перечисленных
ионов металлов характерно «обращение» тиоцианатной группы (см.
табл. 5.11).
Известно, что с Mn (II), Со (II), Ni (II) тиоцианатная группа координи-
руется, как правило, своим жестким концом, но в некоторых соединениях,
например М [Cr (NH3)2 (NCS)4]2, эти ионы могут присоединять SCN-rpynny
и посредством атома серы [247, 248]. В подобных комплексах, содержащих
мостиковую SCN-группу, инверсия М—N -> М—S является «вынужденной»,
ибо в наличии имеется как мягкий, так и жесткий металл.
Большой интерес представляют те случаи инверсии связи в тиоцианатных
соединениях, когда в составе последних содержится только один металл-
комплексообразователь. Наибольшее число случаев изомерии связи встре-
чается в комплексах типа ML2 (SCN)2 (М = Pd, Pt и некоторые металлы по-
граничной области).
Влияние лиганда на инверсию связывания было впервые отмечено в ра-
боте [521] при изучении ИК-спектров ряда соединений: К2 [М (SCN)4],
[М (NH3)2(SCN)2] и [ML2(SCN)2] (М = Pd, Pt; L = Et3P). Несомненно,
что донор но-акцепторные свойства лиганда L являются главным фактором,
определяющим тип связи в комплексах одного и того же металла. С этой точ-
ки зрения было сделано наибольшее число попыток объяснить причину пере-
хода от S- к N-связыванию и выявить общие закономерности этого обращения
[271, 507, 521—524, 546].
Вполне убедительно выглядит объяснение инверсии М—S -> М—N в сме-
шанных комплексах палладия и платины [521] как следствие влияния фосфи-
нов, арсинов и других подобных лигандов, которые благодаря л-дативному
эффекту понижают электронную плотность центрального атома и тем препят-
ствуют участию атома серы SCN-лиганда в образовании новой л-связи. Сле-
довательно, присутствие в комплексе л-акцепторного лиганда дестабилизи-
рует л-дативную связь М—SCN. И чем сильнее л-акцепторная конкуренция
лиганда L, тем больше шансов, что инверсия М—S -> М—N будет иметь
место.
Пользуясь представлениями о «жесткости» и «мягкости», Бурмейстер
объясняет существование устойчивого изотиоцианатного комплекса [Rh •
• (NH3)5NCS]2+ [525] в отличие от тиоцианатного [Rh (SCN)6]3~ [258, 259,
494]. При этом он считает радикал [Rh (NH3)5] «жесткой» кислотой, в то
время как Rh3+ относится к пограничной области. Подобным образом мож-
но объяснить обращение SCN-группы в карбонильных соединениях неко-
торых металлов. Понижение заряда на металле в комплексе [Сг (CO)5NCS1
(связь Сг—N) объясняют изменение в способе координации при переходе к
[Mn (CO)5SCN] (связь Мп—S), так как считают одновалентный марганец
«мягкой кислотой» [271].
Замена части молекул СО, являющихся слабыми л-акцепторами, на ли-
ганды с более выраженными л-акцепторными свойствами приводит к обраще-
нию связи Мп—SCN в Мп—NCS. Например, в транс-[Мп (CO)3(Ph3P)2 •
211
• NCS] тиоцианатная группа координируется через азот, а в [Мп (СО)4 •
• (Ph3P) SCN] — посредством серы.
Различие в способе координации SCN-группы для комплексов [Со •
• (NH3)5NCS] (NO3)2 [588] и К3 [Со (CN)5SCN] [525] можно объяснить с по-
зиций «химического симбиоза» [458], когда CN-лиганд с высокой донорной
функцией обусловливает более прочное закрепление SCN-лиганда (т. е.
через серу), в то время как NH3 способствует присоединению NCS-иона че-
рез азот.
Согласно правилу «антисимбиоза» Пирсона [526], два «мягких» лиганда во
взаимном транс-положении оказывают дестабилизирующее влияние друг на
друга, т. е. присоединение «мягкого» лиганда-основания к «мягкой» кислоте-
металлу уменьшает сродство последнего к другому «мягкому» лиганду.
Поэтому в транс-положении к сильноковалентному лиганду более выгодно
находиться «жесткому» лиганду. Так, в комплексах [Pt (Et3P)2 (NCS)2] и
и [Pd (Et3P)2 (NCS)2] вместо M—S-связей образуются М—N-связи [526].
Пользуясь правилом «антисимбиоза», можно объяснить также наличие
М—SCN- и М—NCS-координации в комплексах палладия [Pd (PN) (NCS) •
• (SCN)] [527, 528] и [Pd (PAs) (NCS, (SCN)] [527]. В соединении [Pd (PN) •
• (NCS) (SCN)] Pd—NCS находится в транс-положении к фосфору лиганда
PN, a Pd—SCN — к азоту PN, поскольку «антисимбиотический» эффект
уменьшается с понижением «мягкости» лигандов в ряду СНГ, Н~ > СО >
фосфины, арсины > галогены, О- и N-содержащие донорные лиганды [526].
С этих же позиций можно, вероятно, объяснить инверсию связей в комп-
лексах CuNCS • L, 2CuNCS • 3L (L = Ph2PfeCPPh2) [526, 5291, а также
смешанную координацию в некоторых комплексах меди (II) [151] и в соеди-
нениях золота (I) типа AuLSCN (L = Ме3Р, Ph3P, Ph3As и др.) [163]. Для
комплексов золота рассчитаны относительные содержания N- и S-изомеров.
Аналогичные [521] соображения были высказаны и в работе [522]. На
основе рентгеноэлектронного исследования комплекса [Pd (Ph3As)2 (NCS)2]
было показано, что эффективный положительный заряд на атоме палладия
выше в случае Pd—NCS, чем Pd—SCN [523].
Подобный переход от S- к N-связыванию наблюдается и в соединениях
платины. С трифенилфосфином и трифениларсином тиоцианат платины (И)
образует Pt—NCS-изомеры. В то же время в [Pt (Ph3Sb)2 (SCN)2] проявля-
ется связь Pt—SCN, так как л-акцепторяые свойства возрастают в ряду:
стибины < арсины < фосфины [522, 524]. Такое же структурное отличие
обнаруживается и для комплексов палладия [Pd (Ph3Sb)2 (SCN)2] и [Pd •
• (Ph3As)2 (SCN)2L Подобным образом можно объяснить переход М—S ->
->М—N в ряду аминов: [Pd (NH3)2 (SCN)2] [521], [PdPy2 (SCN)2], но
[Pd Dipy (NCS)21 (желтая форма) [507].
Влияние стерического фактора на способ координации SCN-группы ча-
сто трудно оценить, так как и электронный, и стерический факторы дейст-
вуют почти одновременно [4]. Правда, он может оказаться решающим в вы-
боре способа координации SCN-группы, как, например, в соединениях
[Pd (dien) SCN]+, [Pd (Et4dien) NCS]+ и др. [530—532]. Для последнего со-
единения в растворе изучена кинетика превращения [Pd (Et4dien) SCN И —>
[Pd (Et4dien) NCS]+, определены АТ/ и AS этого превращения, а также при-
ведены значения соответствующих частот (у (CN) 2125 см~ для S-изомера
и 2060 см-1 для N-изомера; v (CS) 710 см-1 для S-изомера и 830 см-1 для
N-изомера) [533].
Еще более убедительно влияние стерического фактора проявляется
в комплексах [PdL (CNS)2] (L = Ph2P (CH2)rtPPh2). Удлинение углеродной
212
цепи органического лиганда приводит к обращению Pd—SCN-координации
(п = 1) в Pd—NCS (п = 3). При п = 2 наблюдается смешанная Pd—S- и
и Pd—N-координация SCN-группы [534]. С точки зрения стерических за-
труднений рассматривается Pd—NCS-координация в [Pd (Ph3P)2 (NCS)2]
и Pd—SCN-связь в [Pd ((PhO)3P)2 (SCN)2] при замене громоздкого Ph3P
меньшим (PhO)3P [535], а также в других комплексах тиоцианата палладия
со смешанной N-, S-координацией [527].
Кроме стерического фактора, на способ координации SCN-группы оказы-
вают существенное влияние растворитель и характер внешнесферного иона.
Так, в солях Na4 [Cd (SCN)6], Na2Ba [Cd (SCN)6] и др. осуществляется
Cd—SCN-координация, в (NH4)2Cs2 [Cd (SCN)6] и Cs2Ba [Cd (SCN)6] предпо-
лагаются как Cd—SCN-, так и Cd—NCS-связи. Считают, что легкополяри-
зуемый катион цезия создает благоприятные условия для более полярной
связи Cd—N [73].
К3 [Со (CN)5CNS] может существовать в виде двух изомеров: К3 [Со •
• (CN)5NCS] и Кз [Со (CN)5SCN] [508, 525, 536]. Замена катиона калия или
цезия тетрабутиламмоний-ионом способствует быстрой инверсии Со—SCN->
-> Со—NCS [536, 904]. Об изменении способа координации SCN-группы под
влиянием внешнесферного аниона сообщалось в работах [537, 904].
Изомерия связывания тиоцианат-группы в среде различных раствори-
телей, а также при растворении кристаллического вещества наблюдается
сравнительно часто. Например, в водном растворе соли Na2 [Cd (SCN)4]
осуществляется тиоцианатная, а в метанольном —изоцианатная структура
[487]. Аналогичная картина в тех же растворителях наблюдается для
KCs [Cd (SCN)4 ] • Н2О и Cs4 [Cd (SCN)el • H2O : Cd—SCN (вода) и Cd—NCS
(метанол). Но в ацетоновом растворе три- и тетратиоцианатокадматов
одновременно присутствуют и Cd—SCN и Cd—NCS, а для гексатиоцианато-
кадматов только Cd—NCS [538]. Этот факт, по мнению авторов работы [538],
можно объяснить граничным положением кадмия в ряду жестких и мягких
кислот, а также тем, что менее полярные растворители способствуют образо-
ванию более полярной связи Cd—NCS. Образование полярных связей М—N
в малополярных растворителях (С6Н6, СС14, СНС13, СН2С13 и др.) и ковалент-
ной М—S в растворителях с относительно высокой диэлектрической прони-
цаемостью (PhCN, MeCN, DMFA, DMSO) наблюдается для некоторых комп-
лексов [MLn (CNS)21 (n = 1,2) палладия и платины [357, 539, 890]. Довольно
детально исследовалась изомеризация связи в комплексе [СоРу (DH)2SCN]
и ему подобных как в высокополярных (DMFA, DMSO, FA), так и в низко-
полярных (СНС13) растворителях [540, 541, 905, 906]. Показано, что равно-
весие
[CoL (DH)2 SCN] [CoL(DH)2NCS]
(L = Py или другой монодентатный лиганд) сдвигается вправо в растворителях
с высоким значением f и влево в низкополяриых растворителях или их сме-
сях. Кпометого, в растворе возможно образование мостиков Со—NCS—Со
[5411.
Примером инверсии связи, наблюдаемой при переходе от кристалличе-
ского состояния к раствору, может служить обращение [Mn (CO)5SCN]
(кристалл) —>- [Мп (CO)5NCS] (ацетонитрил). Значение частот v (CS) и v (CN)
соответственно 670 и 2160 см-1; 800—810 и 2113 см-1 [542]. К числу анало-
гичных примеров можно отнести изомерию связи в комплексах [Со (NH3)4 •
• SO3SCN] и [Со (NH3)4SO3NCS] • 2Н2О причем изотиоцианатная структура
реализуется в водном растворе, а роль кристаллизационной воды в последнем
соединении сводится к образованию Н-связей с NCS-группой [509].
213
Кроме приведенных выше примеров М—S-, М—N-, а также смешанной
М—S- и М—N-структур, для некоторых тиоцианатов известны случаи суще-
ствования всех трех возможных изомеров, как, например, в соединениях
[Cu (tripyam) (SCN)21 (темно-зеленый), [Cu (tripyam) (SCN) (NCSJ (желто-
зеленый) и [Cu (tripyam) (NCS)2] (коричневый), а также аналогичного соста-
ва и цвета изомеров с фенилди-2-пиридиламином [151]. Случай подобной изо-
мерии установлен и для [Ph4As] [Со (DH)2(SCN)2] [543], который в дихлор-
этане изомеризуется в [Ph4As] [Со (DH)2(SCN) (NCS)] и [Ph4As] [Со (DH)2 •
• (NCS)2]. Инверсия Со—SCN в Со—NCS в дихлорэтане еще раз подтвержда-
ет отмечавшийся выше факт, что в малополярных растворителях преоблада-
ет М—N-координация [538, 539, 906]. Ряд интересных примеров подобного
типа приведен также в обзоре [4] и в работе [532]. Известны случаи изомерии
связи мостиковой SCN-группы [545], инверсия связи FeNCS2+ -> FeSCN2-r
при изменении кислотности раствора [544], а также при изменении темпера-
туры в некоторых соединениях родия [892].
Изомерия связывания тиоцианатной группы и других псевдогалоген ид-
ионов с позиций теории ЖМКО, влияния стерических и электронных фак-
торов, а также эффекта растворителя рассмотрена в обзорной работе
[907].
Помимо ИК-спектрального критерия, для оценки типа координации
SCN-группы иногда применяют рентгеноэлектронную [902, 903] и ЯМР
(ПМР)-спектроскопию [574, 583, 908, 909]. Так, на основании рентгеноэлект-
ронных спектров простых и комплексных тиоцианатов металлов было уста-
новлено, что разность энергий Cis—S2pi/2, 3/2 при координации SCN-груп-
пы через атом азота увеличивается по сравнению с таковой в KNCS, а при
координации через атом серы эта разность уменьшается [902]. В последнем
случае в спектре появляется узкий длинноволновый максимум, интенсив-
ность которого понижается, если SCN-группы являются мостиковыми. Он
исчезает при координации SCN-группы через атом азота или если с металлом
она образует ионную связь [903]. ЯМР-спектроскопия с использованием лан-
таноидных сдвигающих реагентов применялась при исследовании соедине-
ний CpFe (CO)2SCN и CpFe (CO)2NCS, Pd (Ph3As)2 (SCN)2 и Pd (Ph3As)2 •
• (NCS)2 и др.
Величина химического сдвига для М—SCN-связи значительно больше,
чем для М—NCS-связи [908].
5.9.3. Рентгеноструктурное исследование
Полагают, что к 1967 г. число тиоцианатных соединений с частично или
полностью расшифрованными структурами составляло около 60 [3]. В по-
следние годы не только возросло число рентгеноструктурных исследований,
но и повысилась надежность и точность полученных результатов. Возрос
также интерес к структурам соединений, которые содержат в своем составе,
кроме SCN-группы, и органические полидентатные лиганды.
Неравноценность химических связей в разнолигандных комплексах,
а также амбидентатность SCN-группы приводят к усложнению структуры
соответствующих соединений. Это требует осторожности в проведении ана-
логии между структурно изученными и неисследованными комплексами од-
нотипного состава для различных металлов. Например, [Cuen3 (NCS)] С1О4
[520] и [Nien2 (NCS)] • С1О4 [589] не являются изоструктурными, ибо послед-
214
нее соединение имеет димерный характер с двумя мостиками Ni—NCS—Ni,
.Cu
а в первом реализуется формальный мостик SCN<f
^Cu
Анализ данных о стереохимии простых и комплексных тиоцианатов ме-
таллов показывает, что почти всегда ион металла принимает наиболее харак-
терное для себя окружение, если нет веских причин к его изменению, на-
пример, стерического или другого характера. Так, Pd (II), Pt (II),
как правило, имеют квадратную, Со (III), Pt (IV) октаэдрическую,
а Со (II) и Zn (II) тетраэдрическую или реже октаэдрическую конфигу-
рации и т. д.
Повышение точности рентгеноструктурного анализа, а также применение
других физических методов исследования вещества (ИК-спектры, магнит-
ная восприимчивость и др.) позволили в одних случаях исправить явно оши-
бочные толкования о структурах, а в других — уточнить некоторые харак-
терные их особенности. К примеру, для К3 [Сг (NCS)6] • 4Н2О и К4 [Ni •
• (NCS)6]- 4Н2О предполагалось, что координация SCN-группы осуществля-
ется через серу [590] по аналогии с координацией этой группы в соединении
К2 [Pt (SCN)6I. Анализ ИК-спектров этих соединений показал, что в них
имеет место NCS-координация. Подобные ошибки встречаются в работах
последних лет (например, трактовка типа координации SCN-группы в
[NiPn2 (NCS)21) [592, 5931.
В табл. 5.12 приведены следующие структурные данные тиоцианатов:
а) форма полиэдра комплексного иона; б) способ присоединения тиоцианат-
ной группы и в) кристаллографические характеристики тиоцианатного сое-
динения.
В ряде случаев трудно оценить функциональную роль SCN-группы в
структуре соединения, так как расстояния М—S и М—N часто больше
длины обычных ковалентных связей на несколько десятых ангстрема. Так,
в CuPn2 (SCN)2 [594] расстояния Си о.. SCN обеих SCN-групп, занимающих
аксиальные позиции, равны 3,154, А, что отвечает лишь слабому взаимо-
действию между атомами меди и серы, а в соединении Cuen2 (NCS) С1О4
[520] расстояния контактов Си ... N составляют 2,37 А, что также значитель-
но больше длины обычной ковалентной связи Си—N приблизительно на
0,7 А. Подобные различия наблюдаются и в параметрах соли К2 [Cd (SCN)4] •
• 2Н2О [492, 595], где расстояния Cd ... N равны 2,45 А, т. е. являются
завышенными на ~0,4 А. Действительно, в ИК-спектрах последних двух
соединений не обнаруживаются высокочастотные компоненты v (CN), ко-
торые указывали бы на наличие мостиковых SCN-групп. Поэтому вопрос,
являются ли подобные контакты M...S или M...N координационными или
нет, решается в каждом конкретном случае отдельно, исходя из общей карти-
ны структуры, геометрии координационного полиэдра соединения, его изо-
структурности с другими подобными соединениями и т. п.
Угловые линейные характеристики в табл. 5.12 за редкими исключе-
ниями, оговоренными в тексте, не приводятся. Укажем лишь, что в подавля-
ющем большинстве случаев угол MNC находится в пределах 170—180° и
только в отдельных случаях составляет менее 160° [596, 597 и др.]. Повтор-
ные рентгеноструктурные исследования позволили исправить ошибочные
значения валентных углов атома азота в сторону их увеличения. Так, для
комплекса [Pt2Cl2 (Рг3Р)2 (SCN)2] в первоначальном варианте для его сс-
изомера угол PtNC был найден равным 147,7° [598], что позволило высказать
предположение [3] о возможных стерических препятствиях, обусловленных
участием обеих SCN-групп в образовании мостиков между атомами платины.
215
Таблица 5.12. Основные структурные и кристаллографические параметры тиоцианатных
Соединение Коорди- национ- ное число Координационный полиэдр Способ координации SCN-группы
[Zn (N2H4)2 (NCS)2] 6 Октаэдр: 4NL, 2N Zn—NCS
[Zn (NH2C6H5)2 (NCS)2J * 4 Тетраэдр: 2NL, 2N Zn—NCS
K2 [Zn (NCS)J -3H2O 4 Тетраэдр: 4N Zn—NCS
[Zn (tren) NCS] SCN 5 Тригональная бипирамида: Zn—NCS
4Nl, N SCN-ион
[Zn en (NCS) Cl] 4 Искаженный тетраэдр: 2Nl, N, Cl Zn—NCS
[Zn2en4 C2O4] [SCN)2 6 Октаэдр: 4N£, 2OL SCN-ион
[Zn2Pn4C2O4] (SCN)2 6 Октаэдр: 4N^, 2OL SCN-ион
[Cdtu4] [Zn (NCS]4 * 4 Тетраэдр: 4N Zn—NCS
[Cdu2 (NCS).,] 4-J-2 Искаженный октаэдр: 20^, 2N, 2S Cd—NCS—Cd
[Cd (Etu)2 (SCN)2] 4 4-2 Искаженный октаэдр: 2SL, 2S, 2N Cd—NCS—Cd
[Cd (Et2N2O) (NCS)2] 44-2 Искаженный октаэдр: NL, 0L, 2N + 2S Cd—NCS—Cd
[Cd en2 (NCS)2] 6 Искаженный октаэдр: 4NL, 2N Cd—NCS
[Cd en (NCS) (C204)o 5] [Cd en2 (NCS) Cl] 5 + 1 Искаженный октаэдр: 2N£, N, 2Ol + Ol Cd—NCS
5 + 1 Искаженный октаэдр: 4NL) N + Cl Cd—NCS
[CdPn (NCS)2]
4 + 2 Искаженный октаэдр: (две Cd—NCS—Cd
34-3 независимые молекулы: 2Nl, 3N, S и 2Nl, N, 3S)
K2 [Cd (SCN)4].2H2O 4 + 2 Искаженный октаэдр: Cd—SCN
4S 4- 2N Cd—SCN—Cd
Cd (NCS.,) Hg 4 Тетраэдр: 4N Cd—NCS—Hg
Hg (SCN)2 Hg (SCN) Cl * 2 +(4) Линейная: 2S Октаэдр. 2S 4- 4N Hg—SCN . . . Hg
2 +(4) Искаженный октаэдр: S, Cl + 2N 4- 2С1 Hg—SCN . . . Hg
Hg (SCN) Br *
2 +(4) Искаженный октаэдр: S, Br + 2N + 2Вг Hg—SCN . . . Hg
EtO(CH3)2C-
2 Линейная: С, S Hg—SCN
C (CH3)2 CH2HgSCN * MeO (CH3)2 C- 2
Линейная: С, S Hg—SCN
•C(CH3)2CH2HgSCN *
UHC-[Hg (Phen)2 (SCN)2] 6 Искаженный октаэдр: 4N, Hg—SCN
[Hg (Ph3As) (SCN)2] 3 + (2) Плоский искаженный тре- угольник: 2S, As Hg—SCN
[Hgtu2 (SCN)2] Искаженная тригональная бипирамида: 2S, As + 2N Hg—SCN . . . Hg
2 + 4 Искаженный октаэдр: 2S + 4Sl Hg—SCN
Hg(SCN)4Ni (H2O)3 4 Тетраэдр: 4S Hg—SCN—Ni
K[Hg(SCN)3] 2 + 2 Искаженный тетраэдр: Hg—SCN
Cs [Hg (SCN)3] 2S + 2S SCN-ион
3+1 Искаженный тетраэдр: Hg—SCN—Hg
3S+ N Hg—SCN
(3) (Плоский искаженный треугольник: 3S) (Hg—SCN)
216
соединений
а, А ь, А 1 с, А а ₽ V Z Прост- ранствен- ная группа Литера- тура
7,141 14,576 4,214 ,— 105° 30' 2 P2i/a [663]
14,56 9,10 12,7 — — — 4 Ima2 [664]
11,10 13,00 5,41 — — — 2 Р212121 [662]
12,888 16,466 13,633 — — — 8 РЬса [628, 629]
11,25 9,80 7,36 — — — 4 Рпа2х [665]
11,61 12,20 11,70 — — 132,5° 4 P2i/b [666]
6,56 17,03 14,11 — — 106,0° 4 P2i/b [667]
17,207 17,207 4,16 — — — 2 14 [646]
8,63 13,49 8,66 — — 98° 4 P2/b [668]
15,60 8,17 11,51 — 95° 41' — 4 C2/c [644]
9,735 16,202 10,640 — — — 4 P2i/c [669]
9,27 10,30 8,15 — — 123° 2 P2i/b [670, 671]
9,54 7,90 5,97 111,0° 99,8° 89,3° 2 Pl [639]
12,40 11,07 9,30 — — — 4 P21212J [639, 671]
11,73 18,94 12,09 — — 122,0° 8 P2i/b [672]
12,96 5,71 18,85 — 97° 30' — 4 C2/c [492, 595]
11,48 — 4,33 — — — 4 14 [651, 652]
10,878 4,042 6,435 — 95,28° — 4 C2/m [673]
10,16 4,23 10,40 — — — 4 P212121 [492]
6,24 4,28 8,74 — 91° 20' — 2 P2JH1 [492]
15,62 13,33 6,23 — — — 4 P212121 [674]
20,32 7,82 7,48 — 96° 36' — 4 P2x/a [675]
13,252 11,077 8,443 105,20° 83,25° 90,92° 2 Pl [676, 677]
10,290 21,199 10,719 — 112,0° — 4 P2i/c [678, 932]
8,63 9,35 15,92 — — — 4 Pbc2i [679]
13,23 5,27 18,53 — 92,9° — 4 C2/c [680]
11,081 4,07 10,915 — 114° 45' — 2 P2x/m [3,65, 492]
9,543 10,899 19,965 — — — 8 P2x/c [682]
217
Продолжение табл. 5.12
Соединение Коорди- национ- ное число Координационный полиэдр Способ координации SCN-группы
NH4[Hg (SCN),] ** 2 + 2 Искаженный тетраэдр: 2S + 2S Hg—SCN SCN-ион
]Hgen (SCN)2J 4 Искаженный тетраэдр: 2NZ, 2S Hg—SCN
[Hg (Ph3P)2 (SCN)2] 4 Искаженный тетраэдр: 2Р^, 2S Hg—SCN
[Ph4P]2 [Hg (SCN)4] 4 Искаженный тетраэдр: 4S Hg—SCN
[Cuen2] [Hg (SCN)4] 4 Тетраэдр: 4S Hg-SCN
K2 [Hg (SCN)J *** 4 Тетраэдр: 4S Hg—SCN
Hg (SCN)4 Co 4 Тетраэдр: 4S Hg—SCN—Co
Hg (SCN)4Cu 2+2 + + (2) Искаженный тетраэдр: 2S+ 2S Искаженный октаэдр: 2S + 2S + (2S) Hg—SCN—Cu Hg—SCN—Cu Cu
Hg2 (SCN)6 Co - C6H6 2 + 2 Искаженный тетраэдр: 2S + 2S Hg—SCN—Co Hg—SCN—Co HgZ
моноклинная (CH3)2T1NCS 6 Искаженный октаэдр: 4N, 2С и 4S, 2С TI TI
ромбическая 6 Искаженный октаэдр: 4N, 2S, 2С NCS^ П TI
[(СбН^ SiNCS] 4 Искаженный тетраэдр: ЗС, N Искаженный тетраэдр: ЗС, N Si—NCS
[(CH3)3 SnNCS] 4 Sn—NCS . . . Sn
{(CH3)2 Sn (NCS)2] ’ 4 Искаженный тетраэдр: 2С, 2N Sn—NCS . . . Sn
[(C6H5)a SnNCS] 4+1 Искаженная тригональная бипирамида: ЗС, N + S Sn—NCS . . . Sn
{[(CH3)2 SnNCS]2Ob2 Sn (2) = 5 Sn(l) = = 5+1 Искаженная тригональная бипирамида: 2С, 20, N Искаженный октаэдр: 2С, 0, N + N + (Sn) Sn—NCS Sn—NCS Sn''
Pb (SCN)2 8 Восьмивершинник: 4S, 4N Pb Pb SCN Pb 'pb
Rb [Bi (SCN)4] * 4 «Качели»: 4S Bi—SCN
KSe (SCN)3-i/2H2O 4 Квадрат: 4S Se—SCN Se NCS< ^Se
Se(SCN)2 2 Уголковая: 2S Se—SCN
[Me^N] [C6H5Te (SCN)2] 3 Плоский треугольник: С, Те—SCN
[CuPy2 (NCS)2] * fCu-(PyNH NHPy) NCS] SCN 4 + 2 2S Тетрагональная бипирами- да: 2N£, 2N + 2S Cu—NCS . . . Cu
а-форма 5 Искаженная тригональная Cu—NCS
P-форма 5 бипирамида (обе формы): 4N£, N SCN-ион
218
a, A ь, A c. A a ₽ ? z Прост- ранствен- ная группа Литера- тура
11,185 4,08 10,935 — 114° 55' — 2 P21/m [3,65, 492]
12,30 11,33 7,11 — — — 4 Pna2i [681]
17,385 10,581 19,304 — 91,41° — 4 P2i/c [684]
12,859 13,494 16,583 89,68° 76,78° 61,59° 2 Pl [935]
7,51 18,03 14,20 — 96° 32' — 4 P2i/c [600]
11,04 9,22 13,18 — 106° 30' — 4 C2/c [492, 595]
11,109 — 4,379 — — — 2 14 [512, 649]
9,03 7,68 15,15 — — — 4 Pbcn [513]
7,59 8,37 8,87 98,31° 101,01° 95,50° 1 Pl [514]
14,036 4,317 14,617 — 93,2° — 4 C2/m [937]
10,51 4,34 14,56 — — — 4 Pcma
9,402 18,735 11,350 — 118,98° — 4 P2x/c [921]
13,20 10,28 12,01 — — — 8 Pbca [563]
9,82 7,87 5,57 — — — — Pmmn или [562]
P2!mn
19,02 11,67 15,49 — 95,64° — 8 P2t [685]
21,73 7,88 10,96 — 134,7° — 4 C2/m [657]
9,661 6,544 8,253 — 92,37° — 4 C2/c [564, 565]
7,65 11,24 6,52 _ 2 P2A2, [138]
8,775 15,067 8,956 119,64° 101,03° 99,62° 4 Pl [9191
9,87 13,03 4,44 4 Pnma [687]
15,747 9,219 23,153 — 100,58° — 8 C2/c [920]
9,17 14,31 5,65 — 108° — 2 C2/m [688]
16,63 7,88 9,11 124,4° 83,0° 91,6° 2 Pl [689]
26,75 7,62 19,08 — 90,36° — 8 B2x/c
219
Продолжение табл. 5.12
Соединение Коорди- национное число Координационный полиэдр Способ координации SCN-группы
[CuLNCS] L=C6H4OC (=0) nn=chc5h4n 4+1 Квадратная пирамида: 0L, 2Nl, N + S Cu—NCS... Си
[Cu (NH3)2 (NCS)2] 4 + 2 Тетрагональная бипирами- да: 2Nl, 2N + 2S Си—NCS ... Си Си—NCS
Cu (NH3)4 (SCN)2 4 + 2 Тетрагональная бипирами- да: 4Nt + 2S SCN-ион
Cu en2 (SCN)2 4 + 2 Тетрагональная бипирами- да: 4NL + 2S SCN-ион I j
[Cu2 (NH3)3 (CNS)3] II I 4 Cu (I) Искаженный тетраэдр: 2S, 2N Cu—SCN—Си I
[Cu (NH3)3 NCS] [Cu (SCN)2] 4 + 1 + + 1 Cu (II) Искаженная тетрагональ- ная бипирамида: 3N£, N + + s + s I .Си Си—NCS( II 4 Си II II Си—NCS ... Си
Cu en2 (NCS) C1O4 4 или 4+2 Квадрат или искаженный октаэдр; 4NL или 4N£ + + 2N SCN-ион
Cu [CH.,NH (CH2)2 NHCH+- • (NCS)*2 4 + 2 Октаэдр: 4N^ + 2N SCN-ион
{Cu [CH2NH (CH2)2 NH2]2 NCS} • •SCN 4+ 1 Тетрагональная пирамида: 4Nl + N Cu—NCS... Си SCN-ион
CuPn2 (SCN) 2 4 + 2 Тетрагональная бипирами- да: 4Nl + 2S SCN-ион
[Cu (tren) (NCS)]2 (BPh4)2 5 Искаженная тригональная бипирамида: 4NL, N Си—NCS
[Cu (tren) NCS] SCN 5 Искаженная тригональная бипирамида: 4NL, N Си—NCS SCN-ион
[Cu (trien) SCN] SCN 4 + 1 Искаженная тетрагональ- ная пирамида: 4NL, S Си—SCN SCN-ион
Cu (NCS)4 Hg 4 + 2 Тетрагональная бипирами- да: 4N + 2S Си—NCS—Hg /Hg Си—NCS< ‘•Hg
[Cu (Et2N—C2H4O) NCS]2 4 + (1) Искаженная тетрагональ- ная пирамида: 4О£, N£, Си—NCS ... Си
[CuPn3 (NCS)] C1O4 5 N + S Искаженная тригональная бипирамида: 4N£, N Си—NCS
[Cu (dien) (NCS)2] 5 Тетрагональная пирамида: 3Nl, 2N Си—NCS
[€u (C5H10N2O) (NCS) DMFA]2 4+1 Искаженная тетрагональ- ная пирамида: 2NL, OL, N* Odmfa Си—NCS
[Cu (dpt) (NCSy 4+1 Тетрагональная пирамида: 3Nl, n + n Си—NCS
{[Cu (dpt) NCS] CIO4}2 5 + (l) Искаженная тетрагональ- ная бипирамида: SN^, N, S + Ощ Си—NCS—Си
220
a, A b, A c, A a ₽ V z Прост- ранствен- ная группа Литера- тура
16,46 15,72 5,63 — 92,4° — 4 P2i/n [928]
13,94 6,01 8,81 — — — 4 Pnma [690, 691]
6,85 7,78 9,52 — 90,5° — 2 I2/m [513]
7,352 9,364 6,585 86° 56' 113° 23' 125° 8' 1 Pl [692]
8,40 7,17 19,52 — 97,3° — 4 PSj/c [655, 656]
15,60 7,63 10,82 — — — 4 Pnam или Pna2j [520]
6,566 14,741 8,582 — 95,09° — 2 P2J/C [596]
12,038 14,080 8,805 — 98,98° — 4 P2i/n [693]
13,01 6,89 16,12 — 92,3° — 4 C2/c [594]
14,977 9,764 21,226 — 98,79° — 2 P2i/c [930]
9,158 14,000 11,285 — — — 4 P212121 [627]
10,803 9,381 13,815 — — — 4 P212121 [493]
9,03 7,68 15,15 — — — 4 Pbcn [513, 683]
12,058 7,821 11,316 — 101,57° — 4 P2i/n [694]
13,75 — 14,55 — — — 6 P63 [695]
11,24 14,02 7,32 — 97,5° — 4 P2x/n [695]
15,312 7,445 12,227 — 99,067° — 2 P2j/a [696]
7,58 14,12 13,58 — 113,2° — 2 P2x/c [929]
13,94 12,39 15,85 —- — — 8 Pbca [697]
221
Продолжение табл. 5.12
Соединение Коорди- национное число Координационный полиэдр Способ координации SCN-группы
{[Cu (dien) NCS] С1О4}2 i 5-Ь (1) Искаженная тетрагональ- ная бипирамида: 3NL, N, s + °С1О4 Си—NCS—Си
Си (Py-CH2-CH2NH2)2 (NCS)2 4 + 2 Тетрагональная бипирами- да: 4Nl + 2N SCN-ион
Си (2-окси Pn)2 (SCN)2 4 + 2 Тетрагональная бипирами- да: 4Nl + 2S SCN-ион
AgSCN 2 + 2 + + 2 Уголковая: (-cSAgN = = 165°) Искаженный тетраэдр: N, S + 2S Искаженный октаэдр: N, S + 2S + 2Ag Ag—SCN—Ag
AgSCN NH4SCN 1+3 Искаженный тетраэдр: S+ + 3S Ag—SCN SCN-ион
[Ag (Pr3P) NCS] 2+2 Искаженный тетраэдр: Р, N+2S Ag—NCS( X Ag
[Ag (PhgP)2SCN]2 4 3+1 Искаженный тетраэдр: 2Р, S, N Искаженная тригональная пирамида: 2SL, N + S Ag—SCN—Ag
{Ag [SC (NH2) NH (NH2)]2 SCN) з+1 + + (3) Искаженная пентагональ- ная бипирамида: 2SL, N + + S + (S + 2N) Ag—SCN . . . Ag
[Pr (H2O)6 (NCS)3]-H2O 8+1 Искаженная тетрагональ- ная антипризма: 3N, 50 + + о Pr—NCS
MNd (NCS)4 (H2O)4J (SCN)3 8 Додекаэдр: 4N, 40 Nd—NCS SCN-ион
[NdNO3 (NCS) (H2O)6] SCN • 2Urt • -2H2O**** 9 Т ригранецентрированная тригональная призма: 6Он2о> Ono3> nncs Nd—NCS SCN-ион
K4[Eu (NCS)4 (H2O)4J (SCN)3- -2H2O 8 Додекаэдр: 4N, 40 Eu—NCS SCN-ион
[Dy (H2O)6 (NCS)3] H2O 9 Т ригранецентрированная тригональная призма: 3N, 60 Du—NCS
[Bu4N]3 [Er (NCS)fi] 6 Октаэдр: 6N Er—NCS
[ErNO3 (NCS) (H2O)6] SCN-2Urt- •2H2O 9 Тригранецентрированная тригональная призма: 60н2о ®no3’ NNcs Er—NCS SCN-ион
Cs3[UO2(NCS)5] 7 Пентагональная бипирами- да: 20, 5N U—NCS
Cs4 [U (NCS)8] 8 Квадратная антипризма: 8N U—NCS
[Et4N]4 [U (NCS)8] 8 Куб: 8N U—NCS
(NH4)2 [VO (NCS)4 H2O]-4H2O 1+4 + + 1 Искаженный октаэдр: 4N, О’ Он2о V—NCS
[Ph4As]2 [NbO (NCS)5] 6 Искаженный октаэдр: 5N, q Nb—NCS
NH4 [Cr (NH3)2 (NCS)4].2/3H9O 6 Октаэдр: 2NL, 4N Cr—NCS
[PyH] [Cr(NHs)2 (NCS)J.H2O 6 Октаэдр: 2N^, 4N Cr—NCS
Choline [Cr (NH3)2 (NCS)4] • H2O 6 Октаэдр: 2NL, 4N Cr—NCS
222
a, A b, A e, A a 3 7 z Прост- ранствен- ная группа Литера- тура
11,81 7,48 7,48 70,9° 89,7° 105,4° 2 Pl [698]
8,398 14,751 7,834 — — 102,35° 2 P2t/c [699]
8,612 10,926 15,725 — — — 4 Pbca [700]
8,74 7,96 12,32 — 138,6° — 8 C2x/c [701]
4,02 23,86 7,23 — 96,08° — — P2j/n [702]
6,32 14,00 18,24 — 114° — 4 P^/fc [515]
13,210 13,470 10,346 88,14° 79,37° 113,10° 1 pT [704]
11,44 14,96 6,57 — — — 4 Pna2, [705]
14,55 15,25 7,76 — — — 4 Pna2j [553, 916]
25,25 20,62 6,86 — — 122,8° 4 B2/b [706, 918]
17,51 16,90 10,35 — — 105° 4 P2i/n [707]
25,41 20,32 6,52 — — 122,0° 4 B2/b [706, 918]
8,76 13,53 7,20 — — 117,0° 2 P2i [554, 916]
22,63 16,73 18,82 87,97° 89,08° 92,47° 4 Al [191]
17,30 16,78 10,18 — — 105° 4 P2i/n [707, 917]
13,629 13,249 11,556 — — — 4 Pnma [204]
11,958 — 11,170 — — — 2 P4/n [922]
11,64 — 23,01 — — — 2 I4/mmm [197]
16,50 6,82 16,32 — 94,5° — 4 P2x/n [708, 709]
12,43 37,59 13,17 — 119,5° — 4 P2i/c [710]
13,25 — — — — — 6 Im3m [602]
15,52 7,62 14,64 — 102° 24' — 6 A2/a [602]
12,69 22,80 6,75 — — 4 Ibma [602]
223
Продолжение табл. 5.12
Соединение Коорди- национ- ное число Координационный полиэдр Способ координации SCN-группы
[Но (C5H4NCO2H)3 (Н2О)2]. -[Cr (NCS)6]-2H2O 6 Октаэдр: 6N Cr—NCS
К3 [Мо (NCS)6] • Н2О • СН3СООН 6 Октаэдр: 6N Mo—NCS
[РуН]3 [Мо2О4 (СН3СОО) (NCS)J 6 Искаженный октаэдр: 2N, 30, oL Mo—NCS
[Mn (Et2N2O)2 (NCS)2] 6 Октаэдр: 2OL, 2NL, 2N Mn—NCS
[Me4N] [Тс (NCS)6] * 6 Тригонально растянутый октаэдр: 6N Tc—NCS
a-[FePy4 (NCS)2J 6 Октаэдр: 4NL, 2N Fe—NCS
[Fe (Dipy)2 (NCS)2] 6 Искаженный октаэдр: 4NL, 2N Fe—NCS
Co (NCS)4 Hg 4 Тетраэдр: 4N Co—NCS—Hg
K2[Co(NCS)4]-3H2O 4 Тетраэдр: 4N Co—NCS
Na2 [Co (NCS)4]-8H2O 4 Тетраэдр: 4N Co—NCS
[HgtuJ [Co(NCS)4] 4 Тетраэдр: 4N Co—NCS
(C2eH17N4) [Co (NCS)4] 4 Тетраэдр: 4N Co—NCS
[Co (N3S) (NCS)2] -1/2BuOH 5 Искаженная тригональная бипирамида: 3NL, 2N Co—NCS
[Co (N2P) (NCS)2] 5 Искаженная тригональная бипирамида (-> тетраэдр): 2Nl, Pl, 2N Co—NCS
[Co (N2PCH3) (NCS)J 5 Искаженная тригональная бипирамида (-> квадратная пирамида): 2NL, PL, 2N Co—NCS
[CoPy4 (NCS)2] 6 Октаэдр: 4NL, 2N Co—NCS
[Co (винил-Ру)4 (NCS^J 6 Октаэдр: 4NL, 2N Co—NCS
Co (NCS)6 Hg2 - C6H6 6 Октаэдр: 6N Co—NCS /Hg Co—NCS( XHg
a-[CoPy2 (NCS)2] 6 Октаэдр: 2NL, 2N, 2S Co—NCS—Co
NH4 [Co (DH)2 (SCN)2]-2H2O 6 Октаэдр: 4NL, 2S Co—SCN
[Co en2 (NCS) SO3] • 2H2O 6 Октаэдр: 4N£, N, Sso Co—NCS
[Co en NH3 (NCS) (NO2)2] 6 Октаэдр: 2NL, NL> Nncs, 2^no2 Co—NCS
[Co (tren) (NCS)2J SCN-H2O 6 Октаэдр: 4NL, 2N Co—NCS SCN-ион
[Co (NH3)5 SCN] C12-H2O 6 Октаэдр: 5NL, S Co—SCN
[Co (NH3)5 NCS] Cl2 6 Октаэдр: 5N£, N Co—NCS
[Co (NH3)5 NCS] (NO3)2 6 Октаэдр: 5NL, N Co—NCS
[(NH3)b Co-NCS-Co (CN)6] • H2O @ Октаэдр: 5NL, Nncs и 5Ccn. Sscn Co—NCS—Co
[SCNCo (NCS)2L9CoNCS] (L = C19H16N6O) 6 Искаженный октаэдр: SN^, 2N, S Co—NCS—Co Co—NCS
[Ni (N3As) NCS] BPh4 4 Квадрат: SN^, N Ni—NCS
[Ni (P2NO) NCS] PF6 5 Искаженная квадратная пирамида: 2Pr, Nr, Or, N Ni—NCS
[Ni (N3As) (NCS)2] 5 Искаженная квадратная пирамида: 3NL, 2N Ni—NCS
[Ni (tren) (NCS)2j 6 Искаженный октаэдр: 4NL, ,2N Ni—NCS
224
a, A b, A c. A a ₽ 7 z Прост- ранствен- ная группа Литера- тура
9,578 25,954 15,784 — 108,48° 4 P2T/c [711]
13,547 8,568 9,660 — — — 2 Pmmm [712, 713]
8,515 20,975 18,620 — 116,19° — 4 P2]/C [923]
9,332 7,193 11,159 113,9° 96,8° 105,7° 1 Pl [714]
8,84 — — 82° 56' — — 2 R3 [715]
12,25 13,18 16,46 — 117,9° — 4 C2/c [716]
13,17 16,50 10,08 — — — 4 Pcnb [717]
11,109 — 4,379 — — — 2 14 [512, 649]
11,116 12,981 5,354 — — — 2 P21212 [915]
19,00 — 5,47 — — — 4 P4 [718]
17,27 — 4,27 — — — 2 14 [647]
23,441 10,557 19,065 — 106,90° — 4 C2/c [660]
7,615 13,927 25,442 — 97° 59' — 4 P2i/c [624, 625]
15,896 8,786 9,402 111,3° 94,6° 90,5° 2 РГ [626]
13,485 13,503 16,034 — 116,22° — 4 Cc или C2/c [626]
12.48 12,9 16,5 — 118° 30' — 4 C2/c или Cc [728]
17,05 — 25,79 — — — 8 I4r/a [924]
7,59 8,37 8,87 98,31° 101,01° 95,50° 1 Pl [514]
9,09 14,60 5,66 — 111° — 2 C2/m [688]
9,98 12,07 8,48 — 95° 20' — 2 P2r/c [718]
9,06 6,59 22,84 — 95,4° — 4 P2i/c [719]
8,34 10,42 12,49 — 104° — 4 P2i/c [720]
15,615 16,22 13,63 — — — 8 Iba2 [931]
8,74 8,74 14,52 — — — 4 P2A2! [721]
10,16 — — — — — 4 Fm3m [588, 721]
10,687 — — — — — 4 Fm3m [588]
14,166 14,549 15,187 — — — 8 Pbca [.925]
10,58 15,66 27,58 — 111,9° — 4 P2i/c [722]
12,529 13,589 14,120 95,12° 103,68° 99,38° 2 Pl [723]
17,642 11,370 17,914 — 113°31' — 4 P2i/c [724]
15,550 14,552 18,114 — 127° 39' — 4 P2i/c [634, 635]
10,821 14,719 8,620 — — — 4 P212121 [631]
8 2473
225
Продолжение табл. 5.12
Соединение Коорди- национ- ное число Координационный полиэдр Способ координации SCN-группы
(Ni (Me6tren) NCS] SCN -Н2О 5 Искаженная тригональная бипирамида: 4NL, N Ni—NCS
Ni (H2O)2 (NCS)4 Hg 6 Октаэдр: 20, 4N Ni—NCS Ni—NCS—Hg
[Ni (trien) (NCS),] 6 Искаженный октаэдр: 4NL, 2N Ni—NCS
(NH4)2 [Ni (NH3)2 (NCS)4]-H2O * 6 Октаэдр: 2N£, 4N Ni—NCS
NH4 [Ni (NH3)3 (NCS)3] 6 Октаэдр: 3N£, 3N Ni—NCS
[Ni (NH3)3(NCS)2] 5+1 Искаженный октаэдр: 3NL, 2N+S Ni—NCS Ni—NCS—Ni
[Ni (NH3)4 (NCS)2] 6 Октаэдр: 4NL, 2N Ni—NCS
[NiPy4(NCS)2] 6 Октаэдр: 4NL, 2N Ni—NCS
[Ni (4-MePy)4 (NCS)2] 6 Октаэдр: 4NL, 2N Ni—NCS
[Ni (NH2-CSNH-NH2)2 (NCS)2] 6 Октаэдр: 2SL, 2NL, 2N Ni—NCS
[Nitu, (NCS)2] 6 Октаэдр: 4SL, 2N Ni—NCS
[NiEtu2 (NCS),] 6 Октаэдр: 2SL, 2N, 2S Ni—NCS—Ni
{Ni [SC (NHEt)2]4 (NCS)2j 6 Октаэдр: 4S^, 2N Ni—NCS
[Ni (DMAA)4 (NCS),] 6 Октаэдр: 40L, 2N Ni—NCS
[Ni (CH3CSNH2)2 (NCS),] 6 Октаэдр: 2SL, 2N, 2S Ni—NCS—Ni
[NiPn2 (NCS)2] 6 Искаженный октаэдр: 4NL, 2N Ni—NCS
[Nien2 (NCS)2] 6 Октаэдр: 4N£, 2N Ni—NCS
[Nien2 (NCS) (NO2)] 6 Октаэдр: 4Nf, NNOi> N Ni—NCS
[Nien2 (NCS) Cl] 5+1 Искаженный октаэдр: 4N^, N + CI Ni—NCS
[Nien2 H2O (NCS)] NO3 5+1 Искаженный октаэдр: 4N£, N + 0 Ni—NCS
[Ni (C2H4ONH2)2 (NCS)2] 6 Октаэдр: 2N£, 20L, 2N Ni—NCS
[Ni (C,H4ONH2) en (NCS)2] 6 Октаэдр: 3N£, 0L, 2N Ni—NCS
[NiQ2 (H2O)2 (NCS)2]Q2 6 Октаэдр: 2NL, 20L, 2N Ni—NCS
,[Ni2 (C2H4ONH2)2 en2 (NCS)2] •(C1O4)2 5+1 Искаженный октаэдр: 3NL, 0L, N + S Ni—NCS—Ni
[Ni2en4 (NCS)2] I2 5+1 Искаженный октаэдр: 4N£, N + S Ni—NCS—Ni
![Ni2en4 (NCS)2] (C1O4)2 5+1 Искаженный октаэдр: 4N£, Ni—NCS—Ni
[Ir (NH3)5 SCN] (C1O4)2 6 N -1— S Октаэдр: 5NL, S Ir—SCN
[Ir (Ph3P)2 (CO) (CN) Cl (NCS)] 6 Искаженный октаэдр: 2P, 2C, Cl, N Ir—NCS
[Pd (Pr3P)2 (NCS)2] * 4 Квадрат: 2PL, 2N Pd—NCS
{Pd [(Ph3O)3P]2 (SCN)2) 4 Квадрат: 2PL, 2S Pd—SCN
[Pd (Ph2PC=C—t—Bu)2 (SCN)2] 4 Квадрат: 2PL, 2S Pd—SCN
[Pd (PN) (SCN) (NCS)] 4 Квадрат: PL, N£, S, N Pd—SCN Pd—NCS
(Pd (PCP) (SCN)21 4 Квадрат: 2P^, 2S Pd—SCN
[Pd (PC2P) (SCN) (NCS)] 4 Квадрат: 2PL, S, N Pd—SCN Pd—NCS
[Pd(PC3P) (NCS)2] 4 Квадрат: 2PL, 2N Pd—NCS
226
a, A ь, A c, A a 6 V z Прост- ранствен- ная группа Литерату- ра
20,408 13,759 15,002 — — — 8 Pbca (632]
13,23 5,27 18,35 — 92,9° — 4 C2/c [513, 680]
14,14 12,02 8,395 — 106,4° — 4 C2/c [633]
13,41 — — — — — 6 — [725]
10,23 — 11,13 — — — 3 C3121 [725].
11,08 10,48 7,65 — — — 4 Pbnm [726]
11,46 8,18 5,68 — 105° — 2 C2/m [725, 727]
12,55 13,0 16,6 — 119° 45' — 4 C2/c или Cc [728]
16,74 — 22,66 — — — 8 Mi/a [926]
5,300 7,761 16,12 — 92,01° — 2 P2x/c [729, 927)
3,77 7,53 10,07 92,6° 97,9° 103,6° 1 Pl [641-643]
5,592 8,664 8,686 113° 39' 95° 8' 108° 43' 1 Pl [645]
11,14 17,26 9,64 — 100,6° — 2 Р2х/с [730]
9,12 12,35 10,89 — — 110,9° 4 P2jb [668]
9,463 — 12,627 — — » 3 P3j21 [731]
11,21 12,61 10,64 — — — 4 рг^А [593, 732)
10,28 8,26 8,88 — 121° 3' — 2 P2j/a [597]
25,470 6,539 18,828 — 127° 7' — 8 C2/c [637]
8,583 11,864 12,218 — 114° 47' — 4 P2i/c [638]
9,02 12,01 17,61 — — 132,5° 4 P2i/b [732J
8,65 7,61 9,80 — 97,0° 2 P2t/b [732]
8,78 6,61 7,58 92,0° 120.0° 109,0° 1 Pl [681]
12,675 7,700 9,423 95,66° 93,35° 92,49° 1 Pl [733]
15,02 7,13 13,38 — — 94,0° 4 P2i/n [681].
14,601 12,371 6,712 — 93° 30' — 4 P2j/n [640]
14,41 12,98 6,78 — 95° 30' — 4 P2lz/n [589]
7,506 15,744 14,108 — 121,75° — 4 Р2г/с [734]
10,141 11,222 9,381 112,21° 95,02° 64,23° 1 Pl [352]
19,53 8,72 9,36 87° 58' 119°4' 94° 12' 2 Pl или Pl [735]
9,922 10,096 19,334 — 108,46° — 4 P2x/c [535]
12,563 10,460 14,781 — 97,8° — 2 P2T/c [736]
11,684 12,961 14,641 — 110,04° — 4 P2i/c [528, 737]
10,426 29,353 9,884 — 119,86° — 4 P2i/n [534, 933]
17,773 23,212 8,502 — — — 4 P2l2l21 [738, 933]
14,774 9,181 21,182 — 95,48° — 4 I2i/c [534, 933]
8*
227
Продолжение табл. 5.12
Соединение Коорди- национ- ное число Координационный полиэдр Способ координации SCN-группы
{Pd2[(Ph2PO)2H]2(SCN)2} 4 Квадрат: 2PL, S, N Pd—SCN—Pd
К2 [Pd (SCN)4] 4 + (2) Искаженный октаэдр: 4S + 2S Pd—SCN Pd—SCN Pd‘"
цис-fPt (NH3)2 (SCN)21 4 Квадрат: 2NL, 2S Pt—SCN
транс-fPt (NH3)2 (SCN),1 4 Квадрат: 2NL, 2S Pt—SCN
TpaHC-[PtPy2 (SCN)Qj 4 Квадрат: 2N£, 2S Pt—SCN
a-[Pt2 (Pr3P)2 Cl2 (SCN)2] 4 Квадрат: PL, Cl, S, N Pt—SCN—Pt
P-[Pt2 (Pr3P)2 Cl2 (SCN)2] 4 Квадрат: P£, Cl, S, N Pt—SCN—Pt
k2 [Pt (SCN)4J * 4 Квадрат: 4S Pt—SCN
K2 [Pt (SCN)e] * 6 Октаэдр: 6S Pt—SCN
fPt (Ph2PC=C-t-Bu)2 (NCS)- •(SCN)] 4 Квадрат: 2PL, N, S Pt—NCS Pt—SCN
K2 [Pt (C2O4)2(SCN)2]-4H9O 4 Квадрат: 20L, 2S Pt—SCN
* Неполное структурное исследование.
** Установлен изоморфизм солей К, Rb, NH4 в серии MHg (SCN)3. Соли Cs и Me4N имеют дру
*** Установлен изоморфизм солей К. Rb, Cs в серии M2Hg (SCN)4 [65].
**** Данные по изоструктурности.
Повторное исследование дало для а- и 0-изомеров значения углов 164,9 и
167,3° [599], что исключает значительные стерические препятствия.
Угол MSC, как правило, составляет 90—120° и только иногда больше или
меньше этих значений.
Рентгеноструктурные исследования дают также информацию о длинах
связей в SCN-группе. Однако судить о кратности связей SC и CN по этим дан-
ным затруднительно, потому что в ранних работах точность межатомных
расстояний в SCN-группе сравнительно невысока. Тем не менее попытки оце-
нить эти параметры по данным рентгеноструктурных исследований предпри-
нимались [563].
Ниже приведены экспериментально определенные межатомные расстоя-
ния SC и CN для некоторых тиоцианатных соединений и соответствующие
им расчетные значения:
NH4NCS:
Н— N-C-S;
СН3—N=C=S:
NH, [Cr (NH3)2 (NCS)J H2O
Вычисленные расстояния:
N—С 1,47 A; N=C
C—S 1,81 A; C=S
NC 1,25 A; CS 1,59 A [601];
NC 1,22 A; CS 1,56 A [28,31];
NC 1,22 A; CS 1,56 A [29];
NC 1,14 A; CS 1,80 A [602].
1,27 A; N=C1,15A;
1,61 A; C=S1,47A [591].
В изоморфных KNCS и T1NCS [490] межатомное расстояние CS равно
2,12—2,27 А, что явно выходит за пределы определяемых ковалентной свя-
зью расстояний. По данным работы [490], NCS- имеет угловую конфигура-
цию с величиной угла при атоме углерода 125—130°, что не отвечает сущест-
вующим представлениям, так как отклонение / NCS от 180° обычно не пре-
228
а, А ь, А с, А 0С Z Прост- ранствен- ная группа Литерату- ра
10,323 13,340 18,392 — 100,14° — 4 Р2т/с [739]
11,11 12,90 4,23 — 98,2° — 2 Р2х/а [658]
13,74 8,06 13,7 — — — 8 Г руппа mmm [740]
7,31 8,27 13,27 — 90° — 4 P2j/n [741]
5,377 10,568 6,82) 96,8° 107,2° 99,7° 1 Pl [936]
7,514 13,583 16,19“ — 112,38° — 2 Р2,/с [599]
8,084 13,409 14,38о 98,29° — 2 P2i/n [599]
4,26 12,76 11,20 — 98,20° — 2 Р21/С [492, 742]
6,73 — 10,26 — — — 1 P31m [492, 742]
18,272 13.287 16,470 — — 4 Pca2t [743]
11,04 7,14 6.66 108,6° 90,3° 101,1° 1 Pl [934]
гие параметры решетки [65].
вышает нескольких градусов. Поэтому трактовку структур этих соединений
можно квалифицировать как ошибочную.
Данные по межатомным расстояниям в SCN-группе и величины углов
MSC и MNC предполагают комбинацию двух одинарных или одной одинар-
ной и одной двойной связи для тиоцианатов
IS-C-- s-c-~ У=с -
/ S /
мм м
и комбинацию одинарной и тройной или двух двойных связей для изотио-
+ +
цианатов М—N=C ... M=N=C ... [3].
Казалось бы, что наиболее надежным критерием при определении крат-
ности связей в SCN-группе являются значения частот валентных колебаний
v (CN) и v (CS), однако и в этом случае искать корреляцию между смещением
этих частот и кратностью связей не всегда представляется возможным. На-
пример, в изотиоцианатах частота v (CN) близка к таковой для иона NCS~
и лежит в области 2010—2090 см-1, а для типичных ионных изотиоцианатов
она составляет 2050 см~3.
В табл. 5.12 не представлены структурные данные ионных тиоцианатов
KNCS, T1NCS [489—491], Ba (NCS)2 • 2Н2О [492], а также аддукта (пири-
дин-2-кар>боксилато)-медн с KNCS [603], аддуктов тиоцианатов рубидия,
цезия, бария и свинца с макроциклическими полидентатными азот- и кисло-
родсодержащими молекулами C18H36N2O6, C20H40N2O7 (так называемые
«криптаты») [604—606] и аддуктов тиоцианатов щелочных металлов, каль-
ция с макроциклическими кислородсодержащими полиэфирами и др.
(С12Н14О6, С1сН24О8, С24Н32О6, С40Не4О12) [607—613, 913], диядерные тиоциана-
ты кобальта (II) [(NH3)5CoO2Co (NH3)5] (SCN)4 [614] и [en2Co (NH2, O2) •
• Coen2] (SCN)3 • H2O [615]. Структура координационных соединений с
229
= б, пространственная
Рис. 5.3. Вероятные позиции трехдентат-
ного лиганда в тригонально-бипирамидаль-
ных комплексах
макроциклическими пентадентатными азотсодержащими лигандами железа
(II) и железа (III) состава Fe (NCS)2 • L и [FeL (NCS)2J СЮ4 исследована в
работах [883, 914]. В этих соединениях железо имеет пентагональное окру-
жение: в экваториальной плоскости располагаются пять атомов азота ма-
кроцикла, аксиальные позиции занимают изотиоцианатные группы.
Из криптатов представляет интерес криптат бария (Q .,H40N2O7) Ва
• (NCS)2 • 2Н2О [6051 (а = 11, 580; b = 16,030; с = 19,563Л; 0 = 121,15°;
Z = 4; пространственная группа Р24/с), в котором ион бария имеет координа-
ционное число 11, т. е. координирует девять атомов молекулы лиганда и два
атома кислорода двух молекул воды. Криптат кобальта [CoC16H32N2Os] •
• [Со (NCS)4] имеет ромбическую сингонию: а = 22,661; b = 16,092; с =
группа Pbca. Ион кобальта в анионе
тетраэдрически окружен четырьмя
NCS-группами, а в катионе — се-
мью атомами органического лиган-
да (2N + 50), образующими ис-
каженную пентагональную бипи-
рамиду [616]. Высокие координа-
ционные числа проявляет ион свин-
ца в криптатах [Pb (C12H26N2O4) •
• (SCN)2] (к. ч.= 8) [617] и [РЬ •
• (C18H36N2O6) (SCN)2] (к. ч.= 10)
[606].
К соединениям, структуры ко-
торых не приведены в табл. 5.12,
можно отнести 6-изотиоцианатоде-
каборан 6-B10H13NCS [618], NH3 • BH2NCS [619], тример изотиоци-
анатофосфорнитрила [NP (NCS)2]3 [620]" и N3P3C14 (NCS)2 [621], ко-
торые являются представителями тиоцианатных соединений неметал-
лов. Последние соединения — это аналоги известного трифосфорнитрил-
хлорида и также имеют циклическое строение с двумя NCS-группами у фос-
фора, координируемыми посредством азота. Электронографическому изуче-
нию была подвергнута структура соединения Me3SiNCS [622]; найден угол
SiNC, равный 154°, в отличие от соединения Si (NCS)4, где он равен 180° [623].
Наибольший интерес представляют соединения, содержащие три- и
теградентатные лиганды. Чаще всего центральный атом тиоцианатов двух-
валентного металла в этих соединениях проявляет координационное число,
равное 5, а координационный полиэдр имеет конфигурацию тригональной
бипирамиды или тетрагональной пирамиды. Для тригональной бипирамиды
с тридентатными лигандами возможно несколько изомеров, но обычно реали-
зуются изомерысиб (рис. 5.3), причем идеальная тригональная бипирамида
может быть искажена по разным причинам: неравноценность углов в эквато-
риальной плоскости, смещение атома металла из этой плоскости в сторону
монодентатного лиганда; нелинейность оси бипирамиды. Изомер б реали-
зуется в структуре комплекса [Со (N3S) (NCS)21 1624, о625], в котором атом
серы лиганда N3S не связан с металлом (Co...S = 5,83 А). Различие в длинах
связи Со—N органического лиганда (2,148; 2,158 и 2,224 А) и Со—Nncs
(1,978 А) приводит к некоторому искажению бипирамиды.
Значительное отклонение от линейности оси бипирамиды может привести
к возникновению конфигурации, промежуточной между тригональной бипи-
рамидой и тетрагональной пирамидой, как это имеет место в комплексе
[Со (N2PCH3) (NCS)21. Более того, предполагается, что пятикоординацион-
ный [Со (N2P) (NCS)2] искажен в сторону тетраэдра [626].
230
Из комплексов с тетрадентатными лигандами определенный интерес пред-
ставляют комплексы с tren и trien. Так, структурно исследованы [Cu (tren) -
• NCS] SCN [627] и [Zn (tren) NCS] SCN [628, 629]. Ион металла в тригональ-
но-пирамидальном катионе окружен тремя атомами азота первичных ами-
нов, лежащих в экваториальной плоскости, третичным атомом азота и ато-
мом азота NCS-группы, находящимися в аксиальном положении. Бипирамида
несколько искажена за счет неравенства углов NMN экваториальной плос-
кости.
Согласно электронным спектрам, аналогичный комплекс кобальта имеет
подобную конфигурацию [630]. Однако Ni (NCS)2 образует с tren октаэдри-
ческие аддукты [631], конфигурация которых сохраняется и в растворе
[348]. В соединении [Ni •
(Me6tren) NCS] SCN [632]
ион никеля имеет тригональ-
но-бипирамидальное окруже-
ние. Цис-се-октаэдрическую
конфигурацию атом никеля
имеет и в комплексе с trien
[633].
Тиоцианат меди (II) с trien
образует тетрагонально-пира-
мидальный комплекс, причем
ион меди расположен над
плоскостью основания пира-
миды на 0,37 А (расстояние
Cu—Nncs = 2,607 А) [493].
Аналогичное смещение ато-
ма металла с плоскости осно-
вания тетрагональной пира-
миды в сторону монодентатно-
Рис. 5.4. Конфигурация комплекса Nien2(NCS)ClO.
[638]
го NCS-лиганда наблюдается
и в комплексе [Ni (N3As) (NCS)2], в котором атом никеля приподнят над
плоскостью основания на 0,34 А (расстояние Ni—Nncs — 1,97 А) [634, 635].
Другой интересной группой соединений являются комплексы состава
[Men2 (NCS) А], которые наиболее полно представлены соединениями нике-
ля. Как отмечается в работе [636], в кристаллах комплексов [ML2A2] с двумя
бидентатными лигандами при относительной слабости поля центрального ато-
ма монодентатные лиганды имеют тенденцию занимать транс-положение
в октаэдрических комплексах. В противоположность транс-октаэдрическо-
му [Nien2 (NCS)2] [597] стабильные высокоспиновые комплексы, какими ока-
зались, в частности, [Nien2 (NCS)A] [637, 638], имеют цис-октаэдрическое
строение. К соединениям, имеющим цис-конфигурацию, можно отнести
[Cden2 (NCS) Cl] [639].
Замена хлорид-, бромид- и нитрит-ионов в [Nien2 (NCS) А], с которыми
никель образует малоустойчивые комплексы, индифферентными перхлорат-
и иодид-ионами приводит к тому, что [Nien2 (NCS)] СЮ4 [589] и [Nien2 •
♦ (NCS)] I [640] образуют катионные димеры с двумя мостиковыми NCS-
группами.
С точки зрения изоструктурности соединений следует отметить комплек-
сы состава: [Mtu2 (SCN)21, [Mtu4] [М' (SCN)4] и Hg (SCN)4M. Первый тип
соединений представлен структурно исследованным комплексом [Nitu2 •
• (NCS)2J [641]. Структура состоит из октаэдрических узлов, причем роль
мостиков выполняет не SCN-группа, а атомы серы тиомочевины, NCS-группы
231
являются концевыми. Уточненные параметры решетки этого соединения,
приведенные в табл. 5.12, взяты из работы [642]. Аналогичные по состав;
соединения кобальта и кадмия также кристаллизуются в триклинной син-
гонии и имеют следующие кристаллографические данные [642]: для [Cotuo •
• (NCS)21: а = 3,80 А; b = 7,53 А; с = 10,13 А; « = 91,8°; J = 99,5°~и
Т = 103,6°; для [Cdtu, (NCS)2]: а = 4,04; b = 7.68: с = 10,10 А; « = 90,8°;
Р = 99,7°; у = 105,8°.
Как и в соединениях никеля, структура этих комплексов полимерная и
состоит из цепочек октаэдров, a NCS-группа в [Cdtu3 (NCS)2] также присо-
единена к металлу своим жестким концом (у (CN) — 2068 см~’, 2058 см-1 и
v (CS) — 804 см"1, 792 см-1) [642].
Структура tHgtu2 (SCN)2] также представляет собой цепочку октаэдров,
связанных между собой мостиковыми атомами серы органического лиганда,
однако он не изоструктурен с [Nitu2 (NCS)21. Что же касается [Zntu2 (NCS)2],
в нем цинк, вероятно, имеет тетраэдрическое окружение (а = 7,76 А; b =
= 27,94 А; с = 11,83 А; пространственная группа C222J [643].
В соединениях никеля и кадмия с этилентиомочевиной [MEtu2 (NCS)2]
мостиковой является SCN-rpynna [644, 645].
Соединения состава [Hgtu4] [М (NCS)4] (М = Zn, Mn, Со, Ni), а также
[Cdtu4] [Zn (NCS)4] имеют тетрагональную решетку [646—648]. Все они имеют
близкие значения параметров кристаллической решетки (а = 17,25 А,
с = 4,16 А) и принадлежат к пространственной группе 14. Ион металла
в анионе тетраэдрически окружен атомами азота NCS-групп. Из перечис-
ленных соединений структурному исследованию подвергались только со-
единения [Cdtu4] [Zn (NCS)41 [646] и [Hgtu4] [Со (NCS)41 [647].
