/







































































































































































































































































































































Текст
A. M. ГОДЭН
лотация
• Перевод с английского
под общей редакцией проф. докт. техн, наук О. С. БОГДАНОВА
и канд. техн, наук Е. В. ДАНИЛОВОЙ
ГОСУДАРСТВЕННОЕ
НАУЧНО-ТЕХНИЧЕСКОЕ ИЗДАТЕЛЬСТВО
ЛИТЕРАТУРЫ ПО ГОРНОМУ ДЕЛУ
Москва 1959
АННОТАЦИЯ
Второе издание книги Годэна «Флотация», опу-
бликованное в США в 1957 г., значительно отлича-
ется от первого издания, русский перевод которого
в СССР вышел в 1934 г.
Во второе издание вошли многие работы автора и
его сотрудников, опубликованные в последние годы.
В книге дается изложение теории флотации, под-
крепленное большим экспериментальным материалом,
В ряде случаев приводятся новые оригинальные объ-
яснения отдельных вопросов флотации.
Книга представляет значительный научный и прак-
тический интерес для широких кругов специалистов-
обогатителей, а также может являться ценным по-
собием для студентов обогатительных специальностей
втузов.
Издательство просит читателей направ-
лять свои отзывы и пожелания по этой книге
по адресу:
Москва, Грузинский вал, 35.
Г осгортехиздйт
ОГЛАВЛЕНИЕ
От редакции русского перевода ................................ 8
Предисловие .................................................. 9
Глава 1. исторический обзор (перев. А. Б. Рогач, ред. канд. техн,
наук Е. В. Даниловой) ....................................... 11
Литература ........................................ 19
Глава 2. фазы во флотационных системах (перев. И. А. Вайн-
шенкера, ред. В. Я. Хайнмана) ............................... 20
Газовая фаза ..................................... 29
Жидкая фаза ...................................... 30
Кристаллические фазы ............................ 40
Литература ........................................ 63
Глава 3. поверхность раздела газ — раствор (перев. В. Я. Хайн-
мана, ред. Г. В. Панишевой) ............................ .... 65
Литература ........................................ 91
Глава 4. поверхности раздела твердое —газ, твердое —жид-
кость (перев. В. Я. Хайнмана, ред. Г. В. Панишевой) .... 95
Литература ................................................. 119
Глава 5. ЭЛЕКТРИЧЕСКИЕ явления на поверхностях раздела фаз
(перев. Л. А. Шнейдер, ред. А. Н. Долженковой и канд.
техн, наук А. Я. Поднек) ................................. 122
Электрокинетические явления на поверхности раздела твер-
дое — жидкость ................................... 124
Электрокинетические явления на поверхности раздела
жидкость — газ ...............................•••• 139
Литература ....................................... 149
Л. А. Шнейдер, ред. В. Я. Хайнмана и И. С. Михай-
ловой) ......................................... 152
Литература .......................................... 176
> Глава 7. краевые углы (перев. канд. техн, наук Г. С. Стрельцина,
ред. проф. докт. техн, наук О. С. Богданова) .......... 178
Литература ....................................................... 214
Глав а 8. собиратели (перев. С. И. Горловского, ред. канд. техн, наук
п. А. Янис) .................................................... 218
Оксгидрильные собиратели ............................ 220
Сульфгидрильные собиратели ......................... 227
Катионные собиратели ................................ 259
Различные собиратели ............................... 264
Литература .......................................... 265
6
ОГЛАВЛЕНИЕ
Глава 9. механизм действия собирателей(перев. канд. техн, на-
ук Н. А. Янис, ред. канд. техн, наук А. К. Поднек) ........... 273
Действие ксантогенатов на галенит ..................,. 275
Действие ксантогенатов на халькозин ............... 283
Действие ксантогенатов на пирит ................... 292
Действие ксантогенатов и других тиосоединений на различ-
ные минералы ................................... 293
Действие жирных кислот и мыл ......,............... 303
Действие аминов . . .............................. 307
Детали структуры поверхностных пленок собирателя..... 321
Литература ........................................ 328
Глава 10. действие модификаторов флотации (перев. канд.
техн, наук Е. В. Даниловой, ред. канд. техн, наук А. К-
Поднек и канд. техн, наук Н. А. Янис) ........................ 333
Регулирование флотации изменением концентрации водород-
ных ионов ...................................... 333
Гидросульфид-ион как депрессор для сульфидных минерало! 340
Ион цианида как депрессор для сульфидных минерало! 349
Сульфиты и другие сульфоксидные соединения как депрес-
соры для сульфидных минералов ..................... 361
Активация сфалерита медью ......................... 366
Активация кварца .................................. 380
Активация окисленных свинцовых минералов сульфидным
ионом ............................................. 382
Литература ........................................ 384
' Глава И. пены (перев. канд. техн, наук Т. М. Мягковой и И. И. Ва-
неева, ред. канд. техн, наук Б. В. Кизевальтера) .......... 387
Пенообразование . . 388
Введение газа . ................................... 400
Прикрепление твердых частиц к пузырькам газа ...... 402
Минерализованные пузырьки и пены ................. 419
Литература ........................................ 434
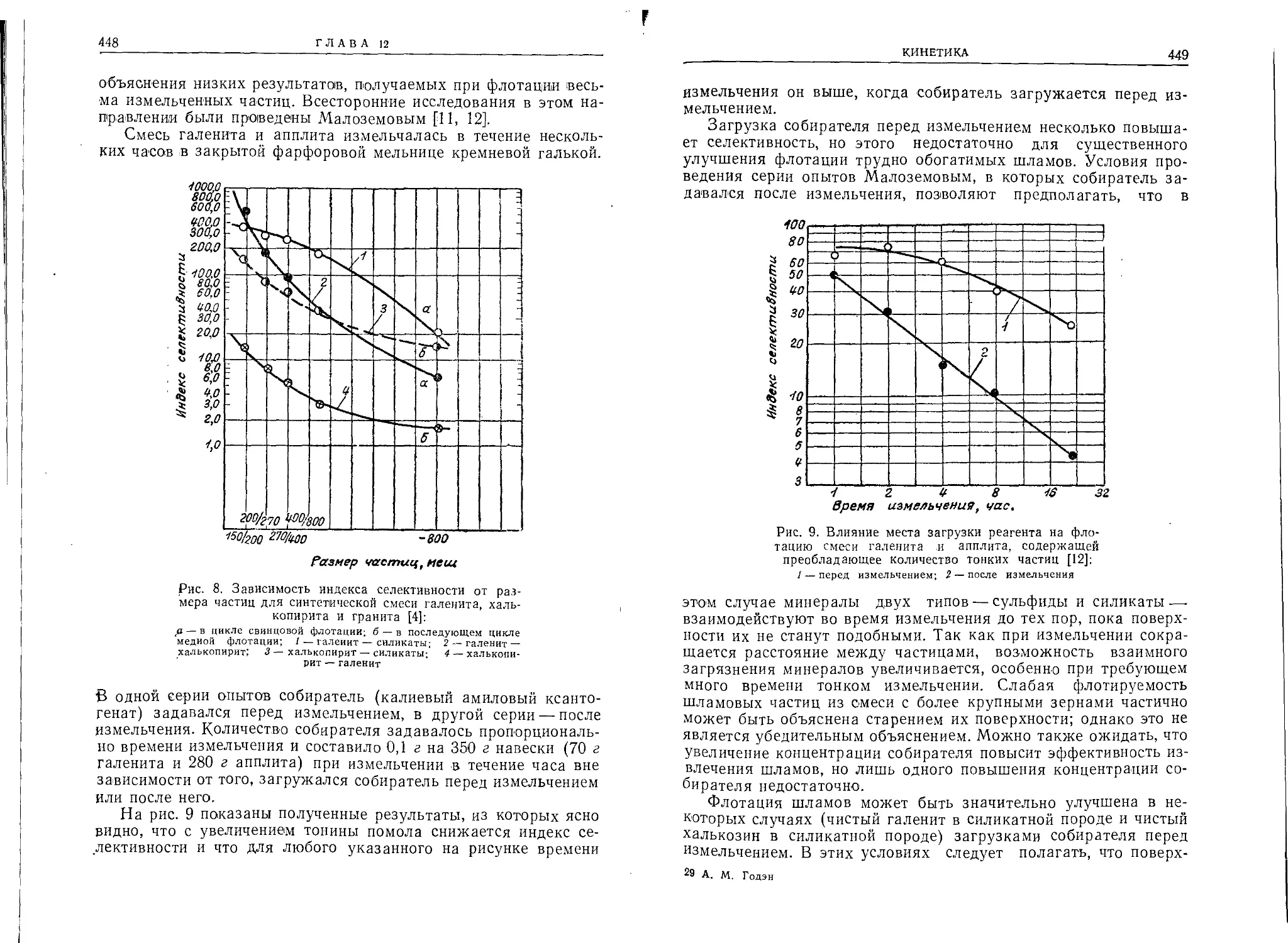
Глава 12. кинетика (перев. Г. М. Корсаковой, ред. проф. докт.
техн, наук О. С. Богданова) .................................. 436
Литература ........................................ 460
Глава 13. минералогия, сростки и их освобождение (перев.
Ю. С. Бадеева, ред. канд. техн, наук Е. В. Даниловой и
Т. Е. Колтуновой) ............................................ 462
Текстура, структура и генезис руды ................ 462
Раскрытие минералов ............................... 475
Литература ........................................ 484
Глава 14. флотационные машины (перев. А. И. Ескина, ред.
Г. В. Панышевой) .................................. 486
Типы машин ...................................... 490
Литература ........................................ 501
Глава 15. сульфидные руды (перев. И. А. Фонякова, ред. канд.
техн. наук. А. С. Конева) .................................... 503
Сульфидные медные руды ............................ 503
Сульфидные свинцово-цинковые руды ................. 512
Прочие сульфидные руды ............................ 527
Литература ........................................ 541
О ГЛАВЛЕНИЕ
7
Глава 16.руды окисленных минералов (перев. А. И. Какори-
на, ред. С. Д. Суховольской) ................................. 544
Окисленные руды меди, свинца и цинка .............. 545
Окисленные минералы щелочноземельных металлов .... 553
Окисленные минералы многовалентных металлов........ 562
Кремнезем и силикаты .............................. 576
Литература ........................................ 593
Глава 17. соли (перев. канд. техн, наук Г. С. Стрельцина, ред.
канд. техн, наук Ю. И. Еропкина и Т. Е. Колтуновой) .......... 600
Малорастворимые соли ............................ 601
Сильвиниты . ..........,........................... 604
Прочие соли ....................................... 614
Литература ........................................ 619
Глава 18.аполярные минералы (перев. канд. техн. наук
10. И. Еропкина, ред. канд. техн, наук Г. С. Стрельцина) 624
Сера ......................................................... 624
Графит....................................••...... 625
Алмаз . , ......................................... 627
Угли ........,..................................... 627
Литература ........................................ 641
Глава 19. различные органические вещества (перев. И. А.
Фонякова, ред. канд. техн, наук 4. С. Конева) ......644
Нефть, битумы, смолы, каучук ...................... 644
Семена и продукты растительного происхождения ..... 646
Бумажные пульпы ................................... 647
Продукты текстильной промышленности ............... 648
Продукты животного происхождения и молочные ....... 649
Очистка вод ....................................... 650
Промышленные и хозяйственно-фекальные сточные воды 650
Растворимые вещества .............................. 651
Литература ...............................,........ 651
ОТ РЕДАКЦИИ РУССКОГО ПЕРЕВОДА
Предлагаемая читателям книга проф. Массачусеттского тех-
нологического института А. М. Годэна «Флотация» опубликова-
на в США вторым изданием в 1957 г. Это издание книги резко
отличается по содержанию и расположению материала от перво-
го издания, выпущенного в свет в США в 1932 г. и в русском
переводе в СССР в 1934 г.
Автор книги хорошо известен советским специалистам в
области обогащения полезных ископаемых как по оригиналь-
ным работам, публикуемым в различных зарубежных периоди-
ческих изданиях, так и по книгам, переведенным на русский
язык («Основы флотации», «Основы обогащения полезных
ископаемых» и упомянутое первое издание книги «Флотация»).
Во второе издание книги вошли многие работы автора и его
сотрудников, опубликованные в последние годы. В ряде случаев
автор дает новые объяснения различным вопросам флотации на
основе анализа экспериментальных данных и общих рассужде-
ний. Книга несомненно представляет значительный научный и
практический интерес.
Надо поставить в большую заслугу автору стремление не
только констатировать те или другие закономерности или поло-
жения, но и объяснить их. Во многих случаях эти объяснения
оригинальны и вполне убедительны, однако встречаются и спор-
ные положения. В русском переводе в примечаниях приводятся
лишь некоторые замечания редакторов разделов.
Кроме того, видимо, в США мало знакомы с советской лите-
ратурой в области обогащения руд, чем и объясняется незначи-
тельное количество ссылок в книге А. М. Годэна на работы совет-
ских исследователей. Тем не менее, необходимо подчеркнуть, что
предлагаемая читателям книга имеет несомненную ценность и
ее издание в русском переводе вызовет положительный отклик
среди широких кругов обогатителей Советского Союза.
Перевод и редакция отдельных глав книги осуществлены
большим коллективом научных сотрудников института Меха-
нобр. При переводе и редактировании книги встретились значи-
тельные трудности ввиду своеобразия стиля изложения автора,
а также ввиду различной интерпретации тех или других поло-
жений и различия в терминологии у советских и американских
авторов.
Проф. докт. техн, наук О. С. Богданов
Канд. техн, наук Е. В. Данилова
Апрель 1958 г.
ПРЕДИСЛОВИЕ
Огромные изменения в искусстве флотации за последние 25
лет, прошедшие со времени выхода в свет данной книги, вызва-
ли необходимость полностью переработать ее. Новый текст бо-
лее сжат и значительно глубже вскрывает научные основы про-
цессов.
Расположение и изложение материала тоже изменены с та-
ким расчетом, чтобы возбудить у читателя интерес к исследова-
нию неразрешенных научных и технических проблем.
Автор указывает использованные им источники как в тексте,
так и в библиографических перечнях. Отмечая большую пользу,
принесенную этими источниками, автор выражает признатель-
ность многим лицам, с которыми он работает сейчас и с которы-
ми работал ранее в Массачусеттском технологическом институте
и в промышленности.
Особую благодарность автор приносит следующим лицам,
непосредственно помогавшим в той или иной форме завершению
данной работы: Вильяму Блокеру (Америкен Сайанамид Ко);
докт. Р. Б. Бусу (директору рудообогатительной лаборатории
Америкен Сайанамид Ко); докт. Д. Дж. Брауну (начальнику
секции пенной флотации отдела обогащения угля Исследова-
тельского института Национального департамента угля в Анг-
лии); проф. М. Бюргеру (Массачусеттский технологический ин-
ститут); Дж. С. Карру (Мельбурнский университет в Австра-
лии); докт. Ч. С. Чангу (Кемикл Констракшн Ко); докт. Р. Дж.
Чарльзу (Дженерал Электрик Ко); докт. А. С. Элму (главному
химику-органику исследовательского отдела Нью-Джерси Цинк
Ко); докт. В. Фрейбергеру (Исследовательский отдел Нью-
Джерси Цинк Ко); М. Л. Фуллеру (управляющему отдела ис-
следования полезных ископаемых Нью-Джерси Цинк Ко);
В. Гриффитсу (Нью-Джерси Цинк Ко); докт. Мохамеду Халфа-
ви (Высшее педагогическое училище в Багдаде, Ирак); проф.
Д. Гаррису (Висконсинский университет); проф. Р. Т. Хукки
(Государственный институт технического исследования в Хель-
синки, Финляндия); Р. В. Лиси (Уитлэнд Тьюб Ко); докт.
О. Меллгрену (Нчанга Консолидейтед Коппер Майнес Лтд. в
Северной Родезии); докт. Дж. Морроу (Оттава, Канада); проф.
Дж. Т. Овербику (Утрехтский университет, Нидерланды);
X. А. Пулу (инж. по флотации Консолидейтед Майнинг энд Сме-
лтинг Ко Лтд. в Канаде); проф. Р. Шуману (Пюрдюиский уни-
10
ПРЕДИСЛОВИЕ
верситет); проф. С. С. Смиту (директору Института металлов
Чикагского университета); X. Р. Спеддену (директору группы
исследования полезных ископаемых Юнион Карбайд энд Кар-
бон Корпорэйшн); докт. Ж. Турнесаку (Институт де Сан Го-
бэн, Париж); докт. И. В. Уорку (начальнику отдела промышлен-
ной химии CSIRO в Мельбурне, Австралия); докт. Е. С. Уилеру
(исследовательский отдел Атлантик Рифайнинг Ко).
Коллеги автора по Массачусеттскому технологическому ин-
ституту проф. П. Л. де Брюэн и Д. В. Фюрстенау затратили
очень много времени на чтение и обсуждение рукописи. Автор
приносит им особую благодарность, так же как и группе аспи-
рантов, особенно Ф. Эплану, Дж. Брауну, Р. Харду, И. Ивасаки,
X. Ли, Г. Мао и Г. Парксу.
Автор выражает глубокую признательность Б. Лиси (сотруд-
нице библиотеки Нэйшнл Лед Ко, Винчестер, Массачусетт) и
С. Уоррен (ранее работавшей в отделе металлургии библиотеки
Массачусеттского технологического института), которые пре-
красно провели работу по проверке библиографических источ-
ников.
Рукопись книги печатали М. Гиббс и А. Маршалл, А. Ску-
фоз и Л. Уотерс, которых автор горячо благодарит.
Автор также приносит благодарность издателям (помимо
Мак-Гроу-Хилл) за разрешение воспроизвести иллюстрации и
некоторые данные, которые отмечаются в тексте.
А. М. Годэн
Глава 1
ИСТОРИЧЕСКИЙ ОБЗОР
Флотация представляет собой процесс разделения тонкоиз-
злельченных частиц, применяемый для обогащения руд различ-
ных металлов, твердого топлива и неметаллических полезных
ископаемых. Кроме того, флотационный способ может быть
использован для извлечения твердых частиц из жидкостей и
для разделения твердых частиц, не являющихся минеральным
сырьем.
При применении флотации для обогащения полезных иско-
паемых выделяется продукт, не содержащий полезных минера-
лов, называемый «хвостами», и концентраты, в которых сосре-
доточиваются полезные минералы. Концентраты в некоторых
случаях могут быть использованы в качестве конечных готовых
продуктов, но большей частью они требуют дальнейшей обра-
ботки (например, плавки).
Перед обогащением руду измельчают, для того чтобы каж-
дая отдельная частица по возможности состояла из одних цен-
ных минералов или же из минералов пустой породы. Эта опера-
ция называется раскрытием минеральных зерен.
При флотации разделение происходит в воде, в которой ча-
стицы твердого находятся во взвешенном состоянии. Осуществ-
ляется оно в результате прилипания некоторых твердых частиц
к пузырькам газа, образующимся в пульпе или введенным в
нее, в то время как другие твердые частицы смачиваются водой
и не прикрепляются к пузырькам, оставаясь в пульпе по-преж-
нему во взвешенном состоянии. Твердые частицы, прикрепивши-
еся к пузырькам воздуха, всплывают на поверхность пульпы,
образуя пенный продукт, отличающийся по своему составу от
исходной пульпы.
Ранее термин «флотация» относили ко всем процессам обо-
гащения, при которых происходило всплывание на поверхность
воды частиц, более тяжелых, чем вода. Процессы, при которых
частицы твердого собирались в слое масла или на поверхности
раздела масло —вода, назывались масляной флотацией. Про-
цессы, при которых частицы, попадая на свободную поверхность
воды, оставались на ней, образуя слой толщиной в одну части-
цу, назывались пленочной флотацией. Процессы, при которых
частицы удерживались на поверхности пульпы в слое пены тол-
12
ГЛАВА I
щиной в несколько сантиметров, назывались пенной флотацией.
Из всех этих процессов в настоящее время в технике применяет-
ся только пенная флотация.
Первые попытки применения флотации. Из сказанного сле-
дует, что газ является одной из необходимых фаз при процессе
флотации. Но на ранних стадиях развития процесса флотации
роли газа не придавали должного значения.
Одним из наиболее ранних указаний о применении флотации
можно считать описание в персидской рукописи пятнадцатого
века четырех обогатительных процессов [31. В двух из них пре-
дусматривалось перемешивание с маслом подлежащего разде-
лению материала и последующее перемешивание с водой омас-
ленной массы. При этом частицы, избирательно смачивавшие-
ся водой, отделялись от частиц, покрытых маслом.
Такой метод флотации применялся еще в 1890 г. [11 при про-
изводстве ультрамарина и был описан Эверсон в американском
патенте 348157/1885.
При масляной флотации, к которой относятся указанные
процессы, происходит селективное омасливание ценных частиц
при перемешивании исходного материала с маслом; затем эти
частицы всплывают на поверхность воды, так как масло имеет
меньший удельный вес, чем вода.
Из процессов масляной флотации следует особо отметить
процесс Вильяма Гейнеса, описанный в британском патенте
488/1860, и второй процесс Эверсон.
Процесс масляной флотации Эльмора (американские патен-
ты 653340; 676679 и 689070/1901 и 692643/1902) был испытан в
производственных условиях [9а].
Кроме масляной флотации, требовавшей большого расхода
масла, можно отметить упоминавшийся ранее процесс пленоч-
ной флотации, при котором разделение’сухих частиц материала
при их соприкосновении со свободной поверхностью воды про-
исходит вследствие различия в их смачивании.
Аппарат, применявшийся при этом процессе, описан Нибели-
усом (американский патент 486495/1892), Мак Квистеном (аме-
риканский патент 865194—5/1907) де Бавэем (американский па-
тент 864597/1907) и Вудом (американский патент 1088050/1914).
Применение газа для подъема твердых частиц на поверх-
ность пульпы. Братья Бессель еще в 1877 г. использовали газ в
качестве среды, способствуюгцей всплыванию частиц минерала
на поверхность пульпы (немецкий патент 42, класс 22) [7]. Од-
нако действительное значение газа при флотации было отчет-
ливо понято ими лишь в 1886 г. (немецкий патент 39369).
Они проводили испытания с графитом, используя в качестве
реагентов различные масла и жиры, добавляемые в количест-
ИСТОРИЧЕСКИЙ ОБЗОР
13
вах от 1 до 10%. Флотация осуществлялась при кипячении в
воде или при образовании пузырьков газа в результате реак-
ций, происходящих при воздействии кислоты на карбонаты. О
важном значении прилипания частицы минерала к пузырьку га-
за для всплывания ее на поверхность пульпы было известно, су-
дя по работам французских физиков Пти [81 и А. Дюпре и
Р. Дюпре [2], еще в восемнадцатом веке, но до исследования,
проведенного братьями Бессель, это, по-видимому, не нашло
практического применения.
Работа братьев Бессель не получила признания ни в Англии,
ни в США, и эти же самые методы (кипячения и создания пу-
зырьков газа в результате химических реакций) были «откры-
ты» позднее.
При флотационных процессах у Фромента (британский па-
тент 12778/1902), Дельпра (американский патент 735071/1903) и
Поттера (американский патент 776145/1904) пузырьки газа об-
разуются в результате химических реакций, происходящих
вследствие действия кислоты на взвешенные частицы сульфидов
и карбонатов.
Эльмор запатентовал способ получения пузырьков газа
электролизом воды (британский патент 13578/1904), но его ме-
тод не нашел применения в технике.
Образование пузырьков газа под вакуумом, запатентован-
ное также Эльмором (британский патент 17816/1904), имело
большой успех в промышленности (этот метод не потерял своего
значения и по настоящее время) [61.
Непосредственное введение газа в пульпу с помощью вра-
щающегося в ней импеллера было описано Салмэном, Пикка-
ром и Балло (американский патент 835120/1906), а также
Гувером (американский патент 953746/1910).
Схематический чертеж аппарата Гувера показан на рис. 1.
В этом аппарате имеются все основные* детали флотационной
машины агитационного типа.
Другими вариантами метода непосредственного введения га-
за в пульпу является подача газа через пористое днище (Кел-
лоу, американский патент 1104755/1914) и через погруженные в
пульпу патрубки (машины Форрестера и Хунта).
Уменьшение расхода масла. При использовании газа в каче-
стве среды, выносящей частицы твердого на поверхность, было
установлено, что нет необходимости применять масло в больших
количествах, так как избыток масла вреден для успешного хода
флотации.
В американском патенте 835120 указывается, что расход мас-
ла следует ограничить количеством, меньшим 1% от веса обра-
батываемой среды. Это было первым крупным шагом к сниже-
нию потребления масла при флотации.
14
ГЛАВА 1
Если выразить норму загрузки масла, рекомендованную аме-
риканским патентом, в современных единицах измерения расхода
реагентов, то она составит 10 кг/т. В настоящее время даже на
тех фабриках, где масло все еще применяется в количествах,
больших, чем это необходимо для нормального хода процесса
(0,15—1 кг/т), расход его во много раз ниже.
Рис. 1. Флотационная машина:
/ — агитационная камера, разделенная на три части; 1а — перегородки,
разделяющие агитационную камеру, 16 — зазор между перегородкой н
дном агитационной камеры для прохода пульпы из одного отделения ма-
шины в другое; 1в— выпускная щель для прохода пульпы из агитацион-
ной камеры в шпицкастен; 2 —- нмпеллерьь 2а — вал импеллера; 3 — эле-
ватор для подачи пульпы; 4 и 5 —реагентные питатели; 6 — шпнцкастеи;
7—желоб для концентрата; 7а — труба для отхода концентрата; Я —
слнвной порог шпицкастена; 9— отбойник; 10— выпускная труба для хвос-
тов
После признания целесообразности снижения расхода масла
было установлено, что не все масла оказывают одинаковое дей-
ствие при флотации.
Гринвэй, Салмэн и Хиггинс (американский патент
962678/1909) рекомендовали применять растворимые вспенива-
тели, как например кетоны, альдегиды и эфиры, являющиеся
главными компонентами сосновых масел.
П рименение химических собирателей вместо масел. Начиная
с 1915 и до 1922 г. широкое распространение получил термин
ИСТОРИЧЕСКИЙ ОБЗОР
15
«собирающие масла», или «собиратели», для обозначения масел,
не образующих пену, но обладающих способностью собирать в
пену определенные частицы минерала. К этому же периоду от-
носится важное открытие Перкинса и Сэйера (американские
патенты 1364304/1921 и 1364307/1921), установивших, что неко-
торые немаслянистые органические соединения, в состав ко-
торых входят трехвалентный азот и двухвалентная сера, обла-
дают собирательными свойствами. Это открытие касалось весь-
ма слабо растворимых органических соединений, но оно не от-
носилось к ксантогенатам, которые позднее стали стандартны-
ми собирателями при флотации сульфидных минералов.
В 1925 г. появился патент Келлера (американский патент
1554216), в котором указывается, что ксантогенаты щелочных:
металлов, представляющие собой весьма легко растворимые со-
ли, являются эффективными собирателями. Таким образом, Кел-
лер опроверг существовавшее ранее мнение о том, что в каче-
стве собирателей необходимо употреблять только нерастворимые
вещества, точно так же, как в свое время Перкинс и Сэйер оп-
ровергли представление о том, что собирателями должны слу-
жить маслянистые вещества.
Вслед за введением ксантогенатов было предложено приме-
нять в качестве собирателей дитиофосфаты (Витворт, амери-
канский патент 1593232/1926), которые до сего времени являют-
ся главными конкурентами ксантогенатов при флотации суль-
фидных минералов.
Начиная приблизительно с 1925 г., наблюдается более широ-
кое применение мыл для извлечения окисленных минералов
(Салмэн и Эдсер, американский патент 1492904/1924). Эти реа-
генты, если не по потребляемому количеству, то по своему зна-
чению не менее важны, чем ксантогенаты и дитиофосфаты.
Позднее для флотации различных минералов, включая си-
ликаты, стали употреблять амины и родственные им органиче-
ские соединения, образующие положительно заряженные ионы,
содержащие углеводородные радикалы, благодаря чему эти со-
биратели и получили название катионных. Эти соединения соз-
дают большие возможности для гибкой регулировки процесса и
до сего времени считаются весьма полезной группой собира-
телей.
Неорганические реагенты. Некоторые замечательные рабо-
ты, проводимые сейчас в области флотации, стали возможны-
ми только благодаря применению неорганических солей, что в
значительной степени усовершенствовало флотацию окисленных
минералов, а также разделение сульфидных минералов и поз-
волило обогащать сульфидные цинковые руды методом флота-
ции.
Кроме кислот, рекомендованных Эверсон в 1886 г., и соли
16
ГЛАВА 1
NaHSO4, предложенной Дельпра в 1903 г., неорганические соли
приблизительно в это же время использовал Шварц (амери-
канский патент 807501/1905). который рекомендовал до флота-
ции обрабатывать окисленные руды цветных металлов серни-
стыми или многосернистыми солями.
В 1913 г. в Австралии Брэдфорд нашел, что медный купорос
увеличивает флотационную активность сфалерита [10]. Открытие
Брэдфорда является одним из наиболее значительных в обла-
сти флотации, но в США оно не было запатентовано.
В 1912 г. Лоури и Гринвэй установили, что двухромовокис-
лые соли препятствуют флотации свинцового блеска (австра-
лийский патент 5065/1912).
В это же время начали использовать щелочные среды. Бла-
годаря меньшему разъедающему действию по сравнению с кис-
лыми средами их начали широко применять при флотации суль-
фидных минералов. Проведение флотации в щелочной среде
привело к открытию депрессирующего действия извести на пи-
рит и сделало возможным использование таких эффективных
селективно действующих реагентов, как сернистокислые соли
(Палланч, американский патент 1486297/1924), цианиды (Ше-
ридан и Гризволд, американские патенты 1421585/1922,
1427235/1922) и сернистые соли (Гельстранд, американский па-
тент 1469042/1923). Селективно действующие реагенты позво-
ляют флотировать одни минералы и депрессировать другие, бла-
годаря чему осуществляется их разделение. В случае их при-
менения флотация называется селективной, или дифференциаль-
ной, в отличие от коллективной, при которой все полезные мине-
ралы собираются в один концентрат.
Возможно, что пользоваться этими терминами вообще из-
лишне, поскольку все флотационные процессы по своему суще-
ству являются селективными операциями, обусловливающими
отделение полезных минералов от минералов пустой породы ли-
бо разделение сульфидных или окисленных минералов.
В данной книге, за исключением тех случаев, когда бывает
желательно подчеркнуть качество процесса, для обозначения
всех флотационных процессов принят термин «флотация».
Признание важного значения характера среды, в которой про-
текает флотация, т. е. является ли среда щелочной или кислой,
привело к введению систематического определения pH. В насто-
ящее время прямой или косвенный контроль'Щелочности среды
применяют и при флотационных испытаниях, и во флотационных
циклах на обогатительных фабриках.
Данные об основных этапах развития процесса флотации
приведены в табл. 1.
Флотогравитация. Для разделения крупного материала счи-
тается рациональным применять процесс Чапмэна и Литлфорда
ИСТОРИЧЕСКИЙ обзор
17
Таблица 1
Важные этапы в развитии процесса флотации
Прибли- зительная дата Автор Изобретение
1491 Мэнсюр Использование различия в смачиваемости минералов маслами и водой при обогащении ультрамарина и азурита
1731 Пти Использование явления прикрепления частиц твердого к пузырькам воздуха для всплывания находящихся в пульпе частиц твердого
1860 Гейнес Процесс масляной флотации
18// Бессель Процесс кипячения для извлечения гра- фита
1885 Бессель Процесс химического производства пу- зырьков газа для извлечения гргфита
1883 Эверсон Применение подкисленной пульпы
10>,2 Фромент, Поттер, Дельпра Применение газа в качестве выносящей среды для флотации сульфидных руд
1905 Шварц Применение сернистого натрия для извле- чения окисленных руд цветных металлов
1906 Салмэн, Пиккар и Б 1лло Уменьшение расхода масла. Введение газа методом энергичного перемешивания
1909 1 рннвэй, Салмэн и Хиггинс Введение растворимых вспенивателей
1913 Брэдфэрд Применение сернистого газа для депрес- сии сфалерита
1913 Брэдфорд Применение медного купороса для акти- вации сфалерита
1921 Перкинс и Сэйер Применение специальных органических собирателей
1921 Флотация в щелочной среде
1922 Шеридан и 1 риз- волд Применение цианистых солей для депрес- сии сфалерита и пирита
1924 Салмэн и Эдсер Применение мыл для флотации окислен- ных минералов
1925 Келлер Применение ксантогенатов в качестве со- бирателей z
1929 Годэн Введение контроля щелочности среды
1929 Дженпрост Флотация сильно растворимых солей
1933 Несслер Разделение при помащи флотации сме- сей растворимых в воде химических солей
1934 Чапмэн и Литл- форд Флотогравитация
1934 Применение щелочных сульфатов в ка- честве собирателей
1935 Применение катионных собирателей
2 А. М. Годэн
18
ГЛАВА 1
(американский патент 1968008/1934). Этот процесс заключается
в соответствующей подготовке материала для образования фло-
кул, состоящих из частиц минерала, слабо связанных с пузырь-
ками воздуха, и отделения этих флокул на деке качающегося
концентрационного стола от нефлокулированных частиц; при
этом используются принципы гравитационного процесса и пле-
ночной флотации.
Процесс Чапмэна и Литлфорда несколько сходен с процес-
сом, неудачно испытанным приблизительно сорок лет назад на
Центральном руднике Брокен-Хилл в Австралии [5], так же как
и процессы, предусматривающие отделение флокул, состоящих
из аэрированных и омасленных частиц, от неаэрированных и не-
омасленных частиц посредством классификации или грохочения
под водой.
Современные направления в области флотации. Применение
флотации позволило удовлетворительно разрешить проблемы
обработки полиметаллических свинцово-цинково-железных и
медно-железных сульфидных руд. Дальнейшие усовершенство-
вания в области обработки этих руд будут направлены главным
образом на достижение более полного раскрытия зерен минера-
лов, на повышение извлечения при обработке весьма крупных
и очень мелких классов и на улучшение разделения при обра-
ботке наиболее тонких частиц.
Проблема обогащения сульфидных медно-цинковых и суль-
фидных медно-свинцовых руд разрешена еще не полностью.
В этом случае, так же как и при обработке свинцово-цинко-
вых и медно-железных сульфидных руд, нахождение более се-
лективно действующих собирателей и более совершенных моди-
фикаторов приведет к дальнейшему усовершенствованию фло-
тационного процесса.
В последнее время широко применяют флотацию при обога-
щении несульфидных руд. Удовлетворительные результаты по-
лучаются при флотации фосфоритов, цементных пород, мине-
ральных солей (как например, калиевых солей), а также загряз-
ненных флюоритовых и баритовых руд.
Некоторый успех достигнут при применении флотации для
разделения силикатов и для обработки окисленных руд, зале-
гающих над сульфидными рудами. В первом случае дальнейший
прогресс затрудняется химическим сходством силцкатов, ослож-
няющим их успешное разделение, в случае же флотации окис-
ленных руд—сложностью структурных прорастаний, а также
сложностью химического состава этих минералов.
Весьма перспективна флотация бедных железных руд для
выделения из них богатых концентратов.
Все более широко применяют флотацию для обогащения уг-
ИСТОРИЧЕСКИЙ ОБЗОР
19
ля. Предполагается, что в этой области перед флотацией откры-
ваются также большие возможности.
Кроме того, флотация иногда применяется для разделения
различных твердых материалов и для выделения малых коли-
честв взвешенных частиц твердого материала из больших объе-
мов водных растворов.
ЛИТЕРАТУРА
1. Church А. Н. The Chemistry of Paints and Painting, p. 181, Seeley and
Co., Ltd., London, 1890.
2. Dupre Anathase. Theorie mecanique de la chaleur, pp. 290—318, Gau-
thier-Villars & Cie, Paris, 1869.
3. G a u d i п A. M. Mineral concentration by oil adhesion in the XVth cetnury,
Eng. Mining J., 141, (10), 43—44, 1940.
4. G a u d i п A. M. The influence of hydrogen- ion concentration on reco-
very in simple flotation systems, Mining and Met., 10, 19—20, 1929.
5. Hebbard James. Evolution of Minerals Separation process at Central Mi-
ne, Proc. Australasian Inst. Min. Engrs., 10, 86—87, 105, 1913.
6. H о о v e r T. J, Concentrating ores by flotation, Mining Mag. (London),
120—133, 1914.
7. M a у e r Erwin W. and Hubert Schranz. Flotation, S. Hirzel Verlag,
Leipzig, 1931.
8. Petit M. De 1’adherence des parties de 1’air entre elles, et de leur ad-
herence aux corps qu’ elles touchent, 72—98. section Memoires, in Histoire
de I’Academie Royale des Sciences (de Paris), 1731. Selected reprinting by
Pierre Mortier, Amsterdam, 1735.
9. Taggart Arthur F. Handbook of Mineral Dressing, (a) pp. 12—49, John
Wiley & Sons, Inc., New York, 1945.
10. W a i n w r i g h t W. E. The development of processes for the treatment of
crude ore, accumulated dumps of tailing and slime at Broken Hill, New
South Wales, Empire Mining Met. Congr. Proc., 3d Congr., South Africa,
1930.
11. Wood Henry E. Early efforts in flotation of dry minerals, Eng Mining
J., 126, 571—573, 1928.
2*
Глава 2
ФАЗЫ ВО ФЛОТАЦИОННЫХ СИСТЕМАХ
Флотационное разделение различных твердых тел происхо-
дит благодаря разному сродству их поверхностей к пузырькам
воздуха и воде.
В случае необходимости эта селективная гидрофобность и
гидрофильность могут быть изменены с помощью реагентов.
Среди различных фаз газовая фаза является наиболее про-
стой, а твердая — наиболее сложной. Жидкая фаза, хотя и ка-
жется очень простой, в действительности все же довольно сло-
жна. Чтобы облегчить последующее изучение флотационного
процесса, полезно дать краткий обзор основных понятий о струк-
туре этих фаз.
Вещество имеет атомную структуру и может быть делимо
без резкого изменения свойств только до известного предела.
Атомы могут объединяться, образуя молекулы. Грамм-атом лю-
бого элемента (т. е. количество элемента, вес которого в грам-
мах равен атомному весу) содержит одно и то же число атомов,
приблизительно равное 6,02 1023. Аналогично грамм-молекула,
или моль, содержит такое же число молекул. Это число извест-
но как число Авогадро. С его помощью, если известен удельный
вес и атомный вес элемента, можно определить объем прост-
ранства, приходящийся на один атом. Так, объем, занимаемый
одним атомом твердого свинца, равен объему куба с ребром
3,1 • 10’8 см, или 3,1 А.
Строение атома [18]. В атоме можно выделить две части. Од-
на часть обладает положительным электрическим зарядом и
известна как атомное ядро; другая образована одним или не-
сколькими одинаковыми отрицательными электрическими заря-
дами— электронами. Каждый атом электрически нейтрален.
Следовательно, положительный заряд ядра, численно равный
атомному номеру Z, равен полному отрицательному заряду, соз-
данному всеми электронами. Атомный номер райен числу элек-
тронов. Ядро занимает центральную часть атома. Оно чрезвы-
чайно мало по сравнению с размерами атома, но обладает по-
давляющей частью его массы. Электроны заполняют ряд обо-
лочек, находясь на которых они совершают периодическое дви-
жение вокруг ядра. Каждая оболочка может содержать различ-
ное число электронов, но не выше определенного максимально-
ФАЗЫ ВО ФЛОТАЦИОННЫХ СИСТЕМАХ
21
го числа. Так, на первой оболочке (К) могут располагаться
один или два электрона, но не может быть трех. Вторая обо-
лочка (L) может содержать до восьми электронов, а третья
(М) —до восемнадцати.
Будучи удалены от. ядра, все электроны тождественны. Внут-
ри атома они занимают положения с различной энергией отно-
сительно ядра. Каждую оболочку (или определенные орбиты)
можно охарактеризовать определенным энергетическим уров-
нем. Наибольшая работа требуется для удаления от ядра элек-
тронов с внутренних орбит. При переходе от элементов с ма-
лым атомным номером к элементам с большим атомным номе-
ром происходит заполнение электронных оболочек. Оболочки
(а также различные орбиты) заполняются в определенном по-
рядке. Однако у многоэлектронных атомов разница в энергиях
электронов, находящихся на внешних оболочках (орбитах),
становится относительно малой, так что заполнение более уда-
ленной от ядра оболочки (например, N) начинается дс того, как
заканчивается заполнение предыдущей оболочки (М). Это ве-
дет к большой сложности химических свойств переходных ме-
таллов, к наличию у них переменной валентности, а также к
малой разнице в химических свойствах между элементами с
соседними атомными номерами, как например между редкозе-
мельными элементами.
Химические свойства зависят от числа электронов, которые
нужно добавить к атому или оторвать от него, чтобы оставшая-
ся оболочка была заполнена. Так, например, кислород, приоб-
ретая два электрона, будет иметь законченную оболочку неона;
кальций, отдавая два электрона, будет иметь законченную
оболочку аргона. Очевидно, что кислород имеет валентность
—2, а кальций +2.
Периодическая таблица. Повторяемость химических свойств
элементов, расположенных согласно возрастающим атомным
весам, привела Менделеева к мысли расположить элементы со-
гласно их атомным весам и свойствам. В течение столетия, про-
шедшего с тех пор, как Менделеев выдвинул свое предложение,
периодическая таблица совершенствовалась, пока не преврати-
лась в наиболее важное обобщение химии. Табл. 1 представля-
ет такого рода классификацию. Элементы в ней располагаются
согласно атомному номеру Z, а не по дробным атомным весам.
Таблица дает для каждого элемента символ, атомный номер и
атомный вес согласно последним данным [47].
Начиная со второго столбца, мы имеем три категории ато-
мов, которые разделяются двумя линиями: а) верхняя группа
благородных газов, содержащая гелий, неон, аргон, криптон,
ксенон и радон; б) средняя группа неметаллов, таких как азот,
сеРа, бром; в) большая нижняя группа металлов. У элементов,
22
ГЛАВА 2
Таблица 1
Периодическая система элементов
♦
Редкие
земли
Зе-эз] РГ'59- Nd-бо- Ргп-бН
140,131L1^23
Еи-63- Gd-64- T’j-65- Dy-бб- Но-67- Ег-68- Tm-89- Yb-70- Lu-71-
f52,o| f 56,9 <58,93 [ J62.46j )6Ч,9Ч] 187,2 168,f73,H 174,99
152.0
**АКТИНИЛЫ Th-SO- Pa-91- U-92' tfp-93- Pu-94- Am-95 Cm-96- 6K-97- Cf-98-
m И»ПДР*| 23£pj| 23t И 238-0711 7Z37)|| (242)1| (243) ll (2451||(245)|| (2й8)1
ФАЗЫ ВО ФЛОТАЦИОННЫХ СИСТЕМАХ
'3
соединенных тонкой чертой, заметно сходство химических
свойств. Так, группа бериллий, магний, кальций, стронций, ба-
рий, радий образует семейство щелочноземельных металлов. Ана-
логично фтор, хлор, бром, иод и астатин образуют семейство га-
логенов.
Благородные газы — это элементы с заполненной электрон-
ной оболочкой. Каждый атом представляет законченную систе-
му, так что у атомов практически отсутствует стремление объе-
диняться. Таким образом, каждый атом можно рассматривать
как законченную молекулу.
Элементам, расположенным непосредственно под благород-
ными газами, не хватает одного электрона во внешней оболочке
(или оболочке «валентных электронов»). Эти элементы обнару-
живают сильное стремление присоединять один электрон, в ре-
зультате чего они приобретают единичный отрицательный заряд.
Таким образом, фтор, хлор, бром и иод обычно находятся в ви-
де соответствующих анионов. Аналогичным образом кислород,
сера и селен присоединяют два электрона, азот и фосфор — три,
а углерод и кремний—-четыре. Эти элементы образуют группу
неметаллов.
Атомы элементов, расположенных в основании каждого
столбца, имеют один электрон на внешней оболочке. Этот элек-
трон они легко отдают, заряжаясь положительно на величину
единичного заряда. Таким образом, литий, натрий, калий, руби-
дий и цезий обычно находятся в виде одновалентных катионов.
Аналогичным образом остальные металлы, отдавая один, два,
три или больше электронов, превращаются в катионы с соот-
ветствующим числом единичных зарядов и с электронной струк-
турой, характерной чертой которой является заполненная элек-
тронная оболочка или, по крайней мере, заполненная электрон-
ная орбита. Элементы,, которые расположены вблизи разделяю-
щей линии между металлами и неметаллами, обладают более
или менее одинаковой способностью к отдаче или присоедине-
нию электронов, что проявляется в двойственности их свойств.
Химическая связь. Связь между атомами в молекулах, жид-
костях и кристаллах является прямым следствием стремления
атомов отдавать или присоединять электроны. Можно отметить
три основных типа химической связи:
1) ковалентная — между атомами, каждый из которых явля-
ется акцептором
2) ионная — между атомами, один из которых является доно-
ром, а второй — акцептором;
1 Атомы, отдающие свои электроны, называются «донорами», а атомы,
присоединяющие дополнительные электроны, называются «акцепторами».
Прим. ред.
24
ГЛАВА 2
3) металлическая — между атомами, каждый из которых яв-
ляется донором.
В газах при обычных температурах связь только ковалент-
ная. В жидкостях и твердых телах встречаются связи всех трех
ТИЛОВ.
Помимо этих трех типов химической связи, существует чет-
вертый тип, так называемая остаточная связь. Она встречается
в веществах, находящихся в твердом и жидком состоянии, и
проявляется в когезии (сцеплении), удерживающей различные
молекулы одну около другой '. Эта связь гораздо слабее любого
из трех основных видов связи, так что иногда ее лучше рассмат-
ривать как «физическую», а не как «химическую». Остаточная
связь весьма важна во флотационных явлениях: действительно,
можно показать, что флотируемость наблюдается только при
таком строении поверхности твердого тела, когда основные
типы связи с окружающей жидкостью проявляются слабо.
Ковалентная связь. Типичная ковалентная связь наблюдает-
ся при взаимодействии одинаковых атомов. Так, если взаимо-
действуют два атома хлора, при образовании молекулы хлора
обобществляются два электрона (по одному от каждого ато-
ма). Реакция образования молекулы может быть представлена
следующим образом:
:С1 . ,С1 : —> : Cl : С1 :
Атом Атом Молекула
В этой записи буквенный символ обозначает атомный оста-
ток, т. е. ядро плюс внутренние электроны, а точки — валентные
электроны.
В молекулярном хлоре два обобществленных электрона в
равной мере принадлежат обоим атомам. Согласно Паулингу
[33], такое обобществление можно рассматривать как проявление
резонанса между двумя структурами или большим их числом.
Так, два электрона, которые в предыдущей реакции показаны
находящимися на равном расстоянии от обоих ядер, могут счи-
таться колеблющимися между положениями, близкими к левому
и правому атомным остаткам. Энергия связи, образованной па-
рой электронов, является энергией резонанса, соответствующей
быстрому обмену электронов между обоими ядрами.
Если каждый из связанных атомов может присоединить толь-
ко по одному электрону, то максимальной первичной ассоциа-
цией из этих атомов будет пара, образующая двухатомную мо-
лекулу. Если же каждый из связанных атомов может присоеди-
1 Молекулы веществ, находящихся в твердом и жидком состояниях, могут
удерживаться и другими видами связи, например водородной. Прим. ред.
ФАЗЫ ВО ФЛОТАЦИОННЫХ СИСТЕМАХ
25
нить по два электрона, как например сера, то существует не-
сколько возможностей:
1) два атома обобществляют каждый по два электрона (все-
го обобществляется четыре электрона);
2) атомы образуют длинную цепочку, причем каждый атом
обобществляет по одному электрону с каждым из двух своих
соседей;
3) образование замкнутой структуры, являющейся вариан-
том случая, рассмотренного в п. 2.
Особым случаем является относительно нестабильная реак-
ционно способная двойная связь, присущая органическим соеди-
нениям.
Сера образует «молекулы» всех трех типов: S = S (в частно-
сти в газообразном состоянии), — S — S — S — S — и Ss. «Моле-
кула» Ss имеет форму восьмиугольника, в котором атомы попе-
ременно слегка отклоняются то в
одну, то в другую сторону от цен- ♦
тральной плоскости [45].
Интересным следствием кова- ——
лентной связи между двухвалент- •
ными атомами является возмож- ,
ность образования структуры, не
ограниченной в одном измерении, ’ • у Т
тогда как в случае одновалент- .. «, I I I
ных атомов возникающие струк- •—
туры обязательно должны быть ® *
ограничены во всех измерениях. рис । Расположение атомов в ал-
Аналогичным образом, если свя- мазе (С. W. Stillwell. Crystal
занные атомы могут присоеди- Chemistry, 1938)
нять для связи по три электрона
каждый (как например, мышьяк), может возникнуть двухмерная
бесконечная структура. Наконец, если связанные атомы могут
присоединять для связи по четыре электрона, возможно образо-
вание трехмерной бесконечной структуры. Такова, например, за-
мечательная структура алмаза (рис. 1).
Для ковалентной связи не требуется, чтобы связанные атомы
были одинаковыми. Например, ковалентная связь имеется в со-
единениях углерода с водородом и серы с кислородом. Особенно
интересен случай соединения углерода с водородом. Благодаря
тому что углерод многовалентен, а водород одновалентен, могут
осуществляться различные структуры углеводородов. Атомы уг-
лерода, кроме того, могут ковалентно соединяться с серой, кис-
лородом и азотом, которые в свою очередь могут соединяться
с другими атомами при помощи связи, более или менее близкой
к ионной. Все это и приводит к необычайному многообразию ор-
ганических соединений.
26
ГЛАВА 2
Ионная связь. Если один из атомов легко присоединяет,
а другой легко отдает электрон, то электрон переходит от одного
атома к другому и возникает структура из двух ионов. Реакция
Na- + -Cl:-^Na+ +[:С1:Г
Атом Атом Ион Ион
должна протекать очень легко. Фактически, однако, получить
металлический натрий и хлор в виде отдельных атомов и затем
сблизить их один с другим настолько трудно, что эта реакция не
может быть продемонстрирована при несложном эксперименте.
Аналогично, если один из атомов легко присоединяет более
одного электрона, а другой легко отдает электроны, возникает
структура из многовалентных ионов. Примером может служить
окись кальция.
Ионная связь отличается от ковалентной прежде всего тем,
что любой катион будет притягивать любой анион и отталкивать
любой другой катион, т. е. эта связь не объединяет определенные
атомы, а существует между любым анионом и любым катионом.
Таким образом, данный катион будет притягивать любой анион
по любому направлению, что делает возможным образование
трехмерных структур как текучих (жидкости), так и твердых
(кристаллы). В качестве различия между ковалентной и ионной
связью иногда подчеркивается анизотропность или векторность
ковалентной связи и изотропность ионной. Ионная связь приво-
дит к упорядоченному расположению ионов противоположной
полярности; в твердых телах при этом не возникает объедине-
ния каких-нибудь определенных анионов с какими-нибудь опре-
деленными катионами. В жидких телах отсутствует упорядочен-
ное расположение ионов, хотя большинство ионов преимущест-
венно окружено ионами противоположного знака. При этом
каждый ион имеет почти полную свободу движения. Определен-
ные молекулы, образов-анные исключительно посредством ионной
связи, постоянно не существуют.
Ионное твердое тело или жидкость могут иметь неограничен-
ную протяженность по всем направлениям. На их поверхностях
условия существенно отличаются от условий внутри тела, что
приводит к специфическим свойствам поверхностей ряда кри-
сталлических веществ.
Металлическая связь. Ранее было указано, что металлы лег-
ко отдают электроны. Это свойство атомов металлов отдавать
свои электроны определяет силы связи между атомами в твер-
дых и жидких металлах и сплавах. Число электронов, исполь-
зуемых для связи, может составлять всего один на атомный
остаток или даже меньше, как это следует из статистики. Одна-
ко, обладая свободой передвижения в твердых и жидких метал-
ФАЗЫ ВО ФЛОТАЦИОННЫХ СИСТЕМАХ
27
лах, электроны занимают все положения, которые им необходи-
мо занять для обеспечения сцепления. Они занимают эти поло-
жения не одновременно, а в очень быстрой последовательности.
Металлическая связь подобна ионной; при этом электроны игра-
ют роль анионов в ионном кристалле. Характерной особенностью
здесь является чрезвычайно высокая подвижность отрицательно
заряженного вещества. У металлов, которые имеют более чем
один валентный электрон на атом, связь становится крепче, а
кристаллы прочнее. В таких металлах, однако, меньше свобод-
ных электронов, так что связь отчасти приобретает характер
ковалентной. Например, в металлах какого-либо одного периода
периодической таблицы ковалентность связи усиливается по ме-
ре возрастания атомного номера.
Известно, что у металлических элементов обнаруживается
переход от чисто металлической связи (щелочные металлы)
к ковалентной (мышьяк, сурьма, теллур). Связь, переходная
между металлической и ионной, наблюдается в интерметалличе-
ских соединениях и у некоторых сульфидов металлов. Таким
образом, в области неорганической химии неоднократно встре-
чается связь, переходная между ионной и ковалентной. Для из-
учения этих вопросов читателю может быть рекомендован ряд
современных работ, как например книги Слэтера [39], Паулин-
га [33], Сиджвика [37, 38], Мотта и Герни [30] и Уэллса [46].
Остаточная связь. В наиболее чистом виде остаточная связь
проявляется у благородных газов, находящихся в жидком и
твердом состояниях. Так как атомы этих элементов не стремятся
присоединять или отдавать электроны, то силы, которые удер-
живают их внутри жидкости или кристалла, не зависят ни от
передачи, ни от обобществления электронов, ни от наличия сво-
бодных электронов. Здесь желательно напомнить, что атом не
является недеформируемой частицей материи, а представляет
собой сложную систему движущихся в пустоте электрических
зарядов. Предположим, что один атом гелия сближается с дру-
гим. Притяжение между ядром одного атома и электронами
второго и наоборот, а также отталкивание ядер и электронов
приведут к небольшому изменению во взаимном расположении
отдельных частей атомов и к некоторому ограничению свободы
их движения. По мере того как ядра сближаются, потенциальная
энергия пары атомов вначале уменьшается, а затем возрастает
в результате совместного действия двух противоположных эф-
фектов. При температуре, равной абсолютному нулю, атомы
сближаются, продолжая, однако, находиться на некотором рас-
стоянии один от другого, соответствующем минимуму потенци-
альной энергии. По мере возрастания температуры кинетическая
энергия каждого атома (тепловая энергия) быстро становится
больше разности потенциальных энергий атомов в состоянии
23 ГЛАВА 2
соприкосновения и до начала взаимного притяжения (т. е. при
достаточно больших расстояниях между атомами). При этом
остаточная связь между атомами уничтожается. Этот пример
показывает, что остаточная связь возникает в результате неболь-
шого перераспределения электронов в соприкасающихся атомах
(иначе никак не связанных), которое соответствует переходу
в положение минимума потенциальной энергии.
Остаточная связь обнаруживается у многих газов в жидком
и твердом состоянии, например у кислорода, азота, водорода,
двуокиси углерода. Молекулы, о которых идет речь, обладают
значительным сходством с молекулами благородных газов, хотя
они являются многоатомными, а не одноатомными. Остаточная
связь в этих газах, конечно, имеет место между атомами сопри-
касающихся молекул, а не между атомами внутри одной и той же
молекулы.
Иногда говорят, что остаточная связь возникает на основе
сил Ван-дер-Ваальса. В этом названии также содержится указа-
ние на эффект взаимной атомной поляризации, связанной с от-
клонением от закона Бойля для идеального газа1:
PV = RT, (1)
где Р, V, Т — соответственно давление, объем и температу-
ра газа;
R—универсальная газовая постоянная.
Эти отклонения, весьма заметные в сжатых газах, впервые
были изучены голландским физиком Ван-дер-Ваальсом. Введя
в закон идеального газа поправки, учитывающие молекулярное
притяжение и собственный объем молекул, Ван-дер-Ваальс по-
лучил уравнение состояния газа в виде
Р + — nb) = nRT, (2)
где Р — давление;
V — объем;
п — число молей газа в объеме V;
R— газовая постоянная;
Т —абсолютная температура;
а — постоянная, характеризующая данный газ;
b — уменьшение эффективного объема, обусловленное «соб-
ственным» объемом молекул газа.
В табл. 2 приведена сводка данных, касающихся различных
типов связи между атомами.
• 1 Термином «остаточная связь» автор обозначает неполярную связь,
обусловленную дисперсионным и поляризационным эффектами. Прим. ред.
ФАЗЫ ВО ФЛОТАЦИОННЫХ СИСТЕМАХ
29
Таблица 2
Типы связи между атомами
Тип Способ обра- зования связи Главные особенности Примеры
Ковалентная Обмен элек- тронами Очень сильная, локали- зованная, ориентирован- ная Кислород — кислород' хлор — хлор; углерод — углерод (алм з)
Ионная Передача электронов Сильная, нелокализо- ванная, неориентирован- ная Каменная соль (твердая или жидкая); известь (твердая.)
Металличе- С помощью Сильная, нелокализо- Ртуть (жидкая): медь
ская свободных электронов ванная, неориентирован- ная (твердая)
Остаточная Слабое сме- щение элек- тронов Слабая, нелок?лизован- ная, неориентированная Жидкие или кристал- лические благородные газы; постоянные газы; углеводороды
Газовая фаза
Газовая фаза играет очень большую роль во флотации, что
связано с плавучестью пузырьков воздуха, с помощью которых
минеральные частицы переносятся в пену. Возможно, однако,
что эта роль преувеличивается. Это станет понятным, если
учесть, что воздух или другой газ при давлении, равном атмос-
ферному или близком к нему, существенно не отличается от ва-
куума Г Так, 1 л воды весит примерно в 800 раз больше, чем 1л
воздуха; 1 моль пара при 100" и 760 мм рт. ст. занимает в 1700
раз больший объем, чем 1 л воды. Иными словами, сродство,
которым вода обладает по отношению к какому-либо веществу,
должно в гораздо большей мере быть заметным в жидкой фазе,
нежели в газовой. Это может быть продемонстрирсвано при фло-
тации с парой в качестве газовой фазы и прокипяченной, свобод-
ной от воздуха водой в качестве среды для пульпы. Эксперимент
свидетельствует о том, что при соответствующей обработке по-
верхность минералов прилипает к пузырькам пара в присутствии
воды, доказывая этим, что прилипание к пузырькам зависит пе
только от химического состава обеих текучих сред (газа и жид-
кости), но также от концентрации молекул в них. Аналогичность
результатов, полученных в экспериментах с паром и воздухом,
показывает, что состав газовой фазы имеет небольшое значение.
Хотя состав газа, рассматриваемого в качестве фазы, играет
небольшую роль, газ может значительно влиять на флотиру-
1 В данном случае автор подчеркивает, что существенное значение имеет
лишь малая плотность газа по сравнению с жидкостью. Прим. ред.
30
ГЛАВА 2
емость минералов, если имеют место реакции между молекула-
ми газа и поверхностью минерала, а также между молекулами
газа и растворенным реагентом или самой водой. Так, кислород
играет важную роль в качестве окисляющего агента; сероводород
является сульфидизирующим агентом и будучи растворенным в
воде, и при прямом действии на минеральные частицы. Двуокись
углерода и сероводород, кроме того, влияют на концентрацию
водородных ионов в водной фазе.
Жидкая фаза
Жидкая фаза во флотации всегда является водным раство-
ром (обычно содержащим растворенные вещества в небольших
концентрациях). Это обусловлено тем, что вода почти всегда
имеется в распоряжении обогатителей, что она дешева и с ней
удобно работать. Конечно, a priori нет причин, по которым другие
жидкости (например, жидкий аммиак, расплавленные соли или
легкоплавкие металлы) не могли бы быть использованы в спе-
циальных флотационных процессах при обработке продуктов
химического или металлургического производства, когда высо-
кая ценность продуктов позволяет применять дорогие флота-
ционные среды. Однако, кроме флотации в нитробензоле [22],
других работ по осуществлению этой заманчивой идеи, по-види-
мому, не велось.
Структура воды. Вода является настолько распространенным
веществом и ее состав — водород и кислород в атомной пропор-
ции 2: 1 — известен так давно, что о ее необычных свойствах
часто забывают. Между тем вода имеет интересные свойства,
выделяющие ее из других веществ, а именно: 1) максимальная
плотность в жидком состоянии при температуре, немного пре-
вышающей точку замерзания; 2) высокая диэлектрическая по-
стоянная; 3) высокая растворяющая способность; 4) низкая
электропроводность, несмотря на большую ионизирующую спо-
собность '; 5) сильная ассоциация молекул.
В течение долгого времени вода в жидком состоянии рассмат-
ривалась как смесь мономерных и полимерных молекул, в кото-
рой соотношение молекул Н2О, (Н2О)2, (Н2О)3 и т. д. меняется
то' мере изменения температуры. Известно, что вода диссоции-
рует на ион водорода и ион гидроксила согласно уравнению:
Н2ОЖН+ + ОН“. (3)
Равновесие в ионизированном состоянии определяется урав-
нением (4), в котором величины, взятые в скобки, означают мо-
лярные концентрации, т. е. количества ионов, выраженные в
грамм-молях и находящиеся в 1 л раствора при 25°:
[Н+] • [ОН-] = 10~14. (4)
1 Речь идет о диссоциации различных соединений на ионы при растворе-
нии их в воде. Прим. ред.
ФАЗЫ ВО ФЛОТАЦИОННЫХ СИСТЕМАХ
31
Рис. 2. Структура молекулы воды [15]:
нЗ-_ ядро водорода; О — ядро кислорода:
О центр отрицательного заряда и молеку-
лы
Ассоциированный характер воды и ее самопроизвольная дис-
социация на ионы были сопоставлены Паннекуком [34], который
пришел к выводу, что эти свойства связаны с асимметричным,
или полярным, характером молекул воды. Это лучше всего за-
метно. если рассматривать строение молекулы воды, исходя из.
структуры электронных оболочек кислорода и водорода.
Атом кислорода имеет два электрона на внутренней или /<-
оболочке и шесть электронов на следующей А-оболочке. Для
образования наиболее вы-
годной структуры инертного
газа неона не хватает двух
электронов. В молекуле во-
ды атомы водорода пере-
дают свои электроны в рас-
поряжение кислородного
ядра. В свою очередь, по од-
ному из электронов А-обо-
лочки атома кислорода по-
ступает в распоряжение
каждого водородного ядра.
Таким образом, три ядра
удерживаются вместе благо-
даря резонансу пары элект-
ронов между кислородом и
каждым из атомов водоро-
да. Относительно центра си-
стемы два водородных ядра
образуют тетраэдрический
угол в 109°30'. Согласно
Эвансу [14], ссылающемуся
на труды Бернала и Фаулера 15, 15], водородные ядра глубоко
проникают внутрь атома кислорода, так что молекула воды лишь
немного больше атома кислорода.
На рис. 2 изображена модель молекулы воды по Берналу
и Фаулеру. Предельный радиус молекулы составляет 1,38 А.
Радиус отсчитывается из центра отрицательного заряда молеку-
лы О~. Ядро кислорода расположено в точке О на расстоянии
0,15 А от О- и 0,96 А от каждого водородного ядра. В этой мо-
дели водородные ядра располагаются на расстоянии 1,50 А
одно от другого.
Можно полагать, что в молекуле воды имеются две области
с избытком положительного заряда (в которых расположены
два водородных ядра) и две области с избытком отрицательного
заряда. По отношению к центру молекулы О~ эти четыре об-
ласти расположены тетраэдрически. С точки зрения статистики
32
ГЛАВА 2
электронная пара преимущественно находится в области с избыт-
ком отрицательного заряда.
Молекулы воды, находящейся в жидком состоянии, связаны
одна с другой таким образом, что положительно заряженная
область одной молекулы располагается против отрицательно
заряженной области другой. Благодаря этому образуется цепоч-
ка из нескольких молекул. Сцепление молекул в воде часто рас-
сматривается как обусловленное наличием водородной связи,
о которой будет сказано далее. Цепочки молекул НгО не прямо-
линейны, а имеют зигзагообразную форму. Они располагаются
таким образом, что у воды в жидком состоянии сохраняется тет-
раэдрическое расположение водородных ядер относительно кис-
лородных и создается возможность гексагонального расположе-
ния в структуре льда. При низких температурах эти цепочки
значительно увеличиваются; при более высоких температурах
преобладают одиночные молекулы Н2О или пары молекул. Как
будет показано в дальнейшем, лед обладает очень рыхлой струк-
турой с значительными пустотами. Если бы лед и вода состояли
из плотно упакованных молекул диаметром 2,76 А) расстояние
между центрами двух ближайших молекул льда), их плотность
составляла бы 1,84 вместо обычных 1,0 и 0,91. То, что у воды в
жидком состоянии плотность меньше 1,00, показывает, что ее
структура далека от плотной упаковки, свойственной твердым
металлам, или от приблизительно плотной упаковки, присущей
жидким металлам. Это согласуется с представлением о том, что
тетраэдрическое расположение молекул НгО, характерное для
льда, сохраняется в воде, находящейся в жидком состоянии. На-
ряду с этим у воды возможно наличие несколько долее плотной
структуры и соответственно’ большей плотности, чем у льда.
Одно из наиболее полных описаний структуры воды как раз-
рушенной структуры льда дано Морганом и Уорреном [29] на ос-
новании рентгеноструктурных исследований воды в жидком со-
стоянии. Эти работы подтвердили высказывания Бернала и Фау-
лера о тетраэдрической связи молекул в воде. Морган и Уоррен
пришли к выводу, что при обычных температурах вода преиму-
щественно состоит из димеров. Это условие дополняется тем,
что каждая молекула воды окружена в среднем четырьмя-пятью
ближайшими соседями, расположенными в среднем на расстоя-
нии от 2,9 до 3,0 А. Это среднее расстояние несколько увеличи-
вается при повышении температуры от близкой к температуре
замерзания до близкой к температуре кипения.
Не следует считать, что такие молекулярные группировки в
воде являются постоянными; они склонны разрушаться и вновь
образовываться из других партнеров. Продолжительность су-
ществования определеных Н2О-полимеров в жидкой воде
ФаЗЫ ВО флотационных системах
33
оценивается в промежутке от 1 микросекунды до 1 микромикро-
секунды (от 106 до 10~12 сек.). Однако, как это следует из зако-
нов статистики, для данной температуры и давления существу-
ет некоторое распределение различных ассоциаций по времени
существования.
Подобно тому как резонанс электронной пары между атома-
ми кислорода и водорода обусловливает связь атомов в моле-
куле воды, резонанс протонов обусловливает связь между мо-
лекулами воды. В процессе перестройки молекулярных ассоци-
аций в жидкой воде часть образующихся обломков имеет из-
быток или недостаток протонов. Эти обломки представляют со-
бой ионы, фигурирующие в уравнениях (3) и (4).
Рис. 3. Возможная структура льда с изображением расположения
атомов водорода. Структура «образуется накладыванием двух изо-
браженных слоев [5]:
а — верхний слой; б — нижиий слой;------- истинная элементарная ячей-
ка; -----------------------псевдоэлементарная ячейка
Лед. Тетраэдрическая ассоциация молекул воды в жидком
состоянии более полно развивается у льда, как это можно видеть
на рис. 3, где изображена структура соседних слоев льда. Каж-
дый атом кислорода соединен по тетраэдрическим направлениям
с четырьмя другими атомами кислорода. Расстояние между их
Центрами равно 2,76 А. Атомы водорода располагаются на ли-
нии О—О. Молекулы образуют гексагональный рисунок, в ко-
тором три молекулы располагаются несколько выше, а три —
несколько ниже каждой плоскости основания. При этом тетра-
эдрический угол 109°30' проектируется на базисную плоскость
в виде угла в 120°, как это и требуется для получения правиль-
ного гексагонального рисунка. «Твердый» лед, очевидно, не очень
тверд, так как описанная структура весьма рыхлая. По-видимо-
3 А- М. Годэн
34
ГЛАВА 2
му, лед является неионным твердым телом, если не рассматри-
вать поверхности кристалла.
Водородная связь. Подобно электронному резонансу, обус-
ловливающему ковалентную связь, обмен протона между двумя
атомами кислорода приводит к образованию резонансной связи
этих атомов. В воде атомы кислорода находятся на расстоянии
2,76 А один от другого (в твердом состоянии) и 2,90 А (в жид-
ком). Протоны расположены на расстоянии 0,96 А от одного кис-
лородного ядра и на остающемся расстоянии от другого. По-
скольку протон гораздо тяжелее электрона, а резонансная энер-
гия должна сохранять тот же самый порядок величин, частота
резонанса протона должна быть много меньше частоты резонан-
са электрона. На самом деле резонансная энергия протона дол-
жна быть еще меньше, так как энергия водородной связи мень-
ше энергии ковалентной. Однако обмен протонами все еще про-
исходит чрезвычайно быстро. По оценке Паулинга, двухэлек-
тронная связь в молекулярном водороде обладает частотой
7 • 1014 пер/сек. Если амплитуда колебаний протона в водород-
ной связи в три раза больше амплитуды колебаний электронной
пары в ковалентной связи, его масса в 900 раз больше, а энер-
гия водородной связи составляет одну десятую энергии ковалент-
ной, то частота резонанса должна быть приблизительно в 300 раз
меньше, т. е. равняться примерно 2 1012 пер/сек. Энергия про-
тонной связи, хотя и меньше энергии ковалентной, но все же ве-
лика по сравнению с энергией остаточной, или ван-дер-Вааль-
совой, связи. Водородная связь отличается от остаточной тем,
что, подобно ковалентной, она анизотропна.
Помимо воды, водородная связь имеется во многих органи-
ческих жидких и кристаллических соединениях. Она играет важ-
ную роль в пластмассах и живых тканях. Так, например, моле-
кулы спиртов или жирных кислот соединяются в бимолекуляр-
ные или двухслойные полимолекулярные группы с помощью во-
дородных мостиков:
R R R R
О ООО
П Н или Н Н Н Н Н
о ^О
7? R
Кроме того, гидролиз протеинов, столь важный в процессе
жизнедеятельности организмов, можно рассматривать как заме-
ФАЗЫ ВО ФЛОТАЦИОННЫХ СИСТЕМАХ
35
щение одной водородной связи другой водородной связью. Одна
водородная связь заменяется другой и при построении протеинов
из аминокислот. Поскольку молекулы, содержащие группы
—ОН, могут ассоциировать через водородные мостики, необхо-
димо помнить, что эти мостики весьма непрочны и что обломки,
образующиеся при их разрушении, сильно реакционно способны.
Гидратация ионов в растворе. Рассматривая уравнение (3),
мы указывали, что вода распадается на ионы водорода и гидро-
ксила. Это происходит при спонтанном разрушении Н20-полиме-
ров, когда у одной из частей образуется избыток положительного
заряда, а у другой — точно такой же избыток отрицательного.
Эти обломки обычно представляют собой не Н+и ОНд а скорее
(«Н2О • Н)+ и (6Н2О • ОН)'. Негидратированный водородный
ион — одиночный протон — в жидкой воде практически не су-
ществует. Определение предельных размеров, до которых гидра-
тируется гидроксильный ион, все еще является спорным.
В водных растворах, помимо гидроксила и водорода, имеется
ряд других гидратированных ионов. Действительно, гидратация
ионов может быть одной из основных причин, обусловливающих
высокую растворяющую способность воды. Например, негидра-
тированные ионы каменной соли в процессе растворения будут
следующим образом реагировать с водой:
(m + m') Н2О + iNa+ + Cl~5- (Na • т Н2О)+ +
+ (С1 • т'Н2О)-. (5)
Представление о более или менее сильно гидратированных
ионах важно для полного понимания флотации: гидратирован-
ные ионы прилипают к поверхности минерала, которая таким
образом оказывается покрытой водой. Однако степень гидрата-
ции у различных ионов неодинакова. Причиной этого, вероятно,
являются различия в их размерах.
Для оценки гидратации ионов в растворе может быть ис-
пользовано несколько методов. Один из них заключается в том,
чтобы вывести заключение о степени гидратации из числа и
расположения молекул воды в кристаллогидратах. Недостатком
этого метода является предположение, что условия, имеющиеся в
жидкой воде, воспроизводят условия в твердом кристалле.
Фактически гидратация, рассчитанная таким образом, дол-
жна быть минимальной, ибо кристаллы часто образуются с не-
достатком воды, как например при естественном росте из водной
среды. Так (2, 4], в кристаллическом NiSO4 6Н2О шесть моле-
кул воды координируются с ионом никеля, то же самое справед-
ливо в отношении шести молекул воды из семи в гептагидрате
NiSO4 • 7Н2О. В пентагидрате сульфата меди CuSO4 • 5Н2О,
представляющем обычную форму гидратированного суль-
фата меди [3], катионом является (Си • 4Н2О)2+, а анионом
з*
36
ГЛАВА i
(SO4 • Н2О)2~. Из этого можно сделать только тот вывод, что
в воде катион никеля будет гидратирован не менее чем шести-
кратно, а катион меди — не менее чем четырехкратно.
Во втором методе одновременно используется скорость пере-
носа электрических зарядов ионами и подвижность ионов, ко-
торая, согласно закону Стокса, зависит от внутреннего трения
(вязкости) и размера частиц. Существует решение уравнения
Стокса для произвольного радиуса гидратированного иона [12].
Первым шагом в этом методе является оценка переносимого ко-
личества различных анионов и катионов. Возможность проведе-
ния такой оценки не вызывает сомнений, но ее интерпретация
и особенно предположение, что закон Стокса приложим к «дис-
персным» частицам любых размеров, включая частицы, образую-
щие дисперсионную среду, требуют еще количественного подтвер-
ждения.
В третьем методе используется влияние растворенного элек-
тролита на удельный вес воды; при этом учитывается вклад
дегидратированных ионов в удельный объем раствора [И]. Это-
му методу, вероятно, присущи значительные ошибки, особенно
при вычислении гидратации ионов, размеры которых в негидра-
тированном состоянии малы, поскольку при расчете по этому
методу вода рассматривается как непрерывная фаза; дискрет-
ность ее структуры во внимание не принимается.
В четвертом методе, примененном Ван Райвеном [44], исполь-
зуется корреляция между ионизацией концентрированного рас-
твора, рассчитываемой по его электропроводности, и ионизаци-
ей,, рассчитываемой по понижению давления пара над раствором
по сравнению с давлением -пара над чистым растворителем. При
этом предполагается, что имеются эталонные электролиты, ионы
которых не гидратируются. В качестве такого эталона применя-
ется азотнокислое серебро; поскольку значения ионизации этой
соли, полученные из данных электропроводности и рассчитан-
ные по понижению давления насыщенного пара, одинаковы, по-
стольку Ag+и NO^ должны быть дегидратированы; так, если
гидратация Na + NOrna пару ионов оказывается равной 8, то все
восемь молекул воды должны быть связаны с Na+ и ни одна
с NO7.
Пятый метод, примененный Реми [36] и затем изученный
Свинджедоу [40], основывается на прохождении воды через по-
ристые диафрагмы. В основе этого метода лежит предположение,
что диафрагма негидратирована и неподвижна относительно
обоих электродов (если бы этого не было, вода автоматически
протекала через диафрагму, что ошибочно приписывалось бы
действию изучаемых ионов). Значения гидратации, полученные
методом, примененным Свинджедоу, в несколько раз больше, чем
ФАЗЫ ВО ФЛОТАЦИОННЫХ СИСТЕМАХ
37
результаты, полученные другими методами, что заставляет со-
мневаться в их справедливости. Типичные результаты, получен-
ные различными методами, приведены в табл. 3.
Таблица 3
Гидратация ионов: число молекул воды на ион
Катионы и анионы Метод оценки
координа- ция в гид- ратирован- ных крис- таллах* 1 общая координа- ция2 волюмет- рически8 волюметриче- ски в комби- нации с изме- рением по- движности ионов4 корреляция между удель- ной электро- проводностью и давлением пара8 перенос воды че- рез мем- брану*
Катионы
L1+ — 6 6 2 13 12,6
Na + — 4 8 1 8 8,4
К+ — 4 0 0 4 4,0
Pb+ — 4 0 — 3 —
Cs+ — 4 0 0 — —
Ag+ — » — — 0,3 0 —
nh4+ — — — — 5 4,4
Mg2+ — 6 6 6,6 .— 32
Ca2+ 2 6 6 5,2 — 28
Ba2+ (8) 6 4 —. —
Zn2+ 6 6 — 6,7 — —
Co2+ 6 6 — 8,0 — —
Mi2+ 6 6 — 7,6 — .—
Cu2 + 4 6 — 7,0 .— —
Al3 + 6 . 6 30
Анионы
F~ — 4 1 0,4 — —
ci- — 4 0 0 3 3,0
Br~ — 4 0 0 .— —
J- — 4 0 0 3 3,7
SO42- — .— — 0,5 — —
NO3- — — — 0 о —
1 См. книги по кристаллохимии.
2 Бернал и Фаулер [5].
2 Бернал и Фаулер [5].
* Дармуа [12].
6 Ван Райвен [44].
8 Реми [36].
В величинах гидратации, получаемых разными методами,
Для одних и тех же ионов наблюдаются большие отклонения.
Однако можно считать установленным следующее:
1) поливалентные катионы чаще всего гидратируются шестью
или восемью молекулами воды;
38
ГЛАВА 2
2) гидратация катионов щелочных металлов уменьшается с
увеличением размера ионов от Li + к Cs +;
3) анионы обычно гидратированы гораздо меньше, а часто,
возможно, совсем не гидратированы.
Связь ядер гидратированных ионов с молекулами воды в
растворе непостоянна. Гидратированные ионы отщепляют и
вновь присоединяют молекулы воды. Так, например, весьма энер-
гично протекает следующая реакция [19, 35]:
[Ni (Н3О)6]2+ + * (Н2О)-> [Ni (Н2О)5*(Н2О)]2+ + Н2О. (6)
Структура электролита, такого, например, как хлористый
магний, напоминает разломанную структуру льда с ионами гид-
рония, оксония, гексагидратом магния и ионом хлора, причем
наблюдается быстрый обмен молекулами воды и электронами
между гидратными слоями и водной фазой.
Вещества, молекулярно растворимые в воде. Эти вещества
при растворении в воде не ионизируются, хотя большое число
их, вероятно, до некоторой степени гидратировано. Неионизи-
рованные растворы образуют атмосферные газы и некоторые
органические соединения. Среди кислород- или азотсодержа-
щих органических соединений иногда одна часть молекулы бо-
лее склонна к гидратированию, чем другая. В частности, это
относится к спиртам, которые в чистом виде представляют ассо-
циированные жидкости, очень сильно растворяющиеся в воде
с выделением тепла; хорошая растворимость и выделение тепла
связаны с гидратацией ОН-групп спиртов.
Концентрация водородных ионов. Одним из наиболее значи-
тельных факторов, характеризующих флотационную -систему,
является концентрация водородных ионов в пульпе. Как извест-
но, в результате ионизации воды образуются гидратированные
ионы водорода и гидроксила. Произведение концентраций этих
ионов — величина постоянная. Если в воде имеется вещество,
при ионизации которого возникает много водородных ионов,
полное число водородных ионов увеличится, а полное число
гидроксильных ионов уменьшится в соответствии с уравнени-
ем (4). Такие растворы носят название кислых. В свою очередь,
растворы с увеличенной концентрацией гидроксильных ионов
и ввиду этого уменьшенной концентрацией водородных ионов
носят название щелочных растворов.
Крайними пределами концентрации водородных ионов в воде
будут приблизительно 10+1 и 1СП15. Первое соответствует пол-
ностью диссоциированному 10-м. раствору одноосновной кисло-
ты, а второе,— столь, же концентрированному раствору одноос-
новной щелочи. Во флотационных пульпах концентрация водо-
родных ионов обычно меняется в более ограниченных преде-
лах-— между 10-3 и 10-12.
ФАЗЫ ВО ФЛОТАЦИОННЫХ СИСТЕМАХ
39
Соренсен ввел специальную шкалу для характеристики кис-
лотности и щелочности. Это так называемая рН-шкала [23, 25],
в которой численное значение pH равно взятому с обратным
знаком десятичному логарифму концентрации водородных ионов:
pH = - lg10 [Н+]. (7)
В растворах, близких к нейтральным, небольшая добавка
вещества, обладающего кислотными или щелочными свойства-
ми, вызывает значительное изменение pH. Это легко можно за-
метить, например, рассматривая концентрацию водородных ио-
нов, необходимую для того, чтобы pH чистой воды изменить от
7 до 6. Большое изменение pH в тщательно очищенной воде при
добавлении малого количества кислоты или щелочи является
причиной, из-за которой бывает трудно получить дистиллиро-
ванную воду с pH = 7. Углекислота, содержащаяся в воздухе,
растворяясь в воде, образует угольную кислоту и обычно вызы-
вает такое дополнительное увеличение концентрации водород-
ных ионов, что pH становится меньше 6. Можно подсчитать, что
углекислота, содержащаяся в 4 мл «чистого» воздуха, способ-
на в два раза увеличить концентрацию водородных ионов в
100 мл «чистой» воды. С другой стороны, требуется значительное
количество кислоты, чтобы изменить pH от 1 до 0 или от 14 до 13.
Большинство солей не дает нейтральных растворов, т. е. рас-
творов, у которых значение pH точно равно 7, даже в дистилли-
рованной воде. Это происходит потому, что они реагируют с
водой (гидролиз), образуя слабокислый или слабощелочной
продукт вместе с сильнощелочным или сильнокислым продук-
том. Так, раствор хлористого алюминия сильнокислый:
А1С13 + ЗН2О7±А1(ОН3) + ЗН+ + ЗСГ, (8)
в то время как раствор карбоната натрия щелочной:
Na2CO3 + Н2От±2Na+ ф НСО? + ОН~. (9)
Находясь в воде, такие соли, как хлористый алюминий или
карбонат натрия, препятствуют резкому изменению pH при до-
бавлении других веществ. Добавление соляной кислоты в рас-
твор, уже содержащий хлористый алюминий, не вызовет такого
эффекта, какой был бы достигнут в растворе с тем же pH, но по-
лученным с помощью одной соляной кислоты. Аналогично добав-
ление щелочи к раствору хлористого алюминия будет способ-
ствовать его гидролизу, и pH раствора получится ниже, чем при
одной щелочи. Способностью «смягчать» действие кислот и щело-
чей обладают все соли, не дающие нейтральных растворов. Это
Их свойство носит название буферного действия.
40
ГЛАВА 2
Кристаллические фазы
Флотационные свойства кристаллической фазы определяют-
ся характером ее поверхности в гораздо большей степени, неже-
ли объемными свойствами. Однако для оценки поверхностных
свойств кристаллов полезно сначала рассмотреть внутреннее
строение этих кристаллов, полагая их бесконечными по всем на-
правлениям. Свойства бесконечных кристаллов будут рассмотре-
ны в данном разделе, свойства их поверхностей — в главе 4.
Кристаллы могут быть классифицированы по типу преобла-
дающей в них связи. Такая классификация приводится в табл. 4.
Таблица 4
Классификация кристаллов
Структура опреде- ляется остаточной связью Существование других типов связи Протяженность ионной струк- туры Типы кристаллов
ионная I СВЯЗЬ 1 1 свобод- ные элек- троны ковалентная связь
(три измере- ния) Пя (два измере- д ния) (одно изме- рение) Нет1 Нет » » ' » Есть Нет Нет » » Есть / Нет | Есть » Нет Есть (только внутри ионов) Нет Конечная протяжен- ность Бесконечный в одном из- мерении Бесконеч- ный в двух измерениях Бесконеч- ный в трех измерениях Атомный Молекулярный Волокнистый Слоистый Алмазоподоб- ный Простой ионный Сложный ИОН- НЫЙ Волокнистый Слоистый Блоковый2 Металлический
» За исключением
некоторых
слоистых кристаллов, у которых по одному направле-
нию чередуются ионная и остаточная связи.
2 Термин «кристалл с блоковой структурой» заимствован из статьи Г. С. Стрельцина
(см. сноску иа стр. 41). Прим. ред.
Если принять во внимание табл. 2, то видно, что четыре типа-
связи и их совместные комбинации позволяют выделить один-
ФАЗЫ ВО ФЛОТАЦИОННЫХ СИСТЕМАХ
41
надцать категорий кристаллов. Такая классификация является
идеальной. Реальные кристаллы не совпадают с этими типами,
располагаясь между различными категориями
В табл. 4 кристаллы прежде всего классифицируют, исходя
из того, определяется ли их структура остаточной связью или
не определяется. Остаточная связь существует во всех случаях,
но она легко перекрывается другими видами связи. Таким обра-
зом, можно сказать, что в начальной стадии классификации
основное — выяснить, определяется ли структура преимуществен-
но одним из главных типов связи или остаточной связью. На
второй стадии классификации устанавливается присутствие или
отсутствие ионной, металлической и ковалентной связи. В кри-
сталлах, в которых остаточная связь не является преоблада-
ющей, металлическая связь отсутствует, а имеется ионная
связь. Третья стадия классификации заключается в определе-
нии протяженности ионной структуры в различных измерениях.
Атомные кристаллы. Атомные кристаллы встречаются толь-
ко среди благородных газов в твердом состоянии. За исключе-
нием гелия, благородные газы кристаллизуются в кубической
системе с плотной упаковкой [48]. Каждый атом имеет двенад-
цать соседей, расположенных на одинаковом расстоянии от него.
Четыре из них лежат в плоскости, проходящей через атом, и по
четыре лежит в каждой из двух равноудаленных параллельных
плоскостей, расположенных по обе стороны от него. Расстояние
между ближайшими соседями довольно велико (3,18 и 3,83 А
для неона и аргона при —253°, и 4,02 и 4,40 А для криптона и
ксенона при —184°). Гелий [21] кристаллизуется в гексагональ-
ной системе с плотной упаковкой, в которой каждый атом также
имеет двенадцать равноудаленных соседей; шесть из них распо-
ложено в плоскости, проходящей через атом, и по три в каждой
из равноудаленных плоскостей по обе стороны от него. Меж-
О
атомное расстояние в гелии при —271° составляет 3,57 А.
Важно отметить, что межатомные расстояния относительно
большие, а силы связи в кристаллах так малы, что плавление
или сублимация происходят при очень низких температурах.
Так, например, температура кипения гелия —268,9°, а темпера-
туры плавления неона, аргона, криптона и ксенона соответствен-
но —249, —189, —157 и —112°. Закристаллизованные благород-
ные газы представляют такую редкость, что мало кому при-
шлось наблюдать их в таком состоянии. Это единственные кри-
Р 1 Ранее и более подробно подобная классификация была разработана
• С. Стрельциным (см. Труды II научно-технической сессии института Me-
Хаиобр, Металлургиздат, 1952). Прим. ред.
42
ГЛАВА 2
сталлы, атомы которых удерживаются исключительно при помо-
щи остаточной связи.
Молекулярные кристаллы. Молекулярные кристаллы подоб-
ны атомным за тем исключением, что место атомов в них зани-
мают молекулы. Ряд органических и неорганических соедине-
ний образует молекулярные кристаллы. Примером таких орга-
нических соединений могут служить алифатические или аромати-
ческие углеводороды, так же как и полностью или частично за-
мещенные галогенопроизводные углерода, например, четыреххло-
ристый углерод, хлороформ и фтористый углерод. Из неоргани-
ческих соединений молекулярные кристаллы образуются посто-
янными газами, серой и иодом.
Рис. 4. Кристалл иода (R. W. G. Wyckoff. Crystal
Structures, I, New York, 1948):
ci — проекция орторомбической структуры твердого иода на кри-
сталлографическую грань Ь; б — упаковка, соответствующая схема а
Азот, например, образует гексагональные кристаллы с плот-
ной упаковкой, у которых расстояние между центрами молекул
равно 4,04 А, в то время как расстояние между ядрами азота в
одной и той же молекуле составляет 1,06 А. Иод образует ром-
бическую гранецентрированную решетку, как это показано на
рис. 4. В кристалле иода два атомных ядра внутри одной моле-
кулы удалены одно от другого на 2,70 А, в то время как
ФАЗЫ ВО ФЛОТАЦИОННЫХ СИСТЕМАХ
43
наименьшее расстояние между центрами атомов, принадлежа-
щих разным молекулам, составляет 3,54 А.
Двуокись углерода кристаллизуется в кубической системе.
Атомы углерода образуют плотно упакованную структуру с рас-
стоянием между центрами атомов 3,98 А. Внутри молекулы яд-
ро углерода удалено только на 1,07 А от ядра кислорода, а два
кислородных ядра удалены одно от другого на 2,15 А. Наимень-
шее расстояние между атомами кислорода в соседних молеку-
лах равно 3,23 А, а наименьшее расстояние между атомом угле-
рода одной молекулы и атомом кислорода другой составляет
3,25 А.
Сера кристаллизуется подобным же образом. Кристалличе-
ская решетка образуется молекулами S8 145]; в состав элемен-
тарной ячейки входит шестнадцать молекул. На рис. 5 показано
расположение атомов и молекул серы. В каждой молекуле каж-
дый атом серы имеет двух ближайших соседей на расстоянии
2,11 А. Ближайшие атомы серы в разных молекулах удалены
на 3,27 А.
Метан СН4 кристаллизуется в гранецентрированной кубиче-
ской системе 127] с расстоянием между углеродными атомами
4,49 А. Внутри молекулы расстояние между углеродом и водо-
родом всего лишь 1,093 А 133а). Атомы водорода в этих кристал-
лах не имеют своих определенных мест и могут занимать раз-
личные положения на сфере с центром в атоме углерода.
Бензол, структура которого хорошо изучена, состоит из ко-
лец, образованных шестью атомами углерода, с каждым из ко-
торых связан один атом водорода. Внутри молекулы расстояние
углерод — углерод составляет 1,42 А; это значение располага-
ется между величиной 1,54 А, полученной для одиночной связи
С—С, и 1,33 А для двойной связи С = С в алифатических органи-
ческих соединениях. Расстояние С—Н также несколько меньше,
чем в метане, и составляет 1,08 вместо 1,093 А. С другой сторо-
ны, кратчайшее расстояние между атомами углерода в сосед-
иих молекулах составляет около 3.8 А. Расположение молекул
обладает орторомбической симметрией (рис. 6). Нафталин, ан-
тРацен, хризен, дифенил и многие углеводороды с комплексами
колец обладают аналогичной структурой.
Парафины образуют длинные зигзагообразные цепочки уг-
о
неродных атомов, располагающихся на расстоянии 1,54 А один
Рис. 5. Структура орторомбической серы. Числа означают коорди-
наты атомов в градусах; некоторые из 16 молекул, входящих в эле-
ментарную ячейку, здесь не изображены, чтобы не затемнять кар-
тину [45]
Рис. 6. Структура бензо-
ла СеНб. Заштрихован-
ные молекулы смешены
по вертикали на полови-
ну высоты элементарной
ячейки; Се кольца изо-
бражены с торца, и по-
этому видны только три
атома углерода (R. С.
Evans. Ап Introducti-
on to Crystal Chemisrty.
New York, 1939)
ФАЗЫ ВО ФЛОТАЦИОННЫХ СИСТЕМАХ
45
от другого и образующих между собой характерные тетраэдри-
ческие углы в 109o30z. Каждый атом углерода связан с двумя
атомами водорода, в свою очередь расположенными тетраэдри-
чески по отношению к направлению связи С—С. Расстояние
между атомами углерода и водорода составляет около 1,09 А.
На рис. 7 изображена зигзагообразная цепочка парафина С18Нз8,
а на рис. 8 показано совместное расположение таких парафино-
вых цепочек внутри кристалла. В кристалле парафина (напри-
мер, «С2дНбо [31]) расстояние между крайними атомами углерода
у молекул, расположенных на одной линии, равно ~4,0 А, а рас-
стояние между центрами атомов углерода соседних молекул —
не менее 3,6—3,9 А.
Рис. 7. Отдельная молекула парафина Cishas. Внут-
ренние кружки представляют «сферы ковалентной
связи» углеродных ядер внутри молекулы, а внешний
контур — «зону влияния» цепи по отношению к дру-
гим подобным цепям
При изучении большого числа кристаллов типа С„Н2п+2
(п в пределах 5—30) Мюллер [32] нашел, что средний размер мо-
лекул по оси с (вдоль по длине слегка наклоненных молекул)
возрастает на 1,25 А каждый раз, когда длина молекулы уве-
личивается за счет присоединения группы СН2. Молекулы могут
кристаллизоваться в двух несколько различных формах. Соеди-
нения С18Н38 и С20Н42 были обнаружены в обеих формах. Мюл-
лер нашел, что при нагревании от температуры жидкого возду-
ха до температуры плавления параметры кристаллов очень ма-
ло возрастают по направлению с и значительно возрастают в
направлениях а и Ь. Наблюдаемые изменения параметров решет-
ки свидетельствуют о том, что межатомные расстояния внутри
молекулы очень мало изменяются с температурой, в то время как
межмолекулярные расстояния изменяются значительно. Эти на-
блюдения еще раз подтверждают, что остаточные силы, которые
удерживают молекулы внутри кристалла, очень слабы по срав-
нению с ковалентными силами внутри молекул.
На рис. 9 показана зависимость молекулярных размеров по
оси с в насыщенном парафине (приблизительно длина молеку-
лы) от числа углеродных атомов. Видно, что существуют две
слегка различные кристаллические формы (А и В).
Рис. 8. Структура нормального парафина. Цепи видны с торца; мо-
лекулы А и В взаимно смещены по вертикали на половину высоты
элементарной ячейки [31]
fj - число атамов С
Рис. 9. Размены молекул по оси с в насыщенных
парафинах, имеющих кристаллическую структу-
ру Л и В [32]
ФАЗЫ ВО ФЛОТАЦИОННЫХ СИСТЕМАХ
47
Данные, приведенные в этом и предыдущих разделах, сум-
мируются в табл. 5, в которой сравниваются внутримолекуляр-
ные межатомные расстояния. Представляет интерес малое изме-
нение внутримолекулярных расстояний даже при очень больших
изменениях атомного веса (например, от Не, до Хе) или при
сравнении таких различных веществ, как аргон, иод, углекисло-
та и бензол. Также имеет большое значение то, что внутримо-
лекулярные расстояния много меньше межмолекулярных рас-
стояний.
Таблица 5.
Межатомные расстояния в атомных и молекулярных кристаллах
Кристалл
Расстояние между
атомами внутри
молекулы
А
Расстояние между
атомами в ближай-
ших соседних мо-
лекулах
о
А
Атомные кристаллы:
гелий............................
неон.........................
аргон .......................
криптон .....................
ксенон ......................
Неорганические молекулярные кри-
сталлы:
азот...............................
ИОД..........................
углекислота .................
сера.........................
Органические молекулярные кри-
сталлы:
метан .......................
бензол ......................
Парафины с длинными цепями . .
То же..........................
3,57 (—271°)
3,18 (—253°)
3,83 (—253°)
4,02 (—184°)
4,40 (—184°),
1,06
2,70
1,07 (С—О)
2,15 (О—О)
2,11
4,04
3 64
3^23 Ю—О)
3,25 (С—О)
3,98 (С—С)
3,27
1,09 (С—Н)
1,42 (С—С)
1,(8 (С—И)
1,54 (С—С)
1,09 (С—Н)
4,49 (С—С)
3,8 (С—С)
3,7 (С—С сбоку)
4,0 (С—С между
концами)
Большинство веществ, образующих атомные и молекуляр-
ные кристаллы (неполярные), имеет настолько низкую темпера-
туру плавления, что, например, многие из них находятся в газо-
образном состоянии уже при температуре замерзания воды. Спи-
сок неполярных кристаллов, представляющих интерес с точки
зрения флотации, ограничен и включает главным образом орга-
нические соединения с относительно большим молекулярным
весом.
Волокнистые и слоистые кристаллы. Наиболее характерные
примеры волокнистых кристаллов дают кристаллы двухвалент-
иых элементов: серы, селена и теллура. Наряду с молекулами:
48
ГЛАВА 2
в виде восьмиатомных колец сера может также образовывать
цепочки из атомов, связанных между собой ковалентными свя-
зями. Такие молекулы серы образуют волокнистую структуру,
обладающую ограниченной упорядоченностью в направлениях,
перпендикулярных оси молекул. Правильное расположение мо-
лекул в такой структуре наблюдается лишь в напряженном со-
стоянии [281.
В кристаллах селена [48а] атомы располагаются на бесконеч-
ных спиралях. Межатомное расстояние внутри одной спирали со-
ставляет 2,32 А. Соседние спирали параллельны, и наименьшее
расстояние между атомами соседних цепочек 3,46 А. Кристал-
лы теллура имеют аналогичное строение с внутримолекулярным
расстоянием в 2,86 А и расстоянием между молекулами не ме-
нее чем 3,74 А.
В трех рассмотренных случаях волокнистые кристаллы об-
наруживают некоторую неполярность в двух направлениях,
по которым расстояния между молекулами заметно больше
внутримолекулярных. Однако эта разница не очень велика, и не-
полярность выражена нерезко.
Подобно тому как молекулярные кристаллы образуются од-
новалентными неметаллами, а волокнистые кристаллы двухва-
лентными неметаллами, можно было бы ожидать, что слоистые
кристаллы будут образовываться трехвалентными неметаллами
и металлами, располагающимися в одной и той же группе пери-
одической системы с ними, т. е. азотом, фосфором, мышьяком,
сурьмой и висмутом. Азот, однако, кристаллизуется только в ви-
де молекулярных кристаллов. Фосфор образует или молекуляр-
ные кристаллы, как Р4, — белый фосфор, или модификацию, на-
зываемую черным фосфором. В последнем случае каждый атом
связан с тремя другими (как следовало бы ожидать от кова-
лентно связанных трехвалентных атомов), но эти атомы не лежат
в одной плоскости (рис. 10). В этой структуре расстояние меж-
ду ковалентно связанными атомами составляет 2,17 А (два) и
2,20 А (одно), в то время как другие ближайшие соседи удалены
на 3,87 А.
В мышьяке, сурьме и висмуте атомы образуют слои, упако-
ванные один параллельно другому. Наименьшее расстояние
между атомами внутри одного слоя составляет для этих трех
элементов соответственно 2,51; 2,87 и 3,10 А, а расстояние меж-
ду каким-нибудь атомом одного слоя и ближайшим к нему ато-
мом другого слоя — соответственно 3,15; 3,37 и 3,47 А. Очевид-
но, можно рассматривать слои как индивидуальные молекулы,
ФАЗЫ ВО ФЛОТАЦИОННЫХ СИСТЕМАХ
49
бесконечные в двух измерениях и ограниченные в третьем (пер-
пендикулярном плоскости слоев). Это, однако, становится ме-
нее справедливым при рассмотрении структуры мышьяка и осо-
бенно висмута, так как висмут обнаруживает преимущественно
металлические свойства.
Лучший пример слоистого кристалла, образованного одним
элементом, представляет графит. Углерод, как хорошо известно,
Рис. 10. Идеальная кристаллическая структура черного фосфора
(часть слоя) (A. F. Well s. Structural Inorganic Chemistry,
New York, 1945)
кристаллизуется также в виде алмаза с межатомным рассто-
янием 1,54 А. В графите (рис. 11) атомы располагаются в плос-
костях, удаленных одна от другой на 3,40 А, в то время как рас-
стояние С—С внутри одного слоя составляет только 1,42 А. Яс-
но, что связь между отдельными атомами внутри одного слоя
более прочна, чем связь С—С в алмазе, в то время как связь
между слоями гораздо слабее. Расстояние между атомами
внутри одного слоя практически такое же, как и в бензоле; каж-
дая связь внутри углеродного слоя эквивалентна 4/з ординарной.
Структура молибденита является примером иного типа сло-
истых кристаллов. Можно считать, что этот минерал состоит из
параллельно расположенных пакетов сульфида молибдена, а
каждый пакет состоит из двух бесконечно протяженных слоев
серы толщиной в один атом, и слоя молибдена между ними так-
же толщиной в один атом, заполняющего промежутки между ато-
мами серы (рис. 12). Расстояние между соседними атомами серы
внутри одного слоя пакета составляет 2,98 А, а расстояние меж-
ду атомами серы и молибдена в пакете 2,35 А. Наименьшее рас-
стояние между атомами серы в соприкасающихя пакетах гораздо
больше, а именно, 3,66 А. В этом случае атомы серы, по крайней
мере частично, связаны ковалентно внутри одного слоя, а атомы
молибдена связаны с атомами серы или ковалентно или частич-
4 А. м. Годэн
50
ГЛАВА 2
но ковалентно. Связь между атомами серы в соприкасающихся
пакетах вряд ли является ионной; она определенно не является
металлической и не более чем частично ковалентной (в основном
это должна быть остаточная связь).
Внешне похожие слоистые кристаллы образуются иодидом
кадмия, который относится к большой группе [48с] слоистых
кристаллов типа МХ2, включающей MgBr2, PbJ , Cd (ОН) ,
Рис. 11. Структура графита
Рис. 12. Молибденит MoS2 (R. W. G.
Wyckoff, Crystal Structures, I,
New York, 1948):
a — проекция; 6 —упаковка, соответ-
ствующая схеме а
Мп(ОН)2, NiGl2. В этих кристаллах галогены или гидроксилы
образуют гексагональную или кубическую плотную упаковку, а
атомы металла заполняют промежутки между противолежащи-
ми слоями галогенов или гидроксилов. Одно важное различие
между структурами CdJ2* и MoS2 состоит в том, что перпенди-
кулярно к базисной плоскости идет последовательность слоев
...Cd—J—Cd—J... вместо... S—Mo—S—S—Mo..: Другое важ-
ное различие заключается в том, что расстояние J — J будет
практически одинаковым независимо от того, находятся атомы
иода в одном и том же слое или в разных. Этого не наблюдает-
* Структура CdJ2 описана Годэном неправильно. Структура CdJ2 пред-
ставляет одну из разновидностей слоистых решеток, в которой в направлении
гексагональной оси следуют пакеты, состоящие из слоя ионов кадмия, окру-
женного с двух сторон слоями ионов иода. Таким образом, чередование слоев
будет не ...Cd — J — Cd — J..., a ...J — Cd — J — J —Cd... (см. например:
П а у л и н г «Природа химической связи» и ван Аркель «Химическая
связь»). Прим. ред. , ь .
ФАЗЫ ВО ФЛОТАЦИОННЫХ СИСТЕМАХ
51
ся для расстояния S — Sb молибдените. Расстояние Cd — J бу-
дет одним и тем же в любой паре слоев. Таким образом, мы
должны исключить CdJ2 из группы слоистых кристаллов, поме-
щая взамен этого кристаллы, обладающие такого рода структу-
рой, между простыми ионными кристаллами. В то же время
молибденит мы оставляем среди слоистых кристаллов.
Кристаллы со структурой алмаза. Типичным представителем
кристаллов со структурой алмаза является алмаз, в котором
каждый атом углерода ковалентно связан по тетраэдрическим
направлениям с четырьмя атомами углерода, находящимися
на расстоянии 1,54 А от него (см. рис. 1). Представляет интерес,
что это расстояние совпадает с расстоянием между атомами
углерода в углеводородной цепи, но несколько больше расстоя-
ния между ближайшими атомами углерода в бензольном коль-
це или в графите (1,42 А). Кремний, германий и серое олово
образуют алмазоподобные кристаллы, изоморфные с алмазом.
В алмазе каждый кристалл можно рассматривать как отдель-
ную молекулу. С этой точки зрения молекула углерода беско-
нечна по всем направлениям.
В известном смысле лед тоже является алмазоподобным
кристаллом. Кислородные атомы в структуре льда распола-
гаются тетраэдрически, приблизительно как углеродные атомы
в алмазе, а атомы водорода занимают положения между атома-
ми кислорода, образуя резонансную водородную связь. По-
скольку водородная связь обладает свойствами как ковалент-
ной, так и ионной связи, лед по своему характеру практически
занимает промежуточное положение между алмазоподобными
и ионными кристаллами.
Другую группу кристаллов, напоминающих алмаз, пред-
ставляют три формы кристаллического кремния: кварц, кристо-
балит и тридимит. В каждом из этих кристаллов каждый из
атомов кремния Связан с четырьмя атомами кислорода, и каж-
дый атом кислорода связан с двумя атомами кремния. Это очень
похоже на структуру льда, в которой, однако, роль водорода иг-
рает кислород, а роль кислорода кремний. Кислородные атомы
из-за их относительно большой массы и размера не обладают та-
кой подвижностью, как ядра водорода в структуре льда. Поэтому
вместо резонанса кислородных атомов между атомами кремния
связь обусловливается резонансом двух электронных пар между
атомом кислорода и двумя атомами кремния, являясь частично
ковалентной и частично ионной. Кислород при этом является
анионом. Если бы структура кварца и других форм кристалли-
ческого кремния была образована ионами Si4+n О? с радиуса-
ми соответственно 0,41 и 1,40 А, то расстояние Si— О было бы
52
ГЛАВА 2
равно 1,81 А. Фактически, однако, это расстояние равняется
1.58—1,62 А в кристобалите и кварце. Во всех случаях, когда
наблюдается ковалентная связь, обнаруживаются межатомные
расстояния, уменьшенные по сравнению с теми, которые име-
лись бы, если бы вместо атомов были ионы. В данном случае
межатомные расстояния значительно меньше, чем межатомные
расстояния, рассчитанные в предположении ионной связи. Мож-
но, по-видимому, рассматривать кристаллические формы крем-
ния как имеющие характер промежуточных между кристаллами
со структурой алмаза и ионными кристаллами. Для удобства в
табл. 6 приводятся соотношения между различными типами
связи.
Таблица 6
Соотношения между различными типами связи
Связь Резонирующий элемент Частота коле- баний в се- кунду
Ионная Нет
Одинарная кова- Электронная па- 5-10х*
лентная Связь О—Н—О ра Протон IO42
(водородная) Si—О—Si Кислородные Возможно
Металлическая ядра Две электронные пары Нет 101о 5-101*
Простые ионные кристаллы. Типичной для простых ионных
кристаллов является каменная соль. Каменная соль состоит из
ионов натрия и хлора, образующих кубическую решетку, изо-
браженную на рис. 13. Каждый ион натрия удален на 2,814 А от
каждого из шести ионов хлора. Поскольку натрий и хлор одно-
валентны, можно считать, что связь между ионом хлора и каж-
дым из шести ионов натрия составляет одну шестую единичной
связи. Однако неправильно предполагать, что каждый опреде-
ленный ион натрия связан с определенным ионом хлора. В ион-
ных кристаллах не существует специфической связи ион — ион.
Например, у соли не существует специфической одновалентной
связи одного натрия с одним хлором в твердом состоянии. Фак-
тически каждый Na+ притягивает каждый С1“ в кристалле и от-
талкивает все другие Na+ . Это электростатическое притяжение
и отталкивание происходят в соответствии с законом Кулона.
Для галита и других ионных кристаллов оценка этого притяже-
ФАЗЫ ВО ФЛОТАЦИОННЫХ СИСТЕМАХ
53
ния и отталкивания была проведена Борном и др. [6]. Для раз-
личных ионных структур были рассчитаны некоторые постоян-
ные (константа Маделунга), которые вместе с основными пара-
метрами решетки позволяют оценить энергию связи кристал-
ла [141.
Решетка хлористого натрия встречается у ряда галогенидов
щелочных металлов. Она встречается также у многих окислов
и некоторых сульфидов. В галените
расстояние между- центрами атомов
заметно меньше нежели сумма радиу-
сов катиона свинца и аниона серы,
найденных в истинно ионных кристал-
лах [48, Ь]. Это свидетельствует либо о
ковалентной, либо о металлической
связи между свинцом и серой. Андер-
сон и Ричардс Ш считают, что несовер-
шенный ионный характер галенита
связан с дефектами решетки. Галенит
обнаруживает электропроводность по-
лупроводникового типа, он не прозра-
чен для света и обладает свойствами
выпрямителя тока. Все это свидетель-
ствует о том, что связь в галените не
чисто ионная. Этих данных достаточ-
но для утверждения, что связь не яв-
ляется полностью ионной, но их недо-
статочно для окончательного выясне-
1-1--1---1----i_I о
О 1 2 3 4 5А
Рис. 13. Структура камен-
ной соли. Два типа кружков
соответствуют двум типам
ионов (W. L. Bragg. Ato-
mic, Structure of Minerals,
1937)
ния характера связи.
В этом разделе, посвященном простым ионным кристаллам,
в общих чертах была рассмотрена только одна атомная решет-
ка, а именно, решетка хлористого натрия. Следует напомнить,
что существует несколько других ионных структур.
Комплексные ионные кристаллы.. Эти кристаллы по структу-
ре и свойствам подобны простым ионным кристаллам. Различие
заключается в том, что один или оба иона содержат несколько
ядер. В хлористом натрии структурная единица обладает од-
ним ядром. Это ядро натрия и хлора с числом электронов, со-
ответствующим ионам натрия и хлора. В сульфате натрия
структурными единицами являются одноядерный катион и
полиядерный анион SOi~. Ионные кристаллы, в которых анион
является полиядерным, многочисленны и разнообразны. К ним
относятся, например, ортосиликаты, бораты, фосфаты, сульфа-
ты, хроматы, карбонаты. Ионные кристаллы с полиядерным ка-
тионом не столь разнообразны. В основном катион в них обра-
зуется атомом металла и несколькими примыкающими к нему
молекулами аммония или воды.
54
ГЛАВА 2
В ангидрите (безводный сульфат кальция) около каждого
ядра серы на равных расстояниях (1,56 А) тетраэдрически рас-
положено четыре кислородных ядра. Ближайшими катионами
являются два иона кальция на расстоянии 3,11 А и два других
на расстоянии 3,48 А от каждого ядра серы. Атомов кислорода,
ближайших к атому кальция, насчитывается четыре на расстоя-
нии 2,42 А, в то время как четыре других удалены на 2,51 А. Все
атомы кислорода 11] занимают одинаковое положение относи-
тельно серы и [2] близки к сере, но не так близки к кальцию.
Чертежа структуры CaOSO3 нет1. Весьма важно, что расстоя-
ние между атомами кислорода и серы много меньше суммы ион-
ных радиусов и приблизительно на 0,14 А меньше суммы одно-
валентных ковалентных радиусов. Это подтверждает наличие
двойной ковалентной связи между серой и кислородом. Другие
безводные сульфаты (такие, как барит, целестит и англезит)
имеют аналогичную структуру, образованную БОд-тетраэдра-
ми (расстояние S — О около 1,5 А) и катионами металлов.
Среди ионных кристаллов с комплексным катионом типич-
ным может считаться кобальтхлоридгексаммин Co(NH3)6C12* *.
В этом кристалле катионы [Co(NH3)6]2+ располагаются вместе с
ионами хлора в кубической гранецентрированной решетке. Эта
решетка в точности обратна решетке гексахлорплатината ка-
лия, изображенной на рис. 14. Роль катионов калия играют
анионы хлора в аммине, а роль аниона гексахлорплатината
играет кобальтамминокатион. В кобальтхлоридгексахлорам-
мине каждый атом кобальта окружен шестью молекулами
аммония; расстояние Со — N составляет 2,5 А, рас-
стояние Со — О 2п! 4,27 А и расстояние N — С1 2п! 3,49 А. В
данном случае, как и в ангидрите, расстояния между центрами
ядер в полиядерном ионе много меньше, чем расстояния между
центрами ближайших соседних ядер в различных ионах. С дру-
гой стороны, нужно подчеркнуть, что >в данном случае кристалл
не представляет собой СоС1а'6ЫН3 в большей степени, нежели
ангидрит СаО • SO3.
В некоторых кристаллогидритах вода часто ведет себя так
же, как аммоний в амминах. В типичном случае тетрагидрата
1 Структура ангидрита изображена, например, в книге Б. Ф. Ормонтг
«Структуры неорганических веществ». Прим. ред.
* Термин «аммин» означает наличие в составе кристалла молекулярного
NH3; он ничего общего не имеет с термином «амин», обозначающим молекулу
аммония, в которой один или более атомов водорода замещены углеводород-
ными цепями (в русской литературе употребляется термин «аммиакат».
Прим, ред.).
ФАЗЫ ВО ФЛОТАЦИОННЫХ СИСТЕМАХ
55
сульфата бериллия структура образована ионами [Ве(Н2О)4]2+
и SO?-, которые расположены приблизительно таким же обра-
зом, как ионы цезия и хлора в простом ионном кристалле хло-
ристого цезия [46]. В некоторых кристаллогидратах число мо-
лекул воды достаточно для того, чтобы можно было предпола-
гать полное окружение центрального металлического ядра в
Рис. 14. Гексахлорплатинат калия (Р. Р. Ewald,
С. Hermann. Structurbericht, I, fig. 194, 1931)
полиядерном ионе; это наблюдается, например, у тетрагидрата
сульфата бериллия. В других случаях центральное металличес-
кое ядро связано с атомами кислорода так же, как в гипсе (ди-
гидрат сульфата кальция). В исключительных случаях число
молекул воды превосходит координационную способность цен-
трального металлического ядра (например, в гептагидрате суль-
фата никеля).
Волокнистые, слоистые и блоковые ионные кристаллы. До
сих пор рассматривались примеры комплексных ионных крис-
таллов с ионами, ограниченными в пространстве; таковы ионы
в кристаллах, содержащие одно ядро. Однако нет причины, по
которой полиядерные ионы должны были бы быть ограничен-
56
ГЛАВА 2
ними, если входящие в их состав ядра могут образовывать ко-
валентные связи более чем по одному направлению. Если ядра
одного вида могут быть ковалентно связаны по четырем на-
правлениям, как в случае кремния, и если ядра другого вида
могут быть ковалентно связаны по двум или больше направле-
ниям, как в случае кислорода, то появляется возможность обра-
зования неограниченно протяженных ионов. Если этот процесс
будет происходить в одном измерении, то мы получим волокни-
стые кристаллы, если в двух — слоистые, в трех — кристаллы,
ионы которых не ограничены в трех измерениях.
В химии пластмасс существенное значение имеют некоторые
принципы, на основе которых происходит образование кристал-
лов с бесконечно протяженными ионами. Однако упорядочен-
ность структуры в твердом состоянии в пластмассах либо сла-
бая, либо отсутствует.
Химия силикатов, достигшая ненормальной сложности на ос-
нове старых идей о многочисленных кремневых кислотах, теперь
чрезвычайно упростилась [7, 26]. Основным является то, что
атом кремния ковалентно связан с четырьмя атомами кислоро-
да и что атом кислорода может быть ковалентно связан с двумя
атомами кремния или может оставаться связанным только с
одним атомом кремния. В основных структурах, которые при
этом образуются, кислород может быть связан с кремнием сле-
дующими способами:
1) каждый атом кислорода ковалентно связан только с од-
ним атомом кремния;
2) две трети атомов кислорода ковалентно связаны только с
одним атомом кремния;
3) шесть одиннадцатых атомов кислорода ковалентно связа-
ны только с одним атомом кремния;
4) одна треть атомов кислорода ковалентно связана только
с одним атомом кремния;
5) ни один атом кислорода не имеет ковалентной связи толь-
ко с одним атомом кремния.
Анионом, в котором каждый атом кислорода связан только с
одним атомом кремния, будет (SiO4)4-; ортосиликаты, среди
которых в качестве примера можно назвать оливин, состоят из
этих ионов. Они принадлежат к классу с комплексными ионами,
о котором писалось в предыдущем разделе.
Если две трети атомов кислорода связаны только с одним
атомом кремния, то анион представляет собой бесконечную
одиночную цепь (SiOj-^ (рис. 15, а). Такую структуру имеют
пироксены: энстатит MgSiO3, диопсид CaMg(SiO3)2 и сподумен
LiAl (SiO3)2.
Если шесть одиннадцатых всех атомов кислорода ковалент-
но связаны только с одним атомом кремния, то анион представ-
ФАЗЫ ВО ФЛОТАЦИОННЫХ СИСТЕМАХ
57
ляет собой бесконечную двойную цепь (Si^fi)^ . Типичными
здесь являются амфиболы, такие, например, как тремолит,
[(ОН)2 Са2 Mg 5'1+][(Si4O)M~l. На рис. 15, б изображена мо-
дель двойной цепи атомов кремния и кислорода.
Когда одна треть атомов кислорода ковалентно связана
только с одним атомом кремния, анионы представляют беско-
нечные листы , наблюдаемые в глинах, тальках и
а.
б
Рис. 15. Кремнекислородные ионные цепи (A. F. Wells.
Structural Inorganic Chemistry, New York, 1945)
a — в пироксенах; б — в амфиболах
других силикатах, расщепляющихся на тонкие слои. В каолине,
тальке и пирофиллите бесконечные кремнийсодержащие листы
состоят только из кремния и кислорода. В слюдах листы состо-
ят из кремния, алюминия и кислорода, причем кремний частич-
но замещен алюминием. Так, во флогопите и мусковите четверть
атомов кремния статистически замещена алюминием, в то вре-
мя как в хрупкой слюде (маргарите) замещена половина
атомов кремния. Так как алюминий трехвалентен, в то время
как кремний четырехвалентен, это замещение изменяет заряд
ионов листа с (Si4Oio")^. на (513А1О^У)Х или на (Si2Al2O1Г)Х.
Структура каолина образуется слоями гиббсита (тригидрат
алюминия), связанного со слоями (S^Ot?).,.. Связь осущест-
вляется при помощи тех атомов кислорода кремнекислородного
Слоя, которые связаны только с одним атомом кремния. Эти
атомы кислорода, осуществляющие связь между слоями, соеди-
няются с одним из гидроксилов гиббситового слоя.
В тальке или пирофиллите гиббситовый слой располагается
58
ГЛАВА 2
между двумя слоями (Si4010")^. В случае каолина кристалл (
состоит из наложенных одна на другую половин таких пакетов,
а в случае пирофиллита наложены один на другой целые
пакеты.
В слюдах, помимо слоев пирофиллита или талька, имеются
еще слои катионов (они отсутствуют у пирофиллита и талька),
посредством которых осуществляется связь между соседними
пакетами. Наличие катионных слоев вызвано необходимостью .
нейтрализовать увеличившийся заряд силикатных слоев. Это
увеличение заряда силикатных слоев является результатом т
частичного замещения в них четырехвалентного кремния трех-
валентным алюминием. Таким образом, в мусковите
KAl2(OH)2Si3AIOio имеется два «типа» атомов алюминия, так
же как и два «типа» атомов кислорода.
В том случае, когда нет атомов кислорода, которые были бы
связаны только с одним атомом кремния, т. е. если каждый
атом кислорода связан с двумя атомами кремния, возникает
нейтральная бесконечная блоковая структура SiO2. Такие кри-
сталлы относятся к кристаллам алмазного типа, о которых го-
ворилось выше. Типичным примером кристалла с блоковой
структурой является кварц. Однако возможна замена четырех-
валентного кремния трехвалентным алюминием (на одну чет- (
верть или на одну вторую), в результате чего блоковая струк-
тура будет состоять из ионов (SiaAlOsL или (Si2Al20s )у. При
этом требуются компенсирующие катионы. Такие катионы
(К+, Са2+, Na+ или Ва2+) располагаются в различных «пусто- .
тах» структуры.
Примером кристаллов такого рода могут служить полевые
шпаты [41—431 типа ортоклаза: IK+L [(AlSisOs)?*], в которых л
катионы относительно велики (г = 1,33 А), или типа плагиокла-
зов от [Na+b [(AlSi3O8)7x] до [(Ca2+)y][(Al2Si2O8)~2j'] ], в которых
катионы имеют меньшие размеры: г = 0,95 до 0,99 А.
Цеолиты представляют другой пример кристаллов с блоко-
вой структурой. Для них характерна настолько рыхлая структу-
ра, что они пропускают и адсорбируют молекулы воды, а также
молекулы некоторых газов и многие ионы [71. Благодаря этому
свойству их применяют в качестве ионообменников. В известном
смысле ионообменные смолы, созданные недавно из органичес-
ких полиядерных ионов, представляют собой, подобно цеоли-
там, твердые пористые ионные остовы. Однако в отличие от
цеолитов они не обладают кристаллической структурой. 4
Гетерополярные слоистые кристаллы. Многие органические
соединения с гетерополярными молекулами, образующие слои-
стые кристаллы, играют значительную роль в поверхностных
ФАЗЫ ВО ФЛОТАЦИОННЫХ СИСТЕМАХ
59
явлениях и флотации. Соединения, молекулы (или ионы) кото-
рых имеют только одну полярную группу, обычно кристалли-
зуются в виде двойных слоев, состоящих из молекул или ионов.
Связь между слоями, образующими двойной слой, осуществляет-
ся за счет взаимодействия полярных частей молекул. Например,
додециловый спирт, пальмитиновая кислота, солянокислый
октадециламин и олеат калия —все кристаллизуются соглас-
но этому общему правилу. В каждом слое молекулы распола-
гаются одна около другой, причем в боковом направлении меж-
ду их углеводородными частями действуют силы остаточной
связи. Основная связь осуществляется полярными частями мо-
лекул согласно различным координационным схемам. Углево-
дородные цепи располагаются перпендикулярно или наклонно
к плоскости слоя.
В кристаллах спиртов группы ОН допускают образование
водородной связи между молекулами соседних слоев. При этом
каждый атом кислорода связан с несколькими атомами кисло-
рода другого слоя парциальными водородными мостиками.
Для натриевого мыла насыщенных жирных кислот известны
орторомбическая (альфа) и моноклинная (бета) формы 1201.
Эти формы отражают незначительные относительные изменения
в расположении молекул, которые связаны полярными группа-
ми—С—О—с помощью ионов натрия и молекул воды. В этих
II
О
соединениях внутри элементарного молекулярного кристалла
толщиной от 30 до 60 А имеется ионный слой (образованный
карбоксильными группами, окружающими ионы натрия). Этот
слой окружен неполярным углеводородным веществом толщи-
ной 12—27 А. Многие так называемые нейтральные мыла в дей-
ствительности являются гидратированными мылами [8, 10, 161,
часто с 0,5 моля воды на 1 моль дегидратированного мыла. В
других гидратированных мылах отношение воды к безводному
мылу меньше. Были найдены значения 1:4, 1:6, 1:8. Во всех этих
гидратированных мылах вода располагается в центральном слое
мыльного пакета, т. е. таким же образом, как атомы металла.
Предполагается, что вода образует водородную связь между
противолежащими слоями, частично выполняя ту же функцию,
что и ионы металла.
Кислые мыла обычно не гидратированы. Можно полагать,
что водородная связь, осуществляемая атомами водорода кис-
лоты, делает излишним участие воды в кристалле, даже если
кристалл образовался из водной среды.
Фактически крупные кристаллы состоят из описанных выше
элементарных молекулярных кристаллов, наложенных один на
Другой. Обычно эти кристаллы относительно плоски и тонки, что
60
ГЛАВА 2
отражает их стремление расти вдоль пакета быстрее, чем пер-
пендикулярно ему. '
В качестве типичного примера может быть приведен кислый
пальмитат натрия, представляющий моноклинный кристалл,
кристаллизующийся в так называемой альфа-форме. Каждый
элементарный пакет его имеет толщину в 45,7 А. Другие разме-
ры ячейки составляют 9,97 и 7,38 А (для четырех молекул кис- .
лого пальмитата натрия NaHP2). Таким образом, каждая угле-
водородная цепь с одним катионом в среднем имеет длину в ?
: 2 = 22,8 А и покрывает поверхность в —(7,38X9,97) =
°2
= 18,4 А.
Металлические кристаллы. Металлы обычно кристаллизуют-
ся, образуя относительно простые структуры с высокой симмет-
рией. Большое количество металлов, сплавов и интерметалли-
ческих соединений образует разнообразные структуры. Метал-
лические кристаллы являются проводниками электричества, , *
отражают свет, обладают ковкостью. Способность образовы-
вать твердые растворы является их характерной особенностью.
Хотя металлические кристаллы были практически неизвестны
пятьдесят лет назад, но в настоящее время сделано очень мно-
го, и в литературе по физике металлов накопилось большое ко-
личество данных о структурах таких кристаллов. В задачу этой
книги не входит подробное рассмотрение этого материала.
Металлические кристаллы можно рассматривать как массу j
катионов, более или менее плотно упакованных и погруженных ’ >
в электронный газ. В совершенном кристалле при температуре,
равной абсолютному нулю, металлические катионы должны
занимать все узлы решетки, а между ними будут находиться
освобожденные валентные электроны. При таких условиях по-
ложение металлических катионов жестко фиксировано, а элек-
троны обладают огромной подвижностью. Эта подвижность сво-
бодных электронов обусловливает электро- и теплопроводность;
с наличием свободных электронов вероятно связан механизм
скольжения, двойникования, пластической деформации и рекри-
сталлизации, свойственных металлам.
В кубических плотно упакованных структурах (один из наи-
более распространенных типов структур металлов) каждый
атом окружен двенадцатью ближайшими соседями, располо-
женными на равных расстояниях от него (четыре атома распо-
лагаются в плоскости грани куба, содержащей рассматривае-
мый атом, и четыре в каждой из двух плоскостей, параллельных
плоскости грани куба). В гексагональных плотно упакованных
структурах, тоже часто встречающихся у металлов, каждый
атом, как и в кубических структурах, окружен двенадцатью
ФАЗЫ ВО ФЛОТАЦИОННЫХ СИСТЕМАХ
61
ближайшими соседями, расположенными на равных расстояни-
ях от него' (шесть из них в одной из плоскостей, проходящих че-
рез рассматриваемый атом, и по три <в каждой из двух плоско-
стей, параллельных этой базисной плоскости и расположенных
по обе стороны от нее).
Межатомные расстояния в металлах относительно малы. В
меди, например, расстояние между центрами ядер составляет
2,55 А. При этом следует помнить, что атомы упакованы так
плотно, как только возможно. Вариации межатомных расстоя-
ний в металлах относительно малы. Железо обладает одним из
наименьших межатомных расстояний (2,48 А), а цезий — наи-
большим (5,24 А). Межатомные расстояния в конструкционных
металлах колеблются в пределах от 2,5 до 3,5 А.
Полупроводниковые кристаллы. Многие минералы, пред-
ставляющие особый интерес во флотации, попадают в группу
кристаллов, условно объединенных вместе под названием «полу-
металлических», или полупроводников. Среди полупроводников
имеются кристаллы очень сложные и простые. Примерами мо-
гут служить сульфиды и сульфосоли многих металлов (так же,
как арсениды, карбиды, бориды и пр.).
Эти кристаллы отличаются от ионных кристаллов тем, что в
них связь между ядрами, образующими структуру, не чисто ион-
ная. Эта связь также не является ни полностью ковалентной, ни
металлической. Многие из этих кристаллов, например, облада-
ют электропроводностью, но значительно меньшей, чем метал-
лы. Величина электропроводности полупроводников отличается
от электропроводности металлов на несколько порядков. В то
же время полупроводники несравненно лучше проводят элек-
трический ток, нежели изоляторы и, что имеет большое значе-
ние, их электропроводность меняется как функция напряжения,
частоты и т. д. Некоторые полупроводниковые кристаллы обна-
руживают свойство пропускать электрический ток только в од-
ном направлении. Например, свойство односторонней проводи-
мости галенита использовалось в первых (детекторных) радио-
приемниках.
Халькозин и пирит представляют два интересных сульфида
среди этого класса соединений. Халькозин или, скорее, дигенит
19], по-видимому, состоит из анионов серы, соприкасающихся
один с другим, и сравнительно более мелких катионов меди,
расположенных в промежутках. Этим можно объяснить элек-
тропроводность минерала, а также и значительно большую диф-
фузию меди в кристаллах [17] по сравнению с серой. Оптичес-
кие свойства халькозина исключают предположение, что связи
в нем имеют лишь ионный характер.
62
ГЛАВА 2
Структура пирита похожа на структуру хлористого натрия,
с ковалентно связанными парами атомов серы вместо ионов
хлора и железом вместо натрия; она (рис. 16) довольно плотно
• Рис. 16. Пирит (R. W. G. Wyckoff. Crystal Structures, I, New York, 1948):
a—проекция элементарной ячейки пирита иа грань куба; б—упаковка, соответ-
ствующая схеме а
Рис. 17. Сфалерит. Перспективный
рисунок, изображающий атомы цин-
ка и серы в структуре ZnS в предпо-
ложении, что они имеют размеры,
принятые для ионов. Если же (что
более вероятно) связь частично ко-
валентная, то кружки должны быть
более близкими по размерам
(R. W. G. Wyckoff. Crystal Struc-
tures, I, New York, 1948)
упакована; расстояние между атомами серы в одной паре со-
ставляет 2,14 А, между атомами железа и ближайшими атома-
ми серы 2,26 А и между соседними атомами железа 3,82 А. Для
сравнения можно указать, что расстояние РЬ—РЬ в галените
4,18 А, а расстояние Zn — Zn в сфалерите (структура цинковой,
обманки) 3,83 А.
Сфалерит обладает структурой (рис. 17), которая похожа
на структуру алмаза. В отличие от алмаза здесь одну половину
атомов представляют атомы серы, а другую — атомы цинка.
ФАЗЫ ВО ФЛОТАЦИОННЫХ СИСТЕМАХ
63.
Неясно, до какой степени эти атомы ионизированы и до какой
степени они ковалентно связаны. Вероятно, атомы резонируют
между ионной и ковалентной связями. «Металлические» свойст-
ва в большой мере отсутствуют в сфалерите, но дело ослож-
няется в изоморфных или почти изоморфных сульфидах, таких
как станнин или халькопирит.
ЛИТЕРАТУРА
1. Anderson J. S. and J. R. Richards. The self-diffusion of lead in
lead sulphide, J. Chem. Soc., 1946, (1), p. 537—541.
2. Beevers C. A. and H. Lipson. The crystal structure of nickel sulfate
hexahydrate, NiSO4-6H2O, Z. Krist., 83, 123—135, 1932.
3. В e e v e г s C. A. and H. Lipson. Crystal structure of CuSO4 • 5H2O, Proc,
Roy. Soc. (London), A146, 570—582, 1934.
4. В ее vers C. A. and С. M. Schwartz. The crystal structure of nickel
sulfate heptahydrate, NiSO4-7H2O, Z. Krist., 91, 157—169, 1935.
