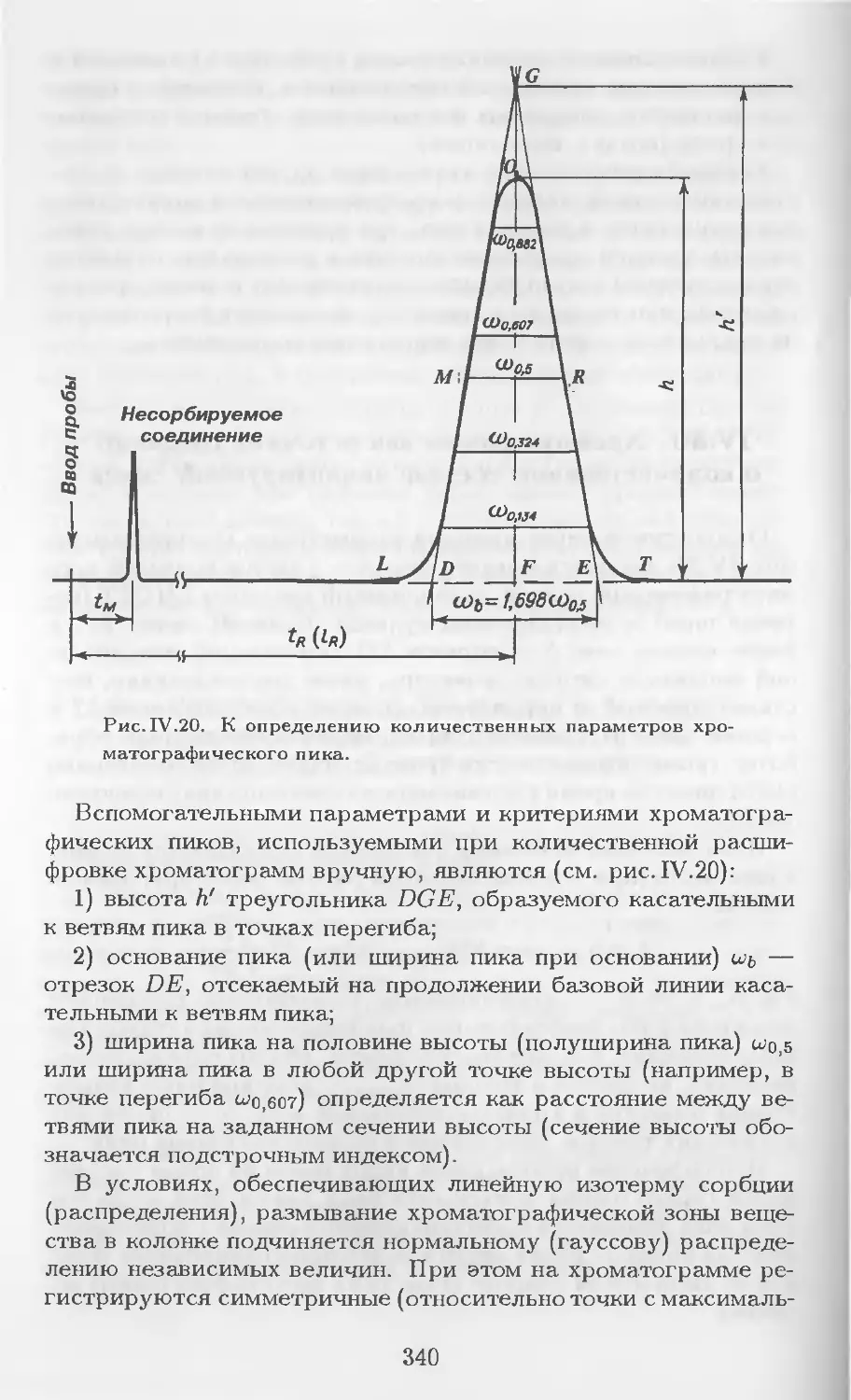
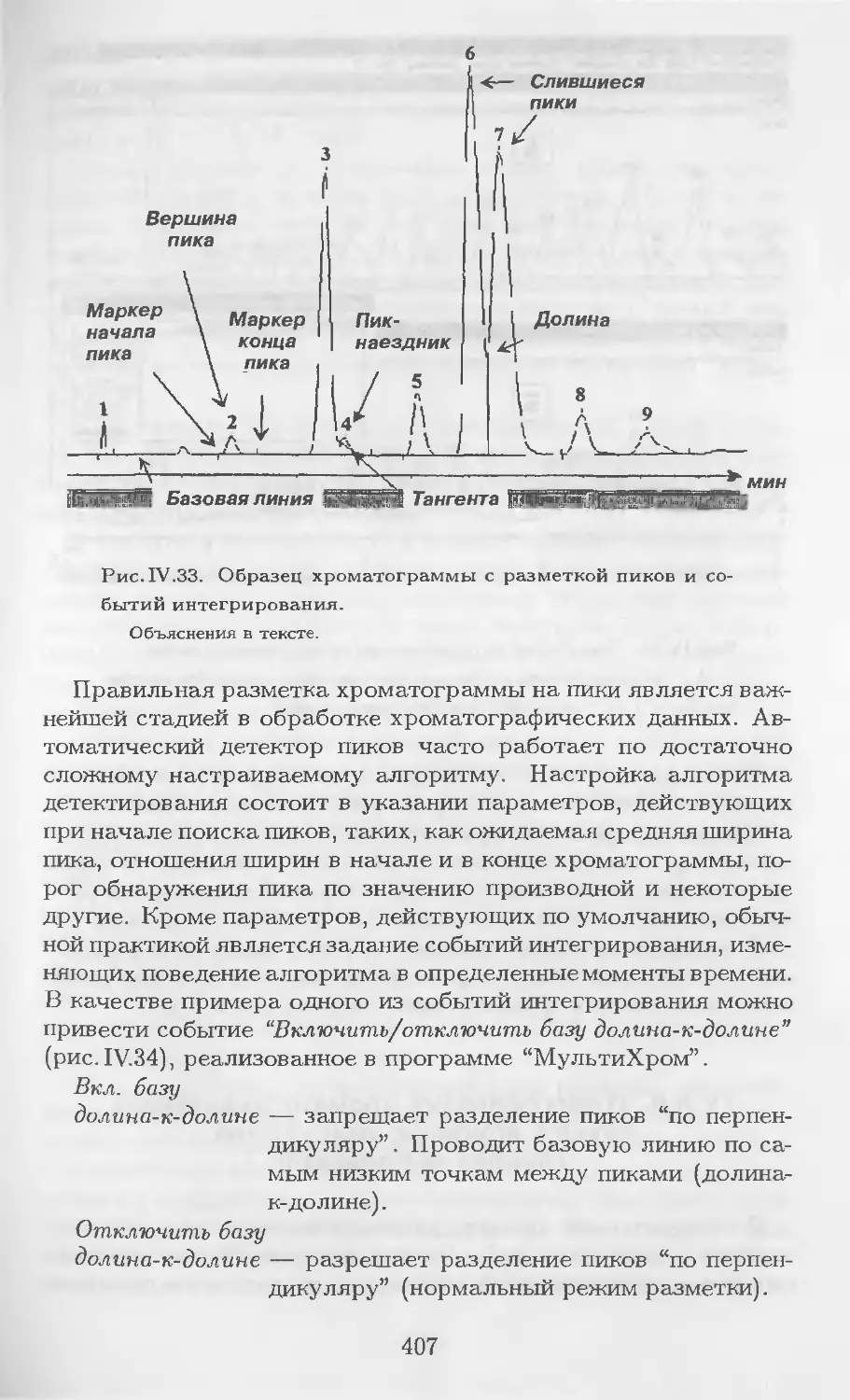
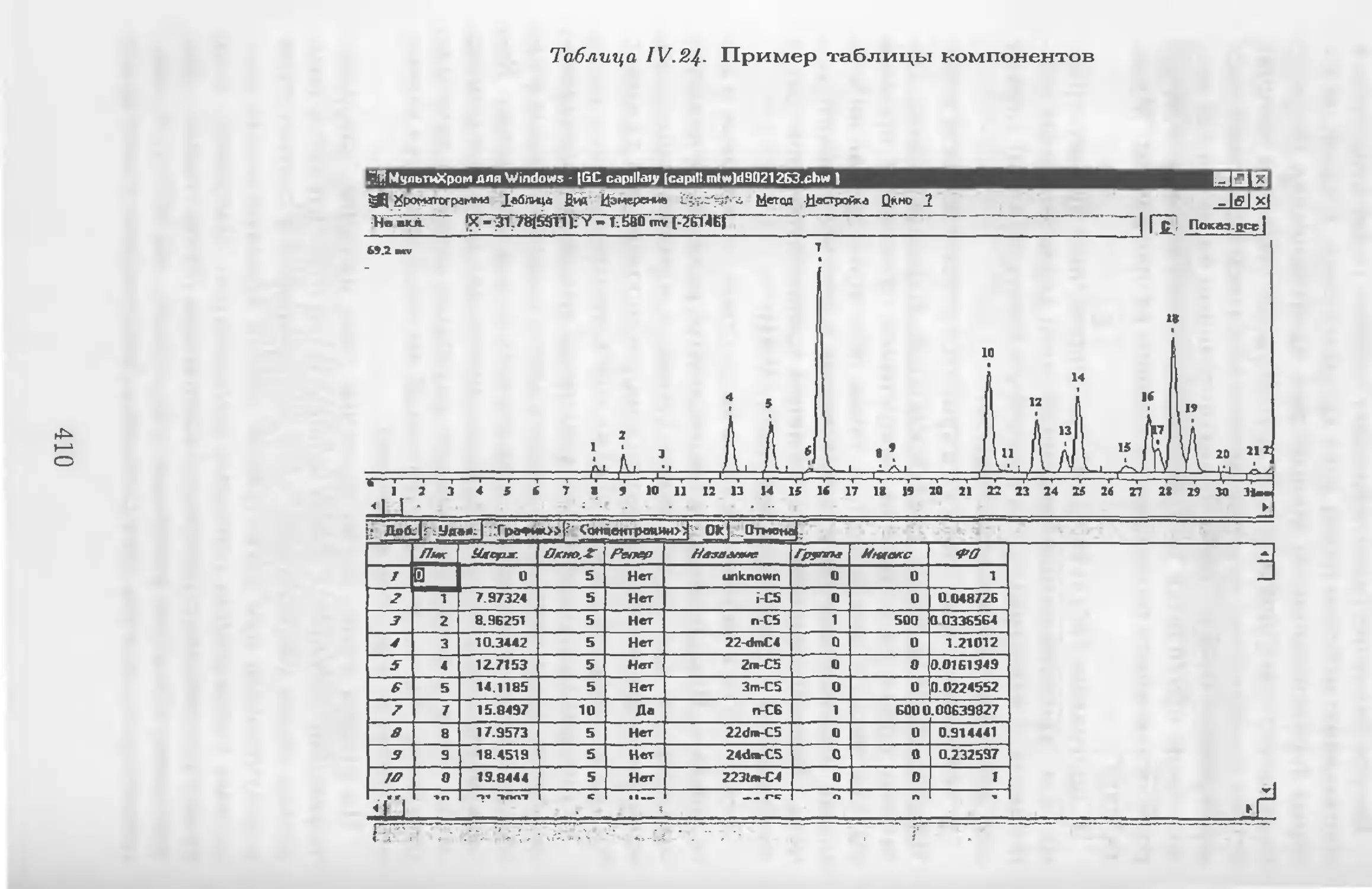
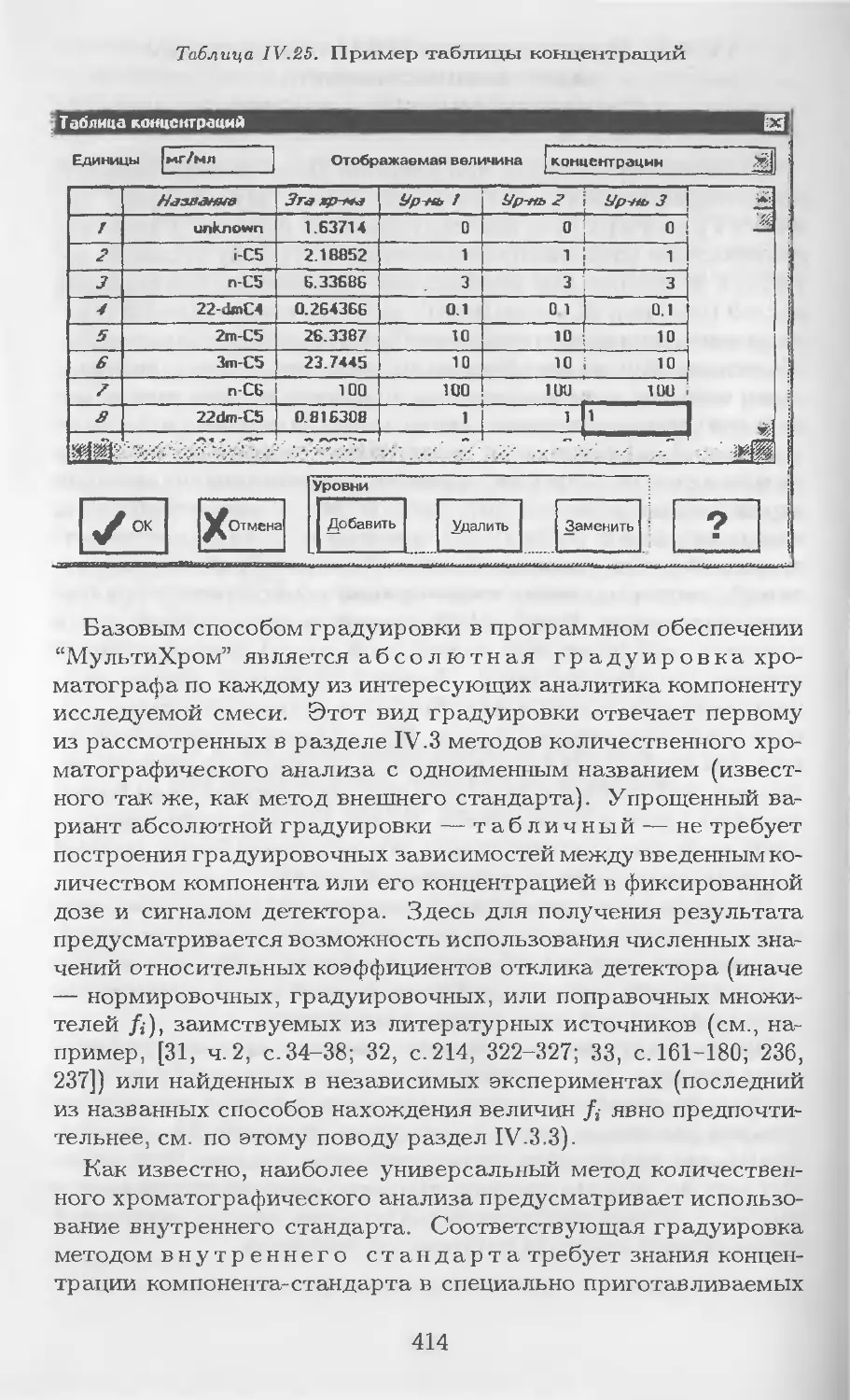
/














































































































































































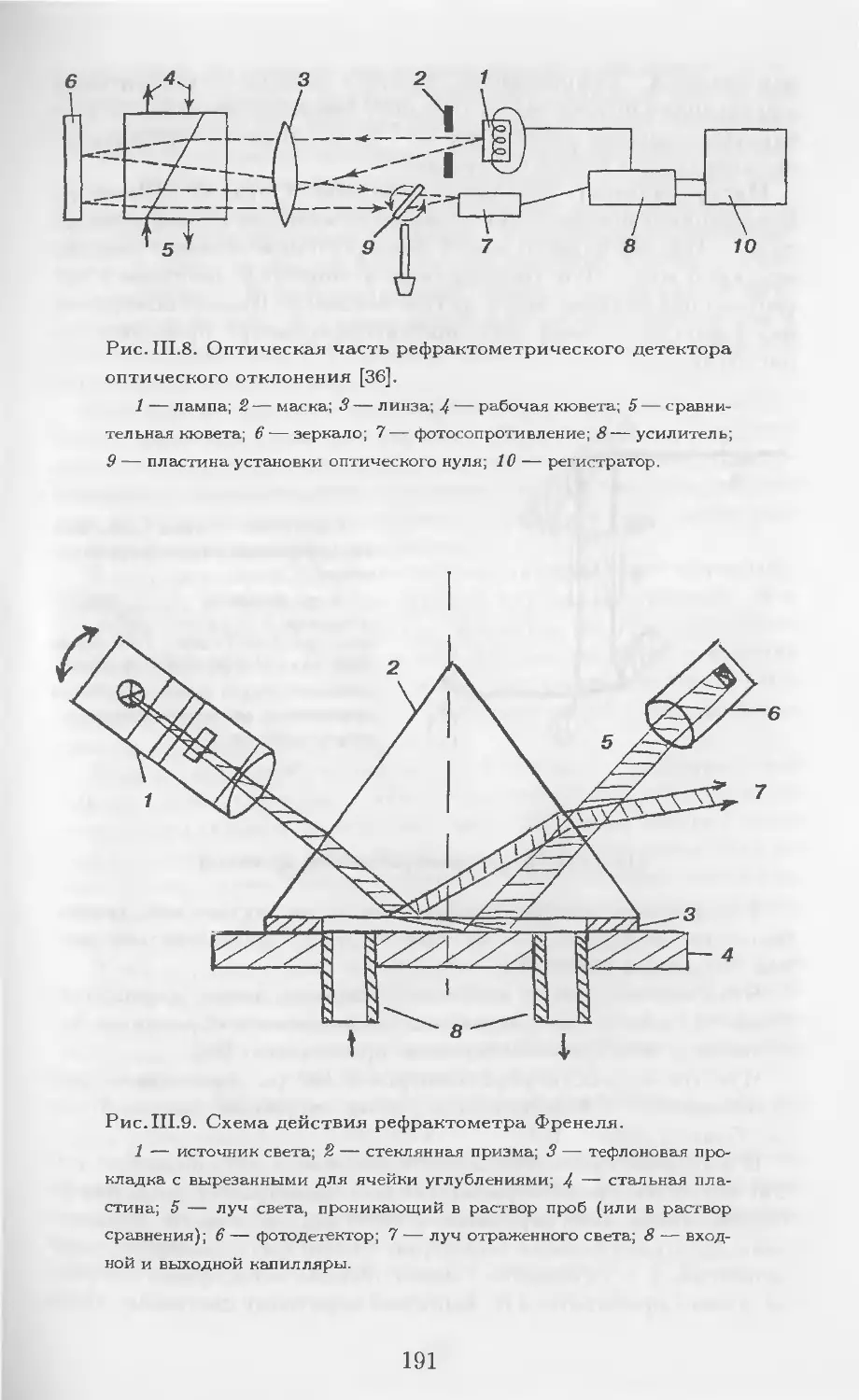





















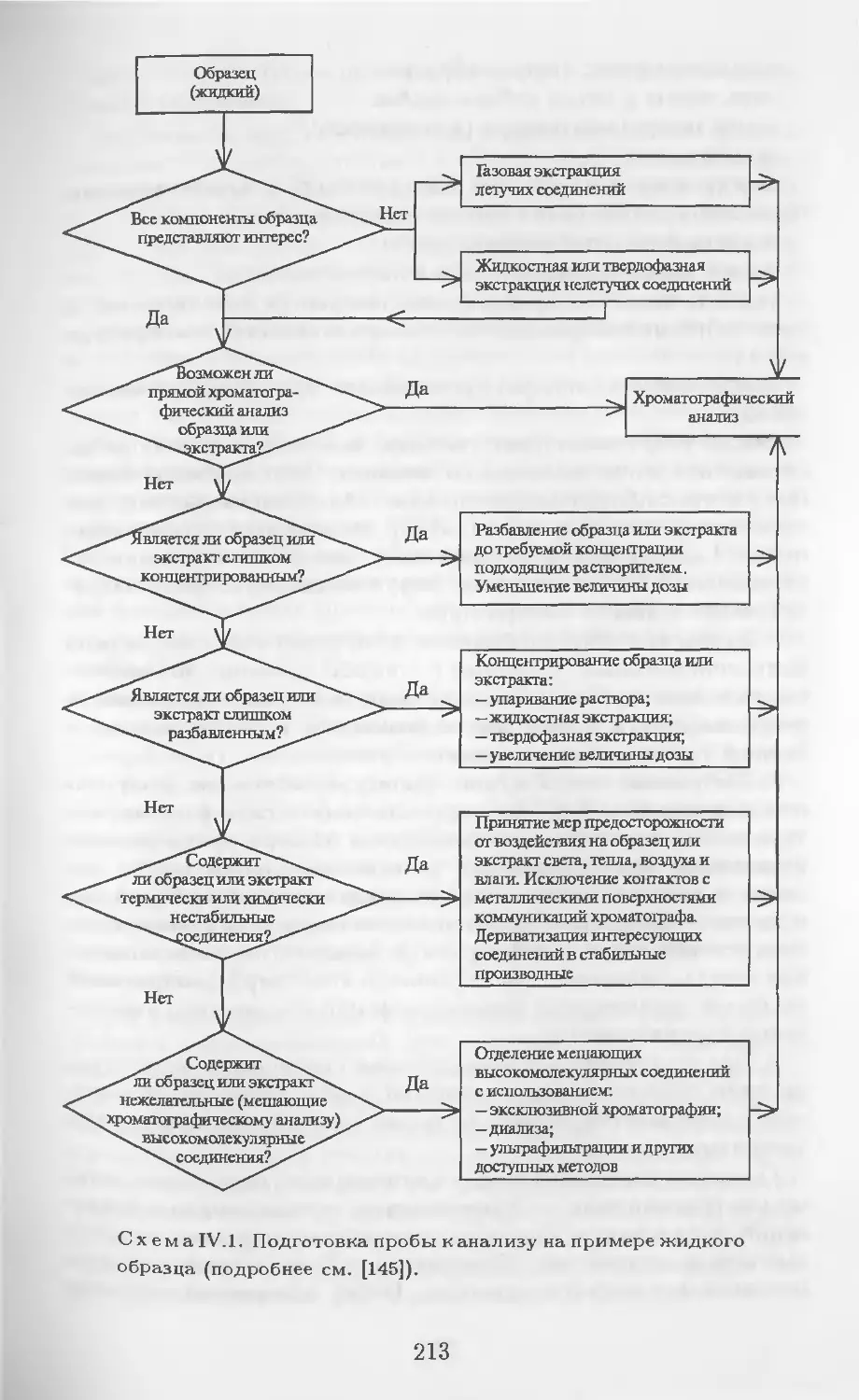


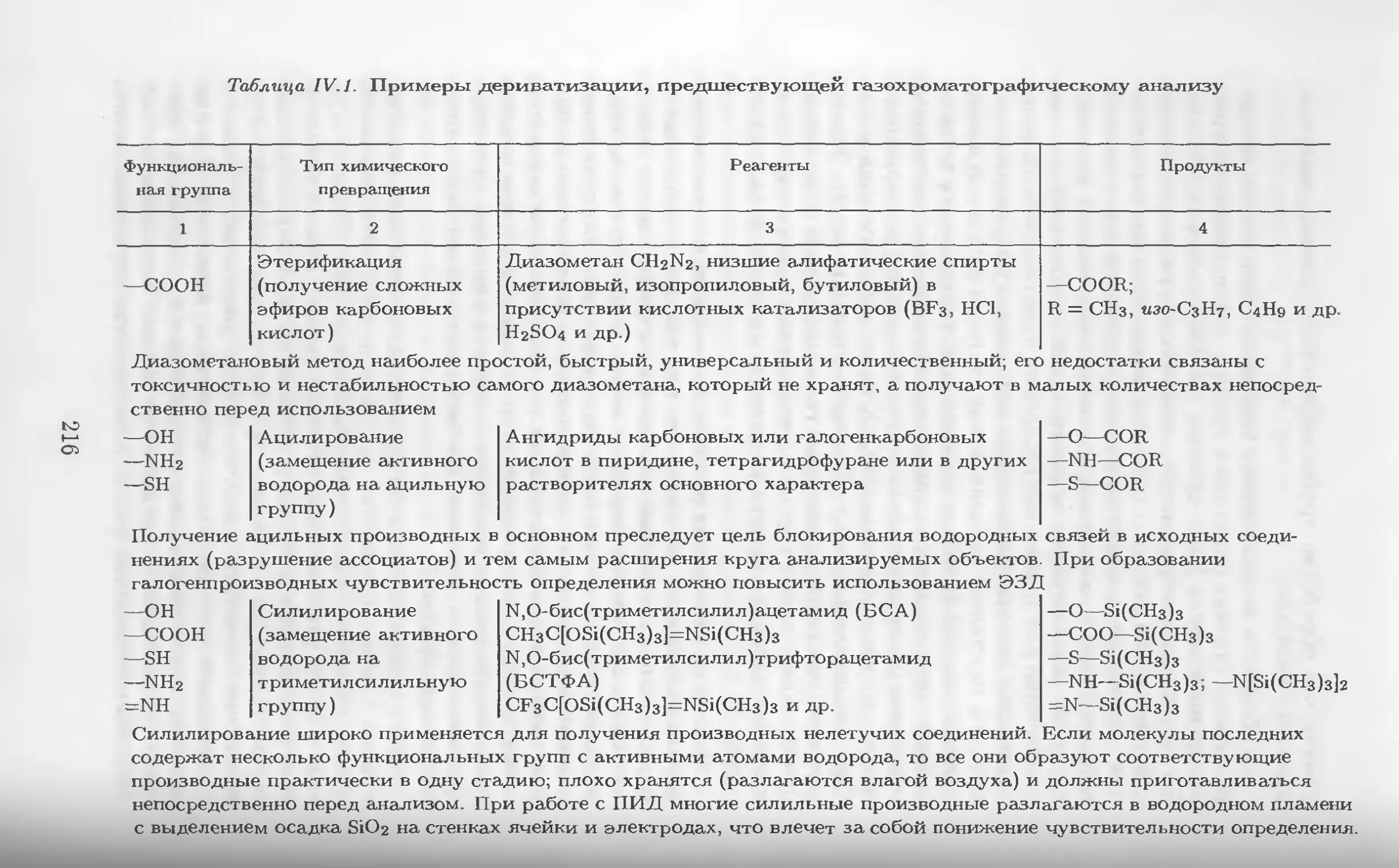



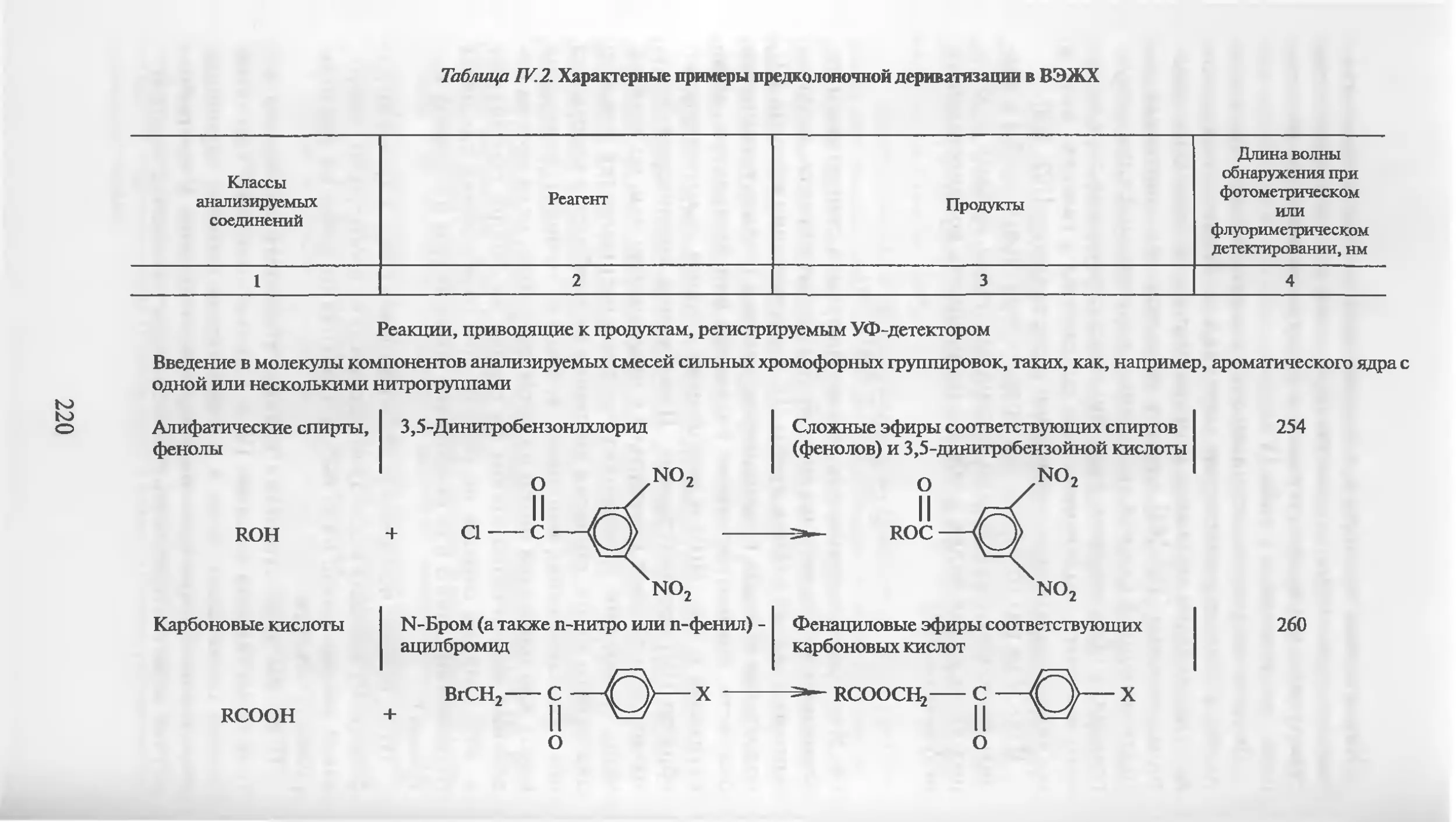
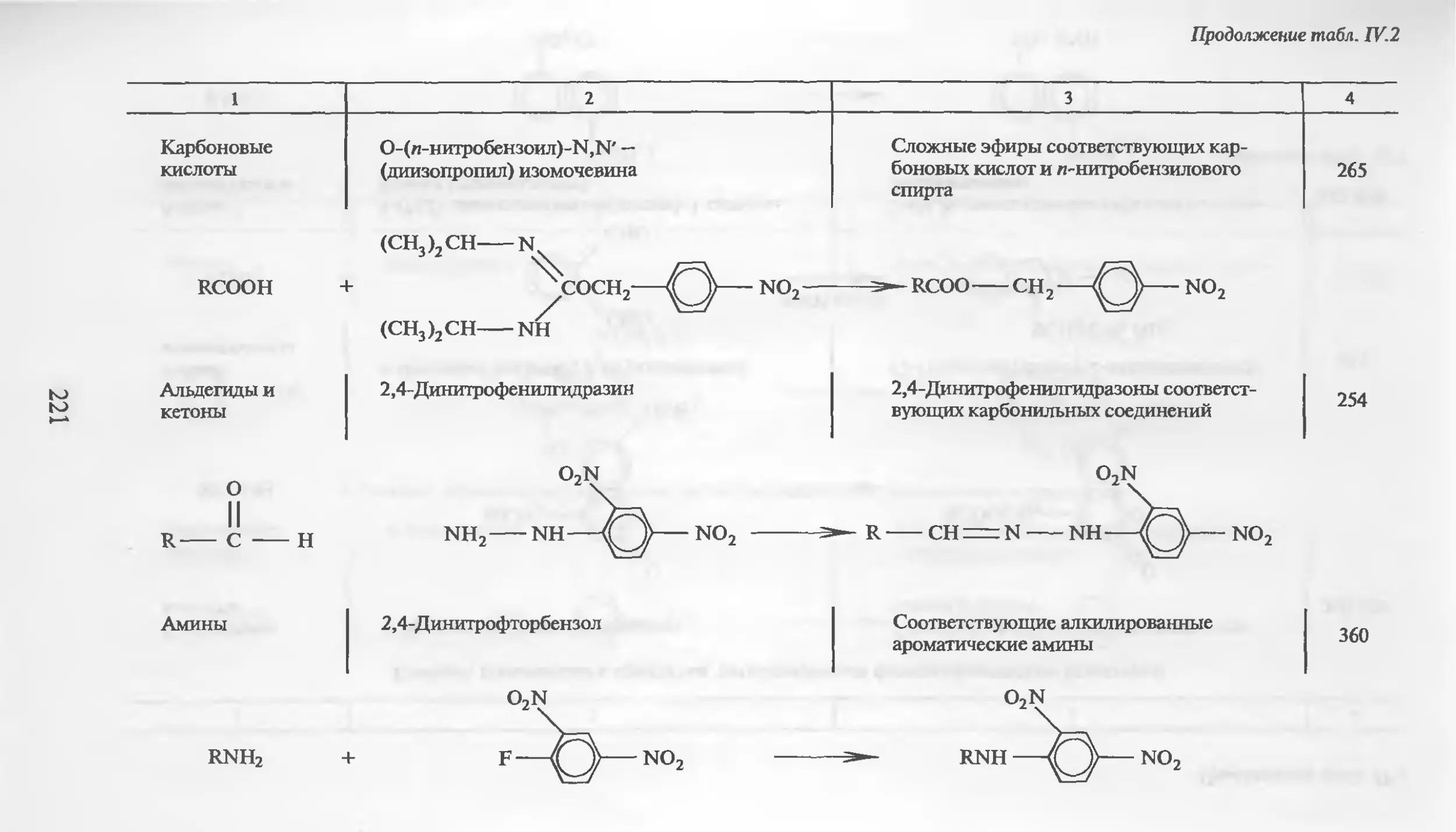
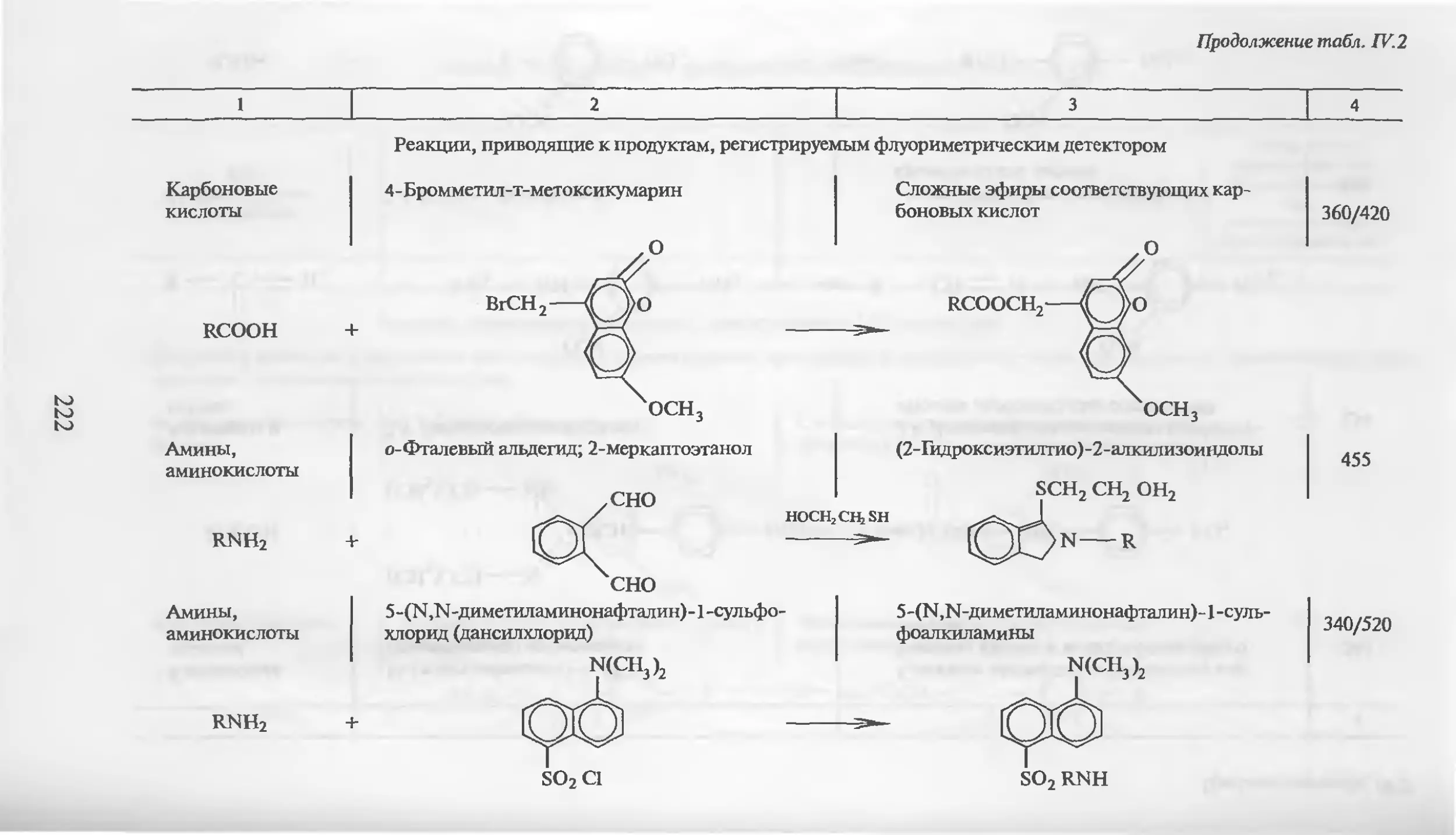
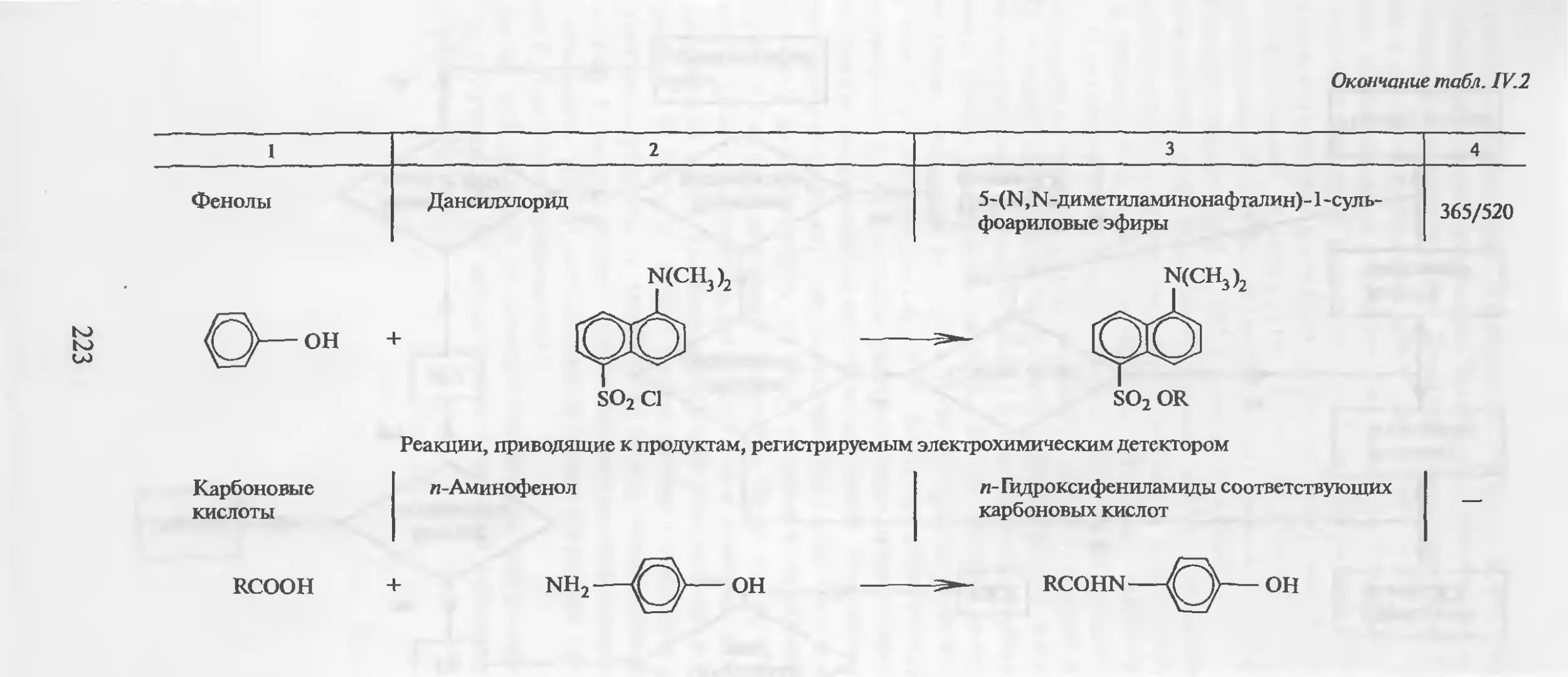
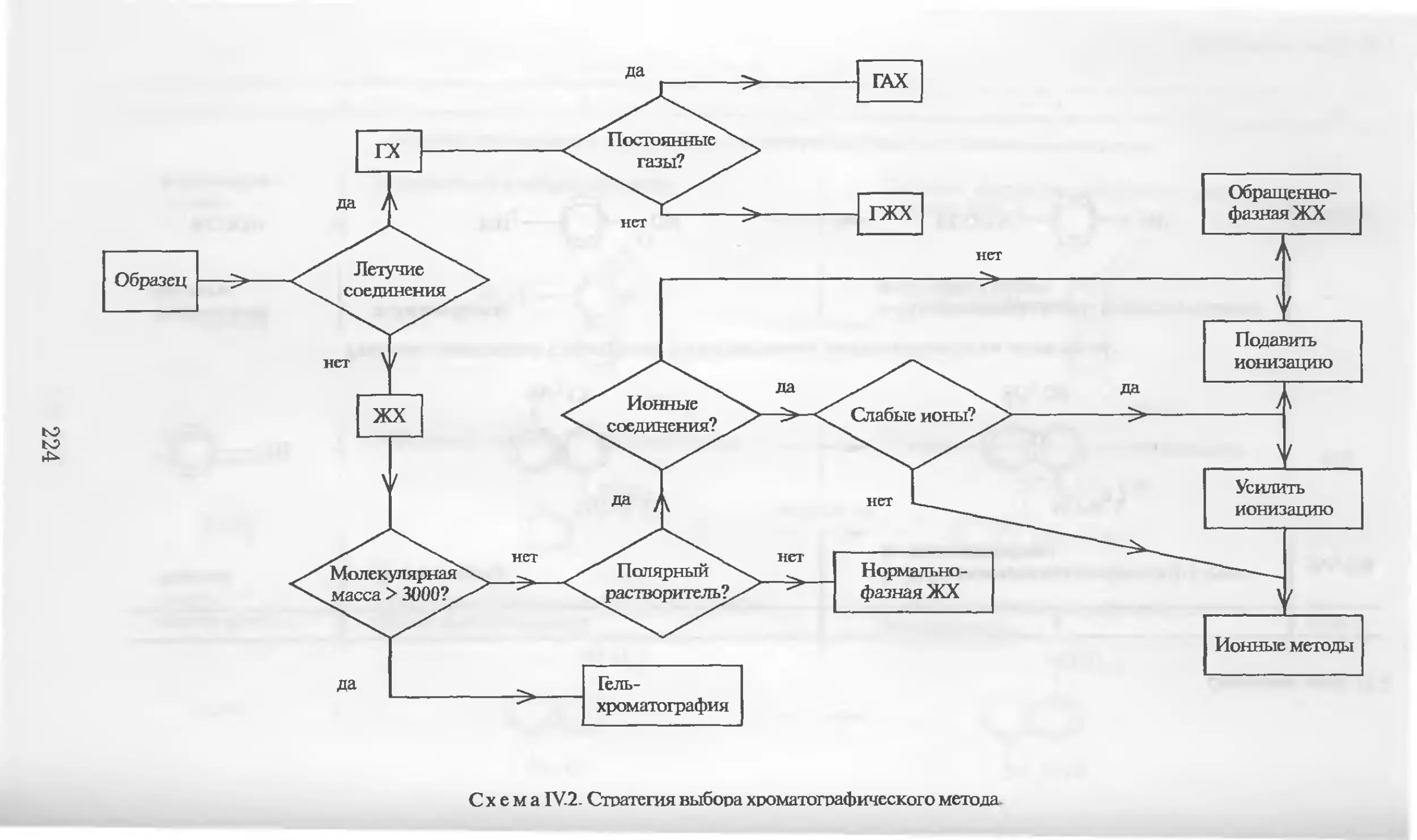



























































































































































































































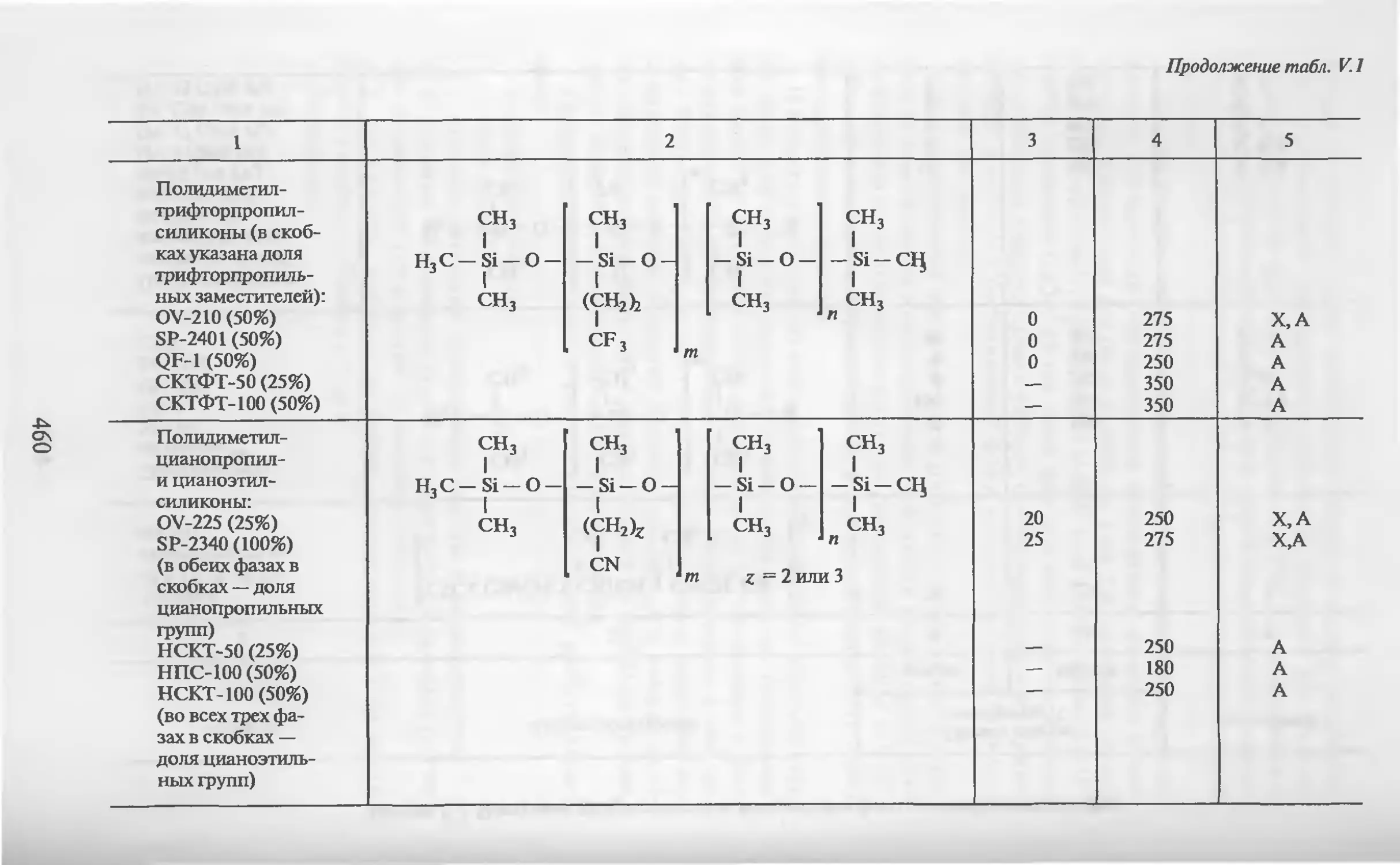



































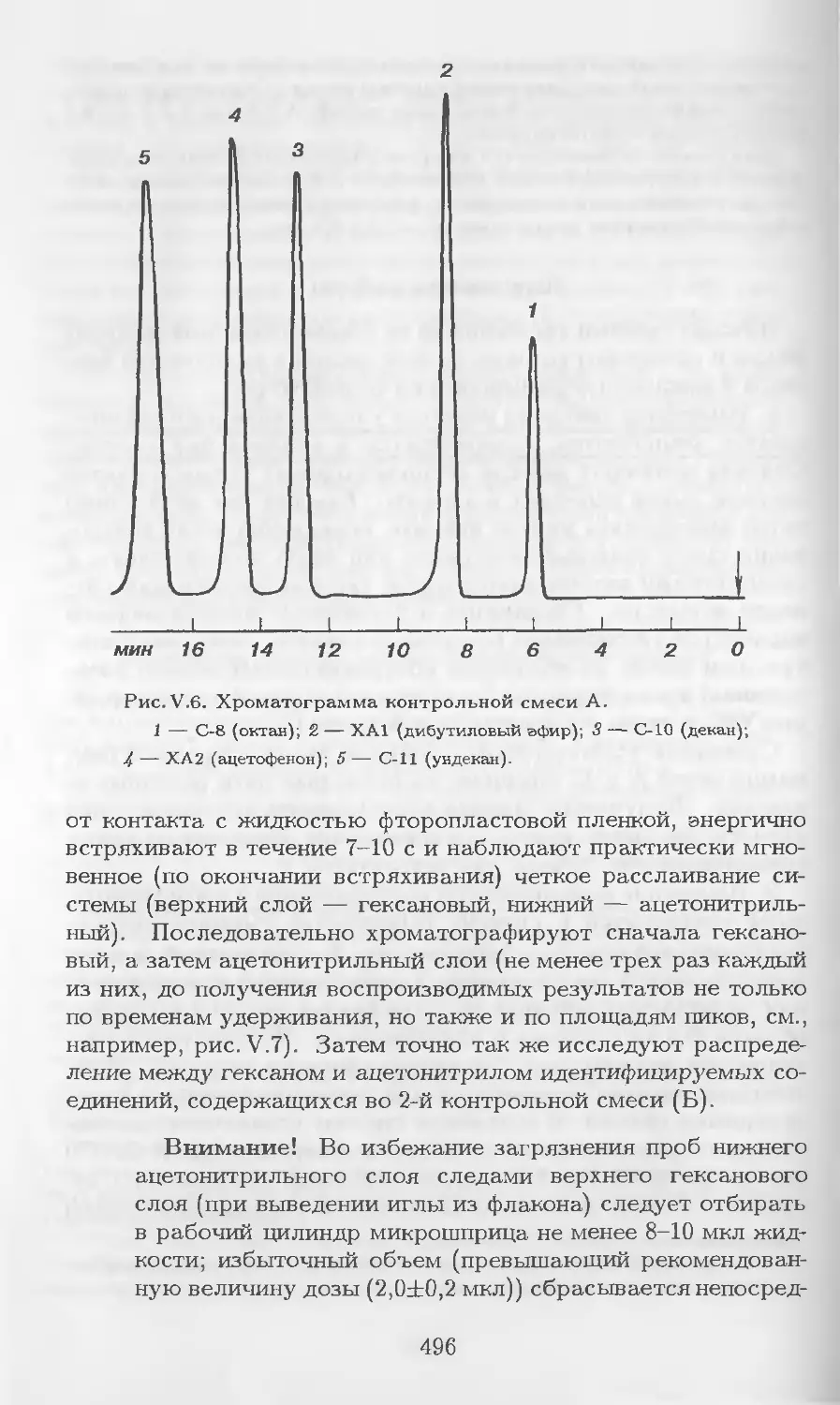
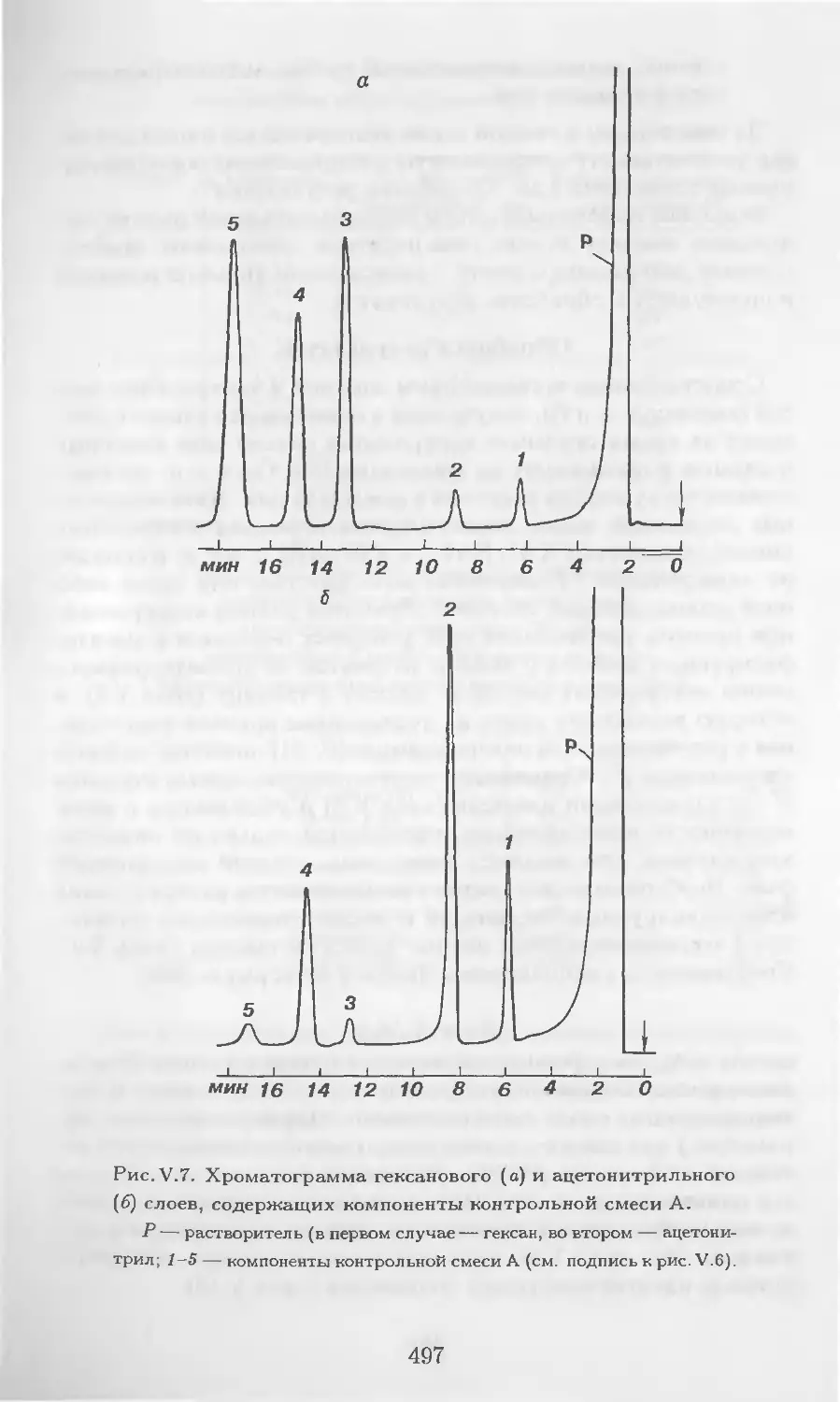
















































































































Автор: Столяров Б.В. Савинов И.М.. Витенберг А.Г. Карцова Л.А.
Теги: другие физико-химические методы анализа (кроме оптических) аналитическая химия химия хроматография
ISBN: 5-288-01938-X
Год: 2002




Текст
УДК 543.544
ББК 24.46
П 69-’
Авторы: Б. В. Столяров, И. М. Савинов, А.Г.Витенберг,
Л.А.Карцова, И. Г. Зенкевич, В. И. Калмановский,
Ю. А. Каламбет.
Рецензенты: кафедра аналит. химии хим. ф-та МГУ; (зам.
зав. кафедрой, д-р хим. наук О. А. Шпигун); д-р
техн, наук Ю. И. Холъкин (СПбГЛА).
Печатается по постановлению
Редакционно-издательского совета
С.-Петербургского университета
Практическая газовая и жидкостная хроматогра-
П69 фия: Учеб, пособие / Б. В. Столяров, И. М. Савинов,
А. Г. Витенберг и др. — СПб.: Изд-во С.-Петербург ун-
та, 2002. 616 с.
ISBN 5-288-01938-Х
В учебном пособии изложены общие приемы и методы работы
на газовых и жидкостных хроматографах, подробно описано при-
менение газовой и жидкостной хроматографии для аналитических
целей, рассматриваются специальные методы идентификации и
установления структуры органических соединений.
Дана характеристика серийно выпускаемых отечественных га-
зовых и жидкостных хроматографов, в том числе оборудования,
материалов и реагентов для тонкослойной хроматографии. Рас-
смотрены приемы и методы количественной расшифровки хрома-
тограмм, включая применение компьютеризованных систем реги-
страции и обработки сигналов детекторов газовых и жидкостных
хроматографов.
Книга предназначена для студентов химических и медицинских
факультетов университетов, химико-технологических и медицин-
ских академий и институтов; она может быть полезна преподавате-
лям и аспирантам вузов, научным сотрудникам, инженерам и лабо-
рантам исследовательских и аналитических лабораторий, исполь-
зующих в своей работе хроматографические методы или только
приступающих к их освоению.
Тем. план 1998 г., № 112
Библиотек,
инв. № Д
ISBN 5-288-01938-Х
ББК 24.46
© Б. В. Столяров,
И. М. Савинов,
А. Г. Витенберг и др.,
2002
© Издательство
С.-Петербургского
университета, 2002
ПРЕДИСЛОВИЕ
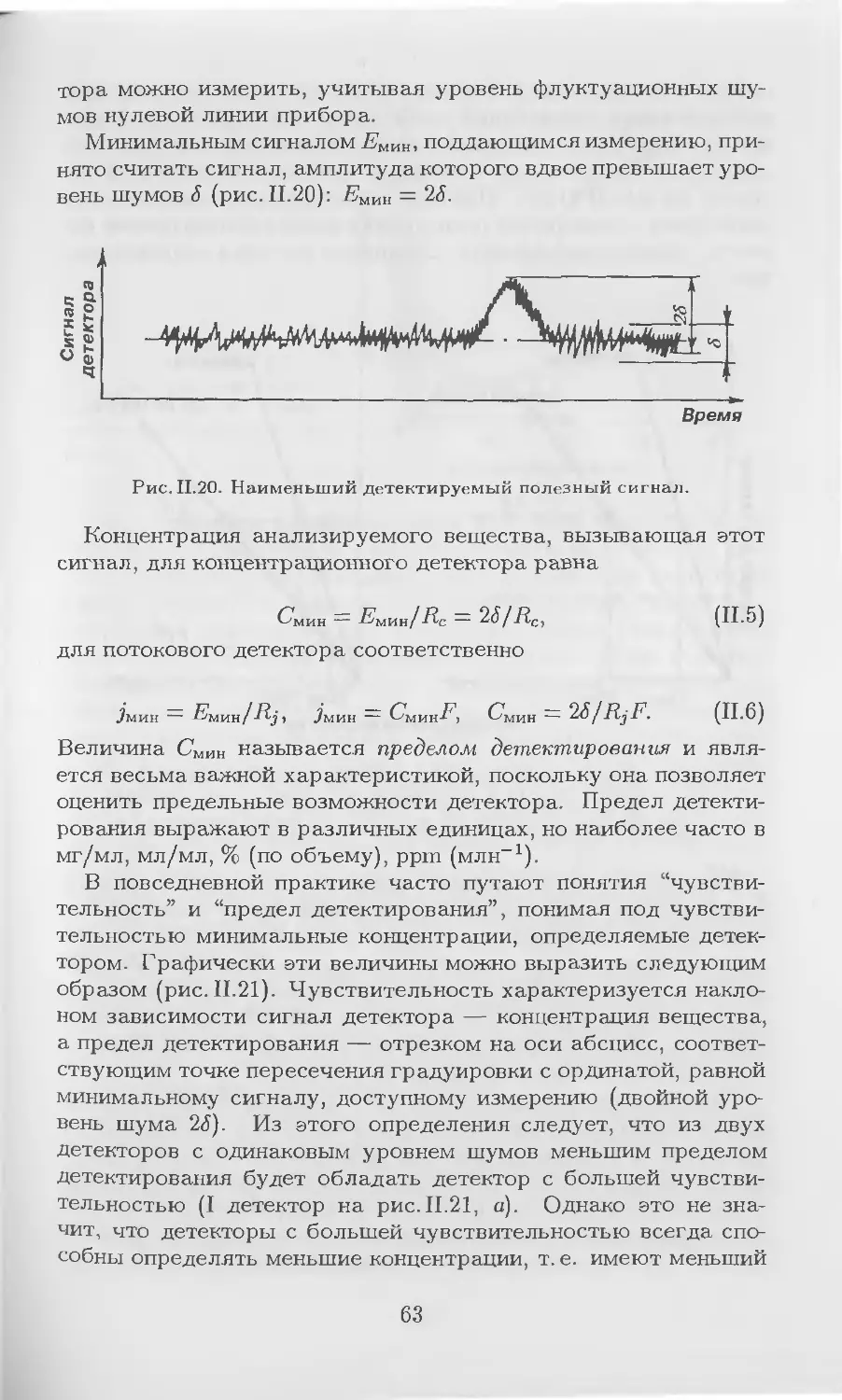
Уходящий XX век ознаменован множеством ярчайших науч-
ных открытий, среди которых хроматография занимает одну из
лидирующих позиций. Хроматографические методы исследова-
ния остро необходимы всем естествоиспытателям, в первую оче-
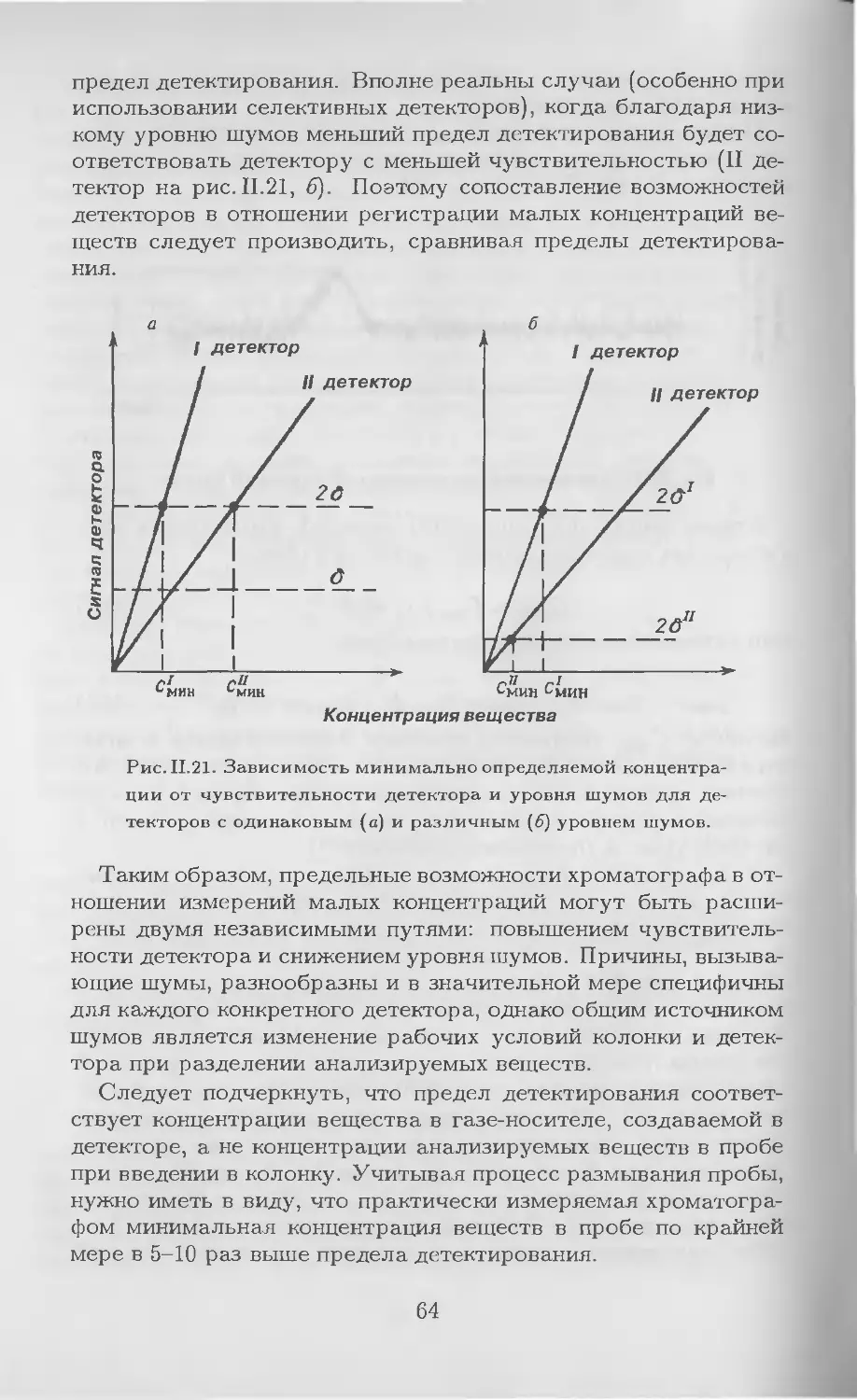
редь, химикам, физикам, биологам, медикам, геологам, а также
специалистам других смежных областей науки и промышленно-
сти (энергетикам, нефтяникам, криминалистам). Этот список
можно было бы продолжить.
Широчайшая сфера применения, надежность и экспрессность
получения информации, сравнительная простота аппаратурно-
го оформления— вот главные причины того, что газовая и жид-
костная хроматография отодвинули на второй план известные
методы разделения и исследования состава сложных многоком-
понентных смесей химических веществ.
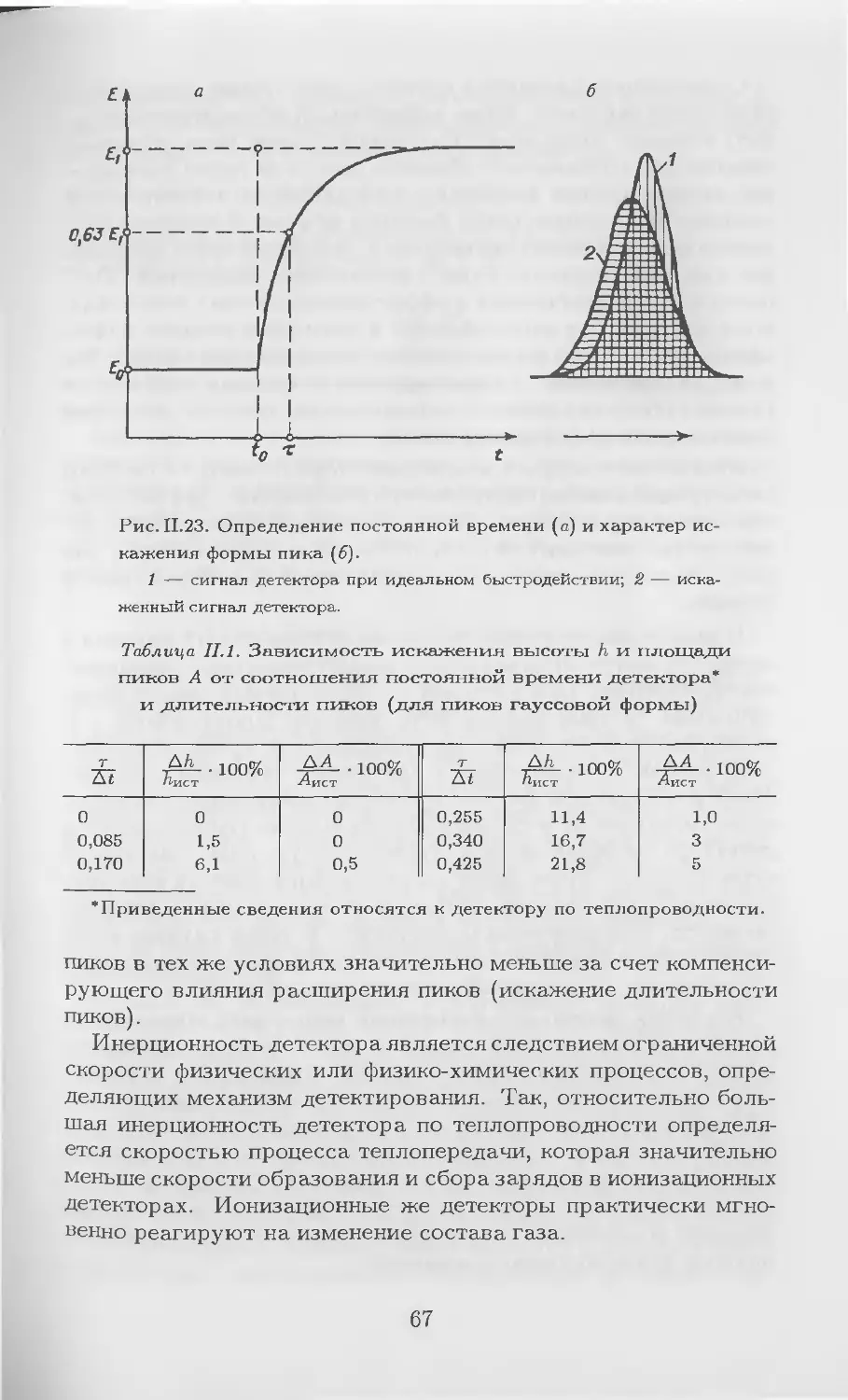
Всеобщий интерес к газовой и жидкостной хроматографии
вызвал появление множества научных статей, специальных
сборников, монографий и справочников. Однако при этом ощу-
щается острый недостаток учебных руководств, пригодных для
использования студентами в жестких рамках учебных планов и
содержащих достаточно подробное и, вместе с тем, лаконичное
описание современной серийной аппаратуры.
При подготовке данной книги частично использованы сведе-
ния из давно уже ставшего библиографической редкостью “Ру-
ководства к практическим работам по газовой хроматографии”
(авторы— Б. В. Столяров, И. М. Савинов, А. Г. Витенберг; на-
учный редактор — проф. Б. В. Иоффе), выпущенного Л О “Хи-
мия” 3-м изданием в 1988 г. Особенностью настоящего учебного
пособия, не имеющего аналогов в отечественной учебной лите-
ратуре, является объединение и изложение материала, охваты-
вающего аппаратурное оформление и методологию выполнения
качественных и количественных измерений как в газовой, так
3
и в жидкостной хроматографии с единых авторских позиций.
При этом основные выработанные многолетней мировой прак-
тикой экспериментальные приемы качественной и количествен-
ной газовой и жидкостной хроматографии проиллюстрированы
отдельными лабораторными работами, сопровождаемыми по-
дробными описаниями.
Практическая направленность книги оправдывает отсутствие
в ней глав с детальным изложением теоретических основ раз-
личных хроматографических методов (как уже отмечалось, эти
вопросы многократно освещались как в научной, так и в учеб-
ной литературе, в том числе изданной относительно недавно).
В необходимых случаях в тексте приведены ссылки на исполь-
зованные литературные источники, опубликованные в разное
время, вплоть до 1998 г., список которых весьма внушителен
(более 400 названий).
Веянием времени в современной аналитике являются актив-
ное использование гибридных методов исследования состава
сложных образцов, компьютерная обработка и метрологиче-
ская аттестация результатов анализа. Без освещения этих
аспектов практической газовой и жидкостной хроматографии
книга была бы явно не полной.
Изложение материала каждой главы книги, бесспорно, отра-
жает личный опыт авторов, одни из которых принимали уча-
стие в создании серийной отечественной аппаратуры для га-
зовой и жидкостной хроматографии (приборы серии “Цвет”) и
программного обеспечения для обработки хроматографических
данных в режиме on-line на ЭВМ (система “МультиХром”), а
другие — в испытании и длительной эксплуатации этих при-
боров и программных средств в научной и учебной лаборато-
рии хроматографии С.-Петербургского университета. Лабора-
торией этой в течение 25 лет с момента основания руководил д-р
хим. наук профессор Б. В. Иоффе, оказавший огромное влияние
на становление хроматографической науки и хроматографиче-
ского образования в нашей стране. К великому огорчению, эта
книга выходит уже без него.
Главы I и IV (за исключением нескольких названных ниже
разделов) и работа 13 написаны совместно канд. хим. наук
доцентами Б. В. Столяровым и Л. А. Карцевой, которые явля-
ются также авторами главы III, разделов IV.2.6; IV.4 и главы VI
(Л. А. Карцева), а также разделов II.1.4.9, IV.2 5, IV.2.7, IV.2.9,
IV.2.10 и главы V (Б. В. Столяров). Глава II написана глав-
ным метрологом ОАО “Цвет” канд. хим. наук И. М. Савиновым
и ведущим научным сотрудником НИИ Химии СПбГУ д-ром
хим. наук, профессором А. Г. Витенбергом, являющимся также
4
автором раздела IV.3.5. При изложении теории детектирования
использованы материалы лекций главного метролога Дзержин-
ского регионального центра экомониторинга д-ра техн, наук,
профессора В. И. Калмановского, им же написан раздел IV.6.
Автором раздела IV.2.8 и Приложения является руководитель
лаборатории газовой хроматографии НИИ Химии СПбГУ д-р
хим. наук И. Г. Зенкевич. Раздел IV.5 написан руководителем
АО “Амперсенд”, канд. физ.-мат. наук Ю. А. Каламбетом. В
процессе подготовки рукописи к печати материалы отдельных
глав обсуждались всем авторским коллективом, который выра-
жает глубокую признательность заместителю генерального ди-
ректора ОАО “Цвет” д-ру хим. наук, профессору Я.И. Яшину
за предварительное ознакомление с описанием аппаратуры по
газовой и жидкостной хроматографии и канд. хим. наук, ст.
научн. сотр. А. Н. Мариничеву за полезную дискуссию по раз-
делу IV.5 и их ценные советы.
Книга родилась по инициативе В. Д. Пиастро, способствовав-
шего на посту главного редактора Ленинградского отделения
издательства “Химия” выходу в свет 2-го и 3-го изданий упо-
мянутого выше прототипа настоящего руководства. Оно заду-
мано, в первую очередь, как учебное пособие к лабораторным
работам для студентов химических, химико-технологических,
медицинских и экологических специальностей вузов, однако ав-
торы надеются, что книга будет востребована также дипломи-
рованными специалистами — аспирантами, научными сотруд-
никами, инженерами и лаборантами исследовательских и при-
кладных аналитических лабораторий, использующих в своей
работе хроматографические методы анализа или только при-
ступающих к их освоению.
При подготовке лабораторных работ и их проведении в Учеб-
но-научном центре химии С.-Петербургского государственного
университета неоценимую помощь авторам оказали и продол-
жают оказывать ст. лаборант А. Г. Еникеева, ст. инженеры
Е. Г. Румянцева и Т Н. Пичугина, ведущие инженеры Э. П. Буб-
нов и Л. И. Макаров.
Авторский коллектив выражает глубокую благодарность
редактору Издательства С.-Петербургского университета
Л. П. Макаренковой за кропотливый, высокопрофессиональный
труд, способствовавший безусловному улучшению стиля и
структуры книги в целом, а также руководству ОАО “Цвет”
за финансовую поддержку настоящего издания.
По поручению авторского коллектива
Б. В. Столяров
5
Глава 1
ВОЗНИКНОВЕНИЕ И ОСНОВНЫЕ ЭТАПЫ
РАЗВИТИЯ ХРОМАТОГРАФИИ.
КЛАССИФИКАЦИЯ МЕТОДОВ ГАЗОВОЙ
И ЖИДКОСТНОЙ ХРОМАТОГРАФИИ
Хроматография как эффективный метод анализа и исследова-
ния веществ и их смесей родилась в начале XX века и к насто-
ящему времени сформировалась в самостоятельную научную
дисциплину, изучающую распределение химических веществ в
системе двух контактирующих несмешивающихся фаз, из кото-
рых, как правило, одна подвижна и перемещается относительно
другой, неподвижной.
В 1902-1903 гг. русский ученый М. С. Цвет* после серии пред-
варительных экспериментов достиг разделения сложной смеси
растительных пигментов при пропускании петролейно-эфирной
вытяжки из листьев растений через стеклянную колонку, запол-
ненную порошкообразным карбонатом кальция. При этом воз-
никал ряд окрашенных зон, по числу которых можно было су-
дить о сложности состава анализируемой смеси. В докладе «О
новой категории адсорбционных явлений и о применении их к
биохимическому анализу», прочитанном 8 (21) марта 1903 г. на
заседании отделения биологии Варшавского общества естество-
испытателей, М. С. Цвет отмечал: «...Еслипетролейно-эфирный
раствор хлорофилла профильтровать через столбик адсорбента
(я применяю для этого, главным образом, углекислый кальций,
плотно набитый в узкие стеклянные трубки), то пигменты по
расположению их в адсорбционном ряду отличаются отдель-
*Михаил Семенович Цвет (1872-1919) — приват-доцент Варшавского
университета, впоследствии преподаватель ботаники и микробиологии
Варшавского политехнического института, с 1917 г. — профессор Юрьев-
ского университета, член Петербургского общества естествоиспытателей.
Материалы о жизни и научной деятельности М. С. Цвета, об истории от-
крытия и развития различных методических вариантов хроматографи-
ческого метода анализа см. в [1—3], а также в монографиях: Сенченкова
Е.М. Михаил Семенович Цвет. М.: Наука, 1973. 306 с.; Сенченкова Е.М.
М. С. Цвет — создатель хроматографии. М.: Янус-К, 1997. 439 с.
6
ними окрашенными зонами по столбику сверху вниз, благо-
даря тому, что пигменты с более сильно выраженной адсорб-
цией вытесняют книзу слабее удерживаемые. Это разделение
становится практически совершенным, если после пропускания
вытяжки пигментов сквозь столбик адсорбента его промывать
струей чистого растворителя. Как лучи света в спектре, в стол-
бике углекислого кальция закономерно располагаются различ-
ные компоненты смеси пигментов, давая возможность своего ка-
чественного и количественного определения. Получаемый та-
ким образом препарат я называю хроматограммой, а предлага-
емую методику — хроматографической» [1].
Так впервые прозвучало слово «хроматография», произве-
денное ученым от греч. урмрато — цвет, окраска и урасрсд —
пишу, описываю.*
Пропуская через колонку растворители различной природы,
оказалось возможным регулировать степень распределения зон
по слою адсорбента: сдвигать или раздвигать их, способствуя
большей селективности их разделения и повышению точности
последующего качественного и количественного анализа. Вы-
деляя условия, при которых осуществимо разделение компонен-
тов этим методом, ученый отмечает: «Для того, чтобы два на-
ходящихся в растворе вещества могли быть разъединены ад-
сорбционным методом, необходимо, чтобы они занимали неоди-
наковый ранг в адсорбционном ряду. Так как расположение ве-
ществ в адсорбционном ряду зависит от природы растворителя,
то крайне ничтожной является вероятность, чтобы два вещества
были в нескольких растворителях равносильными по адсорбци-
онному сродству».
Не оцененные по достоинству современниками исследования
М. С. Цвета, более 20 лет остававшиеся невостребованными, по-
служили мощным стимулом к дальнейшему развитию хромато-
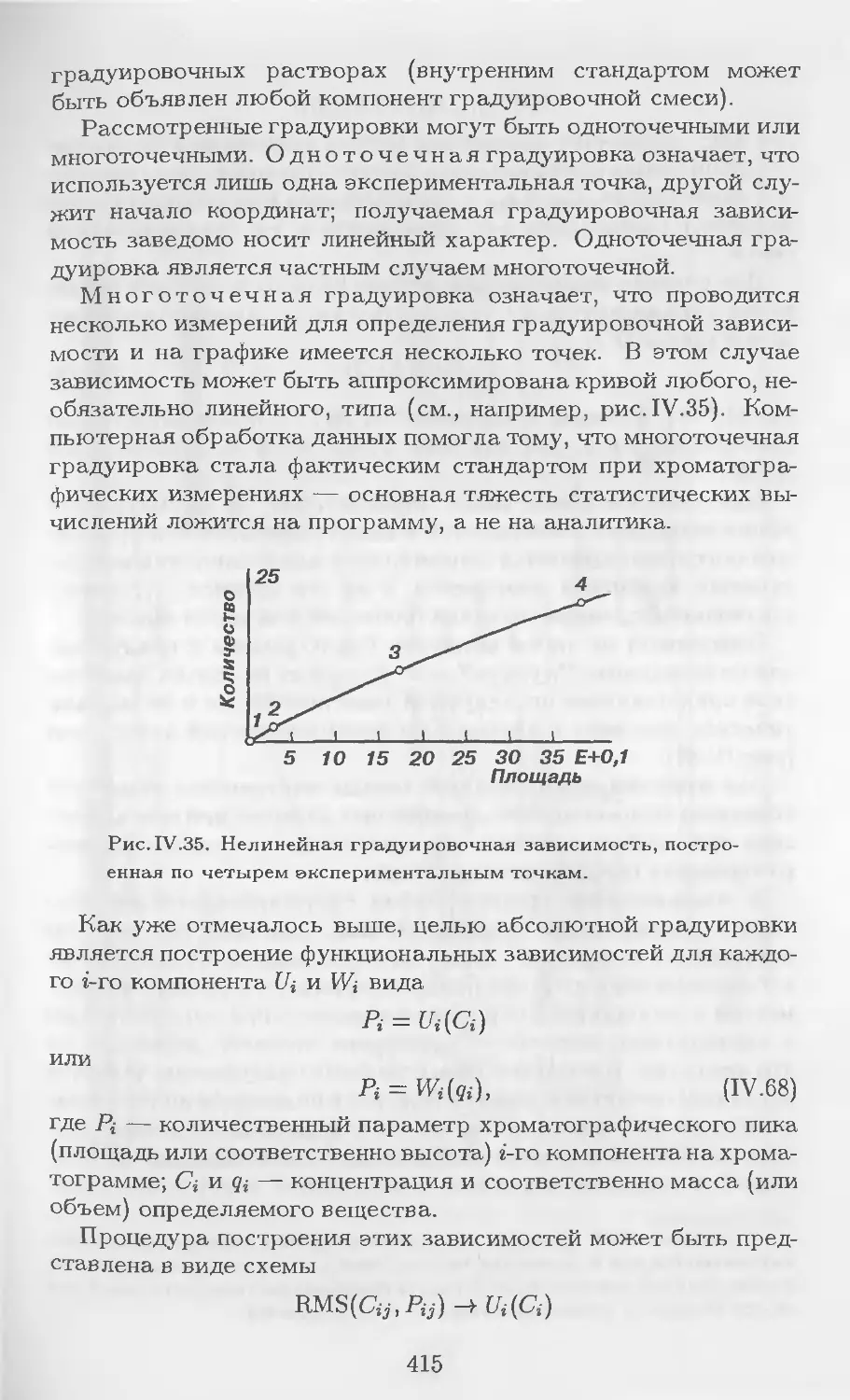
графии на протяжении всего XX века** (табл. 1.1).
С позиций современного знания место хроматографии в це-
лом и отдельных ее разновидностей в ряду других методов раз-
*Впоследствии к разряду хроматографических стали относить различ-
ные методики, объединяемые общим принципом разделения (различное
распределение компонентов смеси между двумя несмешивающимися фа-
зами), но не обязательно приводящие к появлению окрашенных зон, полос
или пятен отдельных компонентов.
**Краткая история хроматографии в «доцветовский» период предста-
влена в [4], а за последующие 90 лет — в обзоре [5], который завершается
словами: «М. С. Цвет любил повторять изречение французского физио-
лога Клода Бернара: “Всякий научный прогресс есть прогресс метода”.
Судьба распорядилась так, что именно М. С. Цвет создал такой метод,
который в наибольшей степени способствовал научному прогрессу».
7
Таблица 1.1. Основные этапы становления
и развития хроматографических методов
Годы Авторы События и комментарии
1 2 3
1902-1903 М. С. Цвет Зарождение хроматографии как общего метода разделения веществ [1]. Откры- тие жидкостно-адсорбционного хрома- тографического анализа
1931 Р.Кун, А. Винтерштайн, Э. Ледерер Возрождение хроматографического ме- тода М. С. Цвета. Исследование при- родных пигментов из моркови, яичных желтков и др. [6]
1938 Н. А. Измайлов, М. С.Шрайбер Создание тонкослойной (плоскостной, или планарной) хроматографии. Разде- ление алкалоидов, содержащихся в ле- карственных растениях, на стеклянной пластинке с незакрепленным слоем ок- сида алюминия [7]
1941 Г. Брокман, Г. Шоддер Стандартизация оксида алюминия (вве- дение пяти степеней активности в зави- симости от процентного содержания во- ды) как неподвижной фазы [8]
1941 А. Лж.П. Мартин, Р. Л.М.Синг Создание жидкостно-жидкостной (рас- пределительной) хроматографии. Раз- деление аминокислот и их ацетильных производных на силикагеле, смоченном водой (неподвижная фаза), в потоке ор- ганического растворителя (хлороформ, ©тилацетат) [9]
1944 Р Консден, А. Гордон, А. Мартин Разработка распределительного вари- анта жидкостной хроматографии на бу- маге (бумажная хроматография) [10, 11]. Разделение смеси аминокислот на по- лоске бумаги («Ватман № 1» размером 1,5x20-56 см) в трубчатой вертикальной камере, подвижная фаза — органические растворители, частично смешивающи- еся с водой (такие, как трет-пентило- вый спирт, бензиловый спирт и др.)
1951 Д.Х. Кемпбел, Е. Люшер, Л. С. Лерман Формулирование принципов аффинной (биоспецифической) хроматографии [12]. Химической сшивкой белка и диазотиро- ванной n-аминобензилцеллюлозы осу-
8
Продолжение табл. 1.1
1 2 3
ществлен синтез сорбента, способного к высокоизбирательному связыванию пеп- тидных и белковых соединений. Новый принцип хроматографического разделе- ния реализован на примере выделения и очистки антител, содержащихся в сыво- ротках
1952 А. Т. Джеймс, А. Дж. П. Мартин Создание газо-жидкостной хроматогра- фии. Разделение и количественное опре- деление летучих жирных кислот (от му- равьиной до додекановой) на колонке, заполненной диатомитовым носителем, смоченным силиконовым маслом, моди- фицированным стеариновой кислотой; подвижная фаза — азот [13]
1952 — Присуждение Нобелевской премии А. Дж. Мартину и Р. Л. М. Сингу за от- крытие распределительной хроматогра- фии и применение ее в биохимии
1956 Э. Шталь Стандартизация метода тонкослойной хроматографии (подготовка сорбентов и пластинок, оценка результатов разде- ления) [14]
1956 С. Мур, В. X. Штейн Создание аминокислотного анализатора, работающего по принципу ионообменной хроматографии (реализованы отличи- тельные особенности инструментальной жидкостной колоночной хроматографии в современном восприятии: градиентное элюирование, постколоночная деривати- зация, фотометрическое детектирование) [15]
1957-1958 М. Ж. Э.Голей Возникновение капиллярной газовой хроматографии. Разработана теория хроматографии для узких (диаметр 0,25-0,5 мм) и длинных (до 90 м) медных и стальных колонок, внутренние стенки которых покрыты тонким слоем непод- вижной фазы. Даны первые рекоменда- ции по технике приготовления подоб- ных колонок и первые примеры их прак- тического использования [16, 17]
9
Окончание табл. 1.1
1 2 3
1959 Дж. Порат, П. Флодин Открытие гель-хроматографии (эксклю- зионной хроматографии). Разделение смеси глюкозы и фракций полисахари- дов (декстранов), различающихся моле- кулярной массой, на слое гидрофиль- ного геля (сефадекса). Подвижная фа- за — вода [18]
1961 Ф.Гельферих Создание лигандообменной хроматогра- фии. Предложен новый метод анализа и препаративного разделения смесей сое- динений (лигандов), способных образо- вывать нестойкие комплексы с ионами некоторых металлов, введенных в состав неподвижной фазы. Первым примером практической реализации нового метода стало разделение растворенных в воде аммиака и органических диаминов на слое катионообменника, заряженного ионами Си2+ или Ni2+ [19]
1962 Е. Клеспер, А. X. Корвин, Д. А. Тернер Зарождение сверхкритической флюид- ной хроматографии. Осуществлено раз- деление порфиринов никеля с использо- ванием в качестве подвижной фазы фре- она в сверхкритическом состоянии [20]
1982-1983 Л. Н. Москвин, А. И. Горшков, М. Ф. Гумеров; Дж. С. Г иддингс, М. Н. Майерс Открытие жидкостно-газовой хромато- графии. Разделение растворенных в воде газов при фильтрации их водного раствора через колонку с гидрофобным сорбентом — политетрафторэтиленом [21-23; 24, с. 168]
деления, основанных на различном перераспределении веществ
между несмешивающимися фазами, и естественная логическая
взаимосвязанность всех названных методов обсуждаются в [24,
25].
Значительной вехой в развитии хроматографических мето-
дов явилось создание в 1938 г. варианта тонкослойной хро-
матографии [7]. Широкое распространение этот вид хрома-
тографии получил благодаря, в первую очередь, исследова-
ниям Э. Шталя, разработавшего методы стандартизации наибо-
лее распространенных сорбентов, технику подготовки пластин,
всех этапов хроматографического анализа, обобщенных в из-
10
вестной монографии (немецкое издание вышло в 1962 г.; на рус-
ском языке она опубликована в 1965 г.) [14]. С современными
возможностями обсуждаемого метода можно познакомиться в
книгах [26-30].
В 1941 г. А. Дж. П. Мартином и Р. Л. М. Сингом сообщается о
создании распределительной (жидкостно-жидкостной) хро-
матографии [9]. В отличие от хроматографии в вышеназванных
системах жидкость — твердое тело (адсорбент), где разделение
обусловлено различием в молекулярной адсорбции компонен-
тов анализируемых смесей на поверхности зерен адсорбента, в
жидкостно-жидкостной хроматографии определяющими стано-
вятся различия в распределении веществ между двумя несме-
шивающимися жидкостями, одна из которых полярна, а другая
неполярна. Если полярность неподвижной фазы превосходит
полярность подвижной, говорят о нормально-фазовом хромато-
графическом режиме. Если же соотношение полярностей кон-
тактирующих фаз иное, имеют дело с обращенно-фазовым вари-
антом жидкостной хроматографии.
По общим оценкам каждые два из трех выполняемых методом
жидкостно-жидкостной распределительной хроматографии ана-
лизов относятся к обращенно-фазному режиму (~70%), для ко-
торого разработаны и внедрены сорбенты на основе силикагеля
с химически привитыми алкильными радикалами (от С 2 до С22) •
Чаще всего используются сорбенты с Се-, Сщ- и Cjg-группами.
Привитые фазы на основе силикагеля стабильны в водных рас-
творах в интервале pH 2-8. Этим объясняется эффективное их
использование при анализе биологических объектов. Равнове-
сие в системе колонка — элюент в случае обращение-фазных
сорбентов устанавливается достаточно быстро.
Мартину и Сингу, создателям нового варианта хроматогра-
фического разделения, уже тогда (в 1940-1941 гг.) было ясно
(о чем прямо говорится в их статье [9]), что в качестве подвиж-
ной фазы можно использовать не только жидкость, но также пар
или газ. Выполненная Джеймсом и Мартином 11 лет спустя об-
стоятельная экспериментальная проверка этой идеи дала бле-
стящие результаты [13] . Семейство хроматографических мето-
дов обогатилось еще одним — газовой хроматографией, бы-
стро завоевавшим всеобщее признание и получившим большое
распространение.* Методу газовой хроматографии посвящена
*Исторически первые работы по газо-адсорбционной хроматографии
были выполнены Лж.Е. Хессе в Германии и опубликованы в 1941 г., од-
нако по понятным причинам они долгое время оставались малоизвест-
ными [5].
11
обширная литература, сошлемся здесь лишь на четыре книги,
недавно выпущенные на русском языке [2, 31-33].
Рожденная задолго до газовой хроматографии жидкостно-
жидкостная хроматография длительное время развивалась до-
вольно медленно, что было связано, главным образом, с трудно-
стями в решении многих чисто технических проблем инструмен-
тального оформления метода. Перелом в темпах развития жид-
костной хроматографии произошел примерно в середине 70-х
годов. Возможности современной высокоэффективной жидкост-
ной хроматографии отражены в книгах [2, 34-37]. Анализ тен-
денций развития инструментальных методов разделения за пе-
риод 1952-1993 гг. представлен в обзоре [38].
Существенным этапом для развития хроматографических ме-
тодов анализа, повышения эффективности разделения слож-
ных многокомпонентных смесей послужили выполненные
М.Ж. Э. Голеем в 1957-1958 гг. исследования по капиллярной
хроматографии [16, 17].
В работах Голея было дано теоретическое обоснование и
экспериментальное подтверждение повышенной эффективности
разделения хроматографических зон при использовании вме-
сто так называемых насадочных колонок (относительно корот-
ких и широких трубок (1-3 м х 2-6 мм), заполненных мелко-
диспергированным сорбентом-насадкой) узких и длинных по-
лых капилляров (25-50 м х 0,25-0,5 мм) с неподвижной фазой,
закрепленной на их внутренних стенках. Довольно продолжи-
тельный период развитие капиллярной хроматографии сдер-
живалось трудностями в отработке технологии приготовления
капиллярных колонок с воспроизводимыми характеристиками
[39]. В настоящее время на долю капиллярных колонок прихо-
дится подавляющее количество всех выполняемых газохромато-
графических анализов [40, 41].
Высокие темпы развития капиллярной газовой хроматогра-
фии стимулировали рождение и последующий прогресс высоко-
эффективной капиллярной жидкостной [42, 43] и сверхкритиче-
ской флюидной хроматографии [20, 44, 45]. Флюидом принято
называть особое агрегатное состояние вещества, находящегося
при температуре и давлении, превышающих критические. При-
влекательность применения флюидов в качестве подвижных фаз
обусловлена тем, что их плотность (при низкой вязкости, почти
такой же, как и у газов) в 102—103 выше, чем у газов, а скорость
диффузии молекул хроматографируемых соединений во флюиде
примерно в 102 раз выше, чем в жидкости. Эти свойства флюи-
дов открывают принципиальную возможность выполнения хро-
матографических анализов смесей весьма высококипящих со-
12
единений, низкая летучесть которых не позволяет проводить
их разделение методом газовой хроматографии. В сравнении
с высокоэффективной жидкостной хроматографией сверхкрити-
ческая флюидная капиллярная хроматография обеспечивает го-
раздо большую эффективность разделения (за счет использо-
вания колонок большей длины (5-25 м)), а при разделении на
укороченных колонках позволяет существенно сократить про-
должительность анализа. Интерес к практическому использо-
ванию этих потенциальных возможностей метода резко возрос
в начале 80-х годов с расширением рынка доступных высоко-
эффективных кварцевых капиллярных колонок и с началом вы-
пуска приборов для сверхкритической флюидной хроматогра-
фии [44, 45].
Особое место занимают методы жидкостной хроматографии,
используемые для разделения и анализа высокомолекулярных
соединений, синтетических и природных полимеров (эксклю-
зионная, или гель-хроматография), очистки, селективного ана-
лиза белков ферментов (аффинная, или биоспецифическая, хро-
матография) .
В эксклюзионной хроматографии молекулы веществ раз-
деляются по размеру (устаревшее название метода — ситовая,
или молекулярная хроматография; гель-хроматография) за счет-
их различной способности проникать в поры неподвижной фазы.
При этом первыми выходят из колонки наиболее крупные моле-
кулы (с большой молекулярной массой), последними — веще-
ства с малыми размерами молекул, свободно проникающие в
поры сорбента. Разделение анализируемых веществ происхо-
дит за счет перераспределения молекул между растворителем,
находящимся
внутри пор сорбента, и растворителем, протекающим между
его частицами. Таким образом, удерживание молекул в экс-
клюзионной (от exclusion — исключение) колонке зависит от со-
отношения размеров анализируемых молекул и пор сорбента.
Молекулы или ионы, размеры которых находятся между макси-
мальным и минимальным диаметром пор геля, разделяются на
отдельные зоны. Этот метод нашел наибольшее распростране-
ние в биохимических исследованиях [46] и в химии полимеров
(в том числе и в варианте тонкослойной хроматографии) [47].
Аффинная хроматография основана на исключительной
способности биологически активных веществ связывать специ-
фически и обратимо другие вещества, называемые в общем слу-
чае лигандами, или аффинантами. Научные основы метода (см.
табл. 1.1) были целенаправленно заложены в 1951 г. в США [12].
В 1953 г. аффинная хроматография была использована для вы-
13
деления ферментов (получение тирозиназы на колонке с целлю-
лозой, соединенной эфирными связями с остатками резорцина).
Основной предпосылкой для проведения аффинной хроматогра-
фии является образование специфического комплекса между вы-
деляемым ферментом и аффинным лигандом, связанным с нерас-
творимым носителем. С разработкой аффинной хроматографии
стали возможными анализ и разделение биологически активных
веществ [48].
Лигандообменная хроматография открывает возможность
аналитического и препаративного разделения нейтральных со-
единений (лигандов), содержащих в качестве донорных гетеро-
атомы N, S, О и способных образовывать нестойкие комплексы
(иначе — аддукты) с ионами металлов, введенными в неподвиж-
ную фазу. Первооткрыватель метода Ф. Гельферих в качестве
потенциально приемлемых сорбентов использовал ионообмен-
ники, содержащие комплексообразующие ионы следующих ме-
таллов: Cu2+, Cu+, Ni2+, Ag+, Со3+ и др. [19]. Нейтральные
молекулы или анионы, способные образовывать с названными
ионами металлов координационные связи, сорбируются из жид-
кой или газообразной подвижной фазы на свободных активных
центрах сорбента или замещают другой, ранее сорбированный
лиганд, предварительно образовавший менее прочный комплекс
с ионом металла. Круг лигандов, которые могут быть выделены
из сложных смесей или разделены, достаточно широк: аммиак,
органические амины, непредельные углеводороды, моно- и по-
лиатомные спирты, фенолы, карбоновые кислоты, серосодержа-
щие органические соединения и др.; в роли лиганда может вы
ступать также вода.
В последнее время лигандный обмен стали осуществлять не-
посредственно в жидкой неподвижной фазе, несущей в себе
ионы металла. В этом случае в качестве неподвижной фазы
используют уже не ионообменник, а типичный для современ-
ной обращенно-фазной высокоэффективной жидкостной хрома-
тографии неполярный сорбент, например силикагель С-18. Как
отмечается в [49], если при этом лиганды образуют незаряжен-
ные комплексы, то последние избирательно распределяются ме-
жду подвижной и неподвижной фазами, что обусловливает их
разделение.
Замечательной особенностью лигандообменной хроматогра-
фии является ярко выраженная зависимость прочности образу-
ющихся комплексов от структурных особенностей (геометрии
молекул) лигандов, на чем основано, в частности, разделение
оптических изомеров аминокислот, квалифицируемое в [49] как
самое серьезное достижение метода.
14
Многообразие рассмотренных выше хроматографических ме-
тодов и решаемых ими задач чрезвычайно осложняет попытки
предложить исчерпывающее определение хроматографии в ее
триединстве (наука — процесс — метод); в развернувшейся по
этому вопросу дискуссии [50-52] последнее слово, возможно,
еще не сказано. В недавно опубликованной разработанной На-
учным советом по хроматографии РАН номенклатуре [53] даны
следующие формулировки основных понятий.*
Хроматография — 1) наука о межмолекулярных взаимодей-
ствиях и переносе молекул или частиц в системе несмешиваю-
щихся и движущихся относительно друг друга фаз;
2) процесс дифференциального многократного перераспреде-
ления веществ или частиц между несмешивающимися и движу-
щимися относительно друг друга фазами, приводящий к обо-
соблению концентрационных зон индивидуальных компонентов
исходной смеси этих веществ или частиц;
3) метод разделения смесей веществ или частиц, основанный
на различии в скоростях их перемещения в системе несмешива-
ющихся и движущихся относительно друг друга фаз.
Подвижная фаза — поток жидкости, флюида или газа, пере-
мещающий компоненты разделяемой смеси вдоль неподвижной
фазы.
Неподвижная фаза — твердый сорбент или несмешивающаяся
с подвижной фазой жидкость, на которых осуществляется диф-
ференциальное удерживание и разделение компонентов смеси.
Сорбент — твердое вещество, жидкость или их смеси, спо-
собные поглощать или удерживать газы, пары или растворен-
ные вещества и используемые в хроматографии в качестве не-
подвижной фазы.
Адсорбент — твердый сорбент, концентрирующий на своей
поверхности газы, пары или растворенные вещества.
Абсорбент — твердый или жидкий сорбент, растворяющий в
своем объеме газы, пары или компоненты жидких смесей.
Сорбат — вещество, удерживаемое сорбентом (в хроматогра-
фии — компонент разделяемой смеси).
Элюент — жидкость, флюид или газ, используемые в каче-
стве подвижной фазы.
*Приводятся здесь выборочно. В документе [53] содержится и пред-
лагаемая классификация методов хроматографии по агрегатному состоя-
нию фаз хроматографической системы, по способу перемещения сорбата,
по конфигурации разделяющей системы, по относительной полярности
подвижной и неподвижной фаз, по механизму разделения веществ, по
цели, по химическому превращению сорбата и по способу детектирова-
ния; к отдельной группе отнесены электрофорез и электрохроматографи-
ческие методы.
15
Элюат— выходящий из колонки поток подвижной фазы с ком-
понентами разделяемой смеси.
Благодаря возможности объединения процесса высокоселек-
тивного разделения с последующим высокочувствительным де-
тектированием, хроматография стала самым распространен-
ным методом анализа сложных смесей, позволяющим опреде-
лять до 1000 веществ в одной пробе с пределом обнаружения
на нанограммовом и фемтограммовом уровнях. В современной
аналитической химии 75-80 % всех выполняемых анализов свя-
заны с использованием хроматографических методов. Возника-
ющие новые хроматографические варианты взаимообогащают
друг друга и стимулируют их дальнейшее развитие. Ниже да-
дим определения лишь основных сложившихся к настоящему
времени хроматографических методов.
В зависимости от агрегатного состояния подвиж-
ной фазы хроматографию подразделяют на газовую (подвиж-
ная фаза — газ), жидкостную (подвижная фаза — жидкость) и
сверхкритическую флюидную, где в качестве подвижной фазы
используется флюид.
Понятие (термин) «газовая хроматография» (ГХ) объеди-
няет все методические варианты, в которых подвижная фаза га-
зообразна (находится в состоянии газа или пара).
Газо-адсорбционная хроматография (ГАХ) включает все
варианты газовой хроматографии, в которых неподвижной фа-
зой является активное дисперсионное твердое тело (адсорбент):
древесный уголь, силикагель, цеолиты, пористые полимерные
сорбенты и др.
К газо-жидкостной хроматографии (ГЖХ) относятся все
методические варианты газовой хроматографии, в которых в
качестве неподвижной фазы используется слой жидкости, нане-
сенный на поверхность твердого носителя (зернистый мелкодис-
персный материал или внутренние стенки хроматографической
колонки).
Жидкостная хроматография — хроматографический ме-
тод, в котором подвижной фазой является жидкость. Основ-
ными разновидностями жидкостной хроматографии являются
жидкостно-адсорбционная (разделение соединений происхо-
дит за счет их различной способности адсорбироваться и де-
сорбироваться с поверхности адсорбента с развитой поверхно-
стью, например силикагеля), и жидкостно-жидкостная (раз-
деление компонентов анализируемых смесей осуществляется за
счет различной растворимости в подвижной фазе (элюенте) и в
неподвижной фазе, химически привитой к поверхности твердого
16
сорбента или физически сорбированной этой поверхностью из
раствора в летучем растворителе) *
По механизму разделения веществ выделяют также
ионообменную жидкостную хроматографию. Здесь разде-
ление достигается за счет обратимого взаимодействия ионизи-
рующихся веществ с ионными группами сорбента-ионита; ион
введенного образца, взаимодействуя с фиксированным зарядом
сорбента, обменивается с противоионом.
В основу ион-парной разновидности жидкостной хромато-
графии положены принципы классической экстракции из вод-
ной в органическую фазу ионных веществ в виде ионных пар —
прекрасный метод разделения веществ, склонных к диссоциа-
ции в режиме обращенно-фазной хроматографии. В подвижную
фазу добавляется противоион, способный вступать в селектив-
ное комплексообразование с формированием ионной пары.
Основные преимущества ион-парной хроматографии заклю-
чаются в том, что при анализе могут быть использованы обра-
щенно-фазные системы, и нет необходимости в использовании
ионообменников. При этом одновременно могут быть проана-
лизированы кислоты, основания и нейтральные соединения.
Если на протяжении всего хроматографического анализа со-
став подвижной фазы сохраняется, такой режим называется изо-
кратическим, если же нет, такой вариант анализа называется
градиентным.
Градиентный режим позволяет более эффективно осущест-
вить разделение веществ различной полярности. При умень-
шении размера частиц сорбентов, используемых в жидкостной
хроматографии, до 3-10 мкм резко возрастает эффективность
хроматографического анализа. На основе этого критерия выде-
ляют высокоэффективную жидкостную хроматографию как
в колоночном, так и в плоскостном варианте.
Сверхкритическая флюидная хроматография — хромато-
графический метод, в котором подвижной фазой является веще-
ство, находящееся в сверхкритическом (или субкритическом)
состоянии (флюид).
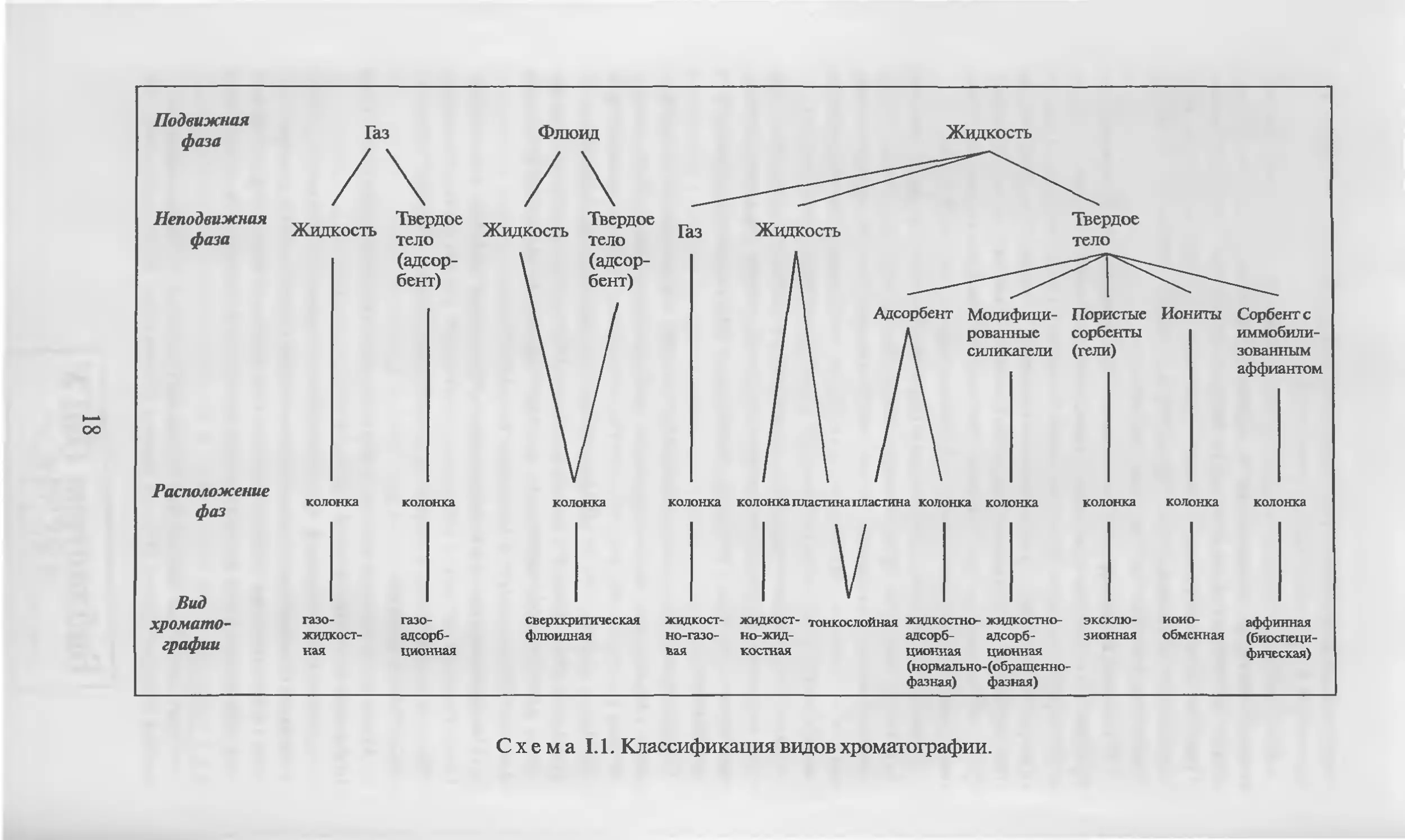
Иерархия всех названных выше хроматографических методов
представлена на схеме 1.1 [25, 54, 55].
С помощью газовой хроматографии можно выполнять каче-
ственное и количественное определение компонентов смесей лю-
бых органических и неорганических газов, жидкостей, твердых
тел, давление пара которых при температуре колонки находится
*Противоточная жидкостная хроматография и ее разновидность —
пенная хроматография [53] — в данном руководстве не рассматриваются.
Подвижная
фаза
Неподвижная
фаза
Расположение
фаз
Вид
хромато-
графии
газо- газо-
жидкост- адсорб-
ная ционная
сверхкритическая жидкост- жидкост- тонкослойная жидкостно- жидкостно- эксклю-
флюидная но-газо- но-жид- адсорб ад сор б- зионная
Ьая костная ционная ционная
(нормально-(обращенно-
фазная) фазная)
ионо-
обменная
аффинная
(биоспеци-
фическая)
Схема 1.1. Классификация видов хроматографии.
в диапазоне 0,133-133 Па (0,001-1 мм рт. ст.), т. е. перегоняю-
щихся без разложения в области температур до 400-500 °C, или
более высококипящих соединений, для которых, однако, отра-
ботана методика воспроизводимого термического разложения.
Методом газовой хроматографии могут также анализироваться
и такие соединения, которые хотя и не попадают в очерченные
границы, но могут быть превращены в летучие производные для
последующего газохроматографического анализа.
Жидкостная хроматография в ее классическом варианте (при
атмосферном давлении) и высокоскоростная, или высокоэффек-
тивная, жидкостная хроматография (ВЭЖХ) при повышенном
давлении позволяют анализировать химические соединения,
ионы, радикалы, вирусы в широком диапазоне молекулярных
масс — от 50 до 10е. Это оптимальный метод анализа хими-
чески и термически нестойких молекул, высокомолекулярных
веществ с пониженной летучестью, что объясняется особой ро-
лью подвижной фазы: в отличие от газа-носителя, элюент в
жидкостной хроматографии выполняет не только транспортную
функцию, способствуя перемещению анализируемых веществ по
слою сорбента; природа и строение компонентов подвижной
фазы контролируют хроматографическое поведение разделяе-
мых веществ.
Среди объектов жидкостной хроматографии белки, нуклеи-
новые кислоты, аминокислоты, красители, полисахариды,
взрывчатые вещества, лекарственные препараты, метаболиты
растений и животных. При выборе условий разделения (при-
роды подвижной и неподвижной фаз) необходимо учитывать мо-
лекулярную массу компонентов образца, их полярность и при-
роду сопутствующих соединений, от которых необходимо отде-
лить исследуемое вещество.
Если молекулярная масса компонентов образца превышает
2000 у. е., то метод, к которому следует обратиться, — эксклю-
зионная хроматография. Для разделения смесей органических
веществ в ионизированной форме и, в первую очередь, сме-
сей неорганических анионов и катионов целесообразно приме-
нение ионной хроматографии с кондуктометрическим детекти-
рованием.
Во всех остальных случаях решение задачи разделения мно-
гокомпонентной смеси возможно, в принципе, либо в нормально-
фазном, либо в обращенно-фазном варианте. Нормально-фаз-
ный вариант предпочтителен, если анализируемые вещества со-
держат функциональные группы. При этом имеют значение ко-
личество функциональных групп, способность анализируемых
соединений к образованию водородных связей, строение (на-
19
пример, цис, транс-изомерия) и т.д. Если вещества обладают
слабым сродством к неподвижной фазе, то используют активные
слои сорбента и слабополярные растворители.
К обращенно-фазовой хроматографии прибегают для хрома-
тографического разделения гомологов, имеющих одну и ту же
функциональную группу, а также неполярных соединений, срод-
ство которых к полярной неподвижной фазе незначительно.
Объекты флюидной хроматографии — хелаты металлов, порфи-
риновые системы, конденсированные ароматические углеводо-
роды. В качестве подвижной фазы используются фреоны, СО2,
пентан, диэтиловый эфир и другие летучие жидкости или легко
сжижаемые газы в сверхкритическом состоянии.
В самых сложных случаях прибегают к комплексному ис-
пользованию газовой и жидкостной хроматографии в едином ап-
паратурном оформлении. Этот подход, подробно освещаемый
в специальной монографии [56], позволяет исследовать состав
сложных смесей как высоко-, так и низкомолекулярных соеди-
нений, проводить групповое и покомпонентное разделение (при
этом колонка жидкостного хроматографа, размещаемая перед
газохроматографической, может выполнять функции своеобраз-
ной системы пробоподготовки). Гибридные приборы подобного
назначения, еще несколько лет назад собираемые в лаборато-
риях из имеющихся разрозненных блоков, в настоящее время
стали выпускаться серийно (сошлемся, например, на т. н. «двой-
ной» — газовый и жидкостный хроматограф фирмы Fisons). В
литературе отмечаются и заманчивые перспективы сочетания
жидкостной хроматографии и капиллярного электрофореза [57].
Наиболее распространенная методика выполнения анализов
смеси веществ при использовании проявительной (иначе — элю-
ентной) колоночной газовой или высокоэффективной жидкост-
ной хроматографии сводится к следующему.
Перед началом анализа хроматографическую колонку, со-
держащую неподвижную фазу, непрерывно промывают подвиж-
ной фазой (в газовой хроматографии подвижную фазу часто на-
зывают газом-носителем, в жидкостной — элюентом) и в этот
поток подвижной фазы на входе в колонку вводят небольшую
порцию анализируемой смеси компонентов, например А, В и С
(при выполнении газохроматографических анализов жидкие и
твердые вещества должны быть предварительно переведены в
парообразное состояние; в жидкостной хроматографии анали-
зируемые образцы предварительно растворяют в элюенте или
в другом подходящем, смешивающемся с ним растворителе).
Вследствие специфических различий в сорбции или раство-
римости при движении через слой неподвижной фазы компо-
20
центы группируются в зоны, отделенные друг от друга подвиж-
ной фазой (рис. 1.1, а). Практически формирование зон проявля-
емых компонентов наблюдается на всем пути их следования, т. е.
на протяжении всей длины колонки. Из-за диффузионных про-
цессов в подвижной и неподвижной фазах границы зон размы-
ваются, так что максимальная концентрация компонента оказы-
вается сосредоточенной в центре зоны.
а
Рис. 1.1. Проявительная хроматография.
а — участок колонки со сложившимся распределением хроматогра-
фических зон; б — хроматограмма.
Если на выходе из колонки регистрировать изменение во вре-
мени какого-либо свойства потока подвижной фазы (так назы-
ваемое дифференциальное детектирование), то выходная хрома-
тографическая кривая — хроматограмма — запишется в виде
более или менее острых пиков, возвышающихся над нулевой
(базовой) линией, уровень которой по окончании анализа, как
правило, соответствует исходному ее положению до начала ана-
лиза (рис. 1.1, б). Таким образом, сразу же по окончании одного
21
анализа колонка автоматически оказывается подготовленной к
выполнению следующего.
Времена выхода компонентов, отсчитываемые от момента
ввода пробы до момента регистрации вершины пика, или, иначе,
объемы подвижной фазы, затраченные на перенос через колонку
каждого компонента, дают качественную характеристику ана-
лизируемых веществ. Сопоставление площадей (или высот)
хроматографических пиков позволяет с высокой точностью вы-
полнять количественные определения. Кроме качественных и
количественных определений рассмотренная методика позво-
ляет проводить препаративное выделение и очистку любого
содержащегося в анализируемом образце вещества, поскольку
имеется принципиальная возможность осуществить полное раз-
деление всех компонентов смеси.
Одним из недостатков хроматографического анализа при по-
стоянных температуре, составе и скорости подвижной фазы
является то, что, анализируя смесь компонентов, сильно разли-
чающихся по характеристикам удерживания, трудно выбрать
оптимальные температуру колонки и скорость газа-носителя
или элюента.
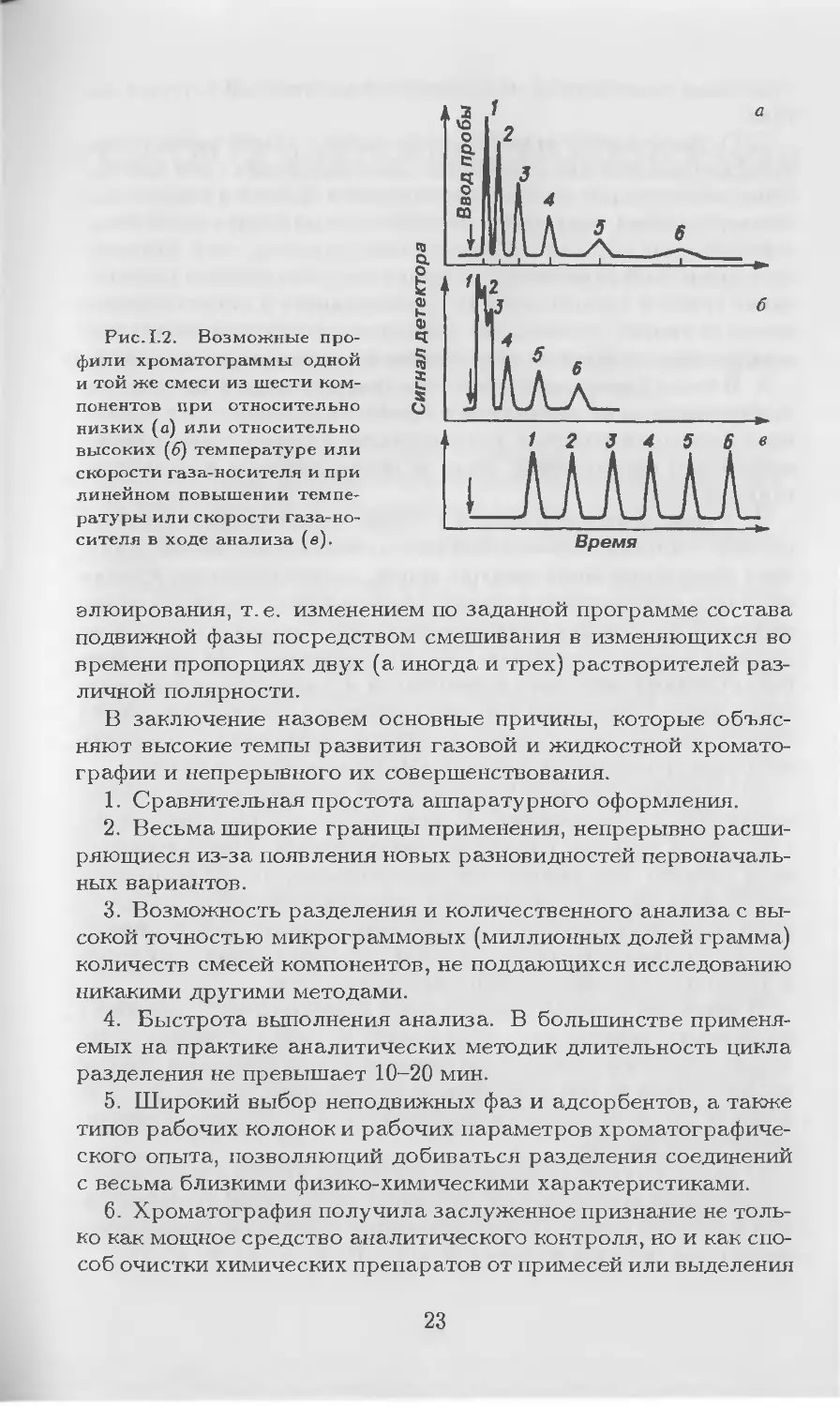
В газовой хроматографии при невысокой температуре ко-
лонки (или небольшой скорости газа-носителя) лишь пики пер-
вых (как правило, наиболее летучих) компонентов пробы бу-
дут резко очерчены на хроматограмме. Хроматографические
зоны последующих компонентов будут все более и более раз-
мываться потоком газа-носителя, что приведет к регистрации
на хроматограмме уширяющихся по оси времени пиков, кото-
рые в пределе могут слиться с нулевой линией. Общая продол-
жительность анализа при этом составит довольно значительное
время.
При повышенной температуре колонки или достаточно боль-
шой скорости газа-носителя удобную для качественных и ко-
личественных измерений форму приобретут на хроматограмме
пики последних выходящих из колонки веществ. Общее время
анализа будет небольшим, однако наиболее летучие (наиме-
нее удерживаемые) компоненты выйдут из колонки частично
или полностью неразделенными. Вид подобных хроматограмм
представлен на рис. 1.2. К настоящему времени найдены пути
технического решения проблемы увеличения температуры или
скорости газа-носителя в ходе анализа по заданной программе,
что намного расширило границы практического использования
газовой хроматографии.
В жидкостной хроматографии рассмотренные проблемы ре-
шаются, как правило, с использованием градиентного режима
22
Рис. 1.2. Возможные про-
фили хроматограммы одной
и той же смеси из шести ком-
понентов при относительно
низких (а) или относительно
высоких (6) температуре или
скорости газа-носителя и при
линейном повышении темпе-
ратуры или скорости газа-но-
сителя в ходе анализа (в).
1 2 J 4 5 6 в
iJWlW.
Время
элюирования, т. е. изменением по заданной программе состава
подвижной фазы посредством смешивания в изменяющихся во
времени пропорциях двух (а иногда и трех) растворителей раз-
личной полярности.
В заключение назовем основные причины, которые объяс-
няют высокие темпы развития газовой и жидкостной хромато-
графии и непрерывного их совершенствования.
1. Сравнительная простота аппаратурного оформления.
2. Весьма широкие границы применения, непрерывно расши-
ряющиеся из-за появления новых разновидностей первоначаль-
ных вариантов.
3. Возможность разделения и количественного анализа с вы-
сокой точностью микрограммовых (миллионных долей грамма)
количеств смесей компонентов, не поддающихся исследованию
никакими другими методами.
4. Быстрота выполнения анализа. В большинстве применя-
емых на практике аналитических методик длительность цикла
разделения не превышает 10-20 мин.
5. Широкий выбор неподвижных фаз и адсорбентов, а также
типов рабочих колонок и рабочих параметров хроматографиче-
ского опыта, позволяющий добиваться разделения соединений
с весьма близкими физико-химическими характеристиками.
6. Хроматография получила заслуженное признание не толь-
ко как мощное средство аналитического контроля, но и как спо-
соб очистки химических препаратов от примесей или выделения
23
отдельных компонентов из смесей (препаративный вариант ме-
тода) .
7. Осуществление химических реакций в самой хроматогра-
фической колонке или в реакторах, составляющих с ней единую
коммуникационную систему (реакционная газовая и жидкостная
хроматография), открывает дополнительные возможности каче-
ственного анализа смесей неизвестного состава. Для химиков-
органиков особый интерес представляет препаративная реакци-
онная газовая хроматография, совмещающая в одностадийном
процессе синтез (с выходами, близкими к количественным) раз-
нообразных соединений и выделение их в индивидуальном виде.
8. В последние годы все более расширяются не ан а литические
применения колоночной хроматографии, связанные с исследова-
нием физико-химических характеристик хроматографируемого
вещества и неподвижной фазы, а также кинетики каталитиче-
ских реакций.
9. Совмещение достоинств газовой и жидкостной хромато-
графии и других современных инструментальных методов ана-
лиза (некоторые виды спектроскопии, рефрактометрия, кулоно-
метрия) в едином аппаратурном оформлении открывает неогра-
ниченные перспективы качественного и количественного иссле-
дования весьма сложных по составу соединений. Из таких ком-
бинированных методов, являющихся в настоящее время наи-
более информативными при качественном анализе сложнейших
смесей неизвестного состава, следует выделить два — хромато-
масс-спектрометрию и хромато-ИК-фурье-спектроскопию.
10. Методы газовой и жидкостной хроматографии хорошо
поддаются автоматизации. В этом их неоспоримое преимуще-
ство перед другими современными приемами физико-химичес-
кого анализа для химической промышленности. В настоящее
время цеха крупных химических заводов-комбинатов оборудо-
ваны десятками газовых и жидкостных хроматографов, связан-
ных со специализированными ЭВМ для оперативного контроля
и управления производственными процессами.
В наши дни газовая и жидкостная хроматография оказывает
неоценимую помощь не только химии, но также медицине, био-
логии, геологии, охране окружающей среды и многим другим
направлениям науки и техники, включая такие разные по сути
области применения, как криминалистика и освоение космиче-
ского пространства.
С более полным освещением основных этапов становления,
современных возможностей газовой и жидкостной хроматогра-
фии и оценкой перспектив их дальнейшего развития можно озна-
комиться в обзорах и книгах [2-5, 24-37, 40 -43, 54-56, 58-62].
24
Глава II
аппаратура для газовой хроматографии
п.1. ПРИНЦИПИАЛЬНАЯ СХЕМА, ОСНОВНЫЕ СИСТЕМЫ
И УЗЛЫ ГАЗОВОГО ХРОМАТОГРАФА
Газовый аналитический хроматограф представляет собой со-
вокупность взаимодействующих систем, предназначенных для
проведения анализа в оптимальном режиме хроматографиче-
ского разделения исследуемой смеси с целью определения ее
состава.
Принципиальная (функциональная) схема аналитического
лабораторного газового хроматографа представлена на
рис. II.1. Установка, стабилизация и очистка потоков газа-
носителя и дополнительных газов (если они необходимы для пи-
тания детектора) выполняются системой подготовки газов. До-
зирующее устройство позволяет вводить в поток газа-носителя
непосредственно перед колонкой определенное количество ана-
лизируемой смеси в газообразном состоянии. В колонке осуще-
ствляется разделение смеси на отдельные составляющие ком-
поненты. Последние в смеси с газом-носителем подаются в де-
тектор, который преобразует возникающие изменения физиче-
ских или физико-химических свойств бинарных смесей компо-
нент — газ-носитель (по сравнению с чистым газом-носителем)
в электрический сигнал. Величина сигнала зависит как от при-
роды компонента, так и от содержания его в анализируемой
смеси. Детектор с соответствующим блоком питания соста-
вляет систему детектирования.
Требуемые температурные режимы колонки, детектора и до-
зирующих устройств достигаются помещением их в термоста-
ты, управляемые терморегулятором. Если необходимо повы-
шать температуру колонки в процессе анализа, используют про-
грамматор температуры термостата. Терморегулятор с про-
грамматором составляет систему термостатирования,
в которую может также входить устройство для измерения тем-
пературы.
25
Рис. IL1. Принципиальная схема газового хроматографа.
1 — система подготовки газов; 2 — устройство для дозирования
пробы; 3 — колонка; 4 — усилитель; 5 — терморегулятор; 6 — блок
питания детектора; 7 — усилитель; 8 — регистратор; 9 — интегратор
или компьютеризованная система обработки сигнала детектора; 10 —
измерители параметров режима анализа (расходов газов, температур,
электрического питания детекторов). Газовые функциональные связи
показаны двойной линией, электрические— одинарной, термостатиру-
емые элементы заключены в штриховой контур.
Сигнал детектора, преобразованный усилителем, записыва-
ется в виде хроматограммы аналоговым пишущим потенциоме-
тром или (как в приборах последних лет выпуска; см. раздел
IV.4) поступает на регистрацию в ЭВМ, оснащенную аналого-
цифровым преобразователем (АПП). Для некоторых детекто-
ров сигнал может быть записан без предварительного усиления.
Количественную обработку хроматограмм выполняют вруч-
ную или с помощью инструментальных средств, измеряющих
параметры хроматографических пиков и производящих расчет
результатов анализа.
Все функциональные системы хроматографа взаимосвязаны,
поэтому работа прибора может быть удовлетворительной лишь
при условии четкой и правильной работы каждой системы в
отдельности.
26
II.1.1. Система подготовки газов
Система подготовки газов предназначена для установки, ста-
билизации и измерения скорости потоков газа-носителя и до-
полнительных газов, питающих некоторые детекторы, а также
для очистки газов. Особенно важное значение имеют уста-
новка и стабилизация оптимального для данного анализа рас-
хода газа-носителя, оказывающего непосредственное влияние
на параметры удерживания и размеры пиков анализируемых
веществ. Важно также исключить влияние колебаний расхо-
дов газа-носителя и дополнительных газов на чувствительность
детекторов, чтобы не допустить связанного с этим неконтроли-
руемого изменения параметров пиков. Кроме того, недостаточ-
ная стабильность газовых потоков часто является причиной не-
устойчивости нулевой линии, что затрудняет количественную
обработку хроматограмм.
Установка и необходимая стабилизация газовых потоков осу-
ществляются совокупностью нескольких элементов, основными
из них являются дроссель, регулятор давления и регулятор рас-
хода.
Рис.П.2. Схема дросселя.
А — входная камера; J5 — выходная
камера. 1 — задающий элемент (винт);
2 — исполнительный элемент; 3— пру-
жина исполнительного элемента.
Дроссель представляет собой устройство, позволяющее
изменять расход (объемную скорость) газа путем изменения аэ-
родинамического сопротивления канала, по которому проходит
газ. Схема конструкции дросселя приведена на рис. II.2. Вход-
ная и выходная камеры сообщаются каналом, в котором нахо-
дится исполнительный элемент, жестко связанный с задающим
элементом с помощью пружины. Изменение сопротивления ка-
27
нала, по которому газ перетекает из входной камеры в выход-
ную, достигается перемещением исполнительного элемента, от-
крывающего или закрывающего канал. Для каждого дросселя
имеется определенная зависимость расхода газа от величины
перемещения исполнительного элемента (его характеристика)
при постоянном входном давлении газа.
Для дросселей I вида эта характеристика близка к линейной
(рис. П.З), при этом в широком диапазоне обеспечивается плав-
ность установки расходов. Обычно в таких дросселях, называ-
емых вентилями тонкой или плавной регулировки, в качестве
исполнительного элемента используется коническая игла, пере-
мещающаяся в цилиндрической втулке. Выдвижение иглы из
втулки увеличивает сечение канала (открытие дросселя), вы-
зывая увеличение расхода. Для плавности установки расхода
задающее устройство снабжается резьбой с малым шагом, что
позволяет осуществлять малые перемещения исполнительного
элемента.
Степень открытия дросселя
Рис. П.З. Характеристики дросселей I и II вида.
Дроссели II вида, напротив, имеют весьма крутую характе-
ристику при начальных смещениях исполнительного элемента
и область ’’насыщения” при больших смещениях (см. рис.П.З).
Подобные дроссели используются в схемах автоматического ре-
гулирования давления или расхода, где малые смещения испол-
нительного механизма должны вызывать заметные изменения
расхода газа. Обычно в качестве исполнительного элемента в
таких дросселях применяется шарик или плоская заслонка, за-
крывающие отверстие в диафрагме (седле).
Дроссель не стабилизирует давление на выходе, он лишь сни-
жает входное давление. Использование дросселя для установки
28
расхода возможно только при постоянном входном давлении
газа (например, при питании от индивидуального баллона).
Роль стабилизатора давления выполняет регулятор дав-
ления, значительно снижающий изменение давления на входе в
колонку, вызванное колебаниями внешнего давления газа. Кон-
структивно регулятор давления (рис. II.4) аналогичен дросселю
с той лишь разницей, что в нем отсутствует жесткая связь ме-
жду задающим элементом и исполнительным, в качестве кото-
рого используется дроссель II вида. Мембрана в регуляторе
давления воспринимает изменение давления газа и передает со-
ответствующее смещение исполнительному элементу.
Рис. П.4. Схема регулятора рас-
хода газа.
1 — задающий элемент (винт);
2 — пружина задающего элемента;
3 — мембрана; 4 — исполнитель-
ный элемент (дроссель); 5 — пру-
жина дросселя.
При открывании регулятора давления задающий механизм
через пружину действует на мембрану и отжимает дроссель от
седла, соединяя входную камеру регулятора с выходной. В вы-
ходной камере устанавливается заданное давление, при котором
усилие, создаваемое давлением газа на мембрану, равно про-
тивоположному усилию пружины задающего устройства. При
этом через дроссель устанавливается расход, поддерживающий
необходимое давление на выходе регулятора.
При уменьшении давления газа во внешней линии (например,
при включении в ту же линию других потребителей газа) не-
сколько уменьшается давление газа под мембраной, что вызы-
вает перемещение мембраны и дросселя вниз. При этом про-
исходит дополнительное открытие дросселя, и увеличивается
скорость перетекания газа в выходную полость, пока давление
под мембраной и расход через регулятор не восстановятся прак-
тически до первоначальных значений при изменившемся вход-
29
ном давлении. Так как расход газа в дросселе имеет крутую
характеристику, необходимое открытие достигается весьма ма-
лым изменением давления под мембраной, поэтому восстановле-
ние первоначального выходного давления регулятором произ-
водится быстро и практически полностью. Аналогичным обра-
зом работает регулятор и при увеличении внешнего давления.
В некоторых случаях, например при программировании тем-
пературы колонки, необходимо поддерживать постоянный рас-
ход газа-носителя через колонку, когда ее сопротивление изме-
няется в процессе анализа. Для этой цели используется регу-
лятор расхода.
Регулятор р асх од а (рис. II.5) имеет три камеры: вход-
ная и промежуточная камеры разделены мембраной и сообща-
ются регулирующим дросселем I вида, выходная и промежуточ-
ная камеры также сообщаются регулирующим дросселем! вида,
связанным с мембраной, воспринимающей разницу входного и
промежуточного давлений.
Газ
Рис. II.5. Схема регулятора рас-
хода газа.
А — входная камера; Б — промежу-
точная камера; В — выходная камера.
1 — задающий элемент; 2 — устано-
вочный дроссель; 3 — мембрана; 4 —
регулирующий дроссель
Необходимый расход газа задается открытием установоч-
ного дросселя. При постоянном входном давлении регулятор
поддерживает расход, заданный суммой сопротивлений обоих
дросселей и хроматографической колонки. Сопротивление
установочного дросселя определяется его начальным положе-
нием и не изменяется. Регулятор расхода реагирует на измене-
ние сопротивления регулирующего дросселя, так что их сумма
всегда остается постоянной и расход не меняется. Например,
30
при увеличении сопротивления колонки с повышением темпера-
туры растет давление в выходной камере и частично под мем-
браной. Это приводит к смещению мембраны вверх и к допол-
нительному открытию регулирующего дросселя, что облегчает
перетекание газа из промежуточной камеры в выходную. В ре-
зультате давление под мембраной снижается практически до
первоначального уровня, а в выходной камере (на входе в ко-
лонку) оно возрастает и расход газа восстанавливается.
Поскольку регулятор расхода реагирует на изменение вы-
ходного давления, время его реакции зависит от объема га-
зовых линий после регулятора: чем меньше этот объем, тем
быстрее меняется в нем давление до порога срабатывания ре-
гулятора. Для уменьшения инерционности регулятора следует
стремиться к сокращению этого объема, который складывается
из объемов фильтров, манометров, дозаторов, соединительных
линий и колонки.
Инерционность регуляторов расхода газов приводит к неудо-
влетворительной стабилизации расхода через колонку. Откло-
нение текущего значения расхода от первоначального из-за тем-
пературного изменения сопротивления колонки может доходить
до нескольких процентов, а восстановление расхода занимает
десятки секунд. Совершенствование конструкции этих регуля-
торов не позволяет существенно улучшить их динамические ха-
рактеристики.
Эффективное решение задачи установки и стабилизации рас-
хода газа достигается с помощью схем, составленных из уни-
фицированных элементов пневмоавтоматики. На этой основе
созданы оригинальные газовые блоки, обеспечивающие точную
установку и высокую стабильность расходов.
Принципиально иным подходом к решению проблемы явля-
ется использование для управления расходами газов микропро-
цессоров в совокупности с соответствующими измерительными
и исполнительными устройствами. Созданная на этой основе
автоматизированная система регулирования обладает необхо-
димой универсальностью и достаточным быстродействием (см.
раздел II. 1.8).
Очистка газовых потоков от пыли, влаги и органических со-
единений выполняется с помощью фильтров, заполненных до-
статочно активными адсорбентами (силикагель, уголь, молеку-
лярные сита). Чистота газов особенно важна при работе с вы-
сокочувствительными ионизационными детекторами (пламенно-
ионизационным, электронозахватным, аргоновым, гелиевым),
где примеси могут являться дополнительным источником иска-
жений нулевой линии. Допустимый уровень загрязнения газов
31
зависит от стабильности газовых потоков, так как колебания
расхода газа могут фиксироваться детектором в виде перемен-
ного фонового потока, создаваемого примесями, и приводят к
нарушению устойчивости нулевой линии.
Измерение скорости газовых потоков производится с помо-
щью мыльно-пленочных измерителей, реометров и ротаметров
(рис. 11.6). В современных приборах расход газов измеряется с
помощью тепловых измерителей с цифровой индикацией.
10"
9-
6-
7-
6-
5-
Рис.П.6. Измерители расхода газа.
а — мыльно-пленочный; б — реометр; в — ротаметр. Стрелками
показано направление потока газа-носителя; пунктиром обозначено ис-
ходное положение мыльной пленки (а), уровня рабочей жидкости (6) и
поплавка (в).
Мыльно-пленочный измеритель определяет расход
газа по времени прохождения мыльной пленкой известного объ-
ема градуированной стеклянной трубки. Он не может быть
встроен в хроматограф и обеспечивает лишь периодическое из-
мерение расхода на выходе из колонки или из детектора.
Для измерения расходов в сравнительно узком интервале мо-
гут быть применены реометры с близкой к линейной зави-
симостью разницы уровней рабочей жидкости от расхода газа
32
через встроенный в реометр дроссель. Реометр удобен отно-
сительной простотой градуировки и наглядностью показаний.
Однако введение его в линию газа для получения непрерывных
измерений нежелательно, так как пары рабочей жидкости рео-
метра загрязняют газ и не исключена возможность переброса
жидкости в газовую линию.
Ротаметр, представляющий собой коническую градуиро-
ванную трубку, в которой уровень поднятия поплавка зависит
от скорости газа, позволяет проводить лишь ориентировочные
измерения.
Расход газа часто измеряют косвенным путем — по давлению
на входе хроматографической колонки, которое всегда доступно
измерению с заданной точностью при использовании маноме-
тров соответствующего класса. Если аэродинамическое сопро-
тивление колонки и дросселя постоянно, манометр может быть
отградуирован непосредственно в значениях расхода.
В современных приборах применяются устройства, позволя-
ющие сочетать достаточную точность с непрерывностью про-
цесса измерения и автоматической цифровой записью (или ин-
дикацией) результатов. Эти устройства чаще всего построены
по принципу теплового расходомера, т. е. на использовании за-
висимости температуры чувствительного элемента от скорости
омывающего его газового потока. Изменение температуры чув-
ствительного элемента преобразуется в электрический сигнал,
величина которого пропорциональна расходу газа.
Дзержинским ОКБ А (ныне ОАО “Цвет”) серийно изготавли-
вается измеритель расхода газа, действие которого основано
на этом принципе. Прибор предназначен для измерения рас-
хода азота (аргона), гелия и воздуха до 100 мл/мин. Основная
погрешность измерения 1,5%. Результат измерения расхода в
мл/мин (приведенный к нормальным условиям) выводится на
цифровой индикатор. Так как показания измерителя не зависят
от давления в газовой линии, прибор может быть включен в лю-
бой участок газовой схемы. Подобные устройства позволяют не
только измерять расход газа, но и оценивать стабильность по-
тока газа или динамику его изменения (например, при работе в
условиях программирования расхода в колонке).
Соединения элементов газовых линий в хроматографах
обычно выполняются с помощью трубок малого диаметра (0,5—
2 мм) из нержавеющей стали. Уплотнения соединений осуще-
ствляются плоскими или фигурными прокладками: металличе-
скими (медь, алюминий) или графитовыми для соединений в го-
рячей зоне и мягкими (резина, полимерные материалы) для со-
единений элементов, работающих при комнатной температуре.
33
Принципиальная схема линии газа-носителя в лабораторном
хроматографе представлена на рис. П.7. Сочетание в одной ли-
нии регуляторов давления и расхода позволяет поддерживать
расход постоянным при изменении внешнего давления газа и со-
противления колонки. Манометры необходимы для контроля
режима работы регуляторов (перепада давления на регулято-
рах) и давления на входе в колонку. Входной фильтр обеспе-
чивает первичную очистку газа от пыли и влаги, выходной —
окончательную его очистку от примесей.
Рис. П.7. Принципиальная схема линии газа-носителя.
1 — входной фильтр; 2 — регулятор давления; 3 — манометр; 4 —
регулятор расхода; 5— контрольный манометр; 6— выходной фильтр.
Герметичность газовой схемы проверяют по отсутствию
уменьшения давления в линии, закрытой на входе и выходе.
Если в газовую схему включен тепловой измеритель расхода, он
может быть использован для проверки ее герметичности. Для
этого выход измеряемого участка схемы закрывают, и после за-
полнения его газом до заданного давления при полной герметич-
ности проверяемых линий измеритель должен показать отсут-
34
ствие расхода. При наличии утечек газа измеритель покажет
их величину.
II.1.2. Дозирующие устройства
Дозирующие устройства (дозаторы) предназначены для вве-
дения в хроматографическую колонку определенного количе-
ства анализируемой смеси (пробы).
При введении пробы должно выполняться несколько общих
требований.
1. Состав пробы, введенной в колонку, должен быть иден-
тичен составу анализируемой смеси, за исключением некото-
рых специальных случаев, когда не требуется определять пол-
ный состав смеси. Если при дозировании не удается достичь
полной идентичности количественного состава анализируемой
смеси и введенной пробы, важно обеспечить его постоянство
при многократном дозировании. В этом случае можно избежать
ошибки анализа, связанной с искажением состава пробы, путем
выбора соответствующего метода градуировки прибора. Нару-
шение идентичности состава пробы и анализируемой смеси мо-
жет быть вызвано многими причинами, в частности: наличием
в дозаторе непродуваемых (“мертвых”) объемов, потерей ча-
сти пробы при введении ее в колонку, химическими реакциями
между компонентами пробы или термодеструкцией их, вызван-
ными высокой температурой дозатора и каталитическим дей-
ствием материалов дозатора, имеющих контакт с пробой.
2. При многократном введении одной и той же пробы в посто-
янных условиях величина пробы (ее объем или масса) должна
изменяться лишь незначительно в заданных пределах (обычно
1-3%), т. е. она должна воспроизводиться. Изменение вели-
чины пробы обусловливается недостатками конструкции до-
зирующего устройства, непостоянством условий дозирования
и субъективной ошибкой оператора, производящего дозирова-
ние. Требования к воспроизводимости могут существенно раз-
личаться в зависимости от выбранных способов градуировки
хроматографа и обработки хроматограмм, а также от требуе-
мой точности анализа.
3. При введении пробы ее размывание и разбавление газом-
носителем должны быть минимальными, так как в общем случае
чем меньшую начальную часть колонки занимает проба, тем
большей эффективности разделения можно достичь. Размыва-
ние пробы зависит от конструкции дозатора и температурного
35
режима. Последний фактор имеет значение при введении жид-
ких и твердых проб и связан с необходимостью их быстрого ис-
парения. Однако температура дозатора не должна быть слиш-
ком высокой, чтобы исключить возможность термической де-
струкции различных веществ.
4. Введение пробы не должно вызывать изменения устано-
вившегося режима систем хроматографа (зашкаливания нуле-
вой линии в момент введения, резкого изменения давления газа-
носителя и температуры дозатора), что может быть обусло-
влено разгерметизацией системы при введении слишком боль-
шой пробы, изменением сопротивления линии газа-носителя при
дозировании.
5. Величина пробы выбирается с учетом сорбционной емко-
сти колонки так, чтобы не вызвать перегрузки колонки (ограни-
чение максимальной величины) и с учетом чувствительности
детектора, который должен четко зарегистрировать соответ-
ствующие количества разделенных веществ (ограничение мини-
мальной величины пробы). На практике приходится иметь дело
с пробами, разнообразными как по величине, так и по агрегат-
ному состоянию, однако универсального дозирующего устрой-
ства, позволяющего эффективно вводить большие и малые га-
зообразные, жидкие и твердые пробы, не имеется.
Существует несколько типов дозаторов, конструкции кото-
рых определяются агрегатным состоянием вводимых проб.
Для дозирования газообразных смесей используют газо-
вые краны-дозаторы, позволяющие включать градуированную
емкость, предварительно заполненную анализируемой газовой
смесью, в поток газа-носителя, который переносит дозу в виде
газовой “пробки” в хроматографическую колонку.
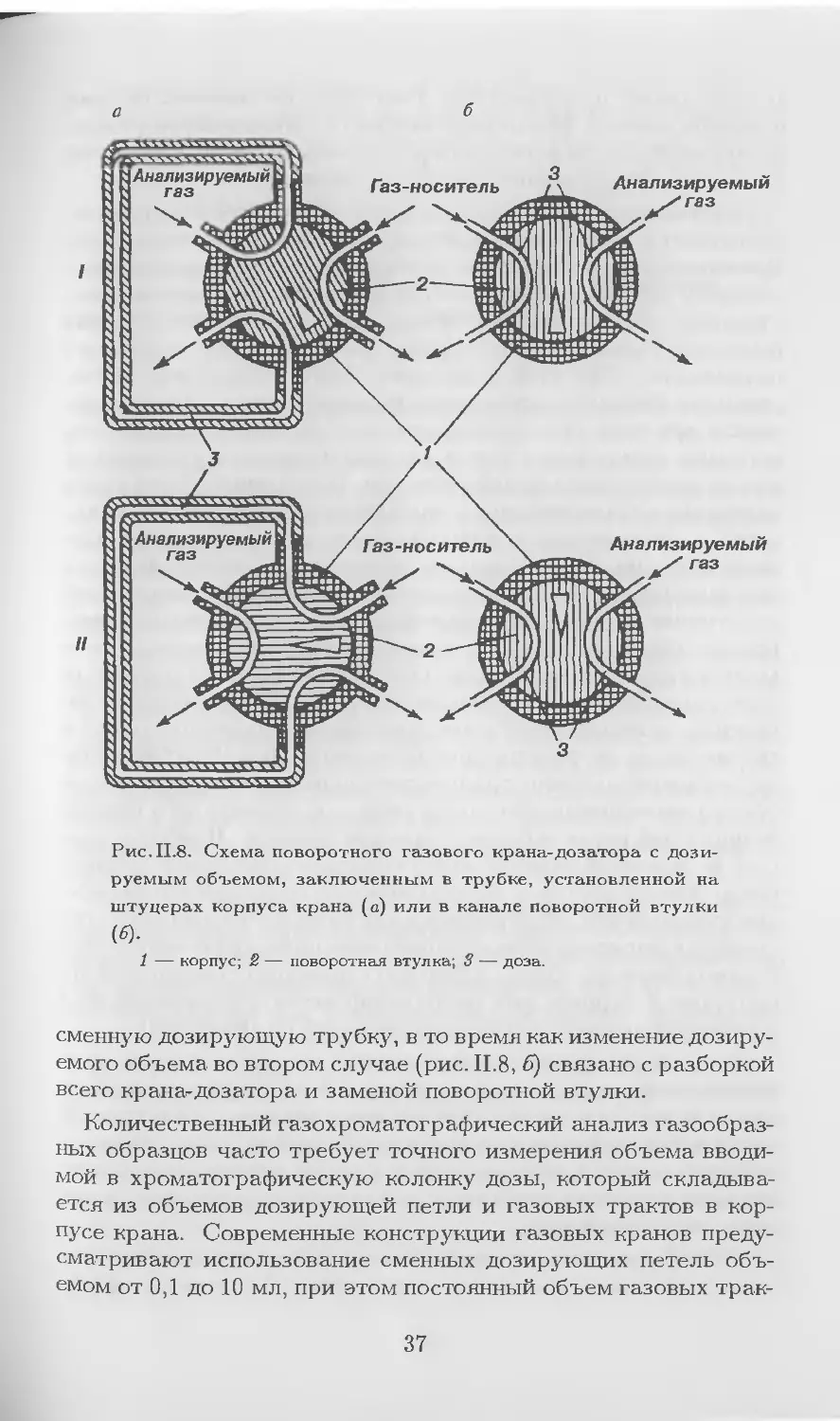
Поворотный газовый к р ан (рис. II.8) состоит из кор-
пуса (неподвижной детали) со штуцерами, через которые под-
водятся газ-носитель и анализируемый газ, и подвижной де-
тали — поворотной втулки с каналами, соединяющими линии
газа-носителя и анализируемого газа. Величина пробы опреде-
ляется объемом градуированной трубки (дозы), которая может
устанавливаться на штуцерах корпуса (рис. II.8, а). Иногда гра-
дуированный объем (доза) заключен в самой поворотной втулке
(рис. II.8, б). В одном из фиксированных положений (положение
I) поворотной втулки происходит заполнение дозы анализируе-
мым газом, после поворота втулки (положение II) газ-носитель
перемещает анализируемый газ в колонку. Для повторного до-
зирования необходимо вернуть кран в положение I и вновь за-
полнить дозу анализируемым газом. Для измерения величины
пробы в первом случае (рис. II.8, а) достаточно лишь заменить
36
a
б
Рис.П.8. Схема поворотного газового крана-дозатора с дози-
руемым объемом, заключенным в трубке, установленной на
штуцерах корпуса крана (а) или в канале поворотной втулки
(6).
1 — корпус; 2 — поворотная втулка; 3 — доза.
сменную дозирующую трубку, в то время как изменение дозиру-
емого объема во втором случае (рис. II.8, б) связано с разборкой
всего крана-дозатора и заменой поворотной втулки.
Количественный газохроматографический анализ газообраз-
ных образцов часто требует точного измерения объема вводи-
мой в хроматографическую колонку дозы, который складыва-
ется из объемов дозирующей петли и газовых трактов в кор-
пусе крана. Современные конструкции газовых кранов преду-
сматривают использование сменных дозирующих петель объ-
емом от 0,1 до 10 мл, при этом постоянный объем газовых трак-
37
тов составляет от 0,2 до 1 мл. Учет этого постоянного объема
в количественных измерениях диктуется соотношением объема
дозирующей петли и постоянного объема, а также требуемой
точностью определения дозируемого объема.
Градуировка сменных дозирующих петель особого труда не
составляет и обычно проводится путем заполнения измеряемой
емкости жидкостью (ртутью или водой) с последующим опре-
делением объема или массы этой жидкости. Определение по-
стоянного объема газового крана несколько сложнее, так как
измерение газовых трактов крайне затруднено или чаще всего
невозможно. Для этой цели может быть использован метод,
сущность которого состоит в проведении серии хроматографи-
ческих анализов газа постоянного состава при последователь-
ной смене дозирующих петель, предварительно отградуирован-
ных по массе заполняющей жидкости. Постоянный объем крана
определяется графически в координатах площадь пика (или вы-
сота) — объем петли экстраполяцией на нулевое значение пло-
щади пика (рис. II.9). Главным достоинством метода является
его простота, так как для выполнения необходимых определений
достаточно располагать газовой смесью постоянного (можно
неизвестного) состава и в процессе измерений газовая схема
хроматографа не изменяется. Основной недостаток метода со-
стоит в необходимости смены петель, что может привести к из-
менению дозируемого объема даже при использовании одной и
той же петли за счет различной ее центровки. Этот недоста-
ток устраняется, если градуировка производится измерением
объема вытесненного из крана газа, т. е. суммарного объема
дозирующей петли и газовых трактов корпуса. Наиболее про-
стой и надежный вариант этого метода — заполнение дозиру-
емого объема азотом с последующим вытеснением его диокси-
дом углерода ГбЗ]. Измерение малых количеств азота после по-
глощения диоксида углерода щелочью проводят в азотометре
(газовая бюретка объемом 2-10 мл) с помощью стандартной ап-
паратуры и техники работы, надежно зарекомендовавшей себя
в элементном анализе (определение азота по Дюма [64]).
При использовании газовых кранов вполне достижима вос-
производимость результатов анализа 0,2-0,5%, при этом необхо-
димо следить за постоянством давления анализируемой газовой
смеси и температуры градуированной трубки (дозы). Исполь-
зование газовых кранов-дозаторов требует наличия достаточно
больших количеств анализируемой газовой смеси для промыва-
ния и заполнения дозы.
Способ включения дозирующего крана в газовую схему хро-
матографа может оказывать существенное влияние на эффек-
38
Рис.П.9. Определение постоянно-
го объема газового крана методом
стандартных добавок.
тивность хроматографической колонки из-за размывания пробы
в элементах газовой схемы до колонки. Поэтому включать
газовый кран следует таким образом, чтобы выход крана со-
единялся кратчайшим путем с хроматографической колонкой и
при этом объем соединительных элементов был наименьшим.
Весьма нежелательно между краном и колонкой включать дру-
гие элементы газовой схемы (например, испаритель жидких
проб), что может привести к диффузионному размыванию вво-
димой пробы или образованию “хвостов” у пиков.
Описанные газовые краны используются не только для до-
зирования, но также для переключения потоков газа-носителя,
когда требуется изменить последовательность соединения хро-
матографических колонок, порядок их соединения с детектором,
направление потока газа-носителя в колонках (варианты обрат-
ной продувки). Эти схемы, составленные с помощью четырех-,
шести- и других многоходовых кранов, применяются для сокра-
щения времени анализа или эффективного разделения сложных
многокомпонентных смесей.
Введение жидких смесей в колонку производят специаль-
ными шприцами через термостойкое резиновое уплотнение ис-
парителя. Испаритель представляет собой нагреваемый до
определенной температуры металлический блок (рис. 11.10) с
каналом для ввода и испарения жидкой пробы. В канал по-
дается поток предварительно нагретого газа-носителя. С од-
ной стороны канал закрыт пробкой из термостойкой резины, с
другой стороны к нему присоединена хроматографическая ко-
лонка. Иглу шприца с анализируемой жидкостью вводят через
термостойкое уплотнение в канал испарителя, введенная проба
быстро испаряется и переносится потоком газа-носителя в ко-
лонку. Для эффективного введения пробы очень важно, чтобы
в канале испарителя не было непродуваемого объема, так как
попадание части пробы в такой объем приводит к длительному
39
Рис. II. 10. Испаритель.
1 — корпус; 2 — канал испа-
рения пробы; 3 — термостойкое
уплотнение из силиконовой рези-
ны; 4 —" прижимная гайка.
диффузионному вымыванию ее в колонку, что вызывает значи-
тельную асимметрию пиков.
Температура испарителя влияет на начальную ширину по-
лосы пробы в колонке и эффективность разделения. Обычно
температура испарителя выбирается равной или на 30-50°С
выше температуры кипения наиболее высококипящих компонен-
тов смеси, чтобы обеспечить быстрое испарение. Если повы-
шение температуры испарителя приводит к увеличению эффек-
тивности разделения, значит, первоначально выбранная темпе-
ратура испарителя мала. Однако излишнее ее повышение неже-
лательно, так как оно может вызвать термодеструкцию анали-
зируемых компонентов или перегрев начальной части колонки,
если последняя присоединена непосредственно к испарителю.
При высокой (более 300°С) температуре термостойкое рези-
новое уплотнение канала испарителя постепенно теряет эла-
стичность, нарушается герметичность испарителя. Для предо-
твращения этого обычно принимают меры по охлаждению “го-
ловки” испарителя, что позволяет сохранить необходимые ка-
чества уплотнения. К увеличению “хвостов” пиков и сниже-
40
нию эффективности разделения смеси приводит попадание ре-
зиновой крошки в канал испарителя. При появлении подоб-
ных симптомов канал испарителя следует тщательно промыть
и продуть газом-носителем или воздухом. Предпочтительны
конструкции испарителей, в которых функции канала испаре-
ния пробы выполняет сменный вкладыш, изготовляемый из ме-
талла (нержавеющей стали) или из стекла. Стеклянные вкла-
дыши имеют следующие преимущества: 1) обеспечивается рав-
номерный прогрев всей зоны испарения; 2) стекло (в сравнении
с другими материалами — металлами) обладает наименьшей
способностью к сорбции на поверхности полярных соединений
(например, воды, аммиака, карбоновых кислот), что обусловли-
вает повышение эффективности хроматографического разделе-
ния; 3) вероятность термодеструкции анализируемых соедине-
ний в испарителе значительно снижается (хотя и не исключа-
ется полностью); 4) прозрачность стекла позволяет легко и опе-
ративно контролировать образование налета кокса на внутрен-
них стенках вкладыша и своевременно заменять его на чистый.
Большое значение имеет внутренний диаметр вкладыша, оп-
ределяющий объем канала испарения пробы. Нетрудно пред-
ставить, что, если объем канала испарения будет намного пре-
вышать объем паров введенной пробы, произойдет нежелатель-
ное размывание паров анализируемых соединений и, как след-
ствие, снизится эффективность разделения. С другой стороны,
если объем канала испарения недостаточен для размещения в
нем паров введенной жидкой пробы, избыточная часть паров пе-
реместится в зазор между внешней стенкой вкладыша (началь-
ного участка хроматографической колонки) и корпусом испа-
рителя, что приведет к еще более замедленному вводу пробы
и существенному снижению эффективности разделения, чем в
рассмотренном выше случае.
Следует также иметь в виду, что степень размывания, или,
иначе говоря, степень замедления процесса переноса паров про-
бы из испарителя в колонку, обратно пропорциональна ско-
рости газа-носителя. Поэтому конструкции испарителей луч-
ших современных моделей газовых хроматографов предусма-
тривают возможность изменения объема испарения пробы в за-
висимости от дозы, типа используемой колонки и условий ана-
лиза (скорости газа-носителя). Изменение объема испарения
пробы достигается использованием вкладышей различного вну-
треннего диаметра в пределах 1-4 мм.
В испаритель жидкие пробы вводят специальными шприцами,
позволяющими дозировать пробы объемом от 0,1 до нескольких
микролитров. Обычно используют шприцы с номинальными
41
объемами 1, 10 и 50 мкл. Наибольшее распространение полу-
чили микрошприцы объемом 10 мкл (рис. 11.11, а). В этих шпри-
цах в качестве поршня используется калиброванная проволока
из вольфрама или нержавеющей стали. Поршень и цилиндр вза-
имно притерты и не требуют смазки. Герметичность шприца
достигается за счет самой дозируемой жидкости, смачивающей
сопряженные поверхности цилиндра и плунжера. Недостатком
такого шприца является относительно большой объем иглы (до
2 мкл). Часть жидкости, заключенная в игле, не выдавливается
из шприца при полном введении поршня в цилиндр, но частично
испаряется при введении иглы в испаритель. Количество извле-
каемой из иглы жидкости зависит от глубины ввода и продол-
жительности пребывания иглы в испарителе и не поддается не-
посредственному контролю, что является дополнительным ис-
точником невоспроизводимости величины пробы. Кроме того,
большой объем иглы ограничивает минимальный объем вводи-
мой пробы (1-2 мкл).
Этого недостатка не имеют микрошприцы, в которых дози-
руемый объем заключен в самой игле (рис. 11.11, б). Плунжер
в виде вольфрамовой или стальной проволоки передвигается в
игле, герметичность шприца обеспечивается фторопластовым
уплотнением, помещенным в основании иглы. С помощью таких
микрошприцев можно вводить пробы от 0,05 до 1 мкл.
При использовании микрошприцев и наличии определенного
опыта оператора воспроизводимость введения жидких проб
можно довести до 1,5-2,5%. Общий недостаток микрошприцев —
малая механическая прочность, что вызывает необходимость
весьма осторожного обращения с ними. При переходе от одной
дозирующей смеси к другой микрошприцы должны быть тща-
тельно промыты соответствующими растворителями.
Микрошприцы позволяют вводить довольно малые пробы (до
0,05 мкл), однако в ряде случаев (при работе с капиллярными
колонками или с высокочувствительными специфическими де-
текторами) необходимо вводить значительно меньшие объемы
жидких проб (10“3-10“4 мкл). Уменьшение пробы достигается
разделением потока газа-носителя перед вводом в колонку с по-
мощью делителя потока (рис.II.12, а): большая часть по-
тока газа-носителя и содержащейся в нем испаренной пробы на-
правляется на сброс в атмосферу, а значительно меньшая часть
попадает в колонку. При этом одновременно решаются две за-
дачи: уменьшение величины пробы и обеспечение необходимой
компактности (эффективности ввода) этой пробы. Нужное от-
ношение деления пробы достигается соответствующим отноше-
нием сопротивления хроматографической колонки и сопроти-
42
Рис. IL 11. Микрошприцы с дозиру-
емым объемом, заключенным в ци-
линдре (а) и в игле (6).
1 — игла: 2 — корпус; 3 — поршень;
— шток.
а б
вления линии сброса, регулируемого вентилем сброса. Отно-
шение деления рассчитывают как отношение объемных скоро-
стей через колонку и линию сброса (обычно для капиллярных
колонок оно выбирается в интервале от 1 : 50 до 1 : 500). Дели-
тель может составлять единое целое с испарителем и колонкой
или включаться дополнительно между испарителем и колонкой.
43
a
Проба в потоке
газа-носителя
б
Газ-носитель
Колонка
носитель газ
Рис.11.12. Схема делителя потока паров пробы (а) и испарите-
лей с делителем потока (фирмы Perkin-Elmer), используемых
при работе с металлическими (6) и стеклянными (в) капил-
лярными колонками.
1 — стеклянный вкладыш (канал испарения пробы); 2 — корпус
испарителя; 3 — термостатируемая зона; 4 — колонка; 5 — накидная
гайка; 6 — камера перемешивания паров пробы и газа-носителя; 7 —
пружина; 8 — сменный градуированный металлический капилляр (по-
стоянный дроссель); 9 — шторный затвор; 10 — накидная гайка; 11 —
термостойкое уплотнение из силиконовой резины; 12— остеклованный
изнутри металлический капилляр, соединяемый с колонкой; 13, 15 —
стекловата; 14 — наполнитель канала испарения пробы.
Для выполнения количественных определений важно, чтобы
конструкция узла испарителя-делителя обеспечивала эффек-
тивное перемешивание и полную гомогенизацию паров компо-
нентов пробы и газа-носителя по сечению газового потока с тем,
чтобы происходило равномерное (так называемое линейное, или
пропорциональное) деление паров каждого компонента анали-
зируемой смеси, даже если они и очень сильно различаются
по температурам кипения. Перемешивание паров компонен-
тов пробы и газа-носителя осуществляется либо в специальной
термостатируемой камере-змеевике достаточно большого объ-
ема (одновременно играющей роль буферной емкости, смягчаю-
щей газодинамический толчок при испарении введенной пробы,
I
44
рис. 11.12, 6), либо непосредственно в канале испарения пробы
(рис. 11.12, е). В последнем случае для повышения эффективно-
сти перемешивания канал испарения пробы рекомендуется за-
полнять стекловатой, стеклянным бисером или инертным твер-
дым носителем, смоченным небольшим количеством (1-3% по
массе) термостабильной неподвижной фазы типа силиконового
эластомера SE-30. Желательно, чтобы линия сброса имела тем-
пературу, равную или близкую температуре испарителя. В
противном случае возможная конденсация паров высококипя-
щих компонентов может привести к постепенному изменению
соотношения расходов газа-носителя через колонку и линию
сброса, вплоть до полной закупорки линии сброса.