Изоструктурная группа комплексов Hg (SCN)4M представлена соедине-
ниями кобальта, марганца, цинка [649, 650] и кадмия [651]. Структура
Hg (SCN)4Co детально изучена в работах [512, 649]. И ртуть, и кобальт тет-
раэдрически окружены соответственно четырьмя атомами серы (Hg—S =
= 2,558 А) и азота (Со—N = 1,921 А). Комбинация двух типов тетраэдров
HgS4 и CoN4 приводит к весьма необычному расположению атомов в струк-
туре, где атомы ртути и кобальта участвуют в образовании четырех спиралей,
каждая из которых содержит по четыре мостиковые SCN-группы. Вероятно,
аналогичное строение имеют соединения других вышеупомянутых металлов,
например Hg (SCN)4Cd [651, 652]. Что же касается сходного по формуле
Hg (SCN)4Cu, то можно предположить, что медь образует искаженный окта-
эдр за счет дополняющих атомов серы, находящихся на расстоянии 3,02 А,
[513]. Тетраэдрическое окружение для иона меди (II) в структурах тиоциа-
натных комплексов пока не обнаружено.
Следует рассмотреть структурные особенности некоторых смешанных
комплексов. Так, для [Cu (NH3)2 (NCS)2] получены две модификации (а и
Р) и их промежуточная фаза. В ИК-спектре «-модификации имеются полосы
2103 и 2120 см"1, что свидетельствует об одновременном существовании мос-
тиковой и монодентатной SCN-групп. Сосуществование двух модификаций
может быть обусловлено либо различием способа объединения молекул в
слои, либо отклонением атомов в [^-модификации от кристаллографической
плоскости гп (в пространственной группе Ршгпа), в которой расположены
атомы меди «-модификации [653, 654].
Комплекс [Cu2 (NH3)3 (CNS)3] [655, 656] содержит в себе как Cu (I),
так и Cu (II). Установлено, что Си (I) имеет тетраэдрическую координацию
(2S + 2N из SCN-групп), а полиэдр иона Cu (II) представляет собой окта-
эдр, имеющий общий атом серы с тетраэдром Cu (I) (рис. 5.5). Для этой
232
структуры установлено существование трех типов SCN-групп: а) мостико-
вой SCN-группы, которая с Cu (II) образует прочную ковалентную связь че-
рез атом азота (Си—N 1,987 А) и слабую — посредством атома серы (Cu...S
3,286 А); б) SCN-групп, которые связаны только с Си (I) (Си—N 1.996 и
2,006 А; Си—S 2,376 А и 2,470 А); в) мостиковой SCN-группы, также образу-
Рис. 5.5. Структура комплекса Cu2 (NH3)3(NCS)3 (проекция на
плоскость (010) [656])
ющей ковалентную связь с Cu (I) (Си—N) и слабую с Си (II) (Cu...S), причем
атом серы одновременно связан с Си (I), т. е. образуется одна из разновид-
/М
ностей мостиковой связи типа NCS\
ХМ
Структура соединения Cuen2 (NCS) С1О4, о котором упоминалось выше,
интересна тем, что ее можно рассматривать как необычный пример с мости-
,Си
ковой SCN-группой, в которой формально реализуется мостик SCN\
xCu
Это приводит к образованию цепочной структуры с мостиковыми NCS-
группами (рис. 5.2). Ион меди находится в тетрагональношскаженном окта-
эдрическом окружении (Си—Ne„ 2,02 А, Си—Nncs 2,73 А). Однако часто-
ты ИК-спектра этого соединения v (CN) 2025, 2085 см--1; (CS) 758 и 6 (NCS)
490 см"1 близки к таковым свободного иона.
К числу оригинальных можно отнести структуру координационного сое-
динения Hg2 (SCN)6 Со С,,Н6 [514] (рис. 5.6). Известно, что, в отличие от
никеля с характерным для него октаэдрическим окружением (табл. 5.12),
233
в тиоцианатных соединениях кобальта преобладает тетраэдрическая коор-
динация. Координационные соединения кобальта с шестью NCS-группами
до сих пор получить не удалось. Поэтому комплекс, приведенный на
рис. 5.6, может служить одним из немногочисленных примеров, где ион
СО (II) октаэдрически окружен шестью изотиоцианат-лигандами.
Довольно сложной представляется структура димера [(Me2SnNCS)2O]2
[657]. В нем имеются мостиковые и концевые NCS-группы, но первая имеет
/М
формальный характер и относится к типу SCN\ . Расстояние Sn...NCS
ХМ
Рис. 5.6. Структура комплекса Hg2 (SCN)6 • Со С6Н6 (проекция на плос-
кость (100) [514])
равно 2,82 А, втовреомя как для концевой NCS-группы межатомное расстоя-
ние составляет 2,06 А. Неудивительно, что подобный мостик не обнаружи-
вается в ИК-спектре этого соединения. Аналогичный тип мостика, но толь-
/М
ко через серу NCS\ , обнаружен в К2 [Pd (SCN)4J [658]. Последнее сое-
ХМ
динение — одно из немногочисленных, в котором реализуется такого типа
мостик (см. табл. 5.12). Образование его возможно лишь потому, что иска-
женные квадраты [Pd (SCN)4]2— накладываются друг на друга таким образом,
что на атом палладия одного аниона проектируются атомы серы соседних
ионов, дополняя координацию атома металла до искаженного октаэдра.
Расстояния Pd—S в квадрате равны 2,312 и 2,392 A, a Pd...S — 3,36 А.
Еще более интересными являются структуры Pb (SCN)2 и (СН3)2 T1NCS.
В них каждая SCN-группа удерживает по четыре атома металла, образуя
234
необычный и еще не встречавшийся ранее мостик [565, 937],
М М
SCN^
м м
Структура К2 [Со (NCS)4] • ЗН2О, по данным [659], оказалась ошибоч-
ной, ибо углы CoNC в тетраэдре [Со (NCS)4]2— слишком малы (106 и 109)°.
Было показано [3], что в действительности они близки к 180°. Это согласует-
ся со значением углов CoNC в других солях, содержащих анион [Со (NCS)4]2—
[616, 647, 660]. Позже эти же авторы [661] уточнили координаты
атомов в К2 [Со (NCS)4] • ЗН2О и К3 [Zn (NCS)4J • ЗН2О, подтвердив их
изоструктурность. Повторное определение структуры К2 [Со (NCS)4] •
- ЗН2О [915] показало правильность выводов работы [31: углы CoNC в
анионе [Со (NCS)4]2~ равны 164,6 и 166,8° а не 106 и 109°, как сообщалось
в работе [659].
5.10. ПРИМЕНЕНИЕ ТИОЦИАНАТНЫХ КОМПЛЕКСОВ
5.10.1. Тиоцианаты в химическом анализе
Тиоцианатные комплексы одними из первых были применены в фотомет-
рическом анализе металлов. Так, по интенсивности окраски растворов в при-
сутствии тиоцианат-ионов определяют содержание кобальта, железа, вис-
мута, молибдена, вольфрама, рения [746]. В последние годы в химическом
анализе все чаще используются смешанные комплексы, в состав которых
вместе с тиоцианат-ионом входит и органический лиганд [744]. Образова-
ние таких комплексов, которые плохо растворяются в воде и хорошо —
в органических растворителях, предоставляет большие возможности для
использования их в различных вариантах химического анализа: спектрофо-
тометрическом, экстракционно-фотометрическом и гравиметрическом. Под-
бирая различные экстрагенты, можно регулировать чувствительность и
избирательность реакций. Перспективность и практическая ценность смешан-
ных тиоцианатных комплексов требуют дальнейшего всестороннего их изу-
чения.
Тиоцианатные комплексы экстрагируются по различным механизмам.
Наиболее часто извлекаются ионные ассоциаты, содержащие комплексный
анион [9,745]. Некоторые элементы экстрагируются в виде смешанных и
нейтральных сольватокомплексов [М (SCN)„ - mSolvl или же в виде внешне-
сферных комплексов [М (Solv)m (SCN)J, как, например, тиоцианаты щелоч-
ноземельных металлов [9].
Большой размер экстрагирующихся комплексных тиоцианатов по сравне-
нию с хлоридными приводит к уменьшению гидратации первых и к лучшей
их извлекаемости органическими растворителями из водного раствора. Этот
факт, а также амбидентатность тиоцианат-иона позволяет создать условия
для экстракции большинства металлов. Однако при этом наблюдается сни-
жение избирательности экстракционно-фотометрического метода.
Несмотря на многочисленные исследования в области применения тио-
цианатных комплексов в химическом анализе [7—9, 744, 746, 938], все еще
остается много неясностей, в особенности в вопросах состава и структуры
используемых в анализе соединений. Так, природа тиоцианатов рения и
235
молибдена, образующихся в растворе в условиях анализа, оставалась предме-
том дискуссии в течение длительного времени. К сожалению, и в работах
последних лет этим вопросам не уделялось надлежащее внимание. Например,
не обсуждался состав комплекса в новом варианте тиоцианатного метода оп-
ределения рения 1747].
Применение в различных вариантах анализа окрашенных тиоцианатных
комплексов кобальта, железа, молибдена, ниобия хорошо известно [7,
746, 938]. Однако по данным различных исследователей по вопросу о составе
окрашенных комплексов наблюдаются существенные расхождения. Интен-
сивно окрашенному красному комплексу молибдена (V) приписывается
состав [Мо (NCS)5] [2, 748], [МоО (NCS)5]2~ и [МоО (NCS)6]3' [749]. В работе
[750] отмечается, что при различных pH раствора в присутствии KNCS
Мо (V) может образовывать три комплекса, а согласно [751, 752] состав тио-
цианатного комплекса молибдена [V] с пирамидоном [751] и тетраэтиламмо-
нием [752] не зависит от кислотности среды. В обоих случаях образуются
комплексы с одинаковым составом аниона: [РугНЬ [МоО (NCS)5] и [Et4N]2 •
• [МоО (NCS)51, которые можно использовать для гравиметрического опре-
деления молибдена. Тиоцианатный метод определения молибдена дает хо-
хорошие результаты при фотохимическом восстановлении Мо (VI) до Мо (V)
[761].
В аналогичном состоянии находится вопрос о тиоцианатных комплексах
вольфрама (IV), вольфрама (V), состав которых, вероятно, зависит от при-
роды восстановителя, pH среды и других факторов Ранее отмечалось, что
ReOF может восстанавливаться до Re (IV) или Re (V). В зависимости от
способа восстановления образуются тиоцианатные комплексы Re (IV) или
Re (V). Даже при одном и том же восстановителе могут сосуществовать тио-
цианаты рения (IV) и (V) [Re (SCN)6]2~ и [Re (SCN)6]~ [288]. Большинство
исследователей предполагают, что в растворе образуются оксотиоцианат-
ные комплексы рения [293, 753, 754], причем сам иоцианат-ион может в
присутствии соли меди восстанавливать ReQT до [ReO2 (SCN)4]3~ [754].
Окрашенные комплексы с тиоцианат-ионом, пригодные для фотометри-
ческого определения металлов, образуют висмут [746, 755], ванадий (III)
[756], рутений (III) [757], уран [758, 759].
Значительное место в фотометрическом анализе занимают смешанные
комплексы. Так, тиоцианатный комплекс титана i IV) с диантипирилмета-
ном [DiantpH]2 [Ti (NCS)6J имеет молярный коэффициент поглощения
6 • 104, что превышает чувствительность других известных реакций на ти-
тан [760, 762]. С этим же лигандом образуют интенсивно окрашенные комп-
лексы тиоцианаты молибдена [763, 764], ниобия [765]. Смешанные тиоциа-
натные окрашенные комплексы применяются для фотометрического опре-
деления титана [766, 767] и тория [768], ванадия [769] и вольфрама [770],
кобальта [771] и марганца [772].
Несомненно, что наиболее важными для аналитической химии являются
окрашенные тиоцианатные комплексы, которые применяются для фотомет-
рического определения металлов. Однако и бесцветные комплексы, чаще
всего смешанные с азотсодержащими лигандами, могут быть использованы
для концентрирования элемента или гравиметрии.
Рассмотрим некоторые примеры по применению однородных и смешанных
тиоцианатных комплексов металлов в химическом анализе. Так, медь опре-
деляют аргентометрическим титрованием тиоцианат-иона, связанного в
пиридинтиоцианатном комплексе [CuPy2 (NCSE1 [773] или амперометри-
чески в ее тиоцианатном комплексе с хинолином [774]. Для фотометрическо-
236
го метода анализа меди предложены дихлорэтановые растворы три- и тет-
ратиоцианата с нитроном [NitH] [Cu (NCS)31 и [NitH]2 [Cu (NCS)4J [775]
и растворы [CuL2 (NCS)21 в хлороформе [772].
Неокрашенные простые и смешанные тиоцианатные комплексы могут
быть использованы для гравиметрического определения ряда металлов:
скандия [776] и индия [777] с антипирином и пирамидоном [ML3 (NCS)3],
галлия — [Bu4N13 [Ga (NCS)6] [7781, скандия [Et4N]3 [Sc (NCS)61 [7791, спект-
рофотометрического [780, 781] и комплексонометрического [782] опреде-
ления цинка, химико-спектрального определения следов сурьмы в хроме
[783] и т. д. Подробнее с вопросами применения тиоцианатных комплексов
в анализе можно познакомиться в обзорных работах [7, 8, 744, 938].
5.10.2. Тиоцианаты в технологии некоторых металлов
Тиоцианатные соединения широко используются в технологии редких
и цветных металлов, для их разделения или концентрирования. Для этих
целей часто применяется экстракция. Посредством экстракции тиоцианатов
осуществляется разделение циркония и гафния, редкоземельных элементов.
В 1947 г. Фишер [784] указал на возможность разделения циркония и
гафния путем экстракции из растворов, содержащих NH4NCS. При этом в
органическую фазу преимущественно переходит гафний. Разделение про-
водят в кислых растворах. При использовании сульфатно-тиоцианатных
растворов коэффициенты разделения значительно больше [786—788], чем
в случае солянокислых [7871; в последних каждый элемент извлекается зна-
чительно полнее [787, 789]. Экстракцией гафния из солянокислых раство-
ров пользуются для отделения циркония от гафния [789].
С целью отделения от тория и титана [790] скандий экстрагируют из
тиоцианатных растворов метилизобутилкетоном, а с помощью трибутил-
фосфата его отделяют от тория и редкоземельных элементов [791].
Экстракционным методом из солянокислых растворов тиоцианатов мож-
но отделить галлий от алюминия [792], тантал от ниобия [793], торий от
лантана [794]. При экстракции трибутилфосфатом смешанных комплексов
[ROx (NCS)2J удается разделить лантаноиды на ряд фракций, а также полу-
чить спектрально чистый лантан [796].
Анионный обмен тиоцианатных водно-спиртовых растворов использу-
ется для извлечения трансплутониевых элементов из редкоземельных ме-
таллов [795]. В обзоре [9] рассматриваются методы разделения ниобия и
тантала, молибдена и рения и других близких по свойствам металлов.
Возможны и другие области применения тиоцианатных соединений.
Например, тиоцианаты ниобия (V) и тантала (V) являются катализаторами
в реакции Фриделя — Крафтса [939], соединения кобальта и никеля с уро-
тропином находят применение в качестве термоиндикаторов [842], тиоциа-
натокомплексы двухвалентных Зй-металлов типа [ML4 (NCS)21 (L = гетеро-
циклический ароматический амин) образуют с ароматическими соединения-
ми клатраты, которые используются для разделения изомеров, например
орто- и пара-ксилола [942]. Разнообразно применение органотиоцианатов,
однако сведения о них выходят за рамки настоящей главы.
СПИСОК ЛИТЕРАТУРЫ
1. Williams Н. Е. Cyanogen Compounds. 2nd Ed. London, 1948.
2. Бабко А. К.— Успехи химии, 1956, 25, с. 872.
3. Порай-Кошиц М. А., Цинцадзе Г. В.— В кн.: Итоги науки. Кристаллохимия. 1965.
М., 1967, с. 168.
4. Beck W., Fehlhammer W. Р.— МТР Int. Rev. Sci. Inorg. Chem., 1972, Ser. 1, p. 253.
5. Pearson R. G.— J. Am. Chem. Soc., 1963, 85, p. 3533.
6. Pearson R. G.— J. Chem. Educ., 1968, 45, p. 581, 643.
7. Rozycki C.— Chem. analit. (PRL), 1966, 11, p. 447.
8. Rozycki C.— Chem. analit. (PRL), 1970, 15, p. 3.
9. Султанова Э. X., Чучалин Б. К., Иофа Б. 3., Золотов Ю. А.— Журнал аналит.
химии, 1973, 28, с. 413.
10. Soderback Е.— Ann., 1919, 419, р. 217.
И. Сингх М. М., Варшавский Ю. С.— Журнал неорган. химии, 1969, 14, с. 2434.
12. Kerstein Н., Hofmann R.— Вег., 1924, 57, S. 491.
13. Martinez С., Calandra A. JArvia A. J.— Elektrochim. acta, 1972, 17, р. 2153.
14. Kaufmann Н. Р., Kogler F.— Вег., 1925, 58, р. 1553.
15. KerridgeD. Н., Mosley М.— Chem. Communs., 1969, р. 429.
16. Новоселова А. В., Почкаева Т. И., Тамм Н. С., Томащик А. Д.— Журнал неорган.
химии, 1969, 14, с. 605.
17. Ohlberg S. М., Van der Maulen Р. А.— J. Am. Chem. Soc., 1953, 75, p. 997.
18. Lecher H., Wittwer M.— Ber., 1922, 55, S. 1481.
19. Lecher H., Goebel A.— Ber., 1922, 55, S. 1483.
20. Bond P. A., Weaver G. A.— J. Am. Chem. Soc., 1938, 60, p. 2614.
। 21. Forbes G. S., Anderson H. H.— J. Am. Chem. Soc., 1943, 65, p. 2271.
22. Jander G., UllmannD.— Z. anorg. Chem., 1936, 230, S. 405.
23. Klason P.— J. prakt. Chem., 1888, 38, S. 383.
24. Dixon A. E.— J. Chem. Soc., 1901, 79, p. 541.
25. Rosenheim A., Levy R. — Ber., 1907, 40, S. 2166.
26. Ruck U., Steinmetz H.— Z. anorg. Chem., 1912, 77, S. 51.
. 27. Birkenbach L., Buchner E.— Ber., 1940, 73, S. 1153.
28. Beard С. I., Dailey В. P.— J. Chem. Phys., 1947, 15, p. 762.
29. Beard С. I., Dailey В. P.— J. Chem. Ph vs., 1950, 18, p. 1437.
30. Goubeau J., Gott 0,— Ber., 1940, 73B, S. 127.
31. Kewley R., Sastry К- V. L. N., Winnerwisser M.— J. Mol. Spectrosc., 1963, 10,
p. 418.
32. Howell J. M., Absar 1., Van Waser J. R.— J. Chem. Phys., 1973, 59, p. 5895.
33. Klement R.— Z. anorg. Chem., 1949, 260, S. 268.
34. Fujimoto M.— Bull. Chem. Soc., Japan, 1953, 26, p. 353.
35. Варшал A. M., Сенявин M. M.— Журнал аналит. химии, 1962, 17, с. 903.
36. Синегрибова О. А., Ягодин Г. А.— Журнал неорган. химии, 1971, 16, с. 1184.
37. Голуб А. М., Билык О. Г.— Журнал неорган. химии, 1957, 2, с. 2723.
38. Schriver L.— Bull. Soc. chim. France, 1973, p. 1882.
39. Boughton J. H., Keller R. N.— J. Inorg. Nucl. Chem., 1966, 28, p. 2851.
40. Morgan T. D., Phillips E. D., Stedman G.— J. Chem. Soc., 1969, A, p. 2318.
41. Lee D. A.— Inorg. Chem., 1964, 3, p. 289.
42. Glavas M., Ribar T.— Glasnik hem. i tehnol. Bih, 1968, 16, s. 123.
43. Luehrs D. C., Kohut J. P.— J. Inorg. Nucl. Chem., 1974, 36, p. 1459.
44. Giggenbach W.— Inorg. Chem., 1971, 10, p. 1308.
45. Михеева Л. M., Комиссарова Л. H., Елфимова Г. И.— Журнал неорган. химии,
1970, 15, с. 3215.
46. Trammer А.— J. chim. phys., 1962, 59, р. 232.
47. Bohland Н., Hanay W.— Z. Chem., 1974, 14, S. 286.
48. Почкаева Т. И.. Михеева Л. М., Григорьев А. И., Ганем А.— Журнал неорган.
химии, 1970, 15, с. 87.
49. Харитонов Ю. Я., Цинцадзе Г. В., Цивадзе А. Ю.— Журнал неорган. химии, 1970,
15, с. 390.
50. Скопенко В. В., Галицъка С. М.— Втник КиТв. ун-ту. Сер. xiMi’i, 1973, № 14, с. 35.
51. Ahuja /. S., Garg А.— J. Inorg. Nucl. Chem., 1972, 34, p. 1929.
52. Ahuja I. S., Singh R.— J. Inorg. Nucl. Chem., 1974, 36, p. 1505.
53. Цинцадзе Г. В.— Журнал неорган. химии, 1971, 16, с. 1160.
54. Rath Р. С., Mohapatra В. К.— J. Indian Chem. Soc., 1974, 51, р. 834.
55. Скопенко В. В., Тряшин А. С., Зуб В. Я-— Укр. хим. журн., 1974, 40, с. 792.
56. Ahuja I. S., Singh R.— Inorg. Nucl. Chem. Lett., 1974, 10, p. 421.
238
57. Цивадзе А. Ю., Харитонов Ю. Я-, Цинцадзе Г. В. и др.— Журнал неорган. химии,
1970, 19, с. 2621.
58. Davies A. R., Murphy С. JPlane R. А.— Inorg. Chem., 1970, 9, р. 423.
59. Jain S. С.— J. Inorg. Nucl. Chem., 1973, 35, p. 413.
60. Cristini A., Ponticelli G., Preti C.— J. Inorg. Nucl. Chem., 1974, 36, p. 2473.
61. Ahuja I. S., Singh R. — Spectrochim. acta, 1974, A30, p. 2055.
62. Lorant B., Bojtor M.— J. Therm. Anal., 1973, 5, p. 379.
63. Ciampolini M., Cristini M., Diaz A., Ponticelli G.— Inorg. chim. acta, 1973, 7,
p. 549.
64. Харитонов Ю. Я-, Цинцадзе Г. В., Цивадзе А. Ю.— Журнал неорган. химии,
1970, 15, с. 949.
65. Larbot A., Beauchamp A. L.— Rev. chim. miner., 1973, 10, р. 465.
66. Forster A. D., Goodgame D. M. L.— Inorg. Chem., 1965, 4, p. 715.
67. Bohland H., Milhle E.— Z. anorg. allg. Chem., 1970, 379, S. 273.
68. Forster A.D., Goodgame D. M. L.— Inorg., Chem., 1965, 4, p. 823.
69. Makhija R., Pazdernik L., Rivest R.— Can. J. Chem., 1973, 51, p. 438.
70. Makhija R., Pazdernik L., Rivest R.— Can. J. Chem., 1973, 51, p. 2987.
71. Makhija R., Rivest R.— Spectrochim. acta, 1974, A30, p. 997.
72. Singh P. P., Shukla U. P., Rivest R., Makhija R.— J. Inorg. Nucl. Chem., 1975,
37, p. 679.
73. Харитонов Ю. Я-, Цинцадзе Г. В., Цивадзе А. Ю.— Журнал неорган. химии,
1970, 15, с. 1811.
74. Харитонов Ю. Я-> Цинцадзе Г. В., Цивадзе А. Ю.— Журнал неорган. химии,
1970, 15. с. 1196.
75. Cocksedge Н. Е.— J. Chem. Soc., 1908, 93, р. 2177.
76. Pohland Е.— Z. anorg. allg. Chem., 1931, 201, S. 282.
77. Sowerby D. B.— J. Am. Chem. Soc., 1962, 84, p. 1831.
78. Patel S. J.— J. Inorg. Nucl. Chem., 1971, 33, p. 17.
79. Rosenheim A., Cohn R.— Ber., 1900, 33, S. 1111.
80. Schmitz-Dumont 0., Ross B.— Angew. Chem., 1964, 76, S. 647.
81. Hajek B., Petru F., Holeckova E.— Coll. Czech. Chem. Comm., 1967, 37, p. 1988.
82. Елфимова Г. И., Михеева Л. М., Комиссарова Л. Н.— Вестник Моск, ун-та. Хи-
мия, 1970, 11, с. 363.
83. Михеева Л. М., Григорьев А. И., Тарасова А. И.— Журнал неорган. химии, 1973,
18, с. 85.
84. Михеева Л. М., Ауэрман Л. Н., Тарасова А. И., Комиссарова Л. Н.— Журнал
неорган. химии, 1971, 16, с. 2111.
85. Михеева Л. М., Тарасова А. И.— Журнал неорган. химии, 1975, 20, с. 365.
86. Михеева Л. М., Тарасова А. И.— Журнал неорган. химии, 1975, 20, с. 541.
87. Patel S. J., TuckD. G.— Can. J. Chem., 1969, 47, p. 229.
88. Михеева Л. M., Елфимова Г. И., Тарасова А. И., Комиссарова Л. Н.— Вестник
Моск, ун-та. Химия, 1970, 11, 481.
89. Михеева Л. М., Григорьев А. И., Тарасова А. И.— Журнал неорган. химии, 1974,
19, с. 2337.
90. Голуб А. М., Самойленко В. М.— Укр. хим. журн., 1963, 29, с. 472.
91. Goggin Р. L., McColm I. J., Shore R.— J. Chem. Soc., 1966, A, p. 1314.
92. Patel S. J., Sowerby D. B., TuckD. G.— J. Chem. Soc., 1967, A, p. 1187.
93. Schmidtke H.-H., Garthoff G.— Z. Naturforsch., 1969, A24, S. 126.
94. Habeed J. J., Tuck D. G.— J. Chem Soc. Dalton, 1973, p. 96.
95. Мальцева В. С., ЕреминЮ. Г.— Журнал неорган. химии, 1974, 19, с. 2323.
96. Patel S. J.— J. Inorg. Nucl. Chem., 1970, 32, p. 3708.
97. Бацанов С. С., Серебрянная Н. Р.— Изв. вузов СССР. Химия и технология, 1960,
6, с. 980.
98. Wells Н. L.— Am. Chem. J., 1902, 28, р. 245, 270.
99. Taimni I. К-—J. Chem. Soc., 1931, р. 2433.
100. Goddard А. Е.— J. Chem. Soc., 1921, 119, р. 672.
101. Hatton J. V.— J. Chem. Phys., 1964, 40, p. 933.
102. Bertazzi N., Stocco G. C., Pellerito L., Silvestri A.— J. Organometal. Chem., 1974,
81, p. 27.
103. Challenger F., Richards О. V.— J. Chem. Soc., 1934, p. 405.
104. Kumar N..Kalsotra B. L., Multani R. K-—J. Inorg. Nucl. Chem., 1974, 36, p. 1157.
105. Miquel P. — Ann. chim. phys., 1877, 11, p. 341.
106. Sowerby D. B.— J. Inorg. Nucl. Chem., 1961, 22, p. 205.
107. Green B. S., Sowerby D. B., Wihksne K- J— Chem. and Ind., 1960, p. 1306.
108. Anderson H. H.— J. Am. Chem. Soc., 1951, 73, p. 5439.
109. Anderson H. H.— J. Am. Chem. Soc., 1956, 78, p. 1692.
239
110. Scaife D. E.— Inorg. Chem., 1967, 6, p. 625.
111. Миронов В. Ф., Кравченко А. Л.— Изв. АН СССР. Сер. хим., 1965, с. 1026.
112. Srivastava Т. N., Tandon S. К.— J- Inorg. Nucl. Chem., 1968, 30, р. 1399.
113. Цинцадзе Г. В., Кверзели Э. К., Лежава А. П.— Журнал неорган. химии, 1975,
20, с. 1552.
114. Chamberlain В. R., Mozer W. —J. Chem. Soc., 1969, А, р. 354.
115. Donaldson J. D., Nicholson D. G.— J. Chem. Soc., 1970, A, p. 145.
116. Donaldson J. D., Nicholson D. G., Senior B. J.— J. Chem. Soc., 1968, A, p. 2928.
117. Голуб A. M., Самойленко В. M.— Укр. хим. журн., 1963, 29, с. 789.
118. Alleston D. L., Devies A. G.— J. Chem. Soc., 1962, р. 2050.
119. Srivastava Т. N., AgarvalM. P.— J. Inorg. Nucl. Chem., 1970, 32, p. 3416.
120. Wada M., Okawara R.— J. Organometal. Chem., 1967, 8, p. 261.
121. Holloway J. H., McQuillan G. P., Ross D. S.— J. Chem. Soc., 1969, A, p. 2505.
122. Mullins M. A., Curran C.— Inorg. Chem., 1968, 7, p. 2584.
123. Czakis-Sulikowska D. M., Radwanska-Dczekalska J., Gorecka A.— Rocz. chem.,
1973, 47, p. 2345.
124. Otto R. J. A., Audrieth L. A.— J. Am. Chem. Soc., 1958, 80, p. 5894.
125. Tesi G., Otto R. J. A., Sherif F. G., Audrieth L. M.— J. Am. Chem. Soc., I960, 82,
p. 528.
126. Rosenheim A., Vogelsang W.— Z. anorg. Chem., 1906, 48, S. 205.
127. Bender G.— Ber., 1887, 20, S. 723.
128. Жумабаее А. Ж-, Скоробогатько E. П.— Укр. хим. журн., 1972, 38, с. 14.
129. Lodzinska-Kulinska Н.— Rocz. chem., 1974, 48, p. 333.
130. Goel R. G., Ridley D. R.— Inorg. Nucl. Chem. Lett., 1971, 7, p. 21.
131. Goel R. G., Masiowsky E., Senoff С. V.— Inorg. Chem., 1971, 10, p. 2572.
132. Cyganski A.— Rocz. chem., 1965, 39, p. 193.
133. Cyganski A.— Rocz. chem., 1965, 39, p. 1735.
134. Cyganski A.— Rocz. chem., 1967, 41, p. 1429.
135. Cyganski A.— Rocz. chem., 1967, 41, p. 1635.
136. Cyganski A.— Rocz. chem., 1967, 41, p. 1161.
137. Cyganski A.— Rocz. chem., 1966, 40, p. 975.
138. Galdecki Z., Golinski B., Ptaszynski P.— Rocz. chem., 1967, 41, p. 1141.
139. Скопенко В. В., Жумабаее А. Ж-— Укр. хим. журн., 1971, 37, с. 639.
140. Kaufmann Н. Р., Liepe J.— Вег., 1924, S. 923.
141. Long С., Skoog D. А.— Inorg. Chem., 1966, 5, р. 206.
142. Orszagh I., BazsaG., Beck M.— Magyar chem. Folyoirat., 1972, 78, p. 190.
143. Toeniskoetter R. H., Solomon S.— Inorg. Chem., 1968, 7, p. 617.
144. Macarovici C. G.,Neamtu M., Grecu I.— Ann. chim. France, 1972, 7, p. 365.
145. Gaughan A. P., Ziolo R. F., Dori Z.— Inorg. chim. acta, 1970, 4, p. 640.
146. Ziolo R. F., Thich J. A., Dori Z.— Inorg. Chem., 1972, 11, p. 626.
147. Mohapatra В. K-—J. Indian Chem. Soc., 1974, 51, p. 705.
148. Massacesi M., Ponticelli G.— J. Inorg. Nucl. Chem., 1974, 36, p. 2209.
149. Melnik M., Kabesova M., Obert TGazo J.— Proc. 4th Conf. Coord. Chem. and 2nd
Sem. Crystallochem. coord, and metallorg. compounds. Bratislava, 1973, p. 41.
150. Maccbkova L., Kabesova M., Garaj J., Gazo J.— Monatsh. Chem., 1973, 104, S. 1473.
151. Kulasingam G. C., McWhinnie W. R.— J. Chem. Soc., 1968, A, p. 254.
152. Patel К- C., Goldberg D. E.— Inorg. Chem., 1972, 11, p. 759.
153. Barbucci R., Paoletti P., Campbell M. J.— Inorg. chim. acta, 1974, 10, p. 69.
154. Dickson I. R., Robson R.— Inorg. Chem., 1974, 13, p. 1301.
155. Raymond K- NBasolo F.— Inorg. Chem., 1966, 5, p. 1632.
156. Slade R. C., Tomlinson A. A. G., Hathaway B. J., Billing D. E.— J. Chem. Soc.,
1968, A, p. 61.
157. Barbucci R., Paoletti P., Ponticelli G.— J. Chem. Soc., 1971, A, p. 1637.
158. Cristini A., Ponticelli G.— J. Inorg. Nucl. Chem., 1973, 35, p. 2691.
159. Misra M. K-, Ramana RaoD. V.— Indian J. Chem., 1972, 10, p. 757.
160. Dash R. N., Ramana RaoD. V.— Z. anorg. allg. Chem., 1972, 393, S. 309.
161. Dash R. N., Ramana RaoD. V.— J. Indian Chem. Soc., 1974, 51, p. 787.
162. Cox J. L., Howatson J.— Inorg. Chem., 1973, 12, p. 1205.
163. Burmeister J. L., Melpolder J. B.— Chem. Commun., 1973, p. 613.
164. Manolov K- R-—Z. anorg. allg. Chem., 1963, 322, S. 101.
165. Scovell W. M., Stocco G. C., Tobias R. C.— Inorg. Chem., 1970, 9, p. 2682.
166. Cassol A., Seminaro A., Paoli G.— Inorg. Nucl. Chem. Lett., 1973, 9, p. 1163.
167. Crawford N. P., Melson G. A.— J. Chem. Soc., 1969, A, p. 427.
168. Crawford N. P., Melson G. A.— J. Chem. Soc., 1969, A, p. 1049.
169. Cac T. M., Комиссарова Л. H., Анацкая H. И.— Журнал неорган. химии, 1971,
16, с. 87.
240
170. Комиссарова Л. Н., Сас Т. М.. Красноярская А. А., Гагарина В. А.— Журнал
неорган. химии, 1970, 15, с. 2952.
171. Комиссарова Л. Н., Гулиа В. Г., Сас Т. М.— Изв. вузов СССР. Химия и хим. тех-
нология, 1969, 12, с. 1315.
172. HartF. A., Laming F. Р.— J. Inorg. Nucl. Chem., 1964, 26, р. 579.
173. Hart F. A., Laming F. P.— J. Inorg. Nucl. Chem., 1965, 27, p. 1605.
174. Hart F. A., Laming F. P.— J. Inorg. Nucl. Chem., 1965, 27, p. 1825.
175. Голуб A. M., Олевинский M. И., Жигулина H. C.— Журнал неорган. химии, 1966,
11, с. 1574.
176. Perrier М., Vicentini G.— J. Inorg. Nucl. Chem., 1973, 35, p. 555.
177. Еремин Ю. Г., Бондаренко Г. И.— Журнал неорган. химии, 1973, 18, с. 1715.
178. Скопенко В. В., Лубяный В. Е., ТряшинА. С.— Укр. хим. журн., 1974, 40, с. 453.
179. Комиссарова Л. Н., Сас Т. М., Гулиа В. Г.— Журнал неорган. химии, 1971, 16,
с. 2091.
180. Condorelli G., Seminara A., Musumeci А.— J. Inorg. Nucl. Chem., 1974, 36, p. 3763.
181. Vicentini G., Perrier M., Zinner L. B., Amin M. I.— J. Inorg. Nucl. Chem., 1974,
36, p. 771.
182. Сас T. M., Комиссарова Л. H., Гулиа В. Г., Григорьев А. И.— Журнал неорган.
химии, 1967, 12, с. 2072.
183. Еремин Ю. Г., Катечкина В. С., Комиссарова Л. Н.— Журнал неорган. химии
1970, 15, с. 1248.
184. Еремин Ю. Г., Каточкина В. С., Комиссарова Л. Н.— Журнал неорган. химии,
1970, 15, с. 2067.
185. Мальцева В. С., Еремин Ю. Г.— Журнал неорган. химии, 1975, 20, с. 325.
186. Rossmanith К-—Monatsch. Chem., 1962, 93, S. 1121.
187. Цинцадзе Г. В., Цивадзе А. Ю.. Борщ А. Н.— Сообщ. АН ГрузССР, 1969, 56,
с. 585.
188. Голуб А. М., БорщА. Н.— Укр. хим. журн., 1966, 32, с. 923.
189. Cousins D. R., Hart F. А.— J. Inorg. Nucl. Chem., 1968, 30, p. 3009.
190. Zinner L. B., Vicentini G.— An. Acad, brasil, cienc., 1973, 45, p. 223.
191. Martin J. L., Thompson L. C., Radonovich L. J., Glick M. D.— J. Am. Chem. Soc.,
1968, 90, p. 4493.
192. Bailey R. A., Kozak S. L., Michelsen T. M., Mills W. N.— Coord. Chem. Rev.,
1971, 6, p. 407.
193. Молодкин A. K-, Скотникова Г. A.— Журнал неорган. химии, 1962, 7, с. 1548-
194. Молодкин А. К-, Скотникова Г. А.— Журнал неорган. химии, 1964, 9, с. 60.
195. Маркое В. П., Траггейм Е. Н.— Журнал неорган. химии, 1961, 6, с. 2316.
196. Grey 1. Е., Smith Р. W.— Austral. J. Chem., 1969, 22, р. 311.
197. Countryman R., McDonald W. S.— J. Inorg. Nucl. Chem., 1971, 33, p. 2213.
198. Brintzinger H., Hatanarat C.— Z. anorg. Chem., 1935, 223, S. 106.
199. Марков В. П., Траггейм Е. Н.— Журнал неорган. химии, 1960, 5, с. 1467.
200. Марков В. П., Траггейм Е. Н.— Журнал неорган. химии, 1960, 5, с. 1493.
201. Марков В. П., Траггейм Е. Н.— Журнал неорган. химии, 1961, 6, с. 1244.
202. Andra К-, Bohland Н.— Z. Chem., 1965, 5, S. 145.
203. Sato T., Kotani S., Good M. L.— J. Inorg. Nucl. Chem., 1974, 36, p. 451.
204. Арутюнян Э- Г., Порай-Кошиц M. A.— Журнал структурной химии, 1963, 4.
с. НО.
205. Ahuja I. S., Singh R.— J. Inorg. Nucl. Chem., 1973, 35, p. 561.
206. Majumdar A. K-, Bhattacharyya R. G.— Sci. and Cult., 1969, 35, p. 271.
207. Hart F. A., Newbery J. E.— J. Inorg. Nucl. Chem., 1966, 28, p. 1334.
208. Sutton G. J.— Austral. J. Chem., 1959, 12, p. 122.
209. Schmitz-Dumont O., Simons PBroja C.— Z. anorg. allg. Chem., 1949, 258, S. 307.
210. Lenz W., Schlafer H. L., Ludi A.— Z. anorg. Chem., 1969, 365, S. 55.
211. Bohland H., Steinecke Z.— Z. Chem., 1974, 14, S. 65.
212. Rosenheim A., Cohn R.— Z. anorg. Chem., 1901, 28, S. 167.
213. Сыч A. M., Мрихина P. И.— Укр. хим. журн., 1973, 39, с. 558.
214. Тананаее И. В., Розанов И. А.— Журнал неорган. химии, 1962, 7, с. 1854.
215. Giddings S. А.— Inorg. Chem., 1967, 6, р. 849.
216. Burmeister J. L., Deardorff E. D., Jensen A., Christiansen V. H.— Inorg. Chem.,
1970, 9, p. 58.
217. Coutts R. S. P., Wailes P. C.— Austral. J. Chem., 1966, 19, p. 2069.
218. Jensen A., Christiansen V. H., Hansen J. H. a. o.— Acta chem. scand., 1972, 26,
p. 2898.
219. Голуб A. M., Сергунькин В. H., Калибабчук В. А.— Укр. хим. журн., 1964, 30,
с. 441.
220. Голуб А. М., Лишко Т. П.— Журнал неорган. химии, 1970, 15, с. 1527.
241
221. Голуб А. М., Лишко Т. ПЦинцадзе Г. В., Скопенко В. В.— Журнал неоргаи-
химии, 1972, 17, с. 2115.
222. Сыч А. М., Демяненко В. П.— Журнал неорган. химии, 1971, 16, с. 3000.
223. Тананаев И. В., Розанов И. А., Колгушкина А. Г.— Журнал неорган. химии, 1963
8, с. 1013.
224. Липатова И. П., Семенова Е. И.— Изв. АН СССР. Сер. хим., 1967, № 1, с. 6.
225. Харитонов Ю. Д., Розанов И. А.— Изв. АН СССР. Отд. хим. наук, 1962, с. 402.
226. Харитонов Ю. Д., Розанов И. А., Тананаев И. В.— Изв. АН СССР. Отд. хим. на-
ук, 1963, с. 596.
227. Sala-Pala JGtterchais J. Е.— Bull. Soc. chim. France, 1974, p. 2683.
228. Schmitz-Du Mont O-, Ross B.— Angew. Chem., 1964, 76, S. 304.
229. Сыч A. M., Богатырь H. И.—Журнал неорган. химии, 1974, 19, с. 2691.
230. Schmitz-Du Mont 0., Ross B.— Z. anorg. allg. Chem., 1966, 342, S. 82.
231. Bailey R. A., Michelsen T. W., Nobile A. A.— J. Inorg. Nucl. Chem., 1970, 32,
p. 2427.
232. Тананайко M. M., Царенко Г. Ф.— Укр. хим. журн., 1965, 31, с. 530.
233. Bohland Н., Malitzke Р.— Z. Naturforsch., 1965, В20, S. 1126.
234. Bohland И., Malitzke Р.— Z. anorg. allg. Chem., 1967, 350, S. 70.
235. Doyle G., Tobias R. S.— Inorg. Chem., 1968, 7, p. 2479.
236. Selbin J., Holmes L. H.— J. Inorg. Nucl. Chem., 1962, 24, p. 1111.
237. Bohland H., Niemann E.— Z. anorg. allg. Chem., 1973, 373, S.217.
238. Голуб A. M., Кострова P. A.— Укр. хим. журн., 1963, 29, с. 128.
239. Wasson G. R.— J. Inorg. Nucl. Chem., 1968, 30, p. 171.
240. Голуб A. M., Сыч A. M.— Журнал неорган. химии, 1964, 9, с. 1085.
241. Vuletic N., Djordievic C.— J. Chem. Soc. Dalton, 1972, c. 2322.
242. Smith J. N., Brown T. M.— Inorg. Chem., 1972, 11, p. 2697.
243. Brown T. M., Knox G. F.— J. Am. Chem. Soc., 1967, 89, p. 5296.
244. Knox G. F., Brown T. M.— Inorg. Chem., 1969 , 8, p. 1401.
245. Bohland H., Zenker E.— J. Less-Common Metals, 1968, 14, p. 397.
246. Bohland H., Tiede E., Zenker E.— J. Less-Common Metals, 1968, 15, p. 89.
247. Oki H., Kyuno E., Tsuchiya R.— Bull. Chem. Soc. Japan, 1968, 41, p. 2357.
248. Oki H., Kyuno E., Tsuchiya R.— Bull. Chem. Soc. Japan, 1968, 41, p. 2362.
249. Bennett M. A., Clark R. J. H., Goodwin A. D. H.— Inorg. Chem., 1967, 6. p. 1625.
250. Ghosh S. P., Mishra A.— J. Inorg. Nucl. Chem., 1971, 33, p. 4199.
251. Contreras G., Schmidt R.— J. Inorg. Nucl. Chem., 1970, 32, p. 1295.
252. Mathur P. K.—J- Inorg. Nucl. Chem., 1974, 36, p. 943.
253. Syamal A.— Z. Naturforsch., 1974, B29, S. 492.
254. Ganescu I., Varhelyi C., Papa I.— Z. anorg. allg. Chem., 1974, 409, S. 121.
255. Patel S. J.— Bol. Soc. chilena quim., 1971, 17, p. 61.
256. Wasson J. R., Trapp C.— J. Inorg. Nucl. Chem., 1968, 30, p. 2437.
257. Bucknall W. R., Carter S. R., Wardlaw W.— J. Chem. Soc., 1927, p. 512.
258. Schmidtke H.-H., GarthoffD.— Helv. chim. acta, 1967, 50, p. 1631.
259. Lewis J., Nyholm R. S., Smith P. W.— J. Chem. Soc., 1961, p. 4590.
260. Gnosn S. PPrasad К- M.— J. Less-Common Metals, 1974, 36, p. 223.
261. Horn C. J., Brown T- M.— Inorg. Chem., 1972, 11, p. 1970.
262. Улъко H. В., Билда О. I.— В1сник Ки!'в. ун-ту. Cep. xiMi'f, 1973, № 14, с. 31.
263. Funk H., Bohland H.— Z. anorg. allg. Chem., 1963, 324, S. 168.
264. Mitchell P. С. H.— Coord. Chem. Rev., 1966, 1, p. 315.
265. Маров И. H., Дубров Ю.Н.. Ермаков А. Н., Мартынова Г. Н.— Журнал неорган.
химии, 1968, 13, с. 3247.
266. Гарифьянов Н. С., Каменев С. Е.— Журнал физ. химии, 1969, 43, с. 1087.
267. Grail J.-M., Kergoat R., Guerchais J. E.— C. r. Acad, sci., 1968, C267, p. 1410.
268. Еремин Ю. Г., Колпикова E. Ф., Некрасова Г. В.— Журнал неорган. химии, 1971,
16, с. 2426.
269. Улько Н. В., Савченко Р. В. —Журнал неорган. химии, 1967, 12, с. 328.
270. Bohland Н., Niemann Е.— Z. anorg. allg. them., 1965, 336, S. 225.
271. Woicicki A., FaronaM. F.— J. Inorg. Nucl. Chem., 1964, 26, p. 2289.
272. Sloan T. E., Wojcicki A.— Inorg. Chem., 1968, 7, p. 1268.
273. Ruff J. K.— Inorg. Chem., 1968, 8, p. 86.
274. Behrens HUhlig D., Lindner E.— Z. anorg. allg. Chem., 1972, 394, S. 8.
275. Behrens H., Schwab R., HerrmannD.— Z. Naturforsch., 1966, B21, S. 590.
276. Behrens H., HerrmannD.— Z. Naturforsch., 1966, B21, S. 1234, 1236.
277. Treichel P. M., Douglas W. M.— J. Organometal. Chem., 1969, 19, p. 221.
278. Эристави Д. И., Цинцадзе Г. В., Кереселидзе Л. Б.— Труды Груз, политехи, ин-та,
1968, № 7 (127), с. 39.
279. Grossmann Н., Hiinseler Е.— Z. anorg. Chem., 1905, 46, S. 361.
242
' 280. Цинцадзе Г. В. Смешанные псевдогалогенидо-аминные соединения некоторых
металлов. Тбилиси, 1974.
281. Asmussen R., Soling Н.— Acta chem. scand., 1957, 11, p. 1331.
282. Forster D., Goodgame D. M. L.— J. Chem. Soc., 1965, p. 268.
283. Le Baccon M., Youinou M. T., Guerchais J.-E.— Bull. Soc. chim. France, 1972,
p. 4525.
284. Волков С. В., Евтушенко H. И., Яцимирский К- Б.— Теорет. и эксперим. химия,
1974, 10, с. 271.
285. Schwochau К.. Pieper Н. Н.— Inorg. Nucl. Chem. Lett., 1968, 4, p. 711.
286. Schwochau K-, Astheimer L., Schenk H. J.— J. Inorg. Nucl. Chem., 1973, 35, p. 2249.
287. Bailey R. A., Kozak S. L.— Inorg. Chem., 1967, 6, p. 419.
288. Bailey R. A., Kozak S. L.— Inorg. Chem., 1967, 6, p. 2155.
289. Cotton F. A., Robinson W. R., Walton R. A.. Whyman R.— Inorg. Chem., 1967, 6,
p. 929.
290. Okubo T., Koshimura H.— J. Chem. Soc. Japan, Chem. and Ind. Chem., 1974, 9,
p. 1649.
291. Nelson С. M., Boyd G. E., Smith Wm. T.— J. Am. Chem. Soc., 1954, 76, p. 348.
292. Wajda S., Pruchnik F.— Rocz. chem., 1967, 41, p. 1169.
293. Рябчиков Д. И., Назаренко И. И.— Журнал неорган. химии, 1962, 7, с. 931.
294. Freni М., Giusto D., Romiti Р.— Gazz. chim. ital., 1969, 99, p. 641.
295. Cotton F. A., Goodgame D. M. L., Goodgame M., Sacco A.— J. Am. Chem. Soc.,
1961, 83, p. 4157.
296. Clark R. J. H.. Williams C. S.— Spectrochim. acta, 1966, 22, p. 1081.
297. Burmeister J. L., O’Sullivan T. P., Johnson K- A.— Inorg. Chem., 1971, 10, p. 1803.
298. Green R. W., Bernice Bell.— Austral. J. Chem., 1973, 26, p. 1663.
299. Ercolani C., Quagliano J. V., Vallarino L.— Inorg. chim. acta, 1973, 7, p. 413.
300. Chow К. K-, McAuliffe C. A.— Inorg. chim. acta, 1974, 10, p. 197.
301. Pecile C.— Inorg. Chem., 1966, 5, p. 210.
302. Eggli R.. Ludwig W.— Inorg. chim. acta, 1973, 7, p. 697.
303. Jona E., Sramco T., Jesondk V.— J. Therm. Anal., 1973, 5, p. 315.
304. Jona E., Jesenak V., Sramko T., Gaio J.— J. Therm. Anal., 1973, 5, p. 389.
305. Eller P. G., MeekD. W.— Inorg. Chem., 1972, 11, p. 2518.
306. BertaccoA., Mazzi U.. OrioA. A.— Inorg. Chem., 1972, 11, p. 2547.
307. Boschi T., Nicolini M., Turco A.— Coord. Chem. Rev., 1966, 1, p. 269.
308. Rigo P., Bressan M., Turco A.— Inorg. Chem., 1968, 7, p. 1460.
309. Morassi R., Sacconi L.— J. Am. Chem. Soc., 1970, 92, p. 5241.
310. Sacconi L., Morassi R.— J. Chem. Soc., 1969, A, p. 2904.
311. Ciampolini M., Paolritti P.— Inorg. Chem., 1967, 6, p. 1261.
312. Bertini I., Ciampolini M., Gatteschi D.— Inorg. Chem., 1973, 12, p. 693.
313. Dori Z., Gray H. B.— Inorg. Chem., 1968, 7, p. 889.
314. Ciampolini M., Nardi N.— Inorg. Chem., 1967, 6, p. 445.
315. Malik M. A., PhillipsD. J.— J. Inorg. Nucl. Chem., 1974, 36, p. 2229.
316. Bond M., Martin R. L., Roos I. A. G.— Austral. J. Chem., 1974, 27, p. 2307.
317. Nelson S. M., Shepherd T. M.— J. Inorg. Nucl. Chem., 1965, 27, p. 2123.
318. Dash К- C., Das S. N.— Z. anorg. allg. Chem., 1973, 400, S. 78.
319. Puglisi C., Levitus R.— J. Inorg. Nucl. Chem., 1967, 29, p. 1069.
320. Flint C. D., Goodgame M.— Inorg. Chem., 1969, 8, p. 1833.
321. Allan J. R., Brown D. H., Lappin N.—-J. Inorg. Nucl. Chem., 1970, 32, p. 2287.
322. Харитонов Ю. Я., Цинцадзе Г. В., Порай-Кошиц М. А.— Журнал неорган. хи-
мии, 1965, 10, с. 35.
323. Reedu J.— J. Inorg. Nucl. Chem., 1973, 35, p. 239.
324. Brzozowski S., Broniarek M.— Rocz. chem., 1974, 48, p. 1213.
325. Solacolu 1., Sandulescu D., Dragulescu C.— Rev. roum. chim., 1974, 19, p. 415.
326. Patel К- C., Goldberg D. E.— J. Inorg. Nucl. Chem., 1972, 34, p. 637.
327. Takemoto J. H., Hutchinson B.— Inorg. Chem., 1973, 12, p. 705.
328. Akabori K-, Matsuo H., Yamamoto Y.— J. Inorg. Nucl. Chem., 1973, 35, p. 2679.
329. Konig E., Watson K- J-—Chem. Phys. Lett., 1970, 6, p. 457.
330. Georgescu I., CiomartanD., Teodorescu G. a. o.— Rev. roum. chim., 1973, 18, p. 1159.
331. Schafer M., Uhlig E.— Z. anorg. allg. Chem., 1974, 407, S. 23.
332. ХаритоновЮ. Я., Мачхошвили P. И.— Журнал неорган. химии, 1971, 16, с. 1203.
333. Харитонов Ю. Я-. Мачхошвили Р. И., Цинцадзе Г. В. и др.— Журнал неорган.
химии, 1974, 19, с. 1337.
334. Харитонов Ю. Я-, Мачхошвили Р. И., Цинцадзе Г. В. и др.— Журнал неорган.
химии, 1974, 19, с. 2769.
335. Харитонов Ю. Я-> Мачхошвили Р. И., Генералова Н. Б., Щелков Р. Н.— Журнал
неорган. химии, 1975, 20, с. 693.
243
336. Harris С. M., McKenzie Е. D.— J. Inorg. Nucl. Chem., 1967, 29, p. 1047.
337. Bacci M., Midollini S., Stoppioni P., Sacconi L.— Inorg. Chem., 1973, 12, p. к j|
338. Urbach F. L., Busch D. H.— Inorg. Chem., 1973, 12, p. 408.
339. Campbell T. G., Urbach F. L.— Inorg. Chem., 1973, 12, p. 1836.
340. Chand P. R., Singh C. R., Kumar B. R., Gurdev S.— J. Inorg. Nucl. Chem., 19“-
36, p. 3703.
341. Dickinson R. C., Long C. J.— J. Inorg. Nucl. Chem., 1974, 36, p. 1235.
342. Rosenheim A., Cohn R.— Z. anorg. Chem., 1901, 27, S. 280.
343. Pralhakaran С. P., Patel С. C.— Indian J. Chem., 1972, 10, p. 438.
344. David P. G.— J. Inorg. Nucl. Chem., 1973, 35, p. 1463.
345. King-Wen Kuo, Miller W. V., Madan S. K-—J. Inorg. Nucl. Chem., 1973, 35,
p. 957.
346. Maki Nobufumi, Sakuraba Shukichi.— Bull. Chem. Soc. Japan, 1969, 42, p. 579
347. Маров И. H., Панфилов А. Т., Иванова Е. К-, Ермаков А. Н.— Координационная
химия, 1975, 1, с. 165.
348. Asmussen R. W., Bostrup О.— Acta chem. scand., 1957, 11, p. 1097.
349. Carbacho H., Ungerer B., Contreras G.— J. Inorg. Nucl. Chem., 1970, 32, p. 579.
350. Sabatini A., Bertini I.— Inorg. Chem., 1965, 4, p. 959.
351. De Haas K- S.— J. Inorg. Nucl. Chem., 1973, 35, p. 3231.
352. Iberas J. A., Hamilton D. S., Baddley W. H.— Inorg. Chem., 1973, 12, p. 229
353. Patel S. J.— Bol. Soc. chilena quim., 1972, 19, p. 13, 15.
354. Кукушкин Ю. H., Данилина Л. И.— Журнал неорган. химии, 1974, 19, с. 1349.
355. Hill W. E., McAulliffe C. A.— Inorg. Chem., 1974, 13, p. 1524.
356. Abbadi A. R. M., Shehadeh M. A., Kingson L. V-—J. Inorg. Nucl. Chem., 1974
36, p. 1173.
357. Levason W., McAulliffe C. A.— J. Chem. Soc. Dalton, 1974, p. 2238.
358. Bjerrum J., Schwarzenbach G., Sillen L. G. Stability Constants of Metal-ion Com-
plexes. II. Inorganic Ligands. London, 1958.
359. Яцимирский К- Б., Васильев В. П. Константы нестойкости комплексных соедине-
ний. М., 1959.
360. Sillen L. G., Martel А. Е. Stability Constants of Metal-ion Complexes. Spec, publi-
cation No 17. London, 1964.
361. Sillen L. G., Martel A. E. Stability Constants of Metal-ion Complexes. Spec, publi-
cation No 25. London, 1971.
362. Bent H. E., French C. L.— J. Am. Chem. Soc., 1941, 63, p. 568.
363. Ramakrishna R. S., Thuraisingham R.— J. Inorg. NucL Chem., 1973, 35, p. 2805.
364. TuckD. G., Woodhouse E. J., Carty P.— J. Chem. Soc., 1966, A, p. 1077.
365. Radhakrishnan T. P., Sundaram A. K-—J- Electroanalyt. Chem., 1963, 5, p. 124.
366. Momoki K-, Ogawa H.. Sato H.— Analyt. Chem., 1969, 41, p. 1826.
367. Яцимирский К- Б., Тетюшина В. Д.— Журнал неорган. химии, 1969, 2, с. 320.
368. Das R. С., Dash А. С., Mishra J. Р.— J. Inorg. NucL Chem., 1968, 30, р. 2417
369. Васильев В. П.— Журнал неорган. химии, 1962, 7, с. 1788.
370. Persson Н.— Acta chem. scand., 1971, 25, р. 543.
371. Lodzinska А.— Rocz. chem., 1967, 41, p. 1007.
372. Ciavatta L., Grimaldi M.— Inorg. chim. acta, 1970, 4, p. 312.
373. Irving H., Williams R. J. P.— J. chem. Soc., 1953, p. 3192.
374. Яцимирский К- Б., Федорова Т. И.— Изв. вузов СССР. Химия и хим. технология,
1958, № 3, с. 40.
375. NancollasG. Н., Torrance К-— Inorg. Chem., 1967, 6, с. 1567.
376. Яцимирский К- Б., Кораблева В. Д.— Журнал неорган. химии, 1958, 3, с. 339.
377. Tanaka N., Takamura Т.— J. Inorg. Nucl. Chem., 1959, 9, p. 15.
378. Новоселова А. В., Михеева Л. М., Почкаева Т. И., Тамм Н. С.— Вестник Моск,
ун-та. Химия, 1966, с. 46.
379. Голуб А. М., Иванченко Г. Д.— Журнал неорган. химии, 1958, 3, с. 333.
380. Ahrland S., Kuliberg L.— Acta chem. scand., 1971, 25, p. 3692.
381. Ружицки Ц., Соловьев Ю. Б., Миронов В. Е.— Журнал неорган. химии, 1973,
18, с. 57.
382. Alexandrides G., Cummiskey С.— J. Inorg. Nucl. Chem., 1966, 28, p. 2025.
383. Gerding P., Johansson B.— Acta chem. scand., 1968, 22, p. 2255.
384. Leden I.— Z. phys. Chem., 1941, A188, S. 160.
385. Туръян Я. И., Макарова Л. М., Сирко В. Н.— Журнал неорган. химии, 1974,
19, с. 1778.
386. Sunden N.— Svensk Kern. Т., 1954, 66, р. 50.
387. Федоров В. А., Робов А. М., Исаев Д. И., Миронов В. Е.— Журнал неорган. химии,
1971, 16, с. 970.
388. Bell R. Р., George J. Н. В.— Trans. Faraday Soc., 1953, 49, р. 619.
244
389. Braibanti A., Chierici I.— Gazz. chim. ital., 1958, 88, p. 793.
390. Голуб A. M., Огняник С. C.— Укр. хим. журн., 1961, 27, с. 283.
391. Голуб А. М., Самойленко В. М.— Укр. хим. журн., 1957, 23, с. 17.
392. Миронов В. Е., Кульба Ф. fl., Трифонов О. И.— Журнал неорган. химии, 1963,
8, с. 2113.
393. Федоров В. А., Самсонова Н. П., Миронов В. Е.— Журнал неорган. химии, 1969
15, с. 3264.
394. Федоров В. А., Калош Т. Н., Черникова Г. Е., Миронов В. Е.— Журнал физ. хи-
мии, 1971, 45, с. 1364.
395. Williams Т.— J. Inorg. Nucl. Chem., 1962, 24, р. 1215.
396. Leden INilsson R. N.— Z. Naturforsch., 1955, A10, S. 67.
397. CaveG. C., HumeD. N.— J. Am. Chem. Soc., 1953, 75, p. 2893.
398. Kiehl C. — Z. phys. Chem. (Leipzig), 1966, 232, S. 384.
399. Cac T. M., Гагарина В. А., Комиссарова Л. H., Гулиа В. Г.— Журнал неорган.
химии, 1970, 15, с. 1255.
400. Кумок В. Н., Серебренников В. В.— Журнал неорган. химии, 1964, 9, с. 2148.
401. Rao С. L., Shachani С. JMatchew К- А.— Inorg. Nucl. Chem. Lett., 1968 4,
p. 655.
402. Sekine T.— Acta chem. scand., 1965, 19, p. 1519.
403. Khopkar P. K-, Mathur J. N.— J. inorg. NucL Chem., 1974, 36, p. 3819.
404. Kinard W. F., ChoppinG. R.— J. Inorg. Nucl. Chem., 1974, 36, p. 1131.
405. ChoppinG. R., Ketels J.— J. Inorg. Nucl. Chem., 1965, 27, p. 1335.
406. Ahrland S., Larsson R.— Acta chem. scand., 1954, 8, p. 137.
407. Day R. A., Wilhite R. N., Hamilton F. D.— J. Am. Chem. Soc., 1955, 77, p. 3180.
408. Sillen L. G., Warnqvist B.— Acta chem. scand., 1968, 22, p. 3032.
409. Harmon H. D., Peterson J. R., McDowell W. J., Coleman C. F.— J. Inorg. Nucl.
Chem., 1972, 34, p. 1381.
410. Malin J. M., Swinehart J. H.— Inorg. Chem., 1968, 7, p. 250.
411. Orhanovic M., Po H. N., Sutin N.— J. Am. Chem. Soc., 1968, 90, p. 7224.
412. Baker B. R., Sutin N., Welch T. J.— Inorg. Chem., 1967, 6, p. 1948.
413. Kruse W., ThusiusD.— Inorg. Chem., 1968, 7, p. 464.
414. Schlund A., Wendt H.— Ber. Bunsenges. Phys. Chem., 1968, 72, S. 652.
415. Postmus C., King E. L.— J. Phys. Chem., 1955, 59, p. 1208, 1218.
416. Perrin D. D.— J. Am. Chem. Soc., 1958, 80, p. 3540.
417. Васильев В. П., Мухина П. С.— Журнал неорган. химии, 1964, 9, с. 1134.
418. Васильев В. П., Мухина П. С.— Журнал неорган. химии, 1963, 8, с. 1895.
419. Миронов В. Е., Рутковский Ю. И.— Журнал неорган. химии, 1965, 10, с. 1069.
420. Миронов В. Е., Рутковский Ю. /4.—Журнал неорган. химии, 1965, 10, с. 2670.
421. Prasad В.— J. Indian Chem. Soc., 1968, 45, р. 1037.
422. Tribalat S., Caldero I.-M.— Compt. rend., 1964, 258, p. 2828.
423. Турьян fl. И.— Журнал неорган. химии, 1959, 4, с. 813.
424. Турьян fl. И., Бондаренко Н. И.—Журнал неорган. химии, 1959, 4, с. 1070.
425. Турьян fl. И., Чеботарь Н. Г.— Журнал неорган. химии, 1959, 4, с. 599.
426. Irving Н. М., Rossotti Н. S.— Acta chem. scand., 1956, 10, р. 72.
427. Аблов В. В., Назарова Л. В.—Журнал неорган. химии, 1961, 6, с. 2043.
428. Аблов А. В., Назарова Л. В.—-Журнал неорган. химии, 1959, 4, с. 2480.
429. Голуб А. М.— Докл. АН СССР, 1958, 120, с. 1255.
430. Мигаль П. К-, Гринберг Н. X.— Журнал неорган. химии, 1962, 7, с. 1309.
431. Голуб А. М., Самойленко В. М.— Укр. хим. журн., 1963, 29, с. 590.
432. Гутман В. Химия координационных соединений в неводных растворах. М..
Мир, 1971.
433. Голуб А. М., Романенко Л. И.— Журнал неорган. химии, 1960, 5, с. 1085.
434. Голуб А. М., Самойленко В. М.— Журнал неорган. химии, 1964, 9, с. 70.
435. Самойленко В. М., Гринюк И. И.— Укр.хим. журн., 1972, 38, с. 8.
436. Miezis A.— Acta chem. scand., 1974, А28, р. 407.
437. Саксин Е. В., Малявинская О. Н., Турьян fl. И.— Журнал физ. химии, 1969
43. с. 517.
438. Gutmann V., Bohuovsky О.— Monatsh. Chem., 1968, 99, S. 751.
439. Gutmann V., Bardy H.— Z. anorg. allg. Chem., 1968, 361, S. 213.
440. Kratochvil В., Long R.— Can. J. Chem., 1970, 48, p. 1414.
441. Самойленко В. M., Ляшенко В. И.— Журнал неорган. химии, 1973, 18, с. 2402.
442. Pool К-—J. Polarogr. Soc., 1967, 13, р. 23.
443. Самойленко В. М.— Журнал неорган. химии, 1968, 13, с. 79.
444. Самойленко В. М., Жуков М. П.— Укр. хим. журн., 1970, 26, с. 765.
445. Самойленко В. М., Ляшенко В. И.— Журнал неорган. химии, 1974, 19, с. 2984.
446. Matsui Y., Date Y.— Bull. Chem., Soc., Japan, 1970, 43, p. 2052.
245
447. Breant M., Georges J., hnbert J., Schmitt D.— Ann. chim., 1971, 6, p. 445.
448. Foil A., Le Demezet M., Courtot-Coupez J.— Bull. Soc. chim. France, 1972, p. 1_
449. Parker A.— Quart. Rev., 1962, 16, p. 163.
450. Aihara M., Misumi S.— Bull. Chem. Soc. Japan, 1973, 46, p. 1674.
451. Бек M. Химия равновесных реакций комплексообразования. М., Мир, 1973.
452. Фридман Я- Д. Окислительно-восстановительные свойства комплексных соед
ний металлов и их устойчивость в растворах. Фрунзе, Илим, 1966.
453. Устойчивость смешанных комплексных соединений в растворах. Фрунзе, И.
1971.
454. Яцимирский К. Б.— Журнал неорган. химии, 1971, 16, с. 585.
455. Marcus Y., Eliezer I.— Coord. Chem. Rev., 1969, 4, p. 273.
456. Burger K-— Kem. Kozl., 1966, 26, p. 363.
457. Белеванцее В. И., Пещевицкий Б. И.— Журнал неорган. химии, 1972, 17, с. 2»
458. Jorgensen С. К.— Inorg. Chem., 1964, 3, р. 1201.
459. Gaizer F., Muray L., Beck M. T.— Acta chim. Acad. sci. hung., 1971, 67, p. 25
460. Yalman R. G.— J. Am. Chem. Soc., 1961, 83, p. 4142.
461. Lister M. N., Rivington D. E.— Can. J. Chem., 1955, 33, p. 1591.
462. Федоров В. А., Исаев И. Д., Робов А. М. и др.— Журнал неорган. химии, 19“_
17, с. 951.
463. Grossmann Н.— Z. anorg. Chem., 1903, 37, S. 411.
464. SpacuG., Spacu P.— Chem. Zentr., 1931, S. 1425.
465. Galinos A. G.— J. Inorg. Nucl. Chem., 1962, 24, p. 1555.
466. Цинцадзе Г. В., Голуб А. М., Манагадзе А. С.— Труды Груз, политехи, ин-га
1970, № 1 (136), с. 52.
467. Цинцадзе Г. В., Голуб А. М., Манагадзе А. С., Цивадзе А. Ю.— Труды Груз, по-
литехи. ин-та, 1972, № 1 (149), с. 10.
468. Цинцадзе Г. В.— Труды Груз, политехи, ин-та, 1970, № 3 (138), с. 81.
469. Квезерели Э. А., Цинцадзе Г. В., Манагадзе А. С., Цивадзе А. К).— Труды Грхз.
политехи, ин-та, 1972, № 5 (153), с. 29.
470. Фридман Я- Д-, Сарбаев Д.— Изв. АН КиргССР. Сер. естеств. и техн, наук, 1963,
№ 5, с. 1.
471. Яцимирский К. Б., Тухлое Б. Д.— Журнал общ. химии, 1956, 26, с. 356.