5. Bernal J. D. and R. H. Fowler. A theory of water and ionic solution,
with particular reference to hydrogen and hydroxyl ions, J. Chem. Phys.,
I, 515—548, 1933.
6. Born Max. Atomic Physics 4th ed., Hafner Publishing Company, New’
York, 1947.
7. В r a g g W. L. Atomic structure of Minerals, Cornell University Press,
Ithaca, N. Y., 1937.
8. Buerger M. J. Soap Crystals, Am. Mineralogist, 30, 551—571, 1945.
9. Buerger M. J. and Newton W. Buerger. Low chalcocite and high
chalcocite, Am. Mineralogist, 29, 55—65, 1944.
10. Buerger M. J., L. B. Smith, A. de Bretteville and F. V. Ryer.
The lower hydrates of soap. Proc. Natl. Acad. Sci. U. S., 28, 526—529, 1942.
11. Darmois Eugene. Mobility and hydration of ions, Compt. Rend., 221,
220—292, 1945,
12. Darmois Eugene. Mobility of ions and viscosity of saline solutions,
J. Chem. Phys., 43, 1—20, 1946.
13. Eastman E. D. and G. K. R о 11 e f s о n. Physical Chemistry, 422—426,
McGraw-Hill Book Company, Inc., New York, 1947.
14. Evans R. C. An introduction to crystal chemistry, p. 299, Cambridge Uni-
versity Press, New York, 1939.
15. Fowler R. H. and J. D. Bernal. Note on the pseudo crystalline structu-
re of water, Trans. Faraday Soc., 29, 1049—1056, 1933,
16. Gardiner K. W., M. J. Buerger and L. B. Smith. The hydrate
nature of soap, J. Phys. Chem., 49, 417—428, 1945.
17. Gau din A. M. and К. C. Vincent. Self-diffusion in minerals, par-
ticularly copper sulphides, AIMME Tech. Publ. 1663, 1944, or Trans. 169,
340—346, 1946.
18. Harrison George R., Richard C. Lord and John F. Loofbourow.
Practical Spectroscopy, Prentice-Hall, Inc., New York, 1948.
19. Hunt John P. and Henry Taube. The exchange of water between hydra-
ted cations and solvent, J. Chem. Phys., 19, 602—609, 1951.
20. T h i e s s e n P. A. and J. Stauff. Feinbau und Umwandlungen kristalli-
sierter Alkalisalze langkeittiger Fettsauren, Z. physik. Chem., 176, 397—429,
1936; quoted in Strukturber. 4, 288, 1936.
21. Keesom W. H. and K. W. Ta con is. On the structure of solid helium,
Physica, 5, 161—169, 1938.
22. Knoll Alexander and Dwight L. Baker. Nature of the adsorption
of fatty acids from organic solvents by inorganic lead compounds, AIMME
Tech. Publ. 1314, 1941.
64
ГЛАВА 2
23. LaMotte Chemical Company, «The ABC of hydrogen-ion determinations»,
Baltimore, 1928.
24. M a с I n n e s D. A. and L. G. Longsworth. Transference numbers by
the method of moving boundaries, Chem. Revs, 11, 171—230, 1932.
25. Clark W. Mansfield. The determination of hydrogen ions, 3ed., The
Williams & Wilkins Company, Baltimore, 1928.
26. Marshall Charles Edmund. The colloid chemistry of the silicate mine-
rals, Academic Press, Inc., New York, 1949. .
27. M c L e n n a n J. C. and W. G. Plummer. The crystal structure of
solid methane, Nature, 122, 571—572, 1928.
28. Meyer Kurt and Y. G о Sur le soufre filiforme et sa structure, Helv.
Chim. Acta, 17, 1081—1093, 1934.
29 Morgan J and В. E Warren. X-ray analysis of the structure of wa-
ter, J. Chem. Phys., 6, 666—673, 1938.
30. Mott N. F. and R. W. Gurney. Electronic processes in ionic crystals,
(a) pp. 26—63, Oxford University Press, New York, 1940.
31. Muller Alex. A further X-ray investigation of long chain compounds
(zi-hydro-carbons), Proc. Roy. Soc. (London), A120, 437—459, 1928.
32. Muller Alex. The crystal structure of the normal paraffins at temperatu-
res ranging from that of liquid air to the melting points, Proc. Roy. Soc.
(London), A127, 417—430, 1930.
33. Pauling Linus. The nature of the chemical bond, (a) p. 168, table 21—5,
Cornell University Press, Ithaca, N. Y., 1940.
34. PennycuickS W. The structure of water, J. Phys. Chem., 32, 1681—
1696, 1928.
35. Plane Robert A., and Henry Taube. Observations of the kinetics of
the exchange of water between Сг(НгО) |+ and solvent, J. Phys. Chem.,
56, 33—38, 1952.
36. Remy Heinrich. The electrolytic transference of water and its significance
for the theories of aqueous solutions, Fortschr. Chem. Physik, u. physik,
Chem., 19, № 2, 5—72, 1927.
37. Sedgwick Nevil Vincent. The electronic theory of valency, Oxford
University Press, New York, 1929.
38. Sedgwick Nevil Vincent. Chemical elements and their compounds, Ox-
ford University Press, New York, 1950.
39. Slater J. C. Introduction to chemical physics, (a), p. 183 MacGraw-Hill
Book Company, Inc., New York, 1939.
40. Swyngedauw Jean. New researches on hydration of ions, J Phys
Radium, (8), 3, 117—120, 1942. '
41. Taylor W. H. The structure of analcite (NaAlSi2O6 • H2O), Z. Krist., 74,
1_ — 19, 1930.
42. Т а у 1 о r W. H. The nature and properties of alumino-silicate framework
structures, Proc. Roy. Soc. (London), A145. 80—103 1934.
43. Taylor W. H. and R. Jackson. Structure of edingtonite, Z Krist., 86
53—64, 1933.
44. Van Ruyven В. H. The degree of ionization of some binary electrolytes
and the hydration of their ions, Rec. Trav. Chim., 56, 1111—1132, 1937.
45. Warren В. E. and J. T. Burwell. Structure of rhombic sulfur, J. Chem
Phys., 3, 6—8, 1935.
46. Wells A. F. Structural inorganic chemistry, Oxford University Press,
New York, 1945.
47. W i c h e r s Edward, Report of the Committee on Atomic Weights of the
American Chemical Society. J. Am. Chem. Soc., 76, 2033—2035, 1954,
48. Wyckoff Ralph W. G. Crystal structures, (a), chap. II, pp. 18—20, (b)
chap. Ill, p. 7 (c), (c) chap. IV, tables 6 and 9, Interscience Publishers, Inc.,
New York, 1948.
Глава 3
ПОВЕРХНОСТЬ РАЗДЕЛА ГАЗ — РАСТВОР
В главе 2 мы рассмотрели бесконечную газообразную фазу
и бесконечную трехмерную фазу, состоящую из водного рас-
твора.
В настоящей главе подробно описывается структура поверх-
ности раздела между этими фазами; электрическим свойствам
поверхности раздела здесь внимания не уделяется, так как они
будут рассмотрены в главе 5.
На поверхности жидкости, находящейся в контакте с атмо-
сферой, плотность вещества резко изменяется приблизительно
в тысячу раз. В настоящее время считается, что это изменение
плотности происходит в пограничном слое толщиной примерно
10 А. Можно сказать, что наличие этого пограничного слоя де-
лает изменение плотности менее резким.
Чистая жидкость в контакте с собственным паром. Простей-
шим случаем поверхности раздела является поверхность раз-
дела на границе жидкости с ее собственным паром. На поверх-
ности жидкости, состоящей из неассоциированных молекул (на-
пример, четыреххлористого углерода), находящейся в контакте
с собственными парами, молекулы жидкости испаряются и кон-
денсируются молекулы газа. В газообразную фазу могут перей-
ти лишь те молекулы, которые, приближаясь к поверхности раз-
дела, приобретают вполне определенную кинетическую энергию.
При равновесии процесс испарения в точности компенсируется
процессом конденсации, поэтому положение поверхности раз-
дела остается без изменения.
Для воды, молекулы которой ассоциированы в жидком со-
стоянии и не ассоциированы в газообразном состоянии, переход
молекул из жидкости в газ сопряжен также с изменением связи
между атома-ми. Это согласуется с исключительно высоким
значением скрытой теплоты испарения воды.
В случае веществ, подобных спиртам, аминам, органическим
кислотам и т. д., содержащим молекулы, обладающие гетеро-
полярными свойствами, что локализует их способность к ассо-
циации в жидкой фазе, процессы испарения и конденсации в
значительной степени зависят от ориентации молекул в поверх-
ности раздела. И наоборот, молекулы на поверхности не могут
быть расположены хаотично: их ориентация отражает стремле-
5 А. М. Годэн
66
ГЛАВА 3
ние к ассоциации. Таким образом, вероятность того, что моле--
кула спирта, ударившись о поверхность жидкого спирта своим
углеводородным концом, станет частью жидкости, невелика, в
то время как молекула, ударившаяся о поверхность жидкости
гидроксильным концом, по всей вероятности перейдет в жидкую
фазу. Молекула жидкости, достигающая поверхности раздела
и обладающая большой кинетической энергией, должна повер-
нуться так, чтобы при испарении ее гидроксильный конец был
направлен в сторону жидкой фазы. Согласно этим качествен-
ным представлениям, в поверхности жидкости, состоящей из ге-
терополярных молекул, в основном находятся углеводородные
группы.
Поверхностное натяжение и поверхностная энергия. Поверх-
ностью раздела между жидкостью и ее парами может считаться
место приложения силы, действующей в плоскости поверхности
раздела и называемой поверхностным натяжением. Эта сила,
являющаяся характеристикой поверхности раздела, измеряется
в динах на сантиметр. Свободная энергия, обусловленная этой
силой, является одной из форм потенциальной энергии. Так как
потенциальная энергия любой системы стремится к минимуму,
то свободная поверхностная энергия системы жидкость—-газ
(выражаемая в эргах на сантиметр в квадрате) также самопро-
извольно стремится к минимуму; это значит, что поверхность
раздела стремится уменьшиться.
Можно рассчитать изменение потенциальной энергии, вы-
званное изменением величины поверхности раздела. Например,
положим, что свободная поверхностная энергия равна у эрг/см2.
Если п капель жидкости диаметром D сольются в одну каплю,
то убыль потенциальной энергии будет равна
2
ЛЕ = гЕ2(/1-п3)р
В среднем жидкость изотропна. Это значит, что связь любо-
го атома, молекулы или иона со своими соседями в среднем оди-
накова во всех направлениях. Но это несправедливо для поверх-
ности, молекулы которой испытывают различные притяжения со
стороны жидкости и газа. В результате того что часть энергии
связи молекул, находящихся на поверхности, не компенсирова-
на, молекулы поверхностного слоя обладают избыточной энер-
гией по сравнению с энергией молекул, находящихся внутри
объема жидкости. Избыточная энергия молекул поверхностного
слоя и есть поверхностная энергия, равная сумме свободной
поверхностной энергии, рассчитанной .на основании измерений
поверхностного натяжения, и связанной поверхностной энергии,
рассчитанной по температурной зависимости поверхностного»
ПОВЕРХНОСТЬ РАЗДЕЛА ГАЗ — РАСТВОР
67
.m
натяжения. Адам [1а] дает следующее уравнение для полной
поверхностной энергии е:
(1)
о 1
где f—свободная поверхностная энергия [численно равная
поверхностному натяжению);
Т — абсолютная температура.
Представим себе, что прямоугольный параллелепипед жид-
кости размером 2x1X1 см разделен на два кубика жидкости с
ребрами, равными 1 см. Работа, необходимая для такого раз-
деления, представляет собой работу, затраченную на разрыв
связей между молекулами жидкости; в результате этого разде-
ления молекулы, находящиеся вблизи вновь образованной по-
верхности площадью, равной 2 см2, уже не испытывают симмет-
ричного взаимодействия со всех сторон.
Эта работа равна 2у. Образование новой поверхности таким
способом приводит к охлаждению всей системы. Если образо-
вание новой поверхности осуществляется «изотермически», а
не «адиабатически», то затраченная энергия равняется 2е
вместо 2 Y.
Интересно сравнить величину поверхностной энергии с энер-
гией, выделяемой при обычных химических реакциях, большин-
ство которых имеет энергию, примерно равную одному элек-
трон-вольту на атом (1,6- 1СН2 эрг).
Для воды при 20° Т = 293°, у = 72,8 эрг/см2, —0,150,
е = 116,7 эрг/см2. Количество молекул в 1 мл воды равно 6,02 X
X Ю23: 18,0; следовательно, средний объем, занимаемый одним
атомом кислорода, равен 30 А3, если принять атомы водорода.за
точки, и средняя площадь, занимаемая атомом кислорода на
поверхности раздела, равна 9,6 А2.
Средняя энергия, необходимая для изотермического отделе-
ния атома кислорода, расположенного на вновь образованной
поверхности, равна 116,7-9,6- 10~16 эрг или 0,07 электрон-вольта.
Эта величина сравнима с изменениями энергии при обычных
химических реакциях.
Поверхностное натяжение различных жидкостей. Поверх-
ностное натяжение различных жидкостей изменяется от величи-
ны, равной примерно 1 дин!см в случае конденсированных бла-
городных газов вблизи критической температуры, до 1000 и
больше дин)см для расплавленных металлов. Большинство ор-
ганических соединений имеет поверхностное натяжение от 20 до
30 дин!см, вода — около 70, расплавленные соли — от 70 до 200,
Жидкая ртуть 450—500; другие расплавленные металлы имеют
еще более высокие значения поверхностного натяжения.
5*
68
ГЛАВА 3
Таблица 1
Значения поверхностных натяжений жидкостей1
Вещества Температура °C Поверхност- ное натяже- ние, дин см Вещества Температура °C Поверхност- ное натяже- ние, OuhIcm
Благородные и посто- глицерин .... 20,0 63,4
янные газы; вода 20 72,45
гелий —270 0,24 Ионные соединения:
неон —248 5,5 иодистый цезий 654 73,1
водород .... —255 2,31 » рубидий 673 79,4
кислород .... — 193 15,7 » натрий Точка 93,9
азот .... — 193 8,27 п лавле-
Органические и дру- НИЯ
гие неассоцииро- бромистый нат-
Ванные жидкости, рий Точка 102,8
не имеющие водо- плавле-
родных связей: НИЯ
бензол 20 28,88 хлористый нат-
сероуглерод . . 20 32,33 рий 803 113,8
четыреххлори- фтористый нат-
стый углерод . 20 26,95 рий 1010 199,5
н-гексан .... 20 18,43 хлористый сви-
Органические иец 490 138,0
жидкости, имею- Металлы:
щие водородную натрий .... 100 206,4
связь: ртуть 0 480,3
ацетилсалицило- серебро .... 970 800,0
вая кислота 25,9 60,06 платина .... 2000 1819
1 Данные взяты из книги С. D. Hodgman и др. «Handboock Chemistry and Physics»,
изданной Chemical Publishing Company Inc., New York, 1947).
-----------------------------------!-----------------------------r ----------------
В табл. 1 приведены значения поверхностных натяжений для
жидкостей с различными типами связей.
Из этой таблицы видна зависимость между поверхностным
натяжением жидкости и типом связей между молекулами. Пол-
ная поверхностная энергия жидкости также зависит от типа
связи между ее молекулами, однако, эту зависимость труднее
установить, так как не имеется достаточно точных значений
производной поверхностного натяжения по температуре.
Измерение поверхностного натяжения. Существует много
методов измерения поверхностного натяжения на границе раз-
дела жидкость — газ и на границе двух жидкостей [13]. Все эти
методы делятся на статические и динамические. При примене-
нии статических методов измеряются силы, обусловленные не-
подвижной «состарившейся» поверхностью; при применении ди-
ПОВЕРХНОСТЬ РАЗДЕЛА ГАЗ — РАСТВОР
69
намических методов измеряются силы, обусловленные движу-
щейся поверхностью. В последнем случае поверхностный слой
жидкости непрерывно обновляется [П.
Статические и динамические методы могут давать одни и те
же или различные результаты в зависимости от скорости, с ко-
торой устанавливается равновесие между поверхностью и объ-
емной фазой. При измерении поверхностного натяжения разбав-
ленных растворов сильно поверхностно активных веществ эти
методы дают существенно различные результаты.
Один из классических методов измерения поверхностного на-
тяжения заключается в измерении высоты подъема жидкости в
стеклянных капиллярах. При этом поверхностное натяжение
определяется уравнением
7 =(2)
где g — ускорение силы тяжести;
D и d —плотности жидкости и газа;
г — радиус капилляра;
h — высота подъема жидкости в капилляре.
Это уравнение справедливо только при условии полного
смачивания поверхности капилляра поднимающейся жидкостью.
Вторым методом является метод максимального давления
пузырьков. При этом методе пузырек выдувается из трубки,
вертикально погруженной в жидкость. По мере роста пузырька
давление в нем возрастает и достигает максимума в момент,
когда пузырек принимает форму полусферы. В этот момент из-
быточное давление внутри пузырька по сравнению с давлением
жидкости на конце трубки равно
Р = ^~, (3)
г
где Р— давление;
у — поверхностное натяжение;
г — радиус пузырька.
Метод достаточно точен, если радиус трубки настолько мал,
что выдуваемый пузырек является шаровым сегментом.
Третьим методом, применяемым для измерения поверхност-
ного натяжения на границе между двумя жидкостями или
жидкостью и газом, является метод счета капель. Метод осно-
ван на том, что вес капель, вытекающих из узкого отверстия, яв-
ляется функцией поверхностного натяжения. Харкинс и Браун
[36] показали, что поверхностное натяжение, вес капли mg, ра-
диус трубки г и параметр F связаны следующим уравнением:
I = f (4)
70
ГЛАВА 3
Параметр F, в свою очередь, зависит от отношения объема
капли к кубу радиуса трубки V/r3. На рис. 1 даны значения па-
раметра F как функции V/r3, найденные экспериментально Хар-
кинсом и Брауном. Фотографии падающих капель, полученные
Эджертоном, Хозером и Тэккером [23], показывают, что процесс
Рис. 1. Зависимость параметра F и отноше-
ния V/r3:
V —объем капли; г —радиус трубки, из которой вы-
текает жидкость
отрыва капли жидкости является сложным и что, кроме основ-
ной капли, при отрыве образуются маленькие капельки. Экспе-
риментальный параметр, предложенный Харкинсом и Брауном,
позволяет статистически учесть сложность протекающих явле-
ний и вычислить поверхностное натяжение.
Четвертым методом, широко применяемым для измерения по
верхностного натяжения, является метод «отрыва кольца». Он
заключается в погружении металлического кольца в жидкость
и измерении величины сил, препятствующих отрыву кольца из
жидкости. Если R — радиус кольца, q — сила, препятствующая
ПОВЕРХНОСТЬ РАЗДЕЛА ГАЗ — РАСТВОР
71
(5)
отрыву кольца, то поверхностное натяжение может быть найде-
но из следующего уравнения:
Т = Е'—?—
4- К
В этом уравнении F' — параметр, изменяющийся от 0,75 до
1,45 [26, 33, 38, 39а, 40] и являющийся функцией R3/V и R/V, где
V — объем жидкости, поднимаемой кольцом, a R — радиус про-
волоки, из которой сделано кольцо. Интересным вариантом мето-
да «отрыва кольца» является метод «отрыва ножа» [24, 68], пер-
воначально примененный Вильгельми.
В последние годы метод «отрыва кольца» получил более ши-
рокое признание, однако он не дает статических значений по-
верхностного натяжения, так как поверхность жидкости дефор-
мируется в момент измерения.
Пятым методом можно назвать метод «закрепленного пу-
зырька» [69, 74]. Этот метод весьма точный и дает истинно стати-
ческое значение поверхностного натяжения. Метод заключается
в закреплении пузырька под плоскостью, измерении с помощью
микроскопа или микрофотографии соответствующих параметров
формы «состарившегося» пузырька и расчета по этим парамет-
рам поверхностного натяжения.
Шестой метод— метод висящей капли в комбинации с уско-
ренной киносъемкой — использовали Хозер и Эджертон [7,23,41,
65] для измерения поверхностного натяжения и его изменения
во времени. Этот метод заключается в точном измерении гори-
зонтального сечения висящей капли по экватору и произвольно
выбранного второго сечения. Если у— поверхностное натяжение,
g—ускорение силы тяжести, Д— разность удельных весов жид-
костей, de— диаметр сечения капли по экватору, ds— диаметр
произвольного сечения капли и параметр Н, как функция djde,
найденная Андреасом, Хозером и Тэккером [7], то поверхностное
натяжение может быть найдено из уравнения
g^d2e
Т Н
Динамические методы измерения поверхностного натяжения
в настоящее время имеют меньшее значение. Среди них можно
отметить метод поверхностных волн лорда Кельвина 171] и метод
колебаний струй и капель [52], используемый Ленардом. Хоро-
ший обзор методов измерения поверхностного натяжения дан
в книгах Адама [1] и Бэрдона [13].
Поверхностное натяжение растворов. В большинстве случаев
поверхностное натяжение смеси органических жидкостей мало
отличается от средневзвешенного значения поверхностных натя-
жений чистых жидкостей. С другой стороны, поверхностное натя-
жение водных растворов органических соединений сильно откло-
(6)
72
ГЛАВА 3
няется от упомянутого выше правила аддитивности. Наблюдае-
мые значительные отклонения поверхностного натяжения от ад-
дитивных значений являются важными опытными данными, сви-
детельствующими о том, что на поверхности растворов имеется
слой, состав которого отличается от состава объемной фазы.
На рис. 2 даны характерные кривые зависимости поверхност-
ного натяжения водных растворов нескольких простых органи-
ческих соединений от концентрации растворенного вещества. Все
эти кривые имеют один и тот же характер: вначале добавка ма-
лых количеств вещества, обладающего меньшим поверхностным
Рис. 2. Поверхностное натяжение водных растворов ти-
пичных органических соединений:
/ — муравьиная кислота, 30°; 2 — этиловый спирт, 25°;
3 — н.-масляная кислота, 25°; 4— дипропиламин, 20°
натяжением, приводит к непропорционально большому пониже-
нию поверхностного натяжения воды; последующие добавки вли-
яют все слабее и слабее.
Поверхностное натяжение водных растворов неорганических
солей мало отличается от поверхностного натяжения воды. Так
как поверхностное натяжение расплавленных солей больше, чем
у воды, естественно ожидать, что растворы солей будут повы-
шать поверхностное натяжение воды. Небольшое повышение
поверхностного натяжения наблюдается при больших и средних
концентрациях растворенных солей, однако наблюдаемое повы-
шение поверхностного натяжения значительно меньше, чем мож-
но было бы ожидать, если исходить из аддитивности поверхност-
ного натяжения. Рассмотрим в качестве примера влияние нитра-
та натрия. Расплавленная соль имеет поверхностное натяжение
119,7 дин/см при температуре 322°, 111,8 дин!см при 466° и
99,4 дин!см при температуре 656° [431. Экстраполируя эти данные
ПОВЕРХНОСТЬ РАЗДЕЛА ГАЗ — РАСТВОР
75
к комнатной температуре, получим значения поверхностного на-
тяжения соли (если бы она могла существовать в жидком состо-
янии), равное 140 дин!см, что примерно на 70 дин/см превышает
поверхностное натяжение воды. Исходя из предположения об ад-
дитивности поверхностного натяжения, рассчитаем поверхност-
ное натяжение четырехмолярного (32% по весу) раствора нитра-
та натрия: 0,32-70, т. е. поверхностное натяжение раствора
должно быть на 22 дин^см больше, чем у воды. Наблюдаемое
возрастание поверхностного натяжения составляет лишь
4,6 дин/см 139Ы.
Интересно отметить, что уменьшение поверхностного натяже-
ния воды, вызванное добавкой веществ, уменьшающих его, непро-
порционально количеству добавленного вещества. Однако ве-
щества, увеличивающие поверхностное натяжение растворов, по-
вышают его в первом приближении примерно пропорционально
добавленному количеству. Данные, относящиеся к нитрату нат-
рия, подтверждают сделанное заключение [39с].
Адсорбционное уравнение Гиббса. С помощью тщательных
теоретических исследований Виллард Гиббс [I Ь, 28, 31] устано-
вил зависимость между составом поверхностного слоя раствора
и составом самого раствора. Эта зависимость выражается урав-
нением
где Г — избыток растворенного вещества (адсорбция) в поверх-
ностном слое, .мо.гь/слг2;
а — активность растворенного вещества в объеме раствора;,
у — поверхностное натяжение, дин)см-,
R— универсальная газовая постоянная, эрг/град (R—
= 8,32- 107);
Т — абсолютная температура;
ау
— — частная производная поверхностного натяжения по ак-
тивности.
В этом уравнении Г представляет собой избыток растворен-
ного вещества в поверхностном слое. При выражении адсорбции
в виде избыточной двухмерной концентрации оказывается более-
удобной [32, 53] другая -форма уравнения Гиббса, а именно, по-
лученная Гуггенгеймом.
В большинстве случаев активность а можно заменить кон-
центрацией с. При этом уравнение Гиббса примет следующий
вид:
74
ГЛАВА 3
так как
Например, для 10°/о-ного раствора этилового спирта в воде
(см. рис. 2) Г = 5,45• 10~10 лголь/слг2, или 2,5- 10-8 г!см2. Так как
удельный вес спирта равен 0,78, а молекулярный вес 46, средний
объем, приходящийся на одну молекулу, равен 98 • 10~24 мл. Мо-
лекулярный слой (если принять молекулы кубическими) имеет
толщину 4,61 А и содержит 3,6 • 10~8 г'см2 спирта. При этом ус-
ловии адсорбция этилового спирта из 10%-ного водного раствора
составляет 69% мономолекулярного слоя. Эти расчеты дают зна-
чительно лучшее приближение, чем приведенные выше. Про-
делав аналогичные вычисления для 2,5; 5; 10; 20 и 40%-ных рас-
творов этилового спирта в воде, при тех же предположениях по-
лучим величины адсорбции спирта, составляющие 34; 51; 69; 66 и
58% мономолекулярного слоя. Учтя содержание спирта в объеме
раствора, получим содержание спирта в поверхностном слое, рав-
ное 36; 54; 73; 75; 78%.
Измерения Спикмэна [66] являются одним из наиболее тща-
тельно проведенных определений адсорбции поверхностно ак-
тивных веществ из водных растворов на границе с воздухом при
атмосферном давлении. Он исследовал водные растворы анили-
на (C6H5NH2) при 20°. При этом измерялись не только поверх-
ностные натяжения при различных концентрациях анилина в
растворе, но также парциальное давление паров анилина и воды.
Измерения давления паров анилина позволили вычислить зна-
чения активности а более строго, чем это можно было сделать,
исходя из концентрации анилина в растворе. Таким образом про-
верялось более общее уравнение Гиббса (7). Результаты этих
опытов приведены на рис. 3.
Измерения Спикмэна показали, что предельному значению
адсорбции отвечает 0,0427 молекулы анилина на 1 А2. При насы-
щении адсорбционного слоя можно предполагать, что образуется
сплошное мономолекулярное покрытие растворенного вещест-
ва, после чего избыток этого вещества будет образовывать новую
фазу. Из этого следует, что площадь, приходящаяся на 1 молеку-
лу, равна 23,4 ±0,ЗА2, что хорошо согласуется с данными рент-
геноструктурного анализа.
Иногда удобно записать уравнение Гиббса в таком виде:
Г ------!----, (9)
RT д\па
д 1п а= — да.
а
Если построить y как функцию In а (или In с), то наклон этой
кривой, взятый с противоположным знаком, пропорционален ад-
сорбции Г. Зависимость поверхностного натяжения водных ра-
створов анилина от логарифма концентрации показана на рис. 4.
Рис. 3. Зависимость адсорбции и
поверхностного натяжения водно-
го раствора анилина от концент-
рации. Адсорбция аиилииа рассчи-
тана Спикманом по уравнению
Гиббса
Рис. 4. Поверхностное натяжение водного раствора
анилина как функция логарифма его концентрации
(по Спикману)
76
ГЛАВА 3
Из данных, приведенных на этом рисунке, видно, что для кон-
центраций, больших, чем 0,2 моль/л, у почти линейно зависит от
In с. Следовательно, адсорбция, достигнув максимального значе-
ния, остается практически постоянной.
Следует иметь в виду, что уравнение Гиббса выведено для
бесконечно делимой жидкости, обладающей идеальной или «гео-
метрической» поверхностью. Такая поверхность раздела, разуме-
ется, не имеет объема, но так как в поверхности раздела может
адсорбироваться некоторое количество растворенного вещества,
необходимо допустить, что адсорбированное вещество находится
не в геометрической поверхности, а в некотором поверхностном
слое жидкости, имеющем конечную толщину. Поэтому неудиви-
тельно, что не имеется полного совпадения между результатами,
вычисленными по уравнению Гиббса, и опытными данными1. ,
Причиной большинства расхождений являются эксперименталь- ' '•
ные трудности проверки уравнения, часть же расхождений выз- f
вана предпосылками, принятыми при выводе уравнения Гиббса, •,
согласно которым реальной физической фазе, состоящей из от- *
дельных частиц, приписываются свойства математического кон-
тинуума. Р
Один из методов экспериментальной проверки уравнения Гиб- .
бса [54, 55] заключается в пропускании пузырьков через наклон- :
ную трубку, заполненную водным раствором поверхностно актив-
ных веществ (например, п-толуидина, н-амилового спирта). Под-
нявшиеся пузырьки подсчитывают и собирают в специальный .
приемник, по окончании опыта анализируют жидкость в трубке ;;
и в сборнике пузырьков. Эти данные позволяют рассчитать плот- f
ность адсорбции на стенках пузырьков.
Вторым методом экспериментальной проверки уравнения Гиб-
бса является метод микротома, также предложенный Мак-Беном
и его учениками [56, 58]. В этом методе с поверхности жидкости,,
находящейся в равновесии с объемной фазой, с помощью быстро-
движущегося ножа срезают слой жидкости толщиной 0,05—
0,1 мм. Срезанный слой жидкости собирают и подвергают анали-
зу. Результаты, полученные методом микротома, хорошо согла-
суются с теорией Гиббса, в то время как метод пузырьков часто
дает значение «адсорбции», в несколько раз больше, чем требуе-
мое теорией.
Время, в течение которого устанавливаются равновесные зна-
чения поверхностного натяжения. Вновь образованная поверх-
ность водного раствора поверхностно активного вещества (на-
пример, спирта) содержит меньшее количество поверхностно ак-
1 Автор несправедливо утверждает, что при выводе уравнения Гиббса
поверхность раздела рассматривается как математическая поверхность. На
самом деле при выводе уравнения рассматривался поверхностный слой жид-
кости, отличный по своему составу от объема жидкости. Прим. ред.
ЬЪ I
ПОВЕРХНОСТЬ РАЗДЕЛА ГАЗ — РАСТВОР
77
дивного вещества, чем поверхность, достигшая равновесия, в ре-
зультате чего возникает диффузия поверхностно активного ве-
щества к поверхности раздела, а воды—в объем раствора.
Представляет интерес выяснить, насколько быстро устанавли-
вается равновесие между вновь образованной поверхностью и
объемом раствора.
Очень трудно теоретически оценить скорость, с которой уста-
навливается равновесие, так как помимо необходимости приме-
нять строгое уравнение диффузии, в которое должны входить не
концентрации, а активности ионов, требуется оценить и величину
обратной диффузии.
Для этого случая строгим является уравнение Фика 125, 70],
в котором концентрация заменена активностью:
ds =- — Dq— dt, (10)
dx
где D — коэффициент диффузии;
q —сечение, через которое рассматривается диффузия;
ds — количество вещества, прошедшее через сечение за
время dt;
da
—----градиент активности.
dx
Этот вопрос рассматривали Уорд и Тордей [73], а также
Лэнгмюр и Шеффер [51] и Дос 121 и 22].
Из всех этих работ следует, что равновесие должно устанав-
ливаться весьма быстро — в течение малой доли секунды. Одна-
ко, по мнению Доса, Гофмана, Бойда и Ральстона [42], Мак-Бена
и Шарпа [57], Блоера [8], Натинга, Лонга, Харкинса [60] и др.,
эксперименты не подтверждают этого вывода.
На рис. 5 изображена зависимость поверхностного натяжения
растворов (цетилсульфата натрия)соли, содержащей углеводо-
родный радикал, от времени. Андреас, Хозер и Тэккер [7] также
нашли, что поверхностное натяжение водных растЬоров стеарата
натрия сильно уменьшается со временем.
Данные об изменении во времени поверхностного натяжения
0,025%-ного водного раствора, измеренном методом «висячей
капли» в тщательно очищенной атмосфере, приведены в табл. 2.
Особенно существенно сказывается влияние времени в том
случае, когда в растворе находятся ионы или способные ионизи-
роваться соединения, содержащие длинный насыщенный углево-
дородный радикал.
Однако уже поверхностное натяжение водного раствора окти-
лового спирта во времени существенно не изменяется, как это
видно на рис. 6.
Следует обратить внимание на то, что теоретически рассчи-
танное время диффузии (без учета обратной диффузии), необхо-
Рис. 5. Изменение во времени поверхностного натяжения водных
растворов цетилсульфата натрия различных концентраций при
40° [60]
Рис. 6. Влияние «старения»
иа поверхностное натяже-
ние. «Возраст поверхно-
сти» [3]:
1 — равновесная кривая; 2 —
ОД сек.; 3 — 0,06 сек.; 4 —
0,04 сек.; 5 — 0,02 сек.; б —
0,01 сек.
ПОВЕРХНОСТЬ РАЗДЕЛА ГАЗ - РАСТВОР
79
Таблица 2
Изменение во времени поверхностного натяжения 0,025%-ного
раствора стеарата натрия в воде при 25°[7]
Время, сек. Поверхностное на- тяжение, дин/СМ Время, сек. Поверхностное на- тяжение, дин,см
5 71,5 240 51,1
10 71,7 480 46,5
15 68,5 960 42,7
30 63,2 1800 39,1
60 57,6 3600 35,9
120 54,5
димое для установления статического поверхностного натяжения;,
имеет порядок 10~3, 10-2 сек., тогда как экспериментально опре-
деленное время диффузии для недиссоциирующих веществ с-
коротким углеводородным радикалом имеет порядок 10-1 сек.
(по данным Эдисона); в случае же диссоциирующих соедине-
ний с длинным углеводородным радикалом необходимое для
достижения равновесия время имеет порядок 104—105 сек. Эта<
расхождение между теоретическими и расчетными данными до-
сих пор не объяснено, несмотря на очевидную важность и непо-
средственную связь этого явления с устойчивостью пены.
Влияние длины углеводородного радикала растворимых ор-
ганических соединений на поверхностное натяжение их водных-
растворов. Увеличение длины углеводородных радикалов в го-
мологических рядах органических соединений приводит к боль-
шему понижению поверхностного натяжения водных растворов
этих соединений при одних и тех же концентрациях растворов.
Иначе говоря, для одинакового понижения поверхностного натя-
жения при увеличении радикала на одну группу СН2 необходима
в три раза меньшая концентрация растворенного вещества по.
сравнению с концентрацией раствора предыдущего члена гомоло-
гического ряда. Эта закономерность носит название правила
Траубе [72].
В качестве примера в табл. 3 приведены концентрации пер-
вичных предельных спиртов, снижающих поверхностное натяже-
ние воды при температуре 20° от 72,8 до 60 дин!см.
Спирты с разветвленными цепями радикалов несколько менее
эффективно снижают поверхностное натяжение, чем спирты тога
же молекулярного веса, но с прямой цепью, как это видно из кри-
вых на рис. 7, где приведены зависимости поверхностного натя-
жения водных растворов от концентрации различных нормаль-
ных спиртов, кончая октиловым.
.80
ГЛАВА 3
Таблица 3
Концентрации спиртов, необходимые для снижения поверхностного натяжения водных растворов при 20° до 60 дин/см [3]
Спирты Концен- трация моль! л Концен- трация г 1л Отношение молярных концентраций сосед- них членов гомоло- гического ряда
Метиловый 1 ,74 56
Этиловый 0,63 29 2,7
н-Пропиловый 0,174 10,4 3,6
н-Бутиловый 0,063 4,7 2,7
н-Амиловый 0,0174 1,53 3,6
н-Гексиловый 0,0062 0,62 2,8
н-Гептиловый 0,00182 0,21 3,4
н-Октиловый 0,00056 0,073 3,2
Рис. 7. Поверхностное натяжение водных растворов спиртов [3]:
/ — нормальный октиловый; 2 — вторичный октиловый; 3 — нормальный гептило-
вый; 4— нормальный гексиловый; 5 — третичный гексиловый (1 : 1 диметилбутило-
еый); 6 — нормальный амиловый; 7— изоамиловый; 8 — нормальный бутиловый;
9 — нормальный пропиловый; 10 — этиловый; 11 — метиловый
Экстраполируя данные табл. 3 для спиртов с более длинными
радикалами, получаем, что даже небольшие концентрации этих
спиртов могут снизить поверхностное натяжение водных раство-
ров до 60 дин/см..
Так, например, понижение поверхностного натяжения водного
раствора до 60 дин!см должно отвечать концентрации додецило-
вого спирта 0,9 мг/л и гексадецилового спирта 0,01 мг/л.
ПОВЕРХНОСТЬ РАЗДЕЛА ГАЗ — РАСТВОР
81
Очевидно, состав объемной фазы и равновесный состав по-
верхностного слоя для столь разбавленных растворов резко раз-
личаются, и для того, чтобы вновь образованная поверхность
пришла в равновесие с раствором, необходимо длительное время,
в течение которого растворенное вещество из толстого слоя рас-
твора может продиффундировать к поверхности. Мало вероятно,
чтобы в условиях практической флотации поверхности растворов
спиртов с длинными радикалами находились в равновесии с
объемом. В качестве примера рассмотрим растворы гомологиче-
ского ряда спиртов, концентрации которых таковы, что поверхно-
стное натяжение каждого раствора при 20° равно 60 дин/см. При
этом условии количество адсорбированных молекул спирта со-
ставляет половину предельного значения адсорбции, а отношение
концентрации спирта в поверхностном слое к его концентрации в
объеме раствора приблизительно равно для этилового спирта 17,
для амилового 300 и октилового 6900.
Найденные экстраполяцией значения этого отношения для
додецилового и гексадецилового спиртов равны 550 тыс. и 45 млн.
Учитывая длину молекул октилового, додецилового и гексадеци-
лового спиртов, концентрирующихся в поверхностном слое, мож-
но рассчитать, что для достижения равновесия между поверх-
ностью растворов соответствующих спиртов и их объемами не-
обходимо, чтобы в поверхностном слое сосредоточились все мо-
лекулы спиртов, первоначально находившиеся в слое раствора
толщиной 0,008; 0,7 и 50 мм.
Согласно уравнению диффузии Эйнштейна [5], время, необ-
ходимое для того, чтобы установилось равновесие, пропорцио-
нально квадратам этих чисел. Если время, в течение которого
установится равновесие в растворе октилового спирта, равно
примерно 10 2 сек., то для додецилового спирта это составит
1 мин., а для гексадецилового спирта—несколько дней. Очевид-
но, для практических целей требуется различная степень прибли-
жения к равновесию адсорбции веществ, которые способны столь
сильно концентрироваться в поверхностном слое, что их едва ли
можно рассматривать как водорастворимые.
Растекание по поверхности воды нерастворимых поверхност-
но активных веществ. Если на чистую поверхность воды нанести
небольшое количество некоторых (жидких или твердых) органи-
ческих соединений (таких, как жирные кислоты с длинными цепя-
ми, амины, спирты, кетоны, простые эфиры, сложные эфиры и
т- п.), то они быстро растекутся по поверхности воды, образуя на
ней пленку. Для удобства наблюдений следует наполнить (до
краев) водой фотографическую кювету и нанести на поверхность
воды небольшое количество поверхностно активного вещества,
используя в качестве дозатора микробюретку.
6 А. м. Годэн
82
ГЛАВА 3
Для того чтобы проследить растекание пленки, на поверх-
ность воды наносят небольшое количество тонкого порошка таль-
ка, служащего индикатором, легкое сдувание которого позволяет
определить границы распространения пленки. Используя этот
метод, как это было показано Покелсом [61], Дево [17 — 20]
и Лэнгмюром [46], можно оценить толщину пленки. Предполо-
жив, что пленки мономолекулярны, можно, зная число Авогадро,
по этим данным найти поперечное сечение углеводородной цепи.
Для поверхности, полностью покрытой молекулами поверхност-
но активного вещества, найдены значения, близкие к 20 А2, что
хорошо согласуется с данными рентгеноструктурных исследова-
ний кристаллов углеводородов.
Простые опыты, описанные в предыдущем параграфе, могут
быть проведены значительно точнее, если использовать более со-
вершенные приборы, которые были применены Лэнгмюром при
измерении поверхностных давлений [lei. Наиболее ценно то, что
эти приборы измеряют разность поверхностных давлений чистой
жидкости и жидкости, поверхность которой покрыта пленкой по-
верхностно активного вещества различной плотности.
Как показали опыты, поверхностное давление, обусловленное
пленкой поверхностно активных веществ, изменяется от 50—
55 динасы, при максимальной плотности пленки до весьма малых
величин порядка 0,01 дин^см или даже меньших. Была найдена
интересная зависимость между величиной площади, приходящей-
ся на одну молекулу А, поверхностным давлением F, температу-
рой, характером полярной группы молекулы, длиной молекулы, ее
формой и числом углеводородных групп в ней. Все мономолеку-
лярные пленки можно разделить на несколько типов, аналогич-
ных различным фазам в трехмерном пространстве. При этом в
пленке наблюдаются фазовые переходы и критические явления.
Подробно эти явления описаны в работе Адама Ш; ниже будут
приведены лишь краткие выводы в объеме, необходимом для по-
яснения строения поверхности раздела жидкость —газ и жид-
кость— твердое. Пленки поверхностно активных веществ могут
быть разделены на следующие типы:
1) газообразные пленки, аналогичные трехмерным газам;
2) конденсированные двухмерные пленки, аналогичные обыч-
ным твердым и жидким телам;
3) растянутые жидкие пленки (этот тип пленок не имеет трех-
мерного аналога).
Газообразные пленки. В газообразных пленках молекулы по-
верхностно активных веществ очень слабо притягиваются одна
к другой и ведут себя аналогично молекулам обычного газа.
Отличие заключается в том, что молекулы двухмерного газа
ПОВЕРХНОСТЬ РАЗДЕЛА ГАЗ — РАСТВОР
83
должны оставаться на поверхности раздела вода—газ. С по-
мощью кинетической теории можно показать, что
FA = kT, (П)
где k — постоянная Больцмана (равная 1,372, если Л выражать
в A, F — в динах на сантиметр и Г —в градусах Кель-
вина) ; при комнатной температуре для идеального двух-
мерного газа значение А теоретически должно быть
равно 400.
Уравнение (11) аналогично уравнению состояния трехмерно-
го газа
PV — RT, (12)
где Р — давление;
V — объем;
R— универсальная газовая постоянная;
Т — абсолютная температура.
Шофилд и Райдил вывели уравнение (12) из уравнения Гиб-
бса, показав применимость этого уравнения (12) и для пленок
поверхностно активных веществ, слабо, но заметно растворимых
в воде. В этом случае площадь, приходящаяся на одну адсорби-
ровавшуюся молекулу, обратно пропорциональна поверхностной
концентрации или приблизительно обратно пропорциональна ад-
сорбции.
Более того, так же, как Ван-дер-Ваальс дал более строгое
уравнение состояния газа в виде
(р +-^j(v-v0)=RT, (13)
где а — постоянная; Го— объем, занимаемый молекулами газа
в твердом или жидком состоянии, Семенов 164] для поверхност-
ных пленок предложил уравнение
(F + ^-UA-A^kT. (14)
\ A J
В этом уравнении а характеризует силы притяжения между
адсорбированными молекулами и является аналогом постоянной
Ван-дер-Ваальса а; Ао — это площадь, занимаемая молекулами
в наиболее конденсированном состоянии.
Очевидно, состояние газообразных пленок лишь приблизи-
тельно описывается уравнением (11) и значительно более точно
уравнением (14). Действительно, необходимо было значительно
улучшить приборы, измеряющие поверхностное давление, преж-
де чем с их помощью можно было начать изучать столь разжи-
женные пленки, для которых уравнение (И) строго применимо
129, 30].
Сходство между двухмерными и трехмерными явлениями ил-
люстрируется рис. 8, на котором даны отношения FA/kT и PVIRT
6*
84
ГЛАВА 3
как функции F или Р для трехмерного углекислого газа (по дан-
ным Амага [6]) и пленок различных органических кислот с от-
носительно небольшими радикалами (по данным Шофилда и
Райдила [62,- 631).
В случае применимости уравнений (11) и (12) отношения
FAlkT и PVIRT должны быть равны единице и все кривые на
рис. 8 должны слиться в одну прямую линию с ординатой, рав-
ной 1,0. Минимум в начале кривых обусловлен членами a/V2 или
а/А2, возрастание при больших значениях Р и F—величинами
Ао и Ео согласно уравнениям (13) и (14).
Данные, приведенные на рис. 8, убедительно показывают, что
поведение пленок гомологов жирных кислот на воде при постоян-
ной температуре очень похоже на поведение углекислого газа
при различных давлениях и температурах. Изменение длины уг-
леводородной цепи, грубо говоря, эквивалентно изменению тем-
пературы углекислого газа. Как и следовало ожидать, изменение
температуры сильнее сказывается на зависимости отношения
FAlkT от F для данной жирной кислоты, чем на зависимости от-
ношения PVIRT от Р для газа.
На абсциссе рис. 8 нанесено давление (в атм) или поверх-
ностное давление (в дин/см). Кривые одинаковой формы полу-
чаются, если масштабы выбраны так, что 10 дин/см и 400 атм
выражены равными отрезками. Это эквивалентно тому, что тол-
щина пленки на поверхности воды равна 2,5 А, т. е. приблизи-
тельно равна диаметру атома кислорода или молекулы воды.
Приведенные данные свидетельствуют также о том, что расстоя-
ние, на котором действует притяжение между атомами, близко
совпадает с размером атома.
Конденсированные пленки. Если начертить F как функцию А,
то согласно уравнению (11) должна получиться равнобочная ги-
пербола и согласно уравнению (14) — несколько более сложная
кривая. В действительности результаты значительно отличаются
от указанных зависимостей, как это видно на рис. 9, взятом из
книги Адама [1]. Миристиновая кислота является в данном слу-
чае типичным примером. С уменьшением А значение F возраста-
ет, но значительно медленнее, чем это следует из уравнения (11).
Начиная с площади, приходящейся на одну молекулу, равной
800 А2, когда F = 0,20 дин/см, уменьшение значения А не приво-
дит к возрастанию величины F. После того как А достигнет зна-
чения 50 А2 на одну молекулу, F начинает сильно возрастать
при небольшом изменении А, пока не достигнет своего макси-
мального значения, приблизительно равного 50 дин)см. Такая
зависимость величины У2 от А может быть объяснена образова-
нием новой фазы при F = 0,20 дин/см и А = 800 А2 на одну моле-
Рис. 8. Зависимость
FAlkT от F для жирных
кислот и PV/RT от Р
для углекислоты:
1 — н-масляная; 2 — н-ва-
лерьяновая; 3 — н-капроно-
вая; 4 — СО2 при темпера-
туре 100°; 5 — н-каприловая;
6 — н-лауриновая; 7 — СО
при температуре 31,5° (кри-
_ со? при
температуре 0°; 9— н-кап-
риновая
Рис. 9. Результаты измерений поверхностного давления пленок на-
сыщенных жирных кислот [Id]:
/ — fa = 396; 2 — тридециловая 14,5°; 3 — лауриновая, 14—16°; 4—мири-
стиновая, 14,5°; 5—пеитадециловая, 14,5°; 6 — пальмитиновая, 12°
86
ГЛАВА 3
кулу. В пределах изменения А от 800 до 50 (при F = 0,20) плен-
ка содержит две фазы; при этом миристиновая кислота нахо-
дится как в негазообразном, так и в газообразном состоянии.
При значениях А, меньших 50 А2, существует только негазб-
образная пленка.
Поверхностное давление паров миристиновой кислоты при
температуре 14,5° равно 0,20 дин/см.
Приняв эффективную толщину поверхностного слоя равной
2,5 А, получим, что этому поверхностному давлению соответ-
ствует давление 8 атм.
Измерения потенциала поверхности, рассматриваемые в гл. 5,
показывают, что двухфазный характер пленки при средних зна-
чениях ее плотности приводит к нестабильным значениям потен-
циала поверхности, в то время как при измерениях потенциала
поверхности как газообразных, так и негазообразных пленок на-
блюдались по существу вполне определенные значения.
Является негазообразная пленка твердой или жидкой, можно
решить на основании ее сжимаемости и поверхностной вязкости,
которую особенно удобно изучать с помощью специального вис-
козиметра [44, 45, 591.
Растянутые жидкие пленки. Детальное изучение зависимости
F от А для негазообразных пленок показало, что найденные зави-
симости напоминают фазовые переходы [14, 15, 16, 35, 37, 67].
На рис. 10 даны эти зависимости для миристиновой кислоты при
различных температурах. На этом рисунке, который сравним с
рис. 9, масштаб по оси абсцисс сильно растянут. Например, при
температуре 14,1° значение F постепенно возрастает с уменьше-
нием величины А от 50 до 35, А2; при этой степени сжатия пленки
резко замедляется возрастание значения F с уменьшением А, од-
нако, из приведенных данных не следует, что F не меняется при
уменьшении А. При дальнейшем возрастании плотности пленки
и уменьшении А до 25 А2 (и даже более малых значений) вели-
чина F возрастает все быстрее и быстрее, пока не достигнет зна-
чения 50—52 дин)см при А = 20,8 А2.
Достигнув такой плотности, которой соответствует давление
4000 атм (если принять эффективную толщину пленки, равную
2,5 А), пленка сморщивается и образуется несколько слоев по-
верхностно активного вещества.
Лэнгмюр и Адам рассматривают растянутые жидкие пленки
как конденсированные, в которых углеводородные цепи не имеют
вполне определенной зигзагообразной формы, а находятся в по-
стоянном движении. Более правильно было бы сказать, что сво-
бодно могут вращаться отдельные углеводородные группы отно-
сительно каждого атома углерода. Энтропия системы в таком со-
Рис. 10. Влияние температуры на зависимость поверхностного давления
пленки миристиновой кислоты на слабых водных растворах минеральной
кислоты от площади, приходящейся на одну молекулу [Id]
© " ~ ) С ' —© © J © ..............................-J
Ц-ОЙ R среЗнем
6
Рис. 11. Пленка миристиновой кислоты на поверхности воды:
а — конденсированная пленка, F = 30 дин/см\ площадь, приходящаяся на молекулу,
равна 20,5 А2; б — растянутая жидкая пленка; F = 2 дин!см; площадь, приходя-
щаяся на молекулу, равна 42,2 А2; в — газообразная пленка, F = 0.16 дин/см\
площадь, приходящаяся на молекулу, равна 1600 А2.
88
ГЛАВА 3
стоянии больше, чем в случае жестких зигзагообразных молекул.
При возрастании плотности пленки расстояния между молекула-
ми уменьшаются настолько, что можно предположить, что моле-
кулы принимают вполне определенную (зигзагообразную) фор-
му. Поэтому они занимают меньшую площадь, благодаря чему
достигается большая плотность пленки. Таким образом, можно
представить появление новой конденсированной фазы. На рис. 11
приведены схемы различных типов поверхностных пленой на
воде.
Ориентация молекул в мономолекулярных слоях. Краткое опи-
сание кристаллической структуры соединений, содержащих пара-
финовые цепи, было дано в гл. 2. Напомним, что эти вещества об-
разуют кристаллы, состоящие из двойных слоев, концы молекул
в которых направлены один к другому, обычно под углом к пло-
скости, отделяющей эти слои. Такие кристаллы легко раскалы-
ваются, не разрушая при этом двойных слоев.
Пальмитиновая и стеариновая кислоты обе кристаллизуются
в двух аллотропных формах, расположение молекул в которых от-
личается значением углов между продольной осью молекулы и
плоскостью спайности. Во всех четырех случаях величину по-
перечного сечения молекулы можно рассчитать из соответствую-
щих данных кристаллической решетки [75]. Эта величина колеб-
лется от 18,3 до 18,9 А2. Под величиной поперечного сечения по-
нимают площадь сечения, перпендикулярного продольной оси
молекулы. Если молекулы этих кислот расположены перпенди-
кулярно поверхности воды, то минимальная плошадь, занимае-
мая ими в конденсированной твердой пленке, равна 18,5 А2.
Величина сечений этих же молекул плоскостями, параллель-
ными основным плоскостям спайности, равна по данным трех из-
мерений для a-кристаллов (угол наклона молекул около 50°) от
23,4 до 23,8 А2, для [3-кристаллов (угол наклона около 6Г) от
21,0 до 21,2 А2. Если молекулы этих кислот на поверхности во-
ды образуют наклонную конденсированную твердую пленку, пре-
дельное значение площади, приходящейся на одну молекулу,,
равно 21 А2.
Пленка пальмитиновой кислоты на поверхности слабо под-
кисленной воды может конденсироваться благодаря увеличению
поверхностного давления до тех пор, пока площадь, занимаемая
каждой молекулой жирной кислоты, не станет равной 20,5 А2,,
величина этой площади хорошо согласуется с данными рентгено-
структурного анализа. Эти данные позволяют сделать оконча-
тельный вывод, что твердые конденсированные пленки пальмити-
новой или стеариновой кислот мономолекулярны, а молекулы.
ПОВЕРХНОСТЬ РАЗДЕЛА ГАЗ — РАСТВОР
89
длина которых почти в пять раз больше ширины, ориентированы
в пленках почти перпендикулярно поверхности воды.
При образовании конденсированных пленок других жирных
кислот площадь, приходящаяся на одну молекулу, имеет то же
значение, а толщина пленки может быть больше или меньше в
зависимости от длины молекул жирных кислот. Аналогичные
результаты были получены и для других жирных кислот при ус-
ловии, что эти соединения имели только одну полярную группу
в молекуле. В отличие от этих соединений углеводороды не рас-
текались по поверхности воды; это также свидетельствует о том,,
что в твердых конденсированных пленках полярные группы мо-
лекул направлены к поверхности воды. Более подробно этот воп-
рос рассмотрен в книге Адама [1] и в работах Лэнгмюра [46, 47,
49], к которым и отсылаем читателя.
При соответствующих условиях молекулы, находящиеся на по-
верхности, могут поворачиваться и ориентироваться в энергети-
чески более выгодном направлении. Подобная перемена ориента-
ции наблюдалась Лэнгмюром [50] и Блоджет [10—12] в основ-
ном при изучении особенностей полимолекулярных слоев, обра-
зуемых из мономолекулярных слоев.
Имеются доказательства того, что молекулы, составляющие
газообразную пленку, лежат (или почти лежат) на поверхности
раствора. Для случая растворимых соединений также применимо
правило Траубе, согласно которому увеличение радикала на од-
ну группу СН2 приводит к возрастанию поверхностной активно-
сти в три раза. Энергия адсорбции при увеличении радикала на
группу СН2 возрастает на постоянную величину [Id]. Это может
произойти лишь в том случае, если каждая группа СН2 располо-
жена относительно поверхности так же, как и все остальные груп-
пы СН2 углеводородной цепи, что может быть только в случае,,
если углеводородный радикал параллелен поверхности раздела.
Вторым доказательством такой ориентации молекул является
расчет энергии изолированной молекулы, различным образом
расположенной на поверхности раздела вода — газ, наименьшая
энергия отвечает случаю молекулы, лежащей в поверхности раз-
дела [48].
Наслаивание мономолекулярных слоев. Если стеклянную-
пластинку вынуть из разведенного водного раствора соли бария
(концентрации 10~4—10-5 моль/л), на поверхности которого на-
ходится конденсированный мономолекулярный слой жирной кис-
лоты, то при движении пластинки через пленку вода стечет и
часть мономолекулярной пленки покроет ее. Если затем пластин-
ку вновь погрузить в раствор, то второй мономолекулярный слой
задержится на ней при погружении, третий слой покроет пла-
стинку при вторичном извлечении ее из раствора и т. д. Этот при-
’90
ГЛАВА 3
ем позволяет получить полимолекулярные слои последователь-
ным добавлением мономолекулярных слоев [9—191.
Гидрофобность пластинки, покрытой мономолекулярным сло-
ем, показывает, что ориентация молекул не меняется при пере-
ходе мономолекулярного слоя с поверхности воды на стекло.
Если пленку жирной кислоты переносить с поверхности очень чи-
стой (не проводящей тока) воды на очищенную стеклянную пла-
стинку, то пленка не увлекается ею, а легко смывается водой.
Как указывалось выше, этого не наблюдается, если в воде, на ко-
торой образуется пленка жирной кислоты, присутствуют ионы
бария. На поверхности металла молекулы стеарата бария и сте-
ариновой кислоты, закрепляясь, ориентируются так же, как мо-
лекулы стеарата бария на стекле, т. е. углеводородная группа
направлена от поверхности твердого.
Во всех случаях, когда твердое тело находится в воздухе, са-
мый верхний слой молекул ориентирован так, что углеводород-
ные цепи направлены в газовую фазу.
Если стеклянный движок покрыт мономолекулярным слоем
стеарата бария, то следующий слой молекул стеариновой кисло-
ты располагается поверх этого слоя. Молекулы стеарата бария
во всех случаях располагаются перпендикулярно или почти пер-
пендикулярно поверхности, в то время как молекулы стеариновой
кислоты наклонены к поверхности твердого под углом 60°, как
это было установлено при исследовании дифракции электронов
Джермером [27]. Фотографии Джермера также показывают, что
пленка стеариновой кислоты толщиной всего лишь в три молеку-
лярных слоя определенно обладает кристаллическими свой-
ствами.
Пленки стеариновой кислоты на поверхности воды, содержа-
щей небольшую концентрацию ионов бария, частично превра-
щаются в бариевое мыло; состав образующейся при этом пленки
является средним между составом мыла и жирной кислоты.
Эти пленки могут выщелачиваться бензином, растворяющим
жирную кислоту; при этом остается пористая кристаллическая
структура, имеющая такую же, как и до выщелачивания, тол-
щину, но обладающая иными оптическими свойствами.
Процесс наслаивания позволяет создать на поверхности ме-
талла или стекла покрытия различной толщины, причем разница
в толщинах покрытия может быть сделана равной двум или не-
скольким длинам молекул. Прецизионное исследование интер-
ференции монохроматического света на пластинках с такими по-
крытиями позволяет рассчитать длину молекул, если известен
угол их наклона и коэффициент преломления пленок.
ПОВЕРХНОСТЬ РАЗДЕЛА ГАЗ — РАСТВОР
91
ЛИТЕРАТУРА
1. Adam N. К. The physics and chemistry of surfaces, 3d ed., (a), p. 12, (b)
pp. 107—113, (c) p. 25, (d) p. 121, (e) pp. 27ff Oxford University Press,
New York, 1941.
2. Adam N. K. and G. Jessop. The structure of thin films. VII. Critical
evaporation phenomena at low compressions, Proc. Roy. Soc. (London),
A110, 423—441, 1926.
3, A d d i s о n С. C. The properties of freshly-formed surfaces. Part IV. The
influence of chain length and structure on the static and dynamic sufrace
tensions of aqueous alcoholic solutions, J. Chem. Soc., 1945, 98—106.
4. Alexander A. E. The structure of the surfaces of solutions, Trans.
Faraday ioc., 38, 54—63, 1942.
5. A 1 e x a n d e r A. E. and P. Johnson. Colloid Science, (a), I, pp. 17, 256,
Oxford University Press, New York, 1949.
6. Am a gat E. H. Memoire sur I’elasticite et la dilatabilite des fluides jusqu’
aux tres hautes pressions. Ann. Chim. et Phys (6), 29, 68—136, 1893.
7. A n d r e a s J. M., E. A. Hauser and W. B. Tucker. Boundary tension
by pendant drops. J. Phys. Chem., 42, 1001—1019, 1938.
8 Blair Charles M„ Jr. Time-dependence of surface tension of solutions,
J. Chem. Phys., 16, 113—116, 1948.
9 . Blodgett Katharine B. Monomolecular films of fatty acids on glass,
J. Amer. Chem. Soc., 56, 495, 1934.
10. Blodgett Katharine B. Films built by depositing successive monomole-
cular layers on a solid surface, J. Amer. Chem. Soc., 57, 1007—1022, 1935.
11. Blodgett Katharine B. Properties of built-up films of barium stearate,
J. Phys. Chem., 41, 975—984, 1937.
12. Blodgett Katharine B. and Irving Langmuir. Built-up films of barium
stearate and their optical properties, Phys. Rev., 51, 964—982, 1937.
13. Burdon R. S. Surface tension and the spreading of liquilds, 2d ed., Cam-
bridge University Press, New York, 1949.
14. Copeland L. E., William D. Harkins and G. E. В о у d. A superliquid
in two dimensions and a first-order change in a condensed monolayer, 11,
J. Chem. Phys., 10, p. 357—365, 1942.
15. D e r v i c h i a n D. G. Changes of phase and transformations of higher
order in monolayers, J. Chem. Phys., 7, 931—948, 1939.
16. Dervichian D. G. Transformations in monolayers, J. Chem. Phys., 8,
347, 1940.
.17 . Deva их H. E. Sur 1’epaisseur critique des solides et des liquides re-
duits en lames tres minces, J. Phys. Radium, 3, № 4, 450—453, 1904.
18. Deva их H. E. Recherches sur les lames d’huile etendues sur Г eau,
J. Phys. Radium, 2, № 5, 699—719, 1912.
19. DevauxH. E. Sur un procede de fixation des figures d’evolution de
1’huile sur Г eau et sur le mercure, J. Phys. Radium, 2, № 5, 891—898,
1912.
20. Deva их H. E. Les lames tres minces et leurs proprietes physiques,
J. Phys. Radium 2, № 7, 237—271, 1931.
2L Doss K. S. Gururaja. Aging of surface of solutions, Current Sci. (India),
4, 405, 1935.
22. Doss K. S. Gururaja. Alterung der Oberflachen von Losungen, III, Kol-
loid-Z., 84, 138—143, 1939.
23. Edgerton H. E., E. A. Hauser and W. B. Tucker. Studies in drop
formation as revealed by the high-speed motion picture camera, J. Phys
Chem., 41, 1017—1028, 1937.
24. F a h r e n w a 1 d A. W. Surface-energy and adsorption in flotation. Mining
Sci Press, 123, 227—234, 1921.
25. Fi ck Adolph. Ueber Diffusion, Pogg. Ann. Physik, 94, 59—86, 1855.
92
ГЛАВА 3
26. Freud В. В. and Н. Z. Freud. A theory of the ring method for the de-
termination of surface tension, J. Am. Chem. Soc., 52, 1772—1782, 1930.
27. Germer L. H. Arrangement of molecules in unimolecular and multimole-
cuiar layers, in F. R. Moulton, ed., «Recent Advances in Surface Chemi-
stry and Chemical Physics», pp. 47—53, American Association for the Ad-
vancement of Science, Washington, D. C., 1939.
28. Gibbs Willard. Collected Works, I, pp. 219—237, Yale University Press,
New Haven, Conn., 1948.
29. Guastalla Jean. Les faibles pressions superficielles, Cahiers Phvs.,
10, 30—42, 1942.
30. Guastalla Jean. Couches monomoleculaires proteiques, J. Chim. Phys.,
42, 71—72, 1945.
31. Guggenheim E. A. An elementarv deduction of Gibbs’ adsorption theo-
rem, J. Chem. Phys., 4, 689—695, 1936.
32. G u g g e n h e i m E. A. The thermodynamics of interfaces in systems of
several components, Trans. Faraday Soc., 36, 397—412, 1940.
33. Harkins William D. Electrical relations at surfaces, the spreading of
liquids, the thickness of surface films and the drop-weight and ring me-
thods for the determination of surface tension, in Harry Bayer Weiser, ed.,
Colloid Symposium Monograph, 6, 17—40, especially 38—40, Chemical
Catalog Company, Inc., New York, 1928.
34. Harkins William D. and Thomas F. An d e r s о n. A simple accurate film
balance of the vertical tvpe for biological and chemical work, J. Am. Chem.
Soc., 59, 2189—2197, 1937.
35. Harkins William D. and Edward Boyd. The states of monolayers,
J. Phys. Chem., 45, 20—43, 1941.
36. Harkins William D. and F. E. Brown. The determination of surface
tension and the weight of falling drops, J. Am. Chem. Soc., 41, 499—524.
1919.
37. Harkins William D. and L. E. С о p e 1 a n d. A superliquid in two dimen-
sions and a first-order change in a condensed monolayer, I, J. Chem. Phvs.,
10, 272—286, 1942.
38. Harkins William D. and Hubert F. J о r d a n. A method for the deter-
mination of surface and interfacial tension from the maximum pull on a
ring, J. Am. Chem. Soc., 52, 1751—1772, 1930.
39. Harkins William D. and T. F. Young. International critical tables.
IV, (a), p. 435, (b), p. 465, (c) p. 466, MacGraw-Hill Book Company, Inc.,
New York, 1928.
40. Harkins William D., T. F. Young and Lang Hua Cheng. The ring
method for the determination of surface tension, Science, 64, 333—336,
1926.
41. Hauser E. A., H. E. Edgerton, В. M. Holt and J. T. С о x. Jr. The
application of the high-speed motion picture camera to research on the
surface tension of liquids, J. Phys. Chem., 40, 973—988, 1936.
42. Hoffman Everett J., G. E. Boyd and A. W. Ralston. Studies on
high molecular weight aliphatic amines and their salts. V. Soluble and
insoluble films of the amine hydrochlorides, J. Am. Chem. Soc., 64, 498—
503, 1942.
43. J a e g e г F. M. Ueber die Temperaturabhangigkeit der molekularen freien
Oberflachenenrgie von Flussigkeiten im Temperaturbereich von —80 bi<
+ 1650°C, Z. anorg. u. allgem. Chem., 101, 1—214, 1917.
44. Joly Maurice, Viscosite des couches monomoleculaires de corps gras
J. Chim. Phys., 44, 206—212, 1947. '
45. Joly Maurice. Application des mesures de viscosite superficielle a I’etude
des couches monomoleculaires, J. Chim. Phys., 44, 213—220, 1947.
46. Langmuir Irving. Fundamental properties of solids and liquids. II Li-
quids, J. Am. Chem. Soc., 39, 1848—1906, 1917.
ПОВЕРХНОСТЬ РАЗДЕЛА ГАЗ — РАСТВОР
93
47. Langmuir Irving. The mechanism of the surface phenomena of flota-
tion, Trans. Faraday Soc., 15 (pt. 3), 62—74, 1920.
48. Langmuir Irving. The distribution and orientation of molecules, in Har-
ry N. Holmes, ed., Colloid Symposium Monograph, 3, 48—75, Chemical
Catalog Company, Inc., New York, 1925,
49. Langmuir Irving. Molecular layers, Proc. Roy. Soc. (London), A170,
1—39, 1939.
50. Langmuir Irving. Overturning and anchoring of monolayers, in F. R.
Moulton, ed., «Recent Advances in Surface Chemistry and Chemical Phy-
sics», pp. 9—18, American Association for the Advancement of Science,
Washington D. C. (1939).
51. Langmuir Irving and Vincent J. Schaefer. The effect of dissolved
salts on insoluble monolayers, J. Am. Chem. Soc., 59, 2400—2414, 1937.
52. Lenard Philipp. Ueber die Schwingungen Fallender Tropfen, Wiedemann
Ann. der Physik, 30, 209—243, 1887.
53. Livingston Herbert K. Adsorption and the free surface energy of
solids, Ph. D. thesis, University of Chicago, 1941.
54. McBain James W. and George P. Davies. An experimental test of the
Gibbs adsorption theorem: A study of the structure of the surface of ordi-
nary solutions, J. Am. Chem. Soc., 49, 2230—2254, 1927.
55. McBain James W. and Robert D u В о i s. Further experimental tests of
the Gibbs adsorption theorem. The structure of the surface of ordinary so-
lutions, J. Am. Chem. Soc., 51, 3534—3549, 1929.
56. McBain James W. and C. W. Humphreys The microtome method
of the determination of the absolute amount of adsorption J. Phys. Chem.,
36, 300—311, 1932.
57. McBain James W. and Evert R. Sharp. A comparative study of sur-
faces of solutions with the film balance and surface tension equipment,
J. Am. Chem. Soc., 63, 1422—1426, 1941.
58. McBain James W. and Robert C. Swain. Measurements of adsorp-
tion at the air-water interface by the microtome method, Proc. Roy. Soc.
(London), A154, 608—623, 1936.
59. Nutting George C. and William D. Harkins. The viscosity of mono-
layers: a test of the canal viscosimeter, J. Am. Chem. Soc., 62, 3155—3161,
1940.
60. N u 11 i n g G. C., F. A. Long and William D. Harkins. The change
with time of the surface tension of solutions of sodium cetyl sulfate and
sodium lauryl sulfate, J. Am. Chem. Soc., 62, 1496—1504, 1940.
61. Pockels Agnes. Surface tension, Nature, 43, 437—438, 1891.
62. Schofield Robert Kenworthy and Eric Keightly R i d e a 1. The Kine
tic theory of surface films. I. The surfaces of solutions, Proc. Roy. Soc.
(London), A109, 57—77, 1925.
63. Schofield Robert Kentworthy and Eric Kieghtly R i d e a 1. The kine-
tic theory of surface films. II. Gaseous expanded and condensed films,
Proc. Roy. Soc. (London), Al 10, 167—177, 1926.
64. Semenov N. A theory of condensation and adsorption, Z. physik. Chem.,
B7, 471—479, 1930.
65. Smith Grant W. and Leonard V. S о г g. The measurement of boundary
tension by the pendant drop method. I. The aliphatic alcohols, J. Phys,
chem., 45, 671—681, 1941.
66. Speakman James C. The surface tensions and partial vapor pressures
of aqueous aniline solutions, J. Chem. Soc., 1935, 776—779.
67. Stenhagen Stina Staellberg and Einar Stenhagen. Phase transi-
tions in condensed monolayers of normal chain carboxylic acids, Nature,
156, 239—240, 1945.
68. Taggart Arthur F. and A. M. G a u d i n. Surface tension and adsorption
phenomena in flotation, Trans. A1MME, 68, 479—535, 1923.
94 Г Л А В A 3
69. Tartar Н. V., V. S i v е г t z and R. E. R e i t m e i e r. The surface tension
of aqueous solutions of some paraffin chain colloidal electrolytes, including
a comparison of the capillary rise and sessile bubble methods of measure-
ment, J. Amer. Chem. Soc., 62, 2375—2380, 1940.
70. Taylor H. S. Treatise on physical chemistry, 2d ed., II, p. 1022, D. Van
Nostrand Company, Inc., New York, 1930.
71. Thomson William. Hydrokinetic solutions and observations. Part III.
The influence of wind on waves in water supposed frictionless, Phil. Mag.,
(IV), 42, 368—374, 1871.
72. T г a u b e J. Ueber die Capillaritaetsconstanten organischer Stoffe irs
waesserigen Loesungen, Ann. Chem. Justus Liebigs, 265, 27—55, 1891.
73. W a r d A. F. and L. T о r d a i, Time-dependance of boundary tensions of
solutions, J. Chem. Phys., 14, 453—461, 1946.
74. Wheeler O. L., H. V. Tartar and E. C. Lingafelter. The deter-
mination of surface tension by the sessile bubble method. J. Am Chem.
Soc., 67, 2115—2119, 1945.
75. Wyckoff Ralph W. G. Crystal structures, vol. II, chap. 13, Interscience
Publishers, Inc., New York, 1951.
Глава 4
ПОВЕРХНОСТИ РАЗДЕЛА ТВЕРДОЕ — ГАЗ,
* ТВЕРДОЕ — ЖИДКОСТЬ
Поверхности раздела между твердыми частицами и газом ш
между твердыми частицами и водными растворами имеют боль-
шое значение во флотации. Вначале будет рассмотрен более про-
стой случай — поверхности раздела твердое — газ, а затем по-
верхности раздела твердое — жидкость. В настоящей главе не
рассматриваются электрические явления на поверхности. Они
будут рассмотрены в гл. 5.
Поверхность раздела кристалл—пар. Наиболее простым при-
мером границы раздела твердой и иной фазы является граница
раздела между кристаллом и паром того же вещества. Границы
такого рода целесообразно рассматривать в свете классификации
кристаллов,.приведенной в табл. 4 гл. 2.
При раскалывании атомного или молекулярного кристаллов
разрываются только остаточные связи. При раскалывании во-
локнистых или слоистых кристаллов подавляющее количество-
разорванных связей также составляют остаточные связи. Следо-
вательно, в этих случаях свойства атомов, находящихся в поверх-
ностном слое, должны мало отличаться от свойств атомов, нахо-
дящихся внутри фазы, что и подтверждается исследованиями
дифракции рентгеновых лучей и о чем сообщалось в гл. 2.
Однако у большинства других кристаллов положение иное.
Возьмем случай, когда на поверхности алмаза разрывается
связь между атомами углерода (С — С). Мы не можем ответить,
на вопрос, приводит ли раскалывание к тому, что один из элект-
ронов, образующих пару, остается у каждого поверхностного
атома углерода или один из двух атомов получит пару электро-
нов, но большинство доказательств приводит к тому, что по-
верхность алмаза может быть хорошо описана резонансом двух
структур 2—0 и 0—2 (где число 2 или 0 обозначает число валент-
ных электронов, остающихся у ядра атома после раскалывания
кристалла). Поэтому можно ожидать, что атомы, находящиеся
на поверхности, будут склонны присоединять ионы водорода и
гидроксильные ионы. В обычных условиях (при атмосферном
давлении) в течение очень короткого времени поверхность алма-
за должна покрыться посторонними атомами. Барер [6] приводит
данные, показывающие, что алмаз легко адсорбирует водород и
96
ГЛАВА 4
кислород; при этом на его поверхности образуется слой адсор-
бированных атомов кислорода или водорода. Способность алма-
зов прилипать к маслу [44], возможно, связана со склонностью
поверхности алмаза к различным реакциям. Подвижность элект-
ронов в алмазе, подтверждаемая тем, что кристаллы алмаза ис-
пользуются в качестве счетчиков ядерного излучения, показыва-
ет, что анионные и катионные места на поверхности раскола
весьма подвижны.
Чтобы разломать кристаллы кварца, необходимо разрушить
определенное число связей Si: О; при этом электроны, по всей
вероятности, образуют пары у одного или у другого ядра. В ре-
зультате при раскалывании образуется ионная поверхность, со-
держащая на каждом участке число ионных мест, равное числу
разорванных связей. Было подсчитано [24], что при образовании
23,4 А2 поверхности раскола разрываются две связи Si: О. Можно
предположить, что на поверхности происходит также смещение
ядер атомов кислорода, позволяющее взаимно замкнуть разор-
ванные связи поверхностных атомов. Такое перераспределение
можно наглядно представить, если атомы кремния, связанные с
тремя атомами следующего от поверхности слоя атомов, считать
неподвижными, а поверхностные атомы кислорода, связанные
лишь с одним атомом кремния, считать обладающими некоторой
свободой. Рассматриваемое смещение приводит к нарушению
тетрагонального расположения электронных пар относительно
ядра атома кремния или линейного распределения пар валент-
ных электронов относительно ядра атома кислорода (возможно,
что происходит и то и другое). Можно предположить, что подоб-
ное смещение атомов отражает временное распределение атомов
на поверхности, которое легко нарушается, как только возника-
ют благоприятные для этого условия, например появляются по-
сторонние атомы или молекулы.
Свежерасколотая или очищенная поверхность ионных кри-
сталлов должна состоять из ионов, заряды которых неполностью
компенсированы окружающими ионами. Например, у кристалла
хлористого натрия ионы расположены так, что каждый ион нат-
рия окружен шестью ионами хлора, расположенными в ортого-
нальных направлениях на равных расстояниях от иона натрия.
Кратчайшее расстояние между соседними ядрами атомов равно
2,814А. Поверхностный ион натрия окружает четыре иона хло-
ра, находящихся в плоскости поверхности, и один, находящийся
в следующем от поверхности слое, шестой ближайший соседний
ион хлора отсутствует вместе с остальными ионами как притяги-
вающими, так и отталкивающими ион натрия, расположенными
в отколотой части кристалла. Из-за нарушения баланса сил при-
тяжения и отталкивания расстояние между ядрами атомов по-
ПОВЕРХНОСТИ раздела твердое —газ, твердое — жидкость
97
верхностного и следующего за ним слоя атомов может изменить-
ся; более того, могут измениться орбиты валентных электронов
и электронов внутренних слоев электронной оболочки. Эти на-
рушения в целом называются поляризацией поверхностных ио-
нов. Значение поляризации поверхностных ионов для химии по-
верхностных явлений подчеркивали разные авторы [43]. Можно
предположить, что в большинстве случаев расстояние между яд-
рами атомов поверхностного и следующего за ним слоя умень-
шается, а расположение ионов в поверхностном слое не изменяет-
ся. Можно также допустить, что анионы и катионы поверхност-
ного слоя смещаются несколько различно, так что «поверхность»
не является плоскостью даже для идеально гладкой грани кри-
сталла. Более того, вероятно, что поляризация поверхностных
ионов индуцирует поляризацию следующего слоя ионов, так что
нарушение кристаллической решетки вблизи поверхности распро-
страняется на некоторую поддающуюся оценке величину, — по
всей вероятности, на величину пяти атомных диаметров.
Подводя итоги, можно с уверенностью сказать, что какие бы
изменения ни происходили из-за раскалывания или разламыва-
ния, они легко заменяются более устойчивыми изменениями,
как только для этого представляются благопритные условия.
Можно предположить, что структура поверхности металличе-
ских кристаллов (в случае их контакта с собственным паром)
мало отличается от структуры внутри кристалла, как это следу-
ет из приведенных выше представлений, согласно которым ме-
талл рассматривается как плотная упаковка одновалентных ка-
тионов, пронизанная быстродвижущимися электронами. Воз-
можны количественные различия в плотности электронов или
расположении ядер катионов в поверхностном слое, однако в на-
стоящее время не имеется достаточного количества данных по
этому вопросу.
Слоистые кристаллы, где слои связаны с соседними слоями
только остаточными связями, обязательно раскалываются, раз-
рывая остаточные связи, так как сцепление, обусловленное ими,
наиболее слабое. Эти кристаллы на границе с собственным па-
ром образуют преимущественно неполярные поверхности. Слоис-
тые кристаллы так же, как и атомные и молекулярные и менее
распространенные волокнистые и чешуйчатые кристаллы, обра-
зуют естественные неполярные поверхности. Остальные твердые
тела образуют естественные ионные или полярные поверхности.
Высказанные замечания сведены в табл. 1
Физическая адсорбция на границе твердое —газ. Прежде все-
го рассмотрим простой опыт. Представим себе стеклянную кол-
бу, частично заполненную тонкоизмельченным кристаллическим
веществом, например кварцем; допустим, что колбу откачали
при умеренном нагревании (при температуре кипения воды) до
7 А. М. Годэн
98
ГЛАВА 4
Таблица 1
Классификация поверхностей кристаллов
Структура определяется остаточными связями Полярность чистой неизмененной поверхности Типы кристаллов1
В трех измерениях В двух измерениях 3 одном измерении Ни в одном Неполярна Неполярна Неполярна Полярна (Атомные) Молекулярные (Волокнистые) Однослойные Некоторые многослойные (Алмазоподобные) Простые ионные Сложные ионные Большинство сложных Решетчатые Металлические
*Типы, отмеченные круглыми скобками, весьма редко встречаются или вообще не
встречаются в условиях обычной флотационной практики.
высокого вакуума (10-5—10~6 мм рт. ст.) и выдержали под от-
качкой в течение продолжительного времени. Затем колбу от-
ключили от насоса и охладили до комнатной температуры, после
чего в нее впустили чистый азот в небольшом количестве, но до-
статочном для того, чтобы давление в колбе установилось рав-
ным 1—10 см рт. ст. Если колбу охладить до температуры ки-
пения азота (—191°) и измерить давление, то1 оно окажется ни-
же, чем вычисленное согласно газовым законам. Понижение дав-
ления можно объяснить прикреплением молекул азота к поверх-
ности кристаллов, но не конденсацией газа (так как давление
внутри колбы ниже давления насыщенных паров газа при тем-
пературе опыта). Такая физическая адсорбция обусловлена оста-
точными связями между молекулами химически неактивного
газа (азота) и поверхностью порошка. Существует мнение, что
физическая адсорбция должна всегда быть на поверхности кри-
сталл— газ, при этом она имеет большее или меньшее значение
в зависимости от условий. В некоторых случаях физическая ад-
сорбция может маскироваться другими явлениями.
По разности между измеренным и рассчитанным давлением
газа в опыте, рассмотренном выше, можно рассчитать количест-
во газа, адсорбированного поверхностью твердого. Было найде-
но, что' количество адсорбированного газа возрастает с ростом
поверхности твердого и ростом давления, однако при увеличении
давления количество адсорбированного газа растет непропорци-
онально давлению. На рис. 1 показано, насколько различно мо-
жет протекать физическая адсорбция (111
ПОВЕРХНОСТИ РАЗДЕЛА ТВЕРДОЕ — ГАЗ. ТВЕРДОЕ — ЖИДКОСТЬ
99
Измерение поверхности методом адсорбции газов. На явлении
газовой адсорбции основаны лучшие из применяемых в настоя-
щее время методов оценки величины удельной поверхности по-
рошков и других измельченных твердых тел. Образец, удель-
ную поверхность которого желательно определить, взвешивают
и помещают в стеклянную ампулу, которую приваривают или
Давление, мм
Рис. 1. Изотермы адсорбции различных газов иа силикагеле [11]:
/ — азот при температуре — 195,8°; 2 — кислород при — 183°: 3 — аргон при
— 183°; 4 — окись углерода при —183°; 5 — углекислый газ при —78°: 6 — азот
при —183°; 7 — сернистый ангидрид при 0°; 8 — бутаи при 0°
Из ампулы, в которой помещен образец, тщательно откачива-
ют воздух (при слабом нагревании), после чего охлаждают
До комнатной температуры. Впуская в ампулу некоторое
количество газа, измеряют объем системы. Затем температуру
образца и газа понижают до стандартного значения (такого, на-
пример, как точка кипения азота) и измеряют давление газа над
образцом. Это давление р, конечно, должно быть ниже давления
насыщенных паров адсорбата р0. По величине измеренного дав-
ления р и количеству газа, введенного в ампулу, содержащую
образец, можно рассчитать объем газа V, поглощенного образ-
цом. Согласно методу, разработанному Брунауэром, Эмметом и
7*
100
ГЛАВА 4
Теллером, сокращенно называемому методом БЭТ, при построе-
1 р ,
нии зависимости — -------- от р/р0 для различных добавляе-
г Ро р
мых при постоянной температуре количеств газа получается
прямая линия (рис. 2). По начальной ординате и углу наклона
этой прямой можно найти величину Vm — количество адсорбата,
необходимое для образования плотного мономолекулярного по-
крытия поверхности адсорбента. Если известен размер молеку-
лы, то, используя значение Vm, можно сразу же определить по-
верхность образца. Зная вес этого образца, можно найти его *
удельную 'поверхность [201.
Другой метод оценки поверхности порошков предложен
Харкинсом и Джурой [261. Они используют те же самые дан-
ные, что также приводит к прямым линиям, получаемым, однако, -
в других координатах. Здесь наклон прямой определяет поверх-
ность образца (рис. 3). Интересно отметить, что не имеется
строго математического доказательства каждого из этих мето-
дов. Мало этого, совершенно не доказано, что, получив прямые
линии по одному из этих методов, их обязательно удастся по-
лучить, применяя второй метод. Однако данные об удельных
поверхностях, полученные двумя методами, хорошо согласуются
[231. Можно считать, что каждый из этих методов дает прибли- •
женное значение действительной поверхности, и, так как изме-
рения производятся при специально подобранных наиболее
удачных значениях относительных давлений, результаты, полу-
ченные обоими методами, согласуются.
Для измерения малых поверхностей Вутен и Браун [451 пред-
ложили заменить измерение адсорбции азота в интервале дав-
лений 1—10 см рт. ст. измерением адсорбции этана при давле-
ниях примерно 0,1 мм рт. ст. Этот метод оказался полезным при
изучении процесса дробления [22]. В последнее время усиленно
предлагается заменять азот криптоном [71. При этом лучшие ре-
зультаты должны получаться благодаря сферической форме од-
ноатомной молекулы криптона. Криптон может оказаться осо-
бенно полезным при изучении проблем обогащения [9].
Зависимость физической адсорбции газа от давления. С по-
мощью физической адсорбции можно не только оценивать ве-
личину поверхности адсорбентов. Характер изотермы адсорбции
позволяет получить интересные данные и о рельефе поверхно-
сти. Из данных рис. 1 видно, что, хотя количество адсорбирован-
ного газа возрастает с ростом давления адсорбента, все же не
наблюдается линейной или другой простой зависимости, или ка-
кой-либо общей для всех газов зависимости между количеством
поглощаемого газа и давлением. Эта зависимость в одних слу-
чах изображается вогнутой (по отношению к оси давлений) кри-
вой, в других выпуклой. Известно несколько причин, влияющих
Рис. 2. Изотерма адсорбции азота иа образце гематита,
построеииая по методу Бруиауэра, Эммета и Телле-
Рис. 3. Изотерма адсорбции азота иа шести продуктах из-
мельчения, построенная по методу Харкинса и Джура [23]
102
ГЛАВА 4
на закономерности адсорбции; в некоторых же случаях причи-
ны недостаточно выяснены; часто влияние нескольких факторов
значительно усложняет наблюдаемые закономерности. Рассмот-
рение этих причин выходит за рамки настоящей книги; интере-
сующегося читателя отсылаем к книгам по адсорбции (например,
см. НИ). Однако полезно отметить, что диаграмма адсорбция —
давление газа в зависимости от ее формы может быть отнесена
к одному из нескольких типов. Наиболее важными являются:
тип I, соответствующий мономолекулярной адсорбции Лэнгмю-
ра, и тип II, соответствующий полимолекулярной адсорбции.
Изменение поверхностного натяжения твердых тел, обуслов-
ленное адсорбцией газов. Выше было показано, что адсорбцию
на поверхности жидкости можно найти, пользуясь производной
поверхностной энергии по активности и применяя уравнение
Гиббса (9) из гл. 3
г . 1
RT din а
Аналогично можно вычислить изменение поверхностной сво-
бодной энергии по адсорбции, проинтегрировав это же уравне-
ние. Такую возможность использовали Бангем и его ученики
13—5] и Харкинс со своими учениками НО, 27, 311, связав уравне-
ние Гиббса с уравнением Юнга для краевого угла (см. гл. 7).
Отметим, что активность в уравнении Гиббса при условии ма-
лых концентраций можно заменить концентрацией, не сделав
при этом значительной ошибки. Тогда
бу = — Г/?Тб1пс;
X
Ь = То —RT’J-y- дс,
о
где у0 — поверхностная энергия при нулевой концентрации ад-
сорбата.
Это уравнение может быть записано в несколько более удоб-
ном виде
X
Ъ — То= — дс' (1>
о
Интегрирование нетрудно осуществить графически, при усло-
вии, что известны точные значения Гис для малых концентра-
ций адсорбата.
В измерениях Ливингстона [31], проведенных для адсорбции
различных газов на различных твердых телах, концентрация ад-
сорбата пропорциональна давлению газа р, так что отношение
Г /с пропорционально g/p (g—адсорбция газа в миллиграммах
на грамм адсорбента), дс пропорционально приращению давле-
ПОВЕРХНОСТИ РАЗДЕЛА ТВЕРДОЕ — ГАЗ, ТВЕРДОЕ — ЖИДКОСТЬ 103
ния др. На рис. 4 приведены типичные зависимости g от р до д/р
от р (или, что то же самое, Г /с от с). Площадь под последней
кривой, ограниченная ординатой с = х, дает величину ух = -у0.
80
Рис. 4. Адсорбция паров воды на анатазе [31]
Мономолекулярная адсорбция по Лэнгмюру. Лэнгмюр предпо-
ложил [30], что молекулы газа или пара непрерывно1 конденси-
руются на поверхности твердого и испаряются с нее. Время за-
паздывания между этими событиями и обусловливает адсорб-
цию. Если столкновение молекулы газа при попадании ее на
покрытый молекулами этого же газа участок поверхности имеет
характер упругого удара, а при попадании на свободный уча-
сток— неупругого удара, то скорость конденсации будет про-
порциональна выражению
а(1-0)рЛ, (1)
104
ГЛАВА 4
где а — коэффициент конденсации;
6 — доля поверхности адсорбента, покрытой адсорбатом;
Р —давление;
А — величина всей поверхности твердого.
С другой стороны, скорость испарения адсорбата пропорцио-
нальна покрытой им поверхности, т. е. QA.
В случае равновесия справедливо равенство
6 А = а (1 — 9) рА
откуда
9 = -3g—.
1 + а Р
Из уравнения Лэнгмюра следует, что величина адсорбции
(которая пропорциональна 0) при малых значениях р растет про-
порционально давлению, затем рост адсорбции с давлением за-
медляется и при значениях ар, больших по сравнению с едини-
цей, величина адсорбции стремится к постоянному значению.
При исследовании процесса адсорбции Лэнгмюр полагал, что
молекулы закрепляются на поверхности твердого до тех пор, по-
ка на ней имеются свободные участки, на которых молекулы газа
могут закрепляться и удерживаться в течение определенного вре-
мени. Если вся поверхность полностью покрыта адсорбированны-
ми молекулами, то дальнейшая адсорбция невозможна. Это до-
пущение равносильно предположению, что сродство молекулы к
свободному и занятому (молекулой газа) участкам поверхно-
сти совершенно различно. Поэтому нам кажется, что теория
мономолекулярной адсорбции Лэнгмюра должна лучше описы-
вать явление хемосорбции, чем явление физической адсорбции.
Полимолекулярная адсорбция БЭТ. Основной предпосылкой
этой теории (Па, 121 является допущение, что силы, обусловли-
вающие физическую, или вандерваальсову, адсорбцию, те же,
что и силы, обусловливающие капиллярную конденсацию. Одна-
ко используемый ими метод рассуждений является обобщением
и развитием метода Лэнгмюра, примененного им при исследова-
нии мономолекулярной адсорбции.
Примем, что So, Si, S2... St — площади поверхности, покры-
тые соответственно 0, 1, 2... i слоями адсорбированных молекул.
При равновесии величина So должна оставаться постоянной, т. е.
скорость конденсации на свободной поверхности и скорость ис-
парения молекул из первого слоя должны быть равны, что при-
водит к уравнению
a1pS0 = 61S1expf—
L
(3)
где Qi и—постоянные для данного адсорбента;
р —равновесное давление адсорбата;
ПОВЕРХНОСТИ РАЗДЕЛА ТВЕРДОЕ - ГАЗ, ТВЕРДОЕ — ЖИДКОСТЬ 105
Е\ — молярная теплота адсорбции первого слоя молекул;
R — универсальная газовая постоянная;
Т —абсолютная температура.
При равновесии величины So, Sb S2... St также rife должны
меняться; поэтому для каждого слоя получается ряд уравнений,,
аналогичных уравнению (3), которые можно записать в следую-
щем виде:
Г г.
, = b .S. ехр — —. (4)'
Уравнение (3) может быть записано в таком виде:
S^ySv, (5>
где
р = М-р ехр -i- .
\ °i / L К*
Остальные уравнения можно упростить, приняв, что-
а21Ь2 = a2lb2=... at/b( и Е2 = Е3 = Е4... E;=EL равно теп-
лоте конденсации. Это допущение справедливо в случае, если,
силы, удерживающие молекулы, действуют лишь на малых рас-
стояниях. Приняв это допущение и введя обозначения
/ \
х = —- р ехр
\ ^2 /
• -
RT
перепишем уравнение (4) в виде
(6)-
Комбинируя уравнения (5) и (6), получим
— ух So.
Обозначив у= Сх, перепишем уравнение (6) в виде
St = CxlS0, (7>
где
При этих предположениях полная поверхность адсорбента
равна
со
Л = (9>
о
а объем поглощенного поверхностью адсорбата
со
К = (Ю>
о
106
ГЛАВА 4
где V — объем адсорбата, необходимый для покрытия 1 см2
поверхности мономолекулярным слоем.
Отношение количества адсорбированного газа к количеству,
необходимому для образования плотного мономолекулярного
’покрытия, равно
СО со
CSa^ixi
Vsz s0(i + c£?)
0 1
Сумма, стоящая в знаменателе дроби, есть геометрическая
прогрессия и равна она х/(1—х); сумма, стоящая в числителе,
.дроби, может быть упрощена следующим образом
со
1 1 — X ___ X
dx Х dx (1 — х)2
:и окончательно:
Сх
2 _ (1 — х)2 __Сх_____
j , с х (1— х)(1— х+Сх)
1 — х
При давлении газа, равном давлению насыщенных паров ад-
сорбата, на поверхности адсорбента образуется бесконечное чи-
сло слоев адсорбированных молекул. При этом х должен быть
равен 1, как это следует из уравнения (13). Поэтому постули-
руем, что в общем случае
х = -^~. (14)
Ро
Подставляя это значение х в уравнение (13), получаем
V = ----------------------, (15)
(Po-P)[l+(C-1)—1
L Ро J
тде Vm—объем адсорбата, необходимый для образования мо-
номолекулярного слоя.
Уравнение может быть переписано в виде
----Р------= —+ _С - _Р_. (16)
У (р0 Р) У nfi У nfi Ро
ПОВЕРХНОСТИ РАЗДЕЛА ТВЕРДОЕ — ГАЗ, ТВЕРДОЕ - ЖИДКОСТЬ 107
Это и есть линейная форма уравнения БЭТ, используемая
при измерении поверхности порошков, о чем говорилось выше.
При выводе уравнения БЭТ были сделаны следующие пред-
посылки:
1) теплоты адсорбции молекул во всех слоях (за исключени-
ем первого) равны между собой;
2) относительные возможности закрепления и испарения из
любого слоя (за исключением первого) равны;
3) возможно образование бесконечно толстого слоя конден-
сированного газа при давлении адсорбата, равном давлению его
насыщенных паров:
4) все участки поверхности адсорбента идентичны;
5) адсорбированные молекулы не влияют на свойства сосед-
них молекул или участков поверхности.
Хотя эти предположения не могут строго выполняться, они
являются логически обоснованным первым приближением при
описании физической адсорбции. Таким образом, мы считаем
очевидным, что поверхности всех твердых тел, находящихся в
контакте с таким газом, как воздух, частично покрыты молеку-
лами газа. Физическая адсорбция таких постоянных газов, как
азот и кислород, при комнатных температурах и атмосферном
давлении, по-видимому, невелика. Конденсируемые газы (та-
кие, как углекислый газ, пары воды, сернистый газ) адсорби-
руются в значительно больших относительных количествах.
Плотность адсорбированного покрытия возрастает с увеличени-
ем давления, уменьшается с повышением температуры и не яв-
ляется постоянной величиной, так как зависит от равновесия ме-
жду твердым телом и окружающей средой.
Хемосорбция на поверхности раздела кристалл — газ. До сих
пор при рассмотрении процесса адсорбции принималось, что' ха-
рактер связи адсорбированных атомов или молекул один и мо-
жет быть точно определен. Однако имеются и такие случаи, ко-
гда характер адсорбции изменяется. Так, например, кислород
при низких температурах адсорбируется древесным углем без
изменения структуры молекулы. Адсорбция его в этом случае
аналогична адсорбции аргона на древесном угле. При более
высоких температурах происходит перераспределение валентных
связей, в результате чего каждый атом кислорода оказывается
-связанным ковалентной связью с атомом углерода. Если образу-
ющиеся при этом ассоциации атомов углерода и кислорода не
испаряются с поверхности, хемосорбция будет продолжаться до
тех пор, пока вся поверхность не покроется парами атомов угле-
род— кислород. Если ассоциации атомов углерод—кислород ис-
паряются в виде окиси углерода, обнажая при этом свободную
поверхность угля, хемосорбция будет протекать со скоростью,
определяемой скоростью подачи кислорода и скоростью удале-
108
ГЛАВА 4
ния окиси углерода. Результат такой хемосорбции аналогичен
горению угля. Такой процесс можно рассматривать и как хемо-
сорбцию кислорода и как процесс горения.
В тех случаях, когда газовая фаза состоит из двух или не-
скольких газов, весьма возможно, что для протекания нужной
реакции между этими газами необходимо, чтобы их молекулы в
течение некоторого времени находились в непосредственной бли-
зости. Реакции не могут протекать в газовой фазе, так как вре-
мя столкновения молекул газов очень мало, если же молекулы
этих газов адсорбированы, то время, в течение которого они на-
ходятся в непосредственной близости, может увеличиться в мил-
лионы раз, хотя время пребывания молекул на поверхности будет
составлять лишь доли секунды. Если реакция протекает в при-
сутствии адсорбента, но он участия в ней не принимает или при-
нимает, но возвращается в первоначальное состояние, то такое
явление называется гетерогенным, или контактным, катализом.
Поверхность раздела кристалл — вода. Влажность газов в
значительной степени влияет на поверхность раздела кри-
сталл-— газ. Влияние это связано с тем, что многие минералы
являются окислами и при взаимодействии с водой превращаются
в гидроокиси. Так, например, кварц хемосорбирует пары воды,
в результате чего поверхность его гидролизуется.
В большинстве случаев возможны сложные изменения по-
верхности минералов при контакте минерала с водой. Эти из-
менения не ограничиваются образованием твердых поверхност-
ных оснований или кислот, а также связаны с переходом веще-
ства из твердой фазы в жидкую. Такой переход атомов или
ионов вызван тем, что поверхностные атомы или ионы испыты-
вают притяжение не только со стороны соседних атомов или
ионов твердого, но и со стороны проходящих вблизи поверхности
молекул или ионов жидкой фазы. Конечное состояние системы
отвечает равновесию между силами притяжения со стороны
жидкой и твердой фазы; при этом с точки зрения статистики не
будет происходить никаких изменений в системе, однако это не
значит, что условия, в которых находится каждый атом или ион,
не меняются.
Ионный кристалл (как простой так и сложный) состоит из
расположенных в определенной повторяющейся последователь-
ности катионов и анионов. Свежерасколотая поверхность такого
кристалла полярна, места с избытком положительного заряда
чередуются с местами избыточного отрицательного заряда. По-
верхность состоит из атомных ядер и электронов, иначе распо-
ложенных один относительно другого-, чем они располагались
бы внутри твердого кристалла [36, 42, 43]. В присутствии поляр-
ной жидкости (такой, как вода) ионы, диполи или просто груп-
пы молекул жидкости неизбежно будут задерживаться вблизи.
ПОВЕРХНОСТИ РАЗДЕЛА ТВЕРДОЕ — ГАЗ, ТВЕРДОЕ — ЖИДКОСТЬ 109
определенных участков поверхности. Адсорбированные таким
образом ионы и молекулы будут притягивать другие молекулы
и ионы, в результате чего образуется пограничный слой, отлича-
ющийся по своей структуре от объемной фазы жидкости, анало-
гично тому как вблизи поверхности кристалла образуется по-
верхностный слой, обладающий иной структурой, чем структура
внутренних участков кристалла. По мере удаления от поверх-
ности твердого структура жидкости постепенно меняется, пока
не перестанет отличаться от структуры внутренних слоев жид-
кости. Конечно, адсорбированные молекулы воды, водородные
или гидроксильные ионы не удерживаются все время на актив-
ных местах поверхности, однако есть основание считать, что
время их пребывания на этих местах или вблизи них в жидкой
фазе велико по сравнению со временем пребывания в заранее
выбранном элементе объема внутри жидкости.
При растворении ионов кристалла происходит процесс, по су-
ществу, аналогичный описанному. Если поместить чистый флюо-
рит в чистую воду, то ионы кальция и фтора будут переходить в
воду до тех пор, пока не установится максимальная концентра-
ция этих ионов в воде. Когда такое состояние будет достигнуто,
минерал перестанет растворяться, вернее, скорость растворения
станет равной скорости кристаллизации.
Во многих случаях ионы поверхности кристалла, погружен-
ного в водный раствор, гидратируются и в таком виде перехо-
дят в жидкую фазу. Растворенные негидратированные ионы на-
ходятся в равновесии с гидратированными, так что процесс гид-
ратации может происходить как на поверхности твердого, так и
в объеме жидкости. Процесс гидратации и растворения до неко-
торой степени аналогичен также реакции газа с твердой поверх-
ностью, при условии, что продукты реакции удаляются с этой
поверхности.
Молекулярные кристаллы менее склонны к смачиванию и ра-
створению в воде, чем ионные. Молекулы веществ, образую-
щие такие кристаллы, и молекулы воды при растворении про-
сто перемешиваются; поэтому растворение таких веществ в воде
можно уподобить процессу испарения.
Скорость растворения твердых тел. Скорость растворения
твердых тел в ненасыщенных растворах зависит от градиента
концентраций в непосредственной близости к поверхности твер-
дого и от величины этой поверхности. Такая зависимость, иног-
да называемая законом Венцеля [33], была использована Грос-
сом и Циммерли [25] для измерения поверхности частиц квар-
ца по скорости их растворения в слабом водном растворе плави-
ковой кислоты.
Первые серьезные исследования скорости реакций, протекаю-
щих на поверхности раздела твердое — жидкое, проведенные
110
ГЛАВА 4
Нойсем и Уитни [32, 39а], привели к следующему уравнению:
Я/»
-^- = m(Cj-C), (17)
at
где с— концентрация
cs— концентрация
т— коэффициент
в объеме раствора в момент времени /;
насыщенного раствора;
пропорциональности, величина которо-
го остается постоянной только в том случае, если во
время опыта не изменяются условия (особенно важ-
но, чтобы не изменялась поверхность твердого и тол-
щина вязкой пленки, смежной с поверхностью твер-
дого) .
Удобно выражать [41] изменение концентрации во времени
de
— в виде функции константы скорости растворения К и отно-
шения объема раствора к величине действующей поверхности
= (18)
S
Это уравнение после замены переменных
примет следующий вид:
К =-- — In -с*—с-’-,
S 6 cs — с2
и интегрирования
(19)
где с,ис2 —исходная и конечная концентрации;
cs— концентрация насыщенного раствора;
6 — время опыта, мин.
В этом уравнении S выражается в см2; V — в см3; К — в
см/мин и все концентрации в любых одинаковых единицах, на-
пример в г/л.
Интересно отметить, что в уравнениях (18) и (19) не учиты-
вается зависимость величины константы скорости растворения
от перемешивания. Для случая перемешивания раствора вра-
щающимся импеллером со скоростью 400 об/мин при условии,
что растворяющаяся поверхность кристалла расположена гори-
зонтально под импеллером, в International Critical Tables да-
ются значения К от 0,18 для йодистого калия до 0,021 для вод-
ного сернокислого кальция (гипса).
Интересно также оценить скорость процесса растворения.
Так, для специального опыта с гипсом (К = 0,021 см/мин) при
условии, что 10 г минерального порошка с удельной поверх-
ностью 4000 см2/г взвешено в 1 л воды, первоначально не содер-
жавшей гипса, уравнение (19) может быть записано в виде
, In (1 — 1/)
123
0,84
ПОВЕРХНОСТИ РАЗДЕЛА ТВЕРДОЕ — ГАЗ, ТВЕРДОЕ — ЖИДКОСТЬ НГ
где у—отношение с^с,. , т. е. доля насыщенной концентрации»,
достигаемая во время опыта.
Время t как функция у принимает следующие значения: при
у = 0,1 t =7,6 сек.; при у = 0,5 i = 49 сек.; при у = 0,9 t =
= 2 мин. 44 сек.; при у = 0,95 t = 3 мин. 34 сек.; при у — 0,99'
t = 5 мин. 29 сек.; при у = 0,999 t = 8 мин. 13 сек. По этим дан-
ным можно судить, что процесс растворения происходит далеко
не мгновенно.
В других случаях процесс может протекать и значительно
быстрее. Например, для того чтобы увеличить концентрацию
500 с.и3 раствора от 99 до 99,9% предельного значения в присут-
ствии 1000 г порошка с удельной поверхностью 200 см2]г (К =
= 0,105), требуется приблизительно 3 сек.
По-видимому, возможно вывести уравнение, аналогичное
уравнению (19), описывающее кинетику поглощения реагента,
минералом из раствора. Однако его до настоящего времени не
выводили, хотя получить такое уравнение было бы весьма ин-
тересно.
Ранее отмечалось, что постоянная диффузия зависит от тол-
щины относительно неподвижной пленки жидкости, окружаю-
щей твердые частицы. Даже если жидкость совершенно непо-
движна (так же, как и твердые частицы), диффузия будет осу-
ществляться благодаря движению молекул в согласии с урав-
нением, приведенным выше. Однако численное значение посто-
янной диффузии, найденное экспериментально, в этом случае
будет меньше. Если поток жидкости ламинарный, вследствие
чего различные ее слои не перемешиваются, то в основном вы-
полняются классические условия диффузии. Если же жидкость
движется турбулентно, т. е. перемешивается во время движе-
ния, конвекция помогает молекулярной диффузии в переносе
вещества. Однако даже в случае турбулентного перемешивания
имеется, конечно, слой, непосредственно прилегающий к ча-
стицам твердого 18], в котором доминирует ламинарное течение
жидкости. Толщина этого неперемешивающегося слоя зависит
от условий опыта и обычно равняется 20—50 и 139, 131. В основ-
ном толщина неподвижного слоя обратно пропорциональна ско-
рости в степени, меньшей, чем единица (равной, например, 2/з.
или 4/s). Несмотря на важность этого явления для флотации,
им пренебрегали, и только Туайнер и Корман [40] по данным
своих опытов вычислили толщину неподвижного слоя жидкости,
при условиях относительно интенсивного и слабого перемешива-
ния. В первом случае, согласно их данным, толщина неподвиж-
ного слоя жидкости равнялась 41 и, во втором 147 ц. В частно-
сти, эти исследователи пришли к выводу, что процесс подготов-
ки материала к флотации включает гетерогенную поверхност-
ную реакцию, при которой диффузия собирателя через прослой-
112
ГЛАВА 4
ку жидкости у поверхности твердого протекает с измеримой ско-
ростью. Ряд соображений (в том числе и это) привел их к вы-
воду, что флотационная пульпа не достигает равновесия даже
в стадии разделения.
Поверхность раздела кристалл — водный раствор. Характер
поверхности раздела твердое—водный раствор в основном оп-
ределяет химию флотационного процесса. Поверхности это-
го типа будут неоднократно рассматриваться в последующих
главах. Поэтому в настоящем разделе будут даны лишь основ-
ные черты явлений обменной адсорбции, которая характерна
для границ раздела такого типа.
Рис. 5. Схематическое изобра-
жение связи кислород — крем-
ний в кварце и адсорбции Н+
и ОН- ионов на поверхности
расколотого под водой кварца.
Так как связи показаны в пло-
скости, на рисунке представ-
лены лишь три валентности
кремния из четырех [24]
Выше отмечалось, что свежерасколотая поверхность кварца
подвергается гидролизу. На рис. 5 показан двухмерный аналог
[24] трехмерной решетки кварца (в связи с чем на этой схеме
атом кремния показан окруженным тремя атомами кислорода
вместо четырех). Атомы водорода и гидроксильные группы по-
верхности непрерывно обмениваются с ионами водорода и гид-
роксильными ионами воды. В среднем в нейтральной воде коли-
чество ионов водорода, покинувших свои места на поверхности,
больше, чем количество гидроксильных ионов, покинувших по-
верхность, поэтому поверхность кристалла заряжена отрица-
тельно, а жидкость— положительно. Избыток положительных
ионов, находящихся в жидкости, стремится сконцентрироваться
вблизи поверхности частиц. Поэтому поверхность твердого мо-
жет рассматриваться как покрытая двойным слоем (подробнее
см. в гл. 5). В нашем примере анионы закреплены на поверх-
ности твердого, а катионы движутся вблизи них.
ПОВЕРХНОСТИ РАЗДЕЛА ТВЕРДОЕ — ГАЗ, ТВЕРДОЕ — ЖИДКОСТЬ 113
Наличие такого двойного слоя приводит к движению частиц
относительно жидкости в тех случаях, когда через суспензию
пропускается электрический ток.*. Если воду заменить водным
раствором, например раствором хлористого бария, скорость,
с которой/частицы движутся в электрическом поле, изменит-
ся. Может оказаться, что частицы будут Двигаться даже в об-
ратном направлении, что свидетельствует об изменении струк-
туры двойного слоя ионов, а следовательно, и структуры по-
верхности раздела.
Рис. 6. Зависимость адсорбции бария на кварце из
раствора нитрата бария от его концентрации при
pH = 8 [14]
Измерения адсорбции ионов бария кварцем из раствора
нитрата бария показывают, что адсорбция бария возрастает с
увеличением концентрации ионов бария и pH (по крайней ме-
ре до pH = 10), как это видно на рис. 6 и 7. Для определения
адсорбции бария использовали радиоактивный изотоп Ва140 [14].
Аналогичные измерения адсорбции додециламина на кварце
дали приблизительно те же результаты (рис. 8). Эти данные бы-
ли получены с помощью радиоактивного углерода С14, которым
был мечен додециламин [18, 19, 21]. Удивительно, что между по-
ведением сферического двухвалентного катиона и крайне несим-
метричного одновалентного катиона наблюдается такое сходст-
во. Но, для того чтобы построить более или менее полную тео-
рию, необходимо в данной области провести значительно боль-
шее число исследований. Тем не менее надо иметь в виду, что
систематическая теория адсорбции электролитов должна быть
тесно связана с теорией электролитов. Поверхности адсорбентов
при этом могут рассматриваться как гигантские полифункцио-
нальные ионы, конкурирующие с ионами того же знака, находя-
щимися в жидкости, и прочно связанные с ионами противопо-
ложного знака.
8 А- М. Годэн
Рис. 7. Влияние pH на адсорбцию ба-
рия кварцем из раствора нитрата
бария [14]-.
1 — Ва2+, концентрация 560 • 10-4 — 588Х
Х1О-4 моль/л', 2— Ва 2Ч-, концентрация
483 • !0-5—577 - 10-5 люль/л
Рис. 8. Адсорбция лауриламинацетата на кварце. Точка М соответствс-
ет образованию мономолекулярного покрытия и критической концент-
рации мицеллообразования [19]
ПОВЕРХНОСТИ РАЗДЕЛА ТВЕРДОЕ - ГАЗ, ТВЕРДОЕ — ЖИДКОСТЬ 115
Если состав водного раствора, окружающего минеральную
частицу, изменяется благодаря добавлению растворимых ре-
агентов, изменение состава поверхности, которое должно после-
довать за этим, определяется не только диффузией нового ин-
гредиента к поверхности, но и одновременно происходящей диф-
фузией замещаемого ингредиента от поверхности раздела в объ-
ем жидкости. Так как градиенты концентраций не обязательно
должны быть равны друг другу, то скорость перестройки поверх-
ности может определяться или скоростью поступления нового
вещества, или скоростью удаления вытесняемого. Это представ-
ляет превосходный случай для появления многих совершенно
неожиданных эффектов.
Обратимость обменной адсорбции. Лишь в немногих случа-
ях имеются прямые доказательства обратимости ионной ад-
сорбции. Так, например, при адсорбции додецил амин ацетата
кварцем катионы додециламмония, закрепившиеся на поверхно-
сти, обмениваются с этими же катионами, находящимися в
жидкости, и наоборот [191. Этот обмен можно заметить, если
ионы, находящиеся в жидкости или на поверхности твердого,
пометить радиоактивным изотопом (например, углеродом С14)
и измерить радиоактивность твердого и жидкого после обмена.
Однако очень мало известно о скорости, с которой протека-*
ет этот обмен. Измерение скорости обмена прольет свет на ко-
эффициент диффузии и «коэффициенты диссоциации» адсорби-
рованных ионов. Но измерить эти величины как методом радио-
активных индикаторов, так и другими методами нелегко.
Естественная флотируемость. С точки зрения флотации
имеется существенная разница между кристаллами, на поверх-
ности которых находятся ионы (вне зависимости от того, явля-
ются кристаллы ионными или нет) и на поверхности которых
ионы отсутствуют. Первые не обладают естественной флотируе-
мостью, вторые в той или иной степени ею обладают
В табл. 2 приведена классификация кристаллов согласно
их естественной флотируемости (данные взяты из табл. 4 гл. 2
и табл. 1 настоящей главы).
Из таблицы видно, что встречающиеся в природе минералы
не обладают естественной флотируемостью. Исключениями яв-
ляются: природные углеводороды, сера, графит, борная кисло-
та, тальк и подобные тальку силикаты. В действительности
возникали сомнения, обладают ли графит и сера естественной
флотируемостью [381. Таггарт и его школа утверждают, что ес-
ли эти минералы тщательно очистить от органических загряз-
1 Влияние характера ненасыщенных связей поверхности на естественную
флотируемость минералов раньше и значительно более подробно описано
в работе Г. С. Стрельцина, помещенной в Трудах II научно-технической сессии
института Механобр (Металлурпиэдат, 1952) Прим. ред.
Ь*
116
ГЛАВА 4
Таблица 2
Естественная флотируемость кристаллов в воде
Тнп кристаллов Примеры Естественная флотируемость
Атимные Молекулярные. Волокнистые и слои- стые Типа алмаза Простые ионные Сложные ионные Волокнистые ионные Слоистые ионные Каркасные Металлические Полупроводники Гелий, аргон а. Иод, сера б. Парафин, нафталин Графит Алмаз Поваренная соль, флю- орит Кальцит, оливины Пироксен а) Слюды б) Тальк в) Жирные кислоты г) Борная кислота Полевой шпат, кварц Медь Галенит, пирнТ Некоторая! Высокая Значительная По всей вероятности не обладает Не обладает > > » » » » Значительная Высокая » Не обладают » » » »
Не существует при температурах выше 0°.
нений, они не будут флотироваться. Эти авторы также отмеча-
ют, что очистить графит и серу весьма трудно. Указанным
утверждениям, базирующимся на измерении краевых углов на
графите и сере, можно противопоставить данные практических
флотационных опытов, проведенных с этими минералами. Кро-
ме того, применяемая перед измерением краевых углов обра-
ботка графита и серы, заключавшаяся в их очистке и после-
дующей шлифовке гидрофильными порошками под водой, мог-
ла привести к загрязнению поверхности.
Опыты с исключительно чистым графитом, содержащим
9,1О'4'7о золы, проводились в Массачусеттском технологическом
институте (графит был приготовлен Россом в лаборатории Car-
bon and Carbide Chemical Corp). Очистка заключалась в на-
гревании уже очищенного графита в потоке азота и хлора при
температурах, изменяемых от 2000 до 2500°, в графитовой печи,
подобной описанной Гартландом [17]. После дробления и из-
мельчения графита под водой результаты флотационных опы-
тов, проведенных в цилиндре и во флотационной камере, пока-
зали, что графит флотировался.
Сера, графит, молибденит и уголь значительно лучше сма-
чиваются углеводородными жидкостями, чем водой. Это при-
водит к тому, что органические масла прилипают к поверхности
ПОВЕРХНОСТИ РАЗДЕЛА ТВЕРДОЕ - ГАЗ, ТВЕРДОЕ - ЖИДКОСТЬ 117
минеральных частиц, находящихся в воде, в результате чего
на поверхности этих частиц может образоваться большой крае-
вой угол. Желательно было бы иметь количественные данные,
особенно, если сравниваются свойства очищенных и неочищен-
ных поверхностей твердого.
Убедиться в том, что борная кислота обладает природной
флотируемостью, очень легко. Для этого достаточно пригото-
вить горячий насыщенный раствор борной кислоты, профильтро-
вать его, если желательна очистка, и, дав возможность раство-
ру охладиться для того, чтобы образовались кристаллы борной
Рис. 9. Структура кристалла бор-
ной кислоты. Показаны атомы,
расположенные в одном слое.
Большие кружки изображают ато-
мы кислорода, маленькие затем-
ненные— бор, маленькие незатем-
ненные — водород [88, стр. 150—
161]
кислоты, отфлотировать их. При флотации без реагентов по-
лученных таким образом кристаллов, образуется сильно на-
груженная шелковисто-белая пена, подобная наблюдаемой при
флотации молекулярных органических кристаллов [1, 2, 28, 351
Высокая естественная флотируемость борной кислоты может
быть объяснена тем, что соседние слои в кристалле притягива-
ются исключительно остаточными связями. Внутри слоя каждая
группа ВО3 связана через шесть атомов водорода с тремя со:
седними группами ВО3. Таким образом, шесть атомов водорода
образуют двойные водородные мостики между каждой парой
ближайших групп ВО3 [46]. как это показано на рис. 9. Меж-
атомные расстояния внутри каждого слоя малы, а расстояния
между слоями велики. Так, расстояние между атомами кисло-
рода и водорода, расположенными в одном и том же слое, в
среднем равно 1,355 А, а расстояние между атомами бора и кис-
лорода одного и того же слоя равно 1,36 А. Расстояние между
атомами кислорода и атомами бора или водорода соседних сло-
ев равно 3,18 А, причем атомы кислорода расположены почти
над иЛи под атомами бора и водорода двух соседних слоев.
Естественная флотируемость иода так же легко может быть
продемонстрирована в лаборатории. Естественная флотируе-
мость таких различных неорганических кристаллов, как борная
кислота, иод и пирофиллит [29], показывает несостоятельность
гипотезы, утверждающей, что для флотации необходимо присут-
118
ГЛАВА 4
ствие углеводородных радикалов. Действительно, большинство
твердых тел, обладающих естественной флотируемостью, содер-
жит углеводородные радикалы.
В то же время приведенные выше примеры показывают, что
естественной флотируемостью обладают также некоторые кри-
сталлы, не содержащие углеводородных цепей радикалов. В по-
давляющем большинстве случаев минералы не обладают естест-
венной флотируемостью. Если же минералы, за исключением
перечисленных выше, флотируются без добавления реагентов,
то это может быть вызвано или загрязнением их поверхности
органическими соединениями, или преднамеренным изменени-
ем поверхности минералов.
Естественная флотируемость органических соединений. Кри-
сталлы органических соединений можно разделить на кристал-
лы, состоящие из молекул, удерживаемых в кристалле остаточ-
ными связями, и на кристаллы, состоящие из групп радикалов,
связанных между собой ионной или водородной связью.
В результате раскалывания твердых тел первого типа обра-
зуется неполярная поверхность. Эти вещества обладают естест-
венной флотируемостью. Примерами таких соединений явля-
ются углеводороды и их полные или неполные галогенпроизвод-
ные. То, что полные галогена рои вводные обладают естественной
флотируемостью, может служить еще одним доказательством
того, что не только углеводородные радикалы обусловливают
флотируемость.
Твердые тела второго типа могут проявлять ионные или во-
дородные связи в двух или трех измерениях.
Разницу между этими структурами можно пояснить, рас-
смотрев некоторые характерные примеры.
Жирные кислоты, н-первичные амины, мыла, соли аминов,
первичные спирты и многие другие одноосновные органические
соединения образуют слоистые кристаллы, на что уже обра-
щалось внимание в гл. 2. Для этих кристаллов характерно, что
их ионные свойства локализованы в плоскостях, параллельных
основным плоскостям. Эти плоскости чередуются с плоскостя-
ми, по которым соединяются концы молекул, не содержащие
ионных или водородных связей. Так как ионные и водородные
связи значительно прочнее остаточных, кристаллы этого типа
раскалываются, обнажая преимущественно слабые остаточные
связи. Как и молекулярные кристаллы, они образуют неполяр-
ные плоские грани. Торцовые грани этих кристаллов имеют как
полярные, так и неполярные обнаженные связи, но так как для
образования единицы поверхности таких граней требуется за-
тратить больше энергии, чем для образования той же поверх-
ности плоских граней, разрывы по этим граням составляют не-
значительную часть всей поверхности. Иными словами, при
ПОВЕРХНОСТИ РАЗДЕЛА f ВЕРДОЕ — ГАЗ, ТВЕРДОЕ - ЖИДКОСТЬ 119
дроблении слоистых кристаллов основная часть образующейся
поверхности неполярна и лишь небольшая часть ее полярна.
Кристаллы органических соединений, в которых каждая
«молекула» обладает двумя или большим числом полярных
групп, образуют бесконечные структуры. Каждый радикал та-
кой молекулы, обладая двумя или большим числом полярных
групп, связан по трем измерениям ионной или водородной
связью. Кристаллы этого типа имеют более высокую твердость,
не обладают совершенной спайностью слоистых кристаллов, и
их раскалывание невозможно без разрушения ионной или во-
дородной связи. При образовании поверхности твердого в за-
висимости От относительного числа разрушенных ионных или
водородных и неполярных связей поверхность этих кристаллов
в большей или меньшей степени флотационно активна.
ЛИТЕРАТУРА
1. Андреева А. И. Отделение борной кислоты из смеси солей, Совет-
ский патент 53403, 30 июня 1938 г.
2. Андреева А. И. и С. А. Кузин. Флотация борной кислоты и бора
'Из продуктов обработки индерских борацитов, Журнал прикладной хи-
мии, 10, 845—852, 1937, (
3. Bangham D. Н. The Gibbs adsorption equation and adsorption on
solids, Trans. Faraday Soc., 33, 805, 1937.
4. Bangham D. H. and R. I. Razouk. The wetting of charcoal and the
nature of the adsorbed phase formed from saturated vapours, Trans. Fara-
day Soc., 33, 1463—1471, 1937.
5. Bangham D. H. and R. I. Razouk. The swelling of charcoal. V. The
saturation and immersion expansions and the heat of wetting Proc. Roy
Soc. (London), A166, 572—586, 1938.
6. В a r r e r Richard N. Sorption processes on diamond and graphite, J. Chem.
Soc., 1936, 1256—1268.
7. Beebe Ralph A., John B. Beckwith and Jurgen M. H о n i g. The de-
termination on small surface areas by krypton adsorption at low temperatu-
res, J. Am. Chem. Soc., 67, 1554—1558, 1945.
8. Binder R. C. «Fluid Mechanics», 2d ed., pp. 91—93, Prentice-Hall, Inc.,
New York, 1949.
9. Bloecher, F. W., Jr. A new surface measurement tool for mineral engi-
neers, Mining Eng., 3, 255—259, 1951.
10. Boyd G. E. and H. K. Livingston. Adsorption and energy changes
at crystalline solid surfaces, J Amer. Chem. Soc., 64, 2383—2388, 1942.
11. Brunau er, Stephen. Physical Adsorption, I, The adsorption of gases
and vapors (a) pp. 151—155, Princeton University Press, Princeton N. Y.,
1943.
12. В r u n a u e r, Stephen, P. H. Emmett and E. Teller. Adsorption of
gases in multimolecular layers, J. Amer. Chem. Soc., 60, 309—319, 1938.
13. Brunner E. Reaktiongeschwendigkeit in heterogenen Systemen, Z. Phy-
sik. Chem., Bd. 47, 56—102, 1904.
14. Chang C. S. Adsorption of barium and laurate ions on quartz, thesis
for the doctorate, Massachusetts Institute of Technology, 1951.
15. Clemmer J. Bruce, Richard W. Smith, В. H. Clemmons, and
R. H. Stacey. Flotation of weathered Alabama graphite schists for cru-
cible flake, Geol. Survey Alabama Bull., 49, pp. 71—91, 1941.
120
ГЛАВА 4
16. Gandrud В. W., G. D. G о e, C. S. Benefield and I. N. Skelton.
The flotation of Alabama graphite ores, U. S. Bur. Mines, Rept. Invest.
3225, 1934.
17. Gartland J. W. A high temperature electric tube furnace, Trans. Ele-
ctrochem. Soc., 88, 121—132, 1945.
18. G a u d i n A. M., P. L. de Bruyn, F. W, Bloecher and C. S. Chang.
Radioactive tracers in flotation, Mining and Met., 29, 432—435, 1948.
19. Gaud in A. M. and F. W. Bloecher. Concerning the adsorption of
dodecylamine on quartz, Trans. AIMME, 187, 499—505, 1950.
20. Gaud in A. M. and F. W. В о w d i s h. Surface measurement by van der
Waals adsorption, Trans. AIMME, 169, 95—102, 1946.
21. Gau din A. M. and P. L. de Bruyn. Radioactive tracers.in mineral en-
gineering problems and particularly in flotation, Can. Mining Met. Bull.,
42, 331—337, 1949.
22. Gaudin A. M. and R. T. Hukki. Prinsiples of comminution-size and
surface distribution, Trans. AIMME, 169, 67—87, 1946.
23. Gaudin A. M. and Gustav S. P r e 11 e r. Surface areas of flotation con-
centrates and the thickness of collector coatings, Trans. AIMME, 169, 248—
258, 1946.
24. G a u d i n A. M. and Alfonso R i z o-P a t r 6 n. The mechanism of activa-
tion in flotation, AIMME Tech. Publ., 1453, 1942.
25. Gross John and S. R. Z i m m e r 1 e y. Crushing and grinding. I. Surface
measurement of quartz particles, Trans. AIMME, 87, 7—26, 1930.
26. Harkins, William D. and George Jura. Surfaces of solids, XII, XIII,
J. Am. Chem. Soc., 66, 1362—1366, 1944, 1366—1373, 1944.
27. Harkins William D. and George Jura. The surfaces of solids and li-
quids and the films that form upon them, III, in Jerome Alexander, ed.
«Colloid Chemistry», VI, I—76, Reinhold Publishing Corporation New
York, 1946.
28. Knickerbocker R. G. and F. K. Shelton. Beneficiation of boron
minerals bv flotation as boric acid. U. S. Bureau of Mines, Rep Invest
3525, pp. 3—13, 1940.
29. Lamb Frank D. and John Ruppert. Flotation of a North Carolina
pyrophyllite ore, U. S. Bur. Mines, Rept. Invest. 4674, 1950.
30. Langmuir Irving. The adsorption of gases on plane surfaces of glass,
mica and platinum, J. Am. Chem. Soc., 40, 1361—1403. 1918.
31. Livingston Herbert Klossner. Adsorption and the free surface energy
of solids, thesis for the doctorate, University of Chicago, 1941.
32. Noyes Arthur A. and Willis R. Whitney. Lieber die Auflosungsge-
schwindigkeit von festen Stoffen in ihren eigenen Losungen, Z Physik.
Chem., 23, 689—692, 1897.
33. Ostwald Wolfgang. Introduction to Theoretical and Applied Colloid
Chemistry, traslated by Martin Henry Fisher, John Wiley and Sons Inc
New York, 1917.
34. Ralston Oliver C. Flotation and agglomerate concentration of non-
metallic minerals, U. S. Bur. Mines, Rept. Invest., 3399, 1938.
35. Shelton F. K. Autoflotation of boric acid, as for separation from colema-
nite ore mineral, U. S. Patent 2317413/1943.
36. S t r a n s к i I. N. and K. Moliere. Ueber die Strukturabweichungen in
der Oberflachen von lonenkristallen, Z. Physik. 124 421—428 429—440-
127, 168—186, 1950. ’ ’
37. T a g g a r t A. F. Handbook of mineral dressing, Sec. 12, pp. 129—430,
John Wiley and Sons, Inc., New York, 1945.
38. Taggart Arthur F., G. R. M. del G i u d i с e. A. M. Sadler and
M. H a s s i a 1 i s. Oil-air separation of non-sulphide and non-metal mi-
nerals, Trans. AIMME, 134, 180—206, 1939.
ПОВЕРХНОСТИ РАЗДЕЛА ТВЕРДОЕ — ГАЗ, ТВЕРДОЕ — ЖИДКОСТЬ 121
39. Taylor Hugh S. A Treatise on Physical Chemistry, 11(a), pp. 1028—
1032, (b) pp. 1030—1031, D. van Nostrand Company, Inc., New York, 1930.
40. Tu winer S. B. and S. Korman. Flotation of chalcocite, Trans. AIMME
187, 217—222, 1950.
41. Van Name R. G. In International Critical Tables, V, pp. 55—60,
McGraw-Hill Book Company, Inc., New York, 1929.
42. V e r w e у E. J. W. Lattice structure of the free surface of alkali halide
crystals, Rec. Trav. Chim., 65, 521—528, 1946.
43. Weyl W. A The role of ionic deformation in surface chemistry, Trans
N. Y. Acad. Sci., 12, 245—256, 1950.
44. Williams Alpheus F. The Genesis of the Diamond, pp. 11—13, Er-
nest Benn, Ltd, London, 1932.
45. Wooten L. A. and C. Brown. Surface area of oxide coated cathodes by
adsorption of gas at low pressures, J. Am. Chem. Soc., 65, 113—118, 1948.
46. Z a c h a r i a s e n W. H. The crystal lattice of boric acid BO3H3, Z. Krist..
88, 150—161, 1934.
Глава 5
ЭЛЕКТРИЧЕСКИЕ ЯВЛЕНИЯ НА ПОВЕРХНОСТЯХ
РАЗДЕЛА ФАЗ
До сих пор мы рассматривали адсорбцию, не принимая во
внимание изменение электрических свойств поверхности. Одна-
ко, если адсорбция имеет ионный характер, то преимуществен-
ная концентрация ионов на поверхностях раздела вода —воздух
или вода — минерал должна обусловливать появление электри-
ческого заряда на поверхности раздела фаз. Так, например, на
поверхности раствора бутирата натрия должны адсорбировать-
ся ионы бутирата, который обладает значительно меньшим по-
верхностным натяжением по сравнению с водой и находится в
растворе в основном в ионизированном состоянии. Это означа-
ет, что поверхностный слой раствора заряжен отрицательно.
По мере увеличения концентрации растворенного вещества и
его адсорбции величина заряда этого слоя возрастает до опре-
деленного значения. Отсюда следует, что катионы концентри-
руются в слое жидкости, который непосредственно примыкает
к ее поверхности. Рис. 1 дает качественное представление о рас-
пределении ионов у поверхности раздела фаз.
Если раствор содержит поверхностно активные ионы, т. е.
такие ионы, которые могут положительно адсорбироваться на
поверхности раздела вода — воздух, то концентрация как этих
ионов, так и каких-либо противоионов вблизи поверхности бу-
дет выше, чем в объеме раствора. Наоборот, если находящиеся
в растворе ионы не являются поверхностно активными, то кон-
центрация ионов в поверхностном слое будет ниже, чем в объ-
еме
Те же рассуждения, что и в случае поверхности раздела во-
да — воздух, можно применить и для случая поверхности раз-
дела вода — минерал, но при этом появляется дополнительное
усложнение, которое состоит в том, что возможен обмен ионов
между поверхностью минерала и раствором, причем обменива-
ются либо ионы кристаллической решетки, либо ионы, ранее ад-
сорбированные твердой фазой.
