/




























































































































































































































































































Текст
основы
ТЕХНОЛОГИИ
нефтехимического
СИНТЕЗА
П.С.Белов
П. С. Белов
основы
ТЕХНОЛОГИИ
нефтехимического
СИНТЕЗА
ИЗДАНИЕ ВТОРОЕ,
ПЕРЕРАБОТАННОЕ
Допущено
Министерством нефтеперерабатывающей
и нефтехимической промышленности СССР
в качестве учебника
для средних специальных учебных заведений
МОСКВА, «ХИМИЯ», 1982
6П7.50
Б 435
УДК 66.09(075)
Белов П. С.
Основы технологии нефтехимического синтеза.
2-е изд., перераб.—М.: Химия, 1982 г. — 280 с., ил.
Данный учебник (первое издание вышло в 1965 г.) пере-
работан в соответствии с новой учебной программой этого
курса.
В книге кратко рассмотрены современные промышлен-
ные процессы получения различных углеводородов, их га-
логен- и кислородсодержащих производных как сырья для
нефтехимических процессов. Рассмотрены процессы получе-
ния моющих средств, а также важнейших полимеров, исполь-
зуемых в производстве пластических масс и синтетических
волокон. Для всех процессов даны основные сведения по
экономике, технике безопасности и охране окружающей
среды.
Книга предназначается для учащихся средних специальных
учебных заведений по специальности «Химическая технология
нефти и газа». Она представляет интерес и для преподавате-
лей средних специальных учебных заведений, а также для ин-
женерно-технических работников нефтеперерабатывающих и
нефтехимических предприятий.
280 с., 70 рис., 4 табл., список литературы. 11 ссылок.
Рецензент — зав^ лаоораториеи ВНИИ НП доктор хими-
ческих наук, профам**. "Корсакова.
^2803010000-056
050(01)-82
56.82
© Издательство «Химия», 1982 г.
СОДЕРЖАНИЕ
Предисловие 6
Введение 7
Глава I. Производство углеводородного сырья и его подготовка к хими-
ческой переработке 10
§ 1. Основные направления химической переработки углеводородно-
го сырья . 10
1.1. Переработка газообразных парафиновых углеводородов 10
1.2. Переработка ненасыщенных углеводородов « 13
1.3. Переработка ароматических углеводородов 16
§ 2. Производство углеводородного сырья . . 17
2.1. Основные источники углеводородного сырья 17
2.2. Производство низших олефинов 18
2.3. Производство высших олефинов 22
2.4. Производство ацетилена 24
2.5. Производство алкилбензолов . 28
2.6. Производство углеводородов изостроепия 38
2.7. Производство алкилфенолов .... 44
§ 3. Подготовка углеводородного сырья к химической переработке 51
3.1. Требования к качеству сырья 51
3.2. Очистка газообразных углеводородов 52
3.3. Осушка газообразных углеводородов 55
3.4. Разделение газообразных углеводородов 57
3.5. Разделение жидких углеводородов 68
Глава II. Производство виниловых мономеров и диенов 76
§ 1. Производство углеводородных мономеров дегидрированием 76
1.1. Теоретические основы процессов дегидрирования .... 76
1.2. Производство бутадиена двухстадийным дегидрированием
«-бутана 78
1.3. Производство бутадиена одностадийным дегидрированием
н-бутана . . 84
1.4. Производство изопрена двухстадийным дегидрированием изо-
пентана . . . 86
1.5. Производство изопрена из изобутилена и формальдегида 88
1.6. Производство изопрена из пропилена 91
1.7. Производство стирола и а-метилстирола ... 92
§ 2. Производство виниловых мономеров с функциональными груп-
пами 95
2.1. Производство винилхлорида 96
2.2. Производство акрилонитрила 98
2.3. Производство винилацетата 101
2.4. Производство акриловой и метакриловой кислот и их эфиров ЮЗ
Глава III. Производство кислородсодержащих продуктов 106
§ 1. Основные представления о процессах окисления углеводородов 106
1.1. Механизм процесса . . . Ю6
1.2. Закономерности окисления углеводородов в газовой и жид-
кой фазе 109
§ 2. Производство кислородсодержащих продуктов окислением выс-
ших парафинов 115
2.1. Производство высших жирных кислот 115
2.2. Производство высших жирных спиртов 118
§ 3. Производство кислородсодержащих продуктов окислением цик-
лических углеводородов . 119
3.1. Основные закономерности окисления циклических углеводо-
родов . 119
3.2. Производство фенола и ацетона через изопропилбензолгидро-
пероксид 122
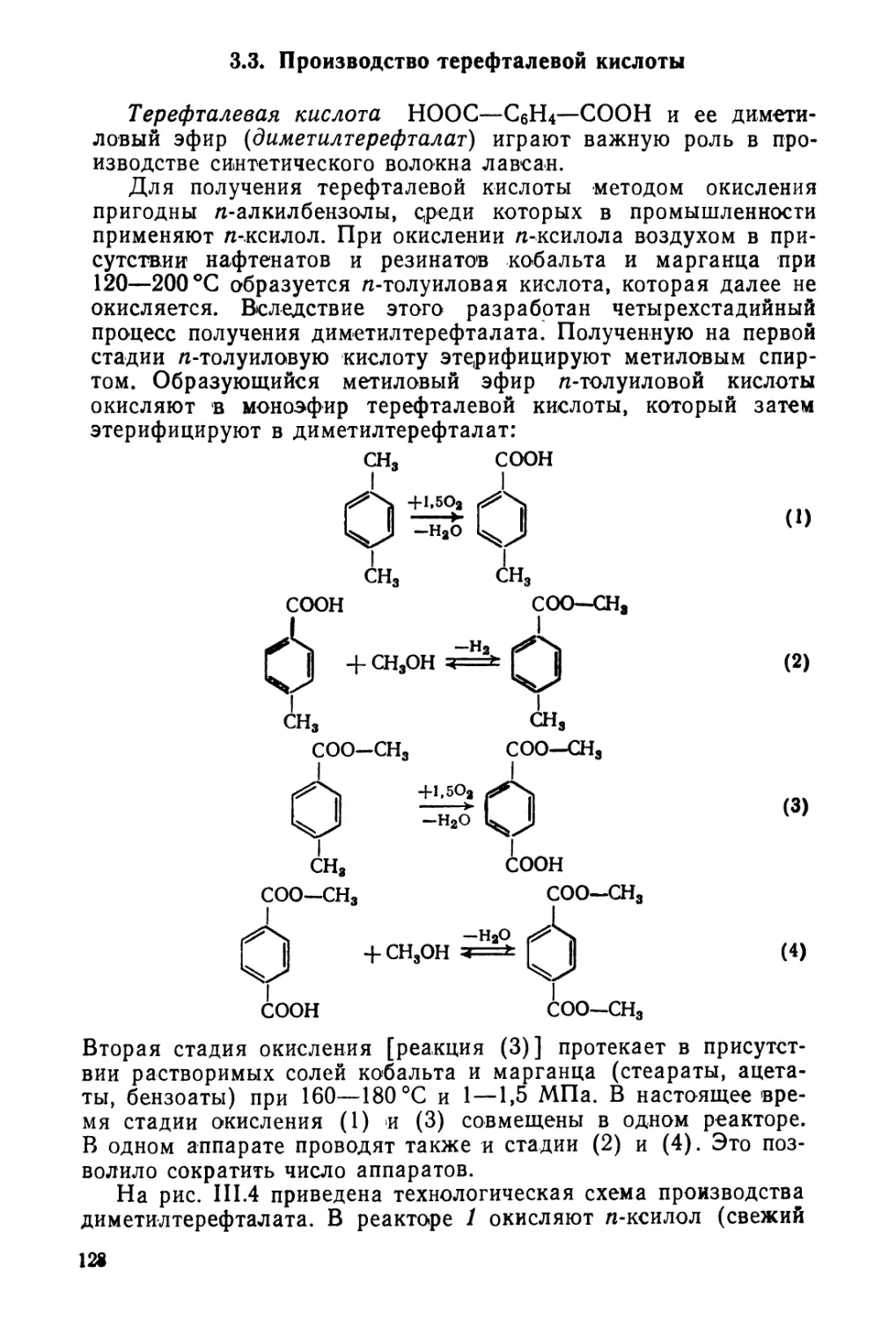
3.3. Производство терефталевой кислоты 128
§ 4. Производство кислородсодержащих продуктов окислением оле-
финов 131
4.1. Особенности окисления олефинов 131
4.2. Производство этиленоксида 134
4.3. Производство акролеина 138
4.4. Производство ацетальдегида . . 139
§ 5. Производство кислородсодержащих продуктов реакциями гидра-
тации 142
5.1. Теоретические основы процессов гидратации . . . . 142
5.2. Производство этилового спирта сернокислотной гидратацией
этилена 145
5.3. Производство изопропилового и изобутиловых спиртов сер-
нокислотной гидратацией пропилена и «-бутиленов . 150
5.4. Производство этилового спирта прямой гидратацией эти-
лена . . ..... 152
5.5. Производство изопропилового и трег-бутилового спиртов
прямой гидратацией пропилена и изобутилена . 155
§ 6. Производство спиртов по реакциям присоединения . 156
6.1. Производство высших жирных спиртов гидрированием кар-
боновых кислот и их эфиров . . 156
6.2. Производство высших жирных спиртов из альдегидов 158
6.3. Производство высших жирных спиртов алюминийорганиче-
ским синтезом . . 161
6.4. Производство метилового спирта из синтез-газа 162
6.5. Производство спиртов оксосинтезом 170
6.6. Другие синтезы из СО и Н2 . 175
6.7. Производство этилен- и пропиленгликоля 177
6.8. Производство глицерина 179
Глава IV. Производство синтетических моющих веществ 182
§ 1. Применение и классификация поверхностно-активных веществ 182
1.1. Применение поверхностно-активных веществ . 182
1.2. Физико-химия действия ПАВ в водных и неводных средах 184
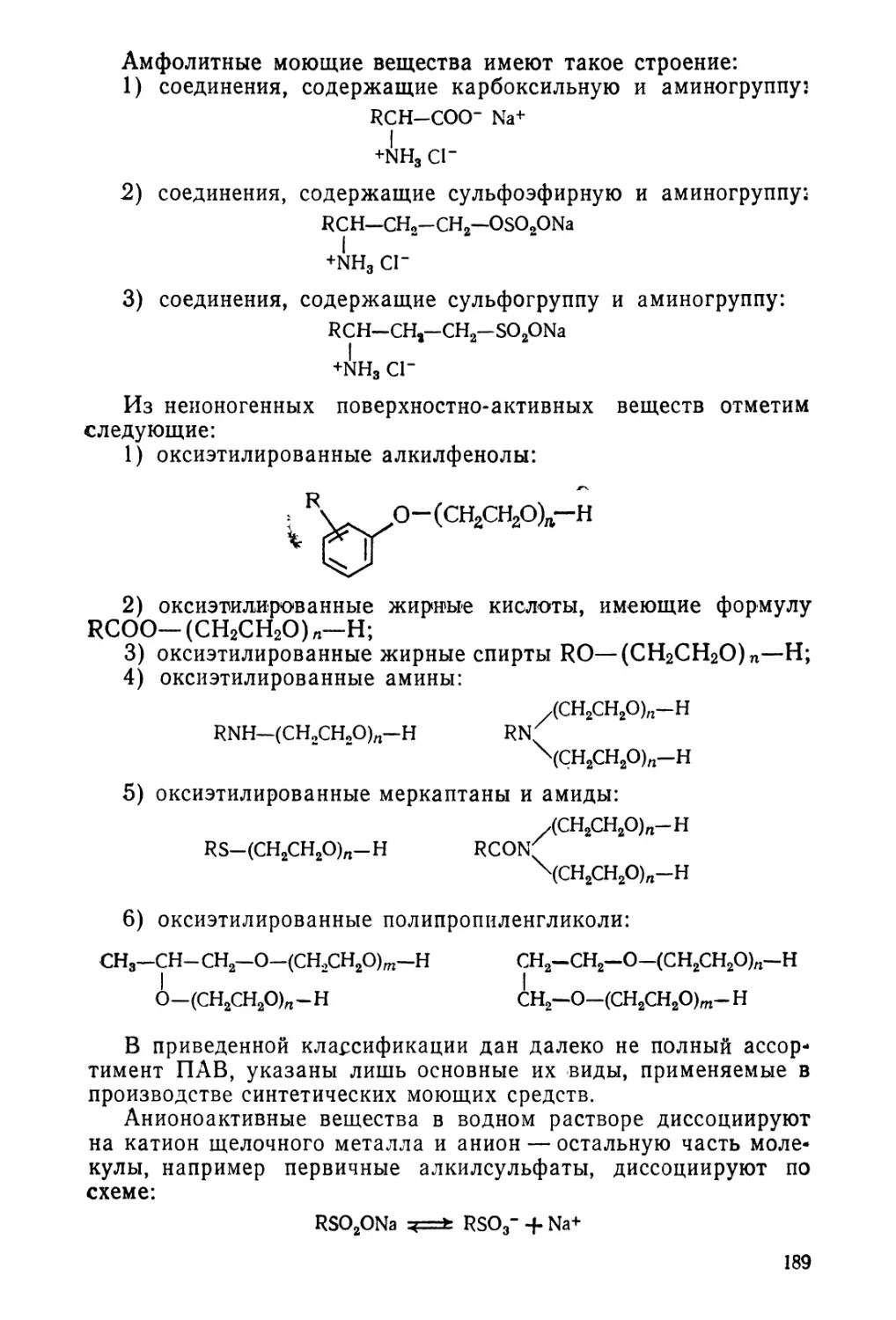
1.3. Классификация поверхностно-активных веществ 187
§ 2. Производство анионоактивных моющих веществ . 190
2.1. Теоретические основы процессов сульфирования, сульфохло-
рирования и сульфоокисления 190
2.2. Производство алкилсульфатов 194
2.3. Производство алкилсульфонатов 196
2.4. Производство алкилбензолсульфонатов 198
2.5. Производство а-олефинсульфонатов 201
§ 3. Производство неионогенных поверхностно-активных веществ 203
§ 4. Повышение качества синтетических моющих веществ . 207
4 .1. Способы повышения моющего действия синтетических мою-
щих веществ . 207
4 2 Перспективные типы ПАВ 208
4
Глава V. Производство полимеров 210
§ 1. Общие сведения о полимерах 210
1.1. Основные понятия и классификация 210
1.2. Химическое строение и структура полимеров 211
1.3. Методы синтеза полимеров 213
1.4. Полимерные материалы . 220
§ 2. Производство полимеров реакциями полимеризации 223
2.1. Производство полимерных углеводородов 223
2.2. Производство галогенсодержащих полимеров . . 238
2.3. Производство полимеров эфиров непредельных кислот и их
производных . 242
2.4. Производство поливинилового спирта и его производных 244
2.5. Производство простых полиэфиров . 246
§ 3. Производство полимеров реакциями поликонденсации 247
3.1. Производство фенолоформальдегидных полимеров 247
3.2. Производство сложных полиэфиров 249
3.3. Производство эпоксидных полимеров 251
3.4. Производство полиамидов 252
3.5. Производство полиуретанов . 254
3.6. Производство кремнийорганических полимеров 255
Глава VI. Производство неорганических продуктов 257
§ 1. Производство серы . 257
§ 2. Производство серной кислоты 259
$ 3. Производство водорода 262
§ 4. Производство аммиака 265
5. Производство карбамида 267
Литература 271
Предметный указатель 272
ПРЕДИСЛОВИЕ
С момента выхода в свет первого издания этой книги прош-
ло более семнадцати лет. За истекший период в технологии
нефтехимического синтеза были достигнуты большие успехи —
созданы новые рациональные процессы, существенно усовер-
шенствованы действующие. Технический прогресс в этой обла-
сти, а также непрерывно продолжающийся процесс химизации
народного хозяйства обусловили необходимость создания учеб-
ника, отражающего современное состояние технологии нефтехи-
мического синтеза.
Для удобства изложения принята систематизация материа-
ла по основным химическим процессам. Это позволило в нача-
ле каждой главы дать теорию и технологию группы сходных
процессов. Учебник написан в соответствии с программой курса.
Материал книги сокращен по сравнению с первым издани-
ем. Исключены сведения по производству синтетических каучу-
ков и по применению ионизирующих излучений. В книгу вклю-
чен ряд новых технологических схем и переработаны имеющие-
ся, с тем чтобы они легче читались. При подготовке настоящей
рукописи частично учтены рекомендации и пожелания читате-
лей. При систематизации материала, основанной на общности
химических процессов, возникли затруднения, которые, вероят-
но, не везде успешно преодолены.
Автор благодарит рецензента доктора химических наук
И. С. Корсакову за ценные замечания и будет признателен
всем, кто пришлет отзывы и критические замечания. В оформ-
лении рукописи большую помощь оказали К. Д. Коренев
и О. А. Белова, которым автор выражает искреннюю благодар-
ность.
ВВЕДЕНИЕ
Нефтехимическая промышленность в качестве сырья ис-
пользует продукты переработки нефти. Задачи этой отрасли —
обеспечение многих органических производств углеводородным
сырьем (олефинами, ацетиленом, ароматическими углеводоро-
дами, синтез-газом), получение многотоннажных органических
веществ и синтетических полимерных материалов.
К многотоннажным органическим продуктам относятся мо-
номеры, спирты, кетоны, кислоты, эфиры, оксиды олефинов,
моющие вещества.
Синтетические полимерные материалы являются крупнейши-
ми потребителями нефтехимического сырья. Без преувеличения
можно сказать, что в настоящее время полимеры (пластические
массы, каучук и резина, волокна, пленки, лаки, клеи и т. д.)
применяются почти во всех отраслях народного хозяйства. Вот
почему очень важно рассмотреть теоретические основы синтеза
полимеров и технологические методы их получения.
Комплексный подход к переработке сырья, повышение эко-
номической эффективности предприятий, рост дефицита в при-
родных ресурсах, интересы охраны окружающей среды приве-
ли к организации на нефтехимических предприятиях собствен-
ного производства некоторых неорганических веществ из газа,
нефти и продуктов их переработки. В ряде случаев эта необхо-
димость диктуется технической и экономической целесообраз-
ностью.
Например, водород обычно используют там, где его произ-
водят (из-за трудностей транспортирования). Водород получают
конверсией метана и других углеводородных газов и потребля-
ют в процессах синтеза аммиака и в различных гидрогенизаци-
онных процессах.
Сероводород, извлекаемый из природных газов и газов пе-
реработки сернистых нефтей, используют как сырье для произ-
водства свободной серы и серной кислоты. Сера является сырь-
ем для производства серной кислоты и в больших количествах
используется для вулканизации синтетических каучуков. Сер-
ная кислота широко применяется в производстве минеральных
удобрений, этилового спирта, моющих веществ, присадок к
маслам и др.
Аммиак используют в основном в производстве карбамида
(эффективное удобрение и добавка к кормам) и азотной кис-
7
лоты. В виде водных растворов аммиак применяют как удобре-
ние и как консервант кормовых продуктов (например, силоса).
Сказанное характеризует нефтехимический синтез как про-
изводство огромного числа веществ различных химических
классов, многообразие химических процессов их получения с
применением катализаторов разной химической природы
и свойств. Для нефтехимической промышленности характерно
динамичное развитие с непрерывным обновлением ассорти-
мента выпускаемой продукции, освоением новых процессов и
аппаратов, широким внедрением более совершенной техноло-
гии. Создаются производства большой единичной мощности,
при разработке которых используются методы математиче-
ского моделирования и оптимизации, химической кибернетики
и вычислительной техники. Управление рядом технологических
процессов (особенно непрерывных) осуществляется с помощью
ЭВМ.
В СССР нефтехимическая промышленность выросла в круп-
ную отрасль и продолжает развиваться. Это определяется тем
влиянием, которое оказывает нефтехимия на народное хозяй-
ство, являясь поставщиком сырья для получения различных
продуктов и материалов, находящихся в природе в ограничен-
ном количестве или вообще отсутствующих. XXVI Съезд КПСС
постановил увеличить выпуск продукции химической и нефте-
химической промышленности; поставлена также задача разра-
ботать высокоэффективные процессы с новыми каталитически-
ми системами, повысить производительность технологического
оборудования.
Большая часть многотоннажных нефтехимических произ-
водств базируется на переработке природных и попутных газов,
газов крекинга и пиролиза, на переработке жидких фракций
нефти и продуктов термического и каталитического крекинга,
твердых и мягких парафинов. Несмотря на огромный ассорти-
мент соединений получают их при помощи сравнительно не-
большого числа реакций — это алкилирование, гидрирование и
дегидрирование, пиролиз, окисление, гидратация, полимериза-
ция, поликонденсация.
Нефтехимические установки, появившиеся на основе нефте-
переработки, часто соседствуют с нефтепереработкой или выде-
ляются в самостоятельные предприятия, расположенные рядом.
Это повышает рентабельность предприятия, сокращает энерге-
тические, эксплуатационные, транспортные и прочие расходы.
Развитие производства нефтехимических продуктов существен-
но влияет на схемы нефтеперерабатывающих заводов. В ряде
случаев в схему завода включают процессы пиролиза, чтобы
обеспечить низкомолекулярными олефинами производство по-
лиэтилена и полипропилена, этил- и изопропилбензола, этило-
вого спирта и т. д. Поэтому характерная особенность современ-
ных предприятий — комплексная переработка сырья на топлив-
ные продукты и продукты нефтехимического синтеза.
8
Опыт, приобретенный нефтепереработкой по ароматизации
сырья и получению алкилароматических углеводородов для вы-
пуска высокооктановых бензинов, позволил переориентировать
установки по производству ароматических соединений на полу-
чение их для целей нефтехимии. Повышается роль попутных га-
зов в химической переработке, в частности роль углеводородов
С4 и С5 для производства бутилена, бутадиена, изопентана, изо-
амиленов и изопрена. Развиваются синтезы «а основе оксида
углерода и водорода.
Важнейшей задачей нефтехимической промышленности яв-
ляется повышение селективности процессов за счет применения
более эффективных катализаторов. Повышение селективности
до 90—96% резко снизит расход сырья на 1 т целевой продук-
ции. Очень важно также повышать глубину превращения ре-
агентов за однократный проход через реактор, что увеличивает
рентабельность и снижает эксплуатационные расходы.
Нефтепереработка и нефтехимия потребляет большие коли-
чества тепла. Утилизация энергии, создание энерготехнологиие-
ских процессов должны быть характерными особенностями от-
расли на ближайшее время. Это нужно сочетать с разработкой
безотходной или малоотходной технологии, с ликвидацией сточ-
ных вод и выбрасываемых в атмосферу отходящих газов, что
существенно улучшит экологическую обстановку в районах раз-
витой нефтехимии.
ГЛАВА I
ПРОИЗВОДСТВО УГЛЕВОДОРОДНОГО СЫРЬЯ
И ЕГО ПОДГОТОВКА
К ХИМИЧЕСКОЙ ПЕРЕРАБОТКЕ
§ 1. ОСНОВНЫЕ НАПРАВЛЕНИЯ ХИМИЧЕСКОЙ
ПЕРЕРАБОТКИ УГЛЕВОДОРОДНОГО СЫРЬЯ
Отличительная особенность нефти и природного газа как
сырья для химической переработки в сравнении с другими го-
рючими ископаемыми (углем, сланцем, торфом) заключается
в том, что нефть и природный газ состоят в основном из угле-
водородов.
Углеводороды нефти, газа и нефтепереработки, использу-
емые в качестве сырья в нефтехимической промышленности,
содержат: насыщенные углеводороды с нормальной или раз-
ветвленной углеродной цепью; ненасыщенные углеводороды с
одной или двумя двойными связями или с тройной связью; аро-
матические углеводороды, алициклические углеводороды с на-
сыщенными или ненасыщенными кольцами (в основном пяти- и
шестичленными). Эти углеводороды неодинаковы по своей ре-
акционной способности, благодаря чему из них можно получить
практически любые химические вещества и организовать их
производство по наиболее рациональным комплексным схемам.
1.1 Переработка газообразных
парафиновых углеводородов
Метан (СН4) в настоящее время используется в основном
для производства хлор- и нитропроизводных — метилхлорида,
метиленхлорида, хлороформа, четыреххлористого углерода,
нитрометана, применяемых в качестве растворителей в различ-
ных областях техники. При пиролизе метана образуются ацети-
лен, сажа и водород, имеющие важное самостоятельное значе-
ние. Перспективно использовать метан в процессах окисления
для производства формальдегида, метанола и ацетальдегида
(схема I), а также как сырье при микробиологическом синтезе.
Этан (С2Нб) по направлениям переработки во многом схо-
ден с метаном. На основе этана получают аналогичные продук-
ты (схема II): этилхлорид, применяемый для изготовления тет-
раэтилсвинца— антидетонатора бензина, нитроэтан — раство-
ритель, используемый там же, где и нитрометан. Важным
направлением использования этана является его пиролиз с по-
лучением этилена —одного из главных углеводородов для неф-
техимического синтеза.
Пропан (С3Н8). При окислении пропана получают ацеталь-
дегид, формальдегид, уксусную кислоту, ацетон. При пиролизе
10
Схема I. Химическая переработка метана
пропана образуются этилен и пропилен. Пропан наряду с мета-
ном и этаном можно использовать и для получения ацетилена.
Нитрованием пропана получают нитрометан, нитроэтан и нит-
ропропан (схема II).
н-Бутан (н-С4Ню) используют в качестве сырья для произ-
водства олефинов — этилена, пропилена, бутиленов, а также
ацетилена и бутадиена (схема III). При окислении бутана об-
разуются ацетальдегид, уксусная кислота, ацетон и другие про-
дукты. При нитровании получают 1- и 2-нитробутаны. При изо-
меризации н-бутан превращается в изобутан.
Схема II. Химическая переработка этака и пропана
11
Изобутан (изо-С4Ню) применяют в процессах алкилирова-
ния для получения высокооктановых компонентов моторных
топлив. При дегидрировании изобутана образуется изобути-
лен— сырье для получения бутилкаучука, полиизобутилена, по-
лимербензина; кроме того, изобутилен является широко распро-
страненным алкилирующим агентом.
Изопентан (^зо-С5Н12) практически полностью перерабаты-
вают (дегидрированием) в амилены, которые изомеризуют, де-
гидрируют и используют для получения изопренового каучука.
Циклогексан (СбН12) из всех нафтеновых углеводородов ис-
пользуют в нефтехимическом синтезе наиболее широко. Он слу-
жит исходным сырьем для производства адипиновой кислоты,
на основе которой осуществляется синтез полиамидного волок-
на найлон. Из циклогексана получают циклогексанон, который
используют в синтезе капролактама, применяемого для синте-
за волокна капрон. Циклогексан получают гидрированием бен-
зола или выделяют из нефтяных фракций.
Высшие парафины (СюН22—С40Н82) по агрегатному состоя-
нию подразделяют на жидкие (т. пл. «25 °C) и твердые (т. пл.
52—55°C). Области применения жидких и твердых парафинов
различны.
Жидкие парафины (СюН22—С2оН42) подвергают окислению
для производства высших жирных кислот и спиртов, применя-
емых для получения синтетических моющих веществ. Для той
же цели используют продукты хлорирования жидких парафи-
нов — хлорпарафины. Кроме того, жидкие парафины являются
сырьем для микробиологической промышленности, производя-
щей бел ково-вита минные концентраты (БВК) —ценную добав-
ку к кормам.
Твердые парафины (С20Н42—С40Н82) при окислении также
дают высшие жирные кислоты. При термическом крекинге
твердых парафинов образуются высшие а-олефины — важное
Схема III. Химическая переработка «-бутана
12
Схема IV. Химическая переработка жидких и твердых парафинов
сырье для производства синтетических моющих веществ (вто-
ричных алкилсульфатов), а также алкилбензолов и алкилфено-
лов для производства различных водо- и маслорастворимых по-
верхностно-активных веществ, в том числе присадок к смазоч-
ным маслам (схема IV).
1.2. Переработка ненасыщенных углеводородов
Этилен (СН2=СН2) является наиболее универсальным неф-
техимическим сырьем: из него получают этиловый спирт, эти-
леноксид, этиленгликоль, стирол, дихлорэтан и 'многие другие
химические продукты (схема V). Однако в наибольшем коли-
честве этилен используется для производства полиэтилена.
Перспективен процесс олигомеризации этилена до высших оле-
финов, применяемых в качестве алкилирующих агентов в про-
изводстве различных продуктов.
Пропилен (СН3—СН = СН2) служит сырьем для синтеза изо-
пропилового спирта, глицерина и эпихлоргидрина, пропиленок-
сида, -а-метилстирола, изопропилбензола (кумол), акролеина и
других веществ (схема VI). Возрастает роль олигомеров пропи-
лена, а также полипропилена, в ряде областей применения об-
ладающего лучшими свойствами по сравнению с полиэтиленом.
Получают распространение в качестве эластомеров и сополиме-
ры этилена с пропиленом.
Новое перспективное направление использования пропиле-
на— синтез изопрена. Сначала получают димер пропилена
13
(в основном 2-метилпентен-1)
2СН3—СН=СН2 ----► СН2=С-СН2-СН2—СН3
О Z । & 4 о
СН3
который затем изомеризуют в 2-метилпентен-2 и подвергают
крекингу в изопрен с отщеплением метана:
СН3—С=СН-СН2-СН3 -----► СН2=С-СН=СН2 + сн4
СНз СН3
Бутилены. (С4Н8) играют весьма важную роль в нефтехими-
ческой промышленности; в основном они используются для син-
теза каучуков. Наиболее массовым сырьем для производства
Пропилен
Полине- Алкили- Гидра- Окис- Хлориро- Оксихло- Оксо- Полимери-
ризация робание тация ление Оание риробание синтез зация
Поли про - пилен Купол Изопропи- ловый спирт Акролеин Алл ил - хлорид Пропилен- хлор- гидрин к-Бути- ловый альдегид Изодути- лобый альдегид Нонилен Додеци - лены
I
Оксосинтез
1 . 1 “* Ацитн гидрЛгм- ,| || ЯтСтрт 1^1 1 Зпихлор- гидрин Схема VI. Химическая переработка пропилена т ♦ н-Бутило- Изодутило Дециловый Додецил- вый спирт вый спирт спирт бензол 1 Пропилен- Моющее уксид вещество сулыронол Пропилен- гликоль
14
синтетических каучуков являются бутен-1 и бутен-2 — промежу-
точные продукты, образующиеся при производстве бутади-
ена-1,3. Сополимеризацией изопрена и изобутилена получают
специальный сорт синтетического каучука — бутилкаучук. На
основе бутиленов, содержащихся в бутан-бутиленовой фракции
крекмнг-газов, осуществляют промышленное производство ал-
килата — высокооктановой добавки к моторному топливу.
В промышленном масштабе выпускают низко- и высокомоле-
кулярные полиизобутилены, применяемые как загустители
нефтяных смазочных масел и для других целей. Бутилены ис-
пользуют также для промышленного производства вторичных
и третичных бутиловых спиртов и других продуктов (схема
VII).
Важное промышленное значение приобрел процесс получе-
ния изопрена из изобутилена и формальдегида (стр. 88):
СН3—С=СН2 + 2НСНО
СН3
- Н2О;
-НСНО
сн2=с—сн=сн2
СНз
Пентены (С5Н10) в настоящее время применяют главным
образом для производства изопренового каучука, идентичного
по свойствам натуральному каучуку. В некоторых случаях пен-
тены олигомеризуют с получением олигомеров, используемых
в качестве алкилирующих агентов.
Бутадиен-1,3 (СН2=СН—СН = СН2) также относится к
числу важнейших нефтехимических продуктов. Он служит
сырьем для синтеза различных каучуков: бутадиенового, бута-
диен-стирольного, бутадиен-метилстирольного, бутадиен-нит-
।— । I Изобутилен
Алкила- Гидра- Сопели- Полимера-
рода- тация мери- зация
н~ Бутилен
Дегидриро-
вание
Гидрата-
ция
Схема VII. Химическая переработка бутиленов
15
Схема VI!'. Химическая переработка ацетилена
рильного, хлоропренового. Известен ряд синтезов на основе бу-
тадиена — получение адипиновой кислоты, циклооктана и цик-
лододекана.
Изопрен [СН2=С(СН3)—СН = СН2] полимеризуют с полу-
чением изопренового каучука, по производству которого СССР
вышел на первое место в мире. Кроме того, в промышленности
изопрен сополимеризуют с изобутиленом, получая бутилкаучук.
Ацетилен (СН = СН) применяется для синтеза винихлори-
да, ацетальдегида, винилацетата, акрилонитрила, хлорэтиленов
и др. (схема VIII). Сейчас идет разработка процессов получе-
ния тех же продуктов из более доступных этилена и пропилена,
однако химическая переработка ацетилена, несмотря на много-
стадийность многих процессов, развита достаточно широко.
1.3. Переработка ароматических углеводородов
Бензол (СбН6) служит сырьем для производства циклогек-
сана, этилбензола, изопропилбензола, высших алкилбензолов
и других ценных продуктов (схема IX).
Толуол (СбН5—СНз) подвергают гидродеалкилированию с
получением бензола. Толуол применяют также в производстве
бензойной кислоты и бензальдегида. Из него 'можно получать
ксилолы. Бензол- и толуолсульфокислоты являются катализа-
торами при алкилировании фенола олефинами.
Ксилолы [СбН4(СН3)2]. Имеется три изомера ксилолов —
орто-, пара- и мета-. n-Ксилол в основном перерабатывают в те-
рефталевую кислоту, которая идет на производство синтетиче-
16
скоро волокна лавсан, окислением о-ксилола получают фтале-
вый ангидрид, окислением ж-ксилола — изофталевую кислоту,
идущую на производство алкидных полимеров.
Этилбензол (СбНб—С2Н5) служит сырьем в производстве
стирола, идущего на производство полистирола -и синтетическо-
го каучука при сополимеризации стирола с бутадиеном.
Изопропилбензол [СбН5—СН(СН3)2] (кумол) идет на про-
изводство «-метилстирола, являющегося, как и стирол, мономе--
ром для синтетического каучука, и на производство фенола и
ацетона через промежуточное образование изопропилбензол-
ги дроп ер оксида.
Высшие алкилбензолы с длиной алкильной цепи Сю—С18
являются исходными веществами для получения алкилбензол-
сульфо'натов — моющих веществ, а также сульфонатных приса-
док к смазочным маслам, обладающих моющими свойствами.
§ 2. ПРОИЗВОДСТВО УГЛЕВОДОРОДНОГО СЫРЬЯ
2.1. Основные источники
углеводородного ;Сырья
Главным углеводородным сырьем для нефтехимических син-
тезов являются смеси газообразных, жидких и твердых угле-
водородов.
Природные газы состоят в основном из метана и других на-
сыщенных углеводородов; в них также присутствуют инертные
(азот, диоксид углерода) и редкие газы (аргон, ксенон). При-
родные газы добывают при разработке газовых и газоконден-
Схема IX. Химическая переработка бензола
2-254
17
сатных (глубоко залегающих газовых, с высоким пластовым
давлением) месторождений.
Попутные нефтяные газы получают как побочный продукт
при добыче нефти. Эти газы растворены в пластовой нефти
и при ее добыче выделяются вследствие снижения давления.
Состав газов колеблется в значительных пределах [32—
90% (об.) СН4] и зависит от типа месторождения и условий до-
бычи нефти.
Газы нефтепереработки образуются в процессах крекинга,
коксования, риформинга; их также отбирают на установках
стабилизации и прямой перегонки нефти. В зависимости от ха-
рактера этих процессов состав получаемых газов меняется в
широких пределах. Например, газ каталитического риформин-
га содержит до 60% водорода; остальное — насыщенные угле-
водороды. Газы крекинга и коксования состоят из насыщенных
и ненасыщенных углеводородов.
Газы стабилизации нефти отличаются высоким содержанием
пропана, бутана, пентана и изопентана, что делает их ценным
сырьем для получения бутадиена и изопрена.
Газовые бензины обычно выкипают в интервале 30—120 °C;
они содержат бутан, пентан, изопентан, а также углеводороды
С6 и С? нормального и изостроения.
Газовые конденсаты выкипают в интервале 40—360 °C.
В них содержится 15—30% ароматических углеводородов, 25—
40% нафтенов и 20—60% парафинов (в зависимости от место-
рождения).
Жидкие дистилляты и нефтепродукты, образующиеся при
различных процессах переработки нефти, тоже используют как
исходное сырье в нефтехимических процессах, точнее как источ-
ник для выделения тех или иных групп углеводородов.
Так, из продуктов каталитического риформинга выде-
ляют ароматические углеводороды, из продуктов термического
и каталитического крекинга — олефины, из продуктов депара-
финизации дизельного топлива — парафины. При этом произ-
водство сырья для нефтехимического синтеза тесно переплета-
ется с выпуском топливных нефтепродуктов. Вместе с тем вы-
работка химического сырья в общем объеме нефтепереработки
постоянно повышается.
2.2. Производство низших олефинов
Этилен и пропилен, широко используемые в многообразных
процессах и синтезах, являются крупнотоннажными продукта-
ми. Их производят в настоящее время пиролизом нефтяного
сырья. В качестве побочных продуктов их получают при термо-
каталитических процессах.
Пиролиз представляет собой процесс глубокого разложения
углеводородов при высокой температуре. Продуктами пиролиза
являются газообразные и жидкие углеводороды. В газах пре-
18
обладают олефины С2—С4, в жидких продуктах — ароматичес-
кие углеводороды. Сырьем для пиролиза служат низшие пара-
фины (этан, пропан, бутан), газовые и низкооктановые бензи-
ны, деароматизированный рафинат каталитического риформин-
га. Пиролизу стали подвергать средние и тяжелые нефтяные
фракции, а также сырую нефть.
При пиролизе наряду с разложением, в результате которого
образуются водород, метан, этилен, пропилен и другие олефи-
ны, протекают реакции гидрирования и дегидрирования олефи-
нов с образованием парафинов, диенов, ацетилена и его произ-
водных; идут также конденсация, циклизация, уплотнение.
Условия пиролиза различного сырья и выход продуктов
приведены в табл. 1.1. Наибольший выход низших олефинов да-
ет пиролиз парафиновых углеводородов нормального строения.
При пиролизе этан почти нацело превращается в этилен. Мало
побочных продуктов образуется и при пиролизе пропана и бу-
тана. С утяжелением сырья выход этилена падает, а выход оле-
финов ?>С4 растет; уменьшается соотношение этилена и пропи-
лена. Изопарафины пиролизуются труднее и дают меньший вы-
ход олефинов. Ароматические углеводороды дают большой вы-
ход смолы и кокса. Нафтены образуют значительные (до 15%)
количества бутадиена. Деароматизированные рафинаты плат-
форминга при пиролизе образуют много кокса, их лучше под-
вергать пиролизу в смеси с прямогонным бензином. С утяже-
лением сырья не только снижается выход этилена, но и уве-
личивается выход кокса. Для снижения коксообразования вво-
дят водяной пар: 0,6 кг на 1 кг бензина и 0,2—0,3 кг на 1 кг
этана или пропана. Время контакта при пиролизе составляет
0,5 с; при большем времени контакта выход этилена снижа-
ется.
Таблица 1.1. Условия процесса и выход продуктов
при пиролизе различного сырья
Показатели Этан Пропан н-Бутан Бензин Легкий газойль Тяжелый газойль
Температура, °C 850—900 800-850 800-850 800—900 750—800 700-800
Количество пара, 20-25 20—25 20-25 60 100 100
добавляемого в сырье, % Степень конверсии 60 70—90 90 85-90
сырья за один проход, % Выход продуктов, % (масс.) н2+сн4 15,3 30,0 20,6 18,0 11,9 11,8
с2н4 76,9 42,0 36,4 31,4 26,4 24,4
С3Н6 2,8 16,2 20,5 14,7 14,2 13,9
с4н6 1,3 3,1 3,0 3,7 3,9 3,4
с4н8 0,6 1,4 12,6 4,1 4,6 4,3
С 5 и выше 2,8 6,0 4,0 24,2 19,5 17,7
2*
19
Пиролиз 'протекает по радикально-цепному механизму. Пред-
ставить первичные реакции образования этилена на примере
пиролиза этана можно следующим образом. Первая стадия —
инициирование цепи через образование метильных радикалов
при высокой температуре:
СН3-СН3---► 2СН3
Вторая стадия—развитие цепного процесса:
СН3 + СН3-СН3 -► СН4 + СН3-СН2
СН3—СН2 ---► сн2=сн2 + н.
Н. +СНз-СН3 ---► Н2+СН3-СН2
Третья стадия — обрыв цепи за счет рекомбинации радикалов
или диспропорционирования:
Н. 4- СН3—СН2 -> СН3-СН3
сн3 + СН3-СН2--► СН3-СН2-СН3
2СН3-СН2 ► СН2=СН2 + СН3-СН3
Для пиролиза пропана совокупность первичных реакций
можно изобразить схемой:
I—С2Н4 -|- сн4
Сзн8 - 2С3Н8 --> С3Нв + С2Нв + СН4
—С3нв -|- Н2
Аналогичные реакции протекают и при пиролизе бутана:
СН4 + С3Нв
гн С2Нв + С2Н4
410 -^2С2Н4 + Н2
— с4н8 + н2
В промышленности наибольшее распространение получил
пиролиз в трубчатых печах. Принципиальная схема технологи-
ческой установки приведена на рис. 1.1. Жидкое сырье поступа-
ет в сепаратор 1, а оттуда в испаритель 2. Паро-жидкостная
смесь вновь идет в сепаратор; пары сырья через перегрева-
тель 3 поступают в трубчатую печь 4. На входе в печь к сырью
добавляют водяной пар. Сырье в печи нагревается до 550—
600 °C в конвекционной секции и до 750—850 °C в радиантной
секции. На выходе из печи газ пиролиза подвергают «закалке»
в аппарате 5 путем впрыскивания воды; при этом снижается
температура газа (до 700 °C, если тепло газа используют для
получения водяного пара, и до 200°C, если на установке нет
котлов-утилизаторов). Из аппарата 5 газ поступает ’в котел-
утилизатор 6, где охлаждается до 350 °C, отдавая тепло дито-
20
Рис. 1.1. Технологическая схема пиролиза жидкого сырья:
1 — сепаратор; 2 — испаритель; 3 — перегреватель; 4 — печь; 5 — «закалочный» аппарату
6 — котел-утилизатор; 7 — конденсатор; 8 — пенный аппарат; 9 — холодильник; 10—про-
мывной аппарат.
лилметану, кипящему при 300 °C. Дитолилметан конденсирует-
ся в конденсаторе 7, отдавая тепло воде; при этом в конденса-
торе образуется пар с давлением 1,2 МПа. Дальнейшее охлаж-
дение газа до 100—150°С происходит в пенном аппарате 8,
орошаемом циркулирующей водой; одновременно происходят
отмывка газа от сажи и кокса, конденсация паров тяжелых уг-
леводородов смолы и водяного пара. После пенного аппарата
газ в холодильнике 9 охлаждается до 40 °C, а окончательное
охлаждение до 20 °C и отмывка от частичек кокса осуществля-
ются в промывном аппарате 10.
Газ выводят на компримирование, а водно-омоляные конден-
саты и смоляную воду направляют в отстойники. Легкую и тя-
желую смолу после отстаивания раздельно отправляют на
склад, а воду возвращают в «закалочный», пенный и промыв-
ной аппараты. Избыток водного конденсата поступает на очист-
ку (флотация и отпаривание углеводородов), после чего его
сбрасывают в канализацию.
В печах применяют трубы из жаропрочных сталей, содер-
жащих до 25% хрома и 20% никеля. Температура наружных
стенок труб в радиантной секции достигает 1150°C. Имеются
установки пиролиза в присутствии теплоносителя, установки
окислительного пиролиза (когда кислород вводят вместе с
сырьем в зону реакции) и установки гомогенного пиролиза (ког-
да в качестве теплоносителя используют перегретый до 900 °C
и выше водяной пар).
Получение этилена высокой чистоты. Современные газо|раз-
делительные установки позволяют получать этилен и пропилен
99,6—99,9%-ной чистоты. Содержание примесей исчисляется
миллионными долями %. Качество этилена, например, характе-
ризуется следующими данными [в % (об.)]:
с2н4 99,8-99,9 СО (8-10)10-»
СзН6 0,03—0,05 СО2 (3—8) 10-»
сн4 0,08-0,1 О2 . . . (54-8)10-6
С2Н2 (10—15)10-6 Н2О (пар) (Юн-15) 10-«
21
Рис. 1.2. Реакторный блок очистки этилена
селективным гидрированием ацетилена:
1,2 — реакторы.
Если этилен получен автотермическим дегидрированием
(окислительным пиролизом) этана, то содержание ацетилена
в газе достигает 1% (об.) и выше. Тогда возникает необходи-
мость в гидрировании ацетилена в этилен, для чего пропускают
газ через реактор с хромо-никелевым катализатором (96%
Сг2О3 и 5% Ni). Температура 200°C, скорость подачи газа
примерно 800 л на 1 л катализатора в час. Гидрирование осу-
ществляется водородом, содержащимся в газе. Газ перед гид-
рированием нагревают до 150—200°C (в зависимости от содер-
жания в нем водорода) и подают в один из двух связанных
друг с другом реакторов (рис. 1.2). Для газов, содержащих ма-
ло водорода, скорость их подачи меньше, а температура гидри-
рования выше.
Катализатор в реакторах расположен в несколько слоев.
Вводя газ, освобожденный от ацетилена, в разные точки по вы-
соте реактора, поддерживают в аппарате постоянную темпера-
туру, что и обеспечивает селективность гидрирования ацетиле-
на. После гидрирования газ идет на разделение. Этим методом
удается нацело освободиться от ацетилена. В качестве катали-
затора можно применить сульфид молибдена на оксиде алюми-
ния, палладий на силикагеле, кобальт-молибденовый катализа-
тор.
2.3. Производство высших олефинов
Во многих нефтехимических процессах (алкилирование бен-
зола .и фенола, производство моющих веществ и присадок к ми-
неральным маслам, производство высших жирных спиртов ок-
сосинтезом) применяют жидкие непредельные углеводороды с
прямой цепью и разветвленные. Разветвленные олефины
Се—Cis (димеры изобутилена, тримеры, тетрамеры и пентамеры
пр опилен а>) используют как алкилирующие агенты и в оксосин-
тезе. В последние годы широкое применение получили а-оле-
фины — непредельные прямоцепочечные углеводороды с двой-
ной связью в начале цепи. Получают высшие а-олефины разны-
ми методами — термическим крекингом парафина и олигоме-
ризацией этилена в присутствии катализаторов Циглера.
_22
Производство а-олефинов термическим крекингом парафи-
нов. Для производства а-олефинов используют мягкие парафины
(т. пл. 22—26°C), получаемые при депарафинизации дизель-
ных топлив, и твердые (т. пл. 52—64°C), получаемые при депа-
рафинизации масел, выкипающих в интервале 350—500 °C. Для
получения а-олефинов с хорошим выходом требуется очищать
масла и дизельные топлива от смол и ароматических соедине-
ний.
Крекинг парафинов ведут в газовой фазе при 550°C в при-
сутствии водяного пара в трубчатых печах. Степень конверсии
парафина за один проход составляет 25—30%, поэтому прихо-
дится вести процесс с рециркуляцией парафина. Продуктами
крекинга являются фракции олефинов от Q до гомологов>
с т. кип. 240—320°C. Содержание олефинов во фракциях со-
ставляет 75—90% и зависит от качества и вида сырья, напри-
мер в продуктах крекинга мягких парафинов содержание оле-
финов достигает 92—96%. Парафины превращаются в газооб-
разные и жидкие олефиновые углеводороды на 70%. Состав^
продуктов крекинга таков [% (масс.)]:
Компоненты Крекинг мягкого парафина (т. пл. 23 °C) Крекинг твердого парафина (т. пл. 62 °C)
Олефины С2—С4 (газ) 22,4 22,1
Фракция и. к. —40 °C 3,4 1,0
40—140 °C 18,5 13,7
140—180 °C 12,0 7,3
180—240 °C 13,3 10,8
240—320 °C — 15,1
Крекинг-остаток 28,5 29,0
Кокс+потери 1,9 1,0
Производство линейных а-олефинов олигомеризацией этиле-
на. Процесс протекает в присутствии триэтилалюминия. Пер-
вую стадию — собственно олигомеризацию этилена — проводят
при 100 °C и 10 МПа до получения олигомеров с длиной угле-
родной цепочки не выше С20:
^(СНгСН^х-СзНз
2(C2H5)3A1 + (x + z/ + z)CH2=CH2-► Al—(СН2СН2)у—С2Нб + Al(C2Hj)3
\сн2сн2)2-с2н5
Достигнув заданной степени олигомеризации, повышают темпе-
ратуру до 200—300 °C. При этом протекает реакция замещения,,
т. е. происходят регенерация триэтилалюминия и образование
высших а-олефинов с различной длиной цепи:
ДСН2СН2)Х—С2Н6
Al— (СН2СН2)У—С2Н5 _(c2h5)3ai
\(СН2СН2)2-С2Н5
С^НСН^Н^-СН^СН,
-> С2Н5—(СН2СН2)!/_1-СН=СН2
С2НЬ^(СН2СН2)2^СН=СН2
Вторую стадию можно проводить и каталитически — в присут-
ствии 0,01% коллоидного никеля.
2&
Смесь олигомергомолопов имеет широкий фракционный со-
став (от С4 до С20). Наиболее ценными являются олефины
Сю—Сю, выход которых составляет в сум'ме 65—68%. Так как
процесс получения олигомеров с узким молекулярномассовым
распределением осуществить не удается, необходимо разрабо-
тать комплексные методы использования олефинов с различной
длиной цепи, в частности олефинов С6 (выход до 10%) и Cs
(выход до 16%).
2.4. Производство ацетилена
В настоящее время ацетилен получают двумя методами: из
карбида кальция и пиролизом низкомолекулярных газообраз-
ных алифатических углеводородов.
При обработке карбида кальция водой вначале образуется
оксид кальция (известь)
СаС2 + Н2О ----► СаО-|-С2Н2 ДЯ =—61,8 кДж/моль
который тоже вступает в реакцию с водой:
СаО-|-Н2О -----► Са(ОН)2 ДЯ = —68,1 кДж/моль
Суммарная реакция протекает по уравнению:
СаС2 -f- 2Н2О -► Са(ОН)2 + С2Н2 ДЯ = —129,9 кДж/моль
Карбид кальция получают взаимодействием извести и кокса в
электротермических печах при 1700—1800°C:
СаО 4- ЗС — » СаС2 + СО ДЯ = 464,8 кДж/моль
Недостатком процесса является высокий расход электроэнергии
(10—11 кВт-ч на 1 кг ацетилена).
В зависимости от сорта карбида кальция из 1 кг СаС2 полу-
чается разное количество ацетилена. Количество ацетилена в
литрах (измеренное при 20°C и атмосферном давлении), обра-
зующееся из 1 кг карбида кальция, называют литражом (или
литражностью) карбида. Практически литраж 1 кг СаС2 состав-
ляет 230—300 л С2Н2.
Ацетилен, полученный из карбида кальция, имеет чистоту
99,5% и содержит примеси NH3, РН3 и H2S. Если .ацетилен идет
на химическую переработку, его очищают хромовой кислотой
(при этом РН3 и H2S окисляются в серную и фосфорную кис-
лоты). Применяют для очистки и гипохлориты.
Ацетилен — бесцветный газ, обладающий в чистом виде сла-
бым эфирным запахом. Сжижается при —83,6 °C. В жидком ви-
де он может сохраняться лишь при повышенном давлении. Аце-
тилен растворим в воде: при 18 °C и атмосферном давлении в
1 объеме воды растворяется 1 объем ацетилена. Наличие трой-
ной связи в молекуле ацетилена обусловливает его высокую
24
реакционную способность; он широко применяется во многих
промышленных синтезах.
Ацетилен является интересным с термодинамической точки
зрения. В отличие от других углеводородов, реакции образова-
ния ацетилена из элементов и из метана являются сильно эндо-
термическими:
2С + Н2 С2Н2 ЛЯ = 226,2 кДж/моль
2СН4 С2Н2 -f- ЗН2 ЛЯ = 380,3 кДж/моль
Выход ацетилена увеличивается при повышении температуры
и при понижении давления: в интервале 1000—2400 К выход
ацетилена возрастает при 0,01 МПа от 4,8 до 99,99%, а при
0,1 МПа он увеличивается от 1,5 до 99,87%.
При температуре выше 1500 °C метан можно практически
полностью превратить в ацетилен. При этом контактный газ
должен содержать 25% ацетилена и 75% водорода. Однако при
такой температуре ацетилен нестабилен и разлагается на эле-
менты. Эту реакцию замедляют, понижая давление и сокращая
время пребывания ацетилена в зоне высоких температур до-
0,01—0,001 с с последующим мгновенным охлаждением газа.
Ацетилен имеет как бы два порога стабильности — при 1600 °C
и выше и при пониженных температурах (менее 200°C). Прак-
тически реакцию ведут при 1400—1500 °C, но при этом прово-
дят быструю «закалку» газов, которая предупреждает разло-
жение ацетилена на углерод и водород.
Все больше развивается пиролиз низкомолекулярных али-
фатических углеводородов с целью получения ацетилена. Мето-
ды пиролиза различаются способом подвода тепла к реагиру-
ющей газовой смеси. Высокие температуры, требуемые для
разложения углеводородов, достигаются тремя способами:
1) при помощи электрической дуги (электрокрекинг), 2) пря-
мым или регенеративным нагревом (собственно пиролиз) г
3) сжиганием части сырья (окислительный пиролиз).
Электрокрекинг заключается в быстром пропускании угле-
водородов через электрическую дугу, с помощью которой соз-
дается высокая температура в зоне реакции. Электродуговая
печь состоит из верхней цилиндрической камеры (диаметр
1000 мм, высота 400 мм) и реакционной трубы (диаметр 95 мм,
длина 1000 мм). На камере установлен медный катод (гильза),
анод находится на 'реакционной трубе ближе к камере. Катод-
ная гильза и реакционная труба имеют рубашки для охлажде-
ния. Газ при давлении 0,05 МПа поступает тангенциально в ка-
меру, приобретает вихревую скорость примерно 100 м/с от пе-
риферии к реакционной трубке и попадает в зону действия
электрической дуги, нагреваясь до 1600°C при температуре ду-
ги 2000 °C.
Электрическая дуга создается постоянным током напряже-
нием 7000—8000 В. Продукты пиролиза проходят через охлаж-
25
даемую водой реакционную трубу со скоростью 600—1000 м/с
и охлаждаются до 600 °C; на выходе они подвергаются «закал-
ке» за счет впрыска воды, охлаждаются до 150 °C и после этого
поступают на разделение.
Электрическая печь приведенных размеров имеет мощность
по метану примерно 2800 м3/ч, что дает производительность по
ацетилену 15 т в сутки. Наряду с ацетиленом при электрокре-
кинге образуются побочные продукты: водород, сажа, этилен
и высшие ацетиленовые углеводороды (винилацетилен, метил-
ацетилен и др.). Степень конверсии метана за один проход че-
рез реактор достигает 45—50%. При работе на природном газе
расход электроэнергии составляет 9,5—10 кВт-ч на 1 кг ацети-
лена. Продукты реакции содержат 13—14% (масс.) ацетилена,
1% (масс.) этилена, 30—35%'(масс.) метана и 50—55% (масс.)
водорода. Недостаток процесса — большой выход сажи (до
50 кг и более на 1 т ацетилена), хотя сажа получается высоко-
качественной и является товарным продуктом. Из 1000 м3 ме-
тана при электрокрекинге образуются 300 кг ацетилена, 26 кг
этилена, 21 кг сажи и 1170 м3 водорода. Производство ацетиле-
на электрокрекингом обходится дешевле, чем при карбидном
методе.
Основное затруднение промышленного осуществления пиро-
лиза углеводородов до ацетилена заключается в создании печи
такой конструкции, в которой газ в течение весьма малого -вре-
мени мог бы нагреваться до 1400—1500°C. На практике ока-
залось возможным осуществить этот процесс в регенеративных
печах периодического действия (способ Вульфа, США). В этом
процессе используется принцип рекуперации тепла — попере-
менное нагревание огнеупорной насадки при сжигании газооб-
разного топлива и пропускание через раскаленную насадку га-
зов, подвергаемых пиролизу. Насадка выполнена из чистого
оксида алюминия в виде параллельно расположенных горизон-
тальных фасонных пластин, между которыми для прохода газа
образуются цилиндрические каналы диаметром 6 мм.
Печь работает периодически. Вначале насадка печи разо-
гревается за счет продуктов сгорания, образующихся при сжи-
гании природного газа в топке (фаза разогрева). Затем через
разогретую насадку пропускают газ (фаза пиролиза). Между
фазами разогрева и пиролиза продувают насадку паром.
Лучшие результаты получаются при пиролизе пропана, так
как для пиролиза метана требуется более высокая температура,
а максимальная температура в печи не может быть выше 1250—
1300°C. Состав продуктов пиролиза метана таков:
Содержание, % (об.) Пиролиз метана Пиролиз пропана Содержание, % (об.) Пиролиз Пиролиз
метана пропана
сн4 28,3 15,0 СО 7,8 6,9
С2Н2 7,1 10,0 Н2 49,0 56,8
с2н4 0,9 3,8 n2 5,2 5,2
СО2 1,3 1,6 02 0,4 0,7
26
При окислительном пиролизе метан расщепляется за счет
тепла, выделяющегося при сжигании части газа с кислородом,
подаваемым в реактор. Подвод тепла и пиролиз углеводородов
осуществляются в факеле горения, что значительно упрощает
теплообмен между источником тепла и газом. Вначале идет
экзотермическая реакция неполного окисления метана:
СН4 + 0,5О2 СО + 2Н2 ДН = —25,4 кДж/моль
Параллельно протекает образование ацетилена при разложе-
нии метана:
2СН4 4—>. С2Н2 + ЗН2 ДЯ = —380,3 кДж/моль
Одновременно идут и побочные реакции:
СН4 + 2О2 СО + 2Н2О ДЯ = —885,5 кДж/моль
СО + Н2О СО2 + Н2 ДЯ = —41,8 кДж/моль
Для получения максимальных выходов ацетилена требуют-
ся применение 98%-ного кислорода и его подогрев до 400—
600 °C. Соотношение кислорода и метана равно (0,64-0,64) 1,
температура процесса 1450—1500 °C, время пребывания газа
в зоне реакции 0,004—0,006 с. Чтобы предотвратить обрыв пла-
мени, в реактор вводят небольшое количество кислорода; это-
обеспечивает поджигание газа, т. е. стабилизацию факела.
В этом процессе применяют одно- и многоканальные реакто-
ры. Скорость газовых потоков в многоканальных реакторах
40—60 м/с, в одноканальном 100—350 °C. Скорость подвода га-
зов должна быть выше скорости распространения пламени. Газ,
выходящий из реактора, имеет следующий состав: 8% (об.)
С2Н2, 0,5% (об.) С2Н4, 4% (об.) СО2, 26,5% (об.) СО, 54% (об.)
Н2, 3% (об.) N2 и 4% (об.) СН4. Содержание сажи и смолы 1 —
3 г/см3. Степень конверсии метана в ацетилен составляет 30—
32%.
Технологическая схема окислительного пиролиза метана
приведена на рис. 1.3. Метан (природный газ) дросселируют
до 0,15 МПа, пропускают через фильтр 3 для улавливания ме-
ханических примесей и направляют в печь 4. В конвекционной
части печи газ нагревается до 350—380°C, в радиантной до
600 °C. Кислород турбогазодувкой под давлением 0,15 МПа по-
дается через фильтр 1 в печь 2 и, пройдя конвекционную и ра-
Рис. 1.3. Технологическая схема
окислительного пиролиза метана:
1, 3 — фильтры; 2, 4 — печи; 5 — сме-
ситель; 6 — реактор; 7, 9~ гидравли-
ческие затворы; 8 — скруббер.
27
диантную секцию, нагревается до 400 °C. Газовые потоки смеши-
ваются и проходят в зону реакции. После зоны реакции газ
пиролиза подвергают «закалке» водой; при этом он охлаждает-
ся до 80 °C.
Реактор 6 имеет прямоугольное сечение (1530X1780 мм).
Внутри размещена горелочная плита с 780 отверстиями диа-
метром 8 мм, состоящая из 26 керамических блоков. В горелоч-
ной плите расположены два коллектора для подачи воды (че-
рез форсунки) с целью охлаждения газа. Из реактора газы
поступают в скруббер 8, орошаемый водой. Там они очищаются
•от сажи и охлаждаются до 30°C; после этого газы идут на раз-
деление. Ацетилен выделяют из газовой смеси селективными
растворителями, лучшим из которых является диметилформ-
амид.
Для получения 1 т ацетилена окислительным пиролизом ме-
тана требуется 1000 м3 природного газа (600 м3 на процесс
и 400 м3 на подогрев) и 3600 м3 кислорода (98%-ного). При
этом в качестве побочного продукта образуется 10300 м3 син-
тез-газа. Расход электроэнергии составляет 1570 кВт-ч на 1 т
ацетилена, т. е. меньше, чем при карбидном методе и электро-
крекинге. Окислительный пиролиз метана является более эко-
номичным, а потому и более предпочтительным методом полу-
чения ацетилена. Однако при наличии дешевой электроэнергии
электрокрекинг выступает как конкурентоспособный процесс.
Разработан и осуществлен в промышленности процесс одно-
временного получения этилена и ацетилена окислительным пи-
ролизом газового бензина. В качестве побочных продуктов об-
разуются синтез-газ и метан. Процесс проводят при 1200 °C
в присутствии водяного пара, бензин предварительно подогре-
вают в печи до 500—600 °C. Для получения 1 т ацетилена тре-
буется 6,4 т бензина. Выход этилена *в 2,5 раза превышает вы-
ход ацетилена. Недостатки процесса — сложность разделения
продуктов и сравнительно низкий выход ацетилена.
2.5. Производство алкилбензолов
Основные представления об алкилировании бензола. Реак-
ция алкилирования ароматических соединений была открыта в
1877 г. Фриделем и Крафтсом, предложившими в качестве ка-
тализатора хлорид алюминия. С тех пор процессы алкилирова-
ния ароматических соединений получили большое промышлен-
ное значение. В качестве катализаторов используют много ве-
ществ кислотного характера — серную и фосфорную кислоту,
фтористый водород, алюмосиликаты, иониты, хлориды алюми-
ния, цинка, титана и др. Галогениды металлов обычно применя-
ют в присутствии промоторов; галогениды образуют с ними
комплексы, являющиеся сильными протонными кислотами.
В качестве алкилирующих агентов можно применять хлорпара-
фины и спирты, но наиболее предпочтительны олефины.
28
Во всех случаях реакция протекает по карбонийионному ме-
ханизму, однако в случае кислот карбоний-ион образуется за
счет протона кислоты
H2SO4 ^=±: Н+ + HSO4-
КСН=СН2 + Н+ RCH—СН3
а в случае хлорида алюминия в образовании комплекса участ-
вует промотор (хлористый водород):
А1С13 + НС1 -► Н+ С1-. А1С13
Н+ СГ-А1С13 + КСН=СН2 RCH—СН3 + С1- Л1С13
Ион карбония или комплекс взаимодействует с ароматическим
ядром и через л- и о-комплексы превращается в алкилбензол,
отщепляя протон:
+ RCH—СН
Л-комплекс
d-комплекс
Неодинаковая стабильность ионов карбония (третичные >
>1вторичные>,пер1вичные) предопределяет преимущественное
образование алкилбензолов с разветвленной боковой цепью.
Особенно заметна изомеризация алкильных цепей при алкили-
ровании бензола и его гомологов высшими олефинами или ал-
килгалогенидами, когда продукты являются смесью большого
числа изомерных (главным образом втор-алкилароматических)
соединений:
RCH2—СН2—СН2—СН=СН2 + [|| -► RCH2—СН2—СН2—СН—СН3 4-
б
-PRCH2-CH2—СН—СН2—СН3 + КСН2—СН-СН2-СН2-СН3
Изомеризация протекает на стадии образования иона кар-
бония и идет с перемещением реакционного центра, но без
деструкции и изомеризации углеродного скелета. При алкили-
29
ровании разветвленными олефинами образуются преимущест-
венно трет-алкилбензолы (за счет образования более стабиль-
ных третичных ионов карбония).
Обычно при алкилировании образуются не только моно-, но
также ди- и триалкилбензолы. Для снижения выхода полиал-
килбензолов применяют 5—10-кратный избыток бензола.
При высоких температурах алкилирования на алюмосилика-
тах и цеолитах и при катализе хлоридом алюминия наблюда-
ются обратимые реакции переалкилирования или диспропор-
ционирования с межмолекулярной миграцией алкильных групп:
Q$B3R3 -f- С6Н6 < ...х CeH5R -|- CeH4R2
C6H4R2 + CeH6 2C6H6R
Идет также обратимая изомеризация с внутримолекулярной
миграцией алкильных групп, поэтому среди диалкилбензолов
преобладает пара-изомер, а среди триалкил бензолов—1,3,5-
изомер:
При алкилировании возможны деструкция или полимеризация
алкилирующих агентов, образование смолообразных побочных
продуктов.
В зависимости от природы алкилирующего агента и ката-
лизатора осуществляют процесс алкилирования в жидкой или
газовой фазе. В качестве примера ниже сопоставлены усло-
вия алкилирования бензола этиленом на хлориде алюминия
(А), фосфорной кислоте на те (В): кизельгуре (Б) и на алюмосилика-
Показатели А Б В
Мольное соотношение бен- зола и этилена 2:1 4:1 6,5:1
Температура, °C 80—120 280—325 450
Давление, МПа . . . 0,1—0,5 4—6 0,5
Концентрация этилена, % (об.) 30-100 90—100 90-100
Степень превращения этиле- на за один проход, % • 95—98 80—100 70—98
Расход катализатора, кг на 1 т этилбензола 25—20 4 —
Съем этилбензола с 1 м3 ре- актора в час, кг 100—200 200—300 150
Состав продуктов, % (масс.) бензол 57,0 76,0 85,0
этилбензол 35,0 20,5 12,5
полиалкилбензолы 8,0 3,5 2,5
30
Как видно из этих данных, алкилирование бензола этиленом в
присутствии хлорида алюминия протекает в более мягких усло-
виях — при более низких температуре и давлении, а для ал-
килирования можно использовать этан-этиленовую фракцию.
В случае же фосфорной кислоты условия значительно жестче.
Поэтому в промышленности этилбензол производят в присутст-
вии хлорида алюминия. Что же касается процесса в присутствии
алюмосиликатов, он изучен еще недостаточно.
Алкилирование бензола пропиленом протекает гладко в
присутствии многих катализаторов: хлорида алюминия (А),
фосфорной кислоты на кизельгуре (Б), алюмосиликата (В),
88—90%-ной серной кислоты (Г) и комплекса ортофосфорной
кислоты и фтористого бора (Д):
Показатели А Б В Г д
Мольное соотношение бен- зола и пропилена 2,5:1 4:1 3:1 4:1 1,2:1
Температура, °C 70—90 250 320 35—40 50
Давление, МПа 0,1-0,5 1,5-2,5 2,0 0,4—1,0 2,0
Концентрация пропилена, % (об.) . 35-80 35 35-80 35-80 35-40
Степень превращения про- пилена за один проход, % (масс.) 99 70—90 50-70 99
Расход катализатора, кг на 1 т изопропилбензола 20 4—6 10 120-150 10
Съем изопропилбензола с 1 м3 реактора в час, кг Состав продуктов, % (масс.) бензол 150-250 150—300 — — —
61,6— 76,0 72,0 73,6 45,0
изопропилбензол 70,6 26-31 21,0 24,4 24,2 44,0
полиалкилбензолы 3,4—5,8 3,0 3,6 2,2 н,о
В промышленности для алкилирования бензола пропиленом
применяют хлорид алюминия, фосфорную кислоту на кизельгу-
ре и серную кислоту. При выборе катализатора следует учиты-
вать качества, требуемые при дальнейшей химической перера-
ботке получаемого изопропилбензола. Изопропилбензол, полу-
ченный в присутствии фосфорной кислоты, содержит некоторое
количество непредельных углеводородов. Он не пригоден для
получения фенола и ацетона, так как плохо окисляется. Для
окисления применяют изопропилбензол, полученный в присутст-
вии А1С13. Однако следует учитывать, что при использовании
А1С1з требуется тщательно очищать пропан-пропиленовую
фракцию от примесей влаги, диоксида углерода и др. При ис-
пользовании кислотных катализаторов требуется кислотостой-
кая аппаратура.
При выборе способа производства изопропилбензола следу-
ет учитывать не только его целевое назначение, но и характер
других нефтехимических процессов данного предприятия. Удоб-
нее использовать один и тот же катализатор для нескольких
процессов.
31
Так же хорошо, как и алкилирование пропиленом, протека-
ет алкилирование бензола бутиленом с образованием втор-бу-
тилбензола. ewp-Бутилбензол может быть окислен до гидро-
пероксида, который при разложении дает фенол и метилэтилке-
тон:
ООН
СН3-СН-С2Н5
I
СН3—С—С2Н5
он
+О2
СН3-СО-С2Н6
Этот 'метод, однако, не имеет распространения в промышленно-
сти, так как бутилены идут на производство бутадиена.
В СССР алкилирование бензола полимерами пропилена для
получения алкилбензолов осуществляют в присутствии хлори-
да алюминия. Для алкилирования используют фракцию 175—
260 °C, состоящую главным образом из тетрамеров пропилена
с примесью тримеров и пентамеров. Основные показатели про-
цесса таковы:
Мольное соотношение бензола и олефинов (фрак-
ция 175—260 °C) 7,5:1
Температура, °C 60
Давление Атмосферное
Длительность реакции, мин . . 50
Расход А1С13, % на взятые олефины . 6,8
Выход целевой фракции алкилбензолов (260—
360 °C), % на взятые олефины 98
Алкилирование осуществляют в двухступенчатом реакторе из
нержавеющей стали емкостью 1,4 м3. Хлорид алюминия обра-
зует с алкилбензолами комплекс (в мольном соотношении
1 1,3). Комплекс после отстаивания вновь подают в реактор
вместе со свежим хлоридом алюминия или с его суспензией в
бензоле.
В целевой фракции алкилбензолов более 75% алкилбензо-
лов имеют боковую цепь с 10, 11 и 12 углеродными атомами.
Диалкилбензолы практически не образуются. Обычно после пе-
регонки алкил бензолы очищают 95—98 %-ной серной кислотой
от непредельных соединений. Расход кислоты составляет 15%
от массы алкилата. Присутствие олефинов в алкилбензолах не-
желательно, потому что при последующем получении сульфоно-
ла (моющее средство) олефины легко сульфируются, полимери-
зуются и ухудшают цвет моющего вещества. Этилен и пропи-
лен, применяемые для алкилирования, должны быть тщательно
очищены от альдегидов, оксида углерода, эфиров, кислорода,
бутадиена, ацетилена. При наличии примесей уменьшается сте-
пень конверсии олефина и увеличивается расход хлорида алю-
миния. С кислородсодержащими соединениями хлорид алюми-
ния образует комплексные соединения, вследствие чего ката-
лизатор довольно быстро теряет активность.
32
К бензолу, применяемому для алкилирования, также предъ-
являются жесткие требования по чистоте, особенно по содержа-
нию органических соединений серы (их должно быть менее
0,1%). Каменноугольный бензол сначала очищают серной кис-
лотой, потом подвергают четкому фракционированию (при
этом уходят сероуглерод и легкие компоненты), а после этого
подвергают осушке. Осушку в промышленности осуществляют
методом азеотропной перегонки, основанной на свойстве бен-
зола образовывать с водой азеотропную смесь (8,9% воды), ки-
пящую при 69,25 °C. При конденсировании смесь расслаивает-
ся на водный и бензольный слой. Содержание влаги в бензоле
снижается до 0,003—0,006%. Такой бензол вполне пригоден для
алкилирования.
Производство этилбензола. При алкилировании бензола эти-
леном наибольшие трудности в промышленной технологии воз-
никают при осуществлении непрерывной и равномерной подачи
хлорида алюминия в реакционную зону. На некоторых зару-
бежных установках хлорид алюминия шнеком подают через
люк в верхней крышке реактора. Такой способ подачи неудо-
бен тем, что в шнек попадают пары бензола и вызывают комко-
вание хлорида алюминия. Непрерывную подачу катализатора
удобнее осуществлять, когда он находится в виде жидкого ка-
тализаторного комплекса. Комплекс готовят в сосуде с мешал-
кой при 60—70 °C, куда подают диэтилбензол, хлорид алюми-
ния, этилхлорид и бензол. Этилхлорид взаимодействует с бен-
золом, образуя этилбензол и хлористый водород. Хлористый во-
дород является промотором хлорида алюминия. Таким обра-
зом, использование комплекса не только упрощает его подачу
в реактор, но и снижает расход хлорида алюминия за счет про-
мотирующего действия хлористого водорода. Промотор влияет
также на соотношение этилбензола и полиалкилбензолов.
Алкилирование бензола этиленом представляет собой экзо-
термический процесс:
ДЯ = —108,6 кДж/моль
Выделяющееся тепло частично расходуется на поддержание
нужной температуры; избыток снимают через рубашку реакто-
ра, а также испаряющимся бензолом.
Технологическая схема алкилирования бензола этиленом
приведена на рис. 1.4. Катализаторный комплекс из емкости 1,
бензол и этилен поступают в нижнюю часть реактора 2. Алки-
лирование проводится в реакторе колонного типа (без мешал-
кц) при атмосферном давлении. Реактор, состоит из четырех
царг, эмалированных или футерованных керамикой. Для луч-
шего контакта в реакторе имеется насадка. Каждая царга
снабжена рубашкой для разогрева при пуске реактора и для
отвода тепла при нормальном режиме. Температура низа
3-254
33
Диэтипбензоп
Рис. 1.4. Технологическая схема получения этилбензола:
/ — емкость; 2 — реактор; 3 — обратный холодильник; 4, 5 — скрубберы; 6—отстойник; 7, 8, 9 — промывные колонны; 10 — нагреватель; 11, 14,
//—«ректификационные колонны; 12, 15, 18 — конденсаторы-холодильники; 13, 16, 19— кипятильники; 20 — вакуум-приемник; 21 — холодильник.
100 °C, температура верха 90—95 °C. С верха реактора отходя-
щие газы и пары бензола поступают в обратный холодильник 3.
Бензол конденсируется и стекает в реактор, а отходящие газы
направляются в скруббер 4. орошаемый диэтилбензолом, для
улавливания бензола, далее в водный скруббер 5 и затем ухо-
дят в атмосферу. Диэтилбензол с абсорбированным бензолом
подают в реактор. Алкилат через сливной штуцер поступает
в отстойник 6. Катализаторный комплекс отстаивается там
и его возвращают в реактор, а алкилат идет последовательно
в колонны 7, 8 и 9 водной, щелочной и водной промывки, по-
сле чего его направляют на ректификацию.
Алкилат, предварительно высушенный кристаллическим ед-
ким натром в сушильной колонне (на схеме не показана), пода-
ют через нагреватель 10 в колонну 11, имеющую 45 тарелок
и работающую с флегмовым числом 1 : 2,5. С- верха колонны
при 80°C отбирают бензол (содержащий 0,25% этилбензола).
Этилбензол отбирают при 136 °C в колонне 14, имеющей 60 та-
релок и работающей с флегмовым числом 1 1. Этилбензол по-
лучается высокой чистоты — он содержит не более 0,04% ди-
этилбензола. Кубовый остаток колонны 14 поступает в колон-
ну 17, в которой отгоняют диэтилбензол при остаточном давле-
нии 1,33 кПа. При 60 °C отгоняют смесь изомеров диэтилбен-
зола, частично возвращаемых в реактор (где они подвергаются,
деалкилированию), а частичка используемых в качестве высо-
кооктановых добавок к топливам, так же как и полиалкилбен-
золы, которые отводят с низа колонны 17. Полиалкилбензолы
могут быть подвергнуты деалкилированию на отдельной уста-
новке при 200 °C в присутствии хлорида алюминия.
Выход этилбензола составляет 90—95% в расчете на бен-
зол и 93% в расчете на этилен. Расход на 1 т этилбензола со-
ставляет: 0,297 т этилена, 0,770 т бензола и 25—30 кг хлорида
алюминия. Количество рециркулирующего бензола равно
1,185 т.
Производство изопропилбензола. В случае применения хло-
рида алюминия процесс алкилирования бензола пропиленом
может быть осуществлен на установках, аналогичных установ-
кам по производству этилбензола.
Существуют промышленные установки, на которых в качест-
ве катализатора применяют серную кислоту и фосфорную кис-
лоту на кизельгуре. Реакция протекает по уравнению:
Й 4- СН3—СН=СН2 -► ГТ АЯ = —97’8 КДЖ/МОЛЬ
Наряду с изопропилбензолом (кумол) образуются ди- и три-
изопропилбензолы. Алкилирование проводят пропиленом или.
пропан-пропиленовой фракцией. Последняя более доступна в
условиях нефтехимического производства и поэтому применяет-
ся чаще.
3*
35
Рис. 1.5. Технологическая схема получения изопропилбензола на фосфорно-
кислотнОхМ катализаторе:
1 — теплообменник; 2 — подогреватель; 3 — реактор; 4, 7, 10, 13 — ректификационные ко-
лонны; 5, 6, 8, 9, 11, 12, 16 — кипятильники; 14 — конденсатор-холодильник; 15 — ва-
куум-приемник; 17 — холодильник.
Процесс алкилирования осуществляют по следующей схеме
(рис. 1.5). Бензол и пропан-пропиленовую фракцию смешива-
ют перед входом в теплообменник /, затем еще подогревают
и подают в реактор 3. Избыточное давление на нагнетательных
линиях насосов равно 2,5 МПа. К свежему бензолу примешива-
ют возвратный бензол. Реактор по устройству напоминает
теплообменный аппарат. В трубках находится «катализатор,
а по межтрубному пространству для отвода выделяющегося
тепла циркулирует масло, которое отдает свое тепло в подогре-
вателе 2 поступающему сырью, охлаждается в холодильнике
и вновь возвращается в реактор. В зоне реакции поддержива-
ют температуру 250—300 °C.
Продукты реакции поступают в теплообменник /, где они
частично конденсируются, а затем на ректификацию. Давление
на входе в колонну 4 снижается до 1,2—1,5 МПа. Пропан ухо-
дит с верха колонны. Бензол отгоняют в -колонне 7 и возвра-
щают в процесс; в третьей колонне 10 отгоняют изопропилбен-
зол при 152 °C. Четвертая колонна 13 работает при разрежении
и служит для выделения диизопропилбензола. Кубовый оста-
ток колонны 13 представляет собой смесь высококипящих поли-
алкилбензолов, которые охлаждают и отводят. Диизопропил-
бензол примешивают к сырью для сохранения равновесного со-
става алкилата.
Катализатор является весьма активным — с 1 кг можно по-
лучить 800 л кумола. Средняя продолжительность работы ката-
лизатора 700 ч. В процессе алкилирования образуется незначи-
тельное количество полимеров пропилена. В реактор подают
сырье в следующем соотношении: 52,5% бензола (18% свежего
и 34,5% возвратного), 47,5% пропан-пропиленовой (с 20—30%
пропилена) фракции. Мольное соотношение бензол : про-
пилен равно 3:1. Степень конверсии пропилена составляет
90—95%.