Наиболее универсальными представляются конструкции ис-
парителей, предусматривающие возможность дозирования про-
бы в капиллярную колонку как с делением, так и без деления
потока (соответствующие названия этих способов дозирования
на английском языке — split и splitless). В последнем случае ввод
пробы производят при закрытой линии сброса. Подвижная фаза
поступает в капиллярную колонку с достаточно большой ско-
ростью (10—15 мл/мин или большей), что обеспечивает обра-
зование в начальном участке колонки компактной пробки паров
компонентов анализируемой смеси. Спустя некоторое время (от
5 с до 1 мин), после того как существенная доля паров компо-
нентов анализируемой смеси войдет в колонку, их избыточное
количество быстро удаляют из камеры испарителя струей про-
дувочного газа, линия которого снабжена соленоидным клапа-
ном, связанным с таймером. Начиная с момента открытия соле-
ноидного клапана расход подвижной фазы через капиллярную
колонку резко падает, вплоть до обычных для режима с деле-
нием потока 0,5—5 мл/мин.
В узлах ввода пробы подобного образца, как правило, при-
сутствует и магистраль обдува резиновой пробки испарителя,
препятствующего появлению ложных пиков (т.н. “пиков-духов”)
на хроматограммах, вызванных попаданием в колонку лету-
чих ингредиентов резины. Это обстоятельство довольно часто
осложняет работу химика-аналитика, особенно при использова-
нии режима программирования температуры.
Среди приборов последних лет выпуска весьма совершен-
ной конструкцией испарителя обладает газовый хроматограф
модели 6890 фирмы Hewlett-Packard. Наиболее распространен-
ные серийные отечественные приборы (“Цвет-500” и модель
3700 Московского завода “Хроматограф”) позволяют дозиро-
вать пробу в капиллярную колонку лишь по способу с делением
потока в его наипростейшем варианте (в качестве делителя по-
45
46
тока используется обычный тройник). Существенно расширяет
приборные возможности отечественных хроматографов устрой-
ство для присоединения капиллярных колонок (модели ИКК-
1.1 и ИКК-1.2), разработанное и выпускаемое фирмой ’’Экрос”
(С.-Петербург). Это устройство (рис. 11.13), состоящее из соб-
ственно корпуса испарителя-делителя потока и блока управле-
ния, позволяет вводить пробу как с делением, так и без деления
потока. Предусматривается возможность
— установки и регулирования давления газа-носителя на
входе в колонку*;
— установки и регулирования расходов газа в линиях обдува
резиновой пробки и деления потока;
— установки и индикации режимов работы испарителя-дели-
те ля;
— установки и индикации временных интервалов открытия и
закрытия соленоидного клапана (для режима ввода пробы без
деления потока, см. выше).
Газовые пробы иногда вводятся через испаритель. При этом
могут быть использованы обычные медицинские или специаль-
ные газовые шприцы, причем испаритель не обогревается.
Еще один путь введения проб в колонку — это парофазное
дозирование проб. Парофазный анализ — одно из быстро раз-
вивающихся направлений газовой хроматографии. Сущность
метода состоит в том, что анализу подвергается не исследуе-
мый жидкий или твердый объект, а контактирующая с ним га-
зовая фаза. Наиболее ответственной операцией, определяющей
точность анализа, является дозирование в хроматограф газа,
*Для стабилизации расхода газа-носителя при программировании
температуры колонки устройство необходимо доукомплектовать регуля-
тором расхода газа (врезается в линию газа-носителя между блоком упра-
вления и испарителем).
Рис. 11.13. Устройство для присоединения капиллярных колонок фир-
мы “Экрос” [65].
с — пневматическая схема устройства: 1 — термостат хроматографа; 2 — детектор;
3 — тройник для подсоединения линии поддувочного газа; 4 — капиллярная ко-
лонка; 5, 9— фильтры; 6—-испаритель; 7—выносной кнопочный выключатель;
10 — блок управления; 11 — электронная схема; 12 — дроссель для регулировки
расхода газа-носителя на линии обдува резиновой прокладки; 13 — дроссель для ре-
гулировки расхода газа-носителя на линии сброса; 14 — соленоидный клапан; 15 —
образцовый манометр; 16 — регулятор давления; 17 — блок подготовки газов.
6 — испаритель-де лите ль потока: 1, 10, 15 — накидные гайки; 2 — уплот-
нительный вкладыш; 3 — прокладка из термостойкой резины; 4 — крышка; 5 —
втулка; 6 — стеклянный вкладыш; 7, 9, 14 — графитовые прокладки; 8 — корпус
испарителя-делителя; 11 — уплотнительная втулка; 12 — вставка; 13 — капил-
лярная колонка; 16 — гайка; 17 — шайба; 18 — термоизоляция; 19 — термостат
испарителя-делителя; 20 — термометр сопротивления; 21 — кожух. А — вход газа-
носителя; Б — выход линии обдува резиновой прокладки; В — выход линии сброса.
47
находящегося в равновесии с конденсированной фазой. Этот
процесс отличается от обычных способов введения в хромато-
граф газовых проб и требует специальной техники.
Обязательное условие подготовки пробы к анализу — поддер-
жание постоянной и хорошо воспроизводимой температуры в
сосуде для установления равновесия между исследуемым образ-
цом и газовой фазой. Кроме того, процесс дозирования дол-
жен гарантировать сохранение достигнутого значения концен-
трации вещества в газовой фазе сосуда с пробой. Поэтому кон-
струкция применяющихся устройств должна обеспечивать воз-
можно меньшее время подготовки пробы к анализу, а выбран-
ный способ введения газа — исключать вероятность выхода
гетерогенной системы из равновесия и потери анализируемых
веществ. Особое внимание должно быть уделено тому, чтобы
предотвратить возможность сорбции или конденсации паров в
соединительных элементах газовой схемы.
Многочисленные приспособления и устройства для парофаз-
ного дозирования проб можно разделить на две основные груп-
пы. В устройствах, принадлежащих к первой группе, для уста-
новления равновесия используются сосуды с постоянным объ-
емом, пробы из которых отбираются при переменном давлении.
Другая группа устройств предусматривает применение систем
с переменным объемом газовой фазы и отбор проб при постоян-
ном давлении. Устройствам каждой группы присущи определен-
ные особенности, которые необходимо учитывать при выборе
методики подготовки пробы, варианта количественного анализа
и способа дозирования газа в хроматограф.
Прямое введение равновесного газа в хроматограф из сосу-
дов с постоянным объемом может производиться несколькими
способами.
1. Отбор проб с помощью шприцев и последующее введение в
испаритель хроматографа. Температура шприца должна быть
равной или выше поддерживаемой в сосуде для установления
равновесия. В противном случае могут произойти конденсация
паров растворителя и резкое изменение концентрации анализи-
руемых компонентов в газе. Для того чтобы давление газа в со-
суде и шприце после отбора дозы оставалось равным атмосфер-
ному, шприц предварительно заполняют газом-разбавителем,
объем которого равен объему отбираемого из сосуда газа.
Главное преимущество этого способа — простота и возмож-
ность использования стандартной газохроматографической ап-
паратуры без каких-либо изменений газовой схемы. К весьма
существенным недостаткам его следует отнести потери опреде-
48
ляемых веществ за счет сорбции на стенках шприца и особенно
на поверхности эластичных резиновых уплотнений. Кроме того,
при дозировании газа шприцем трудно получить хорошую вос-
производимость высот (или площадей) пиков на хроматограм-
мах.
2. Введение в хроматографическую колонку пробы с помо-
щью газового крана, дозируемый объем которого заполняется
за счет изменения давления в системе. На рис. 11.14 приведен
случай, когда дозируемый объем заполняется путем кратковре-
менного и многократного повышения давления в системе и роль
насоса выполняет медицинский шприц, соединенный с выходным
штуцером газового крана. Последовательная прокачка шприца
приводит к заполнению дозирующей петли крана газом из со-
суда. Использование дозатора, термостатируемого при высо-
ких температурах (до 200°С), существенно снижает (но не ис-
ключает совсем) сорбционные потери вещества и позволяет до-
биться хорошей воспроизводимости введения в колонку газовых
проб.
Рис. 11.14. Схема заполнения дозируемого объема газового
крана равновесным газом с помощью шприца путем кратко-
временного и многократного увеличения давления в сосуде.
1 — сосуд для установления равновесия; 2 — газовый кран; 3 —
дозирующая петля газового крана; 4 — шприц.
3. Пневматическое парофазное дозирование проб, осущест-
вляемое созданием перепада давления между сосудом с анали-
зируемым образцом и хроматографической колонкой. Соедине-
ние газового пространства сосуда с испарителем хроматографа
49
обеспечивает импульсное введение пробы, объем которой регу-
лируется главным образом разностью давлений в сосуде и в га-
зовой схеме хроматографа. Обеспечение постоянства перепада
давления позволяет получить воспроизводимость дозирования
проб на уровне десятых долей процента.
В практике применения парофазного анализа используются
два варианта пневматического дозирования. Первый преду-
сматривает создание перепада давления в момент дозирова-
ния пробы за счет кратковременного перекрывания потока газа-
носителя, поступающего в хроматографическую колонку
(рис. 11.15, а). Во втором перепад давления создается заранее
путем задания в сосуде с пробой давления большего, чем в ис-
парителе хроматографа (рис. 11.15, 6). В отличие от первого,
второй вариант пневматического способа позволяет точно из-
мерять перепад давления и тем самым определять долю (или
массу) отобранного из сосуда с пробой вещества [66, 67]. По-
дробные рекомендации по практическому использованию
устройства для проведения парофазного анализа с пневмати-
ческим отбором проб, выполненного в виде приставки к хрома-
тографам “Пвет-500”, приведены в разделе II.2.4.
Рис. 11.15. Принципиальная схема парофазного пневматиче-
ского дозирования проб в хроматограф с перепадом давления,
создаваемым в момент дозирования (а) и задаваемым зара-
нее (б).
Сплошной линией показано положение, при котором кран 1 открыт
(давление в сосуде с пробой и в испарителе одинаково), штриховой ли-
нией — когда кран 1 закрыт и происходит дозирование пробы.
50
Особенность отбора проб из емкостей с постоянным объемом
газового пространства состоит в изменении концентрации опре-
деляемого вещества в растворе и в газе над ним. Поэтому воз-
можность повторного дозирования равновесного газа с первона-
чально установленной концентрацией вещества ограничивается
количеством отбираемого вещества и параметрами анализиру-
емой системы (значением коэффициента распределения и соот-
ношения объемов фаз).
Использование статических систем с переменным объемом
газового пространства позволяет отбирать пробы без измене-
ния концентрации вещества в контактирующих фазах. Поэтому
число повторных измерений содержания определяемого веще-
ства в газе лимитируется только его объемом. Такое устрой-
ство [68], изготовленное на основе стандартного медицинского
шприца вместимостью 50-100 мл, в сочетании с газовым кра-
ном позволяет многократно вводить в хроматографическую ко-
лонку равновесный с раствором газ независимо от коэффици-
ента распределения анализируемого вещества. Это достигается
тем, что при сокращении рабочего объема сосуда (за счет пе-
ремещения поршня) происходит вытеснение равновесного газа
практически без изменения давления в системе. Отбор пробы
при постоянном давлении позволяет проводить нужное число
дозирований (ограничивающееся лишь объемом газовой фазы)
без дополнительного разбавления равновесного газа, а следова-
тельно, не выводя систему из термодинамического равновесия.
На рис. II.16 показана схема дозирования в хроматографиче-
скую колонку равновесного с жидкостью газа из устройства с
переменным объемом. Проба газа вводится в хроматографи-
ческую колонку термостатируемым краном Б. Заполнение до-
зирующей петли 11 газом, приведенным в равновесие с жид-
костью в шприце А, производится перемещением поршня 1.
Объем пропущенного через петлю газа контролируется мыльно-
пленочным измерителем 12, который используется также для
определения момента установления в дозирующей петле давле-
ния, равного атмосферному.
Следует отметить, что дозирование — одна из ответственных
операций — осуществляется вручную, и ошибка при выполне-
нии этой операции составляет, как правило, большую часть по-
грешности анализа. Успешное выполнение дозирования зависит
не только от конструкции дозирующей системы хроматографа,
но и от практических навыков оператора.
В последнее время сделаны успешные попытки автоматиза-
ции дозирования жидких проб: созданы различные типы микро-
Дозаторов, позволяющие существенно повысить точность и вос-
51
Рис. 11.16. Схема дозирования в хроматографическую колонку
равновесной газовой фазы.
А — прибор с переменным объемом; Б — газовый кран; В — хро-
матографическая колонка; Г — детектор. 1 —- поршень стеклянного
медицинского шприца; 2 — эластичные резиновые прокладки; 3 — ци-
линдр шприца; 4 — канюля шприца; 5 — фторопластовая трубка; 6 —
эластичное резиновое уплотнение; 7 — стальная или фторопластовая
капиллярная трубка, соединяющая шприц с газовым краном; 8 — при-
жимной колпачок; § — корпус уплотнительного устройства; 10 — ру-
башка из плексигласа для подачи термостатирующей жидкости; 11 —
дозирующая петля газового крана; 12 — мыльно-пленочный измери-
тель расхода газа.
производимость дозирования. Наиболее распространены ав-
томатические манипуляторы микрошприцем с электрическим
или пневматическим приводом, а также ампульные дозаторы,
в которых анализируемые пробы заранее помещены в открытые
или герметично запаянные ампулы. Управление микродозато-
52
ром осуществляется автоматически, по временной программе,
специальными устройствами на основе микропроцессорной тех-
ники.
11.1.3. Хроматографические колонки
Различают три основных типа аналитических колонок — на-
садочные (наполненные), микронасадочные и капиллярные. На
первых этапах развития газовой хроматографии ввиду про-
стоты приготовления и возможности применения детекторов
средней чувствительности наибольшее распространение полу-
чили насадочные колонки. Эти колонки до сих пор изго-
тавливаются из металлических (нержавеющая сталь), стеклян-
ных или фторопластовых трубок с внутренним диаметром от 2
до 4 мм и длиной от 0,5 до 3 м, которым в настоящее время чаще
всего придается спиральная форма (рис. 11.17). Микронаса-
дочные колонки отличаются от насадочных только диаме-
тром трубок, равным 0,8-1,0 мм. Длина таких колонок обычно
Рис. 11.17. Хроматографические колонки.
Насадочные: 1,2 — стеклянные, S — фторопластовая, 4 — сталь-
ная; капиллярные: 5 — стальная, 6 — кварцевая; микронасадочная;
7 — стал ьная.
53
не превышает 2 м. Форма насадочных и микронасадочных ко-
лонок практически не влияет на эффективность разделения и
определяется размерами термостата.
Чаще всего хроматографы комплектуются колонками из не-
ржавеющей стали, антикоррозионные свойства которой допус-
кают длительную эксплуатацию без специальной обработки и
очистки металла. Для разделения неустойчивых к каталити-
ческому действию металлов соединений или корродирующих
веществ применяют стеклянные колонки. В этом случае кон-
такт с поверхностью металла должен исключаться также и в
других элементах газовой схемы хроматографа, с которыми со-
прикасаются анализируемые вещества (испаритель, детектор).
Колонки из фторопласта исключительно хорошо зарекомендо-
вали себя при выполнении анализов на содержание малых при-
месей высокополярных соединений (вода, аммиаки т. п.), однако
фторопластовые колонки нельзя использовать при температу-
рах выше 90-100 °C.
Заполнитель насадочных и микронасадочных колонок в лите-
ратуре на русском языке принято называть насадкой, иногда —
сорбентом (соответствующий английский термин — packing)
Важнейшие приемы приготовления насадки (сорбента) и запол-
нения насадочных колонок рассмотрены в лабораторной ра-
боте 1. Практические вопросы, связанные с выбором неподвиж-
ной фазы и твердого носителя для конкретных целей хромато-
графического разделения, приготовлением насадки и заполне-
нием колонок различной формы, а также с присоединением ко-
лонок к элементам газовой схемы хроматографа, подробно рас-
смотрены в книге [69], с которой рекомендуется ознакомиться
каждому начинающему хроматографисту.
Капиллярные колонки изготавливают обычно из труб-
ки (нержавеющая сталь, стекло или кварц) внутренним диаме-
тром 0,25-0,5 мм и длиной от 10-20 до 100-200 м. Отличаются
они от насадочных и микронасадочных колонок тем, что непо-
движная фаза (в виде тонкой пленки нелетучего растворителя
или тонкого слоя адсорбента) не заполняет всей внутренней
полости, а покрывает лишь внутреннюю поверхность трубки,
остающейся, по существу, “полой” и после нанесения неподвиж-
ной фазы. Эта особенность капиллярных колонок подчеркива-
ется и в принятом для них в литературе на английском языке
названии “open tubular columns”, часто используемом в послед-
ние годы вместо первоначального термина “capillary columns”.
Поток газа движется по такой колонке с большой линей-
ной скоростью, не встречая значительного сопротивления. По-
этому, несмотря на большую длину (десятки и даже сотни ме-
54
тров), для обеспечения необходимых расходов газа-носителя
через капиллярную колонку оказывается достаточным пример-
но такое же входное давление, что и при работе с насадочными
колонками.
Для газо-жидкостного варианта хроматографии предназна-
чены капиллярные колонки с неподвижной фазой, покрываю-
щей собственно поверхность внутренней стенки трубки (так на-
зываемые классические капиллярные колонки, wall-coated open
tubular columns, или WCOT columns), и капиллярные колонки с
неподвижной фазой, покрывающей слой мелкодисперсного по-
ристого носителя, закрепляемого на внутренней поверхности
трубки (так называемые твердослойные капиллярные колонки,
support-coated open tubular columns, или SCOT columns). Колонки
последнего типа открывают возможность работы в капилляр-
ном варианте хроматографии с гораздо большим ассортимен-
том неподвижных фаз и допускают повышенные нагрузки, од-
нако воспроизведение их рабочих характеристик даже опыт-
ными фирмами пока еще оставляет желать лучшего.
В газо-адсорбционной хроматографии очень хорошо зареко-
мендовали себя кварцевые капиллярные колонки, на внутрен-
ние стенки которых нанесен слой адсорбента (например, оксида
алюминия, модифицированного неорганическими солями, или
какого-либо пористого синтетического сорбента) [70]. В ан-
глоязычной литературе их называют porous-layer open tubular
columns, или PLOT columns.
Замечательной особенностью капиллярных колонок является
весьма высокая эффективность (до нескольких тысяч тарелок на
метр длины), хотя приготовление высокоэффективных и воспро-
изводимых по разделяющей способности капиллярных колонок,
особенно с полярными неподвижными фазами, все еще требует
определенных практических навыков. Эффективность работы
капиллярной колонки в значительной мере определяется чисто-
той и однородностью внутренней поверхности капилляра. Од-
ним из первых относительно инертных материалов для изгото-
вления капиллярных колонок стали применять нержавеющую
сталь.
В 70-е годы большое распространение получили стеклянные
капиллярные колонки, позволяющие анализировать термически
и каталитически неустойчивые, а также высокополярные соеди-
нения. Стекло — наиболее дешевый и доступный материал. Ко-
лонки из натриевых и боросиликатных стекол более инертны
и стабильны, чем металлические, и могут быть изготовлены
практически в любой лаборатории. Однако успешное нанесе-
ние неподвижной фазы на внутреннюю стенку капилляра воз-
55
можно лишь после обязательной специальной подготовки (тра-
вления) стекла. Многочисленные практические рекомендации
по проведению этой процедуры содержатся в монографиях [39-
41]. Правда, стеклянным капиллярным колонкам свойственны
и недостатки, например сравнительно высокая остаточная ад-
сорбционная активность поверхности (особенно по отношению
к полярным веществам), сильно зависящая от типа стекла и спо-
соба его обработки. Кроме того, низкая механическая проч-
ность стеклянных капилляров значительно затрудняет работу с
ними.
В конце 1970-х годов в капиллярной хроматографии сфор-
мировалось новое направление — использование тонкостенных
кварцевых капиллярных колонок, имеющих внутренний
диаметр 0,05-0,32 мм, толщину стенки 40-70 мкм, толщину за-
щитного слоя, придающего колонке механическую прочность,
15-30 мкм. Кварцевые капилляры сочетают низкую остаточную
адсорбционную активность с исключительно высокой механи-
ческой прочностью на изгиб. Высокая инертность внутренней
поверхности кварцевой колонки обусловлена в первую очередь
химической чистотой исходного материала, в качестве кото-
рого используется очищенный кварц или синтетический плавле-
ный диоксид кремния. Долговечность и гибкость тонкостенных
кварцевых капилляров достигается за счет нанесения на них не-
посредственно после вытяжки слоя лака (например, полиимид-
ного), защищающего поверхность от внешних воздействий и до-
пускающего длительную работу при температурах 250-300°C.
Гибкость кварцевых капиллярных колонок такова, что они до-
пускают изгиб на диаметр менее 10 мм. Достигнуть более высо-
ких рабочих температур и больших сроков службы при эксплуа-
тации в жестких температурных режимах позволяют разрабо-
танные в конце 80-х — начале 90-х годов технологии нанесения
на капилляр тонкого внешнего металлического (алюминиевого
или стального) покрытия, оставляющего колонке большой запас
гибкости.
Инертность и высокая эффективность кварцевых капилляр-
ных колонок позволяют разделять сложные смеси соединений,
принадлежащих к различным классам, в том числе карбоновые
кислоты, алифатические и ароматические амины.
Важное ограничение в использовании т. н. классических ка-
пиллярных колонок малого диаметра (0,25-0,32 мм) с тонкими
(~1 мкм ) пленками неподвижных фаз для определения примес-
ных компонентов в сложных матрицах — их невысокая емкость
(требующая деления паров пробы на входе в колонку или ис-
пользования альтернативных приемов исключения перегрузки)
56
в последние годы успешно преодолевается все большим распро-
странением широких (внутренний диаметр ~0,53 мм) капилляр-
ных колонок (wide bore capillary columns) с толстыми слоями (до
5-8 мкм) привитых неподвижных фаз. Рабочая емкость колонок
этого типа приближается к таковой для насадочных колонок, с
ними можно работать без деления паров пробы, а по эффектив-
ности они занимают промежуточное положение между насадоч-
ными и классическими капиллярными колонками.
Из направлений развития газовой хроматографии, рассчи-
танных на далекую перспективу, отметим использование па-
кетов параллельно работающих капилляров малого диаметра
(~30 мкм) и сравнительно небольшой длины (<С1 м) общим чи-
слом до LOGO и более в качестве единой поликапиллярной ко-
лонки (ПКК) [71]; соответствующие зарубежные аналоги полу-
чили название multicapillary GC columns. Колонки этого типа
создают возможность экспрессного и высокоэффективного раз-
деления сложных многокомпонентных смесей при достаточно
низких температурах, что особенно привлекательно при работе
с термически лабильными соединениями. Дополнительным до-
стоинством является высокая загрузочная емкость ПКК, что по-
зволяет осуществлять дозирование относительно больших пор-
ций анализируемых образцов, как и при работе с насадочными
колонками, без сброса.
Из двух известных методов заполнения капиллярных коло-
нок неподвижной фазой — статического и динамического [39-
41] — предпочтение, по-видимому, следует отдать первому, как
обеспечивающему наибольшую воспроизводимость, хотя и бо-
лее трудоемкому*.
Общие вопросы теории и практики капиллярной газовой хро-
матографии подробно освещены в книгах [2, 31, 33, 39-41].
II.1.4. Детекторы
II.1.4.1. Общие сведения
Хроматографический детектор представляет собой устрой-
ство, предназначенное для обнаружения и количественного оп-
*Недавно, в развитие процедуры нанесения неподвижной фазы на вну-
тренние стенки капиллярных колонок динамическим способом, изложен-
ной в [72], опубликована методика ускоренного приготовления и проверки
работоспособности капиллярных колонок, адаптированная к условиям
стУДенческого практикума (выполняется за два-три лабораторных заня-
тия) [73].
57
ределения выходящих из колонки в потоке газа-носителя ком-
понентов анализируемой смеси. Регистрация вещества осуще-
ствляется за счет преобразования в электрический сигнал из-
менений химических или физико-химических свойств газового
потока, выходящего из хроматографической колонки.
Для газовой хроматографии предложено большое число де-
текторов — около 50, однако на практике применяются только
некоторые из них. Комплект современного универсального хро-
матографа включает 4-6 детекторов. Комплектация хромато-
графов несколькими детекторами вызвана тем, что универсаль-
ных детекторов, удовлетворяющих в полной мере предъявляе-
мым к ним требованиям, не существует. Поэтому в каждом слу-
чае следует выбирать такой детектор, характеристики которого
в наибольшей степени соответствуют цели анализа и условиям
его проведения.
Наибольшее распространение в силу универсальности, пре-
восходных характеристик и высоких эксплуатационных качеств
получили пламенно-ионизационный детектор и детектор по те-
плопроводности, входящие в состав почти всех хроматографов.
Кроме того, широко используются селективные детекторы, по-
зволяющие определять в сложных смесях только соединения
определенного состава. К ним в первую очередь относятся де-
текторы электронозахватный, термоионный и пламенно-фото-
метрический, использование которых упрощает расшифровку
хроматограмм, повышает чувствительность, значительно со-
кращает время анализа и объем пробы исследуемой смеси. Та-
кие преимущества селективных детекторов являются основной
причиной их широкого применения при анализе сложных смесей
биологического или природного происхождения и загрязнения
окружающей среды.
Детекторы подразделяются на интегральные и дифференци-
альные. Интегральный детектор регистрирует измене-
ние во времени суммарного количества выходящих из колонки
компонентов, например их общий объем или количество титру-
ющего раствора, израсходованного на нейтрализацию анали-
зируемых веществ. Хроматограмма (рис. 11.18, а) представляет
собой ряд ступеней, высота h каждой из которых пропорцио-
нальна количеству данного компонента разделенной смеси, про-
шедшему через детектор за время — Д. Из-за низкой чув-
ствительности, большой инерционности и недостаточной уни-
версальности эти детекторы имеют весьма ограниченное при-
менение и в дальнейшем не рассматриваются.
Дифференциальный детектор измеряет мгновенную
концентрацию или массовую скорость вещества в потоке газа-
58
Рис.П.18. Интегральная (а) и дифференциальная (б) хромато-
граммы.
носителя. Хроматограмма, полученная с таким детектором,
представляет собой ряд пиков (рис. 11.18, б), причем количество
каждого компонента пропорционально площади А соответству-
ющего пика.
При детектировании возможны два принципиально различ-
ных варианта взаимодействия молекул анализируемого веще-
ства с чувствительным элементом детектора: 1) процесс, раз-
рушающий молекулы при регистрации (делающий повторные
взаимодействия невозможными), и 2) процесс, в результате ко-
торого не утрачивается возможность повторной (многократной)
регистрации тех же молекул. Если заключить некоторое коли-
чество вещества в замкнутый (непроточный) объем детекторов с
однократной и многократной регистрацией, то сигнал первого
детектора быстро уменьшается, так как процесс регистрации
уменьшает количество вещества в детекторе. Сигнал второго
детектора остается постоянным как угодно долго, поскольку при
регистрации вещество не расходуется и каждая молекула может
быть зарегистрирована неограниченное число раз (рис. 11.19).
Рис. 11.19. Зависимость сигнала
потокового (1) и концентрацион-
ного (2) детекторов от времени в
замкнутом объеме.
59
В детекторе с однократной регистрацией сигнал Ej опреде-
ляется мгновенным значением количества вещества q, достига-
ющего чувствительного элемента детектора в момент времени t,
т. е. массовой скоростью j (потоком) вещества через детектор:
Ej = Rjj, j = ~- (П-1)
Детекторы такого типа принято называть потоковыми.
В детекторе с многократной регистрацией сигнал Ес опреде-
ляется текущей концентрацией вещества С в объеме V чувстви-
тельного элемента детектора:
Ес = RcC, С=-^. (11.2)
av
Такие детекторы называют концентрационными. Rj, Rc —
коэффициенты, определяющие чувствительность потокового и
концентрационного детекторов. Для упрощения дальнейших
выводов будем считать Rj и Rc независимыми от величины по-
тока и концентрации т. е. примем, что детекторы работают
в линейной области (подробное определение линейности см.
ниже).
Если известны коэффициенты чувствительности, сигнал де-
тектора во время выхода пика позволяет рассчитать количество
вещества, прошедшее через детектор с момента /j начала до мо-
мента /2 конца пика. Для потокового детектора
для концентрационного детектора
EC = RC^-, dq — (EcdV.
Civ LEq J
Поскольку поток вещества и его концентрация в детекторе
связаны через объемную скорость F газа-носителя, имеем
dq
dt
dV = Fdt,
3 = сЕ,
EcFdt.
Если скорость газа-носителя постоянна,
ti
60
Выражение f*2 Edt есть площадь кривой Е = f(t), т. е. площадь
соответствующего хроматографического пика, поэтому
/ 1 \ / F \ / <7 \
Ч — ( ) Ai' Aj = Rj4t Ч — ( ту I Ас, Ас = Rc ( — ) .
Из сказанного выше можно сделать вывод, что площадь пика
потокового детектора не зависит от скорости пропускания ана-
лизируемого вещества и определяется только его количеством,
а площадь пика концентрационного детектора обратно пропор-
циональна скорости газа-носителя. Физический смысл этого со-
стоит в том, что при использовании потокового детектора все
количество анализируемого компонента успевает однократно
зарегистрироваться вне зависимости от скорости пропускания,
тогда как в концентрационном детекторе от скорости зависит
число актов регистрации каждой молекулы и чем больше эта
скорость, тем меньшее число актов регистрации успевают пре-
терпеть молекулы анализируемого вещества при одном и том
же числе молекул.
Следует отметить, что зависимость чувствительности де-
текторов от скорости газа-носителя в соответствии со специ-
фикой механизмов работы конкретных детекторов несколько
усложняет описанный характер зависимости от скорости газа-
носителя, однако в каждом случае характерные черты потоко-
вых и концентрационных детекторов проявляются достаточно
четко.
Типичным примером потокового детектора является пламен-
но-ионизационный детектор (ПИЛ), в котором происходит сго-
рание органических соединений. Детектор по теплопроводно-
сти, в котором процесс отвода теплоты от чувствительных эле-
ментов не разрушает молекул анализируемых веществ, — ти-
пичный концентрационный детектор. При пропускании одной и
той же порции вещества через эти детекторы с увеличением ско-
рости газа-носителя площадь пика ПИД лишь незначительно
изменяется (это связано с изменением Rj), тогда как уменьше-
ние площади пика детектора по теплопроводности происходит
пропорционально увеличению скорости газа-носителя при со-
хранении Rc. При измерении площадей пиков потоковые детек-
торы более предпочтительны из-за независимости их показаний
от колебаний давления и расхода.
II.1.4.2. Характеристики детекторов
Основными характеристиками хроматографических детекто-
ров являются чувствительность и предел детектирования, ли-
нейность, быстродействие, селективность.
61
Чувствительность является одной из важнейших характери-
стик детектора, поскольку она связывает его сигнал с изме-
ряемой концентрацией и в значительной мере определяет ана-
литические возможности хроматографа в целом. От чувстви-
тельности детектора зависят выбор величины пробы и возмож-
ность использования различных типов хроматографических ко-
лонок. Так, применение микронасадочных и капиллярных коло-
нок возможно лишь с высокочувствительными детектирующими
устройствами, а при работе с обычными насадочными колон-
ками могут использоваться и детекторы средней чувствитель-
ности — по теплопроводности и по плотности. Применение вы-
сокочувствительных детекторов весьма желательно, так как по-
зволяет значительно уменьшить величину вводимой пробы, что
в большинстве случаев (особенно в газо-адсорбционном вари-
анте) улучшает качество разделения компонентов анализируе-
мой смеси. Однако в газо-жидкостном варианте, в особенности
при высоких температурах хроматографических колонок, в не-
которых случаях затруднительно применение детектора высо-
кой чувствительности ввиду значительного фона, создаваемого
за счет летучести жидкой фазы.
Значения чувствительности детектора могут быть вычисле-
ны непосредственно из условий проведения и результатов хро-
матографического анализа. В соответствии с приведенными
выше определениями сигнала концентрационного и потокового
детекторов (см. уравнения (II.1) и (П.2)) чувствительность кон-
центрационных детекторов (мВ • мл/мг) определяется выраже-
нием
Rc = AvF/qw, (П.З)
где А — площадь пика, см2; и — чувствительность самопишу-
щего потенциометра, мВ/см; F — скорость газа-носителя, мл/с;
q — масса компонента, мг; w — скорость протягивания ленты,
см/с.
Чувствительность потоковых детекторов (мВ-с/мг) описыва-
ется формулой
Rj = Avjqw. (II.4)
Весьма важной величиной, характеризующей предельную
чувствительность детектора, является минимальная концентра-
ция анализируемого вещества в потоке газа-носителя, которая
может быть зарегистрирована. Для ее характеристики недо-
статочно располагать данными о чувствительности R, необхо-
димо также знать, какое наименьшее значение сигнала детек-
62
тора можно измерить, учитывая уровень флуктуационных шу-
мов нулевой линии прибора.
Минимальным сигналом Емин, поддающимся измерению, при-
нято считать сигнал, амплитуда которого вдвое превышает уро-
вень шумов 8 (рис. 11.20): Еыиа = 28.
Время
Рис. П.20. Наименьший детектируемый полезный сигнал.
Концентрация анализируемого вещества, вызывающая этот
сигнал, для концентрационного детектора равна
Сми„ = EMliK/Rc = 28/Rc, (II.5)
для потокового детектора соответственно
./мин — ^Мин/Ну, Умин — СМИНК, СмиН — 28/RjF.
(П.6)
Величина Смин называется пределом детектирования и явля-
ется весьма важной характеристикой, поскольку она позволяет
оценить предельные возможности детектора. Предел детекти-
рования выражают в различных единицах, но наиболее часто в
мг/мл, мл/мл, % (по объему), ppm (млн-1).
В повседневной практике часто путают понятия “чувстви-
тельность” и “предел детектирования”, понимая под чувстви-
тельностью минимальные концентрации, определяемые детек-
тором. Графически эти величины можно выразить следующим
образом (рис. 11.21). Чувствительность характеризуется накло-
ном зависимости сигнал детектора — концентрация вещества,
а предел детектирования — отрезком на оси абсцисс, соответ-
ствующим точке пересечения градуировки с ординатой, равной
минимальному сигналу, доступному измерению (двойной уро-
вень шума 28). Из этого определения следует, что из двух
детекторов с одинаковым уровнем шумов меньшим пределом
детектирования будет обладать детектор с большей чувстви-
тельностью (I детектор на рис. 11.21, а). Однако это не зна-
чит, что детекторы с большей чувствительностью всегда спо-
собны определять меньшие концентрации, т. е. имеют меньший
63
предел детектирования. Вполне реальны случаи (особенно при
использовании селективных детекторов), когда благодаря низ-
кому уровню шумов меньший предел детектирования будет со-
ответствовать детектору с меньшей чувствительностью (II де-
тектор на рис. 11.21, б). Поэтому сопоставление возможностей
детекторов в отношении регистрации малых концентраций ве-
ществ следует производить, сравнивая пределы детектирова-
ния.
Рис. П.21. Зависимость минимально определяемой концентра-
ции от чувствительности детектора и уровня шумов для де-«й
текторов с одинаковым (а) и различным (5) уровнем шумов.
Таким образом, предельные возможности хроматографа в от-
ношении измерений малых концентраций могут быть расши-
рены двумя независимыми путями: повышением чувствитель-
ности детектора и снижением уровня шумов. Причины, вызыва-
ющие шумы, разнообразны и в значительной мере специфичны
для каждого конкретного детектора, однако общим источником
шумов является изменение рабочих условий колонки и детек-
тора при разделении анализируемых веществ.
Следует подчеркнуть, что предел детектирования соответ-
ствует концентрации вещества в газе-носителе, создаваемой в
детекторе, а не концентрации анализируемых веществ в пробе
при введении в колонку. Учитывая процесс размывания пробы,
нужно иметь в виду, что практически измеряемая хроматогра-
фом минимальная концентрация веществ в пробе по крайней
мере в 5-10 раз выше предела детектирования.
64
Рис. 11.22. Зависимость сиг-
нала детектора от концен-
трации анализируемого ве-
щества.
Концентрация вещества
Весьма важной характеристикой детектора является линей-
ность показаний, от которой в значительной мере зависит точ-
ность количественного анализа. Под линейностью детектиру-
ющих устройств понимается пропорциональность между кон-
центрацией анализируемого вещества в потоке газа-носителя
на выходе из колонки и сигналом детектора. 'Если измерить
зависимость сигнал детектора — концентрация вещества, т. е.
снять градуировочную кривую (рис. 11.22), то прямолинейный
участок этой зависимости определяет линейную область детек-
тора. Отклонение от прямой свидетельствует об отклонении
от линейности. Диапазон линейности представляет собой ин-
тервал концентраций от предела детектирования до концентра-
ции, при которой наблюдается заметное (3-5%) отклонение от
пропорциональности, и определяется отношением максималь-
ной концентрации к пределу детектирования. В пределах диа-
пазона линейности* чувствительность детектора не зависит от
концентр ации.
При проведении количественного анализа условия хромато-
графирования следует выбирать такими, чтобы сигнал детек-
тора не выходил за пределы диапазона линейности. В против-
ном случае точность анализа снижается. Сказанное можно про-
иллюстрировать следующим образом.
Для любого детектора чувствительность к анализируемому
соединению определяется как угловой коэффициент R = tg<p
(см. рис. 11.22). На линейном участке кривой он одинаков для
любой точки. За пределами линейной части кривой в зависи-
*В литературе “диапазон линейности” иногда заменяют термином “ли-
нейный динамический диапазон”.
65
мости от концентрации вещества в потоке газа-носителя R при-
нимает различные значения, так как представляет собой тан-
генс угла наклона касательной к кривой, т. е. для нелинейного
участка кривой R = f(C), а это означает, что значение пло-
щади пика будет изменяться непропорционально количеству ве-
щества.
Количественный анализ в условиях нелинейной работы де-
тектора требует более детальной градуировки в диапазоне ра-
бочих концентраций анализируемых веществ. Такая градуи-
ровка связана со значительными экспериментальными трудно-
стями — точной дозировкой пробы и тщательным воспроизве-
дением условий хроматографического анализа.
Быстродействие, или инерционность, характеризует способ-
ность детектора реагировать на быстрое изменение концентра-
ции вещества в потоке газа-носителя, проходящего через де-
тектор. Количественно быстродействие обычно оценивается
по значению постоянной времени, характеризующей время ре-
акции детектора на изменение концентрации (время отработки
сигнала): чем меньше постоянная времени, тем быстрее реаги-
рует детектор (малая инерционность) и тем меньшие искажения
появляются при записи хроматограммы.
Если в детекторе в момент времени t0 скачкообразно изме-
нить (например, увеличить) концентрацию, его сигнал должен
соответственно измениться от начального значения Ео до зна-
чения Ei, отвечающего новой концентрации (рис. 11.23, а). Как
правило, это изменение сигнала происходит не мгновенно, а с
некоторым запаздыванием согласно закону
Е= Е1(1-е-‘/т),
где Е — текущий сигнал детектора; t — время с момента из-
менения концентрации; т — постоянная времени. За время t,
равное т, сигнал достигает значения
Et = Ei(l- е-1) = 0,63Е1.
Таким образом, постоянная времени соответствует времени до-
стижения 63% полностью установившегося значения сигнала.
Естественно, искажения сигнала детектора из-за его ограни-
ченного быстродействия (инерционности) проявляются сильнее
при записи узких и высоких пиков (быстрое изменение концен-
трации в детекторе) и практически отсутствуют при регистра-
ции широких пиков. Характер искажений показан на рис. 11.23, 6.
Постоянная времени должна быть мала по сравнению с длитель-
ностью пика. Как следует из табл. II.1, наибольшие искажения
претерпевают высоты пиков, в то время как изменения площадей
66
Рис. П.23. Определение постоянной времени (а) и характер ис-
кажения формы пика (6).
1 — сигнал детектора при идеальном быстродействии; 2 — иска-
женный сигнал детектора.
Таблица II. 1. Зависимость искажения высоты h и площади
пиков А от соотношения постоянной времени детектора*
и длительности пиков (для пиков гауссовой формы)
St • 100% Лист • 100% Лист St • 100% Лист 100% Лист
0 0 0 0,255 11,4 1,0
0,085 1,5 0 0,340 16,7 3
0,170 6,1 0,5 0,425 21,8 5
* При веденные сведения относятся к детектору по теплопроводности.
пиков в тех же условиях значительно меньше за счет компенси-
рующего влияния расширения пиков (искажение длительности
пиков).
Инерционность детектора является следствием ограниченной
скорости физических или физико-химических процессов, опре-
деляющих механизм детектирования. Так, относительно боль-
шая инерционность детектора по теплопроводности определя-
ется скоростью процесса теплопередачи, которая значительно
меньше скорости образования и сбора зарядов в ионизационных
детекторах. Ионизационные же детекторы практически мгно-
венно реагируют на изменение состава газа.
67
Существенное влияние на четкость регистрации результатов
разделения оказывает также эффективный объем чувствитель-
ного элемента детектора. Последний должен быть возможно
меньшим по сравнению с объемом газа, в котором распреде-
лен анализируемый компонент, выходящий из аналитической
колонки. При относительно большом объеме чувствительного
элемента в нем может происходить дополнительное размыва-
ние или даже смешение полос разделенных компонентов. Осо-
бенно жесткие требования к эффективному объему предъявля-
ются при работе с капиллярными и микронасадочными колон-
ками в силу весьма малых объемов анализируемых проб. По-
этому, в частности, с капиллярными колонками используют
главным образом пламенно-ионизационный детектор, имеющий
весьма малый эффективный объем.
Иногда постоянную времени детектора (а чаще — системы
регистрации в целом) искусственно увеличивают для исключе-
ния записи короткопериодных выбросов или флуктуаций нуле-
вой линии, если длительность пиков достаточно велика и они
могут быть записаны без искажений при большой постоянной
времени.
В зависимости от цели газохроматографического анализа к
детектору могут предъявляться диаметрально противополож-
ные требования. При изучении состава сложных смесей детек-
тирующая система должна быть наиболее универсальна, т. е.
регистрировать все вещества, выходящие из хроматографиче-
ской колонки, и желательно с одинаковой чувствительностью.
Наоборот, если цель анализа состоит в определении отдельных
веществ, входящих в состав сложных смесей, универсальность
детектора начинает играть отрицательную роль, так как на
фоне большого числа пиков сопутствующих веществ выделить
анализируемые соединения бывает не только затруднительно,
но иногда и практически невозможно. В таких случаях необ-
ходимо применять селективные детекторы, избирательно реги-
стрирующие определенный класс или группу веществ.
Без селективного детектирования немыслимы современный
газохроматографический анализ и идентификация индивиду-
альных компонентов сложных смесей. Применение селектив-
ных детекторов расширяет возможности анализа микроприме-
сей, причем в ряде случаев это единственная возможность от-
деления от посторонних веществ, даже когда они имеют близкие
или совпадающие параметры удерживания. С помощью селек-
тивных детекторов можно провести предварительную класси-
фикацию компонентов исследуемой смеси и получить данные о
природе анализируемых соединений.
68
Селективность S характеризуют одним из двух способов. В
первом случае исходят из сравнения чувствительностей двух
детекторов Ri и Л2 к одному и тому же веществу:
Si = /?1/Л2.
Во втором случае сравнивают чувствительность одного и того
же детектора к двум веществам Ra и Rt>:
S2 = R<jRb>
при этом детектор считается селективным, если его чувстви-
тельность для двух веществ отличается не меньше чем на поря-
док.
Для качественного анализа, в частности для групповой клас-
сификации или классификации веществ, т. е. возможности изби-
рательного детектирования определенной группы веществ, бо-
лее предпочтителен второй способ. Для характеристики
свойств селективного детектора чаще всего применяются дан-
ные, полученные на основе первого способа.
II.1.4.3. Детектор по теплопроводности
Принцип действия детектора по теплопроводности (ДТП)
основан на изменении температуры нагретых нитей (чувстви-
тельных элементов) в зависимости от теплопроводности окру-
жающего газа, которая в свою очередь определяется его со-
ставом. Нагретые электрическим током нити отдают теплоту
главным образом за счет принудительной конвекции и тепло-
проводности газа. Теплопередача принудительной конвекцией
нежелательна, так как зависит в большей степени от скорости
и теплоемкости газа, а не от его теплопроводности. Наиболее
предпочтительна теплопередача за счет теплопроводности газа.
Этот вид теплообмена можно увеличить, повышая теплопровод-
ность газа-носителя.
Детектор по теплопроводности (катарометр) измеряет раз-
личие в теплопроводности чистого газа-носителя и
смеси газа-носителя с веществом, выходящим из хроматогра-
фической колонки. Поэтому наибольшая чувствительность мо-
жет быть получена в том случае, когда теплопроводность ана-
лизируемого вещества сильнее отличается от теплопроводно-
сти газа-носителя. Большинство органических веществ имеют
низкую теплопроводность (табл. II.2), и для их анализа целесо-
образно использовать газы-носители с возможно более высокой
теплопроводностью. Такими газами являются водород и гелий,
но на практике водород ввиду его взрывоопасности применяется
69
значительно реже гелия. Так как гелий является довольно де-
фицитным и дорогим газом, а работа с водородом небезопасна,
в некоторых случаях в качестве газа-носителя может использо-
ваться азот, аргон, углекислый газ или воздух, однако харак-
теристики детектора по теплопроводности (чувствительность,
линейность) при работе с этими газами значительно ухудша-
ются. Кроме того, при анализе веществ с большей теплопровод-
ностью, чем у газа-носителя, появляются отрицательные пики
Таблица II.S. Теплопроводность газов-носителей
и некоторых органических веществ
Соединение Тепло- проводность при 100°С, х-103 Вт/(м«К) Тепло- проводность по отношению к гелию, % Соединение Тепло- п ровод ность при 100°С, х-103 Вт/(м-К) Тепло- проводность по отношению к гелию, %
Газы-носители Изобутан 24,3 14,0
Водород 223,6 128,0 Нонан 18,8 10,8
Гелий 174,2 100,0 Циклогексан 17,6 10,1
Азот 31,4 18,0 Бензол 17,2 9,9
Диоксид 22,2 12,7 Ацетон 16,7 9,6
углерода Этанол 22,2 12,7
Аргон 21,8 12,5 Этил ацетат 17,2 9,9
Хроматографируемые вещества Хлороформ 10,5 6,0
Этан 30,6 17,5 Метилиодид 7,9 4,6
Бутан 23,4 13,5
Детектор по теплопроводности представляет собой массив-
ный металлический блок, в цилиндрические отверстия (камеры)
которого помещены чувствительные элементы— металлические
спирали из тончайшей проволоки, закрепленные в кронштейне.
Камеры детектора через входной и выходной каналы продува-
ются газом-носителем.
Ячейка детектора состоит из чувствительного элемента, по-
мещенного в камеру блока детектора. Ячейки бывают про-
точными, диффузионными и полудиффузионными (рис. 11.24). В
проточной ячейке газовый поток омывает чувствительные эле-
менты, в диффузионной — газовая смесь поступает к чувстви-
тельным элементам за счет диффузии через специальный канал.
Полудиффузионная ячейка является промежуточной между про-
точной и диффузионной. Детектор с диффузионной ячейкой
обладает малой чувствительностью к изменениям скорости по-
тока газа, но уступает детектору с проточными ячейками по
70
чувствительности и быстродействию. В современных универ-
сальных аналитических хроматографах в основном применя-
ются детекторы по теплопроводности с полудиффузионными
ячейками. Диффузионные детекторы по теплопроводности ис-
пользуются в препаративных хроматографах.
Рис. 11.24. Типы ячеек детектора по теплопроводности,
а — проточные; б — диффузионные; в — полудиффузионные.
Стрелками показано направление потока газа-носителя.
Чувствительные элементы чаще всего изготовляются в виде
спиральных нитей диаметром 0,025-0,125 мм из материала с вы-
соким температурным коэффициентом сопротивления (вольф-
рам, платина). Нити нагреваются постоянным током до темпе-
ратуры, превышающей температуру блока. Например, при ис-
пользовании гелия в качестве газа-носителя и силе тока 200 мА
температура нитей сопротивлением 50 Ом примерно на 100 °C
выше температуры блока детектора.
71
Чувствительными элементами могут служить также терми-
сторы, изготовленные из оксида марганца, никеля или кобальта
в виде остеклованной бусинки диаметром 0,4 мм. Температур-
ный коэффициент сопротивления термисторов примерно в 10 раз
выше температурного коэффициента сопротивления платино-
вых или вольфрамовых нитей накала. При комнатной темпера-
туре термисторы значительно чувствительнее проволочных со-
противлений но при повышении температуры их чувствитель-
ность заметно понижается (при нагревании на 30°C — в 2 раза),
поэтому при температуре выше 100 °C рекомендуется использо-
вать детекторы по теплопроводности с металлическими нитями
накала.
Для получения дифференциального сигнала через одну ка-
меру детектора (измерительную) проходит газ, выходящий из
хроматографической колонки, через другую (сравнительную) —
чистый газ-носитель. Нагретые чувствительные элементы в
сравнительной и измерительной камерах обдуваются потоком
газа-носителя, и их сопротивление приобретает определенное
значение. При прохождении через детектор бинарной смеси,
состоящей из газа-носителя и определяемого компонента с от-
личающейся от чистого газа-носителя теплопроводностью, в из-
мерительной ячейке нарушается теплообмен. При изменении
условий теплообмена изменяется температура чувствительного
элемента и, как следствие, его сопротивление. Различие сопро-
тивлений чувствительных элементов является функцией мгно-
венной концентрации компонента в газовом потоке и измеря-
ется с помощью моста Уитстона (рис. 11.25). Когда температура
и, следовательно, сопротивления чувствительных элементов R[
и Дг одинаковы, мост сбалансирован и на регистрирующий
прибор подается нулевой сигнал (рис. 11.25, а). При прохожде-
нии через измерительную ячейку определяемого компонента со-
противление чувствительного элемента /?2 изменяется, а значе-
ние сопротивления R± остается неизменным. Схема моста при
этом выходит из равновесия, и между точками А и В возникает
разность потенциалов, которая преобразуется в сигнал, непре-
рывно регистрируемый автоматическим потенциометром.
Большая часть современных хроматографов комплектуется
четырехплечевым детектором по теплопроводности, который
отличается от описанного выше двухплечевого тем, что в срав-
нительную и измерительную линии введены по два чувстви-
тельных элемента (рис. 11.25, 6). Такое устройство повышает
чувствительность и стабильность показаний детектора. При
выборе рабочих условий анализа для достижения возможно
большей чувствительности необходимо повышать силу тока мо-
72
Рис.П.25. Схема включения двух- (а) и четырехплечевого (6)
катарометров в измерительный мост.
ста, понижать температуру блока детектора и выбирать газ-
носитель с наивысшей теплопроводностью. Увеличение тока
моста существенно повышает чувствительность детектора, так
как при этом возрастают температура чувствительных элемен-
тов и их сопротивление. Предельно допустимым током моста
считается такой, который при выбранных условиях работы де-
тектора (природа газа-носителя, температура блока детектора)
нагревает чувствительные элементы до 600-700°С. Поэтому с
73
повышением молекулярной массы газа-носителя (понижением
теплопроводности) и температуры блока детектора значение
предельно допустимого тока становится меньше.
Кроме описанного традиционного способа питания мостовой
схемы постоянным током иногда используется вариант поддер-
жания на заданном уровне постоянной температуры нитей чув-
ствительных элементов. В этом случае схема питания постро-
ена таким образом, что она обеспечивает сохранение темпера-
туры (сопротивления) нитей во время выхода анализируемого
вещества соответствующим изменением тока или напряжения на
мостовой схеме, что и является сигналом детектора.
Подобные схемы питания эффективно защищают чувстви-
тельные элементы от перегрева и благодаря этому увеличи-
вают срок эксплуатации детектора.
II.1.4.4. Принципы ионизационного детектирования
Ионизационные методы детектирования обеспечивают наи-
большую чувствительность и широко применяются для опреде-
ления малых количеств анализируемых веществ. В основе этих
методов лежит зависимость электрической проводимости иони-
зированной газовой среды от ее состава. Сигналом ионизаци-
онных детекторов является изменение ионного тока*, вызванное
введением в детектор анализируемого вещества.
Ионный ток возникает в детекторе под действием какого-либо
источника ионизации (радиоактивного изотопа, пламени, раз-
ряда, фотоионизации, электронной и ионной эмиссии) и элек-
трического поля (разности потенциалов) между электродами
детектора. В любой момент времени в детекторе достигается
равновесие, характеризующееся тем, что скорость образования
заряженных частиц (ионов, электронов) равна сумме скоростей
рекомбинации и сбора заряженных частиц на электродах детек-
тора. Скорость сбора определяет ток детектора. В ионизаци-
онных детекторах создаются такие условия, при которых либо
плотность (концентрация) заряженных частиц, либо скорость
переноса их в электрическое поле зависит от состава газа.
Зависимость силы тока I в газовой среде от напряжения ме-
жду электродами U, так называемая вольтамперная характе-
ристика, в общем случае состоит из трех различных участков
(рис. 11.26), каждый из которых может использоваться для раз-
личных способов детектирования. На участке I (слабое поле)
*Ионным током условно назван электрический ток, создаваемый ме-
жду электродами детектора всеми заряженными частицами в газе (ио-
нами и электронами).
74
реализуется режим неполного сбора заряженных частиц, и зна-
чительная часть их успевает рекомбинировать. При постоян-
ной скорости образования и рекомбинации заряженных частиц
в детекторе, работающем на этом участке характеристики, и
постоянном напряжении на электродах ток детектора опреде-
ляется скоростью переноса заряженных частиц в направлении
поля. Скорость зарядов в направлении поля характеризуется
так называемой подвижностью, которая численно равна скоро-
сти, приобретаемой зарядом в поле напряженностью 1 В/см.
Подвижность пропорциональна величине заряда и обратно про-
порциональна массе частиц.
Рис. 11.26. Вольтамперная харак-
теристика ионизационных детек-
торов.
I — участок неполного сбора за-
ряженных частиц; II— участок насы-
щения; III — участок вторичной ио-
низации.
Если введение анализируемого вещества вызывает увеличе-
ние рекомбинаций или существенное уменьшение подвижности,
ток детектора падает, и это уменьшение тока регистрируется
на хроматограмме как пик данного вещества. На этом прин-
ципе основана работа электронозахватного детектора.
Ионизация газа-носителя в этом детекторе приводит к образо-
ванию положительных ионов и электронов малой энергии (ме-
дленных электронов). Почти весь ток, возникающий в детек-
торе, переносится электронами, так как их подвижность бла-
годаря малой массе примерно на 3 порядка выше подвижности
ионов.
При введении в детектор молекул анализируемых веществ,
обладающих большим сродством к электрону (веществ, содер-
жащих атомы галогенов, азота, кислорода и др.), медленные
электроны захватываются ими с образованием соответствую-
щих отрицательных ионов. При этом подвижность захваченных
электронов резко падает и вместе с тем уменьшается плотность
заряженных частиц за счет рекомбинации ионов, которая проте-
кает значительно быстрее, чем электрон-ионные рекомбинации.
Эти эффекты вызывают уменьшение тока детектора, пропорцио-
нальное (в линейной области) количеству анализируемого ком-
понента (рис. II.27, а).
75
Рис. П.27. Зависимость сигнала ионизационных детекторов на
первом (а), втором (6) и третьем (в) участках вольтамперной
характеристики от концентрации вещества.
В правой части показан характер изменения сигнала детектора (пик)
при изменении концентрации анализируемого вещества в газе-носителе
от Со до Смаке при фиксированном напряжении питания детектора.
Подобный процесс может быть реализован только при срав-
нительно малой напряженности поля детектора, когда энергия
электронов невелика и электроны доступны для захвата, а зна-
76
чительная часть ионов успевает рекомбинировать, не достиг-
нув электродов (режим неполного сбора зарядов). Началь-
ный (фоновый) ток детектора должен быть относительно вы-
соким, чтобы его уменьшение (сигнал детектора) было доста-
точно большим и находилось в диапазоне токов, доступных для
измерения электрометром. Поэтому наличие примесей в газе-
носителе, обладающих сродством к электрону и снижающих
фоновый ток детектора, нежелательно. Зависимость сигнала
от напряжения диктует необходимость стабильного электри-
ческого питания детекторов, работающих на участке I вольт-
амперной характеристики.
За участком неполного сбора следует участок насыщения
II, для которого характерны отсутствие рекомбинаций и пол-
ный сбор всех образовавшихся зарядов. В этом случае ион-
ный ток определяется только скоростью образования зарядов.
Сигналом детекторов, работающих на участке II, является уве-
личение тока, вызванное значительным возрастанием скорости
образования заряженных частиц вследствие ионизации анали-
зируемых компонентов, поступающих в детектор. При этом
ионизация чистого газа-носителя, как правило, должна отсут-
ствовать и уровень фонового тока может быть весьма малым.
Типичным примером такого детектора является пламенно-
ионизационный детектор (а также его вариант — термо-
ионный детектор), в котором водородное пламя служит ис-
точником ионизации органических соединений; ток насыщения
возрастает пропорционально количеству вещества, поступаю-
щему в детектор (см. рис.II.27, 6).
Детекторы, работающие в области насыщения, наиболее
удобны, поскольку их сигнал не зависит от напряжения в ши-
роком диапазоне второго участка характеристики, а также не
зависит от подвижности и рекомбинации заряженных частиц.
Кроме того, при малом уровне фонового тока легче добиться
его достаточной стабильности, что позволяет измерять весьма
малые сигналы, т. е. регистрировать очень малые количества
веществ.
На участке III вольтамперной характеристики при высокой
напряженности поля насыщение возрастает за счет размноже-
ния зарядов (вторичной ионизации) при введении в детектор
анализируемых веществ. В этой области работают аргоновый
и гелиевый ионизационные детекторы. Здесь так же, как и на
первом участке вольтамперной характеристики, при ионизации
газа-носителя обеспечивается постоянная скорость образова-
ния зарядов: А —> А+ + е. Освободившиеся электроны малых
энергий разгоняются сильным полем и при соударениях с ато-
77
мами газа-носителя сообщают им энергию, переводящую их в
возбужденное (метастабильное) состояние: А + е —> А*. Полный
сбор электронов и ионов, возникающих в результате первичной
ионизации газа-носителя, создает фоновый ток детектора.
Вероятность перехода возбужденных атомов аргона или ге-
лия в первоначальное энергетическое состояние значительно
увеличивается при введении в детектор веществ, имеющих близ-
кие или меньшие потенциалы ионизации (энергию отрыва элек-
трона), чем энергия возбужденного состояния: А* + М —> А +
+М+ + е. Образовавшиеся в результате ионизации заряды со-
здают дополнительный ток, являющийся сигналом детектора на
введенное количество вещества (рис. 11.27, в).
Чтобы детектор, работающий по этому принципу, был уни-
версален, необходимо применение газов-носителей с высокими
значениями энергии метастабильного состояния. Такому усло-
вию отвечают, в частности, гелий и аргон, энергии метаста-
бильных состояний которых довольно высоки (19,6 и 11,6 эВ) и
превышают потенциалы ионизации большинства веществ. Од-
нако для поддержания достаточной концентрации метастабиль-
ных атомов газы-носители должны иметь высокую чистоту. По
этой причине, а также из-за сравнительно малого диапазона ли-
нейности, неустойчивости работы, необходимости стабильного
высоковольтного питания эти детекторы (особенно гелиевый) не
получили широкого применения.
Наиболее часто в практике газовой хроматографии использу-
ется пламенно-ионизационный детектор; в последнее время по-
лучили распространение детекторы электронозахватный, пла-
менно-фотометрический, фотоионизационный и термоионный.
II.1.4.5. Пламенно-ионизационный детектор
К преимуществам пламенно-ионизационного детектора
(ПИД)*, по сравнению с другими ионизационными детекто-
рами, относятся высокая чувствительность к органическим со-
единениям, широкий линейный диапазон, сравнительно малая
зависимость рабочих параметров от конструкции и внешних
условий, безынерционность и отсутствие жестких требований
к стабильности электрического питания.
*В настоящее время нормативными документами узаконено именно
такое название ©того детектора (в англоязычной литературе его повсе-
местно называют flame ionisation detector (FID), тогда как в литературе на
русском языке довольно часто используется название “детектор иониза-
ционно-пламенный” (ЛИП)).
78
Детектор переставляет собой камеру (рис. 11.28), в которой
поддерживается водородное пламя, являющееся источником ио-
низации. В камеру вводятся необходимые для поддержания
пламени водород и воздух: водород подается в детектор в смеси
с газом-носителем через канал горелки, а воздух — через дру-
гой канал, и вся смесь равномерно распределяется диффузором.
Горелка является одним из электродов, она изолирована от кор-
пуса детектора и соединена с источником стабилизированного
напряжения*. Второй электрод, называемый часто коллекто-
ром, расположен над горелкой. Во внешнюю цепь электрода
детектора включен электрометр, измеряющий ток между элек-
тродами детектора.
Рис. П.28. Схема пламенно-ионизационного детектора.
1 — электрод-кол лектор; 2 — горелка; 3 — изолятор электрода-
коллектора; .4 — изолятор горелки; 5 — диффузор; 6 — изолятор пи-
тания; 7—электрометр.
Поскольку в пламени чистого водорода число ионов мало, со-
противление межэлектродного газового пространства очень ве-
лико (1014-1013 Ом) и ток детектора весьма мал (10-12-10-11 А).
Этот ток, возникающий за счет ионизации примесей, содержа-
щихся в газе-носителе, водороде и воздухе, является постоян-
ным фоновым током детектора. При внесении с газом-носителем
из колонки анализируемых органических веществ число ионов
*Иногда применяется система двух параллельных электродов, в этом
случае горелка не является электродом и не соединена с источником пи-
тания. Поскольку этот вариант используется довольно редко, далее он
не рассматривается.
79
в пламени резко увеличивается, сопротивление пламени падает
и во внешней цепи детектора регистрируется соответствующее
возрастание ионного тока.
Механизм ионообразования при введении органических ве-
ществ в пламя водорода достаточно хорошо изучен. В ниж-
ней части пламени (у среза горелки) происходит термическая
деструкция органических молекул. Окисление продуктов де-
струкции сопровождается хемиионизацией, при которой вся
энергия химической реакции окисления не распределяется в
окружающей среде, нагревая ее, а направлена только на иони-
зацию:
СН + О СНО+ + е.
Масс-спектрометрическими измерениями доказано, что ос-
новными носителями положительных зарядов в пламени явля-
ются ионы гидроксония, которые образуются при взаимодей-
ствии ионов СНО+ с водой:
СНО+ + Н2О Н3О+ + СО.
Именно ионы гидроксония обусловливают электрическую про-
водимость пламени.
Приведенный механизм ионообразования (термодеструкция
с последующей хемиионизацией) хорошо согласуется с фактом
пропорциональности молярной чувствительности ПИД числу
атомов углерода в углеводородах. Особенно точно эта про-
порциональность выполняется в пределах гомологических ря-
дов углеводородов. Сигнал детектора на единицу массы при-
мерно одинаков для различных членов ряда. Механизм ио-
нообразования объясняет пониженную чувствительность к ор-
ганическим соединениям, содержащим уже окисленные атомы
углерода. Чувствительность же к соединениям, имеющим толь-
ко окисленные атомы углерода, мала или вообще отсутствует
(например, ПИД весьма слабо реагирует на муравьиную ки-
слоту).
Относительную молярную чувствительность ко многим орга-
ническим веществам можно характеризовать с помощью так на-
зываемого эффективного углеродного числа (ЭУЧ), которое по-
казывает, сколько атомов углерода содержит линейный алкан,
соответствующий по чувствительности анализируемому веще-
ству. Вклад каждого углеродного атома в ЭУЧ зависит от его
связи с атомами углерода, водорода, кислорода, хлора и т.д-
Вклад любого атома углерода в молекуле алкана принят рав-
ным единице. Ниже приведены вклады в ЭУЧ атомов углерода,
80
кислорода и хлора в зависимости от характера их связи с дру-
гими атомами в молекуле:
Атом Тип соединения Вклад в ЭУЧ
С Алифатические 1.0
С Ароматические 1.0
С Олефиновые 0,95
С Ацетиленовые 1,30
С Карбонильные 0,0
С Нитрил ьный 0,3
О Эфирный -1,0
О Первичный спирт -0,6
О Вторичный спирт -0,75
О Третичный спирт -0,25
С1 Два или более на один -0,12 (на
алифатический С каждый)
С1 На один олефиновый С +0,05
Поскольку процесс ионизации каждого углеродного атома
мало зависит от ионизации соседних, ЭУЧ вещества аддитивно
составляется из вкладов каждого углеродного атома в моле-
куле с учетом характера его связи с другими атомами. Так,
emop-бутиловый спирт, содержащий 4 атома углерода, один
из которых связан с ОН-группой, должен иметь ЭУЧ, равное
3,25 (4 + (—0,75)). Экспериментально измеренное значение со-
ставляет 3,22.
Эффективное углеродное число позволяет рассчитать чув-
ствительность к веществу, если известна его чувствительность
к какому-либо нормальному алкану. Так, если молярная чув-
ствительность к гептану равна /?гепт, то чувствительность к вто-
ричному бутиловому спирту должна быть Rcn = Дгепт • 3,25/7.
Следует, однако, иметь в виду, что этот способ может быть
применим лишь для ориентировочных оценок, так как точные
значения ЭУЧ зависят от конструкции детектора и режима его
работы. Точность метода заметно снижается по отношению к
сложным органическим соединениям, содержащим различные
гетероатомы (азот, серу, металлы и т.п.). Как правило, чув-
ствительность к разветвленным углеводородам несколько выше,
чем к их линейным изомерам.
Чувствительность ПИД обычно характеризуют его иониза-
ционной эффективностью, которая является абсолютной харак-
теристикой, так как не зависит от качества измерительной схе-
мы. Ионизационная эффективность Ид определяется током I,
возникающим в детекторе при прохождении через него единицы
потока j вещества: Яд = //j(А • с/мг). Она имеет смысл количе-
ства электричества, образующегося в детекторе при внесении
единицы массы вещества, и может быть выражена в кулонах
81
на миллиграмм (массовая чувствительность) или в кулонах на
моль (молярная чувствительность).
Ионизационная эффективность находится в пределах от 10-6
в худших конструкциях до 2 • 10-5 А с/мг для самых опти-
мальных конструкций ячейки ПИД. Столь существенная раз-
ница объясняется особенностями работы системы сбора ионов.
Часто ионизационную эффективность определяют как отноше-
ние количества электричества, получаемого при детектирова-
нии 1 моля вещества, к количеству электричества, соответству-
ющему его полной ионизации (9,65 • 104 Кл). Среднее значе-
ние выраженной таким образом ионизационной эффективности
ПИД равно 10~5.
Предел детектирования ПИД при малых уровнях флуктуа-
ций фонового тока и при использовании современных электро-
метров, способных измерять токи до 10~12 А на полную шкалу,
может достигать 10-9 мг/с. Это соответствует минимальным
содержаниям вещества в детекторе до 5 • 10-8% (по объему) при
скорости газа-носителя около 30 мл/мин и молекулярной массе
вещества около 100.
Ионизационная эффективность большинства детекторов,
устанавливаемых на серийных хроматографах, составляет око-
ло 5 • 10-6 А с/мг. При использовании шкалы электрометра
10-11 А и уровне флуктуаций не более 0,5% шкалы минималь-
ный измеряемый сигнал 1М1т составляет 10~13 А (1% шкалы);
этому соответствует предел обнаружения (мг/с)
Jmhh = I^/Rr = Ю-13/(5 • 10-6) = 2 • 10-8.
Таким образом, содержание вещества в детекторе на уровне
10“6% (по объему) дает еще отличимый от шума сигнал. Если
принять, что размывание пробы при дозировании и разделении
в колонке уменьшает ее первоначальную концентрацию в 5 раз,
то минимальное содержание вещества, поддающееся измерению
хроматографом с этим детектором, ориентировочно составляет
5 10-6% (по объему). Следует иметь в виду, что при измере-
нии столь малых сигналов ошибка может достигать 100% из-
за трудностей выделения сигнала на уровне сравнимых с ним
флуктуаций нулевой линии. В обычной практике можно уве-
ренно измерять сигнал, начиная с нескольких процентов шкалы
регистраторов.
Основным фактором, резко снижающим реальную чувстви-
тельность хроматографа, являются флуктуации фонового тока
детектора, как правило, возрастающие с увеличением фонового
тока. Электрометр позволяет полностью скомпенсировать до-
вольно существенные значения фоновых токов (до 10-9 А), по-
82
этому большой фоновый ток сам по себе не создает помех в ра-
боте, однако случайные короткопериодные изменения фонового
тока в любом случае проявляются как флуктуации нулевой ли-
нии, а медленное одностороннее его изменение вызывает соот-
ветствующий дрейф нулевой линии. Увеличение фона и рост
флуктуаций фонового сигнала заставляют переходить на менее
чувствительные шкалы электрометра, поэтому практически в
большинстве случаев не удается реализовать предельную чув-
ствительность ПИД.
Основным источником фона является присутствие паров ор-
ганических веществ в потоках водорода и газа-носителя, вы-
званное загрязнением соответствующих газовых линий и лету-
честью используемой жидкой фазы при высокой температуре
колонки.
Быстрое удаление загрязнений газовой линии обычно осуще-
ствляется прокаливанием участков линии во время продувки.
Однако если от загрязнений газовых линий избавиться сравни-
тельно легко, то исключить или существенно снизить загрязне-
ние газа-носителя парами жидкой стационарной фазы удается
далеко не во всех случаях. Даже при использовании относи-
тельно малолетучих органических жидких фаз, имеющих, на-
пример, давление паров при максимальной рабочей темпера-
туре не более 1,33 Па, фоновый ток при скорости газа-носителя
около 66 мл/мин составляет примерно 3 • 1О-10 А. При этом не-
стабильность расхода газа-носителя вызывает пропорциональ-
ные флуктуации нулевой линии. Так как давление паров органи-
ческих жидкостей изменяется на несколько процентов при изме-
нении температуры на 1 °C, другим серьезным источником изме-
нения фона и появления соответствующих флуктуаций является
колебание температуры колонки.
Обычно преобладающая часть загрязнений вносится в детек-
тор с потоком газа-носителя. Чувствительность к органическим
примесям в воздухе, подаваемом в детектор для поддержания
горения водорода, приблизительно в 100-1000 раз ниже, чем к
тем же примесям, поступающим в детектор с газом-носителем
или водородом. Причина столь низкой чувствительности со-
стоит в том, что примеси в воздухе, как правило, не достигают
зоны ионизации.
Для снижения уровня шумов (с целью работы на более чув-
ствительных шкалах) необходимо принимать меры по стаби-
лизации рабочих параметров, влияющих на уровень фонового
тока, и прежде всего температуры колонки и расходов газов.
Другими путями снижения предела обнаружения могут быть
Уменьшение температуры колонки, использование стационар-
83
ных фаз с малым давлением пара при рабочей температуре или
переход на газо-адсорбционные хроматографические колонки
(последний вариант далеко не всегда возможен, особенно при
анализе полярных веществ из-за резко нелинейного характера
изотермы адсорбции).
ПИЛ обладает наибольшим по сравнению с другими детек-
торами линейным диапазоном (106-107).
Высокое быстродействие ПИД объясняется весьма малым
объемом зоны ионизации детектора и большой скоростью сбора
ионов в режиме насыщения. Постоянная времени собственно
детектора составляет около 10-3 с, что значительно меньше по-
стоянной времени системы усиления сигнала (0,01-0,5 с) и его
регистрации (0,5-0,2 с).
Весьма низкая инерционность ПИД делает его вполне при-
годным для проведения экспресс-анализов и при работе с ка-
пиллярными колонками.
Устойчивость режима работы и максимальная чувствитель-
ность ПИД обеспечиваются правильным выбором расходов во-
дорода, газа-носителя и воздуха. Оптимальные расходы газов и
их соотношения несколько зависят от конструкции детекторов,
однако для большинства конструкций наибольшая чувствитель-
ность и стабильность работы достигаются при соотношении
расходов газа-носителя, водорода и воздуха, близком к 1 : 1 : 10,
при этом расход водорода и газа-носителя должен быть в пре-
делах 2-3 л/ч. Значительное увеличение расхода газа-носителя
по сравнению с рекомендуемым интервалом обычно приводит
к снижению чувствительности и утрате стабильности горения
(колебания и даже срыв пламени). На рис. II.29 показана типич-
ная зависимость чувствительности ПИД от изменения расходов
водорода, газа-носителя и воздуха. Как видно из рисунка, су-
ществует довольно широкая область расходов, в которой чув-
ствительность детектора практически постоянна. Тем не менее
для точных количественных измерений в общем случае следует
строго воспроизводить значения расходов.
Приведенные здесь рекомендации по режиму газового пита-
ния детектора относятся к диффузионному пламени, т. е. к тому
случаю, когда воздух подается снаружи пламени и кислород
проникает в активную зону за счет диффузии. Если подавать
воздух внутрь пламени вместе с водородом (гомогенное пламя),
то при некотором оптимальном соотношении расходов водорода
и воздуха можно достичь несколько большей чувствительности.
Однако резкая зависимость сигнала от изменения соотношения
водорода и воздуха, снижение линейности и высокая чувстви-
тельность к примесям, содержащимся в воздухе, вынуждают от-
84
* £
со о
t
«J S 90
ф x
4 g 80
a “ ____i__i----1__i---1--1
s о 40 60 80
° Расход водорода, мл/мин
Рис. 11.29. Зависимость чувствительности пламенно-иониза-
ционного детектора от скорости водорода и воздуха.
Скорость газа-носителя (азота), мл/мин: 1 — 5; 2 — 14; 3 — 40;
4 — 75.
казаться от гомогенного пламени. Существующие конструкции
ПИД рассчитаны на работу только с диффузионным пламенем,
и даже в этом случае следует избегать использования воздуха
в качестве газа-носителя.
Конструкция детектора должна обеспечивать устойчивость
пламени водорода при изменении расходов газа-носителя в ши-
роких пределах и возможно более полный сбор зарядов.
Часть электрических зарядов не участвует в образовании
сигнала (ионного тока) из-за утечки зарядов на корпус детек-
тора и зажигающий элемент. Наиболее полный сбор зарядов
достигается при наибольшей напряженности поля у среза го-
релки в зоне ионизации. Этому условию отвечает применение
электрода-коллектора в форме цилиндра, когда плоскость его
нижнего среза на 1-2 мм выше горелки, расположенной по оси
цилиндра. Такая система электродов обеспечивает не только
высокую чувствительность, но и наиболее широкий линейный
диапазон (увеличение максимальной концентрации). Излиш-
нее приближение коллектора к горелке может вызвать перегрев
электрода и эмиссию положительных ионов с его поверхности.
Для исключения этого на коллектор должен быть подан отрица-
тельный потенциал. С другой стороны, отрицательный потен-
циал на горелке препятствует рекомбинации положительных ио-
нов и обеспечивает их полный сбор При оптимальном выборе
конструкции и положения электродов ток насыщения практиче-
ски одинаков при любой полярности электродов.
Минимальное напряжение на горелке для работы в области
насыщения должно быть тем больше, чем больше расстояние
между электродами и измеряемая концентрация. Рабочее на-
пряжение обычно устанавливается в пределах 150-300 В, и, так
85
как на широком участке насыщения вольтамперной характери-
стики изменение напряжения мало сказывается на чувствитель-
ности детектора, нет необходимости в жесткой стабилизации
напряжения горелки.
Ответственным элементом конструкции ионизационного де-
тектора является изолятор электрода, соединенного с электро-
метром. Для исключения утечки тока изолятор должен иметь
сопротивление на 2-3 порядка выше, чем сопротивление вход-
ной измерительной цепи электрометра. Изолятор обычно изго-
тавливают из фторопласта или специальной керамики, сохра-
няющих высокое сопротивление при рабочей температуре де-
тектора.
Влияние внешней температуры на работу ПИД незначитель-
но, необходимо лишь прогревать штуцер детектора, к которому
присоединяется колонка, для исключения конденсации анализи-
руемых веществ. Конденсация паров воды, образующейся при
горении водорода, весьма нежелательна, особенно на изоляторе
электрода-коллектора, так как это приводит к нарушению изо-
ляции высокоомной входной цепи электрометра и неустойчиво-
сти нулевой линии хроматографа. Как правило, прогрева де-
тектора от пламени водорода достаточно для исключения кон-
денсации воды внутри ячейки.
Особенности эксплуатации пламенно-ионизационного детек-
тора связаны с необходимостью использования водорода для
поддержания пламени. Надо очень тщательно следить за герме-
тичностью водородной линии не только для того, чтобы обеспе-
чить стабильную работу детектора, но и чтобы предотвратить
возможный взрыв воздушно-водородной смеси при значи-
тельной утечке водорода. После работы с прибором нужно
обязательно закрыть вентиль (или запорный кран) на линии во-
дорода на входе в прибор.
Пламя чистого водорода практически бесцветно. Если пламя
водорода имеет желтоватый цвет, это является свидетельством
загрязнения газовых потоков, подаваемых в пламя. Контроль
за чистотой газов следует осуществлять по уровню фонового
тока. Измерение фонового тока выполняется по изменению
сигнала электрометра при прекращении подачи водорода (ту-
шении пламени). Например: включена шкала электрометра
1О-10 А, при этом сигнал самопишущего потенциометра соста-
вляет 60% шкалы; после прекращения подачи водорода сигнал
уменьшается до 20%. Это означает, что фоновый ток составляет
40% от 1О~10 А, т. е. 4 • 10-11 А.
Значение фонового тока ПИД, вызванного летучестью непо-
движной фазы (испарением и выделением летучих продуктов
86
термодеструкции), может быть использовано для выбора опти-
мального режима работы колонки. В частности, легко оценить
скорость уноса (интенсивность убыли) жидкой фазы (мг/с)
3 — /фон/ 7/ц
(/фон — фоновый сигнал ПИД, А; Дд — чувствительность ПИД,
А - с/мг) и эффективную концентрацию (суммарное содержание
летучих веществ) в потоке газа-носителя (мг/мл)
С = j/F,
где F — скорость газа-носителя, мл/с.
Поддается прогнозированию срок эксплуатации колонки
(время жизни) до достижения максимально допустимой убыли
первоначального количества жидкой фазы (НЖФ) в колонке:
г = Am/j,
Am — абсолютная допустимая убыль, мг. Поскольку фоновый
сигнал по мере эксплуатации колонки постепенно уменьшается,
для более точного расчета необходимо учитывать динамику его
изменения, что осуществляется его периодическим измерением.
Полагая для малых допустимых изменений количества НЖФ
скорость уноса постоянной, можно по первым же измерениям
заранее ориентировочно оценить срок жизни колонки.
Если удерживание определяется только растворением анали-
зируемого вещества в НЖФ, относительная скорость уменьше-
ния количества НЖФ в колонке Kt = j/rn (с-1) позволяет оце-
нить значение приведенного объема удерживания Vt (мл) через
общее время t (с) от начала эксплуатации колонки в заданном
режиме:
V = Цо(1 - Ktt),
где Vt0 — начальное значение объема удерживания, мл.
Кроме того, значение фонового тока может быть успешно ис-
пользовано в качестве критерия при определении максимальной
рабочей температуры неподвижной фазы (см. рис. II.39).
В качестве примера приведем оценку летучести и срока экс-
плуатации колонки, содержащей около 320 мг апиезона М (ко-
лонка 100 х 0,3 см, 15% апиезона на хроматоне, скорость газа-
носителя 2 л/ч, чувствительность ПИД 5 10-6 А с/мг). При
температуре колонки около 200°C развивается фоновый сигнал
10~9 А, что соответствует концентрации летучих веществ в газе-
носителе 3,6 • 10-4 мг/мл и скорости уноса апиезона из колонки
0,72 мг/ч. Если принять уровень допустимой убыли жидкой
фазы в колонке 10% от начального количества, предельный срок
87
ее эксплуатации в этих условиях не превышает 45 ч. Как пока-
зывают сопоставления с результатами прямых хроматографи-
ческих измерений, погрешность подобных оценок не превышает
10-15%.
Очень слабая реакция ПИД на воду и отсутствие чувстви-
тельности к неорганическим соединениям, инертным газам и
водороду делают его незаменимым при анализах примесей ор-
ганических веществ в воздухе промышленных предприятий и
атмосфере, сточных и природных водах, а также в биологи-
ческих водных системах. Однако примесь паров воды в га-
зах, питающих детектор, снижает чувствительность ПИД к ор-
ганическим веществам. Изменение содержания воды в преде-
лах (1,6 ± 0,6) 10-3% вызывает изменение чувствительности
ПИД в пределах 1%. Считается, что такой эффект связан с
уменьшением температуры пламени вследствие увеличения те-
плоемкости газа. Кроме того, в присутствии паров воды в пла-
мени образуются малоподвижные гидратированные ионы НзО+
(НзО+ Н2О; НзО+ 2Н2О), из которых не все достигают коллек-
торного электрода детектора.
Сравнительно слабая зависимость чувствительности детек-
тора от изменения расходов газов и температуры, с одной сто-
роны, и строгая пропорциональность сигнала детектора ко-
личеству вещества в широких пределах — с другой, создали
пламенно-ионизационному детектору репутацию лучшего коли-
чественного детектора.
II.1.4.6. Электронозахватный детектор
Электронозахватный детектор (ЭЗД) по частоте использо-
вания занимает одно из ведущих мест. Универсальные га-
зовые хроматографы, как правило, комплектуются этим де-
тектором наравне со стандартными детекторами — пламенно-
ионизационным и по теплопроводности. Столь быстрое и широ-
кое распространение ЭЗД получил в связи с необходимостью
измерения весьма малых количеств хлорсодержащих пестици-
дов в продуктах растительного происхождения. Он успешно
применяется для определения малых концентраций галоген-,
кислород- и азотсодержащих веществ, некоторых металлоорга-
нических соединений и других веществ, содержащих атомы с
явно выраженным сродством к электрону,
Система детектирования по захвату электронов включает ио-
низационную камеру (ячейку детектора) и источник поляризу-
ющего напряжения (блок питания).
88
По характеру влияния на сигнал расхода газа-носителя де-
тектор в зависимости от конструкции и условий газового пи-
тания может быть отнесен либо к концентрационному, либо к
потоковому.
Для устойчивой работы детектора необходимо прежде всего
обеспечить постоянную скорость образования свободных элек-
тронов в ионизационной камере, что достигается помещением
в нее радиоактивного источника*. В качестве газа-носителя
используются азот, аргон, гелий и другие электронодонорные
газы, способные ионизироваться под воздействием радиации с
освобождением электрона:
N2 -> N+ + е.
Образование электронов происходит в электрическом поле
между электродами детектора. Напряженность поля недоста-
точна для сбора всех зарядов, и начальный (фоновый) ток де-
тектора формируется в основном электронами, подвижность ко-
торых на три порядка выше, чем подвижность ионов. Вклад ио-
нов в ток детектора невелик, так как значительно большая часть
их успевает рекомбинировать, не доходя до соответствующего
электрода. При появлении в детекторе молекул анализируе-
мых веществ, обладающих сродством к электрону, происходит
захват ими свободных электронов:
М + е -> М~
Хотя в результате этого процесса общее число заряженных
частиц в ионизационной камере не меняется, эффективная по-
движность связанных электронов резко падает и они не уча-
ствуют в процессе переноса тока между электродами. Это
приводит к соответствующему снижению фонового тока детек-
тора. Таким образом, полезным сигналом детектора является
уменьшение начального тока, однозначно связанное (в ра-
бочем диапазоне концентраций) с количеством анализируемого
компонента.
Образовавшиеся отрицательные ионы регистрируемых моле-
кул (или их частей, если захват сопровождается разрушением)
легко рекомбинируют с ионами азота
М“ +NJ -> M + N2,
что вносит дополнительный вклад в уменьшение тока детектора.
*Иногда в качестве источника электронов используется газовый раз-
ряд (так называемые нерадиоактивные ЭЗД).
89
Электронозахватный детектор обладает высокой ионизаци-
онной эффективностью*, доходящей в лучших конструкциях при
оптимальных условиях работы до 0,05 Кл/мг; обычно значение
ионизационной эффективности на один-два порядка ниже.
Основной трудностью в достижении малого предела детекти-
рования при работе с ЭЗД является большой уровень флукту-
аций, связанный со значительным током детектора. Этот фоно-
вый ток неизбежен, так как для получения высокой чувствитель-
ности и пропорциональных сигналов от сравнительно больших
количеств пробы необходима высокая концентрация свободных
электронов. По той же причине недопустимо присутствие в
газе-носителе примесей (например, кислорода), снижающих ко-
личество электронов или их подвижность. Обычно уровень фо-
нового тока составляет (1 — 5) • 10~9 А, при этом уровень шума
трудно уменьшить ниже 10“13 А. Значение предела детекти-
рования ЭЗД находится в интервале 5 • 10-1° — 10-11 мг/мл,
что в среднем на два порядка ниже предела детектирования
пламенно-ионизационного детектора и позволяет фиксировать
нано- и даже пикограммовые количества веществ, обладающих
большим сродством к электрону (например, CCI4, CeHeCle).
Существенным недостатком ЭЗД является малый диапазон
зависимости сигнала от концентрации вообще и очень узкий ли-
нейный диапазон, в частности. Ограничение диапазона сверху
(по максимальной концентрации) связано, прежде всего, с са-
мим принципом механизма детектирования. Очевидно, полез-
ный сигнал детектора вообще перестает изменяться, начиная
с того момента, когда в детектор вводится столько вещества,
что оно способно связать больше электронов, чем образуется в
ионизационной камере под воздействием радиоактивного источ-
ника ограниченной мощности. Принимая во внимание лишь “чи-
стый” механизм захвата электронов и возможности измеритель-
ной схемы, можно рассчитать, что диапазон концентраций, для
которых имеется однозначная зависимость между сигналом и
количеством вещества, не должен существенно превышать трех
порядков, начиная с концентрации, отвечающей пределу детек-
тирования. Линейный динамический диапазон обычно соста-
вляет не более 103, т. е. всего лишь несколько рабочих шкал
прибора**.
*Понятие ионизационной эффективности по отношению к ЭЗД приме-
няется условно, так как введение анализируемых веществ не вызывает
образования дополнительных заряженных частиц.
**Линейный динамический диапазон может быть несколько расширен
использованием оригинального способа включения детектора в измери-
тельную схему по напряжению при заданном постоянном токе детектора.
90
Чувствительность детектора сложным образом зависит от
природы анализируемых веществ, вида и числа атомов, обла-
дающих сродством к электрону, структуры веществ. Относи-
тельная чувствительность ЭЗЛ к ряду соединений приведена
ниже:
Хлорметан 1 Фторбензол 0,3
Дихлорметан 11 Хлорбензол 1
Хлороформ 4 - 105 Вромбензол 10
Четыреххлористый 5 106 Иодбензол 3- ю4
углерод
Из приведенных данных видно, что чувствительность ЭЗЛ
резко и неравномерно возрастает с увеличением числа атомов
галогенов в молекуле, а также в ряду фтор-, хлор-, бром- и иод-
содержащих аналогичных соединений. Как правило, не удается
найти какой-либо регулярный характер этой зависимости даже
для близких по составу соединений. Например, в то время как
по отношению к CCI4 достигается рекордная чувствительность,
чувствительность к аналогичному соединению, в котором два
атома хлора заменены на атомы фтора (CCI2F2), в 106 раз ниже
и находится на уровне чувствительности детектора по тепло-
проводности.
Любопытно сравнить пределы детектирования (мг/мл) ЭЗЛ
и ПИЛ для хлорбензолов с различным числом атомов хлора:
ЭЗД ПИД
Хлорбензол 110~4 3 • ю-8
Дихлорбензол 210~8 3,6 10-8
Трихлорбензол 510“9 7,7 10“8
Тетрахлорбензол 1 • 10“9 1 • 10-7
Гексахлорбензол 1,5 • 1О“10 3 • 10~7
Бензол (для Практически не 3 10”8
сравнения) регистрируется
В то время как чувствительность ЭЗЛ быстро нарастает с уве-
личением числа атомов хлора, чувствительность ПИЛ лишь ме-
дленно снижается, оставаясь на довольно высоком уровне. По-
этому во многих случаях, когда для детектирования подобных
смесей не нужна высокая селективность электронозахватного
Детектора, удобнее применять пламенно-ионизационный детек-
тор.
Линейный динамический диапазон ЭЗЛ также зависит от
природы вещества. Чем меньше чувствительность, тем шире
линейная область концентраций. Это объясняется относитель-
но меньшим числом свободных электронов, связываемых еди-
ницей массы вещества с малым сродством к электрону. Так,
линейный динамический диапазон для CCI2F2 составляет 104,
91
тогда как для CCl.-i в тех же условиях он не превышает несколь-
ких десятков. Учитывая очень узкий рабочий диапазон кон-
центраций и не поддающуюся априорному определению зави-
симость чувствительности от природы анализируемых веществ,
ЭЗЛ следует очень осторожно применять для количественных
работ, проводя в каждом конкретном случае детальную гра-
дуировку по каждому компоненту. Часто для выполнения та-
ких аналитических работ электронозахватный детектор исполь-
зуют одновременно с количественным пламенно-ионизационным
детектором.
Рабочие характеристики ЭЗЛ во многом зависят от кон-
струкции камеры, режимов газового и электрического питания,
природы и интенсивности используемого радиоактивного ис-
точника, способа включения детектора в измерительную схему,
поэтому приводимые здесь характеристики следует рассматри-
вать как ориентировочные.
Лля того чтобы обеспечить полное участие всех частиц, ис-
пускаемых радиоактивным источником, в процессе образования
свободных электронов, расстояние между электродами выби-
рают довольно большим (для использования полного пробега
частицы). Это вызывает увеличение объема детектора и допол-
нительное размывание пробы в нем. Лля уменьшения инерцион-
ности детектора иногда используют дополнительный поток газа
через детектор (продувку). Продувка не только сохраняет эф-
фективный объем детектора, увеличивая скорость прохождения
через него анализируемых веществ, но и способствует достиже-
нию максимальной чувствительности без увеличения скорости
газа-носителя.
Чувствительность ЭЗЛ в общем случае изменяется с изме-
нением температуры в пределах рабочего диапазона темпера-
тур, поэтому детектор необходимо жестко термостатировать.
Максимальная рабочая температура ЭЗЛ определяется типом
радиоактивного источника. Лля целей детектирования в газо-
вой хроматографии применяются только так называемые закры-
тые источники, в которых исключена утечка радиоактивного ве-
щества. Наиболее широко используются два типа источников:
тритиевый (3Н) и никелевый (63Ni).
Тритиевый источник изготавливается насыщением тритием
слоя титана, нанесенного на подложку из молибдена, вольфрама
и т.п. Тритиевый источник является “чистым” /3-излучателем
со средней энергией частиц 6 кэВ и пробегом частиц в воздухе
2,5 мм. Период полураспада 3Н 12,5 лет, обычно используемые
активности 0,1-0,2 Ки. Максимальная рабочая температура де-
тектора с тритиевым источником по нормам, принятым в Рос-
92
сии, — не более 150°C (за рубежом допускается температура
до 220°C).
Никелевый источник также является /3-излучателем со сред-
ней энергией частиц около 20 кэВ и пробегом частиц в воздухе
8 мм. Период полураспада 63Ni 125 лет, применяемые активно-
сти 0,1-0,2 Ки. Активный слой никеля обычно наносится на мед-
ную подложку. Максимальная рабочая температура до 300°C.
Для выбора оптимального режима работы ЭЗД очень важна
зависимость эффективности захвата электронов (т. е. в конеч-
ном счете — чувствительности) от их энергии, которая опре-
деляется прежде всего напряженностью электрического поля
между электродами. Характер зависимости обусловливается
природой анализируемых веществ. Для многих веществ кри-
вая зависимости чувствительности от напряжения на электро-
дах имеет максимум. Восходящая ветвь кривой связана с увели-
чением сбора заряженных частиц при повышении напряженно-
сти поля, спад чувствительности объясняется превалирующим
влиянием уменьшения эффективности захвата электронов при
чрезмерном увеличении их энергии с повышением напряжения.
Обычно напряжение питания детектора составляет 3-10 В.
В общем случае на сигнал ЭЗД. обусловленный главным
образом процессом захвата электронов, влияют некоторые дру-
гие процессы, приводящие к образованию или рекомбинации
заряженных частиц в ионизационной камере (например, иониза-
ция метастабильными атомами, электронами повышенных энер-
гий).
Процесс формирования сигнала (образование и сбор заря-
женных частиц) искажается появлением объемного заряда в не-
посредственной близости к электродам детектора и контактной
разностью потенциалов. Кроме того, средняя энергия электро-
нов может изменяться под действием самих анализируемых ве-
ществ, Эти побочные явления иногда приводят к искажению
формы пиков, неожиданным смещениям нулевой линии, сниже-
нию воспроизводимости результатов анализа.
Некоторые нежелательные явления могут быть несколько
снижены при переходе от постоянного к импульсному питанию,
состоящему в том, что большее (до 50 В) напряжение подается
на электроды детектора в течение короткого времени (0,5 мкс)
с периодом следования импульсов 100-1000 мкс. Такой способ
поляризации ионизационной камеры препятствует образованию
объемного заряда и уменьшает долю сигнала, не связанную с
процессом захвата электронов, т. е. делает механизм работы
Детектора более чистым. В частности, эффективность захвата
электронов в течение большей части цикла (в промежутке ме-
93
жду импульсами) остается постоянной, так как средняя энергия
свободных электронов не изменяется в отсутствие поля.
Для предотвращения образования метастабильных атомов в
благородных газах при повышенном напряжении в импульсе
обычно используют соответствующие добавки в газ-носитель.
Например, часто применяется в качестве газа-носителя аргон с
5-10% (по объему) метана или 1-5% водорода и диоксида угле-
рода. За рубежом широко используется ЭЗЛ, работающий в
импульсном режиме. Однако, если принять во внимание все пре-
имущества и недостатки обоих вариантов поляризации ЭЗЛ,
разница между ними невелика. Видимо, для одной конструк-
ции камеры больше подходят условия постоянного питания, в
то время как оптимальные характеристики другой достигаются
при работе в импульсном режиме.
Работу ЭЗЛ существенно ухудшает загрязнение поверхно-
сти источника анализируемыми веществами или парами жид-
кой фазы, так как это уменьшает мощность излучения и меняет
фоновый ток детектора.
Существуют разнообразные конструкции ЭЗЛ. Первый де-
тектор, описанный Ловелоком (рис П.30, а), имел вид конденса-
тора с плоскопараллельными электродами, на одном из которых
был размещен источник. Примером другой типичной конструк-
ции является коаксиальный детектор (рис. II.30, б), в котором
один электрод выполнен в виде цилиндра с источником на вну-
тренней поверхности, а другой — в виде стержня, расположен-
ного на оси цилиндра; характеристики обоих типов детекторов
довольно близки. Несколько позднее был предложен (Грегори)
более совершенный вариант конструкции (рис. II.30, в), в кото-
рой зона ионизации продувочного газа конструктивно отделена
от зоны захвата электронов молекулами пробы. Катод имеет
форму цилиндра, на поверхность которого прикреплен радио-
активный источник. Продувочный газ обтекает катод, подвер-
гаясь ионизации в зоне катода. Газ-носитель из колонки посту-
пает через сетку анода, выполненного в виде стержня с осевым
каналом. Эффективная зона захвата расположена в непосред-
ственной близости от анода. Такая схема имеет ряд преиму-
ществ, состоящих в том, что ионизируется только продувочный
газ, а анализируемые вещества не подвергаются действию ра-
диации. Радиоактивный источник всегда находится в потоке
чистого газа, и его загрязнения исключены.
Несмотря на серьезные недостатки и трудности эксплуата-
ции ЭЗЛ, его высокая чувствительность и селективность вызы-
вают постоянный интерес со стороны аналитиков.
94
t
Газ-носитель
Рис. II 30. Схема электро-
нозахватного детектора
Ловелока (с), коаксиаль-
ного (б) и Грегори (в).
1 — электроды; 2—радиоак-
тивный источник.
Применение детектора не ограничивается веществами с резко
выраженными электроноакцепторными свойствами. Метод мо-
жет быть успешно распространен на нейтральные вещества,
если к ним предварительно химическим путем привить функ-
циональную группу с большим сродством к электрону.
II.1.4.7. Термоионный детектор
Детектор ионизации пламени со щелочным металлом, извест-
ный под названием “термоионный”, “натриевый” или “фосфор-
ный”, является модификацией пламенно-ионизационного детек-
тора. Предложен для использования в газовой хроматографии в
1964 г. До настоящего времени это один из наиболее высокочув-
ствительных и селективных детекторов на фосфорорганические
вещества. Кроме того, получают все большее распростране-
ние варианты термоионного детектора, проявляющие высокую
чувствительность и селективность к азот- и галогенсодержащим
веществам.
95
Действие термоионного детектора (ТИД) основано на увели-
чении ионизации солей щелочных металлов в пламени водорода
при попадании в него элементорганических соединений. Од-
нако сходство термоионного и пламенно-ионизационного детек-
торов ограничивается чисто внешними признаками, поскольку
механизм ионизации и процессы сбора ионов в этих детекторах
различны. Процессы ионизации в ТИД сосредоточены в зоне
самого пламени (рис. 11.31), тогда как ионизация в ПИД про-
исходит у среза горелки. В упрощенном виде механизм иони-
зации можно представить следующим образом. При введении
нейтральных молекул соли щелочного металла в пламя водо-
рода происходит их ионизация, в результате чего наблюдается
резкое увеличение фонового тока (на 2-3 порядка больше, чем
у ПИД). Анализируемая молекула в пламени водорода разру-
шается с образованием радикалов с гетероатомами, взаимодей-
ствие которых с заряженными комплексами солей щелочных ме-
таллов приводит к резкому увеличению скорости образования
ионов, что в итоге вызывает дополнительное образование ионов
элементорганическими соединениями. Лимитирующим процес-
сом в таком механизме является скорость введения в водородное
пламя паров соли щелочного металла. Поэтому для получения
устойчивых и воспроизводимых показаний ТИЛ этот процесс
должен быть тщательнейшим образом стабилизирован. Крите-
рием постоянства потока паров соли щелочного металла, вводи-
мого в пламя, является значение фонового тока, которое, по су-
ществу, определяет чувствительность регистрации элементор-
ганических соединений.
Рис. 11.31. Схема термоионного детектора.
96
Конструкции детекторов различаются в основном способом
размещения и нагревания соли щелочного металла, а также гео-
метрией детектора, причем все эти различия оказывают весьма
существенное влияние на его характеристики — стабильность,
чувствительность, селективность. Соль щелочного металла в
виде таблетки или нанесенная на какой-либо удобный для этой
цели держатель, выполненный из пористого металла или кера-
мики в виде спирали, сетки или петли, может нагреваться либо
водородным пламенем, либо электрическим током.
Значение сигнала ТИЛ для данного соединения зависит от
ряда параметров. На чувствительность детектора оказывают
влияние природа соли щелочного металла, расход газов, пита-
ющих детектор, температура ячейки, напряжение и расстояние
между электродами.
В качестве источника ионов щелочного металла пригодны по-
чти все его соли и гидроксид. Сравнение чувствительности
детектора для различных соединений Na, К, Rb, Cs показало
некоторое повышение эффективности с увеличением атомного
номера щелочного металла. Изменение аниона мало влияет на
чувствительность ТИЛ.
Требования к стабильности газового питания детекторов, в
особенности водородом, могут быть снижены почти до уровня
ПИЛ, если использовать ячейку ТИЛ, имеющую независимый
подогрев солевого источника с помощью электронагревателя.
Температура таблетки, а значит, и чувствительность в этом
случае меньше зависят от расходов газов, питающих детектор,
правда, конструкция ячейки ТИЛ с независимым подогревом
солевого источника много сложнее.
Электрическое питание и схема измерения сигнала ТИЛ в
основном совпадают с таковыми для пламенно-ионизационного
детектора. Однако различие состоит в том, что с целью сниже-
ния уровня шумов в ТИЛ применяются противоположные по-
лярности электродов: «+» на горелке и «—» на коллекторном
электроде. Кроме того, важную роль в определении оптималь-
ного режима работы термоионного детектора играет правиль-
ный выбор положения электродов, так как при изменении рас-
стояния между коллекторным электродом и поверхностью соле-
вого источника чувствительность ТИЛ проходит через макси-
мум.
Термоионный детектор проявляет довольно высокую чув-
ствительность и селективность определения соединений фос-
фора, азота, мышьяка, галогенов (кроме фтора), олова и серы.
Наибольшее отношение сигналов ТИЛ к сигналам ПИЛ наблю-
дается для соединений фосфора, достигая 103-104. При этом
97
минимально определяемые содержания этих веществ в иссле-
дуемых объектах находятся на уровне 10-5%, что соизмеримо с
чувствительностью пламенно-ионизационного детектора к угле-
водородам. Такой результат, на первый взгляд, кажется пара-
доксальным, так как ионизационная эффективность фосфорор-
ганических веществ в термоионном детекторе на 2-3 порядка
выше, чем углеводородов в пламенно-ионизационном. Однако
возможности ТИЛ в отношении определения малых концентра-
ций существенно снижаются из-за более высокого уровня шу-
мов, который на 2-3 порядка выше, чем у ПИЛ. Поэтому мини-
мальное поддающееся обнаружению количество веществ у ТИЛ
сопоставимо с аналогичным для пламенно-ионизационного де-
тектора.
Селективность в определении азотсодержащих веществ у тер-
моионного детектора проявляется для всех щелочных металлов,
за исключением натрия. Молярная чувствительность практи-
чески не зависит от структуры соединения и примерно в 50-100
раз выше, чем у ПИД. Чувствительность ТИД к галоген- и се-
росодержащим соединениям в значительной степени зависит от
режима газового питания детектора и природы применяемого
солевого источника.
В табл. II.3 приведены данные по ионизационной эффектив-
ности некоторых фосфор-, азот-, галоген-, серо- и мышьяксо-
держащих соединений, которые характеризуют чувствитель-
ность ТИД к этим соединениям и его возможности в отноше-
нии селективного определения элементорганических веществ на
фоне других соединений. Ориентировочно соотношение чув-
ствительности ТИЛ к соединениям, содержащим As, Cl, S, N,
P, без учета содержания гетероатома в молекуле составляет
1:10:50:50: 1000.
Наименьшее достигнутое значение предела детектирования
ТИЛ — 5 • 10-11 мг/с, типичное значение (1 — 5) 10-9 мг/с. Ли-
нейный динамический диапазон составляет 103-104 от предела
детекти ров ания.
Несмотря на отмеченные недостатки термоионного детекто-
ра, заключающиеся в резкой зависимости чувствительности от
многих параметров, этот детектор получил довольно широкое
распространение, главным образом, потому, что он обладает
высокой чувствительностью к фосфорорганическим веществам.
Поэтому наибольшее применение он нашел в анализе пестици-
дов, инсектицидов и других биологически активных веществ.
Для определения других классов элементорганических соеди-
нений ТИЛ использовался в меньшей степени, но в литературе
имеется немало примеров его применения, в основном как селек-
98
Таблица U.S. Чувствительность термоионного
детектора к соединениям различных классов
Соединение Иониза- ционная эффектив- ность, Кл/моль Селектив- ность, сигнал ТИД сигнал ПИД Соединение Иониза- ционная эффектив- ность, Кл /моль Селектив- ность, сигнал ТИД сигнал ПИД
Триэтил- арсин 0,23 10 Пиридин 4,6 —
Хлор- бензол 1,4 — Нитробензол — 250
Бром- бензол 2,4 — Акрилони- трил 4,8 —
4-Вром- толуол 2,5 — Анилин — 308
/3- Бро гн- ети рол 2.2 — N-Метил- анилин •— 233
Тиофен -6,0 — Трифенил- фосфин — 1100
Гекс и л- меркаптан -6,1 — 1 Диизопропил- метилфосфат 220 —
Примечание. Поскольку чувствительность ТИД сущест-
венно зависит от его конструкции, приводимые ориентировоч-
ные значения не могут быть использованы в качестве рабочих
и подлежат измерению в процессе градуировки детектора.
тивного детектора на азот- и галогенсодержащие соединения. В
настоящее время различными средствами достигнута необхо-
димая стабильность работы ТИД, что способствует его более
широкому использованию.
II.1.4.8. Пламенно-фотометрический детектор
Селективный пламенно-фотометрический детектор (ПФЛ) на
фосфор- и серосодержащие вещества предложен для использо-
вания в хроматографии в 1966 г. Принцип действия его осно-
ван на измерении свечения водородного пламени при сгорании в
нем фосфор- и серосодержащих соединений. Различие условий
сжигания в пламенно-фотометрическом детекторе и пламенно-
ионизационном состоит в том, что в ПФД пламя обогащено во-
дородом, в то время как в ПИД оно обогащено кислородом
Конструктивно он представляет собой сочетание ячейки ПИЛ
с оптической схемой измерения светового потока. Оптическая
схема ПФЛ изображена на рис. П.32.
99
1
Рис. 11.32. Оптическая схема пламенно-фотометрического детектора.
1 — кварцевая трубка; 2 — зона световой ©миссии; 3 — интерфе-
ренционный фильтр; 4 — фотоумножитель.
Регистрация интенсивности излучения пламени происходит
следующим образом. Световой поток сначала проходит интер-
ференционный фильтр, который поглощает фоновое излучение
пламени, после чего поступает на чувствительный элемент фо-
тоумножителя. Полученный таким образом фототок направля-
ется в электрометрический усилитель и далее на самопишущий
потенциометр.
Выбор измеряемой длины волны определяется характером
эмиссионного спектра пламени фосфор- и серосодержащих со-
единений, имеющего максимум соответственно при 526 и 394 нм.
Спектральное выделение этих полос производится интерферен-
ционными светофильтрами с шириной полосы пропускания
±(5 — 10) нм. Ширина пропускания светофильтра определяет
чувствительность и селективность ПФЛ- Применение фильтров
с более узкой полосой пропускания повышает селективность, но
существенно снижает чувствительность детектора, так как ин-
тенсивность светового потока пропорциональна квадрату ши-
рины пропускаемой полосы.
Защита оптических фильтров от высокой температуры обес-
печивается специальной насадкой в виде кварцевой или пирек-
совой трубки, размещаемой над горелкой в зоне водородного
пламени, или увеличением с помощью световодов расстояния
между зоной пламени и фотоумножителем.
Введение в ПФЛ стандартного коллекторного электрода по-
зволяет регистрировать сигнал пламенно-ионизационного де-
тектора, а при использовании двухканального усилителя и са-
мописца записывать на одной ленте сигналы ПФЛ и ПИЛ. Ис-
пользование двух светофильтров и фотоумножителей, располо-
женных по разные стороны от горелки, позволяет одновременно
регистрировать серо- и фосфорсодержащие соединения, при-
сутствующие в этой смеси.
100
Og+Nj O2 N2 Hj
Рис. 11.33. Варианты смешивания газов, питающих пламенно-
фотометрический детектор.
Режим газового питания ПФД принципиально отличается
от режима ПИД и может быть реализован разными путями
(рис. 11.33) — предварительным смешением азота и кислорода
(а), азота и водорода (б), а также водорода и воздуха (в). Во
многих случаях вместо чистого кислорода могут быть исполь-
зованы смесь кислорода с азотом или воздух. Иногда по цен-
тральной трубке подают водород (г), но это повышает уро-
вень шумов. Недостатком схемы а является необходимость
применения больших скоростей водорода (до 300 мл/мин), так
как в противном случае при высоких концентрациях вещества
в потоке газа-носителя снижается эмиссия пламени. Схема в
также требует больших расходов водорода для поддержания
нормального режима горения пламени. Столь большие рас-
ходы водорода создают яркое свечение пламени, что вызывает
необходимость экранирования светового потока, поступающего
на фотоумножитель. Предварительное смешение водорода с
газом-носителем (азотом) допускает небольшие расходы водо-
рода (30 мл/мин) и не требует ослабления светового потока,
но при введении в хроматографическую колонку больших проб
(несколько микролитров), особенно при выходе узких зон рас-
творителя, пламя может гаснуть.
Соотношение скоростей газов в любых режимах питания
ПФД оказывает большое влияние на устойчивость его работы
и зависит от конструкции. Оптимальные расходы при детекти-
ровании серо- и фосфорсодержащих соединений различаются.
Пламенно-фотометрический детектор обладает низким пре-
делом детектирования фосфор- и серосодержащих веществ, зна-
чение которого для лучших конструкций находится на уровне
10~9 мг/с для серосодержащих и 10“10 мг/с для фосфорсодержа-
101
щих соединений. Селективность относительно углеводородов
для серо- и фосфорсодержащих веществ равна соответственно
103—104 и 105-107, линейный диапазон 102-103 и 103-104.
Процессы, протекающие при сжигании серо- и фосфорсодер-
жащих веществ в ПФЛ, еще полностью не исследованы, и в на-
стоящее время для оценки факторов, влияющих на характери-
стики ПФЛ, можно пользоваться эмпирическими закономерно-
стями, главными из которых являются следующие [74]:
1) чувствительность ПФЛ к серо- или фосфорсодержащим ве-
ществам тем больше, чем выше содержание этих элементов в
молекуле анализируемого соединения;
2) чувствительность детектирования серосодержащих ве-
ществ зависит от степени окисленности атома серы в молекуле
анализируемого вещества;
3) присутствие атома фосфора в сероорганических вещест-
вах, детектируемых по каналу 394 нм, может искажать сигнал
ПФЛ, так же как наличие серы в фосфорорганических веще-
ствах, детектируемых по каналу 526 нм;
4) сигнал фосфорорганических соединений пропорционален
концентрации их в потоке газа-носителя;
5) сигнал серосодержащих веществ пропорционален лога-
рифму потока вещества.
Последнее обстоятельство следует особо учитывать при ко-
личественном анализе и определении предела детектирования
ПФЛ по серосодержащим веществам. В самом деле, если сигнал
детектора пропорционален квадрату потока вещества (обычно
показатель степени колеблется в интервале 1,5-2), то уменьше-
ние времени удерживания вещества в хроматографической ко-
лонке в два раза у симметричного пика увеличивает его вы-
соту Н в четыре раза, а не в два, как это имеет место в случае
фосфорсодержащих веществ (рис. II.34). Хроматографический
пик у логарифмического детектора не отвечает гауссову рас-
пределению, а его площадь зависит от времени удерживания.
Например, ширина пика при расчете площади у квадратичного
детектора должна измеряться не на 1/2 высоты, как у линей-
ного, а на 1/4. Аналогично уменьшение вводимой пробы в два
раза, при прочих равных условиях, понижает высоту пика ана-
лизируемого вещества в четыре раза.
Кроме высокой селективности и чувствительности к серо- и
фосфорсодержащим веществам достоинством ПФЛ является его
быстрый выход на режим при изменении температуры колонки и
скорости газа-носителя. Высокая селективность детектора по-
зволяет работать с программированием температуры при боль-
ших скоростях без существенного смещения нулевой линии.
102
Рис. 11.34. Схема, иллюстрирующая связь высоты пика Н с
временем удерживания у линейного (а) и логарифмического
(6) детекторов.
Характерной особенностью ПФД является зависимость чув-
ствительности к серо- и фосфорсодержащим соединениям от
присутствия в пламени других веществ. Так, наличие в пламени
углеводородов, выходящих из колонки одновременно с серо- и
фосфорсодержащими веществами, может понизить или полно-
стью подавить пики этих элементорганических веществ, хотя
эмиссия от углеводородов сама по себе не детектируется.
Серьезным недостатком большинства конструкций ПФД яв-
ляется гашение пламени детектора дозами элюируемого веще-
ства, характерными для насадочных колонок (несколько микро-
литров). Резкое снижение по этой причине максимально вводи-
мой в хроматографическую колонку дозы повышает предел де-
тектирования ПФД. Для его снижения применяются сложные
системы выброса зоны растворителя или поддержания горения
пламени.
Известны конструкции двухканального ПФД для одновремен-
ной регистрации сигналов по Р- и S-каналам.
103
II.1.4.9. Фотоионизационный детектор
Фотоионизационный детектор (ФИЛ) относится к высокочув-
ствительным детекторам универсального назначения с регули-
руемой селективностью. Предложен для использования в хро-
матографии в конце 50-х годов, в течение двух последующих де-
сятилетий в первоначальную (не слишком удачную) конструк-
цию фотоионизационного детектора вносились существенные
улучшения, и лишь в конце 70-х годов освоен серийный выпуск
газовых хроматографов с ФИЛ, работа над совершенствова-
нием которого не прекращается до сих пор [75-77]. За рубе-
жом лидерство в разработке и производстве фотоионизацион-
ных детекторов давно уже принадлежит американской фирме
HNU Systems, Inc., Newton, Massachusetts, в России (в настоящее
время) — Бюро аналитического приборостроения “Хромдет”
(Москва) (рис. 11.35).
Принцип действия ФИЛ заключается в ионизации молекул
элюируемых из хроматографической колонки веществ под дей-
ствием вакуумного УФ-излучения и измерении возникающего
ионного тока. Можно выделить 4 стадии ионизации.
1. Прямая ионизация молекул регистрируемых веществ (АВ):
АВ -|- hv —АВ+ 4- е.
2. Ионизация в результате взаимодействия молекул АВ с
возбужденными молекулами газа-носителя С:
С + hv —> С*,
АВ + С* —> АВ+ + е + С.
3. Ионизация фотонами с промежуточным переходом молекул
АВ в возбужденное состояние:
АВ + hv —-> АВ*,
АВ* —> АВ+ + е.
4. Как всегда, процессы ионизации сопровождаются побоч-
ными реакциями, из которых главными являются ответственные
за рекомбинацию заряженных частиц:
АВ+ + е —»• АВ,
С + е —>С",
АВ+ + С- —»• АВ + С.
104
a
б
Рис. 11.35. Фотоионизационные детекторы.
а — разработки фирмы HNU Systems [75]: 1 — окно для вывода из-
лучения; 2 — вакуумная УФ-лампа; 3 — корпус лампы; 4 — электрод;
5 — ионизационная камера; 6 — уплотнение Kalrez: 7 — графитовое
уплотнение; 8 — измерительный электрод; 9 — поляризующий элек-
трод; 10 — нагреваемый блок; 11 — вход газа; 12 — выход газа.
6 — разработки фирмы “Хромдет” [77]: 1 — входной канал; 2 —
основание детектора; 3 — ионизационная камера; 4 — коаксиальные
электроды; 5 — УФ-лампа; 6 — защитный кожух; 7— изоляторы; 8 —
канал для поддувочного газа.
Вероятность реализации этих процессов и относительный
вклад каждого обусловливаются совокупным влиянием спек-
тральных характеристик носителей энергии (фотонов), а также
свойствами облучаемых соединений и подвижной фазы (газа-
разбавителя), однако в любом случае энергия фотонов должна
быть равна или больше потенциала ионизации молекул-объек-
тов анализа.
Приведенная схема стадий ионизации и рекомбинации ионов
в рабочей камере детектора не накладывает жестких ограни-
чений на выбор подвижной фазы хроматографической системы,
поскольку потенциалы ионизации обычно используемых газов-
105
носителей существенно выше энергии УФ-облучения (незави-
симо от типа лампы):
Газ-носитель Потенциал ионизации, эВ
Гелий 24,59
Аргон 15,76
Азот 15,58
Водород 15,43
Диоксид углерода 13,79
Тем не менее, в качестве подвижной фазы предпочтительнее ис-
пользовать аргон, так как, по имеющимся экспериментальным
данным, относительный отклик ФИЛ на равные количества ре-
гистрируемого вещества (конкретно — бензола) снижается при-
мерно в 1,5 раза при замене аргона на гелий или азот и при-
мерно в 5 раз при переходе к СОг- Лля объяснения этих фак-
тов обычно ссылаются на повышенную скорость электронов в
среде аргона, чем в других газовых средах (что обусловливает
снижение потерь заряженных частиц из-за рекомбинации), и на
меньшее поглощение молекулами аргона УФ-излучения, однако
главные причины отмеченных наблюдений, вероятно, еще не
раскрыты.
В качестве источников излучения применяются УФ-лампы
аргонового, криптонового, ксенонового и водородного напол-
нения, испускающие фотоны с энергией 9,5, 10,0, 10,2, 10,9 и
11,7 эВ. Лампы взаимозаменяемы, но чаще всего используется
водородная лампа, из спектра которой выделяют полосу с дли-
ной волны 121,6 нм (10,2 эВ). Этот источник УФ-излучения со-
здает наиболее интенсивный пучок фотонов, что особенно важно
при определении микропримесей, а также при работе с капил-
лярными колонками, требующими малых нагрузок.
УФ-радиация проникает в ионизационную камеру, имеющую
весьма малый объем (от 175 — 150 до ~40 мкл), через оптиче-
ские окна (изготавливаемые из галогенидов, чаще всего фтори-
дов щелочных и щелочноземельных металлов), обладающие до-
статочно высокой механической прочностью в широком интер-
вале давлений и температур и необходимыми границами про-
пускания в области вакуумного УФ-излучения (LiF — 105 нм,
MgF2 — 112 нм, СаРг — 122 нм, SrF2 — 128 нм).
Наиболее распространенная водородная лампа (10,2 эВ)
укомплектовывается окошком из фтористого магния, выдержи-
вающим температуру до 275 °C (при длительной работе) и даже
до 300 °C и выше (в течение непродолжительных периодов экс-
плуатации) .
Корпус ионизационной камеры со встроенными электродами
(AU ~ 200 В) изготавливают из термически прочных низкопори-
106
стых материалов с высоким удельным сопротивлением, таких,
как, например, корундовая керамика.
Герметичность ячейки ФИД (если она предусматривается,
см. рис. 11.35, а) достигается использованием специальных высо-
котемпературных уплотняющих элементов (перфторированная
резина, специальные диффузионнотвердеющие припои и др.).
Недостаточный ассортимент подобных материалов и возника-
ющая при их использовании потенциальная опасность загряз-
нений оптических окон и газовой среды внутри рабочей камеры
послужили стимулом к разработке негерметичных ячеек ФИД,
в которых элюат выводится через зазор между источником ио-
низации и корпусом детектора (см. рис. 11.35, б) [77]. Как видно,
конструкция данной ячейки (впрочем, и многих других [76]) пре-
дусматривает поддув в рабочую камеру вспомогательного по-
тока газа с целью уменьшения эффективного объема ионизаци-
онной камеры и подавления размывания поступающей в детек-
тор хроматографической полосы, что особенно оправдано при
работе с “узкими” капиллярными колонками.
Многочисленными экспериментальными исследованиями по-
казано, что ФИД — детектор концентрационный и недеструк-
тивный, обладающий весьма широким линейным динамическим
диапазоном (~ 107). Абсолютные сигналы ФИД (площади пи-
ков) могут быть подвержены влиянию природы газа-носителя
(см. выше) и возрастают при снижении потока газа через ио-
низационную камеру; относительные отклики сложным обра-
зом зависят от нескольких факторов: от энергии УФ-облучения
(типа лампы) и разности между этой энергией и потенциалом
ионизации, а также от особенностей состава и строения моле-
кул вещества.
Достаточно высокие потенциалы ионизации некоторых неор-
ганических и органических соединений (табл. II.4), превышаю-
щие энергии УФ-облучения выбранных типов ламп, исключают
возможность обнаружения соответствующих молекул (так, SO2
и метан не регистрируются даже с применением наиболее высо-
коэнергетичной лампы hv = 11,7 эВ). Однако отмеченные при-
меры носят единичный характер, и подавляющее большинство
веществ (особенно органических) детектируется с весьма высо-
кой чувствительностью (на уровне пикограммовых количеств),
причем селективность их обнаружения возрастает по мере сни-
жения энергии лампы. В этой связи отметим, что ФИД с наи-
более широко распространенной лампой hv = 10,2 эВ не дает
отклика на такие часто встречающиеся органические раство-
рители, как метанол, хлороформ, четыреххлористый углерод,
ацетонитрил. Это обстоятельство освобождает аналитика от
107
Таблица II.4- Первые потенциалы ионизации (ПИ)
некоторых неорганических и органических веществ [78]
Вещество ПИ, эВ Вещество ПИ, эВ
Неорганические соединения
Гелий 24,59 Вода 12,61
Аргон 15,76 Диоксид серы 12,34
Азот 15,58 Оксид серы 12,10
Водород 15,43 Кислород 12,08
Оксид углерода 14,01 Сероводород 10,47
Диоксид углерода 13,79 Аммиак 10,15
Органические соединения
Метан 12,98 Дихлорфторметан 12,40
Ацетилен 11,41 Четыреххлористый 11,47
Этилен 10,51 углерод
Пропан 11,07 Хлороформ 11,42
Бутан 10,63 Хлористый винил 10,00
Пентан 10,33 Т рихлорэтилен 9,45
Гексан 10,17 Метанол 10,85
Этанол 10,47
Декан 9,95 Фенол 8,50
Додекан 9,93 Ацетон 10,00
Бензол 9,25 Ацетон итри л 12,12
Толуол 8,82 Бензонитрил 9,71
Изопропилбензол 8,69 Этиламин 8,86
Бутилбензол 8,69 Анилин 7,70
о- Ксилол 8,56 Муравьиная кислота 11,05
Нафталин 8,12 Уксусная кислота 10,37
1-Метилнафталин 7,96 Нитрометан 11,23
Пи цен 7,80 Нитропропан 10,81
3,4-Бензпирен 7,19 Тиофен 8,86
Хлорбензол 9,07 Мети л меркаптан 9,44
Фторбензол 9,20 Пропил меркаптан 9,19
о-Фторхлорбензол 9,15 Диметилсульфид 8,46
Тетраметилсилан 9,98
поиска условий хроматографического разделения зон раство-
рителя и определяемых соединений, что особенно удобно при
анализе микропримесей. Напротив, не могут быть использо-
ваны в качестве растворителей проб ацетон, гексан, аромати-
ческие углеводороды, к которым ФИЛ обладает очень высокой
чувствительностью.
Формально в ряд недетектируемых растворителей следовало
бы включить и воду (потенциал ионизации 12,61 эВ), однако по-
падание ее паров в рабочую камеру вызывает многочисленные
побочные явления, связанные со способностью молекул воды за-
хватывать электроны (всегда присутствующие в ионизирован-
108
ной газовой среде), поглощать значительную долю фотонов, пе-
реходить в возбужденное состояние и сенсибилизировать иони-
зацию неотделенных в колонке компонентов пробы, поступаю-
щих в ячейку ФИЛ совместно с зоной воды. Из-за отмеченных
обстоятельств, а также из-за вредного воздействия паров воды
на оптические окна ячеек ФИЛ анализировать водные растворы
с данным детектором не рекомендуется.
С наибольшей отдачей ФИЛ применяют при исследовании
качественного и количественного состава сложных многоком-
понентных композиций нефтепродуктов и примесей различных
приоритетных экотоксикантов, содержащихся в объектах окру-
жающей среды: в воздухе, в почвах, в донных отложениях, в
природных и сточных водах (не следует забывать о нежела-
тельности прямых газохроматографических анализов вод-
ных образцов!).
ФИЛ с лампой hv = 10,2 эВ обладает более высокой в сравне-
нии с “базовым” пламенно-ионизационным детектором чувстви-
тельностью ко многим органическим соединениям, и даже по
отношению к углеводородам (наиболее благодарным объектам
для ПИД) его относительный отклик превышает отклик ПИЛ в
5-10 раз, в зависимости от наличия в молекуле детектируемого
углеводорода р- и тг-связей и числа бензольных колец.
Надежные экспериментальные данные, свидетельствующие о
том, что относительный сигнал ФИЛ обусловливается, в пер-
вую очередь, не числом атомов углерода в молекуле углево-
дорода, а его принадлежностью к тому или иному гомологи-
ческому ряду, позволили разработать ряд методик по опреде-
лению группового состава различных фракций нефтепродуктов
(элюат из колонки поступает в два параллельно или последо-
вательно сочлененных с выходом из колонки ФИЛ и ПИЛ; при
последовательном соединении детекторов ячейка ФИЛ должна
быть герметичной). Экспериментально исследуется отношение
нормированных сигналов двух детекторов (Sn) на каждый из
регистрируемых углеводородов:
рФИД , рПИД
Q __ г i__ г ст
°N ~ рПИД . рФИД ’
1 г 1 ст
где Pi, Рст — высоты или площади пиков идентифицируемых со-
единений и веществ, выполняющих функции стандартов. Най-
денные численные значения Sn сравниваются со справочными
(литературными или полученными экспериментально в приня-
тых условиях анализа) величинами (см. ниже). Аналогичные
зависимости найдены для различных групп хлор- и серосодер-
жащих пестицидов, различающихся по принадлежности к про-
109
изводным алифатических насыщенных (I), алифатических нена-
сыщенных (II) и ароматических (III) углеводородов
Класс углеводородов .S'n
Алканы < 2
Алкены 2-4
Ароматические 5—10
Группа пестицидов
I
II
III
S'n
< 2
5-10
12-16
углеводороды
По отношению к представителям других классов органиче-
ских соединений установлено увеличение относительного от-
клика ФИД в ряду: алканы < спирты метиловые эфиры <
этиловые эфиры < альдегиды < 2-кетоны < 3-кетоны. В преде-
лах каждого ряда разветвление углеродного скелета или нали-
чие двойных (тройных) связей и ароматических колец способ-
ствует снижению предела обнаружения, а присутствие нитро-
групп — его повышению.
Отмеченные наблюдения, безусловно, могут быть полезны в
практической работе с ФИД, хотя они, конечно, не исчерпы-
вают всего многообразия состава и строения молекул органиче-
ских соединений, встречающихся в анализируемых образцах*,
и поэтому при количественных определениях необходимо все-
гда опираться на результаты собственных экспериментальных
данных, полученных при градуировке прибора в регламентиро-
ванных условиях анализа (см. лабораторные работы 8 и И).
Границы и количественного, и качественного анализа с ФИД
существенно расширяются при его комплектации несколькими
лампами с различными энергиями эмиттирующих фотонов. В
этом случае открывается возможность направленного регули-
рования селективности детектирования и в ряде случаев суще-
ственного снижения предела обнаружения. В качестве нагляд-
ного примера можно сослаться на возможность обнаружения
с ФИД-11,7 эВ одного из наиболее опасных экотоксикантов —
формальдегида — при его содержании в воздухе ~ 10-8% (за-
метим, что многие другие детекторы вообще не дают заметного
отклика на формальдегид, а катарометр способен регистриро-
вать его в концентрациях, не меньших чем ~ 10-3%).
Напротив, использование менее мощных источников (в част-
ности, ламп с hv = 9,5 или 8,3 эВ) приводит к заметному сниже-
нию чувствительности ФИД при количественных определениях,
но увеличивает селективность его отклика на представителей
*Представительная сводка литературных сведений по относительной
чувствительности ФИД-10,2 эВ к многочисленным органическим соеди-
нениям различных классов приведена в обзоре [75].
110
различных гомологических серий при групповой идентифика-
ции.
Конструкция одной из модификаций ФИЛ, разработанной и
серийно выпускаемой Бюро аналитического приборостроения
“Хромдет” [77], позволяет влиять на селективность обнаруже-
ния тех или иных соединений не только путем замены ламп, но
и изменением температуры оптического окна лампы (при этом
изменяется состав спектра УФ-облучения, проникающего через
это окно), а также посредством установки дополнительных опти-
ческих фильтров (изготавливаемых из флюорита, фтористого
бария или лейкосапфира). Так, например, резко неодинако-
вое влияние температуры оптического окна лампы (криптоно-
вого наполнения, в диапазоне 70-160°С) на относительный от-
клик ФИЛ к углеводородам разных классов может быть исполь-
зовано в качестве дополнительного параметра при групповой
идентификации низкомолекулярных парафиновых и ароматиче-
ских углеводородов. Применение фильтра из лейкосапфира по-
зволяет (хотя и с существенно меньшей чувствительностью) ре-
гистрировать алкилнафталины и бициклические арены и прак-
тически полностью подавляет сигнал ФИЛ на насыщенные али-
фатические и алициклические углеводороды, а также на низшие
алкилбензо лы.
В заключение следует еще раз отметить, что серийно выпус-
каемые ФИЛ являются концентрационными, не деструктивными
и практически нетребовательными к природе газа-носителя де-
текторами.
Возможность регулирования селективности отклика ФИЛ
при использовании ламп с различными энергиями УФ-облуче-
ния открывает широкие возможности применения этих детек-
торов в качественном групповом газохроматографическом ана-
лизе. Весьма высокая чувствительность ФИЛ к широкому кру-
гу неорганических и органических соединений и впечатляющий
линейный динамический диапазон (~ 107) обусловливают все
возрастающее использование этих детекторов в количествен-
ных газохроматографических анализах (в первую очередь —
при исследованиях степени загрязненности различными экото-
ксикантами объектов окружающей среды).
П.1 .5. Система термостатирования
Система термостатирования необходима для поддержания
оптимального температурного режима хроматографических ко-
111
лонок, детекторов, дозирующих устройств. Условия термоста-
тирования этих элементов различны. Наиболее жесткие тре-
бования предъявляются к системе термостатирования хромато-
графических колонок, поскольку из многих факторов, влияющих
на разделение, воздействие температуры особенно велико.
Влияние температуры на объем удерживания Vr проявляется
главным образом через коэффициент Генри Г(Гд = VLS), зави-
сящий от температуры по экспоненциальному закону
Г = Aexp(Q//?T),
где А — коэффициент; Q — теплота сорбции, кДж/моль; R — га-
зовая постоянная, равная 8,4 Дж/(мольК); Т— температура, К.
Логарифмируя и дифференцируя выражение для Г, получаем
зависимость относительного изменения объема удерживания от
изменения температуры:
AVr _ Q ат
VR ~ RT' Т ‘
Так как теплоты сорбции для широкого круга анализируемых
веществ находятся в пределах 12,6-50,4 кДж/моль, можно вычи-
слить относительное изменение объема удерживания, соответ-
ствующее изменению температуры колонки на 1 °C для рабочих
температур 27°C (комнатная) и 227°C: 1,7-6,7 и 0,6-2,4% соот-
ветственно Аналогично можно подсчитать изменение темпера-
туры, вызывающее изменение объема удерживания на 1%: 0,6—
0,15°С (при27°С) и 1,7-0,4°С (при227°С). Практически для из-
мерения абсолютных объемов удерживания с погрешностью 1%
необходимо установить температуру колонки с погрешностью,
не превышающей 0,2-0,3°С. При измерении относительных объ-
емов удерживания требование к точности установки темпера-
туры резко снижается и погрешность в 2-3 °C вполне допустима.
При работе с малыми пробами анализируемых смесей и близ-
ком соответствии формы пиков гауссовому распределению для
высот пиков справедлив тот же характер зависимости от тем-
пературы, что и для объема удерживания.
Влияние температуры на работу детектора зависит прежде
всего от его типа. Для детекторов по теплопроводности и по
плотности часто необходимо более стабильное термостатиро-
вание, чем для колонки (максимальные колебания температуры
не более ±(0,02-0,05)°С). Пламенно-ионизационный и некоторые
другие детекторы могут вполне устойчиво работать без специ-
ального термостатирования, в этом случае необходимо лишь
предотвратить конденсацию анализируемых веществ во время
их перехода из колонки в детектор, нагревая соответствующий
газовый тракт до температуры колонки.
112
Температура дозирующих устройств, в которых происхо-
дит испарение жидкостей или возгонка твердой пробы, должна
быть, как правило, на 30-100°С выше температуры колонки, при
этом изменения температуры в пределах нескольких градусов
не сказываются заметно на результатах анализа.
Система, предназначенная для обеспечения изотермического
режима работы колонок, состоит из термостата, в который за-
ключены колонки и детекторы, требующие термостатирования,
и автоматического терморегулятора, по шкале которого уста-
навливается температура анализа. Диапазон температур коло-
нок современных газовых хроматографов обычно составляет от
— 100 до 450°C, однако наиболее употребительная область ра-
бочих температур — от комнатной до 200-250 °C.
Погрешность установки температуры (разность между тем-
пературой, заданной по шкале терморегулятора, и фактиче-
ски установившейся средней температурой) обычно находится в
пределах ±(2-4)°С. При работе на хроматографе большое прак-
тическое значение имеет воспроизводимость одной и той же ра-
бочей температуры колонки, обычно не выходящей за пределы
0,2-0,5°C, что вполне приемлемо для аналитических целей.
Мерой стабильности термостатирования является макси-
мальное мгновенное отклонение текущей температуры от сред-
него установившегося значения. Как правило, с увеличением
температуры стабильность ухудшается, т. е. возрастают коле-
бания температуры. Современные хроматографы характери-
зуются стабильностью термостатирования в пределах ±(0,05-
0,1)°С при температурах от комнатной до 200°C и (0,1-0,2)°С
при температурах до 400 °C.
Точные средства для измерения температуры термостата
включаются в состав системы термостатирования довольно
редко. Обычно используется система индикации (приблизи-
тельного измерения) температуры в нескольких узлах прибора
(колонка, дозатор, детектор), обладающая значительно боль-
шей погрешностью, чем погрешность задания температуры.
В настоящее время большинство отечественных и зарубеж-
ных хроматографов снабжаются воздушными термостатами.
Воздушный термостат (рис. II.36) представляет собой камеру
с двойными стенками, пространство между которыми запол-
нено теплоизоляционным материалом. Форма камеры может
быть разной, однако чаще используются вертикальный шкаф
для работы с прямыми или U-образными колонками и шкаф
кубической формы для спиральных колонок, расположенных
в вертикальной или горизонтальной плоскости. Вентилятор
создает интенсивный направленный поток воздуха (теплоноси-
113
Рис. 11.36. Система воздушного термостата.
1 — корпус; 2 -— колонка I-го канала; 3 — испаритель I-го канала;
4 — детектор I-го канала; 5 — вентилятор за защитной сеткой (на ри-
сунке не показана); 6 — детектор II-го канала; 7 — испаритель II-го
канала; 8 — колонка П-го канала; 9 — защитный экран, за которым
находятся нагревательные элементы; 10 — теплоизоляция.
теля), проходящий через секции с электрическими нагревате-
лями и прогревающий внутреннюю камеру термостата и тер-
мостатируемые элементы (колонки, детекторы).
Температурный режим термостата устанавливается регули-
рованием мощности нагревателей либо по принципу двухпози-
ционного (релейного) регулирования, либо по типу пропорцио-
нального регулирования. Двухпозиционное регулирование тем-
пературы состоит в том, что мощность, подаваемая на электри-
ческий нагреватель термостата, меняется скачкообразно
(рис. 11.37, а) от нуля до полного значения (две позиции) в зави-
симости от того, достигнута ли заданная температура. В уста-
новившемся режиме нагреватель работает, периодически вклю-
чаясь и выключаясь, при этом выделяемая им средняя мощ-
ность поддерживает температуру термостата около заданного
значения. Включается и выключается нагреватель с помощью
реле, ламповых и полупроводниковых приборов. При пропор-
циональном регулировании (рис. 11.37, б) в области температур,
близких к заданной (в зоне пропорциональности), мощность на-
гревателя меняется плавно от нуля до полного значения, про-
порционально разности достигнутой и заданной температур. В
установившемся режиме текущее значение выделяемой нагре-
вателем мощности меняется незначительно около некоторого
среднего значения, необходимого для поддержания достигнутой
температуры. Мощность нагревателя изменяется чаще всего с
помощью полупроводниковых приборов.
114
Время
Рис. 11.37. Изменение мощности нагревателя и температуры
термостата при двухпозиционном (а) и пропорциональном (6)
регулировании.
Рср — средняя потребляемая мощность; ТСр — средняя установив-
шаяся температура.
Пропорциональное регулирование имеет значительные пре-
имущества перед двухпозиционным, так как обеспечивает боль-
шую стабильность термостатирования. Кроме того, плавное
изменение мощности нагревателя позволяет избежать послед-
ствий резкого изменения сетевой нагрузки. Система пропорци-
онального регулирования обладает большей гибкостью и уни-
версальностью, обеспечивает как изотермический вариант ра-
боты, так и режим программирования по любому закону возра-
стания температуры термостата и поэтому широко применяется
в современных хроматографах.
Роль датчика температуры выполняет термометр сопроти-
вления (ТСП), представляющий собой калиброванное сопроти-
вление из тонкой платиновой проволоки. Сигнал ТСП подается
на компаратор, управляющий работой нагревателей в соответ-
115
ствии с заданным режимом. При температуре термостата ниже
заданной выделяемая мощность имеет максимальное значение
и вызывает быстрый нагрев термостата. Когда температура
термостата достигает значения на несколько градусов меньше
заданной температуры, начинается плавное уменьшение мощ-
ности нагревателя, пока не будет достигнуто равновесие, при
котором выделяемая нагревателем мощность лишь компенси-
рует тепловые потери термостата во внешнюю среду, не вызы-
вая дальнейшего увеличения температуры.
Управление температурой может осуществляться средства-
ми вычислительной техники, при этом регулирующая система
постоянно воспринимает различие между заданной и фактиче-
ской температурами и по определенной программе, учитываю-
щей характеристики термостата и значение температуры, вы-
рабатывает соответствующее управляющее воздействие, при-
водящее к выделению нагревателем необходимой мощности (см.
раздел II.2.3).
II.1.6. Программирование температуры колонок
Разделение смесей с широким интервалом температур кипе-
ния компонентов вызывает необходимость повышения темпера-
туры хроматографических колонок в процессе анализа.
Закон изменения температуры термостата колонок задается
программатором, работающим совместно с терморегулятором.
Чаще всего используется линейный закон (постоянная скорость
повышения температуры) или линейно-ступенчатый режим, при
котором участки повышения температуры чередуются с изотер-
мическими ступенями. Программирование температуры осуще-
ствляется электронной системой, изменяющей задание темпера-
туры (напряжения) с установленной скоростью и обеспечиваю-
щей динамическое управление мощностью нагревателей в соот-
ветствии с программой изменения температуры. Использова-
ние для этой цели средств вычислительной техники позволяет
достичь наиболее гибкого и точного управления температурой
по линейному или более сложному закону, лучшей воспроизво-
димости длительности изотермических ступеней, автоматиче-
ского охлаждения термостата с выведением на начальную тем-
пературу анализа.
Термостаты, предназначенные для работы с программирова-
нием, обладают меньшей теплоемкостью, так как должны бы-
стро прогреваться при ограниченной мощности нагревателей.
116
Поэтому они часто имеют малый объем, допускающий работу
лишь со спиральными колонками.
Система программирования характеризуется определенным
диапазоном скоростей повышения температуры и их погрешно-
стью. Обычно диапазон скоростей нагрева составляет от 0,5 до
25, иногда до 40°С/мин, однако скорости больше 15~20°С/мин
не используются из-за существенного и не поддающегося непо-
средственному контролю отставания температуры насадочных
колонок от заданного закона повышения температуры вслед-
ствие плохой теплопроводности сорбентов.
Погрешность скорости повышения температуры измеряется
как разность между заданной и фактической скоростями. На
точность количественного анализа с программированием темпе-
ратуры оказывает непосредственное влияние лишь та часть по-
грешности скорости, которая определяется случайными откло-
нениями от среднего значения (без систематической ошибки).
Воспроизводимость скорости повышения температуры колонок
может составлять несколько десятых долей процента от устано-
вленного значения.
Изменение температуры колонки в процессе анализа отри-
цательно влияет на стабильность газохроматографического ре-
жима. В литературе, описывающей преимущества и возмож-
ности газовой хроматографии с программированием темпера-
туры, недостаточно освещены трудности, сопровождающие
практическую реализацию метода. Поэтому ниже мы дадим
анализ дестабилизирующих факторов и описание инструмен-
тальных средств, предназначенных для обеспечения устойчи-
вости режима анализа с программированием температуры.
Основные процессы, отрицательно влияющие на стабиль-
ность газохроматографического режима, следующие.
1. При работе в газо-жидкостном варианте, а также с поли-
мерными адсорбентами возрастает эффективная летучесть не-
подвижной фазы (концентрация паров и летучих продуктов де-
струкции). Высокая летучесть ведет к постепенному уменьше-
нию количества неподвижной фазы, изменению разделительной
способности и параметров удерживания, размеров и формы пи-
ков, сокращению срока службы колонки. Введение в детек-
тор с потоком газа-носителя паров неподвижной фазы вызы-
вает появление значительного фонового сигнала, непроизводи-
тельно занимающего часть линейного диапазона детектирова-
ния. Вместе с тем летучесть стационарной фазы может суще-
ственно ухудшать качество нулевой линии. Искажения нулевой
линии ведут к повышению предела обнаружения и увеличению
погрешности измерения параметров (высот, площадей) пиков,
117
т. е. к дополнительной погрешности анализа. Высокая концен-
трация детектируемых паров в газе-носителе усугубляет про-
явление допустимых колебаний скорости газа или температуры
колонки.
2. С увеличением температуры изменяются вязкость и плот-
ность газа-носителя. Вызываемое этим отклонение его скоро-
сти от первоначального значения влияет на удерживание, раз-
мывание и размеры пика, а также сопровождается изменением
чувствительности и искажением нулевой линии.
3. Изменение температуры колонки может оказать влияние
на температурный режим детектора, с чем обычно связано из-
менение чувствительности и появление дрейфа нулевой линии.
Кроме того, отставание температуры участка газовой линии
колонка—детектор может привести к частичной потере высо-
кокипящих веществ и искажению формы пиков.
4. Повышение температуры колонки вызывает десорбцию ра-
нее сорбированных веществ (примесей из газа-носителя или
остатков анализируемых веществ). Эти десорбционные полосы
регистрируются детектором в виде горбов, ступеней или в виде
пиков, близких по форме к обычным хроматографическим пи-
кам (ложные пики). Подобные искажения нулевой линии могут
вызвать ошибки в идентификации и измерении пиков, снижают
чувствительность и точность анализа. Эти явления наиболее
характерны для газо-адсорбционного варианта газовой хрома-
тографии с программированием температуры.
Сфера отрицательного влияния этих факторов распространя-
ется на метрологические характеристики и аналитические воз-
можности метода, ограничивая прежде всего его применение
для определения микропримесей и оценки чистоты индивиду-
альных веществ.
Основными проблемами обеспечения стабильности, решае-
мыми инструментальными средствами, являются прежде всего
подавление нелинейно возрастающего фонового сигнала, вы-
званного летучестью неподвижной фазы, поддержание постоян-
ства скорости газа-носителя и сохранение оптимального тепло-
вого режима детектора.
Для исключения прогрессирующего дрейфа нулевой линии
используется сочетание двухколоночной газовой схемы с ком-
пенсационным детектированием. Сущность его состоит в од-
новременном использовании двух идентичных хроматографиче-
ских колонок, работающих в одном режиме и соединенных с
двумя одинаковыми камерами детектора. В процессе повыше-
ния температуры в каждой из колонок происходит одинаковое
по величине и по времени нарастание концентрации паров не-
118
подвижной фазы в потоках газа-носителя. Введение этих пото-
ков в детекторы одинаковой чувствительности вызывает равные
сигналы. Схема включения этих детекторов такова, что они ра-
ботают как своеобразный нуль-инструмент, компенсируя фоно-
вые сигналы. Хроматограмма анализируемой смеси, введенной
в одну из колонок (аналитическую), записывается детектирую-
щей системой как нескомпенсированный сигнал одного из де-
текторов. Другая колонка (сравнительная) необходима только
для создания идентичного потока газа-носителя с аналогично
изменяющимся количеством паров неподвижной фазы. Детекти-
рование этого потока создает сигнал, компенсирующий дрейф
нулевой линии, имеющий место при использовании только од-
ной аналитической колонки.
В подавляющем большинстве случаев при программирова-
нии температуры используется ПИД или детектор по теплопро-
водности. Так как последний представляет собой симметрич-
ную конструкцию с двумя равноценными измерительными ка-
налами, его включение по компенсационной схеме не требует
каких-либо дополнительных изменений (рис. 11.38). При работе
с ПИД используются две ячейки детектора с противоположной
поляризацией электродов (горелок) и общей цепью измерения
сигнала (рис II.39). Это приводит к образованию равных по ве-
личине, но обратных по знаку сигналов.
Рис. 11.38. Компенсационная схема с двумя колонками и детек-
тором по теплопроводности.
1, 2— колонки; 3, — каналы детектора; 5 — термостат колонок;
6 — термостат детектора.
Исключение дрейфа нулевой линии обеспечивается специ-
альным балансированием системы, имеющим целью достижение
тождественности колонок и условий их работы, а также равен-
ство чувствительности используемых детекторов.
Идентичность сорбента в колонках достигается использо-
ванием одной партии сорбента. Для сохранения идентично-
сти сорбента в течение всего срока эксплуатации колонок они
119
Рис. 11.39. Компенсационная схема включения ПИД с двумя
ячейками и одноканальным усилителем.
lf 2 — горелки детекторов; 3, 4 — источник питания; 5 — усили-
тель; 6 — регистратор.
должны работать одинаковое время и попеременно выполнять
роль аналитической и сравнительной колонок, что обеспечи-
вает одинаковое воздействие на сорбент анализируемых ве-
ществ. Влияние на баланс схемы различий в скорости газа-
носителя, температуре колонок, количестве неподвижной фазы
в колонках и чувствительности детектирования зависит от типа
применяемого детектора.
Наибольшие трудности встречаются при подготовке к работе
системы с пламенно-ионизационным детектором. Специальные
измерения зависимости фонового сигнала ПИД от малых изме-
нений названных условий позволили экспериментально оценить
максимально допустимые значения расхождения рабочих пара-
метров, каждое из которых независимо приводит двухколоноч-
ную схему на грань разбаланса. В табл. II.5 приведены условия
достижения предельного разбаланса не более 5 • 10~п А при фо-
новом токе 5 • 10-9 А, что соответствует допустимому дрейфу
5% за цикл программирования при работе на шкале 10-9 А.
При температуре колонок, отвечающей общему уровню фоно-
вого сигнала, приведенные значения допустимых расхождений
параметров не зависят от природы неподвижной фазы. Исклю-
чение составляет лишь допустимая разность температур коло-
нок, так как наклон зависимостей фонового сигнала от темпе-
ратуры несколько отличается для неподвижных фаз различной
термостойкости (рис. II.40).
Хотя двухколоночная схема дает принципиальную возмож-
ность полной компенсации фоновых сигналов, тем не менее дан-
ные табл. II.5 показывают, что практическая реализация этих
возможностей достигается весьма тщательным уравниванием
120
Таблица II.5. Значения предельно допустимых
расхождений параметров двухколоночной
компенсационной схемы
Параметр Значение допуска
относительное, % абсо л ютное *
Температура о,1 О,25°С
Скорость газа-носителя 1,0 0,3 мл/мин
Чувствительность 1,0 5 • 10-8А • с/мг
Количество фазы 1,7 5 мг
*Для колонки, содержащей 300 мг апиезона на хроматоне, при
скорости газа-носителя 2 л/ч, конечной температуре програм-
мирования 25О°С и чувствительности ПИД 5 • 10—6 А -с/мг.
Рис.II.40. Зависимость фонового тока от температуры для ко-
лонок со скваланом (^), полиэтиленгликольадипинатом (2),
полиэтиленгликолем 20М (3), апиезоном М (4), силиконом Е-
301 (5), полидиметилсилоксаном OV-1 (6), силоксановым кау-
чуком SE-30 (7) и дексилом 300 (3).
параметров каждой из половин схемы. Это предъявляет жест-
кие требования к газохроматографической аппаратуре, предна-
значенной для работы в режиме программирования.
Для обеспечения идентичности теплового режима колонок
используются воздушные термостаты с возможно меньшим гра-
диентом температуры в зоне расположения колонок. Идентич-
ность газового режима достигается подбором элементов уста-
новки и регулирования расходов с близкими динамическими ха-
рактеристиками. Чувствительность ячеек ПИД уравнивается
121
соответствующей корректировкой расходов водорода в каждой
горелке. Наконец, равенство количества неподвижной фазы в
колонках обеспечивается одинаковой геометрией колонок и кон-
тролем массы (а не объема) сорбента при заполнении колонок.
Подготовка двухколоночной схемы к работе должна заканчи-
ваться балансированием по результатам записи нулевой линии
в “холостом” (без введения пробы) цикле программирования
температуры. Оно состоит в таком направленном изменении ра-
бочих параметров (главным образом, расхода газа-носителя в
сравнительной колонке), которое приводит к уменьшению сиг-
нала разбаланса при конечной температуре цикла. При тща-
тельном балансировании схемы возможна работа на шкалах
10“10 А и выше (до максимальных температур неподвижных
фаз).
Следует отметить, что компенсационная схема, существенно
улучшая нулевую линию, не оказывает никакого стабилизиру-
ющего воздействия на параметры удерживания и степень раз-
деления анализируемых веществ, а также не увеличивает время
эксплуатации колонки. Решение этих проблем возможно лишь
на основе методов, приводящих к снижению летучести непо-
движной фазы.
Вследствие влияния температуры на вязкость и плотность
газа массовая скорость газа-носителя быстро уменьшается, ес-
ли давление на входе в колонку поддерживать постоянным. Для
колонки размером 100 х 0,3 см, заполненной сорбентом с диа-
метром зерен 0,15-0,25 мм, повышение температуры на 100°С
сопровождается уменьшением расхода в 1,5-1,7 раза. Такой
режим можно считать допустимым лишь в отдельных случаях
при использовании потоковых детекторов, для которых площадь
пиков анализируемых веществ не зависит от скорости газа и
определяется только массой компонента. Кроме того, необхо-
димо, чтобы изменение скорости не вызывало существенного
дрейфа нулевой линии. Этому условию в первом приближе-
нии может отвечать лишь ПИД, причем только в узком интер-
вале расходов газа-носителя (например, 1,5-2,5 л/ч). Эксплуа-
тация детектора по теплопроводности в этих условиях оказыва-
ется совершенно невозможной. Таким образом, режим постоян-
ной скорости газа-носителя во всех отношениях более предпо-
чтителен, а для достижения приемлемой точности анализа —
единственно возможный. Для поддержания постоянного рас-
хода в процессе повышения температуры колонки используются
рассмотренные выше регуляторы расхода, которые непрерывно
восстанавливают первоначальный расход, увеличивая соответ-
ствующим образом давление на входе в колонку.
122
Изменение температуры термостата колонок при программи-
ровании отражается на тепловом режиме детекторов, что ока-
зывает влияние на чувствительность и нулевой сигнал (поло-
жение нулевой линии). Поскольку глубина этого влияния для
разных типов детекторов резко различна, отличаются и аппа-
ратные средства защиты этих детекторов. Вследствие силь-
ного влияния температуры на чувствительность детектора по
теплопроводности и особенно на нулевой сигнал, его помещают
в индивидуальный термостат, в котором поддерживается посто-
янная температура, близкая к конечной температуре програм-
мирования. В этом случае площади пиков не зависят от тем-
пературной программы колонок, и обеспечивается наибольшая
устойчивость нулевой линии. Термостатирование при более
низкой температуре для достижения большей чувствительности
может привести к конденсации паров неподвижной фазы, что от-
рицательно сказывается на записи нулевой линии. Вынужден-
ное расположение колонки и детектора в различных термоста-
тах вызывает необходимость в удлинении перехода колонка —
детектор. Иногда существенные искажения формы пиков высо-
кокипящих веществ при работе с большими дозами могут быть
вызваны конденсацией анализируемых веществ в газовом пере-
ходе колонка — детектор. Подобные искажения встречаются и
при работе в изотермическом режиме. Для исключения этих ис-
кажений применяется специальный подогрев газового перехода
колонка — детектор.
Рабочие параметры пламенно-ионизационного детектора в
значительно меньшей степени подвержены влиянию темпера-
туры. Изменение чувствительности несущественно (около 0,1%
на 1°С), а смещение нулевой линии ПИД, как правило, наблю-
даемое даже при незаполненной колонке, объясняется косвен-
ными причинами, например увеличением фонового сигнала за
счет поступления в пламя органических веществ, загрязняющих
газовый тракт или отдельные элементы конструкции детектора.
Этот эффект устраняют периодической очисткой газового ка-
нала и горелки ПИД от загрязнений органического происхо-
ждения.
Индивидуальное термостатирование ПИД не является необ-
ходимым., и в большинстве хроматографов детектор устанавли-
вают на крышку внутренней камеры термостата колонок, так
что конечная часть штуцера, к которому присоединяется ко-
лонка, находится внутри термостата. При таком расположе-
нии детектора за счет недостаточного прогрева его основания
при программировании температуры возможна конденсация вы-
сококипящих веществ в наиболее холодной части канала газа-
123
носителя (под горелкой). Это приводит к искажению хромато-
граммы и загрязнению детектора. Введение дополнительного
подогрева основания детектора позволяет полностью исклю-
чить это явление (рис. 11.41).
Рис. 11.41. Хроматограмма смеси алкана С20 (2) и 4,4/-диамино-
дифенилового эфира (3) в ацетоне (1) при программировании
температуры от 120 до 25О°С с ПИД.
а — без подогрева основания ПИД (120 ° С) 6 — с его подогревом
(230°С).
Для работы в режиме программирования температуры, осо-
бенно с высокочувствительным пламенно-ионизационным детек-
тором, характерно возникновение ложных пиков и других ис-
кажений нулевой линии вследствие десорбции при повышении
температуры колонки ранее сорбированных веществ, не являю-
щихся анализируемыми компонентами.
Насыщение колонки посторонними веществами— примесями
в газе-но сите ле, летучими выделениями из резиновой пробки
испарителя или мембран регуляторов — происходит перед ци-
клом программирования, когда колонка выдерживается при от-
носительно низкой начальной температуре. Наиболее простым
124
способом идентификации ложных пиков является запись нуле-
вой линии при программировании температуры без введения
анализируемой пробы, но при строгом соблюдении тождествен-
ности рабочему аналитическому режиму. Природа ложных пи-
ков подтверждается пропорциональной зависимостью площади
пиков от времени выдержки колонки перед включением про-
граммирования и обратной зависимостью от начальной темпе-
ратуры колонки, что также может быть использовано для их
распознавания.
Для уменьшения величины ложных пиков необходимо сокра-
щать время начального изотермического периода и не допус-
кать большего охлаждения колонок, чем это необходимо для
стабилизации начальной температуры программы.
Эффективным средством исключения ложных пиков является
очистка газа-носителя, поступающего в колонку, до низкого
уровня фонового сигнала ПИД (менее 10-11 А). Это достига-
ется установкой дополнительного адсорбционного фильтра ме-
жду газовым блоком хроматографа и испарителем. Важно при
этом исключить влияние изменения температуры на фильтр, так
как оно может вызвать существенные искажения нулевой линии.
Сложнее исключить загрязнения газа-носителя в горячей зо-
не тракта, в частности в испарителе. Чаще всего источником
загрязнения является пробка испарителя из силиконовой ре-
зины, выделяющая летучие вещества при температурах выше
220-250°C. Предварительная обработка силиконовой резины
(выдерживание при 250-300 °C под вакуумом в течение 8-10 ч)
позволяет резко сократить выделение летучих фракций по срав-
нению со “свежей” резиной.
При анализе высококипящих соединений в испарителе, как
правило, накапливаются остатки анализируемых веществ —
продукты осмоления под действием высокой температуры, а
также продукты превращений, катализируемых материалом до-
затора. Эти продукты также могут быть источником загрязне-
ния газа-носителя. Для снижения этого влияния следует вы-
бирать минимальную температуру испарителя, обеспечиваю-
щую достаточно быстрое испарение анализируемой смеси, не
допуская необоснованного перегрева. Применение стеклянного
или кварцевого испарителя с введением пробы с помощью сте-
клянного капилляра предохраняет от частичного разрушения
веществ, склонных к каталитическому превращению в контакте
с металлами, обеспечивая чистоту узла ввода пробы. Наконец,
во многих случаях допускается введение пробы без специаль-
ного испарителя, непосредственно на начальный слой сорбента
в колонке.
125
Таким образом, использование соответствующих инструмен-
тальных средств в значительной степени решает проблему за-
щиты от многих характерных для программирования темпера-
туры возмущений газохроматографического режима. Это спо-
собствует повышению реализуемой чувствительности, прибли-
жая ее к чувствительности изотермического анализа, и суще-
ственно снижает погрешность, доводя ее до значений, характер-
ных для режима работы при постоянной температуре колонки.
Общей особенностью работы в режиме программирования
температуры является существенный рост объема подготови-
тельных операций, выполняемых аналитиком в соответствии с
характером аналитической задачи и требованиями к чувстви-
тельности и точности анализа. Наиболее полно проблема ста-
билизации режима анализа может быть решена только при це-
лесообразном сочетании аппаратурных средств и соответству-
ющих методических приемов.
II.1.7. Регистрация сигналов детекторов
Сигналы детекторов по теплопроводности и по плотности за-
писываются непосредственно с помощью стандартных автома-
тических компенсационных потенциометров общего назначения
со шкалой 1-10 мВ и временем пробега шкалы пером 0,5-0,2 с.
Для согласования величины сигнала со шкалой потенциоме-
тра используются прецизионные делители (аттенюаторы), со-
ставленные из точных сопротивлений, позволяющие направить
на регистрацию лишь часть сигнала детектора (деление сиг-
нала). Обычно делители обеспечивают несколько ступеней де-
ления (загрубления чувствительности), например 1:2, 1:5, 1:10,
1:20, 1:50, 1:100. Коэффициенты 2, 5, 10, 20, 50, 100 предста-
вляют собой отношения номинальных значений шкал по напря-
жению и часто называются множителями шкал, или коэффи-
циентами аттенюации. Общий диапазон измерения сигнала
редко превышает 102—103. Для регистрации сигнала иониза-
ционных детекторов необходимо использовать усилители (элек-
трометры), преобразующие весьма малый ток детекторов в про-
порциональное напряжение, соответствующее шкале применяе-
мого автоматического потенциометра. Наиболее жесткие тре-
бования к электрометру предъявляются при работе с пламенно-
ионизационным детектором. Это связано прежде всего с необ-
ходимостью измерения токов до 10-14 А для реализации пре-
дельных возможностей ПИД при сравнительно широком диа-
126
Рис. 11.42. Схема регистрации сигнала ионизационных детек-
торов.
1 — детектор; 2 — источник питания детектора; 3 — переключатель
входных измерительных сопротивлений; 4 '— усилитель; 5 — переклю-
чатель выходного делителя; 6 — регистратор.
пазоне измеряемых ионных токов (максимальное значение тока
до 10-6 А, диапазон 10-6 А : 10“14 А = 10s), необходимостью
использования высокого быстродействия детектора.
Упрощенная схема системы регистрации сигнала ионизаци-
онных детекторов показана на рис. 11.42. Измерение тока детек-
тора осуществляется косвенным методом — по падению напря-
жения, создаваемому током на входном измерительном сопро-
тивлении. В этом случае электрометр выполняет роль вольт-
метра с высоким входным сопротивлением (до 1013 Ом). По-
скольку самопишущий потенциометр имеет узкий диапазон ре-
гистрируемых сигналов (102 — от 1 до 100% шкалы), а измеряе-
мые токи могут меняться в широком диапазоне, для согласова-
ния сигнала электрометра со шкалой потенциометра применя-
ется выходной делитель с диапазоном 10210;i. Для регистрации
полного диапазона токов используется, кроме того, переключе-
ние входных измерительных сопротивлений, при этом диапазон
расширяется еще на 3-4 порядка. Табл.II.6 иллюстрирует это
положение на примере электрометра, применяемого в хромато-
графах “Пвет-500”.
Многие электрометры, в том числе электрометры, исполь-
зуемые в хроматографах “Цвет”, имеют раздельное включе-
ние входных измерительных сопротивлений и шкал по напря-
жению. Поэтому для включения необходимого предела измере-
ния (шкалы) по току следует одним переключателем установить
127
Таблица II.6. Пределы измерения тока (А) ионизационных
детекторов, регистрируемого электрометром Б14Д-З6
Положение выключателя выходного делителя* Положение переключателя измерительных сопротивлений**
1О10 109 10® 107
1 1 • 10“12 1 • IO-11 1 1О~10 1 - ю-9
2 2 - 10—12 2- 10_11 2 • 1О-10 2 • IO-9
4 4 • 10—12 4- 10~11 4 IO”10 4 -10-9
8 8 • 10“11 8-10~" 8 1О“10 8 - IO-9
16 1,6- ю-11 1,6- 1О~10 1,6- io-9 1,6 - IO-8
32 3,2 Ю-11 3,2 • 1О~10 3,2 10~9 3,2 • IO-8
64 6,4- 10-11 6,4 • 10~10 6 410~9 6,4 IO-8
128 1,28- 1О-10 1,28- IO-9 1,28- IO-8 1,28-10-7
256 2,56- 1О~10 2,56- 10~9 2,56 • 10-® 2,56 - Ю-7
512 5,12 • 1О“10 5,12- Ю-9 5,12 - IO-8 5,12 - IO-7
1024 1,024-10-9 1,024- 10“8 1,024- 10“7 1,024 10-6
* Соответствует шкалам по напряжению от 10 мВ до 10,24 В.
**Соответствует номинальным значениям измерительных со-
противлений в омах.
входное сопротивление, а другим — выходным делителем уста-
новить шкалу по напряжению, при этом измеряемый ток опре-
деляется по закону Ома: /дет = UmK/RK3M.
Сопротивления, составляющие делитель, имеют значения в
пределах 10-1000 Ом и могут быть изготовлены довольно точно,
поэтому обеспечивается соответствие действительных значений
коэффициентов аттенюации их номиналам. При переключении
входного сопротивления значительно сложнее добиться доста-
точно точного соответствия коэффициентов аттенюации их но-
миналам, так как трудно изготовить точно высокоомные со-
противления (1011—109 Ом) и их значения могут существенно
меняться в процессе эксплуатации. Кроме того, переключе-
ние входных сопротивлений иногда сопровождается появлением
временных шумов и непропорциональным смещением нулевой
линии.
Если в ходе количественного анализа необходимо изменить
чувствительность электрометра, для исключения дополнитель-
ных ошибок при пересчете сигнала на другую шкалу следует
избегать переключения входных сопротивлений, а пользоваться
только выходным делителем. Для этого входное сопротивле-
ние должно выбираться таким, чтобы обеспечивался требуемый
128
диапазон измеряемых токов. Так, для измерения сигнала детек-
тора от 5 10“11 до 3 • 10“9 А надо (см. табл. II.6) включить из-
мерительное сопротивление 108 Ом и работать в пределах шкал
по напряжению от 20 до 500 мВ.
Довольно часто измеряемый электрометром полезный сигнал
(пик) значительно меньше уровня сигнала детектора, образо-
ванного фоновым током.
Пример. Необходимо измерять сигнал 5 10“11 А над уровнем фоно-
вого тока 2 • 10—9 А. При включении шкалы по току 5 • 10—9 А, на которой
фоновый сигнал составляет 40%, измеряемый полезный сигнал соответ-
ствует 1% шкалы, т. е. практически не может быть точно измерен. Для
проведения измерений на достаточно чувствительных шкалах в электро-
метрах предусматривается возможность подавления фонового сигнала с
помощью схемы компенсации, дающей ток, противоположный току сиг-
нала. Схема компенсации должна обеспечивать возможность установки
тока компенсации в достаточно широких пределах, необходимую плав-
ность его изменения при установке нуля и, главное, высокую стабиль-
ность установленного значения тока компенсации (изменения тока ком-
пенсации проявляются как обычные флуктуации нулевой линии). После
того, как фоновый сигнал будет скомпенсирован, выбирается шкала элек-
трометра, в пределах которой должен регистрироваться измеряемый сиг-
нал; для нашего примера может быть использована шкала 10-10-10 А, при
этом сигнал (5 10—11 А) составит 50% шкалы.
Схемы компенсации, включаемые в состав электрометров,
обеспечивают стабилизацию тока компенсации до 0,01%. Даль-
нейшее улучшение стабильности достигается значительным ус-
ложнением схемы компенсации и, как правило, не применяется.
Существуют две категории электрометров: электрометры
прямого усиления постоянного тока и электрометры с преобра-
зованием тока детектора в переменный, усилением по пере-
менному току и обратным преобразованием в постоянный сиг-
нал (модуляция — усиление — демодуляция). Последний ва-
риант сложнее, но позволяет получить малый уровень шума
и практически исключить дрейф при высокой чувствительно-
сти электрометра Как правило, схема электрометра предста-
вляет сочетание электрометрической лампы или полевого тран-
зистора на входе и полупроводникового усилителя. Современ-
ные электрометры, специально предназначенные для использо-
вания в газовых хроматографах, обладают чувствительностью
до 10“12 А на полную шкалу регистратора и весьма малым уров-
нем шума — до 5 10“15 А, при этом постоянная времени элек-
трометра обычно не превышает 0,5 с.
В хроматографах, обеспечивающих одновременную и незави-
симую работу двух детекторов, используются два индивидуаль-
но
ных потенциометра или двухканальные, двухперьевые автома-
тические потенциометры, записывающие сигналы двух детекто-
ров на одной ленте чернилами разного цвета. Для измерения и
регистрации сигнала применяются вычисляющие интеграторы
и микропроцессорные системы обработки хроматографической
информации.
II.2. ЛАБОРАТОРНЫЕ ГАЗОВЫЕ ХРОМАТОГРАФЫ
П.2.1. Хроматографы ОАО “Цвет”
В 1997 г. ОАО “Цвет” (бывшее Дзержинское ОКБ А), про-
должая выпуск существенно модернизированных хроматогра-
фов “Пвет-500М”, освоило изготовление приборов новой серии
“Цвет-800” и постепенно наращивает их производство. При мо-
дернизации хроматографов “Цвет-500М” и разработке серии
“Цвет-800” основные усилия были направлены на техническое
совершенствование и повышение надежности функциональных
систем управления режимом хроматографа и обработки хрома-
тографической информации на базе использования микропро-
цессорной техники и персональных компьютеров.
Развитие аналитических возможностей хроматографов про-
ведено в направлении расширения номенклатуры детекторов и
дополнительных аналитических устройств (приставок), в рав-
ной мере применимых к приборам обеих серий. При этом со-
хранена общая аналитическая база, что обеспечивает использо-
вание в хроматографах “Цвет-500М” и “Пвет-800” одинаковых
аналитических узлов и принадлежностей (дозаторов, колонок,
детекторов и приставок), а также создает удобство практиче-
ского совмещения в одной лаборатории старых и новых прибо-
ров.