В частном случае, изображенном на рис. 1, слой жидкости у
поверхности раздела вода — воздух имеет избыток отрицатель-
ных ионов, тогда как слой жидкости под ним имеет избыток по-
ложительных ионов. Безусловно, расположение ионов получит-
ЭЛЕКТРИЧЕСКИЕ ЯВЛЕНИЯ НА ПОВЕРХНОСТЯХ РАЗДЕЛА ФАЗ
123
ся обратным, если углеводородный радикал будет входить в со-
став не аниона, а катиона.
Даже простейшая система минерал — водный раствор, со-
стоящая из одной частицы, окруженной раствором, включает
Концентрация ионов
Воздух ^Поверхность
Поверхностный спой
Расстояние
от поверхнос-
ти
Концентрации
в объеме
Адсорбированные ионы
(напри мер., бутирата)
Противоионы (например, натрий)
Рис. 1. Качественная схема поверхности раздела раствор — воз-
дух для водного раствора бутирата натрия
двойной электрический слой, который содержит адсорбирован-
ные ионы и их противоионы в слое жидкой фазы вблизи поверх-
ности. Электрические свойства суспензии, в состав которой вхо-
дит большое число частиц одного и того же минерала, совер-
шенно одинаковых по форме, размеру и строению, более слож-
ны, поскольку здесь уже имеется множество идентичных двой-
ных слоев. Если же мономинеральная суспензия состоит из ча-
стиц минерала, отличающихся по форме и размеру, то воз-
никает дальнейшее усложнение системы электрических слоев,
так как в этом случае двойные слои скорее похожи, чем тожде-
ственны.
В полиминеральной суспензии, состоящей из частиц разных
размеров (например, измельченная руда), переплетаются раз-
нообразные электрические двойные слои, которые изменяются
при переходе от частицы к частице, даже если система достигла
состояния равновесия. Наконец, система электрических слоев
в водной суспензии руды, в которую вводятся воздушные пу-
зырьки с постоянно обновляемой поверхностью, усложняется в
еще большей степени благодаря разнообразию и взаимодейст-
вию различных двойных слоев.
124
ГЛАВА 5
Электрокинетические явления на поверхности раздела
твердое — жидкость
Вследствие существования двойного электрического слоя на
поверхности раздела фаз при наложении на систему электриче-
ского поля возникает движение дисперсной фазы относительно
дисперсионной среды или наоборот. Это движение может при-
нимать различные формы. Если дисперсная фаза представлена
частицами, капельками или пузырьками, находящимися в воде,
то перемещаться будет дисперсная фаза либо к аноду, либо к
катоду в зависимости от знака заряда. Это явление известно
под названием электрофореза, или катафореза. Если дисперс-
ная фаза представляет собой диафрагму, имеющую капилляр-
ные отверстия, то перемещаться будет жидкость относительно
диафрагмы. Это явление носит название электроосмоса. В обо-
их случаях наблюдается перемещение закрепленных на поверх-
ности раздела ионов относительно жидкости, причем в случае
катафореза дисперсная фаза (твердая, жидкая или газообраз-
ная) движется вместе с закрепленными ионами.
Так как жидкая фаза обладает вязкостью, то сила трения
противодействует электрической силе. В результате быстро
устанавливается постоянная скорость движения. Практически
скорость движения достигает постоянного значения в начале
наблюдений.
Противоионы находятся в диффузной части двойного элек-
трического слоя, которая в случае разбавленных растворов име-
ет толщину порядка нескольких молекулярных слоев, так что не
вся электродвижущая сила приложена на одинаковом расстоя-
нии от поверхности раздела.
В результате градиент скорости внутри двойного слоя не яв-
ляется постоянным, как это должно было бы быть, если исхо-
дить из предположения, что все противоионы концентрируются
на вполне определенном расстоянии от поверхности раздела. В
действительности градиент скорости в двойном слое изменяется
постепенно при удалении от поверхности раздела в бесконеч-
ность (т. е. в объем раствора). Практически бесконечностью
можно считать расстояние от 50 до 5000 А от поверхности раз-
дела (в зависимости от ионной силы электролита). На рис. 2
эти качественные выводы представлены в виде диаграммы.
Вполне понятно, что градиентом скорости является наклон кри-
вой, выражающей зависимость скорости от расстояния до по-
верхности раздела.
В стеклянном капилляре или в промежутках между части-
цами песка размеры каналов обычно в значительной степени
превосходят толщину двойного электрического слоя, в связи с
чем скорость течения жидкости между стенками канала, обус-
ЭЛЕКТРИЧЕСКИЕ ЯВЛЕНИЯ НА ПОВЕРХНОСТЯХ РАЗДЕЛА ФАЗ
125
ловленную электрическим полем, можно Считать постоянной.
Однако подобное допущение является лишь первым приближе-
нием.
Если жидкость продавливается через пористую диафрагму,
состоящую из твердых частиц, или через капилляр, то между
электродами, помещенными у концов диафрагмы, возникает
разность потенциалов. Эта разность потенциалов известна под
Рис. 2. Зависимость электрокинетической скорости от расстояния до
границы раздела фаз
оо
названием потенциала протекания. Если же твердая фаза осе-
дает в жидкости, то возникает потенциал седиментации. Эти че-
тыре явления 14а] — электрофорез, электроосмос, потенциал
протекания и потенциал седиментации — зависят от одного и то-
го же фактора, а именно, — от избирательного распределения
ионов у поверхности раздела фаз.
Двойной слой. На рис. 3 схематически изображена сфериче-
ская частица небольшого размера, находящаяся в водном раст-
воре. Мы можем различить твердую частицу (зона ОА), слой
закрепленных ионов и гидратный слой (зона АВ), водный слой,
содержащий диффузно распределенные противоионы (зона
ВС), и объем жидкости (зона С). Плоскость скольжения меж-
ду частицей и жидкостью находится не у точки Л, а у точки В.
Потенциал ф , определяющий работу переноса единицы заряда
из бесконечности (из объема раствора) в точку, находящуюся
вблизи частицы, равен нулю в точке С, но быстро возрастает по
мере приближения к точке В. Потенциал в этой точке известен
под названием электрокинетического, или ^-потенциала.
126
ГЛАВА 5
Считается, что -слой закрепленных ионов по толщине должен
быть моноионным. Если не рассматривать органические ионы
Рис. 3. Зависимость потенциала от
расстояния до поверхности раздела
для сферической коллоидной части-
цы
с длинными углеводородными
цепями, то толщина этого слоя
О
составляет приблизительно 3 А.
Диффузная часть двойного
слоя имеет значительную тол-
щину, особенно в случае раз-
бавленных растворов. Алек-
сандер и Джонсон [4Ь] отожде-
ствляют толщину диффузной
части двойного электрического
слоя 8 со средним радиусом
ионной атмосферы, окружаю-
щей каждый ион в растворе
электролита. Эта величина
равна обратной величине функ-
ции. Дебая—Гюккеля %, кото-
рая, как обычно считают, явля-
ется одной из основных вели-
чин, широко используемых в
современной теории электроли-
тов:
б
1000 DRT
8тг e2N2
(I)
где е — заряд электрона, равный 4,77 • 10~10 абсолютных элек-
тростатических единиц;
Л/ — число Авогадро, равное 6,023 - Ю23;
с — ионная сила или концентрация одно-одновалентного
электролита, лоль/л;
D — диэлектрическая постоянная, равная примерно 80 для
воды при комнатной температуре;
7? — газовая постоянная (8,314 107);
Т — абсолютная температура ° К (Т = 298 при 25° С).
Функцию Дебая — Гюккеля х находят, применив законы ста-
тической механики к ионной атмосфере, окружающей рас-
сматриваемый ион в растворе электролита [19, 44, 481. Вывод
уравнения (1) можно найти в книге Харнеда и Оуэна [34а].
Подставив численные значения в уравнение (1), мы получим
уравнение, определяющее 6 в ангстремах, при концентрации в
миллимолях на литр, в виде
. 97
(1а>
ЭЛЕКТРИЧЕСКИЕ ЯВЛЕНИЯ НА ПОВЕРХНОСТЯХ РАЗДЕЛА ФАЗ
127
Следует отметить, что 6 изменяется обратно' пропорциональ-.
но корню квадратному из ионной силы раствора; соответствен-
но этому в концентрированных растворах двойной слой очень,
тонок, в то время как в разбавленных растворах он распростра-
няется на относительно большие расстояния. В молярном раст-
воре 6~ 3 А или приблизительно равняется диаметру молеку-
лы воды. В миллимолярных растворах, которые обычно ветре-
О
чаются во флотационных системах, 6 = 97 А. В микромолярных
растворах, т. е. в растворах, приближающихся к дистиллирован-
ной воде, 6 = 3050 А. Этот простой пример показывает, что тол-
щина диффузного слоя может изменяться от 1 до 1000 атомных
диаметров, причем толщина эта значительно увеличивается по.
мере разбавления раствора.
Уместно упомянуть, что отождествление величины 6 с «тол-
щиной» двойного слоя в значительной степени является вопро-
сом определения понятия толщины. Первым ввел понятие двой-
ного электрического слоя Гельмгольц 135], который использовал
концепцию плоско-параллельных обкладок, подобных обклад-
кам конденсатора. Гуи и Чапмэн [17] показали, что такое пред-
ставление является слишком упрощенным. Вместо концепции
Гельмгольца они ввели понятие о диффузной части двойного
электрического слоя. Под толщиной двойного электрического,
слоя (в том смысле, в котором этот термин используется здесь)
понимается расстояние между обкладками плоско-параллельно-
го конденсатора той же емкости, что и реальный электрический
слой. Уместно также второе предположение, заключающееся
в следующем: если плоские поверхности или большие сферы
окружены относительно тонким двойным электрическим слоем,
то характер падения потенциала по мере увеличения расстояния
должен быть иным, чем при малых сферах, окруженных тол-
стыми двойными электрическими слоями. Именно последний
случай рассматривается в развитой Дебаем и Гюккелем теории
электролитов, где центральный ион представляется в виде точеч-
ного заряда. Коллоидные системы приближаются к ионным, так
как частицы дисперсной фазы в них весьма малы немеют сфе-
рическую форму. Этого нельзя сказать о минеральных суспензи-
ях, отклонения которых от предпосылок Дебая — Гюккеля весь-
ма велики. Уменьшение плотности заряда двойного электриче-
ского слоя по мере удаления от поверхности грубодисперсных
частиц происходит медленнее, чем в случае мелких частиц или
точечных зарядов 111.
Концентрация ионов в диффузной части двойного слоя. Вы-
ше уже было показано качественное изменение плотности ионов
в диффузном слое. Количественное решение этого вопроса было
предложено Абрамсоном Hal. Это решение основано на исполь-
128
ГЛАВА 5
зовании уравнения Пуассона, согласно которому оператор Лап-
ласа для потенциала V • V Ф пропорционален плотности заря-
да р и обратно пропорционален диэлектрической постоянной D:
V ?Ф= —
4тгр
То
(2)
Предполагая, что частица настолько велика по сравнению с
толщиной диффузной части двойного слоя, что ее можно рас-
сматривать как частицу с плоской поверхностью, Абрамсон на-
шел следующее выражение для плотности ионов:
D , Г х 1
р =-----------% ехр----------,
4тг82 [ 8 J
(3)
где — эквивалентная толщина двойного слоя;
%— потенциал у границы поверхности;
х — расстояние от этой границы до данной точки в жид-
кости.
Плотность ионов р экспоненциально уменьшается с х, стано-
вясь равной Me части значения при х = 6 или также при х = 0.
Если х= 26, то р уменьшается в 1/е2 раз, как и при х = 0.
Из уравнения (3) видно, что вблизи поверхности, покрытой
закрепленными ионами, плотность противоионов наибольшая
Однако не мало противоионов находится и в жидкости. Так, да-
же на расстоянии х — 46 концентрация противоионов составля-
ет еще около 2% от концентрации вблизи поверхности частицы
(л‘ = 0). Очевидно, 5 не представляет собой расстояние, внутри
которого заключены все противоионы; это есть некоторое эффек-
тивное расстояние, физический смысл которого рассмотрен
выше.
Для случая сферических частиц, размер которых мал по
сравнению с 5, уравнения будут более сложными (см. у Абрам-
сона). Детальное рассмотрение плоского и сферического двойно-
го электрического слоя можно найти в заслуживающей внима-
ния книге Фервея и Овербика 156].
Электрокинетический, или ^-потенциал. Если частицу (см.
рис. 3) поместить в электрическое поле, то она начинает переме-
щаться в определенном направлении, диффузная же часть двой-
ного слоя — в противоположном. Однако вокруг частицы все
время будет образовываться новый диффузный слой, поэтому и
в состоянии движения, как и в состоянии покоя, она будет окру-
жена аналогичным диффузным слоем. Вязкость жидкости ока-
зывает противодействие приложенному электрическому полю,
так что эти две силы уравновешиваются. В однородном поле
электрокинетическая скорость постоянна. Численное значение
этой скорости («) можно получить, используя те же рассужде-
ния, что и при выводе уравнения Стокса для оседания частиц
[27а, 52]. Вывод уравнения для оценки электрокинетического по-
ЭЛЕКТРИЧЕСКИЕ ЯВЛЕНИЯ НА ПОВЕРХНОСТЯХ РАЗДЕЛА ФАЗ
129
тенциала приводится во многих книгах 11, 4, 12]. Наиболее удач-
но этот вопрос изложен у Гортнера [ЗОЬ].
Если предположить, что все ионы диффузной части двойного
слоя расположены на расстоянии 6 от слоя закрепленных ионов
(см. пунктирную линию на рис. 2), то сила трения на единицу
площади поверхности раздела равна иц/6, где и — объемная
скорость течения жидкости относительно частиц, а р—вязкость
жидкости. Движущая сила на единицу поверхности раздела
равна оЕ, где о — заряд на единицу поверхности1 * * * * * * * 9, а Е — прило-
женная разность потенциалов на единицу длины, или градиент
потенциала в направлении движения.
В условиях равновесия эти силы равны:
(4)
Если двойной слой подобен электрическому конденсатору с
емкостью С на единицу площади, то
С = -р (5)
где 5— электрокинетический потенциал.
С можно выразить также через диэлектрическую постоян-
ную среды D и расстояние между обкладками конденсатора 6:
с = -^—. ’ (6)
4л8
Объединяя уравнения (5) и (6), получаем
: = 7)
D '
или, объединяя оба эти уравнения с уравнением (4), получаем:
1 Следует отметить, что заряд на единицу поверхности о отличается от
заряда на единицу объема в диффузной части двойного слоя ₽, оценка кото-
рого дана в предыдущем параграфе и который определяется уравнением (3).
Величина о, введенная в настоящем разделе, является плотностью заряда
прочно закрепленных ионов. Разумеется, эти величины связаны между собой.
В случае электронейтральной системы
х=со
А о = J р dV,
х=0
где. А — площадь;
Р — объем.
Предполагается, что интегрирование производится с учетом геометрии,
системы.
9 А. XI. Годэн
130
ГЛАВА 5
Все величины в этих уравнениях должны быть выражены
в одной системе единиц (например, в электростатических едини-
цах). Если и выражено в сантиметрах в секунду, jx—в пуазах
(или в дин!сек на 1 см2), то £ и Е будут выражены в абсолют-
ных электростатических единицах потенциала и абсолютных
электростатических единицах на сантиметр соответственно. Если
£ и Е должны быть выражены в вольтах и вольтах на сантиметр
соответственно, то следует их значения умножить на 300 [27b, 54а].
Если \ выразить в милливольтах, и — в микронах в секунду
и подставить обычные значения D для разбавленных водных рас-
творов при температуре 20° (D = 80 и) ji = 0,01, то получим
С =141— . (8а)>
Е
Потенциал поверхности и ^-потенциал. Как установили Фер-
вей и Овербик 156а], причиной возникновения двойного электри-
ческого слоя является равновесное распределение потенциал
определяющих ионов между поверхностью частицы и жид-
костью. Это было найдено благодаря тщательной и скрупулез-
ной работе с золем йодистого серебра [10].
Положим, что Со — концентрация потенциал определяющих,
ионов (в случае йодистого серебра — ионов серебра или иода),
отвечающая нулевому значению термодинамического потенциа-
ла тро, а % — термодинамический потенциал, соответствующий
концентрации сп. Хорошо известно, что
= —1п^, (9)
е сп
где с—концентрация;
k — постоянная Больцмана;
Т — абсолютная температура;
е — заряд электрона;
ф — термодинамический потенциал в абсолютных электро-
статических единицах.
В случае, когда значение с0 выбрано таким, что = 0, урав-
нение примет следующий вид:
= 0,058 1g в. (10)
Может возникнуть искушение отождествить величины ч>я и
К сожалению, это совершенно неправильно. Величины эти не
равны, хотя и связаны между собой, причем абсолютная вели-
чина £ меньше абсолютной величины %. Фервей и Овербик по-
лагают, что различие между £ и обусловлено образованием
у самого основания двойного электрического слоя (рядом со
слоем потенциал определяющих ионов) слоя закрепленных про-
тивоионов (таких же, как ионы, находящиеся в диффузной ча-
ЭЛЕКТРИЧЕСКИЕ ЯВЛЕНИЯ НА ПОВЕРХНОСТЯХ РАЗДЕЛА ФАЗ 131
сти двойного слоя). Это согласуется с точкой зрения Штер-
на [51] и схематически изображается на рис. 4. Некоторые по-
лагают, что это различие можно связать с наличием гидратного
слоя, в основном обусловливающего теплоту смачивания твер-
дых тел. Вопрос о связи и ? относится к вопросам коллоид-
ной химии, и на него необходимо обратить внимание.
Рис. 4. Схематическое изображение
структуры двойного слоя в растворе
электролита и распределение потенциа-
ла в двойном слое [28]:
А— потенциал определяющие ионы; В — слой
прочно закрепленных противоионов — слой
Штерна; С — диффузный слой противоионов
(слой Гун); / — потенциал определяющий
ион; 2 — гидратированный противоион; 3 —
противоионы противоионов
Электрический момент двойного слоя. Булл и Гортнер [14]
указывают, что весьма характерной величиной является произ-
ведение плотности заряда в закрепленном слое ст на толщину
двойного слоя 6, отождествляемую со средним радиусом ион-
ной атмосферы, как упоминалось об этом выше. Эту величину
<т6 можно рассматривать как удельный электрический момент
двойного слоя.
Уравнение (7) показывает, что электрический момент пря-
мо пропорционален ^-потенциалу. Чтобы выразить электриче-
ский момент посредством измеряемых и известных величин,
уравнение (4) можно переписать следующим образом:
□3 = 2LL
Е
(Н)'
Гугенгейм [32] заходит настолько далеко, что считает про-
изведение об более важной характеристикой электрокинетиче-
ских явлений, чем ^-потенциал. Еще более настойчив в этом от-
ношении Вуд [57], хотя он и расходится во мнениях с Гугенгей-
мом при толковании этого вопроса.
Все формулы для электрокинетического потенциала содер-
жат величину диэлектрической постоянной. В водной среде, где
концентрация ионов вблизи поверхности может быть- относи-
тельно высокой, использование диэлектрической постоянной во-
ды вместо значения диэлектрической постоянной в двойном слое
9*
ГЛАВА 5
IM
до некоторой степени сомнительно, особенно если значение д
мало. Однако никакой иной выбор величины D не может быть
рправдан. Формулы для электрического момента и для оценки
плотности заряда поверхности не содержат диэлектрической по-
стоянной и благодаря этому менее сомнительны.
Плотность ионов в прочно закрепленном слое. Так как об
можно рассчитать из уравнения (11), а 6 оценить из уравнения
(1а), то поверхностную плотность заряда в закрепленном слое
можно рассчитать из уравнения.
g = 3,25 • 1Q7 “ с- , (12)
где величина и выражена в см/сек, и — в пуазах, с — в моль/л,
Е — в абсолютных электростатических единицах на 1 см, а о—
в'абсолютных электростатических единицах количества элект-
ричества на 1 см2. Е обычно измеряется в вольтах на санти-
метр, и если 6 должно выражать количество одновалетных
ионов в закрепленном слое, то необходимо использовать множи-
тели 300 и 1/(4,77 • 10~10). Тогда уравнение примет следующий
вид:
g — 2,04 • Ю19 “ с электронных зарядов на 1 см2. (12а)
ГН- Е
• В частном случае, когда и — 0,002 см!сек, ц = 0,01 пуаз,
Е'-^ 10 в!см, с = 10—2-м., то 6 = 4,08 1012 одновалентных ионов
на 1- см2, что составляет приблизительно Г% моноионного слоя,
е^л'и считать, что на каждый ион приходится площадь 25 А2.
В том случае, когда данные уже выражены через К и с,
удобнее рассчитывать <т из этих величин. Комбинируя уравне-
ния (8) и (12), получаем
: g=l,42- lOi^j/T, (13)
рде,величина £ выражена в вольтах, с — в моль/л, а ст—в еди-
ницах заряда электрона на 1 см2.
.Согласно предварительным данным Сана (531, полученным
методом микроэлектрофореза (см. ниже), для кварца в 10~2-м.
растворе NaOH 5 = —52 мв. Следовательно, <т = 7,4 • 1012 заря-
дов на 1 см2. Для образования на поверхности моноионного слоя
требуется гораздо большее количество зарядов на квадратный
сантиметр (скажем, 4 • 1014). Не настаивая на точности пред-
варительных данных Сана, все же отметим, что рассчитанное
количество закрепленных на поверхности ионов составляет не-
значительную часть тех ионов, которые могли бы разместиться
на.этой поверхности. Возможно, что на границе раздела места
ЭЛЕКТРИЧЕСКИЕ ЯВЛЕНИЯ НА ПОВЕРХНОСТЯХ РАЗДЕЛА ФАЗ 133
занимают ионы разных знаков в почти одинаковых количествах,
и полученные данные относятся только к избытку ионов одного
знака над ионами другого знака, в противном случае математи-
ческая обработка включает в себя множество необоснованных
предположений. Этот вопрос представляет собой обширное по-
ле для дальнейших исследований. В таблице представлены не-
которые результаты, основанные на данных Сана [53а].
Плотность зарядов в закрепленном слое на пирите в растворах соляной '
кислоты и едкого натра различной концентрации [55]
Величина PH С-потенциал Концентрация ионов с (рассчитана из pH) ст заряд исна/см2 Кажущееся заполнение ионного слоя %
3 +31 ю“3 + 14O-1O10 0,3
5 +9 ю“5 4-4-Ю10 0,01
7
6,3 0 5-10 0 0
— 7 „ „10
7 — 14 10 -0,6-10 0,001
9 —20 10 “5 -8-Ю10 0,02
11 —24 10 “3 —100-ю10 0,2
Методы определения электрокинетического потенциала и
удельного заряда. Для определения величины Ъ или <т можно
использовать любое из четырех основных электрокинетических
явлений. ' :
При электрофорезе и электроосмосе измеряют обусловленные
определенным электрическим полем механические эффекты
(скорость движения частиц в суспензии или скорость протека-
ния жидкости через диафрагму). Методы потенциала протека-
ния и потенциала седиментации основаны на измерении элект-
рической разности потенциалов, возникающей за счет механи-
ческих эффектов (протекание раствора через диафрагму, со-
стоящую из частиц, оседание частиц в растворе). В настоящее
время первый метод пользуется большим вниманием химиков-
коллоидников, работающих обычно с гораздо меньшими по раз-
меру частицами, чем обогатители. Для обогатителей же наибо-
лее удобным методом при изучении свойств минеральных частиц
является метод потенциала протекания [26, 28].
Метод электрофореза. Типичным примером применения ме1-
тода электрофореза является метод Бертона с U-образной труб-
кой [16]. В эту трубку с находящейся в ней суспензией или кол-
134
ГЛАВА 5
Рис. 5. Общий вид макроэлектрофо-
ретической ячейки Тизелиуса (по
Александеру и Джонсону):
1 и 2 — стеклянные трубки; 3 и 4 --
электроды; 5 и б — краны
лоидным раствором осторожно с обоих концов вводят раствор
того же состава, что и электролит, содержащийся в суспензии.
При пропускании электрического тока граница между суспен-
зией и залитым раствором электролита перемещается. Скорость
перемещения границы принимается за электрофоретическую ско-
рость и используется в уравнениях для расчета электрокинети-
ческого потенциала или плотности заряда.
Метод макроэлетрофореза неоднократно совершенствовал-
ся и достиг высокой степени развития в приборе Тизелиуса [55].
Этот прибор, схематически изо-
браженный на рис. 5, детально
описывается в довольно рас-
пространенных книгах Ис, 30с1.
Он состоит из маленькой U-об-
разной трубки, прямоугольной
в сечении и сделанной так, что
верхняя часть ее может сме-
щаться по шлифованной по-
верхности нижней части труб-
ки в горизонтальном направле-
нии. U-образная трубка соеди-
няется с другой частью прибо-
ра, содержащей обычные хлор-
серебряные электроды, при по-
мощи которых к трубке можно
подвести желаемую разность
потенциалов, вплоть до 500 в.
Вначале суспензию вводят
в нижнюю часть U-образной
трубки с таким расчетом, что-
бы уровень ее поднялся выше
плоскости, по которой эта
часть может смещаться. Затем
нижнюю часть U-образной трубки смещают и верхние ответвле-
ния прибора промывают. После промывки верхние части напол-
няют электролитом, соприкасающимся с исследуемой суспен-
зией и с раствором хлористого калия, в который опущены элек-
троды. При этом для сохранения гидростатического равновесия
следует соблюдать некоторые предосторожности. После того как
установится температурное равновесие (весь прибор термоста-
тируется), приводят все его части в рабочее положение, сдвигая
в прежнее положение нижнюю часть U-образной трубки.
Прибор имеет много интересных особенностей. Для точной
оценки положения границы раздела применяется специальный
оптический метод, позволяющий наблюдать границу раздела
соприкасающихся сред с различными коэффициентами прелом-
ЭЛЕКТРИЧЕСКИЕ ЯВЛЕНИЯ НА ПОВЕРХНОСТЯХ РАЗДЕЛА ФАЗ
135
ления. Это особенно важно в случае белковых систем, которые
изучал Тизелиус, так как белковые суспензии прозрачны и не
окрашены.
Преимуществом метода микроэлектрофореза является его
простота. На микроскопе монтируется маленькая электрофоре-
тическая трубка. На ее концах располагаются обратимые элект-
роды (например, Си — CuSO4 или Ag — AgCl), отделенные от
.ее главной части пористыми перегородками (рис. 6). Движение
частиц наблюдают под микроскопом.
Рис. 6. Цилиндрическая микроэлектрофоретическая
ячейка (по Александеру и Джонсону):
1 — место наблюдения; 2 — обратимый электрод; 3 и 4 — стек-
лянная вата
Однако этот метод вызывает некоторые возражения. Дело в
том, что жидкость, которая движется при наложении электри-
ческого поля в одну сторону, должна каким-либо способом воз-
вращаться обратно, в результате чего в трубке возникает цир-
куляция жидкости. Поэтому, чтобы получить точные результа-
ты, необходимо производить измерения в строго определенной
части сечения электрофоретической трубки. Для трубки прямо-
угольного сечения этот уровень отстоит от верхней грани на
расстоянии, равном 21% высоты; для трубки круглого сече-
ния — на расстоянии 14% диаметра от стенки трубки. Даже
в случае идеальной трубки с плоскими отшлифованными стенка-
ми трудно выполнить точные измерения. Имеются и другие за-
труднения 129]. Одно из них связано с оптическими измерениями,
другое — с невозможностью устранить тепловые конвекцион-
ные потоки; и, наконец, твердые частицы, размерами даже 2—
3 р, достаточно быстро осаждаются и таким образом уходят из
области, пригодной для измерения.
Метод измерения потенциала протекания. Для изучения
систем, встречающихся в обогащении, особенно удобным ока-
зался метод измерения потенциала протекания. Этот метод в
основном был развит Крейтом, Бриггсом, Гортнером и Буллом.
Крёйт и Фрейндлих [21] разработали метод определения потен-
циала протекания для отдельных стеклянных капилляров.
Крёйт нашел, что отношение наблюдаемого потенциала проте-
кания Н к гидростатическому давлению Р, проталкивающему
раствор через капилляр, является постоянной величиной.
136
ГЛАВА 5
Бриггс [9] и Булл и Гортнер [13] распространили этот метод на
диафрагмы, состоящие из плотно упакованных твердых частиц.
Уравнение (8), выражающее зависимость электрокинетиче-
ского потенциала от скорости движения частиц, вязкости жид-
кости и градиента потенциала, можно переписать так, чтобы вы-
разить ^-потенциал посредством величин, измеряемых методом
потенциала протекания.
Уравнение примет следующий вид:
~ 4тгр. VA .
DI ' v
Как видно, уравнение выражает зависимость £ от некоторых
новых величин, а именно, от V — объема жидкости, проходящего >
через диафрагму в единицу времени, X—удельной электропро-
водности жидкости в порах диафрагмы и -силы тока I.
Иной вид уравнения получен Бриггсом:
£__ 4тгр. Е А
~ Бр ’
где Р — гидростатическое давление;
Е — наблюдаемый потенциал протекания.
Наиболее удобным для измерения потенциала протекания
является прибор Фюрстенау [26, 281, представляющий собой ви-
доизменение применявшихся ранее приборов [8, 39, 40]. Схема-
тически он изображен на рис. 7. Исследуемый электролит про-
пускают через диафрагму адсорбента (такого, например, как
порошок минерала) под заданным постоянным давлением. Раз-
ность потенциалов, возникающая между платиновыми электро- 7
дами, измеряется при помощи вибрационно-струнного электро- :
метра.
После дополнительного измерения электропроводности мож-
но рассчитать электрокинетический потенциал.
Хотя вывод уравнений (14) и (15) и указывает на то, что
электрокинетический потенциал не должен зависеть от размера
капилляров, имеются данные, противоречащие этому. В самом
деле, Булл и Гортнер [13] пришли к выводу, что существует зави-
симость ^-потенциала от размера частиц, из которых состоит '
диафрагма. Другие авторы нашли, что потенциал протекания
зависит от плотности упаковки диафрагмы. Этот фактор, оче-
видно, для жестких минеральных частиц менее важен, чем для
пластичных органических материалов. По-видимому, при выво- j
де уравнений были сделаны некоторые упрощающие предполо-
жения, которые впоследствии были забыты. Например, следует
напомнить, что в том случае, когда поры диафрагмы малы по
сравнению с характеристической толщиной 6 Дебая — Гюккеля,.
поток, протекающий через капилляры, кроме обычного гидроди-
намического сопротивления, обусловленного вязкостью, будет
ЭЛЕКТРИЧЕСКИЕ ЯВЛЕНИЯ НА ПОВЕРХНОСТЯХ РАЗДЕЛА ФАЗ 137
5
испытывать также электрокинетическое сопротивление. При вы-
воде уравнений этим фактором безусловно пренебрегли, хотя
уравнения и были предназначены для оценки электрокине-
тических явлений [11, 15, 30а]. Подробный анализ метода изме-
рения потенциала протекания был сделан Вудом [57].
Некоторые результаты по определению значений ^-потенциа-
ла. Несмотря на то, что проделано не мало определений ^-потенци-
ала, экспериментальные данные имеют-
ся для удивительно небольшого числа
твердых фаз. Большинство экспери-
ментов проводилось со стеклом или
плавленым кварцем. Кроме того, ре-
зультаты различных экспериментато-
ров расходятся, так что возникают
сомнения о возможности 'определения
абсолютных значений ^-потенциала.
Например, Фрейндлих [20а] сообщает,
что, согласно данным Квинке, ^-потен-
циал стекла в воде равен —53 мв, по
Терешину он равен —46 мв, а для
кварца в воде —44 мв. Определения
Лакса X. и И. Бичика [38] дали для
плавленого кварца —146 мв, а Вуда
[58] —177 мв. Все эти вещества доста-
точно сходны между собой, и для них
можно было бы ожидать большей со-
гласованности результатов определе-
ний.
Лучше согласуются между собой
результаты, полученные при примене-
нии одних и тех же приборов с одними
и теми же твердыми веществами, но с
различными электролитами. Обычно
^-потенциал уменьшается с увеличени-
ем концентрации электролита. Однако
кло'нений. Замечено, что эти отклонения связаны со специфиче-
ским взаимодействием иона электролита с ионами твердого ве-
щества, ^-потенциал которого определяется. Делаются попытки
найти связь между свойствами ионов (массой, размером, харак-
тером гидратации ионов), с одной стороны, и ^-потенциалом, —
с другой, при эквивалентных концентрациях этих ионов. Однако
по этому вопросу до сих пор нет ясных представлений.
Последние измерения Фюрстенау [28] дали несколько более
четкие результаты. На рис. 8 и 9 показано изменение £-потен-
йиала кварца с изменением природы и концентрации электро-
лита. В дистиллированной воде кварц заряжен отрицательно и
Рис. 7. Схема ячейки для-
изучения погтенциала про-
текания [26]:
1— литровая колба; 2—• 18/91
шарик и шарнирное соединение
иа трубках; 3 — край; 4 — 24/40
шлиф; 5 — диафрагма; 6 — сет-
чатый платиновый электрод;
7 — платиновая проволока, 8 —
вольфрамовый ввод
было найдено много от-
Рис. 8 ^-потенциал кварца в растворах четырех
различных электролитов [28]
Рис. 9. i^-потенциал кварца в растворах хлори-
стого натрия при различных рначениях pH:
7—pH = 3,74; 2-pH = 7; 3 - pH = 10 [28]
ЭЛЕКТРИЧЕСКИЕ ЯВЛЕНИЯ НА ПОВЕРХНОСТЯХ РАЗДЕЛА ФАЗ
139
имеет S-потенциал не ниже —72 лв; при отсутствии поверхност-
ной проводимости S-потенциал кварца может превосходить да-
же —150 мв; при pH = 3,7 он равен нулю, и в более кислых сре-
дах кварц становится положительно заряженным. Добавление
соли первичного амина с длинной цепью (например, соляно-
кислого лауриламина) вызывает очень сильное изменение S-по-
тенциала, который даже меняет свой знак уже при малых кон-
центрациях этого реагента. Измерение потенциала протекания
для корунда 1 показывает, что изоэлектрическая точка корунда
находится примерно при pH = 9,4, и, по-видимому, корунд во-
обще ведет себя совершенно иначе, чем кварц.
Электрокинетические явления на поверхности раздела
жидкость — газ 4
Электрокинетические явления на границе раздела жид-
кость — газ были известны много лет назад — по крайней мере
со времени опытов Квинке (1861 г.). Электрокинетические яв-
ления на этой границе раздела исследованы менее полно, чем
на границе раздела твердое — жидкость, ибо трудности экспери-
мента в этом случае весьма велики.
Метод движущегося пузырька. В ранних опытах Мак-Таггар-
та 141, 42] изучалось движение пузырьков в зависимости от
приложенной разности потенциалов. Прибор Мак-Таггарта пред-
ставляет собой цилиндрическую трубку около 1 дюйма в диамет-
ре, в которую вводится пузырек воздуха. Трубка довольно бы-
стро вращается, так что пузырек перемещается по направлению
к ее оси. Вдоль этой оси прикладывают разность потенциалов
и при помощи микроскопа с небольшим увеличением измеряют
скорость смещения пузырька вдоль оси трубки. Эта работа бы-
ла продолжена Олти [5, 6, 18], который использовал аналогич-
ный прибор.
Ранние опыты Мак-Таггарта показали, что скорость движе-
ния пузырьков под действием электрического поля не зависит
от их размера. Но это первоначальное заключение было сдела-
но на основании опытов, проводившихся с пузырьками прибли-
зительно одинаковых размеров (диаметр 0,6—0,16 мм). После-
дующие эксперименты опровергли этот вывод. Кроме того, не-
которая нечеткость получаемых результатов вызвана изменени-
ем размера пузырьков во время самого опыта: в одних случаях
пузырьки уменьшаются вследствие растворения газа в жидко-
сти, в других — увеличиваются вследствие выделения газа из
жидкости. До сих пор неясно, действительно ли наблюдаемая за-
висимость изменения скорости движения пузырька от его раз-
личное сообщение X. И. Моди, Массачусеттский институт технологии.
140
ГЛАВА 5
мера определяется именно размером пузырька. Возможно, что
она обусловлена «старением» поверхности пузырька или невы-
полнением условий движения по Стоксу, что и могло быть в не-
которых опытах. Не исключена возможность загрязнения по-
верхности пузырьков поверхностно активными веществами типа
жиров. Таким образом, вопрос количественной оценки этих экс-
периментов остается открытым.
Несмотря на это, были получены довольно интересные ре-
зультаты. Во-первых, пузырьки воздуха в дистиллированной
воде всегда заряжены отрицательно. Во-вторых, добавление
электролита изменяет заряд вплоть до того, что при некоторых ♦
условиях меняется его знак. Оказывается, что в электролитах,. *
изменяющих знак заряда пузырьков, при концентрациях, близких
к изоэлектрической точке, ^-потенциал зависит от размера пу-
зырька, причем с изменением величины пузырька может ме-
няться даже знак ^-потенциала.
В последние годы метод движущегося пузырька, напоминаю-
щий метод электрофореза при определении ^-потенциала па
границе раздела твердое—жидкость, был забыт. Ему предпочли,
методы измерения потенциала у плоской поверхности жидкости.
Из них в основном применяются три метода: 1) динамический
с концентрической колонкой; 2) статический ионизационный;
3) статический с вибрирующей пластинкой, или метод конден-
сатора. Эти методы позволяют измерять иные величины по
сравнению с методом движущегося пузырька, а именно: не '
электрокинетический (S-потенциал), а разность между потен-
циалами поверхностей (ip-потенциалами) двух растворов, один г
из которых используется в качестве эталона. Соотношение
между ?- и ip-потенциалом на границах раздела твердое — жид-
кость и жидкость—газ показано на рис. 10.
Динамический метод концентрической колонки. Сущность,
этого метода заключается в измерении разности потенциалов
между поверхностями двух растворов, текущих параллельно, а
именно, раствора эталона и изучаемого раствора. Один из рас-
творов течет вертикальной струей по оси трубки, а другой —
вдоль стенки этой же цилиндрической трубки. Оба раствора по-
средством солевых мостиков и платиновых электродов соеди-
няются с квадрантным электрометром (рис. 11). Этот метод
был впервые использован Биша и Блондло [7] и доведен до вы-
сокой степени совершенства Кенриком [37]. Метод широко при- ...”
менялся Фрумкиным и его школой [23, 25, 36].
Метод концентрической колонки дает не действительную
разность потенциалов на границе раздела раствор — воздух, а
скорее изменение разности потенциалов (Мр) при переходе от
стандартной (эталонной) поверхности к исследуемой. Как и при
использовании статических методов, которые будут описаны да-
ЭЛЕКТРИЧЕСКИЕ ЯВЛЕНИЯ НА ПОВЕРХНОСТЯХ РАЗДЕЛА ФАЗ
141
лее, посредством этого метода измеряют относительную величи-
ну — поверхностный потенциал.
Метод концентрической колонки имеет значительное преи-
мущество перед статическими методами: поверхность жидкости
Воздух Я
I Зле ктракине- у Потенциал
\тический илотповерхности Ф
Г £ -патенциалХ
Твердое тело
Рис. 10. Схематическое изображение соотношения с-
и Ар-потенциалов на
Рис. 11. Динамический метод
измерения Ар-потенциала иа
границе раздела газ — жид-
кость с помощью концентриче-
ской колонки [37, fig. 1]
поверхностях раздела
постоянно обновляется, благодаря чему снижается возможность
ее загрязнения. Но, с другой стороны, результаты измерения
зависят от электропроводности воздуха, которая должна быть
весьма малой. Это требование иногда трудно выполнить.
142
ГЛАВА 5
Результаты, полученные при измерении поверхностного по-
тенциала растворов динамическим методом. Интересны резуль-
таты, полученные Кенриком и в особенности Фрумкиным. На
основании их можно сделать следующие заключения:
1) неорганические соединения оказывают незначительное
влияние на потенциал раздела газ — жидкость; некоторые соли
и большинство кислот дают незначительный положительный
потенциал поверхности; некоторые же соли и большинство ще-
лочей дают отрицательный потенциал;
2) поверхностно активные органические соединения оказы-
вают большое влияние на потенциал поверхности, если они не
диссоциированы; в общем они дают большой положительный
потенциал (например, спирты, жирные кислоты, амины, эфиры,
фенолы); галогенсодержащие органические соединения, осо-
бенно полигалогенидные жирные кислоты, являются исключе-
нием: они дают значительный отрицательный потенциал;
3) органические соединения, способные диссоциировать, но
находящиеся в недиссоциированном состоянии, дают высокие
значения потенциала; в случае же их диссоциации не только
уменьшается абсолютная величина потенциала, но иногда даже
меняется его знак;
4) растворимые в воде органические соединения при увели-
чении их концентрации вызывают увеличение поверхностного
потенциала, причем максимальное значение потенциала дости-
гается в насыщенных растворах; для данной концентрации, не
соответствующей насыщению^ по мере увеличения длины угле-
водородной цепи влияние различных членов гомологического
ряда возрастает, что указывает на аналогию с правилом Траубе;
5) в случае насыщенных растворов различия во влиянии от-
дельных типов органических соединений на поверхностный по-
тенциал можно объяснить разницей их структур; различие же
между поведением соединений одного и того же гомологическо-
го ряда незначительно.
Судя по рис. 12, можно прийти к заключению, что органи-
ческие соединения оказывают более сильное влияние на поверх-
ностный потенциал по сравнению с неорганическими. На
рис. 13 приведены данные, характеризующие влияние различ-
ных членов одного и того же гомологического ряда на потенци-
ал раздела жидкость — газ.
Интересно сделать некоторые замечания по поводу выводов,
приведенных выше. Поскольку недиссоциирующие и диссоци-
ирующие органические соединения в условиях, исключающих
диссоциацию, дают примерно одинаковые значения поверхност-
ного потенциала, его существование не может быть обусловле-
но присутствием ионов. Однако это затруднение можно разре-
шить, если вспомнить, что гетерополярные молекулы на поверх-
Рис. 12. Зависимость поверхностного потенциала вод-
ных растворов концентрации [37]:
1— камфара; 2 — масляная кислота; 3 — амиловый спирт; 4 —
этиловый спирт; 5 — уксусная кислота; 6 — муравьиная кисло-
та; 7 — сериал кислота
Рис. 13. Зависимость потенциала поверхности (в
милливольтах) от концентрации (в молях на литр)
для насыщенных жирных кислот [24];
/—уксусная кислота; 2—пропионовая кислота; 3— масляная
кислота; 4 — валериановая кислота; 5 —капроновая кислота
144
ГЛАВА 5
ности водных растворов адсорбируются ориентированно. Эти
молекулы можно рассматривать как диполи. Положим, что
каждый из полюсов диполя имеет заряд, равный заряду элект-
рона, и полюсы находятся на расстоянии б' один от другого.
Пусть каждый диполь имеет угол наклона 0° относительно
нормали к поверхности жидкости. Тогда дипольный момент мо-
лекулы равен eb', нормальная же составляющая момента на
1 см2 равна eb'/A cos 0, где А — поверхность, занятая одной мо-
лекулой, а потенциал поверхности [см. уравнение (7)] равен
, 4~e8'cos9
h - —55------• (16)
Если допустить, что поверхностный потенциал раствора срав-
нения (эталона) равен нулю, то можно принять равным из-
меренной разности потенциалов Дтр, что позволит оценить рас-
стояние между полюсами диполя. Для этого в первую очередь
необходимо знать величину диэлектрической постоянной. Для
Длр = 300 мв, А = 20 А2, е = 4,77 • 10-10, 0 = 0 получаем
6' = 0,034 D А в случае плотной нормально ориентированной
пленки. Любой приемлемой величине D (не более 80 для воды
и 1 для воздуха) соответствует б' порядка доли атомного диа-
метра.
Кроме того, можно отметить, что поверхностный потенциал
растворов и катионных и анионных органических веществ по-
ложителен. Этот результат является неожиданным, и его можно
Рис. 14. Схема измерения потенциала поверхности при по-
мощи радиоактивного воздушного электрода (по Алексан-
деру и Джонсону):
/—потенциометр; 2 — полуэлемент; 3— воздушный электрод; 4__
переключатель; 5 — ламповый электрометр; 6—гальванометр
объяснить лишь тем, что ядро обычно смещается в сторону уг-
леводородной группы, а электроны к противоположному концу
гетерополярных молекул, независимо от того, каким образом эти
молекулы будут ионизироваться в тех или иных условиях.
Статические методы. Статический ионизационный метод,
развитый Гийо [33], широко использовался в Англии Райдилом,
Шульманом и Адамом [2, 3, 49, 50]. Прибор (рис. 14) состоит из
ЭЛЕКТРИЧЕСКИЕ ЯВЛЕНИЯ НА ПОВЕРХНОСТЯХ РАЗДЕЛА ФАЗ
145
воздушного электрода, помещенного над кюветой, содержащей
электролит. Кювета заземляется через хлорсеребряный обрати-
мый электрод и потенциометр. Воздушный электрод представ-
ляет собой хорошо изолированную металлическую проволоку,
кончик которой отстоит от поверхности жидкости па расстоянии
~2 мм. Пространство между воздушным электродом и поверх-
ностью становится проводящим вследствие того, что на проволо-
ке осаждают или прикрепляют к ней у-активный радиоизотоп.
Сначала измеряют потенциал чистой поверхности стандартного
раствора. Для этого воздушный электрод присоединяют к сетке
электрометрической лампы и поданный таким'образом на сетку
потенциал изменяют с помощью потенциометра до тех пор, пока
Рис. 15. Схема, используемая для измерения потенциала
поверхности по методу колеблющегося электрода Цисмана
(по Александеру и Джонсону):
/—потенциометр; 2 —микрофон; 3— усилитель; 4 — динамик
ток в анодной цепи, согласно показаниям гальванометра, не до-
стигнет той величины, которая бывает при заземленной сетке.
При этом потенциал поверхности раствора равен по величине
и противоположен по знаку потенциалу, регистрируемому по-
тенциометром. Измерения повторяются либо с другими раство-
рами, либо со стандартным раствором после нанесения на его
поверхность слоев каких-либо нерастворимых веществ. В резуль-
тате таких измерений находят изменение потенциала поверхно-
сти Ац при различных условиях.
В статическом методе Цисмана [23, 43, 45—47, 59—61] кюве-
та с раствором заземляется через обратимый электрод и потен-
циометр, а нормально поверхности раствор^ вибрирует пластин-
ка из проводящего материала. Эта пластинка соединяется че-
рез усилитель со звуковым или каким-либо другим детектирую-
щим устройством. Изготовляют ее обычно из золота или золо-
ченой латуни. Эта пластинка образует с поверхностью раство-
ра конденсатор переменной емкости. Потенциал, подаваемый
таким образом на обкладки конденсатора, при помощи потенци-
ометра изменяют до тех пор, пока выходной сигнал не достиг-
нет минимума. Ослабление сигнала до минимума означает, что
потенциалы на обкладках конденсатора равны, а поверхностный
потенциал соответствует показаниям потенциометра (рис. 15).
10 А. М. Годэн
146
ГЛАВА 5
Из статических методов наиболее широко используется ио-
низационный. Однако, по-видимому, конденсаторный метод
допускает дальнейшие усовершенствования. Каждый из этих
Рис. 16. Потенциал поверхности водных растворов гептаде-
циламина и стеариновой кислоты при различных
значениях pH [45]
методов может быть использован при изучении и объяснении
поведения флотационных реагентов, особенно в случае пенооб-
разователей.
Результаты измерений потенциала поверхности в присутствии
монослоев нерастворимых веществ. Для объяснения и подтверж-
дения идей о строении монослоев нерастворимых веществ был
использован статический ионизационный метод измерения по-
тенциала поверхности. Следует напомнить, что ранее было пай-
ЭЛЕКТРИЧЕСКИЕ ЯВЛЕНИЯ НА ПОВЕРХНОСТЯХ РАЗДЕЛА ФАЗ
147
дено интересное соотношение между поверхностным давлением
и площадью, занимаемой молекулой на поверхности. Подобное
же соотношение обнаруживается между потенциалом поверхно-
сти и площадью, занимаемой одной молекулой пленки нерас-
творимого вещества, что можно видеть на рис. 16. На рисунке
видно также, каким большим может быть потенциал поверхно-
сти раздела жидкость — газ (несколько сот милливольт по
Рис. 17. Схема моментов поверхности недиссо-
циированных и диссоциированных анионных и ка-
тионных соединений:
а — жирная кислота; б — амии
сравнению с несколькими десятками милливольт на поверхности
раздела твердое — раствор). Кроме того, судя по рисунку, весь-
ма значительна зависимость потенциала от pH, причем для ами-
на потенциал увеличивается с увеличением pH, а для жирной
кислоты — уменьшается. Это согласуется с уже упоминавшимся
выше фактом, состоящим в том, что потенциал поверхности
раствора значительно уменьшается по мере роста диссоциации
органического соединения. Наконец, на рис. 16 видим, что для
амина потенциал поверхности значительно больше, чем для
жирной кислоты. Это вполне согласуется с тем, что потенциал
поверхности воды в присутствии амина возрастает не только
благодаря дипольному характеру молекулы амина, но также и
10*
148
ГЛАВА 5
вследствие ориентационного эффекта соседних молекул воды.
В случае же жирной кислоты эти факторы противодействуют
один другому. Электрическое действие анионных и катионных
поверхностно активных веществ качественно представлено на
рис. 17. Из приведенной на этом рисунке схемы видно, что в
Рис. 18. Соотношение между потенциалом поверхности и поверх-
ностным давлением для пленок стеариновой кислоты на воде при
pH = 7,0 и гептадециламина на воде при pH = 7,7 [45]:
1 — поверхностное давление для стеариновой кислоты; 2 — поверхностное
давление для амина; 3 — потенциал для амина; 4 — потенциал для стеари-
новой кислоты
диссоциированном состоянии молекулы имеют значительно
большие электрические моменты, причем молекулы катионных
поверхностно активных веществ обладают большими положи-
тельными моментами, чем анионных.
Наличие взаимосвязи между потенциалом поверхности и по-
верхностным давлением ясно видно на рис. 18 (график постро-
ЭЛЕКТРИЧЕСКИЕ ЯВЛЕНИЯ НА ПОВЕРХНОСТЯХ РАЗДЕЛА ФАЗ
149
ен на основании данных Портера) [45, 46]. Пленки стеариновой
кислоты и гептадециламина в условиях опытов находятся в
конденсированном состоянии. При более высоких температурах
пленки переходят в газообразное состояние, как это описано в
гл. 4. При этих условиях потенциал поверхности почти пропор-
ционален поверхностному давлению. Таким образом, можно ви-
деть, что данные и по потенциалу поверхности и по поверхност-
ному давлению могут быть использованы совместно для уста-
новления характера двухмерного состояния (газообразное,
конденсированное) пленок нерастворимых веществ.
ЛИТЕРАТУРА
1. Abramson Harold A. Electrokinetic Phenomena and their Application
to Biology and Medicine, (a), pp. 105—106, Chemical Catalog Company,
Inc., New York, 1934.
2. Adam, Neil Kensington. The Physics and Chemistry of Surfaces 3d ed.,
pp. 36—39, 58—63, 72—74, 88, 133—136, Oxford University Press, New York,
1941.
3. Adam N. K. and J. B. Harding. The structure of surface films. XVI.
Surface potential measurements on fatty acids on dilute hydrochloric acid,
Proc. Roy. Soc. (London), A138, 411—430, 1932.
4. Alexander A. E. and P. Johnson. Colloid Science, I, (a), pp. 11—
12, (b) 297—298, (c) pp. 319—321, Oxford University Press, New York, 1949.
5. A i t у Thomas. The cataphoresis of gas bubbles in water, Proc. Roy. Soc.
(London), A106, 315—340 (1924).
6. Alty Thomas. The origin of the electrical charge on small particles in
water, Proc. Roy. Soc. (London), Al 12, 235—251, 1926.
7. Bichat E. and R. В 1 о n d 1 о t. Mesure de la difference de la potentiel
des couches electriqucs qui recouvrent deux liquides au contact, J. Phys.,
[2] 2, 533—551, 1883.
8. Briggs David R. Surface conductance, in Colloid Svmposium Monograph,
6, 41—42, Chemical Catalog Company, New York, 1925.
9. Briggs David R. Determination of the zeta potential of cellulose — a
method, J. Phys. Chem., 32, 641—675, 1928.
10. De Bruyn H. Die Struktur der Phasengrenze bei hydrophoben Kolloiden,
Rec. Trav. Chim., 61, 5—28, 189—200, 1942.
11. Bull Henry B. Die Bedeutung der Kapillarenweite fur das Stromungs-
potential, Kolloid Z., 60, 130—132, 1932.
12. Bull Henry B. Physical Biochemistry, John Wiley and Sons, Inc. New
York, 1943.
13. Bull Henry B. and R. A. G о r t n e r. Electrokinetic potentials. X. The
effect of particle size on the potential, J. Phys. Chem., 36, 111 —119, 1932.
14. Bull Henry B. and Ross Aiken Gortner. Electrokinetic potentials. XI.
The effect of sodium soaps on the electric moment of the double layer at
an aqueous-cellulose interface, Physics, 2, 21—32. 1932.
15. Bull Henry B. and Laurence S. Moyer. Electrokinetics. XVI. Streaming
potentials in small capillaries, J. Phys. Chem., 40, 9—20, 1936.
16. Burton E. F. On the properties of electrically prepared colloidal solu-
tions, Phil. Mag., [6], 11, 425—447, 1906.
17. Chapman, David Leonard. A contribution to the theory of electrocapil-
larity, Phil. Mag., [6], 25, 475—481, 1913.
18. C u r r i e B. W. and T. Alty. Adsorption at a water surface, Part I, Proc.
Roy. Soc. (London), A122, 622—633, 1928.
'150
ГЛАВА 5
19. Debye Р. and Е. Hiickel. Zur Theorie der Elektrolyte, Physik. Z., 24,
185—206, 305—325, 1923.
20. Freundlich Herbert. «Colloid and Capillary Chemistry» translated by
H. Stafford Hatfield, (a), pp. 243, 256—257, Methuen and Co., Ltd, Lon-
don, 1926.
21. Freundlich H. and P. R о n a. Ueber die Beziehungen zwischen dem
elektrokinetischen Potentialsprung und der elektrischen Phasengrenzkraft,
Sitzber. preuss. Akad, Wiss., 20, 397—402, 1920.
22. Frost Arthur A. and Victor R. H u г к a. Adsorption of vapors at solid
surfaces and the change of surface electrical potential, J. Am. Chem. Soc.,
62, 3335—3340, 1940.
23. Frumkin A. Phasengrenzkraefte und Adsorption an der Trennungsflaeche ’
Luft-Loesung anorganischer Elektrolyte, Z. Physik. Chem., Bd. 109, 34—48, у
1924. ;
24. Frumkin A. Phasengrenzkraefte an der Trennungsflaeche gasfoermig-
fluessig, Z. Physik. Chem., Bd. Ill, p. 190—210, 1924.
25. F r u m к i n; A., A. Donde, and R. Kulvarskaya. Phasengrenzkraefte
an der Trennungsflaeche gasfoennig-fluessig, Z. Physik Chem., Bd. 123,
321—338, 1926.
26. F u e r s t e n a u D. W. Streaming potential studies on quartz. Thesis for
the doctorate, Massachusetts Institute of Technology, 1953.
27. G a u d i n A. M. Principles of Mineral Dressing, (a), pp. 166—168, (b)
p. 322, McGraw-Hill Book Company Inc., New York, 1939.
28. G a u d i n A. M. and D. W. F u e r s t e n a u. Quartz flotation with anionic
collectors, Mining Eng., 66—72 (January 1955); Trans. AIMME, 202, 66—
72, 1955.
29. G a u d i n A. M. and S. C. S u n. Correlation between mineral behavior in
cataphoresis and in flotation, Mining Technol. Techn. Publ. 2005, May, 1946.
30. Gortner Ross Aiken. «Outlines of Biochemistry», 3d ed., (a) pp. 133—
: 135, (b), pp. 110—112, (c) pp. 117—118, John Wiley and Sons, Inc., New
York, 1949.
i31. Go uy M. Sur la constitution de la charge electrique a la surface d’un
electrolyte, J. phys., [4], 9, pp. 457—468, 1910.
32. Guggenheim E. A. The helmholtz, Trans. Faraday Soc., 36, 139—144,
. 1940.
33. G u у о t M. J. Effect Yrolta metal-electrolyte et couches monomeleculaires,
Ann. phys., 2, № 10, 506—638, 1924.
34. Harned Herbert S. and Benton B. Owen. «The Physical Chemistry of
Electrolytic Solutions», (a) pp. 24—28, Reinhold Publishing Corporation,
New York, 1943.
35. Helmholtz, Hermann Ludwig Ferdinand von. Studien ueber elektrische
Grenzschichten, Ann. Physik, Bd. 7, № 4, 337—382, 1879.
36. J о f a S., A. Frumkin and P. Tschugunoff. Phasengrenzkraefte an
der Trennungsflaeche gasfoermig-fluessig V, Acta Physicochim U.R.S.S.,
I, 883—900, 1934.
37. Kenrick Frank B. Die Potentialspruenge zwischen Gasen und Fluessig-
keiten, Z. physik. Chem., Bd. 19, 625—656, 1896.
38. Lachs Hilary and Joseph Biczyk. Das elektrokinetische Potential nach
der Methode der Stroemungspotentiale, Z. physik. Chem., 148A, 441—463,
1930.
39. Lauffer Max A. and Ross Aiken Gortner. Electrokinetics. XX. The
electric moment of aliphatic alcohols, acids and esters at cellulose and
aluminium oxide interfaces, J. Phys. Chem., 42, 641—656, 1938.
40. Martin William McKinely and Ross Aiken Gortner. Studies on elec-
trokinetic potentials. V. Cellulose-organic liquid interfaces, J. Phys. Chem.,
34, 1509—1539, 1930.
ЭЛЕКТРОЛИТИЧЕСКИЕ ЯВЛЕНИЯ НА ПОВЕРХНОСТЯХ РАЗДЕЛА ФАЗ 151
41. МсТа EEart Н. A. The electrification at liquid-gas surfaces Phil Mag.,
27, № 6, 297—314, 1914; 28, № 6, 367—378, 1914.
42. McTaggart H. A. On the electrification’ at the boundary between a li-
quid and a gas, Phil. Mag., 44, № 6, 386—395, 1922.
43. Norton Francis J. Electrical characteristics ’ of molecular films J Am
Chem. Soc., 61, 3162—3168, 1939.
44. О ns a ger Lars. Theories of concentrated electrolytes, Chem Revs. 13
73—89, 1933.
45. Porter Eliot F. Monomolecular films of a-aminostearic acid, stearic acid
and heptadecylamine, J. Am. Chem. Soc., 59, 1883—1888, 1937.
46. Porter Eliot F. and Jeffries Wyman Jr. Contact potentials of stearate
films on metal surfaces, J. Am. Chem. Soc., 60, 1083—1094, 1938.
47. R о s e n f e 1 d S. and W. M. Hoskins. A modified Zisman apparatus
for measuring contact potential differencess in air, Rev. Sci. Instr., 16,
343—345, 1945.
48. Scatchard George. The coming of age of the interionic attraction
theory, Chem. Revs, 13, 7—27, 1933.
49. Schulman J. H. and A. H. Hughes. On the surface potentials of
unimolecular films. IV. Effect of the underlying solution and transition
phenomena in the film, Proc. Roy. Soc. (London), A138, 430—450, 1932.
50. Schulman J. H. and Eric K. R i d e a 1. On the surface potentials of
unimolecular films of long chain fatty acids, Proc. Roy. Soc. (London),
A130, 259—294, 1931.
51. Stern O. The theory of the electrolytic double layer, Z. Elektrochem.,
30, 508—516, 1924.
52. Stokes G. C. On the effect of the internal friction of fluids on the mo-
tion of pendulums, Trans. Cambridge Phil. Soc., 9 (pt. Il), 8—106, espe-
cially 48—57, 1851.
53. Sun S. C. Correlation between mineral behavior in cataphoresis and flo-
tation. (a) p. 75, Thesis for the doctorate, Massachusetts Institute of Tech-
nology, 1945.
54. Thomas Arthur W. «Colloid Chemistry», (a) pp. 225—228, McGraw-Hill
Book Company Inc., New York, 1934.
55. Tiselius Arne. A new apparatus for electrophoretic analysis of colloi-
dal mixtures, Trans. Faraday Soc., 33, 524—531, 1937.
56. V e r w e у E. J. W. and J. Th. G. Overbee k. «Theory of the Stability
of Lyophobic Colloids», (a) pp. 46—49, Elsevier Press, Inc., Houston,
Tex., 1948.
57. Wood Lloyd A. An analysis of the streaming potential Method of measu-
ring the potential at the interface between solids and liquids, J. Am. Chem.
Soc., 68, 432—437, 1946.
58. Wood Lloyd A. The measurement of the potential at the interface between
vitreous silica and pure water, J. Am. Chem. Soc., 68, 437—441, 1946.
59. Y a m i n s H. G. and W. A. Zisman. A new method of studying the
electrical properties of monomolecular films on liquids, J. Chem. Phys., 1,
656—661, 1933.
€0. Zisman W. A. A new method of measuring contact potential differen-
ces in metals, Rev. Sci. Instr., 3, 367—370, 1932.
'61 . Zisman W. A. and H. G. Y a m i n s. Experiments on the contact poten-
tial of zinc crystals, Physics, 4, 7—9, 1933.
Глава 6
УСТОЙЧИВОСТЬ МИНЕРАЛЬНЫХ СУСПЕНЗИЙ
Суспензии тонкоизмельченных минералов в воде содержат
или отдельные частицы минерала или частицы минерала, объе-
диненные в относительно большие флокулы. Для правильного
выбора процесса обогащения необходимо знать, в каком состоя-
нии находится суспензия — в диспергированном или флокули-
рованном. Так, для классификации пульпы должны быть
диспергированными, а для сгущения и фильтрации — флокули-
рованными.
Если пульпа не светлеет при стоянии, она'представляет собой
устойчивую суспензию. И наоборот, флокулированная пульпа,
светлеющая при стоянии, представляет собой неустойчивую сус-
пензию. Флотируемость минеральных суспензий связана с их
устойчивостью.
Шульман [31] и Эдсер [7] пришли к выводу, что флокуляция
является обязательным условием флотации. По их мнению, ус-
ловия образования флокул, состоящих только из минерала, и
флокул, состоящих из минерала и пузырьков газа, одни и те
же [9]. Но если даже ограничить понятие флокуляции случаем
твердого вещества, суспендированного в жидкости при отсутст-
вии пузырьков газа, все же уместна взаимосвязь флокуляции
и флотации. Экспериментально показано, что флотации не про-
исходит, если частицы диспергированы и наблюдается Броунов-
ское движение [34].
Причины, вызывающие диспергацию и флокуляцию. Иссле-
дованием сил, обусловливающих диспергацию и флокуляцию,
занимались многие ученые в течение ряда лет.
Результаты исследования этой проблемы описаны в превос-
ходной монографии Фервея и Овербика [40]. Прежде чем обсу-
дить эти результаты полезно остановиться на различных факто-
рах, принятых во внимание этими авторами.
Твердый частицы, диспергированные в жидкой фазе, постоян-
но сталкиваются с молекулами и ионами этой фазы. Каждая
молекула обладает в среднем энергией, равной 3/2 kT (k — по-
стоянная Больцмана, Т—абсолютная температура), что со-
ставляет при комнатной температуре 6 • 10'14 эрг. Эта энергия
вместе с той, которой обладала частица до столкновения, распре-
деляется между участниками столкновения. В результате кине-
УСТОЙЧИВОСТЬ МИНЕРАЛЬНЫХ СУСПЕНЗИЙ
153
тическая энергия минеральных частиц суспензии независимо от
их размера и массы в среднем становится равной 3/2 kT. Факти-
чески частица в каждый момент времени обладает различными
значениями кинетической энергии, однако среднее значение ки-
нетической энергии равно 3/2 kT. Распределение частиц по ско-
ростям дается кривой Максвелла (рис. 1). Вероятность того,
что частица обладает энергией от Е до Е + ДЕ, равна отноше-
нию заштрихованной площади под кривой и полной площади,
ограниченной всей кривой.
Несмотря на то, что средневзвешенная энергия частицы рав-
на 3/2 kT, значительное число частиц обладает энергией, в не-
сколько раз превосходящей это среднее значение. Уравнение
кривой может быть записано в следующем виде [17]:
%У X е-х
kT У~У
(О
где х — энергия частицы, выраженная в единицах kT
, Е \
(х = —;
kT )
fEdx —доля частиц, энергия которых лежит между Е и
Е + dE.
Согласно Ною и Шериллу [28а], доля молекул газа при тем-
пературе 0°, обладающих энергией, превышающей среднее зна-
чение в пять раз (5 • 3/2 kT), составляет 0,18%, доля молекул с
энергией, в десять раз превышающей среднее значение,
154
ГЛАВА 6
0,00014% и, наконец, в двадцать раз — только 0,59 10-10%. Из
Приведенных расчетов видно, что частицы, обладающие столь
большой энергией по сравнению со средней, встречаются край-
не редко или обладают такой энергией в течение лишь очень
короткого промежутка времени.
Из сказанного выше следует, что перемешивание, вызван-
ное тепловым движением, является важным фактором, обус-
ловливающим сближение и разделение частиц, однако следует
иметь в виду, что за счет энергии броуновского или теплового
движения частицы практически не могут преодолеть потенци-
альный барьер, превышающий 50—100 • 10~14 эрг на каждую
частицу.
Можно ожидать, что различие в скоростях осаждения частиц
разного размера или удельного веса вызовет флокуляцию. Одна-
ко, хотя эти факторы, так же как и другие, могут вызвать об-
разование больших флокул из маленьких, этим нельзя объяс-
нить, почему одни пульпы флокулируют, а другие нет '.
Поэтому факторами, определяющими устойчивость пульпы,
могут быть лишь электростатический эффект двойного слоя и
силы притяжения между атомами и молекулами, действующие
на малых расстояниях.
Электростатический характер двойного слоя, обусловливаю-
щий диспергацию. При недостаточно хорошем знакомстве с до-
стижениями термодинамики и статической механики может ка-
заться, что действие двойных электрических слоев двух одинако-
вых частиц заключается только в отталкивании независимо от
размеров частиц, их формы, диэлектрических свойств и концент-
рации ионов в растворе. Хотя некоторые абстрактные математи-
ческие работы показывают, что это не так [20—26], авторы боль-
шинства исчерпывающих теоретических работ последних лет
[36—40] придерживаются общепринятой точки зрения. I
Фервей и многие другие исследователи рассчитывали вели-
чину сил отталкивания между двумя частицами для сфериче-
ских и плоских частиц. Решение этой Задачи для сферических >
частиц может, строго говоря, применяться в случае взаимодей-
ствия капелек жидкости в воде или в случае взаимодействия пу-
зырьков газа. Это решение может быть использовано для частиц
примерно одинакового размера, у которых толщина двойного
слоя велика по сравнению с размером самой частицы.
Примером взаимодействия частиц с плоскими гранями мо-
жет служить взаимодействие частиц кристаллических материа-
лов, чешуйчатых частиц всех размеров и вообще всех частиц,
1 Сферическая частица диаметром 2,0 ц и удельным весом 4,0, падаю-
щая в воде под влиянием своего веса, приобретает кинетическую энергию,
.равную только 3,6 • 10-18 эрг, или 6 • 105- в единицах 3/а kT.
УСТОЙЧИВОСТЬ МИНЕРАЛЬНЫХ СУСПЕНЗИЙ
155
размеры которых велики по сравнению с толщиной двойного
слоя.
Формулы, с помощью которых Фервей и его школа вычис-
ляют силы взаимодействия двойных электрических слоев, весь-
ма сложны, особенно в тех случаях, когда не вводится прибли-
жение, упрощающее решение задачи. Поэтому результаты этих
расчетов обычно выражают в графической форме.
На рис. 2 схематически изображено взаимодействие двух
плоско-параллельных двойных слоев. По оси абсцисс отложено
расстояние между двумя пластинами в единицах, обратных па-
раметру Дебая — Гюккеля, по оси ординат — потенциал. Потен-
циалы твердых поверхностей приняты равными ф0. Потенциал
жидкости в середине между плоскостями (расположенными
одна от другфй на расстоянии, равном 2d) обозначим На
бесконечности значение потенциала полагаем равным нулю.
Фервей и Овербик в своих расчетах исходили из уравнения (2)
в гл. 5, которое было записано в следующей форме [40а]:
В этом уравнении: £ = хх, х—расстояние от пластины до
точки с потенциалом ip, y — ze^/kT, где z — валентность иона
в диффузном слое, е — заряд электрона. Все другие величины
сохраняют те же значения, что и в гл. 5. Обозначение shy есть
гиперболический синус дуги у, равный (е —е~у )/2, где е —
основание натуральных логарифмов.
Уравнение (2) интегрируется при граничных условиях
ге ф,/ dy п ,
У = Уа = и ~ - 0 для х = i
Интегрирование дает:
= - ]/2chy-2chyd, (3)
где
Чтобы найти значение потенциала <bd , уравнение (5) вновь
интегрируют в пределах от = хх = 0 до = хх = xd, что при-
водит к эллиптическому интегралу первого рода, значения кото-
рого про'табулированы. Окончательное решение весьма сложно
140е] и результаты его лучше изобразить графически. Кривые
на рис. 3 построены на основании расчетов Фервея и Овербика
[40Ь]. Эти кривые показывают зависимость потенциала tyd от
расстояния между пластинами d. График на рис. 3 построен в
безразмерных координатах и заслуживает более подробного
объяснения. По оси абсцисс отложена половина расстояния
Рис. 2. Схематическое
изображение электриче-
ского потенциала двой-
ного слоя между двуми
пластинами в сравнении
с электрическим потен-
циалом двойного слоя
одной пластины (по Фер-
вею и другим)
УСТОЙЧИВОСТЬ МИНЕРАЛЬНЫХ СУСПЕНЗИЙ
157
между плоскостями, выраженная в безразмерных единицах
nd (d— расстояние между плоскостями, %—параметр Дебая —
Гюккеля, имеющий размерность, обратную расстоянию). На оси
ординат отложен безразмерный параметр ze^dlkT (z— число
единиц заряда на ион; е — заряд электрона; фй —потенциал;
k— постоянная Больцмана; Т—абсолютная температура).
Рис. 3 обладает теми же преимуществами и недостатками, кото-
рые присущи диаграммам зависимости коэффициента сопротив-
ления от числа Рейнольдса [8а]1.
Например, если в случае одно-одновалентного электролита
потенциал поверхности ф0 равен 256 мв, то zetyQ[kT — 10, и ^d
будет меняться с расстоянием так, как показывает верхняя кри-
вая. Потенциалам поверхности, равным 76,8 и 25,6 мв, соответ-
ствуют средняя и нижняя кривые.
Важно отметить, что, поскольку величина zetydkT существен-
но влияет на характер зависимости фй от d, интервал измене-
ния ф0 от нескольких милливольт до 200—250 мв охватывает
большинство случаев.
Другими словами, большинство интересующих нас случаев
будет попадать между крайними кривыми (см. рис. 3)
С помощью рис. 3 попытаемся оценить энергию, необходимую
для сближения пластин из бесконечности до расстояния 2d —
о I о
= 970 А. Предположим, что фо = 256 мв, —= 970 А (ионная
сила 10~5 моль/л). Отсюда vx = 0,5. Потенциал, соответствую-
щий точке В на рис 3, фй = 3,6 (kT/ze) = 92 мв. Работа сбли-
жения должна быть 0,092 эв для каждого элементарного заря-
да на поверхности пластины.
В случае, когда требуется сблизить пластины, находящиеся
на расстоянии 2910 А, до расстояния 970 А, работа зависит
от изменения потенциала tyd с изменением расстояния
kT
Фя — = (3,6— 1,5)—— = 54 мв.
12 ге
На рис. 4 в графической форме в логарифмических коорди-
натах показана зависимость разности потенциалов поверхности
пластины и потенциала в середине между пластинами от рас-
стояния между плоскими поверхностями. Точки, обозначенные
А' и В' на рис. 4, соответствуют точкам А и В на рис. 3. Орди-
наты точек А' и В' являются разностью между ординатами то-
чек С и А и С и В соответственно на рис. 3.
Электростатическая работа сближения. Интегрированием
ф</’ о между соответствующими пределами можно найти работу
сближения двух пластин. Поскольку заряд о каждой поверхно-
1 Диаграмма Рэлея. Прим. ред.
158
ГЛАВА 6
сти меняется в зависимости от расстояния между пластинами,
произведение трДч не является правильной оценкой работы
сближения пластин. По Фервею и Овербику [40] [см. в их книге
уравнение (35) на стр. 77], изменение свободной энергии AF,
приходящееся на 1 см2 двойного слоя (при полном сближении
Паладина. расстояния между пластинами, Юн
Рис. 4. Зависимость разности между потенциалом
(изолированной пластины) и потенциалом tya (средней
точки между двумя плоско-параллельными пластинами)
от расстояния между плоскими поверхностями
пластин от начального расстояния 2d), определяется следующим
уравнением:
Д F =-----Г— (ЗеУа— 2 — е~У(‘) +
х 2
тот
/---------------- " е~У - е~Уй
+ 2 у 2ch у0 — 2ch yd + 2 f ~ : длг
J V 2ch j/— 2ch yd
yd
dy .
(4)
В этом уравнении п — общее число ионов в миллилитре рас-
твора в точке, бесконечно удаленной от суспендированного твер-
дого тела. Изменение свободной энергии двойного электрическо-
го слоя АТ7,*, , приблизившегося к такому же электрическому
слою от бесконечности до столкновения, дается уравнением
. г, —BnkT / , //„ ,\
« =
а изменение свободной энергии двух электрических слоев пло-
щадью 5 см2, сблизившихся от бесконечности до расстояния 2d,
определится уравнением
11^ = 25(ДР-AFJ эрг. (6)
УСТОЙЧИВОСТЬ МИНЕРАЛЬНЫХ СУСПЕНЗИИ
15$.
Фервей и Овербик [40f] из уравнения (6) рассчитали электро-
статическую работу сближения эрг)см2 и представили полу-,
ченные результаты в таблице. Чтобы ограничить размеры табли-
цы, результаты они выразили через безразмерную величину %d,
которую можно представить как расстояние между пластинами
в единицах 1/х, а произведенная работа сближения выражена в
единицах z2/x (размерность этой величины — эрг/см или дины).
На рис. 5 приведены результаты Фервея и Овербика для че-
тырех значений zety0/kT, между которыми заключены наиболее
вероятные значения этой величины (гр0 изменяется от 25 до
256 мв в растворах одно-одновалентного электролита).
Предположим, например, что электролит одно-одновалент-
ный, ^0 = 76,8 мв, %= 108 Дг1 (величина, приблизительно экви-
валентная с= 1 ммоль/л). Следует определить работу, которая
требуется для сближения двух параллельных пластин площадью
в 1 см2 на расстояние 2d, равное 40А. В данном случае ze^/kT =
= 3. Следовательно, для вычисления нужно воспользоваться
третьей кривой сверху. Точка С соответствует d = 20 • Ю 8 см,
v,d = 0,2. Из графика видно, что z2 1Ил/й=3,45 • 10~7. Отсюда
= 0,345 эрг/см2. В случае кубов с ребром в 1 ц W R= 0,345 •
• 10-8 эрг/см на каждую пару частиц (величина порядка 105 kT).
В случае, если = 25,6 мв, х = 105 (с = 0,01 ммоль/л), точка
D соответствует работе, требующейся для сближения частиц,
находящихся на расстоянии 2000 А. При этих условиях WR=.
= 10-3 эрг/см2. Для кубических частиц с ребром 10 5 см работа
равна 10~!3 эрг/см2 на пару частиц, т. е. примерно 2kT на ча-
стицу.
Из этих двух примеров видно, что для различных условий
работа может изменяться от величины порядка тепловой энер-
гии на частицу до величины, в несколько тысяч раз большей.
На рис. 6 показана зависимость электростатической работы
сближения (в логарифмических координатах) от расстояния
между частицами. Кажется непонятной почти линейная зависи-
мость 1g WR от xd. В некоторых случаях этим рисунком удобнее
пользоваться для вычислений, чем рис. 5. На этом графике так-
же изображены не работа и расстояние как таковые, а простые
функции этих величин.
Силы притяжения Ван-дер-Ваальса — Лондона, обусловли-
вающие флокуляцию. Как было показано выше, взаимодействие
двойных электрических слоев, так же как и гравитационное,
взаимодействие, не приводит к силам притяжения, обусловли-
вающим флокуляцию.
Де Бур [31 и Гамакер [11] предположили, что флокуляцию,
обусловливают силы, аналогичные когезионным [18]. Фервей ц
УСТОЙЧИВОСТЬ минеральных СУСПЕНЗИЙ
161
его школа это предположение количественно обосновали. В па-
мять работы Ван-дер-Ваальса, посвященной исследованию га-
зов, в которой было постулировано существование этих сил, рас-
сматриваемые силы были названы именем этого великого гол-
ландского физика и химика.
Если две дипольные молекулы находятся достаточно близко,
то они взаимно влияют на пространственную ориентацию друг
друга, что обусловливает среднее притяжение [4, 5]. Более того,
каждая молекула индуцирует дополнительный диполь другой
молекулы. И эта взаимная поляризация усиливает притяжение
[15, 16]. Силы когезии действуют также между неполярными
молекулами, они действуют даже между одноатомными молеку-
лами благородных газов. Величину этих сил высчитали Лондон
[27] и Слэтер и Кирквуд [30] с помощью волновой механики.
В первом приближении потенциал сил притяжения между
двумя сходными молекулами изменяется обратно пропорцио-
нально шестой степени расстояния между ними:
“Д—0
где X — коэффициент, зависящий от свойств рассматриваемого
атома или молекулы.
Были сделаны попытки оцепить значение Z,. Согласно расче-
там Лондона,
A = ^-a2/zv0, (8)
где a — поляризуемость атома;
йч0 — количество энергии, соответствующее основной ха-
рактеристической квантово-механической частоте vo,
взятой из уравнения рассеивания рассматриваемого
атома.
Вообще говоря, К — величина порядка 10-59. Так, значение А
для воды, полученное из формулы Лондона, равно 3,2 • 10 59,
из формулы Слэтера 5,4 • 10~59, а из более новой формулы
Нейбауэра 5,1 • 10~59.
Если бы притяжение между частицами уменьшалось с рас-
стоянием так же быстро, как в случае взаимодействия отдель-
ных атомов, рассчитывать на значительное притяжение, особен-
но для частиц с очень малым значением л, было бы весьма труд-
но. Но, как было показано Гамакером [11, 12], соотношение
резко меняется в случае взаимодействия между группами мно-
гих атомов. Это является следствием основных квантово-механи-
ческих представлений, согласно которым все силы, действующие
между атомами, аддитивны. Так, притяжение двух параллель-
ных пластин толщиной 0, находящихся одна от другой на рас-
11 А. М. Годэн
162
ГЛАВА 6
стоянии 2d, будет определяться следующим уравнением:
Коэффициент А этого соотношения был получен Гамакером
из атомной плотности и атомного параметра Лондона X
А — ~2q2 к, (10)
где q — число атомов в миллилитре.
Коэффициент А изменяется от 2 10~13 до 2 • 10 ~12.
Для очень тонких пластин допустимо следующее приближе-
ние:
= _ ем
А 32 тс d4
Как видно, величина обратно пропорциональна четвер-
той степени расстояния между пластинами. Для пластин, тол-
щина которых велика по сравнению с толщиной двойного слоя,
полезно еще одно приближение, а именно:
В этом случае величина WA обратно пропорциональна квад-
рату расстояния между пластинами. Потенциал притяжения
между минеральными частицами меняется не обратно пропор-
ционально шестой степени расстояния между ними [см. уравне-
ние (7)], а обратно пропорционально квадрату расстояния для
прямоугольных частиц или даже обратно пропорционально пер-
вой степени в случае сферических частиц (см. уравнение (14)].
Это позволяет считать, что силы притяжения относительно
дальнодействующие и их действие не обязательно ограничивает-
ся межатомными или межмолекулярными расстояниями.
Казимир и Польдер [2] дали квантово-механический расчет
«замедления», или относительную поправку, которую нужно вне-
сти в уравнение притяжения Лондона — Ван-дер-Ваальса. Из их
расчетов видно, что написанные выше уравнения верны для рас-
стояний вплоть до 500 А. Для расстояний же, больших 1000—
1500 А, силы притяжения, рассчитанные из уравнений (9) и (12),
получаются весьма завышенными. Казимир и Польдер указы-
вают, что причина этих расхождений заключается во времени,
которое требуется для передачи электрического поля от одного
из взаимодействующих атомов до другого и обратно. Эта пере-
дача не мгновенна, и если время составляет существенную часть
периода квантово-механической волны (описывающей состояние
УСТОЙЧИВОСТЬ МИНЕРАЛЬНЫХ СУСПЕНЗИЙ
163
электронов взаимодействующих атомов), то силы притяжения
значительно уменьшаются. Для относительно больших расстоя-
ний эффект «запаздывания» становится настолько большим, что
притяжения вообще не будет, так как различные члены уравне-
ния сокращаются. Поскольку значения расстояний, интересных
для проблемы устойчивости коллоидов, лежат в области от 10 до
500 А, поправки, вызванные «запаздыванием», можно отбросить
как незначительные.
Из уравнения (9) видно, что при d = 0 значение WA беско-
нечно велико. Это уравнение получено интегрированием и яв-
ляется приближенным. На самом деле'взаимодействие атомов,
находящихся на малых расстояниях, следует определять непо-
средственным суммированием при расстояниях между пластина-
ми, меньших, чем два-три атомных диаметра. Поэтому, если d
меньше 5 А, следует воздержаться от применения этого уравне-
ния.
Рассматривая когезию как причину флокуляции, Гамакер и
его последователи обращают внимание на то, что притяжение
двух гидратированных частиц можно представить как сумму
взаимодействия между собой двух частиц, двух гидратных
слоев, а также взаимодействия частицы с гидратным слоем, т. е.
А = Ап А22 — 2АХ2, (13)
Согласно Гамакеру, значение А всегда положительно для
частиц сходных материалов; наоборот, для двух сильно отли-
чающихся дисперсных фаз коэффициент Гамакера должен
быть отрицательным. При этом, если потенциалы поверхностей
двух дисперсных фаз имеют одинаковые знаки, неизбежна дис-
пергация. Данные этой главы будут использованы при рассмот-
рении диспергации, вызванной силами Ван-дер-Ваальса, при
наличии электростатического притяжения.
Величина А обычно меньше, чем Ли или А2г, ее значение
можно принять равным 5 • 1(Н;з (меньше, чем значение, предло-
женное Фервеем и Овербиком). Однако для более точной оцен-
ки коэффициента Гамакера потребуется провести значительные
исследования.
Совместное действие электростатических и вандерваальсовых
сил. Поскольку заряд поверхности вызывает отталкивание, а си-
лы когезии — притяжение, совместное действие этих сил может
привести к самым различным результатам. Так, в случае, если
заряд поверхности равен нулю, взаимодействие частиц на всех
расстояниях сводится к притяжению. Подобная система должна
флокулировать и будет оставаться сфлокулированной.
Если заряд поверхности весьма велик по абсолютной вели-
чине или коэффициент Гамакера очень мал, то наблюдается
11*
164
ГЛАВА 6
только отталкивание частиц на всех расстояниях. Системы это-
го типа будут диспергированными.
Наконец, в ряде случаев, на одних расстояниях будет преоб-
ладать отталкивание, а на других — притяжение, так что систе-
ма сможет находиться в нескольких устойчивых состояниях.
Переход из одного устойчивого состояния в другое будет зави-
сеть от относительной высоты потенциального барьера и тепло-
вой энергии, которую может хотя бы на мгновение иметь части-
ца, молекула или флокула. В зависимости от высоты потен-
циального барьера переход от диспергированного к сфлокули-
рованному состоянию (или наоборот) может быть быстрым или
медленным. Кинетика быстрой и медленной флокуляции была
ранее рассмотрена многими исследователями.
Наиболее интересна количественная оценка совместного
действия электростатических сил и сил когезии.
В табл. 1 приведены характерные численные значения энер-
гии притяжения и отталкивания в мэрг/см2 для заряженных
Таблица I
Энергия сил притяжения и отталкивания, требу ющаяся для соединения
двух частиц, погруженных в водный раствор1
Половина расстояния Энергия сил притяже- ния, мэрг/см2 Энергия лектростатического отталкивания2, мэрг/смг
нами d о А а Ъ с d е
0 8,0 50 НО 220 ' 1010
5 1320 7,9 47 102 199 905
10 330 7,8 44 94 176 800
15 148 7,4 41 88 165 700
20 82 7,0 38 82 154 610
25 52 6,7 36 78 140 540
30 37 6,4 34 71,5 130 490
40 20 5,9 29 63 109 400
50 13 5,4 25 54 91 310
60 9,3 4,7 22 45,5 78 250
80 5,2 3,6 15 33 54 160
100 3,3 2,7 10,5 23,5 38 НО
120 2,4 1,8 7,4 15,5 25,5 74
150 1,47 1,0 4,3 9 14 41
200 0,80 0,35 1,6 з,з 5,0 15
250 0,52 0,14 0,45 1,2 1,9 6,0
‘Принято А = 5 • 1 0 [согласно формуле (12)] для расчета энергии сил притяжения.
‘Принято х= 10® , что в случае одно-одновалентного электролита соответствует
концентрации 10 — ^моль/л:
а — если ф0 = 1 2,8 мв, то ze ii0/kT = 0,5;
Ь — если фо = 25,6 мв, то ze = 1,0;
с — если ф0 = 38,4 мв, то ze =1,5;
d — если ф„ = 5 1,2 мв, то ze ^0/kT = 2,0;
е — если фо = 102,4 мв, то ze ipofk.T = 4,0;
УСТОЙЧИВОСТЬ МИНЕРАЛЬНЫХ СУСПЕНЗИЙ
165
плоско-параллельных толстых пластин в растворе электролита.
Разность между этими энергиями представляет собой работу,
требующуюся для сближения пластин от бесконечности до при-
веденного в таблице расстояния.
В качестве примера приведем расчет случая, в котором зна-
чение коэффициента Гамакера принято равным А = 5 • 10-13, па-
раметр Дебая— Гюккеля к = 106, что соответствует концентра-
ции ионов около 10~3-м., заряд иона z = 1. При расчете исполь-
зовалось уравнение (12).
Рис. 7. Зависимость работы флокуляции для крупных частиц от рас-
стояния между частицами 2 d. Потенциал поверхности равен фо, кон-
станта Дебая—Гюккеля % = 10«;
1 — % — О Мв; 2 — 'р0 = 12.8 мв; J - • -10 — 25,6 мв; 4 —% = 38,4 мв; 5 — %= 51,2 М3
На рис. 7 приведены значения суммы энергий притяжения и
отталкивания. Очевидно, что в случаях, когда заряд поверхно-
сти равен нулю или не превышает 12,8 мв, на всех расстояниях
между частицами будет преобладать сила притяжения. Для
Фо = 25,6 мв при расстоянии между пластинами от 450 до 65 А
доминирующим является отталкивание. Высота энергетического
барьера равна 12 мэрг/см?. Для частиц размером 1 р высота
этого барьера должна быть равна 1,2 • 10-10 эрг, или около
3000 kT на частицу. Практически — это непреодолимое препят-
ствие для флокуляции. Но для частиц диаметром 0,1 ц энерге-
166
ГЛАВА 6
тический барьер должен быть равен 30 kT, а для частиц диамет-
ром 0,03 ц— около 3 kT. Такой барьер может быть преодолен
частицами, обладающими энергией примерно в два раза боль-
шей, чем средняя. Частицы, которые имеют такую энергию,
встречаются часто. Итак, частицы микронных размеров в этих
условиях не могут флокулировать, тогда как частицы субмик-
ронных размеров флокулируют. В случае гр0 = 38,4 мв или
51,2 мв энергетический барьер еще выше, однако качественно
условия не будут отличаться от условий для гр0 = 25,6 мв.
При расчетах взаимодействия тонких пластин вместо уравне-
ния (12) использовалось уравнение (11). Для этого случая по-
тенциал сил отталкивания такой же, как и для толстых пла-
стин, но потенциал сил притяжения несколько отличается от по-
тенциала сил притяжения толстых пластин. Если пластины
достаточно тонкие (величина 0 очень мала), энергетический
барьер может возникнуть между пластинами при значении
гр0 = 12,8 мв.
Рис. 8 относится только к пластинам толщиной 100 А. Он
хорошо иллюстрирует сказанное выше. При потенциале поверх-
ности, равном 12,8 мв, высота энергетического барьера равна
1,2 мврг/см2, а потенциальный барьер лежит на расстояниях
130—560 А от поверхности. Для квадратных пластин со сторо-
ной 1000 А и толщиной 100 А барьер при 12,8 мв равен
12 • 10“14 эрг, или около 3 kT. Вообще говоря, в случае толстых
пластин одинаковый потенциальный барьер наблюдается при
больших значениях потенциала, чем в случае тонких пластин.
Аналогичные результаты были получены Фервеем и Овербиком
[39] для сфер. Дальнейшим обобщением подобных расчетов яв-
ляются кривые, аналогичные кривым, приведенным на рис. 7 и 8,
но для частиц сферической формы.
Величина энергии флокуляции. Величина энергии флокуля-
ции обычно равна 1—100 мэрг!см?, т. е. в 100—10 000 раз мень-
ше поверхностной энергии. Например, в случае пластин толщи-
ной 100 А при 1р0 = 38,4 мв, V. — 10°, потенциальный барьер
флокуляции равен 50 мэре^м2. Энергия на границе раздела во-
да— минерал может быть примерно равна 100 эрг^м2, т. е.
энергия поверхностного натяжения в 2000 раз больше, чем вы-
сота потенциального барьера флокуляции.
До сих пор при рассмотрении электрических эффектов на
границе раздела мы не принимали во внимание атомарный ха-
рактер поверхности на границе раздела минерал — вода. По-
верхность смоченного водой минерала рассматривается нами
как полностью покрытая адсорбированными ориентированными
молекулами воды. Молекулы воды Н2О, (Н2О)2 или (Н2О)з
УСТОЙЧИВОСТЬ МИНЕРАЛЬНЫХ СУСПЕНЗИЙ
167
ориентируются так, что отрицательно заряженная часть этих
молекул, состоящая из ядра атома кислорода, окруженного
четырьмя парами электронов, находится на минимальном рас-
стоянии от катионов поверхности и на максимальном от анио-
нов. Если принять диаметр молекулы равным 3 А и предполо-
жить, что слой адсорбированной воды состоит из молекул Н2О,
(Н2О)2 и (Н2О)з, то поверхности сфлокулированных частиц не
могут сблизиться на расстояние, меньшее 10 А.
+ -105
JQO 200 300 а ООО 500
Расстояние 2d, fl
Рис. 8. Работа флокуляции для тонких пластин. Пластины имеют тол-
щину 0 = 100 А . Работа флокуляции дана для различных значений по-
тенциала поверхности фо (константа Дебая — Гюккеля %, = 106) в за-
висимости от расстояния между пластинами:
1 —ф„= 0 мв-, 2 — % = 12,8 мв; 3 — ф„ = 25.6 мв; 4 —ф0 = 38,4 мв; 5 —ф0 =51,2 мв
С помощью формулы (12) можно оценить энергию, освобож-
дающуюся при флокуляции. Если коэффициент Гамакера при-
нять равным А = 5 10-13, то при 2d = 10 A = 1,32 эрг)см2-.
Это максимальное значение энергии флокуляции. Фактически
всегда существует некоторый потенциал сил отталкивания, ко-
торый ее снижает. Кроме того, геометрическая форма реальных
частиц препятствует их идеальному расположению, которое бы-
ло принято при расчете.
168
ГЛАВА 6
Сравним энергии взаимодействия сфер и пластин. По Фер-
вею и Овербику [40с], для сфер радиусом г и минимальным
расстоянием между сферами, равным 2d, потенциальная энер-
гия притяжения сферической частицы равна
W.=-------— • (14)
А 24d v
В табл. 2 приведены результаты такого расчета. Из этих дан-
ных видно, что у сфер энергия их притяжения много меньше,
чем у кубов, особенно, если сферы не очень малы. Реальные ча-
стицы минерала имеют различные формы граней, углы и, конеч-
но, не будут так плотно упаковываться, как сферы, а тем более
кубы. Поэтому наиболее вероятно, что энергия флокуляции
реальных частиц меньше, чем энергия флокуляции сфер. Для
частиц размером примерно нескольких микрон АЕ ~ 1 мэрг/см2
(или даже меньше) на 1 см2 общей поверхности дисперсной фа-
зы. В случае чешуйчатых минералов (таких, как глины) изме-
нение энергии, по-видимому, будет большим не только потому,
что частицы меньше, но и потому, что форма этих частиц по-
зволяет им плотнее прилегать друг к другу. В подобных случаях
АЕ может превосходить 10 мэрг/см2.
Таблица
Типичные значения потенциальной энергии притяжения частиц,
находящихся на расстоянии 10 А одна от другой1
Диаметр2 о А Потенциальная энергия притяжения, мкмк/эрг
для пары сфер для пары кубов для сферы с 12 ближайшими соседями для сферы с 6 ближайшими к соседями
50 0,1 0,081 о,6 0,24
100 0,2 0,325 1,2 0,975
200 0,4 1,3 2,4 3,9
500 1,0 8,1 6,0 24,4
ЮООз 2,0 32,5 12,0 97,5
20003 4 130 24 390
1 Используется коэффициент Гамакера А = 5-10“ .
2 Диаметр сферы, ребро куба.
3 Для таких больших размеров, как эти, видимо, следует вводить поправку Казими-
ра—Польдера.
Из сказанного выше следует, что энергия, требуемая для дис-
пергации сфлокулированной пульпы, много меньше той, которая
требуется для раздробления твердого минерала (по всей веро-
ятности она в десятки миллионов раз меньше).
УСТОЙЧИВОСТЬ МИНЕРАЛЬНЫХ СУСПЕНЗИЙ
169
На рис. 9 показана энергия сближения для кубических час-
тиц в растворе при А = 5- 1013, х = 106 и различных значениях
фо (от 0 до 153,6 мв). Кривые А и В для 2d > 25 А уже были
показаны на рис. 7 (крайние кривые), но в другом масштабе,.
Рис. 9. Зависимость потенциальной энергии взаимодей-
ствия кубических частиц от расстояния между их бли-
жайшими гранями (2d). Приведены кривые для раз-
личных значений <р0 при А =5 10~13; и = 10е в раство-
ре одно-одновалентного электролита:
А—фо=0 мв; В —=51,2 мв; С— ф0 = 102,4 мв; О — ф„=
= 153,6 мв
На рис. 9 кривые даны вплоть до значений d=5 А. Энергия,
требуемая для того, чтобы оторвать адсорбированную молекулу
воды, находящуюся на столь малом расстоянии от поверхности,
много больше той энергии, значения которой мы рассматри-
вали до сих пор, и сравнима с энергией химической реакции
170
ГЛАВА 6
(порядка одного или нескольких электроновольт). Поэтому
с уменьшением расстояния потенциальная энергия очень быстро
увеличивается для значений d, меньших, чем 5 А, и компенсирует
уменьшение энергии за счет сил притяжения. Хотя определение
критического расстояния в 5 А может вызвать некоторые сомне-
ния, имеется какое-то определенное значение d, для которого
справедливо все сказанное выше.
Из рис. 9 можно найти высоту потенциального барьера, пре-
пятствующего флокуляции. Величина этого барьера определяет-
ся разностью между высотой барьера и нулевой ординатой. По-
тенциальный барьер, противодействующий диспергации, опреде-
ляется расстоянием от вершины барьера до нижней точки потен-
циальный ямы. Так, в случае кривой В потенциальный барьер,
противодействующий флокуляции, близок к 100 мэрг/см2 (см.
также рис. 7), в то время как высота потенциального барьера,
противодействующего диспергации, около 1100 мэрг!см2. Потен-
циал поверхности может быть так низок, что потенциального
барьера флокуляции вообще не будет (кривая А), или так вы-
сок, что не будет потенциального барьера диспергации (кри-
вая D). В общем, однако, имеются оба потенциальных барьера
(кривые В и С), так что возможны оба состояния. Одно из этих
состояний стабильное, другое—метастабильное, причем потен-
циальные барьеры для диспергации и флокуляции будут иметь
различные высоты.
В случае, представленном на рис. 9 кривой В, для перехода
от диспергированного к сфлокулированному состоянию доста-
точно понизить фо до 15 мв (см. рис. 7). Чтобы совершить обрат-
ный переход (от сфлокулированного к диспергированному состо-
янию), потенциал поверхности должен превысить значение, при
котором отсутствует флокуляция. Это значение лежит в интер
вале 102,4—153,6 мв, как видно из кривых С и D. Возможно, что
оно равно 130 мв. После того как достигнуто состояние диспер-
гации, можно вновь понизить потенциал поверхности почти до
18 мв, но это' еще не приведет к заметной флокуляции частиц.
Действие множества частиц. Любая суспензия состоит из
множества частиц. Поэтому обычно расстояние между частица-
ми мало. Так, для суспензии, содержащей 1012 частиц в 1 мл
(для минеральных пульп это значение слишком высоко; в сред-
нем эти пульпы имеют 1010 частиц в 1 мл), расстояние между
частицами примерно равно 10-4 см, т. е.. d 5000 А. Как видно
из рис. 7 и 9, это расстояние лежит за пределами рассматривае-
мых значений. Поэтому проблемы флокуляции и диспергации
можно рассматривать на примере взаимодействия двух изолиро-
ванных частиц.
Но такое приближение будет необоснованным для керамиче-
УСТОЙЧИВОСТЬ МИНЕРАЛЬНЫХ СУСПЕНЗИЙ
171
ских суспензий, где в 1 мл могут находиться 1015—1018 частиц
и d будет мало (50—500 А).
Природа и физические свойства флокул. Две сферы могут
флокулировать только одним способом благодаря изотропному
характеру, связанному с особой геометрической формой сферы.
Если сфер много, то они могут образовывать различные струк-
туры — от плотнейшей кубической упаковки до открытой струк-
туры алмаза.
Для многогранных частиц (таких, как кубы или тетраэдры)
совершенно не обязательно, чтобы частицы, образующие фло-
кулы, соприкасались гранями. Энергетический барьер, препят-
ствующий флокуляции, будет меньше в случае соприкосновения
ребра с ребром или угла с углом, чем в случае соприкосновения
граней. Так, кубы, связанные через углы своих больших диаго-
налей, образуют структуру, у которой энергия притяжения
и энергия отталкивания на много меньше. Подобная структура
может являться промежуточной при преодолении потенциально-
го барьера или метастабильной, обладающей меньшей потенци-
альной энергией, чем энергия частиц в диспергированном состо-
янии, если переходу в стабильное состояние (случай флокуляции
при сближении граней частиц) препятствует непреодолимый по-
тенциальный барьер.
Экспериментально установлено, что флокулы представляют
собой рыхлые агрегаты частиц, которые обычно беспорядочно
ориентированы относительно друг друга.
Кинетика быстрой флокуляции. Рассмотрим вопрос о скорос-
ти, с которой флокулируют суспензии. Эта скорость выражается
в единицах времени 0, которое требуется для уменьшения числа
частиц (или флокул) на единицу объема от J до J/2.
Если на всех расстояниях взаимодействие между частицами
сводится к притяжению, что бывает в случае отсутствия потенци-
ального барьера диспергации (см. две нижние кривые рис. 7), то
говорят, что налицо имеется быстрая коагуляция. Кинетика это-
го процесса была изучена Смолуховским, который нашел, что
е = —!— ,
8~ DrJ
(15)
где D — коэффициент диффузии;
г — радиус частицы;
J— число частиц на 1 мл в начальный период.
Согласно Эйнштейну, коэффициент диффузии может быть вы-
ражен через kT и сопротивление трения, оцененное Стоксом,
D~-^-
6~р г
(16)
172
ГЛАВА 6
(17) '
Таким образом,
9 = —?
4kTJ
Уравнение (17) проверено экспериментально. Например,
Кройт и ван Аркель [19] установили его применимость. Для
fi=0,01 пуаза ^Г=4-10-14, 7= 1012, 0 «= 0,2 сек.
На самом деле, J = 1012 — слишком высокое значение для
флотационных пульп. Так, в случае суспензии, содержащей ку-
бические частицы размером 2 р, и состоящей из твердой фазы бо- *
лее, чем на 25°/0, с удельным весом, равным 5, число частиц сос-
тавит 3,9-1010 на 1 мл. В случае суспензии, содержащей 10%
твердого по объему, с частицами размером от 20 А до 60 ц, рас- у
пределенными по крупности согласно кривой распределения с
тангенсом угла наклона, равным 1,00 на диаграмме Шумана [29],
J = 5,2 Ю10. В этих примерах 0 ~ 4,8 и 3,6 сек. соответственно.
Кинетика медленной флокуляции. Если взаимодействие меж-
ду частицами на малых расстояниях сводится к притяжению, а
на средних — к отталкиванию, то частицы должны преодолеть
энергетический барьер для того, чтобы попасть внутрь потенци-
альной ямы, которая соответствует сфлокулированому состоя-
нию. Для частиц малого размера (меньше 1000 А диаметром)
величина энергии взаимодействия часто соответствует тепловой
энергии 3/2 kT, особенно для сферических частиц или частиц,
близких по форме к сферическим. В случае больших частиц
(крупнее 1000 А) тепловая энергия обычно мала по сравнению с
энергией взаимодействия. Следовательно, в случае больших ча- , 1
стиц любая система является диспергированной или же она бы-
стро флокулирует, как было показано выше. Для малых частиц ' ‘
получаются различные результаты. Тепловая энергия посредст-
вом диффузии будет вызывать флокуляцию, но со скоростью, в . t
W7 раз меньшей, чем при быстрой флокуляции. Фервей и Овербик ?
рассчитали это отношение и нашли, что для потенциального У.
барьера, равного 15 kT, W — 105, а для барьера, равного 25 kT,
W = 109. Последнее значение, по их мнению, обеспечивает ста-
бильность золя. Казалось бы, если тепловая энергия способству- ‘
ет и диспергации и флокуляции, эти процессы будут взаимно ком- а *
пенсироваться. Однако высоты потенциальных барьеров у них
неравны, так что практически медленная флокуляция и медлен-
ная диспергация протекают в данной суспензии с разными скоро-
стями.
Поведение сравнительно крупных частиц. В этом случае мож-
но ожидать, что кинетическая энергия частиц, определяемая их
размером и весом, повлияет на флокуляцию или диспергацию.
Качественно этот фактор можно оценить, сравнивая кинетичес-
УСТОЙЧИВОСТЬ МИНЕРАЛЬНЫХ СУСПЕНЗИИ
173
кую энергию частицы, осаждающейся в воде, по Стоксу, со сред-
ней кинетической энергией молекулы в воде, равной 3/2 kT =
= 6 КС14 эрг. Кинетическая энергия равна '/2 mv2, где т— масса
частицы, v — ее скорость.
Например, для сферической частицы с удельным весом 4, диа-
метром 2 I* скорость падения в воде v = 6,5 10~4 см!сек, а кине-
тическая энергия 3,6 ICC18 эрг, что составляет около' */20000 кине-
тической энергии молекулы. Ясно, что в данном случае этой ве-
личиной можно пренебречь по сравнению со средней энергией
частицы в броуновском движении. Но, так как кинетическая
энергия осаждающейся частицы растет пропорционально седь-
мой степени размера, для частиц диаметром выше 10ц кинетиче-
ская энергия падения становится больше энергии броуновского
движения, а при диаметре 20 ц превосходит ее в 600 раз. Следо-
вательно, кинетическая энергия осаждения частиц размером
больше 10 ц может влиять на их флокуляцию.
В случае, если действие центробежного поля превышает дей-
ствие гравитационного поля, размеры частиц, на флокуляцию
которых может влиять их энергия осаждения, нужно уменьшить
в корень седьмой степени раз из отношения центробежной силы
к силе веса.
Слабое перемешивание, способствующее росту флокул. Вы-
ше было приведено уравнение скорости быстрой флокуляции.
Величина 0 (входящая в него), т. е. время, необходимое для того,
чтобы число частиц в коллоидной системе уменьшилось в два
раза, может быть вычислена по уравнению (17). Так, если вяз-
кость равна 0,01 пуаза, первоначальное число частиц /0 = Ю12,
то время, требующееся для уменьшения числа частиц до J/2,
равно 0,2 сек.
Однако из этого не следует, что уменьшение числа частиц до
J/i потребует еще 0,2 сек. Поскольку частицы теперь находятся
на больших расстояниях одна от другой, вероятность их столк-
новения в результате диффузии уменьшается. Смолуховский
дал следующее решение этой задачи для числа частиц J( при
времени t :
= —. (18)
1+т
Если частицы соединяются во флокулы, состоящие в среднем из
1000 частиц каждая, то Jt = 1012/103 = 109, так что t « 1000 0. В
рассматриваемом случае t ~ 3 мин.
В случае, если суспензия содержит малое число частиц, фло-
куляция только за счет одного процесса диффузии будет продол-
жаться слишком долго. Как можно подсчитать по уравнению
(18), для того чтобы J t стало равным 1000, необходимо 6 лет!
174
ГЛАВА 6
Процесс флокуляции при расстоянии между частицами, боль-
шем, чем толщина относительно неподвижного слоя Стокса, '
окружающего частицу в жидкости (примерно (10—50 ц), уско-
ряется при перемешивании суспензии. В начале процесса умень-
шение числа частиц в основном происходит за счет столкновений,
вызванных тепловой диффузией. Этот процесс будет доминиро-
вать до тех пор, пока число частиц не достигнет значения
0,12 • Ю9 (при толщине неподвижного слоя, равной 10 ц). Этот
этап займет около 30 мин., далее скорость процесса будет опре-
деляться перемешиванием, и число частиц уменьшится до 1000
за сравнительно короткое время. В действительности оба про-
цесса, приводящие к флокуляции, протекают одновременно.
Шламовые покрытия. Интерес к образованию «шламовых по-
крытий» на частицах минерала во время флотации возник до
некоторой степени случайно. Обычно частицы флотируемого ми-
нерала чисты и свободны от шламовых покрытий. Наоборот, ча-
стицы тех же самых минералов, потерявших способность флоти-
роваться, часто покрыты шламами. Самые ранние работы в этой
области принадлежат Тэккеру и Хиду [35]. Эти работы были про-
должены Таггартом, Тейлором и Инсом [33], Инсом [13], Дель-
Джудиче [10] и Саном [32].
Тэккер и Хид считали, что они обнаружили на некоторых
сульфидных минералах, обработанных флотационными реа-
гентами, видимые под микроскопом слои флотационных реаген-
тов или продуктов их взаимодействия с минералом.
Однако, как показали Таггарт, Тейлор и Инс, мнение этих
исследователей о природе пленки было ошибочным.
В обеих последних работах Таггарт и другие окончательно
установили, что ни галенит, ни сфалерит, ни пирит не обнаружи-
вают никаких признаков изменения поверхности даже после про-
должительного перемешивания в растворах любых испытуемых
органических или неорганических реагентов. Тем не менее, рабо-
та Тэккера и Хида все же интересна, так как она привела к об-
наружению шламовых покрытий.
Шламовые покрытия можно определить, как относительно
плотно прикрепленные частицы тонкоизмельченного минерала
на поверхности гранулированных или сравнительно крупных
частиц.
На рис. 10 изображены две частицы галенита в одном и том
же флотационном опыте. Одна из этих частиц флотируется, тог-
да как другая не способна к флотации. Эта разница во флотиру-
емости может зависеть от непосредственного различия в шламо-
вом покрытии, но более вероятно, что она обусловлена каким-ли-
бо химическим отличием поверхности частиц.
Инс в своей работе о шламах попытался связать наличие
шламовых покрытий со знаком ^-потенциала крупных и о’шламо-
УСТОЙЧИВОСТЬ МИНЕРАЛЬНЫХ СУСПЕНЗИЙ
175-
ванных частиц минерала. Вообще говоря, он нашел, что положи-
тельно заряженный шлам должен прилипать к отрицательно за-
ряженному минералу и наоборот. В случае одинаковых зарядов.
Рис. 10. Частицы галенита одного и того же флотационного опыта [10]:.
а — из концентрата; б — из хвостов
Рис. 11. Частицы галенита [10]:
а— частицы галенита в пульпе, содержащей кальцитовые шламы; б — в
пульпу с кальцитовым шламом добавлен силикат натрия (жидкое стекло)
прилипания не будет. Результаты его опытов казались недоста-
точно убедительными; не было уверенности в том, что при прили-
пании частиц знаки зарядов сохраняются теми же, какими они
были для каждой частицы в отдельности. Это возражение было
развито Саном.
Дель-Джудиче дал четкую химическую гипотезу, которая объяс-
няет действие добавляемых реагентов на образование шламовых
покрытий. Он постулировал, что шламовые покрытия образуются
в результате реакции аниона минерала с катионом шлама (ил»
176
ГЛАВА 6
наоборот). В результате этой реакции образуется соль, менееs
растворимая, чем та, которая раньше существовала на повер-
хности минерала и шлама. Таким образом, создается «цемент».
Поэтому предотвращать образование шламов будет реагент,
уменьшающий растворимость соли на поверхности флотируемо-
го минерала. Эта растворимость должна стать меньше, чем рас-
творимость цементирующего соединения, в то же время она дол-
жна быть выше растворимости соединения, образуемого ионами
минерала с ионами собирателя.
На рис. 11 представлены интересные данные о галените и
кальцитовом шламе; показано необычное действие силиката на-
трия на прикрепление кальцита к галениту. Этот вопрос еще не-
достаточно изучен; превосходная работа Дель-Джудиче заслу-
живает внимания, особенно в связи с рядом исключений, заме-
ченных позднее (таких, как галенитовые покрытия на галените),
которые, возможно, будут объяснены в результате развития тео-
рии поверхностной химии.
ЛИТЕРАТУРА
1. Alexander A. F. and Р. Johnson. Colloid Science, (а) II, рр. 575—
576, Oxford University Press, New York, 1949.
2. C a s i m i r H. B. G. and D. Polder. The influence of retardation on
the London—Van der Waals forces, Phys. Rev., 73, 360—372, 1948.
3. D e Boer J. H. Influence of Van der Waals forces and primary bonds
on binding energy, strength and orientation with special refernce to some
artificial resins, Trans. Faraday Soc., 32, 10—38, 1936.
4. Debye P. Van der Waals cohesion forces, Physik. Z., 21, 178—187, 1920.
5. Debye P. Molecular forces and their electrical interpretation Physik.
Z„ 22, 302—308, 1921.
6. Derjaguin B. On the repulsive forces between charged colloid partic-
les and on the theory of slow coagulation and stability of lyophobe sols,
Trans. Faraday Soc., 36, 203—215, 1940.
7. E d s e r Edwin. The concentration of minerals by flotation, Brit. Assoc.
Advance. Sci., Fourth Rept. on Colloid Chemistry and its General and
Industrial Applications, pp. 263—327, H. M. Stationery Office London,
1922.
8. G a u d i n A. M. «Principles of Mineral Dressing», (a) Fig. 101, p. 177,
McGraw-Hill Book Company, Inc., New York, 1939.
9. Gaud in A. M. The greg family, Mining and Met., 25, 347, 1944.
10. Del Giudice, Guido R. M. A study of slime coatings’ in flotation
Trans. AIMME, 112, 398—412, 1934.
11. Hamaker H. C. The London—Van der Waals attraction between sphe-
rical particles, Physica, 4, 1058—1072, 1937.
12. Hamaker H. C. London—Van der Waals forces in colloidal systems,
Rec. trav. chirm, 57, 61—72, 1938.
13 I n с e C R. A study of differential flotation, Trans. AIMME, 87, 261—
284, 1930.
14. Jahnke, E. and F. E m d e. «Tables of Functions», 2d ed., pp. 124 ff.,
Teubner, Verlagsgesellschaft, Leipzig, 1933.
15. Keesotn W. H. The second virial coefficient for rigid spherical molecu-
les whose mutual attraction is equivalent to that of a quadruplet placed
at its center. Akad. Wetenschap., Amsterdam, Proc., 18, 636—646, 1916.
16. Keesom W. H. Quadrupole moments of the oxygen and nitrogen mole-
УСТОЙЧИВОСТЬ МИНЕРАЛЬНЫХ СУСПЕНЗИЙ
177
cules, Proc. Acad. Sci. Amsterdam, 23, 939—942, 1920; Verslag. Akad.
Wetenschap. Amsterdam, 29, 718—721, 1921.
17. К e n n a r d E. H. Kinetic Theory of Oases, McGraw-Hill Book Co, Inc., 1938.
18. Keyes Frederick G. Chemistry and intermolecular force, Richards'
medal address, American Chemical Society, in Nucleus (June, 1942).
19 Kruyt H. R. and A. E. van Arkel. The velocity of flocullation of se-
’ lenium sol, Rec. trav. China., 39, 656—671 (1920), 40, 169—191, 1921.
20 Levine S. Stability in hydrophobic colloidal solutions, Proc. Roy. Soc.
(London), A170, 1, 145—164, vol. 2, 165—182, 1939.
21. Levine S. On the interaction of two colloidal particles, using the com-
plete Debye—Hiickel equation, J. Chem. Phys., 7, 831—848, 1939.
22. Levine S. The interaction of colloidal particles. I. Particular applica-
tion to parallel plates, Trans. Faraday Soc., 4 42B, 102—117, 1946.
23. Levine S. and G. P. Dube. Interaction between two hydrophobic col-
loidal particles using the approximate Debye—Hiickel theory, Trans. Fa-
radav Soc., 35 (I), 1125—1140, (II), 1141—1156 (1939).
24. L e v i n e S. and G. P. Dube. On the mutual electrical energy of two
colloidal particles, Phil. Mag., 29, № 7, 105—128, 1940.
25. Levine S. and G. P. Dube. Stability properties in hydrophobic sols;
application of the mutual energy of two particles. Trans. Faraday Soc., 36,
215—229, 1940.
26. L e v i n e S. and G. P. Dube. Stability properties of hydrophobic colloi-
dal solutions, J. Phys. Chem., 46, 239—280, 1942.
27. L о n d о n F. Zur Theorie und Systematik der Molekularkraefte, Z. Phy-
sik, Bd. 63, 245—279, 1930.
28. Noyes, Arthur A. and Miles S. Sherill. «Chemical Principles» 2d, ed., rew-
ritten, (a) Table V, p. 165 The MacMillan Company, New York, 1938.
29. Schuhmann. Reinhardt. Ir. Principles of comminution, size distribu-
tion and surface calculations, AIMME. Tech. Publ. 1189, 1940.
30. Slater John C. and John G. Kirkwood. The van der Waals forces
in gases, Phys. Rev., 37, 682—697, 1931.
31. Sulm an H. Livingston. A. contribution to the study of flotation, Trans.
Inst. Mining Met. (London), 29, 44—204, 1920,
32. Sun, Shiou-Chuan, The mechanism of slime-coating, Trans. AIMME, 153, 1943.
33. Taggart Arthur F., T. C. Taylor and C. R. Ince. Experiments witl
with flotation reagents, Trans. AIMME, 87, 285—368, 1930.
34. Taggart Arthur F„ T. C. Taylor and A. F. Knoll. Chemical reac-
tions in flotation, Trans. AIMME, 87, 217—260, especially 238—246, 1930.
35. T u с к e r E. L. and R. E. Head. Effect of cyanogen compounds on flota-
bility of pure sulphide minerals. Trans. AIMME, 73, 354—371, 1925.
36. Verwey E. J. W. Electrical double layer and stability of emulsion,
Trans. Faraday Soc., 36, 192—203, 1940.
37. Verwey E. J. W. On the stability of lyophobic colloids, Philips Research
Repts, 1, 33—49, 1945.
38. V e r w e у E. J. W. Theory of the stability of lyophobic colloids, J. Phys,
and Collod. Chem., 51, 631—636, 1947.
39. Verwey E. J. W. and J. Th. G. Overbee k. Long distance forces ac-
ting between colloidsl particles, Trans. Faraday Soc., 42B, 117—131, 1946.
40. Verwey E. J. W. and J. Th. G. Overbee k. Theory of the stability
of lyophobic colloids, (a), pp. 24—25, (b) table 18, p. 68, (c) Eq. (86),
p. 160, (d) pp. 137—140 (especially eq. (56), p. 139, (e) p. 69, (f) Table XI,
p. 82. Elsevier Press, Inc., Houston, Tex., 1948.
41. Von Smoluchowski M. Drei Vortrage liber Diffusion Brownsche Mo-
lekularbewegung und Koagulation von Kolloidteilchen, Physik, Z., Bd. 17, 1916.
42. Von Smoluchowski M. Versuch einer mathematischen Theorie der Koa-
gulationskinetick Kolloider Loesungen, Z. Phys. Chem., 92, 129—168, 1918.
t2 A. M. Годэи
Глава 7
КРАЕВЫЕ УГЛЫ
Две фазы образуют контакт вдоль двухмерной поверхности
раздела, свойства которой являлись основным объектом рассмот-
рения в гл. 3—6. При соприкосновении трех фаз образуется ли-
нейный .контакт. Свойства этой линии контакта определяются
краевым углом, образующимся в плоскости, перпендикулярной
к линии контакта.
Так как вода является наиболее распространенной жидкой
фазой не только во флотационном процессе, но и в других естест-
венных или промышленных системах, то краевой угол принято
измерять через воду. В случае безводных систем требуется спе-
циальное указание о том, как измеряется краевой угол.
Рис. 1. Краевой угол на границе раздела
ртуть — вода — воздух
Уравнение равновесия. На рис. 1 изображен краевой угол на
границе контакта ртуть — вода — воздух. На рисунке видно, что
величина краевого угла составляет около 100°. Стремление пу-
зырька воздуха, закрепленного на ртути, всплыть вверх является
причиной поднятия уровня ртути под пузырьком.
При равновесии системы силы поверхностных и межфазных
натяжений, действующих на точку О, должны точно уравнове-
шиваться:
^рт ж 4“ ^рт Г 4“ ®ЖГ = 0- ( 1 )
В этом основном уравнении силы должны суммироваться
векториально. Уравнение (1) может быть заменено двумя ска-
лярными уравнениями (2):
КРАЕВЫЕ УГЛЫ
179
отг
Минерал
Рис. 2. Краевой угол на границе
раздела минерал — вода — воздух
той же плоскости. Взаимоот-
арт ж cos а + ажг cos (Q — а) — стрт r cos р = 0; (2a)
арт ж sin а 4- ажг sin (6 — а) — <грт,. sin р = 0. (26)
Первое из этих скалярных уравнений, которое можно назвать
уравнением косинусов, определяет работу закрепления, а второе,
уравнение синусов, определяет подъемную силу пузырька возду-
ха. Углы аир, приведенные
в уравнениях (2а) и (26), изо-
бражены на рис. 1. Фокс [28,
29] сделал обзор явлений, свя-
занных с краевыми углами в
системе, состоящей из двух
жидкостей и газа.
Контакт между поверхно-
стью твердого вещества и дву-
мя подвижными фазами также
определяется краевым углом.
В этом случае обе поверхности
(твердое — газ и твердое —
жидкость) находятся в одной и
ношение между фазами показано на рис. 2.
В этом примере аир равны нулю, и поэтому уравнение ко-
синусов может быть выражено как
cos 0 = -g . (3)
^ЖГ
Уравнение синусов вызывает затруднение, если не постули-
ровать существование новой силы, направленной вертикально.
Эта сила закрепления F А равна
Fa = a.Krsin6. (4)
Когда вместо твердого тела имеется жидкость (например,
ртуть, см. рис. 1), то молекулы жидкости перемещаются одна
относительно другой так, что в жидкости не остается внутренне-
го напряжения. Такой .перегруппировки молекул в твердом теле
не происходит; можно предположить, что в этом случае относи-
тельное положение ядер и электронов в атомах несколько изме-
няется; возможно даже, что межатомные расстояния увеличива-
ются в одном, в двух или нескольких атомных слоях. Такие рас-
тяжения можно рассматривать как причину, вызывающую
напряжение или силу притяжения по линии контакта с твердым
веществом.
В уравнениях (1) — (4) поверхностное и межфазное натяже-
ния выражены в динах на сантиметр. FА является силой за-
крепления или удержания, приходящейся на каждый сантиметр
линии контакта. Эта сила также выражена в динах на сантиметр,..
12*
180
ГЛАВА 7
Значение коаевого угла при флотации. Когда краевой угод
на минерале равен нулю, это означает, что вода смачивает мине-
рал предпочтительнее, чем воздух, и что в воде.не может обра-
зоваться контакт минерал — воздух. Нулевое значение краевого
угла указывает на отсутствие флотируемости.
Наоборот, наличие краевого угла 180° указывает на смачи-
ваемость минерала воздухом и отсутствие смачивания водой.
Не известно ни одно твердое вещество, обладающее в системе
твердое — вода — воздух краевым углом более 110°; поэтому
экспериментально невозможно получить краевой угол 180°.
Рис. 3. Характер взаимодействия твердого тела
с жидкостью
Краевой угол 90° указывает на индифферентность твердого
.тела в отношении обеих подвижных фаз1. В случае флотации
краевой угол 90° обозначает индифферентность в отношении сма-
чивания водой или воздухом. Необходимо определить термин
«индифферентность», что лучше объяснить на примере.
Предположим, что длинный цилиндр частично погружен вер-
тикально в воду, и рассмотрим случаи, когда краевой угол
меньше, равен и больше 90° (рис. 3). Если исключить действие
1 Разграничивать смачивание водой и смачивание воздухом неправильно.
Также нельзя считать правильным и утверждение о том, что при значении
краевого угла 0 = 90° взаимодействие твердого тела с водой и воздухом оди-
наково. Во всех случаях реальное тело будет взаимодействовать с водой зна-
чительно сильнее, чем с воздухом, насыщенным парами воды, а тем более
с воздухом, не насыщенным парами воды (например, при нахождении капли
воды на поверхности вещества). Полное или частичное смачивание твердого
тела жидкостью является функцией как взаимодействия тела с жидкостью,
так н взаимодействия молекул жидкости между собой. Если энергия взаимо-
действия тела с жидкостью больше пли равна энергии взаимодействия моле-
кул жидкости друг с другом, то тело полностью смачивается жидкостью, т. е.
0 = 0°. Значение краевого угла 0 = 90° отвечает случаю, когда энергия взаи-
модействия твердого тела с жидкостью равна половине энергии взаимодейст-
вия молекул жидкости между собой.
При отсутствии взаимодействия твердого тела с жидкостью краевой угол
0 = 180°. Но так как взаимодействие твердого тела с жидкостью как будто
бы всегда имеет положительное значение (дисперсионное взаимодействие), то
в природе не наблюдается краевых углов, равных 180° [62]. Прим. ред.
краевые углы
181
гравитационных сил, то при подъеме цилиндра из жидкости при
0<9О° необходимо затратить работу. В случае когда 0 (>90°,
цилиндр самопроизвольно выталкивается в воздух; когда 0 = 9СГ,
не требуется затрачивать работу ни при подъеме, ни при опуска-
нии цилиндра. При переходе твердого тела из водной фазы в воз-
душную вместо поверхности раздела твердое — жидкость обра-
зуется поверхность твердое — газ; на образование новой поверх-
ности АЛ необходимо затратить работу ;( отг—<»тж) АЛ* или,
согласно уравнению (3), ажгсоз0АЛ. При погружении или
всплывании крупных предметов влияние гравитационных сил на-
столько велико, что величину краевых углов можно не принимать
во внимание. Наоборот, при смачивании тканей и при флотации ,
влияние поверхностных сил, определяемых значениями краевых »
углов, преобладает под гравитационными.
Пузырек воздуха самопроизвольно закрепляется на твердом
теле даже тогда, когда краевой угол меньше 90°. Это явление
всегда наблюдается на флотационных фабриках, где флоти-
руемый минерал редко образует краевой угол в 90°. Доказатель-
ство того, что это явление должно иметь место, излагается
в разделе «Работа закрепления». Прежде чем детально рассмот-
реть работу закрепления, желательно проверить значение урав-
нений (1) — (4) с точки зрения термодинамики. Бессистемное
применение этих уравнений приводит, как показал Харкинс [37],
к неправильным заключениям. Поэтому необходимо кратко рас-
смотреть процесс растекания жидкости по жидкости и жидкости
по твердому телу.
Растекание жидкости по жидкости. Если на спокойную по-
верхность жидкости А нанести каплю жидкости Б, то возможны
три случая:
1) капля жидкости Б смешивается с жидкостью А благодаря
поверхностной и объемной диффузии, образуя раствор жидкости
Б в А (например, капля спирта на воде); обычно скорость расте-
кания значительно больше, чем скорость диффузии в большом
объеме жидкости;
2) капля растекается по поверхности жидкости А, образуя
тонкую пленку, называемую двойной пленкой вещества Б на А;
создавшееся переходное нестабильное состояние в конце концов
приводит к разрушению пленки с образованием монослоя Б на А
и одной или нескольких линз вещества Б, плавающих на моно-
слое и образующих с ним краевой угол; так, бензол образует
на воде чрезвычайно долго существующую двойную пленку;
олеиновая кислота, по-видимому, проходит через те же стадии,
что и бензол, но двойная пленка олеиновой кислоты существует
* В этом случае поверхностные натяжения заменены свободными поверх-
ностными энергиями.
182
ГЛАВА 7
в течение такого краткого периода времени, что образование
монослоя и линз происходит мгновенно;
3) капля сразу образует плавающую линзу; это происходит
с жидким парафином (ньюжел) на воде.
Второй случай наиболее интересен. В нем раскрываются яв-
ления, связанные с краевым углом, и возникает возможность
применить закономерности, наблюдаемые в системе жид-
кость — жидкость — газ, к системам, содержащим твердое ве-
щество. Харкинс определил двойную пленку как фазу, настолько
тонкую, что ее свойства не зависят от силы тяжести, но доста-
точно толстую, чтобы могли существовать две не зависимые одна
от другой свободные межфазные энергии с обеих сторон двойной
пленки, равные по величине свободным межфазным энергиям
объемной жидкости. Согласно этому определению монослой не
может считаться двойной пленкой.
При последовательном рассмотрении растекания бензола по
воде можно прийти к выводу, что'при растекании двойной плен-
ки уменьшается свободная поверхностная энергия. Процесс
образования двойной пленки эквивалентен замене поверхности
вода — воздух поверхностью бензол—воздух плюс поверхность
вода—бензол, имеющих одинаковую площадь. Уменьшение по-
тенциальной энергии на площади 1 см2 при постоянной темпера-
туре и давлении составляет
A F1 = ®жг ^жб ®бг’
где ж, б и г —вода (жидкость), бензол и воздух (газ). Так как
ажг — 72,8 эрг/см2-, <гж6 = 35,0 эрг/см2, а <тбг = 28,8 эрг/см2, то
АЛ = 9,0 эрг/см2.
Двойная пленка самопроизвольно разрушается, образуя лин-
зы и обычно невидимый монослой; поэтому такой процесс тоже
должен приводить к уменьшению потенциальной энергии. Изме-
рением поверхностных давлений установлено, что образование
линз бензола на воде с сопутствующим монослоем уменьшает
поверхностное давление на 10,7 дин/см. Таким образом, самопро-
извольное разрушение двойной пленки приводит ко вторичному
уменьшению свободной поверхности энергии АЛ, много меньше-
му, чем при образовании двойной пленки,
АЛ = ю,7 — 9,0 = 1,7 эрг/см2.
Для других веществ АЛ может быть значительно больше,
a AFi намного меньше, поэтому скорость образования и распада
двойной пленки может быть весьма различна.
У некоторых веществ величина АЛ может быть весьма незна-
чительной. Для нормального парафина с шестью атомами угле-
рода (n-гексана) АЛ = 3,3 эрг/см2, тогда как для парафина
с восемью атомами углерода (n-октана) А/7 = 0,2 эрг/см2. Угле-
КРАЕВЫЕ УГЛЫ
183
водороды с еще более длинной цепью имеют отрицательное зна-
чение Afi; поэтому они не растекаются по воде, образуя на ее
поверхности линзы вместо двойной пленки.
Другие вещества (такие, как сероуглерод и иодистый мети-
лен) образуют монослой и лин-
зы. В случае применения этих ве-
ществ образование двойной
пленки не приводит к уменьше-
нию свободной поверхностной
энергии, тогда как при образова-
нии монослоя свободная поверх-
ностная энергия слегка уменьша-
ется. На рис. 4 приведена диа-
грамма свободной поверхностной
энергии при растекании по воде
некоторых типичных масел.
В табл. 1 дается классифика-
ция систем жидкость — вода.
Ясно, что краевые углы могут
образоваться на границе разде-
ла чистых жидкостей и жидко-
стей, покрытых монослоем адсор-
бированного вещества (рис. 5).
А <00
$ 90
| 80
iv/7
0^
с §
У
i 30
| 20
V ^0
й Г '6йГг^9,9
~Вода, 72j
Wi
70,3— i .
2 '
2 ЗЩБ
.28,8 \&F2=5.9
У
-н-гептило-
вый спирт Бензол
С$г
Рис. 4. Свободная поверхностная
энергия при растекании двойных
пленок и монослоев по поверхно-
сти воды [37):
1— монослой; 2 — двойная пленка
Поведение жидкостей на воде
Таблица 1
Классы Видимое растекание Образование пленок Вещества
1 Есть Нет слоев, нет линз Спирт
2 Вначале образуется двой- Бензол, олеиновая кис-
пая пленка, затем монослой лота
и линзы
3 Нет Монослой и линзы Сероуглерод
4 Только линзы Ньюжел
Характерная особенность построения рис. 5, я и 5, б заклю-
чается в том, что поверхность нижней фазы изображена горизон-
тально, как если бы нижней фазой было твердое тело. На
рис. 5, а нижняя фаза непосредственно находится в контакте с
воздухом без промежуточного монослоя, тогда как на рис. 5, б
такой слой образуется самопроизвольно. Поверхностные натяже-
ния, входящие в уравнение косинусов, для случая, приведенного
184
ГЛАВА 7
на рис. 5, а, состоят из поверхностного натяжения жидкости, по-
верхностного натяжения нижней фазы и межфазного натяжения
между жидкостью и нижней фазой. Этот случай рассматривается
дальше. На рис. 5, б поверхностное натяжение нижней фазы,
покрытой монослоем, принимается за поверхностное натяжение
самой нижней фазы, тогда как на самом деле они различны. Ве-
щества, ведущие себя подобно жирным кислотам, нанесенным
на подкисленную воду, или подобно сероуглероду на воде, ха-
рактерны для случая б, тогда как поведение ньюжела на воде
характерно для случая а.