472. Czakis-Sulikowska D. М.— Zesz. nauk. Politechn. Lodzk., 1966, No 80, s. 33.
473. Jozefowicz E., Ladzinska-Kulinska H.— Rocz. chem., 1965, 39, p. 1175.
474. Swinarski A., Danilczuk E.— Rocz. chem,. 1966, 40, p. 737.
475. Фридман Я- Д-, Сарбаев Д.— Журнал неорган. химии, 1959, 4, с. 1849.
476. Сагбаев Д.— В кн.: Химия комплексных соединений редких и сопутствующих
элементов. Фрунзе, Илим, 1970, с. 31.
477. Яцимирский К- Б., Панова В. Е.— Журнал общ. химии, 1952, 22, с. 1284.
478. Бирюков А. А., Шленская В. И. —Журнал неорган. химии, 1967, 12, с. 2579.
479. Бирюков А. А., Шленская В. И., Алимарин И. П.— Изв. АН СССР. Сер. хим.,
1966, № 1, с. 3.
480. Яцимирский К. Б. Термохимия комплексных соединений. М., Изд-во АН СССР.
1951.
481. Jones L. Н.— J . Chem. Phys., 1956, 25, р. 1069.
482. Wagner Е. L.— J. Chem. Phys., 1965, 43, р. 2728.
483. Садовский А. П., МазадовЛ. Н., Гужавина Т.П. и др.— Журнал структурной хи-
мии, 1973, 14, с. 667.
484. Di Sipio L., Oleary L., De Michelis L.— Coord. Chem. Rev., 1966, 1, p.7.
485. Ahrland S., Chatt J., Davies N. R.— Quart. Rev-, 1958, 12, p. 265.
486. Taylor K. A., LongT. V., Plane R. A.— J.Chem. Phys., 1967, 47,p. 138.
487. Howarth W., Richards R. E.. Venanzi L. M.— J. Chem.Soc., 1964, p. 3335.
488. Hester R. E., Krishnan K-— J. Chem. Phys., 1968, 48, p. 825.
489. Klug H. R.— 7. Kristallogr., 1933, 86, S. 214.
490. Russem W., Gilnter P., Tubin K-— Z. phys. Chem., 1934, 24, S. 1.
491. Strada M.— Cazz. chim. ital., 1934, 62, p. 5261.
492. Жданов Г. С., Звонкова 3. В.— Успехи химии, 1953, 22, с. 3.
493. Marongiu G., Lingafelter Е. С., Paoletti Р.— Inorg. Chem., 1969, 8, р. 2763.
494. Mitchell Р. С. Н., Williams Р. J. Р.— J. Chem. Soc., 1960, р. 1912.
495. Накомото К- Инфракрасные спектры неорганических и координационных соеди-
нений. М., Мир, 1966.
496. Gordon D. J., Smith D. F.— Spectrochim. acta, 1974, A30, p. 1953.
497. Харитонов Ю. Я., Цинцадзе Г. В., Порай-Коинщ М. А.—Журнал неорган.
химии, 1965, 10, с. 792.
498. Ellestad О. Н., Klaeboe Р., Tucker Е. Е., Songstad J.— Acta chem. scand., 1972,
26, p. 1721.
246
499. Gerrard W., Lappert M. F., Pyszora H., Wallis J. IV7.— J. Chem. Soc., 1960, p. 2182.
500. Применение спектроскопии в химии. Сборник, М., 1959.
501. Durig J. R., Wertz D. W.— J. Chem. Phys., 1967, 46, p. 3069.
502. Pauling L. The Nature of the Chemical Bond. 3rd ed. N. Y., 1960, p. 228, 234,
260, 274.
503. Михеева Л. M., Елфимова Г. И., Комиссарова Л. Н.— Журнал неорган. химии,.
1971, 16, с. 3377.
504. Goodgame D. М. L.. Malerbi В. W.— Spectrochim. acta, 1968, А24, р. 1254.
505. Keller R. N., Johnson N. B., Westmoreland L. L.— J. Am. Chem. Soc., 1968, 90,
p. 2729.
506. Curtis N. F.— J . Chem. Soc. Dalton, 1975, p. 91.
507. Burmeister J. L., Basolo F.— Inorg. Chem., 1964, 3, p. 1587.
508. Stotz IWilmarth W. K-, Haim A.— Inorg. Chem., 1964, 3, p. 1587.
509. Siebert H., WittkeG.— Z. anorg. allg. Chem., 1973, 399, S. 52.
510. Burmeister J. L., Patterson S. D., Deardorff A. E.— Inorg. chim. asta, 1969, 3, p. 105.
511. Цивадзе А. Ю., Харитонов Ю. Я-, Цинцадзе Г. В.— Журнал неорган. химии,
1970, 15, с. 2123.
512. Jeffrey J. W.— Acta Crystallogr., 1963, 16, р. А66.
513. Порай-Кошиц М. А.— Журнал структурной химии, 1963, 4, с. 584.
514. Grbnback R., Dunitz J. D.— Helv. chim. acta, 1964, 47, p. 1889.
515. Panattoni C., Frasson E.— Acta Crystallogr., 1963, 16, p. 1258.
516. Beck W., Lottes K-— Z. anorg. allg. Chem., 1965, 335, S. 258.
517. Homan J. M., Kawamoto J. M., Morgan G. L.— Inorg. Chem., 1970, 9, p. 2533.
518. Dehnicke K-— Angew. Chem., 1967, 79, S. 942.
519. Mason W. R., Berger E. R., Johnson R. C.— Inorg. Chem., 1967, 6, p. 248.
520. Cannas M., Carta G., Marongiu G.— J. Chem. Soc. Dalton, 1973, p. 251.
521. Turco A., Pecile C.— Nature (London), 1961, 191, p. 66.
522. Sabatini A., Bertini I.— Inorg. Chem., 1965, 4, p. 1665.
523. Holsboer F., Beck W.— Z. Naturforsch., 1972, B27, S. 884.
524. Beck W., Holsboer F.— Z. Naturforsch., 1973, B28, S. 511.
525. Burmeister J. L.— Inorg. Chem., 1964, 3, p. 919.
526. Pearson R. G.— Inorg. Chem., 1973,12, p. 712.
527. Meek D. W., Nicpon P. E., Meek V. I.— J. Am. Chem. Soc., 1970, 92, p. 5351.
528. Clark G. R., Palenic G. J.— Inorg. Chem., 1970, 9, p. 2754.
529. Carty A. J.. Efraty A.— Inorg. Chem., 1969, 8, p. 543.
530. Basolo FBaddley W. H., Burmeister J. L.— Inorg. Chem., 1964, 3, p. 1202.
531. Lauer J. L., Peterkin M. E., Burmeister J. L. a. o.— Inorg. Chem., 1972, 11,
p. 907.
532. Burmeister J. L., Hassel R. L., Johnson K- A., Lim J. C.— Inorg. chim. acta, 1974,
9, p. 23.
533. Basolo F., Baddley W. H., Weidenbaum K. J-— J. Am. Chem. Soc., 1966, 88, p. 1576.
534. Palenik G. JSteffen W. L., Mathew M., Li M., Meek D. W.— Inorg. Nucl. Chem.
Lett., 1974, 10, p. 125.
535. Jacobson S., Wong Y. S., Chieh P. C., Carty A. J.— Chem. Commun., 1974, p. 520.
536. Gutterman D. F., Gray H. B.— J. Am. Chem. Soc., 1969, 91, p. 3105.
537. Burmeister J. L., Lim J. C.— Chem. Commun., 1968, p. 1346.
538. Цивадзе А. Ю., Харитонов Ю. Я-. Цинцадзе Г. В.— Журнал неорган. химии,.
1972, 17, с. 2912.
539. Burmeister J. L., Hassel R. L., Phelan R. J.— Inorg. Chem., 1971, 10, p. 2032.
540. Hassel R. L., Burmeister J. L.— Chem. Commun., 1971, p. 568.
541. Norbury A. H., Shaw P. E., Sinha A. I. P.— Chem. Commun., 1970, p. 1080.
542. FaronaM. F., Woicicki A.— Inorg. Chem., 1965, 4, p. 857.
543. Epps L. A.. Marzilli L. G.— Inorg. Chem., 1973, 12, p. 1514.
544. Po H. N., Wong W.-K., Chen K. D.— J. Inorg. Nucl. Chem., 1974, 36, p. 3872.
545. Buckley R. C., Wardeska J. G.— Inorg., Chem., 1972, 11, p. 1723.
546. Miezis A. — Acta chem. scand., 1973, 27, p. 3746.
547. Goubeau J., Reyhing J.— Z. anorg. allg. Chem., 1958, 294, S. 96.
548. Харитонов Ю. Я-, Цинцадзе Г. В., Цивадзе А. Ю.— Журнал неорган. химии,
1970, 15, с. 710.
549. Харитонов Ю. Я., Цинцадзе Г. В., Цивадзе А. Ю.— Журнал неорган. химии,
1970, 15, с. 1513.
55O. Dutta R.L.,De Dhrubananda.— J. Indian Chem. Soc., 1969,46, p. 1.
551. Павленко Э. С., Кумок В. H., Серебренников В. В.— Труды Томск, ун-та, 1973,
237, с. 104.
552. Цинцадзе Г. В., Квазерели Э. А., Доксопуло Э. П., Лежава А. П.— Труды Груз,
политехи, ин-та, 1972, № 1 (149), с. 5.
247
553. Лазарев П. И., АслановЛ. А., Порай-Кошиц М. А., Иванов В. И.— Журнал стр •
турной химии, 1972, 13, с. 543.
554. Лазарев П. И., Порай-Кошиц М. А., Асланов Л. А., Иванов В. И.— Жхг
структурной химии, 1972, 13, с. 740.
555. Николаев А. В., Григорьев В. А.— Журнал неорган. химии, 1965, 10, с. 281.
556. CyganskiA., Galdecki Z. — Rocz. chem., 1968, 42, p. 747.
557. Chatt J., Duncanson L. A.— Nature, 1956, 178, p. 997.
558. Ratho T., Patel T.— Indian J. Chem., 1969, 43, p. 166.
559. DeStefano N., Burmeister J. L.— Inorg. Chem., 1971, 10, p. 998.
560. Dash R. N., Ramana Rao D. V.— J. Indian chem. Soc., 1974, 51, p. 211.
561. McColm 1, J.— J. Inorg. Nucl. Chem., 1970, 32, p. 1461.
562. Chow Y. W.— Inorg. Chem., 1970, 9, p. 794.
563. Border R. A., SheldrickG. M.— Chem. Commun., 1969, p. 1125.
564. Macuolu J. A. A., Speakman J. C.— Chem. Commun., 1966, p. 25.
565. Macuolu J. A. A., Speakman J. C.— Acta Crystallogr., 1975, B31, p. 172.
566. Farado M. E., James J. M.— Inorg. Chem., 1965, 4, p. 1706.
567. Pecile C., Turco A., PizzolittoG.— Ricerca scient., 1961, 1, p. 247.
568. Ichinose T., Nishida Y., Okawa H., Kida S.— Bull. Chem. Soc. Japan, 1974, 47
p. 3045.
569. MachinD. J., Sullivan J. F.— J. Less-Common Metals, 1969, 19, p. 413.
570. Granifo J., Milller N.— J. Chem. Soc. Dalton, 1973, p. 1891.
571. Харитонов Ю. fl., Цинцадзе Г. В., Цивадзе А. К).— Журнал неорган. химии
1970, 15, с. 1563.„
572. Jona Е., Sramco Т.. Gazo J.— Chem. Zwesti, 1973, 27, p. 145.
573. Vallarino L.— J. Chem. Soc., 1957, p. 2473.
574. TibbettsD. L., Brown T. L.— J. Am. Chem. Soc., 1969, 91, p. 1108.
575. Fereidum H.— J. Appl. Chem. and Biotechnol., 1973, 23, p. 205.
576. Youinon M,-T., Gouerchais J,-E.— J. Mol. Struct., 1973, 18, p. 93.
577. Chamberlain M. M., Bailar J. C.— J. Am. Chem. Soc., 1959, 81, p. 6412.
578. Falk L. C., Linck R. G.— Inorg. Chem., 1971, 10, p. 215.
579. Bailey R. A., Michelsen T. W.— J. Inorg. Nucl. Chem., 1972, 34, p. 2671.
580. FaronaM. F.,Wojcicki A.— Inorg. Chem., 1965, 4, p. 1402.
581. Johnson В. C., Meyer T. J., Winterton N.— Inorg. Chem., 1971, 10, p. 1673.
582. Buckingham D. A., Creaser 1. I., Sargeson A. M.— Inorg. Chem., 1970, 9, p. 655.
583. Marzilli L. G., Stewart R. C., Epps L. A., Allen J. B.— J. Am. Chem. Soc., 1973,
95, p. 5796.
584. Schmidtke H.-H.— J. Am. Chem. Soc., 1965, 87, p. 2522.
585. Schmidtke H.-H.— Inorg. Chem., 1966, 5, p. 1682.
586. Basolo F.. Burmeister J. L., Poe A. J.— J. Am. Chem. Soc., 1963, 85, p. 1700.
587. Синицын H. M., Борисов В. В., Пшеничникова Л. А.— Журнал неорган. химии,
1974, 19, с. 3050.
588. Самусь И. Ц., Белов Н. В.— Докл. АН СССР, 1967, 173, с. 83.
589. Швелашвили А. Е., Порай-Кошиц М. А., Анцышкина А. С. и др.— Журнал струк-
турной химии, 1966, 7, с. 810.
590. Жданов Г. С., Звонкова 3. В., Глушкова В. П.— Журнал физ. химии, 1953, 27,
с. 106.
591. Звонкова 3. В.— Журнал физ. химии, 1957, 31, с. 2074.
592. Швелашвили А. Е., Квиташвили А. И., Тавберидзе М. Г., Щедрин Б. М.— Изв.
АН ЛатвССР. Сер. хим., 1971, с. 495.
593. Швелашвили А. Е., Квиташвили А. И., Тавберидзе М. Г., Щедрин Б. М.— Изв.
АН ЛатвССР. Сер. хим., 1973, с. 376.
594. Andreetti G. D., Cavalca L., Sgarabotto P.— Gazz. chim. ital., 1971, 101, p. 483
595. Звонкова 3. В.— Журнал физ. химии, 1952, 26, с. 1798.
596. Corventranta J., Paiunen А.— Suomenkem., 1970,43, р. 119.
597. Brown В. W., Lingafelter Е. С.— Acta Crystallogr., 1963, 16, р. 753.
598. Owston G., Rowe J. M.— Acta Crystallogr., 1960, 13, p. 253.
599. Gregory U. A., Jarvis J. A. J., Kilbourn В. T., Owston P. G.— J. Chem. Soc., 1970,
A, p. 2770.
600. Scouloudi H.— Acta Crystallogr., 1953, 6, p. 651.
601. Звонкова 3. В., Жданов Г. С.— Журнал физ. химии, 1949, 23, с. 1495.
602. Takeuchi YSaito Y.— Bull. Chem. Soc. Japan, 1957, 30, p. 319.
603. Stephens F. S.— J. Chem. Soc., 1970, A, 2377.
604. Moras D., MetzB., Weiss R.— Acta Crystallogr., 1973, B29, p. 388.
605. MetzB., Moras D., Weiss R.— Acta Crystallogr., 1973, B29, p. 1388.
606. MetzB., Weiss R.— Inorg. Chem., 1974, 13, p. 2094.
607. Dunitz J. D., Seiler P.— Acta Crystallogr., 1974, B30, p. 2750.
248
608. Dobler M., Dunitz J. D., Seiler P.— Acta Crystallogr., 1974, B30, p. 2741, 2744.
609. Dobler M., Phizackerley R. P.— Acta Crystallogr., 1974, B30, p. 2748.
610. Dunitz J. D., Dobler M., Seiler P., Phizackerley R. P.— Acta Crystallogr., 1974,
B30, p. 2733.
611. Dobler M., Phizackerley R. P.— Helv. chim. acta, 1974, 57, p. 664.
612. Layton A. JMallinson P. R., Parsons D. G., Truter M. R.— Chem. Commun., 1973,
p. 694.
613. Phillips S. E. V., Truter M. R.— J. Chem. Soc. Dalton, 1974, p. 2517.
614. Fronczek F. R., Schaefer W. P., Marsh R. E.— Acta Crystallogr., 1974, B30, p. 117.
615. Thewalt IJ.— 7L. anorg. allg. Chem., 1972, 393, S. 1.
616. Mathieu F., Weiss R.— Chem. Commun., 1973, p. 816.
617. Metz B., Weiss R.— Acta Crystallogr., 1973, B29, p. 1088.
618. KendallD. S., Lipscomb W. N.— Inorg. Chem., 1973, 12, p. 2915.
619. Kendall D. S., Lipscomb W. N.— Inorg. Chem., 1973, 12, p. 2920.
620. Faught J. B., Moeller TPaul I. C.— Inorg. Chem.,‘1970, 9, p. 1656.
621. Dieck R. L., Moeller T.— J. Inorg. Nucl. Chem., 1973, 35, p. 75.
622. Kimura K-, Katada K-, Bauer S. H.— J. Am. Chem. Soc., 1966, 88, p. 416.
623. CarlsonG. R.— Spectrochim. acta, 1962, 18, p. 1529.
624. Dapporto P., Di Vaira M.— J. Chem. Soc., 1971, A, p. 1891.
625. Dapporto P., Di Vaira M., Sacconi L.— Chem. Commun., 1969, p. 153.
626. Orlandini A. B., Calabresi C., Ghibardi C. A. a. o.— J. Chem. Soc. Dalton, 1973,
p. 1383.
627. Jain P. C., Lingafelter E. C.— J. Am. Chem. Soc., 1967, 89, p. 6131.
628. Jain P. C., Lingafelter E. C., Paoletti P.— J. Am. Chem. Soc., 1968, 90, p. 519.
629. Andreetti G. D., Jain P. C., Lingafelter E. C.— J. Am. Chem. Soc., 1969, 91r
p. 4112.
630. Ciampolini M., Paoletti P.— Inorg. Chem., 1967, 6, p. 1261.
631. Rasmussen S. E.— Acta chem. scand., 1959, 13, p. 2009.
632. Bertini I., Ciampolini M., Dapporto P., Gatteschi D.— Inorg. Chem., 1972, 11,
p. 2254.
633. Clausen A., Hazell A. C.— Acta chem. scand., 1970, 24, p. 2811.
634. Di Vaira M., Sacconi L.— Chem. Communs., 1969, p. 10.
635. Di Vaira M. —J. Chem. Soc., 1971, A, p. 148.
636. Порай-Кошиц M. А., Швелашвили A. E., Анцышкина А. С., Миначева Л. X.—
Журнал неорган. химии, 1968, 13, с. 1245.
637. Шеелашвили А. Е., Порай-Кошиц М. А., Анцышкина А. С.— Журнал структур-
ной химии, 1965, 6, с. 168.
638. Швелашвили А. Е., Порай-Кошиц М. А., Анцышкина А. С.— Журнал структур-
ной химии, 1968, 9, с. 646.
639. Швелашвили А. Е., Саришвили Л. П.— Журнал неорган. химии, 1973, 18, с. 3133.
640. Швелашвили А. Е., Порай-Кошиц М. А., Анцышкина А. С.— Журнал структур-
ной химии, 1969, 10, с. 650.
641. Nardelli М., Gasparri G. F., Battistini G. G., Domiano P.— Acta Crystallogr., 1966,
20, p. 349. ' -
642. Domiano P., Manfredotti A. M., Grossoni G. a. o.— Acta Crystallorg., 1969, B25,
p. 591.
643. Nardelli M., Cavalca L., Braibanti A.— Gazz. chim. ital., 1957, 87, p. 917.
644. Cavalca L., Nardelli M., Gasparri G. F.— Acta Crystallogr., 1960, 13,Ip. 125.
645. Nardelli M., Gasparri G. F., Musatti A., Manfredotti A.— Acta Crystallogr., 1966,
21, p. 910.
646. Korczynski A.— Rocz. chem., 1967, 41, p. 1197.
647. Korczynski A., Poraj-Koshits M. A.— Rocz. chem., 1965, 39, p. 1567.
648. Owczarek A., Soloniewicz R.— Rocz. chem., 1973, 47, p. 397.
649. Jeffery J. WRose К- M.— Acta Crystallogr., 1968, B24, p. 653.
650. Frasson E., Turco A., Panattoni I. C.— Gazz. chim. ital., 1961, 91, p. 750.
651. Masatatsu I., Toshio S.— Z. Kristallogr., 1968, 126, p. 376.
652. Федоров П. M., Андреянова Л. С., Пахомов В. И.— Координационная химия,
1975, 1, с. 252.
653. Kabesova М., Langfelderova Н., Garaj J., Gazo J.— Chem. Zvesti, 1967, 21, p. 887.
654. Kabesova M., Garaj J., Gazo J.— 2nd Conf. Coordinat. Chem. Bratislava, 1969,
s. a. 133.
655. Garaj J.— Acta Crystallogr., 1966, 21, p. A141.
656. Garaj J.— Inorg. Chem., 1969, 8, p. 304.
657. Chow Y. M.— Inorg. Chem., 1971, 10, p. 673.
658. Mawby A., Pringle G. E.— Chem. Commun., 1970, p. 385.
; 659. Жданов Г. С., Звонкова 3. В.— Журнал физ. химии, 1950, 24, с. 1339.
249
660. Cerrini S., Colapietro M., Spagna R., Zambonelli L.— J. Chem. Soc., 1971 <
p. 1375.
661. Звонкова 3. В., Жданов Г. С., Поветьева 3. П.— Кристаллография, 1969. 1
с. 691.
662. Порай-Кошиц М. А., Цинцадзе Г. В.— Труды Груз, политехи, ин-та. 19г'
4 (102), с. 61.
663. Ferrari A., Braibanti A., Bigliavdi G., Lanfredi А. М.— Acta Crystallogr.. 1Г* '
18, р. 367.
664. Shepherd Т. М., Woodward J.— Acta Crystallogr., 1965, 19, p. 479.
665. Швелашвили A. E., Порай-Кошиц M. A.— Координационная химия, 1975. L
c. 463.
666. Швелашвили A. E., Порай-Кошиц M. А., Квиташвили А. И., Щедрин Б. Al.—
Журнал структурной химии, 1974, 15, с. 310.
667. Швелашвили А. Е., Чантурия Л. М., Пирихалава Н. И. и др.— Сосг
АН ГрузССР, 1974, 75, с. 597.
668. Цинцадзе Г. В., Цивцивадзе Т. И., Орбеладзе Ф. В.— Журнал структурной х.
мии, 1975, 16, с. 319.
669. Bigoli F., Braibanti A., Pellinghelli А. М., Tiripicchio А.— Acta Crystallogr 197
В28, р. 962. ’
670. Швелашвили А. Е., Квиташвили А. И., Саришвили Л. П., Щедрин Б. М.— И
АН ЛатвССР. Сер. химии., 1972, с. 118.
671. Швелашвили А. Е., Порай-Кошиц М. А., Квиташвили А. И. и др.— Журн
структурной химии, 1974, 15, с. 315.
672. Швелашвили А. Е., Квиташвили А. И., Щедрин Б. М. и др.— Из
АН ЛатвССР. Сер. хим., 1974, с. 655.
673. Beauchamp A. L., GoutierD.— Can. J. Chem., 1972, 50, р. 977.
674. Пахомов В. И.— Кристаллография, 1960, 5, с. 800.
675. Пахомов В. И.— Кристаллография, 1962, 7, с. 456.
676. Beauchamp A. L., Saperas В., Rivest R.— Can. J. Chem., 1971, 49, р. 3579.
677. Beauchamp A. L., Saperas В., Rivest R.— Can. J. Chem., 1974, 52, р. 2923.
678. Makhija R. С., Beauchamp A. L., Rivest R.— Chem. Commun., 1972, р. 1043.
679. Korczynski A.— Rocz. chem., 1966, 40, p. 547.
680. Чжоу Ко-сянь, Порай-Кошиц М. А.— Кристаллография, 1960, 5, с. 462.
681. Швелашвили А. Е. Докт. дис. Тбилиси, 1974.
682. Thiele G., Bauer R., Messer D.— Naturwissenschaften, 1974, 61, S. 215.
683. Friebel C., Gonzalez G. M. 1.— Z. Naturforsch., 1975, 30b, S. 883.
684. Makhija R. C., Beauchamp A. L., Rivest R.— J. Chem. Soc. Dalton, 1973, p. 2447.
685. Domingous A. M., Sheldrick G. M.— J. Organometal. Chem., 1974, 67, p. 257.
686. Сыч A. M., Богатырь H. И.— Журнал нрикл. спектроскопии, 1974, 21, с. 277.
687. Ohlberg S. M., Vaughan P. A.— J. Am. Chem. Soc., 1954, 76, p. 2649.
688. Порай-Кошиц M. А., Тищенко Г. H.— Кристаллография, 1959, 4, с. 239.
689. Bailey К. A., McKenzie Е. D.— J. Chem. Soc. Dalton, 1972, р. 1566.
690. Garaj J.— Chem. Zvesti, 1967, 21, p. 865.
691. Гарай Ц.— Журнал структурной химии, 1966, 7, с. 727.
692. Brown В. W., LingafelterE. С.— Acta Crystallogr., 1964, 17, р. 254.
693. Pajunen A., Hamalainen R.— Suomen kern., 1972, 45, p. B122.
694. Pajunen A., Smolander K-— Finn. Chem. Lett., 1974, p. 99.
695. Cannas M., CartaG., Marongiu G.— J. Chem. Soc. Dalton, 1974, p. 550, 553.
696. Yoshio A., Nowacki W.— Z. Kristallogr., 1974, 139, S. 337.
697. Cannas M., CartaG., Marongiu G.— Gazz. chim. ital., 1974, 104, p. 581.
698. Cannas M., CartaG., Marongiu G.— J. Chem. Soc. Dalton, 1974, p. 556.
699. Kozlowski D. L., HodgsonD. J.— J. Chem. Soc. Dalton, 1975, p. 55.
700. Smolander K-—Finn. Chem. Lett., 1974, p. 199.
701. Lindqvist I.— Acta Crystallogr., 1957, 10, p. 29.
702. Lindqvist I., Standberg B.— Acta Crystallogr., 1957, 10, p. 173.
703. Бабу Рам С. Сенгал, Гопал Нараин.— Журнал общ. химии, 1975, 45, с. 339.
704. Howatson J., Morosin В.— Cryst. Struct. Commun., 1973, 2, p. 51.
705. Capacchi C. L., Gasparri G. F., Ferrari M., Nardelli M.— Chem. Commun., 1968,
p. 910.
706. Лазарев П. И., Ионов В. M., Асланов Л. А., Порай-Кошиц М. А.— Журнал струк-
турной химии, 1973, 14, с. 168.
707. Лазарев П. И., Асланов Л. А., Порай-Кошиц М. А.— Журнал структурной химии,
1973, 14, с. 169.
708. Hazell А. С.— J. Chem. Soc., 1963, р. 5745.
709. Hazell A. G.— Acta Crystallogr., 1964, 17, р. 1155.
710. Kamenar В., Prout С. К-— J. Chem. Soc., 1970, А, р. 2379.
250
711. Kay J., Moore J. M., Glick M. D.— Inorg. Chem., 1972, 11, p. 2818.
712. Eriks К.» Knox J. R.— Acta Crystallogr., 1966, 21, p. A140.
713. Knox J. R., Eriks K.— Inorg. Chem., 1968, 7, p. 84.
714. Bigoli F., Braibanti A., Pellinghelli M. A., Tiripicchio A.— Acta Crystallogr., 1973,
B29, p. 39.
715. Hauck J., Schwochau K-> Bucksch R.— Inorg. Nucl. Chem. Lett., 1973, 9, p. 927.
716. Sotofte I., Rasmusen S. E.— Acta chem. scand., 1967, 21, p. 2028.
717. Presinger A.— Tscherm. Mineral. Petrogr. Mitteilung, 1953, 3, S. 376.
718. Аблов А. В., СамусьИ. Д,— Докл. АН СССР, 1962, 146, с. 1071.
719. Baggio S., Becka L. M.— Chem. Commun., 1967, p. 506.
720. Биюшкин В. HПопа Э. В., Аблов А. В., Белов Н. В.— Докл. АН СССР, 1970,
191, с. 1282.
721. SnowM. R., BoomsmaR. F.— ActaCrystallogr., 1972, B28, p. 1908.
722. MangiaA., Nardelli M., Pelizzi G.— Acta Crystallogr., 1974, B30, p. 487.
723. Di Vaira M., Orlandini A.— J. Chem. Soc. Dalton, 1972, p. 1704.
724. Bianchi A., Ghilardi С. M.— J. Chem. Soc., 1971, A, p. 1096.
725. П орай-Кошиц M. А., Юхно Э. К., Анцышкина A. С., ДикареваЛ. М.— Кристалло-
графия, 1957, 2, с. 371.
726. Порай-Кошиц М. А.— Труды Ин-та кристаллографии, 1954, 10, с. 117.
727. Юхно Э. К-, Порай-Кошиц М. А.— Кристаллография, 1957, 2, с. 239.
728. Порай-Кошиц М. А., Анцышкина А. С.— Кристаллография, 1958, 3, с. 686.
729. Garaj JDanaj-Jurco М.— Chem. Commun., 1968, р. 518.
730. Atnzel L. M., Baggio S., Becka L. N.— J. Chem. Soc., 1969, A. p. 2066.
731. Capacchi L., Gasparri G. F., Nardelli M., Pelizzi G.— Acta Crystallogr., 1968, B24,
p. 1199.
732. Швелашвили A. E., Порай-Кошиц M. А., Квиташвили А. И. и dp.— Журнал
структурной химии, 1974, 15, с. 313.
733. Durcanska Е., Garaj J.— Proc. 4th Conf. Coord. Chem. and 2nd Semin. Crystallo-
chem. coord, and metallogr. compounds. Bratislava, 1973, s. a. 30.
734. Flack H. D„ Parthe E.— Acta Crystallogr., 1973, B29, o. 1099.
735. FranssonE., Panattoni C., Turco A.— Nature, 1963, И9 4895, p. 803.
736. Beran G., Carty A. J., Chieh P. C., Patel H. A.— J. Ch:m. Soc. Dalton, 1973,
p. 488.
737. Clark G. R., Palenik G. J., MeekD. M.— J. Am. Chem. Soc., 1970, 92, p. 1077.
738. Beran G., Palenik G. J.— Chem. Commun., 1970, p. 1354.
739. Naik D. V., Palenik G. J., Jacobson S., Carty A. J.— J. Am. Chem. Soc., 1974, 96,
p. 2286.
740. Блейделис Д. Д.— Кристаллография, 1957, 2, с. 278.
741. Блейделис Д. Д., Бокий Г. Б.— Кристаллография, 1957, 2, с. 281
742. Звонкова 3. В.— Журнал физ. химии, 1952, 26, с. 1804.
743. WongY. S., Jacobson S., Chieh P. C., Carty A. J.— Inorg. Chem., 1974, 13, p. 284.
744. Пилипенко A. T., Тананайко M. M.— Журнал аналит. химии, 1973, 28, с. 745.
745. Даймонд Р. М., Так Д. Г. Экстракция неорганических соединений. Госхимиздат,
1962.
746. Бабко А. К., Пилипенко А. Т. Колориметрический анализ. Госхимиздат, 1951.
747. Акимов В. К., Клиопг Л. ДБусев А. И.— Журнал аналит. химии, 1973, 28,
с. 118.
748. Бабко А. И.— Журнал общей химии, 1947, 17, с. 642.
749. Бусев А. И. Аналитическая химия молибдена. М., Изд-во АН СССР, 1962, с. 20.
750. Souchay Р., Cadiot М., Dahameaux М.— С. г. Acad, sci., 1965, 260, р. 186.
751. Еремин Ю. Г., Колпикова Е. Ф, Гусева К- С.— Журнал аналит. химии, 1972, 27,
с. 1293.
752. Еремин Ю. Г., Колпикова Е. Ф., Дорфман А. Е.— Журнал аналит. химии, 1972,
27, с. 1297.
753. Тараян В. М., Мушегян Л. Г., Эмикян М. К.— Изв. АН АрмССР, 1964, 17, с. 296.
754. Лазарев А. И., Лазарева В. И.— Журнал аналит. химии, 1967, 22, с. 1210.
755. Rozycki С., Maksjan J.— Chem. analit. (PRL), 1970, 15, p. 391, 1019.
756. Crouthamel С. E., Hjelte В. E., Johnson С. E.— Anal. Chem., 1955, 27, p. 507.
757. Шленская В. И., Пискунов В. М., Хвостова В. П.— Вестник Моск, ун-та. Химия,
1964, с. 62.
758. Clinch JGuy М. J.— Analyst, 1957, 82, р. 800.
759. Crauthamel С. Е., Johnson С. Е.— Anal. Chem., 1952, 24, р. 1780.
760. Бабко А. И., Тананайко М. М.— Журнал неорган. химии, 1962, 7, с. 562.
761. Немодрук А. А., Безрогова Е. В.— Журнал аналит. химии, 1968, 23, с. 884.
762. Тананайко М. М., Небылицкая С. Л.— Заводская лаборатория, 1962, 28, с. 263.
763. Танямашсо М. М., БлуккеЛ. А.— Укр. хим. журн., 1963, 29, с. 974.
251
764. Тананайко М. М.— Журнал неорган. химии, 1964, 9, с. 608.
765. Бабко А. К-, Тананайко М. М.— Журнал неорган. химии, 1966, 11, с. 548.
766. DuttN. К-, Seshardi Т.— Indian J. Chem., 1968, 6, p. 741.
767. Тананайко M. M., ЛозовикА. C.— Журнал аналит. химии, 1969, 24, с. 844.
768. Живописцев В. ППятосин Л. П.— Уч. зап. Перм. ун-та, 1968, №
с. 168.
769. Гордеева М. И., Козловская И. М.— В кн.: Применение органических реактиво. »
аналит. химии. Л., Изд-во ЛГУ, 1969, с. 64.
770. Ильина Л. И.— В кн.: Физико-хим. методы анализа металлов и сплавов Ч
1969, с. 120.
771. Adamiec I.— Chem. analit. (PRL), 1969, 14, p. 115.
772. AyresG. H., Baird S. S.~ Taianta, 1960, 7, p. 237.
773. Tettamanzi A.— Rassegna chim., 1969, 21, p. 27, 285.
774. Rao A. L. J., Puri B. K-— Z. anal. Chem., 1969, 247, S. 18.
775. Бабенко А. С., Толмачев В. И.— Укр. хим. журнал, 1962, 28, с. 659.
776. Еремин Ю. Г., КаточкинаВ. С.— Журнал аналит. химии, 1970, 25, с. 1641.
777. Каренман И. М., Туманова А. А., Янаева В. Я •— Труды по химии и хим. техноло-
гии. Горький, 1960, с. 86.
778. Михеева Л. И., Тарасова А. И., Ауэрман Л. И., Румер И. А.— Журнал аналит
химии, 1973, 28, с. 1007.
779. Еремин Ю. Г., Каточкина В. С., Комиссарова Л. И.— Журнал аналит химии
1970, 25, с. 1102.
780. Rozycki С.— Chem. analit. (PRL), 1969, 14, р. 459, 755.
781. Minczewski J., Rozycki C.— Z. anal. Chem., 1968, 239, S. 158.
782. Graffmann G., Jackweth E.— Z. anal. Chem., 1968, 244, S. 391.
783. Бабко A. K-, Данилова В. И., Каплан М. Л.— Укр. хим. журн., 1966, 32, с. 1009.
784. Fischer W., Chalybaeus W.— Z. anorg. Chem., 1947, 255, p. 79.
785. Malatesta L.— Gold Bull., 1975, 8, p. 48; РЖхим, 1975, 19Б, c. 405. n
786. Винаров И. В., Ковалева Е. И., Бык Г. И.— Укр. хим. журн., 1968, 34, с. 62.
787. Винаров И. В., Герценштейн Э. Ш., Бык Г. И., Ковалева Е. И.— Укр. хим. журн.,
1968, 34, с. 153.
788. Винаров И В., Русин И. Ф., Ильченко Л. И.— Журнал прикл. химии, 1969, 42,
с. 1664.
789. Hoshio Y.— Japan Analyst, 1962, 11, р. 1032, 1050.
790. Ross J. R., Schack С. H.— U. S. Bureau of Mines. Report of Invest., 1965, No 6580.
791. Yoshida H.— Japan Analyst, 1963, 12, p. 169.
792. Голуб A. M., Фам Ван Ча, Самойленко В. М.— Журнал неорган. химии, 1971,
16, с. 534.
793. Голуб А. М., Сыч А. М.—- Журнал прикл. химии, 1966, 39, с. 2400.
794. ГолубА. М., Дабека Р. В., КовальВ. Т.— В кн.: Комплексообразование и экстрак
ция актиноидов и лантаноидов, 1974, с. 97.
795. ГусеваЛ. И., Тихомирова Г. С.— Радиохимия, 1968, 10, с. 246.
796. Голуб А. М., А Ван Лонг.— Журнал прикл. химии, 1972, 45, с. 1776.
797. Bullpitt М., KUching 117.— J. Organometal. Chem., 1972, 34, р. 321.
798. Wizemann T., Miiller H., Seybold D., Dehnike Ц.— J- Organometal. Chem., 1969,
20, p. 211.
799. Dehnike К.— J. Organometal., 1967, 9, p. 11.
800. Forster J. E., Vargas M., Miiller U.— J. Organometal. Chem., 1973, 59, p. 97
801. LiottaD., Engel E.— Can. J. Chem., 1975, 53, p. 907.
802. Zemlicka JTrnka J.— Krist. und Techn., 1975, 10, S. 715.
803. KarayannisN. M., Mikulski С. M., Pytlewski L. L., Labes M. M.— J. Inorg. Nucl.
Chem., 1972, 34, p. 3139.
804. Савицкий В. H., Скопенко Г. Ф.— Укр. хим. журн., 1975, 41, с. 1038.
805. Желнов А. А., Гаврилин Г. Ф.— Журнал общей химии, 1972, 42, с. 1893.
806. Цивадзе А. ЮХаритоновЮ. ЯЦинцадзе Г. В. и др.— Журнал неорган. химии,
1975, 20, с. 725.
807. Гогоришвили П. В., Нагребашвили С. Ш., Швелашвили А. Е., Мачхошвили Р. И.—
Журнал неорган. химии, 1975, 20, с. 1526.
808. Schilt A. A., Fritsch К-— J - Inorg. Nucl. Chem., 1966, 28, р. 2677.
809. Colombini J., Preti C.— J. Inorg. Nucl. Chem., 1975, 37, p. 1159.
810. Ahuja I. S., Singh R.— J. Coord. Chem., 1975, 4, p. 181.
811. Diaz A., Massacesi M., Ponticelli J., Paschina J.— J. Inorg. Nucl. Chem., 1975,
37, p. 2469.
812. CollinsL. W., Gibson E. K-, WendlandtW. W.— Termochim. acta, 1975, 11, p. 177.
813. Ahuja I. S., Rao K- S.— Indian J. Chem., 1975, 13, p. 413.
814. Singh P. P., Khan S. A., Pal R. B.— Inorg. Nucl. Chem. Lett., 1975, 11, p. 807.
252
815. Цивадзе А. Ю., Цинцадзе Г. В., Гонгадзе Н. П., Харитонов Ю. Я — Координа-
ционная химия, 1975, 1, с. 1212, 1221, 1385.
816. Харитонов Ю. Я-, Мачхошвили Р. И., Гогоришвили П. В., Нагебашвили С. Ш.—
Координационная химия, 1975, 1, с. 2630.
817. Савицкий В. Н., Скопенко В. В., Жумабаев А. Ж-, Трачевский В. В.— Укр. хим.
журн., 1975, 41, с. 903.
818. Dutta R. L., Banerjee S. Р., De Dhrubananda.— J. Indian Chem. Soc., 1975, 52,
p. 582.
819. Gheorghiu C., Marulescu C.— Rev. room, chim., 1975, 20, p. 345.
820. ТевзадзеЛ. А., Пирцхалава H. И., Гарновский А. Д.— Сообщ. АН ГрузССР, 1975,
77, с. 81.
821. Hajek В., Hradilova J., Makarina G.— VSCHT Praze, 1974, 18, S. 107.
822. Madeja K-, Wilke W. Schmidt S.— Z. anorg. allg. Chem., 1966, 346, S. 306.
823. Пирцхалава H. ИГиоргадзе К. П.— Журнал неорган. химии, 1975, 20, с. 3252.
824. GraddonD. Р., Капа В. А.— J. Organometal. Chem., 1976, 105, р. 51.
825. Singh Р. Р., Sharma S. В.— J. Indian Chem. Soc., 1975, 52, р. 617.
826. Duggan D. M., Hendricson D. N.— Inorg. Chem., 1974, 13, p. 2929.
827. Albertin G., Bordignon E., Orio A. A.— Inorg. Chem., 1975, 14, p. 1411.
828. Еремин IO. Г., Бондаренко Г. И.— Журнал неорган. химии, 1976, 21, с. 387.
829. Николаенко Л. НТолмачева Н. С., Коткова Г. В. и др.— Координационная
химия, 1975, 1, с. 849.
830. BhattacharyyaR. G., BeraD. С.— J. Indian Chem. Soc., 1975, 52, p. 373.
831. Sala-Paia J., Guerchais E. J.— Z. anorg. allg. Chem., 1975, 412, S. 281.
832. Скопенко В. В., Голуб А. М.— Укр. хим. журн., 1976, 42, с. 196.
833. Tahmina В., Djordjevic С.— Croat, chem. acta, 1975, 47, р. 79; РЖХим, 1976, 9Г
150.
834. Гусейнов И. К-, Азимов Я. А., Албендов А. А., Рашидов К. Дж.— Азерб. хим.
журн., 1975, № 2, с. 100.
835. Ganescu Г, Varhelyi Cs. OprescuD.— J. Inorg. Nucl. Chem., 1975, 37, p. 324.
836. LodzinskaA., Zawadski H., Kita P.— Rocz. chem., 1975, 49, p. 1239.
837. Коттон Ф., Уилкинсон Дж. Современная неорганическая химия. М., Мир, 1969,
ч. 3, с. 232.
838. Трачевский В. В., Голуб А. М., Улько Н. В.— Журнал неорган. химии, 1975, 20,
с. 2994.
839. Еремин Ю. Г., Колпакова Е. Ф.— Журнал неорган. химии, 1975, 20, с. 1587.
840. Еремин Ю. Г., Колпакова Е. Ф., Родионова Т. В.— Журнал неорган. химии, 1975,
20, с. 3287.
841. Rastogi М. К-, Multani R. К-— J. Inorg. Nucl. Chem., 1975, 37, р. 1995.
842. Баличева Т. Г., Пологих И. В., Селивестрова И. В.— Журнал неорган. химии,
1975, 20, с. 978.
843. Пирцхалава Н. И., Харитонов Ю. Я , Брегвадзе М. В.— Журнал неорган. химии,
1975, 20, с. 1852.
844. Цивадзе А. Ю., Цинцадзе Г. В., П етриашвили Ж. Д., ХаритоновЮ. Я-— Коорди-
национная химия, 1975, 1, с. 1472.
845. Цивадзе А. Ю., Цинцадзе Г. В., Гогнадзе Н. ПХаритоновЮ. Я— Координаци-
онная химия, 1975, 1, с. 1084.
846. Ghosh S. Р., Mishra А.— J. Indian Chem. Soc., 1975, 52, р. 791.
847. Нариманидзе А. П., Цивадзе А. Ю., Харитонов Ю. Я-. Джашнашвили Т. К.—
Координационная химия, 1975, 1, с. 936.
848. Пирцхалава Н. И., ХаритоновЮ. Я, Брегвадзе М. В.— Координационная химия,
1975, 1, с. 1672.
849. Collin R. J., Larkworthy L. F.— J. Inorg. Nucl. Chem., 1975, 37, p. 334.
850. StultsB. R., Marianelli R. S., Day V. W.— Inorg. Chem., 1975, 14, p. 722.
851. Singh P. P., Seth J. N.— Inorg. chim. acta, 1975, 15, p. 227.
852. Massacesi M., Ponticelli G., Preti C.— J. Inorg. Nucl. Chem., 1975, 37, p. 1641.
853. Devote M., Ponticelli G., Preti C.— J. Inorg. Nucl. Chem., 1975, 37, p 1635.
854. Dash К- C., Pujari P.— J. Inorg. Nucl. Chem., 1975, 37, p. 2061.
855. Желнов А. А., Гаврилин Г. Ф.— Журнал общей химии, 1966, 36, с. 2073.
856. Shigemat Т., Sasaki Y.— Bull. Inst. Chem. Res. Kyoto Univ., 1974, 52, p. 658.
857. Benettin M., Sindellari L., Vidali M., Ros R — J. Inorg. Nucl. Chem., 1975, 37,
p. 2067.
858. Coronas J. M., Rosseli 0., Sales J.— J. Organometal. Chem., 1975, 97, p. 473.
859. Coronas J. M., Sales J.— J. Organometal. Chem., 1975, 94, p. 107.
860. BressanM., Rigo P.— Inorg. Chem., 1975, 14, p. 38.
861. Levason W., McAuliffe C., Watson D.— J. Coorg. Chem., 1975, 4, p. 173.
862. Baracco L., Halfpenny T. M., McAuliffe C. A.— J. Chem. Soc. Dalton, 1975, p. 1945.
253
863. Gerloch М., McMeeking R., White A. M.— J. Chem. Soc. Dalton, 1975, p. 2452.
864. Hennig H., BenedixR., Hempel K-, Reinhold J.— Z. anorg. allg. Chem., 1975, 41_
S. 141.
865. Nappier E. T., Feltham R. D., Enemark J. H. a. o.— Inorg. Chem., 1975, 14, p. S.-
866. Riley D. P., Merell P. H., Stone J. A., Busch D. H.-— Inorg. Chem., 1975. К
p. 490.
867. Curtis N. F., Milestone T. N.— Austral . J. Chem., 1975, 28, p. 275.
868. ArmstrongL. G., Lindoy L. F.— Inorg. Chem., 1975, 14, p. 1322.
’ 869. JonaE., Sramko T., Gazo J.— J. Therm. Anal., 1975, 7, p. 551.
870. Савицкий В. H., Авраменко Л. Ф., Скопенко В. В., Починок В. Д.— Укр. хим
журн., 1975, 41, с. 1238.
871. Саруханов М. А., Вальдман С. С., Парпиев Н. А.— Журнал неорган. химия
1975, 20, с. 2396.
872. Szabo-Akos Zs., Simon J., Beck M. T.— J. Therm. Anal., 1975, 8, p. 155.
873. Dabrowiak J. C., Busch D. H.— Inorg. Chem., 1975, 14, p. 1881.
874. Ng F. T. T., Henry P. M.— Can. J. Chem., 1975, 53, p. 3319.
875. Паладе Д. M., ПоповЮ. ЛЧудакова Г. В.— Журнал неорган. химии, 1975, 20.
с. 2754.
876. Шафранский В. Н., Харитонов Ю. Я., Солкан Т. Н., Бубуруз Д. Д.— Координа-
ционная химия, 1975, 1, с. 628.
877. Шафранский В. НФусу И. Л., Дранка И. В. и др.— Координационная химик
1975, 1, с. 1031.
878. Шафранский В. Н., Бубуруз Д. Д.— Журнал физ. химии, 1975, 49, с. 194'
879. Шафранский В. Н., Бубуруз Д. Д., Солкан Т. Н.— Журнал неорган. химии, 19“'
20, с. 1991.
880. Varhelyi С., Finta Z., Kiss S.— Acta chim. Acad. sci. hung., 1975, 106, p. 659.
881. Hay R. W., Lawrence G. A.— J. Chem. Soc. Dalton, 1975, p. 1466.
882. Bosnich B., Harrowfield J., Mac B.— Inorg. Chem., 1975, 14, p. 847,. 861.
883. Drew M. G. B., Hamid 0. A., McIlroy D. A. P.— J. Chem. Soc. Dalton, 1975,
p. 2507.
884. Nelson S., Busch D. H.— Inorg. Chem., 1970, 8, p. 1859.
885. Троицкая А. Д., Орлова И. A.— Журнал общей химии, 1975, 45, р. 951.
886. Winkaus G., Ziegler Р.— Z. anorg. allg. Chem., 1967, 350, S. 51.
887. Dixon K- R., Moss К- C., Smith M. A. R.— J. Chem. Soc. Dalton, 1975, p. 990.
888. Beck W., Schorpp K-— Chem. Ber., 1975, 108, p. 3317.
889. Троицкая А. Д., Сентемов В. В., Антропова Е. И.— Журнал неорган. химик.
1975, 20, с. 3131.
890. Levason W., McAuliffe С. А.— Inorg. chim. acta, 1976, 16, р. 167.
891. Jenkins J. M.„ Shaw В. L.— J. Chem. Soc., 1966, A, p. 1407.
892. Brookes P. R., Shaw B. L.— J. Chem. Soc., 1967, A, p. 1079.
893. Kubota M., PhillipsD. A.— J. Am. Chem. Soc., 1975, 97, p. 5637.
894. Victori L., Tomas X., Celorio J.— Afinidad, 1975, 32, N 331, p. 961; РЖХим, 1976,
14B176.
895. Самойленко В. M., Ляшенко В. Н., Полторацкая Т. В.— Журнал неорган. химии,
1975, 21, с. 3274.
896. Самойленко В. М., Ляшенко В. И.— Журнал неорган. химии, 1976, 21, с. 1530.
897. Боков Н. Т., Лобов Б. И., Добычин Д. П.— Журнал неорган. химии, 1975, 20,
с. 180.
898. Рагулин Г. К., Александрова Н. Е.— Журнал неорган. химии, 1975, 20, с. 2144.
899. Cabon J.-Y., L'Her М., Demezet М.— С. г. Acad, sci., 1975, С280, р. 819.
900. AiharaM., Misumi S.— Bull. Chem. Soc. Japan, 1975, 48, p. 684.
901. Smith D. F.— J. Mol. Spectrosc., 1975, 57, p. 447.
902. Burger K-, Liptay Cy., Varhelyi Cs.— Acta chim. Acad. sci. hung., 1975, 83, p. 315,
1974.
903. BhatN. V., SyamalA.— J.Mol. Struct., 1976, 30, p. 161.
904. Melpolder J. B., Burmeister J. L.— Inorg. chim. acta, 1975, 15, p. 91.
905. Norbury A. H., Raghunathan S.— J. Inorg. Nucl. Chem., 1975, 37, p. 2133.
906. Norbury A. H., Shaw P. E., Sinha A., Ishwari P.— J. Chem. Soc. Dalton, 1975,
p. 742.
£07. BalahuraR. J., LewisN. A.— Coord . Chem. Rev., 1976, 20, p. 109.
908. Anderson S. J., Norbury A. H.— Chem. Commun., 1975, p. 48.
909. Anderson S. J., Goodfellow R. J.— Chem. Commun., 1975, p. 443.
910. CrumblissA. L., GausP. L.— Inorg. Chem., 1975, 14, p. 2745.
911. PinnaR., Ponticelli G.— J. Inorg. Nucl. Chem., 1975, 37, p. 1681.
912. Singh P. P., Seth J. N.— Inorg. Nucl. Chem. Lett., 1975, 11, p. 525.
913. Hasek J., Huml K-— Z. Kristallogr., 1975, 141, p. 158.
254
914. Drew M. G. В., Hamid 0. A., Hill W. E. a. o.— Inorg. chim. acta, 1975, 12, p. 125.
915. Drew M. G. B., Hamid 0. A.— Acta Crystallogr., 1975, B31, p. 613.
916. Лазарев П. И., Асланов Л. А., Порай-Кошиц М. А.— Координационная химия,
1975, 1, с. 979.
917. Лазарев П. И., Асланов Л. А., Порай-Кошиц М. А.— Координационная химия,
1975, I, с. 706.
918. ЛазаревП. И., АслановЛ. А., Порай-Кошиц М. А., Ионов В. М.— Координацион-
ная химия, 1975, 1, с. 710.
919. Hauge S., Henriksen Р. А. — Acta chem. scand., 1975, А29, р. 778.
920. Hauge S., Vicane 0.— Acta chem. scand., 1975, A29, p. 755.
921. Scheldrick G. M., Taylor R.— J. Organometal. Chem., 1975, 87, p. 145.
922. Bombieri G., Moseley P. T., Brown D.— J. Chem. Soc. Dalton, 1975, p. 1520.
923. Glowiak T., SabatM., SabatH., Rudolf M.— Chem. Commun., 1975, p. 712.
924. Andreetti G. D., Sgarabotto P.— Cryst. Struc. Commun., 1975, 1, p. 55.
925. Fronczek F. R., Schaffer W. P.— Inorg. Chem., 1975, 14, p. 2066.
926. Andreetti G. D., Bocelli G., Sgarabotto P.— Cryst. Struct. Commun., 1975, 1, p. 51.
927. Dunaj-Jurco M., Garaj J., Sirota A.— Collect. Czech. Chem. Commun., 1974, 39,
p. 236.
928. Domiano P., Musatti A., Nardelli M. a. o.— J. Chem. Soc. Dalton, 1975, p. 2357.
929. Cannas M., Carta G., Cristini A., Marongiu G.— J. Chem. Soc. Dalton, 1974,
p. 1278.
930. Laskowski E. J., DugganD. M., HendricksonD. N.— Inorg. Chem., 1975, 14, p. 2449.
931. Kundell F. A., Hazell K- G., Rasmussen S. E.— Acta Crystallogr., 1975, B31, p. 2879.
932. Hubert J., Beauchamp A. L., Rivest R.— Can. J. Chem., 1975, 53, p. 3383.
933. Palenik G. J., Mathew M., Steffen W. L., Beran G.— J. Am. Chem. Soc., 1975, 97,
p. 1059.
934. Villa A. C., Manfredotti A. G., Giacomelli A. a. o.— Inorg. Chem., 1975, 14, p. 1654.
935. Sakhri A., Beauchamp A. L.— Acta Crystallogr., 1975, B31, p. 409.
936. Caira M. R., Nassimbeni L. R.— Acta Crystallogr., 1975, B31, p. 581.
937. Chow Y. M., BrittonD.— Acta Crystallogr., 1975, B31, p. 1922.
938. Rozycki C.— Pr. nauk. Warsz., 1975, N 15, p. 3.
939. Bohland H., Harke E.— Z. anorg. allg. Chem., 1975, 413, S. 102.
940. Goher M. A. S.— Can. J. Chem., 1975, 53, p. 2657.
941. Norbury A. H.— Advances Inorg. Chem. and Radiochem., 1975, 17, p. 231.
942. Gawalek G. Einschlussverbindungen. Additionverbindungen. Clatrate. Berlin, VEB
Deutscher Verlag der Wissenshaften, 1969.
943. Bohland H., Schneider F. M.— Z. anorg. allg. Chem., 1972, 390, S. 53.
944. Bohland HTiede E.— J. Less-Common Metals, 1967, 13, p. 244.
6. СЕЛЕНОЦИАНАТНЫЕ СОЕДИНЕНИЯ
6.1. ДИСЕЛЕНОЦИАН И ЕГО СВОЙСТВА
Диселеноциан (SeCN)2 был впервые синтезирован при действии на селено-
цианат серебра эфирным раствором иода [1]
2AgSeCN + I2 -> 2AgI + (SeCN)2. (6 ])
Позже диселеноциан был получен действием KNCSe на тетраацетат свинцг
[26], при окислении SeCN~ пентафторидом иода [3], анодным окисление^
SeCN~ [2,4].
Диселеноциан проставляет собой желтый порошок, растворимый в бен-
золе, хлороформе, тетрахлориде углерода. В твердом состоянии хорошо
сохраняется в вакууме. В ацетонитриле (SeCN)2 диспропорционирует на
Se (SeCN)2 и Se (CN)2 [4].
Селеноциан может образовывать соединения с металлами и неметалла-
ми. Например, упомянутое выше соединение Se (SeCN)2 представляет собой
желтый порошок, который плавится с разложением при 133° С [3, 5, 6];
оно содержит неразветвленные цепи с тремя атомами селена ов середине и
CN-группами на концах, расстояние Se—Se составляет 2,33 A; Se—С 1,83
А; С—N 1,05 А. Валентный угол SeSeSe равен ЮГ. Определены также па-
раметры кристаллической решетки этого соединения: а = 10,07; b = 13,35;
с = 4,48 А [5, 61. При действии Se (SeCN)2 на водные растворы цианидов
щелочных металлов получены соединения MSe (SeCN)3 • 0,5Н2О (М = К,
Rb, Cs) [8]. Для соединения KSe (SeCN)3 • 0,5 Н2О определены параметры
кристаллической решетки: а = 9,170; b = 13,373: с = 9,057 А [9].
В эфирном растворе (SeCN)2 реагирует с ЫВН4 с образованием не-
устойчивых соединений типа LiBH4_n • (SeCN)n (n = 1 или 2) [7].
При действии эфирным раствором диселеноциана на спиртовый раствор
селеноцианата цезия был получен Cs (SeCN)3, представляющий собой крас-
но-коричневые моноклинные призмы, довольно устойчивые на воздухе
[10]. Cs (SeCN)3 можно получить также окислением водного раствора CsNCSe
бензольным раствором брома [81. Структурными исследованиями показано,
htoCs (SeCN)3 относится к пространственной группе С2/с; Z = 4; ZlSeSeSe =
=’178,3°; параметры решетки: а = 7,969; оЬ = 21,156; с = 5,593 А; р =
= 98,84°. Расстояние Se—Se равно 2,650 А [11].
При потенциометрическом титровании KNCSe раствором иода образуют-
ся KSeCNI2, К (SeCN)2I, К (SeCN)3 [10]. Последнее соединение выделено
в виде кристаллогидрата К (SeCN)3 0,5 Н2О при окислении концентри-
рованного водного раствора KNCSe бензольным раствором брома [12]. Ана-
логичным путем было получено соединение Rb (SeCN)3 0,5 Н2О [8].
Детально изучена структура К (SeCN)3 • 0,5 Н2О [12, 131. Определены
256
параметры моноклинной решетки: а = 16,988; b = 4,4-3; с = 23,590 А;
Р = 95,7°; Z = 8; пространственная группа F2. Группы Se—Se—Se линейны,
линейны также все три SeCN-группы. Атом калия окружен пятью атомами
азота и атомом кислорода, образующими неправильный шестивершинник.
6.2. ХИМИКО-АНАЛИТИЧЕСКИЕ ОСОБЕННОСТИ SeCN-ИОНА
Влияние различных ионов металлов на устойчивость SeCN-ионов в вод-
ных растворах изучено Лодзинской [39]. При этом показано, что введение
ионов Na+, К+, Mg2+, Са2+, Ва2+ не влияет на pH разложения селеноциа-
нат-ионов. В присутствии этих катионов SeCN~ начинает разлагаться от
введения соляной кислоты при pH 5,5—4,9. В присутствии же катионов
Cd2+, Zn2+, Pb2+, Hg2+ разложение наступает несколько раньше — при
pH >• 6. Катионы Ni2+, Со2+, Cu'+, Fe2+ вызывают разложение селеноциа-
нат-иона еще скорее, а именно при pH > 7.
Количественный анализ растворов селеноцианатов можно осуществить
в виде обычного аргентометрического титрования с дифенил карбазоном [14],
а также методом потенциометрического титрования соответствующих раство-
ров нитратом серебра [15] или перхлоратом ртути [16].
Недавно предложен метод амперометрического титрования смеси SeCN~-
и СЬП-ионов [54], а также SeCN-- и SCN~-ионов [55] раствором AgNO3.
При этом SeCN- с концентрациями 0,01—0,00001 г-ион/л [54] можно опре-
делять в присутствии цианид-ионов на фоне KNO3. При совместном нахож-
дении SCN-- и $еС№-ионов последние определяют на фоне 0,1 М NH3 и
0,1 М KNO3 [55]. В качестве индикаторного электрода применяют платино-
вый вращающийся электрод, электрод сравнения — Hg | Hgl2.
Для анализа малых количеств селеноцианатов хорошие результаты дает
метод аргентометрического определения селена [17]. Концентрацию селено-
цианатов, растворимых и даже слаборастворимых в воде, удобно устанав-
ливать по селену. При этом селеноцианаты разлагают соляной кислотой
SeCN~ + Н+ Se + HCN. (6.2)
Выделившийся селен определяют весовым или колориметрическим ме-
тодом [18]. Чувствительность последнего метода составляет 5 мкг SeCN~ в
1 мл раствора [18].
В последние годы разработаны высокочувствительные методы определе-
ния селена с применением органических реактивов (см., например, [19—
22]). Этими реактивами можно пользоваться и при анализе селеноцианатов.
6.3. СЕЛЕНОЦИАНАТНАЯ КИСЛОТА И ЕЕ СОЛИ
Селеноцианатная кислота получена Куксом при действии сероводорода
на суспензированный в воде селеноцианат свинца [23]
Pb (SeCN)2 + H2S -> PbS + 2HNCSe. (6.3)
Селеноцианатная кислота известна лишь в растворе; эти растворы отно-
сительно неустойчивы, разлагаются с выделением селена. По силе HNCSe
близка к HNCS. Она растворяет железо и цинк с выделением водорода, вы-
тесняет угольную кислоту из ее солей. Определена эквивалентная электро-
9 2473
257
проводность водных растворов HNCSe при 20° С, которая оказалась близь-:
к таковой для HNCS [24] (табл. 6.1).
Раньше других селеноцианатов был синтезирован KNCSe при сплавлени-
селена с желтой кровяной солью [251. Белые кристаллы KNCSe получаются
при выщелачивании сплава водой. Несколько чище продукт, если вести вы-
щелачивание абсолютным спиртом [23].
Позднее был предложен метод получения KNCSe посредством растворе-
ния селена в концентрированном водном растворе KCN [26, 271 или же его
сплавлением с цианидом калия [28] с последующим выщелачиванием водой.
Исследованию влияния избытка селена, концентрации KCN, температуры на
цианида калия посвящена работа
[29]. Реакция сплавления селена
с цианидом калия может быть
использована для разделения
селена и серы. При сплавлении
смеси Se и S с цианидом калия
образуются KNCSe и KNCS. Ес-
ли сплав выщелочить водой и
обработать соляной кислотой,
выделяется селен, а в растворе
остается сера в виде тиоцианат-
иона [33].
растворение селена в водных растворах
Таблица 6.1. Эквивалентная
электропроводность HNCSe и HNCS
при разных разбавлениях
Разбавление, л/моль 128 256 512 1024 2048
HNCSe, Ом"1 • см2 365 374 380 386 386
HNCS, Ом-1 - см2 392 396 403 405 405
Очень чистый KNCSe был получен при кипячении суспензии селена и циа-
нида калия в абсолютном спирте [301. Однако вследствие взаимодействия се-
леноцианат-иона со спиртами и образования летучих дурнопахнущих алкил-
селенидов последний метод не получил широкого распространения. Поэтому
для синтеза и выщелачивания удобнее пользоваться ацетоном [31]. Вполне
чистый препарат KNCSe был получен и при взаимодействии селена с циани-
дом калия в жидком аммиаке [32].
В литературе имеются сведения о получении селеноцианата натрия ки-
пячением красного селена и цианида натрия в абсолютном спирте [30].
С хорошим выходом образуется селеноцианат натрия при сплавлении селена
с цианидом натрия [31]. Если выщелачивание NaNCSe вести ацетоном, пос-
ле отгонки растворителя выделяются иглообразные прозрачные кристаллы
NaNCSe - 2Act. Сольватированный ацетон легко удаляется при нагревании
соли до 60—70° С.
Растворение селена в водных растворах NaCN протекает по реакции пер-
вого порядка с константой скорости при 40° С 1,73 • 10~4 с-1 (для аморфного
селена) и 2,03 • 10-4 с-1 (для кристаллического селена). Энергия активации
реакций растворения аморфного и кристаллического селена соответственно
равна 54,47 и 15,83 кДж [34].
Для селеноцианатов натрия и калия определена плотность, которая рав-
на соответственно 2,490 и 2,347 г/см3 [30]. KNCSe при нагревании на воздухе
плавится с разложением около 100° С. В воде растворяется очень хорошо,
реакция раствора щелочная.
Для водных растворов селеноцианата калия изучена также электропро-
водность [24]. По данным Биркенбаха [241, эквивалентная электропровод-
ность водного раствора KNCSe при бесконечном разбавлении равна 119,5
(18° С) и 138,2 (25° С). Напряжение разложения KNCSe равно 1,10 В
[10, 401.
Кристаллическая структура KNCSe определена сравнительно недавно
[41]. Она содержит линейные SeCN-ионы (^/ SeCN = 178 ± 2,5°). Расстоя-
ние С—N, равное 1,117 ± 0,046 А, близко к длине тройной связи feN,
а расстояние С—-Se (1,825 ± 0,025 А) значительно короче обычной ординар-
ной связи.
Вокруг каждого атома селена расположены четыре иона калия, образуя
искаженную пирамиду соселеном в вершине. Расстояния Se ... К колеблются
в пределах от 3,4 до 4,0 А. Каждый атом азота имеет около себя три иона ка-
лия на удалении 2,6—3,1 А (рис. 6.1).
В ИК-спектре поглощения линейный SeCN-ион имеет три основные коле-
бательные частоты: две частоты линейных валентных колебаний и одну
(дважды вырожденную) частоту деформационных колебаний. На основании
вычисления частот и форм колебаний селеноцианат-иона показано [42, 43],
что в валентных колебаниях де-
формируются обе связи CN и
CSe, однако в колебании с выс-
шей частотой деформируется
преимущественно связь CN, а в
колебаниях с низшей частотой —
связь CSe. В соответствии с этим
большую частоту валентных ко-
лебаний мы будем в дальнейшем
обозначать через v (CN), а мень-
шую — через v(CSe). Частоту
деформационных колебаний, при
которых изменяется угол SeCN,
будем обозначать через 6 (SeCN).
Морган [44] изучил ИК-спек-
тры поглощения кристаллическо-
го KNCSe при 77 и 300 К,
спектр комбинационного рассеяния
Рис. 6.1. Структура KNCSe [41]
водного раствора KNCSe при 300 К
и провел следующее отнесение найденных
в спектре образца при 77 К v (CN) 2072; v(C13N)
основных частот (см !):
2027; v(CSe) 559; 6 (SeCN)
426 и 417; в спектре образца при 300 К v(CN) 2070; v(C13N) 2026; v(CSe)
558; 6 (SeCN) 424 и 416; в спектре комбинационного рассеяния v (CN) 2075
и v (CSe) 558. К составным частотам и обертонам Морган отнес (в ИК-погло-
щении): v (CN) + v (CSe) 2632; v (CN) + 6 (SeCN) 2491 и 2484; 26 (SeCN)
832; 834; 847 и 849 см-1. Расщепление вырожденной частоты, составных ча-
стот и обертона объяснено влиянием кристаллического состояния. Из най-
денных частот Морган вычислил силовые константы в предположении про-
стейшего валентно-силового поля и нашел: Kcn = 15,1; Kcse = 3,88 Н/см,
где Kcn и Kcse — силовые константы CN и CSe. Несколько позже ИК-
спектры соли KNCSe, включающей изотопы С12 и С13, изучены в работе
[121].
В ИК-спектре кристаллического NaNCSe • пН2О найдены частоты (см *):
414 6 (SeCN); 559v(CSe); 822 26 (SeCN); 1623 6 (Н2О); 2079 v(CN); 3100 -
3700 v(OH) [45].
Хотя о CsNCSe упоминалось еще в работе [10], синтез и свойства этого сое-
динения описаны лишь недавно [8]. Селеноцианат цезия получен обменной
реакцией между CsCl и KNCSe в ацетоне с последующей обработкой твердо-
го осадка (после отгонки ацетона) метанолом [8]. Ромбические кристаллы
этой соли имеют плотность 3,53 г/см3 и плавятся при 228° С; параметры ре-
шетки: а = 8,17; b = 6,42; с = 8,55 А [8]
Аналогично селеноцианату цезия был получен RbNCSe [8,35]. В ацето-
нитрильных растворах селеноцианатов натрия, калия, рубидия или цезия
9
259
в присутствии LiC104 установлено образование ассоциатов LiNCSe
Li+ + SeCN” <=> LiNCSe. (6 л
Константа равновесия этой реакции равна (2,5 ± 0,5) • 102 [35]. В твердев
состоянии селеноцианат лития до сих пор не получен.