Поскольку газовые хроматографы “Цвет” по-прежнему со-
ставляют значительную часть парка приборов, эксплуатирую-
щихся на предприятиях Российской Федерации и стран СНГ
и Балтии, представляется целесообразным подробно описать
хроматографы серии “Пвет-500М”, акцентируя внимание на
наиболее существенных изменениях и дополнениях в их кон-
струкции, реализованных в модели 560-02, а также познакомить
читателя с принципиальными отличиями хроматографов “Цвет-
800” по состоянию на конец 1997 г.*
*Подробное описание блоков, не входящих в состав современного ис-
полнения хроматографов “Цвет-500М”, читатель может найти в [79].
130
Кроме хроматографов универсального назначения ОАО
“Цвет” разработало специализированный капиллярный хрома-
тограф с оригинальной системой криофокусирования “Цвет-
700КФ”, который может найти эффективное применение при ана-
лизе многокомпонентных смесей сложного состава, а также мо-
жет использоваться в качестве хроматографического модуля в
составе хромато-масс-спектрометрической системы.
Для обеспечения удобства эксплуатации любого хромато-
графического оборудования ОАО “Цвет” производит вспомо-
гательную аппаратуру (например, генератор водорода для пи-
тания пламенных детекторов, автономные измерители расходов
газов, средства метрологического обеспечения, микрошприцы
и т.п.). Аналитические принадлежности хроматографов и до-
полнительно^оборудование описаны в отдельном разделе (см.
раздел П.2.4)
Следует отметить, что приводимое здесь описание газохро-
матографического оборудования имеет целью общее ознакомле-
ние с назначением, устройством и некоторыми особенностями
приборов, но не заменяет заводских инструкций и рекоменда-
ций для практической эксплуатации того или иного изделия.
Вследствие непрерывного совершенствования и изменения кон-
струкции приборов приводимые сведения могут несколько отли-
чаться от фактического состояния поставляемой аппаратуры.
II.2.2. Хроматографы серии “Цвет-500М”
II.2.2.1. Общие сведения
Универсальные газовые лабораторные хроматографы “Пвет-
500М” построены по блочно-модульному принципу, позволяю-
щему различные по аналитическим возможностям модели (ис-
полнения) составлять из набора блоков и узлов (модулей), вы-
полняющих преимущественно одну определенную функцию.
Модули, как правило, функционально взаимозаменяемы и не
требуют дополнительной настройки при введении в состав хро-
матографов, что позволяет наиболее рационально и без не-
оправданной избыточности формировать рабочие модели в со-
ответствии с характером аналитической задачи. При этом но-
менклатура и значения выходных характеристик хроматографа
определяются типовыми техническими параметрами составля-
ющих блоков и узлов.
Схема П.1 поясняет блочно-модульный принцип построения
хроматографов из блоков и узлов, выполняющих следующие
131
Управление режимом 1
БПГ-1Б Формирование БПГ-2 всех газовых БПГ-167 потоков БПГ-175 Измерение расходов БУ-125 Установка, регул иро- ПУ-013* вание, индикация температур и времен- ных интервалов
Детектирование анализируемых веществ III «9
ПИД органических ДТП любых ДПР обладающих (ЭЗД) сродством к элек- трону ТИД фосфор- и азот- содержащих ор- ганических ПФД серо- и фосфор- содержащих ор- ганических
1
Термостатирование. Дозирование. II Подготовка пробы. Разделение
БА-95 БА-109 БА-121 Аналитический блок (термостат)
УК-84 Устройство термостатирования при отрицательных температу- рах
КН Готовые несадочные колон- ки (стальные, стеклянные)
кк Готовые квврцевые капилляр- ные колонки Автоматическое введение
БДГ-115 газовых проб
Автосамплер жидких проб Ручное введение
КД-234 газовых проб
БДГО-171 (с обратной продувкой)
МДЖ-163 жидких проб
“Фазе" Парофазное дозирование
УАТМ-133 Анализ газов в трвнсформвтор- ном масле
УКФ-1 Криофокусирующве устройство
УВП-1 Устройство выдувания и накоп- ления
УО-89 Накопление и дозирование при- месей
Усиление, измерение, обработка сигналов детекторов IY
БИД-36 (БИД-39) Питание и усиление сигналов ионизационных детекторов
БПД-56 Питание и усиление сигналов ДТП
CAA-D6-D3 (с принтером) Измерение параметров пиков, градуировка, расчет концентраций,
АПК-01 (с компьютером) регистрация хроматограмм, распечатка результатов
‘Только для исполнения 560-02.
Схема П.1. Блочно-функциональная схема газовых хромато-
графов “Цвет”.
основные функции: управление рабочим режимом хроматогра-
фа — формирование и измерение газовых потоков, задание,
регулирование, индикация температур во всех термостатируе-
мых зонах, включение дискретных устройств по временной про-
грамме (I), подготовка и введение анализируемых проб, хро-
матографическое разделение, термостатирование колонок, до-
заторов и детекторов (II), детектирование анализируемых ве-
ществ различной природы (III), усиление, измерение, преобра-
зование, обработка сигналов детекторов и регистрация резуль-
татов анализа (IV). В группу II условно включено разнообраз-
132
ное дополнительное аналитическое оборудование для предва-
рительного преобразования объектов анализа.
В настоящее время ОАО “Цвет” отказалось от жесткого фор-
мирования состава моделей и предоставило возможность при-
обретать приборы со свободным набором блоков, а также от-
дельные блоки, узлы и принадлежности из состава хроматогра-
фов “Пвет-500М” для расширения, обновления или замены име-
ющейся хроматографической аппаратуры.
Минимальный набор основных блоков, который рассматри-
вается как индивидуальный экземпляр хроматографа и полу-
чает паспорт при выпуске, включает аналитический блок с од-
ним пламенно-ионизационным детектором, блок или пульт упра-
вления, усилитель для ионизационных детекторов и газовый
блок, а также соответствующий набор принадлежностей для
работы хроматографа и комплект эксплуатационной докумен-
тации.
Наиболее полная модель соединяет в себе все возможности
серии — в ее состав входят все типы детекторов. Наличие двух
систем обработки САА-06-03 или двухканальной системы АПК-
01 с ПЭВМ позволяет одновременно и независимо эксплуатиро-
вать два детектора, что создает дополнительные возможности
идентификации анализируемых соединений и проведения слож-
ных анализов.
Все хроматографы комплектуются стальными и стеклянными
насадочными колонками длиной 1, 2 и 3 м с внутренним диаме-
тром 3 мм (оболочки). Поставка заполненных насадочных коло-
нок и готовых к употреблению кварцевых капиллярных колонок
вместе с комплектом монтажных принадлежностей для работы
с обычной ячейкой ПИД или электронозахватным микродетек-
тором осуществляется по специальному заказу с учетом харак-
тера конкретной задачи.
Аналитический блок, основу которого составляет термостат
колонок, в сочетании с блоком (пультом) управления позволяет
работать с температурами в интервале от —100 до 400 °C как
в изотермическом режиме, так и при программировании темпе-
ратуры. Работу в области температур ниже комнатной обеспе-
чивает криогенное устройство, хладоагентом является жидкий
азот.
Дозирование жидких проб осуществляется микрошприцами
через два идентичных испарителя, термостатируемых незави-
симо в интервале температур от 50 до 450 °C блоком (пуль-
том) управления. Он же управляет температурой детекторов по
теплопроводности и электронозахватного, устанавливаемых на
термостат колонок в индивидуальных терморегулируемых ко-
133
Рис. 11.43. Газовый хроматограф “Цвет-560-02” (система обра-
ботки сигналов на фотографии не представлена).
жух ах, а также температурой переходной камеры, на которую
устанавливаются все пламенные детекторы (ПИД, ТИД, ПФД).
Питание мостовой схемы и усиление сигнала ДТП осуще-
ствляется блоком БПД-56. Усиление сигналов ионизационных
детекторов (включая ПФД) производится блоком БИД-36.
В аналитический блок может быть установлен термостати-
руемый до 150 °C кран-дозатор газовых проб. Он либо входит в
состав устройства дозирования газов и обогащения примесей и
допускает только ручную работу, либо устанавливается в виде
отдельного блока БДГ-115 и имеет пневматический привод для
работы в автоматическом режиме. Возможно использование ме-
ханических дозаторов жидкости.
Установка и стабилизация расходов газа-носителя в двух не-
зависимых линиях (в хроматографах “Цвет-500М” сохранена
традиционная двухколоночная схема), водорода и воздуха вы-
полняются блоками БПГ-1Б, БПГ-2 или БПГ-167, БПГ-175.
Контроль за фактическими значениями температур всех тер-
мостатируемых зон и расходов газа-носителя и водорода про-
изводится в цифровой форме.
Автоматизированная обработка сигналов детекторов (хрома-
тограммы) выполняется специализированной микропроцессор-
134
ной системой САА-06-03 или с помощью аппаратно-програм-
много комплекса АПК-01 (соединенного с ПЭВМ) по жестким
алгоритмам градуировки и расчета концентраций. Представле-
ние выходной информации (результатов анализа) возможно в
двух вариантах: либо в виде аналоговой хроматограммы, запи-
сываемой с помощью автоматического потенциометра, напри-
мер КСП-4 (в состав хроматографа не входит), подключаемого
к разъемам на блоках БИД-36 или БПД-56, либо в дискретной
форме на экранах САА-06-03 или ПЭВМ и с помощью принтера,
входящего в состав САА-06-03 или в состав ПЭВМ.
Особенностью последней из модернизированных моделей се-
рии “Пвет-500М” — исполнения 560-02 — является использо-
вание компактного пульта управления ПУ-013 (вместо БУ-125),
задающего и регистрирующего температурный режим хромато-
графа, газового блока БПГ-175 с возможностью цифровой инди-
кации расходов всех газов и давления газа-носителя перед ко-
лонкой, а также согласованного с пультом управления аналити-
ческого блока БА-121. Внешний вид хроматографа исполнения
560-02 приведен на рис. 11.43 (без системы обработки).
Подробное описание хроматографов “Цвет-500М” целесооб-
разно построить по функционально-модульному принципу, рас-
смотрев последовательно конструкцию и работу отдельных
функциональных систем и блоков.
П.2.2.2. Аналитический блок
Узлы аналитической системы хроматографа, выполняющие
функции дозирования проб, газохроматографического разде-
ления и детектирования разделенных веществ, сосредоточены
в аналитическом блоке. Основу конструкции аналитического
блока составляет термостат колонок, на котором размещены до-
заторы и детекторы со своими элементами термостатирования.
К аналитическому блоку присоединяется в виде приставок и до-
полнительное аналитическое оборудование для предваритель-
ной подготовки пробы.
Собственно термостат колонок представляет собой камеру
размерами 380 х 275 х 210 мм с принудительной циркуляцией
воздуха. По сторонам камеры в вертикальной плоскости уста-
навливаются две колонки, концы которых непосредственно при-
соединяются к испарителям (или газовому дозатору) и детек-
торам. Температура в камере может поддерживаться от 50 до
400°C со стабильностью на уровне ±(0,05-0,15)°С в зависимо-
сти от заданной температуры. Погрешность установки темпе-
ратуры не превышает ±2,5% от задаваемого значения, а макси-
135
мальное отклонение от средней температуры при многократном
задании (воспроизводимость) не выходит за пределы ±0,25°C.
Линейное программирование температуры возможно во всем
диапазоне со скоростью от 1 до 25°С/мин с относительным от-
клонением скорости не более ±5%. Возвращение к начальной
температуре после цикла программирования осуществляется
автоматически. Для исключения перегрева термостата при
температурах, близких к комнатной, предусмотрено поступле-
ние в термостат холодного воздуха путем автоматического при-
открывания двери. Для предотвращения аварийного перегрева
используется предохранитель, отключающий нагреватель при
температуре около 420°С.
Работа с температурами ниже комнатной (от —100 °C) дости-
гается подачей в термостат жидкого азота через электромаг-
нитный клапан, устанавливаемый на аналитический блок при
необходимости. В верхней части аналитического блока (над
камерой термостата колонок) расположены два идентичных ис-
парителя, один пламенно-ионизационный детектор и свободное
гнездо для другого пламенного детектора, детектор по тепло-
проводности или электронозахватный (или свободное гнездо
для их установки), клеммные колодки для электрических со-
единений испарителей и детекторов, трубки для подвода газа-
носителя, водорода и воздуха к испарителям и пламенным де-
текторам. На задней стенке аналитического блока находятся
разъемы для кабельных соединений с другими блоками хрома-
тографа, штуцеры для подключения газовых линий и сетевой
шнур.
На передней панели аналитического блока расположен кно-
почный выключатель СЕТЬ. В модификации БА-121 (исполне-
ние 560-02), кроме того, имеются клавиша АНАЛИЗ для вклю-
чения цикла программирования температуры и индикаторы ра-
боты нагревателей.
Каждый испаритель представляет собой металлический
блок, обогреваемый плоскими нагревателями, содержащий ка-
меру испарения (рис. II.44), закрытую сверху термостойким ре-
зиновым уплотнением. Испарение жидкой пробы, введенной ми-
крошприцем, происходит внутри удлиненной начальной части
насадочной колонки, вводимой в испаритель до упора. Для ра-
боты с капиллярными колонками к штуцеру испарителя при-
соединяется специальная камера для деления пробы с потоком
газа-носителя, при этом начало капиллярной колонки вводится
в камеру, как показано на рис. 11.45. Выходная трубка сброса
камеры делителя пробы выводится из термостата и присоеди-
няется к предварительно установленному на его правой стенке
136
Рис. II.44. Испаритель жидких проб для насадочных колонок.
1 — термопреобразователь ЭСП-01; 2 — нагреватель; 3 — втулка;
4 — гайка; 5 — прокладка из термостойкой резины; 6 — шайба; 7 —
колонка; 8 — трубка подвода газа-носителя; 9 — слой сорбента.
Дросселю, управляющему соотношением деления потока газа-
носителя с пробой. Для сохранения оптимального для детек-
тора расхода газа-носителя при работе с капиллярной колонкой
с помощью специального тройника через свободный испаритель
осуществляется дополнительный поддув газа-носителя в детек-
тор.
Кран-дозатор для введения газовых проб при необходимо-
сти монтируется на правой стенке аналитического блока таким
137
Рис. 11.45. Ввод пробы в капиллярную колонку.
а — монтаж капиллярной колонки в термостате хроматографа: 1 —
штуцер пламенно-ионизационного детектора; 2 — камера термостата
(верхняя стенка); 3 — винт М4; 4, 5 — скобы; 5 — вкладыш испари-
теля; 6— графитовая конусная муфта; 7 — конусная муфта; 8 — гайка;
10 — капиллярная колонка;
6 — вкладыш испарителя для капиллярных колонок: 1 — линия
подвода газа-носителя; 2 — линия сброса; 3 — капиллярная колонка.
образом, что дозирование осуществляется только в правую ко-
лонку. При этом газовая проба подается в колонку непосред-
ственно, минуя испаритель. Положение крана в блоке дозирова-
ния газов БДГ-115 вместе с пневмоприводом и клапанами пока-
зано на рис. II.46. На передней панели блока имеется тумблер
ручного включения привода крана. Автоматическое управле-
ние краном по временной программе осуществляется системой
САА-06-03 или пультом управления ПУ-013.
Газовый дозатор представляет собой кран (рис. II.47) с пово-
ротной головкой, на которой установлена сменная доза, проду-
ваемая в одном положении крана анализируемым газом, в дру-
гом — газом-носителем. Дозы объемом 0,125; 0,25; 0,5 и 1,0 см3
проградуированы с погрешностью не более ±4%, а доза объ-
емом 2,0 см3 — ±2%.
138
5
6 7
Рис. П.46. Блок дозирования газов.
1 — газовый кран-дозатор; 2 — сменная доза; 3 — переходная
трубка кран—колонка; 4, 9 — элемент термостатирования; 5 — пнев-
моцилиндр; 6 — поршень; 7 — пневмопривод, 8 — электропневмопре-
образователь.
На задней стенке блока БДГ-115 имеются штуцеры для ввода
газа-носителя, анализируемого газа и воздуха для управления
краном-дозатором. Термостат крана соединяется кабелем с со-
ответствующей клеммной колодкой на аналитическом блоке для
включения в канал термостатирования.
Пламенные детекторы различных типов устанавливаются на
общее основание (переходную камеру), термостатируемое в ин-
тервале температур от 50 до 400 °C. При этом возможны сле-
дующие сочетания: ПИД—-ПИЛ, ПИД—ТИД и ПИД—ПФД.
Обязательная в каждой модели ячейка ПИД находится справа,
на посадочное место слева монтируется вторая ячейка ПИД или
ТИД, или ПФД. При использовании двух ячеек ПИД сигнал
может быть снят с соединенных между собой электродов детек-
торов или с каждого детектора отдельно. На аналитический
блок может быть установлен, кроме того, или ДТП, или ЭЗД
(ДПР) со своим термостатом.
Колонки всех типов при соединении со штуцерами дозато-
ров и детекторов уплотняются с помощью конусных графито-
139
Рис. 11.47. Кран-дозатор.
1 — поворотная головка; 2 — ушко; 3 — фланец; 4 — прокладка;
5 — корпус; 6 -— пружина; 7 — вал; 8, 11 —- штифты; 9 — кольцо;
10 — гайка; 12 — доза.
вых муфт, обеспечивающих герметичность во всем интервале
температур. Графитовые прокладки не требуют значительных
усилий при затягивании, хорошо сохраняют герметичность, но,
как правило, допускают лишь одноразовое использование, т.е.
подлежат замене при перестановке колонок. При работе в ре-
жиме программирования температуры может быть рекомендо-
вано легкое подтягивание соединений колонок между циклами
программирования, если разница между начальной и конечной
температурами достаточно велика.
П.2.2.3. Блок управления
В хроматографах серии “Цвет-500М” (кроме исполнения 560-
02) функции управления температурами всех термостатируе-
мых зон и цифрового контроля температуры и расходов газов
(кроме воздуха) выполняет микропроцессорный блок БУ-125.
Пять независимых изотермических каналов предназначены для
термостатирования испарителей, детекторов и переходной ка-
меры в диапазоне температур от 50 до 450 °C и крана-дозато-
ра — от 50 до 150 °C. При дискретности задания 1°С погреш-
ность установки составляет ±2,5% при случайной составляю-
щей погрешности установки не более ±1°С. Термостатирова-
ние колонок осуществляется программным каналом со следую-
щими характеристиками: задание начальной температуры от
140
— 100 до 400 °C и конечной от 0 до 400 °C с дискретностью 1°С;
время выдержки начальной и конечной температур от 0 до 9999
секунд с дискретностью 1 секунда; скорость повышения темпе-
ратуры от 1 до 25°С/мин с дискретностью задания 1°С/мин.
Режим программирования может включать два участка повы-
шения температуры с различными скоростями. В качестве дат-
чиков температур применены элементы платиновых или пле-
ночных термометров сопротивления (ТСП или ТРП, гр.ЮОП).
Силовыми элементами, непосредственно управляющими мощ-
ностью нагревателей, являются оптронные тиристоры, находя-
щиеся в аналитическом блоке.
Общение аналитика с блоком управления с целью задания
и контроля режима хроматографа происходит с помощью кла-
вишной панели, содержащей цифровые и командные (адресные)
клавиши, и многоразрядного цифрового индикатора, отобража-
ющего заданные и фактические значения температур, времени
в цикле программирования температуры колонок, а также теку-
щие значения расходов газов непосредственно в °C, секундах и
см3/мин. Кроме того, имеющиеся на панели световые индика-
торы позволяют оператору контролировать состояние блока и
облегчают работу с ним. Индикаторы разделены на ряд полей
по функциональному признаку: общее состояние системы, тем-
пература колонок, температуры прочих термостатируемых зон,
измерение расходов, сигналы управления.
Общее состояние системы отражается тремя индикаторами:
тесты, авария и готовность. Свечение индикатора ГОТОВ-
НОСТЬ свидетельствует о том, что температура колонок не
отличается от заданного значения и блоком управления мо-
жет быть выполнена команда пуска режима программирования
температуры. При свечении индикатора АВАРИЯ оператор
должен предпринять предписанные инструкцией на БУ-125 дей-
ствия. Как правило, они состоят во введении нулевых значений
во все задаваемые параметры (обнуление) или подключении к
разъему ТСП на задней стенке блока всех термометров сопро-
тивления и устранении их разрывов. В ситуации “авария” блок
управления не выполняет рабочих функций.
В состоянии ТЕСТЫ, вызываемом оператором, по опреде-
ленным командам выполняется тестирование электронных под-
систем блока управления, а результаты отображаются на ци-
фровом индикаторе.
Кроме электронных подсистем в составе БУ-125 имеются
три датчика расходов газов термоанемометрического типа (см.
Раздел П.1.1): два для индикации расходов газа-носителя (ка-
ждый из них для азота и гелия) и один для индикации рас-
141
хода водорода. Диапазон регистрируемых расходов — от 0 до
100 см3/мин с дискретностью 0,1 см3/мин Показания расхо-
домеров не зависят от давления газа в линии, так как датчики
ориентированы на измерение массовой скорости газа. Расходо-
меры подключаются к соответствующим газовым потокам шту-
церами на задней стенке блока в разрыве линий между газовым
и аналитическим блоками. Подстройка показаний расходоме-
ров в БУ-125 осуществляется с помощью резисторов, выведен-
ных под шлиц под верхней крышкой блока. Кроме того, уста-
новить нулевые значения сигналов расходомеров можно про-
граммным путем с помощью соответствующих команд опера-
тора, вводимых клавишами на панели блока. Пользуясь этим
приемом, можно “обнулить” показания расходомеров в уста
новившемся рабочем режиме расходов, после чего на БУ-125
будут регистрироваться только отклонения от расходов газов,
условно принятых за нулевые значения.
В хроматографах исполнения 560-02 функции блока управле-
ния (кроме измерения и индицирования расходов газов) выпол-
няет компактный переносной пульт ПУ-013, соединенный кабе-
лем с разъемом “ЭВМ” аналитического блока ВА-121. Вместе
с контроллером, находящимся в аналитическом блоке, пульт
предназначен для задания и регулирования температурного ре-
жима колонок, испарителей, переходной камеры и детектора, а
также для управления по временной программе дискретными
устройствами (например, автоматическим дозатором). Термо-
статирование газового крана может осуществляться по свобод-
ному каналу (например, по каналу испарителя 1 или 2).
Пульт ПУ-013 представляет собой сочетание жидкокристал-
лического экрана с клавишной панелью, соединяющей цифро-
вые и командные клавиши. Выводимая на экран информация
содержится на пяти листах.
Лист 1 используется для задания температуры в зонах (по-
строчно) детектора, испарителя 1, испарителя 2, переходной ка-
меры и колонки и содержит информацию о соответствующих
температурах и о состоянии каналов управления дискретными
устройствами. Нижняя строка информирует о времени анализа
от момента нажатия клавиши АНАЛИЗ и состоянии готовности
по температуре колонок или о текущей стадии режима програм-
мирования температуры.
Лист 2 содержит информацию о заданном режиме линейного
программирования температуры раздельно (построчно) по изо-
термическим ступеням и времени их выдержки, а также по ско-
рости роста температуры.
142
Листы 3, 4 и 5 предназначены для задания временных интер-
валов работы дискретных устройств.
Пульт включается клавишей СЕТЬ аналитического блока.
Пуск режима программирования температуры колонок, а также
исполнения программы работы дискретных устройств осуще-
ствляется клавишей АНАЛИЗ на аналитическом блоке только
при индицировании на пульте управления готовности началь-
ной температуры колонок. При ошибочном задании на пульте
значения, большего, чем максимальное для данного параметра
(температура детектора 350 °C, испарителей 450 °C, переход-
ной камеры и детектора 400°C, колонки 400°C), автоматически
устанавливается предельное значение.
Параметры точности задания температуры и стабильности
термостатирования не зависят от варианта задания (блоком или
пультом управления), однако надежность системы с использо-
ванием пульта управления существенно выше.
II.2.2.4. Детекторы
Пламенно-ионизационный детектор. Ячейка детектора
(рис. П.48) представляет собой цилиндрическую камеру из ста-
ли, состоящую из основания 1 и корпуса /. В основание вве-
дены трубки для питания водородом и воздухом. Центральный
канал основания служит для присоединения колонки, его верх-
няя часть оканчивается горелкой 5, установленной на керами-
ческом изоляторе. Через колодку 2 на горелку упругим кон-
тактом подается постоянное напряжение питания. В верхней
части ячейки на круглом керамическом изоляторе установлен
конический полый электрод-коллектор 3 с выводом для присо-
единения сигнального кабеля. Сверху корпус закрыт крышкой
с отверстием для выхода газов и продуктов горения. На кор-
пусе ячейки имеются винты для крепления центральной части
к основанию, изолятора электрода в корпусе и винт для земля-
ного провода. Зажигание пламени производится электрической
зажигалкой, которая вводится в детектор через крышку и по-
лый электрод. Питание детекторов осуществляется от общей
колодки ПИТАНИЕ ДЕТЕКТОРОВ на крышке термостата ко-
лонок. Сигнал подается на электрометрический усилитель тер-
мостойким и виброустойчивым коаксиальным кабелем.
Чувствительность к углеводородам в оптимальном режиме
работы может быть близка к 1 • 10~5 Кл/мг, что при минималь-
ном уровне шума не более 2 10~14 А соответствует пределу
Детектирования на уровне 4 -10-9 мг/с.
143
Рис. 11.48. Пламенно-ионизационный
детектор.
I — основание; 2 — колодка; 3 — элек-
трод; 4 — корпус; 5 — горелка.
Соответствующая минимальная определяемая концентрация
в детекторе при скорости газа-носителя 0,5 мл/с составляет
около 8 • 10~9 мг/мл.
Линейный динамический диапазон детектора доходит до 107,
т. е. искажения сигнала вследствие нелинейности начинаются
при концентрации веществав детекторе, составляющей несколь-
ко процентов.
Максимальная чувствительность и наименьший предел де-
тектирования достигаются при определенном соотношении рас-
ходов газа-носителя, водорода и воздуха, которое ориентиро-
вочно составляет 1:1: 10. Однако в случае количествен-
ных определений следует экспериментально установить зависи-
мость чувствительности данного экземпляра детектора от соот-
ношения расходов газов, ориентируясь на конкретное значение
расхода газа-носителя, продиктованное условиями аналитиче-
ской задачи (см. лабораторную работу 3). Это необходимо для
определения той области расходов, где их случайные колебания
не ведут к заметному изменению чувствительности и поэтому не
вносят дополнительной погрешности.
144
Для работы в режиме программирования температуры це-
лесообразно использовать дифференциальный детектор, состо-
ящий из двух ячеек, включенных по компенсационной схеме с
противоположной поляризацией горелок и общей цепью коллек-
торных электродов. Если это необходимо для анализа, сигнал
каждой ячейки можно регистрировать независимо.
Начиная с исполнения 560-02, в хроматографах “Пвет-500М”
(а также “Цвет-800”) введена измененная конструкция детек-
тора. Вывод сигнала коллекторного электрода произведен че-
рез вваренную в крышку детектора трубку из нержавеющей
стали с высокоомным разъемом на конце трубки. Это позво-
лило несколько снизить уровень шумов нулевого сигнала детек-
тора, благодаря экранированию сигнального провода, умень-
шению механических вибраций и снижению температуры высо-
коомного разъема.
Термоионный детектор. Конструкция детектора показана
на рис. 11.49. На основание 2 со штуцером для ввода хромато-
графической колонки установлен корпус 1 детектора с крышкой
3. Внутри расположены основные элементы детектора: коллек-
торный электрод 4 и поляризующий электрод 5 на керамиче-
ских изоляторах 6 и 7, горелка 8 с наконечником (таблеткой) 9
из соли щелочного металла (бромистый цезий) и фторопласто-
вой втулкой 10. которая предохраняет таблетку от излишнего
нагревания. Одновременно втулка служит диффузором для по-
тока воздуха, подаваемого в отверстие а; через отверстие б в
детектор вводится водород.
Поляризующее питание подается от общей колодки в соответ-
ствии с ее маркировкой, а коллекторный электрод высокоомным
кабелем соединяется с входом усилителя БИД-36.
Термоионный детектор рассчитан на два режима работы: для
селективного детектирования фосфорсодержащих органических
соединений и для детектирования азотсодержащих органиче-
ских веществ. Режимы отличаются расходами газов, которые
влияют на рабочую температуру солевой таблетки. Как пра-
вило, чувствительность детектора по фосфору в метафосе выше
1-10“2 А-с/мгипо азоту в азобензоле выше 5-10“4 А-с/мгпри се-
лективности по отношению к нормальным углеводородам около
104. Уровень шумов термоионного детектора примерно на по-
рядок выше, чем для ПИД, что позволяет достигать пределов
детектирования 1 • 10-1° мг/см3 по фосфору и 1,5 • 10~9 мг/см3 по
азоту. Наиболее низкий предел обнаружения фосфорорганиче-
ских веществ достигается при расходах азота около 30 см3/мин,
водорода 15-16 см3/мин, воздуха 160-180 см3/мин; для азотор-
ганических веществ при расходах азота около 35 см3/мин, во-
145
3
Рис. 11.49. Термоионный детектор.
а — отверстие для подачи воздуха, б — отверстие для подачи во-
дорода. 1 — корпус; 2 — основание; 3 — крышка; 4 — коллекторный
электрод; 5 — поляризующий электрод; 6,1 — изоляторы; 8 — го-
релка; 9 — солевой наконечник; 10 — втулка.
дорода 14-15 см3/мин, воздуха 160-180 см3/мин, при этом тем-
пература переходной камеры должна быть выше 300 °C.
Чувствительность детектора можно повысить умеренным
увеличением расхода водорода (ростом температуры солевой
таблетки), однако при этом резко возрастают флуктуации нуле-
вого сигнала и уменьшается срок эксплуатации таблетки вслед-
ствие более быстрого ее истощения. Оптимальные условия экс-
плуатации детектора заключаются в обеспечении высокой ста-
бильности расходов водорода и воздуха и более высокой тем-
146
пературы основания детектора по сравнению с температурой
колонки.
Чувствительность детектора может снижаться из-за налета
соли на электродах. Лля восстановления его характеристик
следует промывать детали детектора (кроме таблетки) водой
и затем спиртом.
Детектор постоянной скорости рекомбинации. В хрома-
тографах “11вет-500М” эксплуатируется оригинальная система
детектирования по захвату электронов, основное отличие кото-
рой от известных состоит в том, что детектор работает в режиме
постоянного ионного тока.
Как указывалось выше, в ионизационных детекторах в лю-
бой момент времени сумма скоростей образования, рекомбина-
ции и сбора заряженных частиц равна нулю. В общем случае
появление отрицательных ионов, вызванное введением в ЭЗД
анализируемых веществ, должно приводить к возрастанию ско-
рости рекомбинации (что имеет место в “классическом” вари-
анте ЭЗД). Однако при искусственно поддерживаемой постоян-
ной силе тока детектора скорость рекомбинации должна также
оставаться постоянной, что возможно лишь при соответству-
ющем росте напряженности поля, препятствующем рекомбина-
ции. Увеличение напряженности поля происходит за счет воз-
растания напряжения на электродах детектора. Таким образом,
полезным сигналом детектора является увеличение напряжения
на детекторе, однозначно связанное с количеством вещества, по-
ступающим в детектор. В соответствии с принципом работы
этот детектор получил название детектора постоянной скоро-
сти рекомбинации (ДПР).
Оба варианта детектора (“классический” ЭЗД и ДПР) в ко-
нечном счете имеют общий механизм образования сигнала, сво-
дящийся к уменьшению электрической проводимости (увеличе-
нию сопротивления) газового промежутка между электродами
детектора за счет связывания свободных электронов молеку-
лами электроноакцепторных веществ. При этом в ЭЗД фик-
сируется уменьшение силы тока при постоянном напряжении,
а в ДПР — увеличение разности потенциалов на электродах
при постоянной силе тока детектора. Вместе с тем детектор по-
стоянной скорости рекомбинации обладает рядом преимуществ
перед классическим вариантом ЭЗД, среди которых следует на-
звать в первую очередь некоторое расширение линейного ди-
намического диапазона по сравнению с той же конструкцией в
режиме измерения силы тока. Это достигается как за счет уве-
личения верхней границы Л ДД, так и за счет снижения предела
Детектирования. Линейный динамический диапазон ДПР соста-
147
вляет IO3- 1О4. Весьма важно также, что повышение напряжен-
ности поля при введении анализируемого вещества в ДПР пре-
пятствует образованию объемного заряда и устраняет влияние
контактной разности потенциалов на процессы сбора заряжен-
ных частиц, тем самым обеспечивая более устойчивую работу
детектора и отсутствие искажений сигнала.
В состав ДПР входят высокотемпературная камера ВК, явля-
ющаяся собственно ячейкой детектора, к которой присоединя-
ется выход колонки, и выносной блок ВБ, содержащий сопроти-
вления Ri, Н2 и R„, участвующие в формировании электриче-
ского сигнала. Блок-схема, поясняющая включение детектора
и измерение сигнала, приведена на рис. II.50. Блок питания БП
(БИД-36) осуществляет подачу постоянного отрицательного на-
пряжения на выносной блок, который формирует стабильный
электрический ток в пределах (1,5 — 2,0) • 10-9 А. При изме-
нении концентрации анализируемого вещества в ячейке детек-
тора ВК изменяется электрическое сопротивление, и на входе
резисторов Ri и R2 возникает соответствующее изменение паде-
ния напряжения. Резисторы использованы для преобразования
напряжения в электрический ток, который является сигналом
детектора и измеряется с помощью электрометрического уси-
лителя. Сила тока детектора всегда должна быть значительно
меньше силы тока выносного блока. Для этого при определе-
нии малых концентраций анализируемых веществ используется
резистор Ri, включенный в цепь с выходом СИГНАЛ 1, а при
определении относительно высоких концентраций следует поль-
зоваться резистором R2, т. е. разъемом СИГНАЛ 2.
Рис. 11.50. Схема включения детектора постоянной скорости
рекомбинации.
Конструкция ячейки ДПР показана на рис. 11.51. Основу кон-
струкции составляет камера 1 из высокотемпературного изоля-
тора с отверстием для ввода газа-носителя из колонки по трубке
148
10. К внутренней цилиндрической стенке камеры прикреплен
пружинным кольцом 6 источник ионизирующего излучения 63Ni
(7). Торцы камеры закрыты сетками, одна из которых (5) со-
единена с источником и является катодом, а другая (5) прижата
к корпусу 9 и служит анодом. Газ выводится из камеры через
трубку Jt. Регистрация сигнала производится через коаксиаль-
ный кабель 12 и разъем 13, через который ВК соединяется кабе-
лем с выносным блоком. Геометрический объем камеры, в кото-
ром происходят процессы ионизации и формирования сигнала,
не превышает 0,6 см3. ДПР эксплуатируется без поддува до-
полнительным потоком газа-носителя. Ячейка детектора оплом-
бирована и не подлежит разборке в процессе эксплуатации у
потребителя. ВК помещена в собственный термостат контакт-
ного типа, его максимальная температура ограничена 350 ° С, а
в цепи нагревателей имеется соответствующий термопредохра-
нитель. Устанавливается на крышке термостата колонок вместо
Детектора по теплопроводности. Провода термостата ДПР при-
соединяются к клеммной колодке термостата ДТП. Электри-
ческое питание ВБ осуществляется от общей колодки питания
ионизационных детекторов в соответствии с маркировкой. ВК и
ВБ соединяются между собой коротким кабелем, а ВБ соединен
высокоомным сигнальным кабелем с электрометром БИД-36.
149
При работе с детектором следует иметь в виду, что его харак-
теристики выполняются только с чистым бескислородным азо-
том или аргоном с 5% метана. Чувствительность ДПР обратно
пропорциональна расходу газа-носителя. Селективность к на-
сыщенным галогенсодержащим углеводородам составляет не
менее 105. Рекомендуется поддерживать температуру ВК при-
мерно на 30-50 °C выше, чем температура колонки, чтобы ис-
ключить возможность конденсации веществ в детекторе и за-
грязнения радиоактивного источника 63Ni. Существенное значе-
ние для достижения высокой чувствительности и низкого пре-
дела детектирования ДПР имеет тщательная тренировка ана-
литической колонки до получения уровня фонового сигнала не
более (3 —5) • 10-11 А. В оптимальном режиме работы достижим
предел детектирования на уровне 4 • 10-11 мг/см3 по контроль-
ному веществу линдану (7-гексахлорциклогексан).
В связи с использованием в ДПР радиоактивного источника
должны строго соблюдаться положения инструкции “Основные
санитарные правила работы с радиоактивными веществами и
другими источниками ионизирующих излучений” (ОСП 72-80).
Пламенно-фотометрический детектор. Состоит из трех
функциональных частей (рис. II.52): ячейки детектора, пламя
в которой является источником эмиссионного излучения, ин-
терференционного светофильтра и преобразователя эмиссион-
ного излучения в электрический сигнал. Ячейка детектора со-
стоит из основания 1 со штуцером для присоединения колонки
и корпуса 5, в котором поддерживается водородное пламя. Газ-
носитель и водород подаются через центральный канал основа-
ния. Верхняя часть основания с втулкой 2 образует камеру сго-
рания а эмиссионная камера ограничена корпусом 5, заглушкой
3 и защитным стеклом 7. Пламя зажигается через отверстие в
корпусе, закрываемое пробкой Воздух подается между втул
ками 2 и 6, газы выводятся через канал в заглушке 3.
Интерференционный светофильтр 8 в оправке вставлен во
втулку 9, которая имеет резьбовое соединение с корпусом ячей-
ки. Преобразователь эмиссионного излучения состоит из фото-
умножителя 11 и блока делителя напряжения 12, заключенных
в кожух 10, который соединяется по резьбе с втулкой 9 и ячей-
кой детектора. На блоке делителя установлены высокоомные
разъемы для подачи электрического питания на делитель и для
вывода сигнала детектора.
Конструкция детектора одноканальная: селективность детек-
тора к серо- или фосфорсодержащим соединениям обеспечива-
ется применением сменных интерференционных светофильтров
с полосой пропускания 394 или 526 нм соответственно. Визу-
150
ально фильтр с полосой 394 нм пропускает фиолетовые лучи,
а фильтр с полосой 526 нм — зеленые. При выпуске хрома-
тографа в ПФД устанавливается фильтр на серу, а фильтр на
фосфор входит в комплект принадлежностей к детектору.
ПФД устанавливается потребителем на аналитический блок
вместо левой ячейки ПИД. Электрическое питание (+600 В)
подается кабелем к разъему ПИТАНИЕ от общей колодки пи-
тания ионизационных детекторов. Сигнал детектора подается
на вход электрометра высокоомным кабелем от разъема СИГ-
НАЛ. Трубки для подачи водорода и воздуха в ПФД подклю-
чаются к соответствующим газовым линиям аналитического
блока. Любое открывание пробки (например, для зажигания
пламени) следует делать только при отключении напряжения
питания детектора во избежание необратимой засветки фото-
умножителя.
Оптимальный режим газового питания ПФД, при котором
достигается наибольшее отношение сигнал/шум, следующий:
расход водорода 65 ± 5, воздуха 100-110, газа-носителя (45 ±
5) см3/мин. При условии низких значений шумов нулевого сиг-
нала (3 • 10-12 А) достижим предел детектирования (1,5 — 2,5) х
х Ю~9 мг/см3 серы и фосфора в контрольном веществе метафосе.
Для ПФД характерна логарифмическая зависимость сигнала от
концентрации серосодержащих органических веществ в диапа-
зоне около 103. Диапазон линейности для фосфорсодержащих
органических соединений составляет не менее 103 от предела
детектирования.
Питание и усиление сигналов ионизационных детекто-
ров. Электрическое питание ионизационных детекторов осу-
ществляется блоком ионизационного детектирования БИД-36,
в состав которого входит источник стабилизированного посто-
151
янного напряжения. На выходе источника формируются напря-
жения +300 В и —300 В с погрешностью ±5% и пульсациями
не более 10 мВ. Включение источника производится нажатием
клавиши ЛИП на лицевой панели блока. Основной функцией
блока является усиление сигналов всех ионизационных детек-
торов. Электрический усилитель обеспечивает измерение силы
тока в диапазоне от 1 • 10~14 до 1,024 • 10~6 А. Измеряя падение
напряжения на одном из четырех переключаемых входных рези-
сторов с номиналами 107, 10s, 109 и 1010 Ом, находят силу тока.
Выходной делитель позволяет изменять сигнал, поступающий
на аналоговый регистратор (например, КСП-4), с коэффициен-
тами деления (множителями шкал), кратными 2": 1, 2, 4, 8, 16,
..., 1024. Относительная погрешность коэффициентов деления не
превышает ±2%.
Уровень флуктуационных шумов электрометра при измери-
тельном резисторе 1010 Ом составляет не более 100 мкВ, что со-
ответствует силе тока 10-1 А (1% от наиболее чувствительной
шкалы по току 10-12 А). Электрометр содержит схему подавле-
ния (компенсации) фонового тока на входе усилителя в пределах
от —9 10-1° А до +9 10-1° А. Установка тока компенсации про-
изводится ручкой многооборотного потенциометра на передней
панели блока, схема подавления фона включается и выключа-
ется клавишей КОМПЕНСАЦИЯ. Если возможностей компен-
сационной схемы электрометра не хватает для подавления фо-
нового сигнала, надо перейти к работе на менее чувствительных
шкалах (включить меньшее входное сопротивление).
Сигнал на вход усилителя подается от ионизационного де-
тектора высокоомным кабелем через разъем на задней панели
блока. На передней панели кроме переключателей измеритель-
ных сопротивлений и коэффициентов деления находятся кла-
виша включения сети с индикатором и клавиша переключения
полярности сигнала, подаваемого на аналоговый регистратор.
Одна десятая часть выходного сигнала (до 1 В) усилителя, не-
зависимо от установленного значения коэффициента выходного
делителя, подается на разъем САА, к которому следует под-
ключить систему автоматической обработки САА-06 или АПК-
01. Полный сигнал (до 10 В) усилителя можно снять с разъема
ВЫХОД, что иногда может оказаться полезным для регистра-
ции малых сигналов или при низком уровне флуктуационных
шумов.
В БИД-36 используется одна из двух альтернативных схем
усиления: схема с преобразованием постоянного сигнала в пе-
ременный ток, усилением и обратным преобразованием; в мо-
дернизированном варианте заменена на схему прямого усиле-
152
ния постоянного тока. Выходные характеристики блока в обоих
вариантах практически идентичны.
При отдельной поставке блока с рабочими принадлежно-
стями вне состава хроматографа он имеет обозначение БИЛ-39.
Детектор по теплопроводности. В четырехплечевом
(рис. 11.53) ДТП чувствительные элементы в гильзах из ковара
помещены в вертикальные каналы. Спираль элемента диаме-
тром 20 мкм изготовлена из вольфрама, выводы спирали гер-
метизированы стеклянной заливкой. Сопротивление спиралей
при температуре 20°C составляет 30±0,2 Ом.
Рис.II.53. Детектор по теплопроводности.
1 — чувствительный элемент; 2 — корпус;
S, 4 — выводы чувствительных элементов.
Чувствительность катарометра по пропану в гелии при силе
тока моста 220 мА и температуре блока детектора 100 °C не
ниже 5 103 мВ-см3/мг. Шумы выходного сигнала не превышают
10~3 мВ. В этих условиях предел детектирования составляет
2 10~6 мг/мл пропана в гелии
Ячейка детектора находится в собственном контактном тер-
мостате. Температура корпуса детектора поддерживается по-
стоянной на уровне сотых долей °C. Обычно температура де-
тектора в изотермическом режиме должна быть близка к темпе-
ратуре колонки, а при программировании — на уровне конечной
температуры колонок. С увеличением температуры детектора
приходится уменьшать силу тока моста для сохранения прие-
млемого уровня флуктуаций. Вместе с тем уменьшается раз-
ница температур спирали чувствительного элемента и корпуса,
что приводит к быстрому падению чувствительности. Увеличе-
ние силы тока моста до максимального значения, при котором
153
достигается наибольшая чувствительность, сопровождается не-
пропорционально большим ростом флуктуаций, вследствие чего
повышается предел детектирования и сокращается срок службы
элементов. Рекомендуемые значения рабочей силы тока не-
сколько ниже максимально допустимой, они выбраны в зави-
симости от температуры детектора и теплопроводности газа-
носителя.
Температура термостата
детектора, °C................
Сила тока моста, мА
рабочая :
гелий......................
азот......................
максимально допустимая :
гелий.................. . .
азот......................
50 150 300 400
320 220 110 90
160 120 80 70
350 250 120 100
200 150 90 80
Использование в качестве газа-носителя воздуха не рекоменду-
ется, особенно при повышенных температурах детектора.
Для устойчивой работы катарометра следует обеспечивать
равенство скоростей в сравнительной и измерительной линиях
газа-носителя. Включать питание мостовой схемы можно толь-
ко после подачи газа-носителя в детектор.
Питание мостовой схемы катарометра производится блоком
питания детектора БПД-56, позволяющим устанавливать ста-
билизированный ток от 10 до 390 мА цифровым путем с помо-
щью кодового переключателя на панели блока. Дискретность
задания силы тока 10 мА. Блок осуществляет усиление сигнала
ДТП в 10 раз, а также передачу его для регистрации на систему
обработки через разъем САА и через выходной делитель на-
пряжения на аналоговую запись через разъем РЕГИСТР. Вы-
ходной делитель позволяет изменять записываемый аналоговый
сигнал в диапазоне 1-2048 единиц двенадцатью ступенями, крат-
ными двум (последняя ступень не рекомендуется для использо-
вания). БПД-56 обеспечивает защиту катарометра от разба-
ланса мостовой схемы более ±1 В и от перегрева чувствитель-
ных элементов при отключении газа-носителя или аварийном
повышении температуры термостата детектора. В блоке име-
ются компенсационная схема установки нуля и переключатель
полярности регистрации аналогового сигнала.
II.2.2.5. Газовые блоки
Установка и стабилизация всех необходимых для работы хро-
матографа газовых потоков осуществляются или пневмоавтома-
тическими блоками БПГ-1Б и БПГ-2, описанными в [79], либо
154
разработанными позднее блоками БПГ-167 и БПГ-175. Но-
вые блоки (167 и 175) имеют одинаковое назначение и харак-
теристики, за исключением того, что в БПГ-175 предусмотрено
устройство для измерения и цифровой индикации расходов и
давлений газов.
Каждый блок формирует два независимых потока газа-носи-
теля с максимальным расходом не менее 100 мл/мин при давле-
нии на входе в блок 0,3 МПа, один поток водорода с расходом до
70 мл/мин при входном давлении 0,1 МПа и один поток воздуха
с расходом до 400 мл/мин при давлении 0,14 МПа. Изменение
расходов газов, вызванное изменением давления на входе в блок
на ±10%, не превышает ±1 мл/мин для водорода и газа-носителя
и ±10 мл/мин для воздуха. Изменение расхода газа-носителя,
вызванное изменением сопротивления колонки в 2 раза (напри-
мер, при программировании температуры), составляет не более
2% от начального значения. Влияние изменения внешней темпе-
ратуры на расход газов — не более ±10% на каждые 10 °C.
Для установки и стабилизации расходов используются тра-
диционные регуляторы прямого действия, описанные в разде-
ле II.1.1.
Формирование потоков осуществляется в две ступени: уста-
новка и стабилизация давления с помощью регуляторов давле-
ния и установка и поддержание стабильного расхода с помощью
регуляторов расхода или игольчатых дросселей.
Линия газа-носителя состоит из общего регулятора давле-
ния, манометра и двух регуляторов расхода, позволяющих раз-
дельно устанавливать расходы в двух каналах. Кроме того, по-
сле регулятора давления выведен третий канал для работы с
капиллярными колонками при использовании делителя потока
(паров пробы), требующего большого расхода газа-носителя
Линии водорода и воздуха включают регулятор давления,
манометр и игольчатый дроссель для плавной установки рас-
ходов. Характеристики дросселей для водорода и воздуха не-
сколько различаются проходным сечением в паре элементов
игла—втулка. Манометры и ручки регуляторов и дросселей
выведены на переднюю панель блоков. Подключение входных
и Межблочных газовых линий производится через штуцеры на
задней панели. Блоки имеют вертикальную компоновку и мо-
гут быть установлены вплотную к левой стороне аналитиче-
ского блока для удобного размещения хроматографа на рабо-
чем столе.
Установку расходов газов и последующую их корректировку
Для оптимизации режима анализа следует выполнять, ориенти-
руясь на показания цифровых индикаторов расходов газа. Так
155
как в БПГ-167 нет измерителей расхода, он может эксплуати-
роваться в комплекте с блоком управления БУ-125, имеющим
индикацию расходов газа-носителя и водорода.
Ввиду отсутствия в исполнении 560-02 хроматографа блока
БУ-125 он комплектуется газовым блоком БПГ-175 с тремя не-
зависимыми встроенными измерителями расхода. Они рас-
считаны на непрерывную цифровую индикацию расходов газа-
носителя (азота и гелия) до 100 мл/мин, водорода до 100 мл/мин
и воздуха до 300-400 мл/мин. Погрешность измерения не пре-
вышает ±5% от верхнего предела диапазона расходов, а измене-
ние показаний при изменении внешней температуры на 10 °C не
более ±2,5%. Кроме того, в обеих линиях газа-носителя уста-
новлены тензометрические датчики с электрическим выводом,
позволяющие измерить давление газа на входе в колонку (в ана-
литический блок) до 0,3 МПа с погрешностью не более 3%.
Основу измерителя расхода составляет датчик теплового ти-
па в виде тонкостенной стальной трубки с четырьмя медными
обмотками, представляющими мостовую схему с постоянным
питанием около 3 В. Каждый датчик помещен в индивидуальный
активный термостат с температурой 50 °C. При прохождении
газа через трубку возникает разбаланс мостовой схемы, про-
порциональный расходу в пределах от 0 до 12 мл/мин. Датчики
расхода имеют делители потока (байпас), позволяющие напра-
влять на измерение постоянную малую часть общего расхода
в пределах зоны пропорциональности. Напряжение разбаланса
усиливается и подается на общий цифровой вольтметр.
В верхней части блока БПГ-175 имеется цифровое табло с
клавишным переключателем-селектором, на которое выводятся
показания либо измерителей расходов различных газов, либо
датчиков давления в линиях газа-носителя. Измерители расхо-
дов подключаются к соответствующим газовым линиям блока с
помощью штуцеров на задней панели. При необходимости одно-
временной работы с двумя линиями газа-носителя измеритель
расхода водорода может быть включен во вторую линию газа-
носителя, при этом его показания могут отличаться не более
чем на 2% для азота и на 30% для гелия. Показания измерите-
лей расхода практически не зависят от давления газа (до 0,4-
0,5 МПа), так как датчики градуируются в расчете на расход,
приведенный к нормальным условиям газового состояния (20 °C,
760 мм рт. ст.).
Измерители расхода могут быть использованы не только для
установки необходимых потоков, диктуемых конкретной мето-
дикой проведения анализа, но и для оценки стабильности рас-
ходов во время проведения анализа (например, в режиме про-
156
граммирования температуры), а также для удобного контроля
герметичности газовых линий БПГ-175 и аналитического блока.
Следует также иметь в виду, что манометры измеряют да-
вление газа-носителя после регулятора давления (перед регу-
лятором расхода), а цифровые датчики давления — после ре-
гулятора расхода. Это позволяет устанавливать оптимальный
режим работы (перепад давления) регуляторов расхода, что
особенно важно при использовании программирования темпе-
ратуры колонки.
П.2.2.6. Система обработки хроматографической информации
Конечная цель анализа —- получение аналитической инфор-
мации в виде сведений об удерживании компонентов пробы не-
подвижной фазой и их концентрации или количестве — в хрома-
тографах “Пвет-500М” достигается использованием микропро-
цессорных средств вычислительной техники, при этом исклю-
чается необходимость вручную обрабатывать хроматограмму,
записываемую аналоговым регистратором на бумаге, или об-
рабатывать значения времен удерживания и площадей пиков,
измеряемых интегратором. Хотя возможность записи анало-
гового сигнала предусмотрена (имеются соответствующие вы-
ходы на блоках БИЛ и БПД), основным вариантом количе-
ственного анализа является получение аналитической инфор-
мации непосредственно в цифровой форме.
В хроматографах “Пвет-500М” эту функцию выполняет либо
одноканальная система обработки САА-06-03, описанная в [79],
либо аппаратно-программный комплекс АПК-01, позволяющий
одновременно и независимо принимать, преобразовывать и на-
правлять в персональный компьютер сигналы двух хроматогра-
фических детекторов (или двух эксплуатируемых рядом хро-
матографов). АПК представляет собой сочетание аппаратной
части, включающей аналого-цифровой преобразователь и мно-
гоканальный коммутатор, с программным обеспечением
GICHROM, в соответствии с которым ПЭВМ ведет обработку
и управляет коммутатором. Оператор (аналитик) общается
с АПК с помощью клавиатуры компьютера, наблюдая за хо-
дом хроматограммы, отображаемой на экране компьютера в
реальном времени (или повторно). Программное обеспечение
GICHROM позволяет проводить автоматически градуировку и
расчет концентраций всеми общепринятыми и наиболее упо-
требимыми методами: абсолютную градуировку с примене-
нием метода наименьших квадратов (МНК), абсолютную гра-
дуировку, проходящую через нуль координат параметр пика—
157
концентрация, внутренний стандарт с нулевой точкой, а также
простую нормализацию. Программа минимизирует количество
параметров обработки, определяемых аналитиком (порог обна-
ружения и полуширина пика) и предоставляет возможность руч-
ного или автоматического измерения их значений. Обработка
проводится либо по площади, либо по высоте пиков, при этом
возможно назначение для обработки любых интересующих ана-
литика пиков или всех пиков на хроматограмме (но не более
десяти).
Предусмотрена возможность запоминания хроматограммы,
ее повторных вызовов на экран с целью выполнения аналити-
ком маркировки пиков и предварительной ручной разметки ха-
рактерных точек пиков (начала, максимума, конца) или коррек-
тировки автоматической разметки для последующего повторе-
ния обработки той же хроматограммы с другой разметкой или
иными значениями параметров обработки, а также с использо-
ванием других методов градуировки. Аналитик может просма-
тривать фрагменты хроматограммы с изменением масштаба по
временной и амплитудной осям, производить фильтрацию иска-
жений нулевой линии, осуществлять вычитание нулевой линии
и проведение ее по своему усмотрению. Предоставляется воз-
можность выбирать или самостоятельно формировать таблицу
результатов и состав распечатываемого протокола анализа с
включением сведений о характере, размере пробы и месте ее от-
бора, методических условиях анализа, времени его проведения
и другой информации. Программа обеспечивает запоминание
действий оператора, осуществляемых с помощью клавиатуры,
для выполнения сервисных функций в однотипных анализах, а
также запоминание данных по градуировочным коэффициентам.
Все это позволяет аналитику оперативно участвовать в обра-
ботке информации и вводить адаптирующие коррективы в авто-
матический процесс определения состава анализируемой смеси.
На передней панели блока АПК расположены переключатели
управления двумя каналами обработки сигналов хроматографа
с индикаторами состояния этих каналов и включения сетевого
питания. На задней панели блока находятся выключатель сети
и разъемы для подключения АПК к источникам аналоговых сиг-
налов (БИЛ или БПЛ) и к ПЭВМ. После соединения АПК с
компьютером первичное введение в действие производится пу-
тем записи в ПЭВМ программного обеспечения GICHROM с
прилагаемой дискеты.
Обработка хроматографической информации в аналитиче-
ском режиме начинается с момента нажатия кнопки АНАЛИЗ
соответствующего канала на панели блока АПК и заканчива-
158
ется при повторном ее нажатии. Запись хроматограммы и рас-
печатка протокола (таблицы) результатов анализа осуществля-
ются принтером, входящим в состав ПЭВМ. АПК преобразует
в дискретную форму аналоговый сигнал с амплитудой в диапа-
зоне от 1 до 106 мкВ с дрейфом не более 50 мкВ/ч. Собственные
шумы АПК (приведенные к входу) не превышают 0,5 мкВ.
В хроматографах “Пвет-500М” (включая модернизированные
исполнения) предоставлен выбор из двух возможностей реги-
страции сигнала детектора: в аналоговой форме самопишущим
потенциометром со шкалой 1 мВ и в цифровой форме систе-
мами обработки САА-06 или АПК-01. Характеристики хрома-
тографа, связанные с выходным сигналом, также официально
нормируются двояко: в электрических единицах сигнала (%
шкалы регистратора) и в единицах счета высоты (площади)
пика системы обработки. Между этими величинами существует
вполне определенная связь, задаваемая следующими соотноше-
ниями: по амплитуде пика — 1 единица счета (ед. сч.) высоты
САА и АПК соответствует 1 10“15 А для сигналов ионизаци-
онных детекторов и 1 • 10~7 В для сигнала детектора по тепло-
проводности; по площади пика — 1 единица счета площади со-
ответствует 1 10-15 А • с и 1 • 10-7 В с. Приведенный размер
единицы счета для ионизационных детекторов реализуется при
включении системы обработки в разъем САА блока БИЛ-36 и
входном сопротивлении 1О10 Ом.
Например, уровень флуктуационных шумов нулевого сигнала пламен-
но-ионизационного детектора, измеренный в испытательном режиме с по-
мощью САА и АПК, нормируется равным 20 ед. сч. высоты. Это соответ-
ствует 20-10-15 = 2-10~14 А, что при аналоговой записи нулевой линии на
КСП-4 при RBX = 1О10 Ом и коэффициенте выходного делителя 4 должно
отвечать ширине записи 0,5% на шкале 4 • 10“12 А.
Сигналы, измеренные в ед. сч. высоты и площади при вход-
ных сопротивлениях БИЛ-36 109, 108 и 107 Ом, приводятся к
общему масштабу умножением на коэффициенты 10, 100 и 1000
соответственно.
Эти сведения необходимы для оценки уровня шумов и пре-
делов детектирования при проведении периодической поверки
хроматографов “Цвет-500М”, допускающей измерение параме-
тров как в аналоговой, так и в цифровой форме.
159
II.2.3. Хроматографы “Цвет-800”
Хроматографы “Цвет-800” являются универсальными газо-
выми лабораторными хроматографами, продолжающими тра-
диционную для ОАО “Цвет” концепцию блочно-модульного по-
строения и сохраняющими разумную преемственность в кон-
струкции аналитической системы. Тем не менее, приборы
“Цвет-800” относятся к новой серии, принципиально отличаю-
щейся тем, что функции управления режимом хроматографа и
обработки выходного сигнала выполняются с использованием
персонального компьютера (ПК). Хроматографы “Цвет-800” не-
льзя включать и эксплуатировать без ПК, являющегося необ-
ходимой составной частью приборов. Использование возмож-
ностей компьютера вполне отвечает мировой тенденции разви-
тия сложной аналитической техники, оно позволяет более раци-
онально и экономно организовать функционирование аналити-
ческой системы хроматографа.
В состав простейшей (базовой) модели входят (рис. 11.54):
Рис. 11.54. Газовый хроматограф “Цвет-800”.
— аналитический блок БА-121-01 с пламенно-ионизационным
детектором;
— газовый блок БПГ-175;
— блок питания и усиления сигналов ионизационных детек-
торов БИЛ-36*;
— персональный компьютер IBM PC/AT/DX2 с процессором
не ниже 386 и принтером EPSON LX-300**.
*Возможен вариант, когда усилитель встроен в аналитический блок.
*’По согласованию с потребителем ПК может не поставляться.
160
Базовая модель может наращиваться до получения необходи-
мых аналитических возможностей с использованием всего суще-
ствующего в ОАО “Цвет” набора аналитических принадлежно-
стей и дополнительного оборудования (см раздел П.2 4).
Исполнение аналитического блока БА-121-01 отличается на-
личием в нем контроллера системы терморегулирования и уп-
равления дискретными устройствами и, главное, наличием
встроенного аналого-цифрового преобразователя сигналов де-
текторов, имеющего два идентичных и независимых канала пре-
образования напряжения в частоту (ПНЧ). В аналитический
блок могут устанавливаться все описанные ранее детекторы, а
также новый тип — фотоионизационный детектор (ФИЛ).
11.2.3.1. Фотоионизационный детектор
Этот детектор имеет ряд существенных преимуществ, в чи-
сле которых, прежде всего, следует назвать универсальность
при очень высокой чувствительности и низком пределе детекти-
рования — примерно на два порядка лучше, чем для пламенно-
ионизационного детектора (по бензолу). Линейный диапазон со-
ставляет 2 • 104 (по бутану). Селективностью можно управлять,
меняя тип используемого источника фотоионизации (ультрафи-
олетовой лампы). В зависимости от вида конструктивного ис-
полнения — “Ф” (фторопласт) и “К” (керамика) — максималь-
ная рабочая температура детектора достигает 150 или 300 °C.
ФИЛ может эксплуатироваться как с насадочными, так и с
капиллярными колонками. Его следует отнести к концентраци-
онному типу детектора, в котором основная доля анализируе-
мого вещества не разрушается в процессе детектирования (не-
деструктивный детектор). Принцип действия ФИЛ основан на
ионизации анализируемых веществ АВ в потоке газа-носителя
интенсивным ультрафиолетовым излучением
АВ + hv —> АВ+ + е
и сбором зарядов на электродах ионизационной камеры с фор-
мированием токового сигнала I. При этом ионизация посторон-
них примесей в газе-носителе и элементов материала камеры
создает постоянный фоновый сигнал.
В пределах линейной области детектирования соблюдается
следующая зависимость сигнала ФИД от концентрации анали-
зируемого вещества:
I = jStrNLC,
161
где j — плотность потока фотонов на входе в ионизационную
камеру; S — площадь, через которую поступает поток фотонов;
а — сечение фотоионизации анализируемого вещества; А — чи-
сло Авогадро; L — высота ионизационной камеры; С — концен-
трация анализируемого вещества.
Для заданной конструкции и режима детектора его чувстви-
тельность (ионизационная эффективность) к веществам различ-
ной природы определяется главным образом сечением фотоио-
низации, которая зависит от энергии фотонов и потенциала ио-
низации анализируемых веществ. Источником ионизации слу-
жит высокочастотная резонансная ультрафиолетовая лампа, за-
полненная ксеноном (КсРВ) или криптоном (КрРВ). Лампа
представляет собой безэлектродную стеклянную трубку, с тор-
ца которой вклеено окно из кристалла MgFz толщиной 1 мм
для пропуска жесткого ультрафиолетового излучения. Средняя
энергия УФ-излучения составляет 9,5 и 10,2 эВ для ксеноновой
и криптоновой ламп соответственно, что позволяет регистриро-
вать вещества с потенциалом ионизации молекул до 12 эВ.
Предел детектирования ФИД, нормируемый по бензолу
(стандартное контрольное вещество), составляет не более 4 х
х10~9 мг/мл с лампой КсРВ и 5 • 10-11 мг/мл с лампой КрРВ.
Фотоионизационный детектор состоит из двух узлов: иони-
зационной камеры с УФ-лампой и индукционной катушкой и
собственного блока питания, являющегося генератором высо-
кочастотного напряжения. Общий вид детектора показан на
рис. 11.55.
Ионизационная камера выполнена в виде цилиндра диаме-
тром 8 мм и высотой 5 мм. Внутренняя поверхность камеры
ограничена фторопластовыми прокладками или сделана из ке-
рамики. Верхний торец камеры закрыт окном УФ-лампы. Два
кольцевых электрода из нержавеющей стали расположены в ка-
мере параллельно на расстоянии 2 мм. Нижний электрод (ка-
тод) соединен с разъемом СИГНАЛЫ. К аноду подведено на-
пряжение (300±30) В через разъем ПИТАНИЕ. УФ-лампа зажи-
гается с помощью индукционной катушки с резонансной спира-
лью. Электрическое питание катушки осуществляется высоко-
частотным напряжением (27 В, 80 МГц) от блока питания ФИД
через разъем ПИТАНИЕ ВЧ на крышке корпуса катушки.
Детектор устанавливается в аналитический блок на переход-
ную камеру вместо левой ячейки от пламенно-ионизационного
детектора, а его блок питания монтируется на заднюю стенку
аналитического блока. Поток газа-носителя, в качестве кото-
рого может использоваться азот, гелий, аргон или очищенный
воздух, поступает в детектор через центральный штуцер в осно-
162
Рис. 11.55. Фотоионизационный детектор с блоком питания.
вании, к которому присоединяется аналитическая колонка. Сиг-
нал детектора подается непосредственно на вход БИД-36.
Фотоионизационный детектор сертифицирован и зарегистри-
рован в Госреестре средств измерения. Он рекомендуется для
детектирования весьма низких концентраций примесей разно-
образных органических, металлоорганических (например, те-
траэтилсвинец) и ряда неорганических (сероводород, сероугле-
род, аммиак, арсин, фосфин и т.д.) веществ.
П.2.3.2. Программное обеспечение хроматографов “Цвет-800”
Программное обеспечение выполняет двоякую роль. Во-пер-
вых, оно позволяет задавать и контролировать температурный
163
режим термостата колонок (изотермический и программный),
испарителей и крана-дозатора, индивидуальных термостатов
детекторов ДТП и ДПР, оснований ионизационных детекто-
ров (включая ФИД), а также задавать общее время анализа
и временную программу автоматической работы дискретных
устройств в пределах необходимого количества циклов анализа.
Во-вторых, программное обеспечение позволяет проводить пол-
ную обработку сигналов детекторов одновременно по двум ка-
налам. При этом градуировка и расчет концентрации анализи-
руемых веществ выполняются по алгоритмам наиболее употре-
бимых в аналитической практике методов:
— простая нормализация (интегрирование);
— абсолютная градуировка, проходящая через ноль коорди-
нат параметр пика — концентрация;
— абсолютная градуировка по нескольким точкам с приме-
нением метода наименьших квадратов;
— с использованием внутреннего стандарта с нулевой точкой;
— с использованием внутреннего стандарта по нескольким
точкам.
Программное обеспечение рассчитано на работу с многока-
нальным коммутатором и аналого-цифровыми преобразовате-
лями в составе аналитического блока БА-121-01. Программное
обеспечение TSVET 800 работает в операционной системе MS
DOS. Оно поставляется на гибком диске и рассчитано на ис-
пользование компьютера, отвечающего следующим минималь-
ным требованиям:
— программная совместимость с IBM PC/AT, с сопроцессо-
ром;
— объем оперативной памяти не менее 640 кбайт;
— наличие дисковода для флоппи-диска (3,5");
— параллельный порт для подключения принтера;
— DOS версии 3,0 и выше.
По своим возможностям и порядку действий оператора про-
грамма обработки близка к программе GICHROM, реализован-
ной в комплексе АПК-01. Существенным удобством практиче-
ской работы с программой TSVET 800 является то, что она по-
зволяет ограничиться только двумя первичными параметрами
обработки, определяемыми оператором, — порогом обнаруже-
ния и начальной шириной пика. Обработка может проводиться
как по площади, так и по высоте любых или всех пиков на хро-
матограмме (но не более 10 в режиме градуировки).
Предусмотрена возможность запоминания хроматограммы,
ее повторных вызовов на экран с целью выполнения аналити-
ком маркировки пиков и ручной разметки характерных точек
164
(начала, максимума, конца пика) или корректировки автома-
тической разметки для повторения обработки той же хрома-
тограммы с другой разметкой или новыми значениями пара-
метров порога и ширины. Можно повторить обработку с ис-
пользованием иного метода градуировки, просматривать фраг-
менты воспроизведения хроматограммы в измененном масштабе
по времени или по высоте пиков, осуществлять вычитание базо-
вой линии или изменять ее положение.
Программа предоставляет возможность выполнять одно- или
многоступенчатую фильтрацию базовой линии, что позволяет
снизить уровень помех и более надежно выделять полезный сиг-
нал (пик анализируемого вещества) на фоне флуктуаций базо-
вого сигнала (нулевой линии). Дополнительные аналитические
удобства обеспечиваются возможностью сложения или вычита-
ния хроматограмм, записанных на разных каналах обработки,
группирования и переноса информации на один канал. При не-
обходимости начальный вид хроматограмм может быть восста-
новлен и воспроизведен.
Аналитик может самостоятельно формировать содержание
условий проведения и результатов анализа для распечатки про-
токола принтером по окончании анализа.
В программу заложены сервисные функции по оказанию по-
мощи оператору в порядке и последовательности его действий,
по информированию о состоянии системы и запоминанию его
команд в однотипных анализах с целью их последующего авто-
матического исполнения. Это позволяет аналитику адекватно
участвовать в обработке информации, оперативно вводя необ-
ходимую коррекцию в автоматический процесс определения со-
става анализируемой смеси.
Программа работает на русском языке. Вся информация
по своему характеру и назначению сведена в соответствующие
файлы. Взаимодействие с оператором осуществляется в виде
спускающихся “меню” и редактируемых таблиц, позволяющих
формировать наборы параметров в зависимости от назначения
и условий работы. Графический интерфейс дает возможность
воспроизвести хроматограмму на экране в заданном масштабе
Для оценки полноты разделения, проведения разметки и коррек-
тировки нулевой линии.
Программное обеспечение TSVET 800 позволяет оператору
в процессе анализа и обработки результатов непрерывно или
периодически отслеживать на экране текущие значения пара-
метров температурного режима хроматографа с погрешностью
±0,25 °C и готовность термостата колонок к проведению очеред-
ного цикла программирования температуры.
165
Здесь не представляется возможным и целесообразным по-
дробно описывать последовательные действия оператора при
задании режима и обработке результатов на компьютере. Ал-
горитм этих действий приведен в виде оперативной справочной
информации в составе программного обеспечения TSVET 800 и
изложен в инструкции завода-изготовителя хроматографа.
В хроматографах “Пвет-800” возможна регистрация аналого-
вого сигнала и в виде хроматограммы (при наличии записыва-
ющего потенциометра), поскольку аналоговые выводы в блоках
БИ Д-36 и БПД-56 сохранены. Этим можно пользоваться для от-
работки режима разделения в процессе подготовки к анализу.
Однако официально признаваемые (особенно в сфере государ-
ственного надзора за измерениями) аналитические результаты
могут быть получены только с использованием штатной ком-
пьютерной системы обработки. Это связано с тем, что (в от-
личие от “Пвет-500М”) характеристики сигналов в аналоговой
форме не нормированы. Высоты и площади пиков измеряются
и выражаются только в единицах счета системы обработки.
При этом имеют место следующие соотношения для выраже-
ния величины сигнала в электрических единицах (для сопоста-
вления различных систем обработки и типов хроматографов)
1 ед. сч. высоты соответствует 5,2 10~15 А для сигналов ио-
низационных детекторов (БИД-36 включен в разъем САА при
Двх = 1О10 Ом) и 5,2 10-7 В для сигналов детектора по тепло-
проводности, 1 ед. сч. площади соответствует 5,2 10~15 А с и
5,2 10~7 В с.
Например, уровень флуктуационных шумов нулевого сигнала пламен-
но-ионизационного детектора в стандартном испытательном режиме нор-
мируется равным 2 ед. сч. высоты. Это соответствует 5.2 10~15 - 2 =
1,04-10-14 А, что вдвое меньше, чем в хроматографах “Цвет-500М”.
При периодической поверке хроматографов “Пвет-800” изме-
рения выходных сигналов должны выполняться только в цифро-
вой форме.
В хроматографах “Цвет-800” для большинства детекторов
(ПИД, ТИД, ДТП) нормированы более низкие пределы детек-
тирования, что достигнуто уменьшением уровня регистрируе-
мых шумов, благодаря использованию возможностей компью-
терной программы фильтрации искажений нулевого сигнала.
Точностные характеристики (сходимость высот, площадей, вре-
мени удерживания и их временная стабильность) хроматогра-
фов “Цвет-500М” и “Пвет-800” находятся на одном уровне.
166
II.2.4. Аналитические принадлежности и
дополнительное оборудование
Для расширения аналитических возможностей универсаль-
ных хроматографов “Пвет” всех моделей и обеспечения удоб-
ства их эксплуатации разработан и постоянно развивается ком-
плекс принадлежностей и пополнительного аналитического обо-
рудования. Элементы этого комплекса могут быть приобретены
дополнительно или независимо по индивидуальным заказам, ис-
ходя из конкретных аналитических задач и условий работы в
хроматографической лаборатории. Ниже приводятся перечень
и некоторые характеристики оборудования (принадлежностей,
приставок, самостоятельных приборов) из этого комплекса
Хроматографические аналитические колонки.
Насадочные колонки (КН) из стекла и стали, длиной 1-6 м, вну-
тренним диаметром 2 и 3 мм, внешним диаметром 4 и 6 мм,
заполненные сорбентами, содержащими различные неподвиж-
ные фазы. Капиллярные кварцевые колонки (КК) длиной 15-
50 м, внутренним диаметром 0,1-0,4 мм, с привитыми стацио-
нарными фазами SE-30, SE-301, OV-17, OV-101, ПЭГ-20М, ПЭГА,
лукопрен Г-1000 и др., а также специальные колонки для ана-
лиза пестицидов в комплекте с микродетекторами электронного
захвата. При необходимости кварцевые колонки могут поста-
вляться вместе с комплектом принадлежностей для присоедине-
ния к хроматографу (включая регулируемый делитель пробы).
Каждая колонка снабжена паспортом и тестовой хроматограм-
мой контрольной смеси с указанием эффективности, коэффици-
ента емкости и числа разделений.
Криогенное устройство УК-84 позволяет распростра-
нить термостатирование колонок на область отрицательных
температур от —100 до +50°C. Оно включает линию связи с
сосудом Дьюара и регулирующий клапан для паров жидкого
азота.
Блок дозирования газов БДГ-115 с термостатиру-
емым до 150 ° С краном-дозатором позволяет работать в авто-
матическом режиме при анализе газов и газопаровых смесей.
Управление краном производится по временной программе, за-
даваемой САА-06, пультом управления ПУ-013 или ПЭВМ.
Ручное дозирование газов можно осуществлять нетермоста-
тируемым краном-дозатором КД-234, конструкция которо-
го аналогична описанному выше. Для анализа природного газа
рекомендуется применять специализированный блок БДГО-171,
содержащий термостатируемый шестиходовой кран-дозатор и
четырехходовой кран, используемый для включения обратной
167
продувки колонки с целью быстрого освобождения от тяжелых
компонентов.
Для автоматического дозирования жидкостей используется
автосамплер, выполняющий последовательно по заданной
программе операции подготовки (промывки) микрошприца, от-
бора нужного объема пробы из ампул, установленных в кру-
глую кассету, введения пробы в испаритель, повторения этих
операций нужное число раз, перехода к следующей пробе (ам-
пуле) и т. д. Автосамплер состоит из манипулятора микрошпри-
цем с кассетой ампул, монтируемых на аналитическом блоке, и
собственного блока управления.
Микродозатор жидкости МДЖ-163 используется для
ручного дозирования низкокипящих жидких смесей через испа-
ритель хроматографа. Дозирование производится краном, со-
держащим фиксированные объемы доз в виде каналов в подвиж-
ном диске. Объемы доз 1, 2, 4 и 8 мкл выполнены с погрешно-
стью не более 4% от номинала.
Устройство обогатительное У0-89 предназначено
для извлечения и накопления примесей из газового потока с
последующей десорбцией и дозирования в аналитическую ко-
лонку. Накопление примесей осуществляется в обогатитель-
ной колонке, охлаждаемой жидким азотом или другим хладо-
агентом. Десорбция примесей производится нагреванием обо-
гатительной колонки электропечью. Переключение стадий на-
копление — десорбция — анализ выполняется вручную шести-
ходовым краном, имеющим дополнительное положение (сред-
нюю точку), в котором обогатительная колонка запирается на
время проведения десорбции. Перевод крана в положение АНА-
ЛИЗ направляет десорбированные примеси в аналитическую
колонку. Применение обогатительного устройства позволяет на
1-2 порядка снизить предел обнаружения примесей по сравне-
нию с прямым анализом.
К рио ф оку сир у ю щее устройство УКФ-1* позволяет
на хроматографе с капиллярной колонкой проводить определе-
ние примесей, загрязняющих воздух (например, ацетон, бензол,
толуол, о-, м-, п-ксилол, этилбензол, хлорбензол), на уровне
ПДК и ниже. Криоловушка охлаждается жидким азотом, ре-
жим управления анализом ручной.
Метод криофокусирования заключается в пропускании до-
статочного объема анализируемой среды с потоком газа-носи-
теля через охлажденную короткую начальную часть капилляр-
*Кроме приставок к универсальным хроматографам в ОАО “Цвет” раз-
работан специализированный капиллярный хроматограф “Цвет 700КФ” с
встроенной криофокусирующей системой.
168
ной колонки или предколонку малого объема. При этом высо-
кокипящие примеси конденсируются или сорбируются и оста-
ются в холодном участке, а более легкие основные компоненты
проходят с потоком газа-носителя. После завершения накопле-
ния примесей в малом объеме охлажденного участка последний
подвергается быстрому нагреву (термоудару), вследствие чего
примеси поступают в поток газа-носителя в виде компактной по
объему пробы, что является решающим условием для их успеш-
ного разделения на капиллярной колонке и последующей реги-
страции чувствительными ионизационными детекторами. Та-
ким образом, метод криофокусирования решает одновременно
две задачи: извлечение примесей из большого объема анализи-
руемой среды и их сосредоточение в малом объеме (концентри-
рование) и эффективное введение пробы (дозирование) в капил-
лярную колонку. Сочетание высокой разрешающей способно-
сти капиллярной колонки с предварительным преобразованием
пробы криофокусированием существенно расширяет аналити-
ческие возможности хроматографов при определении токсич-
ных примесей в атмосфере предприятий и городов.
Устройства для концентрирования методом вы-
дувания-накопления. Этот метод за рубежом известен под
названием “purge and trap”, он используется для определения со-
держания летучих примесей загрязнителей в водах различного
происхождения. Устройство УВП-1 предусматривает продува-
ние инертного газа через анализируемую жидкость, накопление
выдуваемых примесей в сорбционной ловушке, последующую
термодесорбцию их и криофокусирование и, наконец, разделе-
ние на капиллярной колонке.
Разработаны и выпускаются в виде приставок к хроматогра-
фам несколько вариантов устройств для парофазного
доз ирования, реализующих описанные в разделе II. 1.2 прин-
ципы. Среди этих устройств общего назначения можно назвать
УРП-82 и УРП “Фаза”. Последнее представляет собой упро-
щенный вариант УРП в виде отдельного блока, устанавлива-
емого на правую стенку термостата колонок хроматографа и
использующего обе линии газа-носителя и свободные каналы
термостатирования (например, крана-дозатора).
В основу устройства “Фаза” положен комбинированный спо-
соб дозирования в хроматографическую колонку парогазовых
проб. Пневматическим способом равновесным газом из флакона
с исследуемым образцом заполняют дозирующий объем газо-
вого крана. Введение пробы в хроматографическую колонку
производится с помощью газового крана.
169
Ампула с анализируемым образцом подключается к газовой
схеме накалыванием на иглу, установленную на панели блока
“Фаза”. Переключающий кран с соответствующими переходни-
ками термостатируется в диапазоне от 50 до 150°C.
Те же принципы анализа равновесного пара реализованы в
специализированном устройстве УАТМ-133 для определения
содержания растворенных в трансформаторном масле газов.
Это устройство в виде дополнительного оборудования к хро-
матографам “Цвет” с детекторами ПИД и ДТП позволяет из
одной пробы масла объемом около 5 см3 определять СО, СОг,
СН4, СгНб, С2Н4 и С2Н2 с пределом обнаружения до 10~4 и во-
дород — до 10-2% по объему. При этом оксиды углерода пре-
вращаются в СН4 с помощью метанатора, входящего в состав
устройства.
Основу конструкции составляет блок подготовки пробы, осу-
ществляющий дозирование выделенных из масла газов в анали-
тические колонки хроматографа. Предусмотрены необходимые
технические меры для быстрого достижения равновесного со-
стояния газовой фазы над поверхностью масла в пробоотборной
ампуле и количественной передачи пробы газов на анализ. В со-
став УАТМ по специальному заказу может входить интерфейс
для персональной ЭВМ с программой расчета хроматографи-
ческих данных и анализа состояния трансформатора.
Для введения жидких проб в испаритель хроматографа ис-
пользуются микрошприцы трех типов с соответствующим
диапазоном дозируемых объемов и ценой деления шкалы: МШ-
1М (0,1-1,0 мкл; 0,02 мкл); МШ-10М (1,0-10,0 мкл; 0,2 мкл); МШ-
50 (1,0-50,0 мкл; 1,0 мкл). В минимальный комплект поставки
(коробка) входят два микрошприца с паспортом и мандреном.
Генератор водорода является источником чистого
(99,99% по объему) водорода, получаемого электрохимическим
разложением дистиллированной воды (бидистиллята) в элек-
тролизной ячейке с твердополимерным электролитом. Макси-
мальной производительности электролизера (70 мл/мин) доста-
точно для питания двух пламенно-ионизационных детекторов и
обеспечения их стабильной работы на самых чувствительных
шкалах. Максимальное давление водорода на выходе генера-
тора равно 2 кг/см2. Колебания давления, связанные с процес-
сом регулирования, не превышают ±1,5%. Одной заливки воды
в емкость генератора хватает на 200 ч непрерывной работы. Во-
дород, поступающий из камеры электролизера, проходит через
два последовательных фильтра и предохранительный клапан.
Давление устанавливается мембранным регулятором давления
и контролируется манометром. Далее поток водорода разделя-
170
ется на две линии с независимой установкой расходов с помо-
щью игольчатых дросселей. Кислород из электролизера выво-
дится в атмосферу.
Генератор выполнен в одном блоке (рис. 11.56), на передней
панели которого размещены указатель уровня воды, манометр,
ручки регулятора давления и дросселей, сетевой включатель
и световые индикаторы режима работы, а на задней панели —
выходные штуцеры водорода.
Рис. 11.56. Генератор водорода.
Для оперативного измерения потоков газов при выполне-
нии хроматографических работ, а также для измерения ма-
лых расходов чистых газов в других областях техники и науч-
ных исследованиях весьма эффективны автономные цифровые
измерители расходов г аз ов (ИРГ). В ОАО “Ивет” раз-
работаны и готовятся к серийному выпуску три типа измерите-
лей (ИРГ-10, ИРГ-100 и ИРГ-1000) с верхними пределами изме-
рений расходов 10, 100 и 1000 мл/мин и отсчетной возможностью
0,01; 0,1 и 1 мл/мин.
Предел основной погрешности измерения не превышает 5% от
верхнего предела измерения, а дополнительная температурная
171
погрешность — не более 5% на каждые 10 °C изменения окру-
жающей температуры. Время установления показаний (до 95%
конечного значения) не превышает 20 с, а время выхода на рабо-
чий режим составляет 20 мин после включения. Максимальное
давление газа в точке включения измерителя 0,4 МПа.
Градуировка измерителей производится по расходу, приве-
денному к нормальным условиям, т. е. показания измерителей
пропорциональны массовому расходу и в этом смысле не зави-
сят от давления газа.
Измерители могут регистрировать расходы до 19,99, 199,9 и
1999 мл/мин, однако погрешность при этом может составлять
10-15%. При расходах больше указанных значений на табло
высвечивается только 1 старшего разряда (переполнение). В
случае возникновения обратного потока через измеритель на
табло появляется знак ” (не рекомендуемый режим).
При необходимости измерения расхода газа, по которому
ИРГ /не градуирован при выпуске, может быть выполнена спе-
циальная градуировка потребителем при включении клавиши
СВ. ГРАД (свободная градуировка).
Следует иметь в виду, что градуировки по азоту, водороду
и кислороду близки к заводской градуировке с отклонением по
азоту на 1-2% (аналогично для гелия и аргона).
Существенно, что ИРГ удобно использовать для определе-
ния герметичности газовых линий хроматографа или иных тех-
нических устройств. Для этого измеритель включается между
источником давления газа и испытуемым объектом, а выход из
последнего заглушается. При наличии утечки показания ИРГ
снижаются не до нуля, а до величины утечки. ИРГ-10 позволяет
фиксировать утечки до 0,02-0,03 мл/мин, если предварительно
был тщательно установлен нуль измерителя в отсутствие по-
тока газа (при замкнутых входном и выходном штуцерах).
ИРГ по принципу действия относятся к тепловым расходо-
мерам неконтактного типа (см. раздел II.2.1.2). Каждый из них
выполнен в виде отдельного блока. На передней панели при-
бора расположены цифровое табло, переключатель рода изме-
ряемого газа, клавиши включения сети и регулировочный винт
свободной градуировки. На задней панели находятся штуцеры
ВХОД и ВЫХОД и регулировочные винты резисторов уста-
новки нуля заводской градуировки.
Для метрологической поддержки хроматографических изме-
рений используется специальное устройство ПОТОК-А, вы-
полненное в виде приставки к хроматографу. Оно позволяет
создавать поток парогазовой смеси анализируемых компонен-
тов с температурой кипения от —50 до +160 °C в инертном газе-
172
разбавителе (носителе). Смесь приготовляется путем дина-
мического смешения потоков газа-разбавителя и паров компо-
нента, диффундирующего через стенки термостатируемой фто-
ропластовой ячейки. Газовое питание и термостатирование в
диапазоне 50-200°C до пяти ампул осуществляется от соответ-
ствующих каналов управления хроматографа.
С помощью устройства ПОТОК-А могут решаться две за-
дачи: приготовление градуировочной смеси с заданной концен-
трацией анализируемого компонента (погрешность 5-10%) и на-
копление в сорбционной ловушке заданной массы компонента
(погрешность 2-3%).
По специальному заказу возможны заполнение ампул указан-
ными аналитиком веществами и метрологическая аттестация с
выдачей соответствующего свидетельства.
173
Глава III
АППАРАТУРА ДЛЯ ЖИДКОСТНОЙ
ХРОМАТОГРАФИИ
III.1. ПРИНЦИПИАЛЬНАЯ СХЕМА,
ОСНОВНЫЕ СИСТЕМЫ И УЗЛЫ
ЖИДКОСТНОГО ХРОМАТОГРАФА
Как и газовый, жидкостный аналитический хроматограф
представляет собой совокупность взаимодействующих систем,
предназначенных для проведения анализа в оптимальном ре-
жиме хроматографического разделения. Блок-схема прибора
представлена на рис. III.1. Резервуар с подвижной фазой и си-
стема подачи элюента, а также насос, который должен обес-
печивать поток подвижной фазы со скоростями от нескольких
мкл/мин для колонок малого диаметра до 10 мл/мин для на-
полненных колонок, обычно объединены в один блок. Насос по-
дает подвижную фазу в колонку через кран-дозатор с объемом
сменных дозирующих петель от 0,1 до 100 мкл и более. Раз-
работаны модели с автоматизированной системой ввода пробы.
На входе в колонку, как правило, устанавливается дополнитель-
ный узел ввода пробы для дозирования порции анализируемого
образца микрошприцем типа МШ-10. В состав многих моде-
лей жидкостных хроматографов последних лет выпуска входят
системы термостатирования колонок. Выход колонки соединен
с детектором и коллектором фракций. Особенностью жидкост-
ной хроматографии является то обстоятельство, что она почти
всегда сочетает разделение с препаративным выделением раз-
деленных фракций.
Коллектор фракций включает узел распределения, блок пе-
ремещения пробирок, кассету с пробирками и систему автома-
тизации всего процесса сбора фракций [81]. При выполнении
анализа через заданные временные интервалы происходит пе-
ремещение пробирок для сбора фракций. Число пробирок в
различных коллекторах фракций составляет 100—200.
Сигнал детектора в аналоговой форме подается на регистра-
тор (самописец) и независимо на аналого-цифровой преобразо-
ватель, блок передачи данных и затем на компьютер. К ком-
пьютеру обычно подключен принтер.
174
Рис. Ш.1. Блок-схема жидкостного хроматографа [36].
1 — резервуар с элюентом; 2 — насос; 3 — манометр; 4 — фильтр;
5 — демпфер; 6 — термостат; 7 — устройство для ввода пробы (ин-
жектор); 8 — колонка; 9 — детектор; 10 — регистратор.
Блок-схема жидкостного хроматографа может также вклю-
чать устройство для градиентного элюирования как с одним,
так и с двумя насосами (рис. III.2) [35], а в случае проведения
постколоночной дериватизации — реактор, расположенный ме-
жду хроматографической колонкой и детектором и снабженный
устройством подачи реагента.
Рис.П1.2. Схема устройства для градиентного элюирования.
1, 2 — резервуар с первым и со вторым элюентом соответственно;
3, 4 — насосы для подачи первого и второго элюентов; 5 — смеситель;
6, 7 — контроллеры скорости подачи элюентов 1 и 2 соответственно;
8 — контроллер состава; 9 — программатор градиента.
175
В настоящее время многими приборостроительными фирма-
ми выпускается оборудование для микроколоночных жидкост-
ных хроматографов с наполненными (устаревшее название —
набивными) колонками (диаметром 0,5-1,0 мм) [42].
III. 1.1. Подготовка подвижной фазы
Рекомендуется приготавливать и использовать растворитель
в количестве, необходимом на день работы.
Дегазация. Перед началом работы подготовленную подвиж-
ную фазу надо дегазировать — освободить от растворенных пу-
зырьков воздуха, которые нарушают нормальную работу хро-
матографа, вызывают дрейф нулевой линии, снижают уровень
абсорбции.
Дегазацию осуществляют одним из следующих способов: ки-
пячением, продувкой током гелия (или азота), перемешиванием
с использованием вакуума водоструйного насоса и, наконец,
ультразвуковым перемешиванием.
Кипячение обычно проводят в течение 5-10 мин, используя
обратный холодильник. Этот способ эффективен для индивиду-
альных элюентов и нежелателен для смешанных растворителей,
содержащих компоненты со значительной разницей в темпера-
турах кипения.
Дегазация продувкой током инертного газа основана на низ-
кой растворимости этого газа в жидкостях. При барботирова-
нии через слой растворителя продувочный газ достаточно бы-
стро захватывает и уносит из системы растворенный воздух.
Этот способ особенно предпочтителен при использовании легко
окисляющихся растворителей [36].
При ультразвуковом перемешивании сосуд с растворителем
помещают на 5-10 мин в ультразвуковую ванну, заполненную
водой. Эффективность процесса возрастает, если сосуд подклю-
чить к источнику умеренного вакуума. Без использования ва-
куума данный вариант дегазации рекомендуется применять для
смесирастворителей с заметно различающейся летучестью, по-
скольку он практически не изменяет их состава.
Очистка растворителя. Требования к чистоте раствори-
теля прежде всего определяются выбранным детектором. Так,
незначительная примесь олефинов в алканах не сказывается
на работе рефрактометрического детектора, но практически не
позволяет использовать УФ-детектор с поглощением на длине
волны 260 нм. При этом добавка гептана к гексану не влияет на
работу УФ-детектора.
176
При анализе в режиме адсорбционной хроматографии необ-
ходима тщательная осушка растворителя, поскольку влага в
подвижной фазе заметно изменяет фактор удерживания и сте-
пень разделения компонентов. При градиентном элюировании
требования к чистоте растворителей сильно возрастают При-
меси в элюенте могут концентрироваться в начальном участке
колонки и при увеличении полярности подвижной фазы элюи-
роваться, что является причиной появления на хроматограмме
ложных пиков.
Безусловно должны быть удалены и механические примеси,
для этого подвижную фазу пропускают через фильтры с разме-
ром пор 0,5 мкм.
Адсорбционную очистку проводят методом классической ко-
лоночной хроматографии, используя адсорбенты (оксид алюми-
ния или силикагель) с большой удельной поверхностью и раз-
мером зерен 0,1-0,5 мм. Сами сорбенты предварительно сушат
при 250-300 °C несколько часов. Наилучшие результаты дости-
гаются на колонках с двумя слоями сорбента: нижний — AI2O3,
верхний — силикагель. При массах сорбентов 100 г каждого в
зависимости от их активности можно очистить 300-600 мл не-
полярных растворителей и в 1,5-2 раза меньше растворителей
средней полярности (хлороформ, тетрагидрофуран). Качество
очистки проверяют по УФ-спектрам. Растворители с поляр-
ностью, превышающей в элюотропном ряду этилацетат, таким
методом очистить нельзя из-за очень большого сродства их к
сорбентам.
Элюенты, представляющие собой алканы, очищают от не-
предельных углеводородов концентрированной серной кисло-
той с последующей отмывкой дистиллированной водой до ней-
тральной реакции и осушкой Высушенные растворители затем
подвергают перегонке. Олефины можно независимо удалить ко-
лоночной хроматографией на силикагеле, пропитанном нитра-
том серебра (10% по массе).
X лор с оде р ж ащие элюенты часто имеют в качестве при-
меси НС1, образующийся при их хранении. Под действием хло-
роводорода корродируют металлические детали, разрушаются
адсорбенты. НС1 удаляют адсорбционной очисткой на щелоч-
ном оксиде алюминия. Для исключения возможного, иници-
ируемого светом образования в хлороформе фосгена раство-
ритель стабилизируют 0,5-1%-ной добавкой этилового спирта.
Очистку хлороформа проводят так: его промывают водой и
перегоняют, отделяя основное количество воды в виде азео-
тропной смеси. В перегнанный препарат добавляют 5-10% (по
177
массе) активного цеолита СаА; перед использованием его вновь
перегоняют.
Ацетонитрил очищают от примесей, поглощающих в УФ-
свете, кипячением с перманганатом калия и последующей пере-
гонкой. В колбу емкостью 2 л помещают 1,5 л ацетонитрила и
30 г перманганата калия, кипятят 1 ч с обратным холодильни-
ком и перегоняют с дефлегматором.
Изопропиловый спирт часто используется в качестве
модификатора подвижной фазы. Самая нежелательная при-
месь — вода, с которой он образует азеотропную смесь, вы-
кипающую при т. кип. 80,38°С (содержание воды в этой смеси
отвечает 12,1% (по массе)). При небольшом количестве воды ее
удаляют отгонкой, при содержании воды более 5-6% раствори-
тель сушат над сульфатом натрия и выдерживают над цеолитом
NaA.
При очистке метанола наиболее трудно отделяемой при-
месью является ацетон, который лучше всего удаляется обра-
боткой гипоиодитом натрия: 25 г иода в 1 л метанола вливают
при перемешивании в 500 мл 1 М раствора гидроксида натрия и
добавляют 150 мл воды. Через 6-10 ч отфильтровывают обра-
зовавшийся йодоформ, кипятят фильтрат с обратным холодиль-
ником (до исчезновения запаха йодоформа). Большинство при-
месей удаляют перегонкой на ректификационной колонке. Для
получения сухого продукта метанол сушат над цеолитом NaA
или КА.
В качестве возможных примесей вводе могут присутство-
вать различные соли, микроколичества углеводородов. Наи-
лучший путь очистки — деионизация на ионитах с последую-
щей перегонкой в кварцевой посуде либо пропускание деиони-
зованной воды через слой активированного угля. Для достиже-
ния высшей степени очистки воду пропускают через обращенно-
фазный сорбент, который потом регенерируют промывкой мета-
нолом, ацетонитрилом, тетрагидрофураном. Эффективно также
жесткое УФ-облучение [36].
III.1.2. Насосы
Основное требование к насосам сводится к обеспечению бес-
пульсационной подачи постоянного и регулируемого потока
растворителя через хроматографическую систему и к обеспе-
чению воспроизводимости скорости потока.
178
С помощью насосов можно реализовать два различных ре-
жима работы хроматографа изократический (постоянство со-
става элюента) и градиентный (увеличение элюирующей силы
подвижной фазы). Различают два принципиально различных
типа насосов: насосы постоянного давления и постоянного рас-
хода, поддерживающие стабильный поток элюента в диапазоне
расходов от 0,01 до 10,0 мл/мин. Основными характеристи-
ками насоса являются создаваемое им максимальное давление,
диапазон расходов, точность поддержания расхода (отсутствие
пульсаций).
Для рабочей колонки длиной 5-25 см, диаметром 2-10 мм,
заполненной сорбентом с размером частиц 5-15 мкм, входное
давление может составлять от 0,5 до 30 МПа. Если размер ча-
стиц сорбента 3 мкм требуется большее давление — 50 МПа.
При этом отметим, что в реальных условиях редко используется
входное давление, превышающее 20 МПа. С целью сглаживания
пульсаций предусмотрены демпфирующие устройства.
Насосы постоянного расхода подразделяются на шприцевые,
поршневые и мембранные. В отечественном микроколоноч-
ном хроматографе “Милихром” используется насос шприцевого
типа. Его максимальное давление 5 МПа, точность подачи
элюента ~1%, расход от 2 до 600 мкл/мин. Такие насосы ха-
рактеризуются относительной независимостью скорости потока
подвижной фазы от давления и вязкости растворителя, отсут-
ствием флуктуаций, легкостью замены элюента.
III.1.3. Системы ввода элюента
и анализируемой пробы
Смешение растворителей и системы формирования гра-
диентного элюирования. В жидкостной хроматографии часто
подвижная фаза представляет собой смесь двух и более компо-
нентов, соотношение которых в процессе анализа меняется по
определенной программе. Таким образом, в системе подачи по-
движной фазы необходимо предусмотреть контроль за соотно-
шением растворителей.
Как правило, даже незначительное изменение состава элю-
ента в условиях изократического режима приводит к заметному
изменению времен удерживания. В случае градиентного элюи-
рования отклонение действительного состава подвижной фазы
от запрограммированного оказывает еще большее влияние на
изменение параметров удерживания. Требования к точности
179
воспроизведения состава элюента возрастают с уменьшением
длины колонки, уменьшением крутизны градиента и увеличе-
нием скорости потока подвижной фазы.
Для обеспечения градиентного элюирования используют два
способа.
1. Каждый из растворителей поступает с помощью специаль-
ного насоса в смесительную камеру небольшого объема. Для
того чтобы соотношение растворителей в элюенте строго вы-
держивалось, необходимо, чтобы контроллер объемной скоро-
сти был отградуирован.
2. Растворители последовательно подаются насосом через
определенные интервалы времени.
Системы ввода пробы. В ВЭЖХ они делятся на две группы
в одних приборах проба вводится микрошприцем с использова-
нием принципа шлюзового устройства, в других — дозирующим
краном со сменными калиброванными петлями.
Простейший узел ввода пробы в ВЭЖХ представляет собой
тройник с примыкающими транспортными магистралями, свя-
зывающими его с насосом и колонкой (первая из них снабжена
запирающим краном). Третий “свободный” штуцер тройника
закрыт заглушкой. Непосредственно перед дозированием запи-
рающим краном перекрывают поток подвижной фазы, снимают
заглушку с фторопластовой прокладкой, в открывающийся ка-
нал вводят иглу микрошприца с отобранной пробой, плунже-
ром выталкивают пробу на фильтр колонки, извлекают микро-
шприцы из шлюза, закрывают вход в него снятой ранее заглуш-
кой и, наконец, восстанавливают поток подвижной фазы. Ис-
пользование вместо фторопластовых резиновых прокладок, про-
калываемых иглой микрошприца при дозировании, в принципе,
должно было бы исключить необходимость в остановке потока
элюента для дозирования того или иного образца, сократить
число ручных операций, улучшить воспроизводимость параме-
тров удерживания. Однако набухание резиновых прокладок в
некоторых растворителях, потеря герметичности системы (из-
за выкрошивания резины) после некоторого числа проколов, вы-
деление резиной загрязняющих подвижную фазу соединений и,
как следствие, появление на хроматограмме ложных пиков, а
также шумов на нулевой линии образуют длинный список недо-
статков этого способа дозирования, пожалуй, перевешивающий
ранее названные преимущества.
На рис. III.3 представлена более совершенная система ввода
пробы. Порцию анализируемой смеси отбирают медицинским
шприцем и затем заполняют ею сменную дозирующую петлю
шестиканального (иначе — шестиходового) поворотного крана.
180
a
Рис.111.3. Кран-дозатор с изменяемым объемом сменной петли
(дозы).
а — отбор пробы; 6 — дозирование. 1 — узел ввода пробы; 2— слив
избыточного количества пробы; 3 — сменная петля (доза); / — линия
подачи элюента в кран; 5 — линия, связывающая кран с колонкой.
Эта процедура выполняется при атмосферном давлении. Рас-
творитель, находящийся ранее в петле, замещается пробой и
через специальный капилляр вытекает в слив. При повороте
ручки крана в положение ДОЗИРОВАНИЕ подвижная фаза вы-
талкивает содержимое петли крана в хроматографическую ко-
лонку. Перед каждым новым вводом анализируемого образца
петлю следует промывать избытком пробы, чтобы избежать ее
разбавления элюентом. Для выполнения этой процедуры кран
переводится в начальное положение ОТБОР ПРОБЫ. В це-
лом, работа устройства базируется на тех же принципах, что и
работа газовых кранов-дозаторов (см. раздел II. 1.1).
181
Давление или скорость потока в разделительной колонке при
введении пробы не должны нарушаться. Как уже отмечалось,
в жидкостной хроматографии предъявляются более строгие
требования к прокладкам из полимерных материалов: они не
должны набухать в растворителях. Более всего удовлетворяют
этим требованиям фторированные эластомеры. Система ввода
пробы может быть полностью автоматизирована. Все совре-
менные жидкостные хроматографы снабжены автосамплерами
[81].
III.1.4. Колонки
Колонка — наиболее важная часть любой хроматографиче-
ской системы, поскольку результат хроматографического ана-
лиза в существенной степени определяется ее разделительной
способностью.
В практической ЖХ используют обычные аналитические ко-
лонки диаметром 3-5 мм и длиной 5-25 см и микроколонки диа-
метром 0,5-1 мм и длиной от 5 до 10 см. Большинство стандарт-
ных колонок длиной 25-30 см заполнены сорбентом с размером
частиц 5-10 мкм, число теоретических тарелок в них может до-
стигать (5 — 10) • 103 по неудерживаемому компоненту.
Колонку заполняют и закрывают с обеих сторон металло-
керамическими фильтрами (фритами) для предотвращения вы-
сыпания сорбента. Один из вариантов заполнения колонок за-
ключается в создании суспензии подходящего сорбента. Чтобы
полностью удалить суспендирующую жидкость и уплотнить на-
садку, колонку некоторое время промывают элюентом при по-
вышенном давлении. Этот метод получил название “метод сба-
лансированной плотности”. Особенно сложно приготовление
обращенно-фазных колонок, и главная опасность при этом —-
неоднородность упаковки. При заполнении колонок такими сор-
бентами возникает большой электрокинетический потенциал, а
так как привитые сорбенты являются изоляторами, то на их по-
верхности скапливается избыточный заряд, “расталкивающий”
частицы. Лля снятия заряда в суспензию добавляется электро-
лит (ацетат натрия).
Между дозатором и аналитической колонкой помещается
предколонка, заполненная тем же сорбентом, что и основная,
и предохраняющая основную колонку от влаги и сильносорби-
рующихся примесей (это особенно важно при анализе биологи-
ческих объектов).
182
Разрешение и скорость разделения в жидкостных хромато-
графических колонках могут быть существенно увеличены за
счет оптимизации геометрических параметров колонки, усло-
вий хроматографического анализа, характеристик сорбента,
скорости потока подвижной фазы, величины пробы. Для по-
лучения максимальной эффективности колонки рекомендуется
использование сорбента с возможно меньшим размером частиц.
В основном хроматографические колонки для жидкостной
хроматографии рассчитаны на комнатную температуру. Од-
нако в ряде случаев, например при анализе полимерных систем,
необходимо работать при повышенных температурах (для повы-
шения растворимости полимеров в подвижной фазе).
Во всех современных жидкостных хроматографах предусмо-
трено термостатирование. Максимальная рабочая темпера-
тура для силикагелевых колонок составляет 120 °C, а для мо-
дифицированных сорбентов на основе силикагеля — 80 °C. В
последнее время успешно развивается метод высокотемператур-
ной жидкостной хроматографии (от 100 до 200°C), налажен вы-
пуск соответствующей аппаратуры.
III.1.5. Детекторы
III.1.5.1. Основные характеристики детектора
В ходе развития жидкостной хроматографии было испытано
более 20 типов различных детекторов [35, 36, 80-84, 86].
Детектор — это специальный блок хроматографической си-
стемы, реагирующий на различие в составе подвижной фазы,
не содержащей компонентов разделяемой смеси, и подвижной
фазы с разделенными компонентами, выходящими из колонки.
Сигнал детектора после необходимого усиления подается на ре-
гистрацию пишущим потенциометром или компьютерными си-
стемами типа “МультиХром” (см. раздел IV.5).
Детекторы подразделяют на селективные и универсальные.
Селективные детекторы избирательно реагируют на конкрет-
ный тип соединений, которые имеют одно общее свойство, от-
сутствующее у других соединений, либо фиксируют изменение
какого-либо свойства выходящего из колонки растворителя, об-
условленное наличием в нем анализируемых веществ. Это мо-
жет быть изменение оптических свойств элюента (в ИК-, УФ-
или видимой области), его показателя преломления, способно-
сти флуоресцировать, окисляться и т. д. Хотя в разработке се-
лективных детекторов в настоящее время достигнут значитель-
183
ный прогресс, идеальных детекторов с приемлемым пределом
обнаружения в жидкостной хроматографии нет.
Универсальный детектор реагирует на все соединения. Ти-
пичным примером является рефрактометр.
Идеальным детектором был бы такой, который совмещал бы
в себе следующие свойства [36].
1. Отклик на все вещества с прогнозируемым пределом обна-
ружения.
2. Быстрый отклик на любое вещество при отсутствии ре-
акции на изменение свойств подвижной фазы (скорость потока,
состав, температура).
3. Высокая чувствительность.
4. Широкий линейный диапазон (105 и более).
5. Недеструктивность.
6. Возможность проведения количественного анализа.
7. Простота в использовании и дешевизна.
Важнейшие из названных характеристик на примере детек-
тирующих устройств, используемых в газовой хроматографии,
рассмотрены в разделе II. 1.4.
Критерии, по которым выбирают детекторы в жидкостной
хроматографии, связаны со свойствами элюента и анализиру-
емых веществ, принципом детектирования и конструктивными
особенностями того или иного детектирующего устройства.
По данным [83], в 1993-1995 гг. выпускались следующие де-
текторы для жидкостной хроматографии (в скобках указано ко-
личество фирм): спектрофотометрические (73); флуоресцент-
ные (60); рефрактометрические (53); спектрофотометричес-
кие — на фильтрах или с фиксированными длинами волн (49),
на диодной линейке и сканирующие (42); электрохимические
(45); кондуктометрические (41); амперометрические (22); хе-
милюминесцентные (18); масс-спектрометрические, микроколо-
ночные и светорассеивающие (17); хиральные и массовые (по
испарению) (13); инфракрасные, ультразвуковые и радиоактив-
ные (12); пламенно-ионизационные (10).
Большинство химических соединений имеют интенсивные по-
лосы поглощения в диапазоне длин волн 200-800 нм. Это обусло-
вливает наибольшую распространенность фотометрических ме-
тодов детектирования. По меньшей мере 65% всех образцов,
исследуемых методом ВЭЖХ, содержат компоненты, погло-
щающие УФ-излучение на длине волны 254 нм (к ним отно-
сятся ароматические соединения, содержащие подвижные тг- и р-
электроны, функциональные группы С = О, С = S, N = О, N = N,
а также системы сопряженных связей), тогда как свыше 90%
объектов анализа характеризуются поглощением и в других
184
областях УФ-спектра [87]. Использование подходящих раство-
рителей, прозрачных в указанном выше диапазоне, создает при-
влекательность фотометрическим методам, особенно при гради-
ентном элюировании. УФ-детекторы характеризуются высокой
чувствительностью для поглощающих свет соединений, боль-
шим линейным диапазоном (около 5 порядков), малым рабочим
объемом ячеек (~ 1 мкл). Они недеструктивны, относительно
нечувствительны к колебаниям потока подвижной фазы и изме-
нениям температуры, обеспечивают работу в градиентном ре-
жиме. К недостаткам УФ-детектора можно отнести его неуни-
версальность и непрозрачность некоторых подвижных фаз по
отношению к УФ-излучению в интересующей аналитика обла-
сти длин волн.
III.1.5.2. Фотометрический детектор
Фотометрические детекторы подразделяют на:
— детекторы с фиксированной длиной волны в диапазоне 190—
380 нм (фотометры, кратко обозначаемые также аббревиатурой
УЛФ [2]);
— детекторы с дискретно изменяемой с помощью оптиче-
ских фильтров длиной волны (иначе — фильтровые фотометры,
ФУФЛ);
— спектрофотометрические детекторы с плавно изменяемой
длиной волны, предназначенные для регистрации поглощения в
определенной области УФ-спектра (принятая аббревиатура —
СПФ);
— спектрофотометрические детекторы на фотодиодных ли-
нейках (иное название — спектрофотометрические детекторы на
диодных матрицах, СПФ ЛМ, или проще, ДМЛ).
Фотометры, наиболее дешевые и простые (рис. III.4), приме-
няются в ВЭЖХ для выполнения массовых анализов. Источ-
ником света является ртутная лампа низкого давления; детек-
тирование проводится на длине волны 254 нм, которой соответ-
ствует 90% энергии излучения. Свет от источника излучения
проходит через проточную ячейку, в которую из хроматографи-
ческой колонки поступает поток элюента. Лля аналитических
колонок диаметром 4-6 мм, заполненных сорбентом с размером
частиц 5-7 мкм обычно применяются ячейки с длиной оптиче-
ского пути 10 мм, диаметром светового канала около 1 мм и
рабочим объемом 8 мкл. Увеличение рабочего объема кюветы
может привести к нежелательному размыванию хроматографи-
ческих пиков.
185
Рис.III.4. Принципиальная оптическая схема фотометра [36].
1 — фотоприемник; 2 — рабочая микрокювета; 3 — фильтр; 4 —
ртутная лампа; 5 — микрокювета сравнения.
Фильтровой спектрофотометр с четырьмя интерференцион-
ными фильтрами 217, 254, 263 и 279 нм перекрывает область
200-300 нм. Рабочая длина волны (А) выбирается как компро-
мисс между свойствами анализируемых соединений и подвиж-
ной фазы. Для данного вещества отношение высот хроматогра-
фических пиков при двух различных длинах волн есть величина
постоянная. Изменение этого отношения указывает на то, что
интересующий нас пик не соответствует индивидуальному ве-
ществу. В фильтровых спектрофотометрах применяют источ-
ники света, имеющие сплошной спектр.
Спектрофотометр с перестраиваемой длиной волны (напри-
мер, СПФ микроколоночного жидкостного хроматографа “Ми-
лихром-5”) состоит из источника света, монохроматора и фото-
приемника (рис. III.5). В качестве источника света используется
дейтериевая лампа ДДС-30 с непрерывным спектром от 190 до
600 нм. Необходимую спектральную полосу выделяют с помо-
щью либо дифракционных решеток, имеющих 1000-3000 штри-
хов на 1 мм, либо интерференционных фильтров с заданной ши-
риной спектральной полосы. Монохроматический пучок света
поочередно проходит через рабочую и сравнительную проточ-
ные кюветы. Одно из перспективных направлений в использо-
вании фотометрических детекторов связано с применением фо-
тодиодной линейки [88-94]. В 1982 г. фирма Hewlett-Packard вы-
пустила модель HP 1040 А — первый в мире изготовленный се-
рийно спектрофотометр на фотодиодной линейке.
Традиционные спектрофотометрические детекторы для
ВЭЖХ позволяют осуществлять мониторинг элюируемых ком-
понентов пробы по их максимумам поглощения. Сканирование
спектра требует в большинстве случаев остановки потока и за-
нимает несколько секунд или даже минут в зависимости от диа-
пазона длин волн. При использовании фотодиодной линейки
проба сканируется каждые несколько миллисекунд, т. е. спек-
тральная информация выдается практически постоянно.
186
Рис.Ш.5. Оптическая часть УФ-детектора с перестраиваемой
длиной волны [88].
1 — рабочий фотодиод; 2 — проточная ячейка; 3 — фотодиод срав-
неия; 4 — дифракционная решетка; 5 — зеркало; 6 — оптический
фильтр; 7 — дейтериевая лампа.
Непрерывное излучение от источника проходит через про-
точную рабочую ячейку и попадает на дифракционную решет-
ку. Луч отклоняется и фокусируется на плоскости, где рас-
положена фотодиодная линейка (рис. III.6). В таком детекторе
интенсивность излучения в диапазоне длин волн 190-600 нм из-
меряется в течение 10 мс посредством фотодиодной матричной
электроники. Каждый диод (а их 2048) предназначен для изме-
рения узкой полосы (шириной 50 мкм) спектра. Спектральная
информация выдается практически постоянно. Длина волны ис-
пользуется как третья координата измерения дополнительно к
координатам времени и степени поглощения (трехмерное пред-
ставление результатов, рис. III.7). Применяя детектор на ди-
одной линейке, можно выбрать весь диапазон длин волн и за
один хроматографический цикл анализа идентифицировать все
соединения, поглощающие в этом диапазоне [88-92].
В этих условиях можно определить спектр каждого пика и
вычислить максимальное поглощение в специфической области
длин волн. Детекторы на диодной матрице позволяют анали-
зировать пробу более чем на одной длине волны. Это целесо-
образно, когда длины волн максимумов поглощения компонен-
тов анализируемой пробы различны. Параметры, с помощью
которых оптимизируется выходной сигнал, — это ширина по-
лосы поглощения, стандартная ширина полосы длин волн, по-
стоянная времени сигнала.
187
1
Рис.Ш.6. Оптическая часть детектора с фотодиодной линейкой [88].
1 — дейтеривая лампа; 2 — линзы; 3 — заслонка; 4 — проточная
ячейка; 5 —диодная матрица; 6 — дифракционная решетка.
Замечательная особенность детекторов на фотодиодной ли-
нейке состоит в том, что они позволяют проводить количествен-
ные оценки, даже если хроматографические пики совсем не раз-
деляются и перекрываются на всех длинах волн [89, 90]. Это
возможно осуществить с помощью техники “подавления пика”
при использовании дополнительной длины волны сравнения.
Преимущество детекторов на фотодиодной линейке заключа-
ется также в их высокой селективности. Многие хроматографи-
чески неразделенные пики можно выделить количественно вы-
читанием стандартного сигнала из сигнала пробы.
Поскольку детекторы данного типа измеряют сигнал одно-
временно на нескольких длинах волн спектра, появляется воз-
можность выделить “внутренний стандарт” для повышения чув-
ствительности. Интенсивность стандартного сигнала посто-
янно вычитается из интенсивности анализируемого сигнала.
Этот прием позволяет исключить влияние незначительных ко-
лебаний в составе подвижной фазы, а также на дрейф базовой
линии. К тому же применение внутреннего стандарта эффек-
тивно для ослабления дрейфа базовой линии при градиентном
элюировании.
Спектральные возможности детекторов на диодной линейке
позволяют применять их для решения ряда других проблем,
например для автоматической оптимизации расхода раствори-
теля в ВЭЖХ.
188
Рис.П1.7. Трехмерные графики [88].
а — полная спектрохроматограмма экстракта антибиотика урдами-
цина; б — спектрохроматограмма в диапазоне времен удерживания от
9,1 до 9,6 мин (подтверждение неоднородности хроматографической по-
лосы со временем удерживания 9,2 мин).
III. 1.5.3. Рефрактометрический детектор
Рефрактометрический детектор является универсальным де-
тектором в жидкостной хроматографии и совместим практи-
чески с любой подвижной фазой. Это недеструктивный кон-
центрационный детектор. Он непрерывно регистрирует изме-
нения показателя преломления элюата на выходе из колонки.
В свою очередь, показатель преломления меняется пропорцио-
нально изменению концентрации растворенного вещества. Ре-
189
фрактометр широко применяется в хроматографическом ана-
лизе и особенно полезен в тех случаях, когда анализируемые ве-
щества не поглощают в УФ-области спектра, не обладают флу-
оресценцией и электрохимической активностью. Поскольку ре-
фрактометр реагирует на весь поток в целом, он обладает мень-
шей чувствительностью по сравнению со спектроскопическими
или электрохимическими детекторами. Это детектор средней
чувствительности. Чувствительность может быть повышена за
счет правильного выбора подвижной фазы, а именно — при ис-
пользовании элюента с очень высоким или очень низким пока-
зателем преломления (таким может оказаться как сильнополяр-
ный элюент, так и неполярный).
Показания рефрактометрического детектора в существенной
степени зависят от колебаний параметров, влияющих на свой-
ства подвижной фазы: температуры, концентрации анализируе-
мого вещества, пульсаций потока подвижной фазы (флуктуации
потока не должны превышать 0,01 мл/мин на выходе из колонки),
давления. Чувствительность к изменениям температуры для
разных элюентов составляет от 5 • 10-4 до 5 • 10-5 единиц пока-
зателя преломления (е.п.п.) на 1°С, а к изменению давления —
(1 — 5) • 10~4 е.п.п./мПа. Колонка должна работать в условиях
термостатирования.
Существуют рефрактометрические детекторы трех типов
[35, 36].
Рефрактометр оптического отклонения (наиболее распро-
странен). Луч света проходит через кювету, содержащую две
ячейки (рабочую и сравнительную, рис. III.8), заполненные жид-
костями с различными показателями преломления, при этом
луч отклоняется на угол, пропорциональный разности этих по-
казателей преломления. Если же показатели преломления жид-
костей в обеих ячейках кюветы (рабочей и сравнительной) оди-
наковы, отклонения луча не происходит. Преимуществами де-
тектора являются широкий диапазон линейности и малый объем
ячеек (~10 мкл); порог чувствительности 5 • 10~8-2 • 10~7 е.п.п.
Рефрактометр Френеля. Луч света падает на поверхность
раздела двух оптических сред (рис. III.9). Количество света, от-
раженного от поверхности раздела двух фаз (жидкость/стекло),
пропорционально разности показателей преломления и углу па-
дения света на поверхность раздела. Если угол падения подо-
брать таким образом, чтобы угол проникания был ~ 90°(т. е.
близок к полному углу отражения), то небольшие изменения
показателя преломления приведут к значительным изменениям
интенсивности отраженного луча. Вместимость кювет соста-
вляет 3-5 мкл, т. е. возможна работа при небольших расхо-
190
Рис. III.8. Оптическая часть рефрактометрического детектора
оптического отклонения [36].
1 — лампа; 2— маска; 3— линза; 4 — рабочая кювета; 5— сравни-
тельная кювета; 6 — зеркало; 7 — фотосопротивление; 8— усилитель;
9 — пластина установки оптического нуля; 10 — регистратор.
Рис.III.9. Схема действия рефрактометра Френеля.
1 — источник света; 2 — стеклянная призма; 3 — тефлоновая про-
кладка с вырезанными для ячейки углублениями; 4 — стальная пла-
стина; 5 — луч света, проникающий в раствор проб (или в раствор
сравнения); 6 — фотодетектор; 7 — луч отраженного света; 8 — вход-
ной и выходной капилляры.
191
дах элюента. Рефрактометр Френеля наиболее чувствителен
к пульсациям потока, имеет меньший линейный диапазон, порог
чувствительности составляет 10-7 е.п.п. Высокие требования
предъявляются к чистоте стекла.
Интерферометр (выпускается фирмой “Оптилаб”, Швеция).
Коэффициент преломления измеряется методом интерференции
света. Интерферометр имеет две проточные кюветы вмести-
мостью 5 мкл. Чувствительность и линейный диапазон в не-
сколько раз больше, чем у других моделей. Предел обнаруже-
ния 3 мкг/мл. Схема действия интерферометра приведена на
рис. III.10.
Рис. III. 10. Схема действия
интерференционного рефракто-
метра.
1 — фотодиод; 2 — раствор
сравнения; 3 — раствор пробы; 4 —
источник света (лазер); L — длина
ячеек для раствора пробы и раствора
сравнения; пПр и пср — показатели
преломления раствора пробы и рас-
твора сравнения.
III.1.5.4. Флуориметрический детектор
Флуориметр находит применение в экологической химии,
биологии, медицине, при анализе пищевых продуктов, витами-
нов, стероидов [35, 85, 91].
В тех случаях, когда сам по себе образец не флуоресцирует,
можно до или после разделения исследуемого образца на ко-
лонке получить соответствующие производные [80].
Чувствительность флуориметров в 100 раз превышает чув-
ствительность УФ-детекторов, однако линейный диапазон для
них более узкий — 104.
В флуориметрических детекторах можно использовать лю-
бой источник света с фильтром или монохроматором, важно
только, чтобы фокусирующая оптика удовлетворяла требова-
ниям, предъявляемым к геометрии ячейки флуориметрического
детектора, т. е. освещение данного объема исследуемого образ-
ца должно проводиться по наиболее короткому световому пути.
192
В качестве источников излучения обычно используются га-
зоразрядные лампы: ртутная (254 нм), цинковая (214 и 308
нм), кадмиевая (229, 326 нм; предел обнаружения составляет
5 • 10”10 г), источник /3-частиц от 63М1-радиоактивного источ-
ника. Перспективным является флуориметр с лазерным воз-
буждением, который позволяет обнаружить пикограммовые ко-
личества анализируемого вещества. Разработаны детекторы,
которые могут использоваться и как спектрофотометры, и как
флуориметры. В флуоресцентной спектроскопии нашли при-
менение детекторы на основе линейных гребенок фотодиодов и
полупроводниковых фотоумножителей, применяемые также и в
жидкостной хроматографии.
При флуоресцентном детектировании имеют дело с доста-
точно разбавленными растворами. Количество флуоресцент-
ного света, излучаемого возбужденными молекулами, пропор-
ционально интенсивности источника возбуждающего излуче-
ния, объему освещаемого раствора пробы, концентрации рас-
твора обнаруживаемого соединения.
Поглощениев УФ-области проводят при длине волны мак-
симального поглощения для данной группы соединений. И з-
лучение измеряют на выходе фильтра, не пропускающего
лучи возбуждения. Длина волны флуоресцентного излучения
должна быть больше длины волны поглощенного света. По-
движная фаза не должна поглощать свет ни на длине поглоще-
ния, ни на длине волны излучения.
Длину волны возбуждающего излучения рекомендуется вы-
бирать таким образом, чтобы образовывалось максимальное
количество молекул в возбужденном состоянии, возврат кото-
рых в основное состояние происходил бы в результате излуче-
ния. Этому также благоприятствует использование мицелляр-
ных растворов для индуцирования флуоресценции при комнат-
ной температуре.
В тех случаях, когда необходимо существенно понизить пре-
дел обнаружения, следует применять фильтр, дающий макси-
мально возможное светопропускание на выбранной длине волны
возбуждающего излучения, и максимально снизить интенсив-
ность излучения с большей длиной волны.
Отметим некоторые факторы, способные искажать резуль-
таты измерения флуоресценции.
1. Примеси, содержащиеся в элюенте, и, в первую очередь,
растворенный кислород, способны вызывать гашение флуорес-
ценции. Такое же действие оказывают кислородсодержащие
растворители.
193
2. Рассеянный свет от источника возбуждающего излучения,
который возникает из-за наличия в элюате крупных молекул
других рассеивающих излучение частиц. Такое светорассея-
ние можно обнаружить по сокращению диапазона линейности и
возрастанию уровня шумов.
Перед началом работы следует убедиться в отсутствии фо-
новой флуоресценции.
III.1.5.5. Электрохимические детекторы
Различают электрохимические детекторы, которые реаги-
руют либо на изменение свойств элюента, либо на конкрет-
ное анализируемое соединение. К первому типу относится кон-
дуктометрический детектор, ко второму — амперометрические.
Большинство электрохимических детекторов работают в ампе-
рометрическом режиме при котором поддерживается постоян-
ное напряжение между двумя электродами, погруженными в по-
ток элюента, и регистрируется зависимость силы тока от вре-
мени.
Кондуктометрический детектор. Принцип работы его по-
строен на том, что при создании разности потенциалов ионы, на-
ходящиеся в растворе, начинают перемещаться по направлению
к электродам. Проводимость зависит от числа заряженных ча-
стиц в растворе — именно эта зависимость и положена в основу
количественной оценки в кондуктометрии. Таким образом, для
получения количественных результатов должна быть постоян-
ной молярная проводимость. При применении детекторов этого
типа следует избегать протекания электрохимических реакций
на поверхности электродов, поэтому используют источники пе-
ременного тока с частотой от 50 до 1000 Гц и напряжением от 5
до 10 В. Измерения проводятся с применением моста сопроти-
влений Уитстона.
Детекторы данного типа наиболее пригодны для определения
заряженных соединений в элюате, предпочтительны при ана-
лизе малых концентраций ионов.
Вольтамперометрический детектор. Вольтамперометри-
ческое детектирование заключается в измерении электриче-
ского тока в ячейке, возникающего при окислении (восстановле-
нии) регистрируемого вещества на поверхности рабочего элек-
трода при подаче на него определенного напряжения. Этот
детектор обладает очень высокой чувствительностью, сравни-
мой с чувствительностью детекторов по флуоресценции. Кроме
того, он высокоселективен, поскольку не все вещества легко оки-
сляются или восстанавливаются [35].
194
Вольтамперометрический детектор используется при анали-
зе веществ, обладающих электрохимической активностью, т. е.
способностью при определенном потенциале окисляться или
восстанавливаться. В водном растворе этот интервал соста-
вляет от 4-1,2 до —0,8 В (электрод сравнения — хлорсеребря-
ный). Детектирование при потенциалах более 4-1 В или менее
— 1 В (относительно хлорсеребряного) с использованием уголь-
ных или ртутных электродов приводит к уменьшению чувстви-
тельности и селективности. Вещества, содержащие фенольную,
индольную или альдегидную группу, способны окисляться при
низких потенциалах (0,4-0,7 В). В качестве рабочего электрода
используются электроды из платины, золота, углеродной па-
сты, графита. Электрод, на котором протекает интересующая
нас реакция, и является рабочим электродом: катод (реакция
восстановления) или анод (реакция окисления). Электрохими-
ческие детекторы работают только с проводящей водной по-
движной фазой, поэтому они наиболее подходят для обращенно-
фазной или ионообменной хроматографии. Для увеличения
проводимости подвижной фазы в водный элюент добавляют ни-
трат калия, перхлорат натрия, а в органический — перхлорат
тетраэтиламмония.
Между насосом и дозирующим устройством в линию подачи
растворителя часто помещают скруббер — ячейку большей ем-
кости. Потенциал ячейки устанавливают таким, чтобы все ме-
шающие детектированию соединения либо окислялись, либо
восстанавливались до момента ввода пробы.
Для каждого класса соединений характерно свое напряжение
разложения, следовательно, меняя напряжение, можно сделать
детектор высокоселективным.
На практике легче осуществляется окисление, так как не надо
удалять предварительно растворенный кислород. Измеряя за-
висимость силы тока от приложенного напряжения для электро-
активного соединения в статических условиях, получают вольт-
амперограмму (рис.III.11). Характерная особенность вольтам-
перограммы — наличие участка, на котором сила тока после
резкого увеличения практически не меняется с изменением при-
ложенного напряжения. Этот ток называется предельным. В
величину предельного тока входит вклад электролита подвиж-
ной фазы (остаточный, или фоновый, ток).
Следует отметить, что фоновые токи в электрохимических
Детекторах для ВЭЖХ могут в 1000 и более раз превосходить
Диффузионные (диффузионный ток представляет собой разность
между предельным и фоновым токами).
195
Ток, мкА
Рис. III.11. Вольтамперограмма электроактивного соединения [35].
В ячейке детектора создается такая разность потенциалов,
при которой достигается предельный ток для определенного
класса исследуемых веществ.
Регистрируемые соединения в вольтамперометрических де-
текторах претерпевают минимальные электрохимические пре-
вращения. Степень превращения составляет от 1 до 3% (если
степень превращения приближается к 100%, то детектор рабо-
тает как кулонометрический).
Из-за адсорбции подвижной фазы и продуктов окислительно-
восстановительной реакции на поверхности электродов возмож-
на их пассивация, что, в свою очередь, ведет к снижению чув-
ствительности и в конечном итоге может привести к опреде-
ленным затруднениям в количественном анализе. В связи с
этим рекомендовано применять внутренние стандарты. Пасси-
вирование электродов вызывает и постепенный дрейф нулевой
линии, поэтому необходима тщательная очистка поверхности
электрода от посторонних веществ.
В настоящее время разработаны многоканальные электрохи-
мические детекторы и детекторы по флуоресценции, аналогич-
ные УФ-детектору с диодной линейкой. В электрохимическом
детекторе потенциал электрода может быстро и периодически
изменяться, при этом регистрируется зависимость электрохи-
мического тока от напряжения — т. н. вольтамперометрия с
196
быстрым сканированием, что аналогично записи поглощения в
зависимости от длины волны в УФ-детекторах с диодной линей-
кой.
Ш.2. ЛАБОРАТОРНЫЕ ХРОМАТОГРАФЫ
III.2.1. Хроматограф “Цвет-3006”
В основу действия прибора (рис. III.12) положен метод ион-
ной хроматографии, который сочетает в себе принцип ионооб-
менной хроматографии с детектированием по электропроводно-
сти и с компенсацией электропроводящего фона элюента. В ка-
честве элюентов используются электролиты, обладающие вы-
сокой электропроводностью. Для подавления фона служит вто-
рая ионная колонка (подавительная, или компенсационная), сто-
ящая после разделительной. Вторая колонка превращает элю-
ент в раствор с низкой электропроводностью после того, как
произошло разделение компонентов пробы в рабочей колонке.
Таким образом, срок службы компенсационной колонки опреде-
ляется концентрацией ионов в элюенте.
Рис.III.12. Хроматограф “Цвет-3006”.
197
Хроматограф “Цвет-3006” состоит из отдельных блоков.
Блоки подачи жидкости. Основной блок подачи жидкости
создает регулируемый поток элюента через предварительную,
аналитическую и компенсационную колонки. При необходимо-
сти концентрирования пробы используется концентрирующая
колонка, через которую с помощью шприца или дополнитель-
ного насоса прокачивается необходимое количество пробы. По-
сле колонок поток элюента поступает в ячейку детектора по
электропроводности. Разделенные компоненты пробы меняют
электропроводность элюента, что фиксируется блоком измере-
ния электропроводности, сигнал с которого поступает на вход
системы обработки информации САА-06.
Дополнительный блок подачи жидкости БПЖ-58 использу-
ется при регенерации подавительной колонки. Он служит также
для подачи пробы в случае работы с концентрирующей колон-
кой. Блок формирует стабильный поток, величина которого за-
дается цифровым кодовым переключателем на передней его па-
нели. Постоянство потока обеспечивается электронной схемой
управления.
В приборе имеются два насосных блока с электронными схе-
мами управления, два датчика давления, два демпфера. Тем
самым создаются два стабильных потока растворителей. Рас-
ход каждого задается кодовым переключателем. На передней
панели блока жидкости расположены два съемных насосных
корпуса, кодовые задатчики расхода, два цифровых индикатора
рабочего давления, клавиша СЕТЬ с сигнальным светодиодом,
клавиши задания минимального и максимального давлений в си-
стеме с сигнальными светодиодами. С помощью клавиши Ртах
устанавливается ограничение рабочего давления в системе. На-
сос отключается, если давление в системе превысит значение
давления ограничения (Ртах)> и включается вручную после его
понижения. С помощью специальной клавиши Pmin устанавли-
вается ограничение давления на случай внезапной разгермети-
зации жидкостной линии. Включение насоса возможно только
при отжатой клавише ввода ограничения ВКЛ.
Насос. В комплект входят электродвигатель ДПР-72, редук-
тор, насосный блок и компенсатор сжимаемости. Компенсатор
осуществляет предварительное сжатие элюента при переходе
подвижного плунжера из режима всасывания в режим нагнета-
ния. На компенсаторе нанесены цифры условной компенсации:
в положении “0” предварительное сжатие максимальное, в по-
ложении “4” — минимальное.
Насосный блок включает два плунжера, входной клапанный
узел, перепускной клапанный узел и выходной штуцер. Под пе-
198
репускным клапанным узлом установлен фильтр, который за-
щищает пару седло/клапан от засорения мелкими частичками
фторопласта, появляющимися от трения плунжера с манжетным
уплотнением. Признаком засорения фильтра является замедле-
ние или остановка насосного блока при движении его вправо,
а также течь в манжетном уплотнении подвижного плунжера,
которая не устраняется при ее затяжке.
Демпфер. Служит для сглаживания пульсаций давления от
насоса. Он представляет собой трубку из нержавеющей стали,
прокатанную через вальцы и свернутую в спираль. На концах
спирали установлены два штуцера: один для подсоединения ли-
нии, другой — заглушается после заполнения демпфера элюен-
том.
Аналитический блок БА-110. Служит для размещения
термостата колонок, элементов управления жидкостными пото-
ками, устройства для ввода пробы и системы измерения элек-
тропроводности. На передней его панели размещаются: шту-
цер ввода элюента, штуцер ввода регенерирующего раствора,
ручка устройства ввода пробы при дозировании микрошпри-
цем; штуцер ввода пробы при дозировании петлей крана; отвер-
стие для вывода капилляра, по которому сливаются излишки
пробы при петлевом дозировании. На задней панели располо-
жены штуцеры сброса элюента и регенерирующего раствора.
Внутри аналитического блока размещен термостат колонок и
кондуктометрическая ячейка, предназначенная для измерения
электропроводности проходящего через нее раствора. Напря-
жение, подводимое к электродам, составляет 0,5 В. Кондукто-
метрическая ячейка связана с блоком измерения электропро-
водности, который является частью кондуктометрического де-
тектора и находится в блоке БА-110.
Системы ввода пробы. Имеется дозирующий кран со смен-
ными петлями. Предусмотрена также возможность дозирования
пробы микрошприцем через шлюзовое устройство (см. раздел
П.1.2). В комплект прибора входят микрошприцы МШ-10М.
Колонки. В приборе используются четыре типа колонок (с
внутренним диаметром 6 мм): разделительная длиной 100 мм,
подавительные (или компенсационные) длиной 200 мм, концен-
трирующая и дросселирующая длиной 50 мм, предварительная
длиной 100 мм.
Для разделения анионов и катионов необходимы различные
типы колонок с различными ионообменниками. Для катионов
нужна катионообменная разделительная колонка малой емко-
сти, но достаточно большой эффективности (размер частиц не
Должен превышать 20 мкм). В катионной системе подавитель-
199
нал колонка заполняется анионообменником достаточно боль-
шой емкости (2-4 мг-экв/г) — чем больше емкость, тем дольше
служит колонка до регенерации. Признаком насыщения пода-
вительной колонки является резкий рост электропроводности.
При анализе смеси анионов используется анионообменная
разделительная колонка малой емкости и катионообменная по-
давительная колонка большой емкости. В приборе предусмо-
трено использование двух подавительных колонок: одна дей-
ствует постоянно и соединена последовательно с разделитель-
ной колонкой, другая в это время либо регенерируется, либо
находится в запасе. Регенерация — перевод катионита в Н+-
форму, анионита в ОН_-форму — осуществляется путем прока-
чивания через колонку с помощью насоса для регенерации ли-
бо азотной кислоты (0,5 М) , либо раствора гидроксида натрия
(0,5 М) в течение примерно одного часа. Средний срок службы
подавительной колонки 8-10 ч.
Необходимо учесть, что при работе в режиме разделения ани-
онов на подавительной колонке происходит процесс образова-
ния угольной кислоты, распадающейся на СОг и воду. Для ис-
ключения выделения пузырьков углекислого газа из воды после
ячейки устанавливается дросселирующая колонка, заполненная
любым адсорбентом (10-20 мкм).
Для предотвращения быстрого отравления разделительных
колонок (в случае использования воды недостаточно высокого
качества) между насосом и дозатором устанавливают предко-
лонку, заполненную при работе по катионной схеме катионитом
KPC-8II или КУ-2, а при работе по анионной схеме — аниони-
том АРА-12П или АВ-17. Регенерацию предколонок проводят
раз в неделю по следующей схеме: через катионную предко-
лонку прокачивают 200 мл 2%-ного раствора трилона Б (ЭДТА)
в 0,1 М растворе аммиака, затем 100 мл 1 М азотной кислоты,
после чего промывают 200 мл дистиллированной воды; через
анионную — прокачивают 200 мл 0,5 М раствора карбоната на-
трия в 15%-ном этаноле, затем 100 мл 0,5 М раствора гидроксида
натрия и окончательно промывают 200 мл дистиллированной
воды.
Рекомендуемые элюенты. Для определения анионов: стан-
дартный буфер — 0,003 М NаНСОз, 0,0024 М КагСОз (1 гКаНСОз
и 1 г КагСОз в 4 л деионизованной воды); фосфатный буфер (для
определения фосфатов) — 0,001 М NaOH, 0,002 М КагСОз (0,16 г
NaOH и 0,848 г ИагСОз в 4 л воды); боратный буфер — 0,005 М
Na2B40y • ЮН2О (7,63 г Na2B40y • IOH2O в 4 л деионизованной
воды).
200
Для определения катионов: а) стандартный буфер — 0,003 М
HNO3; б) буфер для определения щелочноземельных метал-
лов — 0,001 М раствор дихлоргидрата п-фенилендиамина
(0,724 г дихлоргидрата n-фенилендиамина в 4 л деионизованной
воды) или 0,002 мг этилендиамина и 0,003 М раствор HNO3.
III.2.2. Хроматограф портативный двухканальный
“Цвет-403” (ХПИ-3)
Принцип действия хроматографа “Пвет-403” (первоначаль-
ное название этой модели ХПИ-3, рис.Ш.13) основан на хро-
матографическом разделении веществ методами ионной или
ион-парной жидкостной хроматографии и детектировании вы-
ходящих компонентов пробы по их электропроводности или по
поглощению в УФ-области. Предназначен для анализа ион-
ных и молекулярных соединений, поглощающих на длине волны
254 нм. При сочетании с кондуктометрическим и фотометри-
ческим детекторами возможно использование как одноколоноч-
ной, так и двухколоночной схемы работы.
Рис.Ш.13. Хроматограф “Цвет-403”.
По двухколоночной схеме используют аналитическую ко-
лонку малой емкости (0,1-0,4 мг-экв/г) и подавительную (ком-
пенсационную) большой емкости (5 мг-экв/г). Подавительную
201
колонку после ее насыщения следует регенерировать Призна-
ком насыщения является резкий рост электропроводности, фик-
сируемый на табло экрана монитора, а также смещение вправо
пера самописца. Подавительную колонку отсоединяют от ана-
литической и подвергают регенерации. При анализе смеси ани-
онов она регенерируется 0,5 М азотной кислотой, при разделе-
нии катионов — 0,5 М раствором гидроксида натрия. Перед
прокачиванием кислоты или щелочи, а также после их пропус-
кания необходимо промывать колонки дистиллированной водой
(в первом случае 160-200 мл, во втором — 100 мл).
Прибор укомплектован незаполненными колонками и специ-
альным устройством для их заполнения.
Предел детектирования по иону калия составляет 5 • 10-9г/см3
при использовании кондуктометрического детектора и 5 • 10-6
мг/мл по бензолу при использовании УФ-детектора. Время вы-
хода на режим не более 40 мин, максимальное давление насосов
не менее 8 МПа (80 кгс/см2) с безклапанной головкой и не ме-
нее 15 МПа (150 кгс/см2) с клапанной головкой насоса. Масса
хроматографа не более 20 кг.
Хроматограф “Цвет-403” (ХПИ-3) состоит из двух блоков:
БА-118 и АПК-01. Блок аналитический БА-118 содержит два
насоса, два детектора (кондуктометрический и фотометриче-
ский 254 нм), две независимые системы ввода пробы. На пе-
редней панели расположены также тумблеры управления и ре-
гулировки. Прибор может работать как два независимых хро-
матографа — по параллельной схеме или по последовательной.
На передней же панели расположены кнопки включения СЕТЬ
насосов Н1 и Н2, кодовые задатчики расхода элюента, ручка
БАЛАНС ( компенсация фонового сигнала и сдвига нулевой
линии), кнопки настройки фотометра, переключения полярно-
сти. На правой половине передней панели находятся держа-
тели колонок, манометры для элюентов 1 и 2, узлы ввода пробы
и ручки кранов-дозаторов, переключающиеся в положения ОТ-
БОР и АНАЛИЗ.
Принцип работы хроматографа следующий. Насос создает
регулируемый поток элюента через аналитическую и компен-
сационную колонки (если используется двухколоночная схема).
Разделенные компоненты пробы поступают в ячейку кондукто-
метрического детектора, что вызывает изменение электропро-
водности. При прохождении через ячейку ультрафиолетового
детектора происходит изменение световой проницаемости про-
ходящего раствора. Возникающий сигнал поступает на АПК-01
и, после преобразования, на ПЭВМ, где производятся запись,
обработка и выдача на печать времен удерживания компонен-
202
тов, численных значений измеренных высот и/или площадей пи-
ков, а также рассчитанных (по одному из методов, предусматри-
ваемых программным обеспечением) концентраций компонентов
в пробе.
Последовательное соединение кондуктометрического и фо-
тометрического детекторов позволяет поочередно регистриро-
вать сигналы каждого из них при использовании какой-либо од-
ной системы ввода пробы. При этом вторая система ввода со
вторым насосом может быть в это время использована для реге-
нерации колонок. Кроме того, она может быть востребована для
ввода реагента дериватизации при проведении постколоночных
реакций.
Ультрафиолетовый детектор представляет собой высокоча-
стотный генератор, внутри которого помещается ультрафиоле-
товая лампа, излучающая в основном на длине волны 254 нм.
УФ-свет направляется в ячейку, перед ней установлен свето-
фильтр УФС-1. Ячейка ультрафиолетового детектора офор-
млена в виде параллелепипеда, который, в принципе, может
быть выполнен из любого инертного материала (в данном слу-
чае из стали), имеет два оптических канала, в каждом из кото-
рых предусмотрены вход и выход жидкостных линий. Ячейка
имеет защитное стекло, закрывающее с обоих концов оптиче-
ский сигнал. Свет, проходя через рабочую кювету ячейки, по-
глощается пробой. Разность световых сигналов, прошедших че-
рез рабочую и сравнительную кюветы, фиксируется фотоприем-
никами. Сигнал с фотоприемников обрабатывается и усилива-
ется.
III.2.3. Хроматограф “Цвет-3110”
Жидкостный хроматограф “Цвет-3110” (рис. III.14) предна-
значен для качественного и количественного определения в ла-
бораторных условиях следующих веществ: афлатоксинов, дан-
силпроизводных аминокислот, полиядерных ароматических со-
единений, в частности 3,4-бензопирена, обладающего канцеро-
генной активностью, лекарственных препаратов и других ве-
ществ с флуоресцентной активностью. Хроматограф может
быть использован в режимах изократического и градиентного
элюирования для выполнения анализов как в нормально-, так и
в обращенно-фазном режиме. В качестве сорбента для обращен-
но-фазной хроматографии используют силасорб С-18 зернением
5—10 мкм, для нормально-фазной— силасорб 600 (5-10 мкм).
203
Рис.III.14. Хроматограф “Цвет-3110".
В составе хроматографа имеются следующие блоки: блок
аналитический БА-106, блок измерений и питания БИП-143, ис-
точник питания лазера ЛГН-409, система автоматизации ана-
лиза САА-08. Используемая система автоматизации анализа
САА-08 обеспечивает управление параметрами насосов (рас-
ход, текущее, максимальное и минимальное давление в жидкост-
ной линии, градиентное элюирование), управление работой ав-
тосамплера (отбор пробы из заданной ампулы и дозирование) и
количественную обработку данных, поступающих с лазерного
флуориметрического детектора. Возможна замена САА-08 на
ПЭВМ. Спектральный диапазон лазерного возбуждения 325 нм
флуоресценции охватывает область от 360 до 750 нм (обеспечи-
вается набором интерференционных фильтров). Время выхода
хроматографа на режим не более 2 ч. Предел относительного
среднего отклонения выходного сигнала хроматографа соста-
вляет 1% по высотам пиков и временам удерживания и 2% по
площадям пиков.
Максимальное рабочее давление насосов не менее 30 МПа,
диапазон расходов подвижной фазы от 0,01 до 1 см3/мин. Воз-
можны три режима работы насосов: при постоянном расходе,
при постоянном давлении и с электронным демпфированием.
Автоматический дозатор содержит 20 ампул (объем каждой ам-
пулы 4 см3). Температура термостата колонок регулируется в
пределах от 40 до 100 °C.
204
Принцип работы хроматографа следующий. Пробы, подле-
жащие анализу, помещаются в ампулы блока автоматического
дозатора БАЛ-38. С помощью перистальтического насоса У5
проба заполняет петлю крана-дозатор а, излишки пробы сбра-
сываются на слиз.
В режиме градиентного элюирования в работе одновременно
находятся оба насоса, заполненные различными растворите-
лями. Смешение растворителей производится в тройнике. Пе-
ременный по составу растворитель подается на разделитель-
ную колонку. Изменение состава растворителя производится
путем изменения объемного расхода в каждой линии, при этом
суммарный расход остается постоянным.
В положении ОТБОР ПРОБЫ анализируемая жидкость из
ампулы прокачивается перистальтическим насосом через дози-
рующую петлю и поступает на сброс. При переводе крана в
положение АНАЛИЗ элюент переносит пробу из петли в ко-
лонку. В зависимости от внутреннего диаметра и длины петли
дозируемый объем может изменяться от 10 до 100 мкл и более.
Флуориметр Ф-00 осуществляет измерение флуоресценции рас-
твора, индуцируемой ультрафиолетовым излучением газового
лазера ЛГН-409, в спектральном диапазоне 360-750 нм.
III.2.4. Хроматограф “Цвет-404” (ХПЖ-1)
В основу действия прибора положено сочетание обращенно-
фазной, ион-парной или ионной хроматографии с электрохи-
мическим (вольтамперометрическим) детектированием. Пре-
дел детектирования составляет 5 • Ю~10г/см3 по гидрохинону
и 5 • 10-9г/см3 по фенолу.
Поскольку в хроматографе ХПЖ-1 используется ячейка ма-
лого объема (менее 1 мкл), то время прохождения молекул ве-
щества на поверхности электрода составляет всего лишь мил-
лисекунды, т. е. намного меньше, чем требуется для их полного
превращения (окисления, восстановления) на поверхности, по-
этому степень превращения составляет всего лишь от 1 до 5%.
При этом чувствительность детектора остается очень высокой
из-за малых шумов (10-12 А).
Ток, измеряемый в электрохимической ячейке, представляет
собой сумму нескольких токов: 1) фонового тока, образующе-
гося вследствие окисления или восстановления примесей, вхо-
дящих в подвижную фазу; 2) сигнального тока, возникающего
при окислении или восстановлении регистрируемого вещества
205
и 3) зарядного (емкостного) временного тока, который возни-
кает при включении ячейки или изменении потенциала. В срав-
нительно широком диапазоне потенциалов относительно элек-
трода сравнения Ag/AgCl (от —0,8 до +1,1 В) фоновый ток незна-
чителен, но за его пределами резко повышается. Пределы этого
диапазона определяются процессами окисления и восстановле-
ния подвижной фазы на электродах, материалом электродов.
Например, предел отрицательного потенциала для стеклоугле-
рода определяется процессом восстановления кислорода, рас-
творенного в подвижной фазе:
2Н+ + 2е + О2 —> Н2О2,
2Н++ 2е + Н2О2 —> 2Н2О.
Если от кислорода избавляться путем дегазации, то предел от-
рицательного потенциала будет определяться процессом вос-
становления ионов водорода:
2Н+ + 2е ->Н2.
Предел положительного потенциала определяется процессом
окисления самой подвижной фазы:
2Н2О -+ 4Н+ + О2 + 4е,
2СН3СООН -> 2СО2 + С2Н6 + 2Н+ + 2е,
2СГ -4- С12 + 2е.
Хорошие результаты (незначительный фоновый ток) дают та-
кие буферные растворы, как фосфатные, цитратные и ацетат-
ные. Широко используются в качестве электролитов перхло-
раты. Для обращенно-фазной хроматографии применяются та-
кие типичные растворители, как спирты, ацетонитрил, тетра-
гидрофуран. Для стабильной работы колонки необходимо со-
блюдать требования к ограничению pH (обычно от 2 до 7,5) и
концентрации ионов ((5 — 10) • 10-3 моль/л); желательно, чтобы
вода была деионизована. По составу элюент обычно предста-
вляет собой смесь водного буферного раствора (pH 2-7,5) и ор-
ганического модификанта в соотношении от 10 до 90%. Пригото-
вленный буферный раствор в течение 5 мин дегазируют. Затем
готовят смесь буферного раствора с органическим раствори-
телем в нужном соотношении, смесь фильтруют через фильтр
Шотта и вновь дегазируют в течение 1 мин.
Хроматограф ХПЖ-1 выполнен в малогабаритном варианте
и размещен в едином корпусе. Он представляет собой аналити-
ческий блок, в состав которого входят система питания, система
обработки и управления, насос.
206
Насос. Максимальное рабочее давление насоса не менее
20 МПа, диапазон расходов 0,02-1,00 см3/мин. Возможны два
режима работы насоса — при постоянном расходе и постоян-
ном давлении. Насос создает регулируемый поток элюента, на-
правляемый через дозирующий кран в аналитическую колонку
и электрохимическую ячейку. Кран-дозатор предназначен для
отбора и последующего ввода в колонку порции анализируемой
смеси (объемом около 1 мкл). Пульсации от насоса сглажива-
ются механическим демпфером. Задание режимов работы на-
соса производится с помощью клавиатуры и соответствующих
кнопок пульта управления насосом, контроль режимов осуще-
ствляется с помощью цифрового индикатора.
Колонки. В приборе используется микронаполненная (уста-
ревшее название — микронабивная) колонка внутренним диа-
метром 2 мм и длиной 100 мм. В колонках этого типа для
обращенно-фазной хроматографии применяется адсорбент си-
ласорб С-18 зернением 5-7,5 мкм. Предусматривается вторая
колонка, устанавливаемая после электрохимической ячейки для
создания небольшого давления в ячейке (около 3-4 кгс/см2) с
целью уменьшения влияния растворенного в элюенте газа на
рабочий электрод электрохимической ячейки. Длина колонки
50 мм, внутренний диаметр 6 мм; заполняется тем же сорбентом.
При подготовке к работе колонку промывают этиловым спир-
том (20 мл) и просушивают воздухом. Заполняют суспензией
сорбента в изопропиловом спирте, объем сорбента должен быть
на 15-20% больше расчетного. Перед употреблением колонку
промывают элюентом.
Системы ввода пробы. Имеется дозирующий кран со смен-
ными петлями. Предусматривается также возможность дозиро-
вания пробы микрошприцами через шлюзовое устройство (по-
дробнее см. раздел III.1.4).
Электрохимическая ячейка. Конструктивно оформлена в
виде металлического блока из нержавеющей стали, в который
через специальное сопло подается элюент (элюат) из колонки.
На расстоянии 0,1 мм от сопла расположен электрод с рабочей
поверхностью из стеклоуглерода. На выходе ячейки устана-
вливается сравнительный хлорсеребряный электрод. Контакт
с элюентом осуществляется через фторопластовую ионообмен-
ную мембрану. В качестве вспомогательного электрода исполь-
зуется сам корпус ячейки. Кроме того, предусмотрены сменная
электрохимическая ячейка, конструкция которой отличается от
описанной выше лишь взаимным расположением электродов, а
также двухэлектродная ячейка — корпус с тремя одинаковыми
рабочими электродами с рабочей поверхностью из стеклоугле-
207
рода. Рабочая поверхность электрода должна быть абсолютно
чистой и отполированной. Если поверхность электрода загряз-
нена, то необходимо очистить ее путем полировки порошком
оксида алюминия, или полировальным порошком из корунда
Ml, или пастой ПОЛ ИИТ на бязи. После полировки для удале-
ния полировочного порошка электрод надо промыть этиловым
спиртом и обтереть.
Электрохимический детектор. Состоит из электрохимиче-
ской ячейки с одним рабочим электродом, ячейки с двумя ра-
бочими электродами или системы двух одноэлектродных элек-
трохимических ячеек. Для регенерации поверхности рабочих
электродов при их загрязнениях требуется задать с помощью
клавиатуры потенциал электрода +3,0 В. При этом на рабочий
электрод подается напряжение переменной полярности, равное
1,3 В, с периодом 32 с Через 10 периодов режим РЕГЕНЕРА-
ЦИЯ выключается.
Для управления работой электрохимического детектора име-
ется плата интерфейса.
Блок обработки и управления выполняет следующие функ-
ции: автоматическое обнаружение хроматографического пика,
измерение высоты, площади и воемени удерживания, автома-
тическую коррекцию нулевой линии под хроматографическим
пиком в зависимости от метода коррекции, заданного операто-
ром, расчет калибровочных (градуировочных) коэффициентов,
вывод результатов на печать и индикацию режимов и результа-
тов анализа.
Предусмотрены удаление (стирание) , а также сохранение и
запоминание заданных параметров режима выполнения и ре-
зультатов анализа после отключения сети.
III.2.5. Микроколоночные хроматографы
В микроколоночных хроматографах возможно применение
мало используемых для других видов ВЭЖХ детекторов: масс-
спектрометрического, пламенно-ионизационного и пламенно-
эмиссионного [42, 43, 95].
Типичная скорость потока в этих приборах 0,1-100 мкл/мин.
Уменьшение объема элюента позволяет использовать капил-
лярные колонки с детекторами, для которых поступление рас-
творителя в больших количествах неприемлемо, например масс-
спектрометрический.
208
Хроматографы такого типа оснащены шприцевыми насосами,
обеспечивают более стабильную подачу растворителя. Приме-
ром такого хроматографа может служить ХЖ-1309 — микро-
гельхроматограф для определения молекулярно-массового рас-
пределения полимеров. Детектор — лазерный рефрактометр.
Время анализа 5-10 мин, расход подвижной фазы 100 мкл/мин.
Отечественный микроколоночный жидкостный хроматограф
“Милихром” имеет шприцевой насос (емкостью 2500 мкл), из
упрочненного стекла, контакт жидкости происходит только с
высокоинертными материалами: фторопластом, стеклом, тан-
талом. Можно использовать высокоагрессивные растворители
и добавки. Насос рассчитан на давление 5-7 МПа. Диапазон
подачи растворителя 1-6 мкл/мин. Прибор снабжен спектрофо-
тометрическим детектором с диапазоном длин волн 90-360 нм.
Время сканирования от 0,15 с. Сканирование осуществляется
без остановки потока. Объем микрокюветы ~ 1,5 мкл при длине
оптического пути 1,5 мм. Отбор пробы от 0,1 мкл и более осу-
ществляется засасыванием исследуемого образца в иглу путем
регулируемого хода шагового двигателя, управляющего насо-
сом.
Прибор снабжен колонками двух типов: стеклянными (диа-
метром 1-1,5 мм), рассчитанными на давление 1,5 МПа, и из
нержавеющей стали (2 мм) длиной 60 и 120 мм.
209
Глава IV
ХРОМАТОГРАФИЧЕСКИЙ АНАЛИЗ
IV.1. ПОДГОТОВКА ПРОБЫ К АНАЛИЗУ.
ВЫБОР ХРОМАТОГРАФИЧЕСКОГО МЕТОДА
Подготовка пробы к хроматографическому анализу включает
операции, позволяющие повысить чувствительность и улуч-
шить метрологические характеристики определения, а также
расширить область применения метода. Сюда входят следу-
ющие процедуры:
— отбор порции анализируемого материала, при необходи-
мости — консервация ее на заданное время и транспортировка
в аналитическую лабораторию;
— удаление мешающих веществ, выделение и концентрирова-
ние определяемых соединений;
— превращение определяемых соединений в более удобные
аналитические формы (получение соответствующих производ-
ных — дериватизация);
— гомогенизация пробы (при последующем анализе методом
ВЭЖХ необходимо подобрать растворитель, по элюирующей
силе не превосходящий подвижную фазу; оптимальным явля-
ется растворение подготовленного образца в подвижной фазе);
— введение дозы подготовленного образца в испаритель (до-
затор) хроматографа или непосредственно в хроматографиче-
скую колонку.
Необходимость выполнения всех или только некоторых из пе-
речисленных процедур обусловливается природой анализиру-
емого объекта и свойствами содержащегося в нем доминиру-
ющего вещества и/или растворителя, характером имеющейся
предварительной информации и задачей анализа (качественное
или количественное определение).
Так, например, водные образцы, такие, как плазма, пища,
объекты окружающей среды, не могут быть непосредственно
проанализированы в нормально-фазном режиме ВЭЖХ, так как
даже небольшие количества воды будут негативно влиять на
воспроизводимость результатов. Таким образом, анализиру-
210
емые вещества должны быть предварительно проэкстрагиро-
ваны соответствующим растворителем.
Следует отметить, что для различных вариантов хромато-
графического анализа требования к подготовке пробы могут от-
личаться, например для тонкослойной хроматографии они су-
щественно ниже, чем для анализа методом ВЭЖХ.
Независимо от намеченного плана решения конкретной по-
ставленной задачи, подготовка пробы к анализу является на-
чальным и одним из самых ответственных этапов любой ана-
литической методики. Как справедливо отмечается в моногра-
фии [96], «... Весь процесс выделения и концентрирования по-
лон опасностей, и можно без преувеличения сказать, что изме-
нения, произошедшие на этих ранних этапах анализа, никогда
нельзя исправить на более поздних его стадиях... Ни новей-
шее аналитическое оборудование, ни лучшие из разработанных
способов ввода пробы, ни самые инертные высокоэффективные
колонки или сложнейшее оборудование по обработке данных не
могут дать корректную информацию, если проба подготовлена
для анализа неправильно».
В связи с этим приведем лишь один пример. Если в хроматографи-
ческую колонку ввести разбавленный спиртовой раствор смеси органи-
ческих веществ, существенно различающихся по летучести, то пик рас-
творителя (спирта) перекроет, замаскирует сигналы детектора на многие
летучие соединения, подлежащие определению, а нелетучие компоненты
пробы, оставаясь длительное время в колонке, могут послужить при-
чиной ложных результатов при обработке последующих хроматограмм.
Поэтому при исследовании такого рода объектов необходимо предвари-
тельно удалить все нелетучие вещества и основную часть растворителя,
причем проделать это так, чтобы относительные концентрации летучих
соединений не изменились.
Для отделения пробы от ее матрицы с целью очистки и кон-
центрирования интересующих соединений используют методы
адсорбции и абсорбции, твердофазной, жидкофазной и газовой
экстракции (статический и динамический макро- и микровари-
анты, в том числе стимулируемой микроволновым излучением)
[97], сверхкритической (флюидной) экстракции, дистилляции,
вымораживания, причем часто прибегают к комбинированию
отдельных названных методов и их разновидностей, включая
обработку порции анализируемого материала специфическими
химическими реагентами для обеспечения селективности опре-
деления уже на стадии пробоотбора и повышения чувствитель-
ности последующего хроматографического анализа. С отличи-
тельными особенностями подготовки проб к анализу, связан-
ными с различиями в природе анализируемых объектов и ха-
211
рактером поставленной задачи, требующими соблюдения опре-
деленного регламента выработанных процедур и использования
специального оборудования, можно познакомиться в специаль-
ных монографиях [96, 98-107], официальных нормативных доку-
ментах [108-112], аналитических библиографических [113-117] и
тематических обзорах [118-144].
Довольно часто при подготовке пробы, содержащей компо-
ненты различной природы (кислотные, основные, нейтральные),
для последующего хроматографического анализа приходится
использовать комплекс методических приемов и рекомендаций.
Фрагмент последовательности операций, иллюстрирующий
стратегию подготовки пробы жидкого образца, представлен на
схеме IV. 1.
В любом случае, независимо от происхождения, агрегатного
состояния и химической природы исследуемого образца, необ-
ходимо руководствоваться следующими основными правилами.
1. Отобранная проба должна быть представительной (т. е.
не отличаться по качественному и количественному составу от
анализируемого материала); процедура отбора пробы должна
быть легко воспроизводимой и, по возможности, рассчитанной
на использование легкодоступного (стандартного) оборудова-
ния. Например, для отбора и хранения проб воды или водных
растворов различного происхождения, а также биологических
жидкостей удобно и вполне надежно использовать медицинские
шприцы вместимостью от 1 до 100 мл, снабженные герметизи-
рующими колпачками.
На начальных стадиях подготовки к анализу проб гетероген-
ных объектов (эмульсий, суспензий, пересыщенных растворов
и т. п.) проводят их гомогенизацию, добиваясь полного рас-
творения в подходящем растворителе или прибегая к жидкост-
ной или газовой экстракции. Точно так же (приготавливая рас-
творы, жидкие или газовые экстракты) поступают с твердыми
материалами, анализируемыми на содержание летучих соеди-
нений. При этом следует использовать и растворители, и газы-
экстрагенты, и всю посуду (включая контейнеры для хранения
и транспортировки проб) гарантированной чистоты.
Отметим, что при газохроматографическом анализе проб, со-
держащих большое количество воды, могут иметь место разру-
шение (гидролиз) некоторых неподвижных фаз и осложнения в
детектировании (ДЭЗ). При ВЭЖХ-анализе водные элюенты
не годятся при работе с силикагелевыми колонками. Особое
внимание следует уделить тщательному заполнению паспорта
на каждую отобранную пробу (или на серию однотипных проб),
в отдельные графы которого вписываются:
212
Схема IV.1. Подготовка пробы к анализу на примере жидкого
образца (подробнее см. [145]).
213
название и номер (шифр) образца;
дата, время и место отбора пробы;
номер (шифр) контейнера (контейнеров);
цель анализа;
информация о первичных манипуляциях с анализируемым
образцом, проведенных на месте отбора пробы;
подпись лица, отобравшего пробу;
дата и время доставки пробы в лабораторию;
условия хранения пробы в лаборатории (в холодильнике, в
эксикаторе, на лабораторном столе при комнатной температуре
и т.п.);
использованная аппаратура и условия хроматографического
анализа;
способ разрушения (уничтожения) неиспользованной пробы,
оставшейся после выполнения анализов (что особенно важно
при работе с образцами материалов, обладающих высокой ток-
сичностью), или маршрут (адрес) дальнейшего перемещения
пробы в другой аналитический центр для дополнительного ис-
следования с использованием оборудования (приборов), отсут-
ствующих в данной лаборатории.
2. Время хранения отобранных проб перед анализом должно
быть минимальным, а условия и способы хранения должны ис-
ключать неконтролируемые потери легколетучих соединений за
счет испарения и любые другие физические и химические изме-
нения в составе анализируемого образца.
3. Выбранные способ и/или процедура собственно дозирова-
ния подготовленной пробы в хроматографическую колонку, ма-
териал, конструктивные особенности и температурные режимы
испарителя (крана-дозатора), узла деления паров пробы, ко-
лонки и всех соединительных газовых коммуникаций должны
исключать возможные изменения качественного и количествен-
ного состава смеси анализируемых компонентов из-за возмож-
ных потерь, связанных с испарением в атмосферу, необратимой
сорбцией, термическим разложением или какими-либо химиче-
скими превращениями.
4. Все необходимые промежуточные операции по концентри-
рованию определяемых соединений и отделению их от меша-
ющих компонентов должны быть как можно более простыми и
унифицированными.
С особым вниманием следует относиться к получению хими-
ческих производных — дериватизации — определяемых соеди-
нений, имея в виду воспроизводимое достижение максимальных
выходов и исключение образования побочных продуктов при
минимальных затратах времени. Выбор конкретной методики
214
химической обработки пробы должен быть основательно про-
думан и обоснован.
Особенности использования химических методов на предва-
рительных стадиях подготовки пробы к анализу подробно рас-
смотрены в книгах [70, с.418-428; 146-150] и обзорах [151-153].
В газовой хроматографии это, прежде всего, расширение обла-
сти применения метода (становится возможным анализ нелету-
чих соединений, ускоряется анализ умереннолетучих соедине-
ний), улучшение разделения анализируемых веществ и количе-
ственных характеристик аналитических определений (за счет
исключения или подавления адсорбции ряда компонентов на по-
верхностях газохроматографической аппаратуры, твердого но-
сителя и на границе раздела между твердым носителем и непо-
движной жидкой фазой), повышение чувствительности детекти-
рования производных по сравнению с исходными соединениями,
изменение полярности анализируемых веществ (разделяемые в
нормально-фазном режиме ВЭЖХ после дериватизации могут
быть проанализированы в обращенно-фазном режиме). Некото-
рые наиболее распространенные типы химической трансформа-
ции функциональных групп молекул органических веществ для
последующего газохроматографического анализа представлены
в табл. IV .1.
Многие из химических реакций, приводящих к селективному
и количественному образованию тех или иных производных
определяемых соединений в сложных матрицах, могут выпол-
няться в одну стадию непосредственно в испарителе газового
хроматографа (т.н. on-column дериватизация). К числу таких
реакций можно отнести метилирование и бензилирование низ-
ших жирных и ароматических кислот, фенолов, бензимидазолов,
сульфонамидов, тиоурацилов и др. с использованием недавно
предложенных новых реагентов — ацетата и цианида триметил-
фениламмония и фторида 3,5-бис(трифторметил)бензилдиме-
тиламмония [155-157].