36
Промышленное алкилирование бензола пропиленом в при-
сутствии серной кислоты осуществляют в жидкой фазе при
30—40 °C. Концентрацию кислоты изменяют в зависимости от
температуры: при 20°C применяют 90%-ную кислоту, а при
40°C 88%-ную. Технология процесса аналогична алкилирова-
нию изопарафинов олефинами. Длительность процесса 20—
30 мин. После реактора смесь отстаивают от серной кислоты,
а затем нейтрализуют водной щелочью для уменьшения корро-
зии дистилляционной аппаратуры.
Производство высших алкилбензолов. Алкилирование бен-
зола высшими (жидкими) олефинами проводят, как правило,
только на хлориде алюминия. В реактор загружают бензол и
А1С1з или катализаторный комплекс (10—20% от объема- реак-
ционной массы), после чего при перемешивании добавляют
жидкие олефины (или хлорпарафины), поддерживая темпера-
туру 40—60 °C.
Непрерывный процесс алкилирования осуществляют по
двум вариантам. В первом случае используют трубчатый реак-
тор (рис. 1.6, а), в нижней части которого находится мешалка.
Исходные реагенты и катализаторный комплекс подают в ниж-
нюю часть аппарата и эмульгируют с помощью мешалки. Реак-
ционная масса поднимается по трубкам, одновременно охлаж-
даясь за счет подачи воды в межтрубное 'пространство. Выхо-
дящая из реактора масса расслаивается в сепараторе на угле-
водородный слой и катализаторный комплекс, который возвра-
щают в процесс. Другой способ непрерывного алкилирования
состоит в применении каскада из двух-четырех реакторов с ме-
шалками (рис. 1.6,6). В первый аппарат подают бензол и оле-
фины, а также некоторое количество свежего хлорида алюми-
ния. Реакционная масса перетекает в следующий реактор, про-
ходя через сепаратор, где отделяется катализаторный комп-
лекс, возвращаемый в первый реактор.
Рис. 1.6. Реакционные узлы алкилирования бензола высшими олефинами:
а — трубчатый реактор; б — каскад из двух реакторов.
37
Получаемый алкилат нейтрализуют щелочью и промывают
водой для отделения следов хлорида алюминия. Промытый ал-
килат подвергают ректификации, отбирая непрореагировавший
бензол, парафиновые углеводороды, содержащиеся в исходных
олефинах, и целевую фракцию моноалкилбензолов (260—
360°C). Кубовый остаток, представляющий собой смесь моно-
и диалкилбензолов, используют для получения сульфонатных
присадок к смазочным маслам, а также в качестве низкоза-
стывающих синтетических смазочных масел.
Отличительная особенность производства высших алкилбен-
золов на основе неразветвленных а-олефинов — значительный
выход полиалкилбензолов, которые также могут быть исполь-
зованы для получения сульфонатных присадок.
2.6. Производство углеводородов изостроения
Основные представления о процессе. Изомеризация углево-
дородов— это внутримолекулярная химическая реакция, в ре-
зультате которой изменяется структура углеводородов при со-
хранении их молекулярной массы. У олефинов помимо изоме-
ризации углеродного скелета может происходить перемещение
двойной связи, но этот процесс здесь подробно не рассматрива-
ется.
Промышленным катализатором изомеризации является хло-
рид алюминия, промотированный хлористым водородом (8—
13% в расчете 'на углеводород).
В нефтеперерабатывающей промышленности давно освоена
изомеризация н-бутана в изобутан, а также н-пентана в изопен-
тан. Промышленное значение этих процессов состоит в том, что
получаемые углеводороды изостроения — изобутан и изопен-
тан — являются ценными продуктами. Изобутан алкилируют
изобутиленом с образованием изооктана — высокооктановой
добавки к авиационным бензинам. Изопентан сам по себе яв-
ляется высокооктановым компонентом, но он также служит
сырьем для получения изопрена — мономера, используемого
для получения ценного изопренового каучука.
Представляет интерес и изомеризация углеводородов выше
С5 как способ повышения октанового числа бензинов, посколь-
ку, как известно, парафиновые углеводороды с разветвленной
цепью имеют более высокие октановые числа, чем у углеводо-
родов нормального строения.
Изомеризация углеводородов выше С5 сопровождается ярко
выраженными реакциями крекинга. Для их предотвращения
ведут процесс под давлением водорода, зависящим от темпера-
туры. Так, в случае изомеризации пентана при 160°C давление
составляет 1,0—1,5 МПа, а при 200°C оно равно 6,5 МПа. Од-
нако при значительном повышении давления снижается актив-
ность катализатора.
38
Процесс крекинга при изомеризации можно подавить так-
же, вводя органические добавки — бензол, толуол, циклогек-
сан и др. Лучшим ингибитором крекинга является трет-бутил-
бензол — в его присутствии при изомеризации пентана выход
изопентана составляет 72% (мольн.). Активным ингибитором
оказался и дифениловый эфир. Количество добавок зависит от
природы углеводорода: бензола добавляют 0,25—0,5% (об.),
циклогексана 5—10% (об.).
На процесс изомеризации заметно действуют олефины, со-
держащиеся в сырье в самых ничтожных количествах. Так,
изомеризация н-бутана в изобутан при введении хлорида алю-
миния не идет даже в присутствии промотора, а добавление
0,01% олефинов вызывает изомеризацию. Это свидетельствует
в пользу карбонийионного механизма реакции, так как для об-
разования карбоний-иона необходимо присутствие олефина:
ЦСН=СН2 + НА1С14 --► RCH-СН3 + А1С14-
Образующийся при взаимодействии олефина и катализатора
ион карбония взаимодействует с н-бутаном; при этом появляет-
ся новый ион карбония, который изомеризуется:
цен—*сн3 + сн3—сн2—сн2—сн3
—- к ип п з
4“ +
---► СН3-СН-СН2-СН3 ----► (СН3)3С
Образовавшийся ион трет-бутилкар-бония насыщается водоро-
дом из новой молекулы «-бутана и дает изопарафин:
+ +
(СН3)3С + СН3-СН2—СН2—СН3 ► (СН3)3СН + сн3—сн-сн2—сн3
Олефин может образоваться во время изомеризации за счет
крекинга, но лучшие результаты получаются, если олефин до-
бавить к сырью заранее.
Увеличивающийся спрос на изобутилен можно удовлетво-
рить за счет изомеризации бутилена, получаемого дегидрирова-
нием н-бутана. Изомеризацию н-бутилена в изобутилен прово-
дят на фосфорной кислоте, нанесенной на шамот, при 300 °C.
Выход изобутилена за один проход через реактор достигает
50%. Во избежание полимеризации изобутилена проводят изо-
меризацию в присутствии водяного пара.
Кроме хлорида алюминия в качестве катализаторов изоме-
ризации применяют бромид алюминия, хлорид цинка, сульфид
молибдена, платину. В большинстве случаев при изомеризации
углеродного скелета олефина наблюдается и изомеризация (пе-
ремещение) двойной связи, а также другие побочные реакции.
Оксид алюминия, фосфорная кислота, оксид тория, алюмоси-
ликат катализируют изомеризацию углеродного скелета, сили-
кагель и активный оксид алюминия вызывают перемещение
двойной связи, а серная кислота, хлорид алюминия и другие
39
катализаторы наряду с изомеризацией углеродного скелета вы-
зывают и полимеризацию олефина.
При изомеризации н-гексена наиболее активной оказалась
фосфорная кислота, а наихудшие результаты получены в при-
сутствии промышленного алюмосиликата. По-видимому, боль-
шое значение имеет кислотность катализатора.
Процесс изомеризации олефинов не получил промышленно-
го развития, так как октановое число бензина при этом про-
цессе повышается всего на 3—4. Широко .используемые в про-
мышленности низшие изоолефины — изобутилен и изоамилен —
получают не изомеризацией, а термическим и каталитическим
крекингом нефтяного сырья. Однако исследования изомериза-
ции олефинов сыграли большую роль в уточнении химизма
этого процесса, а также каталитического крекинга, сопровож-
дающегося реакциями изомеризации.
Алкилароматические углеводороды 'могут подвергаться раз-
личным изомерным превращениям.
1) Изомеризация с изменением положения заместителей:
2) Изомеризация с укорочением боковой цепи и введением
второго заместителя:
3) Изомеризация боковой цепи:
40
4) Миграция алкильных радикалов с одних колец на дру-
гие (переалкилирование):
Наибольший интерес для промышленности представляют
превращения первого и последнего вида. Известно, что если
смесь ксилолов подвергнуть воздействию хлорида и бромида
алюминия, фтористого водорода или алюмосиликатов, то основ-
ным продуктом реакции будет лексилол — в равновесной сме-
си ксилолов содержится до 60% тиета-изомера.
В промышленности способность ксилолов к изомеризации
используют для увеличения выхода n-ксилола, являющегося
наиболее ценным изомером. n-Ксилол выделяют из смеси вы-
мораживанием с применением четыреххлористого углерода в
качестве растворителя. Выкристаллизовавшийся при охлажде-
нии раствора п-ксилол отфильтровывают. От фильтрата, содер-
жащего смесь ксилолов, отгоняют четыреххлористый углерод,
а смесь вновь поступает на изомеризацию; в результате опять
образуется равновесная смесь ксилолов, которую также разде-
ляют вымораживанием. Таким образом о- и ти-ксилолы превра-
щаются в п-ксилол.
В последнее время особый интерес приобретают реакции
переалкилирования. Они протекают в двух направлениях: при
действии на моноалкилбензол хлорида алюминия получается
смесь моно-, ди-, три- и полиалкилбензолов, например)
а при нагревании смеси полиалкилбензолов в присутствии хло-
рида алюминия с бензолом образуется только моноалкилбен-
зол.
Реакция переалкилирования имеет и практическое значение
как способ получения ксилолов из полиалкилбензолов. Для
этой же цели можно использовать изомеризацию моноалкил-
бензолов с длинной боковой цепочкой в диалкилбензолы (изо-
меризация с укорочением боковой цепочки, чаще всего реакция
метилирования). Например, изомеризацией этилбензола полу-
чают смесь ксилолов. Ее легко отделить от этилбензола дистил-
41
ляцией, а из смеси ксилолов выделить пара-изомер уже извест-
ным методом — сочетанием изомеризации и низкотемператур-
ной кристаллизации.
Производство изобутана. Катализатором промышленного
процесса изомеризации н-бутана является хлорид алюминия на
носителе или без него, промотированный хлористым водородом.
Носителем служит боксит, пемза, силикагель. Носитель увели-
чивает срок службы катализатора, повышает его активность
и снижает смолообразование.
Процесс можно осуществлять в жидкой фазе при 80—90 °C
и в газовой при 130—140 °C. При жидкофазном процессе при-
меняется 8—12%-ный раствор хлорида алюминия в трихлори-
де сурьмы, в котором растворяются бутан и хлористый водо-
род. В присутствии трихлорида сурьмы уменьшается раствори-
мость хлорида алюминия в углеводородах, благодаря чему
снижаются его потери. Активность катализатора при этом уве-
личивается, а его агрессивное действие на аппаратуру умень-
шается. Процесс проводится при 85 °C, 2,1 МПа и объемной
скорости подачи сырья 2,5 ч-1; содержание НС1 в сырье равно
5%. Степень конверсии н-бутана составляет 45—50%. Аппара-
туру изготовляют из специальных никелевых сталей.
Существующие процессы изомеризации н-бутана в газовой
фазе различаются способом подачи сырья и катализатора в ре-
актор. Хлорид алюминия может попадать в реактор вместе с
сырьем или в виде заранее приготовленного комплекса с бута-
ном. В последние годы разработан новый процесс изомеризации
н-бутана на платиновом катализаторе при 320—420 °C и малом
времени контакта. Степень конверсии н-бутана в изобутан до-
стигает 95,4% (мольн.). Общая степень конверсии н-бутана за
один проход равна 40% (мольн.). Непрореагировавший н-бу-
тан возвращают в процесс.
В СССР разработан процесс изомеризации н-бутана на
сульфидном катализаторе (сульфиды молибдена или вольфра-
ма) при 400—420 °C, 5 МПа и объемной скорости подачи сырья
0,5—1,0 ч-1. Процесс ведут в присутствии водорода при моль-
ном соотношении водорода и бутана от 1 1 до 2 : 1. Выход изо-
бутана за один проход примерно 30% (масс.), а в расчете на
превращенный бутан это составляет 92—96% (масс.).
На рис. 1.7 приведена технологическая схема газофазной
изомеризации н-бутана на стационарном слое катализатора.
Катализатор загружают в трубки реактора, по конструкции
напоминающего теплообменник; по межтрубному пространству
циркулирует масло для отвода выделяющегося тепла и регули-
рования температуры в реакторе. Катализатор готовят следую-
щим образом. Носитель (боксит) загружают в трубки, а хлорид
алюминия наносят на него до желаемого содержания, для чего
нагревают осушенный н-бутан и пропускают через емкость
с хлоридом алюминия. Горячий н-бутан увлекает с собой часть
хлорида алюминия и переносит его на боксит.
42
Рис. Л.7. Технологическая схема изомеризации «-бутана:
/ — теплообменник; 2 — нагреватель; 3 — реактор; 4, 6, 7, 12, 13 — холодильники; 5 — ад-
сорбер; 8 — сборник; 9 — скруббер; 10 — отгонная колонна; // — кипятильник; 14 — сме-
ситель; 15 — емкость.
н-Бутан, подогретый в теплообменнике 1 и нагревателе 2,
смешивается с хлористым водородом и поступает в верхнюю
часть реактора 3. Циркулирующее через реактор масло охлаж-
дается в холодильнике 4. Продукты с низа реактора направля-
ют в адсорбер 5, заполненный оксидом алюминия или чистым
бокситом, для улавливания частиц катализатора. Газовый по-
ток после адсорбера охлаждается в водяном 6 и пропановом 7
холодильниках до —10 °C и идет в сборник S, из которого лег-
кие газы уходят через скруббер 9, орошаемый освобожденной
от НС1 смесью бутанов. Жидкие продукты из сборника 8 по-
дают в колонну 10, где отгоняют хлористый водород, возвра,-
щаемый в процесс.
Кубовая жидкость колонны 10 проходит теплообменник /,
холодильник 12 и поступает на защелачивание в емкость 15.
Для лучшей промывки перед емкостью установлен смеситель 14,
где смешиваются н^бутан, изобутан и щелочь. Бутаны после от-
стаивания от щелочи подают на дистилляцию, откуда н-бутан
возвращают в процесс.
С течением времени активность катализатора падает. Сни-
жение активности компенсируют, постепенно повышая темпера-
туру и увеличивая количество хлористого водорода с 3% в на-
чале работы до 23% в конце. Срок службы катализатора 1 —
1,5 мес. Температура процесса 100—140 °C, давление 1,05—
1,8 МПа. Объемная скорость подачи сырья 0,7 ч-1. Выход изо-
бутана за один проход через реактор составляет 40—45%. Ре-
акционную аппаратуру изготавливают из углеродистой стали.
43
2.7. Производство алкилфенолов
Основные представления об алкилировании фенолов. Нали-
чие в молекуле фенола сильной электронодонорной гидрок-
сильной группы увеличивает электронную плотность в пара-
и орго-положениях ароматического ядра и тем самым повышает
реакционную способность фенола при взаимодействии с олефи-
нами по сравнению с бензолом. По той же причине промежу-
точный ион карбония, образующийся при алкилировании фе-
нола, значительно более стабилен, что приводит к его пере-
группировке в нейтральный алкилциклогексадиенон и далее в
алкилфенол:
он он о он
Эта особенность механизма алкилирования фенола обусловли-
вает легкость протекания обратной реакции (деалкилирование
алкилфенолов), а также их изомеризации и диспропорциониро-
вания.
В промышленности алкилфенолы получают алкилированием
фенола спиртами и олефинами.
Первым катализатором алкилирования фенола спиртами
была серная кислота. В дальнейшем наряду с серной начали
применять фосфорную кислоту, хлориды цинка и алюминия;
фторид бора. Процесс преимущественно проводили в жидкой
фазе при 60—140 °C. В газовой фазе алкилирование вели на
гумбрине, оксиде алюминия, алюмосиликате. Выход и состав
продуктов реакции зависит от строения применяемого спирта,
от катализатора и условий реакции. Независимо от вида ката-
лизатора наиболее легко реагируют третичные спирты, затем
вторичные и первичные.
В присутствии большинства катализаторов чаще всего1 об-
разуется смесь орто- и пара-изомеров, диалкилпроизводных и
эфиров фенолов, т. е. в результате алкилирования получается
сложная смесь продуктов:
+ROH
ОН ОН ОН
44
При эквимольных количествах реагирующих веществ в присут-
ствии фосфорной кислоты можно получать более высокий вы-
ход моноалкилфенолов, чем в присутствии других катализато-
ров. Больше всего образуется пара-изомеров. Более низкая
температура способствует образованию орто-изомера, который
при нагревании можно изомеризовать в пара-изомер.
Серная кислота вызывает изомеризацию алкильного ради-
кала, вступающего в реакцию. Это проявляется при алкилиро-
вании фенола изобутиловым спиртом — вместо 2-метилпропил-
п-фенола образуется п-трет-бутилфенол:
ОН ОН
I СН3 |
Q+ch3-ch-ch2oh Q|
с(сн3)3
Наиболее простым по оформлению и наиболее доступным
по исходным веществам является алкилирование фенола оле-
финами. Процесс протекает в присутствии катализаторов,
в преобладающем большинстве случаев сильных кислот или
других веществ кислотного характера.
Одним из первых катализаторов, нашедших применение для
алкилирования фенола олефинами (как и при алкилировании
фенола спиртами), является серная кислота. Оптимальное ко-
личество концентрированной кислоты составляет 8—10% от
массы фенола.
Характерной особенностью алкилирования фенола олефи-
нами в присутствии серной кислоты является ее изомеризующее
действие на алкильные группы: первичные превращаются во
вторичные, вторичные — в третичные. Эта перегруппировка обу-
словлена перемещением водородных атомов без изомеризации
углеродного скелета. Требуемое соотношение фенола и олефина
зависит от характера целевого продукта: при эквимольном со-
отношении исходных веществ, как правило, в основном образу-
ется моноалкилфенол, а при избытке олефина получаются ди-
и триалкилфенолы. При относительно низкой температуре, на-
пример близкой к комнатной, наряду с алкилфенолами в зна-
чительном количестве образуются фениловые эфиры, из-за чего
выход замещенных фенолов при низких температурах невысок.
Сдвигу равновесия в смеси фениловый эфир^алкилфенол, об-
разующейся при алкилировании, в сторону алкилфенола благо-
приятствует повышение температуры.
Место вступления алкильной группы при алкилировании фе-
нолов олефинами определяется строением исходного фенола
(в случае алкилирования крезолов), температурой, а также осо-
бенностями применяемых катализаторов.
Катализатором алкилирования фенола олефинами могут
быть и другие кислоты — фосфорная, фтористоводородная,
45
хлорная. Обычно их применяют меньше, чем серную, из-за ма-
лой доступности и в некоторых случаях из-за меньшей ката<
литической активности. Однако вследствие специфичности дей-
ствия эти кислоты являются иногда незаменимыми. Так, в ре-
зультате алкилирования фенола изобутиленом при 25 °C в при-
сутствии фторсульфоновой кислоты образуется почти исключи-
тельно п-трет-бутилфенол; применение хлороксида фосфора
РОС13 позволяет получать орто-изомер в значительно большем
количестве, чем пара-изомер.
При алкилировании фенола высшими олефинами в присут-
ствии-хлорида алюминия алкильный радикал получаемого ал-
килфенола может изомеризоваться или даже расщепляться:
При использовании фтористого бора могут образоваться
алкилфенолы и эфиры фенолов. Кроме того, фтористый бор
вызывает полимеризацию олефинов даже при сравнительно
низкой температуре. Если необходимо получить алкилфенол с
длинной боковой цепью, сначала полимеризуют олефин, а за-
тем полимером алкилируют фенол. Когда полимеризация неже-
лательна, реакцию проводят при большом избытке фенола.
В присутствии фтористого бора получается большой выход ор-
то-изомеров. Высокий выход алкилфенолов достигается при ис-
пользовании эфирата фтористого бора (С2Н5)2О-ВЕз и молеку-
лярного комплекса BF3 с фосфорной кислотой.
В качестве катализаторов алкилирования фенола олефина-
ми применяют также алюмосиликаты, бензол- и толуолсульфо-
кислоту, однако при использовании алюмосиликатов требуют-
ся повышенные температуры (130°C), а сульфокислоты вызы-
вают полимеризацию олефинов и способствуют значительному
выходу нежелательных диалкилфенолов.
В последнее время в промышленность внедрен новый ката-
лизатор этой реакции — ионообменные смолы, лучшим из кото-
рых является катионит КУ-2. Его получают сополимеризацией
стирола с дивинилбензолом, последующим сульфированием со-
полимера и омылением сульфированного сополимера щелочью
или водой. КУ-2 представляет собой монофункциональный силь-
нокислотный катионит с активными группами SO3H. Выпуска-
ется он в натриевой и водородной форме. В натриевой форме
катионит не активен, и перед алкилированием его нужно пере-
вести в водородную форму, обработав 10%-ной серной или со-
ляной кислотой, затем промыть до нейтральной реакции и вы-
46
сушить. Строение катионита можно изобразить следующим об-
разом:
Катионит выпускают в виде зерен размером 0,3—2 мм. Этот
катализатор позволяет проводить алкилирование в сравнитель-
но мягких условиях, он не способствует образованию эфиров
фенолов, деструктивному алкилированию и полимеризации оле-
фина; в качестве основного продукта получается моноалкилфе-
нол.
Помимо того что алкилирование фенолов в присутствии тех
или иных кислотных катализаторов вызывает коррозию аппара-
туры, процесс этот имеет еще один, весьма существенный не-
достаток. Полученный алкилат нужно промывать водой для
удаления катализатора. При этом вместе с катализатором от-
мывается и некоторое количество фенола. Так образуются фе-
нолсодержащие сточные воды, которые без специальной очист-
ки нельзя сбрасывать в реки и водоемы (такие воды уничто-
жают флору и фауну водоемов, делают воду непригодной для
питья). Эти недостатки полностью исключаются, если в каче-
стве катализатора алкилирования используют ионообменные
смолы. Преимуществом их является, кроме того, возможность
проводить процесс непрерывно и автоматизировать его.
Важнейшей областью использования алкилфенолов является
производство на их основе поверхностно-активных веществ для
моющих, смачивающих, эмульгирующих и деэмульгирующих
средств. Для получения неионогенных моющих средств приме-
няют изооктил-, изононил- и изододецилфенолы. Синтез осно-
ван на их конденсации с этиленоксидом.
Не менее важной областью применения алкилфенолов явля-
ется производство присадок, добавляемых к маслам и топли-
вам с целью улучшения их характеристик. Наилучшей анти-
окислительной присадкой к энергетическим маслам и топливам
47
является ионол
(2,6-ди-трет-бутил-4-метилфенол):
ОН
С(СН3)3
Присадки на основе алкилфенолов предотвращают окисле-
ние масла, препятствуют отложению нагара, а также лакообра-
зованию на стенках цилиндра и поршня, т. е. обладают мою-
щим действием, уменьшают коррозию металлических поверх-
ностей в системе смазки. Имеются присадки, обладающие де-
прессорными свойствами, — они понижают температуру засты-
вания масла.
Некоторые производные алкилфенолов являются стимулято-
рами роста растений, а также гербицидами и фунгицидами.
n-трет-Бутилфенол и п-трет-амилфенол применяют в синтезе
формальдегидных смол; при этом получают маслорастворимые,
так называемые 100%-ные смолы, используемые в лакокрасоч-
ной промышленности. Лаки на основе этих смол образуют проч-
ные пленки на различных поверхностях, высыхающие уже на
воздухе и обладающие хорошей атмосферостойкостью.
п-трет-Бутилфеноло-формальдегидный полимер использу-
ют в производстве специальных клеев, применяемых для склеи-
вания твердых материалов (сталь, дерево и др.) с резиной.
Сульфиды п-трет-бутилфенола и п-трет-амилфенола употребля-
ют как антиокислители каучуков. При полимеризации бутил-
винилфенола, образующегося в результате взаимодействия
n-трет-бутилфенола и ацетилена, получается полимер — коре-
зин,, добавляемый к синтетическому каучуку для повышения
его клейкости. Некоторые замещенные фенолы используют в
производстве душистых веществ.
Бутил-п-крезол, получаемый алкилированием n-крезола изо-
бутиленом, широко применяют для получения эффективных бис-
фенольных антиокислителей с метиленовым или сульфидным
мостиком:
ОН ОН ОН
(CHgJgCs^^^jL (CHgJgC^^L ^CHgx^^L^^^Hg) з
| (ИЛИ | I
CH3 CH3 СН3
Производство п-трет-бутилфенола. Существующее производ-
ство основано на алкилировании фенола изобутиловым спиртом
в присутствии серной кислоты. Недостатки этого метода — пе-
риодичность процесса и коррозия аппаратуры, поэтому необхо-
димо изготавливать основные технологические аппараты из
спецстали либо использовать кислотостойкую футеровку. Кро-
48
ме того, в процессе неизбежно образование фенолсодержащих
сточных вод; выход же целевого продукта весьма мал.
В последние годы в производстве м-грет-бутилфенола вместо
серной кислоты применяют катионит КУ-2. Процесс проводят
в автоклавах с мешалками, а по окончании алкилирования от-
качивают реакционную массу в кристаллизатор, не промывая.
Чтобы предотвратить унос катализатора, на линии откачивания
алкилата установлены сетки. Из кристаллизатора суспензию
м-трет-бутилфенола подают на центрифуги. Жидкие продукты,
содержащие изобутиловый спирт, фенол, о-трет-бутилфенол,
м-трет-бутилфенол и 2,4-ди-трет-бутилфенол, возвращают в ре-
актор. Там протекают изомеризация о-трет-бутилфенола в ма-
ра-изомер и диспропорционирование 2,4-ди-трет-бутилфенола.
Содержащийся в маточном растворе изобутиловый спирт также
вступает в реакцию. Благодаря применению КУ-2 ликвидиро-
ваны сточные воды и повысился выход м-трет-бутилфенола.
Разработан и внедрен в промышленность процесс производ-
ства м-трет-бутилфенола алкилированием фенола изобутиленом
в присутствии ионообменных смол. Технологическая схема про-
цесса аналогична схеме производства высших алкилфенолов
(рис. 1.8). Разница в том, что м-трет-бутилфенол либо подвер-
гают кристаллизации и получают в виде чешуек, либо пере-
кристаллизовывают из растворителя.
При алкилировании фенола образуется некоторое количест-
во побочного продукта — о-трет-бутилфенола. Он отгоняется
вместе с фенолом и возвращается в процесс — в реакторе про-
исходит его изомеризация в мара-изомер. Выход орто-изомера
зависит от температуры, например при 120—130°С выход со-
ставляет 2—5%.
Ди-трет-бутилфенюл можно также возвращать в процесс;
при этом протекает реакция переалкилирования:
ОН он он
фГДр-ф
с(сн,)3 (!:(СНз)з
Благодаря этому выход м-трет-бутилфенола достигает 90% на
превращенный фенол и более.
Производство высших алкилфенолов. В промышленности
высшие алкилфенолы получают алкилированием фенола ноли-
мердистиллятом (н. к. 60—70°C, к. к. 205—215СС) или фракци-
ей а-олефинов (180—240°C). Такие алкилфенолы используют
для получения неионогенных моющих веществ и присадок к
минеральным маслам (алкилфенольных и алкилсалицилатных).
Полимердистиллят на 75—80% состоит из диизобутиленов, а
алкилфенол, получаемый при алкилировании фенола, на 80—
90% состоит из о- и м-трет-октилфенолов.
4—254
49
Рис. 1.8. Технологическая схема получения высших алкилфенолов:
1 — емкость; 2 — подогреватель; 3 — реактор; 4 — фильтр; 5 — атмосферная колонна;
6, /0 — конденсаторы-холодильники; 7, // — приемники; 8, /2 — кипятильники; 9 — ва-
куумная колонна; 13 — холодильник.
На рис. 1.8 приведена принципиальная технологическая схе-
ма получения высших алкилфенолов. Смесь фенола и полимер-
дистиллята готовят в емкости 1 заранее, чтобы она была одно-
родной (при обычной температуре фенол плохо растворяется
в полимердистилляте). Сырьевую смесь, в которой мольное
соотношение фенола и олефинов равно 1 (1,24-1,5), насосом
через подогреватель 2 подают в реактор 3. Реактор заполнен
катионитом, а внутри имеется небольшой змеевик для отвода
выделяющегося тепла. Температура в аппарате 125—140 °C,
давление 0,1—0,15 МПа. Сырье проходит слой катализатора
снизу вверх, продукты реакции уходят сверху и через фильтр 4
поступают в атмосферную колонну 5, в которой отгоняются фе-
нол и частично полимердистиллят. В нижней и верхней части
реактора имеются сетки для предотвращения уноса частиц ка-
тализатора; отдельные вынесенные частицы задерживаются на
фильтре.
Отогнанные в колонне 5 фенол и полимердистиллят возвра-
щают в сырьевую емкость /, а продукт с низа колонны подают
в вакуумную колонну 9. Здесь отгоняют фенол, не вступивший
в реакцию, и полимердистиллят. Их также возвращают в про-
цесс, а алкилфенолы с низа колонны 9 через холодильник 13
отводят с установки в цех синтеза присадок.
Содержание свободного фенола в алкилате 5—15%. Ско-
рость подачи сырья 800—1500 кг на 1 кг катионита в час. Тем-
пература в кубе атмосферной колонны 170—175°С, кратность
орошения 0,2—0,4. В вакуумной колонне остаточное давление
0,66—2,6 кПа, а вакуум создается пароструйным трех- или пя-
тиступенчатым эжектором.
На некоторых заводах отгонку фенола и полимердистилля-
та осуществляют полупериодически в кубах с колоннами. Та-
кой процесс затруднен, так как тепловая мощность кубов не-
значительна.
50
§ 3. ПОДГОТОВКА УГЛЕВОДОРОДНОГО СЫРЬЯ
К ХИМИЧЕСКОЙ ПЕРЕРАБОТКЕ
3.1. Требования к качеству сырья
Углеводородное сырье для нефтехимических производств
должно отвечать высоким требованиям, предопределяемым спе-
цификой дальнейших химических превращений углеводородов.
Одно из основных требований к углеводородному сырью —
минимальное содержание или даже полное отсутствие веществ
иной химической природы. Например, при окислении жидких и
твердых парафинов до спиртов и кислот необходимо, чтобы в
исходном сырье содержалось минимальное количество (до
0,5%) нафтеновых и ароматических углеводородов, тормозящих
окисление. Не менее важно отсутствие фенолов, азотистых и
сернистых соединений, обрывающих цепь окисления. В связи
с этим требования по содержанию серы в ароматических угле-
водородах являются жесткими (не более 0,02%) и допустимое
ее содержание постоянно понижается.
Тщательная очистка от сероводорода необходима для этиле-
на, направляемого на гидратацию, поскольку ректификацион-
ная аппаратура для выделения получаемого этилового спирта
из реакционной смеси быстро корродирует и выходит из строя.
По той же причине этилен не должен содержать ацетилена.
Присутствие ацетилена в этилене недопустимо и при получении
полиэтилена полимеризацией при высоком давлении. Этилен
должен иметь 99,99%-ную чистоту.
Присутствие ацетилена и сернистых примесей в этилене
отравляет серебряный катализатор, используемый *в процессе
окисления этилена, а образующийся ацетиленид серебра -взры-
воопасен. Примесь диенов в олефинах приводит к развитию
смолообразования при изомеризации и алкилировании. Ограни-
чения по влажности, содержанию оксидов углерода, аммиака и
т. п. для углеводородного сырья очевидны.
В ряде случаев необходима очистка углеводородного сырья
от изомеров и гомологов той же химической природы. Так, ес-
ли в парафинах имеются углеводороды изостроения, продукты
последующего окисления содержат повышенное количество низ-
комолекулярных кислот, а также изокислот с крайне неприят-
ным запахом. Загрязненность n-ксилола лгета-изомером приво-
дит к получению терефталевой кислоты с примесью изофтале-
вой, что снижает прочность получаемого лавсанового волокна.
При содержании более 0,1% пропилена в этилене, идущем
на гидратацию, нельзя получить этиловый спирт требуемого ка-
чества. Нечеткое разделение изобутилена и изоамилена в про-
цессе олигомеризации изобутилена резко расширяет изомерный
и фракционный состав образующегося полимердистиллята, что
ухудшает его ценность как алкилирующего агента и как сырья
для производства высокооктанового топлива. Наличие в выс-
4*
51
ших а-олефинах непредельных соединений с разветвленной
структурой приводит (при использовании этого сырья для ал-
килирования бензола или фенола) к получению моющих ве-
ществ с пониженной степенью биохимического разложения в
сточных водах.
Приведенные примеры иллюстрируют необходимость тща-
тельной подготовки углеводородного сырья, и значение такой
подготовки постоянно повышается.
3.2. Очистка газообразных углеводородов
Очистка от механических примесей. Нефтезаводские газы не
содержат твердых механических примесей. Газы, с обессерива-
ющих, обезвоживающих и газофракционирующих установок
могут содержать капельки сконденсировавшихся углеводородов
и смазочных масел, унесенных из компрессоров. Природные га-
зы содержат уносимые из скважины частички грунта, ржавчи-
ну, капельки жидких углеводородов.
Наиболее распространенный метод очистки газа от механи-
ческих примесей — барботирование его через слой жидкости
(масло) или орошение газа жидкостью в абсорберах. Чтобы
предотвратить унос капелек жидкости, в верхней части абсор-
беров устанавливают отбойные тарелки.
Очистка от химических соединений. Очистку осуществляют
главным образом для удаления сероводорода, который помимо
коррозии аппаратуры вызывает в некоторых процессах (плат-
форминг, оксосинтез и др.) отравление катализатора. При
очистке газов от сероводородов удаляется и диоксид углерода,
который также вызывает коррозию и может быть причиной за-
бивания аппаратуры, например газофракционирующих устано-
вок, работающих при низких температурах.
В настоящее время широко распространена очистка газо-
образных углеводородов растворителями (этаноламины, фосфа-
ты, алкациды и др.). При этом выделяют концентрированный
сероводород, который затем перерабатывают в серу, серную
кислоту, сероуглерод и др. (см. гл. VI).
При очистке этаноламинами применяют 15—30%-ные вод-
ные растворы моноэтанол'амина NH2CH2—СН2ОН, диэтанол-
амина NH(CH2—СН2ОН)2 и триэтаноламина N(СН2^СН2ОН)3.
Процесс очистки газов этаноламинамиоснован на адсорбцион-
но-десорбционном принципе с регенерацией реагента. При этом
способе удаляют не только сероводород, но и диоксид углерода:
NH2CH2—CH2OH+H2S h2s-nh2ch2-ch2oh
NH(CH2—CH2OH)2 + H2S H2S NH(CH2-CH2OH)2
N(CH2-CH2OH)3 + H2S H2S N(CH2-CH2OH)3
NH2CH2-CH2OH 4-CO2 + H2O H2CO3-NH2-CHo-CH2OH
При 25—40°C реакции идут слева направо, при температуре
выше 105°C—справа налево.
52
Рис. 1.9. Технологическая схема
очистки газа этаноламином:
1 — абсорбер; 2, 6 — холодильники; 3 —
теплообменник; 4 — десорбер; 5 — кон-
денсатор; 7 — сепаратор; 8 — кипятиль-
ник.
Моноэтаноламин является наиболее сильным основанием
по сравнению с другими этаноламинами и поэтому имеет наи-
большую поглотительную способность. Он химически стабилен,
растворяется в воде в любых соотношениях и не растворяется
в углеводородах, что позволяет легко выделять его из водных
растворов, содержащих примеси. Вместе с тем моноэтаноламин
обладает относительно высоким давлением насыщенного пара
(т. кип. 170°C), что обусловливает его потери за счет испаре-
ния. Основной недостаток моноэтаноламина—образование не-
регенерируемых соединений с серооксидом углерода и с серо-
углеродом, присутствующим в небольших количествах в очи-
щаемых газах. Кроме того, он реагирует и с СО2, и с H2S (в по-
следнем случае с несколько большей скоростью). Большую се-
лективность в отношении сероводорода проявляет триэтанол-
амин, но его общая поглотительная способность в 2,5 раза ни-
же, чем у моноэтаноламина. Поэтому чаще всего используют
моноэтаноламин, позволяющий снизить содержание сероводо-
рода до 0,005 г в 1 м3 газа.
Принципиальная схема очистки газа этаноламином приведе-
на на рис. 1.9. Газ поступает в нижнюю часть абсорбера /. Рас-
твор этаноламина подается вверх и стекает вниз, противотоком
к газу. Температура абсорбции 25—40°C. Очищенный газ ухо-
дит сверху. Раствор этаноламина, насыщенный сероводородом,
уходит с низа абсорбера, нагревается до НО °C в теплообмен-
нике 3 за счет тепла регенерированного раствора, выходящего
из десорбера 4, проходит конденсатор 5 и поступает в верхнюю
часть десорбера. Давление в десорбере 0,25 МПа, температура
низа примерно 130 °C (поддерживается при помощи выносного
кипятильника <?). С верха десорбера смесь паров воды, серово-
дорода и диоксида углерода, имеющая температуру 120—
125°C, уходит в аппарат 5, где конденсируются -пары воды, за-
тем охлаждается в холодильнике 6 и поступает в сепаратор 7,
где газы отделяются от конденсата. Конденсат насосом подают
в десорбер, а отходящий газ, состоящий в основном из серово-
дорода, направляют на производство серной кислоты или серы.
Регенерированный раствор этаноламина из десорбера 4 по-
ступает в теплообменник 3, где охлаждается до 40—50 °C, за-
53
тем в холодильник 2, где охлаждается до 25—30 °C, и после
этого поступает в верхнюю часть абсорбера.
Абсорбер и десорбер могут быть тарельчатыми или наса-
дочными. Абсорбер обычно изготавливают из нержавеющей
стали с 15—20 тарелками или насадкой из колец Рашига (слой
насадки высотой 10—12 м). Десорбер также изготовляют из
нержавеющей стали, так как при 120—130 °C наблюдается се-
роводородная коррозия (сами этаноламины не вызывают кор-
розию).
Этаноламинный способ не обеспечивает тонкой очистки от
сероводорода. Кроме того, в растворе постепенно происходят
накопление шлама (сульфид железа, свободная сера) и образо-
вание солей за счет взаимодействия этаноламинов с примесями
кислот в газе (муравьиная, уксусная). Это приводит к дезакти-
вированию этаноламинного раствора, к его вспениванию (за
счет присутствия солей) и перебросам. Регенерацию этанол-
аминного раствора осуществляют следующим образом. Весь
раствор или основную часть его фильтруют, а примерно 3%
отводят в перегонный аппарат периодического действия. Амины
и вода отгоняются в вакууме при 150—190 °C, а шлам и высо-
кокипящие примеси остаются в кубовом остатке. Регенериро-
ванный раствор возвращают на установку очистки газов.
В тех случаях когда требуется очистка газов от СО2, газы
промывают растворами этаноламинов, а также водными щело-
чами или водой под давлением. При 0,5 МПа в 1 объеме воды
растворяется 5,34 объема СО2, а-при 3 МПа — 25,5 объема.
Установки по промывке газа водой работают при давлении в
абсорберах 2,5—3 МПа. Количество циркуляционной воды со-
ставляет 100—200 л на 1000 'м3 газа. Обычно при отмывке газа
от диоксида углерода снижается и содержание сероводорода в
газе. При снижении давления и повышении температуры про-
исходит отдувка диоксида углерода, который выводят в атмо-
сферу или сжижают для получения сухого льда. Установки по
очистке газа раствором щелочи работают при небольшом дав-
лении.
При обессеривании газов фосфатом калия диоксид углеро-
да не извлекается-
К3РО4 + H2S «=> К2НРО4 + KHS
зато сероводород получается более чистым и является целевым
сырьем для производства серы и серной кислоты. Абсорбция
идет при 35—40 °C, десорбция — при 105—118°C. Схема про-
цесса идентична этаноламинной. Высокая селективность при
поглощении сероводорода, малая летучесть рабочего раствора,
несмешиваемость его с жидкими углеводородами делают метод
очистки газов фосфатом калия наиболее перспективным. Метод
позволяет снизить содержание сероводорода до 0,007% при его
концентрации в исходном газе примерно 8%.
54
В ряде стран для очистки газа от кислотных компонентов
H2S и СО2 применяют обессеривание алкацидами. Они пред-
ставляют собой соли алкилзамещенных аминокислот. Взаимо-
действие протекает по схеме:
(CH3)2NCH2—COOK 4- H2S <=> (CH3)2N-CH2-COOH + KHS
(CH3)2NCH2—COOK + co2 + H2O 4—* (CH3)2NCH2—COOH + KHCO3
Алкациды применяют в виде водных растворов концентрацией
30—35%. Разные алкациды имеют неодинаковую селективность
по отношению к H2S и СО2. Так, приведенный на схеме алкацид
сорбирует СО2 значительно медленнее, чем алкацид состава
СНз—'ЬШСН(СНз)—COOK. Это свойство алкацидов использу-
ют для селективного извлечения H2S и СО2 из газов. Односту-
пенчатая очистка дает 95 %-ное извлечение H2S. При повышен-
ной температуре алкациды весьма коррозионны, поэтому требу-
ется изготавливать очистную аппаратуру из алюминия, хромо-
никелевой стали или футеровать обычные аппараты.
3.3. Осушка газообразных углеводородов
Обезвоживанию подвергают природный газ, транспортиру-
емый на далекие расстояния, а также нефтезаводские газы, раз-
деляемые при низких температурах.
Газы с водой образуют гидраты — белые кристаллические
соединения, например СН4-7Н2О, С2Н6-7Н2О, которые образу-
ются под давлением при температуре выше нуля. Так, метан
при 10 МПа образует гидраты при 12,5°C, а при 4 МПа—при
4 °C. Гидраты этилена появляются под давлением 3,3 МПа при
75 °C. Пропан и другие газы дают гидраты еще легче.
Содержание влаги в газе зависит от его температуры и дав-
ления. Концентрация влаги снижается при повышении давле-
ния. При понижении температуры газа водяной пар становится
насыщенным при точке росы, а при дальнейшем охлаждении
выделяется вода. Чем выше давление (например, в процессе га-
зоразделения), тем более глубокая требуется осушка, так как
давление способствует повышению точки росы.
Водный конденсат и гидраты, выпавшие из газа, забивают
трубопроводы, сокращают их пропускную способность. Но гид-
раты образуются только при наличии избыточной влаги, по-
этому, чтобы исключить их образование, следует проводить
осушку газа до точки росы, когда давление насыщенного водя-
ного пара меньше, чем давление паров гидрата при температу-
ре газа.
В промышленной практике последних лет в качестве осу-
шителей применяют жидкости (гликоли) и твердые адсорбен-
ты типа боксита, синтетических гелей алюминия и кремния,
а также цеолиты. Жидкие осушители дают меньшее снижение
точки росы газа, чем твердые (т. е. достигается меньшая сте-
пень осушки), поэтому для более полной осушки при низкотем-
55
Рис. 1.10. Технологическая схема осушки газа твердыми адсорбентами:
а— осушка; б — регенерация адсорбента; 1, 2, 3 — адсорберы; 4 — конденсатор-холо-
дильник; 5 — газосепаратор; 6 — печь.
пературных процесса-х применяют твердые адсорбенты. Из гли-
колей используют ди- и триэтиленгликоли, обладающие вы-
сокой влагоемкостью. Они не токсичны, не вызывают коррозии
аппаратуры и применяются в виде концентрированных (95—
99%-ных) растворов. Триэтиленгликоль при концентрации рас-
твора 99% обеспечивает большее снижение точки росы, чем
диэтиленгликоль.
При адсорбционном методе осушки углеводородных газов
применяют глины типа бентонита, синтетические алюмогели,
силикагель, цеолиты. Влагоемкость глин 2,5—4%, влагоем-
кость алюмогелей 5—8%. Поглотительная способность твердых
адсорбентов обычно сильно уменьшается при длительной экс-
плуатации— они как бы устают поглощать влагу. Так, силика-
гель вначале поглощает 22% воды от своей массы, а через год
работы—только 11%. Осушку ведут при 25—30°C. Адсорбер
работает периодически: после рабочего цикла его отключают
для регенерации. Глины регенерируют при 140°C, оксид алюми-
ния при 300 °C.
Схема осушки включает три адсорбера (рис. 1.10): в двух
(аппараты 1 и 2) идет осушка газа, в третьем (аппарат 3) про-
исходит регенерация адсорбента. Газ проходит адсорберы свер-
ху вниз. Адсорбер заполнен несколькими слоями насадки вы-
сотой по 0,6—1 м, что обеспечивает равномерное и 'многократ-
ное распределение газа по всему сечению после каждой секции,
создает лучшие условия осушки.
Регенерацию адсорбента осуществляют сухим топливным га-
зом. Он нагревается в трубчатой печи 6, а затем проходит ко-
лонну с регенерируемым адсорбентом (в данном случае — ап-
парат 3) снизу вверх. Пары воды и газ охлаждаются в конден-
саторе-холодильнике 4\ вода отделяется от газа в газосепара-
торе 5 и уходит в канализацию, а газ ‘вновь используют для ре-
генерации или сбрасывают в топливную сеть завода. Точка ро-
сы газа, осушаемого активным оксидом алюминия, силикагелем
и глинами, достигает 60°C.
56
Недостаток применения оксида алюминия и силикагеля для
осушки газов, содержащих непредельные углеводороды, заклю-
чается в полимеризации этих углеводородов на поверхности
адсорбента. В последние годы для глубокой осушки газов ис-
пользуют цеолиты — Са-, Na- или К-содержащие кристалличе-
ские алюмосиликаты с размером пор от 0,4 до 1 нм. Цеолиты
имеют высокую влагоемкость (10% и более) даже при 100°C,
когда другие адсорбенты теряют функцию осушителя. При
осушке газов цеолитами остаточное содержание влаги в газе
достигает 0,001%, а его точка росы от —75 до —100 °C. Уста-
новки с твердыми адсорбентами просты в эксплуатации и по-
зволяют достичь высокой степени осушки газа, но они требуют
расхода большого количества тепла; возможно также отравле-
ние и разрушение адсорбента.
3.4. Разделение газообразных углеводородов
Одно из важнейших условий подготовки сырья для нефте-
химических синтезов — разделение углеводородных газов на
фракции. Необходимая четкость разделения определяется ус-
ловиями дальнейшей переработки каждой фракции. Раньше на
нефтеперерабатывающих заводах с газофракционирующих
установок получали фракции С3 и С4. Теперь, в связи с тем что
в нефтехимическом синтезе широко используют этилен и пропи-
лен, требуется выделять пропан-пропиленовую и этан-этилено-
вую фракции с высокой концентрацией олефинов, а иногда вы-
делять олефины и в чистом виде.
В настоящее время в промышленности применяются следу-
ющие методы разделения природных газов: 1) компрессионный,
2) абсорбционный, 3) адсорбционный, 4) низкотемпературная
ректификация, 5) комбинированные методы, 6) хемосорбция,
7) экстрактивная дистилляция.
Компрессионный метод используют в сочетании с другими
способами. Самостоятельное значение он имеет лишь для га-
зобензиновых заводов, на которых из попутного газа комприми-
рованием и охлаждением выделяют конденсат (сырой газовый
бензин).
Абсорбционный метод широко применяется для извлечения
бензина и сжиженных газов из природных и попутных газов.
При этом методе газ орошают абсорбентом (соляровые, кероси-
новые и лигроиновые дистилляты), который извлекает тяжелые
углеводороды (их отгоняют потом в.десорбере).
Углеводороды разделяют двумя путями: либо последова-
тельно (по мере уменьшения их летучести), либо выделяют
смесь углеводородов и в дальнейшем фракционируют ее в от-
дельных колоннах (рис. 1.11). Глубина извлечения углеводоро-
дов возрастает с ростом давления. Одновременно повышается
в абсорбенте и содержание несконденсированных углеводоро-
дов — метана и этана, что вызывает затруднения при регенера-
57
Бутаны
Рис. 1.11. Технологическая схема разделения газообразных углеводородов мас-
ляной абсорбцией:
/ — абсорбер; 2 — подогреватель; 3, 7, 11, 15, 19 — теплообменники; 4 — десорбер; 5, 9,
13, 17, 2/— кипятильники; 6, 10, 14, 18, 22 — конденсаторы-холодильники; S—бутановая
колонна; 12—пропановая колонна; 16 — пентановая колонна; 20 — изопентановая ко-
лонна.
ции растворителя. При разделении методом масляной абсорб-
ции можно получить углеводороды высокой чистоты: 98%-ный
пропан, 95%-ные н-бутан, изобутан и изопентан. Полнота из-
влечения пропана 70—80%, пентанов 100%.
Характерной тенденцией процессов газоразделения являет-
ся применение низких температур. Абсорбция при низких тем-
пературах позволяет снизить кратность циркуляции и приме-
нить более легкие масла, в которых растворяется большее ко-
личество газа. Чаще абсорбцию сочетают с низкотемператур-
ной ректификацией (см. далее).
Адсорбционный метод используют для разделения природ-
ных, попутных и нефтезаводских газов. Метод основан на не-
одинаковом (селективном) поглощении углеводородов тверды-
ми адсорбентами, подобно тому как дистилляция основана на
их разной летучести. Преимущества адсорбции — высокая сте-
пень извлечения отдельных компонентов даже из бедного
сырья. Из газовой смеси, содержащей 6% этилена, удается по-
лучить этилен 98%-ной чистоты.
В качестве адсорбентов применяют активный уголь, силика-
гель, цеолиты. Активный уголь обладает высокой способностью
к поглощению легких углеводородов; он поглощает их в коли-
честве, во много раз большем, чем в случае такого же расхода
масляного абсорбента. Адсорбированные углеводороды легко
удалить при продувке адсорбента инертным газом или водяным
паром. Адсорбцию применяют для извлечения этилена из бед-
ных газов, для разделения природного газа, для очистки водо-
рода, выделяющегося при гидрокрекинге, при концентрирова-
нии ацетилена, получаемого окислительным пиролизом газово-
го бензина, и в других случаях (осушка и очистка газов). Ад-
сорбцию осуществляют в аппаратах периодического и непре-
рывного действия.
Процесс разделения газов на движущемся слое адсорбента
носит название гиперсорбции. Этот метод широкого распростра-
58
нения в промышленности не получил, так как адсорбенты мало
селективны по отношению к углеводородам с близкой молеку-
лярной массой. Кроме того, из разделяемой газовой смеси тре-
буется предварительно удалять тяжелые углеводороды, так как
во время последующей десорбции они с трудом выделяются из
адсорбента.
Низкотемпературная ректификация широко применяется
для выделения этилена из продуктов пиролиза. Метод позволя-
ет получать этилен 99,9%-ной чистоты. Принципиальная схема
установки низкотемпературной ректификации приведена на
рис. 1.12.
Газ пиролиза после компримирования, очистки и осушки
охлаждают в холодильниках 1 до минус 55—минус 60 °C и по-
дают в деметанизатор 2, имеющий 20 тарелок. Охлаждение осу-
ществляется за счет вводимого извне хладоагента, а также за
счет продуктов разделения. Метан, Н2, 'Ы2 и СО уходят с верха
деметанизатора вместе с небольшими количествами этилена и
этана. При давлении в деметанизаторе 3,5 МПа для конденса-
ции орошения требуется температура минус 95 — минус 100°C;
такое охлаждение достигается за счет использования этилена,
испаряющегося при минус 100 — минус 105°C. Аппараты, рабо-
тающие при низких температурах (ниже —30°C), изготовляют
из сталей, содержащих 35% никеля, так как обычная сталь
при низких температурах становится хрупкой.
С низа деметанизатора остаток при 18°C поступает в деэта-
низатор 5, работающий при 2,8 МПа и имеющий 35—40 таре-
Этан
Пропилен
сн4+н2
Рис. 1.12. Технологическая схема разделения газов пиролиза низкотемпера-
турной ректификацией:
1 — холодильники; 2 — деметанизатор; 3, 6, 9 — конденсаторы-холодильники; 4, 7, Ю —
кипятильники; 5 — деэтанизатор; 9 — этиленовая колонна.
59
лок. С верха уходит этан-этиленовая фракция. Она конденси-
руется при минус 10 — минус 12 °C и частично идет на ороше-
ние колонны 5, а основное ее количество поступает в этилено-
вую колонну 8.
Температура в нижней части деэтанизатора равна 70°C. От-
туда отбирают пропилен. В этиленовой колонне, имеющей
60 тарелок и работающей при 2,1 МПа, происходит разделение
этилена и этана. Орошение этиленовой колонны конденсируется
за счет холода, получаемого от испарения пропана в конденса-
торе. Температура в кипятильнике —7 °C. Этилен отбирают
с верха колонны и направляют на переработку, а этан выводят
с низа, и он может быть снова возвращен на пиролиз.
В качестве хладоагентов применяют потоки разделяемого
газа (внутренний холодильный цикл) или используют независи-
мые от технологической схемы разделения холодильные циклы
(внешний холодильный цикл). В системах с внешним холодиль-
ным циклом циркулирующий газ (например, этилен) не смеши-
вается с технологическим потоком. Во внутреннем холодильном
цикле этилен, подаваемый в систему холодильным компрессо-
ром, постоянно смешивается с технологическим потоком, часть
которого непрерывно забирают вновь в систему холодильного
цикла.
Выбор холодильного цикла и параметров его работы опре-
деляется расходом холода, температурой, давлением в системе
технологического потока и холодильного цикла и другими фак-
торами.
Схема внешнего холодильного цикла 'приведена на рис. 1.13.
Пары газа-хладоагента компримируются и поступают в кипя-
тильник низа колонны, где отдают свое тепло. После дроссели-
рования хладоагент поступает в верхнюю часть колонны, созда-
ет там орошение за счет конденсации паров верхнего потока.
После этого пары хладоагента вновь поступают на комприми-
рование.
Схема внутреннего холодильного цикла приведена на
рис. 1.14. Часть паров этилена с верха колонны компримируется
Рис. 1.13. Внешний
холодильный цикл.
Рис. 1.14. Внутренний
холодильный цикл.
60
Рис. 1.15. Технологическая схема разделения газообразных углеводородов методом абсорбции в сочетании с низкотемпера-
турной ректификацией:
/-абсорбер-десорбер; 2 — дебутанизатор; 3 — депентанизатор; 4 — осушитель; 5 — деметанизатор; 6 — деэтанизатор; 7 — этиленовая колон-
на; 8 — депропанизатор; 9 — конденсаторы; 10 — холодильники; // — кипятильники; 12— компрессор.
и поступает в кипятильник низа колонны, подводя тепло. После
дросселирования пары этилена поступают в верхнюю часть ко-
лонны, охлаждают пары и образуют флегму. Схемы с внутрен-
ним холодильным циклом проще, требуют меньших поверхно-
стей теплопередачи.
Комбинированные методы. Газы более широкого фракцион-
ного состава, получаемые с заводов топливно-химического про-
филя, на которых имеются установки термического и катали-
тического крекинга, лучше разделять комбинированным мето-
дом, сочетая абсорбцию с низкотемпературной ректификацией
(абсорбционно-ректификационный метод).
Фракции С4 и выше отделяют в депропанизирующих абсорб-
ционно-отпарных колоннах без применения вводимых извне
хладоагентов; это позволяет сократить затраты холода, а также
уменьшить затраты на осушку, поскольку, как известно, с утя-
желением газа необходимая глубина его осушки возрастает.
Схема комбинированной абсорбционно-ректификационной
установки приведена на рис. 1.15. На такой установке можно
из нефтезаводских газов получать сухой газ (Н2+СН4), этан,
этилен, фракции С3, С4 и С5. Крекинг-газ при 0,8 МПа поступа-
ет в депропанизирующий абсорбер-десорбер /, где углеводоро-
ды ^С3 абсорбируются остатком из депентанизатора 3. Насы-
щенный абсорбент поступает в дебутанизатор 2, с верха кото-
рого отбирают фракцию С4. Остаток подают в депентаниза-
тор 3, где отбирают фракцию С5. Непоглощенный газ из абсор-
бер а-десор'бера 1 компрессором 12 сжимают до 3,5 МПа, осу-
шают и подают в деметанизатор 5 низкотемпературной секции,
принцип работы которой изложен выше. В настоящей схеме
низкотемпературная секция имеет четыре колонны — в четвер-
той пропан отделяется от унесенной газом фракции С4.
Есть и другой тип комбинированных установок, в которых
фракционированная конденсация сочетается с низкотемператур-
ной ректификацией. Сущность такого конденсационно-ректифи-
кационного метода заключается в охлаждении газа под давле-
нием. Углеводороды С2 и выше конденсируются, а метан и во-
дород остаются в газовой фазе. Этот метод отличается от аб-
сорбционно-конденсационного способом перевода углеводоро-
дов в жидкую фазу — при абсорбции это достигается поглоще-
нием их жидкими растворителями, при конденсационном мето-
де — охлаждением. Основным аппаратом в первом методе явля-
ется абсорбционно-отпарная колонна, во втором — конденсаци-
онно-отпарная, Кроме того, при абсорбционно-конденсацион-
ном методе требуется более глубокое охлаждение — примене-
ние этанового, а иногда и метанового холодильного цикла.
Очень важное значение при комбинированных методах га-
зоразделения приобретает очистка газа от СО2, Н2О, H2S
и С2Н2. Диоксид углерода и влага, выделяясь в твердом виде,
забивают трубопроводы, сероводород вызывает коррозию, аце-
тилен же взрывоопасен (особенно при наличии медных деталей).
62
Сравнивая комбинированные методы разделения газов по
экономическим показателям, следует отметить, что для абсорб-
ционно-ректификационного метода требуется больше пара, чем
для конденсационно-ректификационного. Однако в последнем
случае нужны более сложные теплообменная аппаратура и
схема утилизации тепла и холода. Эксплуатационные затраты
и капиталовложения примерно одинаковы.
Хемосорбция. Когда разделение газов дистилляцией эконо-
мически невыгодно, можно выделять олефины из газовых сме-
сей селективными растворителями (хемосорбция). Этилен, на-
пример, связывается раствором однохлористой меди в моноэта-
ноламине. Степень поглощения некоторых непредельных угле-
водородов таким раствором при 20 °C приведена ниже:
Степень поглощения, л/кг
Углеводород при 0,1 МПа при 0,5 МПа при 1 МПа при 2 МПа
Этилен 8,4 15,8 21,2 24,1
Пропилен 1,1 4,3 6,1 —
«-Бутилен 1,0 — — —
Бутадиен 11,0 — — —
Как видно из этих данных, этилен при 2 МПа с помощью рас-
твора Си2С12 в моноэтаноламине можно' легко отделить от дру-
гих олефинов, которые при этих условиях в нем не растворя-
ются.
Все описанные выше методы позволяют разделить газ на
фракции по числу атомов углерода, а в отдельных случаях раз-
делить парафины и олефины с одинаковым числом атомов уг-
лерода: этилен отделить от этана, пропилен от пропана. Одна-
ко во фракции С4 разделить парафины и олефины перегонкой
не удается из-за близости их температур кипения (см. стр. 66),
но это необходимо, потому что углеводороды С4 широко при-
меняются для разных целей. Изобутилен используют для алки-
лирования фенола и для полимеризации, бутан и бутилены
идут на дегидрирование, бутадиен расходуется для синтеза кау-
чуков. Во всех случаях необходимо получать концентрирован-
ные углеводороды, так как на дегидрирование идут 95%-ный
бутан и 78%-ный бутилен, а для синтеза каучуков требуется
бутадиен концентрации 95%.
Изобутилен из бутан-бутиленовой фракции легко выделить
65%-ной серной кислотой. Экстракцию проводят при 35—40°C;
при этом другие олефины с кислотой не реагируют. Бутан-бути-
леновая фракция поступает в абсорбер 1 (рис. 1.16), где встре-
чается с серной кислотой, уже частично насыщенной изобути-
леном в абсорбере 2 и стекающей противотоком к газу. Кисло-
та, насыщенная изобутиленом, поступает в десорбер 3, где изо-
бутилен отдувают паром, а регенерированную кислоту (ее кон-
центрацию с 45% доводят до 65%) передают в абсорбер 2, где
она извлекает изобутилен и перетекает в абсорбер 1. Газ, очи-
щенный от изобутилена, уходит с верха абсорбера 2.
63
Рис. 1.16. Технологическая схема
извлечения изобутилена серной
кислотой:
1, 2 — абсорбционные колонны; 3 — де-
сорбер.
Известно, что диеновые углеводороды, а также ацетилено-
вые углеводороды Сз и С4 и частично олефины образуют комп-
лексные соединения с медными солями. Например, бутадиен
образует комплекс с аммиачным раствором ацетата однова-
лентной меди:
(CH3COO)2Cu2(NH3)4 + C4He C4He (NH3)3Cu2(CH3COO)24 NH3
В некоторой степени сорбируется этим поглотительным раство-
ром и бутилен. В поглотительных растворах, применяемых в
промышленности, комплексы бутадиена с медной солью раство-
ряются хорошо, а комплекс олефина растворяется незначитель-
но, за счет чего и достигается избирательное извлечение бута-
диена.
Аппаратурное оформление процесса выделения бутадиена
может быть различным. Бутадиен поглощается либо в экстрак-
ционной батарее, состоящей из смесителей и отстойников (семи
и более агрегатов), в которых обеспечивается противоток угле-
водородов и поглотительного раствора, либо на установках с
абсорберами колонного типа.
Принципиальная схема выделения бутадиена раствором аце-
тата одновалентной меди в колонных абсорберах показана на
рис. 1.17. Хемосорбция протекает в трех последовательных про-
тивоточных колоннах (/, 2 и 3) при температуре от 0 до—10 °C.
Бутилен-бутадиеновую фракцию охлаждают в рассольном холо-
дильнике 7 до —8 °C и подают в абсорбер 3. Поглотительный
раствор, охлажденный в рассольном холодильнике 10, подают
в верхнюю часть абсорбера 1, а оттуда уходят бутилены. По-
глотительный раствор, перетекая из колонны в колонну, погло-
щает бутадиен из движущейся противотоком бутилен-
бутадиеновой фракции и, нагреваясь в теплообменнике 9 и по-
догревателе 8 до 38 °C, поступает в сепаратор 4 для предвари-
тельной десорбции. Выделенный бутадиен возвращают в абсор-
бер 3, а поглотительный раствор поступает на окончательную
десорбцию в десорбер 5. С верха десорбера уходит бутадиен;
его освобождают от полимеров в ректификационной колонне 6
и отводят с установки.
Поглотительный раствор из десорбера 5 возвращают в аб-
сорбер 1. С течением времени в поглотительном растворе на-
64
капливаются ацетиленовые углеводороды с тремя и четырьмя
атомами углерода (чистый ацетилен не растворяется в этом
растворе); они образуют ацетилениды меди, которые могут
взрываться. Поглотительный раствор освобождают от ацетиле-
новых углеводородов следующим образом. Раствор из десорбе-
ра 5 еще горячим (при 90°C) поступает в емкости, где он на-
ходится длительное время. Ацетиленовые углеводороды поли-
меризуются и выпадают в осадок. Раствор фильтруют и возвра-
щают в процесс.
Очень ценным мономером для синтетического каучука явля-
ется и другой диен — изопрен СН2=С(СН3) — СН=СН2 (жид-
кость, т. кип. 34 °C). Из этого углеводорода получают
синтетический каучук, идентичный натуральному и даже пре-
восходящий его по некоторым свойствам. Одним из источников
получения изопрена должен стать изопентан, выделяемый из
газового бензина или получаемый изомеризацией н-пентана.
Дегидрирование изопрена протекает подобно дегидрированию
бутана при 525—535 °C и может быть осуществлено как в одну,
так и в две стадии.
Изопрен выделяют так же, как бутадиен, — методом экст-
рактивной дистилляции с применением диметилформамида в
качестве третьего компонента. Можно применять водный аце-
тон (с 5% воды), водный фурфурол и метилформиат. Азеотроп-
ная смесь содержит до 92% изопрена. Для его концентрирова-
ния применяют хемосорбцию.
Товарный изопрен требуется тщательно очищать от ацети-
леновых углеводородов, так как они при содержании выше
0,01% сильно замедляют процесс полимеризации изопрена и
снижают качество каучука. Очистку проводят селективным гид-
рированием метано-водородной фракцией на никелевом ката-
лизаторе при 350 °C.
Экстрактивная дистилляция. Сущность экстр активной дис-
тилляции заключается в том, что к разделяемой смеси добав-
ляют третий компонент — растворитель, образующий с компо-
Рис. 1.17. Технологическая схема выделения бутадиена из бутилен-бутадиено-
вых смесей:
/, 2, 3— абсорберы; 4 — сепаратор; 5 — десорбер; 6 — ректификационная колонна;
7, 10 — холодильники; 8 — подогреватель; 9 — теплообменник; 11, 12 — кипятильники;
13, 14 — конденсаторы.
5-254 65.
центами смеси раствор, в котором относительная летучесть
компонентов значительно отличается от летучести этих компо-
нентов без растворителя (это возможно, разумеется, только в
случае, если раствор не подчиняется законам идеальных рас-
творов). Относительная летучесть (а) компонентов выражает-
ся соотношением давлений их насыщенных паров (Ра и Рб)
при одной и той же температуре
a s Рд/Рб
и показывает, насколько легче или труднее испаряется одна
жидкость по сравнению с другой.
Экстр активной дистилляцией можно разделить только те
смеси, которые имеют разную полярность. Эффективность при-
менения третьего компонента — экстрагента — зависит от его
селективности, летучести и легкости отделения компонентов от
растворителя. Для увеличения селективности к экстрагентам
добавляют воду. Чем выше содержание воды в экстрагенте,
тем меньше растворимость углеводородов в нем и тем выше се-
лективность. В. качестве экстрагентов обычно применяют веще-
ства, по температуре кипения значительно превышающие разде-
ляемые компоненты. Это в дальнейшем облегчает выделение
экстрагента.
В табл. 1.2 приведена относительная летучесть углеводоро-
дов С4 и той же смеси с добавкой фурфурола, содержащего
5% воды. Как видно из таблицы, летучесть насыщенных угле-
водородов повышается значительно больше, чем для непре-
дельных; в результате происходит их разделение. В промыш-
ленности в качестве третьего компонента применяют водный
фурфурол и водный ацетон. н-Бутилены отделяют от н-бутана
экстрактивной дистилляцией с фурфуролом.
После выделения изобутилена серной кислотой (стр. 63)
другие компоненты бутан-бутиленовой фракции можно разде-
лить следующим путем (рис. 1.18). В ректификационной колон-
не 1 предварительно разделяют газовую смесь: сверху уходят
изобутан и бутен-1, а н-бутан и бутены-2 выводят снизу. Далее
Таблица 1.2. Относительная летучесть углеводородов С4 при 54 °C
Углеводород Т. кип., ®С Относитель- ная лету- честь Относительная летучесть в присутствии фурфурола
Изобутан —11,7 1,209 2,600
я-Бутан —0,6 0,871 2,020
Бутен-1 —6,3 1,046 1,718
Изобутилен —6,9 1,070 1,666
транс-Бутен-2 0,9 0,843 1,190
ЧИс-Бутен-2 -3,5 0,776 1,065
Бутадиен —4,5 1,000 1,000
66
Рис. 1.18. Технологическая схема разделения бутанов и бутиленов экстрак-
тивной дистилляцией с фурфуролом:
/ — ректификационная колонна; 2, 4 — колонны экстрактивной дистилляции; 3, 5 —от-
парные колонны.
потоки идут в колонны 2- и 4 на экстрактивную дистилляцию.
Бутен-1 остается в фурфуроле, а изобутан отгоняется. Бутен-1
отгоняют от фурфурола в колонне 3. Смесь н-бутана и буте-
нов-2 разделяют тоже экстрактивной перегонкой. С фурфуро-
лом из колонны 4 уходит бутен-2, который отгоняют в колон-
не 5, а с верха колонны 4 отбирают н-бутан. Таким образом,
сложная по составу фракция С4 легко разделяется.
Экстрактивную дистилляцию с применением водного ацето-
на в качестве третьего компонента используют для выделения
концентрированного бутилена из контактного газа с установок
дегидрирования бутана. Вначале из газа выделяют бутан-бути-
леновую фракцию методом абсорбции с низкотемпературной
ректификацией и получают смесь с 60% бутана. Экстрактивной
дистилляцией можно получить из этой смеси бутиленовую
фракцию с 5% бутана и бутановую фракцию с 5% бутилена.
Бутан возвращают на дегидрирование, а бутилен используют
для производства бутадиена.
Бутан-бутиленовая фракция поступает в колонну 1 экстрак-
тивной дистилляции (рис. 1.19), в которой противотоком к фрак-
ции движется водный ацетон (18% воды и 82% ацетона). Бу-
тановую фракцию выводят с верха колонны, отмывают в колон-
не 2 от ацетона и направляют в парк. Ацетон с растворенным
бутиленом выводят с низа колонны 1 и подают в отгонную ко-
лонну 4. С верха этой колонны отводят бутилены, отмывают их
в колонне 5 от ацетона1 и отводят в парк. Ацетон регенерируют
в колонне 3, оттуда он снова идет в колонну /, а воду с низа
колонны 3 используют для промывки бутана и бутилена в ко-
лоннах 2 и 5.
Контактный газ, поступающий с установки дегидрирования
бутилена, разделяют вначале методом абсорбции с низкотемпе-
ратурной ректификацией. В качестве абсорбента используют
тяжелые углеводороды, получаемые в процессе дегидрирования
бутилена, так же как при разделении газа после дегидрирова-
5'
67
Рис. 1.19. Технологическая схема выделения бутилена экстрактивной дистил*
ляцией с ацетоном:
J — колонна экстрактивной дистилляции; 2, 5 — промывные колонны; 3, 4 — отгонные ко-
лонны; 6 — теплообменники; 7 — холодильники; 8 — конденсаторы; 9 — кипятильники.
ния бутана используют фракцию тяжелых углеводородов, по-
лучаемых при дегидрировании бутана. При отсутствии этих
фракций в период пуска установки используют бензино-лигрои-
новую фракцию.
3.5. Разделение жидких углеводородов
Четкая ректификация. Основным фактором, влияющим на
полноту разделения компонентов и чистоту получаемых продук-
тов, а также на энергозатраты и эксплуатационные расходы,
является различие в температурах кипения разделяемых ком-
понентов. Чем ближе температуры кипения, тем труднее раз-
делить компоненты при ректификации. Для хорошего разделе-
ния нужны колонны с большим числом тарелок; при этом сле-
дует вести процесс с большой кратностью орошения.
С увеличением четкости ректификации число тарелок в ко-
лонне и кратность орошения возрастают. Необходимо увеличи-
вать диаметр ректификационной колонны; при этом растет рас-
ход тепла и охлаждающей воды, увеличивается поверхность ки-
пятильника и конденсаторов.
Четкое разделение сложных смесей осуществляют на уста-
новках непрерывного действия с большим числом ректификаци-
онных колонн (на единицу меньше числа получаемых продук-
тов). Смесь углеводородов, состоящую из н-пентана, изогекса-
на, н-гексана, изогептана и н-гептана (фракция 75—110 °C бен-
зина прямой перегонки нефти), можно разделить на установке,
схема которой приведена на рис. 1.20. Из природного газа и га-
зового бензина выделяют индивидуальные углеводороды на
$8
установках четкой ректификации. Число тарелок в колоннах
50—100 (а иногда 150), высота одной колонны 30—45 м и бо-
лее.
При большом числе тарелок (150—300) на установке дела-
ют две или три последовательные колонны. Так, бинарную
смесь изогептана и н-гептана можно разделить на установке,
приведенной на рис. 1.21. В каждой колонне имеется 50 таре-
лок. Обогреваются колонны кипятильниками; колонны работа-
ют с четырех- или шестикратным холодным орошением и горя-
чим орошением такой же кратности. Сырье поступает нагретым
до 56 °C; температура верха колонны 83 °C; температура низа
136 °C. Головной продукт охлаждают вначале до 50 °C в кон-
денсаторе смешения 3, а затем до 40 °C в поверхностном холо-
дильнике 4. На установке извлекается изогептан 95%-ной чи-
стоты.
Особую роль играет четкая ректификация при отделении
смеси ксилолов от этилбензола. В этом случае ректификацию
осуществляют на трех последовательных колоннах. В каждой
из них 100 тарелок, высота колонны примерно. 60 м.
Азеотропная перегонка используется для разделения арома-
тических углеводородов. Процесс идет в присутствии третьего
компонента, образующего азеотропную смесь с одним или не-
сколькими компонентами разделяемой смеси. Азеотропные сме-
си могут быть с минимумом или максимумом температуры ки-
пения. При разделении бинарной смеси могут получаться азео-
тропные смеси с минимальной температурой кипения, но одна
Рис. 1.20. Технологическая схема четкой ректификации фракции С5 — С?:
1» 2, 3, 4, 5 — ректификационные колонны.
н-Гептан
Рис. 1.21. Технологическая схема четкой
ректификации гептанов:
/ — ректификационные колонны; 2 — кипятиль-
ники; <?—конденсатор смешения; 4 — холо-
дильник.
69
из азеотропных смесей обычно кипит ниже, чем вторая. Раство-
ритель может образовывать гомогенные азеотропные смеси
(полная взаимная растворимость растворителя и выделяемого
компонента) и гетерогенные азеотропные смеси. Примером по-
следней служит тройная смесь бензол+этанол+ вода (74,1%
бензола, 18,5% спирта и 7,4% воды) с т. кип. 64,9 °C, образую-
щаяся при обезвоживании этилового спирта бензолом.
К растворителю предъявляются следующие требования: он
должен быть доступным и термически стойким, легко отделять-
ся, не вызывать коррозии. Температура его кипения должна
быть на 10—40 °C ниже температуры кипения компонентов сме-
си. Азеотропная смесь легко разделяется, когда растворитель
не смешивается с углеводородом при комнатной температуре.
В противном случае растворитель приходится регенерировать
(экстрагировать водой или каким-либо другим дешевым рас-
творителем) и выделять перегонкой. Несмотря на то что тео-
рия азеотропной перегонки довольно хорошо Известна, оконча-
тельный выбор растворителя чаще всего осуществляют эмпири-
ческим путем.
Для выделения ароматических углеводородов в промышлен-
ных процессах азеотропной перегонки применяют в качестве
растворителей (азеотропообразователи) метанол и метилэтил-
кетон.
Подробнее опишем выделение толуола. Исходным сырьем
для этого служат бензиновые фракции прямой перегонки неф-
ти, термического и каталитического крекинга, гидроформинга.
Из продуктов гидроформинга выделяют толуольную фрак-
цию (98—122 °C) и подают ее в колонну 1 для азеотропной пе-
регонки с метанолом (рис. 1.22). Азеотропная смесь метанол+
+неароматические компоненты, содержащая 55% метанола,
уходит с верха колонны, а концентрат толуола с низа колонны
смешивают с дополнительным количеством метанола и подают
в колонну 2, с верха которой отбирают парафино-нафтеновую
Рис. 1.22. Технологическая схема выделения толуола азеотропной перегонкой
с метанолом:
1, 2 — колонны азеотропной перегонки; 3 — экстракционная колонна; 4 — ректификацион-
ная колонна; 5 — кипятильники; 6 — холодильник; 7 — конденсаторы-холодильники.
70
5
и нафтены
Рис. 1.23. Технологическая схема выделения бензола экстрактивной дистил-
ляцией с фенолом:
/ — колонна экстрактивной дистилляции; 2 — бензольная колонна; 3 — теплообменник;
4 — холодильник; 5 — конденсаторы; 6 — кипятильники.
фракцию с 69% метанола (азеотропная смесь). Дистилляты ко-
лонн 1 и 2 поступают в колонну 3 для отмывки метанола во-
дой. С верха колонны 3 уходят парафино-нафтеновые углево-
дороды, а с низа водный метанол (примерно 70% метанола и
30% воды). Он поступает в колонну 4, где отделяется от воды.
Оттуда метанол возвращают в колонну 1 азеотропной перегон-
ки, а воду — в колонну 3 для отмывки метанола. С низа колон-
ны 2 уходит 96—99%-ный толуол, который для очистки от не-
предельных углеводородов обрабатывают серной кислотой, про-
мывают щелочью и водой и разгоняют.
Азеотропная перегонка в две стадии более выгодно, чем од-
ностадийная, из-за меньшего расхода метанола.
В качестве третьего компонента при азеотропной перегонке
применяют и мети л этил кетон, содержащий 10—15% воды. Тех-
нологическая схема установки аналогична схеме с применением
метанола, с той разницей что азеотропную перегонку осуществ-
ляют в одной колонне, концентрат толуола, содержащий до
5% метилэтилкетона, подают в ректификационную колонну для
отгонки кетона, возвращаемого в цикл, а толуол направляют
на очистку и вторичную перегонку. Более сложна и регенера-
ция метилэтилкетона из азеотропной смеси, которая проходит
четырехступенчатую экстракцию водой. Рафинат парафино-
нафтеновых углеводородов идет на ректификацию, а верхний
погон, представляющий собой азеотропную смесь парафино-
нафтеновых углеводородов' и метилэтилкетона, направляют на
повторную экстракцию водой. Водные растворы для регенера-
ции метилэтилкетона идут на ректификацию. После ректифика-
ции кетон возвращают в колонну азеотропной перегонки, а во-
ду вновь подают на экстракцию.
71
Рис. 1.24. Технологическая схема экстракции ароматических углеводородов
диэтиленгликолем:
/ — экстракционная колонна; 2 — промывная колонна; 3 — отпарная колонна; 4 — от-
стойник.
Экстрактивная дистилляция применяется для выделения
бензола. Схема выделения бензола экстрактивной дистилляци-
ей с фенолом приведена на рис. 1.23. Узкая бензольная фрак-
ция поступает в колонну 1. С верха ее при 79 °C отбирают па-
рафино-нафтеновые углеводороды, а с низа — смесь фенола с
бензолом, которую направляют в колонну 2. С верха колонны 2
бензол^сырец идет на кислотно-щелочную очистку и вторичную
перегонку, а фенол с низа колонны 2 возвращают в колонну 1
через теплообменник 3 и холодильник 4.
Экстракция. Этот метод, применяемый в промышленности,
основан на использовании растворителей, обладающих высокой
селективностью к ароматическим углеводородам. Осуществля-
ют противоточную экстракцию, как наиболее эффективную. Ра-
финатный и экстрактный растворы разделяют затем в колонне,
растворитель регенерируют, а выделенные углеводороды на-
правляют на очистку и ректификацию. Для повышения четкости
разделения вводят два растворителя: один хорошо растворяет
извлекаемые компоненты, второй — неизвлекаемые. Четкость
разделения можно повысить, применяя орошение экстракцион-
ных колонн.
Для выделения ароматических углеводородов в качестве
растворителя еще в 1907 г. был предложен диоксид серы. В на-
стоящее время его применяют для очистки керосина и как
охлаждающий агент (его т. кип. —10°C). Сырьем для выде-
ления ароматических углеводородов являются фракции катали-
тического риформинга. Экстракция жидким диоксидом серы
протекает при минус 20 — минус 30 °C. Чтобы избежать силь-
ной коррозии аппаратуры, требуется тщательно осушать сырье
и обезвоживать циркулирующий экстрагент.
Для экстракции ароматических углеводородов применяют и
водо-гликолевые смеси (8—10% воды), осуществляя процесс
в условиях противотока. Разделяемую смесь направляют в сред-
нюю часть экстракционной колонны 1 (рис. 1.24). Рафинатный
раствор, выходящий с верха колонны /, отмывают от диэтилен-
72
гликоля водой в колонне 2, а экстрактный раствор отделяют
от растворителя в отпарной колонне 3. С верха колонны 3 аро-
матические углеводороды, часть воды и этиленгликоля уходят
в отстойник 4, где эта смесь разделяется. С низа промывной и
отпарной колонн 2 и 3 диэтиленгликоль поступает на верх
экстракционной колонны 1. Она работает при 160—175 °C и
0,7—0,8 МПа, что обеспечивает ведение процесса в жидкой фа-
зе. Количество циркулирующего растворителя составляет 500—
600% на исходное сырье.