В ряду простейших селеноцианатов целесообразно рассмотреть также
соль аммония, которую впервые получил Крукс нейтрализацией селеноциа-
натной кислоты аммиаком [23]. Позднее селеноцианат аммония получили
Камерон и Дэви обменной реакцией между KNCSe и (NH4)2SO4 в абсолютно-
спирте [36]. Авторы отмечают крайне малую устойчивость этой соли.
Значительно более устойчивыми являются селеноцианаты тетраалкил-
аммония [R4N] NCSe (R = СН3, С2Н5, С4Н9), которые можно получить из
метанольных или этанольных растворов обменной реакцией между [R4N] Cl
и KNCSe. Соли [R4N] NCSe лучше, чем KNCSe или NaNCSe, растворимы в
органических растворителях, поэтому их удобней использовать при синтезе
координационных соединений [37].
Получен также селеноцианат гидрозония N2H4HNCSe [38], для которого
характерна реакция изомеризации
N2H4HNCSe H2N—CSe—NH—NH2. (6.5)
Равновесие этой реакции быстро достигается в присутствии ацетона и отве-
чает 35% превращения селеноцианата гидрозония в селеносемикарбазон.
О селеноцианатах магния и щелочноземельных металлов упоминал еще
Крукс [23], не приводя, однако, никаких сведений об их составе и свойствах.
Селеноцианаты рассматриваемых металлов получены из смеси ацетон_
диоксан (1 : 1) [46]. Для их получения смешивали ацетоновые растворы
Mg (СЮ4)2 или М (С1О4)2 - 2 Diox (М = Са, Sr, Ва) и KNCSe, взятые в соот-
ношении 1 : 2. После отделения выпавшего КС1О4 к фильтрату приливали
диоксан и выделяли прозрачные бесцветные кристаллы М (NCSe)2-2Diox -
• (Sr, Ва); MgNCSe - 4 Diox и Са (NCSe)2-3 Diox получены в виде порошков
белого цвета. Эти соединения хорошо растворимы в воде, спирте, ацетоне,
на воздухе со временем разлагаются с выделением селена.
Изучение ИК-спектров рассматриваемых соединений показало, что
Mg (NCSe)2-4 Diox содержит мостиковые SeCN-группы. В солях Ва (NCSe)2-
2 Diox и Sr (NCSe)2 - 2 Diox SeCN-группы имеют ионную приро-
ду. В Са (NCSe)2 • 3 Diox предполагаются контакты Са NCSe [46]. Не-
давно выделены селеноцианатные соединения бериллия: [BeCl (DMFA)3] -
• (SeCN) • 3,5 DMFA; [Be (NCSe) (DMFA)31 SeCN; К [Be (NCSe)3 (DMFA)l-
• 4 DMFA и K2 [Be (NCSe)J 7 DMFA [229].
6.4. СЕЛЕНОЦИАНАТЫ p- И d10 -МЕТАЛЛОВ
Раньше других были получены селеноцианаты металлов, слабораствори-
мые в воде. Метод их получения сводится в основном к действию раствора
селеноцианата калия на соответствующие соли. Так, Круксом [23] был полу-
чен селеноцианат серебра — белый, труднорастворимый в воде продукт.
Позднее AgSeCN выделили и другие авторы [26, 30], которые нашли, что
плотность AgSeCN равна 4,602 г/см3 [30], а произведение растворимости со-
ставляет 4,0 • 10“16 при 18—20° С [40].
Из ацетонового раствора, содержащего KCu (SeCN) Cl, выделяется при
добавлении воды белый труднорастворимый осадок CuSeCN. Произведение
растворимости CuSeCN в воде при 20° С равно 1,8 • 10 [471. Селеноциа-
260
нат меди (I) получен также из водного раствора при действии на сульфат меди
селеноцианатом калия в присутствии Na2S2O3 [194]. Если к охлажденному
раствору KNCSe прибавлять раствор нитрата меди, образуется плохо
растворимая в воде соль Cu (SeCN)2 [194].
Из водного раствора обменной реакцией между нитратом кадмия и
KNCSe получен Cd (SeCN)2 [48]. Произведение растворимости Cd (SeCN)2
в воде при 20° С равно 1,93 • 10~5 [48].
Селеноцианат ртути в виде белого аморфного осадка синтезирован при
действии KNCSe на раствор ацетата ртути [36], а также из HgCl2 и KNCSe
Таблица 6.2. Колебательные частоты SeCN-групп, см 1 [45]
Соединения v (CN) v (CSe) ст (SeCN) 2 о (SeCN) Соединения V (CN) v (CSe) z о Ф co О 26 (SeCN)
AgSeCN 2147 578 ^400 764 791 Pb (SeCN)2 2100 2142 542 582 ^400 789
Cd (SeCN)2 2109 2150 588 ^400 790 TISeCN 2050 (2086) 548 408 798 814
Hg (SeCN)2 2145 537 ^400 777 Hg (SeCN)Cl (2125) 2144 537 ^400 —
[49] . Из водных растворов получен также Hg (SeCN) Cl [49]. При взаимодей-
ствии селеноцианата калия с Hg2 (NO3)2 получен весьма неустойчивый
Hg2 (SeCN)2 [36].
Из простых селеноцианатов элементов третьей группы периодической
системы известен лишь желтоватый селеноцианат таллия (I), который был
получен при действии селеноцианата калия на водно-спиртовый раствор
ацетата таллия (I) [30].
Плотность TISeCN равна 5,462 г/см3 [30], а произведение растворимости
при 20° С в воде — 6,4 • 10—5, в водно-ацетоновом растворе (50%) — 3,6 X
X 10~5, в диметилформамиде — 1,3 • 10“3 [50].
Подобно соли таллия был получен селеноцианат свинца [30, 51] в виде
мелкокристаллического бледно-желтого порошка с плотностью 4,694 г/см3
[30]. При хранении на солнечном свету он краснеет. При нагревании раз-
лагается несколько выше 100° С, не достигая температуры плавления.
Произведение растворимости в воде при 20° С равно 3,34 • 10“ [51]. Синте-
зирована также соль РЬ (ОН) SeCN [52].
Как видно из табл. 6.2, в ИК-спектрах селеноцианата серебра [45, 53],
кадмия, ртути, свинца [45] компонента v (CN) расположена около 2145 см--1,
что указывает на мостиковый характер SeCN-групп. Очевидно, в AgSeCN,
Hg (SeCN) Cl и Hg (SeCN)2 присутствуют только мостиковые SeCN-группы.
Наличие в ИК-спектрах Pb (SeCN)2, Cd (SeCN)2 полос v (CN) около 2110
и 2145 см-1 вызвано, вероятно, присутствием как мостиковых, так и кон-
цевых SeCN-групп. Соединение TISeCN содержит SeCN-ионы.
6.5. ИЗУЧЕНИЕ СЕЛЕНОЦИАНАТНЫХ КОМПЛЕКСОВ
В РАСТВОРЕ
6.5.1. Комплексообразование в водных
и смешанных растворах
Первая работа по изучению комплексных селеноцианатов в водных рас-
творах относится к 1956 г. [56]. В. Ф. Торопова [56] потенциометрически об-
наружила комплексы Ag (SeCN)3— (Р3 = 7,95 - 1013). Этот комплекс с близ-
ким значением константы устойчивости доказан также в работе [63]. Увели-
чение концентрации KNCSe приводит к образованию Ag (8еСЫ)1~-комплек-
сов [63]. В условиях применимости метода растворимости в водных растворах
было установлено образование комплексов Ag (SeCN)? [63].
Методом растворимости в водном растворе показано образование смешан-
ных комплексов Ag (SeCN) (SCN)?-, Ag (SeCN)2I2-, Ag (SeCN)3I3-, Ag •
• (SeCN)3 (SCN)3-, Ag (SeCN) I3- [65], общие константы устойчивости кото-
Таблица 6.3. Зависимость прочности селеноцианатных комплексов серебра от концентрации неводной добавки при 20°С рых при 20° С (ионная си- ла 2,0) соответственно рав- ны: 5,9 • 1013; 2,6 1015; 5,3 • 1014; 2,5 - 1013; 4,5 • • 1014 [65]), обнаружены так- же Ag (SeCN)2 ’(SCN)2- и Cu (SeCN)2 (SCN)2-, Cu • • (SeCN) (SCN)- [64]. Селеноцианатные комп-
Растворитель Состав комплекса lg₽ Литера- тура i
Вода — метанол (35 об.%) Ag (SeCN)2" 13,86 [66]
Вода — метанол (56%) » 14,25 лексы серебра были изуче-
Вода — метанол (74%) 14,60 » ны в водно-метанольных
Вода — метанол (83%) Ag(SeCN)3~ 15,61 » [66], водно-ацетоновых и
Вода — метанол (92%) » 16,45 » водно-диоксановых раство-
Вода — диоксан (30 об. %) Ag (SeCN)2" 14,05 [68] рах [68]. При этом показа-
Вода — диоксан (50%) » 14,26 » но, что введение неводной
Вода — диоксан (70%) Ag (SeCN)3- 15,55 » добавки влияет на состав
Вода — диоксан (80%) Ag (SeCN)|— 16,00 комплексов и вызывает их упрочнение. В качестве при-
мера в табл. 6.3 приведена
часть полученных данных.
В смешанных водно-диоксановых растворах, содержащих 50; 70;
75 об. % диоксана, установлено образование комплексов Ag (SeCN)f- и
Ag (SeCN)4 . Дальнейшее увеличение концентрации неводной добавки вызы-
вает «упрощение» комплексов. В растворах, содержащих 80; 85 об. % диок-
сана, обнаружены только Ag (ЭеС^з--комплексы [68]. Очевидно, это обу-
словлено тем, что при высоком содержании диоксана последний координиру-
ется возле центрального атома, вызывая тем самым «упрощение» комплексов
серебра.
Селеноцианат серебра AgSeCN растворим в водных растворах AgNO3
или AgC104 с образованием комплексов Ag2SeCN^, Ag3SeCN2+ и даже
Ag4SeCN3+, общие константы устойчивости которых соответственно равны
(для AgCIO4 — AgSeCN — Н2О) 5,0 - 1011; 1,7 • 1012; 2,1 - 1012 [69].
Полярографически в водном растворе еще в 1956 г. был обнаружен
комплекс Cd (SeCN)?- с константой устойчивости 4,0 • 103 [56]. Потенцио-
262
метрическое исследование водных и водно-ацетоновых растворов привело к
доказательству наличия ряда комплексов Cd (SeCN)^-" с п — 1 -т- 6 [48].
В отличие от кадмия для цинка в водном растворе не выявлены селено-
цианатные комплексы вследствие быстрого разложения SeCN-ионов [70].
В водно-ацетоновых растворах с 50 об. % и более органического растворителя
потенциометрическим методом (t = 20° С, р = 1,0 — NaClO4) обнаружены
комплексы от ZnNCSe+ до Zn (NCSe)r [70] (табл. 6.4).
Весьма прочные селеноцианатные комплексы в водном растворе образу-
ет ртуть. Методом полярографии на фоне 0,3 М KNO3 при 25° С установлено
образование комплексов Hg (SeCN)2 и Hg (SeCN)C (lg ₽2 = 21,4; lg £3 =
= 25,6) [71]. Еще раньше было показано образование в водных растворах
комплексов Hg(SeCN)r (lg Р3 = 26,4) и
Hg (SeCN)I-(1g р4 = 29,5) [56].
Комплексы Hg (SeCN) Cl, Hg(SeCN)2,
Hg(SeCN)r и Hg (SeCN)4— были обна-
ружены при кондуктометрическом тит-
ровании водного раствора HgCl2 раство-
ром селеноцианата калия [72]. Изучено
также образование смешанных иодидо-
селеноцианатных комплексов ртути
Hg (SeCN) IT (lg P = 28,0), Hg (SeCN)^-
(lg ₽ = 29,4), Hg (SeCN) if” (lg 0 = 29,7)
[73, 74].
В водных и водно-ацетоновых рас-
творах сравнительно легко образуются
комплексы свинца Pb (SeCN)!- [59], а также кобальта и никеля: Со
• NCSe+ (Pi = 15,9) [60, 61], Ni (H2O)5NCSe+ [60, 62]. В водно-ацетоновом
(50 об.%) растворе обнаружены комплексы Со (Н2О)5 NCSe+ (Pi = 33,3) и
Со (NCSe)4~ (Р4 = 6,2 • 103) [61], Со (DH)2 (SeCN)f^ и Fe (DH)2 (SeCN)i~.
В последних, судя по данным ИК-спектров, связь SeCN-групп с центральным
атомом осуществляется через селен [75]. В водном и водно-ацетоновых рас-
творах, содержащих 30; 50; 75 об. % неводной добавки при 20° С и ионной силе
(NaClO4), равной 2,0, потенциометрически обнаружены малопрочные комп-
лексы TI (NCSe)F и TI (NCSe)!- (для водных растворов 1g р2 = 1,74; 1g рз =
= 1,55 [50]). Позднее появилось сообщение [58] о полярографическом изу-
чении селеноцианатных комплексов таллия в водных растворах при 27° С
и ионной силе 2,0 (NaNO3). При этом обнаружены комплексы TINCSe;
TI (NCSe)? (lg р2 = 0,88); TI (NCSe)3~ (lgP3 = 1,24) [58]. Различие в кон-
стантах здесь вызвано, очевидно, влиянием индифферентного аниона, а так-
же температурными условиями.
Таблица 6.4. Общие константы
устойчивости селеноцианатов цинка
в водно-ацетоновых растворах [70]
Содержа- ние аце- тона, об. % ₽2 ₽3
50 1,4 - 102 4,4 - 102 6,3 • 103
60 2,0 102 3,1 - 103 3,2 • 104
75 5,5 • 102 1,1 • 104 1,0 • 105
90 1,8 • 104 3,5 - 105 3,2 - 10е
(Н2О)5
6.5.2. Метанольные растворы
Потенциометрическим методом при 20° С обнаружены комплексы
Ag (SeCN)4~ (04 = 9,1 • 1016) [66]. Кобальт (II) пои малом избытке SeCN-
ионов образует CoNCSe+ и Со (NCSe)2, которые, судя по электронным спект-
рам поглощения, имеют октаэдрическую конфигурацию за счет сольвата-
ции растворителя. При увеличении концентрации SeCN-ионов в растворе
263
образуются тетраэдрические комплексы Со (NCSe)4 . Общие констант
устойчивости этих комплексов равны [61]: = 4,6 • 103; р2 = 2,4 • 104,
р3 = 5,5 • 104; р4 = 1,2 • 105.
Для никеля установлено образование комплексов от NiNCSe+ до
Ni (NCSe)4~ с константами: рх = 5,0 • 102; р2 = 2,3 • 104; р3 = 5,0 • 105;
Р4 = 5,5 - 106 [62].
Из электронных спектров следует, что в метаноле за счет присоединения
растворителя селеноцианатные комплексы никеля имеют октаэдрическою
структуру. Кинетика образования комплекса Ni (СН3ОН)5 NCSe+ из
Ni (СН3ОН)б+ изучена в работе [195].
В метаноле изучена также кинетика следующих реакций замещения
[87, 88, 186]:
транс- [PtL2Cl2] 4- Х~ -+ транс- [PtL2ClX] + СГ (6.6)
(L = Р (С2Н5)3; As (С2Н5)3, пиперидин, X = SCN~ или SeCN~);
транс- [PtL2NO2X] ф Y-стране- [PtL2NO2Y] ф X” (6.7)
(X = СГ, Вг“, N07, N?; Y = СГ, ВГ, Г, SCN~, SeCN"; L=пиперидин).
Показано, что реакции замещения проходят быстрее для SeCN, чем для
SCN-ионов [87, 88].
Кондуктометрически изучены селеноцианаты кадмия в метаноле и пока-
зано образование комплексов Cd (SeCN)r [48].
6.5.3. Диметилформамидные растворы
Как видно из табл. 6.5, в диметилформамиде изучены комплексы боль-
шого числа металлов и определены их константы образования.
Исследование ИК-спектров поглощения растворов Co(NCSe)4~ [77]
и VO (NCSe)4~ [83] показало, что SeCN-группы связаны с центральным ато-
мом через азот.
Взаимодействие хлоридов висмута [81], ванадия (III) [82], железа (II)
[79] с селеноцианатом натрия ведет к образованию комплексов с соотноше-
нием хлорид металла : селеноцианат, равным 1 : 1 и 1 : 2. Тетрахлорид то-
рия с селеноцианатами щелочных металлов образует комплексы с соотно-
шением Th : SeCN от 1 : 1 до Г. 6 [84].
Если в раствор МоС15 в DMFA вводить KNCSe, то посредством электрон-
ных, И К- и спектров ЭПР можно установить образование комплексов
[МоОСЦ—п (NCSe)J, где n = 1 4 4 [85]. Константы равновесия реакций об-
разования этих комплексов равны: 6, = 5 • 102: 62 = 2 • 105: = 3 • 107:
₽4 = 4 • 109 [851.
При изучении взаимодействия МС14 (М = Zr, Hf) с KNCSe методами спек-
трофотометрии и кондуктометрии обнаружены комплексы МС12 (NCSe)2 и
М (NCSe)t~. Общие константы равновесия реакций образования М (NCSe)|“~
оказались равными 1,3 • 108 (цирконий), 7,7 - 107 (гафний). В ИК-спектрах
поглощения диметилформамидных растворов М (NCSe)!" идентифицирова-
ны узкие и интенсивные полосыт (CN), расположенные в области 2065—
2070 см-1. Полосы проявляются при 625 см-1. Таким образом, из данных
ИКС следует, что SeCN-лиганд связан с цирконием или гафнием через азот
[86].
264
Таблица 6.5. Общие константы устойчивости комплексов в диметилформамидных
растворах
Комплексный ион Темпера- тура, °C Ионная сила Метод Pfl Литера- тура
MnNCSe+ 18—20 Переменная Спектрофото- 2,0 • 102 [76]
метрня
Mn (NCSe)2 » » » 1,8 • 104 »
Mn (NCSe)^ » » » 6,8 • 105 »
Mn (NCSe)2- » » 1,6 • 107 »
Mn (NCSe)|~ » » 3,1 - 10» »
Mn (NCSe)g— » 5,2 109
CoNCSe+ » » » 1,0 - 103 [77]
Со (NCSe)2 » » » 8,0 • 104 »
Со (NCSe)^ » » 5,0 • 105 »
Со (NCSe)J“ » » 6,0 - 10е
NiNCSe+ » » » 1,8 • 102 [78]
Ni (NCSe)2 » » 1,2 • 104 »
Ni (NCSe)^- » » » 3,5 105
Ni (NCSe)J- » » » 8,3 • 106 »
ZnNCSe+ 20 l.o Потенциомет- 4,0 • 102 [70]
рия
Zn (NCSe)2 » » » 2,0 • 104 »
Zn (NCSe)~ » » 7,9 • 104 »
CdNCSe+ 3 25 1,0 » 3,2 - 102
Cd (NCSe)2 » » » 1,1 • 104
Cd (NCSe)j- » » 1,9 • 105 »
Cd (NCSe)2~ » » » 1,0 • 106 »
In (NCSe)2+ » » 1,0 • 107 [80]
In (NCSe)+ » » » 5,6 • 108 »
In (NCSe)3 » » 3,1 • 1010 »
TI (NCSe)~ 20 1,7 18,0 [50]
6.5.4. Диметилсульфоксидные растворы
До сих пор в диметилсульфоксиде изучены лишь селеноцианатные комп-
лексы индия [80], висмута [81], кобальта и никеля [125].
Потенциометрически при 25° С установлено, что перхлорат индия взаи-
модействует с NaNCSe, образуя комплексы In (NCSe)2+ (Рх = 2,1 • 105) и
In (NCSejt (₽2 = 7,5 • 105) [80].
Взаимодействие BiCl3 с NaNCSe изучено спектрофотометрически. При
этом установлено образование комплексов с соотношением Bi : SeCN, рав-
ным 1 : 1 и 1 : 2. Общие константы равновесия реакций образования этих
комплексов равны: (3L = 28,5; р2 = 1,9 • 102 [81].
265
Спектрофотометрически в DMSO обнаружены комплексы Со (DMSO)5 •
• NCSe+, Со (DMSO)4 (NCSe)2 и Ni (DMSO)5 NCSe+, общие константы устой-
чивости которых соответственно равны 55,5; 7,1 • 102; 2,6 • 102 [125].
6.5.5. Ацетоновые растворы
Так как в этом растворителе многие соли, особенно хлориды, слабо дис-
социируют, определение констант образования комплексов имеет слишко1
приближенный характер. Для многих систем приходится говорить лишь об
условных константах равновесия реакций образования комплексов.
При взаимодействии перхлоратов марганца [76], кобальта [61], никеля
[62], цинка [70] с селеноцианатом натрия образуются весьма прочные комп-
лексы от MNCSe+ до М (NCSe)e~ (М = Mn, Ni) или от MNCSe+ до М (NCSe)t~
(М = Со, Zn).
Исследование электронных спектров поглощения показало, что кобальт
(II) образует в ацетоновых растворах тетраэдрические комплексы Со (NCSe)4~
(₽4 = 1,4 - 1013) [61, 90], а никель (II) — октаэдрические Ni (NCSe)e~ (|36 =
= 2,0 • 1013) [62]. Как и кобальт, цинк (II) в ацетоне также образует тет-
раэдрические комплексы Zn (NCSe)i~ ф4 = 7,1 • 1011) [70].
В системе In (С1О4)3 — NaNCSe — ацетон потенциометрически обнару-
жены весьма прочные комплексы In (NCSe)^-" cn = 1 -? 6 [80].
При изучении растворимости Pb (SeCN)2 в ацетоновых растворах селено-
цианата калия установлено образование комплекса Pb (SeCN)V [51].
Хлорид ванадия (III) взаимодействует с KNCSe с образованием комплек-
сов, в которых соотношение V : NCSe равно 1 : 1; 1 : 2 и 1 : 3. Судя по элек-
тронным спектрам поглощения, ванадий (III) в растворе имеет октаэдриче-
ское окружение [82]. В ацетоне обнаружены также комплексы ванадия (IV)
VO (NCSe)4~ и VO (NCSe)2 [831.
В системе BiCl3 — NaNCSe — ацетон согласно спектрофотометрическим
данным образуются комплексы с соотношением Bi : SeCN от 1 : 1 до 1 : 6
[81].
Тетрахлорид тория при взаимодействии с KNCSe образует, очевидно,
смешанные комплексы с соотношением Th : SeCN от 1 : 1 до 1 : 4 [84].
Комплексы смешанного типа обнаружены также для меди (I). При рас-
творении CuCl в растворах KNCSe установлено образование Cu (SeCN) СП
[47], а в системе Cu (SCN)2 — KNCSe—ацетон, по мнению авторов работ
[64, 89], образуются смешанные комплексы Cux (SCN)^ (SeCN)2.
При растворении МоС15 в ацетоне образуется НМоОС14, которая взаимо-
действует с KNCSe, образуя комплексы [MoOCh—п (NCSe),;Solv], где п =
= 14-5. Общие константы равновесия реакций образования этих комплек-
сов равны: & = 3 • 104; 02 = 1 • Ю8; ₽3 = 1 - 10й; ₽4 = 1 - 1014; |35 =
= 1 • 1017. Спектры ЭПР ацетонового раствора МоО (NCSe)!"- при 295—
220 К описываются изотропным спиновым гамильтонианом с параметрами:
S = 1/2; 7 = 0 (для четных изотопов); J — 5/, (для нечетных изотопов);
g = 1,937 ± 0,002; А = (47 ± 3). При 77 К спектр анизотропен и описыва-
ется спиновым гамильтонианом для аксиальной симметрии с параметрами:
= 1,927 ± 0,005; = 1,945 ± 0,005; А „ = (74 ± 5)э; Д± = (33 ±
± 5)э. Параметры спектров ЭПР селенопианатного комплекса оказались
близкими к таковым для аналогичного тиоцианатного комплекса МоО •
266
• (NCS)? , в котором доказана связь Мо—NCS. Это позволило сделать вы-
вод о том, что в МоО (NCSe)I— связь SeCN-групп с молибденом осуществля-
ется через азот. Этот вывод находится в соответствии с данными ИК-спек-
троскопии [85].
6.5.6. Ацетонитрильные растворы
В ацетонитриле установлено образование селеноцианатных комплексов
ряда металлов (табл. 6.6).
В ИК-спектрах поглощения ацетонитрильного раствора М (NCSe)?-
(М = Mn, Ni) найдены v (CN) около 2070 см-1 и v (CSe) 620 см-1, что может
Таблица 6.6. Селеноцианатные комплексы металлов в ацетонитриле
Комплексный ион Темпера- тура, °C Ионная сила Метод Литера- тура
Bi (SeCN)g~ Комнатная Переменная Спектрофото- метрия 1,2 - I012 [81]
Cd (NCSe)+ 25 1,0 Потенциомет- рия 2,5 1010 [70]
Cd (NCSe)2 » » » 1,9 1012 В
Cd (NCSe)- » » » 4,0 • 10“ »
Cd (NCSe)?“ » » » 5,8 • 101& »
Co (NCSe)+ Комнатная Переменная Спектрофото- метрия 8,6 103 [77]
Co (NCSe)2 » » » 2,8 • 10е »
Co (NCSe)^- » » 4,7 109
Co (NCSe)?~ в 6,5 - 1012
In (NCSe)|— 25 1,0 Потенциомет- рия 3,1 • 1024 [80]
Mn (NCSe)+ Комнатная Переменная Спектрофото- метрия 4,1 - Ю2 [91]
Mn (NCSe)2 » » » 1,0 • 105 »
Mn (NCSe)^" » 4,5 • 106
Mn (NCSe)?— » » » 7,3 - 107 »
NiNCSe+ » » 0,7 • 104
Ni (NCSe)2 » » 1,7 . 107 »
Ni (NCSe)- » 2,1 - 1010 »
Ni (NCSe)?- » » 1,8 • 1013 »
ZnNCSe+ 20 1,0 Потенциомет- рия 2,0 • 107 [70]
Zn (NCSe)2 » » » 2,5 • 10® »
Zn (NCSe)^- 5> 5> » 6,0 • Ю10 >
Zn (NCSe)?~ » » » 1,8 • Ю12 >
267
служить указанием на связь SeCN-групп с никелем или марганцем через
азот [91].
В ацетонитриле могут образовываться и хлоридоселеноцианатные комп-
лексы металлов, особенно в отсутствие избытка селеноцианат-ионов. По-
видимому, смешанные комплексы образуются в системе VC13 — KNCSe —
— CH3CN, так как спектрофотометрически установлены комплексы с соот-
ношением V : SeCN, равным 1 : 1 и 1 : 2 [82]. Судя по электронным спек-
трам поглощения, в рассматриваемой системе ванадий (III) имеет октаэдри-
ческую конфигурацию [82].
6.5.7. Влияние природы растворителя на реакции
комплексообразования с участием SeCN-групп
Как указывалось выше, в водных растворах возможно существование
или очень прочных селеноцианатных комплексов (такие комплексы образу-
ют серебро, ртуть, платина), или комплексов средней прочности, но с учас-
тием слабогидролизующихся катионов (селеноцианаты свинца, кадмия).
Ионы же других металлов-комплексообразователей хотя и образуют се-
леноцианатные комплексы, однако в водном растворе их обнаружить прак-
тически нельзя.
Поскольку введение неводного растворителя, способного смешиваться
с водой, ослабляет протекание реакций гидролиза, для некоторых металлов
уже в смешанных растворителях можно установить селеноцианатные комп-
лексы. Так, цинк в водно-ацетоновом растворе (50 об. % ацетона) образует
ряд комплексов вплоть до Zn (NCSe)7-
В смешанных растворителях с ростом концентрации неводной добавки
прочность селеноцианатных комплексов металлов всегда возрастает. При
этом рост концентрации неводной добавки оказывает более заметное влия-
ние на комплексы с М—NCSe-связью, чем на комплексы с М—SeCN-связью.
Наиболее прочные селеноцианаты образуются в ацетоне и ацетонитри-
ле — растворителях со слабой сольватирующей способностью. В этих же
растворителях наиболее легко образуются селеноцианатные комплексы
анионного типа. Например, в ацетонитриле весьма легко образуются комп-
лексы марганца, кобальта, никеля, цинка:
ЙкГ M"(NCSe)2- Со (NCSe)J~ Ni (NCSe)^ Zn (NCSe)2“
lg₽4 7,86 12,82 13,26 12,25
Ряд устойчивости M (NCSe)^-- в ацетонитриле совпадает с известной после-
довательностью Ирвинга — Вильямса. В других изученных растворителях
эта последовательность нарушается, что, очевидно, обусловлено образова-
нием смешанных комплексов с вхождением в координационную сферу моле-
кул растворителя, в среде которого изучались реакции комплексообразова-
ния, или же хлорид-ионов, если в качестве исходной соли металла брался
хлорид.
Сравнение прочности селеноцианатных комплексов различных металлов
показывает, что подобно тиоцианатам (см. раздел 5) комплексы со связью
М—SeCN прочнее комплексов со связью М—NCSe. Это объясняется увели-
чением доли ковалентности связи при переходе от изоселеноцианатных
(М—NCSe) к селеноцианатным (М—SeCN) комплексам. Помимо различия в
сг-донорных свойствах N и Se группы SeCN, при ее координировании сущест-
268
венную роль играет и различие в л-акцепторных свойствах этого лиганда
в зависимости от природы центрального атома и способа связывания SeCN-
группы. Комплексообразователи с nd-электронами при наличии вакантных
nd-орбиталей селена образуют дативные л-связи, что приводит к дальней-
шей стабилизации связи металл — лиганд и к еще большему повышению
прочности соответствующих комплексов.
Как и в других псевдогалогенидах, чем выше сольватирующая актив-
ность растворителя, тем труднее образуются селеноцианатные комплексы
с большим числом ацидолигандов и тем ниже их прочность. Это играет
весьма существенную роль при синтезе координационных соединений.
6.6. СПОСОБ КООРДИНАЦИИ SeCN-ГРУПП
Селеноцианат-ион является амбидентатным лигандом, что хорошо со-
гласуется с квантово-механическими расчетами распределения зарядов
на атомах, входящих в этот лиганд [92] (см. табл. 1.1).
Характер координирования SeCN-групп сказывается на изменении ча-
стот v(CN) и v(CSe) в ИК-спектрах поглощения [43, 45, 93] (табл. 6.7). Как
видно из табл. 6.7, если координационная связь осуществляется через атом
селена, то частота v (CSe) понижается, а частота v (CN) несколько повышается
по сравнению с частотами иона. Если координационная связь осуществля-
ется через атом азота, то частота
v (CSe) повышается, а частота v(CN)
иногда повышается, а иногда не-
сколько понижается по сравнению
с частотами иона. Если SeCN-rpyn-
па осуществляет мостиковую функ-
цию, то частоты v (CN) повышены
как по сравнению с ионом SeCN"-»
так и по сравнению с двумя пре-
дыдущими случаями: частоты v (CSe)
повышаются или понижаются по
сравнению с v (CSe) иона.
Увеличение v (CN) и уменьше-
ние v (CSe) по сравнению с дан-
Таблица 6.7. Приближенные частотные
интервалы для валентных колебаний
SeCN-групп в неорганических
селеноцианатах, см-"1
Способ координации v (CN) v(CSe)
SeCN~ -2050—2100 549—563
М—NCSe -2000—2110 600—680
М—SeCN -2080—2125 520—545
М—SeCN—М -2130—2155* 525—640
* Одновременно возможно присутствие и более
низкочастотных полос.
ными частотами SeCN~ для комп-
лексов со связью М—SeCN вызвано одновременным ростом силовой
константы связи CN и уменьшением силовой константы связи CSe.
Изменения частот v (CN) и v (CSe) изосел еноцианатов объясняются в
основном изменением механики колебаний системы при переходе от SeCN
к группам М—NCSe, а не изменением силовых констант связей CN и CSe.
Повышенный, по сравнению с монодентатными группами, рост v (CN) у мо-
стиковых групп М—SeCN—М обусловлен как некоторым увеличением сило-
вой константы связи CN, так и изменением механики колебаний системы.
Частота v (CSe) у мостиковых SeCN-групп может или уменьшаться, или
возрастать по сравнению с SeCN"- в зависимости от того, какой из двух про-
тивоположно действующих факторов преобладает — изменение механики
колебаний или уменьшение силовой константы связи [43, 93].
Ценную информацию о способе координации SeCN-групп можно полу-
чить, определяя интегральную интенсивность поглощения полосы v (CN).
Как было показано Бурмайстром и Вильямсом [162], интегральная интенсив-
269
i ность при связи М—SeCN лежит в пределах (0,5 4- 1) • Ю4 моль 1 • см' .
> при N-связывании— в пределах (5 ~ 10) • 104 моль-1 • см-2.
В зависимости от способа координации SeCN-групп в ИК-спектре наблю-
даются также некоторые изменения в частотах деформационного колебания
6 (SeCN). Для SeCN-иона 6 (SeCN) наблюдается в области ~405 — 420 см-
для изоселеноцианатных групп — в области ~410— 450 см-1, а для селе-
ноцианатных— в области ~360—410 см-1.
В последнее время ИК-спектроскопия широко используется для иссле-
дования селеноцианатных соединений, а выводы о способе координации
SeCN-групп по данным И КС за редким исключением сомнения не вызывают.
6.7. КООРДИНАЦИОННЫЕ СОЕДИНЕНИЯ СЕЛЕНОЦИАНАТОВ
С АМИНАМИ
6.7.1. Соединения, содержащие пиридин
и его производные
Соединения рассматриваемого типа впервые были получены Г. Спаку
и его сотрудниками: [МРу4 (NCSe)2] (М = Mn, Fe, Со, Ni); МРу2 (NCSe),
(М = Zn, Cd) и NiPy6 (NCSe)2 [94]. Последняя соль получена также пере-
кристаллизацией NiPy4 (NCSe)2 в хлороформе в присутствии избытка пири-
дина [95]. Соединение СоРу4 (NCSe)2 получено Нельсоном, который отмеча-
ет, что при выдерживании в вакууме при 70° С оно переходит в СоРу2 (NCSe)2
При кристаллизации ацетонового раствора V (NCSe)3 в присутствии пи-
ридина выделяется темно-фиолетовый порошок [VPy6] (NCSe)3 [98], а для
индия в тех же условиях удается получить [InPy3 (NCSe)3] • Ру [991. Ана-
логично получены зеленые кристаллы VOPy4 (NCSe)2, которые при выдержи-
вании в эксикаторе легко теряют часть пиридина, превращаясь в VOPy3 •
- (NCSe), 1100].
Из пиридина выделено соединение [СгРу3 (NCSe)3] • 2Ру, которое при на-
гревании до 130° С в вакууме переходит в желтое мелкокристаллическое ве-
щество [СгРу3 (NCSe)3] [97].
При насыщении пиридина селеноцианатом свинца (при 80—90° С) с по-
следующим охлаждением и выдерживанием раствора в эксикаторе полу-
чены светло-желтые кристаллы [PbPy2 (NCSe)2] [101].
Для никеля, кроме [NiPy2 (NCSe)2], получены другие селеноцианатные
соединения состава NiL4 (NCSe)2 (L = 3-метил- или 4-метилпиридин) [102].
Описаны также соединения [CoL2 (NCSe)2l (L = хинолин [103], 2,6-диметил-
пиридин, 2,3,4-замещенные пиридина [104]), СоА4 (NCSe)2 (А = изохино-
лин) [103]. При действии на ацетоновый раствор Zn (NCSe)2 хинолином полу-
чен в виде бесцветных блестящих кристалликов Zn (Q)2 (NCSe)2 [115].
Почти все упомянутые здесь соединения изучены методом ИК-спектро-
скопии, а для некоторых из них получена также рентгенографическая харак-
теристика. Особенностью рассматриваемых здесь соединений является то,
что для большинства из них идентификация v (CSe) затруднена из-за наличия
пиридина, полосы которого в ИК-спектре перекрывают v (CSe). Поэтому для
выяснения способа координации SeCN-групп в этих соединениях, кроме ме-
тода ИК-спектроскопии, желательно применение других методов исследо-
вания.
270
Как видно из табл. 6.8, в ИК-спектрах всех рассматриваемых соединений,
кроме PbPy2 (NCSe)2, v (CN) идентифицируются в области ~2040—2110 см-1.
Следовательно, ни одно из соединений не содержит мостиковых SeCN-групп.
В ИК-спектрах соединений МРу4 (NCSe)2 (М = Мп, Fe, Со, Ni, Zn, Cd),
ZnPy2 (NCSe)2, CdPy2 (NCSe)2 не найдено никаких полос ни около 500 см-1,
т. е. в области, в которой обычно присутствуют v (CSe) для М—SeCN-свя-
зей, ни около ~550—560 см-1 — в области v (CSe) для SeCN-иона. Следо-
Таблица 6.8. Основные колебательные частоты SeCN-грунп, см'
Соединение v (CN) v (CSe) о (SeCN) Тип координации SeCN-группы Литера- тура
CdPy2 (NCSe)2 2052, 2112 590 403, 402 Cd—NCSe [129]
CdPy4 (NCSe)2 2040, 2110 588 405, 415 » »
CoPy4 (NCSe)2 2073 >620 — Co—NCSe [45], [90]
CoQ2 (NCSe)2 2037, 2070 — — » [90]
CrPy3 (NCSe)3 2055, 2065, 2075 —. — Cr—NCSe [97]
FePy4 (NCSe)2 2065 >600 Fe—NCSe [H7]
[InPy3 (NCSe)3J Py 2058 — — In—NCSe [99]
MnPy4 (NCSe)2 2061 >620 — Mn—NCSe [45]
NiPy4 (NCSe)2 2088 664 427 Ni—NCSe [95]
NiPy4 (NCSe)2 • 2Py 2088 664 436, 426,416 » »
PbPy2 (NCSe)2 2075,2110,2140 565, qxo 415 Pb—NCSe—Pb [101]
VPy6 (SeCN)3 2060 — SeCN [98]
VOPy3 (NCSe)2 2048, 2085 — — V—NCSe [100]
ZnPy2 (NCSe)2 2079, 2095 >640 — Zn—NCSe [45]
ZnQa (NCSe)2 2050, 2087 — 1 — » [115]
вательно, в рассматриваемых соединениях нет ни селеноцианат-ионов, ни
лигандов, координированных через атом селена. Остается принять, что во
всех соединениях содержатся изоселеноцианатные группы.
Этот вывод подтверждается данными рентгенографии, показывающими,
что соединение [СоРу4 (NCSe)2] изоструктурно аналогичному тиоцианату,
в котором SCN-группы, занимая транс-положение, связаны с кобальтом через
азот [901. Позднее была установлена изоструктурность октаэдрических
комплексов МРу4 (NCSe)2 (М = Mn, Fe, Со, Ni, Zn, Cd) [117, 129, 138]. Изу-
чение магнитных свойств солей кобальта и никеля состава МРу4 (NCSe)2
также указывает на их октаэдрическое строение (см. табл. 6.11). Полоса
поглощения (~20 000 см-1) в электронном спектре [СоРу4 (NCSe)2] (значе-
ние 10D<7 ~ 9700 см~1) также свидетельствует об октаэдрической конфигу-
рации кобальта в этом соединении [90].
Октаэдрическую конфигурацию со связями Ni—NCSe имеет никель не
только в !NiPy4 (NCSe)2J, но и в соединениях NiPy6 (NCSe)2, Ni (]3-Pic)4-
• (NCSe)2 и Ni (y-Pic)4 (NCSe)2. Интересно, что в соединении NiPy6 (NCSe)2
октаэдрическое окружение никеля достигается не за счет шести молекул
пиридина, а за счет координации NCSe-групп [95]. Укажем также, что в
электронном спектре поглощения NiPy4 (NCSe)2, Ni (|3-Pic)4 (NCSe)2,
Ni (y-Pic)4 (NCSe)2 обнаружено по три полосы около ~22 700, ~ 17 200
и ~ 10 500 см-1, указывающие на октаэдрическую конфигурацию никеля. Об
этом свидетельствуют также величины 10Dq, которые для рассматриваемых
соединений соответственно равны: 10 580; 10 540 и 10 580 см-1 [102]. Поло-
271
жение полос в спектре отражения NiPy2 (NCSe)2 (23 260, 16 530, 13 550 см-1)
также свидетельствует об октаэдрическом строении этого соединения [102].
Октаэдрическая конфигурация принята для СоРу2 (NCSe)2, так как маг-
нитный момент этого соединения равен 5,1 М. Б. [96] (см. табл. 6.11).
В отличие от никеля и кобальта в МРу2 (NCSe)2 цинк и кадмий имеют
тетраэдрическую конфигурацию, а SeCN-группы связаны с центральным ато-
мом через азот (см. табл. 6.8). Тем не менее рассматриваемые соединения не
изоструктурны между собой, что можно, вероятно, объяснить разным
расположением молекул пиридина в их кристаллах [129]. В аналогичном по
составу соединении свинца PbPy2 (NCSe)2, судя по данным ИК-спектров,
содержатся мостиковые SeCN-группы [101].
Для СгРу3 (NCSe)3 получено две модификации: желтая и красная [971,
ИК-спектры обеих форм аналогичны и указывают на связь SeCN-групп с
хромом через азот, а дебаеграммы различны. Очевидно, в СгРу3 (NCSe)3
имеет место координационная изомерия [139].
Как видно из табл. 6.8, в [VOPy3 (NCSe)2[ группы SeCN связаны с вана-
дием через азот. Наличие в ИК-спектре полосы v (VO) при 972 см-1 указы-
вает на присутствие «изолированных» V—О-групп [100]. Следовательно,
ванадий в [VOPy3 (NCSe)2l имеет октаэдрическую конфигурацию. Изучение
соединения 1пРу4 (NCSe)3 методами ИК-спектроскопии [991 и рентгеногра-
фии [140] позволило установить координационную формулу этого соедине-
ния: НпРуз (NCSe)3] • Ру.
Изоселеноцианатные группы присутствуют и в соединениях ZnQ2 (NCSe)2
и CoQ2 (NCSe)2 (табл. 6.8), а центральный атом имеет тетраэдрическую кон-
фигурацию [90, 103, 115].
6.7.2. Соединения, содержащие дипиридил
и фенантролин
В последние годы синтезировано достаточно много координационных селе-
ноцианатов, включающих 2,2'-дипиридил или 1,10-фенантролин. Так, из
ацетонового раствора получены [MDipy2 (NCSe)2] (М = Mg, Са, Sr, Ва),
[MPhen2 (NCSe)2] (М = Mg, Са, Ва), [MPhen3] (NCSe)2 (М = Sr, Ва) [2051.
Соединения [MPhen2 (NCSe)2] (М = Mn, Fe, Co, Ni) и [MDipy2 (NCSe)2]
(M = Mn, Co, Ni) получены из спиртовых растворов [76, 117], a [Fe Phen2 •
• (NCSe)2] — из пиридина [118].
При действии на водный раствор соли [FePhen3] С12 • 7Н2О селеноциана-
том калия получено соединение [Fe Phen3] (SeCN)2 [118]. Его можно полу-
чить, действуя на спиртовый раствор Fe (NCSe)2 избытком фенантролина
[117]. Аналогичным образом получено FeDipy3 (SeCN)2 [117].
Из водных растворов выделены CuDipy (NCSe)2, CuPhen (NCSe)2, а так-
же CuDipy2 (NCSe)2 [131].
Если к ацетоновому раствору Zn (NCSe)2, полученному обменной реакци-
ей между нитратом цинка и селеноцианатом калия, приливать раствор Dipy
или Phen, сразу же выпадают желтоватые порошки [Zn Dipy2 (NCSe)2J
или [Zn Phen2 (NCSe)2] [115].
Весьма прочные дипиридильные соединения состава [MDipy (SeCN)2]
образуют палладий и платина. Оба соединения получены из водных раство-
ров при действии селеноцианата калия на MDipy (NO3)2 [111].
Из водного раствора выделено соединение PdL2 (SeCN)2 (L = 5-нитро-
фенантролин) [1471.
272
Из ацетонового раствора синтезированы [InDipy3] (SeCN)3 [99],
[InPhen3] (SeCN)3 [119], BiPhen (SeCN)3, BiDipy3 (SeCN)3 [120], а из тетрагид-
рофуранового— аналогичные солям индия соединения галлия [119].
Синтез сине-черных [VDipy3] (SeCN)3 и [VPhen3] (SeCN)3 осуществлен из
водно-диоксановых растворов [98].
Из неводных растворов (ацетон, метанол или этанол) получены коорди-
национные соединений РЗЭ, урана [122—124, 127, 204] (табл. 6.9), а из тет-
рагидрофурана— MDipy2(NCSe)4 и MPhen3 (NCSe)4 • Thf (М = Zr, Hf) [86].
Из ацетонитрильных растворов синтезированы соединения NbDipy2 •
• (NCSe)4 и Nb (dmDipy)2 (NCSe)4 (dmDipy = 4,4'-диметил-2,2'-дипири-
дил), а из дихлорэтана выделено TaDipy (NCSe)5 [126].
Исследование соединений MPhen2 (NCSe)2 (M=Mn, Fe.Co, Ni, Zn) методом
рентгенографии показало их изоструктурность, а построение кривых ради-
ального распределения атомной плотности — наличие связи М—NCSe
[76, 117], что находится в соответствии с данными ИК-спектроскопии
(табл. 6.9). Можно полагать, что в октаэдрических комплексах MPhen2(NCSe)2
группы SeCN занимают скорее цис-положение вследствие возможных про-
странственных затруднений, вызываемых двумя молекулами Phen, которые
находились бы в одной плоскости в случае транс-изомерии MPhen2
(NCSe)2.
Для [FePhen2 (NCSe)2] высокотемпературный предел рэф составляет
5,10 ± 0,05 М. Б. при 440 К. При 232 К магнитный момент этого соединения
резко уменьшается, достигая (0,84 ± 0,01) М. Б. при 77,2 К. По мнению ав-
торов, этот факт можно объяснить существованием термического равновесия
между состояниями xAig и 5T2g [137].
Учитывая значения v (CN) и состав соединений, можно принять, что в
соединениях ML3 (SeCN)n (М = Fe24*, In34*, Y34*, VO24*; L = Dipy, Phen)
SeCN-группы находятся во внешней сфере. В соединении BiPhen (SeCN)3
высокочастотная компонента v (CN), расположенная при 2105 и 2120 см-1,
может служить указанием на связь Bi—SeCN. Наличие такого типа свя-
зи не исключено и для BiDipy3 (SeCN)3 (см. табл. 6.9).
Соединения MDipy2 (NCSe)2 (М = Со, Mn, Ni, Zn) изоструктурны, а
связь М с SeCN-группами осуществляется через азот [117].
В селеноцианатных соединениях меди (II), как и в аналогичных тиоциа-
натах, наблюдается стремление центрального атома к тетрагонально-бипи-
рамидальному окружению. В Cu (NH3)4 (SeCN)2 [141] и CuEn2 (SeCN)2
[ПО], которые изоструктурны аналогичным тиоцианатам, SeCN-группы при-
мыкают к квадрату меди (CuN4) со стороны атомов селена, образуя через атом
азота водородные связи с молекулами амина [130]. Поскольку расстояния
Си—Se в этом случае равны ~3,0—3,3 А, состояние SeCN-групп в этих со-
единениях близко к ионному. Очевидно, такую же конфигурацию имеет медь
и в других соединениях (за исключением CuDipy2 (NCSe)2), представленных
в табл. 6.9. В соединениях CuPhen (SeCN)2, CuDipy (SeCN)2, Cu (CH3NH2)2 •
- (SeCN)2 октаэдрическая конфигурация меди достигается за счет мостиковых
SeCN-групп. В соединении CuDipy2 (SeCN)2 медь (И), судя по данным ЭПР,
имеет координационное число 5 (4NDipy + INncsc) [131].
Для циркония и гафния известны до сих пор только соединения MPhen3 •
• (NCSe)4 • Thf и MDipy2 (NCSe)4 [134]. Соединения MPhen3 (NCSe)4 Thf
изоструктурны аналогичным тиоцианатам, изоструктурными являются также
ZrDipy2 (NCSe)4 и HfDipy2 (NCSe)4. Последние соединения не изострук-
турны аналогичным тиоцианатным соединениям [134]. Данные рентгеногра-
фии и ИК-спектроскопии указывают на наличие в рассматриваемых соеди-
нениях связи М—NCSe. Таким образом, этим соединениям можно припи-
273
Таблица 6.9. Основные колебательные частоты SeCN-групп, см 1
Соединение v (CN) v (CSe) 6 (SeCN) Тип координации SeCN-групп Литераторе
BaDipy2 (NCSe)2 2085 575 420 Ba—NCSe [205]
BaPhen2 (NCSe)2 2090 575 412 »
BaPhen3 (NCSe)2 2065 552 —. SeCN“
BiDipy3 (NCSe)3 2075,2110,2118 — — » П201
BiPhen (SeCN)3 2060,2105,2120 — — Bi—SeCN
CaDipy2 (NCSe)2 2080, 2090 588 433 Ca—NCSe [2051
CaPhen2 (NCSe)2 2075 587 — »
CoDipy2 (NCSe)2 2073 >630 — Co—NCSe [1171
CoPhen2 (NCSe)2 2072, 2082 >620 — »
CuPhen (SeCN)2 2110 —. 415 Cu—SeCN—Cu [131]
CuDipy (SeCN)2 2096, 2117 — 432 » »
CuDipy2 (NCSe)2 2062, 2073 — 443 Cu—NCSe »
2113 SeCN-
FeDipy3 (SeCN)2 2050 — 422 SeCN~ [117]
FePhen3 (SeCN)2 2060 559 415 » »
FePhen2 (NCSe)2 2060, 2075, 2100 >600 417 Fe—NCSe [117, 132,
HfDipy2 (NCSe)4 2015 — Hf—NCSe 133] [134]
HfPhen3 (NCSe)4 2015, 2060 — — »
InPhen3 (SeCN)3 2050 — — SeCN” [H9]
InDipy3 (NCSe)3 2062 550 — SeCN- '[99]'
LnDipy3 (NCSe)3 2035—2085 — — La—NCSe [124]
(Ln = Sc, Y, La, Ce,
Pr, Nd, Sm, Eu, Gd)
LnDipy2 (NCSe)3 2033—2093 — »
2CH3OH (Ln = Sc, Y,
La, Ce, Pr, Nd, Sm,
Eu, Gd, Lu)
LnPhen3 (NCSe)3 2040—2060 — — J>
(Ln = Sc, Y, La, Ce,
Pr, Nd, Sm, Eu, Gd,
Lu)
MgDipy3 (NCSe)2 2075 >630 —. Mg—NCSe [205]
MgPhen2 (NCSe)2 2076 625 — » «
MnDipy2 (NCSe)2 2052, 2068 >625 — Mn—NCSe [76]
MnPhen2 (NCSe)2 2070 >640 — » »
NbDipy2 (NCSe)4 2050, 2025 698 468 Nb—NCSe [126]
Nb (dmDipy), (NCSe)4 2085, 2055 695 450 » »
2040
NiDipy2 (NCSe)2 2060, 2095 >630 —. Ni—NCSe [117]
NiPhen2 (NCSe)2 2087 >620 — » ) »
PdDipy (SeCN)2 2116, 2112 523 — Pd—SeCN [111]
518
PtDipy (SeCN)2 2135, 2125 523 — Pt—SeCN »
SrDipy2 (NCSe)2 2075, 2105 568, 580 — Sr—NCSe [205]
SrPhen3 (NCSe)9 2060 552 — SeCN- »
TaDipy (NCSe)5 1930 697 465 Ta—NCSe [126]
UO2Dipy2 (NCSe)2 2048 — — U—NCSe, [127]
NCSe-
UO2Phen2 (NCSe)2 2051 — — » »
VDipy3 (SeCN)3 2055 — 415 SeCN [98]
VPhen3 (SeCN)3 2060 560 » »
VODipy3 (SeCN)g 2060 — — » [100]
VOPhen3 (SeCN)3 2060 — » »
ZnDipy2 (NCSe)2 2088, 2080 610 412 Zn—NCSe [115]
ZnPhen2 (NCSe)2 2071, 2087 620 418 » »
ZrDipy2 (NCSe)4 2010 — — Zr—NCSe [134]
ZrPhen3 (NCSe)4 1190, 2060 — —. » »
274
Таблица 6.10. Основные колебательные частоты SeCN-групп, см'
Соединение v (CN) v (CSe) 6 (SeCN) Тип координации SeCN-групп Литера- тура
Cd (CH3NH2)2 (NCSe)2 2090 580 410 Cd —NCSe [113]
Cd (C2H6NH2)2 (NCSe)2 2105 585 405 » »
Cd (C3H7NH2)2 (l\CSe)2 2090 577 407 » »
Cd (C4H9NH2)2 (NCSe)2 2085 — 407 »
Cd (Bia)2 (NCSe)2 2102 — — » [206]
Cd (MBia)2 (NCSe), 2112, 2125 — — Cd—NCSe —Cd »
Cd (DMBia) (NCSe)2 2096, 2112, — — » »
2130
[Co (NH3)4 (CN) NCSe] Cl 2105 607 — Co — NCSe [111]
[Co (NH3)5 NCSe] (NO3)2 2116 624 — » »
[Co (NH3)b NCSe] Br2 2131 — — » [112]
[Cren3] (SeCN)3 2050, 2070 555 410 SeCN“ [97]
[Cr (NH3)b NCSe] Br2 2095 — — Cr — NCSe [И2]
Cu (NH3)4 (SeCN)2 • H2O 2062, 2105, 560 418 Cu—SeCN [130]
2132
Cuen2 (SeCN)2 2055, 2008 — — » [HO]
CuPn2 (SeCN)2 2053, 2028 — — » »
Cu (N,N'-dimen)2 (SeCN)2 2074. 2024 560 — » »
Cu (N,N-dieten)2 (SeCN)2 2072 530 — » »
Cu (CH3NH2)2 (SeCN)2 2115 — — Cu — NCSe — Cu [131]
GaUrt3 (NCSe)3 2082, 2070 643 — Ga — NCSe [116]
[HIr (Pip)4 NCSe] SeCN 2080, 2050 641, 483 Ir —NCSe, SeCN [197]
LnUrt2 (NCSe)3 • HCH3OH 2068—2092 — [124]
(Ln — Y, La, Ce, Pr, Md,
Sm, Eu, Gd)
Nien2 (NCSe), 2070, 2024 — — Ni — NCSe [HO]
NiPn2 (NCSej2 2096, 2083 692 — » »
NiBn2 (NCSe) 2 2102, 2058 — — » »
Ni (N,N'-dimen)2 (NCSe)2 2096 — — »
Ni (N,N'-dieten)2 (NCSe)2 2081, 2037 — — »
NiPn3(SeCN)2 2069, 2052 520 — SeCN" »
Ni (C4H9NH2)4 (NCSe)2 2085, 2110 580 425 Ni — NCSe [135]
[Ni (Et4dien) NCSe] Ph4B 2094 — 420 » [218]
[Pd (Et4dien) SeCN] Ph4B 2024 533 408 Pd — SeCN [136]
[Pd (Et4dien) NCSe] Ph4B 2085 618 — Pd — NCSe »
Znen3 (SeCN) 2 2075 550 412 SeCN [115]
сать координационные формулы [MDipy2 (NCSe)4] и [MPhen3 (NCSe)2] • (SeCN)2
[134].
Аналогичное строение имеют, вероятно, и селеноцианатные соединения
ниобия (IV) [NbDipy2 (NCSe)4l (см. табл. 6.9). Что касается соединения тан-
тала, то в нем центральный атом имеет координационное число 7 ([TaDipy -
• (NCSe)5J), поскольку ИК-спектры указывают на наличие только изоселе-
ноцианатных групп.
Рассмотрим координационные селеноцианаты лантаноидов. Как видно
из табл. 6.9, в ИК-спектрах полосы v (CN) лежат в области 2033—2093 см-1,
характерной для М—NCSe или ионных селеноцианатов. Рентгенографиче-
ские исследования показали, что аналогичного состава соединения
LnPhen3 (NCSe)3, LnDipy3 (NCSe)3, LnDipy2 (NCSe)3 • 2CH3OH (Ln = La,
Ce, Pr, Nd) изоструктурны [124].
Построение кривых радиального распределения атомной плотности по
данным рентгенографии показало, что в рассматриваемых соединениях при-
сутствуют группы Ln—NCSe [124]. Это дает основание считать, что к. ч.
Ln в селеноцианатных соединениях больше 6.
275
Из данных рентгенографии следует, что изоселеноцианатные группы при-
сутствуют также в соединениях иттрия и скандия YDipy2 (NCSe)3 • 2CHSO
и ScUrt2 (NCSe)3 • 2Act [123]. Палладий (II) и платина (II) предпочитают
М—SeCN-координацию, хотя для палладия и обнаружена изомерия связи
[1361 (табл. 6.10).
В соединениях UO2Dipy2 (NCSe)2 и UO2Phen2 (NCSe)2 уран имеет коор-
динационное число 7 ([UO2Dipy2NCSe] SeCN), на что указывают данные ИКС
и результаты изучения молярной электропроводности метанольных раство-
ров этих солей [127].
Судя по данным ИКС, в соединениях MDipy2 (NCSe)2 (М = Mg, Ca.
Sr, Ba), MPhen2 (NCSe)2 (M = Mg, Ca, Ba) также присутствуют изоселено-
цианатные группы, что подтверждается результатами рентгенографического
исследования [205]. В то же время SrPhen3 (SeCN)2 и BaPhen3 (SeCN)2
содержат SeCN-ионы [205].
6.7.3. Другие соединения селеноцианатов с аминами
К рассматриваемому ряду соединений следует отнести М (NCSe)2 • Urt •
• 4Н2О (М = Mn, Fe, Со, Ni, Zn) [94]. При приливании метанольного раст-
вора Urt к ацетоновому (In (NCSe)3) или тетрагидрофурановому (Ga (NCSe)3)
растворам выделяются довольно устойчивые соединения GaUrt3 (NCSe)3 и
InUrt4 (NCSe)3 [116]. Из неводных сред удалось синтезировать ряд соедине-
ний лантаноидов, содержащих уротропин (см. табл. 6.10) [124].
Большое количество селеноцианатов получено с этилендиамином, аммиа-
ком, первичными аминами, бензидином, толуидином, например: [Ni (NH3)6] •
- (S CN)2, [Cd (NH3)4 (SeCN)2l, [Co (NH3)6] (SeCN) Cl, Mn (Bzd)3 (SeCN)3 -
• H.,O, Co (Tlid) (SeCN). • 6H2O и др. [52, 94, 106—109), [Znen3] (SeCN)2
[115], Cd (RNH2)2 (NCSe)2 [113], Zn (RNH2)2 (NCSe)2 [114] (R = CH3, C2H5,
C3H7, C4H9), [Cren3 (NCSe)3l [52].
При действии селена на цианиды металлов в жидком аммиаке были по-
лучены координационные соединения: Mg (NH3)4 (NCSe).,, Zn (NH3)4 •
• (NCSe)2, Al (NH3)5 (NCSe)3 [32].
Описаны также [Co (NH3)5 NCSe] (NO3).„ [Co (NH3)4 NCSe (CN)1 Cl [111],
[M (NH3)5 NCSe] Br2 (M = Co, Cr) [111], [Cr (NH3)5 NCSe] (C1O4)2 [112] и
[Ru (NH3)5 SeCN] X2 (X = С1ОГ, Г, PF^) [196].
Соединения PdL2 (SeCN). (L = Bu3P, 4-аминопиридин, 2Ь-этилендиа-
мин) получены по схеме [137]
[PdL2ClJ + 2AgNO3 да- [PdL2(H2O)2p+ + 2NOr PdL2(SeCN)2.
(6-8)
Достаточно устойчивыми оказались на воздухе соединения NiL2 (NCSe)2,
CoL2 (NCSe)2 • 2 (CH3)2CO [128], CdA2 (NCSe)2, CdB (NCSe).3 (L = 3-фенил-
5-пиридил(2)-пиразол; А и В-бензимидазол и его производные [206]).
Из табл. 6.10 следует, что в соединениях кадмия аминного типа преобла-
дает Cd—NCSe-координация. Интересно отметить, что в присутствии аммиа-
ка SeCN-группы образуют связь через азот с кобальтом (III). В соединении
иридия (III), для которого характерна связь Ir—SeCN, присутствие коор-
динированного пиперидина и связанного с центральным атомом водорода
вызывает обращение SeCN-групп [197].
276
Как указывалось выше, в селеноцианатах меди (II), включающих аммиак,
фенантролин, наблюдается стремление центрального атома к тетрагонально-
бипирамидальному строению. Такое же строение имеют соединения меди,
включая этилендиамин и его производные.
Сравнительно недавно были получены соединения CuPn2 (SeCN) NO3,
Cu (Dien)2 (SeCN) NO3 (Dien = диэтилендиамин) [216], [Cu (Dben)2 (NCSe) •
• X] (X = NO3 или C1O4, Dben = N.N'-дибензилэтилендиамин) [219],
в которых, судя по электронным спектрам, для меди сохраняется указанная
выше конфигурация. Напротив, в [Cu (Et4dien) NCSe] РЬ4Вмедь имеет плос-
костное квадратное окружение [218].
Таблица 6.11. Магнитные свойства селеноцианатных соединений
Соединение цэф. M. Б. Литера- тура- Соединение цЭф, м. Б. Литера- тура
СоРу2 (NCSe)2 5,1 [96] FePhen2 (NCSe)2 5,13 [118, 137]
CoQ2 (NCSe)9 4,39 [90] NbDipy2 (NCSe)4 1,72 [126]
СоРу4 (NCSe)2 5,1 [96] Nb (dmDipy)2 (NCSe)2 1,86
5,06 [90] Nien2 (NCSe)2 3,24 [ПО]
Со (IQ)4 (NCSe)2 5,02 [ЮЗ] NiPy4 (NCSe)2 3,20 [102]
Cuen2 (SeCN)2 1,79 [ПО] Ni (0-Pic)4 (NCSe)2 3,10 »
Cu (CH3NH2)2 (SeCN)2 1,82 [131] Ni (y-Pic)4 (NCSe)2 3,06 »
CuDipv (SeCN)2 1,93 » NiPv2 (NCSe)2 3,28 »
CuPhen (SeCN)2 1,93 » Ni (CH3NH2)4 (NCSe)2 Ni (C3H7NH2)4 (NCSe)2 3,16 3,17 [135] »
CuDipy2 (SeCN)2 2,02 » Ni (C4H9NH2)4 (NCSe)2 3,12 »
Как видно из табл. 6.10, для селеноцианатов никеля характерна
Ni—NCSe-координация. Никель в рассматриваемых соединениях имеет,
как правило, октаэдрическую конфигурацию. Данные ИК-спектроскопии
хорошо согласуются с результатами исследования магнитных свойств этих
соединений (табл. 6.11).
Интересно отметить, что в присутствии пиперидина никель (И) может
образовывать не только октаэдрические (Ni (Pip)4 (NCSe)2), но и пентакоор-
динационные соединения (Ni (Pip)3 (NCSe)2) [217], а соединение [Ni (Et4 •
• dien) NCSe] Pl^B имеет плоскостное квадратное строение [218].
Изучение ИК-спектров поглощения соединений лантаноидов свидетель-
ствует о связи SeCN-групп с центральным атомом через азот (см. табл. 6.10).
6.8. СОЕДИНЕНИЯ С НЕЙТРАЛЬНЫМИ ЛИГАНДАМИ,
СОДЕРЖАЩИМИ ДОНОРНО-АКТИВНЫЙ КИСЛОРОД
Очевидно, к числу первых селеноцианатных соединений этого типа при-
надлежат соли кобальта и никеля, полученные при действии 1,4-диоксана
на ацетоновые растворы М (NCSe)2 : М (NCSe)2 • 6Diox (М = Со, Ni) и
Со (NCSe)2 - 4Diox [60—62]. Позднее были получены соединения, содержа-
щие в своем составе диметилформамид и диметилсульфоксид (табл. 6.12).
Эти соединения выделяются в виде хорошо образованных кристаллов при
кристаллизации ацетоновых растворов соответствующих селеноцианатов в
присутствии DMFA или DMSO или же при растворении селеноцианатов ме-
таллов (Cd, Pb) в DMSO. Описаны также соединения М (DMAA)4 (NCSe)2
(М = Mn, Со, Ni, Zn; DMAA = диметилацетамид) [207].
277
Таблица 6.12. Основные колебательные частоты SeCN-групп, см‘
Соединение v (CN) v (CSe) 6 (Se- CN) Тип координации SeCN-групп Лите- ратура
[Bi (DMSO)el (SeCN)3 2050, 2110 553 SeCN" [81]
Bi (DMFA)3 (SeCN)3 - 2DMFA 2110, 2123 545 — Bi — SeCN »
Cd (DMSO)? (NCSe)? 2115, 2131 580 — Cd — SeCN — Cd [142]
Co (DMFA)4 (NCSe)2 2033, 2047, 2092 612 413, 427 Co — NCSe [130]
Co (DMSO)4 (NCSe)2 2095 610 — » [142]
Fe (DMFA)4 (NCSe)2 2042, 2088 606 — Fe — NCSe [H7]
Fe (DMSO)4 (NCSe)2 2081, 2030 610 — » [142]
In (DMFA)3 (NCSe)3 2070 618 — In — NCSe [119]
In (DMSO)3 (NCSe)3 Mn (DMFA)4 (NCSe)2 2065, 2080 615 — »
2037, 2080 606 413 Mn — NCSe ИЗО]
Mn (DMSO)4 (NCSe)2 2075 610 — » [142]
Ni (DMFA)4 (NCSe)2 2025, 2034, 2059, 2105 614 415 430 Ni — NCSe » [130]
Ni (DMSO)4 (NCSe)2 2098 618 — [142]
Pb (DMSO)2 (NCSe)2 2096, 2141 543, 582 403 Pb — NCSe — Pb [101]
Th (DMFA)4 (NCSe)4 2070 678 — Th — NCSe [85]
UO2 (DMSO)3 (NCSe)? 2046, 2069 630 — U — NCSe [127]
Zn (DMSO)4 (NCSe)2 2075 647 430 Zn — NCSe [П5]
6.8.1. Соединения, содержащие диметилсульфоксид
и диметилформамид
Соединения М (DMSO)4 (NCSe)2 (М = Mn, Fe, Со, Ni, Zn) изоструктурны
[142] и содержат изоселеноцианатные группы (табл. 6.12). Частоты v (SO)
координированного DMSO проявляются в области 955—1020 см-1, что ука-
зывает на координацию DMSO через кислород. Таким образом, в М (DMSO)4 •
• (NCSe)2 центральный атом имеет октаэдрическую конфигурацию MO4N2,
вероятно, с транс-положением SeCN-группы [142].
Кристаллы [Cd (DMSO)2 (NCSe)2] относятся к ромбической сингонии с
параметрами элементарной ячейки: а — 14,59; b = 8,41; с = 12,25 А. Пик-
нометрическая плотность соли 2,18 г/см3 [198]. Из данных ИКС следует, что
соединение содержит координированный через кислород DMSO и мостико-
вые SeCN-группы [142]. Атомы кадмия соединяются в бесконечные цепи двой-
ными мостиковыми SeCN-группами. Молекулы DMSO дополняют полиэдр
до октаэдра. Расстояния равны: Cd—Se 2,773; Cd—N 2,26; Cd—О 2,30 A;
углы SeCN и CdNC соответственно равны 171,5 и 172° [208]. Аналогичное
строение имеет, вероятно, и [Pb (DMSO)2 (NCSe)2l [101].
В ИК-спектре [Bi (DMSO)6J (SeCN)3 найдены полосы только координи-
рованного DMSO, а значения v (CN) и v (CSe) близки к таковым SeCN-иона
[81].