а 6
Рис. 5. Положение поверхностей раздела в трехфазной системе
Следует отметить, что наличие плотного монослоя и отсут-
ствие слоя адсорбированных молекул на нижней фазе являются
идеальными крайностями. В большинстве случаев адсорбирует-
ся некоторое промежуточное количество молекул жидкости,
образуя частично заполненный монослой. Эти вопросы для слу-
чая, когда нижней фазой являлись твердые тела, были рассмот-
рены Харкинсом и Ливингстоном [40], Ливингсоном [46], Фоксом
и Харкинсом [27], Бойдом и Ливингстоном [15].
Например, если графит поместить в атмосферу водяного
пара, то на нем будут адсорбироваться молекулы воды, хотя и
в меньшей степени, чем на поверхности флотационно неактив-
ных твердых веществ (таких, как кварц или анатаз). Таким
образом, поверхность графитовой частицы во влажном воздухе
должна покрываться в большей или меньшей степени адсорби-
рованными молекулами воды, что видно из данных Ливингстона
145] и схематически приведено на рис. 6.
Работа закрепления. Если система состоит из пузырька воз-
духа и твердой частицы, находящихся в воде, то закрепление
пузырька воздуха на частице приводит к изменению свободной
поверхностной энергии системы. Это изменение можно опре-
делить, исходя из трех межфазных энергий и из изменения пло-
щадей трех поверхностей раздела.
Вводя экспериментальное значение краевого угла, можно
исключить неизвестные свободные поверхностные энергии ми-
нерал— жидкость и минерал — газ. В этом случае изменение
краевые углы
185
энергии системы можно определить, зйая величину свободной
поверхностной энергии жидкости, краевого угла и изменения
площадей трех поверхностей раздела.
Особое внимание следует обратить на системы, содержащие
минералы с плоскими поверхностями, при условии, что: 1) пузы-
рек имеет малые размеры по сравнению с частицей; 2) частица
имеет малые размеры по сравнению с пузырьком.
Рис. 6. Положение поверхностей раздела в си-
стеме графит •— вода — влажный воздух
Случай малого пузырька на большой части-
це1. Предположим, что пузырек воздуха, имеющий радиус г,
самопроизвольно закрепляется в воде на плоской минеральной
Рис. 7. Работа прилипания малых пузырьков к по-
верхности минерала:
* а — начальное условие; б — конечное условие
поверхности площадью А, образуя краевой угол 0 вдоль границы
закрепления сферического сегмента (с радиусом 7?) на поверх-
ности минерала. Уменьшение свободной поверхности энергии
системы (см. рис. 7) определяется разностью поверхностных
энергий до и после закрепления. Радиус площади прикрепления
пузырька b = R sin 0, высота сферического сегмента h = /?(! +
, , „„ 1 4- cos О
+ cos 0), а площадь закрепления — 4 я/?2-------.
1 Это явление рассмотрено Шепардом, Ребиндером с сотрудниками, а бо-
лее подробно Стрельциным и Кизевальтером. Прим. ред.
186
ГЛАВА 7
Убыль свободной поверхностной энергии системы:
LW = (Дсттж + 4тсг2стжг)— (Д — тс Ь2) сттж 4-
+ гсЬ2 сгтг + 4т- ft* .* n~2CCS °—стЖ1.
(5)
После сокращения и замены атг — стж на ажг cose [см. урав-
нение (3)] имеем
Д«7 - -.R-sin’вeosе+ (
Упрощая дальше уравнение, заменяем R = fr, sin2 0 на
1 — cos20 и, вынося за скобки 4л-г2 (первоначальную площадь
поверхности пузырька) и ожг, имеем
Д W = 4л г2 ажг _-/2(1-_S2s26)^° + 1_^l + ^-gL.l .
L 4 2 J
После преобразования получаем
Д W = 4тг г2ажг Г^ЬТТТ9-±-сс s.3A-_2 ~ ^9L + 11 =
[4 J
, 2 / , 24-3 cos 0 — cos3 9 f2 \
= 4тс Г2 Ожг 1--;-----------— г .
\ 4 /
Значение f определяется, если предположить, что перво-
начальный объем пузырька не изменяется после закрепления.
Как известно, объем сферического сегмента с радиусом R равен
'/зяЛ^ЗТ?— h), где /г = /?(1 + cos0). Из равенства объемов
первоначального пузырька и сферического сегмента следует, что
Атсгз = —тс£2(1 4-cos О)2 [37? —7? (1 + cos 9)].
3 3
Упрощая и заменяя R=fr, получаем
2 Ц-3 cos О — cos3 0
Следовательно,
ДГ = 4кг2ажг(1-Т), (6)
где
1
„ 1 / 2 + 3 cos 6 — cos3 0 \ 3
F~T = (----------4--------) ' (7)
Таким образом, работа закрепления определяется произве-
дением площади поверхности первоначального пузырька 4яг2,
свободной поверхностной энергии жидкости стЖ[. и параметра
1 — F, который является функцией только краевого угла 0.
краевые углы
187
Этот параметр положителен для всех значений 0—от 0 до
180°; отсюда следует, что изменение энергии благоприятствует
самопроизвольному закреплению для всех значений краевого уг-
ла, не равного нулю.
Числовое значение 1—F приведено во второй колонке табл. 2.
Эта таблица показывает, что 1—F, а следовательно, и AIF
уменьшается очень быстро с уменьшением 0, в особенности для
углов меньше 15°. Так, например, для 0= 15° значение Д117 со-
ставляет '/is от значения Д1Г для 0 = 30° и 7?оо от значения Д1Р
при 0 = 90°, но оно в 100 раз больше, чем значение ДW
для 0 = 5°.
Таблица 2
Уменьшение потенциальной энергии при закреплении
малого пузырька с первоначальным радиусом
на большой плоской поверхности твердого при
различных значениях краевого угла 91
е град. Фактор 1 - F 9 град. Фактор 1 - F
1 0,000000006 45 0,020
2 0,00000008 60 0,055
5 0,0000036 90 0,206
10 0,000057 120 0,461
15 0,00029 180 1,000
30 0,0044
1 Изменение энергии выражено в единицах 4 г.гг аЖт.
Рассчитано, что если (тжг = 70 эрг/см2, а г=0,1 мм, то Д1^=
= 0,088 (1—F) эрг. Для 0 = 60° работа закрепления равна
0,005 эрг, тогда как для 0 = 5° она составляет 3 • 10' эрг, а для
6 = 1° — около 10-9 эрг.
Очень быстрое изменение работы закрепления малого пузырь-
ка на частице в зависимости от величины краевого угла наводит
на мысль о том, что если краевой угол и пузырек будут достаточ-
но малы, то работа закрепления может достигнуть значения, при-
мерно равного величине тепловой энергии молекул (3/2 kT, или
около 6 10-14 эрг). В условиях, когда работа закрепления рав-
на 10 kT или меньше, можно ожидать, что закрепление будет не-
прочным, и его легко нарушить. Это согласуется с условиями,
приведенными в левой верхней части табл. 3.
Случай большого пузырька и малой частицы.
Работу закрепления во флотационной системе можно прибли-
женно определить, исходя из другого положения. Предположим,
188
ГЛАВА 7
Таблица 3
Вычисленная работа закрепления малых пузырьков на большой
поверхности твердых частиц2
Краевой угол 9 град. Диаметр пузырька, у.
1 10 100 1000
0,5 1 2 5 10 15 30 90 з-ю-3 3,4-10~2 0,44 21,0 3,2-102 6-Ю2 10* 1,2-10» 0,3 3,4 44 2,1-10з 3,2-10* 6-10* 10» 1,2-10s 30 3,4-102 4,4-103 2,1-105 3,2-10® 6-Ю6 IO® 1,2-101° З-Ю3 3,4-10* 4,4-105 2,1- Ю7 3,2-108 6-10» 101° 1,2-1012
1 Выражено в единицах 10 kT.
что тонкая частица (диаметром 5 ц) находится в контакте с
относительно большим пузырьком воздуха (диаметром 2 мм).
Для такой малой частицы поверхность пузырька можно считать
плоской. Закрепление частицы не изменяет объем пузырька, поэ-
тому поверхности раздела вода — воздух и минерал — вода
уменьшаются на S, а поверхность раздела минерал — воздух
увеличивается на S, где S является площадью контакта. Работа
закрепления в этом случае равна
ДГ'==5(-атг4-а1Ж + <тжг). (8)
Применяя уравнение (3) и делая подстановку, получаем
ДГ' = 5<тжг(1 — cos0). (9)
Уравнение (6) выражает работу закрепления малого пузырь-
ка на большой частице, зависящую от произведения поверхно-
сти малого пузырька 4®г2, свободной поверхностной энергии рас-
твора 3,Кг и фактора 1— F, значения которого приведены в
табл. 2. Аналогично этому уравнение (9) выражает работу за-
крепления малой частицы на большом пузырьке, зависящую от
произведения площади контакта У, свободной поверхностной
энергии раствора сжг и фактора 1 — cosQ.
В случае малых пузырьков работа закрепления уменьшается
с уменьшением размера пузырька и краевого угла, а в случае
малых частиц она уменьшается с уменьшением размера частиц
и краевого угла. Это видно из табл. 4.
КРАЕВЫЕ УГЛЫ
189
Таблица 4
Величина работы закрепления кристаллической частицы на относительно
большом пузырьке воздуха1
Краевой угол 9 град. Диаметр частиц, у.
1 10 100 1000
1 10-]3 10~Н ю~9 ю-7
2 ю 12 ю~10 ю'8 ю-6
5 ю-12 ю~10 ю~8 . ю-6
15 10~11 ю~9 ю~7 10' 5
30 10-10 ю~8 ю~6 ю-4
60 ю~9 ю~7 ю~5 10~3
90 ю~э кг7 ю~5 10~3
1 Энергия закрепления выражена в эргах.
В этой таблице приведены расчетные значения, основанные
на том, что поверхность контакта частицы с пузырьком состав-
ляет ‘/io грани правильного куба того же размера. Ясно, что
размер частиц имеет большое значение и что здесь краевой угол
опять является важным фактором.
Обычно во флотационных системах и частицы и пузырьки
малы, поэтому контролирующим фактором при данном краевом
угле может являться и размер пузырьков и размер частиц.
Общий случай. В приведенных выше рассуждениях
предполагалось, что пузырек после закрепления имеет форму
сферического сегмента. Судя по аналогии с измерениями поверх-
ностного натяжения, выполненными методом лежащих капель,
это предположение не совсем верно. Кроме того, при закрепле-
нии несколько изменяется давление, а поэтому изменяется и
объем пузырька. С другой стороны, нет уверенности в том, что
краевой угол остается одинаковым при деформации пузырьков
всех размеров. Например, на основе уравнения (8) изменение ра-
боты закрепления в зависимости от краевого угла [d(&W)/dQ] в
системе с постоянным составом, температурой и давлением дол-
жно при равновесии равняться нулю, или
d(AW)
de
_ /_ । _ _________ \ dS , d (сттж — gTr)__ _ q
гжг °т>к в-гг) . „ ~Г ‘-> . н
d и а и
(10)
190
ГЛАВА 7
Точный анализ проблемы должен основываться на примене-
нии второго закона термодинамики. Со временем этот метод
будет использован при изучении флотационных явлений вместо
приближенного метода, приведенного выше. ,
Возражения другого характера будут приведены при рас-
смотрении удельной работы закрепления как функции о(1—
— cosQ). Некоторые из этих возражений [51] касаются деталей
структуры поверхностей раздела. Хотя при существующем со-
стоянии наших теоретических и практических знаний по этому
вопросу можно оставить без внимания эти возражения, все же в
дальнейшем они будут рассмотрены детально.
Силы, удерживающие частицу при контакте пузырек — ча-
стица. Уравнение косинусов (2а) позволяет определить работу
закрепления, тогда как уравнение синусов (4) позволяет опре-
делить удерживающую силу, зависящую от поверхностного
натяжения, или силу закрепления. Если периметр контакта /,
то сила закрепления равна
f = 1ажг sin а дин, (11)
где а—краевой угол между горизонталью и поверхностью раз-
дела жидкость — газ по линии контакта [в простейшем
случае, когда большие частицы закрепляются на пу-
зырьке воздуха, а может быть равен (но не более!) 0].
В табл. 5 приведено числовое значение максимальной удер-
живающей силы для различных значений краевых углов и частиц
диаметром до 1 мм (значение ожг принято равным 70 дин/см).
Таблица 5
Силы закрепления (в динах) частиц, имеющих форму диска,
на пузырьках воздуха
Краевой угол 9 град- Диаметр частиц, мм
0,0001 0,001 0,01 0,1 1
1 0.00004 0,0004 0,0038 0,038 0,38
2 0 00008 0,0008 0.0076 0,076 0,76
5 0,00019 0.0019 0.019 0,19 1,9
15 0,00057 0 0057 0,057 0,57 5,7
30 0,(01 1 0,011 0,11 Ы 11,0
60 0 0019 0.019 0,19 1,9 19,0
90 0,0022 0,022 0.22 2,2 22,0
120 0,0019 0,019 0.19 1,9 19,0 Г-
Из таблицы следует, что удерживающая сила возрастает
медленно с увеличением краевого угла и что максимальная
удерживающая сила соответствует 90°.
КРАЕВЫЕ УГЛЫ
191
Приведенный расчет сил, удерживающих частицы, слишком
упрощен и для больших частиц не отвечает действительности.
Так как замена двух поверхностей раздела (жидкость — газ и
минерал — жидкость) одной поверхностью раздела (минерал —
газ) приводит к уменьшению потенциальной энергии [см. урав-
нение (8)], то площадь контакта большого пузырька будет рас-
пространяться самопроизвольно, насколько позволяет поверх-
ность минерала. В результате контакт будет распространяться
до достижения линии, отвечающей резкому изменению ориента-
ции поверхности раздела твердое — жидкость.
На рис. 8 показана зависимость краевого угла 0 от угла а
(угла наклона поверхности жидкости к горизонту у острого края
поверхности частицы). У линии контакта определяются направ-
ления векторов поверхностных натяжений минерал — воздух и
жидкость — воздух. Но вектор межфазного поверхностного
натяжения минерал — вода может иметь любое направление
между поверхностью минерала, находящегося в воде, и продол-
жением поверхности раздела минерал — воздух1. В результате
фактический краевой угол может принимать любые значения
между 0! и 02 (рис. 8). Ниже будет показано, что для флотации
это весьма важно.
Если предположить, что минерал имеет такой же удельный
вес, как и жидкость, или частица настолько мала, что можно
пренебречь силой тяжести, то проявление межфазного поверх-
ностного натяжения будет таким, как об этом можно судить по
рис. 9 для различных типичных значений краевого угла (напри-
мер, 15, 45 и 90°). Этот рисунок показывает, что внешний вид
контакта не отражает изменения значения краевого угла: при
любых краевых углах до 90° системы будут иметь одинаковый
внешний вид.
Если минерал обладает большим удельным весом, чем жид-
кость, и размеры его настолько велики, что нельзя пренебречь
силой тяжести, то система должна иметь вид, показанный на
рис. 10. Частица погружается на глубину Н от горизонтальной
поверхности жидкости. Равновесие устанавливается согласно
закону Архимеда, т. е. вес жидкости, вытесненной воздухом и
частицей, должен равняться весу воздуха в вытесненном объеме
и весу частицы. При приближенных вычислениях можно пре-
небречь весом воздуха, и поэтому суммарный вес вытесненной
воды, объем которой равен объему кольца XYZX'Y'Z' плюс
объем призмы YZY'Z', равен весу частицы. Вес воды, вытеснен-
ной из объема кольца, равен произведению вертикальной состав-
1 Этот вопрос подробно рассмотрен Волковой [67]; она предполагает, что
направление межфазного поверхностного натяжения минерал — вода зависит
от микрограней на ребрах частицы. Прим. ред.
Рис. 8. Неопределенное значение краевого угла у ост-
рого края твердой частицы (направление OD для меж-
фазного поверхностного натяжения минерал — вода,
может быть, более вероятно, чем крайние направления
ОВ и OF]
Рис. 9. Контакт на границе раздела минерал — во-
да — воздух для кубических частиц, имеющих не-
значительный вес, при краевых углах 15, 45 и 90°
Рис. 10. Провисание частицы, обладающей доста-
точным весом, на границе раздела вода — воздух;
образовывается видимый краевой угол (а<0)
КРАЕВЫЕ углы
193
ляющей поверхностного натяжения воды на периметр
ажг / sin а. Объем призмы равен SH, где S — площадь контакта
минерал — воздух.
Следовательно,
mg = tn'g + SHg + <тжг/ sin а, (12)
где т — масса частицы;
т' — масса вытесненной воды;
g — ускорение силы тяжести.
Минимальный краевой угол, необходимый для удержания
частицы. В табл. 5 приведены данные о силе закрепления
сжг/з1п0. Эти данные можно рассматривать как равные макси-
мальному значению I сжгз1па при предельном значении краево-
го угла (т. е. при а = 0). Если для случая малых частиц прене-
бречь значением члена SHg, то
S41 “мин . > (13)
тжг ь
где 6МИН — наименьший краевой угол, при котором удержи-
вается частица (интересно оценить значение 0
как функции размера для частиц различного ти-
па).
Предположим, что частицы имеют форму цилиндра (высота
равна диаметру) с удельным весом 6; тогда уравнение примет
следующий вид:
sin 0мин= —~ 1)g-t/2. (13а)
4ТЖГ
Отсюда ясно, что 9МИН будет очень быстро изменяться с из-
менением размера частицы.
Для частиц цилиндрической формы различного удельного
веса и различных размеров минимальный краевой угол, необхо-
димый для удержания частиц, показан на рис. 11 (в логариф-
мических координатах); две из приведенных на этом рисунке
кривых отвечают условиям, при которых отрывающей силой яв-
ляются не сила тяжести, а центробежные силы, имеющие уско-
рение в 10 и 100 раз большие, чем сила тяжести. Расчетные
данные для этого случая были получены при применении урав-
нения (136), в котором вместо ускорения силы тяжести g вве-
дена величина ng, являющаяся ускорением центробежных сил:
sin8„ = (5—(136)
На рис. 11 показано, что даже частицы тяжелых минералов,
подобных галениту, при малых размерах могут легко удержи-
13 А. м. Годэн
194
ГЛАВА 7
ваться благодаря поверхностному натяжению, если образовав-
шийся краевой угол имеет значение, равное нескольким мину-
там. В лабораторной флотационной камере при 1800 об/мин
импеллера с радиусом 2,5 см центробежные силы на периферии
импеллера достигают максимума и превышают силу тяжести
в 90 раз. Даже при максимальной центробежной силе, которую
40 400 4000 40000ju
Размер частиц
Рис. 11. Величина минимального краевого угла, не-
обходимого для удержания частиц, в зависимости
от размера, удельного веса и ускорения:
/ -5 = 75 • 100 g; 2-8 «75 10 3 - 8 = 75 g;
4 — 8 = 3 g; 5 — 3 = 1,25 g
возможно создать во флотационной камере, краевой угол, рав-
ный 10°, вполне достаточен для удержания частиц галенита раз-
мером около 100 ц. При слабом перемешивании или в статиче-
ских условиях на пузырьке воздуха могут удерживаться также
и большие частицы.
Размер частиц, удерживающихся на пузырьках, увеличи-
вается с уменьшением их удельного веса или при закреплении
на частице других пузырьков, которые уменьшают удельный вес
комплекса частица — пузырек.
Максимальный размер флотирующихся частиц. Если в урав-
нении (13) принять угол 0 равным 90° и решить это уравнение
относительно d, то приближенное значение максимального раз-
мера флотирующейся частицы будет определяться следующим
образом:
d = [. 4<7*г ..1г/2 /1ДГ
макс [(8—l)g] • (14'
КРАЕВЫЕ УГЛЫ
195
Например, для золота dMaKC = 1,26 мм. Для других минера-
лов получаются большие значения dMaKC, но обоснованность
приближенных значений, вычисленных без учета члена SHg, при
этом уменьшается [см. уравнение (12)]. Для определения точно-
го значения dMaKC необходимо учитывать этот член уравнения.
Вычисление значений глубины погружения Н в зависимости
от диаметра флотирующейся частицы произвели Б. Фрейд и
X. Фрейд [30]. На основе полученных ими данных [32] составле-
на табл. 6.
Таблица 6
Максимальное значение глубины погружения дисксв,
приготовленных из минерала, в 31 висимости от
диаметра погружаемого диска при краевом угле 90°
Диаметр диска см Глубина погружения см Диаметр диска см Глубина погружения см
0,001 0,003 0,200 0,175
0,002 0,006 0,300 0,212
0,004 0,011 0,500 0,258
0,010 0,023 1 ,о 0,306
0,020 0,039 2,0 0,335
0,040 0,066 8,0 0,372
0,080 0,105 ОС 0,378
0,140 0,145
Критический размер относительно больших частиц, имеющих
форму цилиндра с высотой, равной диаметру, определяется
уравнением (12), которое для этого случая может быть дано в
виде
(8-1)^-^-4ожг = 0. (15)
Это уравнение не может быть решено непосредственно для
d, потому что величина Н зависит от d. Но оно может быть ре-
шено методом приближенных вычислений. Так, например, если
б = 1,25 и g = 980, имеем
d- — ^Hd —1,14 = 0. (15а)
Приняв Н = 0, получим в первом приближении значение
d\ = 1,069 см, для которого соответствующее значение Н\ =
= 0,311. Подставляя это. значение в уравнение (15а), имеем
d2—l,244cZ—1,14 = 0, откуда d2 = 1,859. Для второго прибли-
женного значения d Н2 = 0,331; отсюда d3= 1,919; третье при-
ближение дает значение Н3 = 0,333, откуда d< = 1,925. В этом
13*
196
ГЛАВА 7
крайнем случае максимальный диаметр флотирующейся частицы
почти вдвое больше, чем диаметр, вычисленный на основе при- 4
ближенной формулы [уравнение (14)].
На рис. 12 показана зависимость максимального размера
флотирующейся частицы от ее удельного веса в воде. Если при-
менять упрощенную формулу (14) (нижняя кривая), то значе-
ние dMaKC пропорционально При применении более точ-
ного уравнения (15) зависимость dMaKC от удельного веса ста-
новится более сложной.
Рис. 12. Зависимость максимального размера
флотирующейся частицы от удельного веса в
воде:
1 — с учетом деформации поверхности жидкости; 2 —
без учета деформации поверхности жидкости
Измерение краевых, углов. Для измерения краевых углов
применяется несколько методов. К ним относятся как прямые
измерения углов, основанные на проектировании контура на
экран или на фотографировании изображения, так и косвенные
определения, при которых краевой угол рассчитывают, исходя
из объема или массы закрепившихся пузырьков или капель.
В принципе простейший метод состоит в следующем: погру-
жают в воду пластинку и наклоняют ее до тех пор, пока при
подъеме или опускании пластинки мениск у линии контакта ста-
нет незаметным. Этот метод был применен Адамом и Джессо-
пом [3] и усовершенствован Харкинсом и Фоксом [39]. Особен-
ности метода и детальное описание аппаратуры изложены в
книге Александера «Коллоидная химия» [4]. После того как ис-
чезнет мениск и поверхность жидкости у линии контакта с пла-
краевые УГЛЫ
197
стинкой станет плоской, угол, образованный пластинкой и гори-
зонтальной плоскостью, равен краевому углу.
Второй простой метод заключается в измерении с помощью
оптического гониометра краевого угла капли жидкости, поме-
щенной на полированной поверхности в воздухе, или пузырька
воздуха, помещенного под полированной поверхностью, погру-
женной в водный раствор [11]. Гониометр поворачивают до тех
пор, пока одна из пересекающихся нитей окуляра не будет па-
раллельной поверхности твердого. Затем вторую нить вращают,
пока она не станет касательной к поверхности раздела вода —
воздух. Угол, образованный двумя пересекающимися нитями,
непосредственно определяется на шкале инструмента.
Краевой угол можно измерять и без применения оптическо-
го гониометра. На экран проектируют изображение капли или
закрепленного пузырька и оконтуривают изображение, после
чего угол измеряют транспортиром. Можно также фотографиро-
вать пузырек или каплю, что обеспечивает высокую точность
измерения.
Метод принудительного контакта пузырька с поверхностью
минерала, примененный Таггартом, Тейлором и Инсом [56], яв-
ляется оригинальным вариантом метода оконтуривания. Он от-
личается от других методов тем, что пузырек помещается свер-
ху, а не снизу полированной поверхности. Удерживающие си-
лы поверхностного натяжения соединяют пузырек воздуха с по-
верхностью минерала так же, как при флотации. Было показа-
но, что краевой угол остается неизменным, закрепляется ли пу-
зырек одновременно на держателе и на поверхности минерала
или только на минерале 160]. В этом методе в качестве держа-
теля для пузырька используют стеклянную палочку с углублени-
ем на конце или тонкую стеклянную трубочку. Контролируя по-
дачу газа через тонкую трубочку, можно регулировать размер
пузырька.
В связи с усовершенствованием прибора Таггарт, Тейлор и
Инс предложили некоторые коэффициенты для характеристики
закрепления пузырьков на полированных минеральных поверх-
ностях. Эти коэффициенты включают размеры пузырьков до и
после закрепления. Уорк и Кокс воспользовались этим методом
как одним из возможных способов исследования химических
закономерностей флотационного процесса и при своих первона-
чальных исследованиях предложили ограничиться только изме-
рениями краевых углов указанным методом. Затем они упрости-
ли метод, ограничив его непосредственными измерениями крае-
вых углов в условиях, воспроизводящих обычные условия фло-
тации.
Общепринятая конструкция прибора для принудительного
контакта пузырьков показана на рис. 13. Этот прибор состоит
1Э8
ГЛАВА 7
из прочной подставки, на которой укреплен держатель, плоской
прямоугольной стеклянной кюветы, установленной на столике
микроскопа и источника света. Изображение проектируется на
экран или фотографируется посредством фотонасадки, надетой
на тубус микроскопа.
На рис. 14 приведены характерные краевые углы на чистом
галените.
Примененный Нином и Бентоном [44] метод нанесения капли
на поверхность минерала противоположен методу принудитель-
ного контакта пузырька. Как видно по фотографиям и данным
Тиссена и Шуна [57], оба метода позволяют получить одинако-
вые результаты.
Бартелл и Кардуэлл [8] применяли этот метод для определе-
ния свойств поверхности металла в контролируемой атмосфере,
а Эллефсон и Тейлор [24] — при высокой температуре. В опы-
тах Бартелла и Кардуэлла металл и жидкость, находились в
закрытой камере, но, к сожалению, эти предосторожности не до-
стигли цели, потому что анулировались условиями предваритель-
ной обработки. В способе Эллефсона и Тейлора сильное свечение,
создаваемое раскаленным металлом, обеспечивало освещение об-
ласти контакта.
Другие методы измерения краевого угла основаны на урав-
нении капиллярности, в которое одновременно входят значе-
ния краевого угла и поверхностного натяжения; поэтому для
определения значения краевого угла необходимо независимое
измерение поверхностного натяжения. Среди этих методов име-
ются такие, которые основаны на измерении высоты поднятия
жидкости в капилляре [9, 53] или измерении размера пузырька,
закрепленного на верхней или нижней поверхности минерала
и др. [13, 14, 48, 49.]
Метод, предложенный Маком, применим при измерении кра-
евых углов, образованных при закреплении очень малых пу-
зырьков, которые он рассматривал как сферические. Этот метод
заключается в определении высоты и диаметра сферической
вершины капли или пузырька, спроектированной на экран
или сфотографированной, и в определении расчетом краевого
угла 0 на основе уравнения
O = 2tg~'--, (16)
X
где h — наибольшая высота капли или пузырька;
х — радиус основания сегмента.
Таким образом, краевой угол определяется косвенно на осно-
ве геометрических измерений.
По методу Бикермана определяют вес капли, а краевой угол
рассчитывают по удельному весу жидкости и вычисленному объе-
му всей сферы.
Рис. 13. Аппаратура для изучения при-
нудительного прикрепления пузырька
Г501
а ~ галенит в чистой воде; б— галенит в растворе этилового ксаи-
тогената (краевые углы соответственно равны 0 и 60°)
200
ГЛАВА 7
Обзор существующих методов определения краевых углов
сделал Аллан Фергуссон [25].
Применение прибора для принудительного контакта пузырь-
ка. При работе с прибором для принудительного контакта пу-
зырька требуется, чтобы поверхности минералов были полиро-
ваны (не шлифованы) и свободны от загрязнений-
Приготовлять образцы надо очень тщательно. Обычно образ-
цы минералов закрепляют в бакелитовых брикетах и шлифуют
мокрым абразивом. Затем поверхность минералов полируют на
стеклянном диске, покрытом тонким сукном, в струе воды. Что-
бы предохранить рабочую поверхность минерала от загрязне-
ния руками, необходимо работать в резиновых перчатках. Надо
принимать специальные меры предосторожности [56, 59, 60] и
желательно следить за тем, чтобы применяемые перчатки, по-
суда, стеклянные диски, сукно и абразивы не являлись источ-
никами загрязнения.
Если приняты все меры предосторожности, то большинство
минералов будет обладать нулевым краевым углом в чистой
воде. Это справедливо для различных окисленных минералов,
для большинства сульфидных минералов и неблагородных ме-
таллов. Некоторые исследователи идут дальше и утверждают,
что каждый незагрязненный минерал обладает нулевым крае-
вым углом. Но при рассмотрении этого вопроса необходимо учи-
тывать, что это утверждение не доказано в отношении некото-
рых твердых веществ, таких как графит, уголь, сера, тальк, пи-
рофиллит и борная кислота. Естественная флотационная актив-
ность этих веществ, рассматриваемая в гл. 4, свидетельствует о
том, что на поверхности этих веществ образуется краевой угол,
отличный от нуля.
Если вместо воды применить водный раствор собирателя, то
краевой угол отличается от нуля. Пузырек воздуха, поднесен-
ный к поверхности минерала в растворе собирателя, закреп-
ляется на ней, тогда как в чистой воде если и существует кон-
такт, то весьма неустойчивый. Измерение краевого угла с обеих
сторон профиля пузырька дает различные результаты. Напри-
мер, с одной стороны краевой угол может быть равен 56°, а с
другой 62°. Среднее значение краевого угла 59° можно отнести
ко всей поверхности в целом, в особенности, если оно совпадает
со средним значением измерений, проведенных в различных точ-
ках поверхности. Наблюдаемые отклонения значений краевого
угла от среднего заслуживают дальнейшего изучения в связи с
явлением гистерезиса, которое рассматривается ниже. Обычно
краевой угол увеличивается при повышении концентрации соби-
рателя постепенно, о чем можно судить по данным рис. 15. Ре-
зультаты измерений краевого угла меньше 20° ненадежны, а са-
мо измерение трудно выполнимо. Это отчасти вызвано тем, что
КРАЕВЫЕ УГЛЫ
201
образование малых краевых углов связано с небольшим измене-
нием энергии системы частица — пузырек при закреплении час-
тицы на пузырьке, отчасти же малой величиной вертикальной
составляющей поверхностного натяжения жидкости (удерживаю-
щей силы), которая может маскироваться различными случай-
ными изменениями. Кроме этого, ненадежность измерения ма-
Концентрацая ацетата лауриламина, мг/л
Концентрация ацетата лауриламина, мкмоль/л
Рис. 15. Зависимость величины краевого угла от концентрации аце-
тата лауриламина [50]:
1 — гематит; 2 — сфалерит; 3 — кварц
лых краевых углов обусловливается также и тем. что при малых
концентрациях собирателя краевые углы устанавливаются мед-
ленно из-за незначительных градиентов концентраций собирателя
и, следовательно, медленной диффузии реагента к поверхности.
Трудность измерения малых краевых углов привела Уорка и
Кокса к разработке метода качественного определения свойств
полированной поверхности минерала при различных реагентных
режимах флотации. Они лишь устанавливали, возможен контакт
пузырька с минералом или нет. Этим (нулевым) методом можно
получить двухмерную диаграмму совместного действия двух пе-
ременных величин, например, pH и количества добавляемого эти-
лового ксантогената натрия. Диаграмма показывает, что сущест-
202
ГЛАВА 7
вуют две области: одна соответствует условиям, при которых
контакт возможен, а другая — условиям, при которых контакт не-
возможен. На рис. 16 приведена обычная диаграмма такого типа.
При исследовании влияния трех переменных величин две из
них изменяли, оставляя третью постоянной, и для ряда фикси-
рованных значений третьей переменной получали линии, подоб-
ные изображенной на рис. 16.
Рис. 16. Построение кривых прикрепления частиц к пузырьку на
примере халькопирита в растворе этилового ксантогената калия
25 мг/л [60]:
1 — прикрепление возможно; 2 — прикрепление невозможно
Гистерезис краевого угла. Если не принимать специальных
мер предосторожности, видимые значения краевого угла не бу-
дут одинаковы в различных точках линии трехфазного контак-
та, причем значения краевого угла могут заметно различаться.
Салмэн [54] и Эдсер [21] предполагали, что разность между мак-
симальным и минимальным значениями краевого угла может
служить мерой флотационной активности минерала. Максималь-
ную величину разности между значениями краевого угла они на-
звали гистерезисом. Сейчас становится понятным почему пионе-
ры в теории флотации делали ошибку, считая, что гистерезис яв-
ляется мерой флотационной активности. Несомненно, больший
КРАЕВЫЕ УГЛЫ
203
краевой угол отвечает большей флотационной активности и, воз-
можно, что больший краевой угол создает больший гистерезис.
Таким образом, между гистерезисом и флотируемостью имеется
некоторая непрямая связь, но важны также и другие факторы.
Одним из факторов, вызывающих гистерезис, является ше-
роховатость поверхности. Шероховатая поверхность влияет на
образование видимого краевого угла, хотя она не влияет на ис-
Рис. 17. Влияние шероховатости поверхности на ве-
личину краевого угла при перемещении пузырька
вдоль поверхности
тинный равновесный краевой угол. Если поверхность минерала
не является плоской и в разрезе представляет последовательный
ряд возвышений и углублений, то видимый краевой угол может
значительно отличаться от равновесного краевого угла и значи-
тельно изменяться на различных участках поверхности. Это вид-
но из рис. 17: на рис. 17, а показано, что равновесный краевой
угол равен 45°, на рис. 17, б показано, что видимый краевой угол
может достигнуть 75° или уменьшится до 15°; разница в вели-
чине краевого угла при этом составляет около 60°.
Даже в случае отсутствия шероховатости «натекающий» и
«стекающий» краевые углы при скольжении капли или пузырь-
ка имеют различное значение. Это можно заметить, наблюдая
стекание капли воды по загрязненному оконному стеклу или
по полированной стеклянной пластинке, предварительно обра-
ботанной октадециламинацетатом. Если пластинке сообщить
наклон, достаточный для того, чтобы капля начала стекать, то
будет наблюдаться большая разница между углами «натека-
204
ГЛАВА 7
ния» и «стекания». В связи с этим интересно отметить, что при
одинаковом наклоне для малых капель различие между крае-
выми углами «натекания» и «стекания» меньше, чем для боль-
ших.
С точки зрения физики равновесие капли проанализировано
Макдугаллом и Окрентом [47]. Рассматривая каплю, находящую-
ся на наклонной плоскости с углом наклона а, обозначим краевые
Рис. 18. Краевые углы капли,
находящейся на наклонной
плоскости
углы «натекания» и «стекания»
через 0н и 0С (рис. 18).
Если через центр тяжести
капли провести вертикальную
плоскость, перпендикулярную
наклонной плоскости, то сумма
сил, действующих на тонкий
слой жидкости толщиной dl и
площадью А, равна нулю:
<s dl cos 0H — ? dl cos 9С — A dl 7 g sin a, (17)
где g—ускорение силы тяжести;
у — удельный вес жидкости.
Следовательно,
cos9H — cos9c = ^-S- A sin a. (18)
6
Здесь коэффициент yg/a характеризует жидкость, а Asina—
размер и наклон капли.
Макдугалл и Окрент экспериментально показали, что 0„ име-
ет постоянное значение, тогда как 0С может изменяться от зна-
чения, равного 0Н (когда а = 0), до минимального значения, ко-
торое отвечает условиям стекания капли. Из данных этих ис-
следователей можно сделать вывод, что даже на совершенно
гладкой поверхности имеется два значения краевого угла: 0,,,
равное значению, получаемому на горизонтальной твердой по-
верхности, и 0смин, соответствующее условиям стекания.
Бикерман [12] также ясно показал роль силы тяжести в ис-
кажении капель или пузырьков, закрепленных на негоризон-
тальных поверхностях. Давно известно, что краевые углы «на-
текания» и «стекания» не равны. Аблетт [1] экспериментально
показал при применении парафинированного стеклянного цили-
ндра, помещенного в сосуд с водой, что краевой угол 0 в стацио-
нарных условиях равен 104°34'. При вращении цилиндра всякий
раз, когда периферическая скорость парафинированного цилин-
дра превышала 0,5 мм/сек, углы «натекания» и «стекания» ста-
новились равными 113°09' и 96°20' соответственно. Результаты,
полученные Аблеттом, хорошо согласуются с данными МакДу-
галла и Окрента.
КРАЕВЫЕ УГЛЫ
205
Краевые углы «натекания» и «стекания» различаются не
только из-за шероховатости поверхности, влияния силы тяже-
сти и силы трения, но, по-видимому, также из-за неравновесного
состояния системы, что неизбежно в условиях эксперимента.
Даже в системе парафин—азот—вода могут отсутствовать
равновесные условия на поверхности. Это можно заключить из
термодинамического анализа Грегга [36] и из ранних работ Хар-
кинса и Дальстрома [38], Бангема и Разука [7] и Бангема [5, 6].
Несомненно далее, что даже при такой простой системе, как га-
ленит— воздух—водный раствор ксантогената, достижение рав-
новесных условий может оказаться затруднительным. Допустим,
что минерал, смачивающийся чистой водой, погружают в водный
раствор ксантогената. Предположим также, что до введения пу-
зырьков воздуха система водный раствор ксантогената — мине-
рал находится в равновесии, т. е., что поверхность галенита в
этих условиях по крайней мере частично покрыта адсорбирован-
ными группами ксантогената. Если пузырек воздуха приблизить
к поверхности, двойной слой, окружающий галенит и пузырек,
может измениться существенным образом. При распространении
контакта между пузырьком и минералом невозможно предска-
зать останется ли сорбированный слой ксантогената неподвиж-
ным или он будет двигаться опережая трехфазную линию кон-
такта. Вполне вероятно', что свойства поверхности галенита на
границе с водой и воздухом не одинаковы.
Предположим, что смоченная водой поверхность галенита
высушивается в чистом воздухе (экспериментально это трудно
выполнимо), а затем на эту поверхность наносится капля вод-
ного раствора ксантогената. Капля раствора будет стремиться
распространиться по всей поверхности минерала. Так как
ксантогенат начинает ориентированно закрепляться на поверх-
ности минерала (см. главу 9), то поверхность должна стать ме-
нее смачиваемой. При этом на границе трех фаз образуется оп-
ределенный краевой угол, который составляет приблизительно
90° для раствора, содержащего 25 мг/л амилового ксантогената
калия. В целом данное явление не должно отличаться от явле-
ния образования двойной пленки жидкости с последующим ее
разрушением и образованием монослоя и линз, рассмотренных
выше в этой главе.
Понятие о равновесном краевом угле основывается на пред-
посылке, что имеется достаточное время для того, чтобы уста-
новилось равновесие между соприкасающимися фазами. Хотя в
примере с галенитом минерал и находится в равновесии с рас-
твором, но это еще отнюдь не означает, что трехфазная систе-
ма в целом может достигнуть равновесия в приемлемый проме-
жуток времени [22].
”06
ГЛАВА 7
Это явление недавно изучали де Брёйн, Овербик и Шуман
[16]. С их точки зрения для каждой пары фаз имеются равно-
весные условия, относящиеся к соответствующей поверхности
раздела. У линии контакта трех поверхностей соприкасаются
три адсорбированных слоя, отличающиеся один от другого. Так,
в случае контакта пузырька воздуха с полированной поверх-
ностью минерала состав адсорбированного слоя на границе
твердое—газ отличается от состава адсорбированного слоя на
поверхности раздела твердое—жидкость. Любое боковое пере-
мещение пузырька связано с необходимостью перемещения ад-
сорбированных слоев, а так как это не происходит мгновенно,
то «натекающий» и «стекающий» краевые углы (которые не яв-
ляются в этом случае равновесными) должны быть различны.
Суммируя все, что известно о гистерезисе краевых углов, мож-
но сказать, что имеются, по меньшей мере, две главные причи-
ны гистерезиса — большая шероховатость и трение на плоской
поверхности; третья причина может заключаться в том, что рав-
новесие между тремя фазами достигается не мгновенно. По-ви-
димому, гистерезис не является причиной флотационной актив-
ности, но, вероятно, всегда ей сопутствует.
Смачивание кристаллических и пористых поверхностей.
Краевые углы представляют собой главный фактор, определяю-
щий смачивание или несмачивание пористых пространственных
структур, в которых промежутки между повторяющимися эле-
ментами структуры малы (около 1 мм и менее). 'Гак, например,
несмачиваемость тканей [55, 61], поверхности растений или кожи
животных {17—19] и т. д. определяется величиной краевого угла
и геометрией повторяющихся структурных элементов. Эти же
факторы в некоторых случаях влияют и на фильтрацию [2]. Они
также играют основную роль при флотации многих минералог.
На рис. 19 схематически показано влияние величины краевого
утла на положение призм квадратного сечения на поверхности
воды. Если значение краевого угла находится в пределах от 0 до
90°, то' у призмы имеется одна сухая грань, а остальные— смоче-
ны водой. При величине краевого угла от 90 до 180° только одна
грань призм смочена водой, а остальные — сухие. Когда 0 = 0 или
180°, теоретически все грани призм соответственно должны сма-
чиваться или оставаться сухими.
В случае трехмерного повторения структуры, указанной на
рис. 19, а, при 0=0 получаются результаты, приведенные на
рис. 20, а; результаты, соответствующие 0 = 180°, даны на
рис. 20, б и при промежуточном значении краевого угла — на
рис. 20, в.
Все, что было сказано выше о расположении элементов приз-
матической формы, приложимо с небольшими изменениями к
краевые углы
20?
обладающие
протекания
Воздух
структурам, состоящим из элементов цилиндрической, сфери-
ческой, плоской или неправильной формы.
В природе встречаются поверхностные слои,
структурой, которая обусловливает возможность
жидкости главным образом
в одну сторону, подобно од-
носторонне действующему
клапану [41].
В биологии [10, 26, 411
встречается много веществ,
структура которых обуслов-
ливает указанные выше яв-
ления, а именно, смачива-
ние и несмачивание, протека-
ние жидкости через твердые
тела и течение жидкости
лишь в одну сторону. Так,
например, утки легко пла-
вают по воде, потому что
пух, покрывающий их кожу,
образует как бы простран-
Жидкость
а
Возду*
Рис. 19. Влияние величины краевого-
угла на структуру, состоящую из одно-
го ряда параллельных призм квадрат-
ного сечения:
а - - в< 90°; б — 0> 90°
ственную сетку с малыми, достаточно часто повторяющимися
элементами, через которые вода не может проникнуть и смочить
Рис. 20. Влияние величины краевого угла на трехмерную повторя-
ющуюся структуру:
а — 0 ~ 0°; б — 0 ~ 180°; в — 0~ 90°
кожу птицы. У некоторых насекомых на ногах имеется гидро-
фобное покрытие, благодаря которому в воде создается боль-
шой краевой угол, что позволяет таким насекомым свободно
скользить по воде. Растения в засушливых местностях имеют
строение, которое позволяет им испарять воду в минимальных
количествах, и в то же время помогает этим растениям усилен-
но впитывать воду из окружающей среды. Растения, развиваю-
щиеся во влажном климате, имеют совершенно другое строение:
капли дождя или росы легко стекают с них.
;08
ГЛАВА 7
Уместно сопоставить флотационные свойства пористых час-
тиц со свойствами кристаллов, обладающих большими плоски-
ми гранями. Предположим, что структурные элементы пористой
частицы имеют форму куба, площадь грани которого равна
1 ц2. Предположим далее, что кристаллическая частица имеет
форму параллелепипеда с площадью наибольшей грани, рав-
ной 100 ц2 и высотой 1 ц; предположим также, что пористая и
кристаллическая частицы образуют в воде одинаковый краевой
угол, например в 30°. Удерживающая сила, приходящаяся на
один элемент пористой частицы, будет равна примерно
0,014 дин, тогда как для крупной кристаллической частицы эта
сила будет в 100 раз больше и равна 1,4 дин. Этот расчет сделан
из предположения, что пузырек воздуха, придя в соприкоснове-
ние с гранью, будет быстро распространяться по плоской по-
верхности, доходя точно до края грани. В то же время, измене-
ние свободной поверхностной энергии системы при закреплении
кристаллической частицы будет в 10 000 раз больше, чем при
закреплении элемента пористой частицы [см. уравнение (9)].
Таким образом, расчет показывает, что закрепление более ве-
роятно и более эффективно, когда минеральные частицы пред-
ставлены крупными кристаллами, а не пористыми частицами.
Если пористый минерал так обработан углеводородным мас-
лом, что образует краевой угол в воде значительно больше 90°,
то масло в виде капель, покрывающих минерал, будет прони-
кать непосредственно в пористую структуру, как это показано
на рис. 20, б (только в данном случае масло занимает место
газа) и закрепление пористых частиц на пузырьке газа станет
аналогичным закреплению непористой масляной поверхности
на пузырьке. Краевой угол на границе масло — вода — воздух
также может быть больше, чем на границе минерал — вода —
воздух, и поэтому флотация будет протекать успешно. Возмож-
но, что широко распространенное применение углеводородных
масел при флотации фосфорита объясняется этим явлением
[331, графически изображенным на рис. 21.
Зависимость краевого угла от адсорбции. Очевидно, что меж-
ду величиной краевого угла и плотностью адсорбированного слоя
поверхностно активного вещества должна существовать зависи-
мость. Например, галенит в чистой воде имеет краевой угол, рав-
ный нулю, но в растворе, содержащем 20 мг/л (или более) эти-
лового ксантогената калия, краевой угол равен 60°. При проме-
жуточных концентрациях между 0 и 20 мг/'л краевой угол имеет
значения от 0 до 60°. Также известно, что галенит извлекает ксан-
тогенат из раствора. Зависимость между количеством поглощае-
мого галенитом ксантогената и концентрацией раствора пока
точно не определена.
Интересные данные о зависимости между значением крае-
КРАЕВЫЕ УГЛЫ
209
вого угла и адсорбцией поверхностно активного вещества на
минерале были получены Морроу на гематите при применении
лауриламина [34, 50]. Графически эта зависимость приведена
на рис. 22, на котором видно, что как адсорбция, так и краевой
угол увеличиваются по мере увеличения концентрации собира-
теля, но что между ними не существует простой зависимости.
Рис. 21. Взаимодействие пузырька воздуха с гидрофобным порис-
тым телом:
а — поры не заполнены маслом; б — поры заполнены маслом
Основываясь на измерениях адсорбции и краевого угла, Мор-
роу рассчитал работу закрепления гематита и кварца на пу-
зырьке воздуха в водном растворе лауриламина как функцию
поверхностного покрытия. На рис. 23 приведены расчетные дан-
ные. Так как для полной флотации минерала достаточно, чтобы
адсорбция была меньше плотного мономолекулярного слоя, то
особый интерес для флотации представляют лишь левые ветви
кривых. Из полученных данных Морроу также рассчитал энер-
гию адсорбции в калориях на моль (табл. 7).
Из табл. 7 следует, что вначале энергия адсорбции очень ве-
лика, но с возрастанием плотности покрытия она постепенно
уменьшается и даже принимает отрицательные значения при
приближении адсорбционного слоя к мономолекулярному.
В системе, состоящей из пузырька, закрепленного на твер-
дой частице, газовая фаза должна быть насыщена парами воды
или близка к насыщению ими. Участок поверхности частицы,
находящийся под пузырьком, должен адсорбировать пары во-
ды. Плотность адсорбированной пленки воды на поверхности ча-
стицы зависит от свойств самой частицы. Дос и Рао [20] утвер-
ждают, что когда краевой угол равен нулю, то поверхность ча-
стицы полностью покрыта адсорбированными молекулами воды.
Но когда краевой угол больше нуля, адсорбционное покрытие
и А. М. Годэн
краевые углы
211
Таблица 7
Энергия адсорбции (изменение роботы закрепления с изменением
плотности адсорбционного слоя собирателя)
Минерал Концентрация раствора Плотность адсорбцион- ного слоя % монослоя Свободная поверхностная энергия эргом2 Краевой угол 9 град. Энергия адсорбции кал;моль
мг;л мкмоль-л
0,8 3,2 2 72,2 4 —
Кварц 4,3 17,5 5 71,3 12 2690
17,3 70,0 10 70,2 20 1460
80,0 318,0 25 64,9 28 504
135,0 550,0 50 61,4 34 301
240,0 980,0 100 56,7 40 97
292.0 1190,0 125 55,0 42 —20
4,6 18,8 2 71,3 15 3830
21,0 86,0 5 69,9 22 2920
Гематит 66,0 268,0 10 66,2 29 1985
132,0 540,0 15 61,5 35 1410
252,0 1030,0 25 56,3 40 240
550,0 2250,0 50 48,9 44 —60
980,0 4С00,0 100 42,5 45 —60
поверхности молекулами воды не является сплошным и умень-
шается с увеличением значения краевого угла. Согласно
Досу и Рао, существование краевого угла можно рассматри-
вать как доказательство того, что покрытие поверхности водой
из воздуха, насыщенного парами воды, не является полным (см.
рис. 6). М. А. Кук и его сотрудники указывали на существова-
ние линейной зависимости величины краевого угла от плотно-
сти адсорбционного покрытия [56]. С. Р. Б. Кук указывает, что
существует линейная зависимость косинуса краевого угла от
плотности адсорбционного покрытия 152]. Данные, полученные
в Массачусеттском технологическом институте, количественно
не подтверждают ни одной из указанных зависимостей, но в то
же время подтверждают в общих чертах, что зависимость меж-
ду адсорбцией и краевым углом существует.
Фокс и Харкинс 127] рассчитали адсорбцию на поверхности
минерала поверхностно активного вещества из раствора, при-
меняя оригинальные комбинации различных измерений.
Они определяли следующие величины:
1) поверхностное натяжение в зависимости от концентрации
раствора;
2) значение краевого угла в зависимости от концентрации
14*
212
ГЛАВА 7
раствора в системе минерал — раствор — воздух (воздух насы-
щен парами воды).
Из замеров поверхностного натяжения при различной кон-
центрации поверхностно активных веществ и из уравнения Гиб-
бса была определена адсорбция на поверхности раздела жид-
кость— газ. Сопоставление данных по величине краевого угла и
поверхностного натяжения позволило рассчитать адсорбцию на
поверхности раздела твердое — жидкость. Поверхностное дав-
ление на поверхностях раздела твердое — жидкость такое же,
Число атомов С в алкильной
группе
Рис. 24. Зависимость величины
краевого угла от длины угле-
водородной цепи [60]:
— О —изоксаитогенаты;-------нор-
мальные ксантогенаты
как и на поверхностях раздела жидкость — газ. При адсорбции
на некоторых веществах (таких, как парафин, стибнит, графит
и тальк) бутиламина, бутилового спирта, масляной кислоты и
подобных ей жирных кислот с короткой цепью адсорбирован-
ные пленки обладают свойствами двухмерных идеальных
газов.
Зависимость величины краевого угла от длины углеводород-
ной цепи поверхностно активных веществ. Классические экспе-
рименты в этой области были проведены Уорком и Коксом [60],
которые установили, что при условии предельной адсорбции
минеральной поверхностью собирателя имеется зависимость
между значением краевого угла и длиной углеводородной цепи
собирателя.
На рис. 24 приведены результаты, полученные с различными
сульфидными минералами и с различными ксантогенатами. Из
рисунка видно, что максимальный краевой угол с увеличением
длины углеводородной цепи увеличивается сначала быстро, за-
тем все медленнее, приближаясь к значению краевого угла, ха-
рактерного для твердого парафина.
Обычно максимальный краевой угол, по-видимому, одина-
ков для различных сульфидных минералов (галенита, сфа-
лерита, пирита, борнита, халькопирита, халькозина, пирротина
и др)., когда применяется один и тот же собиратель (табл. 8).
Интересно также отметить, что величина краевого угла, по-
видимому, не зависит от полярной группы собирателя. Экспери-
КРАЕВЫЕ УГЛЫ
213
Таблица 8
Независимость максимальных значений краевых углов от природы
сульфидных минералов при применении различных собирателей [59]
Минерал Натриевый диэтилдитио* карбамат Бензнлмер- каптан Метиловый ксантогенат калия Цетиловый ксантогенат калня
Галенит 59 71 50 100
Сфалерит 59 71 51 97
Пирит 58 69 49 95
Халькопирит 59 72 52 94
Борнит 62 72 51 95
ментальные данные приведены в табл. 9. Эти факты рассматри-
ваются как доказательство того, что покрытие собирателем
идентично по структуре и ориентации на различных минералах.
Таблица 9
Значение максимального краевого угла в град., полученного при
применении собирателей с различной структурой полярной группы
для сульфидных минералов [59]
Тип собирателя Углеводородная цепь Тип собирателя Углеводородная цепь
этиловая бутиловая этиловая бутиловая
Дитио карбамат 60 77 Дитиофосфат . . 59 76
Меркаптан . . . 60 74 Тритиокарбонат 61 74
Ксантогенат . . 60 74 Монотио карбонат 61 73
Зависимость величины краевого угла от потенциала поверх-
ности и поверхностного давления. Наряду с зависимостью меж-
ду значением краевого угла и адсорбцией на поверхности твер-
дого, должна существовать зависимость между значением крае-
вого угла и потенциалом поверхности.
Интересные работы в этой области были проведены Фрум-
киным и его сотрудниками [31, 35, 43] на жидкой ртути и твер-
дой платине. Типичные результаты приведены в табл. 10. Одна-
ко убедительных данных, доказывающих, что поверхности ме-
таллов были свободны от загрязнений, отсутствуют. Поэтому
приведенные данные могут оказаться не вполне воспроизводи-
мыми. Но они показывают систематическое изменение значения
краевого угла в зависимости от потенциала поверхности.
214
ГЛАВА 7
Таблица 10
Изменение значения краевого угла в град., пузырька воздуха на
поверхности ртути и платины в зависимости от потенциала
поверхности [Ь7] и |55]
Потенциал поверхности b Ртуть Платина
н. Na2SO4 9 1 0 -и. Na2S04 и. Na2S04 н. Na2S04 0,1-н. H2S04
—0,8 43 42
—0,6 — — 47 48
—0,4 — — 55 51
-0,2 — 70 63 62
0 85 90 68 68
+0,2 + 0,4 97 97 65 65
100 100 56 55
+0,6 101 100 47 45
+0,8 98 98 39 • —
+ 1,0 92 89 32 —-
+1,1 — 82 — —
+ 1,2 80 — — —
+ 1,4 56 —- — —
+ 1,6 18 — — —
Джура и Харкинс [42], исходя из подобных же рассуждений,,
пришли к заключению, что должна существовать зависимость
между краевым углом на пленке, полученной при определен-
ном поверхностном давлении, и давлением пленки.
ЛИТЕРАТУРА
1. Ablett R. Ап investigation on the angle of contact between paraffin
wax and water, Phil. Mag., 46, № 6, 244—256, 1923.
2. Adam N. K. Principles of penetration of liquids into solids, Discussion
Faraday Soc., 3, 5—11, 1948.
3. A d a m N. K. and G. Jessop. Angles of contact and polarity of solid
surfaces, J. Chem. Soc., 127, 1863—1868, 1925.
4. Alexander Jerome ed. Colloid Chemistry, VI, pp. 59—60, Reinhold
Publishing Corporation, New York, 1946.
5. В a и g h a m, D. H. The Gibbs adsorption equation and adsorption on so-
lids, Trans. Faraday Soc., 33, 805—811, 1937.
6. Bangham, D. H. The recognition of phase transitions in adsorbed films.
J. Chem. Phys., 14, 352, 1946.
7. Bangham D. H. and R. I. R a z о u k, Adsorption and the wettability
of solid surfaces, Trans. Faraday Soc., 33, 1459—1463, 1937.
8. В a r t e 11 R. E. and Paul H. Cardwell. Reproducible contact angles
on reproducible metal surfaces, J. Am. Chem. Soc., 64, 494—497, 1530—
1534, 1641—1643, 1942.
9. Bartell R. E. and A. D. Wooley. Solid-liquid-air contact angles and
their dependence upon the surface conditions of the solid, J. Am. Chem.
Soc., 55, 3518—3527, 1933.
краевые углы
215
10. В е a m е п t J. W. L. The role of wax layers in the waterproofing of insect
cuticle and egg-shell, Discussions Faraday Soc., 3, 177—182, 1948.
11. Bigelow W. C., D. L. Pickett and W. A. Zisman. Oleophobic mo-
nolayers. I. Films adsorbed from solution in non-polar liquids, J. Colloid
Sci., 1, 513—538, 1946.
12. Bikerman J. J. On the formation and structure of multilayers, Proc.
Roy. Soc., (London), A170, 130—144, 1939.
13. Bikerman J. J. Contact angles of built-up monolayers Trans. Faraday
Soc., 36, 412—417, 1940.
14. Bikerman J. J. Measuring contact angles, Ind. Eng. Chem., Analyt.
Ed., 13, 443—444, 1941.
15. Boyd G. E. and H, K. Livingston. Adsorption and the energy chan-
ges at crystalline solid surfaces, J. Amer. Chem. Soc., 64, 2383—2388, 1942.
16. De Bruyn P. L., J. Th. G. Overbee k, and R. Schuhmann. Flota-
tion and the Gibbs adsorption equation, Mining Eng., 6, 519—523, 1954:
17. Cassie A. B. D. Contact angles, Discussions Faraday Soc., 3, 11—16,
1948.
18. C a s s i e A. B. D. and S. Baxter. Wettability of porous surfaces, Trans.
Faraday Soc., 40, 546—551, 1944.
19. Cassie A. B. D. and S. Baxter. Large contact angles of plant and
animal structures, Nature, 155, 21—22, 1945.
20. Doss K. S. Gururaja, and Basrur Sanjiva Rao. A theory of contact
angles, Proc. Indian Acad. Sci., 7A, 113—117, 1938.
21. Eds er Edwin. The concentration of minerals by flotation, Brit. Assoc.
Advance, Sci., Fourth Rept. on Colloid Chemistry and its general and in-
dustrial applications, pp. 263—326, H M. Stationery Office, London, 1922.
22. Eichborn. Johann-Ludwig von. Zur Frage des gegenseitings Haftens
raumlich nicht mischbarer Stoff, I, Kolloid Z., 107, 107—128, 1944.
23. Eichborn Johann-Ludwig von. Zum gegenseitiger Haften raumlich nicht
mischbarer Stoffe, II. Kolloid-Z., 109, 62—78, 1944.
24. Ell ef son Bennett S. and Nelson W. Taylor. Surface properties of
fused salts and glasses, J. Am Ceram. Soc., 21, 193—205, 1938.
25. Ferguson Allan. The measurements of contact angles, Proc. Phys. Soc.,
(London), 53, 554—565, 1941.
26. Fogg G. E. Adhesion of water to the external surface of leaves, Discus-
sions Faraday Soc., 3, 162—169, 1948.
27. Fowkes, Frederick M. and William D. Harkins. The state of mono-
layers adsorbed at the interface of solid-aqueous solutions, J Am Chem
Soc., 62, 3377—3386, 1940.
28. Fox William. Contact angles at liquid-liquid-air interface J. Chem. Phy-
sics, 10, 623—628, 743, 1942.
29. Fox William. Equilibrium relationships between fluid, interfaces: the sy-
stem methylene iodide-water-air, J. Am. Chem. Soc., 67, 700—703, 1945.
30. F r e u d В. B. and H. Z. Freud. A theory of the ring method for the
determination of surface tension, J. Am. Chem. Soc., 52, 1772—1782, 1930.
31. Ф p у м к и н А., А. Г о p о д е ц ка я, Б. Кабанов и Н. Некрасов.
Электрокапиллярные явления и смачивание металлов в растворах элек-
тролитов, Ж. Ф. X., 351, 1932.
32. G a u d i и А. М. «Principles of mineral dressing», p. 352, McGraw-Hil'
Book Company, Inc., New York, 1939,
33. G a u d i и A. M. The flotation process, in Jerome Alexander, ed., «Col-
loid Chemistry», VI, pp 493—495, Reinhold Publishing Corporation,
New York, 1946.
34. Gaudin A. M., and J. G. Morrow. Adsorption of dodecylammonium
acetate on hematite and its flotation effect Mining Eng., 6, 1196—1202,
1954.
35. Городецкая А. и Б. Кабанов. Электрокапиллярные явления и
216
ГЛАВА 7
смачивание металлов в растворах электролитов /К Ф X., 5, 418—431,
1934.
36. Gregg S. J. Hysteresis of the contact angle, J Chem. Phys., 16, 549—
550, 1948.
37. Harkins William D. A general thermodynamic theory of the sprea-
ding of liquids to form duplex films and of liquids or solids to form mul-
tilayers, J. Chem. Phys., 9, 552—568, 1941.
38. Harkins William D. and R, Dahl strom. Wetting of pigments and
other powders, Ind. Eng. Chem., 22, 897—902, 1930.
39. Harkins William D. and Frederick M. Fowkes. The state of mono-
layers adsorbed at the interface solid-aqueuos-solution, J. Am. Chem. Soc.,
62, 3377—3386, 1940,
40. Harkins William D. and H. K. Livingston. Energy relations of
the surfaces of solids. II. Spreading pressure as related to the work of ad-
hesion between a solid and a liquid, J. Chem. Phys., 10, 342—356, 1942.
41. Hurst H. Assymmetrical behaviour of insect cuticle in relation to water
permeability, Discussions Faraday Soc., 3, 193—210, 1948.
42. Jura George and William D. Harkins. The contact angle between wa-
ter and a monolayer of egg albumin on glass as a function of film pres-
sure, J. Colloid Sci., I, 137—140, 1946.
43. Кабанов Б. и H. Иванищенко. Электрокапиллярные явления и сма-
чивание металлов. III. Влияние поверхностноактивных веществ на смачи-
вание. Свойства многомолекулярных слоев, Acta Physicochim URSS, 6,
701—718, 1937, Известия АН СССР, серия химическая, 5, 755, 1936.
44. Kneen Eric and W. W. Benton, A simplified technic for the determi-
nation of contact angles and its application to studies of wetting. J. Phys.
Chem., 41, 1195—1203, 1937.
45. Livingston Herbert K. Adsorption and the free surface energy of so-
lids, Ph. D. thesis, University of Chicago, 1941.
46. Livingston Herbert K. Contact angles and adsorption on solid surfa-
ces. J. Phys. Chem., 48, 120—124, 1944.
47. M a c d о u g a 11 G. and C. Ockrent. Surface energy relations in liquid-
solid systems. I. The adhesion of liquids to solids and a new method of
determining the surface tension of liquids, Proc. Roy. Soc. (London), A180,
151—1173, 1942.
48. Mack Guilford L. The determination of contact angles from mesurements
of the dimensions of small bubbles and drops. I. The spheroidal segment
method for acute angles, J. Phys. Chem., 40, 159—167, 1936.
49. Mack Guilford L. and Dorothy A. Lee. The determination of contact an-
gles from measurements of the dimensions of small bubbles and drops. IL
The sessile drop method for obtuse angles. J. Phys. Chem., 40, 168—176,
1936.
50. Morrow John George. Adsorption of dodecylammonium acetate on hema-
tite and sphalerite, cfissertation for the doctorate, Massachusetts Institute
of Technology, 1952.
51. Pease Daniel C. The significance of the contact angle in relation to the
solid surface, J. Phys. Chem., 49, 107—110, 1945.
52. Philipoff W„ S. R. Cooke and Donald E. Caldwell. Contact an-
gles and surface coverage. Mining Eng., 4, 283—286, 1952.
53. Richards Theodore W. and Emmett K. Carver. A critical study of
the capillary rise method of determining surface tensions, with data for
water, benzene, chloroform, carbon tetrachloride, ether and dimethylani-
line, J. Am. Chem. Soc., 43, 827—847, 1921.
54. S u 1 m a n H L. A contribution to the study of flotation, Trans. Inst. Min.
Met. (London), 29, 44-138, 1920.
КРАЕВЫЕ УГЛЫ
217
55. Shuttleworth R. and G. L. J. Bailey. The spreading of a liquid
over a rough solid, Discussions Faraday Soc., 3, 16—22, 1948.
56. T a g g a r t A. F., T. C. Taylor and C. R. Ince. Experiments with flo-
tation reagents, Trans. AIMME, 87, 285—386, 1930,
57. Thiessen P. A. and Elfriede Sc ho on. Work of adhesion on surfaces
of solid organic compounds. Z. Electrochem., 46, 170—181, 1940.
58. Wadsworth Milton E., Robert G. Conrady and Melvin. A. Cook..
Contact angle and surface coverage for potassium ethyl-xanthate on gale-
na according to the free-acid collector theory, J. Phys. Chem., 55, 1219—
1230, 1951.
59. Wark Ian William. «Principles of Flotation», Australasian Institute of
mining and metallurgy, Melbourne, Australia, 1938.
60. Wark Ian William and Alwyn Birchmore Cox. Principles of flotation..
I. An experimental study of the effect of xanthates on contact angles at
mineral surfaces, Trans. AIMME, 112, 189—244, 1934,
61. Wakeham Helmut, Winston B. Strickland and Evald L. Skan.
The water repellency of textile fabrics, Am. Dyestuff Reptr., 34, 178—182,
1945.
Глава 8
СОБИРАТЕЛИ
Флотационными реагентами называются такие вещества,
прибавление которых в рудную пульпу делает возможным или
облегчает процесс флотации. Без флотационных реагентов об-
ласть применения процесса была бы ограничена рудами, содер-
жащими естественно флотирующиеся минералы.
Из флотационных реагентов наибольшее значение имеют со-
биратели, которые, закрепляясь на минералах, не обладающих 1
естественной флотируемостью, придают им подббную углеводо- .
родам поверхность, способную прилипать к пузырькам газа.
В настоящей главе рассматриваются флотационные реагенты-со-
биратели и их химические свойства. Механизм действия соби- .
рателей обсуждается в гл. 9.
Так как целью флотационного процесса является отделение
одного минерала от другого, в ряде случаев желательно изме-
нить действие собирателей, прибавляя соответствующие реаген-
ты, которые обеспечили бы наиболее эффективное разделение. .
Такие реагенты называются модификаторами; вопросы, связан-
ные с их действием во флотации, рассматриваются в гл. 10.
Собиратели и модификаторы действуют на поверхность ми- '
нералов. Третий класс флотационных реагентов, называемых
пенообразователями, действует в основном на поверхность раз- >
дела газ — жидкость. Эти реагенты рассматриваются в гл. 11 и
12.
Собиратели и пенообразователи являются органическими
соединениями, знание химических свойств которых помогает их
успешному использованию. Следовательно, желательно знание -
органической химии.
Обычно при изучении органической химии в первую очередь
прибегают к общеизвестным книгам таких авторов, как Уит-
мор, Каррер, Рихтер или Физер и Физер [48, 79, 167]. В некото-
рых случаях, однако, следует обращаться к библиографическому
справочнику по органической химии Бейльштейна (Handbuch
der organischen Chemie).
, Характеристики собирателей. Естественной флотируемостью
обладают только такие твердые тела, у которых поверхность
имеет неионный характер (гл. 4). Для практических целей во
флотации имеет значение создание поверхности, аналогичной
СОБИРАТЕЛИ
219
поверхности углеводородов. Следовательно, хотя бы часть мо-
лекулы собирателя должна состоять из углеводородных или
аналогичных им групп. Но этого недостаточно. Неионный, неак-
тивный характер всех чистых углеводородов обусловливает не-
возможность их закрепления на твердых веществах, имеющих
ионную гидрофильную поверхность. Поэтому вторым требова-
нием, предъявляемым к молекулам собирателей, является то,
что они должны иметь активную группу, способную закреплять-
ся на поверхности, и быть достаточно гидрофобными, для того,
чтобы обеспечить закрепление минерала на пузырьке воздуха.
Третьим требованием является необходимость того, чтобы эта
активная часть молекулы была достаточно избирательна к по-
верхностям минералов. Это позволит получить хотя бы очень не-
значительное разделение. Для практического применения важ-
ными свойствами являются легкая диспергируемость в воде,
аналогичная растворимости, и возможно низкая стоимость.
Органические кислоты, состоящие из одной углеводородной
цепи, к которой присоединена одна анионная полярная группа,
могут быть использованы в качестве собирателей, если им при-
сущи такие свойства, как специфическая реакционная способ-
ность, диспергируемость; кроме того, они должны быть неде-
фицитны. Аналогично этому органические основания, состоящие
из одной углеводородной цепи, к которой присоединена одна
катионная полярная группа, становятся пригодными в качестве
флотационных реагентов, если им присущи те же свойства.
Соли органических кислот или оснований также пригодны
для применения в качестве собирателей. Обычно они являются
даже лучшими реагентами вследствие большей диспергируемо-
сти. Эти три класса органических соединений, а именно, кисло-
ты, основания и их соли, являются наиболее распространенны-
ми собирателями. В исключительных случаях в качестве соби-
рателей применяются некоторые неионогенные реагенты. Но ис-
пользование таких реагентов ограничивается, по-видимому, од-
ним-двумя типами соединений. В этих случаях реагенты либо
переходят в ионную форму в результате таутомерии, восстанов-
ления, гидролиза или других преобразований их молекул, либо
закрепляются на легко омасливаемой (негидрофильной) поверх-
ности, придавая ей способность прилипания к пузырьку воздуха.
В большинстве случаев собиратели монофункциональны.
Это значит, что углеводородная часть молекулы имеет одну
присоединенную к ней полярную группу. Если же в такой мо-
лекуле имеются две (или более) полярные группы, то такое
соединение может быть собирателем при условии, что полярные
группы находятся на одном конце молекулы, а неполярные —
на другом.
Классификация собирателей. Анионные собиратели, т. е. та-
220
ГЛАВА 8
кие собиратели, для которых характерно наличие присоединен-
ной к органическому радикалу кислотной группы, имеют наи-
большее значение. Они и их соли могут быть разделены на два
класса. К первому классу относятся такие, у которых «кислот-
ный» водород (или эквивалентный ему металл) присоединен к
углеводородному радикалу через атом кислорода; ко второму
классу относятся такие, у которых он присоединен через атом
серы.
Анионные собиратели первого класса могут быть названы ок-
сигидрильными соединениями. Они включают карбоксилсодер-
жащие соединения, кислые алкилсульфаты и сульфонаты.
Анионные собиратели второго класса могут быть названы
сульфгидрильными соединениями. К ним относятся меркапта-
ны, тиокарбонаты (ксантогенаты), тиомочевина, дитиокарбама- .
ты и дитиофосфаты. В табл. 1 приведены примеры анионных
оксигидрильных собирателей, анионных сульфгидрильных и ка-
тионных собирателей.
В карбоксилсодержащих собирателях полярная группа
—С—О—1 присоединена одним концом к углеводородному ради-
О
калу К, а другим — к атому металла или водорода. В других
оксигидрильных собирателях аналогичным образом присоеди-
О О
II II
йены группы — О — S — О — в алкилсульфатах и —S—О—
П II
о о
в сульфонатах.
Следует отметить, что в сульфонатах атом серы присоединен
непосредственно к углеводородному радикалу, а в алкилсуль-
фатах— через кислород.
У всех сульфгидрильных собирателей атом «кислотного» во-
дорода присоединен к двухвалентному атому серы, но атомом,
соединяющим тиольную группу и неполярный радикал, может
быть как сера (в тиолах), углерод (в тиокарбонатах, тиокарба-
матах и тиомочевинах), так и фосфор (в тиофосфатах). Раз- ч,
личные полярные группы приведены в табл. 1.
У катионных собирателей, которые в настоящее время пред-
ставлены аминами, полярная группа состоит из атома азота, к
которому присоединено от одного до четырех углеводородных
радикалов.
Оксгидрильные собиратели
Карбоксилсодержащие реагенты. Жирные кислоты и соли
металлов и жирных кислот — мыла являются наиболее важны-
ми представителями большого числа соединений, содержащих
СОБИРАТЕЛИ
221
Таблица 1
Классы собирателей
Собиратели | Формула
О кс гидри льные анионные Карбоксилсодержащие соединения Алкилсульфат Сульфонаты Сульфгидрильные анионные Тиолы (меркаптаны) . . .' Тиокарбонаты Тиокарбаматы Монозамещеиные тиомочевины . . . Тиофосфаты Катионные Амины** Я-С—О н 1! О О II Я—О-S—О—н II О о II Я-S-О н II О Я—S . -Н R_-O*-C—S—н II R— S* 1 R-N-C-S Н II S* н я | N\ b; я7 я-о \p-S—н Я'-О II S* Я' я он Я" Я'"
* Кислород и сера, которые могут замещать друг друга, отмечены звездочками.
R'" могут быть водороды вместо углеводородных радикалов.
222
ГЛАВА 8
карбоксильную группу—С—О— Другими типами монокарбо-
II
О
ксилсодержащих соединений являются нафтеновые кислоты
[48а], у которых к углероду карбоксильной группы присоединен
полициклический углеводородный радикал, и смоляные кисло-
ты [48b, 175], имеющие частично гидрогенизованные цикличе-
ские углеводородные радикалы, присоединенные к углероду
карбоксильной группы.
Строение основных представителей группы карбоксилсодер-
жащих кислот и их мыл приведено в табл. 2. Соединения, при-
веденные в этой таблице, подобраны для иллюстрации различ-
ных видов кислот и не связаны с их практическим назначением.
Таблица 2
Формулы представителей карбоксилсодержащих кислот
Соединение Формула Особенности структуры *
н-Валериановая кислота Изовалериановая кисло- та Лауоат натрия Олеиновая кислота Нафтойная кислота С4Н„ — С (О) он С4НЭ — С (О) он СцН28 — С (О) ONa С17Н33 -С(О) он С10Н7 — С (О) он Прямая углеводородная цепь Разветвленная углеводородная цепь Мыло прямоцепочечной кисло- ты Двойная связь в середине уг- леводородной цепи Углеводородный радикал из двух шестичленных колец с пятью двойными связями и
3-фенилпропионовая ки- слота С3н, - С (О) ОН Углеводородный радикал при- соединен в положении 2 к шестичленному кольцу стре- мя двойными связями
Абиетиновая кислота1 С10Н29-С(О)ОН Углеводородный радикал, со- стоящий из трех шестичлен- ных колец, к которым при- соединены две метильные (СН3) н одна изопропильная (С3Н7) группы
1 Абиетиновая кислота имеет ряд изомеров.
Жирные кислоты и их мыла. Насыщенные жирные кислоты
не содержат двойных связей. В молекулах ненасыщенных жир-
ных кислот может быть одна, две или большее число двойных
связей; они соответственно называются олефиновыми, диоле-
финовыми или полиолефиновыми жирными кислотами. Если
кислоты содержат одну или более тройных связей, то в этом слу-
собиратели
223
чае они называются ацетиленовыми. В природе жирные кисло-
ты находятся в виде глицеридов—эфиров глицерина и жирных
кислот. В глицеридах на одну молекулу глицерина приходится
три молекулы жирных кислот, причем обычно они имеют сме-
шанный характер, т. е. в каждой молекуле глицерина имеется
несколько различных жирных кислот. В большинстве случаев
природные жирные кислоты содержат четное число атомов уг-
лерода. Общеизвестными их представителями являются насы-
щенные кислоты с 12, 14, 16 и 18 атомами углерода (лаурино-
вая, миристиновая, пальмитиновая и стеариновая) и кислоты
олефинового, диолефинового и триолефинового рядов с 18 ато-
мами углерода (олеиновая, линолевая и линоленовая).
Жирные кислоты обычно находятся в природе в очень слож-
ных смесях, ввиду чего чистые кислоты получают в лаборатор-
ных условиях либо синтезом, либо выделением из смесей кис-
лот. Чистые насыщенные кислоты могут быть весьма легко по-
лучены фракционной кристаллизацией или фракционной ди-
стилляцией специально синтезированных метиловых эфиров
этих кислот. В то же время изготовление олеиновой кислоты
(даже относительной чистоты) представляет собой очень труд-
ную задачу. Методы очистки, синтеза и физические свойства
жирных кислот приведены в превосходной книге Ральстона
[128]. Некоторые данные о кристаллической структуре жирных
кислот также известны, однако большинство деталей кристал-
лической структуры до сего времени не выяснено [17’0].
Жирные кислоты на воде, имеющей нейтральную или кис-
лую реакцию, располагаются в виде мономолекулярного слоя
(см. гл. 3). Это дает возможность рассчитать площадь, занима-
емую молекулой, находящейся в плотном слое, в котором поляр-
ные группы и водород, по-видимому, гидратированы. Можно-
принять, что в плотном мономолекулярной слое каждая моле-
кула занимает площадь 20 А2. В кристаллах жирных кислот,
где углеводородные радикалы расположены перпендикулярно
оси, площадь, занимаемая молекулой, несколько меньше 20 А2.
Растворимость жирных кислот в воде в большой степени
зависит от длины углеводородного радикала. Согласно прави-
лу Траубе, при возрастании углеводородной цепи нормального
строения на один атом углерода растворимость в воде пони-
жается в 3—3,2 раза. Следовательно, при каждом увеличении
цепи на два атома углерода растворимость уменьшается на-
один порядок.
Мыла жирных кислот и щелочных металлов растворимы в
воде даже в тех случаях, когда длина углеводородного радика-
ла так велика, что кислота практически нерастворима. Однако,
они обладают рядом специфических свойств. Так, например, мы-.
.224
ГЛАВА 8
.ла жирных кислот при определенных концентрациях образуют
мицеллы; для достижения постоянства поверхностных свойств
растворов мыла требуют большего времени. Если мыла гидро-
лизуются, то кислоты, образующиеся при гидролизе, обычно на-
ходятся на поверхности раздела газ — вода, а ионы — предпоч-
тительно в объеме жидкости. Водные растворы мыл щелочных
металлов «высаливаются» при добавлении в раствор неоргани-
ческих солей. Это свойство используется в мыловарении.
Двухвалентные металлы, в основном щелочноземельные, об-
разуют весьма мало растворимые мыла, однако имеющиеся ко-
личественные данные по растворимости этих соединений в воде
недостаточно надежны.
Нещелочноземельные двухвалентные металлы также дают
малорастворимые мыла, но их относительные растворимости не-
известны. Трех- и четырехвалентные металлы образуют только,
кислые мыла [42].
Выпускаемые промышленностью мыла щелочных металлов
обычно состоят из смеси нейтрализованных жирных кислот с
небольшим избытком щелочи, карбонатов, силикатов или фос-
фатов щелочных металлов. Мыла поливалентных металлов, вы-
пускаемые промышленностью, являются смесью более или ме-
нее полностью нейтрализованных различных жирных кислот и
в основном применяются как смазки.
Диолефиновые и более ненасыщенные жирные кислоты окис-
ляются кислородом воздуха. При окислении триолефиновых
кислот благодаря способности каждой молекулы присоединять-
ся к двум другим через кислородные мостики образуется проч-
ная пленка. Например, такая пленка образуется при окислении
льняного масла, которое в значительной части является глице-
ридом триолефиновой линоленовой кислоты и применяется в
красках в качестве связующего вещества.
Прогорклость, приобретаемая маслом и жирами в процессе
хранения, по-видимому, в большой степени обусловлена окис-
лением моноолефиновых групп.
Применение карбоксилсодержащих собирателей. К карбо-
ксилсодержащим кислотам, применяющимся при флотации, от-
носятся кислоты, получаемые гидролизом глицеридов, содер-
жащихся в животных жирах и маслах, специально синтезиро-
ванные или частично реконструированные (т. е. гидрогенизован-
ные) жирные кислоты растительного и животного происхожде-
ния, нафтеновые кислоты, получающиеся при очистке нефти,
•смоляные кислоты [147] и кислоты, получающиеся при производ-
стве целлюлозы.
Представителями смоляных кислот являются различные изо-
меры абиетиновых кислот С2оН3002. Абиетиновая кислота имеет
СОБИРАТЕЛИ
225
две двойные связи. Такой циклический радикал, как у нее, об
наружен у большинства смоляных кислот.
Талловое масло, являющееся отходом бумажного производ-
ства, благодаря своей дешевизне находит все более широкое
применение в качестве анионного собирателя. Большей частью
оно состоит из абиетиновых кислот. Его свойства специально
изучали Т. Гассельстром, Е. Е. Флекк и Л. Ружичка. Как пра-
вило, при флотационном обогащении ненасыщенные кислоты
дают наилучшие результаты. Примером являются очищенные
жирные кислоты кукурузного масла и отходов рыбьего жира,
которые по своим собирательным свойствам не уступают тал-
ловому маслу. Почему ненасыщенные кислоты позволяют полу-
чить лучшие результаты при флотации, остается неясным.
Химия алкилсульфатов и алкилсульфонатов. Общеизвестен
интерес, проявляемый к вопросам синтеза и химическим свойст-
вам короткоцепочечных алкилсульфатов и сульфонатов. Их
свойства описаны в известной монографии [150]. Однако сведе-
ния об интересующих нас длинноцепочечных соединениях (та-
ких, как додецилсульфат натрия, пентадецилсульфат натрия и
др.), которые широко применяются в качестве моющих средств,
весьма скудны и разбросаны. Известны флотационные свойства
этих соединений, однако они также недостаточно исследованы.
Надо полагать, что этот вопрос в дальнейшем несомненно бу-
дет изучен.
Алкилсульфаты щелочных металлов являются солями ал-
килсерных кислот. Эти кислоты можно рассматривать как кис-
лые эфиры спиртов и серной кислоты. Они могут быть получены
взаимодействием серной кислоты и спиртов по следующей ре-
акции: _______
я; ой + _н; oso3h poso3h. (i)
Вместо серной кислоты можно применять другие сульфиру-
ющие агенты (такие, как S03, олеум, хлорсульфоновая кислота
CISO.H, или такие продукты, как сульфаминовая кислота
NH2SO3H).
Другим способом производства алкилсерных кислот являет-
ся взаимодействие олефинов (углеводородов, содержащих
двойные связи) с серной кислотой:
Н R~
#C = CR~+ НО-SO3H->₽C — С — OSO3H. (2)
НН НН
В этой реакции, где для образования алкилсульфатов ис-
пользуются двойные связи, углеводородный радикал имеет раз-
ветвленную структуру.
15 А. М. Годэн
226
ГЛАВА 8
В промышленности давно применяется обработка серной кис-
лотой ненасыщенных спиртов, ненасыщенных жирных кислот и
гидроксилсодержащих жирных кислот и их эфиров. Получаю-
щиеся при этом продукты широко известны как соединения ти-
па так называемого турецкого красного масла и сульфирован-
ного касторового масла. Ничего определенного о составе этих
реакционных смесей неизвестно, за исключением того, что он
сложен и зависит от технологии приготовления. Некоторые из
таких продуктов запатентованы как флотационные реагенты,
однако их поведение при флотации изучено недостаточно.
Выше было указано, что в алкилсульфатах атом серы сое-
динен с углеводородным радикалом через кислород, в то время
как в сульфонатах он непосредственно присоединен к углево-
дородному радикалу. Это вероятно может обусловить некоторые
различия в их свойствах, но детально вопрос еще не изучен.
Среди этих соединений сульфонаты наиболее распростране-
ны. Вообще они очень широко применяются, но наибольшее рас-
пространение имеют как вспомогательные вещества в производ-
стве красителей и при крашении. Соединения этого типа по су-
ществу не испытаны во флотационной практике. Следует отме-
тить, что, очевидно, имеется прямая связь между механизмом
прикрепления красителя к волокну и закреплением собирателя
на минерале.
Комплексообразующие свойства оксгидрильных собирате-
лей. В карбоксилсодержащих соединениях оба атома кислорода
практически равноценны, и водород карбоксильной группы или
атом металла координируется с двумя атомами кислорода.
Тогда карбоксильную группу правильнее изображать так:
•—С , а не так: —С—О—, хотя обычно ее изображают
\, И
О----- о
в этой менее правильной форме [167а].
Такую аналогию можно также провести и между тремя ато-
мами кислорода алкилсульфонатов и между тремя атомами кис-
лорода, не присоединенными к алкильному радикалу, в алкил-
сульфатах. Однако этот вопрос, очевидно, следует изучить тща-
тельнее.
Применение оксгидрильных собирателей при флотации. Сре-
ди всех собирателей оксгидрильные анионные собиратели име-
ют наибольшее применение. Ими пользуются при флотации
сульфидных и окисленных минералов тяжелых металлов, мине-
ралов щелочноземельных металлов, солей ’и ряда силикатов.
Это не означает, что все минералы названного типа одинаково
хорошо флотируются каждым из этих реагентов или что все они
СОБИРАТЕЛИ
227
флотируются реагентами этого класса. Однако при применении
оксгидрильных собирателей, подо-брав условия флотации, мож-
но улучшить селекцию и получить требуемое разделение мине-
ралов. Из всех этих собирателей наиболее широко использует-
ся неочищенная олеиновая кислота (красное масло) и другие
неочищенные карбоксилсодержащие кислоты, такие как, на-
пример, полученные в качестве отхода при производстве дре-
весной целлюлозы (талловое масло), из рафинированных куку-
рузного и хлопкового масел и из очищенного рыбьего жира.
Сульфгидрильные собиратели
Тиолы содержат SH-группу, присоединенную к органическо-
му радикалу.
Если сульфгидрильная группа присоединена к углеродному
атому углеводородного радикала, то такие тиолы называются
меркаптанами. Сульфгидрильная группа может быть присоеди-
нена к углеводородному радикалу через атом углерода, не яв-
ляющийся частью этой углеводородной цепи. Если этот атом уг-
лерода с прикрепленной к нему сульфгидрильной группой с од-
ной стороны присоединен непосредственно к углеводородному
радикалу, а с другой — к атому кислорода или серы, то такие
тиолы называются тиокислотами. Если присоединяющий эту
группу атом углерода прикрепляется к углеводородному ради-
калу через кислород или серу, то такие соединения называются
тиоугольными кислотами, а если через атом азота, то тиокарба-
миновыми кислотами (один атом азота) или тиомочевинами
(если к этому углероду присоединено два атома азота). В слу-
чае, когда тиольная группа присоединяется к углероду через
атом фосфора, такие соединения называются тиофосфорными
кислотами. Различные тиолы даны в табл. 3.
Термин «тиолы» является относительно новым. Он был офи-
циально принят Chemical Abstracts/
Обсуждение термина «тиолы» и всей номенклатуры органи-
ческих соединений было проведено по обычно принятой форме
1123].
Меркаптаны. Меркаптаны являются простейшими тиолами.
Они могут рассматриваться как спирты ДОН, в которых кисло-
род замещен серой, в результате чего и получены соединения
типа ДЗН. Действительно, меркаптаны могут быть получены из
спиртов, но обычно используют другие методы синтеза. Меркап-
таны получили свое наименование из-за того, что они образуют
со ртутью нерастворимые соединения 1122]. Они были описаны
ранее (в 1833 г.) Цейсе как corpus mercurium captans. Меркап-
таны, имеющие углеводородный радикал алифатического ряда,
представляют собой жидкости, слабо растворимые в воде. Сое-
15*
228
ГЛАВА 8
Таблица 3
Классификация тиолов
Тиолы Монотиосоединения Дитиосоединения Тритиосоеди- нения
Меркаптаны /г— sh
Тиокислоты K-C-SH II О fl-C-SH II S
Тиоугольные кислоты 7?О—С—SH II О RO—С—SH II S (ксантогеновые кислоты) -=? я
Тиокарбами- новые кислоты К X СО СО и =О 3 =о А А ас х R ^N-C-SH н II м S R \n—C-SH о/ II К S
Тиомочевина Хз Хз Д Хз z Z Z 1 п- 1 z=<p со СО г д
Тиофосфор- ные кислоты R0 'у P-SH к о S
динения с относительно коротким прямым углеводородным ра-
дикалом имеют отвратительный запах. Меркаптаны являются
слабыми кислотами. Физические свойства некоторых алифати-
собиратели
229
ческих меркаптанов описаны Клэссоном [19] и Эллисом и Ридом
[43].
Меркаптиды. Меркаптаны образуют водонерастворимые сое-
динения со многими металлами, причем обычно с теми, кото-
рые дают водонерастворимые сульфиды. Таким образом, мер-
каптиды ртути, меди, свинца и серебра нерастворимы. В боль-
шинстве случаев свойства и строение этих солей детально не
изучены. Однако в литературе, посвященной вопросам синтеза
лекарственных органических соединений, часто приводятся све-
дения о свойствах этих солей.
Меркаптиды ртути содержат двухвалентную ртуть. Кристал-
лическая структура некоторых из этих соединений изучена [1591.
При нагревании меркаптиды ртути разлагаются до металличе-
’ ской ртути и алкилдисульфида [121]:
Hg (S/?)2 -> Hg + 7?S — ST?.
Иначе ведут себя меркаптиды свинца, которые дают сульфид
свинца и алкилсульфид. Согласно Отто [121], хлористая ртуть в
спиртовом растворе образует двойные соли с этилдисульфидом
или с этилмоносульфидом. Эти координационные соединения
были выделены из этилмеркаптида ртути, приготовленного в
водном растворе взаимодействием меркаптида щелочного ме-
талла и соли ртути. По данным Клэссона [84], метилмеркаптид
ртути нерастворим во всех растворителях, за исключением раз-
лагающих его. Клэссон также описал методы приготовления
метилмеркаптидов свинца, висмута и серебра.
Значительное число исследований было проведено с меркап-
тидами свинца. Отчасти это объясняется тем, что соединения
свинца применяются для удаления меркаптанов из углеводоро-
дов. Исследования проводились с меркаптидами свинца двух
типов — нейтральными и основными [118]. Как нейтральные, так
и основные соли имеют относительно непостоянный состав [10].
образуясь (в той или иной степени) одновременно; кроме того,
они способны взаимодействовать с серой и серу содержащими сое-
динениями [119, 120].
Вертгейм согласился [165] с Отто и Ридом, что при осажде-
нии меркаптанов свинцом получаются меркаптиды, имеющие
непостоянный состав. Новые данные по некоторым длинноце-
почечным меркаптидам свинца сообщают Фор и Бост [49].
Меркаптиды никеля являются объектом нового исследова-
ния [77]. В этих соединениях никель двухвалентен. Меркаптиды
никеля не растворимы в воде и не являются «координационны-
ми» соединениями, как это утверждалось ранее, так как они
Диамагнитны. Олово образует только четырехвалентные мер-
каптиды.
Мэнчот и Галл [109] утверждают, что железо образует мер-
каптиды двухвалентного железа, которые при комнатной тем-
230
ГЛАВА 8
пературе. представляют пирофоры. Другие исследователи уста-
новили, что хлорное железо окисляет многие меркаптаны до ди-
сульфидов.
Существуют меркаптиды одновалентной меди. Они образу-
ются как с алифатическими меркаптанами, так и с соединения-
ми, имеющими более сложное строение, например с серусодер-
жащими аминокислотами, подобными цистеину 1125].
Как утверждают Рид и его сотрудники, меркаптиды меди
реагируют с сероуглеродом. Металлическая медь или сульфид
меди также реагируют с меркаптанами, растворенными в угле-
водородах [145J. При реакции с меркаптанами алифатического
ряда нормальных алифатических углеводородов продукты ре-
акции остаются на поверхности твердой фазы; в случае вторич-
ных (имеющих разветвленный радикал) меркаптанов продукты
реакции переходят в растворитель. Алкилсульфиды и дисуль-
фиды не ведут себя подобно меркаптанам. Медь вначале обра-
зует соли, содержащие одно- и двухвалентную медь, но соеди-
нения двухвалентной меди неустойчивы и разлагаются, образуя
Смесь дисульфидов и солей меди
2Cu(S7?)2-»Cu2(S7?)2 + /?SS7?. (3)
С гидроксилэтилмеркаптаном золото дает устойчивые золо-
тосодержащие соединения, а платина — платину содержащие
соединения, хотя, как сообщают, соединения платины устойчи-
чивы только при определенных условиях. Германн [70] указыва-
ет на образование маркаптидов золота в координации с хло-
ром, типа Au-C1S2£2 и Au-CU'S/?. Букнайт и Смит [11] уста-
новили, что AU CI2 SC2H5 является комплексным соединением.
В этом случае золото может рассматриваться как трехвалент-
ное. Золото и платина образуют комплексные соли с меркапта-
нами, в которых у металла, как считает Рей, проявляются раз-
личные валентности [131]. Чат и Мэн [18] изучали образование
комплексов палладия с галоидом [X] и меркаптидной группой
(SEt), например, следующего типа:
Et
X ZS\ /X
> Pd < > Pd <
X7 7S7 7Х
Et
Двойные соли ртути с меркаптанами и галоидами, карбо-
натами и нитратами были описаны Саксом [139], который, кро-
ме того, изучил гидролитическое разложение этих двойных солей
в щелочном водном растворе до обычных меркаптидов [140].
Еще более сложные соединения образуются при взаимодей-
ствии железа или никеля с алифатическими меркаптанами и
СОБИРАТЕЛИ
231
окисью азота. Эти соединения изучались [74, 109, ПО, 132] и име-
ют строение соответственно ON • NiSC2H5 и (ON) 2Fe SC2H5. До
некоторой степени аналогичные соединения были получены [71]
из окиси углерода, железа и тиофенола; по-видимому, они род-
ственны карбонилам железа [44]. Хотя Манхот и его сотрудники
считают, что металлы в этих комплексных соединениях явля-
ются одновалентными, но мнение о том, что в соединениях
ON Ni S/? и (ON) 2 Fe • S Д никель двухвалентен, а железо трех-
валентно, не противоречит здравому смыслу. Селеновые аналоги
маркэптанов были изучены >в незначительной степени [154]. Они,
как указывается, относительно неустойчивы и имеют отврати-
тельный запах. Данных о селеномеркаптидах тяжелых металлов
не имеется.
Из этих кратких сведений о меркаптидах видно, что вопрос
весьма сложен и не полностью изучен и что в литературе мо-
жет быть найдено большое число противоречий. Однако, сум-
мируя, можно заключить, что:
1) щелочные и другие металлы, образующие водораствори-
мые сульфиды, дают водорастворимые меркаптиды;
2) металлы, образующие водонерастворимые сульфиды, да-
ют водонерастворимые меркаптиды;
3) некоторые водонерастворимые меркаптиды являются
двойными солями, другие—внутрикомплексными соединения-
ми;
4) валентность металлов в меркаптидах часто удвоена;
5) растворимость меркаптидов в воде изучена недостаточ-
но;
6) о кристаллическом строении меркаптидов имеется мало
сведений;
7) недостаточно изучены вопросы устойчивости и пути раз-
ложения меркаптидов.
Окисление меркаптанов. Меркаптаны, наряду со способно-
стью давать водонерастворимые соединения с металлами, имеют
и другое весьма важное свойство — легкую окисляемость до ди-
сульфидов:
2Z?SH->/?SS/? + 2Н+ + 2е. (4)
Эта реакция обратима. Она может быть применена для коли-
чественного определения меркаптана в образце, причем конт-
роль производится по изменению окраски иода при переходе в
иодид. Для повышения точности анализа в конце определения
может быть применен неводный растворитель, в котором иод
сильно окрашен, или водный раствор крахмала:
2/?SH + J2->^SS Д+2J-+ 2Н+. (5)
Дисульфиды, полученные из алкилмеркаптанов, — бесцвет-
ные или окрашенные в бледно-желтый цвет масла с неприят-
232
ГЛАВА 8
ным запахом. Они относительно трудно растворимы в воде, ес-
ли не имеют в своем составе групп, способствующих раствори-
мости. Количественных данных по растворимости дисульфидов
имеется весьма немного, однако ориентировочно можно счи-
тать, что они менее растворимы в воде, чем соответствующие
меркаптаны.
В щелочных растворах меркаптаны окисляются особенно
легко [171].
Тиолдисульфидная реакция широко изучалась в примене-
нии к биологически важным веществам, содержащим сульф-
гидрильные группы. Наибольшее значение из этих веществ име-
ет тиогликолевая кислота и цистеин. Структуру их можно по- .
нять при сравнении с метиламином, уксусной кислотой и ала-
нином. Метиламин является простейшим амином, уксусная кис-
лота— одна из простейших карбоновых кислот, аланин — обра- >.
зец одной из простейших аминокислот, тиогликолевая кисло- ’
та — простейшая тиолкарбоновая кислота и цистеин — про-
стейшая тиоламинкислота:
I Н3С —NH3
О
II Н3С —С^
'он
Метиламин
Уксусная кислота
Н О
I //
III Н3С — С — С
I \
nh2 он
н2 о
IV HS —С —с
\)Н
Аланин
Тиогликолевая кислота
Н2 Н О
I I
V HS —с —с —с
nh2 'он
Цистеин
Соединения IV и V соответственно двух- и трехфункциональ-
ные. Они полностью растворяются в воде и, очевидно, могут
СОБИРАТЕЛИ
'33
вступать в реакции со многими веществами, причем различны-
ми путями. Многообразием возможных путей взаимодействия и
объясняется их большое значение в биологических процессах..
Изобразив молекулу цистеина как HSCz/s, окисление его мож-
но представить следующим образом:
2HS Cys^Cys SS Cys + 2Н+ + 2е. (6)
Цистин является продуктом окисления цистеина. В противо-
положность другим продуктам окисления, например сравнитель-
но инертному веществу дисульфиду, получающемуся окислением
этилового меркаптана, цистин может взаимодействовать еще и
как аминокислота. Он может вступать в реакции в четырех на-
правлениях, в двух — по аминогруппам и в двух—шо карбо-
ксильным группам.
Тиолдисульфидные системы были изучены различными ме-
тодами [6, 7, 138, 168J, включая полярографию и потенциомет-
рическое титрование. Гош [58] и его сотрудники нашли, что сво-
бодная энергия реакции окисления тиолов не зависит от приро-
ды радикала, к которому присоединен атом серы сульфгидриль-
ной группы. Но это, по-видимому, также нуждается в подтвер-
ждении.
В связи с изучением окислительно-восстановительных реак-
ций тиолдисульфидных систем было найдено, что следы неко-
торых металлов ускоряют достижение равновесия [66].
Добавление цианидов предотвращает каталитическое дейст-
вие, но мало вероятно, чтобы это происходило за счет образо-
вания комплексов с катализирующими металлами. Изменение
катализирующего действия щелочами изучал Версии [5], кото-
рый установил, что по уменьшению активности в щелочном ра-
створе металлы могут быть расположены в следующий ряд:
мышьяк, медь, сурьма, цинк, кадмий, серебро, железо, никель.
Образование комплексов металлов с серусодержащими ами-
нокислотами было также изучено Розенталем и Войглиным [1301,
которые нашли, например, что ион меди реагирует в присутст-
вии пирофосфата иначе, чем в среде, свободной от пирофосфата.
Предполагалось [113, 142], что тиоловые или аминотиоловые
группы образуют комплексы с металлами. Указанные комплек-
сы в большинстве случаев неустойчивы в присутствии кисло-
рода, что и приводит к жидкофазному катализу, описанному
выше.
Шуберт провел сравнение аланина, не содержащего тиоло-
вых групп, с тиогликолевой кислотой и цистеином. Внутрикомп-
лексные соединения, получавшиеся с аланином, по-видимому,,
были другого характера, и возможно, что более устойчивые*
комплексы давали тиогликолевая кислота и цистеин:
.234
ГЛАВА 8
II II II
— С—С —С—С —С—с
I II I II II
NO SO S N
V3 Со 1/3Со V3Co
Комплексы кобальта с аланином, тиогликолевой кислотой и цистеином
Интересно отметить, что соединения никеля с цистеином
являются «закисными», соединения кобальта в основном «окис-
ными» (хотя «закисные» соединения, как сообщается, могут
быть получены, но они неустойчивы). В то же время железо в
зависимости от условий реакции дает как «окисные», так и «за-
кисные» соединения. Таким образом, железо-цистеиновые сое-
динения являются катализаторами окисления, что открывает
пути к объяснению роли железа и цистеина в биологических
процессах.
Разложение меркаптанов. Меркаптаны могут быть разруше-
ны не только окислением, но и другими способами. Так, при
нагревании (например, до температуры 400°) молекулы меркап-
танов разлагаются на сероводород и олефины [107]. В одном из
продуктов за счет этого появляется двойная связь:
C„H2„+I SH-^C„H2„ + H2S. (7)
Эта реакция может быть ускорена [40, 41] металлическими
катализаторами, причем она обратима. Меркаптаны с ненасы-
щенным радикалом весьма неустойчивы и полимеризуются даже
при комнатной температуре.
Тиофенолы. Аналогично тому как фенолы подобны спиртам
(хотя они и обладают более ярко выраженными кислотными
свойствами), так тиофенолы аналогичны меркаптанам. Харак-
терные их свойства, обусловленные ионизованной SH-группой,
подобны свойствам обычных меркаптанов.
Тиоэфиры. Моносульфиды, или тиоэфиры, имеют только по-
верхностное сходство с дисульфидами. Их образование требует
разложения меркаптанов другим способом, сопровождающим-
ся отщеплением сероводорода, а именно:
+ H2S. (8)
Получение тиоэфиров из меркаптанов и регенерация тиолов
из них протекают труднее, чем приведенные выше окислитель-
но-восстановительные реакции.
Химию меркаптанов начали серьезно изучать двадцать лет
назад [108]. Имеются обширные обобщения по этому вопросу,
однако наши знания в этой области все еще недостаточны. Осо-
бенно недостаточны сведения по соединениям металлов с меркап-
СОБИРАТЕЛИ
235
танами, представляющие интерес для всех работающих в обла-
сти флотационного обогащения.
Производные угольной кислоты, содержащие азот и серу.
В табл. 3 приведены четыре класса тиолов, в которых сера
сульфгидрильной группы различным образом присоединена' к
производным угольной кислоты. Некоторые из этих соединений,
представляющие интерес для флотации, более детально рассмат-
риваются в настоящем разделе.
В табл. 4 приведены соединения с различной степенью за-
мещения трех атомов кислорода угольной кислоты атомами азо-
та, серы или того и другого элемента. В ней даны соединения не
только с различной степенью замещения кислорода угольной
кислоты, но и таутомерные и изомерные формы, которые возмож-
ны для многих из этих продуктов. Производные, имеющие угле-
водородный радикал, получаются заменой в соединениях, при-
веденных в табл. 4, одного или более атомов водорода (только
Не атомов водорода карбоксильной группы) алкильным или
арильным радикалом.
Ксантогеновые кислоты и ксантогенаты щелочных металлов.
Ксантогенаты являются наиболее важными собирателями для
•сульфидных минералов и окисленных руд свинца и меди. Поэ-
тому целесообразно произвести обзор их свойств.
Ксантогенаты щелочных металлов впервые были получены
около 100 лет назад Цейсе [174], но их применение в промыш-
ленности началось не ранее 30—40 лет назад. Внедрение ксан-
тогенатов во флотационную практику датируется 1921 г., что в
значительной степени обусловлено работами Келлера (80]. Ме-
тоды приготовления были описаны Фостером [50].
Ксантогенаты являются солями ксантогеновых кислот, кото-
рые можно рассматривать как продукты присоединения серо-
углерода к спиртам.
Теоретически реакция получения ксантогеновой кислоты
может быть представлена как
кон + cs2->kocsh
II (9)
S
и аналогична реакции получения эфиров взаимодействием
спирта с ангидридом кислоты.
Ксантогеновые кислоты неустойчивы даже при относительно
низкой температуре, и обычно реакция протекает в противопо-
ложном направлении 163, 81]. Особенно интересна работа фон
Гальбана и Кирша [64]. Они нашли, что скорость разложения
ксантогеновой кислоты, растворенной в органических раствори-
телях, меняется в зависимости от температуры и природы ра-
створителя. Если начальную скорость разложения в сероугле-
236
ГЛАВА 8
Классификация производных угольной кислоты,
содержащих азот и серу
Таблица 4
Группа со- единений Распределение связей от централь- ного углерода Примеры соединений
1 4 к кислороду /ОН Угольная кислота О—(У хон Двуокись углерода О=С=О
2 3 к кислороду 1 к азоту /ОН Карбаминовая кислота H2N—
3 4 2 к кислороду 2 к азоту 1 к кислороду 3 к азоту 1 , ZNH2 Изоциановая О=С—NH о=С/ кислота J t ^NH2 Мочевина •! 1t nh2 н-о-с/ Циановая Н—О—C=N NH кислота
5 4 к азоту NH Гуанидин HN=C/ 2 Цианамид xnh2 n=c—nh2
6 7 3 1 2 2 к кислороду Монотио- угольная кислота h-s-c^° и Хон /ОН s=c< Х)Н
к к к сере кислороду сере
Дитиоугол ьиая /SH О=С(
8 1 к кислороду кислота и XSH
3 к сере (ксантогеновая кислота) /ОН S=C(
V \SH
СОБИРАТЕЛИ
237
Продолжение табл. 4
Группа со- единений Распределение связей от централь- ного углерода Примеры соединений
9 4 к сере Сероуглерод s=c=s
Тритиоугольная кислота ,SH s=c< \SH
10 11 3 к сере 1 к азоту 2 к сере 2 к азоту Дитиокарбаминовая кислота s H2N-C^ XSH и Г ZSH HN=C( XSH I
12 1 к сере 3 к азоту Тиомочевина S н =C/NH* xnh, znh2 -S-C< XNH
13 2 к кислороду 1 к азоту
1 к сере 0= ,SH XNH,
14 1 к кислороду 1 к азоту Монотиокар- баминовая кислота НО т ^-С //s 'NH2
15 2 к сере 1 к кислороду 2 к азоту но t -С ZSH 4NH
1 к сере
238
ГЛАВА 8
роде принять за единицу, то при той же температуре скорость .
разложения в гексане равна 1,5, в хлороформе 3,6, в бензоле
5, нитробензоле 315, в этиловом эфире около 500, в ацетоне 2600,
в спирте 1 млн. и в воде около 1 млн. Эти данные характерны
только для начальной скорости разложения, так как выделяю-
щийся из ксантогеновой кислоты спирт ускоряет разложение, и,
таким образом, реакция является самоускоряющейся. При рас-
смотрении результатов указанной выше работы становится оче- -
видным, что возможность существования ксантогеновой кисло-
ты в воде исчезающе мала. Влияние температуры на начальную
скорость разложения может быть показано на примере бензоль-
ного раствора, содержащего 0,02-н. НА' и 0,174-н. спирта. Ско-
рость разложения возрастала от 0,00084 при 4,7° до 0,0029 при
25°, 0,008 при 45° и 0,020 при 65°.
В основном ксантогенаты изготовляют из насыщенных одно-
атомных спиртов (таких, как этиловый, бутиловый или амило- 1
вый). Однако они также могут быть синтезированы из более
сложных и многоатомных спиртов 197], например целлюлозы,'
ксантогенаты которой применяются при получении искусствен-
ного шелка вискозным методом).
Соли ксантогеновой кислоты и щелочных металлов легко
приготовляются из алкоголятов щелочных металлов и сероуг-
лерода или соответственно из гидроокисей щелочных металлов, '
спирта и сероуглерода:
R ОН + CS3 + КОН -> R ОС SK + Н2О. (10) .
S
Короткоцепочечные ксантогенаты щелочных металлов хоро-
шо растворимы в воде и ацетоне, но мало растворимы в эфире и
нефтяных углеводородах. Эти различия в растворимости исполь-
зуются при их очистке. Водные растворы ксантогенатов щелоч-
ных металлов достаточно устойчивы, их можно применять в ла-
бораториях и на фабриках, но они портятся 122, 141] вследствие
значительного гидролиза и последующего разложения ксанто-
геновой кислоты.
Свежие растворы чистых ксантогенатов щелочных металлов
практически нейтральны. Это свидетельствует о том, что ксан-
тогеновые кислоты относительно сильные. Хранившиеся неко-
торое время растворы ксантогенатов, в которых они частично
разложились, являются щелочными. Работа Шаума, Зидлера и
Вагнера 1141] показала, что очень небольшая часть водного
раствора ксантогената разлагается в результате гидролиза.
Они считают, что во флотационных условиях в растворе ксанто-
генатов щелочных металлов почти не имеется ксантогеновой 4
кислоты. Это! также соответствует данным, полученным фон
Гальбаном и Гехтом 163], о том, что ксантогеновые кислоты яв-
СОБИРАТЕЛИ
239
ляются сильными кислотами с константой диссоциации около.
3 • К)-2, т. е. почти на два порядка выше, чем у угольной кис-
лоты.
Ксантогенаты щелочных металлов несколько влияют на по-
верхностное натяжение воды [33—35]. Однако понижение поверх-
ностного натяжения при практически применяющихся концен-
трациях короткоцепочечных гомологов настолько незначительно»
что их действием на поверхность раздела газ — жидкость можно,
пренебречь. Обычно понижение поверхностного натяжения, вы-
зываемое ксантогенатами щелочных металлов, аналогично по-
нижению поверхностного натяжения, получающемуся при рас-
творении жирных кислот с такой же длиной углеводородного
радикала, в то время как флотационное действие этих коротко-
цепочечных соединений резко отличается. Таким образом, ко-
роткоцепочечные ксантогенаты и короткоцепочечные мыла дей-
ствуют аналогично на поверхностях раздела раствор — газ, на
различно на поверхностях раздела твердое—раствор.
Ксантогенаты тяжелых металлов. Щелочноземельные метал-
лы дают водорастворимые ксантогенаты. Ксантогенаты железа»
марганца и цинка несколько растворимы в воде, но ксантоге-
наты других тяжелых металлов нерастворимы или очень мало-
растворимы. Ксантогенаты тяжелых металлов также существен-
но отличаются по растворимости в различных органических рас-
творителях. Соединения, содержащие три органических радикала
на атом металла, хорошо растворимы, соединения, содержащие
два радикала, умеренно растворимы, и соединения, содержа-
щие только один радикал, по существу нерастворимы. Ксанто-
генаты тяжелых металлов образуют двойные соли и координа-
ционные комплексные соединения. Желтый осадок ксантогената
одновалентной меди образуется при прибавлении солей двухва-
лентной меди к водным растворам ксантогенатов щелочных ме-
таллов. Осадок этот содержит эквимолекулярное количество
дисульфида ксантогеновой кислоты. При определенных условиях
осаждения некоторых ксантогенатов солями меди в течение ко-
роткого времени можно наблюдать коричневый осадок, который
теряет коричневую окраску через несколько секунд. Изменение
цвета осадка связано с неустойчивостью ксантогената меди.
Реакция идет по следующему уравнению:
Cu2+ + 2R S~ Си (ST?)2 -- - CuS /? + 0,5 7?SS R. (11)
Если желтый осадок (.в котором удерживается дисульфид)
экстрагировать органическим растворителем, то дисульфид пе-
рейдет в раствор, а чистый ксантогенат меди останется в осадке.
Этиловый ксантогенат меди нерастворим во всех растворителях;
по крайней мере, растворимость его так низка, что он не окра-
шивает растворитель, несмотря на то что ксантогенаты полу-
•240
ГЛАВА 8
чили свое наименование из-за специфической окраски [по-
гречески ^avioa (ксантос)—желтый].
Длинноцепочечные ксантогенаты также нерастворимы в боль-
шинстве органических растворителей, хотя они несколько рас-
творимы в горячем бензоле и обладают значительной раствори-
мостью в пиридине [56, 57] с образованием координационных
комплексов, состоящих из одной молекулы ксантогената меди
и одной молекулы пиридина [39].
Камби и Коризелли [16] изучили относительную устойчивость
дитиосолей меди. Результаты их исследований сведены в табл. 5.
Таблица 5
Свойства дитиосолей меди [/б]
Кислоты, с которыми получены соли Соединения одновалентной меди Соединения двухвалентной меди
Дитиокислоты (R-C—SH) II S Нестабильный Устойчивые коричне- вые не растворимые в ор- ганических растворителях соединения
Дитисугольные .(ксантогеновые кислоты) (R—О—С—SH) II S Неустойчивы Устойчивые желтые со- единения; производные длинноцепочечных кислот растворимы в органиче- ских растворителях
Монозам ещенные дитиокарбаминовые кислоты / Я. \ / )N-C—SH I HZ || \ s у Неустойчивые соедине- ния, оливково-желтого цвета Устойчивые желтые растворимые в органиче- ских растворителях со- единения
Дизамещенные дитиокарбаминовые кислоты / Яч \ XN-C-SH I Я'/ II 1 \ s / Стабильные оливково- зеленые соединения, растворимые в органи- ческих растворителях Устойчивые, но окис- ляющиеся, желтые, ра- створимые в оргаю’че- ких растворителях со- единения
Из данных табл. 5 видно, что в некоторых случаях стабильны
соединения одновалентной меди, в других же — соединения двух-
валентной меди (в зависимости от природы атомной связи меж-
СОБИРАТЕЛИ
241
ду серой сульфгидрильной группы и углеводородным радика-
лом, так же как и в зависимости от числа углеводородных ра-
дикалов). н
Серебро и золото дают ксантогенаты, подобные ксантогена-
там меди [31, 99, 133]. Так как золото обычно применяется в фор-
ме золотохлористоводородной кислоты HAuC14, в которой оно
является трехвалентным, образование ксантогената золота со-
провождается переходом 2/3 ксантогената в дисульфид. По дан-
ным Микенса, ксантогенаты серебра являются весьма неустой-
чивыми твердыми соединениями, но, ввиду того что Макене про-
верял их свойства в кислой среде, к этому заключению следует
отнестись с осторожностью. Ван Гетерен (Массачусеттский тех-
нологический институт, 1954 г.) приводит приблизительное про-
изведение растворимости продукта взаимодействия серебра и
ксантогената, равное 5-10-19.
Были приготовлены [14, 130] ксантогенаты палладия и пла-
тины. Соединения палладия устойчивы даже при слабом нагре-
вании. Установлено, что они могут применяться для определе-
ния малых количеств сероуглерода.
Соединения палладия и соединения платины растворимы в
органических растворителях. Считают, что металл и четыре ато-
ма серы образуют внутрикомплексное соединение следующего
строения:
ZS. । S х
Я ОС Pd СО/?.
Молекулярный вес соединений платины, растворенных в бро-
мистом этилене, соответствует формуле Pt(S/?)2.
Ксантогенаты свинца, по-видимому, не интересовали хими-
ков, но для инженеров-обогатителей они представляют интерес
в связи с флотацией галенита и окисленных минералов свинца.
Ксантогенаты свинца—палево-желтые соединения, получаю-
щиеся смешением водных растворов реагирующих компонентов.
Они растворимы в ацетоне и бензоле, но мало растворимы в
воде.
В щелочном растворе суспензия ксантогената свинца быстро
разлагается. Наибольший интерес представляет изучение рас-
творимости ксантогената свинца в воде. Арвид Тунаес [153] на-
шел, что в 1 л дистиллированной воды (pH =5,9) растворяется
приблизительно 0,27 кг этилового ксантогената свинца. Эти ре-
зультаты были получены измерением концентрации ионов свин-
ца в растворе, приготовленном перемешиванием очищенного
ксантогената свинца в воде. Норман Мак-Леод [112] измерил
концентрацию иона ксантогената в водно-ацетоновом растворе
ксантогената свинца и, экстраполировав к нулевому содержа-
16 А. м. Годэн
242
ГЛАВА 8
нию ацетона в растворе (рис. 1), установил, что растворимость
этилксантогената свинца составляет около 0,7 -10-6 моль/л, или'
0,32 мг/л.
Растворимости метилового и н-амилового ксантогенатов
свинца были также найдены Мак-Леодом. Они составляли
2 • 10'6 и 2 • 10~8 моль/л. Таггарт и Хассиалис [151], определявшие
произведение растворимости этилового ксантогената свинца, по-
лучили значение 6,7-10-16, что соответствует растворимости
2,5 мг/л. Недавно проведенные ван Гетереном измерения дали
для этилксантогената свинца произведение растворимости
6,8-10~17 и растворимость в воде 2,6- 10-6 моль/л. Следует отме-
тить, что результаты указанных выше измерений (учитывая
трудность проведения опытов) хорошо согласуются между со-
бой.
Тунаес и Абель проводили определение иона ксантогената в
щелочном растворе, полученном перемешиванием ксантогената
свинца с известью, содой и едким натром. Результаты их опытов
приведены на рис. 2.
На рис. 2 видно, что концентрация иона ксантогената, на-
ходящегося в равновесии с ксантогенатом свинца, значительно
изменяется с повышением щелочности системы. Как было уста-
новлено ранее, в сильнощелочной среде ксантогенат свинца пе-
реходит в тиокарбонаты, а в конце концов — в сульфид свинца.
Следовательно, измеренное содержание иона ксантогената
при pH > 12 скорее представляет количество ионов, находящих-
ся в растворе, чем количество, соответствующее равновесному
состоянию, которое не может быть достигнуто при данных усло-
виях.
Впервые изучение ксантогената цинка было проведено в
1935 г. Андреа Джордани [59]. Ксантогенат цинка легко обра-
зуется смешением реагирующих компонентов в водном растворе, .
давая белый шелковистый осадок. Аналогично соединениям
свинца он легко разлагается в щелочном растворе. Джордани
дает растворимость в воде 0,23 г/л для этилсоединений и 0,09 г/л
для н-амилсоединений. Ван Гетерен (Массачусеттский техноло-
гический институт, 1954 г.) нашел, что растворимость этилксан-
тогената цинка составляет 0,27 г/л, а произведение раствори-
мости 5,3 10-9.
Ряд исследователей изучал ксантогенаты никеля, кобальта
и железа [29, 39, 90]. Было установлено, что ксантогенаты двух-
валентного никеля, трехвалентных кобальта и железа являются
устойчивыми соединениями. Ксантогенаты трехвалентных ко-
бальта и железа очень хорошо растворимы в углеводородах и се-
роуглероде. Так, например, в 1 л циклогексана растворяется ч
около 400 г этилового ксантогената кобальта, а в 1 л бензола —
около 200 г. Ксантогенат трехвалентного железа хорошо раство-
16*
ГЛАВА 8
: 244
; рим в бензоле, из которого легко осаждаются большие черные
глянцевые кристаллы. Бензольный раствор ксантогенатов желе-
' за и кристаллы, выделенные из него, пахнут продуктами разло-
, жения ксантогенатов. Одновременно следует отметить, что на
кристаллах может быть замечен налет гидроокиси железа. Воз-
можно, что ксантогенаты железа разлагаются за счет окисления,
однако не исключаются и другие пути прохождения этого про-
' цесса, который нуждается в дальнейшем изучении.
Дюбоск [39] и Делецин [25—30] интересовались пиридиновыми
комплексами ксантогенатов. Ксантогенат трехвалентного железа
образует комплекс, состоящий из трех молекул пиридина и од-
. ной молекулы ксантогената железа. При высушивании кристал-
лов на воздухе пиридин улетучивается, Fe(S/?)3 • 3(C5HsN) пере-
ходит в Fe(S/?)3.
Ксантогенаты трехвалентных кобальта и хрома легко раство-
ряются в пиридине, но не образуют с ним комплексных кристал-
лических соединений. В то же время следует отметить, что при
поверхностном рассмотрении формулы их кажутся аналогичны-
ми формулам ксантогенатов железа. Ксантогенаты двухвалент-
ного никеля и цинка образуют с двумя молекулами пиридина
координационные соединения, но они менее устойчивы, чем ком-
плексное соединение ксантогената железа и пиридина.
Ксантогенат двухвалентной меди образует комплексное сое-
динение с пиридином при соотношении компонентов 1:1. Ксан-
тогенаты ртути и свинца комплексных соединений не образуют.
Суммируя, можно заключить, что в тех случаях, когда ксан-
тогенаты тяжелых металлов образуют комплексные соединения
с пиридином, на каждую ксантогенатную группу приходится
одна молекула пиридина и что только некоторым из ука-
занных металлов свойственна способность к комплексообразо-
ванию.
Как сообщалось в литературе [86, 100, 115], существуют ксан-
тогенаты молибдена и других металлов шестой группы. Они,
по-видимому, обычно представляют собой основные или ком-
плексные соли [такие, как (/?S)4Mo203]. Трехвалентный мышьяк
образует ксантогенаты типа As(S/?)3, которые не ассоциированы
в органических растворителях [152].
Результаты ряда ранее проведенных исследований по изуче-
нию кристаллической структуры ксантогенатов различных ме-
таллов опубликованы во многих изданиях [9 32, 32а, 37].
Окисление и другие пути разложения ксантогенатов. Окисле-
ние ксантогенатов щелочных металлов приводит к получению
соединений, имеющих структуру дисульфидов [17, 78, 83], кото-
рые являются производными ксантогеновых кислот и в то же
время подобны алкилдисульфидам, полученным из меркаптанов:
СОБИРАТЕЛИ
245
C2H6OCSH -> C2H6OCSSCOC2H6+2H+2e.
II S S (12)
Продуктам окисления присваивались различные наименова-
ния (например, диксантогены, алкилпроизводные дисульфидов,
диалкилдисульфодикарботионаты и др.). Реакция, при которой
образуются диксантогены, идет количественно и используется
для аналитических определений. Для окисления могут быть ис-
пользованы различные окислители, включая медь, иод, хлор-
амин-Т, азотную кислоту.
Этилдиксантоген является летучим соединением, перегоняю-
щимся без разложения при низком давлении 1141. При повышен-
ном давлении он разлагается до этилового эфира этилксантоге-
новой кислоты С2Н5ОС—SC2H5. Разложение этилдиксантогена
II
S
сопровождается потерей группы COS2. Однако до настоящего
времени не установлено, в виде каких продуктов выделяются
указанные элементы (высказываются предположения, что в ви-
де COS и элементарной серы).
Этиловый эфир этилксантогеновой кислоты обладает слабым
запахом, напоминающим запах порея, и перегоняется без раз-
ложения при низком давлении.
Очевидно, алкилэфиры ксантогеновых кислот могут иметь с
разных сторон различные углеводородные радикалы; предпола-
гают, что атом серы при двойной связи может обменяться ме-
стом с атомом кислорода (давая эфир S-дитиоугольной кисло-
ты), и, таким образом, для каждого соединения возможно нали-
чие трех изомеров:
R—OC-S/C ОС—S/? и ^-SC—S/?-
II II II
S S О
В определенных условиях эти изомеры переходят один в дру-
гой.
При нагревании ксантогенатов металлов до разложения по-
лучается ряд продуктов, состав и свойства которых зависят от
свойств металла и наличия кислорода. Среди образующихся при
нагревании продуктов обнаружены спирты [68, 1431, элементар-
ная сера,' диксантогены, алкилэфиры ксантогеновых кислот,
меркаптаны, меркаптиды, сульфиды металлов. Очевидно, что при
этом происходит большое число различных реакций.
Применение ксантогенатов в аналитической химии. Весьма
большое число водонерастворимых ксантогенатов и различия в
их свойствах (таких, как цвет, растворимость в органических
растворителях и т. д.) навело на мысль, что анализ катионов
246
ГЛАВА 8
может быть основан на применении иона ксантогената, подобно
тому, как применяется в аналитической химии сульфидный ион
(47, 160, 1611. Методы анализа промышленных ксантогенатов
рассмотрены Гирскиндом [72].
Применение ксантогенатов в промышленности. Ксантогенаты
Находят применение не только при флотации. С помощью ксан-
тогенатов осуществляется растворение целлюлозы — процесс,
положенный в основу производства искусственного шелка ви-
скозным методом. В течение продолжительного времени ксанто-
генаты и их производные используются в качестве ускорителей
при производстве резины. Производные ксантогенатов добав-
ляли к смазочным маслам, чтобы удлинить срок их службы и
уменьшить коррозию движущихся частей.
Получение производных тиокарбаматов и тиомочевин. Ксан-
тогеновую кислоту можно рассматривать как кислый эфир ди-
тиоугольной кислоты:
/?ОН + HOC—SH -» R ОС — SH+H2O.
II II (13)
S S
В результате полной этерификации с разрушением SH-rpyn-
пы образуется нейтральный эфир типа этилового эфира этил-
ксантогеновой кислоты C2H.5OC(S)SC2H5’.
7? OCSrH+HOj7?->7? OCS 7? + Н3О.
(И)
S
Кислые эфиры (ксантогеновые кислоты) —наиболее важные
флотационные собиратели, в то время как область применения
нейтральных эфиров до сего времени не установлена. Аналогич-
но этому [23] сероуглерод (который можно рассматривать как
ангидрид дитиоугольной кислоты) взаимодействует с аминами,
имеющими подвижный атом водорода, т. е. первичными 7?NH2
R'
или вторичными NH аминами. Если нейтрализация ами-
R7
ном происходит при избытке кислотного компонента — дитио-
угольной кислоты, то получается кислый дитиоамид, аналогич-
ный соответствующей ксантогеновой кислоте. Если нейтрализа-
ция проводится полностью, может быть получен нейтральный
тиоамид
NH + HOC- NC—SH+H2O;
R 7 " R / "
о о
(15)
СОБИРАТЕЛИ
247
R~\ /К~ R~\ /R~
NC-SH + HN -> N-C-N +H2S. (16)
R I R r/ « \ 7?
О
Следует отметить, что указанные реакции со спиртами при
первичном аминировании идут с отщеплением воды, в то время
как при вторичном аминировании — с выделением сероводорода.
Кислые тиоамиды называются тиокарбаминовыми кислотами, а
нейтральные — тиомочевинами.
В зависимости от соотношения реагирующих компонентов и
условий, способствующих удалению сероводорода, сероуглерод
взаимодействует с аминами с образованием дитиокарбаминовых
кислот или тиомочевины 154]. Получать тиокарбаминовую кис-
лоту удобнее в щелочном растворе [25], так как при этом повы-
шается устойчивость продукта:
Н
7?NH2+ CS2+NaOH^7?NC — SNa + Н2О.
II ( >
S
Так как аммонийный ион часто может замещать ионы натрия
или калия, нетрудно получить аммонийные соли дитиокарбами-
Н
новой кислоты 7?NC— SNH4.
II
S
Аминогруппа также замещается аммонийной группой, что
приводит к образованию алкиламмонийной соли алкилдитио-
карбаминовой кислоты:
Н Н Н Н < 18)
II ........Н2 II н2
S S
Если в этом соединении R аналогичен R~, то соль, несомнен-
но, может быть отнесена к соответствующей тиомочевине как
вещество, которое было получено по реакции нейтрализации (16)
без выделения сероводорода.
Если нейтрализацию дитиоугольной кислоты проводят ами-
ном или спиртом, то получают тиоуретан или аминоэфир
N — CS.H гНб.^- N —С—ST? + H2O. (19)
R7 J ................... R7 s
О
248
ГЛАВА 8
Дитиокарбаминовые кислоты, тиомочевины и тиоуретаны, а
также ксантогеновые кислоты могут рассматриваться как про-
дукты взаимодействия дитиоугольной кислоты со спиртами и
аминами (при реакции выделяется вода или сероводород). Пред-
полагается, что в присутствии воды реакция может идти в об-
ратном направлении.
Следует отметить, что может быть осуществлен переход от
одного класса указанных соединений к другому.
Амфотерный характер тиомочевины. Свойства тиомочевины
H2NCNH2 (простейшего соединения этого класса) привлекают
II
S
большое внимание не только из-за того, что ее формула ана-
логична формуле мочевины H2NCNH2 (соединению, синтезиро-
II
О
ванному Велером из неорганических исходных веществ), но и
из-за ее отличия от мочевины.
Тиомочевина образует со многими тяжелыми металлами ком-
плексы своеобразного строения. В настоящее время преобладает
точка зрения, что тиомочевина может существовать в различных
формах. Некоторые из них обладают кислыми свойствами, а
другие — основными:
h2nx h.2nx /NH3 rH*N\
C = S~T C—SH;tHN=C I ?±+ CS
H2N/ HN'^ \s h2n^
Тиомочевина
Изотиомочевнна
(дает если с тяжелы-
ми металлами)
Внутренний ком-
плекс (Вернер)
Двуполярный
иен (Лешер)
Тиомочевина и ее гомологи всегда имеют группу SH или S
и, таким образом, могут связывать ионы металлов, имеющие
сродство к сере. Они также могут связывать и анионы, имеющие
сродство к аммонийной группе.
В этом проявляется амфотерно-ионный характер указанных
соединений, который лучше всего виден из структуры двуполяр-
ного иона, предложенной Лешером и его сотрудниками.
Наиболее типичной солью, у которой проявляется амфотер-
ный характер тиомочевины, является соединение Hg^Ch (где
Т — ион тиомочевины):
Hg2++2Cr+4+[7T^Cl Hg ci. (21)
\т/ \т/
Была измерена [124] константа диссоциации серебрянотио-
мочевинного комплекса [AgT'al + . Она составляет 1,4 10-14.
СОБИРАТЕЛИ
249
Содержащей углеводородный радикал тиомочевиной, нашед-
шей применение во флотации, явилась дифенилтиомочевина, или
тиокарбанилид C6H5N (H)CN (Н)С6Н5. Это соединение плохо
II
S
растворимо в воде и может рассматриваться как обладающее
такими же амфотерными свойствами, что и обычная тиомоче-
вина. Некоторого различия можно ожидать вследствие влияния
относительно большой фенильной группы на подвижность атома
водорода, присоединенного к тому же самому атому азота. Ди-
тиокарбаминовые кислоты аналогично тиомочевинам дают тау-
томерные соединения и имеют амфотерные свойства, но в не-
сколько иной степени. У свободной дитиокарбаминовой кислоты
в отличие от тиомочевины более заметно преобладание кислот-
ных свойств над основными. Однако различия в свойствах этих
соединений до настоящего времени нельзя считать достаточно
изученными.
Внутрикомплексные соединения с тиомочевинами и тиокарба-
матами. Как тиомочевины, так и дитиокарбаматы являются сое-
динениями, обусловливающими возможность образования вну-
трикомплексных соединений с атомом металла М, дающим че-
тырехатомное кольцо с тиомочевиной
= N.....С =
М.....+S
а также с дитиокарбаматами
s—
М...S
С дитиокарбаматами может наблюдаться и образование мно-
гочленного комплексного соединения такого типа
I
-N.....S
S
:250
ГЛАВА 8
Дитиокарбаматы металлов. Дитиокарбаминовые кислоты да-
ют водонерастворимые соединения с рядом тяжелых металлов.
Многие из этих соединений растворимы в органических раство-
рителях; в ряде случаев они кристаллизуются с растворителем,
образуя кристаллы различных типов. В выпускавшейся в тече-
ние ста лет литературе по органической химии встречаются от-
дельные сообщения о солях дитиокарбаминовых кислот, причем
только ,в нескольких случаях они являлись основным объектом
исследования. Так, например, Шинцель провел сравнительное
изучение этих соединений, меркаптидов и комплексных соедине-
ний гидразина [1441. Камби и Коризелли [16] сравнивали продук-
ты взаимодействия дитиокислот с окисными и закисными соеди-
нениями меди. Делепин изучил дитиокарбаматы меди, кобальта,
железа, никеля, цинка, свинца и серебра [25—28], а Компин под-
робно исследовал соли никеля и кобальта.