Адсорбция и кристаллизация. Один из эффективных спосо-
бов разделения углеводородных смесей — метод адсорбционно-
го или, иначе, хроматографического разделения. Этот метод,
отличающийся высокой четкостью и позволяющий разделять
смесь на компоненты разной химической структуры, широко
распространен в лабораторной практике.
При адсорбции вещество поглощается поверхностью адсор-
бента за счет возникновения сил притяжения между молекула-
ми адсорбента и адсорбируемого вещества. В качестве адсор-
бентов применяют твердые пористые материалы с сильно раз-
витой поверхностью. Одна из основных характеристик адсор-
бента — его емкость или активность, т. е. относительное коли-
чество поглощенного вещества по отношению к массе адсор-
бента.
На силикагелях, алюмосиликатах и других адсорбентах вна-
чале адсорбируются ароматические углеводороды, а затем наф-
теновые; наименьшая адсорбируемость у парафиновых углево-
дородов. Адсорбционная способность возрастает и при переходе
от моноциклических к би- и полициклическим ароматическим
углеводородам, что позволяет разделять их на отдельные груп-
пы. Для углеводородов одного и того же гомологического ряда
адсорбируемость растет с увеличением их молекулярной массы.
После полного насыщения адсорбента осуществляют его де-
сорбцию, а затем регенерацию. Десорбцию проводят раствори-
телями, которые адсорбируются лучше, чем извлекаемые угле-
водороды. Иногда применяют два растворителя — первый вы-
тесняет парафино-нафтеновые углеводороды, а второй — аро-
матические. Второй растворитель должен обладать высокой
адсорбционной способностью. Это — либо высокомолекулярный
ароматический углеводород, либо полярный растворитель
(спирт, кетон).
Регенерация адсорбента, т. е. подготовка его к следующему
циклу адсорбции, заключается в удалении второго растворите-
ля испарением при повышении температуры и понижении дав-
ления. Если температуры кипения адсорбированных компонен-
тов высоки и удалить их трудно даже полярными растворителя-
ми, применяют окислительную регенерацию, т. е. выжигают
адсорбированные вещества, подавая воздух в адсорбер.
В последние годы предложены способы адсорбционного
разделения в движущемся слое адсорбента. По конструктивно-
73
му оформлению процесс напоминает установку каталитическо-
го крекинга. Адсорбция идет в перколяторе (адсорбер) непре-
рывно, десорбцию осуществляют в промывной колонне, а окис-
лительную регенерацию проводят в отдельном аппарате—реге-
нераторе, выполненном по типу регенератора каталитического
крекинга. После регенера'ции возвращают адсорбент в адсорбер.
Для разделения углеводородов сейчас все чаще используют
цеолиты, обладающие большой адсорбционной емкостью и по-
вышенной термостойкостью.
Метод кристаллизации применяют в тех случаях, когда дру-
гие способы не эффективны. Например, азеотропной перегонкой
и экстрактивной дистилляцией из дистиллятов каталитического
риформинга получают бензол, толуол, а также фракцию Се,
состоящую из этилбензола и смеси ксилолов. Разделить фрак-
цию Се трудно, так как температуры кипения углеводородов Се
очень близки:
Компонент т. кип., °C Т. пл., °C
о-Ксилол 144,4 —25,2
jw-Ксилол 139,1 —47,8
л-Ксилол 138,4 13,3
Этилбензол 137,2 — 95,0
Смесь разделяют следующим образом (рис. 1.25): четкой
ректификацией в колонне 1 выделяют о-ксилол, а в колонне 2—
этилбензол. Эти колонны имеют большое число тарелок (в ко-
лонне 1 100 тарелок, колонна 2 имеет 300 тарелок и состоит из
трех последовательных колонн по 100 тарелок в каждой). Вы-
Рис. 1.25. Технологическая схема разделения смеси ксилолов и этилбензола:
/, 2 — ректификационные колонны; 3 — осушитель; 4, 11, 13 — холодильники; 5, 10,
/4 — емкости; 6, 9, /5 — центрифуги; 7, 12 — отгонные колонны; 8 — нагреватель.
74
сота колонн 1 и 2 равна 60 -м, диаметр их 2,7 м. Кратность
орошения колонн от 60 до 100.
Смесь п- и jw-ксилолов разделяют низкотемпературной крис-
таллизацией. С низа колонны 2 смесь проходит осушитель 3,
холодильник 4, где охлаждается до минус 40 — минус 57 °C,
и поступает в емкость 5 для образования кристаллов*, которые
отделяют на фильтрующей центрифуге 6. Твердая фаза содер-
жит 75—80% n-ксилола. Для получения более чистого продук-
та и-ксилол расплавляют, повторно кристаллизуют и центри-
фугируют (на схеме показана одна ступень), после чего полу-
чают и-ксилол 95—98%-ной чистоты.
Фильтрат после отделения кристаллов и-ксилола еще содер-
жит некоторое его количество (8—9%). Для лучшего выделе-
ния и-ксилола смешивают фильтрат с четырехх лор истым угле-
родом, охлаждают до —79 °C в холодильнике 11 и направляют
в емкость 10, где происходит кристаллизация. л-Ксилол обра-
зует с четыреххлористым углеродом комплексное кристалличе-
ское соединение (плавящееся при —40 °C и при перегонке вы-
деляющее четыреххлористый углерод). Комплекс отделяют на
центрифуге 9, затем расплавляют, нагревают в аппарате 8 и
направляют в колонну 7. Четыреххлористый углерод отводят с
верха колонны и возвращают на кристаллизацию, а концентрат
n-ксилола в смеси с л!-ксилолом рециркулируют в сырье.
Фильтрат после отделения кристаллического комплекса по-
ступает в колонну 12 для отгонки оставшегося четыреххлори-
стого углерода, а остаток (концентрат ж-ксилола в смеси с
л-ксилолом) охлаждают в холодильнике 13 до —73°C, кристал-
лизуют в емкости 14 и разделяют на центрифуге 15. Твердую
фазу, содержащую 80—95% jw-ксилола, выводят как целевой
продукт, а фильтрат (смесь ксилолов) возвращают в сырье, ли-
бо используют в качестве растворителя или компонента бен-
зина.,
Разделять смесь м- и л-ксилолов можно ее сульфированием
при 80 °C. При этом сульфируется только ле-ксилол. Сульфиро-
ванный ле-ксилол переходит в кислотный слой; его отделяют и
подвергают гидролизу, в процессе которого и выделяется ле-кси-
лол.
В рассмотренном процессе кристаллизация идет полунепре-
рывно — с перерывами на загрузку и выгрузку центрифуг.
Предложены непрерывные противоточные колонны для крис-
таллизации, дающие высокоэффективное разделение.
С развитием нефтехимической промышленности резко воз-
рос спрос на чистые продукты, что вызвало необходимость раз-
работки более совершенных методов разделения. При получе-
нии многих изомеров (например, n-ксилола) кристаллизация
является единственным методом их выделения.
Промышленные процессы разделения кристаллизацией раз-
виваются и совершенствуются. Они пока обходятся дороже, чем
экстракция или экстрактивная дистилляция. Требуется усовер-
75
шенствовать методы выращивания кристаллов и их выделения
из маточных растворов. Усовершенствование процесса кристал-
лизации, его большие потенциальные возможности позволят в
дальнейшем шире применять его в промышленной практике.
В заключение скажем, что процессы подготовки углеводо-
родного сырья для химической переработки,, процессы выделе-
ния углеводородов в чистом виде требуют больших затрат. Од-
нако на это приходится идти, так как нужное качество товар-
ной продукции чаще всего обеспечивается высоким качеством
исходного сырья. Процессы подготовки углеводородного сырья
очень разнообразны, что определяется разными требованиями
и регламентированием качества, различием физико-химических
свойств углеводородов и выбором тех процессов их подготовки,
которые обеспечивали бы требуемое качество сырья по самым
разным показателям.
Процессы подготовки углеводородного сырья непрерывно
совершенствуются. Наряду с широко известными промышлен-
ными способами идет поиск новых методов, реагентов, техноло-
гических решений и конструкций аппаратов.
ГЛАВА II
ПРОИЗВОДСТВО ВИНИЛОВЫХ МОНОМЕРОВ
И ДИЕНОВ
§ 1. ПРОИЗВОДСТВО УГЛЕВОДОРОДНЫХ МОНОМЕРОВ
ДЕГИДРИРОВАНИЕМ
Из углеводородов важнейшими мономерами в производстве
синтетических каучуков являются бутадиен, изопрен, стирол,
а-метилстирол и изобутилен. В промышленности все они полу-
чаются дегидрированием. Изопрен, кроме того, производят из
изобутилена и пропилена.
Технология получения мономеров существенно влияет на
экономику производства синтетических каучуков, так как их
себестоимость на 60—70% определяется стоимостью мономеров.
1.1. Теоретические основы процессов
дегидрирования
Важной особенностью реакций дегидрирования является их
обратимость. Они проходят при высоких температурах с погло-
щением тепла. Для получения олефинов, диенов, арилолефинов
76
требуется температура 500—650°C, что предопределяет прове-
дение процессов в газовой фазе. Дегидрирование протекает с
увеличением объема- газов и, следовательно, степень конверсии
углеводородов увеличивается при понижении давления.
Термодинамическая вероятность дегидрирования возрастает
с повышением температуры и молекулярной массы парафина.
При отщеплении одной молекулы водорода константа равнове-
сия имеет следующую зависимость от общего давления (Р)
и равновесной степени конверсии (х):
х2
Поскольку степень конверсии возрастает при понижении давле-
ния, процесс ведут либо при атмосферном давлении, либо в ва-
кууме. Так, для дегидрирования этилбензола при 595 °C
и 0,1 МПа равновесная степень конверсии составляет 40%,
а при 0,01 МПа она возрастает до 80%. Наряду с вакуумом
применяют разбавление реакционной массы паром, что тоже
приводит к снижению парциальных давлений реагентов и росту
равновесной степени конверсии. При разбавлении паром, кроме
того, уменьшается образование побочных продуктов.
Реакции дегидрирования являются каталитическими. Ис-
пользование катализаторов преследует цель снизить температу-
ру, чтобы уменьшить выход побочных продуктов, в первую оче-
редь продуктов крекинга (метан, этилен, пропилен и др.). При-
менение катализаторов способствует разрыву связей С—Н
и предотвращает разрыв связей С—С. Это очень важно, так
как вероятность реакций разрыва цепи выше, чем для реакций
дегидрирования: энергия связи С—С составляет 263 кДж, а
энергия связи С—Н равна 358 кДж.
В качестве катализаторов дегидрирования применяют окси-
ды металлов (Сг2О3, MgO, Fe2O3, ZnO, МоО3 и др.). Их ак-
тивность зависит от состава и способа приготовления, они чув-
ствительны к ядам, содержащимся в сырье (органические со-
единения серы и др.). При дегидрировании наряду с основной
протекают другие, последовательные и параллельные реакции
(крекинг, полимеризация,* гидрогенолиз под действием выде-
лившегося водорода, циклизация и дегидроконденсация), при-
водящие к образованию смолистых веществ и кокса. Регенера-
цию катализаторов проводят, выжигая кокс при температурах,
исключающих их оплавление и дезактивирование.
Селективность процесса определяется рядом факторов —
природой катализатора, температурой, временем контакта.
Обычно селективность составляет 75—80% при степени конвер-
сии 20—40% (однократный проход через реактор) и времени
контакта в несколько секунд.
Важная роль в гетерогеннокаталитических реакциях дегид-
рирования отводится хемосорбции углеводородов на активных
77
центрах катализатора (А)
RH4-A A—H---R 4=t AH-j-AR
(или А—R---H)
т. е. в адсорбированной молекуле ослабляются или разрывают-
ся связи С—Н. Далее протекает взаимодействие двух хемо-
сорбированных частиц или взаимодействие хемосорбированной
частицы с физически адсорбированной или с налетающей из
объема; при этом каждая элементарная стадия является обра-
тимой:
СН3
I -f-A —ЗА
AH-f-A-CHR <=> А---Н2 + GH2—CHR CH2=CHR+H2
При дегидрировании изобутана образуется только один уг-
леводород—изобутилен, при дегидрировании н-бутана образу-
ются два изомера — бутен-1 и бутен-2, из изопентана получа-
ется смесь трех изомеров, а образующиеся бутилены и амилены
способны к дегидрированию до диенов.
Значительную роль при дегидрировании изопентана и изо-
амиленов играют реакции изомеризации, результатом которых
является образование н-пентанов и н-пентенов, превращающих-
ся в пиперилен (стр. 87).
1.2. Производство бутадиена двухстадийным
дегидрированием н-бутана
В настоящее время важнейшим способом получения бутади-
ена-1,3 является каталитическое дегидрирование н-бутана и
н-бутилена, получаемых из природного газа и газов с нефтепе-
рерабатывающих заводов. Существуют два способа получения
бутадиена из н-бутана: 1) дегидрирование н-бутана до н-бути-
лена (первая ступень), который затем дегидрируют до бутади-
ена (вторая ступень); 2) дегидрирование в одну стадию — н-бу-
тан сразу дегидрируют до бутадиена.
Дегидрирование н-бутана в бутилены идет с поглощением
тепла:
-н2
н-С4Н10 —- СН3—СН2—СН=СН2 ДЯ = 132 кДж/моль
бутен-1
5697,2
1П Кр = — 2,013IT -0,0459
-Н2
н-С4Н10 СН3—СН=СН— СН3 ДЯ = 117 кДж/моль
цис-бутен-2
5591 5
In Кр = —— 2,0131Т — 0,0459
78
«-C4H10 -* л CH3—CH=CH—СНЯ ДЯ = 115 кДж/моль
шранс-бутея-2
5484,2
In Кр = —— 2,01317' — 0,0459
Основные факторы процесса — температура, давление, вре-
мя контакта, содержание примесей в сырье и материал реакто-
ра. Дегидрирование протекает с хорошим выходом уже при ат-
мосферном давлении, причем при пониженном давлении требу-
ется более низкая температура. Процесс обычно проводят при
530—600°C. Время контакта зависит от температуры. Для каж-
дой температуры имеется свое оптимальное время контакта
(1—2 с), а с повышением температуры оно уменьшается. При
550—575 °C оптимальное время контакта составляет 2 с, что
соответствует объемной скорости подачи сырья 300—500 ч-1.
Из-за большого теплового эффекта при дегидрировании «-бута-
на необходимо интенсивно подводить тепло в зону реакции. Это
осуществить довольно трудно, из-за чего степень конверсии
«-бутана в промышленных условиях не превышает 50%.
В качестве промышленных катализаторов дегидрирования
«-бутана применяют хром-алюминиевые катализаторы, продо-
тированные едким кали, а также оксидами магния, цинка, бе-
риллия и циркония. Оксид алюминия является носителем.
В процессе реакции на катализаторе отлагается кокс (за 1 ч
до 20% углерода от массы катализатора), поэтому требуется
его регенерация. Кокс выжигают при подаче воздуха или ды-
мовых газов, разбавленных воздухом. В результате активность
катализатора полностью восстанавливается.
Дегидрирование бутиленов также идет с поглощением
тепла:
-на
СН3-СН2—СН=СН2 СН2=СН—СН=СН2
бутен-1
ДЯ =110 кДж/моль
-На
СН3—СН=СН-СН, СН2=СН—СН=СНа
цис-бутен-2
ДЯ =117 кДж/моль
-На
СН3-СН=СН-СН3 СН2=СН— СН=СН2
транс-бутен-2
ДЯ = 122 кДж/моль
Тепло подводят, либо нагревая заполненные катализатором
трубки, либо при помощи теплоносителя — водяного пара, вво-
димого в реактор в большом избытке (тогда наружный подо-
грев не требуется). В любом случае катализатор должен быть
стойким к действию водяного пара.
79
Выход бутадиена зависит от температуры и давления: в ин-
тервале 500—700 К концентрация бутадиена в газовой смеси
меняется от 5 до 26% при 0,1 МПа и от 14,5 до 43,5% при
0,01 МПа (степень конверсии бутиленов соответственно равна
5,5—35,5 и 17—77%). Для достижения больших выходов бута-
диена требуется проводить реакцию при высоких температурах
и низких давлениях. Парциальное давление углеводородов
снижают, подавая водяной пар; он одновременно препятствует
протеканию побочных реакций крекинга и полимеризации бута-
диена. Кроме того, при большом избытке водяного пара умень-
шается отложение кокса на катализаторе. Обычно в реактор
подают перегретый до 700 °C водяной пар в количестве 10—
15 моль на 1 моль бутиленов, которые нагревают до 530 °C.
Для дегидрирования бутиленов в промышленности применя-
ют катализатор, состоящий из оксидов железа, хрома и калия.
Он долго работает без регенерации из-за незначительного от-
ложения кокса, на нем можно получать бутадиен в количестве,
превышающем 85% на разложенное сырье. Для регенерации
катализатора прекращают подачу сырья, но продолжают пода-
вать водяной пар. При соприкосновении с горячим коксом на
поверхности катализатора образуется водяной газ. Реакция
идет с поглощением тепла, поэтому температура не повышает-
ся. После регенерации вновь ведут дегидрирование. За рубежом
применяют катализатор следующего состава: 72,4% MgO, 18,4%
Fe2O3, 4,6% К2О и 4,6% CuO; в СССР применяют катализатор
К-16.
В последние годы для дегидрирования бутиленов применя-
ют кальций-никель-фосфатный катализатор, представляющий
в основном фосфаты никеля и кальция общей формулы
Ca8Ni(PO4)6 с добавкой примерно 2% оксида хрома. Катали-
затор содержит 32% кальция, 5% никеля, 18,7% фосфора и
7% воды. Его готовят в виде таблеток и применяют в непо-
движном слое. Дегидрирование проводят при объемной скоро-
сти подачи сырья, равной 125—175 ч”1, а также в присутствии
водяного пара при разбавлении им сырья в мольном соотноше-
нии 20 1. Степень конверсии бутиленов составляет 42—49%,
селективность равна 90—94%. Катализатор, как и К-16, тре-
бует периодической регенерации. Время контакта 15—30 мин,
регенерация длится тоже 15—30 мин. Активность никель-каль-
ций-фосфатного катализатора снижается с течением времени.
В первый период в реакторе поддерживают температуру 540—
550 °C, а затем постепенно повышают ее до 670 °C. Общая про-
должительность службы катализатора около 4 лет.
Производство бутадиена из бутана — многостадийный про-
цесс, наряду с дегидрированием включающий стадии разделе-
ния контактных газов и выделения бутадиена. При дегидриро-
вании бутана идет побочная изомеризация бутиленов в изо-
бутилен и бутана в изобутан, который при дегидрировании так-
же превращается в изобутилен. Источником изобутилена явля-
ьо
ется и изобутан, содержащийся в сырье (бутан, поступающий
с газобензиновых заводов, содержит до 5% изобутана). Изобу-
тилен следует выводить из системы, так как он, накапливаясь
в системе, загружает аппаратуру и снижает производительность
установок, а его присутствие в бутадиене недопустимо, потому
что изобутилен снижает скорость полимеризации бутадиена.
Поточная схема производства бутадиена из н-бутана приведе-
на ниже.
Технологическое оформление дегидрирования сопряжено с
трудностями, обусловленными экзотермичностью процесса, вы-
сокой температурой, кратковременностью периодов контакти-
рования. В СССР дегидрирование н-бутана осуществляют в ре-
акторах с псевдоожиженным слоем катализатора, а дегидриро-
вание бутиленов ведут в стационарном слое катализатора. Схе-
ма установки дегидрирования н-бутана приведена на рис. II.1.
к-Бутановая фракция поступает вначале в испаритель 9, за-
тем в теплообменник 8, где нагревается за счет тепла контакт-
ного газа, а оттуда направляется в печь 14 с излучающими
стенками, где нагревается до 550 °C. После этого фракция по-
ступает в реактор 7. В' реакторе сырье непрерывно контакти-
рует с катализатором, и при 580 °C идет дегидрирование. Обра-
зующийся газ уходит через двухступенчатый циклон 6 в верх-
ней части реактора. Улавливаемая пыль по спускным трубкам
циклона возвращается в слой катализатора.
Контактный газ охлаждается в теплообменнике 8, испари-
теле 9 и поступает в скруббер' 10, орошаемый водой. В скруб-
бере газ охлаждается и дополнительно очищается от пыли, за-
тем охлаждается в холодильнике 11 и после освобождения от
тяжелых углеводородов в сепараторе 12 уходит на разделение.
Отработанный катализатор в отпарной секции, расположен-
ной в нижней части реактора, освобождают от углеводородов
(продувают азотом) и по стояку подают в линию, по которой
к - Бутан
контактного___|
газа L
♦
Разделение
Бутан - Бутиленовых
смесей
Сухой газ
I
Извлечение
изоБутилена
Дегидрирование
Разделение
.контактного
Тяжелые углеводороды^^ ^аза
Легкие углеводороды
Выделение.
бутадиена
В
бутиленовая
фракция
Схема X. Переработка я-бутана в бутадиен
| Бутадиен^
6-252
81
Рис. II. 1. Технологическая схема дегидрирования н-бутана:
1 — электрофильтр; 2 — увлажнитель; 3 — котел-утилизатор; 4, 6 — циклоны; 5 — репе»
нератор; 7—реактор; 8 — теплообменник; 9 — испаритель; 10— скруббер; 11 — холодиль-
ник; /2 — сепаратор; 13 — топка; 14 — печь.
катализатор пневматически транспортируется подогретым воз-
духом в нижнюю часть регенератора. 5. В регенераторе выжи-
гают кокс горячим воздухом, подаваемым из топки 13, и нагре-
вают катализатор до нужной температуры (650°C). Регенери-
рованный катализатор непрерывно отводят по стояку в реак-
тор, а дымовые газы через двухступенчатый циклон 4 направ-
ляют в котел-утилизатор 3, в котором получают водяной пар.
Охлажденные в котле-утилизаторе до 300 °C дымовые газы
идут в увлажнитель 2, орошаемый водным конденсатом. Окон-
чательная очистка газа от катализатора проводится в электро-
фильтре 1. Катализаторную пыль возвращают (воздухом) в ре-
генератор, а дымовые газы выбрасывают в атмосферу.
Чтобы восстановить каталитическую активность катализато-
ра, образующийся неактивный оксид шестивалентного хрома
восстанавливают в оксид трехвалентного хрома. С этой целью
регенерированный катализатор проходит зону восстановления
в нижней части регенератора. При восстановлении выделяется
вода, неблагоприятно влияющая на процесс, поэтому после зо-
ны восстановления катализатор проходит зону десорбции (азо-
том) продуктов восстановления и поступает в напорный стояк,
по которому транспортируется в реактор.
Температурный режим реактора регулируют, изменяя коли-
чество поступающего катализатора и температуру нагревания
сырья в печи. В регенераторе поддерживают нужную темпера-
туру, подавая воздух.
82
Суммарный выход н-бутиленов и бутадиена составляет 35%
на пропущенное сырье и 85% на разложенное; в том числе об-
разуется 78,5% н-бутиленов и 6,5% бутадиена.
При разделении контактного газа из бутан-бутиленовой
фракции извлекают легкие углеводороды (Ci—Сз), водород и
СО с помощью абсорбента — смеси пентана, бензола, толуола
и ксилола. Водород, углеводороды Ci—С3 и СО частично ис-
пользуют для транспортирования катализатора, а остальное
количество этих газов передают в топливную сеть.
После десорбции бутан^бутиленовая фракция с содержанием
бутана примерно 60% идет на экстрактивную дистилляцию с
ацетоном (эти процессы рассмотрены в § 3); в результате выде-
ляют н-бутан, возвращаемый на дегидрирование, а концентри-
рованные бутилены направляют на производство бутадиена.
Дегидрирование бутиленов в промышленности осуществля-
ют в реакторах адиабатического типа; тепло, необходимое для
дегидрирования, отнимают от поступающих в эти аппараты во-
дяного пара и углеводородов. Цикл контактирования (6 ч) сме-
няется циклом регенерации катализатора (1 ч). Срок службы
катализатора К-16 равен 900 ч.
Сырье (к-бутилены) подают в испаритель 1 (рис. II.2) и
после перегревателя 2 направляют в печь 5, где оно нагревает-
ся до 450 °C. Водяной пар из котла-утилизатора 5 перегревают
в печи до 750 °C и смешивают с сырьем на входе в реактор 4
(мольное соотношение пара и бутиленов равно И 1). Темпе-
ратура паро-бутиленовой смеси перед слоем катализатора рав-
на 630 °C; смесь проходит слой катализатора сверху вниз. Объ-
емная скорость подачи бутиленовой фракции 800 ч-1. На выхо-
де из реактора контактный газ, имеющий температуру 585 °C,
подвергается «закалке» — быстрому охлаждению до 530 °C пу-
тем впрыска воды (это необходимо для предотвращения по-
бочных реакций разложения и уплотнения). Затем газовая
Вода
Рис. II.2. Технологическая схема дегидрирования бутиленов:
I — испаритель; 2 — перегреватель; 3 — печь; 4 — реактор; 5 — котел-утилизатор; 6, 8 —
скрубберы; 7 — конденсатор.
я
61
83
смесь охлаждается до 250 °C в котле-утилизаторе 5, куда посту-
пает очищенная на угольных фильтрах вода. Получаемый пар
направляют в трубчатую печь 3 вместе с паром из заводской
магистрали.
Паро-газовая смесь после котла-утилизатора охлаждается
в скруббере 6 до 105 °C оборотной водой. В первом скруббере
еще не происходит конденсации водяного пара. Он конденсиру-
ется в конденсаторе 7, откуда возвращается на «закалку» кон-
тактного газа. Окончательное охлаждение газа (до 45 °C) про-
исходит во втором скруббере 8, тоже орошаемом оборотной во-
дой, после чего контактный газ уходит на разделение.
Выход бутадиена составляет 17% на пропущенные бутилены
и 80% на разложенные бутилены. Степень конверсии исходных
бутиленов равна 22%.
Разделение контактного газа сводится к выделению бути-
лен-бутадиеновой фракции, из которой бутадиен извлекают ам-
миачным раствором нитрата одновалентной меди, а бутилены
возвращают на дегидрирование.
Двухстадийный процесс производства бутадиена является
сложным, а процессы разделения контактных газов громоздки;
кроме того, при этом методе расходуется большое количество
водяного пара. Выход бутадиена теоретически должен состав-
лять (80-85) 100 = 68%; практически же из-за неполного из-
влечения бутилена и бутадиена из их смесей выход не превы-
шает 50—56% на израсходованный бутан.
1.3. Производство бутадиена одностадийным
дегидрированием н-бутана
Процесс одностадийного дегидрирования проводят в реакто-
рах со стационарным слоем оксидного хромового катализатора.
Разбавление сырья водяным паром недопустимо, так как при
этом отравляется катализатор, поэтому процесс ведут в ваку-
уме. Гранулированный катализатор, содержащий 18—20% ок-
сида хрома, смешивают с инертным теплоносителем (шарики
из плавленого оксида алюминия) в соотношении 1 :3 и загру-
жают в реактор слоем высотой 0,9—1,2 м. Теплоноситель дол-
жен иметь высокие теплоемкость и плотность.
Одностадийное дегидрирование — регенеративный процесс.
Вследствие его экзотермичности температура в период дегидри-
рования понижается на 20—40 °C. В период регенерации потери
тепла восполняются за счет выжигания кокса. Продолжитель-
ность дегидрирования определяется, следовательно, количест-
вом тепла, аккумулированным во время регенерации катализа-
тора. В действительности тепла, выделяющегося при выжига-
нии кокса, не хватает для восполнения потерь, поэтому в пери-
од регенерации в реактор подают и часть топлива; тепло от
сгорания кокса и топлива аккумулируется в основном инерт-
84
Рис. П.З. Технологическая схема одностадийного дегидрирования «-бутана
1 — подогреватель; 2 — печь; 3 — топка; 4, 5, 6 — реакторы; 7 — котел-утилизатор:
8 — стабилизационная колонна; 9, // — кипятильники; 10 — десорбер; 12, /5—холодиль-
ники; 13 — абсорбер; 14 — турбокомпрессор; 16 — масляный скруббер; 17 — «закалоч-
ный» аппарат.
ным носителем. Катализатор должен быть активным, прочным
и стабильным в работе, иметь хорошую регенерационную ха-
рактеристику. Эти требования предъявляются жесткими усло-
виями чередующихся окислительно-восстановительных циклов
дегидрирования и регенерации.
Реакторный блок состоит из трех или более аппаратов. Если
в первом идет дегидрирование, то во втором — регенерация,
а третий находится на переключении. Продолжительность каж-
дой операции 5—10 мин. Переключение реакторов с одной опе-
рации на другую осуществляется автоматически с помощью
таймерного устройства. Реакторы — цилиндрические горизон-
тальные аппараты диаметром 6 м и длиной 12—14 м, они из-
готовлены из углеродистой стали и футерованы огнеупорным
кирпичом. Решетки для катализатора изготовлены из керами-
ческих перфорированных плит.
На рис. II.3 приведена схема установки одностадийного де-
гидрирования н-бутана. Сырье через подогреватель 1 поступа-
ет в печь 2, откуда при 600—620 °C идет в один из реакторов
(4 или 5). Из реакторов контактный газ поступает на «закал-
ку» в аппарат 17, а затем в скруббер 16, где охлаждается хо-
лодным маслом. Охлажденный контактный газ турбокомпрессо-
ром 14 сжимают до 1,3 МПа и подают в абсорбер 13. С верха
абсорбера уходит водородсодержащий газ (топливный газ),
а абсорбированные углеводороды отделяются в десорбере 10.
С верха стабилизационной колонны 8 уходит фракция С3, а с
низа фракция С4 с 11 —13% бутадиена. Ее направляют на вы-
деление бутадиена.
85*
Средняя температура в реакторах 600 °C: в начале дегидри-
рования 610 °C, в конце 590 °C. Остаточное давление 0,133—
0,2 кПа. Содержание бутиленов в сырье 25—30%; степень кон-
версии примерно 20%; выход бутадиена 12—14% (на сырье);
селективность 50—57%. Расход н-бутана на 1 т бутадиена ра-
вен 1,9—2,2 т. По окончании цикла дегидрирования переклю-
чают поток сырья на другой реактор, а из первого отдувают уг-
леводороды и затем проводят регенерацию катализатора то-
почными газами с небольшим содержанием кислорода, пода-
ваемыми из топки 3. Тепло газов регенерации используют в
котле-утилизаторе 7. Продукты сгорания отсасывают паро-
струйными эжекторами, а в реактор вновь направляют поток
сырья.
Продолжительность операции: дегидрирование 5—9 мин,
продувка водяным паром 1,5—1,8 мин, регенерация катализа-
тора 5—9 мин, эвакуация регенерационных газов 1,5—2,7 мин.
Полный цикл длится 15—21 мин. Для характеристики процес-
са введена степень одностадийности. Степень одностадийности
выражают в процентах, она представляет собой отношение бу-
тиленов, содержащихся в контактном газе, к их количеству в
сырье и обычно составляет 100—102%.
Процесс практически не требует пара, а тепло, выделяюще-
еся при выжигании кокса, используют для проведения эндо-
термической реакции дегидрирования в адиабатическом регене-
ративном цикле, благодаря чему упрощается конструкция ре-
актора. К недостаткам процесса следует отнести невысокий вы-
ход бутадиена, применение сложной запорной арматуры и
средств автоматики.
1.4. Производство изопрена двухстадийным
дегидрированием изопентана
Способы каталитического дегидрирования бутана и бутиле-
нов можно применить и для дегидрирования изопентана и пен-
тенов с целью получения изопрена — ценнейшего мономера для
синтетического каучука. Изопреновый каучук является анало-
гом натурального каучука и даже превосходит его по некото-
рым свойствам. Изопрен применяют также как компонент сопо-
лимеризации при получении бутилкаучука.
Изопентан можно выделять из газового бензина, из бензина
прямой перегонки нефти и бензина каталитического крекинга.
Главным источником изопентенов служит соответствующая
фракция продуктов крекинга и пиролиза нефтяного сырья.
Дегидрирование изопентана в изопрен можно осуществить
в одну и в две стадии, подобно дегидрированию к-бутана.
В промышленности в настоящее время получают изопрен двух-
стадийным дегидрированием изопентана. Процесс одностадий-
ного дегидрирования пока не внедрен. При дегидрировании изо-
86
пентана образуется смесь трех изопентенов:
сн,
—► сн,=сн-сн-сн,
—н2 2 3
З-метилбутен-1
(т. кип. 20 °C)
СН3 СН3
СН3-СН-СН,-СН3--------->- СН,=С-СН,—сн3
—н2
2-метилбутен-1
(т. кип. 31,05 °C)
сн3
—-* сн3-с=сн-сн3
—“Я
2-метилбутен-2
(т. кип. 38,49 °C)
Разделить продукты дегидрирования изопентана довольно>
сложно, так как в изопентане имеется некоторое количество
н-пентана, а в продуктах (наряду с не вступившими в реакцию
«-пентаном и изопентаном) содержатся три изопентена, три
«-пентена (пентен-1, ч«Сппентен-2, транс-пентен-2), два изомера
пиперилена (цис- и транс-), изопрен и циклопентадиен.
Алюмо-хромовый катализатор, как было сказано ранее, об-
ладает изомеризующими свойствами, благодаря чему происхо-
дит частично изомеризация изопентана в н-пентан и изопенте-
нов в «-пентены. Они на второй стадии дегидрирования дают
пиперилен, который дегидроциклизуется в циклопентадиен:
СН3-СН2—СН2-СН=СН2 — । -на
СН3-СН=СН-СН2-СН3 —I
сн3-сн=сн-сн=сн2
Выход пиперилена достигает 10—15% от количества диенов.
Изопрен необходимо очищать от пиперилена, а также от ацети-
леновых углеводородов, так как все они уменьшают выход изо-
прена и впоследствии замедляют его полимеризацию.
Первая стадия дегидрирования проводится в псевдоожижен-
ном слое катализатора при 550—560 °C и скорости подачи жид-
кого сырья 1,5—2,0 ч*1. Выход изопентенов составляет 28—32%
на пропущенное сырье при селективности 66—71%. Для выде-
ления изопентенов из изопентан-пентеновой смеси применяют
экстрактивную дистилляцию с диметилформамидом. Все три
87
изопентена при дегидрировании дают изопрен (2-метилбута-
диен):
СН2=СН-СН-СН3 4= =fc СН2=С—СН=СН2 + н2 ДЯ = 123 кДж/моль
<!н3 сн2=с-сн2-сн3 СН3 СН2=С—СН=СН2 + н2 ДЯ = 132 кДж/моль
ii3 СН3-С=СН-СН3 4= <!н3 СН3 =* СН2=С—СН=СН2+Н2 it, ДЯ = 134 кДж/моль
Вторую стадию проводят в реакторах адиабатического типа
с неподвижным слоем катализатора при разбавлении сырья во-
дяным паром (мольное соотношение 1 :20). Используют катали-
заторы на основе оксида железа или кальций-никель-фосфатный
(КНФ). Температура 540—560°C (на КНФ 580—600°C), выход
изопрена до 33% (масс.) при селективности 80%.
Изопрен очень склонен к полимеризации, поэтому применяют
быстрое охлаждение контактных газов, сокращают время пре-
бывания изопентенов в реакторе, а при выделении изопрена
применяют ингибиторы (например, трет-бутилпирокатехин).
..Расход изопентана составляет 2 т на 1т получаемого изопрена.
В настоящее время ведутся поиски новых эффективных ката-
.лизаторов дегидрирования и более эффективных экстрагентов
для разделения контактного газа с первой ступени дегидрирова-
ния.
1.5. Производство изопрена из изобутилена
и формальдегида
Технология получения изопрена из изобутилена и формаль-
дегида через 4,4-диметилдиоксан-1,3 разработана в СССР.
Процесс двухстадийный. Первая стадия — взаимодействие
изобутилена и формальдегида в присутствии разбавленной сер-
-ной кислоты с образованием 4,4-диметилдиоксана-1,3:
С(СН3)а
Н,^ О
СН3—С=СН2 +2НСНО ------ £
сн3 Н2 \ /СН2
О
'На второй стадии диметилдиоксан разлагают на гетерогенных
фосфатных катализаторах при 360—380 °C:
С(СН3)2
Н2С О Ме2НР04
| I ------------>- СН2=С—СН=СН2 + НСНО + Н2О
Н2С СН2 I
\ / СН3
•38
Наряду с основной идут побочные реакции, в частности образов
вание гликоля, который может превращаться в диоксан:
сн3-с=сна + НСНО + Н2О
сн,
С(СН,),
он он / \
I | ч-нсно НаС О
сн3-с—сн2-сн2--------->- | |
I Нас сна
СН3 \/
З-метилбутандиол-1 а3
Побочными продуктами и в этом процессе являются пиперилен
и циклопентадиен. Последний образуется, если в сырье присут-
ствует бутадиен:
СН2
+2НСНО / уПк
СНа=СН-СН=СН2----------► СН2=СН-НС СН2
А А -н4,°
X
4-винилдиоксан-1,3
Сырьем для процесса является изобутилен-изобутановая
фракция после дегидрирования изобутана. При этом расход изо-
бутилена на 1 т изопрена составляет 1,23 т, а расход формаль-
дегида равен 0,83 т. Присутствие н-бутиленов увеличивает вы-
ход побочных продуктов: а-бутилен превращается в пиперилен,
а 0-бутилен — в смесь изопрена и изовалерианового альдегида:
СНа
+2НСНО CjHj—НС СН2
сн3—сна—сн=сн2------> И
---► СН3—СН=СН-СН=СН2 + НСНО
С(СН3)2 сн3
+2НСНО н2</ \ |—* СН2=С-СН=СН2
СН3-СН=СН-СН3------> | | =нсно 2
н2с сн2 ---> СН3-СН2-СН-СНО
V сн3
Поскольку циклопентадиен получается из бутадиена, содержа-
ние последнего в .изобутиленовой фракции не должно превы-
шать 0,5% (масс.).
Реакцию изобутилена с формальдегидом проводят при 100 °C
и 1—1,5 МПа. В этих условиях степень конверсии составляет
для изобутилена 88—92%, для формальдегида 92—96%. Выход
диоксана равен 66—68% на прореагировавший изобутилен. Ре-
актор представляет собой вертикальный трубчатый аппарат,
в межтрубное пространство которого подают воду для отвода
89
выделяющегося тепла. Изобутилен и водный формальдегид по-
дают противотоком.
Реакторный блок установки по получению диметилдиоксана
приведен на рис. II.4. Два реактора соединены последователь-
но. В каждую трубку подают изобутилен через сопло. К фор-
мальдегиду добагвляют 1—2% серной кислоты и подают его
в реактор. Реакционную смесь направляют на нейтрализацию и
промывку в колонну 3, а затем в отстойник-сепаратор 4. Вод-
ный и органический слои перерабатывают на двух раздельных
установках. Узел ректификации органического слоя состоит из
нескольких колонн, на которых последовательно выделяют изо-
бутилен (его возвращают в процесс), разные побочные продук-
ты и, наконец, диметилдиоксан, который подают на разложение.
Из водного слоя отгоняют сначала летучие, а затем формаль-
дегид в виде 8%-ного раствора, который после концентрирова-
ния до 35%-ного содержания НСНО возвращают в процесс.
На рис. II.5 изображен реакторный блок разложения диме-
тилдиоксана. Реакция эндотермична (тепловой эффект равен
125 кДж/моль); ее осуществляют в адиабатических горизон-
тальных реакторах с неподвижным слоем катализатора (на ос-
нове кислых и средних фосфатов кальция) при 370—390 °C. Раз-
ложение диметилдиоксана проводят в присутствии водяного
пара и с подпиткой фосфорной кислотой. Степень разбавления
диметилдиоксана паром равна 1 (2,0-?2,5), скорость подачи
диоксана 0,7 ч-1. Выход изопрена составляет 43—46% на про-
пущенный диоксин (47—50% на разложенный), степень конвер-
сии диметилдиоксана 90—95%.
Контактный газ из реакторов конденсируется в холодильни-
ках (водных и рассольных) и в отстойнике разделяется на два
слоя. Из водного слоя отпаривают органические продукты и
возвращают их в органический слой. Из органического слоя
выделяют изопрен-сырец, подвергаемый водной промывке и
азеотропной осушке. В колонны выделения изопрена и азеотроп-
Рис. П.4. Реакторный блок для получения диметилдиоксана:
1, 2 — реакторы; 3 — промывная колонна; 4 — отстойник-сепаратор.
S0
Рис. II.5. Реакторный блок для раз-
ложения диметилдиоксана в изо-
прен:
/, 2, 3 — реакторы; 4 — холодильник; 5 —
отстойник-сепаратор.
Фосфаты Пар
Пар
Фосфаты
Фосфаты
Пар
Органический
слой
Водный
слой
ной осушки вводят ингибиторы (нитрит натрия, фурфурол) для
предотвращения полимеризации изопрена.
Катализатор разложения диоксана постепенно зауглерожи-
вается, поэтому через 3—4 ч работы приходится выжигать кокс.
Еще один существенный недостаток процесса — большие за-
траты на переработку водного слоя. Процесс можно значитель-
но усовершенствовать, если применять концентрированный фор-
мальдегид. Это позволит исключить побочные реакции гидроли-
за и этерификации, отказаться от переработки водного слоя. За-
дача заключается в разработке стабилизаторов концентриро-
ванных водных растворов формальдегида, из которых формаль-
дегид выделяют в виде твердого полимера.
1.6. Производство изопрена из пропилена
Процесс получения изопрена из пропилена — трехстадийный.
Первая стадия — димеризация пропилена в 2-метилпентен-1, а
вторая — изомеризация 2-метилпентена-1 в 2-мет.илпентен-2:
2СН3—СН=СН2 ► СН2=С-СН2—СН2-СН3 -------► СН3-С=СН-СН2-СНЭ
О А £ । а А о О । & V
СН3 СН3
На третьей стадии проводят деметанизацию 2-метилпентена-2 с
получением изопрена (2-метилбутадиена-1,3):
СН.-С=СН-СН,—сн3 —»- сн2=с-сн=сн2
I ~СН1 I
сн, сн3
В промышленности димеризацию пропилена проводят в при-
сутствии триизобутил- или три-к-пропилалюминия при 150—
200 °C и 20—30 МПа. В этом случае образуется преимуществен-
91
но 2-метилпентен-1 (97%) при степени конверсии пропилена
70—85%. Процесс ведут в двухступенчатом трубчатом реакторе
с рециркуляцией катализатора. Масса из реактора поступает
в колонну однократного испарения. Там отделяют катализатор
и пропилен, возвращаемые в процесс.
2-Метилпентен-1 подвергают изомеризации в 2-метилпен-
тен-2 при 200 °C в присутствии катализаторов кислотного
характера и комплексных катализаторов (алюмосиликат,
фосфорная кислота на носителе, диизобутилалюминийхло-
рид + ди-трет-бутилбензоат никеля). Степень конверсии 46—
60%, селективность 95% •
Деметанизацию (пиролиз) осуществляют в трубчатой печи
при 700 °C, времени контакта 0,3—0,5 сив присутствии водя-
ного пара. Процесс можно инициировать, вводя в сырье, на-
пример, бромистый водород, но он сильно корродирует аппа-
ратуру. Степень деметанизации димера пропилена в изопрен
в условиях неинициированного пиролиза составляет 35%, а
селективность равна 40—46% (мольн.). В случае добавки 0,05—
0,5 моль НВг на 1 моль димера селективность повышается до
65%. Разделение продуктов пиролиза является сложной зада-
чей, так как помимо изопрена и метана в них содержатся
углеводороды С4 и С5, в том числе пиперилен, гексадиен и бо-
лее тяжелые. После «закалки» водой эту смесь разделяют.
Первые две стадии не являются лимитирующими, идут с вы-
сокими выходом и селективностью. Поэтому для совершенство-
вания получения изопрена этим методом нужен подбор эффек-
тивных инициаторов пиролиза с целью повышения селективно-
сти этой стадии.
1.7. Производство стирола и а-метил стирол а
Важнейшими «ароматическими» мономерами для синтетиче-
ского каучука являются стирол и а-метилстирол, получаемые
дегидрированием соответственно этилбензола и изопропилбен-
зола.
Аналогичным путем при дегидрировании этилтолуола можно
получить винилтолуол. При дегидрировании диэтилбензола об-
разуется дивинилбензол
СН2=НС—'^^-СН=СН2
применяемый в синтезе ионообменных смол.
Каталитический процесс дегидрирования этилбензола вклю-
чает собственно дегидрирование, а затем выделение и очистку
стирола. Наряду с основной реакцией
СвН5-С2Н5 —> СвН5-СН=СН2
—н2
92
идут побочные, приводящие к бензолу, этилену, толуолу и ме-
тану:
СвН5—С2Н5 —
--->
+Н2
С6Нв + СН2=СН2
С6Н5-СН3 + СН4
Кроме того, образуются замещенные стильбены
СН3—СН2-СвН4—сн=сн—С6Нб
и другие соединения. Осаждаясь на поверхности катализатора,
они снижают его активность.
При дегидрировании изопропилбензола аналогично протека-
ют основная и побочные реакции:
С6Н5—СН(СН3)2 -
С6Н5-СН=СН2 + СН4
С6Н5-СН3 + СН2=СН2
СвНв + сн3—сн=сн2
Реакции дегидрирования сопровождаются увеличением объ-
ема, поэтому им благоприятствует понижение давления. Чтобы
уменьшить парциальное давление углеводородов, в систему вводят
водяной пар. При 630 °C и атмосферном давлении в равновесной
смеси содержится 25—30% стирола. При снижении парциально-
го давления углеводородов до 0,01 МПа и подаче пара (2,6 кг
на 1 кг этилбензола) выход стирола повышается до 70—80%.
Пар не только снижает парциальное давление углеводородов,
но и является теплоносителем. *
В качестве основных компонентов катализаторов при дегид-
рировании этилбензола применяют оксиды магния, железа, цин-
ка, меди, бериллия и др. Общим для всех этих катализаторов
является небольшая добавка солей калия (например, К2СО3),
присутствие которых ускоряет взаимодействие воды и углерода,
отлагающегося на катализаторе, с образованием СО2, т. е. одно-
временно идет и регенерация катализатора. Применение таких
саморегенерируемых катализаторов позволяет ввести процесс
непрерывно. Пригодны для этой цели и катализаторы, приме-
няемые для дегидрирования бутиленов.
В промышленности для дегидрирования этилбензола получил
распространение катализатор, называемый стирол-контакт и
имеющий следующий состав: 85% ZnO, 5% СаО, 5% K2SO4,
3% КгСгО4, 2% КОН. Этот катализатор требует регенерации.
Катализатор на основе оксида железа, состоящий из 72,4% MgO,
4,2% Fe2O3 и 4,8% CuO (остальное — оксид алюминия), не тре-
бует регенерации. Хорошие результаты дает катализатор из 80%
ZnO, 5—7% СаО, 10% А12О3, 2—3% К2О и 0,5—0,7% Сг2О3.
Срок службы этого катализатора 8—12 мес. На свежем катали-
заторе процесс ведут при 560—580 °C, а к концу срока службы
катализатора повышают температуру до 600 °C.
Дегидрирование этил- и изопропилбензола ведут в реакторах
адиабатического и трубчатого типа. Последние по принципу
93
Рис. II.6. Технологическая схема дегидрирования этилбензола:
/ — реактор; 2 — перегреватель; 3 — испаритель; 4 — котел-утилизатор; 5 — конденсатора
6 — сепаратор; 7 — отстойник; 8 — осушитель; 9 — аппарат с мешалкой.
действия напоминают адиабатические реакторы, используемые
при дегидрировании к-бутиленов. Достоинство реакторов этого
типа — большая производительность.
При выделении и очистке стирола возникает много трудно-
стей, обусловленных близостью температур кипения этилбензола
и стирола и склонностью стирола к самопроизвольной полиме-
ризации. Фракционирование должно быть очень четким, так как
стирол, бензол и толуол должны быть выделены в чистом виде.
Стирол перегоняют при большом разрежении (остаточное дав-
ление 0,4 кПа; при этом т. кип/стирола 54 °C) в колоннах спе-
циальной конструкции. Для предотвращения полимеризации
к стиролу добавляют стабилизатор — гидрохинон.
Стирол представляет собой бесцветную прозрачную жид-
кость с приятным запахом (т. кип. 145,2 °C при атмосферном
давлении).
В отличие от стирола, а-метилстирол (т. кип. 161—162 °C
при атмосферном давлении) не способен к полимеризации, но
сополимеризуется с бутадиеном и другими виниловыми мономе-
рами.
Дегидрирование изопропилбензола с целью получения а-ме-
тилстирола осуществляют в тех же условиях и на тех же ката-
лизаторах, что и при дегидрировании этилбензола. Такая же
степень превращения изопропилбензола достигается при более
низкой температуре. Аппаратурное оформление обоих процессов
практически одинаково.
Парциальное давление углеводорода в реакторе равно
0,01 МПа; мольное соотношение водяного пара и углеводорода
составляет 17 1. Продукты дегидрирования этилбензола содер-
жат 37% стирола. В случае дегидрирования изопропилбензола
примерно таков же выход а-метилстирола.
Технологическая схема дегидрирования этилбензола приве-
дена на рис. II.6. Этилбензол подают в испаритель 3 вместе
с водяным паром, поступающим из котла-утилизатора 4. Нагре-
94
тая до 150 °C смесь идет в перегреватель 2, откуда при 520—
530 °C направляется в адиабатический реактор 1. Тепло, необ-
ходимое для реакции, подводят перегретым водяным паром.
Реакционные газы подают вначале в перегреватель 2, где их
теплом нагревают водяной пар. Затем газы охлаждают в испа-
рителе 3 и в котле-утилизаторе 4. Водяные пары конденсируют-
ся в конденсаторе 5. Несконденсированные газы отделяются в
сепараторе 6 и поступают в топливную сеть, а водный и углево-
дородный конденсаты идут в отстойник 7. Вода уходит из от-
стойника в ловушку, а углеводородный слой осушают хлоридом
кальция в осушителе S, смешивают в аппарате 9 с гидрохино-
ном (ингибитор) и направляют на ректификацию. При регене-
рации катализатора вместе с паром подают воздух. Газы, обра-
зующиеся при регенерации, сбрасывают в атмосферу.
Основные показатели процессов производства стирола и
а-метилстирола таковы:
Показатель Дегидрирование
стирола
Катализатор
Температура, °C
Объемная скорость подачи
сырья, ч-1................
Мольное соотношение уг-
леводорода и водяного
пара ...
Степень конверсии сырья,
% •
Селективность, %
Выход, % (масс.) на про-
пущенное сырье
Стирол-контакт
600-615
0,35-0,5
1:15
38-40
90—92
34—36
Дегидрирование
изопропил-
бензола
К-12
550-580
0,5
1:20
42-43
90-92
37—39
Стирол выделяют в вакуумных колоннах при остаточном
давлении 0,4 кПа, вводя в систему ингибитор для подавления
самопроизвольной полимеризации стирола. Установки по выде-
лению и очистке для стирола и а-метилстирола одинаковы.
§ 2. ПРОИЗВОДСТВО ВИНИЛОВЫХ МОНОМЕРОВ
С ФУНКЦИОНАЛЬНЫМИ ГРУППАМИ
Здесь рассмотрены процессы производства таких мономеров,
которые наряду с винильной группой содержат те или иные
функциональные группы, придающие впоследствии получаемым
полимерам специфические свойства. К таким мономерам отнесе-
ны винилхлорид, акрилонитрил, винилацетат, акриловая кисло-
та и ее эфиры. Все они играют огромную роль в производстве
полимерных материалов реакциями полимеризации, т. е. высту-
пают в роли виниловых мономеров. Поэтому производство ви-
нилацетата, акриловой кислоты и ее эфиров и рассмотрено
здесь, а не в гл. III, посвященной кислородсодержащим продук-
там.
95
2. 1. Производство винилхлорида
Винилхлорид (СН2 = СНС1) при обычных условиях представ-
ляет собой газ (т. конд. —13,9°C). Применяется для получения
поливинилхлорида — одного из самых распространенных пла-
стиков.
В промышленности винилхлорид получают гидрохлорирова-
нием ацетилена и дегидрохлорированием дихлорэтана.
Гидрохлорирование ацетилена— экзотермический процесс:
СН=СН + НС1 СН2=СНС1 ДЯ = —109 кДж/моль
Его можно осуществить в газовой и жидкой фазе.
Процесс в газовой фазе проводят при 120—180 °C на актив-
ном угле, пропитанном сулемой (хлорид двухвалентной ртути).
Очищенный и осушенный ацетилен смешивают с осушенным
хлористым водородом и подают смесь в угольный фильтр 1 для
отделения примесей хлора, содержащихся в НС1 (рис. 11.7). Из
фильтра газы поступают в реактор 2 и проходят сверху вниз
по трубкам, заполненным катализатором. По межтрубному про-
странству циркулирует теплоноситель (масло) при 120 °C. На
свежем катализаторе гидрохлорирование протекает при 120 °C,
а по мере потери активности постепенно поднимают температу-
ру до 200 °C. Отработанный катализатор выгружают и заменя-
ют свежим (при потере активности в контактных газах появ-
ляется ацетилен). Срок службы катализатора 3—4 мес. Хлори-
стый водород берут в небольшом избытке (5—10%) по отноше-
нию к ацетилену. Газы не должны содержать ни хлора, так как
он реагирует с ацетиленом со взрывом, ни влаги, приводящей к
образованию ацетальдегида. В качестве побочного продукта об-
Рис. II.7. Технологическая схема получения винилхлорида лз ацетилена:
1 — фильтр; 2 —реактор; 3, 7, 11, /3 — холодильники; 4—адсорбер; 5, б — промывные
колонны; 8 — осушительная колонна; 9 — ректификационная колонна; 10 — конденсатор;
12 — отпарная колонна.
96
разуется небольшое количество 1,1-дихлорэтана:
CHa=CHCl-f-HCl ► СН3—CHClj
Паро-газовая смесь из реактора проходит водяной холодиль-
ник 3 и поступает в заполненный активным углем адсорбер 4.
Там улавливаются пары сулемы, которую вновь используют для
приготовления катализатора. Смесь, выходящая из реактора,
содержит 93% винилхлорида, 5% хлористого водорода, 0,5%
ацетилена, 0,3% 1,1-дихлорэтана и примерно 0,3% ацетальдеги-
да. Степень конверсии ацетилена за один проход через реактор
составляет 97—98%. Хлористый водород поглощают водой и
колонне 5 (с получением концентрированной соляной кислоты),
а остатки его улавливают щелочью в колонне 6, где отмывают
и ацетальдегид. Газовый поток охлаждается в рассольном холо-
дильнике 7; при этом влага либо конденсируется, либо вымора-
живается. Доосушку газов осуществляют в колонне 8 твердой,
щелочью.
С верха ректификационной колонны 9 уходят винилхлорид и
ацетилен. В рассольном конденсаторе 10 винилхлорид конденси-
руется и частично идет на орошение колонны 9, а основная мас-
са поступает в отпарную колонну 12 вместе с ацетиленом. С ни-
за колонны 9 выводят дихлорэтан через холодильник 11. Верх-
няя часть колонны 12 представляет собой рассольный холо-
дильник, где конденсируется уносимый ацетиленом винилхло-
рид. Ацетилен и инертные газы отводят с верха колонны, а
с низа через холодильник 13 отбирают очищенный жидкий ви-
нилхлорид.
Расход на 1 т винилхлорида составляет: 0,45 т ацетилена,
0,67 т хлористого водорода, 0,2—0,5 кг хлорида ртути.
При жидкофазном процессе гидрохлорирования ацетилена
в качестве катализатора применяют раствор хлорида однова-
лентной меди C112CI2 и хлорида аммония в 12—15%-ной соля-
ной кислоте. Процесс проводят при 60 °C.
При промышленном получении винилхлорида дегидрохлори-
рованием дихлорэтана процесс проводят либо при помощи спир-
товой щелочи
60-70 °с:
СНаС1-СНаС1 + NaOH-----СНа=СНС1 + NaCl +Н2О
либо пиролизом при 480—520 °C на активном угле:
СН,С1-СН2С1 ——> СН2=СНС1
Процесс со щелочью периодический; расход щелочи 0,82 т на
1 т винилхлорида. Пиролиз осуществляют как непрерывный
процесс в трубчатом реакторе. Выход винилхлорида 88%.
Известны также способ окислительного хлорирования этиле-
на хлористым водородом
375—425 °C; СиО
СНа=СНа + НС1 + 0,5Оа---_н о СНа=СНС1
9Т
7—254
и способ высокотемпературного аномального хлорирования эти-
лена
СН2=СН2 —> СН2=СНС1
—пи
когда выход винилхлорида существенно зависит от температу-
ры. Так, при 280°C он составляет 50%, а при 500°C повышается
до 93%.
Разработан сбалансированный по хлору процесс получения
винилхлорида из этилена, включающий хлорирование этилена,
пиролиз 1,2-дихлорэтана и окислительное хлорирование этилена.
2.2. Производство акрилонитрила
Акрилонитрил (винилцианид, нитрил акриловой кислоты)
CH2=CHCN—бесцветная жидкость (т. кип. 78,5 °C, т. затв.
—82°C). Акрилонитрил вызывает небольшую коррозию стали.
Он хорошо растворяется в органических растворителях и в воде
(растворимость в воде при 20 °C составляет 7,3%, раствори-
мость воды в акрилонитриле 3,1%). С водой образует азеотроп-
ную смесь, содержащую 12% воды и кипящую при 71 °C.
Акрилонитрил является ценным мономером, имеющим боль-
шое практическое значение в производстве сополимерных бута-
диен-нитрильных каучуков и синтетического волокна нитрон.
В связи с этим повышается интерес <к способам его производ-
ства.
Например, его получают из этилена многостадийным процес-
сом:
4-0,5Оа +HCN
СН2=СН2 ---> Н2С СН3 -----> НОСН2—CH2CN CH2=CHCN
этиленциангидрин]
Существует и прямой синтез акрилонитрила, заключающий-
ся во взаимодействии ацетилена с синильной кислотой:
CH=CH + HCN ---► CH,=CHCN
Реакцию проводят в присутствии катализатора, представляю-
щего собой водный раствор хлорида одновалентной меди и хло-
рида щелочного металла, подкисленный соляной кислотой.
Кроме основной реакции протекают и побочные:
а) гидратация ацетилена в ацетальдегид, дающий с синиль-
ной кислотой лактонитрил (нитрил молочной кислоты):
+Н2о +HCN
СН=СН------► СН3-СНО ----> GH3—CHCN
ОН
98
б) димеризация ацетилена в винилацетилен и его дальней-
шее уплотнение с образованием полимеров:
2СН=СН
Gisela
сн2=сн-с=сн
4-сн=сн
------>
--► СН2=СН—С=С—СН=СН2 " * Полимеры
в) взаимодействие винилацетилена с хлористым водородом
и синильной кислотой с образованием хлоропрена и 1-*цианбута-
диена-1,3:
4-нс1
--->- СН2=СС1—СН=СН2
СН2=СН-С=СН-
+HCN
NC— СН=СН-СН=СН2
Все эти продукты, взаимодействуя между собой, образуют смо-
лообразные вещества, в присутствии -которых активность ката-
лизатора понижается.
Прямой синтез акрилонитрила проводят в вертикальном ци-
линдрическом реакторе диаметром 1,7 м и высотой 7,5 м. Внут-
реннюю поверхность аппарата гуммируют, а поверх резины фу-
теруют кислотоупорными плитками. Температура процесса 80—
90 °C, давление 0,02—0,04 МПа. Выход акрилонитрила состав-
ляет 75% на ацетилен и 85% на цианистый водород.
Осуществлен также процесс получения акрилонитрила из
ацетальдегида и синильной кислоты, протекающий в две стадии.
Вначале при 10—18 °C получают лактонитрил
СН3—СНО + HCN ► СН3—CHCN
Ан
а затем подвергают его дегидратации. Для этого смешивают
лактонитрил с 80—85%-ной фосфорной кислотой в мольном со-
отношении 3: 1 и впрыскивают смесь в реакционную зону, где
за 1 с она нагревается до 600—700 °C. На выходе из реактора
контактный газ подвергают «закалке» рециркулирующим пото-
ком 30%-.ной фосфорной кислоты. В этих условиях примерно
75% лактонитрила превращается в акрилонитрил:
СН3—CHCN ——CH2=CHCN
А-н3о
н
Выход акрилонитрила составляет 90% на прореагировавший
ацетальдегид и 92% на синильную кислоту.
Наибольший интерес представляет получение акрилонитрила
окислительным аммонолизом пропилена
СН2=СН—СН3 + NH3 + 1,5Оа —* CH2=CHCN
осуществляемым в одну и в две стадии. В двухстадийном про-
цессе вначале окисляют пропилен в акролеин
СН2=СН-СН3 + О2 СН2=СН-СНО
7*
99
а затем, действуя совместно аммиаком и кислородом, превра-
щают его в акрилонитрил:
СН2=СН—СНО + NH3 4-0,50. ——> CH2=CHCN
— 2Н«|О
В обоих случаях (в одно- и в двухстадийном процессе) синтез
проводят при низких давлениях (0,1—0,3 МПа) в присутствии
катализаторов — оксидов вольфрама, кальция, магния, молиб-
дена. В промышленности нашел применение фосформолибдат
висмута на носителе. Температура 450 °C, время контакта 1—
10 с. На состав продуктов реакции, особенно при одностадийном
процессе, существенно влияет соотношение исходных компонен-
тов. Рекомендуется строго поддерживать стехиометрические со-
отношения, т. е. С3Н6: NH3: О2 = 1 1:1,5. Помимо акрилони-
трила образуются побочные продукты — ацетонитрил, циани-
стый водород, акролеин и др. Часть пропилена сгорает до СО2,
я некоторое количество расщепляется:
СН2=СН—СН3 + NH3 + 2,5Оа -► CH3CN + СОа + ЗНаО
СНа=СН—СН8 + Оа ---► СН2=СН-СНО + Н2О
СНа=СН-СН8 + NH3 + 2Оа--► HCN + СН4 + СО2 + 2НЯО
СН2=СН—СНя + 4,5Оа--► ЗСО2 + ЗН2О
Реакции окисления, протекающие при синтезе акрилонитрила,
экзотермичны, поэтому возникают технологические трудности,
обусловленные необходимостью отводить тепло из зоны реак-
ции.
Окислительный аммонолиз можно проводить в реакторах
со стационарным и с псевдоожиженным слоем катализатора. Из
реактора с псевдоожиженным слоем проще отводить тепло (за
счет змеевиков, установленных в реакторе) и обеспечивать изо-
термичность процесса. Однако селективность процесса и степень
конверсии пропилена снижаются вследствие внутриреакторного
’перемешивания и проскока пропилена.
Технологическая схема процесса с псевдоожиженным слоем
катализатора приведена на рис. II.8. Пропилен, аммиак, воздух
и водяной пар подают в реактор 1 под распределительную
решетку. Температура 450 °C; объемная скорость подачи сырье-
вой смеси 500—600 ч-1; давление до 0,3 МПа. Газы из реакто-
ра охлаждаются в котле-утилизаторе 2 и поступают для улавли-
вания аммиака в абсорбер 3, орошаемый концентрированным
раствором сульфата аммония, содержащим свободную серную
кислоту. В колонне 4 газы промывают водой и подают в отпар-
ную колонну 5. С верха этой колонны уходит акрилонитрил-сы-
рец, а из куба отводят водный ацетонитрил, который выделяют
в колонне 6. Воду из колонны 6 возвращают в колонну 4. От
акрилонитрила-сырца в колонне 7 отгоняют синильную кислоту,
а окончательную очистку от примесей проводят в колонне 3, с
верха которой отводят акрилонитрил с концентрацией основного
вещества не менее 99%.
100
Рис. 11.8. Технологическая схема получения акрилонитрила окислительным
аммонолизом пропилена:
/ — реактор; 2 — котел-утилизатор; 3 — абсорбер; 4 — промывная колонна; 5 — отпарная
колонна; 6 — колонна выделения ацетонитрила; 7 — колонна выделения синильной кис-
лоты; 8 — колонна выделения акрилонитрила,
Степень конверсии пропилена на фосформолибдате висмута
достигает 60% при селективности 75%. На 1 т пропилена по-
лучают 1,01 т акрилонитрила, 0,09—0,11 т ацетонитрила и
0,08—0,13 т синильной кислоты. Процесс окислительного аммо-
нолиза протекает при концентрации кислорода, близкой к взры-
воопасной. Поэтому необходимо тщательно контролировать со-
держание кислорода в сырье и не нагревать сырьевую смесь
перед входом в реактор выше 200 °C, чтобы избежать самовос-
пламенения.
2.3. Производство винилацетата
Винилацетат СН2 = СН—О—СО—СН3 — легкокипящая жид-
кость (т. кип. 73 °C) с эфирным запахом. Он применяется в ос-
новном для получения поливинилового спирта и поливинилаце-
талей, а также в производстве лаков, красок, клеев. Его сополи-
меры с винилхлоридом идут на изготовление грампластинок.
В промышленности винилацетат получают газофазной реак-
цией ацетилена с уксусной кислотой в присутствии катализатора
(ацетат цинка на активном углеД:
СН=СН + СН3-СООН ---► СН2=СН-ОСО-СН3
На свежем катализаторе ведут процесс при 170—180 °C, а по
мере снижения активности повышают температуру до 210—
220 °C. Процесс экзотермический, проводится в трубчатом ре-
акторе.
В реактор 3 (рис. П.9) подают ацетилен и уксусную кисло-
ту в соотношении (3,54-5) 1. Свежий и возвратный ацетилен
101
Ацетилен
Рис. II.9. Технологическая схема получения винилацетата из ацетилена
и уксусной кислоты 3
/ — испаритель; 2 — теплообменник; 3— реактор; 4 — утилизатор 5 —циклон;
6, 7, 8 — холодильники; 9 — сборник.
смешивают с уксусной кислотой, направляют в испаритель 1
и в теплообменник 2, а оттуда смесь поступает в реактор. Выде-
ляющееся тепло отводят в утилизаторе 4, Реакционные газы
отдают часть своего тепла в теплообменнике 2, освобождаются
от катализаторной пыли в циклоне 5 и конденсируются вначале
в водяном (ап. 6), а затем в рассольных холодильниках 7 и 8.
Ацетилен из сепараторов возвращают в процесс, а жидкие про-
дукты поступают в сборник 9 и оттуда на ректификацию. При
ректификации в смесь вводят ингибитор полимеризации (гидро-
хинон). Сначала отгоняют низкокипящие продукты (ацетальде-
гид, ацетон), а затем отгоняют винилацетат от уксусной кис-
лоты.
Представляет большой практический интерес получение ви-
нилацетата из этилена и уксусной кислоты. Катализатор может
быть в виде раствора или твердым.
Эффективным жидким катализатором оказался хлорид пал-
ладия, который применяют растворенным в уксусной кислоте
с добавлением щелочных металлов (калий, натрий, литий). Про-
цесс идет так:
PdCl2+CH3COONa
СН2=СН2 + СН3-СООН-------------►
---> СН2=СН—О—СО—СН3 + Pd + 2НС1
Палладий непрерывно окисляется в хлорид палладия, чему спо-
собствуют добавки хлорида двухвалентной меди:
Pd + 2CuCl2 -► PdCl2 + Cu2Cl2
Cu2Cl2 + 0,5О2 + 2НС1 > 2CuC12 + H2O
Побочными продуктами являются винилидендиацетат и ацеталь-
дегид (последний образуется при взаимодействии палладиевого
102
комплекса с водой и гидролизе винилацетата):
СН2=СН-О-СО-СН3 + СН3-СООН ------► СН3—СН(ОСОСН3)2
+н2о
C2H4.PdCl2 --> СН3-СНО + Pd + 2НС1
сн2==сн—О—СО—СН3 + н2о —► сн3—сно + сн3—соон
Кроме того, в побочных продуктах обнаружены хлорпроизвод-
ные, ацетаты, щавелевая кислота. Процесс проводят в присут-
ствии кислорода. При 130 °C и 3 МПа его максимальная кон-
центрация не должна превышать 5,5% (об.). Количество воды
в реакционной смеси не должно превышать 10%, так как при
большем содержании воды существенно увеличивается выход
ацетальдегида.
Гетерогеннокаталитический процесс проводят на твердом
катализаторе (0,1—0,2% палладия на оксиде алюминия или
силикагеле). Производительность катализатора 150—200 кг ви-
нилацетата с 1 л катализатора в час/В качестве побочных про-
дуктов образуется 0,5—2% ацетальдегида и 0,5—2% полиме-
ров. В промышленности проводят процесс при 160 °C и 0,6 МПа
в трубчатых реакторах с неподвижным слоем катализатора.
Степень конверсии за один проход через реактор составляет
10—45% для этилена и 15—30% для уксусной кислоты. Высо-
кая селективность является существенным преимуществом это-
го способа.
2.4. Производство акриловой и метакриловой
кислот и их эфиров
Акриловая кислота СН2=СН—СООН — бесцветная жид-
кость с резким запахом (т. кип. 141 °C, т. пл. 13°C). Акриловую
кислоту и ее эфиры (акрилаты) применяют для получения син-
тетических акриловых волокон, лакокрасочных покрытий, клеев,
а также вспомогательных материалов для текстильной, бумаж-
ной и кожевенной промышленности.
В СССР акриловую кислоту получают в промышленности
двумя методами.
Циангидринный метод включает две стадии: 1) взаимодейст-
вие этиленоксида с синильной кислотой, 2) одновременные гид-
ролиз CN-группы и дегидратацию p-гидроксипропионовой кис-
лоты:
Н2С---CH2 + HCN ---► НОСН2—CH2CN
этиленциангидрин
4-Н*О
hoch2-ch2cn ——> НОСН2-СН2-СООН —СН2=СН-СООН
Этиленоксид взаимодействует с синильной кислотой при 55—
65 °C и 0,25 МПа в присутствии аминов. Гидролиз проводят 80—
85%-ной серной кислотой, чаще в присутствии спиртов: при
103
этом одновременно происходит этерификация с образованием
эфиров акриловой кислоты.
Акриловую кислоту можно также получать гидролизом акри-
лонитрила, протекающим через промежуточное образование
акриламида. Если гидролиз проводят в присутствии спирта,
целевым продуктом является эфир акриловой кислоты:
+н2о
CH2=CHCN ----►
CH2=CH-CONH2
4-ROH; H2SO4
>
---► CH2=CH—COOR + nh4hso4
Метод, однако, пока не является промышленным.
Все большее значение приобретает метод получения акрило-
вой кислоты селективным окислением пропилена. Процесс идет
через промежуточное образование акролеина и сопровождается
выделением большого количества тепла (594 кДж/моль):
4-оа 4-оа
сн2=сн—сн8—►сн2=сн-сно—►сн2=сн-соон
Побочно образуются ацетальдегид, ацетон, уксусная кислота,
СО и СОг. В качестве катализаторов применяют оксиды висму-
та и молибдена с различными промоторами. Окисление ведут в
присутствии водяного пара.
Одностадийное окисление проводят при 400—420 °C, соотно-
шении пропилена, воздуха и водяного пара, равном 1 : 10:9, и
времени контакта 4—5 с. Выход акриловой кислоты составляет
35% на пропущенный пропилен.
Двухстадийный процесс осуществляют в более мягких усло-
виях. На первой стадии температура равна 330—370 °C. Пропи-
лен, воздух и водяной пар в соотношении 1 :7 : 12 поступают
в реактор 1 первой ступени (рис. 11.10) со стационарным слоем
катализатора (оксиды висмута, молибдена, ванадия, железа).
Выделяющееся тепло используют для получения водяного пара
в котле-утилизаторе 2. Продукты реакции, содержащие акро-
Рис. 11.10. Технологическая схема получения акриловой кислоты двухстадий-
ным окислением пропилена:
lt 3 — реакторы; 2, 4 — котлы-утилизаторы; 5 — абсорбер; 6 — экстрактор; 7 — колонна
регенерации экстрагента; 8, Р — колонны выделения акриловой кислоты.
104
леин и акриловую кислоту, поступают в реактор 3 второй сту-
пени, в котором идет доокисление при 250—300 °C на катализа-
торе (оксиды ванадия и молибдена). Из реактора 3 газы по-
ступают в абсорбер 5, орошаемый водой. В аппарате 6 из вод-
ной фазы экстрагируется акриловая кислота и вместе с экстра-
гентом (вода) уходит в колонну 8 для разделения. Рафинат,
содержащий органические вещества, воду и примесь экстраген-
та, уходит с низа колонны 6 в колонну 7. Органические веще-
ства и экстрагент возвращают в колонну 6, а воду отводят с ни-
за колонны 7. В колонне 8 происходит регенерация экстрагента.
В колонне 9 отгоняют низкокипящие примеси (в основном ук-
сусную кислоту) и из куба отводят товарную акриловую кисло-
ту. Выход акриловой кислоты составляет 80% в расчете на про-
пущенный пропилен.
К числу методов получения акриловой кислоты и ее эфиров,
представляющих интерес для промышленности, относится взаи-
модействие ацетилена с оксидом углерода в присутствии воды
и спиртов. В первом случае образуется акриловая кислота, во
втором — ее эфиры:
сн==сн+СО + Н2О —► сн2=сн-соон
CHsCH + со + ROH---* СН2=СН—COOR
Первая реакция проводится в присутствии тетракарбонила ни-
келя Ni(CO)4 при обычном давлении и 40—45 °C. Вторая реак-
ция протекает при 170 °C и 3 МПа в присутствии бромида нике-
ля и алкилбромида, связанных трифенилфосфитом в комплекс.
Алкильный радикал бромида соответствует спирту, применяемо-
му в реакции.
Большое промышленное значение имеют также метакрило-
вая кислота СНг=СН(СНз)—СООН и ее эфиры, особенно ме-
тилметакрилат, идущий на производство органического стекла
(полиметилметакрилат). Высшие эфиры метакриловой кислоты
применяют как загущающие присадки к минеральным смазоч-
ным маслам.
Метакриловую кислоту или метакрилат в промышленности
получают из ацетона через ацетонциангидрин, который обраба-
тывают 100%-ной серной кислотой, а затем водным метанолом:
+h2so4
СН3-СО—СН8 + HCN --► (СН3)2С—CN ----->
ОН
---► (CHS)2C—C=NH -► (CH3)2C-CONH2 --►
н<5> oso2oh iso3H
+сн3он
---► CH2=C-CONH2 H2SO4 --—* СН2=С-СО-ОСН3 + NH4HSO2
I —H2O I
CH3 CH3
105
Метакриловую кислоту (аналогично получению акриловой
кислоты из пропилена) можно получить окислением изобутилена
через промежуточное образование метакролеина:
4-Оа Ч~О2
сн2=с—сн8 ——> сн2=с-сно —>- сн2=с—соон
I -нао ! .
сн, сн3 сн3
Процесс пока не реализован в промышленности. На стадии
окисления метакролеина в метакриловую кислоту идут процес-
сы их полимеризации, поэтому требуется разработка новых, се-
лективных катализаторов. В принципе этот процесс является
перспективным, так как базируется на доступном сырье, не
требует применения серной кислоты и утилизации сульфата ам-
мония.
ГЛАВА III
ПРОИЗВОДСТВО КИСЛОРОДСОДЕРЖАЩИХ
ПРОДУКТОВ
§ 1. ОСНОВНЫЕ ПРЕДСТАВЛЕНИЯ О ПРОЦЕССАХ
ОКИСЛЕНИЯ УГЛЕВОДОРОДОВ
1.1. Механизм процесса
Окисление углеводородов представляет собой сложный про-
цесс. В ходе реакции образуются нестабильные промежуточные
соединения, претерпевающие дальнейшие превращения с обра-
зованием большого числа продуктов.
Химические реакции, как правило, не являются результатом
непосредственного взаимодействия исходных молекул, а идут
в несколько стадий. Для взаимодействия углеводорода с кисло-
родом требуется большая энергия активации, расходуемая на
разрыв связей С—Н (314—418,6 кДж/моль) и 0=0
(488 кДж/моль) или на разрыв одной связи в молекуле кисло-
рода с образованием частицы —О—О— (335 кДж/моль), т. е.
реакция окисления углеводородов энергетически невыгодна.
Молекулярный механизм не может объяснить особенности окис-
ления; не применим и ионный механизм, так как предельные
углеводороды преимущественно являются неполярными соеди-
нениями, из которых образование ионов затруднительно.
Многие химические процессы протекают через промежуточ-
ное образование свободных радикалов, играющих роль актив-
ных центров. Свободные радикалы взаимодействуют с валент-
нонасыщенными молекулами, для чего достаточно малой энер-
106
гии активации. В результате этого взаимодействия вместо ис-
чезнувшего первичного радикала обязательно возникает новый
свободный радикал. Он взаимодействует с исходной молекулой
и дает начало третьему радикалу и т. д. Возникает цепная реак-
ция, в которой со значительной затратой энергии связано обра-
зование только первичного радикала (зарождение цепи), а каж-
дое последующее звено превращений (продолжение цепи) про-
исходит уже с небольшим расходом энергии.
Цепной механизм окисления углеводородов молекулярным
кислородом подтвержден экспериментально. Установлено, на-
пример, что скорость реакции значительно возрастает при до-
бавлении в систему веществ, легко разлагающихся на радика-
лы (пероксиды, гидропероксиды, азосоединения), а также в ре-
зультате действия света или ионизирующих излучений, способ-
ствующих образованию свободных радикалов. Автоускорение
процесса, наблюдаемое после некоторого индукционного перио-
да, связано с накоплением гидропероксидов, разлагающихся в
ходе окисления на свободные радикалы. Еще один характерный
признак цепного процесса — торможение окисления ингибито-
рами (фенолами, нафтолами, аминами).
Зарождение цепи является начальной стадией, приводящей
к образованию свободных радикалов из валентнонасыщенных
молекул углеводорода. Свободные радикалы могут образовы-
ваться из исходных веществ в результате их разложения или
взаимодействия, например, с кислородом:
RH —► R- + Н- или RH + О2-----------► R- + НОО.
Иногда реакции зарождения цепей (инициирование) могут про-
текать на стенках реактора.
Реакциями продолжения цепи называют стадии цепной реак-
ции, связанные с расходованием исходных веществ и образова-
нием продуктов превращений. Идут они с сохранением свобод-
ных радикалов:
R. + О2---► ROO. RH + ROO. ---------► ROOH + R-
Обрыв цепи приводит к исчезновению свободных радикалов.
Он может происходить в результате «захвата» свободного ради-
кала стенкой реактора (скорость обрыва цепи увеличивается
при уменьшении диаметра реактора), при взаимодействии ра-
дикала с молекулой ингибитора или примеси. Например, при
окислении углеводородов в присутствии дифениламина перок-
сидный свободный радикал ROO* взаимодействует с ним и об-
разует малоактивный свободный радикал, который не может
продолжать цепь и погибает в результате рекомбинации с ка-
ким-либо другим свободным радикалом:
+R’
ROO. + NH(CeHs)2 _ROQH> (CeH5)2N- -> RN(CeH5)2
107
Обрыв цепи может происходить и при взаимодействии двух сво-
бодных радикалов.
В основе современного механизма окисления углеводородов
лежит теория цепных процессов с вырожденным разветвлением.
Особенность таких реакций (как в цепных разветвленных про-
цессов вообще) заключается в их самоускорении. Это связано
с тем, что промежуточные вещества, образующиеся при разви-
тии основной цепи, способны давать свободные радикалы, при-
чем с большей легкостью, чем исходные вещества. Процесс на-
чинает развиваться и протекать со значительной скоростью. На-
пример, при окислении углеводорода основная цепь развивается
до образования гидропероксида:
+RH
R. + оа --► ROO. ---► ROOH + R.
Гидропероксид в основном превращается в неактивные конеч-
ные продукты, но он может и диссоциировать на радикалы
ROOH ---► RO. + -ОН или 2ROOH----------► RO- + Н2О 4- ROO-
которые вызывают новые цепи окисления исходного углеводо-
рода — цепи вырожденного разветвления. Вырожденное развет-
вление происходит уже после того, как основная цепь, его соз-
давшая, успела развиться и оборваться. Поэтому самоускорение
вырожденноразветвленных реакций «растянуто» во времени,
т. е. скорость реакции намного меньше, чем в случае обычного
разветвления цепи. Это объясняет длительный период индукции
вырожденноразветвленных реакций. Приведенные схемы разло-
жения гидропероксидов являются стадиями зарождения вырож-
денноразветвленных цепей.