В соединениях [In (DMSO)3 (NCSe)3] и [UO2 (DMSO)3 (NCSe)2] содержат-
ся изоселеноцианатные группы и молекулы DMSO, связанные с центральным
атомом через кислород (см. табл. 6.12). Рассмотренные только что соединения
очень напоминают диметилформамидные комплексы, среди которых первыми
были получены М (DMFA)4 (NCSe)2 (М = Mn, Со, Ni) [1411. Несколько позд-
нее осуществлен синтез аналогичной соли железа (И) [79]. Эти соли оказа-
278
Рис. 6.2. Структура Ni (DMFA)4 (NCSe)2 (проекция на плоскость
(001))
лись изоструктурными [117, 141]. В них SeCN-группы, занимая транс-по-
ложение, связаны с центральным атомом через азот, а молекулы DMFA —
через кислород (см. рис. 6.2). Некоторые свойства этих соединений представ-
лены в табл. 6.13, а детальное описание структуры Ni (DMFA)4 (NCSe)2 дано
в работе [1461.
Во всех других диметилформамидных соединениях, представленных в
табл. 6.12, координированный DMFA связан с металлом через кислород,
а SeCN-группы (за исключением соли висмута) — через азот. В соединении
[Bi (DMFA)3 (SeCN)3l • 2DMFA группы SeCN связаны с висмутом через
селен.
В ИК-спектрах аналогичных по составу с М (DMFA)4 (NCSe)2 соединений
марганца, кобальта, никеля и цинка, содержащих N-метилформамид (M(MFA)4*
Таблица 6.13. Некоторые свойства кристаллов М (DMFA)4 (NCSe)2
Соединение М= Мп М= Ni м = с®
Параметры элементарной ячейки, А а 12,70 12,696±0,012 12,83
b 12,30 12,380 ±0,003 12,30
с 7,35 7,364 ±0,026 7,40
Углы а 90° 90° 6'±0,6' 90°
(3 90° 90° 49'± Г 90°
у 92° 30' 92° 25'±4' 92° 30'
Число формульных единиц 2 2 2
Показатели преломления Np 1,587 1,588 1,594
Nm 1,610 1,614 1.611
Nc 1,615 1,641 1,641
Магнитный момент, М. Б. 6,04 3,22 5,07
Плотность, г/см3 1,63 1,61 1,60
Температура плавления, °C 91 124 разл. 72
279
• (NCSe)2), полосы v(CN) и v (CSe) лежат в областях 2075—2115 и 600—
610 см””1 соответственно, что указывает на связь SeCN-групп с центральнь •
атомом через азот. Молекулы MFA координированы через кислород [183].
В изоструктурных М (DMAA)4 (NCSe)2 (М = Mn, Со, Ni, Zn) диметил-
ацетамид связан с М через кислород, а SeCN-группы — через азот. Для моно-
клинных кристаллов Ni (DMAA)4 (NCSe)2 определены параметры решетки:
а = 7,67; b = 10,40; с = 9,74 А; у = 114° [207].
6.8.2. Соединения, содержащие
гексаметилфосфортриамид
Селеноцианатные соединения, содержащие НМРА, легко образуются при
действии названным реагентом на ацетоновые или спиртовые растворы селе-
ноцианата соответствующего металла. В ИК-спектрах жидкого НМРА
v (РО) лежит при 1208 см-1, в спектрах соединений v (РО) наблюдается
около 1190—ИЗО см-1, что указывает на связь НМРА с катионами метал-
лов через атом кислорода.
Как видно из табл. 6.14, значения v (CN) и v (CSe) для всех соединений,
кроме кадмия и никеля, указывают на наличие связи М—NCSe. Из рентгено-
графических данных следует, что соединения М (НМРА)2 (NCSe)2 (М =
= Mn, Со, Zn) изоструктурны [143]. В электронном спектре поглощения
нитрометанового раствора соли кобальта отмечается максимум около
16 000 см~‘ (переход 4А2 —> 4Т (Р)), что характерно для тетраэдрической кон-
фигурации кобальта. Значение магнитного момента (4,65 М. Б. [2201) для
этой соли хорошо согласуется с тетраэдрической конфигурацией кобальта.
Учитывая это, результаты ИК-спектроскопии, а также изоструктурность сое-
динений марганца, кобальта и цинка, можно утверждать, что в М (НМРА)2 •
• (NCSe)2 центральный атом имеет тетраэдрическое окружение [143, 220].
Для Сг (НМРА)3 (NCSe)3 определено значение магнитного момента)
равное 3,69 М. Б. В электронном спектре поглощения (в видимой области,
раствора этого соединения в хлористом метилене наблюдаются две полосы
Таблица 6.14. Основные колебательные частоты SeCN-групп и РО-групп, см-1
Соединения v (PO) v (CN) v (CSe) 6 (SeCN) Тип координации SeCN-групп Лите- ратура
Bi (НМРА)3 (NCSe)3 1190 2100 >640 Bi — NCSe [144]
Cd (НМРА) (SeCN)2 1195 2125, 2080 575 — Cd-SeCN-Cd Cd — NCSe [143]
Со (НМРА)2 (NCSe)2 1192 2082 650 425 Co — NCSe »
Cr (НМРА)3 (NCSe)3 1190 2080, 2095, 2110 >620 405, 442 Cr — NCSe [144]
In (НМРА)3 (NCSe)3 1195 2075, 2095 650 432 In — NCSe »
La (НМРА)3 (NCSe)3 1195 2070 605 426 La — NCSe »
La (HMPA)4 (NCSe)3 1195 2070 605, 645 410, 426 » »
Mn (HMPA)2 (NCSe), 1192 2065 655 420 Mn — NCSe [143]
Ni (HMPA)2 (NCSe)2 1195 2138, 2110 620, 560 425 Ni —NCSe—Ni Ni — NCSe »
Ni(HMPA)2(H2O)2(NCSe)2 1192 2110, 2070 632 482 Ni — NCSe [220]
Sc (HMPA)3 (NCSe)3 1195 2060, 2085 650 440 Sc — NCSe [144]
Y (HMPA)3 (NCSe)3 1190 2050, 2060, 2070 635 436 Y — NCSe »
Zn (HMPA)2 (NCSe)2 1195 2090 650 425 Zn — NCSe [143]
280
около 22 500 и 15 400 см-1. Следовательно, хром в рассматриваемом соедине-
нии имеет симметрию Оь [144].
Вероятно, во всех других соединениях М (НМРА)3 (NCSe)3 координацион-
ное число центрального атома равно 6, а в La (НМРА)4 (NCSe)3 — 7, по-
скольку в ИК-спектрах обнаружены полосы только координированных
SeCN-групп и НМРА [144].
Несколько неожиданной является Bi—NCSe-координация, поскольку
для висмута более характерно образование связей Bi—SeCN [37]. Очевидно,
обращение SeCN-групп в данном случае вызвано стерическими препятствия-
ми, создаваемыми НМРА. Из-за присутствия НМРА связь Bi—SeCN (угол
BiSeC должен быть 90—120°) становится менее выгодной, чем связь
Bi—NCSe, поскольку в этом случае угол BiNC должен быть близким к линей-
ному [37, 1581.
В ИК-спектрах Ni (НМРА)2 (NCSe)2 и Cd (НМРА) (NCSe)2 присутству-
ют высокочастотные компоненты v (CN), расположенные соответственно при
2138 и 2125 см-1 (табл. 6.14). Это указывает на наличие в этих соединениях
мостиковых SeCN-групп. Для соли никеля полосы v (CN) и v (CSe) расщеп-
лены на две компоненты, что, по-видимому, обусловлено наличием как мости-
ковых, так и немостиковых SeCN-групп. Соль никеля не изоструктурна ана-
логичным солям кобальта, цинка, марганца. С другой стороны, значение маг-
нитного момента, равное 3,6 М. Б., исключает плоскостное строение
Ni (НМРА)2 (NCSe)2. Вероятно, никель в рассматриваемом соединении за
счет мостиковых SeCN-групп имеет координационное число 5. На воздухе
соединение Ni (НМРА)2 (NCSe)2 легко присоединяет воду, превращаясь в
дигидрат. В последнем, судя по электронным спектрам поглощения и маг-
нитному моменту (3,02 М. Б.), никель имеет октаэдрическую конфигурацию,
а NCSe-группы связаны с центральным атомом через азот [220].
В ИК-спектре соединения кадмия обнаружены две полосы v (CN) и одна
v (CSe). Можно полагать, что в Cd (НМРА) (NCSe)2 имеются мостиковые и
изоселеноцианатные SeCN-группы с тетраэдрическим окружением кадмия
[(СН3)2 N]3 РО SeCN NCSe
CdZ Cd^
SeCNZ ^NCSe^ ^OP [N (CH3)2]3
6.8.3. Другие селеноцианаты
В последние годы получено большое число селеноцианатных соединений,
содержащих диантипиринметан. При этом синтез соединений лантаноидов
осуществлен из этанольных [124], цинка, индия, урана — из ацетоновых или
метанольных, галлия — из тетрагидрофурановых растворов.
Применение рентгенографического метода показало, что соединения ана-
логичного состава (Ln (Diantp)3 (NCSe)3, Ln (Diantp)4 (NCSe)3 (Ln = La,
Ce, Pr, Nd)) изоструктурны. На кривой радиального распределения атомной
плотности для La (Diantp)3 (NCSe)3 имеется четкий максимум при 5,0 А,
что соответствует связи Le—NCSe. Наоборот, в соединении La (Diantp)4 •
• (NCSe)3 такой максимум отсутствует [124]. Таким образом, в соединениях
Ln (Diantp)2 (NCSe)3, Ln (Diantp)3 (NCSe)3 присутствуют изоселеноцианат-
ные группы, что находится в соответствии с данными ИК-спектроскопии
(табл. 6.15). Поскольку из ИК-спектров следует, что Diantp занимает два коор-
динационных места, то это означает, что в рассматриваемых соединениях
281
Таблица 6.15. Основные колебательные частоты SeCN-групп, см'
Соединение v (CN) v (CSe) 6 (SeCN) Тип коорди- нации SeCN-групп Литера- ! тури
Со {OP (Ph3)}2 (NCSe)2 2066 — Co — NCSe [901
[CrU6j (SeCN)» • H2O 2065, 2095 — — SeCN- [97]
Ga (Atp)3 (NCSe)3 2070 625 — Ga —NCSe [119]
Ga (Diantp) (NCSe)3 2062, 2082, 2100 620 — » »
Ga {OP (C3H7)3}3 (NCSe)3 2070, 2110 617,650 432 » [1161
Ga {(OP) Ph3}3 (NCSe)3 2070, 2055 618 — » »
Cu (OH?CPy) NCSe 2110 625 442 Cu — NCSe [145]
In (Diantp)3 (NCSe)3 2060 628 — In — NCSe [119]
In (Atp)3 (NCSe)3 In {OP (Ph)3}3 (NCSe)3 2070 650 — » [99]
2062 618 —- » [119]
In {OP (C3H7)3}3 (NcSe)3 2090, 2060 615,650 427 » [116]
Ln (Diantp)2 (NCSe)3 2050—2060 — — Ln — NCSe [124]
(Ln = Ce, La, Lu, Nd) Ln (Diantp), (MCSe)3 2060—2080 —
(Ln = Ce, Pr, Eu, La, Lu, Gd, Nd, Sm) Ln (Diantp)4 (NCSe)3 2060—2065 SeCN- »
(Ln = Ce, Pr, La, Lu, Nd) Sc (Diantp)3 (NCSe)3 Sc (Diantp)4 (NCSe)3 2060 2052, 2062 Sc—NCSe SeCN- » »
UO2 (Diantp)2 (NCSe)2 2060, 2070 — —. U—NCSe- [127]
UO2Atp3 (NCSe) 2 2055 592 SeCN- U—NCSe В
UO9Py3 (NCSe)2 2060 — — » »
Y (Diantp)2 (NCSe)3 2065 — —• Y—NCSe [124]
Y (Diantp)3 (NCSe)3 2070, 2085 — —- » »
Y (Diantp)4 (NCSe)3 2065 — — SeCN- »
Zn Atp2 (NCSe)2 2072 655 428 Zn—NCSe [115]
к. ч. Ln больше 6. В соединениях [Sc (Diantp)4] (SeCN)3, [Y (Diantp)J •
• (SeCN)3 и [Ln (Diantp)J (SeCN)3 селеноцианатные группы находятся во
внешней сфере.
В соединениях индия, галлия, урана Diantp также занимает два места,
координируясь через кислород, о чем свидетельствует понижение v (СО)
координированного Diantp (~ 1600 см-1) по сравнению с v (СО) реактива
(1656 см-1). С другой стороны, данные ИКС указывают на наличие изоселе-
ноцианатных групп в рассматриваемых соединениях (табл. 6.15). Таким об-
разом, строение этих соединений можно представить как [In (Diantp)2 •
• (NCSe)2] NCSe, [UO2 (Diantp)2 NCSe] NCSe и [Ga (Diantp) (NCSe)2] NCSe
или [Ga (Diantp)2] [Ga (NCSe)6L Аналогично Diantp пирамидон и антипи-
рин образуют с центральным атомом связи через кислород, занимая при этом
одно координационное место. Соединение хрома с мочевиной — единствен-
ное (среди приведенных в табл. 6.15), полученное из водного раствора. Оно
содержит SeCN-ионы и молекулы мочевины, координированные посредством
кислорода.
Соединение Со [OP (Ph3)}3 (NCSe)2 выделяется в виде синих кристаллов
после прибавления спиртового раствора Со (NCSe)2 к раствору ОР (СеН5)3
в этилацетате и последующего охлаждения полученной смеси. Оно оказалось
изоструктурным с аналогичным тиоцианатом, для которого установлено тет-
раэдрическое строение [90].
282
В соединениях галлия, индия с фосфороксидами центральный атом
имеет октаэдрическое окружение, при этом связь органического лиганда
осуществляется через кислород, SeCN-групп — через азот.
6.9. СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ СВЯЗИ
МЕТАЛЛ — ФОСФОР, МЕТАЛЛ — УГЛЕРОД
Если к раствору хлорида диарилталлия в пиридине приливать ацето-
нитрильный раствор KSeCN, образуются соединения R2TlSeCN (R-фенил,
о-, т-, р-толил), в которых, судя по данным ИКС, SeCN-группы связаны
с таллием через селен [221]. Соединение Ph2Tl (SeCN) может быть получено
из бензольных или хлороформных растворов при взаимодействии Ph3Tl
и Se (SeCN2) [222]. Аналогичным образом получены соединения R3PbSeCN
(R = Ph, Me, Et) [222]. Взаимодействием Ph3ECl c KSeCN (E = Pb, Sn)
получены Ph3ESeCN [222]. Селеноцианатные группы в этих соединениях
связаны со свинцом через селен, а с оловом — через азот [222].
Обменной реакцией между соответствующими хлоридными соединениями
и KSeCN из ацетоновых растворов получены транс-Rh {Р (С6Н5)3}2 (СО) NCSe,
Ir {Р (С6Н5)3}2 (СО) (Act) NCSe, AuL (SeCN)(L = (C6H5)3P или (C6H5)3As),
в которых SeCN-группа связана с родием и иридием через азот, а с золотом —
через селен [223].
Осуществлен синтез и изучены свойства соединения Pd {Р(С6Н5)3}2 (SeCN)2.
По мнению авторов работы [224], в твердом состоянии, а также в DMSO и
DMFA оно имеет связи Pd—SeCN, в то время как в хлороформе, дихлор-
метане часть SeCN-групп связана с палладием через азот.
6.10. КООРДИНАЦИОННЫЕ СЕЛЕНОЦИАНАТЫ
АНИОННОГО ТИПА
6.10.1. Препаративные данные
Одними из первых среди селеноцианатов этого типа были получены соли
ртути KHg (SeCN)3 и K2Hg (SeCN)4 [36, 49, 53]. При действии на водные
растворы K2Hg (SeCN)4 растворами солей металлов (II) образуются трудно-
растворимые соединения М [Hg (SeCN)J(M = Ni, Zn, Co, Pb, Cu, Cd)
[49, 72]. По данным авторов работы [148], растворимость солей MHg (SeCN)4
в воде, ацетоне и водно-спиртовых растворах уменьшается в ряду Cd >» Со >
>> Zn. Произведение растворимости CuHg (SeCN)4 в воде равно 1,49 • 1О~10
[149].
Из водных растворов выделены К [Hg (SeCN) (CN)2], К [Hg (SeCN) Hal]
(Hal = Cl, Br, I) [36], К [Hg (SeCN)2 (CN)] [150], M [Hg (SeCN)3]2(M =
= Zn, Cu) [72].
Соли кобальта, никеля, цинка, свинца, кадмия и меди при взаимодействии
с водными растворами NaSeCN, насыщенными иодидом ртути, образуют
окрашенные осадки. В чистом виде получены соединения кобальта и никеля
MHg7 (SeCN)uI7, образование которых может служить для качественного
определения этих металлов [151].
При насыщении водных растворов KSeCN селеноцианатом серебра была
выделена соль KAg2 (SeCN)3 [67]. В других концентрационных условиях
из водного раствора получены KAg (SeCN)2, K«Ag (SeCN)3, K3Ag (SeCN)4
[67], а также KAg2 (SeCN)2 (SCN) и K2Ag (SeCN) (SCN)2 [64].
283
При насыщении водно-ацетонового раствора KSeCN селеноцианатов
свинца и последующей кристаллизации раствора удалось получи’:
K4Pb (SeCN)6 [51 ]. Из водно-спиртовых растворов, содержащих соль кобаль-
та, диметилглиоксим и KSeCN, выделена соль К [Со (DH)2 (SeCN)2] • ЗН2'
[152]. Раствор ее образует характерные осадки с катионами NH4", Cd‘~
Fe2+, Со2+, Ni2+, в присутствии кислоты выделяет Н [Co(DH)2 (SeCN)2l,
а при действии щелочи — [Со (DH)2 SeCNH2O] Н2О [152].
Окислением кислородом воздуха водно-спиртового раствора ацетата ко-
бальта, KSeCN и 1,2-циклогександиолдиоксима с последующей обработ-
кой раствора соляной кислотой выделена плохо растворимая в вод
Н [Со (NiOxH)2 (NCSe)2 Н2О. Получены также многочисленные соли это’
кислоты [153].
Давно известен селеноцианат платины (IV) K2Pt (SeCN)6 • 2Н2О, полу-
ченный из водного раствора обменной реакцией между K2PtCl6 и KSeCN
[154]. Эта соль растворима в воде, спирте, причем при перекристаллизации
или получении из спирта выделяется безводная соль [155, 156]. В виде
черных ромбических кристаллов получена также аммонийная соль
(NH4)2Pt (SeCN)6 [157].
При сливании спиртовых растворов селеноцианата калия и хлорида зо-
лота образуется осадок, состоящий в основном из селена. Из фильтрата мож-
но получить темно-красные призматические кристаллы, которым приписан
состав KAu (SeCN)2 [155]. Из-за быстрого разложения соли точно установить
ее состав не удалось [155].
В последние годы получено много соединений рассматриваемого типа.
Большинство из них содержит крупные катионы [57, 158, 159]. Так, для
синтеза соединения [Ph4As] [Au (SeCN)4] в хлористом метилене смешивали
КАиС14 и KSeCN, а затем к полученной смеси добавляли [Ph4 As] Cl [161].
Из ацетона обменной реакцией между KSeCN и транс- [(CH3)4N ] [Au (CN)2C12]
выделено соединение TpaHc-[(CH3)4N] [Au (CN)2(SeCN)2] [160]. Из спиртовых
растворов выделены соли [(С2Н3)4 N]2 [Zn (NCSe)4] [172], [(C4H9)4N]2 •
• [Zn (NCSe)4], [(C4H9)4N]2 [Cd2 (SeCN)6l [162]; из ацетоновых —
KCd (SeCN)3 Act, K2Cd (SeCN)4 • 2Act, K2Cd (SeCN)4, K4Cd (SeCN)6, а также
соли, содержащие в своем составе, кроме SeCN-группы, также I- или SCN-
группы [163]. Описан синтез соли [(С16Н33) (CH3)3N] [Hg (SeCN)4] [161].
Из ацетоново-диоксанового раствора получена соль Na2 [Zn (NCSe)4] • 6Diox
[115].
Долгое время не было никаких сведений о селеноцианатах элементов III
группы. Только в 1966 г. появилось сообщение о синтезе из этанола соеди-
нения [(C4H9)4N]3 [Y(NCSe)6] путем взаимодействия нитрата иттрия с
[(C4H9)4N] SeCN [162]. Вслед за этим была получена аналогичного состава
соль празеодима [164], а затем соли [(C4H9)4N]3 [Ln (NCSe)6] (Ln = Nd, Sm,
Dy, Ho, Er) [165]. Примерно в это же время были получены из спиртовых
или ацетоновых растворов соли индия Kat3 [In (NCSe)6] (Kat = (CH3)4N‘,
(QH5!,N+, (C4H„)4N+, (С„Н5)4А»+) [991, a из тетрагидрофурановых — соли
галлия [(C4H9)4N]3 Ga (NCSe)6 [119], Ba [Ga(NCSe)4]2 • 6Thf, [(C6H5)4 As]3
[Ga(NCSe)6] [116].
Из диметилформамидных растворов удалось осуществить синтез солей
тория Na2Th (NCSe)6 • 3DMFA, K2Th (NCSe)6 • 4,5 DATA и K4Th (NCSe)8 •
• DMFA [84], которые оказались весьма неустойчивыми на воздухе.
Из металлов IV группы соединения рассматриваемого типа известны
лишь для титана и свинца. Кроме K4Pb (SeCN)6, из ацетонового раствора
получена в виде шелковистых игл соль KPb (SeCN)3 [58]. Из этанольных
284
растворов в виде темно-красного порошка получена соль [(C4H9)4N]3 •
. Ti (NCSe)6 [1661.
Значительно лучше изучены селеноцианаты элементов V группы. Уже
известны соли ванадия (III) ([R3V (NCSe)6] (R = (CH3)4N+, (СгН6)4Н+),
K3V (NCSe)6 7Diox [98], [(C4H9)4N]3 [V(NCSe)6] [166, 199], [(C6H5)4 As]3 •
• [V(NCSe)6] [199]), ванадия (IV) [(C4H9)4N]2 [VO (NCSe)4J [166]; K2VO •
• (NCSe)4-6Diox и Na2VO (NCSe)4 • 2Py [100]. Из ацетонитрильных раство-
ров выделены соединения К [М (NCSe)e] (М = Nb, Та), [(С6Н5)4 As] [Та •
• (NCSe)6], а также K2Nb (NCSe)6 [167].
Из ацетона получены также соединения висмута: K2Bi (SeCN)5,
Kat3 [Bi (SeCN)6] (Kat = K+, (CH3)4N+, (C2H5)4N+, (C4H9)4N+) [120],
Na3Bi (SeCN)6, Li3 [Bi (SeCN)6] - 3Act, KBa [Bi (SeCN)6] - 2Act [168]. Из
ацетоново-диоксанового раствора выделены соединения Li2Bi (SeCN)5 •
• 3Diox и M3 [Bi (SeCN)6] • n Diox (M = Mg, Ca, Sr, Ba, Mn; n = 5 4- 10)
[168]. Синтезированы также соединения смешанного типа К3 [Bi (SeCN)3 •
• (SCN)s], K2Bi (SeCN)2 (SCN)3, KBi (SCN)3 (SeCN) [169].
Из элементов VI группы наиболее изученными являются селеноциа-
наты хрома. Первыми из них были синтезированы из этанола соединения
I(CH3)4N]3 [Сг (NCSe)J и [(CH3)4N]2 [Сг (NCSe)5 Н2О] [170], затем осуществ-
лен синтез KsCr (NCSe)6 3Diox, Na3 [Сг (NCSe)6]-nAct, Ag3 [Cr (NCSe)6] •
• 5H2O, [Cren3] [Cr (NCSe)6] [97]. Кроме хрома, селеноцианаты анионного
типа образует молибден. До сих пор известны лишь три соединения: желтые
соли [(CH3)4N]3 [Мо (NCSe)6] [161], [(C2H5)4N]3 [Мо (NCSe)6] [200] и красно-
фиолетовая [(C2H5)4N ]2 [МоО (NCSe)5] [85].
Недавно появилось сообщение о синтезе селеноцианатного соединения
рения [(C4H9)4N ]2 Re2 (NCSe)8, полученного при взаимодействии [(C4H9)4N ]2 •
• Re2 С18 с селеноцианатом натрия. Судя по величине v (CN), равной
2050 см-1, SeCN-группы связаны с рением через азот [171].
Из ацетонового раствора получено соединение [Re (NCSe)4Cl ] • 0,5 Act,
в котором SeCN-группы, судя по данным ИК-спектров, мостиковые [209].
Интересно отметить, что в ацетоне пентахлорид рения образует в присутст-
вии SeCN-ионов комплексы полимерного строения также за счет мостиковых
SeCN-групп [209].
Из элементов VII группы лучше всего изучены селеноцианаты марганца.
Так, из этанольных растворов синтезированы соли [(C4H9)4N]4 [Mn (NCSe)6]
1162] и [(CH3)4NI4 [Мп (NCSe)6] [172]; соединение Kat2+Mn (NCSe)4 получено
при действии ацетонового раствора Mn (NCSe)2 на раствор Kat2+ (NCSe)2
в нитрометане (Ка12+ = ксилилен-бис(трифенилфосфоний)-ион) [172]. Из
смеси ацетон-дноксан (1 : 1) получена соль K4Mn (NCSe)6 • 6Diox [76].
В твердом состоянии выделены также [Nien3]2 Мп (NCSe)6 и [Cuen2] •
• [Mn (NCSe)g] [108].
Сравнительно хорошо изучены координационные селеноцианаты ко-
бальта, никеля, железа. Из смеси ацетон — диоксан выделены соедине-
ния К2Со (NCSe)4 • 3Diox, Na2Co (NCSe)4 • 7Diox [61], из ацетона —
Na4Ni (NCSe)6 • 2Act [62]. При действии ацетонового раствора М (NCSe)2
на раствор Kat2+ (NCSe)2 в нитрометане получены соли Kat2+ М (NCSe)4,
из этанольного раствора выделены [(CH3)4N]4M (NCSe)6 (М = Fe, Со, Ni).
Получены также соли [(C2H5)4N]2 Со (NCSe)4 и [(C2H5)4N]4Ni (NCSe)6 [172],
l(C2H5)4N]4Co (NCSe)e и [(C2H5)4N]3 Fe (NCSe)5 • H2O [200]. Из этанола
осуществлен синтез [(C6H5)4N]2 Со (NCSe)4 [53, 90, 105]. Для получения
селеноцианатов рассматриваемых металлов использована также соль
[(C4H9)4N ] NCSe. При этом из этанольных растворов были выделены соли
285
Kat2Fe (NCSe)4, Kat3Fe (NCSe)6, Kat4Ni (NCSe)6 (Kat=тетрабутил аммо-
ний-ион) [162]. Соль Kat3Fe(NCSe)6 описана также в работе [161]. Из ацетона
получены соли K4[Fe(NCSe)6] • 2Act [117] и [(C4H9)4N]3 [Fe (CN)fiNCSe] [225].
При взаимодействии К2 [Со (CN)5J с К2 [Hg (SeCN)4] из водного раствор
в атмосфере азота получена соль К3 [Со (CN)5 NCSe] [111]. Авторы считают,
что желто-коричневое вещество К3 [Со (CN)6 NCSe] образуется по схеме
Co(CN)s~ + Hg(SeCN)?- -> [(NCSe)s Hg—SeCN—Co (CN),]-5 ->
-* [Co (CN)6NCSeF- + Hg2 (SeCN)2. (6.9)
Кроме рассмотренных выше селеноцианатов платины, из неводных сред
синтезированы [(CH3)4N]2 Pd (SeCN)4 [172], Kat [M (SeCN)4] (M = Pd, Pt),
Kat3Rh (SeCN)6 (Kat = (C4H9)4 N+) [162], [(C2H5)4 N]2 Pd (SeCN)4,
[(C2H5)4 N]2Pt (SeCN)6, [(CHS)4 N]2Pt (SeCN)6, Cs2Pt (SeCN)6 [173],
[(C2H5)4 N]2Pt (SeCN)4 [161, 201].
Все описанные выше соединения можно разделить на три группы: изо-
селеноцианаты, селеноцианаты и соединения, содержащие мостиковые
SeCN-группы.
6.10.2. Анионные комплексы с мостиковыми
SeCN-группами
Среди известных селеноцианатов анионного типа мостиковые SeCN-
группы содержат преимущественно комплексы ртути и кадмия (табл. 6.16).
Так, в соли K2Cd (SeCN)4 кадмий окружен четырьмя атомами селена, до
октаэдрической координации дополняется еще двумя атомами азота сосед-
них SeCN-групп. Здесь, по-видимому, наблюдается такая же картина, как
и в K2Cd (SCN)4 • 2Н2О [175, 176].
Мостиковые SeCN-группы присутствуют и в [(C4H9)4N2] • [Cd2 (SeCN)6L
По мнению авторов работы [162], кадмий в этом соединении имеет тетраэд-
рическую конфигурацию CdN3Se за счет двух мостиковых SeCN-групп и двух
концевых SeCN-групп, связанных с кадмием через азот.
Соединение Ag3Cr (NCSe)6 • 5Н2О является полиядерным комплексом,
в котором хром образует связи с SeCN-группами через азот, а серебро —
через селен.
Из соединений висмута, в которых реализуется мостиковая координация
NCSe-лигандов, можно назвать пентаселеноцианатовисмутит лития и гекса-
селеноцианатовисмутит магния (табл. 6.16).
Сравнение рентгенограмм соединений М [Hg (SeCN)3]2 (M=Cu, Pb) ука-
зывает на их неизоструктурность [174], а из данных ИК-спектроскопии
следует, что они содержат мостиковые SeCN-группы.
Кристаллы CoHg (SeCN)4 имеют игольчатую форму с квадратным сече-
нием и относятся к тетрагональной сингонии [177]. Установлено, что это
соединение изоструктурно такому же тиоцианату [90, 178]: CoHg (SeCN)4:
а = b = 11,29 А; с = 4,67 A; CoHg (SCN)4: а = b= 11,09 А; с = 4,37 А-
Это означает, что CoHg (SeCN)4 содержит мостиковые SeCN-группы, при-
чем атом кобальта окружен по тетраэдру четырьмя атомами азота, а каждый
атом ртути — четырьмя атомами селена. Рассматриваемые соединения сход-
ны друг с другом по растворимости в воде, окраске, величине магнитного
момента 4,32 М Б. для CoHg (SeCN)4 и 4,34 М Б. для CoHg (SCN)4 [90].
На наличие мостиковых SeCN-групп в соединении CoHg (SeCN)4 четко ука-
зывают и данные ИК-спектроскопии (см. табл. 6.16). Рентгенографическое
286
Таблица 6.16. Колебательные частоты SeCN-групп в некоторых координационных соедине-
ниях, см1
Соединение v (CN) v (CSe) 6 (SeCN) 26 (SeCN) Лите- ратура
Li2Bi (SeCN)5 • 3Diox 2085, 2130 545, 573 403, 415 [184] »
Mgs [Bi (SeCN)6]2 - 9Diox 2131 — 406 —
K2Cd (SeCN)4 2096, 2112, 2125, 2131 548, 537 420 755, 792 [129]
l(C4H9)4 N2] Cd2 (SeCN)6 2125, 2109 589, 582 417, 408 — [162]
Cd [Hg (SeCN)4] 2109, 2126, 2141 606 407 790, 818 [45]
Co [Hg (SeCN)4] 2146 637 403, 417 810, 840 [45, 90] [45]
Cu [Hg (SeCN)4] 2148 563, 597 405 780
Cu [Hg (SeCN)3]2 2141, 2160 570,592,697 407, 417 773, 795 [174]
Kat2+ Ni (SeCN)4 2075, 2092, 2145 653 429 — [172]
Pb [Hg (SeCH)4] 2122 545,564,582 400 772, 810 [174]
Pb [Hg (SeCN)3]2 2124, 2140, 2158 535, 545 405 810 [174, 210] [45]
Zn [Hg (SeCN) J • H2O 2100, 2155 627 407, 417 802, 833
Ag3 [Cr (NCSe)6] • 5H2O 2140 550, 655 430 782, 850 [97]
KCd (SeCN)3 2096, 2113, 2122, 2132 540, 547 405, 420 772, 787, 808, 831 [210]
KHg (SeCN)3 2122, 2142 535, 543 — 760, 800
KPb (SeCN)3 2080, 2118, 2140 532, 579 420 780, 860 [101]
исследование соли ZnHg (SeCN)4 • Н2О показало, что удаление воды не на-
рушает ее структуру. Установлено, что соль ZnHg (SeCN)4 изоструктурна
описанной выше соли кобальта [179, 180]. Параметры кристаллической ре-
шетки ZnHg (SeCN)4 равны: а = b = 11,2; с= 4,6 А. Из кривой радиаль-
ного распределения атомной плотности определены расстояния: Hg — Se
2,6 A; Hg—Hg 4,7 A; Zn—Se 5,0 A; Zn—N 2,1 А. Углы NCSe и ZnNC
близки к 180°, угол HgSeC 109° [180].
Строение аналогичной по составу соли кадмия существенно отличается
от строения цинкового соединения [181] (а = 8,19; с = 4,62 А; расстояния:
Hg—Se 2,7 A; Cd—Se 4,9 A; Cd—N 2,2 А; углы CdNC 170°; HgSeC 105°)
[181]. В CdHg (SeCN)4 кадмий, по-видимому, имеет октаэдрическую конфи-
гурацию. Дополняющими лигандами здесь могут быть атомы селена SeCN-
групп.
Мостиковые группы HgSeCNPb присутствуют также в соединении
PbHg (SeCN)4, хотя, судя по значению v (CN), связь Pb—NCSe несколько
ослаблена по сравнению со связью М—NCSe в аналогичных солях кобальта,
меди, цинка (табл. 6.16).
Соединения CuHg (SeCN)4 и CuHg (SCN)4 изоструктурны [174], следова-
тельно, селеноцианатное соединение имеет строение, аналогичное зеленому
CuHg (SCN)4, в котором атом меди окружен по квадрату четырьмя атомами
азота; дополнение до октаэдра осуществляется двумя соседними SCN-
группами, связанными с медью через атом серы [182]. Для меди как в
CuHg (SCN)4, так и в CuHg (SeCN)4 реализуется искаженный октаэдр, а для
ртути — некоторое промежуточное состояние между sp3- и d2sp3-ra6pHflH3a-
циями. Параметры решетки CuHg (SeCN)4 (а = 9,32; b = 6,97; с = 15,37 А;
Z = 4) [174] близки к таковым для CuHg (SCN)4 [182].
Весьма интересен тот факт, что в желтом соединении Kat2+ Ni (NCSe)4
присутствуют мостиковые SeCN-группы, следовательно, никель имеет окта-
287
эдрическую конфигурацию, а аналогичное тиоцианатное соединение по-
лучено в виде двух изомеров — тетраэдра и октаэдра [172]. В электронном
спектре поглощения Kat2+Ni (NCSe)4 наблюдаются полосы при 25 000.
~ 18 800, 13 800 и 6300 см-1, что характерно для комплексов никеля cfv-
метрии Оь- На октаэдрическую конфигурацию никеля в этом соединении ука-
зывает и величина магнитного момента (3.17 М. Б. при 290 К) [172].
Рассмотрим соли состава KM(SeCN)3 (М = Cd, Pb, Hg). Согласно рен~-
генографическим данным они изоструктурны [101, 129, 174]. Соль KHg -
• (SeCN)3 изоструктурна также соответствующему тиоцианату. Пара-
метры решетки KHg (SeCN)3 (а = 11,20; b = 4,30; с = 11,4 А; р = 115е
пространственная группа Р 21/m; Z = 4) [174] близки к таковым KHg (SCN),
[188]. В ИК-спектрах КМ (SeCN)2 обнаружены высокочастотные компонент
v (CN), относящиеся к мостиковым SeCN-группам (табл. 6.16).
6.10.3. Анионные селеноцианатные комплексы
Как видно из табл. 6.17, к рассматриваемой группе соединений относятся
селеноцианаты платиновых металлов, висмута, серебра, золота, ртути и час-
тично кадмия.
Кристаллы KAg (SeCN)2, принадлежащие к ромбической сингонии
(а = 19,71; b = 10,37; с = 7,20 А) [187], судя по данным ИК-спектроскопии,
содержат только селеноцианатные группы [130]. Такой же характер связи
SeCN-групп сохраняется в соединениях золота (III) (см. табл. 6.17).
Данные ИК-спектроскопических и рентгенографических исследований
М3 [Bi (SeCN)6j (М = Na+ [184] или К+ [140]) свидетельствуют о наличии
связи Bi—SeCN, причем расстояние Bi—Se в этих солях равно 2,7 А.
Выше было показано, что в селеноцианатных соединениях кадмия амин-
ного типа присутствуют изоселеноцианатные группы. В соли же К4 [Cd •
• (SeCN)6l лиганды связаны с кадмием через селен. В области 2000—
2200 см-1 был снят спектр концентрированного ацетонового раствора этой
соли [129]. В нем отмечаются интенсивная узкая полоса при 2070 см-1, две
полосы меньшей интенсивности при 2110 и 2145 см-1. Первая полоса отно-
сится к SeCN-иону и обусловлена диссоциацией Cd (SeCN)o~, вторая свиде-
тельствует о связи Cd—SeCN. Наконец, полоса при 2145 см-1 может ука-
зывать на наличие мостиковых SeCN-групп, например в Cd (SeCN)2~, воз-
никших вследствие диссоциации исходной соли.
Из приведенных выше данных видно, что ртуть даже в соединениях
с мостиковыми SeCN-группами всегда образует связи Hg—Se. Не являются
исключением и две соли, ИК-спектральные данные которых представлены
в табл. 6.17. Укажем также, что соль K2Hg (SeCN)4 изоструктурна K2Hg •
• (SCN)4 [174, 175]. Ртуть в тетраселеноцианатомеркурате калия имеет тет-
раэдрическое окружение HgSe4. Параметры о кристаллической решетки
K2Hg (SeCN)4 (а = 11,36; b = 9,75; с = 13,40 А; [3 = 107°; пространствен-
ная группа С2/с) близки к параметрам K2Hg (SCN)4 [175].
Для кобальта (III) известна только одна структура NH4 [Со (DH)2 •
• (SeCN)2] • ЗН2О [190], в которой координационный полиэдр атома кобаль-
та имеет форму незначительно искаженного октаэдра, образованного четырь-
мя атомами азота из двух остатков диметилглиоксима и двумя атомами се-
лена из SeCN-групп, нах< дящихся в транс-положении. Расстояние Со—Se
288
Таблица 6.17. Основные колебательные частоты SeCN-групп, см 1
Соединение v (CN) V (CSe) 6 (SeCN) Литерату- ра
KAg (SeCN)o 2197, 2105 540 398, 419 [130]
[Ph4As] [Au (SeCN)J 2118 516 — [161]
Li3 [Bi (SeCN)e] • 3Act 2110 545 405 [184]
Na3[Bi (SeCN)6J 2073, 2085, 2110 535, 548 405 »
K3 [Bi (SeCN)6] 2090 544 <400 [120]
Ca3 [Bi (SeCN)6]2 • lODiox 2112 —. <400 [184]
Sr3 [Bi (SeCN)6]2 • 8Diox 2100 532 <400 »
Ba3 [Bi (SeCN)e]2 • 5Diox 2092, 2105 532, 557 <400
Mn3 [Bi (SeCN)6]2 • lODiox 2120 530 <400 »
KBa [Bi (SeCN)6] . 2Act 2093 545, 557 <400 »
[(CH3)4 N]3 [Bi (SeCN)e] 2058, 2065, 2090, 544 415 [120]
2100
[(C2H5)4N]3 [Bi (SeCN)6] 2072, 2100 538 417 »
[(C4He)4 N]3 [Bi (SeCN)e] 2070, 2100 535 <400 »
K4 [Cd (SeCN)6] 2078, 2098, 2125 548 415 [129]
l(C16H33) (CH3)3 N]2[Hg (SeCN)4] 2060, 2110 527 342, 380, 392 [161]
— 545 335, 394,410 »
K2 [Hg (SeCN)J 2098 543 389, 407 [53, 185]
2081, 2110 542 406 [174]
[(CH3)4 N]2 [Pd (SeCN)4] 2105, 2114 521 374, 410 [172]
[(C2H5)4 N]2 [Pd (SeCN)4] 2068, 2116 526 381,404,418 [161, 172]
[(C4H9)4 N]2 [Pd (SeCN)J 2060, 2107 519 — [162]
K2 [Pt(SeCN)6] 2080, 2124 520 370,379, 392 [53, 185]
K2 [Pt (SeCN)6J 2130 519 367, 379, 390 [173]
K2 (Pt (SeCN)6J 2116 526 383, 394,419 [161]
Cs2 [Pt (SeCN)6] 2132 522 363, 376, 387 [173]
[(CH3)4 N]2 [Pt (SeCN)6] 2123 528 373, 398 »
f(C2H5)4 N]2 [Pt (SeCN)6] 2127, 2133 524 371,397,418 »
[(C4H9)4 N]2[Pt (SeCN)g] 2116 526 370, 398, 422 [161]
[(C4H9)4 N]2 [Pt (SeCN)4] 2060, 2105 516 — [162]
[(C2C5)4N]2 [Pt (SeCN)4] 2067, 2117 519 375, 400,418 [161]
[(C4H9)4 N]3 [Rh (SeCN)g] 2100 — 392, 435
[(C4H9)4N]3 [Rh (SeCN)g] 2071, 2104 515 — [162]
равно 2,38 А; С—Se 1,80 ± 0,01 А; С—N 1,25 ± 0,02 А; угол SeCN равен
179°, длина SeCN-группы 3,0 А [190].
Методом радиального распределения атомной плотности изучена соль
К2 [Pt (SeCN)6] [189]. Установлено, что она изоструктурна К2 [Pt (SCN)6]
и относится к гексагональной сингонии. Для К2 [Pt (SeCN)6] найдены следу-
ющие расстояния: Pt—Se 2,8 ±0,1 А; С—N 1,1 А; С—Se 1,8 А [1891.
Изучены электронные спектры поглощения [(C4H9)4N]3 [Ru (SeCN)6J,
[(C2H5)4N]2 [M (SeCN)4] (M = Pt, Pd), [(C4H9)4N]2. [Pt (SeCN)6] в CH2C12
[202]. Полосы переноса заряда в рассматриваемых комплексах лежат в
области 31 200—-39 200 см-1 [202].
6.10.4. Анионные изоселеноцианаты
Рассмотрев структуру NH4 [Со (DH)2 (SeCN)2] • ЗН2О, можно сделать
вывод, что кобальт (III) в отличие от кобальта (II) всегда должен образовы-
вать с SeCN-группами связь Со"'—SeCN. Однако это не так. Исследование
соединения К3 [Со (CN)5NCSe] однозначно указывает, что здесь присутству-
ют изоселеноцианато-группы [111]. К удивлению, в аналогичном тиоциа-
натном соединении SCN-группы образуют с кобальтом (III) связь через атом
Ю 2473
289
серы [111]. Судя поданным работы [153], в кислоте Н [Со (NiOxH). •
• (NCSe)2] Н2О и в ее солях осуществляется связь Со—NCSe. Однако этот
вывод нуждается в дальнейшей проверке. Обращает на себя внимание то,
что это соединение устойчиво к действию минеральных кислот, как и
NH4 [Со (DH)2 (SeCN)J, в котором, как известно, доказана связь Со—SeC\
[190].
Из табл. 6.18 видно, что в соединениях хрома имеет место Сг—NCSe-
координация. В электронном спектре поглощения соединения [(CH3)4N]3 •
• [Cr (NCSe)6] отмечаются два максимума поглощения при 18 000 и
29 200 см-1 [170]. Последний максимум обусловлен, очевидно, переносох
заряда, а первый —отвечает переходу 4A2g -> 4T2g, характерному для ок-
таэдрических комплексов хрома. Величина 10Dq, равная 18 000 см-1,
показывает, что SeCN- создает несколько более сильное поле, чем тиоциа-
нат-ион (10Dq = 17 800 см-1) [170].
Октаэдрические изоселеноцианаты образуют также ванадий (III) и ти-
тан (III) (см. табл. 6.18). Для солей Kat3V (NCSe)6 (Kat = (CH3)4N+ и
(C2H5)4N+) из рентгенографических данных оценено расстояние V—N, ко-
торое оказалось равным 2,0—2,1 А [192]. В электронном спектре ацетоно-
вого раствора [(C2H5)4N]3 [V (NCSe)6] полосы переноса заряда лежат при
29 200 и 21 400 см-1 [200].
В ИК-спектре K2VO (NCSe)4 • 6Diox обнаружена полоса v (VO) =
= 970 см-1, следовательно, в рассматриваемом соединении присутствуют
«изолированные» (не мостиковые) связи V=O [100]. Вероятно, в K2VO •
• (NCSe)4 • 6Diox ванадий имеет координационное число 6 (lOVo +
+ 4N Ncse + IOdiox)- Изоселеноцианатные группы присутствуют и в
[(C4H9)4N]2 [VO (NCSe)4] (см. табл. 6.18).
По способу координации NCSe-групп к Cr3+, V3+ близко примыкают
In3+, Ga3+, а также Sc3+, Y3+, лантаноиды (см. табл. 6.18).
Пока что трудно проследить, как влияет степень окисления молибдена,
ниобия на характер координации SeCN-групп. Весьма немногочисленные
данные, которые сейчас имеются, показывают, что с названными металлами,
независимо от их степени окисления, SeCN-группы связаны через азот.
В электронном спектре поглощения раствора соли [(CH3)4N]3 [Мо (NCSe)6]
в СН2С12 обнаружены полосы переноса заряда при 26 400 и 37 600 см-1 [202],
для Мо (NCSe)|— величина 10Dq равна 21 700 см-1 [200].
Из табл. 6.18 видно, что в соединениях (UrtH)2 М (NCSe)4 (М = Со,
Ni) SeCN-группы связаны с никелем, кобальтом через азот. Все же за счет
катиона достигается псевдооктаэдрическое окружение центрального атома
[191]. Так, в спектре отражения соли кобальта найдены две полосы при 19 700
и 18 350 см-1, что характерно для кобальта симметрии Оь- Для (UrtH)2 •
• Со (NCSe)4 определены параметры кристаллической решетки [191]: а =
= 6,47 ± 0,02; 5= 11,14 4= 0,02; с = 17,86 ± 0,03 А; а = у = 90°; [3 =
= 107° 30; пространственная группа Р21/с; Z = 2.
В других селеноцианатах анионного типа, представленных в табл. 6.18,
кобальт (II) имеет преимущественно тетраэдрическую конфигурацию.
Исключением является соль [(CH3)4N]4 [Со (NCSe)6]. Электронный спектр
отражения этой соли типичен для комплексов кобальта симметрии Оь
(табл. 6.19).
Поскольку оказалось, что соли Kat4 [М (NCSe)6] (Kat = (CH3)4N+,
М = Fe, Со, Ni) изоструктурны [172], следовательно, марганец, железо,
290
Таблица 6.18. Основные колебательные частоты SeCN-групп в изоселеноцианатах, см'
Соединение v (CN) v (CSe) 6 (SeCN) Литерату- ра
[(СН3)4 N]4 [Со (NCSe)6] 2087 606, 623 426 [172]
[(С2Н5)4 N]2 [Со (NCSe)J 2053 666 417, 433 »
Kat2+ Со (NCSe)4 2053 672 417, 433 [53]
2062, 2073 654 430 [172]
К3 [Со (NCSe) (CN)6] 2072 664 — [111]
[(С6Н5)4 As] 2 Со (NCSe)4 2062 — — [90]
2060 — — [53]
(UrtH)9 Со (NCSe)4 2083, 2095 608 424, 433 [191]
Н [Со (NiOxH)2 (NCSe)2] • H2O 2120 590 — [153]
[(СН3)4 N]2 Co (NCSe)4 2075 — — [90]
[(CH3)4 N]2 [Cr (NCSe)6] K3 [Cr (NCSe)6] - 3Diox 2041, 2067 663, 668 — [170]
2090 668 442 [97]
Na3 [Cr (NCSe)6] • 3Act Kat2+ Fe (NCSe)4 2100 668 420 »
2062, 2049 646, 654 428 [172]
[(CH3)4N]4Fe (NCSe)6 2083 606 422 »
l(C4H9)4 N]2 Fe (NCSe)4 2055, 2064 666, 673 432 [162]
[(C4H9)4 N]3 Fe (NCSe)6 2049, 2100 658 429 [161]
2055, 2067 666, 673 431 [162]
K4Fe (NCSe)6 Act 2095 592 420 [П7]
l(C4Hfi)4 N]3 Ga (NCSe)6 2075 615, 635 — [H9]
[(CcC5)4 As]3 Ga (NCSe)6 2060 612 — [П6]
Ba [Ga (NCSe)4]9 6Thf [(CH3)4 N]s In (NCSe)6 2075 612 440 »
2040, 2068 2088 640 425 [99]
[(C2H5)4 N]3 In (NCSe)6 2078 649 430 »
[(C4H9)4 N]3 In (NCSe)6 2078, 2095 650 425 »
[(C6H5)4As]3 In (NCSe)6 2070 622, 650 425 »
[(C4H9)4 N]3 Ln (NCSe)6 Ln = Dy 2047 644 434 [165]
Er 2050 646 435 »
Nd 2058 615 428 »
Pr 2058 613 425 [164, 165]
Y 2067, 2030 634 429 [162]
Kat2+ Mn (NCSe)4 Ho 2048 642 433 [165]
2049, 2058 639, 649 429 [172]
[(CH3)4N]4Mn (NCSe)6 2072 623 420 »
[(C4H9)4 N]4 Mn (NCSe)6 K4Mn (NCSe)6 • 6Diox 2070, 2082 2097 617, 640 424 [162]
2090 595 425, 442 [76]
[(C4H9)4 N]3 Mo (NCSe)6 2050 653 443 [161]
[(C2H-)4 N]9 MoO (NCSe)5 K.Nb (NCSe)6 2060 650 470 [85]
1985, 2070 710, 740 490, 445 [167]
KNb (NCSe)6 1918, 1961, 1997, 720, 772 449, 459 [167]
2058, 2062
(UrtH)2 Ni (NCSe)4 2098, 2105 606 426, 431, 438 [191]
[(C2H5)4N]4 Ni (NCSe)6 2096 606 422 [172]
[(CH3)4 N]4 Ni (NCSe)6 2096 606 423 »
[(C4He)4 N]4 Ni (NCSe)6 2102, 2118 625 430 [162]
KTa (NCSe)6 1925,, L972, 2001, 763, 779 449, 455 [167]
2075
I(C6H6)4As] Ta (NCSe)fi 1925, 1981, 2035 749 468 »
[(C4H9)4 N]3 Ti (NCSe)6 2050, 2067 663, 675 441 [166]
[(C4H9)4 N]3 V (NCSe)6 2064, 2072 670, 685 443 »
[(C2H5)4 N]3V(NCSe)6 2062 665 445 [98]
[(CH3)4 N]3 V (NCSe)6 2060, 2075 663 443 »
[(C6H5)4 As]3 V (NCSe)6 2059 — 442 [199]
K3 [V (NCSe)6] • 7Diox 2060, 2070 660 440 [98]
[(C4H9)4 N]2 VO (NCSe)4 2051, 2072, 2083 595, 680 478, 495 [166]
K2VO (NCSe)4 • 6Diox 2080 660 440 [100]
[(C2H5)4 N]2Zn (NCSe)4 2066 660 415, 432 [172]
[•(C4H9)4N]2Zn (NCSe)4 Na2Zn (NCSe)4 • 6Diox 2087 2092 661 665 | 429 428 [162] [115]
10:
291
никель в рассматриваемых соединениях также имеют октаэдрическую конф"
гурацию. Как видно из табл. 6.19, об этом свидетельствуют и спектрально?
данные.
Соли Kat2+M (NCSe)4 (М = Mn, Fe, Со) также изоструктурны [172 .
а спектральные данные однозначно указывают на тетраэдрическое строение
этих соединений (см. табл. 6.19).
Таблица 6.19. Спектральная и магнитная характеристики селеноцианатов
Соединение Полосы в спектрах отражения, см * 10Dq, см 1 V (MN), см 1 Вэф> М. Б. Лите- ратур.-
[(CH3)4N]4 Fe (NCSe)6 9760, 11 150, 34 500* 205 [172]
[(CH3)4 Nj4 (Jo (NCSe)6 9100, 20 000, 29 000* 9700 214 — У>
[(C2H5)4 N]4 [Co(NCSe)6] 16 200, 19 500, 20 400 9200 — •— [200]
[(CH3)4N]4Ni (NCSe)6 10 000, 13 800, 16 600, 26 400, ~ 22 300 10 000 236 — [172]
Kat2+ Mn (NCSe)4 16 000, 19 400, 21 600, 22 700, 25 400, 26 900 * — 243 6,05 »
Kat2+ Fe (NCSe)4 4750, 6050 5400 249 5,30 »
Kat2+ Co (NCSe)4 7700, 8730, 16 300 — 251 —
[(CH3)4 N]2 [Co(NCSe)4] 8000, 15 900 4710 — 4,45 [90]
[(C2H6)4 N]? [Co (NCSe)4] 16 000 — 260 [172] 4,41 [53]
[(C6H5)4 As]2 [Co (NCSe)4] 8000, 15 670 4720 4,55 [90]
* Полосы переноса заряда [202].
Симметрия Td проявляется у кобальта (II) в соединениях Kat^Co (NCSe)4
(Kat+=(CH3)4N+,(C2H5)4N+ и (C6H5)4As+). Об этом свидетельствуют значе-
ния 10Dq и рЭф, а также положение полос в спектрах отражения этих со-
единений (табл. 6.19).
Тетраэдрическую конфигурацию имеет цинк в соединении [(C2H5)4N]2 Zn-
• (NCSe)4, поскольку эта соль изоструктурна аналогичной соли кобальта
[172]. Исследование ИК-спектров раствора этой соли в нитрометане позво-
лило оценить силовые константы: Kcn = 15,67; Kcse = 4,055; Kmn =
= 1,5 Н/см [203].
В электронных спектрах поглощения селеноцианатов железа (II), ко-
бальта (II), никеля (II), цинка (II) анионного типа обнаружены полосы пе-
реноса заряда (29 000—34 500 см-1) [193, 202] и полосы внутр ил игандных
переходов (~43 000—49 000 см-1) [200].
6.10.5. Разнолигандные псевдогалогениды
Среди рассматриваемых соединений наиболее изучены соли кадмия и
ртути. Кроме того, получены тиоцианато-селеноцианаты некоторых 3d-
металлов.
Синтез К2 [Cd (SeCN)2 (SCN)2], К4 [Cd (SeCN)4 (SCN)21 • 0,5Act, K2 ICd-
• (SeCN)2 Hal2] (Hal - СГ, Br-), K4 [Cd (SeCN)4I2] • 0,5Act, K2 [Cd-
•(SeCN)3I] • Act осуществлен кристаллизацией ацетоновых растворов,
в которых CdX2 (X = SCN-, Cl-, Br-, I-) и KNCSe находились в соотно-
шении 1 : 2 или 1 : 4 [210, 211].
Аналогичным способом были получены соли К2 IHg (SeCN)2A2] (А = Br,
I, NCS) и K2 [Zn (NCSe)2 (NCS)2] • 4Diox [211].
Соединения M [Hg (SeCN)2 (SCN)2] синтезированы растворением
292
Таблица 6.20. Волновые числа максимумов полос поглощения ацидогрупп
в ИК-спектрах соединений смешанного типа; см—1
Соединение SeCN-группы SCN- или другие ацидогруппы Ли- тера- тура
v (CN) v (CSe) 6 (Se- CN) v (CN) v (CS) 6 (SCN)
{(СН3)4 N] [Au (CN)a (SeCN)2] 2139 533 2176 [1601
K2Cd (SeCN)г Cl2 2095, 2110, 2119, 2122, 2151 534, 550 403 — — — [210]
K2Cd (SeCN)2 Br2 2109, 2151 545, 582 404 — — — »
K2Cd (SeCN)2 (SCN)2 2113, 2123 2135 543, 552 413, 422 2090, 2094 722, 729 438, 444, 458,473 »
K4Cd (SeCN)4 (SCN)2.0,5Act 2113, 2122, 2132 540, 551 415, 425 2050, 2091 722, 730, 750 438, 446, 460 »
K2Cd (SeCN)3I-Act 2096, 2112, 2121, 2132 534, 632 403 — — — »
K4Cd (SeCN)4 I2-0,5Act 2074, 2111 540 418 — — — »
l(C6H5)4P]2 [Cd {N (CN)2}2 -(NCSe)2j 2080 590 410 2165, 2225 — — 1212]
K4 [Cd {N (CN)2}4 (NCSe)2] 2105, 2120 — 405 2160, 2220 — — »
K2 [Cd {C (CN)3}2 (NCSe)2] 2125, 2150 590 <400 2170, 2220, 2240 — — »
K4 [Cd {C (CN)3}4 (NCSe)2J 2080, 2110 594 415 2180, 2205, 2240 — — »
K2 [Hg (SeCN)2 Br2] 2125, 2138 531, 539 425 — — — [214]
K2 [Hg (SeCN)2 I2] 2118 545, 621 413 — — — »
K2 [Hg (SeCN)2 (SCN)2] 2110 548 424 2110 728 — 1 »
Ba [Hg (SeCN)2 Br2] Act 2107, 2119, 2138, 2143, 2154 545 410 — — — »
Ba [Hg (SeCN)2 (SCN)2]-2Act 2120 553, 570 403, 457, 470 2080 747 — »
Mn [Hg (SeCN)2 (SCN)2]-2Act 2120, 2135 622 402, 420 2120, 2135 790, 805 — [215]
Co [Hg (SeCN).3 (SCN)2[ 2139, 2146 639 426, 436 2139, 2146 801 — »
Ni [Hg (SeCN)2 (SCN)2]-2Act 2160 609 413, 425 2160 768 — »
Zn [Hg (SeCN)2 (SCN)2] 2158 630 404, 420 2158 730, 798 — »
Cu [Hg (SeCN)2 (SCN)]2 2160, 2180 525, 623 405, 427 2160, 2180 760, 830 —
Hg (SeCN)2 в ацетоновых растворах М (SCN)2 (М = Мп, Со). Аналогично
были получены М [Hg (XCN)2A2] (М = Mn, Со, Ni, Zn, Cu; X = S или Se;
A = СГ, Br~, SCN~) [211].
Для кадмия получены соединения, включающие наряду с SeCN-группами
нелинейные N (CN)7- и С (CN)rrpynnbi (см. табл. 6.20). Синтез этих соеди-
нений осуществлен из метанола [212].
293
При кристаллизации ацетоново-диоксановых растворов, содержащих
М (NCS)2 и KNCSe или NaNCSe, выделены соединения К2 [М (NCS), •
- (NCSe)2] • nDiox (М = Mn, Со, Zn) и Na2 [Со (NCSe)2 (NCS)2 ] • 5,5Diox,
в которых, судя по данным ИК-спектроскопии, XCN-группы связаны с
центральным атомом через азот. В электронном спектре отражения для солей
кобальта отмечена одна полоса около 16 000 см-1, указывающая на тетра-
эдрическую конфигурацию кобальта [213].
Обменной реакцией транс-[(CH3)4N] [Au (CN)2C121 и KNCSe в ацетоновом
растворе получен транс-[(CH3)4N] [Au (CN)2 (SeCN)2] [160].
В ИК-спектре К2 [Cd (SeCN)2 (SCN)2] полосы v (CN) обнаруживаются как
в низкочастотной (2090—2120 см-1), так и в высокочастотной области (око-
ло 2135 см-1), что указывает на наличие как мостиковых, так и немостико-
вых XCN-групп. Учитывая большую склонность SeCN-групп, чем SCN,
образовывать мостики, можно считать, что высокочастотные компоненты
v (CN) в спектре рассматриваемого соединения относятся к мостиковым
SeCN-группам, а низкочастотные — к SCN-группам. Полосы v (CS) обнару-
живаются около 720—730 см-1, что указывает на координирование SCN-
групп через атом серы. Таким образом, в К2 [Cd (SeCN)2 (SCN)2] имеются
как концевые (Cd—SCN), так и мостиковые (Cd—SeCN—Cd) связи [210].
Мостиковые SeCN-группы присутствуют также в К2 [Cd (SeCN)2Hal2],
в то время как в соединении К4 [Cd (SeCN),I2] группы SeCN связаны с цент-
ральным атомом через селен [210, 211] (см. табл. 6.20).
Судя по данным ИК-спектроскопии, во всех соединениях кадмия, содер-
жащих нелинейные N (CN)F- и С (CN3)—-группы, кроме К2 [Cd {C(CN)3}2 •
• (NCSe)2], селеноцианатные лиганды координированы посредством атома
азота. Соединение К2 [Cd {C(CN)3}2 (NCSe)2l содержит мостиковые SeCN-
группы (табл. 6.20).
В соединениях К2 [Hg (SeCN)2A2] (А = Br, I, SCN), Ва [Hg (SeCN)2 •
• (SCN)2] • 2Act, присутствуют SeCN-группы, связанные с ртутью через
селен, хотя в случае К2 [Hg (SeCN)2Br2] можно говорить о наличии SeCN-
мостиков, имея в виду полосу при 2138 см-1 [214]. Мостиковые группы при-
сутствуют и в соединении Ва [Hg (SeCN)2Br2J • Act.
Как видно из табл. 6.20, в ИК - спектрах всех соединений типа М [Hg •
• (SeCN)2 (SCN)2] (М = Mn, Со, Ni, Zn) присутствуют высокочастотные ком-
поненты v (CN), расположенные в области 2135—2160 см-1, свидетельст-
вующие о наличии в этих соединениях мостиковых XCN-групп. Мостиковые
XCN-группы присутствуют и в соединении Cu [Hg (SeCN)2 (SCN)2] [215].
Сравнительно недавно описан ряд смешанных комплексов хрома (III)
и кобальта (HI): [Bu4N]3 [Сг (CN)5SeCN], Cs3 [Со (CN)5SeCNl, [Со (NH3)fJ •
- (Со (CN)5SeCN] [226], [Bu4N]3 [Cr (CN) (NCSe)6], [Bu4N]3 [Cr (CN)2 •
- (NCSe)4], [Bu4N]3 [Cr (CN)3 (NCSe)3] и [Bu4N]3 [Cr (CN)4 (NCSe)2l [227].
Комплексы [Me3N]3 [Co (CN)5NCSe] и [Me3N]3 [Co (CN)5SeCN] могут слу-
жить примером изомеров связи [226]. Для ванадия (III) описаны комплексы
типа М3 [V (NCSe)3 (NCS)3] • nL [228].
СПИСОК ЛИТЕРАТУРЫ
1. Birckenbach L., Kellermann К»— Ber., 1925, 58, S. 782.
2. Мельников H. И., Черкасова Е. Н.— Журнал общей химии, 1946, 16, с. 1025.
3. Aynsley Е. Е., Greenwood N. N., Sprague М. J.— J. Chem. Soc., А, 1964, р. 704.
‘ 4. Cauguis G., Pierre G.— Bull. Soc. chim. France, 1972, 3, p. 1225.
5. Aksnes O., Foss O.— Acta Chem. Scand., 1954, 8, p. 702.
294
6. Aksnes 0., Foss О.— Acta Chem. Scand., 1954, 8, p. 1787.
7. Klanberg F.— Z. anorg. allg. Chem., 1962, 316, S. 197.
8. Hauge Sverre.— Acta Chem. Scand., 1971, 25, p. 3081.
9. Hauge Sverre.— Acta Chem. Scand., 1971, 25, p. 1135.
10. Birckenbach L., Kellermann K.— Ber., 1925, 58, S. 2377.
11. Hauge Sverre.— Acta Chem. Scand., 1971, 25, p. 1134.
12. Foss 0., Hauge Sverre.— Acta Chem. Scand., 1963, 17, p. 1807.
13. Hauge Sverre, Stetten Jorunn.— Acta Chem. Scand., 1971, 25, p. 3094.
14. Spacu G., Macarovici C. G.— Z. analyt. Chem., 1936, 105, S. 408.
15. Ripan R.— Z. analyt. Chem., 1933, 94, S. 331.
16. Ripan-Tilici R.— Z. analyt. Chem., 1936, 105, S. 410.
17. Hahn H., Viohl U.— Z. analyt. Chem., 1956, 149, S. 40.
18. Bareza L.— Acta chim. Acad, scient Hung., 1965, 45, p. 23.
19. Parker P. A., Harvey L. C.— Analyst., 1961, 86, p. 54.
20. Рябчиков Д. И., Назаренко И. И.— Успехи химии, 1964, 33, с. 108.
21. Назаренко И. И., Ермаков А. Н. Аналитическая химия селена и теллура. М., Наука,
1971.
22. Бабко А. К., Пилипенко А. Т. Фотометрический анализ. Методы определения
неметаллов. М., Химия, 1974.
23. Crookes W.— Ann., 1851, 78, S. 177.
24. Birckenbach L., Huttner K.— Z. anorg. allg. Chem., 1930, 190, S. 38.
25. Berzelius J. J.— Jahzesber., 1822, 1, S. 49.
26. Kaufmann H. P., Kbgler F.— Ber., 1926, 59, S. 178.
27. Kypke 0., Neger J.— Lieb. Ann. Chem., I860, 115, p. 207.
28. Nuthmann W., Schroder E.— Ber., 1900, 33, S. 1765.
29. Lodzinska A., Racka B.— Roczn. Chem., 1961, 35, 1187.
30. Huttner K-, Knappe S.— Z. anorg. allg. Chem., 1930, 190, S. 27.
31. Неорганические синтезы. Сборник. M., ИЛ, 1951, ч. 2, с. 180.
32. Bergstrom F. W.— J. Am. Chem. Soc., 1926, 48, p. 2319.
33. Ерденбаева M. И., Драговцева А. А.— Изв. АН КазССР. Сер. химии, 1960, вып. 1
(17), с. 59.
34. Хамада Сюити.— J. Chem. Soc. Japan, 1962, 83, р. 667. РЖхим, 1963, 24Б 744.
35. Songstad J., Stangeland L. J.— Acta Chem. Scand., 1970, 24, p. 804.
36. Cameron C. A., Davy E. W.— Chem. News, 1881, 44, p. 63.
37. Скопенко В. В.— Укр. хим. журн., 1972, 38, с. 1196.
38. Huis R., Renson М.— Bull Chim. Beiges, 1957, 66, p. 265.
39. Lodzinska A.— Roczn. Chem., 1960, 34, p. 825.
40. Birckenbach L., Huttner K.— Z. anorg. allg. Chem., 1930, 190, S. 1.
41. SwankD. D., Willett R. D.— Inorg. Chem, 1965, 4, p. 499.
42. Харитонов Ю. Д., Цинцадзе Г. В., П орай-Кошиц М. А.— Докл. АН СССР, 1965,
160, с. 1351.
43. Харитонов Ю. Д., Цинцадзе Г. В.— Журнал неорган. химии, 1965, 10, с. 1191.
44. Morgan Н. W.— J. Inorg. Nucl. Chem., 1960, 16, р. 367.
4г> . Харитонов Ю. ИСкопенко В. В.— Журнал неорган. химии, 1965, 10, с. 1803.
46. Скопенко В. В., Цинцадзе Г. В., Аласания Р. М., Глущенко Л. В.— Укр. хим.
журн. 1969, 35, с. 1317.
47. Голуб А. М., Скопенко В. В.— Журнал неорган. химии, 1960, 5, с. 1973.
48. Голуб А. М., Андрейченко О. Е.— Журнал неорган. химии, 1962, 7, с. 549.
49. Rosenheim A., Pritze М.— Z. anorg. allg. Chem., 1909, 63, с. 275.
50. Микитченко В. Ф., Скопенко В. В., Скоробагатько Е. ПВ1сник КиТв. ун-ту.
Сер. xiMii, 1971, № 12, с. 16.
51. Голуб А. М.— Журнал неорган. химии, 1959, 4, с. 1577.