Соли дитиокарбаминовых кислот, полученных из вторичных
аминов, явились предметом специального исследования, прове-
денного Виби и Матчесоном [166], а относительные растворимо-
сти сульфидов, ксантогенатов и тиокарбаматов тяжелых метал-
лов количественно рассматривались Малатеста [102]. Из при-
веденных выше работ следует, что соединения тяжелых метал-
лов с дитиокарбаматами значительно устойчивее, чем соответ-
ствующие ксантогенаты тяжелых металлов. Некоторые из них
(например, соединения кобальта и никеля) при пониженном дав-
лении могут быть перегнаны без диссоциации, аналогично моле-
кулярным. При хранении соединения двухвалентной меди не раз-
лагаются. Согласно Малатеста [101], дитиокарбаматы обычно
дают более прочные комплексные соединения, чем ксантогена-
ты. Бис-монофенилдитиокарбамат свинца Pb[S — С — N — С6Н5]2
S Н
был получен Круллей [89] прямым взаимодействием анилина,
сероуглерода и свежеосажденного гидрата окиси свинца. Соль
олова также была приготовлена, но отличалась от других дитио-
карбаматов тем, что выделяла при нагревании сероводород.
С другой стороны, Карач [55] на основе первичного аромати-
ческого амина не смог синтезировать дитиокарбамат цинка. При
проведении опытов он получал окись цинка и тиомочевину. Со
вторичными аминами может быть получена цинковая соль. Это
приводит к мысли о большей устойчивости солей дитиокарбами-
новых кислот, полученных из вторичных аминов, что находится
в соответствии с наблюдениями Делепина, касающимися лету-
чести солей никеля и кобальта, полученных из вторичных ами-
нов. Из раствора в хлороформе бис-диметилдитиокарбамат ко-
бальта кристаллизуется с двумя молекулами хлороформа. На-
триевые и бариевые соединения, выкристаллизованные из воды,
СОБИРАТЕЛИ
251
содержат от двух до четырех молекул воды. В бензоле или бро-
мистом этилене соединения кобальта и железа не ионизуются и
не полимеризуются, а находятся в растворе в форме СоТ^ и
РеГз (Т — алкилдитиокарбаматная группа).
В органических растворителях, содержащих незначительное
количество воды, растворяются алкилдитиокарбаматы меди, же-
леза, кобальта и никеля, однако провести аналитическое опре-
деление этих металлов не удавалось, хотя свинец дает сульфатs
или сульфид свинца спустя незначительное время после прибав-
ления раствора серной кислоты или сероводорода. Другие ме-
таллы (кроме свинца) ведут себя специфически. Их соединения
с алкилдитиокарбаматами Лей [95) назвал «внутренними ком-
плексами», а недавно им присвоено название хелатов.
Следует отметить, что внутренние комплексы, или хелаты, по-
лученные из различных металлов и алкилдитиокарбаматов, ин-
тенсивно окрашены. Это свойство было положено в основу коло-
риметрического определения малых количеств металлов.
Делепин считает, что даже меньшее 1 • 1О~бО/о меди или же-
леза может быть определено этим способом [27].
>Дитиокарбаматы и родственные им соединения как ускори-
тели в производстве резины. Алкил- или ар.илдитиокарбаматы
тяжелых металлов (наиболее часто соли цинка) применялись
в производстве резины для ускорения горячей вулканизации
смеси серы, окиси цинка и каучука. По мнению Бедфорда и его
сотрудников [2, 3, 4], дитиокарбаматы цинка разлагаются при
нагревании, давая амин, сероуглерод и сероводород, которые,
диффундируя, взаимодействуют с окисью цинка и восстанавли-
вают катализатор. Конечная реакция, по-видимому, будет
H2S + Zn* 0+ Zn (DT)2 _> ZnS + H20 + Zn* (DT)2. (22)
Сероводород частично образуется при взаимодействии серы
с аминокислотами, находящимися в каучуке (в протеинах, об-
волакивающих латекс).
Соли тиомочевины и металлов. Тиомочевина не образует про-
стых солей со щелочными, щелочноземельными и тяжелыми ме-
таллами, но она дает большое число двойных или комплексных
солей. Это является следствием амфотерного характера тиомоче-
вины, о чем говорилось выше. Если атом металла образует связь
с анионной частью амбииона, то некоторая часть аниона должна
ассоциировать с катионной частью амбииона. Многие комплексы
имеют интенсивную окраску и используются в анализах. Юи и
Оверхолсер исследовали возможность применения многих тио-
мочевин и описали качественное их определение [172]. Ден [24]
приготовляет соли гидроокиси меди и замещенных тиомочевин.
Они представляют собой желтые, красные, пурпурные и корич-
невые кристаллы довольно характерной формы. Приготовить та-
252
ГЛАВА 8
кие соли можно, прибавив растворенную в ацетоне тиомочевину
к меди, находящейся в феллинговом (водном) растворе.
Среди этих интересных соединений комплексы, содержащие
медь, хлор и тиомочевину, наиболее известны и изучены. Осо-
бенно много занимались этими продуктами Вальтер, Шторфер
и их сотрудники [148, 156—158]. Как показали исследования, об-
разуются комплексы типа СиГС1, CuT^Cl и СиТ^С!.
Своеобразную флокуляцию, или осаждение комплексов, мож-
но осуществить, добавив ион хлора в достаточной концентрации
(например, КС1); при этом, очевидно, один комплекс присоеди- ।
няется к другому. В водном растворе комплексы, по-видимому,
сильно ионизированы, например на [Си7'з'|+ и С1_. Монотиомо-
чевинный комплекс может быть получен из тритиомочевинного
соединения его окислением перекисью водорода. Это можно рас-
сматривать как образование дисульфида из двух третей тиомо-
чевины методом окисления; возможно, что в этом процессе такая
реакция является промежуточной.
Было исследовано действие хлористой меди на тиокарбани-
лид [87]. Комплексы солей серебра с тиомочевиной известны
при соотношении 3, 2 или 1 моль тиомочевины на 1 моль сереб-
ра. Галоидопроизводные, нитраты и цианиды были изучены Рей-
нольдсом [134]. Недавно Павелка [124] дал значение константы
диссоциации для [AgT3]+.
О комплексных соединениях свинца и галлия сообщили Мар
и Охл [98]. О ртутных соединениях упоминал Гроссман [62], а
соединения цинка и ряд других изучали Вальтер и Шторфер
[158]. Соединения висмута также подвергались исследованию г
[155]. Было, например, установлено, что обычной формой являет- »
ся BiTX3, где X — галоген. Следует отметить, что при взаимо-
действии галогенида висмута с тиофенолом образуется простая
соль Bi(SC6H5)3.
Окисление дитиокарбаматов и тиомочевины. Можно полагать,
что дитиокарбаматы окисляются подобно ксантогенатам, но этот
вопрос, по-видимому, детально не изучен, очевидно, в связи с
рядом трудностей. По окислению же тиомочевины проведено
большое число исследований. Даже незамещенное простейшее
соединение этой группы — тиомочевина H2NCNH2 имеет много
возможностей для окисления [163]. В первой стадии окисления
образуется дисульфид, формамидиндисульфид:
H2N\
CS
HNyT
2
]- HN\ />NH
-> C —S —S —C +2e.
H2N// \nh2
(23)
Этот дисульфид обладает щелочными свойствами (как мож-
но заметить, кислотные функции тиокарбанилида исчезли); он
известен как основание Шторха.
СОБИРАТЕЛИ
253
В щелочной среде формамидиндисульфид неустойчив и вы-
деляет серу.
Это происходит даже в такой слабой щелочи, как умеренно
разбавленный раствор уксуснокислого натрия [53]. В кислой
среде формамидиндисульфид присоединяет водородный ион, об-
разуя большой катион. Таким образом, в растворе иодистово-
дородной кислоты две молекулы Ш присоединяются к
каждой молекуле формамидиндисульфида, образуя катион
(C2S2N4H8)2+. Получены соли этого иона не только с иодисто-
водородной, азотной и хлористоводородной кислотами, но и с
анионами пикриновой и платино-хлористоводородной кислот.
При нагревании в щелочном растворе формамидиндисульфид
разлагается на серу, цианамид и тиомочевину:
(C2S2N4H8)2+ + 20 Н~-, CSN2H4 + S + CN • NH2 + 2НаО. (24)
Тиомочевина Цианамид
Детальное исследование окисления тиомочевин было прове-
дено Китамура [82], который показал, что пути окисления в кис-
лой и щелочной среде различны.
Два особенно интересных исследования по сравнительному
окислению тиомочевины были сделаны Фрейндлихом и его со-
трудниками [51, 52]. В обеих работах тиомочевина окислялась
кислородом в присутствии древесного угля. В одной — объектом
исследования был тиокарбамид (обычная тиомочевина); в дру-
гой—фенилтиомочевина. В случае фенилтиомочевины реакция
окисления может быть представлена как
Н
N
II
/ X|NCsH5
CeH6N = C-SH+0,5O2->S+CeH6N I + Н2О. (25)
I 1 8
NH2
II
HN
При этом была обнаружена [69] адсорбция на древесном угле
циклического соединения, известного как основание Гектора.
Возможно, имеется аналогия между адсорбцией фенилтиомоче-
вины или тиокарбамида на древесном угле и адсорбцией сульф-
гидрильных собирателей на сульфидных минералах. Этот во-
прос следует изучить.
Ориентированное нарастание тиомочевины на минералах.
Было проведено изучение быстрого нарастания тиомочевины на
некоторых кристаллах. Ройер [137], например, нашел, что на
254
ГЛАВА 8
кристаллах, имеющих ионную (или близкую к ионной) струк-
туру, всегда наблюдается неориентированное нарастание моле-
кулярного органического вещества. Ориентированное нарастание
имеет место в том случае, если соединение является ионным или
частично ионным; это установлено, по крайней мере, в одном
направлении решетки. Одним из характерных примеров являет-
ся наличие решетки тиомочевины на сфалерите. Эти представ-
ления были значительно расширены Нейгаузом [116, 117]. Он
связывал их с явлением окрашивания растущих кристаллов по-
сторонними включениями 194].
Классификация фосфорорганических соединений. Алкил- и
арилдитиофосфорные кислоты и их щелочные соли широко при-
меняются в качестве собирателей сульфидных минералов под
промышленным наименованием «аэрофлоты». Для установления
их структуры целесообразно рассматривать их совместно с дру-
гими фосфорорганическими соединениями. Надо отметить, что
они составляют очень малую часть известных фосфорорганиче-
ских соединений (несомненно менее 1%) [60, 88].
Фосфорорганические соединения можно классифицировать
следующим образом: соединения, в которых атом фосфора при-
соединен непосредственно к углеводородному радикалу, и та-
кие, в которых он присоединен через другой атом (в основном
через кислородный).
В отношении присоединения углеводородного радикала к
«ключевому» атому алкил- и арилдитиофосфорные кислоты при-
надлежат ко второму классу соединений, аналогичному алкил-
серным и ксантогеновым кислотам, но не жирным кислотам или
аминам.
Некоторые химики, очевидно, уделяют недостаточно внима-
ния фосфорорганическим соединениям, у которых фосфор не свя-
зан непосредственно с углеводородной цепью. Так, например, об
этих соединениях не упоминается в книге Годдара [60],
160 страниц которой посвящены описанию 2600 фосфороргани-
ческих соединений.
Соединения, у которых фосфор непосредственно связан с уг-
леводородным радикалом, могут быть сгруппированы как соеди-
нения с трехвалентным и соединения с пятивалентным фосфо-
ром. Различные типы соединений, содержащих трехвалентный
фосфор, приведены в табл. 6.
Из приведенных в таблице соединений наиболее известны
третичные фосфины. Первичные фосфины окисляются кислоро-
дом воздуха; аналогично ведет себя большинство вторичных
фосфинов. Первичные фосфины и первичные фосфониевые кис-
лоты неизвестны, за исключением их производных. Ряд других
кислот, приведенных в этой таблице, также хорошо не изучен.
Фосфины являются основаниями; по свойствам они подобны
СОБИРАТЕЛИ
255
Таблица 6
Органические соединения, содержащие трехвалентный фосфор
А. Соединения без кислорода и серы:
и р К'р (^)гх
m .НзР т , H2Z )Р (R)3P
Ы1 Первичный Н Третичный фосфин
А ‘ д фосфин Вторичный фосфин
Соединения, у
серы)
гяч
)РОН
нх
которых один атом кислорода
присоединен к фосфору:
(/?)2РОН
(и л и
Первичная фосфиновая
кислота
)PSH
Первичная тисфосфинбвая
кислота
Вторичная фосфиновая
кислота
(fl)2PSH
Вторичная тиофосфиновая
кислота
В. Со е д и н е н ия, у которых два атома кислорода
(или серы) п р и с о е д и н е н ы к фосфору:
Г /ОН]
,SH
РР<
Первичная фосфониевая
кислота
Первичная тисфосфониевая
кислота
Г. Соединения, у которых три атома кислорода
(или серы) присоединены к фосфору:
Р4О«
Фосфористый ангидрид
аминам и аммиаку. Увеличение числа углеводородных радика-
лов (от одного до трех) ведет к повышению основности фосфи-
нов. Они .присоединяют галоидоалкилы, образуя галоидные соли
четвертичных фосфониевых оснований. Хотя фосфор связывает
четыре углеводородных радикала, он является трехвалентным,
так как комплекс представляет собой одновалентный катион.
Пятивалентные органические соединения, содержащие фос-
фор, в которых он непосредственно соединен с углеводородным
радикалом, приведены в табл. 7. Соединения расположены в по-
рядке повышения содержания кислорода. Большинство из них
является кислотами, устойчивыми к кислороду воздуха и воде,,
но в качестве флотационных реагентов они не испытывались.
356
ГЛАВА 8
Таблица 7
Органические соединения, в которых пятивалентный фосфор
непосредственно связан с углеводородным радикалом
А, Один атом кислорода (или серы) присоединен
к фосфору:
R3PO Окись триалкилфосфина
R3PS Триалкилфосфинсульфид
Б. Два атома кислорода (или серы и кислорода)
присоединены к фосфору:
Н
I
R—Р -ОН Кетомоиоалкилфосфиновые кислоты
Н
I
R—Р—SH Кетомоно алкилдитиофосфиновые кислоты
II
S
(R)2 = р—ОН Диалкилфосфиновые кислоты
I
(R)2=p_SH Диалкилтиофосфиновые кислоты
II
О
(Т?)2=Р—SH ДиалкилдиТиофосфиновые кислоты
В. Три атома кислорода (или три атома серы
и кислорода, или три атома серы) присоединены к фосфору:
ОН
R-P(
II ХОН
О
Моноалкилфосфиновые кислоты
.ОН
R-P(
II XSH
О
,SH
R-P'
и XSH
о
Моноалкилтнофосфиновые кислоты
Моноалкилдитиофосфиновые кислоты
ySH
R—P<
Мо ноа л ки л т р ит ио фос фи нов ые ки сл от ы
СОБИРАТЕЛИ
257
Различные серусодержащие соединения, особенно диалкил-
дитиофосфиновые кислоты, кажутся достойными изучения. Было
проведено небольшое число работ по изучению этих соединений,
но не в связи с применением их во флотации [1, 73, 85].
Недавно интерес к ним проявился в Италии, где было про-
ведено исследование никелевых солей кислых эфиров дитиофос-
финовых, дитиофосфорных и дитиофосфониевых кислот [103,
104, 106].
Фосфорорганические соединения, в которых фосфор не свя-
зан непосредственно с углеродом, содержат кислород, серу или
азот. Из этих соединений наибольшее значение имеют те, кото-
рые содержат кислород. Они могут рассматриваться как кислые
или нейтральные эфиры различных фосфорсодержащих кислот,
количество которых значительно. Надо полагать, что очень
большое различие будет между алкильными и арильными эфи-
рами как нейтральных, так и кислых фосфорных кислот. Из про-
стейших представителей этих кислот основное внимание обра-
щалось на фосфористую кислоту Н3РО3 и ортофосфорную кис-
лоту Н4РО3. Несмотря на различия в формуле, обе кислоты со-
держат пятивалентный фосфор, но ортофосфористая кислота
имеет два подвижных атома водорода и одну кетоструктуру
ОН
Н—Р . Таким образом, для каждого замещающего угле-
11 \
О ОН
водородного радикала с ортофосфористой кислотой получается
один кислый эфир и один нейтральный. С ортофосфорной кисло-
той возможно образование двух кислых и одного нейтрального
эфира. Очевидно, теоретически возможно получить большое чис-
ло соединений при этерификации метафосфорной кислоты, кото-
рая имеет различные степени полимеризации [173]. В табл. 8 по-
казаны типы эфиров ортофосфористой и ортофосфорной кислот.
Тиофосфорные кислоты. Замещая в фосфорной кислоте кисло-
род серой, получают неорганические тиофосфорные кислоты. Эти
соединения изучал Эфраим с сотрудниками. Наиболее известны
тритиофосфористая H3PS3 и ортотетратиофосфорная H3PS4 кис-
лоты [44а, 45, 46]. Действительно, частичное замещение кисло-
рода серой может происходить аналогично кислотам мышьяка,
для которых известны все степени замещения: H3AsO4, H3AsSO3,
H3AsS2O2, H3AsS3O, H3AsS4. Замещение кислорода серой, но в
различном положении, дает изомеры, имеющие разные свойства.
ОН
Так, О = P(SH)3 отличается от S = Р
17 А. М. Годэн
(SH)a
258
ГЛАВА 8
Таблица 8
Кислота
Первичный
кислый эфир
Вторичный кислый
эфир
Нейтральный эфир
ОрТофос-
фористая
кислота
.ОН
Н—Р<
IIХОН
о
/ОТ?
н-р/
II хон
о
/ОР
Н-р/
II хор-
ОрТофос-
форная
кислота
ОН
НО-р/
II ХОН
о
/ОР
НО—Р<
II ХОН
о
/ОР'
Т?О-Р(
IIхон
о
/ОР
ро-р/ 2_
о
Вторичные кислые эфиры дитиоортофосфорных кислот, в ко-
торых один атом серы двойной связью присоединен к фосфору,
а другой связан с фосфором и водородом, являются только од-
ними из представителей соединений этой обширной группы и,
как показали опыты, они флотационно активны:
Они также применяются в качестве присадки к смазочным
маслам и в производстве резины.
Несмотря на широкое применение этих соединений, весьма
немного известно о способах их приготовления, очистки, о точ-
ной структуре и о химических свойствах. Хотя в области их про-
мышленного производства было много сделано в США, где они
выпускаются как флотационные реагенты под маркой «аэро-
флоты», в научной литературе имеется мало статей, посвящен-
ных кислым эфирам дитиофосфорной кислоты. Пионером работ
по этим соединениям был Пищимука в России [126, 127]. В более
позднее время эти исследования были дополнены работами
Гамби, Малатеста и Пизотти в Италии [15, 105], Мастеном, Нор-
маном и Уилмунстером в США [111].
Кислые эфиры дитиофосфорной кислоты образуют нераство-
римые соединения со свинцом и медью. Соли никеля водорас-
творимы. Двухвалентное железо, по-видимому, солей не обра-
СОБИРАТЕЛИ
259
зует; соли трехвалентного железа хотя и образуются, но они
весьма неустойчивы. Считается, что существуют соединения
двух- и трехвалентного кобальта. Полагают, что кислые эфиры
дитиофосфорных кислот окисляются до соответствующих ди-
сульфидов. Принимая во внимание сравнительную легкость,
с которой изменяются родственные им тиофосфористые соеди-
нения, можно предполагать, что процесс идет дальше образова-
ния дисульфидов. Некоторые соли металлов и эфиров дитио-
фосфорной кислоты сильно окрашены; ряд солей хорошо рас-
творим в органических соединениях, что наводит на мысль об
образовании внутрикомплексных соединений.
Катионные собиратели
В отличие от анионных собирателей, которые являются ио-
низированными органическими соединениями с углеводородным
радикалом в анионе, катионные собиратели, также являющиеся
ионизированными веществами, имеют углеводородный радикал
в катионе. Типичным примером таких соединений является со-
лянокислый додециламин. По структуре иона это соединение
аналогично хлористому аммонию, отличаясь от него тем, что
один водород группы NH4 заменен углеводородным радикалом,
имеющим двенадцать атомов углерода. Солянокислый додецил-
амин можно рассматривать как продукт присоединения хло-
ристоводородной кислоты к основанию додециламина C12H25NH2,
или как соль хлористоводородной кислоты с гидратированным
основанием C12H25NH3OH.
В зависимости от числа углеводородных радикалов, присое-
диненных к азоту, амины классифицируются на первичные, вто-
ричные и третичные. В этом случае, когда углеводородный ра-
дикал замещает четвертый атом водорода, образуются четвер-
тичные аммониевые основания.
Амины также можно классифицировать на алкиламины, арил-
амины и алкилариламины в соответствии с тем, присоединен
ли атом азота к углеводородному радикалу жирного ряда, аро-
матическому углеводороду или к радикалам обоих типов. Кро-
ме аминов, в которых азот не входит в ароматическое кольцо,
хотя и может быть присоединен к ароматическому радикалу
(например, в ариламинах), имеются азотсодержащие соедине-
ния, в которых атом азота является частью циклической струк-
туры. Образцами таких соединений могут служить пиридин и
его гомологи, хинолин и его гомологи, пиперидин и его гомо-
логи. Следует отметить, что указанные примеры не охватывают
все возможные органические соединения.
В табл. 9 приведены примеры первичных, вторичных и тре-
тичных насыщенных алкиламинов, солей первичных аминов, со-
17*
260
ГЛАВА 8
Таблица 9
Представители азотсодержащих катионных соединений
Наименование Формула Тип соединения
н-Амиламин C5HnNH2 Первичный алифатический амин
я-Додециламин Ci2H25NH2 Первичный алифатический амин
Ди-н-амиламин (C5Hu)2NH Вторичный алифатический амин
Три н-амиламин (C6Hn)3N Третичный алифатический амин
’Солянокислый амиламин [C5HltNH3)+Cl- Соль первичного алифатического амина
Тетраметиламмо- ний хлорид [(CH3)4N]+C1- Соль четвертичного аммониевого ос- нования
Анилин C,H5NH2 Первичный ароматический амин
п-Толуидии CH3-CeHrNH2 Первичный ароматический амин
Бензиламин CeH5-CH2-NH2 Первичный алифатический амин
Дифениламин (3-НафТиламин (3-1! тиламин Пиридин C()Hg * N • CgHg H nh2 /\/\ a \/\/ \/\/ nc5h5 Вторичный ароматический амин Первичный ароматический амин Первичный ароматический амин Гетероциклическое азотсодержа- щее соединение (с бензолоподобным циклом)
Солянокислый Y-пиколин [HNCj^CHjl+cr Соль гетероциклического азотсо- держащего соединения (с бензолопо- добным циклом)
Солянокислый хи- нолин [HNC9H7]+C1- Соль гетероциклического азотсодер- жащего соединения (с аналогичным нафталину циклом)
Солянокислый пи- перидин [HNC5H1o]+C1- Соль гетероциклического азотсодер жащего соединения (с аналогичным циклогексану циклом)
СОБИРАТЕЛИ
261
лей четвертичных аммониевых оснований, некоторых первичных
ариламинов, вторичных ариламинов и нескольких солей соеди-
нений пиридинового и хинолинового ряда. Следует отметить,
например, что бензиламин и п-толуидин, которые имеют одина-
ковый атомный состав, существенно отличаются по своим свой-
ствам: первый является алкиламином, а второй ариламином.
Растворимость и основность аминов. Короткоцепочечные
амины хорошо растворимы в воде; с повышением длины углево-
дородного радикала растворимость соответственно понижается,
и начинают в большой степени проявляться коллоидные свой-
ства. В ряду первичных алкиламинов они появляются у соеди-
нений, содержащих восемь углеродных атомов в радикале, и
становятся резко выраженными при С12, С14 и С16. Это также
сопровождается изменением проводимости растворов солей
аминов и числа переноса катионов.
По данным Г. И. Харвуда (частное сообщение), 'раствори-
мость н-додециламина, измеренная по электропроводности рас-
твора, равна 42 мг/л или 0,000227-н. Галоидные соли аминов
более растворимы, чем свободные основания. Однако в этом
случае растворимость длинноцепочечных гомологов также от-
ражает коллоидные свойства, обусловленные наличием длин-
ных радикалов. Так, например, Ральстон и Гоерр показали
[129], что в то время как растворимость солянокислого гексила-
мина в воде постепенно увеличивается с повышением темпера-
туры, растворимость солянокислого додециламина до темпера-
туры 28° остается невысокой, а от 28 до 30° повышается почти в
100 раз (от 0,4 до 26,5%).
В табл. 10 приведены константы ионизации ряда аминов.
Таблица 10
Константы ионизации аминов в воде
Аммиак2 . . . Метиламин2 Диметиламин2 0,18-10“4 4,4.10~4 5,2-10“4 ди-н-Октил- амин3 . . . н-Додецил- амин3 . . . 10,2-10~4 4,3-10“4 Прг 25° 25°
Триметиламин2 н-Амиламин3 0,55-10“4 4,3-10“4 при 25° и-Октадецил- амин3 . . . 4,0-Ю“4 25°
изо-Амиламин3, 4,0-10“4 » 25° ди-н-Октаде- циламип3 9.9-10-4 25°
н-Октиламин3 1 Согласно Гр! 2 Согласно Xaj 4,5-10“4 » 25° 1ньяру [61]. )иеду и Оуену [65]. Анилин1 . . . Фенилметил- амин1 . . . Бензиламин1 3 Согласно Ра * Согласно У 3,5-1О“10 2,6-1О~10 0,23-10“4 льстону [128а]. ат мору [ 167Ь].
262
ГЛАВА 8
Из данных табл. 10 видно, что константы ионизации алкил-
аминов несколько выше констант ионизации аммиака, но кон-
станты ионизации ариламинов значительно ниже. В разбав-
ленных растворах при pH ~ 6—9 растворы алкиламинов в ос-
новном состоят из ионов, в то время как ариламины гидролизо-
ваны. Интересно отметить, что все первичные алкиламины, не-
зависимо от длины цепи, имеют почти одинаковые константы
ионизации (около 4 10 4). Вторичные амины являются несколь-
ко более сильными основаниями (К ~ 10-3), в то время как тре-
тичные более слабыми. Четырехзамещенное аммониевое основа-
ние (как гидрат окиси тетраметиламмония) сильнее (приблизи-
тельно на один порядок) первичных, вторичных и третичных
аминов, однако данных по другим четырехзамещенным аммо-
ниевым основаниям не имеется 1961. Ариламины почти в тысячу
раз более слабые основания, чем первичные алкиламины
(Л7 ~ 10-10), а вторичные ариламины настолько слабые основа-
ния, что практически не образуют устойчивых солей [61].
Соли аминов. Несмотря на то, что основные свойства ами-
нов и растворимость их солей представляют существенный ин-
терес, все еще не имеется достаточного числа данных по этому
вопросу. Известно, например, что некоторые соли аминов (та-
кие, как фосфаты, сульфиды и сульфаты) менее растворимы в
воде, чем галоидные соли; однако количественных данных по
этому вопросу не имеется. По-видимому, это может явиться
плодотворной областью для изучения.
Установлено, что карбонаты стремятся к изомеризации до
карбаматов и что эти последние нерастворимы. Однако следует
подчеркнуть, что это явление еще недостаточно хорошо установ-
лено. Свойства карбонатов аминов или карбаматов кажутся
особенно важными ввиду того, что двуокись углерода широко
распространена в природе. Когда смешиваются водные раство-
ры аммиака и угольной кислоты в стехиометрическом отноше-
нии, образуется раствор карбоната аммония. Карбонат аммо-
ния можно дегидратировать до карбамата аммония, из которо-
го карбонат может быть регенерирован при соответствущих ус-
ловиях:
NH4—СО3—NH^N Н4—С-0 • NH2+Н2О. (26)
II
О
Образование карбамата, имеющего алкильные радикалы,
идет аналогичным образом:
R NH3—СО3—NH3/? flNH;!-C-ON(H) R + H2O. (27)
II
О
СОБИРАТЕЛИ
263
Практически возможно получить кислый карбамат
Н—С—ON(H)7?.
II
О
Алкилкарбаматы можно рассматривать как первую стадию
дегидратации алкиламмонийкарбонатов либо как продукты, по-
лучающиеся прямым присоединением амина к двуокиси углеро-
да. Вторая стадия дегидратации карбонатов, или дегидратация
алкилкарбаматов, дает алкилмочевины.
Например,
7?NH2+CO2->7?N(H)COH
II
О
Кислый алкилкарбамат
2R NH2 + СО2 -> R N (Н) CON (Н)3 R ’
II
О
Нейтральный алкилкарбамат
(28)
Дегидратация карбоната
2 (7? NH3)+ + СО!" -> R N(H)C—ON(H)3 R + H2O. (29)
II
О
Дегидратация карбамата
R N (H)C-ON(H)3 R -> R N (H)CN (H) R + H2O. (30)
II II
О О
Диалкил мочевина
Следует отметить, что при флотации, когда количество амина
и угольной кислоты значительно меньше количества воды, обра-
зование алкилкарбаматов, а тем более алкилмочевин, не должно
происходить; но это является нашим мнением, не основанным на
количественных данных.
Представляет интерес установить взаимосвязи аминов и
карбоксилсодержащих соединений, из которых они могут быть
получены. Карбоновые кислоты являются анионными соедине-
ниями. Аммиачные соли этих кислот при дегидратации дают
амиды и нитриды;
7? СОН + NH4OH 7? С — ONHiН2О. (31)
Аммонийная соль карбо-
новой кислоты
264
ГЛАВА 8
R—C-ONH47±^—C-NH2+H2O. (32)
/?C — NH2“-/<C^N + H,0. (33)
II
о
Алкилиитрил
Последовательный переход аммонийных мыл к амидам и
нитрилам обусловлен только дегидратацией.
Восстановлением нитрила металлическим натрием в спирто-
вой среде можно (почти с количественным выходом) получить
соответствующие амины [67, 128].
Известно большое число двойных солей аминов с тяжелыми
металлами. Из длинноцепочечных аминов такие соли были по-
лучены в безводных условиях [13]. Соединения меди, цинка,,
кадмия, ртути и серебра были получены с гидрохлоридами и
ацетатами н-додецил-, н-октадецил-, и ди-н-октиламинами. Все-
эти соединения содержат две аминогруппы на атом металла.
С аммиаком, как правило, образуются гексамины, в которых
на каждый атом металла координируется шесть атомов азота.
В соединениях металлов с метиламином отношение азота к ме-
таллу также выше, чем у продуктов, полученных с длинноцепо-
чечными аминами, но оно меньше, чем у соединений аммиака
[149]. В системе, содержащей воду и хлороформ (две несмеши-
ваюшиеся жидкие фазы), бис-н-додециламинацетат меди раз-
рушается; амин переходит в хлороформ, а ацетат меди — в вод-
ную фазу. Следовательно, можно заключить, что этот комплекс
не обладает высокой утойчивостью [13].
Различные собиратели
Было найдено, что ряд органических соединений более слож-
ного строения, чем описанные выше, обладает собирательными,
свойствами. К ним относятся некоторые красители. Часть кра-
сителей является катионными соединениями, другие—анион-
ными. Например, бриллиантовый зеленый — катионный реагент
трифенилметанового ряда — может быть применен при флота-
ции гематита; соединения другого типа могут быть использова-
ны для флотации касситерита.
В этой главе уже указывалось, что собиратели являются мо-
нофункциональными соединениями, у которых сила функцио-
нальных групп такова, что они ионизированы в водном раство-
ре. Однако имеются некоторые полифункциональные соедине-
ния, обладающие собирательными свойствами. Так, эмульсол'
Х-1, который является аммонийной солью сульфированного-
н-додецилдиэтиленгликоля, рекомендовался в качестве анион-
ного собирателя, а эмульсол Х-25, представляющий собой, алка-
СОБИРАТЕЛИ
265
ноламиновую соль сульфированного сложного спирта, предла-
гался в качестве катионного реагента. Некоторые красители,
обладающие собирательными свойствами, также полифункцио-
нальны.
Другими образцами полифункциональных органических со-
единений, обладающих собирательными свойствами, являются
«аэрозоли». Среди этих соединений можно назвать «аэрозоль
МА», являющийся натриевой солью бис-(1,3-диметилбутил)
сульфосукцината. Это соединение имеет три полярные группы,
две из которых обусловлены наличием янтарной кислоты, а од-
на является сульфогруппой.
Некоторые реагенты, предложенные в качестве собирателей,,
неионогенны, за исключением случаев, когда они становятся
ионогенными в процессе взаимодействия с минералами. Так,
реагенты «минерек», которые представляют собой продукты
окисления ксантогенатов, являются собирателями для минера-
лов-восстановителей (таких, как сульфиды). По-видимо.му, эти
реагенты вначале восстанавливаются на поверхности минерала
или ионами, выделяемыми минералами. Как тиокарбанилид,
так и тиомочевина, возможно, являются эффективными собира-
телями благодаря своей тиольной таутомерии, о которой уже
говорилось .в настоящей главе.
Последней группой собирателей, которые кажутся неионизи-
рованными (и, по-видимому, остаются неионизированными), яв-
ляются углеводородные масла типа ньюжел. Эти масла, по-ви-
димому, налипают на поверхность некоторых минералов и при-
дают им гидрофобность; такой механизм отличается от дейст-
вия ионогенных собирателей. Связано ли это различие в пове-
дении реагентов с их химической структурой, все еще не уста-
новлено.
Многие природные масла-собиратели представляют собой
смесь нескольких собирателей и вспенивателей или неэффектив-
ных разбавителей. Примерами таких масел-собирателей могут
служить каменноугольный креозот, древесносмоляные масла,
минеральные масла и большое количество разнообразных пре-
паратов.
ЛИТЕРАТУРА
1. Арбузов А. Е. иКамай Г. К. Приготовление тиофосфиновых кислот,
содержащих асимметрический фосфор. Журнал Русского физико-хими-
ческого общества, 61, 2037—2042, 1929; Chem. Abstr., 24, 5736, 1930.
2. В е d f о г d С. W. and Harold Gray. Reactions of accelerators during
vulcanization, (V), Dithiocarbamates, thiuram disulfides and the action
of hydrogen sulfide, Ind. Eng. Chem., 15, 720—724, 1923.
3. Bedford C. W. and Winfield Scott. Reactions of accelerators during
vulcanization, Ind. Eng. Chem., 12, 31—33, 1920.
4. Bedford C. W. and L. B. Sebrell. Reactions of accelerators during
vulcanization. Mechanism of the action of zinc compounds, Ind, Eng.
Chem., 14, 25—31, 1922.
ГЛАВА 8
266
5. В е г s i n Theodor. Acceleration of dehydrogenation of mercapto-compo-
unds by metals, Biochem. Z., 245, 466—472, 1932.
6. В e r s i n Theodor. The importance of the thiol-disulfide system for the
activity of biochemical materials, Sitzber. Ges. Beforder. ges Naturw.
Marburg, 71, 55—70, 1937; of Chem. Abstr,, 32, 5006, 5, 1938.
7. В e r s i n Theodor and Juliane S t e u d e 1. Polarimetric investigations of
the thioldisulfide system, Ber. deut. chem. Ges., 71B, 1015—1024, 1938.
8. В о n a t i S. Crystal structure of nickel xanthate, Atti soc. toscana sci.
nat. (Pisa), Mem., 47, 71—84, 1938.
9. В on at i S, and T. Derenzini. Determination of the crystal structure
of the salts of xanthic acid. Atti soc. toscana sci. nat (Pisa) Processi
verbali, 47, 7—16, 1938.
10. Borgs from P., L. M. Ellis and E. Emmett Reid. Preparation, pro-
perties and reactions of lead mercaptides, J. Am. Chem. Soc., 51, 3649—
3651, 1929.
11. Bouknight Joseph W. and George McP. Smith. Addition products
of metallic salts with certain sulfur and oxygen compounds, J. Am. Chem.
Soc., 61, 28—30, 1938.
12. Braun Julius v.. and Theodor Plate. Polymerization of unsaturated
mercaptans, Ber. deut. chem. Ges., 67B, 281—285, 1934.
13. Broome F. K., A, W. Ralston and M. H. Thornton. Complex for-
mation with high molecular weight amines, I, J. Am. Chem. Soc., 68, 67—
69, 1946; II, ibid. 68, 849—851, 1946.
14. Bulmer Gerald and Frederick G. Mann. Chemistry of xanthic acid
derivatives, J. Chem. Soc., 1945, 666—686.
15. Cam bi Livio. О—O’ Diaryl esters of dithiophosphoric acids, Chimica
e industria (Milan), 26, 97—101, 1944.
16. Cambi Livio and Carla Cor is el li. The dithiosalts of copper, Gazz.
chim. ital., 66, 779—784, 1936.
17. Cambron Adrien and George Stafford Whitby. Oxidation of xantha-
tes and some new dialkyl sulfur and disulfurdicarbothionates, Can, J. Re-
search, 2, 144—152, 1930.
18. Chatt Joseph and Frederick G. Mann. The constitution of complex
metallic salts. VIII. Bridged thioderivatives of palladous halides with
tertiary phosphines, J. Chem. Soc., 1938, 1949—1954.
19. Claesson Peter. Ueber Aethylmerkaptan, J. prakt. Chem. Bd. 15, № 2,
193—218; 1877.
20. С о m p i n Louis. Sur les thiosulfocarbamates et les xanthates de cobalt
et de nickel, Thesis, Ecole superieure de pharmacie de Paris. 1920.
21. Comp in Louis. Sur les thiosulfocarbamates metalliques notammant ceux
de cobalt et de nickel, Bull. soc. chim. France, 27, 464'—469, 1920.
22. Damon W. A. Stability of sodium methyl xanthate, Ann. Rept. Alkali,
etc., Works 71st Rept., 1934, 27—32; of. Chem. Abstr., 30, 3412. 9, 1936.
.23 . Debus H. Ueber die Verbindungen der Sulfocarbaminsaure, Ann. Chem.,
Justus Liebigs, 73, 26—34, 1850.
24. Dehn William M. Additive compounds formed in the desulfurization of
thioureas by copper hydroxide, J. Am. Chem. Soc., 62, 3189—3190, 1940.
25. Delepine Marcel. Metallic solts of dithiocarbamic acids; preparation
of isothiocyanates in the aliphatic series, Compt. Rend., 144, 1125—1127
(1907).
26. Delepine Marcel. Sulfur and nitrogen compounds derived from carbon
disulfide. XII. Metallic thiosulfocarbamates, Bull soc. chim. France 3,
№ 4, 643—652, 1908.
27. Delepine Marcel. Experiments on copper and iron, Bull. soc. chim.
France, 3, № 4, 652—654, 1908.
28. Delepine Marcel. Properties of the metallic salts of dithiocarbamic
acid, Compt. Rend., 146, 981—984, 1908.
СОБИРАТЕЛИ
276
Д. Delepine Marcel, and Louis С о m p i n. Sur les xanthates de cobalt
et de nickel, Bull. soc. chim. Fiance, 27, 469—474, 1920.
30. Delepine Marcel, and Louis С о m p i n. Absorption spectra of dithio-
carbamates and xanthates of cobalt, Bull. soc. chim. France, 27, 474—477,
1920.
31. D e n к о Charles W. and Arthur K. Anderson. The synthesis of
some organic compounds of gold, J. Am. Chem. Soc., 67, 2241, 1945.
32. D e r e n z i n i T. Crystal structure of cobalt xanthate, Atti soc. toscana
sci. nat. (Pisa), Mem., 47, 114—122, 1938.
32a. Derenzini T., and P. Rossoni. Crystal structure of antimony xan-
thate, Atti soc. toscana sci. nat. (Pisa), Process! verbali, 47, 67—73, 1938.
33. DeWitt С. C., and Edwin E. Roper. The surface relations of po-
tassium ethyl xanthate and pine oil, I, J. Am. Chem. Soc., 54, 444—455,
1932.
34. DeWitt С. C. and R. F. Make ns. The surface relations of the com-
ponents of pine oil and of potassium ethyl xanthate, II, J. Am. Chem.
Soc., 54, 455—464, 1932.
35. DeWitt С. C., R. F. Ma kens and A. W. H e 1 z. The surface relations
of the xanthates, J. Am. Chem. Soc., 57, 796—801, 1935.
36. Dixon, Augustus Edward, and John Taylor. The constitution
and reactions of thiocarbamides, J. Chem. Soc., 101, 2502—2528, 1912.
37. D о n a t i s C. d e. Crystal structure of iron xanthate, Atti soc. toscana
sci. nat. (Pisa), Mem., 47, 123—132, 1938.
38. Duncan W. E., Emil О 11, and E. Emmett Reid. Some copper
mercaptides and their reaction with carbon disulfide, Ind. Eng. Chem.,
23, 381—384, 1931.
39. Dub sky J. V. The saturation of the affinities of the principal and se-
condary valences in compounds of higher order, J. prakt. Chem., I, 90,
61 — 118, 1914; II, 93, 142—161, 1916: III, 103, 109—128, 1921.
40. E 1 g i n J. C. Catalytic reactions of sulfur compounds present in petroleum
II. Pure sulfur compounds in hydrocarbon materials in contact with nickel
catalysts, Ind. Eng. Chem., 22, 1290—1293. 1930.
41. E 1 g i n J. C., G. H. W i 1 d e r, and H. S. Taylor. Catalytic reactions of
sulfur compounds present in petroleum. I. High-sulfur naphthas in contact
with nickel and iron catalysts. Ind. Eng. Chem., 22, 1284—1290, 1930.
42. Elliott, Stanley B. The Alkaline-earth and Heavy-metal Soaps, Reinhold
Publishing Corporation, New York, 1946.
43. Ellis L. M., Jr. and E. Emmett Reid. The preparation and proper-
ties of a double series of aliphatic mercaptans, J. Am. Chem. Soc., 54,
1674—1687, 1932.
44. Ephraim Fritz. Inorganic Chemistry 4th ed., translated by P. C. L.
Thorne and E. R. Roberts, (a) p. 737, Nordeman Publishing Co., Ins.,
New York, 1943.
45. Ephraim Fritz, and Etta M a j 1 e r. Some sulfophosphates, Ber. deut.
chem. Ges., 43, 285—288, 1910.
46. E p h r a i m Fritz, and Rebecca Stein. Th’ophosphates and thio-
phosphites, Ber. deut. chem. Ges., 44, 3405—3416, 1911.
47. Ferrer Jaime and Angel del Campo. An analytical application
of some xanthogenates, Anales soc. espan, fis., у quim, 9, 173—174, 1911.
48. F i e s e r, Louis F., and Mary Fieser. «Organic Chemistry» (a)'
pp. 88—90, (b) pp. 901—904, D. C. Heath and Company, Boston, 1944.
49. F о r e Dan, Jr., and R. W. Bos t. Sulfur studies. XIV. Some derivati
ves of certain higher mercaptans, J. Am. Chem. Soc., 59, 2557—2559, 1937.
50. Foster, Laurence S. The preparation of xan’.hates and other orga-
nic thiocarbonates, Utah Eng. Expt. Sta., Tech. Paper 2, 1929.
•51. Freundlich H. and A. В j e г с к e. The kinetics of the accelerated
oxidation of phenylthiourea by charcoal, Z. physik. Chem., 91, 1—45, 1916.
268
ГЛАВА 8
52. Freundlich Н. and А. Н. Fischer. The kinetics of the oxidation of
thiourea on charcoal, Z. physik. Chem., 114, 413—429, 1924.
53. F г о m m, Emil. Ueber ungesattigte Disulfide, Ann. Chem., Justus Lie-
fa gs, 348, 144—160, 1906.
54. Fry H. S. New methods of preparing thiocarbanilides, J. Am. Chem..
Soc., 35, 1539—1546, 1913.
55. G a r a c h J. The formation of zinc diphenyldithiocarbamate, Caoutchouc
guttapercha, 20, 11946—11949, 1923.
56. G a u d i n A. M., Franklin Dewey, Walter E, Duncan, R. A.
Johnson, and Oscar F. Tangel. Jr. Reactions of xanthates with
sulfide minerals, Trans. AIMME, 112, 319—347, 1934.
57. G a u d i n A. M. and Reinhardt Schuhmann, Jr. The action of
potassium amyl xanthate on chalcocite, J. Phys. Chdm., 40, 257—275, 1936.
58. Ghosh J. C., S. N. R а у c h a u d h u r i, and S. C. G a n g u I i. Oxidation-
reduction potential. 1, Cystine, J. Indian Chem. Soc., 9, 43—52, 1932.
59. G i о r d a n a, Andrea. Zinc xanthates. Montana School Mines Rcpts., 1935.
60. Goddard Archibald Edwin. In J. Newton Friend, ed. «Textbook
of Inorganic Chemistry», vol. XI, pt. Ill, Charles Griffin & Co., Ltd.,.
London, 1936.
61. Grignard, Victor, ed. «Traite de chimie organique», by M. Cramer,
E. Fourneau, and M. Lesbre, vol. XII, (a) pp. 48—51, Masson et Cie.
Paris, 1941.
62. Grossmann, Hermann. Thiourea, Chem.-Ztg., 31, 1195—1196, 1907.
63. H alb an H. von, and W. Hecht. Xanthic acids and the kinetics of their
decomposition, Z. Elektrochem., 24. 65—82, 1918.
64. Halban H. von, and Alexander Kirsch. Xanthogenic acid and the kinetics
of its decomposition, Z. physik. Chem., 82, 325—360, 1913.
65. Harned, Herbert S. and Benton B. Owen. The thermodynamic
properties of weak acids and bases in salt solutions, and an exact method
of determining their dissociation constants, J. Am. Chem. Soc., 52, 5079—
5091, 1930.
66. H a r r i s о n D. C. Catalytic action of traces in iron and copper on the
anaerobic oxidation of sulfhydryl compounds, Biochem. J., 21, 335—346,1927.
67. Harwood H. J a m e s. U. S. Patent 2122644/1938.
68. Hebert Alexander. Thermal decomposition qi metallic xanthates,.
Bull. soc. chim. France, (4) 9, 523—532, 1911.
69. H e c t о r D. S. Ueber die Einwirkung von Wasserstoffsuperoxyd und
Monophenylthioharnstoff, Ber. deut. chem. Ges., 22, 1176—1180, 1889.
70. Herrmann F. Ueber Verbindungen des Goldes mit schwefelhaltigere
organischen Radicalen, Ber. deut. chem. Ges., 38, 2813—2825, 1905.
71. Hi eb er W., and C. Scharienberg. Metal carbonyls. XXL The ac-
tion of organic sulfur compounds on the iron carbonyls, Ber. deut. chem.
Ges., 73B, 1012—1021, 1940.
72. Hirschkind W. Rapid method of analyzing xanthate, Eng. Mining J
Press, 119, 968—970, 1925.
73. Hofmann A. W. v. and F. Mahla. Ueber Diaethyldithiosphophinsaure,.
deren Salzen und Derivate, Ber. deut. chem. Ges. 25, 2436—2444, 1892.
74. Hofmann, K. A. und O. F. W i e d e. Weitere Mittelungen uber Nitro-
soverbindungen des Eisens, Z. anorg. Chem. 9, 295—303, 1895.
75. Hugershoff Albert. Acetyl derivatives of thiourea and urea. Con-
stitution of th'ourea. Ber. deut. chem. Ges., 58B, 2477—2487, 1925.
76. Huntress Ernest Hamlin. «А brief Introduction to the Use of
Beilstein’s Handbuch der organischen Chemie», 2d ed., John Wiley & Sons,
Inc., New York, 1938.
77. Jensen, K. A. Nickel mercaptides, Z. anorg. Chem., 252, 227—233, 1944.
78. Jimenez Herrera J. The action of hydrogen peroxide on organic
sulfur compounds, Anales soc. espan. fis. у quim, 33, 877—886, 1935.
СОБИРАТЕЛИ
269
79. Karr er Paul. «Organic Chemistry», translated by A. J. Mee Elsevier
Press, Inc., Houston, Tex. 1938.
80. Keller С. H. U. S. Patent 1554216/1925.
81. King Cecil V., and Emilie Dublon. The rate decomposition of
xanthic acid, J. Am. Chem. Soc., 54, 2177—2186, 1932.
82. Kit a m u r a Ryoichi. Reaction between organic sulfur compounds
and hydrogen peroxide. III. The reaction mechanism. J. Pharm. Sue.
Japan, 55, 300—349, 1935, with abstract in German, 72—82; Cf. Chem.
Abstr., 29, 5810, 8, 1935,
.83 . Kitamura Ryoichi. Reaction between organic sulfur compounds and
hydrogen peroxide. IV. Thioacids, J. Pharm. Soc. Japan, 57, 5—22, 1937.
.84 . Klason Peter. Ueber Darstellung von Sulfhydraten und Sulfiden des Met-
hans und Aethans, Ber. deut. chem. Ges., 20, 3407—3413, 1878,
85. Kohler H., and A. Michaelis. Ueber PnosphenylsuHochlorid und
Derivate, Ber. deut. chem. Ges., 9. 1053—1054, 1876.
86. Koppel J. The detection of molybdenum with xanthic acid, Chem. Ztg.,
43, 777—778, 1919.
87. Кошкин H. В. Действие однохлористой и двухлористой меди и метал-
лического натрия на тиокарбаиилид, Журнал общей химии, 8, 1083—
1090, 1938.
88. Kosolapoff Gennady М. «Organophosphorus Compounds», John
Wiley & Sons, Inc., New York, 1950.
•89. К r u 1 1 a Rudolf. Eine neue Gruppe metallorganischer Verbindungen der
Aromatischen Thioharnstoff-Reihe, Ber. deut. chem. Ges., 46, 2669—2672, 1913.
90. Kutzelnigg Artur. Solubility of cobalt xanthate in organic solvents
and its analytical use, Z. anorg. Chem., 256, 46—48, 1948.
QI. Larsson Erik. The electrolytic dssociation of dibasic acids. IV. The
dissociation constants of some mercaptomonocarboxylic acids, Z. anorg. u.
allgem. Chem., 172, 375—384, 1928.
92. Lecher Hans, Fritz Graf, Claus Heuck, Karl Koeberee,
Fritz Gnaedinger, and Fritz Heydweiller. Constitution of
thiourea and the thiuronium-salts, Il and III, Ann. Chem., Justus Lieb.gs,
445. 35—61, 77—82, 1925.
93. Lecher Hans and Claus Heuck. Constitution of thiourea and the
thiuronium salts, I, Ann. Chem., Justus Liebigs, 438, 169—184, 1924.
94. Lehmann О Uber Kiinstliche Farbung von Krystallen. Z. physik. Chem.
8, 543—553, 1891.
95. Ley H. Cber innere metall-komplex Salze, I, Z. Elektrochem., 10, 954—
956, 1904.
96. Li Ch oh-Hao and T. D. Stewart. Comparison of basicity of ali-
phatic amines in different solvents, J. Am. Chem .Soc., 59, 2596—2599, 1937.
97. L i e s e r T., and W. Nagel, Carbohydrates. I. Xanthate reaction with
lower alcohols and sugars, Ann. Chem., Justus Liebigs, 495, 235—249, 1932.
98. M a h r C„ and Hertha О h 1 e. Thiourea complex salts of lead and
thallium, Ann. Chem., Justus L ebigs, 542, 44—48, 1939.
99. M a k e n s R. F. The silver xanthates, J. Am. Chem. Soc., 57. 405—406, 1935.
100. Malatesto Lambert o. Xanthates of metals of group VI Gazz. chim.
ital., 69, 408—416, 1939,
101. M a 1 a t e s t a L a m b e r t o. The magnetic moments of xanthates and
d'thiocarbamates, Gazz. chim. ital., I, 69. 629—639, 1939; II, 70, 541—553,
1940; 111, 70, 553—559, 1940.
102. Ma I a test a Lambert o, The relation between the solubilities of sul-
fides and xanthates or thiocarbamates of the heavy metals, Chimica e in-
dustria (Milan), 23, 319—321, 1941.
103. M a I a t e s t a Lamberto. The electric moments of the nickel salts of
a and P diethyldithiophosphinic and of other thiophosphoric acids, Gazz.
chim. ital., 76. 182—186, 1946.
104. M a 1 a t e s t a Lamberto. The reaction between phosphorus pentasul-
270
ГЛАВА 8
fide and Grignard compounds, Gazz. chim. ital., 77, 509—517,518—525, 1947.
105. Malatesta Lamberto and Rachele Pizzoti. Nickel, cobalt,
and iron salts of acids containing the radical = P(:S)SH, Chimica e in-
dustria (Milan), 27, 6—10, 1945.
106. M a 1 a t e s t a Lamberto, and Rachele Pizzotti. The reaction1
between phosphorus pentasulfide and Grignard compounds, Gazz. chim..
ital., 76, 167—181, 1946.
107. M a 1 i s о f f W. M., and E. M. Mark s. Thermal behavior of sulfur com-
pounds in hydrocarbon solvents. I Aliphatic mercaptans, Ind. Eng. Chem;,.
23. 1114—1120, 1931.
108. Malisoff William M., Ernest M. Marks, and Fred G. Hess.
Mercaptan chemistry, Chem. Revs., 7, 493—547, 1930.
109. Manchot W., a n d H. G a 11, Univalent iron, cobalt and nickel, Ш,
Ber, deut. chem. Ges., 60, 2318—2322, 1927.
110. Manchot W_, and Fr. Kaess. Univalent iron, cobalt and nickel, IT, :
Ber. deut. chem. Ges., 60B, 2175—2180, 1927.
111. Mastin T. W„ George R. Norman, and E. A. Weil m ue n- '
st er Chemistry of the aliphatic esters of thiophosphoric acids, I, J. Am.
Chem' Soc., 67, 1662—1664, 1945.
112. McLeod Norman, Solubility of lead xanthates in acetone-water
mixtures, Thesis, Montana School of Mines, 1934.
113. Michaelis Leonor, and Maxwell P. Schubert. Cobalt com-
plexes of thioglycol'c ac:d, J. Am. Chem. Soc., 52, 4418—4426, 1930.
114. Mikumo, Jiro, Hiromi Kawai, and Akira Matsu oka. Surfa-
ce-active compounds. IV. Higher sodium xanthates, J. Soc Chem. Ind.,.
Japan, 49, 129—132, 1946. '
115. Montequi Ricardo. The reaction of molybdenum with the xanthates'.
The molybdenum ion, Anales soc. espan. fis. у quim., 28, 479—487, 1930.
116. Neuhaus A. Partially isomorphous organic mixed systems (so-calledi
anomalous mixed crystals), Z. physik. Chem., A191, 359—374, 1943.
117. Neuhaus A. Partial isomorphic systems. VII. Oriented growth of thiou-
rea and urea on inorganic carrier lattices Naturwissenschaften 32 34—
36, 1944.
118. Ott Emil, and E. Emmett Reid. Reactions of some mercaptans
with alkaline sodium plumbite solutions, Ind. Eng. Chem., 22, 878—881, 1930’.
119. Ott Emil H, and E. Emmett Reid. Auto-oxidation of lead mer-
captides, Ind. Eng. Chem., 22, 882—884, 1930.
120. Ott Emil, and E. Emmett Reid. Reactions of lead mercaptides with
sulfur, Ind. Eng. Chem,, 22, 884—886, 1930.
121. Otto Robert. Uber der Verhalten des Ouecksilber und Ble'a^hvlmer-
captides bei hoherer Temperatur, Ber. deut. chem. Ges., 13,1289—1290, 1880.
122. Patterson T. S. The derivation of the word mercaptan Chemistry &
Industry, 43, 196—198, 1924.
123. Patterson. Austin M., Leonard T. Capel 1, and Mary A. Ma-
gill. Thiols (mercaptans), Chem. Abstr., 39, 5946, 1945.
124. Pawelka Franz Georg. Organic silver complexes Z Electrochem.,.
30, 180—186, 1924.
125. Pirie Norman Wingate. Cuprous derivatives of some sulfhydryl
compounds, Biochem. J., 25, 614—628. 1931.
126. P i s t c h i m u к a P. S. Transformations of thio and selenophosphoric
esters, J. prakt. Chem.. 84, 746—760, 1911.
127. Пишимука P. С. Превращения эфиров тио- и селенофосфорных кис-
лот. Журнал русского физико-химического общества. 44, 1406—1554, 1912.
128. Ralston A. W. «Fatty Acids and Their Derivatives», (a) 671, (b),
pp. 645—646, John Wiley & Sons, Inc. New York, 1948.
129. Ralston A. W. and C. W. H о e r r. The solubilities of hexyl— and do-
decylammonium chloride in various dilutions of aqueous ethanol., J Am.
Chem. Soc., 68, 851—854, 1946.
СОБИРАТЕЛИ
271
130. Ramberg Ludwig. Ober die Platosalze einiger schwefelhaltigen orga«
nischen Sauren, Z. anorg. Chem., 50, 439—445, 1906.
131. Ray Prafulla Chandra. Interaction of mercuric and cupric chlori-
des respectively with the mercaptans and potential mercaptans, J. Chem.
Soc., 115, 871—878, 1919; Varying valency of platinum with respect to
mercaptanic radicals, J. Chem. Soc., 123, 133—141, 1923; Varying valency
of gold with respect to mercaptanic radicals, I, Quart. J. Indian Chem.
Soc., I, 63.73, 1924.
132. Reihlen, Hans, Adolf v. Friedolsheim, and Walter Os-
wald. Nitric oxide and carbon monoxide compounds of apparently uni-
valent iron and nickel. Ann. Chem., Justus Liebigs, 465, 72—96, 1928.
133. Re у chler A. Silver xanthate, Bull. soc. chim. Belg., 37, 165—167, 1928.
134 Reynolds J. Emerson. Some silver compounds of thiourea, J. Chem.
Soc., 61, 249—254, 1892.
135. Rosenheim A. and I. Davidsohn. Ober die Bildung von Komplex-
salzen bei Thiosauren die Thioglykolsauren Salze, Z. anorg. Chem., 41,
231, 1904.
136. Rosenthal, Sanford AL., and Carl V о e g 11 i n. Action of heavy
metals on cysteine and on sulfhydryl groups of poteins, U. S. Publik
Health Repts., 48, 347—364, 1933.
137. Royer L. The orientation of organic crystals on minerals with an ionic
structure, Compt. rend., 196, 282—285, 1933.
138. Ry к lan, Lionel R., and Carl L. A. Schmidt. Oxidation poten-
tials of cystine-cysteine and related systems Univ. Calif, Pubis Physiol.,
8, 257—276, 1944.
139. Sachs G. The ethyl mercapto-mercury salz, Z. anorg allgem. Chem.,
135, 273—282. 1924.
140. Sachs G. Mercuric oxyethyl mercaptide, Z. anorg. u. allgem. Chem.,
135, 283—287, 1924.
141. Schaum K-, Ph. S i e d 1 e r, and E. Wagner. Hydrolysis and dis-
sociation of xanthates, Kolloid-Z., 58, 341—348, 1932,
142. Schubert, Maxwell P. Cobalt complexes of cysteine, J. Am. Chem..
Soc., 53, 3851—3861, 1931.
143. Schuhmann Reinhardt, Jr. The nature of the action of potassium
n-amyl xanthate on chalcocite Thesis, Montana School of Mines, 1935.
144. Shinzel Theodore. Preparation, stabilite et action pysiologique
de combinaisons organiques contenant des metaux lourds dans la serie
des acides dithiocarbamiques, des acides thiols et des hydrazines diacylees,
ANC Imprimerie de la cour d’appel, Paris. 1938; cf. Chem. Abstr., 32,
7676, 1938.
145. Slagle, Kenneth H„ and E. Emmett Reid. Action of some mer-
captans in hydrocarbon solution on copper and copper sulfide, Ind. Eng.
Chem., 24, 448—451. 1932.
146. Spedden H. Rush. Flotation in R. E. Kirk and D. F. Othmer, eds.,
«Encyclopedia of Chemical Technology», vol. 6, The Interscience Encyclope-
dia, Inc., New York, 1951.
147. Steele, Laurence L. Abietic acid and certain metal abietates, J. Am.
Chem. Soc., 44, 1333—1341, 1922.
148. S t о г f e r, E. The flocculation of complex trithiourea cuprochloride from
aqueous solution, Monatsh. Chem.. 70. 236—250, 1936.
149. Straumanis and Cirulis. New complex compounds of mercury
and copper halides with aliphatic amines, Z. anorg. u. allgem. Chem.,
230, 65, 1936; A few new complex compounds of mercury halides with the
halides of aliphatic amines, Z. anorg. u. allgem. Chem,, 234, 17, 1937.
150. Suter, Chester Merle. «The Organic Chemistry of Sulfur», John
Wiley & Sons, Inc., New York, 1944.
151. Taggart Arthur F.. and M. D. H a s s i a 1 i s. Solubility product
272
ГЛАВА 8
and bubble attachment in flotation, AIMME, Mining Technol., 10 (5),
Tech. Publ. 2078, 1946.
152. Tarugi N. and F. S о r b i n i. Arsenic xanthogenate in analytical che-
mistry. Bull. chim. farm., 51, 361—370, 1'912.
153. Thunaes Arvid, and Leo Abell. Private reports, Consolidated Mi-
ning and Smelting Company of Canada, Ltd., 1935.
154. Tschugaeff L. Ober selenomercaptane und einige Derivate derselben,
Ber. deut. chem. Ges., 42, 49—54, 1909.
165. Vanino L., and F._ M u s s g n u g. Organic bismuth compounds of the
bismuth halides, Ber. deut. chem. Ges., 50, 21—24, 1917.
156. Walter George. Complex metal thiourea salts and a non-complex
arsenic thiourea compound, Ber. deut. chem. Ges., 64B, 1087—1095, 1931.
157. Walter George, Max Adler and Georg Renner. Complex metal-
thiourea salts, V, Monatsh. Chem., 65, 59—80, 1934.
158. Walter George, and Ernst Storfer. Complex metal thiourea
salts, II, III, IV, Monatsh. Chem., 65, 21—58, 1934.
159. Wells A. F. The crystal structures of mercury n-alkyl mercaptides,
Z. Krist., 96, 435—450, 1937.
160. Wenger P., Z. В e s s o, and R. D u с к e r t. Potassium ethylxanthate as
an analytical reagent, Mikrochemie ver. Mikrochim, Acta, 31, 145—148,
1943; Helv. Chim. Acta, 27, 291—293. 1944.
161. Wenger P., R. D u с к e r t, and E. A n к a j d i. New method of gene-
ral analysis for cations based on the use of potassium ethylxanthate, 11,
Helv. Chim. Acta, 28, 1316—1325; 1592—1609, 1945.
162. Werner Emil Alphonse. The action of n.trous acid on thiocarba-
mide and on formamidine disulfide, A new structural formula for thiocar-
bamide, J. Chem. Soc., 101, 280—291, 1912.
163. Werner Emil Alphonse. The interaction of iodine and thiocarba-
mide. The properties of formamidine disulfide and its salts, J. Chem. Soc.,
101, 2166—2180. 1922.
164. Werner Emil Alphonse. «The Chemistry of Urea», Longmans,
Green & Co., Inc., New York, 1923.
165. Wertheim, E. Derivatives for the identification of mercaptans, J. Am.
Chem. Soc., 51, 3661—3664, 1929.
166. Whitby G. S., and G. L. Matheson. Some heavy-metal salts of
disubstituted dithiocarbamic acids, Trans. Roy. Soc. Can., 18 (sec. 1111,
111 — 114. 1924.
167. Whitmore Frank C. «Advanced Organic Chemistry», (a) p. 285, (b)
p. 746, D. Van Nostrand Company, Inc., New York, 1937.
168. Williams, J. W., and E. M. D r i s s e n. Oxidation-reduction poten-
tials of certain sulfhydryl compounds, J. Biol. Chem., 87, 441—451, 1930.
169. Wuyts Y., and A. Vangindert aelen. The quadrivalence of tin in
its mercaptides. Bull. soc. chim. Belg., 30, 323—329, 1921.
170. Wyckoff Ralph W. G. «Crystal Structures», vol. Il, Interscience
Publishers. Inc., New York, 1951.
171. X a n J., E. A. Wilson, L. D. Roberts, and N. H. Horton. The
absorption of oxygen by mercaptans in alkaline solution, J Am Chem
Soc., 63, 1139—1141, 1941.
172. Y о e J. H., and L. G. Overholser. Reactivity of substituted thioureas
with inorganic ions, Ind. Eng. Chem., Anal. Ed., 14. 435—437, 1942.
173. Yost D о n M._ and Horace Russell, Jr. «Systematic Inorganic
Chemistry of the Fifth and Sixth Group /Metallic Elements», Prentice-
Hall, Inc., New York, 1944.
174. Zeise W. C. Vorlaufige Anzeige einer neuen Classe von Schwefelver-
bindungen, J. Chem. Physik, 35. 173, 1822; Die Xanthogensaure nebs!
einigen Produkten und Verbindungen derselben, J. Chem. Physik, 36, 1, 1822.
175. Zeiss Harold H. The chemistry of the resin acids, Chem Revs., 42,
163—188, 1948.
F
Глава 9
МЕХАНИЗМ ДЕЙСТВИЯ СОБИРАТЕЛЕЙ
Механизм взаимодействия собирателя с минералами являет-
ся основной проблемой теории флотации. В отношении его было
высказано большое число предположений. В последние годы он
стал предметом все возрастающего числа исследований, на ос-
новании которых в недалеком будущем удастся сформулировать
систематизированную теорию. С этой точки зрения представ-
ляет интерес рассмотрение различных гипотез, а также полу-
ченных экспериментальных доказательств.
Краткое изложение предложенных гипотез. Применение на
заре практики флотации некоторых масел, которые способство-
вали прилипанию частицы к пузырьку, получило рациональное
объяснение в предположении, что масло прилипает к частицам
и пузырькам и цементирует их. Согласно этой точке зрения, ма-
сла поглощаются концентратами и не поглощаются хвостами.
Желая объяснить причины закрепления частичек на пузырь-
ках воздуха, Бэйнс [3] предположил, что при перемешивании
благодаря трению развивается статическое электричество.
Против этой теории возражали Фаренволд [12], Ральстон и
Андерсон [1], и она была оставлена.
Толщина слоя собирателя на флотированных частицах была
вначале вычислена Фаренволдом и его учениками [13, 14]. Со-
гласно этим вычислениям, толщина слоя собирателя составляла
от 20 до 100 молекул. В настоящее время хорошо известно, что
эти цифры слишком завышены. В то время еще не были разра-
ботаны методы для измерения поверхностей минеральных по-
рошков и приблизительный подсчет величины удельной поверх-
ности минеральных порошков давал низкие значения из-за
большого числа неучтенных при подсчете факторов.
Более современные представления были введены в теорию
флотации Ирвингом Лэнгмюром [52] и другими [73]. В пределах
точности довольно грубых аналитических определений и изме-
рений величины поверхностей минералов, проведенных трид-
цать лет назад, было сделано предположение, что поверхност-
ные покрытия, образованные собирателем на минералах, пред-
ставляют собой монослои.
В противоположность господствовавшим ранее представле-
ниям о закреплении собирателя на поверхности минерала в ви-
18 А. м. Голэи
274
ГЛАВА 9
де капель или масляных пленок были выдвинуты предположе-
ния о наличии молекулярной формы закрепления, причем боль-
шое значение придавалось растворенной части собирателя.
Процесс, благодаря которому собиратель поглощался минера-
лом, был назван в общем виде «адсорбцией» без уточнения его .
механизма. Возрастающее применение и увеличивающееся зна- I
чение растворимых ионогенных собирателей способствовало пе- . i
реходу от представлений о молекулярной адсорбции к пред- |
ставлениям об ионной адсорбции [17], однако продолжала оста- j
ваться непонятной возможность адсорбции иона одного знака
без соответствующей адсорбции противоиона. j
Таггарт и его сотрудники ясно поняли, что существует ион- , |
ный обмен при взаимодействии между минералами, образую- . j
щими водную суспензию, и ионными или ионизованными в воде
собирателями [72, 74]. Это было большим шагом вперед.
Наличие ионного обменного механизма не обязательно свя-
зано с образованием новой фазы, как это вначале предполагал
Таггарт и его сотрудники. В самом деле, этот механизм отве-
чает представлениям современной теории адсорбции, причем
более вероятна адсорбция и десорбция ионов, нежели молекул
[19, 81].
Достижения в методах измерения поверхностей порошков и ,
в усовершенствовании аналитических методов определения ма-
лых количеств веществ, растворенных в воде, или же распреде-
ляющихся на минеральной поверхности, сделали возможным
измерение плотности покрытия собирателя. Эти измерения яс-
но показали, что в большинстве случаев адсорбированный мо-
нослой далеко не насыщен [21, 23, 33]. При этом было уставов- [
лено, что существует равновесие между двумерной концентра- j
цией собирателя на поверхности минерала и объемной концен-
трацией собирателя в водном растворе.
Было также высказано предположение [8, 57], что эффектив-
ным собирателем является неионная (молекулярная) форма,
получающаяся при гидролизе ионных собирателей. Согласно
этой точке зрения, действие собирателя представляет собой из-
бирательную молекулярную адсорбцию. Однако отсутствуют
какие-либо доказательства в пользу преимущества этой гипоте- ;
зы по сравнению с современной гипотезой ионного обмена. Со- '
временные представления о действии реагентов, основывающие- ’
ся на механизме ионной адсорбции, встречают также известные
затруднения. Обычно они являются результатом игнорирования
структуры двойного слоя. Дальнейшее развитие эти представ-
ления получат, когда будет накоплен количественный и качест-
венный материал, который может быть использован в соответ-
ствии с физико-химическими представлениями о свойствах по-
верхностей раздела твердых тел и жидкостей.
।
МЕХАНИЗМ ДЕЙСТВИЯ СОБИРАТЕЛЕЙ
275
Действие ксантогенатов на галенит
Механизм действия собирателя на минералы, вероятно луч-
ше всего изучить, произведя обзор всего, что известно для си-
стемы галенит — этилксантогенат калия. Выбор этой системы
для рассмотрения сделан на основании имеющихся детальных
исследований ее.
Краевые углы для системы галенит—ксантогенат. На поли-
рованной поверхности галенита в чистой воде краевой угол ра-
вен 0°. В разбавленных растворах ксантогената краевой угол
равен примерно 60°. Это справедливо для всех концентраций,
превышающих, скажем, 2- 10 3 %. В еще более разбавленных
растворах краевой угол также образуется, однако точно изме-
рить его трудно, и требуется большое терпение для получения
воспроизводимых результатов. Время, необходимое для того,
чтобы произошло прилипание, называется «временем индук-
ции». Наличие определенного «времени индукции» свидетель-
ствует о том, что диффузия ксантогената происходит медленно,
или это может также свидетельствовать о передвижении ксан-
тогената вдоль поверхности раздела как относительно поверх-
ности пузырька воздуха, так и твердой поверхности.
На рис. 1 представлены экспериментальные данные Уорка
и Кокса [82], характеризующие зависимость краевого угла от
концентрации ксантогената.
Толщина поверхностных пленок ксантогената на галените.
На рис. 2 [27] приводятся данные, характеризующие зависи-
мость извлечения галенита от количества введенного ксантоге-
ната. При применении этилксантогената калия или реагента с
более длинной углеводородной цепью количество реагента, не-
обходимое для получения, например, 90%-ного извлечения, не-
достаточно для образования на поверхности минерала плотного
монослоя. Так, например, если принять коэффициент формы
равным 4,2 [39] (вместо 1,2, как это предполагалось ранее) и
площадь поперечного сечения, занимаемую группой ксантогена-
та, разной 29 А2 (вычисленной из предположения плотной упа-
ковки), то количество реагента, необходимое для образования
монослоя, равно 46 г/т в случае этилксантогената и 58 г/т в
случае амилксантогената. Для достижения извлечения, равного
90%, требовалось ввести соответственно 41 и 14 г/т, т. е. коли-
чества, значительно меньшие, чем это необходимо для образова-
ния монослоя. Это является одним из наиболее ранних доказа-
тельств того, что эффективное поверхностное покрытие собира-
теля может составить только часть насыщенного монослоя.
Новые измерения Преллера [32] подтвердили, что неполный
монослой собирателя обеспечивает полное извлечение. Чистый
галенит подвергали мокрому измельчению в галечной мельнице
18*
Рис. 1. Краевые углы на галените в растворах этил-
ксантогената калия после 40—60 мин. контакта [82]
Рис. 2. Флотация галенита различными ксантогенатами; минерал
крупностью —100 + 600 меш, флотировался при добавках 0,045 кг/т
терпинеола, 0,450 кг/т едкого натра и ксантогената в количествах,
указанных на оси абсцисс:
I — метилксаитогеиат калия; 2 — этилксантогеиат калия; 3 — гтрогтилксаитоге-
иат калия; 4 — и-бутнлксавтогеиат калия; 5 — изоамилксавтогенат калия
МЕХАНИЗМ ДЕЙСТВИЯ СОБИРАТЕЛЕЙ
277
и флотировали при помощи различных количеств этилксантоге-
ната калия при pH ~ 8,5. Удельная поверхность питания флота-
ции измерялась методом адсорбции газа и была найдена рав-
ной 4100 см2!г.
Если предположить, что применявшийся ксантогенат был хи-
мически чистым, полностью поглощался минералом и распреде-
лялся равномерно по поверхности всего минерала, то получен-
ные результаты будут такими, как показано в табл. 1. Плот-
ность покрытия можно выразить в долях монослоя при условии
плотной упаковки ксантогенатных радикалов, считая, что пло-
щадь поперечного сечения радикала равна 29 А2.
Таблица 1
Зависимость извлечения галенита от плотности покрытия
поверхности минерала ионами ксантогената [52]
Этилксанто- геиат калия Ketm Извлечение галенита % Максимальная величина по- верхностного покрытия % Этилксаито- генат калия кг,т Извлечение галенита % Максимальная величина по- верхностного покрытия %
0,051 7 15 0,172 83 51
0,085 21 25 0,214 90 63
0, 28 49 38 0,258 96,8 76
0,168 74 43 0,300 97,5 89
0,163 80 48
Следует иметь в виду, что процент плотности покрытия по-
верхности минерала собирателем, приводимый в табл. 1, являет-
ся, скорее верхним пределом, чем действительно измеряемой
величиной, так как отсутствуют данные, позволяющие оценить
количество ксантогенатных ионов, оставшихся в растворе.
Явление ионного обмена для системы ксантогенат — галенит.
Экспериментальное доказательство поглощения ксантогената
галенитом из щелочных растворов ксантогената было получено
Таггартом, Тейлором и Ноллом [74]. Результаты исследований,
которые были подробно изложены Тейлором и Ноллом [76], мо-
жно свести к следующему.
Галенит, измельченный сухим или мокрым способом, погло-
щает ион ксантогената из водных растворов этилксантогенатй
натрия или калия. При этом происходит обмен с сульфатными
ионами, сульфоксидными ионами меньших степеней окисления
и с карбонатным ионом. Как можно видеть из табл. 2, баланс
этого обмена удовлетворительно отвечает стехиометрическим
отношениям (если принять во внимание наличие СОз~). Катион
278
ГЛАВА 9
Таблица 2
Обмен ионов между галенитом и ксантогената м1,2
Первоначаль- ный раствор ксантогената8 Количество поглощенных ионов ксантогената Количество выделенных ионов с поверхности минерала
сульф- оксидные ионы низших степеней окисления сульфатные воин карбонатные воны общее количество ионов
16,3 12,5 2,5 10,9
23,2 17,7 2,8 12,6 — —
32,4 23,6 3,1 12,8 — —
65,0 30,2 3,0 12,7 — —
129,0 31,4 з,з 12,9 —
162,6 32,8 3,6 13,1 — —
293,0 40,9 3,5 11,2 — —
200,0 39,04 8,0 5,8 26,0 39,0
200,0 52,35 12,7 12,8 — —
200,0 58,3’ 13,8 16,1 27,2 57,1
200,0 64,8’ 14,1 18,7 25,8 57,6
200,0 88,8’ 19,6 27,1 41 87
1 Согласно Тейлору и Ноллу [76].
8 Ион ксантогената вводился в виде этилксантогената калия; концентрация всех ионов
в этой таблице выражена в миллиграммах на литр этилксаитогеиата калия, или в стехио-
метрических эквивалентах.
4 На каждые 30 г галенита добавлялось 300 мл раствора.
4 Без выдерживания после измельчения и перед обработкой ксаитогенатом.
4 Растворы выдерживались одну неделю.
4 Растворы выдерживались две недели.
7 Растворы выдерживались три недели.
•Растворы выдерживались около месяца; новая навеска галенита.
щелочного металла не участвует в обмене с галенитом. В одном
из опытов при добавлении 20,4 мг этилксантогената натрия к
30 г галенита в 300 мл воды в конечном растворе содержание
ионов натрия было достаточным для образования 20,6 мг ксан-
тогената натрия, тогда как содержание ксантогената отвечало
всего 12 мг соли. В опыте с другой навеской галенита и исход-
ным содержанием этилксантогената калия, равным 60,6 мг, ко-
нечный раствор содержал ионы калия в количестве, достаточ-
ном для образования 61,2 мг соли, и ионы ксантогената — толь-
ко для 25,4 мг соли.
Если через А обозначить анионы, первоначально присутству-
ющие в какой бы то ни было форме на поверхности галенита,
то полученные данные отвечают реакции:
__________А~
Галеиит___X
________Х~
Галеиит 4" •
(1)
МЕХАНИЗМ ДЕЙСТВИЯ СОБИРАТЕЛЕН
279
Из табл. 2 видно, что общее количество анионов А~, участ-
вующих в обмене, возрастает с увеличением концентрации ио-
нов ксантогената в растворе, однако значительно медленнее,
чем их концентрация. Следовательно, количество анионов А~,
участвующих в обмене, увеличивается в зависимости от степе-
ни окисленности галенита.
В самом деле, если минерал измельчать в среде азота, то его
ионообменная емкость понижена (в одном из опытов наблюда-
лось трехкратное понижение ионообменной емкости от 16 до
5,5 мг Х~ на 100 г галенита). В более поздних исследованиях
Херда и Юра [45], проведенных с галенитом, измельченным при
отсутствии кислорода, обменная емкость минерала настолько
уменьшилась, что авторы пришли к заключению о том, что чи-
стый галенит не обнаруживает заметной обменной способности
и не флотируется щелочными ксантогенатами. В основном при-
мерно в одно и то же время такой же вывод был сделан Нол-
лом и Вакером [50].
Условия электронейтральности требуют, чтобы на поверхно-
сти галенита количество катионов соответствовало либо коли-
честву вытесняемых анионов А~, либо поглощаемых ксантогенат-
ных ионов, так что уравнение (1) можно переписать в следую-
щем виде:
Pb2+ 2А
Галеиит
+ 2К+ + 2Х~
Pb2+. + 2Х
Галенит
В растворе
+ 2К+ + 2А~.
В растворе
(2)
Казалось бы, можно предположить, что галенит перед обра-
боткой ксантогенатом покрыт такими солями, как сульфат
свинца, карбонат свинца и сульфит свинца, а после обработки
ксантогенатом — ксантогенатом свинца. Однако это предполо-
жение следует пересмотреть, принимая во внимание несоответ-
ствие растворимости этих «соединений» свинца в присутствии
соответствующих анионов на поверхности галенита.
Этот вопрос исследовали Тейлор и Нолл и рассматривал
Таггарт [70]. Тейлор и Нолл, например, нашли, что при переме-
шивании с водой сильно окисленного галенита раствор содер-
жит 42,8 мг сульфата свинца в 1 л при 23°, что почти точно со-
ответствует растворимости обычного сульфата свинца [65]
(41 мг/л при 20°, 45 мг/л при 25°). Свежеизмельченный галенит
в воде дает раствор, содержащий только 9—10 мг сульфата
свинца в 1 л, однако при обработке галенита этилксантогенатом
калия содержание сульфата в растворе изменялось до коли-
честв, эквивалентных 13—29 жг/л сульфата свинца. Очевидно,
равновесие между минералом и водой было таково, что на по-
верхности имелись ионы свинца и сульфатные ионы в относи-
280
ГЛАВА 9
тельно большом количестве даже в том случае, когда раствор,
находившийся в контакте с минералом, не являлся насыщен-
ным раствором сульфата свинца. Соответственно водная вы-
тяжка содержала только около 8 мкмоль ионов серы низших
степеней окисления, тогда как раствор после обработки ксанто-
генатом содержал до 56 мкмоль указанных ионов.
Более того, галенит может флотироваться при погружении в
ненасыщенный водный раствор ксантогената свинца. Вводя ра-
диоактивный свинец в состав ксантогената свинца для того,
чтобы отличить его от свинца, входящего в состав галенита,
установили, что хотя после выдерживания минерала в растворе
ксантогената свинца и происходит его флотация, он не погло-
щает из водного раствора свинец, но поглощает ксантогенат [43].
Поведение указанной системы минерал—раствор согласует-
ся с гипотезой, согласно которой свежеобразованная поверх-
ность галенита покрыта неполным монослоем адсорбированных
ионов; при этом катионами обычно являются ионы свинца, а
анионами—сульфатные, сульфоксидные и карбонатные (а воз-
можно и гидроксильные) ионы. Указанная система вступает в
ионный обмен с водным раствором ксантогената, в результате
которого на поверхности образуется неполный монослой ксанто-
генатных анионов. Можно предположить, что это покрытие
электростатически нейтрализуется более или менее полным мо-
нослоем катионов, главным образом свинца. С этой точки зре-
ния сильно окисленный галенит состоит из участков галенита,
подобных описанным выше, а также из более или менее плотно
прикрепленных корок псевдоморфных окисленных свинцовых
минералов.
Галенит, обработанный водным раствором щелочной соли
ксантогената, промытый и обработанный растворителем ксанто-
гената свинца, дает раствор ксантогената свинца в этом раство-
рителе. С другой стороны, галенит, не обработанный ксантоге-
натом, поглощает ксантогенат свинца из ненасыщенного рас-
твора ксантогената свинца в органическом растворителе. Эти
наблюдения не служат необходимым доказательством сущест-
вования ксантогената свинца в качестве отдельной фазы на
поверхности галенита, обработанной ксантогенатом, однако они
показывают, что на поверхности имеются компоненты ксантоге-
ната свинца. Интересно отметить также, что халькозин сорби-
рует из раствора бензола ксантогенат меди [33] и что галенит
поглощает свинцовую соль жирной кислоты из раствора органи-
ческого растворителя [51]. Явление обратимости поглощения
соли металла из раствора соли в органическом растворителе,
таким образом, получает общее значение.
Согласно имеющимся данным, невозможно вытеснение суль-
фидного иона с поверхности галенита ионом ксантогената. Реак-
МЕХАНИЗМ ДЕЙСТВИЯ СОБИРАТЕЛЕЙ
281
ция (3) протекает справа налево; аналогично можно ожидать,
что реакция (4) будет происходить также справа налево:
PbS + 2X~^PbX2 + S2";
Pb2+ S2~ Pb2+ 2X~
+ S2“
Галеиит
+ 2Х~
Галенит
(3)
(4)
Критические кривые поглощения реагентов галенитом, и их
значение для механизма собирательного действия. На рис. 1 по-
казано изменение краевого угла в зависимости от концентрации
ксантогената. Особенно следует отметить то, что краевой угол
становится неизмеримо малым при концентрациях около
0,5 мг]л. Это критическая концентрация ксантогената. При из-
менении длины углеводородной цепи возможно изменение вели-
чины критической концентрации. Аналогично можно изменить
критическую концентрацию, изменив pH или добавив другие
реагенты. Эта исключительно важная тема рассматривается де-
тально в гл. 10. В данном случае о ней необходимо упомянуть в
связи с тем, что кривые прилипания, подобные изображенным
на рис. 3, использовались для подтверждения молекулярной
теории собирательного действия, недавно предложенной Куком
и Никсоном [8].
На рис. 3 показана зависимость между критической концен-
трацией реагента и pH раствора при действии этилксантогената
калия и диэтилдитиофосфата натрия на галенит. Эти данные
были получены Уорком и Коксом [82] и были рассмотрены Бар-
ским [4]. Как Уорк и Кокс, так и Барский полагали, что эти ре-
зультаты подтверждают конкуренцию между ионами ксантоге-
ната и гидроксила на поверхности галенита. Эта точка зрения
с некоторыми изменениями принимается автором и развивается
в гл. 10.
Кук и Никсон, а также Уордсворт [8, 801 утверждают, что,
гидрофобные пленки, образованные на твердом, получаются при
адсорбции минералом собирателя в форме кислоты, образую-
щейся при гидролизе соли собирателя. Они приписывают пони-
жение собирательного действия, наблюдающееся при увеличе-
нии щелочности, понижению концентрации ксантогеновой кисло-
ты. Согласно изложенному, данные, представленные на рис. 3,
можно объяснить как с точки зрения Кука и Никсона, так и с
позиций Уорка и Кокса, т. е. данные рис. 3 не могут быть ис-
пользованы для подтверждения справедливости ни той, ни дру-
гой гипотезы.
Подвижность ксантогенатной пленки на галените. Хассиалис
и Майерс [44] сделали новое открытие, связанное с механизмом
собирательного действия. Они экспериментально исследовали
282
ГЛАВА 9
способность собирателя, находящегося на (поверхности раздела
минерал — жидкость, к перемещению в двухмерном простран-
стве, представленном поверхностью раздела, и заключили, что
адсорбированные частицы подвижны и перемещаются автома-
тически, стремясь создать равномерное распределение собира-
теля по всей поверхности минерала. Трудно согласиться с вы-
водами Хассиалиса и Майерса, когда на поверхности минерала
Рис. 3. Кривые прилипания галенита в растворах кса:ь
тогената и дитиофосфата как функция pH [81]; точки,
изображенные треугольниками, по [80]:
1 и 2 — диэтилдитиофосфат натрия; 3 и 4 — этилксаитогенат
калия
образуется твердая полимолекулярная пленка собирателя или
даже когда имеется «твердая» мономолекулярная пленка в том
смысле, как это принято понимать в химии поверхностных явле-
ний (см. гл. 3). Однако это легко понять, если перемещающими-
ся частицами являются адсорбированные ионы (или молекулы
ксантогеновой кислоты, согласно предположениям Кука и Ник-
сона) и если температура такова, что эти адсорбированные ио-
ны и их противоионы ведут себя подобно газу или даже вязкой
жидкости. Может оказаться плодотворным дальнейшее расши-
рение исследований по выяснению влияния изменения pH, дли-
ны углеводородной цепи, общей ионной концентрации, концен-
МЕХАНИЗМ ДЕЙСТВИЯ СОБИРАТЕЛЕЙ
283
трации катиона минерала и температуры. Один из основных
вопросов, представляющих интерес,— это вопрос о том, пере-
двигается ли адсорбат, скользя вдоль поверхности раздела или
благодаря непрерывному процессу адсорбции и десорбции.
Действие ксантогенатов на халькозин
Ранние исследования действия ксантогенатов на халькозин
показали, что они оказывают более сильное собирательное дей-
ствие при флотации халькозина, чем галенита [34].
Протекание гетерогенной реакции между ксантогенатом и
халькозином. Дьюи [11, 25] провел тщательные исследования
для выяснения реакции взаимодействия амилксантогената ка-
лия с очищенным халькозином. Минерал измельчался в галеч-
ной мельнице с реагентом и водой в течение различных проме-
жутков времени. Затем его отфильтровывали и подвергали экс-
трагированию такими растворителями, как метанол, ацетон,
эфир и бензол. Экстракты содержали различные вещества,
обычно ярко окрашенные (желтые, красные, коричневые, чер-
ные), и каждый из них содержал более или менее определенные
количества меди, серы и углеводородных групп. Проводилось
исследование экстрагированных веществ, в результате которо-
го было установлено, что они представляют собой целый ряд
промежуточных соединений — от амилксантогената одновалент-
ной меди и до сульфида одновалентной меди.
Характерно, что в экстрактах были обнаружены следующие
соединения:
Cu2 [SC (S) ОСЬНИ]2;
A: Cu2S • Cu2 [SC (S) ОС6НЦ]2;
A': Cu2S • Cu2 [SC (О) OC^].,;
В: 2Cu2S-Cu2 [SC (S)OC6Hn]2;
C: 3Cu2S-Cu2[SC(S)OC6Hn]2.
Эти соединения образуются в следующем порядке: вначале
ксантогенат одновалентной меди, затем соединения А, В и С. Со-
единение А' образуется в течение того же самого времени из-
мельчения, что и соединение А, но лишь при заметной щелоч-
ности и в присутствии кислорода.
При очень длительном измельчении были получены только
следы смолообразного вещества, а после непродолжительного
измельчения обнаружены следы «масла». Ни в одном случае не
извлекалась элементарная сера.
В табл. 3 показаны результаты выщелачивания минерала,
обработанного ксантогенатом, и результаты определения содер-
жания различных ионов в фильтрате.
284
ГЛАВА 9
Таблица 3
Влияние времени измельчения на содержание продуктов, извлекаемых с
поверхности халькозина (50 г) при обработке 10 ммоль (2,02 г)
н-амилксантогената калия [//]
Время измельчения, час.
1 ,25 2,5 5 10 20 30» 40 96»
Продукты, выщелачиваемые из минерала, мг
Ксантогенат ной меди одновалент- 16702 15242 12242 405 200 95 2 0
Соединения А, А' и В 0 171 246 793 303 60 0 0
Соединение С 0 0 177 519 650 402 20 0
«Масло» Следы Следы Следы Следы 0 0 0 0
«Смола» 0 0 0 0 0 0 0 Следы
Сера . . . 0 0 0 0 0 0 0 0
Продукты в фильтрате, ммоль
он- 5,30 3,43 3,48 1,78 0,75 4 4 0,22
СО|- 2,39 3,86 3,95 5,03 4,59 4 4 5,57
SO2- Ионы низших степеней 0,51 0,65 0,93 1,02 1,48 3,27 3,50 1,47
окисления серы5 . . . 0,00 0,18 0,16 0,12 0,02 4 4 0,02
Остаток Х~ Общее количество анио- 1,77 0,30 0,00 0,00 0,00 0,00 0,00 0,00
НОВ 9,97 8,41 8,52 7,95 6,84 4 4 7,25
Катионы калия 9,73 8,49 6,63 11,4е 6,76 7,72 8,89 8,05
pH 11,8 12,0 12,0 И,6 П,4 4 4 10,6
1 Минерал сильно флотировался.
1 Загрязнен небольшими количествами соединения А.
8 Указано приблизительное время измельчения.
4 Не определялось.
8 Во всех анализировавшихся опытах это в значительной степени были SO|—, ио в
некоторых случаях были найдены также
4 Эта величина, вероятно, завышена.
Из этой таблицы видно, что вначале образуется ксантогенат
одновалентной меди. До тех пор пока в водном растворе имеют-
ся ионы ксантогената (первые два опыта), количество ксантоге-
ната одновалентной меди, экстрагируемое с поверхности мине-
рала, велико. Ксантогенат меди, однако, быстро исчезает по ме-
ре образования соединений А, А', В и С. После 40-час. измель-
чения ксантогенат одновалентной меди полностью исчезает.
После 96-час. измельчения исчезли все последующие соединения
(А, А', В и С), хотя минерал сохранил высокую флотируемость,
что свидетельствовало о том, что его поверхность покрыта со-
бирателем, находящимся в форме, не выщелачиваемой ни од-
ним из применявшихся растворителей, а именно: водой, этано-
МЕХАНИЗМ ДЕЙСТВИЯ СОБИРАТЕЛЕЙ
285
лом, метанолом, эфиром, ацетоном, бензолом и петролейным
эфиром. Примечательно, что общее количество анионов пример-
но равно количеству катионов, находящихся в растворе, но, од-
нако, общее количество катионов и анионов меньше количества
первоначально загруженных ионов (10 ммоль)-, расхождение со-
ставляет примерно 2—3 ммоль, и так как это наблюдается во
всех случаях, то весьма важно его отметить. Такое уменьшение
концентрации ионов в растворе можно обусловить тем, что мине-
рал, покрытый собирателем, адсорбирует ионы обоих знаков.
Если вместо амилксантогената калия берется диксантоген,
то с поверхности обработанного им минерала можно извлечь не
диксантоген, а ксантогенат одновалентной меди после коротко-
го времени измельчения и по мере последовательного увеличе-
ния времени измельчения — соединения А, В и С (но не А');
при дальнейшем увеличении времени измельчения не извлекает-
ся ни одно из этих соединений, хотя минерал при этом сохраня-
ет прекрасную флотируемость.
Если измельчение с ксантогенатом производить в эвакуиро-
ванной мельнице или в бескислородной среде, то количество из-
влекаемого ксантогената весьма заметно уменьшается, в то вре-
мя как количество извлеченного диксантогена не понижается.
В табл. 4 представлены соответствующие данные.
Эти результаты согласуются со следующими выводами:
1. Диксантоген реагирует с халькозином с образованием
ксантогената одновалентной меди. При этом происходит реак-
ция окисления — восстановления:
Cu2S +A2->S° + Cu2X2. (5)
Однако в настоящее время эту реакцию следует рассматри-
вать лишь как предполагаемую. Так как халькозин может со-
держать избыток серы в виде твердого раствора, то образую-
щаяся сера может растворяться в твердом халькозине. Способ-
ность меди легко диффундировать внутри халькозина будет так-
же благоприятствовать этому процессу 17, 371.
2. Ион ксантогената взаимодействует с халькозином; при
наличии заметного избытка ионов ксантогената образуется
ксантогенат одновалентной меди. Для протекания этой реакции
необходим кислород, так как в растворе не найдено сульфидных
ионов.
3. При увеличении поверхности халькозина измельчением и
при удлинении времени контакта образовавшийся ксантогенат
одновалентной меди реагирует затем с халькозином с образова-
нием комплексных соединений, содержащих медь, ксантогенат и
сульфидные группы, при этом образуется пленка, не выщелачи-
ваемая пи одним из следующих растворителей: вода, этанол,
метанол, ацетон, эфир, бензол, гексан.
286
ГЛАВА 9
Таблица 4
Взаимодействие искусственного халькозина1 (50 г) с 10 ммоль
н-амилксантогената калия и 10 ммоль н-амилдиксантогена при измельчении
в течение 20 час. на воздухе и в вакууме (по Дьюи [У/])
Опыты с ксантогенатом Опыты с диксантогеиом
Добавленные реагенты, мэкв: кон 10
СаО 50 — —.
H2SO4 40 -
Атмосфера Воз- Ваку- Воз- Воз- Воз- Воз- Ваку-
Остаток собирателя в вод- ной фазе, ммоль .... ДУХ ум Дух ДУХ ДУХ дух ум
Нет 6,7 Нет Нет Нет Нет Нет
Соединения, выщелоченные из минерала, мг\ ксантогенат одновале- нтной меди .... 200 250 750 310 495 300 600
соединения А, А' и В соединение С . . . . 803 660 10 50 70 270 3591 569) 1029 653 1 10 1 200
Соединения в фильтрате, ммоль: ОН~ 0,75 0,27 0 0,30 3 3 3
СО 32- 4,59 1,05 0,08 2,75 3 3 3
SO. 1,48 0,18 0,27 0,40 3 3 3
Ионы-восстановители2 . . . 0,02 0,00 3 3 3 3 3
Общее количество ионов . 6,84 8,20 0,35 3,45 3 3 3
Катион кадия 6,76 8,47 3 4,69 3 3 3
pH 11,4 3 7,3 н,з 3 3 3
1 Согласно современной номенклатуре, скорее «дигенит*, чем лхалькозии».
г За исключением ксантогенатных.
* Не определялись.
Следует указать на то, что эти опыты ставились с 50 г
минерала и 2 г собирателя, т. е. при соотношении минерал со-
биратель, равном ~25 вместо нескольких тысяч (скажем, 10 000),
отвечающих практическим условиям. Наоборот, тонина измель-
чения в опытах была в 10—200 раз больше, чем на практике.
В связи с этим количество вещества, приходящегося на единицу
поверхности в этих опытах, заметно не отличалось от применя-
ющегося на практике, однако концентрация вещества в процес-
се реакции и время протекания реакции были значительно
больше, чем на практике. Поэтому вполне возможно, как это
было показано недавно, что перечисленные выше интересные
явления в известном смысле являются побочными реакциями.
Протекание реакции между ксантогенатом и халькозином в
МЕХАНИЗМ ДЕЙСТВИЯ СОБИРАТЕЛЕЙ
287
нормальных условиях. Шуман [33, 63], затем Дьюи проводили
исследования в условиях, приближающихся к практическим.
В этом случае минерал измельчали при отсутствии ксантогена-
та в течение непродолжительного времени, затем его перемеши-
вали с водой при доступе воздуха также в течение непродолжи-
тельного времени. Отношение минерала к ксантогенату в опы-
тах возрастало, достигая нескольких тысяч, что соответствует
практическим условиям; при этом концентрация реагентов и
время реакции, хотя и оставались высокими, но были ближе к
принятым на практике, чем в ранее приведенных исследовани-
ях. В этих опытах количество ксантогената возрастало от 0,25
до 12,9 мэкв на 200 г минерала. В экстрактах при этом были
обнаружены только амилксантогенат одновалентной меди и со-
единение А', установленное в опытах Дьюи. Очевидно они об-
разуются при недостатке кислорода. В опытах Шумана количе-
ство соединений А' было также очень мало. Важное значение
имело установленное в опытах наличие невыщелачиваемой с
поверхности части собирателя при небольших загрузках ксан-
тогената калия. Так, хотя, при введении 0,25 мэкв ксантогената
на 200 г минерала происходит поглощение всего ксантогената,
только 97% от этого количества экстрагируется растворителем.
При концентрации 1,1 мэкв примерно половина определяется в
виде ксантогената одновалентной меди, а при концентрации
4,21 мэкв — две трети. Очевидно, вначале образуется «невыще-
лачиваемая» ксантогенатная пленка, которая имеет первосте-
пенное значение в условиях недостатка собирателя. Наимень-
шее количество ксантогената, применявшееся в опытах
(0,25 мэкв), отвечало примерно расходу 0,23 кг/т халькозина —
величине, приближающейся к расходу, применяющемуся на
практике (если расход реагента рассчитать только на флотиру-
емый минерал). Отсюда мы можем заключить, что в практиче-
ских условиях загружаемый ксантогенат полностью поглощает-
ся минералом и не способен растворяться в растворителях
ксантогената одновалентной меди.
На рис. 4 графически показано распределение загружаемого
ксантогената. Количество невыщелачиваемой части ксантогена-
та возрастает с увеличением его загрузок, но значительно мед-
леннее, достигая максимума, отвечающего примерно 1,7 мэкв
на 200 г минерала. В опыте, проведенном с минералом, измель-
ченным в аналогичных условиях, но обесшламленным, было
установлено наличие 0,145 мэкв невыщелачиваемой пленки на
150 г халькозина, соответствующей образованию монослоя
ксантогената.
На основании опытов Шумана может быть высказана следу-
ющая гипотеза относительно механизма собирательного дейст-
вия ксантогената на халькозин:
288
ГЛАВА 9
1. Первоначально адсорбированные анионы на поверхности
халькозина обмениваются с ионами ксантогената водного рас-
твора (в согласии с работой Таггарта, Тейлора и Нолла на га-
лените). При этом на минерале образуется «невыщелачивае-
мое» соединение
_____________________х-
Cu2S +Х~-> Cu2S +Х~. (6)
Мимиэквиваленты введенноео
ксантогената
Рис. 4. Амилксантогенат и халько-
зин. Распределение ксантогената, до-
бавляемого к халькозину, между
ксантогенатом щелочного металла,
остающимся в жидкости, ксантоге-
натом одновалентной меди на халь-
козине и невыщелачиваемой частью
на халькозине (условия опытов:
200 г халькозина измельчали в те-
чение часа и обрабатывали 15 мии.
ксантогенатом) [33, 63]:
1 — избыток ксантогената в фильтре;
2 — ксантогенат одновалентной меди; 3 —
невыщелачиваемое вещество
2
Халькозин окисляется кислородом по следующей реакции:
xCu2S-z/CuS -)-0.5О2 + НгО->
-> (х — 2) Cu2S (у + 2) CuS + 2Cu+ + 2OFT. (7)
В этой реакции халькозин изображен в виде твердого рас-
твора CuS в CU2S, так как хорошо известно, что халькозин со-
держит переменное избыточное количество серы сверх коли-
чества, отвечающего формуле Cu2S [60]. Легко понять, что ис-
ключительная подвижность меди в твердом халькозине может
обусловливать смещение реакции в сторону образования CuS.
3. Образующийся ион одновалентной меди реагирует с ксан-
тогенатом с осаждением ксантогената одновалентной меди,
способного выщелачиваться с поверхности,
2Cu+ + 2Х“->Си2Х2. (8)
При взаимодействии иона одновалентной меди с ксантогена-
том с образованием труднорастворимого ксантогената реакция
сдвигается вправо, так же как и при окислении дисульфида ме-
МЕХАНИЗМ ДЕЙСТВИЯ СОБИРАТЕЛЕЙ
289
ди (см. выше). Протекание этих реакций можно рассматривать
как механизм, катализирующий связывание кислорода сульфи-
дом одновалентной меди. Уравнениями (7) и (8) изображается
образование ионов одновалентной меди вне кристаллической
решетки халькозина. Соответствующие уравнения можно напи-
сать и для иона двухвалентной меди. Их необходимо дополнить
реакцией (9) для того, чтобы связать образующийся диксанто-
ген. Таким образом, собирательное действие ксантогената на
халькозин можно предста!вить рядом следующих уравнений:
А- х-
Cu2S + Х~ -> Cu2S + А~; (6)
xCu2S-yCuS + 0,5О2 + Н2О->
-> (х — l)Cu2S(t/ + l)CuS + Cu2+ + 2ОН“; (7а)
2Cu2+ + 4Х“ Cu2 Х2 + Х2; (8а)
Cu2S + Х2~>CuS + Cu2X2. (9)
Наиболее важной является реакция (6).
Кинетика реакции ксантогената с халькозином. Кинетическая
теория может быть построена на основании приведенных выше
уравнений [63]. Изменение количества ионов ксантогената в вод-
ном растворе А равно сумме изменений количества адсорбиро-
ванного (невыщелачиваемого) ксантогената В и количества
ксантогената одновалентной меди С:
dA dEi . dC
dt dt dt
Изменение количества адсорбированного ксантогената про-
порционально величине поверхности, не покрытой ксантогена-
том, и концентрации ионов ксантогената в растворе (согласно
адсорбционным представлениям Лэнгмюра):
— = /гД1 —x)SA, (11)
dt
где S —поверхность (выраженная в миллиэквивалентах эле-
ментарных площадок);
&i— константа;
А — количество ионов ксантогената в растворе, мэкв;
х — часть элементарных площадок, на которых адсорбиро-
вался ксантогенат ко времени t.
Для определения изменения ксантогената меди во времени,
необходимо предположить, что концентрация Си+ постоянна.
19 А. М. Годэн
290
ГЛАВА 9
Это вполне допустимо, если принять уравнение (7). Изменение
содержания ксантогената меди при этом будет пропорциональ-
но поверхности, покрытой ксантогенатом, xS, и концентрации
иона ксантогената, или
~ = k2xSA. (12)
dt
Подставляя уравнения (11) и (12) в уравнение (10), получаем:
^£ = ^(1— x)SA -\-k2xSA. (13)
dt
Так как изменение адсорбции ксантогената равно изменению
степени покрытия поверхности по определению, то
(14)
dt dt V '
Комбинируя уравнения (14) и (11), имеем
^ = k^\-x)A. . (15)
Деля уравнение (13) на уравнение (15), имеем
=_<?/1 + /<-^Ц, (16)
dx \ 1 — х /
г» &2
где К = — •
«1
Интегрируя, получим
А = (-х + Кх-К)+К1п(1-х). (17)
О
Если через Ло обозначить количество ионов ксантогената,
адсорбированных минералом в конце реакции (Ло = А—At ),
когда часть поверхности, покрытая ксантогенатом, будет рав-
няться xt, то уравнение (17) примет следующий вид:
4- = хД1-К)-К1п(1-хД (18)
О
Для того чтобы проверить справедливость приведенных вы-
ше кинетических представлений, необходимо принять за исход-
ную величину экспериментальную точку, полученную из опытов
адсорбции [33, табл. 1]. Кроме того, необходимо выбрать значе-
ние эффективной минеральной поверхности в условиях опыта.
В качестве такой предельной величины из экспериментальных
данных значение В принято равным 1,75 мэкв [33, табл. 1, стол-
бец 5]. Отсюда числовое выражение для уравнения (18) будет
Ло=1,25 Во-9,1 lg(l--M, (18а)
\ 1,75 /
где Ло— количество мэкв ксантогената, поглощенного 200 г
халькозина;
МЕХАНИЗМ ДЕЙСТВИЯ СОБИРАТЕЛЕЙ
291
Во—! количество мэкв ксантогената, адсорбированного (не-
выщелачиваемого) 200 г минерала.
Экспериментальные данные Шумана и данные, вычисленные
на основании уравнения (18а), изображены на рис. 5. Близкое
совпадение между экспериментальными и расчетными данными
подтверждает справедливость высказанной гипотезы.
йо,мзкв поглощенного ксантогена-
та на 200 а халькозина
Рис. 5. Соотношение между общим поглоще-
нием ксантогената Ло и максимальной адсорб-
цией ксантогената при образовании монослоя
Во [33, 63]:
•—опытные данные; -----------теоретическая кривая
Возражением против предложенной гипотезы может служить
то, что количество ксантогената, адсорбированного в виде не
выщелачиваемых с поверхности соединений, не может быть не-
посредственно экспериментально определено и рассчитывается
по разности (Д—С). Это возражение вполне обоснованно и под-
тверждает необходимость проведения дополнительной работы.
Интересно отметить, что халькозин, обработанный ксантогена-
том, который затем выщелачивается при помощи метанола или
этанола, эфира, ацетона, бензола и гексана, так, что с поверхно-
сти минерала уже ничего не извлекается при помощи этих рас-
творителей, тем не менее способен отдавать невыщелачиваемый
ксантогенат при обработке таким комплексообразующим рас-
творителем, как пиридин (см. гл. 8). Однако количественных
определений с этим растворителем еще не проводилось. Заслу-
живает внимания также то, что не обработанный ксантогенатом
халькозин поглощает ксантогенат меди из раствора бензола.
Можно предположить, что поглощенный реагент образует невы-
19*
:292
ГЛАВА 9
щелачиваемый ориентированный слой, так как благодаря этому
минерал становится несмачиваемым.
Этот результат согласуется с наблюдениями Нолла и Ваке-
ра о поглощении галенитом свинцовых мыл из органических
.растворителей [51].
Действие ксантогенатов на пирит
Ранее [27] было показано, что чистый пирит может быть до-
статочно полно сфлотирован при добавлении различных ксанто-
генатов в количествах, недостаточных для образования полного
монослоя, если даже при этом весь реагент полностью погло-
щается поверхностью минерала. Однако на флотируемость пи-
рита сильно влияет наличие на поверхности минерала участков
окисленных соединений, благодаря чему флотируемость частич-
но окисленных зерен сильно понижается [58].
Первоначальные исследования, проводившиеся для выясне-
ния действия ксантогенатов щелочного металла на пирит, пока-
зали, что углеводородный радикал ксантогената поглощается
минералом, который благодаря этому становится флотационно
активным. Кроме того, они показали, что ксантогенат железа не
может быть извлечен бензолом с поверхности минерала хотя бен-
зол и является превосходным растворителем для ксантогената
окиси железа; с другой стороны, были экстрагированы элемен-
тарная сера и масло неопределенного состава [38].
При измельчении пирита в эвакуированной фарфоровой
мельнице с ксантогенатом калия, водой и бензолом [25, 75] бен-
зольная фаза содержала некоторое количество ксантогената же-
леза. Однако количество ксантогената, пересчитанного на ксан-
тогенат окиси железа, было слишком мало по сравнению с об-
щим количеством поглощенного минералом ксантогената.
Опыты с применением радиоактивных изотопов к системе
ксантогенат-—пирит. Адсорбция этилового ксантогената на пи-
рите дополнительно исследовалась с помощью радиоактивного
ксантогената [30, 55]. Поглощение ксантогената определялось
при помощи метода колонки. Этот метод состоит в следующем:
изготовляют короткий столбик из 5 г минерала и пропускают
через этот минерал большой объем исследуемого раствора до
тех пор, пока поступающий и вытекающий растворы не будут
иметь одинаковый состав. Произведенные затем анализы по-
рошков минерала дают возможность определить адсорбцию
ксантогената, соответствующую исследуемому раствору. В ис-
следованиях Меллгрена применялся ксантогенат, содержащий
S35, а для анализа использовался метод Карра [22].
Было установлено, что необходимо удалять растворенный
кислород из раствора ксантогената, так как в противном случае
МЕХАНИЗМ ДЕЙСТВИЯ СОБИРАТЕЛЕЙ
293
радиоактивность минерального порошка в столбике может не-
определенно возрастать, особенно в верхнем слое, через кото-
рый исходный раствор ксантогената проходит вначале. Это объ-
яснялось образованием капелек диксантогена, способных при-
липать к минералу, но подобное объяснение не является един-
ственно правильным. Когда кислород был удален из исходного
раствора, поглощение ксантогената не превышало монослоя. На
рис. 6 даны кривые адсорбции ксантогената при различных кон-
центрациях, полученные Меллгреном. «Пирит 1» представлял
собой узкую фракцию (—200 + 400 меш) тщательно очищенного
минерала, предохраняемого, насколько' это возможно, от кон-
такта с кислородом. «Пирит 2» и «пирит 3» представляли собой
несколько более тонкие фракции минерала, более тонкой тексту-
ры, также специально очищенного,, и сохранявшегося влажным
под водой, содержавшей сероводород. На рис. 6 видно, что ад-
сорбция ксантогената увеличивается с ростом концентрации,
достигая в случае пирита предельной величины около
6.5- 1СН10 лголб/с.и2. На рисунке видим также, что адсорбция
значительно меньше на пирите, сохранявшемся в присутствии
сероводорода. Экспериментальные данные показывают, что
значение Г изменяется пропорционально С в степени 0,2—0,25.
Максимальное поглощение почти приближается к величине, от-
вечающей монослою.
Меллгрен изучал обратимость адсорбции ксантогената на
пирите при пропускании воды или раствора «немеченого» ксан-
тогената (той же концентрации, что и раствор, применявшийся
при первоначальной обработке минерала ксантогенатом) через
минерал, первоначально покрытый «меченым» ксантогенатом^
Полученные результаты оказались неубедительными. В некото-
рых опытах адсорбция была полностью обратимой, а в других
нет. Очевидно, необходимо продолжать исследования в этом
направлении.
Исследовалось также действие pH. Максимальная адсорб+
ция получалась вблизи нейтральной точки (pH =5—8); адсорб-
ция очень резко падает при pH < 5, достигая нуля при pH < 4.
Это, вероятно, обусловлено разложением ксантогеновой кисло-
ты. При более высокой щелочности адсорбция также понижает-
ся пропорционально увеличению щелочности. На рис. 7 показа-
ны критические значения отношения [X-] : ЮН-].
Действие ксантогенатов и других тиосоединений
на различные минералы
Действие ксантогенатов на сфалерит. Первое время имелись
значительные расхождения во мнениях при рассмотрении соби-
рательного1 действия ксантогенатов на сфалерит; некоторые экс-
Рис. 6. Этилксантогенат и пирит. Плотность адсорбционного слоя
этилксантогената на пирите как функция концентрации этилксанто-
гената калия [30, 55]:
/ — пирит 1: 2 —пирит 2 и 3
Рис. 7. Плотность адсорбционного слоя ксантогената на
пирите как функция отношения концентраций ионов ксан-
тогената к ионам гидроксила [30, 55]:
/ — концентрация ксантогената 6,24' 10-* • моль/л; 2 — 6,24* •
• 10-5 моль!л; 3 — 6.241 10-“ моль/л
МЕХАНИЗМ ДЕЙСТВИЯ СОБИРАТЕЛЕЙ
295
перименты свидетельствовали о сильном собирательном дейст-
вии, другие же — о слабом. В конечном счете это нашло объяс-
нение в непреднамеренной неконтролируемой активации мине-
рала в растворе при помощи солей металлов, в особенности со-
лей меди [18, 20, 59]. Когда эта активация совершенно исключе-
на, низкомолекулярные ксантогенаты не являются собирателя-
ми для сфалерита, тогда как гомологи с более длинным углево-
дородным радикалом показали заметное собирательное дейст-
вие. Это видно на рис. 8. Следует заметить, что ксантогенат ад-
сорбируется в некоторой степени и на неактивированном сфале-
рите, однакс эта адсорбция по величине значительно меньше,
чем на галените, пирите или халькозине; поглощение реагента
минералом возрастает с увеличением длины углеводородной це-
пи. Измерений распределения ксантогената между сфалеритом
и водным раствором не производилось, однако ориентировоч-
ные опыты (Д. Л. Харриса в Массачусеттском технологическом
институте в 1952 г.) со сфалеритом и раствором этилксантогена-
та калия (0,9-10~3%) показали, что около 95% реагента оста-
ется в водном растворе, а поверхностное покрытие на минерале
эквивалентно 2—3% полного монослоя. Кроме того, поверхност-
ное покрытие легко удалялось при промывании водой. Повыше-
ние pH увеличивает концентрацию ксантогената, необходимую
для прилипания пузырька воздуха на неактивированном сфале-
рите. Это показано, например, на рис. 9, не только для амилово-
го ксантогената, но также для трех гомологов дитиокарбаматов:
диэтилового, ди-н-бутилового и ди-н-амилового.
Действие меркаптанов на сфалерит. Меркаптаны, имеющие
углеводородный радикал средней длины, и тиофенолы являют-
ся прекрасными собирателями для неактивированного сфалери-
та. В исследованиях Харриса [28, 42] для выяснения распреде-
ления реагента между минералом и раствором в качестве соби-
рателя применяли н-гексилмеркаптан. Меркаптан был «мечен»
S35. Для его анализа был использован метод Карра. Количест-
во реагента, поглощенного минералом, обычно было значитель-
но меньше, чем это необходимо для образования монослоя
(рис. 10); однако, когда применяется раствор, концентрация ко-
торого составляет несколько сотых процента, толщина слоя со-
бирателя может достигать двух молекул. Это поверхностное по-
крытие собирателя не удаляется при промывании водой. Так
как было найдено, что поглощение S35 (первоначально введен-
ной в виде гексилмеркаптана) происходит необратимо, то мож.
но допустить, что реагент мог измениться во время процесса
поглощения или после него под действием какого-нибудь при-
сутствующего совместно с ним агента. Возможно, кислород игра-
ет при этом ту же роль, что при флотации халькозина и пирита.
•Это должно явиться предметом дальнейших исследований.
Рис. 8. Флотация иеактивироваииого сфалерита
(—100 + 600 меш) различными ксантогенатами:
1—этилксантогеиат калия, pH == 6,5 ± 0,1; 2 — и-амилксанто-
геиат калия, pH = 6,5 ± 0,1; 3 — и-гептилксаитогенат калия,
pH = 6,5 ± 0.1
Рис. 9. Зависимость между концентрациями различ-
ных собирателей и критическими значениями pH
для сфалерита [82]:
1 — амилксаитогеиат; 2 — диэтилдитиокарбамат; 3—ди-и-бу-
тилдитиокарбамат; 4 — ди-и-амилдитиокарбамат
МЕХАНИЗМ ДЕЙСТВИЯ СОБИРАТЕЛЕЙ
297
Действие ксантогенатов и меркаптанов на окисленные мине-
ралы тяжелых металлов. В большинстве случаев окисленные
минералы тяжелых металлов образовались в процессе измене-
ния сульфидных минералов. Образование их происходило в раз-
личных условиях, причем одновременно с этим растворялись
или замещались многие сопутствующие минералы пустой поро-
ды. Поэтому окисленные руды минералогически более сложны,
Рис. 10. Меркаптан н сфалерит. Адсорбция гекснлмер-
каптана сфалеритом при различных его концентрациях
в растворе {28, 42]:
М — монослой из расчета 20,5А2 иа молекулу меркаптана;
О — время адсорбции 1—2 часа; □ — время адсорбции 20 час,
чем первичные, имеют более переменный состав и тонкую струк-
туру. Действительно, очень трудно получить для исследователь-
ских целей даже небольшие количества относительно чистых,
хорошо кристаллизованных окисленных минералов.
Действие ксантогенатов на окисленные минералы свинца.
Имеются лишь отрывочные сведения о флотируемости большин-
ства окисленных минералов свинца, цинка, меди и железа и то
только для незначительного числа образцов. Церуссит (карбо-
нат свинца), англезит (сульфат свинца), вульфенит (молибдат
свинца), крокоит (хромат свинца) и другие окисленные минера-
лы свинца флотировались серусодержащими собирателями, од-
нако имеющиеся данные большей частью неудовлетворительны.
Обычно количество реагента, необходимое для их флотации,
значительно больше, чем для сульфидных минералов I20J
(рис. 11). Так, если для флотации галенита идет от 9 до 23 а со-
бирателя на 1 т сульфидной руды, то для окисленных руд тре-
буется в двадцать (и более) раз большее количество собирате-
ля. Кроме того, поглощение собирателя происходит не настоль-
298
ГЛАВА 9
ко полно и не так быстро и требуются собиратели с более длин-
ной углеводородной цепью.
Эти различия частично могут быть отнесены за счет большей
удельной поверхности микрокристаллических окисленных мине-
ралов, однако они в большей степени обусловлены качественны-
ми различиями минералов. Ионы сульфидных минералов, всту-
пающие в обмен с ионами собирателя, не входят в состав решет-
ки минерала, как это наблюдается для англезита и церуссита.
Поэтому окисленные минералы сильнее поглощают собиратель,
п-тиокрезол, кг/т п-тиокрезол, кг]т
Рис. 11. Разделение церуссита и кальцита. Разделялась смесь (1 :4)
частиц крупностью —100 +600 меш с применением п-тиокрезола и
0,09 кг/т терпинеола. Перемешивание с реагентами 2 мин.; флотация
6 мин.; pH ss 8—8,2; температура 18—21° [20|
чем сульфидные. Размер адсорбционных ячеек на окисленных
минералах отличается от размера ячеек на сульфидных (обычно
они больше вследствие большого размера окисленных ионов),
так что если даже получается покрытие одинаковой плотно-
сти (при расчете на доступный размер адсорбционной ячейки),
то в действительности на окисленных минералах образуется ме-
нее плотное покрытие с точки зрения плотной упаковки углево-
дородных цепей. Наконец, более рыхлая структура поверхност-
ных слоев реагентов на окисленных минералах может допускать
более легкую перекристаллизацию их с образованием мозаич-
ных покрытий из толстых пленок, перемежающихся с участка-
ми, свободными от собирателя.
Измерения поглощения н-амилксантогената церусситом бы-
ли произведены Р. А. Джонсоном [47]. Его данные представлены
в табл. 5.
Исходя из этих опытов, подсчитали, что для образования мо-
нослоя ионов амилксантогената необходимо около 0,04 мг ксан-
тогената калия [25] и что слой ксантогената свинца, образовав-
шийся после 10 мин. перемешивания, отвечает поверхностному
покрытию толщиной в несколько сот ионов. Из таблицы видно,
МЕХАНИЗМ ДЕЙСТВИЯ СОБИРАТЕЛЕЙ
299
Таблица 5
Поглощение церусситом (1г) н-амилксантогената калия
из водного раствора (50 мл) в течение 10 мин. при 20° [47]
№ опыта Количество введенного н-амилксанто- гената мг Количество ксантогената калия, оставше- гося в растворе мг Количество ксаитогеиа- та свинца на церус- сите, выраженное в эквивалентном коли- честве ксантогената калия, мг Расхождение мг
1 2,69 0,17 2 59 +0,07
2 7,01 0,17 6,93 +0,09
3 8,08 0,31 7,78 +0,01
4 10,66 2,13 8,50 —0,03
5 11,13 2,40 8,74 +0,01
6 17,06 6,78 10,26 —0,02
7 21,30 11,10 10,20 0,00
8 27,70 17,00 10,55 —0,15
9 53,25 Не опреде- лялось 10,98 Среднее 0,05
что количество ксантогената, поглощенного из раствора, при-
мерно соответствует количеству ксантогената свинца, извлечен-
ного с твердого при применении соответствующего растворите-
ля, например такого, как бензол.
Если ион ксантогената из раствора закрепляется в форме
ксантогената свинца, то карбонатный ион должен перейти в
раствор, так что реакция может быть представлена обменом
РЬСО3 + 2Х“-^РЬХг + СОГ. (19)
Новая фаза отличается по кристаллохимическим свойствам
от старой твердой фазы; поэтому связь между подкладкой и по-
верхностным покрытием должна быть слабой.
Если рассматривать последовательный ряд ксантогенатов в
порядке увеличения длины углеводородной цепи, то можно ожи-
дать, что равновесие в уравнении (19) будет сдвигаться по мере
увеличения длины цепи в сторону образования ксантогената
свинца. Это предположение основывается на понижении раство-
римости ксантогенатов свинца в воде с увеличением длины угле-
водородной цепи (см. гл. 8). Можно ожидать, что протеканию
реакции будет способствовать поглощение карбонатного иона.
Однако нет уверенности, что фаза ксантогената свинца облада-
ет флотационной активностью.
В самом деле, уравнение (19) можно представить себе как
нежелательную побочную реакцию, которой следует избегать
с точки зрения экономии реагентов, и возможно, что действи-
тельно эффективным является агент, лишь адсорбируемый на
поверхности карбоната свинца.
300
ГЛАВА Ь
Подобное рассуждение чисто умозрительно, так как никаких
измерений адсорбции ионов ксантогената поверхностью окис-
ленных минералов свинца в условиях насыщения пульпы по от-
ношению к ксантогенату свинца не делалось; образование новой
фазы будет происходить до тех пор, пока произведение концент-
раций ионов ксантогената и ионов свинца превышает произве-
дение растворимости ксантогената свинца; старая фаза (карбо-
нат свинца) будет растворяться до тех пор, пока концентрация
ионов карбоната и свинца не достигнет произведения раствори-
мости карбоната свинца.
Параллельно растворению церуссита и образованию ксантоге-
ната свинца происходит также адсорбция ионов ксантогената
(находящихся в растворе в преобладающем количестве) на це-
руссите и адсорбция карбонатных ионов на ксантогенате свинца.
Исходя из атомных размеров, для каждой поверхности раз-,
дела твердое — жидкость в каждый данный момент можно себе
представить образование участков из ионов свинца, карбонат-
ных и ксантогенатных ионов. При равновесии, т. е. когда одно-,
временно присутствуют две твердые фазы в неизменяющихся
количествах, каждая фаза имеет собственную пленку из адсор-
бированных ионов в количестве, отвечающем равновесию. По-
добное адсорбционное равновесие пока еще количественно не
определено. Неизвестно, будет ли адсорбционное равновесие
ксантогената на церуссите таким же, как и на других окислен-
ных минералах свинца; точно так же неизвестно, будет ли при
этом образовываться одинаковый краевой угол. Эксперимен-
тальные исследования в этом направлении могут оказаться весь-
ма полезными.
Практически флотация окисленных минералов происходит
при быстром поглощении ксантогената, что требует большого
избытка концентрации ксантогената по сравнению с равновес-
ной концентрацией в опытах по измерению краевых углов.
Можно ожидать, что краевой угол является функцией време-
ни (при условии, что поверхность минерала достаточна для по-
глощения реагента, как это происходит на практике), возможно^
что эта зависимость сильнее проявляется для окисленных мине-
ралов, чем для сульфидных.
Действие ксантогенатов на окисленные медные минералы.
Как ясно видно на рис. 12, основные карбонаты меди — малахит
и азурит — активно взаимодействуют с растворами щелочного
ксантогената; образующаяся при этом пленка не удерживается
на поверхности и легко отслаивается. Так, при измельчении ма-
лахита в галечной мельнице в течение длительного времени в
присутствии избытка амилксантогената калия минерал лишает-
ся поверхностной пленки, которая скатывается в виде шариков,
налипающих на гальку подобно замазке. Этому способствует
МЕХАНИЗМ ДЕЙСТВИЯ СОБИРАТЕЛЕЙ
301
образование диксантогена совместно с ксантогенатом меди.
Другим фактором, способствующим этому, является несоизме-
римость атомных размеров малахита и ксантогената меди или
малахита и диксантогена. До сих пор при флотации малахита
в качестве собирателя применяется н-амилксантогенат калия,
123
Рис. 12. Пленка, получаемая на азурите при взаимодей-
ствии с амилксантогенатом калия. X 40. Применялся
2%-ный раствор амилксантогената; время реакции 2 мин.
как это видно на рис. 13; с точки зрения теоретических представ-
лений, высказанных ранее, желательно ограничивать время и
Интенсивность перемешивания с собирателем.
При проведении исследований с различными синтетическими
соединениями меди кристаллического характера (таким, как
различные галогениды меди, окись меди, закись меди, фосфаты
меди и др.) могут быть получены необходимые данные, позво-
ляющие установить зависимость между флотируемостью, крае-
вым углом, адсорбцией собирателя и кристаллической структу-
рой.
Действие ксантогенатов на окисленные минералы железа.
Как известно, ни один из ксантогенатов или других сульфгид-
рильных соединений не является собирателем для какого-либо
окисленного железного минерала. К числу не флотирующихся
302
ГЛАВА 9
этими собирателями железных минералов относятся гематит,
магнетит, гетит и сидерит. В сидерите железо двухвалентное,
однако возможно, что оно окисляется на поверхности, так как
Рис. 13. Разделение смеси малахит — кальцит. Флотация различны-
ми ксаитогенатами калия (смесь 1 :4, крупность —100 + 600 меш):
1 — этилксаятогенат калия при 16°; 2 — н-пропилксантогенат калия при 16°;
3—н-бутилксантогенат калия при 18°; 4 — н-амнлксантогенат калия при 18°;
5 — н-гексилксантогенат калия при 18°; 6 — н-гептилксантогенат калия при 20°
этот минерал даже в разбавленных щелочных растворах погло-
щает кислород из воздуха. Отсутствуют какие-либо доказатель-
ства поглощения иона ксантогената этими минералами из его
растворов, но, судя по их поведению при флотации, можно ожи-
дать, что эти минералы не способны прочно связывать ион
ксантогената. В то же время пирит, марказит и пирротин по-
глощают ион ксантогената из раствора и флотируются при его
помощи. Противоречивое поведение сульфидных железных ми-
нералов и окисленных железных минералов по отношению к
сульфгидрильным соединениям непонятно и заслуживает даль-
нейшего изучения.
! МЕХАНИЗМ ДЕЙСТВИЯ СОБИРАТЕЛЕЙ 303
Действие меркаптанов на окисленные минералы цинка.
О флотации окисленных минералов цинка известно еще меньше,
чем о флотации окисленных минералов свинца и меди. Было
показано, что меркаптаны флотируют окисленные минералы
цинка [15]. Измерения, недавно проведенные Харрисом [28, 42],
показали, что гексилмеркаптан поглощается синтетической
окисью цинка и минералами цинкитом, смитсонитом и виллеми-
том.
Действие дитиофосфатов на сульфидные минералы. Несмот-
ря на то что дитиофосфаты широко применяются в качестве со-
бирателей сульфидных минералов, относительно мало известно
о деталях механизма их собирательного действия. Обычно пред-
полагается, что механизм их действия аналогичен действию
ксантогенатов.
Исследования с применением радиоактивных индикаторов
были проведены Симардом и его сотрудниками в исследователь-
ской лаборатории Америкен Цианамид Компани [48, 66, 67].
Проведенные опыты имели две особенности. Одной из них явля-
ется то, что сорбция измерялась на ограниченной площади еди-
ничного красталла (примерно 1 см2); второй особенностью яв-
ляется кинетический подход, благодаря которому было уста-
новлено, что сорбция зависит от времени воздействия собирате-
ля на минерал.
Симард и его сотрудники нашли, что галенит сорбирует из
водного раствора ион дитиофосфата, «меченый» фосфором; это
поглощение уменьшается, если галенит предварительно выще-
лочить для удаления окисленных пленок. Скорость поглощения
собирателя вначале очень велика; затем она замедляется, при-
чем «медленное» поглощение требует присутствия кислорода.
Галенит, покрытый дитиофосфатом, обменивает сорбированные
ионы собирателя с ионами дитиофосфата равновесного раство-
ра, однако скорость обмена исключительно мала.
Действие жирных кислот и мыл
• Действие жирных кислот и мыл на сульфидные минералы.
Несмотря на то что эти реагенты одними из первых были ис-
пользованы при флотации руд, почти нет никаких количествен-
ных данных о механизме их действия. Насыщенные жирные кис-
лоты являются собирателями для галенита [271. Количество ре-
агента, необходимого для флотации, уменьшается с увеличени-
ем длины углеводородной цепи, причем это уменьшение стано-
вится менее заметным по мере увеличения длины цепи. Для
жирных кислот, содержащих больше тринадцати атомов угле-
рода, не наблюдается заметного уменьшения количества реаген-
та, необходимого для флотации, и для того чтобы обеспечить
нужное собирательное действие, необходимо нагревание. Пред-
ав
304
ГЛАВА 9
полагается, что карбоксилсодержащий ион ведет себя по отно-
шению к поверхности галенита таким же образом, как и ксанто-
генатный ион, однако это не подтверждено экспериментально.
Обычно предполагается, что аналогичное поведение наблюдает-
ся и при флотации других сульфидов различными жирными кис-
лотами и мылами, однако это следует рассматривать, не более
как приемлемое предположение.
Действие жирных кислот и мыл на окисленные минералы.
Карбоксилсодержащие ионы поглощаются несульфидными ми-
нералами. Наиболее раннее исследование этого явления припи-
сывается Люйкену и Бирбрауэру [53, 54]. Поглощение собира-
теля вычислялось ими по разности между исходной и остаточ-
ной концентрацией в растворе, путем измерения поверхностного
натяжения этих растворов по методу, применявшемуся в более
ранних исследованиях [13, 73]. Этот метод требует осторожности
при работе с такими гетерополярными соединениями, как мыла,
поверхностная активность которых заметно изменяется с изме-
нением pH. Так как при обработке минерала раствором взаимо-
действующего с ним реагента возможно изменение pH, то необ-
ходима компенсационная регулировка pH перед определением
поверхностного натяжения.
Таггарт, Дель-Джудиче и Цил сообщали о повторении ими
в несколько меньшем объеме опытов Люйкена и Бирбрауэра, а
также о распространении этих опытов на исследование погло-
щения олеиновой кислоты галенитом.
Были произведены измерения краевых углов на карбонате
железа в растворах гептиловой кислоты [36]. Согласно этим из-
мерениям, как видно на рис. 14, максимальный краевой угол
получается равным 62°; с увеличением pH величина краевого
угла падает, причем наблюдается постепенный переход от обла-
сти контакта до области его отсутствия.