Для расширения области применения жидкостной хромато-
графии с целью повышения чувствительности детектирования
и/или улучшения разделения компонентов пробы, преобразуе-
мых в более удобные аналитические формы (обычно содержа-
щие хромофорные или электрофорные группы), дериватизацию
осуществляют как на стадиях, предшествующих анализу (пред-
колоночная дериватизация), так и непосредственно в хромато-
графической системе до или после формирования зон. Для раз-
деления оптических изомеров прибегают к специальным при-
емам дериватизации, предусматривающим использование хи-
Ральных неподвижных фаз, а также введение хирального агента
215
Таблица IV. 1. Примеры дериватизации, предшествующей газох р о мато графическому анализу
Функциональ- ная группа Тип химического превращения Реагенты Продукты
1 2 3 4
ьо о —соон Диазометан токсичность ственно пер —nh2 —SH Получение нениях (раз галогенпрог —ОН —СООН —SH —nh2 =NH Силилировг содержат не производны непосредств с выделение Этерификация (получение сложных эфиров карбоновых кислот) овый метод наиболее пре ю и нестабильностью са ед использованием Ацилирование (замещение активного водорода на ацильную группу) щильных производных в рушение ассоциатов) и т( вводных чувствительное Силил ирование (замещение активного водорода на триметилсилильную группу) шие широко применяется сколько функциональны! е практически в одну стг енно перед анализом. Пр м осадка S1O2 на стенках Диазометан CH2N2, низшие алифатические спирты (метиловый, изопропиловый, бутиловый) в присутствии кислотных катализаторов (BF3, НС1, H2SC>4 и др.) >стой, быстрый, универсальный и количественный; егс мого диазометана, который не хранят, а получают в м Ангидриды карбоновых или галогенкарбоновых кислот в пиридине, тетрагидрофуране или в других растворителях основного характера основном преследует цель блокирования водородных зм самым расширения круга анализируемых объектов ть определения можно повысить использованием ЭЗЛ М,О-бис(триметилсилил)ацетамид (БСА) CH3C[OSi(CH3)3]=NSi(CH3)3 М,О-бис(триметилсилил)трифторацетамид (БСТФА) CF3C[OSi(CH3)3]=NSi(CH3)3 и др. 5 для получения производных нелетучих соединений. < групп с активными атомами водорода, то все они об 1дию; плохо хранятся (разлагаются влагой воздуха) и и работе с ПИД многие силильные производные разл ячейки и электродах, что влечет за собой понижение —COOR; R = СНз, U30-C3H7, С4Н9 и др. недостатки связаны с алых количествах непосред- —О—COR —NH—COR
связей в исходных соеди- При образовании —О—Si(CH3)3 —COO—Si(CH3)3 —S—Si(CH3)3 —NH—Si(CH3)3; —N[Si(CH3)3]2 =N—Si(CH3)3 Если молекулы последних >азуют соответствующие должны приготавливаться агаются в водородном пламени чувствительности определения
Образование оксимов
Гидрохлориды метокси-, бутокси-, пентилокси- или
бензилоксиамина в пиридине
Окончание табл. IV. 1
=C=NOR
Чаще всего получают метоксимы. По сравнению с исходными карбонильными соединениями метоксимы намного более
стабильны, не разлагаются в условиях анализа и могут быть подвергнуты дополнительной химической обработке
(например, силилированию). Образование бутоксимов (пентилоксимов и бензилоксимов) особенно оправдано при хрома-
то-масс-спектрометрическом анализе стероидов ввиду достаточной летучести производных и простоты интерпретации
соответствующих масс-спектров
=С=О Образование гидразонов
Гидразин или его алкил(арил)производные, чаще
других 2,4-динитрофенилгидразин (в подкис-
ленном растворе)
=C=NH2; =C=NHR
Незамещенные гидразоны могут быть количественно восстановлены в соответствующие алканы по реакции Кижнера
непосредственно в хроматографической колонке-реакторе [154]. Получение твердых 2,4-динитрофенилгидразонОв
(2,4-ДНФГ) особенно оправдано для группового выделения карбонильных соединений из сложных смесей. Прямой
газохроматографический анализ 2,4-ДНФГ возможен лишь в жестких условиях и часто осложняется разложением
2,4-ДНФГ в испарителе хроматографа. Возможна регенерация исходных альдегидов или кетонов (перегидразонирование)
непосредственно в хроматографической системе при введении в фор-колонку (сменный реактор) смеси 2,4 ДНФГ и
реагентов с активированной карбонильной группой (например, а-кетоглутаровой кислоты, п-диметиламинобензаль-
дегида в смеси со щавелевой кислотой).
в подвижную фазу. Химические воздействия на компоненты
анализируемой смеси непосредственно в разделительной ко-
лонке жидкостного хроматографа или в колонке-реакторе, раз-
мещаемой между разделительной колонкой и детектором, фор-
мально родственные применяемым в реакционной газовой хро-
матографии, подробнее будут рассмотрены в разделе IV.2.6.
В общем виде можно выделить четыре различных варианта
дериватизации, используемых в жидкостной хроматографии:
— предколоночная off-line (получение производных и сама
процедура хроматографического анализа разделены во време-
ни);
— предколоночная on-line (введение дериватизирующего
агента непосредственно в поток элюента); получаемые произ-
водные в этом случае должны хорошо растворяться в подвиж-
ной фазе, в процессе химической реакции не должен образовы-
ваться осадок или газ*;
— постколоночная off-line (наименее описанная) и соответст-
венно постколоночная on-line дериватизация (в этом случае ис-
пользуют аппаратуру, обеспечивающую полную автоматиза-
цию процессов получения производных и хроматографического
анализа). Обычно введение хромофорных или электрофорных
групп проводится в гомогенных растворах, реже -— с использо-
ванием твердофазных реагентов.
Широко применяемая предколоночная (off-line) дериватиза-
ция обладает большей гибкостью и отличается большим разно-
образием возможных химических превращений отдельных ком-
понентов анализируемой смеси по сравнению с on-line и пост-
колоночной дериватизацией. Это обусловлено следующими об-
стоятельствами.
1. Хроматографическому разделению подвергаются не ис-
ходные вещества, а их производные.
2. Выбранная химическая реакция может идти довольно ме-
дленно или осуществляться при высоких температурах. На
условия ее выполнения, включая аппаратурное оформление, не
накладывается ограничений, связанных с продолжительностью
и степенью хроматографического разделения.
3. Избыток реагента и любые побочные продукты могут быть
удалены либо разделением, либо в процессе хроматографиче-
ского анализа.
*Ион-парная хроматография может рассматриваться как динамиче-
ский вариант такого типа дериватизации: анализируемое вещество пре-
вращается в другую форму (ионная пара) с иными хроматографическими
характеристиками.
218
Характерные примеры предколоночной дериватизации, охва-
тывающие важнейшие реакции получения производных, реги-
стрируемых ультрафиолетовым и электрохимическим детекто-
рами, представлены в табл.IV.2.
Достаточно широко используются химические методы подго-
товки проб и неорганических материалов. Помимо получения
летучих хелатов металлов и органических производных неко-
торых анионов [158-161] следует выделить перспективный ме-
тод реакционной газовой экстракции, включающий химическую
реакцию с образованием газообразного соединения определяе-
мого элемента, выделение этого соединения в газовую фазу и
последующую его идентификацию и определение [162, 163].
Итак, еще раз отметим, что процесс подготовки пробы к ана-
лизу включает, как правило, несколько этапов, каждый из кото-
рых вносит свой вклад в общую погрешность количественного
результата [128]:
S^Sf+Sfj + Sfu + Sfy,
где S — общая погрешность, характеризуемая стандартным от-
клонением результата анализа; Sj-Sjv — погрешности, допуска-
емые на стадиях отбора пробы (I), гомогенизации образца (II),
подготовки образца к выполнению анализа (отфильтровывание
балласта, концентрирование растворов или экстрактов, дери-
ватизация и т. п. (III)) и выполнения анализа подготовленного
образца (IV) соответственно. Приведенное соотношение пока-
зывает, что следует стремиться к исключению или, по крайней
мере, к сведению к минимуму частных погрешностей каждого
отдельного этапа, обращая внимание на наиболее уязвимые из
них. Бессмысленно, например, улучшать точность измерения
высот или площадей пиков на хроматограмме, если потери от-
дельных компонентов пробы на стадии ее отбора составляют
> 50% (случай отнюдь не редкий при определении следовых
содержаний одного или нескольких компонентов в сложных ма-
трицах).
Итак, прежде чем непосредственно приступить к хроматогра-
фическому анализу исследуемых образцов, необходимо решить,
какой именно метод надо выбрать исходя из природы анализи-
руемого объекта.
В общем виде стратегия выбора хроматографического ме-
тода представлена на схеме IV .2. Следует отметить, что такой
подход справедлив, если у исследователя имеется априорная
информация о характере анализируемого образца. В противном
случае может потребоваться комбинация различных методов.
219
Таблица IV.2. Характерные примеры предколоночной дериватизации в ВЭЖХ
Классы анализируемых соединений Реагент Продукты Длина волны обнаружения при фотометрическом или флуориметрическом детектировании, нм
I 2 3 4
Реакции, приводящие к продуктам, регистрируемым УФ-детектором
Введение в молекулы компонентов анализируемых смесей сильных хромофорных группировок, таких, как, например, ароматического ядра с
одной или несколькими нитрогруппами
ROH
Алифатические спирты,
фенолы
Сложные эфиры соответствующих спиртов
(фенолов) и 3,5-динитробензойной кислоты
254
NO2
Карбоновые кислоты
N-Бром (а также n-нитро или п-фенил) -
ацилбромид
Фенациловые эфиры соответствующих
карбоновых кислот
260
RCOOH
1
2
Карбоновые
кислоты
О-(л-нитробензоил)-М,Ь? —
(диизопропил) изомочевина
RCOOH
(СН3)2СН--N
ХСОСН
(СН3)2СН--NlT
NO2---
Альдегиды и
кетоны
О
R--- С
Н
Амины
2,4-Динитрофенилгидразин
2,4-Динитрофторбензол
э
RNH2
Продолжение табл. IV.2
3
Сложные эфиры соответствующих кар-
боновых кислот и л-нитробензилового
спирта
265
2,4-Динитрофенилгидразоны соответст-
вующих карбонильных соединений
Соответствующие алкилированные
ароматические амины
NO2
Продолжение табл. IV.2
Реакции, приводящие к продуктам, регистрируемым флуориметрическим детектором
Карбоновые
кислоты
RCOOH +
Амины,
аминокислоты
RNH2 +
4-Бромметил-т-метоксикумарин
о-Фталевый альдегид; 2-меркаптоэтанол
Сложные эфиры соответствующих кар-
боновых кислот
(2-Гидроксиэтилтио)-2-алкилизоиндолы
HOCH2CH,SH
360/420
455
Амины,
аминокислоты
5-(N,N-диметиламинонафталин)-1 -сульфо-
хлорид (дансилхлорид)
5-(Ь1,М-диметиламинонафталин)~ 1 -суль-
фоалкиламины
340/520
RNH2 +
SO2C1
SO2 RNH
Окончание табл. ГУ.2
1 2 3 4
Фенолы Дансилхлорид 5-(N ,Ь1-диметиламинонафталин)-1 -суль- фоариловые эфиры 365/520
Реакции, приводящие к продуктам, регистрируемым электрохимическим детектором
Карбоновые
кислоты
RCOOH +
л-Аминофенол
л-Гцдроксифениламвды соответствующих
карбоновых кислот
224
Схема IV.2. Стратегия выбора хроматографического метода
На первом этапе надо ответить на вопрос, достаточно ли
летуч образец, чтобы его можно было проанализировать в ре-
жиме газовой хроматографии. Верхний температурный предел
колонок в газовой хроматографии составляет ~ 350 °C. Таким
образом, обязательным условием является необходимая лету-
честь образца при этой температуре. Немаловажным фактором
является и термическая стабильность анализируемых соедине-
ний при выбранной температуре. Если эти условия соблюда-
ются, дальнейший выбор делается между газо-адсорбционной
(ГАХ) и газо-жидкостной (ГЖХ) хроматографией.
В том случае, если исходный образец нельзя проанализиро-
вать газохроматографически, перед выбором конкретного ва-
рианта жидкостной хроматографии необходимо иметь инфор-
мацию о молекулярной массе компонентов исследуемого объ-
екта. Для разделения крупных молекул (с молекулярной массой
> 3000) — синтетических и природных полимеров — использу-
ется эксклюзионная хроматография (гель хроматография).
В том случае, если молекулярная масса меньше указанной,
должен быть поставлен вопрос о природе растворителя, в ко-
тором может раствориться образец. Если анализируемый объ-
ект растворяется в неполярном растворителе, предпочтительно
использование нормально-фазной жидкостной хроматографии.
Если же требуется полярный растворитель (а значит, и поляр-
ный элюент), надо ответить на вопрос, имеют ли компоненты
исследуемой смеси ионогенную природу. Если да, то к какому
типу ионов они относятся: к сильным или к слабым. Сильные
ионы разделяют методами ионообменной или ион-парной хро-
матографии. В случае слабых ионов можно рассмотреть аль-
тернативные варианты, а именно: а) метод обращенно-фазной
жидкостной хроматографии, при этом ионизация подавляется
путем изменения pH; б) метод ионной хроматографии (соответ-
ственно с усилением ионизации).
Если же информация об ионных свойствах анализируемого
объекта отсутствует, то используют, в первую очередь, обра-
щенно-фазную ВЭЖХ.
225
IV.2. КАЧЕСТВЕННЫЙ АНАЛИЗ В ГАЗОВОЙ И
ВЫСОКОЭФФЕКТИВНОЙ КОЛОНОЧНОЙ
ЖИДКОСТНОЙ ХРОМАТОГРАФИИ
IV.2.1. Стратегия выполнения качественного
анализа, типовые задачи и основные
экспериментальные приемы их решения
При выработке стратегии выполнения качественного анализа
конкретного образца учитывают характер и сложность поста-
вленной задачи, имеющуюся в распоряжении аналитика апри-
орную информацию о возможном составе пробы, опыт, нако-
пленный при решении родственных задач, и уровень инструмен-
тальной оснащенности лаборатории.
В наиболее сложных случаях, как, например, в экоаналити-
ческих исследованиях, при исследовании запаха пищевых про-
дуктов или состава летучих выделений растений или микроор-
ганизмов, возникает необходимость индивидуальной или груп-
повой идентификации десятков и даже сотен соединений. В дру-
гих ситуациях, например при подтверждении индивидуальности
вновь синтезированного лекарственного препарата и определе-
нии природы загрязняющих его примесей, круг веществ, под-
лежащих идентификации, может быть существенно меньше, од-
нако это обстоятельство отнюдь не всегда автоматически пере-
водит данную задачу в разряд более простых.
При составлении общего плана выполнения исследования
конкретных объектов весьма полезной может оказаться недавно
сформулированная концепция о разведочном и подтверждаю-
щем анализах, существенно различающихся как по числу и ха-
рактеру выполняемых экспериментальных операций, так и по
способам интерпретации получаемых данных [164, 165]. Прак-
тически любой как качественный, так и количественный ана-
лиз в зависимости от априорной формулировки его цели может
быть классифицирован как разведочный или подтверждающий
(близкую смысловую нагрузку несут специфичные для эколо-
гической экспертизы термины “анализ пробы неизвестного со-
става” и “определение целевых соединений” [143]).
Наиболее существенные методологические особенности и от-
личия этих двух категорий анализов* (в реальной аналитиче-
*В монографии по теории информации в аналитической химии, издан-
ной на английском языке [166], термин, отвечающий русскому “качествен-
ный анализ” (qualitative analysis), трактуется именно как “подтверждаю-
щий анализ”, тогда как смысловая нагрузка термина “разведочный ана-
лиз” передается словосочетанием identification analysis.
226
ской практике возможны промежуточные варианты) сгруппиро-
ваны в табл IV.3. В соответствии с вышеуказанным, в зависи-
мости от характера задачи и наличия или отсутствия априор-
ной информации можно выделить следующие типовые задачи
качественного анализа.
1. Отнесение пиков на хроматограмме к тому или иному со-
единению при исследовании смесей, состав которых полностью
известен (например, в практике работы химиков-синтетиков при
анализе продуктов изученных химических реакций или в завод-
ских лабораториях при разработке технологического контроля
производственных процессов).
2. Подтверждение наличия или отсутствия в исследуемом об-
разце каких-либо конкретных веществ или представителей опре-
деленных классов химических соединений (групповая идентифи-
кация) при анализе смесей известного происхождения.
3. Идентификация индивидуальных соединений или компо-
нентов смесей неизвестного происхождения, т. е. определение
исследуемых индивидуальных соединений или содержащихся в
анализируемых смесях веществ как химических индивидуумов.
Решение первой задачи, требующее наличия эталонных
(стандартных) соединений или литературных данных по их аб-
солютному или относительному удерживанию в выбранных или
заданных условиях анализа, не представляет сколь-нибудь се-
рьезных затруднений и может быть поручено даже студенту,
лаборанту или инженеру-химику, только приступающим к осво-
ению хроматографических методов Параметры удерживания
индивидуальных соединений или компонентов исследуемых сме-
сей напрямую связаны с коэффициентами распределения этих
веществ в конкретных системах жидкость — газ или жидкость —
жидкость (при фиксированной температуре анализа). Поэтому
в выбранных условиях определяемое соединение должно иметь
одинаковый абсолютный или относительный параметр удер-
живания независимо от того, с какими другими веществами
оно совместно дозируется в хроматографическую колонку. По-
пытки решения двух других типовых задач, названных в по-
рядке их усложнения, с помощью одних только данных по хрома-
тографическому удерживанию из-за недостаточной эффектив-
ности используемых хроматографических колонок и возмож-
ного наложения зон часто приводят к неоднозначным результа-
там. Например, могут быть получены данные, свидетельству-
ющие лишь об отсутствии в пробе тех или иных веществ, но не
гарантирующие отсутствия или присутствия других. Несмо-
тря на поразительные успехи, достигнутые к настоящему вре-
мени в развитии разрешающей способности аналитических ко-
227
Таблица IV.3. Важнейшие особенности и отличил
разведочного и подтверждающего анализов [165]
Критерий сравнения Разведочный анализ Подтверждающий анализ
1 2 3
Формулировка аналити- Общие признаки Допускает ©цементы Максимально кон-
ческой задачи неопределенности и кретна
Априорная информация уточнение в ходе ана- лиза Минимальна Определяемые соеди-
об определяемых веществах Число определяемых Неизвестно; опре- нения известны Известно; прочие от-
компонентов делению подлежат все носятся к мешающим
Необходимые затраты соединения в рабочих диапазонах применя- емых методов. Не существует понятия “мешающие компо- ненты” Не нормируются (как Должны быть мини-
времени (произво- правило, невысокая). мальными (макси-
дительностъ анализа) В зависимости от не- мальна). Варьиро-
Оценка стоимости ана- обходи мости возмож- но дублирование ана- лиза в иных условиях и другие дополни- тельные операции Затруднительна, вание заранее ус- тановленных опти- мальных условий не рекомендуется Должна быть мини-
лиза может быть высокой мальной
Требования к квалифи- Максимальны, хими- В зависимости от
кации специалистов ческая подготовка сложности объектов
Официальные требо- обязательна Нет. Возможность анализа При официальном
вания к лаборатории, выполнения анализа представлении ре-
Производящей анализ определяется анали- зультатов необходи-
Характер представ ле- тиками Перечни идентифици- ма сертификация ла- боратории Количественные дан-
ния результатов рованных соединений ные по содержанию
с оценками их содер- жания в образце известных веществ с указанием погреш- ностей определения
228
Продолжение табл. IV.3
1 2 3
Селективность всех стадий анализа Необходимость допол- нительной химичес- кой обработки образ- цов Минимальна Возможна для целей идентификации компо- нентов Максимал ьна Возможна для повы- шения селективности и снижения предела об- наружения
Характеристика экспериментальных операций
Выбор техники эксперимента, в том Не детерминирована; ориентирована на Определена применяе- мой методикой ана-
числе способа отбора и подготовки проб максимально более широкий круг веществ лиза
Условия хроматогра- В газовой хромато- В газовой хромато-
фического разделе- графии капиллярные графии желательно
ния (при определе- колонки с макси- использование наса-
нии органических мальной эффектив- дочных колонок в изо-
соединений) ностью в широком диапазоне темпера- тур; в ВЭЖХ выпол- нение анализов в нормально- и обращен- но-фазном вариантах с привлечением гради- ентного элюирования Идентификация вещест! термических условиях; в ВЭЖХ применение изократического режи- ма с наиболее распро- страненными элюен- тами и сорбентами
Формулировка задачи Идентификация или определение строения неизвестных веществ Подтверждение при- сутствия известных или предполагаемых соединений
Общая характеристи- Наиболее сложная Желательно использо-
ка стадии интерпре- тации результатов стадия анализа вание простейших ме- тодов обработки дан- ных
Проблема идентифи- Выбор из всего Упрощается за счет
кации как выбор из теоретического много- постановки задачи:
некоторого множества образия органических выбор только из под-
объектов соединений лежащих определе- нию веществ
Неоднозначность Неизбежна. Возможна Должна быть исклю-
идентификации от- групповая идентифи- чена. Групповая иден-
дельных компонентов кация (определение класса соединений) тификация допустима только при соответ- ствующей постановке задачи
229
Окончание табл. IV.3
1 2 3
Наличие неразделен- ных компонентов Неизбежно Должно быть исклю- чено
Характер используе- мого информацион- ного обеспечения Базы аналитических данных с учетом межлабораторных погрешностей Стандартные образцы (см. [167]) с учетом только внутрилабора- торных погрешностей
Проблема идентифи- кации веществ, не представленных в базах данных Крайне актуальна Отсутствует
Наиболее опасные ошибки идентифика- ции, подлежащие контролю и устране- нию в первую оче- I рода (пропуск пра- вильных ответов за счет искусственного повышения их одно- значности) II рода (ошибочное опознание ожидаемых веществ за счет мешающего действия других компонентов)
редь
Количественный анализ (см. раздел IV.3)
Предпочтительные Внутреннего или внеш- Внешнего стандарта
методы количест- него стандарта (абсо- (абсолютной градуи-
венного анализа лютной градуировки) ровки) или стандарт- ной добавки (в хро- мато-масс-спектро- метрии — метод изо- топного разделения)
Необходимость в пре- Отсутствует (опреде- Необходимы стандарт-
паратах определя- емых соединений ляемые вещества до анализа неизвестны) ные образцы
Требования к пределу Зависят от применяе- Регламентированы
обнаружения мых методов анализа формулировкой задачи
Оценка системати- ческих погрешностей Затруднител ьна Должны быть мини- мальными
Оценка случайных В “разумных” преде- Должны быть мини-
погрешностей лах мально возможными
Необходимость метро- логической аттестации применяемых методик анализа Отсутствует Крайне актуальна
Примечание. В [165] рассматриваются условия лишь газо-
хроматографических анализов.
лонок, в изучении взаимосвязи сорбционных характеристик со
230
структурой индивидуальных веществ, в конструировании селек-
тивных детекторов и привлечении ЭВМ для обработки резуль-
татов хроматографического эксперимента, приближающаяся к
абсолютно надежной идентификация неизвестных соединений в
смесях любой степени сложности достигается лишь при мно-
гоуровневом комплексном исследовании (см. следующие раз-
делы книги), причем самым результативным является сочета-
ние газовой или высокоэффективной жидкостной хроматогра-
фии (обеспечивающих разделение компонентов смеси и, часто,
их избирательное детектирование) с другими химическими или
инструментальными физико-химическими методами исследова-
ния, такими, как, например, масс-спектрометрия и ИК(фурье)-
спектроскопия.
Основные современные экспериментальные приемы качест-
венного анализа в газовой и высокоэффективной жидкостной
хроматографии следующие.
1. Измерение параметров удерживания идентифицируемых
соединений различными по полярности неподвижными фаза-
ми — сорбционных характеристик, специфичных для каждой
пары индивидуального вещества и выбранного сорбента в газо-
вой хроматографии; в ВЭЖХ представительным является изме-
рение параметров удерживания идентифицируемых соединений
в различных режимах — нормально- и обращенно-фазном.
Наряду с измерением параметров удерживания весьма полез-
ными для дальнейшего совместного использования оказываются
еще три приема.
2. Сопоставление сигналов (откликов) хроматографических
детекторов различного принципа действия (универсальных и
избирательно реагирующих на представителей отдельных клас-
сов химических соединений). К избирательным (селективным)
детекторам в ВЭЖХ относятся электрохимический, флуори-
метрический и ультрафиолетовый. При флуориметрическом
и У Ф-детектировании избирательность легко регулируется на-
стройкой на одну из фиксированных длин волн, предусматрива-
емых конкретной моделью прибора. Использование УФ-детек-
тирования на диодной матрице открывает возможность одно-
временной регистрации поглощения на нескольких длинах волн.
В газовой хроматографии широко используются следующие
селективные детекторы: электронозахватный (обладающий
ультравысокой чувствительностью к галоген- и особенно хлор-
содержащим органическим соединениям), термоионный (высо-
кая чувствительность и селективность определения фосфор-,
азот- и галогенсодержащих (кроме фторсодержащих) веществ),
а также пламенно-фотометрический (избирательная регистра-
231
ция фосфор- и серосодержащих органических соединений). Вы-
пускаемые с 1989 г. фирмой Hewlett-Packard газовые хромато-
графы с атомно-эмиссионным детектором открывают уникаль-
ную возможность одновременной многоканальной избиратель-
ной регистрации органических соединений, в состав молекул
которых входят различные химические элементы (предусматри-
вается возможность настройки детектора на обнаружение чуть
ли не всех элементов таблицы Л. И. Менделеева).
3. Прямая регистрация масс-спектров или ИК-спектров элю-
ируемых из колонки компонентов разделяемых смесей при ис-
пользовании комбинированных приборов, т. н. двойных гибри-
дов хроматограф — масс-спектрометр и хроматограф —
ИК(фурье)-спектрометр, а также тройного гибрида хромато-
граф — ИК(фурье)-спектрометр — масс-спектрометр, с целью
определения молекулярной структуры идентифицируемых со-
единений* .
В тех случаях, когда в масс-спектрах присутствует сигнал
молекулярного иона, появляется дополнительная возможность
определения и молекулярной массы идентифицируемого соеди-
нения.
4. Проведение химических реакций в аналитической газохро-
матографической колонке или специальных микрореакторах,
примыкающих к ней, одновременно и параллельно с процессом
разделения с целью получения из анализируемых веществ ха-
рактерных производных или соединений с характерной окрас-
кой, продуктов пиролитического расщепления и т. п.
IV.2.2. Параметры удерживания
Важнейшие параметры удерживания в колоночной хромато-
графии и их определения в современной трактовке приведены в
[53, 170-172].
*Для сокращенного обозначения таких гибридных методов в совре-
менной англоязычной литературе стали использовать термин “hyphenated
methods” (в буквальном переводе на русский язык — “написанный через
черточку”). Современное состояние и перспективы развития ©тих мето-
дов освещены в специальном выпуске Journal of Chromatography [168]. На-
званные комбинации приборов являются наиболее распространенными,
но не единственно возможными (одним из перспективных направлений
является, например, сочетание жидкостного хроматографа с ЯМР-спек-
трометром [169]).
232
При сохранении в ходе опыта постоянными температуры ко-
лонки (изотермическая хроматография) и скорости подвижной
фазы первичными экспериментально измеряемыми являются
следующие параметры:
— полное время удерживания (total retention time) анализиру-
емого вещества, /д;
— полный объем удерживания (total retention volume), или
полный удерживаемый объем, анализируемого вещества, Vr;
— полное расстояние удерживания (total retention distance)
анализируемого вещества, /r (отрезок временной оси хромато-
граммы, соответствующий времени или объему удерживания).
На численные значения этих параметров оказывают влияние
следующие факторы:
— состав, свойства и количество неподвижной фазы в газовой
и жидкостной хроматографии (в эксклюзионной хроматографии
определяющим является и объем пор сорбента), а также при-
рода твердого носителя в распределительном варианте газовой
и жидкостной хроматографии;
— природа и состав подвижной фазы (в частности, в ВЭЖХ
процентное содержание органического модификатора, ионная
сила, pH, наличие комплексообразующих добавок), а также, в
газовой хроматографии, перепад давления подвижной фазы на
входе и выходе колонки;
— условия проведения опыта (температура колонки и ско-
рость подвижной фазы);
— конструктивные особенности используемой аппаратуры
— свободные (“мертвые”) объемы колонки, испарителя (доза-
тора), детектора и соединяющих их коммуникаций;
— метод дозирования и величина дозы* (опасность пере-
грузки колонки и выхода из области линейной изотермы сорб
ЦИи).
*И то, и другое влияет не только на параметры удерживания, но и на
форму пика (отклонения от симметричной), что позволяет обнаруживать
эти искажающие влияния. Чтобы избежать ошибок при качественных
определениях, необходимо стремиться к компактному (импульсному) до-
зированию возможно меньших количеств анализируемых веществ (требу-
ются высокочувствительные детектирование и регистрация сигнала де-
тектора). В газовой хроматографии, при работе с растворами жидких
или твердых соединений следует весьма продуманно задавать темпера-
туру испарителя. С одной стороны, необходимо обеспечить как можно
более быстрый переход характеризуемых веществ в парообразное состоя-
ние, с другой стороны, не допустить их неконтролируемого термического
Разложения. Как правило, температура испарителя должна быть выше
температуры кипения наименее летучего компонента смеси на 30—50°С.
233
Полное время удерживания (или сокращенно — время удер-
живания) tn — это время от момента ввода пробы в хромато-
графическую колонку до момента выхода из нее максимальной
концентрации анализируемого вещества.
Полный объем удерживания, или полный удерживаемый объем,
(сокращенно — объем удерживания), Vr — объем подвижной
фазы, прошедший через хроматографическую колонку от мо-
мента ввода пробы до момента выхода максимальной концен-
трации анализируемого вещества. В газовой хроматографии
полный объем удерживания (в см3) находят по уравнению
Vr = tRFc, (IV. 1)
где Fc — объемная скорость подвижной фазы, измеренная при
давлении на выходе из колонки и при температуре колонки.
Многие современные модели газовых хроматографов ком-
плектуются специальными блоками (системами) точного зада-
ния и (или) измерения расходов газа-носителя и вспомогатель-
ных газов. При отсутствии таких устройств объемную скорость
газа-носителя приходится измерять с помощью мыльно-пленоч-
ных измерителей расходов газа при температуре окружающей
среды. Рабочей жидкостью в этих измерителях чаще всего
является водный раствор мыла или поверхностно-активного ве-
щества. Поэтому при вычислении удерживаемых объемов сле-
дует использовать значение объемной скорости, исправленное
на температуру колонки и давление водяного пара при темпе-
ратуре измерения:
FC = F~
Ta
(IV.2)
где F — измеренная объемная скорость газа-носителя, мл/мин;
Тс — температура колонки, К; Та — температура окружающей
среды (ambient temperature), К; pw — давление насыщенных па-
ров воды при температуре окружающей среды, Па; ра — атмо-
сферное давление (ambient pressure), Па.
В ВЭЖХ задача измерения объемной скорости подвижной
фазы решается чисто приборными средствами. В современных
жидкостных хроматографах предусматривается возможность
задания расхода подвижной фазы с точностью до 0,1 мл/мин.
Полное время удерживания (в минутах или секундах), как
правило, измеряют с помощью электронных интеграторов или
компьютерных систем обработки результатов хроматографиче-
ского анализа, а при их отсутствии — секундомерами (вруч-
ную).
234
Величине Vr соответствует отрезок ОВ на рис. IV.1, если по
оси абсцисс отложен объем газа-носителя; если по оси абсцисс
отложено время, то отрезок ОВ отвечает полному времени удер-
живания. Полное расстояние удерживания Ir измеряют на хро-
матограмме линейкой от старта до вершины пика.
Рис.IV.1. Типичная хроматограмма индивидуального веще-
ства и несорбируемого соединения.
Названные параметры (объемы, времена, расстояния) удер-
живания могут быть использованы для качественной характери-
стики соединения лишь при проведении анализа в строго задан-
ных условиях на одном и том же приборе. Для сопоставления
получаемых значений удерживаемых объемов с литературными
данными или полученными на другом приборе, или в иных усло-
виях (для той же неподвижной фазы и температуры колонки)
необходимо вводить ряд поправок: на объем подвижной фазы,
не принимающий участия в вымывании компонентов пробы (в
газовой и жидкостной хроматографии), на градиент давления
газа-носителя по слою неподвижной фазы (в газовой хромато-
графии) и на количество неподвижной фазы.
В зависимости от того, какие поправки учитываются, разли-
чают следующие характеристики удерживания.
235
Исправленный объем удерживания (adjusted retention volume)*
анализируемого вещества, VR — полный объем удерживания,
пересчитанный с учетом поправки на объем подвижной фазы,
заполняющий хроматографическую систему (узел дозирования,
колонку, детектор и связывающие их коммуникации) к моменту
начала опыта и не принимающий участия в элюировании ана-
лизируемого соединения, VM:
Vk = vR - VM- (IV.3)
В новых правилах номенклатуры для хроматографии [53,
170-172] параметр Kt определен как объем пребывания (hold-up
volume) в хроматографической системе вещества, концентрация
которого в неподвижной фазе пренебрежимо мала по сравне-
нию с его концентрацией в подвижной фазе Иными словами, Kt
представляет собой объем подвижной фазы, необходимый для
элюирования неудерживаемого вещества (устаревшее, но пока
еще весьма часто встречающееся в литературе название пара-
метра К — мертвый объем хроматографической системы, или
мертвый удерживаемый объем, dead volume [173]). Кроме этого
названия параметра К в англоязычной литературе по ВЭЖХ
многие авторы пользуются определениями:
— column void volume и равнозначное по смысловой нагрузке
kinetic void volume — пустой (незанятый неподвижной фазой)
объем [174, 175];
— thermodynamic void volume — совокупность объемов по-
движной фазы, заключенной в колонке между частицами сор-
бента (interparticle volume of the column) и, кроме того, проника-
ющей во все доступные внутренние поры сорбента [175].
Дополнительный учет объема всех пор неподвижной фазы
особенно важен в эксклюзионной хроматографии, поскольку
здесь объем удерживания VR зависит не только от размера мо-
лекул элюируемого вещества, но и от характера распределения
пор неподвижной фазы по их размерам.
Исправленное время удерживания (adjusted retention time) ана-
лизируемого вещества t'R находят по аналогии с определением
параметра VR по уравнению
t'R = tR - tM, (IV.4)
*В правилах номенклатуры, выработанных Научным советом по хро-
матографии РАН [53], а также в справочнике [173] и некоторых других
авторитетных изданиях исправленный объем удерживания и родствен-
ный ему параметр исправленное время удерживания (см. ниже) названы
как приведенный объем удерживания и приведенное время удерживания
(буквальный перевод с английского соответствующих терминов adjusted
retention volume и adjusted retention time).
236
где tM — время пребывания в хроматографической системе не-
удерживаемого вещества (hold-up time), называемое часто мерт-
вым временем (dead time). Последнее название параметра tM
квалифицируется в [170] как устаревшее.
При невозможности прямого экспериментального измерения
Пм или tM обращаются к косвенным расчетным [176-179] или гра-
фическим [179] методам, в основе которых лежат известные за-
висимости между объемом (временем) удерживания и числом
атомов углерода соединения, принадлежащих к какому-либо го-
мологическому ряду (см., например, лабораторную работу 5).
Несмотря на то, что теория хроматографии — единая, из-
мерение как мертвого объема, так и мертвого времени в жид-
костной хроматографии сталкивается с определенными трудно-
стями. В литературе накоплены многочисленные рекомендации
по определению VM в различных вариантах ВЭЖХ, обобщен-
ные в обзоре [180], библиографический список которого насчи-
тывает 120 названий, и в сокращенном варианте рассмотренные
в книге [173]. Сам факт обилия подобных рекомендаций (число
которых продолжает увеличиваться) свидетельствует о слож-
ности проблемы. Обусловлено это следующими причинами:
— коэффициенты диффузии в жидкой фазе по крайней мере в
104 раз меньше чем в газе;
— вязкость элюента в 100 раз выше для жидкостей, чем для
газа;
— в газовой хроматографии взаимодействие между молеку-
лами подвижной и неподвижной фаз пренебрежимо мало, в жид-
костной хроматографии эти взаимодействия значительны.
В том случае, когда подвижная фаза — жидкость, она может
сорбироваться стационарной фазой, вытесняя молекулы анали-
зируемого вещества. При этом могут существенно изменяться
факторы удерживания (см. ниже).
Важность как можно более строгого определения мертвого
объема (времени) очевидна: от этого зависит точность вычи-
сления фундаментальных параметров, используемых при опре-
делении хроматографических характеристик анализируемых
веществ, основанных на их удерживании неподвижной фазой.
В газовой хроматографии численные значения 1М и VM в опре-
деленных условиях могут быть легко найдены введением веще-
ства, для которого коэффициент распределения очень мал по
сравнению с его значением для других компонентов. Обычно
(при работе с детектором по теплопроводности или по плотно-
сти) для этой цели используют азот, воздух или благородные
237
газы* [173]. При работе с ПИЛ значение VM часто рассчиты-
вают по пику метана, который вводят непосредственно перед
дозированием анализируемого образца.
Использование в качестве характеристики Пм в ВЭЖХ хро-
матографического поведения вещества-маркера (стандарта) оп-
тимально, поскольку позволяет снизить возможные ошибки в
определении мертвого объема. В качестве стандарта рекомен-
дуется выбирать изомерное анализируемым веществам соеди-
нение либо гомолог, поскольку наличие одной и той же функци-
ональной группы обеспечивает идентичность взаимодействий.
Вместе с тем, в литературе отмечается, что значения мертвого
объема часто зависят не только от состава подвижной фазы,
но даже от способа ее приготовления (массовые или объемные
отношения, порядок смешения компонентов подвижной фазы).
В качестве маркеров для определения Пм предлагались раз-
личные соединения: первый выходящий из хроматографиче-
ской колонки компонент (первый пик на хроматограмме); рас-
творы неорганических солей (в режиме обращенно-фазной хро-
матографии) — ионные образцы (нитрат или нитрит натрия,
хлорид или иодид калия, бихромат калия); фруктоза, бензой-
ная кислота, бензоат натрия, ацетон, NjN-димети л формамид и
др. При этом рекомендовано выбирать маркеры таким образом,
чтобы вещества позволяли проводить хроматографический ана-
лиз с использованием наиболее распространенного ультрафио-
летового детектирования. Наряду с неорганическими солями в
режиме обращенно-фазной хроматографии (отметим, что боль-
шинство анализов в ВЭЖХ, около 80%, выполняется именно в
обращенно-фазном режиме) в качестве соединений-маркеров мо-
гут быть рекомендованы тяжелая вода, а также дейтерирован-
ные компоненты подвижной фазы: метанол, ацетонитрил, тетра-
гидрофуран. При работе с рефрактометрическим детектором
предпочтение отдается тяжелой воде D2O.
В литературе также имеются указания на использование в
качестве маркеров для определения мертвого времени репер-
ных веществ, содержащих меньшее число углеродных атомов,
чем компоненты подвижной фазы. В случае нормально-фазного
варианта, где в качестве подвижной фазы могут выступать али-
*Строго говоря, этот экспериментальный прием позволяет найти
мертвый объем хроматографической системы, включающий свободные
(незанятые неподвижной фазой) объемы испарителя, колонки, детектора
и связывающих их коммуникаций. При одновременном синхронном до-
зировании неудерживаемого вещества в узлы ввода пробы, смонтирован-
ные на входе в колонку и соответственно на выходе из нее (на входе в
детектор), возможно определение мертвого времени (объема) собственно
колонки [181].
238
фатические углеводороды, такой подход может себя оправдать,
но он неприемлем для обращенно-фазного варианта, где, напри-
мер, элюентом служит водно-метанольная система.
Для определения мертвого времени в нормально-фазном ре-
жиме (т. е. когда полярность неподвижной фазы превосходит
полярность подвижной) рекомендовано также использование хи-
мических соединений, по элюирующей силе уступающих по-
движной фазе, либо применение дейтерированного раствори-
теля. Так, если в качестве подвижной фазы в нормально-фазном
варианте используется н-гептан и хлористый метилен соответ-
ственно, то в качестве маркеров могут выступать н-гексан и дей-
терированный дихлорэтан (детектор — рефрактометрический);
для работы с УФ-детектором рекомендован тетрахлорэтан.
Известны и косвенные методы определения 17м, основанные, в
частности, на корреляционных зависимостях объема удержива-
ния от каких-либо физико-химических свойств хроматографи-
руемых веществ, например от числа атомов углерода в выбран-
ном гомологическом ряду.
Эти подходы, как отмечалось выше, часто используются в
газовой хроматографии, однако нет никаких принципиальных
ограничений для их применения и в ВЭЖХ, осуществляемой в
изократических условиях [178, 179, 182]. Для всех видов хрома-
тографии, в которых удерживание обусловливается образова-
нием комплексов включения между хроматографируемыми со-
единениями и неподвижной фазой, мертвый объем предложено
находить из графической зависимости между Vr и размерами
молекул веществ [183].
По мере уменьшения размеров молекул элюируемых веществ
снижаются вероятность контакта и степень их взаимодействия
со стенками полости сорбента, и, как следствие, уменьшается
удерживание хроматографируемых соединений данной непод-
вижной фазой. Подобное решение задачи определения мерт-
вого объема оказалось, в частности, весьма эффективным при-
менительно к колонкам, содержащим в качестве неподвижной
фазы /?-циклодекстрин. Для построения указанных корреляци-
онных зависимостей использовали экспериментально измерен-
ные удерживаемые объемы низших алифатических спиртов Сг~
С4 и рассчитанные по справочным данным размеры их молекул
[183].
Отношение исправленного времени удерживания к мертвому
времени {фактор удерживания, retention factor, k) является ха-
рактеристикой продолжительности нахождения молекул анали-
зируемого соединения в неподвижной фазе (в сорбированном
или растворенном состоянии) относительно времени пребыва-
239
ния их в подвижной фазе (в транспортном потоке)*:
к —
м
(IV.5)
В газовой хроматографии учет поправочного фактора j (mobi-
le phase compressibility correction factor), характеризующего сжи-
маемость подвижной фазы**.
3 (р, /Ро)2 - 1
2 (р, /Ро)3 - 1
(IV.6)
(рг- — давление на входе в колонку; ро — давление на выходе из
колонки), приведение затраченного на элюирование вещества,
объема газа-носителя к О °C (273,15 К) и отнесение его к 1 г не-
подвижной фазы дает возможность определить принципиально
новую физико-химическую константу соединения — его удель-
ный объем удерживания
_ 273,15
6 WSTC
ИЛИ
v _ з (/д -/М)Е- 273,15 / _ Pw\ (Рг/Ро)2 - 1
6~ 2 IVsTa pJ (Pi/Po)3-!’
(IV-7)
(IV.8)
где И4 — масса неподвижной фазы (сорбента) в колонке, г.
Если приборное оснащение газового хроматографа позволяет
осуществить прямое измерение объемной скорости газа-носите-
ля Fc при давлении на выходе из колонки и при температуре
колонки, выражение (IV.8) преобразуется к виду
т/ _ 3 7д ~ ^м)Ез • 273,15 (Рг/Ро)2 — 1
g~2’ WSTC ' (р,/Ро)3-1' ( J
Измерение удельных удерживаемых объемов лежит в основе
исследования физико-химических свойств веществ методом га-
зовой хроматографии, так как они являются такими же харак-
терными константами, как температура плавления (кипения),
показатель преломления и плотность.
Однако в практике качественного газохроматографического
анализа Ц; применяются редко, поскольку их расчет возможен
*Использовавшиеся ранее и широко применяемые в периодической ли-
тературе и по сию пору для обозначения параметра к выражения коэффи-
циент, или фактор, емкости [емкостный фактор — capacity factor) по но-
вым правилам номенклатуры для хроматографии ИЮПАК [170] признаны
устаревшими. Единственным рекомендованным символом, представля-
ющим данный параметр, является к (а не к, встречающийся довольно
часто в публикациях по жидкостной хроматографии).
**В жидкостной хроматографии сжимаемость подвижной фазы прене-
брежимо мала.
240
лишь при использовании прецизионной аппаратуры, позволя-
ющей измерять абсолютные значения температуры колонки и
скорости газа-носителя. Кроме того, для их определения необ-
ходимо знать количество неподвижной фазы в колонке, экспе-
риментальное определение которого с высокой точностью при
работе в газо-жидкостном варианте, особенно при использова-
нии капиллярных колонок, встречает практические трудности.
При работе с капиллярными колонками, на стенки которых
неподвижная фаза наносится непосредственно в лаборатории
статическим методом, многие из названных трудностей опре-
деления Vg отпадают. Расчет Ve в этом случае можно прово-
дить по вытекающему из (IV.9) модифицированному уравнению
[184], включающему лишь ограниченное число достаточно про-
сто контролируемых экспериментальных параметров (измеряе-
мые с неминуемо большими погрешностями масса неподвижной
фазы и объемная скорость газа-носителя в это уравнение не вхо-
дят):
_ k ( 2 273,15
6 - С \ тс
(IV.10)
где k — фактор удерживания (коэффициент емкости); С — кон-
центрация неподвижной фазы в растворе, заполняющем под-
готовленную капиллярную трубку при изготовлении колонки
статическим методом, г/мл; Тс — абсолютная температура ко-
лонки, К; р — плотность неподвижной фазы при температуре
колонки, г/мл. Строгое численное значение р, как правило,
неизвестно, однако плотность полимеров и других соединений,
обычно используемых в качестве неподвижных фаз в капилляр-
ной газовой хроматографии, варьирует в довольно узких преде-
лах [185]: от 0,8 до 1,25 при 50°C, от 0,7 до 1,1 при 250°С. Ввиду
доказанного в [184] малого влияния этого параметра на оконча-
тельный результат, авторы уравнения (IV. 10) считают возмож-
ным проводить расчет Vg, принимая р = 1 г/мл (при температу-
рах колонки ~100-150°С) и р = 0,9 г/мл (~ 150-250°С).
В повседневной практике и в газовой, и в жидкостной хрома-
тографии гораздо большее распространение, нежели Vg, полу-
чили такие параметры, как относительное удерживание (relative
retention) г и фактор разделения (separation factor) at
^(st) ^k(st) ^k(st)
(IV. 11)
Величиной г характеризуют удерживание соединений i относи-
тельно какого-либо стандартного вещества (st), исправленный
объем (время, расстояние) удерживания которого (^д(б1)>
241
^fi(st)) может быть больше или меньше соответствующего па-
раметра удерживания интересующего соединения. Отсюда чи-
сленное значение г может быть как больше, так и меньше 1 (или
равно 1)
а = Vfl(2) = *й(2) = Zfl(2) = М (IV.12)
^й(1) ^й(1) ^й(1) ^(1)
Величиной а пользуются для характеристики относительного
удерживания двух соединений (1 и 2), последовательно реги-
стрируемых на хроматограмме. Поскольку, по определению
Пд[2)(<й(2)-") > ^й(1)(^й(1)--)> численное значение а всегда боль-
ше 1. Ранее в литературе фактор разделения, рассчитанный
по уравнению (IV. 12), иногда называли селективностью. Пра-
вилами [170] использование для фактора а этого названия не
рекомендуется.
В качестве стандартных веществ, когда это возможно, при-
меняют 1/-алканы (в газовой хроматографии) и алкан-2-оны, ал-
киларилкетоны или алкил-4-гидроксибензоаты ( в обращенно-
фазной ВЭЖХ). Если неудобно или невозможно использо-
вать эти соединения, следует применять стандартные вещества,
параметры удерживания которых относительно названных из-
вестны*. Еще удобнее и целесообразнее определять параме-
тры удерживания относительно двух стандартов один из ко-
торых имеет меньшее, а другой — большее время удержива-
ния, чем характеризуемое соединение. При этом появляется
возможность представлять данные об относительных величинах
удерживания в форме так называемых интерполяционных пара-
метров удерживания, из которых наибольшее распространение
получили индексы удерживания.
Индекс удерживания I, введенный в 1958 г. Ковачем в прак-
тику газохроматографического анализа, впоследствии (начиная
с 90-х годов) стал применяться и для представления данных по
относительному удерживанию в высокоэффективной жидкост-
ной хроматографии.
Индекс удерживания (Ковача) в изотермической газовой хро-
матографии, характеризующий удерживание вещества непод-
вижной фазой (н.ф) при температуре t (°C) относительно двух
реперных к-алканов с числом атомов углерода z и z + п (п =
1,2,3,...), рассчитывается путем линейной интерполяции лога-
рифмов исправленных параметров удерживания, чаще всего
*Предпочтительны соединения того же класса, что и определяемое. Не
рекомендуется выбирать в качестве стандарта первые члены гомологиче-
ских рядов и вещества с очень малым объемом удерживания в принятых
условиях анализа.
242
времен удерживания,
1“офс(г) = 100
, " ^(г)
2 + ~;—Н-
(IV.13)
при соблюдении условия
^Д(г) ^й(г) tR(z+ny
Использование предложенной Ковачем линейной интерполя-
ции оказалось возможным из-за выявленных к концу 50-х годов
линейных зависимостей между логарифмами исправленных па-
раметров удерживания членов гомологических рядов (и, в пер-
вую очередь, н-алканов) различными неподвижными фазами и
числом атомов углерода z в молекулах хроматографируемых
соединений*:
IgV^ — a + bz, (IV.14)
lg4 = a + bz. (IV.15)
С целью повышения точности определения индексов удержи-
вания следует стремиться использовать в качестве реперных
н-алканов ближайшие гомологи, различающиеся лишь на один
атом углерода (п = 1). Тогда уравнение (IV.13) приобретает
вид
1“ф(г) = 100
(IV.16)
В некоторых случаях (газохроматографический анализ при
высоких температурах колонки и (или) с использованием вы-
сокополярных неподвижных фаз, а также при работе с селек-
тивными детекторами — электронозахватным, фотопламенным)
вместо н-алканов в качестве стандартных применяют вещества,
близкие по химической природе к изучаемым соединениям, на-
пример:
— )/-алканолы, 2-алканоны, н-альдегиды, н-алкилбензолы, ме-
тиловые эфиры жирных кислот с неразветвленной цепью угле-
родных атомов — при использовании полярных неподвижных
фаз;
— углеводороды ароматического ряда с конденсированными
бензольными ядрами (нафталин, антрацен, фенантрен, хризен,
*Подобного рода корреляционные зависимости оставались предметом
пристального изучения и в последующий довольно длительный период
времени, вплоть до середины 80-х годов. Большой объем накопленной
информации позволил установить, что, вообще говоря, эти зависимости
не являются строго линейными, причем отклонения от линейности функ-
ции lgt'R — f(zc) для н-алканов и их функциональных производных носят
различный характер (подробнее об этом см. в разделе IV.2.4.3).
243
пицен) — при анализах сложных смесей (в том числе объектов
окружающей среды) на содержание полициклических аромати-
ческих углеводородов (ПАУ);
— н-алкилбромиды, н-алкилтрихлорацетаты — при работе с
электронозахватным детектором;
— н-алкилсульфиды — при измерении индексов удерживания
серосодержащих соединений по сигналам фотопламенного де-
тектора.
Изначальным достоинством системы индексов удерживания
является её наглядность. Ковач постулировал, что выбранные
им в качестве стандартов (реперов) н-алканы всегда (незави-
симо от природы неподвижной фазы и условий анализа) должны
иметь численные значения индексов удерживания, равные чи-
слу углеродных атомов в молекуле, умноженному на 100 ед. (на-
пример, для метана — 100, для пентана — 500, для декана —
1000). Нулевое значение индекса приписывается водороду. Эти
числа и образуют в шкале индексов удерживания серию фикси-
рованных точек. Из рис IV.2, поясняющего возможность графи-
ческого нахождения значений I, видно, что индекс удерживания
интересующего вещества равен умноженному на 100 ед. числу
атомов углерода z гипотетического н-алкана, имеющего такое
же исправленное время (расстояние или объем) удерживания,
что и вещество г.
Рис. IV.2. Графическое определе-
ние индексов удерживания Ковача.
1, 2, 41 5 — н-алканы (стандарты);
3 — характеризуемое соединение.
Графическое определение индексов удерживания не обеспе-
чивает, однако, необходимой точности результата, и поэтому
при выполнении ответственных анализов рекомендуется нахо-
дить индексы расчетным путем. При этом значительная эко-
номия времени обеспечивается использованием программируе-
мых микрокалькуляторов или персональных компьютеров (см.
Приложение).
244
Весьма привлекательной представляется возможность полу-
чения численных значений индексов удерживания при компью-
теризованной обработке результатов хроматографического
анализа в режиме on-line (см. раздел IV.5).
При необходимости идентификации по индексам удержива-
ния большого числа пиков на густозаселенных хроматограммах
приходится проводить вынужденное раздельное хроматографи-
рование исследуемой смеси и реперных н-алканов. В этом слу-
чае можно воспользоваться следующей удобной формой расчет-
ного уравнения:
/“.Фс(г) = ЮО
(IV.17)
где b — тангенс угла наклона прямой, выражающей зависи-
мость (IV.15). Величина b (так же, как и численное значение
самого индекса удерживания) зависит только от температуры и
свойств неподвижной фазы* и, в принципе, может быть найдена
в отдельных опытах при хроматографировании искусственной
смеси трех-четырех н-алканов с z > 5-6 при строго заданной
температуре колонки и при обеспечении стабильного расхода
газа-носителя**. По результатам этих же опытов можно найти
и усредненное значение и тогда для вычисления индек-
сов удерживания интересующих соединений необходимо и до-
статочно получить в тех же условиях работы прибора хро-
матограммы исследуемой смеси (не содержащей реперных н-
алканов).
Однако даже при использовании современной (серийной) га-
зохроматографической аппаратуры трудно обеспечить абсо-
лютную тождественность условий раздельного хроматографи-
рования (в частности, исключить влияние газодинамического
толчка при дозировании пробы в капиллярную колонку), вслед-
ствие чего более надежными следует признать значения индек-
сов удерживания, найденные при хроматографировании иссле-
дуемого образца, содержащего хотя бы один (первый, т. е. элю-
ируемый до интересующего вещества) реперный н-алкан с чи-
слом углеродных атомов z. Понятно, что при этом все равно
не удается избежать проведения повторных анализов, так как
’Для газо-жидкостного варианта хроматографии ©то утверждение
справедливо лишь при отсутствии заметного влияния адсорбции на по-
верхности раздела неподвижной фазы и твердого носителя. Адсорбция
^-алканов на других поверхностях пренебрежимо мала.
Для исключения случайных ошибок обработку зависимости (IV.15)
рекомендуется проводить по большому числу экспериментальных точек
методом наименьших квадратов.
245
для отождествления на хроматограмме пика этого f/-парафина
(или пиков двух введенных н-парафинов) предварительно не-
обходимо прохроматографировать ‘‘чистую” исходную смесь.
Такое дублирование анализов можно устранить (и тем самым
значительно сэкономить время, особенно при выполнении дли-
тельных анализов на капиллярных колонках), если в качестве
реперов использовать характерные пики любых других компо-
нентов смеси, индексы которых в данных условиях хроматогра-
фирования известны. В таком случае расчетная формула Ко-
вача приобретает более общий вид [186]:
г_а MlgZW) lgZ«(i) т
А — (J2 — Л) 7~----------Г—-------Г 11,
(IV. 18)
где Ц, А и /2 — индексы удерживания характеризуемого и двух
любых известных компонентов, принимаемых за стандарты;
^й(1р 2) — соответствующие исправленные параметры
удерживания.
Методы расчета индексов удерживания для случаев, когда
пик вещества i перекрывается с пиком одного из реперных со-
единений (п-алканов с числом атомов углерода z тт г -J-1) или
регистрируется на хроматограмме после второго из них, изла-
гаются в [187].
Индекс удерживания в изократической высокоэффективной
жидкостной хроматографии, по аналогии с индексами Ковача
в газовой хроматографии, рассчитывается посредством линей-
ной интерполяции логарифмов факторов удерживания (коэффи-
циентов емкости) к
1(г) = 100
1g *(i) - 1g к(г)
2 nig^(z+n) - Igfyz).
(IV.19)
при соблюдении условия
k(z) k(i) k(z+n)'
Воспринятая от газовой хроматографии система индексов удер-
живания достаточно подробно исследована и рекомендована к
широкому использованию в основном для варианта обращенно-
фазной ВЭЖХ с такими подвижными фазами, как метанол —
вода, ацетонитрил — вода, тетрагидрофуран — вода, содержа-
щими различные долевые количества органических модификан-
тов. Принципиальная возможность выполнения интерполяции
по (IV.19) базируется на установленных к началу 80-х годов ли-
нейных зависимостях типа 1g к = a + bz, выполняющихся практи-
чески для всех гомологических рядов при работе с названными
246
выше подвижными фазами (за необходимое для расчета к чи-
сленное значение в рассматриваемых системах подвижных
фаз обычно принимали время выхода из колонки нитрита или
нитрата натрия, дозируемого в нее в водном растворе.
В качестве реперов предлагалось использовать соединения,
представляющие различные гомологические ряды: н-алканы,
1-нитроалканы, алкан-2-оны, алкилбензолы, алкил-п-гидрокси-
бензоаты, алкилфенилкетоны. От н-алканов, традиционно ис-
пользуемых в газовой хроматографии, пришлось отказаться по
следующим причинам: невозможность работать с ними при ис-
пользовании наиболее распространенного УФ-детектирования;
времена удерживания н-алканов на неполярном обращенно-фаз-
ном сорбенте значительно превышают параметры удерживания
многих органических соединений. В настоящее время в прак-
тической работе чаще других реперов используют н-алкилфе-
нилкетоны CeHsCOR (как правило, R = СН3, ...jCyHis, хотя мо-
гут быть карбонильные соединения этого ряда и с большим
числом атомов углерода в алкильном радикале). Именно н-
алкилфенилкетоны полностью удовлетворяют всем критериям,
выдвигаемым при отборе реперов (стандартов) [188, 189]. Этих
критериев четыре.
1. Молекулы стандартов должны обладать хромофорной
группировкой, обеспечивающей их легкое обнаружение в элю-
ате при работе с чаще всего используемым в практике ВЭЖХ
ультрафиолетовым детектором.
2. Численные значения параметров удерживания выбранных
стандартов должны перекрывать численные значения параме-
тров удерживания максимально возможного числа характеризу-
емых соединений (включая наиболее распространенные лекар-
ственные препараты и природные продукты) при использова-
нии наиболее употребительных подвижных фаз: ацетонитрил —
вода, метанол — вода, тетрагидрофуран — вода.
3. Стандарты должны быть доступными для многих лабора-
торий.
4. Стандарты должны быть химически инертными по отноше-
нию к компонентам подвижных фаз и химически устойчивы при
изменении pH элюента.
Первому члену ряда — ацетофенону, содержащему 8 угле-
родных атомов в молекуле, присваивают индекс удерживания,
равный 800 ед., а всем последующим на 100 ед. больше, чем
предыдущему. Экспериментальная проверка показала, что из-
меренные в этой шкале стандартов индексы удерживания ряда
представительных модельных соединений гораздо более устой-
247
чивы к изменению содержания органического модификанта в по-
движной фазе, нежели фактор удерживания k.
Более того, в ходе накопления информации был сделан вы-
вод, что определяемые численные значения индексов удержива-
ния фактически не зависят от точности измерения мертвого вре-
мени (объема) колонки. Последнее заключение особенно важно
ввиду сложности задачи корректного измерения в ВЭЖХ (см.
выше).
Индекс удерживания в газовой хроматографии с программиро-
ванием температуры. В условиях программированного изме-
нения температуры колонки в газовой хроматографии наиболее
представительным абсолютным параметром является темпера-
тура удерживания веществ* — температура, соответствующая
выходу из колонки максимальной концентрации определяемого
вещества (иначе говоря, температура колонки в момент реги-
страции вершины хроматографического пика). На основании
близкой к линейной зависимости температуры удерживания н-
алканов от числа углеродных атомов Ван-ден-Лоолом и Крат-
цем [191] было предложено следующее уравнение для расчета
индексов удерживания в условиях линейного программирова-
ния температуры**:
1Т = 100
(IV.20)
+ z
при соблюдении условия
2д(г) ?й(г) 2ft(z + l),
где 2й(г), Tr(2+i) — температуры удерживания исследу-
емого вещества и н-алканов с числом углеродных атомов z и
z + 1 соответственно. Так как численные значения 7д(,), 7д(г) и
2д(г+1) прямо пропорциональны исправленным временам удер-
живания, а время удерживания можно измерить с большей точ-
ностью, чем температуру, при выполнении расчетов подста-
вляют в уравнение (IV.20) не температуры, а соответствующие
времена удерживания (вносить в них поправку на мертвое время
*Особенности измерения характеристик удерживания в газовой хро-
матографии с программированием температуры подробно рассмотрены в
обзоре [190].
**В номенклатуре Научного совета по хроматографии РАН [5з] — ин-
декс удерживания Ван-ден-Доола и Кратца (Van-den-Dool and Kratz retention
index). Взаимосвязь индексов удерживания, рассчитанных по уравнению
(IV.20), со значениями изотермических индексов при некоторой эквива-
лентной температуре подробнее рассмотрена в [192].
248
нет необходимости, поскольку и числитель, и знаменатель со-
держат разность двух величин):
1Т = 100
tT -tT
.Т _ /Т
Л(г + 1) £й(г)
(IV.21)
z +
Итак, уравнение (IV.21) отличается от приведенного выше
для расчета изотермических индексов уравнения (IV. 16) тем,
что оно включает просто неисправленные времена удержива-
ния, а не логарифмы исправленных времен удерживания. Дру-
гие способы расчета неизотермических индексов удерживания
обсуждаются в [192-194].
Индекс удерживания в высокоэффективной жидкостной хро-
матографии с градиентным элюированием рассчитывается по
уравнению, близкому по форме к (IV.21), поскольку в данном
режиме (как и при программировании температуры в газовой
хроматографии) имеет место зависимость k = а + bz. В связи с
этим обстоятельством уравнение для расчета индекса удержи-
вания в высокоэффективной жидкостной хроматографии с гра-
диентным элюированием приобретает вид
/(г) = 100
кщ - k{z)
z+ п~Г~^----~г—
(IV.22)
при соблюдении условия (п = 1,2,...)
Линейные индексы удерживания J (изотермические) в газовой
хроматографии, предложенные Вигдергаузом [193], рассчитыва-
ются по уравнению, сходному с (IV.21),
J(i) = z + tR{i^ (IV.23)
tR(z+l) — tR(z)
при соблюдении условия
^й(г) С tR(i) iRtz + ip
Как видно, в отличие от процедуры измерения I ( ср. уравне-
ние (IV.16)) при расчете J не требуется предварительного нахо-
ждения мертвого времени (расстояния) и логарифмирования ис-
правленных параметров удерживания хроматографических пи-
ков. Тем не менее, несмотря на очевидную простоту экспери-
ментального измерения, линейные индексы не получили пока
еще столь широкого распространения, как индексы удержива-
ния Ковача.
Обобщенные (линейно-логарифмические) индексы удерживания
GI в газовой хроматографии. Сложности расчета и отсутствие
249
единой общепринятой системы индексов удерживания в режиме
программирования температуры обусловлены невозможностью
характеристики зависимости I(t) линейной функцией, аналогич-
ной зависимости I = algtR + b, лежащей в основе системы изо-
термических индексов Ковача. По этой причине для точных
расчетов индексов в таких условиях чаще всего используют ап-
проксимации нелинейной функции I(t) полиномами различных
степеней. Наибольшее распространение получил способ рас-
чета на основе полиномов третьей степени (так называемый ме-
тод кубических парабол). В этом случае для расчета I анализи-
руемого соединения необходимы времена удерживания не двух
(как в изотермическом режиме), а четырех реперных 1/-алканов.
Другой подход к проблеме расчета индексов удерживания в
режиме программирования температуры, основанный на приве-
дении зависимости от некоторой функции f(t) к линейному виду,
предложен в [194, 195].
Этим открывается возможность применения формул линей-
ной интерполяции, построенных по типу формулы индексов Ко-
вача. В качестве такой функции f(t) целесообразно использо-
вать линейно-логарифмическую функцию вида
IT = at’R + blgt'R + с= a(t'R + qlgt'R) + с. (IV.24)
Коэффициенты a,b,cvtq = b/a этой зависимости могут быть рас-
считаны по временам и стандартным значениям индексов удер-
живания н-алканов, равных по определению 100z.
Из факта существования зависимости вида (IV.24) следует
система обобщенных индексов удерживания:
Д(>) + Ч !g * W - [^(z) + Ч !g*'д(г)] |
[^(z+1) + Ч lg%+1)] - Рад + Ч 1g*'д(г)]
Ее отличие от всех остальных систем — включение в расчет-
ную формулу переменного параметра q, зависящего от условий
анализа (начальной температуры колонки и скорости програм-
мирования). Расчет значений этого параметра может быть про-
веден различными методами, простейший из которых требует
измерения времен удерживания трех последовательно выходя-
щих из колонки реперных компонентов:
’=<,х+'/;?)ТЯ1г-
‘ьгд(2)'1Д(1)1Д(3)1
Для строгого расчета обобщенных индексов (как и индексов
Ковача в изотермических условиях) используются исправлен-
ные времена удерживания, вычисление которых, в принципе,
GI = 100
z(IV.25)
250
+ z , (IV.27)
GI = 100
требует знания мертвого времени колонки tM. Тем не менее, не-
давно было установлено [196], что при практическом использо-
вании системы линейно-логарифмических индексов удержива-
ния как в изотермических условиях, так и при программирова-
нии температуры нет необходимости точно определять или рас-
считывать время пребывания в хроматографичеекой системе
неудерживаемого вещества. Согласно этим рекомендациям, в
уравнения для расчета обобщенных индексов удерживания и пе-
ременного параметра q ((IV.25) и (IV.24)) вместо исправленных
параметров и t'R^z+1y вводят полные (неисправленные)
времена удерживания характеризуемых соединений и реперных
н-алканов:
~ + glg*fl(z)]
[*/?(z+l) + Q lg </?(z+l)] - [tR(z) +?lg<fi(z)]
_ ^fi(z-l) + tR(z+l) ~
^ёРя(г)Ля(г-1)^Н(г+1)]
Наименьшим погрешностям определения GI способствует вы-
полнение следующего соотношения времен удерживания трех
выбираемых для расчета переменного параметра q реперных н-
алканов и идентифицируемых компонентов анализируемых сме-
сей:
tR(z-i) < tR(i) < tR(z) < Ir(z+1)- (IV.29)
Возможность нахождения величин GI по полным временам
удерживания освобождает от необходимости прибегать к слож-
ным (не рассматриваемым здесь) методам определения времени
пребывания в колонке (приборе) неудерживаемого соединения
при программировании температуры [197, 198].
Конкретные практические рекомендации по измерению обоб-
щенных индексов удерживания при идентификации соединений
в многокомпонентных смесях даны при описании лабораторной
работы 5.
Главные преимущества линейно-логарифмических индексов
по сравнению с другими заключаются в их высокой воспро-
изводимости в любых режимах линейного программирования
температуры вне зависимости от выбора реперных компонен-
тов. Причина этого состоит именно в существовании линейной
зависимости (IV.24), вследствие чего ошибки расчета GI по раз-
ным наборам н-алканов минимальны. В табл. IV.4 сравниваются
средние значения и стандартные отклонения индексов линейно-
логарифмических, линейных и вычисляемых методом кубиче-
ских парабол для бензонитрила по временам удерживания раз-
личных реперных н-ал капов. Из приведенных в таблице данных
251
следует, что средние значения индексов во всех случаях совпа-
дают в пределах их стандартных отклонений, но сами стандарт-
ные отклонения резко различны: более 10 для линейных, вдвое
меньше в методе кубических парабол и всего ±1,5 для линейно-
логарифмических индексов. Максимальная воспроизводимость
в последнем случае делает хроматографическую идентифика-
цию по таким параметрам наиболее надежной.
Таблица IV.Средние значения и стандартные
отклонения индексов удерживания Для бензонитрила
(Условия анализа: стеклянная капиллярная
колонка с OV-101, to = 50°С, г = 3°С/мин,
tM = 5,03 мин, ^(CeHgCN) = 20,05 мин)
н-Алкан Время удерживания, мин Индексы удерживания
линейно- логарифмические линейные вычисленные методом кубических парабол
С7 7,80 945,2 (7, 8, 9) 957,0 (8, 9) 942,0 (7, 8, 9, 10)
с8 11,24 946,9 (8, 9, 10) 970,7 (7, 9) 947,2 (8, 9, 10, 11)
с9 16,85 945,8 (9, 10, 11) 944,6 (9, 10) 945,8 (9, 10, 11, 12)
Сю 24,02 947,9 (10, 11, 12) 948,5 (10, 11) 936,9 (7, 8, 9, 11)
Си 31,67 943,9 (8, 10, 12) 937,9 (8, 10) 948,1 (7, 9, 10, 11)
С12 39,34 948,2 (10, 12) 951,7 (7, 9, 11, 12)
Среднее 945,9± 1,5 951,2± 11,4 945,3 ±5,2
Примечание. В скобках указаны числа атомов углерода в
н-алканах, по которым выполнен расчет.
Обобщенные (линейно-логарифмические) индексы удерживания
в высокоэффективной обращенно-фазной жидкостной хромато-
графии, измеренные Зенкевичем [199, 200] для многочисленных,
главным образом, лекарственных веществ различной химиче-
ской природы в режимах изократического и градиентного элю-
ирования (подвижная фаза ацетонитрил — вода) при исполь-
зовании в качестве реперов алкилфенилкетонов, по своей ин-
формативности превосходят газохроматографические индексы
удерживания на стандартных неполярных неподвижных фазах
и рекомендуются для широкого практического использования.
При этом вводятся ограничения на выбор подвижной фазы и
реперных соединений. Оптимальной подвижной фазой является
система ацетонитрил — вода (в различных соотношениях) [199].
Наиболее удобными (стандартизованными) реперами признаны
алкилфенилкетоны. Специально разработанный алгоритм вы-
числения линейно-логарифмических индексов удерживания в
252
обращенно-фазной ВЭЖХ позволяет получать их воспроизво-
димые значения и для веществ с временами удерживания, мень-
шими, чем у первого реперного компонента— ацетофенона [200].
Приведенные выше уравнения для расчета различных ти-
пов индексов удерживания (логарифмических (IV. 13), линейных
(IV.21) и линейно-логарифмических (IV.25)) можно выразить со-
отношением [201, 202]
/(г) = /(г) + [I(z + п) - 1(г)] ~ Л^)) (IV.3O)
J\tR(z+n)) ~ JPfi(z))
где 1(г) = 100z и I(z + n) — 100(z + п) — постулированные зна-
чения индексов удерживания реперных соединений с временами
удерживания и tR(z+n)> удовлетворяющими условию интер-
поляции
Ir(z) <5 1д(г) tR(z+n)-
При подстановке в уравнение (IV.30) функции f(tR) различного
вида приходим к различным системам индексов:
1) f($R) = — логарифмические индексы I Ковача (изо-
термические) ;
2) f (Jr) = tn — линейные индексы I7 (измеряемые в режимах
программирования температуры);
3) j(tR) = tR+qlgtR — линейно-логарифмические
индексы GI, измеряемые в любых температурных условиях га-
зохроматографического анализа, а в обращенно-фазной
ВЭЖХ — в любых условиях элюирования.
IV.2.3. Источники погрешностей при
измерении параметров удерживания
Воспроизводимость и правильность измерения абсолютных
и относительных параметров удерживания как в газовой, так и
в жидкостной колоночной хроматографии обусловливаются со-
вокупным влиянием многочисленных факторов, вклад каждого
из которых в общую погрешность измеряемой величины опре-
деляется классом используемой аппаратуры, техникой дозиро-
вания, типом и степенью перегрузки колонки (ее эффективно-
стью), природой, происхождением и количеством используемых
подвижной и неподвижных фаз, наличием в них нежелательных
примесей, физико-химическими свойствами поверхности твер-
дого носителя (в газовой хроматографии), “возрастом” колонки,
а также квалификацией оператора.
253
При решении типовых задач качественного хроматографиче-
ского анализа всегда следует стремиться исключать (или сво-
дить к минимуму):
1) нестабильность (случайные колебания) температурного
режима колонки, испарителя и скорости газа-носителя, а также
их систематическое отклонение от номинальных значений;
2) превышение максимально допустимой для данной колонки
величины дозы, немгновенность дозирования;
3) несинхронность операций дозирования и включения сред-
ства измерения времени удерживания (секундомера, интегра-
тора, микропроцессора) или нанесения стартовой отметки на
диаграммной ленте; случайные и систематические ошибки
средств измерения времени удерживания, нелинейность скоро-
сти движения диаграммной ленты и систематическое отклоне-
ние ее от номинала (при измерении расстояний удерживания).
В табл. IV.5 приведены погрешности параметров опыта, воз-
никающие при их измерениях на стандартной газохроматогра-
фической аппаратуре, предназначенной для аналитических це-
лей, и на аппаратуре, специально изготавливаемой для физико-
химических исследований. Установлено, что для определения
неисправленного удерживаемого объема Vr с погрешностью 1%
необходимо поддерживать и измерять температуру колонки и
расход газа-носителя с погрешностью не хуже ±0,1°С и ±0,5%
соответственно. Чтобы снизить погрешность определения Vr
до ±0,1%, надо уменьшить погрешность измерений темпера-
туры колонки до 0,01°С, а расхода газа-носителя до ±0,1%, что
трудно достижимо при использовании стандартной аппаратуры
(см. табл. IV.5).
Измерение удельных удерживаемых объемов Vg лимитиру-
ется погрешностью определения массы адсорбента и особенно
неподвижной жидкой фазы в колонке. Даже на прецизи-
онной аппаратуре трудности определения шн.ф приводят к сум-
марной погрешности определения Vzg не менее 0,15%, что не по-
зволило авторам [203] рекомендовать Vg для надежной иденти-
фикации компонентов смеси по параметрам удерживания*.
Главными источниками погрешностей при измерении наибо-
лее представительных относительных газохроматографических
интерполяционных параметров — индексов удерживания — яв-
ляются:
’Отмеченные трудности удается, однако, обойти при использовании
капиллярных колонок и расчете Vg по уравнению (IV.10), включающему,
как уже отмечалось ранее (см. раздел IV.2.2), лишь ограниченное число
надежно контролируемых параметров [204].
254
Таблица IV. 5. Достигнутые наименьшие
погрешности измерения параметров опыта
на стандартной газохроматографической
аппаратуре и на специальном оборудовании [203]
Измеряемый параметр Средство измерения Погрешность измерения
стандартная аппаратура специальная аппаратура
Температура термо- стата поддержание Образцовый ртутный термометр Терморегулятор про- ±0,05°С ±(0,1-0,5)°С ±(0,01-0,02)°С
постоянства градиент вдоль колонки Расход газа-носи- теля (у выхода из колонки) поддержан ие порционального типа Мыльно-пленочный измеритель Термостатируемый мыльно-пленочный измеритель Ртутный измеритель потока Регулятор расхода ±0,1°С ±(0,5-1)% ±0 1% ±1% ±0,07% ±0,25%
постоянства Входное давление Выходное давление Время удерживания Образцовый манометр Барометр Секундомер ±0,16% ±0,1% ±(0,1-0,2)% (с термоста- тированием регулятора)
а) несовершенство техники (процедуры) измерения первич-
ных параметров удерживания вещества г и реперных н-алканов
как в изотермическом режиме, так и при программировании
температуры; недостаточная эффективность колонки, ее пере-
грузка пробой, промахи оператора;
б) погрешности определения мертвого времени (объема);
в) негомогенность и (или) нестабильность неподвижной фазы
в принятых условиях анализа, заметное снижение ее содержа-
ния на твердом носителе при длительной эксплуатации как на-
садочных, так и особенно капиллярных колонок (эффект “ста-
рения”, приводящий к постепенному переходу к новой колонке,
отличающейся по селективности и эффективности от первона-
чально приготовленной); наличие в неподвижной фазе неконтро-
лируемых примесей*;
*К сожалению, поступающие в продажу под одинаковыми торговыми
названиями некоторые (главным образом, полимерные) неподвижные фа-
зы не тождественны.
255
г) различия в свойствах используемых твердых носителей,
проявляющиеся через неодинаковый вклад вторичных адсорб-
ционных явлений в общее удерживание интересующих соедине-
ний и реперных г (-алканов;
д) неточность задания и (или) измерения температуры ко-
лонки, температурные флуктуации; неточность измерения и не-
стабильность расхода газа-носителя.
Наибольшей тщательности требует измерение индексов удер-
живания полярных соединений на неполярных неподвижных фа-
зах и, наоборот, неполярных соединений (включая н-алканы)
на высокополярных неподвижных фазах. Здесь особенно важно
контролировать температуру колонки, стабильность расхода
газа-носителя и снижать величину дозы (следует стремиться
работать в области, где удерживание не зависит от концентра-
ции хроматографируемых веществ в неподвижной жидкой фазе
или на поверхности адсорбента).
В дополнение к сказанному следует добавить рекомендации
[205] о желательности приготовления смесей идентифицируемых
соединений и реперных н-алканов в соотношении ~ 1 : 1 : 1. Ве-
роятно, по тем же причинам (влияние избыточного количества
некоторых компонентов пробы на коэффициенты распределения
в системе газ — неподвижная фаза) низка воспроизводимость
численных значений I при анализах сложных смесей, резко раз-
личающихся концентрациями соединений, имеющих близкие па-
раметры удерживания [206].
Судя по литературным данным последних лет, на серийных
газовых хроматографах и насадочных колонках с достаточно
большой степенью пропитки твердого носителя (более 15-20%
по массе) неполярными или малополярными неподвижными фа-
зами (апиезон L, динонилфталат) межлабораторный разброс
значений I обычно составляет ±(1-3) ед. индекса; при переходе
к полярным неподвижным фазам расхождения, как правило, воз-
растают и, например, для полиэтиленгликоля-400 разных пар-
тий достигают ±(3-5) ед.
Применение капиллярных колонок, помимо существенно уве-
личивающейся эффективности разделения, обеспечивает и
большую надежность значений индексов; в этом случае (при
использовании стандартной аппаратуры и термостабильных, а
также не подверженных химическому окислению неподвижных
фаз) межлабораторная воспроизводимость значений I соста-
вляет ±(1-2) ед. Важно подчеркнуть, что усовершенствование
процедуры нанесения неподвижных фаз на специально подго-
товленную поверхность стеклянного капилляра, последующее
аккуратное кондиционирование колонки, использование газов-
256
носителей, с максимальной тщательностью очищенных от неже-
лательных примесей (кислород, влага и др.), а также обязатель-
ная герметизация (запаивание) концов капилляра при хранении
обеспечивают возможность весьма длительной (1-7 лет) экс-
плуатации колонок без изменения рабочих характеристик [207].
Иногда отклонения экспериментально найденных индексов
удерживания от справочных данных могут быть весьма суще-
ственны (для газо-жидкостной хроматографии и насадочных ко-
лонок — до 40-60 ед. [208, 209]), что обычно объясняют адсорб-
цией анализируемых соединений на поверхностях раздела газ —
жидкость и неподвижная жидкая фаза — твердый носитель [210].
Искажающее влияние адсорбционных свойств этих поверхно-
стей особенно заметно сказывается на значениях абсолютных,
относительных и интерполяционных параметров удерживания
при небольших содержаниях неподвижной фазы на твердом но-
сителе (менее 10-20% по массе). Значительный разброс данных
может наблюдаться не только для разных типов твердых носи-
телей, но и для разных партий одного и того же носителя.
Для идеализированного чисто распределительного варианта
газовой хроматографии справедливо соотношение
t°T« = VgTH = Ki/Ka, (IV.31)
где Ki и Kst — коэффициенты распределения в системе непо-
движная жидкая фаза — газ г-го и стандартного вещества соот-
ветственно.
Реальный сорбент в ГЖХ состоит из двух объемных фаз
(твердый носитель и неподвижная жидкая фаза) и двух поверх-
ностей раздела: твердый носитель — неподвижная жидкая фаза
и неподвижная жидкая фаза — газовая фаза. Для таких слож-
ных систем Березкин и сотр. [210] выявили линейные зависимо-
сти вида
1 1
ri,st = --F = ^st + (IV.32)
где r°st — инвариантная (предельная) величина относительного
параметра удерживания, обусловленная только различным рас-
пределением г-го и стандартного веществ в системе неподвиж-
ная фаза — газ; AjjSt — коэффициент, характеризующий вклад
адсорбции в величину удерживания; Р — параметр, характери-
зующий сорбент (объем, эффективную толщину слоя, содержа-
ние неподвижной жидкой фазы на твердом носителе или фактор
Удерживания стандартного соединения на данной колонке).
Подобные зависимости справедливый для капиллярной газо-
жидкостной хроматографии [211].
257
В качестве стандартных рекомендовано выбирать вещества,
наиболее близкие по химическим свойствам к изучаемым соеди-
нениям. В этом случае различие в удерживании (r,->st) будет об-
условливаться только неодинаковой растворимостью стандарт-
ного и изучаемого веществ в неподвижной жидкой фазе.
Использование инвариантных (предельных) величин не
зависящих от содержания неподвижной фазы и от типа твердого
носителя, существенно улучшает межлабораторную воспроиз-
водимость данных по относительным параметрам удерживания
в газо(жидкостно-твердо)фазной хроматографии [210, 211].
И в капиллярной газовой хроматографии активность вну-
тренней поверхности колонки порой может стать причиной мно-
гих осложнений при трактовке результатов качественного ана-
лиза. Отмечена, например, ярко выраженная зависимость чи-
сленных значений I полярных соединений от величины пробы,
анализируемой на кварцевой несиланизированной капиллярной
колонке с неполярной (полидиметилсилоксановой) неподвижной
фазой: при последовательном увеличении величины пробы на-
блюдается пропорциональное снижение I (разница между экс-
периментально измеренными максимальным и минимальным чи-
сленными значениями индексов удерживания, например, для
октанола-1 составляет 15, а для 2,6-диметилаланина — 12 ед.
[212])-
Применение инвариантных величин удерживания тормозится
(помимо очевидной трудоемкости измерения) бедностью мас-
сива накопленных данных в сравнении с обычными индексами
удерживания Ковача. Поэтому заслуживает внимания предло-
жение об использовании в необходимых случаях (т. е. при об-
наружении систематических отклонений в значениях индексов
удерживания от справочных данных) корреляции между экс-
периментально найденными и наиболее авторитетными литера-
турными значениями I с целью нахождения индексов удержива-
ния для других (отсутствующих в распоряжении эксперимента-
тора) веществ [208, 209].
Следует отметить, однако, что среди множества публикуе-
мых величин логарифмических, линейных и линейно-логариф-
мических индексов удерживания, относящихся к изотермиче-
ским режимам колонки и программированному изменению ее
температуры соответственно и заметно различающихся между
собой по отмеченным объективным причинам, порой очень
трудно (а иногда невозможно) выбрать наиболее достоверные,
с которыми следовало бы сравнивать результаты собственных
измерений. В этой ситуации продуктивной оказывается ста-
тистическая обработка, т.н. “рандомизация” [213] всех имею-
258
щихся литературных данных безотносительно условий их по-
лучения [202, 214], позволяющая отбраковывать заведомо оши-
бочные величины и выявлять “окна”, в которые должны попа-
дать значения индексов идентифицируемых веществ. По дан-
ным [215], средние значения стандартных отклонений рандоми-
зованных индексов удерживания составляют для неполярных
силиконовых фаз около 8 ед. (чаще всего находятся в диапа-
зоне 5 4- 15), а для полиэтиленгликолей около 20 (15 4- 25).
Учитывая названные выше источники погрешности при из-
мерении параметров удерживания, рекомендуется приводить в
публикациях следующие подробности эксперимента:
1) температуру колонки с точностью до 0,1 °C;
2) полную характеристику неподвижной фазы (чистота, фир-
ма-изготовитель, количество фазы на носителе, параметры про-
цесса тренировки колонки);
3) полные данные для твердого носителя;
4) длину, диаметр и материал колонки, для капиллярных ко-
лонок — условия предварительной обработки внутренней по-
верхности стенок колонки;
5) количество вводимой пробы и зависимость времен выхода
от величины пробы;
6) метод измерения мертвого времени системы;
7) данные о природе газа-носителя, его входном и выходном
давлении и скорости;
8) чувствительность регистрации сигналов детектора и шка-
лу самописца;
9) тип и модель хроматографа, а также интегратора или ком-
пьютеризованной системы обработки данных;
10) ошибки определения параметров удерживания.
Желательно также указывать время эксплуатации колонки,
замеченные изменения параметров удерживания при работе ко-
лонки в течение длительного времени, роль адсорбционных эф-
фектов. Целесообразно проводить сравнение полученных дан-
ных с литературными.
В ВЭЖХ основными приборными источниками погрешностей
при измерении параметров удерживания являются нестабиль-
ность температурного режима колонки и непостоянство потока
подвижной фазы. В подавляющем большинстве моделей жид-
костных хроматографов, предназначенных для аналитических
целей, колонка не термостатируется. Изменения температуры
воздуха в лаборатории, наблюдаемые при выполнении экспери-
мента в течение одного рабочего дня ( и еще вероятнее — на про-
тяжении более длительного периода.измерений), могут превы-
шать несколько градусов. Если, например, ограничить темпе-
259
ратурные флуктуации диапазоном ±3 °C, то, согласно [34, с. 529-
562], разброс численных значений измеряемых времен удержи-
вания может превышать 1,5% отн., тогда как оснащение прибора
активным воздушным или пассивным металлическим термоста-
том колонки, способным поддерживать ее рабочую температуру
с точностью не хуже ±0,1 °C, обеспечит воспроизводимость ре-
зультатов измерений на уровне 0,3-0,4%.
Нестабильность потока подвижной фазы (вызываемая в наи-
большей степени техническими возможностями насоса) при ра-
боте на большей части приборов, выпускавшихся в последнее
десятилетие, также составляет ~0,3%.
Более всего на измеряемые абсолютные и относительные па-
раметры удерживания в ВЭЖХ влияют природа и состав по-
движной фазы*. В изократическом режиме это влияние мо-
жет проявляться особенно сильно, если компоненты подвижной
фазы накачиваются автономными насосами из разных емкостей.
Для обеспечения погрешности измерения 1% необходимо под-
держивать состав подвижной фазы, подаваемой в колонку, с точ-
ностью не хуже ±0,1%. Как отмечается в [34, с. 529-562], совре-
менные приборы позволяют выполнить это требование, поэтому
проблема здесь не столько приборная, сколько методическая.
Так, даже при подаче в колонку заранее составленной смеси
растворителей из одной емкости и одним насосом важно, наряду
со строгим соблюдением пропорций в составе элюента, обес-
печить близкую к абсолютной химическую индивидуальность
смешиваемых жидкостей. Наличие в намеченных к использова-
нию растворителях тех или иных нежелательных примесей мо-
жет кардинальным образом повлиять на измеряемые параметры
удерживания.
В градиентном режиме, когда состав подвижной фазы явля-
ется, помимо всего прочего, и функцией времени, ошибки изме-
рения в сравнении с изократическим режимом возрастают.
Типичные ошибки в воспроизведении состава подвижной фазы,
связанные с приборными возможностями, чаще всего соста-
вляют ~ 1% (в изократическом режиме ±0,1%), что должно соот-
ветственно значительно ухудшить воспроизводимость измере-
*В противоположность жидкостной, в газовой хроматографии зави-
симость относительных параметров удерживания от природы подвиж-
ной фазы проявляется весьма слабо [33, ч.1, с. 73]; количественная оценка
недавно установленного влияния природы и давления газа-носителя на
относительные параметры удерживания в капиллярной газо-жидкостной
хроматографии дана в работах В. Г. Березкина и сотр.: Berezkin KG.,
Korolev A. A., Malyukova I. V. // Analusis. 1997. Vol. 25. P. 299—302; J. High
Resol. Chromatogr. 1997. Vol. 20. №6. P. 333-336; J. Microcolumn Separation.
1996. Vol. 8. № 6. P. 389-396.
260
ния параметров удерживания. Строгий анализ ожидаемой вос-
производимости численных значений осложняется, однако,
тем обстоятельством, что на результаты измерений будут вли-
ять не только отклонения от заданного состава подвижной фазы
(как в изократическом режиме), но и диапазон градиента.
Отмеченные трудности в обеспечении стабильности важней-
ших параметров эксперимента в ВЭЖХ (температура колонки,
расход и состав подвижной фазы), не говоря уже о невозможно-
сти абсолютной стандартизации самих разделительных коло-
нок, обусловливают низкую межлабораторную воспроизводи-
мость не только абсолютных, но и относительных параметров
удерживания, в частности факторов удерживания (коэффициен-
тов емкости).
Сложилась такая ситуация, что, по заключению авторитет-
ных специалистов [2, 54], чуть ли не каждый отдельный хрома-
тограф должен быть прокалиброван по индивидуальным стан-
дартам. В связи с этим безусловно оправданным является поиск
представления данных по удерживанию интересующих соедине-
ний, в наименьшей степени зависящих от особенностей исполь-
зуемой аппаратуры и условий анализа и связанных только с
природой характеризуемого вещества, неподвижной и подвиж-
ной фаз. Этим стремлением и объясняется все возрастающий
интерес к представлению данных по удерживанию в ВЭЖХ в
виде индексов удерживания — интерполяционных параметров
удерживания, достаточно прочно укоренившихся в практике га-
зовой хроматографии (см раздел IV.2.2). Как показали ре-
зультаты специальной экспериментальной проверки [199], при
использовании принятой в качестве стандартной системы рас-
творителей ацетонитрил — вода в 16 различных градиентных
режимах и применении в качестве реперов алкилфенилкетонов
CeHgCOR (R = С? — Сб) стандартное отклонение обобщенных
(линейно-логарифмических) индексов удерживания ряда лекар-
ственных препаратов составляет от 2 до 12 ед. при средней
величине Sj = 6,5 ед.
Полученные результаты свидетельствуют о том, что при
представлении данных по удерживанию в обращенно-фазной
ВЭЖХ в форме обобщенных индексов удерживания (при со-
блюдении необходимых условий стандартизации эксперимента,
отмеченных выше) наблюдается весьма высокая воспроизводи-
мость результатов, сравнимая со средней воспроизводимостью
индексов удерживания в газовой хроматографии при использо-
вании стандартных неполярных фаз (5-10 ед. индекса) и суще-
ственно лучшая, чем при использовании полярных фаз (15-25
еД. индекса). Отмеченное обстоятельство открывает возмож-
261
ность решения задач качественного ВЭЖХ-анализа, базирую-
щегося не только на результатах градуировки прибора по ин-
дивидуальным соединениям (веществам сравнения), но и (что
много привлекательнее) на сопоставлении экспериментальных
и надежных литературных (справочных) данных.
IV.2.4. Качественный анализ
по параметрам удерживания
IV.2.4.1. Использование стандартных соединении
(метод метки)
Метод метки заключается в добавлении к проанализирован-
ному образцу известного стандартного соединения (метки) с по-
следующим хроматографированием в тех же условиях и сопо-
ставлением исходной и конечной хроматограмм.
Обычно этот метод применяется в подтверждающих анали-
зах для решения одной из наиболее часто встречающихся в
практике задач, а именно — для отождествления пиков соедине-
ний, присутствие которых в анализируемом образце ожидается.
Однозначный (отрицательный) результат получается при несо-
впадении числа пиков на исходной и конечной хроматограммах
(на последней регистрируется одним пиком больше). Увеличе-
ние высоты (площади) одного из пиков на конечной хромато-
грамме без заметного возрастания его ширины и без изменения
общего числа пиков может быть вызвано и случайным наложе-
нием зоны метки и зоны идентифицируемого соединения в вы-
бранной колонке в принятых условиях.
При газохроматографическом анализе положительный вывод
в этом случае можно сделать более или менее уверенно только
при условии, что такая же картина сохранится при хроматогра-
фировании образца с эталоном в разных температурных режи-
мах, по крайней мере, на трех колонках, содержащих неподвиж-
ные фазы различной природы. Так, методом метки идентифи-
кацию компонентов смеси углеводородов парафинового, олефи-
нового и ацетиленового рядов можно проводить по результатам
хроматографирования на колонках, содержащих следующие не-
подвижные фазы:
а) неполярную — сквалан или метилсиликоновый эластомер;
б) средней полярности — динонилфталат (трикрезилфосфат)
или какой-либо полиэфир;
в) сильнополярную или комплексообразующую — /?, /?'-окси-
дипропионитрил или насыщенный раствор нитрата серебра в
этиленгликоле.
262
С такой же осторожностью следует подходить к положитель-
ным заключениям о тождественности и при косвенной идентифи-
кации анализируемых веществ по литературным данным об их
относительном удерживании какой-либо известной неподвиж-
ной фазой.
Понятно, что надежность положительных заключений воз-
растает с повышением эффективности использованных колонок
(как и с увеличением числа дополнительных “нехроматографи-
ческих” сведений об исследуемом объекте). Тем не менее не все
встречающиеся в практике задачи требуют использования вы-
сокоэффективных колонок и полного разделения всех компонен-
тов смеси. Так, для получения информации о качестве, сорт-
ности или происхождении какой-либо сложной промышленной
композиции, например различных типов бензинов, моторных то-
плив, пищевых продуктов или лекарственных средств (в том
числе наркотического действия), достаточно выявить присут-
ствие (а иногда и относительное количество) в анализируемой
пробе лишь нескольких, так называемых “ключевых” компонен-
тов. Вопрос решается простым сопоставлением стандартной и
контрольной хроматограмм, записанных в одинаковых условиях
на одном и том же приборе и при использовании одной и той же
колонки. С этим приемом качественного анализа, получившим
название “техника отпечатков пальцев”, подробнее можно озна-
комиться в специальных публикациях (см. литературу в [31-33,
193].
Отметим, что решения о тождественности или нетождествен-
ности сравниваемых образцов, принимаемые только на осно-
вании визуального сопоставления хроматограмм контрольных
смесей и исследуемого объекта, вследствие их субъективно-
сти могут оказаться неверными. Надежность экспертных оце-
нок во много раз возрастает при подключении на стадии выра-
ботки решения математического аппарата, разработанного спе-
циально для осуществления процедуры “распознавания образа”
[216, 217].
Введение эталонных (стандартных) соединений в анализи-
руемую смесь и последующее сопоставление хроматограмм,
полученных до и после прибавления метки, широко применя-
ется и в качественном ВЭЖХ-анализе. При подтверждаю-
щих анализах не слишком сложных смесей этот прием явля-
ется основным. Для исключения опасности случайного нало-
жения пиков метки и идентифицируемого компонента анали-
зируемого образца смесь хроматографируют в различных ре-
жимах (обращенно-фазная и нормально-фазная адсорбционная
и/или распределительная хроматография). О достигаемом при
263
этом в ряде случаев существенном изменении порядка выхода
из колонки (и степени относительного удерживания) компонен-
тов смеси можно судить, например, по данным табл. IV.6.
Таблица IV.6. Факторы удерживания к ряда
модельных соединений в обращенно-фазовой
(ОФ) и нормально-фазовой (НФ)
распределительной хроматографии [218]
Соединение ОФ* НФ**
п-Крезол 1,64 11,13
2,6-Ксиленол 2.68 4 81
2,4-Ксиленол 2,95 7,88
2,3,4-Триметилфенол 4,50 7,81
2,4}5-Триметил фенол 4,80 7,06
’Колонка д-Bondapak Cis; подвижная фаза метанол — вода (1 : 1).
“Колонка Partisill 5; подвижная фаза н-гексан — атилацетат (95 : 5).
IV.2.4.2. Использование справочных данных
по удерживанию идентифицируемых соединении
различными неподвижными фазами.
Хроматографические спектры.
Многоступенчатые методы разделения
В газовой хроматографии совокупность данных по удержива-
нию вещества различными неподвижными фазами (общим чи-
слом три-четыре или более) в идентичных условиях является
специфичной для каждого индивидуального соединения*, что
позволяет проводить идентификацию путем прямого сопоста-
вления соответствующего набора экспериментально найденных
численных значений параметров удерживания (чаще всего, ин-
дексов удерживания) с литературными справочными данными.
Надежность идентификации обусловливается рациональным
выбором неподвижных фаз и их комбинаций (важно обеспе-
чить реализацию различных типов межмолекулярных взаимо-
действий фазы с анализируемыми соединениями), эффективно-
стью приготавливаемых колонок и идентичностью условий ана-
лиза исследуемых объектов и образцов сравнения. Отметим,
’Здесь прослеживается аналогия с оптическим спектром вещества.
Это дает основание говорить о своеобразных спектрах сорбционных ха-
рактеристик, или “хроматографических спектрах” [193, 219] (в расшири-
тельном толковании термина “спектр”).
264
что в наиболее общем виде идентификация компонента смеси
или индивидуального соединения по совокупности отличитель-
ных признаков может быть представлена как известная задача
“распознавания образов”, решаемая с помощью ЭВМ [220-223].
Многочисленные источники необходимой информации, пред-
ставленной в форме соответствующих таблиц или атласов, при-
ведены в [224-226] (см. также [227-235]). Однако в настоя-
щее время поиск надежных справочных величин, характеризую-
щих удерживание идентифицируемых соединений различными
неподвижными фазами в газовой хроматографии, рациональ-
нее всего проводить, обращаясь к созданным недавно банкам
(иначе — базам) данных общего [236, 237] или специализирован-
ного [238-241] профиля.
Принципы формирования подобных массивов справочных ве-
личин различны. Автоматизированная база данных (АБД) уни-
фицированной системы хроматографического анализа (УСХА
[242]) “СВЕТОХРОМ-1ЕС” содержит около 200 000 опублико-
ванных за примерно тридцатилетний период (1960-1992 гг.) све-
дений по индексам Ковача, относительному удерживанию и
удельным объемам удерживания 15000 органических веществ на
350 неподвижных фазах в изотермических режимах. Состав со-
зданной АБД и порядок пользования ею подробно изложены в
[236, 237].
База данных компьютерно-хроматографической системы
“ИНЛАН-ГХ” ориентирована на санитарно-экологический кон-
троль воздушных объектов и включает (по состоянию на 1993 г.)
параметры удерживания примерно 270 основных загрязните-
лей воздуха на 8 стандартизованных колонках с неподвижными
жидкими фазами различной полярности при трех температурах
[238, 239].
Разработанный в НИИ Химии СПбГУ банк хромато-масс-
спектрометрических данных для идентификации органических
соединений — основных загрязнителей атмосферного воздуха—
содержит индексы удерживания и сокращенные масс-спектры
(содержащие по 4-8 основных сигналов) примерно 660 (по со-
стоянию на 1992 г.) летучих примесей открытой атмосферы и
воздуха производственных помещений [240, 241]. В этот массив
справочной информации [243] впервые включены статистически
обработанные (рандомизованные, см. раздел IV.2.3) индексы
удерживания на стандартных неполярных полидиметилсилок-
сановых фазах, применимые в любых температурных режимах
(табл. IV.7). Независимость сформированного банка индексов
Удерживания от конкретных условий их определения и откры-
вающаяся возможность комплексной идентификации, основы-
265
Таблица IV.7. Фрагмент базы данных для хромато-масс-спектрометрической
идентификации органических примесей атмосферного воздуха [240, 241]
Названия соединения Восемь главных сигналов масс-спектра* Моле- куляр- ная масса Индекс удер- живания на стандартных неполярных фазах**
цис-1,2- Диметил циклопентан 56(99) 70(83) 41(76) 55(72) 27(33) 39(32) 69(31) 42(30) 98 693
1,3-Диметил бензол (лс-ксилол) 91(99) 106(63) 105(28) 77(14) 51(14) 39(14) 92(8) 27(8) 106 866
1, транс-2, г^ис-З-Тримети л циклопентан 70(99) 55(74) 41(41) 56(37) 69(24) 42(22) 27(22) 39(18) 112 747
транс-1,2-Диметил циклогексан 55(99) 97(95) 41(47) 112(33) 56(30) 42(23) 69(22) 27(22) 112 804
1-Метил-З-этилбензол 105(99) 120(31) 106(9) 91(9) 77(9) 39(8) 79(6) 51(6) 120 951
1,2,4-Т ри мети л бензол 105(99) 120(58) 119(15) 77(11) 39(10) 106(9) 91(9) 51(8) 120 985
(Е)-4-Нонен 55(99) 41(61) 56(39) 70(24) 69(22) 42(22) 27(22) 43(20) 126 893
1,2,3,5-Тетраметил бензол 119(99) 134(56) 91(12) 133(11) 39(11) 120(10) 41(10) 77(8) 134 1106
1,4-Диметил-2-этил бензол 119(99) 134(30) 91(11) 120(10) 39(9) 77(8) 105(6) 65(6) 134 1064
1-Метил-4-изопропилбензол (п-цимол) 119(99) 134(24) 91(16) 120(10) 117(8) 39(8) 77(6) 65(6) 134 1012
1,3-Диэтил бензол 119(99) 105(95) 134(46) 91(21) 77(15) 39(13) 27(12) 120(10) 134 1045
2-Метилнафталин 142(99) 141(76) 115(23) 143(12) 139(10) 71(10) 57(8) 70(7) 142 1309
1,4-Диметилнафталин 156(99) 141(64) 155(29) 157(13) 77(12) 115(11) 153(10) 76(10) 156 1416
2-Этилнафталин 141(99) 156(52) 115(18) 142(12) 157(7) 155(7) 76(7) 70(7) 156 1381
* В соответствии с рекомендациями [244] изомеры положения ароматических углеводородов и цис-, транс-
изомеры алкенов и циклоалканов охарактеризованы одинаковыми усредненными масс-спектрами.
** В рассматриваемой базе данных для всех соединений принята одинаковая межлабораторная погрешность
индексов удерживания, равная 8 ед. индекса.
вающейся на сочетании хроматографической и масс-спектро-
метрической информации, выгодно отличают специализирован-
ную базу данных НИИ Химии СПбГУ от ранее рассмотренных.
К настоящему времени сформирована и опубликована пол-
ная база газохроматографических индексов удерживания гало-
генпроизводных простейших углеводородов Ci и С2 (хладонов,
всего 114 соединений) на стандартных сорбентах (неполярном
порапаке Q и полярном силикагеле силипор 600), включающая
как статистически обработанные экспериментальные, так и рас-
четные значения I [214]. Начался процесс формирования базы
данных по индексам удерживания синтетических лекарствен-
ных препаратов, компонентов эфирных масел растений и других
биологически активных веществ в обращенно-фазной высокоэф-
фективной жидкостной хроматографии [199, 200].
На примере представительной группы веществ различной хи-
мической природы выявлено отсутствие какой бы то ни было
корреляции между величинами индексов удерживания в газо-
вой и жидкостной хроматографии. Это обстоятельство пред-
определяет резкое повышение информативности их сочетания
при идентификации компонентов сложных смесей по сравнению
с теми и другими, привлекаемыми в этих же целях по отдельно-
сти. По прогнозу [199], использование таких комбинированных
баз данных (содержащих численные значения индексов удержи-
вания, измеренных методами газовой и жидкостной хроматогра-
фии) может обеспечить получение правильных однозначных ре-
зультатов идентификации при объеме баз данных до 4000 соеди-
нений.
Одним из наиболее удобных практических приемов быстрого
получения необходимых сведений по удерживанию индивиду-
альных веществ различными неподвижными фазами является
последовательное соединение нескольких колонок и использова-
ние схем газовых коммуникаций, обеспечивающих отвод части
элюата на выходе каждой колонки к детектору (см. лабора-
торную работу 7 в [79]). Применение подобных схем позволяет
решать и обратную задачу — получать информацию об услов-
ной хроматографической полярности и селективности различ-
ных неподвижных фаз по совокупности параметров удержива-
ния и рассчитываемых из них термодинамических характери-
стик сорбции представительного набора стандартных соедине-
ний.
Другие процедурные варианты получения хроматографиче-
ских спектров не только индивидуальных соединений, но и ком-
понентов сложных смесей, включающие использование схем га-
зовых коммуникаций с параллельными, последовательными, па-
267
раллельно-последовательно и крестообразно соединенными на-
садочными колонками с индивидуальными и смешанными сор-
бентами, рассмотрены в [193, 219].
Для получения простейших бинарных хроматографических
спектров идентифицируемых соединений рекомендованы также
простейшие варианты подобных схем, предусматривающие ис-
пользование параллельных капиллярных колонок, связанных
через тройники с общим испарителем и пламенно-ионизацион-
ным детектором и различающихся по полярности и толщине
пленки неподвижных фаз (OV-1 и карбовакс 20М), а также по
длине и внутреннему диаметру колонок [245].
Следует, однако, отметить, что при исследовании не инди-
видуальных соединений, а смесей компонентов задача суще-
ственно осложняется, поскольку далеко не всегда можно уве-
ренно соотнести принадлежащие одному и тому же компоненту
пики на различных хроматограммах. В качестве одного из воз-
можных простых решений проблемы взаимного соотнесения пи-
ков на хроматограммах, получаемых при резком изменении по-
лярности неподвижной фазы, предложено сравнивать индексы
удерживания идентифицируемых соединений, измеряемые на
неполярной колонке (с полидиметилсилоксаном) и на колонке
с бинарной неподвижной фазой (полидиметилсилоксан и кар-
бовакс 20М в соотношении яв 10 : 1). При такой комбина-
ции используемых колонок вероятность изменения порядка вы-
хода веществ уменьшается в 5-15 раз, и, кроме того, обеспе-
чивается существенное расширение интервала индексов удер-
живания, измеряемых на полярных фазах, обладающих мень-
шей термостабильностью в сравнении с полидиметилсилоксано-
выми эластомерами. Практическая реализация такого подхода
сдерживается отсутствием необходимого массива справочных
данных по индексам удерживания на смешанных сорбентах, од-
нако это ограничение может быть преодолено привлечением ве-
личин I, рассчитываемых по специально разработанному алго-
ритму [226].
Разумеется, можно проследить “путь” каждого компонента
при использовании набора нескольких колонок с последова-
тельно изменяющейся условной хроматографической полярно-
стью индивидуальных или смешанных неподвижных фаз [193],
но этот способ требует неоправданно больших затрат рабочего
времени.
Наиболее рациональный прием получения хроматографиче-
ских спектров удерживания компонентов сложных смесей за-
ключается в использовании метода так называемой многомер-
ной газовой хроматографии (multi-dimensional gas chromatogra-
268
phy; простейший вариант метода — димерная газовая хрома-
тография, two-dimensional gas chromatography), предусматриваю-
щего две или большее количество ступеней разделения. С по-
мощью механических или пневматических устройств переклю-
чения газовых потоков производится “вырезание” фракции элю-
ата, покидающей колонку 1-й ступени разделения, и дозирова-
ние этой фракции, состоящей из одного или нескольких компо-
нентов, в колонку 2-й ступени с иной неподвижной фазой (таких
колонок может быть несколько; их число, равно как и число
ступеней разделения, определяется аппаратурными возможно-
стями и характером аналитической задачи). На рис. IV.3 в каче-
стве примера представлены хроматограммы, полученные после
1-й и 2-й ступеней разделения одной из модельных смесей при
Рис. IV.3. Димерная газовая хроматография искусственной
смеси органических соединений разных классов.
а — 1-я ступень разделения (колонка с полифениловым эфиром);
6 — 2-я ступень разделения (колонка с силиконовым эластомером БЕ-
ЗО). 1 — пентан; 2 — гексан; 3 — пропан-1-ол; 4 — гептан; 5 — этил аце-
тат; 6 — дипропиловый эфир; 7 — диметиловый эфир этиленгликоля;
# — бензол; 9 — октан; 10 — пентан-2-ол; 11 — З-метилбутан-1-ол;
12 — нонан.
269
использовании механического переключения потока подвижной
фазы (см. описание лабораторной работы 8 в [79]).
Для достижения предельно высокой эффективности и селек-
тивности разделения требуется использование хотя бы на од-
ной ступени капиллярных колонок. В специально сконструи-
рованных для реализации многомерной газовой хроматографии
приборах переключение газовых потоков производится только
с помощью пневматических клапанов, управляемых специали-
зированными микропроцессорами. В книгах [34, с. 643-668; 41,
с. 164-175; 246] и обстоятельных обзорных статьях, опублико-
ванных в специализированном томе журнала [247], подробно
обсуждаются варианты инструментального оформления соот-
ветствующих схем газовых коммуникаций и рассматриваются
многочисленные примеры успешного применения многомерной
газовой хроматографии для исследования состава таких слож-
ных и разнохарактерных объектов, как, например, бензины и
керосины, смеси диоксинов и полихлорированных бифенилов,
а также композиции органических соединений, создающих аро-
мат цветов некоторых растений, в частности жасмина (этот пе-
речень можно было бы продолжить).
К многоступенчатым методам прибегают при необходимости
разделения особо сложных смесей и в тонкослойной хромато-
графии (ТСХ), и в ВЭЖХ. Димерная ТСХ, например, заключа-
ется в последовательном элюировании рабочей пластины двумя
разными подвижными фазами, направления движения которых
перпендикулярны друг другу (см. раздел IV.4). Различные ва-
рианты схем многоступенчатого разделения, надежно зареко-
мендовавшие себя в практике ВЭЖХ, описаны в [248, 249].
В последние годы активно развиваются гибридные варианты
многоступенчатых методов, в которых на 1-й ступени разде-
ление осуществляется в колонке жидкостного хроматографа, а
затем отдельные фракции элюата могут быть направлены (в
режиме off-line или on-line, рис. IV.4) в насадочную или капил-
лярную колонку газового хроматографа для дальнейшего ис-
следования их состава [56, 247]. Димерная ЖХ—ГХ особенно
оправдана при анализах лекарственных препаратов и объектов
окружающей среды (за счет много большей, чем у жидкостного
хроматографа, чувствительности газохроматографического де-
тектора заметно снижается предел обнаружения идентифици-
руемых веществ, отделенных от мешающих соединений на 1-й
ступени разделения). К ограничениям метода следует отнести
необходимость подбора растворителя (подвижной фазы) для 1-й
ступени разделения, совместимого с газохроматографическими
детекторами и колонками, которые должны содержать преиму-
щественно химически привитые неподвижные фазы.
270
а
б
Рис. IV.4. Простейший вариант сочленения колонок жидкост-
ного (ЖХ) и газового (ГХ) хроматографов для двухступенча-
того разделения через стандартный шестиходовой кран.
а — заполнение дозирующей петли крана фракцией жидкого элю-
ата; б — дозирование отобранной порции элюата в газовый хромато-
граф.
IV.2.4.3. Использование корреляционных
зависимостей параметров удерживания
При решении первых трех типовых задач качественного ана-
лиза (см. раздел IV.2.1) в газовой хроматографии широко ис-
пользуют линейные зависимости между логарифмом исправлен-
ного объема (времени) удерживания или индексом удерживания
членов гомологического ряда и каким-либо их свойством, за-
кономерно изменяющимся в пределах данного ряда, например
числом атомов углерода в молекуле z, температурой кипения
^кип, отношением абсолютной температуры кипения вещества к
абсолютной температуре колонки Ткип/Ткол^
Р = а + bz,
(IV.33)
(IV.34)
Р — а + {’(Т'нип).
Р = а + ЦДип/Ткол),
(IV.35)
где Р — параметр удерживания (1g Vg, Ig^fi, lg& или Л‘> a и & —
коэффициенты, зависящие от условий анализа (природы непо-
движной фазы и твердого носителя, температуры колонки) и
Функциональной группы гомологического ряда.
Подобные зависимости выявлены и в ВЭЖХ (преимущест-
венно в ее обращенно-фазном варианте). Здесь следует еще раз
271
подчеркнуть, что в отличие от газовой, в жидкостной хромато-
графии на удерживание вещества существенно влияет не только
природа и состав неподвижной, но и подвижной фазы (большое
внимание, в частности, следует уделять доли органического
компонента в смешанных водно-органических подвижных фазах,
солевым добавкам, pH среды, температуре колонки).
Стандартизация условий обращение-фазного ВЭЖХ-анализа
по этим параметрам позволила установить линейные корре-
ляции между величиной логарифма фактора удерживания к и
числом атомов углерода z в гомологических сериях алканов,
алкилбензолов, алкил бромидов, алифатических спиртов, али-
фатических и ароматических кетонов, метиловых эфиров жир-
ных кислот, 2-н-алкилпиридинов и др. (что, собственно, и яви-
лось основой для характеристики относительного удерживания
в ВЭЖХ в форме индексов удерживания, см. раздел IV.2.2):
lg fc = a + bz. (IV.36)
Примеры корреляционных зависимостей (IV.33), (IV.34) и
(IV.35) в графической форме представлены на рис. IV.5 и IV.6.
На основе многочисленных экспериментальных данных выве-
дена зависимость 1g к от объемной доли (9?) органического ком-
понента в элюенте [250]
IgA: = Sip + IgA:0,
где к° — значение фактора удерживания в отсутствие органиче-
ского компонента в подвижной фазе; S — коэффициент, отража-
ющий влияние концентрации органического модификатора на
удерживание вещества.
Распределение хроматографируемого соединения между дву-
мя фазами аддитивно и непосредственно определяется его
структурой:
1g к = 1g кр + ДЯ,„ (г),
здесь ^2ДА?т(г) — групповые вклады; 1g кр — характеристика
удерживания “остовного” соединения. В свою очередь, вклады
различных функциональных групп могут быть определены как
разность lg A'rx — lg A'rh, где X — функциональная группа.
В газовой (и отчасти, в жидкостной) хроматографии при
идентификации по индексам удерживания полезно руководство-
ваться следующими ориентировочными правилами, частично
установленными еще Ковачем, но в значительной мере транс-
формированными по мере накопления экспериментального ма-
териала [193].
272
Рис.IV.5. Корреляционные зависимости параметров удержи-
вания.
а — зависимость логарифма исправленного времени удерживания
сополимером стирола и дивинилбензола при 200 ° С представителей
различных гомологических рядов от числа атомов углерода z в мо-
лекулах: 1 —- алканы; 2 — спирты; 3 — алкилацетаты.
6 — зависимость логарифма исправленного времени удерживания
от температуры кипения изомерных пентил бензолов: 1 — 1-фенил-2,2-
диметилпропан; 2 — 2-фенил-З-метилбутан; 3 — 3-фени л пентан; 4 —
2-фен и л-2-ме тил бутан; 5 — 2-фенилпентан; 6 — 1-фенил-2-метилбутан;
7 — 1-фенил-З-метилбутан; 8 — фенилпентан. Колонка капилляр-
ная медная 50 м X 0,35 мм; неподвижная фаза — сквален, темпера-
тура 80°C; скорость газа-носителя (азота) через колонку 3 мл/мин при
сбросе 250 мл/мин.
1. Индексы удерживания любых ближайших гомологов на лю-
бых неподвижных фазах различаются на ~ 100 ед. (встреча-
ются, однако, и исключения, когда эти разности составляют ве-
личины, как существенно большие, так и существенно меньшие,
чем 100 ед. инд.; в отдельных случаях, при хроматографиро-
вании сильнополярных соединений, отмечалась даже инверсия
порядка элюирования гомологов).
2. Индексы удерживания любого соединения на различных
неполярных неподвижных фазах сохраняют постоянство (в пре-
делах нескольких ед. инд.). При хроматографировании на непо-
движных фазах различной полярности сохраняют постоянство
(с теми же допусками) лишь индексы удерживания неполярных
(и неполяризуемых) соединений.
3. Разность индексов удерживания di изомерных соединений
на неполярной неподвижной фазе в большинстве случаев свя-
273
Рис. IV.6. Линейные зависимости в ВЭЖХ фактора удержива-
ния к от числа атомов углерода z в молекулах представителей
различных гомологических рядов [189].
1 —- я-алканы; 5 — н-алкены; 3 — н—ал кил бензолы; 4 — метило-
вые эфиры жирных кислот; 5— алкан-3-оны; 6— 2-н-алкилпиридины;
7— н-алкан-1-олы. Сорбент — нуклеозил 5-100-Ci-PMSCie, подвиж-
ная фаза метанол — вода (90:10).
зана с разностью температур кипения </Ткип этих изомеров со-
отношением
di ~ МТККП.
4. Введение одной и той же функциональной группы в моле-
кулы близкого строения приводит к приращению индексов удер-
живания на одну и ту же величину. Для сильнополярных соеди-
нений численные значения этих инкрементов, характеризующих
вклад функциональной группы в аддитивную величину индекса
удерживания, могут варьировать в пределах 5-10 ед. инд.
5. Соединения, содержащие несколько атомов кислорода
и(или) азота, равно как и вещества, в молекулах которых при-
сутствуют единичные атомы кислорода или азота, включенные
274
в системы сопряженных связей, имеют большие индексы удер-
живания, нежели их структурные аналоги, не имеющие назван-
ных атомов.
6. При необходимости идентификации соединений, принад-
лежащих к нескольким гомологическим рядам, целесообразно
сопоставлять на одном графике параметры удерживания инте-
ресующих веществ неподвижными фазами различной природы.
Так, на достаточно большом экспериментальном материале,
относящемся к гомологическим сериям углеводородов и ки-
слородсодержащих соединений разных классов (алкилбензолы,
спирты, кетоны, альдегиды, сложные эфиры) и нескольким не-
полярным и полярным неподвижным фазам (сквалан, апиезон L,
диоктилфталат, карбовакс 1540 и трицианоэтоксипропан), по-
казана возможность групповой идентификации исследуемых ве-
ществ на основе линейных зависимостей типа индекс — индекс
[209, 251]:
/Ф1=а + Ь/ф2, (IV.37)
где 1Ф1 и I®2 — индексы удерживания одного и того же соеди-
нения неподвижными фазами Ф1 и Ф2 соответственно; а и b —
константы. Линейность зависимостей вида (IV.37) прослежена
на апробированных комбинациях неподвижных фаз в интервале
численных значений индексов удерживания от 500 до 1600 ед.
Подобными корреляционными соотношениями предложено
пользоваться и для т. н. адаптации экспериментально изме-
ренных на какой-либо неподвижной фазе индексов удерживания
/ИКСп к их численным значениям 1пт, заимствуемым из надежных
литературных источников*. Так, например, в результате стати-
стической обработки массивов данных, относящихся к апиезону
L (ап) и карбоваксу 20М (карб), получены следующие уравне-
ния [237]:
Ссп = 1,00ПХ +0,213,
СсРп = 0,999/^ит6 + 3,160.
Надежность групповой идентификации на основе корреля-
ционных зависимостей индекс — индекс возрастает, если воз-
можно образование нестойких комплексов между одной из срав-
ниваемых неподвижных фаз и соединениями того или иного го-
мологического ряда. Так, непредельные углеводороды обра-
зуют непосредственно в колонке газового хроматографа тг-комп-
лексы с растворенными в низкомолекулярных полиэтиленглико-
*Как уже отмечалось в разделе IV.2.3, расхождения индексов удержи-
вания, измеренных в разных лабораториях на насадочных колонках, мо-
гУт быть весьма значительны и достигать 40-60 ед.
275
лях нитратом серебра (при температурах 20-65 °C) и нитратом
таллия (при температурах до 100-130°С). Первичные и вторич-
ные амины в области температур 85-140 °C вступают в донорно-
акцепторные взаимодействия с NaOH, а алкилпиридины спо-
I I
собны образовывать комплексы типа — N ... — Р = О с введен-
I I
ным в состав неподвижной фазы тринатрийфосфатом, разруша-
ющиеся при температурах выше 120-130°С [252].
Комплексообразование как основной фактор, ответственный
за большее или меньшее удерживание компонентов анализиру-
емых смесей, играет решающую роль в жидкостной и, в пер-
вую очередь, в ионообменной и лигандообменной хроматогра-
фии. В ионообменной хроматографии степень удерживания ве-
щества снижается при увеличении ионной силы подвижной фазы
и увеличивается с возрастанием ионообменной емкости сор-
бента. Для проведения лигандного обмена необходимо нали-
чие склонных к координации органических соединений (обычно
это вещества, содержащие донорные гетероатомы — азот, серу,
кислород — или кратные связи) и комплексообразующего кати-
она металла (Cu2+, Со2+, Ni2+, Hg,2+ и т.д.). Их применяют для
разделения энантиомеров аминов, аминокислот, нуклеотидов,
белков, углеводов.
Комплексообразование лежит в основе и жидкостной ион-
парной хроматографии, основанной на принципах ион-парной
экстракции. В качестве ионов-модификаторов используют те-
тр аалкиламмоний (NRJ), алкилсульфонат (RSO3), перхлорат
(СЮ4 ) и т.п. Чаще всего применяют ион-парную хроматогра-
фию на обращенной фазе; подвижной фазой служат водный бу-
ферный раствор и органический растворитель, смешивающийся
с водой. Ион-парный реагент адсорбируется на гидрофобной
поверхности неподвижной фазы, образуя подвижный анионный
слой, в результате поверхность сорбента приобретает свойства
ионообменника.
Природа ионов-модификаторов в значительной степени вли-
яет на удерживание. Чтобы величина к соответствовала опти-
мальному диапазону, для каждого иона-модификатора требу-
ется своя специфическая концентрация. Выбор ионов-модифи-
каторов в основном определяется природой ионного образца.
Так, для образцов анионной природы требуются в качестве ион-
парных добавок катионы и наоборот. Первоначальное доба-
вление в подвижную фазу ион-парных агентов приводит к их
адсорбции неподвижной фазой вплоть до насыщения. Поэтому
влияние изменения концентрации ионов-модификаторов должно
276
быть гораздо заметнее в области низких, чем в области высо-
ких, концентраций.
В последние годы в качестве комплексообразующего агента,
изменяющего хроматографическое поведение сорбата, исполь-
зуют макроциклы: краун-соединения, криптанды. Движущей
силой ассоциации макроцикла и субстрата могут быть некова-
лентные взаимодействия самых разных типов; ион-ионные, ион-
дипольные, диполь-дипольные, дисперсионные; важнейший слу-
чай — образование водородных связей. В газовой хроматогра-
фии краун-эфиры являются компонентами неподвижных фаз, в
жидкостной — подвижной и неподвижной (см. обзоры [253, 254]).
В газовой хроматографии для осуществления групповой
классификации на основе рассматриваемых зависимостей мож-
но воспользоваться также эффектом заметного изменения пара-
метров удерживания представителей различных классов орга-
нических соединений при существенном изменении содержания
неподвижной фазы на твердом носителе. Согласно [210, 211], та-
кой прием, основанный на перераспределении вкладов адсорб-
ции и абсорбции в общую величину удерживания, особенно эф-
фективен при разделении геометрических изомеров непредель-
ных углеводородов на капиллярных колонках.
В качестве независимой аналитической характеристики того
или иного гомологического ряда используют коэффициенты
температурной зависимости индексов удерживания di/dT, ко-
торые в ограниченном интервале температур сохраняют посто-
янство и при рациональном выборе неподвижной фазы не пере-
крываются. В основе идентификации лежат зависимости тем-
пературных коэффициентов от молекулярной структуры анали-
зируемых веществ: от степени насыщенности и разветвления
углеродного скелета, от числа атомов углерода, от положе-
ния ненасыщенной связи, от положения и химической природы
функциональной группы. Например, температурные коэффици-
енты индексов удерживания углеводородов разных классов при
их хроматографировании на неполярных фазах [255] уменьша-
ются в ряду: алкилбензолы > циклогексаны > циклопентаны >
тетра- и триалкилпарафины > моно- и диалкилпарафины.
Существенные различия в температурных коэффициентах ин-
дексов удерживания на различных неподвижных фазах для ши-
рокого круга органических соединений наблюдаются не только
в газо-жидкостном, но и в газо-адсорбционном варианте хро-
матографии. Так, например, по данным [256], при использова-
нии насадочных колонок с силикагелем силипор 600 большин-
ство полифторорганических соединений (фреонов) имеют отри-
цательные численные значения dl/dT, что отличает их от со-
277
единений других классов и может быть использовано при иден-
тификации. ’
Выявленное в последние годы влияние температуры на хро-
матографическое поведение анализируемых веществ в ВЭЖХ
[257] обусловлено созданием новых хроматографических фаз,
реализующих различные механизмы взаимодействий с сорбен-
тами (хиральное распознавание, комплексообразование с уча-
стием макроциклов, ион-парных агентов и т.д.). При этом тем-
пература часто оказывается решающим параметром оптимиза-
ции разделения той или иной конкретной смеси. В общем виде
эта зависимость может быть выражена следующим соотноше-
нием
In к = -&.H/RT + AS//? + In /?,
где АН — изменение энтальпии (доминирующий фактор, осо-
бенно при разделении больших молекул); AS — изменение эн-
тропии; этот параметр (мера беспорядка) начинает играть за-
метную роль, когда неподвижная фаза чувствительна к геоме-
трии хроматографируемых веществ, в частности в случае ли-
гандного обмена или при разделении энантиомеров (большие
различия в изменении энтропии могут приводить к полному раз-
делению структурных изомеров); /? — фазовое отношение.
При повышении температуры уменьшается вязкость подвиж-
ной фазы, изменяются коэффициенты диффузии, ускоряется
массообмен между подвижной и неподвижной фазами, что, в
свою очередь, ведет к снижению размывания хроматографиче-
ских зон и соответственно к повышению эффективности разде-
ления [258].
Влияние температуры на удерживание различных веществ
неодинаково, что сказывается на селективности хроматографи-
ческого анализа. Это особенно четко проявляется в случае
различной геометрии разделяемых молекул [259]. При анализе
высокомолекулярных соединений и использовании колонок не-
большого диаметра (^ 1 мм) оправданным и целесообразным
становится режим программирования температуры (широко ис-
пользуемый в газовой, но все еще сравнительно редко — в жид-
костной хроматографии) [260].
Совместное влияние температуры и состава подвижной фазы
на удерживание описывается уравнением [250]
In к = Ai 99(1 — TcfT) + А2/Т + A3,
где Ai, А2, Аз — константы для данного соединения и конкрет-
ной неподвижной фазы при двух заданных компонентах подвиж-
ной; 9? — объемная доля органического компонента в элюенте;
278
Т — т. н. компенсационная температура, при которой удержи-
вание не зависит от состава неподвижной фазы (при этой тем-
пературе энтропийный и энтальпийный вклады компенсируют
друг друга).
Еще более эффективным для газохроматографического реше-
ния многих аналитических задач, связанных с групповой иден-
тификацией, является привлечение разностей индексов удержи-
вания исследуемых соединений полярной и неполярной непо-
движными фазами.
Напомним, что удерживание любых веществ неполярной фа-
зой определяется, главным образом, универсальными дисперси-
онными силами. При замене фазы на полярную удерживание
полярных (или неполярных, но способных к поляризации) со-
единений, как правило, возрастает из-за проявления ориентаци-
онных, индукционных и донорно-акцепторных взаимодействий
между молекулами анализируемых соединений и неподвижной
фазой.
Таким образом, можно сказать, что разность индексов удер-
живания Д/ исследуемого вещества, измеренных при одинако-
вой температуре на двух неподвижных фазах, из которых одна
полярна (Ф1), а другая неполярна (Фг),
Д/ = /Ф1 -
является мерой количественной оценки названных специфиче-
ских взаимодействий, иначе говоря, характеризует селектив-
ность выбранной хроматографической системы.
Для иллюстрации возможностей групповой идентификации
анализируемых веществ в табл. IV.8 приведены значения Д/ для
15 гомологических рядов, измеренные для трех комбинаций не-
подвижных фаз. По представленным данным хорошо просле-
живается общая тенденция к увеличению разностей индексов
удерживания при последовательном переходе снизу вверх от
одного гомологического ряда к другому, причем наблюдаемые
приращения Д/ в ряде случаев вполне достаточны для группо-
вой классификации. Вместе с тем, отчетливо видны и ограни-
чения метода, вызываемые многочисленными перекрываниями
значений Д/, обусловленными, главным образом, разной энер-
гетической ценой одной единицы индекса удерживания на не-
подвижных фазах различной полярности. Учет этого обстоя-
тельства позволил выработать надежный термодинамический
критерий Д<2 ( энергетический эквивалент разности индексов
Удерживания Д/ [261]), возможности практического использова-
ния которого в качественном газохроматографическом анализе
подробно рассмотрены в [79, с. 186-188] (см. там же лаборатор-
ную работу 7).
279
Таблица IV.8. Разность индексов удерживания
для 15 гомологических рядов и трех
комбинаций неподвижных фаз [261]
№ п/п Гомологический ряд Карбовакс — апиезон Карбовакс — тритон Тритон — апиезон
1 Rm ОН 535 - 569 112- 115 423 - 454
2 RmOC(O)CH3 338 - 377 59-82 269 - 295
3 RmC(O)CH3 363 - 408 72 - 87 285 - 341
4 RmOC(O)H 365 - 403 72-89 283 - 314
5 RmNHCH(CH3)2 228 - 171 67-54 111 - 161
6 RmNHCH2CH(CH3)2 218- 154 63-46 108 - 165
7 RmNH(CH2)2CH(CH3)2 229 - 180 72-55 124- 158
8 RmNHCH(CH3 )2CH2 CH3 206 - 138 65-41 97-141
9 RmNHCH3 287 - 245 51 - 83 162 - 218
10 RmNHC2H5 206 - 256 51 - 71 134 - 205
11 RmNHC3H7 188 - 240 64 - 69 123 - 171
12 RmNH2 290 - 340 55 - 107 190 - 285
13 RmN(CH3)2 119-140 33-47 80 - 94
14 RmN(C2HS)2 80 - 130 23-52 56-78
15 RmN(C3H7)2 48-83 12-23 36-60
Один из приемов, способствующих повышению надежности
групповой идентификации по величинам Д/, заключается в рас-
ширении набора привлекаемых пар разнохарактерных непод-
вижных фаз (вплоть до шести). Понятно, что этот путь при-
водит к существенному повышению трудоемкости эксперимента
и требует компьютерной обработки результатов с использова-
нием ранее составленных для этих колонок банков данных [220].
Альтернативный подход к устранению неоднозначности оце-
нок принадлежности идентифицируемого соединения к тому или
иному гомологическому ряду, получаемых из разностей Д/ при
использовании всего лишь двух или трех неподвижных фаз раз-
личной природы, заключается в направленной химической об-
работке пробы (дериватизация, см. раздел IV.1) с целью из-
менения природы межмолекулярных взаимодействий между не-
подвижной фазой и образующимся производным. Так, напри-
мер, метиловые эфиры жирных кислот на неполярных неподвиж-
ных фазах характеризуются величинами I, тождественными ин-
дексам удерживания н-алканов с эквивалентным числом угле-
родных атомов. Точно так же, как при алкилировании, рез-
кое подавление полярности амино-, гидрокси- и карбоксильных
групп в идентифицируемых соединениях достигается получе-
нием силильных производных, для которых разность Д/ в индек-
280
сах удерживания на полярной и неполярной неподвижных фазах
равна (или весьма близка) нулю [262, 263].
Дополнительная весьма полезная информация может быть
получена из экспериментальных данных по внеколоночным фа-
зовым равновесиям в системе ограниченно смешивающихся рас-
творителей [264, 265]. Наиболее универсальной признана пара
гексан — ацетонитрил; данная система растворителей обеспе-
чивает относительное постоянство параметра а для разных го-
мологических рядов в корреляционном уравнении
lgK = a/ + b, (IV.38)
связывающем коэффициенты распределения К с индексами
удерживания I [265]. Поскольку приближенное среднее значе-
ние а в уравнении (IV.38) может быть принято равным 1 10-3
для любых органических соединений, то информационная на-
грузка о принадлежности характеризуемого вещества к тому
или иному гомологическому ряду ложится на свободный член Ь.
На этом основании для целей групповой идентификации был
предложен новый дифференциальный параметр j, рассчитыва-
емый по данным о межфазовых равновесиях, характеризуемых
коэффициентами распределения К, и по одновременно измеря-
емым индексам удерживания I (на неполярных или полярных
неподвижных фазах) [265]:
j = kI — \gK, (IV.39)
где к — коэффициент, пропорциональный разности свободных
энергий сольватации метиленовой группы СНг в двух использу-
емых несмешивающихся растворителях; для рекомендуемой ге-
терогенной системы гексан — ацетонитрил численное значение
к, сохраняющего постоянство для разных рядов в достаточно
широких температурных пределах, может быть принято 1 • 10-3.
В табл. IV.9 приведены статистически обработанные параме-
тры j для 17 гомологических рядов. Выполненная по данным
табл. IV.9 численная оценка сравнительной информативности R
параметров j и А/ через отношение диапазона их численных
значений к удвоенному стандартному отклонению в этом диа-
пазоне (R(j) = 12 и R (AI) = 18), свидетельствует о том, что
введение дифференциального параметра j обеспечивает груп-
повую идентификацию по эффективности, лишь незначительно
меньшую, чем разность индексов удерживания А/ на полярных
и неполярных неподвижных фазах. Подробные рекомендации
по экспериментальному измерению величин К и j и использо-
ванию их для решения конкретных задач качественного газо-
хроматографического анализа даны при описании лаборатор-
ной работы 6.
281
Таблица IV.9. Статистически обработанные
параметры j zt Sj некоторых гомологических
рядов (выборка из данных [265])
Гомологический ряд
Алканы
Алкены
Цикло- и бициклоалканы
Простые эфиры
Алкилхлориды
Сопряженные енины
Алкилбромиды
Арены
Арилхлориды
Алкилалканоаты
Алкилтрихлорацетаты
втор- Алканолы
Ц и к лоа л кан о л ы
перв-Алканолы
Алкиларилкетоны
Диалкилдикарбоксилаты
Фенолы
J ± Sj
-0,40 ± 0,07
-0,19 ±0,09
-0,08 ±0,11
0,25 ±0,05
0,52 ±0,12
0,53 ±0,04
0,60 ± 0,04
0,70 ± 0,05
0,93 ± 0,04
1,02 ±0,12
1,18 ±0,07
1,39 ±0,19
1,48 ±0,13
1,61 ±0,12
1,69 ±0,08
2,29 ±0,15
2,50 ±0,40
Полная газохроматографическая идентификация существен-
но облегчается выделением еще на стадии подготовки пробы к
анализу смесей веществ с однотипной функциональной группой
или с однотипными структурными фрагментами. Для после-
дующего исследования состава таких смесей используют спе-
циально подобранную систему из трех-четырех колонок, обес-
печивающую оптимальные условия разделения и позволяющую
математически выразить зависимость хроматографического по-
ведения (удерживания) вещества от его физико-химических
свойств. При этом следует иметь в виду, что обсуждавшиеся
ранее корреляционные зависимости типа (IV.33)-(IV.35), строго
говоря, не выполняются для первых членов гомологических ря-
дов (z < 5-7), а также для любой последовательности гомоло-
гов, включающей более 7-8 членов [266].
К настоящему времени установлено, что газохроматографи-
ческое удерживание органических соединений, образующих го-
мологические ряды, достаточно строго описывается нелиней-
ными трех- или четырехпараметрическими корреляционными
уравнениями, коэффициенты которых находят методом наимень-
ших квадратов по данным для гомологов с известными /, t'R, VR
или Ve (см., например, [79, с. 183]. Эти уравнения могут быть ис-
пользованы для расчетов параметров удерживания отсутству-
282
юших гомологов, поэтому-то данные методы идентификации и
были названы бесстандартными [267].
Обсуждению различных способов формализованного пред-
ставления количественных соотношений между структурными
особенностями идентифицируемых соединений и их параметра-
ми удерживания посвящена многочисленная литература, сум-
мированная в обзорах [263, 268].
Оформившиеся в газовой хроматографии к концу 80-х годов
аддитивные схемы прогнозирующего расчета параметров удер-
живания путем суммирования инкрементов, соответствующих
вкладам отдельных структурных фрагментов (атомов, групп
атомов, различных типов химических связей), перенесены и в
ВЭЖХ [269].
Из всех ранее предложенных наиболее простым и физико-
химически обоснованным представляется трехпараметрическое
корреляционное уравнение вида (IV.40), позволяющее прово-
дить априорный расчет индексов удерживания в газовой хро-
матографии с погрешностью, обычно не выходящей за пределы
межлабораторной воспроизводимости экспериментально изме-
ренных величин I на стандартных полидиметилсилоксановых
неподвижных фазах [270, 271]:
lg / = a 1g Ткип 4- ЬА + с, (IV.40)
где Ткип — температура кипения, К; А — значения любых ад-
дитивных свойств гомологов: молекулярные массы, суммарное
число атомов углерода в молекуле и т.п.; а, b и с — константы
корреляционного уравнения.
Следует особо подчеркнуть, что уравнения вида (IV.40) по-
зволяют рассчитывать газохроматографические индексы удер-
живания не только веществ-гомологов, но и соединений, входя-
щих в другие родственные группы, т. е. различающихся друг
от друга по составу не на гомологическую разность СНг, а
на любые другие повторяющиеся фрагменты состава и струк-
туры молекул. Типичным примером могут служить моно-, ди-,
три- и тетрахлор(бром)метаны (здесь варьируется число ато-
мов галогена при единственном атоме углерода). В качестве
другого подобного сообщества формально родственных веществ
можно привести следующий ряд соединений: СгНб, СН3ОС2Н5,
СН3СН(ОСНз)2, СН3ОСН2СН2ОСН3, к которым можно прийти
при последовательной замене атомов водорода в этане на ме-
токсигруппу. Особо следует выделить группы углеводородов и
их производных, разнящихся лишь величиной т.н. формальной
непредельности (ФН), равной сумме числа циклов и кратных
283
связей в молекуле (тройные связи учитываются с коэффициен-
том 2)*.
Широкие возможности априорного расчета газохроматогра-
фических индексов удерживания по уравнениям вида (IV.40) ил-
люстрируются данными табл. IV.10 (произвольная выборка из
работы [271]), из которых следует, что средняя ошибка резуль-
тата не превышает усредненной межлабораторной погрешности
экспериментального измерения численных значений I
Таблица IV, 10. Коэффициенты уравнений
вида (IV.40) и характеристика точности
расчета индексов удерживания для некоторых
родственных групп органических соединений,
различающихся варьируемым параметром А
Группа веществ Число предста- вителей А* a b-103 c |Д1|
С6Н14 —> СбНб 10 ФН 2,2315 1,9361 -2,8711 4
С6Н12С12 -» С6Н4С12 7 ФН 1,4631 1,0592 -0,8797 7
С2Н6 -> С2С16 10 zCL 1,7147 8,3957 -1,5840 6
С2Н6О -» С2Н3ОС13 5 zCL 1,3608 43,865 -0,7456 5
C6H5NO2 -> C6C15NO2 5 zCL 1,1320 21,042 -0,0107 5
C5Hl2 -> С5Н12Оз*‘ 9 zO 2,1261 - 9,4868 -2,5907 9
CyHie —> С7Н16О3** 9 zO 2,2502 - 6,0448 -2,9334 7
С5Н12 -» C5H12S 6 zS 2,1995 -28,576 -2,7775 2
С4Нз —> C4HgS2 5 zS 2,1881 -59,809 -2,7213 4
C6H14 —> СбНцКз 13 zN 1,5282 14,415 -1,0946 7
CgHie —> CgHi8N2 8 zN 2,4034 -10,070 -3,3413 2
*ФН — формальная неп ре дельность; zCl, zO, zS и zN — число
атомов хлора, кислорода, серы и азота соответственно.
** За исключением спиртов, содержащих активные атомы во-
дорода.
IV.2.5. Реакционная газовая хроматография
Аналитическая реакционная газовая хроматография (АРГХ)
предусматривает совместное использование химических и хро-
матографических методов исследования в единой хроматогра-
*Алгоритм нахождения численных значений ФН представлен в [271].
284
фической схеме [272]*. Типичными химическими реакциями,
осуществляемыми в АРГХ, являются: гидрирование и деги-
дрирование, гидрогенолиз, дегидратация и дегидрогалогениро-
вание, этерификация и декарбоксилирование, обмен функцио-
нальными группами между реактантом и реагентом и другие
реакции, приводящие к образованию соединений, заметно от-
личающихся по летучести и параметрам удерживания от ве-
ществ, присутствующих в исходной пробе. Использование этих
реакций для целенаправленного “химического тестирования”
индивидуальных соединений или компонентов сложных смесей
позволяет расшифровать структуры весьма сложных объектов
анализа (например, природных веществ), представленных в ми-
крограммовых количествах. В связи с этим методы АРГХ
особенно ценны при исследовании природы микропримесей и в
функциональном анализе органических соединений.
Самостоятельной областью АРГХ является пиролитическая
газовая хроматография, сочетающая в едином аппаратурном
оформлении процесс пиролиза вещества и хроматографическое
разделение продуктов его термического разложения. Для осу-
ществления пиролиза пробы в динамическом режиме разрабо-
таны и выпускаются промышленностью специальные пироли-
зеры [274]. Недавно была показана возможность проведения
пиролиза образцов горных пород и каменного угля непосред-
ственно в стеклянном вкладыше испарителя газового хромато-
графа, укомплектованного системой программированного повы-
шения температуры от комнатной до 630 °C со скоростью, рав-
ной 480 град/мин** [275].
Осуществляли три последовательных цикла наложения на
образец температурного поля с промежуточным (перед разде-
лением на капиллярной колонке) криоконцентрированием обра-
зующихся продуктов. В первом цикле образец прогревали до
200°C, во втором — до 400 (в отдельной серии опытов — до
*Именно такой принцип сочетания хроматографического разделения
и направленного химического воздействия на компоненты пробы лежит в
основе современной реакционной хромато-масс-спектрометрии [273].
Предложенное позже расширенное толкование понятия АРГХ и включе-
ние в ее сферу контроля и использования химических процессов, осуще-
ствляемых вне хроматографической схемы [148], представляется неоправ-
данным, поскольку вне хроматографической схемы нет и хроматографии.
В настоящее время за рубежом подобные испарители, способные ра-
зогреваться от комнатной до температуры 300 ° С и более за очень ко-
роткое время (не превышающее 1 мин), выпускаются серийно. Их очень
Удобно использовать не только для проведения пиролиза, но и для десор-
бирования примесей на заключительном этапе твердофазной экстракции
как в макро-, так и особенно в ее микроварианте (см., например, обзоры
[ИО, 141]).
285
Pnc.IV.7. Хроматографические схемы в аналитической реак-
ционной газовой хроматографии.
а — принципиальная схема газовых коммуникаций стандартного га-
зового хроматографа; 6 — реактор после дозатора (испарителя) перед
колонкой; в, г — реактор после колонки перед детектором и за детек-
тором соответственно; д — функции реактора выполняет колонка; е —
выполняет испаритель. 1 — устройство ввода проб; 2— реактор; 3—
хроматографическая колонка; 4 — детектор; 5 — регистрирующее и
записывающее устройство.
530 °C), в третьем цикле конечная температура программиро-
ванного прогрева испарителя составляла 600 °C. Приращение
конечной температуры пиролиза в каждом последующем цикле
приводило к десорбированию и последующему газохроматогра-
фическому разделению все новых — более “тяжелых” — продук-
тов углубляющегося термического разложения пробы. В проме-
жутках между циклами испаритель и термостат с капиллярной
колонкой быстро охлаждали жидким азотом до температуры
50°C. Преимущества такого методического подхода заключа-
ются в гибком выборе температуры одно- или многостадийного
термического разложения материала пробы и сведении к мини-
муму возможных потерь продуктов пиролиза, характерных для
более громоздких систем, в которых пиролизер и разделитель-
ная хроматографическая колонка отделены друг от друга до-
вольно длинной магистралью, требующей автономного термо-
статирования.
Реакторами для выполнения названных выше химических
превращений (за исключением пиролиза) в единой хроматогра-
фической системе обычно служат короткие прямые или изогну-
тые стеклянные или стальные трубки (длиной от 1-2 до 25 см и
286
более и диаметром 2-6 мм), включаемые в схему газовых комму-
никаций стандартного газового хроматографа после дозатора
(испарителя) перед колонкой, после колонки перед детектором
или после него. Часто реакцию проводят в самой хроматогра-
фической колонке или во вкладыше, размещенном в испарителе
хроматографа (рис. IV.7)*.
IV.2.5.1. Химические превращения компонентов пробы
на выходе из хроматографической колонки
В простейшем варианте этот вид аналитической реакцион-
ной газовой хроматографии (т.н. химическая индикация) не
требует каких-либо переделок или изменений стандартной га-
зовой схемы хроматографа при работе с детектором по тепло-
проводности**. Следует только предусмотреть подключение к
линии сброса газового потока в атмосферу специальной рас-
пределительной гребенки, связанной с серией стеклянных “ми-
крореакторов” — небольшого размера пробирок или пеницил-
линовых склянок. В каждую пробирку (склянку) перед нача-
лом опыта помещают свежеприготовленный раствор специфи-
ческого группового реагента. Пробирки соединяют с распре-
делительной гребенкой с помощью стальных капилляров (ме-
дицинских игл) таким образом, чтобы при выполнении ана-
лиза поток газа из хроматографической колонки барботиро-
вал через каждый раствор группового реагента. По наблюдае-
мому изменению окраски или выпадению характерного осадка
(табл. IV.11) судят о функциональной природе компонента раз-
деляемой смеси, только что зарегистрированного на хромато-
грамме.
При необходимости выполнения иных химических реакций,
не приводящих к выпадению осадка или изменению окраски, а
также с целью повышения чувствительности определения при-
бегают к микропрепаративному улавливанию компонентов про-
’Указанные варианты размещения реакционных петель (реакторов) не
исчерпывают всех возможных схем в АРГХ (подробнее см. [148]). Пло-
дотворным направлением использования хроматографических реакторов
является применение их в физико-химических исследованиях для изуче-
ния разнообразных каталитических процессов и кинетики реакций, про-
текающих на поверхности адсорбента-катализатора или в растворе непо-
движной жидкой фазы.
’Использование ионизационных (деструктивных) детекторов возмож-
но лишь при разделении газового потока на выходе из колонки на два,
примерно в соотношении 1:100. Меньшая часть газа направляется в де-
тектор, большая — выводится за пределы термостата колонок через обо-
греваемый штуцер.
287
Таблица IV.11. Характерные реакции представителей
различных классов соединений (выборка из [272, с. 16G])
Класс соединений Реагент Окраска в случае положительной реакции
Спирты Бихромат калия + азотная кислота Синяя
Альдегиды и кетоны 2,4-Динитрофенилгидразин Желтый осадок
Альдегиды Флороглюцин N-(4-Аминобен зоил)-п-фе- нилендиамин Оранжево-красный осадок Светло-оранжевая
Кетоны Нитропруссид натрия Бордово-красный осадок
Сульфиды Тот же Изатин Красная Зеленая
Амины Нитропруссид натрия Красная или синяя
Ароматические Формальдегид + серная Темно-красная
углеводороды кислота (винная)
Олефины Тот же Та же
Галогенпроиз- водн ые Нитрат серебра в спирте Белый осадок
бы, элюируемых из колонки, например, в охлаждаемых жидким
азотом стеклянных капиллярах.
Предложенный в [276] очень простой технический прием осу-
ществления этой процедуры, позволяющий улавливать нано-
граммовые количества фракций элюата, иллюстрирует
рис. IV.8.
К установке соответствующего микрореактора между выхо-
дом колонки и детектором газового хроматографа иногда прибе-
гают в количественном газохроматографическом анализе, пре-
следуя цель повышения чувствительности (снижения предела
обнаружения интересующего соединения). В качестве нагляд-
ного примера можно привести метанизацию оксида углерода,
легко осуществляемую с количественным выходом на слое ни-
келевого катализатора при температуре 350-400°С в атмосфере
водорода с последующей регистрацией образующегося метана
пламенно-ионизационным детектором [277]. В качественном ана-
лизе постколоночная дериватизация применяется для повыше-
ния информативности показаний используемых детекторов, да-
же таких мощных, как масс-спектрометр [273] в системах ГХ—
МС (см. раздел IV.2).
288
Рис.IV.8. Микропрепаративное улавливание отдельных фрак-
ций елюата в охлаждаемом жидким азотом стеклянном капил-
ляре и осуществление направленных химических превраще-
ний сконденсированных веществ.
а — сочленение стеклянного капилляра (внутреннее сечение до
1,5 мм) с обогреваемым штуцером выходной линии хроматографа;
6-г — введение в капилляр раствора реагента, образование и извлече-
ние реакционной смеси. 1 —- накидная гайка с внутренней прокладкой
из силиконовой резины; 2 — полиэтиленовая пробка с двумя отверсти-
ями под капилляр, заполняемая жидким азотом; 3 — сконденсирован-
ная фракция элюата; 4 — раствор реагента; 5 -— игла микрошприца с
раствором реагента; 6 — реакционная смесь.
IV.2.5.2. Методика “вычитания” пиков
(образование нелетучих соединений)
Избирательное образование каким-либо из компонентов раз-
деляемой смеси нелетучих соединений с реагентом, помещен-
ным непосредственно в аналитическую колонку или в короткую
трубку (реакционную петлю) до или после аналитической ко-
лонки перед детектором (см. рис.IV.7, б, в), легло в основу
получившей широкое распространение методики вычитания пи-
ков. Вычитание может быть осуществлено либо в результате не-
обратимо протекающих химических реакций, либо вследствие
необратимой сорбции разделяемых компонентов неподвижной
289
Таблица IV. 12. Реагенты и адсорбенты для селективного
удаления представителей различных классов
органических соединений из анализируемой смеси [148]
Удаляемые соединения Реагенты и адсорбенты
н-Парафины, н-олефины, о-олефины Ароматические угле- водороды Молекулярные сита, карбамид Смесь перхлората ртути и хлорной кисло- ты, оксид алюминия, 20% Ь1,Г4-бис(2-циано- этил)формамида на твердом носителе*
Эфиры карбоновых кислот Карбамид, гидроксиды натрия и калия, алюмо- и боргидрид лития, 3% борной кислоты + 10% карбовакса 20М на твердом носителе*, 3% гидроксида лития 10% полиэтилен гл и коля на твердом носителе*
Сопряженные диены Малеиновый ангидрид, хлормалеиновый ангидрид, полиэтиленгликольмалеат
Г алоген производи ые Нитрат серебра (бромалкилы), версамид-900 (хлоралкилы)
Олефины Бром на угле, серная кислота, хлорат ртути, 20% HgSC>4 на твердом носителе*, 4% Ag2SO4 -|- 95% H2SO4 на твердом носителе*
Альдегиды Гидросульфит натрия, о-дианизидин, бен- зидин, раствор гидроксиламина в триэта- ноламине, алюмо- и боргидрид лития
Кетоны Гидросульфит натрия, бензидин, алюмо- и боргидрид лития, тринитрофталевый ангидрид, семикарбазид, гидроксиламин- хлорид
Серосодержащие Спирты Хлорид ртути Борная кислота, тригексилциклоборат, гидрид кальция, алюмо- и боргидрид лития, 3% гидроксида лития + 10% полиэтилен- гликоля на твердом носителе*
Амины Фосфористая кислота
Кислоты Гидроксиды калия и натрия, оксид цинка
Эпоксиды Борная кислота, о-дианизидин (частично), алюмо- и боргидрид лития, фосфорная кислота
Ацетилен Ртутьорганические 5% нитрата серебра на твердом носителе Порошки железа, никеля, алюминия, цинка, свинца
*В качестве твердых носителей чаще всего используются
диатомиты или, в некоторых случаях, силикагели с развитой
поверхностью.
290
Phc.IV.9. Пример использо-
вания методик “вычитания”
и “сдвига” пиков для анализа
смеси углеводородов.
1 — толуол; 2 — метил ци-
клогексан; 3 — изооктен; 4 —
к-гептан; 5 — изооктан; 6 — гек-
сен-1.
Исходная смесь > 34 2 И ч * 56
2 4 5 После поглощения 1 1 1111 H2S°4 _ J (J У J
2 5 После поглощения 11 11 цеолитами II II
1 После I1 — 5 1
5 После поглощения 11 H2SO4 J 1
фазой. Применяемые для этих целей некоторые селективные
реагенты (поглотители) и вычитаемые ими из анализируемой
смеси классы химических соединений представлены в
табл. IV. 12.
Сопоставление хроматограмм, записанных до и после удале-
ния из исходной смеси тех или иных компонентов, позволяет
провести их групповую классификацию (рис. IV.9).
Возможности методики вычитания могут быть расширены по-
следовательным включением в газовую схему нескольких реак-
ционных петель с различными реагентами (что равнозначно за-
полнению одной петли несколькими слоями поглотителей) или
проведением анализа на хроматографе с двумя параллельными
газовыми линиями и детекторами.
Использование химического вычитания в одном из звеньев
хроматографической системы для селективного удаления рас-
творителя, основного вещества или мешающих компонентов,
291
Рис. IV.10. Хроматограммы раствора ароматических углево-
дородов в уксусной кислоте (10~4% (по массе)) без (а) и с вы-
читанием растворителя (6).
Условия анализа: температура 100°С; скорость газа-носителя (ар-
гона) 60 мл/мин; ПИД, шкала электрометра 2 • 10—12 А; доза 1 мкл.
а — фор-колонка 50 X 0,4 см с 10% (по массе) ПЭГ-600 на сферохро-
ме-1 (0,1—0,2 мм); колонка 100 X 0,4 см с 20% (по массе) апиезона L на
сферохроме-1 (0,1—0,2 мм); 6— в состав неподвижной фазы форколонки
введено 20% от массы носителя едкого кали.
маскирующих зоны определяемых соединений, давно уже на-
шло широкое распространение в газохроматографическом ана-
лизе примесей. На рис. IV.10, например, представлены хрома-
тограммы раствора ароматических углеводородов в уксусной
кислоте, зарегистрированные без использования и с использо-
ванием приема вычитания растворителя (уксусной кислоты) ще-
лочью в фор-колонке.
Подробное изложение процедуры вычитания пиков на при-
мерах удаления олефинов, спиртов, алкилиодидов в реакторе
с диатомитовым носителем, смоченным раствором сульфата
ртути в 29%-ной серной кислоте, а также удаления спиртов в
реакторе с борной кислотой дано при описании лабораторной
работы 9 в [79].
IV.2.5.3. Методика “сдвига” пиков
Методика сдвига пиков заключается в том, что необратимо
протекающая химическая реакция используется для превраще-
ния некоторых компонентов разделяемой смеси в производные,
хроматографируемые с иными параметрами удерживания. Со-
292
поставление хроматограмм исходной смеси и той же смеси по-
сле химических превращений некоторых ее компонентов позво-
ляет провести групповую классификацию (см. рис. IV.9).
Одним из наиболее изученных методов превращения отдель-
ных компонентов анализируемой смеси является их гидрирова-
ние и дегидрирование. Эти реакции иногда требуют высоких
температур (150-300 °C) и поэтому, как правило, проводятся в
специальных микрореакторах с автономным обогревом, содер-
жащих слой катализатора.
Изменение степени насыщенности молекул приводит к значи-
тельному изменению времени удерживания соединения на по-
лярных неподвижных фазах. Для проведения реакций гидриро-
вания и дегидрирования предложены платиновые, палладиевые
и никелевые катализаторы, которые в небольшом количестве (1-
3% (по массе)) наносятся на диатомитовые или иные твердые
носители. Длина слоя катализатора при разделении продуктов
на насадочных колонках обычно составляет от 1 до 3 см (иногда
больше) в реакторе диаметром 2-6 мм. Процесс ведут в токе во-
дорода, гелия или водорода и гелия, используемых в качестве
газов-носителей.
Функции реактора может выполнять начальный участок на-
садочной колонки, находящийся в обогреваемой зоне испари-
теля хроматографа. При необходимости использования капил-
лярной колонки для разделения сложных смесей продуктов ги-
дрирования (дегидрирования) и присутствующих в анализиру-
емых образцах не вступающих в эти реакции “балластных” со-
единений слой катализатора размещают в канале испарения
пробы узла делителя потока, как это показано, например [278],
на рис. IV. 11.
При наличии в анализируемом образце функциональных про-
изводных в этих же условиях возможен их гидрогенолиз, т. е.
деструктивное гидрирование с разрывом связей между углеро-
дом и гетероатомом, приводящее к образованию парафиновых
углеводородов с тем же самым или меньшим, чем в исходном
соединении (в зависимости от его класса и структуры), числом
углеродных атомов.
Это направление АРГХ весьма активно развивалось в 60-е -—
80-е годы и в англоязычной литературе даже получило специ-
альное название “carbon skeleton chromatography”, обычно пере-
водимое на русский язык как “определение углеродного ске-
лета” [100, с. 216; 148, с. 119; 279]. Исключение из сложных по со-
ставу и строению молекул органических соединений одной или,
что существеннее, нескольких однотипных или различных функ-
циональных групп приводит к образованию соответствующих
293
Рис-XV.ll. Размещение катализатора в канале испарения про-
бы узла делителя потока капиллярного газового хромато-
графа [278].
1 — капиллярная колонка; 2 — делитель потока паров пробы (из
жаропрочного стекла); 3 — слой катализатора; 4 — линия подачи во-
дорода; 5 — микрошприц; 6 — тампоны из стекловолокна; 7 — корпус
испарителя хроматографа; 8 — вентиль тонкой регулировки.
углеводородов, идентификацию которых осуществить гораздо
легче, нежели исходных веществ. Во-первых, газохроматогра-
фический анализ углеводородов в отличие от их моно- и, осо-
294
бенно, полифункциональных производных не встречает принци-
пиальных затруднений. Во-вторых, в существующих базах дан-
ных наиболее подробная информация относится именно к угле-
водородам. У становление углеродного скелета исследуемых ве-
ществ по продуктам их гидрогенолиза можно рассматривать как
важный начальный этап последующей более подробной иденти-
фикации.
Отмеченные выше и частично иллюстрируемые схемой IV.3
на примере монофункциональных производных наблюдения об
образовании при гидрогенолизе представителей некоторых
классов органических веществ (в первую очередь, это отно-
сится к первичным спиртам и аминам, а также к альдегидам,
карбоновым кислотам, простым и сложным эфирам и др.) не
единственного продукта — алкана, соответствующего по угле-
родному скелету исходному соединению, а смеси гомологов с
возможным преобладанием парафинового углеводорода, содер-
жащего на один (или даже на два атома углерода) меньше, мо-
гут быть использованы как источник дополнительной информа-
ции, облегчающей идентификацию. В самом деле, если, напри-
мер, при гидрогенолизе продукта связывания карбонильного
соединения гидразином зарегистрируется смесь метана, этана,
пропана, бутана и пентана, то однозначно можно заключить,
что гидрогенолизу подвергался гидразон метилпропилкетона.
Методическое решение задачи определения углеродного ске-
лета, объединяющее стадию гидрогенолиза, осуществляемую
непосредственно в испарителе газового хроматографа, и после-
дующую идентификацию продуктов, разделяемых на насадоч-
ных или капиллярных колонках, по индексам удерживания или
с привлечением масс-спектрометрического детектирования за-
рекомендовало себя как весьма информативное при исследова-
нии структуры многочисленных серосодержащих соединений в
нефтях и нефтепродуктах, не менее сложных по строению мо-
лекул кислород- и азотсодержащих веществ [280, 281], полихло-
рированных бифенилов, нафталинов и линейных алканов [282],
Других опасных экотоксикантов и веществ природного происхо-
ждения [100, с. 216; 148, с. 119; 279].
Для исключения возможного совместного элюирования из
колонки однотипных продуктов, образующихся при гидрогено-
лизе различных функциональных производных, одновременно
Дозируемых в слой катализатора, можно рекомендовать при-
менение схем газовых коммуникаций, аналогичных используе-
мым в димерной газовой хроматографии (см. [79, с. 299 и сл.]
и рис. IV 12). При таком аппаратурном оформлении метода ги-
дрогенолизу можно подвергать не одновременно все компоненты
295
н3с - с- сн2 - сн2 - сн3
II
NH
-> сн4
* С2н6
> С3Н8
* С4Н1О
* С3н12
Кетон
Галогенид
Сульфид втор- или трет- О- или N-
производные
О- или N- при первичном атоме углерода:
альдегид R-i-СНО амин R-i-CH2-|-NH2
кислота R-1-COOH амид R-i-C—NH2
II
спирт R-i-CH2-!-OH о
эфир (простой) R-i- СН2 -1-0 -;-СН2 -I- R
эфир (сложный) R-i- COO -i- СН2 R'
исходный
углеводород
_____и (или)
следующий
низший
гомолог
Схема IV.3. Ожидаемые продукты гидрогенолиза некоторых
классов органических соединений [279].
сложных образцов, а лишь отдельные составляющие, предста-
вляющие аналитический интерес.
Заметим, что замена платинового или палладиевого катали-
затора на никель-алюминиевый и повышение температуры реак-
тора до 350-375°C приводят к полному разрыву всех межатом-
ных связей в молекулах органических соединений с образова-
нием исключительно метана и соответствующих продуктов вос-
становления других химических элементов (аммиака, галогено-
водородов, сероводорода, фосфина), присутствующих в составе
молекул подвергаемых гидрогенолизу веществ.
296
Рис. IV.12. Схема газовых коммуникаций хроматографа для
определения углеродного скелета компонентов сложных сме-
сей после их предварительного разделения.
1,10 — испарители; 2, 8 — хроматографические колонки; S — тер-
мостат колонок; 4 — дозирующая петля переключающего крана; 5 —
корпус переключающего крана; 6 — термостат реактора гидрогенолиза;
7— реактор гидрогенолиза; 9, 11 — ячейки пламенно-ионизационных
детекторов.
Названные неорганические гидриды (не регистрируемые наи-
более часто используемым пламенно-ионизационным детекто-
ром) медленно десорбируются с поверхности платино-палла-
диевых и никелевых катализаторов, а некоторые из них (НС1,
H2S) могут даже необратимо сорбироваться и вообще не по-
ступать в колонку. В многочисленных исследованиях возмож-
ностей определения углеродного скелета хроматографические
зоны этих “побочных” продуктов гидрогенолиза не наблюда-
лись. Тем не менее установлено, что в жестких условиях исчер-
пывающего гидрогенолиза, осуществляемого в режиме реакци-
онной газовой экстракции, не только СН4, но и NH3, НС1, H2S
и РН3 образуются и десорбируются с количественными выхо-
дами, что может быть использовано при анализах разнообраз-
ных объектов, в том числе объектов окружающей среды — воз-
духа, воды, почв, растительного сырья — на общий органиче-
ский углерод, азот, серу, галоген (хлор) и фосфор (см. обзор
[283]).
Группа методов направленного химического воздействия на
отдельные компоненты анализируемой смеси, приводящего к
297
продуктам с иными параметрами удерживания, включает также
образование алкилазидов* из соответствующих иод- или бром-
алканов [284], дегидратацию спиртов на поверхности алюмоси-
ликатных адсорбентов-катализаторов или при контакте с плен-
кой минеральной кислоты, нанесенной на подходящую подлож-
ку (возникают пики соответствующих олефинов), этерификацию
карбоновых кислот, например, под воздействием этилсульфата
калия (появляются пики сложных эфиров), термическое декар-
боксилирование двухосновных карбоновых кислот (регистриру-
ются пики монокарбоновых кислот), гидролиз сложных эфиров,
инициируемый едкой щелочью, нанесенной на диатомитовый но-
ситель (возникают пики соответствующих спиртов, а при пра-
вильном выборе колонки и условий анализа можно зарегистри-
ровать и парные спиртам свободные карбоновые кислоты) и т. п.
Подробнее практические возможности методики сдвига пиков
на примерах направленного превращения алкилиодидов (бро-
мидов) в соответствующие алкилазиды, а также фосфорноки-
слотной дегидратации некоторых алифатических спиртов С3-С5
рассмотрены в лабораторной работе 9 [79].
IV.2.6. Постколоночная дериватизация в
высокоэффективной жидкостной хроматографии
Для увеличения чувствительности и селективности анализа в
ВЭЖХ используют т. н. постколоночную дериватизацию**, ко-
торая сродни реакционной газовой хроматографии. Этот ана-
литический прием осуществляется в режиме on-line и заключа-
ется во введении электрофорных или хромофорных групп в мо-
лекулы разделенных исследуемых соединений, выходящих в по-
токе элюента из хроматографической колонки, перед их посту-
плением в ячейку детектора; соответствующая схема прибора
представлена на рис.IV.13 [286]. Для образования поглощаю-
щих УФ-свет или флуоресцирующих производных используют
реакторы трех типов: полые стальные капилляры (0,3-0,5 мм х
10-30 м), заполненные насадкой трубки (с внутренним диаме-
*Эта реакция, осуществляемая при пропускании паров алкилиодидов
или алкилбромидов через колонку-реактор с раствором азида натрия в
полиэтиленгликоле-ЮОО или твине-80, нанесенном на диатомитовый но-
ситель, в рекомендованных в [284] условиях проходит количественно, что
может быть использовано для синтеза необходимых алкилазидов в пре-
паративных целях [285].
**В англоязычной литературе наряду с термином «post-column derivati-
zation» используют также «reaction detection».
298
Рис. IV.13. Стандартное аппаратурное
оформление постколоночной деривати-
зации в ВЭЖХ.
1 — сосуд с элюентом; 2, 6 — насосы; 3 —
узел ввода пробы; 4 — колонка; 5 — сосуд с
реагентом; 7 — камера смешения; 8 — реак-
тор; 9 — детектор.
1
2
тром, сопоставимым с диаметром колонки, и длиной 10-15 см) и
реакторы с делителем потока.
При разработке постколоночных реакторов приходилось
учитывать время пребывания веществ в реакторе, которое
должно соответствовать времени, необходимому для проведе-
ния реакции. Это время можно менять, варьируя длину реак-
тора. Реакторы с насадкой выбирают в том случае, когда тре-
бования к размыванию хроматографических полос достаточно
жесткие, а реакторы с делением потока предпочтительны, если
процесс получения производных сравнительно медленный.
Альтернативным вариантом является постколоночная дери-
ватизация с последующей экстракцией (технические и проце-
дурные возможности осуществления on-line постколоночной экс-
тракции рассматриваются в разделе IV.2.9). Так же, как и в
реакционной газовой хроматографии, получение производных
после выхода компонентов анализируемой смеси из колонки не
приводит к изменению их хроматографических характеристик.
Особенности постколоночной дериватизации заключаются в
том, что хроматографическому разделению подвергаются ком-
поненты исходной смеси, а не образующиеся производные, как
в предколоночном варианте. Продолжительность реакции огра-
ничивается достигнутым разделением анализируемых веществ
(реакция должна быть достаточно быстрой — от 30 с до несколь-
299
ких мин — во избежание заметного внеколоночного размывания
хроматографической полосы). Артефакты при таком способе
получения производных сведены к минимуму.
Несомненным достоинством on-line постколоночной деривати-
зации являются невысокие требования к стабильности получа-
ющихся продуктов, что в случае предколоночного превращения,
наоборот, лимитирует процесс.
Недостатки и ограничения постколоночной дериватизации
следующие:
— для подачи реагента в поток элюента требуется дополни-
тельный насос;
— реагенты и продукты реакции должны быть совместимы с
используемой подвижной фазой, поскольку она выступает и как
растворитель реакционной системы; в свою очередь, подвижная
фаза не должна мешать детектированию и вступать во взаимо-
действие с реагентом дериватизации и компонентами анализи-
руемой смеси;
— степень конверсии должна быть воспроизводимой (чему
способствует сохранение отношения реагент/вещество), при
этом сама реакция может и не доходить до конца; в идеальном
случае результаты конверсии должны быть близки к количе-
ственным;
— избыток реагента не должен мешать детектированию.