При окислении углеводородов вырожденное разветвление
происходит и за счет превращения альдегидов. Например, форм-
альдегид, образующийся при окислении метана, может реаги-
ровать с кислородом и образовывать два свободных радикала
НСНО + О2 --► НОО. + нсо
которые могут начать цепи вырожденного разветвления. Альде-
гиды, образующиеся при окислении высокомолекулярных пара-
финов, могут вести цепь вырожденного разветвления за счет
радикалов, образующихся по другой схеме:
RCHO + О2 --► RCOO. + НО- НО. + RH -------- Н2О + R.
I
CO + RO. RO-+RH-----► ROH + R.
Наряду с цепными реакциями, в которых участвуют проме-
жуточные вещества, в ряде случаев эти вещества без участия
свободных радикалов превращаются в более стабильные про-
дукты (например, спирты и альдегиды при взаимодействии с
молекулярным кислородом превращаются в кислоты).
108
1.2. Закономерности окисления углеводородов
в газовой и жидкой фазе
Процессы окисления углеводородов проводят в газовой и'
жидкой фазе. Жидкофазное окисление протекает при более мяг-
ких условиях (при меньшей температуре и в большинстве слу-
чаев при атмосферном давлении), поэтому оно не вызывает
столь глубокой деструкции, как в случае газофазного окисления.
Окисление углеводородов молекулярным кислородом в жидкой
и газовой фазе чаще всего проводят в присутствии катализато-
ров. Наибольшее распространение получило гетерогенноката-
литическое окисление углеводородов. Основными реакциями яв-
ляются следующие:
1) частичное окисление углеводорода:
RCHS-|-Oa ► RCHO-f-H2O
CH3-CH=CHj + Oj --► СН2=СН-СНО4-НаО
2) прямое присоединение молекулярного кислорода по двой-
ной связи ненасыщенных соединений:
СНа=СН2 + 0,5О2---► Н2С--СН2
3) деструктивное окисление с образованием двух или более
молекул кислородсодержащих соединений:
RCH2-CH2R'4-2,5O2---► RC0OH+R'C00H4-H2O
RCHa—СН=СН2 + О2 --► RCHO-f-CHj—СНО
4) окисление углеводорода с образованием в молекуле не-
скольких групп СО или СООН:
~ Л сн—СО
0—1 >
СН-СО
НООС—(СН2)4—соон
5) глубокое окисление с полным разрушением углеродного
скелета исходного углеводорода:
СпН^+а + 02 --► nCO2 + (n + 1) Н2о
Перечисленные реакции не исчерпывают всего многообразия
окислительных превращений (в частности, совсем не рассмат-
ривается взрывное окисление в пламени); все они относятся
к реакциям медленного окисления.
Реакции окисления экзотермичны. Перегрев катализатора
стимулирует глубокое окисление, что снижает селективность
процесса и его рентабельность. Одна из основных задач при
проведении процесса в заводских условиях — обеспечение изо-
термического режима работы реакторов. Газофазное окисление
углеводородов протекает при высоких температурах (200^
109
600 °C) и сопровождается крекингом; жидкофазное окисление
идет при температуре ниже 200 °C с меньшей деструкцией. Га-
зофазное окисление парафинов выше Сб вести 'нецелесообразно,
так как окисление высокомолекулярных углеводородов при
300—350 °C приводит к сложной смеси продуктов. Жидкофаз-
ное окисление углеводородов является боле^ направленным, оно
приводит в основном к кислотам и спиртам.
Между окислением низких гомологов метана в газовой фазе
и окислением высших парафинов в жидкой фазе имеется много
общего: в определенных условиях оба процесса протекают и без
катализаторов; последние интенсифицируют окисление, но чет-
кой избирательности, как, например, при гидрогенизационных
процессах, достигнуть не удается; окисление высших и низших
парафинов дает сложную смесь продуктов. Эта аналогия явля-
ется результатом того, что окисление в жидкой и газовой фазе
протекает по единому, цепному механизму.
Промышленное осуществление процессов окисления низших
парафинов связано с серьезными трудностями, обусловленными
образованием взрывоопасных смесей углеводородов с воздухом.
Поэтому нужно всегда работать лишь с теми смесями, в кото-
рых концентрация углеводородов либо ниже нижнего предела
взрываемости, либо выше верхнего, т. е. с избытком либо угле-
водорода, либо воздуха. В случае окисления .высших парафинов
воздухом это затруднение отпадает. Пределы взрываемости
смесей паров углеводородов в смеси с воздухом [в % (об.)]:
Метан 5,0—15,0 Бутан 1,8—9,1
Этан 2,9—15,0 Пентан 1,4—7,8
Пропан 2,1—9,5
В большинстве случаев при окислении газообразных парафи-
нов образуется сложная смесь продуктов, разделение которых
связано с большими затратами энергии. Как правило, при окис-
лении побочно образуются СО и СОг, в результате чего степень
полезного’использования углеводорода снижается.
Из газообразных парафиновых углеводородов наиболее
трудно окисляется метан, а при переходе от метана к бутану
скорость окисления возрастает. Метан (как и его гомологи)
реагирует с кислородом в газовой фазе при температуре выше
250 °C. Теоретически из метана можно получить метиловый
спирт, формальдегид и муравьиную кислоту:
4-0,5Оа
4-02
С.Г14 —Н2О
4-1,5О2
-н2о
НСНО —
4-0,5О2
СН3ОН —-> НСНО
—н2о
со + н2
СО + Н2О
СО2+ Н2О
нсоон
по
При атмосферном давлении в процессе окисления метана выход
формальдегида намного выше, чем выход метилового спирта.
При повышении давления и росте соотношения метана и кис-
лорода увеличивается выход метилового спирта. Помимо окси-
дов азота в качестве катализаторов окисления применяли фос-
фаты металлов и хлористый водород, но они не оказывали за-
метного влияния на процесс. Чтобы снизить выход побочных
продуктов, ведут реакцию при небольшой степени конверсии
метана и малом времени пребывания газовой смеси в реакторе.
При 360 °C и 10 МПа в результате окисления метана кислоро-
дом (в соотношении 9:1) образуется смесь, содержащая 17%
(масс.) метилового спирта и 0,6% (масс.) формальдегида;
остальное приходится на СО, СО2 и< Н2О. Если к метану до-
бавить этан, выход метилового спирта сильно повышается (поч-
ти в два раза). Метан в присутствии значительного количества
этана окисляется уже при 300—400 °C и 13—13,5 МПа, в то
время как для окисления чистого метана требуется температу-
ра 520 °C. На процесс благоприятно влияет добавка небольших
количеств оксидов азота.
При окислении этана образуется сложная смесь продуктов,
состав которой в зависимости, от условий реакции резко меняет-
ся. Состав смеси жидких продуктов окисления этана при 10 МПа
в зависимости от температуры и длительности процесса таков
[в % (масс.)];
Продукт При 262 °C и 40 мин При 278 °C и 5 мин
Этиловый спирт 24,3 28,6
Метиловый спирт 15,5 42,3
Ацетальдегид . . 6,3 20,9
Формальдегид и муравьи-
ная кислота 1,6 2,7
Уксусная кислота 52,3 5,5
Пропан окисляется легче этана — при 15 МПа реакция на-
чинается уже при 112 °C, а если желательно вести процесс при
меньшем давлении, нужно повысить температуру. Например,
в продуктах окисления пропана, проводимого при 6 МПа, 252 °C
и соотношении пропан : воздух= 1 : 3,6, обнаружено 6,2% (масс.)
изопропилового спирта и 12,5% (масс.) ацетона. При повыше-
нии давления выход изопропилового спирта и ацетона увеличи-
вается. Вероятно, давление способствует атаке кислорода по
метиленовой группе пропилена.
При окислении пропана и этана в определенных условиях
можно получить пероксид водорода. Так, в результате окисле-
ния пропана кислородом в соотношении 9:1 при 470°C, дли-
тельности реакции 4—5 с и последующем быстром охлаждении
газов образуется «конденсат», в 1 л которого содержится 13,2 г
активного кислорода — 30% из органических пероксидов и 70%
из пероксида водорода. Получение пероксида водорода окисле-
нием пропана представляет интерес и как промышленный спо-
соб.
111
Продукты окисления бутана мало отличаются от продуктов
окисления пропана. Окисление бутана ведут трже при неболь-
шом давлении (3—5 МПа) и 400 °C. Содержание продуктов
окисления зависит от давления: ;
Продукт
Метиловый спирт
Этиловый спирт
Пропиловые спирты
Бутиловые спирты
Альдегиды
Кислоты**
Ацетон ....
Оксид углерода .
Диоксид углерода
Содержание*, %
при 3,3 МПа при 13,3 МПа
15,9 8,4
2,5 6,3
3,2 8,9
0,6 5,5
6,9 15,9
16,7 4,6
1,4 0,7
3,8 9,4
9,4 6,1
* В % кислорода, израсходованного на образование дан-
ного продукта.
** Муравьиная и уксусная в соотношении 1 :6.
Окисление бутана можно проводить и при недостатке кисло-
рода, но в присутствии водяного пара (разбавитель) и при
малом времени пребывания газов в зоне реакции. Так, из сме-
си бутана с воздухом и водяным паром в массовом соотношении
1 : 5 : 34 при 400 °C, 2—3 МПа и времени контакта. 1,15 с полу-
чается следующее количество жидких продуктов (в масс. ч. на
100 масс. ч. бутана):
Метиловый спирт 19,0
Пропиловый спирт 1,0
Бутиловый спирт 0,5
Формальдегид 15,2
Ацетальдегид . . . 19,8
Карбоновые кислоты 11,4
Ацетон . 7,0
.В СССР разработан процесс окисления бутана с высоким
выходом уксусной кислоты. Окисление ведут при 150—160 °C и
давлении примерно 5 МПа; выход кислоты достигает 80% от
теоретического (считая на прореагировавший бутан).
В последнее время важное практическое применение полу-
чают продукты окисления изобутана. Окисление проводят в
присутствии бромистого водорода и кислорода. Процесс харак-
теризуется образованием небольшого числа индивидуальных ве-
ществ. Так, из изобутана, взятого с кислородом и бромистым
водородом в соотношении 10:10:1, при 160 °C можно полу-
чить трет-'бутилгидропероксид с выходом 75 %:
СН(СН3)3 + О2 --► С(СН3)з
(!)ОН
Образуется также некоторое количество изобутилового спирта и
трет-бутилпероксида (СНз)зС—ОО—С(СН3)з. трет-Бутилгидро-
пероксид и трет-бутилпероксид применяют в промышленности
как катализаторы радикальной полимеризации.
112
При окислении газообразных парафинов наблюдается общая
закономерность: чем выше давление, тем больше выход спиртов
и тем меньше выход альдегидов и карбоновых кислот. Высокая
объемная скорость подачи газов благоприятствует получению
спиртов с более длинной цепью, а низкая скорость подачи спо-
собствует образованию низкомолекулярных кислот и спиртов,
воды и СОг.
Иногда окисление низших углеводородов используют как
вспомогательный процесс для получения тепла, например при
окислительном пиролизе метана с целью получения ацетилена,
а также для получения этилена из этана или пропана. Тепло,
образующееся при частичном сжигании газа, используется в
эндотермической реакции пиролиза:
2СН4---► С2Н2 + ЗН, ДЯ = 400 кДж/моль
При окислении высших парафинов образуются высшие жир-
ные кислоты, широко применяемые в мыловаренной промышлен-
ности, и высшие жирные спирты, используемые в производстве
синтетических моющих веществ. Раньше высшие жирные кис-
лоты и спирты вырабатывали из растительных и животных жи-
ров, поэтойу получение синтетических жирных кислот и спиртов
(стр. 115, 118 и 156) позволяет для технических целей заме-
нять ценное и труднювосполняемое природное сырье синтетиче-
скими продуктами.
Окисление высших парафинов в жидкой фазе кислородом
воздуха протекает примерно по тому же механизму, что и окис-
ление низкомолекулярных парафинов в газовой фазе. Но в этом
случае не наблюдается взрывного течения реакции и, кроме
того, первичные продукты реакции окисляются до кислот.
Молекулярный кислород присоединяется к молекуле углево-
дорода с образованием гидропероксида. В случае углеводородов
изостроения кислород присоединяется к третичному углеродно-
му атому, а в случае углеводородов нормального строения —
к метиленовой группе:
4-Oj
СНЭ-(СН2)П-СН2------СН3 ---*- СН,-(СН2)„-СН-----СН,
ООН
Кислород действует на все метиленовые группы в одинаковой
степени, а его присоединения к конечной метильной группе
почти не наблюдается. Образующийся гйдропероксид разлагает-
ся на кетон и спирт:
R-(CH2)„-CH-CH2-CH2-------СН, —►
ООН
---► R—(СН2)П—С—СН, + СН,-----СН2ОН
8-254
113
Кетон окисляется в кетогидропероксид, который разлагается на
кислоту и альдегид:
R'CH2~C—CH—CH2R*----► R'CH2-COOH + R"CH2-CHO
о Аон
В результате протекания основных и побочных реакций при
этом процессе образуется сложная смесь кислородсодержащих
соединений —спиртов, кислот, гидроксикислот, альдегидов и
др.; ее разделение является сложной технологической задачей.
В СССР для окисления используют нефтяной парафин с чис-
лом углеродных атомов от 18 до 44, синтетический парафин,
получаемый по Фишеру — Тропшу, а также жидкие (мягкие)
парафины с установок карбамидной депарафинизации нефтепро-
дуктов. Присутствие в парафине углеводородов изостроения не-
желательно, потому что они образуют низкомолекулярные кис-
лоты (менее 12 атомов углерода) и кислоты изостроения с не-
приятным запахом. В парафине не должно быть нафтеновых и
ароматических углеводородов, маслянистых примесей и других
веществ, тормозящих окисление (фенолов, органических соеди-
нений азота и серы).
Товарными продуктами являются кислоты Сю—С20, для по-
лучения которых требуется парафин, выкипающий при 320—
450 °C. Кислоты Сю—Сю идут на производство туалетного мыла,
а кислоты Си—С20 — на выработку хозяйственного мыла.
В процессе окисления парафина образуются и низкомолекуляр-
ные кислоты. Кислоты С5—Сб идут в парфюмерную промыш-
ленность для получения сложных жиров, а кислоты С?—С9 —
для получения спиртов и пластификаторов на их основе.
На процесс окисления и выход желаемых продуктов влияют
температура, катализаторы и способ подачи кислорода или воз-
духа. В заводских условиях поддерживают температуру 105—
120 °C; продолжительность окисления при этом составляет 20—
24 ч. При 170—180 °C окисление протекает намного быстрее —
за 3—5 ч, но качество кислот значительно хуже, так как обра-
зуются не пригодные для мыловарения гидроксикислоты. Обыч-
но окисление ведут до получения оксидата с кислотным числом
68—70 мг КОН/г (эфирное число равно 50 мг КОН/г). Более
глубокое окисление проводить нерационально, потому что ско-
рость окисления начинает резко падать.
В качестве катализатора наиболее широко применяют мар-
ганцевые соли, например перманганат калия, расход которого
не превышает 0,3% от количества парафина. В присутствии пер-
манганата не только ускоряется окисление, но и разрушаются
ингибиторы, мешающие автоокислению. Перманганат вводят
в парафин, нагретый до 120 °C, в виде водного раствора. Вода
испаряется, а перманганат восстанавливается до МпОг, кото-
рый благодаря хорошему перемешиванию распределяется в объ-
еме в виде тонкой взвеси, что позволяет снизить температуру
114
окисления до НО—120 °C, сохранив приемлемую скорость про-
цесса.
На окисление парафина расходуется только та часть кис-
лорода, которая в нем растворена. Поэтому скорость окисления
будет тем выше, чем больше кислорода растворится в парафи-
не, т. е. чем выше давление. При повышении давления с 1,5 до
6 МПа длительность реакции при одной и той же глубине окис-
ления снижается с 8 до 2 ч. Расход воздуха составляет 40—
60 м3 на 1 т парафина в час. Чем выше колонны окисления, тем
меньше расход воздуха на 1 т парафина. На выход кислот су-
щественно влияет поверхность соприкосновения воздуха с пара-
фином, поэтому подводимый воздух распределяют по сечению
колонны при помощи решеток или насадки из колец Рашига.
§ 2. ПРОИЗВОДСТВО КИСЛОРОДСОДЕРЖАЩИХ ПРОДУКТОВ
ОКИСЛЕНИЕМ ВЫСШИХ ПАРАФИНОВ
2.1. Производство высших жирных кислот
В промышленности процесс окисления высших парафинов
можно проводить периодически и непрерывно. В настоящее вре-
мя преобладают периодические процессы.
Окисление обычно осуществляют в вертикальных колоннах
емкостью 8—12 м3, выполненных из алюминия, поскольку низ-
комолекулярные кислоты вызывают сильную коррозию. Окис-
ление сопровождается выделением большого количества тепла.
Так, при окислениц до кислотного числа оксидата, равного
70 мг КОН/г, выделяется 2095 кДж на 1 кг окисленного пара-
фина. Тепло из колонн отводят через вмонтированные в них
змеевики, охлаждаемые водой. Давление в колоннах атмосфер-
ное. Продолжительность окисления 20—24 ч. Промышленные
установки имеют несколько реакторов для обеспечения задан-
ной производительности.
Полученный при окислении оксидат-сырец обрабатывают со-
дой и щелочью (этот процесс принято называть омылением).
Жирные кислоты быстро нейтрализуются и переходят в мыль-
ный раствор. Неомыляемые-I, представляющие собой неокислен-
ный парафин с примесью кислородсодержащих продуктов, воз-
вращают на окисление. Оксидат-сырец кроме жирных кислот
содержит небольшое количество гидроксикислот, эфиров, лакто-
нов, кетонов и спиртов, которые омыляются только в жестких
условиях. Эти соединения вместе с небольшим количеством
парафина хорошо растворяются в мыльном растворе, обладаю-
щем поверхностно-активными свойствами. Их надо удалять, так
как они ухудшают моющее и пенообразующее действие мыла,
придают ему неприятный запах (особенно гидроксикислоты).
Для этой цели мыльный раствор подвергают термообработке
при 320 °C; в результате происходит «облагораживание» — лак-
8*
115
тоны и кетоны омыляются, а гидроксикислоты дегидратируются:
+NaOH
RCH2—CO-CH2R' --> RCH2—COONa + CHaR'
RCH2— CH—СН,—COOH —RCH=CH—СН,—COOH
2 I 2 —H2O 2
OH
Продукты, не омыленные при термообработке (иеомыля-
емые-П), отгоняют из мыльного раствора перегретым паром,
образующимся в результате испарения воды, находящейся в
мыле, или подаваемым специально. В неомыляемых-П кроме
парафина содержатся в основном спирты. После конденсации
и отделения воды неомыляемые-П возвращают на окисление.
Технологическая схема получения высших жирных кислот
приведена на рис. III. 1. Водный раствор катализатора, приго-
товленный в емкости 1, смешивают с парафином в аппарате 2;
перманганат калия в виде 15%-ного раствора (0,1—0,3% на за-
грузку) вводят в парафин, нагретый до 120 °C. Вода испаряет-
ся, а КМпО4 диспергируется в парафине. Смесь парафина с ка-
тализатором идет в окислительную колонну 3, куда подают очи-
щенный воздух. В колонне поддерживают температуру 105—
115 °C с помощью нагревающих и охлаждающих змеевиков. Во
время индукционного периода (2—3 ч) в колонну подводят теп-
ло, подавая пар в нижний змеевик.
Рис. III.1. Технологическая схема получения высших жирных кислот окисле-
нием парафина:
/, 14, /5 —емкости; 2 — смеситель; 3 — окислительная колонна; 4, 19 — холодильники;
5 —сепаратор; 6 — промывной скруббер; 7 — сборник; 8 — шламоотстойник; 9, 16 — про-
мывные колонны; 10, И — омылители; 12, /« — отстойники; 13 — печь; // — поглотитель-
ная колонна.
116
Таблица Ш.1. Условия дистилляции и выход
высших жирных кислот
№ куба Температура, eG Остаточное давление, Па Фракция кислот Выход кислот, % (масс.) на загрузку
1 160—180 266-400 С6—Се 4-5
2 170-180 26,6 с7-с. 12—13
3 185 20 Сю Си —
4 200 20 С14 С1в 65
5 280 20 С17 С20 —
6 310 20 >С2о 4-5
Пройдя колонну, воздух увлекает с собой пары воды, низ-
комолекулярных кислот и другие летучи^ продукты, которые ох-
лаждаются в холодильнике 4. Конденсат отделяют в сепарато-
ре 5 и направляют в сборник 7. Охлажденный воздух промыва-
ют водой в колонне 6 и подают в печь. Промывную воду из ко-
лонны 6 тоже подают в сборник 7. Смесь расслаивается там:
на два слоя — верхний (масляный) и нижний (иодный). Вод-
ный слой либо собирают, либо направляют на промывку окси-
дата в колонну 9. Этот слой может содержать кислоты Ci—С4.
Оксидат из реактора направляют в шламоотстойник Я, а от-
туда в промывную колонну 9 для извлечения водорастворимых
низших кислот. В колонну можно подавать свежую воду и вод-
ный конденсат из сборника 7. Промытый оксидат направляют*
в омылитель 10, где кислоты нейтрализуют 25%-ным раствором
соды при 90—100°C (первая ступень омыления), а затем пере-
дают в аппарат И, где для омыления используют раствор нат-
риевой щелочи (вторая ступень). Омыленный продукт в отстой-
нике 12 при 70 °C расслаивается на неокисленный парафин (не-
омыляемые-I) и раствор мыла.
Парафин возвращают на окисление, а мыло под давлением
2 МПа подают в печь 13, обогреваемую дымовыми газами. Тем-
пературу в печи поднимают постепенно — вначале до 180 °C, за-
тем до 220 °C и, наконец, до 320 °C. После печи мыло поступает
в испаритель, установленный в кладке; в нем при атмосферном
давлении неомыляемые-П отгоняются с перегретым паром и
проходят в поглотительную колонну 17, орошаемую холодной
водой из отстойника 18. Конденсат и вода поступают в отстой-
ник 18, из которого неомыляемые-П возвращают на окисление.
Сухое мыло разбавляют в емкости 14 водным раствором
сульфата натрия, после чего мыльный клей в емкости 15 разла-
гают 92 %-ной серной кислотой при 100 °C и сильном перемеши-
вании. Смесь жирных кислот и сульфата натрия промывают во-
дой в колонне 16. Промытые кислоты направляют на дистилля-
цию (на схеме не показана). Аппараты, в которых мыльный
клей обрабатывают серной кислотой, футеруют для защиты ок
коррозии. i
11г
Дистилляцию получаемых кислот осуществляют в кубах с
насадочными колоннами. Кубы изготовлены из кислотоупорного
чугуна, а змеевики и колонны — из нержавеющей стали. По ме-
ре увеличения молекулярной массы кислот повышают темпера-
туру и уменьшают остаточное давление в кубах (табл. III.1).
Экономические показатели процесса можно улучшить, ис-
пользуя неомыляемые-П для получения спиртов и выделяя низ-
комолекулярные кислоты из водного конденсата (из скруббера)
и из промывных вод после промывки оксидата.
2.2. Производство высших жирных спиртов
Спирты получают окислением парафина (фракция 275—
320°C). Содержание ароматических углеводородов в нем не
должно превышать 0,5%, иначе процесс окисления не идет.
Наиболее приемлемым сырьем является фракция 275—320 °C
синтина*, так как он не содержит ароматических углеводородов.
Окисление ведут при 165—170 °C азото-кислородной смесью, со-
держащей 3—5% кислорода. Расход окисляющего газа состав-
ляет 500—700 л на 1 кг парафина в час. В присутствии борной
кислоты образующиеся спирты дают эфиры — триалкилбораты
(RO) зВ, т. е. цепь окислительных превращений обрывается на
стадии образования спиртов, чем и достигается селективность
процесса. Расход борной кислоты составляет 5% от парафина;
подают ее в реакционную зону в виде суспензии в углеводоро-
дах.
Реакцию окисления можно представить уравнением:
+о2 +R'H
R-(CH2)n-CH2-CH3 ----> R-(CH2)n-CH-CH3 —* R—(СН2)П—СН—СН3
ООН он
+н3во3
3R—(СН^—СН—СН3 ~1
| —of12U
R—(СН2)П-СН-О] В
CHS
Js
Процесс окисления протекает без разрыва углеродной цепи,
поэтому при получении высокомолекулярных жирных спиртов
можно использовать для окисления мягкие парафины, получае-
мые при депарафинизации дизельных топлив. На процесс окис-
ления, как и в случае окисления до кислот, влияет поверхность
соприкосновения газа-окислителя с парафином, поэтому жела-
тельно как можно более мелко дробить поток газа по сечению
окислительной колонны.
Окисление парафина ведут в колонне 2 (рис. III.2) в течение
3—4 ч при 165—170 °C. В колонну из емкости 1 подают суспен-
зию борной кислоты в парафине, а также парафин и газ-окис-
* Синтин — смесь парафиновых углеводородов, полученных синтезом
из СО и Н2.
118
Рис. III.2. Технологическая схема получения высших жирных спиртовз
1 — емкость; 2 — окислительная колонна; 3 — обратный холодильник; 4 — отстойник?..
5 — нутч-фильтр; 6, 11 — кубы с колоннами; 7, 13 — вакуум-приемники; 8, 9» 10 — аппа-
раты с мешалками; 12 — конденсатор-холодильник.
литель (воздух). Отходящие газы уходят через обратный холо-
дильник 3, где конденсируются уносимые газом капли- парафи-
на. По окончании окисления оксидат поступает в отстойник 4,
где отстаивается избыток борной кислоты; остальную кислоту
отфильтровывают на нутч-фильтре 5. В кубе 6 отгоняют не всту-
пивший в реакцию парафин, который после защелачивания в
аппарате 3, необходимого' для освобождения от кислот, возвра-
щают в процесс. Бораты омыляют сначала водой в аппарате 9,
а затем щелочью в аппарате 10 при НО °C. После этого в ку-
бе 11 от омыленного продукта отгоняют спирты при остаточном
давлении 665 Па до 185 °C, а затем продолжают перегонку с
водяным паром до 275 °C при остаточном давлении 266 Па.
Глубина окисления составляет 35—40%. Получаемые спирты на
80% состоят из вторичных изомеров. Из 1 т синтина можно по-
лучить 670 кг жирных спиртов, 115 кг высших жирных кислот
и 124 кг низкомолекулярных продуктов окисления.
§ 3. ПРОИЗВОДСТВО КИСЛОРОДСОДЕРЖАЩИХ ПРОДУКТОВ
ОКИСЛЕНИЕМ ЦИКЛИЧЕСКИХ УГЛЕВОДОРОДОВ
3.1. Основные закономерности окисления
циклических углеводородов
В настоящее время в промышленности получают большое
число кислородсодержащих продуктов окислением бензола и
его гомологов — толуола, ксилолов, этилбензола, изопропилбен-
зола, циклогексана. Процессы осуществляют в газовой и жидкой
фазе.
Газофазное окисление алкилароматических углеводородов
проводят в присутствии катализаторов, в основном пентоксида
ванадия; оно протекает деструктивно, с изменением структуры
11»
исходного углеводорода. Так получают малеиновый и фталевый
ангидриды. Большое значение приобретает также пиромелли-
товый диангидрид, получаемый газофазным окислением дуро-
ла (тетраметил бензол):
4-Qg; V2Q5
-6Н2О
Жидкофазное окисление алкилароматических углеводородов
.широко применяют для получения гидропероксидов, которые ис-
пользуют как инициаторы радикально-цепных процессов и как
исходные вещества для получения (путем их разложения) цен-
ных кислородсодержащих соединений. Жидкофазным окисле-
нием получают также ароматические карбоновые кислоты. При
окислении до гидропероксидов катализаторы не применяют. Для
снижения индукционного периода окисления вводят те же гид-
ропероксиды. Окисление до карбоновых кислот ведут в присут-
ствии солей металлов переменной валентности (соли кобальта
или марганца). Разложение гидропероксидов протекает по раз-
ным схемам в зависимости от условий. Например, изопропил-
бензолгидропероксид при повышенных температурах (>120 °C)
разлагается на ацетофенон и метанол, а в присутствии кислот —
на фенол и ацетон. В присутствии щелочи образуется диметил-
-фенилкарбинол, причем этот третичный спирт дегидратируется
л а-метилстирол:
Образование гидропероксидов сопровождается получением
побочных продуктов, количество которых увеличивается с повы-
шением глубины окисления углеводорода (для изопропилбензо-
ла ограничивают 25—30%, для этилбензола 10—15%). На раз-
ложение гидропероксидов существенно влияет температура, ко-
торую в конце процесса понижают (в случае этил- и изопропил-
бензолов от 120—130°C в начале до 105—108°C в конце).
Окисление алкилароматических углеводородов можно прово-
дить и азотной кислотой, причем этот окислитель способен окис-
•120
лять сразу две алкильные группы и длинные боковые цепи пря-
мо в карбоновые кислоты:
HNOS
COOH
Однако азотная кислота как окислитель применяется редко, так
как она сильно корродирует аппаратуру. Кроме того, образу-
ются побочные продукты нитрования.
Окисление алкилароматических углеводородов воздухом
имеет ряд особенностей. Толуол окисляется в бензойную кисло-
ту в мягких условиях (при 130—150°C). Ксилолы и другие
полиметилбензолы при жидкофазном каталитическом окислении
вначале образуют монокарбоновые кислоты, которые тормозят
окисление следующей метильной группы (ее можно окислить
только в очень жестких условиях —при 260—280°C и 7 МПа).
Дезактивирующее действие карбоксильной группы можно снять,
переводя ее в сложноэфирную. Так поступают при окислении
n-ксилола в терефталевую кислоту. Известны комбинированные
приемы окисления — вначале используют воздух, а завершают
процесс азотной кислотой (пример —получение пиромеллитовой
кислоты из дурола).
При жидкофазном каталитическом окислении толуола возду-
хом метильная группа окисляется в карбоксильную, при окисле-
нии этилбензола образуется только кетон, трег-алкильные груп-
пы не окисляются. Таким образом, способность к окислению из-
меняется в порядке
-СН(СН3)2 > —сн2-сна-сн3 > — сна-сна
а для образования карбоновых кислот справедлива другая по-
следовательность:
—СН3 > —СНа-СН3 > -СН(СН3)2
Катализатор должен растворяться в углеводороде; используют
его в количестве 0,05—0,2%. Для поддержания углеводорода
в жидкой фазе применяют давление 1—1,5 МПа.
Окисление нафтенов сходно с окислением парафинов. При
жидкофазном каталитическом окислении нафтенов воздухом
можно остановить процесс .на стадии образования спирта и ке-
тона. Для этого окисление, например циклогексана, ведут при
121
420—160 °C и небольшой (10—20%) степени конверсии:
В смеси содержание циклогексанола составляет 30—50%. Что-
бы увеличить его выход, надо снизить концентрацию кислорода
в воздухе. При более глубоком окислении происходит деструк-
ция цикла с образованием дикарбоновых кислот с тем же, что
и в исходном нафтене, числом углеродных атомов: при окисле-
нии циклогексана выход адипиновой кислоты составляет 90% и
более. Побочными продуктами окисления являются гликоли, ке-
тоспирты, дикетоны.
В промышленности окисление ведут в две ступени — сначала
окисляют циклогексан в смесь циклогексанола и циклогексано-
на, а на второй ступени (после отделения непрореагировавшего
циклогексана) окисляют смесь спирта и кетона 50—60%-ной
азотной кислотой. Вначале окисление азотной кислотой ведут
при 60—80 °C, а в конце процесса поднимают температуру до
105 °C:
+3HNO3^
НООС-(СН2)4-СООН + 1,5N2O3 + 1,5Н2О
Газофазное окисление нафтенов в промышленности не при-
меняют, так как идет деструкция цикла с образованием низших
-кислородсодержащих продуктов, диоксида углерода и воды.
3.2. Производство фенола и ацетона (Через
изопропилбензолгидропероксид
Химия и технология этого метода (кумольный метод) разра-
ботаны в СССР П. Г. Сергеевым, Б. Д. Кружаловым, Р. Ю. Уд-
рисом и М. С. Немцовым. Впервые в мире метод внедрен в про-
мышленность в нашей стране в 1949 г. В 1953 г. процесс внед-
рен в Канаде и Франции, в 1954 г. в США и ФРГ.
В процессе имеются следующие стадии: 1) окисление изо-
пропилбензола (кумол) воздухом; 2) выделение изопропилбен-
золгидропероксида; 3) разложение гидропероксида в присутст-
вии серной кислоты; 4) нейтрализация реакционной массы пос-
ле разложения; 5) выделение товарных фенола и ацетона,
который не должен содержать других алкилбензолов, фенола,
Скорость окисления зависит от чистоты изопропилбензола,
122
сернистых соединений и олефинов, тормозящих процесс. Прак-
тически скорость накопления гидропероксида равна 5—7%1
в час. Температуру процесса и глубину окисления подбирают
такими, чтобы содержание продуктов разложения было мини-
мальным. Для инициирования процесса используют сам изопро-
пилбензолгидропероксид, концентрация которого в исходной,
смеси составляет 2,5—3,5%. В реакторе поддерживают темпе-
ратуру НО—120 °C и давление 0,3—0,5 МПа, окисление прово-
дят в щелочной среде (pH 8,54-10,5). Окисление в щелочной4
среде обеспечивает нейтрализацию продуктов кислотного ха-
рактера, в частности муравьиной кислоты, образующейся при
окислении метанола. Окисление в эмульсии (при добавлении
водных растворов соды или щелочи) облегчает отвод тепла за.
счет испарения воды.
Применяют реакторы колонного типа, аппараты с мешалка-
ми и эрлифтные. В колонных аппаратах устанавливают барбо-
тажные тарелки, а над каждой тарелкой располагают змеевик
водяного охлаждения для отвода выделяющегося тепла
(«1970 кДж на 1 кг изопропилбензола). Реакторы с мешалка-
ми и эрлифтные устанавливают каскадом. Колонные реакторы
очень компактны, реакторы с мешалками отличаются простотой
конструкции. Кроме того, ступенчатая подача воздуха позволя-
ет выводить из реактора легкокипящие продукты окисления
(формальдегид и муравьиную кислоту, являющиеся ингибито-
рами окисления) и исключает попадание их в следующие реак-
торы каскада. В колонных реакторах ведут окисление «сухим»
способом, а выделяющееся тепло отводят с помощью змеевиков.
Гидропероксид выделяют из реакционной массы путем концент-
рирования — отгонки (в вакууме) непро-реагировавшего изопро-
пилбензола и воды; концентрированный гидропероксид содер-
жит 90—93% основного вещества.
Изопропилбензолгидропероксид — бесцветная маслянистая
жидкость; при попадании на кожу вызывает ожоги. Разлагается
с заметной скоростью при 145 °C с саморазогреванием. Его мож-
но превратить в твердую натриевую соль, обработав раствором
едкого натра:
^\ХС(СНз)2 ^^С(СН3)2 • 6Н2О
Jj] ООН + Na0H + 5Н2Э * Ц* Jjf OONa
Соль отфильтровывают, растворяют в воде и через раствор про-
пускают диоксид углерода. Изопропилбензолгидропероксид, вы-
деленный таким образом, имеет концентрацию основного веще-
ства 97—98%. Этот метод, однако, менее технологичен, чем пе-
регонка.
В промышленности для разложения изопропилбензолгидро-
пероксида применяют 98%-ную серную кислоту в количестве
0,07—0,1% в расчете на гидропероксид. Другие кислоты (напри-
мер, щавелевая, метафосфорная) активны лишь при 100—
123
’Рис. III.3. Технологическая схема получения фенола и ацетона кумольным
методом:
7 — окислительная колонна; 2 — подогреватель; 3 — конденсатор; 4 — сепаратор; 5 — рек-
тификационная колонна; 6 — вакуум-приемник; 7 — кипятильник; 8 — холодильник; 9 —
колонна разложения изопропилбензолгидропероксида; 10 — рбратный холодильник; 11,
12 — осушители; 13 — промывная колонна.
420 °C. Разложение в (Присутствии серной кислоты ведут при
•50—60 °C.
На рис. Ш.З приведена схема окисления изопропилбензола
<сухим» методом. Окисление проводят кислородом воздуха при
0,4—0,5 МПа и ПО—120 °C в реакторах колонного типа с та-
релками, над которыми вмонтированы холодильники (через них
циркулирует вода для съема выделяющего тепла и для удоб-
ства регулирования температуры по высоте колонны). Реактор
выполнен из нержавеющей стали. В реакционную массу добав-
ляют некоторое количество изопропилбензолгидропероксида
(ИПБГП) для снижения индукционного периода. Средняя ско-
рость окисления составляет 5—7% гидропероксида в час.
Свежий и возвратный изопропилбензол через подогрева-
тель 2 поступают в верхнюю часть колонны 1, а вниз подают
очищенный воздух. В колонне поддерживают температуру
ПО °C при помощи холодильников, расположенных по высоте
аппарата. Отходящие газы охлаждаются в конденсаторе 3, где
уносимый потоком воздуха изопропилбензол конденсируется.
Затем его отделяют от газа в сепараторе 4 и присоединяют к
потоку возвратного изопропилбензола.
Продукты окисления, содержащие 20—25% гидропероксида,
направляют в ректификационную колонну 5, в которой при глу-
боком вакууме (остаточное давление 400 Па) и температуре
85—92 °C отгоняют изопропилбензол. Отогнанный изопропил-
бензол в колонне 13 обрабатывают 5%-ным едким натром,
124
осушают над CaCh в колонне 12, затем осушают твердым
едким натром в колонне 11 и возвращают на окисление, смеши-
вая со свежим изопропилбензолом. Технический изопропилбен-
золгидропероксид с низа колонны 5 поступает на разложение
в пустотелую колонну 9. Для разложения применяют 1%-ный
раствор серной кислоты в смеси фенола и ацетона.
При разложении выделяется огромное количество тепла
(1676 кДж на 1 кг гидропероксида). Во избежание бурного
протекания реакции, которая может принять взрывной харак-
тер, применяют разбавленную кислоту, а выделяющееся тепло
отводят в выносном холодильнике 8, что позволяет поддержи-
вать температуру реакционной массы ;в пределах 40—60 °C. Ко-
лонна 9 имеет обратный холодильник 10, охлаждаемый рассо-
лом. Продукты разложения уходят на ректификацию, которую
осуществляют в многоколонной системе. При этом выделяют
ацетон, фенол и а-метилстирол. Фенол и а-метилстирол осво-
бождают от воды в вакуумных колоннах и подвергают очистке.
Температура по высоте окислительной колонны изменяется
сверху вниз от 115—120 до ПО—112 °C. По отношению к про-
дуктам основной реакции на стадии окисления образуется до
7% побочных веществ. Состав побочных продуктов таков [в %
(масс.)]:
«-Метилстирол .... 15,7 Кумилфенол .... 33,4
Димер а-метилстирола 43,0 Диметилфенилкарбинол 0,9
Ацетофенон 7,0
Продукты разложения изопропилбензолгидропероксида, по-
лучаемого окислением изопропилбензола в водо-содовой эмуль-
сии, имеют такой примерный состав [в % (масс.)]:
Изопропилбензол 76 Ацетофенон 1
Ацетон 8 а-Метилстирол 1
Фенол 14
Фенол является многотоннажным промышленным продук-
том, широко применяемым в производстве неионогенных мою-
щих веществ, присадок к маслам и топливам, пластических
масс и синтетических волокон, пестицидов и т. д. Известны и
другие способы его получения. Одним из первых промышленных
способов является щелочной гидролиз хлорбензола при 350—
370 °C и давлении до 30 МПа:
C,HSC1 + NaOH --► С.Н6ОН + Nad + Н2О
Известен также метод щелочного плавления бензолсульфокис-
лоты, но этот путь многостадиен и нетехнологичен. Сначала
бензолсульфокислоту нейтрализуют сульфитом натрия:
2C„H5SO3H + Na2SO3 -► 2С,H5SO3Na -J- SO2 + H2O
Образующийся бензолсульфонат натрия сплавляют со щелочью
при 320 °C и 30 МПа. При этом выделяется фенолят натрия, ко-
125
торый нейтрализуют диоксидом серы и получают фенол:
CeH6SO8Na + 2NaOH ---► C,H6ONa + Na2SO3 -f- H2O
CeH6ONa + SO2 + H2O -► 2C,H6OH + Na2SO3
Оба эти метода вытеснены более современными. В СССР
более 85% федола получают кумольным методом. Из современ-
ных окислительных процессов получения фенола следует еще
назвать окисление толуола, и прямое окисление бензола. По
первому методу толуол вначале окисляют воздухом в бензойную
кислоту при 150—175 °C в присутствии ацетата кобальта:
Со2+
СвН6-СН3 + 1,5О2 —> CeHs—СООН
—-HgO
Затем окисляют бензойную кислоту в бензоилсалициловую кис-
лоту в присутствии водяного пара и бензоата меди при 220—
245 °C:
СООН
Бензоилсалициловую кислоту гидролизуют водяным паром с об-
разованием бензойной и салициловой кислот, причем салицило-
вая кислота при этих условиях декарбоксилируется в фенол:
Процесс прямого окисления бензола в фенол сопровождается
образованием большого количества, побочных продуктов, что
затрудняет его реализацию в промышленности — образуются
гидрохинон, хинон, диоксид углерода, вода. Чтобы увеличить
селективность, ведут процесс при низкой степени конверсии
бензола, что вызывает рециркуляцию больших количеств сырья.
В промышленности осуществлены два варианта этого процес-
са— в газовой и жидкой фазе. Газофазный процесс ведут при
600—700 °C и 0,05—0,2 МПа, жидкофазный — при 400—500 °C
и 5—7 МПа. Степень конверсии бензола за один проход равна
4—5%, а выход фенола составляет примерно 50% на превра-
щенный бензол.
Ацетон является одним из широко применяемых растворите-
лей. Его используют в производстве химических волокон и ки-
26
нопленки, в экстракционных процессах, при очистке масел. Он
служит сырьем во многих химических синтезах — при получении
метакриловой кислоты и ее эфиров, изопрена, дифенилолпропа-
на (идущего на получение поликарбонатов и эпоксидных поли-
меров), изобутилметилкетона.
Наряду с рассмотренным кумольным методом ацетон в про-
мышленности получают окислительным дегидрированием изо-
пропилового спирта. При газофазном методе процесс ведут при
450—650 °C и невысокой степени конверсии спирта (с целью
снижения суммарного теплового эффекта) в присутствии катали-
заторов — меди, серебра, платины, никеля. Обычно металл
осаждают на носителе или применяют металлические сетки;
окислителем является воздух. Наряду с основной реакцией
-|-0,5О2
(СН3)2СНОН —> СН3—СО—СН3
—Н
протекает полное окисление и образуется уксусная кислота.
Если процесс вести без катализатора, при 300—350 °C и соотно-
шении спирт : кислород, равном 5:1, с выходом до 70% образу-
ется пероксид водорода.
Жидкофазный процесс ведут при 1,5—2,2 МПа. без катализа-
торов. Инициатор процесса — пероксид водорода, вводимый в
систему в количестве 0,5—1% (масс.). Процесс идет через про-
межуточное образование изопропилбёнзолгидропероксида, раз-
лагающегося на ацетон и пероксид водорода:
Ч-0,5Оа +0,502
(СН3)2СНОН ---> (СН3)2СНООН ——> СН3-СО-СН3
Окислительное дегидрирование ведут воздухом или кислородом;
выход пероксида водорода достигает 87% в расчете на взятый
спирт.
Процесс дегидрирования изопропилового спирта в ацетон
проводят при 325—400 °C в присутствии катализаторов — окси-
да цинка, меди и ее солей, солей хрома:
(СН3)2СНОН —> СН3—СО-СН3
—Иг
Ацетон можно также получать прямым окислением пропи-
лена в црисутствии водного раствора хлорида палладия при
90—120 °C и 0,9—1,2 МПа. В катализаторный раствор добавля-
ют хлориды меди и железа. Окислителями могут быть воздух
или кислород. Процесс аналогичен процессу получения ацеталь-
дегида (стр. 139):
PdCI.; CuCla
СН3—СН=СН2 4-0,50,---------»- СН3-СО—СН,
Существуют и другие способы получения ацетона, но они не
имеют большого промышленного значения.
127
3.3. Производство терефталевой кислоты
Терефталевая кислота НООС—С6Н4—СООН и ее димети-
ловый эфир (диметилтерефталат) играют важную роль в про-
изводстве синтетического волокна лавсан.
Для получения терефталевой кислоты методом окисления
пригодны n-алкилбензолы, среди которых в промышленности
применяют л-ксилол. При окислении п-ксилола воздухом в при-
сутствии нафтенатов и резинатов кобальта и марганца при
120—200 °C образуется n-толуиловая кислота, которая далее не
окисляется. Вследствие этого разработан четырехстадийный
процесс получения диметилтерефталата. Полученную на первой
стадии n-толуиловую кислоту этерифицируют метиловым спир-
том. Образующийся метиловый эфир n-толуиловой кислоты
окисляют в моноэфир терефталевой кислоты, который затем
этерифицируют в диметилтерефталат:
(I)
СООН
+1,5Оа
-н2о
(2)
СОО-СН,
I
(3)
СООН
соо—сн3
соо-сн3
—HjO
4-СН,ОН
(4)
СООН
соо—сн3
Вторая стадия окисления [реакция (3)] протекает в присутст-
вии растворимых солей кобальта и марганца (стеараты, ацета-
ты, бензоаты) при 160—180 °C и 1—1,5 МПа. В настоящее вре-
мя стадии окисления (1) и (3) совмещены в одном реакторе.
В одном аппарате проводят также и стадии (2) и (4). Это поз-
волило сократить число аппаратов.
На рис. Ш.4 приведена технологическая схема производства
диметилтерефталата. В реакторе 1 окисляют n-ксилол (свежий
128
Рис. III.4. Технологическая схема получения диметилтерефталата:
/ — реактор; 2, 6, 12, /7 — холодильники-конденсаторы; 3, 7, 13, 18 — сепараторы; 4, /5 —
сборники; 5 — этерификатор; 8 — подогреватель; 9 — отпарная колонна; 10, 14, /Р —ки-
пятильники; 11, 16 — ректификационные колонны.
и возвратный в соотношении 1 :2) и метиловый эфир «-толуи-
ловой кислоты кислородом воздуха. Реактор барботажного ти-
па, по высоте имеет охлаждающие змеевики. Воздух подают в
нижнюю часть аппарата компрессором под давлением 0,6—
1 МПа. Барботируя через реакционную массу, он захватывает
пары «-ксилола, которые конденсируются в холодильнике-кон-
денсаторе 2 и возвращаются на окисление. При периодическом
процессе ведут окисление до остаточного содержания «-ксилола
в реакционной массе 1% и до степени конверсии метил-«-толуи-
лата, равной примерно 50%. Оксидат содержит 20—25% «-то-
луиловой кислоты, 30% ее метилового эфира, 11—15% терефта-
левой кислоты, 20—25% метилтерефталата, а также небольшое
количество диметилтерефталата и смол.
Массу из реактора через сборник 4 насосом подают на этери-
фикацию в колонну 5 (оксидат подают сверху, а метанол —
снизу через подогреватель 8). Этерификация протекает при
250 °C и 2,5 МПа. Спирт подают в избытке; при этом пары ме-
танола уносят 'воду, способствуя увеличению выхода эфира.
Пары метанола и воды конденсируются в холодильнике-конден-
саторе б; водный раствор поступает на регенерацию спирта.
Эфиризат в отпарной колонне 9 отделяют от смол, а в ректифи-
кационной колонне 11 выделяют метиловый эфир л-толуиловой
кислоты. Его собирают в сборнике 15, а оттуда возвращают на
окисление в реактор 1.
В колонне 16 более летучий диметилтерефталат отгоняют
от диметиловых эфиров изомерных дикарбоновых кислот («изо-
фталаты»), С верха этой колонны диметилтерефталат поступает
на дополнительную очистку путем перекристаллизации из мета-
нола (до 99,9 %-ной чистоты). Кубовый остаток колонны 16 так-
9-254 129
же соде,ржит значительное количество диметилтерефталата. Его
извлекают кристаллизацией из метанольных растворов. Диме-
тилтерефталат в метаноле растворяется хуже; этот эфир вы-
деляют в виде кристаллов. Выход диметилтерефталата состав-
ляет 85—90% в расчете на исходный п-ксилол.
Разрабатывается одностадийный процесс окисления п-кси-
лола. Достигается это окислением в уксусной кислоте с приме-
нением смешанных катализаторов на основе солей кобальта и
марганца, промотированных NaBr и NH4Br. Применяют также
кобальтовый катализатор с добавками ацетальдегида или ме-
тилэтилкетона (сопряженное окисление). Окисление ведут в
одном реакторе колонного типа; выделяющееся тепло отводят
за счет циркуляции реакционной смеси через выносной холо-
дильник. Получают терефталевую кислоту, которая после очист-
ки (кристаллизацией из растворителей и возгонкой) имеет чис-
тоту 99,9%. В присутствии смеси солей кобальта и марганца
ведут окисление при 195—205 °C, в присутствии ацетата кобаль-
та с добавкой метилэтилкетона температура равна 130 °C, в
процессе с добавкой ацетальдегида температура составляет 80—
100 °C.
Интересны методы получения терефталевой кислоты из то-
луола. Из многочисленных способов промышленное применение
имеют хлорметилирование толуола и его окисление.
Хлорметилирование толуола осуществляют водным раство-
ром формальдегида в присутствии хлористого водорода. Обра-
зующийся n-хлорметилтолуол гидролизуют до п-метилбензило-
вого спирта, который окисляют в терефталевую кислоту азотной
кислотой:
Производство терефталевой кислоты и ее диметилового эфи-
ра через бензойную кислоту окислением толуола проходит через
четыре основные стадии:
1) окисление толуола в бензойную кислоту:
ru г нон
2) получение бензоата калия:
СООН
COOK
130
3) каталитическое диспропорционирование бензоата калия в
терефталат калия:
COOK COOK
<) -О +О
COOK
4) выделение терефталевой кислоты:
COOK COOH
6 ^>6
I I
COOK COOH
Катализаторами диспропорционирования являются бензоаты и
фталаты кадмия и цинка. Процесс ведут при 440—460 °C и дав-
лении 1—1,5 МПа, создаваемом диоксидом углерода во избежа-
ние декарбоксилирования. Применение катализатора в количе-
стве 5% обеспечивает выход кислоты до 86%. Метод является
перспективным, так как базируется на доступном сырье.
§ 4. ПРОИЗВОДСТВО КИСЛОРОДСОДЕРЖАЩИХ ПРОДУКТОВ
ОКИСЛЕНИЕМ ОЛЕФИНОВ
4.1. Особенности окисления олефинов
При окислении олефина кислород атакует и двойную связь,
и соседний с ней атом углерода. Это приводит к образованию
сложной смеси продуктов (выход целевого продукта неболь-
шой). Поэтому в цромышленной практике чаще осуществляют
гетерогеннокаталитические процессы, которые идут с более вы-
сокой селективностью по сравнению с радикально-цепными. Ге-
терогенный катализ применяют при окислении этилена в эти-
ленокоид
СН2=СН2 + 0,5Оа -> Н2С-СН2
при окислении пропилена в акролеин (окисление по насыщенно-
му атому углерода с сохранением двойной связи)
СН2=СН—СН3 + о2 ——> сн2=сн-сно
и при окислительном аммонолизе олефинов:
СН2=СН-СН3 + NH3 + 1,5О2 CH3=CHCN
Среди катализаторов гетерогенного окисления олефинов
практическое значение приобрели медь, серебро, оксиды пере-
9*
131
ходных металлов (меди, ванадия, молибдена, кобальта). Важ-
ную роль в катализе играет адсорбция реагентов на поверхно-
сти контакта. На металлах кислород адсорбируется очень
быстро, затем идет более медленный процесс его проникания
в приповерхностный слой. У серебряного катализатора происхо-
дит глубокое изменение свойств приповерхностного слоя. В слу-
чае неблагородных металлов при адсорбции на них кислорода
образуются оксиды. Адсорбция углеводородов на металлах
является обратимой, образующиеся связи непрочны. Более силь-
на адсорбция на оксидах, активными центрами которых явля-
ются ионы переходных металлов.
Образование продуктов окисления олефинов можно объяс-
нить взаимодействием молекулы кислорода, хемосорбированной
на поверхности металла, с олефином. Например, окисление про-
пилена на оксидах меди и молибдена можно представить так:
СН2=СН-СН3 + 2Ме+бб ---► СН2=СН—СН2-ООМе+ + Ме+ООН
СН2=СН—СН2—ООМе+ > СН2=СН-СНО + Ме+ОН
Ме+ОН + Ме+ООН --► Ме+бб + Ме+ + Н2О
Все большее значение приобретает процесс эпоксидирования
олефинов с помощью гидропероксидов. В промышленность внед-
рен так называемый халкон-процесс совместного получения
пропиленоксида и стирола из этилбензола:
СвН5-С2Нб + О2--► СвН5-СН-СН3
ООН
СН3-СН=СН2 + сбн5-сн-сн3 -->
ООН
---► СН3—НС---СН2 + СвНб—СН(ОН)-СН3
о | -н2о
СбНб-СН=СН2
Выход пропиленоксида достигает 95% по пропилену и 80—90%
по этилбензолгидропероксиду; выход стирола 95—98% по этил-
бензолу. Вместо этилбензола можно применять, например, изо-
бутан; тогда целевым продуктом будет изобутилен.
Катализаторами эпоксидирования являются соли молибдена,
ванадия и титана, а также комплексы (ацетилацетонаты и кар-
бонилы металлов). Скорость процесса и селективность зависят
от природы -металла и формы, в которой его применяют. В про-
мышленности используют нафтенат молибдена, карбонил и аце-
тилацетонат молибдена. Важное значение имеет и гидроперок-
сид. Так, в случае нафтената кобальта при 100 °C за 15 мин
с пропиленом реагируют: этилбензолгидропероксид на 92%, изо-
Л32
прбпилбензолгидропероксид на 79%, изопентилгидропероксид на
29%. В том же примерно порядке изменяется и селективность.
Основной из побочных реакций является разложение гидропер-
оксида:
ROOH-----► ROH-f-0,5O2
Обычно процесс проводят в жидкой фазе при 2—5-кратном из-
бытке олефина и 80—100 °C. При эпоксидировании легколетучих
олефинов необходимо повышенное давление. Количество ката-
лизатора составляет 0,001—0,005 моль на 1 моль гидроперок-
сида.
Большое практическое значение приобрел процесс окисления
олефинов по двойной связи в присутствии хлорида палладия.
При этом из этилена образуется ацетальдегид, а из других оле-
финов получаются кетоны:
СН2=СН2 + 0,5О2 --► СНз-СНО
СН3—СН=СН2 + 0,5О2 ► СН3-СО—сн3
При взаимодействии олефина с водным раствором PdCh обра-
зуется альдегид (или кетон), а палладий восстанавливается:
PdCl2 4- СН2=СН2 + Н2О -► СН3—СНО + Pd + 2НС1
Вводимый в реактор кислород окисляет палладий, но реакция
идет слишком медленно. Если же добавить в раствор соль двух-
валентной меди, происходит быстрое окисление палладия, а
выделяющаяся одновалентная медь легко окисляется кислоро-
дом, т. е. соль меди является переносчиком кислорода:
+Pd +0,5О2
2Си«+ ---2Си+ -г-?- 2Си«+
_Pd«+ —н2о
Эти реакции протекают в кислой среде; при этом хлорид пал-
ладия находится в форме HjPdCk.
Окислению в присутствии хлорида палладия могут подвер-
гаться и циклоолефины, а также ароматические углеводороды
с двойной связью в боковой цепи:
СН=СН2.
со—сн3
Побочными продуктами являются примеси хлоркетонов (за счет
хлорирующего действия хлоридов меди), а при окислении гомо-
логов этилена — примеси альдегидов; например, при окислении
пропилена образуется пропионовый альдегид:
СН3—СН=СН2
СН3—СО—СН3
СН3—СН2-СНО
133
Из всех продуктов окисления низкомолекулярных алифати-
ческих олефинов наибольшее значение имеют продукты окисле-
ния этилена и пропилена.
4.2. Производство этиленоксида
При окислении этилена образуется этиленоксид
СН2=СН2 + 0,5О2--► Н2С---СН3 ДЯ = —138 кДж/моль
При обычных условиях он представляет собой газ (т. кип.
10,7 °C), имеющий немного резкий залах. Пары этиленоксида
с воздухом взрывоопасны в концентрации 3—80% (об.). Этилен-
оксид токсичен, особенно для насекомых, вследствие чего его
широко применяют в качестве инсектицида при обработке фрук-
товых деревьев. Применяют этиленоксид и для обработки пи-
щевых продуктов с целью их консервации. Удобство' применения
заключается в том, что этиленоксид легко улетучивается с об-
работанных объектов. Чтобы избежать образования взрыво-
опасных смесей, этиленоксид применяют в смеси с диоксидом
углерода в соотношении 1 : 9.
Этиленоксид благодаря своей высокой реакционной способ-
ности широко используется в органическом синтезе. Наиболее
важная реакция этиленоксида — его гидратация в этиленгли-
коль
+н2о
Н2С---СН2 ---> СН2—СН2
\>// он Ан
проводимая либо при 50—100 °C в присутствии следов кислоты,
либо при 200 °C под давлением, но без катализатора.
При взаимодействии этиленоксида с аммиаком образуются
моно-, ди- и триэтаноламины:
+nh8
Н2С—сн2 ----->- NH2CH2-CH2OH
+nh3
2Н2С--СН2 ---► NH(CH2—СН2ОН)2
V
4-NHs
ЗН2С--СН2 ---► N(CH2—СН2ОН)з
V
При реакции этиленоксида с цианистым водородом образуется
134
этиленциангидрин, дегидратация которого дает ценный моно-
мер — акрилонитрил:
+HCN
н2с—сн2 —►- сн2-сн2 > ch2=chcn
\ / II ”"н2°
О ОН CN
Взаимодействие этиленоксида с кислотами, спиртами, алкилфе-
нолами и аминами дает поверхностно-активные вещества с мою-
щими свойствами (см. гл. IV). Наиболее ценными из этих ве-
ществ являются О1ксиэтилированные алкилфенолы (додецил- и
октилфенолы).
Некоторые оксиэтилированные спирты можно применять как
синтетические смазочные масла. Такие масла имеют хорошую
вязкостно-температурную характеристику и отличаются низкой
температурой застывания (до —70°C), их целесообразно при-
менять при высоких температурных режимах работы двигате-
лей— до 320 °C. В качестве смазочных масел могут быть ис-
пользованы не только моноэфиры полиэтиленгликолей
R—(ОСН2СН2)Л—ОН, но и сами полиэтиленгликоли
Н—(ОСН2СН2)Л—ОН, хорошо растворимые в воде.
При взаимодействии этиленоксида с сероводородом получа-
ются монотио- и дитиогликоли:
+h2s
---> СН2-СН2
ОН SH
2Н2С----СН2
СН2—CH2-S-CH2—сн2
он он
Дитиогликоль широко применяют как растворитель в печатном
деле.
Этиленоксид легко реагирует со многими другими соедине-
ниями, имеющими подвижный атом водорода.
В определенных условиях этиленоксид взаимодействует с
бензолом. При 0 °C в присутствии хлорида алюминия этилен-
оксид в избытке бензола с хорошим выходом дает fi-фенилэти-
довый спирт
СН2-СН2ОН
применяемый в парфюмерной промышленности.
Этиленоксид склонен к полимеризации в присутствии осно-
ваний. Образующиеся полиэтиленгликоли растворимы в воде.
В зависимости от молекулярной массы это либо прозрачные
жидкости, либо продукты с консистенцией парафина. Применя-
ются они в качестве смазочных масел, увлажнителей, раствори-
сь
телей в косметической, фармацевтической, резиновой и пищевой
промышленности, а также как вспомогательные вещества в бу-
мажной, нефтяной и других отраслях промышленности.
До недавнего времени этиленоксид получали только двух-
стадийным процессом из этилена через этиленхлоргидрин (хлор-
ный, или хлоргидринный метод). Вначале при пропускании эти-
лена и хлора через воду образуется этиленхлоргидрин:
СН2=СНа + С12 + Н2О -► НОСН2-СН2С1 + НС1
На второй стадии этиленхлоргидрин превращают в этиленок-
сид действием едких щелочей; в промышленности применяют
известковое молоко:
2НОСНа—СН2С1 + Са(ОН)2 ► 2Н2С-СН2 + СаС12 + Н2О
В последние годы преимущественное развитие получил ме-
тод прямого синтеза этиленоксида каталитическим окислением
этилена на серебряном катализаторе при 250—280 °C. Серебро
можно промотировать добавками Ba, Са, Na, Li или К.
Обычно применяют металлическое серебро, нанесенное на
пемзу. Наряду с образованием этиленоксида при этом идет
окисление этилена до СОг и НгО:
СН2=СН2 ЗОа -----► 2СОа -|- 2НаО ДЯ = —2645 кДж/моль
Селективность процесса зависит от способов приготовления ка-
тализатора, его активирования и термообработки. Для повыше-
ния селективности в реакционную смесь добавляют бензол, кси-
лол, этиловый спирт, амины, хлор (следы).
Серебряный катализатор чувствителен к сернистым, фосфор-
ным, мышьяковистым соединениям и к ацетилену, способному
образовывать взрывоопасный ацетиленид серебра. Поэтому эти-
лен и воздух, подаваемые на окисление, необходимо тщательно
очищать. Присутствие высших олефиновых и парафиновых угле-
водородов нежелательно, так как они окисляются до СО2 и Н2О.
Чистота этилена должна быть 96—98%. Процесс можно вести
с воздух-этиленовыми смесями, в которых концентрация этиле-
на либо ниже нижнего предела взрываемости {3% (об.)], либо
выше верхнего [29% (об.)].
Технологически процесс оформляют как непрерывный с не-
подвижным слоем катализатора. Разрабатывают также процесс
в псевдоожиженном слое. Наибольшие технологические трудно-
сти вызывает отвод тепла. Реактор со' стационарным слоем ка-
тализатора напоминает кожухотрубный теплообменник с труб-
ками небольшого диаметра: выделяющееся тепло отводится
циркулирующим по межтрубному пространству даутермом.
Схема прямого окисления этилена приведена на рис. III.5.
Сжатые до 1,5 МПа этилен и воздух, а также рециркулирующий
136
Рис. III.5. Технологическая схема получения этиленоксида прямым окисле-
нием этилена:
1,5 — реакторы; 2, 6, 9 — теплообменники; 3, 7 — холодильники; 4, 8 — скрубберы; 10 —<
отпарная колонна; 11 — колонна выделения СОг; 12 — колонна выделения этиленоксида.
газ поступают в реактор /, в котором регулируют температуру
теплоносителем. Контактные газы охлаждают вначале в тепло-
обменнике 2 рециркулирующим газом, а затем в холодильни-
ке 3 водой и направляют в скруббер 4, где этиленоксид погло-
щают водой.
Часть газов, выходящих из скруббера 4, идет на рециркуля-
цию, а остальное во избежание накопления азота и диоксида
углерода сбрасывают, но не сразу в атмосферу, а через реак-
тор 5, где этилен, оставшийся в газе, полностью окисляется.
Контактные газы из реактора 5 через теплообменник 6 и холо-
дильник 7 направляют в скруббер 8 второй ступени для извле-
чения этиленоксида. Неабсорбированные газы сбрасывают в ат-
мосферу.
Водные растворы этиленоксида из скрубберов 4 и 8 через
теплообменник 9 поступают в отпарную колонну 10, Воду воз-
вращают на орошение скрубберов, а верхний продукт (этилен-
оксид) идет в колонну 11 для отделения СО2. Окончательная
ректификация этиленоксида происходит в колонне 12, С низа
колонны уходят вода и высококипящие продукты, а с верха —
этиленоксид. Чистота его достигает 99,5%.
Температура в реакторе 1 составляет 250—280 °C, время
контакта равно 2—4 с. Производительность катализатора 600—
700 кг этиленоксида с 1 м3 катализатора в час. Выход этилен-
оксида примерно 60% от теоретического; селективность 60—
70%. Состав рециркулирующего газа таков: 3—5% (об.)
С2Н4, 5—7% (об.) О2, 8—10% (об.) СО2, остальное—азот.
Аппаратуру изготавливают из нержавеющей стали.
При окислении этилена в реакторах с псевдоожиженным
слоем катализатора схема установки мало отличается от рас-
смотренной со стационарным слоем катализатора (тоже при-
меняют два реактора с рециркуляцией газа из первого аппара-
та). Не нужно подогревать гае перед реактором, так как в такие
аппараты его можно подавать холодным, однако необходимо
устанавливать циклоны (после реактора или внутри него) для
улавливания катализаторной пыли.
137
4.3. Производство акролеина
Акролеин СНг = СН—СНО используют в производстве алли-
лового спирта, акрилонитрила, глицерина и других продуктов.
Окислением акролеина можно получить акриловую кислоту, а
гидрированием — пропионовый альдегид или н-пропиловый
спирт. Акролеин используют в синтезе инсектицидов и хитЛико-
фа1рмацевтических препаратов. На его основе уже сейчас полу-
чают значительное количество метионина СН3—S—СН2—СН2—
—CHNH2—СООН, добавляемого в корм домашней птицы в ка-
честве стимулятора ее роста. Но основным направлением ис-
пользования акролеина, однако, является синтез глицерина.
Известен метод получения акролеина альдольной конденса-
цией ацетальдегида и формальдегида с последующей дегидра-
тацией ацетальдоля:
СН3—СНО + НСНО ► СН2ОН-СН2-СНО СН2=СН—СНО
Основной промышленный метод производства акролеина —
газофазное каталитическое окисление пропилена:
-4-С>2
СН8—СН=СН2 СН2=СН—СНО
Сырьем является пропилен, очищенный от сернистых соедине-
ний и других олефинов. Можно применять и очищенную про-
пан-пропиленовую фракцию. Окислителем служит воздух или
кислород, разбавленный азотом или водяным паром. Побочно
образуются оксиды углерода, формальдегид, ацетальдегид, ор-
ганические кислоты и полимеры. Катализаторами процесса яв-
ляются закись и оксид меди на носителе (карбид кремния, ке-
рамика, пемза, оксид алюминия); промоторы — иод и селен.
Температура процесса 350—400°C, давление до 1 МПа, время
контакта 0,2—2 с. Наибольший выход акролеина достигнут на
катализаторах такого состава: 0,25% меди на пемзе или 0,1%
меди на .карборунде.
Для подавления полного окисления пропилена ведут про-
цесс при недостатке кислорода и малой степени конверсии.
Объемное соотношение пропилена и кислорода равно (34-4) U
а пропилена и воздуха 1 : (0,754-1,0). Количество водяного пара
составляет 2,5—3 объема на 1 объем пропилена. Водяной пар
снижает выход полимеров. При повышении давления увеличи-
вается производительность катализатора, уменьшается объем
газов, облегчается выделение непрореагировавшего пропилена.
Из реакционных газов, которые подвергают «закалке» водой,
извлекают акролеин водой при 1,8 МПа; при этом концентрация
его в водном растворе составляет 1,7% (масс.).
При разделении контактного газа вначале отгоняют легко
летучие вещества вместе с акролеином. Затем акролеин-сырец
перегоняют в присутствии гидрохинона (ингибитор полимериза-
ции), предварительно удалив из раствора кислород путем крат-
138
повременного (3 мин) нагревания при pH 6—7. Отогнанный
акролеиюсырец содержит 80—90% (масс.) акролеина, 3—10%'
(масс.) ацетальдегида, 2,4—6,0% (масс.) воды, 0,5—3% (масс.)
пропионового альдегида, 2—5% (масс.) ацетона и 1—2%’
(масс), полимеров. Ацетальдегид легко отгоняют ректифика-
цией. Вследствие близости температур кипения акролеина
(52,5 °C) и пропионового альдегида (49 °C) для выделения чис-
того акролеина проводят экстрактивную дистилляцию в присут-
ствии воды. В отпарной колонне выделяют 99,4 %-ный акролеин.
Реакторы окисления пропилена имеют различную конструк-
цию — со стационарным слоем, с восходящим потоком катали-
затора, трубчатые (в межтрубном пространстве циркулирует
теплоноситель). В реакторах с восходящим потоком катализато-
ра степень конверсии пропилена за один проход составляет
50%; выход акролеина примерно 70% считая на пропилен.
Имеются схемы с двумя реакторами со стационарным слоем
разных катализаторов (в первом аппарате—оксид меди, во
втором — молибдат висмута). На второй стадии условия более
жесткие (400—475°C). На обеих стадиях в катализатор вво-
дят промоторы (галогениды мышьяка, селена, теллура). Выход
акролеина составляет 70% считая на пропилен, степень конвер-
сии которого достигает 95%.
4.4. Производство ацетальдегида
Ацетальдегид СН3 — СНО является многотоннажным про-
дуктом и используется как сырье для производства уксусной
кислоты, уксусного ангидрида, надуксусной кислоты. По реак-
ции с синильной кислотой получают циангидрин, последующи-
ми превращениями которого получают молочную кислоту, акри-
лонитрил, эфиры акриловой кислоты. Процессами альдольной
конденсации получают бутан диол-1,3, кротоновый альдегид,
w-бутиловый спирт. Конденсация ацетальдегида с аммиаком да-
ет гомологи пиридина и вини л пиридинов — мономеры для СК.
Получают ацетальдегид разными методами. В 1881 г.
М. Г. Кучеров открыл реакцию взаимодействия ацетилена с во-
дой:
HgSO4
СН=СН + Н2О ----> СН3—СНО ДЯ = —162,8 кДж/моль
Процесс протекает только в присутствии катализаторов — суль-
фата ртути в разбавленной серной кислоте. Процесс ведут при
75—95 °C; количество ртути в кислоте (в расчете на HgO) со-
ставляет 0,5—1%; концентрация кислоты 10—20%. Металличе-
ская ртуть не катализирует процесс. Перевод ее в сульфат пред-
ставляет определенные трудности, так как ацетальдегид вос-
станавливает двухвалентную ртуть вначале до одновалентной,
139
а затем до металлической:
СН3-СНО + 2HgSO4 + Н2О -► HgaSO4+ H2SO4 + СН3—СООН
CH3-CHO + Hg2SO4 + H2O Hg + H2SO4 + СН3—СООН
Регенерацию сульфата ртути (продление срока службы катали-
затора) осуществляют, вводя в раствор сульфат трехвалентного
железа. Он окисляет ртуть, переводя ее в каталитически актив-
ную форму:
Hg + Fe2(SO4)3 HgSO4-f-2FeSO4
В результате окислительно-восстановительных реакций об7
разуется уксусная кислота как побочный продукт. Кроме того,
при альдольной конденсации появляются смолы, кротоновый
альдегид, небольшое количество паральдегида. Для снижения
смолообразования отдувают ацетальдегид из реакционной мас-
сы ацетиленом, который подают в небольшом избытке. Катали-
заторный раствор сильно корродирует аппаратуру, поэтому
реакторы гуммируют, а аппараты в системе регенерации изго-
товляют из нержавеющей стали.
Паральдегид — жидкий тример ацетальдегида:
О
+н+ СНз-НС^ \н-СН3
ЗСН3—СНО
СН3
Реакция образования паральдегида обратима, поэтому во мно-
гих процессах его используют вместо ацетальдегида.
В промышленности ацетальдегид получают и дегидрирова-
нием этилового спирта. Процесс можно осуществить двумя ме-
тодами — окислительным дегидрированием (окисление воздухом
на серебреном катализдторе) и каталитическим дегидрирова-
нием:
СН3—СН2ОН + 0,5О2 ——СН3—СНО ДЯ = —179 кДж/моль
СН3—СН,ОН —*• СН3-СНО ДЯ = 62,7 кДж/моль
—н3
По второму способу образуется меньше побочных продуктов,
поэтому он более распространен. Катализатор процесса дегид-
рирования— медь (с добавками 5% о'ксида кобальта и 2% ок-
сида хрома), .нанесенная на асбест. Температура процесса 275—
300 °C, степень конверсии спирта за один проход 33—50%. По-
бочный продукт — винилацетат (до 10% от ацетальдегида).
В настоящее время в промышленности ацетальдегид полу-
чают окислением этилена в присутствии водного раствора хло-
рида палладия как катализатора. Процесс можно проводить в
одну или в две стадии (окисление этилена и восстановление ка-
тализатора идет в одном аппарате, а окисление катализатора —
140
В стмссферу?
Рис. III.6. Технологическая схема получения ацетальдегида двухступенчатым
окислением этилена:
1 — регенератор; 2 — реактор; 3 — отпарная колонна; 4, 7, 10 — холодильники-конден-
саторы; 5, 8, 6, 9 — ректификационные колонны; 11 — кипятильники.
в другом). В обоих случаях соотношение скоростей самого
синтеза, окисления палладия хлоридом двухвалентной меди и
окисления одновалентной меди зависит от концентрации хлори-
да палладия, хлорида меди и от pH среды. Лучшие результаты
дают растворы, содержащие 0,3—0,5% PdCl2, 12—33%
CuC12-2H2O и 2—3% Си(ОСОСН3)2-Н2О.
Процесс ведут при 100—130 °C и 0,3—1,1 МПа. Среда кис-
лая (pH 0,8-?3) или нейтральная (до pH 7,5). Повышение pH
приводит к выпадению из раствора хлорида одновалентной
меди. Вследствие ограниченной растворимости солей меди кон-
центрация их в растворе невысока, а удельная производитель-
ность невелика. Соотношение олефина и кислорода в целях
взрывобезопасности поддерживают в интервале (2,5-?4) 1. Вы-
ход ацетальдегида составляет 84—98%. При одностадийном
процессе окислителем является кислород, а при двухстадий-
ном — воздух. Катализаторный раствор сильно коррозионный,
поэтому реакторы футеруют титаном.
На рис. III.6 приведена технологическая схема двухступен-
чатого окисления этилена в ацетальдегид. В регенераторе 1 воз-
духом регенерируют катализатор и подают его в реактор 2 на
окисление этилена. Образующийся ацетальдегид вместе с рас-
творенным в нем газом уносит катализатор в отпарную колон-
ну 3. Катализатор с низа отпарной колонны возвращают в ре-
генератор 1. Ацетальдегид в колонне 6 отделяют от воды, а
в колонне 9 — от растворенного газа. В колонне 6 для лучшего
отделения ацетальдегида снижают давление. Выделяющееся
при окислении тепло отводят за счет испарения части воды, со-
держащейся в катализаторном растворе. Поэтому в раствор не-
прерывно подают воду, поддерживая концентрацию катализа-
торного раствора постоянной.
141
Степень конверсии этилена при двухступенчатом процессе
приближается к 100%, что исключает рециркуляцию этилена.
Выход ацетальдегида составляет примерно 95% от теоретиче-
ского. Двухстадийный процесс отличается меньшей взрывоопас-
ностью (этилен не смешивается с воздухом), в нем применяют
более дешевый окислитель. Преимущество одностадийного про-
цесса — меньшие капитальные затраты.
Известен вариант гетерогеннокаталитического процесса, ког-
да PdCl2 наносят на твердый носитель (силикагель, пемза,
уголь). Однако этот процесс сопряжен с трудностями отвода
выделяющегося тепла, а, кроме того, нужно очень точно дози-
ровать кислород и этилен. Процесс не получил промышленной
реализации.
§ 5. ПРОИЗВОДСТВО КИСЛОРОДСОДЕРЖАЩИХ ПРОДУКТОВ
РЕАКЦИЯМИ ГИДРАТАЦИИ
5.1. Теоретические основы процессов
гидратации
В промышленности гидратацию олефинов в спирты осущест-
вляют в двух вариантах: сернокислотную (через промежуточное
образование алкилсульфатов) и прямую (непосредственное при-
соединение воды к олефину в присутствии твердых катализато-
ров) .
Первая стадия сернокислотной гидратации — взаимодейст-
вие олефина с серной кислотой — является реакцией электро-
фильного присоединения по ненасыщенной связи. Протон кисло-
ты, присоединяясь к молекуле олефина, образует карбоний-ион.
Он взаимодействует с кислотой, отщепляя протон и образуя
алкилсульфат:
4-н+ + +Haso4
RCH=CH2 ----> RCH-СНз RCH-CHg
OSOgH
Если в системе присутствует вода, реакция может протекать
иначе — через ион алкоксония, который отщепляет протон и
превращается 'в спирт:
4- 4-НаО
RCH-CHg RCH-CHg ------------+- RCH-CHg
I “Н I
Н2О+ он
Процесс сернокислотной гидратации сопровождается образова-
нием побочных продуктов. При взаимодействии алкилсульфата
с олефином образуются диалкилсульфаты:
RCH-CHg + RCH=CH, ----►
I
OSO3H
FRCHO—I SO
I
CH3 J2
2
142
Из двух молекул спирта при отщеплении молекулы воды обра-
зуется простой эфир:
2RCH—СН3 ——f RCH-O-CHR
। -н2о ( ।
ОН сн3 сн3
При дегидрировании этилового спирта может образоваться
ацетальдегид:
СН3—СН2ОН СН3—СНО
Серная кислота вызывает полимеризацию олефинов:
2RCH=CH2 --► RCH—CH=CHR и т. д.
СН3
Присоединение серной кислоты к олефину протекает по пра-
вилу Марковникова, поэтому первичный спирт получается толь-
ко из этилена. Из олефинов Сз и выше образуются вторичные
или третичные спирты. Олефины проявляют неодинаковую ре-
акционную способность. Например, для олефинов С2—С4 отно-
сительная скорость их поглощения 80%-ной серной кислотой
изменяется в 16 000 раз:
СН2=СН2 СН3-СН=СН2 СН3-СН2-СН=СН2 СН3-С(СН3)=СН2
1 500 1000 16 ооо
Поэтому в целях достижения приемлемой скорости для каждого
олефина отдельно подбирают параметры процесса — давление,
температуру и концентрацию серной кислоты (табл. III.2).
Вторую стадию — гидролиз алкилсульфатов — проводят
обычно острым паром; одновременно происходят отгонка спир-
та и разбавление серной кислоты. При гидролизе в спирты
превращаются и моно-, и диалкилсульфаты:
RCH—СН3 + Н2О RCH—СН3 + H2SO4
OSO3H ОН
•RCHO-1 SO2 +2H2O h 2RCH-CH3 + H2SO4
CH3 _2 OH
Количество диалкилсульфата зависит от избытка олефина.
Чтобы снизить расход серной кислоты, при абсорбции берут
Таблица III.2. Параметры абсорбции олефинов серной кислотой
Олефин Концентрация кислоты, % Давление в аб- сорбере, МПа Температура, °C
Этилен 96-98 2,5 65-75
Пропилен 70 0,8 65-70
Бутилен 75-85 0,3-0,4 45
Изобутилен 65 0,3—0,4 30
143
1,2—1,3 моль олефина на 1 моль кислоты. Степень конверсии
олефинов при сернокислотной гидратации достигает 97%, а се-
лективность по спирту составляет 85—95%.
Среди низкомолекулярных алифатических спиртов, получа-
емых гидратацией олефинов, главным является этиловый спирт.
Наиболее крупный его потребитель — промышленность синте-
тического каучука. Кроме того, этиловый спирт идет на получе-
ние уксусного альдегида, уксусной кислоты, диэтилового эфира,
этил ацетата. Применяется он и как растворитель для разнооб-
разных целей. Производство этилового спирта гидратацией эти-
лена дает экономию миллионов тонн зерна и картофеля (на
каждой тонне синтетического спирта можно сэкономить более
4 т зерна или 12 т картофеля), Себестоимость синтетического
этилового спирта почти в четыре раза ниже, чем себестоимость
спирта, получаемого из пищевого сырья, а затраты труда
в 20 раз меньше.