Для изучения кинетики адсорбции лаурата баритом и искус-
ственно полученными кристаллами сульфата бария Турнесак
[35, 78] применил лаурат натрия, в котором радикал лаурата со-
держал радиоактивный изотоп С14. В этой работе наблюдалось
два интересных явления. Одним из них является то, что адсорб-
ция протекает удивительно медленно и, чтобы приблизиться к
равновесию, требуется не менее 24 час. Результаты могут быть
выражены эмпирическим экспоненциальным уравнением ско-
рости
где Г — адсорбция за время t\
а и а — константы;
е — основание натуральных логарифмов.
Рис. 14. Изотоническая карта для сидерита при приме-
нении гептиловой кислоты и буферных водных раство-
ров [36]
Отношение определенного на опыте произведе-
ния ионов к произведению растворимости лау-
рата бария
Рис. 15. Адсорбция лаурата сульфатом бария в за-
висимости от насыщения раство>ра лауратом бария
при pH =10,85 ±0,1 [35, 78]:
1 — Na2SOt = 7 • 10—5 мг/л; 2 — Na2SO4 =7- 10-5 мг!л
20 А. М. Годэн
306
ГЛАВА 9
Эти результаты трудно интерпретировать.
Второе явление состоит в том, что наблюдается заметная ад-
сорбция даже в том случае, когда произведение концентраций
ионов бария и лаурата, находящихся в растворе, значительно
меньше произведения растворимости лаурата бария. Результа-
ты опытов с искусственным сульфатом бария в среде, содержа-
щей сульфат натрия, показаны на рис. 15. Оказывается, что ад-
сорбция значительна и хорошо воспроизводима в условиях,
когда произведение концентраций ионов, находящихся в раство-
ре, в 100 или даже 1000 раз меньше произведения растворимо-
сти. Как следует из рис. 15, предельная адсорбция составляет
около 3,4 гиббса (3,4-1О-10 моль/см2), что отвечает примерно
двум третям монослоя. Монослой вычисляли, исходя из величи-
ны удельной поверхности, определенной по измерению адсорб-.
ции криптона методом БЭТ, параметров кристаллической ре-
шетки барита (а = 8,86, с = 7,14 А) и предположения, что два
иона лаурата закрепляются на каждом участке поверхности
кристалла, равном а-сА2.
Собирательное действие кислот, обладающих ненасыщенной
связью, на несульфидные минералы. На практике, когда в ка-
честве собирателей необходимо применять карбоновые кислоты
или их мыла, преимущественно пользуются олеиновой кислотой,
или некоторыми другими ненасыщенными кислотами, хотя име-
ются отдельные исключения, как например, в случае флотации-
малахита в Бельгийском Конго, когда необходимо точно регули-
ровать температуру и ненасыщенность собирателя [77]. Преиму-
щества применения ненасыщенных жирных кислот способство-
вали изысканию дешевых источников различных ненасыщенных
жирных кислот (таких, как кукурузное масло, хлопковое масло
и отходы рыбьего жира). Полагают, что эти жирные кислоты
имеют большую растворимость в воде и более низкую точку
плавления, чем насыщенные жирные кислоты, имеющие углево
дородные цепи эквивалентной длины. Некоторые из ненасыщен-
ных кислот также легко окисляются.
В связи с этим представляло интерес исследовать возмож-
ность окисления олеиновой кислоты в процессе собирательного
действия ее на минералы.
С этой целью Коле 124] флотировал флюорит высокой чисто-
ты при помощи тщательно очищенной олеиновой кислоты и пос-
ле неокисляющего слабого подкисления экстрагировал флоти-
рованный минерал при помощи растворителей олеиновой кисло-
ты и исследовал экстракты на содержание двойных связей. Он
заключил, что олеиновая кислота не изменяется, если минерал,
на котором она закрепилась, подвергается даже повторной
флотации; линолевая кислота, имеющая две неконъюгирован--
МЕХАНИЗМ ДЕЙСТВИЯ СОБИРАТЕЛЕЙ
307
ные двойные связи, также заметно не изменяется. Но в отличие
от этого линолевая кислота, имеющая три неконъюгированные
двойные связи, заметно изменяется даже после одной простой
флотационной операции.
Действие аминов
Действие аминов на металлы. Арбайтер, Келлог и Таггарт
[2] исследовали механизм собирательного действия катионных
реагентов на металлы. Они помещали образцы различных ме-
таллов с полированными поверхностями в раствор солянокис-
лого лауриламина (25 мг/л), в котором создавалось значение
pH = 2,88 и 11,0, и замеряли образующиеся краевые углы мето-
дом принудительного контакта пузырька с поверхностью мине-
рала. При этом сравнивались полученные результаты с резуль-
татами измерений краевых углов тех же металлов, помещенных
в растворы этилксантогената калия и олеата натрия, концентра-
ция которых равнялась 50 мг/л. Полученные результаты пред-
ставлены в табл. 6.
Поведение анионных собирателей на поверхностях металлов
можно объяснить обменом анионов, подобно тому, как это
происходит у сульфидных минералов. Это оказывается возмож-
ным благодаря тому, что, как известно, все металлические по-
верхности на воздухе (или в воде) покрыты кислородсодержа-
щими группами. При обработке металла раствором ксантогена-
та или олеата происходит обмен гидроксила, карбоната или
другого аниона на ион ксантогената или олеата следующим об-
разом:
он х~
Металл +Х~-- Металл + ОН-. (21)
Ме+ Ме+
Можно ожидать, что при применении катионных реагентов
катион металла будет обмениваться с ионом амина:
он он
Металл + Ат+
Металл
(22)
Это может происходить только в кислой среде, тогда как в
Щелочной среде ион амина удаляется из раствора, причем это
не сопровождается переходом иона металла в раствор. Это яв-
ление можно объяснить различными причинами: 1) в щелочной
среде ион металла осаждается в виде гидроокиси; 2) обмен
20*
308
ГЛАВА 9
Таблица 6
Краевые углы на металлах, обработанных растворами
этилксантогената калия, олеата натрия и солянокислого
лауриламина [2]
Металл Ксантоге- нат1 Олеат2 Амин
КИСЛЫЙ* нейтраль- ный4 щелочной*
Медь 61 936 Скользящий 81 89
Платина 58 94 6 88 70 89
Серебоо 61 — 50 45 89
Цинк — 88 0 83 89
Никель — 85в 0 76 89
Кадмай .... — 82 0 83 89
Женено 0 936 0 75 89
Олово 0 89 0 65 68
Магний — 90 0 Скользящий 0
Алюминий .... 0 956 0 То же 0
Свинец 43 — 0 » » Скользящий
1 50 мг/л.
2 50 мг л.
8 Частицы перемешивали в растворе солянокислого лауриламина концентрации 25 мг/л,
промывали раствором HCI концентрации 10 ^-м. и исследовали в растворе 10*“2-м. НС1.
4 Частицы перемешивали и исследовали в растворе солянокислого лауриламина кон-
центрации 25 мг!л.
8 Частицы перемешивали в растворе солянокислого лауриламина, концентрации
25 мг/л, промывали раствором 10“*^*м. НС1.
4 Частицы перемешивали в растворе реагента, промывали и изучали в воде.
иона амина с ионом водорода, принадлежащим гидроксилу, бо-
лее вероятен, чем обмен с катионом металла; 3) образуется
комплекс металл — амин; 4) происходит молекулярная адсорб-
'ция свободного амина, образующегося при гидролизе соли
амина.
В конечном счете все эти «причины» равноценны, так как
различными путями они приводят к одинаковым результатам:
адсорбции группы амина без десорбции металла. Арбайтер,
Келлог и Таггарт придерживаются гипотезы образования
комплекса амина. Кук поддерживает представление об адсорб-
ции свободного амина, тогда как другие придерживаются ги-
потезы обмена амина с водородом по реакции
ОН" О 2- Ат +
+ Ат+ Металл + Н+. (23)
Ме+ Ме+
Металл
________________МЕХАНИЗМ ДЕЙСТВИЯ СОБИРАТЕЛЕЙ 309
Флотация сульфидных минералов аминами. В 1920 г. а-наф-
тиламин, являющийся ариламином, применялся в качестве со-
бирателя для сульфидных минералов, однако отсутствуют ка-
кие-либо количественные данные, позволяющие объяснить меха-
низм его действия.
Было установлено, что амиламины — первичные амины с
пятью атомами углерода — являются собирателями для халько-
зина и сфалерита 129, 34]. На рис. 16 показано извлечение этих
минералов (при мономинеральной флотации) как функции pH
1161. Можно видеть, что большее извлечение отвечает значениям
pH, при которых присутствуют примерно одинаковые количест-
ва свободного амина и иона амина, и в поведении обоих суль-
фидных минералов существует большое сходство. Совсем недав-
но действие лауриламина на сфалерит и галенит изучали Кел-
лог и В аскез-Роз ас [49], используя «нулевой» метод и производя
флотацию как руды, так и синтетических смесей сульфидов и
кварца. При измерениях краевых углов они применяли раство-
ры, содержащие 2,5 мг/л солянокислого амина. Результаты, по-
лученные при измерении краевых углов на частицах, помещен-
ных в этот раствор, показаны на рис. 17, а.
Далее они изучали краевые углы частиц в растворе 10-4-м.
NaOH после перемешивания в растворе амина при различных
значениях pH. Полученные результаты показаны на рис. 17, б.
Измерения краевых углов показывают, что все три минерала
флотируют лучше в щелочной среде, чем в кислой. Это под-
тверждается также результатами флотационных опытов.
Совпадение оптимумов pH флотации всех трех минералов
заставляет предполагать, что механизм действия аминов для
всех трех минералов один и тот же, однако это расходится с
представлениями, развиваемыми Келлогом и Васкез-Розасом.
Уместно отметить, что наибольшие краевые углы, так же как и
наибольшие извлечения, получаются при значениях pH, при ко-
торых большая часть реагента находится в форме недиссоции-
рованного амина. Келлог и Васкез-Розас [49] рассчитали содер-
жание различных форм, в которых амин присутствовал в иссле-
дованных растворах. Этот расчет основан на точном определе-
нии константы диссоциации лауриламина [46], однако имеется
еще одно соображение, которое необходимо учитывать, а имен-
но,— растворимость лауриламина в воде. Проведенные недав-
но Брауном 1 * измерения растворимости в воде лауриламина [10]
показали, что при комнатной температуре в воде может нахо-
диться в молекулярном состоянии до 3,7 мг/л лауриламина
(2 • 10“5 моль/л). По этой причине данные Келлога и Васкез-Ро-
1 D. J. Brown. Potentiometric titrations, Massachusetts, Institute of
Technology, 1953.
Рис. 16. Извлечение крупнозернистых сфалерита и
халькозина в присутствии 0,23 кг/г изоамиламина и
0,09 кг/г терпинеола при различных значениях pH:
/ — халькозин; 2 — сфалерит
б
Рис. 17. Краевые углы некоторых
минералов при различных значе-
ниях pH в растворе солянокисло-
го лауриламина [49]:
а — перемешивание в растворе, содер-
жащем 2,5 мг!л и исследование в том
же растворе; б — перемешивание в
2.5 мг!л реагента и исследование в рас-
творе 10-4-м, NaOH [49]; / — сфалерит
2 — галенит; 3 — кварц
МЕХАНИЗМ ДЕЙСТВИЯ СОБИРАТЕЛЕЙ
31!
заса были изменены; распределение реагента в виде ионной и
молекулярной формы в зависимости от pH и общего количества
реагента показано на рис. 18 в форме диаграммы. Роджерс и
Сазерленд 161] показали, что при pH = 10,6—10,7 половина ами-
на находится в ионной форме, а другая половина — в форме ра-
створенного свободного амина. Из рис. 18 следует, что если об-
щее количество реагента превышает количество, способное удер-
жаться в растворенном состоянии, то реагент распределяется в
Рис. 18. Распределение солянокислого лауриламииа
в растворе в зависимости от значения pH:
1 — максимальное количество в растворе; 2—расход
2- Ю-6 моль!л\ 3 — расход 2 • 10-4 моль 1л
виде трех форм: ионно растворенной, молекулярно растворен-
ной и нерастворенной. При высоких значениях pH достигается
предельная величина концентрации не только молекулярно ра-
створенной, но и ионно растворенной формы. Принципиально
диаграммы рис. 18 применимы ко всем умеренно растворимым
солям, способным к гидролизу, при котором один из продуктов
312
ГЛАВА 9
гидролиза значительно меньше растворим, чем нейтральная
соль. Например, они применимы к мылам и алкилсульфатам.
Диаграммы, подобные изображенным на рис. 18, могут быть
полезны, за исключением тех случаев, когда отсутствуют дан-
ные растворимости и гидролиза, определенные с достаточной
степенью точности.
Келлог и Васкез-Розас, используя данные, подобные изобра-
женным на рис. 17, для вычисления распределения солянокисло-
го лауриламина в форме иона и растворенного молекулярного
Концентрация молекулярно растворенного амина, мг/л
Рис. 19. Краевой угол на сфалерите в растворах лауриламина [49]:
/ — в растворе 1 1 • Ю-5 мг/л амина в присутствии NaOH; 2 —в раствора
1,1 • 10-5 мг/л амина в присутствии СаО; 3 — в растворах переменных концен-
траций амина в присутствии NaOH
амина, пришли к заключению, что эффективной формой, обра-
зующей поверхностное покрытие на минерале, является свобод-
ный амин. Графическое изображение полученных ими данных с
внесенной поправкой на распределение вещества между ионной
формой и свободным амином, представлено на рис. 19. Как мож-
но видеть, существует определенная зависимость между краевым
углом и концентрацией в растворе молекулярно растворенного
амина. В связи с этим, однако, следует помнить о том, что от-
ношение иона лауриламина к иону водорода прямо пропорцио-
нально концентрации молекулярно растворенного амина. Это
вытекает из определения константы ионизации
К-
(он~) (rnh+>
(R NHgOH)
(24>
где выражения в круглых скобках означают концентрации соот-
МЕХАНИЗМ ДЕЙСТВИЯ СОБИРАТЕЛЕЙ
313
ветствующих соединений. Так как (Н + ) • (ОН-) = 10-14, то по-
лучаем
------— =(/?NH3OH)-/C', (25)
<н+)
т. е. отношение иона амина к иону водорода пропорционально
концентрации свободного основания (или свободной кислоты в
случаях гидролиза, протекающего в обратном направлении).
Таким образом, согласно данным рис. 19, можно высказать
три гипотезы о механизме собирательного действия солянокис-
лого лауриламина на сфалерит: 1) обмен ионов между ионами
амина и водорода; 2) одновременная адсорбция ионов амина и
гидроксила; 3) молекулярная адсорбция недиссоциированного
амина.
Эти три механизма действия лауриламина возможны; одна-
ко следует иметь в виду, что последний из них наиболее вероя-
тен, так как комплексные соединения цинка с амином [6] анало-
гичны хорошо известным аммиакатам цинка.
Одинаковое поведение кварца, галенита и сфалерита при
действии лауриламина, однако, наводит на мысль, что образо-
вание подобных комплексов не может иметь решающего зна-
чения.
Гидрофобизирующее действие алкиламинов на несульфидные-
солеобразные минералы. Таггарт и Арбайтер измеряли краевые
углы на полированных поверхностях солеобразных несульфид-
ных минералов в растворах солянокислого лауриламина. Опыты
ставились с волластонитом (силикатом кальция), кальцитом,
флюоритом и баритом. В этих опытах установлено, что краевой
угол возрастает с увеличением количества реагента, достигая
1 максимальной величины 70—75°. Гидрофобизирующее действие
амина по отношению к взятым минералам уменьшается от вол-
ластонита к бариту примерно параллельно изменению раство-
римости соли амина с анионом минерала. Кривые изменения
краевых углов показаны на рис. 20.
Таггарт и Арбайтер предположили, что происходит обмен
металла минерала с катионом собирателя. В случае барита, на-
пример, происходит реакция
BaSO4 + 2Am+ -> Ва2+ + Am2SO4. (26)
Для того чтобы произошла эта реакция, должно быть пре-
вышено произведение растворимости сернокислого амина. Из
рис. 20 видно, что флотация барита происходит при почти мак-
симальном значении краевого угла и концентрации реагента
1 ммоль!л (точка А, 221 мг/л). Так как растворимость сульфата:
бария при комнатной температуре составляет около 10-5 моль/лг
то в точке А произведение растворимости (SO4-2) (Am + )2 рав-
но 10-11. По данным Таггарта и Арбайтера, растворимость сер-
314
ГЛАВА 9
нокислого лауриламина составляет 12,5—25- Ю"4 моль/л. Прини-
мая среднее значение растворимости 20-Ю-4, получаем, что про-
изведение растворимости Am2SO4 3,2 • 1Сг8. Другими словами,
:в точке А (рис. 20) произведение концентраций ионов
(SO4~2) • (Ат +)2 составляет менее тысячной доли произведения
растворимости сернокислого амина. Так как на рис. 20 показа-
но образование краевых углов при значительно меньших добав-
ках солянокислого лауриламина, то, очевидно, поглощение ми-
нералом иона амина происходит при концентрациях, весьма да-
Рис. 20. Гидрофобизируюшее действие солянокислого лаурил-
амина на солеобразные несульфидные минералы [71]:
/ — волластонит; 2 — кальцит; 3 — флюорит; 4 — барит
•фазы продукта реакции между катионом собирателя и анионом
минерала. Таким образом, на сульфате бария одинаково прояв-
ляется преимущественная адсорбция как анионов, так и катио-
нов, содержащих углеводородный радикал (см. ранее приведен-
ную работу Турнесака, касающуюся барита и лаурата).
Таггарт и Арбайтер показали, что добавка сульфата натрия
может уменьшить количество солянокислого амина, необходи-
мого для получения такого же краевого угла, как и при отсут-
ствии сульфата, и, наоборот, добавка хлористого бария может
уменьшить краевой угол до исчезающе малой величины. Это со-
гласуется как с представлениями «теории растворимости», так
и с представлениями об адсорбции при ненасыщенном монослое.
Действительно, если допустить, что определенная степень
насыщения монослоя соответствует определенному краевому
углу и если далее допустить, что эта степень насыщения соот-
ветствует также определенному отношению произведений ак-
тивностей ионов (при данной их концентрации в растворе) к
МЕХАНИЗМ ДЕЙСТВИЯ СОБИРАТЕЛЕН
315
произведению растворимости, то должны быть получены резуль-
таты, подобные приведенным выше. Это почти полностью совпа-
дает с определениями Турнесака.
Флотация кварца алкиламинами. Изучение флотации кварца
аминами явилось предметом ряда исследований, широко постав-
ленных в Массачусеттском технологическом институте. В на-
стоящее время этот вопрос настолько продвинулся вперед, что
Концентрация ацетата лауриламина, моль/л
Рис. 21. Ацетат лауриламина и кварц. Зависимость
плотности сорбционного слоя собирателя от концентра-
ции реагента в растворе при нейтральном значении pH
[Ю]
стали доступны пониманию многие его детали. Последние наи-
более законченные исследования были осуществлены Блокером
[5, 21] и Морроу [561. Основные исследования ими были выпол-
нены с уксуснокислым лауриламином в качестве собирателя; в
некоторых случаях применялись также и другие соли первич-
ных н-аминов.
Адсорбция лауриламина кварцем. Результаты многочислен-
ных измерений представлены на рис. 21. Согласно этим данным,
количество адсорбируемого вещества увеличивается с увеличе-
нием концентрации реагента, однако при этом не наблюдается
прямой пропорциональности. Наклон нижней части кривой ра-
вен 0.5; таким образом, можно заключить, что в пределах из-
менения концентрации соли амина от 10~7 до 2-10"4 моль)л
Г = д/7, (27)
где Д = const.
316
ГЛАВА 9
Верхняя часть кривой имеет более крутой наклон, что сви-
детельствует о более быстром возрастании значения Г по срав-
нению с величиной с. При концентрациях, превышающих
10“3 ммоль/л, видимо, происходит полимолекулярная адсорбция;
так, например, при с = 0,004 ммоль/л адсорбированный слой
достигает толщины около 5 молекул.
С точки зрения флотации представляет интерес область из-
менения концентраций от 1,5-10"6 до 4-10-5 моль/л, для которой
применимо уравнение (27). При высоких концентрациях в ра-
створе появляются мицеллы и, возможно, даже в двойном слое»
па собирателем, % монослоя
Рис. 22. Извлечение кварца при
флотации в зависимости от сте-
пени покрытия собирателем. Со-
биратель— ацетат лауриламина;
пенообразователь терпинол
(0,036 кг!ц'}; минерал — специаль-
но очищенный и обесшламленный ।
кварц —200 меш [5, 21]
относительно обогащенном раствором,— «хемимицеллы». Суще-
ствование этих «хемимицелл» было постулировано [261 при изу-
чении потенциала протекания. Они состоят из плоских агрега-
тов ионов и молекул толщиной в один ион или молекулу, при-
чем ионы или молекулы в агрегатах ориентируются таким обра-
зом, что с одной стороны имеются углеводородные радикалы, а -
с другой — полярные группы.
На адсорбцию амина оказывает сильное влияние величина
pH раствора, однако это явление очень сложно и недостаточно
объяснимо. В общем амин адсорбируется больше в щелочной
среде, однако его адсорбция при этом меньше, чем можно было
бы ожидать, исходя из теории ионного обмена.
Необходимая для флотации плотность покрытия кварца ами-
ном. В области флотационных концентраций (от 2 до 20 jwa/л)
адсорбционное покрытие составляет только долю монослоя. Так,
например, при 10 мг]л поверхностное покрытие составляет около
7% полного монослоя.
На рис. 22 показано соотношение между извлечением при
флотации и насыщенностью сорбционного слоя. Приведенные
данные взяты из исследований Блокера. Как следует из этих
данных, при поверхностном покрытии, составляющем 4% моно-
слоя, достигается 90%-ное извлечение; при этом изменение плот- •
ности сорбционного покрытия от 1 до 1,5% вызывает увеличение
извлечения на 15%. Такой значительный флотационный эффект
при столь незначительном изменении плотности адсорбционного.
МЕХАНИЗМ ДЕЙСТВИЯ СОБИРАТЕЛЕЙ
317
«слоя наводит на мысль о способности к передвижению поверх-
ностного слоя собирателя в горизонтальном направлении.
Подобную подвижность можно допустить, если сорбирую-
щееся на поверхности вещество находится в жидком, а не твер-
дом состоянии.
Блокер нашел, что «меченое» покрытие амина на кварце мож-
но частично удалить при промывании водой или раствором не-
радиоактивного амина той же концентрации. Таким образом,
Рис. 23. Кривые критических
значений pH для ацетатов
первичных аминов на кварце
[Ю]:
— ацетат октадециламииа; 2 — аце-
тат тетрадециламииа; 3 — ацетат
лауриламина; 4— ацетат децилами-
иа; 5 — ацетат октиламииа
обменная способность амина, хотя и большая, чем в аналогич-
ных опытах с сульфидными минералами, все еще далеко недо-
статочна с точки зрения кинетики, согласно которой адсорб-
ция — десорбция рассматриваются подобно испарению и кон-
денсации. Можно полагать, что это обусловлено взаимодействи-
ем цепей амина.
Влияние длины, углеводородной цепи при флотации кварца
аминами. Флотационные кривые критических концентраций раз-
личных уксуснокислых аминов от н-октилового до н-октадецило-
вого были получены де Брейном при помощи метода вакуум-
флотации по способу Шумана и Пракаша [64] (рис. 23). Кривые
на рисунке показывают наличие верхнего предельного значения
pH при флотации аминами, которое примерно одинаково для
всех аминов (pH = 12,2—12,4) и выше которого никакие добав-
ки амина не вызовут флотацию. Существует также нижнее пре-
дельное значение pH, однако оно несколько неопределенно и
имеет другой характер. Было установлено также, что для фло-
тации достаточно образование адсорбционного покрытия, отве-
чающего части насыщенного монослоя. Также было установле-
но, что критическая концентрация, необходимая при заданном
3J8 ГЛАВА 9
pH, понижается с увеличением длины углеводородной цепи ами-
на примерно в соответствии с правилом Траубе.
Краевые углы на кварце в растворах уксуснокислого лаурил-
амина. Мо-рроу измерял краевые углы на полированных образцах
кварца, погруженных в растворы уксуснокислого лауриламина.
На рис. 24 показаны результаты определений краевых уг-
лов, причем на оси ординат отложены краевые углы в граду-
сах, а на оси абсцисс — логарифмы концентраций. Этот способ
изображения был принят вследствие медленного увеличения
краевого угла с концентрацией; пользоваться для оси абсцисс
арифметической шкалой неудобно.
Используя данные Блокера и де Брейна, связывающие ад-
сорбцию с концентрацией раствора и величинами краевых уг-
лов (согласно Морроу), можно построить кривую, связывающую
краевой угол с плотностью адсорбционного покрытия. На рис
25 дается соотношение между косинусом краевого угла и плот-
ностью покрытия в процентах монослоя. Например, при адсорб-
ционном покрытии, составляющем 6% монослоя (при котором
минерал полностью флотируется) образуется краевой угол, при-
мерно равный 18° (1 — cos 0 = 0,95).
Из рис. 25 следует, что для образования краевого угла необ-
ходимо поверхностное покрытие определенной плотности. Когда
концентрация раствора превысит предельную величину, обра-
зуется краевой угол, косинус которого изменяется почти линей-
но с плотностью покрытия, выраженной в процентах монослоя
(но не линейно с изменением концентрации собирателя в рас-
творе). Начиная с некоторой плотности покрытия, увеличение
адсорбции все меньше влияет на краевой угол; при довольно вы-
соких концентрациях реагента .значительные изменения концент-
рации вызывают относительно небольшие изменения адсорбции
и почти не влияют на краевой угол.
Работа адгезии в системе кварц—амин. Уместно напомнить,
что согласно уравнению (9) гл. 7 работа адгезии твердого тела
к пузырьку воздуха равна произведению из площади контакта,
поверхностной энергии жидкости у и фактора (1—cos 0), где
0—‘краевой угол. Для единицы площади контакта
W = у (1 — cos 6).
Работу адгезии можно связать с плотностью адсорбционного
покрытия. Эта зависимость изображена графически на рис. 26
в форме кривой, построенной из нескольких точек, выбранных
из числа точек, хорошо укладывающихся на кривой рис. 25.
Интересно отметить, что работа адгезии проходит через макси-
мум при плотности покрытия, составляющей немного больше 2
гиббсов (2-Ю'10 моль/см2), что отвечает примерно 35% насы-
щенного плотно упакованного монослоя. Это более или менее
Концентрация ацетата лауриламина,мг/л
Рис. 24. Краевые углы для системы кварц—аце-
тат лауриламина; pH изменяется от 6 до 7 [31, 5’6]
Степень покрытия минерала реагентом,
% монослоя
Рис. 25. Краевой угол в зависимости от степени по-
крытия реагентом (в процентах монослоя) для сис-
темы кварц — ацетат лауриламина (при построении
кривой использованы данные различных авторов [5,
10, 56]
320
ГЛАВА 9
соответствует излому линии на рис. 21. Можно полагать, nio
образование поверхностного покрытия на кварце включает два
явления; одно из них эндотермическое, а другое экзотермическое.
Это согласуется также с представлениями об образовании на
поверхности раздела кварц — раствор амина двойного электри-
ческого слоя, что подтверждается измерениями потенциала про-
текания.
Рис. 26. Работа адсорбции для системы кварц—ацетат
лауриламина [56]
При изменении концентрации изменяется величина Г (см.
рис. 1); при этом изменяются также значения у и 0 (см. рис. 24).
В результате этого изменяется и величина 1F. Скорость измене-
ния работы адгезии с адсорбцией dW/dV представляет меру
удельной энергии адсорбции или теплоты адсорбции. По этой
причине она является важной термодинамической величиной.
Теплота адсорбции, которую можно изобразить в виде
dW _ rf[Y(l- cos 6)] 2
dr dr ’ ' '
представляет собой наклон кривой на рис. 26. На графике вид-
но, что наклон уменьшается с увеличением значения Г и доходит
до нуля при 35°/0-ном покрытии поверхности и даже приобрета-
ет небольшое отрицательное значение при больших значениях
Г. Количественные даные для системы уксуснокислый лаурил-
амин — кварц приведены в табл. 7-
В начале процесса теплота адсорбции, выраженная в ка-
лориях на моль, значительна (примерно отвечает теплоте неко-
торых химических реакций), но затем заметно уменьшается.
Де Брайен сделал попытку обобщить все исследования, каса-
ющиеся адсорбции аминов на кварце и флотации минерала
аминами. Он пришел к выводу, что все существующие теории
несовершенны и не могут объяснить наблюдаемые явления.
Требуются дополнительные исследования даже этой хорошо
изученной системы, прежде чем удастся создать общую теорию.
МЕХАНИЗМ ДЕЙСТВИЯ СОБИРАТЕЛЕЙ
321
Таблица 7
Адсорбция, краевой угол и теплота адсорбции на образцах
кварца, погруженных в растворы уксуснокислого лауриламина
при рН=6,5 [56]
Концентрация раствора с мг;мл Адс орбцио иная плотность Г % монислоя Поверхностное натяжение при 20° дин/см Краевой угол 9 град. dW d Г кал/моль
0,8 2 72,2 4
4,3 5 71,3 12 2690
17,2 10 70,2 20 1460
80 25 64,9 28 504
135 50 61,4 34 301
240 100 56,7 40 97
292 125 55,0 42 —20
В особенности необходимы данные по кинетике адсорбции, об-
ратимости адсорбции, сравнению адсорбции на поверхностях
раздела газ — твердое и жидкость — твердое и влиянию pH (в
особенности в щелочной области). По мнению де Брейна, наи-
более вероятна ионная теория, однако она еще не ясна во всех
деталях.
Собирательное действие уксуснокислого лауриламина на ге-
матит. Морроу провел исследования с образцами гематита и
«меченым» уксуснокислым лауриламином; он определял адсорб-
цию реагента минералом и измерял краевые углы при различ-
ных концентрациях амина. При этом качественно были полу-
чены такие же выводы, как и для кварца.
Детали структуры поверхностных пленок собирателя
Ориентация и расположение поверхностных пленок собира-
теля. Необходимо доказать, что пленка собирателя ориентиро-
вана неуглеводородной (полярной) частью собирателя в сторо-
ну минерала. Как указывалось ранее в этой главе, количество
собирателя, концентрирующееся на поверхности минерала и
обусловливающее флотацию, образует лишь часть мономолеку-
лярного слоя.
Так, если на кварце образуется пленка додециламина, со-
ставляющая 5% полного монослоя, то a priori существуют сле-
дующие возможности:
1) ионы ориентированы своей длинной осью перпендикуляр-
но поверхности, углеводородным радикалом — в сторону твер-
дого;
21 А. М. Годэн
322
ГЛАВА 9
2) ионы ориентированы своей длинной осью нормально к по-
верхности, углеводородным радикалом — в сторону, противопо-
ложную твердому;
3) ионы лежат плоско или почти плоско на поверхности раз-
дела, однако закреплены на твердом теле концом, противополож-
ным углеводородному радикалу.
Если считать справедливым первое предположение, то сле-
дует допустить наличие собирательной способности у молекул
углеводородов, чего, однако, не наблюдается на практике (в
смысле их способности адсорбироваться из водных растворов
на минералах суспендированных в воде). Кроме того, можно
ожидать, что пленка первого типа должна быть гидрофильной, а
не гидрофобной, а это как раз не является назначением собира-
теля.
Предложения второе и третье кажутся более вероятными, так
как в обоих случаях собиратель закрепляется на минерале при
помощи своей полярной части. Если пленка является тонкой, то,
можно ожидать, что термодинамически более вероятно горизон-
тальное положение закрепленной части собирателя; затем с уве-
личением плотности адсорбции оно постепенно изменяется, пере-
ходя в наклонное, а затем в вертикальное. Так, для упомянутой
ранее системы кварц — амин 5°/0-ное покрытие, подсчитанное
на основе предположения о вертикальном положении молекул,
при горизонтальном положении будет составлять 21% монослоя.
Поскольку сорбция собирателя определяется химическими
свойствами твердого тела и реагента, можно ожидать, что со-
биратель будет закрепляться скорее на каких-то определенных
местах, чем беспорядочно, как это наблюдается при физической
адсорбции. Поэтому максимальная плотность адсорбционного
покрытия определяется площадью, занимаемой атомами и иона-
ми в поверхностном слое адсорбента, а также площадью моле-
кул или ионов адсорбата при плотной упаковке. Так, в случае
галенита, у которого свинец и сера образуют решетку хлори-
стого натрия с расстоянием между атомами свинца, равным
5,93 А (в направлении, параллельном одной из кубических
осей), элемент поверхности, соответствующий каждому адсор-
бированному иону амина, пальмитата, ксантогената или дитио-
фосфата, должен быть простым кратным числом или правильной
дробью от (5,93)2 А2, или 35,1 А2 Другими словами, можно
представить себе, что при адсорбции собирателя на каждые
35 А2 поверхности адсорбируется одна, две или три частицы ад-
сорбата, или, наоборот, — одна частица адсорбата на каждые
две или три единицы поверхности площадью 35 А. Можно легко
представить себе случай, при котором простое целое число ча-
МЕХАНИЗМ ДЕЙСТВИЯ СОБИРАТЕЛЕЙ
323
стиц адсорбата (скажем, четыре) соответствует простому цело-
му числу элементов поверхности (скажем, трем).
В настоящее время площадь поперечного сечения углеводо-
родной цепи может быть рассчитана из данных кристаллической
структуры длинноцепочечных парафиновых углеводородов [83];
она оказывается равной 18,5 А2, что отвечает площади прямо-
угольника, равной 3,73-4,96 А. Эта величина соответствует наи-
более плотной упаковке ионов или молекул с углеводородными
радикалами. При плотной упаковке нормальных насыщенных
карбоновых кислот площадь, приходящаяся на одну молекулу,
равна 20,5 А 2.
На основании размеров радиусов серы и кислорода можно
оценить, что ион ксантогената должен занимать около 29 А2
[32].
н-Дитиофосфат, имеющий две углеводородные цепи, должен
занимать не менее 41 А2.
Понятно, что на каждой элементарной ячейке галенита не мо-
гут закрепиться две частицы собирателя, несмотря на то, что при
закреплении одной частицы собирателя на каждой ячейке оста-
ется еще свободное пространство (за исключением случая сорб-
ции дитиофосфата).
На рис. 27 дается схема наиболее плотной упаковки ксанто-
гената на поверхности галенита. При таком расположении соби-
рателя образуется двухмерный кристалл, для которого воз-
можна нечеткая диффракция электронов. Подобный эффект был
найден в Японии Хагихарой [40, 41]. На основании полученных
им результатов не удается показать образования определенной
кристаллической решетки при обработке галенита ксантогена-
том, хотя некоторое изменение диффракции и наблюдалось.
Для такой короткой молекулы, как у этилксантогената, труд-
но ожидать образования кристаллического поверхностного по-
крытия, учитывая возможность достижения хороших результа-
тов флотации даже при наличии частичного монослоя. Это более
вероятно, когда опыты проводятся в условиях образования
полислоев.
Ранее было показано, что краевой угол, получаемый для дан-
ной пары минерал — собиратель, изменяется по мере изменения
концентрации реагента и величины pH раствора (см. гл. 7). Бы-
ло также показано, что максимальные краевые углы для дан-
ного собирателя на различных минералах идентичны и что су-
ществует большое сходство в величинах краевых углов для раз-
личных собирателей, имеющих одинаковые угловодородные
Радикалы.
В заключение следует напомнить о том, что максимальный
21*
324
ГЛАВА 9
краевой угол для данного минерала и гомологического ряда со-
бирателей возрастает с длиной углеводородной цепи до тех пор,
пека не достигнет величины краевого угла, получаемого для
чистого парафина. Все эти наблюдения согласуются с приведен-
ным здесь изображением ориентации и расположения поверхно-
стного покрытия собирателя.
О 2 U 6 8 40 72 40
Величина pH
Рис. 27. Возможное расположение
ионов ксантогената на поверхности
галенита (маленькими кружками изо-
бражается свинец, а большими — се-
ра; заштрихованными участками
изображаются ионы ксантогената)
[32]
Рис. 28. Зависимость между
значениями pH и концентраци-
ей цетилтриметиламмонийбро-
мида, необходимой для уста-
новления прилипания пузырь-
ка воздуха к поверхности
кварца; пунктирной линией
изображена кривая адсорбции;
температура 10° [62]:
/ — прилипание; 2 — отсутствие при-
липания
Действие избытка собирателя. При применении длинноцепо-
чечных соединений (таких как соли аммония и алкилсульфаты)
наблюдалось уменьшение краевых углов при высоких концен-
трациях реагентов.
Действительно, при достижении определенной концентрации
собирателя краевой угол не образуется, и флотации минерала
не происходит [62]. Это иллюстрируется рис. 28, на котором по-
казано, что прилипание кварца к пузырьку воздуха в растворе
цетилтриметиламмонийбромида возможно в пределах измене-
ния pH от 4 до 12 в том случае, когда концентрация реагента
превышает 0,1 мг/л. При наиболее благоприятных значениях pH
МЕХАНИЗМ ДЕЙСТВИЯ СОБИРАТЕЛЕЙ
325
(pH = 6 — 8) концентрация реагента, превышающая 125 мг[л.
предотвращает прилипание, а в пределах изменения значений pH
от 3 до 6, так же как от 8 до 13, область прилипания еще более
ограничена. Таким образом, прилипание возможно в пределах
1000-кратного изменения концентрации реагента; при низких
концентрациях прилипания не происходит (из-за отсутствия до-
статочной сорбции), так же как при высокой концентрации в
результате избыточной адсорбции, характеризующейся специфи-
ческими особенностями. Роджерс, Сазерленд и Уорк предпо-
лагают, что уменьшение краевых углов при высоких концентра-
циях вызывается не очисткой поверхности минерала собирателем
Рис. 29. Зависимость ме-
жду значениями pH и
концентрацией цетил-
сульфата натрия; темпе-
ратура 35° [62]:
/ — для установления прили-
пания воздуха к поверхно-
сти галенита: 2 — для фло-
тации галенита в цилинд-
ре; 3 — для флотации га-
ленита в пневматическом
аппарате; 4 — прилипание;
5 — отсутствие прилипания
(как это следует по теории Дина и Амброзе [9]), а совместным
влиянием двух факторов: образованием второго слоя, в котором
пленка собирателя обратно ориентирована по сравнению с пер-
вым слоем, т. е. ориентирована полярными группами в сторону
воды, и полимолекулярной адсорбцией (толщиной в два или бо-
лее слоев) на поверхности раздела жидкость — газ. Дальнейшее
доказательство обоснованности представлений, выдвинутых Род-
жерсом, Сазерлендом и Уорком, мы находим на рис. 29, на кото-
ром изображено соотношение между прилипанием и отсутстви-
ем прилипания, флотацией и отсутствием флотации и так назы-
ваемыми кривыми «адсорбции» для галенита в растворе цетил-
сульфата натрия. Кривая прилипания 1 иллюстрирует поведение
галенита при наличии двух новых явлений, а именно, образова-
ния второго гидрофильного слоя на минерале и гидрофильного
слоя на относительно «состарившемся» пузырьке. Кривая фло-
тации в цилиндре 2 показывает поведение минерала при наличии
неполного гидрофильного слоя на пузырьках из-за отсутствия
старения пузырьков. Кривая 3 справедлива для условий пневма-
тической флотации (при наличии большого объема воздуха), ко-
гда гидрофильный слой на поверхности пузырьков в значитель-
ной степени нарушен. Эта кривая аналогична кривой прилипания
326
ГЛАВА 9
воздушного пузырька к полированной минеральной поверхности,
которая предварительно обрабатывалась собирателем опреде-
ленной концентрации и при определенном значении pH и пе-
реносилась после промывания в воду, где измерялось прилипа-
ние. Точки, показанные с внутренней стороны кривых, свиде-
тельствуют о наличии прилипания или флотации.
Образование гидрофильного слоя, по-видимому, связано с
появлением мицелл при применении реагентов в таких количе-
ствах, когда они могут служить детергентами. Энергия адсорб-
ции при образовании второго гидрофильного слоя незначитель-
на, поскольку она обусловлена главным образом вандерваальсо-
выми силами. Поэтому величина адсорбции во втором слое от-
носительно невелика по сравнению с величиной адсорбции в пер-
вом слое.
Приведенные здесь представления об образовании второго
слоя вполне вероятны, однако, так как они не имеют значения
для практики флотации, дальнейшее их рассмотрение не пред-
ставляет интереса.
Собирательное действие и состояние равновесия. Ранее пред-
полагалось, что между минеральной поверхностью и окружаю-
щей водной средой достигается равновесие. Здесь уместно рас-
смотреть, происходит ли это действительно в обычных условиях.
Существует мнение, что равновесное состояние системы ча-
сто достигается в случае применения слабых собирателей, когда
поглощение реагента минералом незначительно. Однако при на-
личии сильных собирателей, распределение которых между ми-
нералом и раствором сильно смещено в сторону твердого, можно
полагать, что обычно в практических условиях равновесие не ус-
танавливается; с большим трудом оно достижимо в лаборатории
и то лишь при специально подобранных условиях.
Большой заслугой Тувинера и Кормана [79] является то, что
они обратили внимание на роль диффузии при установлении рав-
новесия. Тувинер и Корман вначале вычислили толщину непо-
движной пленки, окружающей частицы халькозина, суспендиро-
ванного в воде, в условиях, изменяющихся от спокойного состоя-
ния до интенсивного перемешивания. Толщина пленки, через ко-
торую единственным процессом переноса реагента к поверхности
минерала является диффузия, изменяется от 2000 до 40 ц при из-
менении от спокойного состояния до интенсивного перемешива-
ния. Тувинер и Корман измерили необходимое время контакта
раствора собирателя (этилового ксантогената калия в присут-
ствии извести при pH = 10,5) с халькозином, т. е. время, необхо-
димое для такого изменения характера поверхности, при кото-
ром возможно прилипание частицы к пузырьку воздуха в одно-
пузырьковом аппарате. Некоторые из их данных, представлен-
ные на рис. 30, подтверждают высказанное положение о том, что
МЕХАНИЗМ ДЕЙСТВИЯ СОБИРАТЕЛЕЙ
327
поверхность халькозина не находится в равновесии с раствором
реагента даже после 10 мин. интенсивного перемешивания (или
30 мин. неинтенсивного перемешивания). На основании расхо-
ждения кривых, полученных в «спокойном состоянии» и при «ин-
тенсивном перемешивании», видно, что, для того чтобы достиг-
нуть равновесия в случае прилипания при спокойном состоянии,
недостаточно даже нескольких дней.
Рис. 30. Изменение крити-
ческой концентрации раст-
вора н-бутилксантогената
калия в зависимости от
времени перемешивания его
с халькозином при pH —
= 10,5 при отсутствии воз-
духа [79]:
1 — интенсивное перемешивание;
2— умеренное перемешивание;
3 — спокойное состояние
Концентрация п-бутипксамтогената
калия, ms/л
Время индукции. Свен-Нильсон [68, 69] наблюдал, что для
ясно выраженного прилипания пузырька к полированной поверх-
ности минерала необходимо вполне определенное время сопри-
косновения. Он вызывал колебания пузырька, закрепленного дер-
жателем; частота и амплитуда колебаний контролировались не-
зависимо одна от другой. Он обозначил минимальное время со-
прикосновения, необходимое для прилипания пузырька, как вре-
мя индукции. В обычных условиях время индукции было слиш-
ком мало для того, чтобы его можно было измерить, и только в
случае «голодного» расхода реагента оно могло быть измерено.
Представляло бы интерес связать время индукции с явле-
ниями, определяемыми диффузией, изученными Тувинером и
Корманом. Однако условия опытов Свен-Нильсона были таковы,
что при измерении времени индукции определялась не столько
степень приближения твердой поверхности к состоянию равно-
весия с окружающей жидкостью (ибо перемешивание твердого
в растворе не контролировалось), сколько способность вибри-
рующего пузырька прилипать к минеральной поверхности в ус-
ловиях изменения времени соприкосновения. По-видимому, опы-
328
ГЛАВА 9
ты Свен-Нильсона имели больше общего с процессом адгезии
твердое — газ, чем с химическим механизмом собирательного
действия.
Выводы.. Переходя к выводам, необходимо указать на то,
что в механизме собирательного действия было установлено
следующее:
1. Минералы способны флотироваться при поглощении из вод-:
ных растворов групп, содержащих углеводородные радикалы. \
2. Это поглощение происходит только в том случае, если yr-i
леводородные группы представляют собой ионные или способ-'
ные ионизироваться соединения (имеются исключения, упомяну-
тые в гл 18).
3. Плотность пленки собирателя на флотируемых минералах
обычно значительно меньше насыщенного монослоя.
4. Поглощение собирателя из раствора сопровождается экви-
валентным выделением с поверхности минерала первоначально
адсорбированных ионов.
5. При действии собирателей в случае окисляющихся мине-
ралов и окисляющихся реагентов кислород играет большую (хо-1
тя все еще неясную) роль.
6. Поглощение собирателя возможно из растворов, ненасы-
щенных по отношению к соединению, образующемуся при взаи-
модействии с минералом.
7. Адсорбированный собиратель прикреплен к минералу '
неуглеводородным (полярным) концом. Независимо от того, на-
блюдается перпендикулярная ориентация закрепленного собира-
теля или он лежит плоско, поверхность оказывается покрытой
углеводородными радикалами.
8. Распределение собирателя между раствором и минералом
сильно смещено в сторону минерала.
Эти факты согласуются с представлениями о том, что при
обмене, происходящем с первоначально адсорбированными ио-
нами, ионы собирателя дегидратируются, так что связь между
адсорбатом и адсорбентом осуществляется не через водную про-
слойку, как это происходит с первоначально адсорбированными
ионами.
ЛИТЕРАТУРА
1. Anderson Robert J. The flotation of minerals, Trans. AIMME, 55,
527—546, with discussion by О. C. Ralston, 1916.
2. Arbiter Nathaniel, Herbert H. К e 11 о g, and Arthur F. Tag-
gart. The mechanism of collection of metals and metallic sulfides by
amines and amine salts, Trans. AIMME, 153, 517—527, 1943.
3. В a i n s T. M., J r. The electrical theory of flotation, Mining Sci. Press,
Hl, 824—826, 883—884, 1915.
4. Barsky, G. Discussion of Wark and Cox paper of principles of flotation,
I, Trans. AIMME, 112. 236—237, 1934.
5. Bloecher F. W. Jr. Concerning the adsorption of dodecylamine on
quartz, Thesis, Massachusetts Institute of Technology, 1949.
МЕХАНИЗМ ДЕЙСТВИЯ СОБИРАТЕЛЕЙ
329
6, Broome F. К., A. W. Ralston, and М. Н. Thornton. Complex
formation with high molecular weight amines, I, J. Am. Chem. Soc., 68,
67—69, 1946.
7. Buerger M. J., and Newton W. Buerger. Low-chalcocite and high-
chalcocite, Am. Mineralogist, 29, 55—65, 1944.
8. С о о к Melvin A., and John C. Nixon. Theory of water-repellent
films on solids formed by adsorption from aqueous solutions of heteropolar
compounds. J. Phys. & Colloid Chem., 54, 445—459, 1950.
9. Dean R. S., and P. M. Ambrose. Development and use of certain flo-
tation reagents, U. S. Bur. Mines, Bull. 449, 1944.
10. De Bruyn P. L. Flotation of quartz by cationic collectors Mining Eng.,
7, 291—296, 1955.
11. Dewey Franklin. Reactions of some sulfur-bearing collecting agents
with certain copper minerals, Thesis, Montana School of Mines, 1933.
12. Fa hr en wald A. W. The electro-statics of flotation, Mining Sci. Press,.
112. 375—378, 1916.
13. Fa h re n wald A. W. Surface-energy and adsorption in flotation, .Mi-
ning Sci Press, 123, 227—234, 1921.
14. Fahrenwald A. W. Surface reactions in flotation, Trans. AIMME, 70,
647—739, 1924.
15. Gat у F., H. de Rycker, and H. T h у s s e n. Etude de la flotation des
minerals calaminaires aux laboratoires de 1’institut de metallurgie, Rev.
universelle mines, 89, 155—166, 1946.
16. Gaudin A. M. The influence of hydrogen-ion concentration on recovery
in simple flotation systems, Mining and Met., 10, 19—20, 1929.
17. Gaudin A. M. Flotation mechanism, a discussion of the functions of
flotation reagents, Trans. AIMME, 79, 50—77, 1928.
18. Gaudin A. M. The effect of xanthates, copper sulfate and cyanides on
the flotation of sphalerite, Trans. AIMME, 87, 417—428, 1930.
19. G a u d i n A. M. «Principles of Mineral Dressing», 378—380, McGraw-
Hill Book Company, Inc., New York, 1939.
20. Gaudin A. M., and Arvid E. Anderson. Flotation fundamentals,
V, Utah Eng. Expt. Sta., Tech. Paper 9, 1930.
21. Gaudin A. M., and F. W. Bloecher, Jr. Concerning the adsorption
of dodecylamine on quartz, Trans. AIMME, 187, 499—505, 1950.
22. Gaudin A. M., and J. S. Carr. Analytical procedures for ethyl xan-
thate labeled with radiosulfur, Anal. Chem., 24, 887—890, 1952.
23. G a u d i n A. M., and C. S. Chang. Adsorption on quartz, from an
aqueous solution, of barium and laurate ions, Trans. AIMME, 193, 193—
201, 1952.
24. G a u d i n A. M., and Richard E. Cole. Double-bond reactivity of
oleic acid during flotation, Trans. AIMME, 196, Tech. Note 144-B (in
Mining Eng., 5 (4), 418). 1953.
25. Gaudin A. M., Franklin Dewey, Walter E. Duncan, R. A.
Johnson, and Oscar F. Tangel, Jr. Reactions of xanthates with
sulfide minerals Trans. AIMME, 112, 319—347, 1934.
26. Gaudin A. M., and D. W. Fuerstenau. Streaming potential studies.
Quartz flotation with cationic collectors, Trans. AIMME, 202, 66—72, 1955.
27. Gaudin A. M., Harvey Glover, M. S. Hansen, and C. W. Orr.
Flotation fundamentals I, Utah Eng. Expt. Sta., Tech. Paper 1 (a) Fig. 10,
pp. 19—20, 1928.
28. Gaudin A. M., and Dwight L. Harris. Adsorption of a mercaptan
on zinc minerals. Trans. AIMME, 199, Tech. Publ. 3871-B (in Mining
Eng., 6, 925—928), 1954.
29. Gaudin A. M„ С. B. Haynes, and E. C. Haas. Flotation fundamen-
tals, IV, Utah, Eng. Expt. Sta., Tech. Paper 7, 1930.
30. Gaudin A. M., P. L. de Bruyn, a n d О 1 a v M e 11 g r e n. On the
adsorption of ethyl xanthate on pyrite, Mining Eng., 8, 65—70, 1956,
330
ГЛАВА 9
31. Gau din A. M., and John G. Morrow. Adsorption of dodecylammo-
nium acetate on hematite and its flotation effect, Trans. AIMME, 169 (Mi-
ning Eng., 6, 1196—1202), 1954.
32. G a u d i n A. M., and Gustav S. Prelie r. Surface area of flotation
concentrates and the thickness of collector coatings AIMME Mining Te-
chnol., 10, (3) Tech. Publ., 2002, 1946.
33. Gaudin A. M. and Reinhardt Schuhmann, Jr. The action of
potassium n-amyl xanthate on chalcocite, J. Phys. Chem., 40, 257—275, -
1936.
34. Gaudin A. M., and Paul M. Sorensen. Flotation fundamentals, II, '
Utah Eng. Expt. Sta., Tech. Paper 4, 1928.
35. Gaudin A. M., and J. C. Tournesac. Adsorption du laurate sur le
sulfate de baryum, First World Congress on Detergence, Paris, September,
1954.
36. Gaudin A. M., and Kenneth C. Vincent. Observations on the
magnitude of contact angles and their significance in flotation phenomena,
AIMME Tech. Publ. 1242, 1940.
37. Gaudin A. At., and Kenneth C. Vincent. Self-diffusion in mine-
rats, particularly copper sulfides, AIMME Tech. Publ. 1663, 1944.
38. G a u d i n A. M., and Walter D. Wilkinson. Surface actions of
some sulfur-bearing organic compounds on some finely-ground sulfide
minerals, J. Phys. Chem., 37, 833—845, 1933.
39. Gaudin A. M. and S. Suphi Yavasca. Principles of comminution- 1
size and surface distribution, AIMME Tech. Publ. 1819, 1945.
40. Hagihara Hitosi. Surface oxidation of galena in relation to its flo-
tation as revealed by electron diffraction, J. Phys. Chem., 56, 610—615, 1952.
41. Hagihara Hitosi. Mono and multilayer adsorption of aqueous xantha,te
on galena surfaces, J. Phys. Chem., 56, 616—621, 1952.
42. Harris Dwight L. Hexyl mercaptan and zinc minerals, Dissertation
for the doctorate, Massachusetts Institute of Technology, 1953.
43. H a s s i a 1 i s M. D. Surface-active compounds in flotation ore dressing,
Ann. N. Y. Acad. Sci., 46 (art 6), 495—500, 1946.
44. H a s s i a 1 i s M. D., and C. G. Myers. Collector mobility and bubble
contact, Mining Eng., 3, 961—968, 1951.
45. Herd Harold H., and William Ure. Surface chemistry in the ,
flotation of galena, J. Phys. Chem., 45, 93—106, 1941.
46. Ho err, C. W„ M. R. McCorkle, and A. W. Ralston. Studies on
high molecular weight aliphatic amines and their salts. X. Ionization con-
stants of primary and symmetrical secondary amines in aqueous solution,
J. Am. Chem. Soc., 65, 328—329, 1943.
47. J о h n s о n R. A. The reaction of certain sulfur-bearing collectors with
cerussite and galena, Thesis, Montana School of Mines, 1933.
48. J u d s о n С. M., A. A. L erew, R. B. Booth, J. S. Kennedy, and
G. L. Simard. Radio-tracer studies of the action of dithiophosphate in ;.
the selective flotation of galena and sphalerite with copper sulfate and
sodium cyanide, Trans. AIMME, Tech. Publ. 3306—В (in Mining Eng. 4,
375—380), 1952.
49. Kellog, Herbert H., and Hugo V a s q u e z-R о s a s. Amine flota-
tion of sphalerite-galena ores, Trans. AIMME, 169, 476—504, 1946
50. Knoll Alexander, and Dwight L. Baker. Adsorption of potas-
sium xanthate by galena in oxygen-free atmosphere, AIMME Tech. Publ
1313, 1941.
51. Knoll Alexander, and Dwight L. Baker. Nature of the adsorp-
tion of fatty acids from organic solvents by inorganic lead compounds,
Trans. AIMME, 153, 508—512, 1943.
52. Langmuir Irving. The mechanism of the surface phenomena of flo-
tation, Gen. Elec. Rev., 24, 1025—4033, 1921; The mechanism of the surface
phenomena of flotation, Trans. Faraday Soc., 15, 1—13, 1920.
МЕХ/.НИЗМ ДЕЙСТВИЯ СОБИРАТЕЛЕЙ
331
53. Luyken Walter, and Ernst В i er bra u er. Untersuchungen liber
die Beziehungen zwischen Adsorption Benetzbarkeit und Flotation Metall
u. Erz, 26, 197—202, 1929.
54. Luyken Walter, and Ernst Bierbrauer. Untersuchungen zur
Theorie der Flotation, Mitt. Kaiser-Wilhelm-Inst. Eisenforsch Dtisseldorf
11(3), 37—52, 1929.
55. Mellgren 01 a v. Adsorption of ethyl xanthate on pyrite. Dissertation
for the doctorate, Massachusetts Institute of Technology, 1954.
56. Morrow, John C. Adsorption of dodecylammonium acetate on hematite
and sphalerite, Thesis, Massachusetts Institute of Technology, 1952.
57. N i x о n John C. The mechanism of the action of collectors in the flo-
tation of minerals, Thesis, University of Utah, 1949.
58. Plante Enid C., and K- L. Sutherland. Effects of oxidation of
sulfide minerals on their flotation properties, Trans. AIMME, 183, 160—
188, 1949.
59. Ralston О. С., C. R. King, and F. X. T a r t a г о n. Copper sulfate as
flotation activator for sphalerite, Trans. AIMME, 87, 380—400, 1930.
60. Ramdohr Paul. «Die Erzmineralien und ihre Verwachsungen», Aka-
demische Verlagsgesellschaft m.G.H., Leipzig, 1950.
61. Rogers J., and K. L. Sutherland. Principles of flotation-activation
of minerals and adsorption of collectors, Trans. AIMME, 169, 317—339, 1946.
62. Rogers J., K. L. Sutherland, E .E. Wark, and I. W. Wark.
Principles of flotation-paraffin-chain salts as flotation reagents, Trans.
AIMME, 169, 287—316, 1946.
63. Schuhmann Reinhardt, Jr. The nature of the action of potassium
n-amyl xanthate on chalcocite, Thesis, Montana School of Alines, 1935.
64. Schuhmann Reinhardt, Jr., and Brahm Prakash. Effect of
barium chloride and other activators on soap flotation of quartz, Trans.
AIMME, 187, 591—600, 1950.
65. Seidel Atherton. «Solubilities of Inorganic and Organic Compounds»,
vol. 1, 1940, vol. II, 1941, supplement, 1952, D. Van Nostrand Company,
Inc., New York.
66. Simard G. L., M. Burke, and D. J. Salley. Radiotracer studies on
the interaction of dithiophosphate with galena. The depressant action of
phosphate, Trans. AIMME, 190, 39—44, 1951.
67. S i m a r d G. L., J. C h u p a k, and D. J. Salley. Radiotracer studies
on the interaction of dithiophosphate with galena, Trans. AIMME, 187,
359—364, 1950.
68. Sven-Nilsson I. Effect of contact time between mineral and air bub-
bles on flotation, Kolloid-Z., 69, 230, 1934.
69. Sven-Nilsson I. Effect of contact time between mineral and air bub-
bles on flotation, Ing. Vetenskaps Akad. Handl., No. 133, 1935.
70. Taggart Arthur F. ;Elements of Ore Dressing», pp. 251—253, John
Wiley & Sons, Inc., New York, 1951.
71. Taggart Arthur F., and Nathaniel Arbiter. The chemistry of
collection of nonmetallic minerals by amine-type collectors, Trans. AIMME,
169, 266—271, 1946. . , ,
72. T a g g a r t Arthur F., G. R. M. del G i u d i c e, and O. A. Z i e h 1.
The case for the chemical theory of flotation, Trans. AIMME, 112, 348—381,
1934.
73. Taggart Arthur F., and A. M. Gaudin. Surface tension and
adsorption phenomena in flotation, Trans. AIMME, 68, 479—535, 1923.
74. Taggart Arthur F., T. C. Taylor, and A. F. Knoll. Chemical
reactions in flotation, Trans. AIAIME, 87, 217—260, 1930. .
75. T a n g e 1, Oscar F. The reaction of alkali xanthates with pyrite, Thesis,
Montana School of Mines, 1934.
Z6. TaylorT Clinton and A. F. Knoll. Action of alkali xanthate on
galena, Trans. AIMME, 112. 382—397, 1934.
332
ГЛАВА 9
77. Theys L. L'evolution de la technique de la flotation des minerals oxydes
du Katanga, 1947 Congress, Centenary Alumni Association, University of
Liege, Belgium.
78. TournesacJean. Adsorption of laurate on barium sulfate, Dissertation
for the doctorate, Massachusetts Institute of Technology, 1953.
79. T u win er S. B., and S. Korman. Effect of conditioning on flotation
of chalcocite, Trans. AIMME, 187, 226—238, 1950.
80. Wadsworth Mi It on E. Acid and base adsorption on solids from
aqueous solutions of strong electrolytes, Thesis University of Utah, 1951.
81. Wark Ian William. «Principles of Flotation», Australasian Institute
of Mining and Metallurgy, Melbourne, Australia, 1938.
82. Wark Ian William, and Alwyn Birchmore Cox. Principles
of flotation, I, Trans. AIMME, 112, 189—244, especially 209, (a) p. 218,
1934, III, ibid., 267—302, (b) pp. 271, 288, 1934.
83. Wyckoff Ralph W. G. «Crystal structures», vol. II, chap. XIII, p. 34,
Interscience Publishers, Inc., New York, 1951.
Глава 10
ДЕЙСТВИЕ МОДИФИКАТОРОВ ФЛОТАЦИИ
Основы собирательного действия реагентов при флотации из-
ложены в предыдущей главе. Для обеспечения успешного разде-
ления минералов действие собирателя должно быть селективным
по отношению к различным видам минералов. При флотации
необходимо выбирать и тшательно соблюдать условия, при кото-
рых достигается максимальное извлечение ценных минералов в
пенный продукт при минимальном увлечении в него нежелатель-
ных минералов.
Из рассмотрения вопроса о влиянии концентрации водород-
ных ионов пульпы на флотацию (изложенного в предыдущей
главе) следует, что водородные и гидроксильные ионы играют
главную роль при регулировании действия собирателя. Кроме
водородных и гидроксильных ионов, другие анионы и катионы
также усиливают или ослабляют действие данного собирателя
на тот или иной минерал. Процесс, при котором собирательное
действие усиливается, обычно называется активацией; ослабле-
ние собирательного действия носит название депрессирования.
Когда функция депрессора сводится к уничтожению активации,
то такой процесс часто называют дезактивацией.
Регулирование флотации изменением концентрации
водородных ионов
Влияние на флотацию значительных загрузок кислот или ще-
лочей было давно установлено, как показывает патентная лите-
ратура начала XX века. Влияние на процесс флотации относи-
тельно небольших изменений концентраций водородных и гид-
роксильных ионов было оценено значительно позже [14].
Рис. 1 дает некоторое представление о величине этого эф-
фекта по данным флотационных опытов, поставленных на из-
мельченных чистых минералах.
Этот рисунок показывает, что переход от полной флотации к
полному ее отсутствию может быть достигнут в пределах изме-
нения pH на одну единицу, что отвечает десятикратному изме-
нению концентрации (Н+) или (ОН-). В реальных флотационных
системах влияние концентрации водородных и гидроксильных
ионов проявляется менее резко вследствие присутствия различ-
334
ГЛАВА 10
них минералов, различного размера частиц и наличия сростков.
На рисунке видим, что существуют пределы значений pH, при
которых флотация возможна, а избыток (Н+) или (ОЬГ ) пре-
пятствует действию собирателя.
Рис. 1. Влияние величины pH на флотацию различных чистых мине-
ралов (необходимое значение pH создавалось загрузкой различных
количеств NaOH или НС1):
/ — халькозин (0,09 кг/г терпинеола; 0,014 кг/г амилового ксантогената калия);
2 — пирит <0,09 кг/т терпинеола, 0,014 кг/т амилового ксантогената калия); 3 —
сфалерит (0,09 кг/т терпинеола, 0,045 кг/т амилового ксантогената калия); 4 —
халькозин (0,045 кг/т терпинеола, 3,15 кг/т спирта, 0 045 кг/т тиокарбанилида);
5—халькозин (0,09 кг/т терпинеола, 0,23 кг/т изоамилового амина); 6 — сфале-
рит (0,09 кг/т терпинеола, 0,23 кг/т изоамилового амина); 7 — малахит, извле-
каемый из смеси с кальцитом (0,09 кг/т терпинеола, 1,35 кг/т амилового ксан-
тогената натрия); 8 — микроклиновый полевой шпат (0,09 кг/т олеата натрия,
0,09 кг/т терпинеола); 9 — микроклиновый полевой шпат (0,09 кг/т олеата на-
трия, 0,45 кг/т сульфата меди, 0,09 кг/т терпинеола); 10 — кварц (0,1 кг/т
олеата натрия, 0,1 кг/т терпинеола 0,5 кг/т сульфата меди)
В случае применения сульфгидрильных собирателей избыточ-
ная концентрация водородных ионов может привести к разложе-
нию собирателя (см. гл. 8), в то время как избыточная концен-
трация гидроксильных ионов приводит к их конкуренции с иона-
ми собирателя за адсорбционные точки на поверхностях мине-
ралов. Резкая депрессия пирита, происходящая, как показано
на рис. 1, при pH, близком к 7, обусловливается, по-видимому,
увеличением степени окисления минералов при повышении ще-
лочности.
ДЕЙСТВИЕ МОДИФИКАТОРОВ ФЛОТАЦИИ
335
В случае применения катионных собирателей (аминов), оче-
видно, наблюдается аналогичный эффект, но максимум флоти-
руемости лежит в щелочной области. Причина резкого падения
извлечения при очень высокой щелочности непонятна.
Наблюдающаяся хорошая флотируемость силикатных мине-
ралов мылами в пределах значений pH от 6 до 11, очевидно, в.
большей степени может быть отнесена за счет действия актива-
тора, чем одного лишь собирателя, и, следовательно, здесь мож-
но предполагать совместное действие нескольких факторов.
Эти замечания сделаны для того, чтобы показать, что изме-
нение величины pH может:
1) сводиться к конкуренции ионов Н + или ОН- с собирате-
лем на поверхности минерала;
2) оказывать дополнительный эффект к действию вводимого
в пульпу активатора или депрессора;
3) оказывать дополнительное воздействие в сочетании с та-
ким вездесущим компонентом системы, как кислород.
Депрессирующее действие гидроксильных ионов. Уорк и Кокс
провели обширные исследования депрессирующего действия гид-
роксильного иона иа различные флотационные системы, главным
образом сульфидных минералов с сульфгидрильными собирате-
лями. Эта работа, выполненная на аппарате с принудительным-
контактом пузырька и основывающаяся на измерениях краевых
углов, показала, что в том случае, когда невозможно получить
измеримый краевой угол, также сомнительна возможность фло-
тации. Отсюда было сделано заключение, что условия, при ко-
торых начинает наблюдаться закрепление пузырька на минера-
ле, отвечают практическим условиям флотации. При этом было
бездоказательно принято, что прилипание пузырька воздуха к
минералу всегда происходит при достижении приблизительно
одинаковой плотности адсорбционного слоя собирателя на по-
верхности минерала.
Работа Уорка и Кокса позволила Барскому сделать обобщаю-
щий вывод о том, что при построении диаграммы зависимости
необходимого расхода собирателя от величины pH раствора ли-
ния, отделяющая область, где может образоваться краевой угол,
от области, где это невозможно, отвечает уравнению:
[Н+] • [Х~} = const. (1)
В этом уравнении обозначение [А-] — отвечает концентрации
аниона собирателя.
Уорк подтвердил это соотношение в форме
—PC 1 const.
[ОН—1
;336
ГЛАВА 10
Уравнение, предложенное Барским [45], предполагает конку-
ренцию анионов собирателя и гидроксильных иовов за поверх-
ность минерала. Ниже мы приводим некоторые данные, получен-
ные Уорком и Коксом, для того чтобы проверить, насколько они
отвечают соотношению, установленному Барским.
Уорк [45а] приводит ряд значений критических величин pH,
вызывающих депрессию сульфидных минералов в присутствии
25 мг/л этилового ксантогената калия (табл. 1). Очевидно, что
для достижения одинаковой степени депрессии различных мине-
ралов потребуются различные концентрации гидроксильных
ионов.
Таблица 1
Критические значения pH для сульфидных минералов
в растворе этилового ксантогената калия при концентрации
его 25 мг/л [45а]
Минерал Критическое значение pH при
1 0° комнатной температуре 35»
Сфалерит — 1 —
Пирротин — 6,0 —
Арсенопирит — 8,4 —
ГалениТ 10,8 10,4 9,7
Пирит 10,2 10,5 10,0
Марказит — 11,0 —
Халькопирит 13,0 11,8 10,8
Ковеллин — 13,2 —
Борнит — 13,8 —
Тетраэдрит — 13,8 —
Халькозин —. Около 14,0 —
1 Пузырек воздуха ие прилипает к сфалериту ии при каких значениях pH.
Для различных собирателей величина критического значения
pH также различна (табл. 2).
Проверка правильности соотношения, установленного Бар-
ским. Если бы уравнение Барского было вполне точно, то зави-
симость логарифма критической концентрации иона собирателя
от логарифма концентрации гидроксильного иона (или pH) вы-
ражалась прямой линией с постоянным углом наклона. Однако
было показано, что в действительности в соответствии с рис. 3
указанное выше соотношение должно выражаться следующим
образом:
[X-l=m(OH-f, (2)
где m — коэффициент пропорциональности;
у — другая характеристическая константа.
ДЕЙСТВИЕ МОДИФИКАТОРОВ ФЛОТАЦИИ
337
Таблица 2
Критические значения pH для сульфидных минералов
в водных растворах различных собирателей [4да1
Собиратель Концентра- ция собирателя мг]л Критическое значение pH
сфа- лерит галеиит пирит халько- пирит
Диэтилдитиофэсфат натрия . . 32,5 1 6,2 3,5 9,4
Этиловый ксантогенат калия . 25 1 10,4 10,5 11,8
Диэтилдитиокарбамат натрия . 26,7 6,2 >13 10,5 >13
Амиловый ксантогенат калия . 31,6 5,5 12,1 12,3 >13
Диамилдитиокарбамат калия . 42,3 10,4 >13 12,8 >13
1 Сфалерит не реагирует с этими собирателями
ни при каких значениях pH.
Для двух примеров, показанных на рис. 3, величина у, опре-
деленная для флотации галенита этиловым ксантогенатом калия
или диэтилдитиофосфатом натрия, составляет соответственно
0,65 и 0,52. Величина у при применении ксантогената была уста-
новлена при pH, большем 9, с тем чтобы можно было прирав-
нять концентрацию иола ксантогената к общему количеству
загруженного ксантогената. Высокая степень разведения и от-
носительно сильные кислотные свойства ксантогеновой кислоты
позволяют принять такое допущение (см. гл. 8). Такое же допу-
щение было сделано и в случае применения дитиофосфата, хотя
и с меньшими основаниями, поскольку при этом значение pH
снижалось до 6 и недостаточно известны кислотные свойства ди-
тиофосфорной кислоты.
Дополнительные данные, приводимые в подтверждение урав-
нения Барского, были также получены Уорком и Коксом при по-
мощи измерения величины краевых углов, образуемых на сфале-
рите различными дитиокарбаматами — от диэтилового до нор-
мального диамилового. Если по этим данным построить график
зависимости lg [Х-1 от 1g ЮН-], наклон прямых, определяемый ве-
личинами 0,56 (диэтилтидиокарбамат), 0,72 (дибутилдитиокар-
бамат) и 0,75 (диамилдитиокарбамат), будет отвечать соответ-
ственно крайним значениям pH от 6 до 8, от 7,5 до 10 и от 8 до И.
Это показано на рис. 2, построенном по данным Уорка и
Кокса.
На этом рисунке кружками обозначаются условия, при кото-
рых происходит контакт минерала с пузырьком, крестиками от-
мечаются условия, при которых контакта не происходит. В этом
случае, однако, не учитывалось изменение концентрации иона
22 А. М. Годэн
Рис. 2. Кривые критических концентраций при
прилипании сфалерита в водных растворах диал-
к'илдитиокарбаматов при различных значениях
pH [45]:
9 О—флотация происходит; X — флотации нет; / — по
Уравнению Барского; 2 — диэтилдитиокарбамат натрия;
3 — дибутилдитиокарбамат калия; 4 — диамилдитиокар-
бамат калия; 5 — по уравнению Барского
Рис. 3. Кривые критических концентраций при при-
липания, пирита, галенита и халькопирита при при-
менении диэтилдитиофосфата иатрия [45]:
/ — по уравнению Барского; 2 — пирит; 3 — галеиит; 4 —
халькопирит; 5 — по уравнению Барского
т а 1
ДЕЙСТВИЕ МОДИФИКАТОРОВ ФЛОТАЦИИ
3S9
дитиокарбамата в растворе за счет происходящего гидролиза.’.
Данные для пирита, галенита и халькопирита при действии ди-
этилдитиофосфата натрия получены в интервалах pH от 2 до 6, >
от 6 до 9 и от 8 до 11 соответственно и дают величину у около 0,6
(рис. 3).
Нужно отметить, что для каждого примера величина у, харак-
теризующая наклон прямой, меньше единицы. Этот вывод под-
тверждается также опытами по прилипанию сидерита к пузырь-
ку воздуха в присутствии иона гептилата. В этих условиях ве-
личина у в пределах изменения значений pH от 5 до 10 при той
же величине краевого угла (20°) (примерно отвечающей вели-
чине угла, при котором происходит прилипание) составляет не-
много менее 0,3 (см. гл. 9, рис. 14).
Очевидно, что в этом случае существует постепенный переход
от условий, вызывающих прилипание, к условиям, когда прили-
пания не происходит.
Обобщая, можно сделать вывод, что при применении анион-
ных собирателей наблюдается качественное соотношение между
концентрациями иона-собирателя и гидроксильного иона. Однако
установить существование количественной пропорциональности
между величинами [X-] и [ОН-] для определения условий закреп-
ления не только не удалось, но можно сказать, что даже самое
существование подобной зависимости было опровергнуто. •
Данные по катионным собирателям пока еще недостаточны,
однако и здесь существует качественная зависимость между кон-
центрациями IZ+] и [Н+] при отсутствии строгого количествен-
ного постоянства отношения [Z+] : [Н + ].
Опыты, на основании которых было выведено уравнение (2),
проводились с растворами, имеющими различные суммарные;
концентрации электролитов. Например, при изучении взаимодей-
ствия нормального дибутилдитиокарбамата и сфалерита общая
концентрация ионов изменялась в 1000 раз в испытанном интер-
вале щелочности. Такие большие изменения концентрации были’
обусловлены стремлением оказать наибольшее воздействие на
структуру двойного слоя и таким образом определить причину
изменений адсорбции любых ионов, присутствующих в условиях
низкой суммарной ионной концентрации. Представляло бы ин-
терес повторить эти опыты, доводя ионную концентрацию до
возможно большей величины, скажем, от 0,1 до 1 м. (для этой це-
ли, по-видимому, можно использовать NaCl, NaNO3 или КС1).
В этом случае удалось бы точнее выяснить влияние общей ион-'
ной концентрации, что позволило бы уточнить соотношение, ус-
тановленное Барским [уравнение (1)].
Использование депрессирующего действия гидроксильного
иона при разделении минералов. На рис. 4 приводятся кривые,
изображающие зависимость между критической концентрацией
22*
340
ГЛАВА 10
диэтилдитиофосфата натрия и критическим значением pH для
трех сульфидных минералов — пирита, галенита и халькопирита.
Согласно этим данным, при любой заданной концентрации со-
бирателя возможна флотация лишь одного сульфидного минера-
ла. Так, при расходе собирателя 50 мг)л при pH = 8,5 может фло-
тироваться только халькопирит, а при pH = 6 только один пирит
не будет флотироваться. Это заключение справедливо при ус-
ловии, что минералы не активируются добавленными солями и
не оказывают вредного действия один на другой.
Гидросульфид-ион как депрессор для сульфидных минералов
Почти пятьдесят лет назад [39] стало известно, что примене-
ние сульфидов щелочных металлов может способствовать за-
креплению маслянистых веществ на некоторых окисленных ми-
нералах. В связи с этим давно установлено, что обработка ще-
лочными сульфидами окисленных руд тяжелых металлов может
повысить извлечение их при флотации.
Позднее было выяснено, что добавка сульфидов щелочных
металлов в пульпу сульфидных руд приводит к депрессии суль-
фидов, причем необходимая для этого концентрация во многих
случаях оказывается весьма малой.
Уорк и Кокс исследовали это явление [47], которое может
быть использовано для того, чтобы депрессировать сульфиды
при флотации несульфидных минералов [17].
На рис- 5 показаны кривые прилипания, полученные Уорком и
Коксом для ряда сульфидных минералов при различных значе-
ниях pH и.переменных расходах сернистого натрия. В качестве
собирателя, в этих опытах применялся этиловый ксантогенат ка-
лия (25 мг/л).
На первый взгляд не наблюдается какой-либо простой за-
висимости между величиной pH и различными концентрациями
сернистого натрия, препятствующими закреплению пузырьков
на. сульфидных минералах. Однако, если учесть хорошо извест-
ный гидролиз сернистого натрия и способность к двухступенча-
той диссоциации сероводородной кислоты (на HS- и 52--ионы), то
станет очевидным, что действительным депрессирующим агентом
является гидросульфидный ион HS-, а не сульфид натрия или се-
роводород.
Пользуясь двумя константами диссоциации сероводородной
кислоты Ki и Кг:
[H7J S1"1 = 10~7 для H2S->H+ + HS~, (3)
[ri2oj
, К2 = IH-+.HS2Zj_2 • 10~15 для HS~-»H+ + S2-, (4)
.:' [HS-]
Рис. 4. Зависимость концентрации диэтилди-
тиофосфата от критического значения pH [46]:
1 — пирит; 2 — галенит; 3 — халькопирит
Рис. 5. Зависимость между pH и концентрацией сер-
нистого натрия, необходимого для предотвращения
прилипания сульфидных минералов к воздушным
пузырькам в растворе этилового ксантогената ка-
лия (25 мг/л [47]:
1 — галеиит; 2 — активированный сфалерит; 3 — халькопи-
рит; 4—бориит; 5 — ковеллин; 6 — пирит; 7 — халькозин
342
ГЛАВА 10
полученными Еллинеком и Червинским [23], можно рассчитать
распределение всего количества растворенного сульфида в форме
^H2S, HS- и S2- при различных значениях pH.
; Результаты такого расчета приводятся в табл. 3.
Таблица 3
। Влияние pH на относительное содержание в растворе сульфид-иона,
; гидросульфид-иона и сероводорода
| pH 1 1 На каждый миллимоль NaaS.9H2O* в 1 л
растворенный серо- водород ММОЛЬ,1Л гидросульфид-ион ММОАЬ'Л сульфид-ион ММОЛЪ/Л
1 4 1,0 0,0010 2-10-14
j 4,5 1,0 0,0030 2-10-13
j 5 * 0,99 0,010 2-10-12
i 5,5 0,97 0,030 2-Ю-11
I 6 0,91 0,09 2- КГ10
i 6,5 0,75 0,25 1,6-Ю-9
; 7 0,50 0,50 0,5-10~8 *
7,5 0,25 0,75 6,5-10-8
8 0,09 0,91 2-10~7
8,5 0,030 0,97 7-10- 7
9 0,010 1,00 0,С00002
10 0 0010 1,00 о.оиоэг
11 0,00010 1,00 0,0002
12 0,000010 1,00 0,(02
13 O.COJOOl 0,98 0,02
14 0,0000001 0,80 0,20
; ♦ Количеством, поглощенным минералами или растворенными солями, и количест-
вом, потерянным за счет улетучивания сероводорода, пренебрегаем.
При сопоставлении данных табл. 3 с кривыми рис. 5 становит-
ся очевидным, что для каждого минерала с изменением pH значи-
тельно изменяется величина критической концентрации H2S, так
же как и критическая концентрация S2-. Однако, концентрация
S2 * *~ изменяется обратно пропорционально изменению критической
концентрации H2S.
Концентрация же гидросульфид-иона почти не изменяется.
В табл. 4 приведены типичные детальные данные для халько-
пирита.
Данные табл. 4 показывают, что концентрация иона HS~ око-
ло 0,30 мг/л предотвращает собирательное действие 25 мг/л эти-
лового ксантогената калия. Иными словами, депресоирующее
действие 9 мкмоль/л гидросульфид-иона уничтожает собиратель-
ное действие 156 мкмоль иона этилового ксантогената.
ДЕЙСТВИЕ МОДИФИКАТОРОВ ФЛОТАЦИИ
343
Таблица 4
Концентрация S2~, HS~ и H2S в растворах, содержащих минимальное
количество Na2S, необходимое для предотвращения прилипания пузырька
к поверхности халькопирита [47]
pH Общее количест- во сульфида в виде Na2S • 9НгО мг{л Концентрация, мг/л
H2S HS— s2-
5 150 21 0,21 4-10“11
5,5 52 7,2 0,22 1,4-Ю-10
6,0 21 2,7 0,25 6-10“10
6,5 8 0,8 0,27 1,7-10—9
7,0 4 0,3 0,28 5,2.10-9
8,0 3 0,04 0,37 7,8-10-8
9,0 3 0,004 0,41 7,8-10“7
10,0 2,5 0,00035 0,34 6,5-10-6
11,0 2,5 0,000035 0,34 6,5-10“5
В табл. 5 приводятся концентрации гидросульфид-иона, необ-
ходимые для подавления флотации различных сульфидных мине-
ралов в присутствии 156 мкмоль!л этилового ксантогената калия.
Таблица 5
Концентрация гидросульфид- иона, необходимая для уничтожения
собирательного действия этилового ксантогената калия при расходе,
равном 156 мкмоль/л или 25 мг/л [47]
Минерал [HS-] мг/л [HS-] мкмаль/л Минерал [HS-] мг/л [HS-] мкмоль/л
Галенит 0.007 0,25 Ковеллин . . . 1,7 50
Халькопирит . . 0,30 9 Пирит 2,5 75
Борнит 1,3 40 Халькозин . . . 6,4 190
Данные табл. 5 показывают, что со всеми испытывавшимися
минералами, за исключением халькозина, гидросульфид-ион бо-
лее активно взаимодействует с поверхностью минералов, чем ион
ксантогената; для галенита ион депрессора оказывается в
600 раз «сильнее» иона собирателя.
Соотношение между ионом собирателя и гид росу льфид-ионом.
Проведенные Лестом исследования [28] расширили наши пред-
ставления о критическом соотношении между концентрацией ио-
на собирателя и гидросульфид-иона. В качестве собирателя им
344
ГЛАВА 10
был выбран амиловый ксантогенат калия. Опыты были поставле-
ны на галените.
Метод «закрепления пузырька», разработанный Куком и Диг-
ре [7, 8], был использован для определения соотношения критиче-
ских концентраций растворов собирателя и сернистого натрия,
при которых происходит закрепление пузырька.
Рис. 6. Соотношение концентраций амилового ксантогената ка-
лия, концентрации сернистого натрия и pH при критических
условиях флотации галенита [28]:
/ — pH = 5,7; 2 —pH = 7,2; 3 — pH = 8,8; 4 — pH = 12,0; 5 — pH = 13.3
Во всех случаях общая концентрация электролита поддержи-
валась постоянной и равной 0,2-н.; pH создавался стандартными
буферными электролитами (такими, как НРО42~, Н2РО.г, СОз2~,
КОН, КС1).
Значения pH изменялись от 5,7 до 13,3. Данные, полученные
в пяти сериях опытов при значениях pH — 5,7; 7,2; 8,8; 12,0 и 13,3,
соответственно представлены графически на рис. 6.
Этот чертеж показывает, что все кривые, построенные для
значения pH < 12, проходят через начало координат, что наклон
кривых при значениях pH > 8,8 существенно не изменяется и что
с понижением pH наклон кривых становится значительно круче.
Кривая, отвечающая pH = 13,3, весьма сходна с кривой для
pH = 12, но она смещена по вертикали на высоту, отвечающую
расходу Na2S 9Н2О 18 мг/л.
ДЕЙСТВИЕ МОДИФИКАТОРОВ ФЛОТАЦИИ
345*
Для представления данных Леста в более общем виде необ-
ходимо рассчитать концентрации гидросульфид-иона и ксантоге-
нат-иона по экспериментальным материалам, приведенным в при-
ложении к его работе.
Рис. 7. Соотношения между концентрациями ионов
гидросульфида и амилксантогената при критиче-
ских условиях флотации галенита ’28]:
I — pH = 5,7; 2 —pH = 7.2; 3 — pH = 8,8; 4 — pH =11.0;
5 — pH = 13,3
Концентрация гидросульфид-иона определялась по табл. 3.
Ион ксантогената во всех случаях в пределах испытанных рас-
ходов и pH представляет единственную форму существования
ксантогената. Для определения концентрации иона ксантогената
использовалась константа диссоциации ксантогеновой кислоты,
полученная ван-Хальбаном и Хектом [22].
Как видно на рис. 7, в логарифмических координатах график
зависимости [HS-] и [Х~] для всех значений pH дает прямую ли-
нию с постоянным наклоном. Если отложить в логарифмических
346
ГЛАВА 10
координатах [IIS-] и [Х~] (как показано на рис. 7), то эксперимен-
тальные данные, полученные при различных значениях pH, ло-
жатся на одну прямую, имеющую постоянный наклон. Это пока-
зывает, что отношение [HS~]: [Л~] не зависит от значения pH.
В случае постоянства этого отношения все прямые линии на
рис. 7 должны совпадать. Это и наблюдается в действительности
(как видно из рис. 7), за исключением линии, отвечающей pH =
= 5,7, которая проходит несколько ниже, возможно, вследствие
небольшой систематической ошибки определения pH или вследст-
вие частичных потерь ксантогената за счет его разложения при
таком низком pH. Если экспериментальные данные для pH = 13,3
непосредственно нанести на рис. 7, то они не лягут на прямую,
ибо, как это видно на рис. 6, для образования краевого угла тре-
буется 123 мг/л амилового ксантогената (610 мкмоль/л) при пол-
ном отсутствии HS~.
В связи с тем, что потребовалось бы около 60 мкмоль/л HS',
чтобы вызвать депрессию галенита, эквивалентную действию гид-
роксильных ионов при pH = 13,3, на рис. 7 при нанесении экспе-
риментальных данных, полученных при этом pH, на оси ординат
откладывалось значение [HS~], увеличенное на 60 мкмоль/л. Это
количество может быть названо гидросульфидным эквивалентом
присутствующего гидроксильного иона.
Из рис. 7 видно:
1) существует пропорциональность между критическими кон-
центрациями [X-] и [HS~];
2) коэффициент пропорциональности между концентрацией
амилового ксантогената и концентрацией гидросульфид-иона при-
близительно равен десяти;
3) поскольку концентрация гидроксильного иона при pH =
= 13,1 составляет 200 ммоль/л, а эквивалентная депрессия дости-
гается при 60 мкмоль/л гидросульфид-иона, гидросульфид-ион
можно рассматривать как депрессор, в 3000 раз более эффектив-
ный, чем гидроксильный ион.
Интересно отметить прямолинейную зависимость между от-
ношениями критических концентраций [Х~] и [HS~], В другом слу-
чае при определении зависимости между критическими концен-
трациями [ОН'] и [X'] было установлено, что отношение [ОНИ :
:(Х~] не остается постоянным.
Возможно, это объясняется тем, что эксперименты Леста бы-
ли поставлены при условии постоянства общей ионной концен-
трации.
Уместно указать, что приведенная здесь интерпретация резуль-
татов измерений Леста не является единственной.
Действительно, он утверждает, что молекулярный H2S яв-
ляется депрессором в изучаемой системе. Видимо, в этом случае
на основании работы Кука и Никсона [6] собирательное действие
ДЕЙСТВИЕ МОДИФИКАТОРОВ ФЛОТАЦИИ
347
приписывается ксантогеновой кислоте. Лест допускает, однако,
что если собирателем является ион ксантогената, то его данные
j также подтверждают утверждение Уорка и Кокса о том, что де-
прессия обусловливается в основном гидросульфид-ионом. По
мнению автора, ионная концепция не только более удобна, но и
• больше согласуется со всеми известными фактами флотации.
Относительная поверхностная активность ионов ксантогената,
1 гидросульфида и гидроксила. Ранние исследования Уорка и Кок-
- са, посвященные изучению конкуренции между этиловым ксанто-
генатом. как собирателем и гидросульфид-ионо.м, как депрессо-
I ром при взаимодействии их с галенитом, были проведены при по-
стоянной концентрации собирателя и вследствие этого имеют ме-
I нее общий характер, чем работа Леста.
I, Интересно напомнить, что при расходе этилового ксантогена-
; та калия в количестве 25 мг!л критическая концентрация гидро-
сульфид-иона изменяется от 0,19 до 0,38 (в среднем составляет
0,25 мкмоль1л}.
Поскольку соответствующая концентрация ксантогенат-иона
составляет 156 мкмоль!л, было сделано заключение, что гидро-
сульфид-ион взаимодействует с поверхностью галенита в 600 раз
активнее, чем ион этилового ксантогената.
Эту величину интересно сравнить с соотношением критиче-
ских концентраций ионов гидросульфида и амилового ксантоге-
ната, равным 10.
Иными словами, ион амилового ксантогената является в 60 раз
более эффективным собирателем для галенита, чем ион этилово-
го ксантогената. Полученный результат удовлетворительно со-
гласуется с тем, что относительная растворимость амилового
ксантогената свинца в 30 раз меньше, чем этилового (правило
Траубе).
Далее следует отметить, что в системе галенит-амиловый ксан-
тогенат относительная эффективность депрессирующего действия
гидроксильного иона и гидросульфид-иона составляет 1 :3000, в
то время как для системы галенит — этиловый ктантогенат она
равна 1 : 1000. Это неплохое совпадение, если учесть случайный
характер исходных данных.
Полученное соотношение эффективностей депрессии галенита
ионами ОН~ и HS- (возможно, определяемое различием в адсор-
бируемости этих ионов галенитом) интересно сравнить с раство-
римостями и произведениями растворимости сульфида свинца,
окиси свинца (гидрата окиси), этилового и амилового ксантоге-
натов свинца.
Вопрос о растворимостях и произведениях растворимости
сульфидов рассматривается в критических обзорах Кольтгофа
124] и Равица [38], данные о растворимостях и произведениях рас-
348
ГЛАВА tO
творимости гидрата окиси свинца можно найти в работе Гаррета,
Велленга и Фонтана [13]; данные для ксантогенатов приведены
в гл. 8.
Примечательно качественное согласование между растворимо-
стями и поверхностной активностью галенита (табл. 6).
Таблицаб
Сравнение соотношения растворимостей окиси свинца, сульфида
свинца и ксантогенатов свинца с относительной аффективностью
действия ионов гидроксила, гидросульфида и ксантогената на галенит
Соединение Растворимость мклсоль/л Приблизитель- ная величина pH насыщеи- иого раствора Произведение растворимос- ти Относительная эффективность действия иоиа на галенит
РЬО (сурик)1 . 226 10 5-10“,в 1»
2,6 (Ван ХетереЮ 7-10“'7
РЬ (£/Х)2 . . 0,7 (Мак-Леод) 7 1-10“18 О
РЬ (АтХ)а . . 0,02 (Мак-Леод) 7 8-10“24 300
PbS 0,00004 7 7.1О“30 3000
1 Р:0 + Н,О->РЬ (ОН)+ + ОН- К= 3,46 • 10-8',
РЬ (ОН)+-> РЬ2++ ОН— н= 1,5- 10-8.
* Эффективность действия ОН на галенит условно принята за единицу.
Для установления точной количественной зависимости тре-
буется проведение значительно более тщательных эксперимен-
тальных исследований; при этом необходимо напомнить, что
сульфидный ион может быть депрессором наравне с гидроксиль-
ным ионом и гидросульфид-ионом и даже может являться более
сильным депрессором, чем любой из одновалентных анионов. То,
что это не было установлено, может быть объяснено тем, что ис-
следования проводились в области таких концентраций сульфи-
дов, при которых количество сульфид-иона настолько ничтожно^
что его влияние неуловимо мало по сравнению с действием гид-
росульфид-иона.
Разделение минералов при помощи гидросульфид-иона. На
рис. 5 показана возможность разделения различных сульфидных
минералов при применении в качестве собирателя этилового ксан-
тогената калия и с использованием гидросульфид-иона в качест-
ве селективно действующего депрессора. Так, при концентрации
этилового ксантогената калия 25 мг/л и Na2S • 9Н2О 30 мг/л при
pH = 9 можно флотировать халькозин при полной депрессии всех
других сульфидов. При расходе же Na2S • 9Н2О 5 мг)л вместо
30 мг!л, и при pH = 7 вместо 9 можно флотировать халькозин,
ДЕЙСТВИЕ МОДИФИКАТОРОВ ФЛОТАЦИИ
349
пирит, борнит и ковеллин, причем галенит оказывается депресси-
рованным и не флотируется.
Этот метод регулировки процесса селективной флотации не
применяется в современной практике вследствие трудности кон-
троля малых концентраций гидросульфид-иона, легкой окисляе-
мости ионов сульфида и гидросульфида, а также и потому, что
селективная флотация успешно осуществляется применением ио-
на цианида.
Как показано в предыдущих разделах, на депрессию галени-
та гидросульфид-ионом и гидроксильным ионом оказывает влия-
ние применяемый собиратель, причем заметная разница полу-
чается при замене амилового ксантогената этиловым. Однако это
различие не было изучено достаточно, чтобы установить, могут
ли критические кривые флотации различных минералов раздви-
нуться при замене этилового ксантогената амиловым или наобо-
рот. Заранее можно сказать, что такая замена собирателя смес-
тит все кривые, но, по-видимому, более или менее в соответствии
друг другу, поскольку у амилового и этилового ксантогенатов
имеется одинаковая полярная группа. Однако замена ксантоге-
ната другими сульфгидрильными собирателями может привести
к существенным изменениям, так же как и переход к оксигид-
рильным собирателям. Нужно отметить, что исследования в этом
направлении не производились, хотя их результаты могли бы
представить практический интерес.
Ион цианида как депрессор для сульфидных минералов
Флотация многих сульфидных минералов сульфгидрильными
собирателями ухудшается (часто даже до полного подавления)
при добавке цианида щелочного металла. Медные и железные
минералы в значительной степени подвергаются воздействию
цианида, галенит же к нему нечувствителен. Депрессия сфалери-
та представляет значительные трудности вследствие того, что он
обычно активирован ионами меди, а также из-за присутствия в
нем железа в виде твердого раствора. Действие цианида в боль-
шей степени зависит от значения pH пульпы. Применение циа-
нида при селективной флотации распространено повсеместно.
Этот процесс, первоначально’разработанный Шериданом и Гриз-
волдом [40], сделался основным при селективной флотации свин-
цово-цинковых руд.
Уорк и Кокс произвели обширное исследование воздействия
цианида на прилипание воздушных пузырьков к полированным
шлифам сульфидных минералов. На рис. 8 представлены полу-
ченные результаты.
Этот рисунок показывает, что при отсутствии цианида халь-
копирит можно отделить от галенита при pH от И до 12; обрат-
ный же порядок флотации будет наблюдаться в присутствии
350
ГЛАВА 10
40 .мг/л цианида натрия при pH = 8,5 — 10. Полезность примене-
ния цианида очевидна и при отделении галенита от пирита и при
отделении халькопирита от пирита.
Роль иона цианида. Подобно случаю сернистого натрия дан-
ные рис. 8 могут быть объяснены как результат депрессирующего
Рис. 8. Влияние концентрации
цианида натрия на прилипание
к воздушным пузырькам гале-
нита, пирита и халькопирита
при 10° (с этиловым ксантоге-
натом калия, 25 .иг/л) (по
Уорку и Коксу, AIMME Trans.,
134, 42, 1939):
1 — пнрнт; 2 — халькопирит; 3 —
галеннт
действия ионов модификатора, а именно ионов цианида. Кон-
центрацию иона цианида можно найти, исходя из константы дис-
социации синильной кислоты, определенной при 18°,
HCN -> Н+ 4- СЬГ
К = _ ГНИ 1SN_L.= 4,7- 1СГ10
[HCN]
(5)
В табл. 7 приводится распределение цианида в виде свободно-
го иона и растворенной синильной кислоты при различных зна-
чениях pH.
Таблица 7
Распределение цианида щелочного металла в виде иона (CN )
и растворенного продукта гидролиза (HCN)
pH ммоль'ммоль всего щелоч- ного цнаинда рн ммольммоль всего щелоч- ного цианида
в виде CN в виде HCN в виде CN в виде HCN
6,0 О^ООэ1 0,9995 9,0 0,320 0,680
6,5 0,1,015 0,998 9,5 0.597 0,403
7,0 0,0047 0,995 10,0 0.825 0,175
7,5 0,0146 0,985 11,0 0,979 0,021
8,0 0,045 0,955 12,0 0,999 0,001
8,5 0,129 0,871 13,0 1,000 0,000
1 Для получения концентрации иона CN в миллиграммах на I мг цианистого нат*
рия достаточно цифры этого столба умножить на 0,531.
ДЕЙСТВИЕ МОДИФИКАТОРОВ ФЛОТАЦИИ
35?
Сопоставляя данные табл. 7 и рис. 8, можно установить, что
критическая концентрация иона цианида для каждого минерала
в основном постоянна.
По более детальным данным, при 25° (вместо 10°) и 25 мг/л
этилового ксантогената калия критические концентрации иона
CN~ для различных минералов могут быть расположены в такой
последовательности: халькозин 180 мг/л, ковеллин 36, тетраэд-
рит 20, борнит 6,5, марказит 2,5, халькопирит 0,42, пирит 0,П
Исходные данные для этих цифр взяты из книги Уорка «Прин-
ципы флотации» [451.
Величина pH
i0~7 IO'610"5 <0'* 10-3 10"г -Ю"1 У
Концентрация (ОН-), моль/л
Рис. 9. Критические концентрации иона цианида, пре-
пятствующие прилипанию пузырьков воздуха к различ-
ным сульфидным минералам в водном растворе этило-
вого ксантогената калия (25 мг/л) [45]:
1— пирнт; 2 — халькопирит; 3 — марказит; 4 — борнит; 5 — тет-
раэдрит; 6 — ковеллин; 7 — халькозин
На рис.-9 видим, что если бы в присутствии 25 мг/л этило-
вого ксантогената калия удалось поддерживать концентрацию
иона цианида в пределах значений, лежащих между соседними
критическими концентрациями ряда 180, 36, 20, 6,5, 2,5, 0,42,
и 0,1 мг/л CN-, то можно было бы последовательно разделить
все минералы. Этого, пожалуй, можно достигнуть, но следует
иметь в виду, что эксперименты, результаты которых представ-
лены на рис. 9, были выполнены в идеальных условиях.
Отношение иона собирателя к иону цианида. Уорк и Кокс 146]
определили кривые критических концентраций для халькопири-
та при различных концентрациях этилового ксантогената калия;
расход цианистого натрия и величина pH в этих опытах были
также переменными. Данные, представленные на рис. 10, качест-
венно характеризуют влияние концентрации этилового ксантоге-
:352
ГЛАВА 10
ната на ход кривых прилипания. Те же данные, изображенные в
логарифмических координатах, более наглядны. На рис, II
видно, что ICN“1k1,ht составляет 0,20, 0,43 и 1,8 мг/л при концент-
рации ксантогената соответственно 5, 25 и 625 мг/л.
В трех случаях отношение [Х~]: ICN-] было равно 4,0, 9,4 и 57
(очевидно, это отношение не постоянно). С другой стороны, инте-
ресно отметить, что отношение [X-]: [CN42 (концентраций, выра-
женных в мкмоль/л) более или менее постоянно, составляя 0,52,
!0,57 и 0,82 соответственно загрузкам ксантогената 5, 25 и
625 мг/л. Полученные данные достаточны для того, чтобы сде-
лать определенные выводы.
Уорк и Кокс также измерили критические концентрации ио-
на цианида для различных собирателей при одной той же кон-
центрации их (0,155 ммоль) для одного и того же минерала
(халькопирит). Полученные результаты представлены на рис.
12. На этом рисунке показано, что для каждого собирателя суше-
•ствует своя критическая концентрация иона цианида. В порядке
возрастания собирательного действия испытанные реагенты мо-
гут быть расположены в следующий ряд: диэтилдитиофосфат
натрия, этиловый ксантогенат калия, диэтилдитиокарбамат на-
трия, амиловый ксантогенат калия и диамилдитиокарбамат
калия. Ожидаемое постоянство [CN-] р , характерное для каж-
дого минерала, не всегда соблюдается. Тем не менее в присут-
ствии различных собирателей можно принять следующие значе-
ния для [CN-]крнт (в ммоль/л):
Диэтил дитиофэсфгт . . •.............3 • 10~8
Этиловый ксантогенат....................1,6- 10-5
Диэтилдитиокарбамат...................5 • 10-5
Амиловый ксантогенат ................<3 • 10— 5
Диамиловый дитиокарбамат.............8 • 10“4
При этом концентрация собирателя составляла 0,155-
• }0~3ммоль/л.
Если бы ион цианида был единственным депрессором (как в
случае кривых на рис. 12), то каждая кривая при разных значе-
ниях pH должна бы представлять собой горизонтальную линию.
Однако можно заметить, что все кривые загибаются книзу при
высоком pH и кверху при низких pH. Повышение при низких
значениях pH концентрации [CN_1, требующейся для предотвра-
щения прилипания пузырька воздуха к минералу, возможно за
счет потери цианида в виде HCN, образующейся при перемеши-
вании и аэрации системы. Понижение [CN“]KpHT с повышением
pH, показывает, что ион гидроксила при высоких концентрациях
является дополнительным депрессором.
Роль комплексных цианидов металлов. Хорошо известно, что
металлы подгруппы меди (медь, серебро, золото) образуют не-
Величина pH
Рис. 10. Влияние концентрации
лового ксантогената калия на
вые прилипания для халькопирита
[46]:
1 — 5 мг/л этилового ксантогената калия:
2 — 25 мг/л', 3 — 625 мг/л
Величина pH
Рис. 11. Критическая концент-
рация иона цианида, препятст-
вующая закреплению на халь-
копирите пузырька воздуха в
водных растворах этилового
ксантогената калия различной
концентрации [45]:
/—5 мг/л этилового ксантогената
калия: 2 — 25 мг/л\ 3 — 645 мг/л
эти-
кри-
Рис. 12. Критическая концентрация иона цианида на
халькопирите для различных собирателей (0,155 •
- 10~6-м.) [45]:
1 — диэтилдитиофосфат натрия: 2 — этиловый ксантогенат ка-
лия; 3 — диэтилдитиокарбамат натрия; 4 — н-амиловый ксанто-
генат калия; 5 — н-амиловый днтиокарбамат калия
23 А. М. Годэи
354
ГЛАВА 10
растворимые простые цианиды, растворяющиеся лишь в избытке !
водного раствора цианида с образованием воднорастворимых
«двойных цианидов», в которых тяжелый металл находится в ви- j
де комплексного аниона. Так, серебро [21, 30] образует харак- ।
терный комплексный аргентоцианид калия K+Ag(CN)2_. Кон- i
станта диссоциации комплексного иона [21, 30] порядка 10'21
Ag(CN)? —Ag+ +2CN~ [Ag~] ]2.-= IO-21 . (6) ;
[ Ag (CN)2 j ।
Уравнение (6) показывает, что при большом количестве циа-» I
нида концентрация свободного иона серебра обратно пропорцио- I
нальна квадрату концентрации свободного иона цианида. Кон- i
центрация свободного иона серебра ничтожна, когда имеется из-
лишек свободного иона цианида. Так, если общая концентрация :
серебра равна 10~} и [CN-] = 10~5, то концентрация свободного i
иона серебра будет около 10-15. ;
Подобно этому образуется и комплексный цианид меди, ко- [
' торый можно рассматривать как соединение CuCN с CN-. Как г
сообщают 12, 43], в твердом состоянии имеется еще несколько [
двойных цианистых солей калия и одновалентной меди [таких, [
как К2Си(СП)3 и КзСи(СМ)4], но в водном растворе устойчив *
только одновалентный анион дицианида одновалентной меди [4]. j
Эта точка зрения не согласуется с прежними литературными дан- [
ными [25], в которых говорится о существовании и важном значе- ]
: нии комплексов Cu(CN)2- и Cu(CN)4~ Однако, по-видимому, t
] устаревшие представления еще имеют приверженцев, поскольку |,
их принимает Сиджвик в своей недавно вышедшей в свет кни- Г
ге [41].
Если к раствору соли двухвалентной меди добавить цианид
щелочного металла, то сначала осаждается цианид двухвалент-
ной меди. Но анион вскоре восстанавливает катион, при этом , j
цианид двухвалентной меди переходит в цианид одновалентной I
меди и образуется дициан (CN)2. После добавления большого .
количества цианида щелочного металла осадок растворяется, .
так как образуется комплексный циановый анион одновалентной
меди. В целом реакция может быть написана следующим обра- ;
зом:
Cu2+ + 3CN“ ->Cu (CN)F + 0,5 (CN)2- (7)
Образование растворимых медно-циановых комплексов. При
перемешивании медного купороса с водой и цианистым натрием
в образующемся растворе медь представлена ионами одно- и
двухвалентной меди и медно (1) циановыми ионами, в то время
как группа CN встречается в виде иона цианида, синильной кис-
лоты, комплексного медно (1) цианового иона и дициана. Теоре-
ДЕЙСТВИЕ МОДИФИКАТОРОВ ФЛОТАЦИИ
355
тически, зная константу диссоциации комплексного иона, кон-
станту диссоциации синильной кислоты и окислительно-восстано-
вительный потенциал для реакции взаимодействия одновалент-
ной меди с цианидом, можно показать распределение Си и CN
между различными формами (в которых они встречаются) в за-
висимости от их общего содержания, так же как и в зависимости
| от концентрации водородных ионов. Уравнения эти довольно
[трудно решить, но они могут оказаться полезными для специаль-
|ных случаев и служить иллюстрацией для решения задач подоб-
ного рода, а поэтому ниже приводится их вывод.
Допустим, что А молей меди в виде сульфата прибавляемой
к В молей цианида в виде цианистого натрия и добавляется вода
в количестве, необходимом для получения 1 л раствора. Допу-
стим также, что при этом не образуется твердая фаза, содержа-
щая медь или цианид. Обозначим через х, у и z концентрации
иона одновалентной меди, иона двухвалентной меди и медно (1)
цианового иона [Cu(CN)2~]. Тогда баланс по меди можно напи-
сать так:
A = x + yA-z. (8а)
Далее обозначим через и, v и w концентрации иона цианида,
синильной кислоты и дициана C2N2. Тогда баланс по цианиду
можно написать следующим образом:
В = и + v + 2w + 2г. (86)
Коэффициент 2 перед w и г поставлен потому, что в дициан и
медно(1)циановый ион входит по две группы CN. Так как ион
двухвалентной меди восстанавливается до иона одновалентной
меди и в результате этого восстановления происходит окисление
иона С.\ь до дициана, то количество окислившихся ионов
СН_(2а>) равно количеству ионов двухвалентной меди, вое-'
становленных до ионов одновалентной меди (встречающейся
в виде свободных ионов меди и комплексных анионов), или
2w = х + г. (8в)
В результате гидролиза цианистого натрия получается си-,
нильная кислота, зная константу диссоциации которой [см. урав-
нение (5)], можно написать:
(8г)
v [Н+].
Латимер 129а — с] вычислил, что константа нестойкости мед-
но (1) цианового комплекса, согласно диссоциации по уравнению
Cu(CN)7-^Cu + +2CN~, составляет около 10~16. Это вычисление
основывалось на старых измерениях Спицера, выполненных в
1905 г. Однако, согласно последней работе Владимировой и Ка-
23*
356
ГЛАВА 10
ковского [44], величина константы нестойкости гораздо ниже и
составляет 2-10-24. Эта величина получена в результате тщатель-
но проведенных измерений, поэтому она и принимается для
настоящих расчетов. Отсюда
— = 2-10-24. (8д)
г
Латимер дает следующие значения потенциалов реакций окис-
ления иона одновалентной меди и синильной кислоты:
Cu+ = Си2+ + ё~ Е° =—0,153 в;
HCN = 0,5 (C2N^ + Н+ + ё~ Е° = — 0,37 в,
пользуясь которыми, находим:
Cu2++ HCN = Cu++ Н++ 0,5(C2N2)£ Е° = — 0,217 в.
Это означает, что потенциал при фугативности дициана в 1 атм
и активности всех растворенных компонентов в 1 моль/л равен
—0,217 в. Потенциал Е° можно выразить через константу рав-
новесия для этой реакции в виде
Е° = 0,059 п~' 1g К,
где п — количество чисел Фарадея для данной реакции ( в дан-
ном случае п = 1);
д- [Си+] [Н+] [(CN)2]‘/*
[Cu2+J[HCN]
откуда
—0,217 — 0,059 1g ^ИД*НД1'''
|Cu!+j [HCNj
или
!н+] - = 2,1 . 10~4. (8e)
yv
Как отмечено выше, решение системы из шести уравнений
возможно, но весьма трудоемко. В отдельных случаях с доста-
точным приближением можно получить ответ упрощенным спо-
собом.
Так, при значительном преобладании цианида над медью
можно получить простое выражение для свободных ионов одно-
валентной и двухвалентной меди. При этом В> А, и мы можем
заместить А на z в уравнении (8д), тогда:
х = 2- 10-24-А_. (9а)
Приняв приблизительно А = z и пренебрегая х в сравнении с
г, из уравнения (8в) найдем w = А[2. Подставляя это значение
ДЕЙСТВИЕ МОДИФИКАТОРОВ ФЛОТАЦИИ
357
w, а также х из уравнения (9а) и а из уравнения (8г) в уравне-
ние (8е), получим
у=З.Ю-30^. (96)
и2 '
Если А = 10”4-м., В = 10-2-м. и и = 10~3-м., то х — 10-22 и
у = 10~27, т. е. концентрации свободных ионов одновалентной и
двухвалентной меди исчезающе малы.
Уравнения (9а) и (96) показывают, что в присутствии избыт-
ка цианида концентрация свободного иона одновалентной меди
изменяется пропорционально общей концентрации меди в раст-
воре и обратно пропорционально квадрату концентрации иона
цианида; концентрация иона двухвалентной меди изменяется так
же, как общая концентрация меди, в степени 3/2, и обратно про-
порционально кубу общей концентрации иона цианида.
Значение этих выводов заключается в том, что они позволяют
лучше объяснить результаты флотационных опытов. Так, напри-
мер, можно доказать, что кривые критических концентраций,
рассмотренные выше, в случае медных минералов являются кри-
выми равных концентраций не только иона цианида, но также
и кривыми равных концентраций ионов меди. Уорк и Кокс в сво-
их ранних работах, очевидно, предполагали это, но они не ис-
пользовали своих соображений при рассмотрении вопросов регу-
лирования флотации. Рис. 13, воспроизведенный из их работы,
показывает поразительное сходство между кривыми критических
концентраций и кривыми концентрации ионов меди.
Концентрация ионов меди, по-видимому, определялась с по-
мощью концентрационной цепи, но так как никаких указаний на
это в работе Уорка и Кокса не приводится, то эти данные и метод
должны быть проверены.
Ранее было отмечено, что критические условия для флотации
халькопирита в системе ксантогенат—цианид таковы, что отно-
шение концентрации иона ксантогената к квадрату концентра-
ции иона цианида остается постоянным. Так как мы установили,
что концентрация иона одновалентной меди обратно пропорцио-
нальна квадрату концентрации иона цианида, то при критиче-
ских условиях флотации должно наблюдаться постоянство про-
изведения ионных концентраций [Cu+] [X-]. Это, очевидно, не оз-
начает, что такое произведение должно превышать произведение
растворимости цианида одновалентной меди, но оно должно быть
равно вполне определенной доле произведения растворимости
этого соединения. Другими словами, можно ожидать, что во всех
случаях, когда произведение концентраций ионов будет состав-
лять определенную долю произведения растворимости, плотность
адсорбционного покрытия будет одинаковой.
Интересно сделать приближенную оценку произведения ион-
'358
ГЛАВА 10
ных концентраций. Для халькопирита в присутствии 5 мг/л эти-
лового ксантогената калия критическая концентрация иона циа-
нида равна 0,2 мг/л (см. рис. 11). Это значит, что и = (0,2- 10-3)
26 = 8 -10~6-м., так что при общем содержании меди в растворе
Рис. 13. Сопоставление кривых прилипания пузырь-
ка к халькопириту с кривыми постоянных концент-
раций иона меди (сплошные линии обозначают кри-
вые концентрации иоиов меди; пунктирные линии —
кривые прилипания к халькопириту пузырька возду-
ха) (по Уорку и Kokcv, AIMME Trans., 112, 256,
1934):
/—концентрация Cu2 + = 10—2—то же. 10 3 — то
же. 10 24; 4 — то же. 10 — 23; 5 — то же. 10**27; 6 — то же,
10—30;7 —то же, 10~30-« —то же, 10 ~32;9 — халькопирит
(принятом Уорком для своих опытов), равном 10-7-м., коцентра-
ция свободного иона одновалентной меди должна была со-
ставлять около З-Ю-21. С другой стороны, концентрация ксанто-
гената 5 мг/л приближенно равна 3-10~5-м., и, следовательно,
критическое произведениеконцентраций ионов [Си4*] [Х~] состав-
ляло около 10~25.
Изложенное выше объяснение механизма депрессии сульфид-
ных минералов (и особенно медьсодержащих сульфидных мине-
ралов) цианидами связано главным образом с конкурирующей
адсорбцией на минерале собирателя и иона цианида, а также
с удалением с поверхности твердого адсорбированных ионов ме-
ДЕЙСТВИЕ МОДИФИКАТОРОВ ФЛОТАЦИИ
359
ди и переходом их в раствор в виде комплексного иона. Какое
именно из этих явлений происходит в действительности, не было
установлено. Возможно, что наблюдается и то и другое наряду с
адсорбцией комплекса. Эти проблемы заслуживают дальнейшего
изучения.
Комплексные цианиды других металлов. Хотя действие циа-
нида на сульфидные минералы рассматривалось лишь на приме-
ре сульфидов меди, очевидно, что сделанные выводы могут быть
распространены и на другие металлы, образующие устойчивые
комплексы, а в особенности на железо.
В табл. 8 представлены константы диссоциации комплексных
цианидов различных металлов, а в табл. 9 даны отношения кон-
центрации свободного катиона металла к концентрации комп-
лексного аниона при различных концентрациях цианида в ра-
створах, применяемых на практике. Одновалентных ионов золота,
меди и ртути практически не существует в растворе даже и в том
случае, когда концентрация иона цианида равна Ю"8. Серебро
занимает промежуточное положение. Содержание ионов железа
в растворе сильно зависит от концентрации цианида. При умерен-
ной концентрации иона цианида концентрация свободных ионов
двухвалентного или трехвалентного железа весьма низка, но при
малой концентрации иона цианида она может быть значительной.
Кадмий, цинк и в некоторой степени никель присутствуют преи-
мущественно в виде свободного катиона даже при сравнительно
большой концентрации иона цианида.
Таблица 8
Константы диссоциации комплексных анионов цианидов металлов1
на свободный катион металла и анион цианида
Ag (CN)2 1,8-10“19 Fe (CN)§“ 10-35
Au (CN)^* 5-10“39 Hg (CN)2' 4-10“42
Cd (CN)2- 1,4.10“19 Ni (CN)2- 10“22
Cu (CN)7 2-10“24! Zn (CN)V l,2-10“18
Fe(CN)3- 10“42
1 В основном по Латимеру [29].
2 По Владимировой н Каковскому [44]
Интересно более детально разобрать пример с железом. До-
пустим, имеется сульфидная пиритсодержащая руда, в которой
железный купорос находится в количествах, достаточных для об-
разования в растворе 10-3-м. концентрации иона двухвалентного
360
ГЛАВА 10
Таблица 9
Приблизительное отношение концентраций свободного катиона
металла и комплексного аниона металла как функция концентрации
иона цианида
CN-
Металл
10_8 io-7 10~6 IO—5 1 0-4 10—3
Ag+ io-2 io-4 10-6 10~8 IO-10 10-12
Au+ IO-20 IO-22 IO-24 Ю-26 10-23 IQ-ЭО
Cd2+ Большое IO-3 10-7
Cu+ 10~8 Ю-ю io-12 IO-14 10-16 10~18
Fe2 + Большое 10~5 io-11 IO-17
Fe3+ Большое 1 IO-6 io-12 10-18 10~24
Hg2+ IO-9 io-'2 io-'7 IO-21 10-25 10-29
Ni2 + Большое 10~2 10~6 10-Ю
Zn2+ Большое 1 io-2 IO-6
железа (это эквивалентно загрузке 0,5 ка/г железного купороса
при Ж : Т = 4 : 1).
Из табл. 9 следует, что при [CN~], равной 10-5, отношение
[Fe2+] : [Fe(CN)4-6] = 10-5; следовательно, [Fe2+] составляет около
IO-8.
Произведение растворимости гидроокиси двухвалентного же-
леза равно 1,8 • 10~15; поэтому если [ОН-] не превышает 4- Ю^-м.,
то железо присутствует в растворе преимущественно в виде комп-
лекса. При отсутствии иона цианида гидроокись будет осаждать-
ся в том случае, если [ОН-] превысит 10-6-м.
Если то же самое количество железного купороса окислится
до соли трехвалентного железа, то отношение IFe3+] :
: [Fe(CN)3-6] = 10-12, следовательно, [Fe3+] = 10-15.
Так как произведение растворимости гидроокиси трехвалент-
ного железа [Fe3+] [ОН-]3 = 6-10-38, то, если ЮН-] не превышает
0,93 • 10-7, железо останется полностью в растворе, но не в виде
свободного трехвалентного железа. Эти примеры показывают,
как, изменяя концентрацию иона цианида, можно регулировать
концентрации в растворе ионов двухвалентного и трехвалентного
железа.
Интересно также разобрать пример, когда имеется два метал-
ла, способных образовать различные цианистые комплексы. Важ-
ное практическое значение имеет одновременное присутствие ме-
ди и железа. Так как оба этих металла двухваленты, то задача
осложняется. Поэтому рассмотрим более простые условия, когда
ДЕЙСТВИЕ МОДИФИКАТОРОВ ФЛОТАЦИИ
361
присутствуют серебро и железо, причем железо остается двухва-
лентным. Если общее количество двухвалентного железа в рас-
творе 10'3-м., общее количество серебра 10-4-м. и добавлено
достаточное количество цианида при подходящей щелочности,
чтобы получить концентрацию свободного иона цианида 10~3-м.,
тогда количество' свободного иона железа будет 10-3 • 10-17, или
10—20, в то время как концентрация свободного иона серебра будет
10-4 • 1042, или 10-1е. Тогда отношение [Ag+]: [Fe2+]= 10~16:
: Ю-20 = 10+4. Если же конечная концентрация свободного ио-
на цианида составляет только 10~5-м., концентрация свободно-
го иона двухвалентного железа будет 1Q-3 • 10~5, или 10~8, в то
время как концентрация свободного иона серебра 10-4 • 10~8, или
10-12, так что
1Ag+] = 1Q-4
[Fe1 2+j
Таким образом, 100-кратное изменение [CN-] от 10-3 до 10~5-м.
дает 100 000 000-кратное изменение отношения концентраций сво-
бодных ионов серебра и двухвалентного железа. Даже неболь-
шое изменение pH, небольшое изменение количества добавленно-
го цианида щелочного металла могут привести к значительному
изменению отношения [Ag+] : [Fe2 + ].
Эти рассуждения справедливы и при рассмотрении совместно-
го присутствия меди и железа, однако здесь необходимо учиты-
вать изменения валентностей металлов и происходящие в при-
родных системах окислительные процессы.
Сульфиты и другие сульфоксидные соединения как депрессоры
для сульфидных минералов
Различные сульфоксидные соединения применялись при се-
лективной флотации сульфидных руд для избирательной депрес-
сии некоторых минералов. Механизм их действия с химической
точки зрения не изучен в достаточной мере.
Сульфиты щелочных или щелочноземельных металлов посто-
янно используются для разделения свинцовых и цинковых мине-
ралов на фабрике Мидвэйл Ю. С. Смелтинг, Рифайнинг энд Май-
нинг Компани (Мидвэйл, Юта) 132—35]. Гидросульфиты щелоч-
ных металлов или гидросульфиты цинка (не смешивать с би-
сульфитами или кислыми сульфитами) были предложены в Гер-
мании* и применялись там с некоторым успехом для разделения
свинцовых и цинковых минералов.
В Чапмэн Кэмп (Британская Колумбия) было признано по-
лезным депрессировать двуокисью серы марматит в цинковых
1 По частной информации У. Хиршкиида, вице-президента Доу Кемикл
Компани (Питтсбург, Калифорния).
362
ГЛАВА 10
концентратах, содержащих пирротин и галенит (получаемых
обычным способом с применением сульфата меди и ксантогената
щелочного металла).
Эта операция, осуществляемая при pH = 4, в условиях из-
бытка двуокиси серы и при дополнительной подаче ксантогена-
та дает прекрасное разделение. Попытка осуществить этим ме-
тодом депрессию светлоокрашенного сфалерита из цинкового
концентрата на фабрике Джоплин (не содержащего сульфидов
железа и свинца) не дала положительных результатов. Меха-
низм депрессирования марматита остался невыясненным.
Двуокись серы как восстановитель. Одной из возможных
функций двуокиси серы при депрессировании активированного
и сфлотированного сфалерита является уменьшение концентра-
ции иона двухвалентной меди. Это может быть показано с по-
мощью реакций окисления — восстановления (по Латимеру)
Н2О + H2SO3->SO1“ 4Н+ 4-2е“ £° = —0,170;
Cu+-*Cu2+ + e~ Е° = —0,153.
Вычтем второе уравнение из первого и получим
Н2О + H2SO3 + 2Cu2+ ->SO1~ + 4Н+ + 2Cu+ Е° = —0,017. (10)
Константа равновесия для этого уравнения может быть рассчи-
тана из величины Е° следующим образом:
—0,017 = (0,059) 1g Д,
откуда К = 0,266.
Преобразуя соотношение констант равновесия, получаем:
ГСц+] =0,52_____|Н^Оз11/‘ . (11)
[Си2+] [Н+]2 [SG2~
Поскольку константы диссоциации сернистой кислоты известны,
можно рассчитать концентрации [SO^-], [HSQ^], [H2SOj для
данного общего количества загруженной SO2. Если на 1 л раст-
вора задается 0,03-м. SO2 и если величина pH равна 4, то, обо-
значая через х, у м z соответственно концентрации НгЭОа, H2SO3~
и SO32-, имеем;
0,03 = х + у г; j
10“4 • = 1,25-1 о-2;
х
10-4 • — =5,6 . 10~8
X ’ >
ДЕЙСТВИЕ МОДИФИКАТОРОВ ФЛОТАЦИИ
363
для которых значение х = 0,00024-м. Подставляя эту величину
в уравнение (10) и принимая, что общая концентрация сульфит-
иона равна 4-Ю-2, мы получаем
[Cu+l— = 4 • 10е.
[Си2+]
Очевидно, что концентрация одновалентной меди значительно
превышает концентрацию двухвалентной меди. При этих услови-
ях медь легко выпадает в осадок и таким образом удаляется из
системы. Часть меди может восстанавливаться до металличе-
ской. Это подтверждается равновесием между -двухвалентной,
одновалентной и металлической медью
Cu2+ + Cu-»Cu+ К = (Си+|2 =6,3-10~7. (13)
1Си2+1
Сопоставляя эту константу равновесия с отношением однова-
лентной и двухвалентной меди, найденным для системы, содер-
жащей 0,03-м. двуокиси серы на 1 л, мы получим Си+= 1,6-10~13
и Си2+ = 0,4 • 10-19.
Отсюда может быть сделан вывод, что если исходное содержа-
ние меди в системе значительно превышает 10чз-м., то большая
часть меди должна восстановиться до металлической.
Если в системе не достигнуто значение pH, равное 4, которое
может быть получено, например, за счет реакции пирротина с
сернистой кислотой, и pH не превышает 2, то концентрация иона
двухвалентной меди вследствие осаждения металлической меди
достигает примерно 10-13 вместо 10~19. Наоборот, если pH повы-
шается до 5 или 6 за счет продолжающейся реакции между пир-
ротином; и сернистой кислотой, то конечная концентрация иона
двухвалентной меди падает ниже 10~19.
Эффективность сульфита как депрессора для сфалерита вы-
зывала много сомнений. Новейшие неопубликованные исследова-
ния показали, что этот реагент представляет ценность для разде-
ления галенита и сфалерита в условиях, когда цианид мало эф-
фективен, т. е. для руд, в которых цинковые минералы или сов-
сем не содержат железа или имеют малое его содержание. На-
оборот, для руд с высоким содержанием марматита цианид пред-
ставляется более эффективным депрессором, особенно потому,
что он активно воздействует также и на железные минералы.
Но результаты этих наблюдений противоречат тому, что было
сказано выше о действии двуокиси серы при pH = 4. Возможно,
что изменение pH от 4 до 8 приведет к каким-либо иным химиче-
ским взаимодействиям.
В целом проблема механизма депрессирующего действия
сульфоксидных соединений требует проведения детальных иссле-
дований, которые связаны с большими трудностями.
364
ГЛАВА 10
Сульфоксидные соединения. Составление перечня сульфо-
ксидных соединений может быть полезным [10], так как, помимо
применения в качестве депрессоров, они могут встречаться в
практике флотации вследствие их самопроизвольного образова-
ния в аэрируемых пульпах сульфидных минералов-
Сиджвик [41] называет шесть окислов серы, а именно: SO,
SO2, S2O3, SO3. S2O7 и SO4, из которых второй и четвертый пред-
ставляют общеизвестные химические вещества. Имеется также
по меньшей мере двенадцать кислород-и серусодержащих кис-
лот, как показано в табл. 10.
Кислоты № 2, 5 и 6 общеизвестны. Сульфоксиловая кислота
(№ 1) —настолько сильный восстановитель, что не встречается
в свободном виде и известна лишь в виде нескольких солей. Сер-
новатистая кислота (№ 3), которая может быть получена при
конденсации кислот № 1 и 2, также является сильным восста-
навливающим агентом и используется для этой цели при краше-
нии. Кислота № 4 известна лишь в форме эфиров; соли ее раз-
лагаются сразу же при образовании. Кислоты № 7—10 полу-
чаются обычными способами, но их химические свойства недо-
статочно изучены, вследствие чего эти соединения трудно опре-
делимы. Кислоты № 11 и 12 образуются только в присутствии
сильных окислителей и могут существовать в пульпах сульфид-
ных руд лишь в течение весьма непродолжительного времени.
Комплексные сульфоксидные соединения. Применение суль-
фидов и тиосульфатов может обусловливаться их способностью
к образованию комплексных соединений со многими металлами
[42]. Ниже приводятся уравнения диссоциации комплексного
тиосульфата серебра и комплексного сульфита серебра [29]:
Ag (S2O8 Ag++ 2 (S2O3 )2- К = 6,0- КГ14; (14)
Ag(SO3 )23--> Ag+ + 2 (SO3 )2“ K=3-10-9. (15)
Приведенные величины констант диссоциации свидетельст-
вуют об относительной устойчивости данных комплексов. Сидж-
вик установил, согласно Вернеру [4, 9], что сернистая кислота
образует большое количество комплексных соединений, особен-
но со следующими металлами: Мп, Fe, Со, Ni, Си, Zn, Ru, Rh,
Pd, Ag, Cd, Os, In, Pt, Au, Hg.
Комплексные соединения co ртутью, кобальтом и платиной
весьма устойчивы. Некоторые из них имеют по меньшей мере
внутрикомплексную связь атома металла с серой.
Комплексные тиосульфаты образуются многими металлами,
особенно серебром, свинцом, ртутью, медью (одновалентной, но
не двухвалентной), золотом.
действие модификаторов флотации
365
Таблица 10
Кислород и серусодержащие кислоты
м по пор. Кислота Формула
1 Сульфоксиловая .... он s\ он он.
2 Сернистая . O=s/ он S-OH
3 Серноватистая O=S / он SH
4 Тиосернистая O=s/ он о /ОН
5 6 Серная Тиосерная S ХОН 0^ /SH S о^ 4 он 0 о 1! II НО—S—Sm—S—он*
7—10 Политионовые II II о о но-о о \ //
И Мононадсерная S но ° 0 О О 0 ч //
12 Надсерная S S НО^ ^о-о"7 ХОН
* Здесь т изменяется от 0 до 3.
366
ГЛАВА 10
Известно, что сульфит, задаваемый в пульпы сульфидных
руд, расходуется очень быстро, но на что именно он расходует-
ся, не установлено. Можно предположить, что он окисляется
растворенным кислородом либо непосредственно, либо катали-
тически на поверхности минералов.
Активация сфалерита медью
В противоположность изменению собирательного действия
под влиянием депрессоров, направленному на ухудшение фло-
тируемости минералов, действие собирателя может усиливаться
в присутствии некоторых других реагентов. Типичным примером
этого служит применение сульфата меди для повышения флоти-
руемости сфалерита ксантогенатом калия.
Необходимо напомнить, что относительно крупно измельчен-
ный сфалерит почти не флотирует при загрузке этилового ксд,н-
тогената калия (см. рис. 8). При тонком измельчении сфалерита
достигается несколько большее извлечение минерала, и это
представляет одно из самых больших затруднений при селектив-
ной флотации в промышленных условиях.
Если такая медная соль, как сульфат меди, подается в со-
держащую сфалерит щелочную или нейтральную пульпу и на-
ходится в контакте с минералом в течение нескольких минут,
то флотационная активность минерала сильно возрастает.
При флотации относительно крупных частиц загрузка суль-
фата меди вызывает резкий переход от практически полного от-
сутствия флотации к полной флотируемости. На рис. 14 приве-
дены некоторые экспериментальные данные, полученные много
лет назад при флотации сфалерита крупностью —100 + 150 меш.
Если материал относительно тонко измельчен, то переход от не-
активированного состояния к состоянию активации происходит
более постепенно, но тем не менее изменение флотируемости
остается весьма заметным.
Описанные ниже опыты показывают, что медь поглощается
сфалеритом в обмен на цинк.
При этом происходит следующая реакция:
Cu2+ + |ZHS|2n2+-^|ZHS|Cu2+ + Zrf". (16)
Эта реакция сильно смещена в правую сторону, и можно допу-
стить, что она идет до полного поглощения меди минералом.
Интересно отметить, что имеется количественное соотноше-
ние между числом активных поверхностных точек на минерале
и количеством меди, которое дает резкое возрастание извлече-
ния (см. ось абсцисс на рис. 14). При расчетах принимается,
что вся медь поглощается минеральной поверхностью, что по-
OS I
ДЕЙСТВИЕ МОДИФИКАТОРОВ ФЛОТАЦИИ
367
верхность сфалерита состоит из тетраэдрических плоскостей
(НО) и что удельная поверхность минерала равна 480 с.и2/г.
Эта величина удельной поверхности была выбрана на осно-
вании измерений поверхности двух классов сфалерита крупно-
стью —65 +150 меш. (368 см2/г) и —150 +200 меш. (698 сл2/г),
произведенных по методу БЭТ с применением криптона в каче-
стве адсорбируемого газа.
На единицу поверхности 5,42 • 7,66 ~ 41,6 А2 тетраэдриче-
ской плоскости сфалерита приходится два катионных места
(рис. 10—15).