Большинство используемых реакций хорошо известны в
функциональном органическом анализе, некоторые — специ-
ально разработаны для хроматографии Чаще всего постко-
лоночной дериватизации подвергаются соединения с амино- и
гидроксифункциональными группами, а также содержащие кар-
бонильную, карбоксильную и тиольную группировки.
В качестве объектов исследования выступают плазма, ткани
мозга, моча, сыворотка, в которых определяют аминокислоты,
стероиды, катехоламины [287]. Многие из описанных в лите-
ратуре реакций могут быть использованы как в пред-, так и
в постколоночном режиме; в ряде случаев последний вариант
предпочтителен. Так, например, дериватизация аминогруппы
с применением орто-фталевого альдегида в присутствии вос-
станавливающих агентов этантиола или 2-меркаптоэтанола при
pH > 7 приводит с высоким выходом в течение нескольких ми-
нут к флуоресцирующему нестабильному производному. Для
постколоночного перевода аминокислот в производные распро-
странено использование нингидрина. Реакция заканчивается за
1 мин.
300
IV.2.7. Качественный анализ по
относительному сигналу детекторов
При одновременной работе нескольких (двух-трех, редко —
больше) детекторов и при равенстве количеств вещества, по-
падающих в каждый детектор, на хроматограммах регистри-
руются пики различной высоты (и площади) в зависимости от
природы анализируемого вещества и принципа действия детек-
тора, как показано, например, на рис. IV.14. Отношение сигна-
лов (относительный сигнал или относительный отклик) различ-
ных детекторов на анализируемое вещество является важным
показателем при групповой идентификации компонентов смесей
неизвестного состава.
Рис.1У.14. Схема одновременно-
го использования двух детекто-
ров газового хроматографа с па-
раллельным (а) и последова-
тельным (6) размещением детек-
торов.
1 — колонка; 2, 3 — детекторы;
5 — преобразователи сигналов детек-
торов; 6 — двухканальный регистра-
тор.
Газовая схема хроматографа для экспериментального осуще-
ствления этого приема качественного анализа может быть со-
брана в двух вариантах: с параллельным (рис. IV.14, а) и по-
следовательным (рис. IV.14, 6) размещением детекторов относи-
тельно колонки [288]. Сигнал каждого детектора после усиления
(или ослабления) подается на один двухканальный или на два
одноканальных регистратора.
Очевидно, что вариант б возможен лишь при условии, что де-
тектор, соединяемый непосредственно с колонкой, является не-
деструктивным (как, например, катарометр или ФИЛ). При по-
строении схемы необходимо учитывать возможность дополни-
тельного размывания хроматографических зон в соединитель-
ных линиях после колонки (ухудшение разделения компонентов
смеси, снижение чувствительности детектирования). Следует
также иметь в виду возможные различия в абсолютной чув-
ствительности выбранных детекторов (что определяет требуе-
301
мую чувствительность регистрации сигнала каждого детектора
и различную степень деления газового потока на выходе из ко-
лонки — в первом варианте схемы).
Вариант схемы б предусматривает попадание из колонки в
детекторы одного и того же количества паров анализируемого
соединения. При этом условии численное значение относитель-
ного отклика детектора R& находят непосредственно из хрома-
тограмм соотнесением площадей, высот пиков или других пара-
метров, отражающих реакцию (отклик) каждого детектора на
выход из колонки интересующего г-го вещества* (при заданной
чувствительности регистрации сигнала каждого детектора):
T?D = AM' ~ hi/h'i,
где Ai(hj) и А'(/^) — площади (высоты) хроматографических
пиков, зарегистрированных первым и вторым детектором соот-
ветственно**.
Начиная с середины 60-х годов наиболее употребительными
стали детекторы по теплопроводности и пламенно-ионизацион-
ный. Структурные особенности молекул по относительному
(сравнительному) отклику ДТП и ПИД выявить трудно. И
тем не менее величина относительного сигнала даже этих двух
детекторов в ряде случаев оказывается достаточно информа-
тивной при необходимости отнесения исследуемого вещества
к тому или иному гомологическому ряду [237]. Гораздо боль-
шие возможности функционального качественного анализа (рас-
ширение круга анализируемых объектов) обеспечивает ком-
бинирование катарометра, ПИД или ФИД и какого-либо од-
ного (двух, редко — большего числа) селективного детектора,
например термоионного, электронозахватного или пламенно-
фотометрического (ПФД).
Выбор конкретной комбинации детекторов обусловливается
природой определяемых соединений и веществ, не представляю-
щих аналитического интереса, однако присутствующих в пробе.
Так, например, для групповой идентификации полиароматиче-
ских углеводородов оправдано сопоставление сигналов ПИД
*Абсолютную величину введенной пробы знать необязательно, но важ-
но, чтобы регистрируемые сигналы не выходили за пределы линейного
диапазона каждого детектора. При использовании первого варианта га-
зовой схемы необходимо вводить коэффициенты, учитывающие неравен-
ство количеств веществ, попадающих в детекторы. Эти коэффициенты
определяются отношением расходов газа-носителя через детекторы.
**Строго говоря, отношения площадей А{/А\ и высот hi/h\ будут равны
только при элюировании из колонки симметричных зон исследуемых со-
единений и при отсутствии их неодинакового размывания (искажения
формы полосы) в соединительных линиях, связывающих выходной конец
колонки с каждым детектором.
302
и ФИД [289], тогда как при определении природы различных
пестицидов в пищевых продуктах становится целесообразным
измерение относительных откликов трех высокочувствитель-
ных селективных детекторов: электронозахватного, пламенно-
фотометрического и термоионного [290]. При этом следует
иметь в виду, что некоторые балластные соединения, присут-
ствующие в анализируемых образцах сложного состава, мо-
гут блокировать (маскировать) сигналы селективных детекто-
ров, что приведет к частичной или даже полной потере цен-
ной информации. Это прежде всего относится к пламенно-
фотометрическому детектору, избирательная реакция которого
на серо- или фосфорсодержащие соединения может теряться
при попадании в пламя совместно с указанными веществами не-
которых т.н. тушителей характерной для атомов серы и фос-
фора хемилюминесценции [74]. В роли тушителей чаще всего
выступают углеводороды (в первую очередь — ароматические).
Поэтому, несмотря на то, что вероятность проявления этого
нежелательного эффекта зависит от конструктивных особенно-
стей ячейки ПФД и рабочего режима его эксплуатации [33, ч. 1,
с. 554], возможность проявления пиков серосодержащих соеди-
нений на канале ПФД параллельно с регистрацией пиков угле-
водородов на канале ПИД при исследовании образцов нефти
или продуктов ее переработки, как об этом говорится в [288,
с.50], является сомнительной.
Качественный анализ по относительному сигналу детекторов
требует накопления как можно большего числа эксперименталь-
ных данных. При их обработке достаточно информативной мо-
жет оказаться интерполяционная характеристика — индекс чув-
ствительности [291-293], рассчитываемый (по аналогии с ли-
нейным индексом удерживания) по формуле
, _ 1/Дм(») ~ l/^M(z)
l/^M(z + l) - 1/Km(z)
где 1//£м(«), l/^M(z), l/l^M(z+i) — значения относительной
мольной чувствительности детектора (обычно относительно
бензола) соответственно к г-му компоненту и к стандартам (ве-
ществам сравнения) — нормальным парафиновым углеводоро-
дам с числом атомов углерода в молекулах z и (z + 1), причем
V^M(z+t) > 1/Дм(г) > l/^M(z)-
Сопоставление экспериментально найденных величин, харак-
теризующих относительный отклик детекторов — и(или)
^ч(г), сданными библиотеки хроматографических спектров [292-
294] позволит провести групповую идентификацию с весьма вы-
сокой достоверностью.
303
В ВЭЖХ с У Ф-детектированием при групповой идентифи-
кации органических соединений — представителей различных
групп структурных аналогов — весьма информативным ока-
залось измерение коэффициентов относительного поглощения
Лотн = А(А1)/А(Аг) на разных фиксированных длинах волн [295].
В качестве наиболее универсальных длин волн для разных ве-
ществ рекомендовано выбирать 254 нм (международный стан-
дарт УФ-детектирования) и длину волны в области неселектив-
ного поглощения органических соединений на границе ближнего
ультрафиолета, например 220 нм (прозрачной для многих по-
движных фаз — ацетонитрила, метанола, тетрагидрофурана и
др.). Принципиальных теоретических ограничений на выбор
пары Ai и Аг нет, важно, однако, чтобы разность ДА была не
слишком мала и, лучше всего, отвечала интервалу 30-40 нм.
Кроме названных длин волн удобной для групповой идентифи-
кации органических соединений является пара 280 и 246 нм. В
качестве дополнительной можно выбрать и еще одну — тре-
тью — длину волны (например, 317 нм) с последующим пере-
крестным измерением трех спектральных отношений (для при-
веденных длин волн соответственно А(280/246), Л (317/280) и
А(317/246)).
Установленное в работе [295] постоянство отношений А(А1/Аг)
для структурных аналогов разных групп органических соеди-
нений (например, содержащих бензоильный фрагмент алкилфе-
нилкетонов, бензоиламинов, бензоилгидразинов и др.) свиде-
тельствует о возможности использования коэффициентов Аотн
для выявления тех или иных интересующих аналитика хромо-
форных группировок.
IV.2.8. Хромаю-масс-спектрометрия
IV.2.8.1. Общие сведения
Проблемы установления структуры и идентификации неиз-
вестных органических соединений часто встречаются в разно-
образных исследованиях и относятся к числу наиболее слож-
ных в современной химии. При этом следует иметь в виду не
выбор из сравнительно небольшого числа заранее известных ве-
ществ (подтверждающий анализ), а, в первую очередь, случаи,
когда априорные сведения о природе объектов минимальны и
практически всю информацию об их структурах приходится из-
влекать из совокупностей различных аналитических сигналов
(разведочный анализ). Преобразование таких совокупностей
304
измеряемых параметров в связные графы, представляющие со-
бой структурные формулы органических соединений, и явля-
ется основной задачей идентификации и структурного анализа*.
Хроматографические параметры удерживания (в форме ин-
дексов удерживания — инвариантов, обладающих межлабора-
торной воспроизводимостью при стандартизации неподвижных
фаз) безусловно относятся к числу “средств” решения струк-
турных задач. Однако эти величины являются одномерными,
вследствие чего их информативность не слишком велика. Бли-
жайшей аналогией хроматографических индексов удерживания
при таком рассмотрении оказываются “классические” физико-
химические константы органических соединений — темпера-
туры кипения. Так же, как по значению Гкип, например 80°C,
при атмосферном давлении, без дополнительной информации
невозможно идентифицировать соединение, так и по единствен-
ному значению индекса 653 на стандартной неполярной поли-
диметилсилоксановой неподвижной фазе эта задача не может
быть решена (приведены данные для бензола).
Таким образом, одна из важнейших особенностей хромато-
графии как метода структурного анализа и идентификации со-
стоит в том, что практически любая информация в дополне-
ние к хроматографическим параметрам удерживания резко уве-
личивает надежность и однозначность результатов. Наибо-
лее известными примерами объединения разнородной информа-
ции могут служить применение нескольких колонок с неподвиж-
ными фазами различной полярности [220], привлечение химиче-
ских данных о функциональных группах в составе молекул [148],
информация о внеколоночных фазовых равновесиях (коэффи-
циенты распределения в гетерофазных системах органических
растворителей [264, 265]) и др. Однако наибольшее распростра-
нение получили комбинированные хромато-спектральные ме-
тоды: сочетание газовой хроматографии с масс-спектрометрией
[296], ИК-спектроскопией и, в последнее время, с атомно-эмис-
сионной спектроскопией [297]. Самый известный из. этих мето-
*Под идентификацией соединений следует понимать отождествление
соответствующих наборов аналитических сигналов с данными для ра-
нее охарактеризованных веществ. Термин “структурный анализ” шире
и охватывает еще не охарактеризованные или даже еще не известные
соединения, для установления формул которых привлекают не столько
фактические данные, сколько спектроструктурные корреляции для дру-
гих (известных) соединений сходной природы, часто обобщаемые на бо-
лее крупные таксономические единицы (гомологические ряды, признаки
функциональных групп, фрагментов структуры и др.). В англоязычной
литературе нет подобной дифференциации терминов и их объединяют
словом “identification”.
305
дов — хромато-масс-спектрометрия — рассматривается в на-
стоящем разделе. Особая роль хроматографии в подобных со-
четаниях состоит в том, что предварительное разделение позво-
ляет распространить возможности второго “компонента” ком-
бинированных методов (спектрального) на анализ сложных сме-
сей веществ. Тем самым исключаются длительные и трудоем-
кие процедуры выделения и очистки индивидуальных компонен-
тов таких смесей.
Простейшая и наиболее наглядная оценка информативности
различных одномерных аналитических параметров (А) при ре-
шении задач идентификации возможна на основе отношений
диапазонов вариации значений этих параметров Амакс — Длин
к удвоенным средним стандартным отклонениям, характеризу-
ющим изменения конкретных величин А в этих диапазонах:
\ Амакс Длин
Л(Л) *----2S~A---•
Как правило, оценки внутри- и межлабораторных погрешностей
сильно отличаются, что приводит к неодинаковым оценкам ин-
формативности.
Можно оценить, что двоичный логарифм этого отношения,
т. е. ld/?(A), представляет собой количество информации (в
битах), получаемой за счет измерения рассматриваемой ве-
личины. Сравнительная оценка информативности некоторых
свойств органических соединений представлена в табл. IV.13.
Таблица IV.IS. Сравнительная оценка информативности
некоторых свойств органических соединений
Параметр -Амин Sa Я(Л)
Т’кип (атм. дав л) Индексы удерживания: 20 350 1 ~ 165
на неполярных фазах 500 3500 8 ~ 200
на полярных фазах 700 2000 15 ~ 45
в обращенно-фазной ВЭЖХ 400 1500 17 ~ 30
Молекулярные массовые числа 1 > 500 « 1 Более 500
Если два аналитических параметра не коррелируют между
собой, то оценки информативности их совместного примене-
ния мультипликативны относительно значений Л-критериев ка-
ждого из них, т.е. R(AB) <С /?(А) • R(B). Сравнение приведен-
ных оценок R(A) показывает, что молекулярные массы исследуе-
мых веществ существенно превосходят все остальные по инфор-
306
мативности. Этот факт является одним из теоретических об-
оснований того широкого распространения, которое получила
хромато-масс-спектрометрия как метод структурного анализа и
идентификации неизвестных органических соединений в слож-
ных образцах.
IV.2.8.2. Принципы масс- и хромато-масс-спектрометрии
Масс-спектрометрия как метод исследования предполагает
ионизацию веществ различными способами с последующим де-
тектированием образующихся ионов. Именно за счет реги-
страции заряженных частиц достигается намного большая чув-
ствительность, чем в недеструктивных методах, основанных
на взаимодействии электромагнитного излучения с веществами
(ИК-, УФ- и ЯМР-спектроскопия). Пределы обнаружения до
1О“10 г/с (на уровне ионизационных хроматографических де-
текторов) являются типичными для большинства простейших
моделей хромато-масс-спектрометров; значительная же часть
современных приборов обеспечивает регистрацию до 10“15-
10“14 г/с органических соединений.
Таблица IV. 1/. Основные методы ионизации ионов
Агрегатное состояние веществ
I. Газовая фаза (для летучих
соединений)
II. Жидкости (подвижные фазы в
ВЭЖХ)
III. Конденсированные среды
(нелетучие вещества в специальных
матрицах)
Основные методы ионизации
(принятая аббревиатура)
Электронный удар (EI)
Химическая ионизация (CI)
Термораспыление (TS)
Электрораспыление (ES)
Ионизация быстрыми атомами
(FAB); другие вторично-
эмиссионные методы
Методы генерации ионов связаны с агрегатным состоянием
вещества на стадии ионизации (табл. IV. 14). Чаще всего в масс-
спектрометрии используют процессы, приводящие к образо-
ванию положительно заряженных ионов (катионов и катион-
радикалов), однако при решении специальных задач (сниже-
ние пределов обнаружения, повышение селективности детекти-
рования, исследование труднолетучих веществ и др.) возмо-
жен переход к детектированию отрицательно заряженных ио-
нов (анионов и анион-радикалов). При использовании масс-
спектрометров как детекторов газовых хроматографов методы
307
ионизации обычно ограничены I группой, а в случае жидкост-
ных — II группой. Методы ионизации в конденсированных сре-
дах в хромато-масс-спектрометрии не применяются.
Любой хромато-масс-спектрометр включает следующие
основные конструктивные системы:
Для обеспечения достаточной длины свободного пробега ионов
(соударения с нейтральными молекулами остаточных газов или
стенками приборов приводят к “гибели” заряженных частиц)
источник ионов и особенно масс-анализатор должны быть ваку-
умированы до давлений 10-5 Па и менее.
IV.2.8.3. Системы газовый хроматограф —
масс-спектрометр (ГХ—МС)
Поток газа на выходе из хроматографической колонки нахо-
дится при давлении, близком к атмосферному (105 Па). Давле-
ние же в источнике ионов при ионизации электронами соста-
вляет менее 10-3 Па, а в условиях химической ионизации —
10-100 Па. Поддержание указанной разности давлений только
за счет интенсивной откачки газа-носителя возможно лишь при
работе с капиллярными колонками (расход 0,5-5 мл/мин). При
работе с насадочными колонками (расход 5-50 мл/мин) необхо-
димо применение специальных устройств для отделения боль-
шей части газа-носителя (Не, Щ) без существенных потерь опре-
деляемых компонентов и уширения их хроматографических
зон — молекулярных сепараторов. Описание их конструкций
и принципов действия можно найти во многих руководствах по
хромато-масс-спектрометрии [296, 298].
308
Ионизация потоком электронов, эмиттируемых нагретым ка-
тодом, является стандартным методом ионизации органических
соединений. Энергию электронов можно легко варьировать, из-
меняя разность потенциалов между катодом и вторым электро-
дом-ловушкой. Нижней границей рабочего диапазона явля-
ются энергии ионизации веществ (для органических соедине-
ний — от 7 до 12 эВ), верхняя не превышает 100 эВ (1 эВ =
1,6-10-19 Дж). Международный стандарт энергии ионизации —
70 эВ (1,1 • 10-17 Дж) — является настолько общепризнанным,
что на многих простейших приборах (например, хроматографах
с т. н. масс-селективными детекторами фирмы Hewlett-Packard
HP 5971, HP 5972) предусмотрен только такой фиксированный
режим ионизации.
Характер взаимодействия электронов с молекулами орга-
нических соединений в газовой фазе зависит от их энергии;
при 70 эВ преобладает образование положительно заряженных
катион-радикалов (молекулярные ионы):
ионизация: М + е М+* + 2е;
последующая фрагментация
молекулярных ионов: М+’ -+ R+ + А*, М+* —> В+* + N*.
Распад молекулярных ионов при давлениях менее 10-3 Па в
отсутствие соударений с другими частицами подчиняется кине-
тическим закономерностям реакций первого порядка. Образую-
щаяся совокупность М+* , первичных, вторичных и более “глу-
боких” осколочных ионов после их разделения и регистрации
представляет собой масс-спектр характеризуемого соединения.
Химическая ионизация в отличие от электронного удара ос-
нована на ион-молекулярных взаимодействиях в источнике, да-
вление в котором увеличено до 10-100 Па за счет введения газа-
реагента (до 80-х годов использовали название “газ-реактант”).
Для этих целей применяют более сотни соединений, включая
Ar, Н2О, NO, N2O, СН4, NH3, щ?о-С4Н10, (СН3)2СО, СН3ОСН =
СН2 и др. При взаимодействии с электронами в первую очередь
ионизации подвергается газ-реагент, который далее взаимодей-
ствует с молекулами определяемых соединений:
ионизация газа-реагента:
последующие
ион-молекулярные реакции:
ионизация определяемых
соединений:
R + е R+* + 2е;
R+* + R RH+ + (R - Н)*;
М + R+* -> М+* + R, М + RH+ -> МН+ + R.
Следовательно, при химической ионизации основным типом
ионов оказываются протонированные молекулярные ионы ис-
309
следуемых веществ и, нередко, более сложные ассоциаты. Про-
цессы фрагментации подобных ионов выражены в значительно
меньшей степени, чем при электронном ударе.
IV.2.8.4. Системы жидкостный хроматограф —
масс-спектрометр (ЖХ—МС)
Сочетание ГХ—МС, пригодное для создания серийных при-
боров, было предложено в 1959 г. Первые же удовлетворитель-
ные результаты решения более сложной задачи — соединения
жидкостного хроматографа с масс-спектрометром— относятся
только к 1983 г. В этой комбинации наличие специальных слож-
ных интерфейсов между хроматографической колонкой и ис-
точником ионов, необходимых для удаления значительных ко-
личеств растворителей (типичный интервал расходов элюента
0,1-1 мл/мин), является безусловно необходимым.
В зависимости от pH элюента и природы ион-парных реаген-
тов в обращенно-фазной ВЭЖХ часть органических соединений
в растворе уже может быть частично ионизована за счет прото-
тропных равновесий (положение равновесия определяется кон-
стантами рКА или рКц сорбатов):
S + HA # SH+ + А".
В системах ЖХ—МС на выходе из хроматографической ко-
лонки элюент подвергается максимальному диспергированию
и в виде аэрозоля попадает в обогреваемую камеру для испа-
рения большей части растворителя. Метод ЖХ—МС предъ-
являет очень “жесткие” требования к чистоте растворителей
(метанол, ацетонитрил и их смеси с водой) и к природе ион-
парных реагентов, которые должны быть летучими и не образо-
вывать твердых частиц при удалении растворителя: уксусная,
трифторуксусная кислоты, ацетат аммония, трифторацетат ам-
мония и т. д. Непосредственным продолжением испарительной
камеры является источник ионов, рассчитанный на улавлива-
ние и преобразование потока ионов требуемого заряда. Тогда в
данном варианте ввода проб в масс-спектрометр (термораспы-
ление) дополнительная ионизация органических соединений, пе-
реводимых в газовую фазу из раствора, может быть совсем ис-
ключена. Фактически при этом регистрируются ионы, уже при-
сутствующие в жидкой фазе при данном pH среды.
310
Пример. При регистрации масс-спектров водных растворов солей
металлов (МеХ) наблюдаются совокупности сигналов сольватированных
катионов металлов Ме(Н2О)+, например для солей натрия:
Na+ Na(H2O)+ Na(H2O)+ Na(H2O)+ и т.д.
m/z 23 41 59 77 23 + п • 18
В зависимости от способности анализируемых соединений к
протонированию при использовании метода термораспыления
некоторые вещества вообще не регистрируются, что является
одним из главных недостатков этого способа. Например, при pH
элюента 4,0 можно детектировать анилин (рКд 4,6), имидазол
(рКд 7,0) и другие ароматические и гетероциклические амины,
но пиразол (РКА 2,5), фенол (рКд 9,9) не образуют ионов SH+ в
растворе в заметных количествах. Более универсальным мето-
дом является поэтому т.н. электрораспыление, когда процесс
испарения растворителя осуществляется в электрическом поле
(1-6 кВ). Это приводит к неселективной ионизации практически
любых органических соединений. Вместо электрораспыления
иногда используют сочетание термораспыления с последующей
ионизацией электронным ударом.
Следует подчеркнуть, что системы ЖХ—МС являются весь-
ма дорогостоящими, все еще сложными в обращении и пре-
дусмотрены только на достаточно уникальных приборах (на-
пример, Finnigan MS 80). На самом деле процесс ионизации
в жидкостной хромато-масс-спектрометрии намного более сло-
жен, чем изложенное здесь его схематическое описание.
IV.2.8.5. Масс-анализаторы.
Обработка и представление результатов
хромато-масс-спектрометрического анализа
Назначение масс-анализатора — разделение образующейся
в источнике ионов совокупности заряженных частиц по массам
(точнее — по отношениям mlz, однако в органической масс-
спектрометрии преобладают однозарядные ионы, z = 1). По-
добное разделение ионов возможно различными способами, в
том числе по их импульсам (магнитные анализаторы), кинетиче-
ским энергиям (электростатические) или скоростям (времяпро-
летные). Широкое распространение получили квадрупольные
анализаторы, в которых разделение осуществляется в частотно-
модулированном электрическом поле, и сходные по принципу
действия конструкции типа “ионная ловушка”. В последнем
случае источник ионов практически совмещен с анализатором,
и за счет уменьшения размеров они удобны именно как детек-
торы в хромато-масс-спектрометрии. Следует учитывать, что
311
различия в конструкциях источников ионов и особенно масс-
анализаторов проявляются в вариациях интенсивностей сигна-
лов даже в стандартных масс-спектрах при ионизации электрон-
ным ударом.
Каждый анализатор в отдельности обеспечивает разрешение
прибора по массам регистрируемых ионов порядка (0,5-2) • 103
(так называемая масс-спектроскопия низкого разрешения). В
сочетании с газовым хроматографом такой диапазон спектроме-
тра более чем достаточен для решения большинства задач, так
как соединения с большими массами обладают малой летуче-
стью и обычно не могут быть подвергнуты хроматографиче-
скому разделению. Градуировку шкалы масс при этом про-
водят по целочисленным массовым числам ионов (суммы цело-
численных массовых чисел образующих эти ионы атомов; для
атома массовое число равно сумме числа нуклонов в ядре).
Комбинация двух анализаторов (обычно магнитного и электро-
статического) повышает разрешение прибора до (1-2)-105, что
обеспечивает точность измерения масс ионов в углеродной шка-
ле до 5-6 значащих цифр, т.е. до 0,1-1 мДа. Столь высо-
кая точность измерений позволяет непосредственно устанавли-
вать формулы ионов, что полезно при интерпретации сложных
масс-спектров или (в хромато-масс-спектрометрии) для обеспе-
чения максимальной селективности детектирования. По этой
причине, например, для обнаружения полихлорированных аро-
матических гетероциклических соединений (дибензодиоксины,
дибензофураны и др.) используют только приборы с двойной
фокусировкой.
Пример. В масс-спектрометрии низкого разрешения массовому чи-
слу иона т/г 70 могут соответствовать более двадцати возможных его
составов; при измерении же его массы с точностью до ±0,5 мДа брутто-
формула устанавливается однозначно.
Массовое число: m/z 70 Масса в углеродной шкале:
70,0534 ± 0,0005 Да
Возможные брутто-формулы:
С5Н1О C2H2N2O
с4н6о CN3O
С3Н2О2 C2NO2 Единственный ответ:
c4h8n C4HnB c3h6n2
c3h6n2 c4h3f
c2h4n2 CHF3 Рассчитанное значение m/zz
ch2n4 C3H2S 70,0531
c3h4no c2ns
Для детектирования ионов после их разделения применяют
фотоэлектронные умножители, что же касается обработки и
представления полученной информации, то все современные
хромато-масс-спектрометры оснащены ЭВМ и предусматрива-
312
ют запись масс-спектров с интервалом 1-2 с в течение всего вре-
мени регистрации хроматограммы (требуемый объем памяти
ЭВМ для одного такого анализа составляет 0,5-1 Мбайт). Да-
лее полученная информация может быть представлена в виде
хроматограммы по полному ионному току (TIC) в заданном диа-
пазоне масс (обычно от 40 до 500 и более), хроматограммой
по сигналам ионов с заданными массовыми числами (SIM) и в
виде масс-спектров отдельных компонентов. Дополнительные
сервисные процедуры стандартного программного обеспечения
большинства современных хромато-масс-спектрометров вклю-
чают усреднение масс-спектров, записанных для одного и того
же хроматографического пика, вычитание фона, выявление не-
разделенных компонентов, сравнение спектров с информацией
различных баз данных для идентификации веществ и т. д.
Рис. IV.15 схематически иллюстрирует основные возможно-
сти хромато-масс-спектрометрического анализа. Хроматограм-
ма по полному ионному току (а) может быть дополнена хро-
матограммой по массовому числу m/z 91 (6), которая позво-
ляет быстро выявить из всего многообразия хроматографиче-
ских сигналов пики, принадлежащие ароматическим углеводо-
родам с фрагментом структуры СбН5—СНг—. Каждый из ком-
понентов образца может быть охарактеризован полным масс-
спектром (е).
Интегральные сигналы на хроматограммах по полному ион-
ному току допускают их обычную обработку (измерение пло-
щадей) так же, как и сигналы любого другого хроматографиче-
ского детектора. Линейный диапазон масс-спектрометров сопо-
ставим с линейным диапазоном пламенно-ионизационных детек-
торов (~106), следовательно, они могут применяться для коли-
чественного анализа методами абсолютной градуировки и стан-
дартной добавки. Особую осторожность необходимо соблю-
дать при использовании метода внутреннего стандарта, так как
относительные коэффициенты чувствительности даже к соеди-
нениям близкой структуры в масс-спектрометрии могут варьи-
ровать в очень широких пределах (в зависимости от различной
эффективности ионизации). По этим же причинам такой спо-
соб количественного анализа, как внутренняя нормализация, в
хромато-масс-спектрометрии неприменим.
С другой стороны, возможен интересный вариант метода
стандартной добавки, теоретически требующий только одно-
кратного анализа образца — метод изотопного разбавления (в
подтверждающем анализе). Для определения заранее извест-
ных веществ в образцы до всех операций подготовки проб вво-
дят известные количества определяемых соединений, меченных
313
Phc.IV.15. Иллюстрация типичных возможностей ГХ—МС
анализа.
а — фрагмент хроматограммы смесей углеводородов по полному
ионному току; б — селективное детектирование по массовому числу
т/z 91; в — стандартные масс-спектры компонентов с временами удер-
живания 13,40 мин (*) и 16,33 мин (**).
стабильными изотопами (2Н, 13С, 15N, 18О и др.). По соотноше-
ниям интенсивностей сигналов меченого и немеченого соедине-
ний в масс-спектрах неразделенных хроматографических пиков
определяют исходное содержание аналитов в препаратах.
Интерпретацию масс-спектров производят сочетанием хро-
матографической и масс-спектрометрической информации.
Основным приемом интерпретации масс-спектров низкого
разрешения в практике подтверждающего хромато-масс-спек-
трометрического анализа является библиотечный поиск с це-
лью выявления соединений с наиболее “похожими” признаками.
Современное информационное обеспечение в масс-спектромет-
рии достаточно представительно: так, например, база данных
Wiley/NBS (1989 г.) включает около 112 тыс. масс-спектров
314
[299], a Registry of Mass-Spectral Data (1991 r.) — более 400 тысяч
[300]. Процедуры библиотечного поиска, как правило, приво-
дят к перечням альтернативных вариантов интерпретации (их
искусственно ограничивают 10-20), из которых на заключитель-
ных стадиях отбора по одномерным параметрам — факторам со-
впадения Q (0 Q 1) — для дальнейшего рассмотрения оста-
вляют только соединения с Q > 0,9. Таким образом, формирова-
ние окончательных ответов остается задачей химика-аналитика
и предполагает привлечение любой доступной дополнительной
информации.
В разведочном анализе такой подход к интерпретации масс-
спектров возможен, но не исключает применения различных
спектроструктурных корреляций, аналогичных известным для
других видов спектроскопии. Даже при ионизации электронным
ударом около 85% охарактеризованных масс-спектрами органи-
ческих соединений дают надежно выявляемые сигналы моле-
кулярных ионов. Следовательно, масс-слектрометрия является
наиболее надежным методом установления молекулярных масс
исследуемых веществ. Для интерпретации совокупностей сиг-
налов осколочных ионов рекомендованы специальные таблицы
характеристических массовых чисел и характеристических раз-
ностей. Особенности их применения (см., например, [300]) по
объективным причинам требуют некоторого опыта работы и
“масс-спектрометрической” квалификации аналитика.
Масс-спектрометрия как метод структурного анализа орга-
нических соединений обладает высокой информативностью на
стадии групповой идентификации (отнесение к соответствую-
щим гомологическим рядам), но существенно меньшей — при
установлении строения углеродного скелета молекул. Извест-
ны масс-спектрометрические инварианты гомологических ря-
дов — спектры ионных серий [300], применение которых позво-
ляет формализовать именно стадию групповой идентификации.
Что же касается второй части задачи, то необходимо признать,
что попытки ее решения только средствами масс-спектрометрии
неоптимальны и приводят к большому количеству ошибочных
ответов. Наиболее “естественным” способом повышения на-
дежности и однозначности результатов оказывается сочетание
масс-спектрометрических и хроматографических данных. Од-
нако современное информационное обеспечение для газохрома-
тографических индексов удерживания (ИУ) существенно усту-
пает базам масс-спектров по числу охарактеризованных ве-
ществ.
Целесообразность привлечения хроматографических пара-
метров удерживания при хромато-масс-спектрометрической
315
идентификации можно дополнительно проиллюстрировать дан-
ными табл. IV. 15, содержащей информацию о простейших при-
мерах структурных фрагментов молекул органических соедине-
ний, неразличимых только по масс-спектрам. Перечень подоб-
ных структурных фрагментов включает разветвления углерод-
ного скелета, особенно в положениях, сильно удаленных от ме-
ста локализации заряда молекулярных ионов, изомерию поло-
жения в ароматических и гетероциклических соединениях, изо-
барные фрагменты и структуры (прежде всего, С2Н4—N2—СО с
m/z 28) и изомерию сопряженных систем. Последний случай
относится к наиболее сложным, однако подобный тип изоме-
рии (например, для рядов нафталины — азулены, хинолины —
изохинолины и др.) отчетливее всего проявляется в различиях
электронных спектров поглощения. Таким образом, для иден-
тификации подобных соединений рациональнее всего использо-
вать анализ методом ВЭЖХ с У Ф-детектированием, но при обя-
зательном наличии баз данных для интерпретации полученных
результатов. Другой, не менее важный вывод из изложенного
состоит в том, что ни один из известных аналитических мето-
дов не может гарантировать надежную идентификацию любых
соединений. Проблема выбора оптимальных сочетаний таких
методов характеризует “искусство” химика-аналитика в реше-
нии тех или иных задач.
Использование хроматографических параметров удержива-
ния для идентификации позволяет сократить объем масс-спек-
трометрической информации до массовых чисел молекулярных
и наиболее интенсивных сигналов спектров. Сочетание всего
трех аналитических параметров — М+*, m/z и индексов удер-
живания на стандартных фазах — по критериям однозначности
и правильности результатов превосходит использование пол-
ных масс-спектров, рассматриваемых без привлечения хрома-
тографической информации [241]. Табл.IV.16 включает указан-
ные параметры для изобарных соединений различного состава
с молекулярным массовым числом 80. Из приведенных дан-
ных следует, что все изомерные углеводороды CeHg обладают
настолько похожими масс-спектрами, что об их идентифика-
ции без использования индексов удерживания не может быть
и речи (массовые числа главных сигналов спектров одинаковы
для всех известных соединений этой группы). Даже и в этом
случае некоторые пары изомеров неразличимы (ср., например,
З-метил-З-пентен-1-ин и 1-гексен-5-ин). Однако у соединений с
М = 80, содержащих гетероатомы, диапазоны вариаций как ин-
дексов удерживания, так и значений (m/z)MaKC больше, чем у
углеводородов. Сочетание перечисленных в таблице аналити-
316
Таблица IV. 15. Информативность дополнительных аналитических параметров для повышения надежности и однозначности
интерпретации результатов хромато-масс-спекгромстрического анализа
Фрагменты структуры, неразличимые по масс-спектрам Простейшие примеры
Информативные аналитические параметры Изомерные или (D изобарные соединения (П) Молекулярная массаДш/Ояакс ИУ на стандартных неполярных фазах
(I) (II)
Разветвления углеродного скелета ИУ(ГХ) 134/91 104б±5 997+6
Изомерия положения ИУ(ГХ) фг" 120/105 963 + 8 984 + 8
Изобарные фрагменты структуры ИУ (ГХ, ВЭЖХ), элементный анализ (АЭД), ИК-спектры ^^сосн3 120/105 914 + 6 1041+ 9
NHCH3
Изомерия сопряженных систем УФ-спектральные данные (ВЭЖХ), ИУ(ГХ,ВЭЖХ) 107/106,107 920+8 1041+8
Таблица IV. 16. Аналитические параметры для
хромато-масе-спектрометри ческой идет-ификации
избранных соединений с М = 80: массовые числа
максимальных сигналов спектров и рандомизованные
индексы удерживания на стандартных неполярных
полидиметилсилоксановых неподвижных фазах
Соединение {rnf z)tA3.KC ИУ
Брутто-формула CeHg:
1-метил циклопентадиен 79 635 ±8
2-метил циклопентадиен 79 637 ±8
1,3-циклогексадиен 79 664 ±6
1,4-циклогексадиен 79 708 ± 77
З-метил-З-пентен- 1-ин 79 622 ±6
4-метил-3-пентен-1-ин 79 654 ± 10
2-метил-1-пентен-З-ин 79 674 ±4
1-гексен-З-ин 79 677 ±9
1-гексен-5-ин 79 625 ± 12
1,3,5-гексатриен 79 653 ±8
Брутто-формула СьЩО:
1,4-п ентадиин-3-о л 79 833*
Брутто-формула C4H4N2:
пиразин 80 713 ± 12
пиримидин 80,26 728 ±8
пиридазин 80 897 ± 10
динитрил 1,2-этандикарбоновой кислоты 53 867 ± 15
Брутто-формула CeHeFj:
1,3-дифторпропан 47 481 ± 14
Брутто-формула C2H5OCI:
2-хлорэтанол 31 633 ±8
метилхлорметиловый эфир 45 542 ±5
Брутто-формула C2H2FCI:
1-фтор-1-хлорэтилен 80,45 375 ±6
*Единственное (нерандомизованное) значение индекса данно-
го соединения указано без стандартного отклонения.
ческих параметров обеспечивает их однозначную идентифика-
цию практически во всех случаях.
Пример. Для дополнительной иллюстрации необходимости примене-
ния индексов удерживания в сочетании с масс-спектрами целесообразно
вернуться к рис. IV.15. Два компонента с практически неразличимыми
масс-спектрами не могут быть идентифицированы только средствами
масс-спектрометрии. Однако после определения их индексов удержива-
ния на силиконовой фазе DB-1 — 997(*) и 1045(**) — результаты стано-
вятся однозначными: изобутил бензол и н-бутилбензол (справочные дан-
ные по индексам 998 ±6 и 1047 ± 7 соответственно).
318
В заключение необходимо отметить, что неоднократно пред-
принимавшиеся попытки теоретического предсказания масс-
спектров оказались безуспешными из-за сложности задачи. Од-
нако простейшие общие правила фрагментации молекулярных
ионов во многих случаях позволяют успешно прогнозировать
массовые числа максимальных сигналов спектров. То же от-
носится и к газохроматографическим индексам удерживания,
для которых известны методы расчета как по аддитивным схе-
мам, так и на основании физико-химических констант органи-
ческих соединений. Безусловно актуальным являются еще не
решенные проблемы повышения межлабораторной воспроизво-
димости и расчета индексов удерживания в обращенно-фазной
ВЭЖХ. Одно из главных направлений дальнейшего прогресса
всех хромато-масс-спектрометрических методов исследования
органических соединений связано именно с совершенствованием
методов интерпретации получаемых данных.
IV.2.9. Хромато-ИК(Фурье)-спектроскопия
Возможности использования ИК-спектрометра в качестве де-
тектора в газовой и жидкостной хроматографии неодинаковы,
в первую очередь, из-за существенных различий в физических
свойствах используемых подвижных фаз, в потоке которых
сформировавшаяся в хроматографической колонке зона веще-
ства переносится в измерительную ячейку детектора.
Для структурного анализа ИК-спектры обычно регистри-
руют в интервале частот между 4000 и 650 см-1 (некоторые
приборы, оборудованные более совершенной оптикой, позво-
ляют расширять границы этого диапазона до 4800 см-1 в ко-
ротковолновой и до 400 или даже до 200 см-1 в длинноволно-
вой части спектра). Азот и гелий, чаще всего используемые
в качестве подвижных фаз в газовой хроматографии, практиче-
ски полностью оптически прозрачны в указанной области, тогда
как многие жидкие элюенты характеризуются сильно выражен-
ным собственным поглощением, что может привести к частич-
ной или полной маскировке ИК-спектров молекул анализируе-
мого образца. Дополнительные трудности возникают при необ-
ходимости работы с водными подвижными фазами, поскольку
контакт влаги с оптическими элементами, изготовленными из
бромида или хлорида калия, недопустим.
Рассмотрим, прежде всего, возможности комбинированного
метода ГХ—ИК [301].
319
В ранний период развития газовой хроматографии (60-е —
начало 70-х годов) возможности идентификации разделенных
в хроматографической колонке соединений по их ИК-спектрам
ограничивались недостаточными уровнями чувствительности и
быстродействия обычных спектрометров с диспергирующими
элементами (призма или решетка). По этим причинам при-
ходилось прибегать либо к улавливанию фракций элюата на
выходе аналитической колонки (т. е. осуществлять предвари-
тельное микропрепаративное хроматографическое разделение
компонентов смеси), либо (при пропускании элюата непосред-
ственно через кювету ИК-спектрометра) останавливать поток
газа-носителя на период регистрации спектра интересующего
компонента. Оба эти приема не потеряли своего значения и в
наши дни в относительно простых случаях (достаточно высо-
кое содержание интересующего вещества в образце, малое чи-
сло сопутствующих компонентов, возможность их разделения
на насадочных колонках), однако неприемлемы в сложных слу-
чаях (низкое содержание идентифицируемых соединений, необ-
ходимость использования капиллярных колонок, обладающих
повышенной эффективностью, но малой емкостью).
Успехи в развитии фурье-спектроскопии и вычислительной
техники (середина 70-х — начало 80-х годов) привели к развер-
тыванию работ по созданию комбинированных приборов газо-
вый (или жидкостный) хроматограф — инфракрасный фурье-
спектрометр (ГХ—ИКФС и ВЭЖХ—ИКФС), рассчитанных на
регистрацию ИК-спектров соединений, разделяемых в реаль-
ном масштабе времени (режим on-line). В настоящее время веду-
щие приборостроительные фирмы за рубежом выпускают такие
приборы под сокращенным названием GC—FTIR (Gas Chromato-
graph—Fourier Transform Infrared Spectrometer) и соответственно
HPLC—FTIR (High Performance Liquid Chromatograph—Fourier
Transform Infrared Spectrometer).
Отличительные особенности фурье-спектроскопии как мето-
да детектирования и установления структуры разделенных в
хроматографической колонке соединений заключаются в сле-
дующем [302, 303].
1. Фурье-спектрометр представляет собой интерферометр со
встроенной ЭВМ, обеспечивающей процесс регистрации интер-
ферограммы как суперпозиции кривых поглощения (или отра-
жения), соответствующих каждой из присутствующих в спектре
источника частот, с последующим преобразованием интерферо-
граммы в обычный ИК-спектр.
2. Современные инфракрасные фурье-спектрометры, сочле-
ненные с газовыми или жидкостными хроматографами, охваты-
320
вают диапазон волновых чисел от ~400 до 4500 см-1 с разреше-
нием от 1 до 8 см-1.
3. Время, требуемое для регистрации полного ИК-спектра,
составляет менее 1 с (некоторые приборы позволяют регистри-
ровать до 20 спектров в секунду), что намного меньше времени
элюирования хроматографической зоны из разделительных ко-
лонок любых типов.
Весьма высокие быстродействие и чувствительность интер-
ферометров обусловливаются тем, что за время сканирования
получается информация сразу обо всем спектральном диапа-
зоне. Анализируя все спектральные элементы одновременно
(тогда как обычный дифракционный спектрометр делает это по-
следовательно, по мере попадания узкой спектральной полосы
на выходную щель прибора), фурье-спектрометр обеспечивает
существенный выигрыш в отношении сигнала к шуму в спектре.
Фурье-спектрометр обладает большей светосилой, нежели
дисперсионный спектрометр одинакового с ним разрешения, в
результате чувствительность его возрастает примерно на два
порядка. Так, уже в начале 80-х годов лучшие приборы GC—
FTIR позволяли идентифицировать вещества, содержание кото-
рых в дозируемом количестве образца не превышало 50-300 нг и
доходило даже до 5 нг. Реальная нижняя граница определяемых
содержаний идентифицируемого вещества в дозе сильно зави-
сит от конструктивных особенностей прибора, природы функци-
ональной группы и наличия в молекуле структурных элементов,
обусловливающих необходимый набор характеристических ча-
стот в спектре. По этой причине получаемая информация мо-
жет оказаться недостаточной для однозначной идентификации
вещества, но может послужить основанием для отнесения его к
тому или иному классу органических соединений.
В настоящее время выпускаются три типа приборов GC—
FTIR [304]. Наибольшее распространение получили приборы
(рис.IV.16), в которых элюат из кварцевой капиллярной ко-
лонки, покрытой толстой пленкой иммобилизованной неподвиж-
ной фазы, поступает в обогреваемую кювету-световод (позо-
лоченный изнутри стеклянный капилляр, обычно с внутренним
диаметром 1-2 мм и длиной от 10 до 80 см), торцы которого
закрыты солевыми окошками из бромида калия. Модулирован-
ный луч от источника ИК-излучения фокусируется в световоде,
через который элюируются зоны хроматографируемых соеди-
нений, многократно отражается от внутреннего золотого покры-
тия и достигает охлаждаемого жидким азотом ртутно-кадмиево-
теллуридного детектора. Сигнал детектора (интерферограмма)
поступает в ЭВМ для фурье-преобразования и дальнейшей об-
321
Рис. IV.16. Принципиальная схема
системы газовый хроматограф —
инфракрасный фурье-спектрометр
с кюветой-световодом.
1 — термостат газового хроматогра-
фа; 2 — испаритель; 3 — колонка; 4 —
ПИД; 5 — модулированный ИК-луч;
6— солевое окно; 7— обогреваемый по-
золоченный световод; 8 — ИК-детектор;
9 — внешняя память ЭВМ на дисках;
10 — ЭВМ; 11 — графопостроитель.
работки (сравнения с банком справочных данных и выдачи ре-
зультатов идентификации).
Чем длиннее путь луча в световоде, тем выше чувствитель-
ность прибора; с другой стороны, для предотвращения раз-
мывания хроматографической полосы желательно, чтобы мерт-
вый объем световода был минимальным и не превышал объема
газа-носителя, отвечающего полуширине самого узкого пика на
хроматограмме, Vb,5 [303]. Если объем кюветы-световода ока-
зывается большим, необходимо продувать через нее вспомо-
гательный инертный газ, что приводит к повышению предела
обнаружения ИК-детектора. Напротив, если объем световода
оказывается меньше И)д, в кювету поступает только доля хро-
матографической зоны и предел обнаружения также повыша-
ется. Поэтому экспериментально подбирают такие условия ра-
боты хроматографической колонки (включая и режим програм-
мирования температуры), чтобы Год каждого пика был равен
или слегка превышал объем кюветы-световода, колеблющийся
в пределах от 50 до 300 мкл. Поскольку ИК-облучение не вызы-
вает деструкции вещества, элюат из кюветы-световода может
быть направлен на дополнительное детектирование (пламенно-
ионизационный детектор или масс-спектрометр).
Выпускаются также приборы GC—FTIR, в которых элюиру-
емые из капиллярной или насадочной колонки соединения ула-
вливаются в виде твердой аргоновой матрицы, образующейся
при использовании в качестве газа-носителя смеси гелия и ар-
гона (с последующим удалением гелия в молекулярном сепа-
раторе струйного типа, см. раздел IV.2.8.3) и омывании элю-
атом зеркальной поверхности позолоченной медной пластины,
охлаждаемой в специальном криоколлекторе до температуры
322
12-15 К [305]. Конструкция криоколлектора позволяет произво-
дить замораживание до 32 идентифицируемых соединений в те-
чение одного аналитического цикла. Их последовательное ула-
вливание и регистрация ИК-спектров отражения твердых ма-
триц осуществляются автоматически с помощью встроенного
карусельного механизма, приводимого в действие, как только
концентрация каждой зоны в элюате превысит заданную.
Матричная изоляция обеспечивает большую (примерно на 1-
2 порядка) чувствительность детектирования в сравнении с при-
борами, укомплектованными кюветой-световодом; регистриру-
емые спектры не искажены межмолекулярными взаимодействи-
ями исследуемых соединений (образование ассоциатов), и, на-
конец, исключается опасность термического разложения анали-
зируемых веществ в разогретой до 200-250°C кювете-световоде.
Несмотря на эти преимущества, приборы с матричной изоля-
цией используются довольно редко, главным образом, вслед-
ствие их высокой стоимости, обусловленной сложностью изго-
товления криоколлектора.
В приборах третьего типа применяют криоулавливание (пря-
мое осаждение) идентифицируемых соединений на прозрачных
для ИК-излучения пластинах из селенида цинка. При этом
достигаются пределы обнаружения, сопоставимые с приведен-
ными выше для приборов с матричной изоляцией, однако полу-
чаемые ИК-спектры легче поддаются расшифровке, поскольку
они практически полностью тождественны стандартным би-
блиотечным спектрам веществ, запрессованных в таблетках
КВг. К преимуществам такого способа фиксации идентифици-
руемых соединений следует отнести также возможность его ис-
пользования и при сочетании ИК-спектроскопии (как метода де-
тектирования) с другими хроматографическими методами раз-
деления (жидкостная хроматография, сверхкритическая флю-
идная хроматография).
Информация, получаемая при использовании систем GC—
FTIR, позволяет устанавливать структурные особенности мо-
лекулы: наличие функциональных групп, непредельных связей,
цис, транс-изомерии и т.п. По ИК-спектрам легко удается раз-
личать изомеры (по масс-спектрам— гомологи).
Основные трудности идентификации при использовании при-
боров с кюветой-световодом заключаются в недостаточной из-
ученности ИК-спектров органических соединений в газовой фа-
зе при 200-250 °C. Необходимые для машинной расшифровки ре-
гистрируемых ИК-спектров банки данных, поставляемые фир-
мами-изготовителями приборов, охватывают пока ограничен-
ное число (3000-5000) соединений. Еще более актуальным пред-
323
ставляется пополнение библиотек справочных данных, кото-
рыми комплектуются приборы с матричной изоляцией хрома-
тографируемых веществ.
Первые приборы, предназначенные для регистрации ИК-
спектров соединений на выходе из колонки жидкостного хрома-
тографа, по аналогии с рассмотренными выше системами GC—
FTIR, включали проточную ячейку-кювету, диаметр и толщина
которой варьировались в зависимости от типа (диаметра) раз-
делительной колонки и характера (природы) подвижной фазы
[306].
Наиболее эффективно эти приборы зарекомендовали себя в
практике эксклюзионной (ситовой или гель-проникающей) жид-
костной хроматографии (разделение смеси веществ, различа-
ющихся по размерам молекул и/или по способности прони-
кать в поры неподвижной фазы) при использовании в каче-
стве подвижных фаз низших хлоралканов, а также тетрагидро-
фурана. Оптическая прозрачность указанных растворителей
падает в ряду: четыреххлористый углерод > хлороформ >
хлористый метилен, что накладывает определенные ограниче-
ния на возможности регистрации в каждом конкретном случае
тех или иных участков ИК-спектров. Чувствительность детек-
тирования не слишком высока; при использовании в качестве
подвижной фазы тетрагидрофурана количества идентифициру-
емых соединений, элюируемых из колонки в ячейку-кювету ИК-
детектора, должны быть не менее 1 мг.
В нормально-фазной ВЭЖХ емкость стандартных раздели-
тельных колонок (по величине дозы), как правило, на 1-2 по-
рядка меньше, а большинство используемых подвижных фаз
(алифатические углеводороды Се-Се, иногда модифицирован-
ные добавками полярных растворителей, таких, как этилацетат
или изопропиловый спирт) поглощает ИК-излучение с интен-
сивностью, не уступающей тетрагидрофурану или даже превос-
ходящей ее. По этим причинам И К-детектирование оказыва-
ется возможным лишь в отдельных благоприятных ситуациях,
например при возможности использования в качестве элюента
дейтерированного хлороформа CDCI3. Высокая стоимость дей-
терированных препаратов сдерживает употребление CDCI3 как
подвижной фазы в рутинной практике ВЭЖХ-анализов со стан-
дартными колонками (внутренний диаметр 3-4 мм). Однако это
направление весьма привлекательно при обращении к микро-
колоночной ВЭЖХ [42, 43], требующей в 10-20 раз меньшего
расхода растворителей.
В наиболее распространенной разновидности ВЭЖХ — об-
ращенно-фазной— трудности съемки ИК-спектров идентифици-
324
руемых веществ в проточной кювете-ячейке неизмеримо возра-
стают еще и потому, что используемые подвижные фазы (бинар-
ные или более сложного состава), как правило, содержат воду
в качестве одной из составляющих.
Для преодоления отмеченных ограничений было предложено
удалять мешающий съемке ИК-спектров растворитель.
В варианте нормально-фазной ВЭЖХ прибор оснащается
устанавливаемой на выходе колонки короткой полой обогрева-
емой трубкой, направляющей образующуюся в ней смесь паров
вещества и элюента либо к поверхности мелкодисперсного по-
рошка хлористого калия (заключенного в специальную откры-
тую кювету и обдуваемого снизу для ускорения испарения рас-
творителя струей воздуха или азота), либо на сброс. Переклю-
чение потока в нужные моменты времени осуществляется соле-
ноидным вентилем в соответствии с показаниями У Ф-детектора,
выполняющего функции детектора-индикатора. Кюветы разме-
щаются на карусельном механизме, обеспечивающем возмож-
ность конденсации элюата на поверхности зерен соли и после-
дующей автоматической съемки ИК-спектров отражения необ-
ходимого числа разделенных в колонке соединений (здесь про-
сматривается определенная аналогия с системами GC — FTIR
2-го и 3-го типов, см. выше).
Достигаемая чувствительность позволяет уверенно регис-
трировать неискаженные подвижной фазой ИК-спектры компо-
нентов разделяемой смеси, присутствующие в дозе в количе-
ствах до нескольких мкг. Ограничения этой методологии свя-
заны, во-первых, с тем, что она применима только к соеди-
нениям, молекулы которых имеют хромофорные группировки
(иначе УФ-детектор не зарегистрирует их выход из колонки);
во-вторых, используемые подвижные фазы должны быть абсо-
лютно сухими, так как даже следы конденсирующейся в ячейке
влаги будут растворять зерна хлористого калия, нарушая од-
нородность порошка, и из-за высокого поверхностного натяже-
ния и высокой скрытой теплоты испарения воды будут заметно
увеличивать длительность испарения растворителя, тем самым
удлиняя срок готовности кюветы к съемке спектра.
Применительно к микроколоночной ВЭЖХ—ИКФС рассмо-
тренная рабочая схема была адаптирована следующим обра-
зом. Поток элюата, поступающий из колонки с очень малой
объемной скоростью, подается в виде капель для испарения
растворителя непосредственно на поверхность таблетки бро-
мида калия, обдуваемой теплым азотом и медленно перемеща-
емой по специальным направляющим. Таким образом, поверх-
ность таблетки КВг выполняет функции своеобразного микро-
325
коллектора фракций. Последующее перемещение таблетки осу-
ществляется в направлении, перпендикулярном лучу света от
источника ИК-спектрометра, что и позволяет регистрировать
ИК-спектры каждой собранной «фракции» (в данном случае
ИК-спектры пропускания, а не отражения, как в примере, рас-
смотренном выше).
Необходимость эффективного и надежного удаления воды,
предшествующего съемке ИК-спектра, особенно актуальна для
обращенно-фазной ВЭЖХ, ориентированной на использование
водных систем растворителей. Было предложено три способа
решения этой задачи.
Первый, наиболее универсальный но, пожалуй, и наибо-
лее сложный по аппаратурному оформлению заключается в не-
прерывной экстракции органических составляющих из потока
элюата несмешивающимся с ним органическим растворителем
(обычно, дихлорметаном). Экстракция осуществляется в свер-
нутой в спираль полой трубке, сочленяемой с колонкой через
тройник, связанный с резервуаром, содержащим экстрагент.
На выходе экстрактора верхний (водный) слой отсасывается,
а нижний (органический) направляется в приемник-накопитель.
Дальнейшие операции аналогичны апробированным в установ-
ках для осуществления нормально-фазной ВЭЖХ— ИКФС (см.
выше). Дихлорметановый экстракт из приемника-накопителя
поступает в виде капель в кюветы с порошкообразным хлори-
дом калия. После быстрого испарения экстрагента регистри-
руются ИК-спектры отражения. Для подавления возможной
взаимной растворимости элюата и экстрагента (обеспечения
лучшего разделения слоев и сведения к минимуму образования
эмульсии) предусматривается подача на вход экстрактора до-
полнительного количества воды по резервной линии. По этой
же линии с целью увеличения коэффициента экстракции вме-
сто воды можно ввести в экстрактор разбавленные кислоту или
основание, или соответствующий буферный раствор.
Из изложенного должно быть понятно, что такой способ уда-
ления воды не рассчитан на микроколоночную ВЭЖХ, однако
при использовании стандартных «широких» колонок (с вну-
тренним диаметром 3-4 мм) совокупность перечисленных проце-
дур экстрагирования и концентрирования (которые эффективно
применяются и в ион-парной, и в ионной хроматографии, по-
скольку неорганические модификаторы подвижной фазы не экс-
трагируются дихлорметаном) обеспечивает очень низкий уро-
вень предела обнаружения — доли мкг.
Второй способ менее универсален, но более прост в аппара-
турном оформлении — это химический способ связывания воды
326
при ее взаимодействии с 2,2'-диметоксипропаном (этим прие-
мом иногда пользуются и в реакционной газовой хроматогра-
фии с целью повышения чувствительности детектирования при
определении влаги в органических растворителях; тем самым
открывается возможность использования ионизационных детек-
торов — ПИД или ФИЛ):
Н20 + СН3С(ОСН3)2СН3^4сН3СОСН3 + 2СН3ОН.
Образующиеся ацетон и метанол совместно с избыточным ко-
личеством реагента испаряются с поверхности порошкообраз-
ного хлористого калия, после чего кювета с подготовленным
препаратом подается к оптическому зонду прибора для съемки
ИК-спектров отражения. При этом существует опасность ис-
кажения спектров ионными модификаторами подвижной фазы,
попадающими в кювету вместе с идентифицируемым веществом.
Третий способ базируется на распылении (иногда — термо-
распылении, активно применяющемся также в системах жид-
костный хроматограф — масс-спектрометр, см. раздел IV.2.8.4)
элюата по локальным участкам поверхности диска-коллектора
фракций, покрытого подходящим материалом, например алю-
миниевой или алмазной пудрой. По мере вращения диска реги-
стрируются ИК-спектры отражения идентифицируемых соеди-
нений (рис. IV.17).
Об успешном использовании такого подхода в изучении со-
става сложной смеси стероидов сообщается в [307]. В техни-
ческом отношении данный способ решения проблемы удаления
мешающего съемке ИК-спектров элюента является самым про-
стым из числа рассмотренных выше. К его ограничениям сле-
дует отнести направленность на получение спектральной ин-
формации лишь о нелетучих соединениях, содержащихся в раз-
деляемой смеси (летучие компоненты образца необратимо те-
ряются при распылении). Кроме того, здесь также возможны
искажения регистрируемых ИК-спектров потенциально возмож-
ными нелетучими модификаторами подвижной фазы.
Перспективы использования комбинированного метода коло-
ночная жидкостная хроматография — ИК-спектроскопия до-
статочно широки, однако, как и в случае сочетания жидкост-
ного хроматографа с масс-спектрометром, все еще недостаточ-
ная отработанность отдельных узлов и элементов конструкций
(особенно в случае сверхкритической флюидной хроматогра-
фии [308]) и высокая стоимость созданных приборов сдержи-
вают их внедрение в аналитическую практику.
В тонкослойной хроматографии для осуществления ИК-иден-
тификации предусматривается автоматизированный перенос
327
Рис.IV.17. Одно из возможных технических решений сочле-
нения колонки жидкостного хроматографа и ИК-фурье-спект-
рометра [307].
Пояснение в тексте.
уже разделенных зон с пластинки в прозрачную для ИК-излу-
чения среду. Этот прием, с подробным описанием которого
можно познакомиться в монографии [28], обеспечивает лучшее
качество регистрируемых спектров, нежели съемка в режиме
on-line.
IV.2.10. Атомно-эмиссионное детектирование.
Комплексный подход к решению
задач повышенной сложности
Общий принцип работы группы атомно-эмиссионных хрома-
тографических детекторов (АЭЛ) заключается в деструкции
молекул элюируемых соединений до свободных атомов, обла-
дающих избыточной энергией, и регистрации атомно-эмиссион-
ных элементных спектров при переходе возбужденных атомов в
основное электронное состояние:
328
плазма
C„HmNfcO/...
nC* + mH* + feN* + 10* +... —>
nC + ДА
mH + A A'
ArN + AF"
ZO + AF"'
Атомизация молекул элюируемых соединений осуществля-
ется в создаваемой и поддерживаемой в ячейке детектора плаз-
ме инертного газа, имеющей температуру от 3000 до 10000 К.
Возбужденные атомы каждого присутствующего в молекулах
идентифицируемого вещества химического элемента испускают
свет, характеризуемый набором определенных длин волн в ви-
димой и ультрафиолетовой областях. Интенсивность и поло-
жение полос в образующемся эмиссионном спектре измеряются
оптическим спектрометром.
В качестве среды для получения плазмы в атомно-эмиссион-
ной спектроскопии (АЭС) обычно используют аргон, азот или
гелий. При сочетании АЭС и хроматографии наиболее оправ-
дано применение трех основных способов получения плазмы
(раскаленной смеси электронов, ионов и атомов плазмообразу-
ющего газа): возбуждение под действием электрических раз-
рядов между двумя или несколькими электродами — плазма
постоянного тока; возбуждение под действием энергии высо-
кочастотного переменного тока, передаваемой газу с помощью
магнитной индукции, — индуктивно-связанная плазма; возбу-
ждение под действием сверхвысокочастотного разряда — СВЧ-
плазма. Первые два из названных плазменных источников по
ряду причин удобнее устанавливать на выходе колонок жид-
костного хроматографа. В атомно-эмиссионных детекторах,
предназначенных для газовой хроматографии, наиболее оправ-
данным оказалось применение СВЧ-плазмы, создаваемой в сре-
де высокочистого гелия, используемого обычно в качестве газа-
носителя, расходы которого через ячейку при атмосферном да-
влении могут составлять от 20 до 100 мл/мин [297, 309-311].
Первые попытки применения атомно-эмиссионного спектро-
метра в качестве детектора газового хроматографа относятся
еще к середине 60-х годов, однако потребовалось более 20 лет,
чтобы подобные усовершенствованные тандемные системы ста-
ли серийно выпускаться несколькими ведущими зарубежными
приборостроительными фирмами. Принципиальная схема газо-
вого хроматографа с атомно-эмиссионным детектором НР-5921
фирмы Hewlett-Packard представлена на рис. IV.18 [312].
329
co
о
Рис. IV. 18. Принципиальная блок-схема газового хроматографа с автономно-эмиссионным детектором HP-5921
фирмы Hewlett-Packard.
Пояснения в тексте.
Поток подвижной фазы (гелий сверхвысокой чистоты —
99,9999%), содержащий зоны компонентов анализируемой про-
бы, на выходе из хроматографической колонки смешивается с
вспомогательным газом* и по обогреваемой трубке направля-
ется в ячейку АЭЛ. Здесь под воздействием СВЧ-разряда ге-
лий переходит в плазмообразное состояние, а молекулы попа-
дающих в плазму веществ деструктируют до свободных ато-
мов. Излучение, испускаемое возбужденными атомами соста-
вляющих данную молекулу элементов, фокусируется на вход-
ную щель спектрометра и далее проходит через систему зер-
кал и промежуточную поворотную голографическую решетку.
Диспергированный свет направляется к приемникам излуче-
ния — фотодиодам (оптическим элементам диодной матрицы;
диодно-матричное детектирование активно применяется также
в ВЭЖХ, см. раздел III.1.5.2, способным регистрировать эмис-
сионные линии в спектрах как какого-либо одного, так и одно-
временно нескольких (вплоть до восьми) химических элемен-
тов, потенциально присутствующих в молекулах компонентов
пробы.
Таким образом, атомно-эмиссионный детектор, в противопо-
ложность пламенно-фотометрическому или термоионному (ка-
ждый из которых избирателен лишь по отношению к соедине-
ниям, содержащим фосфор и серу или фосфор и азот соответ-
ственно), является детектором со строго регулируемой селек-
тивностью по отношению к многочисленным химическим эле-
ментам. Так, при настройке на обнаружение в эмиссионных
спектрах органических соединений только линий углерода (по
каналу углерода) регистрируется выходная хроматографиче-
ская кривая, подобная хроматограмме, записываемой по показа-
ниям универсального пламенно-ионизационного детектора. Од-
нако параллельно с детектированием всех углеродсодержащих
веществ АЭЛ позволяет количественно прослеживать выход из
колонки соединений, содержащих наряду с углеродом и водо-
род, кислород, азот, серу, галогены и другие, чуть ли не все хи-
мические элементы таблицы Л. И. Менделеева и их стабильные
*Иногда этот газ называют газом-реагентом, или «отмывающим» га-
зом. Применяется во избежание тушения плазмы полосой растворителя,
основную часть которого обычно отводят от газоразрядной трубки, при-
бегая к обратной отдувке, а остаточное количество дожигают непосред-
ственно в ячейке детектора, добавляя в подвижную фазу в качестве газа-
реагента кислород или водород. Вспомогательный газ необходим также
для исключения перегрузки детектора и предотвращения конденсации
высококипящих веществ на поверхностях соединительных трубок и опти-
ческих элементов. В этих целях вместо водорода или кислорода можно
использовать гелий (ос.ч.).
331
изотопы. Наглядной иллюстрацией к сказанному может слу-
жить рис. IV.19 [313]. В современных приборах настройка детек-
тора на регистрацию эмиссионных линий заданной длины осу-
ществляется компьютеризованной системой в автоматическом
режиме.
АЭЛ обладают к тому же весьмавысокой чувствительностью
и широким диапазоном линейности показаний, что предопреде-
ляет эффективное их использование в эколого-аналитическом
контроле таких сложных объектов, как, например, воздушная
среда полигонов по уничтожению химического оружия [314].
Отклик АЭЛ на фиксированную массу углеродсодержащего ве-
щества (по каналу углерода, С) ~ в 5 раз выше, чем отклик
ПИЛ, и ~ в 10 раз (по каналу S) превосходит отклик пламенно-
фотометрического детектора (обладающего еще и много мень-
шим линейным диапазоном) по серосодержащим соединениям.
Выборка численных значений параметров, характеризующих
аналитические возможности АЭЛ модели 5921А фирмы Hewlett-
Packard [309], приведена в табл. IV.17.
Важно подчеркнуть, что селективность атомно-эмиссионного
детектора по отношению к регистрируемым химическим эле-
ментам близка к абсолютной, иначе говоря, отклик детектора
на единицу мольной массы какого-либо элемента остается прак-
тически постоянным вне зависимости от химической индивиду-
альности детектируемого соединения, содержащего данный эле-
мент*. Это обстоятельство открывает широкие возможности ко-
личественного определения (при регистрации по разным кана-
лам) соединений, не полностью разделенных или даже совсем
не разделенных в хроматографической колонке, выходящих из
нее единой зоной.
В качественном анализе параллельная запись многоканаль-
ных хроматограмм (в первую очередь, по эмиссионным линиям
углерода, водорода, кислорода, азота, серы) открывает возмож-
ность установления количественного соотношения атомов на-
званных элементов в молекулах детектируемых соединений, что
позволяет рассчитывать эмпирические формулы идентифици-
руемых веществ [316-319]. На воспроизводимость и точность ре-
зультатов, получаемых в отдельных опытах, влияют несколько
факторов, таких, как нестабильность плазмы (вследствие воз-
*Отмечаемые в некоторых литературных источниках исключения из
этого общего правила часто объясняются внешними причинами. Так,
например, при детектировании кислородсодержащих соединений по ка-
налу кислорода большую опасность представляют примеси кислорода,
содержащегося в газе-носителе (гелии), во вспомогательном газе (азоте),
а также диффундирующего в систему из воздуха в случае ее неабсолют-
ной герметичности [316].
332
минуты
Рис. IV.19. Параллельная четырехканальная регистрация уг-
лерод-, кремний-, водород- и хлорсодержащих органических
соединений по показаниям атомно-эмиссионного детектора
[313].
А — триметилхлорсилан; В — гексаметилдисилоксан; С — толуол
(растворитель); D — триметилпентилсилан; Е — метилэтилпропилси-
лан; F —- дихлорбензол; G — метилэтилпропилбутилсилан; Н — ун-
декан; I — дихлорметилфенилсилан; J — дихлордипропилсилан; К —
додекан; L — тетрапропилсилан; М — метилпропилдибутилсилан; N —
тридекан; О — метилтрибутилсилан; Р — тетрадекан; Q — дипропил-
дибутилсилан; R -— пентадецен; S — пентадекан; Т — гексадекан; U —
ди п роп и лди пенти леи л ан.
333
Таблица IV. 17. Основные рабочие характеристики
СВЧ-раз рядного АЭД (выборка из [309])
Элемент Ллина волны, нм ПО,* ** пг/с (пг) Сел екти вность по отношению к углероду Л ЛИ"
Углерод 247,9 2,7(12) 1 >1000
Водород 656,3 7,5(22) 160 500
Хлор 479,5 39 25 000 20000
Бром 470,5 10 11400 >1000
Фтор 685,6 40 30000 2 000
Сера 180,7 1,7 150000 20000
Фосфор 177,5 1 5 000 1000
Кремний 251,6 7,0 90000 40000
Кислород 777,2 75 25 000 4000
Азот 174,2 7,0 6000 43000
Алюминий 396,2 5,0 >10 000 >1000
Сурьма 217,6 5,0 19000 >1000
Галлий 294,3 ~200 >10 000 >500
Германий 265,1 1>3(3,9) 7600 >1000
Олово 303,1 (0,5) 30000 >1000
Мышьяк 189,0 3,0 47000 500
Селен 196,1 4,0 50000 >1000
Хром 267,7 7,5 108000 >1000
Железо 302,1 0,05 3500000 >1000
Ртуть 253,7 0,1 3000000 >1000
Ванадий 292,4 4,0 36000 >1000
Титан 338,4 1,0 50000 >1000
Никель 301,2 1,0 200000 >1000
Палладий 340,4 5,0 >10 000 >1000
Марганец 257,6 1,6(7,7) 110000 >1000
* Предел обнаружения при соотношении сигнала и шума 1:3;
реализуется при использовании в качестве газа-носителя ге-
лия особой чистоты (99,99999%). При меньшей чистоте гелия
(~99,99%) чувствительность детектирования некоторых эле-
ментов, естественно, падает (по данным [315] в среднем в 2—6
раз).
** Линейный динамический диапазон.
можных флуктуаций питающего ее электрического разряда),
т. н. стеночный эффект (обусловленный недостаточной химиче-
ской инертностью по отношению к генерируемым атомам вну-
тренней поверхности кварцевой газоразрядной трубки), воз-
можно, неполная фрагментация в плазме присутствующих в
пробе высокомолекулярных соединений [313, 320].
334
В связи с этим при определении эмпирических формул ре-
комендуется сначала оптимизировать рабочий режим детек-
тора для съемки эмиссионных спектров по каждому из наме-
ченных каналов (например, по каналам углерода, водорода,
хлора), а затем подобрать компромиссные условия одновремен-
ного детектирования нескольких элементов. Основными варьи-
руемыми параметрами эксперимента являются мощность элек-
трического разряда, скорость подачи газа-носителя и вспомога-
тельного газа, длины волн регистрируемых эмиссионных линий.
Для получения информации о соотношении числа атомов ин-
тересующих элементов в молекулах идентифицируемых соеди-
нений в анализируемую смесь вводят (в качестве своеобраз-
ного внутреннего стандарта) заведомо известное соединение,
как можно более близкое по физико-химическим свойствам к
определяемому. Тем самым получают возможность при об-
работке результатов анализа учитывать относительную чув-
ствительность АЭЛ к регистрируемым элементам в выбранных
условиях эксперимента. При этом количество введенного в ана-
лизируемый образец стандарта, равно как и количество при-
сутствующего в нем идентифицируемого соединения, знать не-
обязательно. Важно лишь, чтобы и стандарт, и идентифици-
руемое соединение регистрировались в пределах линейного от-
клика АЭД на каждый из контролируемых элементов. Оконча-
тельные расчеты проводят, используя следующее соотношение
[313, 318]:
Е _ Fe(0 PC(,t) AE(st)
С PC(i) Nc.t} ’
где Е/С — вычисляемое отношение числа атомов какого-либо
элемента и углерода в молекуле идентифицируемого вещества;
-Ре(1) и Рсцу — измеряемые сигналы детектора (высоты или пло-
щади пиков) на элемент и углерод при детектировании иден-
тифицируемого вещества; Pc(,t), PE(st) №(st) и Ac(st) — сигналы
детектора (высоты или площади пиков) на элемент и углерод
и соответственно число атомов каждого из них в используемом
соединении сравнения (стандарте).
Иллюстрацией близкого соответствия рассчитываемых та-
ким путем эмпирических формул истинным могут служить дан-
ные, приведенные в табл. IV.18. Ошибки определения Н, С1, Вг,
I, Si и некоторых других элементов, в частности серы, обычно
не превышают 3-6% отн., тогда как для кислорода отклонение
эмпирического индекса от истинного может достигать 10-12%
отн., что понятно по причинам, отмеченным выше.
Установление собственно молекулярной формулы идентифи-
цируемого вещества по результатам выполненного хроматогра-
335
Таблица IV. 18. Результаты расчета эмпирических
формул различных органических соединений
Соединение Эмпирическая формула
истинная рассчитанная
Алканы [313]
Ундекан С11Н24 СцНг4,5
Додекан С12Нгв С12Н25.6
Тридекан С13Н28 С1зНг8,4
Тетрадекан С14Н30 С14Нзо,2
Пентадекан С15Н32 Стандарт
Хлорпроизводные алканов [322]
Хлористый метилен СН2С12 С1Н1>944 С11|968
Хлороформ СНС13 01 H0j987 012,937
Четыреххлористый СС14 01 С1з Э04
углерод Линдан CeHeCle Стандарт
Хлорпроизводные аренов [323]
Дихлорбензол С6Н4С12 О6Н4Д5 Clij96
Трихлорбензол СвНзС1з с6 Н3Д3 CI304
Хлортолуол С7Н7С1 Стандарт
Тетраалкилсиланы [313]
Тетрапропилсилан S1C12H28 SiCiijgH28,o
Метилпропилдибутилсилан SiCi2H28 SiCut7H28,7
Метилтрибутилсилан SIC13H30 SiCi2,9H3o,7
Дипропилдибутилсилан S1C14H32 SiCis.eHss^
М етилотилп ро п и л- бутилсилан SiCioH24 Стандарт
Кислородсодержащие соединения [322]
Метанол СН4О СН3 88 01,08
Этилацетат С4Н8О2 C4H7,8701.76
Нитробензол C6H5NO2 Стандарт
фического эксперимента в некоторых (но далеко не во всех)
случаях возможно с привлечением рассмотренных в разделе
IV.2.4.3 корреляционных зависимостей параметров удержива-
ния, однако гораздо более эффективный путь заключается в
объединении информации, получаемой на хроматографах с
атомно-эмиссионным и масс-спектрометрическим детекторами.
Алгоритм надежного компьютеризованного расчета молекуляр-
ных формул выходящих из хроматографической колонки инди-
видуальных веществ или компонентов смесей, требующий зна-
ния массового числа молекулярного иона и отношения числа
атомов двух-четырех элементов, недавно опубликован в [321].
336
Хроматографическое разделение смеси
Неспецифическое детектирование цри последовательном изменении поляр- ности неподвижных фаз в ГХ и поляр- ности сорбента и состава подвижной фазы вЖХ Неспецифическое детектирование с привлечением РГХ (в ГХ) н деривати- зации (в ГХ и в ЖХ) как вне, так и в хроматотрафи- ческой системе Специфическое детектирование
Селективные детекторы Масс- спектрометрия Фурье-ИК- спектроскопия Атомно- эмиссионное детектирование вГХ
Идентификация по хромато- графическим спектрам Групповая идентификация и установление углеродного остова молекулы Измерение относительного отклика универсального и селективного детекторов Регистрация осколочных ионов; определение молекулярной массы Выявление фу нкционал ь ных групп, характерных структурных элементов Определение элементного состава молекул, вычисление брутто-формулы
Схема IV.4. Совокупность аналитических данных, необходи-
мых для идентификации органических соединений в сложных
смесях.
Все рассмотренные выше на примере газовой хроматографии
специфические особенности и преимущества атомно-эмиссионно-
го детектирования давно уже стимулируют развитие комбини-
рованных систем приборов ВЭЖХ—АЭЛ, однако их коммер-
ческий выпуск и широкое внедрение в практику аналитической
работы сдерживаются несколькимим факторами, и прежде всего
трудностями технического решения задачи необходимого изби-
рательного удаления жидкой подвижной фазы на выходе хрома-
тографической колонки. Чаще всего, как и в гибридных систе-
мах ВЭЖХ—МС и ВЭЖХ—ИКФС, прибегают к методу термо-
распыления, так что остающиеся компоненты анализируемого
образца вводятся в ячейку АЭЛ в форме более или менее одно-
родных аэрозолей. Из-за значительных потерь при формирова-
нии аэрозолей лишь 1-5% дозы образца, введенной в колонку,
достигает плазмы.
В созданных к настоящему времени лабораторных установ-
ках и опытных образцах фирменных жидкостных хроматогра-
фов, укомплектованных атомно-эмиссионными детекторами,
для атомизации молекул идентифицируемых соединений приме-
337
няются все три названных в начале данного раздела типа плаз-
менных источников, однако наиболее оправдано использование
ячеек АЭЛ с индуктивно-связанной плазмой и плазмой посто-
янного тока.
Из-за отмеченных сложностей реализации атомно-эмиссион-
ного детектирования в ВЭЖХ круг исследуемых объектов огра-
ничивается пока, главным образом, высококипящими металло-
органическими соединениями, а наблюдаемые пределы обнару-
жения выше, чем при использовании АЭЛ в газовой хромато-
графии. Тем не менее, перспективность развития и совершен-
ствования комбинированных приборов ВЭЖХ—АЭЛ не вызы-
вает сомнений (см., в частности, обстоятельный обзор [309]).
Несмотря на успехи, достигнутые при исследовании состава
разнообразных объектов синтетического и природного происхо-
ждения гибридными инструментальными (хромато-спектраль-
ными) методами, при решении задач разного уровня сложно-
сти как в разведочных, так и в подтверждающих анализах це-
лесообразно комплексное использование результатов всего ар-
сенала изложенных выше средств и приемов качественного хро-
матографического анализа, как это показано на схеме IV.4.
IV.3. КОЛИЧЕСТВЕННЫЙ АНАЛИЗ В ГАЗОВОЙ
И ВЫСОКОЭФФЕКТИВНОЙ КОЛОНОЧНОЙ
ЖИДКОСТНОЙ ХРОМАТОГРАФИИ
В количественном хроматографическом анализе чаще всего
решаются следующие типовые задачи.
1. Определение содержания одного, нескольких (ключевых)
или всех соединений, присутствующих в искусственно пригота-
вливаемых многокомпонентных смесях или в продуктах слож-
ного состава синтетического или природного происхождения
в сопоставимых концентрациях; примерами подобных объек-
тов могут служить, например, различного рода технологиче-
ские композиции, нефтепродукты (моторные топлива), препа-
раты бытовой химии, лекарства и т. п.
2. Определение суммарного содержания группы из несколь-
ких компонентов, объединяемых каким-либо общим признаком,
относительно присутствующих в смеси остальных соединений —
т.н. определение группового состава (например, определение
общего содержания отдельных классов углеводородов, хлор-
или серосодержащих веществ в нефти и продуктах ее перера-
ботки) .
338
3. Определение содержания малых (или микро-) примесей в
индивидуальных химических соединениях и их смесях, в пище-
вых продуктах, лекарствах и в различных объектах окружаю-
щей среды (воздух, вода, почва).
Успешное решение всех этих и ряда других задач, т. е. до-
стижение высокой точности и воспроизводимости количествен-
ных результатов, возможно лишь при правильном выборе аппа-
ратуры, условий проведения анализа и рационального метода
количественной расшифровки хроматограмм, а также при ис-
ключении или сведении к минимуму возможных погрешностей
на каждой отдельной стадии выполнения эксперимента.
IV.3.1. Хроматограмма как источник сведений
о количественном составе анализируемой смеси
Основными количественными параметрами хроматограммы
(рис. IV.20) являются площадь пика А — участок выходной хро-
матографической кривой, ограниченный контуром LMORT (ве-
твями пика) и продолжением нулевой (базовой) линии LT, а
также высота пика h — отрезок FO, отвечающий максималь-
ной амплитуде сигнала детектора, иначе перпендикуляр, вос-
станавливаемый от продолжения нулевой (базовой) линии LT к
вершине пика. В газовой хроматографии количественную обра-
ботку хроматограмм иногда проводят также по произведениям
высот пиков на время удерживания соответствующих компонен-
тов htp.
Использование названных параметров основывается на име-
ющих место (при условии линейной работы детектора) зависи-
мостях
, ,, ПТ
А = Kq, h = K Стах, htR = К \ —q,
V 2тг
где К, К' и К" — коэффициенты, учитывающие постоянную
детектора и его относительную чувствительность к определяе-
мому веществу; q — количество (масса, объем) определяемого
вещества, введенное в колонку; Стах — максимальная концен-
трация вещества в хроматографической зоне; W — число тео-
ретических тарелок, определяемое по интересующему пику.
Использование произведений высот пиков на время удержи-
вания, строго говоря, допускается лишь для случаев, когда ши-
рина пика изменяется пропорционально времени удерживания,
что, как правило, имеет место при хроматографировании близ-
ких по химической природе веществ на высокоэффективных ко-
лонках.
339
Рис. IV.20. К определению количественных параметров хро-
матографического пика.
Вспомогательными параметрами и критериями хроматогра-
фических пиков, используемыми при количественной расши-
фровке хроматограмм вручную, являются (см. рис. IV.20):
1) высота h! треугольника DGE, образуемого касательными
к ветвям пика в точках перегиба;
2) основание пика (или ширина пика при основании) а>ъ —
отрезок DE, отсекаемый на продолжении базовой линии каса-
тельными к ветвям пика;
3) ширина пика на половине высоты (полуширина пика) wq,5
или ширина пика в любой другой точке высоты (например, в
точке перегиба оддот) определяется как расстояние между ве-
твями пика на заданном сечении высоты (сечение высоты обо-
значается подстрочным индексом).
В условиях, обеспечивающих линейную изотерму сорбции
(распределения), размывание хроматографической зоны веще-
ства в колонке подчиняется нормальному (гауссову) распреде-
лению независимых величин. При этом на хроматограмме ре-
гистрируются симметричные (относительно точки с максималь-
340
ной концентрацией) пики колоколообразной формы (типа пред-
ставленных на рис. IV.20), называемые часто гауссовыми. Кри-
вая Гаусса описывается уравнением, связывающим площадь
пика и ширину его на различных сечениях высоты:
у = Аехр[— ^(а:/о-2)]/о-\/27г, (IV.41)
где у — высота точки на кривой (ордината), измеренная на рас-
стоянии х от ординаты максимума (вершины пика); А — пло-
щадь; ст — стандартное отклонение, или дисперсия, гауссова
хроматографического пика. Стандартное отклонение <т отве-
чает ширине пика в точке, расположенной на расстоянии 0,882Л
от основания (010,882)> и может быть определено также из соот-
ношений, справедливых для гауссовых пиков:
2<Т = О>0,607> 3<т = О>0,324, 4(7 — О>0,134-
В количественном хроматографическом анализе следует
стремиться к получению хроматограмм с гауссовыми пиками.
Надежное измерение площадей асимметричных пиков требует
использования специализированных приборов — электронных
интеграторов или программно-аппаратных комплексов на базе
персональных компьютеров. Однако, несмотря на то, что совре-
менные инструментальные средства обработки сигнала детек-
торов способны с высокой точностью измерять площадь пика
практически любой формы (как симметричных, так и асимме-
тричных), сам факт асимметричности хроматографической по-
лосы свидетельствует о нелинейной изотерме сорбции (распре-
деления), что может служить причиной скрытого недостаточно
полного разделения соседних зон и, как следствие, меньшей точ-
ности результатов количественного анализа. В подобных ситу-
ациях требуется более тщательная (более подробная, жестко
привязанная к методике выполнения измерений) градуировка
прибора (см. раздел IV.3.3), нежели в тех случаях, когда пики
на хроматограмме симметричны (гауссовы).
Для определения принадлежности формы хроматографиче-
ского пика к гауссовой удобно использовать отнесение ширины
пика при основании к полуширине пика. Для истинно гауссовых
пиков должно соблюдаться равенство (критерий Эттре)
Wf, = 1 ,698u>o,5-
В первом приближении можно считать пик гауссовым, если чи-
сленное значение отношения шь/шо 5 находится в пределах 1,67-
1,73.
При больших отклонениях от гауссовой формы выраженную
асимметрию характеризуют с помощью т.н. фактора асимме-
тричности Fas, численное значение которого находят
341
Pmc.IV.21. К определению фак-
тора асимметричности хромато-
графического пика.
(рис. IV.21) по формуле
Fas = А/В,
где Л и В — расстояния от высоты до каждой ветви на уровне
ОДЛ.
IV.3.2. Выбор и измерение основных количественных
параметров хроматографических пиков
В практической работе выбор того или иного параметра для
количественной расшифровки хроматограмм определяется со-
вокупным влиянием нескольких факторов: быстротой и удоб-
ством расчета, формой (широкий, узкий) и степенью асимме-
трии хроматографического пика, эффективностью используе-
мой колонки, полнотой разделения компонентов смеси, нали-
чием необходимых автоматизированных устройств (интеграто-
ров, компьютерных систем обработки данных хроматографиче-
ского анализа).
Предпочтительность расшифровки хроматограмм по площа-
дям или по высотам пиков обусловливается также свойствами
детектора и стабильностью температуры колонки и скорости
подвижной фазы при выполнении анализа (серии анализов). На-
помним, что влияние температуры колонки и скорости подвиж-
ной фазы на высоты и площади хроматографических пиков не-
одинаково для различных типов детекторов. При использова-
нии концентрационных детекторов прогрессирующее изменение
или нестабильность (пульсация) расхода подвижной фазы при-
водят к изменению площадей пиков, но практически не сказыва-
ются на высотах, для потоковых детекторов с повышением ско-
рости подвижной фазы площадь пика не изменяется, а высота
увеличивается (рис. IV.22).
342
Fc
Рис. IV.22. Зависимость высоты и площади пика от расхода
подвижной фазы для концентрационного (с) и потокового (6)
детекторов.
I, 2, 3 — пики одного и того же количества анализируемого веще-
ства при последовательно увеличиваемой объемной скорости подвиж-
ной фазы Fc (мл/мин).
Как известно, в газовой хроматографии из наиболее широко
используемых детекторов типичным концентрационным явля-
ется катарометр, а типичным потоковым — ПИЛ (ЭЗД, равно
как и ЛПР, занимают промежуточное положение). В жидкост-
ной хроматографии большинство используемых детекторов —
концентрационные (см. разделы II.1.4, III.1.1 и [83, 84, 324-326]),
что обусловливает необходимость особо надежного демпфиро-
вания потока элюента при планировании обработки результа-
тов количественного анализа по площадям пиков.
Влияние температуры колонки (независимо от типа детек-
тора) мало проявляется при измерении площадей, но весьма за-
метно — при измерении высот пиков (вследствие температурной
зависимости коэффициентов распределения). Важное практи-
ческое следствие этой закономерности в газовой хроматогра-
фии — невозможность использования высот как количествен-
ного параметра хроматографического пика в режиме програм-
мирования температуры колонки.
При работе в изотермическом режиме невоспроизводимость
задания температуры колонки всего лишь в 1 °C (при выключе-
нии и последующем включении газового хроматографа) приво-
дит к изменению времени удерживания примерно на 3%. Сокра-
щение (или, напротив, увеличение) времени удерживания ком-
пенсируется соответствующим изменением высоты пика. От-
сюда для обеспечения воспроизводимости высот пиков в преде-
лах 1% требуется поддерживать температуру колонки с точно-
стью ±(0,1-0,3) °C.
Не менее важным представляется жесткое термостатирова-
ние колонок и в жидкостной хроматографии. Рекомендуется
343
также предусматривать соответствующий предварительный по-
догрев подвижной фазы до ее поступления в узел ввода пробы и
далее в колонку и детектор. В частности, рефрактометрический
детектор весьма чувствителен не только к изменениям скорости
потока, но и к изменению температуры подвижной фазы.
В жидкостной хроматографии обработка хроматограмм по
высотам пиков требует обеспечения постоянства состава по-
движной фазы. Лля воспроизведения времен удерживания в
пределах 1% пропорции компонентов элюента необходимо под-
держивать неизменными в пределах 0,1%. В связи с этим при не-
обходимости выполнения большого числа однотипных анализов
следует единовременно готовить достаточно большой объем по-
движной фазы и всякий раз при начале работы с новой партией
растворителя заново калибровать колонку. Сосуд с подвижной
фазой должен храниться герметично закрытым.
В газовой хроматографии природа и состав подвижной фазы
(чаще всего — индивидуального газа-носителя, иногда — со-
держащего небольшие количества модифицирующих добавок) в
гораздо меньшей степени влияют на удерживание компонентов
анализируемого образца, нежели в жидкостной*. Доминиру-
ющее влияние здесь принадлежит природе, составу и количе-
ству неподвижной фазы в колонке, которое в газо-жидкостном
варианте, да и при работе с некоторыми полимерными сорбен-
тами подвержено более или менее выраженному естественному
уменьшению. Интенсивность истощения газо-жидкостных хро-
матографических колонок зависит от упругости паров исполь-
зуемых неподвижных жидких фаз (в том числе и некоторых син-
тетических адсорбентов) при рабочей температуре и от жест-
кости задаваемого температурного режима. Если принять до-
пустимым улетучивание 50% общей массы неподвижной фазы
за 2000 ч работы колонки (т.е. примерно за год эксплуатации),
то унос неподвижной фазы за один день составит около 0,2%
от первоначального количества. При этом высоты пиков бу-
дут последовательно увеличиваться также примерно на 0,2% в
*Недавно, однако, обнаружено заметное влияние природы (и давле-
ния) газа-носителя на величины удерживания углеводородов Cj-С4 в ка-
пиллярной газо-адсорбционной хроматографии, причиной которого мо-
жет быть взаимодействие хроматографируемых соединений с модифи-
цированной подвижной фазой поверхностью используемого адсорбента.
Относительное изменение фактора удерживания, например, бутана (ад-
сорбент AI2O3/KCI) при замене газа-носителя гелия на азот и гелия на
диоксид углерода составляет соответственно 13 и 21% [327]. О влиянии
природы и давления газа-носителя на параметры удерживания компонен-
тов пробы в капиллярной газо-жидкостной хроматографии см. сноску на
с. 260.
344
день, что приведет к прогрессирующему завышению результа-
тов при длительном использовании градуировочных графиков
(см. раздел IV.3.3.2), составленных на основании измерения вы-
сот пиков.
Итак, вопрос о предпочтительности использования при ко-
личественной расшифровке хроматограмм высот пиков, пло-
щадей или произведений высот на время удерживания дол-
жен решать сам хроматографист после тщательного всесто-
роннего анализа аппаратурных возможностей, вида хромато-
граммы и допустимой погрешности определения. Дилемма, ко-
торую здесь предстоит решать, заключается в том, насколько
погрешности из-за изменения высот пиков под влиянием пере-
менных факторов будут больше или меньше погрешностей из-
мерения площадей пиков или пропорциональных им величин
в зависимости от сложности формы хроматографического кон-
тура и наличия необходимых специализированных программно-
аппаратных устройств.
Приемы и способы, используемые для нахождения основных
количественных параметров хроматографических пиков, пред-
ставлены в табл. IV.19.
Из двух основных количественных параметров хроматогра-
фического пика — площади или высоты — проще всего и чаще
всего (при работе вручную) с наименьшей затратой времени и с
большей точностью измеряются высоты пиков. Автоматическое
измерение высот хроматографических пиков в ходе анализа вы-
полняется с помощью некоторых моделей электронных инте-
граторов и всеми современными компьютеризованными сред-
ствами обработки результатов хроматографического анализа.
При отсутствии названных приборов оператор вынужден изме-
рять на хроматограмме высоту пиков вручную по линейке. Для
снижения возможных погрешностей измерения соответствую-
щих отрезков на хроматограмме следует пользоваться остро
отточенным карандашом и линейкой с четко нанесенными де-
лениями. Длины отрезков оценивают с точностью до десятых
долей миллиметра.
Для измерения площадей хроматографических пиков пред-
ложено несколько графических и инструментальных приемов,
некоторые из них (см. табл. IV.19) уже устарели и предста-
вляют лишь исторический интерес. В настоящее время в прак-
тике крупных специализированных лабораторий измерение пло-
щадей пиков на хроматограмме производится исключительно
с помощью электронных интеграторов или специализирован-
ных ЭВМ. Тем не менее, студенты и лица, самостоятельно
приступающие к изучению газовой и жидкостной хроматогра-
345
Таблица IV. 19. Приемы и способы измерения
количественных параметров хроматографических пиков
Техника обработки хроматограммы Определяемый параметр Приемы и способы измерения
Определяемый пара- метр хроматографичес- кого пика измеряется оператором на хромато- грамме вручную по окончании цикла разде- ления компонентов ана- лизируемой смеси Площадь пика Высота пика или произве- дение высоты пика на время удержи в ан ия Графические способы, основанные*: а) на гео- метрических приемах из- мерения площади тре- угольника или трапеции; б) на анализе и преобра- зовании уравнения гаус- совой кривой. Инструментальные спо- собы**: а) планиметриро- вание; б) взвешивание вырезанных по контуру хроматографических пиков участков хроматограмм Графический способ — измерение линейкой соот- ветствующих отрезков на хроматограмме. Комбинированный спо- соб — измерение времени удерживания секундомером, а высоты пика — линейкой
Определяемый пара- метр хроматографичес- кого пика измеряется автоматически с по- мощью цифровых вольт- метров, электронных интеграторов или спе- циализированных ЭВМ одновременно с разде- лением компонентов анализируемой смеси в колонке и записью хро- матограммы или по окон- чании регистрации хро- матограммы в процессе интерактивного диалога оператора с ЭВМ Площадь или высота пика Автоматическое изме- рение электронными инте- граторами или програм- мно-аппаратными ком- плексами на базе персо- нальных компьютеров
*06 ограничениях графических способов измерения площадей
пиков см. в [79, с. 217-221].
**Данные способы измерения площадей пиков в настоящее
время практически не используются и представляют лишь
исторический интерес.
346
фии, должны овладеть и навыками определения площадей пиков
вручную с применением простейших графических способов, по-
дробно рассмотренных в [79, с.217-221].
IV.3.3. Основные методы количественного анализа
IV.3.3.1. Общие сведения
Первостепенной задачей количественного анализа является
градуировка прибора, т. е. установление строгой числовой вза-
имосвязи между сигналом детектора и количеством интересу-
ющего вещества. Угловой коэффициент такой зависимости ха-
рактеризует чувствительность детектора газового или жидкост-
ного хроматографа к определяемым соединениям, которая зави-
сит от многих факторов и, прежде всего, от принципа детекти-
рования и конструктивных особенностей того или иного детек-
тора и хроматографа в целом, агрегатного состояния исследу-
емой подвижной фазы (газ или жидкость) и физико-химических
свойств детектируемых веществ.
Формирование полосы (зоны) определяемого соединения в ко-
лонках газового и жидкостного хроматографов, несмотря на не-
которую специфику того и другого метода разделения, в общем
подчиняется единым законам распределения (сорбции). Вме-
сте с тем, коренное отличие газовой и жидкостной хроматогра-
фии как методов анализа заключается в специфике агрегатного
состояния подвижных фаз, что обусловливает различные прин-
ципы детектирования в ГХ и ВЭЖХ.
Относительная чувствительность детекторов газовых хрома-
тографов, наиболее широко используемых при выполнении ко-
личественных анализов (катарометр, ПИЛ, ФИЛ), достаточно
легко прогнозируется. Имеются обширные сводки численных
значений экспериментально измеренных относительных откли-
ков названных детекторов на различные неорганические и орга-
нические соединения, перечни которых в соответствующих та-
блицах [32, 33, 75] или банках данных [236, 237] весьма внуши-
тельны*. При решении некоторых частных задач относитель-
ные отклики детектора к определяемым веществам можно при-
‘Отметим, однако, что пользоваться литературными данными по отно-
сительной чувствительности того или иного детектора к интересующим
веществам следует с большой осторожностью.
347
нять одинаковыми и проводить анализ без градуировки хрома-
тографа*.
В жидкостной хроматографии для количественного опреде-
ления соединений в потоке жидкой подвижной фазы чаще всего
применяют устройства, работа которых строится на принципах
измерения светопоглощенил, относящегося к заданным длинам
волн (УФ-детектор), или светопреломления (рефрактометр), а
также гораздо более селективные детекторы по флуоресценции
и электрохимический. Табулирование относительных откли-
ков этих детекторов не представляется возможным из-за боль-
шого влияния, оказываемого как на абсолютный, так и на от-
носительный сигнал детектора, природы и состава растворите-
лей, образующих подвижную фазу. Лаже незначительные от-
клонения в качественном и количественном составе подвижной
фазы могут, например, изменить коэффициент экстинкции фото-
метрируемого соединения на несколько порядков его численной
величины. Вследствие этого обстоятельства из трех основных
методов количественного хроматографического анализа, преду-
сматривающих градуировку прибора в прямой форме (метод
абсолютной градуировки), либо в косвенной (методы внутрен-
ней нормализации, внутреннего стандарта и их модификации),
в жидкостной хроматографии преимущественно используют ме-
тод абсолютной градуировки и реже два других метода* ** [325].
Суть каждого из них будет рассмотрена ниже.
Прямая или косвенная градуировка предусматривает в каче-
стве начальной и очень ответственной процедуры приготовле-
ние и хроматографирование нескольких искусственных смесей,
содержащих различные количества каждого из определяемых
компонентов, и, в некоторых случаях, дополнительно эталон-
ных (стандартных) веществ.
’Типичные примеры — анализ смесей изомерных органических со-
единений с любым из только что упомянутых детекторов, исследование
количественного состава смесей углеводородов различных классов при
анализе нефтепродуктов с пламенно-ионизационным детектором, опреде-
ление относительного содержания в смесях веществ с достаточно боль-
шой молекулярной массой (в молекулах которых насчитывается более 10
углеродных атомов и вследствие ©того обладающих очень низкой тепло-
проводностью) при детектировании катарометром.
**Отметим, что не так давно была реализована базирующаяся на за-
коне Ламберта—Бугера—Бера привлекательная возможность выполне-
ния количественных анализов в жидкостной хроматографии с УФ-детек-
тированием без какой-либо градуировки при условии, что состав подвиж-
ной фазы жестко закреплен, а коэффициенты экстинкции определяемых
соединений взяты из надежных литературных источников или найдены
опытным путем в специальных экспериментах [328, 329].
348
Смеси твердых соединений обычно растворяют в подходя-
щем растворителе*. Рекомендуется разбавлять подходящим
растворителем и смеси жидкостей, особенно содержащие не-
большое число компонентов в сопоставимых концентрациях.
При этом становится возможным дозирование относительно
больших количеств образца (от нескольких долей мкл до 2-
4 мкл в газовой и до 10-20 мкл в высокоэффективной жидкостной
хроматографии) без риска выйти за границы линейного дина-
мического диапазона детектора; одновременно улучшается вос-
производимость ввода пробы.
Необходимая степень разбавления зависит от чувствитель-
ности используемого детектора к определяемым компонентам.
Известно, например, что в газовой хроматографии для боль-
шинства современных моделей пламенно-ионизационного детек-
тора верхний предел линейного отклика ограничивается концен-
трацией анализируемого вещества в потоке газа-носителя, не
превышающей 5% (по объему)**. Если принять, что размыва-
ние пробы при дозировании и разделении в колонке в принятых
условиях анализа уменьшит ее первоначальную концентрацию
в 10 раз, то углеводородные компоненты, содержание которых
в исходном образце составляет более 50%, будут регистриро-
ваться уже за пределами линейного диапазона ПИЛ.
Опасность выхода за пределы диапазона линейности осо-
бенно велика при использовании некоторых высокочувствитель-
ных селективных детекторов (ЭЗД или ЛПР). В этом случае
необходимые для градуировки прибора искусственные смеси
определяемых соединений и подлежащие анализу образцы не-
известного состава разбавляют растворителем в 103-106 раз.
Напротив, при работе с детекторами меньшей чувствительно-
сти (катарометр — в ГХ, рефрактометр — в ВЭЖХ) предвари-
тельно разбавлять пробу не приходится.
В окончательных расчетах как в газовой, так и в высокоэф-
фективной жидкостной хроматографии введение растворителя
учитывается или не учитывается в зависимости от выбранного
метода количественной интерпретации результатов хромато-
графического анализа (см. ниже). Тем не менее, при анализе од-
нотипных образцов всегда следует разбавлять пробу примерно
одинаковым количеством растворителя, чтобы не вносить за-
*При подготовке к выполнению ВЭЖХ-анализов наиболее оправдано
растворение компонентов градуировочных смесей в подвижной фазе. В
газовой хроматографии принятые условия анализа должны обеспечи-
вать, по возможности, полное разделение выбранного растворителя и
компонентов смеси. Растворитель не должен содержать примесей, на-
кладывающихся на пики определяемых соединений.
**По пропану, этилену и другим легким углеводородным газам.
349
метных изменений в условия работы хроматографической ко-
лонки и детектора.
IV.3.3.2. Метод абсолютной градуировки
Метод абсолютной градуировки, называемый иногда мето-
дом внешнего стандарта, заключается в построении графиче-
ской зависимости одного из количественных параметров хрома-
тографического пика (обычно высоты или площади) от содер-
жания вещества в пробе. На рис. IV.23 представлен типичный
градуировочный график для линейно и нелинейно работающего
детектора. Как правило, по оси ординат откладывают значения
высот hi (мм) или площадей А, (мм2 или мкВ • с) хроматографи-
ческих пиков, а по оси абсцисс — концентрацию г-го вещества в
пробе (Ci, %, мг/мл, ммоль/мл) или его абсолютное количество
(qi, см3, мкл, мкг).
Рис. IV.23. Градуировочная зависимость
между содержанием компонента в пробе
и количественным параметром хромато-
графического пика для линейно (I) и не-
линейно (2) работающего детектора.
В случаях, когда зависимость hi (А) = fi(Ci) линейна, можно
вычислить угловой коэффициент (градуировочный множитель
fi = Ail Ci или fi = hi/Ci) и использовать его численное значе-
ние в дальнейших расчетах. Если и при градуировке прибора,
и при выполнении анализа пробы неизвестного состава вводить
в колонку одинаковые по величине дозы газов, жидкостей или
растворов твердых веществ, то, например, концентрацию г-го
соединения в анализируемом образце легко можно найти непо-
средственно по графику или по простейшей формуле
Ci = Pi/fi,
где Pi — измеряемый параметр хроматографического пика (вы-
сота или площадь).
Если по каким-либо причинам на этапах градуировки при-
бора и выполнения анализов подготовленных образцов в ко-
лонку вводились дозы разной величины, перед расчетом С, из-
меренные в каждом опыте величины аналитического сигнала (Л,-
350
или Л,) необходимо скорректировать на различия в величине
использованных доз.
Важнейшими требованиями к эксперименту при выполнении
количественного анализа по методу абсолютной градуировки
являются точность и воспроизводимость дозирования пробы,
а также строгое соблюдение постоянства (тождественности)
условий хроматографирования при градуировке прибора и при
определении содержания интересующего вещества в пробе неиз-
вестного состава. При использовании новейшего прецизионного
хроматографического оборудования выполнение последнего из
сформулированных обязательных требований не встречает осо-
бых затруднений.
Точность и воспроизводимость дозирования газообразных
образцов в газовой хроматографии обеспечиваются использо-
ванием газового крана-дозатора. Воспроизводимость и соот-
ветственно точность результатов при дозировании жидких проб
как в газовой, так и в жидкостной хроматографии микрошпри-
цами определяются типом шприца, качеством его изготовления
и многими субъективными факторами, присущими оператору.
В газовой хроматографии, особенно при дозировании отно-
сительно малых (^ 1 мкл) объемов жидкостей микрошприцами
с полой иглой и с рабочими объемами 5-10 мкл (типа отече-
ственных МШ-10 и аналогичных моделей ведущих зарубежных
фирм), большое влияние на воспроизводимость и точность ре-
зультатов оказывают, в частности, интенсивность толчка плун-
жера и продолжительность нахождения иглы микрошприца в го-
рячей зоне камеры испарителя. В специальных экспериментах
было установлено [330], что при медленном (осторожном) до-
зировании выдавливаемая жидкость “зависает” на конце иглы,
обволакивая ее внешние стенки, и отрывается только тогда, ко-
гда силы гравитации преодолеют силы когезии (рис. IV.24, а).
Возникает ситуация, провоцирующая дискриминацию состава
пробы, попадающей в колонку, поскольку образующееся в испа-
рителе облако пара обогащается в первую очередь легколету-
чими компонентами.
При энергичном выталкивании столбика жидкости из рабо-
чего цилиндра и иглы микрошприца происходит быстрый от-
рыв капли от остающейся жидкости, однако линия отрыва нахо-
дится не на срезе (как хотелось бы, см. рис. IV.24, б), а внутри
иглы (примерно на глубине от 3 до 12 мм), так что реальная
величина дозы превышает заданную минимум на 0,1-0,2 мкл,
а то и больше, в зависимости от внутреннего диаметра иглы,
вязкости дозируемой жидкости, интервала температур кипения
входящих в смесь компонентов, степени их разбавления раство-
351
Рис.IV.24. Различный характер парообразования при медлен-
ном (а) и быстром (6) вводе жидкой пробы в испаритель хрома-
тографа.
1 — “зависание” жидкости, выдавливаемой из иглы микрошприца;
2 — отрыв выдавливаемой жидкости от иглы микрошприца (зеркало
отрыва — выше среза иглы); 3— начальные участки хроматографиче-
ских колонок.
рителем и т. п. По совокупности признаков быстрое дозирова-
ние предпочтительнее медленного, однако для оценки реальной
величины дозы необходимо заранее измерить рабочий объем
иглы микрошприца Vp (он может составлять от 1,2 до 2 мкл) и
после выполнения каждого ввода пробы измерять объем жидко-
сти, оставшейся в игле, К>ст (для чего следует движением плун-
жера втянуть оставшийся в игле столбик жидкости в рабочий
цилиндр и оценить его объем по шкале микрошприца). Разность
Vp — V<,cT приплюсовывается к объему жидкости, находившейся
в рабочем цилиндре перед дозированием.
Недавно было продемонстрировано [331], что различия в со-
отношении компонентов в дозируемой жидкости и в образую-
щемся в испарителе облаке пара можно устранить, если вме-
сто микрошприцев с полой иглой воспользоваться микрошпри-
цами типа МШ-1 с рабочим объемом, заключенным в самой
игле. Следует, однако, иметь в виду, что данные шприцы вслед-
ствие их конструктивных особенностей (главным образом, из-
за принципиальной невозможности добиться высокого качества
притирки плунжера — тонкой металлической нити — к внутрен-
ним стенкам стальной иглы, а также из-за наличия быстро изна-
шивающегося сальника) неспособны обеспечить высокую вос-
производимость дозирования по абсолютной величине пробы;
разброс может достигать 25-30%. Эти шприцы обладают и го-
352
раздо большей памятью на предшествующий образец, подлежа-
щей обязательному контролю.
В качестве альтернативного приема, устраняющего искаже-
ние состава образующегося в испарителе пара при использо-
вании микрошприцев с полой иглой, давно уже было предло-
жено сначала отбирать в рабочий цилиндр 2-2,5 мкл какого-
либо подходящего растворителя* и уже затем анализируемую
смесь, отделяемую от столбика растворителя зоной воздуха (1-
1,5 мкл). При дозировании слой растворителя полностью вы-
талкивает находящуюся в рабочем цилиндре шприца и его игле
анализируемую смесь в испаритель хроматографа, тем самым
обусловливая достоверность результатов как по абсолютным,
так и по относительным показателям. Активное использование
этой процедуры сдерживается необходимостью подбора усло-
вий работы прибора, исключающих маскировку интересующих
компонентов полосой избыточного растворителя.
В колонку жидкостного хроматографа обычно вводят порции
жидкостей от 1-3 до 20 мкл, для чего используют микрошприцы
с рабочим объемом от 1 до 25 мкл. Дозирование жидкостей
микрошприцами с полой иглой при комнатной или близкой к
комнатной температуре практически не сопровождается отяг-
чающими градуировку эффектами (см. выше), обычно проявля-
ющимися в тем большей мере, чем больше разница между темпе-
ратурой испарителя и температурами кипения отдельных ком-
понентов анализируемых смесей.
Совокупность причин случайного характера, влияющих на
сигнал детектора (в первую очередь, температурные флукту-
ации, нестабильность потока и состава подвижной фазы, не-
воспроизводимость дозирования), обусловливает погрешность
в окончательных результатах анализов жидких проб методами
как газовой, так и жидкостной хроматографии не меньше 10-
25%, а иногда и больше, тогда как при анализах газообразных
образцов методом газовой хроматографии суммарная погреш-
ность редко превышает 10%.
К абсолютной градуировке чаще всего прибегают в тех слу-
чаях, когда есть сомнения в линейной работе детектора, и то-
гда, когда нужно определять не все компоненты анализируемой
смеси, а только некоторые из них, например при анализе приме-
сей к основным веществам. В соответствии с этим условия хро-
матографического анализа должны обеспечивать, по возможно-
сти, полное отделение лишь пиков интересующих компонентов
от соседних пиков на хроматограмме.
*Например, растворителя, использованного при подготовке пробы к
анализу.
353
В практической работе при анализе газов градуировку га-
зового хроматографа проводят, как правило, только по одной
какой-либо смеси постоянного состава с известным содержа-
нием интересующего компонента (например, смеси этого ком-
понента с газом-носителем). При этом путем последовательной
смены дозирующих петель газового крана вводят в колонку ее
разные количества*, а при построении градуировочного гра-
фика наблюдаемый в каждом опыте отклик детектора соотносят
с абсолютным или относительным количеством компонента (мл,
моль и т. п.).
При градуировке по жидким соединениям как газового, так и
жидкостного хроматографа из-за опасности перегрузки колонки
и изменения условий работы детектора предпочтительнее при-
готавливать несколько искусственных смесей (растворов) с раз-
ной концентрацией определяемого компонента и вводить в хро-
матограф одну и ту же по величине дозу.
Следует уделять большое внимание чистоте реагентов и рас-
творителей (газов и жидкостей), использумых для приготовле-
ния искусственных смесей, и учитывать количество примесей
в каждом из них при выполнении градуировки. Относитель-
ные концентрации компонентов, составляющих смеси, необхо-
димо подбирать с расчетом перекрывания ожидаемой концен-
трации интересующих соединений в анализируемых образцах.
Для построения градуировочного графика каждую искусствен-
ную смесь рекомендуется хроматографировать по меньшей мере
три-пять раз с целью исключения случайных промахов и усред-
нения данных.
В некоторых случаях, при полной уверенности в линейной
области показаний детектора и затруднительности приготовле-
ния искусственных смесей с варьированием в них количества
определяемого компонента (анализ микропримесей), допуска-
ется градуировка хроматографа лишь по одной смеси. На гра-
дуировочном графике проводят прямую линию через единствен-
ную экспериментальную точку и начало координат.
IV.3.3.3. Метод внутренней нормализации
Метод внутренней нормализации заключается в отнесении
измеренного количественного параметра хроматографического
пика (площади, высоты или произведения высоты на время
*Этот прием позволяет также “попутно” определить истинный дози-
руемый объем, включающий, как известно, кроме объема сменной дози-
рующей петли постоянный объем самого крана и соединительных комму-
никаций. Подробнее об этом см. раздел П.1.2.
354
удерживания) к суммарному сигналу детектора на все компо-
ненты пробы, присутствующие в анализируемом образце. В со-
ответствии с этим традиционный вариант внутренней норма-
лизации предусматривает измерение количественных параме-
тров всех зарегистрированных пиков*, приведение их к единой
шкале чувствительности детектирования (или регистрации**)
и суммирование полученных значений. Окончательно содержа-
ние компонентов в анализируемой смеси Ci находят по формуле
Ci(%) = 100,
ЕЛ Л-
1
(IV.42)
где Pi — нормируемый параметр хроматографического пика
(площадь, высота или произведение высоты на время удержи-
вания); fi —нормировочный (градуировочный) множитель.
Как уже отмечалось выше, для получения достоверных ре-
зультатов при использовании метода внутренней нормализации
необходимо вводить в расчетную формулу (IV.42) надежно уста-
новленные нормировочные (градуировочные) множители Л, чи-
сленные значения которых лучше всего находить эксперимен-
тально. В высокоэффективной жидкостной хроматографии этот
путь является единственно возможным; в газовой хроматогра-
фии иногда допускается приравнивание численных значений fi
для всех компонентов пробы к 1, а также использование расчет-
ных или литературных данных (см. ниже).
Наиболее распространенный способ экспериментального
определения fi для какого-либо вещества относительно стан-
дартного соединения заключается в хроматографировании ис-
кусственно составленных смесей необходимых компонентов с
выбранным стандартным веществом и последующем расчете по
формуле
Cj/Pj = QP^
CBt/PBt CBtPi'
(IV.43)
*Кроме пика растворителя, введенного на стадии подготовки образца
к анализу. При расчетах количество растворителя (степень разбавления
пробы) никак не учитывается.
**Если во время записи хроматограммы изменялась чувствительность
регистрации пиков отдельных компонентов, то перед нормированием не-
обходимо привести значения измеренных параметров к единой шкале
чувствительности регистрации. Необходимость в этой операции отпа-
дает при измерении площадей (или высот) пиков интегратором, подклю-
ченным непосредственно к блоку тепловых детекторов или к выходу уси-
лителя сигнала ионизационных детекторов, а также при использовании
Для измерения сигналов детектора специализированных компьютеризо-
ванных систем обработки результатов анализа (см. раздел IV.5).
355
При этом принимается, что нормировочный (градуировочный)
множитель для стандартного соединения /st = 1,0. Содержание
компонентов Ci и C'st может быть выражено в % по массе, объ-
ему или молярности; соответственно в тех же единицах будет
определен и состав анализируемого образца.
Надежность получаемых при экспериментальном определе-
нии fi данных зависит от погрешностей не только на стадии
хроматографирования, но и на стадии приготовления искус-
ственных смесей (вследствие возможных потерь из-за неакку-
ратного смешивания компонентов, из-за испарения при отборе
пробы для взвешивания и т. п. операции приготовления искус-
ственных смесей должны проводиться с максимальной тщатель-
ностью!). В литературе, рассматривающей возможности ре-
ализации метода внутренней нормализации в газовой хрома-
тографии, опубликованы обширные сводки экспериментально
найденных значений fi для представителей многих классов хи-
мических соединений относительно различных стандартов при
их регистрации катарометром, пламенно-ионизационным и дру-
гими детекторами*. Эти данные частично включены в неко-
торые учебники. Известны также приближенные расчетные и
графические способы оценки относительной чувствительности
детекторов [33, с. 134-180; 332].
Однако, как показали предпринятые в последние годы специ-
альные критические исследования, полезность опубликованных
экспериментально найденных и расчетных данных по относи-
тельной чувствительности различных детекторов к определя-
емым веществам носит довольно ограниченный характер и ис-
пользовать эти сведения в практической работе следует с боль-
шой осторожностью. Здесь уместно процитировать следующее
заключение, относящееся к детектору по теплопроводности [31,
ч. 2, с. 15]: “... при работе не с водородом или гелием, а с дру-
гими газами специфическая для вещества чувствительность де-
тектора в особенно большой мере зависит от тока моста, ра-
бочей температуры и области концентраций. В этих случаях
поправочные коэффициенты, приводимые в литературе, не при-
годны даже приближенно!”.
Градуировочные множители fi, характеризующие относи-
тельный отклик пламенно-ионизационного, термоионного, элек-
тронозахватного, фотопламенного и многих других детекторов,
значительно изменяются в зависимости от конструктивных осо-
*В некоторых оригинальных статьях и монографиях наряду со значе-
ниями fi (или вместо них) приводятся значения относительного (моляр-
ного или массового) отклика детектора Ri, равного If fi.
356
бенностей и режима их эксплуатации*, а также от состава гра-
дуировочных смесей, степени разделения компонентов и вы-
бранного для нормализации параметра хроматографического
пика. Разброс данных при использовании хроматографов раз-
личных фирм (детектор пламенно-ионизационный с установлен-
ным оптимальным отношением расходов газа-носителя, водо-
рода и воздуха) в одинаковых условиях для одной и той же
смеси бензола, метилциклогексана и октана, растворенных в
ксилоле, достигает 20-40%. Меньший, но все же значительный
разброс экспериментально определяемых величин fi наблюда-
ется и при использовании катарометра, причем не только для
хроматографов различных моделей (типов), но и для разных
экземпляров однотипных приборов.
Таким образом, достижению высокой точности результатов
количественных определений при расшифровке хроматограмм
методом внутренней нормализации будет способствовать вы-
полнение следующих двух условий.
1. Использование не литературных или расчетных, а экспе-
риментально найденных значений градуировочных множителей
fi, определяемых на том же приборе и в том же режиме хро-
матографирования, в котором будет проводиться анализ смеси
неизвестного состава.
2. При определении величин надо исходить из результа-
тов хроматографирования смесей, имитирующих состав анали-
зируемого образца или приближающихся к нему.
Преимущества метода внутренней нормализации в сравнении
с методом абсолютной градуировки заключаются в том, что нет
необходимости в точной дозировке образца и соблюдении то-
ждественности условий анализа при повторных определениях.
В то же время метод имеет ряд существенных ограничений
и недостатков. Использование внутренней нормализации для
определения содержания г-го компонента в смеси с j-м числом
других веществ правомерно лишь при условии, что природа
всех интересующих соединений в смеси известна (иначе невоз-
можно определение /4) и что все они проявляются на хромато-
грамме. Поэтому, приступая к анализу незнакомой смеси, сле-
дует прежде всего путем изменения в широких пределах условий
анализа, специфичных для газовой или жидкостной хроматогра-
*Например, на полезный сигнал ПИД оказывает значительное влия-
ние даже такой редко контролируемый экспериментальный фактор, как
атмосферное давление, причем колебание его в пределах ±27 • 102 Па
(около 20 мм рт. ст.) вызывает отклонение ионизационной эффективности
в пределах 10%. Существенно изменять численные значения коэффици-
ентов fi может и температура детектора [333].
357
фии, убедиться в однозначном определении числа компонентов,
составляющих анализируемую смесь. Окончательно принятые
условия должны обеспечивать линейное детектирование и, по
возможности, полное отделение всех входящих в смесь соедине-
ний друг от друга, поскольку погрешность в измерении площади
или другого количественного параметра хроматографического
пика хотя бы одного компонента (или приписание ошибочного
значения fi хотя бы одному компоненту) приведет к неправиль-
ной оценке содержания в образце всех веществ.
IV.3.3.4. Метод внутреннего стандарта
Метод внутреннего стандарта (эталона) в традиционном ва-
рианте предусматривает прибавление к известному количеству
анализируемого образца известного количества не содерж-
ащегося в нем эталонного соединения (“внутреннего стан-
дарта”) и последующее хроматографирование приготовленной
смеси.
Содержание компонентов в анализируемом образце (% по
массе, по объему или по молярности) находят по формуле
Q (%) = 100, (IV.44)
StffcM
где Pi, Pst — измеряемые параметры хроматографических пи-
ков интересующего и стандартного веществ (площади, высоты
или произведения высот на время удерживания); fi — граду-
ировочный (нормировочный) множитель для определяемого со-
единения относительно стандартного вещества*; 9st, 9см — коли-
чества стандартного вещества и анализируемой смеси (мг, мкл
или ммоль), отобранные и смешанные для выполнения анализа.
Если подготовленную к анализу пробу перед хроматогра-
фированием разбавляют посторонним растворителем, то его
количество (степень разбавления) не должно сказываться на
результатах и расчетной формулой не учитывается. Иногда,
однако, удобнее разбавлять растворителем порции анализи-
руемой смеси и стандартного соединения по отдельности и
смешивать аликвоты приготовленных растворов. В этом слу-
чае необходимо строго учитывать количества растворителя с
тем, чтобы ввести в расчетную формулу коэффициенты, отра-
жающие различную степень разбавления подлежащей анализу
смеси и стандартного соединения.
’Способы нахождения величин fi и случаи, когда их численные значе-
ния можно приравнять 1, рассмотрены выше (см. раздел IV 3.3.3).
358
Преимущество метода внутреннего стандарта состоит в том,
что при его использовании сводятся к минимуму погрешности в
результатах, вызванные случайными изменениями основных па-
раметров хроматографического опыта (температуры колонки,
состава и скорости подвижной фазы и режима работы детек-
тора), поскольку возможные отклонения от заданных условий
должны равным образом влиять на количественные параметры
хроматографических пиков как стандартного, так и анализируе-
мого соединения. Отпадает необходимость дозирования строго
заданных количеств пробы и соблюдения постоянства всех пе-
ременных параметров хроматографирования.
Наряду с этим оказывается возможным получать равноточ-
ные результаты сопоставлением как площадей или произведе-
ний высот пиков на время удерживания компонентов, так и вы-
сот пиков интересующего компонента и стандартного вещества
(при обработке хроматограмм вручную это приводит к значи-
тельной экономии времени).
Поскольку техника расшифровки хроматограмм сводится к
измерению параметров хроматографических пиков интересую-
щего и стандартного соединений, условия хроматографирова-
ния должны обеспечивать, по возможности, полное их разделе-
ние; все остальные составляющие исходной пробы в принятых
условиях анализа могут не отделяться друг от друга или даже
вообще не проявляться на хроматограмме (в этом состоит пре-
имущество метода внутреннего стандарта перед методом вну-
тренней нормализации).
Главные ограничения применения метода внутреннего стан-
дарта заключаются, во-первых, в необходимости специальной
подготовки пробы для анализа (т. е. введения в известное по
объему или массе количество анализируемого образца извест-
ного количества внутреннего стандарта — при этом трудно из-
бежать погрешностей, связанных с возможным изменением со-
става пробы) и, во-вторых, в возникающих иногда затруднениях
при выборе внутреннего стандарта.
На этой стадии обычно руководствуются следующими тре-
бованиями.
1. Стандартное (эталонное) вещество должно полностью сме-
шиваться с компонентами анализируемой смеси (растворяться в
ней) и в химическом отношении должно быть абсолютно инерт-
ным и к компонентам анализируемой смеси, и к используемым
неподвижной и подвижной фазам (а также к твердому носителю
неподвижной жидкой фазы — как в газовой, так и в высокоэф-
фективной жидкостной хроматографии). Желательно выбирать
стандартное вещество из числа соединений, близких к объектам
359
анализа по структуре и молекулярной массе. Это исключает
возможное неодинаковое влияние условий опыта на параметры
пиков стандарта и определяемых соединений.
2. Концентрацию эталонного вещества в анализируемой про-
бе следует подбирать таким образом, чтобы отношение площа-
дей (или других количественных параметров) пиков стандарта
и интересующего компонента было близким к единице.
3. В принятых условиях анализа пик стандартного вещества
должен располагаться на хроматограмме в непосредственной
близости от пиков соединений-объектов анализа, не наклады-
ваясь ни на них, ни на пики других веществ. Если в задачу
анализа входит определение содержания в пробе двух или бо-
лее веществ, значительно различающихся по временам удержи-
вания (или по концентрации), целесообразно использовать два
или более внутренних стандартов*.
Важно, чтобы стандарт (стандарты) не содержал примесей,
пики которых накладываются на пики определяемых соедине-
ний. Поэтому предварительно в тех же условиях контролируют
содержание в стандарте нежелательных примесей. При этом
100%-ная чистота стандарта необязательна, так как присутствие
примесей, пики которых не накладываются на пики определяе-
мых соединений, учитывается градуировкой (значением /;).
Вариантом метода внутреннего стандарта является метод
стандартной добавки. Если выбор вещества-стандарта с учетом
перечисленных выше требований затруднен, возможно и оправ-
дано использование в качестве стандарта соединений, уже при-
сутствующих в анализируемой смеси. В этом случае в одинако-
вых условиях получают две серии хроматограмм: а) исходной
анализируемой смеси и б) порции смеси дсм с добавленным к
ней известным количеством qsi одного из компонентов, играю-
щего роль стандартного соединения. Содержание в исходной
смеси интересующих компонентов и вещества-стандарта (в %
по массе) рассчитывают по формулам
cd%) = ъ—/fPgst/,?p /fP 10°- (IV-45)
C'st(%) = ,p /p fSt-^CM ,p > 1 100, (iv.46)
(n(l)/Pst(l)) (Bst(2)/B,(2)) - 1
*При этом каждый стандарт используется для расчета концентрации
ближайших к нему по времени удерживания компонентов смеси. Недавно
было предложено использовать два или даже больше стандартов для
расчета концентрации каждого определяемого компонента; подробнее об
этом новом методе, получившем название “метод двойного стандарта”,
см. в разделе IV.3.4.
360
где <7См, 1st — количества отобранной для анализа порции смеси
и введенного в нее стандарта; Рцг), ^’(2), ^st(2) — ко-
личественные параметры пиков интересующего или реперного
компонента и вещества-стандарта на хроматограммах первой
и второй серии соответственно; fi — нормировочный (градуи-
ровочный) множитель для интересующего компонента относи-
тельно вещества-стандарта (принимается, что fst = 1). При
условии линейности детектора точность получаемых результа-
тов не хуже, чем при выполнении анализов по основному методу
внутреннего стандарта или по методу внутренней нормализа-
ции.
Как видно из приведенных расчетных уравнений, наиболее
заманчиво использовать в качестве стандартной добавки веще-
ство, подлежащее определению. В этом случае при расчете его
исходной концентрации Cst в анализируемом образце отпадает
необходимость учета каких-либо градуировочных множителей
(как fi, так и fst). В принципе, если обеспечить стабильный
режим работы всех систем газового или жидкостного хрома-
тографа (как этого требует рассмотренный выше метод абсо-
лютной градуировки), то вообще отпадает необходимость в из-
мерении на хроматограмме количественных параметров пиков
каких-либо иных веществ (играющих роль соединения сравне-
ния), кроме подлежащих количественному определению. Ис-
ходное (начальное) массовое или объемное содержание этого
соединения (мкг или мкл) в пробе известного дозируемого объ-
ема (при регистрации сигналов детектора в линейной области)
можно найти уже по простой пропорции, связывающей прирост
площади (высоты) пика на хроматограммах с массой или объ-
емом стандартной добавки, поступившей в колонку (дозы ана-
лизируемых смесей до и после введения стандартной добавки
должны быть одинаковы).
Точность получаемого результата можно повысить (а заодно
и проверить линейность показаний детектора), если к несколь-
ким одинаковым навескам анализируемой смеси прибавить из-
вестные (контролируемые по объему или по массе) последова-
тельно увеличиваемые количества определяемого соединения.
По результатам хроматографирования каждого из приготовлен-
ных таким способом образцов (условие постоянства величины
Дозы сохраняется!) можно построить график в координатах пло-
щадь (высота) пика — масса или объем стандартной добавки
(рис. IV.25). Экстраполяция полученной зависимости до ее пе-
ресечения с продолжением оси абсцисс позволяет достаточно
точно оценить начальное содержание интересующего соедине-
ния в анализируемой смеси.
361
Рис.IV.25. Градуировочная
зависимость между последо-
вательно увеличиваемым со-
держанием стандартной до-
бавки в пробе и высотой (или
площадью) соответствующих
хроматографических пиков.
Более общий случай использования метода стандартной до-
бавки, когда в качестве стандарта применяется вещество, не
только присутствующее в анализируемой смеси, но и содержа-
щее некоторое количество примесей — соединений, также при-
сутствующих в смеси, рассмотрен в работе [334].
IV.3.4. Развитие методов количественной
интерпретации хроматограмм сложных смесей
Использование не одного, а двух или большего числа стан-
дартов для получения информации о содержании в образце
каждого определяемого компонента приводит, по данным [335,
336], к заметному повышению воспроизводимости и правильно-
сти результатов анализа за счет их меньшей подверженности
искажающему влиянию экспериментальных факторов (флуктуа-
циям режима). Проиллюстрируем это на примере метода двой-
ного внутреннего стандарта, заключающегося в том, что в пор-
цию анализируемой смеси вводят два стандартных вещества
(st (1) и st (2)), элюирующихся до и после определяемого со-
единения.
При выборе стандартов помимо соблюдения требований, из-
ложенных в разделе IV.3.3.3, следует стремиться обеспечить ми-
нимальную и, по возможности, одинаковую (или сопоставимую)
удаленность пика определяемого компонента смеси на хромато-
грамме от пиков каждого из стандартных веществ. Проще всего
эта задача решается подбором двух ближайших гомологов (н-
алканов или каких-либо их производных), содержащих z и z + 1
углеродных атомов*.
*В етом случае особенно наглядно прослеживается аналогия с рассмо-
тренными выше методическими приемами измерения индексов удержи-
вания в качественном хроматографическом анализе (см. раздел IV.2.2).
362
По окончании регистрации хроматограмм содержание опре-
деляемых компонентов в анализируемом образце С,- (% по массе
или по объему) находят по формуле
Ci - Pi^ I gst(1) gst(2) • 100
7см у ^st(l)/st(l) ^st(2)/st(2)
Если стандарты и определяемые соединения имеют близкую
химическую природу и близкие молекулярные массы, возмож-
ные небольшие различия в численных значениях нормировоч-
ных (градуировочных) множителей можно не учитывать, что
существенно упрощает выполнение анализа.
В контрольных газохроматографических анализах относи-
тельные стандартные отклонения среднего результата при ис-
пользовании метода двойного стандарта оказались в 1,5-3 раза
меньше, чем при работе с единичным стандартом; примерно на
порядок снизилась и систематическая составляющая погрешно-
сти.
Следует, однако, иметь в виду, что необходимость подбора
и введения в порцию анализируемой смеси нескольких стандар-
тов, удовлетворяющих названным выше условиям, увеличивает
трудоемкость подготовительных стадий, а при отсутствии ин-
тегрирующих устройств — и самой процедуры обработки ре-
зультатов анализа.
IV.3.5. Методы количественного парофазного анализа
IV.3.5.1. Общие сведения
В начале 1960-х годов в литературе появились работы, в ко-
торых газохроматографическому анализу подвергались не ис-
следуемые жидкие или твердые объекты, а газовая фаза над
ними. Этот простой прием применялся при исследовании со-
става летучих соединений, выделяющихся из пищевых продук-
тов, для контроля содержания вредных веществ в воде, поли-
мерных и биологических материалах. Дозирование в хромато-
граф газа вместо жидкости или твердого тела значительно рас-
ширяет возможности газовой хроматографии, так как позволяет
определять летучие компоненты в объектах, прямой ввод кото-
рых в прибор невозможен или нецелесообразен по причине не-
достаточной чувствительности детекторов, присутствия легко
разлагающихся компонентов, загрязнения колонки нелетучим
остатком или нарушения существующего в системе химического
363
равновесия. Такой способ определения летучих веществ в ан-
глийской литературе получил название «Head-Space Analysis» (в
современном написании — «Headspace Analysis»), а в русской —
сначала «анализ равновесного пара», а затем «парофазный ана-
лиз» (ПФА).
В настоящее время ПФА — одно из быстро развивающихся
направлений газовой хроматографии — представляет собой ме-
тод получения информации о составе и свойствах жидкостей
и твердых тел путем анализа контактирующей с ними газовой
фазы. Область применения ПФА достаточно широка и кроме су-
губо аналитических и физико-химических приложений способы
и приемы ПФА оказываются полезными при решении проблем
экологии, медицины, санитарной химии, биологии, криминали-
стики и других отраслей науки и техники.
Парофазный анализ основан на технике и приемах газовой
экстракции. В зависимости от условий применения различают
дискретную газовую экстракцию, осуществляемую отдельными
порциями газа в замкнутой системе, обычно в статике, и непре-
рывную газовую экстракцию потоком инертного газа, проходя-
щего через жидкость или над поверхностью конденсированной
фазы.
IV.3.5.2. Анализ систем
с известными коэффициентами распределения
Простейший вариант ПФА — однократная газовая экстрак-
ция летучих веществ из жидкостей или твердых тел — состоит
в следующем. В герметично закрывающийся сосуд объемом V
помещают исследуемый объект объемом Vl> содержащий опре-
деляемое летучее вещество концентрации С£. Объем газовой
фазы VG при этом оказывается равным V — Vl. После выдержи-
вания системы при постоянной температуре до установления
равновесного распределения летучего вещества газовая фаза
вводится в хроматограф и измеряется абсолютное значение рав-
новесной концентрации определяемого вещества в газовой фазе
над исследуемым образцом Cg- Поскольку в процессе устано-
вления фазового равновесия некоторая часть содержащегося в
исследуемом объекте летучего вещества переходит в газовую
фазу, равновесная его концентрация в конденсированной фазе
будет меньше исходной С£- Количество вещества, перешедшее
в газовую фазу из раствора (CgVg), зависит от отношения объ-
емов фаз г = Vg/Vl и коэффициента распределения К = ClJCg-
Если пренебречь изменением объема жидкости за счет испаре-
ния раствора в процессе установления равновесия, концентра-
364
цию вещества в исходном растворе можно вычислить по его со-
держанию в равновесном газе из уравнения
C^ = CG(K + r). (IV.47)
Эта формула лежит в основе статических вариантов ПФА, а
также элементарных актов динамических вариантов, конечные
расчетные формулы которых несколько сложнее
Из всех методов, используемых в ПФА, варианты, основан-
ные на однократной газовой экстракции, в силу простоты тех-
нического оформления анализа применяются чаще других. К
этим методам относятся абсолютная градуировка (или внешний
стандарт) и внутренний стандарт. Существует два принципи-
ально различных варианта абсолютной градуировки. Первый
связывает площадь или высоту пика на хроматограмме, полу-
ченную в результате дозирования в хроматограф равновесного
газа, с концентрацией вещества в анализируемом образце, т. е.
Ag(Cl), а второй — площадь пика с концентрацией вещества в
равновесном газе — Ag(Cg).
Градуировка Ag(Сц) проводится с помощью специально
приготовленных стандартных растворов с известными концен-
трациями вещества и постоянными, но не обязательно точно из-
вестными соотношениями объемов фаз в сосуде для установле-
ния равновесия и количеством вводимой в хроматограф пробы.
Для единичных измерений достаточно приготовить один стан-
дартный раствор с концентрацией С£1, для которой площадь
пика на хроматограмме Ад оказывается соизмеримой с пло-
щадью пика Ag определяемого соединения в анализируемом
образце. Тогда в соответствии с основным уравнением коли-
чественного ПФА (IV 47) искомая концентрация может быть вы-
числена по простому соотношению
= Q'Ac/Ag. (IV.48)
Если анализ проводится для большого числа образцов в ши-
роком интервале концентраций или в нелинейной области изо-
терм распределения, расчет С£ лучше проводить графически по
предварительно измеренной зависимости Ag(С^) (рис. IV.26). К
преимуществам графического способа следует отнести возмож-
ность проведения количественного ПФА в случаях нелинейной
изотермы распределения, когда К зависит от Сц.
Способ градуировки Ag(C'£) получил широкое распростране-
ние в аналитической практике, но ему все же присущи опреде-
ленные ограничения, которые необходимо учитывать для предо-
твращения грубых промахов и больших ошибок.
1. Особенность использования градуировки Ag(Cl) состоит
в том, что для расчета не требуется знания абсолютного значе-
365
Рис. IV.26. Зависимость пло-
щади пика на хроматограм-
ме от концентрации вещест-
ва в исходном растворе.
гр м гр 1 — углы наклона для
линейной и нелинейной области
изотермы распределения.
ния коэффициента распределения. Но это отнюдь не означает,
что количественный анализ проводится при неизвестном К. По
существу, градуировка Лс(С'£) и решает задачу определения К,
так как угловой коэффициент этой зависимости (см. рис. IV.26)
характеризует чувствительность ПФА, которая является функ-
цией К. Иными словами, при таком способе градуировки коэф-
фициент распределения находят, но в скрытой форме — обычно
в виде суммарного градуировочного коэффициента, учитыва-
ющего не только значение К, но и соотношение объемов фаз,
и чувствительность детектирования. Поэтому свойства стан-
дартных растворов и исследуемых образцов, а также условия
проведения анализа должны быть идентичными. В против-
ном случае изменение К может привести к дополнительным и
трудно выявляемым ошибкам.
2. Указанные причины приводят к тому, что Ag(C'£) может ис-
пользоваться только для объектов с постоянными и не изменя-
ющимися в различных образцах свойствами конденсированной
фазы
3. В процессе приготовления градуировочных растворов лег-
колетучих веществ (с малыми К) следует учитывать возмож-
ность изменения заданного значения С'£’ за счет перехода части
вещества в газовую фазу сосуда, а также испарения в процессе
его заполнения или отбора пробы.
Градуировка -Ag(Cg)> в отличие от описанного метода
учитывает только чувствительность хроматографического де-
тектора к данному веществу. Поэтому расчет исходной кон-
центрации в растворе возможен только при наличии данных по
абсолютным значениям К и г. Если анализируются объекты
с постоянными коэффициентами распределения в различных
образцах, значения К могут быть предварительно измерены од-
ним из известных способов и в дальнейшем использованы при
366
расчетах во всей серии анализов. Когда объектом исследова-
ния являются растворы с изменяющимися свойствами (случаи
переменных К, вызванные изменением состава раствора или
нелинейностью изотермы распределения), процедура анализа
должна включать определение значения К, присущего данному
образцу.
Установление зависимости Ag(Cg) требует приготовления
серии газовых смесей с интервалом концентраций паров анали-
зируемых веществ, наблюдаемым в равновесном с исследуемым
раствором газе. Для этого могут быть использованы известные
методы, в том числе диффузионные. При введении таких сме-
сей в хроматограф дозируемый объем может быть неизвестен,
но должен быть строго постоянным как при градуировке, так и
при анализе исследуемых образцов.
Метод внутреннего стандарта аналогичен тради-
ционному использованию его в хроматографии. В исследуемый
раствор вводится стандартное вещество в концентрации ко-
торая, согласно (IV.47), связана с равновесной концентрацией
стандарта в газовой фазе соотношением
dl = Cg(Kst +r). (IV.49)
Далее равновесный газ вводится в хроматограф, и измеряют
концентрации C'q и Cg в виде соответствующих площадей пиков
на хроматограмме и Ag-
Расчетное уравнение для содержания определяемого веще-
ства в исследуемом растворе методом ПФА с внутренним стан-
дартом имеет вид
Cl = fid4^- • K + r (IV.50)
L J LAg Kst + r’ V ’
где fi — коэффициент, учитывающий относительную чувстви-
тельность хроматографического детектора к анализируемому
веществу по отношению к стандартному.
К преимуществам количественного ПФА с внутренним стан-
дартом следует отнести хорошую воспроизводимость и высо-
кую точность измерения соотношения площадей или высот пи-
ков, не требующую тщательного воспроизведения условий газо-
хроматографического анализа и, что особенно важно, дозируе-
мого объема равновесного газа. Последнее упрощает аппара-
турное оформление парофазного анализа и позволяет использо-
вать в качестве дозаторов не только специальные дозаторы, но
и газовые шприцы.
Все же, касаясь целесообразности применения внутреннего
стандарта при определении примесей в растворах с малыми
367
значениями К (например, газы, углеводороды или галогени-
рованные углеводороды в воде), надо обратить внимание на
сложность и трудоемкость приготовления растворов летучих
веществ с концентрациями на уровне мг/л и менее. Примене-
ние внутреннего стандарта в таких случаях может оказаться
целесообразным лишь при единичных измерениях. Но при се-
рийных анализах использование абсолютной градуировки сле-
дует считать более оправданным, поскольку современная ап-
паратура позволяет с высокой точностью дозировать газовые
пробы и хорошо воспроизводить условия анализа.
IV.3.5.3. Анализ систем
с неизвестными коэффициентами распределения
Особенность методов ПФА, основанных на однократной газо-
вой экстракции, состоит в том. что коэффициент распределения
определяемого вещества в различных образцах анализируемого
объекта должен быть известен заранее в явной или скрытой
форме. Довольно часто, однако, состав исследуемых матери-
алов может колебаться в столь широких пределах, что игно-
рирование зависимости К от содержания других компонентов
становится недопустимым и использование постоянных, одина-
ковых для всех используемых образцов значений К невозмож-
ным. Так, значительные колебания в содержании минеральных
солей в природных водах, анализируемых на следы углеводо-
родов, существенно отражаются на коэффициентах распреде-
ления этих веществ. Аналогичные осложнения возникают при
определении летучих органических примесей в промышленных
стоках в связи с колебаниями общего количества растворенных
веществ. В этих случаях следует использовать варианты коли-
чественного ПФА, не требующие априорного знания численных
значений К и включающие их определение в процедуру ана-
лиза.
Многократная газовая экстракция. Последовательное из-
влечение летучих веществ из жидких или твердых объектов рав-
ными порциями нейтрального по отношению к определяемым ве-
ществам газа в статических условиях является одним из наибо-
лее употребляемых способов анализа объектов с неизвестными
или непостоянными коэффициентами распределения. Актуаль-
ность аналитических задач, для решения которых предлагалось
осуществление процесса повторной и многократной экстракции,
возможность ее использования в сложных системах с неизвест-
ными коэффициентами распределения и отсутствие удовлетво-
рительных альтернативных методов вызвали появление различ-
368
ных модификаций дискретной газовой экстракции. Были пред-
ложены двухкратная экстракция для газохроматографического
определения летучих компонентов растворов [337-339] и много-
кратная экстракция для анализа жидких и твердых материалов
[340-342].
Процедура анализа включает следующие операции. Иссле-
дуемый объект помещается в замкнутую емкость и приводится
в равновесие с газом не содержащим определяемого вещества.
После газохроматографического определения CG производится
полная замена равновесного газа на чистый воздух (или ней-
тральный газ), при этом оставшаяся масса вещества в растворе
(ClVl) вновь распределяется между двумя фазами. Операция
замены равновесного газа на чистый газ повторяется п раз.
Первая экстракция используется для определения абсолютного
значения Cg, а последующие — только для измерения коэф-
фициента распределения. Результаты минимум двух экстрак-
ций, т. е. газохроматографического анализа равновесной газо-
вой фазы до и после ее замены на чистый газ, позволяют рас-
считать содержание летучих компонентов раствора по формуле
0 _ CGr
L 1 - (A’g/Ag)1/" ’
(IV.51)
Значение Cg находят с помощью градуировочного графика, и,
в отличие от других измеряемых параметров (AG, Ag и г), оно
включает погрешности градуировки хроматографа.
Возможности рассматриваемого варианта ПФА ограничива-
ются определением летучих компонентов со средними (порядка
сотни) и малыми коэффициентами распределения. Важно от-
метить также некоторое увеличение предела обнаружения мно-
гократной экстракции в сравнении с однократной экстракцией.
Это связано с тем что к началу проведения последней экстрак-
ции исходное количество вещества уменьшается на долю извле-
ченного в предыдущих.
В аналитической практике получило распространение урав-
нение, позволяющее рассчитать массу определяемого вещества
в исследуемом объекте М(_ по результатам измерения массы ве-
щества в равновесном газе на первой экстракции n/G и отноше-
ния площадей пиков на хроматограммах газовой фазы последу-
ющей (г 4- 1) и предыдущей (г) экстракций:
Мо =-----------—.
1-(A*G+1M’G)
Эта формула— прямое следствие уравнения (IV.51) — не тре-
бует знания соотношения объемов фаз системы и позволяет про-
водить анализ в случаях, когда измерение численного значения
369
г оказывается сложным или невозможным (например, анализ по-
лимерных пленок или кусочков ткани биологического матери-
ала).
Метод добавки определяемого вещества к исследуемому
объекту. Этот метод представляет собой разновидность мно-
гократной, точнее двухкратной (повторной), экстракции. Тех-
ника проведения анализа во многом аналогична варианту с вну-
тренним стандартом, но отличие состоит в том, что содержание
вещества в равновесном газе в виде площади пика на хромато-
грамме определяется два раза — до и после введения добавки.
Отбор для анализа пробы wG из равновесной системы с кон-
центрацией Cg, т. е. массы mv = vgCg, и добавление в систему
анализируемого вещества массой ms приводит к изменению кон-
центрации вещества в газовой фазе до значения CG . Расчет
результатов анализа производится по уравнению
0 _ ms - т, CG _ пт,а - mv AG ,TV
L “ Vl ' C'G - CG ~ VL ' A'g - Ag ’ ( ’
которое позволяет определить исходную концентрацию веще-
ства в растворе, не располагая данными по абсолютным зна-
чениям коэффициентов распределения. Для этого необходимо
кроме отношения площадей пиков на хроматограмме До и AG, а
также объема исследуемого раствора Vl точно измерить массы
введенного в систему и отобранного на анализ веществ. По-
следнее удобно производить по значению AG и предварительно
проведенной градуировке AG(C£).
Важным условием достаточно точного проведения анализа
является правильный выбор соотношения между исходным, вве-
денным и отобранным на анализ количеством вещества. Для
получения приемлемой точности масса введенного вещества ms
должна существенно превышать (хотя бы в несколько раз) ис-
ходное количество вещества Мо и отбираемое из системы rnv.
Преимущество способа в сравнении с вариантами ПФА, осно-
ванными на повторной экстракции, состоит в том, что он так
же, как и метод внутреннего стандарта, не накладывает огра-
ничений на численные значения коэффициентов распределения
анализируемых веществ и предел обнаружения в исследуемом
растворе остается таким же, как и в простейшем варианте —
однократной экстракции. Однако этот способ довольно тру-
доемок, так как включает операции, выполняемые и при абсо-
лютной градуировке, и при введении внутреннего стандарта.
Кроме того, если в методе абсолютной градуировки достаточно
обеспечить только постоянство дозируемого объема газа, то в
рассматриваемом случае надо знать абсолютную величину вве-
370
денной пробы. Этого все же можно избежать, если обеспечить
условие mv .
IV.3.5.4. Количественный ПФА с пневматическим
отбором проб (дискретная газовая экстракция
с неполной заменой газовой фазы)
Изложенные выше основные соотношения дискретной (по-
вторной и многократной) экстракции предусматривают полную
замену газовой фазы на чистый газ. Однако в ряде случаев (при
анализе вязких жидкостей и твердых объектов) полностью вы-
теснить из сосуда с объектом анализа газовую фазу и содер-
жащиеся в ней летучие вещества довольно сложно, а иногда и
невозможно. Более целесообразный путь проведения дискрет-
ной газовой экстракции — неполная замена равновесного газа
на чистый газ. Эта операция легко и с высокой точностью реа-
лизуется путем отбора из сосуда части газа, находящегося под
повышенным давлением, и может сочетаться с пневматическим
дозированием газа из сосуда с пробой в хроматограф [343].
Если из замкнутого объема воздуха или инертного газа, нахо-
дящегося при давлении р' и содержащего массу паров вещества
Mg, отобрать часть газа, то давление в этом объеме снизится до
значения р. Отобранная проба будет содержать долю (р' — р)/р'
от исходной массы Mg, т. е. масса отобранного вещества тп\
составит
mi = Мс(р' - р)/р', (IV.54)
а объем отобранной пробы vg (при давлении р') — эту же долю
от объема газа в сосуде
vg = Vg(p'-р)/р'. (IV.55)
В закрытой гетерогенной системе конденсированная фаза — газ,
содержащей массу Mg летучего вещества, последнее будет рас-
пределено в соответствии со значением коэффициента распре-
деления между жидкостью и газом. После установления равно-
весия в газовой фазе окажется масса Mg вещества, а его доля
от общего содержания вещества в системе будет равна
MG/MG = г/(К + г). (IV.56)
Подставив значение Mg из этого соотношения в уравнение
(IV.54), получим величину mi для случая отбора пробы газа
из гетерогенной системы конденсированная фаза — газ:
mi (IV.57)
К + г р
371
При п-m пневматическом дозировании пробы из одного флакона
отобранная масса составит
тп = Мо
К + г р/р'
К + г
р' ~Р
Р'
(IV.58)
К + г
Воспроизводимость пневматического дозирования равновес-
ного пара в хроматограф при использовании современных ус-
тройств может достигать десятых долей процента. Столь хо-
рошая воспроизводимость в зависимости от характера иссле-
дуемого объекта и его свойств достигается при использовании
любого из описанных выше методов количественного ПФА —
абсолютной градуировки, добавки анализируемого вещества
или метода внутреннего стандарта. Однако точное измере-
ние абсолютных давлений р' и р позволяет рассчитать долю и
массу отбираемого вещества из сосуда с пробой и, таким обра-
зом, открывает дополнительные возможности количественного
определения летучих примесей в системах с известными и не-
известными коэффициентами распределения, с концентрирова-
нием в адсорбционной или криогенной ловушке при однократ-
ном и многократном отборе из сосуда равновесного газа.
Если К и г известны, исходная масса вещества Мо может
быть вычислена по уравнению (IV.57). Для этого достаточно
провести одно дозирование равновесного газа из флакона с про-
бой и по площади пика на хроматограмме и предварительно
проведенной градуировке детектора измерить массу отобран-
ного вещества пц. Условия проведения анализа существенно
влияют на точность измерения Мо- Для снижения вклада по-
грешности измерения давлений р' и р их разность должна быть
возможно больше, а для того чтобы этот вклад не превышал
суммарной погрешности измерения р' и р, необходимо выполне-
ние условия р' 2р.
В случае количественного ПФА с неизвестными К тл г опре-
деление Мо возможно после проведения не менее двух последо-
вательных дозирований газа из одного сосуда с пробой и изме-
рения площадей пиков на хроматограмме Агс и А^)"1, пропорци-
ональных массам отобранного вещества при предыдущем im*G)
и последующем (т^1) введении пробы в хроматограф. Доля
извлеченного из сосуда вещества оказывается равной
m‘r+1 AJA1 г р' —р
тгс Агс К + г р' ’
(IV.59)
и уравнение (IV.57) преобразуется в (IV.52), позволяющее рас-
считать исходное количество вещества в системе по градуи-
ровочной характеристике детектора Ag(hig) и площади пиков
372
на хроматограммах двух последовательных дозирований рав-
новесного газа. Точность такого измерения Мо так же, как и в
случае дискретной газовой экстракции с полной заменой газо-
вой фазы, существенно зависит от значения отношения Ag-1 /А’с:
погрешность анализа тем больше, чем ближе численное значе-
ние этого отношения к единице. Поэтому условия анализа сле-
дует выбирать так, чтобы площадь пика на хроматограмме при
каждом последующем дозировании газа из одного сосуда умень-
шалась не менее чем на 15-20%.
Концентрирование примесей равновесного газа при пнев-
матическом отборе проб. Необходимость промежуточного на-
копления веществ, содержащихся в газовой фазе сосуда с про-
бой до введения в хроматографическую колонку, возникает в
случаях, когда прямое дозирование либо не обеспечивает до-
статочной чувствительности анализа, либо снижает эффектив-
ность разделения, как это имеет место при анализе с капил-
лярной колонкой. Расчеты анализов с однократным отбором
пробы и концентрированием не отличаются от описанных выше
случаев с известными и неизвестными К и г. Когда накопле-
ние примесей в концентраторе производится многократным от-
бором газа из сосуда с пробой, масса отобранного вещества за
п дозирований может быть вычислена по одной из следующих
формул:
= (IV.60)
или
п
£ тп = М0 [1 - (A’G+1/A’G)"] (IV.61)
1
Более точные результаты достигаются при анализе систем с
известными If и г и расчете по формуле (IV.60).
IV.3.5.5. Динамический парофазный анализ
Динамические варианты количественного ПФА базируются
на непрерывной газовой экстракции. В сравнении со статиче-
скими они требуют более сложного оборудования, а практи-
ческая их реализация накладывает более жесткие ограничения
на условия проведения анализа. Так, на обеспечение равно-
весности процесса накладываются ограничения в скорости га-
зового потока, степени распыления газа в жидкости. Возмож-
ность образования тумана или пены существенно усложняет
конструкцию используемого оборудования. Поэтому непрерыв-
ную газовую экстракцию следует применять в тех случаях, ко-
373
гда статические условия принципиально неприемлемы или не-
удобны для использования. К таким случаям относятся анализ
систем с неизвестными и большими коэффициентами распреде-
ления (> 400-500) и концентрирование примесей равновесного
газа для снижения предела обнаружения ПФА.
Сущность этих методов ПФА состоит в определении измене-
ния концентрации летучих веществ в газовой фазе над исследу-
емым раствором после пропускания через этот раствор объема
газа VG- Для наиболее распространенного случая извлечения
газовым потоком летучих примесей из растворов в нелетучем
растворителе исходная их концентрация С£ связана с текущим
значением (Сц), соответствующим объему пропущенного газа
Vg, уравнением
C£ = CLexp[Vg/(AVL)]. (IV.62)
Отношение С*£/Сь в этом уравнении можно заменить отноше-
нием соответствующих концентраций вещества в газовой фазе
C'g/C'q или, в идентичных условиях дозирования в хроматограф
проб равновесного газа, отношением площадей пиков на хро-
матограмме Aq/A'q. Тогда коэффициент распределения может
быть рассчитан по формуле
к W
ln(AG/A'G) ’
(IV.63)
Возможны три основных способа определения летучих при-
месей жидких и твердых тел динамическими вариантами ПФА.
1. Анализ равновесного газа над раствором, выполняемый в
статических условиях, до и после пропускания через него по-
тока газа известного объема.
2. Непосредственный анализ потока газа, прошедшего через
исследуемый раствор.
3. Анализ концентратов примесей.
В первом случае анализ реализуется в три стадии
(рис. IV.27): 1) определение в статических условиях концентра-
ции анализируемой примеси в равновесной газовой фазе (соот-
ветствующей концентрации вещества в растворе С'ь) по пред-
варительно проведенной градуировке (C'g(Ag)); 2) пропускание
через раствор с концентрацией вещества потока газа объема
Vg, при этом концентрация вещества снижается до С*ы; 3) опре-
деление в статических условиях концентрации С'ц по содержа-
нию вещества в равновесной газовой фазе в виде площади пика
Ag, пропорциональной концентрации C'G. Расчетное уравнение
374
Phc.IV.27. Схема распределения
вещества между жидкостью и га-
зом на всех стадиях парофазного
анализа с непрерывной газовой
экстракцией.
1
для определения летучих компонентов растворов в нелетучем
растворителе имеет вид
Cl = CG
Уъ/Уъ
Lln(AGM'G)
(IV.64)
Правильный выбор объемов продуваемого газа во многом
определяет успех проведения анализа, так как слишком малые
объемы приводят к небольшим изменениям концентрации веще-
ства в растворе и, следовательно, к малому различию между
значениями VG и ?1'g и в силу этого к большим погрешностям
анализа. Поэтому критерием обеспечения достаточного объема
газа, продуваемого через исследуемый раствор, может служить
отношение >1G/>1G , которое должно быть не меньше е (основа-
ние натурального логарифма).
Во втором способе, т. е. при анализе газового потока, про-
шедшего через исследуемый раствор, необходимо измерить зна-
чения площадей (или высот) пиков 71g и A'g , пропорциональ-
ных равновесным концентрациям C*G и CG в потоке газа, соот-
ветствующих двум различным объемам пропущенного газа Vg и
Vg (Vg < IV). Полученные данные позволяют рассчитать С£ по
формуле
Cl = CG
VLln(>lGM(3)
V^Ag/A'g)
ехр v-vg
(IV.65)
Так же, как и в предыдущем случае, важным условием успеш-
ного проведения анализа является правильный выбор оптималь-
ного объема пропускаемого газа IV, который должен удовлетво-
рять условию IV ХРц. Следует, однако, иметь в виду, что
чрезмерно увеличивать объем продуваемого газа нельзя, так
как это приведет к уменьшению площади пика A'G, увеличению
погрешности ее измерения и повышению предела обнаружения.
375
В третьем случае, при концентрировании примесей равновес-
ного газа, масса уловленного вещества mG, извлеченного из ис-
следуемого объекта пропусканием через него газа объемам Vg,
согласно (IV.62) определяется уравнением
mG = Мо [1 - ехр(—Vg/KVb)], (IV.66)
которое в системах с известными коэффициентами распределе-
ния позволяет рассчитать исходное содержание вещества в ана-
лизируемом объекте по результатам газохроматографического
измерения mG.
В системах с неизвестными К рассчитать М$ можно, если
после удаления вещества массой mG через тот же образец по-
вторно пропустить равную первой порцию газа Vg> которая из-
влечет уже меньшую массу вещества т'с. Измерив площадь
пика на хроматограмме A'G, пропорциональную массе mG, ис-
ходное количество вещества в образце можно рассчитать по
формуле
(IV-67)
аналогичной уравнению (IV.52).
IV.4. ХРОМАТОГРАФИЯ В ТОНКОМ СЛОЕ
IV.4.1. Общие сведения
Метод хроматографии в тонком слое, которую, в принципе,
можно рассматривать как “открытую колонку”, был открыт в
1938 г. (М. С. Шрайбер, Н. А. Измайлов) [7] и благодаря рабо-
там Э. Шталя по его стандартизации [14, 344, 345] занял одно из
ведущих мест в качественном и количественном анализе слож-
ных смесей химических соединений, фармацевтических препа-
ратов, биологических жидкостей и природных объектов [26-30,
346-356]. В 1964 г. вариант хроматографии в тонком слое при-
знан в качестве официального стандартного метода в фармако-
пеи.
Тонкослойную хроматографию (ТСХ) подразделяют на ана-
литическую и препаративную. С использованием ТСХ воз-
можно осуществлять разделение компонентов сложной смеси,
очистку с последующим препаративным выделением требуе-
мого соединения, идентификацию и количественный анализ.
Хроматографическое разделение анализируемых веществ
основано на различии в их скоростях миграции. Миграционной
376
средой в плоскостной хроматографии для разделяемых веществ
служит сорбент, имеющий форму листа или пленки. Разновид-
ностью метода ТСХ является бумажная хроматография (БХ)
[11], исторически возникшая раньше тонкослойной и характе-
ризуемая специфическими особенностями бумаги как носителя
стационарной фазы.
Особенности ТСХ в сравнении с другими методами хрома-
тографического анализа заключаются в следующем:
— это фактически единственный метод, позволяющий прово-
дить полный анализ неизвестной смеси, поскольку исследова-
тель может выяснить, остались ли на старте неэлюированные
компоненты;
— выбор возможных подвижных фаз в режиме ТСХ суще-
ственно больше, чем в колоночной хроматографии;
— требования к очистке образца менее строгие;
— по производительности метод ТСХ превосходит газовую
и высокоэффективную жидкостную хроматографию, по крайней
мере, на порядок,
— метод имеет широкие возможности для детектирования
любых компонентов, в том числе содержащих радиоактивные
изотопы [357];
— характеризуется малым временем анализа;
— этот вид хроматографии методически прост и сравни-
тельно дешев.
Дополнительным преимуществом ТСХ является то обстоя-
тельство, что можно анализировать одновременно несколько
различных образцов. Это, в свою очередь, экономит время и
позволяет в рамках одного эксперимента сравнивать свойства
различных объектов.
В первоначальном варианте, называемом хроматографией с
незакрепленным слоем, порошок сорбента просто наносили на
горизонтальную поверхность стеклянной пластинки. Развитие
ТСХ ускорилось с 1966 г. с появлением ТСХ-пластинок с пред-
варительно нанесенными (закрепленными) слоями. Высокораз-
решающие пластины стали использоваться в тонкослойной хро-
матографии начиная с 1973 г., когда диаметр частиц сорбента
уменьшился до 11 мкм. Следующий этап в эволюции этого ме-
тода произошел в 1975 г. с введением высокоэффективной тонко-
слойной хроматографии (ВЭТСХ) [29, 30, 349, 358, 359]. Позднее
возник метод проточной ТСХ [360].
Начатый в 70-х годах фирмой Merck серийный выпуск высоко-
эффективных пластинок, а также разработанные фирмой Camag
приборы для многократного проявления пластин позволили уве-
личить эффективность хроматографического разделения в тон-
377
ком слое: на одной пластине стало возможным проведение ана-
лиза десятков и сотен веществ [30].
Работы Р. Кайзера [361, 362], посвященные количественным
методам в ТСХ, и Г. Йорка [363, 364] в области денситометри-
рования ТСХ-хроматограмм приблизили этот метод к ВЭЖХ,
сохранив все преимущества хроматографии в тонком слое. В
1977 г. Р. Кайзер для характеристики эффективности разделе-
ния в тонком слое вводит термин “separation number”. Для высо-
коэффективной тонкослойной хроматографии эта величина со-
ставляет ~10-20, для ВЭЖХ — 20-40. При этом было показано,
что для двумерной ТСХ ( см. ниже) значение этого параметра
может достигать 100-150.
С внедрением высокоэффективной тонкослойной хроматогра-
фии (High-pressure planar liquid chromatography (HPPLC) — тон-
кослойная хроматография высокого давления, High-performance
thin layer chromatography (HPTLC) — высокоэффективная хрома-
тография в тонком слое) говорят о ренессансе этого метода ана-
лиза [365-369]. К настоящему времени БХ и ТСХ практически
утратили свои позиции в анализе неорганических веществ, но
по-прежнему широко применяются при определении органиче-
ских соединений [11]. Использование приемов хроматографии
в тонком слое составляет в фармацевтике 30%, в биохимии и
клинической химии 25%, в анализе пищевых продуктов, парфю-
мерных материалов 10%, исследовании неорганических веществ
5%, в других областях 15% [28, 29, 349].
Для более строгой качественной и количественной интерпре-
тации получаемых результатов хроматография в тонком слоена
современном этапе используется в сочетании с другими анали-
тическими методами: ТСХ—УФ-спектрометрия, ТСХ — масс-
спектрометрия [370-373] и т. д. Тем самым резко снизилось коли-
чество рутинной работы, исключены основные источники оши-
бок, повысилась производительность. При этом аналитик вы-
свобождается для выполнения более важных задач: оценки ре-
зультатов, рассмотрения их достоверности, осуществления кон-
трольных функций.
Как и в других хроматографических методах анализа, ТСХ
включает следующие необходимые этапы:
— пробоподготовку;
— выбор хроматографической системы (подготовку подвиж-
ной фазы, поиск соответствующего сорбента);
— развитие ( или проявление ) хроматограммы ( перенос ве-
щества по слою сорбента, осуществляемый подвижной фазой в
проявительной камере);
— детектирование разделенных компонентов;
378
— качественный и количественный анализ.
Для подготовки пробы при проведении ТСХ-анализа, вклю-
чающей извлечение анализируемых компонентов из исследуе-
мого объекта, находят применение двухфазные слои, которые в
виде готовых пластинок поставляются различными фирмами.
Анализируемые растворы наносят на слой предварительного
концентрирования, а сам хроматографический процесс прохо-
дит на основном слое [346]. При этом необходимо обеспечить
такие условия, чтобы мешающие компоненты не определялись
наряду с анализируемыми веществами. Для этого проводят
предварительную (первичную) очистку пробы (дистилляцию,
экстракцию, сублимацию, осаждение), включающую различные
приемы концентрирования. Следует отметить, что требования
к очистке образца в тонкослойной хроматографии менее жест-
кие, чем в других хроматографических методах. Полученный
образец содержит меньшее число компонентов, но в процессе
концентрирования вследствие близких физико-химических ха-
рактеристик вместе с интересующими исследователя соедине-
ниями могут содержаться и посторонние вещества.
Конечными процедурами очистки и подготовки пробы явля-
ются обычно электрофорез и (или) хроматографическое раз-
деление. Если матрица липофильная, адсорбционная очистка
может быть проведена на силикагеле, оксиде алюминия, фло-
рисиле, полиамидном сорбенте. Липофильную матрицу элюи-
руют неполярным растворителем, при этом анализируемое ве-
щество удерживается в концентрированной форме на колонке и
элюируется позднее. Если далее необходимо провести фрак-
ционирование, то элюенты используют в соответствии с элюо-
тропными рядами, проводя таким образом групповое разде-
ление. Если матрица гидрофильная, все липофильные компо-
ненты, присутствующие в водной матрице, поглощаются липо-
фильной фазой. Для селективной очистки разработаны фазы,
обладающие наряду с липофильными и ионообменными свой-
ствами. Такие сорбенты известны под названием polysphere R.P-
18САТ [30, 374].
Эффективной оказалась комбинация ТСХ с твердофазной
экстракцией, что позволило автоматизировать процедуру про-
боподготовки и повысить чувствительность самого анализа.
Весьма перспективными признаны попытки прямого сочетания
ТСХ со стадией пробоподготовки в режиме on-line. Соответ-
ствующее оборудование выпускается фирмами Tecan, Zymark,
Merck.
379
IV.4.2. Основы метода ТСХ
ТСХ — наиболее простой, экспрессный и универсальный
хроматографический метод разделения, использующий недоро-
гое оборудование. Улучшения разделения компонентов ана-
лизируемой смеси можно добиться, модифицируя состав непо-
движной фазы введением градиентного элюирования, увеличе-
нием числа проявлений, снижением температуры.
В методе ТСХ используются стеклянные, металлические,
пластмассовые пластины, покрытые тонким слоем силикагеля,
оксида алюминия или других сорбентов (100-300 мкм) (см. ни-
же). Если проявленная хроматограмма должна быть подверг-
нута затем воздействию температур выше 150°C, используют
боросиликатные стекла (пластмасса выдерживает температуру
не выше 130°C). Применяют также пластинки, полученные пу-
тем спекания сорбента на поверхности стекла. Преимущество
таких пластин заключается в том, что после соответствуюшей
регенерации их можно использовать повторно. Пластинки на
основе стекловолокна имеют торговую марку “Instant thin-layer
chromatography” (ITLC) [80].