52. Spacu G., Macarovici С. Gh.— Bull. Soc. Stiinte Cluj, 1931, 6, p. 95.
53. Turco A., Pecile C., Nicolini M. — J. Chem. Soc., 1962, p. 3008.
54. Ikeda Sanae, Hirata Junko, Sataki Hiromu.— J. Chem. Soc. Japan, Chem. and Ind.
Chem., 1972, 10, p. 1980.
55. Ikeda Sanae, Hirata Junko.— J. Chem. Soc. Japan, Chem. and Ind. Chem., 1973,
2, p. 425.
56. Торопова В. Ф.— Журнал неорган. химии, 1956, 1, с. 243.
57. Голуб А. М., Скопенко В. В.— Успехи химии, 1965, 34, с. 2098.
58. Ramakrishna R. S., Thuraisinghen R.— J. Inorg. Nucl. Chem., 1973, 35, p. 2805.
59. Голуб A. M., Косматый Ю. E.— Журнал неорган. химии, 1959, 4, с. 1327.
60. Голуб А. М., Скопенко В. В.— Докл. АН СССР, 1961, 141, с. 851.
61. Голуб А. М., Скопенко В. В.— Журнал неорган. химии, 1962, 7, с. 1012.
62. Голуб А. М., Скопенко В. В.— Журнал неорган. химии, 1962, 7, с. 1265.
63. Голуб А. М., Померанц Г. Б.— Журнал неорган. химии, 1959, 4, с. 769.
295
64. Lodzinska A.— Studia Soc. Scient torunensis, 1961, B3, № 4, p. 53; РЖхим., 1962
10B12.
65. Голуб A. M., Скопенко В. В., Померанц Г. Б.— Журнал неорган. химии, 1965,
10, с. 344.
66. Голуб А. М., Скопенко В. В.— Журнал неорган. химии, 1961, 6, с. 140.
67. Голуб А. М.— Научные доклады высшей школы. Химия и химическая технология
1958, 4, с. 685.
68. Скопенко В. В.— Шсник КиТв. ун-ту. Сер. астрономп, ф!зики, xiMii, 1961, 4, вип I
с. 81.
69. Голуб А. М., Скопенко В. В.— Докл. АН СССР, 1961, 138, с. 601.
70. Скопенко В. В., Аласания Р. М., Глущенко Л. В.— Укр. хим. жури., 1970 36,
с. 129.
71. Marayama Teisuke, Takayanagi Akio.— Bull. Chem. Soc. Japan, 1972, 45, № 2
p. 3549. .
72. Swinarsci A., Lodzinska A., Cacha M., Bieniak'K--—1Roczn. Chem., 1959, 33, p. 899.
73. Czakis-Sulikowska M.— Roczn. Chem., 1965, 39, p. 1161.
74. Czakis-Sulikowska M.— Roczn. Chem., 1966, 40, p. 1821.
75. Burger K-, Pinter B.— J. Inorg. Nucl. Chem., 1967, 29, p. 1717.
76. Станчева П., Скопенко В. В., Цинцадзе Г. В.— Укр. хим. жури., 1969, 35, с. 166.
77. Скопенко В. В., Брусиловец А. И.— Укр. хим. журн., 1968, 34, с. 1210.
78. Скопенко В. В., Брусиловец А. И.— Укр. хим. журн., 1964, 30, с. 24.
79. Брусиловец А. И., Скопенко В. В.— В1сник Ки‘1в. ун-ту. Сер. сЫзики та xiMii, 1967,
№ 7, с. 151.
80. Скопенко В. В., Микитченко В. Ф.— Укр. хим. журн., 1972, 38, с. 923.
81. Скопенко В. В., Жумабаев А. Ж.— Укр. хим. журн., 1971, 37, с. 528.
82. Скопенко В. В., Иванова Е. И., Григорьева А. С.— Укр. хим. журн., 1973, 39,
с. 754.
83. Скопенко В. В., Иванова Е. И.— В1сник КиТв. ун-ту. Сер. xiMii, 1969, № 10, с. 24.
84. Голуб А. М., Калибабчук В. А.— Журнал неорган. химии, 1967, 12, с. 2370.
85. Голуб А. М., Гречихина В. А., Грачевский В. В., Улько И. В.— Журнал неорган.
химии, 1973, 18, с. 2119.
86. Голуб А. М., Лишко Т. П., Скопенко В. В.— Укр. хим. журн., 1971, 37, с. 835.
87. Cattalini L., Belluco UMartelli M., Ettorre R.— Gazz. chim. Ital., 1965, 95,
p. 567.
88. Belluco UCattalini L., Graziani M., Martelli M.— Gazz. chim. Ital., 1965, 95,
p. 576.
89. Lodzinska A., BadickaD.— Roczn. Chem., 1961, 35, p. 1195.
90. Cotton F. A., Goodgame D. M. L., Goodgame M., Haas H. E.— Inorg. chem., 1962,
1, p. 555.
91. Скопенко В. В., Брусиловец А. И., Безродная Л. П.— Укр. хим. журн., 1970, 36,
с. 878.
92. Wagner Е. L.— J. Chem. Phys., 1965, 43, р. 2728.
93. Харитонов Ю. Л— В кн.: Колебательные спектры в неорганической химии. М.,
Наука, 1971, с. 161—170.
94. Spacu G., Ripan R.— Bull. Soc. Stiinte Cluj, 1928, 4, p. 3.
95. Norbury A. H., Ryder E. A., Williams R. F.— J. Chem. Soc., A, 1967, № 9, p. 1439.
96. Nelson S. M.— Proc. Chem. Soc., 1961, p. 372.
97. Брусиловец А. И., Скопенко В. В., Цинцадзе Г. В.— Журнал неорган. химии, 1969,
14, с. 467.
98. Скопенко В. В., Иванова Е. И.— Журнал неорган. химии, 1969, 14, с. 742.
99. Скопенко В. В., Микитченко В. Ф., Цинцадзе Г. В.— Журнал неорган. химии,
1969, 14, с. 1790.
100. Скопенко В. В., Иванова Е. И., Цинцадзе Г. В.—Укр. хим. журн., 1968, 34, с. 1000.
101. Скопенко В. В., Цинцадзе Г. В., Труба Н. А.— Вкник КиТв. ун-ту. Сер. xiMii,
1970, № 11, с. 9.
102. Nelson S. М., Shepherd Т. М.— Inorg. Chem., 1965, 4, р. 813.
103. King Н. С., Koros Е., Nelson S. М.— J. Chem. Soc., 1964, р. 4832.
104. Lewer А. В., Nelson S. М.— Chem. Communs., 1965, 9, р. 168.
105. Turco A., Pecill С., Nicolini М.— Prec. Chem. Soc., 1961, р. 213.
106. Spacu G., Armeanu V.— Bull. Soc. Stiinte Cluj, 1930, 5, p. 294.
107. Spacu G., Macarovici C. G.— Bull. Soc. Stiinte Cluj, 1930, 5, p. 169.
108. Spacu G., Armeanu V.— Bull. Soc. Stiinte Cluj, 1931, 6, p. 64.
109. SpacuG., Macarovici C. G.— Bull. Soc. Stiinte Cluj, 1932, 6, p. 401.
110. Farago M. E., James J. M.— Inorg. Chem., 1965, 4, p. 1706.
111. Burmeister J. L., Al-Janabi M. Y.— Inorg. Chem., 1965, 4, p. 962.
М2. Duffy Narman V., Kosel F. G.— Inorg. Nucl. Chem. Letters, 1969, 5, № 7, p, 519.
296
113. Скопенко В. В,, Галицкая С. М.— Укр. хим. журн., 1972, 38, с. 709.
114. Скопенко В. В., Галицька С. М.— ЕИсник Ки1в. ун-ту. Сер. xiMii, 1973, № 14,
с. 35.
115. Аласания Р. М., Скопенко В. В., Цинцадзе Г. В.— Укр. хим. журн., 1969, 35,
с. 568.
116. Микитченко В. Ф., Скопенко В. В., Цинцадзе Г. В.— Укр. хим. журн., 1971, 37,
с. 1292.
117. Скопенко В. В., Брусиловец А. И., Цинцадзе Г. В.— Укр. хим. журн., 1969, 35,
с. 489.
118. Madeja К., Wilke WSchmidt S.— Z. anorg. allg. Chem., 1966, 346, S. 306.
119. Скопенко В. В., Микитченко В. Ф.— Укр. хим. журн., 1971, 37, с. 878.
120. Голуб А. М., Скопенко В. В., Жумабаее А.— Журнал неорган. химии, 1969, 14,
с. 2995.
121. Burger Н., Schmid W.— Z. anorg. allg. Chem., 1972, 388, S. 67.
122. Цинцадзе Г. В., Голуб А. М., Копа М. В.— Журнал неорган. химии, 1969, 14,
с. 2743.
123. Голуб А. М., Копа М. В., Скопенко В. В., Цинцадзе Г. В.— Укр. хим. журн.,
1970, 36, с. 871.
124. Golub А. М., Кора W. V., Skopenko V. W., ZinzadseG. W.— Z. anorg. allg. Chem.,
1970, 375, S. 302.
125. Скопенко В. В., Спивак И. С.— Укр. хим. журн., 1975, 41, с. 657.
126. Smith J. КBrown Т. М.— Inorg. Chem., 1972, 11, р. 2697.
127. Скопенко В. В., Иванова Е. И.— Укр. хим. журн., 1970, 36, с. 16.
128. Скопенко В. В., Хенниг X., Сухан Т. А., Цинцадзе Г. В.— Докл. АН УССР, се-
рия Б, 1973, № 3, с. 268.
129. Аласания Р. М., Цинцадзе Г. В., Скопенко В. В.— Труды Груз, политехи, ин-та,
1968, № 3 (123), с. 28.
130. Харитонов Ю. Я., Цинцадзе Г. В.— Журнал неорган. хим., 1965, 10, с. 35.;
131. Савицкий В. Н., Скопенко В. В., Жумабаее А., Грачевский В. В.— Укр. хим.
журн., 1975, 41, № 9, с. 903.
132. Baker W. A., LongG. J.— Chem. Commun., 1965, 15, р. 368.
133. Konig Е., Madeja К.— Spectrochim. Acta, 1967, 23A, p. 45.
134. Голуб A. M., JIuluko T. П , Цинцадзе Г. В., Скопенко В. В.— Журнал неорган.
химии, 1972, 17, с. 2115.
135. Скопенко В. В., Савицкий В. Н., Стахов Д. А.— Укр. хим. журн., 1974, 40,
№ 11, с. 1129.
136. Burmeister J. L., Gysling Н. J., Lim J. С.— J. Am. Chem. Soc., 1969, 91, p. 44.
137. Konig E., Madeja K.— Inorg. Chem., 1967, 6, p. 48.
138. Цинцадзе Г. В. Канд. дис. Тбилиси, 1964.
139. Цинцадзе Г. В., Скопенко В. В., Брусиловец А. И.— Сообщ. АН ГрузССР, 1968,
50, с. 109.
140. Цинцадзе Г. В.., Скопенко В. В., Микитченко В. Ф., Жумабаее А. — Докл.
АН УССР, серия Б, 1969, № 2, с. 130.
141. Скопенко В. В., Цинцадзе Г. В.— Журнал неорган. химии, 1964, 9, с. 2675.
142. Скопенко В. В., Цинцадзе Г. В., Брусиловец А. И.— Укр. хим. журн., 1970, 36,
с. 474.
143. Тряшин А. С., Скопенко В. В., Стахов Д. А.— Журнал неорган. химии. 1973,
18, с. 2658.
144. Скопенко В. В., Лубяный В. Е., Тряшин А. С.— Укр. хим. журн., 1974, 40,
с. 453.
145. Удовенко В. В., Пилипенко Л. С.— Укр. хим. журн., 1971, 37, с. 115.
146. Цинцадзе Г. В., Порай-Кошиц М. А., Анцишкина А. С.— Журнал структурной
химии, 1967, 8, с. 296.
147. Burmeister J. L., Gysling Н. J.— Inorg. Chim. Acta, 1967, 1. p. 100.
148. Swinarski A., Lodzinska A.— Roczn. Chem., 1958, 32, p. 1053.
149. Lodzinska A., Cichocka H.— Roczn. Chem., 1960, 34, p. 297.
150. Agrawal A. K— Proc. Nat. Acad. Sci. India, 1964, A34, № 4, p. 603.
151. Czakis-Sulikowska M.— Chem. analit. (Polska), 1965, 10, № 6, p. 1189.
152. Аблов А. В., Самусь H. M.— Докл. АН СССР, 1960, 133, c. 1327.
153. Varhelyi Cs., Finta Z., Zsako J.— Z. anorg. allg. Chem., 1970, 374, S. 326.
154. Minozzi A.— Att. Inst. Veneto, 1909/1910, 69, p. 456; Gmelin, 1940, 68, p. 212.
155. Clarke J. W.— Ber., 1878, 11, S. 1325.
156. Peters W.— Z. anorg. allg. Chem., 1912, 77, S. 182.
157. Billow E. — Z. Kryst., 1912, 50, S. 494.
158. Порай-Кошиц M. А., Цинцадзе Г. В.— В кн.: Кристаллохимия. Итоги науки,
М., Изд-во АН СССР, 1967.
297
159. Bailey R. A., Kozak S. L., Mtchelsen T. WMilis W. N.— Coord. Chem. Rev .
1971, 6, p. 407.
160. Melpolden J. B., Burmeister J. L.— Inorg. Chem., 1972, 11, № 11, p. 911.
161. Schmidtke H. H., CarthoffD.— Helv. cheim. Acta, 1967, 50, p. 1631.
162. Burmeister J. L., Williams L. E.— Inorg. Chem., 1966, 5, p. 1113.
163. Андрейченко О. E.— Укр. хим. журн., 1964, 30, с. 1255.
164. Burmeister J. L., Patterson S. D., Deardorff E. A.— Inorg. Chem. Acta, 1969, -
p. 105.
165. Burmeister J. L., Deardorff E. A.— Inorg. Chim. Acta, 1970, 4, p. 97.
166. Burmeister J. L., Williams L. E.— J. Inorg. Nucl. Chem., 1967, 29, p. 839.
167. Brown T. M., Bush B. L.— J. Less-Common Metals, 1971, 25, № 4, p. 397.
168. Скопенко В. В., Жумабаев A. Ж.— Укр. хим. журн., 1969, 35, с. 428.
169. Жумабаев А. Ж., Скоробагатько Е. П.— Укр. хим. журн., 1972, 38, с. 14.
170. Mtchelsen К-— Acta Chem. Scand., 1963, 17, р. 1811.
171. Hendriksma R. R.— Inorg. Nucl. Chem. Letters, 1972, 8, № 12, p. 1035.
172. Forster D., Goodgame D. M. L.— Inorg. Chem., 1965, 4, p. 1712.
173. Sabatini A., Bertini I.— Inorg. Chem., 1965, 4, p. 959.
171. Аласания P. M., Скопенко В. В., Цинцадзе Г. В.— Труды Груз, политехи, ин-та,
1967, № 7(119), с. 21.
175. Звонкова 3. В.— Журнал физ. химии, 1952, 26, с. 1798.
176. Lewis J., Nyholm R. S., Smith P.— J. Chem. Soc., 1961, p. 4590.
177. Саука Я. Ц., Карклинь Ю. А.— Кристаллография, 1961, 6, с. 775.
178. FrassonE., Turco A., Panattoni С.— Gazz. chim. Ital., 1961, 91, p. 750.
179. Цинцадзе Г. В., Скопенко В. В., Швелашвили А. Е.— Сообщ. АН ГрузССР, 1966,
41, № 2, с. 337.
180. Цинцадзе Г. В., Швелашвили А. Е., Скопенко В. В.— Труды Груз, политехи,
ин-та, 1967, № 1 (113), с. 29.
181. Цинцадзе Г. В., Швелашвили А. Е., Скопенко В. В.— Труды Груз, политехи,
ин-та, 1967, № 4 (114), с. 53.
182. Порай-Кошиц М. А.— Журнал структурной химии, 1963, 4, с. 584.
183. Цинцадзе Г. В., Скопенко В. В , Кереселидзе Л. Б.— Сообщ. АН ГрузССР, 1971,
61, № 1, с. 53.
184. Скопенко В. В., Жумабаев А. Ж-, Цинцадзе Г. В.— Укр. хим. журн., 1970, 36,
с. 329.
185. Pecile С., Turco A., Pizzolotto G.— Ricer. Sci., 1961, 1, р. 247.
186. Bolluco IL, Graziani М., Nicolini М., Rigo Р.— Inorg. Chem., 1967, 6, p. 721.
187. Цинцадзе Г. В., Порай-Кошиц М. А., Анцишкина А. С — Журнал структурной
химии, 1964, 5, с. 495.
188. Жданов Г. С., Сакадзе В. В.— Журнал физ. химии, 1952, 26, с. 469.
189. Цинцадзе Г. В., Скопенко В. В., Швелашвили А. Е.— Труды Груз, политехи, ин-та,
1968, № 1 (121), с. 19.
190. Аблов А. В., Самусь И. Д.— Докл. АН СССР, 1962, 146, с. 1071.
191. Вассоп М. Le, Youinos М.-Т., Guerchais J. Е.— Bull. Soc. chim. France, 1972,
12, p. 4525.
192. Цинцадзе Г. В., Скопенко В. В., Иванова Е И.— Сообщ. АН ГрузССР, 1968,
51, с. 107.
193. Day Р.— Inorg. Chem., 1966, 5, р. 1619.
194. Soderback Е.— Acta Chem. Scand., 1974, A28, № 1, p. 116.
195. Pearson R. G., Ellgen P.— Inorg. Chem., 1967, 6, p. 1379.
196. Lim S. WSchreiner A. F.— Inorg. Chim. Acta Rev., 1971, 5, p. 290.
197. Birnbaum E. R.— J. Inorg. Nucl. Chem., 1971, 34, p. 3499.
198. Цинцадзе Г. В., Товбис А. Б., Мамулашвили А. М., Квиташвили А. И.— Труды
Груз, политехи, ин-та, 1969, № 6 (134), с. 13.
199. Schmidtke И. Н., GarthoffD.— Z. Naturforsch., 1969, 24А, № 1, S. 126.
200. Pruchnik F., Wajda S., Kwaskowska-Chec E.— Roczn. Chem., 1971, 45, № 4, p. 537.
201. Schmidtke H. H.— J. Inorg. Nucl., Chem., 1966, 28, p. 1735.
202. Schmidtke H. H.— Ber. Bunsen ges. phys. Chem., 1967, 71, № 9, S. 1138.
203. Forster D., Horrocks W. De W.— Inorg. Chem., 1967, 6, № 2, p. 339.
204. Kona M. B.— Журнал неорган. химии, 1973, 18, с. 2072.
205. Савицкий В. Н., Скопенко В. В.— Укр. хим. журн., 1975, 41, № 10, с. 1038.
206. Скопенко В.В., Зуб Ю.Л., Тряшин А. С. и др. — Укр. хим. жури., 1984, 40,
с. 920.
207. Цинцадзе Г. В., Цивцивадзе Т. В , Орбеладзе Ф. В., Самсонадзе Л. А — Труды
Груз, политехи, ин-та, 1973, № 8 (164); РЖхим, 1974, 14Б458.
208. Цинцадзе Г .В., Мерабова И.А., Квиташвили А. И., Товбис А. Б. Труды
Груз, политехи, ин-та, 1973, № 8 (164), с. 129.
298
209. Голуб А. М., Стерник Б. О., Улько Н. В.— ЕИсник КиТв. ун-ту. Сер. xiMii, 1971,
№ 12, с. 21.
210. Цинцадзе Г. В., Харитонов Ю. Я., Цивадзе А. Ю. и др.— Журнал неорган. химии,
1970, 15, с. 2336.
211. Цинцадзе Г. В., Голуб А. М., Манагадзе А. С.— Труды Груз, политехи, ин-та,
1970, № 1 (136), с. 52.
212. Келер X., Скопенко В. В., Тряхин А. С., Зуб Ю. Л.— Укр. хим. жури., 1974, 40,
№ 10, с. 1121.
213. Цинцадзе Г. В., Голуб А. М., Манагадзе А. С., Квезерели Э. А.— Труды Груз,
политехи, ин-та, 1972, № 5 (153), с. 5.
214. Цивадзе А. К)., Цинцадзе Г. В., Манагадзе А. С.— Сообщ. АН ГрузССР, 1972,
65, № 1, с. 65.
215. Цинцадзе Г. В., Цивадзе А. Ю., Манагадзе А. С.— Сообщ. АН ГрузССР, 1972,
65, № 2, с. 329.
216. Dash R. N., Ramana Rao D. Т.— J. Indian Chem. Soc., 1974,51, p. 211.
217. Eggli R., Ludwig W.— Inorg. Chim. Acta, 1973, 7, p. 697.
218. Burmeister J. L., O'Sullivan Th. P., Johnson K.A.— Inorg. Chem., 1971, 10, p. 1803.
219. Patel К. C., Goldberg D. E.— Inorg. Chem., 1972, 11, p. 759.
220. Coz E. Le, Guerchais J. E., Goodgame D. M. L.— Bull. Soc. chim. France, 1969, 11,
p. 3855.
221. Srivastava T. N., Bajpai К. K-— J. Organometal. Chem., 1971, 31, № 1, p. 1.
222. Aynsley E. E., Greenwood N. N., Hunter G., Sprague M. J.— J. Chem. Soc., A., 1966,
p. 1344.
223. De Stefano N. J., Burmeister J. L.— Inorg. Chem., 1971, 10, № 5, p. 998.
224. Burmeister J. L., Hassel R. L., Phelan R. J.— Inorg. Chem., 1971, 10, № 9, p. 2032.
225. Gutterman D. F., Gray H. B.— Inorg. Chem., 1972, 11, №8, p. 1727.
226. Melpolder I. B., Burmeister I. L.— Inorg. chim. Acta, 1975, 15, p. 91.
227. Botar A., Blasius E., Augustin H.— Z. anorg. allg. Chem. 1975, 417, S. 93.
7. ТРИЦИ АНМЕТ АНИДЫ
7.1. ПСЕВДОГАЛОГЕНИДНЫЙ ХАРАКТЕР
ТРИЦИАНМЕТАНИД-ИОНА
На принадлежность трицианметанид-иона к псевдогалогенидам указал
Биркенбах [1,29], а Маделунг и Керн [1а] показали, что введение цианидных
групп в гидриды неметаллов ведет к образованию гомологов кислот
НЕ (CNUi
ЕН„ = СН4 NH3 OH2(SH2)
НЕ (CN)„_j = НС (CN)3 HN (CN)2 HOCN (HSCN)
Поэтому трицианметанид-ион очень напоминает другие цианидсодержа-
щие псевдогалогениды — дицианамид, N (CN)7, а также цианаты ECN-
(Е = О, S, Se). Эта тесная связь особенно отчетлива, если принять во внима-
ние псевдохалькогенидный характер С (СМ)2-группы [26],
(NC)2C=C=N- o=c=n-
t
Псевдохалькогенидная группа
Открытие трицианметанид-иона относят к 1896 г. [2], когда Шмидтманн
синтезировал трицианметанид калия из натриевой соли динитрила малоно-
вой кислоты и галогенциана. Через три года Ганч и Освальд [3] установили
большую силу псевдогалогенидной кислоты «цианоформа»: «...цианоформ не
только отчетливо выраженная, но и чрезвычайно сильная кислота». Сейчас
мы знаем, что соединение с рКа = —5,1 в безводном состоянии является
не цианоформом НС (CN)3, а дицианкетенимином HNC=C=(CN)2, напоми-
нающим сильные минеральные кислоты [4, 5].
Химия трицианметанид-иона исследована более подробно лишь в послед-
ние годы. При этом установлено, что псевдогалогенидный характер этого
иона проявляется как в образовании плохо растворимой в воде соли серебра,
так и в многочисленных других типично псевдогалогенидных реакциях. На-
пример, трицианметанид аналогично цианату и дицианамйду способен $
разнообразным реакциям комплексообразования и образует устойчивые
элементоорганические трицианметаниды [6]. AgC (CN)3 реагирует с хлором
или бромом, образуя галогентрицианметаниды С1С (CN)3 и ВгС (CN)3 [7, 8!.
В этой связи представляет интерес проведение реакции между AgC (CN)3
и C1CN, позволяющей получить тетрацианметан С (CN)4 (Td-симметрия,
v (CN) = 2273 см-1). Последний можно рассматривать как межпсевдогало-
генидное соединение (цианотрицианметанид), которое расщепляется в щелоч-
ной среде с образованием цианата и трицианметанида [9],
NC—C (CN)3 + 2ОН- -> OCN- + С (CN)J + Н2О. (7Д)
300
Известен также соответствующий трицианметанид-иону псевдогалоген
гексацианэтан [10]. Соединение было получено путем двойного обмена между
пентацианэтанидом натрия и хлорцианом
NaC (CN)2 — С (CN)3 + C1CN -> (NC)3 С — C (CN)3 + NaCl. (7.2)
Гексацианэтан—бесцветные кристаллы, которые сублимируются в ва-
кууме (2 мм, 75° С) и характеризуются частотой v (CN) при 2273 см-1. При
комнатной температуре он медленно разлагается с образованием тетрациан-
этилена, при — 80° С соединение сохраняется без разложения длительное
время. Доказательство псевдогалогенидного характера этого соединения
следует из возможности анодного окисления трицианметанида до гексациан-
этана в неводных средах. Однако реакция обратима: катодное восста-
новление гексацианэтана ведет к разрыву С—С-связи и образованию три-
цианамида [62]
2С (CN)F (NC)3 С — С (CN)3 + 2е~. (7.3)
7.2. СВОЙСТВА И СТРУКТУРА ТРИЦИАНМЕТАНИД-ИОНА
Трицианметаниды щелочных металлов могут быть получены различны-
ми способами. Если в первых синтезах исходили из натриевой соли динит-
рила малоновой кислоты и галогенциана [2, 3, 8], то в последнее время в ка-
честве исходных веществ применяют моноброммалонитрил [12] или дибром-
малонитрил [7]. Для реакции диброммалонитрильного комплекса бромида
калия [59] с цианидом калия в мольном соотношении 1 : 2, ведущей к обра-
зованию КС (CN)3 с хорошим выходом [7], предложен механизм
CN CN CN CN
X—С—X + CN~ -> XCN +“ | С—X — - С + CN" | С—CN. (7.4)
I I Х I I
CN CN CN CN
Кроме того, КС (CN)3 получен путем обменной реакции между 1,1-ди-
циано-2-амино-2-алкоксиэтиленом и КОН [2, 13].
Если NaC (CN)3 можно получить аналогично соли калия [14], то для
получения соли аммония оправдал себя ионообменный метод между раство-
ром КС (CN)3 и ионитом в КН4-форме [15].
Трицианметанид лития, кристаллизующийся, как и другие трицианмета-
ниды щелочных металлов, в форме бесцветных игл, получают реакцией об-
мена между LiCl и КС (CN)3 в ацетонитриле [16], а трицианметаниды комп-
лексных катионов МС (CN)3 (М = Ph4P+, Ph4As+, Ph3MeAs+) можно легко
получить из водного раствора обменом КС (CN)3 с соответствующими оние-
выми хлоридами [16].
Трицианметаниды калия, аммония и натрия не изоструктурны из-за раз-
личия в координации катионов; в то же время трицианметанид-ион имеет
почти плоское строение.
Строгая планарность трицианметанид-иона наблюдается в натриевой
и калиевой солях 114]. Обе соли триклинные и относятся к пространственной
группе Р1. Средние расстояния С—С составляют 1,408 А (трицианметанид
натрия), длина связей С—N варьируется незначительно между 1,145 и 1,16 А,
а углы связи ССС 119,2; 120,1 и 120,5° очень близки к ожидаемому углу
120° для зр2-гибридизации.
301
В аммонийной соли для аниона наблюдаются незначительные отклоне-
ния от планарности: углы связи ССС равны 119°ЗГ, 119° 32' и 119° 1
(Z.CCN = 180°; длина связей С—С 1,40 А; С—N 1,14—1,16 А). Соль моно-
клинна. Пространственная группа Р21/с [151.
Соответствующие соотношения связей в трицианметанид-ионе хорош
передаются мезомерными формами I—III, в соответствии с которыми заряд
аниона в значительной мере локализован на цианидном азоте,
Таблица 7.1. Полосы
поглощения
в ИК-спектре С (CN)^-
V, см ! Отнесение
404 (сл.) т(С-с3)
483 (сл.) б (С—ср
567 (оч. с.) у (С—C=N)
608 (сл.) 6 (С—C=N)
1239 (с.) v (С-С)
1251 (с.)
2176 (оч. с.) v (C=N)
Относительная интенсивность:
сл. — слабая, с. — сильная , оч.
с. — очень сильная.
[20]. Согласно работе
МО-расчеты с помощью расширенного мето-
да Хюккеля показывают, что атом азота циа-
но-группы имеет более отрицательный заряд
(б + зт = —1,114), чем центральный атом уг-
лерода (6 + зт = —0,0374) [17].
ИК- и КР-спектры С (CN)^ хорошо ин-
терпретируются на основе П31а-симметрин.
В табл. 7.1 приведены важнейшие ИК-частоты
для кристаллического КС (CN)3 [18].
Образование ковалентных или комплексных
трицианметанидов ведет к таким значительным
изменениям, прежде всего в области валентных
колебательных v (CN), что их можно привлекать
для выяснения структуры производных трициан-
метанида [19]. УФ-спектр С (CN)r характеризу-
ется очень интенсивной полосой при 47 400 см~
[60], энергия N—1s для С (CN)7* составляет
399,1 эВ (КС (CN)3), а для NCO и NCS она равна 398,5 и 397,8 эВ [61].
73. ТИЛЫ СВЯЗИ В ТРИЦИАНМЕТАНИДАХ И СПОСОБЫ
ИХ ИНТЕРПРЕТАЦИИ
В противоположность устойчивым солеобразным трицианметанидам ще-
лочных и щелочноземельных металлов сильно ковалентные трицианметани-
ды элементов главных подгрупп (описанных лишь в отдельных случаях)
представляют собой малоустойчивые соединения, склонные уже при ком-
натной температуре к полимеризации.
Особенно интересен тот факт, что трицианметанид-ион, несмотря на свою
чрезвычайно малую основность, выступает как сильный комплексообразу-
ющий лиганд. Трицианметанидные комплексы термически довольно, устой-
чивы, что может быть обусловлено высокой степенью резонансной стабили-
зации анионных лигандов. Это соответствует тому, что трицианметанид об-
разует координационные связи, главным образом благодаря своей планар-
302
ности. При этом не последнюю роль играет высокая электронная плотность
на азоте цианидной группы, в результате чего образуются N-связи между
металлом и лигандами (структурные формулы IV и V; С2у-симметрия). Сое-
динения этого типа можно представить как дицианкетениминат (структура
V), в то время как соединениям с линейными М—N—С-группами отвечает
структура ГУ. Установить различие между ними возможно лишь с помощью
рентгеноструктурных исследований.
Z1
M-NsC-C^
Ус
В то время как тип связи IV (монодентатный трицианметанид) во многом
напоминает изотиоцианатные комплексы, например при сравнении силы по-
ля лигандов [21] или транс-эффекта [22], в отличие от цианатов трицианме-
танид проявляет более сильную тенденцию выступать как мостиковый лиганд
и образовывать при этом координационные полимеры [23, 24].
Из-за далеко идущей делокализации заряда все три цианидных атома
азота проявляют в одинаковой мере способность к образованию координа-
ционной связи. Это приводит к тому, что трицианметанид не только функцио-
нирует как монодентатный лиганд, но чаще может проявлять мостиковую
функцию как би- и тридентатный лиганд (структуры VI, VII).
VII
Он также может выступать как тетрадентатный лиганд (структура VIГГ),
что доказано структурным исследованием (СН3)2 TIC (CN)3 [54].
Координируясь посредством азота, трицианметанид выступает как «жест-
кий» лиганд (в терминах ЖМКО) с относительно малой способностью к обра-
зованию 6-связей и очень малой—к формированию зх-связей. Известны еди-
ничные случаи координации С (CN')y через углерод с «мягкими» центральными
303
атомами (структура IX).
N
С
М—С—C = N|
С
N
IX
В комплексах этого типа встречающаяся тетраэдрическая структура лв
ганда (зр3-гибридизация центрального атома углерода) наблюдается также
и в различных производных неметаллов, например в галогентрицианмета-
нидах. Напротив, в комплексах переходных металлов и HNCC (CN)2 домини-
руют структуры IV, V, а в ряде металлоорганических три-
2200 2400см' цианметанидов —структура VI.
О Способ координации трицианметанида сказывается на
i I ц ИК- и КР спектрах соединений, причем особенно заметны
ijh изменения в области частот валентных колебаний v (CN).
I / н Если ИК-спектр свободного С (CN)^ (симметрия D3h) ха-
I J и растеризуется узкой интенсивной полосой v (CN) при
i. I и J/V 2176 см“ , то в спектре N-трицианметанидов металлов (то-
чечная симметрия C2v) можно ожидать три полосы v (CN)
(2А1} EJ, а в С-трицианметанидах металлов (точечная сим-
метрия C3v) — две v (CN) (А1} EJ-частоты. Чаще всего в
спектрах N-трицианметанидов металлов наблюдаются две-
три полосы v (CN) около 2170 и 2230 см“\ а для С-трициан-
метанидов оба v (CN)- колебания проявляются в одинако-
вой области. Надежная интерпретация обоих типов связи
на основании числа и положения v (CN)-4acTOT невозможна,
однако в комбинации с 14N-4MP возможно четкое раз-
граничение М—N- и М—С-связи. C(CN^"-hoh вследствие
эквивалентности трех атомов азота имеет только один
7.1. Сравнение положения полос v(CN) моно-, би- и тридентатных три-
цианметанидных групп в комплексах никеля:
а — Ni (NCC (CN)2)2 Ру4; б — Ni (NCC (CN)2)2 Ру2; в — Ni (NCC (CN)2)2
сигнал (6NOa = +116 м. д.). В С-трицианметанидах металлов этот сигнал
смещен в сторону сильных полей, в то время как в N-трицианметанидах
металлов из-за несохранения эквивалентности атомов азота можно наблю-
дать два сигнала, причем сигнал для координированного азота сдвинут в
слабое поле [25].
В спектре С1С (CN)3 наблюдаются обе ожидаемые для симметрии С3у
полосы v (CN) при 2262 и 2209 см-1. Положение этих полос указывает
на повышение порядка связи CN-rpynn.
Для координированной нитрильной группы наблюдается сдвиг v (CN)
в длинноволновую область. Поэтому в ИК-спектрах трицианметанидных мос-
тиковых многоядерных комплексов обнаруживаются характерные сдвиги
v (СМ)-полос в сторону высоких волновых чисел: в комплексах с бидентат-
ным трицианметанидом (структурный тип VI) — 2185—2200 и 2240—
2250 см-1, а в случае тридентатности С (CN)JT (VII) полосы v (CN) наблю-
304
даются в областях 2200—2220 и 2250—2270 см-1. Рис. 7.1 позволяет
сравнить положение полос валентных колебаний v (CN)-rpynn в ИК-спект-
рах трицианметанидных комплексов Ni (II): NiPy4 (NCC (CN)2)2, NiPy2 •
• (NCC (CN)2)2 и Ni (C (CN)3)2, которые содержат соответственно моно-,
ди- и тридентатный трицианметанид.
7.4. КОВАЛЕНТНЫЕ ТРИЦИАНМЕТАНИДЫ ЭЛЕМЕНТОВ
ГЛАВНЫХ ПОДГРУПП
7.4.1. Дицианкетенимин HNCC(CN)2
Дицианкетенимин, ранее называемый цианоформом (CH (CN)3),— бес-
цветные, не содержащие воды, сублимируемые в вакууме кристаллы, ус-
тойчивые лишь при низких температурах [2, 3]. Получение соединения опи-
сано в работах [2, 3, 5].
Дицианкетенимин является сильной кислотой (рКк = —5,1) [4, 5], что
обусловлено высокой мерой резонансной стабилизации трицианметанид-
иона, и в этом отношении он сравним с другими многочисленными перциан-
углеродными соединениями. Его ИК-спектр характеризуется интенсивной
полосой валентного колебания v (CN) при 2198 см-1. Отношение связей мож-
но описать через мезомерные граничные формы [51
N
- /С
Н—N = C=C<^
ХС
N
H-N=C-C|©
С
С
N
XI
В водном или эфирном растворе дицианкетенимин образует ониевые соли
Н3О+С (CN)-E или R2OH+ С (CN)i~. При реакции с аминами образует соот-
ветствующие аммониевые трицианметаниды, а с AgNO3 — трицианметанид
серебра.
Дицианкетенимин по реакционной способности подобен изоцианатной
кислоте, что не в последнюю очередь обусловлено псевдохалькогенидным
характером С (СИ)2-группы [26]. Как и HNCO, дицианкетенимин вступает
в реакции присоединения с водой, спиртами и галогеноводородами [3, 7,
24, 28]
NC NC NH2
\c=C=NH + HR -> \с=с/
NC NC \
(R=OH, NH2, NHR, Cl, Br). (7.5)
Тесное родство между HNCC (CN)2 и HNCO прослеживается также в реак-
ции изомеризации анилиновой соли дицианкетениминной кислоты
/NH2
[C,H6NH,]+ [С (CN),!- -> (NC), С=С< (7.6)
XNHC6H6,
l/2 Н 2473
305
которая аналогична синтезу Велера мочевины из цианата аммония [28].
На основании псевдогалогенидного характера С (CN)2-rpynnH 1,1-диамино-
2,2-дицианоэтилен рассматривается как гомолог мочевины [13].
7.4.2. Галогентрицианметаны
Реакцией трицианметанида серебра с хлором или бромом в эфире получа-
ют соответствующие галогентрицианметаны [7, 29] в форме бесцветных субли-
мирующихся соединений
AgC (CN)3 + Х2 -> AgX + ХС (CN)3 (X = Cl, Br). (7.7)
Оба вещества имеют тетраэдрическую структуру. ОС (CN)3 — бесцвет-
ные иглы (т. пл. 46—47° С; v (CN) 2262, 2209; v (CC) 857 см-1).
Для BrC (CN)3 характерны реакции расщепления водой и цианидом до
гипобромидной кислоты, бромциана и трицианметанида [29]
BrC (CN)3 + Н2О -> НОВг + Н+ + С (CN)f; (7.8)
BrC (CN)3 + CN- -> BrCN + С (CN)f. (7.9)
Подобно другим галогенпсевдогалогенидам BrC (CN)3 реагирует с метал-
лоорганическими, бромртутьорганическими соединениями и бромидами
металлов, образуя металлоорганические псевдогалогениды или смешанные
трипианметанидбромиды металлов [30]. Эти реакции свидетельствуют о по-
6— <н-
лярности связи С—Вг (С—Вг)
R4Sn + BrC (CN)3 -> R3SnC (CN)3 + RBr; (7.10)
R2M + BrC (CN)3-> RMC (CN)3+RBr; (7.11)
RHgBr+ BrC (CN)3 -> RHgC (CN)3 + Br2; (7.12)
MBr4 + BrC (CN3) -> MBr3C (CN)3 + Br2. (7.13)
Хлорид и бромид фосфора (III) легко присоединяют хлор- или бромтри-
цианметан с образованием желтых или оранжевых соединений фосфора
(V) РС14С (CN)3 [31] и PBr4C (CN)3 [30]. Оба производных имеют строение
NC X
/С=с<^ (Х=С1, Вг).
NC XN=PX3
Эти соединения также получают реакцией обмена РС15 с NaC (CN)3
[64, 65]. Галогентрицианметаны ХС (CN)3 (X = Cl, Вг) реагируют с РЬэР
164], образуя соединения
NC X
\с=с<^
NC XN=PPh3
7.4.3. Металлоорганические трицианметаниды
Металлоорганические трицианметаниды получают различными спосо-
бами. Наряду с реакциями (7.10) — (7.12) возможны реакции обмена метал-
лоорганических галогенидов с трицианметанидом серебра [32, 33]
R„MX + AgC (CN)3 -> RnMNCC (CN)3 + AgX;
R2T1X + AgC (CN)3 -> R2T1NCC (CN)2 + AgX, (7.14)
306
реакции дицианкетенимина с ме-
таллоорганическими гидрокси-
дами [32]
R/ЮН + HNCC (CN)2 ->
R3MNCC (CN)2 + Н2О, (7.15)
а также обмен хлоридов диалкил-
олова с трицианметанидами ще-
лочных металлов [34]
R2Sn2+ + 2С (CNK ->
-> R2Sn(NCC(CN)2)2. (7.16)
Если металлоорганические
трицианметаниды являются до-
Таблица 7.2. Частоты валентных колебаний CN
элементоорганических трицианметанидов
Соединение v (CN). cm *
(СеН5)3 SiNCC (CN)2 (CeH6)2 T1NCC (CN)2 (С6НБ)3 PbNCC (CN)2 (CeH6)3 SnNCC (CN)2 (C2H5)3 SnNCC (CN)2 (CH3)2 Sn (NCC (CN)2)2 (C6H6)2 Sn (NCC (CN)2)2 2162, 2172 (пл.), 2220 2174, 2228 2180, 2193, 2233 2172, 2200 (пл.), 2240 2185, 2203 (пл.), 2255 2185, 2215, 2233 2175—2190 (ш.), 2219
Относительная интенсивность: пл. — плечо; ш. — ши-
рокая полоса.
вольно стабильными соединениями, то единственное до сих пор соедине-
ние кремния Ph3SiC (CN)3 [331 устойчиво лишь при температуре ниже
—20° С. Это соединение спонтанно полимеризуется при комнатной темпе-
ратуре в оранжево-красное ве-
щество.
В известных до сих пор ме-
таллоорганических трицианмета-
нидах связь металл — лиганд
осуществляется через азот, что
подтверждается числом и поло-
жением полос v (CN) (табл. 7.2).
Для трифенилтрицианметанида
олова этот тип координации
С (CN)3-rpynn однозначно следу-
ет также из спектров 14N-HMP
[29] (6NO> = +221, +51 м. д.).
Соединения R3SnNCC (CN)2 и
R2Sn (NCC (CN)2)2 представляют
собой координационные полиме-
ры, в которых металл имеет три-
гонально-бипирамидальную или
октаэдрическую конфигурацию
за счет мостиковой функции би-
дентатных анионных лигандов
(тип структуры VI). Отчасти это
следует из ИК-спектров соедине-
ний, а также из данных спектров
Мессбауэра, согласно которым
особенно высокое квадрупольное
9е ®N
7.2. Структура (СН3)2 TIC (CN)3 [54]
расщепление указывает на координационные числа олова больше 4 (на-
пример, для Ph3SnNCC (CN)2 6 = —1,19; е = 3,50 мм/с относительно SnO2
как стандарта). Подобное строение имеют метилоловодицианамиды [35, 36].
Для MeTINCC (CN)2, полученного из Ме2ТП и AgC (CN)3, выполнено
рентгеноструктурное исследование [54]. В этом соединении (кристаллы ор-
торомбические, а = 26,123; b = 6,264; с = 21,007 A; Z = 16; пространст-
венная группа Fddd) реализуется очень интересный тип структуры, в кото-
ром трицианметанид функционирует как четырехдентатный мостиковый
лиганд, а центральный атом имеет координационное число 6 (рис. 7.2).
Связь в линейной группе СН3Т1СН3 (^Z СТ1С = 178,7°) преимущественно
% П
307
ковалентная. В экваториальной плоскости таллий окружен 4М-атомами,
причем связь таллий — трицианметанид-ион преимущественно ионная. Уг-
лы связи равны: CNT1 169,5; ССС 121,0 и 118,0° CCN 180 и 176,4°. Расстояния:
С—N 1,13 и 1,21 А; С—С 1,42 и 1,33 А. Представляет также интерес соед! -
некие (Me4N)+ [С (CN)3 (А1Ме3)3]~, которое получают реакцией [Me4N] -
• С (CN)3 с Ме3А1. В нем трицианметанид функционирует как тридентатный
лиганд (тип XII), в то время как линейные псевдогалогениды (CN, SCX
в соответствующих комплексах бидентатны [37]
"АКСН3)3 А1(СН3)3
..-.N
^С. .
А1(СН3)3
XU
7.5. ТРИЦИАНМЕТАНИДЫ d-МЕТАЛЛОВ
Трицианметаниды двухвалентных Зс1-металлов М (С (CN)3)2 легко полу-
чают из водных растворов при взаимодействии солей металлов (II) с трициан-
метанидами щелочных металлов [2, 7, 17, 38]
М(Н2О)1+ + 2С (СК -* М (С (CN)3)2. (7.17)
При этом чаще всего образуются безводные или содержащие 0,5 моля
Н2О соединения, не растворимые в органических растворителях малой или
средней донорной силы. Этот факт, а также ПК- и электронные спектры сое-
7.3. Строение полимерно-октаэдрических трицианмета-
нидов металлов М (С (CN)3)2 [17]
динений показывают, что
трицианметаниды металлов
типа [М (С (CN)3)2J — это
координационные полиме-
ры, в которых анионный
лиганд является мостиком.
Поэтому центральные ато-
мы всегда находятся в ок-
таэдрическом окружении
лигандов (рис. 7.3).
Структура, представлен-
ная на рис. 7.3, подтверж-
дается спектроскопически-
ми и магнитными данными
табл. 7.3 (частоты v (CN)
характерны для тридентат-
ных С (CN)3 -групп; удовлетворительное отнесение d—d-полос возмож-
но при принятии октаэдрической симметрии; магнитные моменты сое-
динений никеля и кобальта находятся в типичной для октаэдрических комп-
лексов области). В ИК-спектрах [М (С (CN)3)2] (М = Zn, Cu, Mn, Fe, Со,
Ni) в области 200—320 см 1 появляются полосы средней интенсивности,
308
которые относят к М—N-валентным колебаниям. Их положение зависит от
энергии стабилизации кристаллическим полем. Свободный С (CN)T-hoh в этой
области имеет лишь одну слабоинтенсивную полосу при 210 см-1 [55].
Рентгеноструктурное исследование Си (С (CN)3)2 показывает, что окта-
эдрическая структура этого соединения тетрагонально искажена (эффект
Яна—Теллера). Лиганд, выступающий тридентатным мостиком, в значи-
тельной степени сохраняет свою планарность (углы связи ССС 12Г 24',
Таблица 7.3. Трицианметаниды двухвалентных Sd-металлов
Соединение Цвет Элек- тронный спектр, см—1 Отнесение ^эф v(CN)
Fe (С (CN)3)2- • 0,5Н2О Зеленый 12 100 21 800 5^ —> 5р 12g с g ппз 5,17 2160 2205 (пл.), 2196 (пл.) (оч. с.), 2246 (ср.)
Co(C(CN)3), Светло-ко- ричневый 9600 20 000 21 300 25 000 4у 4у % % 4Т -> 47 (Р) 4g т1& ’ ППЗ 4,79 2160 2220 (пл.), 2208 (оч.' с.) (пл.), 2268 (пл.)
Ni (С (CN)3)2 Светло-го- лубой 9700 16 700 25 500 ад ад О, см — ' S. S. ад СО СО | ф СО ад ад Ф <м <м Ч й СО СО СО 2,86 2170 2225 (пл.), 2218 (оч. с.) (пл.), 2268 (сл.)
Си (С (CN)3)2 Коричне- вый 13 900 21 800 2р -> 2Т £2g '2g ППЗ 1,86 2210 2275 (оч. с.), 2218 (пл.) (ср.)
Zn (C (CN)3)2 Бесцветный — — — 2160 2220 (пл.), 2172 (ср.) (оч. с.), 2267 (ср.)
Относительная интенсивность: пл. — плечо, сл. — слабая, оч. с. — очень сильная, ср. — средняя.
117° 20'). Длины связей С—С и С—N 1,40—1,42 А и 1,15 А, такие же, как
в свободном ионе; в тетрагонально искаженном октаэдре CuN6 длины свя-
зей Си—N колеблются от 1,98 до 2,49 А [39].
ЭСХА-спектры трицианметанидов d-металлов обнаруживают ожидае-
мое повышение N—ls-энергии связи лигандов примерно на 0,2—0,5 эВ
вследствие образования координационных связей металл—лиганд [60].
Трицианметанид ртути как полимернокоординационное соединение (v(CN)
2160 (сл.), 2195, 2210 (оч. с.), 2259 см-1) отличается от других псевдогалоге-
нидов ртути сильной ионной составляющей связи. Это соединение гидроли-
тически разлагается щелочами, в пиридине является сильным электроли-
том типа 1 : 2 [38]. Подобно Hg (SCN)2 соль Hg (С (CN)3)2 разлагается при
нагревании с образованием объемистых коричневых продуктов реакции.
Трицианметанид серебра выпадает из водного раствора ионного трициан-
метанида при добавлении AgNO3 в видео бесцветных кристаллов (ортором-
бические: а = 6,21; b = 10,19; с = 7,98 A; Z = 4; пространственная группа
12cm). AgC (CN)3 -— координационный полимер, в котором С (CN)3-rpynna
является тридентатной. Каждый атом Ag, образуя пирамиду, окружен
тремя N-атомами разных С (CN)3-rpynn. Соединение характеризуется следу-
ющими углами связи: NAgN 99,7 и 123,9°; CCN 172,7 и 172,9°; ССС 118 и
123,5°; CNAg 172 и 151,3°; длина связи Ag—N 2,11 или 2,23 А [58].
11 + */2 2473
309
Таблица 7.4. Частоты валентных колебаний v (CN)
металлоорганических трицианметанидов
[RMNCC (CN)2]n, см-1
[RZnNCC (CN)2]„ [RCdNCC (CN)2jn [RHgNCC (CN)2]n
R=C2H52215 (оч.с.) 2238 (с.) 2270 (ср.) R=C6H5 2150 (пл.) 2215 (оч. с.) 2275 (ср.) 2215 (оч. с.) 2230 (сл.) 2270 (сл.) 2220, 2270 (пл.) 2205 (оч. с.) 2230 (пл.) 2260 (ср.)
Относительная интенсивность: сл. — слабая; с. — сильная;
оч. с. — очень сильная; ср. — средняя; пл. — плечо.
Трицианметаниды трехвалентных Зd-мeтaллoв М (С (CN)3)3 (М = Ti,
V, Сг) получают обменом AgC (CN)3 с соответствующими галогенидами ме-
таллов в ацетонитриле [11, 39]
МХ3 + 3AgC (CN)3 -> М (С (CN)3)3 + 3AgX. (7.18
Для этих соединений также характерны трицианметанидные мостики,
образующие соединения с полимерными структурами: V (NCC (CN)9)3 —
фиолетовый, v (CN) 2210
(оч с.), 2263 (сл.); Ti(NCC-
- (CNj2)3 — серо-белый, v
(CN) 2180 (сл.), 2213 (оч.
с), 2250 (сл.).
Полимерные структуры
предполагаются также для
трицианметанидобромидов
металлов MBr3NCC (CN),
(v (CN) 2170 (пл.), 2212
(оч. с.), 2250 (сл.)) и Nb-
- (С (CN)3)5, WO (С (CN)3)4
[11].
Интересно, что метал-
лоорганические трициан-
метаниды RMNCC (CN)2,
реакцией между BrC (CN)3
упоминавшиеся в 7.2 и получаемые обменной
и металлоорганическими соединениями, также содержат тридентатный три-
цианметамид. Эти соединения, не растворимые в растворителях малой поляр-
ности, обнаруживают полосы CN-валентных колебаний в типичной для три-
дентатного трицианметанида области (табл. 7.4). На рис. 7.4 воспроизведен
вариант строения полимерно-тетраэдрических металлоорганических псевдога-
логенидов. Высокодонорные растворители, например пиридин, расщепляют
типа
7.4. Строение органометаллтрицианметанидов
RMNCC (CN)2 (М = Zn, Cd, Hg)
логенидов. Высокодонорные растворители, например пиридин,
анионные мостики с образо-
ванием комплексов типа
[RM — NCC (CN)2 Ру2].
Анионные трицианмета-
нидные комплексы Зд-ме-
таллов неустойчивы, что,
вероятно, обусловлено сте-
рическими факторами [42,
431. Однако для цент-
ральных ионов большего
объема такие типы комп-
лексов более устойчивы.
Так, для кадмия, наряду
с [Cd (С (CN)3)4]2—, описа-
ны смешанные псевдо-
галогенидокадматы [Cd (С (CN)3)2 (NCS)2]2' и [Cd (С (CN)3)4 (NCS)2]4-
(v (CN) (C (CN)3)“ 2175, 2209, 2240 см-1). При попытке получить [Pd (С •
• (CN)3)4]2- в ходе реакции между PdCl2 и КС (CN)3 удалось синтезировать
лишь смешанный комплекс К2 [Pd (С (CN)3)3C1] (желтые кристаллы, т. пл.
295° С) [57].
Образованию трицианметанидных комплексов в растворе посвящены
лишь единичные исследования. Трицианметанид подобно другим псевдога-
|2-
[44]
310
логенидам реагирует с железом (III) с образованием красного комплекса
[Fe (Н2О)6 С (CN)3]2— [45]. Б системе Си2+ —С (CN)T доказано существо-
вание комплекса [Си (Н2О)5 N (CN)2]+ [43].
Как и следовало ожидать, ввиду малой основности трицианметанид-
иона, устойчивость трицианметанидных комплексов значительно ниже ана-
логичных цианатных комплексов.
7.6. ТРИЦИАНМЕТАНИДНЫЕ КОМПЛЕКСЫ ПЕРЕХОДНЫХ
МЕТАЛЛОВ С ДРУГИМИ ЛИГАНДАМИ
Для получения трицианметанидных комплексов металлов с другими ли-
гандами применяют два метода синтеза: 1) непосредственное взаимодейст-
вие трицианметанидов металлов с подходящими лигандами; 2) из других
комплексов металлов в результате реакции замещения. Первый метод можно
использовать прежде всего для получения трицианметанидных комплексов
с «жесткими» лигандами, второй — преимущественно для синтеза три-
цианметанидных комплексов с «мягкими» лигандами.
7.6.1. Комплексы с N- и О-донорными молекулами
Обменом между солями двухвалентных Зб-металлов в водных растворах
со щелочными трицианметанидами в присутствии пиридина получены пири-
диновые комплексы различных типов, которые при нагревании или пере-
осаждении из пиридина способны к взаимному переходу друг в друга [38].
Комплекс меди (II) [CuPy4 (С (CN)3)2] (голубые иглы) на воздухе отщепляет
две молекулы пиридина и превращается в зеленый [CuPy2 (С (CN)3)2].
Ниже приведен состав некоторых комплексов с пиридином
МРу2 (NCC (CN)2)2 (М - Mn, Со, Ni, Cu, Zn, Cd);
МРу3 (NCC (CN)2)2 (М = Mn, Fe, Со, Zn);
МРу4 (NCC (CN)2)2 (M = Ni, Си, Co, Cd).
Из растворов трицианметанидов ртути (II) или меди (I) в пиридине выде-
лены бесцветные монопиридиновые комплексы HgPy (C(CN)3)2 nCuPyC(CN)3
[38]. Комплекс PdPy2 (NCC (CN)2)2 получен обменной реакцией PdPy2Cl2
с AgC (CN)3 [57].
Согласно электронным спектрам и магнитным измерениям, как тетра-,
так и дипиридиновые комплексы имеют октаэдрическую конфигурацию.
Монодентатность анионных лигандов в тетрапиридиновых комплексах и их
бидентатность в дипиридиновых комплексах подтверждается положением по-
лос v (CN) в ИК-спектрах (табл. 7.5). В противоположность большинству
описанных до сих пор комплексов с пиридином с мостиковым трнцианмета-
нидом, комплекс PdPy2 (NCC (CN)2)2 является планарным, а анионный ли-
ганд монодентатным. На рис. 7.5 приведена предлагаемая структура длясое-
динений полимерного типа.
Трицианметаниды металлов в пиридине ведут себя как слабые или сред-
ней силы электролиты, так что в этом растворителе предполагается частич-
ный обмен лигандов
MPy4(NCC(CN)2)2 -Ь Ру [MPy5(NCC(CN)2]+ + C(CN)^ (7:19)
п + v? зи
Таблица 7.5. Частоты валентных колебаний v (CN) некоторых аминокомплексов, см
Соединение v (CN)
Монодентатные C (CN)3 NiPy4 (NCC (CN2)2 CoPy4 (NCC (CN)2)2 PdPy2 (NCC (CN)2)2 Бидентатные C (CN)3 NiPy2 (NCC (CN)2)2 MnPy2 (NCC (CN)2)2 CuPy2 (NCC (CN)2)2 ZnPy2 (NCC (CN)2)2 Ni (2-CH3C5H4N)2 (NCC (CN)2)2 Ni (2,4- (CH3)2 C5H3N)2 (NCC (CN)2)2 Co (2-CH3C5H4N)2 (NCC (CN)2)2 Co (4-CH;C5H4N); (NCC (CN)2)2 2150 (пл.), 2178 (оч. с.), 2230 (ср.) 2175 (оч. с.), 2229 (ср.) 2180 (с.), 2235 (с.) 2160 (ср.), 2203 (оч. с.), 2245 (ср.) 2160 (пл.), 2195 (оч. с.), 2245 (ср.) 2198 (оч. с.), 2241 (ср.) 2160 (пл.), 2205 (с.), 2250 (ср.) 2200 (пл.), 2212 (оч. с.), 2252 (ср.) 2162 (ср.), 2202 (оч. с.), 2250 (ср.) 2200 (оч. с.), 2250 (пл.) 2190 (оч. с.), 2250 (ср.)
Относительная интенсивность: сл. — слабая: с. — сильная; оч. с. — очень сильная; ср. — средняя;
пл. — плечо.
Как было показано на примере целого ряда диаминокомплексов кобальта
и никеля М (Amin)2 (NCC (CN)2)2 (Amin = хинолин, изохинолин, 2-,3,4-
метилпиридии, 2,3-, 2,4-диметилпиридин), октаэдрическая структура сохра-
няется и тогда, когда в качестве лигандов выступают а-замещенные пириди-
7.5. Строение полимерно-октаэдри-
ческих аминокомплексов типа
[М (NCC (CN)2)2 (Amin)2] (Amin —
= Ру; 2-, 3-, 4-CH,C5H5N; 2,3-,
2,4-(СН3)2 C5HsN; M = Co. Ni)
на 121, 46, 47]. Это заслуживает внимания,
так как в аналогичных комплексах с ли-
нейными псевдогалогенидами такого же
типа М (Amin)2 (NCA)2 замещение пириди-
на на его а-замещенные всегда ведет к изме-
нению полимерно-октаэдрической конфи-
гурации на мономерно-тетраэдрическую
или плоскую.
С гетероциклическими диаминами, как
Dipy или Phen, трицианметаниды никеля
(II) и железа (II) образуют соответству-
ющие трис(диамин)-комплексы [М (Dipy)3] •
• (С (CN)s)2 и [М (Phen)3] (С (CN)3)2 [16].
Описан смешанный диаминокомплекс пал-
ладия, включающий 5-нитро-, 1,10-фенан-
тролин [57]. Для железа (III) описан син-
тез тетрафенилпорфинжелезо (III) трици-
анметанида, полученного из [i-оксо-бис-
(тетрафенилпорфин) аквожелеза (III) и вод-
ного раствора дицианкетенимина [56].
Аналогично пиридину другие растворители с высокой донорной силой,
например диметилсульфоксид [48] или диметилформамид [17], разрывают
лигандные мостики в трицианметанидах переходных металлов. В получен-
ных растворах спектрофотометрически доказано существование катионов
М (DMSO)|+ и М (DMFA)26+. В случае с DMSO были изолированы крис-
таллические соединения типа [М (DMSO)6] (С (CN)3)2 (М = Мп, Со, Си,
Zn, Cd), а также [Ni (DMSO)6] (С (CN)3)2 • 2DMSO (v (CN) в [Mn (DMSO)61 •
• (C (CN)3)2 2176 cm”1).
Трицианметаниды металлов тенденцией к образованию катионов
M (DMSO)!4' похожи на перхлораты металлов. Это является следствием ма-
лой основности трицианметанид-иона, так как сильно основные линейные
псевдогалогениды образуют в этих же условиях псевдогалогенидные комп-
лексы металлов, например [М (NCS)2 (DMSO)J.
Трицианметаниды двухвалентных Зс1-металлов довольно хорошо раство-
римы в гексаметилфосфортриамиде (НМРА). Путем добавления индифферент-
ных растворителей получают комплексы типа М (НМРА)3 (NCC (CN)2)2
(М = Мп, Со, Ni, Zn), а также комплекс меди Cu (НМРА)4 (NCC (CN)2)2
[62]. В большинстве случаев трицианметанид выполняет функцию бидентат-
ного мостика, и только для комплексов Zn (НМРА)2 (NCC (CN)2)2 (тетраэдр)
и Cu (НМРА)4 (NCC (CN)2) (искаженный октаэдр) можно допустить коорди-
национную монодентатность лиганда [63].
Из электронных спектров различных трицианметанидных комплексов
металлов можно установить положение трицианметанид-иона в спектрохими-
ческом ряду. В смешанных комплексах с жесткими лигандами трицианмета-
нид всегда координируется через азот. Поэтому, как следовало ожидать,
в спектрохимическом ряду трицианметанид всегда располагается между дру-
гими азотсодержащими лигандами, но по отношению к изотиоцианату смещен
вправо: Г < Вг~ < СГ < F- < Н2О < NF < NCO- <C5H5N < NCS- ~
- N (CN)7 < SeCN- < NH3 < NCC (CNF < NOF < Phen < CNO- <
< CN-.
Би- и тридентатный С (С^3-лиганд создает несколько более слабое поле,
как это следует из сравнения энергетически слабых d—d-переходов (3A2g ->
-> 3T2g= 10Dq) ряда комплексов никеля: NiPy4 (NCC (CN)2)2 (11 000 см-1),
[NiPy2 (NCC (CN)2)2]„ (10 870 см-1), [Ni (C (CN)3)2]„ (9700 см-1) и Ni (Py6)2+
(9850 cm-1) [49].
7.6.2. Комплексы с С- и Р-донорными молекулами
Псевдогалогенидный характер трицианметанид-иона проявляется также
в его поведении по отношению к гексакарбонилам металлов. При обмене меж-
ду трицианметанидом калия или тетраэтиламмония и гексакарбонилами
М (СО)в (М = Cr, Мо, W) легко образуются соответствующие трицианмета-
нидкарбонилметаллаты
М (СО)6 + КС (CN)3 -> КМ (СО)5С (CN)3 + СО. (7.20)
Устойчивость тетраэтиламмониевых солей, получаемых с очень хорошим
выходом, увеличивается, как и для аналогичных изотиоцианатных комплек-
сов, в ряду Сг—Мо—W [50] (v (CN) [Et, N] [W (СО)5С (CN)3] 2170,
2227 см-1).
Галогенпентакарбонилы марганца и рения реагируют с трицианметани-
дом калия или серебра путем замещения двух молекул СО и образова-
ния полимерных комплексов типа [М (СО)3 NCC (CN)2]n (v (CN) для М = Мп
2218 см-1, для М = Re 2192 см-1) [25]
М(СО)5 X + KC(CN)3 -> [M(CO)3NCC(CN)2] +КХ +2СО. (7.21)
Трифенилфосфин разрушает лигандные мостики: образуются мономерные
соединения [М (СО)3 (Ph3P) NCC (CN)2]. Б то время как в карбонильных
производных марганца и рения осуществляется связь лиганда с металлом
через азот, в комплексе [W (СО)5С (CN)3], который первоначально тоже рас-
сматривался как производное N-трицианметанида, новые данные 14N-HMP-
313
измерении говорят в пользу связей трицианметанида с металлом через угле-
род [52].
Нитрозилгалогениды кобальта и никеля реагируют с КС (CN)3, образуя
полимерные дицианкетениминатные комплексы [Со (NO)2 NCC (CN)2]n (тем-
но-коричневый, v (CN) 2200, 2220 (сл.), 2268 см"1) и [Ni (NO) NCC (CN).,]n
(темно-голубой, v (CN) 2218 см~ ). В обоих случаях в подходящих реакцион-
ных условиях фосфины разрушают трицианметанидные мостики до моно-
или биядерных фосфиновых комплексов: [Со (NO)2 R3 PNCC (CN)2], [Ni (NO) •
•(РзР)г NCC (CN)21 и [Ni (NO) R3PNCC (CN)2]2 [51]. Реакции между цикло-
пентадиенилдикарбонилиодидом железа, а также бис (циклопентадиенил)-
Таблица 7.6. Частоты валентных колебаний v (CN) некоторых фосфиновых комплексов
благородных металлов
Соединение v (CN), см 1
Цис- (Ph3P)2 PtCIC (CN)3 Транс- (Ph3P)„ PtHC (CN)3 (Ph3P2) IrCONCC (CN)2 Транс- (Et3P)2 PtHNCC (CN)2 (Ph3P2) Rh CONCC (CN)2 Транс- (Ph3P)2 Pd (NCC (CN)2)2 (Ph3P) Rh (NCC (CN)2)2 (Ph3As)2 Pt (NCC (CN)2)2 (Ph3P)2 Pd (NCC (CN)2)2 (Ph3As)2 Pd (NCC (CN)2)2 2184 (c.). 2235 (cp.) 2178, 2227 2174, 2192 (cp.), 2230 2175 (c.), 2185, 2220 2174 (c.), 2190, 2227 2175 (c.), 2193 (сл.), 2238 (cp.) 2173 (c.), 2223 (сл.) 2182 (c.), 2190 (пл.), 2239 (cp.) 2170 (c.), 2230 (c.) 2145, 2170 (c.), 2180 (c.), 2235 (cp.)
Относительная интенсивность: с. — сильная; ср. — средняя; пл. — плечо.
дихлоридом титана или дибромидом циркония с трицианметанидом серебра
или калия ведут к образованию комплексов Cp2Fe (СО)2 С (CN)3, а также
Ср2М (NCC (CN)2)9 (v (CN), М - Ti 2177 (оч. с.), 2210 (оч. с.), 2240 (сл.);
М = Zr 2175 (оч. с.), 2240 (ср.) [53]).
Трицианметанид железа (II) реагирует с ар ил изоцианидами с образова-
нием желтых октаэдрических комплексов типа Fe (RNC)4 (NCC (CN)2)2.
В противоположность этому трицианметанид кобальта (II) восстанавливает-
ся фенилизонитрилом. Получены желтые комплексы кобальта (I)
[Со (RNC)5]+ [С (CN)3]~, которые содержат наряду с тригонально-бипирами-
дальным катионом пентафенилизонитрила кобальта (I) ион трицианметанида
(v (CN) С (CN)3 2178 см”1) [16].
Путем обмена трицианметанида калия с рядом фосфиновых комплексов
платиновых металлов, например
[(Ph3P)2 PtHCl] + КС (CN)3 -> (Ph3P)3 PtHC (CH)3 + KC1, (7.22)
можно получить трицианметанидные комплексы состава [МСО (Ph3P)2-
•С (CN)3] (М = Ir, Rh) и [МХС (CN)3L2] (М = Pt, Pd; L = Ph3P, Ph3As;
X — Cl, Br, CN, H, C (CN)3), а также соединения родия (Ph3P)3 RhC (CN)3
[22, 25]. Описаны комплексы палладия типа [Pd (NCC (CN)9)2L2] (L = Ph3P,
Ph3As) [57].