Если допустить, что два расположенных на поверхности
атома цинка могут замещаться медью и что, кроме того, ионы
меди могут закрепляться у атомов серы, находящихся на по-
верхности, возможное количество адсорбированной меди воз-
растет до четырех ионов на каждые 41,6 А2. При таком допуще-
нии при наиболее плотной упаковке один ион меди должен при-
ходиться на каждые 10,4 А2. На основании этого число атомов
меди, необходимое для образования плотного монослоя, состав-
ляет 480/10,4 • 10~15 для каждого грамма минерала. Подставляя
число Авогадро и молекулярный вес пятиводного сульфа-
та меди, определяем величину равной 0,19 мг/г минерала или
0,19 кг/т. На рис. 14 показано, что максимальное извлечение
достигается при покрытии, составляющем приблизительно поло-
вину теоретически рассчитанного плотно упакованного моно-
слоя.
Уравнение (16) было выведено двадцать пять лет назад
[15, 37]. Детальное исследование, произведенное в последнее
время Д. Мао в Массачусеттском технологическом институте,
дало много новых сведений о поглощении меди. Это исследова-
ние также подтвердило наличие стехиометрического соотноше-
ния между поглощенными минералом ионами меди и выделен-
ными в раствор ионами цинка.
Адсорбция меди сфалеритом. Адсорбция меди сфалеритом
была измерена как в опытах с перемешиванием отдельных на-
весок в постоянном объеме раствора, так и в опытах с непрерыв-
ным протеканием медьсодержащего раствора через небольшой
слой сфалерита. В опытах с отдельными навесками навеску
минерала перемешивали с определенным объемом раствора в
течение продолжительного времени, затем минерал отфильтро-
вывали и центрифугировали, после чего определяли содержание
в нем меди.
В опытах с непрерывным протеканием раствора этот раствор
проходил через цилиндрический слой минерала. Прошедший
сквозь слой минерала раствор непрерывно анализировали на
Рис. 14. Флотация чистого сфалерита. Материал
крупностью —100+150 меш флотировался ксантоге-
натом 0,045 кг)т в присутствии терпинеола 0,09 кг/т,
карбоната натрия 0,09 кг/т я сульфата меди в пере-
менных количествах:
/ — флотация в течение 3 мин,; 2 — флотация в течение
8 мин.
Рис. 15. Сечение кристалла сфа-
лерита по плоскости (НО). Пло-
щадь ячейки 41,6 А2 [9]:
I — цинк на поверхности,; 2 — цинк, рас-
положенный в следующем, нижнем, ря-
ду; 3 — сера
ДЕЙСТВИЕ МОДИФИКАТОРОВ ФЛОТАЦИИ
369
содержание меди. Эти опыты показали, что сначала вся медь,
содержащаяся в растворе, полностью поглощалась минералом,
затем поглощение начинало уменьшаться, пока не прекращалось
полностью, что указывало на практически полное насыщение
медью поверхности минерала. Для анализа применялась Си84 —
радиоактивный изотоп с периодом полураспада 12 час. На
рис. 16 представлены типичные результаты, полученные в опы-
тах с отдельными навесками и определенными порциями рас-
твора. На рис. 17 приводятся данные, полученные в опытах с
непрерывным протеканием раствора.
На нижней половине рис. 17, так же как и на рис. 16, пока-
зано максимальное количество меди, которое может адсорбиро-
ваться при данной температуре.
Поскольку измеренная удельная поверхность навески сфале-
рита составляла 698 см21г, для образования плотного монослоя
ионов меди потребуется 698- ионов, или 11,14 •
10'7 г-ион меди на 1 г сфалерита.
Экспериментальные данные показывают, что в обоих случаях
почти половина меди адсорбировалась при условиях максималь-
ного поглощения, что составляло 16,5 10'7 г-ион!г минерала.
Дальнейшие эксперименты установили, что максимальная
величина адсорбции мало зависит от концентрации иона двух-
валентной меди, заметно зависит от температуры и прямо про-
порциональна величине поверхности минерала.
Влияние концентрации иона двухвалентной меди было уста-
новлено в нескольких опытах, поставленных на сфалерите круп-
ностью —65 +150 меш.
Этот материал имел удельную поверхность, равную 368 см2)г,
определенную адсорбцией криптона (по методу БЭТ). Для об-
разования на этой поверхности плотного монослоя потребова-
лось бы 5,9 • 10-7 г-ион меди на 1 г минерала.
Для каждого опыта брались отдельные порции раствора или
его непрерывно пропускали через цилиндрический слой мате-
риала.
В результате было установлено, что максимальное поглоще-
ние меди составило 8,75 • 10~7 г-ион (8,8 • 10~7 в опытах с от-
дельными порциями раствора и 8,7 • 10~7 при непрерывном про-
текании раствора); исходная концентрация раствора сульфата
меди равна 1,2 мг/л при pH = 4,9. В параллельных опытах, в
которых в исходный раствор добавлялся аммиак в количестве,
достаточном для создания общей концентрации аммиака
0,008-м. при pH =10,4, адсорбция меди понижалась до
7,85 • 10~7 г-ион)г минерала (7,8 при опытах с непрерывным
протеканием раствора и 7,9 в опытах с отдельными порциями
24 А. М, Годэи
Равновесная концентрация меди, г+ион/л-
Рис. 16. Адсорбция иона двухвалентной меди на сфале-
рите (—150+200 меш)
Ю6
Рнс. 17. Адсорбция иона
двухвалентной меди на
сфалерите из раствора,
непрерывно протекающе-
го через слон минерала
толщиной Ю см. Исход-
ный раствор содержал
1,22 .иг сульфата меди в
I л; pH = 4,9; скорость
протекания 12,3 мл/мин
через слой с площадью
сечення 0,5 см2
ДЕЙСТВИЕ МОДИФИКАТОРОВ ФЛОТАЦИИ
371
раствора). В опыте же, в котором концентрация аммиака была
увеличена в восемь раз, т. е. до 0.064-м., при непрерывном про-
текании раствора адсорбция понизилась до 6,0 • 10~7 г-ион)г
минерала. Концентрация иона двухвалентной меди в растворе,
насыщенном аммиаком, была, конечно, много ниже, чем в пер-
вом случае, поскольку большая часть меди в этих условиях при-
сутствует в виде комплекса Cu(NH3)4+
Латимер [29е] определил величину константы диссоциаций
медно-аммиачного комплекса, равной 4,7 • 10~15:
Cu(NH3)4+ --Cu2+ + 4NH3(aq). (17)
По величине этой константы можно судить о том, что вслед-
ствие заметной концентрации аммиака в растворе отношение
концентрации меди, находящейся в комплексном соединении, к
свободной меди очень велико.
Так, при [NH3] = 1СН отношение концентрации меди, входя-
щей в комплекс, к концентрации ионов свободной меди превы-
шает 200.
Опыты адсорбции меди, проведенные в присутствии аммиака,
показали, что при этом происходит адсорбция комплексного
медно-аммиачного иона.
Влияние температуры исследовалось предварительно в опы-
тах с отдельными порциями раствора, причем сфалерит раз-
личной крупности обрабатывался раствором сульфата меди при
температурах от 25 до 60°. В этих опытах количество меди,
содержащейся в данном объеме раствора, значительно превы-
шало максимальное количество, которое могло бы быть адсор-
бировано минералом. Отношение между количествами меди,
поглощенными минералом при 25 и 60° (при продолжительном
контакте), по данным пяти серий опытов, поставленных на ми-
нерале крупностью от 65 до 400 меш, получено в среднем рав-
ным 2,17 (минимальная величина этого соотношения 1,99, мак-
симальная 2,24).
Если предположить, что существует два вида адсорбции
ионов меди поверхностью сфалерита, причем один из них не за-
висит от температуры (при образовании плотно упакованного
монослоя), а другой зависит от температуры, то при повышении
температуры от 25 до 60° количество меди, поглощаемой за счет
этого второго вида адсорбции, возрастает в четыре-пять раз.
Линейная зависимость адсорбции меди единицей поверхности
при 25° показана в двух сериях опытов. В одной серии количество
поглощенной меди рассчитывалось на удельную поверхность ми-
нерала, замеренную методом БЭТ с применением криптона в
качестве адсорбированного газа. Результаты этих опытов пока-
заны на рис. 18. В другой серии опытов ряд навесок сфалерита
24*
372
ГЛАВА Ю
крупностью —150 4- 200 меш, имеющего исходную удельную
поверхность 698 см2/г, измельчали в течение различного време-
ни в шаровой мельнице и для каждой навески определяли ве-
личину удельной адсорбции меди. При этом было установлено,
что увеличение количества адсорбированной меди после измель-
чения по сравнению с количеством меди, адсорбированной ма-
териалом исходной крупности, пропорционально продолжитель-
ности измельчения минерала. Этого можно было ожидать на
основании закона измельчения Риттингера.
§ 0,80
t
§ 0,70
0,50
O,W
Is
§*0,30
С а
» 0,20
S
0,10
с
Площадь поверхности Сфалерита смг/г(ВЕТ)
Рис. 18. Зависимость количества поглощаемой меди от
величины поверхности сфалерита (по Халфави, Масса-
чусеттский технологический институт, 1953):
/ — при температуре 25°; 2— при температуре 60э
Активация сфалерита другими катионами. Кроме ионов ме-
ди, ряд других катионов также активирует сфалерит. Этот во-
прос исследовался Ральстоном и Хантером [36], которые устано-
вили, что серебро и ртуть являются активаторами сфалерита,
в то время как свинец и кадмий не оказывают активирующего
действия на сфалерит. Е. Уорк и Н. Уорк [48] пополнили списки
активаторов -сфалерита, включив в него, кроме меди, серебра и
ртути, следующие металлы: платину, золото, висмут, кадмий,
свинец, церий, сурьму и мышьяк. Они сделали общий вывод, что
эффективными активаторами являются металлы, образующие
относительно нерастворимые сульфиды, в то время как метал-
лы, образующие относительно растворимые сульфиды, не оказы-
вают активирующего действия. Они также не могли установить,
является ли растворимость единственным фактором, регулирую-
щим процесс активации.
ДЕЙСТВИЕ МОДИФИКАТОРОВ ФЛОТАЦИИ
373
Детально изучался механизм адсорбции иона серебра сфа-
леритом [9, 19]. Эти исследования проводились по методу непре-
рывного протекания раствора; в опытах относительно большой
объем слабого раствора азотнокислого серебра протекал сквозь
слой минерала. При этом непрерывно производился анализ рас-
твора, прошедшего через слой минерала. Для этой цели при-
меняли радиоактивный изотоп Ag110.
Ион серебра поглощался из раствора минералом сначала
полностью, а затем в значительно меньшей степени. Анализы
прошедшего сквозь минерал раствора показали, однако, что
минерал во всех случаях поглощает некоторое количество се-
ребра из раствора.
На рис. 19 показаны результаты, полученные при различной
температуре (от 17 до 49°) для сфалерита крупностью —48
+ 65 меш. •
Для образования плотного монослоя серебра потребуется
0,39 мкмоль азотнокислого серебра на 1 г минерала (считая, что
один атом серебра занимает место каждого атома цинка на по-
верхности минерала и, кроме этого, один атом серебра распо-
лагается против каждого атома серы, находящегося на поверх-
ности сфалерита).
Очевидно, при более высокой температуре адсорбции сереб-
ра легко превышает в 15—20 раз количество, необходимое для
образования монослоя, и происходит вплоть до полного погло-
щения серебра из раствора. При благоприятных условиях се-
ребросодержащий поверхностный слой может достигать такой
толщины, что вызывает заметное изменение цвета минерала.
При более низкой температуре поглощение серебра замедляется
по мере заполнения монослоя, но при этом не происходит резко-
го изменения характера кривой.
При сравнении поведения ионов серебра и двухвалентной
меди становится ясно, что ион серебра более активно замещает
цинк в сфалерите, по крайней мере после образования монослоя.
Очевидно, что реакция
ZnS + 2Ag+-> Ag2S + Zn2+ (18)
не прекращается с образованием монослоя, в то время как ана-
логичная реакция с ионом двухвалентной меди практически пол-
ностью прекращается по заполнении монослоя.
Некоторое объяснение этого явления было получено в ре-
зультате опыта, в котором азотнокислое серебро пропускалось
сквозь слой сфалерита до резкого уменьшения поглощения се-
ребра. После этого на некоторое (довольно продолжительное)
время пропускание раствора прекращалось, а затем снова про-
должалось. Удивительно, что в этом случае минерал вновь с
прежней жадностью начинал поглощать серебро. Такой полный
Общее количество пропускавшегося серебра,
мкмоль/г ссралерита
Рис. 19. Адсорбция иона серебра на сфалерите из
раствора, непрерывно протекающего сквозь слой
минерала [19]:
1 — при температуре 17°; 2 — при температуре 26°; 3 — при
температуре 49J
Рис. 20. Адсорбция иона серебра на сфалерите из
раствора, протекающего с перерывами сквозь слой
минерала [19]
ДЕЙСТВИЕ МОДИФИКАТОРОВ ФЛОТАЦИИ
375
цикл обработки был повторен несколько раз, как это представ-
лено на рис. 20.
Опыты показали, что в этих условиях происходит дуффузион-
ное перераспределение серебра, в результате чего непрерывно
образуется новая поверхность сульфида цинка. В то время как
покрытый серебром минерал «отдыхает», диффузионное пере-
распределение не прекращается.
Подобное объяснение поведения покрытого серебром сфале-
рита подтверждается большим влиянием температуры на по-
глощение серебра, подобием кристаллической структуры сфале-
рита и аргентита, а также сходством в координации между се-
ребром и серой, с одной стороны, и цинком и серой — с другой.
Возможно, что поглощение меди сфалеритом, зависящее от
температуры, обусловлено подобной же диффузией ионов меди
и цинка, но более затрудненной наличием барьера твердого
сульфида меди.
Предупреждение активации сфалерита медью. Активацию
сфалерита медью можно предупредить, поддерживая .(в окру-
жающем сфалерит растворе) достаточную концентрацию иона,
способного значительно понижать концентрацию иона двухва-
лентной меди в растворе. Так, сульфид-ион и цианид-ион яв-
ляются весьма эффективными для предупреждения активации
сфалерита. Равиц [38] в обзоре растворимостей сульфидов ме-
таллов приводит величину растворимости сульфида двухвалент-
ной меди, равную 3,48 • 10~38, и сульфида одновалентной меди
3,6 10-s0. Это означает, что при легко достигаемой концентра-
ции сульфид-иона 10-1° концентрация иона двухвалентной меди
не может превышать 10~27, а иона одновалентной меди 1О~20.
Поскольку сероводород является восстанавливающим аген-
том, часть двухвалентной меди будет восстановлена до одно-
валентной.
Так, Латимер дает
H2S = S + 2Н+ + 2е~ £° = —0,141 в,
Си+= Си2++ е— Е° = — 0,153 е,
откуда получаем
H2S + 2Cu2+--S + 2H+4-Cu+ £°=+0,012e. (19)
Исходя из этого значения £°, константу равновесия для реак-
ции (19) можно определить равной 2,5, откуда после подстанов-
ки получаем ______
[Си+ ] = V 2,5 jH2S] _ (20)
[Си2+ ] [Н+]
376
ГЛАВА 10
Если в 1 л воды растворен 1 ммоль сульфида натрия и pH =
= 10, то [H2S] равна 10~6-м. (см. табл. 3) и отношение кон-
центрации ионов одновалентной меди, к концентрации ионов
двухвалентной меди составит 1,6 107, т. е. большая часть меди
в растворе будет представлена одновалентной медью. Таким
образом, сульфидный ион понижает до исчезающе малого зна-
чения допустимую концентрацию растворенных (не входящих в
комплекс) ионов меди, причем особенно активно при избытке
двухвалентной меди.
Ион цианида понижает концентрацию ионов одно- и двухва-
лентной меди путем образования комплексного медно-цианового
иона Cu(CN2)_. В примере, детально рассмотренном ранее в
данной главе, было показано, что при общем содержании меди
во всех формах, равном 10-4-м. и общем содержании цианида
1О2-м. (причем [CN-] составляет 10~3-м.), концентрации однова-
лентной и двухвалентной меди составят соответственно 10-22 и
10-27. При связывании меди в комплексный анион медь остает-
ся в растворе (но в форме, не способной к реакции), в то время
как связывание меди осаждением (например, с помощью суль-
фидного иона) удаляет ее из раствора. Естественно, что в слу-
чае связывания меди при образовании комплексного иона ка-
кое-либо существенное изменение состава раствора может мгно-
венно и радиально изменить концентрацию меди в растворе, в
то время как в случае связывания меди осаждением для такого
изменения концентрации меди в растворе потребовалось бы зна-
чительно больше времени.
Действие цианида, обычно применяемого при флотации сфа-
лерита совместно с сульфатом меди и ксантогенатом, показано
на рис. 21. Из этого рисунка следует, что когда цианид приме-
няется в количестве, стехиометрическом количеству меди (три
группы CN на каждый атом меди), критические кривые прили-
пания имеют горизонтальный участок, распространяющийся в
широком диапазоне значений pH. Выше этого горизонтального
участка система имеет избыток цианида; практически в, ней пол-
ностью отсутствует свободная медь в растворе и, следовательно,
здесь отсутствует флотация. Ниже этой линии в системе имеет-
ся избыток медных ионов в растворе, и флотация происходит.
Регулирование концентрации ионов меди при помощи реа-
гентов, образующих внутренние комплексы. Многие комплексо-
образующие агенты применимы для связывания меди. Некото-
рые из них [29е] действуют на одновалентную медь (например,
тиомочевина), другие (например, аммиак, пиридин, дипири-
дил [31], тиоцианат, ортофенантролин) действуют и на однова-
лентную и на двухвалентную медь. Некоторые же (этилендиа-
мин, пропилендиамин, глицин, диэтилентриамин и пирофосфат)
действуют только на ионы двухвалентной меди [3, 5, 26].
ДЕЙСТВИЕ МОДИФИКАТОРОВ ФЛОТАЦИИ
377
Значения констант диссоциации некоторых медных ком-
плексных соединений даны в табл. 11. Обычно комплексная
группа представляет недиссоциированную молекулу. В случае
цианида комплексную группу представляет ион цианида.
Отношение количества свободных ионов меди ср к количе-
ству комплексных ионов сс в растворе может очень сильно ме-
няться вследствие наличия ряда значений констант диссоциа-
ций для .разных комплексов и большого числа различных ком-
плексных групп, содержащих ион меди.
Рис. 21. Влияние концентрации цианида натрия
на кривые прилипания для сфалерита, предвари-
тельно активированного сульфатом меди; концен-
трация этилового ксантогената калия 25 мг/л [46]:
1 — CuSOi • 5Н2О = 0; 2 — CuSO4 • 5НО = 25 мг/л- 3 —
CuSO4 • 5НаО = 150 мг/л
Так, если [NH3] составляет 10~4, то отношение ср 1сс = 47, в
то время как при [NH3] = 10~3 оно составляет лишь 0,0047. При
концентрации этилендиамина, равной 10-3, отношение ср 1сс =
= 1,9 • 10-14. Даже при концентрации этилендиамина, равной
10-6, отношение сР /сс остается все еще очень малым (равным
1,9 10~8). Такие же вычисления могут быть сделаны и для
комплексных соединений, содержащих одновалентную медь.
Отсюда следует, что поскольку в присутствии восстановите-
лей концентрация иона одновалентной меди зависит от концент-
рации иона двухвалентной меди, то применение комплексооб-
разующихся агентов, эффективных для одновалентной меди,
позволяет также контролировать концентрацию двухвалентной
меди, и наоборот.
378
ГЛАВА 10
Таблица 11
Константы диссоциации комплексных соединений
одновалентной и двухвалентной меди
Соединение Число комплексных групп Константа диссо- циации1
I. Комплексные
соединения
двухвалентной
меди
Этилендигмин ..... 2 1,9-Ю-20
Пропилендиамин .... 2 6,8-10“21
Диэтилентриамин . . . 2 1,4-10“21
Глицин 2 7,9.10“16 *
Ди пиридил 3 1,4-1и“18
Аммиак 4 4,7-10“’5
Оксалат 2 4,8-10“11
II. Комплексные
соединения
одновалентной
меди
Дипиридил 2 6.3-10“15
Тиомочевина 4 4,1-10“16
Цианид 2 2,0-10“24
Аммиак 1 6,6.10“7
Аммиак 2 1,3. ю-11
1 Если Ср — концентрация неассоцинроваиного медного иона, скон-
центрация комплекса, т — концентрация комплексной группы н р —
число комплексных групп, приходящихся на один атом меди в комплекс-
ном соединении, то константа диссоциации определится по уравнению
При оценке концентрации активно действующего комплексо-
образователя нужно помнить, что не обязательно все количество
комплексообразующего агента представлено активной формой.
Так, если в раствор добавляется аммиак, то должна быть рас-
считана концентрация свободного NH3, также в случае примене-
ния цианида должна определяться концентрация иона цианида,
а при применении диаминов — концентрация свободного ди-
амина.
ДЕЙСТВИЕ МОДИФИКАТОРОВ ФЛОТАЦИИ
379
Интересно сравнить эффективность взаимодействия различ-
ных комплексообразующих реагентов с ионами двухвалентной
меди lCu2 + J.
Так, согласно уравнению (96), для концентрации иона циа-
нида, равной 10-4, легко достигаемой в умеренно щелочной сре-
де, и общего количества меди, равного 10~4-м. (обычного для ус-
ловий, когда стремятся депрессировать сфалерит), концентра-
ция иона двухвалентной меди составит около 10~24.
Чтобы достичь столь низкой концентрации иона двухвалент-
ной меди при применении этилендиамина, потребовалась бы его
молярная концентрация.
Данные табл. 11 свидетельствуют о том, что такие же труд-
ности встречаются при применении других реагентов, образую-
щих комплексные соединения двухвалентной меди.
Дезактивация активированного медью сфалерита. Если ад-
сорбция меди на сфелерите представляет обратимый процесс (как
и должно быть, по нашему мнению), то уменьшение концентра-
ции иона двухвалентной меди ниже равновесной величины дол-
жно привести к удалению адсорбированной меди, и следова-
тельно, реакция пойдет справа налево. Если активированный
минерал был покрыт (хотя бы частично) анионами собирателя,
находящимися в равновесии с анионами собирателя, присутству-
ющими в растворе, то понижение концентрации активирующего
иона должно привести к удалению с поверхности минерала как
адсорбированного собирателя, так и адсорбированного акти-
ватора.
При подаче цианида в пульпу, содержащую активированный
и покрытый собирателем сфалерит, концентрация иона меди
весьма резко уменьшается, например от 10~’° до 10-25, при одно-
временном очень большом уменьшении количества адсорбиро-
ванного ксантогената.
В этом случае комплексообразующий агент находится на-
столько близко к поверхности минерала, что связывает медные
ионы, как то,лько они отделятся от поверхности или даже когда
они еше связаны с ней. И наоборот, если производят дезактива-
цию активированного медью сфалерита посредством повышения
концентрации сульфид-иона так, чтобы понизить равновесную
концентрацию меди до той же малой величины 10~25, то, по-ви-
димому, сульфидный ион будет замещать ион ксантогената без
удаления меди в жидкую фазу, так что минерал депрессирует-
ся, сохраняя покрытие медью.
В практических условиях кислород, содержащийся в пуль-
пе, будет понижать концентрацию сульфидного иона, вследст-
вие чего ксантогенатное покрытие будет вновь восстанавливать-
ся на уже активированной поверхности минерала.
380
ГЛАВА 10
Активация кварца
Чистый кварц не флотируется жирными кислотами или их
щелочными мылами. Однако минерал легко активируется мно-
гими солями, особенно солями щелочноземельных металлов,
свинца, цинка, меди и железа.
На рис. 22 показана эффективность активирующего действия
соединений трехвалентного железа и двухвалентной меди.
Процесс активации кварца барием и последующей флотации
лауратом щелочного металла детально изучал Чанг. В проведен-
Рис. 22. Влияние величины pH и наличия активатора
на флотацию кварца олеатом натрия; крупность квар-
ца —100 +600 меш; собиратель: 0,09 кг/т олеата на-
трия; пенообразователь: 0,09 кг/т терпинеола; акти-
ватор 0,45 кг/т хлорного железа или 0,45 кг/т азотно-
кислой меди:
1 — активатор не задавался; 2— Fe 3 — Си 24-
НЫХ им опытах достигалось равновесие между пробой тонкоиз-
мельченного кварца, удельная поверхность которого была изве-
стна, и раствором, содержащим барий или одновременно барий
и лаурат, а также измерялись концентрации бария и лаурата в
равновесном растворе и на поверхности минерала.
Для определения сорбции реагентов Чанг применял радио-
активный барий и радиоактивный углерод (входящий в углево-
дородный радикал лауриновой кислоты).
При этом были получены интересные зависимости:
1) кварц адсорбирует барий из раствора; эта адсорбция
медленно возрастает с увеличением концентрации бария и ве-
личины pH;
2) при отсутствии бария или другого активатора кварц ад-
сорбирует небольшое количество лаурата, но эта адсорбция не
вызывает флотационного эффекта;
ДЕЙСТВИЕ МОДИФИКАТОРОВ ФЛОТАЦИИ 381
3) в присутствии бария кварц адсорбирует значительно боль-
ше лаурата, чем без него; адсорбция бария повышает вторич-
ную адсорбцию лаурата; на первичной адсорбции бария благо-
приятно сказывается повышение pH;
4) кварц, адсорбировавший определенное количество бария
и лаурат-иона, становится флотационно активным; это проис-
ходит даже и в том случае, если покрытие, образованное каж-
дым из двух видов ионов, много меньше монослоя.
Флотация происходит в условиях, когда концентрация ио-
нов недостаточна для насыщения раствора лауратом бария.
Существует полная аналогия между этими условиями и ус-
ловиями, вызывающими флотацию ксантогенатом сфалерита,
активированного медью.
В этом случае ион бария адсорбируется в поверхности ми-
нерала, аналогично иону двухвалентной меди' при активации
сфалерита, а водородный ион вытесняется подобно иону цинка.
Затем лаурат-ионы адсорбируются около адсорбционных точек,
покрытых барием, так же как ионы ксантогената адсорбируют-
ся частью поверхности сфалерита, покрытой, медными ионами.
Это сходство проявляется и в том, что в обоих случаях до-
статочно образования неполного монослоя собирателя для того,
чтобы сделать минерал флотирующимся.
Дезактивация кварца. Дезактивация кварца мало изучена.
Можно предположить, что если ионы меди являлись активато-
рами, то активация может быть уничтожена применением циа-
нида или одного из комплексообразующих агентов, о которых
упоминалось в этой главе.
В случаях, когда активатором кварца является не медь, а,
скажем, ион щелочноземельного металла или свинца, дезакти-
вация может осуществляться реагентами, удаляющими соот-
ветствующие активирующие ионы.
На практике, в тех случаях когда кварц или другие силикат-
ные минералы проявляют нежелательную флотационную ак-
тивность, обычно для регулирования процесса применяют каль-
цинированную соду (карбонат натрия), жидкое стекло (силикат
натрия) или фосфаты щелочных металлов.
Хотя функции этих реагентов еще точно не установлены, но
можно с уверенностью сказать, что они имеют определенную
ценность для регулировки процесса.
Другими реагентами, оказавшимися активными в аналогич-
ных случаях, являлись тартраты, цитраты, красители, особенно
гидрохиноновые красители, содержащие относительно большое
число гидроксильных групп.
382
ГЛАВА 10
Активация окисленных свинцовых минералов сульфидным ионом
В начале этой главы было показано, что сульфиды щелочных
металлов и в еще большей степени гидросульфид-ион депресси-
руют сульфидные минералы.
Поэтому вызывало удивление, что сульфиды щелочных ме-
таллов активируют окисленные минералы цветных металлов,
особенно свинцовых минералов.
Активирующее действие сульфидов может быть легко и на-
глядно продемонстрировано на церуссите.
С увеличением загрузки сульфида натрия во флотационный
цикл белый вначале минерал изменяет цвет от кремового до
коричневого. При использовании в качестве собирателя амило-
вого ксантогената извлечение свинца возрастает по мере потем-
нения минерала. Об этом можно судить по данным табл. 12.
Таблица 12
Влияние сульфидизации на флотацию чистого церуссита
(крупностью —100 -[•800 меш)
Реагенты, кг т Извлечение % Цвет пены
терпинеол амиловый ксантогенат сульфид натрия
0,068 13 Бледно-кремовый
0,068 0,045 52 »
0,038 0,045 0,090 94 Очень светло-коричневыц
0,068 0,045 0,180 96 Светло-коричневый
0,068 0,045 0,454 98 Коричневый
0,068 0,018 0,454 96 »
Действие сульфида натрия на флотацию англезита (сульфа-
та свинца) и церуссита (карбоната свинца) исследовалось
Уорком и Коксом [47]. Они установили, что малые концентрации
сульфид-иона оказывают активирующее действие, в то время
как большие концентрации вызывают депрессию. Это показа-
но, например, на кривых прилипания, изображенных на рис. 23.
В области, расположенной внутри замкнутых кривых, про-
исходит контакт минералов с воздушными пузырьками; в об-
ластях, лежащих вне замкнутых кривых, этот контакт отсутст-
вует. Уорк и Кокс показали, что активатором является сульфид-
ион, а в соответствии с действием на сульфидные минералы мож-
но ожидать, что депрессором в данном случае является гидро-
сульфид-ион.
Действие растворимого сульфида на флотацию вторичных
свинцовых минералов явилось объектом нового физико-химиче-
ского исследования М. Г. Флеминга 1121. Он установил, что да-
действие модификаторов флотации
383
же при относительно небольших загрузках сульфида натрия
(например, 10 мг/л) в разбавленные суспензии ванадинита (ва-
надохлорита свинца) и деклуазита (основного ванадата свинца
и цинка) указанные минералы перестают флотироваться, когда
собирателем служит этиловый ксантогенат, и заметно депрес-
сируются, если собирателем является амиловый ксантогенат.
Флеминг также установил, что минералы поглощают сульфид-
ион из раствора.
Рис. 23. Влияние величины pH и концентрации
сернистого натрия на закрепление пузырька
на поверхности церуссита. Закрепление воз-
можно лишь внутри кривых (по Уорку и Кок-
су, AIMME Trans., 134, 13, 1939):
1 — 31,6 мг/л амилового ксантогената калня; 2 —
25 мг/л этилового ксантогената калня
В случае церуссита Флеминг показал, что сульфид-ион по-
глощается из раствора, и минерал при малых концентрациях
сульфида натрия покрывается налетом коричневого цвета, ста-
новясь темно-синим при высоких концентрациях сульфида. Это
с очевидностью подтверждает, что происходит реакция обмена
PbCO3 4- PbS + СОз~ (21)
Константа равновесия этой реакции составляет З-Ю-16.
В этом случае отношение [СО^"~ ]: 1S2-] эквивалентно константе
равновесия, а коэффициенты активности близки к единице. Ясно,
что реакция должна весьма энергично проходить слева напра-
во. Даже если [СО 4 — ] достигает молярной концентрации, рав-
новесная [S2-] будет около1 10-16. По измерениям Флеминга
[S2] находилась в пределах от 10~6 до 10“п, т. е. была во много
больше, чем максимально возможная равновесная величина.
Можно предположить, что реакция полностью не заканчивается
после образования пленки продуктов реакции, но только тормо-
зится за счет замедления диффузии сульфид-ио'на к поверхности
384
ГЛАВА 10
церуссита сквозь образовавшуюся пленку продуктов реакции.
Сульфидизированный и затем промытый церуссит проявляет
оптимальную флотируемость. Эти наблюдения проливают неко-
торый свет на процесс сульфидизации церуссита, но они не объ-
ясняют, почему галенит флотируется ксантогенатами значи-.
тельно легче, чем церуссит.
ЛИТЕРАТУРА
1. Barsky G. Discussion, Trans. AIMME, 112, 236—237, 1934.
2. Bassett Henry, and Alexander Steven Corbet. A phase
rule study of the cupro-, argento-, auro-, and thallo-cyanides of potassium,
J. Chem. Soc., 125, 1660—1675, 1924.
3. Bjerrum Jannik. «Metal Ammine Formation in Aqueous Solution.
Theory of Reversible Step Reactions», P. Haase & Son, Copenhagen, 1941;
abstracted in Chem. Abstr., 35, 6527—6634, 1941.
4. Brigand o, Jeanne. Etude des argento- et des cuprocyanures de
potassium et de leurs acides, Compt. rend., 214, 908—910, 1942.
5. Carlson Gordon A., James P. McReynolds, and Frank H
V e r h о e k. Equilibrium constants in the formation of ammine complexes
with certain metallic ions, J. Am. Chem. Soc., 67, 1334—1339, 1945.
6. С о о k Melvin A., and John C. Nixon. Theory of water-repellent
films on solids formed by adsorption from aqueous solution of heteropolar
compounds, J. Phys. & Colloid Chem., 54, 445—459, 1950.
7. С о о k e S. R. B. The flotation of quartz using calcium ion as activator,
Trans. AIMME, 184, 306—309, 1949.
8. С о о k e S. R. B., and M. D i g r e. Studies on the activation of quartz
with calcium ion, Trans. AIMME, 184, 299—305, 1949.
9. Corriveau Martial. Concerning the action of silver ion in aqueous
solution with sphalerite, Master’s thesis, Massachusetts Institute of Tech-
nology, 1950.
10. E p h r a i m Fritz: «Inorganic Chemistry», translated by P. C. L. Thorne
and E. R. Roberts, pp. 577—594, Nordeman Publishing Co., Inc., New York,
1943.
11. Fenwick Florence. The equilibrium between cupric ion, cuprous ion
and metallic copper, J. Am. Chem. Soc., 48, 860—870, 1926.
12. Fleming M. G. Effects of soluble sulphide in the flotation of secondary
lead minerals, Paper 24, Institution of Mining and Metallurgy, Symposium
on Mineral Dressing, September, 1952.
13. Garrett A. B., Simon V e 1 1 e n g a, and С. M. Fontana. Th-
solubility of red, yellow and black lead oxides (2) and hydrated lead oxide
in alkaline solutions. The character of the lead-bearing ion J Am. Chem.
Soc., 61, 367—373, 1939.
14. Gaudin A. M. The influence of hydrogen-ion concentration on recovery
in simple flotation systems, Mining and Met., 10, 19—20, 1929.
15. Gaudin A. M. The effect of xanthates, copper sulfate and cyanides on
flotation of sphalerite, Trans. AIMME, 87, 417—428, 1930.
16. Gaudin A. M. «Principles of Mineral Dressing», McGraw-Hill Book
Company, Inc., New York, 1939.
17. Gaudin A. M. U. S. Patent 2231265/1941.
18. Gaud in A. M. and C. S. Chang. Adsorption on quartz from an aqueous
solution of barium and laurate ions, Mining Eng., 4, 193—201, 1952.
19. Gaudin A. M., H. R. S p e d d e n, and M. P. Corriveau. Adsorption
of silver ion by sphalerite, Mining Eng., 3, 780—784, 1951.
ДЕЙСТВИЕ МОДИФИКАТОРОВ ФЛОТАЦИИ
385
20. G a u d i п А. М., and Kenneth С. Vincent. Observations on the
magnitude of contact angles and their significance in flotation phenomena,
AIMME Tech. Publ. 1242, 1941.
21. G a u d i n Roland. Etude potentiometrique des proprietes du cyanure
d’argent, J. chim. phys., 42, 28—38, 1945.
22. V о n Ha lb an H., and W. Hecht. Xanthic acids and the kinetics of
their decompositions, Z. Elektrochem., 24, 65—82, 1918.
23. Jellinek K., and J. Czerwinski. Dissociation of hydrogen sulfide,
sodium sulfide and sodium hydrogen sulfide in aqueous solutions, Z. physik.
Chem., Ю2, 438—479, 1922.
24. Kolthoff I. M. Solubilities and solubility products of metallic sulfides
in water, J. Phys. Chem., 35, 2711—2721, 1931.
25. Kun sc her t F. Untersuchungen von Losungen des Kupfers in Cyan-
kaliums, Z. anorg. Chem., 41, 359—376, 1904.
26. Laitinen H. A., and E. I. О n s t о 11. Polarography of copper com-
plexes. III. Pyrophosphate complexes, J. Am. Chem. Soc., 72, 4729—4733,
1950.
27. Laitinen H. A., E. I. О n s t о 11, J. С. В a i 1 a r, Jr., and Sherlock
Swann, Jr. Polarography of copper complexes. I. Ethvlenedia'mine, propy-
lenediamine, diethylenetriamine, and glycine complexes, J. Am. Chem. Soc.,
71, 1550—1552, 1949.
28. Last George A. The mechanism of collector-depressant equilibria at
mineral surfaces, Thesis for the doctorate, University of- Utah, 1951. •
29. Latimer Wendell M. «The Oxidation States of the Elements and
Their Potentials in Aqueous Solutions», 2d ed., (a) p. 186 (b), p. 185, (c)
p. 137, (d) p. 191, (e) p. 187, Prentice-Hall, Inc., New York, 1952.
30. Noyes Arthur A., and Miles S., Sherrill. «Chemical Principles»,
p. 267, The Macmillan Company, New York, 1922.
31. Onstott E. L, and H. A. Laitinen. Polarography of copper complexes.
II. Dipyridyl, orthophenanthroline and thiourea complexes. A double complex
system, J. Am. Chem. Soc., 72, 4724—4728, 1950.
32. Pallanch R. A. The role of sulfites in the differential flotation plants
of the U. S. Smelting, Refining and Mining Co., Trans. AIMME, 79, 78—81,
1928.
33. Pallanch R. A. Factors governing the separation of lead and zinc in
ore by flotation, Mining and Met., 17, 386—389, 392, 1936.
34. P a 11 a n c h R. A. Flotation of ores an individual problem, Mining and
Met., 26, 167—169, 1945.
35. Pallanch R. A. Concentration at the Midvale Mill, Mining and Met.,
29, 544—547, 1948.
36. Ralston Oliver C., and William C. Hunter. Activation of sphale-
rite for flotation. AIMME Tech. Publ. No 248, 1929.
37. Ralston Oliver С., C. R. King, and F. X. T a r t a г о n. Copper
sulfate as flotation activator for sphalerite, Trans. AIMME, 87, 389—400,
1930.
38. Ravitz S. Frederick. The solubilities and free energies of some metal-
lic sulfides, J. Phys. Chem., 40, 61—70, 1936.
39. S c h w a r z, A1 f r e d. U. S Patent 807, 501/1905.
40. Sheridan, Guy E., and George G. Griswold. U S Patents
1421585/1922; 1427235/1922.
41. Sidgwick N. V. «The Chemical Elements and Their Compounds», vol. 1,
p. 133; vol. II, pp. 894—920, 938—943, Oxford University Press, New York.
1950.
42. Taggart A. F. «Elements of Ore Dressing», p. 276, John Wiley & Sons,
Inc., New York, 1951.
43. Truthe Wilhelm. Ueber die binaeren Systeme des Kalium und Natrium-
cyanids mit den entsprechenden Salzen von Ag, Cu, Zn nud mit den Chlo-
riden des Kalium und Natrium, Z. anorg. Chem., 76, 129—160, 1912.
25 A. M. Годэн
386
ГЛАВА 10
44. Владимирова М. Г. и Каковский И. А. Физико-химические кон-
станты, характеризующие образование и состав цианистых комплексов двух-
валентной меди. ЖПХ, 23, 580—598, 1950.
45. Wark, Ian W. «Principles of Flotation», (a) p. 187, Australasian Insti-
tute of Mining and Metallurgy, Melbourne, Australia, 1938.
46. W a r k Ian W., and Alwyn B. Cox. Principles of flotation. III. An
expermental study of influence of cyanide, alkalies and copper sulfate on
effect of sulfur-bearing collectors of mineral surfaces, Trans. AIMME,
112, 267—302, 1934.
47. W a r k Ian W., and Alwyn B. Cox. Prinsiples of flotation. IV. An
experimental study of influence of sodium sulfide, alkalies and copper
sulfate on effect of xanthates at mineral surfaces, Trans. AIMME 134,
7—25, 1939.
48. Wark Elsie Evelyn, and Ian W. Wark. The physical chemistry
of flotation. VIII. The processes of acrivation, J. Phys. Chem., 40, 799—
810, 1936.
49. Werner A. «Neuere Anschauungen auf dem Gebiete der anorganischen
Chemie», 5th ed., revised by P. Pfeiffer, p. 120, Vieweg-Verlag, Brunswick,
Germany, 1923.
Глава 11
ПЕНЫ
В гл. 7—10 рассматривались явления прикрепления минераль-
ных частиц к пузырькам воздуха с точки зрения химии и физи-
ко-химии. В данной главе этот процесс рассматривается с точки
зрения механики и исследуется поведение, а также прочность аг-
регата минерал — воздух.
Закрепление минеральных частиц на поверхности раздела
вода — воздух может осуществляться на поверхности пульпы,
для чего требуется подвести частицы твердого к поверхности
раздела газ — жидкость. Это может иметь место при падении
через воздух на свободную поверхность воды' смеси порошкооб-
разных сухих твердых мелких частиц, из которых одна часть
остается на поверхности раздела вода — воздух, а другая сма-
чивается водой и тонет. Закрепление частиц на поверхности
пульпы может осуществляться и из жидкой фазы, если минера-
лизованная пульпа представлена весьма тонким слоем, в кото-
ром каждая частица имеет возможность достигнуть поверхно-
сти. Для закрепления значительных количеств минерала в обо*
их этих случаях требуется очень большая поверхность
жидкости.
Закрепление минеральных частиц на поверхности раздела
фаз кроме указанных выше случаев, может происходить и внут-
ри пульпы, в которой диспергированный воздух появляется при:
вдувании, механическом перемешивании, кипячении или выде-
лении из раствора. Во всех этих процессах поверхность газ —
жидкость значительно больше при том же объеме пульпы, чем
в том случае, когда она ограничивается лишь свободной поверх-
ностью жидкости без образования пузырьков внутри пульпы.:
Диспергированные воздушные пузырьки, нагруженные минера*:
ламп или без них, обычно поднимаются к поверхности пульпы,
образуя пену. Эта пена состоит из газа, жидкой фазы пульпы,:
сфлотированного минерала и определенного количества не;
флотирующегося, но захваченного механически минерала. При-
сутствие пены настолько характерно для большинства флота-
ционных процессов, что обычно эти процессы не мыслятся без-
нее. По-видимому, целесообразно рассмотреть образование и;
поведение пены, лишенной минералов, и затем перейти к изу-:
чению механизма закрепления твердых частиц на поверхности!
раздела газ — жидкость и поведения минерализованных пен.1
25*
388
ГЛАВА И
Пенообразование
Свойства .поверхности раздела газ — жидкость были рас-
смотрены в гл. 3. В настоящей главе мы несколько более деталь-
но разберем зависимость между поверхностным натяжением
водных растворов и строением поверхностных слоев в согласии
с адсорбционным уравнением Гиббса. В гл. 3 отмечалось, что
для восстановления равновесия между внутренними слоями
жидкости и поверхностью раздела газ — жидкость, которое бы-
ло нарушено, например, вследствие растяжения поверхности
раздела, требуется некоторое время. Время, необходимое для
достижения равновесия, зависит от концентрации растворенного
вещества. При относительно концентрированном растворе, со-
держащем, например, 1% растворенного вещества, скорость, с
которой устанавливается равновесие, зависящее в первую оче-
редь от скорости диффузии, велика. В разбавленном растворе
(например, 0,001%) для достижения равновесия потребуется
значительно больше времени. В случае еще более разбавленного
раствора время, необходимое для достижения равновесия, мо-
жет оказаться настолько большим, что его трудно измерить,
так как за столь продолжительные отрезки времени возникают
различные вторичные явления, мешающие определению поверх-
ностного натяжения.
Чистые жидкости не пенятся. Газ, введенный в такую жид-
кость через погруженную в воду стеклянную трубку, проходит
как поток больших пузырьков, которые быстро поднимаются к
поверхности жидкости без образования пены. Но если в воду
добавлены растворы определенных углеводородов низкой кон-
центрации, то размер пузырьков резко уменьшается и на по-
верхности жидкости образуется пена.
При повышении концентрации пенообразующих веществ, до-
бавляемых к воде, пенообразование увеличивается до некоторо-
го предела. При этом поверхностное натяжение может понижать-
ся на величину, равную примерно 5 дин/см. Дальнейшее повы-
шение концентрации растворенного вещества приводит к умень-
шению количества пены, которая обычно исчезает в насыщен-
ных растворах. Таким образом, чистые жидкости или насыщен-
ные растворы, не пенятся, в то время как разбавленные
растворы могут образовывать пену.
Иными словами, пенообразование можно рассматривать как
результат нарушения равновесия на поверхности раздела сис-
темы жидкость — газ. Если бы пенообразование можно было
измерить, то результаты такого измерения дали бы ясное
представление о скорости, с которой устанавливается равно-
весие.
Пузырьки пены. Плато 1171 провел прекрасную работу с
ПЕНЫ
389
мыльными и мыльно-глицериновыми пленками, а также с масля-
ными капельками, взвешенными в жидкости, имеющей тот же
самый удельный вес. Согласно Плато, образованный внутри
жидкости воздушный пузырек, приближаясь к ее поверхности,
поднимает жидкость вверх так, что тонкая ее пленка остается
на поверхности пузырька.
Пузырьки, находящиеся в неподвижном состоянии вблизи
поверхности жидкости, имеют сферическую форму; при этом
очень большие пузырьки (диаметром около 50 мм) имеют вид
в
Рис. 1. Пузырьки различных размеров на поверх-
ности воды {17]:
а — натуральная величина; б — увеличение 4; в — увеличе-
ние 20
приблизительно полусфер, очень маленькие пузырьки (диамет-
ром 0,5 мм)—полных сфер, а пузырьки средних размеров —
промежуточную форму. Пузырьки находятся в гидростатиче-
ском равновесии и располагаются таким образом, что у боль-
ших пузырьков основание обычно находится на уровне поверх-
ности жидкости, у маленьких же — центр расположен прибли-
зительно на уровне жидкости или даже ниже ее. Это располо-
жение показано на рис. 1, причем пузырьки среднего размера
даны при увеличении 4, мелкие пузырьки — при увеличении 20,
а большие пузырьки — в натуральную величину. Поясок жидко-
сти, который образуется у места соприкосновения пленки пу-
зырьков со свободной поверхностью воды (рис. 1, а, б, в), иног-
да называется слоем Гиббса или слоем Плато. Он свидетельст-
вует о наличии капиллярных сил, подобных силам, поднимаю-
щим жидкость в капиллярной трубке и используемым для изме-
рения поверхностного натяжения. О форме этих слоев указыва-
лось ранее в труде Б. Б. Фрёйда и X. Ц. Фрёйда [6].
При одновременном нахождении на поверхности спокойной
жидкости нескольких пузырьков можно наблюдать, как они
плывут навстречу один другому и образуют группы. Если пу-
390
ГЛАВА 11
зырьки имеют однаковый диаметр, то границы между соседни-
ми пузырьками в каждой такой группе плоские. При наличии
пузырьков разного диаметра граница между ними искривлена,
причем выпуклостью она обращена в сторону больших пузырь-
ков. Радиус кривизны сферической внешней оболочки, кото-
рая образует перегородку между пузырьками с радиусами
Г] и rs, будет
7? = _Ж?... . (1)
Г2 —
Причиной искривления границы между пузырьками являет-
ся различие в давлении внутри пузырьков, которое, в свою оче-
редь, зависит от поверхностного натяжения жидкости и радиу-
са пузырьков. Если на поверхности жидкости сходятся в одной
точке только три пузырька, то касательные к криволинейным
пленкам, находящимся между ними, расположены под углом в
120°. При трехмерной пене и четырех соприкасающихся пу-
зырьках равновесие сохраняется лишь в том случае, когда пере-
городки между ними образуют углы, равные углам между гра-
нями тетраэдра (109° 30'). Если на поверхности жидкости
четыре пузырька на мгновение соприкасаются в одной точке, то
эта система находится в неустойчивом равновесии, которое не
может долго сохраняться. Однако соединение четырех пузырь-
ков всегда устойчиво, если пузырьки одинакового радиуса бу-
дут соединяться так, как это показано на рис. 2, д.
Форма пленки между воздушными пузырьками, близкими по
размерам, более приближается к плоскости, чем поверхность
свободных пузырьков, вследствие чего пена, образованная из
больших пузырьков, является многогранной. Тонкие стенки пу-
зырьков при старении высыхают и настолько утончаются, что
любые их движения приводят к коалесценции, происходящей с
большой скоростью. Пузырьки, образующиеся в результате ко-
алесценции, имеют объем, примерно равный суммарному объе-
му пузырьков, из которых они образовались, но несколько из-
мененные давление и кривизну поверхности. Быстрое изменение
кривизны поверхности вызывает колебания стенок пузырьков,
которые передаются соседним пузырькам и могут способство-
вать дальнейшей коалесценции.
Жидкость, стекающая со стенок пузырьков, собирается в
слое Плато, по которому в основном удаляется в объем пульпы.
При этом количество стекающей жидкости непрерывно умень-
шается как вследствие утончения оболочек пузырьков, так и
вследствие увеличения расстояний, которые должны пройти ча-
стицы жидкости до слоя Плато.
Структура неминерализованной пены. Сначала удобнее рас-
смотреть неминерализованную пену у поверхности жидкости.
‘ ПЕНЫ'
391
Такой слой пены увеличивается благодаря добавлению новых
воздушных пузырьков, поднимающихся снизу, и уменьшается
благодаря исчезновению старых пузырьков в верхнем слое пе-
ны. В равновесном состоянии добавляемые и исчезающие объе-
мы газа равны, но число добавляемых пузырьков больше, чем
Рис. 2. Двух-, грех- и четырех-
пузырьковая пена на поверх-
ности воды [17]:
« — два пузырька одинакового ра-
диуса; б —два пузырька с радиу-
сом г и радиусом г/2; в — два пу-
зырька а радиусом г и г/3; г — три
пузырька различного радиуса; д —
четыре пузырька одинакового ра-
диуса
число исчезающих вследствие
коалесценции пузырьков внутри
слоя. При увеличении размера пу-
зырьков в пене их сферическая .
форма изменяется, и они принима-
ют вид многогранников, опреде-
ленных Плато и описанных Сми-
том [28]. Пена, состоящая из боль-
Рис. 3. Группа правильных четырнадца-
тигранников [27]
ших пузырьков одного и того' же размера, теоретически должна
быть представлена структурой, в которой каждый пузырек име-
ет четырнадцать граней. Образование таких структур, рассмот-
ренное Кельвином [11, 12], является наилучшим способом деле-
ния данного объема на ячейки одного и того же вида и размера,
имеющие минимальную площадь раздела. Эти многогранники по-
казаны на рис. 3, который взят из превосходной монографии
Смита [27] о форме зерен металла. Каждый многогранник имеет
шесть квадратных и восемь правильных шестиугольных граней.
В таком многограннике углы между гранями в точках контакта
четырех граней отличаются от углов между гранями тетраэдра
(109°28'16"). Фактически имеется два угла по 90° и два угла по
120°. Ни один правильный многогранник с плоскими гранями не
392
ГЛАВА 11
имеет правильных углов тетраэдра между гранями, хотя пяти-
угольный двенадцатигранник имеет углы, близкие к этим значе-
ниям (108°). Правильные двенадцатигранники, однако, не могут
целиком заполнить пространство. Таким образом, требование
заполнения пространства пузырьками (многогранниками)
одинаковой формы с наименьшей поверхностью и необхо-
димость, чтобы углы между гранями имели значение 109°28'16",
противоречат одно другому, поэтому форма пузырьков в пене
не могла быть найдена теоретически. Экспериментально
же Метцке [15] нашел, что пузырьки имеют вид мно-
гогранников с несколько различными числом и формой граней.
Число граней у пузырьков в каждом из трех опытов изменялось
от 8 до 18 и в среднем составляло 13,7; основная часть много-
гранника имела 12, 13, 14 и 15 граней. Форма граней была в
основном пятиугольной, хотя встречались также квадратные,
шестиугольные и семиугольные грани. Эти статистические дан-
ные получались для весьма стабильной мыльной лены, которая
образовывалась при выпускании из капилляра по одному пу-
зырьку постоянного объема. В случае одновременного образо-
вания в пене пузырьков различного размера [16] мелкие пузырь-
ки сформировывали многогранники с несколько меньшим чис-
лом граней, чем 13,7, в то время как большие пузырьки имели
значительно большее число граней; при этом пузырьки распола-
гались так, что большие пузырьки были окружены мелкими.
Наличие у граней пузырьков небольшой кривизны еще более
сближает результаты расчетов Плато и Кельвина. На рис. 4 по-
казана типичная монодисперсная пена, полученная Метцке.
Смит указывает, что в среднем число сторон каждой грани
должно быть равно 5’/?. Это совершенно точно подтверждается
измерением Метцке.
При устойчивом состоянии пена в своей нижней части состо-
ит из мелких сферических пузырьков и большого количества
воды, а в верхней части — из больших многогранных пузырьков
и небольшого количества воды (рис. 5). Процесс восходящего
движения газовых пузырьков сопровождается подъемом некото-
рого количества воды, увлекаемого пузырьками на своих стен-
ках. Это явление компенсируется стоком воды вниз по тем же
стенкам пузырьков. Скорость движения вверх пузырьков в пене
сильно уменьшается по сравнению со скоростью в пульпе, а пе-
риод их существования соответственно удлиняется.
Рост, устойчивое состояние и распад пен. Если в жидкость,
обладающую пенообразующими свойствами, вводится с посто-
янной скоростью газ, то она покрывается пеной все возрастаю-
щей толщины, до тех пор, пока коалесценция верхнего слоя
пузырьков с атмосферным воздухом не достигнет той же самой
скорости, с которой вводится газ. «Возраст» пузырька увеличи-
Рис. 4. Типичная монодисперсная пена [15]
1
Рис. 5. Разрез типичной пены:
1 — свободный воздух; 2 — пульпа
394
ГЛАВА 11
вается по направлению от нижнего слоя пены к ее верхнему
слою. Если подача газа прекращается, то пена начинает осе-
дать более или менее постепенно, вплоть до полного исчезнове-
ния. Один из методов определения пенообразующей способно-
сти жидкости состоит в измерении времени, необходимого для
Время, мин.
Рис. 6. Объем пены в зависимости от продолжитель-
ности пропускания газа при постоянной скорости.
Раствор 0,48 г изоамилового спипта в дистиллиро-
ванной воде [25]:
А—скорость воздуха 0,86 см/сек-. В — то же, 1,70; С — то
же, 2,5
исчезновения стандартной пены (под «стандартной» пеной пони-
мают такую пену, которая получается при введении в опреде-
ленный объем жидкости определенного объема воздуха с задан-
ной скоростью). Этот метод определения пенообразующей спо-
собности жидкости называется методом распада пены. Другой
метод оценки пенообразующей способности жидкости заклю-
чается в установлении связи между скоростью роста пены и
временем пропускания через жидкость газа при постоянных ус-
ловиях. При третьем способе определения пенообразующей спо-
собности жидкости оценивается объем устойчивой пены, обра-
зованной при пропускании с определенной скоростью газа через
постоянный объем жидкости, помещенной в стандартный сосуд.
Этот метод измерения пенообразующей способности называется
методом устойчивости пены.
Прежде чем выбрать метод измерения пенообразующей спо-
собности, желательно изучить (хотя бы качественно) весь
процесс — накопление пены, устойчивое состояние и распад как
функцию скорости введения газа. Рис. 6 демонстрирует за-
395
ПЕНЫ '
-------------------------.—.---------------------------
висимость объема пены от времени [25]. Данные, представленные
на этом рисунке, получены для растворов изоамилового спирта
при пропускании газа с тремя скоростями, относящимися одна
к другой приблизительно как 1 :2 : 3. Видно, что устойчивое
состояние пены достигается соответственно после 35 сек.,
1 мин. 45 сек. и 3 мин. 30 сек. (точки Л4); одновременно в
этих опытах измерялся период образования пены Высота у
(пропорциональная объему пены) в статическом состоянии рав-
нялась 10,5, 26,5 и 56 см, а время распада х2 составляло около
12, 20 и 32 сек. Таким образом, приведенными опытами по це-
нообразованию измерены также и величины х2 и у.
Пенообразующая способность. Пенообразующую способность
[2] удобно выражать через параметры, имеющие размерность
времени. Как показано на рис. 6, x'i и х2 имеют размерность
времени; у — устойчивый объем пены, полученный при введе-
нии в единицу времени определенного объема газа; следова-
тельно, у также характеризуется величиной, связанной со вре-
менем. Обычно оценка пенообразующей способности является
сугубо эмпирической: инженеры оценивают ее лишь по внеш-
нему виду пены. Высказывания Сана 131] представляют собой
ь приятное исключение. Интересные сведения по этому вопросу
Bfc имеются также у Росса [21], Росса и Кларка 122] и Бикер-
мана [2].
Пенообразующие вещества. Хотя неорганические вещества
способны вызывать пенообразование жидкости, реальными пе-
МР нообразователями являются органические соединения. Обычно
пенообразующие вещества являются гетерополярными соедине-
ниями, у которых одна часть молекулы является аполярной, а
другая — полярной. Типичными пенообразователями будут:
амиловый спирт С5Н11ОН, крезол СНзСбН4ОН и толуидин
CH3-C6H4-NH2.
Пенообразующие свойства зависят от состава и строения обе-
их частей молекул. В гомологическом ряду веществ (т. е. ве-
ществ, имеющих одинаковое строение и одну и ту же полярную
группу, но различные углеводородные цепи) при концентраци-
ях, отвечающих максимальному ценообразованию, пенообразую-
щая способность повышается с увеличением длины углеводо-
родной цепи до некоторого предела, а затем начинает умень-
шаться, если длина углеводородной цепи становится очень
большой. Это особенно хорошо видно в ряду жирных спиртов,
у которых с увеличением длины углеводородной цепи пенооб-
разующая способность повышается до спирта, содержащего
семь или восемь атомов углерода, а затем понижается. Вещест-
ва, имеющие одну и ту же полярную группу и обладающие од-
ной и той же растворимостью, но различным строением аполяр-
ной части молекулы, иногда имеют одинаковые пенообразующие
396
ГЛАВА II
свойства. Так, гексиловый спирт CeHiaOH или гептиловый
С7Н15ОН примерно эквивалентны терпинеолу, циклическому
терпеновому спирту С!0НпОН.
В установлении пригодности реагента полярная часть моле-
кулы пенообразователя играет такую же важную роль, как и апо-
лярная. Таггарт, Тейлор и Инк [33] пришли к выводу, что хоро-
шие пенообразователи должны обладать только одной полярной
группой, предпочтительно группой, содержащей кислород в виде
гидроксила ОН, карбоксила СООН или карбонила СО, или азот
в виде амина NH2 или нитрила CN.
Однако гетерополярные вещества, у которых ионогенной по-
лярной группой является карбоксильная или аминогруппа, явля-
ются нежелательными пенообразователями по двум причинам.
Во-первых, эти вещества обладают также и собирательными свой-
ствами, а потому их пенообразующие свойства сопровождаются
собирательным действием. Во-вторых, изменение pH резко влияет
на их вспенивающее действие, так как растворимость солей
обычно гораздо выше, чем растворимость кислот и оснований.
Вследствие растворимости в воде щелочных солей длинноцепо-
чечных карбоксилсодержащих кислот и нерастворимости самых
кислот в результате гидролиза мыла образуется недиссоцииро-
ванная кислота, которая стабилизируется мылом. Для всех прак-
тических целей двухфазная система становится трехфазной ста-
билизованной мылом системой, состоящей из воды, жирной кис-
лоты и воздуха; количественный состав системы весьма разли-
чен в объеме пульпы и на поверхности.
Мыла, поверхностно активные сульфонаты и поверхностно
.активные сульфаты, амины, протеин, сапонин дают более устой-
чивую мыльную пену, чем спирты и фенолы, при применении
которых получается хрупкая, быстро исчезающая пена. Проч-
ность пены часто увеличивается в значительной степени, если в
обрабатываемой руде присутствует много тонких частиц. Напри-
мер, высокая стабильность пены получена в случае применения
аминов при обработке глинистых руд.
На рис. 7 показана пенообразующая способность трех мыль-
ных водных растворов, определенная по методу распада пены.
Из прямолинейности графиков, представленных в координатах
логарифм высоты — время распада пены, вытекает экспонен
циальный характер рассматриваемых зависимостей. Углы накло-
на этих трех кривых к оси абсцисс близки между собой. Прини-
мая во внимание, что масштаб времени для данных Кионка
уменьшен в 50 раз против масштаба, принятого для данных
Сауэра и Алдингера, можно сказать, что пенообразующая спо-
собность мыльных растворов более чем в 50 раз сильнее, чем
клеевых. Аналогичных сравнительных данных для флотационных
пенообразователей нет, но, можно полагать, что в присутствии
ПЕНЫ
397
о
&
7. Пеиообразование сольно
Рис,
пенящихся водных растворов:
/ — 1О°/о-ный водный раствор клея [24];
2—1%-ный водный раствор мыла (тор-
говый сорт [13]; 3 — 10%-иый водный
раствор мыла [24]
их будет наблюдаться значительно большая скорость разруше-
ния пены, чем в присутствии растворов клея, — возможно в
100 раз большая, чем представленная на рис. 7.
Хотя можно произвести некоторую классификацию пенообра-
зователей по молекулярному строению, как это сделано в настоя-
щем изложении, все же в этой
области осталось много неизу-
ченного, особенно если учесть,
что в последнюю четверть века
на нее мало обращали внима-
ния.
Механизм пенообразования.
Образование пены можно непо-
средственно приписывать двой-
ственному характеру молекул
пенообразователя, которые
имеют в своем составе поляр-
ную группу, способную взаи-
модействовать с водой, и непо-
лярную;— отталкивающую во-
ду. Функции обеих групп вы-
полняются, если молекулы рас-
положены на поверхности сте-
нок пузырьков таким образом,
что их полярные концы направ-
лены к водной фазе, а аполяр-
ные— в сторону от нее. Таким
образом молекулы пенообра-
зователя концентрируются в
поверхностном слое между
жидкой и воздушной фазами
флотационной системы. В ре-
зультате добавления пенообра-
зователей пузырьки, образо-
ванные внутри жидкости, по-
крыты мономолекулярным сло-
ем молекул пенообразователя.
Это позволяет каждому пу-
зырьку приближаться к друго-
му пузырьку, не сливаясь с
ним. Вначале при добавлении пенообразователя количество обра-
зуемой пены пропорционально загруженному количеству пено-
образователя; дальнейшие добавки пенообразователя менее эф-
фективны; прибавление еще больших количеств пенообразовате-
ля уменьшает его пенообразующие свойства и, наконец, полно-
стью уничтожает пеиообразование Г321. Концентрация, соответ-
398
ГЛАВА 11
ствующая полному исчезновению пены, отвечает насыщенному
раствору данного пенообразователя в воде.
Своеобразное действие растворенных веществ на пенообразо-
вание может быть объяснено следующим образом. Допустим, что
пленка АВ растягивается, причем А—вещество, понижаю-
щее поверхностное натяжение раствора. По мере того как плен-
ка растягивается, жидкость из массы раствора диффундирует к
поверхности, и содержание вещества А на поверхности умень-
шается. Вследствие этого поверхностное натяжение увеличива-
ется и противодействует растягивающей силе. Пленка получает
ту эластичность, которой нет в чистой жидкости, так как поверх-
ностное натяжение жидкости не может увеличиваться при растя-
жении поверхности.
Рассмотрим теперь две пленки, образованные растворами из
вещества А в веществе В, и предположим, что концентрация ве-
щества А в первой пленке меньше, чем во второй. Если при рас-
тяжении пленок их площади удвоятся, то поверхностное натяже-
ние каждой пленки возрастет, согласно рис. 4, гл. 3, где показа-
на зависимость между поверхностным натяжением и логариф-
мом концентрации водного раствора анилина (подобные резуль-
таты были получены и с другими растворами). Как видно из ри-
сунка, для случаев, когда начальная концентрация больше
0,1-м., последовательное уменьшение в два раза концентрации
раствора увеличивает поверхностное натяжение в одно и то же
число раз. Для случаев же, когда начальная концентрация мень-
ше 0,1-м., аналогичное последовательное уменьшение концентра-
ции раствора не приводит к столь существенному увеличению
поверхностного натяжения. Таким образом, можно ожидать, что
устойчивость пены будет увеличиваться при увеличении концен-
трации до 0,1-м., а затем будет оставаться относительно посто-
янной. Однако благодаря диффузии между поверхностным слоем
и пульпой равновесие устанавливается быстрее в тех случаях, ко-
гда присутствует большое количество растворимых молекул. Наи-
большая концентрация растворенного вещества, при которой
достигается максимальное ценообразование, получается в ре-
зультате действия этих двух факторов.
Требования, предъявляемые к пенообразователям. Основные
качества, которыми должны обладать пенообразователи,
имеются у многих веществ. Однако их выбор до известной степе-
ни ограничивается рядом факторов, из которых стоимость, до-
ступность пенообразователя и отсутствие у него собирательных
свойств являются наиболее важными. Из всего сказанного выше
следует, что хороший пенообразователь должен быть органиче-
ским веществом, у которого: 1) молекулы являются гетерополяр-
ными и состоят из одного или нескольких углеводородных ради-
калов с одной полярной группой; 2) растворимость не слишком
ПЕНЫ
399
большая, но и не слишком малая (желательно от 0,2 до 5 г/л);
3) молекулы не распадаются на ионы; 4) стоимость достаточно
низка.
На практике в качестве пенообразователей очень широко при-
меняют сосновое масло, крезиловую кислоту и некоторые синте-
тические спирты. Сосновые масла применяют различных сортов.
Их получают сухой перегонкой, а также и перегонкой с водяным
паром. Сосновое масло, полученное перегонкой с водяным паром,
содержит органические соединения с более низким молекуляр-
ным весом, чем масло сухой перегонки, и меньшее количество ве-
ществ, имеющих собирательные свойства. Сосновое масло, полу-
ченное перегонкой с водяным паром, состоит главным обпазом из.
терпеновых углеводородов, терпеновых кетонов и терпеновых
спиртов, в частности из терпена, пинена, цитронеллола и терпи-
неола. Терпеновые спирты сосновых масел более растворимы &
в воде, чем терпеновые углеводороды, образуют пену с правиль-
ным ячеистым строением и имеют низкие собирательные свойст-
ва. Полученный в промышленности а-терпинеол довольно дешев
и обладает 'более высокими пенообразующими свойствами, чем-
сосновое масло.
Пенообразующую характеристику сосновых масел, а также
определение индекса флотационной активности читатель найдет
в работах Сана (31].
Поступающая в продажу крезиловая кислота содержит [1}
некоторые высшие гомологи фенола CeHsOH, в частности, кре-
золы СН3С6Н4ОН и ксиленолы С2Н5С6Н4ОН или (СНз)гСзНбОН.
В большей части сортов поступающей в продажу крезиловой ки-
слоты преобладают ксиленолы.
В последние годы синтетические спирты (например, октило-
вый) применяются с большим успехом. Эти специально подоб-
ранные реагенты создают благоприятную структуру минерали-
зованной пены.
Среди пенообразователей, имеющих местное распространение,
можно указать на эвкалиптовое, шалфейное и камфарное масла,
низшие ароматические амины и амиловый спирт. Эвкалиптовое
масло, получаемое при переработке эвкалиптового дерева, при-
менялось в течение многих лет в Австралии, где из-за наличия
большого количества эвкалиптовых деревьев оно дешевле сосно-
вого масла. По-видимому, эвкалиптовое масло обладает более
сильными собирательными свойствами, чем сосновое масло, бла-
годаря чему его применяли в течение некоторого времени в каче-
стве единственного органического реагента при обогащении руд.
Брокен-Хилл. В Японии вместо соснового или эвкалиптового ма-
сла пользуются камфарным маслом. Шалфейное масло было ре-
комендовано для употребления с учетом того, что шалфей в боль-
шом количестве произрастает на западе США (но его произвол-
400
ГЛАВА It
ство до сих пор не организовано). Рекомендуется также приме-
нение низших ароматических аминов и амилового спирта, но они
дороги и обладают относительно слабыми вспенивающими свой-
ствами.
Летучесть пенообразователей. Составными частями пено-
образователей являются вещества, способные улетучиваться.
Многие из этих веществ обладают приятным запахом. Они встре-
чаются обычно в эфирных маслах, широко применяемых в пар-
фюмерии. Таким образом, существенная часть пенообразовате-
лей, используемых во флотации, улетучивается, и поэтому при
обороте воды уменьшение расхода пенообразователя не всегда
соответствует добавленному объему воды.
На основании измерения поверхностного натяжения жидкости
в исходном питании, концентрате и хвостах невозможно соста-
вить баланс поверхностно активного вещества, так как благо-
даря летучести пенообразователей поверхностное натяжение
обоих последних продуктов часто бывает выше, чем питания.
Введение газа
Введение газа в пульпу осуществляется одним из четырех ос-
новных способов: 1) образованием пузырьков при химической
реакции между веществами, растворенными в воде, и твердыми
частичками, взвешенными в ней; 2) образованием пузырьков под
вакуумом при уменьшении давления, благодря чему растворен-
ный газ выделяется из раствора; 3) выделением газа при кипе-
нии; 4) введением газа в пульпу механическим способом.
Образование пузырьков газа при химической реакции. Обра-
зование газовых пузырьков в пульпе химическим способом явля-
лось типичным для процесса Поттера — Дельпра [10], в котором
при реакции серной кислоты с частицами карбоната образовы-
вались пузырьки углекислоты. При проведении процесса необхо-
дима температура около 80°, сравнительно большие количества
кислоты (2—3% по весу воды) и густая (желательно обесшлам-
ленная) пульпа. При повышении температуры уменьшается рас-
творимость СО2 в воде и возрастает скорость образования газа;
увеличение концентрации кислоты увеличивает количество обра-
зующегося газа; бо'льшая плотность пульпы способствует прили-
панию пузырьков газа к флотируемым минералам; присутствие
шламов нежелательно, так как слишком ускоряет образование
газовых пузырьков и препятствует их прилипанию к минераль-
ным частицам. Интересно отметить, что в процессе Поттера —
Дельпра прикрепление флотируемых минералов к пузырькам
воздуха осуществляется непосредственным столкновением суль-
фидных частиц с крупными пузырьками, а не выделением на
них газа, так как пузырьки в основном возникают на поверхно-
стях частиц карбоната.
ПЕНЫ
401
Образование пузырьков газа под вакуумом. В вакуумных про-
цессах, типичным представителем которых является процесс
Эльмора (рис. 8), происходит выделение растворенных в воде
газов в в.иде пузырьков и прилипание этих пузырьков к соответ-
ствующим образом подготовленным поверхностям минералов.
Эффективность вакуумного процесса определяется количеством
Рис. 8. Вакуумфлотационная машина Эльмора [101
растворенного воздуха, который может содержаться в воде и вы-
деляться из нее при образовании вакуума.
Процесс образования пузырьков под вакуумом был недавно
предложен Шуманом и Пракошем [26] как один из методов, поз-
воляющих быстро оценивать флотируемость чистых минералов
при добавлении реагентов.
Образование газовых пузырьков при кипении. Пар, образую-
щийся при кипении, может быть также хорошо применен для
флотации, как и воздух или углекислый газ. Химики-аналитики
давно знакомы с флотацией серы, которая происходит при дей-
ствии азотной кислоты на сульфидные руды-
Образование газовых пузырьков механическим способом.
Способы получения для флотационного процесса газовых пу-
зе А. М. Годэи
402
ГЛАВА 11
зырьков при химических реакциях и под вакуумом устарели и
были заменены более простыми механическими методами.
Механическое введение газа в пульпу может быть осущест-
влено следующими способами: 1) введением газа через полый
вращающийся вал с импеллером на конце, погруженным в пуль-
пу и сообщающим центробежное движение пульпе, засасываю-
щей воздух (введение газа субаэрацией); 2) засасывание возду-
ха через воронку, образующуюся при быстром движении импел-
лера в пульпе (введение газа перемешиванием); 3) вдуванием
газа через трубки или через какие-либо пористые тела, погружен-
ные в пульпу (пневматическое введение газа); 4) переливанием
пульпы в виде каскадов, при котором пульпа перемешивается с
воздухом (введение газа каскадным методом).
В настоящее время введение воздуха во флотационные маши-
ны в промышленности осуществляется одним из первых трех ме-
тодов, каскадный же способ совсем не применяется. Однако о
нем следует упомянуть, так как каскадный способ имеет отноше-
ние к другим методам. Некоторое подобие каскадной флотации
можно наблюдать при переливании соответствующим образом
подготовленной пульпы из одного стакана в другой, частично
наполненный той же самой пульпой. Если высота падения струи
достаточная, то в нижнем стакане на поверхности пульпы по-
явятся пузырьки, при этом можно заметить, что на поверхности
каждого пузырька имеются частицы полезного минерала, хотя
прочного слоя пены в этих условиях не образуется. Этот про-
стой опыт показывает, что прилипание минералов ж воздушным
пузырькам есть результат непосредственного соприкосновения
пузырьков и частиц минералов. Можно предполагать, что тот
же самый эффект, но с различной степенью интенсивности по-
лучается при образовании пузырьков субаэрацией, перемеши-
ванием и пневматическим способом введения воздуха.
Прикрепление твердых частиц к пузырькам газа
Выделение газа из раствора. Известно, что на поверхности
минеральных частиц, погруженных в водный раствор, выделяют-
ся пузырьки газа. Это явление можно наблюдать под микроско-
пом, дающим даже небольшое увеличение, на частицах галенита
и кварца, помещенных в раствор ксантогената калия, налитого
в закрытый сосуд, из которого выкачивается воздух. При нали-
чии вакуума на поверхности минеральных частиц появляются
пузырьки газа. При этом на одних твердых частицах их оказы-
вается больше, чем на других. Можно увидеть, что некоторые ча-
стицы как бы висят под пузырьками, в то время как от других
частиц пузырьки, превышающие определенные размеры, отрыва-
ются. Наконец, можно заметить, что некоторые пузырьки исче-
зают и вновь появляются в одних и тех же местах и что выделе-
ПЕНЫ
403
ние пузырьков газа особенно заметно в трещинах, царапинах, на
острых углах или углублениях на поверхности твердого. Эти яв-
ления наводят на мысль, что часть пузырьков выделяется из га-
за, находящегося в порах и трещинах твердого, в то время как
другая выделяется из раствора.
Если размеры твердых частиц достаточно малы, а выделяю-
щиеся на них пузырьки воздуха сравнительно велики, то пу-
зырьки могут поднять частицы на поверхность пульпы. Таким
образом осуществляется флотация, вызванная выделением газа
из раствора.
Этот процесс протекает относительно медленно; для его за-
вершения обычно требуется много времени. Однако легко пред-
ставить, что процесс можно ускорить, специально вводя газ при
повышенном давлении в водные растворы или перемешивая
пульпу. Огромная трудность в применении вакуум-процесса, с
которым связано выделение газа из раствора, заключается в не-
большой растворимости воздуха в воде. При комнатной темпера-
туре и нормальном давлении 1 л воды может содержать пример-
но 18 мл воздуха. Поэтому даже при понижении давления до
0,1 атм объем воздуха, выделившегося из раствора, не может
превысить 180 мл.
Столкновение частиц с пузырьками. Вероятность столкнове-
ния частиц с пузырьками во вращающейся аэрированной пульпе
не определялась. Математически оценка вероятности может быть
произведена для упрощенных, идеальных условий. В дальнейшем
мы познакомимся с экспериментальным доказательством стол-
кновения частиц с пузырьками, проведенном с применением высо-
коскоростной киносъемки [29].
Линии тока воды, при обтекании минеральных частиц и пу-
зырьков газа. Вначале рассмотрим движение воды по отношению
к частицам и пузырькам. Предположим, что жидкость находится
в спокойном состоянии за исключением возмущений, вызываемых
всплывающими воздушными пузырьками или оседающйми части-
цами минералов, которые движутся с постоянной скоростью под
влиянием сил тяжести и трения. Известно, что такую систему
можно изучать, предполагая, что частицы минералов (или пу-
зырьки) неподвижны, а жидкость движется с той же постоянной
скоростью, но в противоположном направлении.
На рис. 9 и 10 показано движение жидкости около цилиндра
в условиях так называемого вязкого и турбулентного течений.
Эти фотографии, взятые из классической работы по механике
жидкости Прандтля и Титьенса [18, 19], получены при добавлении
красителя в струю жидкости в условиях, когда цилиндр и фото-
камера находились в неподвижном состоянии. Продолжитель-
ность экспозиции выбиралась такой, чтобы каждая окрашенная
частица могла пройти короткое расстояние и оставить на фото-
26*
Рис. 9. Вязкое течение вокруг неподвижного ци-
линдра [19]
Рис. 10. Турбулентное течение (жидкость течет сле-
ва) вокруг неподвижного цилиндра [19]
ПЕНЫ
405
графин след в виде небольшой линии. Эти линии показывают
путь жидкости около цилиндра.
При вязком течении потоки жидкости около препятствия плав-
ны, но при повышенных скоростях позади препятствия появляют-
ся завихрения, образующие след довольно значительной длины.
Рис. 11. Зависимость коэффициента сопротивления от числа
Рейнольдса:
Экспериментальные данные
1 — парафиновые шарики в анилине (по Аллену); 2 — воздушные пузырь-
ки в воде (по Аллену); 3 — янтарные и стальные шарики в воде (по Ал-
лену); 4— металлические шарики в сурепном масле (по Арноиду); 5 —
стальные шарики в воде (по Либстеру); 6 — стальные бронзовые и свин-
цовые шарики в воде (по Луннону); 7 — шарики в воздушной трубе (по
Визельбергеру)
У р авнения
8 — по Озину; 9 — по Гольдштейну; 10 — по Стоксу
На рис. 11, на котором представлен график зависимости коэф-
фициента сопротивления Q от числа Рейнольдса Re, показано, что
вплоть до Re = 1 достаточно точно действует закон Стокса, при
котором сопротивление определяется только вязкостью. Этот ха-
рактер течения изображен на рис. 9. При более низких числах
Рейнольдса, например Re = 200, превалирует вихревое сопротив-
ление; этот характер течения изображен на рис. 10. Из кривой
рис. И можно вычислить максимальный размер воздушного пу-
зырька для полностью вязкого течения, используя уравнение,
406
ГЛАВА 11
определяющее число Рейнольдса Re и коэффициент сопротивле-
ния Q:
Q=-~ (2)
31>2 Д' v ’
где г — радиус частицы (или пузырька);
v — их скорость;
Д и Д' — плотность частицы (или пузырька) и воды;
g— ускорение силы тяжести;
П — коэффициент вязкости воды.
Из приведенных уравнений скорость можно исключить, умно-
жив Де2 на Q:
l^2Q| Д'|) .
Зт;2
В случае воздушных пузырьков в воде [ Д — Д'| 1; Д' = 1;
g 103 см/сек2-, -л = IO"2 пуазов, и Re2Q становится равным приб-
лизительно 108г3; таким образом, условиям возникновения турбо-
лентности отвечает диаметр пузырька (2г), приблизительно рав-
ный 0,13 мм (эта величина получена из предположения, что вяз-
кое течение наблюдается при Re < 1. Прим. ред.).
Если вместо воздушных пузырьков рассматривать частицы га-
ленита, то получим, что вязкое течение осуществляется для час-
тицы размером до 0,07 мм. Другие минералы дают приблизи-
тельно такие же величины. Обобщая, можно сказать, что' вязкое
обтекание частиц, перемещающихся под влиянием сил тяжести и
трения, имеет место для частиц диаметром меньше 150—200 меш.
Если на частицы действуют и центробежные силы, как это мо-
жет быть в местах сильного перемешивания, то течение переста-
ет быть вязким и для более мелких частиц. Принимая центро-
бежное ускорение равным qg (<7>1) и учитывая, что постоян-
ство величин Re2Q требует постоянства r2qg, получим, что г будет
обратно пропорционально кубическому корню из q. Если q равно
100, то в условиях вязкого обтекания находятся частицы галени-
та размером только до 15 ц. Во флотационной пульпе большин-
ство частиц движется под действием силы тяжести, в основном
в условиях вязкого обтекания, за исключением зон с очень силь-
ным перемешиванием, где центробежные силы преобладают нац
силой тяжести. Напротив, пузырьки, которые обычно значитель-
но крупнее частиц минералов, движутся главным образом в ус-
ловиях турбулентного течения.
Линии тока при вязком течении. Целесообразно рассмотреть
траекторию элемента жидкости около сферического препятствия
ПЕНЫ
407
(пузырька или частицы) при вязком обтекании. Уравнение этой
траектории может быть представлено в форме
z=j2yz—Зу + — 'j sin2 9, (3)
\ У /
где у—отношение расстояния г между точкой Р траектории
элемента жидкости и центром О сферического препят-
ствия (который выбирается центром системы полярных
координат) к радиусу частицы а;
0 — угол между линией, соединяющей точку Р с О, и осью
ОХ, проведенной в направлении течения (рис. 12).
Рис. 12. Течение вокруг сферического препят-
ствия
Для какой-либо линии тока z является постоянной величиной,
например 0,1; 1; 2; 5.
Это уравнение можно получить из выражения для функции
тока [14а], предполагая, что инерцией жидкости можно прене-
бречь, т. е. учитывая (в данном случае только силу тяжести и
вязкости.
Дамб [14b] показал, что для рассматриваемого течения функ-
ция тока ф имеет следующий вид:
ф = — А и(1 — — + —V2sin29. (4)
' 2 \ 2г 2г3 /
Из уравнения (4) можно получить уравнение (3), принимая
где ф — функция тока;
U — скорость жидкости на бесконечном удалении от обте-
каемого препятствия;
а — радиус этого препятствия.
На рис. 13, а показан характер течения для z = 0,02; 0,1; 0,5;
1; 2. Струи показаны только впереди пузырька, течение же явля-
ется симметричным спереди и сзади. Этот рисунок, достаточно
точно передающий характер течения струй около малых препят-
408
ГЛАВА 11
Z=0,02
-2 = 0,1
г=Й5
2 = 2,0
ствий, показывает, что влияние препятствия может сказываться
на значительном от него расстоянии. Видно также (что очень
важно в связи с расчетами столкновения частиц с пузырьками),
что линии токов жидкости нигде не касаются стенок пузырька.
Линии тока при турбулентом течении. В предшествующем раз-
деле уже рассматривался такой вид течения, при котором влия-
ние сил вязкости было значительным, в то время как силами
инерции можно было пренебречь. Наоборот, можно представить
себе такую жидкость, в
которой вязкостью по сра-
внению с силами тяжести
и инерции можно пренеб-
речь. Такая жидкость
[18а]—идеальная жид-
кость классической гидро-
динамики — была хоро-
шо изучена. Условия те-
чения идеальной жидкос-
ти перед препятствием
приблизительно соответ-
ствуют условиям течения
вязкой жидкости при
больших числах Рей-
нольдса (за исключением
слоя, находящегося около
препятствия). Позади
препятствия условия те-
чения идеальной жидкости значительно отличаются от условий
течения реальной жидкости, так как в идеальной жидкости,
связанной в основном со средними скоростями, не принимается
во внимание турбулентность (последнюю можно рассматривать
как добавление к «инерционному течению» для получения ис-
тинного потока).
Траектория элемента идеальной (невязкой) жидкости около
сферического препятствия может быть представлена уравнением
г = (2у2----------------------— 'j sin2 9, (6)
\ У /
где обозначения те же, что и раньше.
Это уравнение может быть получено из функции тока, дан-
ной Ламбом [14с],
Ф = —£/<о3(1 — —1 , (7)
т 2 \ г3 ' '
Рис. 13. Линии тока жидкости, обте-
кающей сферическое препятствие:
а — вязкая жидкость; б — «идеальная» или
невязкая
z = 0,02 ~
2 = 0,1
2=0,5
г = 1,0
z = 2,0
где U — скорость жидкости на бесконечности;
со = г • sin0.
ПЕНЫ
40?
Преобразование уравнения (7) в уравнение (6) получается-
подстановкой
4ф г « /г>.
z = -—, У= — для у^\. (8)?
иа2, а
Характер течения впереди препятствия представлен на
рис. 13, б для z = 0,02; 0,1; 0,5; 1; 2. Сравнение с рис. 13, а пока-
зывает, что в обеих (вязкой и невязкой) жидкостях линии тока
не сталкиваются с препятствием. Однако в случае идеальной
жидкости линии тока вблизи экватора не расходятся, а собира-
ются и влияние препятствия перестает сказываться на сравни-
тельно малых расстояниях.
Вероятность прохождения линии тока около препятствия.
Существует зависимость между расстоянием линии тока от по-
верхности препятствия' (в случае, когда линия тока близка к
препятствию) и расстоянием линии тока от оси движения на ве-
сьма большом удалении от препятствия.
В случае вязкого течения уравнение (3) может быть решено’
при двух граничных условиях: на бесконечности, когда рассто-
яние линии тока от оси препятствия есть sMHH= у sin 0, и на эква-
торе, где расстояние от оси есть sMaKC = 1+As, причем все значе-
ния $ выражены в единицах а:
на бесконечности
= (2----L 4- Х\ . sin2 0) = 2s^HH ;
\ У у3 /
на экваторе
z0=f 2у-— Зу + — 'j sin2 0 =
\ У /
= [2 + 4 Д s + 2 (Д s)2] - (3 + 3 Д s) + - Д ,
Принимая приближенно 1 : (1 + As)^1—As-]-(As)2, что можно
сделать, разлагая в ряд 1 : 1-J-As и пренебрегая величиной (As)3'
как малой, получим значение для z на экваторе
г0 — 3 Д s2.
Так как значение z для одной и то же линии тока должно быть
одинаковым при всех расстояниях от препятствия и в частном,
случае равняется 3As2, то на бесконечности
3 Д s = 2змин.
В случае турбулентного обтекания при тех же приближениях
получается, что
6 Д 5 = 2Smhh-
•410
ГЛАВА 1)
Рассматривая правильный цилиндр с радиусом основания,
равным единице, и образующей, параллельной оси движения,
получим, что вероятность ф приближения произвольно выбран-
ной линии тока к сфере на расстоянии ближе, чем As, равна
s2BH . Поэтому в случае вязкого течения эта вероятность равна
3/2 (As)2, в случае невязкого, или идеального, течения она рав-
на 3As:
вязкое течение
<p=-|-(As)2; (9а)
турбулентное течение
<р = 3 As. (96)
Иными словами, вероятность того, что линия тока обтекает
данное препятствие ближе чем на расстоянии As, пропорциональ-
на этому расстоянию в случае турбулентного течения и пропор-
циональна его квадрату в случае вязкого течения.
Столкновение твердых частиц с пузырьком в неподвижной
суспензии. Если рассматриваемые частицы настолько малы, что
скорость их осаждения весьма незначительна по сравнению со
скоростью поднимающихся пузырьков и частицы и пузырьки
имеют достаточно малые скорости, чтобы двигаться в условиях
вязкого течения, то вероятность столкновения будет равна нулю.
Очевидно, частица не будет подходить к стенке пузырька, если
она не расположена на оси движения пузырька, а вероятность
такого ее положения равна нулю1.
* Вероятностью какого-либо события называется отношение числа благо-
приятных для наступления данного события случаев к общему числу возмож-
ных случаев. В данных условиях благоприятными будут такие случаи, когда
линии тока иа бесконечность удалены от оси движения на расстояние, мень-
шее Змии Очевидно, они все будут лежать внутри цилиндра, площадь осно-
вания которого равна гс$мин. Следовательно, вероятность того, что частица
жидкости, находящаяся в бесконечности иа расстоянии от оси движения
пузырька, меньшем или равном радиусу пузырька, подойдет к пузырьку бли-
же, чем на расстояние As, будет равна
Прим. ред.
1 Это положение неточно. В действительности, очевидно, частица может
•столкнуться с пузырьком, даже если оиа движется по линии тока (в том
случае, если линия тока проходит около пузырька на расстоянии, меньшем
радиуса частицы). Эта линия тока в бескоиечиости будет отставать от оси
движения пузырька иа некоторое конечное расстояние, определяемое форму-
лами (9а) и (96), и, следовательно вероятность столкновения не будет равна
нулю. Теоретический расчет вероятности столкновения частиц с пузырьком
при указанном предположении был произведен Сазерлендом [34]. Прим. ред.
ПЕНЫ
411
К такому же заключению можно прийти 1 и в случае, когда
нельзя пренебречь скоростью осаждения частицы, если принять,
что возмущение жидкости, вызываемое двумя движущимися
препятствиями (пузырьком и твердой частицей), является точ-
но векторной суммой возмущений, вызываемых каждым препят-
ствием, т. е. пренебрегая возмущениями второго порядка 2.
Если небольшие твердые частицы минералов движутся на-
встречу пузырьку в условиях вязкого обтекания, а пузырек дви-
жется в жидкости в условиях турбулентного обтекания, то в
первом приближении можно принять, что частица твердого сле-
дует вдоль одной из линий токов, обтекающих пузырек. В этом
случае вероятность столкновения частицы с передней частью
пузырька равна нулю3.
В случае, когда частица достаточно велика, так что ее дви-
жение происходит в условиях турбулентного обтекания, но без
вращения, резонно применить вышеприведенное рассуждение, а
именно, суммировать возмущения жидкости, вызываемые двумя
препятствиями. Но если частица является угловатой и враща-
ется или колеблется в потоке, то можно предположить, что один
из ее углов или граней войдет внутрь пузырька при прохожде-
нии частицы около него. Сфера, описываемая при вращении ча-
стиц, может быть, конечно, значительно больше шара, равнове-
ликого по объему частице. По этой причине вероятность встречи
частиц с пузырьком может увеличиться.
Пусть х—радиус шара, равновеликий по объему частице
(выражаемый в долях радиуса пузырька а), а х + Дх —радиус
сферы, образуемой при ее вращении. Тогда столкновение части-
цы с пузырьком произойдет, если центр ее проходит у стенки
пузырька, на расстоянии меньшем x-j-Ax, и если скорость вра-
щения частицы достаточно велика. Столкновение при вращении
частиц эквивалентно столкновению без вращения при условии,
что центр частицы находится на линии тока, проходящей через
1 См. первое издание настоящей книги [стр. 85—86 (по русскому изданию
1934 г.)]. Прим. ред.
2 Рассуждения, приведенные в цитированном выше первом издании книги
автора, являются ие вполне правильными. Скорость частицы относительно
центра пузырька в некоторой точке жидкости можно рассматривать как
сумму двух скоростей: а) скорости жидкости относительно центра пузырька
в данной точке, б) скорости частицы относительно жидкости. Последняя ско-
рость, если пренебречь инерцией частицы, является постоянной величиной.
Автор же определяет скорость частицы относительно пузырька как сумму
скоростей жидкости относительно частицы и относительно пузырька. Теоре
тический расчет вероятности столкновения частицы с пузырьком произвели
О. С. Богданов и Б. В. Кизевальтер; [35], приняв во внимание силу тяжести
частицы, т. е. предположив, что частица двигается относительно жидкости
с постоянной скоростью. Прим. ред.
3 Это утверждение неправильно (см. предыдущее примечание редакции).
Прим. ред.
412
ГЛАВА 11
экваториальную плоскость на расстоянии Ах от стенки пузырька
радиуса (a-)-x) или приближенно— радиуса а (если х является
малым по сравнению с а*).
Это наводит на мысль, что величина Ах может быть отожде-
ствлена с величиной As уравнения (96). Так как для частиц,
имеющих ту же самую форму или конфигурацию, Ax = fex, то
можно написать, что вероятность столкновения частицы с верх-
ней половиной пузырька, поднимающегося в турбулентных усло-
виях, будет
Ф = 3kx, (Ю>
где k — фактор, зависящий от формы (вероятно меньше еди-
ницы) ;
х — отношение размера частицы к размеру пузырька.
Уравнение (10) показывает, что в условиях частично вязкого
обтекания вероятность столкновения пропорциональна диаметру
частицы. Если течение полностью вязкое, вероятность столкно-
вения равна нулю.
В качестве примера мы можем рассмотреть кубическую ча-
стицу галенита (размер грани 100 Н) и сферический пузырек с
радиусом, равным 1 мм. Радиус шара, эквивалентного по объ-
ему кубической частице, будет равен 62 ц, а максимальный ра-
диус шара, полученного при вращении куба (равный половине
диагонали куба), 86,6 ц. В этом случае
_<86.6-62)
62
а х = 0,062 (в единицах а).
Отсюда вероятность столкновения; .
<Р = (3) • (0,39) • (0,062) «= 0,073, или 7,3%.
Вычисленное значение является максимальным, так как для
осуществления столкновения частицы с пузырьком при условии
их сближения необходимо, чтобы она вращалась достаточно
быстро. Полученный результат означает, что при нахождении
частицы в пределах вертикального цилиндра с радиусом, равным
радиусу пузырька, вероятность столкновения не превышает 7%.
При условии нахождения пузырька в стеклянной трубке диамет-
ром 2 см, т. е. в двадцать раз большим диаметра пузырька, и
* Логичнее написать, что столкновение частицы при вращении эквива-
лентно столкновению без вращения при условии, что частицы проходят эква-
ториальную плоскость иа расстоянии х + Дх от пузырька радиусом а, так
как частицы ввиду малости их размера по сравнению с размером пузырька
не должны оказывать существенного влияния иа движение пузырька. Прим,
ред.
1 Учитывая предыдущее примечание, следует считать что величина k
фактически всегда больше единицы. Прим. ред.
ПЕНЫ
413
расположения частицы в любой точке поперечного сечения труб-
ки вероятность столкновения в 202, или в 400 раз, меньше 7%,
т. е. не превышает 0,018%.
Экспериментальное доказательство прикрепления частицы к
пузырьку при столкновении в воде. Спедден и Ханнан [29] полу-
чили экспериментальное доказательство возможности эффек-
тивного столкновения частицы с пузырьком *. Они сыпали час-
тицы галенита размером около 0,2 мм в длинную вертикальную
стеклянную трубку, содержащую разбавленный раствор ксан-
тогената, обеспечивающий получение краевого угла в 60°. Одно-
временно через стеклянный капилляр в трубку впускались пу-
зырьки воздуха. Процесс столкновения фиксировался с помо-
щью высокоскоростной киносъемки, произведенной аппаратом
Кодак, дающим около 3000 кадров в секунду.
В экспериментах Спеддена и Ханнана по изучению столкно-
вения и закрепления частиц на пузырьке не соблюдались усло-
вия вязкого обтекания по следующим причинам: во-первых, ко-
нечные скорости как частицы, так и пузырька были слишком
большими, и потому их движение не могло не быть турбулент-
ным; во-вторых, фотографирование производилось вблизи вы-
пускного отверстия капилляра, вследствие чего1 газовые пузырьки
еще не успевали достигнуть конечной скорости; фактически воз-
душный пузырек диаметром 2 мм должен пройти много санти-
метров, прежде чем сможет достигнуть, например, 99% своей
конечной скорости; в-третьих, пузырьки имели не круглую фор-
му — поверхность их имела выступы и углубления и, кроме то-
го, быстро колебалась. Подобная форма пузырьков вероятно об-
разовалась частично в результате их отделения от струи газа в
капилляре. По этим причинам эксперименты Спеддена и Хан-
нана производились не в тех идеальных условиях, которые рас-
смотрены выше. Однако они значительно ближе подходят к
условиям, встречающимся на практике.
Из результатов экспериментов видно, что частицы отклоня-
ются от пузырька вдоль линии тока, которые впереди пузырька
имеют сходство с вычисленными и показанными на рис. 13. При
прохождении частицы около пузырька наблюдалось ее враще-
ние, а также контакт с передней частью пузырька около эквато-
ра. Неожиданными явились частые эффективные столкновения
частиц с поверхностью пузырька, расположенной за экватором,
По-видимому, частицы всасываются вогнутыми частями пузырь-
ков.
Необходимо повторить эксперименты Спеддена и Ханнана,
но в условиях, более близких к вязкому обтеканию, причем по-
1 Ранее возможность эффективного столкиовеиия частиц с пузырьком
воздуха была экспериментально доказана с помощью высокоскоростной ки-
носъемки О. С. Богдановым и М. Ш. Фплаиовским (36]. Прим. ред.
414
ГЛАВА И
стараться выяснить влияние на процесс столкновения колеба-
ний поверхности пузырька. Место фиксации должно быть выбра-
но на достаточном удалении от капилляра. Необходимо также
исследовать влияние стенок, т. е. влияние расстояния между
пузырьком и стенками трубы.
Форма и скорость пузырьков в воде. Обычно предполагает-
ся, что пузырьки в воде имеют шарообразную форму. Однако это
правильно только в тех случаях, когда они очень малы. Более
крупные пузырьки, движущиеся с конечной скоростью, имеют
вид сплющенных сфероидов с короткой осью в направлении дви-
жения. Наконец, очень крупные пузырьки принимают форму
приплюснутых сфероидов с плоской нижней частью [8, 20]. Мож-
но считать, что движущиеся в воде воздушные пузырьки диамет-
ром более 1,5 мм имеют несферическую форму. По-видимому,
сферичность пузырьков непосредственно зависит от их скорости,
которая может быть определена из диаграммы, связывающей
число Рейнольдса с коэффициентом сопротивления для твердых
шариков. Эта диаграмма (см. рис. 11), устанавливающая указан-
ную связь [уравнение (2)], справедлива при использовании в ка-
честве скорости конечной скорости различных твердых шариков,
в любой безгранично протяженной жидкости. На диаграмме,
построенной на основании многих экспериментальных данных,
нанесены также линии, отвечающие закону Стокса и теоретиче-
ским зависимостям Озина и Гольдштейна.
Скорости больших пузырьков в воде отличаются от получа-
емых по расчету из рис. 11. Это видно на рис. 14, где нанесены
зависимости скорости пузырька от его размера, полученные рас-
четом по данным рис. 11 (кривая 1) и найденные эксперимен-
тально Фюрстенау и Вэйманом. В данный момент кривые 3 и 4
не следует принимать во внимание.
Значения скорости для пузырьков с радиусом от 0,02 до
0,13 см, полученные в опытах, выше, чем вычисленные, вероят-
но, вследствие влияния движения газа внутри пузырька, выз-
ванного движением жидкости вне его [4, 9, 23].
Понижение скорости пузырьков, имеющих радиус больше
0,13 см, по савнению с вычисленной, объясняется влиянием их
несферичности, благодаря которой при подъеме увеличивается
сила сопротивления. Острый выступ на кривой 2 (рис. 14) соот-
ветствует эквивалентному размеру пузырька, при котором фак-
тор деформации начинает превосходить «гидродинамический»
фактор, вследствие чего зависимость скорость — размер изменя-
ется на противоположную.
Форма и скорость пузырьков в растворе пенообразователя.
Учитывая большую разбавленность растворов пенообразователя,
казалось бы, нельзя ожидать, что воздушные пузырьки могут
ПЕНЫ
415-
вести себя в таких растворах иначе, чем в воде. Однако в дей-
ствительности это не так.
На рис. 14 приведены две кривые зависимости конечной ско-
рости пузырьков от их эквивалентного размера, полученные для
растворов терпинеола (линии 3 и 4). Из рассмотрения кривых
следует, что пенообразователь существенно замедляет скорость.
Эквивалентный радиус пузырька г, см
Рис. 14. Скорости подъема воздушных пузырьков в
воде (по Фюрстенау и Вэйману):
} — в дистиллированной воде; 2—в растворе терпинеола
3,7 мг!л\ 3 — в растворе терпинеола 22 мг!л\ 4~ скорость
подъема твердых шариков согласно диаграмме Рэлея
подъема пузырьков. Создается впечатление, что пенообразова-
тель мешает ускорению движения пузырьков, которое должно-
было происходить вследствие движения газа внутри пузырька.
По-видимому, молекулы пенообразователя, находящиеся на по-
верхности раздела газ — жидкость, оказывают тормозящее дей-
ствие. Это явление, обнаруженное Фюрстенау и Вэйманом, ока-
залось неожиданным и заслуживает дальнейшего изучения1.
1 Влияние поверхностно активных веществ на скорость подъема воздуш-
ных пузырьков было изучено теоретически и экспериментально Фрумкиным!
и Левичем [37]. Специально вопрос о влиянии пенообразователей на движе-
ние пузырьков воздуха рассматривается в работе Богданова, Кизевальтера-
и Масловой [38]. Прим. ред.
•416
ГЛАВА 11
Столкновение пузырьков и частиц в турбулентном потоке.
В условиях очень сильного перемешивания и больших градиентов
скорости внутри жидкости должно происходить вращение ча-
стиц, причем, возможно, весьма быстрое. Особенно сложные вра-
щения должны возникать позади движущихся воздушных пу-
зырьков. Можно ожидать также, что при перемешивании воздуш-
ные пузырьки разрушаются. В области больших и изменяющих-
ся градиентов скорости пузырьки, без сомнения, становятся
растянутыми и распадаются на ряд мелких. В связи с этим мож-
но указать, что, согласно Плато [17], продолговатая цилиндричес-
кая капля, находящаяся во взвешенном состоянии в водном
растворе того же самого удельного веса, является устойчивой
в том случае, если ее длина не превышает в четыре раза вели-
чины диаметра.
Неизвестно, в какой степени распад пузырьков способствует
их столкновению с частицами твердого, но он может играть су-
щественную или даже главную роль. Интересные опыты по опре-
делению формы пузырьков, выходящих из насадки, были прове-
дены в Массачусеттском технологическом институте под руко-
водством проф. Фюрстенау. Установка состояла из аппарата, по-
дающего сжатый воздух, прибора для измерения скорости его
истечения, капиллярной трубки, из которой выпускаются пузырь-
ки, и стеклянного сосуда с плоско-параллельными стенками,
наполненного водой или другой исследуемой жидкостью. Ско-
рость истечения газа можно было регулировать, а при желании
и измерять. Поток пузырьков фотографировался при чрезвычай-
но короткой экспозиции — около 10 мксек, осуществляемой с по-
мощью специального освещения.
При низкой скорости истечения газа из капилляра выходят
воздушные пузырьки сферической формы. Они отделяются и
поднимаются в верхнюю часть камеры (рис. 15, а, б). При про-
межуточных скоростях истечения на пузырьках появляются плос-
кие площадки, образующие более или менее прямые углы с на-
правлением течения. Пузырьки, получив неправильную форму
при выходе из капилляра, сохраняют ее также в течение корот-
кого промежутка времени после отделения. При высокой скоро-
сти истечения форма большинства поднимающихся пузырьков
не является сферической (рис. 15, в). Обычно на пузырьках об-
разуются выступы, и поверхность некоторых из них похожа на
разбитый кусок кварца или стекла. Наконец, можно видеть, что
пузырьки обладают различными размерами: наряду с крупны-
ми остроугольными пузырьками имеются и группы небольших.
При детальном исследовании формы пузырьков вблизи от-
верстия, из которого они выходят, можно наблюдать, что при
высокой скорости истечения газа пузырек, имеющий при выходе
из капилляра грушевидную форму, при отрыве от капилляра ча-
Рис. 15. Воздушные пузырьки (по Фюрстеиау и Вэйману):
а, б, в, — в растворе терпинеола 20 мг/л XI.5; количество газа 0,2, 0,7
и 4,6 MAjceK’, г, д, е — в растворе терпинеола 10 мг/л Х4; количество
газа 4 мл/сек
¥1 А. М, Годэн
418
ГЛАВА II
сто принимает цилиндро-коническую или цилиндрическую фор-
му. Это можно увидеть на фотографии, приведенной на рис. 15,
г, д, е. Так, на рис. 15, г показан пузырек грушевидной формы,
следующий за цилиндро-коническим пузырьком, который отры-
вается. На рис. 15, д приведены три пузырька, нижний из кото-
рых имеет грушевидную форму, средний — цилиндрическую и
верхний — линзообразную. На рис. 15, е показана фотография
двух больших пузырьков, один из которых имеет в средней ча-
сти утончение, а второй, верхний, — приблизительно цилиндри-
ческую форму.
Во флотационных камерах механического или субаэрацион-
ного типа расстояния между лопатками импеллера и статором
небольшие. При этом импеллеру сообщается высокая скорость,
в то время как статор остается неподвижным. Если цилиндри-
ческий импеллер диаметром 8 см вращается внутри статора со
скоростью 1800 об/мин, следовательно, с максимальной окруж-
ной скоростью 750 см/сек, и если расстояние между ним и ста-
тором равно 0,6 см, то средний градиент скорости составляет
750/0,6 = 1250 сек~1, что представляет весьма большую величи-
ну. Если импеллер не является строго цилиндрическим, а име-
ет неправильную форму или сделан из стержней (как импеллер'
в камере Фагергрина), то течение при больших градиентах ско-
рости между импеллером и статором безусловно будет сильно
турбулентным.
Скорости жидкости на противоположных полюсах сфериче-
ской частицы диаметром в 20 ц, расположенной между соосным
цилиндрическим импеллером и статором, различаются на
1250 X 0,0020 или на 2,5 см/сек. Вследствие этого частица мо-
жет начать вращаться вокруг своей оси с окружной скоростью:
1,25 см/сек или 200 об/сек.
В действительности скорость вращения несомненно ниже
этой огромной величины, во-первых, вследствие того, что ча-
стица не успевает достичь максимальной скорости вращения до
выхода в спокойную зону камеры, и, во-вторых, потому, что ее
вращение может влиять на движение жидкости, вызывая изме-
нение местных градиентов скорости. Однако совершенно точно
можно сказать, что частица в рассматриваемых условиях будет
вращаться с очень высокой скоростью. Пузырьки воздуха в этой
же зоне должны растягиваться, приобретая, возможно, самые
фантастические несферические очертания, и разрываться. Та-
ким образом, главной причиной дробления газовых пузырьков на
части можно считать наличие градиента скорости движения
жидкости. В условиях весьма быстрого движения и большой ско-
рости вращения частиц при изменении вращения и скорости их
движения, а также при деформировании и дроблении быстро
движущихся пузырьков благоприятные возможности непосредст-
ПЕНЫ
419
венного столкновения частиц с пузырьками, по-видимому, во
много раз увеличиваются по сравнению с возможностями столк-
новения в спокойной воде, находящейся в стеклянной трубке.
Столкновение пузырьков в столбе пены. В столбе пены отно-
сительное движение пузырьков и воды может казаться медлен-
ным, но поскольку пузырьки имеют тонкие стенки, градиент
скорости между пузырьками, однако, может быть весьма суще-
ственным. Поэтому в указанных областях частицы могут начать
вращаться и, как следствие этого, столкнуться с пузырьками.
Кроме того, путь жидкости, стекающей между пузырьками,
весьма извилист, особенно если учитывать выступы, образован-
ные на пузырьках уже прикрепившимися частицами, что увели-
чивает возможность столкновения других частиц с пузырьками.
По мере обезвоживания пены (от нижнего слоя к верхнему) ха-
рактер пены все больше и больше отличается от пульпы, и она
приобретает совершенно другие свойства. Можно предполагать,
что высокоскоростная киносъемка окажется также весьма полез-
ной при исследованиях в этой области.
Выводы из исследования столкновений частиц с пузырьками.
Столкновение частиц с пузырьками экспериментально изучалось
в условиях турбулентного обтекания. Теоретически показано,
что в условиях вязкого обтекания столкновение сферических
частиц с пузырьками невозможно и происходит лишь с частица-
ми угловатой формы. Вероятность столкновения сильно увели-
чивается в условиях турбулентности и высоких градиентов ско-
рости жидкости в зоне импеллера, а также при сильной стеснен-
ности движения жидкости в слое пены.
Минерализованные пузырьки и пены
Выше столкновение пузырька с твердой частицей рассмат-
ривалось как основное условие прикрепления минеральных ча-
стиц или других твердых тел к воздушным пузырькам. Анализ
процесса столкновения производился на основании изучения
столкновения одного пузырька с одной частицей. В рудной пуль-
пе имеется огромное число частиц. Вследствие этого каждый
пузырек воздуха за время его движения в камере флотационной
машины вверх по определенной траектории будет с большой
скоростью перемещаться через какой-то объем, содержащий мно-
жество частиц, способных прилипнуть к пузырьку воздуха.
Попытаемся в некоторой степени количественно изучить вы-
сказываемые положения. Для этого предположим, что все части-
цы имеют одинаковый размер и по объему эквивалентны шари-
кам с радиусом а см, что отличие формы частиц от шарообраз-
ной учитывается коэффициентом формы k (уравнение (10)] и
что все пузырьки воздуха имеют форму шариков с радиусом
27*
'42b
ГЛАВА ))
7? см. Далее предположим, что длина пути пузырьков в пульпе
равна h см и что флотационно активные частицы составляют до-
лю т объема всей суспензии (т <( 1; часто т имеет величину от
0,01 до 0,001 доли единицы).
. Если не учитывать изменение формы пузырька воздуха при
'Подъеме его в пульпе, то объем пульпы, «вытесненный» одним
•пузырьком, будет равен ztR2h. Этот объем содержит nmR2h см3
,, л т R2h ~
флотируемого материала в виде -------- частиц. 1 ак как ве-
4/з~«3
роятность столкновения для определенной частицы, находящейся
в объеме пульпы, вытесняемом пузырьком воздуха при его дви-
величиной <? = 3 k~ {уравнение (10)], то
R
жении, выражается
число частиц, сталкивающихся с поверхностью пузырька, будет
равно (после упрощения)
9 t, R
п = — mhk-----.
4 а2
(Н)
Практически, если а = 2-10~3 см-, Д=0,2 см, т = 0,003; h =
= 100 см; k = 0,2, то п получится равным 6750 частицам.
Поскольку площадь поверхности пузырька определяется ве-
личиной 4 л/?2 см2, число частиц, которые могут разместиться
ща этой поверхности при плотной упаковке, будет равно1
2 /За2
Таким образом, если бы пузырек имел на своей поверхности
число частиц N, то он был бы полностью минерализован слоем
частиц на глубину, равную диаметру одной частицы.
Степень минерализации пузырька, достигающего слоя пены,
будет, равна (после упрощения)
f —. 9 3 mhk Л91
‘ ~~ 8 -R 11
Для этого уравнения характерно то, что в нем отсутствует
размер частиц а. Это показывает, что степень минерализации
/ пузырька, достигающего слоя пены, прямо пропорциональна
коэффициенту формы частиц /?, объему пульпы, занимаемому
флотационно активным минералом т, высоте пульпы h и обратно
пропорциональна размеру пузырьков R.
В процессе анализа предполагалось, что в вихревых потоках,
возникающих в пульпе, отсутствует коалесценция или дробление
1 Приведенное выражение для N справедливо только в том случае, ког-
, да частицы являются шарами радиусом а. Автор же считает, что частицы
имеют неправильную форму и лишь равновелики по объему шарам указан-
•иого радиуса. Прим. ред.
ПЕНЫ
421
пузырьков воздуха. Также предполагалось, что движение в жид-
кости частично минерализованного пузырька такое же, как и не-
минерализованного, и что все столкновения частиц с пузырька-
ми воздуха ведут к закреплению частиц.
В частном случае (уже разобранном), где « = 0,002 см,
k = 0,2 см, h = 6750, N получается равным 36 300 и f = 0,18.
Степень минерализации пузырька в пульпе. Уравнение (12)
показывает, что степень минерализации пузырька не зависит
от размера твердых частиц. Это кажется странным, так кан
степень минерализации определяется разнообразными фактора-
ми, которые, в свою очередь, связаны с размером частиц. Указан-
ный вывод является обоснованным, однако, лишь в том случае,
если предпосылки, на основании которых он был сделан, прием-
лемы. Вообще говоря, на основании сказанного выше можно
полагать, что минерализация пузырьков в пульпе не зависит пи
от ситовой характеристики флотируемого материала, ни от сте-
пени измельчения руды.
Степень минерализации прямо пропорциональна глубине
пульпы, объемной концентрации флотируемой составной части
пульпы и форме твердых частиц (степени угловатости). Зная
удельный вес флотируемого минерала, можно выразить объем-
ную концентрацию флотируемой составной части пульпы в виде
весового содержания в процентах.
Из уравнения (12) можно сделать вывод, что степень мине-
рализации обратно пропорциональна размеру пузырьков. Оче-
видно, чтобы получить полностью минерализованные пузыькп
до того момента, как они достигнут поверхности, необходимо,
чтобы эти пузырьки по возможности были мелкими.
Преполагалось, что наличие некоторой минерализации пу-
зырьков не оказывает влияния на процесс закрепления новых
частиц на двигающихся пузырьках. Это, по-видимому, правиль-
но до тех пор, пока минерализация пузырьков сравнительно не-
велика. Известно, что частица, прикрепленная к пузырьку,
скользит по нему, стремясь к наиболее низкому положению.
Этот процесс скольжения должен происходить очень быстро
из-за отсутствия трения. Действительно, рисунки Спеддена и
Ханнана показывают, что скольжение частиц по пузырьку про-,
исходит самое большее в течение нескольких миллисекунд.
До тех пор, пока пузырек минерализован незначительно, про-
цесс дальнейшей минерализации его, по-видимому, не затруднен.
Когда же большая часть нижней поверхности пузырька оказы-
вается покрытой частицами, дальнейшая минерализация посред-
ством прикрепления частиц к нижней части пузырька неизбежно
сокрашается. Наконец, когда минерализация пузырька превы-
шает 0,5, ухудшается прикрепление частиц к передней части пу-
зырька. Стадия полной минерализации пузырька в пульпе пред-
422
ГЛАВА 11
ставляет собой, таким образом, асимптотический процесс; она
может являться также результатом коалесценции двух или бо-
лее пузырьков, каждый из которых частично минерализован.
Строение минерализованных пузырьков. Строение минерали-
зованных пузырьков можно изучать следующим образом. Пу-
зырьки покрывают крупными частицами минерала и высушива-
ют. Полученный скелет сухого пузырька разламывают, после
чего изучают его строение как внутри, так и снаружи. Подобные
исследования были проведены с пузырками воздуха, покрытыми
свинцовым блеском, церруситом, малахитом и слюдой. На
Рис. 16. Сухой пузырек, покрытый минерализо-
ванными частицами:
а — снимок сфокусирован по грани частиц; б — снимок
сфокусирован на нижнюю часть оболочки скелета пу-
зырька. Х20
рис. 16 изображен пузырек, покрытый зернами малахита и рас-
колотый на две части. Сначала снимок сделан при наведении
фокуса на видимые грани зерен, а затем — на нижнюю часть
оболочки скелета. На снимке видно, что частички минерала по-
крывают пузырек в один слой и что внутренняя поверхность
«скорлупы» сравнительно гладкая. Частицы прилипают к стенке
пузырька гранью, имеющей наибольшую площадь. Это явление
находится в соответствии с требованием второго закона термо-
динамики.
Изменение частично минерализованных пузырьков в пене.
Следует напомнить, что пена, составленная из минерализован-
ных пузырьков, образуется в жидкости из скопления большого
числа сравнительно небольших газовых шариков (нижний слой
пены) и превращается в сетку плоских жидких перегородок
между газовыми многогранниками в верхнем слое пены. Эти
многогранники имеют в среднем около четырнадцати граней
(13, 7), при этом каждая грань имеет пять углов (5, 1) и очень
небольшую кривизну.
ПЕНЫ
423
В минеральной суспензии, в которой частицы могли прили-
пать к поверхности раздела жидкость — воздух, мелкие пузырь-
ки воздуха приближаются к нижнему слою пены, обычно будучи
лишь частично минерализованными. Рост пузырьков в резуль-
тате коалесценции происходит быстро до тех пор, пока они об-
ладают значительными площадями поверхности, не покрытой
минеральными зернами. По мере же увеличения размера пу-
зырьков, пена все больше принимает многогранную форму при
одновременном увеличении минерализации стенок пузырьков.
Во время образования пены процессы слияния свободных
поверхностей пузырьков могут конкурировать с процессами со-
единения пузырьков с помощью минеральных зерен. Таким об-
разом, если частично минерализованный пузырек догоняет более
«старый» пузырек, располагаясь в пене под ним, то свободная
поверхность первого пузырька может соприкасаться с минера-
лизованной нижней частью более «старого» пузырька. В ре-
зультате этого частицы, уже ранее связанные с нижней частью
поверхности «старого» пузырька, прикрепляются к пузырьку,
поступившему в пену позднее. При соединении пузырьков та-
ким способом между ними образуется перегородка из минера-
лов толщиной в одну частицу. С другой стороны, если свободные
участки поверхностей двух пузырьков соприкасаются, то пу-
зырьки сливаются, образуя один пузырек большого размера.
Можно доказать, что пена образуется двумя изложенными вы-
ше способами. Первый способ (слияние пузырьков) преобладает,
если пузырьки слабо минерализованы, второй (образование
между соединившимися пузырьками перегородки из одного
слоя минеральных частиц) преимущественно происходит, если
пузырьки значительно минерализованы. Рассматривая только
первый способ образования пены (коалесценцию), можно отме-
тить, что если пузырьки, прибывающие в слой пены, имеют диа-
метр d и степень минерализации f, то они могут увеличиваться
до тех пор, пока не достигнут диаметра d/f, когда их минераль-
ное покрытие является полным. Когда диаметр пузырьков до-
стигнет величины d/f, дальнейший рост пузырьков прекращается.
Рассматривая только второй способ образования пены, прини-
мая те же предположения относительно размера возникающего
пузырька и степени его минерализации, получим, что максималь-
ный размер пузырьков в многогранной пене равен d/2f. Множи-
тель 2 отражает то, что в пенах, имеющих перегородки толщи-
ной в одну минеральную частицу, обе стороны каждой частицы
минерализуют стенки пузырьков. Таким образом, максималь-
ный размер диаметров пузырьков в верхней части столба будет
изменяться от d/f до d/2f.
При флотации, когда целью является отделение флотирую-
щегося материала от нефлотирующегося, устойчивой пены не по-
424
ГЛАВА 11
лучают. В этом случае часть содержимого аппарата, в котором
происходит пеиообразование непрерывно, удаляется в виде пе-
ны. Независимо от того, в лабораторных или промышленных
условиях производится флотация, устойчивую пену получать
не рекомендуется.
Если величина /? сравнительно невелика, а значение f в пуль-
пе оказывается равным единице, то пузырьки полностью мине-
рализуются во всем слое пены и имеют сравнительно небольшой
размер. Пузырьки такого типа сохраняют шарообразную форму,
придавая переливающейся пене значительную текучесть; по
внешнему виду пена походит на рыбью икру. При этом каждый
минерализованный пузырек свободно движется между сосед-
ними пузырьками. В пене этого типа постоянным элементом
структуры является вода, находящаяся между пузырьками.
При условии, что Ruf невелики, полностью минерализован-
ные пузырьки имеют большой размер и форму многогранников.
Более того, возникает много возможностей образования перего-
родки между газовыми объемами толщиной в один минеральный
слой. Пена в этом состоянии может переливаться из камеры
флотационной машины и имеет определенную пластичность, ко-
торая отсутствует у более высоко минерализованных пен. Здесь
постоянным элементом в структуре пены является слой твердого
материала между пузырьками газа, возможно с небольшим ко-
личеством воды.
В случае, когда значение R сравнительно большое, a f неве-
лико, получается насыщенная воздухом пена, несущая малое
количество минералов. Пузыри в пене имеют большой размер и
легко деформируются. Если значение f очень мало, минерализа-
ция пузырьков недостаточна для предотвращения их слияния со
свободным воздухом на поверхности пульпы. В результате обра-
зуется шипящая обводненная пена с небольшим количеством
минерала. Ее трудно удалить из камеры флотационной машины
до того, как пузырьки, образующие пену, соединятся со сво-
бодным воздухом.
На рис. 17 схематически показано: а — икрообразная чрез-
мерно перегруженная пена; б — многогранная пластичная пена
наиболее благоприятной при флотации структуры; в обводнен-
ная «шипящая» пена, обычно получаемая в конце флотацион-
ного цикла. На рисунке видим агрегат А, состоящий из двух
пузырьков, и двойные стенки В и В', являющиеся переходной
формой между первым и вторым типами структур пены.
Изменения в характере пены в зависимости от времени фло-
тации или от места флотационной операции в схеме. Характер-
но, что пена изменяет внешний вид от начала до конца флота-
ции независимо от того, проводится она в лабораторных усло-
виях или в непрерывно действующей флотационной промышлен-
5
5
Рис. 17. Пена трех типов
426
ГЛАВА И
ной установке. При проведении флотации в камере лабораторной
флотационной машины внешний вид пены изменяется с тече-
нием времени. В условиях флотации на многокамерных флота-
ционных машинах вид пены зависит от места ее съема. Изме-
нение характера пены связано со степенью минерализации пу-
зырьков в нижнем слое пены и с процессом изменения пузырь-
ков в пене. Это изменение характера пены можно попытаться
рассмотреть, исходя из следующих рассуждений. Предположим,
что происходит флотация пульпы, состоящей из трех частей во-
ды и одной части твердого, в котором флотационно активные
зерна составляют пятую часть при удельном весе 5, а нефлоти-
рующиеся частицы имеют удельный вес 3. Это означает, что объ-
емная доля т, занятая флотационно активным твердым, равня-
ется 0,013. Если коэффициент формы k равен 0,4 и высота подня-
тия пузырька h равна 50 см, то по уравнению (12)
f — 0 62
‘ ~ ’ R ~ R ' :
при R = 0,1 см f будет равно 1,612. Если величина т в резуль-
тате флотации сократится от 0,013 до 0,013/1,61, то значение f
станет равным единице. При дальнейшем уменьшении т значе-
ние f будет меньше единицы. Минерализация пузырьков в пуль- |
пе, извлечение минералов за единицу времени, размер пузырь- /
ков и минерализация пузырьков в верхнем слое пены — все из-
меняется с течением времени флотации или в зависимости от
места съема пены, как это графически изображено на рис. 18.
Пена, благоприятная для флотации, похожа на кружева (так
называемая «кружевная» пена). Такая пена имеет верхний слой
пузырей, неполностью покрытых твердыми частицами, и являет-
ся промежуточной между тяжелой многогранной и обводненной
слабо минерализованной пенами.
С течением времени изменяется и форма жидких перегородок
между газовыми фазами пены от почти плоской горизонтальной
до сильно растянутой сферической, когда пузырек предпоследне-
го слоя становится пузырьком верхнего слоя. Это изменение гео-
метрии перегородок происходит согласно уравнению (1); други-
ми словами, оно отражает изменение в давлении на перегородку,
когда она перестает быть перегородкой внутри пены и становит-
ся ее внешней границей.
Размер пузырьков. Значительный интерес у исследователей
вызывает вопрос получения воздушных пузырьков наиболее
эффективного размера. Если предположить, что можно получить
пузырьки любого размера, то вопрос о том, какой величины они
должны быть (большими или малыми), решается на основании
следующих соображений:
В чем сильнее диспергирован взятый объем воздуха, тем
ПЕНЫ
427
больше поверхность образовавшихся пузырьков и тем больше
твердых частиц может разместиться в виде монослоя на этой
поверхности;
2) при диспергировании газа (воздуха) затрачивается энер-
гия; чем сильнее диспергация, тем выше стоимость образо-вания
пузырьков из единицы объема газа; вероятно, после достижения
Рис. 18. Изменение характера
минерализованных пузырьков
и пены с течением времени
флотации или в зависимости
от места съема пены по фрон-
ту флотации:
а — коэффициент минерализации
пузырьков в нижней части слоя
пены; б — скорость извлечения ми-
нералов; в — размер пузырьков
в верхней части слоя пены; г — ко-
эффициент минерализации пузырь-
ков в верхнем слое пены; а'—силь-
но флокулированная пена; б' — мно-
гогранная пена; в' — кружевная
пена; г' — обводненная «шипящая»
пена
некоторой степени диспергации газа в воде стоимость дальней-
шей диспергации его увеличивается настолько, что делает эту
операцию экономически неоправданной;
3) из числа полностью минерализованных пузырьков возду-
ха (т. е. пузырьков, полностью покрытых частицами в один
слой) пузырьки меньшего диаметра имеют больший кажущийся
удельный вес; это объясняется тем, что подъемная сила пузырь-
ков прямо пропорциональна кубу их диаметра, тогда как вес
минеральной нагрузки, препятствующий подъему пузырька, пря-
мо пропорционален квадрату этой же величины; можно пред-
полагать, что существуют минерализованные пузырьки настоль-
ко малого диаметра, что их удельный вес равен удельному весу
окружающей пульпы; такие пузырьки не могут подняться вверх
со своей минеральной нагрузкой.
Подобное явление может возникнуть при механическом пе-
ремешивании пульпы во флотационной машине в условиях очень
428
ГЛАВА И
высокой флотационной активности минералов и значительного
содержания их в пульпе. Однако при прохождении пульпы око-
ло импеллера перегруженные пузырьки разрушаются и теряют
свою минеральную нагрузку.
На рис. 19 графически изображена зависимость между удель-
ным весом пузырьков, полностью покрытых галенитом, и раз-
мером пузырьков и частиц.
Из приведенных графиков следует, что при наличии в пульпе
только очень крупных частиц флотации может не происходить,
Рис. 19. Зависимость между удельным весом пузырь-
ков, минерализованных галенитом, размером пузырь-
ков и размерами частиц минералов:
1~ частицы размером 50 р-; 2 — частицы размером 20 р.;
3 — частицы размером 10 и; 4 — частицы размером 5 р.;
даже если эти частицы будут покрыты пузырьками воздуха. Та-
кое явление, например, наблюдается при флотации фосфатов
гальки крупностью —1,17 +0,3 мм. В этом случае большая часть
пузырьков имеет меньший размер, чем минеральные зерна, в
результате чего вместо прилипания многих частиц к одному воз-
душному пузырьку несколько пузырьков может прикрепляться
к одной частице. Иначе говоря, частицы и пузырьки прилипают
друг к другу, образуя слабосвязанные гроздеобразные скопле-
ния. Эти скопления пузырьков легче отдельных твердых частиц,
но могут быть тяжелее воды. Такое явление наблюдается при
флотогравитации руд на столах.
Поведение нефлотирующихся частиц в пене. Турбулентные
потоки в пульпе, вызываемые движением пузырьков воздуха,
выносят в нижнюю часть пены частицы всех видов (флотирую-
щегося материала и пустой породы). С увеличением крупности
ПЕНЫ
429
Рис. 20. Фильтрующее действие пены
материала это явление наблюдается в меньшей степени, так как
крупные нефлотирующиеся частицы оседают с большой ско-
ростью, не успев задержаться в слое пены. Мелкие же частицы
породы попадают в нижнюю часть слоя пены в количестве, пря-
мо пропорциональном содержанию их в пульпе,
В пене, где нет турбулентных потоков, частицы пустой поро-
ды, находящиеся в водных прослойках между пузырьками, по-
степенно оседают. При
этом они проходят более
или менее извилистый
путь в тонких водных
прослойках. Пузырьки
воздуха до некоторой сте-
пени выполняют роль
фильтра, пропуская из-
быток воды. В результа-
те этого толщина водных
прослоек уменьшается, и
некоторое количество час-
тиц пустой породы как
бы запутывается между
пузырьками (рис. 20).
Очевидно, более круп-
ные частицы пустой по-
роды легче задержива-
ются фильтром из пу-
зырьков в самой верхней
части пены, но они скорее
других частиц оседают
в пульпе, не успев достигнуть верхних слоев пены. Эти два дей-
ствия уравновешиваются, в результате чего в пене задержива-
ются частицы и очень большого и среднего размера. Мелкие
частицы породы оседают в пространствах между пузырьками
с весьма малой скоростью и частично могут быть подняты на
поверхность пены вместе с водой. Следовательно, несмотря на
незначительный размер этих частиц, некоторые из них все же
настолько велики, что задерживаются «пузырьковым филь-
тром». В результате извлечение мелких частиц пустой породы
обычно выше, чем крупных.
Сухие пены. При закреплении воздушного пузырька на по-
верхности минеральной частицы в общем случае краевой угол
меньше 90°, а в условиях процесса флотации, вероятно, еще зна-
чительно меньше. Вследствие этого возможна флотация частиц,
соприкасающихся с воздушным пузырьком лишь одной гранью;
по мере возрастания краевого угла все больше и больше граней
минеральных зерен приходит в соприкосновение с воздушной
430
ГЛАВА 11
фазой; в результате этого пленка воды между двумя слоями
частиц может стать очень тонкой (рис. 21). Этот процесс ведет к
столь значительному обезвоживанию пены, что пленки между
воздушными пузырьками часто легко разрушаются. Получается
настолько хрупкая пена, что становится практически невозмож-
ным снимать ее.
Прочность пены. Прочность пены зависит главным обцазом
от свойств и количества диспергированной в ней твердой (и
жидкой) фазы. При современных методах флотации дисперги-
рованная жидкая фаза (масло) в пене почти отсутствует, так
что прочность пены полностью зависит от количества и свойств
Рис. 21. Два типа расположения частиц на границе раздела вода —
воздух:
а — мокрая пена; б — сухая пена
находящейся в ней твердой фазы. Количество диспергированно-
го твердого, прилипшего к стенкам пузырьков, можно лучше
всего выразить как отношение поверхности, покрытой минераль-
ными частицами, ко всей поверхности пузырька, в процентах.
Качество минеральной нагрузки может быть определено формой
твердых частиц и их крупностью. Опытным путем было установ-
лено, что пена тем прочнее, чем большая площадь поверхности
пузырька покрыта частицами минерала. Это объясняется тем,
что вероятность коалесценции пузырьков можно рассматривать
приблизительно как квадратичную функцию (квадратичную по-
тому, что коалесцируют два пузырька) площади поверхности
пузырьков, не покрытых твердыми частицами и поэтому способ-
ных прилипать к другой подобной же поверхности. Опыт также
показывает, что чем тоньше измельчены твердые частицы, тем
прочнее пена. Это происходит потому, что тонкие твердые части-
цы почти не сдвигаются с тех мест, которые они занимают
на поверхности пузырька, тогда как крупные частицы сдвигают-
ся. Наконец, форма твердой частицы является фактором неопре-
деленного характера, но легко заметить, что плоские частицы,
например, способны создать лучшее покрытие на поверхности
пузырьков, чем частицы угловатой формы, а частицы в форме
куба будут укладываться на поверхностях пузырьков гораздо-
компактнее, чем имеющие округлую форму.
ПЕНЫ
у-
431
Если бы с поверхности пузырков не испарялась влага (для-
насыщения газового пространства внутри пузырьков), то было,
бы возможно неопределенно долгое существование пузырьков,
полностью минерализованных тонкими частицами. В действи-
тельности же пленки воды, толщина которых в результате выте-
кания сокращается до нескольких микронов, легко изчезаюг
вследствие испарения влаги. Если пузырьки сильно минерализо-
ваны и разделяющие их стенки образуют многогранные фигуры,,
то после испарения из них влаги получаются скелеты пен, состоя-
щие из одних только твердых частиц, примыкающих одна к
другой.
Пена, содержащая масло, эластична, обладает чрезвычайной
прочностью, не уступающей сильно минерализованной пене. Это
можно объяснить тем, что капельки масла в состоянии изменять
свою форму, тогда как твердые дисперсные частицы ее не меня-
ют, а только передвигаются одна по отношению к другой. Пену
такого характера очень трудно разрушить, даже добавляя воду
(при помощи обрызгивания и т. п.) или при долгом стоянии; по-
лучаемая из такой пены пульпа очень трудно сгущается и фло-
тируется.