При сернокислотной гидратации пропилена образуется изо-
пропиловый спирт:
+h2so4 +н2о
сн8-сн=сн2--------► сн8-сн-сн8 ———>• сн8-сн—сн3
A— H2SO4 1
SO3H ОН
Изопропиловый спирт идет на производство ацетона и изопро-
пилацетата, используется как растворитель нитратцеллюлозных
лаков, как антифриз (в смеси с метанолом), а также для при-
готовления косметических '.препаратов.
Гидратацией н-бутилена в присутствии H2SO4 получают
втор-бутиловый спирт:
+h2so4 +н2о
СН3-СН2-СН=СН2--------> СН3-СН2— СН-СНз ———>
—H2SO4
SO3H
---► СН3-СН2-СН-СН3
Ан
втор-Бутиловый спирт идет на производство метилэтилкетона
и втор-бутилацетата, применяется как растворитель нитратцел-
люлозных лаков, как компонент гидротормозных жидкостей.
Если гидратации подвергать изобутилен, образуется трет-бу-
тиловый спирт.
Прямая гидратация олефинов протекает -в одну стадию в
присутствии катализаторов:
RCH=CH2 + H2O RCH-CH3
I
ОН
Эффективным катализатором является фосфорная кислота на
твердом носителе с развитой поверхностью (широкопористый
силикагель, алюмосиликат). Так как фосфорная кислота вымы-
144
вается с поверхности контакта и обладает корродирующим дей-
ствием, разработаны катализаторы на основе оксида вольфрама,
а для гидратации изобутилена предложены катионообменные
смолы.
Процесс прямой гидратации олефинов экзотермический и
обратимый. Равновесие смещается вправо лишь при низкой тем-
пературе, т. е. условия равновесия не выгодны для образования
спиртов. Равновесная степень конверсии олефина снижается
с повышением температуры, но если понизить температуру,
реакция идет слишком медленно. Увеличить степень конвер-
сии олефина можно, повышая давление.
Прямая гидратация сопровождается образованием побочных
продуктов, в первую очередь простых эфиров (за счет взаимо-
действия ионов карбония с образовавшимся спиртом):
4- -н+
RCH+RCH-CH3 RCH—СН3 <=* TRCH-TO
СН, ОН НО—CH(CH,)R . СН, J,
Процесс обычно ведут при избытке олефина, поэтому выход
простого эфира не превышает 2%. В качестве побочных продук-
тов образуются также полимеры. На скорость фосфорнокислот-
ной гидратации влияют поверхность носителя и размер пор.
С (ростом парциального давления олефина скорость гидратации
повышается, но водяной пар оказывает обратное действие. Дело
в том, что фосфорная кислота на поверхности носителя обра-
зует тонкую пленку, в которой при каждой данной температуре
устанавливается равновесная концентрация воды, соответствую-
щая парциальному давлению водяного пара. При чрезмерном
повышении этого давления фосфорная кислота- разбавляется и
скорость реакции падает. При снижении парциального давления
водяного пара в жидкой пленке происходят полимеризация оле-
фина и образование простого эфира, что снижает селективность
процесса.
Сульфокатионит КУ-2 обладает большей кислотностью, чем
фосфорная кислота, поэтому на нем температуру гидратации
можно снизить. Но катионит, как уже говорилось, проявляет ак-
тивность только при прямой гидратации изобутилена.
5.2. Производство этилового спирта
сернокислотной гидратацией этилена
В процессе производства этилового спирта сернокислотной
гидратацией этилена имеются следующие стадии:
1) абсорбция этилена серной кислотой с образованием этил-
сульфатов;
2) гидролиз этилсульфата в этиловый спирт с выделением
серной кислоты;
10—254 145
3) отделение этилового /спирта от серной кислоты и его
очистка;
4) регенерация серной кислоты.
Важнейшая стадия — абсорбция этилена серной кислотой.
При этом протекают следующие реакции:
СН2=СН2 + H2SO4 < —СН3—CH2OSO2OH ДЯ = —146 кДж/моль
этилсульфат
СН3—CH2OSO2OH + CH2=CH2 +=± (CH3-CH2O)2SO2
диэтилсульфат
ДЯ = —96 кДж/моль
Диэтилсульфат можег реагировать с серной кислотой и превра-
щаться в этилсульфат
(CH3-CH2O)2SO2 + H2SO4 2СН3—CH2OSO2OH
причем чем выше степень насыщения серной кислоты этиленом,
тем меньше содержится диэтилсульфата в смеси.
Этилен может реагировать с серной кислотой только в рас-
творенном состоянии, т. е. он вначале растворяется в ней, а
затем взаимодействует, образуя' этилсульфат (этилсерная кис-
лота). С образованием этилсульфата скорость поглощения уве-
личивается, так как этилен в нем растворяется лучше, чем в
чистой серной кислоте. Однако этилсульфат разбавляет серную
кислоту, в связи с чем общая скорость реакции снижается. По-
этому по высоте абсорбционной колонны условия для поглоще-
ния этилена неодинаковы.
Скорость абсорбции этилена зависит также от концентрации
серной кислоты, от давления, температуры и интенсивности пе-
ремешивания. Максимальная степень насыщения этиленом
97,5 %-ной кислоты составила 1,4 моль С2Н4 на 1 моль кислоты,
а для 95 %-ной кислоты 1,2 моль С2Н4 на 1 моль кислоты.
Обычно в заводских условиях абсорбцию ведут до степени насы-
щения в верху абсорбера, равной 0,6 моль/моль, что при
97,5%-ной кислоте достигается за 1 ч 15 мин, а при 95%-ной
за 2 ч 15 мин. Поэтому практически концентрация серной кис-
лоты должна быть 97—98%. Температура абсорбции равна 70—
80 °C. При повышении температуры скорость абсорбции увели-
чивается, но при этом усиливается полимеризация этилена и
растет выход диэтилового эфира. Хотя скорость абсорбции эти-
лена серной кислотой и зависит от давления, однако повышение
давления сверх 1,5 МПа не оказывает заметного влияния. На
практике поддерживают парциальное давление этилена пример-
но 1,5 МПа. В случае использования этан-этиленовой фракции
с 50—60% этилена общее давление составляет 2,5—3 МПа. При
хорошей абсорбции этилен поглощается серной кислотой до
остаточного содержания 2—6%, что соответствует его парциаль-
ному давлению 0,15—0,25 МПа. Скорость поглощения этилена
при этих давлениях составляет 0,3—0,8 моль на 1 моль кислоты.
Средняя скорость насыщения этилена при 1,5 МПа равна
146
0,3 моль на 1 моль кислоты в час. Следовательно, длительность
реакции составляет 3 ч.
Чтобы повысить растворимость этилена, к исходной кислоте,
подаваемой в колонну, примешивают некоторое количество кис-
лоты, частично насыщенной этиленом. Выделяющееся тепло
(121 кДж на 1 моль поглощенного этилена) снимают в холо-
дильниках, расположенных внутри абсорбера по его высоте;
обычно применяют абсорберы с 20 холодильниками.
В этиленовой фракции, идущей для производства этилового
спирта, не должно содержаться более 0,1% пропилена; в про-
тивном случае ухудшается качество спирта (примеси изопропи-
лового спирта и полимеров). Не допускается и наличие ацети-
лена, так как он способен образовать взрывоопасный ацетиле-
нид меди при соприкосновении с медной футеровкой аппарату-
ры, используемой для защиты от коррозии.
Вторая стадия получения этилового спирта — гидролиз этил-
сульфатов. При этом протекают следующие реакции:
+н2о
C2H6OSO2OH C2H5OH + H2SO4
+Н2о
C2H5OSO2OH + С2Н5ОН
(C2H5O)2SO2
+с2н5он
С2Нб—О—С2Нб + C2H5OSO2OH
+н2о
C2H5-O-C2H5 + H2SO4
Этилсульфаты при обычной температуре стабильны в присутст-
вии воды, но при нагревании скорость их гидролиза сильно уве-
личивается. Диэтилсульфат гидролизуется примерно вдвое мед-
леннее, чем этилсульфат. В щелочной среде этилсульфат до-
вольно стабилен даже при кипячении.
В качестве побочного продукта при гидролизе, как видно из
уравнений реакции, образуется диэтиловый эфир. Чтобы умень-
шить его выход, поступают следующим образом. Гидролиз ведут
по возможности быстро и затем сразу же отгоняют из смеси
спирт, чтобы его концентрация в растворе была небольшой. При
этом получается 4—7% эфира. Спирт получается в виде 35%-но-
го раствора, а концентрация серной кислоты снижается до 45%.
Можно поступать и иначе. Смесь этилсульфатов разбавляют во-
дой; при этом кислота и этилсульфат переходят в раствор, а
диэтилсульфат выделяется (нижний слой). Его отделяют и
гидролизуют 30%-ной серной кислотой:
(C2H5O)2SO2 + H2SO4
2C2H5OSO2OH
После раздельного гидролиза оба слоя подвергают совместной
переработке. Выход спирта достигает 97% от теоретического,
а выход эфира составляет всего 1—2%. Концентрация отрабо-
танной серной кислоты равна 35%. При гидролизе поддержива-
147
Рис. III.7. Технологическая схема получения этилового спирта сернокислот-
ной гидратацией этилена:
1, 7 — сепараторы; 2 —абсорбер; 3 — гидролизер; 4— отпарная колонна; б — нейтрали-
затор; 6 — конденсатор; 8 — подогреватель; 9, 12 — ректификационные колонны; 10, 13 —
дефлегматоры; 11 — промывная колонна; 14 — холодильник.
ют температуру от 90 до 100 °C. Продолжительность гидролиза
зависит от концентрации кислоты (от степени ее разбавления):
при 40%-ной кислоте гидролиз длится более 10 ч, при 45%-ной
1 ч, при 50%-ной 0,5 ч. После гидролиза смесь подают в отпар-
ную колонну, где водо-спиртовую смесь отгоняют с водяным па-
ром. С низа колонны уходит отработанная серная кислота, за-
грязненная полимерами. Полимеры извлекают зеленым маслом
(газойлем), кислоту упаривают до 88—90%-ной концентрации
и «доукрепляют» олеумом для дальнейшего использования в
процессе.
Технологическая схема процесса приведена на рис. III.7.
Этиленсодержащую фракцию подают в нижнюю часть абсорбе-
ра 2. Свежую серную кислоту смешивают с этилсульфатом, от-
водимым с восьмой тарелки снизу, и подают на орошение абсор-
бера. Соотношение свежей кислоты и экстракта устанавливают
148
таким, чтобы получить смесь со степенью насыщения 0,5—0,5
моль С2Н4 на 1 моль H2SO4.
Абсорбер имеет 20 тарелок; над каждой тарелкой установ-
лен холодильник для отвода выделяющеюся тепла, что позволя-
ет поддерживать температуру 75—80 °C. Изнутри абсорбер ос-
винцован и футерован кислотостойкой плиткой. С низа
абсорбера этилсульфат поступает в гидролизер 3 с фарфоровой
насадкой, смешиваясь на входе с водой: Из гидролизера смесь
идет в отпарную колонну 4, где завершается гидролиз и отпари-
вается спирто-водная смесь. С низа колонны 4 разбавленная
кислота уходит на очистку и упаривание, а пары спирта и воды
направляют в колонну 5, где их нейтрализуют едким натром.
Далее пары конденсируются и через сепаратор 7 и подогре-
ватель 8 уходят на ректификацию. В колонне 9 отбирают диэти-
ловый эфир, который промывают водой в колонне 11 и отводят
в парк. Спирт от эфира отмывают водой, которую возвращают
в колонну 9. Спирто-водная смесь из колонны 9 идет в колон-
ну 12, где отгоняется этиловый спирт. Его отбирают с верха .ко-
лонны, охлаждают и отводят в парк. Температуру верха ко-
лонн 9 и 12 регулируют при помощи дефлегматоров 10 и 13.
Отдувочный газ отводят из системы после предварительного из-
влечения из него паров этилового спирта.
Регулировать режим работы абсорбера довольно сложно,,
так как на каждой тарелке выделяется различное количество
тепла в зависимости от скорости поглощения этилена, изме-
няющейся по высоте колонны. На верхних тарелках, орошаемых
98%-ной серной кислотой, которая очень энергично реагирует
с этиленом, выделяется мало тепла. Это объясняется тем, что
в газе на верхних тарелках содержится мало этилена, так как
он поглотился на нижних тарелках. С другой стороны, раство-
римость этилена, в серной кислоте, еще не содержащей этил-
сульфатов, мала. В верхней зоне колонны свободного этилена
(в растворенном состояний) практически нет. Чем ниже распо-
•части абсорбера выделяется
ложена тарелка в абсорбере, чем больше концентрация этил-
сульфата и тем выше растворяющая способность смеси по отно-
шению к этилену. В это^
шее количество тепла.
К низу абсорбера растворимость этилена увеличивается, но
из-за уменьшения количества свободной кислоты скорость его
поглощения снижается и выделяется меньше тепла. В реакцион-
ной смеси, уходящей с низа абсорбера, содержится много рас-
творенного этилена.
На установках сернокислотной гидратации получают этило-
вый спирт концентрацией 95,6%. Это так называемый спирт-
ректификат—азеотропная смесь с водой (4,4% воды), кипящая
при 78,1 °C.
Безводный этиловый спирт получают азеотропной
дистилляцией с бензолом. Бензол образует с водным спиртом
тройную азеотропную смесь, кипящую при 64,9 °C и содержа-
14£>
щую 74,1% бензола, 18,5% спирта и 7,4% воды. Эта смесь при
отстаивании расслаивается на верхний (бензоло-спиртовый) и
нижний (водо-спиртовый) слои.
5.3. Производство изопропилового и изобутилового
спиртов сернокислотной гидратацией пропилена
и н-бутиленов
Первым синтетическим спиртом, полученным сернокислотной
гидратацией пропилена, был изопропиловый спирт. Процесс
абсорбции пропилена серной кислотой можно осуществить в
двух вариантах: 92%-ной кислотой при 20°C и атмосферном
давлении, либо 80%-ной кислотой при 60°C и 2,5 МПа.
При втором методе в абсорбере одновременно идет и гидро-
лиз изопропилсульфата. Во второй колонне из реакционной сме-
си отгоняют изопропиловый спирт и диизопропиловый эфир, а
серную кислоту возвращают в абсорбер. Регенерация серной
кислоты не требуется. Изопропиловый спирт образует с водой
азеотропную смесь, содержащую 12,3% (масс.) воды и кипящую
при 80,4 °C (чистый изопропиловый спирт кипит при 82,4 °C).
При поступлении реакционной смеси из абсорбера во вторую ко-
лонну и смеси добавляют такое количество воды, чтобы кон-
центрация серной кислоты была равна 60%. При отгонке азео-
тропной смеси изопропиловый спирт+вода концентрация серной
кислоты увеличивается до 70%.
Абсорбцию пропилена 92%-ной серной .кислотой осуществля-
ют в таких же абсорберах, какие применяют для абсорбции эти-
лена. При действии кислоты на пропилен наряду с образова-
нием изопропилсульфатов происходит полимеризация пропилена.
Применяя растворители, инертные к серной кислоте, удает-
ся резко затормозить полимеризацию. Гидратации можно под-
вергать чистый пропилен или пропан-пропиленовую фракцию
с различным содержанием пропилена — 20—24% (из газов тер-
мического крекинга), 40% (из газов каталитического крекинга)
и 80% (из газов пиролиза).
С увеличением парциального давления паров пропилена воз-
растает скорость его поглощения серной кислотой. При исполь-
зовании пропан-пропиленовой фракции для сохранения той же
эффективности поглощения необходимо иметь более высокое об-
щее давление. При парциальном давлении пропилена 1 МПа
для пропан-пропиленовой фракции с 20% пропилена требуется
давление 3,7 МПа, для 40%-ной фракции 2,5 МПа, для 80 %-ной
1,25 МПа. Абсорбцию ведут при линейной скорости потока
в свободном сечении колонны, равной 75 мм/с. При увеличении
скорости подачи пропилена улучшается его контакт с кислотой
и возрастает скорость поглощения. При увеличении скорости до
400 мм/с степень использования пропилена уменьшается.
Решающим фактором процесса является концентрация сер-
ной кислоты: каждой концентрации соответствуют свои опти-
150
мальные температура и скорость поглощения, причем скорость
поглощения пропилена 80%-ной кислотой в 1,5 раза выше ско-
рости его поглощения 70%-ной кислотой. Применение 70%-нои
кислоты предпочтительнее — более концентрированная кислота
лишь незначительно уменьшает продолжительность поглоще-
ния, а при использовании 70%-ной кислоты сильно облегчает-
ся процесс упаривания и снижается выход полимеров.
Температура при гидролизе составляет 70—90°C; при по-
нижении температуры скорость этого процесса падает. Концент-
рация кислоты при гидролизе влияет на выход спирта, на соот-
ношение спирта и эфира. Для получения максимального выхода
изопропилового спирта гидролиз кислотных экстрактов следует
проводить при их разбавлении водой до 35%-ной концентрации.
Длительность гидролиза при этом составляет всего 10 мин.
Сернокислотной гидратацией н-бутиленов получают втор-бу-
тиловый спирт. В качестве сырья .можно использовать бутан-бу-
тиленовую фракцию газов крекинга и бутилены после дегидри-
рования бутана. Присутствие бутадиена нежелательно: он вы-
зывает ряд побочных реакций, а извлекать его затруднительно.
При абсорбции бутиленов наблюдается их полимеризация. Чтобы
избежать значительного образования полимеров (а способ-
ность к полимеризации при обработке олефинов серной кисло-
той растет с увеличением молекулярной массы олефина от эти-
лена к н-бутилену), абсорбцию бутиленов ведут в мягких усло-
виях—при «40 °C, 0,3—0,4 МПа и концентрации кислоты 75—
80%.
Бутилены поглощаются серной кислотой почти нацело. Гид-
ролиз реакционной массы и выделение втор-бутилового спирта
осуществляют по схеме, описанной для этилового спирта. Реак-
ционную смесь -разбавляют водой до 30%-ной концентрации.
Обычно втор-бутиловыи спирт начинает выделяться уже при
разбавлении; его отделяют и перегоняют. Частичный гидролиз
происходит и в случае использования 70%-ной кислоты. При
работе на бутан-бутиленовой фракции газов термического кре-
кинга, после извлечения изобутилена содержащей 35% бутиле-
нов, степень насыщения кислоты не превышает 0,8 моль бутиле-
нов на 1 моль серной кислоты. Обычно же получают экстракты
с 0,5—0,6 моль Н-С4Н8 на 1 моль кислоты.
Очистка втор-бутилового спирта затруднена из-за наличия
в нем примесей. Можно применять следующий прием. К спир-
ту-с.ырцу добавляют диизобутилен и отгоняют тройную азео-
тропную смесь воды, диизобутилена и примесей. После этого
втор-бутиловый спирт подвергают ректификации. Чистый втор-
бутиловый спирт кипит при 99,5 °C.
Если в газах присутствует изобутилен, он легко абсорбиру-
ется 60—65%-ной серной кислотой. При абсорбции изобутилена
получают экстракт, содержащий 1,5 моль изобутилена на
1 моль кислоты. При этом содержание изобутилена в бутан-
бутиленовой фракции снижается с 10 до 1%. После извлечения
151
изобутилена газ идет на получение втор-бутилового спирта.
Лучшие результаты получаются при двухступенчатой абсорбции
изобутилена. На первой ступени при 38 °C изобутилен извлека-
ют кислотой, поступающей со второй ступени и уже имеющей
степень насыщения, равную 0,5 моль/моль. На вторую ступень
поступает свежая кислота, а абсорбция протекает при низкой
температуре (10—20 °C) до степени насыщения, равной
0,5 моль/моль. С первой ступени кислотный экстракт
(1,5 моль/моль) отводят на гидролиз для получения трет-бути-
лового спирта. Смесь разбавляют водой и дают отстояться. От-
гонку с серной кислотой вести нельзя, так как при нагревании
с кислотой спирт дегидратируется. Выделенный при отстаивании
водный слои перегоняют с водяным паром, получая спирт, раз-
бавленный водой, так как при ректификации отгоняется азео-
тропная смесь, содержащая 22% воды и кипящая при 81 °C.
Чистый втор-бутиловый спирт кипит при 82,4 °C. Для получения
безводного спирта можно проводить обезвоживание бензолом
обычным методом.
5.4. Производство этилового спирта
прямой гидратацией этилена
Недостатки метода сернокислотной гидратации этилена (вы-
сокие капиталовложения при сооружении установок, необходи-
мость применения кислотостойкой аппаратуры, большие энер-
гозатраты на упаривание кислоты) вызвали необходимость раз-
работки процесса прямой гидратации. Трудность состояла в том,
что при этой реакции не удавалось достигнуть хорошего вы-
хода этилового спирта. Все предлагаемые катализаторы — вод-
ные растворы минеральных кислот (серной, соляной, фосфор-
ной) и твердые контакты (соли и оксиды металлов) —давали
низкий выход целевого продукта.
Присоединение воды к этилену является равновесной экзо-
термической реакцией:
С2Н4 + Н2О С2НВОН ДЯ = —45,5 кДж/моль
Равновесный выход спирта зависит от условий гидратации —
от соотношения Н2О : С2Н4, температуры и давления. Поэтому
реакцию следует проводить при низких температурах, высоких
давлениях и большом мольном соотношении воды и этилена.
Наибольший выход («18%) получен при 150°C, 8 МПа и соот-
ношении Н2О С2Н4, равном 0,8 : 1.
Наилучшим катализатором является фосфорная кислота на
носителе (кизельгур, силикагель, алюмосиликат). Исследуют
для этой цели и нейтральные катализаторы, например оксид
вольфрама на носителях. В присутствии катализаторов на мел-
копористых носителях, пропитанных фосфорной кислотой, выход
спирта на 20—25% ниже, чем на широкопористых; широкопо-
ристый носитель облегчает диффузионные процессы, т. е. важ-
152
ними показателями при катализе являются скелет и поверх-
ность носителя. Катализ осуществляется свободной кислотой,
находящейся на поверхности носителя в жидком состоянии. Кон-
центрация кислоты, зависящая от давления водяного пара над
поверхностью кислоты, — один из важных показателей активно-
сти катализатора.
При повышении температуры с 240 до 280 °C степень кон-
версии этилена значительно увеличивается. При мольном соотно-
шении воды и этилена, равном 0,65: 1, общем давлении 7 МПа
и объемной скорости -подачи этилена 2000 ч-1 степень конвер-
сии составляет 3% при 260°С и 5% при 280°C. При объемной
скорости подачи этилена 2700 ч-1 его степень конверсии увели-
чивается с 2,2% при 260 °C до 3,7% при 280 °C. При дальней-
шем повышении температуры степень конверсии изменяется не-
значительно. При общем давлении 7 МПа и мольном соотноше-
нии воды .и этилена, равном 0,6 : 1, давление водяных паров со-
ставляет 2,75 МПа. Этому давлению соответствует концентра-
ция кислоты 83% при 2806С и 85% при 290°C.
Если концентрация кислоты на поверхности катализатора
ниже 83%, выход спирта резко уменьшается. Поэтому нельзя
поддерживать большие мольные соотношения воды и этилена,
как это вытекает из термодинамики процесса, так как в этом
случае концентрация фосфорной кислоты становится ниже опти-
мальной, равной 83—85%. На практике мольное соотношение
воды и этилена поддерживают равным (0,64-0,7) 1, время кон-
такта 18—20 с, объемную скорость подачи этилена 1800—
2000 ч-1. Степень конверсии этилена за один проход составляет
4—6%. Низкая степень конверсии и, следовательно, необходи-
мость многократной циркуляции этилена, является существен-
ным недостатком процесса и заставляет применять концентриро-
ванный этилен (не ниже 98%).
Производительность 1 м3 катализатора в час составляет
180—200 кг спирта. Катализатор работает в течение 500 ч, пос-
ле чего его активность снижается вследствие уноса фосфорной
кислоты (0,4—0,5 кг с 1 м3 катализатора ,в час). Для удлинения
срока службы катализатор во время процесса подпитывают фос-
форной кислотой. В этом случае длительность работы катализа-
тора без регенерации увеличивается до 900 ч.
В процессе прямой гидратации этилена имеются следующие
основные стадии: 1) приготовление паро-газовой смеси; 2) ре-
куперация тепла циркуляционных потоков; 3) гидратация эти-
лена; 4) нейтрализация продуктов реакции; 5) очистка цирку-
ляционного газа.
Паро-газовую смесь получают двумя способами: 1) совмест-
ным нагреванием паров воды и этилена в теплообменниках и
в трубчатой печи; 2) смешением этилена с перегретым паром
высокого давления.
Из-за многократной циркуляции этилена экономически важ-
но создать рациональную схему теплообмена. Если паро-газо-
153
Рис. III.8. Технологическая схема получения этилового спирта прямой гидра-
тацией этилена:
1 — печь; 2 — гидрататор; 3 — солеотделитель; 4, 5 — теплообменники-рекуператоры; 6,
8 — сепараторы; 7 — скруббер.
вую смесь получают смешением этилена с паром высокого дав-
ления, схема теплообмена усложняется, так как продукты реак-
ции (пары спирта, не вступивший в реакцию этилен, водяной
пар) после гидрататора вступают в теплообмен только с этиле-
ном, которому нельзя передать все тепло горячего потока. При-
ходится устанавливать котлы-утилизаторы и получать пар низ-
кого давления. При пуске системы необходим дополнительный
теплообменник для нагревания этилена паром высокого давле-
ния. Однако применение пара высокого давления оправдано
только там, где близко расположена ТЭЦ, производящая такой
пар.
Чаще паро-газовую смесь получают первым способом. Эти-
лен и воду нагревают и испаряют в теплообменниках за счет
тепла нейтрализованных продуктов, выходящих из гидрататора,
а далее в печи с огневым- нагревом испаряют оставшееся коли-
чество воды и нагревают смесь до 280—290 °C. Схема эта про-
ста и удобна в эксплуатации (рис. III.8).
Этилен из цеха газоразделения сжимают до 7 МПа, смеши-
вают с циркуляционным газом и вместе с паровым конденсатом
подают в теплообменники-рекуператоры 5 и 4. Смесь нагревает-
ся до 200 °C за счет тепла нейтрализованных продуктов реак-
ции. Далее паро-газовую смесь нагревают до 280—290 °C в
трубчатой печи 1 и подают в гидрататор 2, где она проходит
слой катализатора сверху вниз. В гидрататор .подают также све-
жую фосфорную кислоту (на подпитку катализатора).
На выходе из гидрататора смесь этилового спирта, непро-
реагировавших этилена и водяного пара нейтрализуют едким
натром. Фосфаты отделяют в солеотделителе 3, а паро-газовую
смесь после охлаждения в теплообменниках 4 и 5 направляют
в сепаратор 6 высокого давления. Жидкость дросселируют до
154
0,5—0,6 МПа и подают в сепаратор 8 низкого давления. Газо-
вая фаза из сепаратора 6 поступает в скруббер 7 для отмывки
спирта водой, после чего ее компримируют и возвращают (в про-
цесс. Спирто-водный конденсат из скруббера 7 идет в сепара-
тор 8 для выделения растворенного этилена (за счет снижения
давления до 0,5—0,6 МПа). Этилен с верха сепаратора 8 пода-
ют на компримирование и возвращают в процесс (или подают
в цех газоразделения). Водный спирт направляют на ректифи-
кацию.
Ректификацию осуществляют в одной колонне. При этом по-
лучают этиловый спирт-ректификат с содержанием ацетальде;
гида не выше 2% и до 1% диэтилового эфира. Если спирт идет
на получение бутадиена, ацетальдегид не мешает процессу. Та-
кой спирт пригоден для получения и ацетальдегида, и уксусной
кислоты.
После дополнительной очистки и ректификации синтетиче-
ский этиловый спирт не уступает спирту, получаемому из пище-
вого сырья. Концентрация спирта в водо-спиртовом растворе^
получаемом на установке, составляет 15—16%. Степень кон-
версии этилена за однократный проход равна 4—5%. Общая
степень конверсии этилена распределяется так: 95% в этиловый
спирт, 2% в диэтиловый эфир, 1% в ацетальдегид, 2% в диме-
ры и полимеры.
Метод прямой гидратации этилена весьма перспективен, од-
нако дальнейшее его усовершенствование заключается в под-
боре наиболее активных катализаторов и изыскании путей осу-
ществления жидкофазного процесса; при этих условиях можно
будет снизить температуру и увеличить степень конверсии эти-
лена. Метод позволяет создавать установки большой произво-
дительности, избавляет от необходимости транспортировать и
хранить большие количества серной кислоты. Географическое
размещение заводов определяется лишь наличием этилена.
Сравнение технико-экономических показателей сернокислотной
и прямой гидратации показывает, что себестоимость 1 т спирта
при прямой гидратации на 20% ниже.
5.5. Производство изопропилового
и трет-бутилового спиртов прямой гидратацией
пропилена и изобутилена
При прямой гидратации пропилена, как следует из термо-
динамики процесса, степень конверсии пропилена в изопропи-
ловый спирт с повышением температуры уменьшается (от 15%
при 150°C до 3% при 225°C). Скорость газофазной прямой
гидратации пропилена намного меньше скорости сернокислотной
гидратации. Для получения практически приемлемой скорости
проводят реакцию при высоких температурах, что, однако, ве-
дет к снижению степени конверсии.
За рубежом в качестве катализатора прямой гидратации
пропилена применяют оксид вольфрама WO3, промотированный
155
оксидом цинка. Катализатор и промотор наносят на носитель —
силикагель. Реакция протекает при 230—240 °C, 20—25 МПа и
мольном соотношении воды и пропилена, равном 10:1. При
однократном проходе через реактор степень конверсии пропиле-
на составляет 50%. Спирт получается в виде 20%-ного водного
раствора. Общий выход спирта составляет 95% в расчете на
пропилен.
В Советском Союзе процесс прямой гидратации пропилена
разработан с теми же катализаторами, которые применяют для
прямой гидратации этилена, т. е. на основе фосфорной кислоты.
Так, в присутствии фосфорной кислоты, нанесенной на силика-
гель, при 170 °C, 0,7—0,9 МПа и мольном соотношении воды и
пропилена, равном (0,7-4-1) 1, степень конверсии пропилена со-
ставляет 6%, а съем спирта равен 80 г с 1 л катализатора в час.
Концентрация фосфорной кислоты на поверхности носителя рав-
на 72%, концентрация* получаемого изопропилового спирта со-
ставляет 20%. Проводятся исследования и по подбору нейтраль-
ных катализаторов, в частности оксидов вольфрама и свобод-
ного вольфрама, промотированных оксидом цинка.
В СССР освоен промышленный процесс прямой гидратации
изобутилена в трет-бутиловый спирт на ионообменных смолах.
Например, на катионите КУ-2 процесс протекает в сравнитель-
но мягких условиях — при 90—100°C, 1—4,5 МПа и времени
контакта 100 с. Мольное соотношение воды и изобутилена со-
ставляет от 2 1 до 8 1, степень конверсии изобутилена за про-
ход равна 75—85%, что обеспечивает получение 10—40%-ного
трет-бутилового спирта. Производительность катализатора рав-
на 500 г спиртового раствора с 1 мл катализатора в час. Про-
должительность работы катионита свыше 700 ч.
Регенерация катализатора заключается в его обработке 6—
10%-ной соляной или серной кислотой. Смесь изобутилена и
воды непрерывно подают в раствор, заполненный катионитом.
Получаемые водные растворы спирта перегоняют, причем отго-
няется азеотропная смесь (78% спирта и 22% воды), кипящая
при 81 °C. Азеотропную смесь или водный спирт подвергают
дегидратации на том же катионите КУ-2. В результате обра-
зуется чистый изобутилен. Процесс дегидратации рекомендуется
как метод получения чистого изобутилена, идущего на получе-
ние полиизобутилена. Если вести процесс при температуре выше
100 °C, выход трет-бутилового спирта резко снижается и уве-
личивается выход полимеров изобутилена.
§ 6. ПРОИЗВОДСТВО СПИРТОВ ПО РЕАКЦИЯМ ПРИСОЕДИНЕНИЯ
6.1. Производство высших жирных спиртов
гидрированием карбоновых кислот и их эфиров
В мировой промышленной практике для получения высших
спиртов Сю—С20 широко используют процессы каталитического
гидрирования жирных кислот или их эфиров.
156
Гидрирование карбоксильной группы протекает через обра-
зование альдегидов, спиртов и углеводородов:
+н2 +н2 +н2
RCOOH —f RCHO -------RCH2OH —RCH3
—Н2О —Н2О
Процесс удается остановить на стадии спиртов. Альдегиды гид-
рируются быстрее кислот, поэтому в продуктах реакции они от-
сутствуют. Сложные эфиры гидрируются легче, чем карбоновые
кислоты. Поэтому те спирты, что образуются при гидрировании
кислот, дают с ними эфиры, и процесс гидрирования облег-
чается:
+2На +RCOOH
RCOOH -----> RCH2OH —-> RC-O-CH2R
—Н2О —Н2О ||
О
Однако в практическом отношении гидрирование карбоновых
кислот выгоднее, так как исключаются стадии получения слож-
ных эфиров и регенерации спирта.
Важное значение имеет гидрирование синтетических жирных
кислот или их эфиров. В результате получают смесь первичных
спиртов Сю—С2о нормального строения, идущих на производ-
ство высококачественных ПАВ (алкилсульфатов и оксиэтилиро-
ванных продуктов; см. гл. IV). Для гидрирования применяют
медь-хромитные (СиО*Сг2О3), цинк-хромитные (ZnO-Cr2O3) и
медь-цинк-хромитные -катализаторы (СиО • ZnO • Сг2О3). Реак-
цию проводят при 250—350 °C и давлении 25—35 МПа, необхо-
димом для увеличения скорости и повышения равновесной сте-
пени конверсии. Реакции гидрирования экзотермичны. В одина-
ковых условиях гидрирования степень конверсии кислот состав-
ляет примерно 90%, степень конверсии бутиловых эфиров равна
93—94%, а для метиловых эфиров 97%. При этом в спиртах
содержатся углеводороды: 5—7% в спиртах, получаемых из
кислот, 2—2,5% в спиртах, получаемых из эфиров.
Технологическая схема гидрирования метиловых эфиров
СЖК в неподвижном слое катализатора приведена на рис. II 1.9.
Сырье (метиловые или бутиловые эфиры), нагретое до 150—•
160°C, поступает^ специальную секцию печи 6. Свежий водо-
род смешивают с циркуляционным, нагревают в теплообмен-
нике 5 и печи 6, смешивают с эфирами и нагревают смесь до
300—350 °C. Из печи смесь направляется в реактор 4 (или в
два последовательных реактора) и проходит сверху вниз. Про-
дукты реакции поступают в горячий сепаратор 3. Жидкую фа-
зу, содержащую высшие спирты и метанол (или бутанол), ох-
лаждают и подают в эвапоратор 10.' Газовая фаза из сепарато-
ра 3 в теплообменнике 5 отдает свое тепло водороду, поступаю-
щему на гидрирование, и поступает в холодный сепаратор 7, где
тоже разделяется на жидкую и газовую фазу. Газовая фаза ох-
лаждается в холодильнике 8 и поступает .во второй холодный
сепаратор 9.
Рис. III.9. Технологическая схема получения высших спиртов гидрированием
метиловых эфиров СЖК:
Л 8 — холодильники; 2 — подогреватель; 3, 7, 9 — сепараторы; 4 — реактор; 5 —тепло-
обменник; 6 — печь; 10 — эвапоратор.
Циркулирующий водород, отделенный в сепараторе 9, .сме-
шивают со свежим и возвращают на гидрирование. Жидкую
фазу из сепараторов 7 и 9 объединяют с жидкой фазой из сепа-
ратора 3 и подают в эвапоратор 10. Выделившийся там раство-
ренный водород вместе с метанолом уходит на регенерацию ме-
танола, а жидкая фаза (гидрогенизат) поступает на разделение
высших спиртов. После сепаратора 7 давление понижают с 30
до 1 МПа, а после сепаратора 9 — до атмосферного.
Технология гидрирования сложных эфиров осложняется ста-
диями этерификации карбоновых кислот и регенерации спирта,
поэтому все большее практическое значение приобретают про-
цессы прямого гидрирования жирных кислот до спиртов.
В СССР в промышленную практику внедрен процесс гидрирова-
ния СЖК на медь-хром-бариевом суспендированном катализато-
ре. Степень конверсии кислот при 340—350 °C и 30 МПа дохо-
дит до 99,9% при селективности по спиртам 97,3—98,9%.
6.2. Производство высших жирных спиртов
из альдегидов
В основе метода лежит реакция конденсации альдегидов, от-
крытая в 1872 г. А. П. Бородиным. При получении спиртов этим
методом имеются три стадии:
1) альдольная конденсация:
rch2—CHO + RCH2—СНО —> rch2— сн—сн-сно
(J)H R
158
2) дегидратация альдоля в кислой среде:
rch2-ch~ch-cho-----------►- RCH2-CH=C-CHO
I I “H2° I
OH R R
3) гидрирование замещенного кротонового альдегида до на-
сыщенного спирта:
4-2Н2
rch2-ch=c-cho-----> RCH2-CH2-CH-CH2OH
R R
Конденсацией альдегидов в настоящее время в промышлен-
ности получают н-бутанол и 2-этилгексанол. Стадию конденса-
ции используют в производстве бутадиена-1,3 из этилового
спирта:
+сн3сно
СН3-СН2ОН —-> СНз-СНО -------* сн3-снон—сн2-сно —>
—Н2 —Н2О
+н2
---► СН3—СН=СН— СНО —сн3-сн=сн—сн2он —>
—► сн2=сн—сн=сн2
Реакция альдольной конденсации катализируется щелочами
и кислотами, имеет первый порядок по альдегиду. Под влия-
нием оснований альдегиды ионизуются, а анионы взаимодейст-
вуют с соседними молекулами альдегида:
сн3—+
н
Na+OH
Na+
+ СН,СНО
сн3—сн—сн2—СНО
о"
Образовавшиеся анионы реагируют с протонами альдегида
(или растворителя) или с водой:
СН3—СН-СН2—СНО+Н2О ► СН3-СН—СН2-СНО + НО-
А- <!>н
В случае кислотного катализа вначале образуется катион, ко-
торый взаимодействует с альдегидом в енольной форме и дает
альдоль:
2 СН3—С\
Н
+ н+
--► СНо— СНОН + сн2=сн-о—н
О ь
СН9— сн—СНо-СНО
3 I
он
159
15
Рис. III. 10. Технологическая схема получения «-бутанола из ацетальдегида:
1, 3, 15 — реакторы; 2, 4, 7 — холодильники-конденсаторы; 5, 8, 12 — кипятильники; 5 —
ректификационная колонна; 9, 16 — холодильники; 10 — отстойник; 11 — колонна азео-
тропной перегонки; 13 — нагреватель; 14 — теплообменник; 17 — сепаратор.
Побочно получаются продукты более глубокой конденсации, на-
пример:
СН3—СН-СНа-СНО + СН3-СНО --► СН3—СН-СН2-СН-СН2-СНО
Лн Ан Ан
Альдоли не стабильны и при нагревании выше 80 °C отщепляют
воду с образованием ненасыщенных альдегидов, которые можно
гидрировать селективно—только по двойной связи, только по
карбонильной группе или в обоих направлениях до образова-
ния насыщенного первичного спирта. Применяют никелевые и
медные катализаторы (никель Ренея, медь на пемзе), процесс
проводят в жидкой или газовой фазе.
Избирательность действия катализаторов зависит не только
от их состава, но и от режима гидрирования (температура,
объемная скорость, соотношение водорода и альдегида). При
повышении температуры и количества водорода гидрирование
идет до спирта.
Технологическая схема получения «-бутанола через ацет-
альдоль приведена на рис. III.10. Ацетальдегид смешивают со
щелочью и возвратным ацетальдегидом и подают на альдолиза-
цию в реактор 1, представляющий собой тарельчатую колонну.
В нижнюю часть аппарата подают уксусную кислоту для ней-
трализации щелочи. За счет выделяющегося тепла часть ацеталь-
дегида испаряется и конденсируется; ее возвращают в реактор.
С низа реактора 1 подкислённая смесь альдоля, ацетальде-
гида, воды и ацетата натрия поступает на кротонизацию в ко-
лонный реактор 3. С верха колонны кротоновый альдегид,
ацетальдегид и вода уходят в холодильник-конденсатор 4, а от-
туда в ректификационную колонну 6. С низа реактора 3 уходят
подкисленная вода и ацетат натрия. С верха колонны 6 отгоня-
ют ацетальдегид. Он конденсируется, часть конденсата идет на
160
орошение .колонны, а балансовое количество возвращают на
конденсацию. С низа колонны 6 уходит обводненный кротоно-
вый альдегид, который поступает в отстойник 10.
Нижний слой из отстойника представляет собой кротоновый
альдегид, насыщенный водой; он поступает в колонну 11 на
азеотропную перегонку. Верхний слой из отстойника 10—кро-
тоновый альдегид — поступает на гидрирование. Его смешивают
с водородом, нагревают и подают в реактор 15, заполненный
катализатором. Паро-газовая смесь из реактора отдает свое
тепло водороду, охлаждается и поступает в сепаратор 17. Водо-
род с верха сепаратора возвращают на гидрирование, а н-бута-
нол с низа сепаратора подают на ректификацию для отделения
альдегидов и сложных эфиров.
6.3. Производство высших жирных спиртов
алюминийорганическим синтезом
Производство спиртов через алюминийорганические соедине-
ния включает следующие стадии:
1) получение высших алюминийтриалкилов полимеризацией
этилена с триэтилалюминием при 100—120 °C и 5—12 МПа:
✓(СН2—СН2)Х—
пСН2=СН2 + А1(С2Нб)8 -> А1С(СН2-СН2)У-С2Н5
\(СН2-СН2)г-С2Н5
n=x4-i/+z
2) окисление высших галюминийтриалкилов воздухом или
кислородом при 80—100 °C с образованием алкоголятов алюми-
ния:
/R yOR
Al-R'+ l,5O2---► Al—OR'
\r- \qR"
3) гидролиз алкоголятов до спиртов при 100—150 °C в при-
сутствии кислот или щелочей:
,OR
Al—OR' + ЗН2О --► А1(ОН)з + ROH + R'OH-J- R’OH
\OR*
В промышленности спирты этим методом получают с 1961 г.,
когда был освоен прямой синтез алюминийтриалкилов при 100—
110 °C и 28 МПа:
СН2=СН2 + Al + 1,5Н2--► АЦСД^з
Полимеризацию этилена с триэтилалюминием проводят в
растворителе, что облегчает отвод выделяющегося тепла, но
разбавление не должно снижать концентрацию триэтилалюми-
ния менее 25%. Присоединение этилена протекает ступенчато;
11-254
161
образуется смесь алюминийтриалкилов с длиной цепи С4—Сое.
Степень конверсии этилена при 105 °C и 12 МПа составляет
83—85%. В качестве примесей образуется некоторое количество
(5—10%) а-олефинов за счет реакции вытеснения:
/CH2-CH2R /С2Н5
Al— R' +СН2=СН2 ----► Al—-R' +RCH=CH2
\r* \r„
Возможны также димеризация а-олефинов и образование пара-
финовых углеводородов, если в систему попадает влага:
>R уОН
Al—R'+H2O ----> A1--R' +RH
\r, \r*
При последующем окислении алюминийтриалкилов две ал-
кильные группы окисляются быстро — уже при 20—25 °C за
2 ч. Окисление третьей группы протекает труднее — при 50—
60 °C или в присутствии кислорода. При этом образуются по-
бочные продукты — альдегиды и олефины.
Гидролиз алкоголятов алюминия протекает легко и с теоре-
тическим выходом спиртов. Гидролиз ведут водой или разбав-
ленной серной «кислотой при 20—60 °C в течение 1 ч. При гидро-
лизе образуется смесь нормальных спиртов с четным числом
углеродных атомов, содержащая примесь спиртов изостроения.
Существенные недостатки алюминийорганического синтеза —
его многостадийность, применение огне- -и взрывоопасных ве-
ществ (алюминийтриалкилы), большой расход алюминия (100—
120 кг на 1 т спиртов). Достоинства метода — дешевое и до-
ступное сырье (этилен), возможность получения высших первич-
ных спиртов, идущих на производство синтетических моющих
веществ и пластификаторов.
6.4. Производство метилового спирта
из синтез-газа
Производство синтез-газа. Синтез-газ (смесь оксида углеро-
да и водорода) может быть получен из каменных и бурых
углей, а также из всех других видов топлива, способного к гази-
фикации. Для получения синтез-газа из кокса пропускают водя-
ной пар через слой кокса, раскаленного до 1000 °C. При этом
водяной пар разлагается:
С + Н2О --> СО + Н2 ДЯ = 120 кДж/моль
При более низких температурах реакция протекает с образова-
нием диоксида углерода:
С + 2Н2О --> СО2 + 2Н2 ДЯ = 80 кДж/моль
Для получения 100 м3 синтез-газа этим методом требуется 50—
60 кг кокса и 70 ,кг водяного пара. Соотношение СО и Н2 со-
162
ставляет 1 1. Если синтез-газ предназначен для синтеза угле-
водородов или метанола, к нему добавляют водород, получа-
емый каталитической конверсией оксида углерода:
СО + Н2О ---► СО2 + Н2 ДЯ = —38 кДж/моль
При получении синтез-газа из бурых углей в генераторы вместе
с водяным паром подают кислород в таком -количестве, чтобы
тепла, выделяющегося при горении угла
С 4- О2 -► СО2 ДЯ = —406 кДж/моль
С + 0,5О2 --► СО ДЯ = —122 кДж/моль
было достаточно для разложения водяного пара. Получаемый
газ содержит 25—30% СО2, который отмывают водой под давле-
нием.
В настоящее время для получения синтез-газа широко ис-
пользуют метод конверсии метана. Конверсию можно прово-
дить с водяным паром, с диоксидом углерода и с кислородом
(окислительная конверсия):
СН4 + Н2О СО + ЗН2 ДЯ = 206 кДж/моль
СН4+ СО2 2СО + 2Н2 ДЯ = 248 кДж/моль
СН4 + 0,5О2 СО + 2Н2 ДЯ = 35 кДж/моль
При конверсии метана с водяным паром образующийся ок-
сид углерода претерпевает дальнейшую конверсию до СО2.
Таким образом, процесс конверсии метана с водяным паром
позволяет получать газ любого состава (вплоть до чистого во-
дорода), что достигается изменением соотношения исходных
газов и температуры (см. гл. VI). При повышении температу-
ры равновесие реакции
СН44-Н2О СО + ЗН2
сдвигается вправо. Например, проводя процесс при 835—940 °C,
можно получить газовую смесь с 23,7—24,8% (об.) СО и 71—
74% (об.) Н2. В отсутствие катализаторов конверсия метана
с водяным паром протекает при 1200 °C, а каталитический про-
цесс идет уже при 700—870 °C. Наиболее распространен никеле-
вый катализатор, активированный добавками оксидов кальция,
магния, хрома и др.; носитель — оксид алюминия.
Конверсия метана с диоксидом углерода и окислительная
конверсия протекают на никелевых катализаторах при 800—
1000 °C.
Конверсию метана с водяным паром или с диоксидом угле-
рода проводят в трубчатых печах. Смесь исходных газов нагре-
вают в теплообменниках до 365 °C и пропускают через трубы из
жаропрочной стали, заполненные катализатором и обогрева-
емые снаружи за счет сжигания газа в форсунках печи. Темпе-
ратура в зоне реакции 750—800 °C. Окислительную конверсию
метана проводят в реакторах шахтного типа, заполненных ката-
лизатором.
11* 163
Рис. Ш.Н. Технологическая схема паро-кислородной конверсии углеводород-
ных газов:
J — смеситель; 2 — конвертор; 3 — увлажнитель; 4 — пароперегреватель; 5, 6 — теплооб-
менники.
Наиболее эффективным процессом получения синтез-газа
является конверсия метана паро-кислородной смесью. Тепло,
необходимое для этого процесса, обеспечивается за счет полного
окисления ,части метана. При этом образуются СО2 и Н2О, ко-
торые взаимодействуют с продуктами конверсии и исходным ме-
таном, образуя СО и Н2. При паро-кислородной конверсии не
требуются жаропрочные стали, катализатор не отравляется сер-
нистыми соединениями и требования к сырью менее жесткие
(можно использовать газы любого состава). Капиталовложения
меньше, чем при конверсии ,в трубчатых печах, однако эксплуа-
тационные расходы более высоки из-за относительно высокой
стоимости кислорода. Технологическая схема паро-кислородной
конверсии приведена на рис. Ш.Н.
Природный газ, поступающий при давлении 0,4—0,5 МПа,
дросселируют до 0,08—0,12 МПа и подают в теплообменники 5
и 6. На входе в теплообменники к газу примешивают водяной
пар, перегреваемый с 180 до 350 °C. После смешения паро-газо-
вая смесь имеет температуру 180 °C. В теплообменниках ее на-
гревают до 345 °C и затем направляют в вертикальный кожухо-
трубный смеситель /, куда поступает также кислород. Из смеси-
теля паро-газовая смесь идет в конвертор 2 с никелевым ката-
лизатором ГИАП-3. Там при 800—900 °C и 0,14 МПа протека-
ют реакции конверсии с образованием синтез-газа. Конвертиро-
ванный газ поступает в увлажнитель 5, куда впрыскивают
водный конденсат (при этом температура газа снижается до
650°C). Затем газ проходит трубное пространство пароперегре-
вателя 4, теплообменники 5 и 6 и, охладившись до 500 °C, идет
на конверсию оксида углерода (если требуется получать водо-
род) или охлаждается до комнатной температуры и поступает
в газгольдеры.
Объемное соотношение пара и природного газа на входе в
конвертор равно t: 1. Соотношение между компонентами (СО,
164
C02, Н2 и Н2О) в конвертированном газе соответствует состоя-
нию равновесия при 850 °C на выходе из конвертора. Содержа-
ние метана в сухом конвертированном газе не превышает 0,5%.
Для получения синтез-газа с объемным соотношением СО и
Н2, равным 1 : 2, конверсию метана проводят смесью водяного
пара, диоксида углерода и кислорода. Если нужно повысить со-
держание водорода в синтез-газе, проводят паро-кисло родную
конверсию метана (без СО2) и получают соотношение СО : Н2 =
= 1:3.
Кроме перечисленных способов синтез-газ получается и как
побочный продукт при окислительном пиролизе метана с целью
получения ацетилена и при пиролизе газового бензина, когда
одновременно получают этилен и ацетилен.
Можно подвергать конверсии и другие парафины — этан,
пропан и др.:
С3Н8 + ЗН2О ЗСО + 7Н2
Предварительно парафины необходимо очистить от непре-
дельных соединений, так как они при температуре конверсии
претерпевают пиролиз с выделением углерода
ч — ПС -f- нН2
который оседает на катализаторе и снижает его активность. Пи-
ролиз особенно заметен в присутствии активных никелевых ка-
тализаторов. Однако при 750 °C скорость конверсии возрастает
настолько, что процесс идет без накопления углерода. Пиролиз
олефинов снижается, если к никелю добавить оксид кальция.
Производство метилового спирта. В настоящее время основ-
ным промышленным процессом получения метилового спирта
(метанол) является гидрирование оксида углерода:
СО + 2Н2 СН3ОН ДЯ = —109 кДж/моль
Синтез идет при высоких давлении и температуре. Равновесные
выходы метилового спирта незначительны, поэтому процесс про-
водят с многократной циркуляцией исходной газовой смеси. Сте-
пень конверсии исходной газовой смеси (СО + 2Н2) в метанол
растет с увеличением давления и уменьшается с повышением
температуры. Высокое давление не только способствует увели-
чению степени конверсии, но и уменьшает побочные реакции,
приводящие к метану, формальдегиду, диметиловому эфиру и
высшим спиртам:
СО2 + ЗН2---► СН4 + Н2О
СО + Н2 --НСНО
2СО + 2Н2 --и СН4 + СО2
2СН3ОН----> СН3—О-СН3 + Н2О
СН3ОН + пСО + 2пН2 -> СН3-(СН2)П-ОН + иН2О
СН3ОН + Н2 --> СН4 + Н2О
На промышленных установках степень конверсии газовой смеси
165
в метанол за однократный проход через реактор составляет
5-20%.
Синтез метилового спирта — процесс каталитический, причем
применяемые в промышленности катализаторы активны лишь
при 300—400 °C (в отсутствие катализаторов реакция не идет
совсем). Катализатор должен обладать избирательностью, т. е.
максимально ускорять образование метанола и одновременно
подавлять побочные реакции. Большинство катализаторов в ка-
честве основного компонента содержат оксид цинка, к которо-
му добавлены оксиды хрома, и меди. В присутствии железа и
никеля образование метана ускоряется. Щелочные оксиды
(Na2O, К2О, СаО), наоборот, способствуют образованию выс-
ших спиртов. Катализатор состава 8ZnO-Cr2O3-CrO3 весьма
стоек при высокой температуре, мало чувствителен к контакт-
ным ядам и отравляется обратимо. Он легко регенерируется и
имеет высокую селективность. Готовят такой катализатор, сме-
шивая сухие оксиды цинка и хрома с раствором хромового ан-
гидрида. Полученную пасту состава 8ZnO-Cr2O3-CrO3-H2O
формуют, сушат при ПО—120 °C и восстанавливают тем же га-
зом, который применяют для синтеза (метанола. Медные катали-
заторы активируют добавками Сг2О3, ZnO, V2O5 и др. Эти ка-
тализаторы более активны, чем цинк-хромовые, но менее стой-
ки к высоким температурам и контактным ядам. Отравление
идет необратимо, поэтому исходную газовую смесь следует тща-
тельно очищать. Кроме того, медные катализаторы менее селек-
тивны.
При взаимодействии, оксида углерода со стальными стенками
аппаратов и трубопроводов образуется летучий пентакарбонил
железа, который разрушает стенки (карбонильная коррозия).
Кроме того, пентакарбонил железа разлагается на катализато-
ре с выделением свободного железа, которое катализирует обра-
зование метана. Поэтому внутренние стенки колонн синтеза и
теплообменников облицовывают медью, ее сплавами или изго-
товляют из высоколегированных сталей.
Синтез метанола на цинк-хромовом катализаторе проводят
при 370—400 °C и 25—30 МПа, а на медном [катализаторе — при
300 °C и 15 МПа. Процесс ведут при больших объемных скоро-
стях подачи исходной газовой смеси:
Объемная ско- рость подачи газа, ч-1 Продолжитель- ность контакта, с Выход метанола, г на 1 л ка- тализатора в час
2400 150 170
9000 40 327
18000 20 375
35000 10 750
Видно, что при высоких объемных скоростях производитель-
ность катализатора увеличивается. Обычно синтез метилового
спирта проводят при объемных скоростях 10 000—35 000 4~L
166
Рис. III. 12. Технологическая схема получения метилового спирта из СО и ЬЬ:
/, 2 — компрессоры; 3 — скруббер; 4 — угольный фильтр; 5 — теплообменник; 6 — колон-
на синтеза; 7 — холодильник-конденсатор; 8 — сепаратор; 9 — сборник.
При более высоких скоростях повышается расход электроэнер-
гии на циркуляцию газов.
Процесс осуществляют по следующей схеме (рис. III.12).
Исходную газовую смесь (СО+Н2), очищенную от сернистых
соединений, .сжимают пятиступенчатым компрессором 1 до
25 МПа. Между третьей и четвертой ступенями сжатия газ под
давлением 3 МПа отмывают водой в насадочном скруббере 3
от диоксида углерода. Сжатый до 25 МПа и очищенный от СО2
газ смешивают с циркулирующим газом и подают на фильтр 4
для очистки от пентакарбонила железа активным углем. Затем
газ поступает в трубное пространство теплообменника 5, где
отходящими контактными газами нагревается до 320 °C, и при
этой температуре попадает в колонну синтеза 6.
В этой колонне газ проходит снизу вверх через внутренний
теплообменник, расположенный ниже катализаторной зоны, а
затем идет сверху вниз через катализатор, размещенный на
полках колонны. После этого контактный газ проходит через
трубки внутреннего теплообменника, выходит снизу и посту-
пает в межтрубное пространство теплообменника 5, где ох-
лаждается до 80— 100 °C, а оттуда — в холодильник-конденса-
тор 7, где охлаждаются и конденсируются пары метанола. Газ
вместе с образующимся метанолом при 30 °C подают в сепара-
тор 8 высокого давления, затем направляют в приемную линию
циркуляционного компрессора 2 и возвращают в процесс, а ме-
танол-сырец через дроссельный клапан проходит в сборник 9,
а оттуда идет на ректификацию и химическую очистку.
Метанол-сырец — бесцветная или слегка желтоватая про-
зрачная жидкость (плотность 0,811—0,812 г/см3), имеющая сле-
167
дующий состав [в % (масс.)]:
Метанол . 92—93
Диметиловый эфир . ... 2,8 —4,7
Изобутиловый спирт и другие кислородсодержа-
щие соединения 0,43—0,45
Олефины 0,13
Вода ... . 3,0 —3,5
Пентакарбонил железа 0,001
При синтезе метилового спирта выделяется тепло. Во из-
бежание перегрева катализатора предусмотрена подана холод-
ного газа на каждую полку колонны. Внутри аппарата смон-
тирован электроподогреватель для разогрева газа в пусковой
период. Внутренняя поверхность колонны и теплообменника об-
лицована красной медью для защиты от карбонильной кор-
розии.
В метанол-сырец, поступающий на дистилляцию, добавляют
раствор едкого натра для омыления сложных эфиров и нейтра-
лизации кислот, которые могут вызывать коррозию аппаратуры.
Вначале отгоняют диметиловый эфир, а затем азеотропную
смесь побочных продуктов с водой. После этого метанол прохо-
дит химическую очистку перманганатом калия, который добав-
ляют в виде 2%-ного водного раствора. Метанол и высшие спир-
ты более стабильны к действию перманганата, чем карбониль-
ные соединения (которые переходят в кислоты, нейтрализуемые
едким натром, ранее добавленным к метанолу-сырцу). В этих
условиях пентана рбонил железа разрушается полностью, а
КМпО4 при окислении превращается в МпО2, который отделяют
на фильтр-прессах. Окончательную дистилляцию метанола осу-
ществляют в колонке с большим числом тарелок (примерно 75).
На нижних тарелках собирается «изобутиловое масло» — фрак-
ция, содержащая изобутиловый и высшие спирты, примесь ме-
танола и воду. Изобутиловый спирт отгоняют в другой колонне
и получают в чистом виде как побочный продукт. Диметиловый
эфир (т. кип. —23,7 °C) используют частично в холодильной тех-
нике, а частично как топливный газ.
Очищенный метанол получают 99,5—99,7%-ной чистоты (ос-
тальные 0,3—0,5% приходятся на другие органические кисло-
родсодержащие соединения и воду). Практически на 1 т мета-
нола расходуется 2420—2500 м3 смеси CO-f-H2, т. е. из 1 м3 га-
зовой смеси получают 400—410 г спирта. Общая степень кон-
версии синтез-газа в метанол составляет 84—87%.
Метиловый спирт широко применяется в органических син-
тезах. Основное его количество используют для получения
формальдегида неполным окислением кислородом воздуха на
серебряном катализаторе:
СН3ОН+ 0,5О2 --► НСНО + Н2О ч
В результате взаимодействия метилового спирта с аммиаком
при 450 °C и 5—10 МПа над глиноземными катализаторами об-
168
разуются метиламины:
CHaOH + NH3 CH3NH2 + H2O
2СН3ОН + NH3 (CH3)2NH + 2H2O
С уксусной кислотой метиловый спирт дает метилацетат:
СН3ОН + СН3-СООН СН3-СОО-СН3 + Н2О
Пропуская пары метанола ,и синтез-газа при 450—475 °C и
25 МПа над оксидным цинк-хромовым катализатором с до-
бавкой щелочи, получают смесь насыщенных спиртов изострое-
ния (преимущественно изобутиловый спирт):
СН3ОН + пСО + 2пН2 СП+1Н2П+3ОН
В результате взаимодействия метилового спирта с бензолом при
370 °C и 5 МПа над фосфатом цинка получается толуол:
СН3
“ои+О О
С ароматическими аминами в присутствии серной кислоты (ка-
тализатор) метанол образует диметиламины:
с NH2 N(CH,)2
II I
2сн>он+О о
При взаимодействии метанола с оксидом углерода образуется
метилформиат, применяемый для получения диметилформ-
амида:
+CH3ONa;
80 °C; 3 МПа +NH(CH3)2
CHgOH+CO ----------НСОО-СНз —> HCON(CH3)2
—CH3Url
Диметилформамид является селективным .растворителем, при-
меняемым для выделения ацетилена, при абсорбции диоксида
серы и других газов кислотного характера. Кроме того, диме-
тилформамид является хорошим растворителем для многих смол
и полимеров и применяется в процессах формования волокон
(нитрон и др.).
В различных производствах метанол используют как раство-
ритель, например при очистке бензинов от меркаптанов и как
третий компонент при азеотропной перегонке. Вместе с этилен-
гликолем метанол применяют для экстракции толуола из бензи-
нов, употребляют его также и как реакционную среду для пе-
рекристаллизации. Как растворитель лаков и политур метанол
имеет ограниченное применение из-за его токсичности.
Метанол обладает высоким октановым числом ,и может
быть использован как компонент синтетического моторного топ-
лива, однако он имеет низкую теплоту сгорания (22340 кДж/кг).
169
6.5. Производство спиртов оксосинтезом
При совместном действии оксида углерода и водорода на
алифатические олефины образуются альдегиды:
rch=ch2 + co + h2 -
RCH2—СН2— СНО
-> RCH—СН3
СНО
Эту реакцию называют оксосинтезом, или гидроформилирова-
нием, так как она представляет собой введение карбонильной
(формильной) группы в молекулу олефина. Реакция открыта
в 1938 г. Рёленом, в СССР ее разрабатывали под руководством
Д. М. Рудковского. Если при сернокислотной гидратации олефи-
нов (за исключением этилена) образуются вторичные спирты,
то при гидроформилировании и последующем гидрировании
альдегидов получаются только^ первичные спирты — нормальные
и изостроения.
Образующиеся карбонильные соединения частично восста-
навливаются в спирты. Гидрирование олефинов в насыщенные
углеводороды 'протекает в значительно меньшей степени, хотя
термодинамически реакция гидрирования более вероятна. Одна-
ко на практике гидрирование двойной связи почти полностью
подавляется. Это достигается подбором условий, благоприятных
для основной реакции.
Для промышленности взаимодействие оксида углерода и во-
дорода с олефинами представляет интерес как метод получения
низкомолекулярных альдегидов и высших спиртов (лаурилово-
го, миристилового, гексадецилового), идущих на производство
моющих средств. Необходимые для оксосинтеза додецен, гекса-
децен и другие высокомолекулярные олефины образуются при
крекинге парафина. В принципе, для оксосинтеза можно исполь-
зовать разнообразные олефины. Из этилена вырабатывают про-
пионовый альдегид, из пропилена — бутиловый и изобутиловый
спирты, из гептенов, образующихся при совместной полимери-
зации пропилена и изобутилена, — октиловые спирты. Так же
успешно можно использовать диизобутилен, три- и тетрамеры
пропилена.
Вне зависимости от положения двойной связи в исходном
олефине нормального строения обычно образуется смесь, со-
стоящая из 40—60% нормальных спиртов и 40—60%
спиртов изостроения. В случае высокомолекулярных олефинов
происходит изомеризация двойной связи, в результате чего
образуется смесь спиртов изостроения. К третичному углеродно-
му атому, а также к атому углерода, расположенному рядом
с четвертичным углеродным атомом, карбонильная группа не
присоединяется. Например, из изобутилена образуется только
170
3-метил бутанол-1 :
СН3—С=СН2 + СО + Н2 —► сн3-сн-сн2—сн2он
СН3 сн3
Некоторая особенность этой реакции наблюдается л для этиле-
на — заметно образование диэтилкетона:
2СН2=СН2 + СО + Н2 ► С2Н5-СО-С2Нв
Из а-олефинов, производимых крекингом парафина, в резуль-
тате оксосинтеза получают смесь спиртов нормального и изо-
строения:
СНО
rch=ch2+CO: +На
RCH-СН3
СН2ОН
+н2 - I
---> RCH-CH3
+на
rch2-ch2-cho —> RCHj—сн2—СН2ОН
Оксосинтез является гомогеннокаталитическим процессом.
Протекает он в присутствии комплексных соединений кобаль-
та — дикобальтоктакарбонила и гидрокарбонила кобальта:
+н2
2Со + 8СО----► Со2(СО)8 ----► 2НСо(СО)4
Эти комплексы не стабильны: дикобальтоктакарбонил разлага-
ется выше 50 °C, а гидрокарбонил — ниже 0 °C. Стабильность
они приобретают только при высоком парциальном давлении
оксида углерода. Предполагается, что истинным катализатором
является гидрокарбонил кобальта, поскольку он обладает кис-
лотными свойствами и является акцептором электронов, постав-
ляемых олефинами. При взаимодействии с олефином образует-
ся алкилметаллкарбонил:
RCH2-CH2 Со(СО)4
RCH=CH, + НСо(СО)4 —
I-» RCH—сн8
Со(СО)4
Далее алкилметаллкарбонил присоединяет СО и превращается
в ацилметаллкарбонил:
RCH2-CH2 Со(СО)4 + СО RCHa-CHa—СОСо(СО)4
Завершается катализ регенерацией гидрокарбонила кобальта
и образованием альдегида:
+н2
RCH2—СН2—СОСо(СО)4 =?=* RCH2—СН2—СНО + НСо(СО)4
Присоединение гидрокарбонилов к олефинам протекает по пра-
вилу Марковникова. Образование альдегидов нормального
строения с карбонильной группой, присоединенной против пра-
171
вила Марковникова, объясняется изомеризацией ацилметалл-
карбонилов. Реакция гидроформилирования имеет первый по-
рядок по катализатору и по олефину.
Скорость реакции повышается при увеличении соотношения
Н2: СО. Повышение парциального давления СО до определен-
ного предела повышает скорость реакции, препятствуя разложе-
нию карбонилов кобальта, а при слишком высоком парциальном
давлении СО тормозится переход дикобальтоктакарбонила в
гидрокарбонил кобальта.
Реакция чувствительна к температуре — с повышением ее
растет выход альдегидов изостроения и ускоряется гидрирова-
ние альдегидов в спирты. Практически процесс ведут при 120 —
180 °C. Общее давление в системе, равное 10—30 МПа, склады-
вается из необходимых парциальных давлений СО и Н2. Каж-
дой температуре соответствует определенное давление, выше ко-
торого скоро-сть не зависит от давления. Время контакта при-
мерно 1 ч, чему соответствуют объемные скорости подачи оле-
фина 0,4—2 ч-1. Соотношение СО : Н2 равно (14-‘2) 1. Степень
конверсии олефинов зависит от их молекулярной массы и со-
ставляет 65—80%.
Процесс является экзотермическим; тепловой эффект состав-
ляет примерно 117 кДж/моль и мало меняется при переходе от
одного олефина к другому. Выделяющееся тепло отводят во
внутренних змеевиках, выносных холодильниках или за счет
ввода в реактор холодных рециркулирующих потоков. Крат-
ность циркуляции синтез-газа равна (2ч-3) 1. Олефины, ис-
пользуемые )в оксосинтезе, должны быть тщательно очищены от
диенов, ацетиленовых углеводородов и кислорода. Ацетилено-
вые углеводороды и диены образуют с карбонилами кобальта
неактивные комплексы, а кислород разлагает карбонилы ко-
бальта с образованием СоО.
Особенностью процесса является извлечение карбонилов ко-
бальта из продуктов реакции и возвращение их в процесс. По
способу декобальтизации, существенно усложняющей процесс,
различают: 1) схемы с термическим разложением карбонилов
кобальта, основанные на неодинаковой летучести карбонилов
кобальта и продуктов оксосинтеза, и 2) солевые схемы, основан-
ные на разложении карбонилов кобальта окислителями.
Термическое разложение карбонилов кобальта основано на
обратимости реакции:
+н2
2Со + 8СО =?=> Со2(СО)8 <=* 2НСо(СО)4
Для разложения карбонилов снижают парциальное давление
СО. Чтобы не допустить отложений металлического кобальта на
стенках аппаратуры, в реактор вводят твердый носитель (пем-
за, кизельгур). Применяют реакторы с суспендированным но-
сителем, со стационарным слоем носителя и без него, с тонко-
дисперсным металлическим кобальтом.
172
Рис. II1.13. Двухреакторный блок ок-
сосинтеза:
1, 3—колонные реакторы; 2 — холодильник.
Рис. III.14. Трехреакторный блок
оксосинтеза:
1, 2, 3 — колонные реакторы.
В схемах с суспендированным носителем в реактор подают
заранее приготовленный катализатор, олефин и синтез-газ.
Реакционную массу редуцируют и в сепараторе отделяют газы,,
содержащие карбонилы кобальта. Их улавливают в скруббере,,
орошаемом олефином. Раствор возвращают на приготовление
суспензии катализатора. На этих установках гидрирование аль-
дегидов осуществляют на кобальтовом катализаторе в одном
аппарате, а жидкие продукты реакции передают в другой реак-
тор вместе с носителем, содержащим карбонилы кобальта.
Карбонилы гидрируются до металлического кобальта, на кото-
ром и протекает гидрирование. Гидрогенизат отфильтровывают
от катализатора (кобальт на носителе), который в виде суспен-
зии в продуктах гидрирования возвращают «в процесс оксосин-
теза на приготовление карбонилов.
В схемах со стационарным слоем носителя реакторный блок
представляет собой двух- или трехколонную систему. В двух-
реакторном блоке (рис. III.13) оба реактора заполнены насад-
кой. В колонне 1 происходят образование карбонилов, растворе-
ние их в олефине и гидроформилирование. В колонне 3 (деко-
бальтизер) снижают парциальное давление СО за счет подачи
водорода; при этом карбонилы разлагаются и кобальт осажда-
ется на насадке. После истощения катализатора в колонне 1
направление потоков меняется: гидроформилирование протекает
в колонне 3, а колонна 1 работает как декобальтизер. Выделяю-
щееся тепло отводят в холодильнике 2.
В трехреакторном блоке (рис. III. 14) в колонне 1 идет об-
разование карбонилов кобальта, в колонне 2 — оксосинтез, в
колонне 3— дакобальтизация (триадная схема). В колонне 1
температура равна 150—180 °C, а давление составляет 15—
30 МПа. Периодически направление потоков изменяют. Колон-
на 2 представляет собой своеобразный котел-утилизатор, в кото-
ром за счет выделяющегося тепла образуется водяной пар;
котел работает непрерывно.
173
Рис. III. 15. Технологическая схема оксосинтеза в суспендированном слое но-
сителя:
/ — аппарат для приготовления катализатора; 2, 3 — реакторы; /—холодильник; 5 —
сепаратор.
При солевых схемах (кобальт вводят в виде солей жирных
или нафтеновых кислот) образование карбонилов происходит
в реакторе гидроформилирования:
2(RCOO)2Co+ 8СО + 2Н2 4RCOOH + Со2(СО)8
По окончании реакции гидроформилирования разлагают карбо-
нилы серной кислотой в присутствии пероксида водорода
Со2(СО)8 + 2H2SO4 + 2Н2О2 -> 2CoSO4 + 8СО 4- 4Н2О
а карбоновые кислоты извлекают в виде натриевых солей. Рас-
твор сульфата кобальта используют для получения солей кар-
боновых или нафтеновых кислот, возвращаемых в процесс на.
образование карбонилов .кобальта:
CoSO4 + 2RCOONa --► (RCOO)2Co + Na2SO4
На рис. III. 15 приведена схема окоосинтеза в суспендирован-
ном слое носителя. Суспензию катализатора из аппарата 1 по-
дают в нижнюю часть первого реактора 2, куда поступают так-
же олефин и синтез-газ. Из этого реактора, где степень конвер-
сии олефина достигает 70—75%, смесь переходит во второй
реактор 3. В этот аппарат тоже подают смесь СО+Н2, что пре-
пятствует расслоению реакционной массы. После охлаждения
реакционной массы в холодильнике 4 она поступает в сепара-
тор 5. С низа сепаратора суспензию катализатора в альдегидах
откачивают в блок гидрирования, а катализатор — кобальт
(образовавшийся при гидрировании карбонилов) на носителе —
из блока гидрирования возвращают в реакторы гидроформили-
рования. С верха сепаратора смесь СО + Н2 вместе со свежим
синтез-газом тоже возвращают в процесс. Гидрирование альдеги-
174
дов до спиртов проводят в присутствии гомогенных (карбони*
лы кобальта, родия, иридия) и гетерогенных катализаторов (ко-
бальт на носителях, никель, вольфрам, цинк-хромовые, медь-
хромовые, алюмо-цинк-хромовые).
В приведенном варианте оксосинтеза в суспендированном
слое носителя ведут гидрирование альдегидов на кобальте,
осажденном на кизельгуре. Металлический кобальт недостаточ-
но активен (на нем не гидрируются побочные продукты оксо-
синтеза). Никель не годится в тех случаях, когда есть примеси
сернистых соединений. Наиболее пригодны цинк-хромовые, ни-
кель-хромовые и алюмо-цинк-хромовые катализаторы. Селектив-
ность при их применении достигает 95% при 90%-ной степени
конверсии альдегидов. Выход альдегидов составляет 75—85%
в расчете на олефин. Процесс аналогичен другим гидрогениза-
ционным процессам, его проводят с циркуляцией водорода при
160—300 °C и давлении до 30 МПа.
Промышленное значение метода непрерывно возрастает, мас-
штабы производства продуктов оксосинтеза расширяются. До-
стоинство процесса — гибкость в отношении сырья и получаемых
спиртов (на одном и том же оборудовании можно выпускать
разнообразные спирты).
6.6. Другие синтезы из СО и Н2
Перспективы применения смеси СО+Н2 связаны с разработ-
кой многих других процессов. Например, образование метана
из оксида углерода и водорода гладко протекает на металличе-
ских катализаторах (особенно на никеле) при 200—250 °C:
СО + ЗН2 СН4 + Н2О
В 1923 г. Фишер и Тропш установили, что если добавлять
щелочи к металлическим катализаторам, продуктом этой реак-
ции при высоком давлении является в основном смесь высших
спиртов (синтол):
400—500 °C;
10—15 МПа
пСО 4- тН2 -----> СпН^-хОН
При низких давлениях образуется преимущественно смесь угле-
водородов:
180—200 °C:
0,1—3 МПа
ziCO + (2п 1) Н2 „ * С/гН2/1+2
—ПП2'-'
При совместном действии оксида углерода и водяного пара
на олефины образуются карбоновые кислоты:
RCH=CH2 + СО + Н2О ---> RCH2-CH2-COOH
Этот процесс проводят в жестких условиях — при 300—400 °C
и 50—100 МПа. Так, пропионовую кислоту получают, пропуская
смесь 2% (об.) этилена, 90% (об.) оксида углерода и 8% (об.)
175
паров воды при 325°C и 70 МПа через слой катализаторам
фосфорную /кислоту на активном угле:
СН2=СН2 + СО + Н2О -> СН3-СН2-СООН
Реакция катализируется фосфорной и серной кислотой, фтори-
стым бором. Исследования процесса продолжаются. Появились
сведения о синтезе кислот этим методом при низком давлении.
Вместо олефинов можно брать спирты, так как при высокой
температуре и в присутствии кислотных катализаторов они де-
гидратируются. Пропилен и пропиловые спирты в указанных
выше условиях превращаются в изомасляную кислоту.
Если вместо воды взять метиловый спирт, получаются мети-
ловые эфиры кислот. Например, из этилена, метанола и оксида
углерода образуется метилпропионат:
СН2=СН2 + СН3ОН + СО -► СН3—СН2—ОСО—сн3
Этот синтез подробно исследован в присутствии иодидов ко-
бальта, никеля и железа на силикагеле при 200—350 °C и
15 МПа.
Если метанол заменить высокомолекулярными первичными
или вторичными спиртами, то в первую очередь идет дегидри-
рование спиртов, а выделяющийся водород вступает с олефина-
ми и оксидом углерода в реакцию гидроформилирования. Если,
действовать синтез-газом на спирт в тех же условиях, в кото-
рых проводят оксосинтез, но при более высокой температуре,
образуется спирт, следующий за ним в гомологическом ряду:
RCHaOH + СО + 2На > RCH2-CH2OH
Механизм реакции можно объяснить следующим образом. Вна-
чале спирт дегидратируется, а олефин вступает в реакцию гид-
роформилирования. Образующийся альдегид гидрируется в
спирт. В пользу такой точки зрения говорит то, что третичные
спирты вступают в реакцию наращивания углеродной цепи лег-
че, чем вторичные, а последние — легче, чем первичные. Однако
в эту реакцию вступает и .метиловый спирт, легко превращаю-
щийся в этиловый и бензиловый спирт (превращающийся да-
лее в р-фенилэтиловый), что вышеизложенным механизмом не
объясняется.
При взаимодействии оксида углерода и водяного пара с аце-
тиленом образуется акриловая кислота:
СН=СН + СО + Н2О —► сн2=сн-соон
Этот синтез представляет промышленный интерес. Если вместо
воды взять метанол, можно получить метилакрилат:
СН=СН + СО + сн3он —► сн2=сн—соо-сн3
По этой же схеме можно синтезировать и другие эфиры акрило-
вой кислоты. Процесс проводят при 150—180 °C и 10 МПа на
176
никелевом катализаторе. Собственно катализатором является
тетракарбонил никеля Ni(CO)4; при взаимодействии с ацетиле-
ном и водой он переходит в металлический никель. Так как
реакцию проводят в присутствии кислот (уксусной, соляной,
фосфорной), они связывают никель в водорастворимые соли,
которые в присутствии СО под давлением вновь образуют тет-
ракарбонил никеля.
6.7. Производство этилен- и пропиленгликоля
Двухатомные спирты (гликоли), являющиеся производными
олефинов, содержат в молекуле две гидроксильные группы у
соседних атомов углерода и относятся к классу 1,2-гликолей,
или а-гликолей.
Первый представитель этого класса — этиленгликоль
НОСН2—СН2ОН — жидкость со слабым приятным запахом,
сладкая на вкус. Т. кип. при атмосферном давлении 197,6 °C;
плотность при 20 °C равна 1,1130 г/см3. Он не огнеопасен. Вод-
ные растворы его имеют низкую температуру затвердевания:
40%-ный раствор затвердевает при —21 °C, а 60%-ный при
—75 °C. Благодаря этому свойству он широко применяется как
антифриз. Этиленгликоль вступает во все химические реакции,
характерные для одноатомных спиртов. В /реакции могут всту-
пать одна и две гидроксильные группы.
Моноэфиры этиленгликоля — целлозольвы — широко при-
меняют как растворители нитрата и ацетата целлюлозы, а так-
же как компоненты сложных растворителей. Динитро этиленгли-
ко ль O2NO—СН2—CH2ONO2 представляет собой взрывчатое
вещество, по силе взрыва не уступающее нитроглицерину. Эта-
ноламины имеют сильные основные свойства, широко применя-
ются при очистке углеводородных газов от сероводорода
(стр. 52). Этаиоламиды высших карбоновых кислот, «имеющие
формулу RCONH—СН2—СН2ОН, обладают поверхностной ак-
тивностью :и применяются в композициях синтетических мою-
щих средств.
Важнейшая область применения этиленгликоля — производ-
ство полиэтилентерефталатного волокна лавсан (стр. 249). Он
используется и как растворитель. Сочетание консервирующих
и гигроскопических свойств делает этиленгликоль удобным для
использования в производстве косметических средств, три от-
делке и крашении текстильных изделий.
Этиленгликоль можно получить гидролизом дихлорэтана или
этил ен хл о р гидр ин а:
МагСОз
СН2С1-СН2С1+2Н2О --->- НОСН2—СН2ОН + 2НС1
NaHCO3
HOCH2-CH2C1 + H2o -----> НОСН2—СН2ОН + НС1
12—254
177
Вода
Рис. III.16. Технологическая схема получе-
ния гликолей:
1 — подогреватель; 2 — гидрататор; 3 — сепара-
тор; 4 — холодильник-конденсатор.