Выбор неподвижной фазы. Одним из преимуществ тонко-
слойной хроматографии является то, что она позволяет исполь-
зовать довольно большое число сорбентов [375].
В качестве неподвижной фазы чаще всего применяется сили-
кагель — гидрофильный полярный сорбент с активными по-
верхностными ОН-группами, действующими как доноры прото-
- Si — О — Si —
нов, и с силоксановыми фрагментами \ / , облада-
о
ющими протоноакцепторными свойствами. Наиболее известны
силикагели марок Н, HF 254, HR (Merck). HF 254 содержит неор-
ганический флуоресцентный индикатор. При облучении светом
длиной волны 254 нм для веществ, поглощающих в УФ, появля-
ются розовато-лиловые пятна на зеленом фоне. HR — адсорбент
высокой чистоты — применяется для препаративных целей.
Реже в качестве неподвижной фазы используется оксид
алюминия (у-модификация), который получают дегидрата-
цией соответствующего гидроксида при 500 ° С . Его протоно-
акцепторные свойства выражены гораздо сильнее, чем у сили-
кагеля, за счет присутствия ионов О2-; ионы А13+ проявляют
при удерживании веществ основный характер. На слоях оксида
алюминия возможно проводить хроматографический анализ ки-
слот, предельных и непредельных алифатических спиртов, фе-
нолов, аминопроизводных.
380
В 1972 г. впервые появились силанизированные хрома-
тографические фазы, а с 1978 г. начинается серийный выпуск
обращенно-фазных (ОФ) пластин. Покрытые химически моди-
фицированным силикагелем, они расширяют диапазон селек-
тивности. Лля режима ОФ ТСХ применяют сорбенты С2 — Cg,
в качестве алкильных заместителей — диметил- (RP2), этил-,
октил- (RP8) или октадецильные группы. Гидрофобность слоев
возрастает с увеличением длины цепи алкильного заместителя.
Были разработаны амино-, циано- и диоловые фазы: (-СН2-
CH2-CH2-NH2; -CH2-CH2-CH2-C=N; -СН2-СН2-СН2-О-СН2-
СН(ОН)-СН2(ОН)).
Аминофазы обладают основными свойствами, поэтому воз-
можен анализ аминов, алкалоидов. Кроме того, они могут быть
использованы как слабые анионообменники: в кислой среде
аминогруппы находятся в протонированной форме, и вещества,
заряженные отрицательно, на таком сорбенте удерживаются
сильнее. Таким образом, существовавший разрыв в селектив-
ности на немодифицированных фазах и гидрофобных сорбентах
был заполнен новыми умеренно полярными сорбентами.
Используемая в качестве сорбента целлюлоза (а также
сорбенты на ее основе — триацетатцеллюлоза, циклодекстрины
и их производные) применяется для разделения энантиомеров и
различных гидрофильных соединений (сахара, аминокислоты,
растворимые неорганические ионы, нуклеиновые кислоты).
В тех случаях, когда хроматографическим материалом явля-
ются полисахариды ( бумага, декстрановый гель и т.д.), роль
неподвижной фазы в действительности выполняет вода , и та-
ким образом реализуется принцип распределительной хромато-
графии. Иногда в ТСХ используют материалы, адсорбционные
свойства которых меняются определенным образом в направле-
нии от одного края пластины к другому (градиентные слои).
В настоящее время для хроматографии в тонком слое раз-
работаны некоторые химически модифицированные целлюлозы
в качестве ионообменников [30]: для анионного обмена — АЕ
(аминоэтил), ЛЕАЕ (диэтиламиноэтил), PEL (полиэтиленимин);
для катионного обмена — СМ (карбоксиметил), Р (с введе-
нием фосфора), poly-P (полифосфат); хиральные лигандообмен-
ные фазы.
Флорисил — силикат магния — имеет полярность, проме-
жуточную между силикагелем и оксидом алюминия. Этот сор-
бент особенно подходит при хроматографическом анализе са-
харов, эфиров, спиртов, гликолей.
Часто пластины готовятся с предадсорбентом. Диаметр ча-
стиц слоя предадсорбента несколько больше, либо по своей
381
природе он отличается от основного сорбента. Использование
предадсорбента уменьшает размывание на старте анализируе-
мых компонентов, позволяя провести хроматографический ана-
лиз более эффективно.
Диаметр частиц сорбента в классической ТСХ составляет
от 11 до 15 мкм, в ВЭТСХ — 5-7 мкм; объем пор сорбента 0,8—
1,2 мл/г; толщина слоя 250 мкм, а в ВЭТСХ — 5-7 мкм. Тол-
щина слоя влияет на количество вещества, которое может быть
подвергнуто разделению, а кроме того, определяет время раз-
деления: чем меньше количество исследуемого вещества, тем
тоньше должен быть слой, так как концентрация на единицу пло-
щади достаточно высока и это увеличивает чувствительность
детектирования. В предложенных недавно В. Г. Березкиным и
сотр. новых вариантах реализации микротонкослойной хрома-
тографии используют частицы сорбентов диаметром 2-5 мкм и
толщину сорбционного слоя до 50-70 мкм [376].
Напротив, в препаративной ТСХ толщина слоя сорбента до-
стигает 0,5-2 мм (500—2000 мкм). Толстые слои адсорбируют
большое количество подвижной фазы, и при необходимости мно-
гократного проявления требуется долгая осушка. Серийно вы-
пускаются пластины со слоями 500, 1000, 1500, 2000 мкм. Разра-
ботаны уникальные пластины для препаративной хроматогра-
фии: линейные (фирма Ватман), имеющие полосу предадсор-
бента силикагеля или С8; конусные (фирма Analtech). Сорбент
содержит слои частиц различного диаметра — от 30 мкм у осно-
вания пластины до 1700 мкм у вершины; слой предадсорбента —
700 мкм, скорость передвижения подвижной фазы максимальная
у основания. На такие пластины можно нанести ~5 мг образца
из его 2 - 100%-ного раствора. Аналитическое разделение осу-
ществляетсяна слоях 0,25 мм, препаративное — на слоях 0,50 мм
для образца ~20 мг и 1,00 мм — для ~100 мг.
В качестве связующих при изготовлении пластин с закре-
пленным слоем используют гипс (12-15%), крахмал (3-10%), по-
лиакриламид и полиакриловый эфир (2-10%). Связующий ма-
териал не должен реагировать с анализируемыми вещества-
ми, мешать детектированию, разбухать и сжиматься под дей-
ствием подвижной фазы, не должен растворяться в подвижной
фазе и перемещаться по хроматографической пластинке, погло-
щать свет или флуоресцировать. Молекулы многих неокрашен-
ных веществ обладают свойством поглощать электромагнитное
излучение. Если это поглощение имеет место при А, равной
254 или 366 нм, возможно детектирование на “флуоресцирую-
щих слоях”. При производстве таких пластин (F366 или UV366)
к сорбенту добавляется флуоресцентный индикатор, например
382
пирен, цианиновый краситель, морин или флуоресцеин. Флу-
оресцентный индикатор не должен вступать в химическое вза-
имодействие с анализируемыми веществами, сорбентом и ком-
понентами подвижной фазы. В качестве неорганических флу-
оресцентных добавок используют силикат цинка, активирован-
ный магнием, сульфид цинка, которые возбуждаются до желто-
зеленого свечения коротковолновым УФ-излучением (F254 или
UV25-})- На участках слоя, где присутствуют вещества, по-
глощающие излучение, анергия возбуждения фосфоресценции
меньше, чем в оставшихся частях.
По технологии ИВС РАН (Б. Г. Беленький, Э. С. Панкина)
в России выпускаются высокоэффективные и аналитические
ТСХ-пластинки на лавсане и стекле, а также на алюминиевой
фольге с использованием силиказолевого связующего (перехо-
дящего при нагревании в силикагель).
Возрождение интереса к ТСХ произошло с появлением в
продаже анализатора системы “latroscan” (Япония). Кварце-
вые, медные или стальные стержни диаметром 0,9 мм и длиной
150 мм (“Chromarod”) покрыты слоем силикагеля толщиной 75
мкм. Анализируемую смесь наносят на слой сорбента шприцем
и элюируют, как в обычной ТСХ. После испарения подвиж-
ной фазы стержни медленно с постоянной скоростью вводят в
пламя ПИД. Каждый стержень можно использовать около 100
раз. Они легко регенерируются. Воспроизводимость результа-
тов анализа составляет 1,9%.
В нашей стране налажен выпуск пластин “Силицел” для бу-
мажной ТСХ. Бумагу изготавливают из целлюлозы и силика-
геля (50 мкм). Сорбент имеет нейтральную реакцию (pH 7-7,5).
Нанесение пробы на неподвижную фазу. Если разделе-
ние проводят в аналитических целях, образцы наносят из 0,1-
1%-ных растворов в виде пятен ( стартовые пятна); объем дозы
~2—10 мкл. Если же разделение проводится в препаративных
целях, анализируемые образцы наносят вдоль стартовой линии
в виде полосок, параллельных краю пластины (рис.1У.28). Для
этого обычно используют либо пластинки с концентрирующими
зонами, либо специальные аппликаторы. ВЭТСХ-пластинки с
концентрирующими зонами изготавливаются из двух слоев си-
ликагеля, которые имеют различные свойства. Этот вариант
предпочтителен, когда анализируются пробы большого объ-
ема или непосредственно на пластинку наносятся неочищенные
образцы.
Для нанесения на ВЭТСХ-пластинки вязких проб исполь-
зуют специальное устройство, переносчиком проб в котором
383
Рис. IV.28. Препаративное ТСХ-разделение.
служит фторированная полиэтиленпропиленовая пленка, натя-
нутая над рядом углублений в металлической подставке
Нанесение пробы вещества на неподвижную фазу до насто-
ящего времени все еще является наиболее критической и дли-
тельной по времени процедурой. Анализируемый образец вно-
сят на поверхность слоя сорбента в виде пятна или полосы на
небольшом расстоянии от нижнего края пластинки при линей-
ной хроматографии, либо по окружности на периферии пла-
стинки при антикруговой [366] хроматографии. Стартовые зоны
должны быть минимальны по размерам: диаметр нанесенного
пятна ~2-4 мм; для слоев сорбентов, используемых в ВЭТСХ,
оптимальный размер пятен составляет около 1,0 мм. Размер
стартового пятна сильно зависит от выбранного растворителя.
Центры пятен должны отстоять друг от друга на расстоянии
~10-15 мм Слой сорбента не должен повреждаться при нане-
сении пробы. Используемый растворитель должен иметь мини-
мальную элюирующую силу (неполярные растворители умень-
шают размывание пятна в точке нанесения образца), быть низ-
кокипящим, дешевым, нереакционноспособным. Кроме того, он
должен хорошо растворять пробу, смачивать слой сорбента и
быть достаточно летучим, чтобы его можно было легко уда-
лить с пластины по окончании элюирования. Сорбенты с хими-
чески связанными слоями плохо смачиваются некоторыми рас-
творителями, что в значительной степени препятствует проник-
новению пробы в слой. Это особенно проявляется при работе
с обращенно-фазными сорбентами и анализируемыми пробами,
растворенными в водно-органических системах В таких слу-
чаях в качестве растворителей рекомендованы метанол, ацетон,
ацетонитрил, хлористый метилен.
384
Для нанесения больших объемов используют шприцы емко-
стью до 500 мкл.
Главным источником ошибок в количественном анализе в ре-
жиме ТСХ является воспроизводимость нанесения постоянного
объема раствора на слой сорбента. При нанесении 1 мкл рас-
твора стандартное отклонение достигает ~10%.
Выбор подвижной фазы. При выборе подвижной фазы руко-
водствуются следующими параметрами: низкая стоимость, чи-
стота, низкая температура кипения, инертность по отношению к
разделяемым компонентам и стационарной фазе, нетоксичность,
инертность по отношению к неподвижной фазе и анализируемым
веществам [29, 30, 349].
При разделении сложных смесей редко используют в ка-
честве подвижной фазы индивидуальный растворитель. При
этом компонентный состав подвижной фазы должен быть мак-
симально прост и индивидуальные компоненты подвижной фазы
не должны сильно различаться по полярности во избежание ее
расслоения. Элюирующая сила смеси может быть оценена с
учетом элюирующей силы и объемной доли (9?) индивидуаль-
ных компонентов [250].
Существенное значение в открытой хроматографической си-
стеме имеет давление паров компонентов растворителя. Испа-
рение более летучих компонентов подвижной фазы может из-
менить ее состав до начала хроматографического разделения.
При этом и свойства неподвижной фазы могут слегка меняться в
процессе нахождения пластины внутри проявительной камеры,
поскольку слой сорбента, контактирующий с паровой фазой,
сорбирует из нее молекулы растворителей.
Развитие (или проявление) хроматограммы. Пластины с
нанесенными образцами помещают в проявительную камеру с
жидкой фазой, которая предварительно в течение 5-10 мин на-
сыщается парами подвижной фазы. Элюент перемещается по
слою сорбента под действием капиллярных сил, увлекая за со-
бой анализируемые компоненты.
Скорость элюента зависит от типа и размера частиц сор-
бента, геометрических параметров проявительной камеры, от
пройденного расстояния. По мере увеличения расстояния,
пройденного элюентом, скорость перемещения потока уменьша-
ется, что, в конечном итоге, приводит к ухудшению разделения.
Для обычных пластин ТСХ разделительное рабочее расстоя-
ние не должно превышать 10 см, для ВЭТСХ — 5 см. Кроме
того, силы адгезии между сорбентом и подвижной фазой зави-
сят от вязкости подвижной фазы и поверхностного натяжения.
385
Скорость перемещения элюента мала, если мало поверхностное
натяжение и велика вязкость.
Если скорость подвижной фазы задается внешними устрой-
ствами, как в ТСХ с принудительным потоком, то ограничения,
накладываемые капиллярными взаимодействиями на передви-
жение элюента, снимаются, и длину пробега и скорость подвиж-
ной фазы можно оптимизировать независимо от других экспери-
ментальных условий. Лля этого используют специальные про-
явительные камеры, в которых слой сорбента зажимается ме-
жду нижней стеклянной пластинкой и непроницаемой мембра-
ной, приведенной в тесный контакт со слоем сорбента при по-
мощи гидравлического давления [347, 348]. Различия в нахожде-
нии пластины внутри проявительной камеры могут также ска-
заться на параметрах удерживания, поскольку слой сорбента,
контактирующий с паровой фазой, сорбирует молекулы из па-
ров подвижной фазы.
С уменьшением свободного пространства над слоем сорбента
возрастает скорость элюирования, а картина разделения стано-
вится более четкой и воспроизводимой. Этого можно достичь в
хроматографических камерах специальной конструкции (напри-
мер, в “сэндвич’-камерах или камерах типа Бреннера — Нидер-
визера). Использование “сэндвич’-камер требует меньшего рас-
хода элюента. Новый тип пластин с закрытым слоем сорбента
и простое устройство для реализации ТСХ с принудительным
движением подвижной фазы описаны в [377-379].
Известен также метод ТСХ с программированным измене-
нием состава паров подвижной фазы вдоль поверхности слоя
сорбента.
IV.4.3. Методы проявления хроматограмм
Можно выделить несколько способов проявления хромато-
грамм в ТСХ.
1. Нисходящее проявление (подвижная фаза подается к
верхнему краю вертикально расположенной пластины)
(рис.1У.29, а).
2. Круговое проявление (подвижная фаза медленно вносится
в центр пятна образца, помещенного в середину горизонтально
расположенной пластины). Элюирование происходит от цен-
тра к периферии слоя сорбента (рис.1У.29, в). Проявление осу-
ществляется в виде концентрических окружностей, расположен-
ных на различном расстоянии от центрального пятна. Скорость
386
Pnc.IV.29. Типы пластин для ТСХ высокого давления в зави-
симости от движения элюента.
а — одномерная; 6 — двумерная (элюент движется от центра в
противоположных направлениях); в — круговая; г — двумерная (по-
вторное проявление хроматограммы под углом 90°); д — многослойные
системы.
миграции разделенных зон можно увеличить, вращая пластинку
в горизонтальной плоскости. При антикруговом элюировании
пробу наносят по окружности внешнего круга вблизи краев пла-
стинки, а элюирование осуществляется по направлению к цен-
тру. Уникальной особенностью антикруговой хроматографии
являются высокая скорость и возможность использования боль-
ших нагрузок.
3. Горизонтальное проявление. Пластинку помещают на ме-
таллический блок, покрывают покровной пластиной, предот-
вращающей испарение подвижной фазы из слоя. Растворитель
из склянки Дрекселя через тефлоновую трубку подается в ло-
ток, а затем через бумажный фитиль — на пластину.
4. Многократное проявление. В одном и том же раствори-
теле проявление осуществляют несколько раз (в одном и том
же направлении), между проявлениями пластину высушивают
на воздухе в течение 5—10 мин.
5. Ступенчатое проявление. Применяется для разделения
смесей различной полярности.
387
6. Градиентное проявление. В процессе проявления меняется
состав подвижной фазы; возможно программирование активно-
сти адсорбента по длине пластины (полизональная ТСХ).
7. .Двумерное проявление (для исследования сложных сме-
сей). Образец наносят на один угол пластины (рис. IV.29, г),
смесь проявляют на всю длину обычным способом. Затем
пластинку поворачивают на 90 °и опять проявляют. Для того
чтобы получить существенное увеличение разрешения, необхо-
димо подобрать условия таким образом, чтобы селективность
подвижной фазы была различной при элюировании в обоих на-
правлениях. При этом также можно изменять природу непо-
движной фазы: для этого неиспользованную часть адсорбента
удаляют и покрывают пластину повторно другим адсорбентом.
Так в пределах одного эксперимента удается разделить боль-
шое число компонентов [380]. Высокоэффективная ТСХ эквива-
лентна по числу возможных анализируемых веществ хромато-
графической колонке эффективностью до 50 000 теоретических
тарелок.
Для определения следовых количеств и при анализе слож-
ных смесей используется методика автоматизированного много-
кратного хроматографического проявления (АМХП). Все ста-
дии анализа управляются и контролируются при помощи ком-
пьютера.
Образец и стандартные вещества наносят на ТСХ-пластины,
используя специальные приборы для нанесения проб
(рис.IV.30). При этом предварительно задаются следующие
параметры: объем пробы, скорость нанесения анализируемого
образца, протяженность полосы, разделительный интервал ме-
жду полосами, порядок расположения индивидуальных хрома-
тографических треков.
Приготовленные пластины для ВЭТСХ размещают в камере
прибора АМХП. Затем проводят откачку камеры для удале-
ния следов растворителей, остающихся после нанесения иссле-
дуемых проб. Камеру заполняют газообразным азотом и зака-
чивают насосом первую подвижную фазу. Если для выполне-
ния анализа требуется многокомпонентная подвижная фаза, ее
предварительно готовят в специальной камере смешения. После
прогона первого растворителя (примерно через 1 мин) подвиж-
ную фазу удаляют с помощью высокого вакуума и пластину
высушивают. Камеру прибора вновь заполняют газообразным
азотом и осуществляют второй прогон подвижной фазы, кото-
рый по времени длится несколько больше, чем первый. Во время
каждого последующего прогона элюента расстояние, пройден-
ное фронтом растворителя, увеличивается на 1-3 мм.
388
Рис.IV.30. Устройство для автоматизированного нанесения
образца на стартовую линию (фирма Camag).
Блок АМХП объединяет преимущества ступенчатого и мно-
гократного хроматографического проявления с сохранением
преимуществ, которые дает градиентное элюирование. При
этом, в отличие от градиентной колоночной хроматографии,
при работе блока АМХП подвижная фаза с максимальной по-
лярностью используется в начале работы, на старте. Выбор
такого градиента растворителей (от более полярного к менее
полярному) приводит к тому, что полярные вещества остаются
на старте, а неполярные — перемещаются с фронтом раствори-
теля [381] (рис.1У.31).
АМХП компенсирует размывание хроматографических зон
за счет концентрирования веществ в каждом прогоне подвиж-
ной фазы, улучшает разрешение трудноразделяемых компонен-
тов за счет оптимизации селективности растворителя. Много-
кратное элюирование является важным методом для анализа
389
DPN CMP
* f AMP
CD£Gaump
PAG* •
W^ADGP^ IMP+TPN
UDPG
GDPM
GMP
CDP
_ ADP
UDPGA
II.
UDP |
/DP ।
• GDP
• CTP
UTP*'A™
' •tITPGTP
.flnmrfWwrYnni' n * -J
Рис. IV.31. Разделение смеси мононуклеотидов на целлюлозе
с использованием двумерной тонкослойной хроматографии.
смесей, содержащих вещества с очень различающейся поляр-
ностью, для полного разделения которых не существует какой-
нибудь единой системы растворителей. Каждый шаг процесса
разделения можно оптимизировать независимо, при этом коли-
чественные измерения могут быть выполнены на разных шагах
элюирования.
Для обработки хроматограмм используют сканирующие ус-
тройства, также управляемые ЭВМ. Программное обеспечение
“CATS” поставляется фирмой Camag. Оптические измерения
проводят в режиме поглощения света и диффузного отражения
или флуоресценции. Возможен вариант с подавлением флуо-
ресценции. Используемый спектральный диапазон — от 200 до
800 нм.
IV.4.4. Детектирование разделенных компонентов
По окончании хроматографического разделения возможны
различные способы идентификации и определения концентра-
ции компонентов анализируемой смеси: от непосредственного
визуального их обнаружения (как с использованием процедуры
предварительной обработки соответствующим химическим ре-
агентом, так и без нее), до точных количественных измерений с
помощью сканирующих денситометров [363, 364, 382-384].
390
Окрашенные вещества обнаруживаются визуально.
При проведении полуколичественных измерений обученный
аналитик может делать оценку с точностью до 10%. С по-
явлением вариантов, гарантирующих достоверность результа-
тов анализа, этот метод детектирования стал использоваться
гораздо реже.
Весьма распространенным методом детектирования является
опрыскивание пластин окрашивающим реагентом, т. е. исполь-
зование химических реакций, которые, в принципе, могут про-
водиться как до, так и после хроматографирования [363, 364,
375]. Некоторые из этих реакций идут на холоду, чаще — тре-
буется нагревание. Обработка хроматограмм химическим реа-
гентом приводит к образованию окрашенных или флуоресциру-
ющих зон, позволяющих обнаружить разделенные компоненты
и количественно их оценить. Основное условие при этом, чтобы
интенсивность окраски или флуоресценции оставалась стабиль-
ной в течение ~30 мин.
Описано и практикуется также добавление реагентов в по-
движную или неподвижную фазу (например, использование пла-
стин на основе силикагеля, предварительно обработанного 4%-
ным раствором серной кислоты в метаноле). Поскольку все ре-
акции протекают при высокой температуре быстрее, чем при
низкой, хроматограмму после обработки химическим реагентом
обычно 5-10 мин выдерживают при температуре 100-120°С.
Детектирование может быть универсальным или селектив-
ным.
Для первой оценки достигнутого разделения смеси неизвест-
ного состава обычно применяют универсальные реагенты. Наи-
большее распространение среди них получили иод (пары иода
растворяются в большинстве органических соединений), кон-
центрированная серная кислота (при этом нельзя использовать
в качестве неподвижной фазы оксид алюминия вследствие про-
текания химической реакции), бихромат натрия в серной ки-
слоте (после опрыскивания свободной от растворителя пла-
стины ее недолго нагревают). Далее возможно использование
серии реагентов, специфических для определенных групп соеди-
нений. Информация об индивидуальном веществе складывается
из совокупности результатов различных методов, подтвержда-
ющих присутствие данного вещества. В последнее время появи-
лось немало публикаций об использовании ферментативных ме-
тодов детектирования: для обнаружения биологически актив-
ных веществ используют чувствительные к этим веществам ми-
кроорганизмы (биоавтография).
391
В случае неизвестных образцов детектирование начинается с
использования физических методов, так как они не разрушают
определяемое вещество и позволяют применять независимые ва-
рианты детектирования. Они обычно включают поглощение
или испускание электромагнитного излучения, регистрируемое
фотоумножителем.
После введения Г. Йорком прямой спектрометрической оцен-
ки тонкослойных хроматограмм в видимом и УФ-свете был на-
лажен серийный выпуск хроматографических спектрофотоме-
тров, позволяющих определять флуоресцирующие и поглощаю-
щие ультрафиолетовый свет вещества непосредственно на пла-
стинке в диапазоне длин волн от 200 до 800 нм. Стали возмож-
ными регистрация спектров поглощения разделенных соедине-
ний с хроматограммы на пластинке и сопоставление получен-
ных результатов со спектрами эталонных веществ. Это также
применимо и для in situ спектров ИК [370], рамановских [372] и
масс-спектров [371] (см. также обзор [379]).
Для обнаружения радиоактивных веществ используют ме-
тод радиоавтографии. На фотографической пленке., прижатой
эмульсионным слоем к поверхности хроматограммы, экспони-
руются участки, соответствующие радиоактивным зонам. Об-
работав такую хроматограмму, получают фактически точную
копию хроматограммы, на которой радиоактивным зонам соот-
ветствуют темные пятна. Для детектирования долгоживущих
изотопов могут наряду с авторадиографией использоваться де-
тектирование с помощью сцинцилляционной камеры, различные
сканирующие методы.
Эмпирически предел детектирования определяется путем на-
несения на ТСХ-пластину ряда проб анализируемого вещества
с переменной уменьшающейся концентрацией. Отношение сиг-
нал/шум определяют после проведения сканирования. Обычно
считается, что предел детектирования достигнут, если это от-
ношение равно 3, т. е. высота пика в 3 раза превосходит ампли-
туду шума нулевой линии.
Основным параметром, используемым для определения по-
ложения пятна на ТСХ-хроматограмме, является коэффициент
запаздывания (иногда встречается старое название — параметр
удерживания) Rj — отношение расстояния от стартовой линии
до центра пятна к расстоянию, пройденному растворителем от
стартовой линии до фронта [170]. Для многократного проявле-
ния этот параметр рассчитывают по формуле
nRf =
где ^Rj — параметр удерживания при первом проявлении; п —
число проявлений. Значения Rj линейно не соотносятся ни с
392
факторами, влияющими на хроматографический процесс, ни со
структурой разделяемых соединений, но для логарифмической
функции от Rf эта линейность наблюдается.
Фактор удерживания k и величина Rf связаны следующим
соотношением [385, 386]:
fc = (1 - Rt)/Rf.
На воспроизводимость Rf оказывают влияние многие пара-
метры: качество сорбента (с учетом стандартизации адсор-
бента с известной активностью), толщина слоя, размер образца
(Tfy не зависит от количества образца, если только не пре-
вышена линейная емкость), температура (увеличение темпера-
туры окружающего воздуха на 10 °C обусловливает небольшое
возрастание Rf за счет испарения растворителя), природа элю-
ента и скорость подвижной фазы (в случае восходящего про-
явления на величину Rf оказывает влияние наклон пластины),
тип проявительной камеры, насыщение камеры парами раство-
рителя, объем вводимой дозы и размер начального пятна, рас-
стояние между начальными зонами, правильное определение
фронта растворителя. Элюент надо выбирать таким образом,
чтобы значение Rf находилось в интервале от 0,3 до 0,7. Для
очень полярных образцов, способных к ионизации, например
аминов, кислот, в подвижную фазу добавляют уксусную ки-
слоту или аммиак.
Более удерживаемые компоненты (с низким значением Rf)
определяются в режиме ТСХ с большей чувствительностью,
чем в ВЭЖХ.
Разрешение R двух хроматографических зон определяется
выражением
R_ d
(wi + w2)/2’
где d — расстояние между центрами зон, w — ширина соответ-
ствующей зоны. Теоретически невозможно разделить вещества
с различием в значениях Rf < 0,05.
Наряду с Rf используется величина Rx — отношение рассто-
яния, пройденного растворенным веществом от стартовой ли-
нии до центра пятна, к расстоянию, пройденному стандартом
от стартовой линии до центра пятна.
Поскольку величина Rf всегда меньше 1 и, следовательно,
создается обманчивое впечатление завышенного численного вы-
ражения точности ее измерения, что не отражает реальной си-
туации, в настоящее время в научных работах часто приводят
величину hRf = Rf 100. Обычно следует выбирать такие усло-
вия разделения, при которых величина hRf находилась бы в
интервале от 30 до 80.
393
При поиске оптимальных условий разделения [250, 385] учи-
тывается как природа подвижной и неподвижной фаз, так и ха-
рактер анализируемых веществ: наличие и количество функцио-
нальных групп, способность к образованию водородных связей,
структура (например, цис, транс-изомерия) и т. д.
Если вещество обладает слабым сродством к сорбенту, то ис-
пользуют активные слои сорбента и слабополярные раствори-
тели, стоящие в начале элюотропного ряда (табл. IV.20-IV.22).
Активность сорбента определяется количеством сорбированной
воды. Так, для оксида алюминия различают пять степеней ак-
Таблица IV.20. Элюотропная серия для полярных
адсорбентов
Соединения е°(А12О3) Ткип., °C
Фторалканы -0,25 —
Пентан 0,001 36
Гептан 0,01 98,4
Декан 0,04 174
Ц и клогексан 0,04 81
Циклопентан 0,05 49,3
Сероуглерод 0,15 45
Тетрахлорметан 0,18 76,7
1-Хлорпентан 0,26 180,2
Диизопропиловый эфир 0,28 69
Толуол 0,29 100,6
Хлорбензол 0,30 132
Бензол 0,32 80,1
Диэтиловый эфир 0,38 34,6
Трихлорметан 0,40 61,6
Дихлорметан 0,42 41
Тетрагидрофуран 0,45 65
1,2-Дихлорэтан 0,49 84
Пропанон 0,56 56,2
Диоксан 0,56 104
Этилацетат 0,58 77,1
1-Пентанон 0,61 137,3
Диметилсульфоксид 0,62 190
Анилин 0,62 184
Нитрометан 0,64 100,8
Ацетонитрил 0,65 80,1
Пиридин 0,71 115,5
2-Пропанол 0,82 82,4
Этанол 0,88 78,5
Метанол 0,95 65
Этиленгликоль 1.11 198
Уксусная кислота Высокая 118,5
394
Таблица IV.21. Элюотропные серии для смеси
растворителей на силикагеле
Метанол : диэтиловый эфир Элюирующая сила е° Ацетонитрил : метанол
0,25 : 99,75 0,40
0,75 : 99,25 0,45
1,70 : 98,30 0,50
3,50 : 96,50 0,55
8,00 : 92,00 0,60
18,00 : 82,00 0,65 70,0 : 30,0
42,00 : 58,00 0,70 60,0 : 40,0
100,00 : 0,00 0,73 0,0 : 100,0
Таблица IV. 22. Элюотропные серии для смеси
растворителей на оксиде алюминия
2-Хлорпропан : пентан Элюирующая сила е° Диэтиловый эфир : пентан
8 : 92 0,05 4 : 96
19 : 81 0,10 9 : 91
34 : 66 0,15 15 : 85
52 : 66 0,20 25 : 75
77 : 23 0,25 38 ; 62
тивности (по Брокману) в зависимости от процентного содер-
жания воды: 1-0%, П-3%, Ш-6%, IV—10%, V-15% воды соответ-
ственно.
Для определения степени активности используемого сорбен-
та проводят хроматографический анализ тестовых веществ
(азобензол, n-метоксиазобензол, судан желтый, судан красный
и n-аминоазобензол) и сравнивают значения Rj с табличными.
Если значение Rj на силикагеле выше, чем требуется, необхо-
димо снизить полярность подвижной фазы.
Основным методом идентификации в качественном ТСХ-ана-
лизе является эталонирование. Если в распоряжении исследо-
вателя не имеется необходимых эталонов (стандартов), можно
воспользоваться их ближайшими гомологами, предварительно
измерив инкремент RM величины Rj, отвечающий одной мети-
леновой группе:
= log(-^“ - 1).
Как отмечалось выше, значение Rj линейно не соотносится ни с
факторами, влияющими на хроматографический процесс, ни со
395
структурой разделяемых соединений. Однако для RK наблюда-
ется линейная зависимость от log/fy.
IV.4.5. Количественный анализ в ТСХ
Развитие количественной оценки тонкослойных хромато-
грамм начиналось со сравнения размеров зон. Следующим ша-
гом в этом направлении явилось использование фотометриче-
ских измерений после элюирования хроматографических зон.
Количественная оценка предельно малых концентраций опреде-
ляемых веществ (от нано- до пикограмм) стала доступна после
введения прямого сканирования [359, 369]. Существуют ска-
неры, работающие в аналоговом режиме (без использования
компьютера), и сканеры с компьютерным управлением [27, 29,
387].
Распространенный способ количественного анализа в ТСХ
включает следующие этапы [388-390]: нанесение на старто-
вую линию ТСХ-пластин анализируемых объектов и стандар-
тов (“внешних” стандартов) — растворов, содержащих извест-
ные концентрации вещества, идентичного определяемому; хро-
матографическое разделение проб и стандартов; фото денсито-
метрическое сканирование полученных хроматограмм; расчет
концентрации определяемого вещества в пробе. При этом могут
получиться ошибочные результаты, если часть определяемого
вещества теряется на стадии пробоподготовки. Этих ошибок
можно избежать, используя метод внутреннего стандарта.
Приборы для сканирующей денситометрии имеют много об-
щего. Чтобы охватить весь диапазон УФ- и видимого излу-
чения (200-800 нм), необходимо располагать в качестве источ-
ника излучения различными лампами. Видимое излучение по-
лучают на галогеновых или вольфрамовых лампах, ультрафи-
олетовое — на дейтериевых. Для флуоресцентного анализа
целесообразно использовать ртутные или ксеноновые дуговые
лампы. Выбор требуемой длины волны осуществляется с помо-
щью монохроматора или фильтра.
Во всех современных денситометрах положение луча, скани-
рующего пятно пробы, зафиксировано. Пластинка сканируется
путем установки ее на платформу, передвигаемую при помощи
шаговых двигателей. В большинстве сканирующих денситоме-
тров предусмотрена возможность записи спектров любого чи-
сла пятен непосредственно с пластинки либо автоматически,
либо вручную. В последнем случае пятно сканируют повторно
396
несколько раз при различных длинах волн, отличающихся на
постоянную величину. Отношения величин откликов, получен-
ных при этих характеристических длинах волн, могут быть ис-
пользованы при идентификации компонентов.
При измерении флуоресценции селективность спектра значи-
тельно выше, поскольку для каждого определения используются
две различные длины волны — возбуждения и испускания.
На правильность и воспроизводимость количественных изме-
рений оказывают влияние качество и толщина слоя сорбента,
форма и размер пятна. Стандарты для калибровки должны
хроматографироваться на каждой пластинке, используемой для
анализа. Рекомендуется готовить стандартные растворы с раз-
личными концентрациями эталонных соединений и наносить
на пластинку в постоянном фиксированном объеме раствори-
теля. Для более корректного количественного определения
пятна стандартов и проб должны быть одного размера.
В современной сканирующей денситометрии с использова-
нием ВЭТСХ-пластинок величина относительного стандартно-
го отклонения за счет всех ошибок (воспроизводимость нанесе-
ния пробы, хроматографических условий, результатов измере-
ний) может быть не выше 2%, что делает ее надежным методом
количественного анализа.
Денситометры с компьютерным управлением отличаются от
аналоговых приборов, в первую очередь, тем, что в них меха-
нические методы сглаживания нулевой линии заменяются элек-
тронными (pnc.IV.32).
Рис. IV.32. Схема оптических устройств сканирующих денси-
тометров различных типов [387].
а — однолучевое сканирование; 6 — двухлучевое сканирование; е —
однолучевое сканирование с использованием двух длин волн. 1 — ис-
точник света; 2 — монохроматор; 3 — фотоумножитель; 4 — пластина;
5 — прерыватель.
397
Приборы с микропроцессорным управлением осуществляют
двухволновое сканирование. Первое сканирование хроматогра-
фического трека происходит на длине волны измерения, соот-
ветствующей максимуму поглощения, при етом полученные сиг-
налы записываются и сохраняются в памяти компьютера. По-
сле этой процедуры тот же самый трек сканируется вновь на
другой длине волны, преимущественно в области спектра, где
анализируемое вещество не поглощает. Разница между этими
сканированиями позволяет получить хроматограмму, свобод-
ную от фоновых помех.
IV.5. ОБРАБОТКА РЕЗУЛЬТАТОВ
ХРОМАТОГРАФИЧЕСКОГО АНАЛИЗА С ПОМОЩЬЮ ЭВМ
IV.5.1. Регистрация сигналов детекторов
Для регистрации сигнала хроматографических детекторов
могут применяться аналоговые самописцы, интеграторы или
ЭВМ, оснащенные аналого-цифровым преобразователем (АПП).
Преимуществом аналогового самописца является простота его
использования, недостатком — недостаточная точность измере-
ния сигнала и трудоемкость последующего измерения площадей
или высот пиков. Интегратор представляет собой специализи-
рованную микро-ЭВМ, способную измерять выходной сигнал
детектора, выявлять хроматографические пики и печатать на
встроенном принтере результаты измерения. Особенно широ-
кое распространение интеграторы получили в то время, когда
не было персональных ЭВМ широкого профиля. В настоящее
время интеграторы вытесняются системами на базе персональ-
ных ЭВМ.
При сопряжении устройства-измерителя сигнала с хромато-
графом необходимо согласовывать выходной диапазон источ-
ника аналогового сигнала с входным диапазоном измерителя.
Наиболее сложно это согласование выполнить в случае само-
писцев: их входной диапазон обычно составляет от 1 до 10 мВ,
и все сигналы приходится приводить к этому диапазону. Осо-
бые сложности регистрации хроматограммы на самописце воз-
никают при заметном дрейфе базовой линии хроматограммы
(например, в случае программирования температуры в газовой
хроматографии или градиентного элюирования в жидкостной
хроматографии). При этом зачастую нужно измерять неболь-
шие пики на фоне большого фонового напряжения. Например,
398
при пике высотой 1 мВ на фоне 100 мВ получающаяся высота
пика составляет не более 1% высоты диаграммной ленты, и та-
кой пик не может быть измерен с достаточной точностью. Ча-
стично эту проблему решают схемы компенсации, однако они
требуют регулирования компенсационного тока в процессе за-
писи хроматограммы, что приводит к искажениям записывае-
мого сигнала и возможной потере данных.
В отличие от самописца, интеграторы и аналого-цифровые
преобразователи ЭВМ рассчитаны на гораздо больший дина-
мический диапазон сигналов. Типичные значения измеряемых
АЦП-сигналов лежат в диапазоне 0-5 В или ±2,5 В, при этом
точность измерения зависит от разрядности АЦП. К примеру,
для 20-разрядного АЦП, работающего в диапазоне ±2,5 В, точ-
ность измерения составит ±2,5 мкВ. Для подключения АПП
или интегратора в большинстве хроматографов предусмотрен
выход, называемый ИНТЕГРАТОР, на который выходное на-
пряжение поступает безо всякого деления.
В последнее время все чаще и чаще фирмы-производители
хроматографов вставляют аналого-цифровые преобразователи
внутрь своих приборов, выдавая на ЭВМ измеренное напряже-
ние в цифровом виде (так, например, устроены почти все совре-
менные модели хроматографов фирмы Hewlett-Packard). Такая
схема позволяет исключать искажения и шумы сигнала, воз-
никающие из-за длинных подсоединительных проводов, сокра-
щает общее количество электронных компонентов и тем самым
способствует не только увеличению точности, но и повышению
надежности измерительной системы.
IV.5.2. Аналого-цифровое преобразование
Как правило, детектор хроматографа выдает напряжение или
ток, пропорциональный измеряемому детектором параметру
(аналоговый сигнал). Этот сигнал не может быть напрямую
воспринят компьютером. Для передачи величины сигнала в
компьютер используется аналого-цифровой преобразователь,
переводящий аналоговый сигнал в цифровую форму, которая
уже может быть принята и обработана компьютером. АЦП от-
личаются по динамическому диапазону, скорости сбора данных
и количеству каналов.
Динамический диапазон представляет собой отношение мак-
симального напряжения, измеряемого АПП, к минимальному
изменению сигнала, которое может быть им зарегистрировано.
399
Таблица IV.2S Сравнительные характеристики
томности аналого-цифровых преобразователей
Двоичный диапазон, бит Десятичный диапазон, раз Максимальное отношение высот пиков Минимальное количество примеси, %
12 4000 400 0,25
16 65000 6500 0,015
20 1000000 100000 0,001
24 16000000 1600000 0,00005
Это отношение может измеряться как десятичным числом, к ко-
торому все привыкли, так и двоичным числом. Чем выше ди-
намический диапазон, тем точнее А11П. Табл. IV 23 дает пред-
ставление об АЦП, отличающихся по точности. В таблице при-
ведены предельные значения, которые может обеспечивать пре-
образователь. Высота минимального пика, надежно измеряе-
мого системой, принята за 10 единиц преобразования исполь-
зованного ЛПП, а уровень ошибки преобразования предпола-
гается равным единице. При расчете данных, внесенных в таб-
лицу, не учтены уровни шумов детектора хроматографа и со-
единительных кабелей, так что действительный диапазон изме-
ряемых сигналов в конкретном случае будет заметно ниже (в
таблице приведен теоретический предел).
Следует также учитывать, что многие фирмы приводят дру-
гую характеристику АЦП — разрешающую способность — и
тоже выражают ее в битах. Эта характеристика показывает
число двоичных разрядов, которое АПП выдает в компьютер.
Понятно, что часть разрядов может оказаться шумовыми и
тогда разрешающая способность окажется выше точности. К
примеру, популярная микросхема AD7710 фирмы Analog Devices
имеет разрешение 24 бит, тогда как предельная точность, до-
стигаемая при 10 изм/с, составляет всего 21,5 бит.
При оценке пригодности АЦП для хроматографии следует
обращать внимание на принцип измерения, в частности на время
преобразования измеряемого сигнала. Все АПП можно раз-
бить на два класса: быстрые, измеряющие мгновенное значение
напряжения, и медленные (или интегрирующие), измеряющие
среднее значение сигнала за время измерения. Для хромато-
графии несомненным преимуществом обладают интегрирующие
АПП — они подавляют значительную часть высокочастотных
(импульсных) помех.
400
Скорость измерений должна быть достаточной для того, что-
бы обеспечить от 20 до 30 измерений на полуширину самого
узкого измеряемого пика. Типичная скорость измерений соста-
вляет 10 Гц для газовой и 1 Гц для жидкостной хроматографии.
В экспрессных методах анализа, когда вся хроматограмма за-
писывается за несколько секунд, могут быть необходимы более
высокие скорости сбора данных.
Время преобразования интегрирующих АЦП желательно вы-
бирать кратным периоду частоты сетевого напряжения (в Евро-
пе — 1/50 секунды, в США — 1/60 секунды). Это позво-
ляет уменьшить влияние наводок на подсоединительные про-
вода со стороны окружающих электрических устройств. Самое
популярное время преобразования 10 изм/с пригодно как для
Европы (50 Гц/5), так и для США (60 Гц/6). При выборе вре-
мени преобразования следует также учитывать, что точность
интегрирующих АЦП уменьшается с уменьшением времени пре-
образования. Так, упомянутый выше АЦП AD7710 при частоте
50 изм/с обеспечивает точность лишь 19 бит.
IV.5.3. Использование персональных ЭВМ
в хроматографии
Появление микропроцессоров и компьютеров преобразило
хроматографическое оборудование. Сейчас вряд ли можно
найти хроматограф, где не был бы установлен микропроцессор.
В жидкостной хроматографии микропроцессор может входить в
состав каждого блока хроматографа, такого, как детектор или
насос.
Основные функции микропроцессоров в составе оборудова-
ния — прием вводимых с клавиатуры прибора данных, преобра-
зование и передача их на исполнительные устройства, прием
данных от датчиков, осуществление управляющих воздействий
(регулирование температуры, потока, давления и т. д.) диагно-
стика работоспособности всех составляющих узлов. В функции
микропроцессора в составе прибора входит также связь с упра-
вляющей ЭВМ, которая может передать команды, задающие
значения параметров или программу работы, а также прини-
мать результаты измерений, если прибор оснащен встроенным
АЦП.
В последнее время усилилась тенденция к созданию прибо-
ров, не имеющих собственной клавиатуры. Преимущества та-
кой схемы состоят в уменьшении стоимости прибора и увели-
401
чении его надежности за счет уменьшения числа составных ча-
стей.
Во многих книгах и описаниях программ встречается тер-
мин “реальное время”. “Реальное время” (real time) — термин,
принятый в компьютерном сообществе для обозначения процес-
сов, требующих применения управляющих воздействий в тече-
ние определенного промежутка времени после наступления си-
туации (состояния системы), когда такое вмешательство потре-
бовалось. В качестве примеров процессов реального времени
можно привести управление градиентом температуры в газо-
вой хроматографии (включение—выключение нагревателя тер-
мостата в зависимости от показаний термопары), а в жидкост-
ной хроматографии — управление клапаном слива элюента в
емкость, предназначенную для его рециркуляции, в зависимо-
сти от значения отклика детектора. Применение термина “ре-
альное время” к измерению сигнала детектора не всегда оправ-
дано, поскольку управляющих воздействий при этом не произ-
водится. В этом случае обычно говорят о прямом (on-line) или
отложенном (off-line) приеме данных от измерительного устрой-
ства. Функции контроля времени измерения при этом возлага-
ются на измерительное устройство.
Большинство задач реального времени решаются встроен-
ными процессорами в составе оборудования, а на управляющий
компьютер возлагается в основном обеспечение адекватной ре-
акции хроматографической системы на аварийные ситуации.
Первые серии интеграторов вычисляли только площади и вы-
соты хроматографических пиков. Последующие — стали осу-
ществлять градуировку хроматографической системы и расчет
концентраций компонентов. Параллельно с интеграторами со-
здавались автоматизированные комплексы для хроматографии
на базе АПП и ЭВМ общего назначения, такие, как РДР 11
(СМИ) или VAX
С появлением персональных ЭВМ — Apple, а затем IBM PC —
были разработаны аппаратные средства и программное обеспе-
чение для них. К настоящему времени отмечена тенденция ин-
тенсивного вытеснения интеграторов вычислительными систе-
мами на основе персонального компьютера. Можно назвать не-
сколько причин такого решения.
1. Возможность работы компьютерной системы с несколь-
кими хроматографами одновременно. В условиях современной
аналитической лаборатории на одного химика-аналитика при-
ходится целый парк хроматографов. Таким образом, возникает
необходимость создания рабочего места, обеспечивающего од-
новременную работу со всеми приборами — единый интерфейс
402
пользователя. Это, в свою очередь, позволяет значительно сни-
зить расходы на обучение персонала и сократить число ошибок
оператора при работе с оборудованием. Сложившийся к насто-
ящему времени стереотип основан на использовании графиче-
ского интерфейса операционной системы Windows, что обеспе-
чивает пользователю программ одинаково комфортные условия
при работе с текстовым редактором, электронными таблицами
или программами обработки хроматографических данных.
2. Работа квалифицированного химика-аналитика подразу-
мевает проверку и уточнение результатов лабораторного жур-
нала, подготовку текстовых документов (отчетов). При этом за-
метим, что все это должно быть выполнено в процессе проведе-
ния очередных анализов. Поэтому одним из существенных тре-
бований к программному обеспечению является возможность
обработки ранее сделанных хроматографических анализов па-
раллельно с получением новой хроматограммы.
3. Возросли т; ебования к оформлению отчетной документа-
ции. Сравнительно недавно в США и в большинстве европей-
сих стран были приняты законы-стандарты о Хорошей лабо-
раторной практике (Good Laboratory Practice, GLP), регламен-
тирующие оформление результатов аналитических исследова-
ний. Эти стандарты указывают обязательный перечень сведе-
ний, приводимых в документации, порядок хранения образцов и
исходных (необработанных) данных; определяют круг лиц, от-
ветственных за результаты анализа. Программное обеспечение
для обработки хроматограмм строится таким образом, чтобы
помогать пользователю соблюдать требования стандартов.
Многие крупные фирмы-производители хроматографическо-
го оборудования разрабатывают свое собственное программ-
ное обеспечение для хроматографии, например HP ChemStation
(фирма Hewlett-Packard), TurboChrom (фирма Perkin-Elmer). На-
ряду с ними на рынке программного обеспечения для хромато-
графии работают небольшие фирмы, для которых хроматогра-
фические программы являются основной продукцией, например
фирма Scientific Software, Inc (программа “EZChrom”), Ampersand
Ltd. (программа “МультиХром”).
К настоящему времени сложился де-факто стандарт на ком-
пьютерные системы сбора и обработки данных для хромато-
графии, по крайней мере в тех случаях, когда используются
достаточно простые (одноканальные) хроматографические де-
текторы. Для многоканальных систем, основанных на детек-
торах типа линейки диодов, такому стандарту еще предстоит
появиться.
403
Типовая система основана на аналого-цифровом преобразо-
вателе разрядностью 20 бит или более, обеспечивающем прием
данных от двух или четырех детекторов, работает на IBM PC —
совместимом компьютере с операционной системой Windows 3.1
(Windows-95) — и выполняет следующие функции:
— получение преобразованного аналогового сигнала и ото-
бражение его на экране компьютера;
— запись полученной информации на магнитный диск и со-
здание базы данных выполненных анализов с защитой инфор-
мации от несанкционированного изменения;
— возможность автоматизированной работы с автосампле-
ром и повторной обработки серий ранее проведенных анализов;
— выявление хроматографических пиков;
— градуировка системы, позволяющая получить значение
ожидаемых времен удерживания и градуировочные зависимо-
сти отклика детектора от количества введенного компонента по
смесям известного состава;
— идентификация хроматографических пиков на основе по-
лученных в градуировочных экспериментах ожидаемых времен
удерживания и расчет концентрации анализируемых компонен-
тов с использованием метода внешнего или внутреннего стан-
дарта;
— оценка функционирования хроматографической системы
(SST — System Suitability Test) — оценка качества хроматографи-
ческого разделения компонентов и соответствие этого качества
ожидаемому. Типовые статистические расчеты — статистиче-
ская оценка результатов измерения концентрации в нескольких
анализах (абсолютная и относительная погрешности, довери-
тельный интервал);
— настройка бланков печати на форму, необходимую поль-
зователю, и печать отчета, совмещающего графическое изобра-
жение хроматограммы и результаты расчета концентраций ана-
лизируемых компонентов;
— аннотирование анализа — ввод сопутствующей информа-
ции, которая может потребоваться для повторной оценки ре-
зультаты анализа или для повторения анализа в тех же усло-
виях;
— наконец, управление хроматографическим оборудованием.
Кроме того, в ряде случаев (для газовой хроматографии)
добавляются реализация метода имитированной дистилляции
продуктов переработки нефти, а также расчет молекулярно-
массовых распределений полимеров (эксклюзионная хромато-
графия), выполняемые в виде отдельных программ или модулей
программы.
404
Логическим продолжением систем сбора и обработки хро-
матографических данных являются интегрированные лабора-
торные информационные системы (LIMS, Laboratory Information
Management Systems). Они предназначены для учета резуль-
татов самых разнообразных аналитических измерений в мас-
штабах небольшого предприятия и часто бывают специализи-
рованы для конкретного приложения.
IV.5.4. Фильтрация шумов
Одним из преимуществ применения ЭВМ для обработки хро-
матограмм является возможность фильтрации шумов. Наибо-
лее существенно она помогает при работе с малыми концентра-
циями исследуемых веществ, вблизи их предела обнаружения.
Из всего множества существующих алгоритмов фильтрации шу-
мов мы опишем три наиболее часто используемых фильтра: ме-
дианный, гауссов и фильтр Савицкого—Голея. При использова-
нии любого из этих фильтров хроматограмма обрабатывается
с помощью специального окна с нечетным количеством точек.
Результатом фильтрации является новое значение отклика де-
тектора в точке, соответствующей середине окна.
При медианной фильтрации значения внутри окна сор-
тируются в порядке возрастания и отклик, соответствующий
середине окна, заменяется другим значением, попадающим в
центр отсортированного массива. Этот метод влияет на хрома-
тографические пики в наименьшей степени, хорошо сглаживает
базовую линию, не меняет формы пика на склонах и очень эф-
фективно устраняет отдельные выбросы (в этом случае выброс
заменяется на одну из соседних точек). Однако он слегка “при-
глаживает” вершины пиков и ложбины между пиками и может
изменять как высоту, так и площадь хроматографических пиков.
В случае применения гауссова фильтра вычисляется
средневзвешенное значение всех точек внутри окна с весом, рас-
пределенным по функции Гаусса с центром в середине окна,
результат используется как новое значение отклика детектора.
Высота и форма пиков при этом меняются, тогда как площадь
остается неизменной. Гауссов фильтр, по сравнению с медиан-
ным, дает лучшее визуальное сглаживание собственно пиков, но
меньше сглаживает шумы базовой линии. Пики после сглажива-
ния становятся ниже и шире, их площадь при этом не меняется.
Фильтрация шумов по методу Савицкого—Голея со-
стоит в проведении по нечетному числу точек (5 или более) ку-
405
бического полинома и замене значения в середине диапазона
на значение аппроксимирующего полинома в этой точке. Ока-
зывается, что с математической точки зрения эта процедура,
аналогично гауссову фильтру, эквивалентна вычислению сред-
невзвешенного значения с определенным распределением весов.
Поскольку любые фильтры искажают исходный хроматогра-
фический сигнал, их использование оправдано только при вы-
соких значениях шума. При этом рекомендуется начинать с ми-
нимально возможной степени сглаживания и постепенно увели-
чивать ее до получения удовлетворительного результата.
IV.5.5. Поиск пиков и их интегрирование
Одной из наиболее важных процедур в компьютерной обра-
ботке хроматограммы является разметка ее на пики. Процедура
поиска пиков называется интегрированием. Интегрирование
включает в себя операции определения границ пиков, построе-
ние базовой линии, вычисление таких характеристик пиков, как
время удерживания, высота и площадь. Далее алгоритмы инте-
грирования проиллюстрируем на примере программы “Муль-
тиХром”.
Программа “МультиХром” включает встроенный детектор
пиков для автоматической разметки, дополненный механизмом
событий интегрирования, а также редактор пиков для ручной
коррекции картины разметки хроматограммы на пики.
Величиной, характеризующей содержание компонента в ана-
лизируемой смеси, является площадь или высота пика. Для вы-
числения этих величин необходимо знать его начало, конец и
положение базовой линии (рис.ГУ.ЗЗ). Базовая линия в обла-
сти, расположенной между пиками, совпадает с профилем хро-
матограммы; под пиками используется прямая базовая линия.
В программе “МультиХром” изломы базовой линии возможны
только на границах пиков. Сверху пики ограничены хромато-
графической кривой, а снизу — базовой линией. Смежные или
слившиеся пики (неразделенные у основания) объединяются в
группу, где конец предыдущего пика совпадает с началом сле-
дующего. В этом случае базовая линия начинается в точке,
относящейся к началу первого пика, и заканчивается в точке,
относящейся к концу последнего пика в группе. Смежные пики
разграничиваются вертикальной линией, соединяющей хрома-
тографическую кривую с базовой линией (перпендикуляром).
Так называемый пик-наездник отделяется от пика-носителя ли-
нией тангенциального спуска (тангентой).
406
Рис.IV.33. Образец хроматограммы с разметкой пиков и со-
бытий интегрирования.
Объяснения в тексте.
Правильная разметка хроматограммы на пики является в аж-
нейшей стадией в обработке хроматографических данных. Ав-
томатический детектор пиков часто работает по достаточно
сложному настраиваемому алгоритму. Настройка алгоритма
детектирования состоит в указании параметров, действующих
при начале поиска пиков, таких, как ожидаемая средняя ширина
пика, отношения ширин в начале и в конце хроматограммы, по-
рог обнаружения пика по значению производной и некоторые
другие. Кроме параметров, действующих по умолчанию, обыч-
ной практикой является задание событий интегрирования, изме-
няющих поведение алгоритма в определенные моменты времени.
В качестве примера одного из событий интегрирования можно
привести событие “Включить/отключить базу долина-к-долине"
(рис. IV.34), реализованное в программе “МультиХром”.
Вкл. базу
долина-к-долине — запрещает разделение пиков “по перпен-
дикуляру” . Проводит базовую линию по са-
мым низким точкам между пиками (долина-
к-долине) .
Отключить базу
долина-к-долине — разрешает разделение пиков “по перпен-
дикуляру” (нормальный режим разметки).
407
Рис.IV.34- Различные алгоритмы детектирования пиков.
А — интегрирование по умолчанию (разделение пиков “по перпен-
дикуляру”); Б — включена база “дол ина-к-дол ине”.
Такого рода события разбивают хроматограмму на участки,
на каждом из которых действует свой режим выявления пиков.
Однако настройка алгоритма может оказаться делом трудо-
емким или специфичным только для одного анализа. В таких
случаях вместо тонкой настройки алгоритма применяется руч-
ная разметка на пики с помощью инструмента (редактора пи-
ков), который позволяет изменить картину пиков путем удале-
ния или создания пиков, перемещения точек начала или конца
пика, расщепления существующего пика на два и т. д.
IV.5.6. Идентификация хроматографических
пиков с помощью компьютера.
Таблица компонентов
В компьютерной хроматографии существует два значения
термина “идентификация”. Первое значение (будем называть
его далее идентификацией первого рода) состоит в соотнесе-
408
нии пика на хроматограмме и некоторого химического веще-
ства. Такого рода идентификация выполняется с использова-
нием комплекса процедур и методов, изложенных в предшеству-
ющих разделах этой главы.
Второе значение (идентификация второго рода) относится к
соотнесению наборов пиков двух хроматограмм, у одной из ко-
торых (градуировочной) произведена идентификация (первого
рода) некоторых пиков, а для другой предполагается наличие
тех же компонентов, которые имеются на градуировочной хро-
матограмме. Программам, ориентированным на рутинный ана-
лиз, чаще приходится решать задачу идентификации второго
рода; остановимся подробнее на примере из программы “Муль-
тиХром”.
В программе “МультиХром” градуировочная хроматограм-
ма (т. е. хроматограмма известной смеси анализируемых ком-
понентов, с известными, как правило, концентрациями) служит
для сознания таблицы компонентов.
В таблице компонентов хранятся данные обо всех иден-
тифицированных пиках градуировочной хроматограммы: на-
звание, время удерживания, допустимое отклонение времени
выхода пика от ожидаемого, а также некоторая другая инфор-
мация, необходимая для идентификации и количественного рас-
чета. Для наглядности над таблицей компонентов приводится
изображение хроматограммы (табл. IV.24).
Компоненты в таблице делятся на два типа — реперные и все
остальные. Пики реперных компонентов являются хорошими
ориентирами для продолжения процесса идентификации: их
времена удерживания служат для корректировки ожидаемых
времен удерживания других компонентов.
В программе принят двухступенчатый механизм распознава-
ния пиков. На первом этапе распознаются только реперные ком-
поненты (если они заданы!), на втором — все остальные. Ре-
перный пик ищется в пределах заданного для него временного
окна. Если в окне присутствует несколько пиков, в качестве
реперного обычно выбирается самый высокий пик (хотя можно
использовать и другие критерии).
На втором этапе, когда реперные пики найдены, для иден-
тификации остальных пиков используется относительное ожи-
даемое время удерживания, скорректированное в соответствии
с полученными при регистрации данной хроматограммы вре-
менами удерживания реперных компонентов. Например, если
время удерживания реперного компонента (пусть только один
компонент объявлен реперным) увеличилось на 10%, то и ожи-
даемое время выхода всех остальных компонентов также будет
409
Таблица IV.24- Пример таблицы компонентов
ЙМулыиХром для Windows - [GC capillaiy [capiti.mtw]d90212G3.phw |
SJfJ Йромгггограмма Д_вблща £цд Измерение
Йетод Цастройкл Qkho 2.
] I fi. Показ.дсе |
увеличено на 10%. При заметной вариации времен удерживания
первых пиков можно выбрать 2-3 различных соединения.
Дадим описание столбцов таблицы компонентов, имеющих
отношение к процедуре вторичной идентификации (см.
табл. IV.24).
Пик — номер пика. Соответствует нумерации пиков на
хроматограмме. При вводе номера пика в колонке
Удерж, появляется время удерживания компонента,
курсор при этом устанавливается на вершине пика с
данным номером.
Удерж. — ожидаемое время удерживания компонента. Если
время удерживания компонента установлено равным
нулю, то этот компонент даст название всем неиден-
тифицированным пикам.
Окно — идентификационное окно данного компонента, изме-
ряемое в процентах от его ожидаемого времени удер-
живания. Во время процедуры идентификации поиск
данного компонента будет производиться только вну-
три интервала tr ± tr • w%/100, где tr — время удер-
живания компонента, w% — его идентификационное
окно. Значение идентификационного окна устанавли-
вается в зависимости от стабильности времен удер-
живания, которую обеспечивает конкретная хромато-
графическая система, эффективности колонки, слож-
ности анализируемой смеси и обычно лежит в диапа-
зоне 2-10%. Если в идентификационное окно попадает
несколько пиков, выбирается один из них, ближайший
по времени выхода (или высоте для реперных пиков).
Репер — триггер (Ла/Нетп), показывающий, является ли дан-
ный пик реперным. Как правило, в качестве реперных
выбираются самые большие, отдельно стоящие пики.
Идентификационное окно для реперных пиков обычно
устанавливается вдвое большим, чем для остальных
компонентов.
Название — название компонента.
Таблица компонентов записывается на диск вместе с хрома-
тограммой, и при необходимости повторной обработки она вы-
дается пользователю в том виде, какой она была зафиксирована
в момент измерения.
По завершении заполнения таблицы компонентов, вызвав ме-
тод “Расчет индексов”, пользователь может дать команду на
вычисление логарифмических (Ковача) или линейных индексов
Удерживания I идентифицируемых веществ. При вычислении
411
индексов* используется время удерживания предыдущего и по-
следующего пиков, отвечающих веществам с известными чи-
сленными значениями I. При отсутствии на хроматограмме
пика с известным индексом до или после сигнала детектора на
идентифицируемое соединение программа проводит экстрапо-
ляцию, выбирая два ближайших пика, принадлежащих веще-
ствам с известными индексами (в качестве таковых оправдан-
нее всего использовать стандартные вещества-реперы, напри-
мер н-алканы в газовой хроматографии или алкилфенилкетоны
в ВЭЖХ).
Программа “МультиХром” позволяет строить т. н. внутрен-
нюю и внешнюю шкалы индексов удерживания. При построе-
нии внутренней шкалы используются реальные времена удер-
живания идентифицируемых и реперных соединений, присут-
ствующих в анализируемой смеси. При построении внешней
шкалы принимаются в расчет ожидаемые времена удерживания
компонентов, заимствуемые из градуировочной хроматограммы
(при этом присутствия опорных компонентов в анализируемых
образцах не требуется). При таком подходе, как уже отмеча-
лось в разделе IV.2.3, точность вычисления индексов удержи-
вания может оказаться несколько ниже, чем при использовании
внутренней шкалы.
IV.5.7. Использование ЭВМ для
характеристики колонки и качества
достигнутого разделения
В программе “МультиХром” предусмотрена возможность
расчета факторов удерживания (коэффициентов емкости) к, ко-
эффициента асимметрии хроматографического пика (см. [32,
с. 56-57; 33, с. 26-37]), эффективности используемой (тестируе-
мой) колонки по любому из пиков, выражаемой общим числом
теоретических тарелок (т. т.), а также числом т. т., отнесенных
к 1 м длины колонки, и, кроме того, приведенной высотой, экви-
валентной теоретической тарелке (ПВЭТТ). Наряду с этим по
любой паре соседних пиков может быть рассчитан критерий
разделения R. Для выполнения расчетов названных параметров
необходимо вызвать метод “Тест колонки” и задать длину ко-
лонки, мертвое время и размер частиц сорбента.
*Расчет проводится по формулам, рассмотренным в разделе IV 2.2.
412
IV.5.8. Использование ЭВМ при решении
задач количественного
хроматографического анализа
Начальной процедурой при решении задач количественного
хроматографического анализа в программе “МультиХром” яв-
ляется градуировка, преследующая две цели: получение ха-
рактеристики удерживания и зависимостей между откликом де-
тектора и дозируемым количеством компонента (выражаемым
массой (мкг) или объемом (мкл)), либо его концентрацией (еди-
ницы измерения концентрации могут быть любыми: массовыми,
объемными или массо-объемными, чаще всего для характери-
стики концентрации компонентов в градуировочных смесях ис-
пользуют массо-объемные единицы — мг/л или мкг/мл).
Результаты градуировки представляются в форме таблицы
компонентов (содержит времена удерживания и названия
определяемых веществ, см. табл. IV.24), а также таблицы
концентраций (табл.IV.25), в которую, используя соответ-
ствующий шаблон, вызываемый из “метода обработки резуль-
татов”, вносят заданные концентрации компонентов в градуи-
ровочных смесях. В табл. IV.25 каждой градуировочной смеси
отвечает свой (присущий только этой смеси) градуировочный
уровень (столбец таблицы). Каждый созданный уровень кон-
центрации используется для обработки результатов только од-
ного градуировочного анализа. При желании можно использо-
вать для построения градуировочной зависимости сигналы де-
тектора, полученные в параллельных (двух-четырех или более)
анализах одной и той же смеси. В этих случаях необходимо со-
здать несколько (соответственно два-четыре или более) уровней
с одинаковыми концентрациями компонентов.
По результатам хроматографирования градуировочных рас-
творов компьютер построит градуировочные кривые (см. ниже),
отражающие зависимость между введенным количеством (или
концентрацией) вещества в фиксированной дозе и откликом де-
тектора на каждый определяемый компонент.
Виды градуировок в количественном хроматографиче-
ском анализе. Программное обеспечение “МультиХром” рас-
считано на обработку экспериментальных данных при исполь-
зовании различных видов градуировок хроматографа, необхо-
димых для реализации рассмотренных в разделе IV.3 основ-
ных методов количественного хроматографического анализа, а
именно — метода абсолютной градуировки, метода внутренней
нормализации и метода внутреннего стандарта.
413
Таблица IV. 25. Пример таблицы концентраций
Базовым способом градуировки в программном обеспечении
“МультиХром” является абсолютная градуировка хро-
матографа по каждому из интересующих аналитика компоненту
исследуемой смеси. Этот вид градуировки отвечает первому
из рассмотренных в разделе IV.3 методов количественного хро-
матографического анализа с одноименным названием (извест-
ного так же, как метод внешнего стандарта). Упрощенный ва-
риант абсолютной градуировки — табличный — не требует
построения градуировочных зависимостей между введенным ко-
личеством компонента или его концентрацией в фиксированной
дозе и сигналом детектора. Здесь для получения результата
предусматривается возможность использования численных зна-
чений относительных коэффициентов отклика детектора (иначе
— нормировочных, градуировочных, или поправочных множи-
телей fi), заимствуемых из литературных источников (см., на-
пример, [31, ч.2, с. 34-38; 32, с. 214, 322-327; 33, с. 161-180; 236,
237]) или найденных в независимых экспериментах (последний
из названных способов нахождения величин fa явно предпочти-
тельнее, см. по этому поводу раздел IV.3.3).
Как известно, наиболее универсальный метод количествен-
ного хроматографического анализа предусматривает использо-
вание внутреннего стандарта. Соответствующая градуировка
методом внутреннего стандарта требует знания концен-
трации компонента-стандарта в специально приготавливаемых
414
градуировочных растворах (внутренним стандартом может
быть объявлен любой компонент градуировочной смеси).
Рассмотренные градуировки могут быть одноточечными или
многоточечными. Одноточечная градуировка означает, что
используется лишь одна экспериментальная точка, другой слу-
жит начало координат; получаемая градуировочная зависи-
мость заведомо носит линейный характер. Одноточечная гра-
дуировка является частным случаем многоточечной.
Многоточечная градуировка означает, что проводится
несколько измерений для определения градуировочной зависи-
мости и на графике имеется несколько точек. В этом случае
зависимость может быть аппроксимирована кривой любого, не-
обязательно линейного, типа (см., например, рис.IV.35). Ком-
пьютерная обработка данных помогла тому, что многоточечная
градуировка стала фактическим стандартом при хроматогра-
фических измерениях — основная тяжесть статистических вы-
числений ложится на программу, а не на аналитика.
Площадь
Рис. IV.35. Нелинейная градуировочная зависимость, постро-
енная по четырем экспериментальным точкам.
Как уже отмечалось выше, целью абсолютной градуировки
является построение функциональных зависимостей для каждо-
го i-го компонента Ui и IV) вида
Pi = Ui{Ci}
или
Pi = Wi(qi), (IV 58)
где Pi — количественный параметр хроматографического пика
(площадь или соответственно высота) г-го компонента на хрома-
тограмме; Ci и qi — концентрация и соответственно масса (или
объем) определяемого вещества.
Процедура построения этих зависимостей может быть пред-
ставлена в виде схемы
НМ8(С0-,Ргу-) -> Ui(Ci)
415
или
RMS(<7jj, Pij) -> Ж (ft),
где RMS обозначает применение метода наименьших квадратов
для вычисления коэффициентов соответствующей градуировоч-
ной зависимости, Cij и qij — концентрация или соответственно
количество введенного г-го компонента в j-й градуировочной
смеси.
Для расчета концентрации интересующего аналитика соеди-
нения в анализируемых объектах требуется знание объема вво-
димой пробы (V):
Q = Wf(Pi)/V, (IV.69)
где wf" — функция (“обратная” к W;), с помощью которой
рассчитывается q, для значения Pi по формуле, аналогичной
(IV.68).
Программное обеспечение “МультиХром” предусматривает
оформление этих зависимостей в виде графика, на оси ординат
которого откладываются концентрации или количества интере-
сующего аналитика компонента, а на оси абсцисс — соответ-
ствующие численные значения площадей или высот пиков*.
Выводимый на экран монитора бланк работы с градуиров-
кой по программе “МультиХром” содержит не только графиче-
ское представление исследуемой зависимости, но и ее матема-
тическое описание с элементами метрологической аттестации
(рис. IV.36).
Как известно, использование метода внутреннего стандарта
позволяет компенсировать ошибки при дозировании пробы и по-
тери при пробоподготовке, что существенно улучшает воспро-
изводимость градуировочных точек.
В компьютерной хроматографии сформировалось два под-
хода к установлению градуировочных зависимостей методом
внутреннего стандарта. Один из этих подходов (назовем его
алгоритмом первого рода) предусматривает построение зависи-
мостей в координатах отношение концентраций определяемого
и стандартного веществ — отношение откликов детектора на
эти вещества. Преимуществом подобных градуировок является
наглядное представление результатов в виде коэффициентов от-
носительного отклика детектора /г-. Однако использование дан-
ного алгоритма в случае нелинейных градуировочных зависи-
мостей в широком диапазоне концентраций внутреннего стан-
*В традиционной практике построения подобных зависимостей, сфор-
мировавшейся еще в “докомпьютерную” эпоху, количество введенного ве-
щества (или его концентрацию в градуировочном растворе) откладывают
на оси абсцисс, а отклик детектора — на оси ординат.
416
Рис. IV.36. Бланк из программного обеспечения “МультиХром” для работы с градуировочной зависимостью.
дарта может приводить к ошибочным результатам (что и отра-
жается в описаниях некоторых фирменных программ, например
в описании программы “ChemStation” фирмы Hewlett-Packard).
Иной алгоритм (второго рода) предусматривает построение
только абсолютных градуировочных зависимостей типа (IV.68)
для всех определяемых компонентов и для веществ, выполняю-
щих функции внутреннего стандарта. Учет концентрации стан-
дарта производится на этапе расчета и концентрация соеди-
нений, представляющих аналитический интерес, находится по
формуле
Ci = Cstwf(Pi)/W*(Psi), (IV.70)
где W#(Pi) и Ж,#(Fst) — соответствующие функциональные за-
висимости концентрации определяемого (г) и стандартного (st)
соединений, используемые в формуле (IV.68). Соответствую-
щий столбец в таблице пиков программы “МультиХром” носит
название “Относительная концентрация”.
Преимущество алгоритма второго рода в сравнении с алго-
ритмом первого рода заключается в открывающейся возмож-
ности работы в условиях нелинейной градуировки и в широ-
ком диапазоне концентраций всех определяемых компонентов, в
том числе и стандарта. Безусловно, построение зависимостей
Wi(Pi) и PVst(Fst) методом абсолютной градуировки — дело до-
статочно трудоемкое и требующее постоянного внимания. Од-
нако в случае нелинейных градуировок без построения граду-
ировочной зависимости для всех компонентов, включая стан-
дарт, обойтись, по-видимому, нельзя. В случае линейных гра-
дуировок, проходящих через начало координат, оба алгоритма
дают практически совпадающие результаты.
Следует отметить одну полезную особенность формулы
(IV.70). В нее входит отношение двух функциональных зави-
симостей (градуировок). Если измерения градуировок носят
синхронный характер (т. е. каждая из них домножается на одно
и то же число), то отношение в этой формуле сохранится, и
результат расчета останется тем же. Синхронное изменение
градуировок — достаточно частое явление, вызываемое неизби-
рательными изменениями абсолютной чувствительности детек-
тора. По этой причине повторную градуировку системы в том
случае, если рассчитывается только относительная концентра-
ция, проводить можно гораздо реже, чем при расчете абсолют-
ной концентрации по формуле (IV.69). По необходимой частоте
повторных градуировок разницы между подходами первого и
второго рода нет.
В программе “МультиХром” принят гибридный метод гра-
дуировки Внутренний стандарт, имитирующий алгоритм пер-
418
Генератор отчета
Кудвнапрякиткотчет
Принтер
И
Т «блица пиков
Метод расчета:
Стенд. компонент:
концентрация стенд.:
Нормировка.
Порядок печати:
« Столбцы!
I Заказной
То1
ГоО
По пикам
Файл
: Образец:
I Еаздолигвль; пробел
Паромеры печати в фа1'
; и«я I ~1 _ Клтйлог J
Режим: ♦ Переписать
Кодировка; Windom
Программа
Отчет
Принять
✓j Отчет по доем пикам
ruttian.rU Д
~Я| Табулятор: |0 |
Допоямигь
ЙО5
3
7
Рис. IV.37. Бланк настройки отчета с указанием всех возмож-
ных параметров, рассчитываемых для пиков.
вого рода при неизвестной градуировочной зависимости для
стандартного компонента и строящий зависимости, практиче-
ски неотличимые от абсолютной градуировки, при известной
градуировочной зависимости для стандарта.
На основе выполненных градуировок ЭВМ способна прове-
сти необходимые расчеты и осуществить количественную ин-
терпретацию хроматограммы и по методу внутренней нор-
мализации* с учетом неодинаковых откликов детектора fi на
различные компоненты смеси.
Расчет содержания интересующих компонентов может вы-
полняться как по площадям, так и по высотам пиков. Вве-
дена также возможность обработки результатов анализа по
т.н. заказному методу, позволяющему в пределах одной хро-
*Отметим, что программой “МультиХром” наряду с расчетами по ме-
тоду внутренней нормализации, требующей обязательного использова-
ния факторов отклика детектора, называемых иначе нормировочными,
калибровочными или поправочными множителями, предусматривается
также возможность осуществления нормализации без учета относитель-
ной чувствительности детектора к определяемым веществам; такой спо-
соб обработки экспериментальных данных назван методом нормализации
(прилагательное “внутренняя” в названии данного метода отсутствует).
419
Рис. IV.38. Пример многоканальной хроматограммы, получен-
ной на жидкостном хроматографе “МилиХром-А02”.
матограммы рассчитывать содержание отдельных компонентов
смеси по любому выбираемому (заказываемому) аналитиком па-
раметру пика. Образец бланка формирования отчета с указа-
нием всех возможных параметров, рассчитываемых для пиков,
представлен на рис. IV.37.
Обработка многоканальных хроматограмм. Как правило,
детектор хроматографа выдает только один сигнал, соответ-
ствующий отклику на поступающие в него из колонки компо-
ненты. Этот сигнал воспринимается программой как один ка-
нал данных. Если же детектор может измерять две и более
независимых характеристик компонента в подвижной фазе, то
говорят о многоканальной хроматографической системе, а хро-
матограмма, отображающая эти данные, называется многока-
нальной.
Далеко не любой хроматограф обеспечивает возможность
получения многоканальных хроматограмм. Типичный пример
многоканальной системы — хроматограф “Милихром-А02”, де-
тектор которого позволяет производить параллельное измере-
ние оптической плотности раствора на нескольких длинах волн
(рис. IV.38).
420
Иногда используются также многоканальные хроматографи-
ческие системы, в которых детектирование одной пробы после
разделения на колонке производится одновременно двумя или
более детекторами, например детектором по теплопроводности
и пламенно-ионизационным. Многоканальное детектирование
обеспечивает целый ряд преимуществ, вот некоторые из них:
— увеличенный линейный диапазон определяемых концентра-
ций веществ;
— сравнение спектров и распознавание пиков по спектрам;
— проверка гомогенности хроматографических пиков;
— количественный анализ групп перекрывающихся пиков,
при необходимости учитывающий наличие априорной инфор-
мации о спектрах некоторых компонентов группы.
Использование этих преимуществ становится возможным
только при условии программной поддержки указанных функ-
ций со стороны программного обеспечения. Такая поддержка
обеспечивается, например, модулем спектрального анализа
программы “МультиХром”.
IV.6. МЕТРОЛОГИЯ ХРОМАТОГРАФИЧЕСКОГО
АНАЛИЗА
IV.6.1. Общие сведения
Химики-аналитики, в том числе и профессионалы с большим
опытом, обычно имеют довольно поверхностное представление
о метрологии. Поэтому указания метрологических органов о
выполнении ряда обязательных требований они часто восприни-
мают как необоснованные придирки. Между тем Законом РФ об
обеспечении единства измерений ряд важных применений хими-
ческого анализа (здравоохранение, охрана окружающей среды,
обеспечение безопасности труда и др.) отнесен к сфере Госу-
дарственного метрологического надзора и контроля. Поэтому
в настоящий раздел включено изложение основных принципов
метрологии — науки об измерениях [391] — применительно к
химическому анализу.
Общая схема рассуждений здесь такова:
— объектом измерения является тело (физическая система,
процесс, явление и т.п.), которое характеризуется одной или
несколькими измеряемыми физическими величине ми;
421
— указанные физические величины подлежат измерению*',
— для каждой физической величины устанавливается еди-
ница измерения',
— размер единицы реализуется в виде меры,',
— измерение заключается в сравнении с мерой',
— отклонение результата измерения от истинного значения
измеряемой величины характеризуют погрешностью.
Переходя к химическому анализу, в первую очередь необ-
ходимо определить измеряемую физическую величину Конеч-
ной целью количественного химического (в частности, хрома-
тографического) анализа является измерение содержания од-
ного или нескольких,определяемых компонентов в объекте ана-
лиза**. По определению ИЮ ПАК [392], количественный анализ
вещества — это экспериментальное определение концентрации
химических соединений или их форм в анализируемом веществе,
выраженное в виде границ доверительного интервала или числа
с указанием стандартного отклонения.
Следовательно, в качестве физической величины можно на-
звать массовую концентрацию, т.е. отношение массы какого-
либо компонента, содержащегося в пробе, к общему объему
этой пробы. Это отношение определяет и размерность мас-
совой концентрации — М/L3, а единицей измерения является
кг/м3. Рекомендовано применение кратных и дольных единиц —
кг/дм3, г/см3 и т. п., допускается применение внесистемных еди-
ниц, например кг/л, мг/мл [393].
Однако определенная таким образом концентрация не явля-
ется физической величиной, а представляет собой некоторую
математическую абстракцию, за которой не стоят реальные фи-
зические свойства. Так, назвав значение массовой концентра-
ции, равное 10-3 мг/мл, мы не можем представить себе, идет
ли речь о газообразной, жидкой или твердой пробе. Поэтому
для определения массовой концентрации как физической вели-
чины необходимо указать конкретное вещество, масса которого
содержится в объеме пробы. Растворы различных веществ, на-
пример этанола и ацетона в воде, обладают совершенно различ-
ными физическими свойствами (показатель преломления, вяз-
кость, электропроводность и др.), поэтому их концентрации
являются различными физическими величинами, которые не-
*Результат измерения представляет собой параметр физической мо-
дели объекта измерения.
**Термин “анализ”относится к объекту, а термины “определение со-
держания” или “измерение концентрации” — к определяемому веществу.
Поэтому формулировка “анализ воздуха на содержание оксида углерода”
правильна, а “анализ оксида углерода в воздухе” неправильна.
422
возможно непосредственно сравнить друг с другом. Следова-
тельно, под концентрацией в химическом анализе подразуме-
вают несчетное множество различных физических величин, под-
лежащих измерению.
Под измерением понимают сравнение измеряемой физической
величины с однородной (одноименной) физической величиной,
принятой за единицу. Сама процедура сравнения может быть
сложной, многоступенчатой, иметь опосредованный характер,
но сущность измерительного процесса при этом не меняется:
это всегда сравнение с мерой*. В официальном документе по
метрологии [394] указано, что если принцип измерений — это
физическое явление или процесс, положенный в основу измере-
ний, то метод измерений — это прием или совокупность прие-
мов сравнения измеряемой физической величины с ее единицей
в соответствии с реализованным принципом измерений. Вполне
очевидно, что для каждой физической величины должна быть
установлена своя единица, так как невозможно пользоваться од-
ной единицей для измерения различных физических величин. В
химическом анализе используется несчетное множество единиц,
которые называют одним общим термином “единицы концентра-
ции”.
Особое значение имеют узаконенные единицы, установленные
для применения в стране в соответствии с законодательными ак-
тами. В России Законом “Об обеспечении единства измерений”
введена в действие Международная система единиц СИ, приме-
нение которой является обязательным. Как известно, она вклю-
чает 7 основных единиц для следующих физических величин:
длина (м), масса (кг), время (с), температура (К), сила электри-
ческого тока (А), сила света (кд), количество вещества (моль).
Когерентные производные единицы СИ, как правило, образуют
с помощью простейших уравнений связи между величинами, в
которых числовые коэффициенты приняты равными 1 [393].
Все выполненные измерения должны удовлетворять осново-
полагающему требованию единства измерений [394, 395]. Это
такое состояние измерений, при котором их результаты выра-
жаются в узаконенных единицах, размеры которых в устано-
вленных пределах равны размерам единиц, воспроизводимых
первичными эталонами, а погрешности результатов измерений
известны и с заданной вероятностью не выходят за установлен-
ные пределы. Широко применяемый термин “метрологическое
* Другим весьма распространенным способом получения количествен-
ной информации наряду с измерением является счет. При этом сравнение
с мерой не производится: единицей при счете являются сами подсчиты-
ваемые объекты.
423
обеспечение измерений”, по сути, является синонимом термина
“обеспечение единства измерений”. Понятие единства измере-
ний учитывает два различных аспекта: узаконенные единицы и
известные (в смысле оценки) погрешности.
Лля воспроизведения и хранения размера единицы физиче-
ской величины создается ее эталон. Эталон, обеспечивающий
воспроизведение единицы с наивысшей в стране точностью, на-
зывается первичным. Рабочие эталоны (которые раньше назы-
вали “образцовыми средствами измерений”) предназначены для
передачи размера единицы рабочим средствам измерений.
В России принята централизованная передача размеров еди-
ниц при помощи поверочных схем. Поверочная схема — это
документ, устанавливающий средства, методы и точность пе-
редачи размера единиц от эталонов рабочим средствам изме-
рений. Все без исключения рабочие средства измерений под-
лежат метрологическому контролю и надзору в форме поверки
или калибровки. Содержание этих операций одинаково: уста-
навливается соотношение между значением величины, получен-
ным с помощью данного рабочего средства измерений, и соот-
ветствующим значением величины, определенным с помощью
эталона, с целью определения метрологических характеристик
этого средства измерений и его пригодности к применению. По-
верка выполняется органом Государственной метрологической
службы. В соответствии с Законом “Об обеспечении единства
измерений” поверке подлежат средства измерений, применяе-
мые, в частности, для целей здравоохранения, охраны окружа-
ющей среды, обеспечения безопасности труда.
Перечисленные элементы — единицы, эталоны, поверочные
схемы и метрологический надзор — входят в Государственную
систему обеспечения единства измерений (ГСИ) и должны обес-
печить получение и выражение любого результата измерений в
узаконенных единицах.
В химическом анализе централизованная передача размеров
единиц невозможна или затруднена. Это связано с нецелесо-
образностью, а часто и с невозможностью создания эталонов и
поверочных схем для всех химических соединений, подлежащих
анализу.
Рассмотрим в качестве примера хроматографическое определение эти-
лового спирта в водном растворе*. Будем считать, что имеется удовле-
*Водные растворы с заданной концентрацией этанола могут быть ис-
пользованы, например, при оценке качества различных сортов водки и
других алкогольных напитков, а также для приготовления аттестован-
ных парогазовых смесей с известным содержанием этанола в воздухе или
инертных газах для поверки газоанализаторов, применяемых при поли-
424
творительная методика хроматографического разделения: пик этанола
полностью отделен от остальных пиков хроматограммы, а амплитуда его
достаточна для качественного измерения. Чтобы определить по этому
пику концентрацию этанола, необходимо установить масштаб шкалы в
единицах концентрации этилового спирта. Для этого гравиметрическим
(весовым) методом из чистого этилового спирта готовят раствор и атте-
стуют его по процедуре приготовления. Записывают хроматограмму в
тех же условиях и, сопоставляя площади или высоты пиков на двух хро-
матограммах, рассчитывают содержание этанола в проанализированном
растворе.
Описанная процедура представляет собой простейший вариант граду-
ировки, представляющей собой установление зависимости между значе-
ниями величин на входе и выходе хроматографа. Носителем единицы
здесь является чистый этиловый спирт, использованный для пригото-
вления градуировочного раствора. Качество единицы определяется ка-
чеством исходного этанола. Если в различных лабораториях для градуи-
ровки используют различный этиловый спирт, то и полученные единицы
отличаются друг от друга. Однако существование единого для всей Рос-
сии исходного этилового спирта, используемого для всех определений
этанола в различных объектах, не предусмотрено никакими нормативно-
техническими документами (НТД), поэтому создание общегосударствен-
ной поверочной схемы для измерения концентрации этилового спирта не-
возможно.
В химическом анализе повсеместно принято децентрализо-
ванное воспроизведение размеров единиц. Чаще всего для этого
используют чистые вещества и абсолютные измерения (т. е.
прямые измерения одной или нескольких основных величин и
(или) использование значений физических констант). В приме-
няемых для этой цели чистых веществах должно быть нормиро-
вано содержание основного вещества.
Другой особенностью химического анализа является про-
блема избирательности. В метрологическом плане это означает
отделение определяемой физической величины (или сигнала от
нее) от других физических величин. Весь арсенал методов ана-
литической химии направлен на решение проблем избиратель-
ности и создание избирательных методов анализа. Хромато-
графия — один из таких наиболее удачных методов. В класси-
ческой метрологии такая проблема возникает весьма редко (не-
трудно отделить длину от массы), и там ограничиваются поня-
тием влияющей физической величины — физической величины,
оказывающей влияние на размер измеряемой физической вели-
чины и (или) результат измерения.
Цейском контроле выдоха провинившихся (подозреваемых в нетрезвом со-
стоянии) водителей автотранспорта.
425
Обратимся снова к примеру. При хроматографическом анализе почвы
на содержание хлорорганических пестицидов исходную пробу заливают
смесью гексана и ацетона для экстракции органических веществ. Затем
экстракт фильтруют через слой сульфата натрия, упаривают до 5—7 мл
и вводят в хроматограф с электронозахватным детектором (ЭЗД). Допу-
стим, что в гексане, используемом в качестве экстрагента, присутствует
примесь (содержание которой определяется пятым знаком после запятой),
вызывающая сигнал ЭЗД. Вполне очевидно, что результат анализа бу-
дет неправильным. Следовательно, требования к чистоте веществ, при-
меняемых в анализе, могут быть исключительно высокими, но избира-
тельными: ограничивается содержание примесей с определенными свой-
ствами.
В связи с подобными соображениями возникла квалификация
реактивов, специализированных по методам анализа: “чистый
для спектроскопии”, “чистый для хроматографии” и т.п. Под
“чистым” здесь понимают вещество, не дающее сигнала при
анализе соответствующим методом. Необходимость детальной
спецификации (в НТД на реактив, “чистый для хроматогра-
фии”, необходимо указывать тип детектора, так как “чистый”
для ЭЗД может быть “грязным” для ПИД и наоборот) и расту-
щая чувствительность методов делают эти реактивы не очень
надежными.
Особое место в анализе занимают стандартные образцы (СО)
состава — образцы веществ с установленными в результате
метрологических исследований значениями одной или более
величин, характеризующими состав этого вещества [167, 396].
Основной особенностью СО является то, что он аттестован
строго по определенному назначению, специфика которого яв-
ляется определяющей при его создании и аттестации. Не су-
ществует стандартного образца гексана вообще, но существует
стандартный образец состава гексана ГСО 2583-83, предназна-
ченный для градуировки хроматографов по гексану.
Определение состава СО проводится методами химического
анализа в квалифицированных лабораториях и, по существу,
является результатом экспертной оценки. Наличие СО разного
уровня (государственные стандартные образцы — ГСО, отра-
слевые стандартные образцы — ОСО и стандартные образцы
предприятий — СОП) узаконивает процедуру децентрализован-
ного воспроизведения размеров единиц.
Следующей особенностью химического анализа является не-
обходимость тщательного изучения физической модели измери-
тельной процедуры, которая обычно включает ряд последова-
тельных операций. Известно, что результатом измерения явля-
ется не абстрактная физическая величина, а параметр физи-
426
ческой модели* объекта измерения [397]. Модель часто вхо-
дит в наши рассуждения совершенно незаметно, ее очевидность
как бы сама собой подразумевается. Измеряя штангенцирку-
лем диаметр шариковой ручки, мы подразумеваем, что шарико-
вая ручка — это прямой круговой цилиндр, и эта физическая
модель введена словом “диаметр”. Если сечение ручки пред-
ставляет собой эллипс, то результат неверен, так как найден-
ное значение может лежать в пределах между большой и малой
осями эллипса в зависимости от положения ручки в момент из-
мерения. В этом случае замена штангенциркуля более точным
микрометром не уменьшит погрешности, поскольку она обусло-
влена неадекватностью модели — несоответствием объекта из-
мерения и принятой физической модели этого объекта.
Измеряя плотность алюминиевого бруска гидростатическим
взвешиванием, мы подразумеваем, что брусок состоит из од-
нородного сплошного материала — эта модель введена словом
“плотность”, так как принято считать, что плотность характе-
ризует однородный сплошной материал. Если внутри бруска
имеется раковина, то результат снова неправилен и замена тех-
нических весов аналитическими не уменьшит погрешности. По-
грешность, обусловленная неадекватностью модели, не может
быть уменьшена использованием более точных средств измере-
ния, а требует проведения специальных исследований.
Как видно из приведенного выше примера анализа почвы на
содержание хлорсодержащих пестицидов, физическая модель
анализа может быть достаточно сложной, а правильность ре-
зультата зависит не столько от точности измерения выходного
сигнала, сколько от правильности выбора и тщательности вы-
полнения отдельных стадий и процедур.
В связи с изложенными особенностями химического анализа
в число нормативно-технических документов (НТД) системы
ГСИ входит категория методик выполнения измерений (МВИ),
включающая методики количественного химического анализа
(МКХА) [398]. Необходимость в МВИ возникает в тех случаях,
когда правильность результата измерения зависит не только от
применяемых средств измерений, но и от принятой процедуры.
МВИ — это установленная совокупность операций и пра-
вил при измерении, выполнение которых обеспечивает полу-
чение необходимых результатов измерения с гарантированной
точностью в соответствии с принятым методом. Другими сло-
вами, МВИ — это совокупность метода измерения, технических
*Под физической моделью понимают описание измеряемого объекта
или всех стадий измерительного процесса с полнотой, достаточной для
количественной оценки.
427
средств (мер и измерительных приборов), правил подготовки и
проведения анализа, обработки и представления результатов.
Обычно воспроизведение описанной в литературе методики
или разработка новой выполняется специалистами аналитиче-
ской лаборатории, опирающимися на свой опыт и на имеющи-
еся оборудование, материалы, реактивы и персонал. Вопросы
обеспечения требуемой точности решаются при этом в рамках
задач предприятия, а их корректность остается на совести ана-
литиков. Как правило, наибольшее внимание уделяется внутри-
лабораторной воспроизводимости. Чаще всего эта воспроизво-
димость определяется в процессе выполнения анализа и припи-
сывается полученному результату.
В связи с широким распространением химического анализа в
промышленности, сельском хозяйстве, здравоохранении и дру-
гих видах общественной деятельности требования к анализу
стали постепенно изменяться. На первое место выдвигается воз-
можность сравнения результатов анализа одних и тех же объек-
тов, полученных в различных лабораториях, в разное время и в
различных условиях. Эти требования, по существу, идентичны
требованию единства измерений [394] и поэтому естественно ис-
кать их решение в рамках метрологии, где накоплен большой
опыт решения аналогичных проблем. Речь идет, следовательно,
о методиках, которые должны быть реализованы не в одной, а
в различных лабораториях разных ведомств с различной мате-
риальной базой и разной квалификацией персонала.
В соответствии со ст. 13 Закона Российской Федерации “Об
обеспечении единства измерений” к сфере Государственного
метрологического контроля и надзора относятся следующие
виды деятельности, в которых широко применяется химический
анализ: здравоохранение, ветеринария, охрана окружающей
среды, обеспечение безопасности труда, обеспечение обороны
государства, испытание и контроль качества продукции в целях
определения соответствия обязательным требованиям государ-
ственных стандартов, обязательная сертификация продукции и
услуг и некоторые другие. Методики, применяемые в сфере рас-
пространения государственного надзора, подлежат обязатель-
ной метрологической аттестации. За аттестованными методи-
ками устанавливается государственный метрологический над-
зор.
В заключение необходимо отметить, что за рубежом принято
другое понимание метрологии химического анализа, построен-
ное на концепции неопределенности [399-401]. Это не другая си-
стема терминов и определений, а другая трактовка самих основ
метрологии. В этой концепции приняты следующие постулаты.
428
1. Измерять можно не только физические, но и многие другие
величины, определенные однозначно по установленным прави-
лам в том числе по условию, когда отсутствует точное указание
формы определяемого вешества (например, содержание пара-
финовых углеводородов в бензине) или с помощью процедуры
регламентированного воздействия на объект измерения (напри-
мер, потеря массы при нагревании в заданных условиях).
2. Сравнение с мерой — лишь один из возможных методов
измерения определяемой величины.
3. Сама мера вводится в соответствии с процедурой опреде-
ления измеряемой величины.
4. Наряду с погрешностью вводится новая характеристика
измерения — неопределенность. Не взамен погрешности, и не в
дополнение к ней, а как независимая характеристика.
5. Неопределенность вводится как параметр, связанный с ре-
зультатом измерения и характеризующий рассеяние значений,
которые могут быть с достаточным основанием приписаны из-
меряемой величине.
6. Важнейшими составляющими неопределенности являются
неполное определение измеряемой величины и нечеткое описа-
ние отдельных стадий измерительной процедуры, обусловлива-
ющих правильность результата измерения.
7. Погрешность и неопределенность выступают как взаимоис-
ключающие характеристики измерения. Выбор какой-то одной
из них определяется особенностями измерительной процедуры.
В настоящее время обсуждается возможность гармонизации
отечественных и зарубежных подходов к проблеме химико-ана-
литических измерений.
IV.6.2. Метрологическая аттестация аналитических
методик
IV.6.2.1. Общие сведения
Метрологическая аттестация (МА) МКХА — это исследова-
ние, направленное на определение такой процедуры, условий и
режимов выполнения всех стадий анализа, которые обеспечи-
вают минимальные погрешности, априорную оценку значений
этих погрешностей и оформление документа с указанием по-
лученных результатов. Основная цель аттестации — подтвер-
ждение возможности измерений по рассматриваемой МКХА с
погрешностью, не превышающей указанную. Аттестация явля-
ется одним из основных этапов разработки МКХА и выполня-
429
ется, как правило, лабораторией, разрабатывающей методику.
В с дельных случаях МА может быть выполнена организацией,
аккредитованной Госстандартом на право аттестации МКХА в
определенной области [398].
Для рассматриваемого круга методик важнейшим требова-
нием является возможность “тиражирования”, т. е. воспроиз-
ведения методики с сохранением метрологических характери-
стик в различных лабораториях. Это накладывает весьма жест-
кие требования на текст методики: на полноту и однозначность
описания всех процедур и операций, применяемых средств из-
мерений и других технических средств, стандартных образцов,
материалов, реактивов, методов градуировки, измерения ана-
литического сигнала и расчета концентраций. Однако на прак-
тике полностью удовлетворительные тексты методик встреча-
ются весьма редко и представляют скорее исключение, чем пра-
вило. В большинстве случаев текст методики используют как
рекомендацию, которую корректируют, изменяют или допол-
няют в зависимости от квалификации персонала и материально-
технического состояния лаборатории. Естественно, что эта
практика приводит в ряде случаев к появлению непредвиден-
ных источников погрешностей. Выходом из этого затруднения
является включение в методику правил и процедуры оператив-
ного контроля показателей качества результатов измерений.
Методики, применяемые в сферах распространения государ-
ственного метрологического контроля и надзора, перед утвер-
ждением подлежат экспертизе в одном из Государственных на-
учных метрологических центров (ГИМН), несущих ответствен-
ность за обеспечение единства измерений в области химиче-
ского анализа. В настоящее время таких ГНМП четыре: НПО
“ВНИИМ им. Д. И. Менделеева” (г. Санкт-Петербург), ВНИИ
МС (метрологической службы) (г. Москва), Уральский НИИ
метрологии (УНИИМ) (г. Екатеринбург) и ВНИИФТРИ
(пос. Менделееве Моск. обл.). Методика вводится в действие
приказом метрологической службы Государственного органа
управления (Министерства, ведомства) для лабораторий этого
ведомства.
Метрологическая аттестация МКХА включает следующие
обязательные этапы.
1. Назначение измеряемой физической величины (концентра-
ции химического соединения или его формы с детальным опи-
санием) в соответствии с ГОСТом 8.417-81 ГСИ “Единицы фи-
зических величин”. Во многих случаях необходимо дать де-
тальное (желательно, исчерпывающее) описание измеряемой ве-
430
личины, так как она может быть определена соглашением или
связана с регламентированной процедурой.
2. Воспроизведение единицы измеряемой физической вели-
чины в требуемом диапазоне на основе стандартных образцов
по ГОСТу 8.315-78 либо на основе чистых веществ и абсолют-
ных измерений, либо на основе соглашения или принятой в ме-
тодике регламентированной процедуры.
3. Оценка погрешности воспроизведения единицы измеря-
емой физической величины путем прослеживания до государ-
ственных стандартных образцов или до эталонов основных ве-
личин.
4. Составление или анализ физической модели МКХА, вклю-
чающей все этапы выделения измеряемой физической величины
(пробоотбор, селективная пробоподготовка, хроматографиче-
ское или химическое разделение, спектральное или другое се-
лективное выделение сигнала).
5. Градуировка измерительной системы с обоснованием аде-
кватной модели градуировочной характеристики и оценкой по-
грешности градуировки в принятом диапазоне измерения
МКХА.
6. Поэтапный экспериментальный или расчетно-эксперимен-
тальный контроль адекватности и количественная оценка пре-
дельной систематической погрешности каждого этапа, обусло-
вленной допустимой неадекватностью модели.
7. Оценка случайной составляющей погрешности в принятом
диапазоне измерения МКХА.
8. Определение доверительных границ суммарной погрешно-
сти результата анализа в принятом диапазоне измерения
МКХА.
9. Установление соответствия показателей точности МКХА
заданным нормам точности.
10. Оценка полноты и правильности назначения параметров
оперативного контроля точности МКХА.
Приведенный перечень основных этапов охватывает три раз-
личные составные части разработки любой методики:
— воспроизведение единицы измеряемой физической величи-
ны (п. п. 1-3) и передачу её размера рабочему средству измере-
ния (п.5);
— анализ модели (п. 4), оценку характеристик погрешности
во всём диапазоне (п. п. 5-8) и оценку пригодности МКХА (п. 9);
— оперативный контроль показателей качества результатов
измерений (п. 10).
Воспроизведение единицы концентрации определяемого ве-
щества обычно выполняют путем создания аттестованных сме-
431
сей веществ, для которых известны аттестованное (номиналь-
ное) значение концентрации и его погрешность. Для жидких
и реже для газовых смесей аттестация выполняется на основе
тщательного анализа процедуры приготовления смесей из чи-
стых веществ с учетом их чистоты и погрешностей применяе-
мых средств измерений (весы, мерная посуда, термометры, ма-
нометры и др.). В литературе этот процесс оценки погрешности
полученного значения единицы концентрации на основе приня-
той модели и погрешностей используемых средств измерений
по аналогии с англоязычным термином traceability иногда не
совсем удачно называют прослеживаемостью* (см.п.3). В не-
которых случаях контроль процедуры воспроизведения единиц
содержания компонентов может быть реализован в рамках по-
верочной схемы, опирающейся на Государственный первичный
эталон (ГЭТ) единиц молярной доли и массовой концентрации
компонентов в газовых средах ГЭТ 154-88 [402].
Для создания аттестованных смесей могут быть использо-
ваны две категории измерительных средств: образцовые меры
и стандартные образцы состава, а также чистые вещества с
надёжно установленным содержанием основного вещества. Под
мерой понимают техническое средство,имеющее нормирован-
ные метрологические характеристики, воспроизводящее и (или)
хранящее единицу физической величины, размер которой при-
нимается неизменным (в пределах установленной погрешности)
в течение заданного времени. В качестве образцовых мер мо-
гут быть использованы источники микропотоков газов и паров,
применяемые совместно с термодиффузионными генераторами
[402]. Стандартный образец — это образец вещества (матери-
ала) с установленными в результате метрологической аттеста-
ции значениями одной или более физических величин, характе-
ризующими свойство или состав этого вещества (материала).
Наиболее ответственным моментом является выбор веществ
для приготовления смесей. Для этих веществ обязательно на-
личие сертификата или другого документа, подтверждающего
квалификацию. Наиболее желательно использование ГСО (ко-
гда они имеются).
IV.6.2.2. Оценка характеристик погрешности аналитических
методик
Рассмотрим более подробно оценку характеристик погрешно-
сти МКХА. Наибольший интерес представляют те хроматогра-
*И совершенно недопустимо переводить traceability как единство изме-
рений.
432
фические методики, погрешность которых изучена до их прак-
тического применения, т. е. на этапах разработки и аттестации.
Требования к точности измерений устанавливают в виде преде-
лов допускаемых значений характеристик абсолютной или от-
носительной погрешности. Форма приведенной погрешности из-
мерений не используется [403]. Нормативные документы ГСИ
допускают две формы представления характеристик погрешно-
сти таких методик:
— задание границ интервала, в которых погрешность мето-
дики находится с принятой вероятностью Р;
— указание наибольшего возможного среднего квадратич-
ного отклонения (СКО) случайной составляющей погрешности
методики и задание границ интервала, в которых неисключен-
ная систематическая погрешность методики находится с задан-
ной вероятностью Р.
Первая форма принята для методик, в которых результат
анализа рассчитывают по одному измеренному значению сиг-
нала, вторая — для таких, в которых результат рассчитывают
по среднему значению сигнала из двух или нескольких измере-
ний. В обоих случаях для норм принято указывать пределы до-
пускаемых значений ( соответствующие доверительной вероят-
ности Р = 1), а для приписанных характеристик — наибольшие
возможные значения, соответствующие Р = 0,95. Если характе-
ристики погрешности не являются окончательными, а предна-
значены для совместного использования с другими характери-
стиками, то вместо интервальных характеристик целесообразно
использовать точечные.
Методом выявления основных источников систематических
погрешностей хроматографического анализа является изучение
физической модели МКХА, включающее как эксперименталь-
ные исследования, так и расчетные оценки. Как правило, мето-
дика состоит из ряда операций, таких, как поглощение опреде-
ляемых компонентов в различных ловушках и их последующее
выделение, экстракция, проведение химических реакций, кон-
центрирование и др., и лишь как заключительный этап — хро-
матографическое разделение, детектирование и измерение ана-
литического сигнала.
Например, для сравнительно простой методики определе-
ния оксида углерода в воздухе в диапазоне концентраций 1-
40 мг/м3 методом реакционной газовой хроматографии (конвер-
сия оксида углерода в метан в среде водорода на никелевом
катализаторе) формирование модели основано на анализе сле-
дующих основных операций методики.
433
Анализируемый воздух отбирают в медицинский шприц объ-
емом 100 см3, предварительно несколько раз прокачав шприц
этим воздухом.
Воздухом из шприца продувают дозу газового крана объемом
~ 1 мл, обеспечивая десятикратную продувку.
Вводят воздух из дозирующей петли крана в хроматографи-
ческую колонку с молекулярным ситом СаА и выполняют хро-
матографический анализ, используя в качестве газа-носителя
водород. Выходящий из колонки газовый поток пропускается
через реактор-метанатор, заполненный активированным нике-
левым катализатором ([277], см. также раздел IV 2.5), непре-
рывно продуваемый водородом. В реакторе оксид углерода
конвертируется в метан; конверсии подвергается также диок-
сид углерода. Регистрируют хроматограмму по показаниям
пламенно-ионизационного детектора. Измеряют количествен-
ные параметры пика оксида углерода, детектируемого в форме
метана.
Для количественной оценки содержания СО по измеренной
высоте h заполняют дозирующую петлю газового крана атте-
стованной смесью СО в воздухе с известным содержанием Со
и проводят такой же анализ, регистрируя высоту пика оксида
углерода ho
Расчет концентрации С оксида углерода в анализируемом
воздухе выполняют по формуле
С = С0^. (IV.71)
По
Найденное значение концентрации оксида углерода приводят
к нормальным условиям.
Уравнение (IV.71) основано на следующих предположениях,
составляющих физическую модель измерительного процесса.
1. Отсутствие сорбции или других потерь оксида углерода
на стадиях отбора пробы и ее ввода в хроматограф.
2. Полное отделение пика оксида углерода от пиков дру-
гих веществ: а) способных конвертироваться на никелевом ка-
тализаторе до углеродсодержащих органических соединений;
б) способных детектироваться пламенно-ионизационным детек-
тором.
3. Постоянная степень конверсии оксида углерода в метан во
всем диапазоне концентраций в течение времени, охватываю-
щего анализ исследуемого воздуха и градуировочной смеси.
4. Прямо пропорциональная зависимость между высотой h
пика метана,образованного из оксида углерода, и его концен-
трацией С в детекторе.
434
5. Идентичность условий дозирования газовым краном (объ-
ем дозирующей петли, температура и давление) анализируе-
мого воздуха и градуировочной смеси.
6. Анализируемый воздух рассматривается как идеальный
газ.
Далее необходимо выполнить количественную оценку
погрешности, вносимой в результат анализа каждой стадией
аналитического процесса. Чаще всего такая оценка требует
проведения специального эксперимента. В отдельных случаях,
когда физические закономерности хорошо изучены, возможна
расчетная оценка.
В рассмотренном примере неисключенную систематическую
погрешность (НСП), связанную с п.п.1 и 2 модели, оценивали на
основании анализа литературных данных о разделении летучих
веществ на молекулярных ситах СаА и рассмотрения реальных
хроматограмм, полученных по МКХА. Она оказалась незначи-
мой.
НСП, возникающую вследствие п.п.З и 4 модели, оценивали,
изучая наличие прямо пропорциональной зависимости между С
и h известным методом экспоненциального разбавления (метод
Лавлока) [404]. Он заключается в создании непрерывного газо-
вого потока, в котором концентрация С оксида углерода законо-
мерно убывает во времени по закону, близкому к экспоненциаль-
ному, и в измерении сигнала h как функции времени разбавле-
ния. Найденное значение НСП характеризуется относительным
CKO (ОСКО), не превышающим 0,015.
НСП, возникновение которых связано с п. п. 5 и 6 модели,
оценивали в предположении, что наибольшие изменения баро-
метрического давления за рабочую смену А/о не превышают
4 кПа (~ 30 мм рт. ст.), а наибольшие возможные изменения тем-
пературы в лаборатории АТЬ не превышают 10 К. При этом
относительное значение НСП не превышает 0,05.
Если принять погрешность аттестации образцовой смеси, по
которой определяется градуировочный коэффициент, 0аТт =
0,05, то суммарная неисключенная относительная системати-
ческая погрешность не превышает 0,08. Оценка выполнена по
правилам, изложенным в [405, 406].
Оценку случайной составляющей погрешности выполняют
по обычным правилам, которые изложены во многих руковод-
ствах [407-409]. Для описанной выше методики найдено значе-
ние ОСКО, равное 0,013 для числа степеней свободы f = 18 (из
двух серий по 10 измерений в начале и в конце диапазона гра-
дуировки). Наибольшее возможное значение СКО (оценка на
основании [410]) не превышает 0,02.
435
В экспериментах обязательно должен повторяться весь цикл
анализа, а не только его заключительная стадия, которой ча-
сто является хроматографический процесс. Так, в приведенном
выше примере анализа пестицидов (см. с. 425-426) необходимо
в каждом эксперименте заново проводить извлечение и концен-
трирование пестицидов, а не подвергать многократному хрома-
тографическому анализу один и тот же экстракт.
Следует также помнить, что ценность полученных результа-
тов в решающей степени зависит от того, удовлетворяет ли
МКХА модели стационарного случайного процесса [411], т. е.
сохраняются ли полученные статистические оценки достаточно
длительное время.
IV.6.2.3. Градуировка хроматографической аппаратуры
Рассмотрим теперь занимающую центральное место в хро-
матографическом анализе процедуру градуировки [412-
414], при которой устанавливается действительное значение фи-
зической величины (X), приписанное тому или иному значе-
нию выходного сигнала (У). Она должна предусматривать
проверку адекватности выбранной градуировочной характери-
стики (ГрХ).
В хроматографии часто полагают, что сигнал прямо пропор-
ционален измеряемой концентрации, т. е. ГрХ — это прямая
линия, проходящая через начало координат. На этом предполо-
жении основаны метод нормализации, внутреннего и внешнего
стандарта, стандартной добавки и др. Логической основой та-
кого допущения является мысль о том, что отсутствие концен-
трации означает отсутствие сигнала, так как высота и площадь
пика отсчитываются от нулевой линии между пиками, которая и
есть запись результата “холостого” опыта. В действительности
в присутствии соседних “мешающих” пиков на хроматограмме
и особенно при заметном изменении содержания мешающих со-
единений ГрХ не проходит через нуль, а описывается линейным
уравнением вида
У = AjX + Ад,
где У — регистрируемый сигнал; X — измеряемая концентра-
ция; Ад и Ai — константы.
Для выполнения градуировки обычно готовят ряд аттесто-
ванных смесей, равномерно накрывающих диапазон МКХА, и
поочередно анализируют их, получая совокупность п пар зна-
чений (Xi, Yi).
Процедура построения градуировочной характеристики
включает и оценку ее погрешности — погрешности действитель-
436
ного значения величины X, приписанной каждому значению сиг-
нала Y в результате градуировки.
Градуировочная характеристика может быть представлена
в виде формулы, графика или таблицы. Г рафик зависимости
сигнала от концентрации удобен для расчетов, но не позволяет
оценить погрешность ГрХ. Поэтому чаще всего для построения
ГрХ используют метод наименьших квадратов (МНК). Сама
процедура МНК как метода выбора наилучших значений ко-
эффициентов Ai иАо ГрХ не накладывает никаких ограниче-
ний на вид функций распределения погрешностей входных (X)
и выходных (У) сигналов [413]. Однако назвав частное от де-
ления остаточной суммы квадратов на число степеней свободы
остаточной дисперсией, мы вводим модель: дисперсии сигналов
Y во всем диапазоне градуировки однородны, т е. распреде-
лены нормально и принадлежат одной генеральной совокупно-
сти. Проверка этого обстоятельства выполняется по критерию
Бартлета [409].
Если диапазон градуировки (отношение наибольшей концен-
трации к наименьшей) превышает 10, то обычно условие одно-
родности для абсолютных дисперсий нарушается и оценка по-
грешности является неоправданной. В этом случае необхо-
димо присвоить различные веса измерениям, выполненным при
различных концентрациях, и проверить однородность взвешен-
ных дисперсий. Веса необходимо подобрать таким образом,
чтобы условие однородности выполнялось. На практике в боль-
шинстве случаев (при использовании ПИД и ДТП) однород-
ными оказываются относительные дисперсии Dy/Y2, чему со-
ответствует примерное постоянство относительной погрешно-
сти сигнала ДУ/Y. При этом вес Р = 1/У2. При использова-
нии фотоионизационного детектора однородными оказываются
отношения Dy/Y, чему соответствует примерное постоянство
ДУ/л/У и вес Р= 1/У.
Применение МНК с соответствующей весовой функцией обес-
печивает корректную оценку погрешности градуировки и по-
грешности значения концентрации, найденной по измеренному
значению сигнала.
Для нелинейных ГрХ, характерных для методик с использо-
ванием ЭЗД, ПФД (для определения серы) и ФИД с высоко-
частотным возбуждением источника света, необходимо вначале
подобрать линеаризующее преобразование и применять проце-
дуру МНК к преобразованным значениям сигнала. Например, в
МКХА воды на содержание летучих хлорированных углеводо-
родов, в которой использован ДПР, ГрХ для всех компонентов
имеет вид кривой с плавным переходом от прямой пропорци-
437
ональности к насыщению. Для линеаризации применено пре-
образование измеренной высоты пиков h в новый сигнал Z по
формуле Z = h/(A\h + Ao). Параметры этого уравнения для ка-
ждого компонента найдены МНК, а расчет концентрации С по
измеренному значению h выполняется по “обратной” формуле
С — Aoh/ (1 — A\h).
МНК позволяет оценить адекватность построенных ГрХ на
основе сравнения по критерию Фишера двух дисперсий: относи-
тельной дисперсии сигнала в отдельных точках ГрХ и остаточ-
ной дисперсии, характеризующей разброс точек относительно
прямой МНК [415]. Адекватность ГрХ в указанном смысле
означает, что при данной погрешности измерения выходного
сигнала улучшать найденные коэффициенты уравнения ГрХ не
имеет смысла.
МНК позволяет также сделать обоснованный выбор между
линейкой и прямо пропорциональной градуировочными харак-
теристиками двумя способами: путем оценки значимости коэф-
фициента Ад в уравнении ГрХ по критерию Стьюдента и путем
сравнения дисперсий линейной и прямо пропорциональной ГрХ
по критерию Фишера. Обычно эти оценки дают совпадающие
результаты.
МНК содержит громоздкие вычислительные процедуры с
большим числом значащих цифр, его использование предпола-
гает наличие ЭВМ и специализированного программного обес-
печения, но это окупается надежностью полученных ГрХ и оце-
нок погрешности.
Оценка суммарной погрешности МКХА предполагает сложе-
ние всех составляющих, обусловленных отбором пробы, ее хра-
нением, пробоподготовкой, аттестацией градуировочных сме-
сей и градуировкой, хроматографическим разделением и изме-
рением сигнала. Процедура сложения изложена в литературе
[405] и специальных НТД [406]. Она позволяет отдельно оценить
случайную составляющую погрешности.
Обязательным разделом МКХА является оперативный кон-
троль точности. Он заключается в оценке соответствия ха-
рактеристик погрешности результатов измерений, выполняе-
мых при контроле, установленным нормативам. В роли средств
контроля могут использоваться стандартные образцы, специ-
ально приготовленные пробы с известной добавкой определя-
емого компонента либо рабочие пробы. Процедуры контроля
могут включать методы добавок и разбавления пробы в раз-
личных комбинациях. Норматив контроля устанавливается в
зависимости от его алгоритма на основе известных характери-
стик погрешности МКХА.
438
IV.6.3. Пример метрологической аттестации МКХА.
Анализ измерительного процесса и
физическая модель методики
В этом разделе в качестве примера МА рассмотрим методику
количественного химического анализа питьевых, хозяйственно-
бытовых, поверхностных и очищенных сточных вод на содер-
жание летучих хлорированных углеводородов методом газовой
хроматографии. Она разработана АО “Цвет” (на первоначаль-
ной стадии), Хроматографическим центром Санкт-Петербург-
ского государственного университета и МП “РПем” [416], ат-
тестована НПО ВНИИМ им. Д. И. Менделеева (г. Санкт-Петер-
бург) и УНИИМ (г. Екатеринбург), включена в Реестр мето-
дик, допущенных к применению организациями Госкомэкологии
(ПНД Ф14.1:2:4. 10-95).
Методика устанавливает порядок количественного опреде-
ления летучих хлорированных углеводородов (ЛХУ) в питье-
вых, хозяйственно-бытовых, поверхностных и очищенных сточ-
ных водах и предназначается для экологических лабораторий,
контролирующих качество вод на содержание перечисленных
ниже компонентов в указанных диапазонах концентраций:
Диапазон концентраций, мкг/дм3
Хлороформ (ХФ) 2—350
1,2-Дихлорэтан (ДХЭ) 100-6000
Четыреххлористый углерод (ЧХУ) 0,3—24
Трихлорэтилен (ТХЭ) 2—350
Тетрахлорэтилен (ТеХЭ) 1—160
Доверительные границы погрешности результата определе-
ния содержания ЛХУ при доверительной вероятности 0,95 не
превышают ±25%.
Метод измерения включает отбор равновесной паровой фазы
над пробой анализируемой воды и ее последующий газохрома-
тографический анализ с детектированием по захвату электро-
нов.
Измеренный объем анализируемой воды помещают в пени-
циллиновый флакон известного объема и выдерживают при
фиксированной температуре до установления равновесия. При
этом часть летучих веществ, растворенных в воде, переходит
в паровую фазу. Концентрация каждого компонента в паровой
фазе прямо пропорциональна его первоначальной концентрации
в воде. Коэффициент пропорциональности зависит от коэффи-
циента распределения (связанного с температурой) и от со-
отношения объемов, занимаемых газом и водой во флаконе.
439
Во флаконе создается избыточное контролируемое давле-
ние газа-носителя. В момент соединения флакона с входом хро-
матографа определенная порция газа поступает на вход разде-
лительной хроматографической колонки. Объем этой порции
зависит от объема газовой фазы во флаконе и от перепада да-
влений между флаконом и колонкой.
Масса т? компонента, попавшего в колонку, пропорциональ-
на объему указанной порции газа и его концентрации в паровой
фазе. В процессе хроматографического разделения эта масса не
изменяется. Она рассчитывается по измеренным значениям Y
сигнала детектора постоянной скорости рекомбинации (ДПР)
по нелинейному уравнению. Адекватность этого уравнения и
численные значения коэффициентов устанавливают, измеряя за-
висимость сигнала Y от известной концентрации X определяе-
мого компонента в градуировочной пробе анализируемой воды.
При постоянных значениях всех влияющих факторов мето-
дики анализа (выделенных выше жирным шрифтом) и условий
хроматографического разделения между массой т,- и концен-
трацией X компонента в анализируемой воде существует прямо
пропорциональная зависимость.
На основании приведенного описания можно сформулиро-
вать следующие предположения, составляющие физическую
модель измерительного процесса.
1. Постоянство всех перечисленных влияющих факторов ме-
тодики анализа.
2. Полное отделение пика определяемого компонента от пи-
ков других компонентов пробы.
3. На основе анализа процессов детектирования можно ожи-
дать, что зависимость между сигналом Y и концентрацией X
нелинейна и может быть описана уравнением вида
Ai X + Ао
(IV.72)
В соответствии с описанной моделью программа метрологи-
ческих исследований включает анализ принятых технических
решений по стабилизации параметров аналитического процес-
са. Оценка возможной в рамках МКХА нестабильности влияю-
щих факторов методики проводится путем определения внутри-
лабораторной воспроизводимости. Основное внимание уделено
построению нелинейных ГрХ, доказательству их адекватности
и оценке погрешности градуировки.
Программа проведения исследований с целью выявления и
оценки составляющих погрешности результатов измерений, вы-
полняемых по МКХА, включает следующие разделы.
440
1. Анализ технических решений МКХА, обеспечивающих ста-
бильность перечисленных ниже параметров аналитического
процесса при многократном повторении анализа.
1.1. Объема флакона.
1.2. Объема пробы анализируемой воды во флаконе.
1.3. Давления газа во флаконе и перепада давлений между
флаконом и колонкой.
1.4. Температуры флакона.
2. Оценка погрешностей, связанных с приготовлением атте-
стованных проб воды для градуировки.
2.1. Приготовление аттестованного исходного раствора.
2.2. Оценка погрешности аттестованных градуировочных проб.
3. Оценка составляющих случайной погрешности МКХА.
3.1. Оценка сходимости.
3.2. Оценка внутрилабораторной воспроизводимости.
4. Оценка погрешностей, связанных с процедурой хромато-
графического разделения, градуировкой и измерением анали-
тического сигнала.
4.1. Обоснование вида градуировочной характеристики.
4.2. Построение градуировочной характеристики и оценка ее
адекватности.
4.3. Оценка погрешности градуировки и установление норма-
тивов оперативного контроля процедуры построения ГрХ.
5. Оценка погрешностей, связанных с влиянием неопределяе-
мых компонентов и наложением хроматографических пиков.
6. Оценка доверительных границ погрешности МКХА.
7. Метрологический анализ обоснованности принятых мето-
дов оперативного контроля показателей качества результатов
измерений.
7.1. Оперативный контроль внутрилабораторной воспроизво-
димости.
7.2. Оперативный контроль погрешности.
Ниже в сжатом виде приведены результаты метрологических
исследований, изложенные в соответствии с пунктами приведен-
ной программы.
1. Обеспечение стабильности параметров аналитическо-
го процесса.
1.1. Воспроизводимость объема флакона обеспечивается пу-
тем отбора стандартных пенициллиновых флаконов вместимо-
стью 15 см3, выпускаемых по ТУ 64-2-10-77, с резиновыми проб-
ками по ТУ 38006-108-78. Объем каждого определяется по раз-
ности результатов взвешивания флакона с дистиллированной
водой и пустого флакона. В группу (не менее 10), пригодную
441
для проведения градуировки и анализа, включают флаконы, от-
личающиеся по объему не более чем на 1%. При замене группы
флаконов на другую градуировку проводят заново.
1 2. Воспроизводимость объема Vo (5 мл) пробы анализируе-
мой воды, введенной во флакон, обеспечивается путем отбора
шприцев медицинских комбинированных вместимостью 5 см3,
выпускаемых по ТУ 64-1-863-87. Истинный объем воды, вы-
тесняемой из шприца, определяется по разности результатов
взвешивания сухого бюкса и того же бюкса с вытесненной в него
водой из шприца. К применению в МКХА допускаются шприцы,
у которых относительное отклонение AVo/Vo истинного объема
от номинального (5 мл) не превышает 1%.
1.3. Воспроизводимость установленных значений давлений во
флаконе и на входе хроматографической колонки обеспечива-
ется их контролем при помощи образцовых манометров типа
МО со шкалой 2,5 кгс/см2 класса 04, мод.1120, ГОСТ 6521-60.
Отклонение установленного значения давления от заданного не
должно превышать 1 кПа. Это обеспечивает стабильность пе-
репада давлений между флаконом и колонкой в пределах 2 кПа.
1.4. Воспроизводимость температуры флакона определяется
колебаниями температуры в лабораторном помещении, так как
флакон не термостатируется.
2. Приготовление и аттестация градуировочных смесей.
2.1. Для воспроизведения единиц концентрации ЛХУ гото-
вят исходный уксуснокислый раствор этих соединений методом
смешивания чистых веществ с последующим трехкратным раз-
бавлением в уксусной кислоте. Метод используется для граду-
ировки, метрологической аттестации и оперативного контроля
показателей точности методики.
В очищенную уксусную кислоту, помещенную во взвешенную
мерную колбу вместимостью 50 мл, последовательно добавляют
с помощью пипеток на 1, 5 и 10 мл индивидуальные ЛХУ, взве-
шивая колбу после каждой операции добавки. Свойства исполь-
зованных ЛХУ с указанием нормативной документации приве-
дены в табл.IV.26.
Массовая доля Cj каждого вещества в полученном проме-
жуточном растворе рассчитывается как отношение разностей
масс. Для j-ro компонента
тп{ — тгч-1
mK- m0
(IV.73)
где т2- — масса мерной колбы после добавки определяемого ком-
понента, mi-i — масса мерной колбы перед добавкой определя-
емого компонента. тпк — масса мерной колбы после последней
442
Таблица IV.26. Свойства индивидуальных
веществ, прмпгемпепгых для приготовления
аттестованных растворов [417, 418]
ЛХУ нтд Массовая доля основного вещества, % м, г/моль р. г/см3
ХФ ТУ 6-09-4263-76 99,85 119,38 1,488
дхэ ГСО 6411-92 99,85 98,97 1,257
ЧХУ ГСО 3305-85 99,85 153,82 1,595
ТХЭ ГОСТ 9976-83 98,50 131,39 1,465
ТеХЭ ТУ 6-09-2274-81 98,00 165,82 1,619
СНзСООН ГОСТ 61-75 99,8 60,05 1,049
добавки уксусной кислоты, т0 — масса сухой мерной колбы пе-
ред началом операции.
При последующем разбавлении промежуточного раствора
примерно в 50 раз очищенной уксусной кислотой применяется
та же процедура взвешивания, и коэффициент массового разба-
вления рассчитывается по формуле, аналогичной (IV.73). Опе-
рация разбавления повторяется трижды. В результате полу-
чают аттестованный раствор.
Массовая концентрация каждого ЛХУ получается путем ум-
ножения его массовой доли на плотность р аттестованного рас-
твора, совпадающую с плотностью уксусной кислоты. Так, мас-
совая концентрация Xj компонента j равна
Д- = Ю6 • [мг/дм3], (IV.74)
где Kj, /<2 и 7<з — коэффициенты массового разбавления на
последовательных стадиях процедуры разбавления; Qj — мас-
совая доля основного вещества; р = 1,049 г/см3 — плотность
аттестованного раствора (совпадающая с плотностью уксусной
кислоты).
Применяемая мерная посуда используется для приблизитель-
ного отмеривания смешиваемых веществ, ее погрешность не ока-
зывает влияния на точность аттестации приготавливаемых рас-
творов.
Предельную (т. е. соответствующую доверительной вероят-
ности, равной 1) погрешность аттестованного значения концен-
трации bX-j/X-j находят суммированием модулей отдельных со-
ставляющих.
443
Из (IV.74) следует, что предел относительной погрешности
^•_AQ ДКх ДК2 ДК3 Др 1-Qj nv_
v7--Q- + W + W + ^r + T + -Qr- (v-75)
Структура первых четырех слагаемых, определяемых проце-
дурой взвешивания, одинакова для всех компонентов. Массовая
доля компонента в исходном растворе определяется по формуле
(IV.73). Поэтому
AQ _ Amj + Am,_i Атк + Дт0
Cj rrii — mi-i тк — то ’
где Ат — погрешность отдельного взвешивания.
При взвешивании масс от 50 до 100 г на весах аналитиче-
ских лабораторных общего назначения 2-го класса точности
с наибольшим пределом взвешивания 200 г по ГОСТ 24101-
88 погрешность взвешивания, включающая погрешность весов,
погрешность из-за неравноплечности и наибольшую допусти-
мую погрешность массы гирь (в соответствии с ГОСТ 7328-
73), не превышает 0,5 мг. Погрешность от взвешивания на
воздухе пренебрежимо мала, так как результат (IV.73) пред-
ставляет собой отношение двух разностей масс. Полагая все
Ami » Ат = 0,5 мг, получим
АС,
= 2Дт
1 1
-----------1--------
гщ — mj_i тк — то
(IV.77)
По аналогичным формулам рассчитывают относительные по-
грешности Д7<1//<1, ДК2/-К2 и ДЛ3/К3 коэффициентов массо-
вого разбавления.
Очевидно, что погрешность определяется наименьшей мас-
сой навески (mi — тг-_1) чистого ЛХУ. Массы смешиваемых ве-
ществ, расчетные значения массовых концентраций Xj и относи-
тельных погрешностей XCj/Cj для выбранных навесок ЛХУ и
принятых коэффициентов разбавления приведены в табл. IV.27.
Масса раствора в колбе (тк — то) принята равной примерно
70 г. Это мало влияет на результат расчета, так как второе
слагаемое в (IV. 76) намного меньше первого. В той же таблице
приведена погрешность (1 — обязанная своим происхо-
ждением чистоте используемых веществ Очевидно, что именно
она является доминирующей и в основном формирует значение
предельной погрешности аттестации XXj/Xj, указанной в по-
следнем столбце таблицы. Для три- и тетрахлорэтилена по-
грешность аттестации целиком определяется чистотой исход-
ных веществ (погрешности, вносимые процедурами смешивания
444
Таблица IV.21. Метрологические характеристики
аттестованного раствора для градуировки
ЛХУ Дозируемые bCj/Cj (1 - Qi)/Qi мг /дм3 b-Xj/Xj, %(отн)
объем, мл масса, г
дхэ 2,0 2,514 4,1 • 10-4 1,5-10-3 106 0,3
ХФ 3,0 4,464 2,4-10-4 1,5 • IO-3 3,56 0,4
ЧХУ 0,3 0,4785 2,1 • IO-3 1,5- IO-3 0,382 0,4
тхэ 3,0 4,395 2,4 • IO-4 1,5 • 10~2 3,50 1,5
ТеХЭ 1,0 1,619 6,3 • IO”4 2,0- 10-2 1,29 2,0
и взвешивания, незначимы в сравнении с возможными загрязне-
ниями исходных веществ).
2.2. Градуировочные пробы готовят в отобранных пеницил-
линовых флаконах из полученного аттестованного раствора. В
каждый флакон наливают отобранным медицинским шприцем
Vb = 5 мл очищенной воды. Для получения смесей, соответ-
ствующих разным точкам диапазона градуировки, во флаконы
вводят разные объемы V, (от 2 до 50 мкл) аттестованного рас-
твора с концентрацией Xj. Эти объемы отмериваются и вво-
дятся микрошприцем МШ-10 с номинальным значением дози-
руемого объема 10 мкл и относительной погрешностью 1% от
максимального объема. Следовательно, Д14 = 0,1 мкл. Шприц
используется несколько раз [п = 1, ...,5) в зависимости от тре-
буемого объема Vi вводимой смеси.
Номинальное значение Хгр концентрации компонента в гра-
дуировочной пробе определяют по формуле
(IV.78)
гр И •
Неисключенная относительная систематическая погрешность
аттестации градуировочной пробы равна
2 1
+ 1,12
2
2
4V-'
п——-
ДГо
Го
й = 1,1
(IV.79)
AXj
*3
где п — число дозирований микрошприцем (п — 1,...,5); Ц
объем вводимого во флакон аттестованного уксуснокислого
раствора; И) — объем вводимой во флакон очищенной воды;
XXj/Xj — погрешность аттестации уксуснокислого раствора.
В табл. IV.28 приведены приписанные значения концентраций
компонентов в использованных при аттестации градуировочных
пробах и границы допускаемых относительных погрешностей,
445
рассчитанные по формуле (IV.74). Приняты следующие зна-
чения составляющих: погрешность измерения объема Vo воды
AVo/Vb — 0,01 (определяется принятой процедурой отбора ме-
дицинских шприцев), погрешность дозирования аттестованного
раствора АЦ- = 0,1 мкл, а погрешность его аттестации заим-
ствована из табл. IV.27. Максимальная погрешность имеет ме-
сто для ТХЭ при Vi = 2 мкл и не превышает 6% (отн).
Таблица IV.28. Метрологические
характеристики градуировочных проб
дхэ ХФ ЧХУ ТХЭ ТеХЭ
х, мкг дм3 «i, % X, МКГ дм3 «i, % X, мкг дм3 5», % X, мкг дм3 % X, мкг дм3 г., %
38,5 5,6 1,30 5,6 0,139 5,6 1.27 5,8 0,468 6,0
192 1,6 6,48 1,6 0,694 1 6 6,36 2,2 2,34 2,5
385 1,6 13,0 1,6 1,39 1,6 12,7 2,2 4,68 2,5
673 1,7 22,7 1,7 2,43 1,7 22,3 2,2 8,19 2,6
962 1,6 32,4 1,6 3,47 1,6 31,8 2,2 11,7 2,5
3. Оценка случайной погрешности. Случайная погреш-
ность изучалась в условиях сходимости и в условиях межла-
бораторной воспроизводимости*.
3.1. Сходимость оценивалась по результатам анализа 7 гра-
дуировочных смесей, а также 6 проб реальной водопроводной
воды с добавками определяемых веществ (в виде уксусноки-
слого раствора). Проверка относительных дисперсий единич-
ного измерения на однородность по критерию Бартлета [409]
показала, что величина ОСКО единичного измерения слабо за-
висит от компонента, концентрации и объекта анализа (градуи-
ровочные смеси или реальные пробы). ОСКО единичного изме-
рения сигнала по всем компонентам в рабочих диапазонах кон-
центраций можно оценить величиной S'CJ = 0,05 для 325 степеней
свободы (13 смесей • 5 измерений • 5 компонентов).
*В некоторых случаях обычная статистическая обработка одномерных
массивов, подразумевающая расчет среднеарифметических значений и
стандартных отклонений Хср ± sx> должна быть модифицирована, по-
скольку разброс данных в “разные стороны” от среднего может быть нео-
динаковым. В подобных случаях рекомендуется запись результата в виде
Хср + «(Х; > ХСр) — s(X; < ХСр). Для такой обработки можно использовать
программу 2, приведенную в Приложении. Подробнее об условиях и при-
мерах необходимости подобной асимметрической стандартной обработки
см. [427].
446
3.2. Внутрилабораторная воспроизводимость оценивалась
путем сравнения результатов анализа контрольных проб по ка-
ждому определяемому компоненту в начале и в конце рабочей
смены (по 5 измерений). В качестве контрольных проб исполь-
зовали: 1) градуировочный раствор, приготовленный путем вве-
дения в каждый флакон с очищенной водой 5 мкл аттестованного
раствора; 2) реальную рабочую пробу водопроводной воды с
добавкой градуировочного раствора.
Первый случай характеризует процедуру градуировки, вто-
рой — процедуру анализа рабочих проб. Отличие состоит в
том, что каждый образец градуировочного раствора готовится
независимо и случайная погрешность дозирования уксусноки-
слого раствора микрошприцем МШ-10 по разному влияет на
результат каждого отдельного измерения, а проба водопровод-
ной воды готовится один раз и используется во всех измере-
ниях. Использование в качестве контрольной пробы рабочей
пробы без добавок затруднительно, так как трудно найти рабо-
чую пробу, содержащую все определяемые компоненты в нуж-
ных концентрациях.
Найденные значения ОСКО единичного измерения (характе-
ризующие воспроизводимость результатов) S'bch равны 0,034 и
0,029 для двух указанных случаев и неразличимы по критерию
Бартлета. Примем S'bch = 0,034. Наибольшее возможное значе-
ние ОСКО (1В) оценим в соответствии с [410] как верхнюю гра-
ницу доверительного интервала (Р = 0,95) для СКО:
/в = — Звсп, (IV.80)
Хв
где Хв — параметр хи-квадрат распределения для Р = 0,975,
f — кт — 1. Число серий к = 4, число измерений в серии т — 10,
поэтому f = 39 и значение у2(Р = 0,975, f = 39) = 25,6. Отсюда
предел ОСКО 1В — 1,23 • S'bch = 0,042.
Переходя к процедуре градуировки, примем для ОСКО еди-
ничного измерения значение Sx — 0,05. При этом максимальное
относительное расхождение между результатами двух парал-
лельных определений не должно превышать 2,77 • Sx = 0,14.
4. Оценка погрешности градуировки и измерения концен-
трации.
4.1. Использование детектора постоянной скорости рекомби-
нации (ДПР), основанного на явлении захвата электронов, не-
избежно приводит к нелинейным градуировочным характери-
стикам [419]. Механизм нелинейности связан с ограниченным
числом свободных электронов, стационарная концентрация ко-
447
торых п в отсутствие захватывающих электроны веществ опре-
деляется применяемым радиоизотопным источником излучения.
Примем для ГрХ уравнение
X
А^Х + До
(IV.81)
где Ai и До — эмпирические коэффициенты, подлежащие опре-
делению. ГрХ, описанная этим уравнением, имеет вид кривой,
проходящей через начало координат и плавно переходящей от
прямой пропорциональности к насыщению. Это соответствует
физике процессов в электронозахватных детекторах, к которым
относится ДПР [419].
Для прояснения физического смысла коэффициентов Ау и До
преобразуем (IV.81) к виду
У =--------------т. (IV.82)
Отсюда ясно, что коэффициент До обратно пропорционален чув-
ствительности вблизи нуля, т. е. при малых концентрациях.
Для обеспечения необходимой чувствительности значение До
не должно быть бо ль ше некоторого норматива, величина
которого должна быть установлена в процессе исследования ме-
тодики. Коэффициент А\ определяет кривизну ГрХ, т. е. сте-
пень отклонения от закона прямой пропорциональности.
Градуировочная характеристика для каждого определяемого
компонента устанавливается путем анализа ряда аттестованных
проб. Для использования математического аппарата метода
наименьших квадратов (МНК) [412] введем новую переменную
Z (линеаризующее преобразование)
Z = X/Y. (IV.83)
Из (IV.83) следует, что
Z = AiX + Ао. (IV.84)
В предположении, что аттестованные значения X свободны
от случайных погрешностей, из (IV.83) получим связь между
дисперсиями Dz преобразованных и DY измеренных значений
сигналов в виде
Dz = ^DY. (IV.85)
В том же предположении преобразование (IV.83) не меняет зна-
чений относительных дисперсий, т. е.
Dz/Z2 = Dy/Y2. (IV.86)
448
Для расчета значений концентрации X по измеренным значе-
ниям сигналов У из (IV.83) и (IV.84) следует расчетная формула
'Y=rr£r <IV-87>
4.2. Для определения параметров ГрХ каждого из определя-
емых вешеств анализировали 5 градуировочных проб с аттесто-
ванными значениями концентраций X. Каждую смесь анализи-
ровали 2 раза и вычисляли среднее значение сигнала (высоты
пика) У. Относительное максимальное расхождение dY сигна-
лов У1 и Уг ни в одной серии измерений не превысило устано-
вленного норматива, равного 0,14.
В качестве примера в табл. IV.29 приведены результаты из-
мерения сигнала У и расчета линеаризующей функции Z (урав-
нение (IV.83)) для четыреххлористого углерода в рабочем диа-
пазоне концентраций.
Таблица IV.29. Исходные данные для построения
ГрХ по четыреххлористому углероду
Концентрация X, мкг/дм3 Сигнал У, ед. сч Преобразованный сигнал Z 106
0,147 22212 6,6180
0,737 88698 8,3091
1,47 138336 10 655
2,58 190575 13,538
3,68 226972 16,213
Зависимость Z от X для каждого компонента обрабатывали
взвешенным МНК, приняв весовую функцию Р = 1/Z2, кото-
рая соответствует примерно постоянному ОСКО. Для сред-
него значения У из двух параллельных определений принято
SY = 0,05/72 = 0,035.
Результаты расчета МНК для четыреххлористого углерода
приведены в табл.IV 30, а на рис. IV.39 — график ГрХ в без-
размерных координатах (У/Утах, Х/Хтах), где Утах и Хтах —
сигнал и концентрация ЧХУ в аттестованной пробе с наиболь-
шим содержанием определяемого компонента.
Итоговые значения коэффициентов
Л = (2,8 ± 0,3) 10"6 (ед.сч)"1,
До = (6,3 ± 0,4) 10-6 мкг/(дм3/ед.сч).
449
Таблица IV.SO. Результаты расчета МНК
Параметры ско коэффи ци ентов Случайные погрешности коеффи циентов
Ai = 2,7852 • 10—6 Ао = 6,2726 -10“6 г = 0,9987 Do = 3,9025 • 10“4 = 6,4467-10-13 DAq - 1,3427- 10~14 SA1 = 8,029 10“8 SAo = 1,159 10“7 ДЛ1 = 2,553- 10—7 ДАо = 3,686 • 10~7
Do/Вотн.ср = 0,32 < FKp(0,95; 3,314) = 3,67
Рис. IV.39. Градуировочная
характеристика для четы-
реххлористого углерода.
Адекватность найденной градуировочной характеристики
доказана сравнением остаточной дисперсии ГрХ Dq с относи-
тельной дисперсией £>Отн.ср — Sy среднего значения сигнала
(последняя строка табл. IV.30). Незначимость по критерию Фи-
шера означает, что при достигнутой погрешности измерения
сигнала улучшать ГрХ бессмысленно.
Расчет концентрации ЧХУ по измеренному значению анали-
тического сигнала Y (ед.сч) проводят по формуле
0 3 • У
X [мкг/дм3] = 10б128.у (IV.88)
4.3. Оценка доверительных границ относительной погрешно-
сти найденного значения X концентрации проводится на основа-
нии анализа формулы (IV.87). При этом необходимо принять во
внимание, что коэффициенты А\ и Aq взаимно коррелированы,
и предварительно исключить Aq с помощью соотношения
Zo = AiXq + Aq, (IV.89)
где Zq и Хо — координаты центра тяжести ГрХ.Такая оценка
показывает, что приближенно
450
^ = 5у[Л1/Л0Х + 1]1 (IV.90)
Sy — 0,05 — ОСКО единичного измерения сигнала У. Из этого
выражения очевидно, что поведение Sx/X определяется вели-
чиной первого слагаемого в квадратных скобках. Сравнивая с
выражением (IV.82), нетрудно заметить, что рост погрешности
с ростом концентрации также обусловлен нелинейностью ГрХ.
Доверительные границы относительной погрешности граду-
ировки АХ/Х складываются из относительной случайной по-
грешности Sx/X и относительной погрешности аттестации гра-
дуировочных смесей по закону [420]
(IV.91)
Результаты расчета для ЧХУ в диапазоне градуировки пред-
ставлены на phc.1V.40.
Рис. IV.40. Доверительные
границы относительной по-
грешности градуировки по
четыреххлористому угле-
роду (ЧХУ).
5. Влияние неопределяемых компонентов и неполного
разделения.
В 1993 г. в г. Дзержинске была проведена работа по иссле-
дованию состава летучих органических загрязнителей в при-
родных, питьевых и сточных водах хромато-масс-спектроме-
трическим методом. Было идентифицировано более 100 ин-
дивидуальных соединений и групп изомеров [421, 422]. Более
40% всех идентифицированных соединений составляют углево-
дороды различного строения (парафиновые, олефиновые, наф-
теновые, ароматические), приблизительно 20%—галоидсодер-
жащие соединения.
Применение приемов селективного газохроматографического
определения, а именно — анализа равновесной паровой фазы,
детектора постоянной скорости рекомбинации (селективного к
451
хлорорганическим соединениям), эффективных разделительных
колонок позволило исключить мешающее влияние большинства
органических соединений.
Присутствующие летучие хлорорганические соединения с
температурами кипения до 40 °C, определение которых не пре-
дусмотрено настоящей МКХА (хлористый этил, винилхлорид,
дихлорметан), на колонке с неполярной жидкой фазой (SE-30,
апиезон) выходят до пиков определяемых компонентов. Относи-
тельные времена удерживания других обнаруженных галоген-
органических соединений на колонке с SE-30 в условиях, преду-
смотренных методикой, приведены ниже:
Относительное время
удерживания
Дихлорметан 1,00
Хлороформ 1,83
Дихлорэтан 2,17
Четыреххлористый углерод 2,82
Т рихлорэтилен 3,58
Т рихлорэтан 7,01
Дихлорбромметан 7,00
Тетрахлорэтилен 8,67
Тетрахлорэтан 11,20
Хлорбензол 30,00
Эти данные показывают, что в принятых условиях хроматогра-
фического анализа влияние неопределяемых компонентов незна-
чимо.
Для обеспечения необходимого качества хроматографиче-
ского разделения компонентов в МКХА предусмотрен контроль
качества Применяемой хроматографической колонки по двум
параметрам: эффективности N колонки и степени разделения R,
которые характеризуют селективность и размывание пиков ана-
лизируемых веществ. Эффективность N, измеренная по послед-
нему пику хроматограммы — пику трихлорэтилена, не должна
5ыть меньше 2400 теоретических тарелок, а степень разделения
R наиболее близких друг к другу пиков хлороформа и дихлор-
этана не должна быть меньше 1,3. Принятые значения этих па-
раметров позволяют оценить верхний предел погрешности от
взаимного наложения пиков на хроматограмме величиной, не
превышающей 0,1%(отн.) [423].
6. Оценка доверительных границ погрешности МКХА.
Ввиду незначимости влияния мешающих компонентов получен-
ный ранее результат (IV.91), показанный на рис. IV.40, можно
рассматривать как окончательный. В диапазоне от 0,15 до
3,7 мкг/м3 доверительные границы относительной погрешности
452
(для Р = 0,95) измерения содержания ЧХУ плавно расширя-
ются от 14 до 27%(отн.).
7. Оперативный контроль показателей качества резуль-
татов измерений.
7.1. Норматив оперативного контроля воспроизводимости.
Оперативный контроль случайной погрешности выполняется в
условиях внутрилабораторной воспроизводимости Использу-
ются градуировочные пробы, соответствующие примерно пер-
вой трети диапазона определяемых концентраций. Исполь-
зование в этой процедуре градуировочного раствора вместо
обычно применяемых рабочих проб обусловлено двумя обсто-
ятельствами: 1) рабочая проба, содержащая все определяе-
мые компоненты в нужном диапазоне концентраций, встреча-
ется крайне редко, а планировать ее появление невозможно;
2) контрольная процедура с использованием градуировочного
раствора включает операцию ввода во флакон аттестованного
уксуснокислого раствора, вносящую дополнительные погреш-
ности (дозирование микрошприцем) в сравнении с процедурой
анализа рабочей пробы, и поэтому более полно характеризует
воспроизводимость.
Готовят два флакона с указанным раствором. Один из них
анализируют в начале, а другой — в конце рабочей смены, опре-
деляя значения хроматографических сигналов Yi и У2- Рассчи-
тывают концентрации Xi и А'2.
Максимальное относительное расхождение d между резуль-
татами определяют по формуле
(IV.92)
Норматив d оперативного контроля рассчитан на основании
результатов измерения внутрилабораторной воспроизводимо-
сти сигналов и принят равным 0,14 ( из формулы (IV.87) видно,
что относительное расхождение концентраций не может пре-
вышать относительного расхождения сигналов, так как всегда
4UPmax < 1).
7.2. Норматив оперативного контроля погрешности. Опера-
тивный контроль погрешности выполняют в одной серии с КХА
рабочих проб. Исходными образцами для контроля являются
реальные пробы природных, питьевых или очищенных сточных
вод.
Образцы проб для контроля готовят в соответствии с МКХА
следующим образом. Первый образец — исходная рабочая
проба: во флакон для анализа вводят 5 см3 исходной пробы.
Второй образец — исходная рабочая проба, разбавленная в
453
2 раза: во флакон для анализа вводят 2,5 см3 исходной пробы
и добавляют 2,5 см3 очищенной воды. Третий образец — ис-
ходная рабочая проба, разбавленная в 2 раза (2,5 см3 пробы
+2,5 см3 очищенной воды), в которую вводят микрошприцем
10 мкл аттестованного уксуснокислого раствора. Концентра-
цию С добавки рассчитывают по формуле (IV.78).
Подготовленные образцы анализируют в точном соответст-
вии с МКХА, получая результаты анализа: Xi — результат
анализа исходной рабочей пробы; X? — результат анализа ис-
ходной рабочей пробы, разбавленной в два раза; Хз — резуль-
тат анализа исходной рабочей пробы, разбавленной в два раза,
с добавкой С. (Результаты анализа Xi, Х2 и Хз получают, по
возможности, в одинаковых условиях, т. е. их получает один
аналитик с использованием одного набора мерной посуды, од-
ной партии реактивов и т.д.)
Точность контрольных измерений признается удовлетвори-
тельной, если выполнено неравенство
Аконт = |Хз-^|'2 ~ + |2Х-2-^Х11 < К, (IV.93)
где К — норматив оперативного контроля погрешности.
Таким образом, процедура включает три независимых изме-
рения содержания компонентов в трех специально приготовлен-
ных пробах. Каждое измерение выполняется в соответствии с
МКХА. Рассматривая результат контрольной процедуры как
комбинацию трех независимых измерений с относительной по-
грешностью Аотн каждого из них, получим для относительной
погрешности результата значение погрешности, равное
К = у/З • Аотн = у/З 0,25 = 0,43,
которое и является нормативом оперативного контроля.
Необходимо подчеркнуть две особенности уравнения (IV.93):
1. При отсутствии определяемого компонента в рабочей про-
бе Xi = 0 и Х2 = 0 (содержание компонента ниже диапазона
определяемых концентраций также равносильно его отсутствию
в пробе). Процедура превращается в сравнение результата из-
мерения с аттестованным значением концентрации в образце
для контроля, т.е. сохраняет смысл контрольной операции. Од-
нако норматив К при этом должен быть уменьшен до величины
АОтн = 0,25, так как используется только одно измерение.
2. Значение относительной погрешности в (IV.93) получают
делением на аттестованное значение концентрации С добавки, а
не на концентрацию Xi компонента в исходной рабочей пробе.
Это обосновано тем, что в противном случае при отсутствии
454
Таблица IV.31. Результаты
оперативного контроля погрешности
Характеристика рабочей пробы Компонент С, мкг/дм3 Xi, мкг/дм? х2 мкг/дм3 Xs, мкг/дм3 Аконт
Выход РОС* ХФ 2,13 0,48 0,41 2,70 0,23
(05.05.95) ЧХУ 0,30 0,10 0,05 0,39 0,15
ТХЭ 1,80 0,12 0,13 2,20 0,23
ТеХЭ 1,27 0,10 0,10 1,70 0,34
р. Ока ниже ХФ 3,55 0,17 0,10 3,33 0,10
выпуска ЧХУ 0,51 0 0 0,53 0,04
ПО “Корунд" ТХЭ 2,99 0 0 2,79 0,07
(15.05.95) ТеХЭ 2,11 0 0 2,05 0,03
Водопровод- ХФ 3,55 6,48 3,4 6,00 0,39
ная вода ЧХУ 0,51 0,09 0 04 0,48 0,15
ул. Кирова, 1 ТХЭ 2,99 1,66 0,87 3,10 0,28
(15.05.95) ТеХЭ 2,110 0 0 1,80 0,15
дхэ 180 0 0 160 0,11
Выход РОС* ХФ 2,00 0,47 0,22 2,03 0,11
(27.06.95) ЧХУ 0,22 0,29 0,15 0,40 0,18
ТХЭ 1,96 12,0 6,20 7,80 0,39
ТеХЭ 0,90 0 0 0,98 0,09
*РОС — районные очистные сооружения.
определяемого компонента (Л'х = 0) уравнение (IV.93) вырожда-
ется, так как деление на ноль невозможно.
Некоторые результаты проведения контрольных операций
приведены в табл. IV.31. Они подтверждают обоснованность
установленных нормативов оперативного контроля погрешно-
сти.
455
Глава V
ПРАКТИЧЕСКИЕ РАБОТЫ
ПО ГАЗОВОЙ ХРОМАТОГРАФИИ
Предлагаемые практические работы в подавляющем боль-
шинстве рассчитаны на использование хроматографов “Цвет”
Дзержинского ОКБА, ныне преобразованного в ОАО “Цвет”.
Однако можно применять и другие газовые хроматографы, не
уступающие по своим характеристикам рекомендуемым (напри-
мер, отдельные модели приборов НПО “Манометр”, “Кристалл
2000”).
Успешное выполнение любой практической работы предпо-
лагает обязательное предварительное усвоение теоретического
материала в объеме программы лекционного курса по газовой
хроматографии, ознакомление с аппаратурой и неукоснитель-
ное соблюдение правил ее эксплуатации. Лишь при выполне-
нии этих условий студент не выйдет за рамки отпущенного ему
лимита времени (4-6 ч на каждую работу), а к концу практи-
кума сможет приобрести необходимые навыки по проведению
типовых хроматографических анализов.
Внимание! Студенту разрешается приступать к прак-
тической работе только после согласования с преподава-
телем основных этапов ее выполнения и последовательно-
сти операций: по включению и выведению хроматографа
на рабочий режим; по проведению собственно хромато-
графического анализа; по обработке полученных хрома-
тограмм.
С полученными от лаборанта для выполнения той или иной
работы инструментами и особенно микрошприцами необходимо
обращаться бережно. Внимательно ознакомьтесь с инструкцией
по их эксплуатации. Не забывайте промывать микрошприцы
подходящим растворителем и просушивать их в струе воздуха
от компрессора или в вакууме водоструйного насоса перед ка-
ждым дозированием очередного анализируемого образца. Пре-
небрежение этой процедурой может привести к значительному
искажению результатов анализа.
456
О каждой выполненной лабораторной работе должен быть
составлен отчет в рабочей тетради в соответствии с рекомен-
дуемым планом (см. с. 470). Вместе с отчетом преподавателю
необходимо предъявлять итоговые хроматограммы. При реги-
страции хроматограмм пишущим потенциометром студент дол-
жен обязательно записывать непосредственно на диаграммной
бумаге основные параметры анализа, часто изменяемые в ходе
эксперимента по мере оптимизации условий и качества дости-
гаемого разделения: температуру колонки, испарителя и де-
тектора, расход газа-носителя (и скорости подачи вспомога-
тельных газов), количество (дозу) и название анализируемого
образца, шкалу электрометрического усилителя сигналов ио-
низационных детекторов и множитель шкалы при использова-
нии тепловых детекторов, а также другую важную для достиже-
ния представляемых результатов информацию. Значения всех
измеримых или вычисляемых параметров хроматографических
пиков рекомендуется также записывать непосредственно на хро-
матограмме, как это показано, например, на рис. V I.
Рис-V.l. Образец рабочей хроматограммы.
Хроматограф “Цвет-101”. Колонна стальная 1 м X 3 мм, заполнен-
ная хроматоном N (0,20—0,25 мм) с 15% (по массе) апиезона L. Темпера-
тура колонки 90, испарителя 150°C. Газ-носитель — азот, 44 мл/мин.
Детектор пламенно-ионизационный, шкала электрометра 10~8 А, рас-
ход водорода 30, воздуха 300 мл/мин. Скорость движения диаграммной
ленты 600 мм/ч.
Указанные в работах с использованием пламенно-ионизаци-
онного детектора расходы водорода и воздуха являются ори-
ентировочными. Поскольку экспериментальное определение
оптимальных расходов этих газов обычно не встречает труд-
457
ностей и требует очень небольших затрат времени (в частно-
сти, по методике, описанной в работе 3, вариант Б), целесо-
образно каждый раз при использовании ПИЛ устанавливать
оптимальные расходы водорода и воздуха относительно задан-
ного расхода газа-носителя по.собственным экспериментальным
данным.
Большая часть лабораторных работ рассчитана на исполь-
зование насадочных (стальных или стеклянных) колонок с вну-
тренним диаметром 2-4 мм и длиной от 1 до 3 м. Колонки запол-
няют хроматоном N, хроматоном N-AW или хроматоном N-AW-
HMDS зернения 0,20-0,25 мм (или другим твердым носителем,
см. описания лабораторных работ), смоченным одной из приве-
денных в табл. V.1 неподвижной фаз в количестве, как правило,
10-15% (по массе).
Для выполнения лабораторной работы 5 необходимо распо-
лагать одной-двумя капиллярными колонками (стальными, сте-
клянными или из плавленого кварца размерами 30-50 м х 0,25—
0,35 мм) с неполярной (типа SE-30) и полярной (типа ПЭГ-20М)
неподвижными фазами.
В ряде случаев соблюдение рекомендаций относительно при-
менения той или иной хроматографической колонки (неподвиж-
ной фазы) не является обязательным и, по согласованию с пре-
подавателем, рекомендованная колонка (неподвижная фаза) мо-
жет быть заменена на другую, обеспечивающую решение по-
ставленной задачи. Следует, однако, помнить, что замена коло-
нок (неподвижных фаз), как правило, влечет за собой необходи-
мость изменения условий хроматографирования.
Рекомендуемый план отчета о лабораторной работе вклю-
чает:
1) название работы,
2) цель работы,
3) краткое описание содержания работы и эксперимента,
4) условия выполнения хроматографического анализа (с уче-
том возможных отклонений от рекомендованных),
5) результаты анализа по форме, предложенной в руковод-
стве (графики, таблицы и т.п.),
6) выводы.
458
Таблица V. 1. Некоторые распространенные неподвижные фазы в газовой хроматографии
Название Химическое строение Область рабочих температур, °C Растворитель*
нижняя верхняя
1 2 3 4 5
Сквалан (2,6,10,15,19,23- гексаметилтетра- козан) [(CH,),CH(CH) X СН(СЦ ) СНСЦ СЦ - 20 150 А, Г
1
СИ, СН3 2
Полидиметил- сл силиконы** 40 SE-30 OV-1 OV-101 SP-2100 СКТ сн3 ( Н3С — Si —О — СН3 сн3 1 — Si —О- 1 сн3 СН3 1 — Si—су СН3 п 50 100 0 0 50 350 350 350 350 300 Х,Т х,т,г Х,А X X
Полиметилфенил- силиконы (в скоб- ках указана доля фенильных заместителей): SE-52 (5% Ph) OV-7 (20% Ph) OV-17 (50% Ph) SP-2250 (50% Ph) OV-25 (75% Ph) СН3 1 Н3С- Si-O- сн3 сн3 1 — Si —О- 1 Ph СН3 1 -Si-су сн3 п 50 0 0 0 0 300 350 350 375 350 Х,ТА X, Т, А X, А X X, А
1 2
Полидиметил- трифторпропил- силиконы (в скоб- ках указана доля трифторпропиль- ных заместителей): OV-210 (50%) SP-2401 (50%) QF-1 (50%) СКТФТ-50 (25%) СКТФТ-100 (50%)
СН3 1 Н3С — Si — О — СН3 СН3 1 — Si — О- 1 (CH2h CF3
Полидиметил- цианопропил и цианоэтил- силиконы: OV-225 (25%) SP-2340 (100%) (в обеих фазах в скобках — доля цианопропильных групп) НСКТ-50 (25%) НПС-100 (50%) НСКТ-100 (50%) (во всех трех фа- зах в скобках— доля цианоэтиль- ных групп) СН3 1 Н3С —Si —О — СН3 сн3 1 — Si —О- 1 (CH2)Z CN
Продолжение табл. V.1
3 4 5
сн3 сн3
-Si-O Si — СЦ
СН3 сн3
0 275 X, А
0 275 А
0 250 А
— 350 А
— 350 А
сн3 сн3
-Si-O Si — CH
1 1 сн3 сн3 20 250 X, А
1 3 J„ 3 25 275 ХА
т z = 2илиЗ
250 А
— 180 А
250 А
Окончание табл. V.1
1 2 3 4 5
Полиэтиленглико- ли (карбоваксы) с мол. массой: НО- СН2СЦ -[-ОСЦ СЦ --J -o-cg eg -он
300 20 100 X
600 50 ПО X
1000 60 125 X
1500 60 130 X
. 4000 60 175 X
os 20000 (20 М) 80 225 X
Полиэтиленгли- коля моно(1,1,3,3- тетраметилбутил)- фениловый эфир (тритон Х-305) C8H17QH4O[-OCg сц Чо и 0-20 200-250 А, Me
Полиэтилен- гликольадипат НО-(-СН2 СН2 ОСОСЦ СЦ СЦ Cg COO-J -н 50 200 А
* А — ацетон, Г — гексан, Me — метанол, Т — толуол, X — хлороформ.
** Фирмы-производители: SE — General Electric, OV — Ohio Valley, SP — Supelco, CKT — Реахим.
Работа 1. Нанесение неподвижной фазы на
твердый носитель и заполнение колонки
Цель работы. Ознакомление с различными приемами нане-
сения неподвижной жидкой фазы на твердый носитель и с тех-
никой заполнения насадочной колонки.
Материалы, посуда, оборудование
Твердый носитель — хроматон N (или другой, по выбору преподава-
теля) зернения 0,16-0,20,0,20-0,25 или 0,25-0,315 мм.
Неподвижная жидкая фаза — трикрез ил фосфат (ТКФ), динонилфталат
(ДНФ), полиэтиленгликоль-1000 (ПЭГ-1000) или какая-нибудь другая, по
выбору преподавателя.
Растворитель — ацетон, метанол или мети лэти л кетон.
Весы технические 1-го класса.
Цилиндр мерный на 100 мл.
Воронка стеклянная.
Колба коническая или стакан на 100 мл.
Пипетки стеклянные типа медицинских.
Чашка фарфоровая и шпатель.
Вентилятор или фен.
Стеклянный фильтр №3, желательно диаметром 50—75 мм и высотой
130-150 мм.
Промывная склянка (склянка Тищенко).
Колба Бунзена для фильтрования под вакуумом.
Трехходовой стеклянный кран.
Водоструйный насос.
Баллон со сжатым газом (азот, аргон), оборудованный редуктором.
Сушильный шкаф.
Вибратор.
Набор стандартных сит.
Колонки газохроматографические металлические или стеклянные с во-
ронками для засыпания сорбента.
Выполнение работы
Нанесение неподвижной фазы на твердый носитель. Дан-
ная практическая работа знакомит студентов с тремя спосо-
бами приготовления сорбента (насадки) хроматографической
колонки и выполняется соответственно в трех вариантах.
Вариант А. Получение сорбента при механическом пере-
мешивании и испарении растворителя. По формуле V = nr^h
определяют объем хроматографической колонки заданных раз-
меров.
462
Через воронку порциями по 5-10 мл засыпают твердый но-
ситель* в предварительно взвешенный мерный цилиндр. При
этом следует время от времени постукивать цилиндром о стол
или ставить его на работающий вибраторный столик для того,
чтобы утрамбовать твердый носитель. Его общий объем дол-
жен превышать на 20-25% вычисленный свободный объем ко-
лонки. Взвешивают цилиндр с носителем и определяют общую
массу (г) и насыпную плотность (г/см3) твердого носителя, взя-
того для приготовления насадки.
В коническую колбу или стакан берут необходимую навеску
неподвижной жидкой фазы (в количестве 10-20% от массы твер-
дого носителя, по согласованию с преподавателем). В этой же
посуде приготавливают раствор неподвижной фазы в ацетоне
или другом подходящем растворителе и выливают этот раствор
на носитель, перенесенный в фарфоровую чашку. Объем приго-
товленного раствора неподвижной фазы должен несколько пре-
вышать объем твердого носителя, так чтобы при смешивании в
фарфоровой чашке над поверхностью твердого носителя обра-
зовался слой жидкости высотой около 5 мм. Например, если
взято 24 мл твердого носителя, то следует взять примерно 30
мл раствора неподвижной фазы.
Для испарения растворителя чашку помещают в вытяжной
шкаф на расстоянии 1-1,5 м от работающего вентилятора и
время от времени осторожно, стараясь не дробить частиц твер-
дого носителя, перемешивают содержимое чашки шпателем.
По испарении растворителя, когда приготовленная насадка
станет легкосыпучей и по внешнему виду не будет отличаться
от исходного твердого носителя (исчезнет запах растворителя),
ее необходимо поместить для просушивания в термостат, на-
гретый до 70-100 °C. После просушивания в термостате в те-
чение 3-4 ч* ** приготовленный сорбент (насадку) осторожно от-
сеивают на ситах от пыли и комков*** (диаметр отверстий сит
определяется зернением исходного носителя) и приступают к
заполнению хроматографической колонки (см. ниже).
’Продажный твердый носитель отмучивают дистиллированной водой
от пыли и самых мелких частиц, просушивают в сушильном шкафу при
150-160°С, просеивают и хранят в плотно закрытой таре. В условиях сту-
денческого практикума ©ту подготовительную операпию лаборант проде-
лывает заранее.
**Работа выполняется с перерывом. Освобождающееся время (в тече-
ние которого насадка просушивается) студенты используют для следую-
щей работы.
***Если твердый носитель не обладает достаточной механической
прочностью, отсеивание приготовленной насадки проводить не рекомен-
дуется.
463
Вариант Б. Получение сорбента (насадки) при испарении
растворителя и перемешивании потоком газа. Этот способ
обеспечивает более равномерное нанесение неподвижной жид-
кой фазы на зерна твердого носителя (достигается лучшая вос-
производимость при изготовлении новых партий насадок), по-
зволяет использовать даже весьма хрупкие твердые носители
(исключается их измельчение) и окисляющиеся на воздухе не-
подвижные фазы (исключается контакт неподвижной фазы с воз-
духом).
Стебель стеклянного фильтра № 3 (под тягой) соединяют ре-
зиновыми шлангами (через промывную склянку с концентриро-
ванной серной кислотой) с редуктором баллона со сжатым га-
зом.
Необходимое для приготовления насадки количество твер-
дого носителя (см. вариант А) помещают в фильтр и осторожно
(опасность выбрасывания!) начинают подавать газ, контро-
лируя его расход по пузырькам, барботирующим через серную
кислоту. Выливают на твердый носитель раствор неподвиж-
ной фвзы в соответствующем растворителе (объем раствора
должен на 20-25% превышать взятый объем твердого носителя)
и одновременно регулируют расход газа, стремясь обеспечить
умеренное “кипение” частиц твердого носителя в растворе не-
подвижной фазы (опасность разбрызгивания и переброса!).
При этом частицы твердого носителя, прилипающие к стенкам
фильтра, могут быть сброшены вниз шпателем.
На испарение легкокипящего растворителя (эфир, ацетон)
требуется не более 30 мин. При работе с более высококипящими
растворителями (бензол, четыреххлористый углерод) для уско-
рения испарения фильтр обогревают электронагревателем для
горячего фильтрования. По испарении растворителя пригото-
вленную насадку досушивают в термостате и отсеивают от ком-
ков и пыли (см. вариант А).
Вариант В. Фильтрационный метод получения сорбента
(насадки) с хорошими и воспроизводимыми характеристиками
путем поглощения твердым носителем части раствора жидкой
фазы. Этот метод особенно широко используется при приго-
товлении сорбента с низким содержанием неподвижной жидкой
фазы (0,1-3%).