Окислительное присоединение тетрацианметана или дицианкетенимина
к Pt (Ph3P)4 приводит к соответствующим комплексам Pt (II) [25]
Pt (Ph3P)4 + С (CN)4 -> транс-(РЬ3Р)2 Pt (CN) C (CN)3 + 2Ph3P; (7.23)
Pt (Ph3P)4 + HNCC (CN)2 -> транс-(РИ3Р)2 PtHC (CN)3 + 2Ph3P. (7.24)
314
Для большинства описанных трицианметанидфосфиновых комплексов
благородных металлов связь трицианметанида с металлом осуществляется
через азот. Это подтверждается наличием трех частот CN-валентных коле-
баний. Такая связь для комплекса иридия (Ph3P)2 IrCONCC (CN)2 следует
также из 14N-Я МР-измерений (6No3~ +234), а для (Et3P)2 PtHNCC (CN)2 —
из сравнения положения сигнала протонов в ПМР-спектрах (т = 27,6)
с аналогичными изоцианатными комплексами (для изоцианатных и изотио-
цианатных комплексов этого типа т = 27,7 и 27,6). Эти данные также пока-
зывают, что трицианметанид, как и NCX (X = О, S)-rpynna, обладает лишь
незначительным транс-эффектом [22]. В фосфиновых комплексах благород-
ных металлов трицианметанидные группы могут координироваться через
углерод, что подтверждается наличием лишь двух валентных CN-колебатель-
ных полос (табл. 7.6), а также положением сигнала в спектрах 14N-HMP для
комплексов цис-(РЬ3Р)2 PtCIC (CN)3 (6nos= + 118) и транс-(РН3Р)9 Pt НС •
- (CN)3 (6No3 = +99 м. д.) [52].
СПИСОК ЛИТЕРАТУРЫ
1. Birckenbach L., Huttner К.— Z. anorg. allg. Chem., 1930, 190, S. 1.
la. Madelung WKern E.— Ann. Chem., 1922, 427, p. 1.
2. Schmidtmann H.— Ber., 1896, 29, S. 1168.
3. Hantsach A., Osswald G.— Ber., 1899, 32, S. 641.
4. Boyd R. H.— J. Am. Chem. Soc., 1961, 83, p. 4288.
5. Trofimenko S.— J. Org. Chem., 1963, 28, p. 217.
6. Kohler H.— Z. Chem., 1973, 13, S. 401.
7. Trofimenko S., Little E. L., Jr., Mower H. F.— J. Org. Chem., 1966, 27, p. 433.
8. Birckenbach L., Huttner K.— Ber., 1929, 62, S. 153.
9. Mayer E.— Monatsh. Chem., 1969, 100, S. 462.
10. Trofimenko S., McKusick В. C.— J. Am. Chem. Soc., 1962, 84, p. 3677.
11. Kohler H. Unveroffentlicht.
12. Cox E., Fontaine A.— Bull. Soc. chim. France, 1954, p. 948.
13. Middleton W. J., Engelbardt V. A.— J. Am. Chem. Soc., 1958, 80, p. 2788.
14. Anderson P., Klewe B., Thom E.— Acta chem. Scand., 1967, 21, p. 1530; Nature,
1963, 200, p. 464.
15. Desiderate R., Sass R. L.— Acta Cryst., 1965, 18, p. 1.
16. Kohler H. Habilitationsschrift. Halle, 1967.
17. Enemark J. H., Holm R. H.— Inorg. Chem., 1964, 3, p. 1516.
18. Miller F. A., Baer W. K-—Spectrochim. Acta, 1963, p. 73.
19. Kohler H.-— Z. Chem., 1973, 13, S. 401.
20. Boyd R. H.— J. Phys. Chem., 1963 , 67, p. 737.
21. Kohler H., Hartung HSeifert B.— Z. anorg. allg. Chem., 1966, 347, S. 30.
22. Lenardt M., Baddley W.— J. Organometal. Chem., 1972, 39, p. 217.
23. Kohler H.— Z. anorg. allg. Chem., 1964, 331, S. 237.
24. Kohler H.— Wiss. Z. Univ. Halle. XVIII’69 M, 1969, 33.
25. Beck WSchlopp R., Oetker C. a. o.— Chem. Ber., 1973, 106, p. 2144.
26. Kohler H., Kotte B., Salowski R.— Z. anorg. allg. Chem., 1970, 379, S. 183.
27. Middleton W. J., Little E. L., Coffman D. D., Engelhardt V. A.— J. Am. Chem. Soc.,
1958, 80, p. 2795.
28. Allenstein E.— Chem. Ber., 1963, 96, S. 3230.
29. Birckenbach L., Huttner K-— Ber., 1929, 62, S. 153, 2065.
30. Kohler H., v. Millmann E.— Z. anorg. allg. Chem., 1970, 373, S. 222.
31. Emeleus H. J., Toy A. D. F.— Inorg. Nucl. Chem., 1967, 29, p. 269.
32. Beck W„ Smedal H. S., Kohler H.— Z. anorg. allg. Chem., 1967, 354, S. 69.
33. Kohler H., Maushake R. H.— J. Organometal. Chem., 1967, 14, p. 103.
34. Kohler H., Seifert B.— J. Organometal. Chem., 1968, 12, p. 293.
35. Chow V. K.— Inorg. Chem., 1971, 10, p. 1938.
36. Kohler H., Neef L., Korees L., Burger K.— J. Organometal. Chem., 1975, 90, p. 159.
37. Weller F., Wilson I. 0., Dehnicke K-— J. Organometal. Chem., 30, c. 1.
38. Kohler H.— Z. anorg. allg. Chem., 1964, 331, S. 237.
39. Biondi C., Bonamico M., Torelli L., VaciagoA.— Chem. Commun., 1965, 191.
40. Ley H., Kissel H.— Ber., 1899, 32, S. 1361.
315
41. Kohler H., Laub P.— J. Less-Common Metals (Amsterdam), 1971, 23, p. 171.
42. Kohler H., Seifert B.— Z. anorg. allg. Chem., 1966, 344, S. 63.
43. Kohler H., Seifert B.— Z. Naturforschg., 1967, 22b, S. 238.
44. Келер X., Скопенко В. В., Тряшин А. С., ЗибЮ. Л.— Укр. хим. журн., 1974, 40,
с. 1121.
45. Kohler И., Seifert В,— Z. Chem., 1967, 7, S. 32.
46. Kohler Н. — Z. Chem., 1965, 5, S. 142.
47. Kohler H., Seifert В.— Z. anorg. allg. Chem., 1967, 352, S. 265.
48. Kohler FL— Z. anorg. allg. Chem., 1965, 336, S. 245.
49. Rosenthal M., Drago R. S.— Inorg. Chem., 1965, 4, p. 840.
50. Beck W7., Nitzschmann R., Smedal H. S.— J. Organometal. Chem., 1967, 8, p. 547.
51. Beck W., Hieber W., Heumsir G.— Z. anorg. allg. Chem., 1966, 344, S. 285.
52. Beck W., Fehlhammer W. P.— MTP Internal. Rev. Sci., Inorg. Chem., Serie One,
1973, 2, p. 253.
53. Issleib K-, Kohler FL, Wille G.— Z. Chem., 1973, 13, S. 347.
54. ChowY. M., BrittonD.— Acta Cryst., 1975, B31, p. 1934.
55. Kolbe A., Kohler H.— Z. anorg. allg. Chem., 1977, 428, S. 113.
56. Reich M. F., Cohen LA.— J. Inorg. Nucl. Chem., 1970, 32, p. 343.
57. Likowski T. L., Burmeister I. L.— Inorg. Chim. Acta, 1976, 17, p. 117.
58. Konnert J., BrittonD.— Inorg. Chem., 1966, 5, p. 1193.
59. Middleton W. J., Engelhardt V. A.— J. Am. Chem. Soc., 1958, 80, p. 2788.
60. Salyn Js. W., Nefedow WKohler H.— Z. anorg. Chem., 1977, 433, S. 13.
61. Escard J., Mavel G., Guerchais J. E., Kergoat R.— Inorg. Chem., 1974, 13, p. 695.
62. Matschiner H. Unveroffentlicht.
63. Kohler H., Skirl R., Skopenko V. W.— Z. anorg. allg. Chem., 1977, 428, S. 115.
64. Кухарь В. П., Павленко FL Г., Кирсанов А. В.— Журнал общей химии, 1973, 43,
с. 1896; 1978, 48, 778.
8- ДИЦ^АИАМИДЫ
8.1. ПСЕВДОГАЛОГЕНИДНЫХ ХАРАКТЕР
ДИЦИАНАМИД-ИОНОВ
Дицианамид, N (CN)S , впервые описан в 1922 г. [1]. Авторы работы
[11 подчеркнули генетическую связь этого иона с трицианметанид-и цианат-
ионами, что позволило отнести N (CN)^-hoh к псевдогалогенидам [21.
Тесная связь [N (CN)2]~ с цианатом, а также с трицианметанидом явля-
ется следствием псевдохалькогенидного характера NCN-группы (ср. струк-
туры I и II) [44]
NCN = С == N“ О — С = №
I II
Дицианамиды щелочных металлов получают реакцией обмена цианами-
да с бромцианом в щелочной среде
NaNHCN + BrCN + NaOH -> NaN (CN)2 + NaBr + H2O. (8.1)
Они представляют собой игольчатые кристаллы, которые легко растворя-
ются в воде и умеренно — в спирте и других полярных растворителях. От-
носительно сильная кислота HN (CN)2 в водных растворах неустойчива.
Попытки выделения ее в индивидуальном состоянии до сих пор не привели к
успеху [1, 3].
Дицианамиды щелочных металлов во многих реакциях похожи на циа-
наты. При нагревании они превращаются в соли циануровой кислоты —
гомолога трицианмеламина [4]
NCN 3—
3NaN (CN)2Na3 'с N N II 1 NCN—С С—NCN (8.2)
Подобно цианату и трицианметаниду дицианамиды взаимодействуют со
спиртами, аминами и галогенводородами.
Маделунг и Керн [1] описали взаимодействие дицианамида с одним молем
спирта
NaN(CN)2 + С2Н5ОН -> Na+
NH
II
/С-ОС2Н8
IM
с
N
(8.3)
317
позже было показано, что ионы металлов Zn2+, Си катализируют реакцию
присоединения двух молекул спирта [5]
Н3СО NH ОСН3
N (CN)~ 4- 2СН3ОН + H+(+Zn2+u Jf (8’4)
NH NH
NaN (CN)2 способен присоединять галогеноводороды в мольном соотно-
шении 1:1 и 1 : 3. При этом образуются N-циан-галоген-формамидины —
соединения солевого типа [6, 7]
NH2
NaN (CN)2 + 2НХ -> NaX + ^C-NCN
XX
4-2HX
"NH2 nh2~
\c=N=C<X
XX \\
X -f- NaX.
(8.5)
Амины реагируют c NaN (CN)2 в мольном соотношении 1 : 1 и 1 : 2. При
реакциях, частично катализируемых ионами металлов, образуются произ-
водные гуанидина [8, 9]
NHCH3
NaN (CN)2 + [CH3NH3] Cl NCN=c/ + NaCl; (8.6)
xnh2
NH NH
II II
NaN (CN)2 + 2 [(CH3)2 NH2] X -> (CH3)2N—С C —N (CH3)2+Na+ + H+.
\hZ
Для дицианамидных аммонийных солей характерны реакции превраще-
ния в производные мочевины. В синтезе Веллера гомологов мочевины
дицианамид подобен трицианметаниду аммония в аналогичных реакциях
NHR
]RNH31+ [N (CN)2]~ -> NCN=C<^ ;
NH2
nh2
[NHJ+[N(CN)2]- N=C—N=C<^
'nh2
(8-7)
Типичный псевдогалогенидный характер NCN-группы, вытекающий из
названных здесь параллелей его реакционной способности с цианатом и три-
цианметанидом, находит свое выражение также в образовании труднорас-
творимой в воде соли AgN (CN)2, в ярко выраженной комплексообразующей
тенденции, в существовании характерных соединений переходных металлов
[MN (CN)2]n и ковалентных металлоорганических дицианамидов [10—12].
Соответствующий псевдогалоген, (NC)2N — N (CN)2, тетрацианогидразин
до сих пор не получен.
318
8.2. СВОЙСТВА И СТРУКТУРА ДИЦИАНАМИД-ИОНА
Дицианамиды щелочных металлов можно синтезировать разными спо-
собами. Модифицируя уже известный способ получения MN (CN)2 из
MNHCN и XCN, в качестве исходных препаратов используют Ca (CN)2
и галогеноциан [9].
Дицианамид выделяют из реакционной смеси осаждением труднораство-
римой соли цинка Zn [N (CN)2]2.
Согласно работе [13], NaN (CN)2 получают взаимодействием NaNH2
и BrCN. NH4N (CN)2 получен действием сероводорода на аммиачный раствор
Cu [N (CN)2]2 или при взаимодействии BrCN, растворенного в 1,2-димето-
ксиэтане с жидким аммиаком [14].
Дицианамиды различных щелочных металлов получают реакциями обме-
на Zn [N (CN)2]2 с карбонатом или гидроксидом соответствующего щелочного
металла. Этот способ используется и для получения [Me4N] N (CN)2. Дици-
анамиды [Ph4P] N (CN)2, [Ph4As] N (CN)2 и [Ph3MeAs] N (CN)2 получены об-
менной реакцией соответствующих галогенидов с дицианамидами щелочных
металлов в воде [16].
По данным ИК- и КР-спектров [3, 15], N (CN)r имеет угловую структу-
ру. Кратность связи передается мезомерными граничными формулами
N
С
С
N
Таблица 8.1. Полосы
в ИК-спектре N (CN)2“
V, см 1 Отнесение
529 6 (NCN)
543 6 (NCN)
664 ё (CNC)
930 vc (C-N)
1344 Vac (C-N)
2179 Vc (C=N)
2232 Vac (C=N)
2286 vc + Vac (C—N)
группа Р2г/с;
Расчеты МО [17] дают значительно большую плотность заряда на атоме
азота циано-группы (о + зт = —1,155), чем на амидном атоме азота (о +
+ эх = —0,677), поэтому отдается предпочтение граничным формам III
и IV. Интересно, что плотность заряда на атоме
азота циано-группы значительно отличается от
вычисленной для азота CN-группы трицианметани-
да. Из рентгеновских спектров получены следую-
щие значения N — ls-энергии связи в NaN (CN)2:
N-циано-группы 398,8; N-амидной группы 400,1 эВ
[36]. Экспериментально найденные энергии связи
и вычисленные электронные плотности находятся
в хорошем согласии [36].
Для NaN (CN)2 наблюдается химический сдвиг
сигналов 14N : +391 (для N-амидной группы) и
+225 м. д. (для N-циано-группы) (стандарт —
насыщенный водный раствор NaNO3) [43].
Плоская структура N (CN)^~, установленная из
данных спектроскопии, подтверждена рентгено-
структурным анализом. Для KN (CN)2 (пространственная
а = 3,8701; b = 15,9630; с = 6,8411 А; [3 = 99,73°). Найдены такие пара-
метры: углы связи CNC 120,3° и NCN 172,8°; расстояния С—N в циано-груп-
пах 1,155 и 1,156 А; длина связи: N—С 1,304; 1,309; 1,317 А [18]. При этом
необычными являются отклонения NCN-групп от ожидаемой линейности.
319
Основные частоты в ИК-спектрах N (СЫ)Г (Сгу-симметрия) приведе- .
в табл. 8.1 (для NaN (CN)2). В области ниже 400 см~’ проявляются мало1 -
тенсивные полосы при 202,225 и 250 см-1 [37].
Образование ковалентных или комплексных дицианамидов металлов ве-
дет к характерным изменениям прежде всего в положении полос валентных
колебаний CN-группы, а также к появлению полос валентных колебан; 1
v (М— N) средней интенсивности в области 200—360 см-1. Анализ ИК-
спектров дает важнейшую информацию о характере связи N (CN)2-rpynn з
производных дицианамида.
8.3. ТИПЫ СВЯЗИ В ПРОИЗВОДНЫХ ДИЦИАНАМИДА
И ИХ ИНТЕРПРЕТАЦИЯ
До последнего времени были описаны лишь отдельные ковалентные ди-
цианамиды металлов, главным образом металлоорганические дицианамиды.
В настоящее время известен целый ряд дицианамидов комплексного типа.
Несмотря на малую основность (дицианамид занимает положение между
С (CN)r и NCS-), N (CN)7 является хорошим лигандом. Будучи амбиден-
татным лигандом, он в принципе способен образовывать координационные
связи через азот циано-группы или через амидный азот. Более высокая элек-
тронная плотность на азоте CN-группы приводит к образованию преимуще-
ственно связи М—L через атом азота циано-группы. Как видно из структур-
ных формул Via и VI6, принципиально возможны линейные или искривлен-
ные сочетания М—N—С.
zN=C=N М—N==C—N
Via VI5
Как и трицианметанид, дицианметанид способен выступать в роли мос-
тика
М—N=C—N
Vila
VII б
M-N=C-N
VIII
Изображенные структурные формулы дают представления о типах свя-
зей, не затрагивая проблему возможных углов связей MNC.
320
Поскольку дицианамид-ион имеет четыре свободные электронные пары,
в принципе он может выступать и в качестве тетрадентатного мостикового ли-
ганда (структура IX). Недавно на примере (СН3)2 TIN (CN)2 было показано,
что этот структурный тип также осуществим [35].
м-n/
IX
X
Наконец, принимая во внимание пространственные затруднения, можно
утверждать, что дицианамидные группы не должны образовывать хелат-
ных соединений. Случаи образования связи лиганд — элемент через амид-
ный азот до сих пор единичны и наблюдаются в некоторых комплексах пал-
ладия (табл. 8.2) [19] (см. формулу X).
Проявляемый тип связи в производных дицианамида находит свое отра-
жение в характере ИК-спектров этих соединений [12]. При этом решать во-
прос о типе связи Via (тип симметрии Cs) или X (при принятии планарной
Таблица 8.2. Положение полос валентных колебаний CN-групп дицианамидов
с различным типом связи *
Соединение v (CN) Тип связи Дентатность лиганда
[Со (N (CN)2)4]2~ (раствор) 2185 (c.), 2233 (cp.) VI 1
[Со (N (CN),)O (NCS)2]2~ [Pd (N (CN)2)4]2- 2168 (оч. c.), 2230 (cp.) 2232 (оч. c.), 2298 (c.) X 1
[Me2Sn (N (CN)2)2]„ [NiPy2 (N (CN)2)2]„ 2210—2225 (c.), 2265 2200 (оч. c.), 2255 (cp.) VII 2
[Ni (N (CN)2£-]„ [Ni (N(CN)2)2]„ 2185 (оч. c.), 2250 (cp.) 2210 (c.), 2260 (пл.) VIII 3
* Если не указано отдельно, ИК-спектры получены в нуйоле или КВг.
Относительная интенсивность: с.—сильная; оч. с. —очень сильная; ср. — средняя; пл.—плечо.
М—N (СЫ)2-группы; симметрия С2у) на основании сдвига полос поглощения
невозможно. В обоих случаях можно ожидать 13 собственных колебаний
группировок металл — дицианамид, которые, за исключением А2 колеба-
ний для типа X, являются ИК-активными [40]. Однако здесь возможно
различие в положении полос валентных колебаний CN-группы. В случае
связи М—L через атом азота циано-группы (тип VII) полосы v (CN) наблю-
даются в области 2160—2175; 2230—2235 см-1. При этом различие между
структурами Vila и VI16 можно обнаружить лишь рентгеноструктурным
анализом. До сих пор структурные данные для комплексов с монодентат-
ной N (CN)2-rpynnoft отсутствуют, для полимерных металлоорганических
дицианамидов (структуры VII и IX) углы MNC близки к 180° [20, 35].
Для координационного полимера AgN (CN)2 угол AgNC равен 156,3° [41].
О соединениях, в которых N (CN)2-rpynna координирована через амид-
ный азот (структура X), до сих пор данных мало, все же исследования изо-
мерии связи в комплексах палладия типа [Pd (N (CN)2)il2~ показывают, что
321
2000 2200 2400см''
этот тип координации характеризуется смещением полос
v (CN) в коротковолновую область [45] (см. табл. 8.2).
Если в соединении дицианамид выполняет мости-
ковую функцию, то подобно полимерным трицианме-
танидным комплексам наблюдается сдвиг v (CN) в сто-
рону более высоких волновых чисел. Для типа связи
VII колебания v (CN) наблюдаются в области 2190—
2200 и 2235—2500 см-1, а для тридентатной N (CN)2-
группы—в области 2200—2220 и 2270—2280 см-.
Табл. 8.2 дает представление о положении частот ва-
лентных колебаний CN-групп для отдельных типов свя-
зей, а на рис. 8.1 приведены соответствующие части спек-
тров.
Обе возможности координации дицианамида в качест-
ве бидентатного лиганда (структуры Vila, VI16) нельзя
различить лишь на основании положения полос погло-
щения CN-группы. Для этого необходимо применить
другие источники информации, например данные о сдви-
ге полос v (М—N)b области ниже 400 см-1 (см. 8.5 и 8.6).
К настоящему времени рентгеноструктурные исследо-
Рис. 8.1. Часть ИК-спектров дицианамидных комплексов:
а—(Ph4P)2Co(N(CN)2)2(NCS)2 (монодентатный динианметанид-лиганд); б —
Ni(2v-Pic)2(N(CN)2)2 (бидентатный дицианметанид-лиганд); в — Ni(N(CN)2)2
(тридентатный динианметанид-лиганд)
вания известны лишь для некоторых соединений: Me3SnN (CN)2, Me2Sn(N •
• (CN)2)2 [20] и AgN (CN)2 [45]. Бидентатность анионных лигандов в этих
соединениях соответствует типу связи Vila, в то время как в Me2TlN •
• (CN)2 N (СН)2-лиганд является тетрадентатным (тип связи IX) [35].
8.4. ДИЦИАНАМИДЫ р-ЭЖМЕНТОй
Кроме солеобразных дицианамидов металлов, например Pb (N (CN)2),
и TIN (CN)2, к настоящему времени получены металлоорганические дици-
анамиды элементов IV и III групп. Для их синтеза в большинстве случаев
используют реакции металлоорганических галогенидов с AgN (CN)2 в таких
органических растворителях, как бензол, тетрагидрофуран или метанол
[35, 46],
R3MX + AgN (CN)2 -> R3MN (CN)2 + AgX (M = Si, Ge, Sn, Pb; X = Hal);
(8.8a)
R2T1X + AgN (CN)2 R2T1N (CN)2 + AgX. (8.86)
Me2Sn (N (CN)2)2 также получают прямым взаимодействием катиона
Me2Sn2+ с N (CN)? в водном растворе [46]
Me2Sn2+ + 2N (CN)? -> Me2Sn (N (CN2)2. (8.9)
Совершенно по-другому реагирует с дицианамидами щелочных металлов
РС15. Компоненты вступают в реакцию в мольном соотношении Ь: 1 с обра-
,322
зованием 1,1,3,5-тетрахлор-!-фосфор-2,4,6-триазина (21]
N(CN)2- + РС15
ПЫ^С-Ьк
VL-C!
-®|n=c=nx
CI3P-N^
4
Cl
I
/N-Сх
CI2Pf
Cl
(8.10)
Ковалентные дицианамиды металлов из-за отсутствия резонансной ста-
билизации менее устойчивы по сравнению с ионными и комплексными ди-
цианамидами. Прочность металлоорганических дицианамидов возрастает
с увеличением электрополо-
жительного характера метал-
ла. Кремнийорганические ди-
цианамиды устойчивы только
при низкой температуре, при-
чем их устойчивость очень
сильно зависит от природы
органического остатка. Отно-
сительно устойчивыми явля-
ются Ph3SiN (CN)2 и Ph2Si •
• (N (CN)2)2, которые в про-
тивоположность Me3SiN (CN)2
выше 5° С спонтанно полиме-
ризуются с образованием твер-
дых веществ, окрашенных в
цвета от желтого до оранже-
вого [19]. На основании ИК-
спектроскопических исследо-
ваний (табл. 8.3) можно пред-
полагать в рассматриваемых
соединениях образование свя-
Таблица 8.3. Полосы валентных колебаний
CN-группы некоторых металлоорганических
дицианамидов
Соединение v (CN)
(С6Н5)3 SiN (CN)a 2200 (оч. c.), 2284 (cp.) 2148 (пл.), 2212 (оч. c.), 2266 (cp.)
(C6H5).2Si (N (CN)2)2
(C6H5O)3 SiN (CN)2 2150 (пл.), 2210 (оч. с.), 2267 (ср.)
(CH3)3SiN (CN)2 2154 (пл.), 2214 (оч. с.), 2262 (ср.)
(C6H5)3 GeN (CN)2 2207 (оч. с.), 2255 (с.)
(C6H5)3 SnN (CN)2 2173 (оч. с.), 2200 (оч. с.) 2175—2190 (оч. с.), 2219 (ср.)
(C6H5)3 Sn (N (CN)2)2
(CH3)2 Sn (N (CN)2)2 2217 (оч. с.), 2265 (ср.)
(C6H5)3 PbN (CN)2 2155 (оч. с.), 2225 (ср.)
Относительная интенсивность: с. — сильная; оч. с. —
очень сильная; ср. — средняя; пл. — плечо.
зи кремний — цианамид через амидный азот (тип X), в то время как в ме-
таллоорганических дицианамидах Ph3MN (CN)2 (М = Ge, Sn, Pb) и других
оловоорганических дицианамидах связь М—L осуществляется через азот
CN-группы. Из данных ИК-спектроскопии следует, что в Me2Sn (N (CN)2)2
и Me3SnN (CN)2 присутствуют мостиковые N (СИ)2-группы. В Ph3SnN (CN)2
наличие мостиковых N (CN)2-rpynn подтверждается данными мессбауэров-
ских спектров [22] (табл. 8.4).
Высокие значения квадрупольного расщепления указывают на то, что
олово имеет координационное число больше 4 за счет мостиковой функции
N (CN)2-rpynn.
Для Me3SnN (CN), и Me2Sn (N (CN)2)2 известны рентгеноструктурные дан-
ные, которые показывают, что в обоих соединениях N (CN)2-rpynnbi мости-
323
ковые. В полимерных соединениях атомы олова имеют тригонально-бипи-
рамидальную или слегка искаженную октаэдрическую конфигурацию.
СН3-группы занимают в Me3SnN (CN)2 экваториальное, а в Me2Sn (N •
• (CN)2)2 аксиальное положение. Как видно из рис. 8.2 и 8.3, планарность
Таблица 8.4. Мессбауэровские спектры N (СМ)2-группы сохраняется. Наблюдаемый угол связи только незначительно отличается от
Соединение Изомерный сдвиг 6 Квадруполь- ное расщепле- ние Д£, мм/с предполагаемого для типа связи Vila [20]. Me3SnN (CN)2 крис- таллизуются в орторомбической решетке, пространственная груп- па Pnam, а = 17,644; b = 6,565; с = 7,864 А; дляМе25п (N (CN)2)2
Me9Sn (N (CN)2)2 Ph3SnN (CN)2 —1,07 —1,12 4,47 3,72
найдена моноклинная простран-
ственная группа Р2/С с а =" 6,178; b = 11,265; с = 6,860 А; [э = 99,56°.
Соответственно для Me3SnN (CN)2 и Me2Sn (N (CN)2)9 углы равны: NCN 172,1
и 171,8°;oCNSn 173,7 и 174,0°; CNC 129,6 и 124,2°;‘расстояния: N—С 1,263
и 1,289 А; С—N (CN-rpynna) 1,116 и 1,192 A;Sn—N 2,335 и 2,289 А.
Интересна структура Me2TlN (CN)2 (тип IX). Тетрадентатность мости-
ковых N (СМ)2-лигандов обусловливает искаженную октаэдрическую конфи-
гурацию центрального атома с двумя метильными группами в аксиальном
положении (угол СН3Т1СН3 176°) и атомами азота аниона в экваториальном
положении. Соединение относится к моноклинной пространственной группе
Р2х/с параметрами: а = 6,739; b = 11,830: с = 9,891 А; р = 117,8°; Z = 4
Все атомы азота N (CN)2-rpynn связаны с разными атомами Т1 с расстояния-
ми 2,60 и 2,90 А, 2,79 или 2,81 А [35]. Межатомные расстояния равны:
©W О 5/?
Рис. 8.2. Кристаллическая структура (CH3)2Sn (N(CN)2)2 [20] (проекция
на плоскость (010))
324
©c @/v Q)Sn
Рис. 8.3. Кристаллическая структура (CH3)3SnN (CN)2 (проекция на
плоскость (001))
NaMIW—С 1,31 и 1,26 А; Мциан — С 1,19 и 1,18 А; углы связи: NCN 172,0 и
171,4°; CNC 120,4°; СМцианТ1 156,5, 117,2 и 117,6°; CNaMWXTl 116,0 и 123,5°
135].
8.5. ДИЦИАНАМИДЫ d-МЕТАЛЛОВ
Дицианамиды одно- и двухвалентных металлов MN (CN)2 (М = Ag,
Cu, Hg) и М (N (CN)2)2 (М = Cu, Zn, Cd, Hg, Mn, Fe, Co, Ni) описаны разны-
ми авторами [1, 3, 10, 37]. Их получают реакцией обмена между соответ-
ствующими солями металлов и дицианамидами щелочных металлов в воде или
органических растворителях малой и средней донорной силы или термоли-
зом их дипиридиновых комплексов 110].
Образованием плохо растворимых дицианамидов Ag (I) и Hg (I) дициан-
метанид-ион аналогичен галогенид-ионам, и его можно отнести к типичным
псевдогалогенидам. Образуя в то же время малорастворимые дицианамиды
двухвалентных Sd-металлов по уравнению
М(Н2О)б+4-2N (CN)F -> М (N (CN)2)2 + 6Н2О, (8.11)
дицианамид-ион весьма напоминает трицианметаниды. Подобно трицианме-
танидам образуются координационные полимерные дицианамиды метал-
лов. Если для трицианметанидов наблюдается единый полимерно-октаэдри-
ческий тип соединений, то дицианамиды металлов М (N (CN)2)2 относятся
к двум структурным типам. Наряду с доминирующим полимерно-октаэдри-
ческим типом IX (рис. 8.4) наблюдается полимерно-тетраэдрический [10,
37], в котором N (CN)2-rpynna связана с двумя атомами металла, причем
координация осуществляется, вероятно, через атомы азота CN- и амидо-
групп (структура VI16). Здесь возможна, как, например, в дицианамидах
Со (II) и Mn (II), изомерия связи.
Полимерно-октаэдрические a-формы получают прямым синтезом из вод-
ных растворов по схеме (8.11), а полимерно-тетраэдрические [3-формы —
325
термолизом дипиридиновых комплексов в вакууме
[МРу2 (N (CN)2)J ₽-М (N (CN)2)2 + 2Ру (8.12)
(М = Мп, Со).
Рис. 8.4. Строение полимерно-октаэдриче-
ских дицианамидов металлов М (N (CN)2)2
(М-двухвалентный Зд-металл)
, £-Со (N (CN)2)2 при хранении на воздухе медленно превращается в более
устойчивую a-форму. Из водных растворов получают также полимерно-
октаэдрические дицианамиды никеля (II) и меди (II). Соединения меди из-за
интенсивной зеленой окраски пред-
ложено использовать для качест-
венного обнаружения дицианамида.
В отличие от дицианамидов кобаль-
та, никеля и меди дицианамид цин-
ка осаждается из водного раство-
ра в полимерно-тетраэдрической
форме. На это указывает образова-
ние синих смешанных кристаллов
Со/Zn (N (CN)2)2, а также желтых
Cu/Zn (N (CN)2)2, причем в послед-
них присутствует невыделенный в
свободном состоянии р = Cu (N •
•(CN)2)2. В табл. 8.5 представлены
важнейшие данные для некоторых
дицианамидов металлов. Электронные спектры соединений Ni, Со и Си и маг-
нитные моменты комплексов Со и Ni согласуются с принятым строением
этих соединений.
Частоты v (CN) обоих типов комплексов по сравнению с таковыми сво-
бодного цианамид-иона значительно повышены; практически не наблюдается
никаких изменений в положении частот vac (С—N) по сравнению с NaN (CN)2,
в то время как частоты vc (С—N) преимущественно сдвинуты в более длинно-
волновую область спектра. В области ниже 400 см-' для а- и Р-дицианами-
дов металлов наблюдается сильное расщепление полос в области 200—280
Таблица 8.5. Характеристика дицианамидов некоторых Зй-металлов
Соединение Цвет Электрон- ный спектр, см—1 Переход Иэф» М. Б. v (CN) Структура
Ni(N(CN)2)2 Светло-го- лубой 9700 16 000 26 140 ЗД х ЗТ rt2g 4g ЗД ЗТ a2g 11g 3A2g-3Tlg(P) 3,18 2210 (оч. с.) 2260 (пл.) Полимерно- октаэдриче- ская
а-Со (N (CN)2)2 Розовый 19 200 20 800 4ты -4A2g 4Tig-4Alg(P) 4,98 2210(оч. с.) 2250 (ср.)
cs-Cu (N (CN)2)2 Зеленый 14 080 29 970 4g ППЗ 1,85 2204 (оч. с.) 2220 (пл.)
p-Co (N (CN)2)2 Со/Zn (N (CN)2)2 Cu/Zn (N (CN)2)2 Zn(N (CN)3)2 Т емно-го- лубой Темно-го- лубой Желтый 16 660 16750 11 000 21 500 4А2 -> 2Т1 (Р) 4А2->Ь(Р) аТ2 -> аЕ ппз 4,60 2192 (оч. с.) 2220 (пл.) 2208 (оч. с.) 2281 (ср.) Полимерно- тетраэдри- ческая
Относительная интенсивность; оч. с. — очень сильная; ср. — средняя; пл. —> плечо.
326
и 300—360 см ’. Эти полосы относят к v (М—N). Для р-дицианамидов
металлов полосы v (М—N) лежат выше 300 см-1 и смещены в длинноволно-
вую область [37].
Рентгеновские спектры дицианамидов d-металлов указывают вследствие
образования координационных связей М—L на повышение энергии N—ls-
связи, которая в этом случае составляет 0,2—0,5 эВ [361.
AgN (CN)2 имеет тригональную сингонию (пространственная группа
РЗдЗ!; а = 3,601; с = 2,2868 A; Z = 3) и образует спиралеобразные це-
пи — Ag—NCNCN—Ag—NCNCN— с почти линейной N—Ag—N-связью
(X. NAgN = 172,9°). Расстояния Ag—N, N=C и N—С равны соответствен-
но 2,111, 1,148 и 1,291 А. Найденное расстояние Ag—N соответствует тако-
вому в AgNCO и меньше, чем в AgC (CN)3 (2,16 А). Углы связей CNC и NCN
составляют 127,0 и 170,3° [41]. Относительно большой угол связи CNC сви-
детельствует о том, что, кроме граничных структур III—V, для описания
соотношения связей в лигандах можно принять граничную формулу “IN=
=C=N=NI~ [41].
Как и трицианметанид, дицианметанид ртути (И) изменяет свой цвет при
нагревании с характерным вспучиванием, которое напоминает наблюдаемое
для Hg (SCN)2 образование «фараоновых змей». В пиридине является силь-
ным электролитом типа 1 : 2.
Дицианамиды трехвалентных переходных металлов М (N (CN)2)3 (М =
= V, Ti, Сг) получают обменной реакцией галогенида металла с AgN (CN)2
в ацетонитриле [23]
МХ3 + 3AgN (CN)2 -> М (N (CN)2)3 + 3AgX. (8.13)
Приведенные ниже соединения также являются координационными поли-
мерами: V (N (CN)2)3, фиолетовый, v (CN) 2200 (оч. с.), 2279 (пл.); Ti (N-
• (CN)2)3, светло-серый, v (CN) 2193 (оч. с.), 2242 (пл.). Дицианамидные мос-
тики образуются в соединениях Nb (N (CN)2)5 (желтый) и WO (N (CN)2)4
(красный).
8.6. ДМЦИАНАМИДНЫЕ КОМПЛЕКСЫ d-МЕТАЛЛОВ
8.6.1. Комплексообразование в растворе
Образование дицианамидных комплексов переходных металлов изучено
недостаточно. Подобно другим псевдогалогенидам N (CN)7 реагирует в воде
с Fe3’1' с образованием окрашенного в оранжево-красный цвет раствора, в
котором обнаружены комплексы FeN (CN)2+ и [Fe (N (CN)2)2]+ [24]. Медь
(II) образует с N (CN)F зеленые растворы Cu (N (CN)2)2 [25]. По устойчивос-
ти дицианамидные комплексы располагаются в ряду между тиоцианатными
и трицианметанидными (NCS~ > N (CN)F > С (CN)^).
8.6.2. Анионные комплексы
Подобно другим линейным псевдогалогенидам в органических раство-
рителях средней донорной силы дицианамид образует с рядом d-металлов
анионные комплексы. При этом Sd-металлы образуют соединения типа
327
[M (N (CN)2)4]Z и [M (N (CN)2)3]ra, а для более объемных центральных ионов
(например, Cd2+) возможно образование комплексов [М (N (CN)2)6]4~.
Синтез анионных комплексов осуществляют обменными реакциями в
в ацетоне или ацетонитриле по уравнению
MJ [МХ4] + 4AgN (CN)2 М2 [М (N(CN)2)4] + 4AgX
(Х = С1, Br; М = Со, Cu, Cd, Pd). (8.14)
Кроме того, на сольватированные дицианамиды металлов можно дейст-
вовать избытком дицианамида
M(N (CN)2)2 (сольв.) + 2M'N (CN)2 -> М2 [М (N (CN)2)4J. (8.15)
При этом особенно благоприятное влияние на стабилизацию анионных
комплексов оказывают катионы типа Ph4P+ или Ph4As+.
Таблица 8.6. Структурные типы анионных дицианамидных комплексов
Соединение v(CN) Структура
[Ph3MeAs]2 [Со (N (CN)2]4 (раствор) [Ph3MeAs]2 [Со (N (CN)2]4 (кристаллы) [Ph4As] [Ni (N (CN)2)3] K4 [Cd (N (CN)2)6J K2 [Cd (N (CN)2)4) 2185 (оч. c.), 2233 (c.) 2170, 2195 (оч. c.), 2243, 2257 (cp.) 2185 (04. c.), 2250 (cp.) 2160 (04. c.), 2220 (c.) 2160 (пл.), 2200 (04. сЛ, 2230 (пл.), 2270 (ср.) ' Мовомерно-тетраэдри4еская (дипианамид монодентатный) Полимерно-октаэдринеская (дипианамид моно- и биден- татный) Полимерно-октаэдринеская (дицианамид бидентатный) Мономерно-октаэдринеская (дицианамид монодентатный) Полимерно-октаэдринеская (дипианамид моно- и биден- татный)
Относительная интенсивность: с.— сильная; оч.
с.— очень сильная;
ср.— средняя; пл.— плечо.
Дицианамидные соединения металлов интересны тем, что в зависимости
от природы центрального атома и условий синтеза они различны по струк-
туре. Так, дицианамиды Со (II) проявляют интересную изомерию: в раство-
ре и расплаве они имеют мономерно-тетраэдрическую конфигурацию, а в
кристаллическом состоянии образование дицианамидных мостиков приводит
к полимерно-октаэдрической конфигурации этих соединений [26] (см.
табл. 8.6). Подобные структурные изменения от искаженной мономерно-
тетраэдр'ической конфигурации в растворе к полимерно-октаэдрической в
кристаллическом состоянии наблюдаются и для [Cu (N (CN)2)4]2~ [25].
В растворе [Со (N (CN)2)4]2~ имеет интенсивные полосы поглощения при 606
(1120) и 615 (пл.) нм, а также 1160 (пл.), 1300 (173) и 1430 (пл.) нм, которые
соответствуют переходам 4А2—> 4ТХ (Р) и 4А2 -> 4ТХ (F). Число и положение
полос валентных колебаний CN-групп (см. табл. 8.7) подтверждает моноден-
татность анионных лигандов. Напротив, полимерно-октаэдрическая струк-
тура кристаллических комплексов этого типа подтверждается как окраской,
так и магнитным моментом [Ph3MeAs]2 [Со (N (CN)2)4] (рэф = 4,92 М. Б.),
а также числом и положением наблюдаемых v (СЙ)-полос (табл. 8.7) [26].
Из спиртовых растворов можно выделить полимерно-октаэдрические
комплексы кобальта (II), например [Ph3MeAs] [Со (N (CN)2)3] (рэф = 5,01
М. Б.). Выделенный из спирта [Ph4As] [Ni (N (CN)2)3] (цэф = 3,17 M. Б.)
также имеет полимерно-октаэдрическую структуру [27]. Вероятно, в этих
328
1
Таблица 8.7. Структурные типы смешанных анионных псевдогалогенидных комплексов
Соединение v (CN), cm-1 Структура
Цианат Дицианамид
[Ph4P]2 [Со (N (CN)2)2(NCS)2] 2070 (оч. с.) 2168 (с.), 2230 (ср.) Мономерно-тетраэдриче- ская
[Ph4P]2 [Со (N (CN)2)2(NCO)2] 2195 (с.) 2172 (с.), 2210 (ср.) (лиганды монодентатные)
[Ph4As]2 [Ni (N (CN)2)2(NCS)2] 2100 (с.) 2160 (пл.), 2185 (с.), 2260 (пл.) 2188 (оч. с.), 2252 (ср.) Полимерно-октаэдричес- кая
[Ph4P]2 [Ni (N (CN)2(NCS)2] 2058, 2170 (с.) (дицианамид бидентатный)
K4 [Cd (N (CN)2)4(NCSe)2] 2104,2120 (с.) 2158, 2218 (с.) Октаэдрическая (лиганды монодентатные)
[Ph4P]2[Cd (N (CN)2)2(NCS)2] 2082 (с.) 2165 (с.), 2225 (с.) Мономерно -1 етраэ дричес- кая
(Ph4P)2 [Cd (N (CN)2)2 (NCSe)2] 2085 (с.) 2165 (с.), 2225 (с-) (лиганды монодентатные)
Относительная интенсивность: с. — сильная; оч. с. — очень сильная; ср. — средняя; пл. — плечо.
полимерных комплексах образуются мостики посредством атома азота
CN-группы. В низкочастотной области ИК-спектра для [Ni (N (CN)2)3]~
наблюдается только одна полоса при 267 см-1 (240 пл.), которую относят к
Q)=M(Co.Nc)
= N(CN)-Z
Рис. 8.5. Строение полимерно-октаэдрических комплексов
[M(N(CN)2)3]~
(М = Со. Ni)
валентному колебанию М—Ncn [37]. Рис. 8.5 дает представление о структуре
дицианамидов металлов типа [М (N (CN)2)3]~.
Как и следовало ожидать, [Cd (N (CN)2)6]4— содержат монодентатные
N (CN)2-rpynnbi, а из ИК-исследований следует, что комплексы типа
[Cd (N (CN)2)4]2~ образуют полимерно-октаэдрическую структуру с мости-
ковой функцией части анионных лигандов (табл. 8.7) [26].
В смешанных псевдогалогенидах металлов, например [М (N (CN)2)2 •
• (NCA)2]2~ (А = О, S, Se), во многих случаях реализуется иное строение,
чем в однородных дицианамидах. Так, кобальт в [Со (N (CN)2)2 (NCA)2]2~
и [CoN (CN)2 (NCA)3]2— в твердом состоянии и в растворе имеет пре-
имущественно мономерно-тетраэдрическую конфигурацию, что следует из
12 2473
329
ИК-спектров соединений (табл. 8.7), а также из магнитных и спектрофото-
метрических данных: [Ph4As]2 [Со (N (CN)2)2 (NCS)21: рЭф = 4,58 М. Б.;
d—d-переход 4А2 —4ТХ (Р): 590 (пл.), 622,5 (1800) нм; [Ph4As]2 [CoN
• (CN)2(NCO)3J: рэф = 4,65 М. Б.; 4А2 -> 4ТХ (Р) переход: 575 (380), 612
(680), 637 (760) нм [39].
Никель (И) образует полимерно-октаэдрические комплексы типа
[Ni (N (CN)2)2 (NCS)2]2~, в которых N (СЫ)2-группы проявляют бидентат-
ную мостиковую функцию [27]. Такое же строение имеет [Ph4P]2 [Ni (N •
• (CN)2)2 (NCS)2]: рЭф = 3,31 M. Б. В противоположность этому смешанные
комплексы кадмия типа [Cd (N (CN)2)4 (NCA)2]4~ и несколько неожиданно
также [Cd (N (CN)2)2 (NCA)2]2— (А = S, Se) содержат только монодентатные
псевдогалогениды [28] (табл. 8.7).
8.6.3. Дицианамидные комплексы
с другими лигандами
При взаимодействии дицианамидов металлов с другими лигандами
образуются преимущественно полимерно-октаэдрические комплексы типа
[ML2 (N (CN)2)2], в которых дицианамид выступает как мостиковый
бидентатный лиганд (рис. 8.6).
Обменным взаимодействием солей двухвалентных Зб-металлов с дициан-
амидами щелочных металлов в присутствии пиридина образуется [МРу2-
Рис. 8.6. Строение полимерно-октаэд-
рических комплексов типа [ML2(N •
• (CN)2)2J
(L = C5H6N; 2-,3-,4-CHsCeH4N; 2,3-,
2,4-(CHs)2C5H3N: M = Co, Ni)
• (N (CN)2)2] (M = Mn, Fe, Co, Ni, Cu, Zn). Эти соединения имеют полимер-
но-октаэдрическое строение [10]. Как видно из табл. 8.8, полосы v (CN)
лежат в области, типичной для бидентатных N (CN)2-лигандов. Это строение
сохраняется, если вместо пиридина ввести его а-производные, например
2-метилпиридин, 2,4- или 2,3-диметилпиридин, хинолин или изохинолин.
Так, спектры отражения и магнитные моменты комплексов [NiL2 (N (CN)2)2]
четко указывают на октаэдрическую симметрию соединений [29, 30]
(табл. 8.8 и 8.9). Это также справедливо для аналогичных комплексов ко-
бальта (II) [31].
330
Таблица 8.8. Полосы валентных колебаний CN-группы и тип связи дицианамидных комплексов
ML2 (N (CN)2)2
Соединение v (CN), CM-1 Тип связи
Ni (2-CH3C5H4N)2(N (CN)9)2 Ni (2,4-(CH3)2C5H3N)2(N (CN)2)2 Ni (4-CH3C5H4N)2(N (CN2)2 CoPy2(N (CN)9)2 Co (2-CHsC5H4N)2(N (CN)2)2 Co (2,3-(CH3)2C6H3N)2(N (CN)2)2 Mn (DMSO)2(N (CN)2)9 Ni (DMSO)2(N (CN)9)2 Co (DMSO)2(N (CN)2)2 Ni (HMPA)2(N (CN)2)2 Co (C6H5CH2NC)2(N (CN)2)2 Mn (HMPA)2(N(CN)2)2 2208 (c.), 2254 (cp.) 2199 (оч. c.), 2257 (cp.) 2193 (оч. c.), 2253 (cp.) 2188 (оч. c.), 2246 (c.) 2190 (оч. c.), 2254 (c.) 2191 (оч. c.), 2246 (cp.) 2190 (оч. c.), 2245 (cp.) 2200 (оч. c.), 2363 (cp) 2192 (оч. c.), 2255 (cp.) 2190—2225 (c.), 2300 (cp.) 2179 (c.),2235 (cp.) 2172 (c.), 2236 (cp.) Полимерно-октаэдричес- кая структура, дицианамид в мостиковой функции би- дентатный Дицианамид монодентат- ный
Относительная интенсивность: с.— сильная: оч. с.— очень сильная: ср.— средняя.
Вследствие полимерной структуры комплексы типа [МРу2 (N (CN)2)21
нерастворимы в большинстве органических растворителей, но растворяются
в растворителях с высокой донорной способностью. Пиридиновые растворы
соединений МРу2 (N (CN)2)2 содержат, очевидно, тетрапиридиновые комп-
лексы, поскольку рассматриваемые растворы являются неэлектролитами.
Диметилсульфоксид подобно пиридину разрушает дицианамидные мос-
тики полимерных дицианамидов металлов [М (N (CN)2)2]n. В растворе можно
обнаружить ионы [М (DMSO)6]2+; в кристаллическом состоянии получены
лишь [М (DMSO)2 (N (CN)2)21 (М = Cu, Zn, Mn, Fe, Со, Ni), которые также
имеют полимерно-октаэдрическую структуру, подтвержденную ИК-
(табл. 8.8) и магнитными данными для комплексов кобальта и никеля [32]:
[Ni (DMSO)2 (N (CN)2)2] рэф = 3,09 M. Б.; [Со (DMSO)2 (N (CN)2)2] рэф =
= 4,90 М. Б.
Дицианамиды Зб-металлов М (N (CN)2)2 хорошо растворимы в гексаме-
тилфосфортриамиде (НМРА). Из полученных при этом растворов мож-
но выделить комплексы состава [М (НМРА)2 (N (CN)2)2L Исключение со-
ставляет дицианамид никеля, который почти не реагирует с НМРА. Со-
единение [Ni (НМРА)2 (N (CN)2)21 можно получить добавлением НМРА к
этанольному раствору дицианамида никеля, полученному реакцией между
KN (CN)2 и нитратом никеля. При однотипном составе комплексов с НМРА
для разных металлов реализуются различные структурные типы:
Таблица 8.9. Электронные спектры и магнитные моменты диаминных комплексов
никеля (II) [Ni (Amin )2(N (CN)2)2J
Соединение d — d-полосы, cm-1 Иэф, M. Б
NiPy2(N (CN)2)2 * 10 420 13 300 (пл.) 16 900 27 170 3,14
Ni (4-CH3C,H4N)2(N (CN)2)2 * 10 360 13 500 (пл.) 16 700 26 600 3,09
Ni (C8H7N)2(N(CN)2)2 * 9 710 13 500 (пл.) 16 100 26 300 3,16
Ni (2,4-(CH3)2C5H4N)2(N (CN)2)2 * 9 620 12 900 (пл.) 16 000 25 800 3,22
NiPy4(N (CN)2)2 ** 10 470 12 930 (пл.) 17 210 27 780 *
Переход ЗД _к.ЗТ _2»1Р _^зт -ь-ЗТ ГР) rt2g^ 12g^ ILg Xlg^ 1lgv)
* Отражение.
** Поглощение.
12*
331
мономерно-тетраэдрическая структура с монодентатным N (CN)^ для мар-
ганца и полимерно-октаэдрическая с бидентатным анионным лигандом для
никеля (табл. 8.8) [381.
Диамины, например Dipy и Phen, образуют с дицианамидами никеля
(II) 116] и железа (II) соединения состава ML3] (N (CN)2)2, а арилизоциани-
ды с Со (N (CN)2)2 — тетраэдрические комплексы [Со (CNR)2 (N (CN)2)21
[16]. Дицианамид Fe (II) с арилизонитрилами образует октаэдрические комп-
лексы типа [Fe (RCN)4 (N (CN)2)2L Синтезирован комплекс К [W (CO)6N •
• (CN)2] [331. Ср2МС12 (М = Ti, Zr) реагирует с AgN (CN)2 с образовани-
ем оранжевого комплекса Cp2Ti (N (CN)2)2 (v (CN) 2175, 2245 см-1) и бес-
цветного Cp2Zr (N (CN)2)2 (v (CN) 2175, 2220 см-1). Описаны некоторые
плоские комплексы Pt (II): цис- и транс-lPt (N (CN)2)2 (Bu3P)2] и транс-
[PtH (N (CN)2) (Ph3P)2] [42].
Дицианамид в координационном отношении проявляет сходство с три-
цианметанидом и частично с тиоцианатом. Подобно трицианметаниду он
образует координационные полимеры, а аналогично тиоцианату склонен
образовывать анионные комплексы. В спектроскопическом ряду дицианамид
располагается между тиоцианатом и трицианметанидом (10Dq : С (CN)3 >»
> N (CN)2 >> NCS) (см. 7.6.1). Как и в случае С (CN)^-HOHa, с увеличением
дентатности лиганда наблюдается уменьшение силы создаваемого им поля.
СПИСОК ЛИТЕРАТУРЫ
1. Madelung WKern F.— Ann. Chem., 1922, 427, p. 1.
2. Birckenbach L., Huttner K-— Z. anorg. allg. Chem., 1930, 190, S. 1.
3. Kuhn M., Mecke R. — Chem. Ber., 1961,94, S. 3010.
4. Madelung WKern F.— Ann. Chem., 1922, 427, p. 28.
5. RembarzG., Fischer E., Roeber К- C. a. o.— DDR — Patent, 1970, 70296 (1969), CA73,
66066.
5a. Fischer E., RembarzG.— J. Prakt. Chem., 1969, 311/6, S. 889.
6. Allensiein E.— Z. anorg. allg. Chem., 1963, 322, S. 265.
7. Allenstein E.— Z. anorg. allg. Chem., 1963, 322, S. 276.
8. Slotta К. H., Tschesche R.— Chem. Ber., 1929, 62, S. 1390.
9. Curd F. H. S., Hendry I. B., Kenney T. S. a. o.— J. Chem. Soc., 1948, p. 1632.
10. Kohler H.— Z. anorg. allg. Chem., 1964, 331, S. 237.
11. Kohler H — Wiss. Z. Univ. Halle, 1969, 69, M, S. 33.
12. Kohler H.— Z. Chem., 1973, 13, S. 401.
13. Biechler— C. r. Acad. Sci., 1935, 200, p. 141.
14. Huttner K-, Knape S.— Z. anorg. allg. Chem., 1930, 190, S. 34.
15. Sprague J. WCrazelli I. G., Ritchey W. M.— J. Phys. Chem., 1964, 68, p. 434.
16. Kohler H.— Habilitationsschrift. Halle, 1967.
17. Enemark I. HHolm R. H.— Inorg. Chem., 1964, 3, p. 1916.
18. Jensen H., Klewe B., TjeltaE.— Acta chem. scand., 1977, A31, p. 151.
19. Kohler H., Maushake К- H.— J. Organometal. Chem., 1968, 14, p. 103.
20. Chow Y. M.— Inorg. Chem., 1971, 10, p. 1938.
21. Becke-Goehring M., J ting D.— Z. anorg. allg. Chem., 1970, 372, S. 233.
22. Korecz L., Kohler H., Reef L., Burger K-— J- Organometal. Chem., 1974, 69, p. Ю5.
23. Kohler H., Laub P., Frischkorn A.— J. Less-Common Metals, 1971, 23, p. 171.
24. Kb'zler H., Seifert В — Z. Chem., 1967, 7, S. 32.
25. Kohler H., Seifert B.— Z. Naturforsch., 1967, 22b, S. 238.
26. Kohler H., Seifert В.— Z. anorg. allg. Chem., 1966, 344, S. 63.
27. Kohler- H., Hartung H., Golub A. M.— Z. anorg. allg. Chem., 1974, 403, S. 41.
28. Келер X., Скопенко В. В., Тряшин А. С., ЗубЮ. Л.— Укр. хим. журн., 1974 , 40,
с. 1121.
29. Kohler Н., Hartung Н., Seifert В.— Z. anorg. allg. Chem., 1966, 347, S. 30.
30. Kohler H., Seifert B.— Z Chem., 1965, 5, S. 142.
31. Kohler H., Seifert B.— Z. anorg. allg. Chem., 1966, 344, S. 63.
32. Kohler H.— Z. anorg. allg. Chem., 1965, 336, S. 245.
33. Beck U7. Unveroffentlicht.
332
34. Issleib К-, Kohler H., Wille G.— Z. Chemie, 1973, 13, S. 347.
35. ChowY. M., Britton D.— Acta crystallogr., 1975, B31, p. 1934.
36. Salyn Ja. WNefedow WKohler H.— Z. anorg. allg. Chem., 1977, 433, S. 13.
37. Kohler H., Kolbe A., LuxG.— Z. anorg. allg. Chem., 1977, 428, S. 103.
38. Kohler H., Skirl R., Skopenko V. V. In Vorbereitung.
39. Kohler H., Hartung H., Lisko T. P., Golub A. M.— Z. anorg. allg. Chem., 1974, 403,
40. Kohler H., Beck W.— Z. anorg. allg. Chem., 1968, 359, S. 241.
41. Britton D., Chow Y. M.— Acta crystallogr., 1977, B33, p. 697.
42. Kreutzer P. H., Schorpp К. TBeck WZ. Naturforsch., 1975, 30b, S. 544.
43. Beck W., Bock H., Schlodder R.— Z. Naturforsch., 1974, 29b, S. 75.
44. Колер X.— Координационная химия, 1977, 3, с. 139.
45. Kohler Н., Wusterhausen Н., Skirl R. Unveroffentlicht.
46. Kohler H., Seifert B. — J. Organometal. Chem., 1978, 12, p. 253.
47. Скопенко В. В., Голуб А. А.— Укр. хим. журн., 1976, 13, с. 196.
9. СРАВНИТЕЛЬНАЯ
ХАРАКТЕРИСТИКА ПСЕВДОГАЛОГЕНИДОВ
В этой заключительной главе обобщены некоторые аспекты химии псев-
догалогенидов. Рассмотрены следующие вопросы: структура и кислотные
свойства псевдогалогеноводородов; растворимость псевдогалогенидов ме-
таллов в различных растворителях в сопоставлении с растворимостью гало-
генидов металлов; получение и склонность анионов выступать в качестве
лигандов; возможные пути развития химии псевдогалогенидов.
9.1. КИСЛОТНЫЕ СВОЙСТВА И СТРУКТУРА
ПСЕВДОГАЛОГЕНИДНЫХ КИСЛОТ
Сопоставление констант диссоциации псевдогалогеноводородных кислот
(табл. 9.1) показывает, что в большинстве своем они являются слабыми кис-
лотами. Кислотность псевдогалогеноводородов изменяется от слабых кис-
лот (HCN, HCNO, HNCO, HNO2 и HN3) до кислот средней силы (HNCS и
HNCSe), проходя через соединение HN (CN)2, кислотность которого нель-
зя точно определить из-за неустойчивости, а затем увеличивается к дициан-
кетенимину, HNCC (CN)2, который безусловно можно отнести к сильным
кислотам (рКа — —5,1).
Если не принимать во внимание HNO2, то приведенные в табл. 9.1 псев-
догалогеноводороды можно рассматривать как С—Н- или N—Н-кислоты.
К С—Н-кислотам относятся HCN и HCNO, для которых характерны линей-
ные структуры Н—C==N и Н—C=N—О. В ряду псевдогалогенидных кислот
HCN является самой слабой. Для фульминатной кислоты электронооття-
гивающее действие кислорода ведет к сильной поляризации связи С—Н, что
обусловливает значительное повышение кислотных свойств HCNO по
сравнению с HCN [9].
Псевдогалогеноводороды HN3, HNCO и HNCS (в общем виде HNAX)
имеют аналогичное строение, причем угол связи HNA в данном ряду возрас-
тает от 124 через 126° до 134°, а группировки NAX при относительно малом
различии в длине связи N—А (1,24, 1,207 и 1,216 А соответственно) линейны
или почти линейны. Примечательно, что в отличие от симметричного азид-
иона расстояния N—N в HN3 неодинаковы (1,24 и 1,13 А), поэтому соотно-
шение связей можно описать двумя мезомерными формулами
Н—N-NWN |<->Н—-N=N=N I.
I II
334
Строение халькогеноводородных кислот можно передать граничными фор-
мами
Н—N—С=Х| <->
“ III
Н—N=C=X| <-> Н—N=C—XI.
IV V
Определяющим фактором увеличения
силы кислот в ряду HN3, HNCO, HNCS,
HNCSe является изменение полярности
связи Н—N под влиянием заместите-
лей, каковыми являются N2-, СО- и
CS (СЗе)-группы.
Значительное повышение кислотных
свойств в ряду циансодержащих псев-
догалогеноводородов НЕ (CN)n, HOCN,
HN (CN)2 и НС (CN)3 с увеличением п,
как и в случае оксоаниона ЕО^-, веро-
ятно, происходит в результате роста де-
локализации ионных зарядов, что ведет
к уменьшению электростатического про-
тон-анион-обменного взаимодействия и
к усилению кислотности НЕ (CN)n [12].
Из ИК-спектров трицианметана следует, что этой кислоте соответст-
вует структура дицианкетенимина [6, И]
Таблица 9.1. Константы диссоциации
псевдогалогенидных кислот
Формула Константа диссо- Литера-
КИСЛОТЫ циации тура
HCN 1,3-10—9 [2, 4]
HCNO 1 • 10~5 [9]
HN3 1,2-10-5 [2, 3]
HNCO 2,0 -10~4 [1]
HNCS 1,1-ю-1 »
HNCSe f=?10—1 »
hno2 1,4-IO-5 [2, 5]
HN(CN)2 Неустойчивая [7]
HNCC(CN)2 1,3-105 [6, Н]
N=C=C(CN)2.
VI
Дицианамид в водных растворах неустойчив и до сих пор еще не получен
[7]. Очевидно, это соединение существует в растворе очень короткое время,
и ему можно приписать строение
n=c=ncn.
Н^ VII
9.2. РАСТВОРИМОСТЬ ПСЕВДОГАЛОГЕНИДОВ
И ГАЛОГЕНИДОВ МЕТАЛЛОВ
В литературе наиболее полно представлены данные о растворимости га-
логенидов, а также важнейших псевдогалогенидов щелочных металлов
в воде, а отчасти в спиртах; значительно хуже изучена их растворимость в
других растворителях. При сравнении растворимости солей щелочных ме-
таллов бросается в глаза различие в растворимости солей лития и частично
натрия, с одной стороны, и солей калия, рубидия, цезия — с другой. Рас-
творимость ионных соединений определяется энергией кристаллической ре-
шетки, а также энергией сольватации катиона и аниона [21]. Лучшая рас-
творимость солей легких щелочных металлов объясняется прежде всего от-
носительно сильной сольватацией этих катионов. Сравним энтальпию
гидратации (—А Я) ионов щелочных металлов: Li+ (510), Na+ (397), К+
(318), Rb+ (289), Cs+ (259) (цифры в кДж/моль) [11]. В такой же последова-
335
тельности изменяются энтальпии растворения некоторых солеи щелочных
металлов в воде, метаноле, диметилформамиде (табл. 9.2).
Можно ожидать, что псевдогалогениды лития и в меньшей мере псевдо-
галогениды натрия должны иметь относительно высокую растворимость в
полярных растворителях. Так как энергии гидратации рассматриваемых
анионов, за исключением фторид-иона,
весьма близки (табл. 9.3), можно пред-
положить, что наряду с энергией решет-
ки солей определенное влияние на раст-
воримость галогенидов и псевдогалоге-
нидов в воде оказывает природа катиона.
Наиболее полные данные о раствори-
мости в воде галогенидов и псевдогало-
генидов серебра AgX представлены в
табл. 9.4.
Если сравнить растворимость гало-
генидов и псевдогалогенидов серебра
Таблица 9.2. Энтальпии растворения
некоторых галогенидов щелочных
металлов, кДж/моль (t = 25° С) [14]
Соеди- нение Растворитель
Вода Метанол DMFA
LiCl —370,5 —51,9 —51,1
Nal —7,5 —32,0 —56,9
KI +20,34 +0,71 —32,2
Таблица 9.3. Энергии гидратации галогенидов
и некоторых псевдогалогенидов, кДж/моль [14]
F— ci— Br— I— CN— NCO— NCT NCS—
—486 —352 —318 —280 —347 —389 —410 —310
в других полярных растворителях, то наблюдается аналогичная тенденция
в последовательности изменения произведения растворимости солей сереб-
ра. Растворимость хлорида и, вероятно, аналогичных солей серебра в раз-
личных растворителях не зависит от диэлектрических постоянных послед-
них (табл. 9.5). Так, относительно высокая растворимость AgCl наблюдается
в формамиде и DMSO, наи-
более низкая — в пропи-
ленкарбонате. В полярных
растворителях, приведен-
ных в табл. 9.5 и 9.6, на-
блюдается низкая раство-
римость солей серебра; на-
против, галогениды и псев-
догалогениды серебра хо-
рошо растворимы в пиридине. Причину этого следует искать в образова-
нии стабильных аддуктов пиридина с Ag+. В этой связи можно указать на
хорошую растворимость нитрита серебра в ацетонитриле.
Для слабосольватируемых катионов наблюдается увеличение раствори-
мости галогенидных и псевдогалогенидных солей в полярных апротонных
растворителях в таком ряду: NCO- Т CNO~ < NCXf < Вг~ < NT <
<1 CN < I <; NCS~ <; NCSe~. При этом трицианметаниды и дицианме-
таниды калия, рубидия и цезия должны располагаться в середине ряда.
таниды калия, рубидия и цезия
Таблица 9.4. Произведение
растворимости галогенидов
и псевдогалогенидов серебра
вводе(/ =25° С) [8, 13,73]
AgX —ignp AgX —lg ПР
AgNCO 6,64 AgSCN 12,0
AgC(CN)3 8,34 AgBr 12,3
AgN3 8,54 AgSeCN 15,4
AgN(CN)2 8,85 AgCN 15,92
AgCl 9,79 Agl 16,08
Таблица 9.5. Произведение растворимости
некоторых солей серебра в различных
растворителях (t = 25° С) [20]
Соединение Растворитель
CH3CN DMAA DMFA HMPA CH3OH
AgCl — 12,4 —14,3 —14,5 —11,9 —13,1
AgBr — 12,9 —14,5 — 15,0 —12,3 —15,2
Agl — —14,7 —15,8 — —18,3
AgN3 —9,6 — 10,8 —11,0 —8,5 —H,2
AgSCN —10,0 — 10,5 —11,5 —7,4 —13,9
336
В табл. 9.7 приведена растворимость некоторых солей калия и серебра в
жидком SO2 115]. Растворимость солей калия КХ в ацетоне изменяется в
ряду F- <С С1— < Вг- < Г < NCS- [16, 73]. Как правило, растворимость
солей калия и рубидия в таких полярных растворителях, как ацетон, DMSO,
Таблица 9.6. Сравнение произведения
растворимости AgC! и диэлектрической
проницаемости растворителей
Раствори- тель -1g ПР 8 Раствори- тель -1g ПР е
PDC 19,87 64,4 Метанол 13,1 32,6
Ацетон 16,4 20,7 Ацето-
DMFA 14,5 36,7 нитрил 12,4 38
Диэтило-
вый эфир 14,3 3 НМРТ 11,8 30
DMAA 14,3 37,8 DMSO 10,4 46,4
Этанол 14 25,8 Формамид 9,4 109,5
Таблица 9.7. Растворимость некоторых
галогенидов и псевдогалогенидов калия
и серебра в жидком SO2 при 0° С,
моль/кг[13]
Соеди- нение Раствори- мость Соединение Раствори- тель
КС1 0,0055 AgCl 0,021
КВг 0,040 AgBr 0,00016
KI 2,49 Agl 0,00068
KCN 0,00262 AgCN 0,00142
KNCS 0,502 AgSCN 0,000845
KF 0,0031 1 AgF н. p.
ацетонитрил, пиридин и SO2, растет с увеличением радиуса аниона, очевид-
но, вследствие повышения энергии сольватации с увеличением поляризуе-
мости аниона. Возможно, аналогичная причина вызывает увеличение рас-
творимости солей натрия в метаноле в ряду С1“ < Br- < I- < NCS-
[16, 73].
Наоборот, в протонных растворителях сольватация анионов понижается
с ростом ионного радиуса в последовательности F- > С1— > Вг- > N3” >
> 1“ > NCS- [16, 17].
9.3. ХАРАКТЕРИСТИКА ДОНОРНОЙ АКТИВНОСТИ
ПСЕВДОГАЛОГЕНИД-ЛИГАНДОВ
Проблема однозначного определения донорной активности псевдогало-
генидов довольно сложная. Это объясняется тем, что на свойства лигандов
большое влияние оказывают такие факторы, как природа центрального
атома, амбидентатность некоторых псевдогалогенидных лигандов и влия-
ние растворителя на способ их присоединения к центральному атому.
Теория поля лигандов располагает возможностью установления донор-
ной активности рассматриваемых лигандов. На основе исследования элек-
тронных спектров аналогичных по составу комплексов переходных металлов
галогениды и псевдогалогениды по их способности расщеплять d-термы
(параметр расщепления 10 Dq) образуют такой «спектрохимический ряд»
[18, 26—29]: 1“ < Вг~ < СГ « SCN- « N Д < F- < NCO- < ONO- <
< NCS- « N (CN)? < C (CN)? < NO? < CNO- < CN-.
Нетрудно видеть, что место псевдогалогенид-ионов в спектрохимическом
ряду определяется донорным атомом. Тиоцианат- или нитрит-ион, каждый
из которых может координироваться через азот или серу, азот или кисло-
род, располагается в разных местах этого ряда. Сильные поля, создавае-
мые CNO-- и CN--группами, способствуют образованию с этими лигандами
низкоспиновых комплексов.