Гликоли
Зтилен-
окоид
I Вода
Основной промышленный метод получения этиленгликоля — гид-
ратация этиленоксида
Н2С---СН2 + Н2О
НОСН2-СН2ОН
при 180—195 °C и 1,7—1,8 МПа в присутствии небольших коли-
честв серной кислоты. Реакция не останавливается на образо-
вании моноэтиленгликоля: по мере увеличения его концентра-
ции он начинает реагировать с этиленоксидом, образуя диэти-
ленгликоль и т. д.:
НОСН2—СН2ОН + Н2С--СН2 ---► НОСН2-СН2-О—сн2—СН2ОН
О
Водный раствор этиленгликоля упаривают, обезвоживают в ва-
кууме и подвергают ректификации. Выход этиленгликоля со-
ставляет 84,5%. На 1 т этиленгликоля расходуется 0,85 т
99,5%-ного этиленоксида. В качестве побочных продуктов на
1 т этиленгликоля образуется 114 кг диэтиленгликоля и 13 кг
триэтиленгликоля.
При производстве гликолей (рис. III.16) этиленоксид, вод-
ный конденсат и возвратную воду смешивают перед подогрева-
телем /, нагревают и подают в гидрататор 2. В смеси концент-
рация этиленоксида равна 12—14%. Выделяющееся тепло не от-
водят, поэтому реакционная масса нагревается на 40—50 °C, но
за счет повышенного давления находится в жидком состоянии.
На выходе из гидрататора ее дросселируют и подают в сепара-
тор 3. Водяные пары конденсируются в конденсаторе 4. Воду
возвращают в процесс, а гликоли, содержащие некоторое ко-
личество воды, поступают на упаривание и дистилляцию в ва-
кууме.
На аналогичных установках получают пропиленгликоль и
этаноламины, а также целлозольвы. Последние получают по
реакции этиленоксида со спиртами, причем продукт реакции не
упаривают, а сразу подвергают ректификации.
Гидратация пропиленоксида в пропиленгликоль
СН3—НС---СН2 + Н2О
СН3-СН-СН2ОН
он
178
проходит в тех же условиях, что и гидратация этиленоксида.
В смеси, поступающей на гидратацию, концентрация пропилен-
оксида составляет 15—17%. Наряду с цропиленгликолемобразу-
ются ди- и полипропиленгликоли: в продуктах реакции содер-
жится 85% пропиленгликоля, 13% дипропиленгликоля и 1,5—
2% трипропиленгликоля.
Аэрозоли пропиленгликоля обладают бактерицидным дейст-
вием. Основное же количество пропиленгликоля расходуется на
производство полиэфиров и пластификаторов.
Полипропиленгликоль (т. кип. 187,4 °C при атмосферном дав-
лении) заменяет в ряде случаев этиленгликоль как раствори-
тель и как антифриз (60%-ные водные растворы 'пропиленгли-
коля затвердевают при —70°C). Он практически не токсичен,
поэтому дрименяется в пищевой, фармацевтической и парфю-
мерной промышленности. Он гигроскопичен, поэтому его до-
бавки позволяют сохранять нужную влажность пищевых про-
дуктов и табака при длительном хранении.
Сополимеры этилен- и пропиленгликоля растворяются в во-
де, являются моющими веществами.
6.8. Производство глицерина
Глицерин СН2ОН—СН (ОН) —СН2ОН — важнейший среди
трехатомных спиртов. Это сиропообразная бесцветная жидкость,
сладкая на вкус. Глицерин смешивается во всех отношениях
с водой и этиловым спиртом, он растворяется в дцетоне, но не
растворяется в бензине, бензоле, жирах, хлороформе. Глицерин
очень гигроскопичен — поглощает до 40% влаги. При атмосфер-
ном давлении кипит при 290 °C с частичным разложением; в ва-
кууме перегоняется без разложения.
Вследствие наличия в молекуле трех гидроксильных групп
из глицерина могут образовываться три ряда производных, при-
чем моно- и дипроизводные способны существовать в двух
структурно изомерных формах, а дипроизводные СН2Х—СНХ—
—СН2ОН и монопроизводные СН2Х—СНОН—СН2ОН содержат
асимметрический атом углерода и существуют в двух оптически
изомерных формах. Глицерин может давать моно-, ди- и тригли-
цериды металлов. Сложные эфиры глицерина и органических
кислот составляют большую и важную группу производных, сре-
ди 'которых особое место занимают жиры.
Глицерин применяется в различных областях: в производстве
алкидных полимеров, в пищевой, бумажной, текстильной, фар-
мацевтической, лакокрасочной промышленности, в радиотехни-
ке, производстве взрывчатых веществ, при изготовлении целло-
фана и др., а также в сельском хозяйстве.
Значительную часть глицерина получают гидролизом рас-
тительных и животных жиров, однако из года ц год растет роль
синтетических методов производства глицерина. Синтетический
12* 179
глицерин получают из пропилена двумя способами — хлорным
и бесхлорным.
При хлорном методе хлорируют пропилен при высокой тем-
пературе и получают хлористый аллил:
500 °с
СН2=СН-СН3 + С12 —t СН.=СН—СН2С1
—-НС1
Побочно образуются изомерные моно- и дихлорпропилены,
1,2-дихлорпропан. Чтобы увеличить выход хлористого аллила,
ведут хлорирование при мольном соотношении пропилена и хло-
ра, равном 5:1. Степень конверсии пропилена за один проход
25%. Степень конверсии хлора 100%. Выход хлористого аллила
80—85%. Реакция экзотермична (112 кДж/моль).
Из хлористого аллила глицерин получают двумя путями:
1) гидролизом хлористого аллила 5%-ным раствором едкого
натра в аллиловый спирт, гипохлорированием аллилового спир-
та до монохлоргидринов глицерина и гидролизом монохлоргид-
ринов:
+NaOH
СН2=СН-СН2С1 СН2=СН-СН2ОН
СН2=СН-СН2ОН
4-нсю
ОН ОН С1
I I I
—► СН2— CH—СН2 — |+NaOH
СН2-СН-СН2 _J-NaC1
ОН С1 С1
сн2—сн-сн2
ОН <!)Н он
2) гипохлорированием хлористого аллила в дихлоргидрин
глицерина, гидролизом дихлоргидрина водной щелочью с обра-
зованием глицерина или действием известкового молока с обра-
зованием эпихлоргидрина, который также может быть подверг-
нут гидролизу в глицерин:
+нсю
СН2=СН-СН2С1 --->
4-NaOH
СН2-СН-СН2 —t сн2-сн-сн2
I I I -Nacl I I I
ОН С1 С1 он он он
снас1-нс—сна
Xb//
Практический интерес представляют оба пути синтеза, так
как промежуточные продукты — аллиловый спирт и дихлоргид-
рин — имеют важное промышленное значение. Эпихлоргидрин
применяют -в синтезе эпоксидных полимеров, а аллиловый спирт
благодаря реакционной способности, придаваемой ему гидрок-
180
силом и двойной связью, используют в ряде синтезов (в том чис-
ле при получении сложных эфиров, применяемых в производстве
пластических масс), а при действии -пероксида водорода из него
можно получить глицерин. Большой расход хлора, превращение
его в трудно утилизируемые продукты является существенным
недостатком хлорного метода.
Бесхлорные методы базируются главным образом на превра-
щении аллилового спирта в глицерин. Спирт может быть -полу-
чен разными методами. Из акролеина аллиловый спирт получа-
ют восстановлением в газовой фазе при 350—450 °C, атмосфер-
ном давлении и в присутствии доноров водорода (первичные
или вторичные спирты) над магний-цинковыми катализаторами
или в жидкой фазе в присутствии изопропилата алюминия:
СН2=СН—СНО + СН3—СНОН-СНз ► СН2=СН-СН2ОН + СН3-СО-СН3
Аллиловый спирт получают также изомеризацией пропилен-
оксида:
СН3—НС—сн2 -----► СН2=СН-СН2ОН
'''о7
Гидроксилирование аллилового спирта ведут пероксидом во-
дорода при катализе вольфрамовой кислотой; при этом эпокси-
дирование аллилового спирта и гидратация глицидола протека-
ют в одном аппарате:
hwo4 Ч-н2о
СН2=СН-СН2ОН + Н2О2 —> н2с—сн-сн2 —> сн2— сн-сн2
он он Ан он
глицидол
Процесс получения глицерина из пропиленоксида, по-видимо-
му, является наиболее перспективным. Из пропиленоксида полу-
чают аллиловый спирт, эпоксидируют его в глицидол, который
гидролизуют в глицерин. Аллиловый спирт можно эпоксидиро-
вать не только пероксидом водорода, но и надуксусной кислотой.
Гидратацию глицидола проводят, не выделяя его из реакционной
массы, при 100 °C в присутствии кислотных или щелочных ката-
лизаторов (H2SO4, Na2CO3). Выход глицерина составляет 85—
87% в расчете на вступивший в реакцию аллиловый спирт. Пер-
спективность метода обусловлена тем, что необходимый для гид-
роксилирования пероксид водорода может быть получен при
окислении изопропилового спирта
СН3—СНОН—СН3 + О2 --► СН3-СО-СН3 + Н2О2
т. е. производство глицерина обеспечивается одним сырьем —
пропиленом.
ГЛАВА IV
ПРОИЗВОДСТВО синтетических
МОЮЩИХ ВЕЩЕСТВ
§ 1. ПРИМЕНЕНИЕ И КЛАССИФИКАЦИЯ
ПОВЕРХНОСТНО-АКТИВНЫХ ВЕЩЕСТВ
1.1. Применение поверхностно-активных веществ
Среди поверхностно-активных веществ (ПАВ) наиболее мно-
готонмажным является производство синтетических моющих
средств (СМС) — композиций, состоящих из активного моюще-
го вещества (ПАВ), вспомогательных компонентов и наполни-
телей, усиливающих моющий эффект всей композиции. Моющие
средства выпускают в виде порошков, паст и жидкостей.
Моющие средства бытового назначения и компоненты, входя-
щие в них, должны удовлетворять ряду требований. Они не
должны вызывать коррозии металлических поверхностей сти-
ральных машин, кухонной утвари, поэтому к ним добавляют
ингибиторы коррозии — жидкое стекло, соли бериллия и др.
Стиральные порошки должны быть негигроскопичными, иссле-
живающимися и обладать способностью сохранять белизну тка-
ней; они не должны вредно действовать на ткани и изменять их
окраску. Моющие средства не должны действовать' на кожу.
Иногда к моющим составам добавляют оптические отбеливате-
ли. Моющие композиции для шерсти, грубого и тонкого белья,
изделий из шерсти, шелка и синтетических тканей различаются
характером моющего вещества и природой активных добавок.
Поверхностно-активные и моющие вещества применяют в
текстильной промышленности при процессах крашения, обесцве-
чивания и специальных видах отделки тканей.
ПАВ используют при изготовлении различных косметиче-
ских .средств, туалетной воды (лосьоны), препаратов для ухода
за кожей и волосами, средств для бритья, зубных паст, туалет-
ных мыл, а также специальных составов, применяемых для
очистки рук в производственных условиях и в медицинских уч-
реждениях.
Поверхностно-активные вещества применяют и как абразив-
ные составы для чистки металлов. Эти вещества хорошо уда-
ляют жировые загрязнения, причем их применяют не только при
специальных операциях очистки и травления металлов перед
окраской или эмалированием, но и в более крупных масштабах,
например при мытье отсеков танкеров. Употребляют поверхност-
но-активные вещества и для защиты металлов от коррозии (они
улучшают смачивание металла и адгезию покрытий), а также
.182
как смазочно-охлаждающие жидкости при холодной обработке
металлов.
Находят применение поверхностно-активные вещества и при
обработке кожи. При дублении кожи для облегчения ее смачи-
вания к дубителям добавляют вещества, повышающие раствори-
мость дубителей и препятствующие их выпадению из дубящего
раствора при длительном процессе. ПАВ применяют также при
крашении кожи и меха; при этом обеспечиваются лучшее про-
никание красителя и равномерность окраски.
ПоВерхностно-активные вещества со стабильным пенообра-
зованием применяют при изготовлении пенобетонов. ПАВ упо-
требляют также в производстве сухой штукатурки, кровельных
и обшивочных материалов. Они повышают смачивающую спо-
собность красок, применяемых при малярных работах. При мо-
щении дорог эти вещества используют как смачиватели гравия
и песка, обеспечивающие плотное прилипание асфальта и биту-
ма к покрытию. Они улучшают текучесть цемента, повышают
скорость его схватывания, что особенно важно для строитель-
ных работ под водой.
При флотации руд также важны поверхностно-активные ве-
щества. В смесь измельченной руды и воды вводят флоторе-
агент и продувают смесь воздухом. При этом порода или мине-
рал прилипает к пузырькам воздуха под действием поверхност-
ных сил, выносится вверх и уходит с пеной. Остальные компо-
ненты руды становятся гидрофильными и оседают на дно.
В производстве каучуков и пластических масс поверхностно-
активные вещества применяют при эмульсионной полимериза-
ции ,и как эмульгаторы полимеров. Как вспениватели, поверх-
ностно-активные вещества употребляют при получении губчатой
резины из вспененного латекса. ПАВ применяют также для ста-
билизации латексов, для улучшения смешения каучука с пиг-
ментами и наполнителями.
Широко используют поверхностно-активные вещества в сель-
ском хозяйстве. Так, чтобы облегчить нанесение и удержать на
опрыскиваемой поверхности инсектициды (средства для уничто-
жения насекомых), фунгициды (вещества для протравливания
семян) и гербициды (средства для борьбы с сорняками), к ним
добавляют поверхностно-активные вещества. Некоторые мою-
щие вещества сами обладают высокой инсектицидной способ-
ностью. Прибавление некоторых поверхностно-активных веществ
в корм цыплятам и поросятам увеличивает их вес, т. е. ускоряет
рост.
Поверхностно-активные вещества применяют и в нефтяной
промышленности, например при добыче нефти. При промывке
скважин соляной кислотой добавляют к кислоте смачиватели,
облегчающие проникание кислоты в породу. Если в песчаных
породах к устью скважины поступает обводненная нефть, для
снижения поверхностного натяжения водной фазы п увеличения
притока нефти обрабатывают устья скважины растворами ПАВ.
183;
При деэмульгировании нефти на промыслах и нефтеперера-
батывающих заводах используют поверхностно-активные веще-*
ства для облегчения разрушения нефтяных эмульсий.
Поверхностно-активные вещества употребляют и в пищевой
промышленности, например в производстве маргарина, в хлебо-
печении, при изготовлении кондитерских изделий и мороженого.
Поверхностно-активные вещества используют и при получе-
нии противопожарных пен, а также как пенообразователи в про-
изводстве бродящих напитков (квас, пиво).
Приведенный далеко не полный перечень областей примене-
ния ПАВ подтверждает их важное значение для народного хо-
зяйства. Вместе с тем становится очевидным, что для столь мно-
гообразных областей применения необходимы различные ПАВ.
Свойства ПАВ и эффективность их использования тесно связа-
ны со строением молекул ПАВ.
В настоящее время жировое мыло почти полностью (за ис-
ключением туалетного) вытесняется синтетическими моющими
средствами. Быстрое развитие СМС и вытеснение ими жирового
мыла обусловлено следующими преимуществами СМС:
1) по эффективности СМС в 2—4 раза превосходят жировое
мыло, что снижает расход ПАВ для достижения такого же мою-
щего эффекта;
2) исходным сырьем для производства СМС являются про-
дукты переработки нефти и газа, что сокращает расход пище-
вых жиров;
3) СМС не образуют нерастворимых солей кальция и магния
в жесткой воде, тогда как 30—35% жирового мыла расходу-
ется на умягчение воды;
4) СМС не гидролизуются и не создают щелочной среды,
благодаря чему обладают мягким моющим действием, не раз-
рушают синтетические ткани;
5) максимальная моющая способность СМС достигается при
значительно более низкой температуре, чем у жирового мыла.
Это позволяет добиться очищающего эффекта при температуре
не выше 40 °C, что также способствует увеличению срока служ-
бы изделий из шерстяных, шелковых и синтетических тканей’
6) трудовые затраты на производство 1 т СМС в 14—15 раз
ниже, чем на производство 1 т сырья для жирового мыла.
1.2. Физико-химия действия ПАВ в водных
и неводных средах
Большинство технологических процессов, а также явлений
в природе протекает на границе раздела двух и более фаз: жид-
кость — газ, жидкость — твердое тело, жидкость — твердое те-
ло — газ и т. п. Способность растворенного вещества самопроиз-
вольно адсорбироваться на поверхности раздела фаз и умень-
шать при этом поверхностное натяжение служит мерой поверх-
ностной активности данного вещества.
484
Проявление поверхностной активности предопределяется ди-
фильным строением веществ, т. е. наличием в их молекуле двух
частей, противоположных по природе и свойствам и представ-
ляющих в общем случае сильнополярные и неполярные группы.
Сильнополярная группа — гидрофильная — способна к межмоле-
кулярным взаимодействиям, обусловливающим растворимость
молекул ПАВ в воде и других полярных средах, а также проч-
ную адсорбцию на границе раздела фаз. Неполярная (или сла-
бополярная) — гидрофобная — группа является носителем ли-
пофильных (маслолюбивых) свойств. В большинстве случаев это
углеводородные цепочки, которые определяют сродство ПАВ
к газовой фазе, углеводородным и другим средам.
В зависимости от соотношения полярной и неполярной ча-
стей молекулы ПАВ, от так называемого гидрофильно-липо-
фильного баланса (ГЛБ) различают водо- и маслорастворимые
ПАВ. К водорастворимым ПАВ относят синтетические моющие
вещества, эмульгаторы жиров, деэмульгаторы водо-нефтяных
эмульсий, добавки для интенсификации добычи нефти и т. д.
Маслорастворимые ПАВ, в частности присадки к смазочным
маслам, смазкам и смазочно-охлаждающим жидкостям, а так-
же ингибиторы коррозии, получают все большее применение.
Ориентированно адсорбируясь на границе раздела фаз, на-
пример на границе вода — воздух, молекулы ПАВ образуют на
этой поверхности прочную мономолекулярную пленку, изменяю-
щую ее первоначальные свойства (повышается поверхностная
вязкость и, как следствие, снижается поверхностное натяже-
ние). Снижение поверхностного натяжения на границе раздела
фаз понижает потенциальный барьер между ними, облегчает их
взаимопроникание. Раствор ПАВ легко смачивает гидрофобные
поверхности, например при обработке тканей в водных средах
(крашение, стирка) или при вытеснении нефти из нефтесодер-
жащих пород. Вследствие хорошего смачивания твердых мате-
риалов и проникания раствора ПАВ в поры и капилляры сни-
жаются затраты на измельчение и диспергирование этих мате-
риалов. Образование па границе раздела фаз адсорбционной
пленки из молекул ПАВ обусловливает пенообразование рас-
творов.
С увеличением концентрации ПАВ в растворе происходит на-
сыщение поверхностного слоя. Молекулы ПАВ начинают агре-
гировать в мицеллы, обращаясь друг к другу неполярными или
полярными группами, в зависимости от полярности самого рас-
творителя. Возникновение мицелл ПАВ изменяет и объемные
свойства раствора, придавая ему способность поглощать ранее
не смешивающуюся с ним фазу и образовывать стабильные дис-
персные системы. Одним из важнейших видо<в таких систем яв-
ляются эмульсии. В них эмульгируемая фаза находится внутри
мицелл, внешние оболочки которых вследствие отталкивания
друг от друга защищают эмульсию от разрушения. Если ПАВ
с большой гидрофильностью (с высоким ГЛБ) добавить в
18&
Рис. IV. 1. Механизм моющего действия ПАВ:
а —частица загрязнения на ткани; б, в—адсорбция молекул ПАВ на частице; г —
частица, переведенная в раствор при помощи ПАВ.
эмульсию типа «вода в масле», то ПАВ вытеснит исходный
эмульгатор из капелек воды и они вновь объединятся в единую
фазу. Такой процесс называется деэмульгированием.
Если в молекуле ПАВ углеводородный радикал небольшой
(С2—С3), такое вещество полностью растворяется в воде и прак-
тически не обладает поверхностной активностью. При длине ра-
дикалов Сю—Сю полного растворения ПАВ не происходит—в
растворе содержатся неорганические и органические ионы, а
также мицеллы — агрегаты из 50—100 плотно упакованных мо-
лекул ПАВ, т. е. образуется полуколлоидная система. Образо-
вание мицелл начинается при достижении критической концент-
рации ПАВ и наблюдается в узкой области концентраций. При
этом меняются плотность, электропроводность, поверхностное
натяжение и моющее действие этих растворов. Разные ПАВ
имеют разные критические концентрации мицеллообразования
(ККМ).
Механизм моющего действия ПАВ заключается в следую-
щем: 1) смачивание загрязненной ткани водным раствором
ПАВ; 2) удаление загрязнения с поверхности ткани; 3) удержи-
вание загрязнений в объеме раствора. Вначале (рис. IV.1) про-
исходит адсорбция ПАВ на частицах загрязнений (гидрофобные
части молекул ПАВ направлены в сторону загрязнения, гидро-
фильные— в раствор). На границе раздела загрязнение — рас-
твор благодаря адсорбции ПАВ резко снижается поверхностное
натяжение, за счет чего в зазор между частицей загрязнения и
тканью лучше проникает моющий раствор, создавая адсорби-
рованный слой ПАВ. Возникает расклинивающий эффект, части-
ца загрязнения отрывается от поверхности ткани и переходит в
раствор. Чаще всего загрязнения бывают жирового характера,
поэтому эффективность моющего действия зависит от солюбили-
зирующей способности ПАВ. Молекулы солюбилизируемого ве-
щества входят внутрь мицеллы, располагаясь между гидрофоб-
ными концами молекул ПАВ. Смачивание ткани или другого
твердого тела — процесс вытеснения из них воздуха или жид-
кости раствором ПАВ. Чем меньше поверхностное натяжение
на границе раздела фаз, тем быстрее смачивается ткань.
186
Эмульгирующая способность растворов ПАВ — способность
переводить жидкие частицы загрязнений с очищаемой поверх-
ности в раствор в виде водных эмульсий. Чтобы повысить ста-
бильность эмульсий, в растворы ПАВ добавляют эмульгаторы и
стабилизаторы.
Диспергирующее действие растворов ПАВ — способность
дробить и переводить в водный раствор твердые частицы загряз-
нений в виде суспензии, предотвращая при этом агломерацию
частиц. Способность удерживать загрязнения в растворе опре-
деляет эффект стирки. Улучшить эту способность можно путем
введения антиресорбционных добавок, -например натриевой со-
ли карбоксиметилцеллюлозы.
Пенообразующая способность — результат ориентированной
адсорбции молекул ПАВ на границе раздела вода — воздух:
гидрофильные части молекул ПАВ направлены в воду, а гидро-
фобные—в сторону воздуха. Поверхностное натяжение снижа-
ется, и при диспергировании воздуха образуется пена. Пенооб-
разование непосредственно не связано с моющим действием.
Хотя накапливание в пене загрязнений и играет определенную
роль, но эта роль косвенная, так как, например, неионогенные
моющие вещества при незначительном ценообразовании обла-
дают хорошими моющими свойствами.
Маслорастворимые ПАВ проявляют поверхностную актив-
ность в основном на границах раздела жидкость — твердое те-
ло, а также на границах раздела с водой. Маслорастворимые
ПАВ образуют коллоидную мицеллярную структуру с фазовыми
границами раздела мицелла — масло. Число молекул, агреги-
рованных в мицелле, и критическая концентрация мицеллообра-
зования ПАВ в нефтепродуктах тесно связаны с функциональ-
ными свойствами ПАВ в объеме. Поверхностная активность на
границе с металлом или водой определяет функциональные свой-
ства ПАВ на поверхности — проявление защитных, противокор-
розионных, противоизносных, антифрикционных и других
свойств. Противоизносные и антифрикционные свойства масел с
присадками объясняют адсорбцией присадок на поверхности ме-
талла; то же относится и к ингибиторам коррозии. Существуют
водомаслорастворимые ПАВ, применяемые в системах нефтепро-
дукт+вода. Гидрофильные части молекул обеспечивают им
растворимость в воде, а гидрофобные придают растворимость
в углеводородах (в топливах и маслах). Водомаслорастворимые
ПАВ применяют в качестве деэмульгаторов, эмульгаторов, сма-
зочно-охлаждающих жидкостей, ингибиторов коррозии.
1.3. Классификация поверхностно-активных веществ
По растворимости в тех или иных средах ПАВ можно разде-
лить на три основные группы: водорастворимые, водомаслорас-
творимые и маслорастворимые. Самую многочисленную группу
187
составляют водорастворимые ПАВ. Их классификация основана
на химическом строении. Если ПАВ диссоциируют в воде на
ионы, их относят к ионогенным веществам, если ПАВ не диссо-
циируют, — к неионогенным.
Ионогенные ПАВ подразделяют на анионоактивные и катио-
ноактивные в зависимости от того, каким ионом (катионом или
анионом) обусловлена поверхностная активность. Имеются и
амфолитные ПАВ; они содержат в молекуле две (или более)
функциональные группы, которые в кислой среде проявляют ка-
тионоактивные свойства, а в щелочной — анионоактивные.
К анионоактивным относятся моющие вещества следующих
типов:
1) алкилкарбонаты (мыла) RCOONa;
2) алкилсульфаты — первичные RCH2O—SO2ONa и вторич-
ные RR'CHO—SO^ONa;
3) сульфаты жирных кислот, имеющие формулу
RCH (OSOoONa) — (СН2)COONa;
4) алкилсульфонаты— первичные RSO2ONa и вторичные
RR'CHSO2ONa;
5) сульфонаты жирных кислот, имеющие формулу
RCH (SO2ONa) — (СН2) п—COONa;
6) алкиларилсульфонаты — алкилбензолсульфонаты и ал-
килнафталинсульфонаты:
SO20Na R
SO2ONa
7)( а-олефипсульфопаты RCH = CH—SO2ON;
8) сульфаты оксиэтилированных алкилфенолов:
О— ( СН2СН2О)/—SO2ONa
К катионоактивным моющим веществам относятся:
1) аммониевые соли первичных, вторичных и третичных ами
+ + +
нов (RNH3 С1-, R2NH2 С1-, R3NH Cl~);
2) четвертичные аммониевые основания RNR3 С1~;
3) соли бензиламмония:
ff у_сн2—NR3 С1-
188
Амфолитные моющие вещества имеют такое строение:
1) соединения, содержащие карбоксильную и аминогруппу:
RCH-COO- Na+
+NH3 СГ
2) соединения, содержащие сульфоэфирную и аминогруппу;
RCH—СН2—СН2—OSO2ONa
+NH3 CI-
3) соединения, содержащие сульфогруппу и аминогруппу:
RCH—СН2—СН2—SO2ONa
+NH3 Cl-
Из неионогенных поверхностно-активных веществ отметим
следующие:
1) оксиэтилированные алкилфенолы:
= R 0-(CH2CH2O)ft-H
Ч
2) оксиэтилированные жирные кислоты, имеющие формулу
RCOO— (СН2СН2О) п—Н;
3) оксиэтилированные жирные спирты RO—(СН2СН2О)П—Н;
4) оксиэтилированные амины:
/(СН2СН2О),1-Н
RNH—(СН„СН„О)„—Н Rb/
\(СН2СН2О)„-Н
5) оксиэтилированные меркаптаны и амиды:
/(CHaCHjOJn-H
RS-(CH2CH2O)„-H RCObZ
\(СН2СН2О)П-Н
6) оксиэтилированные полипропиленгликоли:
СН3-СН-СН2-О-(СН2СН2О)т-Н
О-(СН2СН2О)„-Н
СН2-СН2-О-(СН2СН2О)„-Н
СН2—О—(CH2CH2O)m—Н
В приведенной классификации дан далеко не полный ассор-
тимент ПАВ, указаны лишь основные их виды, применяемые в
производстве синтетических моющих средств.
Анионоактивные вещества в водном растворе диссоциируют
на катион щелочного металла и анион — остальную часть моле-
кулы, например первичные алкилсульфаты, диссоциируют по
схеме:
RSO2ONa ^=t RSO3- + Na+
189
Катионоактивные ПАВ хорошо ионизуются в растворах, а чет-
вертичные аммониевые основания ионизуются полностью.
Важнейшими представителями катионоактивных ПАВ явля-
ются триэтилцетиламм'онийхлорид и цетилпиридинийбромид,
которые из-за легкой ионизации изображают в ионной форме:
[(C2H5)3N—С1вН33] Cl" CieH33-N^2)> Br'
Катионоактивные вещества применяют как моющие и бактери-
цидные препараты или как бактерицидные добавки к моющим
средствам. Они применяются также в медицине при стерилиза-
ции и, дезинфекции.
Неионогенные моющие вещества обладают хорошими мою-
щими свойствами; лучше, чем другие моющие вещества, удер-
живают загрязнения в растворе, предотвращая их обратное осе-
дание на ткань; способны к биохимическому разложению. Их
получают при взаимодействии алкилфенолов, жирных кислот,
спиртов с этиленоксидом, причем число оксиэтильных групп в
ПАВ может быть от 4 до 20, а иногда и больше. Вместо эти-
леноксида можно применять пропиленоксид.
Таким образом, свойства синтетических моющих веществ
можно широко варьировать за счет изменения их структуры,
функциональных групп, соотношения полярных и неполярных
групп. В настоящее время известно более 200 видов синтетиче-
ских ПАВ и более 3000 торговых наименований различных ком-
позиций из них. Тем не менее основой для СМС и ПАВ техниче-
ского назначения служат всего несколько десятков препаратов,
выпускаемых промышленностью.
§ 2. ПРОИЗВОДСТВО АНИОНОАКТИВНЫХ
МОЮЩИХ ВЕЩЕСТВ
2.1. Теоретические основы процессов сульфирования,
сульфохлорирования и сульфоокисления
Наибольшее распространение среди анионоактивных моющих
веществ имеют соли карбоновых кислот (алкилкарбонаты нат-
рия, мыла), а также соли сульфокислот, получаемых при суль-
фировании высших жирных спиртов, олефинов, парафинов и ал-
килароматических углеводородов.
В зависимости от химической природы сырья при сульфиро-
вании образуются алкиловые эфиры серной кислоты (алкил-
сульфаты) RCH2OSO2ONa, в которых углеродный атом липо-
фильной группы связан с серой через кислородный атом, или
алкил (арил) сульфонаты RSO2ONa, углеродный атом которых
связан с атомом серы непосредственно.
Сульфирующими агентами служат 98%-ная серная кислота,
олеум (раствор триоксида серы в серной кислоте) и жидкий
или газообразный триоксид серы. Взаимодействие спиртов с сер-
190
ной кислотой протекает обратимо:
ROH + H2SO4 ROSO2OH + H2O
Глубина сульфирования спиртов возрастает при уменьшении
содержания воды в реакционной смеси и увеличении избытка
серной кислоты. Однако с повышением температуры и концент-
рации кислоты, а также при использовании олеума образуется
заметное количество побочных продуктов, в частности диалкил-
сульфатов
ROH + ROSO2OH roso2or + Н2О
которые не обладают поверхностной активностью в водной сре-
де; появляются также олефины, 1простые эфиры, альдегиды, ке-
тоны и смолистые вещества.
Чтобы усовершенствовать процесс и повысить качество ал-
килсульфатов, в последнее время для сульфирования спиртов
используют газообразный триоксид серы или его комплексы с
карбамидом и аминами. В этом случае реакция становится не-
обратимой и достигается высокая глубина сульфирования при
небольшом избытке сульфирующего агента. При использовании
SO3 тоже образуются побочные продукты, особенно при сульфи-
ровании вторичных спиртов, и поэтому здесь важно проводить
процесс в мягких условиях.
Сульфирование олефинов серной кислотой рассмотрено выше
(гл. III, § 2). Отметим, что в случае олефина с достаточно длин-
ной углеродной цепочкой промежуточные ионы карбония спо-
собны к изомеризации с перемещением реакционного центра
вдоль цепи, т. е. частично идет скелетная изомеризация. Вслед-
ствие этого из а-олефинов нормального строения образуется
заметное количество р- и у-алкилсульфатов (иногда с развет-
вленной алкильной группой), обладающих пониженной моющей
способностью. Кроме того, возможны побочная полимеризация
олефинов, образование спиртов, простых эфиров и диалкилсуль-
фатов.
Чтобы снизить выход побочных продуктов, сульфирование
олефинов проводят 92—95%-ной серной кислотой при низкой
температуре (0—20°C). Другие сульфирующие агенты (олеум и
SO3) приводят не к алкилсульфатам, а к а-олефинсульфонатам:
R—(СН2)П—СН=СН2 + SO3 -> R—CH=CH-CH2SO2OH
Однако в последнее время установлено, что а-олефинсульфонаты
представляют собой ценные моющие вещества (стр. 201), отли-
чительной особенностью которых является легкое биохимическое
разложение в сточных водах (практически такое же, как у ал-
килсульфатов). Поэтому для сульфирования олефинов иногда
используют газообразный триоксид серы, но только разбавлен-
ный воздухом или диоксидом серы, а также в виде растворов
(в диоксане, метиленхлориде и др.). В качестве побочных про-*
дуктов образуются сультоны (стр. 202).
191
С парафинами ни один из указанных выше сульфирующих
агентов не реагирует, поэтому были разработаны методы суль-
фохлорирования и сульфоокисления парафинов.
При сульфохлорировании обрабатывают парафины смесью
диоксида серы и хлора при УФ-облучении; при этом образу-
ются алкилсульфохлориды:
hv
rh + so2 + ci2 —> RSO2C1
—1
Реакция протекает по радикально-цепному механизму. Цепь
зарождается за счет гомолитического расщепления молекулы
хлора иод действием УФ-облучения:
hv
С12 ---> 2CI- R- +so2 > RSO2
Cl- +RH----► R- 4-HC1 RSO2 + C12--► RSO2C1 + Cl-
При сульфохлорировапии параллельно протекает фотохимиче-
ское хлорирование исходного парафина, подавить которое мож-
но, используя избыток диоксида серы по отношению к хлору.
Однако полностью подавить таким путем хлорирование олефи-
нов и ароматических углеводородов не удается, из-за чего ис-
ходное парафиновое сырье необходимо очищать от этих при-
месей. Активно протекает также побочное хлорирование изопа-
рафинов, поэтому самым подходящим сырьем для сульфохлори-
рования являются «-парафины С12—Ci8.
По аналогии с сульфохлорированием протекает реакция
сульфоокисления парафинов, приводящая к непосредственному
образованию алкансульфокислот:
hv
RH + SOa + 0,5Оа -»- RSO2OH
При действии инициаторов образуется алкильный радикал, ко-
торый взаимодействует с SO2, образуя алкилсульфонрвый ра-
дикал. Он реагирует с кислородом и превращается в алкилсуль-
фопероксидный радикал, который, взаимодействуя с углеводоро-
дом, дает новый радикал и надкислоту. Поскольку радикалы
появляются в процессе самой реакции, сульфоокисление может
протекать автокаталитически:
hv
RH ----► R- + Н. RSOO- 4- О2------► RSO2OO.
R. +SO2 ---► RSOO. RSO2OO. +RH ---------> R- + RSO2OOH
Надкислота при взаимодействии с диоксидом серы и водой дает
алкансульфокислоту и серную кислоту:
RSO2OOH + so2 + Н2О --> RSO2OH + H2SO4
Механизм реакции сульфоокисления был доказан на приме-
ре циклогексана. Оказалось, что серная кислота образуется
всегда, даже при использовании совершенно безводных веществ.
192
Вода образуется за счет дегидрирования циклогексана в цикло-
гексен при его взаимодействии с надкислотой:
RSO2OOH +
Циклогексен реагирует с надкислотой и образует побочные про-
дукты:
RSOjOOH 4-
RSO2O-i
Существует несколько вариантов процесса сульфоокисления
парафинов: в присутствии воды, метанола, уксусного ангидрида.
Если сульфоокисление проводят в присутствии воды, надкисло-
та сразу же восстанавливается диоксидом серы, и новые ради-
калы не образуются. Для осуществления такого процесса тре-
буются постоянное облучение, подача озона или пероксидов.
Алкансульфокислоты и серная кислота образуются в эювимоль-
ных количествах. Непрерывное облучение нужно и в том слу-
чае, если процесс проводят в присутствии метанола, так как
надкислота тоже восстанавливается:
RSO2OOH + soa + СН,ОН -► RSO2OH + CHsOSO2OH
При сульфоокислении в присутствии уксусного ангидрида пер-
вичным продуктом также является надкислота; она образует с
уксусным ангидридом пероксид, который сам по себе является
источником свободных радикалов, инициирующих реакцию:
+ (СН3СО)2О
RSO2OOH RSO2-OO—CO-CHj'—► rso2o. +CHj— coo.
—CH3COOH
RSO3O- 4. RH-► R- 4- RSOaOH
CHS-COO. 4-RH----► CHS—COOH4-R-
Пероксид ацетилалкилсульфонила стабилен: его можно вы-
делять и применять для инициирования цепных реакций, в том
числе сульфоокисления. Он, однако, разлагается легче, чем пер-
оксид бензоила, и поэтому более удобен. Процесс ведут при
20—30 °C, диоксид серы и кислород подают в соотношении 2:1.
Образующиеся при сульфохлорировании и сульфоокислении про-
дукты нейтрализуют щелочью и получают алкилсульфонаты, об-
ладающие хорошим смачивающим и пенообразующим, но отно-
сительно невысоким моющим действием.
Алкилбензолсульфонаты получают сульфированием высших
алкилбензолов серной кислотой, олеумом или триоксидом серы.
Наиболее активным сульфирующим агентом является триоксид
серы, применяемый в газообразном и в жидком состоянии (в по-
следнем случае — в растворе SO2, алкилгалогенидов, пиридина,
диоксана и т. п.). Сильным сульфирующим агентом оказался
13-254 193
олеум. Активна и серная кислота, но при ее взаимодействии с
алкилбензолами выделяется вода, тормозящая процесс. По этой
причине сульфирование серной кислотой обычно проводят при
повышенной температуре (80—100°C и более), а олеум и диок-
сид серы активны при комнатной и даже при более низких тем-
пературах.
В качестве побочных продуктов возможно образование
сульфонов
СлН2Л+1—СвЩ—SO2OH + СлН2Л+1—СвН5--►
н2о> слн2Л+1—CeH4 SO2 СвН4 СлН2Л+$
ангидридов сульфокислот
+so3
2СлН2Л+1—CeH4—SO2OH ——> (СлН2Л+1-СвН4-5О2)2О
а также кетонов, карбоновых кислот и смолистых веществ. Вы-
ход побочных продуктов можно уменьшить, 'вводя растворители,
изменяя порядок смешения реагентов, поддерживая оптималь-
ную температуру и т. д.
Сульфирование тр-иоксидом серы является необратимой и
сильно экзотермической реакцией. Если для сульфирования при-
меняют олеум, тепловой эффект уменьшается в связи с затра-
той энергии на разрыв связей избыточного SO3 с H2SCU
В отличие от реакций с триоксидом серы и олеумом, сульфи-
рование серной кислотой имеет обратимый характер. Обратная
реакция десульфирования протекает обычно при повышенных
температурах, и ее можно провести почти нацело, отгоняя аро-
матический углеводород с перегретым водяным паром. Ввиду об-
ратимости процесса, а также из-за значительного уменьшения
сульфирующей способности серной кислоты (при ее разбавле-
нии выделяющейся водой) сульфирование прекращается еще
задолго до исчерпания всей кислоты. Потеря активности кисло-
ты происходит при разной ее концентрации в зависимости от ис-
ходного ароматического углеводорода.
На практике при сульфировании олеумом или серной кис-
лотой реакционная масса обычно является гетерофазной, при-
чем химическая реакция происходит в слое сульфирующего
агента. Поэтому на общую скорость процесса влияют раствори-
мость ароматического углеводорода в серной кислоте или олеу-
ме, а также скорость диффузии и поверхность контакта фаз. Два
последних фактора очень существенны при сульфировании олеу-
мом, когда химическая реакция протекает быстро. Для процес-
са с серной кислотой химическая реакция является медленной
стадией, определяющей скорость всего процесса.
2.2. Производство алкилсульфатов
Алкилсульфаты получают сульфированием спиртов и а-оле-
финов. При этом используют либо первичные высшие жирные
спирты, либо смесь первичных и вторичных спиртов, образую-
194
Отходящие
газы
Рис. IV.2. Технологическая схема получе-
ния первичных алкилсульфатов:
1, 3, 5, 6 — мерники; 2 — абсорбер; 4 — реактор;
7 — нейтрализатор; 8, 9 — емкости.
щуюся при окислении парафинов в
присутствии борной кислоты (гл. III,
стр. 118). Лучшими моющими свой-
ствами обладают первичные алкил-
сульфаты Си—С17- При перемеще-
нии сульфатной группы к середине
алкильной цепи моющая способ-
ность алкилсульфатов снижается.
В последние годы производство ал-
килсульфатов увеличивается из-за
их хороших моющих свойств, а глав-
ное из-за легкой биохимической
окисляемости в сточных водах. Не-
маловажное значение при-этом име-
ет доступность исходных спиртов и
а-олефинов С12—Ci8.
Сульфирование первичных жирных спиртов проводят 98% -
ной серной кислотой (70—75 масс. ч. кислоты на 100 масс. ч.
спирта) при 30—50°C и перемешивании (рис. IV.2). В реактор-
сульфуратор 4 с мешалкой и охлаждающим змеевиком загру-
жают серную кислоту из емкости 9 через мерник 3. При непре-
рывном перемешивании в кислоту тонкой струей подают спирт
из обогреваемой емкости 8 через мерник 1. Реакцию продол-
жают в течение нескольких часов и считают законченной, если
проба сульфомассы полностью растворяется в воде. По оконча-
нии сульфирования реакционная масса перетекает в аппарат 7,
где ее нейтрализуют 20—40%-ным едким натром при 50—60°C.
Газообразные продукты из сульфуратора, а также SO2 отсасы-
вают через абсорбер 2. Алкилсульфаты, нейтрализованные и от-
беленные гипохлоритом натрия, в виде пасты направляют на
сушку или сливают в тару.
Сульфирование вторичных спиртов и а-олефинов протекает
с меньшим выходом, чем сульфатирование первичных спиртов,
и сопровождается образованием побочных продуктов (диалкил-
сульфаты, полимеры). Для повышения выхода и улучшения ка-
чества получаемых вторичных алкилсульфатов (техническое на-
звание— типолы) ведут сульфирование 90—98%-ной серной
кислотой при 5—10 °C и минимальном времени контакта (при-
мерно 1 мин).
Очень важным аппаратом установки сульфирования являет-
ся реактор. Он должен быть таким, чтобы обеспечивать кратко-
временность контакта и быстрый отвод тепла. Обычно применя-
ют реакторы, представляющие собой корпус с рубашкой, внутри
которого вращается цилиндрический ротор. Зазор между корпу-
13
195
Рис. IV.3. Технологическая схема получения вторичных алкилсульфатов:
/ — реактор; 2 — нейтрализатор; 3 — гидролизер; 4 — сепаратор; 5 — экстрактор; 6 —
разгонный куо; 7, 9 — конденсаторы; 8 — колонна для отгонки спирта.
сом и ротором небольшой, что обеспечивает малое время пре-
бывания реакционной смеси в аппарате — менее 30 с. Серную
кислоту и олефины подают в нижнюю часть реактора /, корпус
и ротор которого охлаждают кипящим хлористым метилом или
водой (рис. IV. 3.) Ротор вращается со скоростью 120 об/мин.
Из реактора смесь поступает в нейтрализатор 2, где эфиры сер-
ной кислоты нейтрализуют 18%-ным едким натром. Диалкил-
сульфаты гидролизуют в аппарате 3 в течение 1 ч при 70—
80°C; при этом образуются вторичные спирты и вторичные ал-
килсульфаты:
4-NaOH
RCH—-О—SO2—О—CHR --> RCHOH + RCHOSCLONa
I III
CH3 CH3 CH3 CH3
Далее смесь обрабатывают 20%-ным этиловым спиртом. В нем
олефины и жирные спирты не растворяются и уходят с верха
сепаратора 4, а спиртовой раствор вторичных алкилсульфатов
экстрагируют лигроином при 60 °C для >более полного извлечения
органических веществ. Лигроин уходит с верха экстрактора 5,
его отгоняют от извлеченных им углеводородов в кубе 6 и воз-
вращают в нижнюю часть экстрактора. Водно-спиртовой рас-
твор вторичных алкилсульфатов направляют в колонну 8, где
из него отгоняют спирт. Алкилсульфаты отводят /в виде пасты.
2.3. Производство алкилсульфонатов
Технические алкилсульфонаты по моющим свойствам хуже,
чем первичные алкилсульфаты и алкиларилсульфонаты, они
менее стабильны в жесткой воде и резко снижают в ней пено-
образование. Поэтому их применяют в смеси с другими моющи-
196
ми веществами и мылом как моющие композиции в быту и тек-
стильной промышленности.
Производство алкилсульфонатов RSO2ONa основано на фото-
химическом сульфохлорировании и сульфоокислении парафинов
С12—Cis нормального строения, очищенных от олефинов, изопа-
рафинов и ароматических углеводородов. Сульфохлорирование
очищенного парафина (когазин) осуществляют в стальных ко-
лоннах, покрытых изнутри поливинилхлоридом, бакелитовым
лаком или выложенных фарфоровыми плитками. Лампы распо-
ложены внутри реактора вертикально по всей высоте в защит-
ных трубках. Диоксид серы и хлор (рис. IV.4) смешивают и на-
правляют в нижнюю часть реактора 1 через перфорированный
коллектор. Парафин подают в верхнюю часть аппарата.
Для отвода выделяющегося тепла реакционная смесь циркули-
рует через выносной холодильник 3; этим достигается и пере-
мешивание реакционной массы.
Для получения продуктов сульфохлорирования с низкой кон-
центрацией дисульфохлоридов ведут процесс до 30%-ной степе-
ни конверсии парафина при подаче 35 м3 смеси SO2+C12 на
10 м3 парафина в час. Длительность реакции примерно 16 ч. На
установке имеются 5—6 реакторов, работающих последователь-
но. В отходящих из реакторов газах содержится преимуществен-
но хлористый водород, который абсорбируют водой в аппара-
те 6; при этом образуется 36—38%-ная соляная кислота. Чтобы
снизить в кислоте содержание диоксида серы, который тоже по-
глощается водой, его отдувают воздухом (SO2 присутствует в
отходящем газе, потому что ее берут в избытке по отношению к
хлору). В продуктах сульфохлорирования содержатся также
хлористый водород и диоксид серы. Эти, газы отдувают возду-
хом в колонне 4. С низа колонны продукты сульфохлорирования
Рис. IV.4. Технологическая схема получения алкилсульфонатов:
J — реактор; 2 — трубки для ламп; 3 — оросительный холодильник; 4 — отдувочная ко-
лонна; 5 — емкость; 6 — абсорбер.
197
отводят в промежуточную емкость 5, откуда они поступают на
омыление 10%-ным раствором NaOH.
Не вступившие в реакцию углеводороды и алкилхлориды
(хлор в алкилхлоридах при этих условиях не омыляется) отде-
ляют в отстойнике и подают на дегидрохлорирование в присут-
ствии катализаторов — оксидов металлов. При этом алкилхло-
риды дегидрохлорируются до олефинов. Затем смесь подвергают
каталитическому гидрированию и возвращают на сульфохлори-
рование. При нейтрализации образуется хлорид натрия, кото-
рый при охлаждении солей сульфокислот уходит в нижний вод-
ный слой (его сбрасывают в канализацию). Раствор алкилсуль-
фонатов (верхний слой) упаривают, а горячий расплав подают
на барабанный кристаллизатор, где он застывает тонкой плен-
кой на поверхности барабана. Ее срезают ножом и подают в
бункер, а оттуда на упаковку.
Кроме того, алкилсульфохлориды, являющиеся реакционно-
способными соединениями, могут быть использованы для полу-
чения сложных эфиров, амидов и др. При нагревании выше
100 °C алкилсульфохлориды разлагаются:
rch2-ch2so2ci —
RCH=CH2 + НС1 + so2
RCH2—СН2С1 + so2
Первая реакция идет при 200—300 °C, а разложение алкилсуль-
фохлоридов с отщеплением SO2 и образованием алкилхлоридов
протекает при 140—150 °C. При более низких температурах
диоксид серы отщепляется очень медленно, а при более высоких
в заметных количествах образуются олефины. При облучении
десульфирование до алкилхлоридов заметно уже при 100 °C. Эту
реакцию можно использовать для получения моно- и диалкил-
хлоридов, которые прямым хлорированием углеводородов синте-
зировать весьма трудно. Сами по себе алкилсульфохлориды мо-
гут быть применены в качестве инсектицидов, дубителей и сма-
зочных масел, работающих при высоких давлениях.
По технологическому и аппаратурному оформлению процесс
сульфоокисления в значительной степени сходен с сульфохлори-
рованием. Парафин обрабатывают смесью 70% диоксида серы
и 30% кислорода при УФ-облучении. Газовая смесь циркулирует
в системе (так как ее степень конверсии за один проход невели-
ка) и по мере расходования пополняется свежей.
2.4. Производство алкилбензолсульфонатов
Алкилбензолсульфонаты в промышленности производят суль-
фированием нефтяных фракций, содержащих алкилбензолы и
другие алкилароматические углеводороды, а также сульфирова*
198
нием высших алкилбензолов, специально синтезированных для
этой цели путем алкилирования бензола олефинами.
Алкилароматические углеводороды в составе нефтяных фрак-
ций очень разнообразны по строению (по длине и числу алкиль-
ных групп и наличию нафталиновых и других колец), поэтому
получаемые из них алкиларилсульфои1а1ты являются сложной
смесью веществ. В зависимости от средней молекулярной массы
исходного сырья образующиеся алкиларилсульфонаты могут
быть водо- или маслорастворимыми. Например, сульфированием
керосиновых и газойлевых дистиллятов получают водораство-
римые нефтяные сульфонаты под названием контакт Петрова и
ДС-РАС (детергент советский; рафинированные алкилсульфо-
наты). У нефтяных сульфонатов поверхностная активность ни-
же, чем у синтетических, но зато нефтяные дешевле. Они обла-
дают неприятным запахом и окрашены в темный цвет. Поэтому
их применение для приготовления моющих средств ограничено,
но они широко используются в технике как смачиватели, эмуль-
гаторы и деэмульгаторы, флотореагенты, а также как катализа-
торы кислотного характера. Алкиларилсульфонаты, полученные
сульфированием нефтяных 'масел, в виде кальциевых и бари-
евых солей являются важными моющими присадками к мотор-
ным смазочным маслам и производятся в значительных мас-
штабах.
Синтетические алкилбензолсульфонаты натрия (сулъфонолы)
производят на основе а-олефинов Сю—Си, хлорпарафинов
Сю—Сю или тетрамеров пропилена. Предпочтительным является
применение а-олефинов, поскольку получаемые из них линей-
ные алкилбензолсульфонаты обладают удовлетворительной ско-
ростью биохимического разложения в сточных водах, вследствие
чего при их использовании не загрязняются водоемы и не нару-
шается работа очистных сооружений. Сульфонол, получаемый
на основе тетрамеров пропилена, не разлагается в воде за 25 су-
ток и лишь на 65% за 60 суток. Сульфонол, производимый из
хлорпарафинов, занимает промежуточное положение по степени
биохимического разложения. В связи с этим производство суль-
фонолов на основе тетрамеров пропилена и хлорпарафинов по-
степенно сокращается.
Ключевым процессом в производстве алкилбензолсульфона-
тов натрия является сульфирование, которое реализуют в про-
мышленности в основном по описанным далее схемам.
Сульфирование алкилбензолов проводят в аппаратах с ме-
шалками— сульфураторах (рис. IV.5). Изододецилбензол или
другой алкилбензол подают в сульфуратор 2 первой ступени,
куда одновременно подают олеум. Реакционную массу тщатель-
но перемешивают при 34—45 °C. Выделяющееся тепло отводят
в выносном холодильнике 1. Из сульфуратора 2 реакционная
смесь поступает в сульфуратор 3 второй ступени для заверше-
ния сульфирования. В аппарате 4 сульфомассу разбавляют во-
дой при 60—75 °C (при более высокой температуре масса тем-
199
Рис. IV.5. Технологическая схема получения алкилбензолсульфонатов:
J, 5 — холодильники; 2, 3 — сульфураторы; 4—аппарат для разбавления; 6, 7 — нейтра-
лизаторы; 8 — отстойник.
иеет, а при более низкой плохо отстаивается сергаая кис-
лота).
В отстойнике 8 слои разделяются. Нижний слой содержит
75—78%-ную серную кислоту с примесью алкилбензолов, верх-
ний слой представляет собой ал кил бензолсульфокислоты с при-
месью серной кислоты (10—15%). Серную кислоту используют
или «доукрепляют», а сульфокислоты подают в нейтрализатор 6
первой ступени (на приемную линию насоса, перекачивающего
пасту через холодильник 5). Сульфокислоты разбавляются ней-
трализованной пастой, которая поглощает часть выделяющего-
ся тепла. На приемную линию того же насоса подают 15—
17%-ную щелочь. Соотношение сульфокислот и пасты составляет
1 : (30-?40). Температура при нейтрализации равна 45—55 °C.
Концентрация щелочи ниже 15% не допускается, так как паста
получается жидкой и пенится, а при концентрации щелочи выше
17% паста очень густая и это затрудняет отвод тепла и пере-
качку пасты.
В нейтрализаторе 7 второй ступени доводят pH до 7,5-j-8,
если нужно, отбеливают гипохлоритом натрия и вводят наполни-
тели. .Паста содержит 40—50% воды и 50—60% твердых ве-
ществ (83—85% алкилбензолов, 13—15% сульфата натрия,
1,5—2% непросульфированных углеводородов). После нейтрали-
затора 7 пасту можно направить в дополнительный аппарат, где
при интенсивном 1перемешивании вводят в пасту наполнители,
улучшающие моющие, отбеливающие и другие свойства синтети-
ческих моющих веществ. При повышении вязкости разбавляют
пасту водой, поддерживая ее вязкость на заданном уровне.
Пасту сушат в башнях горячим восходящим потоком газов,
в который пасту вводят через форсунки в верхней части башни.
Топочные газы, используемые для сушки, имеют температуру на
входе 250—350 °C, на выходе 105—120 °C в зоне распыления
температура равна 160—200 °C. Унесенные газовым потоком
частицы СМВ задерживаются в циклонах и фильтрах. Совре-
200
менные сушильные агрегаты задерживают до 99,8% порошка
СМВ.
Более совершенной является технология сульфирования ал-
килбензолов триоксидом серы, имеющая следующие преимуще-
ства по сравнению с использованием олеума:
1) меньшая стоимость сульфирующего агента;
2) отсутствие отработанной серной кислоты (и, следователь-
но, сложностей, связанных с ее утилизацией);
3) повышение содержания активного вещества в алкилбен-
золсульфонатах за счет отсутствия сульфата натрия;
4) сокращение расхода едкого натра на 12—15% на стадии
нейтрализации.
В то же время при сульфировании триоксидом серы возни-
кают сложности при его транспортировании и хранении: повы-
шается тепловыделение, побочно образуются ангидриды алкил-
бензолсульфокислот, продукты окисления, деструкции и осмоле-
ния алкилбензолов. Однако эти недостатки в значительной мере
можно уменьшить с помощью определенных технологических
приемов.
В настоящее время все методы сульфирования триоксидом
серы можно разделить на две группы по его фазовому состоя-
нию: 1) газообразным SO3 в смеси с инертными газами, возду-
хом или газообразными парафиновыми углеводородами; 2) жид-
ким SO3 в смеси с жидкими инертными растворителями.
Сульфирование углеводородов газообразным триоксидом
серы проводят при 45—50 °C. Концентрацию SO3 поддерживают
от 6—7 до 15—20%. Сульфураторы представляют собой либо
аппараты с мешалками, либо колонные реакторы пленочного
типа. Сульфирование газообразным триоксидом серы, разбав-
ленным воздухом, применяют при производстве алкилбензол-
сульфонатов из линейных алкилбензолов (сульфонол НП-3).
Перспективный метод сульфирования жидким триоксидом
серы в растворе диоксида серы разработан в СССР. Процесс
проводят при температуре кипения SO3 (—10°C). Жидкий
SO2 снижает вязкость реакционной смеси, а за счет его испаре-
ния отводится выделяющееся тепло. После конденсации диоксид
серы возвращают в процесс. При таком низкотемпературном
сульфировании можно избежать потемнения сульфомассы.
2.5. Производство а-олефинсульфонатов
Производство а-олефинсульфонатов (АОС) сульфированием
а-олефинов триоксидом серы относительно несложно по техно-
логии и перспективно по используемому сырью. Им служат
а-олефины С14—Си, производимые термическим крекингом па-
рафина (стр. 23) и олигомеризацией этилена в присутствии
триэтилалюминия (стр. 23). При сульфировании триюксид серы
разбавляют воздухом или инертным газом; применяют также
его растворы в диоксиде серы и диоксане. Продукты сульфиро-
201
конструкции
в пленочном
вания обычно имеют темный цвет, поэтому их отбеливают гипо-
хлоритом натрия. Качество пасты зависит и от
сульфуратора — более светлый продукт получают
реакторе.
При сульфировании перемещается двойная
а-олефина и образуются алкенилсульфокислоты,
лучаются 1,3- (а) ’и 1,4-димерные сультоны (б):
+SOg
I---> R'CH=CH-(CH2)rt-SO2OH
связь в цепи
Побочно по-
RCH=CH2
I R'-(CH2)„_1-CHCH2CH2 + R'-(CH2)„_2-CHCH2CH2CH2
О-----------------------so2 о-------sb2
a 6
Нейтрализация продуктов реакции щелочью дает а-олефинсуль-
фонаты и гидроксиалкилсульфонаты натрия:
Ч-NaOH
R'CH=CH-(CH2)„-SO2OH ——> R'CH=CH— (CH2)„-SO2ONa
—H2O
ct-олефинсульфонат
4-NaOH
R'-(ch2)„_1-chch2ch2 * R'-(CH2)n_1-CH-CH2-CH2
I I -H2° I I
О----SO2 HO NaOOzS
4-NaOH
R'-(CH2)„_.-CHCH2CH2CH2 —► R'-(CH2)n_2-CH-CH2-CH2-CHa
I I -H2° I I
О--------SO2 HO NaOO2S
При производстве а-олефинсульфонатов (рис. IV.6) жидкий
триоксид серы испаряют в аппарате /, смешивают с воздухом и
Рис. IV.6. Технологическая схема получения а-олефинсульфонатов:
1 — испаритель; 2 — реактор; 3 — сепаратор; 4 — нейтрализатор; 5 — нагреватель; 6
эвапоратор; 7 — холодильник; 8 — отстойник; 9— аппарат с мешалкой.
202
подают в реактор 2 пленочного типа, куда поступают и олефи-
ны. Температура в реакторе 35—40 °C, мольное соотношение
олефинов я SO3 равно 1 (1,14-1,5), содержание SO3 в смеси
составляет 15—20% (масс.). Смесь проходит реактор сверху
вниз и поступает в сепаратор 3, где отделяются газы. Жидкие
продукты в аппарате 4 нейтрализуют водной щелочью при 40—
50 °C до pH 84-9. Омыление сультонов осуществляют за счет на-
гревания пасты в нагревателе 5 до 95—100 °C. В эвапораторе 6
углеводороды в виде азеотропной смеси с водой испаряются и
конденсируются в холодильнике 7. Олефины отделяют от воды
в отстойнике 8. В аппарате 9 пасту доводят до товарных кон-
диций (если нужно, добавляют щелочь и отбеливают пасту).
§ 3. ПРОИЗВОДСТВО НЕИОНОГЕННЫХ
ПОВЕРХНОСТНО-АКТИВНЫХ ВЕЩЕСТВ
Неионогенные ПАВ по объему производства находятся на
втором месте после алкилбензолсульфонатов. Широкое развитие
неионогенных ПАВ обусловливают их следующие преимущест-
ва перед анионоактивными:
1) повышенная способность удерживать загрязнения в мою-
щем растворе;
2) проявление оптимального комплекса смачивающих, эмуль-
гирующих и моющих свойств в воде любой жесткости, вплоть до
морской;
3) хорошая совместимость с ПАВ других типов и с наполни-
телями;
4) простота технологии получения при возможности широко
варьировать ассортимент продуктов и их свойства.
Синтез основного типа неионогенных ПАВ — оксиэтилирован-
ных соединений — осуществляют последовательным взаимодей-
ствием любых гидрофобных молекул, имеющих подвижные ато-
мы водорода, с этиленоксидом в присутствии щелочного катали-
затора:
ОН (ОСН2СН2)„-ОН
R R
ROH + лН2С--СН2 ---► R—(ОСН2СН2)Л-ОН
RCOOH + nH2C---СН2 --> RC-(OCH2CH2)„-OH
RSH + пН2С----СН2
RS—(СН2СН2О)П—Н
2ЭЗ
/(СН2СН2О)п-Н
RCONH2 + 2hH2C--СН2 --► RC—N\
\ / II X(CH2CH2O)n-H
О о
/(СН2СН2О)П_Н
RNH24-2nH2C---CH2 --► RN\
\ / дСН2СН2О)п—H
о
Взаимодействие всех этих протонодонорных веществ с этилен-
оксидом происходит по типу автокаталитической поликонденса-
ции, в результате которой молекулярномассовое распределение
полимергомологов подчиняется среднестатистическому, т. е. наи-
большее количество прореагировавшего вещества присоединяет
среднее число молей этиленоксида, но определенная часть про-
дукта содержит полимергомологи с меньшим и большим, чем
среднее, числом оксиэтильных групп.
Растворимость оксиэтилированных соединений в воде объяс-
няется образованием водородных связей между молекулами во-
ды и атомами кислорода оксиэтильной цепочки. Отсюда ясно,
что чем длиннее оксиэтильная цепочка, тем лучше растворяется
данное вещество в воде. При повышении температуры водород-
ные связи разрываются и растворенное неионогенное ПАВ «вы-
саливается» — образует самостоятельную фазу. При этом рас-
твор сильно мутнеет. Чем длиннее оксиэтильная цепочка, тем
выше температура помутнения раствора (до 100°C и более).
Для проявления высоких поверхностно-активных свойств
гидрофильная оксиэтильная цепочка должна уравновешивать-
ся достаточно длинной гидрофобной алкильной группой
(С12—С18). Поэтому в качестве исходного сырья для синтеза
неионогенных ПАВ используют высшие жирные кислоты и их
производные (амиды, этаноламиды), а также высшие спирты,
меркаптаны, амины и алкилфенолы. В случае алкилфенолов в
число углеродных атомов гидрофобной части входят четыре уг-
леродных атома ароматического ядра. Оксиэтилированные ал-
килфенолы имеют торговое название ОП, например ОП-7, ОП-10
(цифры означают число присоединенных молей этиленоксида).
За рубежом они носят название игепалей.
Как уже отмечалось, особенностью неионогенных ПАВ яв-
ляется лучшая, чем у анионоактивных веществ, способность (без
специальных добавок) препятствовать обратному осаждению
загрязнений на ткань. Так, оксиэтилированный нонилфенол по
этому показателю превосходит додецилбензолсульфонат натрия
почти в два раза, а первичный лаурилсульфат — примерно в
полтора раза. Неионогенные ПАВ без добавок обладают высо-
кой моющей способностью по отношению к хлопчатобумажным
тканям. Однако при введении добавок (полифосфаты натрия,
натриевая соль карбоксиметилцеллюлозы) моющая способность
анионоактивных веществ увеличивается гораздо больше, чем для
204
Кислоти.
Азот
Рис. IV.7. Технологическая схема получения неионогенных ПАВ типа ОП:
1 — мерник; 2 — реактор; 3, 4 — емкости.
неионогенных, поэтому в итоге СМС на основе' анионоактивных
ПАВ более эффективны.
Неионогенные ПАВ имеют важное значение как моющие ве-
щества для техники, как эмульгаторы пестицидных препаратов,
а также как добавки к промывочным растворам для увеличения
нефтеотдачи пласта.
Производство неионогенных ПАВ осуществляют по единой
технологической схеме для всех исходных гидрофобных веществ.
Основные трудности технологического процесса заключаются в
отводе большого количества тепла (922 кДж на 1 моль этилен-
оксида), соблюдении обычного молекулярномассового распреде-
ления полимергомологов (при нарушении которого могут резко
ухудшиться свойства продукта), а также в соблюдении взрыво-
безопасности.
Для преодоления указанных трудностей, а также в связи с
тем что реакция юкс'иэтил1ирования идет с большой скоростью,
процесс обычно проводят по периодической схеме, хотя извест-
ны установки и непрерывного действия. На рис. IV.7 приведена
схема периодической установки для производства неионогенных
ПАВ типа ОП из алкилфенолов. В стальной реактор 2 с мешал-
кой и рубашкой загружают алкилфенол из емкости 3 и нагре-
вают при перемешивании до 130—150 °C. Сырье не должно со-
держать воды, так как ее присутствие приводит к образованию
полигликолей, плохо растворяющихся в конечном продукте и
вызывающих его помутнение. Если сырье обводнено, его осу-
шают в реакторе. По окончании обезвоживания добавляют 0,1—
0,5% безводной щелочи (катализатор) и в реактор подают эти-
леноксид из емкости 4 под давлением азота. Этиленоксид дол-
жен быть 99,5%-ной концентрации и содержать не более 0,01%
ацетальдегида, так как в присутствии ацетальдегида получается
окрашенный продукт. Во время реакции температура поднима-
ется до 200—210 °C и реакционную массу приходится охлаждать
до 160—180 °C, подавая воду в рубашку.
205
Рассчитанное количество этиленоксида подают порциями;
давление в реакторе поддерживают в пределах 0,08—0,12 МПа.
По завершении оксиэтилирования (2—8 ч, в зависимости от ко-
личества присоединяемого этиленоксида) продувают реактор
азотом для удаления следов этиленоксида и нейтрализуют ката-
лизатор, содержащийся в реакционной массе, соляной, уксусной
или фосфорной кислотой. Продукт выгружают из реактора и
фильтруют.
Несмотря на единую технологию получения неионогенных
ПАВ, продукты, вырабатываемые из разного сырья, различа-
ются по составу и свойствам.
В частности, оксиэтилированные карбоновые кислоты вслед-
ствие склонности сложных эфиров к реакциям алкоголиза и пе-
реэтерификации представляют собой равновесную смесь поли-
гликолей и их моно- и диэфиров:
RC-(OCH2CH2)n-H + RC—(OCH2CH2)m—Н T=fe
RC—(OCH2CH2)n—О—CR + НО-(СН2СН2О)^-Н
Кроме того, оксиэтилированные карбоновые кислоты легко омы-
ляются в щелочной среде и поэтому находят лишь ограничен-
ное применение (главным образом в составе текстильно-вспомо-
гательных средств для крашения и отделки тканей).
Оксиэтилированные жирные спирты, напротив, обладают вы-
сокой химической стойкостью в любых средах. Большим их пре-
имуществом является также легкая биохимическая окисляемость
в сточных водах. Однако из-за того что оксиэтилированные
спирты обладают более высокой реакционной способностью, ко-
нечные продукты всегда содержат то или иное количество не-
прореагировавшего спирта, а также могут иметь необычное мо-
лекулярномассовое распределение полимергомологов, что сни-
жает их поверхностно-активные свойства.
Для оксиэтилированных n-алкилфенолов картина обратная:
исходный алкилфенол наиболее реакционноспособен и практи-
чески не присутствует в продуктах оксиэтилирования. Среди
неионогенных ПАВ оксиэтилированные алкилфенолы обладают
наилучшим сочетанием свойств, что при известном дефиците
высших спиртов ранее содействовало их преимущественному
производству. Однако оксиэтилированные эфиры алкилфенолов
очень медленно подвергаются биохимическому разложению в
сточных водах и в перспективе будут совсем вытеснены окси-
этилированными спиртами (если за это время не будут разра-
ботаны принципиально новые методы очистки сточных вод от
ПАВ).
206
§ 4. ПОВЫШЕНИЕ КАЧЕСТВА СИНТЕТИЧЕСКИХ
МОЮЩИХ ВЕЩЕСТВ
4.1. Способы повышения моющего действия
синтетических моющих веществ
Недостатки всех синтетических моющих веществ — их плохая
способность удерживать загрязнения в растворе и высокая обез-
жиривающая способность. Поэтому в настоящее время в чистом
виде их не применяют. Имеются два способа существенно повы-
сить моющее действие: 1) применять композицию из двух-трех
ПАВ и более и 2) вводить в композицию органические и неор-
ганические добавки различного назначения.
Установлено, что многие ПАВ в смеси обладают более высо-
ким моющим действием, чем исходные компоненты в отдель-
ности. Например, композиции сульфонола НП-1 с первичными
алкилсульфатами в соотношении 70 : 30 обладают моющим дей-
ствием, вдвое превосходящим моющее действие любого компо-
нента смеси.
Подбор оптимального состава композиций в зависимости от
назначения осуществляют эмпирическим путем. Встречаются
случаи, когда моющее действие композиции ниже по сравнению
с индивидуальными компонентами, например в композиции
сульфонол НП-1 с жировым мылом при стирке ib жесткой воде.
Смесь же сульфонола с неионогенными моющими веществами,
особенно с оксиэтилированными алкилфенолами, обладает вы-
сокой эффективностью. Создание композиций еще не решает
проблему получения эффективных СМС; для этого в них (в пас-
ту, до сушки) вводят различные активные добавки, в основном
минеральные соли. Это в первую очередь — карбонат и бикар-
бонат натрия, силикаты натрия nNa2O -mSiC^, пирофосфат нат-
рия Na4P2O7, триполифосфат натрия Na5P3Oi0 и гексаметафос-
фат натрия (МаРОз)б.
Роль карбоната, бикарбоната и силикатов натрия сводится
к тому, что они в водных растворах гидролизуются, за счет чего
увеличивается щелочность среды. Волокна ткани в щелочной
среде набухают и частицы загрязнений легче отрываются от них.
Кроме того, ионы натрия адсорбируются на частицах загрязне-
ний и гидрофилизируют их; в результате частицы загрязнений
удерживаются в растворе и не оседают обратно на ткань, по-
скольку образование адсорбционных слоев заряженных ионов
препятствует слипанию частиц. Силикаты натрия, кроме того,
повышают пенообразование, а при сушке СМС способствуют по-
лучению гранулированного порошка.
Фосфаты натрия хорошо умягчают воду за счет образования
комплексов с солями жесткости:
Na5P3O10 + СаС12 -Na3CaP3O10 + 2NaC!
Образующиеся соли не выпадают в осадок, а следовательно, не
оседают на ткань, что способствует снижению ее износа. При
207
стирке мылом зольность ткани повышается за счет образовав-
шихся и осевших на ткани нерастворимых солей кальция и маг-
ния. В случае синтетических моющих средств зольность можно
увеличить за счет взаимодействия солей жесткости с карбона-
том или бикарбонатом натрия. Триполифосфат натрия подав-
ляет эти реакции. Вводимый в СМС сульфат натрия увеличивает
моющую способность, предотвращает слеживание и комкование.
В качестве отбеливающих средств в синтетические моющие
вещества вводят перборат натрия NaBO2-H2O2-3H2O (10,4%
активного кислорода), перкарбонат натрия Na2CO3-H2O2-2H2O
(9% кислорода), перпирофосфат натрия Na4P2O7-2H2O2-2H2O
(9% кислорода). При разложении этих веществ выделяется ак-
тивный кислород, который и отбеливает ткань. Количество пер-
юксидных добавок равно 0,5—1%. При большем количестве мо-
жет происходить взаимодействие кислорода с тканью, что по-
высит ее износ. Общее количество минеральных добавок состав-
ляет 65—70%, причем большая их часть приходится на фосфаты
и сульфат натрия.
Из органических добавок (вводимых в СМС в количестве
5—10%) на первом месте стоит натриевая соль карбоксиметил-
целлюлозы (2—4%). Ее назначение — предотвратить обратное
оседание загрязнений из моющего раствора на ткань и тем уве-
личить степень белизны ткани. Добавки алкилоламидов (2—
4%) увеличивают пенообразование. Они эффективны ib смеси с
натриевой солью карбоксиметилцеллюлозы. В составе высоко-
качественных СМС содержатся добавки оптических отбеливате-
лей — сложных органических соединений, относящихся к кра-
сителям. Адсорбируясь на ткани, эти добавки поглощают
УФ-лучи и испускают видимый свет голубовато-синего оттенка.
Кроме названных добавок в СМС вводят и комплексообра-
зующие вещества, которые, как и триполифосфат натрия, умяг-
чают воду. Например, тетранатриевая соль этилендиаминтетра-
уксусной кислоты
СН2—N(CH2-COONa)a
СН2—N(CH2—COONa)2
образует водорастворимые комплексы с солями жесткости.
К СМС добавляют также так называемые отдушки, придающие
моющим порошкам приятный запах.
4.2. Перспективные типы ПАВ
Завершая рассмотрение производства ПАВ, кратко опишем
некоторые перспективные продукты, получаемые пока в относи-
тельно небольших масштабах, но обладающие ценными поверх-
ностно-активными и другими эксплуатационными свойствами.
К числу таких ПАВ можно отнести сульфаты оксиэтилиро-
ванных спиртов и оксиэтилированных алкилфенолов, обладаю-
208
щие свойствами как неионогенных, так и анионоактивных ПАВ.
Сульфирование и нейтрализация протекают так же легко, как
и в случае первичных жирных спиртов, а полученные продукты,
содержащие в среднем 3—4 оксиэтильные группы, равноценны
первичным алкилсульфатам по моющему действию, причем оно
не уменьшается и в жесткой воде. По способности к биохимиче-
скому разложению эти продукты также приемлемы, поскольку
после отщепления сульфатной группы они теряют поверхност-
ную активность. Сульфаты неионогенных ПАВ представляют
большой интерес, так как на их основе можно получать СМС,
не содержащие фосфатных добавок. Необходимость производ-
ства таких СМС выявилась в последнее время из-за усиленного
роста водорослей под влиянием фосфатов в сточных водах.
В то же время эфиры фосфорной кислоты и неионогенных
ПАВ обладают высокими эмульгирующими, моющими и анти-
статическими свойствами при хорошей способности к биохими-
ческому разложению, а их токсичность еще ниже, чем у исход-
ных неионогенных ПАВ. Синтез таких эфиров осуществляют
взаимодействием оксиэтилированных спиртов и алкилфенолов
с пентоксидом фосфора:
R—(OCH2CH2)n—Н
О
+Р2О5RHOCH2CH2)n-OP(OH)t
I— [R—(ОСН2СН2)Л-О]2РОН
о
На основе спиртов или оксиэтилированных алкилфенолов
получают динатриевые соли сульфоянтарной кислоты:
НС-СО
ROH + ||
НС—СО
СН2—COONa
Н+ НС—COOH 4-NaHSO3 |
---> || ------► CH-COOR
HC-COOR |
SO3Na
Эти соли обладают высоким моющим действием, не токсичны,
не раздражают кожный покров и подвержены быстрому биохи-
мическому разложению в сточных водах. Разновидностью синте-
зированных соединений являются натриевые соли моноамида
сульфоянтарной кислоты, получаемые по схеме:
Н<ГС° +rnh2 HC-CONHR
„А /с. НС-СООН ~Н2°
H.C-CONHR
I
НС—COONa
I
SO3Na
Натриевые соли моноамида сульфоянтарной кислоты проявляют
высокие моющее действие и пенообразование, они не токсичны.
Это позволяет использовать их для изготовления шампуней.
Традиционный компонент шампуней — продукт конденсации
14—254 209
жирных кислот с диэтаноламином:
RCOOH + HNX ---►
\СН2-СН2ОН ~н2°
RCN(CH2-CH2OH)2
II
О
Оксиэтилированием этих соединений можно получить высоко-
эффективные эмульгирующие и моющие вещества, более стойкие
к щелочному гидролизу, чем полигликолевые эфиры карбоновых
кислот.
Большинство ПАВ, содержащих атом азота в молекуле, спо-
собно диссоциировать в водных растворах и, следовательно, про-
являть катионоактивный характер. Катионоактивные ПАВ об-
ладают обычно не только поверхностно-активными, но и бакте-
рицидными свойствами. Особую активность в этом отношении
проявляют четвертичные аммониевые соли, получаемые на осно-
ве третичных аминов:
RN(CH3)2 + С1Н2С—\ > RN(CH3)2-H2C-/~A Cl-
В СССР помимо веществ указанного типа производят катионо-
активные ПАВ (катапин А) по такой схеме:
Синтез еще одной перспективной группы катионоактивных
ПАВ может быть осуществлен на основе имидазолинов:
Н2С-СН2
b/ СН2—СН2ОН
H-RX
С—С1бН33
Н2С-СН2
^NR(CH2CH2OH) X-
с—с1вн33
Эти катионоактивные ПАВ помимо поверхностной активности
обладают бактерицидными, антиокислительными, антикоррози-
онными, антистатическими и другими ценными свойствами.
ГЛАВА V
ПРОИЗВОДСТВО ПОЛИМЕРОВ
§ 1. ОБЩИЕ СВЕДЕНИЯ О ПОЛИМЕРАХ
1.1. Основные понятия ;и классификация
К высокомолекулярным соединениям, или полимерам, отно-
сят вещества с молекулярной массой 5 000 и более. Полимеры с
молекулярной массой от 500 до 5000 занимают промежуточное
210
положение между высокомолекулярными и низкомолекулярны-
ми соединениями и называются олигомерами.
Полимеры и олигомеры состоят из многократно повторяю-
щихся группировок — остатков мономеров, называемых элемен-
тарными звеньями. Большая молекула, составленная из элемен-
тарных звеньев, называется макромолекулой или полимерной
цепью.
Важной характеристикой полимеров является степень поли-
меризации, равная числу элементарных звеньев в макромоле-
куле.
По форме макромолекул полимеры делятся на линейные,
разветвленные и сетчатые. В линейных полимерах каждое эле-
ментарное звено связано только с двумя другими звеньями, об-
разуя длинную цепь без боковых ответвлений. Разветвленные
полимеры состоят из основной цепи и боковых ответвлений. Сет-
чатые полимеры построены из полимерных цепей, соединенных
друг с другом поперечными связями.
В зависимости от состава основной цепи различают карбо-
цепные и гетероцепные полимеры. Основные цепи карбоцепных
полимеров построены из углеродных атомов. К ним относятся
полиолефины, полидиены. Атомы водорода в макромолекулах
могут быть замещены различными функциональными группами:
—С1, —ОН, —NH2, —CN и другими. Основные цепи гетероцеп-
ных полимеров кроме углерода содержат атомы кислорода, азо-
та, серы. К гетероцепным полимерам относятся полиэфиры, по-
лиамиды, полиуретаны, эпоксидные полимеры.
Полимеры, построенные из одинаковых элементарных звень-
ев, называются гомополимерами. Полимеры, полученные из раз-
ных мономеров и содержащие в цепи неодинаковые элементар-
ные звенья, называются сополимерами.
1.2. Химическое строение и структура полимеров
Если в цепи полимера наблюдается монотонное чередование
звеньев, то полимер построен регулярно. Нарушение этого по-
рядка приводит к нерегулярности строения. Нерегулярность
цепи может обусловливаться различным способом последова-
тельного присоединения друг к другу одних и тех же мономер-
ных звеньев. Так, элементарные звенья в цепи могут соединять-
ся по схеме I или по схеме II:
^СН2—СН—СН2—СН—СН2—СН-^ (I)
XXX
^СН2- СН—СН—СН2-СН2—СН- СН—СН2^ (II)
XX XX
Соединение по первой схеме называется «голова к хвосту», по
второй — «голова к голове». Полимеры, у которых все замести-
14* 211
тели в элементарных звеньях расположены в определенном по-
рядке в пространстве, называются стереорегулярными.
В зависимости от степени упорядоченности расположения ма-
кромолекул различают аморфное и кристаллическое состояния
полимеров. Аморфное состояние полимеров характеризуется
беспорядком во взаимном расположении молекул. Кристалличе-
ское состояние характеризуется сохранением упорядоченности
на большом участке цепи.