Экспериментально установлено, что при пропускании (“филь-
тровании”) раствора неподвижной фазы в подходящем раство-
рителе через слой твердого носителя концентрация раствора
не изменяется; вместе с тем на слое зерен носителя задержива-
ется определенное количество раствора. Последующее прину-
дительное испарение растворителя приводит к получению на-
464
садки с массой неподвижной фазы, пропорциональной ее кон-
центрации в растворе. Найденные опытным путем графические
зависимости между содержанием неподвижной фазы в растворе
и в насадке для ряда комбинаций неподвижная фаза/раствори-
тель — твердый носитель, представлены на рис. V.2.
Phc.V.2. Зависимость меж-
ду содержанием неподвижной
фазы в растворе и в насадке.
1 — полиэтиленгликоль-400/ме-
танол — хроматон N-AW; 2— дино-
нилфталат/метилэти.ч кетон — сфе-
рохром-1; 3 — 1,2,3-трис(2-циано-
этил)амин/ацегон — сферохром-1
(зернение твердых носителей 0,25—
0,315 мм).
Содержание неподвижной фазы
в растворе, % (по массе)
Неподвижная фаза, растворитель, твердый носитель, его зер-
нение, необходимое количество насадки и содержание в ней не-
подвижной фазы задаются преподавателем.
Приготавливают раствор неподвижной фазы в растворителе
в количестве, вдвое превышающем заданный объем сорбента
(необходимую концентрацию раствора находят по одному из
графиков, представленных на рис. V.2). С этой целью рас-
считанную навеску неподвижной фазы взвешивают непосред-
ственно в конической колбе или стакане, куда затем вводят от-
меренный цилиндром или пипеткой объем растворителя.
Для нанесения неподвижной фазы на твердый носитель ис-
пользуют устройство, изображенное на рис.У.З. На стеклян-
ный фильтр помещают заданное количество твердого носителя
и заливают его приготовленным раствором неподвижной фазы
(перед этим втулка трехходового крана должна быть устано-
влена в положение /). Спустя 5-10 с, т. е. после того как весь
твердый носитель успеет пропитаться раствором неподвижной
фазы, поворачивают втулку трехходового крана в положение
II и отсасывают в вакууме водоструйного насоса раствор не-
подвижной фазы до тех пор, пока со стебля фильтра не пере-
станут капать отдельные капли и не исчезнет обильная пена.
Затем возвращают втулку трехходового крана в положение I,
465
Рис. V.3. Устройство для нанесения
неподвижной фазы на твердый носитель
фильтрационным способом.
1 — стеклянный фильтр; 2 — колба Бун-
зена; 3 — трехходовой кран.
отсоединяют систему от вакуумного насоса, снимают фильтр
и сливают фильтрат в заранее подготовленную для его хране-
ния тару*. Вновь устанавливают фильтр с еще непросушенным
сорбентом на колбу Бунзена, переводят втулку трехходового
крана в положение II и продолжают просасывание воздуха до
тех пор, пока сорбент не станет легкосыпучим и по внешнему
виду неразличимым с исходным твердым носителем**.
Приготовленный сорбент просушивают в воздушном термо-
стате при 70-100°С в течение 3-4 ч и заполняют им газохрома-
тографическую колонку.
Заполнение U-образной колонки приготовленной насад-
кой (сорбентом). Предварительно промытую органическими
растворителями и высушенную колонку, пропустив через коль-
ца штатива, ставят вертикально на рабочую пластину вибра-
тора. Накидными гайками закрепляют на концах колонки (и
узловом отверстии или отверстиях) металлические или пласт-
массовые воронки для засыпания насадки.
Включают вибратор и равными порциями начинают засы-
пать предварительно взвешенную насадку в колонку до полного
ее наполнения. Заполненную колонку выдерживают на вибра-
торе не более 1 мин; в случае слишком плотной упаковки ча-
стиц при работе возникнет большой перепад давлений на входе
в колонку и выходе из нее, что приведет к снижению эффектив-
ности разделения и создаст осложнения при дозировке образ-
цов. Оставшуюся в воронках насадку и небольшое ее количе-
ство (примерно по 0,5 мл) из концов колонки осторожно ссы-
пают.
Концы колонки закрывают маленькими пробочками (тампо-
нами) из шнурового асбеста, стекловолокна или металлической
сетки. Важно, чтобы после отсыпания насадки расстояние от
границы слоя сорбента до верхнего фланца колонки составляло
*Этот раствор неподвижной фазы в дальнейшем можно использовать
многократно.
’"'Отдельные конгломераты слипшихся вначале зерен твердого носи-
теля должны полностью разрушиться, что является признаком заверше-
ния данной операции.
466
не более 5 мм. Тампоны, предотвращающие высыпание насадки,
должны быть поставлены “заподлицо” с фланцем колонки.
Взвешиванием (по разности масс) определяют количество
сорбента, засыпанного в колонку. Свежезаполненная колонка
должна пройти тренировку (кондиционирование, см. ниже).
Заполнение спиральной колонки. Один из концов промы-
той органическими растворителями и высушенной колонки за-
крывают тампоном из шнурового асбеста, стекловолокна или
тонкой металлической сетки и подключают к водоструйному
насосу. Колонку закрепляют в штативе таким образом, чтобы
спиральные витки располагались горизонтально. К открытому
концу колонки прикрепляют воронку для засыпания насадки.
Создав разрежение, начинают осторожно присыпать отсеянную
от пыли и комков насадку, одновременно постукивая по колонке
деревянной палочкой* для достижения более равномерного за-
полнения. О заполненности колонки судят по прекращению
убыли насадки в воронке. Сняв воронку, закрывают открытый
конец колонки тампоном, как описано выше.
Свежезаполненная колонка нуждается в тренировке.
Тренировка (кондиционирование) колонок**. Устанавли-
вают свежезаполненную колонку в термостате хроматографа
(не забывайте о прокладках!), не соединяя выход из колонки с
детектором, и в течение 3-4 ч продувают колонку азотом, про-
пуская его со скоростью 40-60 мл/мин при ступенчатом или
непрерывном (режим программирования) повышении темпера-
туры примерно от 75 °C до температуры на 20-30 °C выше пред-
полагаемой рабочей (но не выше максимально допустимой для
данной неподвижной фазы). Затем охлаждают колонку и со-
единяют ее выходной конец с детектором (не забудьте по-
ставить прокладки!). Проверяют герметичность газовой ли-
нии прибора, выводят хроматограф на рабочий режим и прове-
ряют стабильность нулевой линии на хроматограмме. Шумы и
дрейф сигнала свидетельствуют о необходимости продолжить
кондиционирование (при подготовке колонки к анализу следо-
вых количеств компонентов пробы, регистрируемых на макси-
мально чувствительных шкалах прибора, кондиционирование
может продолжаться долго — десятки часов). Иногда, при ра-
*Конечно, можно воспользоваться вибратором, но и указанный прием
достаточно эффективен для обеспечения равномерности заполнения.
Г5 программу практикума не входит и приводится здесь для ознако-
мления студентов с этой обязательной при самостоятельной работе про-
цедурой. По возможности, после кондиционирования студентам поруча-
ется исследовать рабочие характеристики приготовленной колонки (см.
лабораторную работу 2).
467
боте с силиконовыми эластомерами, рабочие характеристики
сорбента улучшаются, если свежеприготовленная колонка про-
гревается первые 8-10 ч в отсутствие газа-носителя.
При оформлении протокола ответьте на следующие вопросы.
1. Какие растворители (из числа альтернативных, приводи-
мых в справочниках) следует предпочтительно выбирать для
нанесения неподвижной фазы на твердый носитель по вариан-
ту А? Чего следует опасаться при реализации этой процедуры?
Какие открываются преимущества (и возникают ограничения)
при использовании для упаривания раствора роторного испа-
рителя?
2. В каких случаях приготовление сорбента (насадки) по ва-
риантам Б и В предпочтительнее варианта А?
3. К чему может привести использование в этой работе для
смачивания неподвижной фазой изначально неотмученного и не-
рассеянного твердого носителя?
4. Какие цели преследует стадия кондиционирования свеже-
заполненной колонки?
Работа 2. Влияние скорости газа-носителя на
эффективность работы хроматографической
колонки
Цель работы. Исследование влияния скорости газа-носите-
ля, изменяемой в широких пределах, на эффективность разделе-
ния компонентов бинарной смеси.
Хроматографируют искусственную смесь двух близкокипя-
щих углеводородов при последовательном изменении скорости
газа-носителя На основании полученных данных определяют
число теоретических тарелок (т. т.) (по пику второго компо-
нента для каждого из режимов хроматографического разделе-
ния) s находят высоту, эквивалентную теоретической тарелке
(ВЭТТ), строят зависимость ВЭТТ от скорости газа-носителя
и рассчитывают значения критерия разделения R компонентов
анализируемой смеси.
Аппаратура, условия, объекты хроматографирования
Хроматограф с детектором по теплопроводности, сила тока моста 80 мА,
множитель шкалы 10.
Колонки стальные (2 м X 3 мм), заполненные хроматоном N (0,20-
0,25 мм) с 10% (по массе) динонилфталата или трикрезилфосфата.
Температура термостата колонок 60, испарителя 115—125, детектора
90°С.
468
Газ-носитель — гелий, расход 12, 18, 25, 40 и 60 мл/мин.
Скорость движения диаграммной ленты 1800 мм/ч.
Объект хроматографирования — бинарная смесь бензола и циклогек-
сана ~1 : 1.
Дозирование осуществляется микрошприцем МШ-10, доза 2,0±0,1 мкл.
Выполнение работы
Устанавливают колонки* в термостате хроматографа, про-
веряют и обеспечивают герметичность газовых линий. Затем
задают расход гелия через обе колонки около 12 мл/мин** и
выводят хроматограф на рекомендованный режим работы. По
установлении стабильной нулевой линии на диаграммной ленте
приступают к хроматографированию смеси бензол — циклогек-
сан. Если из-за несколько затянутой во времени процедуры до-
зирования и низкой чувствительности детектирования на пер-
вых пробных хроматограммах перо автоматически не пропишет
начало анализа, при повторных вводах пробы немедленно по-
сле дозирования мягко толкните перо самописца рукой — про-
ставьте стартовую отметку.
Движением плунжера микрошприца МШ-10 переводят остав-
шуюся в игле после дозирования жидкость в рабочий канал сте-
клянного корпуса и оценивают с возможно большей точностью
количество смеси, введенное в колонку. Записывают устано-
вленную реальную величину дозы на хроматограмме против
стартовой отметки.
Высоты пиков бензола и циклогексана на первых пробных
хроматограммах должны достигать 80-90% ширины диаграмм-
ной ленты. Если зарегистрируются значительно меньшие или,
наоборот, “зашкаленные” пики измените соответствующим об-
разом дозу или чувствительность регистрации сигнала детек-
тора.
Получив несколько (не менее трех) воспроизводимых хрома-
тограмм, удовлетворяющих этому условию, увеличивают ско-
рость пропускания гелия (одновременно через рабочую и срав-
нительную колонки) примерно до 18 мл/мин и по прекращении
дрейфа нулевой линии вновь хроматографируют смесь бензола
и циклогексана несколько раз, однако уже не изменяя окон-
чательно принятые в первом рабочем цикле величину дозы и
*Рабочей колонкой должна служить одна из названных, сравнитель-
ной, вообще говоря, может быть любая другая (выдается лаборантом).
**Эту и последующие большие скорости пропускания газа-носителя
через обе колонки можно устанавливать с некоторым отклонением от ре-
комендованных (реперных) значений, в диапазоне ±2 мл/мин. Важно,
однако, знать, при какой именно скорости газа (с точностью до 0,5—
1 мл/мин) проводится хроматографирование каждой дозы.
469
чувствительность регистрации сигнала. Постоянное значение
этих важных для данной работы параметров опыта поддер-
живают и в следующих циклах хроматографирования при по-
следовательно увеличиваемых расходах гелия (около 25, 40 и
60 мл/мин). Получив полный комплект хроматограмм, выклю-
чают прибор (помните о необходимости выключения в пер-
вую очередь тока моста катарометра!), срезают диаграмм-
ную ленту и приступают к обработке результатов.
Обработка результатов
Число т. т. N (по пику бензола) и ВЭТТ (Я) рассчитывают
по формулам
N = 5,54(<я/ш0,5)2 и H = L/N,
где /д — расстояние на хроматограмме от стартовой отметки
до вершины пика бензола, см; и>о,5 — полуширина пика, см; L —
длина колонки, см.
Используя полученные данные, строят график, откладывая
по оси абсцисс скорость движения газа-носителя через колонку
(мл/мин), а по оси ординат—усредненные значения ВЭТТ (Б).
По графику находят оптимальную скорость газа-носителя, обес-
печивающую наибольшую эффективность разделения.
Анализируя полученные хроматограммы, находят также зна-
чения критерия разделения R для каждого из режимов разделе-
ния по формуле
R = AZ/(u>oj5(i) + Wo,5(2)),
где AZ — расстояние между вершинами пиков на хроматограм-
ме, см; индексы 1 и 2 при символах wq 5 относятся к пикам ци-
клогексана и бензола соответственно.
Полученные данные приводят в виде таблицы (табл. V.2).
При оформлении протокола ответьте на следующие вопросы.
1. Чем вызвана необходимость как можно более строгого вос-
произведения величины дозы при выполнении всех анализов?
2. Изменится ли эффективность использованной колонки при
снижении величины дозы в 5-10 раз?
3. Какие дополнительные параметры режима работы хрома-
тографа потребовалось бы контролировать и, возможно, после-
довательно изменять при использовании для регистрации сиг-
нала детектора не катарометра, а ПИД?
4. Назовите самый простой, по вашему мнению, способ за-
метного увеличения достигнутой эффективности колонки.
5. Насколько увеличится эффективность колонки, характери-
зуемая числом т.т., при увеличении длины колонки в 2 (4 или
6) раз?
470
Таблица V.2. Экспериментальные данные по
изучению влияния скорости газа-носителя на
эффективность работы хроматографической колонки
Компоненты смеси* Скорость газа-носителя, мл/мин Ir, СМ дг, см ^0,5, СМ N Н R
Бензол (циклогексан) 12
Средние зна^ Бензол (циклогексан) чения 15
Средние значения
‘Расчет числа теоретических тарелок ведут по пику бензола, для рас-
чета критерия R принимают во внимание необходимые параметры пика
циклогексана.
Работа 3. Зависимость чувствительности
пламенно-ионизационного детектора от
относительных расходов газа-носителя,
водорода и воздуха (определение
оптимальных условий работы)
Цель работы. Освоение различных экспериментальных при-
емов определения оптимальных условий работы пламенно-иони-
зационного детектора.
Известно, что максимальная чувствительность и линейность
показаний ПИД проявляются лишь при правильно выбранном
соотношении расходов воздуха, водорода и газа-носителя. По-
следовательное увеличение расхода воздуха от нескольких де-
сятков до сотен мл/мин при каком-либо постоянном соотноше-
нии скоростей водорода и газа-носителя приводит сначала к
резкому, а затем к более плавному увеличению чувствитель-
ности детектора. Как правило, разумно устанавливать расход
воздуха в пределах 300-450 мл/мин.
Оптимальные соотношения расходов водорода и газа-носите-
ля при заданном расходе воздуха в большой мере зависят от
внутреннего диаметра колонки и сопла горелки детектора, а
также от конфигурации и взаимного расположения электродов
в ячейке детектора. В литературе чаще всего рекомендуется
поддерживать расходы газа-носителя, водорода и воздуха в со-
471
отношении около 1:1:10, например 30:30:300 мл/мин. Однако эту
рекомендацию следует рассматривать как ориентировочную.
Целесообразно устанавливать такую скорость газа-носителя,
которая обеспечивает максимальную эффективность разделе-
ния, т. е. минимальное значение ВЭТТ (для насадочных ко-
лонок в пределах 20-50 мл/мин). Однако в целях сокращения
продолжительности анализа иногда расход газа-носителя уве-
личивают вплоть до 80-100 мл/мин. Каждой конкретной скоро-
сти газа-носителя в этом диапазоне соответствует оптимальная
подача водорода в ячейку детектора, причем даже небольшие (в
пределах ±5 мл/мин) отклонения от оптимума могут приводить
к значительному снижению чувствительности ПИД (на 10- 25%
и более).
Для обеспечения максимальной чувствительности и точно-
сти следует проводить предварительное экспериментальное оп-
ределение оптимального соотношения расходов газа-носителя и
водорода применительно к типам используемых колонок (наса-
дочных, микронасадочных, капиллярных).
Аппаратура, условия, объекты хроматографирования
Хроматограф с пламенно-ионизационным детектором*, шкала электро-
метра изменяется при выполнении разных этапов работы в пределах
10“9—10“11 А. Расходы газа-носителя (азота), водорода и воздуха в ходе
выполнения работы изменяют в пределах 15—60 для газа-носителя и для
водорода и 150-600 мл/мин для воздуха.
Колонка стальная (3 м X 3 мм), заполненная хроматоном N (0,20-
0,25 мм), смоченным ди нон и л фтал атом, трикрезилфосфатом или сквала-
ном в количестве 10-20% (по массе).
Температура термостата колонок комнатная (вариант А); 90-100 ° С (ва-
риант Б); температура испарителя комнатная.
Скорость движения диаграммной ленты 240 (вариант А) или 1800 (ва-
риант Б) мм/ч.
Объект хроматографирования — искусственная смесь азота и метана
(или бытового газа) в соотношении 10:1 (5:1).
Дозирование осуществляется газовым кран ом-дозатором, доза** 0,1-
0,5 см3.
*При эксплуатации двухъячеечного ПИД желательно оптимизиро-
вать работу каждой ячейки в отдельности. Независимое задание опти-
мального режима каждой ячейке ПИД особенно важно при использовании
компенсационной схемы регистрации сигнала в режиме программирова-
ния температуры. Для удобства выполнения эксперимента на рабочих
линиях водорода и газа-носителя (азота) устанавливают дополнитель-
ные образцовые манометры.
**Помимо рабочего объема установленной на кране сменной петли
доза определяется еще и заданным соотношением метана и азота в смеси.
472
Внимание! Успешное выполнение данной работы воз-
можно только при полной герметичности газовых маги-
стралей в блоке подготовки газов и в термостате хромато-
графа, а также при полной герметичности газового крана-
дозатора.
Выполнение работы
Вариант А. 1. Определение оптимального соотношения
потоков газа-носителя и водорода при заданном расходе воз-
духа. Пользуясь графиком давление—расход*, задают скорость
газа-носите ля через колонку 40 мл/мин и подают в ячейку ПИД
водород со скоростью 60 мл/мин, воздух — 400 мл/мин. Спустя
1-2 мин поджигают водород, включают пишущий потенциометр,
регулируют положение пера на диаграммной ленте и выжидают
несколько минут до воспроизведения ровной нулевой линии.
С помощью газового крана-дозатора вводят в хроматограф
смесь бытового газа (метана) и азота и получают 2-3 пробные
хроматограммы. При этом чувствительность регистрации под-
бирают таким образом, чтобы высота пика составляла 50-70%
ширины диаграммной ленты**. Для обеспечения лучшей вос-
производимости петлю (дозу) газового крана промывают ка-
ждый раз одинаковым количеством рабочей газовой смеси (че-
рез дозирующую трубку достаточно пропускать 5-10 мл газо-
вой смеси, контроль по мыльно-пленочному измерителю).
Затем снижают скорость водорода до 55 мл/мин и повто-
ряют цикл дозирования стандартного количества смеси быто-
вого газа (метана) и азота***. В дальнейшем, после выполнения
очередного цикла операций, каждый раз снижают скорость по-
дачи водорода примерно на 5 мл/мин (и так до тех пор, пока не
погаснет пламя в ячейке детектора). При таком порядке выпол-
нения работы пики метана на хроматограмме со снижением по-
дачи водорода будут вначале возрастать, а затем уменьшаться.
Для того чтобы выявить оптимальную скорость водорода бо-
лее строго, полезно повторить хроматографирование, изменяя
*Графики строятся лаборантом заранее; для газа-носителя — приме-
нительно к устанавливаемой в приборе хроматографической колонке.
**Если это условие будет выполнено, при дальнейшем хроматографи-
ровании того же количества газовой смеси при меньших расходах водо-
рода, очевидно, не потребуется изменять чувствительность регистрации.
***Цикл включает 2—3-кратное дозирование стандартного количества
данной газовой смеси, причем последующий ввод ее в хроматограф произ-
водят только после регистрации на хроматограмме пика метана от пре-
дыдущего ввода. Для экономии времени, по согласованию с препода-
вателем, можно ограничиться однократным дозированием стандартного
количества искусственной газовой смеси
473
скорость подачи водорода в детектор в обратном порядке, т. е.
последовательно увеличивая ее от минимальной, обеспечиваю-
щей горение, до 50-60 мл/мин. При этом вблизи предполага-
емого оптимального расхода водорода подачу его следует из-
менять на очень небольшую величину (примерно 2 деления по
шкале образцового манометра).
Аналогичным образом находят (по согласованию с препода-
вателем) оптимальные расходы водорода при скоростях газа-
носителя около 15 и 60 мл/мин, начиная хроматографирование
стандартного количества газовой смеси при исходной подаче
водорода в ячейку ПИД 30 и 80 мл/мин соответственно.
На основании полученных экспериментальных данных строят
график в координатах: по оси абсцисс — расход водорода
(в мл/мин), по оси ординат — высота пиков* метана (в % от мак-
симальной**, отвечающей оптимальному соотношению расхо-
дов газа-носителя и водорода). Построенный график наглядно
иллюстрирует влияние соотношения расходов газа-носителя и
водорода на чувствительность ПИД для каждой из трех испы-
танных скоростей газа-носителя.
2. Определение влияния расхода воздуха на чувствитель-
ность пламенно-ионизационного детектора при заданном соот-
ношении потоков газа-носителя и водорода. Измерения прово-
дят при одном из двух заданных оптимальных соотношений,
например 15 и 17,5 или 60 и 30 мл/мин азота и водорода со-
ответственно***. В каждом из этих режимов цикличное хро-
матографирование смеси бытового газа (метана) и азота начи-
нают при расходе воздуха около 150 мл/мин. Чувствитель-
ность регистрации подбирают таким образом, чтобы высота
пика метана на хроматограмме составляла 50-70% ширины диа-
граммной ленты. Затем повторяют циклы хроматографирова-
ния, постепенно увеличивая подачу воздуха каждый раз еще
на 50 мл/мин, вплоть до достижения расхода воздуха
600 мл/мин****.
Полученные экспериментальные данные оформляют в гра-
фической форме, откладывая по оси абсцисс скорость воздуха
(в мл/мин), а по оси ординат высоту пиков метана (в % от макси-
*Если проводилось 2—3-кратное дозирование стандартного количества
газовой смеси бытового газа (метана) и азота, то предварительно находят
усредненное значение высоты пика метана для каждого расхода водорода.
**С учетом возможной записи пиков метана при различной чувстви-
тельности регистрации сигнала детектора.
***Указанные оптимальные соотношения азота и водорода условны.
Следует ориентироваться на собственные экспериментальные данные.
****В процессе работы, возможно, придется загрублять чувствитель-
ность регистрации сигнала детектора.
474
мальных значений, полученных в опыте) — с учетом масштаба
переключения чувствительности.
Вариант Б отличается от варианта А тем, что не требует
последовательного хроматографирования стандартного коли-
чества газообразных образцов при различных соотношениях
газа-носителя и водорода, поступающих в ячейку ПИД.
1. Определение оптимального соотношения потоков газа-но-
сителя и водорода при заданном расходе воздуха. Задав какую-
либо из рекомендованных преподавателем скоростей газа-носи-
теля через колонку (например, 40 или 60 мл/мин), разогревают
термостат хроматографа примерно до 100 °C для увеличения
концентрации паров неподвижной фазы в потоке газа-носителя.
Далее подают в ячейку ПИД воздух со скоростью 350 мл/мин и
водород, расход которого вначале устанавливают в соотноше-
нии 1:1 к расходу газа-носителя. Спустя 1-2 мин поджигают
водород, включают пишущий потенциометр и, подбирая со-
ответствующую чувствительность регистрации фонового сиг-
нала, выводят перо на уровень 30-50% ширины диаграммной
ленты. Выжидают несколько минут до воспроизведения устой-
чивой базовой линии и увеличивают подачу водорода в ячейку
ПИД до зашкаливания пера самописца.
Затем начинают медленно и, по возможности, равномерно
снижать расход водорода вплоть до затухания пламени. Перо
самописца должно при этом начать более или менее плавное
“сползание” влево, однако при приближении к оптимальному
для данной скорости газа-носителя расходу водорода направле-
ние движения пера изменится на обратное (слева направо) и по
прохождении максимума перо вновь изменит направление сво-
его движения. В итоге на диаграммной ленте запишется кривая
с пологими ветвями, разделенными максимумом. Давление во-
дорода в линии в момент прохождения пером самописца макси-
мума и будет обеспечивать оптимальный расход его для задан-
ной скорости газа-носителя. Поэтому в ходе выполнения экспе-
римента следует заметить это оптимальное давление водорода
и записать его.
С целью исключения случайного промаха и достижения боль-
шей точности измерения вновь задают расход водорода, соот-
носимый с расходом газа-носителя, поджигают водород и по-
вторяют описанную процедуру, причем при повторении целесо-
образно после поджигания водорода установить давление водо-
рода, отвечающее только что зарегистрированному максимуму,
и плавно изменять его в небольших пределах в большую и мень-
шую стороны.
475
Эксперимент может быть выполнен иначе. После установле-
ния рекомендованных расходов газа-носителя, водорода и воз-
духа, поджигания водорода, выведения пера на уровень 30-50%
ширины диаграммной ленты и воспроизведения устойчивой ба-
зовой линии фонового сигнала начинают снижать расход водо-
рода не плавно и непрерывно, а дискретно — так, чтобы стрелка
образцового манометра перемещалась каждый раз примерно на
2-3 деления.
После каждого снижения подачи водорода выжидают 3-4 мин,
наблюдая за постоянством смещенного уровня фонового сиг-
нала (ступеньки на хроматограмме). Так же, как и при плавном
изменении расхода водорода, перо самописца вначале должно
двигаться влево, затем вправо и вновь влево. В ходе работы
рекомендуется записывать прямо на диаграммной ленте про-
тив каждого нового уровня положения пера устанавливающееся
давление водорода в линии, считываемое с образцового мано-
метра. Сдвиги уровней фонового сигнала при изменении пор-
циями скорости водорода, вначале значительные, при прибли-
жении к области оптимального расхода водорода начнут зату-
хать. Соответственно следует уменьшать размер каждой по-
следующей порции. В итоге на диаграммной ленте запишется
ступенчатая кривая, по которой легко определить оптимальный
расход водорода, соответствующий заданной скорости газа-
носителя. Очевидно, оптимальный расход обеспечивается да-
влением водорода в линии, при котором была зарегистрирована
ступенька-вершина промежуточного максимума кривой.
Эта техника, хотя и требует несколько больших затрат вре-
мени, имеет некоторое преимущество перед ранее описанной,
поскольку здесь исключаются случайные промахи, связанные с
инерционностью работы элементов газовой схемы.
Далее по построенному заранее графику давление—расход
находят оптимальную скорость подачи водорода для задан-
ной скорости газ а-но сите ля. Сопоставляют полученные резуль-
таты с данными, полученными при выполнении работы по вари-
анту А.
2. Определение влияния расхода воздуха на чувствитель-
ность пламенно-ионизационного детектора при заданном соот-
ношении потоков газа-носителя и водорода. Как и при выполне-
нии работы по варианту А, измерения проводят при одном из
двух оптимальных заданных соотношений, например 15 и 17,5
или 60 и 30 мл/мин азота и водорода соответственно.
Установив заданные скорости азота и водорода, подают воз-
дух в ячейку ПИД с минимальной скоростью, т.е. около
150 мл/мин, и поджигают водород.
476
Выводят перо самописца на отметку, близкую к 10% ширины
диаграммной ленты, и выжидают несколько минут до воспро-
изведения устойчивой линии фонового сигнала. Затем, руко-
водствуясь графиком давление—расход, начинают увеличивать
подачу воздуха в ячейку ПИД порциями по 50 мл/мин. По-
сле каждого повышения расхода воздуха 1-2 мин наблюдают за
постоянством смещенного уровня фона. Значительное вначале
повышение уровня фонового сигнала, обусловливаемое увели-
чением расхода воздуха, по мере приближения к максимальным
расходам 500-600 мл/мин заметно затухает. Как и всегда при
подобных измерениях, всю процедуру повторяют еще по мень-
шей мере дважды.
Обработка результатов
На основании полученных экспериментальных данных строят
график в координатах: по оси асбцисс — расход воздуха
(в мл/мин), по оси ординат — высота уровня фонового сигнала
(в % от максимальной).
При аккуратном выполнении эксперимента по вариантам А и
Б получают близкие результаты.
Приведенными рекомендациями пользуются при выполнении
последующих лабораторных работ.
При оформлении протокола ответьте на следующие вопросы.
1. Изменится ли установленное вами оптимальное соотноше-
ние расходов газа-носителя, водорода и воздуха при
— заметном увеличении (снижении) абсолютного расхода
газа-носителя (например, в полтора-два раза);
— сохранении расходов всех газов, но при замене колонки;
— замене горелки в ячейке ПИД на другую (с иным диаме-
тром сопла);
— замене подвижной фазы на другую, например азота на ге-
лий, азота на водород?
2. Каковы сравнительные преимущества и ограничения при-
менения для установления оптимального газового питания ПИД
изложенных в работе процедур (вариант А и вариант Б)?
Работа 4. Зависимость чувствительности
детектора по теплопроводности от тока моста
и скорости газа-носителя
Цель работы. Проследить изменение сигнала детектора по
теплопроводности на заданное количество анализируемого ве-
щества при изменении тока моста и скорости газа-носителя.
477
400
Температура детектора,°C
Pmc.V.4. Зависимость между током моста, сигналом детек-
тора и сроком жизни чувствительных элементов.
I — область повышенной чувствительности и непродолжительного
срока жизни чувствительных элементов (максимальные шум и дрейф
сигнала); II — область малой чувствительности и продолжительного
срока жизни чувствительных элементов (минимальные шум и дрейф
сигнала); III— повышение чувствительности, сокращение срока жизни
чувствительных элементов; IV — понижение чувствительности, удли-
нение срока жизни чувствительных элементов.
Детектор iio теплопроводности (катарометр) — типичный
концентрационный детектор, и поэтому его сигнал существенно
зависит от скорости газа-носителя (см. раздел II. 1.4.3). Другой
причиной наблюдаемой зависимости чувствительности катаро-
метра от скорости газа-носителя является нарушение условий
теплообмена в ячейке детектора (изменение расхода подвиж-
ной фазы приводит к изменению температуры и, следовательно,
электрического сопротивления чувствительных элементов).
При постоянной скорости газа-носителя сигнал (отклик) ка-
тарометра на заданное постоянное количество анализируемого
вещества определяется, прежде всего, разностью теплопровод-
ностей газовых сред в сравнительной и измерительной ячейках
детектора и перегревом его чувствительных элементов относи-
тельно корпуса. Температура чувствительных элементов воз-
растает с увеличением подаваемой на них электрической на-
грузки (тока моста).
В практической работе важно помнить, что, обусловливая
увеличение чувствительности детектирования, повышение тока
моста приводит вместе с тем к сокращению срока жизни чув-
ствительных элементов и может вызвать преждевременный вы-
ход их из строя (перегорание). Поэтому при выборе тока моста
катарометра нельзя превышать предельно допустимые значе-
478
ния устанавливаемые заводом-изготовителем для разных газов-
носителей и рабочих температур (соответствующие таблицы
приводятся в инструкциях к приборам). Зависимость между то-
ком моста, сигналом детектора и сроком жизни чувствительных
элементов в общем виде представлена на рис. V.4.
Скорость газа-носителя и величина тока моста являются наи-
более легко и часто изменяемыми экспериментальными пара-
метрами, существенно влияющими на чувствительность детек-
тора. Это обстоятельство всегда следует иметь в виду при про-
ведении количественного газохроматографического анализа, в
особенности методом абсолютной градуировки (внешнего стан-
дарта).
Аппаратура, условия, объекты хроматографирования
Хроматограф с детектором по теплопроводности, укомплектованный
интегратором.
Колонки*: а) стальная или стеклянная (3 м X 3 мм), заполненная хе-
засорбом или динохромом Н (0,25-0,315 мм), смоченным скваланом или
метилсиликоном типа SE-30 (25% по массе); б) стальная или стеклянная
(2 м X 3 мм), заполненная свежепрокаленным цеолитом СаА (0,20-0,25мм).
Ток моста детектора при выполнении различных этапов этой работы
устанавливают в пределах от 80 до 350 мА; множитель шкалы — от 1 до
20 или 30 соответственно.
Температура термостатов колонок и детектора комнатная.
Газ-носитель — гелий или водород; при выполнении различных эта-
пов этой работы расход подвижной фазы устанавливают в пределах от
10 до 100 мл/мин.
Скорость движения диаграммной ленты 200 или 600 мм/ч (при ре-
гистрации сигнала программно-аппаратным комплексом типа “Мульти-
Хром” на базе ПК пишущий потенциометр вообще может быть отключен).
Объекты хроматографирования: при использовании колонки а) — раз-
бавленный газом-носителем бытовой газ (метан или пропан-пропилен);
при использовании колонки б) — разбавленный газом-носителем азот
(или аргон).
Дозирование осуществляется газовым краном-дозатором, рабочий
объем сменной дозирующей петли крана ~0,25 см3.
Выполнение работы
Знакомятся с собранной в термостате хроматографа газовой
схемой, проверяют и обеспечивают ее герметичность.
Внимание! При выполнении данной работы предъ-
являются повышенные требования к герметичности газо-
вого крана-дозатор а и всех остальных газовых линий.
*Рабочей колонкой может служить одна из названных, сравнительной
любая другая.
479
1. Определение зависимости чувствительности катарометра
от скорости газа-носителя при заданной величине тока моста
(200 мА). Задают расход газа-носителя через обе колонки около
10 мл/мин* и выводят хроматограф на рекомендованный ра-
бочий режим. По установлении стабильного фонового сиг-
нала производят пробное дозирование заранее приготовленной
в стальном баллоне вместимостью до 0,5 л или в резиновой
(волейбольной) камере газовой смеси (разбавленного подвиж-
ной фазой бытового газа или азота (аргона) — в зависимости
от используемой колонки). Сразу после дозирования дают ко-
манду интегратору на автоматическую регистрацию сигнала
детектора. На хроматограмме должен зарегистрироваться пик,
по высоте достигающий 70-90% шкалы регистратора. При не-
обходимости (зашкаленный или слишком малый пик) коррек-
тируют соответствующим образом чувствительность регистра-
ции (множитель шкалы), величину дозы или состав (степень раз-
бавления подвижной фазой) дозируемой газовой смеси. В вы-
бранных условиях проводят еще по меньшей мере два-три ана-
лиза, стремясь к получению воспроизводимых результатов; при
отсутствии воспроизводимости следует обратиться к препода-
вателю. В дальнейших опытах (при последовательном увели-
чении расхода газа-носителя) состав анализируемой газовой
смеси не изменяют!
Затем увеличивают скорость газа-носителя примерно на
10 мл/мин, дожидаются стабилизации потока подвижной фазы
в рабочей и сравнительной линиях (контроль и строгое изме-
рение — по цифровой индикации имеющихся у современных
приборов соответствующих электронных устройств, либо по
показаниям мыльно-пленочного измерителя, поочередно под-
ключаемого к выходным штуцерам рабочей и сравнительной
ячеек детектора по теплопроводности), дожидаются стабили-
зации фонового сигнала, приближают его уровень к нулевой
отметке шкалы регистратора и вновь несколько раз (не менее
трех) записывают хроматограммы бытового газа или азота (ар-
гона) в подвижной фазе, помечая точное значение скорости газа-
носителя, при которой проводится анализ.
Получив воспроизводимые результаты, переходят к хромато-
графированию при следующей скорости газа-носителя (около
30 мл/мин) и так далее, вплоть до его расхода, отвечающего
~100 мл/мин.
Завершив намеченный цикл анализов, увеличивают ток мо-
ста катарометра примерно до 300 мА, снижают скорость газа-
*См. сноску на с.479.
480
носителя до начальной (~10 мл/мин) и, дождавшись воспроиз-
ведения устойчивого фонового сигнала, проводят аналогичный
цикл измерений*.
2. Определение зависимости сигнала катарометра от тока
моста для заданной скорости газа-носителя (около 30 мл/мин).
Задают ток моста катарометра примерно 80 мА и дожидаются
исчезновения дрейфа фонового сигнала, после чего производят
пробное дозирование заранее приготовленного (см. выше) рас-
твора бытового газа или азота (аргона) в подвижной фазе. В
последующих анализах подбирают численное значение множи-
теля шкалы, обеспечивающее запись сигнала детектора с ам-
плитудой в пределах 50-90% шкалы регистратора.
При выбранных условиях работы прибора записывают не ме-
нее трех хроматограмм (до получения воспроизводимых резуль-
татов). Затем увеличивают ток моста примерно до 130-150 мА,
дожидаются исчезновения дрейфа фонового сигнала, при необ-
ходимости смещают его к нулевой отметке шкалы регистратора
и вновь несколько раз (не менее трех) записывают сигналы де-
тектора на одно и то же количество бытового газа или азота (ар-
гона), растворенных в газе-носителе. Получив воспроизводи-
мые результаты, переходят к хроматографированию при следу-
ющем по величине токе моста (около 200 мА) и так далее, вплоть
до достижения предельно допустимого его значения (~350 мА),
последовательно загрубляя чувствительность регистрации при
возможном зашкаливании пиков.
В ходе выполнения этого этапа экспериментальной работы
необходимо периодически контролировать постоянство задан-
ной вначале скорости газа-носителя и отмечать точное ее зна-
чение, а также точные значения периодически изменяемых тока
моста и других параметров опыта в паспорте на хроматограмму
(или непосредственно на диаграммной бумаге задействованного
пишущего потенциометра).
Завершив намеченный цикл анализов, увеличивают скорость
газа-носителя до 50-70 мл/мин, снижают ток моста примерно
до 100 мА, дожидаются стабилизации фонового сигнала и про-
водят аналогичный цикл измерений. Закончив эксперименталь-
ную часть работы, выключают хроматограф (в первую оче-
редь отключают ток моста!) и приступают к обработке ре-
зультатов. В отчете о выполненной работе следует сформули-
*По согласованию с преподавателем, на этом экспериментальная часть
работы может быть закончена. В таком случае выключают хроматограф
(в первую очередь отключают ток моста!) и приступают к обработке
результатов.
481
ровать практические выводы, вытекающие из анализа получен-
ных закономерностей.
Обработка результатов
Измеренные интегратором площади пиков на хроматограм-
мах приводят к единой шкале чувствительности* в пределах 1-й
и/или 2-й серий анализов.
Усредняют найденные площади пиков для каждой из исполь-
зованных скоростей газа-носителя и для каждого значения тока
моста и выражают их в процентах от максимальной зарегистри-
рованной величины сигнала (площади пика).
Зависимости величины отклика катарометра на одинаковое
количество вещества при различных скоростях газа-носителя
и различных величинах тока моста выражают в графической
форме, откладывая по оси ординат относительный сигнал де-
тектора (площадь пика) в % от максимального значения, а по
оси абсцисс в первом случае скорость газа-носителя (мл/мин),
а во втором — ток моста (мА).
При оформлении протокола ответьте на следующие вопросы.
1. Можно ли при решении поставленных в этой работе задач
использовать в качестве объекта хроматографирования модель-
ную смесь, составленную из жидкостей? На что следовало бы
обратить первоочередное внимание при таком изменении мето-
дики выполнения эксперимента?
2. Какая опасность грозит аналитику при задании тока мо-
ста детектора по теплопроводности, превышающего по вели-
чине разрешенную заводом-изготовителем для выбранных вами
условий (природа газа-носителя, температура корпуса детек-
тора)?
3. Какими соображениями следует руководствоваться при
задании рабочей температуры детектора по теплопроводности?
Работа 5. Качественный анализ по индексам
удерживания при использовании
капиллярных колонок
Цель работы. 1. Определение индексов удерживания Кова-
ча (изотермических) I и/или обобщенных индексов удержива-
ния GI (наиболее удобные относительные параметры для каче-
ственной характеристики компонентов анализируемых смесей в
*Если сигнал детектора измерялся интегратором, подключенным не-
посредственно к блоку детектора (а не к клеммам самописца), эту опера-
цию опускают.
482
режиме программирования температуры) и идентификация со-
единений, содержащихся в контрольных образцах, сопоставле-
нием экспериментально найденных численных значений I и/или
GI с надежными справочными данными.
2. Освоение косвенных методов измерения времени пребыва-
ния в колонке (приборе) неудерживаемого соединения tM (т. н.
мертвого времени колонки (прибора)) по полным (неисправлен-
ным) временам удерживания трех ближайших гомологов (на-
пример, н-алканов с числом атомов углерода z 6) при выпол-
нении качественного газохроматографического анализа в изо-
термических условиях.
3. Ознакомление с отличительными особенностями схемы га-
зовых коммуникаций в термостате хроматографа, применяемой
при дозировании пробы в капиллярную колонку с делением по-
тока; приобретение навыков задания необходимых абсолютных
и относительных расходов газа-носителя через колонку и линию
сброса.
Работа выполняется в двух вариантах: в изотермическом ре-
жиме работы хроматографической колонки и в режиме програм-
мирования температуры. Общим для обоих вариантов является
начальное хроматографирование в рекомендованных ниже усло-
виях при использовании “классической” капиллярной колонки
типа WCOT со стандартной неполярной полидиметилсилокса-
новой неподвижной фазой опорных смесей н-алканов, например
Сб-Сэ (в изотермическом режиме) или Сб~Сц (в режиме про-
граммирования температуры). Далее в тех же условиях хро-
матографируют одну-две (по согласованию с преподавателем)
искусственные смеси известного состава использованных ранее
н-алканов и углеводородов других классов или их простейших
функциональных производных (кетонов, спиртов, сложных эфи-
ров). На заключительном этапе работы исследуют состав кон-
трольных образцов, содержащих реперные н-алканы и одно-два
или большее число неизвестных студентам соединений.
По результатам хроматографирования находят индексы
удерживания Ковача 1 и/или обобщенные индексы удержива-
ния GI компонентов искусственных смесей (известного состава)
и неизвестных соединений, содержащихся в контрольных образ-
цах. Сравнивают найденные 1 и/или GI компонентов искус-
ственных смесей и контрольных образцов с приведенными в
справочниках [227, 228] или рандомизованными величинами ин-
дексов удерживания, заимствуемыми из базы данных НИИ Хи-
мии СПбГУ ([243], табл. V.3), и оценивают надежность выполня-
емой таким путем идентификации (число возможных вариантов
483
Таблица V.3. Рандомизованные индексы удерживания
некоторых соединений на стандартных неполярных
полидиметилсилоксановых неподвижных фазах
Соединение Индекс удерживания GI ± СКО Соединение Индекс удерживания GI ± СКО
Бензол 657 ±9 Метил бутаноат 709 ± 5
Циклогексан 668 ±8 Изопропилбутаноат 826 ± 5
Изооктан 690 ±2 Изобутил пропаноат 851 ± 7
Толуол 760 ±9 Изобу тил бутаноат 940 ± 5
Этилбензол 854 ±9 2-Пентан он 670 ±8
az-Ксилол 865 ±8 2-Гексанон 768 ±9
Изопропилбензол 914 ±6 4-Гептанон 851 ±4
Пропилбензол 945 ±9 Циклогексанон 873 ± 14
Бутилбензол 1047 ± 7 2,6-Диметил-4- 954 ±3
гептанон
ответа на поставленный вопрос о природе соединений, содержа-
щихся в проанализированных композициях).
Аппаратура, условия, объекты хроматографирования
Хроматограф с пламенно-ионизационным детектором, укомплектован-
ный интегратором или компьютеризованной системой обработки резуль-
татов анализа. Шкала электрометра переменная, в диапазоне 5 • 10—10—
5 • 10'11 А. Расход водорода и воздуха около 30 и 350 мл/мин соответ-
ственно.
Колонка капиллярная (кварцевая) с внутренним диаметром 0,25—
0,35 мм и длиной 30—60 м со стандартной неполярной полидиметилсилок-
сановой неподвижной фазой (типа SE-30 или DB-1).
Температура термостата колонок в изотермическом режиме 60°С,
в режиме программирования температуры от 60 до 100 или 120°С
(2 или 3 град/мин в зависимости от состава исследуемых смесей — по
указанию преподавателя); температура испарителя 175°С, температура
детектора (задается при работе на хроматографах типа “Цвет-500”, пре-
дусматривающих такую возможность) 150 ° С.
Газ-носитель — азот, расход через колонку ~1 мл/мин, сброс в атмо-
сферу -100 мл/мин; поддув в детектор по обводной линии 25-30 мл/мин.
Скорость движения диаграммной ленты* 600 мм/ч.
Объекты хроматографирования. 1. Изотермический режим.
Опорная смесь: 1) н-алканы Се—Cg. Искусственные смеси: 2) н-алканы
Ос—Cg, бензол, циклогексан, изооктан, толуол, этилбензол, м- и п-ксило-
лы; 3) н-алканы Cg—Cg, 2-пентанон, 3-пентанон, 2-гексанон, 3-гептанон;
4) изобутиловый, бутиловый, mpem-пентиловый, изопентиловый, пенти-
ловый и гексиловый спирты; 5) н-алканы Се—Cg, изопропилацетат, про-
*При использовании для регистрации хроматограмм электронных пи-
шущих потенциометров.
484
пилацетат, изобутилацетат, бутилацетат, изопентил ацетат. Контроль-
ные смеси н-алканов Cg—Cg и нескольких соединений из других назван-
ных гомологических рядов.
2. Режим программирования температуры. Опорная смесь:
6) м-алканы Се-Сц. Искусственные смеси: 7) н-алканы Cg—Сц, циклогек-
сан, изооктан, ароматические углеводороды, входящие в искусственную
смесь 2, и, дополнительно, пропил- и изопропилбензолы; 8) н-алканы Се—
Сц, спирты С4— Се, входящие в искусственную смесь 4, и, кроме того,
2-гептанол, 1-гептанол, 2-октанол, 1-октанол; 9) н-алканы Се-Сц, кетоны
С5-С7, входящие в искусственную смесь 3, и, дополнительно, циклогек-
санон и 2-октанон; 10) н-алканы Cg—Сц, алкилацетаты С3—С5, входящие в
искусственную смесь 5, и, кроме того, пентилацетат и гексилацетат. Кон-
трольные смеси н-алканов Cg—Сц и нескольких соединений из других
названных гомологических рядов.
Дозирование осуществляется микрошприцем МШ-1, величина дозы
0,1-0,5 мкл.
Выполнение работы
Знакомятся с собранной в термостате хроматографа схемой
газовых коммуникаций для работы с капиллярными колонками,
включающей, во-первых, делитель потока паров пробы и линию
сброса их избыточного количества в атмосферу и, во-вторых,
обводную линию газа-носителя, соединяющую вторую камеру
испарителя хроматографа с коммутирующим тройником, уста-
навливаемым на выходе капиллярной колонки для исключения
размывания элюируемых из хроматографической колонки хро-
матографических зон в коммуникации, связывающие выход ко-
лонки с детектором (см. рис. 11.13, а).
При подготовке хроматографа к работе прежде всего задают
давление газа-носителя на выходе из 1-го и П-го каналов блока
подготовки газа, обеспечивающее рекомендованные выше рас-
ходы азота через обводную линию, линию сброса и капилляр-
ную колонку. Скорость газа-носителя на выходе колонки кон-
тролируют с помощью миниатюрного мыльно-пленочного изме-
рителя с рабочим объемом 5-10 мл; скорости газа по линиям
поддува и сброса измеряют отдельными мыльно-пленочными
измерителями с рабочим объемом 20-50 и 50-100 мл соответ-
ственно. Выставив необходимые расходы азота, герметично со-
единяют капиллярную колонку с коммутирующим тройником 3
так, как показано на рис. 11.13, а (важно, чтобы струя подду-
ваемого азота омывала выходной конец колонки, а не про-
ходила над ним !), и выводят хроматограф на согласованный
с преподавателем режим работы — изотермический или с про-
граммированием температуры колонки.
При выполнении работы на приборе “Цвет-500” температур-
ную программу задают с помощью блока управления БУ-125.
485
При использовании хроматографа “Ивет-100” на температур-
ной шкале лимба программатора устанавливают верхний и ниж-
ний ограничители на отметки 120 (или 100, в зависимости от
состава исследуемых смесей) и 60 °C соответственно. Переклю-
чатель скорости нагрева устанавливают в положение 3°С/мин
(при этом расположенная под ним клавиша кратности “xl, х2”
должна быть отжата). Вращением шкалы прижимают ограничи-
тель нижнего предела программы к кнопке микровыключателя и
нажимают клавишу НАГРЕВ. Термостат хроматографа разо-
гревается до 60°C за 5-7 мин, прогрев испарителя и детектора
до рекомендованных выше температурных значений требует 20-
25 мин. В этот период времени задают расходы вспомогатель-
ных газов, спустя 5-10 мин поджигают водород в ячейке ПИД,
выводят фоновый сигнал детектора на уровень шкалы регистра-
тора, близкий к нулевой отметке, и контролируют в течение 3-
5 мин стабильность базовой линии хроматограммы.
Убедившись в готовности прибора по всем заданным параме-
трам к выполнению анализов в изотермических условиях или
с программированием температуры колонки, производят проб-
ное дозирование опорной смеси реперных н-алканов и неме-
дленно включают: 1) интегратор или программно-аппаратный
комплекс “МультиХром”, заранее подготовленный к измерению
параметров удерживания компонентов анализируемых образ-
цов; 2) программирование температуры термостата — если
оно необходимо! Для этого при использовании хроматографа
“Пвет-500” нажимают кнопку ПРОГРАММА на лицевой па-
нели блока БУ-125; при выполнении работы на хроматографах
“Цвет-100” нажимают клавишу ПУСК на лицевой панели про-
грамматора. В тех случаях, когда хроматограмма записыва-
ется пишущим потенциометром, на диаграммной бумаге каран-
дашом или фломастером наносят стартовую отметку (допуска-
ется “мягко” толкнуть перо самописца) и помечают величину
дозы и заданную шкалу электрометра. При регистрации хро-
матограммы компьютеризованной системой обработки данных
соответствующие сведения записывают в рабочий блокнот для
последующего внесения в память ЭВМ.
Наблюдают за ходом регистрации хроматограммы и выпол-
нением температурной программы (если она задавалась). По
окончании цикла программирования термостат хроматографа
” Пвет-500” автоматически охладится и через несколько минут
будет готов к повторному анализу. При работе на приборе
“Цвет-100” по окончании программирования температуры и за-
вершении регистрации хроматограммы клавишу НАГРЕВ—
ОХЛАЖДЕНИЕ переводят в положение ОХЛАЖДЕНИЕ и
486
открывают дверку термостата. Охлаждение производят около
10 мин. За это время шкала программатора должна автоматиче-
ски возвратиться в исходное положение, а температура (контро-
лируемая ртутным термометром) снизиться до 30-35 °C. Закры-
вают дверку термостата и выжидают 1-2 мин (время, необходи-
мое для выравнивания температуры внутри термостата), после
чего вновь переводят клавишу НАГРЕВ—ОХЛАЖДЕНИЕ в
положение НАГРЕВ. Через 5-7 мин устанавливается темпера-
тура 60°C; прибор готов к очередному циклу программирова-
ния.
Опорные, а затем искусственные и контрольные смеси (из
числа названных выше) в выбранном температурном режиме
аналогичным образом хроматографируют по несколько раз (по
меньшей мере дважды) до получения воспроизводимых резуль-
татов по параметрам (временам) удерживания, измеряемых ин-
тегратором или компьютеризованной системой обработки дан-
ных. В рекомендованных условиях выполнения анализа на хро-
матограммах запишутся пики, занимающие по высоте 30-90%
шкалы регистратора. Если по каким-либо причинам на хрома-
тограммах будут записываться слишком малые или зашкален-
ные пики, следует изменить соответствующим образом чувстви-
тельность регистрации ранее неудачно записанных сигналов де-
тектора или уменьшить дозу.
Внимание! Увеличивать дозу при работе с капилляр-
ными колонками опасно: это может привести к захлебыва-
нию колонки, снижению эффективности разделения и смы-
ванию неподвижной фазы со стенок колонки.
Хроматограммы опорных смесей необходимы для расчета
времени пребывания в колонке (приборе) несорбируемого веще-
ства, а также для опознания пиков н-алканов на хроматограм-
мах искусственных и контрольных смесей.
Обработка результатов
Расчет времени пребывания несорбируемого соединения
по параметрам удерживания трех гомологов. Напомним, что
при хроматографировании в изотермических условиях соеди-
нений, принадлежащих к какому-либо гомологическому ряду,
имеет место линейная зависимость между 1g и числом z ато-
мов углерода в молекуле (подробнее см. раздел IV.2.4.3):
lg t'R = a +bz,
где а и b — константы.
487
Рис. V.5. Гипотетические хроматограммы трех последова-
тельно элюируемых ближайших гомологов.
Обозначим на гипотетической хроматограмме трех последо-
вательно выходящих из колонки ближайших гомологов испра-
вленные расстояния до вершин пиков через Х±, Х2 и Хз соответ-
ственно (рис. V.5, а). Если расстояния до вершин пиков гомоло-
гов измерять не от вершины пика несорбируемого соединения
(X), а от произвольно выбранной точки (У), то расстояния У,
У2 и Уз будут отличаться от Xi, Х2 и Хз на величину ё:
Xi =Yi+ 6.
Так как численное значение логарифма Xi пропорционально
z, что можно представить в виде
1ё(У + 6)ooz,
то при условии Z2 — zi = Z3 — Z2 (т. е. для трех ближайших гомо-
логов) можно записать
Y2+S = У3+£
У +£ “ У2+<5’
откуда
У22-УзУ1
Уз + У —2У2’
Зная ё, нетрудно найти Хм (расстояние на хроматограмме от
стартовой отметки до вершины пика несорбируемого соедине-
ния): если ё положительна, Хм = Уо — ё; если ё отрицательна,
Хм = Уо +|<5|. Лля упрощения расчетов за Уо принимают рассто-
яние от стартовой отметки до вершины пика среднего гомолога
(см. рис. V.5, 6). В этом случае У2 = 0, а У1 становится отрица-
тельной величиной, и тогда
5 = УзУ/(Уз-У).
488
Окончательно
XM = Y0- [У3У1/(У3 - У)].
Величины Yq, Yi и Уз находят из неисправленных (полных)
времен удерживания соответствующих гомологов, измеряемых
в ходе выполнения эксперимента интегратором или програм-
мно-аппаратным комплексом типа “МультиХром”. Результаты
измерений и расчетов заносят в таблицу (табл. V.4).
Таблица V.^. Экспериментальные данные, необходимые
для расчета времени пребывания несорбируемого соеди-
нения в колонке (приборе) по полным (неисправленным)
временам удерживания трех последовательных гомоло-
гов ( я-a л каков)______________________________
№ хроматограммы Го У1 Уз ° *3-Yl iM (текущее) tM (среднее)
1 2 3
В режиме программирования температуры время пребыва-
ния несорбируемого соединения в колонке (приборе) tM зависит
от скорости программирования и от начальной температуры
анализа. В связи с недавно показанной возможностью расчета
обобщенных индексов удерживания (наиболее удобных пара-
метров для качественной характеристики различных органиче-
ских соединений в режиме программирования температуры) по
полным (неисправленным) временам удерживания [196] извест-
ные (и достаточно сложные) методы, предложенные для расчета
tM при программировании температуры [197, 198], здесь не рас-
сматриваются.
Определение индексов удерживания Ковача соединений,
содержащихся в искусственных и контрольных смесях. Со-
поставлением хроматограмм опорной смеси и искусственной
или контрольной смеси, полученных в одинаковых условиях
изотермического режима работы колонки, опознают на послед-
них пики н-алканов и приписывают им индексы удерживания,
равные числу атомов углерода, умноженному на 100 ед. Вно-
сят поправку на время пребывания в колонке (приборе) неудер-
живаемого соединения tM в параметры (полные времена) удер-
живания tp_ всех компонентов, зарегистрированных на хрома-
489
тограмме искусственной или контрольной смеси*, усредняют
исправленные времена удерживания /.'R, найденные по несколь-
ким последовательно записанным хроматограммам, и логариф-
мируют численные значения
Далее строят графическую зависимость между lg н-алка-
нов (по оси ординат) и приписанными им индексами удержива-
ния (по оси абсцисс).
Индексы удерживания соединений, присутствующих в искус-
ственных или контрольных смесях, находят либо по построен-
ному графику, либо, что намного точнее, по формулам (IV.13)
или (IV.16).
Напомним, что с целью повышения точности определения ин-
дексов удерживания Ковача следует стремиться использовать
в качестве реперных н-алканов ближайшие гомологи, различа-
ющиеся лишь на один атом углерода (n = 1).
Расчет численных значений индексов удерживания Ковача
можно выполнить на микрокалькуляторе или ПК по програм-
мам, приведенным в Приложении. Возможность расчета индек-
сов удерживания в автоматическом режиме заложена и в про-
граммное обеспечение компьютеризованной системы обработки
результатов хроматографического анализа “МультиХром” (см.
раздел IV.5).
Результаты идентификации компонентов искусственных и
контрольных смесей по индексам удерживания Ковача при изме-
рении параметров удерживания электронными интеграторами,
не связанными с ПК, заносят в таблицу (табл. V.5).
Определение обобщенных индексов удерживания соеди-
нений, содержащихся в искусственных и контрольных сме-
сях. Сопоставлением хроматограмм опорной смеси и искус-
ственной или контрольной смеси, полученных в одинаковых
условиях (начальная и конечная температуры и скорость про-
граммирования), опознают и выбирают на последних пики трех
ближайших последовательно элюируемых >(-алканов, времена
удерживания которых соотносятся с временем удерживания ха-
рактеризуемого г-го соединения и между собой согласно нера-
венству (IV-29). По полным (неисправленным) временам удер-
живания выбранных н-алканов, согласно уравнениям (IV.27) и
(IV.28), рассчитывают параметр q и обобщенные индексы GI.
Расчет переменных (для разных характеризуемых соедине-
ний и групп н-алканов, удовлетворяющих неравенству (IV.29))
параметров q и соответствующих обобщенных индексов удер-
*Для повышения точности результатов следует рассчитывать по
этой же хроматограмме.
490
Таблица V.5. Результаты идентификации
компонентов искусственной (контрольной)
смеси по индексам удерживании Ковача*
Кв
хромато-
граммы
Ка-
пика
tR1
(текущее)
(среднее)
ДО
Название
компонента
смеси**
*В таблицу заносят данные и по опознанным н-алканам.
**При обработке хроматограмм контрольной смеси названия иден-
тифицируемых соединений устанавливаются сопоставлением найденных
индексов удерживания Ковача I с обобщенными индексами GI, приведен-
ными в табл. V.3.
живания GI можно выполнить на персональном компьютере по
специальной программе (см. Приложение).
Результаты первичной обработки хроматограмм и последу-
ющих расчетов заносят в таблицу (табл. V.6).
Таблица V.6. Результаты идентификации
компонентов искусственной (контрольной)
смеси по обобщенным индексам удерживанил*
№ хромато- граммы Кв пика 9 GI (текущее) GI (среднее) Название компонента смеси
1 2 1
1 2 2
И т.д.
*См. сноски к табл V.5.
491
Сравнивая найденные численные значения I и/или GI для
соединений, содержащихся в искусственных смесях известного
качественного состава, с представленными в выборке из базы
данных НИИ Химии СПбГУ (см. табл.У.З), оценивают пра-
вильность проведенного эксперимента и надежность выполнен-
ных измерений и расчетов. Если расхождения между найден-
ными и сравниваемыми численными значениями I и/или GI не
превышают окна рандомизации (±8 ед.), сопоставлением экспе-
риментально найденных и приведенных в табл. V.3 значений ин-
дексов удерживания проводят идентификацию соединений, со-
держащихся в контрольных смесях. Результаты идентификации
представляют в таблицах (табл.У.б и/или V.6).
При оформлении протокола ответьте на следующие вопросы.
1. Какие цели преследует предусматриваемое в данной ра-
боте деление потока паров пробы на выходе испарителя хрома-
тографа?
2. Оцените эффективность работы колонки по одной из ваших
хроматограмм числом теоретических тарелок. Как повлияет на
эффективность разделения увеличение доли паров смеси, сбра-
сываемых в атмосферу (например, при задании соотношения
расходов газа-носителя через колонку и линию сброса ~ 1:200
или ~1:300 вместо рекомендованного ~1:100)?
3. Изменится ли эффективность разделения при замене по-
движной фазы (например, при использовании в качестве газа-
носителя не азота, а гелия или водорода)?
4. Чем вызвана целесообразность измерения индексов удер-
живания по результатам хроматографирования смеси, содержа-
щей не только идентифицируемые соединения, но и вещества-
реперы? В каких ситуациях оправдано раздельное хромато-
графирование образцов с характеризуемыми соединениями и
веществ-реперов?
5. Какие задачи решает поддув газа-носителя в ПИЛ через
тройник на выходе капиллярной колонки? При каких обстоя-
тельствах этого можно было бы избежать?
6. Каковы главные преимущества системы обобщенных ин-
дексов удерживания в сравнении с системами индексов удер-
живания Ковача (изотермических) и индексов Ван-ден-Доола и
Кратца (предложенных для режимов с программированием тем-
пературы)?
492
Работа 6. Идентификация органических
соединений в сложных смесях по индексам
удерживания и коэффициентам распределения
в системе ограниченно смешивающихся
растворителей
Полная (групповая и индивидуальная) идентификация ор-
ганических соединений по параметрам удерживания при га-
зохроматографическом исследовании смесей неизвестного со-
става требует, как правило, привлечения дополнительных све-
дений, получаемых, например, при направленном химическом
воздействии на образец на стадии подготовки пробы или непо-
средственно в процессе осуществления анализа, а также (что
наиболее эффективно) при регистрации разделяемых компонен-
тов с помощью таких мощных информативных детекторов, как
атомно-эмиссионный, ИК(фурье)- или масс-спектрометр. Из-
вестны, однако, и другие, весьма простые и не требующие
применения дорогостоящего оборудования приемы идентифика-
ции, предусматривающие, в частности, использование наряду с
параметрами (индексами) удерживания данных о внеколоноч-
ном распределении компонентов исследуемых смесей в системе
двух ограниченно смешивающихся растворителей (см. раздел
IV.2.4.3). В этой работе описывается методика именно такого
эксперимента [424].
Цель работы. Идентификация неизвестных соединений, при-
надлежащих к разным классам органических веществ, в много-
компонентной смеси по результатам измерений индексов удер-
живания на колонке со стандартной неполярной полидиметил-
силоксановой неподвижной фазой и коэффициентов распределе-
ния в системе гексан—ацетонитрил.
Идентификация органических соединений в сложных смесях
только по их “хроматографическим спектрам”, т.е. по набору
численных значений индексов удерживания каждого вещества
на нескольких (от трех до шести) колонках с неподвижными фа-
зами, отличающимися по условной хроматографической поляр-
ности, часто приводит к неоднозначным результатам, в первую
очередь, потому, что далеко не всегда можно уверенно соот-
нести принадлежащие одному и Лму же компоненту пики на
разных хроматограммах. Определение коэффициентов распре-
деления идентифицируемых веществ между двумя ограниченно
смешивающимися растворителями (по данным хроматографи-
ческого анализа каждого из образующихся растворов) обеспе-
чивает получение весьма ценной дополнительной информации,
493
часто достаточной для решения поставленной задачи при усло-
вии параллельного измерения индексов удерживания компонен-
тов смеси всего лишь на одной колонке (с полярной или непо-
лярной неподвижной фазой). К настоящему времени устано-
влено, что из возможных комбинаций ограниченно смешиваю-
щихся растворителей наиболее универсальной является пара
гексан — ацетонитрил. Данная система растворителей обеспе-
чивает относительное постоянство параметра а для разных го-
мологических рядов в корреляционном уравнении (IV.38), свя-
зывающем коэффициенты распределения Кр с индексами удер-
живания I.
Располагая экспериментально измеренными Кр и I, по урав-
нению (IV.39) можно рассчитать численные значения дифферен-
циального параметра j, позволяющие, как это уже рассматрива-
лось в разделе IV.2.4.3, достаточно надежно проводить группо-
вую идентификацию компонентов сложных смесей. Совместное
использование измеренных коэффициентов распределения Кр и
рассчитанных дифференциальных параметров j позволяет вы-
явить природу содержащихся в сложных смесях органических
соединений как химического индивидуума.
Аппаратура, условия, объекты хроматографирования
Хроматограф с пламенно-ионизационным детектором и интегратором
или компьютеризованной системой обработки данных хроматографиче-
ского анализа*. Шкала электрометра переменная, в диапазоне 3 • 10~8—
3- 10“9 А. Расход водорода 40 мл/мин, воздуха 400 мл/мин.
Колонка стеклянная (3 м X 2 мм), заполненная динохромом П (0,20-
0,25 мм) с 10% (по массе) метилсиликона OV-101.
Температура термостата колонок в режиме программирования** из-
меняется от 55 (30 с) до 170 ° С со скоростью 10 град/мин; температура
испарителя 180°C, температура детектора 150°С.
Газ-носитель — азот, расход 40 мл/мин.
Скорость движения диаграммной ленты*** 200 или 600 мм/ч.
Объекты хроматографирования: 1) опорная смесь реперных н-алканов:
гептан, октан, нонан, декан, ундекан; 2) две контрольные многокомпонент-
ные смеси А и Б (В и Г и т.п.), включающие подлежащие идентификации
органические соединения разных классов и необходимые для измерения
*По возможности, обработку данных хроматографического анализа
проводят с применением программно-аппаратного комплекса “Мульти-
Хром.”
**Так как неподвижная фаза OV-101 обладает низкой упругостью па-
ров, анализы с программированием температуры в рекомендуемых усло-
виях можно выполнять при использовании т.н. одноколоночной схемы
газовых коммуникаций.
***При использовании компьютеризованной системы регистрации и
обработки данных хроматографического анализа пишущий потенциометр,
по согласованию с преподавателем, может быть отключен.
494
индексов удерживания реперные н-алканы*; 3) гексановый и, отдельно,
ацетонитрильный слои двухфазной системы гексан — ацетонитрил после
распределения компонентов контрольных смесей (А и Б или В и Г и т.п.)
между гексаном и ацетонитрилом.
Дозирование осуществляется микрошприцем МШ-10; величина дозы
опорной и контрольных смесей при измерении индексов удерживания —
0,2 мкл, величина дозы гексанового и ацетонитрильного слоев при изме-
рении коэффициентов распределения — 2,0 ± 0,2 мкл.
Выполнение работы
Выводят газовый хроматограф на рекомендованный рабочий
режим и проверяют готовность всех систем к выполнению ана-
лизов в режиме программирования температуры.
1. Измерение линейных индексов удерживания идентифици-
руемых компонентов, содержащихся в контрольных смесях.
Сначала получают две-три воспроизводимых хроматограммы
опорной смеси реперных н-алканов. Каждый раз необходимо
четко фиксировать начало анализа, немедленно после дозиро-
вания смеси включая интегратор или иную подключенную к
хроматографу автоматизированную систему регистрации сиг-
налов детектора. Убедившись в устойчивом воспроизведении
параметров удерживания реперных н-алканов, записывают две-
три (или более, до получения воспроизводимых времен удер-
живания) хроматограммы 1-й контрольной смеси (например, А;
рис. V.6), а затем и 2-й контрольной смеси (Б).
Сравнивая хроматограммы опорной смеси с хроматограм-
мами смесей А и Б, опознают на последних пики реперных н-
алканов. Полученные данные в дальнейшем используют для
расчета линейных индексов удерживания идентифицируемых
компонентов (см. “Обработка результатов”).
2. Измерение коэффициентов распределения идентифициру-
емых компонентов в системе ограниченно смешивающихся
растворителей гексан — ацетонитрил. В стандартный флакон
вместимостью 7 мл с помощью автоматической микропипетки
или медицинских шприцев на 1 мл вводят строго одинаковые
объемы (0,5 мл) гексана и ацетонитрила. Используемые рас-
творители должны быть “хроматографически чистыми”. Аце-
тонитрил заранее высушивают над свежепрокаленными моле-
кулярными ситами. В созданную систему ограниченно смеши-
вающихся растворителей с помощью микрошприца на 50-100
мкл вводят 25-30 мкл 1-й контрольной смеси (например, А), за-
крывают флакон пробкой из силиконовой резины, защищенной
""Контрольные смеси могут содержать не все промежуточные реперные
н-алканы, входящие в опорную смесь.
495
2
Pmc.V.6. Хроматограмма контрольной смеси А.
1 — С-8 (октан); 2 — ХА1 (дибутиловый афир); 3 — С-10 (декан);
4 — ХА2 (ац| тофенон); 5 — С-11 (ундекан).
от контакта с жидкостью фторопластовой пленкой, энергично
встряхивают в течение 7-10 с и наблюдают практически мгно-
венное (по окончании встряхивания) четкое расслаивание си-
стемы (верхний слой — гексановый, нижний — ацетонитриль-
ный). Последовательно хроматографируют сначала гексано-
вый, а затем ацетонитрильный слои (не менее трех раз каждый
из них, до получения воспроизводимых результатов не только
по временам удерживания, но также и по площадям пиков, см.,
например, рис. V.7). Затем точно так же исследуют распреде-
ление между гексаном и ацетонитрилом идентифицируемых со-
единений, содержащихся во 2-й контрольной смеси (Б).
Внимание! Во избежание загрязнения проб нижнего
ацетонитрильного слоя следами верхнего гексанового
слоя (при выведении иглы из флакона) следует отбирать
в рабочий цилиндр микрошприца не менее 8-10 мкл жид-
кости; избыточный объем (превышающий рекомендован-
ную величину дозы (2,0±0,2 мкл)) сбрасывается непосред-
496
a
Phc.V.7. Хроматограмма гексанового (а) и ацетонитрильного
(б) слоев, содержащих компоненты контрольной смеси А.
Р — растворитель (в первом случае— гексан, во втором — ацетони-
трил; 1-5— компоненты контрольной смеси А (см. подпись к рис. V.6).
497
ственно перед дозированием пробы ацетонитрильного
слоя в хроматограф.
По измеренным в каждой серии экспериментов площадям пи-
ков рассчитывают коэффициенты распределения идентифици-
руемых соединений (см. “Обработка результатов”).
Выполнив намеченный объем экспериментальной работы, ох-
лаждают колонку в токе газа-носителя, выключают прибор,
срезают диаграммную ленту с записанными хроматограммами
и приступают к обработке результатов.
Обработка результатов
Сопоставлением хроматограмм опорной и контрольных сме-
сей (например, А и Б) полученных в одинаковых условиях, опо-
знают на хроматограммах контрольных смесей пики реперных
н-алканов и обозначают их символами С-7. С-8 и т. п. соответ-
ственно числу атомов углерода в каждом из них. Пики неизвест-
ных соединений, выявленных на хроматограммах контрольных
смесей, обозначают ХА1, ХА2... и ХБ1, ХБ2 ... и т. д. в порядке
их элюирования. Измеренные интегратором или какой-либо
иной использованной системой обработки данных неисправлен-
ные времена удерживания всех реперных н-алканов и иденти-
фицируемых веществ в каждом из опытов по хроматографиро-
ванию контрольных смесей А заносят в таблицу (табл. V.7), в
которую вписывают также их усредненные времена удержива-
ния и рассчитанные из них по формуле (IV.21) линейные индексы
удерживания 1Т. Сравнивают полученные численные значения
1Т со справочными данными (табл. V.8) и убеждаются в неод-
нозначности идентификации, проводимой только по индексам
удерживания, относящимся всего лишь к одной неподвижной
фазе. Необходимые для расчета коэффициентов распределения
идентифицируемых соединений и дифференциального параме-
тра j экспериментальные данные заносят в таблицу (табл.У.Э).
Коэффициенты распределения находят по формуле [265]
Ар = Аг/Аац,
где Аг и Аац — усредненные значения площадей пиков иденти-
фицируемых соединений на хроматограммах гексанового и аце-
тонитрильного слоев соответственно. Дифференциальные па-
раметры j для каждого идентифицируемого соединения рассчи-
тывают по формуле (IV.39). Сравнивая полученные величины
j с приведенными в табл. IV.9, проводят групповое отнесение
и, вновь обращаясь к справочным данным по индексам удер-
живания (см. табл. V-8), приходят к окончательным выводам о
природе идентифицируемых соединений (табл. V. 10).
498
Таблица V.7. Параметры удерживания
компонентов двух контрольных смесей [424]
Смесь Компонент Время удерживания*, мин Линейный индекс удерживания I?
А С-8 6,04 800
ХА1 8,48 871
С-10 12,90 1000
ХА2 14,57 1047
С-11 16,47 1100
Б С-7 3,51 700
ХБ1 5,92 778
ХБ2 8,38 858
С-10 12,75 1000
ХБЗ 14,51 1049
* Усреднен ное из данных трех опытов.
Таблица V.8. Линейные индексы удерживания
некоторых органических соединений
полидиметилсилоксаном OV-101,
измеренные в рекомендованном режиме
программирования температуры колонки [424]
Соединение 1т
Бутилацетат 778
Гексан-1-ол 845
п-Ксилол 857
Ци клогексанол 858
Дибутиловый эфир 871
Октан-2-ол 988
Ацетофенон 1047
н-Бутилбензол 1049
При оформлении протокола ответьте на следующие вопросы.
1. Каковы преимущества использованной вами при выпол-
нении данной работы методологии в сравнении с возможно-
стями идентификации неизвестных веществ по их хроматогра-
фическим спектрам?
2. Какую иную (альтернативную) систему ограниченно сме-
шивающихся растворителей можно было бы использовать для
определения коэффициентов распределения идентифицируемых
веществ?
499
Таблица V.9. Коэффициенты распределения
идентифицируемых соединений в системе
гексан — ацетонитрил, дифференциальные
параметры j и экспериментальные данные,
необходимые для их расчета [424]
Смесь Компонент Площадь, пиков* 1<р 1g кр 3
Л-Г Лац
А С-8 279,7 20,99 13,33 1,12 -0,32
ХА1 207,1 49,16 4,21 0,62 0,25
С-10 403,8 19,35 20,87 1,32 -0,32
ХА2 45,4 324,5 0,14 -0,85 1,89
С-11 504,8 21,4 23,63 1,37 -0,27
Б ХБ1 47,4 98,8 0,48 -0,32 1,10
ХБ2 46,0 286,7 0,16 -0,79 1,64
С-10 362,9 18,6 19,53 1,29 -0,29
ХБЗ 324,9 131,9 2,46 0,39 0,65
С-11 579,8 27,7 20,94 1,32 -0,22
*в условных единицах.
Таблица V.10. Результаты идентификации
неизвестных соединений в контрольных смесях
Идентифи- цируемое соединение Найдено Результаты идентификации
1т Ар 3 групповая индивидуальная
ХА1 871 4,21 0,25 Простой эфир Дибутиловый эфир
ХА2 1047 0,14 1.89 Алкиларилкетон Ацетофенон
ХБ1 778 0,48 1,10 Ал кил алканоат Бутил ацетат
ХБ2 858 0,16 1,64 Циклоалканол Циклогексанол
ХБЗ 1049 2,46 0,65 Арен н-Бути л бензол
индексы удерживания внесенных в
при использовании для их измерения ко-
3. Изменятся ли
табл. V.10 соединений
лонки с неподвижной фазой большей условной хроматографи-
ческой полярности (например, с карбоваксом 20М)?
500
Работа 7. Количественный анализ воздуха
на содержание кислорода и азота
Цель работы. Ознакомление с возможностями газо-адсорб-
ционного варианта хроматографии на примере разделения ос-
новных компонентов атмосферного воздуха.
При хроматографировании воздушных проб на колонках, за-
полненных молекулярными ситами (цеолитами) NaX или СаА
в качестве адсорбента, содержащиеся в воздухе кислород и
азот могут быть полностью разделены. Получаемые хромато-
граммы легко расшифровываются количественно методом вну-
тренней нормализации площадей пиков.
Аппаратура, условия, объекты хроматографирования
Хроматограф с детектором по теплопроводности, по возможности,
укомплектованный интегратором.
Колонка* стальная или стеклянная (2 м X 3 мм), заполненная свеже-
прокаленным цеолитом СаА (0,20-0,25 мм).
Ток моста детектора 160 мА.
Чувствительность регистрации 20 мВ.
Температура колонки и детектора комнатная.
Газ-носитель — гелий или водород, 30 мл/мин.
Скорость движения диаграммной ленты 200 или 600 мм/ч ( при авто-
матическом измерении сигналов детектора интегратором); 1800 или 2400
мм/ч (при измерении площадей пиков вручную).
Объект хроматографирования — лабораторный воздух, дозируемый
объем ~0,5 см3.
Дозирование осуществляется газовым краном-дозатором. Сменная до-
зирующая петля заполняется лабораторным воздухом, поступающим из
медицинского шприца на 50 (или 100) см3 или из заранее заполненной им
волейбольной камеры.
Выполнение работы
Устанавливают колонку (колонки) в термостате хроматогра-
фа, проверяют и обеспечивают герметичность газовых линий и
выводят прибор на рекомендованный режим работы.
Убедившись в регистрации стабильной нулевой линии, до-
зируют первую пробу воздуха и (при наличии интегратора)
’Сравнительная колонка может быть заполнена либо теми же моле-
кулярными ситами, либо другим сорбентом, например полисорбом-1 или
порапаком Q. В этом случае появляется возможность контроля лабора-
торного воздуха на содержание углекислого газа (почему такая возмож-
ность не обеспечивается рабочей колонкой с цеолитом СаА?).
501
дают команду на автоматическое измерение сигналов детек-
тора. На хроматограмме должны зарегистрироваться два пол-
ностью разделенных пика — азота и кислорода с аргоном*, при-
чем больший из них (пик азота) по высоте обычно занимает 75-
90% ширины диаграммной ленты. В случае, если по каким-либо
причинам на хроматограмме запишутся меньшие пики (высота
пика азота составит менее 50% шкалы самописца) или, наобо-
рот, пики окажутся зашкаленными, изменяют соответствующим
образом величину дозы или чувствительность регистрации сиг-
нала детектора и проверяют воспроизводимость показаний при-
бора, анализируя воздух последовательно еще 2-4 раза.
Выполнив намеченный цикл анализов, выключают прибор (в
первую очередь отключают ток моста детектора!), срезают
диаграммную ленту с записанными хроматограммами и присту-
пают к обработке результатов.
Обработка результатов
Количественную расшифровку хроматограмм проводят ме-
тодом внутренней нормализации площадей пиков, измеренных
интегратором и/или найденных одним из известных графиче-
ских (предпочтительно трапецеидальным — почему?) приемов,
по согласованию с преподавателем. Нормирование площадей
пиков азота и кислорода с аргоном проводят при допущении
одинаковой чувствительности катарометра к этим газам, т. е.
принимают, что /; = 1 и для азота, и для кислорода с аргоном.
Полученные при расчете каждой хроматограммы данные ус-
редняют, заносят в таблицу (табл. V.11) и сравнивают их с ис-
тинным содержанием азота и кислорода в воздухе по массе и
объему (табл. V.12).
В протоколе необходимо объяснить предполагаемые причи-
ны расхождений между известным содержанием азота и кисло-
рода в воздухе, выраженным в массовых и объемных процентах,
и найденным в опыте.
При оформлении протокола ответьте на следующие вопросы.
1. Чем вызвана характерная для газо-адсорбционной хрома-
тографии асимметрия пиков? Предложите практические меры
по ее устранению.
"Из-за близкой поляризуемости молекул кислорода и аргона эти газы
в указанных условиях не разделяются, а все другие составляющие воз-
духа из-за малой концентрации не проявляются на хроматограмме. Так
как содержание аргона в сухом воздухе тоже невелико (см. табл. V.12),
присутствием его в хроматографической зоне кислорода в данном случае
можно пренебречь.
502
Таблица И 77. Результаты количественного анализа лабораторного воздуха на содержание основных компонентов
хромато- граммы Определяемые компоненты воздуха Найдено (I) нормиро- ванием площадей пиков, измеренных интегратором*, % Задано, % Ошибки определения, %
по массе (П) по объему (III) II — I [(П —1)/1] • 100 Ш-1 ЦШ- D/I] • юо
1 Ln Азот Кислород + аргон 75,6 24,4 78,1 21,9
2 Азот Кислород + аргон 75,6 24,4 78,1 21,9
Среднее Азот Кислород + аргон 75,6 24,4 78,1 21,9
’Аналогичная таблица составляется для представления результатов анализа воздуха нормированием площадей пиков, измеренных вручную
одним из рекомендованных преподавателем графических приемов.
Таблица V.12. Усредненный состав сухого воздуха
Газ Содержание, %
по массе по объему
Азот 75,60 78,08
Кислород 23,10 20,95
Аргон 1,25 0,93
Диоксид углерода и примеси других видов 0,05 0,04
2. Как изменится вид полученных вами хроматограмм, если
вместо гелия (или водорода) попытаться использовать азот
(или аргон)?
3. К каким единицам концентрации (в процентах по массе или
по объему) вы приходите при нормировании площадей (или вы-
сот) хроматографических пиков, не скорректированных на от-
носительную чувствительность детектора к названным компо-
нентам смеси? Какой (не предусматриваемой при выполнении
данной лабораторной работы) стадией анализа следовало бы
воспользоваться для устранения неоднозначности ответа на по-
ставленный вопрос?
Работа 8. Основные методы количественного
газохроматографического анализа
Цель работы. 1. Определение содержания всех или неко-
торых составляющих в 3-4-компонентных смесях известного ка-
чественного состава методами внутренней нормализации, вну-
треннего стандарта и/или стандартной добавки с предвари-
тельным экспериментальным определением градуировочных
множителей.
2. Выполнение метрологической оценки результатов количе-
ственных определений.
В данной лабораторной работе рассматриваются все этапы
количественных определений с использованием методов вну-
тренней нормализации, внутреннего стандарта и стандартной
добавки. Предлагаются формы представления и аттестации ре-
зультатов анализа, согласованные с надежными литературными
источниками (см. раздел IV.6).
504
Аппаратура, условия, объекты хроматографирования
Хроматограф с детектором по теплопроводности или пламенно-иониза-
ционным детектором, укомплектованный интегратором или компьютери-
зованной системой обработки результатов хроматографического анализа.
Сила тока моста ДТП 200 мА, множитель шкалы (в зависимости от со-
става смеси и величины дозы) в пределах от 2 до 10. Шкала электрометра
ПИД переменная, в диапазоне от 2 X 10“8 до 2 X 10-9 А. Расход водорода
и воздуха 40-50 и 450 мл/мин соответственно*.
Колонка стальная или стеклянная (2 м X 3 мм), заполненная хромато-
ном N-AW (0,20-0,25 мм) со смешанной неподвижной фазой: апиезон L и
полиэтиленгликоль-4000 (1:1, 20% по массе), или стеклянная (2 м X 3 мм)
с индивидуальной неполярной неподвижной фазой: 10% (по массе) поли-
диметилсилоксана OV-101 на динохроме П (0,20-0,25 мм).
Температура термостата колонок 70-90°С, термостата детекторов 100-
130°C, испарителя 150-175°С.
Газ-носитель при работе с ДТП — гелий или водород, при использо-
вании ПИД — азот, расход 40—50 мл/мин.
Скорость движения диаграммной ленты 200 или 600 мм/ч (при автома-
тическом измерении сигналов детектора интегратором или программно-
аппаратным комплексом на базе ПК — в последнем случае пишущий по-
тенциометр вообще может быть отключен, а также при измерении вы-
сот пиков вручную); 1800 или 2400 мм/ч (при измерении площадей пиков
вручную методом триангуляции).
Объекты хроматографирования. 1. Искусственные градуировочные
смеси известного качественного и количественного состава: а) бензол, то-
луол, пропилацетат, 1-бутанол; б) метилпропилкетон, октан, 1-пентанол,
Ai-ксилол; в) четыреххлористый углерод, 1-бутанол, толуол, л-ксилол и
др. 2. Контрольные смеси известного качественного, но неизвестного
студентам количественного состава, приготавливаемые из тех же компо-
нентов, что и перечисленные выше искусственные смеси.
Дозирование осуществляется микрошприцем МШ-10, доза 0,8—1 мкл
(при работе с ПИД) и 1-2 мкл (при работе с катарометром)-
Внимание! Во избежание выхода за линейный диапа-
зон детекторов каждую используемую смесь разбавляют
(примерно 1:10) подходящим растворителем (хлорбензол,
циклогексанол или др.).
Выполнение работы
Собирают в термостате колонок газовую схему, предусма-
тривающую использование катарометра или пламенно-иониза-
ционного детектора (по согласованию с преподавателем), или
знакомятся с собранным лаборантом заранее соответствующим
ее вариантом. Проверяют и обеспечивают герметичность схе-
*Предварительно лаборантом проводится оптимизация режима газо-
вого питания ПИД применительно к конструкции используемой ячейки
и выбранному расходу газа-носителя (см. описание лабораторной ра-
боты 3).
505
мы, задают необходимые расходы газа-носителя* через сравни-
тельную и измерительную колонки и выводят хроматограф на
рабочий режим.
Убедившись в регистрации устойчивого фонового сигнала,
приступают к пробным анализам.
1. Определение градуировочных множителей Одну и
ту же выданную преподавателем искусственную градуировоч-
ную смесь известного качественного и количественного состава
хроматографируют несколько раз, стремясь к получению вос-
производимых результатов по временам удерживания и количе-
ственным параметрам пиков. В пробных опытах изменяют, если
потребуется, температуру колонки, расход газа-носителя, чув-
ствительность регистрации сигнала детекторов** и величину
дозы таким образом, чтобы записывались незашкаленные пики
с амплитудой, превышающей уровень шума по меньшей мере на
1-2 порядка. На хроматограммах не должно быть не полностью
разделенных пиков.
Выполнив намеченный цикл анализов, т. е. получив по 5-7
воспроизводимых хроматограмм градуировочной смеси и убе-
дившись в воспроизводимости показаний интегратора (в том
случае, если именно интегратор используется для измерения
сигналов хроматографического детектора), выключают хрома-
тограф, срезают диаграммную ленту с записанными хромато-
граммами и приступают к обработке результатов.
Обработка результатов. Градуировочные множители fi рас-
считывают по уравнению (IV.40), используя в качестве пара-
метров Pi высоты и (или) площади хроматографических пиков
(по согласованию с преподавателем), измеренные интегратором
*При включении хроматографа расходы газа-носителя (и вспомога-
тельных газов) задают по заранее составленным графикам расход — да-
вление. По достижении рабочей температуры термостата колонок рас-
ходы газов уточняют либо по цифровой индикации имеющихся у совре-
менных приборов соответствующих электронных устройств, либо (при
их отсутствии или сомнениях в достоверности их показаний) с помощью
мыльно-пленочных измерителей, подключаемых к выходным штуцерам
детектора теплопроводности или непосредственно к горелке пламенно-
ионизационного детектора (в последнем случае мыльно-пленочный изме-
ритель покажет суммарный расход газа-носителя и водорода; для измере-
ния расхода только газа-носителя подачу водорода временно перекрыва-
ют).
**В смесях компоненты могут находиться как в сопоставимых количе-
ствах, так и в резко различных. В последнем случае при записи хрома-
тограммы пишущим потенциометром следует переключать чувствитель-
ность регистрации сигнала детектора на отдельные компоненты (под-
бирается опытным путем), о чем студенты могут быть предупреждены
заранее.
506
(непосредственно в ходе выполнения хроматографических ана-
лизов) или компьютеризованной системой обработки данных по
завершении регистрации всех хроматограмм*.
При выполнении данной лабораторной работы для унифика-
ции расчетов Д в качестве стандартного соединения выбирают
компонент смеси, элюируемый из колонки первым. Найден-
ные по результатам обработки нескольких (5-7) хроматограмм
одной и той же смеси значения fi представляют в табличной
форме (см., например, табл. V.13).
Таблица V.13. Результаты первичной обработки экспери-
ментальных данных при вычислении характеризую-
щих относительный отклик*
№ хромато- граммы Компо- ненты смеси Задано, % по массе Измеренные** количественные параметры пиков Градуировочные множители fi
высота I площадь II I II
1 А Б В
2 А Б В
*Продолжить название таблицы и назвать используемый детектор.
**У казать, каким способом: вручную, с использованием интегратора
или компьютеризованной системы обработки сигналов хроматографиче-
ского детектора.
Сомнительные (выпадающие из ряда) значения fi отбрасы-
вают, пользуясь критическим параметром ткр (для уровня зна-
чимости Р = 0,01) [213, с. 100] или Q-тестом [425]. Оставшиеся
достоверные величины Д усредняют** и для характеристики
*Студентам может быть предложено также найти численные значения
ft по высотам или площадям пиков, измеренных вручную. При обработке
хроматограмм вручную площади пиков находят умножением высот пиков
на их полуширину или на ширину пиков, измеренную на расстоянии 1/3
высоты от основания. Более подробные рекомендации по измерению ко-
личественных параметров хроматографических пиков вручную см. в [79,
с. 217-221].
**Число п остающихся достоверных величин единичных определений
507
воспроизводимости результатов рассчитывают [213, 425, 426]
выборочное стандартное отклонение s величины fi и относи-
тельное стандартное отклонение sr :
sr = s/x, (V.2)
где n — число измерений- Xi — текущее значение величины ,
найденное в отдельных опытах; х — среднеарифметическое зна-
чение fj.
Расчеты по формулам (V.1) и (V.2) рекомендуется выполнять
на микрокалькуляторе (по программе, приведенной в [198]) или
с использованием ПК (по программе, приведенной в Приложе-
нии).
Правильность найденных значений /г- оценивают [213, 425,
426] доверительными границами (интервалами), fi±(ta,n -s/y/n),
в пределах которых среднее значение fi соответствует его ис-
тинному значению на заданном уровне доверительной вероят-
ности (обычно Р = 0,95)*. Сводку данных о воспроизводимо-
сти и правильности значений fi оформляют в виде таблицы
(табл. V.14).
2. Количественный анализ контрольных смесей методом
внутренней нормализации. Следуя данным выше рекомен-
дациям, получают 5-7 воспроизводимых и удобных для после-
дующей обработки хроматограмм контрольной смеси (смесей).
Измеренные на каждой хроматограмме количественные пара-
метры пиков, рекомендованные преподавателем, представляют
в табличной форме (например, табл. V.15). Нормируя согласо-
ванные с преподавателем количественные параметры хромато-
графических пиков, относящиеся к каждому отдельному опыту
(каждой отдельной хроматограмме), находят состав контроль-
ной смеси по формуле (IV.42) без учета и с учетом градуи-
ровочных множителей /, и заносят его в ту же таблицу (см.
табл. V.15).
должно быть не меньше 4-5. В противном случае исследуют и устраняют
вероятные причины невоспроизводимости результатов и повторяют цикл
анализов.
*В некоторых случаях обычная статистическая обработка одномер-
ных массивов подразумевает расчет среднеарифметических значений и
стандартных отклонений х + s(a?t > т) — < х). Для такой обработки
можно использовать программу 2, приведенную в Приложении. Подроб-
нее об условиях и примерах необходимости подобной асимметрической
стандартной обработки см. в [427].
508
Таблица V.14- Воспроизводимость и правильность
найденных значений градуировочных множителей
ft (п = -----) при измерении площадей* пиков**
Компоненты
смеси
fi ± (ta,n s/y/n)
Детектор по теплопроводности
А
Б
В
Детектор пламенно-ионизационный
А
Б
В
*См. сноску** к табл. V.13.
** Аналогичные таблицы составляют (по согласованию с преподава-
телем) для представления результатов метрологической обработки чи-
сленных значений Д, полученных при измерении высот пиков.
Сомнительные (выпадающие из ряда) значения Ci отбрасы-
вают, как и при выбраковке Д. Оставшиеся достоверные ве-
личины С, усредняют и для характеристики воспроизводимо-
сти и правильности результатов анализа рассчитывают s, sr
и С> ± (ta>n s/y/n) для доверительной вероятности Р = 0,95
(табл. V.16).
Дополнительно правильность результатов выражают абсо-
лютной и относительной погрешностями. В нашем случае абсо-
лютная погрешность результата ДСг- равна разности найденной
средней Ci, и заданной С\ концентраций компонентов в анали-
зируемой смеси (в % (по массе)):
ДС = Ci - С?.
Относительная погрешность ДСОТн (величина безразмерная,
обычно выражается в процентах) равна отношению абсолютной
погрешности к заданной концентрации компонента
ДСОТН = (ДС,/С?) 100.
3. Количественный анализ контрольных смесей методом
внутреннего стандарта. Подготовка пробы к анализу. В спе-
циальный сосуд с узкой шейкой вместимостью около 2 мл стек-
509
Таблица V. 15. Количественные параметры хроматогра-
фических пиков*, измеренные**, и ре-
зультаты анализа контрольной смеси методом внутрен-
ней нормализации при использовании пламенно-иониза-
ционного детектора***
№ хроматограммы Компоненты смеси Высоты (или площади) пиков Найдено % (по массе)
без учета fi с учетом fi
1 А Б В
2 А Б В
*В названии таблицы уточнить, какие именно -— высоты или площади.
** Назвать способ измерения количественных параметров пиков.
*** Аналогично таблицу составляют при представлении результатов
анализа с детектором по теплопроводности.
лянными пипетками (или чистыми медицинскими шприцами,
снабженными длинными иглами) отбирают порции контрольной
смеси (приблизительно 800 мг) и рекомендованного преподава-
телем подходящего внутреннего стандарта — вещества, отсут-
ствующего в анализируемой контрольной смеси (около 200 мг).
Массы взятых навесок определяют на аналитических весах. Со-
суд быстро герметизируют накидной гайкой с эластичной проб-
кой из силиконовой резины и аккуратно перемешивают содер-
жимое так, чтобы жидкость не попала на внутреннюю поверх-
ность пробки.
Выполнение анализов. Получают сначала 1-2 хроматограм-
мы исходной контрольной смеси, а затем — первую хрома-
тограмму приготовленного образца контрольной смеси с вне-
сенным в нее внутренним стандартом. К повторным анали-
зам можно приступить только после уверенного опознания на
хроматограмме пиков, отвечающих всем компонентам исходной
контрольной смеси и внесенного стандартного вещества. Затем
регистрируют еще 5-7 воспроизводимых хроматограмм, при-
годных для последующей количественной расшифровки.
510
Таблица V.16. Воспроизводимость и правильность результатов количественного анализа контрольной
смеси (п — - ) методом внутренней нормализации* с пламенно-ионизационным детектором по
площадям пиков, измеренным**----------------------------------------------------------
Компоненты смеси Задано С?, % (по массе) Найдено Ci, % (по массе) ns, % (по массе) £ Ci ± (ia.n nS/y/n) (Р = 0,95), % (по массе) ДС{, % (по массе) (ACi/C?) 100, %
Без учета
А
Б
Си В С учетом fi
А
Б
В
* Аналогичные таблицы составляют и для представления результатов метрологической аттестации численных зна-
чений Ci, получаемых при использовании других апробированных методов (внутреннего стандарта и/или стандартной
добавки), другого детектора (ДТП), другого количественного параметра (высот) пиков.
** Назвать способ измерения количественных параметров пиков.
Измеренные площади и (или) высоты пиков и найденные по
формуле (IV.44) без учета и с учетом градуировочных мно-
жителей концентрации компонентов представляют в табличной
форме (см. табл. V.15). В эту же таблицу вносят результаты
метрологической оценки выполненных анализов (см. п. 2)
4. Количественный анализ контрольных смесей методом
стандартной добавки. Подготовка пробы к анализу. Отлича-
ется от изложенной в п. 3 лишь тем, что в навеску контрольной
смеси (около 800 мг) вносят одно из согласованных с препода-
вателем веществ (приблизительно 100 мг), присутствующих в
контрольной смеси.
Выполнение анализов. Получают две серии из 5-7 воспроиз-
водимых и пригодных для последующей количественной расши-
фровки хроматограмм исходной контрольной смеси и контроль-
ной смеси с внесенной в нее стандартной добавкой. Измеренные
на парных хроматограммах одинакового порядкового номера I и
II серий площади и (или) высоты пиков и концентрации опреде-
ляемых компонентов, найденные по формулам (IV.45) (без учета
и с учетом градуировочных множителей) и (IV.46), заносят в
таблицу (табл. V.17).
Метрологическую аттестацию результатов проводят так же,
как и при выполнении количественного анализа методом вну-
тренней нормализации или внутреннего стандарта. Получае-
мые характеристики представляют в виде табл. V.17.
5. Подведение итогов. Сопоставление метрологических по-
казателей, характеризующих каждый из рассмотренных мето-
дов анализа (см. табл. V. 16), должно привести к заключению,
что при аккуратном выполнении всех рекомендованных проце-
дур, при обеспечении практически полного разделения опреде-
ляемых компонентов и использовании при расчетах надежных
численных значений fi (экспериментально найденных в тожде-
ственных условиях при применении одного и того же прибора
и однотипных приемов измерения сигнала детектора) метода
внутренней нормализации, внутреннего стандарта и стандарт-
ной добавки по воспроизводимости и правильности результа-
тов равноценны. Сравнивают использованные методы и по дру-
гим параметрам: трудоемкости, длительности, иным преимуще-
ствам и недостаткам. Формулируют краткие вывода по проде-
ланной работе.
При оформлении протокола ответьте на следующие вопросы.
1. Каковы главные причины неодинаковой чувствительности
детектора по теплопроводности и пламенно-ионизационного де-
тектора к различным химическим веществам?
512
Таблица V.17. Количественные параметры хроматографических пиков*, измеренные -__________
и результаты анализа контрольной смеси методом стандартной добавки при использовании пламенно-
ионизационного детектора
Компоненты смеси № хроматограммы I серии Высоты (или площади) пиков № хроматограммы II серии Высоты (или площади) пиков Найдено Ci, % (по массе)
без учета fi с учетом fi
А Б В 1 1
сс & Б В 2 2
См. сноски к табл. V. 15.
2. К каким соединениям различия в относительной чувстви-
тельности детектора по теплопроводности и пламенно-иониза-
ционного детектора несущественны?
3. Почему использование заимствуемых из литературных ис-
точников численных значений нормировочных (градуировоч-
ных) множителей не гарантирует получения достоверных ре-
зультатов?
4. Какими соображениями следует руководствоваться при
выборе внутреннего стандарта?
5. Какова самая ответственная (и, одновременно, самая уяз-
вимая) стадия количественного анализа методом внутреннего
стандарта?
Работа 9. Количественный анализ
многокомпонентной смеси с
программированием температуры
Цель работы. Сопоставление возможностей газохромато-
графического анализа в изотермическом режиме и с програм-
мированием температуры на примере многокомпонентной смеси
н-алканов. Определение количественного состава контрольной
смеси методом внутренней нормализации площадей пиков.
Аппаратура, условия, объекты хроматографирования
Хроматограф, предусматривающий использование компенсационной
двухколоночной газовой схемы, с двойным пламенно-ионизационным де-
тектором, укомплектованный интегратором или компьютеризованной си-
стемой обработки результатов хроматографического анализа. Шкала
электрометра 2-10~8—5-10—9 А. Расходы водорода 20, воздуха 350 мл/мин
в каждой ячейке.
Две идентичные колонки (стальные или стеклянные 2 м X 3 мм), запол-
ненные хроматоном N-AW (0,20-0,25 мм) с 5% (по массе) метилсиликона
SE-30.
Газ-носитель — азот, расход 20 мл/мин через каждую колонку.
Температура термостата в режиме программирования изменяется от
50°C (60 с) до 170°C (30 с) со скоростью 4 град/мин*, в изотермическом
режиме — 130°С.
Температура испарителя 250—280°С.
Скорость движения диаграммной ленты 200 или 600 мм/ч (при автома-
тическом измерении сигналов детекторов интегратором или программно-
аппаратным комплексом на базе ПК — в последнем случае пишущий по-
тенциометр вообще может быть отключен); 1800 или 2400 мм/ч (при из-
мерении площадей пиков вручную методом триангуляции; при обработке
*При использовании приборов устаревших конструкций задание изо-
термических площадок определенной длительности в начале и в конце
цикла программирования можно опустить.
514
хроматограмм вручную площади пиков находят умножением высот пи-
ков на их полуширину или на ширину, измеренную на расстоянии 1/3
высоты от основания; более подробные рекомендации по измерению ко-
личественных параметров хроматографических пиков вручную см. в [79,
с. 217-221]).
Объект хроматографирования — искусственная смесь н-парафинов,
включающая 5-7 гомологов с числом атомов углерода от 7 до 13.
Дозирование осуществляется микрошприцем МШ-10, доза 0,6-1,2 мкл.
Выполнение работы
Знакомятся с собранной в термостате хроматографа двухко-
лоночной газовой схемой, задают одинаковые расходы азота,
водорода и воздуха в обеих линиях (контроль — по цифровой
индикации имеющихся у современных хроматографов электрон-
ных устройств, либо по заранее сос ьвленным лаборантом гра-
фикам расход—давление). Выводят прибор на рабочий режим,
отвечающий нижт °й рекомендованной температуре термостата
колонок. Поджигают водород в обеих ячейках детектора.
На пульте программатора температуры термостата колонок
хроматографа задают продолжительность изотермического пе-
риода, отвечающего нижней границе температурного диапазона
(60 с), скорость программирования (4 град/мин), верхнюю тем-
пературную границу (170 °C) и продолжительность изотермиче-
ского периода, отвечающего верхней границе температурного
диапазона (30 с) (см. сноску на с. 514).
Выждав около 20-25 мин (время, необходимое для прогрева
электрометра и разогрева испарителя), включают самописец,
интегратор (или программно-аппаратный комплекс типа
“МультиХром”) и проверяют стабильность нулевой линии.
Убедившись в ее стабильности, производят пробное дозирова-
ние анализируемой смеси и немедленно включают программи-
рование температуры термостата клавишами ПУСК или ПРО-
ГРАММА, а также дают команду интегратору на автоматиче-
ское измерение сигналов детектора.
По окончании программирования и завершении регистрации
хроматограммы термостаты современных хроматографов авто-
матически охлаждаются до начальной нижней границы темпе-
ратурного диапазона. При использовании приборов, конструк-
циями которых такая возможность не предусмотрена (ЛХМ-80,
“Пвет-100”), клавишу НАГРЕВ—ОХЛАЖДЕНИЕ переводят в
положение ОХЛАЖДЕНИЕ и открывают дверцу термостата.
Охлаждают колонки около 10 мин до достижения температуры
воздуха внутри термостата 30-35 °C (желателен контроль по
ртутному термометру). За это время шкалу программатора
возвращают вручную (или она автоматически возвращается) в
515
исходное положение. Закрывают дверцу термостата и выжи-
дают 1-2 мин (время, необходимое для выравнивания темпера-
туры внутри термостата), после чего вновь переводят клавишу
НАГРЕВ—ОХЛАЖДЕНИЕ в положение НАГРЕВ. Через 3-
4 мин устанавливается температура 50°C; прибор готов к оче-
редному циклу программирования.
По полученной пробной хроматограмме судят о необходимо-
сти корректировки при последующих анализах величины дозы
и (или) чувствительности регистрации пиков отдельных компо-
нентов. На окончательных хроматограммах не должно быть как
зашкаленных, так и слишком малых пиков (желательно, чтобы
амплитуда сигналов детектора на отдельные компоненты пре-
вышала уровень шума на менее чем на 1-2 порядка).
Записав 3-4 воспроизводимых хроматограммы анализируе-
мой смеси в режиме программирования температуры, присту-
пают к анализу той же смеси в изотермическом режиме при тем-
пературе колонок 130 °C. Величина дозы и чувствительность
регистрации такие же, что и в режиме программирования тем-
пературы. Записывают 1-2 хроматограммы смеси углеводоро-
дов в изотермическом режиме и сравнивают их с полученными
ранее хроматограммами.
Обработка результатов
Для определения количественного состава смеси используют
хроматограммы, полученные при программировании темпера-
туры анализа. Расчет производят методом внутренней норма-
лизации площадей, измеренных умножением высоты на полу-
ширину пиков или интегратором (программно-аппаратным ком-
плексом типа “МультиХром”), принимая чувствительность де-
тектора одинаковой ко всем компонентам смеси. Усредненные
результаты анализа представляют в таблице, по форме близкой
к табл. V.16. Заданный состав смеси, необходимый для оценки
абсолютных и относительных погрешностей определения, сооб-
щается студентам по выполнении ими эксперимента и расчетов.
В протоколе работы необходимо, используя собственные экс-
периментальные наблюдения, обсудить преимущества и ограни-
чения программирования температуры по сравнению с изотер-
мическим анализом смеси углеводородов, выкипающих в широ-
ком интервале температур.
При оформлении протокола ответьте на следующие вопросы.
1. Чем вызвана необходимость использования двухколоноч-
ной дифференциальной схемы газовых коммуникаций при вы-
полнении анализов в режиме программирования температуры?
516
2. Почему при обработке результатов данной работы нельзя
в качестве количественных параметров пиков использовать их
высоты?
3. Почему при обработке результатов данной работы можно
нормировать площади пиков, нескорректированные на неодина-
ковую чувствительность детектирования?
4. При анализе какого типа объектов оправдан режим про-
граммирования температуры колонок?
Работа 10. Газохроматографическое
определение фенолов в водных объектах с
предварительной дериватизацией
В промышленных сточных водах содержатся многие вредные
органические соединения. В группу наиболее опасных для жи-
вых организмов входят фенол, крезолы и хлорфенолы. Меры,
принимаемые для очистки сточных вод от содержащихся в них
в больших концентрациях токсичных и канцерогенных веществ,
не всегда оказываются эффективными. Из-за несовершенства
очистных сооружений вредные вещества попадают в реки, озера
и другие открытые природные водоемы, а оттуда — в водопро-
водную сеть (применяемые на станциях городских водопрово-
дов системы водоподготовки также зачастую не обеспечивают
требуемого качества питьевой воды). Лаже ничтожно малое ко-
личество фенола в воде придает ей неприятный запах и вкус.
Предельно допустимая концентрация (ПДК) фенола в питьевой
воде составляет 0,001 мг/л, в рыбохозяйственных водоемах —
0,05 мг/л, в сточных водах, сбрасываемых в нерыбные водо-
емы, — 1,0 мг/л. Крезолы оказывают токсичное действие на
теплокровных животных, раздражают кожу и слизистые обо-
лочки, являются аллергеном. ПЛК м- и n-крезолов для откры-
тых водоемов — 0,004 мг/л. При хлорировании воды, поступа-
ющей на станции водоподготовки городских водопроводов, фе-
нол и крезолы превращаются в хлорфенолы, которые обладают
еще более резким запахом и, будучи сами по себе довольно ток-
сичными соединениями, могут участвовать в образовании по-
лихлорированных диоксинов и дибензофуранов — чрезвычайно
опасных веществ, разрушающих иммунную систему организма.
Вредное воздействие фенолов проявляется и в нарушении ки-
слородного режима водоемов. Легко окисляясь, фенолы потре-
бляют большое количество растворенного в воде кислорода,
что приводит к гибели населяющих водоемы живых организмов.
517
При необходимости определения фенолов в водах на уровне
предельно допустимых и более низких концентраций прибегают
к их предварительному концентрированию методами жидкост-
ной или твердофазной экстракции с последующим исследова-
нием состава экстрактов (десорбатов) методами газовой или
высокоэффективной жидкостной хроматографии, а также с при-
влечением комбинированных хромато-масс-спектральных мето-
дов.
Наиболее эффективными экстрагентами фенолов и хлорфе-
нолов из водных сред признаны бутилметилкетон, метилэтил-
кетон, н-бутилацетат, а также органические N-содержащие ок-
сиды (растворенные в толуоле), особенно триоктиламиноксид.
Международный стандарт ИСО 1-8165 предусматривает экс-
трагирование фенолов и их хлорпроизводных более доступным
растворителем — диэтиловым эфиром.
Сорбционное концентрирование осуществляется на химиче-
ски модифицированных кремнеземах (С-18 или С-16) или на ор-
ганических синтетических сорбентах типа сополимера акрило-
нитрила и дивинилбензола. Накопленные фенолы десорбируют
метанолом, этилацетатом (при последующем анализе сконцен-
трированных фенолов методом ВЭЖХ — акрилонитрилом или
его смесью с водой). Для достижения низких пределов об-
наружения и повышения селективности определения получен-
ный экстракт (десорбат) упаривают (иногда досуха), после чего
сконцентрированные фенолы обрабатывают дериватизирую-
щим агентом. Хорошо отработаны, например, методики получе-
ния триметилсилиловых или пентафторбензоильных производ-
ных, не склонных к образованию асимметричных зон в процессе
хроматографического разделения, что часто наблюдается при
анализе самих исходных фенолов; при наличии в молекуле про-
изводного электроотрицательных заместителей и использова-
нии электронозахватного детектора становится возможным об-
наружение пикограммовых количеств исходных фенолов.
Альтернативный вариант подготовки пробы к анализу преду-
сматривает выполнение дериватизации непосредственно в вод-
ной фазе. В качестве дериватизирующих реагентов хорошо за-
рекомендовали себя, например, уксусный ангидрид [428, 429] и
диметилсульфат [430] (с более подробной библиографией можно
познакомиться в [431]). Образующиеся ацетаты извлекают из
водной среды жидкофазной экстракцией, а метиловые эфиры
фенолов определяют методом парофазного анализа, основан-
ного на газовой экстракции. В излагаемой ниже лаборатор-
ной работе дается подробное описание выполнения измерений
по обеим этим методикам.
518
Цель работы. Определение содержания нескольких фенолов
в контрольной водной пробе в форме уксуснокислых или мети-
ловых эфиров с применением модифицированного метода вну-
треннего стандарта, предусматривающего использование со-
единения сравнения.
Выполнение работы
Приготовление подлежащих анализу водных растворов
фенолов. В мерную колбу на 500 мл, заполненную примерно на
две трети рабочего объема свежеперегнанной дважды дистил-
лированной водой, с помощью медицинских шприцев на 1-5 мл
вносят порции (0,5-3 мл, по указанию лаборанта) заранее приго-
товленных трех-пяти концентрированных индивидуальных вод-
ных растворов фенолов (в этих растворах могут присутство-
вать фенол, о-крезол, n-х лор фенол и другие алкил- и хлорза-
мещенные фенолы; студентам сообщаются качественный состав
приготавливаемой пробы и концентрация лишь одного из рас-
творов, содержащего т.н. соединение сравнения, см. ниже). Со-
держимое колбы тщательно перемешивают и доводят до метки
дистиллированной водой.
Ацилирование фенолов в водных растворах уксусным ан-
гидридом. В делительную воронку на 200-250 мл вносят 100 мл
контрольной пробы и добавляют 4 г кристаллического гидро-
карбоната натрия. Плавным покачиванием перемешивают со-
держимое воронки. Закрепляют воронку в лапке штатива и
медицинским шприцем на 1 мл вводят в воронку 0,6 мл уксус-
ного ангидрида, моментально растворяющегося в воде. Закры-
вают воронку притертой стеклянной пробкой, вынимают из шта-
тива и аккуратно встряхивают в течение 5 мин (этого времени
достаточно для количественного связывания присутствующих
в водном растворе фенолов в соответствующие уксуснокислые
эфиры).
Продукты ацилирования дважды экстрагируют дихлормета-
ном (порциями по 5 мл). Объединенные органические вытяжки
помещают в коническую колбочку емкостью ~25 мл, перено-
сят ее в вытяжной шкаф и под руководством преподавателя
или лаборанта начинают пропускать над зеркалом экстракта
струю газообразного азота или чистого воздуха со скоростью
80-100 мл/мин. По испарении примерно 2/3 исходного объема
экстрагента пропускание газа приостанавливают.
Остаток экстракта, по возможности, количественно перено-
сят в специальный градуированный флакон с конусообразным
днищем и возобновляют пропускание струи газа над зеркалом
жидкости до тех пор, пока ее объем не составит 200-300 мкл
519
(для ускорения испарения хлористого метилена допускается по-
догрев колбы и флакона в бане с теплой водой). Оставшийся во
флаконе экстракт подвергают газохроматографическому ана-
лизу.
Метилирование фенолов в водных растворах диметил-
сульфатом. В сухие и чистые пенициллиновые флаконы (об-
щим числом от двух до шести) емкостью 15 мл вносят одина-
ковые навески (6,0 ± 0,1 г) карбоната калия (х.ч.) и пробы по
5 мл одного или двух-трех (по согласованию с преподавателем)
анализируемого водного образца; каждую пробу дублируют.
Флаконы герметизируют с помощью специальных накидных
оправок, защищая внутреннюю поверхность стандартных ме-
дицинских пробок из самоз втягивающейся резины фторопла-
стовой пленкой, и медицинским шприцем (на 1 мл, прокалы-
вая пробки флаконов иглой шприца) вводят диметилсульфат
(0,1 мл).
Внимание! Диметилсульфат очень ядовит. Следует
работать в резиновых перчатках при включенной тяге.
Для осуществления исчерпывающего метилирования и урав-
новешивания жидкой и паровой фаз содержащие реакционные
смеси флаконы помещают в малогабаритную воздушную баню,
заранее разогретую до 80 °C, и энергично встряхивают (на ви-
браторе) в течение 1 ч, после чего первый из них переносят
в обогреваемую рубашку (разогретую до 80 °C) приставки для
парофазного анализа [66] газового хроматографа, заранее выве-
денного на рабочий режим (см. ниже).
Основные побочные продукты реакции — метанол и димети-
ловый эфир, образующиеся в больших количествах, не должны
мешать регистрации пиков целевых метиловых эфиров фено-
лов, хлорфенолов и алкилфенолов. Другие возможные побоч-
ные продукты реакции (алкилхлориды) сами выступают в каче-
стве метилирующих агентов и также не должны мешать опреде-
лению целевых метиловых эфиров.
Приведенные условия метилирования обеспечивают конвер-
сию приоритетных фенолов до 90% от исходного содержания в
диапазоне от 0,01 до 10 мг/л.
Ограничения. Методика не подходит для определения фе-
нолов в водах, содержащих низкомолекулярные спирты и хло-
риды, так как при этом диметилсульфат в первую очередь будет
реагировать с ними с образованием большого количества лег-
колетучих продуктов (возможен взрыв флакона!).
520
Газохроматографическое определение
фенолов в водных растворах
в форме уксуснокислых эфиров
Аппаратура, условия, объекты хроматографирования
Хроматограф серии “Цвет-500” с пламенно-ионизационным или фото-
ионизационным детектором. Шкала электрометра переменная, в диапа-
зоне от 1,6-10“11 до 2,56-10“10 А. Расход водорода и воздуха (при работе
с ПИЛ) 40 и 400 мл/мин соответственно.
Колонка стеклянная (2 м х 3 мм), заполненная хроматоном N-cynep
(0,16-0,20 мм) с 5% (по массе) силиконового эластомера OV-225*.
Температура термостата колонок 60 ° С (2 мин) с последующим про-
граммированием со скоростью 5 град/мин до 180°C.
Температура детектора (переходной платы) 150°C, испарителя 250°C.
Газ-носитель — азот, расход 30 мл/мин.
Скорость движения диаграммной бумаги 600 мм/ч (при подключении
к выходу измерителя малых токов персонального компьютера для авто-
матической регистрации и последующей обработки сигналов детектора
по специальной программе, например по программе “МультиХром”, за-
писывать хроматограмму пишущим потенциометром необязательно).
Объекты хроматографирования — один или два (по согласованию
с преподавателем) образца сконцентрированных экстрактов уксусноки-
слых эфиров фенолов, подготовленных согласно процедурам, описанным
выше.
Дозирование осуществляется микрошприцем МШ-10, величина дозы
~2 мкл.
Выполнение анализа
Знакомятся с собранной в термостате хроматографа схемой
газовых коммуникаций и выводят прибор на заданный рабочий
режим. (Подготовку хроматографа к работе целесообразно про-
вести параллельно с процедурой испарения избыточного объ-
ема экстрагента.)
Внимание! При работе с фотоионизационным детек-
тором вначале включают электропитание электрометра
БИЛ-36 и лишь затем подают напряжение на водород-
ную ультрафиолетовую лампу
Спустя 20-30 мин контролируют и, при необходимости, кор-
ректируют установившиеся температуры детектора (переход-
ной платы), испарителя и начальную температуру колонки (ко-
лонок), а также расходы газа-носителя и водорода. Поджигают
водород в ячейке (при использовании двухколоночной схемы —
*При возможности работать с двойным ПИД целесообразно устано-
вить в термостате хроматографа две идентичные колонки — рабочую и
сравнительную.
521
в ячейках) детектора. Включают пишущий потенциометр и про-
веряют стабильность нулевой линии на шкале электрометра
1,6 -10-11 А.
Если прибор подключен к ПК, то для проверки стабильно-
сти нулевой линии следует нажать кнопку микровыключателя
на кабеле, связывающем хроматограф с ПК, заранее настроен-
ным под руководством преподавателя или лаборанта на прием
хроматограммы. Убедившись в стабильности фонового сигнала
детектора, вновь нажимают эту же кнопку.
Проводят пробное дозирование анализируемого образца и
немедленно дают команды на программирование температуры
(для чего необходимо нажать клавишу ПУСК ПРОГРАММЫ
на блоке БУ-125) и на начало регистрации сигнала компьюте-
ризованной системой (см. выше). При записи хроматограммы
пишущим потенциометром на диаграммной ленте мягким каран-
дашом или фломастером наносят стартовую отметку (можно
“мягко” толкнуть пишущий узел) и здесь же приводят сведения
о величине введенной дозы и шкале электрометра.
По окончании рабочего цикла программирования термостат
колонок автоматически охлаждается до начальной температу-
ры, о возможности выполнения повторного анализа судят по ин-
дикации ГОТОВНОСТЬ на блоке БУ-125. При выполнении по-
вторных анализов корректируют, при необходимости, величину
дозы или рабочую шкалу электрометра. Для каждого анализи-
руемого образца необходимо записать не менее трех воспроиз-
водимых хроматограмм.
Газохроматографическое определение фенолов
в водных растворах в форме метиловых эфиров
Аппаратура, условия, объекты хроматографирования
Хроматограф серии “Цвет-500” с пламенно-ионизационным детекто-
ром, укомплектованный устройством для пневматического дозирования
порций равновесной паровой фазы (обогреваемой приставкой для паро-
фазного анализа) и термостатирующим стаканом-рубашкой для флаконов
с пробами. Шкала электрометра переменная, в диапазоне от 1,6-10“11 до
2,56-Ю~10А.
Колонка стеклянная (2 м X 4 мм), заполненная динохромом П (0,25—
0,315 мм) с 10% (по массе) полидиметилсилоксана OV-101.
Газ-носитель — азот, расход 40 мл/мин.
Температура термостата колонок 100°С (1 мин) с последующим про-
граммированием со скоростью 5 град/мин до 170°C.
Температура испарителя 200°С, детектора (переходной платы) 150°C.
Объекты хроматографирования — равновесные паровые фазы, образу-
ющиеся при метилировании в приведенных выше условиях водных рас-
творов определяемых фенолов.
522
Дозирование осуществляется с помощью приставки для ПФА, реко-
мендуемое превышение давления азота в линии накачки относительно
давления на входе в колонку ~0,1 атм.
Выполнение анализа
Знакомятся с собранной схемой газовых коммуникаций и вы-
водят прибор на заданный рабочий режим. (Подготовку хрома-
тографа к работе целесообразно провести параллельно с про-
цедурой метилирования.) На период подготовки прибора к ра-
боте и при последующей проверке памяти устройства пневма-
тического дозирования на иглу приставки для ПФА (рис. V.8)
устанавливают пустой резервный пенициллиновый флакон. Из-
начально переключающий кран 6 приставки должен находиться
в положении НАКАЧКА, в дальнейшем в этом же положе-
нии крана производят замену флаконов — резервного пустого
на флакон с образцом № 1, флакона с образцом № 1 на фла-
кон с образцом №2 и т.д. С помощью регуляторов давления
блока подготовки газа-носителя задают рекомендованный рас-
ход азота через колонку (контроль индикации на блоке управле-
ния БУ-125) и необходимое давление в линии накачки приставки
для ПФА (контроль по показаниям образцового манометра 1J,
см. рис. V.8).
Используя возможности блока управления БУ-125, задают
рекомендованные температурные режимы. Спустя 20-30 мин
контролируют и, при необходимости, корректируют установив-
шиеся температуры детектора (переходной платы), испарителя
и начальную температуру колонки (колонок), а также расход
газа-носителя через колонку, его давление на входе в колонку и
давление азота в линии накачки.
Включают пишущий потенциометр и проверяют стабиль-
ность нулевой линии на шкале электрометра 1,6-10-11 А. Если
прибор подключен к ПК, то для проверки стабильности нуле-
вой линии следует нажать кнопку микровыключателя на кабеле,
связывающем хроматограф с ПК, заранее настроенным под ру-
ководством преподавателя или лаборанта на прием хромато-
граммы. Убедившись в стабильности фонового сигнала детек-
тора, вновь нажимают эту же кнопку.
Перед первым дозированием анализируемой паровой фазы
проверяют “память” приставки для ПФА на предшествующие
образцы (анализировавшиеся в предыдущие дни). Для этого
переводят кран 6 в положение ДОЗИРОВАНИЕ и немедленно
дают команду на программирование температуры (для чего не-
обходимо нажать клавишу ПУСК ПРОГРАММЫ на блоке БУ-
125) и на начало регистрации сигнала компьютеризованной си-
523
Pnc.V.8. Схема приставки для пневматического дозирования
при выполнении парофазного анализа.
а — создание повышенного давления; 6 — стадия дозирования па-
ровой фазы в хроматограф. 1 — стеклянный (пенициллиновый) фла-
кон; 2 — резиновая пробка; 3 — вентиль тонкой регулировки; 4 —
линия сброса; 5 — подача вспомогательного газа (линия “накачки”);
6 — двухпозиционный шестиходовой газовый кран; 7—- стальная игла
с боковым отверстием; 8 — обжимное резиновое кольцо; 9 — термоста-
тирующая рубашка; 10 — хроматографическая колонка; 11 — подача
газа-носителя; 12 — испаритель; 13 — соединительная муфта; 14 —
съемный образцовый манометр.
стемой (см. выше). При записи хроматограммы пишущим по-
тенциометром на диаграммной ленте мягким карандашом или
фломастером наносят стартовую отметку (можно “мягко” толк-
нуть пишущий узел) и здесь же записывают заданное превы-
шение давления азота в линии накачки относительно давления
газа-носителя на входе в колонку, что определяет величину вве-
денной дозы газа из флакона, и шкалу электрометра. При по-
явлении неожиданных и достаточно заметных сигналов детек-
тора (проявление “памяти” приставки), способных исказить ре-
зультаты последующих анализов подготовленных образцов, пе-
реводят ручку крана 6 в положение НАКАЧКА, заменяют пу-
стой пенициллиновый флакон на аналогичный флакон с 5 мл
дистиллированной воды и попеременно (пять-десять раз) пере-
ключают кран из положения НАКАЧКА в положение ДОЗИ-
РОВАНИЕ и обратно (временной интервал между переключе-
524
ниями крана ~10-15 с). Чередующееся пропускание водяного
пара через внутренние коммуникации приставки позволяет до-
вольно быстро избавиться от мешающей “памяти” и приступить
к анализам равновесной паровой фазы во флаконах с подгото-
вленными образцами, заведомо содержащими метиловые эфиры
фенолов.
Удостоверившись в том, что кран 6 находится в положении
НАКАЧКА, заменяют пустой пенициллиновый флакон или фла-
кон с дистиллированной водой на первый из флаконов с образ-
цами прометилированных фенолов и немедленно надевают на
него заранее разогретый стакан-рубашку 9, закрепляя ее лап-
кой фиксатора 8.
Через 1-3 мин переключают кран 6 в положение ДОЗИРО-
ВАНИЕ и записывают пробную хроматограмму равновесной
паровой фазы, выполняя все описанные выше (см. проверку
“памяти”) операции. При выполнении повторных анализов кор-
ректируют, при необходимости, величину дозы или рабочую
шкалу электрометра (только если хроматограммы записыва-
ются пишущим потенциометром). Для каждой анализируемой
равновесной паровой фазы необходимо получить не менее трех
воспроизводимых хроматограмм. При переходе к анализам
каждого последующего образца рекомендуется проверять “па-
мять” приставки на предыдущий образец.
Обработка результатов
Концентрацию определяемых фенолов в контрольных образ-
цах рассчитывают по формуле
С(% по массе) = AAGt/Ast/st,
где А, и Ast — приведенные к единой рабочей шкале электроме-
тра площади пиков уксуснокислого или метилового эфира опре-
деляемого фенола и соединения сравнения; fi и At — градуиро-
вочные множители, учитывающие неодинаковый отклик детек-
тора на уксуснокислые или метиловые эфиры различных фено-
лов (в последнем случае эти множители учитывают также раз-
личия в коэффициентах распределения метиловых эфиров опре-
деляемых фенолов и фенола, выбранного в качестве соединения
сравнения, табл. V. 18); Gt — известная концентрация соедине-
ния сравнения в контрольном образце (% по массе).
Текущие и усредненные результаты, полученные при обра-
ботке двух-четырех хроматограмм каждого контрольного об-
разца, представляют в виде табл. V. 19. Заданное содержа-
ние определяемых фенолов в проанализированных контрольных
525
Таблица V.18. Экспериментально
найденные градуировочные множители /j
Эфиры фенолов ПИД ФИД
Уксуснокислые:
фенилацетат 1,0 1,0
2-метилфенилацетат 1,27 1,57
4-метилфенилацетат 1,14 1,14
3,4-диметилфенилацетат 1,33 1,69
4-хлорфенилацетат 1,92 1,18
2-изопропил-5-метил- 1,69 3,04
фенилацетат
Метиловые:
анизол 1,0
2-метиланизол 1,44
4-метиланизол 2,43
3,4-ди мети л ан изо л 6,27
4-хлоранизол 9,18
2-изопропил-5~метиланизол 9,57
образцах, необходимое для оценки абсолютных и относитель-
ных погрешностей результатов (обычно не превышающих 10-
20% отн.), сообщается студентам по выполнении ими экспери-
ментов и расчетов.
При оформлении протокола ответьте на следующие вопросы.
1. Чем обусловливается целесообразность определения в
водных растворах низких содержаний фенолов в форме их раз-
личных производных? Возможно ли проводить анализ водных
объектов на содержание фенолов без их предварительной дери-
ватизации? При положительном ответе уточните формулировку
задачи.
2. В чем заключаются преимущества ацилирования фено-
лов непосредственно в водной среде с последующими жидкост-
ной экстракцией арилацетатов, концентрированием экстрактов
и их газохроматографическим анализом в сравнении с методи-
кой, предусматривающей начальное экстрагирование фенолов
из анализируемого водного объекта подходящим растворите-
лем и концентрирование экстракта, с последующей деривати-
зацией и газохроматографическим анализом производных?
3. С какой целью при осуществлении ацилирования (мети-
лирования) фенолов, содержащихся в анализируемой воде, в
отобранную пробу вносят гидрокарбонат натрия (карбонат ка-
лия)?
526
Таблица V.19. Результаты газохроматографического
определения фенолов в воде в
форме уксуснокислых эфиров
Определяемое соединение* Задано (I), мг/л Найдено (II), мг/л [(П - 1)/1] • 100 %
Для уксуснокислых эфиров (ФИД)
о-Крезол 2,94 3,00 1,9
1,47 1,47 0
п-Крезол 2,29 2,28 0,5
1,15 1,08 6,1
2,4-Ксиленол 5,09 6,14 2,5
2,99 3,39 13,2
3,4-Ксиленол 3,79 3,89 2,5
1,90 2,03 7,1
п-Хлорфенол 4,62 4,49 2,9
2,31 2,23 3,7
2-Изопропил- 6,00 6,25 4,1
5-метилфенол 3,00 2,46 18,0
(тимол)
Для уксуснокислых эфиров (ПИД)
о-Крезол 0,77 0,78 1,3
2,4-Ксиленол 1,00 1,02 2,0
3,4-Ксиленол 1,53 1,68 10,4
Для метилов! six эфиров (ПИД)
о-Крезол 40,90 39,72 2,9
20,45 21,72 6,2
п-Крезол 45,90 45,85 0,1
3,4-Ксиленол 44,30 42,89 3,2
22,15 24,70 11,5
п-Хлорфенол 78,60 77,77 1,1
2-Изопропил- 46,60 45,64 2,1
5-метилфенол
(тимол)
* Вещество сравнения — фенол; средний результат 2-4 анализов.
4. Каковы ожидаемые побочные продукты метилирования
фенолов диметилсульфатом? Велика ли опасность наложения
их хроматографических полос на зоны определяемых производ-
ных? При анализе каких водных объектов методика определе-
ния фенолов в форме метиловых эфиров неприемлема?
527
Работа 11. Газохроматографическое определение
содержания хлороформа в питьевой
(водопроводной) воде
Контроль за содержанием примесей органических соедине-
ний в питьевой воде, обладающих токсическим (например, кан-
церогенным или мутагенным) действием, является актуальной
задачей в санитарно-химическом анализе. К настоящему вре-
мени установлено, что в обычной водопроводной воде обнару-
живаются примеси многих химических веществ: фенолы, хлор-
фенолы, галогенсодержащие соединения других классов, аро-
матические углеводороды, альдегиды, кетоны, спирты, эфиры,
нафтены, парафины и другие — всего более 70-80 потенциаль-
ных экотоксикантов, содержание которых необходимо контро-
лировать. Наиболее опасными для здоровья человека являются
низшие хлоралканы (хлороформ, четыреххлористый углерод),
появляющиеся в водопроводной воде в результате ее хлори-
рования на водоочистительных станциях. Содержание хлоро-
форма в водопроводной воде различных крупных городов мира
(~20 мкг/л) примерно на порядок превышает содержание четы-
реххлористого углерода (~1 мкг/л), поэтому именно на разра-
ботку простых и надежных методик определения хлороформа в
питьевых водах направлено внимание специалистов санитарно-
химических служб. В большинстве стран ПДК хлороформа в
питьевых водах установлена на уровне 30-100 мкг/л, следова-
тельно, необходимо иметь возможность контролировать и зна-
чительно меньшее его содержание в анализируемом образце
(вплоть до 1-5 мкг/л).
Химическая природа низших галогенированных углеводоро-
дов обусловливает использование для их обнаружения и ко-
личественного определения в различных матрицах электроно-
захватных детекторов (ЭЗД и ДПР, см. раздел П.1.4.6). Про-
стейшие методы прямого ввода в хроматографическую колонку
больших водных проб не получили, однако, широкого распро-
странения из-за снижения чувствительности детектора под дей-
ствием паров воды. Предварительное концентрирование лету-
чих галогенуглеводородов с помощью жидкостной экстракции
также не находит применения из-за необходимости тщательной
очистки экстрагентов (н-октана, толуола, других углеводород-
ных растворителей), содержащих, как правило, недопустимо
высокие количества мешающих примесей.
528
Ниже излагаются два варианта решения поставленной за-
дачи*, предусматривающие экстракцию хлороформа (и сопут-
ствующих ему родственных соединений) газом (воздухом) в
статических условиях с последующим газохроматографическим
анализом равновесной паровой фазы над жидкостью. Напо-
мним, что в аналитической химии многие современные вари-
анты газовой экстракции объединяются термином “Парофаз-
ный анализ” (ПФА) [98].
Цель работы. Определение содержания хлороформа в водо-
проводной воде методом абсолютной градуировки и по одной
из версий метода стандартной добавки с использованием при-
емов статического парофазного газохроматографического ана-
лиза и избирательного детектирования галогенсодержащих со-
единений по захвату электронов.
Аппаратур.- , условия, объекты хроматографирования
Хроматограф “Пвет-500” с детектором постоянной скорости рекомби-
нации, ДПР (рабочие шкалы электрометра 1 • 10“Х1-4 • 10“12 А), уком-
плектованный приставкой для парофазного анализа и интегратором или
программно-аппаратным комплексом на базе ПК типа “МультиХром”.
Колонка стальная, стеклянная или фторопластовая (3 м X 2 мм), за-
полненная хроматоном N (0,20-0,25 мм), смоченным полиэтиленгликолем
ПЭГ-20М (0,1% (по массе)) и метилсиликоном SE-30 (5% (по массе)).
Температура колонки 65°C, испарителя, приставки для парофазного
анализа и детектора 150°C.
Газ-носитель — азот (в.ч.), расход через колонку 30 мл/мин.
Скорость движения диаграммной ленты 200 или 600 мм/ч.
Объекты хроматографирования: 1) лабораторный воздух, используе-
мый в качестве газа-экстрагента; 2) паровая фаза над градуировочными
растворами (их общее число — от трех до пяти) с известным (по проце-
дуре приготовления) содержанием хлороформа в воде; 3) паровая фаза
над водой, отобранной из водопроводной сети; 4) паровая фаза над водо-
проводной водой с внесенной в нее добавкой хлороформа.
Дозирование пневматическое, осуществляемое с помощью приставки
для парофазного анализа**; вспомогательный газ — азот (в.ч.), превы-
шение давления в сосуде с образцом над давлением в испарителе хрома-
*Варианты адаптированы к условиям студенческого практикума, пред-
ставляя лишь часть материала разработанной в НИИ Химии СПбГУ
и официально аттестованной методики определения низших галогениро-
ванных углеводородов в питьевой воде [416].
Для реализации принципа пневматического дозирования из обеих
газовых линий в блоке подготовки подвижной фазы необходимо исклю-
чить регуляторы расхода газа и оставить только надежные (“неперепуска-
ющие”) регуляторы давления (см. раздел П.1.1). Все элементы собранной
газовой схемы блока подготовки и самой приставки (особенно линии на-
качки, обеспечивающей создание повышенного давления газа во флаконе
перед дозированием) должны быть с особой тщательностью прове-
рены на герметичность. Наиболее удобный способ проверки герметич-
529
тографа (на входе в колонку) ~0,2 атм; расход газа через линию сброса
1—3 мл/мин.
Выполнение работы
Подготовка проб градуировочных растворов и водопро-
водной воды к анализу. Все названные выше объекты анализа
помещают в стандартные чистые пенициллиновые флаконы ем-
костью 15 мл, герметизируемые с помощью пробок из самоза-
тягивающейся “медицинской” резины и специальных накидных
оправок. Альтернативный способ герметизации флаконов —ис-
пользование стаканов-рубашек из органического стекла с завин-
чивающимися крышками, прижимающими пробки к фланцу фла-
конов; в крышках заранее должно быть высверлено отверстие
(диаметр ~1,6 мм) под иглу дозирующего устройства. Лля ис-
ключения возможной эмиссии вовнутрь флаконов летучих орга-
нических веществ из резины нижнюю поверхность пробок экра-
нируют дисками из тонкой фторопластовой пленки.
Градуировочные растворы, охватывающие диапазон
ожидаемого содержания хлороформа в водопроводной воде (от
~10 до ~100 мкг/л), приготавливают разбавлением в мерных
колбах на 50-100 мл свежепрокипяченной и охлажденной до ком-
натной температуры дистиллированной водой отбираемого ка-
либрованными пипетками или медицинскими шприцами базо-
вого раствора*, имеющего в 100-200 раз большую концентра-
цию. Все операции, связанные с отбором порций водных раство-
ров хлороформа и водопроводной воды, следует проводить, не-
возможности, быстро во избежание искажения их состава вслед-
ствие свободного массообмена между жидкостью и окружаю-
щим воздухом.
ности линии накачки с уже установленным на игле дозирующего устрой-
ства флаконом с образцом — включение в газовую магистраль электрон-
ного измерителя расхода газов (типа ИРГ-110). При полной герметично-
сти системы прибор должен показать нулевой или близкий к нему расход
газа. При отсутствии герметичности надеяться на получение воспроиз-
водимых результатов нет решительно никаких оснований.
*Базовый раствор готовят заранее в два этапа: 1) растворяют в мер-
ной колбе на 25—50 мл необходимую навеску хлороформа (квалификации
“для хроматографии”) в ледяной уксусной кислоте; 2) аликвоту этого рас-
твора смешивают с прокипяченной и охлажденной до комнатной темпе-
ратуры дистиллированной водой в мерной колбе на 100—250 мл; хранят в
холодильнике в плотно закрытых, желательно узкогорлых, заполненных
доверху флаконах. По мере расходования раствора и увеличения доли
свободного внутреннего пространства возрастает вероятность искажения
концентрации раствора из-за перераспределения хлороформа между во-
дой и контактирующим с ней воздухом. При строгом подходе подобные
градуировочные растворы должны исключаться из дальнейшей работы
уже после первого отбора пробы.
530
С помощью медицинского шприца на 5 мл, снабженного длин-
ной иглой широкого сечения, переносят строго одинаковые пор-
ции каждого раствора (5 мл, по шаблону) в стандартные чистые
пенициллиновые флаконы и немедленно герметизируют их из-
ложенным выше способом. Подготовленная партия с пробами
каждого градуировочного раствора должна включать не ме-
нее трех флаконов. Уравновешивание жидкой и паровой фаз
производится при комнатной температуре в течение 12-15 мин
при периодическом аккуратном покачивании флаконов вручную
(попадание воды на фторопластовую пленку, защищающую па-
ровую фазу внутри флакона от прямых контактов с резиной
пробки, нежелательно).
Образцы водопроводной воды приготавливают следу-
ющим образом. Открывают водопроводный кран и в течение
25-30 мин спускают “застойную” воду достаточно энергичной
струей. Несколько ослабив напор, подставляют под струю ко-
ническую колбу ла 250 мл и заполняют ее водой практически
доверху (кран оставляют открытым). С помощью медицинского
шприца объемом 5 мл переносят строго одинаковые пробы воды
(по 5 мл) в заранее подготовленные 3-4 пенициллиновых фла-
кона, строго соблюдая регламент всех необходимых процедур,
подробно изложенных выше.
Для внесения стандартной добавки хлороформа в
образцы водопроводной воды используют заранее приготовлен-
ный вспомогательный раствор хлороформа в ледяной уксус-
ной кислоте (сообщаемая студентам концентрация хлороформа
в этом растворе не должна превышать 1% по массе).
Подставляют под струю воды, вытекающую из крана, мер-
ную колбу емкостью 2 л и заполняют ее водой до уровня, на 1-
5 см превышающего положение риски; избыточное количество
воды (контроль — по риске) удаляют медицинским шприцем на
2-10 мл с толстой длинной иглой.
Микрошприцем МШ-10 вносят в колбу 10 мкл (или дважды по
10 мкл, по согласованию с преподавателем) базового раствора.
Объем внесенного раствора оценивают с точностью до 0,1 мкл.
При выполнении этой операции опускают корпус микрошприца
в горловину колбы так, чтобы игла оказалась в непосредствен-
ной близости к зеркалу водной поверхности (при “сбросе” ми-
кроколичеств раствора хлороформа в воду желательно избе-
жать разбрызгивания его по стенкам колбы). Закрывают колбу
пробкой и тщательно перемешивают содержимое. Затем, как
описано выше, отбирают порции воды с внесенной в нее добав-
кой хлороформа (5 мл, по шаблону) в подготовленные стандарт-
ные пенициллиновые флаконы. Флаконы тотчас же герметизи-
531
руют и, периодически помешивая, выдерживают при комнатной
температуре до установления межфазового равновесия (не ме-
нее 10-15 мин). Приготовленную партию флаконов используют
при определении содержания хлороформа в водопроводной воде
по варианту Б.
Вариант А. Определение содержания хлороформа в водо-
проводной воде методом абсолютной градуировки. Знакомятся
с вариантом схемы газовых коммуникаций, используемым при
выполнении данной работы, и выводят хроматограф на задан-
ный рабочий режим*.
На период подготовки прибора к работе и при последующей
периодической проверке памяти дозирующего устройства, как
об этом уже говорилось в описании работы 10, на иглу при-
ставки для ПФА (см. рис. V.8) устанавливают резервный пени-
циллиновый флакон, заполненный окружающим воздухом. Из-
начально переключающий кран 6 должен находиться в положе-
нии “накачка”, в дальнейшем в этом же положении крана про-
изводят замену флаконов — резервного пустого на флаконы с
градуировочными растворами или отобранной на анализ водо-
проводной водой.
По установлении стабильного (без заметного дрейфа) фоно-
вого сигнала детектора приступают к анализам порций воздуха
лаборатории с целью проверки:
— воспроизводимости дозируемого объема газовой фазы (по
отклику ДПР на содержащийся в воздухе кислород);
— отсутствия в воздухе заметных количеств хлороформа и
других летучих галогенированных растворителей (в противном
случае использование лабораторного воздуха в качестве газа-
экстрагента исключается);
— памяти приставки для ПФА на хлороформ и сопутствую-
щие ему низшие галоидные алкилы в предшествующих (анали-
зировавшихся в предыдущие дни) градуировочных растворах и
исследуемых водных образцах.
Для записи первой хроматограммы лабораторного воздуха
резко переводят кран 6 приставки из положения НАКАЧКА
в положение ДОЗИРОВАНИЕ, немедленно наносят фломасте-
ром или мягким карандашом стартовую отметку на диаграмм-
ной бумаге пишущего потенциометра (можно “мягко” толкнуть
пишущий узел самописца) и дают команду на начало реги-
страции сигналов детектора интегратором или программно-
аппаратным комплексом “МультиХром” (при использовании по-
*Для выхода хроматографа на рекомендованный рабочий режим обыч-
но требуется 1—1,5 ч. Для экономии студенческого времени прибор может
быть включен лаборантом заранее.
532
следнего пишущий потенциометр может быть отключен). При-
мерно через минуту наблюдают появление зашкаленного пика
кислорода воздуха, над шлейфом которого со временем удер-
живания, не превышающим 5-7 мин, могут проявиться пики
хлоралканов, представляющих аналитический интерес (хлоро-
форм, четыреххлористый углерод, бромдихлорэтан и др.).
Опасными (требующими принятия мер по борьбе с “памятью”
дозирующего устройства — соответствующие рекомендации да-
ны в описании лабораторной работы 10) являются ложные пики
с амплитудой, не менее чем в 3-5 раз превышающей уровень
шума фонового сигнала.
Через 8-10 мин после дозирования регистрацию показаний
ДПР в холостом опыте прекращают, возвращают кран при-
ставки для ПФА в положение НАКАЧКА, устанавливают на
иглу 7 приставки новый герметично закрытый пенициллиновый
флакон, содержащий лишь лабораторный воздух, выжидают
примерно 2 мин для уравновешивания давления газа в системе
и повторяют холостой опыт, соблюдая все приведенные выше
рекомендации.
При воспроизведении результатов первичной проверки и от-
сутствии симптомов проявления нежелательной “памяти” до-
зирующего устройства можно приступать к градуировке при-
бора по искусственно приготовленным водным растворам хло-
роформа, последовательно переходя от более разбавленного к
более концентрированному.
Каждый градуировочный раствор анализируют, по меньшей
мере, трижды, ограничиваясь однократным отбором порции
равновесной паровой фазы из соответствующего флакона (по-
чему?). При записи хроматограмм пишущим потенциометром
по результатам первого анализа корректируют чувствитель-
ность регистрации сигналов детектора (шкалу электрометра)
или величину дозы (определяемой превышением давления газа
в линии накачки относительно его давления на входе в колонку)
с тем, чтобы высота пиков хлороформа составляла 20-50% ши-
рины диаграммной бумаги. В дальнейшей работе принятую
величину дозы равновесного пара не изменяют! По мере
увеличения концентрации хлороформа в градуировочных рас-
творах (равно, как и в исследуемых образцах водопроводной
воды) лишь кратным образом загрубляют шкалу электрометра,
обязательно записывая ее непосредственно против соответству-
ющего сигнала детектора (учет возможных различий в масшта-
бах чувствительности детектирования потребуется при постро-
ении градуировочной зависимости, характеризующей аналити-
ческие возможности вашего прибора, и при ее использовании
533
для определения содержания хлороформа в лабораторной во-
допроводной воде).
Закончив градуировку хроматографа, исследуют равновес-
ную паровую фазу над водопроводной водой в каждом из не ме-
нее трех ранее заготовленных пенициллиновых флаконов. Полу-
чив воспроизводимые результаты ПФА, устанавливают на иглу
дозирующего устройства с краном, находящимся в положении
НАКАЧКА, пустой резервный пенициллиновый флакон, выклю-
чают хроматограф (не забудьте отключить индивидуальное
электрическое питание печки пневматического дозатора!)
и приступают к обработке результатов.
Вариант Б. Определение содержания хлороформа в водо-
проводной воде методом стандартной добавки. Отличается от
варианта А тем, что после проверки чистоты лабораторного
воздуха и контроля “памяти” дозирующего устройства сразу же
анализируют паровую фазу над пробами водопроводной воды,
строго соблюдая все процедурные рекомендации, данные выше
(см. с. 532). Затем приступают к столь же внимательному хро-
матографированию паровой фазы флаконов, содержащих водо-
проводную воду со стандартной добавкой хлороформа. Полу-
чив не менее трех воспроизводимых результатов (по площадям
и/или по высотам пиков) в каждой серии анализов — водопро-
водной воды и водопроводной воды с добавкой известного ко-
личества хлороформа, выключают прибор и приступают к об-
работке результатов.
Обработка результатов
Вариант А. Приведенные к единой шкале чувствительно-
сти регистрации сигналов детектора площади или, по согласо-
ванию с преподавателем, высоты пиков хлороформа (их теку-
щие Ai или hi и усредненные, соответственно Аср или /гср, чи-
сленные значения), найденные по результатам анализа градуи-
ровочных растворов, вписывают в таблицу, близкую по форме
к табл. V.13, против соответствующих заданных концентраций
CHCI3 в водной фазе, Схл. Полученные данные используют для
построения градуировочного графика, откладывая по оси орди-
нат величины Аср (или hcp), а по оси абсцисс Схл в соответству-
ющих растворах. Пользуясь построенным графиком, находят
содержание хлороформа в отобранных пробах водопроводной
воды, оценивают разброс данных от среднего значения, сопо-
ставляют полученный результат с имеющимися у преподава-
теля сведениями о среднестатистической концентрации хлоро-
534
форма в водопроводной воде, характерной для данного региона
и для данного времени года.
Вариант Б. У средняют измеренные интегратором или най-
денные в диалоге с программно-аппаратным комплексом “Муль-
тиХром” высоты и/или площади пиков хлороформа (по согла-
сованию с преподавателем), относящиеся к 1-й и ко 2-й сериям
анализов соответственно. Содержание хлороформа в водопро-
водной воде Мд находят по формуле
М° Р2/Р!-!’
где Fi и .Рг — количественные параметры пиков хлороформа
(высоты или площади пиков) на хроматограммах 1-й и 2-й серий
соответственно; Мд — масса добавленного хлороформа, рас-
считываемая из сообщаемых студентам данных о концентрации
хлороформа в уксусной кислоте, степени разбавления внесен-
ного хлороформа водой в мерной колбе и объема водного рас-
твора, отбираемого на анализ.
Окончательно содержание хлороформа в воде выражают в
массообъемных единицах концентрации (мкг/л) и сопоставляют
полученные результаты с найденными по варианту А. При акку-
ратной работе оба метода (абсолютная градуировка и исполь-
зование стандартной добавки) должны привести к близким зна-
чениям Мо-
При оформлении протокола ответьте на следующие вопросы.
1. Чем обусловлена целесообразность определения хлоро-
форма (и сопутствующих ему низших галогенированных угле-
водородов С1-С4) в воде именно методом статического паро-
фазного анализа?
2. Какие преимущества открывает при решении названной
задачи использование электронозахватного детектора (ДПР)?
Можно ли было воспользоваться каким-либо другим детекто-
ром, при каких условиях?
3. Почему при обработке результатов данной работы непри-
меним метод внутренней нормализации?
4. На что, в первую очередь, следует обращать внимание при
абсолютной градуировке газового хроматографа с электроно-
захватным детектором (равно как и с ДПР)?
5. Почему при градуировке прибора приготовленные градуи-
ровочные растворы анализируют в порядке увеличения содер-
жания в них определяемых соединений, а не в обратном или
произвольном порядке?
535
Работа 12. Газохроматографическое
определение содержания тиофена в бензоле с
пламенно-фотометрическим детектированием
Газохроматографический анализ с использованием селектив-
ного пламенно-фотометрического детектора (ПФД) является в
настоящее время основным методом качественного и количе-
ственного определения летучих фосфор- и серосодержащих со-
единений в различных объектах окружающей среды (атмосфер-
ный воздух, почвы, природные и сточные воды), а также в про-
дуктах химической переработки нефти, каменного угля и дре-
весины.
Цель работы. Определение содержания тиофена в образ-
цах каменноугольного бензола и/или в контрольных образцах
искусственно приготовленных смесей бензола и тиофена ме-
тодом абсолютной градуировки с использованием пламенно-
фотометрического детектора.
Одним из сохраняющих до сих пор важное значение источни-
ков промышленного получения бензола является каменноуголь-
ная смола, содержащая свыше ста различных соединений, боль-
шей частью ароматического характера. Легкая фракция (т. кип.
до 170 °C) перегонки смолы состоит преимущественно из бен-
зола и его гомологов. Их выделяют при повторных тщательных
фракционированиях. Однако в бензоле, получаемом из камен-
ноугольной смолы, всегда содержится примесь (до 0,05%) тио-
фена, от которого очень трудно освободиться* (т. кип. бензола
80,1 °C, т. кип. тиофена 84°C). Близость физических свойств
обусловливает трудности и газохроматографического разделе-
ния бензола (основного компонента) и тиофена (примеси). Учи-
тывая способность ароматических углеводородов тушить ха-
рактеристическую для серосодержащих соединений световую
эмиссию (394 нм) при их одновременном элюировании из ко-
лонки в горелку ПФД (см. раздел II. 1.4.8), при отработке усло-
вий газохроматографического анализа необходимо обеспечить
как можно более полное разделение хроматографических зон
тиофена и бензола (растворителя).
В настоящей работе возможности селективного детектиро-
вания серосодержащих веществ с помощью ПФЛ иллюстриру-
*Очищают бензол от тиофена встряхиванием с концентрированной
серной кислотой — тиофен сульфируется легче и растворяется в кислоте
быстрее, чем бензол. Другой способ извлечения тиофена основан на его
способности легко меркурироваться: при обработке сырого бензола аце-
татом ртути меркурируется только тиофен.
536
ются на примерах количественного определения примеси тио-
фена в каменноугольном бензоле и в искусственно приготовлен-
ных смесях бензола и тиофена.
Аппаратура, условия, объекты хроматографирования
Хроматограф с пламенно-фотометрическим детектором, укомплекто-
ванный интегратором или программно-аппаратным комплексом типа
“МультиХром”. Рабочие шкалы электрометра (1-10)- 10—9А.
Колонка стальная или стеклянная (2 м х 2 мм), заполненная хромато-
ном N-AW (0,16-0,20 мм) с 10% (по массе) ПЭГ-400.
Температура термостата колонок 50°C, испарителя и детектора 130°C.
Газ-носитель — азот, расход 40 мл/мин; вспомогательные газы — во-
дород, расход 75 мл/мин: воздух, расход 110 мл/мин.
Скорость движения диаграммной ленты 240 или 600 мм/ч.
Объекты хроматографирования: 1) пять градуировочных растворов с
известным (по процедуре приготовления) содержанием тиофена в чистом
бензоле*; 2) один-два контрольных раствора тиофена в чистом бензоле*;
3) один-два контрольных образца каменноугольного бензола.
Дозирование осуществляется микрошприцем МШ-10, величина дозы
2,0 ± 0,2 мкл.
Выполнение работы
Выводят хроматограф на заданный рабочий режим и, убе-
дившись в регистрации устойчивого фонового сигнала, присту-
пают к градуировке прибора по искусственно приготовленным
растворам тиофена в чистом бензоле**.
Внимание! Во избежание необратимой засветки фото-
умножителя поджигание водорода в ячейке ПФД сле-
дует производить при отключенном электрическом
питании (соответствующая клавиша на передней панели
блока электрометрического усилителя БИД-36 должна
быть отжата). Электрическое питание детектора вклю-
чают, лишь убедившись в устойчивом горении водорода
в ячейке ПФД, закрытой пробкой (см. рис. 11.52).
В процессе выполнения всех анализов градуировочных рас-
творов, контрольных смесей тиофена в бензоле и образцов ка-
менноугольного бензола необходимо тщательно контролиро-
вать постоянство важнейших инструментальных параметров
*Для приготовления градуировочных растворов и контрольных образ-
цов используют бензол, получаемый из бензиновых фракций нефти (про-
цесс “платформинга”), не содержащий тиофена по происхождению.
**Подробная градуировка предполагает получение 5—7 эксперимен-
тальных точек, накрывающих диапазон ожидаемых содержаний тиофена
в контрольных образцах.
537
(температура колонки, расход газа-носителя и вспомогатель-
ных газов) и, при необходимости, своевременно их корректи-
ровать.
Сначала хроматографируют самый разбавленный раствор,
а по получении не менее 3-7 воспроизводимых результатов
переходят к анализам ближайшего более концентрированного
(при обратном порядке выполнения градуировки существует
опасность возможного нежелательного проявления “памяти” ко-
лонки, газовых коммуникаций и даже микрошприца). Каждый
градуировочный раствор хроматографируют по меньшей мере
3-7 раз, стремясь к получению воспроизводимых по количе-
ственным параметрам пиков результатов. Как обычно, при
возможной регистрации на первоначальных хроматограммах
слишком малых или, напротив, зашкаленных сигналов ПФД из-
меняют соответствующим образом чувствительность регистра-
ции (шкалу электрометра). При выполнении дозирования оце-
нивают объем раствора, испарившийся из иглы микрошприца, и
записывают в рабочем блокноте или непосредственно на хрома-
тограмме против стартовой отметки реальную величину дозы
и окончательно выбранную чувствительность регистрации.
Проанализировав все искусственные растворы тиофена в бен-
золе известного состава и получив, таким образом, необхо-
димый для построения градуировочного графика массив экс-
периментальных данных, приступают к анализам контрольных
образцов, состав которых сообщается студентам только по
представлении результатов определения. Все приведенные вы-
ше практические рекомендации действительны и при выполне-
нии этого этапа работы.
Обработка результатов
Измеряют на всех хроматограммах высоты и/или площади
пиков тиофена*, пересчитывают их относительно единой, вы-
бранной для всех опытов величины дозы (например, относи-
тельно дозы, равной строго 2,2 мкл), усредняют отвечающие ка-
ждому градуировочному раствору, а также каждому контроль-
ному образцу численные значения согласованных с преподава-
телем количественных параметров пиков**, приводят их к еди-
ной шкале чувствительности регистрации сигнала детектора,
*При комплектации прибора интегратором или программно-аппарат-
ным комплексом “МультиХром” измерение параметров хроматографиче-
ских пиков производится в автоматическом режиме одновременно с реги-
страцией хроматограмм.
**Сомнительные значения высот и/или площадей пиков отбрасывают,
пользуясь рекомендациями, содержащимися в работе 8.
538
находят логарифмы скорректированных величин /г или А и за-
носят все эти данные в таблицу против соответствующих из-
вестных содержаний тиофена.
Полученные данные используют для построения градуиро-
вочного графика, откладывая на оси ординат 1g h (или IgA), а по
оси абсцисс — концентрации тиофена в соответствующих гра-
дуировочных растворах. Пользуясь построенным графиком, на-
ходят содержание тиофена в контрольных образцах и сверяют
найденные величины с имеющимися у преподавателя контроль-
ными цифрами; оценивают абсолютную и относительные по-
грешности определения.
При оформлении протокола ответьте на следующие вопросы.
1. Известно, что ПФЛ отличается от других ионизационных
детекторов повышенной чувствительностью и селективностью к
фосфор- и серосодержащим веществам. Как увязать это спра-
ведливое утверждение с отмеченной выше необходимостью при
определении примесей тиофена в бензоле обеспечить как можно
более полное разделение хроматографических зон обоих компо-
нентов?
2. Пламя водорода в ячейке ПИД иногда называют окисли-
тельным. Каков характер пламени водорода в ячейке ПФЛ?
3. Каков линейный динамический диапазон пламенно-фото-
метрического детектора7
4. С какими из эмпирических закономерностей процессов
сжигания в водородном пламени различных индивидуальных
органических соединений вы знакомы?
5. Как изменится высота пика тиофена на хроматограмме
при вдвое меньшем (по сравнению с начальным) расходе газа-
носителя, если принять во внимание, что ПФЛ — логарифмиче-
ский детектор по отношению к серосодержащим веществам?
Работа 13. Анализ пищевого спирта на
содержание вредных примесей
Цель работы. Количественное определение примесей вред-
ных органических соединений в пищевом этиловом спирте мето-
дом внутреннего стандарта. Освоение программно-аппаратного
комплекса “МультиХром”.
Пищевой этиловый спирт, из которого изготавливаются раз-
личные сорта водок и других алкогольных напитков, может
содержать ряд весьма опасных для здоровья человека приме-
сей, количество которых, согласно действующим ГОСТам, не
должно превышать указанные пределы: метанол 0,03-005 (%
539
по объему); альдегиды (в сумме) 2-15 мг/л; сложные эфиры
(в сумме) 18-50 мг/л; сивушные масла 2-15 мг/л. Тем не ме-
нее, по данным [432], только лишь в Кемеровской обл. до 30%
поступающих в продажу водок и других алкогольных напитков
содержат вредные примеси в количествах, существенно превы-
шающих названные.
Прогрессирующее проникновение даже в систему государ-
ственной торговли фальсифицированных продуктов питания
требует организации на федеральном уровне и в районирован-
ных органах санэпиднадзора службы оперативного и надежного
контроля за качеством алкогольных напитков [433].
Большую помощь в решении этой задачи может оказать хро-
матографический анализ. Заметим, что методы определения
токсичных примесей в спиртах различной квалификации, реко-
мендуемые действующими ГОСТами, трудоемки, длительны и
не могут применяться для систематических измерений [432].
В предлагаемой лабораторной работе излагается методика
газохроматографического анализа пищевого этилового спирта
на содержание вредных примесей, ориентированная на исполь-
зование насадочных колонок и режим программирования темпе-
ратуры. Анализ выполняется методом внутреннего стандарта,
в качестве которого в принятых условиях удобно использо-
вать вторичный гексиловый спирт (4-метилпентан-2-ол). Реги-
страция сигнала детектора и интерпретация результатов каче-
ственного и количественного газохроматографического анализа
осуществляются с привлечением программно-аппаратного ком-
плекса “МультиХром”.
Аппаратура, условия, объекты хроматографирования
Хроматограф “Цвет-500” с пламенно-ионизационным детектором, уком-
плектованный программно-аппаратным комплексом “МультиХром”; ра-
бочие шкалы электрометра 1,28- 10“9—1,6* 10“10 А.
Колонка стеклянная, 2 м X 2 мм, заполненная хромосорбом W (30-
60 меш), смоченным тритоном Х-ЗО5 (20% (по массе)).
Температура колонки повышается от 40 ° С (200 с) до 70 °C со скоро-
стью 4 град/мин; температура испарителя 200°C, детектора (переходной
платы) 150 ° С.
Газ-носитель — азот, расход 20 мл/мин; вспомогательные газы — во-
дород и воздух, расход 30 и 400 мл/мин соответственно.
Объекты хроматографирования: 1) один-три градуировочных рас-
твора известного качественного и количественного состава, содержащих
в абсолютном этиловом спирте (основной компонент) рекомендуемый
внутренний стандарт — вторичный гексиловый спирт (4-метилпентан-2-
ол) — и обычно присутствующие в качестве примесей в пищевом этило-
вом спирте следующие органические соединения (от 0,1 до 1% каждого):
диэтиловый эфир, ацетальдегид, ацетон, метиловый, этиловый (основ-
ной компонент), пропиловый, изобутиловый, н-бутиловый, вторичный
540
гексиловый (4-метилпентан-2-ол — внутренний стандарт), изопентило-
вый спирты (перечислены в порядке выхода из колонки); 2) образец ана-
лизируемого пищевого спирта; 3) образец того же пищевого спирта с вве-
денным внутренним стандартом.
Дозирование осуществляется микрошприцем типа МШ-10, величина
дозы 1,0 ±0,2 мкл.
Выполнение работы
Выводят хроматограф на рабочий режим и подготавливают
программно-аппаратный комплекс “МультиХром” к приему и
обработке сигналов детектора. Для этого включают персональ-
ный компьютер (ПК), связанный через аналого-цифровой пре-
образователь (АЦП) с электрометрическим усилителем ПИД
хроматографа и под руководством преподавателя запускают
программные средства (ПС) “МультиХром”.
Настройка программы на определенный метод сбора и об-
работки данных. Прежде всего необходимо создать ту часть
метода, которая отвечает за сбор данных (настройку програм-
мы на обработку поступающей информации по тому или иному
алгоритму можно сделать позднее, уже во время регистрации
сигналов детектора). Наилучший способ решения поставлен-
ной задачи — выбрать подходящий прототип из имеющихся
в ПС “МультиХром” шаблонов нескольких стандартных ме-
тодов, ориентированных на определенный тип используемого
АПП, и, при необходимости, модифицировать его. Обраща-
ются к опции Измерение/Открыть метод и запустить из глав-
ного меню (появляющегося сразу же после запуска ПС) и в
раскрывающеемся окне ОТКРЫТЬ ФАЙЛ находят перечень
возможных названий файлов Выбирают название* 2com2.mtw,
щелкают по нему дважды левой клавишей мыши (в дальней-
шем тексте для краткости будет использоваться словосочета-
ние “щелкают мышкой”) или, высветив название файла, щел-
кают по кнопке [ОК] (можно также нажать клавишу [Enter]).
Откроются незаполненное окно хроматограммы и диалоговое
окно ЗАПУСК АНАЛИЗА. Пользуясь клавиатурой компью-
тера, впечатывают в соответствующую строчку название ана-
лизируемой смеси и намечаемый объем пробы (позже, в про-
цессе выполнения хроматографических анализов, всякий раз по-
сле дозирования пробы в хроматограф этот параметр необхо-
*Это название отвечает 2-му каналу используемой в лаборатории га-
зовой хроматографии химического факультета СПбГУ системы прибо-
ров газовый хроматограф — выносной (внешний) АЦП — ПК. Название
должно быть изменено при смене канала приема хроматографической ин-
формации, а также при использовании АЦП иного типа.
541
димо корректировать с учетом объема жидкости, остающейся в
игле микрошприца).
Щелкают мышкой по кнопке [Измерение] и в соответству-
ющих полях открывающегося окна с таким же названием ука-
зывают ожидаемую продолжительность анализа (в нашем слу-
чае — 15 мин)* и необходимую периодичность записи поступа-
ющей информации на диск (каждые 3-5 мин)**. В строке Запуск
должно быть записано Внешний (ориентация машины на прием
хроматографической информации по сигналу от внешнего ми-
кровыключателя) .
Щелкают по кнопке [ОК] или нажимают клавишу [Enter] для
подтверждения заданных параметров и возврата в диалоговое
окно ЗАПУСК АНАЛИЗА. Теперь следует раскрыть новое
окно ОБРАБОТКА ДАННЫХ, что достигается щелчком по
кнопке [Параметры обработки]. Здесь нужно активировать
поля Выбросы (указание на способ фильтрации данных), За-
писать (чтобы регистрируемая хроматограмма по окончании
анализа автоматически была записана на диск) и Показать все
(для показа сразу всей хроматограммы по окончании ее реги-
страции). Закрывают окно ОБРАБОТКА ДАННЫХ и щел-
кают мышкой по кнопке [Запуск] в диалоговом окне ЗАПУСК
АНАЛИЗА. Система переходит в состояние ожидания сигнала
внешнего запуска (машина готова к приему хроматограммы).
Регистрация первой (пробной) хроматограммы градуиро-
вочного раствора и корректировка метода сбора и обработки
данных. Вводят в испаритель хроматографа порцию (1,0±
0,2 мкл) 1-го градуировочного раствора и немедленно после до-
зирования нажимают кнопку микровыключателя на подведен-
ном к прибору кабеле (о приеме команды свидетельствует из-
менение цвета экрана — молочного на зеленый). В процессе
регистрации сигналов детектора необходимо дополнить и уточ-
нить ранее внесенные в окно ЗАПУСК АНАЛИЗА начальные
сведения о пробе и условиях хроматографирования, иначе го-
воря, создать паспорт хроматограммы. С этой целью вызы-
вают пункт меню Метод/Паспорт. В открывающееся окно
ПАСПОРТ перейдет часть уже заполненных вами строк из
окна ЗАПУСК АНАЛИЗА. В незаполненные резервные строки
впечатывают сведения о детекторе, колонке и ее температуре,
расходе газа-носителя. Отдельная строка предусмотрена для
фамилии аналитика, выполняющего анализ (пользователя ПС
*В обычной практике ожидаемая продолжительность анализа устана-
вливается в предварительных опытах без использования компьютерной
обработки хроматографической информации.
**Это поможет избежать случайной потери данных.
542
“МультиХром”), Щелкают по кнопке [Коммент.] и вводят в
расширенную версию паспорта другие полезные сведения об
условиях анализа и характере анализируемой пробы, не вклю-
ченные в рубрики окна ПАСПОРТ.
Программа автоматически заканчивает регистрацию хрома-
тограммы по истечении указанной ранее в строчке окна ИЗМЕ-
РЕНИЕ продолжительности анализа (15 мин). На экране мони-
тора вы увидите хроматограмму, над обработанными пиками
которой появятся порядковые номера (это означает, что в па-
мяти компьютера имеются данные о площади, высоте и времени
удерживания каждого пронумерованного пика). Для удобства
просмотра можно изменить масштаб графического представле-
ния результатов анализа по любой оси, воспользовавшись либо
мышкой, либо клавиатурой ПК. Оба способа во многом допол-
няют друг друга, причем все операции по масштабированию
изображения хроматограммы действуют и во время приема дан-
ных.