Рост параметра 10Dq при переходе от галогенидов, стоящих в начале
спектрохимического ряда, к N- и С-донорным лигандам нельзя сводить к
337
увеличению ковалентности связи. Хотя ковалентная составляющая связи
и оказывает влияние на величину 10Dq, доминирующим является электро-
статический эффект.
Из анализа электронных спектров следует, что вследствие нефелок-
сетического эффекта расстояние между термами комплексных ионов метал-
ла оказываются меньшими, чем у ионов металла в газообразном состоянии.
Положение лигандов в «нефелоксетическом ряду» F" < NCS- < С1— ~
~ CN— <С Вг~ <С I- отражает их возрастающую тенденцию к образованию
ковалентных связей [18].
Данные о степени ковалентности или электровалентности координацион-
ной связи в псевдогалогенидных комплексах можно получить из сравнения
их групповых или ионных электроотрицательностей. Принимая во внима-
ние различный способ координации псевдогалогенидных групп, на основе
расчетов [34] их можно разместить по возрастающей электроотрицательности
в ряд: CN (3,84) < SCN (3,91) < NCS (4,17) < NO2 (4,33) < N3 (4,42) <
< ONO (4,43) < NCO (4,46) < OCN (4,66).
Клиффорд [35] предлагает следующий ряд возрастающих электроотри-
цательностей: CN (2,6) < CNO (2,9) < NCS (2,9) < N (CN)2 (2,9) < С (CN)3
(2,95) <Z N3 (2,95) <С NCO (3,05) <С NO2 (3,2). Расчет из положения полос
переноса заряда МХ6-хромофоров дает ряд «оптической электроотрицатель-
ности» [191: 1“ (2,6) ~ SCN“ (2,6) ~ CNO (2,6) < CN (2,8) ~ Вг“ (2,8) <
< NCS~ (2,9) < СГ (3,0) < F~ (3,9).
Исследование положения полос v (СО) в комплексах типа транс-
[МХ (СО) (PPh3)2] (М = Ir, Rh) в зависимости от лиганда X указывает на
уменьшение л-акцепторной силы этих лигандов в ряду [941: I > Вг >> CN >>
> Cl > NO2 > NCS ~ N3 > NCO.
Изменение интенсивности полосы v (СО) в плоских комплексах транс-
[МХ (СО) (PPh3)2] в зависимости от X указывает на увеличение л-донор-
ной способности в ряду: CN < I < Br < Cl < N (CN)2 < С (CN)3 <Z
< NCO < NCS < N3 [95].
Интересна возможность классификации аналогичных галогенидных и
псевдогалогенидных лигандов, исходя из изучения реакционной способ-
ности планарных комплексов платины; кинетика реакций замещения
лигандов в комплексах PtXL3, находящихся в транс-положении к лигандам
X, обусловлена «транс-эффектом» доноров X: X — СГ Вг~ < SCN~
« Г < NO7 <. CN~. Положения лигандов в этом ряду определяются,
с одной стороны, их поляризуемостью, и с другой — способностью к обра-
зованию л-связей [87, 88].
Если реакцию замещения в квадратных комплексах в общем виде пред-
ставить уравнением
[PtL„C14_ra] 4- Х~ [PtLnCl3-nX] + СГ,
(9.1)
то по нуклеофильной силе, т. е. по возрастающей величине констант скоро-
сти замещения СГ на Х~, получаем ряд [78, 88]: F~ <СГ < ВГ < NO^ <
< Nr < Г ~ SCN~.
Сравнение рядов лигандов по их транс-эффекту и нуклеофильности для
комплексов Pt (II) показывает, что лиганды с сильным лабилизирующим дей-
ствием на группы в транс-положении являются одновременно и потен-
циальными партнерами для реакций замещения в комплексах Pt (II) Упо-
мянутый выше ряд хорошо коррелирует с возрастающей нуклеофильностью
галогенидов и псевдогалогенидов относительно С-соединений [17]: F~ <;
338
< Cl- < Вг~ < NT < NCS- < I- < SCN-. Одновременно он отражает в
некоторой степени общую способность этих лигандов к координации мяг-
кими кислотами Пирсона.
Согласно классификации Пирсона [24, 74], псевдогалогенид-ионы можно
разделить на три группы: а) мягкие лиганды (С№, CNO-); б) жесткие ли-
ганды (Nr, NCO-, N (CN)7) и в) амбидентатные (жестко-мягкие) лиганды,
имеющие разные (мягкие и жесткие) донорные атомы (NCS-, NCSe- и
С (CN)3).
Например, тиоцианат-ион образует с мягкими металлами S-тиоцианаты,
а с жесткими металлами координируется предпочтительно через азот.
В разнолигандных комплексах, напротив, поведение рассматриваемого иона
усложняется (см. раздел 5). Для объяснения влияния других лигандов
привлекались различные явления (симбиоз лигандов, стерические препят-
ствия, образование л-дативной связи). Норбури [84] объясняет координа-
ционное поведение тиоцианат-иона с альтернативной жесткой (N)- или мяг-
кой (5)-координацией посредством введения четырех параметров мягкости.
В соответствии с этим тиоцианат в присутствии жестких лигандов образует
с жесткими кислотами М—NCS-связи, в то время как тот же центральный
атом в присутствии мягких лигандов координирует тиоцианат через серу.
Напротив, для мягкого металл-иона предполагается N-координация, если
он окружен мягкими лигандами, и S-координация в присутствии жестких
лигандов.
В этой связи интересен тот факт, что селеноцианатные комплексы (связь
М—SeCN) прочнее соответствующих тиоцианатных (связи М—SCN). Изо-
селеноцианатные комплексы (ЛА—NCSe) менее прочны, чем изотиоцианаты
(М—NCS) [25, 30]. Для трицианметанида известны лишь немногие примеры
координации этого лиганда через углерод [26].
Важные сведения о донорной активности рассматриваемых здесь анио-
нов можно получить из термодинамических характеристик реакций комплек-
сообразования, в частности из ступенчатых констант и энтальпий образо-
вания комплексов.
Так как комплексообразование в растворе, с одной стороны, представ-
ляет собой конкурентные реакции между молекулами растворителя и ли-
ганда (комплексообразование затрудняется с ростом донорной способности
растворителя) и так как, с другой стороны, донорная активность лиганда
зависит от его сольватации (сильная сольватация уменьшает его донорную
активность), необходимо при сравнении реакций комплексообразования
в растворе учитывать природу растворителя. По Гутману [39], меру донор-
ной активности растворителя целесообразно выражать через донорное
число DN (DN = —A/7SbCl5 • D), а меру акцепторной активности — через
акцепторное число AN (определяется химическим сдвигом в 31Р-ЯМР-спект-
рах Et3PO — растворитель) [92].
На основании спектрофотометрических исследований равновесных реак-
ций
[VO (Асас)2]+ ф- L [VO (Асас)2 L] (9.2)
в ацетонитриле можно получить относительные донорные числа анионных ли-
гандов. Установлено, что донорная способность галогенид- и псевдогалоге-
нид-ионов относительно сильного акцептора VO (Асас)+ повышается в ряду
I- < Вг— < С1— <2 NCS- <L (F—, CN—) < Nr [И, 36]. Этот ряд сохраня-
ется при определении донорности анионных лигандов относительно жестких
или промежуточных кислот. Так, при изучении реакций обмена в ацетоне
339
и DMFA
MoOCl? + nX~ MoOCl4_nX~ 4- пСГ (9.3)
получили подобный ряд донорной активности лигандов относительно Мо (V)
[38]: Г < Вг~ < СГ < NCSe" < NCS" < NCO" < N^-
При сравнении молярного количества лиганда Х~, необходимого для
превращения Со2+ в СоХ4" в различных растворителях, во многих случаях
наблюдаем определенную корреляцию между донорной активностью рас-
творителя и комплексообразующей способностью лиганда [11]. Устойчивость
тиоцианатов Ni (II) в неводных средах также зависит от донорных свойств
растворителя [41].
Мерой электродонорной функции интересующих нас анионов может слу-
жить величина потенциала напряжения разложения, определенная в 30-х
годах Биркенбахом в водных и этанольных растворах солей калия и нат-
рия [8,22]: TeCN" < SeCN" < Г < SCN" < CN" < Br" < Nr <
«С Cl" c OCN" < FT Этот ряд возрастающего напряжения разложения
совпадает с рядом активности лигандов, установленным по работе [11].
Сравнение устойчивости галогенидных или псевдогалогенидных комп-
лексов МХ”+ жестких или промежуточных металлов по классификации
Пирсона (М = Fe3+, Cr3+, UO|+, In3+, Со2+, Ni2+, Zn2+) в воде показыва-
ет, что их прочность понижается в ряду F" >- NT > NCS" > СГ > Вг" >
> Г [131.
Идентичный с рядом донорной активности анионных лигандов Гутмана
ряд наблюдается, например, и для комплексов индия в формамиде [40]
и воде [191: NCS" > СГ > Вг" > Г.
Сравнительно мало данных имеется о термодинамической устойчивости
дицианамидных и трицианметанидных комплексов. Изучением систем
Fe3+ — X" — Н2О и Си2+ — X" — Н2О установлено уменьшение стабиль-
ности комплексов МХп+ в ряду NCS" > N (CN)T > С (CN)T [27].
Тип растворителя существенно влияет на устойчивость комплексов.
Если в CH3CN устойчивость комплексов FeX+ лишь незначительно умень-
шается в ряду X = СГ, Br", NCS", Г [31], то устойчивость аналогичных
комплексов олова и свинца в более донорном растворителе DMSO уменьша-
ется в ряду СГ >» Вг" > Г > NCS" довольно отчетливо [32, 33, 37].
По отношению к типичным мягким кислотам, как и следовало ожидать,
наблюдается обращение ряда донорной активности лигандов. Например,
для комплексов ртути (II) в воде [13] донорность лигандов уменьшается в
в ряду С№ >> Г ~ SeCN" > Вг" >» SCN" >» СГ, т. е. наиболее проч-
ными являются комплексы с мягкими анионами. Такую же закономерность
можно наблюдать и при сопоставлении величин произведения раствори-
мости солей серебра AgX; наименьшие значения произведения растворимости
найдены для солей «мягких» анионов (Г, SeCN", CN~) (см. 9.2) [8, 13, 73].
Изменение положения СГ и NCS" в ряду комплексообразующей актив-
ности лигандов относительно мягких кислот в некоторых неводных раство-
рах можно видеть на примере комплексов серебра (растворитель DMFA)
[43], ртути (DMSO) [44] и кадмия (ацетонитрил) [45].
Исследование стабильности комплексов HgX4" (X = Г, Вг-, СГ
SCN") в Н2О, DMSO, DMFA и DMAA является интересным примером влия-
ния растворителя на их относительную устойчивость. Несмотря на прибли-
340
зительно одинаковые донорные числа растворителей, в ряду DMSO —
DMFA — DMAA наблюдается четкое увеличение устойчивости комплек-
сов [37, 44]. Это повышение прочности, очевидно, является результатом
уменьшения акцепторного числа растворителей (Н2О 54,8; DMSO 19,3;
DMFA 16,0; DMAA 13,6), вследствие чего уменьшается сольватация и повы-
шается донорная сила анионов.
9.4. РАЗНОЛИГАНДНЫЕ КОМПЛЕКСЫ ЛИНЕЙНЫХ
И НЕЛИНЕЙНЫХ ПСЕВДОГАЛОГЕНИДОВ
Разнолигандные комплексы, образуемые линейными псевдогалогенида-
ми и галогенидами или различными линейными псевдогалогенидами, изу-
чены достаточно полно [30, 46, 48]. Отдельные главы монографии содержат
многочисленные примеры соединений этого класса. Разнолигандкые же комп-
лексы с участием как линейных, так и нелинейных псевдогалогенидных групп
только начинают изучаться.
Образование разнолигандных комплексов иногда удается обосновать
с учетом так называемого «химического симбиоза» [46]. Полагая, что в коор-
динационной сфере мягкие лиганды могут сосуществовать с мягкими, а
жесткие с жесткими, во многих случаях можно предсказать образование
и стабильность разнолигандных комплексов. Хотя в ряде систем такой под-
ход оказался полезным, тем не менее он не является всеобъемлющим, так
как само понятие «мягкости — жесткости» не получило глубокого обосно-
вания. Например, вопреки ожиданию комплекс [CuICl] оказался прочнее,
чем [Cui (SCN)] [47], в соединениях К2 [Cd (SeCN)2Hal2] прочность связи
Cd—SeCN уменьшается в ряду С1~ > Вг~ > 1~ [49], хотя мягкость рас-
сматриваемых лигандов в этом ряду усиливается, а координированный че-
рез селен селеноцианат-ион может рассматриваться как мягкий донор [24].
В общем можно исходить из того, что устойчивость разнолигандного комп-
лекса [AAA„Bm] тем выше, чем прочнее связи М—А и М—В. Разнолиганд-
ный комплекс [MA„Bml более устойчив, чем MAn+m или МВте+п, тогда,
когда
[47’50]- <9-4>
Взаимное влияние разных лигандов в комплексе может повысить ли-
бо понизить совместимость их. В этом существенную роль играет и природа
растворителя, в среде которого происходит образование разнолигандных
комплексов.
Разумеется, различные галогенид- и псевдогалогенид-ионы тем легче
будут координироваться (образуя разнолигандные комплексы), чем ближе
их донорная активность [51, 72]. Это свойство лиганда является постоянной
величиной, и зависит оно от природы растворителя и центрального атома.
В соответствии с этими соображениями из водного раствора относитель-
но легко можно получить смешанные иодидоселеноцианатные комплексы
ртути и палладия [52]. Изучение ИК-спектров показало, что в К2 [Zn (NCS)2
Hal2] (Hal = Cl~ Br~) связь Zn—NCS более прочная в присутствии Cl ,
чем Br~ [48].
Можно полагать, что правило совместимости лигандов с близкой до-
норной активностью справедливо и при образовании разнолигандных комп-
лексов с нелинейными псевдогалогенидами. Как было показано в разделе
8, N (CN)r, например, как лиганд весьма напоминает тиоцианат-ион. По-
341
этому оба эти иона должны относительно легко образовывать разнолиганд-
ные комплексы. Подобно этому возможно образование раз но лигандных N-
трицианметанидоцианатных комплексов. При координации трицианметани-
да через углерод возможно образование разнолигандных комплексов с этим
лигандом и цианид-группами. До сих пор имеются лишь отдельные работы
об образовании разнолигандных комплексов с линейными и нелинейными
псевдогалогенид-лигандами.
Так, в ацетоне реакцией обмена Со (N (CN)2)2 и KNCX (X = О, S) были
получены тетраэдрические комплексы К2 [Со (N (CN)2)2 (NCX)2], а посред-
ством реакции
Kat2 [CoBr4] + 2AgN (CN)2 + 2AgNCO ->
-*Kat2 [Co (N (CN)2)3 (NCO)J + 4AgBr (Kat+ = Ph4P+, Ph4As+). (9.5)
выделены в твердом виде [53]. Аналогичные комплексы никеля Kat2 [Ni •
• (N (CN)2)2 (NCS)2], которые получены из Ni (NCS)2 и KatN (CN)2 (Kat+ =
= Ph4P+, Ph4As+) в этаноле, имеют полимерно-октаэдрическую структуру
[54]. Изучено образование разнолигандных комплексов кадмия [Ph4P]2 •
• [Cd (N (CN)2 (NCX)21; K2 [Cd (C (CN)3)2 (NCX)2]; K, [Cd (N (CN)2)4 (NCX),];
K4 [Cd (C (CN)3)4 (NCX)2] и K2 [Cd (N (CN)2)2 (NCX)2] (X = S, Se) [551.
ИК-спектры показывают, что во всех случаях, как и следовало ожидать,
координация XCN-групп осуществляется через азот. В К2 [Cd (С (CN)3)2 •
• (NCSe)2l присутствуют мостики Cd—SeCN—Cd [551.
Получены также разнолигандные псевдогалогенидонитритные комплек-
сы. Кроме комплексов платины К2 [Pt (CN)n (NO2)4_„1 и Na2 [Pt (NCS)n •
• (NO2)6_ra] [56, 57], синтезированы соединения свинца K3 [Pb (NO2)4SCN]
и K2 [Pb (NO2)3SCN] [62].
В растворе доказано образование комплексов [Hg (SCN)2NO2]~, [HgCl2 •
• (NO2)2]2~, [HgCl2NO2r, [CdCl2NO2r, [CdCl2 (NO2)2]2~ [35, 58—60], a
также [Cu (SCN) NO2]“ [61].
Известные до сих пор результаты исследования смешанных псевдогало-
генидных комплексов подтверждают высказанное предположение, что для
совместимости линейных и нелинейных псевдогалогенидов в этих соедине-
ниях определяющим фактором является сопоставимость донорных свойств
лигандов.
9.5. ПЕРСПЕКТИВЫ РАЗВИТИЯ ХИМИИ
ПСЕВДОГАЛОГЕНИДОВ
Рассмотрение обширного материала химии псевдогалогенидов, особен-
но линейных — азидов, цианидов, а также халькогеноцианатов — подво-
дит к вопросу о перспективах дальнейшего развития химии псевдогалоге-
нидов.
Весьма актуальными являются исследования псевдогалогенидов лантано-
идов и актиноидов в неводных средах, которые с успехом были использо-
ваны при синтезе селеноцианатов [63, 76, 77]. Кроме того, представляет
интерес исследование образования ковалентных псевдогалогенидов элементов
III—VI главных подгрупп реакциями обмена между элементгалогени-
дами и псевдогалогенидами металлов.
Необходимо продолжать работу по синтезу разно лигандных псевдогало-
генидных комплексов различных металлов с целью изучения их устойчи-
342
вости и взаимного влияния лигандов внутри координационной сферы, обра-
тив особое внимание на лиганды с близкими донорными характеристиками.
Не исключено, что разнолигандные псевдогалогенидные комплексы также
могут иметь практическое значение.
Интересную проблему затрагивает сравнительное изучение псевдогало-
генидов как лигандов в растворе и в изолированных соединениях. Новые
возможности заключены в изучении реакционной способности координи-
рованных псевдогалогенидных лигандов. Не решена проблема термодинами-
ческой устойчивости псевдогалогенидных комплексов в различных раствори-
телях, в том числе смешанных. Представляется необходимым определение
термодинамических характеристик простых и комплексных соединений в
растворах. Поэтому до сих пор не существует отчетливого и удовлетворитель-
ного критерия для предсказания растворимости различных псевдогалоге-
нидов металлов в неводных растворителях или оптимальных условий син-
теза новых соединений.
Одной из важных задач дальнейшего развития химии псевдогалогени-
дов следует считать поиск новых общих методов синтеза псевдогалогенид-
ных соединений, как это имеет место, например, в случае реакций псевдо-
галогенидов с карбонилами металлов [64, 651
М (СО)в + [Me4N] NCS -> [Me4N] [М (CO)5NCS] + СО. (9.6)
Новые результаты можно ожидать в химии теллуроцианат-иона NCTe~,
сведения о котором очень ограничены вследствие его малой стабильности
[22, 66].
Наконец понятие «псевдогалогенид» будет расширяться с открытием но-
вых галогеноподобных анионов. Можно представить общий принцип обра-
зования целого ряда псевдогалогенидов путем замещения атомов водорода
гидрид-анионов ХН— (X = О, S, Se), NHF и СНГ на CN--группу (см. 1.7)у
а также расширением спектра электроотрицательных заместителей [12,
27]. Так, замещение в SH~ водорода С6Р5-группой ведет к образованию
CeF5S~, псевдогалогенидный характер которого проявляется, например,
в образовании тетраэдрического аниона [Со (SC6F5)4]2“ или при синтезе ряда
псевдогалогенокарбонилов металлов IM (CO)5SC6F5]“ (М — Cr, Мо, W)
[79—81]. 1,2,3,4-тиотриазол-8-тиолат [97, 98], образующийся при взаимо-
действии азида с CS2, может рассматриваться как псевдогалогенид [96] в
ряду потенциальных S-донорных псевдогалогенидных лигандов.
Многочисленные нелинейные псевдогалогениды типа NXp и СХГ (X =
электроотрицательный заместитель) дают возможность предположить, что
N (CN)r и С (CN)p можно рассматривать как представителей этой большой
группы соединений, в которых вместо X наряду с CN могут выступать NO,
NO2, RCO, RSO2, R2PO [82, 83].
Среди соединений этого типа псевдогалогенидный характер может иметь,
например, триэтилсульфурилметанид [EtO2S]3C~ [83]. Этот анион, имеющий
тригонально-планарную С£3-конфигурацию [42], образует с переходными
металлами комплексы типа [М {С (SO2R)3}2 • СН2О и [М {С (SO2R)3}2Py4],
в которых лиганд координирован через кислород.
К классу псевдогалогенидов типа СХГ следует также отнести тринитро-
метанид-ион [С (NO2)3]~ [93]. Так как тринитрометаниды взрывоопасны, до
сих пор они изучены относительно мало.
Псевдогалогенидный характер нелинейных псевдогалогенидов NXF и
СХТ (см. 7.8) особенно сильно выражен, когда X = CN. Он четко ощутим.
343
если CN-группы постепенно замещать другими электроотрицательными
группами типа NO, NO2, RCO, RSO2, R2PO. Лиганды типа С (CN)2X~ и
NCNX- очень интересны, поскольку по своим свойствам напоминают как
классические псевдогалогениды, так и оксоанионы ХО— (NO7, NCXf, RCO7,
RSOr, R2POD [12, 67, 69, 88, 92].
В настоящее время ведутся интересные исследования реакций с участием
нитрозоцианометанид-иона NOC (CN)7- Соответствующую кислоту полу
чают взаимодействием динитрила малоновой кислоты с азотистой кислотой
[68]
СН2 (CN)2 + HNO2 -> HONC (CN)2 + H2O. (9.7)
Желтая соль серебра этой кислоты плохо растворима в воде, но растворя-
ется подобно AgNO2 в ацетонитриле. Псевдогалогенидные свойства
NOC (CN)^T проявляются, например, при образовании комплексов типа
MPy4(ONC(CN)2)2 (M=Mn, Fe, Со, Ni, Cu,Zn), в которых, по данным ИК-спект-
ров, координация лиганда осуществляется через азот нитрозо-группы
[67, 69], а в металлоорганических соединениях типа Me2Sn (ONC (CN)2)2
и Cp2Ti (ONC (CN)2)2, вероятно, через кислород [12, 86].
Характер связей в нитрозодицианметанидных комплексах соответствует
таковому в нитро- или нитритокомплексах
О
И—NC м—о—N\
C(CN)2 "NC(CN)2
VI VII
Заметное сходство ионов NOC (CN)2 и NO2 проявляется в окислительно-
восстановительных реакциях, а также в строении этих ионов. Если NOF
полярографически неактивен в нейтральной среде, то в кислом растворе он
восстанавливается до гидроксиламина. Ион NOC (CN)7 восстанавливается
до NH2OH уже в нейтральной среде с одновременным образованием динит-
рила малоновой кислоты [70].
Окисление KNOC (CN)2 ведет к образованию аналога нитрата — нитро-
дицианметанида NO2C (CN)7 [71]. Соль AgNO2C (CN)2 плохо растворима
в воде. В комплексах типа МРу4 (O2NC (CN)2)2 нелинейный лиганд коорди-
нируется металлом через кислород [88].
Отношения связей планарных анионов NOC (CN)^" и NO2C (CN)7 (это
принципиально справедливо и для других псевдогалогенидов метанидного
или амидного типов) позволяет с учетом делокализованной системы л-свя-
зей предположительно описать их строение [67, 71]
VIII
IX
Рентгенографическое определение параметров обоих ионов согласуется
с предложенным их строением [12, 89—-91]. Наконец можно сослаться на
интересное предложение перенести понятие «псевдогалоген» в химию пере-
344
ходных металлов и рассматривать карбонилметалло-ионы типа Mn (CO)s
или Со (СО)? как псевдогалогениды, а биядерные карбонилы типа
Мп2 (СО)10, Со2 (СО)8 — как псевдогалогены.
В общем химия координационных соединений, как классических линей-
ных, так и новых нелинейных псевдогалогенидов типа СХ^ и NX7, пред-
ставляет интерес и широкое поле для дальнейших исследований.
СПИСОК ЛИТЕРАТУРЫ
1. Boughton Н., Keller R. N.— J. Inorg. Nucl. Chem., 1966, 28, p. 2851.
2. Голуб A. M. Загальна та неоргашчна xiMia. Ч. I, II, Кшв, 1968, 1971.
3. Evans В. L., Loffe A. D., Gray Р.— Chem. Rev., 1959, 59, р. 51£.
4. Izatt R. М., Christensen J. J., Pack R. Т., Bench R.— Inorg. Chem., 1962, 1, p. 828.
5. Крестов Г. А. Термодинамика ионных процессов в растворах. Л., Химия, 1973.
6. Boyd R. Н.— J. Am. Chem. Soc., 1961, 83, р. 4288.
7. Kuhn М., Mecke R.— Chem. Вег., 1961, 74, S. ЗОЮ.
8. Birckenbach L., Huttner K.— Z. anorg. allg. Chem., 1930, 190, S. 1.
9. Beck IF., Swoboda P., Feldl K., Tobias R. S.— Chem. Ber., 1971, 104, S. 536.
10. Trofimenko S. — J. Org. Chem., 1963, 28, p. 217.
11. Gutmann V.— Chemische Funktionslehre. Wien, 1971.
12. Келер X.— Координационная химия, 1977, 3, с. 139.
13. Sillen L. G., Martell A. E. Stability Constants. London, 1964.
14. Мищенко К- ППолторацкий Г. М. Вопросы термодинамики и строения водных и
неводных растворов электролитов. Л., Химия, 1976.
15. Неводные растворители. Под ред. Т. Баддингтон. М., Химия, 1971.
। 16. Miller JParker A. G.— J. Am. Chem. Soc., 1961, 83, p. 117.
17. Паркер А. Дж.— Успехи химии, 1963, 32, с. 1270.
18. Schlafer Н. L., Gliemann G. Einfuhrung in die Ligandenfeldtheorie. Leipzig, Verlag
Geest und Portig, 1967.
19. Joergensen К- C. Inorganic Complexes. London, Academic Press, 1963.
20. Батлер Дж.— В кн.: Электрохимия металлов в неводных растворах. М., Мир,
1974, с. 201.
21. Яцимирский К. Б. Термохимия комплексных соединений. М., Изд-во АН СССР.
1951, с. 24.
22. Birkenbach L., Kellermann К.— Вег., 1925, 58, S. 786, 2377.
23. Ahrland S., Chatt J., Davies N.R.— Quart. Rev. (London), 1958, 12, p. 265.
24. Пирсон P. Дж.— Успехи химии, 1971, 40, с. 1259.
25. Цинцадзе Г. В. Докт. дис. Тбилиси, 1973.
26. Beck W., Fehlhammer W. Р., Pollmann Р. а. о.— Chem. Вег., 1967, 100, S. 2335.
27. Kohler Н.— Wiss. Z. Univ. Halle, 1968, 18, 69М, S. 33.
28. Kohler H.— Z. Chem., 1973, 13, S. 4016.
29. Бальхаузен К- Введение в теорию поля лигандов. М., Мир, 1964.
30. Голуб А. М., Скопенко В. В.— Успехи химии, 1965, 34, с. 2098.
31. Kratochvil В., LongR.— Can. J. Chem., 1970, 48, р. 1414.
32. Самойленко В. М., Ляшенко В. И.— Журнал неорган. химии, 1973, 18, с. 2402.
33. Pool К.— J. Polarogr. Soc., 1967, 13, р. 23.
34. Huheey J. F.— J. Phys. Chem., 1966, 70, p. 2086.
35. Clifford A. F.— J. Phys. Chem., 1959, 63, p. 1227.
36. Gutmann V., Mayer H. — Monatsh. Chem., 1968, 99, S. 1383.
37. Самойленко В. M., Ляшенко В. И.— Журнал неорган. химии, 1974, 19, с. 2984.
38. Голуб А. М., Грачевский В. В., Улько И. В., СамовскаяН. Д.— Журнал неорган.
химии, 1975, 20, с. 2704.
39. Гутман В. Координационная химия в неводных растворах. М., Мир, 1971.
40. Самойленко В. М., Гринюк И. И.— Укр. хим. журн., 1972, 38, с. 8.
41. Miezis А.— Acta Chem. Scand., 1974, А28, р. 407.
42. Silverton J. V., Gibson D. T., Abrahams S. C.— Acta Crystallogr., 1965, 19, p. 651.
43. Breant M., Georges J., Imbert J., SchmittD.— Ann. chim., 1971, 6, p. 245.
44. Foil A., Le Demezet M., Courtot-Coupez I.— Bull. Soc. chim. France, 1972, p. 1207.
45a . Aihara M., Misumi S.— Bull. Chem. Soc. Japan, 1973, 46, p. 1674.
456. Misumi S., Aihara M.— Bull. Chem. Soc. Japan, 1974, 47, p. 127.
46. Joergensen С. K-— Inorg. Chem., 1964, 3, p. 1201.
47. Фридман Я • Д. Устойчивость смешанных комплексных соединений в растворах.
Фрунзе, Илим, 1971.
345
48. Цинцадзе Г. В.— Труды Груз, политехи, ин-та, 1972, № 8, с. 230.
49. Цинцадзе Г. В., ХаритоновЮ. ЯЦивадзе А. Ю. и др.— Журнал неорган. химии,
1970, 15, с. 2336.
50. Marcus YEliezer I.— Coord. Chem. Rev., 1969, 4, c. 273.
51. Голуб A. M., Андрейченко О. E.— Журнал неорган. химии, 1962, 7, с. 549.
52. Голуб А. М., Померанц Г. Б., Иванова С. А.— Журнал неорган. химии, 1969,
14, с. 2826.
53. Kohler Н., Lischko Т. Р., Hartung Н., Golub А. М.— Z. anorg. allg. Chem., 1974,
403, S. 35.
54. Kohler H., Hartung H., Golub A. M.— Z. anorg. allg. Chem., 1974, 403, S. 41.
55. Келер X., Скопенко В. В., Тряшин А. С., Зуб Ю. Л.— Укр. хим. журн., 1974, 40,
с. 1121.
56. Черняев И. И.— Известия сектора платины и других благородных металлов, 1955,
31, с. 5.
57. Дершибер Г. В., Евстафьева О. Н., Бабаева А. В.— Журнал неорган. химии, 1974,
5, с. 1433.
58. Czakis-Sulikovska М., Zaidler А.— Roczn. Chem., 1973, 47, р. 3.
59. Czakis-Sulikovska М., Swinarski А.— Rocz. Chem., 1962, 36, р. 389.
60. Гринберг А. А. Введение в химию комплексных соединений. М., 1966.
61. Swinarski A., Danilczuk Е., Gogolin R.— Roczn. Chem., 1966, 40, р. 737.
62. Голуб А. М., Акмырадов Р.— Журнал неорган. химии, 1966, 11, с. 2347.
63. Golub А. М., Кора М. V., Skopenko V. V., Cincadze G. V.— Z. anorg. allg. Chem.,
1Q7O Ч7Ч 4 309
64. Ruff J. K.— Inorg. Chem., 1969, 8, p. 86.
65. Wojcicki A., Farona M. F.— J. Inorg. Nucl. Chem., 1968, 26, p. 2289.
66. Austad T., Songstad JAse K.— Acta Chem. Scand., 1971, 25, p. 331.
67. Kohler H., Seifert B.— Z. anorg. allg. Chem., 1968, 360, S. 137.
68. Longo J.— Gazz. chim. ital., 1931, 61, p. 575.
69. Kolbe A., Kohler H.— Z. anorg. allg. Chem., 1970, 373, S. 230.
70. Matschiner H., Kohler H., Matuschke R.— Z. anorg. allg. Chem., 1971, 380, S. 287.
71. Kohler H., Eichler B., Kolbe A. Z.— Chem., 1970, 10, p. 154.
72. Голуб A. M.— Химический сборник Киев, ун-та, 1955, № 6, с. 61.
73. Справочник химика, т. 3. М., 1964, с. 229—230.
74. Pearson R. J., Songstad 1. J.— Am. Chem. Soc., 1967, 89, p. 1827.
75. Morris D. F. C.— Acta Crystallogr., 1961, 14, p. 547.
76. Скопенко В. В., Иванова Е. И.— Укр. хим. журн., 1976, 42, с. 1011.
77. Голуб А. М.— Успехи химии, 1976, 45, с. 961.
78. Коттон Ф., Уилкинсон Дж. Современная неорганическая химия. Ч. 1. М., Мир,
1969.
79. Beck WFehlhammer W. Р., Stetter К- Н., Tadros S.— Chem. Ber., 1967, 100, S. 3944.
80. Beck Stetter К- H., Tadros S., SchwarzhansК• F.— Chem. Ber., 1967, 100, S. 3944.
81. Beck WTadros S.— Z. anorg. allg. Chem., 1970, 375, S. 231.
82. Kohler H., Eichler B., Salewski R.— Z. anorg. allg. Chem., 1970, 370, S. 183.
83. Beck WJohansen 0., Fehlhammer W. P.— Z. anorg. allg. Chem., 1968, 361, S. 147.
84. Norbury A. H.— J. Chem. Soc., 1971, A, p. 1089.
85. Kohler H., Seifert B.— Z. anorg. allg. Chem., 1970, 379, S. 1.
86. Kohler H., Lange U., Eichler B.— J. Organometal. Chem., 1972, 35, p. C17.
87. Черняев И. И.— Известия ин-та платины, 1926, 4, с. 243, 261.
88. Basolo F., Pearson R. G.— Progr. Inorg. Chem., 1962, 4, p. 381.
89. BreantM., Nicolas J.-P., Alam S., Lavergne M.— C. r. Acad. Sci., 1973, C277, p. 855.
90. ChowY. M., Britton D.— Acta Crystallogr., 1974, B30, p. 147.
91. Chow Y. M., Britton D.— Acta Crystallogr., 1974, B30, p. 1117.
92. Gutmann V.— Coord. Chem. Rev., 1976, 18, p. 225.
93. Hantzsch A., Rirkenberger A.— Ber., 1899, 32, S. 628.
94. Vaska L., Peone J.— Chem. Commun., 1971, p. 418.
95. Schlodder R., Vogler S., Beck W.— Z. Naturforsch., 1972, 27, b, S. 463.
96. Audrieth L. F.— Chem. Rev., 1934, 15, p. 169.
97. Seel F., Nogradi J.— Z. anorg. allg. Chem., 1951, 264, S. 311.
98. Neves E. A., Franco D. W.— J. Inorg. and nucl. Chem., 1974, 37, p. 277; 1975, 36,
p. 3851.
ПРИНЯТЫЕ СОКРАЩЕНИЯ
Асас CH3—C=CH—C—CH3 1 II о о
Act CH3COCH3
As As _/^As(CH3)2 Q^As(CH3)2
As2P As3P Atp [(C2H6)2 As—CH2—CH2—-CH2]2 p—c6hf [(CH3)2 As—CH2—CH2—CH2]3 p CH3—C=CH 1 1 H3C—N C=O 1 C6Hb
Bia 1 T Jch
Bn hn2—ch2—chnh2—ch2—ch3
BTPP Bu /-Bu [(CcH6)3 P-(CH2)3-P(CeH5)3J2+ CH3—CH2—CH2—CH2 (CH3)3C
Bzd NH2-£^H^y-NH2 H H ZC=CH
Cp Co ►u |C( 1 XC=CH H
Dben dh2 C6H6 -CH2—NH—(CH2)2—NH—CH2—CeH6 H3C—C=NOH 1 H,C—C=NOH О II il /С C—CH2—c—cx
Diantp H5Cfi-N( II || )NC6Hb \N с c NZ II II CH3 CH3 CH3 CH3
dien hn/<ch2)2-nh2 X(CH3)2-NH2
N, N'-dieten C2H6—NH—(CH2)2—NH—C2H6
N, N'-dimen CH3—NH—(CH2)2—NH—CH,
Diox /CH2—CH2X XCH2-СН/
Dipy f 1-0
dm DMAA Чгн 4,4'-dimethyl-2,2' CH3CON (CH3)2
DMBia CH3 V-Ln^c-ch3
DMFA DMSO HCON (CH3)2 OS(CH3)2
GO
►U
00
Dpb
dpt
dppa
en
Es
NH2—CH2—CH2—nh2
CH2-CH2
О о
Et
EtC2Et
Et4dien
Et2N2O
C2H5
(С2НБ)2 P—CH2—CH2—P (С2НБ)2
(С2НБ)2 N—(СН2)2—N—(СН2)2—N (С2НБ)2
:?X/CON(C2H5)2
Etphos
Etu
FA
I
C2H5
SC (NHCH2)2
hconh2
F
FDP F-fV-PlCeHsh FA>P|C6H5IZ F
Hal Галогенид
НМРА /N (CH3)2 OP<-N (CH3)2 XN(CH3)2
IQ OCn H ।
MBia Qc^e-снз
Me CH3
MeC2Me MeC3Me Me4en Mephen (CH3)2 P—CH2—CH2—P (CH3)2 (CH3)2 P—CH2—CH2—CH2—P (CH3)2 (CH3)2 N—CH2— CH2—N (CH3)2 2=N N=^ CH3
MFA Mephos HCONH—CH3 CH3
mrt ,CH2—CH2X NH< 2 У хСНа-СН/
Meetrien
Meetren
N2AS2
N3As
N2P
N3P
n2p2
N3S
Ox
(CH3)2N- (CH2)2 —N— (CH2)2 —N— (CH2)2 —N (CH3)2
CH3 CH3
N [CH2—1CH2—N (CH3)2]3
{(CeH5)2 As- (CH2)2}2 —N— (CH2)2 —N (C2HB)2
{(C2HB)2 N - (CH2)2}2 —N— (CH2)2 - As (C6HB)2
(C2H5)2 N— (CH2)2 -NH- (CH2)2 -P (C6HB)2
{(C2H6)2 N - (CH2)2}2 —N— (CH2)2 - P (CeH6)2
{(С2НБ)2 P- (CH2)2}2 N - (CH2)2 - N (C2H5)2
{(C2H5)2 N - (CH2)2}2 —N— (CH2)2 -S-CH3
349
PAs
^3^2
PCCP
PCnP
PDC
P2P
P3P
Ph
Pic
^^AsICbHsk
{(CeH6)2 PCH2}3 C CH3
(CeHB)2 P-CH=CH-P (CeHB)2
(C6HB)2 P- (CH2)n -P (CeHB)2 (n = 1 - 4)
CH,—CH—CH3
A A
xcz
о
{(C6HB)2 P- (CH2)2}2 P (C6HB)
{(CeH6)2 P (CeH4)}3 P
СеНб
К
Pip
Phen
PN (CeH6)2P-(CH2)2-N(CH3)2
Pn NH2— (CH2)3—nh2
POPh P(CfiHB)(OC2H5)2
P2NO {(C6H6)2 P (CH2)2}2 n- (CH2)2 och3
Pr C3H7
Py
Py—NH—
—NH—Py
РУг^г
Py2CG
Q
гы Н2С—СН2 Н2С СН2
Tlid сн3—с6н4—nh2
ТМР OP (ОСН3)3
tmso Н2С—сн2 1 1 Н2С сн2 S 11 О
tmu oc/N (CH3)2 XN (CH3)2
tren trien N (CH2-CH2-NH2)3 NH2— (CH2)2 -NH (CH2)2 - NH (CH2)2 —NH2
tripyam 0
tu SC(NH2)2
u Urt oc (NH2)2 (CH2)6 n4
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Азид-радикал 21, 22
Азидная кислота 17
Азоимид 15, 16
Актиноиды 34, 35, 64, 134, 177, 191,
192, 205, 222, 236, 237 266, 274,
276, 278, 282, 284
Алюминий
— азид 30
— селеноцианат 276
— тиоцианат 168, 204
— цианид 54
Алкил цианаты (-изоцианаты) 126, 147,
153, 154
Амбидентатность 8—10, 13, 52, 53,
132, 200, 269, 302—304, 320—323
Аммоний
— азид 23, 24
— дицианамид 318
— селеноцианат 260
— тиоцианат 164
— трицианметанид 301
— цианат 125
— цианид 48, 49
Анализ 22, 235—237
Арилцианаты (-изоцианаты) 126, 147,
154
Арсен
— тиоцианат 171, 204
— цианат 127
— цианид 55
Барий, см. Щелочноземельные метал-
лы
Бериллий
— азид 25, 26
— селеноцианат 260
— тиоцианат 164, 190, 204
— цианид 50
Бор
— азид 27—29
— тиоцианат 162, 168, 204, 230
— цианат 126
— цианид 54
Ванадий
— азид 35
— дицианамид 327
— селеноцианат 264, 266, 271, 273,
274, 285, 391
— тиоцианат 179, 192, 222
— трицианметанид 310
— цианид 65, 66, 94, 98, 99, 013, 104
Висмут
— селеноцианат 265—267, 273 278,
285, 287
— тиоцианат 162, 171, 172, 190, 198,
204, 218
— цианид 55, 58
Влияние растворителя на образование
псевдогалагенидных комплексов
188—195, 262—269, 339
Вольфрам
— азид 35, 36
— дицианамид 432
— тиоцианат 181, 205, 207, 236
— трицианметанид 313
— фульминат 130
— цианат 137
— цианид 67—70, 94, 98, 99, 103, 104
Галлий
— азид 30
— селеноцианат 275, 282, 284
— тиоцианат 168, 169, 190, 204, 237
— цианат 126, 132, 133
Галоген псевдогалогениды 28, 29, 35, 55,
56, 90, 105, 107, 128, 172, 306
Гафний
— тиоцианат 178, 205, 237
— цианат 136, 149
Гексациан 45
Германий
— азид 39
— дицианамид 322, 323
— тиоцианат 170, 204
— цианид 54, 58
Гремучая кислота 130
Гремучая ртуть 130, 131, 151, 153
Гремучее серебро 131, 145, 151
Диоксоциан 123
Диселеноциан 256
Дитиоциан 161
Дициан 45, 46, 104, 107
Донорная активность псевдогалогени-
дов 337—341
351
Железо
— азид 53
— дицианамид 325, 327, 330—332
— селеноцианат 270—274, 276 , 286,
290—292
— тиоцианат 183—187, 192, 198, 207,
224, 235, 236
— трицианметанид 308, 309, 311, 314
— фульминат 139—141, 151
— цианат 133, 135, 137, 138, 149
— цианид 73, 82, 89, 94, 95, 102,
105, 107
Золото
— азид 33
— селеноцианат 284, 289
— тиоцианат 174, 191, 206, 212
— фульминат 140, 150
— цианид 59—63, 87, 91, 95, 97—100,
106
— цианат 137, 149, 150
Изомерия
— цианат-фульминат 123
— псевдогалогенидных соединений 54,
70, 80, 81, 96, 103, 105, 149, 184,
185, 207, 208, 321
Изоцианид 54, 96
ИК-спектральные данные
— азид-ион 25—27, 34, 35
— дицианамид-ион 319, 321—323,
326—329, 331
— селеноцианат-ион 259, 269—271, 274,
275, 278, 280—282, 287, 289, 291 —
294
— тиоцианат-ион 201—214
— трпцианметанид-ион 302, 304, 305,
307, 309, 310, 312—314
— фульминат-ион 146, 150—152
— цианат-ион 147—150
— цианид-ион 92—96
Инверсия связи тиоцианатной группы
207, 208, 211—214
Индий
— азид 30
— селеноцианат 265, 267, 274, 276,
278, 280, 282, 291
— тиоцианат 168, 169, 190, 194, 207
— цианат 126, 132, 134
— цианид 54
Иод-азидная реакция 22
Иридий
— азид 37
— селеноцианат 275
— тиоцианат 187, 188, 226
— трицианметанид 314
— фульминат 140, 151
— цианат 137, 138, 149
— цианид 83, 85, 102, 104
Кадмий
— азид 33, 34
— дицианамид 325, 329
селеноцианат 216, 265, 267, 270,
— 271, 275, 278, 280, 281, 284, 286,
287, 292—294.
— тиоцианат 165—167, 190, 197, 206,
216, 231
— трицианметанид 310
— фульминат 140, 151
— цианат 133, 135, 136, 142, 143
— цианид 53, 54, 56—58, 87—89, 91,
97, 106
Калий, см. Щелочные металлы
Кальций, см. Щелочноземельные ме-
таллы
Кислоты псевдогалогенидные 17,46—
48, 87, 107, 123, 130, 163, 257,
334, 335
Клатраты 54, 74, 79, 84, 98, 99, 101
Кобальт
— азид 36
— дицианамид 321, 325, 326, 328—331
— селеноцианат 263—267, 283, 285—
290
— тиоцианат 183—187, 195, 207, 212—
214, 224, 230, 232, 235
— трицианметанид 308, 309, 311—313
— фульминат 140, 151
— цианат 129, 133—135, 138, 142, 143,
149, 150
— цианид 73—76, 78—82, 89—92,
94—96, 98, 99, 102—107
Комплексообразование в растворе 75,
86—92, 188—198, 262—269, 310,
311, 327, 328, 339—342
Лантан и лантаноиды
— азид 34
— селеноцианат 274—276, 280—282,
284, 291
— тиоцианат 175, 176, 191, 204, 205,
222, 237,
— цианат 133, 134, 149
— цианид 64
Лиганды псевдогалогенидные
— распределение разряда 11, 12,50—
52, 131, 132, 200, 302, 319
— сила поля 337, 338
— характеристика 6—13, 18—21, 50—
53, 123, 199, 200, 300—302, 319
Литий, см. Щелочные металлы
Магний, см. Щелочноземельные ме-
таллы
Марганец
— азид 36
— дицианамид 325—326, 330, 331
— селеноцианат 265—268, 270, 271,
274, 276, 278, 280, 285, 291
— тиоцианат 181, 192, 205, 207, 213,
224
— трицианметанид 308, 311—313
— фульминат 141, 151
— цианат 135, 138, 142, 149, 150
— цианид 70—73, 94—96, 102—104,
106
Медь
— азид 32—34, 38
— дицианамид 325—328, 330
— селеноцианат 260, 261, 266, 272—
275, 277
352
— тиоцианат 172—174, 190, 198, 204,
206, 207, 218—222
— трицианметанид 308, 309, 311—313
фульминат 140, 141, 151
— цианат 129, 133, 135, 136, 144,
149, 150
— цианид 45, 59—63, 87, 88, 90—92,
95, 97—102, 104, 106
Молибден
— азид 35
— селеноцианат 180, 181, 205, 207,
224, 236
— фульминат 140, 151
— цианат 133, 137, 144
цианид 67—70, 94, 98, 99, 103—105
Моноциан 46
Натрий, см. Щелочные металлы
Никель
— азид 36
— дицианамид 325, 328—331
— селеноцианат 264—268, 270—281,
991__________993
— тиоцианат 183—186, 192, 195, 203,
207, 208, 210, 214, 215, 224—226,
231, 232
— трицианметанид 308, 309, 311—314
— фульминат 139, 140, 151, 153
— цианат 129, 135, 136, 142, 145, 149,
150
— цианид 73—75, 78, 79, 82, 89, 91,
93, 98—102, 104—107
Ниобий
— селеноцианат 285, 291
— тиоцианат 179, 202, 205, 207, 222,
237
— цианид 66
Нитродицианметанид 344
(три-) нитрометанид 343
Нитрозодицианметанид 344
Оксициановодород 123
Олово
— азид 31, 34
— ди циан амид 322—324
— селеноцианат 283
— тиоцианат 170, 218
— трицианметанид 306, 307 х
— цианат 132
— цианид 55, 91, 92
Органометаллпсевдогалогениды 54, 55,
96, 100
Осмий
— тиоцианат 187
— цианид 83—86, 94, 102
Палладий
— азид 36, 37
— дицианамид 321
— селеноцианат 274, 275, 276, 283,
286, 289
— селеноцианат 274, 275, 276, 283,
286, 289
— тиоцианат 187, 188, 206, 208, 211 —
213
— трицианметанид 312
— фульминат 139, 141, 151
— цианат 133, 136—139, 149, 150
— цианид 83, 84, 86, 93, 98—102, 104
— парациан 45
Платина
— азид 37
— дицианамид 321
— селеноцианат 274, 276, 284, 286,
289
— тиоцианат 187, 188, 206, 208, 211,
212
— трицианметанид 312
— Фульминат 140, 141, 151
— цианат 133, 136, 137, 149, 150
— цианид 83—86, 93—96, 98—102,
107
Применение псевдогалогенидов в ана-
лизе 105, 106, 235—237
Произведение растворимости псевдо-
галогенидов серебра 336
Разложение азидов 21, 22
Растворимость псевдогалогенидов ме-
таллов 335—337
Реакционная способность координи-
рованных псевдогалогенидов 37,
137, 138, 172, 317, 318
Рений
— азид 36
— селеноцианат 385
— тиоцианат 182, 183, 205, 207, 235,
236
— трицианметанид 313
— цианат 133, 149
— цианид 70—72, 98, 102
Рентгеноструктурные данные
— азидные соединения 33
— дицианамидные 321, 324, 325, 327
— селеноцианатные 256—258, 278—
280
— тиоцианатные 214—235
— трицианметанидные 302, 307—309
— фульминатные 144—146
— цианатные 142—144
— цианидные 97—105
Рентгеноэлектронные спектры псев-
догалогенидов 132, 302, 319
Роданидная кислота, см. Тиоцианат-
ная кислота, псевдогалогенидные
кислоты
Родий
— азид 37
— селеноцианат 283, 289
— тиоцианат 187, 188, 206, 208
— трицианметанид 312
— фульминат 140
— цианат 137, 138, 149
— цианид 83, 85, 86, 95, 102
Ртуть
— азид 34
— дицианамид 327
— селеноцианат 261, 263, 283, 284,
286—288
— тиоцианат 165—167, 190, 206—208,
216—218, 231—233, 234
— трицианметанид 309, 311
353
— цианат 129, 133, 136, 152, 153
— цианид 45, 53, 54, 56, 57, 87—91,
95, 97—102, 106
Рутений
— азид 37
— селеноцианат 289
— тиоцианат 187, 188, 203, 205, 207
— цианат 140
— цианид 83—85, 94, 98, 102
Свинец
— азид 31, 34, 38
— селеноцианат 261, 263, 266 271
283, 286
— тиоцианат 170, 171, 190, 206, 218,
234
— цианат 132, 133
— цианид 55, 58, 90—92
Серебро
— азид 33, 48
— дицианамид 325, 327
— селеноцианат 260—262, 283
— тиоцианат 174, 190, 206, 210, 222
— трицианметанид 309, 310
— фульминат 131, 139—141 144 146
151, 153
— цианат 129, 142, 144, 145, 150, 153
— цианид 59—63, 87—92, 95, 97, 104
106
Селен 55, 90, 162, 218
Селеноцианатная кислота 257, 258
334, 335
Сера 55, 90, 162
Силиций
— азид 29
— дицианамид 322, 323
— тиоцианат 170, 204, 218
— трицианметанид 307
— цианат 126, 127, 147
— цианид 55
Синильная кислота 46—48
Скандий
— селеноцианат 274, 280, 282
— тиоцианат 175, 176, 191, 204, 237
Спектрохимический ряд (место псев-
догалогенидов) 20, 337
Сурьма
— азид 28
— тиоцианат 171
— цианат 128, 147
— цианид 55, 58
Таллий
— азид 30, 31, 34, 38
— дицианамид 322
— селеноцианат 261, 263, 265
— тиоцианат 169, 190, 206, 218, 229
— трицианметанид 306, 307
— фульминат 131
— цианат 126, 142
— цианид 54, 58
Тантал
— азид 35
— селеноцианат 274, 285, 291
— тиоцианат 179, 202, 205, 207, 237
Теллур 55, 90
Тетрацианометан 55
Технеций 72, 94, 182, 205, 224
Тиофульминаты 80
Тиоцианатная кислота 162, 163, 258,
334, 335
Тиоцианатоводород 162, 163
Торий
— селеноцианат 266, 278, 284
— тиоцианат 177, 205, 236, 237
— цианид 64
Устойчивость псевдогалогенидных
комплексов
— в воде 75, 189—193, 262. 310, 311,
327, 328
— в смешанных растворителях 88,
193—195, 262, 263, 268
— в неводных 86—90, 193—195, 263—
— 267, 339—342
— в солевых расплавах 90—92
Уран '
— азид 34, 35
— селеноцианат 274, 276, 278, 282
— тиоцианат 177, 191, 205, 222
— цианат 134
— цианид 64
Фосфор
— азид 30
— тиоцианат 171, 204, 230
— трицианметанид 306
цианат 127, 128, 147
— цианид 55, 58, 90
Хром
— азид 35—36
— дицианамид 327
— селеноцианат 270—272, 275, 276,
280, 282, 285, 290, 294
— тиоцианат 180, 181, 192, 207,
222—224, 228
— трицианметанид 310
— фульминат 140, 151
— цианат 133, 134, 137, 144
— цианид 66, 67, 69, 70, 90, 94, 96,
98, 99, 102—104
Цианатная кислота 123—125, 334, 335
Цианизоцианат 56
Циановодород 46—48
Цианоформ 305
Цинк
— азид 33, 34
— дицианамид 325, 330
— селеноцианат 263—268, 270—272,
274, 276, 278, 280, 282, 283, 291
— тиоцианат 165—167, 190, 198, 206,
207, 216
— трицианометанид 308, 309, 311, 312
— фульминат 140, 141, 151
— цианат 133, 135, 136, 149
— цианид 53, 56—58, 90, 91, 95,
97—100, 104, 106
Цирконий
354
— селеноцианат 264, 273, 274
— тиоцианат 178, 205, 237
— цианат 136, 137, 149
— цианид 65
Щелочные металлы
— азид 23—28
— дицианамид 317, 318
— селеноцианаты 258—260
— тиоцианат 163—165, 204, 229
— трицианметанид 301, 302
— фульминат 131, 144, 151
— цианат 125
— цианид 48—50, 87, 92, 105—107
Щелочноземельные металлы
— азид 25—28
— селеноцианат 260, 272, 274, 276
— тиоцианат 164, 190, 204, 229
— фульминат 131
— цианат 126
— цианид 48—50
Экстракция тиоцианатных комплексов
235—237
Электроотрицательность псевдо-
галогенидных групп 338
ЯМР-данные псевдогалогенидов и их
производных
— дицианамид 319
— тиоцианит 207
— трицианметанид 304
— цианат 132
— цианид 48, 52, 54, 61, 79, 90
ОГЛАВЛЕНИЕ
Предисловие к русскому изданию................................................ 3
Предисловие к немецкому изданию .............................................. 4
1. Введение
1.1. Понятие «псевдогалоген». Псевдогалогениды. Псевдогалогены................ 6
1.2. Номенклатура псевдогалогенидных соединений .............................. 7
1.3. Характеристика псевдогалогенидов ........................................ 8
1.4. Классификация псевдогалогенидных соединений ...............;............ 10
Список литературы........................................................ 14
2. Неорганические азидные соединения
2.1. Водородные соединения азота........................................... 15
2.2. Азидоводород и азидная кислота ......................................... 15
2.2.1. Газообразный и жидкий HN3 (азоимид) .................................. 15
2.2.2. Азидная кислота ...................................................... 17
2.3. Аналитические и структурные свойства азидной группы..................... 18
2.3.1. Порядок связей и структура............................................ 18
2.3.2. Структура азидного радикала и нитренной группировки................... 21
2.3.3. Аналитическая химия азидных соединений ............................... 22
2.4. Классификация азидов ................................................... 23
2.5. Азиды s-элементов....................................................... 23
2.5.1. Азиды щелочных металлов............................................... 23
2.5.2. Азиды щелочноземельных металлов ...................................... 25
2.5.3. Свойства азидов щелочноземельных и щелочных металлов.................. 25
2.6. Азиды р-элементов....................................................... 28
2.6.1. Азиды неметаллов ..................................................... 28
2.6.2. Азиды алюминия, галлия, индия и таллия ............................... 30
2.6.3. Азиды олова, свинца, сурьмы и висмута................................. 31
2.7. Азиды элементов побочных подгрупп ...................................... 32
2.7.1. Азиды элементов I и II побочных подгрупп.............................. 32
2.7.2. Азиды элементов III—VII побочных подгрупп............................. 34
2.7.3. Азиды элементов VIII побочной подгруппы............................... 36
2.8. Применение азидов ................................................ 37
Список литературы ................................................ 39
3. Цианиды
3.1. Получение и свойства дициана............................................ 45
3.2. Циановодород и его кислотные свойства................................... 46
3.2.1. Общие свойства HCN.................................................... 46
3.2.2. Получение циановодорода и его строение................................ 47
3.2.3. Химические свойства HCN............................................... 47
3.3. Цианиды аммония, щелочных металлов и металлов подгруппы бериллия .... 48
3.3.1. Получение солей ...................................................... 48
3.3.2. Общие свойства ионных цианидов........................................ 48
3.3.3. Свойства отдельных соединений элементов I и II главных подгрупп....... 49
3.4. Цианид-ион и его комплексообразующая способность ....................... 50
3.4.1. Электронное строение и донорная способность........................... 50
3.4.2. Акцепторная способность .............................................. 51
3.4.3. Амбидентатность цианид-иона .......................................... 52
3.5. Цианиды р- и й10-элементов.............................................. 53
3.5.1. Простейшие цианиды и координационные соединения несолевого типа .... 53
356
3.5.2. Анионные комплексы р- и с110-элементов............................. . . 56
3.6. Простые и координационные цианиды d- и f-металлов ...................... 58
3.6.1. Цианиды металлов I побочной подгруппы................................. 59
3.6.1.1. Простые цианиды..................................................... 59
3.6.1.2. Однородные комплексные цианиды Cu, Ag, Аи .......................... 59
3.6.1.3. Смешанные соединения металлов подгруппы меди........................ 61
3.6.2. Цианиды металлов III побочной подгруппы .............................. 64
3.6.2.1. Цианиды лантаноидов................................................. 64
3.6.2.2. Цианиды актиноидов ................................................. 64
3.6.3. Цианидные комплексы металлов IV побочной подгруппы.................... 64
3.6.4. Цианиды металлов V побочной подгруппы................................. 65
3.6.5. Цианиды металлов VI побочной подгруппы................................ 66
3.6.6. Цианиды металлов VII побочной подгруппы............................... 70
3.6.7. Цианиды семейства железа ............................................. 73
3.6.7.1. Простые цианиды .................................................... 73
3.6.7.2. Комплексные цианиды ................................................ 74
3.6.8. Цианиды платиновых металлов .......................................... 83
3.6.8.1. Простые цианиды .................................................... 83
3.6.8.2. Комплексные цианиды ................................................ 83
3.7. Цианидные комплексы в неводных средах................................... 86
3.7.1. Цианокомплексы металлов I и II побочных подгрупп...................... 87
3.7.2. Цианокомплексы металлов семейства железа.............................. 89
3.7.3. Исследование цианидов неметаллов ..................................... 90
3.7.4. Исследование цианокомплексов в солевых расплавах...................... 90
3.8. Структура цианидов ..................................................... 92
3.8.1. Применение колебательных спектров для выяснения строения цианидных комп-
лексов ...................................................................... 92
3.8.2. Изучение кристаллической структуры цианидных соединений............... 97
3.9. Применение цианидов ....................................................105
3.9.1. Цианиды в химическом анализе..........................................105
3.9.2. Цианидные соединения в цветной металлургии и гальванотехнике ..... 106
3.9.3. Каталитические процессы ..............................................106
3.9.4. Другие области применения цианидов....................................107
Список литературы .....................................................107
4. Цианаты и фульминаты
4.1. Диоксициан .............................................................123
4.2. Цианатная кислота. Структура и свойства.................................123
4.3. Получение и свойства цианатов аммония, щелочных и щелочноземельных металлов 125
4.4. Изоцианаты элементов III—VII главных подгрупп...........................126
4.5. Цианаты d- и f-металлов ................................................129
4.6. Гремучая кислота .......................................................130
4.7. Фульминаты металлов ....................................................130
4.8. Структура и способ координации цианат- и фульминат-ионов................131
4.9. Цианатные и изоцианатные комплексы......................................132
4.9.1. Анионные соединения ..................................................132
4.9.2. Координационные соединения цианатов с другими лигандами...............134
4.9.3. Реакции изоцианатных комплексов.......................................138
4.10. Фульминатные комплексы ................................................139
4.11. Структура цианатных и фульминатных соединений .........................142
4.11.1. Рентгеноструктурные исследования ....................................142
4.11.2. ИК-спектральные исследования ........................................146
4.11.2.1. ИК-спектральные исследования цианатов .............................147
4.11.2.2. ИК-спектральные исследования фульминатов ..........................150
4.11.3. 14М-ЙМР-исследования цианатов и фульминатов..........................152
4.12. Применение цианатов и фульминатов......................................153
Список литературы......................................................154
5. Тиоцианатные соединения
5.1. Получение и свойства дитиоциана.........................................161
5.2. Тиоцианатоводород и тиоцианатная кислота ...............................162
5.3. Тиоцианаты s-металлов и аммония.........................................163
5.4. Получение и свойства тиоцианатных соединений р- и d10^eMeHTOB ...... 165
5.4.1. Тиоцианаты металлов II побочной подгруппы.............................165
5.4.2. Тиоцианаты элементов III главной подгруппы ...........................168
357
5.4.3. Тиоцианаты элементов IV главной подгруппы............................170
5.4.4. Тиоцианаты элементов V—VII главных подгрупп..........................171
5.5. Тиоцианаты переходных металлов.........................................172
5.5.1. Тиоцианаты металлов I побочной подгруппы.............................172
5.5.2. Тиоцианаты редкоземельных элементов..................................175
5.5.3. Тиоцианаты актиноидов................................................177
5.5.4. Тиоцианаты металлов IV побочной подгруппы............................178
5.5.5. Тиоцианаты металлов V побочной подгруппы.............................179
5.5.6. Тиоцианаты металлов VI побочной подгруппы............................180
5.5.7. Тиоцианаты металлов подгруппы марганца..............................181
5.5.8. Тиоцианаты металлов семейства железа ...............................183
5.5.9. Тиоцианаты платиновых металлов......................................187
5.6. Образование тиоцианатных комплексов в водном и неводном растворах .... 188
5.6.1. Состав и устойчивость тиоцианатных комплексов в водном растворе......189
5.6.2. Состав и устойчивость тиоцианатных комплексов в смешанных и неводных рас-
творителях .................................................................193
5.7. Координационные тиоцианатогалогенидные соединения и их устойчивость в рас-
творе ..................................................................... 196
5.8. Характеристика комплексообразующей способности тиоцианат-иона..........199
5.9. ИК-спектральное, электронографическое и рентгеноструктурное исследования
тиоцианатных соединений.....................................................201
5.9.1. ИК-спектральный критерий способа координации SCN-группы..............201
5.9.2. Инверсия связи SCN-группы............................................211
5.9.3. Рентгеноструктурное исследование.....................................214
5.10. Применение тиоцианатных комплексов....................................235
5.10.1. Тиоцианаты в химическом анализе ......................*.............235
5.10.2. Тиоцианаты в технологии некоторых металлов..........................237
Список литературы....................................................238
6. Селеноцианатные соединения
6.1. Диселеноциан и его свойства ...........................................256
6.2. Химико-аналитические особенности SeCN-иона ............................257
6.3. Селеноцианатная кислота и ее соли .....................................257
6.4. Селеноцианаты р- и ё10-металлов .......................................260
6.5. Изучение селеноцианатных комплексов в растворе.........................262
6.5.1. Комплексообразование в водных и смешанных растворах..................262
6.5.2. Метанольные растворы ................................................263
6.5.3. Диметилформамидные растворы .........................................264
6.5.4. Диметилсульфоксидные растворы .......................................265
6.5.5. Ацетоновые растворы .................................................266
6.5.6. Ацетонитрильные растворы ............................................267
6.5.7. Влияние природы растворителя на реакции комплексообразования с участием
SeCN-групп..................................................................268
6.6. Способ координации SeCN-групп .........................................269
6.7. Координационные соединения селеноцианатов с аминами....................270
6.7.1. Соединения, содержащие пиридин и его производные.....................270
6.7.2. Соединения, содержащие дипиридил и фенантролин.......................272
6.7.3. Другие соединения селеноцианатов с аминами...........................276
6/8. Соединения с нейтральными лигандами, содержащими донорно-активный кисло-
род .....................................................................277
6.8.1. Соединения, содержащие диметилсульфоксид и диметилформамид...........278
6.8.2. Соединения, содержащие гексаметилфосфортриамид.......................280
6.8.3. Другие селеноцианаты ................................................281
6.9. Соединения, содержащие связи металл — фосфор, металл — углерод ........283
6.10. Координационные селеноцианаты анионного типа .........................283
6.10.1. Препаративные данные ...............................................283
6.10.2. Анионные комплексы с мостиковыми SeCN-группами......................286
6.10.3. Анионные селеноцианатные комплексы .................................288
6.10.4. Анионные изоселеноцианаты ..........................................289
6.10.5. Разнолигандные псевдогалогениды ....................................292
Список литературы.................................................. 294
7. Трицианметаниды
7.1. Псевдогалогенидный характер трицианметанид-иона........................300
7.2. Свойства и структура трицианметанид-иона ..............................301
7.3. Типы связи в трицианметанидах и способы их интерпретации ..............302
358
ЧА. Ковалентные трицианметаниды элементов главных подгрупп............. , , . 305
7.4.1. Дицианкетенимин HNCC (CN)2 ................................. 305
7.4.2. Галогентрицианметаны............................................. . 306
7.4.3. Металлоорганические трицианметаниды............................... » . 306
7.5. Трицианметаниды d-металлов ............................................308
7.6. Трицианметанидные комплексы переходных металлов с другими лигандами ... 311
7.6.1. Комплексы с N-и О-донорными молекулами...............................311
7.6.2. Комплексы с С- и Р-донорными молекулами..............................313
Список литературы ....................................................315
8. Дициаг амиды
8.1. Псевдогалогенидный характер дицианамид-ионов...........................317
8.2. Свойства и структура дицианамид-ионов .................................319
8.3. Типы связи в производных дицианамида и их интерпретация ............320
8.4. Дицианамиды р-элементов.............................................322
8.5. Дицианамиды d-металлов..............................................325
8.6. Дицианамидные комплексы d-металлов ................................327
8.6.1. Комплексообразование в растворе .......................................327
8.6.2. Анионные комплексы ..................................................327
8.6.3. Дицианамидные комплексы с другими лигандами...........................330
Список литературы .......................................................332
9. Сравнительная характеристика псевдогалогенидов
9.1. Кислотные свойства и структура псевдогалогенидных кислот................- 334
9.2. Растворимость псевдогалогенидсв и галогенидов металлов....................335
9.3. Характеристика донорной активности псевдогалогенид-лигандов...............337
9.4. Разнолигандные комплексы линейных и нелинейных псевдогалогенидов .... 341
9.5. Перспективы развития химии псевдогалогенидов .............................342
Список литературы ........................................................ 345
Принятые сокращения ...........................................................347
Предметный указатель ..........................................................351
Андрей Матвеевич Голуб, Хельмут Кёлер,
Виктор Васильевич Скопенко и др.
ХИМИЯ ПСЕВДОГАЛОГЕНИДОВ
Редактор А. Е. Балясная
Переплет художника Т. Т. Кузнецовой
Художественный редактор Т. С. Преснякова
Технический редактор Н. Н. Бабюк
Корректоры 3. П. Чиркова, О. Г. Малышевская, А. Ф. Пасечный
ИБ 6888
Сдано в набор 13.10.80. Подп. в печать 27.04.81. БФ 11083. Формат 70X100/16. Бумага типогр.
№ 1. Лит. гарн. Выс. печать 29,02 усл. печ. л. 29,83 уел. кр.-отт. 30,59 уч.-изд. л. Тираж
2000 зкз. Изд. № 1462-к. Зак. № 2473. Цена 5 р.
Издательство при Киевском государственном университете издательского объединения
«Вища школа», 252001, Киев-1, Крещатик, 4.
Головное предприятие республиканского производственного объединения «Полиграфкнига»
Госкомиздата УССР, 252057, Киев-57, ул. Довженко, 3.