Кристаллические полимеры не являются полностью кристал-
лическими веществами. Обычно они содержат как кристалличе-
скую, так и аморфную фазы. Соотношение кристаллической и
аморфной фаз в полимере носит название степени кристаллич-
ности, Высокая степень кристалличности характерна только для
строго регулярных линейных полимеров, например для поли-
этилена:
^СН2-СН2—СН2—СН2—СН2— СН2^
Нерегулярно построенные полимеры могут кристаллизовать-
ся только при небольшом содержании боковых ответвлений (за-
мещающих групп). С увеличением числа боковых ответвлений
полимер теряет способность к кристаллизации.
К кристаллическим полимерам относятся полипропилен, по-
ликапроамид, полиэтилентёрефталат и другие полимеры.
Аморфные полимеры могут находиться в трех физических со-
стояниях: стеклообразном, высокоэластическом и вязкотекучем.
Полимеры в стеклообразном состоянии обладают прочностью
твердых тел: при сжатии, растяжении, изгибе они деформиру-
ются незначительно. Это объясняется тем, что в стеклообразном
состоянии молекулы связаны наиболее прочно и, как следствие,
они наименее гибки. Чем ниже температура в области стеклооб-
разного состояния, тем меньшей подвижностью обладают мак-
ромолекулы, и при определенной температуре, называемой тем-
пературой хрупкости, стеклообразные полимеры разрушаются
без деформации.
В определенном интервале температур полимеры переходят
из стеклообразного состояния в высокоэластическое (и обрат-
но). Средняя температура перехода называется температурой
стеклования. Высокоэластическое состояние обусловлено способ-
ностью макромолекул непрерывно изменять свою форму благо-
даря большой их гибкости. При нагревании полимера, находя-
щегося в высокоэластическом состоянии, до определенной темпе-
ратуры силы сцепления, фиксирующие отдельные участки мак-
ромолекул, исчезают и молекулы приобретают возможность
перемещаться друг относительно друга. Это соответствует вязко-
текучему состоянию полимера. Температура перехода 'из высо-
коэластического в вязкотекучее состояние называется темпера-
турой текучести.
При вытягивании образцов аморфных полимеров происходит
ориентация макромолекул, которая иногда сопровождается крис-
212
таллизацией. Процессу ориентации препятствует перепутанность
макромолекул и их подвижность. Поэтому ориентация никогда
не происходит по всей длине макромолекулы, и кристаллические
участки перемежаются с аморфными.
Более упорядоченную структуру, а следовательно, и лучшие
механические свойства полимеру можно придать ориентацией,
возникающей вследствие приложения к нему нагрузки. Ориента-
ция облегчается, если полимер находится в вязкотекучем состоя-
нии, в котором подвижность цепей высока. Ориентация проис-
ходит и при медленном растяжении полимеров, находящихся в
кристаллическом или высокоэластическом состоянии («холод-
ная» вытяжка), при этом происходит распрямление цепей и их
взаимное упорядочение. Полимер может быть ориентирован как
в одном направлении (одноосная ориентация), так и в несколь-
ких -направлениях (многоосная ориентация).
Прочность полимера в ориентированном состоянии значи-
тельно больше, чем в неориентированном, причем в случае од-
ноосной ориентации прочность в направлении ориентации -боль-
ше, чем в перпендикулярном направлении.
При переходе из аморфного в кристаллическое состояние
свойства полимера изменяются — возрастают плотность, твер-
дость, жесткость, механическая прочность, снижается упругость.
Переход к полностью аморфному полимеру от двухфазного
кристаллическо-аморфного состояния наблюдается при темпера-
туре плавления. При повышении температуры соотношение фаз
постепенно изменяется. Полимер плавится не при одной какой-
либо температуре, а в интервале 10—20 °C, причем этот интер-
вал может смещаться в зависимости от степени предваритель-
ной ориентации и скорости повышения температуры.
Температура стеклования лишь условно разграничивает об-
ласти проявления упругих и высокоэластических свойств. Для
кристаллических полимеров температура стеклования всегда ле-
жит ниже их температуры кристаллизации.
Температуры стеклования и кристаллизации, а также ско-
рость кристаллизации зависят от химического строения полиме-
ра и гибкости макромолекул. С повышением гибкости макро-
молекулы температуры переходов снижаются, интервал между
ними увеличивается и скорость кристаллизации возрастает.
Полиэтилен имеет высокую скорость кристаллизации, поэтому в
аморфном виде его получить нельзя.
1.3. Методы синтеза полимеров
Основными методами синтеза полимеров являются полимери-
зация (цепная и ступенчатая) и поликонденсация.
Полимеризация. Полимеризацией называется реакция обра-
зования высокомолекулярных веществ (полимеров) путем соеди-
нения нескольких молекул мономера, которая не сопровождав
213
ется выделением побочных продуктов*. Элементарные звенья
образующегося полимера обычно не отличаются по составу от
исходных мономеров.
В реакцию полимеризации могут вступать соединения, имею-
щие кратные связи. Простейшим примером таких реакций явля-
ется полимеризация моно- и диолефинов. Полимеризация олефи-
нов может протекать под действием тепла, света, ионизирующе-
го излучения, минеральных кислот и солей (H2SO4, Н3РО4,
А1С13, BF3, HF), пероксидов, алюминийорганических соединений
(часто в смеси с хлоридами титана), силикатов, гидросили-
катов.
В промышленности в качестве катализатора при полимериза-
ции (олигомеризации) низших олефинов чаще всего применяют
серную и фосфорную кислоты на носителях; широкое распрост-
ранение получила олигомеризация этилена в присутствии триал-
килалюминия.
Полимеризация может протекать по механизму цепных реак-
ций— цепная полимеризация или ступенчатых процессов — сту-
пенчатая полимеризация.
Цепной полимеризацией называется процесс получения высо-
комолекулярных соединений, при котором макромолекула обра-
зуется путем последовательного присоединения молекул мономе-
ров к растущей цепи. При цепной полимеризации молекулярная
масса образовавшегося полимера очень высока, причем она на-
растает не постепенно, а достигается почти мгновенно.
Цепная полимеризация является наиболее распространенной
разновидностью реакций полимеризации. Цепная полимеризация
может протекать через промежуточное образование свободных
радикалов, и тогда она называется радикальной, или через про-
межуточное образование ионов, и тогда она называется ионной.
В зависимости от заряда иона различают катионную и анион-
ную полимеризацию.
Реакция полимеризации может также быть стереоспецифиче-
ской, т. е. приводить к образованию полимеров с регулярным
расположением замещающих групп во всех звеньях макромоле-
кул относительно плоскости основной цепи (стереорегулярные
полимеры).
Ступенчатая полимеризация протекает при ступенчатом при-
соединении молекул мономера с постепенным наращиванием мо-
лекулярной массы полимера. Образующиеся промежуточные про-
дукты на любой стадии полимеризации являются устойчивыми
соединениями. Ступенчатая полимеризация осуществляется пу-
тем миграции (перемещения) атома водорода от одной молеку-
лы к другой и иногда называется миграционной полимериза-
цией. Например, при ступенчатой полимеризации изобутилена в
* Термин «полимеризация», используемый в химии моторных топлив,
подразумевает получение не высокомолекулярных соединений, а олигомеров.
214
присутствии серной кислоты миграция водорода происходит сле-
дующим путем:
? СН3 СН3 СН3 СН3
* I I I I
СН2=С +СН2=С —> сн3—с—сн=с
II II
сн3 сн3 сн3 сн3
Каждый новый акт присоединения требует затраты энергии.
Молекулярная масса конечных продуктов реакции обычно не
очень высока. Ступенчатая полимеризация применяется для по-
лучения компонентов моторных топлив, промежуточных продук-
тов для синтеза моющих веществ, присадок к маслам и др.
Термодинамически реакция полимеризации, как и любая хи-
мическая реакция, протекает с уменьшением энергии Гиббса.
Вероятность протекания реакции можно оценить по изменению
AG в ходе реакции в стандартных условиях:
ДО = — RTlnKp
где Кр— константа равновесия реакции.
Если AG имеет отрицательное значение при постоянных темпе-
ратуре и давлении, то, в принципе, реакция может протекать
самопроизвольно. Зависимость энергии Гиббса от температуры
в стандартных условиях выражается уравнением
где ДЯ° — изменение энтальпии (теплосодержания) системы, равное тепло-
вому эффекту реакции с обратным знаком при постоянном давлении; Д5° —
изменение энтропии системы.
При полимеризации олефинов происходят разрыв одной двой-
ной связи С = С (энергия связи равна 607 кДж/моль) и образо-
вание двух простых связей С—С (энергия связи равна
352 кДж/молъ). Разность энергий этих связей представляет
собой тепловой эффект реакции:
2 • 352 — 607 = 97 кДж/моль
Для большинства непредельных соединений действительная
теплота полимеризации ниже теоретической:
ДЯ, кДж/моль
Этилен
Изобутилен
Стирол
Изопрен
Бутадиен
94 Хлоропрен
53 Метилстирол
69 Метилметакрилат
74 Винилацетат
72 Винилиденхлорид
ДЯ, кДж/моль
68
35
54
89
60
Вероятно, это объясняется потерей энергии сопряжения при пе-
реходе от мономера к полимеру и потерей энергии, связанной с
возникновением напряжений при образовании полимерной цепи
из замещенных олефинов вследствие взаимодействия боковых
групп.
215
Таблица V.L Дипольные моменты некоторых
непредельных соединений
Мономер
Формула
Дипольный
момент, Д
Этилен
Пропилен
Изобутилен
транс-Бутен-2
Стирол
Винилхлорид
Акрилонитрил
Бутадиен-1,3
Изопрен
Хлоропрен
СН2=СН2
СН3-<Н=СН2
(СН3)2С = СН2
СНз—СН=СН—СН3
СбН5-СН = СН2
СН2=СНС1
ch2=chcn
сн2=сн—сн=сн2
сн2=с—сн=сн2
I
сн3
СН2=С—сн = сн2
I
С1
0,0
0,35
0,49
0,0
0,37
1,44
3,88
0,0
0,38
1,42
Кинетика полимеризации различных олефинов зависит от их
строения. Применение катализаторов приводит к увеличению
скорости реакции, ускоряет достижение равновесия, но не сме-
щает его, так как ДО и в присутствии, и в отсутствие катализа-
торов не изменяется.
При цепной полимеризации олефинов скорость процесса за-
висит от степени поляризации мономера или легкости смещения
электронной плотности в молекуле, или, что то же самое, поля-
ризуемости молекулы.
Молекула этилена неполярна, ее дипольный момент равен
нулю. У молекул пропилена и изобутилена двойная связь поля-
ризована (табл. V.1). Метильная группа является электронодо-
норным заместителем — она отталкивает электроны к углерод-
ному атому незамещенной метиленовой группы:
H^c^-ch=Qh2
Поляризуемость двойной связи олефина увеличивается при вве-
дении в молекулу полярных атомов или групп атомов (галоге-
на, нитрильной, карбоксильной группы), которые оттягивают на
себя электроны:
СН2=СН—С1
СН2=СН—CN
216
В результате поляризации один атом углерода приобретает по
вишенную электронную плотность, другой — пониженную:
f . CHQ
5+ 6- 3\6+ 6-
сн3—сн=сн2 V=ch2
снз
Неполярные молекулы этилена и бутадиена могут, поляризо-
ваться под влиянием полярных веществ:
6-
сн2=сн2 —> сн2=сн2
С повышением степени поляризации мономера возрастает его
реакционная способность.
Ионная полимеризация протекает в присутствии кислых и
основных катализаторов. Катализаторами катионной полимери-
зации являются сильные минеральные кислоты. Чем сильнее
кислота, тем легче катион водорода присоединяется к атому уг-
лерода с повышенной электронной плотностью, образуя катион
карбония:
ен2=8сн8)а + н+ СН3~С(СН3)2
Такой катион присоединяется к другой поляризованной молекул
ле с образованием нового катиона, который присоединяется к
следующей молекуле, и т. д.:
4. <5— д+ /СНз
СН3-С(СН3)2 + СН2=(/
ч2Н3
СН3
I +/СНз
сн3—с—сн2-с(
сн, с"’
При полимеризации неполярных мономеров в систему вводят
соединения, способные в определенных условиях расщепляться
на радикалы, например:
СвНб-СОО-ООС-СвН5 ► 2СвН6-соо.
Такие радикалы реагируют с мономерами с образованием актив-
ных центров, которые инициируют полимеризацию. Соединения,
способные к разложению на радикалы, называются инициато-
рами. В качестве инициаторов могут использоваться органиче-
ские пероксиды, пероксид водорода, натрия, соли неорганиче-
ских перкислот (персульфаты, пербораты, хроматы и др.).
Некоторые виниловые мономеры могут полимеризоваться в
присутствии окислительно-восстановительных систем. Этилен мо-
жет полимеризо'ваться в присутствии тетрахлорида углерода,
который в присутствии пероксидов способен образовывать ра-
дикалы (реакции теломеризации).
Способность олефинов к полимеризации зависит от их строе-
ния. При замене всех атомов водорода и углерода при двойной
217
связи на алкильные группы полимеризации не происходит. Боль-
шую склонность к полимеризации проявляют монозамещенные
и несимметричные дизамещенные этилена, так как заместители
поляризуют связь и делают молекулу более реакционноспособ-
ной. Симметричные дизамещенные этилена R—СН=СН—R'
полимеризуются с трудом.
В промышленности полимеризация осуществляется в блоке,
в растворе, в эмульсии и суспензии.
Поликонденсация. Поликонденсацией называется реакция об-
разования высокомолекулярных веществ (полимеров) путем сое-
динения нескольких молекул, сопровождающаяся одновремен-
ным выделением какого-либо низкомолекулярного вещества
(воды, спирта, аммиака, хлористого водорода и др.). В отличие
от продуктов полимеризации состав элементарного звена поли-
мера, полученного в результате реакции поликонденсации, не
соответствует элементному составу исходного мономера.
В реакцию поликонденсации могут вступать соединения, со-
держащие в своем составе различные функциональные группы
(—ОН, —СООН, —NH2 и др.). Для образования полимера не-
обходимо, чтобы каждый мономер содержал не менее двух
функциональных групп. Процесс поликонденсации, в котором
участвуют однородные молекулы, называется гомополиконден-
сацией. Примером такой реакции может служить поликонденса-
ция аминокислот с образованием полиамидов:
H2N~R—COOH + H2N-R—соон —
—Н2О
H2N—R—CONH—R—СООН
+nH2N-R—СООН
--► Н—[—NH—R—СО—|п+2—ОН + пН2О
Процесс поликонденсации разнородных молекул носит назва-
ние гетерополиконденсации. Например, реакции диаминов с ди-
карбоновыми кислотами с образованием полиамидов:
nHaN—R—NH2 4- nHOOC—R'—СООН ►
---► Н—[—HN—R—NHCO—R'—СО—]п—ОН 4- (2п — 1) Н2О
Поликонденсация может быть равновесной и неравновесной.
Равновесная поликонденсация характеризуется сравнитель-
но небольшой константой равновесия, а для неравновесной
поликонденсации характерны высокие скорости реакции. Приве-
денные выше реакции являются примерами равновесной поли-
конденсации. Примером неравновесной поликонденсации служит
реакция образования полиамидов при межфазной поликонден-
сации хлорангидридов дикарбоновых кислот с диаминами (см.
ниже). Равновесная поликонденсация обратима. Течение про-
цесса и характер образующихся продуктов реакции зависят от
функциональности исходных соединений, а также скорости и
218
полноты удаления низкомолекулярных продуктов реакции. Если
исходные мономеры содержат по две функциональные группы,
то образуются линейные полимеры:
па—А—а + nb—В—b -► а—[—А—В—]п—b 4- (п — 1) ab
Если одна из исходных реагирующих молекул имеет более двух
функциональных групп, образуется полимер разветвленного или
сетчатого строения:
где а и b — функциональные группы исходных веществ; ab — молекула вы-
деляющегося низкомолекулярного вещества.
Реакция поликонденсации носит ступенчатый характер. Каж-
дый акт взаимодействия функциональных групп приводит к на-
ращиванию цепи на одно звено: сначала образуется димер, за-
тем тример, тетрамер и т. д. Наращивание цепи происходит не
только в результате взаимодействия молекул мономера с диме-
ром, тримером, но и в результате взаимодействия низкомолеку-
лярных полимеров (олигомеров) между собой. Одновременно с
ростом цепи полимера протекают побочные реакции — деструк-
ция полимера и сшивание макромолекул. Деструкцию, проте-
кающую под действием низкомолекулярных продуктов реакции
(воды, аммиака и др.), можно уменьшить путем тщательного
удаления их из зоны реакции или проведением реакции в ат-
мосфере азота.
Скорость реакции поликонденсации можно регулировать из-
менением температуры реакционной среды. В равновесных ус-,
ловиях скорость образования полимера равна скорости его де-
струкции. Для того чтобы сдвинуть реакцию в сторону образо-
вания полимера, следует повысить (до определенного предела)
температуру или понизить давление (до глубокого вакуума).
При этом уменьшается также вязкость среды, что способствует
удалению низкомолекулярных продуктов.
Поликонденсацию проводят с эквимольными количествами
исходных веществ. При избытке одного из мономеров наруша-
ется соотношение между разноименными функциональными
группами, и после израсходования мономера, взятого в мень-
шем количестве, процесс поликонденсации прекращается.
Катализаторами поликонденсации служат минеральные кис-
лоты, кислые соли, оксиды металлов, щелочные металлы и аро-
219
магические сульфокислоты (для полиэтерификации); при кон-
денсации формальдегида с фенолами и карбамидом — щелочи и
кислоты.
Поликонденсацию чаще всего осуществляют в расплаве при
температуре 200—280°C (если мономеры устойчивы при этих
температурах). Процесс проводят в среде инертного газа и за-
канчивают в вакууме для более полного удаления побочных
продуктов.
Поликонденсацию можно проводить также в растворе и на
поверхности раздела двух фаз. Поликонденсация на Границе
раздела фаз протекает при комнатной температуре, благодаря
чему исключается возможность деструкции полимера и, кроме
того, отпадает необходимость поддержания эквимольного соот-
ношения мономеров (оно устанавливается автоматически на
границе раздела фаз). Так, если к раствору хлорангидрида в
метиленхлориде прибавить раствор диамина в воде, то на гра-
нице раздела фаз сразу образуется полиамид, который «можно
подвергнуть формованию. Выделяющийся хлористый водород
диффундирует в водный слой и нейтрализуется специально до-
бавляемой щелочью.
В промышленности поликонденсация осуществляется следую-
щими основными способами: в расплаве, в растворе, на грани-
це раздела фаз, в твердой фазе.
1.4. Полимерные материалы
Полимерные материалы состоят из собственно полимеров и
различных добавок, придающих им определенные свойства.
Важнейшими полимерными материалами являются пластиче-
ские массы (пластмассы), каучуки (эластомеры) и волокна.
Пластические массы. Пластические массы, полученные на
основе полимеров, обладают способностью формоваться и в
обычных условиях сохранять приданную им форму (в виде го-
товых изделий). Кроме полимеров в состав пластических масс
входят наполнители, пластификаторы, стабилизаторы, красители
и другие добавки.
Наполнители вводят для улучшения физико-механических
свойств пластмасс, уменьшения усадки и снижения стоимости
полимерного материала. Некоторые пластмассы (например, фе-
нопласты, аминопласты) могут содержать до 60% наполнителя.
В качестве наполнителей применяют древесную муку, бумагу,
хлопчатобумажную ткань, слюду, тальк, каолин, стеклянное во-
локно (порошковые, волокнистые, слоистые наполнители).
Пластификаторы придают пластмассам гибкость и эластич-
ность, уменьшают жесткость и хрупкость, улучшая процесс фор-
мования/ Однако при введении больших количеств пластифика-
торов снижается теплостойкость пластмасс и их прочность. В ка-
честве пластификаторов применяют дибутилфталат, олеиновую
кислоту, стеарин, камфору, глицерин и др.
220
Для повышения стойкости полимеров к действию тепла,
света, кислорода воздуха и других агентов к ним добавляют спе-
циальные добавки — стабилизаторы (противостарители, антио-
кислители, термостабилизаторы и др.), способствующие дли-
тельному сохранению свойств полимерных материалов в усло-
виях эксплуатации.
Красители вводят в пластмассу с целью придания ей нужно-
го цвета.
В зависимости от поведения при нагревании полимеры де-
лятся на термопластичные и термореактивные.
Термопластичные полимеры, или термопласты, при нагрева-
нии размягчаются и становятся пластичными, а при охлаждении
снова затвердевают. Размягчение и отверждение можно прово-
дить многократно. К термопластам относятся полиэтилен, поли-
пропилен, поливинилхлорид, полистирол, полиметилметакрилат,
полиамиды, фторопласты и др.
Термореактивные полимеры, или реактопласты, з начале тер-
мообработки размягчаются, становятся пластичными и прини-
мают заданную форму. Однако при дальнейшем нагревании они
теряют пластичность и переходят в неплавкое и нерастворимое
состояние, обусловленное сшиванием макромолекул полимера и
образованием сетчатой структуры. К реактопластам относятся
фенопласты, аминопласты и др.
Каучуки. Каучуки представляют собой высокомолекулярные
соединения с повторяющимися структурными единицами (эле-
ментарными звеньями). Поведение и свойства каучуков опреде-
ляются их составом, строением, молекулярной массой.
Отличительными особенностями каучуков (независимо от их
происхождения) являются: 1) большая длина макромолекул и
соответственно их большая молекулярная масса (104—106);
2) высокая эластичность; 3) способность вулканизоваться и об-
разовывать при этом пространственную сетку.
При вулканизации (превращении в резину)4 каучук подвер-
гают обработке серой при температуре 140—170 °C и давлении
1,5—2 МПа. При этом макромолекулы каучука связываются
между собой мостиками из серы, или сшиваются. Получаемая
в результате вулканизации резина приобретает более высокую
механическую прочность по сравнению с каучуком: она обла-
дает меньшим разрывным удлинением вследствие резкого умень-
шения или полного устранения пластических деформаций, боль-
шей эластичностью, меньшим набуханием и растворимостью в
органических растворителях и большей теплостойкостью.
Для придания требуемых свойств перед вулканизацией к ка-
учуку добавляют различные вещества — ингредиенты. Для по-
вышения прочности, твердости, сопротивления истиранию в кау-
чук вводят технический углерод или диоксид кремния (белую
сажу), а также наполнители — мел и каолин. Для повышения
пластичности в резиновую смесь вводят пластификаторы (мягчи-
тели).
221
Одним из существенных недостатков резиновых изделий яв-
ляется понижение их эластичности и возрастание хрупкости при
эксплуатации и хранении, что обусловлено процессом старения
резины.
Для замедления процесса старения и удлинения срока служ-
бы резиновых изделий в резиновую смесь вводят противостари-
тели (антиоксиданты): фенолы, амины, в частности фенил-р-
нафтиламин (неозон Д).
Волокна. Особую группу высокомолекулярных соединений
составляют волокнообразующие полимеры, из которых полу-
чают волокна.
Все волокна можно разделить на два больших класса: нату-
ральные и химические. Химические волокна получают в основ-
ном из природных или синтетических высокомолекулярных сое-
динений. В зависимости от природы исходного полимера химиче-
ские волокна подразделяются на искусственные и синтетиче-
ские.
Искусственные волокна вырабатывают из природных высоко-
молекулярных соединений — целлюлозы и белков.
Синтетические волокна вырабатывают из полимеров, полу-
чаемых в промышленности путем синтеза из различных хими-
ческих соединений. В зависимости от строения макромолекул
эти волокна в свою очередь делятся на гетероцепные и карбо-
цепные.
Из гетероцепных волокон в промышленности вырабаты-
ваются полиамидные, полиэфирные и полиуретановые во-
локна.
Из карбоцепных волокон в промышленном масштабе выра-
батываются полиакрилонитрильные и полипропиленовые во-
локна.
Существует два основных метода получения волокон из во-
локнообразующих полимеров. По первому методу полимеры на-
гревают до температуры плавления и образовавшуюся вязкую
жидкость продавливают через мельчайшие отверстия — филье-
ры. Полученные тонкие струйки расплавленного материала за-
тем охлаждают. Этот процесс называется формованием из рас-
плава. Второй метод заключается в растворении полимера в ра-
створителе и продавливании раствора через фильеры. Это —
метод сухого формования. Вариантом этого метода является ме-
тод мокрого формования, при котором раствор полимера в
растворителе поступает в осадительную ванну, и полимер
высаживается в виде тонкой нити, а растворитель вымыва-
ется.
Применение полимерных материалов в технике дает огром-
ный экономический эффект — повышает производительность
труда, снижает себестоимость, улучшает качество продукции, об-
легчает условия труда, способствует повышению культуры про-
изводства, высвобождает миллионы рабочих рук и огромные де-
нежные средства.
222
§ 2. ПРОИЗВОДСТВО ПОЛИМЕРОВ
РЕАКЦИЯМИ ПОЛИМЕРИЗАЦИИ
2.1. Производство полимерных углеводородов
Олигомеры, этилена
Олигомеры этилена широко применяют в качестве сырья для
различных химических процессов (алкилирования бензола, фе-
нола, производства бутиленов, диенов, моющих веществ и т. д.).
Этилен димеризуется в бутилен в присутствии гидрида алю-
миния или литийалюминийгидрида при 180—200 °C и небольшом
давлении. Вначале гидрид алюминия образует с этиленом три-
этилалюминий, который выполняет функцию катализатора. При
добавлении в систему металлического никеля реакция останав-
ливается на стадии образования димера; при этом высшие по-
лимеры образуются в незначительном количестве:
А1Н3 + ЗСН2=СН2---► А1(С2Н5)з
гидрид триэтил-
алюминия алюминий
А1(С2Н5)з + СН2=СН2 -> (С2Н5)2А1-СН2-СН2-СН2-СН3 -и
----► (С2Н5)2А1Н + сн2=сн-сн2—сн3
При добавлении в систему хлоридов титана образуется высоко-
молекулярный твердый полиэтилен.
При олигомеризации этилена в присутствии концентриро-
ванной фосфорной кислоты при 300 °C и 3,5 МПа получают бу-
тилен, изобутан и жидкие углеводороды, содержащие парафи-
ны, циклопарафины, олефины и ароматические углеводороды,
т. е. происходит смешанная полимеризация, в результате кото-
рой получается бензин с октановым числом 82. Жидкие угле-
водороды можно получить и олигомеризацией этилена при ком-
натной температуре и давлении 5 МПа в присутствии фтористо-
го водорода.
В присутствии безводного хлорида алюминия под давлением
2—4 МПа и температуре 100—150 °C этилен олигомеризуется с
образованием низко- и высококипящих углеводородов сложного
состава (парафины нормальные и изостроения, циклические
углеводороды — циклопентан' и циклогексан). Высококипящие
углеводороды применяют в качестве смазочных масел, обладаю-
щих высоким индексом вязкости.
Для полимеризации применяют чистый этилен, не содержа-
щий примесей кислорода, сероводорода, СО и Н2. Реакция про-
водится в растворителях, например в изооктане. Расход хлорида
алюминия составляет 0,5% на полученное масло. Вязкость го-
тового масла регулируется температурой полимеризации (для
получения масла с низкой вязкостью процесс проводят при бо-
лее высокой температуре).
223
Еще более жесткие требования к чистоте 'исходного этилена
предъявляются при регулируемой полимеризации в присутствии
металлоорганического катализатора — триалкилалюминия. По-
лучение олигомеров этилена (высших а-олефинов С6—С4о) рас-
смотрено в гл. I.
Полиэтилен
Полиэтилен удачно сочетает такие свойства, как химическая
стойкость, механическая прочность, морозостойкость, хорошие
диэлектрические свойства, стойкость к радиационным излуче-
ниям, низкие газопроницаемость и влагопоглощение. Его при-
менение определяется комплексом физико-механических, хими-
ческих и диэлектрических свойств. Из полиэтилена изготовляют
трубопроводы, емкости для хранения химических реагентов. Он
применяется в качестве электроизоляционного материала, в
строительстве, в сельском хозяйстве, в качестве антикоррозион-
ных покрытий для производства упаковочной пленки, посуды,
для пропитки тканей, бумаги, древесины и т. д.
Существуют три основных промышленных метода получения
полиэтилена — полимеризация этилена при высоком, среднем и
низком давлении. Наибольшее распространение получили два
метода — полимеризация этилена при высоком и низком давле-
нии.
Свойства полиэтилена определяются в основном строением
полимерной цепи. Макромолекула полиэтилена высокого дав-
ления (ПЭВД) представляет собой длинную метиленовую цепь
с небольшим числом метильных групп (2—3 группы СН3 на
каждые 100 метиленовых групп). Длина макромолекулы поли-
этилена и количество боковых групп зависят от условий полиме-
ризации. Присутствие боковых групп в макромолекулах нару-
шает упорядоченное расположение молекул и приводит к обра-
зованию наряду с кристаллическими участками и аморфных
областей. С повышением температуры кристаллическая структу-
ра исчезает, а количество аморфной фазы увеличивается; при
температуре плавления весь полиэтилен становится аморфным.
Охлаждение полиэтилена сопровождается кристаллизацией.
Однако быстрое охлаждение (закалка) приводит к получению
большого количества аморфного продукта. Соотношение кри-
сталлической и аморфной части определяет многие свойства по-
лиэтилена. С уменьшением размеров кристаллов и степени кри-
сталличности полимер становится более гибким и эластичным.
При комнатной температуре полиэтилен не растворяется ни в
одном из растворителей; при 70 °C он набухает и растворяется
в тетрахлориде углерода, трихлорэтилене, толуоле, ксилоле. При
охлаждении полимер выпадает из раствора.
Полиэтилен низкого давления (ПЭНД) характеризуется
меньшим количеством боковых ответвлений в макромолекуле.
Он отличается от полиэтилена высокого давления более высоки-
224
ми плотностью и прочностью, а также более высокой темпера-
турой размягчения.
Основные свойства полиэтилена, полученного разными спо-
собами, приведены ниже:
Показатель
Степень кристалличности, %
Плотность, кг/м3
Температура плавления, °C
Молекулярная масса ....
Разрушающее напряжение при
растяжении, МПа
Относительное удлинение при
разрыве, %
Температура хрупкости, °C
Диэлектрическая проницае-
мость при 106 Гц
Электрическая прочность,
кВ/мм
/пэвд
55-67
920
108—110
15000-30000
12,0-16,0
150—600
Около 70
2,2-2,3
45-60
пэнд
80—90
940
120—130
70000—350000
22,0-35,0
200—900
Около 70
2,1-2,4
45-60
Производство полиэтилена при высоком давлении. Полиме-
ризация этилена при высоком давлении протекает по радикаль-
ному механизму. Она проводится в реакторах специальной кон-
струкции при давлении 150—170 МПа и температуре 180—
240°C. Инициатором полимеризации служат кислород или пер-
оксиды. Реакция сопровождается выделением огромного коли-
чества тепла (3600 кДж на 1 кг полиэтилена). Конверсия эти-
лена составляет 8—15%; при более высокой конверсии затруд-
няется отвод тепла и поддержание постоянной температуры
процесса.
Процесс сопровождается побочными реакциями разложения
этилена, протекающими с выделением тепла:
СН2=СН2 ---> СН4 + С ДЯ = —127 кДж/моль
СН2=СН2 ——> 2С 4- 2Н2 ДЯ = 47 кДж/моль
Крайне нежелательной реакцией, ингибирующей полимериза-
цию, является образование формальдегида:
С2Н4 + О2 -> 2НСНО
Вследствие низкой конверсии этилена за один проход, а
также за счет использования газа рециркуляции в рабочем газе
накапливаются примеси, и концентрация этилена снижается.
В результате уменьшается скорость полимеризации. Для под-
держания определенной скорости следует увеличивать количе-
ство инициирующего кислорода, что приводит к снижению мо-
лекулярной массы полимера. Поэтому концентрация этилена в
исходном газе должна быть не менее 99,9% (об.), чтобы в ра-
бочем газе она составляла 99% (об.).
Полимеризацию проводят в аппаратах с мешалкой или в
трубчатых реакторах змеевикового типа. Реакторы с перемеши-
вающим устройством более производительны, что обусловлива-
ется как конструкцией реактора, позволяющей вводить этилен
15-254
225
Рис. V.l. Технологическая схема получения полиэтилена при высоком дав-
лении:
/ — компрессор первого каскада; 2—компрессор второго каскада; 3 — реактор; 4, 5,
6 — сепараторы; 7 — холодильник; 8 — колонна сухой очистки возвратного этилена.
и инициатор по зонам, так и возможностью применения более
эффективных пероксидных инициаторов. Однако трубчатые, ре-
акторы более просты по конструкции и в эксплуатации.
На рис. V.I. приведена технологическая схема получения
полиэтилена на установке с трубчатым реактором. Этилен, со-
держащий 0,01% кислорода, компрессором первого каскада 1
сжимается до 32 МПа и компрессором второго каскада 2 ком-
примируется до 150 МПа. Затем его подают в реактор 3. Реак-
тор представляет собой теплообменник «труба в трубе» (диа-
метр внутренних труб 32 мм), разделенный на три зоны. Этилен
поступает в реактор пр«и 35 °C и в зоне предварительного нагре-
ва нагревается до 150 °C, затем во второй зоне дополнительно
нагревается до 185 °C и в зоне реакции полимеризуется при
185—240 °C. Тепло реакции снимается горячей водой, имеющей
температуру около 200 °C. Охлаждение холодной водой не при-
меняется, так как на внутренних стенках труб при температуре
ниже 175 °C образуются полимеры, что затрудняет дальнейший
отвод тепла.
Из реактора продукты реакции через дроссельный клапан,
в котором давление снижается до 32 МПа, поступают в обогре-
ваемый сепаратор высокого давления 4. Не вступивший в реак-
цию этилен направляется в циклонные обогреваемые сепара-
торы 6, в которых отделяется пыль полимера. Обычно устанав-
ливают несколько сепараторов и при переходе из одного в дру-
гой газ проходит через трехступенчатый холодильник 7. Из се-
параторов газ проходит колонну сухой очистки 8 (от СОг и
226
НСОН), после чего охлаждается (температура в колонне 50 °C)
и поступает на компрессию. Сжатый до 150 МПа газ возвра-
щается в процесс. Из сепаратора высокого давления 4 полимер
подается в сепаратор низкого давления 5 через дроссель, при
этом давление уменьшается до 0,1—0,6 МПа. При дросселиро-
вании температура снижается с 220 до 180—200°C. Этилен из
сепаратора низкого давления поступает на компримирование и
возвращается в процесс.
Из сепаратора низкого давления жидкий полиэтилен посту-
пает на грануляцию. Грануляция заключается в продавливании
полиэтилена при 220—250 °C и давлении 0,2—0,8 МПа через
насадку (головку) с калиброванными отверстиями, в результате
чего получаются жгуты или прутки. Жгуты проходят через ван-
ну с проточной водой и подаются на режущее устройство для
нарезки гранул цилиндрической формы или в форме чечевицы.
Нарезка может производиться непосредственно у плоскости го-
ловки вращающимся ножом со сбросом гранул в воду для ох-
лаждения и устранения слипания. Затем гранулят отделяется от
воды, высушивается и поступает в бункеры. Полученный грану-
лят имеет насыпную плотность 400—250 кг/м3.
При нарушении теплового режима реактора автоматически
срабатывает предохранительный клапан и сбрасывает газ в ат-
мосферу. Периодически производится сдувка газа (сброс части
газа на газоразделение) с целью поддержания концентрации
этилена на заданном уровне и во избежание накопления при-
месей.
Перед пуском установки всю аппаратуру продувают азотом
и промывают толуолом.
Поскольку конверсия этилена за один проход невелика, доля
газа рециркуляции составляет около 90%. Это вызывает большой
расход энергии, оборотной воды и пара. На 1 т полиэтилена
расходуется примерно 2,4 т пара, 325 м3 оборотной воды,
1800 кВт-ч электроэнергии.
Полимеризация в реакторах с мешалкой проводится при
тех же условиях. Реактор представляет собой толстостенный
сосуд с наружным диаметром 550—650 мм и внутренним 250—
350 мм, высотой 5—6 м. Производительность одного аппарата
составляет 6000—7000 т полиэтилена в год, в то время как про-
изводительность трубчатого реактора составляет 3000—4000 т
в год. Подвод жидкого инициатора по зонам обеспечивает его
одинаковую концентрацию в реакционном объеме и, следова-
тельно, получение более однородного продукта. Производитель-
ность реактора лимитируется теплосъемом.
В трубчатом реакторе отвод тепла осуществляется с по-
мощью циркулирующей через рубашку воды. Реактор имеет два
подвода горячей воды. Первый поток с температурой 155 °C под
давлением 2,0 МПа подается в зону подогрева, откуда направ-
ляется в зону охлаждения и при 153°C возвращается на стан-
цию горячей воды. Второй поток при температуре 229°C и дав-
15*
227
лении 5 МПа подается в зону реакции, нагревается до 236°C,
поступает в зону дополнительного нагрева и возвращается на
станцию горячей воды.
В реактор с мешалкой можно подавать этилен при сравни-
тельно низкой температуре (30—50°C), так как температура
вследствие перемешивания быстро выравнивается. В результа-
те этого часть тепла реакции расходуется на нагрев сырья.
Благодаря перемешиванию устраняются местные перегревы и
улучшаются условия отвода теплоты через стенку. Степень кон-
версии этилена достигает 20—25%.
Производство полиэтилена при среднем давлении. По этому
способу полимеризацию проводят в присутствии оксидных ката-
лизаторов, нанесенных на активный носитель. Метод не требует
дорогостоящей аппаратуры высокого давления, компрессоров,
реакторов и т. д. Процесс осуществляется на более простых и
дешевых катализаторах, безопасных при хранении и примене-
нии. Недостатком процесса является трудность удаления взве-
шенных частиц катализатора из раствора полимера.
Полимеризация этилена проводится при температуре 130—
170 °C и давлении 3,5 МПа в среде инертного растворителя (пен-
тан, гексан, гептан, бензол, толуол и др.). В качестве катализа-
тора применяется шестивалентный оксид хрома, нанесенный на
алюмосиликат. Технологические условия процесса зависят от
природы применяемого оксида.
Производство полиэтилена при низком давлении. Полимери-
зацию этилена при низком давлении проводят в присутствии
металлоорганического катализатора в среде инертного раство-
рителя (гептан, гексан, циклогексан, бензин). Катализатором
полимеризации служит комплексная система, состоящая из тет-
рахлорида титана и алкилов алюминия. В качестве катализато-
ров реакции могут использоваться и другие комплексные сое-
динения алкилов металлов первой, второй и третьей групп с
солями металлов переменной валентности. Однако на практике
чаще всего применяют алкилы алюминия (этил, пропил, бутил)
с галогенами титана (TiCl4 и TiCl3). Полимеризация этилена на
этих катализаторах протекает по анионному механизму.
Этилен вводят в реактор, в котором он полимеризуется с
большой скоростью с образованием суспензии высокомолекуляр-
ного полиэтилена. Процесс проводят практически при атмосфер-
ном давлении (или давлении 0,2—0,5 МПа) при 60—70°C. Реак-
ция сопровождается выделением значительного количества
тепла.
Полиэтилен, полученный в присутствии каталитического ком-
плекса, подвергается тщательной очистке от катализатора. Для
этого комплекс разлагается и продукты разложения отмываются
сначала спиртом, а потом водой. Катализатор разлагается спир-
тами, смесью спиртов с углеводородами и водой. Очистка по-
лимера водой связана с большими трудностями, так как вода
вызывает гидролиз соединений титана и алюминия с частичным
228
переводом их в нерастворимые соединения. Кроме того, вода
не смачивает полимер. Зольность полимера, отмытого сначала
спиртом, а потом водой, составляет 0,01—0,1%. При повышен-
ной зольности изделия окрашиваются в светло-кремовый цвет.
Молекулярная масса полимера зависит от соотношения три-
этилалюминия и тетрахлорида титана в катализаторе. Обычно
мольное соотношение этих компонентов равно 1 1 — 1 1,2, что
обеспечивает получение полиэтилена с молекулярной массой
70 000—350 000. При соотношении триэтилалюминия и тетрахло-
рида титана, равном 2 1, образуется жесткий полиэтилен с мо-
лекулярной массой более 1 000 000, а при соотношении, равном
1 2, получаются низкомолекулярные (ниже 30 000) хрупкие
полимеры. При замене триэтилалюминия диэтилалюминийхло-
ридом молекулярная масса также понижается.
Процесс полимеризации может осуществляться периодически
и непрерывно. Реакцию проводят в обычном вертикальном ци-
линдрическом аппарате колонного типа или -в реакторе с пере-
мешивающим устройством. Процесс состоит из следующих ста-
дий: приготовления катализатора, полимеризации, разложения
каталитического комплекса спиртом, выделения полиэтилена,
промывки, сушки И гранулирования.
Особенность технологического процесса полимеризации за-
ключается в том, что при его осуществлении необходимо полное
исключение из системы воды, кислорода, спирта и т. п., так как
их присутствие приводит к разложению компонентов каталити-
ческой системы и, следовательно, к замедлению или прекраще-
нию полимеризации.
В связи с этим процесс полимеризации проводят в атмосфе-
ре азота. Полимер получается в виде суспензии.
Отвод тепла из реактора через стенку затруднен из-за того,
что в процессе полимеризации стенки реактора покрываются
коркой полимера. Циркуляция суспензии через выносные холо-
дильники приводит к быстрому обрастанию стенок холодильни-
ка полимером и выходу их из строя через 2—4 ч работы.
Отвод тепла производится путем отдувки части этилена с
парами бензина (или другого растворителя), конденсации паров
бензина и охлаждения потока этилена и возвращения их в ре-
актор. По окончании процесса полимеризации каталитический
комплекс разлагают спиртом. При разложении водой образу-
ются гидроксиды алюминия и титана, которые не растворяются
в воде и не вымываются из полимера. Разложение проводят аб-
солютированным спиртом (чаще пропиловым) при 50—70 °C.
Концентрация суспензии полимера равна 70—80 кг/м3. Для раз-
ложения используют 5%-ный избыток спирта (при большем из-
бытке спирта могут высаживаться высокомолекулярные фрак-
ции полиэтилена). Промывка полиэтилена смесью спирт — бен-
зин обеспечивает получение полиэтилена с зольностью не более
0,1%. Промывку и разложение проводят в атмосфере азота.
Фильтрация полимера вызывает большие трудности, так как при
16-254
229
Ъстборшпель
Рис. V.2. Технологическая схема получения полиэтилена при низком дав-
лении:
1, 2 — емкости для компонентов катализатора; <3 — смеситель для получения каталитиче-
ского комплекса; 4 — реактор; 5 — каплеотбойник; 6 — холодильник; 7 —газодувка; 8 —
аппарат для разложения каталитического комплекса.
получении полиэтилена с большой молекулярной массой обра-
зуются труднофильтруемые растворы.
После разложения каталитического комплекса спирт и бен-
зин подвергают регенерации, которая заключается в ректифи-
кации смесей с получением либо азеотропной смеси, либо
раздельно спирта и бензина. Смесь перед перегонкой нейтрали-
зуют спиртовым раствором алкоголята натрия, и образующийся
хлорид натрия отделяют на фильтр-прессах. Полиэтилен сушат
в камерных сушилках в кипящем слое. Сушку проводят подо-
гретым азотом. После сушки порошок полиэтилена поступает на
грануляцию.
На рис. V.2 приведена Принципиальная технологическая схе-
ма получения полиэтилена низкого давления непрерывным спо-
собом. Растворы триэтилалюминия и тетрахлорида титана из
емкостей 1 и 2 дозировочными насосами подаются в смеси-
тель 3, в котором происходит образование 'каталитического
комплекса. Время пребывания продуктов в смесителе 15 мин;
температура 25—30 °C поддерживается с помощью горячей во-
ды, подаваемой в рубашку. Катализатор обычно готовится в
виде 1%-ного раствора.
Из смесителя 3 суспензию каталитического комплекса непре-
рывно подают в реактор-полимеризатор 4, в который одновре-
менно поступают бензин и этилен. При производительности ре-
актора 500 кг/ч полимера объем растворов компонентов состав-
ляет 600 л/ч. Температура в реакторе регулируется за счет цир-
230
куляции парогазовой этилен-бензиновой смеси и конденсации
части бензиновых паров. Парогазовая смесь из верхней части
реактора поступает в циклонный каплеотбойник 5, в котором
капли бензина отделяются от газа и возвращаются в реактор,
а парогазовая смесь охлаждается в холодильнике 6 и газодув-
кой 7 возвращается в нижнюю часть реактора, благодаря чему,
а также эрлифтному устройству, осуществляется перемешива-
ние. Температуру регулируют количеством циркулирующей па-
рогазовой смеси. Давление в реакторе 0,2—0,5 МПа. Из ниж-
ней части реактора суспензия полимера непрерывно дроссели-
руется в аппарат S, в котором с помощью азеотропной смеси
спирта с бензином проводится разложение каталитического
комплекса. При. смешении суспензии полимера из реактора, на-
гретой до 70 °C, и холодного азеотропа с температурой 20 °C
устанавливается температура около 50 °C. При температуре
ниже 50 °C в рубашку подают пар. Аппарат 8 снабжен про-
пеллерной мешалкой и диффузором, что обеспечивает эффек-
тивное перемешцванме. Суспензия полимера, содержащая про-
дукты разложения катализатора, непрерывно перекачивается
насосом на центрифугирование. Промывка полиэтилена может
проводиться периодически и непрерывно; сушка — периодически.
Часть этилена после реактора непрерывно сдувается для пре-
дотвращения накопления инертных примесей в сырье.
Для производства 1 т полиэтилена низкого давления при-
мерно расходуется 1,1 т этилена, 2,4 т триэтилалюминия, 4,8 т
тетрахлорида титана.
Способ получения полиэтилена низкого давления имеет ряд
преимуществ по сравнению со способом получения полиэтилена
высокого давления: отсутствие необходимости применения аппа-
ратов высокого давления; меньшие капиталовложения; высокая
(почти 100%-ная) конверсия этилена за один проход.
Однако при получении полиэтилена высокого давления не
требуются катализаторы и растворители. Кроме того, он имеет
очень высокие элекроизоляционные характеристики, обусловлен-
ные пониженной плотностью и отсутствием в нем следов продук-
тов разложения катализатора.
Сополимеры этилена
Сополимеры этилена и пропилена обладают ценными техни-
ческими свойствами. Изменяя соотношение этилена и пропилена,
можно варьировать эти свойства в широких пределах. Сополи-
меры имеют хорошую тепло- и морозостойкость, более высокую
по сравнению с полиэтиленом температуру плавления (137°C).
Сополимеризацию проводят в присутствии алкилов алюми-
ния с хлоридами титана, оксида хрома, а также оксидов вана-
дия, молибдена, вольфрама.
Сополимеризация этилена с пропиленом протекает с меньшей
скоростью, чем полимеризация этилена. Температура процесса
оказывает значительное влияние на свойства сополимеров. Чем
16
231
выше температура, тем ниже молекулярная масса, прочность и
относительное удлинение. Плотность и температура плавления
сополимеров зависят от соотношения исходных мономеров.
Одним из важных свойств сополимеров этилена и пропилена
является их стойкость к старению, что объясняется насыщенным
характером макромолекул. Однако такие сополимеры с трудом
вулканизуются.
Большое практическое значение приобретают тройные сопо-
лимеры этилена, пропилена и бутадиена. В тройных сополиме-
рах, состоящих в основном из звеньев этилена и пропилена,
звенья бутадиена (0,1 — 1 моль/кг) содержат одну двойную
связь. Такие сополимеры вулканизуются обычным методом.
При вулканизации сополимеров в присутствии вулканизую-
щих агентов образуются каучукоподобные материалы — этилен-
пропиленовые качуки (СКЭП).
Полипропилен
Полипропилен — высокомолекулярный термопластичный
кристаллический полимер пропилена общей формулы:
^СН2— СН—СН2—СН—СН2—СН^
(!н8 сн3 сн3
Технологический процесс получения полипропилена аналоги-
чен процессу получения полиэтилена низкого давления в присут-
ствии металлоорганических катализаторов. По своим свойствам
полипропилен близок к полиэтилену. Полипропилен имеет более
высокую температуру плавления (164—170°C), чем полиэтилен,
однако значительно уступает полиэтилену по морозостойкости.
Он является более жестким материалом, чем полиэтилен. Поли-
пропилен нерастворим в органических растворителях при ком-
натной температуре. Полипропилен очень чувствителен к дейст-
вию кислорода, особенно при повышенных температурах. Для
предотвращения окисления в полимер вводят стабилизаторы.
Ниже приведены показатели основных свойств полипропи-
лена:
Плотность, кг/м3 . . 900—910
Температура хрупкости, °C . —10
Разрушающее напряжение, МПа
при растяжении 30—35
при сжатии . . . 60—70
Относительное удлинение при разрыве, % 500—700
Диэлектрическая проницаемость 2,0—2,1
Электрическая прочность, кВ/мм 30—32
Полипропилен находит широкое применение в производстве
труб, фитингов, шестерен, деталей станков, пылесосов, холо-
дильников, ванн, баков, емкостей для химических реагентов,
пленок, канатов, различных предметов бытового назначения.
232
Из полипропилена получают синтетическое волокно, не усту-
пающее по свойствам полиамидным волокнам.
Пленки из полипропилена имеют меньшую влаго- и газопро-
ницаемость, чем полиэтиленовые, и широко использутся в каче-
стве упаковочного материала. Хорошие диэлектрические свойст-
ва позволяют применять полипропилен в электротехнической
промышленности.
Низкая морозостойкость несколько ограничивает области
применения полипропилена.
Полипропилен получают анионной полимеризацией в присут-
ствии комплексных катализаторов триэтилалюминия или ди-
этилалюминийхлорида и трихлорида титана. Процесс проводит-
ся в углеводородных растворителях. Технология производства
поли про пи лен а ана логи чн а т ехнол оги и про из вод ств а полиэти-
лена при низком давлении, но имеет некоторые особенности.
Применение TiCl3 вместо Т1СЦ дает высокий выход стереорегу-
лярной фракции (до 95%).
При полимеризации пропилена наряду со стереорегулярным
кристаллическим полипропиленом образуется атактический
аморфный полимер. Кристаллическую и аморфную фазы можно
разделить путем экстракции различными растворителями.
В эфире растворяется атактический полипропилен с молекуляр-
ной массой 20 000—30 000, в гептане — в основном атактический
и частично стереорегулярный полимер с молекулярной массой
30 000—40 000. Нерастворяющаяся в гептане при комнатной
температуре фракция представляет собой стереорегулярный кри-
сталлический полипропилен с молекулярной массой 40 000.
В промышленности полимеризацию пропилена проводят в
бензине или пропане при температуре 60—70 °C и давлении
1 — 1,2 МПа.
В качестве исходного сырья применяются чистый пропилен
(99%) и пропан-пропиленовая фракция (с содержанием 30—
50% пропана), тщательно очищенная от примесей воды и вла-
ги. Растворитель (бензин или гептан) не должен содержать не-
предельных углеводородов. Содержание серы в нем не должно
превышать 0,001 %, воды — 0,006%.
Полимеризацию проводят по периодической и непрерывной
схемам. Мольное соотношение триэтилалюминия и трихлорида
титана равно (3 1)-т-(2 1). При непрерывном ведении про-
цесса пропан-пропиленовую фракцию и растворитель подают в
таком соотношении, чтобы обеспечить концентрацию полимера
в растворителе, равную 140—170 кг/м3. Важной особенностью
полимеризации пропилена (в отличие от полимеризации этиле-
на) является меньшая адгезия полимера к стенке реактора и
меньший тепловой эффект реакции (58,6 кДж/моль). Это об-
легчает отвод тепла, позволяет строить установки большой про-
изводительности. Тепло отводится путем конденсации смеси па-
ров пропан-пропиленовой и бензиновой фракций в выносном
конденсаторе с возвратом конденсата в реактор.
233
При использовании в качестве сырья пропан-пропиленовой
фракции процесс можно проводить без растворителя — его за-
меняет жидкий пропан. Для получения высококачественного по-
лимера необходима тщательная очистка пропан-пропиленовой
^фракции от других олефинов, в частности от бутиленов. Поли-
меризацию осуществляют на таких же установках, что и поли-
меризацию этилена при низком давлении. При применении чи-
стого пропилена концентрация его должна быть не менее 99%.
Для производства 1 т полипропилена расходуется 1,1 т
пропилена (100%-ного), 25 кг диэтилалюминийхлорида
(100%-ного), 12 кг трихлорида титана (100%-ного), 0,3 т бен-
зина.
Полиизобутилен
Полиизобутилен получают путем полимеризации изобутиле-
на при комнатной и пониженной температурах:
СН3 “ СН3
I I
пС=СН2 —> -с~сн2—
I I
снз _ СН3 _ п
Полимеризация изобутилена сопровождается выделением боль-
шого количества тепла (41,8—83,6 кДж/моль). В результате
реакции получают полимеры с различной молекулярной массой.
Полиизобутилены с молекулярной массой до 50 000 представ-
ляют собой вязкие жидкости, с молекулярной массой около
200 000 — твердые, каучукоподобные материалы.
Полиизо1бутилен растворяется в нефтепродуктах или смеши-
вается с ними. Он применяется в качестве присадки к смазоч-
ным маслам, улучшающей их вязкостные свойства. Для загу-
щения маловязких масел обычно применяется полиизобутилен с
молекулярной массой 20 000—40 000.
Полиизобутилены обладают хорошей морозо- и теплостой-
костью, эластичностью (высокомолекулярные сорта), стой-
костью к окислению и действию химически агрессивных сред,
хорошими электроизоляционными свойствами.
Предельный характер полиизобутилена не позволяет прово-
дить его вулканизацию. Резина из полиизобутилена характери-
зуется низкой прочностью и ползучестью (течет на холоду), в
связи с чем ее применение ограниченно. Поэтому в резиновой
промышленности полиизобутилен смешивают с каучуками—на-
туральным, бутилкаучуком, бутадиеновым, бутадиенстирольным
и др. В зависимости от содержания полиизобутилена в смеси
можно получить различные сорта резины — от самой мягкой до
твердой.
Полиизобутилен применяется в качестве электроизоляцион-
ного материала для пропитки изоляционной бумаги или волок-
нистых материалов. При добавлении в низкомолекулярные сор-
234
та полиизобутилена наполнителей — смолы, воска, парафинов—
получают высококачественный герметичный материал. Высоко-
молекулярные полиизобутилены применяются в качестве доба-
вок к изоляционным лакам для улучшения их электроизоляци-
онных и адгезионных свойств, а также для повышения влаго-
стойкости. Полиизобутилены могут быть использованы для по-
лучения клеев, защитных покрытий, в качестве пластификато-
ров для синтетических материалов (полистирола, поливинилхло-
рида и др.), как вяжущее средство в печатных пастах и краси-
телях и т. д.
Высокомолекулярные полимеры изобутилена получают в при-
сутствии кислых катализаторов типа Фриделя — Крафтса. Чаще
всего применяют фторид бора или хлорид алюминия. Полиме-
ризация протекает почти мгновенно (цепной процесс) с выделе-
нием большого количества тепла.
При проведении полимеризации при комнатной температуре
(20 °C) вследствие интенсивного течения процесса затрудняется
быстрый отвод тепла, что приводит к повышению температуры и
деструкции полимера (при 300 °C полимер полностью деполи-
меризуется до мономера) , и в результате получается низкомо-
лекулярный полиизобутилен. Для получения высокомолекуляр-
ного полимера растворитель и мономер предварительно охлаж-
дают до низких температур. Выделяющееся при полимеризации
тепло расходуется на нагрев реакционной смеси, благодаря чему
удается избежать большого повышения температуры и предот-
вратить деструкцию полимера. При температурах от 20 до 30 °C
образуется полиизобутилен с молекулярной массой 5 000—8 000,
при —80°С и ниже — с молекулярной массой 100000—150 000.
Молекулярная масса полимера регулируется введением ус-
корителей или замедлителей процесса. Как правило, ускорители
добавляют к реакционной смеси в небольших количествах (от
0,001 до 1%). При применении хлорида алюминия в качестве
регулятора вводят НС1. Для получения полиизобутилена с высо-
кой молекулярной массой необходима очистка изобутилена от
бутенов-2 путем сверхчеткой ректификации. В присутствии дие-
нов молекулярная масса полимера также снижается. Исполь-
зуемый растворитель не должен содержать ингибирующих при-
месей: ароматических углеводородов и олефинов. Ингибитора-
ми полимеризации являются также меркаптаны, сульфиды и
другие органические соединения серы.
Полистирол
Полистирол представляет собой продукт полимеризации сти-
рола:
^СН-СН2-СН—СН2—СН-СН2^
235
Фенильные группы препятствуют упорядоченному расположению
макромолекул и формированию кристаллических образований.
Полистирол — жесткий аморфный полимер с невысокой ме-
ханической прочностью при растяжении и изгибе и весьма низ-
кой прочностью при ударе. Он легко деформируется при отно-
сительно невысоких температурах (80°C).
Изотактический полистирол плавится «при 220°C. По сравне-
нию с аморфным полистиролом он имеет повышенные хруп-
кость и 'плотность. Кристаллический полистирол не растворя-
ется в углеводородах при комнатной температуре, 'но полностью
растворяется в кипящих ароматических углеводородах и метил-
этилкетоне.
Полистирол в промышленности получают полимеризацией
стирола в блоке, эмульсии или суспензии. Полимеризацию осу-
ществляют периодическим или непрерывным способом.
Блочную полимеризацию стирола проводят в реакторах-фор-
полимеризаторах. В них процесс продолжается около 60 ч при
75—85 °C и завершается получением продукта, содержащего при-
мерно 30% полимера в стироле. Такой продукт поступает для
окончательной полимеризации в полимеризационные колонны,
состоящие из 6—8 царг. В -каждой царге поддерживается свой
температурный режим: в верхней — в пределах 80—85°C, в ниж-
ней— 212—215 °C. Теплота реакции полимеризации стирола со-
ставляет около 84 кДж/моль. Через рубашки царг циркулирует
дифенильная смесь. Расплавленный полистирол выдавливается
из колонны с помощью шнека в виде непрерывного стержня, ох-
лаждается в ванне и после измельчения в грануляторе посту-
пает на расфасовку. Продолжительность полимеризации 25—
30 ч. Производительность одной колонны 1 т/сут.
Более широко применяется эмульсионная полимеризация сти-
рола — латексная и суспензионная, которые различаются разме-
ром частиц полученного полимера. Инициаторами эмульсионной
полимеризации служат пероксид водорода, персульфаты калия
и аммония, эмульгаторами — мыла, некаль и другие соли суль-
фокислот.
Эмульсионную полимеризацию проводят в эмалированных
полимеризаторах емкостью 3—5 м3, снабженных паровой рубаш-
кой, рамной мешалкой и обратным холодильником (рис. V.3).
Водную фазу (pH = 3,5—7) из аппарата 1 подают в аппа-
рат 2, где перемешивают со стиролом. Полученную эмульсию
нагревают до 40 °C в нагревателе 3 и подают в систему после-
довательно работающих полимеризаторов 4. Полимеризацию
проводят при 95—96 °C в течение 7—8 ч до содержания мономе-
ра в полимере около 0,5%. Готовый латекс коагулируют при
120 °C в коагуляторе 6, после чего полимер осаждают, отделяют
на центрифуге 7, промывают водой, сушат горячим воздухом в
сушилках и отправляют на упаковку.
Суспензионную полимеризацию проводят аналогично эмуль-
сионной. Гранулы полимера образуются уже в процессе полиме-
236
Рис. V.3. Технологическая схема получения эмульсионного полистирола:
/ — аппарат для приготовления раствора эмульгатора; 2—аппарат для приготовления
эмульсии; 3 — нагреватель; 4 — полимеризатор; 5 — сборник латекса; 6 — коагулятор;
7 — центрифуга.
ризации. В качестве эмульгатора применяется поливиниловый
спирт. Полимеризацию проводят при 80 °C в течение 5 ч. Полу-
ченный полимер отделяют от маточника на центрифугах.
Полистирол является хорошим диэлектриком и поэтому ши-
роко используется для изготовления деталей радио-и электроап-
паратуры, а также оболочек высокочастотных кабелей. Благо-
даря высокой водо-, щелоче- и кислотостойкости из него «изго-
товляют лабораторную посуду, аккумуляторные баки, а также
различные изделия бытового назначения.
В строительстве, холодильном машиностроении и других от-
раслях широко применяется легкий материал — пенополисти-
рол, который получают двумя методами. По первому методу в
полимер вводят легко разлагающиеся вещества — порофоры, при
распаде которых выделяется газ и вспенивает материал. По
второму методу полимер растворяют в легколетучем раствори-
теле, и раствор (нагревают под давлением. После снятия давле-
ния масса вспенивается вследствие испарения растворителя.
В качестве порофоров применяют азотсодержащие соедине-
ния, например карбонат аммония, динитрил азобисизомасляной
кислоты и др.
Стирол легко вступает в сополимеризацию с другими моно-
мерами — бутадиеном, акрилонитрилом и т. д. Сополимеры сти-
рола с дивинилбензолом используются для получения ионооб-
менных смол.
При сополимеризации стирола с бутадиеном получается син-
тетический бутадиенстирольный каучук СКС, сополимеризацией
с акрилонитрилом — синтетический каучук СКН, применяемые
в качестве конструкционного материала при отделке кабин са-
молетов и автомобилей.
Большой интерес представляет тройной сополимер акрило-
нитрила, бутадиена, стирола (пластик АБС), называемый ударо-
237
прочным полистиролом. Он обладает высокой ударной проч-
ностью, повышенной жесткостью и химической стойкостью.
Ударопрочный полистирол получают прививкой стирола к
синтетическим каучукам, обычно к бутадиеновому или бутади-
енстирольному. Вначале готовят раствор каучка в стироле, кото-
рый подают в полимеризатор колонного типа (подобно процес-
су полимеризации стирола блочным методом). С низа колонны
расплавленный полимер непрерывно выдавливается шнеком,
проходит водяную ванну и поступает в гранулятор.
АБС-сополимер обладает повышенной прочностью к удару,
а также повышенной (по сравнению с блочным и эмульсионным
полистирол а мп) эластичностью.
2.2. Производство галогенсодержащих полимеров
Поливинилхлорид
Исходным сырьем для получения поливинилхлорида явля-
ется винилхлорид. Полимеризацию винилхлорида в промышлен-
ности осуществляют в основном двумя методами — блочным и
эмульсионным. Наибольшее распространение получил эмульси-
онный метод (латексный и суспензионный).
Полимеризация протекает по радикальному механизму. В ре-
зультате полимеризации звенья винилхлорида соединяются пре-
имущественно по типу «голова к хвосту»:
^СН2— СН—сн2—сн—сн2— сн^
I I I
С1 С1 С1
По мере протекания процесса скорость полимеризации винил-
хлорида постепенно увеличивается и по достижении 30—
40%-ного превращения мономера становится постоянной. К кон-
цу процесса, когда степень превращения винилхлорида состав-
ляет 75—80%, скорость заметно падает. Это явление автоката-
лиза носит название гель-эффекта. Возрастание скорости поли-
меризации объясняется появлением твердой фазы полимера, на
поверхности которого образуются активные центры, способные
продолжать реакцию. Полимер не растворяется, но набухает в
мономере. Это также оказывает влияние на скорость реакции,
ъак как в набухших частицах полимера подвижность молекул
мономера велика, а скорость передвижения макрорадикалов и
вероятность их столкновения и обрыва цепи мала: макроради-
калы играют роль активных центров. При высокой степени пре-
вращения мономеров вязкость системы увеличивается, что при-
водит к уменьшению скорости реакции.
Инициаторами полимеризации винилхлорида являются пер-
сульфаты калия, натрия и аммония, пероксид водорода, перок-
сид бензоила и др. Пероксид бензоила при нагревании до 30—
238
40°C легко разлагается с образованием свободных радикалов:
(С6Н5-СОО)2 ------------> с6Н5-СОО. +со2+ .С6Н5
Радикалы взаимодействуют с винилхлоридом, образуя растущий
радикал
СбН5—соо.+СН2=СНС1 ----> С6Н5—СОО—СН2—СНС1
который активирует другие молекулы винилхлорида, обеспечи-
вая рост цепи.
Суспензионная полимеризация проводится в присутствии пер-
оксида бензоила, не растворимого в мономере. При полимери-
зации получаются довольно крупные частицы полимера. Латекс-
ная полимеризация проводится в присутствии водораствори-
мых инициаторов — персульфатов и пероксида водорода. Поли-
мер получается в виде тонкой дисперсии. В качестве эмульгато-
ров латексной полимеризации используют некали (натриевые
соли алкилнафталинсульфокислоты), алкилсульфонаты и др.
При полимеризации используют дистиллированную или очищен-
ную ионитами воду.
Процесс проводят при температуре 30—60 °C и давлении
0,5—0,7 МПа. С повышением температуры средняя молекуляр-
ная масса полимера понижается. Процесс может проводиться
по периодическому или непрерывному способу. Периодическая
полимеризация проводится в горизонтальных вращающихся ав-
токлавах емкостью 10—15 м3, снабженных мешалкой и рубаш-
кой для обогрева и охлаждения. Коагуляцию латекса проводят
электролитами, полимер отделяют на центрифугах от маточни-
ка и сушат. Полимер можно сушить в сушильных камерах —
эмульсию после коагуляции впрыскивают через форсунки в ка-
меру, в которую подают горячий воздух при 150 °C для испа-
рения воды. Сухой порошок полимера пневмотранспортом на-
правляют в бункер для расфасовки.
На рис. V.4 приведена схема непрерывной полимеризации
винилхлорида. Водная фаза из емкости 1 непрерывно подается
в полимеризационные колонны 2 и 3, работающие последова-
тельно. Колонны снабжены лопастными мешалками и рубашка-
ми для отвода тепла реакции (около 92 кДж/моль). Первая
колонна охлаждается рассолом, вторая — водой. После колонн
латекс собирают в коагулятор 4, куда для коагуляции добав-
ляют раствор электролита. Эмульсию полимера подают в рас-
пылительную сушилку 5. Горячий воздух вместе с полимером на-
правляется в циклон. В циклоне полимер отделяется и поступа-
ет в бункер 7 для расфасовки, а воздух проходит через рукав-
ный фильтр 8 и выпускается в атмосферу. Рукавный фильтр
имеет приспособление для встряхивания. Ссыпающийся поро-
шок полимера поступает в бункер.
Поливинилхлорид — белый тонкий аморфный порошок, рас-
творимый при нагревании в хлорированных углеводородах.
239
Рис. V.4. Технологическая схема получения эмульсионного поливинилхло-
рида:
/ — емкость для приготовления эмульгатора; 2, 3 — полимеризаторы; 4 — коагулятор;
5 — сушилка; 6 — циклон; 7 — бункер для полимера; 8 — фильтр.
Поливинилхлорид применяется для изготовления деталей ма-
шин, кислотоупорных труб, футеровочных плит, пленок, электро-
изоляционных покрытий, заменителей кожи, водопроводных
труб, кровельного материала, настила для полов и облицовки
стен и др. Для технических целей поливинилхлорид выпускает-
ся неокрашенным. Декоративный пластикат (пластифицирован-
ный полимер) окрашивают в разные цвета.
В зависимости от назначения поливинилхлорид применяется
в пластифицированном и непластифицированном виде. В каче-
стве пластификаторов используются эфиры фталевой и себаци-
новой кислот. Все гибкие материалы — кабельную изоляцию,
пленку, искусственную кожу, обувь и т. д. изготовляют из плас-
тифицированного поливинилхлорида. Недостатком поливинил-
хлорида является низкий температурный предел эксплуатации
изделий (до 65 °C) и заметная ползучесть материала, находя-
щегося под нагрузкой.
Для повышения температуры плавления винилхлорид сопо-
лимеризуют с винилиденхлоридом СН2 = СС12. Сополимер ви-
нилхлорида с винилиденхлоридом имеет температуру размяг-
чения 185—200 °C, но уже при 150 °C наблюдается деструкция
сополимера с выделением НС1. Он плохо растворяется в орга-
нических растворителях. Переработка его в изделия затрудне-
на. Меняя соотношение мономеров, можно получить сополиме-
ры с различной температурой размягчения. Сополимер саран
с содержанием 70% винилиденхлорида используется для изго-
товления труб, жестких пленок, а также волокон для сетей,
канатов, материалов для обивки мебели.
Для получения антикоррозионных лаков и эмалей поливи-
нилхлорид подвергают дополнительному хлорированию. Кроме
240
того, из полученной перхлорированной смолы изготовляют во-
локно хлорин; для этого смолу растворяют в ацетоне и форму-
ют по мокрому способу. Хлорин применяется для изготовления
фильтрующих материалов (волокно не поглощает влаги, не
гниет, хорошо выдерживает действие кислот и щелочей), ков-
ров, а также медицинского белья. Недостатком хлорина являет-
ся его способность размягчаться при 75 °C.
Фторсодержащие полимеры
Политетрафторэтилен получается полимеризацией тетрафтор-
этилена при температуре 60 °C и давлении около 5 МПа в при-
сутствии инициаторов — персульфатов, пероксида водорода и
органических пероксидов. При полимеризации выделяется боль-
шое количество тепла (197 кДж/моль). Для облегчения отвода
тепла реакцию обычно проводят в водноэмульсионном слое, по-
лимер получается в виде порошка.
Политетрафторэтилен
F F F F
1111
С—с—
1111
F F F F
относится к числу термостойких полимеров; деструкция полиме-
ра наблюдается при 350—360 °C и сопровождается выделением
фтора.
Вследствие симметричного строения макромолекул и малого
размера атома фтора политетрафторэтилен имеет упорядочен-
ную структуру. Степень кристалличности полимера достигает
80—90%. При нагревании до 327 °C кристаллическая фаза рас-
плавляется, и полимер переходит в аморфное состояние. При
охлаждении происходит усадка полимера — плотность его по-
вышается с 1830 до 2300 кг/м3. Соотношение кристаллической и
аморфной фаз зависит от скорости охлаждения. Медленное ох-
лаждение приводит к повышенной кристалличности. Практи-
чески закалку осуществляют охлаждением нагретого до 350—
380 °C полимера в холодной воде.
Полимер представляет собой рыхлый порошок, спекающийся
при 360—380 °C. Изготовление из него изделий сильно затрудне-
но, так как политетрафторэтилен не растворяется ни в одном из
известных растворителей. Обычно для его переработки приме-
няются следующие методы: холодное таблетирование порошка,
спекание таблеток в стержни и охлаждение стержней, механи-
ческая обработка на станках.
Политетрафторэтилен используется при температурах от 300
до —200 °C. Он обладает исключительной химической стой-
костью. Политетрафторэтилен применяется как электроизоля-
ционный материал для высокочастотных кабелей, эксплуатируе-
мых в условиях действия повышенных температур (до 250°C).
241
Из него 'изготовляют трубы, прокладки, сальниковые набивки,
манжеты, детали насосов, фильтрующие перегородки, вкладыши
подшипников и др. Недостатками политетрафторэтилена явля-
ются ползучесть и малая твердость.
Политрифторхлорэтилен получают суспензионной полимери-
зацией трифторхлорэтилена CF2=CFC1. Он может эксплуати-
роваться в температурном интервале от 100 до —195°C. По хи-
мической стойкости он лишь немногим уступает политетрафтор-
этилену— при повышенной температуре разлагается хлорсуль-
фоновой кислотой и расплавами едких щелочей. При нагревании
политрифторхлорэтилен растворяется или набухает в ксилоле и
бензоле, что облегчает его переработку в изделия. Применяется
он для тех же целей, что и политетрафторэтилен. Кроме того, из
политрифторхлорэтилена получают лаковые покрытия путем на-
несения суспензии полимера в растворителе на металлы и дру-
гие материалы, которые выдерживают нагрев до 270 °C.
2.3. Производство полимеров эфиров
непредельных кислот и их производных
Полиметилметакрилат
Полиметилметакрилат представляет собой полимерный эфир
непредельной метакриловой кислоты.
Полимеризация метилметакрилата протекает под действием
света, пероксидов, персульфатов, гидропероксидов при нагрева-
нии:
пСН9=С
" I
соо—сн3
В промышленности для полимеризации метилметакрилата
применяются блочный и эмульсионный способы. Наибольшее
распространение получила блочная полимеризация. По этому
методу мономер смешивают с инициатором и проводят форполи-
меризацию (частичную полимеризацию) до получения сиропа,
который затем разливают в формы. Процесс проводят при 70 °C.
Формы изготовляют из стекла или металла. Расстояние между
стеклами равно толщине получаемого блока. Форму оклеивают
бумагой и в нее заливают (до краев) форполимер, после чего
полимеризацию завершают в термокамерах. Температура в тер-
мокамерах поддерживается от 50 до 100 °C. Чем ниже темпе-
ратура, тем медленнее протекает полимеризация, но тем боль-
ше молекулярная масса получаемого полимера. Полимеризация
сопровождается выделением тепла (545 кДж/кг). Скорость ре-
акции непостоянна, она возрастает при 20%-ной степени пре-
вращения мономера (гель-эффект). Вследствие этого полный от-
вод теплоты реакции затруднен — происходит резкое повышение
242
температуры, образование полимера с низкой молекулярной
массой и возникновение пузырей из-за местных перегревов.
При получении толстых листов и крупных блоков при поли-
меризации применяют различные окислительно-восстановитель-
ные системы, позволяющие проводить процесс при более низких
температурах.
Листы полимера после обрезки краев подвергают полировке.
Обрезки и брак используются для изготовления изделий мето-
дом литья под давлением.
Полиметилметакрилат применяют в качестве органического
стекла (оргстекло, плексиглас) для остекления автомобилей, са-
молетов, изготовления прозрачных деталей приборов, оптических
линз и других изделий. Полимер не имеет запаха и физиологи-
чески безвреден; он обладает высокой светопрозрачностью —
пропускает 73% ультрафиолетовых лучей. Детали из полиметил-
метакрилата свариваются при 200—225 °C в токе горячего воз-
духа. Недостатки его — небольшая поверхностная твердость
(стекло легко процарапывается) и невысокая теплостойкость —
до 100—110 °C. При нагревании до 300°C полиметилметакрилат
деполимеризуется с образованием мономера. Это свойство ис-
пользуется при утилизации полимерных отходов.
Полиакрилонитрил
Полимеризацию акрилонитрила проводят в присутствии пер-
оксидов, диазосоединений, персульфата аммония:
пСН2=СН
CN
СН8—СН—'
CN ]„
При полимеризации выделяется 73 кДж/моль тепла. Полиме-
ризацию акрилонитрила можно инициировать также металличе-
ским натрием, у-излучением, бутиллитием. Процесс осущест-
вляется в растворе толуола или в водном растворе. Для сокра-
щения индукционного периода в систему добавляют небольшое
количество нитрата серебра. Молекулярная масса получаемого
полимера 35 000—70 000.
Акрилонитрил растворяется в воде (7% при 20°C). На этом
основан метод полимеризации в водной эмульсии, растворах ми-
неральных солей.
В водной эмульсии процесс проводят при 30—75 °C и атмос-
ферном давлении. В качестве инициаторов используются пер-
сульфаты, пероксид водорода, окислительно-восстановительные
системы; в качестве эмульгаторов — соли сульфокарбоновых
кислот; регуляторами являются альдегиды, кеторы, меркапта-
ны. Степень превращения мономера в полимер 80—85%.
243
Полимеризацию акрилонитрила можно проводить в водных
растворах минеральных солей — хлоридов цинка, кальция, же-
леза и др. Регулирование роста цепи осуществляется введением
тиогликолевой кислоты. Процесс ведут до 70%-ной конверсии
мономера. Получаемый полимер растворяется в водном раство-
ре солей (роданида натрия). Из раствора можно формовать во-
локна и изготовлять пленки. Молекулярная масса полимера
40 000—45 000.
Полиакрилонитрил представляет собой аморфный полимер
с температурой размягчения 220—230 °C. Он не набухает и не
растворяется в обычных органических растворителях, но рас-
творяется в диметилформамиде. При нагревании до 270 °C про-
исходит его разложение с выделением аммиака и цианистого
водорода. Полимер используется для получения волокна нит-
рон, которое идет на изготовление тканей, трикотажных изде-
лий, искусственного меха. Изделия из нитрона сохраняют проч-
ность до 180—200 °C, они огнестойки и не поддаются гниению.
Акрилонитрил легко взаимодействует с другими мономерами.
В технике широко применяются его сополимеры с бутадиеном—
нитрильный синтетический каучук СКН, сополимеры с метилме-
такрилатом (пластик МС), сополимеры с винилацетатом, трой-
ные сополимеры с бутадиеном и стиролом (пластик АБС).
2.4. Производство поливинилового спирта
и его производных
Поливинилацетат
Винилацетат легко полимеризуется в жидкой и газовой
фазе при нагревании и облучении УФ-светом в присутствии ини-
циаторов и катализаторов с образованием полимера линейного
строения:
^СН2—СН-СН2—СН^
ОСОСНз ОСОСНз
Реакция сопровождается выделением большого количества теп-
ла (89 кДж/моль).
В промышленности полимеризацию проводят в растворе или
в эмульсии. Полимеризацию в растворе проводят в присутствии
инициаторов — пероксидов или порофора N, в качестве раство-
рителей применяют спирты, бензол, ацетон, этилацетат. Полу-
ченный раствор полимера используют как^таковой или из него
выделяют поливинилацетат. Процесс проводится периодическим
и~ непрерывным способами.
244
Поливиниловый спирт
Поливиниловый спирт получают омылением спиртового рас-
твора поливинилацетата щелочью:
сн3он
NaOH
^СН2-СН-СН2—сн^
I I
ОСОСНз ОСОСНз
СН2—СН-СН2-СН^ + СН3—COONa + СН3—СОО-СН3
ОН ОН
Поливиниловый спирт является водорастворимым полиме-
ром. Его водный раствор используется для производства ацета-
лей. При добавлении в раствор дибутилфталата получают пла-
стифицированные поливинилацетатные дисперсии, которые при-
меняют для нанесения покрытий, изготовления водных красок,
для пропитки бумаги, ткани, изготовления искусственной ко-
жи и т. д.
Твердый поливинилацетат имеет низкую водостойкость, не-
высокие твердость, морозостойкость и теплостойкость, изделия
из него обладают ползучестью. Он применяется в производстве
лаков, клеев. В пластифицированном виде поливиниловый спирт
обладает высокой эластичностью и применяется для изготовле-
ния бензо- и маслостойких шлангов.
Поливиниловый спирт применяется в основном в производ-
стве синтетического волокна. Для придания водостойкости во-
локно обрабатывают формальдегидом. В результате такой обра-
ботки происходит ацеталирование и частичное сшивание цепей.
Волокно из полимера получают методом мокрого прядения —
продавливанием спиртового раствора через ванну с водным рас-
твором сульфата натрия. Поливиниловый спирт физиологически
безвреден, широко используется в пищевой, фармацевтической
промышленности, в медицине. Применяется в качестве стабили-
затора эмульсий в процессах полимеризации.
Поливинилацетали
Важное промышленное значение имеют ацетали поливинило-
вого спирта — продукты его конденсации с альдегидами (фор-
мальдегидом, ацетальдегидом и масляным альдегидом) — поли-
винилформаль, поливинилэтилаль и поливииилбутираль:
СН2
+RCHO ^СН2—НС СН-СН2—СН^
^СН2-СН-СН,-СН-СН2-СН^ —-> | | |
I I 2 I -н2° О О он
он он он \ /
CHR
R=H, сн3, с3н7
245
Практически в поливинилацеталях содержатся ацетильные,
гидроксильные и ацетатные группы. Их соотношение и молеку-
лярная масса поливинилового спирта определяют свойства по-
ливинилацеталей. Поливинилацетали применяются для изготов-
ления лаков, эмалей, пленок (при добавлении пластификато-
ров). Наибольшее применение имеет поливинилбутираль, кото-
рый применяется для склеивания силикатных и органических
стекол, в производстве многослойного безосколочного стекла
«триплекс», для изготовления клеев БФ.
2.5. Производство простых полиэфиров
Полиформальдегид
Полиформальдегид относится к простым полиэфирам, мак-
ромолекулы которого имеют линейное строение:
^СН2-О-СН2-О-СН2-О^
Наибольший интерес представляет собой полимер со степенью
полимеризации выше 1000.