Чтобы изменить масштаб с помощью мыши, устанавливают
ее курсор в левый нижний угол интересующей области, нажи-
мают левую клавишу и, удерживая ее, перемещают курсор в
правый нижний угол области, очерчиваемой пунктирной рам-
кой. Отпустите левую клавишу. В окне появляется увеличен-
ное изображение выделенного участка хроматограммы. Сдви-
гать изображение влево-вправо можно с помощью линейки про-
крутки, расположенной в нижней части экрана монитора. Щел-
чок по кнопке [Показать все] восстановит исходный вид хрома-
тограммы.
Широкие возможности по управлению масштабом изображе-
ния хроматограммы предоставляет клавиатура. Так, например,
чтобы увеличить масштаб по оси Y следует нажать (можно не-
сколько раз) клавишу [ф]; соответственно для уменьшения мас-
штаба — клавишу [J.]. Чтобы растянуть хроматограмму по
оси X, нажимают (можно несколько раз) клавишу [—>]; соот-
ветственно для сжатия хроматограммы — клавишу [<—] (о дру-
гих многочисленных возможностях масштабирования хромато-
граммы с использованием клавиатуры см. «Руководство поль-
зователя “МультиХром” для Windows»).
Для исключения случайной потери поступившей в ПК инфор-
мации записывают хроматограмму на диск, выбрав пункт меню
Хроматограмма/Записать. Теперь можно приступить к на-
стройке алгоритма интегрирования и редактированию резуль-
татов первоначальной обработки хроматограммы, выполненной
ПС “МультиХром” автоматически. Прежде всего следует про-
верить разметку пиков и положение базовой линии под ними.
543
Для корректировки вызывают пункт меню Метод/Разметка и
заменяют выбранное машиной численное значение (в секундах)
параметра Ширина (ожидаемая ширина у основания самого пер-
вого из подлежащих интегрированию, как правило, наиболее уз-
кого пика) на измеренное вами непосредственно по записанной
хроматограмме. Аналогичным образом исправляют числен-
ное значение параметра Уширение (показывающего, во сколько
раз последний интегрируемый пик на хроматограмме шире пер-
вого) .
Разумная величина параметра Порог должна находиться в
пределах от 0,5 до 5. Меньшее численное значение этого пара-
метра обеспечивает большее количество обрабатываемых пи-
ков. Лучшими могут оказаться значения 2 или 3 (выбираемые
машиной по умолчанию). Если окажутся размеченными неко-
торые нежелательные пики, задают величину параметра Мин.
Высота чуть больше, чем высота пиков, которые вы хотели бы
исключить из рассмотрения. Для того чтобы увидеть резуль-
тат, щелкают по кнопке [Размечать].
Особое внимание следует уделить заданию области интегри-
рования, ограничиваемой под пиками базовой линией. Щел-
кают мышкой по кнопке [События] (имеются в виду события
интегрирования) в окне ПАРАМЕТРЫ РАЗМЕТКИ. Появля-
ется новое окно с незаполненным (по умолчанию) списком со-
бытий интегрирования. Скрытые пока от пользователя обусло-
вленные ПС события позволяют настроить процесс разметки в
соответствии с особенностями данной серии хроматограмм, за-
давая для некоторых участков хроматограммы индивидуальные
параметры и правила разметки.
Для ознакомления с предусмотренным ПС списком событий
интегрирования и выбора одного из них щелкают мышкой по
кнопке [Добавить] или [Менять]. Развернется окно СОБЫ-
ТИЕ ИНТЕГРИРОВАНИЯ. Шелкая по кнопке прокрутки в
правой части поля Событие, выбирают требуемое событие из
списка и вводят его как дополнительный параметр разметки,
щелкнув по кнопке [ОК] (предварительно, при необходимости, в
строке параметр задают численное значение момента времени,
начиная с которого ПС начнут учитывать вашу команду).
Выбирают событие Вкл. базу долина-к-долине По этой ко-
манде машина проведет ломаную базовую линию по самым низ-
ким точкам между пиками (при отключенной базе “долина-к-
долине” интегрирование площадей пиков осуществится “по пер-
пендикуляру” , что при недостаточном разделении зон примес-
ных компонентов приведет к значительным погрешностям ре-
зультатов количественного анализа).
544
Щелкнув мышкой по кнопке [Размечать], наблюдают резуль-
тат корректировки. Для сохранения оптимизированных параме-
тров разметки для будущих анализов перезаписывают создан-
ный метод сбора и обработки данных, вызывая пункт меню Ме-
тод/Сохранить; для сохранения достигнутой разметки переза-
писывают хроматограмму (Хроматограмма/Записать).
Градуировка хроматографа. Первым этапом градуировки яв-
ляется создание Таблицы компонентов, которая должна содер-
жать данные по всем компонентам анализируемого раствора:
их названия, времена удерживания, градуировочные коэффици-
енты и т. п. Создание такой таблицы — первый шаг как для
идентификации соединений, так и для последующего получения
градуировочных зависимостей и расчета концентраций компо-
нентов анализируемых образцов. Таблица компонентов созда-
ется на базе градуировочной хроматограммы.
Загружают с диска полученную на предварительном этапе
градуировочную хроматограмму (Хроматограмма/Открыть),
выбирают пункт меню Метод/Градуировка/Компоненты— в
нижней половине окна появляется шаблон таблицы компонен-
тов, содержащий строку с указаниями столбцов, которые надо
сформировать [Пик; Удерж; Окно, %; Репер; Название и т. д.).
Шелкают по кнопке [Добавить] столько раз, сколько компонен-
тов в выбранной градуировочной смеси. Так формируется кар-
кас таблицы компонентов.
В столбец Пик вводят номер пика на хроматограмме. Нажи-
мают клавишу [—>]. Курсор скачком перемещается на вершину
выбранного пика, а в столбце Удерж, появляется его время
удерживания. В столбец Окно, % вводят ширину окна идентифи-
кации. Значение этого параметра обычно задают в пределах от
2 до 10% в зависимости от конкретного состава смеси. В столбец
Название вводят название компонента. Аналогичным образом
заполняют строки для всех остальных компонентов. По окон-
чании заполнения каждой строки нажимают клавишу [Enter].
Закрывают таблицу компонентов, щелкая по кнопке [ОК].
Вызывают диалоговое окно ПАРАМЕТРЫ ГРАДУИРОВ-
КИ (Метод/Градуировка/Установки). Параметры в данном
диалоговом окне сгруппированы по назначению: Идентифика-
ция, Количественный расчет и Градуировка. Задают метод гра-
дуировки Внутренний стандарт и метод количественного рас-
чета Внутренний стандарт с указанием, какое именно соеди-
нение используется в качестве внутреннего стандарта (в нашем
случае 4-метилпентан-2-ол). По окончании задания всех требу-
емых параметров щелкают по кнопке [ОК].
545
Записывают дополненный вами метод сбора и обработки ин-
формации на диск под новым названием (например, ALCOHOL),
для чего используют пункт меню Метод/Сохранить. Вызвав
пункт меню Хроматограмма/Записать, перезаписывают на
диск и хроматограмму. Осуществляют проверку записи всей
необходимой информации в паспорте, включая название метода
(Метод/ Паспор т).
Следующим этапом является подготовка Таблицы концентра-
ций, которой отводится особое окно с одноименным названием.
Эта таблица содержит концентрации компонентов во всех гра-
дуировочных смесях, которые используются для градуировки,
и создается всегда на базе уже существующей Таблицы ком-
понентов. Каждой градуировочной смеси соответствует один
столбец таблицы, называемый также Градуировочным уровнем.
В процессе градуировки из градуировочных хроматограмм на
каждый уровень должны заноситься данные по площадям и вы-
сотам пиков компонентов.
Сначала необходимо создать шаблон таблицы концентраций,
содержащей столько градуировочных уровней, сколько будет
градуировочных хроматограмм. Открывают окно ТАБЛИЦА
КОНЦЕНТРАЦИИ через пункты меню Метод/Градуировка/
Компоненты/Кондентрации. Шелкают мышкой по кнопке
[Добавить]. В появившееся поле вводят номер градуировоч-
ного уровня и нажимают кнопку [ОК]. Программа создаст но-
вый столбец Ур-нъ 1, соответствующий созданному первому гра-
дуировочному уровню. Заносят в этот столбец значения кон-
центраций для каждого компонента, находящегося в первом гра-
дуировочном растворе Аналогичным образом создают уровни
для последующих градуировочных растворов.
Закрывают окно ТАБЛИЦА КОНЦЕНТРАЦИИ, щелкая
мышкой по кнопке [ОК]. Перезаписывают метод (Метод/Сох-
ранить) и хроматограмму (Хроматограмма/Записать).
Далее анализируют 2-й градуировочный раствор, а затем
и 3-й, выполняя аналогичные процедуры по обработке резуль-
татов хроматографирования. Вызывают пункт меню Измере-
ние/Перезапустить — ЭВМ готова к приему следующей хро-
матограммы*. В конечном итоге надо записать три градуиро-
вочные хроматограммы, повторив все этапы, которые были опи-
саны ранее.
*При градуировке допускается вместо хроматографирования одинако-
вых порций нескольких градуировочных растворов, различающихся по
концентрациям компонентов, записывать хроматограммы одного и того
же единственного градуировочного раствора при последовательном из-
менении строго оцениваемой величины дозы.
546
Для визуального контроля правильности идентификации ком-
понентов градуировочных растворов устанавливают опцию
Вид/Метка пика/Имя компонента. В этом случае над ка-
ждым идентифицированным пиком будет проставлено его назва-
ние.
Имея записанные градуировочные хроматограммы, можно
перейти к обработке результатов градуировки, т. е. к построе-
нию градуировочных зависимостей для каждого компонента и
расчету градуировочных множителей, иначе — факторов от-
клика (ФО), учитывающих различную чувствительность детек-
тора к интересующим примесным компонентам и внутреннему
стандарту. Для выполнения этой расчетной процедуры це-
лесообразно поставить записанные хроматограммы в очередь.
Выбирают пункт меню Хроматограмма/Открыть, высвечи-
вают градуировочные хроматограммы и щелкают по кнопке
[В очередь] В поле Имя вводят название очереди (1) и щел-
кают по кнопке [ОК]. Тем самым создают очередь с назва-
нием 1.QUE, о чем свидетельствует появление Таблицы очереди,
где в столбце Гр. Точка необходимо проставить номера уров-
ней градуировочных хроматограмм. Далее щелкают по кнопке
[Пересчет]. В диалоговом окне ПЕРЕСЧЕТ выбирают гра-
дуировочные хроматограммы и щелкают по кнопке [Градуи-
ровать]. После этого извлекают из архива первую градуи-
ровочную хроматограмму (Хроматограмма/Открыть) и в от-
крывшемся окне высвечивают требуемую строку. Шелкают
мышкой по кнопке [ОК] и выводят на экран монитора соот-
ветствующую хроматограмму. Обращаются к пунктам меню
Метод/Градуировка/Компоненты/Графики— на экране по-
являются представленные в графическом виде градуировочные
зависимости для каждого компонента и численные значения
градуировочных коэффициентов.
Если какая-либо точка градуировочной зависимости для ка-
кого-либо компонента “выпадает”, ее можно исключить. Данная
операция затронет только этот компонент, а не градуировоч-
ный уровень в целом. Для исключения точки нужно из списка
уровней в левой нижней части окна выбрать мышкой строку с
номером исключаемой точки и щелкнуть по кнопке [Использо-
вать?]. При этом индикатор использования уровня (Да/Нет)
изменит свое состояние. Здесь же высвечивается и численное
значение градуировочного множителя для данного компонента
раствора. Используя меню Метод и соответствующее подменю
Градуировка/Записать в метод записывают результаты гра-
дуировки на диск. Закончив все операции по градуировке, при-
ступают к анализу реального объекта.
547
Анализ образца пищевого спирта на содержание вредных
примесей. Подготавливают ЭВМ к приему новой хроматограм-
мы (Измерение/Перезапустить). Вводят в хроматограф пор-
цию (1,0 ± 0,2 мкл) спирта с неизвестным составом примесных
компонентов. Получаемая хроматограмма не обрабатывается,
а нужна исключительно для того, чтобы подтвердить, что ком-
понент, выбранный в качестве внутреннего стандарта — вто-
ричный гексиловый спирт, отсутствует в реальном объекте.
Убедившись в этом, готовят реальный объект к количествен-
ному хроматографическому анализу методом внутреннего стан-
дарта.
На аналитических весах отбирают навеску образца пищевого
спирта (500-800 мг) и вводят в нее с помощью микрошприца на
100 мкл порцию (50-100 мкл) раствора внутреннего стандарта в
этиловом спирте (необходимое для последующих расчетов со-
держание стандарта в растворе в % (по массе) уточняют у пре-
подавателя или лаборанта). Количество введенного раствора
стандарта следует проконтролировать на аналитических весах.
Подготавливают ЭВМ к приему новой хроматограммы кон-
тролируемого этилового спирта с внутренним стандартом (Из-
мерение/Перезапустить). На экране появляется бланк пас-
порта, который необходимо заполнить, назвав новый объект
анализа. Вводят пробу в испаритель хроматографа и неме-
дленно, как и ранее, нажимают кнопку внешнего микровыклю-
чателя. По окончании заявленного времени машина завершит
прием хроматограммы и проведет ее обработку в соответствии
с созданным методом.
Полученную хроматограмму проанализированного пищевого
спирта записывают на диск (Хроматограмма/Записать), а за-
тем приступают к ее количественной обработке (Обработка/
Выдать отчет). На экране появляется диалоговое окно ГЕНЕ-
РАТОР ОТЧЕТА, в котором предусмотрены различные вари-
анты выдачи отчета: экран или принтер. Мышкой отмечают
нужный вариант.
Обработка результатов
Отчет о количественном определении примесей в контроль-
ном образце пищевого этилового спирта предъявляют препо-
давателю И сличают полученные результаты с данными неза-
висимого анализа, выполненного лаборантом заранее. Прини-
мая эти данные за арбитражные, характеризуют воспроизво-
димость и погрешность своих результатов согласно рекоменда-
циям, изложенным в описании лабораторной работы 8.
548
При оформлении протокола ответьте на следующие вопросы.
1. В чем заключаются главные преимущества метода вну-
треннего стандарта перед методом абсолютной градуировки
при количественном газохроматографическом определении?
2. Каковы основные требования, предъявляемые к внутрен-
нему стандарту?
3. Каковы основные преимущества компьютерной обработки
результатов хроматографического анализа?
549
Глава VI
ПРАКТИЧЕСКИЕ РАБОТЫ
ПО ЖИДКОСТНОЙ ХРОМАТОГРАФИИ
Работа 14. Разделение смеси геометрических
изомеров азобензола методом препаративной
адсорбционной тонкослойной хроматографии
Цель работы. Ознакомление с методическими особенно-
стями осуществления тонкослойной хроматографии в препара-
тивном варианте. Приготовление пластин для препаративной
ТСХ с незакрепленным слоем адсорбента. Разделение смеси
цис- и транс-изомеров азобензола методом препаративной ад-
сорбционной ТСХ с использованием в качестве неподвижной
фазы оксида алюминия, выделение индивидуальных изомеров и
установление количественного состава исходной смеси грави-
метрическим (весовым) методом.
Оборудование, реактивы, условия
и объекты хроматографирования
Пластины для препаративной ТСХ размером 24x24 см с насыпным
слоем оксида алюминия (III—IV степень активности по Брокману) толщи-
ной 1,5 мм.
Металлический станок с валиком для приготовления пластин с неза-
крепленным слоем адсорбента.
Проявительная камера, специально предназначенная для пластин с не-
закрепленным слоем сорбента, имеющая лунку для подвижной фазы и
кронштейн для удерживания пластины в рабочем положении.
Столик с феном для просушки пластин после их проявления.
Калиброванные пипетки (или медицинские шприцы) вместимостью
5 см3.
Микроаспиратор.
Стеклянные фильтры Шотта №4 (2 шт).
Колбы круглодонные емкостью 25—50 мл.
Весы аналитические.
Растворители: петролейный эфир (гексан, гептан или циклогексан ква-
лификации х.ч.) и диэтиловый эфир (“медицинский”)- Требуемые ко-
личества растворителей для одного препаративного цикла — 60 и 4 мл
соответствен но.
550
Объект хроматографирования — раствор азобензола в петролейном
эфире (5% по массе).
Выполнение работы
Ознакомившись с подготовленным рабочим местом, присту-
пают к выполнению эксперимента.
На матовую сторону стеклянной пластины, размещенной на
станке для нанесения сорбента, насыпают (по возможности, рав-
номерно по всей площади пластины) оксид алюминия, толщина
слоя которого должна составить ~2 мм. Разравнивают слой
сорбента с помощью специального валика, прокатывая его от
центра к краям. Качество подготовленного слоя сорбента оце-
нивают визуально (не должны обнаруживаться какие-либо ше-
роховатости и складки).
Подготовленную пластину переносят на поверхность рабо-
чего стола, покрытую фильтровальной бумагой. Калиброван-
ной пипеткой, содержащей 5 мл раствора азобензола в петро-
лейном эфире, порциями (~0,2 мл) наносят анализируемый об-
разец в виде полосы на стартовую линию (дожидаются испаре-
ния растворителя во избежание размывания хроматографиче-
ской полосы). Образующаяся полоса должна отстоять от боко-
вых сторон пластины и от нижнего края примерно на 1,5 см и
по ширине не превышать 0,5 см.
В лунку проявительной камеры, предназначенную для по-
движной фазы, вводят петролейный и диэтиловый эфиры в соот-
ношении 60:4, в элюент опускают полоску фильтровальной бу-
маги 2x24 см и сразу же закрывают камеру стеклянной крыш-
кой. Через 15-20 мин атмосфера камеры насыщается парами
подвижной фазы.
Подготовленную пластину помещают в проявительную ка-
меру на специальный кронштейн так, чтобы ее нижний край ка-
сался подвижной фазы. Закрыв камеру крышкой, наблюдают
проявление хроматограммы После того, как визуально бу-
дет зафиксировано образование двух окрашенных полос цис- и
транс-изомеров азобензола ( из которых верхняя полоса при-
надлежит транс-, а нижняя — цис-форме), и расстояние между
ними составит не менее 1 см, пластину извлекают из камеры и
для удаления подвижной фазы ее переносят на рабочий столик
с феном в вытяжной шкаф.
С помощью микроаспиратора, подключенного к водоструй-
ному насосу, отделяют последовательно полосы сорбента с раз-
деленными изомерами и собирают каждый из них на стеклянные
фильтры Шотта.
551
Индивидуальные изомеры азобензола смывают с сорбента
тремя порциями (общий объем 10 мл) диэтилового эфира в
предварительно взвешенные колбы-приемники; после отсасыва-
ния эфира в вакууме водоструйного насоса до образования су-
хого остатка колбы вновь взвешивают и по разности масс нахо-
дят количественное соотношение цис- и транс-изомеров. Выде-
ленные индивидуальные соединения сдают лаборанту.
Обработка результатов
В протоколе необходимо подробно описать все этапы работы
и дать объяснение следующим фактам.
1. Почему в условиях эксперимента транс-изомер удержива-
ется слабее, чем цис-?
2. Почему полоса, принадлежащая транс изомеру, более ин-
тенсивно окрашена?
3. Как повлияло бы на результаты разделения изменение со-
става элюента (подвижная фаза — диэтиловый эфир; подвиж-
ная фаза — петролейный эфир)?
Работа 15. Разделение смеси моно-, ди- и
триэтаноламинов методом лигандообменной
жидкостной хроматографии
Цель работы. Ознакомление с методом лигандообменной
хроматографии, с градиентным элюированием в жидкостной
хроматографии, с новыми вариантами детектирования разде-
ленных компонентов. Разделение смеси моно-, ди- и триэта-
ноламинов методом колоночной хроматографии. Установление
количественного состава смеси.
Лигандообменная хроматография основана на образовании
координационных связей между сорбентом и разделяемыми ио-
нами или молекулами. В качестве стационарной фазы в ли-
гандной хроматографии используют ионообменники с противо-
ионом — металлом-комплексообразователем. В свою очередь,
объектами лигандообменной хроматографии могут являться со-
единения, содержащие кратные связи или донорные атомы (се-
ра, кислород, азот), способные координироваться с ионами ме-
таллов (Cu2+, Ag+, Fe2+, Cu2+, Ni2+, Hg2+).
Разделение анализируемых соединений осуществляется за
счет обратимого обмена лигандов. Характерной особенностью
любого из видов лигандной хроматографии являются селектив-
ность самого процесса комплексообразования и высокая ско-
рость обмена лигандами.
552
Если А и В — разделяемые компоненты, М — ионы металла-
комплексообразователя, R — сорбент, С — исходный лиганд
у иона комплексообразователя и одновременно элюент, тогда
процессы, протекающие на границе двух фаз, можно описать
следующим образом:
1) при внесении вещества в колонку
RMC + A#RMA + C,
RMC + В # RMB + С;
2) при прохождении элюента через колонку
RMA + С # RMC + А,
RMB + C#RMC + B.
В равновесных условиях лиганды А и В распределяются ме-
жду подвижной и неподвижной фазами в соответствии со ста-
бильностью образующихся комплексов, т. е. в зависимости от
значений констант комплексообразования: чем больше эти раз-
личия, тем выше селективность разделения, которая в общем
случае может быть оценена как отношение констант нестойко-
сти разделяемых соединений.
С использованием таких комплексов было проведено успеш-
ное разделение аминов, аминокислот, пептидов, белков, нуклео-
тидов, углеводов и др.
Лигандообменная хроматография предъявляет высокие тре-
бования к геометрии сорбируемых соединений, являясь уни-
кальным методом разделения изомеров. Одним из перспектив-
ных направлений этого варианта хроматографического анализа
является возможность расщепления и разделения рацематов ор-
ганических соединений. Первыми в этом направлении стали ра-
боты по разделению оптически активных аминокислот с исполь-
зованием полистирольного сорбента, ковалентно связанного с
оптически активной природной аминокислотой — Z-пролином
и ионом Си2+. Прочно координируясь с сорбентом, ион двух-
валентной меди имеет две валентные позиции для связывания
подвижного лиганда молекулы D- или /.-аминокислоты.
Оборудование, реактивы, условия
и объекты хроматографирования
Хроматографическая разделительная стеклянная колонка (15x2 см),
заполненная сефадексом G-25 в форме аммиаката меди и соединенная по-
следовательно с колонкой-ловушкой (3x2 см).
Коллектор фракций, снабженный таймером для последовательного от-
бора порций элюата.
553
Хроматографические пластины силуфол (ЧССР ) с закрепленным сло-
ем силикагеля.
Подвижная фаза — 0,15, 1 М и 3 М водные растворы аммиака.
Проявительная смесь для ТСХ — ацетон : вода : 25%-ный раствор
аммиака (6:2: 1), общий объем 45 мл.
Водный раствор кристаллической соды Na2CC>3 • 10 ЩО (2%-ный).
Водный раствор перманганата калия (1%-ный).
Титрованный раствор соляной кислоты.
Объект хроматографирования — смесь моно-, ди- и триэтаноламинов.
Выполнение работы
Для разделения анализируемой смеси, состоящей из моноэта-
ноламина HOCH2CH2NH2, диэтаноламина (НОСНгСНг^МН и
триэтаноламина (НОСНгСН^зМ, в качестве неподвижной фазы
используют слабокислотную смолу СМ сефадекс G-25 (размер
частиц 40-120 мкм) в форме аммиаката меди. Для этого ее сна-
чала переводят в медную форму, пропуская через колонку, за-
полненную смолой, последовательно 0,5 М водный раствор мед-
ного купороса CUSO4 • 5НгО и дистиллированную воду, а за-
тем переводят в медно-аммиачную форму, пропуская 1 М рас-
твор аммиака (указанные процедуры осуществляются лаборан-
том заранее).
Приготовленную таким образом разделительную колонку со-
единяют с помощью тефлоновой трубки последовательно с ко-
лонкой-ловушкой, содержащей сефадекс G-25 в аммиачной фор-
ме. В ловушке происходит улавливание катионов меди.
Исследуемую смесь растворяют в 1 мл дистиллированной
воды и вносят в колонку при закрытой ловушке; затем ее от-
крывают и дают раствору впитаться в смолу. Вновь закрывают
ловушку и вносят стеклянной пипеткой 5—6 мл дистиллирован-
ной воды для предохранения слоя смолы от перемешивания при
подаче элюента.
Анализируемую смесь элюируют 0,15 М водным раствором
аммиака. С помощью коллектора фракций элюат последова-
тельно отбирают порциями по 5 мл. Коллектор, снабженный
таймером, позволяет контролировать сбор требуемых порций
элюата. Время между отдельными перемещениями пробирок
для сбора элюата составляет ~2,5 мин.
Контроль за составом элюата осуществляют методом тонко-
слойной хроматографии на пластинках силуфол. Состав про-
явительной смеси ацетон : вода : 25%-ный раствор аммиака (6 :
2:1), общий объем проявительной смеси 45 мл.
Детектирование проводят смесью 2%-ного водного раствора
кристаллической соды ЫагСОз • ЮНгО и 1%-ного водного рас-
твора перманганата калия (1 : 1 (по объему)). Анализируемые
554
соединения обнаруживаются в виде желтых пятен. Первым из
колонки элюируется триэтаноламин. После выхода диэтанол-
амина (общий объем израсходованного элюента к этому вре-
мени достигает ~90 мл) производят замену подвижной фазы на
более концентрированный (3 М) водный раствор аммиака. Ис-
пользование градиентного элюирования необходимо для сокра-
щения времени анализа и увеличения его эффективности. Ин-
дивидуальные этаноламины собирают в процессе элюирования
в приемники (плоскодонные стеклянные колбы на 25 мл), рас-
творитель (аммиак) упаривают на водяной бане и проводят ти-
трование раствором соляной кислоты для установления коли-
чественного состава исходной смеси.
Обработка результатов
При оформлении протокола необходимо дать подробное опи-
сание всех этапов работы и ответить на следующие вопросы.
1. Почему при разделении этаноламинов потребовалось гра-
диентное элюирование?
2. Чем объяснить такой порядок элюирования анализируе-
мых компонентов?
Работа 16. Разделение смеси органических
веществ методом нормально-фазовой
ВЭЖХ с использованием УФ-
и рефрактометрического детекторов
Цель работы. Проведение качественного анализа смеси ор-
ганических соединений по параметрам удерживания с исполь-
зованием рефрактометрического детектора. Количественная
оценка эффективности хроматографического разделения. Срав-
нение относительной чувствительности УФ- и рефрактометри-
ческого детекторов к различным соединениям анализируемой
смеси.
Современная высокоэффективная жидкостная хроматогра-
фия — один из эффективных методов анализа и разделения
многокомпонентных сложных смесей. В адсорбционной хро-
матографии разделение веществ, входящих в смесь и движу-
щихся по колонке в потоке растворителя, происходит за счет
их различной способности адсорбироваться на поверхности и
десорбироваться с поверхности адсорбента. Достигаемое при
этом разделение зон компонентов обусловливается их различ-
ным взаимодействием как с растворителем, так и с адсорбен-
том. Если подвижная фаза менее полярна, чем используемая
555
неподвижная, то такой вариант хроматографического анализа
называется нормально-фазовым. В случае нормально-фазовой
хроматографии удерживание в основном определяется специ-
фическими взаимодействиями адсорбат — адсорбент. Для ха-
рактеристики вещества, как и в любой другой хроматографии,
помимо времени и объема удерживания используют также чи-
сленные значения факторов удерживания и относительного от-
клика: отношение сигнала одного детектора к сигналу другого
при условии, что оба они работают в своем линейном диапа-
зоне.
Как уже упоминалось, в ВЭЖХ детекторы фиксируют из-
менение какого-либо свойства элюата, связанное с наличием в
нем анализируемых веществ. Универсальный рефрактометри-
ческий детектор непрерывно регистрирует изменение показа-
теля преломления элюата на выходе из колонки. Сигнал ли-
нейно возрастает с увеличением количества растворенного ве-
щества. Селективный УФ-детектор способен зафиксировать ин-
тересующие исследователя вещества, имеющие специфические
свойства на фоне других соединений. Он обнаруживает разде-
ленные компоненты анализируемой смеси, поглощающие в УФ-
области, с большей чувствительночтью в сравнении с рефрак-
тометрическим. Кроме того, этот детектор обеспечивает воз-
можность работы в градиентном режиме.
Оборудование, реактивы, условия
и объекты хроматографирования
Жидкостный хроматограф (производство ЧССР), укомплектованный
УФ- и рефрактометрическим детекторами, а также считающим интегра-
тором (модель CI-100). Шкала регистрации сигналов детекторов при за-
писи хроматограммы самописцем в диапазоне от 16 до 128 мВ. Скорость
диаграммной бумаги 0,3 см/мин.
Колонка стеклянная двухсекционная, состоящая из двух последова-
тельно соединенных колонок, каждая размером 150x3 мм, заполненных
сепароном CGX (гидроксилированный силикагель, 5 мкм).
Подвижная фаза — гептан (ч.д.а.), содержащий 1% 2-пропанола квали-
фикации ч.д.а.; расход 0,3 мл/мин*.
Объекты хроматографирования. 1. Искусственная смесь А раство-
ренных в подвижной фазе органических веществ следующего примерного
состава (в % (по массе)): бензол (~ 18), лс-ксилол (~ 18), n-ксилол 18),
додекан (~36), фенилацетилен (~ 10). Все вещества квалификации “для
хроматографии”. 2. Индивидуальные градуировочные растворы Б, В, Г,
Д, Е каждого из вышеназванных веществ в подвижной фазе (~ 10-15% (по
массе)). 3. Один-два контрольных образца неизвестного студентам ка-
чественного состава, приготавливаемые растворением в подвижной фазе
*Дегазацию подвижной фазы проводят в вакууме водоструйного на-
соса при перемешивании магнитной мешалкой в течение 15 мин непосред-
ственно перед выполнением хроматографических анализов.
556
нескольких органических соединений из числа вышеназванных (а воз-
можно, и иных) веществ.
Дозирование осуществляется микрошприцем МШ-10,доза 1,6±0,2мкл.
Выполнение работы
Качественный анализ по параметрам удерживания методом
метки. Выводят хроматограф с установленным рефрактометри-
ческим детектором на рабочий режим. Включают прибор в сеть
и на блоке питания и управления нажимают клавишу ON.
Подготовленный элюент через специальный фильтр и тефло-
новую трубку нажатием клавиши FILL подают в резервуар на-
соса при открытом верхнем переднем вентиле. Первоначаль-
ное заполнение резервуара ~20-30 мл. Периодическая прокачка
этой порции элюента в обратном направлении (нажатие кла-
виши EMPTY) при открытом верхнем заднем вентиле (передний
при этом закрыт) через выходной металлический капилляр на
задней панели прибора способствует избежанию попадания пу-
зырьков воздуха в резервуар насоса. Закачивают в резервуар
насоса дополнительное количество подвижной фазы (ее общий
объем должен составлять 80-100 мл). Оба вентиля закрывают
и нажимают клавишу START (подвижная фаза через соедини-
тельный стальной капилляр начинает поступать в хроматогра-
фическую колонку).
Далее приступают к заполнению кюветы сравнения детек-
тора, что может быть выполнено либо путем последовательного
соединения с рабочей кюветой, либо с помощью шприца. Когда
пузырьки воздуха в соединительных капиллярных трубках пол-
ностью изчезнут, оба стальных капилляра кюветы сравнения
соединяют тефлоновой трубкой, а элюент вместе с разделяе-
мыми компонентами поступает в рабочую кювету и далее на
слив (скорость подачи фазы устанавливают равной 0,3 мл/мин).
Включают электропитание интегратора (нажимают клавишу
ON на передней панели прибора). После прогрева электронной
схемы загорается светодиод STANDBY (режим ожидания после-
дующих команд). Нажимают клавишу St, тем самым перево-
дят прибор в режим MONITOR (загорается одноименный свето-
диод) для прослеживания и запоминания интегратором уровня
шумов и последующей коррекции нулевой линии. О готовности
интегратора к обработке сигналов детектора свидетельствуют
звуковой сигнал и загорание светодиода INJECT.
Теперь можно приступать к выполнению хроматографиче-
ских анализов. Включают самописец, задают шкалу чувстви-
тельности регистрации фонового сигнала, равную 16 мВ, и до-
жидаются регистрации устойчивой (без дрейфа) нулевой ли-
557
нии. Не прерывая потока элюента, вводят в узел дозирования
пробу смеси А органических соединений известного состава и
сразу же нажимают клавишу INJECT на лицевой панели инте-
гратора. По завершении интегрирования сигнала детектора на
последний (пятый) из ожидаемых компонентов нажимают кла-
вишу RES, отдавая тем самым команду на распечатку отчета
с результатами выполненного анализа. На термографическую
бумагу печатающего устройства выводятся времена удержива-
ния, площади хроматографических пиков и содержание в про-
анализированной смеси каждого вещества (в процентах), рас-
считываемое интегратором нормализацией площадей зареги-
стрированных пиков, без учета относительной чувствительно-
сти детектора к компонентам смеси. Если на хроматограмме
выявятся зашкаленные или слишком малые пики, при повтор-
ных анализах изменяют соответствующим образом шкалу ре-
гистрации сигнала детектора и, при необходимости, величину
дозы, которую в дальнейших опытах сохраняют неизменной.
Записав две-три воспроизводимых (по времени удерживания
и по площадям пиков) хроматограммы смеси А, переходят к по-
следовательному хроматографированию в тех же условиях гра-
дуировочных растворов Б — Ес целью отождествления пиков
всех компонентов смеси А. Расхождение во временах удержива-
ния отдельных соединений на хроматограммах смеси А и хро-
матограммах градуировочных растворов Б-Е обычно не превы-
шает 10% отн., что обеспечивает идентификацию пиков соеди-
нений, образующих смесь А. В сомнительных случаях запра-
шивают у преподавателя или лаборанта штатив с чистыми и
сухими микропробирками, в которых с помощью медицинских
пипеток смешивают равные (по числу капель) порции анали-
зируемой смеси А и соответствующего градуировочного рас-
твора. Увеличение площади какого-либо из пиков на хромато-
граммах смешанной пробы служит дополнительным критерием
правильности идентификации, выполняемой по совпадению вре-
мен удерживания компонентов смеси А и градуировочных рас-
творов при их раздельном хроматографировании.
Наконец, в тех же самых условиях анализа записывают две-
три воспроизводимых хроматограммы каждого контрольного
образца.
Выявление возможности выполнения анализа смеси А с
УФ-детектированием (А = 254 нм). По завершении этапов ра-
боты, изложенных выше, заменяют под наблюдением препода-
вателя или лаборанта универсальный рефрактометрический де-
тектор на селективный ультрафиолетовый и 2-3 раза хромато-
графируют в принятых ранее условиях исходную пятикомпо-
558
нентную смесь, а затем и контрольный образец, регистрируя в
каждом опыте с помощью интегратора времена удерживания и
площади всех пиков (если в предварительных опытах с новым
детектором на хроматограммах запишутся зашкаленные или,
наоборот, слишком малые пики, изменяют шкалу регистрации
сигнала, сохранив прежней величину дозы).
Обработка результатов
Измеренные в каждом из выполненных опытов “текущие” вре-
мена удерживания и площади пиков и их усредненные числен-
ные значения заносят в таблицу (табл. VI. 1), в которую вписы-
вают также рассчитываемые по уравнению (IV.43) величины от-
носительного отклика рефрактометрического и ультрафиолето-
вого детекторов на компоненты смеси А.
Таблица VI.1. Экспериментальные
данные и результаты идентификации
№ хромато- граммы № пика Время удержи- вания Площадь пика (I)*, мкВ -с Площадь пика (II)** ***, мкВ-с Относи- тельный отклик Я”’ Название соедине- ния
теку- щее сред- нее теку- щее сред- нее теку- щее сред- нее
Искусственная смесь А Градуировочный раствор Б Градуировочный раствор В Градуировочный раствор Г Градуировочный раствор Д Бензол .м-Ксилол п-Ксилол Додекан
Фенил-
ацетилен
Градуировочный раствор Е
Контрольный образец 1
* По показаниям рефрактометрического детектора (I).
** По показаниям УФ-детектора (II).
*** Отношение площадей пиков II/I.
Для выполнения количественной оценки эффективности ра-
боты использованной двухсекционной колонки рассчитывают
559
число теоретических тарелок N по каждому компоненту, заре-
гистрированному на хроматограммах по показаниям либо ре-
фрактометрического, либо ультрафиолетового детектора (по
согласованию с преподавателем или лаборантом). Расчет вы-
полняют по известной формуле
N = 5,54(ir/w0,5)2,
где tr — измеряемое интегратором полное (неисправленное)
время удерживания; и?о,5 — измеряемая по линейке вручную
(с точностью до ±0,5 мм) ширина пика на половине высоты.
При оформлении отчета по данной работе следует исходя из
полученных данных обсудить установленный порядок элюиро-
вания компонентов анализируемой смеси с учетом их химиче-
ской природы и режима разделения (нормально-фазная хрома-
тография), а также ответить на вопросы.
1. Какие факторы влияют на эффективность работы колонки
жидкостного хроматографа и как можно достигнуть ее повыше-
ния?
2. Чем объясняется меньшее количество пиков на хромато-
граммах смеси А при использовании ультрафиолетового детек-
тора? Можно ли было предвидеть это обстоятельство a priori?
3. Каковы причины неодинакового относительного отклика
рефрактометрического и ультрафиолетового детекторов на за-
регистрированные каждым из них компоненты смеси А? Какой
детектор обладает большей чувствительностью к зарегистри-
рованным компонентам?
Работа 17. Разделение смеси органических
соединений с различными молекулярными
массами методом эксклюзионной хроматографии
Цель работы. Ознакомление с методом эксклюзионной хро-
матографии и с новыми методами детектирования с использо-
ванием химических реагентов. Разделение полиэтиленглико-
лей с молекулярными массами 20000 и 1000 и диэтиленгликоля
этим методом; расчет коэффициента распределения для диэти-
ленгликоля.
В качестве стационарной фазы в эксклюзионной (гель) хро-
матографии применяются природные и синтетические пористые
материалы со свойствами молекулярных сит. К таким матери-
алам относятся, например, декстрановые гели (коммерческие
названия “сефадекс” и “молселект”), полиакриловые гели, био-
гели.
560
Разделение осуществляется за счет различного распределе-
ния веществ между стационарной (частицы набухшего геля) и
подвижной (элюент) фазами. Молекулы, размеры которых пре-
вышают величину пор набухшего геля, не могут в эти поры про-
никнуть и элюируются первыми в свободном объеме колонки.
Молекулы меньших размеров, которым поры геля полностью
или частично доступны, выходят позднее.
Таким образом, порядок элюирования определяется молеку-
лярными массами — от больших к меньшим.
Оборудование, реактивы, условия
и объекты хроматографирования
Стеклянная колонка (60x3 см), заполненная набухшим гелем (сефадекс
G-50).
Элюент — дистиллированная вода.
Коллектор фракций с чистыми пробирками.
Хроматографические пластинки силуфол с закрепленным слоем сили-
кагеля.
Подвижная фаза для ТСХ — хлороформ, ©танол и ацетонитрил в со-
отношении 4:2:1 соответственно (по объему).
Стеклянные капилляры.
Калиброванный шприц на 1 мкл для нанесения анализируемых образ-
цов в режиме ТСХ.
Пульверизатор.
Нагревательный столик.
Смесь для детектирования: 2,25 г уксусной кислоты (ч.д.а.), 1,25 г три-
хлоруксусной кислоты (ч.д.а.), 0,5 г диметиланилина (х.ч.) и 23 г дистил-
лированной воды.
Объект хроматографирования — смесь полиэтиленгликолей.
Выполнение работы
Стеклянную колонку диаметром ~3 см и высотой 60 см закре-
пляют вертикально на штативе. В узкую часть помещают там-
пон из ваты и надевают резиновый шланг с заглушкой. Приго-
товленную таким образом колонку заполняют на 1 /3 дистилли-
рованной водой. Предварительно приготовленную суспензию
набухшего геля (35-40 г сефадекса G-50 в 500-600 мл дистилли-
рованной воды) переносят в подготовленную колонку. В тече-
ние 3-4 ч через колонку пропускают элюент (дистиллированная
вода) без внесения анализируемой смеси.
При дозировании исследуемого объекта важно не нарушить
слой геля. Для этого колонку закрывают заглушкой и избы-
ток растворителя над слоем геля удаляют пипеткой, оставив
над поверхностью столбик растворителя высотой ~1 мм. За-
тем осторожно, по стенкам колонки, вносят 300 мг смеси поли-
этиленгликолей с молекулярными массами 20 000, 1000 и диэти-
ленгликоля, растворенных в 1,5 мл воды. Снимают заглушку
561
и дают раствору впитаться в гель, вновь закрывают колонку и
пропитывают верхний слой геля дистиллированной водой (1,5
мл). После этого колонку наполняют растворителем (дистилли-
рованная вода) и последовательно отбирают фракции. Первую
фракцию объемом около 90 мл отбирают отдельно в цилиндр,
последующие 25-30 фракций объемом около 10 мл собирают в
пробирки.
Из каждой фракции, содержащей полимеры и димер, вносят
по 5-10 мкл на пластинку силуфол, куда одновременно поме-
щают и стандартные вещества. Подготовленные пластины опус-
кают в проявительную камеру, содержащую подвижную фазу
следующего состава: хлороформ, этанол и ацетонитрил в соот-
ношении 4 : 2 : 1. Высушенные после проявления пластины
опрыскивают из пульверизатора смесью для детектирования
(см. выше) в вытяжном шкафу и нагревают на нагревательном
столике. Полиэтиленгликоли проявляются в виде белых пятен
на синем фоне, димер — в виде белого кольца.
Обработка результатов
Используя экспериментальные данные, рассчитывают коэф-
фициент распределения К диэтиленгликоля по формуле
К = (Vi - V0)/(Vg - Vo),
где Vo — свободный объем колонки, Vi — объем удерживания
диэтиленгликоля, V& — объем колонки, заполненной гелем. Сво-
бодный объем колонки определяется объемом удерживания по-
лиэтиленгликоля с молекулярной массой 20000.
При оформлении рабочего протокола необходимо описать
ход работы и ответить на следующие вопросы.
1. Почему соединение с наибольшей молекулярной массой
элюируется первым?
2. Какие значения может принимать коэффициент распреде-
ления в эксклюзионной хроматографии?
Работа 18. Использование ионообменных
смол для обессоливания
Цель работы. Ознакомление с методом колоночной ионооб-
менной хроматографии и с одним из приемов регенерации коло-
нок. Отделение аминокислоты от неорганических солей.
Для обессоливания высокомолекулярных соединений в на-
стоящее время успешно применяют гельхроматографию. Од-
нако при обессоливании низкомолекулярных соединений этот
562
метод может оказаться неэффективным из-за совпадения вре-
мен удерживания органических соединений и неорганических
солей. В этом случае для обессоливания применяют ионооб-
менную хроматографию, причем подбирают условия разделе-
ния таким образом, чтобы органическое соединение задержива-
лось на стационарной фазе. Так, например, при обессоливании
аминокислот используют в качестве стационарной фазы кати-
онит в Н+-форме. Тогда на ней происходит протонирование
аминогруппы, что обеспечивает взаимодействие аминокислоты
со стационарной фазой по реакции
RSO3H+ + RCH(NH2)COOH -» RSO3NH3CHCOOH.
R
Соли, например фосфаты, хлориды, элюируются из колонки
в первую очередт (подвижная фаза — вода). Для выделения из
колонки аминокислоты используют 1 М раствор аммиака. Ами-
нокислоты (бифункциональные соединения) могут быть очи-
щены от минеральных солей и на анионите в ОН-форме, в каче-
стве подвижных фаз в этом случае применяют последовательно
дистиллированную воду и 1 М раствор уксусной кислоты.
Оборудование, реактивы, объекты хроматографирования
Стеклянная колонка (14x2 см), заполненная катионитом КРС-2.
Хроматографическая бумага.
Нагревательный столик.
Стеклянные капилляры.
1 М раствор водного аммиака.
Дистиллированная вода.
1%-ный раствор молибдата аммония.
1%-ный раствор хлорида олова (II).
10%-ный раствор соляной кислоты.
0,2%-ный спиртовой раствор нингидрина.
Объект хроматографирования — смесь аланина, хлорида и фосфата
натрия (~100 мг).
Выполнение работы
Смесь, состоящую из 20 мг а-аланина, 35-40 мг хлорида на-
трия и 30-35 мг фосфата натрия, растворяют в 2 мл дистиллиро-
ванной воды и вносят в верхнюю часть колонки, заполненную
катионитом КРС-2 в Н+-форме. Смола КРС-2 является силь-
нокислотным катионитом, полученным на основе полистирола,
активная группа — SO3H. Смесь вносят при закрытой колонке,
затем колонку открывают и дают раствору впитаться в смолу,
вновь закрывают колонку и вносят 5-8 мл дистиллированной
563
воды для предохранения слоя смолы от перемешивания при по-
даче на колонку подвижной фазы — системы растворителей.
В качестве подвижной фазы используют последовательно
дистиллированную воду и 1 М раствор аммиака. Отбирают
фракции по 4-5 мл. Очень быстро (через 3-4 фракции) начи-
нают выходить сначала соляная кислота (качественная реакция
на ионы хлора), затем фосфорная кислота (качественная реак-
ция на фосфат-ионы).
Во фракции последовательно добавляют 5 капель 1%-ного
водного раствора (NH4 ЯЙМоС^ и по 3 капли 1%-ного раствора
SnCh и 10%-ной НС1. После того, как pH выходящего элюента
станет равным 7 (проверку осуществляют с использованием ин-
дикаторной бумаги), производят замену элюента на 1 М рас-
твор аммиака для разрушения комплекса аминокислоты с ионо-
обменной смолой и элюирования <т-аланина.
Для детектирования аминокислоты используют 0,2%-ный
раствор нингидрина в этаноле. Вышедшие из колонки фрак-
ции собирают по 5 мл в пробирки и проверяют наличие ами-
нокислоты. Для этого на полоску хроматографической бумаги
капилляром (или микрошприцем) вносят пробы из каждой про-
бирки и после подсушивания на нагревательном столике хрома-
тографической бумаги с нанесенными анализируемыми компо-
нентами добавляют с помощью стеклянной пипетки в каждую
из стартовых точек спиртовый раствор нингидрина (по 1-2 ка-
пле). После нагревания полоски бумаги с образцами в течение
1-3 мин на нагревательном столике аминокислота проявляется
в виде розово-фиолетовых пятен.
По окончании работы колонку с катионитом необходимо ре-
генерировать — вернуть в Н+-форму. Для этого сначала ее
промывают дистиллированной водой, а потом раствором соля-
ной кислоты.
Обработка результатов
При оформлении протокола надо указать основные этапы ра-
боты, записать качественную реакцию с нингидрином на пер-
вичную аминогруппу и ответить на следующие вопросы.
1. Удастся ли решить данную задачу, используя метод экс-
клюзионной жидкостной хроматографии?
2. Можно ли провести обессоливание аминокислоты, исполь-
зуя в качестве неподвижной фазы не катионит, а анионит?
564
Работа 19. Разделение и количественный
анализ смеси органических соединений
при использовании обращенно-фазового
варианта ВЭЖХ
Цель работы. Ознакомление с порядком элюирования со-
единений, принадлежащих к классам фенолов, ароматических
и алифатических углеводородов, в режиме обращенно-фазовой
хроматографии. Разделение смеси модельных соединений и
проведение количественного анализа методом внутреннего стан-
дарта.
В ВЭЖХ примерно 60-70% всех аналитических разделений
осуществляется методом обращенно-фазовой хроматографии
(ОФХ). Работа в режиме ОФХ характеризуется использова-
нием неполярного адсорбента и полярного элюента. Удержи-
вание разделяемых веществ преимущественно определяется не-
специфическими (вещество — адсорбент) и специфическими (ве-
щество — элюент) взаимодействиями.
Применение химически связанных неподвижных фаз обеспе-
чивает постоянство их состава, повышает воспроизводимость
хроматографического анализа, исключает потери фазы. По-
движные фазы (ацетонитрил, вода, метанол), используемые в
ОФХ, позволяют проводить детектирование в широком УФ-
диапазоне, относительно легко растворяют практически все
важнейшие соединения, входящие в состав биологических объ-
ектов, лекарственных препаратов, гербицидов и т. д.
Порядок выхода соединений в режиме ОФХ соответствует
обращению элюотропного ряда для адсорбционной хромато-
графии. Наиболее сильные элюенты в системе ОФХ — алифа-
тические углеводороды. Удерживание компонентов пробы уве-
личивается с уменьшением их полярности. Соединения с раз-
ветвленным углеродным скелетом всегда элюируются раньше,
чем с неразветвленной углеродной цепью.
Оборудование, реактивы, условия
и объекты хроматографирования
Жидкостный хроматограф (производства ЧССР) с рефрактометриче-
ским детектором, укомплектованный считающим интегратором.
Стеклянная колонка (150x3 мм), заполненная неподвижной фазой сепа-
рон CGX-C18; температура колонки комнатная.
565
Подвижная фаза — метанол (фирмы Мегк), содержащий 0,1% гептана
(квалификации “для хроматографии”)*; скорость подачи элюента
0,2 мл/мин.
Шкала регистрации сигнала детектора (при записи хроматограммы
самописцем) в диапазоне от 128 до 16.
Скорость движения диаграммной ленты 0,3 см/мин.
Объекты хроматографирования. Искусственные градуировочные сме-
си известного качественного и количественного состава: 1) раствор фе-
нола (х.ч.), бензола, к-октана, кумола, псевдокумола (все вещества квали-
фикации “для хроматографии”) в элюенте; 2) раствор фенола в элюенте;
3) раствор фенола и н-октана в элюенте; 4) раствор фенола, н-октана и
кумола в элюенте; 5) раствор фенола, н-октана, кумола и псевдокумола
в элюенте. Контрольный раствор в элюенте фенола, бензола, н-октана,
кумола, псевдокумола неизвестного студентам количественного состава.
Дозирование осуществляется микрошприцем МШ-10, величина дозы
1—1,5 мкл.
Выполнение работы
Получение информации, необходимой для идентифика-
ции пиков на хроматограммах и расчета градуировочных
множителей. Включают хроматограф и интегратор и выво-
дят их на рабочие режимы. Убедившись в записи устойчивой
(без дрейфа) нулевой линии при наивысшей в рекомендован-
ном интервале шкал чувствительности регистрации фонового
сигнала, не прерывая поток элюента, вводят в узел дозирова-
ния порцию градуировочного раствора 1 и записывают первую
пробную хроматограмму. При обнаружении зашкаленных или
слишком малых пиков при повторных анализах изменяют соот-
ветствующим образом величину дозы или масштаб шкалы реги-
страции зон отдельных компонентов. В предварительных опы-
тах оптимизируют и режим работы интегратора, обращая, в
первую очередь, внимание на такой параметр, как полуширина
самого первого (наиболее узкого) пика. Важно, чтобы интегри-
рование каждого пика начиналось при однотипном отклонении
пера самописца от уровня фонового сигнала и заканчивалось
при соответствующем приближении к нему. В уточненных усло-
виях записывают одну-две (или большее количество, по согла-
сованию с преподавателем) пробных и от трех до шести рабочих
хроматограмм градуировочного раствора 1, пики на которых по
высоте должны быть не менее 30 и не более 90% ширины диа-
граммной ленты.
Результаты обработки интегратором поступившей хромато-
графической информации выводятся на печать: времена удер-
*Дегазацию подвижной фазы проводят в вакууме водоструйного на-
соса при перемешивании магнитной мешалкой в течение 15 мин непосред-
ственно перед выполнением хроматографических анализов.
566
живания, площади хроматографических пиков и процентный со-
став анализируемой смеси без учета коффициентов относитель-
ной чувствительности детектора. Все эти сведения вносят в
таблицу вида табл. VI.2 в порядке возрастания времен удержи-
вания зарегистрированных компонентов.
Таблица VI.2. Экспериментальные данные,
необходимые для идентификации компонентов смеси:
неизвестных соединений методом эталонирования и
количественной интерпретации результатов
эксперимента методом внутренней нормализации
без учета градуировочных множителей
Компоненты градуи- Время удерживания, Площади пиков, Содержание,
ровочных растворов мин мкВ • с % (по массе)
Градуировочный раствор 1
1
2
3
Градуировочный раствор 2
Фенол | |
Градуировочный раствор 3
Фенол
н-Октан
И т.д.
Данные, необходимые для идентификации каждого пика и
расчета градуировочных множителей, учитывающих неодина-
ковую чувствительность рефрактометрического детектора к
компонентам исследуемых смесей в принятых условиях анализа,
получают, последовательно хроматографируя названные выше
искусственные градуировочные растворы (не менее двух раз ка-
ждый). Результаты измерения интегратором времен удержива-
ния и площадей пиков известных компонентов вписывают в таб-
лицу, близкую по форме к табл. V.13.
Количественный анализ контрольной смеси методом
внутреннего стандарта. Общая схема выполнения анализа со-
ответствует ранее рассмотренной последовательности стадий
газохроматографического анализа методом внутреннего стан-
дарта (см. работу 8).
Выбор внутреннего стандарта. Получив от лаборанта или
преподавателя контрольный образец неизвестного качествен-
ного и количественного состава, хроматографируют его в при-
567
нятых ранее условиях работы прибора с целью идентификации
(по временам удерживания) входящих в него компонентов и вы-
явления отсутствующих (из числа внесенных в табл. VI.2, обра-
зующих смесь 1 соединений), по которым проводилась граду-
ировка хроматографа. Любое из этих отсутствующих в кон-
трольном образце (по сравнению со смесью 1) веществ может
быть использовано в качестве внутреннего стандарта. Для ре-
шения этой частной задачи достаточно записать две воспроиз-
водимые хроматограммы.
Подготовка пробы к анализу В специальный сосуд (жела-
тельно с узкой шейкой, типа пикнометра) или стандартный ме-
дицинский флакон емкостью от 2 до 5 мл, закрываемый проб-
кой из самозатягивающейся силиконовой резины, на аналитиче-
ских весах, пользуясь пипетками или медицинскими шприцами с
длинными и широкими иглами, отбирают навеску контрольной
смеси (около 1 г) и вносят в нее навеску соединения, выбран-
ного в качестве внутреннего стандарта (~200 мг). Содержимое
закрытого пробкой сосуда аккуратно и тщательно перемеши-
вают.
Выполнение анализов контрольного образца со стандартом.
Записывают сначала одну-две пробные хроматограммы кон-
трольного образца с внесенным стандартом, корректируют, ес-
ли потребуется, величину дозы или чувствительность регистра-
ции отдельных компонентов, а затем регистрируют еще три-
пять воспроизводимых хроматограмм, пригодных для последу-
ющей количественной расшифровки.
Обработка результатов
Расчет численных значений градуировочных множителей fi
проводят по формуле (IV.43), используя в качестве параметров
Pi площади пиков, измеренные интегратором. Найденные по ре-
зультатам всех градуировочных смесей величины fi предста-
вляют в табличной форме (см., например, табл.V. 14). Полу-
ченный массив данных подвергают метрологической обработке
согласно рекомендациям, приведенным в работе 8.
Рассчитанные по формуле (IV.44) без учета и с учетом экс-
периментально найденных градуировочных множителей концен-
трации компонентов контрольного образца представляют в виде
таблицы, близкой по форме к табл. V.16. В эту же таблицу вно-
сят результаты метрологической оценки выполненных анализов
(см. раздел IV.6 и описание работы 8).
При оформлении работы следует обсудить установленный
порядок элюирования идентифицированных компонентов с уче-
том их химической природы и условий хроматографического
568
анализа (обращенно-фазная хроматография) и ответить на сле-
дующие вопросы.
1. Как изменился бы вид хроматограммы, если бы вместо ре-
фрактометрического детектора использовался ультрафиолето-
вый?
2. Где на хроматограмме мог бы оказаться пик, принадлежа-
щий ундекану?
3. Чем вызвана неодинаковая чувствительность рефрактоме-
трического детектора к компонентам градуировочных смесей?
4. Какие из стадий выполненного количественного анализа в
наибольшей степени влияют на точность получаемых результа-
тов?
Работа 20. Определение содержания
2,4-динитрофенилгидразонов формальдегида
и ацетальдегида в растворах ацетонитрила
методом абсолютной градуировки с
использованием обращенно-фазной ВЭЖХ
Цель работы. Построение градуировочных зависимостей
между содержанием 2,4-динитрофенилгидразонов (2,4-ДНФГ)
формальдегида и ацетальдегида в искусственно приготовлен-
ных растворах в ацетонитриле и количественными параметрами
(площадями и/или высотами) соответствующих хроматографи-
ческих пиков, регистрируемых по показаниям УФ-детектора.
Определение содержания 2,4-ДНФГ формальдегида и ацеталь-
дегида в контрольных ацетонитрильных растворах методом аб-
солютной градуировки хроматографа по высотам и/или площа-
дям пиков.
Все возрастающее загрязнение объектов окружающей среды
диктует острую необходимость непрерывного совершенствова-
ния существующих и создания новых высокочувствительных и
надежных методов аналитического контроля качества воздуха,
воды и почвы Одними из приоритетных загрязняющих веществ
для атмосферного воздуха, воздуха производственных помеще-
ний, водных объектов являются низшие алифатические альде-
гиды С1-С5, из которых наибольшую опасность представляют
формальдегид, в меньшей степени — ацетальдегид и другие аль-
дегиды с большим числом атомов углерода (Сз-Сб). Наиболее
распространенный метод определения следовых количеств низ-
ших карбонильных соединений в объектах окружающей среды
569
заключается в их хемосорбционном концентрировании с обра-
зованием соответствующих 2,4-динитрофенилгидразонов с по-
следующим определением этих соединений методом высокоэф-
фективной жидкостной хроматографии [434].
В излагаемой ниже лабораторной работе иллюстрируется
возможность количественного определения 2,4-ДНФГ формаль-
дегида и ацетальдегида в растворах ацетонитрила (обычно ис-
пользуемого для экстрагирования этих гидразонов с поверхно-
сти хемосорбента) методом абсолютной градуировки жидкост-
ного хроматографа.
Аппаратура, условия, объекты хроматографирования
Жидкостный хроматограф с ультрафиолетовым детектором (365 нм),
укомплектованный электронным интегратором или программно-аппарат-
ным комплексом “МультиХром”. Шкала регистрации сигнала детектора
при записи хроматограммы самописцем (множитель шкалы) в диапазоне
от 16 до 128 мВ. Скорость диаграммной бумаги 400-600 мм/ч.
Стальная колонка, 150 x3 мм, заполненная неподвижной фазой сепарон
С-18 зернения 7 мкм.
Подвижная фаза — бинарная система ацетонитрил/вода (65 : 35 по
объему), расход 0,6 мл/мин.
Объекты хроматографирования. 1. Четыре-пять градуировочных рас-
творов 2,4-ДНФГ формальдегида и 2,4-ДНФГ ацетальдегида в ацетони-
триле с известным содержанием каждого из них. 2. Один-два контроль-
ных ацетонитрильных раствора названных гидразонов.
Дозирование осуществляется микрошприцем МШ-50или “Гамильтон”
с рабочим объемом 25 мкл, доза 20 мкл.
Выполнение работы
Абсолютная градуировка хроматографа. Градуировку хро-
матографа выполняют по искусственно приготовленным аце-
тонитрильным растворам с концентрациями 2,4-ДНФГ форм-
альдегида в диапазоне от 1 до 20 мг/л и 2,4-ДНФГ ацеталь-
дегида — от 0,3 до 3 мг/л соответственно*. В процессе граду-
ировки хроматографа необходимо получить минимум 4-5 экс-
периментальных точек, накрывающих названные диапазоны со-
держаний обоих гидразонов в ацетонитрильных растворах.
Работу начинают с подготовки подвижной фазы. Смешан-
ные в указанном объемном соотношении ацетонитрил (квалифи-
кации “для спектроскопии”) (65 мл) и дистиллированную воду
(35 мл) помещают в толстостенную однотубусную (со шлифом
29-го размера) кругло донную колбу на 250 мл, опускают в колбу
*Указанные градуировочные растворы трижды перекристаллизован-
ных 2,4-ДНФГ формальдегида и ацетальдегида приготавливаются лабо-
рантом заранее и хранятся в плотно закрытой таре в холодильнике.
570
остеклованный магнитик, устанавливают колбу на столик маг-
нитной мешалки и соединяют тубус колбы с водоструйным на-
сосом. Включают мотор магнитной мешалки и аккуратно со-
здают вакуум (не менее 100 мм рт. ст.), наблюдая вначале ак-
тивное, а затем затухающее, вплоть до полного прекращения,
удаление микропузырьков растворенного в подвижной фазе воз-
духа. Процесс дегазации подвижной фазы обычно требует 20-
30 мин, по его окончании аккуратно соединяют систему с атмо-
сферой (снимают вакуум), закрывают водоструйный насос и за-
гружают подвижную фазу в резервуар насоса. Включают при-
бор, а также интегратор или программно-аппаратный комплекс
типа “МультиХром” и выводят их на рабочий режим.
Убедившись в записи устойчивой (без дрейфа) нулевой ли-
нии при наивысшей чувствительности регистрации фонового
сигнала, записывают пробную хроматограмму первого (наибо-
лее разбавленного) из подготовленных градуировочных раство-
ров. Этот и все последующие растворы (в порядке увеличе-
ния концентрации 2,4-ДНФГ формальдегида и ацетальдегида
в каждом из них) хроматографируют несколько раз (не менее
четырех-шести), изменяя, если потребуется, чувствительность
регистрации сигнала детектора так, чтобы на хроматограммах
каждого раствора записывались пики с высотой в пределах 50-
90% шкалы регистратора.
Внимание! Величина дозы градуировочных растворов
должна сохраняться постоянной как при выполнении гра-
дуировки, так и при последующих анализах контрольных
образцов!
Всякий раз немедленно после дозирования в узел ввода пор-
ции градуировочного раствора (начиная с первого анализа)
включают интегратор и по завершении интегрирования 2,4-
ДНФГ ацетальдегида (второй пик на хроматограммах, образец
которых представлен на рис. VI.1) распечатывают результаты.
Численные значения измеренных площадей (или высот) пиков
заносят в заранее подготовленную в рабочей тетради таблицу
(см. табл. VI.3).
При использовании для количественной интерпретации хро-
матограмм программно-аппаратного комплекса типа “Мульти-
Хром” в распечатке результатов интегрирования по окончании
каждого отдельного анализа нет необходимости — они оста-
ются в памяти ЭВМ; в программном обеспечении подобных ком-
плексов предусматривается возможность итоговой обработки
полного массива накопленной информации по завершении вы-
полнения всех намеченных анализов (см. раздел IV.5).
571
1
Рис. VI.1. Образец хроматограм-
мы одного из градуировочных аце-
тонитрильных растворов.
1 — 2,4-ДНФГ формальдегида; 2 —
ацетал ьдегида.
Анализ контрольных образцов. Записывают пробную хрома-
тограмму первого из них, подбирая при повторных анализах
чувствительность регистрации (см. выше), обеспечивающую
амплитуду сигналов детектора в диапазоне 50-90% шкалы ре-
гистратора*.
Получив несколько (не менее четырех-шести) воспроизводи-
мых хроматограмм с распечаткой результатов каждого анализа
(при использовании интегратора), приступают к хроматографи-
рованию второго контрольного образца по аналогичной схеме,
после чего выключают хроматограф и приступают к обработке
результатов.
*Контрольные образцы могут содержать 2,4-ДНФГ формальдегида и
ацетальдегида в резко различных концентрациях, о чем студенты могут
быть информированы заранее. В этом случае целесообразно переключать
чувствительность регистрации сигналов детектора (множителя шкалы)
непосредственно в ходе записи хроматограммы.
572
Таблица VI.3. Экспериментальные данные, необходимые для построения зависимостей отклика УФ-детектора
(365 им) на различные содержания 2,4-ДНФГ формальдегида и ацетальдегида в градуировочных растворах
градуи- ровоч- ного раствора Заданные концентрации 2,4-ДНФГ, Cf, мг/л Количественные параметры пиков 2,4-ДНФГ, приведенные к единой шкале чувствительности
Площадь пиков Высота пиков
формальдегида ацетальдегида формальдегида ацетальдегида
формальдегида ацетальдегида текущее среднее текущее среднее текущее среднее текущее среднее
1 2
Обработка результатов
Обработку результатов начинают с построения градуиро-
вочных зависимостей вида
Ai(hi) =
где Ai(hi) — площади или соответственно высоты пиков 2,4-
ДНФГ формальдегида и ацетальдегидана хроматограммах (из-
меренные интегратором или вручную, по согласованию с препо-
давателем); С, —концентрации 2,4-ДНФГ формальдегида и/или
ацетальдегида в градуировочных растворах.
На лист миллиметровой бумаги стандартного размера А4
наносят прямоугольные координаты и, подобрав удобный мас-
штаб, откладывают по оси абсцисс заданные концентрации ги-
дразонов, а по оси ординат отвечающие им, внесенные в
табл. VI.3, усредненные величины площадей (или высот, по со-
гласованию с преподавателем) соответствующих хроматогра-
фических пиков*. Перед усреднением найденных в отдельных
анализах “текущих” значений А, и hi сомнительные (выпадаю-
щие из ряда) значения этих параметров отбрасывают, пользу-
ясь критическим параметром ткр (для уровня значимости /3 =
0,01) [213, с. 100] или Q-тестом [425].
По построенному графику определяют концентрации 2,4—
ДНФГ формальдегида и/или ацетальдегида в проанализиро-
ванных контрольных образцах. Сравнивают найденное содер-
жание гидразонов с заданным (навести справку у лаборанта или
преподавателя) и характеризуют свои результаты абсолютной
и относительной погрешностями. Студентам может быть также
предложено провести более подробную метрологическую атте-
стацию экспериментальных данных (см. раздел IV.6).
Окончательно результаты анализов контрольных образцов
представляют в виде таблицы, по форме близкой к рекомендо-
ванной в работе 12.
При оформлении протокола ответьте на следующие вопросы.
1. Чем обусловлена высокая чувствительность У Ф-детектора
к 2,4-динитрофенилгидразонам?
2. Какова сравнительная чувствительность УФ-детектора к
2,4-ДНФГ формальдегида и ацетальдегида?
3. Какие условия необходимо соблюдать при выполнении ко-
личественного анализа методом абсолютной градуировки?
*В программном обеспечении “МультиХром” предусмотрена иная на-
грузка по осям координат: по оси X автоматически откладываются пло-
щади пиков (или их высоты — по команде пользователя), а по оси Y — за-
данные концентрации соединений, по которым выполняется градуировка
(в данном случае — гидразонов), в градуировочных растворах.
574
ПРИЛОЖЕНИЕ
ПРОГРАММЫ ДЛЯ ОБРАБОТКИ
ЭКСПЕРИМЕНТАЛЬНЫХ ДАННЫХ И РАСЧЕТА
ХРОМАТОГРАФИЧЕСКИХ ПАРАМЕТРОВ
Приведенные ниже программы на языке QBasic могут быть
использованы на всех современных компьютерах в операцион-
ной системе MS-DOS и предназначены для выполнения про-
стейших статистических расчетов (вычисления средних зна-
чений и стандартных отклонений, параметров линейных ре-
грессионных уравнений), определения индексов удерживания и
обработки данных количественного газохроматографического
анализа методами абсолютной градуировки, внутреннего стан-
дарта и стандартной добавки.
В настоящее время программные средства для различных
статистических расчетов легко доступны, так как включены,
например, в некоторые приложения Windows (Excel, Origin и
др.). Известны многочисленные разновидности готовых про-
грамм для обработки хроматографических данных с помощью
как ЭВМ, так и программируемых интеграторов. В связи с
этим следует отметить, что целесообразность приведения здесь
простейших программ на языке QBasic, не относящемся к языкам
высокого уровня, обусловлена исключительно потребностями
практики хроматографического анализа. Многообразие реаль-
ных аналитических задач не может быть адекватно отражено в
рамках “жестко” фиксированных раз и навсегда алгоритмов и
программ. Достаточно часто возникает необходимость в моди-
фикации способов обработки данных в зависимости от условий
получения и характера исходной информации или совершен-
ствования методов ее обработки. По этой причине всегда жела-
тельно сохранить возможность внесения изменений в программ-
ное обеспечение непосредственно его пользователями, для чего
программы, написанные на различных вариантах Bas с, оста-
ются вне конкуренции из-за их наглядности, легкости восприя-
тия, простоты интерпретации и внесения желаемых дополнений.
С полным перечнем программ для обработки хроматографи-
ческой информации, а также с новыми версиями существующих,
часть из которых приведена ниже, можно ознакомиться, обра-
тившись в лабораторию газовой хроматографии НИИ Химии
СПбГУ.
575
Все программы, по возможности, содержат непосредственные
указания на ввод требуемой для расчетов исходной информа-
ции, вследствие чего их детальное описание является излишним.
Приведена только краткая характеристика их особенностей и
в озм ожно стей.
Программа 1 (1-DIM STAT) предназначена для расчета сред-
неарифметических значений и стандартных отклонений, но до-
полнительно включает контроль выборок данных на присут-
ствие явно ошибочных значений по критерию “3<т”.
REM: PROGRAM 1-DIM STAT
REM: program for statistical processing of single-dimensional data
REM: calculation of average values and standard deviations with 3S-test
PRINT : PRINT ’’INPUT INITIAL VALUES”;
PRINT ’’NOTHING OR ZERO - THE SYMBOL OF THE END”
DIM 1(200): S = 0: FOR К = 1 TO 200: PRINT ”N(”: K; ”) =”; : INPUT I(K)
IF 1(1) = 0 THEN 2
IF I(K) = 0 THEN 1
L = K: S = S + I(K): NEXT К
1 : S = S I L: D = 0: IF L = 1 THEN 6
FOR К = 1 TO L: D = D + (S - I(K)) ' 2: NEXT K: D = SQR(D / (L - 1))
PRINT ’’NUMBER OF POINTS L
PRINT ’’AVERAGE VALUE =”; S, ’’STANDARD DEVIATION =”; D: PRINT
PRINT ’’TEST 3-SIGMA”: PRINT : A = 0: FOR К = 1 TO L
IF I(K) > S + 3 * D THEN 3
IF I(K) < S - 3 * D THEN 4
GO'7'© 5
3 : PRINT ’’THE VALUE”; I(K); ”IS TOO MUCH, PLEASE EXCLUDE” A = 1:
GOTO 5
4 : PRINT ’’THE VALUE"; I(K); "IS TOO SMALL, PLEASE EXCLUDE”
A = 1
5 : NEXT K: IF A = 1 THEN 2
PRINT ”NO ERRONEOUS DATA”: GOTO 2
6 : PRINT ’’ONE POINT, NO STATISTICAL PROCESSING”
2 : PRINT ’’END”: END
Алгоритм программы 2 является оригинальным и предназна-
чен для так называемой асимметрической статистической об-
работки одномерных массивов данных (а:,). Результатом ее ра-
боты является расчет обычного среднеарифметического значе-
ния х и двух стандартных отклонений вместо одного. Первое из
них характеризует разброс величин Xi, превышающих среднее
значение, т. е. s(xi > ж), тогда как второе — значений, меньших
среднего s(xi < х). Необходимость в таком видоизменении “тра-
диционной” статистической обработки может возникать, напри-
мер, в случае усреднения величин, близких к пределу обнару-
жения метода анализа, когда их разброс “снизу” объективно
ограничен значением X — 0, а “сверху” подобных ограничений
не существует и, как правило, > х) > s(xi < х). Другим
576
примером является усреднение данных, содержащих случайные
экспериментальные погрешности, которые по сути аналитиче-
ского метода оказываются погрешностями одного знака. Это
наблюдается, например, при статистической обработке (ран-
домизации) межлабораторных данных по индексам удержива-
ния.
Результаты статистической обработки с помощью програм-
мы 2 дополнительно характеризуются значениями фактора
асимметрии стандартных отклонений As = s(xi > x)/s(xi < х), а
также стандартными отклонениями в обычном смысле.
REM: PROGRAM for asymmetrical statistical processing
REM: Simplest version (without correction of initial data):PRINT
PRINT "Asymm. stat, processing:”: DIM A(100): PRINT "Number of data”;
INPUT N: IF N = 0 OR N = 1 OR N = 2 THEN 7:
FOR I = 1 TO N: PRINT I; : INPUT A(I)
NEXT I: В = 0: FOR I = 1 TO N: В = В + A(I)
NEXT I:B = B/N:D = L = M = 0: 0 = 0: FOR I = 1 TO N: IF A(I) = В
THEN 3
IF A(I) < В THEN 2
C = C + 1:L = L + (A(I) - B) ' 2: GOTO 3
2:D = D+1:M = M+ (A(I) - B) * 2
3 : NEXT I: S = L + M: IF C > 1 THEN 5
PRINT "number of data more than average value are not enough": GOTO 4:
IF D > 1 THEN 5
PRINT "number of data below than average value are not enough”: GOTO 4
5 : S(l) = SQR(L / (C - 1)): S(2) = SQR(M / (D - 1))
S = SQR(S / (N - 1)): PRINT : PRINT "AVERAGE VALUE =”; В
PRINT "ST.DEV.(Total) =”; S: PRINT
PRINT "ST.DEV.(Up) =”; S(l), ”N(Up) ="; C
PRINT "ST.DEV.(Down) ="; S(2), ”N(Down) =”; D
PRINT "ASYMM (ST.DEV.) =”; .01 * INT(100 * S(l) / S(2) + .5): PRINT
GOTO 4
7 : PRINT "NUMBER OF DATA IS TOO SMALL; NO PROCESSING"
4 : PRINT "END”: END
Программа 3 (2-DIM STAT) предназначена для вычисления
параметров линейных корреляционных уравнений вида у = ах +
b и коэффициентов корреляции величин (а?, у). В программе пре-
дусмотрена строка для преобразования переменных х —> /(«) и
У ~* f(y) в случае необходимости
REM: PROGRAM 2-DIM STAT
REM: program for statistical processing of two dimensional data
REM: calculation of parameters of linear dependence у = a*x + b
DIM X(200), Y(200): PRINT : PRINT "INPUT NUMBER OF DATA, N =”;
INPUT N: IF N = 1 THEN 2: PRINT : PRINT "INPUT X(i),Y(i) VALUES”
FOR I = 1 TO N: PRINT ”X(”; I; ”),Y(”; I; ”)”;
INPUT X(I), Y(I): NEXT I: A = 0: В = 0: C = 0: D = 0: E = 0
REM: The fragment of program for transformation x — > f(x), у — > f(y)
FOR I = 1 TO N: X(I) = X(I): Y(I) = Y(I): NEXT I
REM *********************************************
577
FOR 1= 1 TO N: A = A + X(I) / N: В = В + X(I) ' 2 / N
C = C + Y(I) / N: D = D + Y(I) ' 2 / N E = E + X(I) * Y(I) / N: NEXT I
Z = В - A " 2: К = (E - A * C) / Z: L = (B * C - A *E) / Z
IF N = 2 THEN 1
M = (E - A * C) * 2 / Z: M = (D - C ' 2 - M) * N / (N - 2): KI = M / N / Z
K2 = KI * В
1 : R = E-A‘C:P = SQR(Z * (D - C ' 2)): R = R / P: PRINT
PRINT "PARAMETERS OF LINEAR EQUATION у = ax + b"
PRINT ” *****************************************”
PRINT
PRINT " a =”; K; ” s(a) =”; SQR(Kl)
PRINT ” b =”; L; ” s(b) =”; SQR(K2): PRINT
PRINT и *************,|'11!*>|£****’,(*****ж**^**|1!*******”
PRINT
PRINT "General dispersion S(0)2 =”; M
PRINT "Corellation coefficient R =”; R: PRINT
PRINT ” *****************************************”
2 : PRINT "END”: END
Программа (LIN&LOG) позволяет вычислять как линей-
ные, так и логарифмические (Ковача) индексы удерживания,
выбор между которыми определяется используемым темпера-
турным режимом хроматографического анализа (линейное про-
граммирование температуры или изотермические условия со-
ответственно). В обоих случаях для расчета индексов необхо-
димы данные о временах удерживания двух реперных компонен-
тов.
REM: PROGRAM for calculation of Kovats & linear retention indices
PRINT : О = 0: PRINT "INDICATE YOUR GO TEMPERATURE REGIME:
PRINT "1-ISOTERMIC, 2-TEMPERATURE PROGRAMMING”;
INPUT K: IF К = 2 THEN 1
PRINT : PRINT "INPUT DEAD TIME T(O) =”;
INPUT О
1 : PRINT "INPUT NUMBER OF CARBON ATOMS & RETENTION TIMES”;
PRINT ”OF TWO REFERENCE ALKANES”: PRINT : PRINT ”C(1),T(1)";
INPUT C(l), T(l): PRINT ”C(2),T(2)”;
INPUT C(2), T(2): PRINT
PRINT "INPUT RETENTION TIME OF COMPT BEING CHARACTERIZED”;
INPUT X: IF X = 0 THEN 5
IF К = 2 THEN 2
FOR I = 1 TO 2: T(I) = LOG(T(I) - O): NEXT I
X = LOG(X)
2 : R = (C(l) + (C(2) - C(l)) * (X - T(l)) / (T(2) - T(l))) * 100
R = .1 * INT(10 * R + .5) PRINT : PRINT "RETENTION INDEX
IF К = 1 THEN 3
PRINT "(LINEAR) =”; R
GOTO 4
3 : PRINT "(KOVATS = LOGARITHMIC) =”; R
4 : PRINT : PRINT "Continue (1) or exit (0) ”;
INPUT Z: IF Z = 1 THEN 1
5 : PRINT "END”: END
578
Более универсальный вариант предусмотрен в программе 5
(LINLOGRI) для вычисления линейно-логарифмических (обоб-
щенных) индексов удерживания, применимых в любых темпера-
турных режимах анализа. Однако для расчета таких параме-
тров необходимы сведения уже не о двух, а о трех реперных
компонентах.
REM: PROGRAM for calculation of lin-log retention indices
REM: SIMPLEST VERSION
REM: you need data about three reference compounds
PRINT ”****************************************”
PRINT ’’Calculation of lin-log retention indices (from exp. ret. times)”
PRINT "****************************************”• PRINT
PRINT ’’input dead time of your chromatographic system”
PRINT "input zero or nothing, if you don’t know this parameter”
PRINT ”t(0) =”; : INPUT t: PRINT
PRINT ’’input the numbers of carbon atoms and the retention times of’
PRINT ’’three reference n-alkanes”: PRINT ”C(l),t(l)”; : INPUT C(l), t(l)
PRINT ”C(2),t(2)”;: INPUT 0(2), t(2): PRINT ”0(3),t(3)”; : INPUT 0(3), t(3)
t(l) = t(l) - t: t(2) = t(2) - t: t(3) = t(3) - t: PRINT
Q = (0(3) - 0(2)) * t(l) + (0(2) - 0(1)) * t(3) - (C(3) - 0(1)) * t(2)
P = (0(3) - 0(2)) * LOG(t(l)) + (0(2) - 0(1)) * LOG(t(3))
P = P - (0(3) - 0(1)) * LOG(t(2))
Q = -.01 * INT(100 * Q / P + .5): R(l) = t(l) + Q * LOG(t(l))
R(2) = t(2) + Q * L0G(t(2))
PRINT ’’the value of q for regime being used is”; Q: PRINT
2 : PRINT ’’input the retention time of compound being characterized”
PRINT ’’(print 0 for exit)”: PRINT ”t(X) =”; : INPUT X: IF X = 0 THEN 1
X = X-t:X = X + Q* LOG(X)
I = 0(1) + (C(2) - 0(1)) * (X - R(l)) / (R(2) - R(l)): I = 100 * I: PRINT
PRINT ’’RETENTION INDEX is”; .1 * INT(10 * I + .5): PRINT : GOTO 2
1 : PRINT ’’END OF CALCULATIONS": PRINT : PRINT : END
Программа 6 (QUANT-1) предназначена для обработки ре-
зультатов количественного анализа методом абсолютной
градуировкис помощью однопар аметрового регрессионного
уравнения С — К х Р и включает расчет параметров этого
уравнения методом наименьших квадратов и его последующее
использование для вычисления конкретных значений масс или
концентраций определяемых компонентов (С) по эксперимен-
тально измеряемым параметрам хроматографических сигналов
(Р, площади или высоты).
REM: PROGRAM QUANT-1
REM: Program for quantitative analysis by method of absolute calibration
REM: Calculation by LSM with equation C = A*P
DIM C(100), P(100): PRINT ’’NUMBER OF DATA”;
INPUT N: IF N = 0 OR N = 1 THEN 1: PRINT ’’INPUT C(i),P(i)”: PRINT
FOR I = 1 TO N: PRINT ”C(”; I; ”), P(”; I; ”)”;
INPUT C(I), P(I): NEXT I: A = О: В = 0: D = 0: E = 0
FOR I = 1 TO N: A = A + C(I) * P(I): В = В + P(I) ' 2: E = E + C(I) ' 2
NEXT I: К = A / B: FOR I = 1 TO N: D = D + (C(I) - К * P(I)) ' 2: NEXT I
579
SI = D / (N - 1): S2 = SQR(S1 / B)
PRINT ’’PARAMETERS OF CALIBRATION EQUATION C = K*P”
PRINT ”*******************************************
PRINT : PRINT ”K =”; K: PRINT ; ”S(K) =’’; S2
PRINT ’’Relative st.dev. S(K) =”; .1 * INT(1000 * S2 / К + .5); ”%”
2 : PRINT : PRINT "Calculations with calibration equation (nothing - NO)”;
INPUT T: PRINT : IF T = o THEN 1
PRINT ”P(X), S(P(X)). % ”;
INPUT X, Y: PRINT : Z1 = K ‘X: Z2 = SQR((S2 / К) " 2 + (.01 * Y) ' 2)
PRINT ”C(X) =”; Zl: PRINT ; ”S(C(X)) =”; Z2 * Z1
PRINT "Relative standard deviation C(X) =”; .1 * INT(1000 * Z2 + .5);
PRINT "%”: PRINT : GOTO 2
1 : PRINT "END": END
Программа 7 QUANT-2 позволяет вычислять массы или кон-
центрации определяемых компонентов в образцах методом
стандартной добавки. Этот метод количественного ана-
лиза по совокупности экспериментальных операций подготовки
проб для анализа может быть реализован разными способами;
в простейшем варианте изменениями объема или массы анали-
зируемого образца за счет стандартной добавки определяемого
компонента практически можно пренебречь, что дает возмож-
ность использования простейших расчетных соотношений. В
общем же случае это условие может не соблюдаться, что при-
водит к увеличению количества исходной информации для рас-
четов.
REM: PROGRAM QUANT-2
REM: Program for method of standard additives
REM: MODE 1 - The changes in the volume of sample is negligible
REM: MODE 2 - The changes in the volume of sample is large
PRINT "MODE 1(V = const) or MODE 2(V # const)”;
INPUT Z: IF Z = 0 THEN 1 '
IF Z = 2 THEN 2:
PRINT : PRINT "MODE 1: INPUT M(st.add.), M(sample)”;
INPUT M, Ml: PRINT
PRINT "INPUT PEAK AREA BEFORE STANDARD ADDITIVE with st. dev.”;
INPUT Si, Dl:
PRINT "INPUT PEAK AREA AFTER STANDARD ADDITIVE with st. dev.”;
INPUT S2, D2: X = M * Si / (S2 - Si)
XI = SQR(2 * (Dl / Si) ' 2 + (D2 / S2) ‘ 2): PRINT
PRINT "QUANTITY BEING DETERMINED M =”; X
PRINT "ST.DEV S(M) =”; Xl * X
PRINT "CONCENTRATION IN THE SAMPLE, % w/w ”; M / Ml * 100
PRINT "ST.DEV. S(C) =”; Xl * X / Ml * 100
PRINT "Relative st. dev. S(M) and S(C) ”; .1 * INT(1000 * Xl 4- .5);
PRINT : GOTO 1
2 : PRINT "MODE 2”
PRINT "INPUT QUANTITIES (g) OF SAMPLE AND ADDITIVE, Ql, Q2”;
INPUT Ql, Q2: PRINT
PRINT ” INPUT AREAS OF test-compt BEFORE & AFTER ADDITIVE, Pl, P2”;
INPUT Pl, P2: PRINT "Corresponding standard deviations”;
580
INPUT Dl, D2
PRINT "INPUT AREAS OF COMPONENT BEFORE AND AFTER, SI, S2”;
INPUT SI, S2
PRINT ” Corresponding standard deviations”;
INPUT El, E2: M = Q2 / (S2 / P2 - SI / Pl): С = M / QI: PRINT
PRINT ”M(X) =”, M, ”C(X) =”; C * 100; ”% w/w”
Y = Pl * S2 / (S2 * Pl - Si * P2)
Y = SQR((D1 / Pl) ’ 2 + (D2 / P2) " 2 + (El / Si) ' 2 + (E2 / S2) * 2) / Y
PRINT ’’ST.DEV. C(X) =”; Y * C * 100
PRINT ’’ST.DEV. M(X) =”; Y * C * QI
PRINT ’’Relative standard deviation M(X) and C(X)”; .1 * INT(1000 ’ Y + .5);
PRINT
1 : PRINT ’’END”: END
В заключительной программе 8 (QUANT-3) реализован про-
стейший алгоритм обработки количественных данных мето-
дом внутреннего стандарта (по результатам единич-
ных измерений без учета данных по воспроизводимости пара-
метров хроматографических пиков). Такой способ настолько
прост, что вполне может быть реализован даже на калькулято-
рах, однако в практике хроматографического анализа нередко
возникает необходимость оценки нормировочных множителей
вместо их предварительного экспериментального определения.
В приведенной программе предусмотрена возможность получе-
ния подобных оценок на основании брутто-формул аналита и
внутреннего стандарта.
REM: Program QUANT-3
REM: QUANTITATIVE ANALYSIS BY INTERNAL STANDARD METHOD
PRINT ”************************>’
PRINT ’’INTERNAL STANDARD METHOD”
PRINT ” ************************”
PRINT
PRINT ’’INPUT F-FACTOR (Input zero for preliminary estimation of F-factor)”
PRINT "F =”;
INPUT F: IF F > 0 THEN 1
PRINT : PRINT ”MOL. FORMULA OF ANALYTE, C,H,N,O,S,Cl,Br (IN LINE)”;
INPUT Al, A2, A3, A4, A5, A6, A7
PRINT ’’MOLECULAR FORMULA OF INT. STANDARD, C,H,N,O,S,Cl,Br”;
INPUT Bl, B2, B3, B4, B5, B6, B7
Ml = 12 * Al + A2 + 14 * A3 + 16 * A4 + 32 * A5 + 35 * A6 + 80 * A7
М2 = 12 * Bl + B2 + 14 * B3 + 16 * B4 + 32 * B5 + 35 * B6 + 80 * B7
F = (Bl / М2) / (Al / Ml): F = INT(100 * F + .5) / 100: PRINT
PRINT ”F =”; F: PRINT ; ’’CONTINUE (zero if NO)”;
INPUT Y: IF Y = 0 THEN 2
1 : PRINT ’’INPUT QUANTITIES OF SAMPLE AND STANDARD”;
INPUT QI, Q2: PRINT ’’INPUT PEAK AREAS OF SAMPLE fc STANDARD”;
INPUT Pl, P2: M = Pl * F * Q2 / P2: С = M / QI: PRINT
PRINT ”**************WER**************”
PRINT ’’MASS OF ANALYTE IN THE SAMPLE, M =”; M
PRINT ’’CONCENTRATION IN THE SAMPLE, C =”; 100 * C;
581
PRINT ”**********************************”
PRINT : PRINT "REPEATE (zero if NO)”;
INPUT Z: PRINT : IF Z > 0 THEN 1
2 : PRINT ’’END”: END
Программы 6 и 7 для обработки результатов количествен-
ного анализа предусматривают возможность оценки случай-
ных составляющих погрешностей определяемых величин по вос-
производимости экспериментальных данных, что отличает их
от большей части существующего хроматографического про-
граммного обеспечения. Что же касается метода внутренней
нормализации, то для него режим on-line (выполнение расчетов
непосредственно после регистрации хроматограммы) оказыва-
ется наиболее предпочтительным, и возможности программного
обеспечения сбора и обработки хроматографических данных
“МультиХром” (см. раздел IV.5) полностью удовлетворяют
всем практическим потребностям.
582
Л итер атур а
Глава I
1. Цвет М. С. Хроматографический адсорбционный анализ.
Избранные работы / Ред. А. А. Рихтер, Т.А Красносель-
ская. М.: Изд-во АН СССР, 1946. 272 с.
2. Аналитическая хроматография / К. И. Сакодынский,
В. В. Бражников, С. А. Волков и др. М.: Химия, 1993. 462 с.
3. Ettre L. S., Sakodynskii К. I. // Chromatographia. 1993. Vo] 35.
№3/4. Р. 223-231; №5/6. Р. 329-338; Сакодынский К.И. //
Хроматогр. журн. 1993. №1. С. 2-22.
4. Вигдергауз М. С. Цветопись. М.: Химия, 1980. 95 с.
5. Яшин Я. И. //Журн. аналит. химии. 1994. Т.49. №10.
С. 1047-1058.
6. Kuhn R., Wint< istein A., Lederer E. // Z. Physiol. Chem. 1931.
Bd 197. S. 141-160.
7. Измайлов H.A., Шрайбер M. С. // Фармация. 1938. №3. С. 1-
7.
8. Brockmann Н., Schodder Н. // Biochem. J. 1941. Vol. 35. № 1.
Р. 91-121.
9. Martin A. J. Р., Synge R. L. М. // Biochem. J. 1941. Vol. 35. № 12.
Р. 1358-1368.
10. Consden R., Gordon A.H., Martin A.J.P. ff Biochem. J. 1944.
Vol. 38. №3. P. 224-232.
11. Хроматография на бумаге / Под ред. И. М. Хайса, К. Мачека.
Пер. с чешек. Под ред. М. Н. Запрометова. М.: ИЛ, 1962.
851 с.
12. Campbell D.H., Luescher Е., Lerman L. S. ff Proc. Natl. Acad.
Sci. U.S.A. 1951. Vol. 37. №9. P. 575-578.
13. James A.T., Martin A.J.P. // Biochem. J. 1952. Vol. 50. №5.
P.679-690.
14. Хроматография в тонких слоях / Под ред. Э. Шталя. Пер. с
нем. Под ред. К. В. Чмутова. М.: Мир, 1965. 508 с.
15. Moore S., Stein W.H. // J. Biol. Chem. 1954. Vol. 211. №2.
P. 893-906.
16. Golay M. J.E. // Gas Chromatography. A Symposium Held under
the Auspices of the Instrument Society of America. August, 1957
/ Ed. by V. J. Coates, H.J.Noebels, I. S. Fagerson. New York: AP
Inc., 1958. P. 1-13.
17. Голей M. If Газовая хроматография. Сб. докладов на
II Междунар. симп. в Амстердаме и по анализу смесей
летучих веществ в Нью-Йорке / Пер. с англ. Под ред.
583
А. А. Жуховицкого, H. M. Туркельтауба. М.: ИЛ, 1961. С. 39-
60.
18. Porath J., Flodin Р. // Nature. 1959. Vol. 183. №4676. Р. 1657—
1659.
19. Helferich F. // Nature. 1961. Vol. 189. № 4769. P. 1001-1002.
20. Klesper E., Corwin A.H., Turner D. A. // J. Org. Chem. 1962.
Vol. 27. №2. P. 700-701.
21. Москвин Л. H., Горшков А. И., Гумеров M. Ф. // ДАН СССР.
1982. Т.265 №2. С. 378-382.
22. Родинков О. В., Москвин Л.Н. //Журн. аналит. химии. 1996.
Т. 51. №1. С. 102-109.
23. Giddings J. С., Meyers M.N. // J.High Resolut. Chromatogr. 1983.
Vol. 6. №7. P. 381-382.
24. Москвин Л H., Царицына Л. Г. Методы разделения и концен-
трирования в аналитической химии. Л.: Химия, 1991. 255 с.
25. Sewell Р., Clarke В. Chromatographic Separations / Ed. by D.Kea-
ley. New York: Wiley, 1987. 335 p.
26. Шаршунова M., Шварц В., Михалец Ч. Тонкослойная хрома-
тография в фармации и клинической биохимии: В 2 частях /
Пер. со словацк. Под ред. В. Г. Березкина, С. Д. Соколова.
М.: Мир, 1980. 621 с.
27. Березкин В. Г., Бочков Н. С. Количественная тонкослойная
хроматография. Инструментальные методы. М.: Наука,
1980. 183 с.
28. Planar Chromatography in the Life Sciences / Ed. by J. C. Touch-
stone. New York, etc.: Wiley, 1990. 199 p.
29. Fried B., Sherma J. Thin-layer Chromatography. Techniques and
Applications: Third Edition, Revised and Expanded. New York,
etc.: M. Dekker, Inc., 1994. 451 p.
30. Руководство по современной тонкослойной хроматографии /
Под ред. О. Г. Ларионова. М., 1994. 311 с.
31. Руководство по газовой хроматографии: В 2 частях / Под
ред. Э. Лейбница, X. Г. Штруппе. Пер. с нем. Под ред.
В. Г. Березкина. М.: Мир, 1988. 480 с.
32. Голъберт К. А., Вигдергауз М. С. Введение в газовую хрома-
тографию: Изд. 3-е. перераб. и доп. М.: Химия, 1990. 352 с.
33. Гиошон Ж., Гийемен К. Количественная газовая хроматогра-
фия для лабораторных анализов и промышленного контроля:
В 2 частях / Пер. с англ. Под ред. О. Г. Ларионова. М.:
Мир, 1991. 582 с.
34. High Performance Liquid Chromatography / Ed. by P. R. Brown,
R. A. Hartwick. New York, etc.: Wiley, 1989. 688 p.
584
35. Высокоэффективная жидкостная хроматография в биохимии
/ Бауэр Г., Энгельгард X., Хеншен А. и др. Пер. с англ.
Под ред. И. В. Березина. М.: Мир, 1988. 687 с.
36. Стыскин Е. Л., Ициксон Л. Б., Брауде Е. В. Практическая вы-
сокоэффективная жидкостная хроматография. М.: Химия,
1986. 288 с.
37. Киселев А. В., Яшин Я. И. // Адсорбционная газовая и жид-
костная хроматография. М.: Химия, 1979. С. 196-280.
38. Архипов Д Б., Беленький Б. Г. //Журн. аналит. химии. 1995.
Т.50. №8. С. 806-817.
39. Grob К. Making and Manipulating Capillary Columns for Gas
Chromatography. Heidelberg, etc.: Huthig, 1986. 232 p.
40. Тесарик К, Комарек К. Капиллярные колонки в газовой хро-
матографии / Пер. с чешек. Под ред. В. Г. Березкина. М.:
Мир, 1987. 222 с.
41. Высокоэффективная газовая хроматография / Ред. К. Хайвер.
Пер. с англ. Под ред. В. Г. Березкина. М.: Мир, 1993. 288 с
42. Беленький Б. Г., Ганкина S С., Мальцев В. Г. Капиллярная
жидкостная хроматография. Л.: Наука, 1987. 208 с.
43. Введение в микромасштабную высокоэффективную жидкост-
ную хроматографию / Ред. Д. Исиа. Пер. с англ. Под ред.
В. Г. Березкина. М.: Мир, 1991. 240 с.
44. Сверхкритическая флюидная хроматография / Ред. Р.Смит.
Пер. с англ. Под ред. В. Г. Березкина. М.: Мир, 1991. 280 с.
45. Вигдергауз М. С., Лобачев А. Л., Лобачева И. В. и др. //
Успехи химии. 1992. Т.61. №3. С.497-522.
46. Остерман Л. А. Хроматография белков и нуклеиновых ки-
слот. М.: Наука, 1985. 536 с.
47. Беленький Б. Г., Виленчик Л. 3. Хроматография полимеров.
М.: Химия, 1978. 343 с.
48. Аминная хроматография: Методы / Ред. П.Дин, У. Джон-
сон, Ф. Мидл. Пер. с англ. М.: Мир, 1988. 278 с.
49. Даванков В. А., Навратил Дж., Уолтон X. )[ Лигандно-
обменная хроматография / Пер. с англ. Под ред. В. А. Да-
ванкова. М.: Мир, 1989. 294 с.
50. Москвин Л.Н. // Журн. аналит. химии. 1996. Т. 51. №8.
С. 901-902.
51. Березкин В. Г. // Журн. аналит. химии. 1995. Т. 50. №6.
С. 677-678.
52. Вигдергауз М. С. // Журн. аналит. химии. 1993. Т.48. №8.
С. 1291-1295.
53. Хроматография Основные понятия. Терминология / Сбор-
ники научно-нормативной терминологии. Вып. 114. Отв.
ред. В.А. Даванков. Комитет научной терминологии РАН в
585
области фундаментальных наук. Научный совет по хромато-
графии. М., 1997. 48 с.
54. Miller J. М. Chromatography: Contrasts and Concepts. New York:
Wiley, 1988. 297 p.
55. Scott R.P.W. If Chemical Instrumentation: A Systematic
Approach: Third Ed.. New York, etc.: Wiley, 1989. P. 863-895.
56. Grob K. On-line Coupled LC—GC. Heidelberg: Huthig, 1991. 462 p.
57. Беленький Б. Г. //Росс. хим. журн. 1994. Т.38. №1. С. 25-32.
58. Smith R.M. Gas and Liquid Chromatography in Analytical Chemist-
ry. Chichester etc.: Wiley, 1988. 402 p.
59. Ravindranath B. Principles and Practice of Chromatography. Chi-
chester: Horwood, 1989. 502 p.
60. Gas Chromatography: Biochemical, Biomedical and Clinical Appli-
cations / Ed. by R. E. Clement. New York, etc.: Wiley, 1990. 393 p.
61. Ahuia S. Trace and Ultratrace Analysis by High Performance Liquid
Chromatography. New York, etc.: Wiley, 1992. 419 p.
62. Вигдергауз M. С. Физико-химические основы и современные
аспекты газовой хроматографии. Изд-во Самарского ун-та,
1993. 153 с.
Глава II
63. Смирнова С. А., Витенберг А. Г., Столяров Б. В. // Заводск.
лаб. 1978. Т.44. №5. С. 541-542.
64. Методы количественного органического элементного анали-
за / Под ред. Н.Э. Гельман. М.: Химия, 1987. С. 129-134.
65. Устройство для подсоединения капиллярных колонок ИКК-
1.1. Техническое описание и инструкция по эксплуатации.
СПб.: АО “ЭКРОС”, 1997.
66. Витенберг А. Г., Резник Т. Л. //Журн. аналит. химии. 1984.
Т.39. №4. С. 683-691.
67. Витенберг А. Г. //Докл. АН СССР. 1982. Т. 267. №1. С. ИЗ-
117.
68. Витенберг А. Г., Бутаева И. Л., Димитрова 3. Ст. // За-
водск. лаб. 1975. Т.41. №8. С. 931-933.
69. Супина В. Насадочные колонки в газовой хроматографии /
Пер. с англ. Под ред. В. Г. Березкина. М.: Мир, 1977. 256 с.
70. Другое Ю. С., Конопельке Л. А. Газохроматографический ана-
лиз газов М.: МОИМПЕКС, 1995. С. 78-90.
71. Малахов В. В., Сидельников В. Н., Уткин В. А. // Докл. РАН.
1993. Т.329. №6. С. 749-751.
72. Berezkin V.G., Korolev А.А. // J. Chromatogr. 1988. Vol.440.
Р. 323-328.
73. Kominar R. J. //J. Chem. Educat. 1991. Vol. 68. №10. P. A249-
A255.
586
74. Зайнуллин Р. Ф., Березкин В. Г. Обзорн. информ. Сер. “Ав-
томатизация и контрольно-измерительные приборы в нефте-
переработке и нефтехимической промышленности”. Приме-
нение пламенно-фотометрических детекторов в хроматогра-
фии. М., 1990. 50 с.
75. Будоеич В. Л., Шляхов А. Ф. // Успехи химии. 1989. Т. 58.
№8. С. 1354-1384.
76. Detectors for capillary chromatography / Ed. by H. H. Hill,
D.G. McMinn. New York etc.: Wiley, 1992. P. 51-81.
77. Адамия T.B., Бондаренко И. В., Будоеич В. Л. и др. // За-
водск. лаб. 1993. Т.59. №5. С. 17-20.
78. Энергии разрыва химических связей. Потенциалы ионизации
и сродство к электрону / Отв. ред. В. Н. Кондратьев. М.:
Наука, 1974. Гл. III.
79. Столяров Б. Б., Савинов И.М., Витенберг А. Г. Руководство
к практическим работам по газовой хроматографии: Изд.
3-е., перераб. / Под ред. Б. В. Иоффе. Л.: Химия, 1988. 335 с.
Глава III
80. Хроматография: Практическое приложение метода: В 2 ча-
стях / Ред. Э.Хефтман. Пер. с англ. Под ред. В. Г. Березки-
на. М.: Мир, 1986. 4.1. С.43-51.
81. Rothman L.D. // Analyt. Chem. 1996. Vol. 68. №12. P. 587R-
598R.
82. Melander W.R., Horvath C. // High Performance Liquid Chroma-
tography. Advances and Perspectives / Ed by C. Horvath. New
York: AP, 1983. Vol. 2. P. 192-226.
83. ЯшинЯ.И. // Журн. аналит. химии. 1998. Т.53. №1. С.7-
19.
84. Parris N. A. Instrumental Liquid Chromatography: A practical ma-
nual on high-performance liquid chromatographic methods. 2nd
compL rev. ed. Amsterdam, etc.: Elsevier, 1984. 432 p.
85. Detectors for liquid chromatography / Ed. by E. S. Yeung. New
York etc.: Wiley, 1986. 366 p.
86. Selective detectors: Environmental, industrial and biomedical appli-
cations / Ed. by R.E. Sievers. New York etc.: Wiley, 1995 261 p.
87. Poppe H. // Chromatographia. 1987. Vol. 24. №1. P. 25-32.
88. Хубер Л. Применение диодно-матричного детектирования в
ВЭЖХ / Пер. с англ. Под ред. В. Г. Березкина. М.: Мир,
1993. 92 с.
89. George S.A., Maute А. // Chromatographia. 1982. Vol. 15. №7.
Р. 419-425.
90. Scott R. P. W. Chromatography Detectors. New York: M. Dekker,
Inc., 1997. 536 p.
587
91. Smalley М. В., Mcgown L. В. // Adv. Chromatogr. 1997. Vol. 37.
P. 29-71.
92. Fell A.P., Scott H.P., Gill R. e.a. // J. Chromatogr. 1983.
Vol. 273. P. 3-17.
93. Fell A.P., Scott H.P., Gill R. e.a. // J. Chromatogr. 1983.
Vol. 282. P. 123-140.
94. Gaillard Y., Pepin G. 11 J. Chromatogr. 1997. Vol. 763 №1-2.
P. 149-164.
95. Lucue de Castro M.D. Gamiz-Gracia L. // Analyt. Chim. Acta.
1997. Vol. 351. №1-3. P. 23-40.
Глава IV
96. Дженнингс В., Рапп А. Подготовка образцов для газохрома-
тографического анализа / Пер. с англ. Под ред. В. Г. Берез-
кина. М.: Мир, 1986. 116 с.
97. Кузьмин Н. М. II Журн. аналит. химии. 1996. Т. 51. №2.
С. 202-210.
98. Витенберг А. Г., Иоффе Б. В. Газовая экстракция в хрома-
тографическом анализе. Парофазный анализ и родственные
методы. Л.: Химия, 1982. 280 с.
99. Kolb В., Ettre L. S. Static Headspace — Gas Chromatography.
Theory and practice. New York etc.: Wiley —- VCH, 1997. 298 p.
100. Байерман К. Определение следовых количеств органических
веществ / Пер. с англ. М.: Мир, 1987. 462 с.
101. Water analysis / Ed. by W.Fresenius, К. E. Quentin, W. Schneider.
Berlin etc.: Springer-Verlag, 1988. 804 p.
102. Дмитриев M. T., Казнина Н.И., Пинигина И. А. Санитарно-
химический анализ загрязняющих веществ в окружающей
среде: Справочник. М.: Химия, 1989. 367 с.
103. Концентрирование следов органических соединений / Отв.
ред. Н.М. Кузьмин. М.: Наука, 1990. 280 с.
104. Новиков Ю.В., Ласточкина К. О., Болдина 3. Н. Методы ис-
следования качества воды водоемов. М.: Медицина, 1990.
400 с.
105. Руководство по контролю вредных веществ в воздухе рабо-
чей зоны / С. И. Муравьева, М. И. Буковский, Е К Прохоро-
ва и др. М.: Химия, 1991. 368 с.
106. Исидоров В. А. Органическая химия атмосферы: Изд. 2-е,
перераб. и доп. СПб.: Химия, 1992. С.220-262.
107. Исидоров В. А. Летучие выделения растений. СПб.: Алга-
Фонд, 1994. С. 7-16.
108. Руководство по контролю загрязнения атмосферы. Руково-
дящий документ РД 52.04.186 — 89. М.: Гос. ком. СССР по
гидрометеорологии, 1991. 693 с.
588
109. Руководство по контролю источников загрязнения атмосфе-
ры. Общесоюзный нормативный документ. ОНД-90. Ч. 1 и
2. СПб.. 1992. 200 с.
ПО. Руководство по санитарно-химическому исследованию почвы
(Нормативные материалы). М.: Гос. ком. сан.-эпидемиол.
надзора России, Рос. респ. информ.-аналит. центр, 1993.
130 с.
111. Фомин Г. С., Фомина О. Н. Воздух. Контроль загрязне-
ний по международным стандартам: Справочник / Под ред.
С. А. Подлепы. М.: Протектор, 1994. 228 с.
112. Фомин Г. С. Вода. Контроль химической, бактериологиче-
ской и радиационной безопасности по международным стан-
дартам: 2-е изд. Энциклопедический справочник. М.:
ВНИИстандарт, 1995.
113. Высочин В. И. Диоксин и родственные соединения: Аналит.
обзор. Новосибирск, 1989. 153 с.
114. Кузубова Л. И. Токсиканты в пищевых продуктах: Аналит.
обзор. Новосибирск, 1990. 126 с.
115. Кузубова Л. И., Морозов С. В. Очистка нефтесодержащих
сточных вод: Аналит. обзор. Новосибирск, 1992. 72 с.
116. Кузубова Л. И., Морозов С. В. Органические загрязнители пи-
тьевой воды: Аналит. обзор. Новосибирск, 1993. 166 с.
117. Кузубова Л. И., Кобрина В. И. Химические методы подготовки
воды (хлорирование, озонирование, фторирование): Аналит.
обзор. Новосибирск, 1996. 131 с.
118. Onuska F. I. // J. High Resolut. Chromatogr. & Chromatogr.
Comm. 1989. Vol. 12. №1. P.4-11.
119. Liska L, Krupcik J., Leclercq P. A. // J. High Resolut. Chromatogr.
& Chromatogr. Comm. 1989. Vol. 12. №9. P.577-590.
120. Be Zeeuw R.A. // J. Chromatogr. 1989. Vol.488. № 1. P. 199-213.
121. Теренина M. Б., Головня P. В., Журавлева И. Л. //Журн. ана-
лит. химии. 1989. Т.44. №5. С. 773-793.
122. Namiesnik J., Gorecki Т., Biziuk М. е. а. // Analyt. Chim. Acta.
1990. Vol. 237. №1. P. 1-60.
123. Bishop R. W., Valis R.J. ]] J. Chromatogr. Sci. 1990. Vol. 28.
№11. P. 589-593.
124. Poole S. K., Dean T. A., Oudsema J. W. e. a. ff Analyt. Chim.
Acta. 1990. Vol.236. №1. P.3-42.
125. Lopez-Avila V., Benedicto J., Dodhiwala N. S. e. а. Ц J. Chroma-
togr. Sci. 1992. Vol.30. №9. P. 335-343.
126. Woolfenden E. A., Broadway G. M. // LC & GC Intern. 1992. Vol. 5.
№ 12. P. 28-35.
127. Vannoort R. W., Chervet J.-P., Lingeman H. e.a. // J. Chroma-
togr. 1990. Vol.505. №1. P.45-77.
589
128. Lichon M.J. // J. Chromatogr. 1992. Vol. 624. №1+2. P. 3-9.
129. Symposium on solid-phase extraction in environmental and clinical
chemistry, San-Francisco, CA, April 5-10, 1992 / Ed. by M. S. Mills,
E. M.Thurma //J. Chromatogr. 1993. Vol. 629. № 1. P. 1-93.
130. Lesage S. // J. Chromatogr. 1993. Vol. 642. №1+2. P. 65-74.
131. Merbel van de N.C., Hageman J. J., Brinkman U. A. Th. // J.
Chromatogr. 1993. Vol. 634. №1. P. 1-29.
132. Font G., Manes J., Motto J.C. e.a. Ц J. Chromatogr. 1993.
Vol. 642. № 1+2. P. 135-161.
133. Hennion М.-C., Coquart V. // J. Chromatogr. 1993. Vol.642.
№1+2. P. 211-224.
134. Janda V., Bartie K.D., Clifford А. А. Ц J. Chromatogr. 1993.
Vol. 642. №1+2. P. 283-299.
135. Милюкин M.B., Пилипенко A.T. ff Химия и технол. воды.
1993. Т.15. №6. С. 419-424.
136. Клюев Н.А., Бродский Е. С., Сойфер В. С. и др. // Рос. хим.
журн. 1994. Т 38. №2. С. 9-12.
137. Starostin L., Witkiewicz Z. // Cheni. Anal. (Warsaw). 1994.
Vol. 39. №3. P. 263-279.
138. Delft Van R. J., Doveren A. S. M. J., Snijders A. G. // Fresenius J.
Anal. Chem. 1994. Vol. 350. №10-11. P. 638-641.
139. Charmas B., GierakA., Leboda R. // Chem. Anal. (Warsaw). 1994.
Vol. 39. №1. P.1-11.
140. Zhang Z., Yang M. J., Pawliszyn J. // Anal. Chem. 1994. Vol. 66.
№17. P.844A-853A.
141. Kern H., Penton Z. ff Chromatography. Celebrated Michael
Tswett’s 125th Birthday / Ed. by O. Kaiser, R. E. Kaiser, H. Gunz,
W. Gunther. Dusseldorf: InCom, 1977. P. 153-166.
142. HelmigD. // J. Chromatogr. 1996. Vol. 732. №2. P. 414-417.
143. Крылов А. И. If Журн. аналиг. химии. 1995. Т. 50. №3.
С.230-241.
144. Клюев Н.А. // Журн. аналит. химии. 1996. Т.51. №2.
С. 163-172.
145. Guide to sample preparation // LC & GC Intern. 1997 Vol. 10.
№ 1. Appendix.
146. Handbook of Derivatives for Chromatography / Ed. by K.Blau,
G.S.King. London: Heyden, 1978. 576 p.
147. Knapp D. R. Handbook of Analytical Derivatization Reactions. New
York, etc.: Wiley, 1979. 741 p.
148. Березкин В. Г. Химические методы в газовой хроматографии.
М.: Химия, 1980. 286 с.
149. Drozd J. Chemical Derivatization in Gas Chromatography. Amster-
dam, etc.: Elsevier, 1981. 232 p.
590
150. Crompton T. R. Comprehensive Organometallic Analysis. New York
& London: Plenum Press, 1987. 883 p.
151. Другое Ю.С., Березкин В. Г. // Успехи химии. 1986. Т. 55.
№6. С. 999-1023.
152. Krull I.S., Deyl Z., Lingeman H. // J. Chromatogr. B. 1994.
Vol. 659. №1+2. P. 1-17.
153. Toyo’oka T. // J. Chromatogr. B. 1995. Vol.671. №1+2. P.91-
112.
154. Столяров Б. В., Нагимуллина А. Г., Карцева Л. А. // Журн.
аналит. химии. 1984. Т. 39. №9. С. 1674-1678.
155. Amijee М., Wells R. J. ff J. Chromatogr. A. 1994. Vol. 662. №1.
P. 123-137.
156. Amijee M., Cheung J., Wells R. J. // J. Chromatogr. A. 1996.
Vol. 738. №1. P. 43-55.
157. Amijee M., Cheung J., Wells R.J. // J. Chromatogr. A. 1996.
Vol. 738. №1. P. 57-72.
158. Соколов Д.Н. Газовая хроматография летучих комплексов
металлов. М.: Наука, 1981. 124 с.
159. Шведт Г. Хроматографические методы в неорганическом
анализе / Пер. с англ. Под ред. В. Г. Березкина. М.: Мир,
1984. 254 с.
160. Inorganic chromatographic analysis / Ed. by J. C. Macdonald. New
York, etc.: Wiley, 1985. 450 p.
161. Inorganics (CRS handbook of chromatography) / Ed. by M. Qure-
shi. Boca Raton (FL): CRS press, Vol. 1. 1987. 367 p.
162. Карпов Ю.А., Кузнецов Л. Б., Беляев В. Н. // Заводск. лаб.
1981. Т.47. №3. С. 3-9.
163. Карпов Ю.А. II Рос. хим. журн. 1994. Т. 38. №1. С. 12-15.
164. ЗенкевичИ. Г. //Журн. эколог, химии. 1993. №4. С. 287-
293.
165. Зенкевич И. Г., Максимов Б. Н., Родин А. А. // Журн. аналит.
химии. 1995. Т.50. №2. С. 118-135.
166. Eckschlag К., Danzer К. Information Theory in Analytical Chemis-
try. New York, etc.: Wiley, 1994. 275 p.
167. Шаевич А. Б. Стандартные образцы для аналитических це-
лей. М.: Химия, 1987. 184 с.
168. 4-th International symposium on hyphenated techniques in chroma-
tography and hyphenated chromatography analyzers. Bruges, 7-9
February 1996. //J. Chromatogr. 1996. Vol.750. №1+2. 411 p.
169. Griffiths L. 11 Analyt. Chem. 1995. Vol. 67. №15. P. 4091-4095.
170. Nomenclature for Chromatography (IUPAC Recommendations.
1993) . Prepared for publication by L.S. Ettre // Pure & Appl.
Chem. 1993. Vol. 65. №4. P. 819-872.
591
171. Ettre L. S. // J. High Resolut. Chromatogr. & Chromatogi. Comm.
1993. Vol. 16. №4. P.258-261.
172. Эттре JI.C. // Хроматогр. журн. 1995. №4. С.81-88.
173. Вычисления и величины в сорбционной колоночной хромато-
графии / М. Крейчи, Я. Паюрек, Р. Комере и др. Пер. с англ.
Под ред. В. Г. Березкина. М.: Мир, 1993. С. 46.
174. Malik A., Jinno К. // Chromatographia. 1990. Vol. 30. №3/4.
Р.135-137.
175. Knox J.H., Kaliszan R. // J. Chromatogr. 1985. Vol 349. №2.
P. 211-234.
176. Lebrdn-Aguilar R., Quintanilla-Lopez J. E., Garcia-Dominguez J. A.
// J. Chromatogr. 1997. Vol 760. №2. P. 219-226.
177. Le Vent S. // J. Chromatogr. A. 1996. Vol. 752. № l-f-2. P. 173-181.
178. Fleming P. //Analyst. 1992. Vol. 117. P. 1553-1557.
179. Вигдергауз M.C., Платонов И. А., Лобачев А. Л. и др. //
Журн. физ. химии. 1993. Т.67. №4. С. 857-860.
180. Smith R. J., Nicass G. S., Wainwright M. S. // J. Liq. Chromatogr.
1986. №9. P. 1387-1430.
181. Liu Y., Yun K. S., Parcher J. F. // J. Chromatogr. A. 1994.
Vol. 679. № 2. P. 392-396.
182. Smith R. J., Haken J. K., Wainwright M. S. // J. Chromatogr. 1985.
Vol. 334. №2. P. 95-127.
183. Malik A., Jinno K. Il Chromatographia. 1990. Vol.30. №3/4.
P. 138-143.
184. Lebrdn-Aguilar R., Quintanilla-Lopez J. E., Tello A. Ad. e. a. // J.
Chromatogr. A. 1995. Vol.697. №1+2. P.441-451.
185. Kersten B.R., Poole S.K., Poole C.F. // J Chromatogr. 1989.
Vol. 468. P. 235-260.
186. Иванов А., Эйзен О., Орав A. // Изв. АН Эст. ССР. Сер.
химия, геология. 1971. Т. 20. №1. С. 78-80.
187. Беляев Н. Ф., Вигдергауз М. С. // Журн. аналит. химии. 1986.
Т.41. №1. С 116-120.
188. Smith R. М. ff Advances, in Chromatogr. 1987. Vol. 26. Р. 277-319.
189. Retention and selectivity in liquid chromatography / Ed by
R.M. Smith. Amsterdam, etc.: Elsevier, 1995. 402 p.
190. Sun Y., Zhang R., Wang Q e. a // J. Chromatogr. 1993. Vol. 657.
№1. P.1-15.
191. Van-den-Dool H., Kratz P.D //J. Chromatogr. 1963. Vol. 11 №4.
P. 463-471.
192. Budahegyi M. V., Lombosi E. R., Lombosi T. S. e. a. // J. Chroma-
togr. 1983. Vol.271. №3. P.213-307.
193. Качественный газохроматографический анализ / M. С. Виг-
дергауз, Л. В. Семенченко, В. А.Езрец, Ю. Н. Богословский.
М.: Наука, 1978, 244 с.
592
194. Зенкевич И. Г. // Журн. аналит. химии. 1984. Т. 39. №7.
С.1297-1307.
195. Zenkevich I. G., Ioffe В. V. // J. Chromatogr. 1988. Vol. 439. №2.
Р. 185-194.
196. Зенкевич И. Г., Щепаняк Л. М. //Журн. аналит. химии. 1992.
Т.47. №3. С. 507-513.
197. Головня Р. В., Светлова Н. И. //Журн. аналит. химии. 1988.
Т.43. №5. С. 859-865.
198. Мариничев А.Н., Турбович М.Л., Зенкевич И. Г. // Физико-
химические расчеты на микро-ЭВМ. Л.: Химия, 1990. 262 с.
199. Зенкевич ИГ // Журн. прикл. химии. 1994. Т.67. №11.
С.1877-1882.
200. Зенкевич И. Г. // Журн. прикл. химии. 1995. Т. 68. №8.
С. 1321-1327.
201. Zenkevich I. G., Kuznetsova L. М. // Collect. Czech. Chem. Comm.
1991. Vol. 56. №10. P. 2042-2054.
202. Зенкевич И. Г. fj Журн. эколог, химии. 1994. Т.З. № 1.
С.39-48.
203. Киселев А. В., Пошкус Д.П., Яшин Я. И. Молекулярные осно-
вы адсообционной хроматографии. М.: Химия, 1986. С. 39-
41.
204. Quintanilla-Lopez J. Е., Lebron-Aguilar R., Tello А. М. е.а. // J.
Chromatogr. А. 1996. Vol. 721. №1. Р. 147-155.
205. Зенкевич И. Г., Цибульская И. А. // Журн. аналит. химии.
1989. Т.44. № 1. С. 90-96.
206. Yin Н. F., Zhu Y., Sun Y. L. // Chromatographia. 1989. Vol. 28.
№9/10. P. 502-504.
207. Golovnya R. V., Samusenko A.L., Mistryukov E.A. // J. High
Resohit. Chromatogr. & Chromatogr. Comm. 1979. Vol. 2. №10.
P.609-612.
208. Вигдергауз M. С., Пахомова В. И. // Заводск. лаб. 1986. Т. 52.
№7. С. 19-22.
209. Березкин В. Г., Герасименко В. А., Набивач В. М. // Журн.
аналит. химии. 1996. Т.51. №4. С.410-418.
210. Березкин В. Г. Газо-жидко-твердофазная хроматография. М.:
Химия, 1986. 112 с.
211. Березкин В. Г. // Журн. аналит. химии. 1996. Т. 51. №2.
С. 148-155.
212. Березкин В. Г., Королев А. А. // Журн. аналит. химии. 1993.
Т.48. №8. С. 1355-1360.
213. Чарыков А. К. Математическая обработка результатов хими-
ческого анализа. Л.: Химия, 1984. 168 с.
214 Конюхова С. В., Зенкевич И. Г., Максимов Б. Н. // Журн. ана-
лит. химии. 1994. Т.49. №4. С. 402-409.
593
215. Зенкевич И. Г., Кузнецова Л. М. // Журн. аналит. химии.
1992. Т.47. №6. С. 982-993.
216. Lavine В. К., Mayfield Н., Kromann Р. R. е. а. // Analyt. Chem.
1995. Vol. 67. №21. Р. 3846-3852.
217. Wang Z., Fingas M. // J. Chromatogr. A. 1997. Vol. 774. №1+2.
P. 57-78.
218. Colin H., Guiochon G. // J. Chromatogr. 1978. Vol. 158. P. 183-
205.
219. Вигдергауз M. С., Буланова А. В. // Изв. вузов. Химия и хим.
технол. 1994. Т.37. №7-9. С. 92-97
220. Вигдергауз М. С., Курбатова С. В. //Журн. аналит. химии.
1991. Т.46. №4. С. 683-694.
221. Курбатова С. В., Колосова Е. А. // Успехи газовой хромато-
графии. Изд-во Самарского ун-та, 1992. Вып. 8. С. 57-64.
222. Шаевич А. Б., Мучник В. Л. //Журн. физ. химии. 1993. Т.67.
№4. С. 827-832.
223. Вигдергауз М. С., Ревинская Е. В., Лобачев А. Л и др. // Изв.
вузов. Химия и хим. технол. 1995. Т. 38. №1. С. 35-42.
224. Tarjan G., Nyiredy Sz., Gyor M. e. a. // J. Chromatogr. 1989.
Vol. 472. №1. P.1-92.
225. Takacs J. M. // J. Chromatogr. Sci. 1991. Vol. 29. №9. P. 382-389.
226. Зенкевич И. Г., Шляпникова Ю.Д., Чупалов А. А. Ц Вести.
СПбГУ. Сер. 4. Физика, химия. 1993. Вып.3 (№18). С.66-73.
227. Богословский Ю.Н., Анваер Б. И., Вигдергауз М. С. Хромато-
графические постоянные в газовой хроматографии. Углево-
дороды и кислородсодержащие соединения. М.: Изд-во стан-
дартов, 1978. 192 с.
228. Король А. Н. Неподвижные фазы в газожидкостной хромато-
графии: Справочник. М.: Химия, 1985. 240 с.
229. Yasuhara A., Morita М., Fuuia К. // J. Chromatogr. 1985. Vol. 328.
Р. 35-48.
230. Pfleger К., Maurer FL, Weber A. Mass Spectral and GC Data of
Drugs, Poisons and Their Metabolites. Weinheim: VCH Verlagsge-
sellschaft mbH, 1985. P. 1. 208 p.
231. Gas-Chromatographic Retention Indices of Toxicologically Relevant
Substances on SE-30 or OV-1. 2-nd ed. Weinheim: VCH Verlagsge-
sellschaft mbH, 1985. 175 p.
232. Lora-Tamayo C., Rams M.A., Chacon J.M.R. // J Chromatogr.
1986. Vol. 374. №1. P. 73-85.
233. Шаевич А.Б., Мучник В. Л., Рабинович В. А. и др. Веще-
ства органические. Индексы хроматографического удержи-
вания. Таблицы рекомендуемых справочных данных. М.:
Гос. служба станд. справ, данных, 1991. 47 с.
594
234. Donelly J. R., Abdel-Hamid M.S., Jetter J.L. e. a. // J. Chroma-
togr. 1993. Vol. 642. №1+2. P. 409-415.
235. Еремин C.K., Изотов Б.Н., Веселовская H. В. Анализ нарко-
тических средств. Руководство по химико-токсикологическо-
му анализу наркотических и других одурманивающих
средств / Под ред. Б. Н. Изотова. М.: Мысль, 1993. 259 с.
236. Вигдергауз М.С., Курбатова С. В., Матусов М. И. и др. //
Экспресс-информация. Хим. пром-ть. Сер. “Автоматизация
хим. производств”. М.: НИИТЭХИМ, 1988. Вып.8. С.8-14.
237. Вигдергауз М. С., Колосова Е.А., Курбатова С. В. // Заводск.
лаб. 1993. Т. 59. №6. С. 7-10
238. Сергеев С. К., Прохорова Е. К. // Заводск. лаб. 1993. Т. 59.
№6. С. 1-6.
239. Прохорова Е.К. //Журн. аналит. химии. 1995. Т.50. №10.
С. 1017-1022.
240. Ioffe В. V., Zenkevich I. G., Bureiko A. S. // Collect. Czech. Chem.
Comm. 1991. Vol. 56. №3. P. 590-594.
241. Буреико A.C., Зенкевич И. Г., Иоффе Б. В. ff Вести. СПбГУ.
Сер. 4. Физика, химия. 1992. Вып. 1 (№4). С.57-61.
242. Яшин Я. И. //Журн. аналит. химии. 1985. Т.40 №2. С. 201-
214.
243. Банк хромато-спектрометрических данных для идентифика-
ции органических примесей в атмосферном воздухе // Журн.
эколог, химии. 1993. Т. 2. №1. Рекламная информация на 3-й
с. обложки.
244. Исидоров В. А., Зенкевич И. Г. Хромато-масс-спектрометри-
ческое определение следов органических веществ в атмо-
сфере. Л.: Химия, 1982. 136 с.
245. Gupta Р.К., Nikelly J.G. // Analyt. Chem. 1991. Vol. 63. №13.
P. 1264-1270.
246. Chromatography in petroleum industry/ Ed. by E. R.. Adlard. Am-
sterdam etc.: Elsevier, 1995. P. 231-268.
247. Multidimensionality. Hyphenation and coupled-column techniques/
Ed by U.A.Th. Brinkman, H. Poppe //J. Chromatogr. A. 1995.
Vol. 703. №1+2. P. 1-692.
248. Brinkman U.A. Th. // Analyt. Comm. 1997. Vol 34. №7. P.9H-
12H.
249. Multidimensional chromatography / Ed. by H.I. Coates. New York
& Basel: M. Dekker, Inc., 1990. 357 p.
250. Схунмакерс П. Оптимизация селективности в хроматогра-
фии / Пер. с англ. Под ред. В. А. Даванкова. М.: Мир,
1989. 399 с.
251. Терентьев В. А., Вигдергауз М. С. //Журн. аналит. химии.
1985. Т.40. №9. С. 1670-1674.
595
252. Polanuer В. M., Zhuravleva I.L., Golovnya R. V. Ц J Chromatogr.
1986. Vol. 365. P. 399-404.
253. Карцова Л. А., Столяров Б.В. //Журн. аналит. химии. 1993.
Т.48. №4. С. 582-591.
254. Карцова Л. А., Маркова О. В. //Современные проблемы орга-
нической химии. Изд-во С.-Петербург, ун-та, 1998. Вып. 12.
С. 233-251.
255. Шляхов А. Ф., Анваер Б. И., Золотарева О. В. и др. // Журн.
аналит. химии. 1975. Т.30. №4. С. 788-792.
256. Зенкевич И. Г., Цибульская И. А., Родин А. А. // Журн. ана-
лит. химии. 1991. Т.46. №1. С. 101-110.
257. Ooms В., Spark Н.В. V. // LC & GC Intern. 1996. Vol. 9. №9.
Р. 574-585.
258. Antia F.D., Horvath Cs. // J. Chromatogr. 1988. Vol.435. №1.
P.1-15.
259. Sandler L. C., Wise S. A. // Analyt. Chem. 1989. Vol. 61. №15.
P. 1749-1754.
260. Bowermaster J., McNair H.M. // J. Chromatogr. Sci. 1984.
Vol. 22. №4. P. 165-170.
261. Головня P. В., Григорьева Д.Н. // Журн. аналит. химии.
1985. Т.40. №3. С. 515-526.
262. Peng С. Т. // J. Radioanalyt. & Nuclear Chem. Articles. 1992.
Vol. 160. №2. P. 449-460.
263. Peng С. T. // J. Chromatogr. A. 1994. Vol.678. №2. P. 189-200.
264. Хроматораспределителъный метод / В. Г. Березкин, В. Д. Ло-
щилова, А. Г. Панков, В. Д. Ягодовский. М/. Наука, 1976.
112 с.
265. Зенкевич И. Г., Васильев А. В. //Журн. аналит. химии. 1993.
Т.48. №3. С. 473-486.
266. Головня Р.В., Григорьева Д. Н. // Журн. аналит. химии.
1985. Т.40. №2. С. 316-329.
267 Григорьева Д.Н., Головня Р. В. // Журн. аналит. химии.
1985. Т.40. №10. С. 1733-1760.
268. Набивач В. М., Дмитриков В. П. //Успехи химии. 199,3. Т. 62.
№1. С. 27-38.
269. Smith R.M., Burr С.М. // J. Chromatogr. 1989. Vol.475. Р.57-
74.
270. Зенкевич И. Г., Конюхова С. И., Максимов Б. Н. //Журн. физ.
химии. 1993. Т. 67. № 7. С. 1474-1479.
271. Зенкевич И. Г. // Журн. аналит. химии. 1995. Т. 50. №10.
С. 1048-1056.
272. Березкин В. Г. Аналитическая реакционная газовая хромато-
графия. М.: Наука, 1966. 184 с.
596
273. Заикин В. Г., Микая А. И. Химические методы в масс-спектро-
метрии органических соединений. М.: Наука, 1987. С. 96-165.
274. Алексеева К. В. Пиролитическая газовая хроматография. М.:
Химия, 1985. 256 с.
275. van Lieshout М. Р. М, Janssen H.-G., Cramers С. А. е. а. // J.
Chromatogr. А. 1997. Vol. 764. № 1. Р. 73-84.
276. Attygalle А. В., Morgan E.D. // J. Chromatogr. 1984. Vol. 290.
Р. 321-330.
277. Методика определения концентрации оксида углерода и ме-
тана методом реакционной газовой хроматографии // Сбор-
ник методик по определению концентраций загрязняющих ве-
ществ в промышленных выбросах. Л.: Гидрометеоиздат,
1987. С. 49-54.
278. Kuzmenko Т.Е., Samusenko A.L., Uralets V.P. e.a. // J. High
Resohit. Chromatogr. & Chromatogr. Comm. 1979. Vol. 2. №1.
P. 43-44.
279. Бероза M., Инской M. // Методы-спутники в газовой хрома-
тографии I Пер. с англ. Под ред. В. Г. Березкина. М.: Мир,
1972. С. 108-127.
280. Thompson C.J., Coleman H.J., Hopkins R. L. e.a. // J. Gas
Chromatogr. 1967. Vol. 5. №1. P. 1-10.
281. Андерсон А. А. Газовая хроматография аминосоединений.
Рига: Зинатне, 1982. 374 с.
282. Cooke М., Nickless G., Roberts D. J. // J. Chromatogr. 1980.
Vol. 187. №1. P. 47-55.
283. Столяров Б. В., Карцова Л. А., Филиппова O.B. //Журн. ана-
лит. химии. 1997. Т.52. №5. С 454-468.
284. Kartsova L.A., Balakin I.M., Stolyarov В. V. // Chromatographia.
1978. Vol. 11. №12. P. 720-724.
285. Столяров Б. В., Шаварда А. Л. // Журн. орг. химии. 1978.
Т.14. №2. С. 202-203.
286. Lingeman Н., Brinkman U. A. Th. // Sample preparation for biome-
dical and environmental analysis / Ed. by D. Stevenson, I. D. Wilson.
New York & London: Plenum Press, 1994. P. 21-35.
287. Brinkman U. A. Th., Frei R. W., Lingeman H. // J. Chromatogr.
Biomed. Appl. 1989. Vol. 492. P. 251-298.
288. Баффингтон P., Уилсон M. Детекторы для газовой хромато-
графии / Пер. с нем. Под ред. В. Г. Березкина. М.: Мир,
1993. 79 с.
289. Bemgoard A., Colmsjd А. // J. Chromatogr. Sci. 1992. Vol. 30.
№1. Р. 23-28.
290. Stan H.-J., Heil S. // Fresenius J. Anal. Chem. 1991. Vol. 339.
№1. P. 34-39.
597
291. Арутюнов Ю.И., Вигдергауз М. С. //Журн. аналит. химии.
1994. Т.49. №8. 796-803.
292. Вигдергауз М. С., Арутюнов Ю.И., Курбатова С. В. и др. //
Журн. аналит. химии. 1994. Т.49. №10. С. 1067-1072.
293. Вигдергауз М. С., Арутюнов Ю.И., Колосова Е.А. и др. ))
Журн. аналит. химии. 1995. Т.50. №8. С. 827-833.
294. Курбатова С. В., Колосова Е.А., Вигдергауз М.С. // Журн.
аналит. химии. 1993. Т.48. №5. С.864-869.
295. Зенкевич И. Г., Косман В. М. // Журн. аналит. химии. 1996.
Т. 51. №8. С. 870-875.
296. Карасек Ф., Клемент Р. Введение в хромато-масс-спектро-
метрию / Пер. с англ. М.: Мир, 1993. 237 с.
297. Баффингтон Р. Применение атомно-эмиссионной спектроско-
пии в высокочастотном разряде для газовой хроматографии /
Пер. с англ. М.: Мир, 1994. 78 с.
298. Хмельницкий Р. А., Бродский Е. С. Хромато-масс-спектро-
метрия. М.: Химия 1984. 212 с.
299. McLafferty F. W., Stauffer D. В. Important Peak Index of the Regis-
try of Mass-spectral Data. New York: Palisade Corp., 1991.
300. Зенкевич И. Г., Иоффе Б. В. Интерпретация масс-спектров ор-
ганических соединений. Л.: Химия, 1986. 176 с.
301. Malissa Н., Jr., Kreindl Т., Winsauer К. // Fresenius J. Analyt.
Chem. 1990. Vol. 337. №7. P. 843-847.
302. Белл P. Дж. Введение в фурье-спектроскопию / Пер. с англ.
Под ред. Г. Н. Жижина. М.: Мир, 1975. 380 с.
303. Griffiths P.R., de Haseth J. A., Azarraga L. V. // Analyt. Chem.
1983. Vol. 55. №13. P. 1361A-1387A.
304. Ragunathan N., Krock K.A., Klauun C. e.a. // J. Chromatogr.
1995. Vol. 703. №1+2. P. 335-382.
305. Ragunathan N., Sasaki T.A., Krock K. A. e.a. ff Analyt. Chem.
1994. Vol. 66. №21. P. 3751-3756.
306. Griffiths P. R., de Haseth J. A. // Fourier Transform Infrared Spec-
troscopy. New York: Wiley, 1986. P. 611-647.
307. Dwyer J.L., Chapman A.E., Lin X. // LC & GC Intern. 1995.
Vol. 8. №12. P.704-710.
308. Gurka D. F., PyleS., Titus R. e.a. // Analyt. Chem. 1994. Vol. 66.
№15. P. 2521-2528.
309. Uden P. C. // J.Chromatogr. A. 1995. Vol. 703. №1+2. P. 393-416.
310. Uden P. C. // Selective detectors. Environmental, industrial and
biochemical applications / Ed. by R. E. Sievers. New York: Wiley,
1995. P. 143-169.
311. Anderson J.T., Schmid B. // Fresenius J. Analyt. Chem. 1993.
Vol. 346 №4. P. 403-409.
598
312. Sensitive and selective universal element detection for routine or
research analyses. HP G2350A Atomic emission detector for gas
chromatography. Проспект фирмы Хьюлетт-Паккард. Hewlett-
Packard Co., 1995. 6 p.
313. Slatkavitz K.J., Hoey L.D., Uden P. C. e. a. // Analyt. Chem.
1985. Vol. 57. №9. P. 1846-1853.
314. Степанов А. И., Гехман A.E., Рамендик Г. И. и др. // Рос.
хим. журн. 1995. №4. С. 27-31.
315. Пильдус И. Э., Вехтер Е. П. Опыт использования HP 5921
АЭЛ. Доклад на семинаре “Плазменные газохроматографи-
ческий и масс-спектрометрический анализаторы фирмы HP”.
Москва, 1 марта 1995 г.
316. Ke-Wei Z., Qing-Yu О., Guo-Chuen W. е. а. //Spectrochim. Acta.
1985. Vol.40В. №1/2. P. 349-356.
317. Uden P. C., Slatkavitz K. J., Barnes R. M. // Analyt. Chim. Acta.
1986. Vol. 180. P. 401-416.
318. Valente A.L.P., Uden P. C. // Analyst. 1990. Vol. 115. №5.
P. 525-529.
319. Yieri Huanq, Qingyu Ou, Weile Yu. // J. Chromatogr. Sci. 1990.
Vol. 28. №11. P. 584-588.
320. Webster C., Cooke M. // Anal. Proc. 1994. Vol.31. №8. P.237-
240.
321. Иоффе Б.В., Витенберг Д. A. // Журн. аналит. химии. 1996.
Т. 51. №8. С. 876-881.
322. Пилъдус И.Э., Вехтер Е. П. // Хроматогр. журн. 1995. №4.
С. 63-70.
323. Slatkavitz К. J., Uden Р. С., Hoey W. е. а. // J. Chromatogr. 1984.
Vol. 302. Р. 277-287.
324. Бражников В. В. Детекторы для хроматографии. М.: Маши-
ностроение, 1992. 317 с.
325. Оган К. // Количественный анализ хроматографическими ме-
тодами / Ред. Э.Кэц. Пер. с англ. Под ред. В. Г. Березкина.
М.: Мир, 1990. С. 31-56.
326. Meyer V. Practical High Performance Liquid Chromatography. New
York: Wiley, 1988. 280 p.
327. Rothman L. D. // Analyt. Chem. 1996. Vol. 68. №12. P. 587R-
598R.
328. Torsi G., Chiavari G., Lippolis M. T. // LC & GC Intern. 1992.
Vol. 5. №1. P. 37-39.
329. Torsi G., Chiavari G., Laghi C. e. a. // J. Chromatogr. 1990.
Vol. 518. №1. P. 135-140.
330. Grob K., Bronz M. // J. High Resolut. Chromatogr. 1993. Vol. 16.
№2. P. 121-122.
599
331. French С.Д., Gray C.J., Lehrle U.S. // J. Chromatogr. A. 1997.
Vol. 773. № 1+2. P. 372-376.
332. Jorgensen A.D., Picel K.C., Stamoudis V.C. // Aualyt. Chem.
1990. Vol. 62. №7. P. 683-689.
333. Dressier M., Ciganek M. // J. Chromatogr. A. 1994. Vol. 679. №2.
P. 299-305.
334. Арутюнов Ю.И., Гришин АП.// Заводск. лаб. 1972. Т. 28.
№12. С. 1441-1443.
335. Краузе Н. М. // Хроматографический анализ углеводородов
и их производных. Межвуз. сб. Изд-во Куйбышевского ун-та,
1985. С.126-130.
336. Вигдергауз М. С., Краузе Н.М. //Журн. аналит. химии. 1986.
Т.41. №11. С. 2064-2074.
337. Витенберг А. Г., Столяров Б. В., Смирнова С. А. // Вести.
ЛГУ. 1977. № 16. Сер. физ. и хим. С. 132-139.
338. Ioffe В. V., Vitenberg A.G. // Chromatographia. 1978. Vol. 11.
Р. 282-286.
339. Иоффе Б. В., Столяров Б. В., Смирнова С. А. // Журн. ана-
лит. химии. 1978. Т.ЗЗ. С. 2196-2201.
340. McAulife С. // Chem. Tech. 1971. Vol. 1. Р. 46-51.
341. Kolb В., Pospicil Р. // Chromatographia. 1977. Vol. 10. P. 705-709.
342. Иоффе Б. В., Резник Т.Л. // Журн. аналит. химии. 1981.
Т.36. С.2191-2198.
343. Витенберг А. Г. // Докл АН СССР. 1982. Т. 267. № 1. С. ИЗ-
117.
344. Modern thin-layer chromatography / Ed by N. Grinberg. New York:
M. Dekker, 1990. 252 p.
345. Stahl E., Mangold H. -K. // Chromatography / Ed. by E. Heftmann.
3rd ed. New York etc.: Van Nostrand Reinhold, 1975. P. 164-189.
346. Touchstone J. C, Ed. Techniques and Applications of Thin Layer
Chromatography. New York, etc., 1985. 395 p.
347. Geiss F. Fundamentals of Thin Lauer Chromatography: (Planar
Chromatography). Heidelberg, etc.: Hiithig, 1987. 482 p.
348. Hamilton R. J., Hamilton S. Thin Layer Chromatogaphy / Ed. by
D. Kealey. Chichester, etc.: Wiley, 1987. 129 p.
349. Touchstone J. C. Practice of Thin Layer Chromatography. 3rd ed.
New York, etc.: Wiley & Sons, Inc., 1992. 377 p.
350. Sherma J. // Analyt. Chem. 1994. Vol. 66. №12. P. 67R-83R.
351. Sherma J. // Rev. Analyt. Chem. 1995. Vol. 14. P. 75-142.
352. Sherma J. Ц Analyt. Chem. 1996. Vol. 68. №12. P. 1R-19R.
353. Fried B., Sherma J. Practical Thin Layer Chromatography. A
Multidisciplinary Approach. Boca Raton: CRC Press Inc., 1996.
336 p.
600
354. Kalasz H., Bathori M. LC-GC Intern. 1997. Vol. 10. №7. P. 440-
445.
355. Kalasz H., Ettre L. S., Bathori M. // LC—GC. 1997. Vol. 15. №11.
P. 1044, 1046-1050.
356. Cserhati T., Forgoes E. // J. Chromatogr. Sci. 1997. Vol.35. №8.
P. 383-391.
357. Roberts T. R. Radiochromatography: The Chromatography and
Electrophoresus of Radiolabelled Compounds. Amsterdam, etc.:
Elsevier, 1978. 174 p.
358. Высокоэффективная тонкослойная хроматография / Ред.
А. Златкис, Р. Кайзер. Пер. с англ. Под ред. В. Г. Березкина.
М.: Мир, 1979. 245 с.
359. Planar Chromatography / Ed. by R.E. Kaiser with contrib. by
R.M.Belchamber, K.D.Burger, M. llomotoe.a. Heidelberg, etc.:
Huethig, 1986. Vol. 1. 265 p.
360. Воронцов A.M. //Журн. эколог, хим. 1993. №1. С.33-43.
361. Kaiser R.E. // J. High Resolut. Chromatogr. & Chromatogr.
Comm. 1978. Vol.l. №3. P. 164-168.
362. Kaiser R. E. //J. Planar Chromatogr. 1989. Vol. 2. №4. P. 323-
326.
363. Thin Layer Chromarography: Reagents and Detection Methods.
Vol. la. Physical and Chemical Detection Methods.: Fundamentals,
Reagents I / H.Jork, W.Funk, W. Fischer e.a. Weinheim, etc.:
VCH, 1990. 464 p.
364. Thin Layer Chromatography: Reagents and Detection Methods.
Vol. Ib: Physical and Chemical Detection Methods: Activation
Reactions, Reagents Sequences, Reagents II / H.Jork, W.Funk,
W.Fischer e.a. Weinheim: VCH, 1994. 496 p.
365. Introduction to High-Pressure Thin Layer Chromatography, HPTLC
/ By ed. R.E.Kaiser. Inst. Chromatog.: Bad Duerkheim. 1976.
235 p.
366. Halpaap H., Krebs K.-F. //J. Chromatogr. 1977. Vol. 142. P. 823 -
853.
367. Poole C. F., Schuette S. A. Contemporary Practice of Chromatogra-
phy. Amsterdam, etc.: Elsevier, 1984. P. 619-700.
368. Kaiser R., Reider R. // Planar Chromatography / Ed. by R. E. Kai-
ser with contrib. by R. M. Belchamber, K.D. Burger, M. Domoto
e.a. Heidelberg, etc.: Huethig, 1986. Vol.l. P. 165-191.
369. Kaiser R. Einfiihrung in die HPPLC. Heidelberg, etc.: Hiithig, 1987.
256 S.
370. Zuber G.E., Warren R.J., Begosh P. P. e.a. // Analyt. Chem.
' 1984. Vol. 56. №14. P. 2935-2939.
371. Brown St.M., Schulz H., Busch K.L. // J. Planar Chromatogr.-
Mod. TLC. 1990. Vol.3. №5-6. P. 222-227.
601
372. Koglin E. // J. Planar Chromatogr.- Mod. TLC. 1993. Vol.6. №2.
P. 88-92.
373. Somsen G. W., Morden W., Wilson I.D. // J. Chromatogr. A. 1995.
Vol. 703. № 1 + 2. P. 613-665.
374. Junker-Buchheit A., Jork H. // J. Planar Chromatogr. 1988. Vol. 1.
№3. P. 214-219.
375. Мотл О., Новотный Л. // Лабораторное руководство по хро-
матографическим и смежным методам / Под ред. О. Микеша.
Пер. с англ. Под ред. В. Г. Березкина. М.: Мир, 1982. Ч.П.
С.485-552.
376. Ляхин Д.В., Гимпельсон В. Г., Березкин В. Г. // Журн. ана-
лит. химии. 1997. Т.52. №11. С. 1163-1167.
377. Березкин В. Г., Бузаев В. В. // Изв. АН. Сер. хим. 1996. №5.
С.1143-1146.
378. Березкин В. Г.. Гугля Е.Б. // Изв. АН. Сер. хим. 1996. №3.
С.771-773.
379. Berezkin V. G., Buzaev V. V. // J. Chromatogr. A. 1997. Vol. 758.
№1. P. 125-134.
380. Zakaria M., Gonnord M.-F., Guiochon G. // J. Chromatogr. A.
1983. Vol.271. №2. P. 127-192.
381. Келлер Г. 11 Руководство по современной тонкослойной хро-
матографии / Под ред. О.Г. Ларионова. М., 1994. С. 165-202.
382. Instrumental HPTLC: Proc, of the First Intern. Symp. on
Instrumental High Performance Thin Layer Chromatography
(HPTLC), Bad Durkheim (West Germany), May 18-21,1980. With
contrib. M. Amin, U. A. Th. Brinkman, V. Damman e. a. / Ed by
W.Bertsch e.a. Heidelberg, etc.: Huthig, 1980. 390 p.
383. Poole C.F., Poole S.K. // J. Chromatogr. 1989. Vol.492. P.539-
584.
384. Vovk I., Prosek M. //J. Chromatogr. 1997. Vol. 768. №2. P. 329-
333.
385. Kaiser R.E., Oelrich E. Optimization in HPLC. Heidelberg: Hue-
thig, 1981. 278 p.
386. Nurok D., Kleyle R. M., Hajdu P. e. a. // Analyt Chem., 1995. Vol.
67. №23. P. 4423-4430.
387. Пул К. Ф Хатиб С. 11 Количественный анализ хроматогра-
фическими методами / Ред. Э. Кэц. Пер. с англ. Под ред.
В. Г. Березкина. М.: Мир, 1990. С. 156-209.
388. Ганкина Э. С. // Аналитическая хроматография / К. И.Са-
кодынский, В. В. Бражников, С. А. Волков и др. М.: Химия,
1993. С. 368-373.
389. Ebel S. // J. Planar Chromatogr. — Mod. TLC. 1996. Vol. 9. №1.
P.4-15.
602
390. Janchen D. // Chromatography. Celebrating Michael Tswett’s 125
Birthday / Ed. by O. Kaiser, R. Kaiser. InCom Sonderband, 1997.
P. 241-244.
391. Селиванов M.H., Фридман А.Э., Кудряшова Ж. Ф. Качество
измерений. Метрологическая справочная книга. Л.: Лениз-
дат, 1987. 295 с.
392. Термины, определения и обозначения метрологических ха-
рактеристик анализа вещества // Журн. аналит. химии.
1975. Т.30. №10. С. 2058-2063.
393. Единицы физических величин. Сборник нормативно-техни-
ческих документов. М.: Изд-во стандартов, 1987. 176 с.
394. МИ 2247-93. Рекомендация. ГСИ. Метрология. Основные
термины и определения. СПб.: ВНИИМ им. Д. И. Менделе-
ева, 1994. 61 с.
395. Основные термины в области метрологии. Словарь-справоч-
ник. М.: Изд-тю стандартов, 1989. ИЗ с.
396. ГОСТ 87315-91. ГСИ. Стандартные образцы состава и
свойств веществ и материалов. Основные положения. М.:
Изд-во стандартов, 1991. 20 с.
397. МИ 1317-86. Методические указания. ГСИ. Результаты и
характеристики погрешностей измерений. Формы предста-
вления. М.: Изд-во стандартов, 1986. 30 с.
398. ГОСТ Р 8. 563-96. ГСИ. Методики выполнения измерений.
М.: Изд-во стандартов. 1996. 18 с.
399. Guide to the expression of uncertainty in measurement. Geneva:
ISO, 1993.
400. Количественное описание неопределенности в аналитических
измерениях / Пер. документа ЕВРАХИМ. СПб.: 1997. 93 с.
401. Колмановский В. И. // Законодат. и прикл. метрология.
1998. №2. С. 45-52.
402. Конопелъко Л. А. Разработка и исследование системы метро-
логического обеспечения газоаналитических приборов: Ав-
тореф. докт. дис. ВНИИМ им. Д. И. Менделеева. СПб.,
1997.28 с.
403. Миф Н. П. Методики выполнения измерений. Методический
материал в помощь метрологам. М.: Изд-во ТОО “Тот”,
1996. 35 с.
404. Ловелок Дж. // Газовая хроматография. Труды Третьего
Междунар. симп. по газовой хроматографии в Эдинбурге /
Пер. с англ. Под ред. А. А. Жуховицкого и Н. М. Туркельтау-
ба. М.: Мир, 1964. С. 40-42.
405. Рабинович С. Г. Погрешности измерений. Л.: Энергия, 1978.
262 с.
603
406. ГОСТ 8.207-76. ГСИ. Прямые измерения с многократными
наблюдениями. Методы обработки результатов наблюдений.
М.: Изд-во стандартов, 1981. 12 с.
407. Грановский Б. А., Сирая Т. Н. Методы обработки эксперимен-
тальных данных при измерениях. Л.: Энергоатомиздат, 1990.
288 с.
408. Румшиский Л. 3. Математическая обработка результатов
эксперимента. М.: Наука, 1971. 192 с.
409. Пустпылъник Е. И. Статистические методы анализа и обра-
ботки наблюдений. М.: Наука. 1968. 288 с.
410. Методы обработки результатов наблюдений при измерениях
//Труды метрологических институтов СССР. 1972. Вып. 134
(194). С.31.
411. Алимов Ю.И., Шаевич А. Б. // Журн. аналит химии. 1988.
Т.43. №10. С. 1893-1915.
412. Семенов Л. И., Сирая Т.Н. Методы построения градуировоч-
ных характеристик средств измерения. М.: Изд-во стандар-
тов, 1986. 128 с.
413. Колмановский В. И. // Измерительная техника. 1998. №2.
С. 56-66.
414. Айвазян С. А., Енюков И. С., Мешалкин Л.Д. Прикладная ста-
тистика. Основы моделирования и первичная обработка дан-
ных: Справ, изд. М.: Финансы и статистика, 1983. С. 284.
415. Дёрффелъ К Статистика в аналитической химии / Пер. с нем.
Под ред. и с предисл. Ю.П. Адлера. М.: Мир, 1994. 268 с.
416. Количественный химический анализ вод. Методика выпол-
нения измерений массовой концентрации летучих хлориро-
ванных углеводородов в питьевых, хозяйственно-бытовых и
поверхностных водах методом газо-жидкостной хроматогра-
фии. ПНД. Ф 14.1:2:4. 10-95. Минприроды. М., 1995.
417. Рабинович Б. А., Хавин З.Я. Краткий химический справоч-
ник. / Под общ. ред. А. А. Потехина и А. И. Ефимова. 4-е
изд., стер. СПб.: Химия, 1994. 432 с.
418. Химические реактивы и высокочистые химические вещества:
Каталог. М.: Химия, 1993.
419. Ротин В. А. Радиоионизационное детектирование в газовой
хроматографии. М.: Атомиздат, 1974. С. 114-168.
420. МИ 137-77. Методика по нормированию метрологических ха-
рактеристик, градуировке, поверке хроматографических при-
боров универсального назначения и оценке точности резуль-
татов хроматографических измерений. М.: Изд-во стандар-
тов, 1977.
421. Качественный анализ летучих органических примесей в во-
дах РОС-350, питьевых и поверхностных водах г. Дзержинс-
604
ка. Технический отчет. НИИ полимеров им. В. А. Каргина.
Дзержинск, 1993.
422. Определение состава летучих органических загрязнителей
вод РОС-350. Отчет о НИР (заключительный). НИИ по-
лимеров им. В. А. Каргина. Дзержинск, 1994.
423. Колмановский В. И., Жуховицкий А. А. // Газовая хромато-
графия. Труды III Всесоюз. конф, по газовой хроматогра-
фии / Глав. ред. А. А. Жуховицкий. Дзержинск: Дзержин.
филиал ОКБА, 1966. С. 93-105.
Глава V
424. Еникеева А. Г., Столяров Б. В. // Вести. СПбГУ. Сер. 4.
Физика, химия. 1997. Вып.3 (№18). С.62-68.
425. Фритц Дж., Шенк Г. Количественный анализ / Пер. с англ.
Под ред. Ю. А. Золотова. М.: Мир, 1978. С. 26-43.
426. Представление результатов химического анализа (рекомен-
дации ШРАС 1994 г.) // Журн. аналит. химии. 1998. Т. 53.
№9. С. 999-1008.
427 Зенкевич И. Г. // Вести. СПбГУ. Сер. 4. Физика, химия.
1998. Вып. 2 (№11). С. 84-90.
428. Courts R. Т., Hargescheimer Е. Е., Pasutto F. М. // J. Chromatogr.
1980. Vol. 195. №1. Р. 105-112.
429. Courts R. Т., Hargescheimer Е. Е., Pasutto F. М. // J. Chromatogr.
1979. Vol. 179. №2. P. 291-299.
430. Neu H.-J., Ziemer W., Merz W. // Fresenius J. Anal. Chem. 1991.
Vol. 340. №2. P. 65-70.
431. Еникеева А. Г., Столяров Б. В. //Вести. СПбГУ. Сер. 4.
Физика, химия. 1998. вып. 4 (№25). С.72-80.
Глава VI
432. Гускова В.П., Беляева Р. Ф., Георгиев Е. С и др. // Гигиена
и санитария. 1995. №2. С. 50-51.
433. Золотов Ю.А. // Журн. аналит. химии. 1997. Т.52. №8.
С. 789.
434. Карцова Л. А., Макарова Я. Л., Столяров В. В. // Журн. ана-
лит. химии. 1997. Т.52. №4. С. 380-383.
605
ОГЛАВЛЕНИЕ
Предисловие . . .............. .................. . . . 3
Глава I. Возникновение и основные этапы развития
хроматографии. Классификация методов
газовой и жидкостной хроматографии...........................6
Глава II. Аппаратура для газовой хроматографии .... 25
II.1. Принципиальная схема, основные системы и узлы
газового хроматографа ...................................—
II. 1.1. Система подготовки газов . 27
II. 1.2. Дозирующие устройства .....................35
II.1.3. Хроматографические колонки..................53
II. 1.4. Детекторы..................................57
II.1.4.1. Общие сведения..........................—
II. 1.4.2. Характеристики детекторов ............61
П.1.4.3. Детектор по теплопроводности .... 69
П. 1.4.4. Принципы ионизационного
детектирования .... .74
II.1.4.5. Пламенно-ионизационный детектор .... 78
П.1.4.6. Электронозахватный детектор .88
IL1.4.7. Термоионный детектор....................95
II. 1.4.8. Пламенно-фотометрический детектор . . 99
II. 1.4.9. Фотоионизационный детектор ..........104
II.1.5. Система термостатирования..................111
II. 1.6. Программирование температуры колонок . . 116
II. 1.7. Регистрация сигналов детекторов 126
П.2. Лабораторные газовые хроматографы.................130
П.2.1. Хроматографы ОАО “Цвет”.......................—
II.2.2. Хроматографы серии “Цвет-500М”.............131
II.2.2.1. Общие сведения .... ... ... —
П.2.2.2. Аналитический блок.....................135
II.2.2.3. Блок управления .... ..... 140
II.2.2.4. Детекторы.................. . . 143
II.2.2.5. Газовые блоки.........................154
П.2.2.6. Система обработки хроматографичес-
кой информации .... 157
II.2.3. Хроматографы “Цвет-800”....................160
II.2.3.1. Фотоионизационный детектор .... 161
II.2.3.2. Программное обеспечение хроматогра-
фов “Цвет-800”................................ 163
П.2.4. Аналитические принадлежности и
дополнительное оборудование ... 167
Г лава III. Аппаратура для жидкостной хроматогра-
фии ............................................................174
III. 1. Принципиальная схема, основные системы и
узлы жидкостного хроматографа...................—
III. 1.1. Подготовка подвижной фазы............176
IIL1.2. Насосы................................ 178
606
III. 1.3. Системы ввода элюента и анализируемой
пробы ........................................179
III. 1.4. Колонки ... 182
Ш.1.5. Детекторы............................ 183
III. 1.5.1. Основные характеристики детектора ... —
III.1.5.2. Фотометрический детектор .......185
III. 1.5.3. Рефрактометрический детектор .... 189
III.1.5.4. Флуориметрический детектор......192
Ш.1.5.5. Электрохимические детекторы.......194
III .2. Лабораторные жидкостные хроматографы 197
III.2. 1. Хроматограф “Цвет-3006”...............—
III.2. 2. Хроматограф портативный двухканальный
“Цвет-403” (ХПИ-3)...........................201
Ш.2.3. Хроматограф “Цвет-3110”................203
Ш.2.4. Хроматограф “Цвет-404” (ХПЖ-1) . .... 205
III.2. 5. Микроколоночные хроматографы........208
Глава IV. Хроматографический анализ........................210
IV .1. Подготовка пробы к анализу. Выбор хроматогра-
фического метода .................................. —
I V.2. Качественный анализ в газовой и высокоэффек-
тивной колоночной жидкостной хроматографии . . 226
IV.2.1. Стратегия выполнения качественного
анализа, типовые задачи и основные
экспериментальные приемы их решения .... —
IV.2.2. Параметры удерживания.................232
IV.2.3. Источники погрешностей при измерении
параметров удерживания.......................253
IV.2.4. Качественный анализ по параметрам
удерживания................................. 262
IV.2.4.1. Использование стандартных соедине-
ний (метод метки).......................... . —
IV.2.4.2. Использование справочных данных по
удерживанию идентифицируемых сое-
динений различными неподвижными
фазами. Хроматографические спектры.
Многоступенчатые методы разделения 264
IV.2.4.3. Использование корреляционных зави-
симостей параметров удерживания 271
IV.2.5. Реакционная газовая хроматография .... 284
IV.2.5.1. Химические превращения компонентов
пробы на выходе из хроматографической
колонки....................................287
IV.2.5.2. Методика “вычитания” пиков
(образование нелетучих соединений) . 289
IV.2.5.3. Методика “сдвига” пиков..........292
IV.2.6. Постколоночная дериватизация в высокоэф-
фективной жидкостной хроматографии . - 298
IV.2.7. Качественный анализ по относительному
сигналу детекторов............................301
IV.2.8. Хромато-масс-спектрометрия ....... 304
IV.2.8.1. Общие сведения ...................—
607
IV.2.8.2. Принципы масс- и хромато-масс-спектро-
метрии ................................. . . 307
IV.2.8.3. Системы газовый хроматограф —
масс-спектрометр (ГХ — МС) . . . . 308
IV.2.8.4. Системы жидкостный хроматограф —
масс-спектрометр (ЖХ — МС) . . . . 310
IV.2.8.5. Масс-анализаторы. Обработка и пред-
ставление результатов хромато-масс-
спектрометрического анализа . 311
IV.2.9. Хромато-ИК(фурье)-спектроскопия . . . 319
IV.2.10. Атомно-эмиссионное детектирование.
Комплексный подход к решению задач
повышенной сложности ..........................328
IV.3. Количественный анализ в газовой и высокоэф-
фективной колоночной жидкостной хроматографии 338
IV.3.1. Хроматограмма как источник сведений о
количественном составе анализируемой
смеси .........................................339
IV.3.2. Выбор и измерение основных количествен-
ных параметров хроматографических пиков 342
IV.3.3. Основные методы количественного анализа 347
IV.3.3.1. Общие сведения . —
IV.3.3.2. Метод абсолютной градуировки .... 350
IV.3.3.3. Метод внутренней нормализации . . . 354
IV.3.3.4. Метод внутреннего стандарта........358
IV.3.4. Развитие методов количественной интерпре-
тации хроматограмм сложных смесей .... 362
IV.3.5. Методы количественного парофазного анализа 363
IV.3.5.1. Общие сведения ......................—
IV.3.5.2. Анализ систем с известными коэф-
фициентами распределения....................364
IV.3.5.3. Анализ систем с неизвестными коэф-
фициентами распределения .....................368
IV.3.5.4. Количественный ПФА с пневмати-
ческим отбором проб (дискретная
газовая экстракция с неполной заменой
газовой фазы)................................371
IV.3.5.5. Динамический парофазный анализ . . 373
IV.4. Хроматография в тонком слое ..................376
IV.4.1. Общие сведения ...........................—
IV.4.2. Основы метода ТСХ....................... 380
IV.4.3. Методы проявления хроматограмм..........386
IV.4.4. Детектирование разделенных зон 390
IV.4.5. Количественный анализ в ТСХ.............395
IV.5. Обработка результатов хроматографического
анализа с помощью ЭВМ 398
IV.5.1. Регистрация сигналов детекторов .... —
IV.5.2. Аналого-цифровое преобразование . . 399
IV.5.3. Использование персональных ЭВМ в
хроматографии . . 401
IV.5.4. Фильтрация шумов . . . . . . 405
IV.5.5. Поиск пиков и их интегрирование ........406
608
IV.5.6. Идентификация хроматографических пиков
с помощью компьютера. Таблица компонен-
тов . ....... ... 408
IV.5.7. Использование ЭВМ для характеристики
колонки и качества достигнутого разделения 412
IV.5.8. Использование ЭВМ при решении задач
количественного хроматографического ана-
лиза ............................................—
IV.6. Метрология хроматографического анализа 421
IV.6.1. Общие сведения ...........................—
IV.6.2. Метрологическая аттестация аналитических
методик ................................... 429
IV.6.2.1. Общие сведения .... . —
IV.6.2.2. Оценка характеристик погрешности
аналитических методик .... 432
IV.6.2.3. Градуировка хроматографической
аппаратуры ..................................436
IV.6.3. Пример метрологической аттестации МКХА.
Анализ измерительного процесса и физичес-
кая модель методики.......................... . 439
Глава V. Практические работы по газовой хромато-
графии ................................................ 456
Работа 1. Нанесение неподвижной фазы на твердый
носитель и заполнение колонки . . . 462
Работа 2. Влияние скорости газа-носителя на
эффективность работы хроматографической
колонки ....................................... 468
Работа 3. Зависимость чувствительности пламенно-
ионизационного детектора от относительных
расходов газа-носителя, водорода и воздуха
(определение оптимальных условий работы) 471
Работа 4. Зависимость чувствительности детектора
по теплопроводности от тока моста и
скорости газа-носителя ... 477
Работа 5. Качественный анализ по индексам удержи-
вания при использовании капиллярных
колонок ... ............ .... 482
Работа 6. Идентификация органических соединений в
сложных смесях по индексам удерживания
и коэффициентам распределения в системе
ограниченно смешивающихся растворителей 493
Работа 7. Количественный анализ воздуха на содер-
жание кислорода и азота . . .501
Работа 8. Основные методы количественного газо-
хроматографического анализа.......................504
Работа 9. Количественный анализ многокомпонентной
смеси с программированием температуры . 514
Работа 10. Газохроматографическое определение фе-
нолов в водных объектах с предвари-
тельной дериватизацией............................517
Работа 11. Газохроматографическое определение содержа-
ния хлороформа в питьевой (водопровод-
ной) воде .........................................528
609
Работа 12. Газохроматографическое определение содержа-
ния тиофена в бензоле с пламенно-фотоме-
трическим детектированием....................... 536
Работа 13- Анализ пищевого спирта на содержание
вредных примесей .............................. 539
Глава VI. Практические работы по жидкостной хрома-
тографии ................................................. 550
Работа 14. Разделение смеси геометрических изоме-
ров азобензола методом препаративной
адсорбционной тонкослойной хромато-
графии ............................................-—
Работа 15. Разделение смеси моно-, ди- и триэтанол-
аминов методом лигандообменной жид-
костной хроматографии ...........................552
Работа 16. Разделение смеси органических веществ
методом нормально-фазной ВЭЖХ с ис-
пользованием УФ- и рефрактометричес-
кого детекторов .............................. . 555
Работа 17. Разделение смеси органических соеди-
нений с различными молекулярными
массами методом эксклюзионной хрома-
тографии ........................................560
Работа 18. Использование ионообменных смол для
обессоливания....................................562
Работа 19. Разделение и количественный анализ
смеси органических соединений при
использовании обращенно-фазового вари-
анта ВЭЖХ........................................565
Работа 20. Определение содержания 2,4-динитрофе-
нилгидразонов формальдегида и ацет-
альдегида в растворах ацетонитрила
методом абсолютной градуировки с ис-
пользованием обращенно-фазовой ВЭЖХ 569
ПРИЛОЖЕНИЕ. Программы для обработки эксперименталь-
ных данных и расчета хроматографических
параметров ................................... ...... . . 575
Литература ................................................583
610
Учебное издание
ПРАКТИЧЕСКАЯ
ГАЗОВАЯ И ЖИДКОСТНАЯ
ХРОМАТОГРАФИЯ
Редактор Л. П. Макаренкова
Технический редактор Е. Г. Учаева
Компьютерная верстка А. В. Павлов
Лицензия ЛР №040050 от 15.08.96.
Подписано в печать 08.09.98. Формат 60x90 1/16.
Бумага офсетная. Печать офсетная.
Усл. печ. 38,5. Доп. тираж 1000 ©кз. Заказ 16.
Издательство СПбГУ. 199034, С.-Петербург,
Университетская наб., 7/9.
Типография Издательства СПбГУ.
199061, С.-Петербург, Средний пр., 41.