В промышленности полиформальдегид получают полимери-
зацией формальдегида в среде инертного растворителя в присут-
ствии ионного катализатора (гомополимер)
пНСНО----> [—СН2—О—]п
или сополимеризацией триоксана с диоксоланом в присутствии
эфирата фторида бора в бензоле (сополимер):
Н2С—(X Н2С-СН2
пО\ ^СН2 + т(/ | ---►
HjC—О' Н2С-0
---► [-СН2-О]зп-[-СН2-СН2-О-СН2-О-]т
При увеличении содержания сомономера в сополимере уве-
личивается содержание С—С-связей, повышается термостабиль-
ность, уменьшается степень кристалличности, температура плав-
ления, твердость.
Для получения высокомолекулярного гомополимера вначале
тщательно готовят формальдегид 50—60%-ной концентрации.
Затем из раствора отгоняют газообразный формальдегид и на-
правляют его в вымораживатель, где он освобождается от влаги.
Чистый газообразный формальдегид подают на полимеризацию,
которую проводят в уайт-спирте в присутствии стеарата каль-
ция. Полученную суспензию полимера разделяют на центрифу-
ге и полимер передают на ацетилирование (для предотвраще-
ния старения полимера). После ацетилирования полимер много-
кратно промывают и сушат. Товарные марки полимера содержат
антиокислители (например, дифениламин), стабилизаторы (на-
пример, диоксид титана) и иногда добавки полиамидов.
246
Полиформальдегид широко используется в качестве конст-
рукционного материала для замены цветных металлов и спла-
вов в автомобилестроении, приборостроении, электронике и дру-
гих отраслях промышленности. Литьем под давлением из него
изготовляют втулки, зубчатые колеса, шестерни, корпуса теле-
фонов, детали переключателей, краны, вентили, дверные ручки,
приборные щитки, шпули, катушки, валики для оснастки тек-
стильных машин.
§ 3. ПРОИЗВОДСТВО ПОЛИМЕРОВ
РЕАКЦИЯМИ ПОЛИКОНДЕНСАЦИИ
3.1. Производство фенолоформальдегидных
полимеров
Фенолоформальдегидные полимеры широко используются
для получения термореактивных пластических масс. Эти мате-
риалы отличаются высокой механической прочностью, малым
водопоглощением, хорошими диэлектрическими свойствами,
стойкостью к действию химических реагентов.
При взаимодействии эквимольных количеств фенола и фор-
мальдегида в кислой среде образуются о- и п-гидроксибензило-
вые спирты:
В щелочной среде при избытке формальдегида наряду с
гидроксибензиловыми спиртами образуются ди- и триметилоль-
ные производные фенола:
он он он
Метилольные производные в кислых средах легко взаимо-
действуют между собой и с фенолом, образуя полимер линейно-
го строения. При некотором избытке фенола получается ново-
лачная смола — продукт обшей формулы:
ОН
Новолачные смолы, или новолаки, получают при мольном со-
247
отношении фенола и формальдегида, равном 7 6. Они раство-
ряются в органических растворителях и используются в лако-
красочной промышленности.
При поликонденсации гидроксибензиловых спиртов и смеси
метилольных производных в щелочной среде происходит обра-
зование разветвленных низкомолекулярных полимеров (молеку-
лярная масса 700—1000) —резолов, или резольных смол:
ОН ОН ОН ОН
Поликонденсация протекает ступенчато и ее можно остановить
на любой стадии процесса.
Резольные смолы представляют собой твердую хрупкую мас-
су с температурой размягчения 65—75 °C. Они растворяются в
спирте, ацетоне, бензоле. Применяются для изготовления пресс-
порошков, текстолита и антикоррозионных лаков.
При нагревании резольных смол поликонденсация возобнов-
ляется, и резолы легко переходят в пространственные полиме-
ры — резиты:
Резиты утрачивают способность растворяться в растворите-
лях и плавиться, т. е. происходит переход термопластичных
смол в термореактивные. Резиты сохраняют термостойкость до
280 °C; при более высокой температуре начинается их деструк-
ция.
248
Новолачная смола может переходить в резольную при до-
бавлении формальдегида. Резольная смола может реагировать
с фенолом и переходить в новолачную.
Новолачные и резольные смолы применяются в производст-
ве различных предметов технического и бытового назначения.
Из сухих смол производятся пресспорошки. При приготовлении
пресспорошков из новолачной смолы в нее добавляют гексамети-
лентетрамин. Из пресспорошков изготовляют выключатели, ро-
зетки, канцелярские товары, приборы, корпуса телефонных ап-
паратов и репродукторов и т. д.
Смолы используются для изготовления высокопрочных сло-
истых материалов (текстолита, гетинакса, древесно-слоистых
пластиков, стеклотекстолита).
Для получения текстолита хлопчатобумажную ткань пропи-
тывают смолой, складывают в пакет и прессуют под давлением
8—10 МПа до отверждения смолы. При использовании в каче-
стве наполнителя асбеста получается материал, называемый
фаолитом, который применяется для изготовления кислотостой-
ких труб, химической аппаратуры и т. д.; при пропитке смолой
бумаги получается гетинакс, широко применяемый в радио- и
электропромышленности.
3.2. Производство сложных полиэфиров
Полиэтилентерефталат
Полиэтилентерефталат получают поликонденсацией диметил-
терефталата и этиленгликоля. Процесс протекает в две стадии.
Сначала при 180 °C в присутствии катализатора (РЬО,
Zn(CH3COO)2 и др.) осуществляют переэтерификацию — полу-
чают дигликолевый эфир терефталевой кислоты:
СН3—ООС—^~А~СОО—СН3 + 2НОСН2—сн2он -►
---> НОСН2-СН2—ООС—СОО—СН2-СН2ОН + 2СН3ОН
Затем полученный диэфир подвергают поликонденсации при вы-
сокой температуре (280 °C) в вакууме:
пНОСН2—СНа—ООС—СОО—СН2—СН2ОН -------►
---► I—О-СН2-СН2-ООС-/~Л-СО-1 + пНОСН2-СН2ОН
L \==/ Jn
Расплавленный полиэфир выдавливается через фильеры (по-
лучается нить) или щелевое отверстие (получается пленка) и
охлаждается водой.
Полиэтилентерефталат представляет собой кристаллический
полимер с температурой плавления 264 °C, нерастворимый в
17-254
24»
обычных органических растворителях. Он широко применяется
для изготовления синтетических волокон и пленок. Волокно лав-
сан получается формованием из расплава. Оно подвергается вы-
тяжке, совмещенной с предварительной круткой, затем оконча-
тельной крутке и фиксации (фиксация сводится к выдержке во-
локна при 80—120°C). Волокно лавсан обладает высокой тер-
мической стойкостью, стойкостью к светопогоде, истиранию, дей-
ствию окислителей, кислот и других реагентов (кроме горячих
щелочных растворов). Оно отличается хорошими диэлектриче-
скими показателями w практически не поглощает влагу. Из него
вырабатывают прочные и немнущиеся ткани, трикотаж. В тех-
нике из лавсана изготовляют прочные и долговечные канаты,
рыболовные сети, транспортерные ленты, ремни, брезенты,
фильтрткани и др.
Поликарбонаты
Поликарбонаты представляют собой полиэфиры угольной
кислоты и дигидроксисоединений. Наибольший интерес пред-
ставляют ароматические поликарбонаты, для получения кото-
рых главным образом используют дифенилолпропан.
Синтез поликарбонатов осуществляют поликонденсацией ди-
фенилолпропана с фосгеном
и переэтерификацией эфиров угольной кислоты:
СН3
пНО—С —-ОН + nRO—С— or
\=/ I \=/ II
СН3 О
Поликарбонаты обладают высокой ударной вязкостью, тепло-
стойкостью, хорошими диэлектрическими свойствами. Из них
изготовляют шестерни, втулки, смотровые стекла, катушки
электрообмотки, заклепки, гвозди, скобы, винты, клапаны. Из-
делия легко окрашиваются. Растворы поликарбонатов приме-
няют в качестве термостойких электроизоляционных лаков и
клеев.
250
Ненасыщенные полиэфиры
Из ненасыщенных полиэфиров наибольшее применение
имеют полиэфиры, получаемые полиэтерификацией малеиновой
кислоты с гликолями (моно- или диэтиленгликолем):
пНООС—СН=СН—СООН + мНОСН2—СН2ОН з=>
т—Н— [—ООС—СН=СН—СОО— СН2-СН2—]„
Полученный ненасыщенный линейный полиэфир сшивается по
двойным связям ненасыщенным соединением (например, стиро-
лом) для получения неплавкого полимера сетчатого строения.
Сшивание сопровождается отверждением полимера.
Ненасыщенные полиэфиры широко используются в качестве
связующего в производстве стеклопластиков. Стеклопластики
как конструкционные материалы имеют ряд преимуществ по
сравнению с металлами — они легки, прочны, стойки к вибра-
ционным нагрузкам и обладают хорошими теплоизоляционными
свойствами.
Из стеклопластиков можно изготовлять крупногабаритные
изделия: корпуса шлюпок и катеров, кузова автомобилей, от-
дельные части самолетов (части крыла, отсеки фюзеляжа, топ-
ливные баки и т. д.). В строительстве стеклопластики использу-
ются в качестве полупрозрачной кровли, для изготовления рако-
вин, дверей, оконных переплетов, труб и т.д. Они находят при-
менение также в производстве мебели.
3.3. Производство эпоксидных полимеров
Эпоксидные полимеры получают поликонденсацией эпихлор-
гидрина с дифенилолпропаном. Поликонденсация проводится
при температуре 60—70 °C в присутствии щелочи, добавляемой
для связывания хлористого водорода. В результате получается
полиэпоксид общей формулы:
Н2С СН—СН2— Г— OROCH2-CHCH2—1—ORO—СН2—НС-----СН2
Ан j„
сн,
В зависимости от молекулярной массы температура размяг-
чения получаемых смол изменяется от 20 до 155 °C. Смолы легко
растворяются в ацетоне, толуоле, хлорбензоле, метилэтилкето-
не. Наличие эпоксидных и гидроксильных групп придает им вы-
сокую реакционную способность. Они легко взаимодействуют с
полиаминами. При присоединении аминов быстро возрастает
молекулярная масса смолы, а взаимодействие с гидроксильны-
ми группами соседних макромолекул приводит к образованию
17* 251
пространственной сетки (отверждение). В качестве отвердите*
лей применяются гексаметилендиамин, гексаметилентетрамин,
термореактивные фенолоформ альдегидные смолы. При отверж-
дении диамином получается эпоксидная смола с температурой
стеклования 70—80 °C.
Эпоксидные смолы применяются для получения лакокрасоч-
ных покрытий, клеев, литых и слоистых пластиков и стекло-
пластиков. Эпоксидные лаки и эмали используются как элек-
троизоляционные материалы. Они отличаются высокой адгезией,
эластичностью, твердостью.
3.4. Производство полиамидов
Полиамиды представляют собой гетероцепные высокомоле-
кулярные соединения, содержащие в основной цегГи макромоле-
кулы амидные группы —NHCO—. Наибольшее практическое
применение получили полигексаметиленадипамид (полиамид 6,6,
анид), поликапроамид (полиамид 6, капрон), полигексаметилен-
себацинамид (полиамид 6, 10, найлон 6, 10), полиэнантоамид
(полиамид 7, энант), полидодекаамид (полиамид 12), из кото-
рых в основном получают синтетические волокна. Цифровые
обозначения у полиамидов означают число углеродных атомов
в мономере или в сомономерах.
Полигексаметиленадипамид
Полигексаметиленадипамид получается поликонденсацией
гексаметилендиамина с адипиновой кислотой:
nH2N— (CH2)e— NH2 + лНООС—(СН2)4—СООН ►
---► [_ NH-(CH2)e-NHCO-(CH2)4-COHn+ 2пН2О
Для получения полимера используют строго эквимольные коли-
чества адипиновой кислоты и гексаметилендиамина. Поэтому
сначала получают соль адипиновой кислоты и гексаметиленди-
амина (соль АГ) смешением метанольных растворов адипино-
вой кислоты и гексаметилендиамина. При охлаждении соль АГ
выпадает в виде порошка, который отделяют на центрифуге и
сушат. Температура плавления соли АГ равна 190 °C.
Поликонденсацию соли АГ проводят в расплаве в присутст-
вии небольших количеств уксусной или адипиновой кислоты,
играющих роль стабилизаторов, при 270 °C и давлении 1,5 МПа.
Затем воду удаляют и дальнейшую поликонденсацию проводят
в расплаве полиамида. По окончании поликонденсации расплав-
ленный полимер выдавливают через фильеру с двумя-тремя от-
верстиями. После затвердевания полимер подается на рубиль-
ную машину, где разрезается на мелкие кусочки (крошку).
252
Поликапроамид
Поликапроамид получается полимеризацией капролактама в
присутствии воды в качестве активатора:
н2о
HN-(CH2)5-CO --->- Н—[—NH—(СН2)б—СО-—]п—-ОН
Полимеризацию проводят в расплаве при температуре 250—
260°C и давлении 1,5—2,5 МПа в инертной среде (азоте). Про-
должительность полимеризации 8 ч. Процесс протекает по ме-
ханизму ступенчатой полимеризации.
Технологическая схема получения поликапроамида приведе-
на на рис. V.5. На схеме показан вариант гидролитической по-
лимеризации в присутствии раствора соли АГ.
Капролактам из бункера 1 подают в плавитель 2, где кап-
ролактам плавится и нагревается до 90—95 °C. Далее расплав
поступает в полимеризационную колонну 4. Раствор соли АГ
готовится в аппарате 3, Некоторое количество раствора непре-
рывно подается в колонну 4. Выделяющаяся в процессе поли-
конденсации вода собирается в сборнике 6. Расплавленный по-
лимер из колонны поступает под давлением в фильеру, откуда
его выдавливают через щель на холодную поверхность поливоч-
ного барабана 7. После охлаждения ленты или жгута полиами-
да с помощью 'направляющих валков 8 и тянущих валков 9 он
передается «на измельчение в машину 10, Крошка полимера со-
бирается в бункере 11, а затем поступает в промыватель-
Рис. V.5. Технологическая схема получения поликапроамида:
/—бункер капролактама; 2 — плавитель капролактама; 3—аппарат для приготовления
раствора соли АГ; 4 — полимеризацнонная колонна; 5 — конденсатор; 6 — сборник во-
ды; 7 — поливочный барабан; 8 — направляющие валки; 9 — тянущие валки; 10— реза-
тельная машина; // — бункер для крошки; 12 — промыватель-экстр актор.
экстрактор 12, в котором промывается горячей водой для удале-
ния капролактама и примесей.
Полиамиды представляют собой полимеры линейного строе-
ния с высокой степенью кристалличности. Они характеризуются
высокой прочностью к ударным нагрузкам, эластичностью, хо-
рошей масло- и бензостойкостью. Они не растворяются в обыч-
ных растворителях, а растворяются лишь в фенолах и концент-
рированных минеральных кислотах. При нагревании растворя-
ются в ледяной уксусной кислоте, формалине, бензиловом спир-
те. Стойки к холодным щелочам и органическим растворителям.
По механической прочности и прочности на истирание поли-
амидные волокна превосходят полиакрилонитрильные и поли-
эфирные волокна, но в мокром состоянии их- прочность пони-
жается.
Из полиамидов изготовляют подшипники, шестерни, зубча-
тые передачи, втулки, трубы, детали для радиотехнической ап-
паратуры. Полиамидные волокна используются для производ-
ства тканей, меха, ковров, кордного волокна.
3.5. Производство полиуретанов
Полиуретаны получаются при взаимодействии диизоциана-
тов с диолами:
nOCN—R—NCO + лНО—R'~ОН -----► ^С—NH—R—NH—С—О-R'—
II II
О О
Реакция протекает по механизму ступенчатой полимеризации
и сопровождается интенсивным выделением тепла.
В качестве исходного сырья применяют бутавдиол-1,4 и гек-
саметилендиизоцианат.
Полиуретаны на основе бутандиол а-1,4 являются линейны-
ми кристаллическими полимерами, близкими по свойствам к
полиамидам. Они обладают высокой прочностью, обусловленной
наличием водородных связей между карбонильными и имино-
группами соседних макромолекул. Уретановая группа
—NH—СОО—, входящая в главную цепь полимера, придает им
большую гибкость и снижает температуру плавления полимера.
Полиуретаны более стойки к действию кислот и окислителей,
чем полиамиды. Полиуретаны применяются для получения пле-
нок, волокон и универсальных клеев.
Для производства высокоэластичных пленок, защитных по-
крытий, прочной резины, пенопластов (жестких и эластичных)
применяют полиэфируретаны. Они также являются линейными
кристаллическими полимерами и имеют высокую прочность при
небольшой молекулярной массе.
Синтез полиэфируретанов проводят в две стадии. Вначале
получают низкомолекулярный полиэфир, а затем из этого поли-
эфира и диизоцианата получают полиэфируретан. Исходными
продуктами для синтеза полиэфиров служат адипиновая, себа-
254
циновая и терефталевая кислоты и моно-, ди- и триэтиленгли-
коли. Полиэфиры должны содержать гидроксильные группы по
обоим концам макромолекулы, поэтому реакцию полиэтерифи-
кации проводят в избытке гликоля. Реакция протекает с боль-
шой скоростью. Процесс можно вести даже при комнатной тем-
пературе. При повышении температуры процесса молекуляр-
ная масса полимера увеличивается.
В зависимости от природы исходных реагентов и строения
макромолекул полиуретаны могут быть термопластичными и
термореактивными, пластичными и хрупкими, мягкими и твер-
дыми. Линейные полиуретаны на основе низкомолекулярных
гликолей способны образовывать волокна. Широкое применение
находят полиуретановые эластомеры на основе простых и слож-
ных олигомерных полиэфирполиолов с молекулярной массой
1000—3000. Они отличаются высокой стойкостью к истиранию,
что особенно важно для шин, транспортерных лент, обуви и др.
В промышленности широко выпускаются насыщенные и не-
насыщенные уретановые каучуки. Насыщенные каучуки вулка-
низуют диизоцианатом, ненасыщенные — пероксидами или се-
рой. Полиуретаны широко применяются в производстве пено-
пластов.
Газонаполненные пенополиуретаны получают взаимодействи-
ем ди- или полиизоцианатов с простыми или сложными гидро-
ксилсодержащими полиэфирами в присутствии воды и катали-
затора. Вспенивающим агентом служит выделяющийся диоксид
углерода:
+н2о
RN=C=O -----> RNH—СООН ---► R—NH2 + COa
Пенополиуретаны могут быть эластичными (на основе поли-
эфиров линейного строения) и жесткими (на основе разветвлен-
ных полиэфиров). Плотность пенополиуретанов регулируют с
помощью добавляемой воды.
Распространенным эластичным пенополиуретаном является
поролон, получаемый на основе адипиновой кислоты, диэтилен-
гликоля с добавкой небольших количеств триметилолпропана и
смеси 2,4- и 2,6-толуилен-диизоцианатов (в соотношении
65 : 35), а также воды.
Эластичные полиуретаны применяются в производстве мяг-
кой мебели, губок, ковров, игрушек, сидений в автомашинах,
автобусах, самолетах.
Жесткие пенополиуретаны применяются как тепло- и звуко-
изоляционный материал, в авиа-, авто- и судостроении, холо-
дильном деле и т. д.
3.6. Производство кремнийорганических (полимеров
Кремнийорганические полимеры представляют собой высоко-
молекулярные соединения, содержащие в основной цепи атомы
кремния. Они обладают высокой стойкостью к действию повы-
255
шейных и пониженных температур, а также к действию света
и влаги. Кремнийорганические полимеры применяются в элек-
тротехнической, радиотехнической, авиационной, текстильной,
резиновой и других отраслях промышленности, в медицине,
быту.
Из кремнийорганических полимеров наибольшее практиче-
ское значение имеют полиорганосилоксаны (силиконы), в ко-
торых основная цепь состоит из атомов кремния и кислорода, а
боковое «обрамление» из органических радикалов:
R R R
^Si—О—Si—О—Si—О~^-
I I I
R R R
Исходным сырьем для синтеза кремнийорганических полимеров
являются алкил (арил) хлорсиланы и замещенные эфиры орто-
кремневой кислоты. В зависимости от числа атомов хлора ал-
кил (арил) хлорсиланы делятся на монофункциональные RaSiCl,
бифункциональные R2SiC12 и трифункциональные RSiCU.
Алкил- и арилхлорсиланы легко гидролизуются по схеме:
R R R
I +Н20 I +Н2о |
Cl—Si—С1 ——Г Cl—Si—ОН ——► НО—Si—ОН
। —на । —на ।
R R R
Получаемые силандиолы нестойки и легко вступают в реакцию
ступенчатой межмолекулярной конденсации с образованием ли-
нейных олигомеров и полимеров:
R R R R
I III
пНО— Si— ОН ->- -wSi—О— Si—О—Si— О^
Трифункциональные алкил (арил) хлорсиланы при гидролизе
образуют продукты более сложного состава, из которых образу-
ются неплавкие и нерастворимые полимеры.
В качестве побочных продуктов при конденсации силандио-
лов образуются циклические соединения:
3R2Si(OH)2
SiR
“H2° R2si siR2
V
Кислая среда при гидролизе способствует образованию цик-
лических олигомеров, нейтральная и щелочная среда — линей-
ных полимеров.
256
На практике процессы гидролиза и поликонденсации разде-
лены. Гидролизат промывают и направляют на поликонденса-
цию. Сырьем для получения кремнийорганических полимеров
служат также эфиры ортокремневой кислоты R2Si(OR)2.
Свойства полиорганосилоксанов зависят от длины органиче-
ского радикала и его природы, от соотношения R : Si. При уве-
личении размера R и соотношения R : Si повышается эластич-
ность и водостойкость, но снижается термостойкость и твердость.
Жидкие полимеры применяют в качестве смазок, гидравли-
ческих и гидрофобизирующих жидкостей. Пропитка тканей по-
лиорганосилоксанами придает им гидрофобные свойства.
Лаки применяют в качестве электроизоляционных материа-
лов, смолы — в качестве связующего в производстве стеклопла-
стиков.
Кремнийорганические каучуки эксплуатируются в интервале
температур от —50. до 250 °C. Они отличаются высокой проч-
ностью, обусловленной большой энергией связи Si—О
(374 кДж/моль).
Кремнийорганические полимеры можно модифицировать вве-
дением полиэфирных, фенолоформальдегидных и полиэпокснд-
ных олигомеров. При модификации значительно улучшаются
механические и другиё свойства полиорганосилоксанов.
ГЛАВА VI
ПРОИЗВОДСТВО НЕОРГАНИЧЕСКИХ
ПРОДУКТОВ
§ 1. ПРОИЗВОДСТВО СЕРЫ
Сероводород, полученный при очистке углеводородных газов,
можно непосредственно перерабатывать в серную кислоту либо
сначала получать из него свободную серу.
Для получения свободной серы сероводород сжигают до
диоксида серы (на !/з от общего его количества), который затем»
взаимодействуя с сероводородом, дает серу:
H2S + 1,5О2 ----► Н2О + SO2
SO2 + 2H2S -----►- 2Н2О + 3S
3H2S + 1,5O2 ---► 3H2O + 3S
ДЯ = —518,3 кДж/моль
ДЯ = —104,5 кДж/моль
ДЯ == —622,8 кДж/моль
Окисление сероводорода при 1000 °C протекает в отсутствие
катализатора. При 700 °C окисление происходит на катализа-
торах: силикагеле, глиноземе, боксите.
257
Рис. VI. 1. Технологическая схема получения серы из сероводорода:
/ — печь для сжигания сероводорода; 2— котел-утилизатор; 3, 6 — холодильники; 4 —
нагреватель; 5 — контактная печь; 7 — мультициклоны.
На рис. VI. 1 приведена схема получения свободной серы из
сероводорода. В обжиговую печь 1 подают кислые газы (H2S
и СО2) и воздух для окисления сероводорода. Температура в
печи около 450 °C. Продукты сгорания, содержащие диоксид
серы, сероводород, диоксид углерода, водяные пары, пары серы
и азот, охлаждаются в котле-утилизаторе 2 до 300 °C. Сера,
сконденсировавшаяся в топке печи 1 и котле-утилизаторе, сте-
кает в серопровод. Газы после котла-утилизатора охлаждаются
в холодильнике 3 до 140—160°C и поступают в нагреватель 4
(печь подогрева), в котором нагреваются до 280—340°C за счет
сжигания топливного газа или смеси сероводорода с воздухом.
В последнем случае состав газо-воздушной смеси регулируется
таким образом, чтобы не нарушалось требуемое соотношение
сероводорода и воздуха в основном потоке. После нагревателя
газы поступают в контактный аппарат 5, в котором на боксите
при 300—500 °C происходит образование серы из сероводорода.
При этом температура газов повышается на 10—20 °C.
После охлаждения газов в холодильнике 6 сера конденси-
руется и стекает в серопровод, а сера, остающаяся в газах,
улавливается в мультициклонах 7. Выход серы достигает 85%.
Степень конверсии сероводорода 96—97%. Сера перекачивается
в хранилище или ящики, в которых она затвердевает. Сера вы-
пускается трех сортов. Чистота первого сорта — 99,6%, второ-
го — 98,6%, третьего — 96,5%.
Из мультициклонов отходящие газы направляются в печь
дожита, где при 500—700 °C на бокситовой насадке оставшиеся
сера и сероводород сжигаются до диоксида серы. Дымовые га-
зы, содержащие кроме SO2 еще СО2, азот и водяные пары, вы-
брасываются в атмосферу через дымовую трубу с содержанием
диоксида серы не выше 0,2%. Дымовые трубы устанавливают
высотой 50—80 м во избежание загрязнения диоксидом серы
воздуха в прилегающих к заводу районах. В последние годы
дымовые газы подвергают очистке с целью извлечения кислых
компонентов.
258
§ 2. ПРОИЗВОДСТВО СЕРНОЙ кислоты
Процесс получения серной кислоты из серы включает сле-
дующие основные стадии:
получение диоксида серы:
S + О2 --► SO2 ДЯ = —346 кДж/моль
окисление диоксида серы в триоксид:
SO2 + 0,5О2 -► SO3 ДЯ = —95,7 кДж/моль
поглощение триоксида серы водой (или серной кислотой).
Триоксид серы хорошо растворяется в серной кислоте, по-
этому можно получать олеум с любым содержанием свободно-
го SO3.
Окисление диоксида серы в триоксид протекает медленно.
Поэтому на практике окисление проводят в присутствии катали-
заторов, вследствие чего этот метод производства серной кис-
лоты получил название контактного.
Применение серы вместо флотационного колчедана сущест-
венно упрощает процесс, так как диоксид серы практически не
содержит примесей. Перед подачей в печь серу плавят и подают
в печь через форсунку. Сера испаряется и сгорает в газовой
фазе. Процесс проводят в избытке воздуха; при этом обжиговый
газ содержит около 12% SO2. Температура горения серы в воз-
духе 1240 °C. Окисление диоксида серы в триоксид протекает
при 450—600 °C в присутствии катализаторов — платины и пен-
токсида ванадия, содержащего некоторые добавки, которые по-
вышают активность катализатора, его термическую и механиче-
скую прочность. Более активным катализатором является пла-
тина, однако ванадиевые катализаторы имеют более низкую
стоимость. Сильным ядом для катализатора являются оксиды
мышьяка, так как отравление триоксидом мышьяка необратимо,
а пентоксид мышьяка, накапливаясь на катализаторе, заметно
снижает его активность. Поэтому газ нужно очищать от мышь-
яковистых соединений.
Скорость окисления диоксида серы в триоксид зависит от
температуры процесса, однако при высокой температуре конвер-
сия диоксида серы в триоксид снижается. Поэтому на практике
поступают следующим образом. В контактную камеру вводят
диоксид серы при 450 °C. После первой зоны катализатора газ
нагревают до 600 °C за счет теплоты реакции. Затем газы ох-
лаждают в котле-утилизаторе до 450 °C и пропускают через вто-
рую зону катализатора. При таком режиме время контакта со-
ставляет 4 с; степень конверсии достигает примерно 90% после
первой зоны и 99,5%—99,7% — после второй.
Схема процесса приведена на рис. VI. 2. Сера из бетонного
плавильника 2 подается в форсунки печи 3 для сжигания, куда
259
Рис. VI.2. Технологическая схема получения серной кислоты методом сухо-
го катализа:
/ — сушильная башня для воздуха; 2 — плавильник серы; 3 — печь для сжигания серы;
4, 6 — котлы-утилизаторы; 5 — контактный аппарат; 7 — теплообменник; 8, 12, 14—хо-
лодильники; 9 — олеумный абсорбер; 10 — моногидратный абсорбер; //-—емкость для
моногидрата; 13 — емкость для олеума.
поступает воздух, высушенный моногидратом* в сушильной баш-
не 1. Газы, выходящие из печи, охлаждаются до 450 °C в котле-
утилизаторе 4 и направляются в контактный аппарат 5, Пройдя
первую зону катализатора, газы охлаждаются в выносном кот-
ле-утилизаторе 6, затем проходят вторую зову катализатора,
охлаждаются в воздушном теплообменнике 7 и в холодильни-
ке 8 и поступают вначале в олеумный абсорбер 9, а затем в
моногидратный абсорбер 10. Абсорбция серного ангидрида про-
текает с поглощением тепла, поэтому циркулирующие олеум и
моногидрат охлаждаются в холодильниках 12 и 14. Часть моно-
гидрата циркулирует через сушильную башню 1. Кислота на-
сыщается до заданной концентрации свободного SO3, после че-
го олеум направляется на склад.
Методом сухого катализа (в контактный аппарат не посту-
пают водяные пары) обычно ’получают олеум; для этого уста-
навливается специальный олеумный абсорбер, в котором погло-
щается 40—60% SO3. По мере повышения концентрации олеу-
ма его разбавляют моногидратом, а накапливающийся избыток
отводят на склад. Необходимое количество моногидрата в уста-
новке поддерживается путем ввода его извне, например с сер-
нокислотного производства.
Контактные аппараты с выносными теплообменниками про-
сты и удобны в эксплуатации. Число слоев катализатора, рав-
ное 3—5, обеспечивает высокую степень конверсии SO2.
* В сернокислотном производстве кислоту, орошающую абсорбер, на-
зывают моногидратом, хотя содержание кислоты в ней менее 100%.
260
Для уменьшения потерь SO3 с отходящими газами и защиты
атмосферы от вредных выбросов процесс проводят с большой
степенью поглощения. Полнота абсорбции зависит от концент-
рации серной кислоты. При концентрации кислоты ниже 98,3%
равновесное давление SO3 мало, а для воды — значительно.
Десорбция молекул воды происходит с поверхности серной кис-»
лоты. Основная масса SO3 абсорбируется серной кислотой, но
часть молекул SO3 встречается с молекулами воды, испаривши-
мися с серной кислотой, и образует пары серной кислоты.
Пары конденсируются, образуя туман, который плохо улавли-
вается и уносится с отходящими газами в атмосферу. Наиболь-
шую абсорбционную способность имеет 98,3%-ная серная кисло-
та, что обеспечивает меньшие потери SO3 и выбросы в атмосфе-
ру тумана серной кислоты.
Объем производства серной кислоты из серы в СССР и за
рубежом продолжает возрастать; такая тенденция сохранится и
в будущем. Поскольку в процессе используется чистая сера с
теплотой сгорания 10 000 кДж/кг, на таких установках можно
устанавливать газовые или паровые турбины с генераторами,
которые будут вырабатывать электроэнергию. После использо-
вания тепла газов они поступают на производство серной кис-
лоты. Такое комбинированное производство называется энерго-
технологическим — одновременно получается серная кислота и
«товарная» электроэнергия.
Совершенствование процесса производства серной кислоты
связано с увеличением мощности агрегатов, повышением давле-
ния в печах для окисления SO2. Окисление протекает с умень-
шением объема: увеличение давления способствует сдвигу рав-
новесия в сторону образования SO3. Целесообразно при окисле-
нии вместо воздуха применять кислород, что помогает разгружать
аппаратуру от балластного азота, существенно увеличи-
вает производительность установок, улучшает технико-экономи-»
ческие показатели производства серной кислоты.
Сероводород можно перерабатывать в серную кислоту мето-
дом мокрого катализа, при котором диоксид серы, полученный
сжиганием H2S, окисляется на ванадиевом катализаторе в три-
оксид серы. Газ с диоксидом серы содержит значительное коли-
чество паров воды (отсюда название — метод мокрого ката-
лиза).
Схема производства серной кислоты из сероводорода приве-
дена на рис. VI.3. Сероводород кон центр ацией 80—90% вместе
с воздухом поступает в печь /, в которой сгорает при 900—
1100 °C. Из печи газы поступают в котел-утилизатор 2, где ох-
лаждаются до 450 °C. Окисление диоксида серы в триоксид про-
исходит в четырехслойном контактном аппарате 3.
На второй,, третий и четвертый слои катализатора подают
воздух для обеспечения более полной глубины окисления. Из
контактного аппарата триоксид серы вместе с серной кислотой
направляется в абсорбер 4, орошаемый 90—94%-ной серной
261
Рис. VI.3. Технологическая схема получения серной кислоты методом мокро-
го катализа:
/ — печь для сжигания сероводорода; 2 — котел-утилизатор; 3 — контактный аппарат;
4 — абсорбер; 5 — емкость для серной кислоты; 6 — холодильник; 7 — электрофильтр.
кислотой. Серная кислота образуется за счет воды, поступаю-
щей в контактный аппарат вместе с диоксидом серы. В абсор-
бере триоксид серы превращается в серную кислоту. Основная
масса образующейся кислоты поглощается в абсорбере 4, а кис-
лота, унесенная с газом, улавливается в электрофильтре 7, от-
куда стекает в емкость 5. Из емкости кислота насосом подается
на орошение абсорбера через холодильник 6 для отвода тепла
поглощения и поддержания температуры в абсорбере не выше
60 °C. Часть кислоты отводится на склад.
Капитальные затраты на строительство сернокислотного це-
ха завода, перерабатывающего 6 млн. т сернистой нефти с со-
держанием 1,5% серы, в 2—3 раза ниже, чем затраты на строи-
тельство завода такой же мощности, работающего на колчедане.
§ 3. ПРОИЗВОДСТВО ВОДОРОДА
Водород необходим прежде всего для синтеза аммиака. По-
скольку в стоимости аммиака более половины приходится на
водород, получение дешевого водорода имеет большое зна-
чение.
Поиски новых путей получения водорода обусловлены не
только увеличением производства аммиака и внедрением про-
цесса гидрокрекинга, гидроочистки, но и развитием производст-
ва нефтехимических продуктов, осуществляемым на основе ок-
сида углерода и водорода (метанол, спирты оксосинтеза и
т. д.), а также процессов гидродеалкилирования (с целью полу-
чения бензола и нафталина) и гидрирования бензола (для по-
лучения циклогексана).
262
Наиболее перспективным методом получения водорода явля-
ется конверсия метана. Конверсия метана до водорода осуще-
ствляется в две стадии. Первая стадия — конверсия метана на
никелевом катализаторе при 850—900 °C:
СН4 + Н2О ► СО + ЗН2 ДЯ = 206 кДж/моль
Вторая стадия — конверсия оксида углерода при 410—500 °C на
железо-хромовом катализаторе:
СО + Н2О --► СО2 + Н2 ДЯ = —38 кДж/моль
На промышленных установках первая стадия конверсии осу-
ществляется в трубчатых печах (рис. VI.4). Природный газ на-
гревается в теплообменнике 3 до 300 °C за счет тепла конверти-
рованного газа и поступает в абсорбер /, заполненный железо-
содовой массой и твердым поглотителем* предназначенными
для очистки от серы. После сероочистки газ смешивается с во-
дяным паром в соотношении 1 : 2,2. Паро-газовая смесь с тем-
пературой 365°C 'направляется в печь 2, имеющую 44 радиант-
ные трубы, которые заполнены катализатором. Конверсия про-
текает при 850 °C.
Конвертированный газ поступает в пароперегреватель 4, в
котором он нагревает пар до 400 °C, затем в теплообменник 3
и с температурой 420 °C проходит в конвертор оксида углеро-
да 5. Дымовые газы из печи, нагретые до 900 °C, направля-
ются в котел-утилизатор 6, в котором образуется насыщенный
пар. Пар поступает в пароперегреватель 4. Выходящий из па-
роперегревателя пар с температурой 400 °C используется для
конверсии углеводородных газов. В конвертор для конверсии
СО также добавляется пар с температурой 400 °C.
После конвертора газы поступают в котел-утилизатор 6, ох-
лаждаются до 120 °C и проходят в конденсационную башню,
орошаемую водой; оттуда газ при 30 °C подается на моноэтанол-
Рис. VL4. Технологическая схема получения водорода:
/ — адсорбер; 2 — печь; 3 — теплообменник; 4 — пароперегреватель; 5 — конвертор; 6 —
когел-утилизатор; 7 — конденсационная башня.
263
Рис. VI.5. Технологическая схема получения азото-водородной смеси:
/ — теплообменник; 2 — увлажнитель; 3 — конвертор метана; 4 — конвертор оксида уг-
лерода; 5 — котел-утилизатор; 6 — подогреватель воды; 7 — конденсационная башня;
Л—абсорбер диоксида углерода; 9 — десорбер; 10— холодильник; // — подогреватель
этаноламина.
аминовую очистку от СО2. В результате получается водород
95-^96%-ной чистоты.
При проведении конверсии метана под давлением достига-
ется более полное использование тепла конвертированного газа
в виде пара высокого давления, уменьшаются размеры обору-
дования и энергетические затраты на сжатие водорода.
Если вместо кислорода используют воздух -или кислородо-
воздушную смесь, содержащую 38% кислорода, получается
азото-водородная смесь, пригодная для синтеза аммиака. Кон-
версию можно проводить при повышенном и атмосферном дав-
лении. Схема процесса при атмосферном давлении приведена
на рис. VI.5.
Метан подогревают в теплообменнике 1 за счет тепла кон-
вертированного газа. Перед входом в теплообменник к газу до-
бавляют горячую воду и насыщают его до соотношения газа и
пара 0,9 : 1. Паро-газовая смесь нагревается до 600 °C и прохо-
дит с кислородо-воздушной смесью в конвертор 3. Конверсия
протекает ма никелевом катализаторе при 850—900 °C. Из кон-
вертора газовая смесь поступает в увлажнитель 2, в котором
она охлаждается водой до 700 °C, а затем поступает в теплооб-
менник, где охлаждается до 400 °C. После этого газовая смесь
направляется .в конвертор оксида углерода 4. Температура в
конверторе регулируется путем впрыска различного количества
конденсата в увлажнитель и изменением подачи пара в конвер-
тор. Катализатор (железо-хромовый) помещается в двух сек-
циях конвертора.
Газ из конвертора поступает в котел-утилизатор 5, в кото-
ром получается пар давлением 0,5 МПа. Затем он направляется
в подогреватель воды 6, нагревая воду до 85 °C. Окончательное
охлаждение газа до 30 °C происходит в конденсационной баш-
не 7 путем орошения ее оборотной водой. Охлажденный кон*
264
вертированный газ подается в абсорбер 8 для очистки от диокси-
да серы раствором моноэтаноламина. Азото-водородная смесь
направляется на синтез аммиака. Раствор моноэтаноламина,
выходящий из абсорбера, регенерируется в десорбере 9, пройдя
предварительно подогреватель 11, в котором он нагревается до
100—110 °C. Диоксид углерода сбрасывается в линию инертного
газа или в дымовую трубу, а раствор этаноламина охлаждается
в холодильнике 10 и вновь подается в абсорбер
Представляет интерес процесс конверсии метана в углерод и
водород по уравнению:
СН4 (газ) -► С (тв) + 2Н2 (газ)
Углерод сжигают до СО и СО2 и таким путем получают тепло,
необходимое для процесса. Предполагают, что это процесс ста-
нет источником получения дешевого водорода.
§ 4. ПРОИЗВОДСТВО АММИАКА
Синтез аммиака протекает по уравнению:
N2+3H2 ч=± 2NH3 + Q
Необходимый для производства азот получают из воздуха, либо
используют азото-водородную смесь с установок паро-кислород-
ной конверсии метана.
Синтез аммиака является обратимым процессом, который
протекает с уменьшением объема и выделением тепла. Для
сдвига равновесия в сторону образования аммиака необходимо
повышать давление и понижать температуру. На практике про-
цесс проводят в присутствии катализаторов (железа, вольфра-
ма, марганца, платины и др.) при 400 °C и выше. Активным яв-
ляется катализатор, полученный сплавлением Fe3C>4 с актива-
торами (А120з, К2О, CaO, SiO2) и последующим восстановле-
нием железа до металлического. Он стоек к перегревам и менее
чувствителен к примесям.
В промышленности процесс осуществляется при низком
(10—15 МПа), среднем (25—60 МПа) и высоком (60—
100 МПа) давлениях. Чаще процесс проводят при среднем дав-
лении, при котором обеспечивается достаточно высокая скорость
процесса и значительно упрощается стадия выделения аммиака.
Тепловой эффект реакции при 500 °C и 30 МПа равен
55,8 кДж/моль, при 60 МПа — 61,4 кДж/моль аммиака. Объ-
емные скорости составляют 15 000—30 000 ч-1. Конверсия газов
за проход низкая, поэтому установки работают с рециклом
азото-водородной смеси. Концентрация аммиака в продуктах
синтеза зависит от объемной скорости подачи газов — при
18—254 265
Рис. VI.6. Технологическая схема получения аммиака при среднем давлении:
/ — котел-утилизатор; 2 — колонна синтеза; 3 —водяной холодильник; 4—сепаратор;
5 — турбокомпрессор; 6 — конденсационная колонна; 7—аммиачный холодильник.
500°С увеличение объемной скорости с 15000 до 60 000 ч-1 при-
водит к снижению концентрации аммиака в реакционной смеси
с 20% До 17%, но повышает съем аммиака с 1 м3 катализато-
ра. Для уменьшения содержания инертных примесей в цирку-
ляционном газе часть его выводится из цикла в виде отдувоч-
ного газа.
Технологическая схема получения аммиака при среднем дав-
лении приведена на рис. VI.6. Азото-водородная смесь проходит
вначале через конденсационную колонну 6, откуда подается
сверху вниз в колонну синтеза 2. Теплота реакции отводится за
счет котла-утилизатора /, через который при температуре
400 °C проходит азото-водородо-аммиачная смесь, охлаждается
и возвращается в колонну синтеза. В теплообменнике, смонти-
рованном внутри колонны, смесь охлаждается до 90—100 °C и
поступает в водяной холодильник 3, а далее в сепаратор 4,
В водяном холодильнике при 30 МПа конденсируется только
часть аммиака. Газ турбокомпрессором 5 подается в конден-
сационную колонну 6, часть аммиака из колонны циркулирует
через аммиачный холодильник 7, в котором охлаждается и воз-
вращается в конденсационную колонну, что способствует лучше-
му выделению аммиака из газовой смеси. Жидкий аммиак из
сепаратора и с низа конденсационной колонны поступает на
склад.
При обычных условиях аммиак представляет собой газ с
характерным резким запахом; сжижается он при —33°C. Ам-
миак хорошо растворяется в воде — при 20 °C и атмосферном
давлении в 1 л воды растворяется 700 л газа. В органических
растворителях он растворяется хуже.
Единичные мощности агрегатов достигают 2000 т/сут. Перс-
пективы дальнейшего совершенствования процесса получения
аммиака связаны с разработкой низкотемпературных катализа-
торов, обладающих активностью при 300 °C и ниже и давлении
5—10 МПа.
266
Представляет интерес создание энерготехнологических уста-
новок, на которых тепло процесса применяется для получения
пара, используемого в паровых турбинах для выработки элек-
троэнергии.
§ 5. ПРОИЗВОДСТВО КАРБАМИДА
Карбамид (или мочевина) CO(NH2)2 представляет собой
диамид угольной кислоты. Он является концентрированным
азотным удобрением (содержит 46% азота), хорошо усваивае-
мым растениями. Он сохраняется в почве довольно продолжи-
тельное время, что позволяет его использовать в районах по-
ливного земледелия и обильных осадков; эффективен для овощ-
ных, плодовых культур, хлопчатника, кукурузы. Карбамид об-
разуется в живом организме за счет аммиака, который выделя-
ется при распаде белка и окислении аминокислот. Образова-
ние карбамида предотвращает накопление аммиака в орга-
низме. В промышленности его получают из аммиака и СО2.
Карбамид используют также для получения продуктов фор-
мальдегидной конденсации — смол, лаков, клеев. Он применя-
ется и в синтезе лекарственных веществ, фармацевтических пре-
паратов и косметических средств. В нефтепереработке карбамид
используется при депарафинизации дизельных топлив. Карба-
мид является хорошей добавкой в корм жвачным животным.
Получение карбамида из аммиака и диоксида углерода от-
носится к важнейшим химическим пройзводствам. Процесс про-
текает в две стадии. На первой стадии происходит образование
карбамата аммония
2NH3 + СО2 NH2COONH4 ДЯ = —159 кДж/моль
на второй — отщепление молекулы воды от карбамата с полу-
чением карбамида и воды:
NH2COONH4 Н2О + NH2—СО—NH2 ДЯ = 28,5 кДж/моль
Тепло, выделяющееся при сильно экзотермической реакции об-
разования карбамата аммония, расходуется на проведение эндо-
термической реакции его дегидратации.
Производство включает синтез карбамида, дистилляцию пла-
ва, переработку раствора в кристаллический карбамид. На рав-
новесие и скорость реакции получения карбамида оказывают
влияние давление, температура, состав реакционной среды. Так
как процесс протекает с уменьшением объема, то при повыше-
нии давления увеличивается равновесный выход карбамида.
Избыток аммиака смещает равновесие в сторону образования
карбамида, так как он связывает воду, образующуюся при де-
гидратации карбамата аммония. Кроме того, при избытке ам-
миака уменьшается коррозия аппаратуры. Скорость процесса,
в частности второй, лимитирующей, стадии, резко возрастает с
повышением температуры от 140 до 180 °C.
18’
267
Синтез проводится при температуре 180—200 °C и давле-
нии 18—20 МПа. Осуществление процесса осложняется силь-
ным корродирующим действием реагентов, особенно карбамата
аммония. Поэтому применяется аппаратура, изготовленная из
специальных (преимущественно аустенитных) сталей, содержа-
щих хром, никель и медь.
Реактор для проведения синтеза имеет внутренние стаканы,
которые служат реакционной зоной и препятствуют соприкосно-
вению продуктов реакции со стенками реактора. Стаканы изго-
товляются из высоколегированных сталей, а корпус реактора —
из более дешевой легированной стали. Между стаканом и кор-
пусом существует кольцевое пространство, в которое подается
жидкий аммиак. Пройдя кольцевое пространство, аммиак посту-
пает в стакан, в котором происходит реакция. Благодаря этому
коррозии подвергается только стакан, который периодически
заменяется; таким образом сохраняется корпус реактора.
Степень конверсии реагентов за один проход составляет 35—
40%. Диоксид углерода и аммиак, не вступившие в реакцию,
после их отделения от карбамида и карбамата аммония вновь
возвращают в процесс.
Существуют различные схемы получения карбамида — с
большим и небольшим избытком аммиака, с одно- и двухступен-
чатой дистилляцией плава карбамида, а также с циркуляцией
не вступивших в реакцию газов и без циркуляции — в этом слу-
чае не вступивший в реакцию аммиак поглощается азотной кис-
лотой с образованием аммиачной селитры. При небольшом из-
бытке аммиака установки работают без циркуляции газов.
Избыточный аммиак поглощается азотной кислотой. Выход ам-
миачной селитры составляет 5—5,7 на 1 т карбамида.
Существенным недостатком является получение 60%-ных
растворов карбамида, так как для производства кристалличе-
ской селитры упариванием затрачивается большое количество
тепла.
Процесс с двухступенчатой дистилляцией плава является бо-
лее совершенным, поскольку исходные реагенты используются
более полно. При двухступенчатой дистилляции плава аммиак
после первой ступени возвращается в колонну синтеза, а не-
большая часть ам!миака, выделяющаяся во второй ступени, по-
глощается азотной кислотой. Выход получаемой при этом ам-
миачной селитры составляет 2—2,5 т на 1 т аммиака.
На рис. VI.7 приведена схема получения карбамида из ам-
миака и диоксида углерода с двухступенчатой дистилляцией
плава, с циркуляцией газа и кристаллизацией плава для полу-
чения кристаллического продукта.
Аммиак и диоксид углерода подаются в колонну синтеза 1.
Процесс осуществляется со 100%-ным избытком NH3. В колонне
синтеза поддерживается температура 180—200 °C и давление
18—20 МПа. Давление дросселируется до 1,6 МПа и продукты
поступают в колонну дистилляции первой ступени 2. Газооб-
268
Рис. VI.7. Технологическая схема получения карбамида:
/ — колонна синтеза; 2 — колонна дистилляции первой ступени; 3 — абсорбер; 4— водя-
ной холодильник; 5 — аммиачный холодильник; 6 — колонна дистилляции второй ступе-
ни; 7 — сборник плава карбамида; 6 — фильтр-пресс; 9 — шнековый кристаллизатор;
10 — центрифуга; // — сборник маточного раствора; 12 — выпарной аппарат; 13 — сепа-
ратор; 14—сборник раствора карбамида.
разные продукты с верха колонны поступают в абсорбер 3, оро-
шаемый водой и жидким аммиаком. В абсорбере происходит
конденсация паров воды и части паров аммиака и растворение
в аммиачной воде солей аммония. Водная фаза стекает в ко-
лонну 2, а газообразный аммиак с верха абсорбера поступает
сначала в водяной холодильник 4, а затем в аммиачный холо-
дильник 5. Часть жидкого аммиака подается на орошение аб-
сорбера, а часть возвращается в колонну синтеза. Раствор кар-
бамида и солей аммония из колонны 2 поступает в колонну ди-
стилляции второй ступени 6, работающую под давлением 0,03—
0,05МПа. Плав нагревается глухим и острым паром, в результа-
те чего весь карбамат аммония разлагается. Выделяющиеся
NH3 и СО2 с верха колонны поступают на разделение; аммиак
возвращается в процесс или передается на переработку в суль-
фат аммония или аммиачную селитру.
Водный раствор карбамида — плав (примерно 60%-ный)—
собирается в сборнике 7, откуда через фильтр-пресс 8 поступает
в шнековый кристаллизатор 9. В кристаллизатор подастся хо-
лодная вода и холодный воздух для охлаждения раствора кар-
бамида до 30—40 °C. Карбамид выкристаллизовывается; обра-
зующаяся пульпа поступает на центрифугу 10. Кристаллический
карбамид с 0,4% влаги поступает па упаковку, а маточный
раствор собирается в емкости 11, из которой направляется в
выпарной аппарат 12. Упаренный раствор карбамида собира-
26£>
ется в емкости 14, а затем возвращается в кристаллизатор. Вы-
парной аппарат работает при 80—90 °C и остаточном давлении
80—93 кПа. Повышение температуры недопустимо, так как при
этом может произойти разложение карбамида. Отсасываемые
водокольцевым ротационным компрессором пары воды через се-
паратор 13 (называемый иногда соковой камерой) проходят в
холодильник 15, в котором конденсируются. Вода нейтрали-
зуется и сбрасывается в канализацию.
Процесс производства по замкнутому циклу позволяет полу-
чать карбамид более низкой стоимости. Для получения карба-
мида в гранулированном виде упаренный раствор направляют
в грануляционную башню, в которой с помощью форсунок или
роторного распылителя поток дробится на мелкие капельки и
опускается вниз в восходящем потоке воздуха. Гранулирован-
ный продукт с низа башни по транспортеру подается на склад.
ЛИТЕРАТУРА
Белов И. С. Основы технологии нефтехимического синтеза. М.. Химия, 1966^
378 с.
Паушкин Я. М., Адельсон С. В., Вишнякова Т. П. Технология нефтехимиче-
ского синтеза. М.., Химия, 1973, Ч. 1. 448 с. 1975. Ч. 2. 352 с.
Лебедев Н. Н. Химия и технология основного органического и нефтехимиче-
ского синтеза. 3-е изд. М., Химия, 1981. 608 с.
Юкельсон И. И. Технология основного органического синтеза. М., Химия,.
1968. 847 с.
Лебедев Н. Н., Манаков М. Н., Швец В. Ф, Теория технологических процес-
сов основного органического и нефтехимического синтеза. М. Химия, 1975.
478 с.
Черный И. Р. Производство мономеров и сырья для нефтехимического син-
теза. М., Химия, 1973. 264 с.
Неволин Ф. В. Химия и технология синтетических моющих средств. М., Пи-
щевая промышленность, 1971. 424 с.
Общая химическая технология/Под рел А. Г. Амелина. М., Химия, 1977.
400 с.
Технология пластических масс/Под ред. В. В. Коршака. 2-е изд. М., Химия,
1976. 608 с.
Литвин О. Б. Основы технологии синтеза каучуков. 3-е изд. М., Химия,
1972. 526 с.
Тюряев И. Я- Физико-химические и технологические основы получения ди-
винила из бутана и бутиленов. Л., Химия, 1966. 180 с.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Абсорбционное разделение углеводо-
родов 57, 61
Абсорбционно-ректификационное раз-
деление газов 62
АБС пластик (Ударопрочный поли-
стирол) 237, 238, 244
АГ соль 252
Адипиновая кислота 252
Адсорбенты 58, 73
Адсорбционная осушка газов 55 сл.
Адсорбционное разделение газов 58
Адсорбция 73
Азеотропная (ые)
перегонка 69 сл.
смеси 69
Акрилаты (Эфиры акриловой кисло-
ты) 103 сл., 176
Акриловая кислота 103 сл.» 138, 176
Акрилонитрил 98
гидролиз 104
полимеризация 243 сл.
получение 98 сл., 135
сополимеры 237, 244
Акролеин 99, 100, 138 сл.
Алкансульфокислоты 192
Алкациды 55
Алкиларилсульфонаты 188
Алкилароматические углеводороды
дегидрирование 92 сл.
изомеризация 40 сл.
окисление 119 сл., 128, 133
Алкилбензолсульфонаты 188, 193,
198 сл.
Алкилбензолы 16, 17
высшие 37 сл.
окисление 128
получение 28 сл., 37 сл.
сульфирование 199 сл.
Алкилирование
ароматических углеводородов
28 сл.
олефинами 28 сл., 44 сл.
спиртами 44, 48 сл.
фенолов 44 сл.
Алкилкарбонаты 188, 190
Алкилнафталинсульфонаты 188
Алкилсульфаты 142 сл., 188, 194 сл.
Алкилсульфокислоты 202
Алкилсульфонаты 188, 196 сл.
2 72
Алкилсульфохлориды 192
Алкилфенолы
высшие 49 сл.
оксиэтилированные (ОП) 135,
188, 189, 204, 205, 208, 209
получение 44 сл., 49 сл.
Алкилхлорсиланы 256
Алкоголяты алюминия 161, 162
Аллиловый спирт 180, 181
Альдегиды
конденсация 158 сл., 245
получение 133, 170
Альдоли 159
Альдольная конденсация 138, 139
Алюминийорганический синтез 161 сл.
Алюминийтриалкилы 161, 162
Амиды оксиэтилированные 189
Амилфенол 48
Амины
в синтезах 210
оксиэтилированные 189
Аммиак, получение 265 сл.
Аммонолиз окислительный 99 сл.
Аморфные полимеры 212
Амфолитные моющие вещества 189
Анид 252
Анионная полимеризация 214
Анионоактивные ПАВ 188, 190 сл.
Антиокислители 48
Антиоксиданты (Противостарители)
222
АОС (а-Олефинсульфонаты) 188,
201 сл.
Арилхлорсиланы 256
Ароматические углеводороды
алкилирование 28 сл.
в синтезах 16
выделение 70 сл.
окисление 119 сл., 128, 133
Ацетали поливинилового спирта 245
Ацетальдегид 99, 138 сл., 160
Ацетальдоль 138, 160
Ацетилен 24 сл.
в синтезах 16, 98, 101, 105, 176
гидратация 139
гидрохлорирование 96
димеризация 99
карбидный 24 сл.
получение 24 сл.
Ацетон 126 сл.
в синтезах 105
как экстрагент 67
получение 122 сл.
Ацетонциангидрин 105
БВК (Белково-витаминные концент-
раты) 12
Бензины
газовые 18
пиролиз 19
Бензоилсалициловая кислота 126
Бензойная кислота 121, 126, 130
Бензол
алкилирование олефинами 28 сл.,
33 сл., 35 сл.
в синтезах 16, 17, 135
выделение 71, 72, 74
окисление 126
очистка 33
Бензолсульфокислота 125
Бесхлорные методы синтеза глицери-
на 181
Бисфенолы 48
Блочная полимеризация 218, 236, 238,
242
Бута диен-1,3
в синтезах 15
выделение 64
получение 78 сл., 81, 84 сл., 159
сополимеры 232, 237, 244
к-Бутан
в синтезах 11, 12, 81
выделение 66 сл.
дегидрирование 78 сл., 84 сл.
изомеризация 38, 42 сл.
окисление 112
пиролиз 19
Бутандиол-1,4 254
w-Бутанол 159
грег-Бутилгидропероксид 112
«-Бутилены
алкилирование бензола 32
в синтезах 14
выделение 66 сл.
гидратация 144, 151
дегидрирование 78 сл., 83 сл.
изомеризация 39
получение 78 сл.
Бутилкаучук 15
Бутил-л-крезол 48
Бутиловые спирты 144, 151, 152, 156
трет-Бутилпероксид 112
л-трет-Бутилфенол 48 сл.
Взрывоопасность парафинов ПО
Винилацетат 101 сл.
полимеризация 244
сополимеры 244
Винилацетилен 99
Винилдиоксаны 89
Винилиденхлорид, сополимеры 240
Винилпиридины 139
Винилтолуол 92
Винилхлорид 96 сл.
полимеризация 238 сл.
сополимеры 240
Водомаслорастворимые ПАВ 187
Водорастворимые ПАВ 187
Водород, получение 262 сл.
Волокна, виды 222
Волокнообразующие полимеры 222
Вулканизация каучуков 221
ВырожденноразветвЛенные процессы
108
Высокоэластическое состояние поли-
меров 212
Вязкотекучее состояние полимеров
212
Газовые конденсаты 18
Газы
нефтепереработки 18, 58 сл., 62
осушка 55 сл.
очистка 52 сл.
попутные 18, 58
природный 17, 58, 263 сл.
стабилизации нефти 18
Галогенсодержащие полимеры 238 сл^
Гексаметилендиамин 252
Гексаметилендиизоцианат 254
Гель-эффект 238, 242
Гептаны, разделение 69
Гербициды 48, 183
Гетерополиконденсация 218
Гетероцепные полимеры 211, 222
Гетинакс 249
Гидратация
олефинов 142 сл.
прямая 142, 144, 145, 152 сл.^
155 сл.
сернокислотная 142 сл., 145 сл.г
150 сл.
Гидрирование карбоновых кислот
156 сл.
Гидроксибензиловые спирты 247, 24В
Р-Гидроксипропионовая кислота 103
Гидропероксиды 108, 113, 120, 132
Гидрофобизирующие жидкости 257
Гидроформилирование см. Оксосинтез
Гиперсорбция 58
ГЛ Б (Гидрофильно-липофильный ба-
ланс) ПАВ 185
Гликоли (Спирты Двухатомные)-
177 сл., 251, 254
Глицерин 179 сл.
Глицидол 181
Гомополиконденсация 218
Гомополимеры 211
27J*
Дегидрирование 76 сл.
алкилароматических углеводоро-
дов 92 сл.
окислительное 127, 140
олефинов 79 сл.
парафинов 77 сл.
степень одностадийности 86
Деструктивное окисление 109
Деструкция полимеров 219, 235
Ди алкилсульфаты 142 сл.
Дивинилбензол 92
сополимеры 237
Диизоцианаты 254
Диметиламины 169
Диметилдиоксаны 88, 89
Диметилтерефталат 128, 249
Диметилфенилкарбинол 120
Диметилформамид 169
Динитроэтиленгликоль 177
Диоксолан, сополимеры 246
Диолы см. Гликоли
Диспергирующее действие ПАВ 187
Дифенилолпропан 250, 251
Дихлоргидрин глицерина 180
Дихлорэтан
гидролиз 177
дегидрохлорирование 96 сл.
пиролиз 97
Диэтилбензол 92
Диэтиленгликоль 72
Диэтилсульфат 146 сл.
Добавки к ПАВ 207 сл.
ДС-РАС 199
Дурол (Тетраметилбензол) 120
Изобутан
в синтезах 12
дегидрирование 78
окисление 112, 132
получение 38, 42 сл.
Изобутилен
алкилирование изобутана 38
в синтезах 15, 49, 88 сл.
выделение 63 сл.
гидратация 144, 156
окисление 106
полимеризация 214, 234
получение 132
Изобутиловый спирт 48
Изомеризация углеводородов 38 сл.
алкилароматических 40 сл.
олефинов 39
парафинов 38 сл.
Изооктан 38
Изопентан
в синтезах 12
дегидрирование 86 сл.
получение 38
Изопрен (2-Метилбутадиен-1,3)
выделение 65
очистка 65
Изопрен (2-Метилбутадиен-1,3)
полимеризация 16
получение 13, 15, 86 ел., 88 сл.,
91 сл.
Изопропилбензол (Кумол)
в синтезах 17
дегидрирование 92 сл.
получение 35 сл.
Изопропилбензолгидропероксид 120,
122 сл, 127
Изопропиловый спирт
дегидрирование 127
получение 144, 150, 155 сл.
Изопропилсульфат 150
Имидазолины 210
Инициирование (Зарождение) цепей
107
Инсектициды 183
Ионная полимеризация 214, 217
Ионогенные ПАВ 188
Ионол (2,6-Ди-трет-бутил-4-метилфе-
нол) 48
Ионообменные смолы 46, 47, 237
Капролактам 253
Капрой 252
Карбамид (Мочевина) 267 сл.
Карбоксиметилцеллюлоза, натриевая
соль 208
Карбоновые кислоты
ароматические 120
в синтезах 210
высшие жирные 113, 115 сл,
156 сл.
гидрирование 156 сл.
оксиэтилированные 189, 206
получение 175
Карбоцепные полимеры 211, 222
Катиониты 46, 49
Катионоактивные ПАВ 188 сл, 210
Катионная полимеризация 214
Каучуки синтетические 65, 221 сл,
237
вулканизация 221
кремнийорганические 257
мономеры 12, 14, 15, 65, 92, 98,
139
уретановые 255
этилен-пропиленовые (СКЭП)
232
Кетоны 133
Клеи 245, 246, 250, 251, 254
Когазин 197
Компрессионное разделение углеводо-
родов 57
Конверсия углеводородов 163 сл.
Конденсационно-ректификационное
разделение газов 62
Конденсация альдегидов 158 сл.
Контактный способ получения серной
кислоты 259
274
Корезин 48
Крекинг парафинов 23
Кремнийорганические полимеры
255 сл.
Кристаллизация 73 сл.
Кристаллические полимеры 212
Ксилолы
в синтезах 16
изомеризация 41
окисление 121, 128 сл., 130
разделение 74 сл.
Кумол см. Изопропилбензол
Кумольный метод синтеза фенола
и ацетона 122 сл.
Лавсан 128, 250
Лаки 240, 245, 246, 250, 254, 257
Лактонитрил 98, 99
Латексная полимеризация 236, 238,
239
Летучесть углеводородов 66
Линейные полимеры 211, 219
Литраж карбида кальция 24
Малеиновый ангидрид 120
Малеиновая кислота 251
Маслорастворимые ПАВ 185, 187
Меркаптаны оксиэтилированные 189
Метакриловая кислота 105 сл.
эфиры 242
Метакролеин 106
Метан
в синтезах 10, 11
конверсия 163 сл., 263
окисление ПО
пиролиз 25 сл.
получение 175
Метанол см. Метиловый спирт
Метилакрилат 176
Метиламины 169
Метилацетат 169
п-Метилбензиловый спирт 130
2-Метилбутадиен-1,3 см. Изопрен
3-Метилбутандиол-1,3 89
Метилбутены 87
Метилметакрилат 105, 242
Метиловый спирт (Метанол)
в синтезах 168 сл., 176
как азеотропообразователь 70
получение 162 сл., 165 сл.
Метилпентены 91
а-Метилстирол 92 сл., 94 сл., 120
Метилэтилкетон 71
Метионин 138
Миграционная полимеризация 214
Мономеры
виниловые 76 сл., 95 сл.
для СК 12, 14, 15, 65, 92, 98, 139
с функциональными группами
95 сл.
Монохлоргидрины глицерина 180
Мочевина (Карбамид) 267 сл.
Моющее действие ПАВ 186, 207 сл.
Моющие средства 182
МС пластик 244
Найлон 252
Наполнители пластмасс 220
Натуральные волокна 222
Нафтены, окисление 121, 122
Неионогенные ПАВ 188 сл., 203 сл..
Неомыляемые I и II 116
Нерегулярные полимеры 211
Нефтепродукты 18, 19
Нефтехимия 8, 10 сл.
Нефть
добыча 183
переработка 18
пиролиз 19
Низкотемпературная ректификация
59 сл.
Нитрон 244
Новолачные смолы (Новолаки) 247,..
249
Обрыв цепи 107
Окисление 106 сл.
алкилароматических углеводоро-
дов 119 сл., 128, 133
газофазное 109 сл.
глубокое 109
деструктивное 109
жидкофазное 109 сл.
механизм 107 сл., 132
нафтенов 121, 122
олефинов 131 сл., 133
парафина 114, 115, 118 сл.
парафинов 109 сл., 115 сл.
циклоолефинов 133
частичное 109
Окислительный аммонолиз 99 сл., 131
Оксид углерода
в синтезах 105, 169 сл., 175 сл.
гидрирование 165
Оксиэтилированные соединения 135г
188, 189, 203 сл.
Оксосинтез 170 сл.
а-Олефинсульфонаты (АОС) 188^
201 сл.
Олефины
алкилирование бензола 28 сл.
— фенолов 44 сл.
в синтезах 13 сл., 175
высшие 22 сл., 37 сл.
гидратация 142 сл.
гидроформилирование 170 сл.
дегидрирование 79 сл.
изомеризация 39
низшие 18 сл.
275-
СИефины
окисление 131 сл., 133
окислительный аммонолиз 131
олигомеризация 214 сл., 223 сл.
получение 18 сл., 22 сл.
сульфирование 191, 194 сл.,
201 сл.
полимеризация 143, 214 сл.
эпоксидирование 132
•Олигомеры 211
Органическое стекло (Полиметил-
метакрилат) 105, 243
Осушка газов 55 сл.
Отбеливатели 208
Очистка углеводородов 51 сл.
алкацидами 55
от серы 52 сл.
этаноламинами 52 сл.
ПАВ 152 сл.
гидрофильно-липофильный баланс
{ГЛБ) 185
классификация 187 сл.
моющее действие 186, 207 сл.
физико-химия действия 184 сл.
Паральдегид 140
Парафин нефтяной 114, 118 сл.
окисление 114, 115, 118 сл.
Парафины
в синтезах 10 сл.
взрывоопасность НО
высшие 12, 13, 113
газообразные 10 сл., ПО
дегидрирование 77 сл.
жидкие 12, 13, 114
изомеризация 38 сл.
крекинг 23
мягкие 12, 13, 114
низшие 10 сл.
окисление 109 сл., 115 сл.
пиролиз 19
сульфоокисление 192, 193, 197 сл.
сульфохлорирование 192, 197 сл.
твердые 12, 13
хлорирование 192
Пенообразующая способность ПАВ
187
Пенопласты 254, 255
Пенополистирол 237
Пенополиуретаны 255
н-Пентан, изомеризация 38
Пентены 15
Переалкилирование 41
Переэтерификация 249,. 250
Пероксид водорода
в синтезах 180
получение 111, 127
Пероксиды органические 108
Петрова контакт 199
Пиперилен 87
276
Пиридины 139
Пиролиз 18 сл., 24 сл.
бензинов 19
окислительный 22, 25, 26
печи 21, 25 сл.
разделение продуктов 59 сл.
регенеративный 25 сл.
Пиромеллитовый диангидрид 120
Пластификаторы 220
Пластические массы 220, 221
Пленки 246, 254
Полиакрилонитрил 243 сл.
Полиалкилбензолы 41
Полиамиды 252 сл.
Поливинилацетали 245 сл.
Поливинилацетат 244, 245
Поливинилбутираль 245
Поливиниловый спирт 245
Поливинилформаль 245
Поливинилхлорид 238 сл.
Поливинилэтилаль 245
Полигексаметиленадипамид 252
Полигексаметиленсебацинамид 252
Полидодекаамид 252
Полиизобутилен 234 сл.
Поликапроамид 252 сл.
Поликарбонаты 250
Поликонденсация 218 сл., 247 сл..
251, 252
Полимербензин 223
Полимердистиллят 49
Полимеризация 211 сл.
виды 214, 218, 236
галогенсодержащих мономеров
238 сл.
инициаторы 214, 217
катализаторы 112, 214, 217, 228
механизм 216 сл.
мономеры 215
олефинов 143, 214 сл.
промышленное оформление 218,
236
Полимерная цепь 211
Полимерные материалы 220 сл.
Полимеры 210 сл.
виды 211 сл., 219, 221
деструкция 219
ориентация макромолекул 212 сл.
отверждение 252
получение 223 сл.
стереорегулярные 214
Полиметилметакрилат (Органическое
стекло) 105, 243
Полиорганосилоксаны (Силиконы)
256
Полипропилен 232 сл.
Полипропиленгликоли 179
оксиэтилированные 189
Полистирол 235 сл.
ударопрочный (АБС пластик)
237, 238, 244
Политетрафторэтилен 241
Политрифторхлорэтилен 242
Полиуретаны 254 сл.
Полиформальдегид 246, 247
Полиэнантоамид 252
Полиэтерификация 251
Полиэтилен 224 сл.
Полиэтиленгликоли 135
моноэфиры 135
Полиэтилентерефталат 249 сл.
Полиэфируретаны 254, 255
Полиэфиры 242
ненасыщенные 251
простые 246
сложные 249 сл.
Поролон 255
Порофоры 237
Пресспорошки 249
Присадки к маслам 47, 48
Продолжение цепей 107
Пропан
в синтезах 10, 11
конверсия 165
окисление ПО
пиролиз 19, 26
Пропилен
алкилирование бензола 31, 35 сл.
аммонолиз окислительный 99 сл.
в синтезах 13, 14, 91 сл.
гидратация 144, 150, 155 сл.
димеризация 91
окисление 99, 104, 127, 131, 133,
138
полимеризация 32, 233 сл.
получение 18 сл.
сополимеры 231, 232
Пропиленгликоль 178
Пропиленоксид
гидратация 178
изомеризация 180
получение 132
н-Пропиловый спирт 138
Пропионовая кислота 175
Пропионовый альдегид 133, 138
Радикальная полимеризация 214
Разветвленные полимеры 211, 219
Реактопласты (Термореактивные по-
лимеры) 221
Регулярные полимеры 211
Резины 221, 234, 254
Резиты 248
Резольные смолы (Резолы) 248,’ 249
Саран 240
Сера
вулканизация каучуков 221
окисление 259
оксиды 190, 259 сл.
Сера
получение 257 сл.
удаление 52 сл.
Серная кислота, получение 259 сл.
Сероводород
окисление 257 сл.
удаление 52 сл., 177
Сетчатые полимеры 211, 219
Силиконы (Полиорганосилоксаны)
256
Синильная кислота (Цианистый во-
дород) 98, 99, 103
Синтез-газ 162, 175, 176
Синтетические
волокна 222, 245
моторные топлива 169
моющие вещества 182 сл., 207 сл.
— средства (СМС) 182
смазочные масла 135, 223
Синтин 118
Синтол 175
СКН (Бутадиен-нитрильный каучук)
237, 244
СКС (Бутадиен-стирольный каучук)
237
СКЭП (Каучуки этилен-пропилено-
вые) 232
Сополимеризация олефинов 231 сл.
Сополимеры 211
Спирты
алкилирование 44, 48 сл.
в синтезах 176, 209
высшие жирные 113, 118 сл.,
156 сл., 161 сл.
двухатомные (Гликоли) 177 сл.
оксиэтилированные 135, 189, 208,
209
получение 111 сл., 118сл., 142 сл.,
156 сл., 161 сл., 169, 170 сл.
сульфирование 191, 194 сл.
Стабилизаторы 221
Стеклообразное состояние полимеров
212
Стеклопластики 251, 252
Стеклотекстолит 249
Стереорегулярные полимеры 212
Стереоспецифическая полимеризация
214
Степень
кристалличности полимеров 212
полимеризации 211
Стильбены 93
Стирол 94 сл.
полимеризация 235 сл.
получение 92 сл., 132
сополимеры 237 сл., 244
Стирол-контакт 93
Ступенчатая полимеризация 214
Сультоны 202
277
Сульфаты
оксиэтилированных соединений
208, 209
жирных кислот 188
Сульфирование
алкилбензолов 199 сл.
олефинов 191, 194 сл., 201 сл.
серной кислотой 190 сл., 194 сл.,
198 сл.
спиртов 191, 194 сл.
Сульфонаты жирных кислот 188
Сульфонолы 199
Сульфоокисление парафинов 192, 193,
197 сл.
Сульфохлорирование парафинов 192,
197 сл.
Сульфураторы 199, 200
Суспензионная полимеризация 218,
236, 238, 239
Сшивание макромолекул 219
Текстолит 249
Теломеризация 217
Терефталевая кислота
получение 121, 128 сл.
эфиры 249
Термопластичные полимеры (Термо-
пласты) 221
Термореактивные полимеры (Реакто-
пласты) 221
Тетраметилбензол (Дурол) 120
Тетрафторэтилен 241
Тетраэтилсвинец 10
Технологические схемы
выделения углеводородов 68, 69,
70, 71, 72, 74
дегидрирования «-бутана 82, 85
— бутиленов 83
— этилбензола 94
изомеризации н-бутана 43
конверсии 164
оксосинтеза 173, 174
осушки газов 56
'очистки газов 53
пиролиза 21, 27
получения акриловой кислоты 104
— акрилонитрила 101
— алкилбензолсульфонатов 200
— алкилсульфатов 195, 196
— алкилфеполов 50
— аммиака 266
— ацетальдегида 141
— ацетона 124
— к-бутанола 160
— винилацетата 102
— винилхлорида 96
— водорода 263, 264
— высших жирных кислот 116
— гликолей 178
— диметилдиоксана 90
— диметилтерефталата 129
Технологические схемы
— изопрена 91
— изопропилбензола 36
— карбамида 269
— неионогенных ПАВ 205
— а-олефинсульфонатов 202
— поликапроамида 253
— поливинилхлорида 240
— полиэтилена 226, 230
— метилового спирта 167
— серной кислоты 260, 261
— серы 258
— спиртов 119, 158
— этилбензола 34
— фенола 124
— этиленоксида 137
— этилового спирта 148, 154
разделения газов 58, 59, 61, 64,
65, 67, 68
Тиогликоли 135
Толуол
в синтезах 16
выделение 70, 74
окисление 121, 126, 130
хлорметилирование 130
Триадная схема оксосинтеза 173
Триоксан, сополимеры 246
:Триплекс» 246
Трифторхлорэтилен 242
Углеводородное сырье
газообразное 10 сл., 52 сл.
жидкое 68 сл.
качество 51 сл.
очистка 51 сл.
переработка 10 сл.
производство 17 сл.
разделение 57 сл.
Ударопрочный полистирол (АБС пла-
стик) 237, 238
Удобрения 267
Уксусная кислота 139
в синтезах 101, 169
получение 112
Уксусный ангидрид 139
Фаолит 249
Р-Фенилэтиловый спирт 135
Фенол
алкилирование 44 сл., 48 сл.
в синтезах 247
как экстрагент 71, 72
метилольные производные 247
получение 122 сл.
Фенолоформальдегидные полимеры
247 сл.
Фенолы, алкилирование 44 сл.
Фишера — Тропша синтез 175
Флотация руд 183
278
Формальдегид
в синтезах 15, 88 сл., 138, 247
полимеризация 246
получение 168
Формование волокон 222
Форполимеризация 242
Фталевый ангидрид 120
Фторсодержащие полимеры 241 сл.
Фунгициды 48, 183
Фурфурол 66 сл.
Халкон-процесс 132
Хемосорбция 63 сл.
Химические волокна 222
Хлорбензол 125
Хлорин 241
Хлорирование
аномальное 98
окислительное 97
парафинов 192
Хлористый аллил 180
Хлорметилирование толуола 130
Хлорный метод синтеза глицерина 180
Хрупкость полимеров 212
Целлозольвы 177, 178
Цепная полимеризация 214, 216
Цепные реакции 107
Циангидрины 103, 106, 139
Цианистый водород (Синильная кис-
лота) 98, 99, 103
Циклогексан
окисление 121, 122
получение 223
Циклогексанол 122
Циклогексанон 122
Циклогексен, сульфоокисление 192,
193
Циклоолефины, окисление 133
Циклопентан 223
Четкая ректификация 68, 69
Щелочное плавление бензолсульфо-
кислоты 125
Экстрактивная дистилляция 65 сл.,
71, 72
Экстракция 72
Электрокрекинг 25
Эмали 240, 246, 252
Эмульгирующая способность ПАВ
187
Эмульсионная полимеризация 218,
236, 238, 239
Энапт 252
Эпихлоргидрин 180, 251
Эпоксидирование олефинов 132
Эпоксидные полимеры 251 сл.
Этанол см. Этиловый спирт
Этаноламины 134, 177, 178, 210
очистка углеводородов 52 сл.
Этан
в синтезах 10, 11
конверсия 165
окисление 111
пиролиз 19, 22
Этилбензол
в синтезах 17
выделение 74
дегидрирование 92 сл.
окисление 132
получение 33 сл.
2-Этилгексанол 159
Этилен
алкилирование бензола 30, 33 сл.
в синтезах 13, 14, 98
выделение 59 сл.
гидратация 145 сл., 152 сл.
димеризация 223
окисление 131, 133 сл., 136,
140 сл.
олигомеризация 23, 223 сл.
очистка 21
полимеризация 224 сл.
получение 18 сл., 21 сл.
сополимеры 231 сл.
хлорирование 97, 98
Этиленгликоль 134, 177, 249
Этилендиаминтетрауксусная кислота,
натриевая соль 208
Этиленоксид
в синтезах 103, 134 сл., 203, 204
гидратация 177
полимеризация 135
получение 134 сл.
производные (Оксиэтилированные
соединения) 135, 188, 189,
203 сл.
Этиленхлоргидрин 136, 177
Этиленциангидрин 98, 135
Этиловый спирт (Этанол) 144
в синтезах 159
дегидрирование 140, 143
получение 145 сл., 152 сл.
ректификат 149
Этилсульфаты 145 сл.
Этилтолуол 92 сл.
Эфиры
гидрирование 156 сл.
ортокремневой кислоты 257
простые 143
сложные 156 сл., 176
Петр Степанович Белов
ОСНОВЫ
ТЕХНОЛОГИИ
НЕФТЕХИМИЧЕСКОГО
СИНТЕЗА
ИЗДАНИЕ 2-е,
ПЕРЕРАБОТАННОЕ
Редакторы
Н. И. Урывалова, А. А. Рогайлина
Художник
Е. В. Бекетов
Художественный редактор
Н. М. Биксентеев
Технический редактор
В. В. Лебедева
Корректор
Л. В. Гаврилина
ИБ № 1089
Сдано в наб. 17.04.82. Поди, в печ. 14.06.82.
Формат бумаги бОХЭО’/ю. Бумага тип. № 2.
Гарн. литературная. Печать высокая.
Усл. печ. л. 17,5. Усл. кр.-отт. 17,5.
Уч.-изд. л. 18,72. Тираж 7300 экз. Заказ № 254.
Цена 80 к. Изд. № 1947.
Ордена «Знак Почегга» издательство «Химия».
107076, Москва, Стромынка, 13.
Московская типография Ns 11
Союзполиграфпрома при Государственном
комитете СССР по делам издательств,
полиграфии и книжной торговли.
Москва, 113106, Нагатинская ул., д. 1.