Очистка пены. Пену очищают, удаляя из нее пульпу. Можно
производить очистку, обрызгивая поверхности пены водой. При
этом в пене увеличивается объем жидкости, которая, протекая
между пузырьками, уносит механически захваченные частицы
пустой породы. В результате этого после прекращения ороше-
ния и уменьшения объема воды в пене до первоначального умень-
шается количество содержащегося в ней нефлотирующегося
минерала. В процессе орошения пены разрушается ее верхний
слой, причем минеральная нагрузка пузырьков (которую они
несли до разрушения) переходит к пузырькам нижележащего
слоя пены. При этом минеральные частицы как бы проталки-
ваются между пузырьками и конкурируют за ограниченные сво-
бодные места на их поверхности, в результате чего пена очи-
щается и минерализация ее увеличивается. Подобное же селек-
тивное выделение частиц из пены происходит в результате вто-
ричной флотации. Поэтому механизм очистки в обоих этих слу-
чаях по существу одинаков.
Содержание воды в пене. Количество воды в пене, которое
вместе с концентратом снимается с флотационных камер, раз-
лично. Оно является наибольшим для неполностью минерали-
зованной пены, средним — для сильно минерализованной и наи-
меньшим — для пены, имеющей многогранно-ячеистую струк-
туру.
В табл. 1 приведены данные, показывающие, как изменяется
содержание твердого и воды в пене, а также относительные объ-
емы твердого, прилипшего к пузырькам в процессе флотации.
432
ГЛАВА П
Таблица 1
Изменение характера пены в зависимости от времени флотации
Промежуток времени съема пены, мин.
0-1 1—2 2 — 4 4-8
«Выход слива, г: твердого 102 33 8 6
жидкого . 48 49 90 407
Галенита в сливе, выход ко всему » количеству твердых частиц, % (вес.) Отношение объема пульпы в пене к L. объему твердых частиц, прилип- ших к пузырькам 96 91 68 43
3,7 11,8 113 816
Флотация проводилась на искусственной смеси, составленной из
галенита и гранита, в лабораторной флотационной машине ме-
ханического типа с фракционным съемом пены.
Изменение относительного объема флотируемого материала
(по отношению к воде) во времени соответствует изменениям
эдой же переменной вдоль фронта флотационных машин на не-
прерывно действующих установках.
В случае образования пены, содержащей 50% твердых час-
тиц по весу, при содержании свинца 60% и наличии пустой по-
роды с удельным весом 3, объем воды в пене равен приблизи-
тельно 84%, а объем твердого 16%. В твердом веществе одна
половина объема представлена галенитом, другая — пустой по-
родой. Если допустить, что четвертая часть частиц пустой поро-
ды прикрепляется к стенкам пузырьков, то нетрудно предста-
вить, что около 10% общего объема твердых частиц и жидкости
прилипает к пузырькам газа, а 90% находится между пузырь-
ками пены. Отсюда можно сделать вывод, что среднее расстоя-
ние между пузырьками воздуха в пене приблизительно в 18 раз
больше диаметра частиц, которые удерживаются между пузырь-
ками. Если средний размер сульфидных частиц 10 ц, то средняя
толщина стенок пузырьков приблизительно равна 180 ц. В дей-
ствительности же, конечно, величина расстояния между пузырь-
ками колеблется, и существует большое число мест с малым оаз-
мером отверстий «пузырькового фильтра». По этой причине,
очевидно, только тонкие частички минералов не задерживаются
при прохождении через пену. Извлечение очень тонких частиц
пустой породы должно соответствовать извлечению воды, так
как, имея незначительную скорость оседания, они двигаются
вместе с водой. Это согласуется с экспериментальными наблю-
ПЕНЫ
433
дениями. Если принять, что содержание твердого в пене по весу
составляет 10% всего количества твердых частиц и жидкости,
то средний размер стенок пузырьков будет больше диаметра
частиц в 130 раз, или около 1300 ц. В этом случае, вероятно,
частицы крупностью 100 ц могут фильтроваться между стенка-
ми пузырьков. Указанное рассуждение объясняет, почему при
обрызгивании пены каплями воды пена в значительной степени
очищается.
Разжижение пульпы. Разжижением пульпы называется отно-
шение веса жидкости к весу твердого. Обычно при флотации оно
бывает от 5:1 до 2: 1. Для однотипных руд разжижение изме-
няется в узких пределах, но для руд, резко различающихся по
своему минеральному составу, оно может изменяться значитель-
но. Вообще для руд, содержащих твердую пустую породу, кото-
рая незначительно переизмельчивается, можно применять плот-
ные пульпы, но в случае флотации минерального сырья, содер-
жащего легко шламующиеся минералы пустой породы, требуется,
применение разбавленных пульп. Трудно точно обосновать необ-
ходимость той или иной степени разжижения, хотя экономичнее
пользоваться возможно более плотной пульпой, так как благо-
даря этому: 1) экономится расход свежей воды, подаваемой в
процесс; 2) увеличивается производительность флотационных
машин, определяющаяся количеством твердого, проходящего че-
рез каждую машину в единицу времени; 3) снижается необходи-
мый расход реагентов, особенно пенообразователя, и реагентов,
регулирующих щелочность пульпы, т. е. таких, которые произво-
дят свое действие в жидкой фазе пульпы или на поверхности
раздела фаз газ — жидкое-
С другой стороны, в случае использования плотных пульп
увеличивается количество твердых частиц, механически увлекае-
мых в пену, так как пена получается более загрязненной пустой
породой, чем в случае применения разбавленных пульп. Эго
иллюстрируется данными, приведенными в табл. 2, из которых
можно сделать вывод, что по мере разбавления пульпы извлече-
ние снижается, но более интенсивно из пены удаляется пустая
порода.
Следует обратить внимание на то, что общие чистые потери
металла в хвостах почти не зависят от первоначального количе-
ства металла в пульпе, хотя, конечно, в разбавленных пульпах
теряется больше металла.
Практический вывод, который следует сделать из предыдущих
рассуждений, заключается в том, что в основной (грубой) флота-
ции разжижение пульпы должно быть небольшим, а в перечист*
ной — значительным.
28 А. М. Годэн
434
ГЛАВА И
Таблица 2
Влияние разжижения пульпы на результаты флотации1
Загрузка в камере в начале опыта, г Содержание РЬ, % Действительные потери РЬ в хвостах, г
твердое вода в концентра- те в хвостах
2400 2800 43,4 0,12 2,2
1800 3000 56,6 0,14 1,9
1200 3200 59,0 0,14 1 ,4
900 3300 62,4 0,14 1,1
600 3400 63,9 0,27 1,4
450 3450 68,2 0,45 1,8
300 3500 74,8 0,56 1.5
1 Опыты проводились на искусственной смесн, содержащей По весу 10% чистого гале-
нита и 90% чистого гранита. В качестве реагентов применялись: 1) терпинеол при посто-
янном расходе 1 капля; 2) амиловый ксантогенат 20 г/т.
ЛИТЕРАТУРА
1. Bates W. A., and R. J. Miller Technical applications of cresylic acids
to flotation. Mining Technol., 10 (4), Tech. Publ. 2015, 5 pp., 1946.
2. Biker man J. J. The unit of foaminess, Trans. Faraday Soc., 34, 634—
638, 1938.
3. Bikerman J. J. «Foams Theory and Industrial Applications», Reinhold
Publishing Corporation, New York, 1953.
4. Boussinesq J. Vitesse de la chute lente, devenue uniforme, d’ une
goutte liquide spherique, dans un fluide visqueux de poids specifique
moindre, Ann. chim. phys., 29, 364—372, 1913.
5. Chapman Q. A., and J. W. L i tt 1 e f о r d U. S. Patent 1968008/1934.
6. F r e u d В. B., and H. Z. Freud. A theory of the ring method for the
determination of surface tension. J. Am. Chem. Soc., 52, 1772—1782, 1930.
7. A. M. Годэн. «Основы обогащения полезных ископаемых»: а) глава VIII,
б) рис. 102, 103. ГНТИ, Москва, 1946.
8. Haberman W. L., and R. К. Morton. An experimental investigation
of the drag and shape of air bubble rising in various liquids. U. S. Navy
Dept., David W. Taylor Model Basin, Rept. 802, September, 1953.
9. Hadamard J. Mouvement permanent lent d’une sphere liquide et
visqueuse dans un liquide visquex, Comp, rend., 152, 1735—1938, 1911.
10. Hoover, Theodore J. «Concentrating ores by flotation», Mining Ma-
gazine, London, 101—113, 1912.
11. Kelvin, Lord. On the devision of space with the minimum partitional
area, Phil. Mag., 24, 503—514, 1887.
12. Kelvin, Lord. On homogeneous division of space, Proc. Roy. Soc.
(London), 55, 1—16, 1894.
13. Ki on k a, H. Medicated soaps. Z. deut. 01- u-Fet-Ind., 43, 449—451,
467—469, 483—485, 1923.
14. Г. Л а м б. «Гидродинамика» ГИТТЛ M—Л., 1947. а) стр. 83, б) стр.
750 с) стр. 310.
15. Matzke Е. В. The three-dimensional shape of bubbles in foam, Am.
J. Botany, 33, 58—80, 1946.
16. Matzke E. B., and J. N e s 11 e r. Volume-shape relationships in variant
forms, Am. J. Botany, 33, 130—144, 1946.
ПЕНЫ
435
17. Plateau J. Memoire sur les phenomenes que presente une masse liquide
libre et soustraite а Г action de la pesanteur;' recherches sur les tigures
d’equilibre d’une masse liquide sans pesanteur, Mem. acad. roy. sci Bel-
gique, 16, 23, 30, 31, 33, 36, 37, 1842—1868.
18. Л. Прандтль и О. T и т ь e н с. Гидро- и аэромеханика, т. I, ГТИ
НКТП М—Л, 1935.
19. Л. Прандтль и О. Титьенс. Гидро- и аэромеханика т. II, ГТИ
НКТП, М—Л, 1935.
20. Rosenberg, Benjamin. The drag and shape of air bubbles moving
in liquids, U. S. Navy Dept., David W. Taylor Model Basin, Rept. 727,
September, 1950.
21. Ross, Sidney. Foam and emulsion stabilities, J. Phys. Chem. 47 266—
277, 1943.
22. Ross, Sidney, and G. L. Clark. The measurement of foam stability
with special reference to beer, Wallerstein Labs. Commun. Sci. Practice
Brewing, (6), 46—54, 1939.
23. Rybczynski W. Ueber die fortschreitende Bewegung einer fluessigen
Kugel in einem zachen Medium, Bull. Acad. Sci. Cracovie, ser. A, pp. 40—
46, 1911.
24. Sauer E. and W. A1 d i n g e r. Surface tension and foam formation in
glue solutions, Kolloid-Z„ 88, 329—340, 1939.
25. Scheller Joseph B. Dynamic frothing characteristics for isoamyl
alcohol-water solutions, Thesis, Massachusetts Institute of Technology,
1954.
26. Schuhmann R.,Jr. and Brahm Prakash. Effects of barium chloride
and other activators on soap flotation of quartz, Trans. AIMME, 187, 591—
600, 1950.
27. Smith Cyril Stanley. Grain shapes and other metallurgical applica-
tions of topology, Trans. Am. Soc. Metals, Preprint 37, 42 pp., 1951.
28. Smith Cyril Stanley. The shape of things, Sci. American, 190
(No. 1), 58—64, 1954.
29. Sped den, H. Rush, and William S. Hannan, Jr. Attachment of
mineral particles to air bubbles in flotation, Mining Technol., 12(2),
AIMME, Tech. Publ., 2354, 1948.
30. S t о k e s G. G. On the steady motion of incompressible fluids, Trans.
Cambridge Phil. Soc., 8, 1842.
31. Sun S. C. Frothing characteristics of pine oils in flotation, Mining Eng.
4(1), 65—71, 1952.
32. Taggart Arthur F., and A. M. Gaudin. Surface tension and adsorp-
tion phenomena in flotation, Trans. AIMME, 68, 479—535, 1923.
33. Taggart Arthur F„ T. C. Taylor, and C. R. Ince: Experiments
with flotation reagents, Trans. AIMME, 87, 285—368, 1930.
ДОПОЛНИТЕЛЬНЫЙ СПИСОК ЛИТЕРАТУРЫ К РУССКОМУ ИЗДАНИЮ
34. Sutherland К. S. Kinetics of the flotation progress, Journ. of Phys.
Colloid Chemistry, V, 52, № 3, 1948.
35. Б о г д а н о в О. С., Б. В. К и з е в а л ь т е р. Некоторые итоги изучения
физики флотации. Труды II научно-технической сессии института Меха-
нобр. Металлурпиздат, 1952.
36. Богданов О. С. и М. Ш. Фи ла невский. К вопросу о прикрепле-
нии минеральных частичек к пузырькам воздуха, Журнал физической
химии, т. 14, № 2, 1940.
37. Левич В. Г. Физико-механическая гидродинамика, АН СССР, 1952.
38. Богданов О. С., Б. В. К и з е в а л ь т е р, С. Г. Маслова. Влияние
пенообразователя на содержание воздуха во флотационной пульпе, Из-
вестия АН СССР, ОТН, № 3, 1950.
28*
Глава 12
КИНЕТИКА
Процесс флотации включает не только взаимодействие твер-
дых поверхностей с реагентами для придания соответствующих
свойств твердым поверхностям и прилипания твердых частиц к
пузырькам воздуха, но также и механическое отделение прилип-
ших к пузырькам воздуха твердых частиц от частиц гидрофиль-
ных. В гл. 11 показан статистический характер прилипания.
В этой главе рассматриваются исследования кинетики операций
аэрации и разделения.
Периодическая флотация. Сначала следует провести доста-
точно ясное различие между периодической и непрерывной фло-
тацией. В периодической флотации задаваемая смесь твердых
частиц различных размеров, формы и химического состава под-
вергается химическому воздействию реагентов, затем аэрирует-
ся, после чего происходит разделение ее на флотирующуюся
часть, более или менее непрерывно удаляющуюся из камеры, и
нефлотирующуюся, из которой в данный момент доизвлекается
часть материала. В каждый момент увеличение количества сфло-
тированного материала находится в зависимости от состава
пульпы в камере ниже уровня слоя пены. Поскольку состав
пульпы в камере представляет исходное питание в начале фло-
тации и хвосты в конце флотации, ясно, что увеличение коли-
чества сфлотированного материала зависит от широко меняю-
щегося состава пульпы от начала к концу флотации. Более того,
так как твердые частицы и вода удаляются из камеры, для под-
держания объема пульпы в камеру должна добавляться вода, в
результате чего изменяется отношение твердого к жидкому; та-
ким образом, на извлечение частиц в условиях периодической
флотации влияет не только различное соотношение твердого к
жидкому в пене и пульпе, но и добавка воды в камеру.
Непрерывная флотация. В операции непрерывной флотации
вновь загружаемое питание, предварительно химически подго-
товленное, постоянно поступает в камеру, из которой выходят
два продукта: концентрат (сфлотировавшаяся часть) и хвосты
(нижний продукт). Скорость образования этих двух продуктов,
считая по любому составляющему компоненту, такова, что сум-
ма скоростей выделения одного компонента в эти продукты рав-
на скорости поступления этого компонента с исходным питанием.
КИНЕТИКА
437
Если непрерывная флотация производится в нитке последова-
тельно соединенных камер механического типа и если предполо-
жить, что в каждой камере пульпа перемешивается полностью, то
концентрат флотируется из пульпы, представляющей по составу
хвосты предыдущей камеры.
Если флотационная машина для непрерывной флотации имеет
большое число камер, то состав пульпы значительно меняется
от питания в одном конце до хвостов в другом. Снимаемая пена
также меняется по характеру, так как в начале концентрат фло-
тируется из пульпы, представляющей исходное питание, а в кон-
це— из пульпы, имеющей состав хвостов. Это условие полностью
применимо и для машин пневматического типа, длина которых
может в 20—50 раз превышать ширину.
Различие между длинной пневматической машиной и равно-
объемной машиной механического типа в этом отношении ско-
рее искусственное, так как пневматическую машину всегда мож-
но представить состоящей из сегментов с длиной А/, достаточно
малой для того, чтобы состав пульпы на протяжении длины AZ за-
метно не менялся. Подобное теоретическое рассмотрение пнев-
матической машины позволяет представить ее как нитку камер
механического типа.
В любом случае непрерывной флотации разбавления (в ре-
зультате добавки воды), о котором говорилось выше, не проис-
ходит, так как сокращение объема пульпы за счет удаления кон-
центрата компенсируется понижением уровня сливного порога
в последующих камерах. Изменение отношения твердого к жид-
кому в пульпе по ходу процесса от одной камеры к другой пол-
ностью обусловлено только суммарным эффектом, получаемым
из-за различия отношения твердого к жидкому во флотационных
концентратах, снимаемых в каждой камере, и в пульпе каждой
соответствующей камеры.
По данным Шумана [18], для лабораторной работы можно
сделать камеру механического типа для непрерывной флотации,
которая питается подготовленной пульпой с постоянной ско-
ростью, а механическое удаление пены и хвостов производится
с эквивалентными скоростями для поддержания постоянных ус-
ловий.
Удельная скорость флотации. Пусть с является концентраци-
ей какого-либо компонента в объеме пульпы, выраженной в грам-
мах на литр воды. Если V— объем воды в камере и г—скорость
флотации интересующего нас компонента, выраженная в грам-
мах на единицу времени, то удельная скорость флотации Q мо-
жет быть определена как
(о
eV
4.38
ГЛАВА 12
Физическая величина удельной скорости флотации имеет об-
ратную зависимость от времени /'. Если г выражается в грам-
мах на единицу времени, то Q выражается в мин.-1.
В специальном опыте, который описан Шуманом, скорость
флотации меди (сульфидных минералов) и кварца из кварцево-
сульфидной руды была соответственно rCu=6,0 г[мин, rsx0 =
= 7,2 г!мин. Но, поскольку сСи = 1,05 г/л и cSiOa = 190 г/л, удель-
ная скорость флотации для V= 1,6 л была Q си = 3.57 мин.-1 и
QSio2 =0,024 мин.-1. Значения скорости флотации, которые в
данном случае оказались очень близкими, совпали в результате
огромной разницы в концентрациях и удельных скоростях фло-
тации.
Значение параметра удельной скорости флотации, приве-
денное выше, в основном заключается в том, что этот показа-
тель рассчитывают вне зависимости от многокомпонентности
цульпы. В отличие от других параметров удельная скорость
флотации позволяет сравнивать флотационные свойства состав-
ляющих пульпу компонентов. В специальном примере, приве-
денном выше, удельная скорость флотации медных минералов
в 150 раз больше, чем кварца. Поэтому можно сказать, что в
данных условиях опыта медные минералы в 150 раз флотацион-
но активнее, чем кварц.
Относительная флотационная активность. Вычисление отно-
шения удельных скоростей флотации двух компонентов или от-
носительной флотационной активности Ф, может рассматривать-
ся как наиболее общий метод сравнения поведения этих двух
компонентов при данных условиях флотации:
Важно отметить, что сходные безразмерные величины широ-
ко применяются в химии при описании поведения веществ в та-
ких операциях, как дистилляция, экстракция из раствора, селек-
тивная кристаллизация.
В приведенном выше примере значение Ф было равно 150, но
встречаются много большие и много меньшие значения. Значе-
ние Ф, равное единице, указывает на отсутствие разделения,
но даже такой низкий коэффициент разделения, как 1,01, может
быть полезен, например, при разделении изотопов.
Индекс селективности. В поисках количественных методов
для сравнения различных флотационных операций хотелось бы
иметь однозначную характеристику. Индекс селективности [7]
может быть использован для этой цели. Этот индекс может быть
определен как среднее геометрическое из отношения содержаний
КИНЕТИКА
439
компонентов AR и Вйв концентрате и обратного' отношения со-
держаний этих же компонентов к в хвостах:
Л Дд в}
BR AJ
(3)
Первоначально предполагалось, что индекс селективности
может быть использован и для периодической флотации, и для
непрерывной, причем как для отдельной части каких-либо опе-
раций, так и для какой-либо группы операций.
Однако теперь стало ясным, что следует различать индекс в
данный момент (в периодической флотации) или индекс в дан-
ной точке (в непрерывной флотации), с одной стороны, и общий
индекс, с другой. В самом деле, индекс в данный момент, или
мгновенный индекс ( SJMrH ), тесно связан с величиной Ф, в то
время как общий индекс ( SJоб1Ц ), как будет показано, не име-
ет этой зависимости.
Содержание компонентов в данный момент в концентрате и
соответственно в хвостах (или пульпе) можно выразить сле-
дующим образом:
Ar
— ; br=— ;
X г R Sr
или, пользуясь формулой (3),
о Г -\f ГА СВ _л/~ rA VcB
^мгн — I/ — 1/17 ’
’ ГВ СА V VcA ГВ
или, применяя формулу (1),
/ V»
SJMrH = — =Ф‘\ (4)
\ Чв /
В случае опыта Шумана, о котором упоминалось выше, где
Ф=150, 5/мгн составляет примерно 12,3.
Общий индекс селективности не может быть отождествлен с
Ф"/2. Это объясняется тем, что концентрат представляет собой
сумму многих фракций, получающихся из различной по составу
пульпы.
Предположим, что имеется смесь одинаковых частиц гале-
нита и одинаковых частиц кварца, для которых Ф = 100 (и, сле-
довательно, SJ мгн =10), и что из этой смеси последовательно
извлекается 20; 20; 20; 20; 10; 5; 3 и 1% галенита; тогда полу-
чатся следующие значения для SJкумул : 10,0; 10,7; 11,8; 13,8;
17,0; 21,4; 29,2 и 38,1; последний кумулятивный индекс являет-
ся З/общ .
440
ГЛАВА 12
Практически минеральные частицы не бывают одинаковыми,
так что значение S/MrH не остается постоянным и уменьшается
непрерывно для любой разделяемой пары компонентов. Это
отражается на значении 5/ку,,ул , которое увеличивается вна-
чале, а затем уменьшается. Во всяком случае изменение вели-
чины S/вумул не столь значительно, как в теоретическом слу-
чае разделения, который показывает, что
5^мгн * SJofiui- (5)
Коэффициент минерализации. Коэффициент минерализации
М является следующим критерием, употребляемым в кинетике
флотации. Он представляет отношение концентрации данного
компонента в дезаэрированном необезвоженном концентрате к
концентрации того же компонента в объеме пульпы (включая
воду). Коэффициент минерализации, равный единице, показы-
вает, что компонент ведет себя как вода. Коэффициент больше
единицы означает, что извлечение твердого в концентрат боль-
ше, чем воды, а коэффициент меньше единицы, — что твердое
извлекается в концентрат медленнее, чем вода.
Для примера, рассмотренного выше, при флотации силикат-
но-сульфидной медной руды коэффициент минерализации был
равен 40 и представлял отношение концентрации меди в концен-
трате (42 г/д воды в концентрате) к концентрации меди в не-
флотирующейся пульпе (1,05 г/д). Аналогично коэффициент ми-
нерализации для кварца был равен 0,26, и отношение двух коэф-
фициентов составляло 150, или равнялось относительной фло-
тационной активности.
Таким образом:
Л4 = Спены. . . (5а)
^пульпы
Изменения кинетических индексов с развитием процесса.
Если бы пульпа состояла из частиц совершенно одинаковых по
размерам, форме и свойствам поверхности, то очевидно удель-
ная скорость флотации оставалась бы постоянной от начала и
до конца флотации. В случае флотационно активного твердого
коэффициент минерализации в течение всего процесса должен
уменьшаться, как и отношение твердого к жидкому в пульпе.
С другой стороны, в случае нефлотационно активного твердого,
состоящего из отдельных одинаковых частиц, когда М<^ 1, мож-
но ожидать, что коэффициент минерализации будет постепенно
увеличиваться с продолжением операции, так как отношение
твердого к жидкому в пульпе будет возрастать.
КИНЕТИКА
44f
Однако случай, когда пульпа состоит из одинаковых частиц,
является совершенно идеальным. Даже когда для опытов при-
меняется чистый минерал только определенной крупности,
всегда имеются значительные различия в форме и поверхност-
ных свойствах отдельных частиц; в результате этого изменяет-
ся удельная скорость флотации для различных отдельных час-
тиц. Более флотационно активные частицы флотируют первыми,
а оставшаяся масса частиц в пульпе извлекается в бедные кон-
центраты. Из этого следует, что удельная скорость флотации бу-
дет в общем уменьшаться с развитием операции не только для
частиц флотируемого минерала, но также и для других мине-
ралов.
В несколько идеализированном примере, когда пульпа со-
стоит лишь из свободных частиц, отличающихся не только по-
форме и поверхностным свойствам, но и по размерам, будут
происходить такие же изменения индексов, но в большей степе-
ни. В общем же случае, когда присутствуют также и сростки,
проблема становится более сложной, поскольку нефлотирую-
щиеся частицы извлекаются в концентрат в виде сростков.
Все упомянутые рассуждения позволяют подойти более созна-
тельно к рассмотрению промышленного процесса флотации.
Влияние других переменных на кинетические индексы. Уве-
личение слоя пены увеличивает коэффициент минерализации
без заметного изменения удельной скорости флотации (рис. 1).
Этого можно было ожидать, поскольку коэффициент минерали-
зации характеризует различие в поведении твердого и жидкого,
в то время как удельная скорость флотации статистически опре-
деляется избирательным прилипанием твердых частиц к пузырь-
кам. Если бы пузырьки в слое пены не освобождались от твер-
дых частиц, удельная скорость флотации не зависела бы от тол-
щины слоя пены. И наоборот, если эта независимость экспери-
ментально подтверждается, то этим доказывается, что демине-
рализации в слое пены не происходит.
Увеличение количества пенообразователя незначительно из-
меняет коэффициент минерализации, но сказывается на удель-
ной скорости флотации, возможно, из-за изменения размера
пузырьков.
Повышение расхода собирателя увеличивает извлечение и
удельную скорость флотации; одновременно возрастает значе-
ние коэффициента минерализации. Например, в частном случае
при флотации галенитовой пульпы с увеличением загрузок эти-
лового -ксантогената от 0,05 до 1,5 кг/т извлечение галенита по-
высилось с 40 до 100%, а кинетические индексы возросли более
чем в 100 раз (рис. 2). Таким образом, оба кинетических индек-
са являются гораздо более чувствительными при оценке изме-
нений в процессе флотации, чем извлечение.
442
ГЛАВА 12
Изменение кинетических индексов в зависимости от размера
частиц. Влияние размера частиц на кинетические индексы изу-
чал Шлехтен ИЗ]. Он использовал флотационную машину не-
прерывного действия, по Шуману, в которой флотировал тща-
тельно очищенный, измельченный мокрым способом галенит;
чтобы определить распределение частиц по крупности в продук-
тах флотационных опытов, он применил седиментационный
анализ.
Рис. 1. Зависимость значений
коэффициента минерализации
н извлечения от толщины слоя
пены [18]:
/— коэффициент минерализации, %;
2 — удельная скорость флотации,
мнн-1
Рис. 2, Зависимость удельной
скорости флотации, коэффици-
ента минерализации и извлече-
ния от загрузки собирателя
[18]:
1— удельная скорость флотации.
мин.—1; 2 — коэффициент минера-
лизации; 3 — процент извлечения
Было установлено, что для данной загрузки собирателя
удельная скорость флотации и коэффициент минерализации
уменьшались в линейной зависимости от крупности частиц при
изменении размера частиц от 28 до 4 ц. Для частиц меньше 4ц
было установлено, что эти индексы не зависят от их размера
(рис. 3). Такие же результаты в отношении зависимости индек-
сов от размера частиц крупностью свыше 4 ц были получены и
для всех других испытанных загрузок собирателя (от нуля до
0,6 кг/т).
КИНЕТИКА
443
Для пульпы, состоящей только из галенита и воды и не имею-
щей других твердых частиц, эти результаты можно объяснить
различным поведением зерен галенита. Очевидно, зерна >4 н из-
влекаются в виде отдельных частиц, а частицы <4 ц скорее все-
го флокулируют и флотируются в виде флокул.
Рис. 3. Зависимость удельной скорости флотации и
коэффициента минерализации от размера частиц.
Минерал — галенит; собиратель — 540 г/т этилксан-
тогената калия [13]:
а — удельная скорость флотации;
б — коэффициент минерализации
Как можно судить по рис. 4 и 5, изменение коэффициента
минерализации (и в значительно меньшей степени удельной
скорости флотации) в зависимости от расхода собирателя и
размера частиц может быть очень большим: при указанном на
рис. 5 максимальном изменении загрузок собирателя и крупнос-
ти частиц 400 меш, коэффициент минерализации М возрастает
в 1000 раз.
В условиях, существовавших в пульпе (при данной степени
измельчения минерала), расход собирателя 0,6 кг/т не обеспе-
КИНЕТИКА
445
чивал мономолекулярное покрытие поверхности, но позволял
значительно гидрофобизировать поверхность всех частиц, при-
чем были получены такие же результаты, как и при загрузке со-
бирателя в количестве 0,4 «г/т.
Удельные скорости флотации при загрузке собирателя в
0,6 кг/т минерала и 0,4 кг/т очень близки; это, вероятно, объя-
сняется тем, что в обоих случаях флотация определяется для
каждого размера числом столкновений частиц с пузырьками. В
случае же меньших загрузок собирателя создается условие, ко-
гда вероятность флотации столкнувшихся пузырька и частицы
зависит от плотности пленки собирателя на поверхности части-
цы. Представляют интерес дальнейшие исследования в этом на-
правлении.
Если предположить, что при загрузке собирателя 0,6 кг/т все
столкновения оканчиваются прикреплением, то вид кривой, вы-
ражающей зависимость удельной скорости флотации от разме-
ра частиц, согласуется с уравнением, выведенным из гидроди-
намического анализа [уравнение (10) главы 11].
Зависимость между крупностью частиц и временем флота-
ции. Так как удельная скорость флотации уменьшается с умень-
шением крупности частиц до нескольких микрон в случае хоро-
шо подготовленной пульпы из чистого минерала, можно ожи-
дать, что она также будет уменьшаться с уменьшением крупно-
сти и в случае пульпы, состоящей из смеси флотационно актив-
ного и нефлотационно активного минералов, по крайней мере,
в области до критического размера частиц в несколько микрон.
В области частиц критического размера могут существовать
другие зависимости, например, благодаря образованию смешан-
ных флокул, флотационные свойства которых не выяснены.
В области размеров выше критического (обычно крупнее
5 ц, для большинства минералов) подробные исследования име-
ются в опытах Шлехтена. Так, из опыта периодической флота-
ции при фракционном съеме пены (рис. 6) видно, что для более
крупных частиц (не крупнее 28 меш) за первые отрезки времени
извлечение с увеличением размера частиц значительно быстрее
возрастает, чем за последующие промежутки времени.
Селективность и извлечение крупных частиц. Так как у круп-
ных частиц (до определенного размера) удельная скорость фло-
тации выше, чем у мелких ,в случае гидрофобного твердого, и ни-
же, чем у мелких частиц в случае гидрофильного твердого,
то относительная флотационная активность и индекс селективно-
сти между гидрофобным и гидрофильным твердым будут умень-
шаться с уменьшением размера частиц. Об этом можно судить
по рис. 7.
На этом рисунке изображены две кривые для цикла свинцо-
вой флотации. Нижняя кривая построена для частиц, содержа-
Рис. 6. Извлечение частиц галенита различных раз-
меров в зависимости от времени:
/ — 0,5 мни.; 2—1 мии.; 3 — 2 мин.; 4 — 4 мин.; 5 — 8 мин.
Размер частиц, меш
Рис. 7. Разделение минералов в свинцовом цикле
на фабрике Сулливан (Канада):
Г—только свободные частицы; 2—все частицы
кинетика
447
щих галенит, а верхняя — только для свободных зерен галенита.
Для этой кривой микроскопически определяли число сростков,
исследуя фракции по крупности в концентрате и в хвостах с по-
мощью микроскопа, применяемого в металлографии. Определен-
ные таким образом сростки вычли из общего числа частиц. Кри-
вая, построенная для всех частиц (включая сростки), имеет мак-
симум селективности в пределах крупности от 400 до 560 меш,
тогда как кривая, построенная только для свободных зерен, не
имеет максимума; наибольшая селективность достигается для
самых крупных зерен. Совершенно ясно, что существующий оп-
тимум является результатом общего эффекта кинетики флота-
ции свободных зерен и сростков.
Детальное исследование извлечения как функции от размера
частиц на действующих флотационных фабриках, перерабаты-
вающих сульфидные руды, было проведено Гро и Хендерсо-
ном [10]. Полученные ими результаты, по данным фабрики Сул-
ливан, показаны на рис. 7. В дальнейшем было получено много
данных, позволяющих заключить, что' шламы извлекаются труд-
но, флотация их происходит очень медленно и неселективно. Так-
же было подтверждено, что очень крупные частицы обычно явля-
ются сростками и поэтому флотация их менее селективна.
Интересные исследования влияния размера частиц на флота-
цию свинцово-медной руды провел Брунер [4]. Результаты этих
исследований приводятся на рис. 8. На рисунке видно, что для
испытанных размеров частиц максимума селективности не су-
ществует. Вообще, чем крупнее частицы, тем выше индекс селек-
тивности. Эта зависимость подтверждается при рассмотрении га-
ленитового или халькопиритного циклов флотации. Следует от-
метить к тому же, что в свинцовом цикле халькопирит не так хо-
рошо депрессируется, как силикатная порода. Опыты Брунера
подтвердили данные исследования Гро и Хендерсона о влиянии
сростков при флотации.
Трудно обогатимые шламы. При периодической флотации ис-
кусственной смеси галенита и гранита (с одной перечисткой кон-
центрата основной флотации) технологические результаты пока-
зали, что потери свинца в хвостах основной флотации в различ-
ных классах распределяются следующим образом: +100 меш
0,07%; — 100 + 200 меш 0,47%; —200 +400 меш 1,17% —400 +
+ 800 меш 2,30% и — 800 меш, 14,87% и в хвостах перечистки
соответственно 0,02; 0,07; 0,23; 0,29; 6,33%. ’Из этих данных оче-
видно, что фракция — 800 меш (—9 ц для галенита) относятся
к трудно обогатимому классу.
Дальнейшие исследования подтвердили трудную обогати-
мость шламов и выявили возможное влияние ряда факторов;
один из них связан с трудностью столкновения частиц с пузырь-
ками. Но одного этого фактора, по-видимому, недостаточно для
448
ГЛАВА 12
объяснения низких результатов, получаемых при флотации весь-
ма измельченных частиц. Всесторонние исследования в этом на-
правлении были проведены Малоземовым [11, 12].
Смесь галенита и апплита измельчалась в течение несколь-
Рис. 8. Зависимость индекса селективности от раз-
мера частиц для синтетической смеси галенита, халь-
копирита и гранита [4]:
fl — в цикле свинцовой флотации; б —в последующем цикле
медиой флотации; / — галеиит—силикаты; 2 — галенит—
халькопирит; 3 — халькопирит — силикаты; 4 — халькопи-
рит — галенит
В одной серии опытов собиратель (калиевый амиловый ксанто-
генат) задавался перед измельчением, в другой серии — после
измельчения. Количество собирателя задавалось пропорциональ-
но времени измельчения и составило 0,1 г на 350 г навески (70 г
галенита и 280 г апплита) при измельчении в течение часа вне
зависимости от того, загружался собиратель перед измельчением
или после него.
На рис. 9 показаны полученные результаты, из которых ясно
видно, что с увеличением тонины помола снижается индекс се-
лективности и что для любого указанного на рисунке времени
КИНЕТИКА
449
измельчения он выше, когда собиратель загружается перед из-
мельчением.
Загрузка собирателя перед измельчением несколько повыша-
ет селективность, но этого недостаточно для существенного
улучшения флотации трудно обогатимых шламов. Условия про-
ведения серии опытов Малоземовым, в которых собиратель за-
давался после измельчения, позволяют предполагать, что в
Рис. 9. Влияние места загрузки реагента на фло-
тацию смеси галенита и апплита, содержащей
преобладающее количество тонких частиц [12];
1— перед измельчением; 2— после измельчения
этом случае минералы двух типов — сульфиды и силикаты.—
взаимодействуют во время измельчения до тех пор, пока поверх-
ности их не станут подобными. Так как при измельчении сокра-
щается расстояние между частицами, возможность взаимного
загрязнения минералов увеличивается, особенно при требующем
много времени тонком измельчении. Слабая флотируемость
шламовых частиц из смеси с более крупными зернами частично
может быть объяснена старением их поверхности; однако это не
является убедительным объяснением. Можно также ожидать, что
увеличение концентрации собирателя повысит эффективность из-
влечения шламов, но лишь одного повышения концентрации со-
бирателя недостаточно.
Флотация шламов может быть значительно улучшена в не-
которых случаях (чистый галенит в силикатной породе и чистый
халькозин в силикатной породе) загрузками собирателя перед
измельчением. В этих условиях следует полагать, что поверх-
29 А. М. Годэ и
450
ГЛАВА 12
ность флотационно активных минералов предохраняется от дей-
ствия гидрофильных ионов, отрывающихся с поверхности гид-
рофильных минералов. Также возможно предположить, как это
сделал Малоземов [14], что загрузка собирателя перед измельче-
нием способствует селективной флокуляции сульфидов, по край-
ней мере в некоторых случаях при определенных сочетаниях ми-
нерал— реагент. Кинетические преимущества при флотации
сравнительно небольших по крупности флокул вместо разроз-
ненных весьма тонких частиц являются очевидными и точно со-
гласуются с выводами Шлехтена. В некоторых опытах, в кото-
рых собиратель — амиловый диксантоген — загружался в боль-
ших количествах в мельницу, он как бы цементировал частицы
сульфидного минерала, собиравшегося в компактные шарики,
тогда как частицы пустой породы оставались в диспергирован-
ном виде в пульпе. Такое образование агрегатов частиц являет-
ся’ примером слипания частиц посредством масляного мостика
[9].
Выше рассматривались две основные причины, объясняющие
слабую флотируемость шламов. Одна из них —тонкость частиц,
обусловливающая малую вероятность столкновения частиц с пу-
зырьками воздуха, другая — слабая химическая поверхностная
активность шламов, обусловливаемая воздействием ионов, вы-
деляемых другими частицами системы. Помимо этого, сущест-
вует еще ряд факторов, связанных с характером ионной оболоч-
ки, окружающей частицы и пузырьки.
Пузырьки воздуха в водной суспензии несут электрический
заряд, обычно отрицательный (гл. 5). Этот заряд обусловлива-
ется ионными парами; анионы преобладают главным образом со
стороны газовой фазы поверхности раздела, а катионы располо-
жены в диффузном слое со стороны водной фазы поверхности
раздела. Минеральные частицы также электрически заряжены
парами ионов, имеющими аналогичное расположение. Когда пу-
зырек и частица благодаря сообщенному гидродинамическому
усилию сближаются, то изменяется расположение ионов в двой-
ных слоях. Если двойные слои имеют одинаковый заряд, то для
перестройки их потребуется дополнительное усилие; если двой-
ные слои имеют противоположные заряды, то вероятно, перест-
ройка произойдет самопроизвольно, что будет способствовать
притяжению пузырька и частицы. Описанные выше положения
весьма напоминают закономерности, наблюдаемые при явлениях
флокуляции и диспергации (гл. 6).
Когда частицы имеют относительно крупные размеры (обыч-
но больше 10 ц.), электростатическое притяжение или отталкива-
ние ничтожны по сравнению с силами инерции и вязкостью. Но
для очень тонких частиц (1 и), для которых пузырьки воздуха
будут настолько крупными (обычно 1 мм в диаметре), что могут
КИНЕТИКА
451
рассматриваться как плоскость, электростатические силы, веро-
ятно, играют главную роль при приближении или отталкивании.
Математический анализ этих явлений не производился. Но все
же следует попытаться установить эмпирические зависимости
между величиной и знаком потенциалов протекания для пузырь-
ков воздуха и частиц, определенных в растворе одних и тех же
электролитов, концентрацией ионов и флотационным поведением
частиц.
Хорошо известно, что большинство собирателей анионного
типа в какой-то степени увеличивает отрицательный заряд ми-
нералов, с которыми они взаимодействуют. Эта зависимость со-
ответствует тем условии, при которх шламы не флотируют. В
то же время собиратели катионного типа уменьшают отрица-
тельный заряд минералов и при определенных концентрациях
могут изменить знак заряда на положительный. Однако еще не-
известно, что происходит с пузырьками воздуха в подобных ус-
ловиях. Если существует такая область концентраций собирате-
ля, в которой частицы имеют положительный заряд, а пузырьки
воздуха отрицательный, то это будет способствовать успешной
флотации шламов. Возможно, этим и объясняются некоторые
особенности флотации катионными собирателями, которые иног-
да предпочтительнее флотируют шламы (например, глины), чем
зернистые материалы.
Максимальный размер флотирующихся частиц. В гл. 7 эта
тема рассмотрена с теоретических позиций. Были проведены
опыты для подтверждения этих теоретических положений как
при максимальном покое (пленочная флотация), так и при пе-
ремешивании. На рис. 10 показаны опытные результаты, полу-
ченные для пяти минералов при максимальном покое. Из этого
рисунка можно заключить, что при перемешивании в условиях
обычной флотации частицы в пульпе достаточно тонко измельче-
ны, и отсутствие их флотации трудно объяснить чрезмерной
крупностью; даже если принять во внимание центробежные си-
лы, возникающие при сильном перемешивании, то и тогда опас-
ность отрыва пузырька воздуха от частицы незначительна.
Опубликованные исследования по кинетике флотации. Из чи-
сла опубликованных работ, в которых изучались различные сто-
роны этого вопроса, следует отметить исследования Белоглазова
[2], Богданова [3], Эйгелеса, [5, 6] и Волковой [21, 24] в России,
Гарсиа, Цуниги [25] в Чили, Шумана [18], Мориса [15, 16], Арбай-
тера [1] и Филлипова 117] в США и Сазерленда [9] в Австралии.
Цунига обращает внимание на экспоненциальный характер
изменения извлечения во времени, хотя быстрое снижение извле-
чения флотируемого минерала с увеличением времени уже от-
мечалось ранее [8]. Теория кинетики флотации с точки зрения хи-
мико-механических понятий была выдвинута Шуманом и успеш-
29*
452
ГЛАВА 12
но развита Сазерлендом, который считал, что флотация являет-
ся результатом встречи частиц с пузырьками, и вычислил веро-
ятность столкновения, применяя гидродинамические схемы, по-
добные описанным в главе 11 этой книги. Сазерленд также вво-
дит понятие времени индукции. Он пишет: «... прилипание не яв-
ляется мгновенным действием, поэтому для его осуществления
время контакта должно превышать определенное минимальное
время индукции». Эта гипотеза, которая кажется приемлемой,
MeialS- 200 tfOO 800
•/* 20 28 35 08 65 400 450 280 560 4420
Размер частиц
Рис. 10. Сравнительные размеры минеральных частиц, удержива-
емых силой поверхностного натяжения (приведены данные для
угля, серы, активированного медью сфалерита, халькозина и га-
ленита в растворе ксантогената):
/ — уголь; 2 —сера; 3 — сфалерит; 4—халькозин; 5 — галенит; а — об-
ласть оптимальной селекции в практических условиях: б — область опти-
мальной селекции свободных частиц; в — обычная область флотационных
пульп
используется для внесения поправки к вероятности столкнове-
ния пузырька и частицы, но значительно осложняет этот вопрос.
Конечное уравнение Сазерленда (19) для скорости флотации
показывает, что скорость флотации возрастает линейно с увели-
чением размера частиц [см. его уравнение (18)' на стр. 403] и за-
висит также от размера пузырьков, плотности пульпы и скорости
аэрации.
Сазерленд рассматривает суммарную скорость флотации ми-
нерала, состоящего из зерен различной крупности (обычный слу-
чай), и считает, что эта скорость представляет собой сумму ско-
ростей, каждая из которых является показательной функцией
времени, тогда как суммарная скорость сама по себе не является
точной показательной функцией времени. Он также учитывает
влияние одновременного присутствия пузырьков различных раз-
меров и частиц, имеюших неодинаковое время индукции, и рас-
КИНЕТИКА
453
сматривает влияние степени минерализации пузырька на его
способность к захвату дополнительных частиц.
В заключение Сазерленд приводит несколько опытов, постав-
ленных со стеклянными шариками одинакового состава и равно-
го размера с применением катионного собирателя. Результаты
этих опытов частично подтверждают его теоритические предпо-
сылки.
Весьма интересной является работа Филиппова, в которой
дается экспериментальный метод оценки времени контакта час-
тицы с пузырьком. Измеренное время контакта для относительно
крупных частиц примерно равно 10 мсек. В работе приведены
также вычисления времени контакта на основании теорий упру-
гости и поверхностного натяжения; согласно этим вычислениям,
время контакта изменяется в пределах от 1 мксек для частиц
галенита крупностью 1 ц до 15 мсек для частиц галенита круп-
ностью 1 мм [17, табл. 11. Опытные измерения проводились для
частиц крупностью 0,5—0,7 мм.
Арбайтер в одной из своих работ придерживается эмпиричес-
кого подхода к кинетике флотации; он считает, что контакт меж-
ду пузырьком и частицей может быть реакцией второго порядка.
Представляют интерес дополнительные исследования этого поло-
жения.
Зависимость извлечения и качества продукта от кинетических
индексов. Чтобы наглядно представить себе значение кинетиче-
ских индексов, небезынтересно рассмотреть их зависимость от
извлечения. Употребляя такие же обозначения, как и выше,
предположим, что количество минерала А, сфлотированное за
время dt, есть rdt. Оно равно также количеству минерала, уда-
ленному из камеры,
— Vdc = rdt. (7)
Так как r=QcV, то уравнение (7) примет следующий вид:
или после интегрирования •
1п с — — Qt + а.
Постоянная интегрирования а равна In с0, если принять гранич-
ное условие: при /=0 с = с0. Тогда
In с = — Qt + In со (8)
или
In
Со
454
ГЛАВА 12
При этом предполагается, что все частицы А идентичны и
величина Q остается постоянной в течение всего процесса флота-
ции. Следующее ограничение применимости уравнения (8) обус-
ловлено тем, что при его составлении не принимается во внима-
ние переход в концентрат значительного количества воды. Для
периодической флотации, где уровень воды поддерживается
добавками ее в камеру, это ограничение не существенно, но для
непрерывной флотации, в которой вода, удаляемая с концентра-
том, заменяется пульпой, решение уравнения (8) можно рас-
сматривать лишь в качестве первого приближения.
Время 0, необходимое для того, чтобы значение с сократилось
вдвое, может быть получено подстановкой с0/2 вместо с в урав-
нении (8); тогда
. (9)
Q
Уравнение (8) показывает, что зависимость изменения кон-
центрации компонента в пульпе от времени является экспонен-
циальной. График этой зависимости в полулогарифмических
координатах, в которых по оси абсцисс откладывается время, а
по оси ординат — логарифм концентрации, будет прямой ли-
нией; тангенс угла наклона этой линии к оси абсцисс равен Q.
В качестве примера рассмотрим свинцово-цинковую руду,
состоящую из галенита А, сфалерита В и силикатов D, в которой
все частицы свободны и каждая частица одного и того же мине-
рала имеет одинаковую скорость флотации. Конечно, такой слу-
чай является идеальным. Если QA = 1,2 мин.-1, QB = 0,1, Qd =
= 0,02 и соответствующая скорость удаления воды в пену
(%=0.05, то доля минералов и доля воды, остающихся в пульпе
к концу заданного отрезка времени, например 2 мин., могут
быть легко подсчитаны по уравнению (8). Производя расчет,
получим для галенита 9,01%, сфалерита 81,9%, пустой породы
96,1%, воды 90,5% от исходного количества каждого компо-
нента.
Эти же результаты могут быть получены графически, как по-
казано на рис. 11. При построении этого графика удобнее поль-
зоваться временем 0, за которое извлечение составит 50% под-
считанного при помощи уравнения (9): 0Л = О,577 мин., 0В=
= 6,93 мин., ()д= 34,65 мин., 0^ = 13,86 мин. Доли компонентов,
оставшихся в пульпе через 2 мин., по отношению к исходному
могут быть найдены как ординаты пересечения линий А, В, D с
вертикальной линией от абсциссы t=2\ получаемые таким спо-
собом значения соответствуют значениям, определяемым по
уравнению (8).
Реальные данные опытов флотации не совпадают с идеаль-
ными кривыми, приведенными на рис. 11, ввиду наличия срост-
КИНЕТИКА
455
ков, разного химического состава поверхности минеральных ча-
стиц, разного их размера и формы и изменения концентрации
реагента в пульпе. Все эти переменные факторы обусловливают
Рис. 11. Доля неизвлеченных компонентов из ги-
потетической флотационной пульпы в зависимо-
сти от времени флотации (теоретические расче-
ты):
/ — галенит; 2 — сфалерит; 3 — пустая порола; 4 — вода
частиц; благодаря этому все прямые линии на рис. 11 практи-
чески являются кривыми с вогнутостью, направленной вверх.
Автор не располагал хорошими экспериментальными дан-
ными (хотя они и были бы весьма желательны) в то время,
когда писался настоящий раздел.
Концентрат основной и контрольной флотации. Операция, в
которой флотационно активный минерал флотируется в первый
раз, называется черновой1 флотацией, потому что ее целью
1 В советской литературе более распространено наименование этой опе-
рации «основная» флотация. В дальнейшем в русском переводе используется
это наименование. Прим. ред.
456
ГЛАВА 12
является получение чернового концентрата, относительно незна-
чительно отличающегося от исходного продукта. Главной целью
этой операции является достижение высокого извлечения. Соглас-
но рис. 11 для проведения удовлетворительной основной флота-
ции необходимо лишь продлить время операции, что, однако,
может привести к разубоживанию концентрата из-за флотации
слабо флотирующихся минералов В и D, а также выделения в
Рис. 12. Идеализированная флотация свинцово-
цинковой руды. Оставшееся количество минера-
лов и воды, не извлеченных в основной и конт-
рольной флотации и первой перечистке контроль-
ного концентрата:
/ — вода; 2 — пустая порода; 3— сфалерит; 4— галенит
пену воды. Поэтому очень часто бывает необходимо разграни-
чить концентрат на первую фракцию, называемую черно'вым
концентратом, и вторую фракцию, называемую концентратом
контрольной флотации.
Для теоретического примера, показанного на рис. 11, кон-
центрат основной флотации может быть представлен как кон-
центрат, полученный за 2 мин., а концентрат контрольной фло-
тации за последующие 4 мин. при общем времени флотации
обоих концентратов 6 мин. Из рис. 11 или еще лучше из левой
части рис. 12, в котором ординаты представляют количества
различных составляющих пульпы в различное время, можно
определить, какое извлечение компонентов возможно получить
в черновом и контрольном концентратах и в отвальных хвостах;
эти рассуждения позволяют наметить дальнейшие операции.
При построении рис. 12 принималось, что питание флотации
содержит три части твердого и семь частей воды, и что твердое
КИНЕТИКА
457
состоит из 5% минерала А (галенита), 10% минерала В (сфа-
лерита) и 85% минерала D (силикатов). Было принято, что ве-
личина изменений Q согласуется с теоретическими кривыми,,
приведенными на рис. 11. Наклон линий на рис. 12 является,
следовательно, таким же, как и на графике рис. И, при условии
что эти графики имеют одинаковые шкалы.
Применяя уравнение (8), можно определить количество ми-
нералов, оставшихся в конце контрольной флотации. Зная коли-
чество минералов в питании и в хвостах контрольной флотации,
можно определить количество минералов в концентрате конт-
рольной флотации следующим расчетом: галенита 9,01—0,08 =
= 8,93%, сфалерита 81,9—54,9 = 27,0%, минералов пустой поро-
ды 96,1—88,6 = 7,5% и воды 90,5—74,0=16,5% (приведенные
проценты для каждого минерала представляют разность коли-
чества каждого компонента в питании и хвостах контрольной
флотации);. Из) этих .данных можно определить , содержание?
каждого минерала в концентрате контрольной флотации; гале-
нита 0,9%, сфалерита 5,6%, пустой породы 13,3% и воды
80,17%.
Перечистка контрольного концентрата. Целью получения
контрольного концентрата является обеспечение наиболее пол-
ного извлечения минералов, но такая операция будет малоцен-
на, если концентрат контрольной флотации не сможет быть
перечищен. Часто этот концентрат возвращается в голову фло-
тации.
В идеальном случае, представленном на рис. 12, концентрат
контрольной флотации содержит в шесть раз больше сфалерита,
чем галенита, а поэтому его значительно выгоднее перечистить
один раз, прежде чем соединить с рудой, поступающей в основ-
ную флотацию. Если принять, что химические и другие условия
одинаковы с условиями первичной контрольной флотации, то
четырехминутную перечистку концентрата контрольной флота-
ции можно изобразить так, как показано с правой стороны на
рис. 12. Хвосты этой перечистки имеют состав, определяемый
длиной ординаты LL 1 и содержат 0,83% РЬ, 67,0% Zn, 92,3%
пустой породы и 81,9% воды от содержания их в питании пере-
чистки. Качество хвостов перечистки примерно такое же, как и
хвостов основной флотации свинцового цикла.
Из 100 весовых единиц, поступающих в перечистную опера-
цию (0,9 галенита, 5,6 сфалерита, 13,3 пустой породы, 80,2 во-
ды), 18,22 единицы относятся к концентрату перечистки (0,92 га-
ленита, 1,85 сфалерита, 1,03 пустой породы, 14,42 воды). В этом
продукте отношение свинца к цинку примерно такое же, как и
в питании основной флотации, но содержание пустой породы на-
много меньше, а содержание воды несколько больше, чем в пи-
458
ГЛАВА 12
<00
<ор
*
0)
о У 2 о 11,2
время, мин.
Рис. 13. Идеальная
флотация свинцово-
цинковой руды. Остав-
шееся количество не-
сфлотированного мате-
риала в первой и вто-
рой перечистках черно-
вого концентрата:
./ — вода; 2 — пустая порода;
3 — сфалерит; 4 — галеиит
тании основной флотации. На практике этот концентрат направ-
ляют в основную флотацию.
Перечистка. Черновой концентрат неизменно содержит за-
метные количества нежелательных примесей. Они могут быть
отделены при перечистке концентрата. Обычно эта операция
проводится до того момента, пока соотношение между нефлоти-
рующейся частью продукта и флота-
ционно активным минералом в хвос-
тах перечистки не станет примерно та-
ким же, как и в питании основной
флотации, что позволит соединить эти
продукты.
В идеальном случае, который при-
водится ниже, полученные за 2 мин.
флотации хвосты перечистки (рис. 13)
содержали 1,29 части галенита, 4,67
части сфалерита, 10,0 частей пустой
породы и 63,0 части воды или в об-
щем 78,96 части. Этот продукт доста-
точно близок по качеству к питанию
основной флотации, что позволяет
объединить их; таким образом, выде-
ляется концентрат перечистки, содер-
жащий 13,01 части галенита, 1,03 час-
ти сфалерита, 0,40 части пустой поро-
ды и 6,60 части воды. Очевидно, этот
продукт содержит много сфалерита и
не может быть конечным, но подверг-
нуть его сразу перефлотации нельзя
из-за малого содержания воды. Это
же наблюдается и на практике. Обыч-
но для создания необходимой теку-
чести продуктов в желоба добавляют
воду, благодаря чему продукты само-
теком направляются непосредственно в следующую операцию.
Можно считать, что в большинстве случаев в результате пере-
чисток получается концентрат, содержащий 60% воды, этот кон-
центрат может стать исходным питанием при второй перечистке.
Вторая перечистка будет проводиться до того момента, ког-
да отношение нефлотирующейся части в этой операции к фло-
тационно активному минералу станет примерно таким же, как в
питании первой перечистки. Это легко достигается в течение
1,2 мин. (согласно рис. 13); хвосты перечистки содержат 56,6 ча-
сти воды, 8,52 части галенита, 2,48 части сфалерита и 0,39 части
пустой породы, а концентрат перечистки содержит 27,48 части
галенита, 0,32 части сфалерита, 0,01 части пустой породы и 3,4
КИНЕТИКА
459
части воды. Таким образом, получается очень хороший по каче-
ству продукт, вполне оправдывающий назначение данной опера-
ции. В то же время он содержит очень мало воды, так что
практически вряд ли такой продукт можно будет получить.
Таким образом, уравнение и предположения, из которых сдела-
ны эти расчеты, не являются совершенными.
Ковре питание
100(30’6* 7ОЖ)
Вода
И
6.5
# J5 _
^-перечис- ’ перечист-12
тка г- ка
14,5
I
115,5
; ОСНоЬнаЯ
флотация
104,5 , ТЛ
Контрольная 56,5(26 *62,5 j
флотация ---------------лдссты
К-т
К-т
К-т
11
К-т
16
Концещпр ат
Перечистка контроль- 12,5(2,5те *-10*)
кого концентрат а ---------»
Дополнитесь-
ные хвосты
___________f-V
Рис. 14. Принципиальная схема для цикла свинцовой флотации при обо-
гащении идеальной свинцово-цинковой руды. Показан относительный тон-
наж потока пульпы (общий, твердое и жидкое)
Рис. 14, представляет качественную схему операций, пока-
занных на рис. 12 и 13. На схеме дана взаимосвязь пяти различ-
ных операций. В каждом узле схемы дается относительный тон-
наж для потока пульпы с указанием содержания твердого и
жидкого.
Эти данные приблизительны, так как для замкнутых циклов
предположено, что возвращаемые в операцию продукты имеют
аналогичный состав с питанием данной операции.
Следует отметить, что величина потока пульпы сильно ме-
няется в различных операциях схемы; например, во второй пере-
чистке концентрата поток пульпы равен 6,5 т, а в основной фло-
тации 115 т. Очевидно, что при выборе оборудования для схемы
(подобной той, которая показана на рис. 14) требуются точные
значения потоков пульпы для выдерживания необходимого вре-
мени флотации в каждой операции.
Практические схемы. Схемы, применяемые на практике, во
многом схожи с идеальной схемой, показанной на рис. 14.
Обычно применяются две или три перечистки. Число их будет
460
ГЛАВА 12
больше, когда руда трудно обогатима или когда на концентрат
установлены высокие кондиции. Перечистка концентрата конт-
рольной флотации часто не производится: этот концентрат воз-
вращается либо в голову процесса, либо в середину основной
флотации.
Поскольку в основной флотации часто не бывает полного
раскрытия минеральных зерен, то операцию обычно проводят
таким образом, чтобы выделить наиболее чистый концентрат,
промежуточный продукт, в котором собираются сростки, и
Рис. 15. Принципиальная схема для одноминеральной флотации,
включающая доизмельчение промежуточных продуктов
хвосты. Промежуточный продукт потом доизмельчается в спе-
циальном цикле доизмельчения, обычно мало сходном с первич-
ным циклом. Одно из основных встречающихся затруднений за-
ключается в чрезмерном обводнении продукта, поступающего в
доизмельчение, что делает необходимым применять сгущение;
встречаются также затруднения в операции классификации в
цикле доизмельчения ввиду того, что слив, поступающий во фло-
тацию, весьма разжижен. Схема на рис. 15 является типичной,
включающей доизмельчение промежуточного продукта.
Полезно отметить, что в схеме, показанной на рис. 15, в
окончательный концентрат попадают те частицы, которые были
сфлотированы минимум три раза, часть частиц, которые извле-
каются в концентрат контрольной флотации, сфлотированы че-
тыре раза и много частиц, флотирующихся без сомнения боль-
шее число раз (имеется в виду возвращение различных проме-
жуточных продуктов в голову отдельных операций).
ЛИТЕРАТУРА
1. Arbiter Nathaniel. Flotation rates and flotation efficiency, Min.
Eng., 3, 791—796, 1951.
КИНЕТИКА
461
2. Белоглазов К. Ф. Кинетика флотационного процесса, Цветные ме-
таллы (9), 70—76, 1939.
3. Богданов О. С. и М. Ш. Филановский. К вопросу о прикреплении
минеральных частичек к пузырькам воздуха, Ж.Ф.Х., 14. 244—247. 1940.
4. Brunner J. J. The effect of particle size on flotation, M. S. thesis, Mon-
tana School of Mines, 1931.
5. Эй re лес M. А. Кинетика прилипания минеральных частиц к воздуш-
ным пузырькам во флотационных суспензиях, ДАН СССР, XXIV, 342—
346, 1939.
<5. Э й г е л е с М. А. Кинетика минерализации воздушного пузырька во фло-
тационных суспензиях, Цветные металлы, (2), 39—45, 1940.
7. G a u d i n А. М. Selectivity index: a yardstick of the segregation accomplis-
hed by concentrating operations, Trans. AIMME, 87, 483—487, 1930.
8. Gaudin A. M. «Flotation», Table 13, p. 116, 1st et., McGraw-Hill Book
Company, Inc., New York, 1932.
9. Gaudin A. M. The greg family—Magne, lono, Dermo, Oleo, Aqua, and
Bulla, Mining and Met., 25, 347, 1944.
10. Gaudin A. M., J. O. Groh, and H. B. Henderson. Effect of par-
ticle size on flotation, AIMME Tech. Publ. 414, 1931.
11. Gaudin A. M., and Plato Malozemoff. Recovery by flotation of
mineral particles of colloidal size, J. Phys. Chem., 37, 597—607, 1933.
12. Gaudin A. M., and Plato Malozemoff. Hypothesis for the nonflo-
tation of sulfide minerals of near colloidal size, Trans. AIMME, 112,
303—318, 1934.
13. Gaudin A. M., R. Schuhmann, Jr., and A. W. S ch lech ten. Flo-
tation kinetics. II. The effect of size on behavior of galena particles.
J. Phys. Chem., 46, 902—910, 1942.
14. M a 1 о z e m о f f, Plato. Flotation of sulfides from mineral mixtures
ground extremely fine, I, M. S. thesis, Montana School of Mines, 1932; ibid.,
Report, Montana School of Mines, 1933; ibid. HI, Report, Montana School
of Mines, 1934.
15. Morris, Thomas M. Measurement of equilibrium forces between an air
bubble and an attached solid in water, Trans. AIMME, 187, 91—95, 1950.
16. Morris Thomas M. Measurement of flotation rate as a function of
particle size, Univ. Microfilms, Ann Arbor, Mich., Publ. 1792, 1951.
17. P h 111 p о f f W. Some dynamic phenomena in flotation, Min. Eng., 4, 386—
390, 1952.
18. Schuhmann R., Jr. Flotation kinetics. I, Methods for steady-state study
of flotation problems, J. Phys. Chem., 46, 891—902, 1942.
19. Sutherland K. L. Kinetics of the flotation process, J. Phys. Chem., 52,
394—424, 1948.
20. Volkova Z. Laws of adhesion in flotation, Acta Physicochim., U. R. S. S.,
20, 467—478, 1945.
21. Volkova Z. The bubble mineralization process of flotation, Compt. rend,
acad. sci. U. R. S. S., 51, 449—452, 1946.
22. Volkova Z. The process of bubble mineralization, Acta Physicochim.
U. R. S. S., 21, 171—186, 563—574, 1946.
23. V о 1 k о v a Z. Floatability of solids, J. Phys. Chem. U. R. S. S., 20,
1213—1224, 1946.
24. V о 1 k о v a Z. The laws governing the process of separation of solids of
different floatabilities, Acta Physicochim. U. R. S. S., 21, 1405—1113, 1946,
22, 331—337, 1947.
25. Zuniga, H. Garcia. The efficiency obtained by flotation is an exponen-
tial function of time, Bol. minero Soc пас. minera (Santiago Chile). 47.
83—86, 1935.
Глава 13
МИНЕРАЛОГИЯ, СРОСТКИ И ИХ ОСВОБОЖДЕНИЕ
При теоретических исследованиях процесса флотации обыч-
но обращалось особое внимание на флотационные свойства
твердых частиц однородного строения. Однако при обработке
руд приходится иметь дело с частицами, поверхности которых
неоднородны по своему минералогическому составу, иначе
говоря, со сростками минералов. Флотационные свойства срост-
ков представляют среднее между флотационными свойствами
каждого из компонентов. В то же время у частиц различных раз-
меров способность к флотации неодинакова, даже если их по-
верхности однородны по своему минералогическому составу.
Следовательно, частицы, присутствующие в рудных пульпах,
имеют различную флотационную активность, которая зависит от
химических свойств поверхности и размера частиц. Второстепен-
ными факторами являются форма и угловатость частиц, так же
как их удельный вес.
Текстура, структура и генезис руды
Определить минеральный состав руды в первом приближе-
нии можно с помощью макроскопического исследования. Жела-
тельно, чтобы исследователь обладал знаниями в области ми-
нералогии [8. 12L Однако детальное изучение невозможно без
микроскопического анализа. При микроскопическом исследова-
нии существуют два основных направления: одно из них исполь-
зует оптические характеристики минералов, полученные в про-
ходящем свете при помощи петрографических микроскопов (эта
область знания называется оптической минералогией) [2, 15, 221;
другое направление в исследовании использует отраженный
свет и металлографические микроскопы (эта область исследова-
ния называется минералографией) [13, 16, 19]
Микроскопический анализ руд также показывает, каким
образом ассоциируются различные минералы, что позволяет
определить текстуру и структуру руды [7]. Под термином тек-
стуры руды здесь понимается полуколичественное измерение
размера зерен, ее составляющих. Оценка текстуры руды являет-
ся важным фактором при установлении размера, до которого
должна быть измельчена руда. Для этого используются различ-
МИНЕРАЛОГИЯ, СРОСТКИ И ИХ ОСВОБОЖДЕНИЕ
463
ные микроскопы. Если большая часть минералов прозрачна, то
применяется петрографический микроскоп, с помощью которого
руда исследуется в тонком слое или в виде свободных кристал-
лических частиц, погруженных в жидкую среду, с соответствую-
щим показателем преломления. Для непрозрачных минералов
требуются полированные шлифы. Особенно эффективны иссле-
дования с маломощными объективами, погруженными в масло.
Это происходит потому, что отражательная способность минера-
лов значительно уменьшается в среде с показателем преломле-
ния 1,5 (вместо 1,0 для воздуха), а также потому, что для мине-
ралов с низкой отражательной способностью это уменьшение в
способности к отражению соответственно выше. Это, в свою
очередь, увеличивает разницу во внешнем виде минералов.
К тому же применение маломощных объективов, погруженных
в масло, значительно повышает точность наблюдения ани-
зотропии анизотропных минералов.
Влияние строения (текстуры) руды на выбор метода обога-
щения. Иногда встречаются руды с такой крупной вкраплен-
ностью, что полное освобождение зерен полезных минералов
происходит при дроблении руды до крупности, значительно пре-
вышающей флотационную. Однако таких крупновкрапленных
руд, которые легко обогащаются гравитационными методами,
очень мало, и они постепенно вырабатываются.
На рис. 1 показан участок шлифа свинцово-цинковой руды с
высоким содержанием металлов при сравнительно низком уве-
личении (примерно восьмикратном). В основной массе галенита
видны округленные зерна сфалерита, некоторые из них сдвоены.
Освобождение одного минерала от другого в основном может
быть достигнуто при сравнительно крупном измельчении (до
1 мм). Другие руды настолько тонко вкраплены, что измельче-
ние их до наибольшей крупности частиц, при которой возможна
флотация, не приводит к раскрытию зерен минералов. В таких
случаях, хотя и происходит некоторое освобождение минералов
при измельчении до 48—65 меш, значительная часть более круп-
ных частиц представляет собой сростки. Подобные руды необхо-
димо измельчать значительно тоньше максимально допустимого
размера зерен при флотации. Такие руды встречаются очень
часто, и запасы их являются источником наибольшей части
мировой добычи металлов.
Образец руды, требующей тонкого измельчения, показан на
рис. 2. Здесь, так же как и на рис. 1, показана свинцово-цинко-
вая руда, но при увеличении в 100 раз. Несмотря на более круп-
ное увеличение, размеры зерен на втором рисунке мельче. В этом
случае требуется измельчение до 40 ц вместо 1 мм, чтобы до-
стигнуть одинаковой степени освобождения минералов. В каче-
стве хорошо известного примера можно привести руду место-
Рис. 1. Крупновкрапленная свинцово-цинковая руда. X 8.
Проба руды месторождения Брокен-Хилл, Северная Ро-
дезия. Галенит (белый), сфалерит (серый). Размер ча-
стиц в среднем равен 1,5 мм
Рис. 2. Тонковкрапленная свинцово-цинковая руда. X 100.
Проба руды месторождения Гоппештейн на Лечберге.
Галенит (белый), сфалерит (серый) и пустая порода
(темно-серый). Размер частиц в среднем равен 30 р.
МИНЕРАЛОГИЯ, СРОСТКИ И ИХ ОСВОБОЖДЕНИЕ
455
рождения Сулливан, принадлежащего Канадской объединенной
горнозаводской компании. При макроскопическом осмотре
образцов этой руды, в основном состоящей из пирротина, пирита,
марматита и галенита [3], трудно обнаружить отдельные компо-
ненты; это возможно только при микроскопическом исследова-
нии. На рис. 3 приведен шлиф при увеличении в 800 раз, пока-
зывающий исключительно тонкое взаимопрорастание галенита,
пирротина и марматита. Средний размер зерен минералов при-
мерно равен 10 |i.
Влияние специфических особенностей структуры руды на
необходимую крупность ее измельчения. Некоторые руды, кажу-
щиеся на первый взгляд очень
тонко вкрапленными, требующи-
ми особо тонкого измельчения,
иногда обладают такими благо-
приятными особенностями строе-
ния, что уже при сравнительно
грубом их измельчении возмож-
но отделить значительное коли-
чество пустой породы. Метод от-
деления отвальных хвостов пос-
ле сравнительно крупного из-
мельчения с последующим тон-
ким измельчением и флотацией
лишь концентратов, дает хоро-
Рис. 3. Весьма тонко вкрап-
ленная свинцово-цинковая ру-
да. Х800. Проба руды руд-
ника Сулливан, Британская
Колумбия, Канада. Галенит
(белый), марматит (черный),
пирротин (вкрапленный)
шие экономические результаты,
так как расходы на измельчение
уменьшаются. Такая схема при-
меняется на некоторых обогати-
тельных фабриках, особенно для
руд зоны вторичного обогащения
или для таких руд, в которых
наиболее легко флотирующийся минерал встречается в виде че-
шуек (например, графит или молибденит).
При дроблении руд, состоящих из твердых и мягких или
хрупких и прочных минералов, взаимно тонко прорастающих,
разлом происходит по месту соприкосновения зерен отдельных
минералов или поперек более слабого зерна. Благодаря мяг-
кости и хрупкости некоторых сульфидных минералов они срав-
нительно легко отделяются от жильных минералов. При общем
грубом измельчении руды отдельные частицы сульфидных ми-
нералов получаются настолько мелкими, что приобретают вы-
сокую флотационную способность. Нужно подчеркнуть значение
характерных особенностей строения различных составных ча-
стей руды и воспользоваться ими, чтобы довести расходы на
Разделение составных частей руды дроблением и тонким измель-
30 А. М. Годэн
466
ГЛАВА 13
чением до минимума. Представляется возможным поставить
ряд исследований в этом направлении.
Влияние чрезвычайно тонкой вкрапленности руды на методы
ее обогащения. В некоторых рудах минералы настолько тонко-
вкраплены, что для их раскрытия требуется очень тонкое из-
мельчение. Стоимость такого измельчения может оказаться зна-
чительной, и тогда обогащение руды станет нерентабельным
даже в том случае, если разделение можно будет осуществить
с помощью флотации.
В качестве примера чрезвычайно тонко вкрапленной руды,
которую вследствие этого при современном состоянии техники
невыгодно обогащать, можно привести руду, где халькопирит
вкраплен в сфалерит. Эти включения, образовавшиеся осажде-
нием из твердых растворов вследствие их охлаждения, часто
располагаются вдоль кристаллографических осей, как это мож-
но видеть на рис. 4. Размер таких включений варьирует от долей
микрона до 5 ц при осаждении сфалерита и до 50 ц и более в
случае образования зерен халькопирита на границах нескольких
кристаллов сфалерита. На рис. 5 показан шлиф цинкового кон-
центрата, полученного на фабрике Мидвэйл (Юта). На рисунке
отчетливо видны тонкие включения размером от 6 до 2 и.
Так как медь (равно как и серебро) менее ценна в цинковом
продукте, нежели в свинцовом или медном концентратах, то
включения халькопирита пытаются удалить из цинкового кон-
центрата. Однако доизмельчение и повторная флотация цинко-
вых концентратов, содержащих серебро и медь, невыгодны, даже
если не учитывать трудности флотации очень тонких частиц
после доизмельчения.
Влияние генезиса руды. Генезис определяет строение руд
и, следовательно, сильно влияет на их способность к флотации.
Сульфидные руды, как известно, образовались выделением из
расплавленной магмы или осаждением из подымавшихся из
недр горячих минеральных растворов, насыщенных газами.
Сульфидные руды всегда плотные; несмотря на трещиноватости,
сбросы, сдвиги, которые сопровождают образование этих место-
рождений, пористости не наблюдается.
Окисленные руды образовались под воздействием кислорода
на сульфидные руды. Некоторая наиболее растворимая часть
сульфидной руды выщелачивалась обогащенными кислородом
водами. В результате взаимодействия растворенных рудных
компонентов с веществами, принесенными циркулирующими1
водными растворами, осаждались новые минералы. Окисленные
руды состоят главным образом из окислов, сульфатов, карбона-
тов и силикатов. Кроме того, иногда встречаются хлористые,
фосфорнокислые, хромовокислые и тому подобные соединения..
Окисленные руды имеют землистый вид и не обладают металли-
Рис. 4. Исключительно тесное вкрапление халькопирита в
сфалерите. X 200. Некоторые зерна халькопирита распо-
ложены вдоль кристаллографической оси сфалерита, а
другие — по границам зерен сфалерита
Рис. 5. Шлиф цинкового флотационного концентрата, фрак-
ция 325—400 меш, из рудника Мидвэйл, Юта. X 1000. Ча-
стицы сфалерита или сфалерита с включениями халько-
пирита
30*
468
ГЛАВА 13
ческим блеском. Они характеризуются микрокристаллическим
.строением .и иногда пористы, .в противоположность крупнокри-
сталлическим и плотным сульфидным рудам. Кроме того, окис-
ленные руды исключительно разнообразны по химическому сос-
таву даже в пределах небольшого участка. Это отражает мест-
ные колебания в количестве кислорода и в проникновении воды
Рис. 6. Полированный шлиф угля. X 200. Коксу-
ющийся уголь Рурского месторождения. На шли-
фе видны слои фюзена (сверху), дюрена или ат-
трита (в центре), витрена или антраксилона
(внизу)
сквозь трещины и щели. Землистый внешний вид зависит не толь-
ко от низкого показателя преломления составляющих минера-
лов, но и от размера их зерен, которые могут быть так малы, что
их можно сравнивать с длиной световой волны.
Уголь и осадочные руды, как например фосфаты, подобны
окисленным рудам в отношении величины составляющих ча-
стиц. В обоих случаях, однако, зоны однородного характера мо-
гут быть относительно широки (рис. 6). Рис. 7 и 8, на которых
показана кубинская марганцевая руда, дают некоторое пред-
ставление о чрезвычайно тесной ассоциации слагающих ее
зерен, аналогично встречаемой в рудах окисленных минералов.
На рис. 7 можно увидеть идиоморфные силикаты, вкрапленные
в двуокись марганца. Вмещающая порода выглядит бесструк-
турной, но это лишь результат несовершенства метода наблюде-
1
Рис. 7. Полированный шлиф, богатой марганцем частицы из марганце-
вой руды. Месторождение Кристо, Куба. X 500. Светлые участки — пи-
ролюзит и псиломелан, темные — угловатые включения силикатных
минералов
Рис. 8. Полированный шлиф простой частицы из окислов марганца.
X 200. Месторождение Кристо, Куба. Светлые участки — пиролюзит и
псиломелан, серые — бакелит, цементирующий частицы, темно-серые —
выбоины
470
ГЛАВА 13
ния. Если поверхность шлифа рассматривать в поляризованном
свете, то можно увидеть структурное строение марганцевых ми-
нералов. На рис. 8 (увеличение 1200) можно видеть очень боль-
шую пористость некоторых зерен, содержащих марганец и во-
локна псиломелана (обладающего высокой отражательной
способностью), окруженные бакелитом, в котором минерал был
закреплен перед шлифованием.
Рис. 9. Полированный шлиф частицы, состоящий из халько-
пирита и пирита. X 300. Рудник Метахамбра, Пииар дель
Рио, Куба. Медный флотационный концентрат, класс 65—
100 меш
Влияние последовательности минерализации на структуру и
флотацию сульфидных руд. В значительном числе сульфидных
руд могут быть замечены различные стадии минерализации.
Структура минералов, относящихся к разным стадиям, различ-
на. Более старые минералы в основном разъединены и разруше-
ны, в то время как минералы, образовавшиеся позже, окружают
и цементируют их. Первичные минералы весьма тверды (на-
пример, пирит); вторичные минералы, как правило, мягкие, по-
этому распад зерен при дроблении происходит главным обра-
зом по границам между отдельными зернами. При приготовле-
нии шлифа требуется особенно тщательная полировка, так как
пирит много тверже, чем галенит, халькопирит и халькозин.
В противном случае на полированном шлифе виден только пи-
рит, а более мягкие минералы выкрашиваются и оставляют тем-
ные пятна. На рис. 9 показана увеличенная частица флотацион-
МИНЕРАЛОГИЯ, СРОСТКИ И ИХ ОСВОБОЖДЕНИЕ
471
«ого концентрата средних размеров, состоящая из пирита и
халькопирита. Отсутствие резкой разницы уровней поверхности
на границе зерен пирит — халькопирит является показателем
хорошей полировки. Частицы имеют длину около 200 ц (65
меш); встречаются зерна пирита размером 10—30 ц.
Вторичное обогащение. Вторичное обогащение представляет
собой процесс покрытия поверхности первичных сульфидных
минералов другими минералами, содержащими тот же или
другой металл. Таким образом, малоценные сульфиды, например
пирротин или пирит, могут быть покрыты ценным халькозином;
ценный сульфид — сфалерит может быть покрыт другим цен-
ным сульфидом, таким как аргентит; медьсодержащий суль-
фид— халькопирит — может быть покрыт другим медьсодержа-
щим сульфидом — ковеллином. Толщина покрытий, возникших
в результате вторичного обогащения, значительно колеблется: от
очень тонких, окрашивающих минерал пленок до полного заме-
щения первичного минерала. В тех случаях, когда вторичное обо-
гащение достигает стадии полного замещения первичных сульфи-
дов, характер строения руды такой же, как и у руд различной
стадии первичной минерализации (см. рис. 5). Когда же вторич-
ное обогащение только, начинается, покрытия могут быть на-
столько тонкими, что они не видны (в шлифе) даже под микро-
скопом. Силы сцепления тонкого покрытия с основным минера-
лом могут быть более прочны, нежели толстого. В результате
этого поверхности большого количества первичного сульфида
приобретают свойства, которыми обладает покрывающее вещест-
во. Даже в случае, если сцепление у этих тонких пленок с первич-
ными минералами не прочнее, чем у толстых пленок, все же в
концентрат переходило бы большое количество минералов, обра-
зующих внутреннюю часть зерна. Отсюда очевидно, что тонкие
покрытия вторичных сульфидов на малоценных сульфидах либо
на сульфидах, которые должны быть выделены в концентрат, так
же важны, как и толстые покрытия, иногда целиком замещаю-
щие первичнй минерал. Интересным примером вторичного обога-
щения «окрашивающего» типа является руда месторождения
Игл Майн, Бонанца (Колорадо), описанная Вюншем [23], кото-
рый говорит, что «... большая часть цинковой обманки покрыта
черным, как сажа, тонким слоем вторичного аргентита». На
рис. 10 показано вторичное обогащение в процессе замещения.
Вторичное обогащение медных минералов. Процессы вто-
ричного обогащения сыграли особенно важную роль при обра-
зовании медных руд. Причиной этому, по-видимому, послужила
крайне трудная растворимость сульфидных минералов меди,
которые вместе с сернистыми минералами ртути и серебра яв-
ляются наименее растворимыми сульфидными соединениями
тяжелых металлов.
М2
ГЛАВА 13
В тех рудах, которые содержат очень мало пирита или сов-
сем его не содержат, вторичное обогащение происходит на по-
верхности первичных минералов меди (главным образом халь-
копирита). В этих случаях нет никакой необходимости измель-
чать руду настолько, чтобы отделить минерал, образующий
внешнюю оболочку, от первичных минералов.
В тех случаях, когда вторичному обогащению подвергалась
медно-пиритная руда, ее необходимо измельчать до такой сте-
пени, чтобы удалить пленки халькозина, ковеллина, борнита
Рис. 10. Полированный шлиф руды. X 100. Рудник Tcv-
меб, Юго-Западная Африка. Халькозин заместил
сфалерит
или халькопирита с поверхности зерен пирита и таким образом
отделить сульфиды меди от малоценного пирита. Хотя пленки
вторичных минералов меди образуются почти на всех сульфид-
ных минералах, находящихся в руде, однако, к счастью, наи-
большая часть этих пленок располагается на ядрах, состоящих
из медных сульфидных минералов [1, 18].
Ввиду того что образовавшиеся на пирите пленки серни-
стых минералов меди тонки, количество меди, теряемое в случае
выделения пирита в хвосты флотации, не было бы значитель-
ным. Однако на практике почти невозможно отделить флота-
цией зерна пирита, покрытые пленками сульфидных минералов
меди, от собственно медных сульфидных минералов, так как по-
верхности их обладают одинаковыми свойствами. Удалить
МИНЕРАЛОГИЯ, СРОСТКИ И ИХ ОСВОБОЖДЕНИЕ
473
тончайшие пленки сульфидов меди с частиц пирита можно, при-
менив тонкое измельчение медноколчеданных руд, если вторич-
ное обогащение происходило в ограниченных количествах. Кро-
ме того, удалять сульфидные пленки можно при помощи соот-
ветствующих растворителей.
Влияние гидротермальных изменений пустой породы на
свойства флотационной пульпы. Растворы, образующие отложе-
ния руд, или растворы, близкие к ним, могут изменять горные
породы, через которые они циркулируют. Особенно важны сле-
дующие изменения: окремнение, каолинизация, хлоритизация,,
серитизация. При окремнении изменяются осадочные и извер-
женные породы как за счет растворения некоторых составных
частей породы, так и вследствие отложения цементирующего
кремнезема.
Каолинизация и серцитизация являются процессами измене-
ния полевых шпатов. Каолин и серицит представляют водные
силикаты алюминия, обладающие скрытокристаллическим че-
шуйчатым строением; отдельные кристаллы полевого шпата
в граните или диорите часто замещаются миллионами серици-
товых или каолиновых зерен, образующих чешуйчатые ско-
пления.
Хлоритизация железисто-магнезиальных минералов проис-
ходит таким же путем. Глинистые минералы обладают огром-
ной удельной поверхностью и способностью к ионообменным
процессам. Вместе с серицитом, каолином и хлоритом эти гли-
нистые минералы дают громадное количество тонких частиц,
даже при сравнительно крупном измельчении. Значительная
часть так называемых «первичных шламов» образуется именно-
вследствие наличия в рудах подобных микрокристаллических
минералов [4, 7, 9, 1II.
Окисленные руды. Окисленные руды более сложны, нежели
сульфидные, из которых они образуются. Активными вещества-
ми, участвующими в их образовании, являются прежде всего
кислород, углекислота и вода, а затем соли металлов, способ-
ных иметь различные валентности, например таких, как серно-
кислые соли железа. Возможность одновременного нахождения
в руде различных продуктов окисления объясняется сложным
взаимодействием различных факторов и, кроме того, высокой
концентрацией реагирующих веществ.
Природный процесс образования новых минералов в зоне
окисления не был стабильным, в нем происходили сезонные из-
менения, связанные, с одной стороны, с периодами относитель-
ной сухости и окисления, а, с другой — с периодами большой
влажности и растворения. Эти изменения вместе с понижением
температуры, также влияющей на реакцию окисления, способст-
вовали увеличению количества микрокристаллических и даже
474
ГЛАВА 13
аморфных продуктов окисления. Одновременно это приводило
к осаждению пленок лимонита и образованию глинистых частиц,
которые покрывали поверхность ценных минералов и заполняли
трещинки между ними. Ценные минералы, в свою очередь, могут
быть псевдоморфными по первичным сульфидным минералам.
Все это обусловливает высокий расход реагентов при флотации
окисленных руд.
Рис. И. Полиров,энный шлиф полуокисленной медной
руды. X 250. Рудник Тсумеб, Юго-Западная Африка.
Теннантит (белый) окружен халькозином (серый) азу-
ритом и хризоколлой (черный)
Смешанные руды. Частично окисленные руды еще более
•сложны, нежели окисленные, так как, кроме сложных минера-
лов, входящих в состав сульфидных руд, в них содержится це-
лый ряд окисленных минералов. Так, по исследованиям А. В. Ха-
на, смешанная свинцово-серебряная руда из округа Тинтик
содержит по меньшей мере тринадцать различных форм сереб-
ряных минералов. Понятно, что руды подобного состава очень
трудно обогащаются при флотации. На рис. 11 показан полиро-
ванный шлиф медной руды, содержащей теннантит (медно-
мышьяковистый сульфид) в качестве первичного минерала,
вторично обогащенного халькозином и затем окисленного до
азурита и хризоколлы. Этот шлиф иллюстрирует чрезвычайно
сложный и разнородный по составу минералов характер полу-
окисленных руд.
МИНЕРАЛОГИЯ, СРОСТКИ И ИХ ОСВОБОЖДЕНИЕ
475
Потускнение поверхности минералов. Флотационные свойст-
ва минералов, имеющих свежие и потускневшие поверхности,
весьма различны. Это различие возникает оттого, что на по-
верхностях, значительно потускневших до изменения цвета или
появления радужной окраски (иридесценции), образуется плен-
ка продуктов окисления толщиной в несколько молекул. По
флотационным свойствам минералы с потускневшими поверхно-
стями ближе к окисленным минералам, чем к сульфидам со
свежей поверхностью. Даже в монометаллических рудах от-
дельные зерна сульфидных минералов значительно отличаются
по степени окисления, что может привести к ухудшению резуль-
татов флотации. Образование окисленных пленок может проис-
ходить в недрах земли до вскрытия месторождения; оно проис-
ходит во время добычи при воздействии на руду рудничных
вод и воздуха; наконец, окисление продолжается при хранении
руды. Все эти причины окисления руды (за исключением естест-
венного окисления на месте залегания) можно, вероятно, ча-
стично устранить либо ослабить их действие, изменив методы
добычи, хранения и дробления руды. Однако это выгодно толь-
ко в том случае, если все произведенные дополнительные за-
траты компенсируются резким улучшением результатов обога-
щения.
Раскрытие минералов
Обычно под словами «зерно» или «частица» понимаются не-
большие кусочки твердых веществ. У минералогов выражение
«зерно» применяется для обозначения минеральных отдельно-
стей, видимых под микроскопом в шлифе минерала или горной
породы. В этой книге выражение «зерно» применяется в том
смысле, как им пользуются минералоги, а под выражением «ча-
стица» понимается небольшая часть твердого вещества, отде-
ленная от общей массы. Таким образом, частица может состо-
ять из одного или нескольких зерен. Если каждая частица ка-
кой-либо руды состоит только из одного зерна, то говорят, что
эти частицы свободны и что раскрытие минералов полное. Если
раскрытие неполное, то процент освобождения какого-либо од-
ного минерала есть отношение количества освобожденного ми-
нерала ко всему количеству данного минерала в руде. Частицы,
содержащие зерна нескольких минералов, называются срост-
ками.
Раскрытие частиц какой-либо руды осуществляется дробле-
нием и измельчением. Дробление применяется для раскрытия
относительно крупных частиц, а измельчение — для мелких.
В случае идеального измельчения происходил бы разрыв свя-
зей между соприкасающимися зернами без разрушения самих
зерен. При дроблении большинства руд наблюдается стремле-
476
ГЛАВА 13
ние к разлому между зернами, а не поперек зерен, хотя пос-
леднее также имеет место и наблюдается в большей или мень-
шей степени [5]. Вообще говоря, чем руда тверже, тем больше
вероятность раскола самих зерен и тем труднее использовать
свойство руды распадаться по плоскостям соприкосновения раз-
личных зерен. По мере того, как при дроблении получаются все
более тонкие продукты, раскрытие минералов увеличивается, но
вместе с тем увеличивается и число случаев раскола отдельных
свободных зерен. В результате происходит переизмельчение
свободных частиц. Это переизмельчение сопровождается беспо-
лезной тратой энергии, уменьшением пропускной способности
дробильной аппаратуры и увеличением расходов на ее ремонт
и эксплуатацию. Если подобного рода переизмельчение спо-
собствует улучшению технологических результатов обогащения,
то его безусловно нужно применять, но, к сожалению, переиз-
мельчение обычно ухудшает результаты обогащения вследствие
того, что чрезмерно тонкие частицы обладают слабой флотаци-
онной способностью. С точки зрения экономики весьма жела-
тельно осуществить возможно более полное раскрытие зерен
минералов в руде, но ограничить до самого необходимого мини-
мума их переизмельчение.
Практика дробления и измельчения. Перед тем как перейти
к рассмотрению вопроса раскрытия сростков в дробленой руде,
необходимо сделать краткий обзор схем дробления и измельче-
ния [6, 14, 20, 21].
Было бы желательно производить раскрытие минералов дроб-
лением в один прием, однако обычно это неосуществимо. Наобо-
рот, на практике было установлено, что каждый тип измельчи-
тельных машин дает удовлетворительные результаты только при
небольшой степени измельчения, поэтому необходимо применять
несколько дробильных аппаратов, расположенных последова-
тельно. Подобного рода дробление и измельчение называют ста-
диальным дроблением и измельчением.
В обычной практике для измельчения крупнокусковой руды,
выдаваемой рудником, до размера зерен, пригодных для флота-
ции, применяется от трех до шести стадий. Принято все операции
измельчения разделять на крупное дробление, среднее дробле-
ние и измельчение. Предельные размеры частиц для этих трех
стадий измельчения довольно неопределенны, но в качестве ча-
стиц, лежащих по своей величине как бы на границе, разделя-
ющей отдельные стадии, в среднем принимают 100 и 12 мм.
Для крупного дробления применяют конические и щековые
дробилки. Как в тех, так и в других дробление руды происходит
между неподвижными и подвижными дробящими телами, изго-
товленными из твердого металла; применяются они для дробле-
ния материала крупностью от 1500 до 75 мм.
МИНЕРАЛОГИЯ, СРОСТКИ И ИХ ОСВОБОЖДЕНИЕ
477
Для среднего дробления обычно используют только кониче-
ские дробилки, которые работают по принципу раздавливания,
подобно коническим дробилкам для крупного дробления, и
применяются при дроблении руды от 100 до 12 мм.
Для тонкого измельчения наибольшее применение получили
вращающиеся цилиндрические или конические мельницы, из-
мельчающей средой в которых являются стержни, шары или
галька. В зависимости от измельчающей среды они называются
стержневыми, шаровыми или галечными мельницами. Стержне-
вые мельницы не дают таких тонких продуктов измельчения,
как шаровые, но зато в них можно загружать более крупный
материал. Стержневые мельницы обычно применяются в первой
стадии измельчения. Шаровые мельницы являются стандарт-
ным аппаратом для тонкого измельчения. Шары выполняются
из литого чугуна, литой стали, специальных сталей. Диаметр
шаров от 100 до 20 мм. Вместо стальных шаров иногда поль-
зуются галькой либо кусками твердой руды.
Дробильные аппараты в основном применяются вместе с ап-
паратами для классификации, которые необходимы для выделе-
ния готового продукта из руды и продуктов дробления, направ-
ляемых в следующие стадии дробления. В результате класси-
фикации получается экономия в расходе энергии и уменьшается
переизмельчение руды.
Аппараты для классификации разделяются на два типа: гро-
хоты и классификаторы. На грохотах частицы разделяются по
их действительной величине; они эффективны и экономичны для
классификации частиц крупностью более 1 мм, однако могут
быть использованы и для более тонкой классификации, хотя это
повышает стоимость операции. Классификаторы разделяют ча-
стицы по скорости их осаждения в воде; эта скорость зависит
от величины частиц и их удельного веса. В настоящее время
классификаторы используются для разделения частиц менее
2 мм и крупнее 0,01 мм. Ввиду этого при крупном и среднем
дроблении для классификации применяют грохоты, а при тон-
ком измельчении — классификаторы.
Руда дробится обычно в сухом виде (т. е. с естественной
влажностью), а измельчение производится в водной среде.
Многие дробильные аппараты используются в замкнутом ци-
кле с грохотом или классификатором, т. е. крупный материал, вы-
деленный при классификации, возвращается обратно в дробилку
или мельницу для повторного измельчения.
Типовая схема дробления и измельчения. На рис. 12 приведе-
на типичная схема дробления и измельчения, применяемая для
переработки сульфидных руд. Из приемного бункера 1 руда на-
правляется на колосниковый грохот 2 с щелью около 90 мм; над-
решетный продукт поступает в конусную дробилку для крупного
478
ГЛАВА 13
Рис. 12. Типичная
схема цепи аппа-
ратов в цикле
дробления и из-
мельчения
дробления 3 с загрузочной щелью 500 мм. Дробленый продукт
соединяется с подрешетным продуктом колосникового грохота и
направляется на вибрационный грохот 4 с квадратными отвер-
стиями 19 мм. Надрешетный продукт вибрационного грохота дро-
бится в конической дробилке 5 (разгрузочная щель 10 мм), подре-
шетный продукт вместе с дробленым продуктом складируется в
бункер 6 дробленой руды. Во избежание
простоев в случае аварий и поломок обыч-
но устанавливают резервные грохоты и дро-
билки. Руда из бункера дробленой руды на-
правляется на несколько секций, каждая из
которых оборудована шаровой мельницей
7, работающей в замкнутом цикле с рееч-
ным или спиральным классификатором 8,
которые выдают готовый продукт для фло-
тации крупностью 48 меш. Переработка ру-
ды до бункера дробленой руды ведется
обычно периодически, но после бункера —
непрерывно. Таким образом, дробление ру-
ды может выполняться в течение одной сме-
ны, тогда как отделения измельчения и
флотации работают круглосуточно.
Современные приемы подготовки руды
к флотации. Последние усовершенствова-
ния в схемах измельчения руд характери-
зуются введением стержневых мельниц пе-
ред шаровыми мельницами и выбором бо-
лее компактных аппаратов для классифи-
кации в циклах с шаровыми мельницами.
Другими словами, намечается стремление
упростить схему подготовки руды к флота-
ции.
Почти полное освобождение сульфидных минералов от породы
часто получается при более крупном измельчении, нежели это
необходимо для освобождения сульфидных минералов одного от
другого, поэтому широко распространены схемы, включающие
доизмельчение коллективных концентратов перед последующей
селективной флотацией. Черновые концентраты обычно доизмель-
чаются в отдельном цикле.
Практика измельчения в шаровых мельницах выявила их пре-
имущества по сравнению с другими измельчительными аппарата-
ми; были сделаны попытки применить их для получения возмож-
но большей степени измельчения продуктов, ибо введение селек-
тивной флотации потребовало получения более тонкой пульпы.
Однако оказалось что на крупных фабриках выгодно ограничить
пределы крупности частиц при измельчении в отдельных шаро-
МИНЕРАЛОГИЯ, СРОСТКИ И ИХ ОСВОБОЖДЕНИЕ
479<
вых мельницах. Это было достигнуто: 1) благодаря применению*
нескольких шаровых мельниц, расположенных последовательно,
причем каждая из них наполнена шарами, величина которых со-
ответствует крупности частиц, подвергающихся измельчению в.
данной мельнице; 2) благодаря ограничению величины частиц,,
поступающих в каждую мельницу и выходящих из нее, что дости-
гается работой мельниц в замкнутом цикле с классификаторами.
Циркуляционные нагрузки в цикле измельчения — классифи-
кации. Практика показала, что стоимость измельчения уменьша-
ется при применении значительных циркуляционных нагрузок
для мельниц и классификаторов. В настоящее время стремятся
увеличивать емкость классификаторов по сравнению с теми из-
мельчительными аппаратами, которые работают в замкнутом
цикле с ними, вследствие чего увеличиваются циркуляционные
нагрузки и измельчение протекает более успешно. Это происхо-
дит благодаря более быстрому удалению из мельниц частиц, уже
достаточно тонко измельченных, что сводит к минимуму количе-
ство переизмельченных частиц. Теоретически в стержневых мель-
ницах должны измельчаться только более крупные частицы,
вследствие некоторой классифицирующей способности самой мель-
ницы. Но это препятствующее переизмельчению влияние мельницы
менее совершенно, чем в классификаторе. Применение высоких
циркуляционных нагрузок уменьшает переизмельчение [5].
Классификаторы. В настоящее время практически на всех
фабриках применяются классификаторы двух типов: спиральный-
и реечный. Они дешевы, просты в эксплуатации, требуют мало
энергии и воды. Однако их эффективность сравнительно низка,
и пески, возвращающиеся в мельницу, содержат большое количе-
ство готового продукта. Чашевые классификаторы, которые
специально сконструированы для классификации более тонкого
материала, чем поступающий в обычные реечные классифика-
торы, эффективнее последних, но также не обеспечивают совер-
шенного разделения. Лучшие результаты достигаются в гидрав-
лических классификаторах, но они потребляют настолько боль-
шее количество воды по сравнению с чашевыми, реечными и спи-
ральными, что при их применении необходимо перед флотацией
сгущать разбавленную пульпу.
Степень раскрытия минералов. Микроскопическое исследова-
ние классифицированных продуктов измельчения руды показы-
вает, что степень раскрытия минералов из сростков тем больше,
чем тоньше продукт. Если исследовать крупные классы рудной,
пульпы, то можно получить сравнительную картину раскрытия
минералов. Особенно ясно это показано в разделе IV табл. 1, где
видно, что при постепенном уменьшении крупности частиц в пуль-
пе, равном , раскрытие зерен галенита увеличивается со-
ответственно от 43 до 65, 83, 94 и 99%. Подобно галениту, рас-
480
ГЛАВА 13
крытие зерен сфалерита в этих условиях происходит от 53 до 71,
87, 96 и 99%.
Хотя метод определения степени раскрытия минералов, осно-
ванный на изучении под микроскопом только крупных классов
пульпы, может вызвать сомнение, все же он дает некоторое пред-
ставление о достаточности измельчения. Иногда полезно произ-
вести количественную оценку степени раскрытия минералов для
каждой серии опытов при определенных условиях измельчения.
Для этого необходимо: 1) взять среднюю пробу пульпы; 2) про-
извести отмучивание пробы или грохочение на классы; 3) опре-
делить вес всех полученных фракций; 4) изготовить шлифы из
каждой фракции; 5) определить под микроскопом число свобод-
ных зерен каждого минерала и число сростков, состоящих из этих
минералов.
Зная число частиц каждого типа, можно вычислить весовое
соотношение частиц данного типа, выраженное в процентах.
Для этого необходимо знать удельный вес каждого минерала
и объем частиц каждого типа. Когда выделение различных клас-
сов материала производится осаждением в жидкости, средний
объем частиц каждого типа можно определить по формуле Сток-
са. Степень раскрытия каждого минерала в классах различной
крупности, а также для всей массы твердых частиц в пульпе по-
лучается непосредственно из весового соотношения частиц каж-
дого типа в этих классах крупности. Для разъяснения данных,
приведенных в табл. 1, нужно указать, что разделение двух суль-
фидных минералов является значительно более сложным, нежели
отделение частиц минералов пустой породы от каждого из суль-
фидов.
Отсюда вытекает, что, применяя соответствующую схему,
можно выделить в отвалы пустую породу ранее, чем наступит
взаимное раскрытие сульфидных минералов, и подвергнуть даль-
нейшему измельчению коллективный свинцово-цинковый концен-
трат. Показатель эффективности доизмельчения для раскрытия
минералов приведен на диаграмме (рис. 13), которая показывает
степень раскрытия минералов в зависимости от различной круп-
ности частиц борнита во флотационном (исходный продукт опе-
рации доизмельчения) и доизмельченном концентратах. Эти дан-
ные получены Р. Л. Монкрифом и X. Б. Хендерсоном в Горной
школе в Монтане. На диаграмме по оси абсцисс отложен размер
зерен, а по оси ординат — количество свободного минерала, вы-
раженное в процентах. Процент освобождения минерала из
сростков увеличивается от 45 до 75% по мере уменьшения круп-
ности руды от —65 до —200 меш.
Экономически выгодные пределы раскрытия минералов. На
практике раскрытие минералов не доводят до конца, наоборот,
экономически выгодно найти какую-то наиболее благоприятную
МИНЕРАЛОГИЯ, СРОСТКИ И ИХ ОСВОБОЖДЕНИЕ
481
Таблица 1
Микроскопическое определение освобождения минералов
комплексной свинцово-цинковой Руде1,
после измельчения, подвергнутой отмучиванию
Размер фракций
А В С D Е F
I. Классы, р. галенит 105—75 75-52 52—37 37—26 26—18 — 18
сфалерит .... 150—105 105—75 75—52 52—37 37—26 —26
пустая порода . 210—150 150—105 105—75 75—52 52—37 —37
11. Процентное отно- шение по весу (опре- делено под микроско- пом) 100,0 100,0 100,0 100,0 100,0 100,0
Свободный галенит . . 7,1 8,6 8,6 8,4 8,1 9,32
Свободный сфалерит 10,3 11,6 и,з 10,7 10,3 10,42
Свободная порода . . . 59,2 68,0 75,6 79,9 81,5 80,3
Сростки галенит-сфале- рит (50% галенита) . 16,3 8,3 3,2 0,9 0,1 —
Сростки галенит-порода (25% галенита) . . . 3,7 1,9 0,7 0,1 0,0 —
Сростки сф-лерит-поро- да (25% сфалерита) . 2,4 1,2 0,5 о.о 0,0 —
Сростки галенит-сфале- риг-порода (29% каж- дого сульфида) . . . 1,0 0,4 0,1 0,0 0,0 —
III.Выход фракций по ве- су, % 1,5 7,3 14,6 16,2 9,7 50,7
IV. Процент освобож- денных зерен в каж- дой фракции галенига .... 43 65 83 94 99 1002
сфалерита .... 53 71 87 96 99 1002
породы 92 97 99 100 100 1002
V. Процент освобожден- ных зерен во всей пу- льпе: галенита 91,3 сф лерита 92,8 породы 99,9
1 Руда содержит 8,4% РЬ, 7,7% Zn.
2 Суммарный.
31 А. М. Годэн
48?
ГЛАВА 13
среднюю степень раскрытия, лежащую между полным раскры-
тием и таким, которого явно недостаточно с точки зрения техно-
логии. Это среднее экономически выгодное раскрытие минералов
зависит от содержания полезных минералов в руде, от произво-
дительности данной фабрики и в особенности от размеров вкрап-
лений в данной руде. Чем выше содержание полезных минералов
количество минерала, в каждом классе
Рис. 13. Увеличение степени раскрытия сростков
при доизмельчении промежуточных продуктов фло-
тации с высоким содержанием борнита и большим
количеством сростков
в руде, чем больше производительность фабрики и, наконец,
чем крупнее отдельные зерна минералов в руде, тем более жела-
тельно и возможно полное раскрытие их. Несмотря на то, что
опытных данных очень мало, можно думать, что на большей ча-
сти фабрик среднее раскрытие всех минералов составляет 75%
и выше.
Флотация сростков. Как и можно ожидать, флотационная спо-
собность сростков занимает среднее положение между флота-
ционными способностями каждого составляющего сросток ком-
понента. Так, частицы, состоящие из кварца и галенита, будут
флотироваться медленнее, чем частицы галенита. Флотационная
МИНЕРАЛОГИЯ, СРОСТКИ И ИХ ОСВОБОЖДЕНИЕ
483
активность во многом будет зависеть от строения поверхности
сростков. Если, например, галенит находится внутри частицы,
то такой сросток будет вести себя при флотации подобно кварцу,
но, если хотя бы небольшая часть галенита выходит на поверх-
ность сростка, то флотационные способности его улучшаются,
хотя для полного извлечения потребуется больше времени, чем
Рис. 14. Образцы сростков в
свинцовом концентрате, полу-
ченном из руды месторожде-
ния Кимберлей, Британская
Колумбия. Характерно, что
галенит в основном располо-
жен на внешней поверхности
частиц:
/ — марматит; 2 — галеиит; 3 —
пирротин
Рис. 15. Образцы сростков в
цинковом концентрате, полу-
ченном из руды месторожде-
ния Кимберлей, Британская
Колумбия (характерно, что
марматит располагается в ос-
новном на внешней поверхно-
сти частиц):
1 — марматит; 2 — галенит: 3 —
пирротин
при флотации зерен чистого галенита такого же размера. Таким
образом, в процессе флотации количество свободных зерен в
пульпе уменьшается.
При флотации руды, содержащей галенит, марматит, пирит
и несульфидную породу, свободные зерна галенита флотируют
первыми в свинцовом цикле, затем будут флотировать частицы
(сростки) галенита и других минералов. Характер процесса фло-
тации меняется с течением времени. Это связано с тем, что кине-
тика процесса прилипания минеральной частицы к пузырьку
воздуха — процесс статистический. Количество флотирующихся
сростков (имеющих на поверхности галенит) уменьшается в
пульпе постепенно (в случае опытов с отдельными навесками)
или по мере продвижения пульпы от первой камеры флотацион-
ной машины к последней (в случае непрерывной флотации). По-
31*
484
ГЛАВА 13
скольку существует также некоторое соотношение между объем-
ным содержанием галенита в сростках и площадью, занимаемой
галенитом на их поверхности, количество галенита во флотируе-
мых сростках также постепенно уменьшается в процессе флота-
ции. к концу свинцовой флотации сростки содержат тонковкрап-
ленный галенит, поэтому сфлотированные фракции, содержащие
преимущественно сростки, направляют на доизмельчение. Этот
измельченный материал может содержать больше или меньше
свинца, чем в исходном питании, в зависимости от содержания
его в сфлотированном продукте, но содержание других компо-
нентов может сильно отличаться от исходного материала, так как
состав измельченного продукта в первую очередь зависит от
встречающихся минеральных ассоциаций. При флотации крем-
нистых свинцово-цинково-железистых руд концентрат контроль-
ной флотации может содержать большее количество цинка, чем
исходная руда. Для достижения лучших результатов необходи-
мо, чтобы после основной и контрольной свинцовой флотации
в исходном материале цинкового цикла было мало свинцово-цин-
ковых сростков. Те сростки, которые проходят свинцовый цикл
не флотируясь, обычно содержат включения галенита внутри
марматита; если же включения его и располагаются на поверх-
ности, то они трудно доступны для пузырьков воздуха.
Сростки, которые флотируются в цинковом цикле, состоят
преимущественно из марматита и пирита, подобно тому как в
свинцовом цикле сростки в основном состоят из галенита и сфа-
лерита. На рис. 14 и 15 даны примеры сростков, встречающихся
в свинцовом и цинковом концентратах.
ЛИТЕРАТУРА
1. Beeson J. F. The desseminated copper ores of Bingham Canyon, Utah,
Trans. AIMME, 54, 356—401, 1916.
2. Chamot, Emile Monnin, and Clyde Walter Mason: «Hand-
book of Chemical Microscopy», 2 vols., John Wiley & Sons, Inc., New York,
1938.
3. Diamond R. W. Ore concentration practice of the Consolidated Mining
and Smelting Co. of Canada, Ltd., Trans. AIMME, 79, 95—106, 1928.
4. Eitel W’i’lhelm. «The Physical Chemistry of the Silicates», University
of Chicago Press, Chicago, 1954.
5. Gaudin A. M. An investigation of crushing phenomena, Trans. AIMME,
73 , 253—310, 1926.
6. Gaudin A. M. «Principles of Mineral Dressing» (a) Chap. 4, McGraw-
Hill Book Company, Inc., New York 1939.
7. Grim Ralph E. «Clay Mineralogy», McGraw-Hill Book Company Inc.,
New York, 1953.
7a. Hahn A. W. Silver bearing minerals of some ores from the Tintic Mining
District, AIMME Tech. Publ. 202, 1929.
8. Hurlbut Cornelius S. «Dana’s Manual of Mineralogy», John Wiley
& Sons, Inc., New York, 1941.
9. Kerr Paul F. Problems of clay and laterite genesis, A Symposium, Ame-
rican Institute of Mining and Metallurgical Engineers, New York, 1952.
МИНЕРАЛОГИЯ, СРОСТКИ И ИХ ОСВОБОЖДЕНИЕ
485
10. Lemke С. A. Difficulties met in differential flotation, Mining and Met.,
8, 183—184, 1927.
11. Marshall, C. Edmund. «The Colloid Chemistry of the Silicate Mine-
rals», Academic Press, Inc., New York, 1949.
12. Palache Charles, Henry Berman, and Clifford Fr'ondel.
«Dana’s System of Mineralogy», (1) Sulfides, sulfosats, oxides, (2) Halides,
nitrates, borates, carbonates, sulfates, etc., (3), Silica, silicates, John Wiley
& Sons, Inc., New York, 1944.
13. Ram do hr Paul. «Die Erzmineralien und ihre Verwachsungen,», Aka-
demie-Verlag GmbH, Berlin, 1950.
14. Richards R. H., and С. E. Locke. «Textbook of Ore Dressing» 3d ed.,
McGraw-Hill Book Company, Inc., New York, 1940.
15. Rogers, A u s’t i n F., and Paul F. Kerr. «Optical Mineralogy», 2d ed.,
McGraw-Hill Book Company, Inc., New York, 1942.
16. Schneiderhoehn Hans, and Paul Ramdohr. «Lehrbuch der
Erzmikroskopie», Verlagsbuchhandlung Gebrtider Borntraeger, Berlin, 1931.
17. Schwartz G. M. Classification and definitions of textures and mineral
structures in ores, Econ. Geol., 46, 578—591, 1951.
18. Spencer Arthur C. Chalcocite enrichment, Econ. Geol., 8, 622, 1913,
19. S tach Erich. «Lehrbuch der Kohlenprographie», Verlagsbuchhnadlung
Gerbruder Borntraeger, Berlin. 1935.
20. T a g g a r t A. F. «Handbook of Mineral Dressing», John Wiley & Sons,
Inc., New York, 1945.
21. Taggart A. F. «Elements of Ore Dressing», John Wiley & Sons, Inc.,
New York, 1951.
22. Winchell, Alexander N. «Elements of Optical Mineralogy», 5th ed.,
3 pts., John Wiley & Sons, Inc., New York, 1937.
23. Wu^nsch, C. Erb. Secondary enrichment at Eagle Mine, Bonanza, Colorado,
Trans. AIMME, 69, 99—109, 1923.
Глава 14
ФЛОТАЦИОННЫЕ МАШИНЫ
В общей стоимости оборудования флотационной фабрики
стоимость флотационных машин имеет небольшое значение. На
обогатительных фабриках флотации обычно предшествует дроб-
ление, измельчение, грохочение, классификация, опробование и
складирование руды. Завершающими операциями являются сгу-
щение и фильтрация. Суммарная стоимость оборудования для
этих операций, как правило, значительно выше стоимости флота-
ционных машин.
В самом флотационном отделении флотационным машинам
могут предшествовать буферные чаны и в некоторых случаях
пульпораспределители и контактные чаны; для контроля процесса
применяется аппаратура, при помощи которой измеряют вели-
чину pH, концентрацию различных ионов и др. Контрольная
аппаратура часто связана с питателями реагентов. Пульпа транс-
портируется либо самотеком, либо насосами, либо другими меха-
ническими устройствами. Несмотря на то что оборудование фло-
тационных отделений имеет большое значение для успешного
проведения флотации, в данной книге оно полностью не рассмат-
ривается, и читатель, желающий получить исчерпывающие све-
дения, может обратиться к справочникам [35] или к другим
источникам [11, 20, 21, 28, 31, 36]. Для начинающего обогатителя
на рис. 1 и 2 указаны основные операции обогащения.
Процесс флотации включает в себя несколько операций, вы-
полняемых одновременно, хотя они могут быть выполнены и по-
следовательно одна за другой.
Операции эти следующие: 1) перемешивание твердых частиц,
газа и водных растворов; 2) отделение твердых частиц, прилип-
ших к поверхности раздела вода — газ, от других твердых частиц,
смачивающихся водой; 3) удаление и транспортировка продук-
тов обогащения.
Подготовительная операция. Первая операция (подготови-
тельная) начинается с момента смешения твердого с водой и кон-
чается, когда часть твердого всплывает. В виде исключения мож-
но указать на процесс пленочной флотации, применявшийся ра-
нее и заключавшийся в том, что сухой твердый материал загру-
жался на свободную водную поверхность, после чего гидрофоб-
ный материал оставался на поверхности, а гидрофильный — то-
нул.
ФЛОТАЦИОННЫЕ МАШИНЫ
487
В том случае, когда твердые частицы смешиваются с водным
раствором в отдельной операции, плотность и состав пульпы
могут быть иными, чем при флотации. В качестве примера можно
Исходная руда.
Приемный
бункер
Дробление и
контрольное
грохочение
Взвешивание
опробование
Вода
Бункер сухой
руды
I Реагенты
Реагенты
Измельчение
контрольная
к л а ссисрикация
Воздух
Хв
Опробование
Опробование
В отвал
Рис. 1. Схема флотационной фабрики
Обезвоживание
Конечный
Вода
привести перемешивание фосфатных песков в барабанном смеси-
теле в плотной пульпе. В специальных контактных чанах может
производиться также аэрация пульпы; при этом частицы прили-
пают к пузырькам воздуха.
Операция разделения. Вторая операция или операция раз-
деления, заключается в том, что флотирующиеся твердые частицы
поднимаются к поверхности пульпы, где они образуют минерали-
зованную пену. В некоторых случаях, однако, эта вторая опера-
Сукая дробленая
Рис. 2. Общая схема измельчительного и флотационного отделе-
ний флотационной фабрики
ФЛОТАЦИОННЫЕ МАШИНЫ
489’
ция ограничивается просто использованием изменения физиче-
ских свойств частиц, связанных воздушными пузырьками и обра-
зующих крупные агрегаты твердых частиц; в этом случае отделе-
ние этих агрегатов от остальных частиц происходит так же, как
в гравитационном обогащении, т. е. применяются сотрясательные
столы и разделение в слое воды.
Методы введения газа. Когда операции подготовки и разде-
ления объединяются в одной общей операции, пульпа насыщает-
ся газовыми пузырьками (обычно воздушными), после чего гид-
рофобный материал собирается в верхней части флотационной
машины. Из восьми известных способов введения газа в настоя-
щее время четыре представляют лишь исторический интерес.
Не употребляются теперь (см. главу 1) следующие способы:
1) агитационный (засасывание воздуха в пульпу производится
благодаря весьма большому числу оборотов вала импеллера)
2) каскадный (засасывание воздуха осуществляется при помощи
перепада пульпы); 3) кипячение пульпы; 4) химический (образо-
вание газа в результате химических реакций). Частое применение
находят следующие четыре способа подачи воздуха (газа): суб-
аэрационный, пневматический, вакуумный и введение газа под
давлением.
При субаэрационном способе используют импеллер, основ-
ным назначением которого является поддержание пульпы во
взвешенном состоянии. Воздух вводится через отверстие, распо-
ложенное ниже уровня пульпы в области импеллера. Подача газа
в пульпу может осуществляться как при атмосферном, так и при
более высоком давлении1 2. В настоящее время это наиболее рас-
пространенный способ введения воздуха в пульпу.
Пневматический способ подачи воздуха также представляет
весьма значительный интерес. Введение газа в пульпу произво-
дится под небольшим давлением у дна флотационной машины.
Для введения газа используются трубки, погруженные в пульпу,
пористая ткань или резина, образующие покрытие воздушных
камер.
При вакуумном способе подачи воздуха пульпа, насыщенная
воздухом при атмосферном давлении, поднимается в камере
(вследствие вакуума) выше нормального гидростатического
уровня. Растворенные газы выделяются из пульпы и образуют
1 Судя по дальнейшему изложению автор считает, что агитационный спо-
соб подачи воздуха в машину заключается в засасывании воздуха через во-
ронку в пульпе, образующуюся около вала импеллера и относит к агитацион-
ным машинам лишь машины Минерале Сепарэйшн. Этот тип машин, хотя и
редко, но все же находит применение и в настоящее время. Прим. ред.
2 Автор к этому типу относит все механические флотационные машины
(кроме машины Минерале Сепарэйшн, не имеющей поддува воздуха), рабо-
тающие как с подачей, так и без дополнительной подачи воздуха. Прим. ред.
490
ГЛАВА 14
большое количество очень мелких пузырьков, которые избира-
тельно прикрепляются к гидрофобным твердым частицам.
При введении газа под давлением пульпа насыщается возду-
хом, затем проходит через расширительную насадку. Давление
резко падает, и воздух выделяется.
В двух последних способах разделение гидрофобных и гидро-
фильных частиц происходит в спокойной среде.
Операция удаления материала. В ряде случаев удаление
ефлотированного материала осуществляется естественным пере-
ливом образующейся пены. Таким образом, удаляется пена в
пневматических машинах, в которых на единицу объема пульпы
приходится очень большое количество воздуха.
В субаэрационных машинах, в которые подается относительно
небольшое количество воздуха, пена снимается с помощью ло-
пастей, сгребающих ее через порог камеры. Применение механи-
ческих пеносъемников необязательно в том случае, когда имеется
значительное количество флотационно активного материала. Если
в процессе флотации образуется небольшой объем пены, целе-
сообразнее вести флотацию в машине уменьшенного поперечного
сечения.
В вакуумных машинах и в машинах с подачей воздуха под
давлением, работающих с незначительным расходом воздуха, t
в которых обычно нет многократной циркуляции пульпы и поэто-
му возможность для всплывания материала не повторяется, же-
лательно полностью удалять все всплывшие в данной камере ча-
стицы.
Удаление несфлотировавшей части материала осуществляет-
ся свободной разгрузкой пульпы. При расположении разгрузоч-
ного отверстия в нижней части машины удаление суспензии про-
исходит так же, как в агитационном чане. Таким образом, круп-
ные частицы при разгрузке не задерживаются.
В машинах, где образуются крупные легкие агрегаты из воз-
душных пузырьков и прилипшего к ним материала (например, на
сотрясательных столах, ваннерах и при грохочении под слоем
воды), способы удаления продуктов аналогичны таким же спосо-
бам, которые приняты при гравитационном обогащении.
Типы машин
Агитационные машины. Хотя агитационные машины1 не
имеют теперь промышленного значения (их заменили более
эффективные субаэрационные машины), они сыграли значитель-
ную роль в развитии флотации [10, 15, 17] и исследовании флота-
ционных свойств различных минералов.
1 Машины типа Минерале Сепарэйши. Прим. ред.
ФЛОТАЦИОННЫЕ МАШИНЫ
491
Сильное перемешивание в камере, обусловленное вращением
импеллера, создает в ней потоки пульпы. Пульпа лопастями
импеллера отбрасывается к стенкам камеры, вдоль этих стенок
движется вверх и затем вниз к импеллеру. Кроме этой вертикаль-
ной циркуляции, пульпа имеет горизонтальное круговое движе-
ние вокруг вала импеллера. Потоки пульпы движутся турбу-
лентно [36]. Большие пузыри воздуха, засосанные водоворотом,
разбиваются импеллером на мелкие пузырьки (рис. 3).
Рис. 3. Схематический раз-
рез флотационной машины
агитационного типа
Рис. 4. Флотационная камера
Халлимонда — Эверса (по Са-
зерленду и Уорку)
Пневматические машины. Пневматические машины наиболее
просты по принципу действия.
При необходимости для лаборатории [23, 27] можно изгото-
вить пневматическую камеру, имея стеклянный резервуар, стек-
лянную трубку с капиллярным отверстием и источник нагнета-
ния газа. Чтобы усовершенствовать устройство, вместо капилляр-
ной трубки можно подобрать стеклянный распылитель и исполь-
зовать манометр или другой прибор для регулирования объема
поступающего газа и скорости его потока. Расположить флота-
ционный аппарат надо таким образом, чтобы было возможно
сливать пенный продукт. Это устройство предложено Халлимон-
дом [13, 14], улучшено Прайером и Леу [29] и широко использо-
вано Эвансом [о, 6] и Сазерлендом [34]. На рис. 4 показан аппа-
рат, который можно применять для однограммовых и меньших
навесок минерала. Навеска должна быть представлена частица-
ми узкого класса крупности. Минерал располагается в точке А
492
ГЛАВА 14
непосредственно над аэрирующим капилляром 1. Пузырьки воз-
духа, проходя через постель минерала, минерализуются и вместе
с прилипшими частицами поднимаются к точке В, где вследствие
перепада давления лопаются; частицы же падают в трубку 2,
откуда разгружаются через отверстие 3.
Усовершенствованная камера Халлимонда—Эванса вклю-
чает магнитную мешалку и манометр, обеспечивающие подачу
пузырьков одного размера с постоянной скоростью.
Рис. 5. Пневматическая машина Келлоу
Одна из первых промышленных машин пневматического типа,
известная как машина Келлоу (по имени автора — изобретате-
ля), имела наклонное дно в виде пористой ткани, под которой
находилась воздушная камера (рис. 5).
Эксплуатация машины Келлоу затруднялась по следующим
причинам: 1) трудно было поддержать твердое во взвешенном
состоянии; 2) ткань разъедалась, забивалась и рвалась.
В машине Мак-Интош вращающаяся перфорированная труба
покрывается пористым материалом; труба устанавливается вдоль
дна ванны; ванна имеет V-образное сечение. Это устройство не-
сколько уменьшает те трудности, которые встречаются при
Эксплуатации машин Келлоу, за счет усложнения конструкции
(в связи с введением вращающейся трубы).
Для пневматических машин вместо матерчатых покрытий
предлагались перфорированные резиновые покрытия, а также
ФЛОТАЦИОННЫЕ МАШИНЫ
493
пористые твердые бруски, подобные фильтрам из оплавленного
стекла, применяемым в лабораториях.
В машинах Келлоу и Мак-Интош пористая поверхность отде-
ляет воздушную камеру от пульпы. Это улучшает диспергацию
воздуха в пульпе.
В большинстве современных пневматических машин воздух
во флотационную ванну подается при помощи простого устрой-
ства— трубок, погруженных в пульпу. Так, например, в машине
Рис. 6. Аэролифтная машина Форрестера без
применения тканевого покрытия
Форрестера (рис. 6) в пульпу, заполняющую длинную ванну, сжа-
тый воздух нагнетается через вертикальные трубки диаметром
1,2—2,5 см. В эти трубки, в свою очередь, подается воздух от
продольно расположенного коллектора большого диаметра. Кол-
лектор соединен воздухопроводом с компрессором низкого дав-
ления высокой производительности. Длинная ванна разделена на
несколько отделений продольными перегородками и покрыта
колпаком.
Колпак, находящийся выше аэрирующих трубок, распола-
гается с внешней стороны перегородок, образующих аэролифт-
ную камеру. Такое устройство обеспечивает создание области
аэролифта 1, области разбрызгивания 2, области столба пузырь-
ков 3 и области пульпы 4. Степень аэрации должна быть доста-
точной для того, чтобы создать перемешивание, при котором
твердые частицы находятся во взвешенном состоянии. Питание
494
ГЛАВА 14
производится на одном конце машины через желоб, вставленный
в паз, находящийся ниже уровня пульпы, разгрузка — на другом
конце машины через разгрузочное приспособление.
Пневматические флотационные машины удобны для перера-
ботки руды при большой производительности. Они имеют также
преимущество перед субаэрационными машинами при обработ-
ке песчанистой пульпы, содержащей мало шламов. Целесооб-
разно применять пневматические машины для обработки боль-
ших тоннажей бедных сульфидных медных руд. Следует избегать
применения пневматических флотационных машин в случаях,
когда процесс разделения идет неровно, и пульпа содержит пре-
имущественно тонкий материал, в особенности, если производи-
тельность средняя или малая.
Вакуумные машины и машины, работающие с воздухом, вво-
димым под давлением. Хотя вакуумная флотация известна со
времени возникновения процесса флотации (Эльмор, ранее
1900 г.), вакуумные машины не используют для обработки руд
вероятно потому, что в спокойной среде даже тонкие илы оседают,
преодолевая ее сопротивление. Однако вакуумные машины имеют
преимущество при обработке пульп, в которых твердые части-'
цы, подлежащие удалению, так тонки, что не осаждаются, и от-
деление их по гравитационному принципу затруднительно (на-
пример, при обработке воды, содержащей бумажную массу, или
при очистке сточных вод).
Вакуумная флотационная машина представляет собой срав-
нительно большой герметически закрытый чан. Вакуум создается
при помощи насоса. Подготовленная к флотации пульпа посту-
пает через центральную трубу большого диаметра и разгружает-
ся по периферии. Удаление сфлотированного материала осуще-
ствляется самотеком или вакуумным отсасыванием. В некоторых
случаях оба продукта (и сфлотированный и несфлотированный)
разгружаются через барометрические колена. Приготовление
пульпы заключается в перемешивании руды с водой и реагентами
и насыщении ее воздухом. Для этого применим любой аэратор.
В машине Клеменса (3] (рис. 7), которая предназначена для
осветления загрязненных растворов, исходный раствор разделя-
ется на пенный продукт, осветленный раствор и осажденный от-
стой. Отстой сгребается и разгружается около центра машины
(подобно разгрузке в сгустителе Дорра).
Во флотационной машине Джуля, работающей при введении
воздуха под давлением [16] (рис. 8), пульпа сначала насыщается
воздухом, затем давление понижается настолько, что воздушные
пузырьки выделяются из жидкости и поднимают вверх прикре-
пившиеся к ним частицы твердого, направляемые затем в срав-
нительно большой отстойник. Сфлотированные твердые частицы
ФЛОТАЦИОННЫЕ МАШИНЫ
495
сгребаются бесконечным конвейером, имеющим гребки, и разгру-
жаются в желоб.
С у баэ рационные машины. Как это можно видеть из списка
патентов или промышленных каталогов, субаэрационные машины
имеют наиболее широкое применение.
1
Рис. 7. Принципиальная схема вакуумной флотаци-
онной машины Клеменса:
/ — привод пеногона: 2—пекогон; 3 — камера снижения
давления; 4— выпускное отверстие для пены; 5 — воронка
для питания; 6 — механизмы для удаления песков; 7 — от-
верстие для выпуска песков; 8 — желоб для подачи пуль-
пы; 9 — сборник для выходящей пульпы
Рис. 8. Флотационная машина Джуля, работающая с пода-
чей воздуха под давлением:
/ _ пеногон; 2 — вал. регулирующий отверстие; 3 — центробежный
насос; 4 — насадка для понижения давления
В машине Фаренволда или субаэрационной машине Денвера
(рис. 9) вводимый воздух обеспечивает как перемешивание, так
И аэрацию пульпы. Воздух засасывается в пульпу через трубу,
Окружающую вал импеллера, расположенную в центре камеры, и
496
ГЛАВА 14
разбивается на мелкие пузырьки между импеллером и непод-
вижным диском. Вращательное движение, сообщаемое импелле-
ром пульпе, в значительной мере устраняется с помощью реше-
ток, установленных в камере над импеллером, благодаря чему
пульпа в верхней части остается сравнительно спокойной. Пита-
ние камеры пульпой происходит через трубу, подающую пульпу
на импеллер. Такая подача пульпы увеличивает ее циркуляцию
в камере за счет снижения производительности камеры.
Поперечное сечение по fl,
Концентрата
ВоэВрат
промпроЭукта
Разгрузка
Резиновое покрытие
камеры
Вид спереди. Вертикальный разрез и
сечение по 6
Ч1 ',=•-=
JA I' ’
(lUrkaHue
Рис. 9. Субаэрационная машина Денвера (по
Таггарту и Джону Уайли):
У^ВозЗух
Современные флотационные машины характеризуются тек- ;
сропными приводами, массивными импеллерами, шарикоподшип-
никами, установленными на валах импеллера и мотора, и гу- '•
мированными движущимися частями. Эти усовершенствования
снижают эксплуатационные расходы.
Плоский импеллер с лопатками и неподвижный диск камеры
Фаренволда в машине Фагергрина заменяются вращающимся
импеллером типа «беличьей клетки», помещенным внутри стато-
ра, имеющего такую же форму (рис. 10). В промышленной маши-
не Фагергрина, состоящей из нескольких камер, пульпа подается
по длинному желобу с одного конца и, пройдя через поперечные
перегородки между отдельными камерами, разгружается с дру-
гого конца через разгрузочное отверстие. Зона между статором
и ротором — это зона интенсивной диспергации, в которой воз-
душные пузырьки становятся тонкодисперсными. Широко исполь-
зуется лабораторная модель машины Фагергрина; доступность
осмотра движущихся частей, выполнение деталей из нержавею-
щей стали и стекла и превосходные качества делают лаборатор-
ную машину Фагергрина лучшим лабораторным аппаратом, хо-
тя полученные результаты трудно воспроизводятся в практиче-
ФЛОТАЦИОННЫЕ МАШИНЫ
497
ских условиях. В настоящее время промышленность выпускает
лабораторную машину Фагергрина с усовершенствованием, пред-
ложенным Клеммером и Клеммонсом [4]. Особенностью этой ма-
шины является периодическое возвращение в камеру отфильтро-
ванного от пены раствора. Это способствует поддержанию посто-
янства пульпы, чего нельзя добиться добавлением воды, пода-
ваемой для сохранения объема жидкости в камере.
Рис. 10. Флотационная машина Фагергрина
Ни машина Денвера, ни машина Фагергрина не нуждаются
в воздуходувке. Машина Денвера также не требует насосов для
транспортирования пульпы, тогда как машина Фагергрина ну-
ждается в этом дополнительном оборудовании, если камеры не
установлены каскадом от головного конца к хвостовому. Насосы,
устанавливаемые для возврата промежуточных продуктов, все-
гда необходимы при работе на машине Фагергрина и иногда при
работе на машине Денвера.
Субаэрационные машины Минерале Сепарэйшн и Аджитейр
должны иметь воздуходувку. В машину Минерале Сепарэйшн
(рис. 11), работающую с небольшим числом оборотов импелле-
ра, воздух подается через отверстие, расположенное на дне ка-
меры непосредственно против центра импеллера. Машина Аджи-
32 А. М. Годэи
498
ГЛАВА 14
тейр отличается от машины Фагергрина формой статора и рото-
ра и от субаэрационной машины Минерал Сепарэйшн использо-
ванием воздуха, вводимого под низким давлением.
Рис. 11. Субаэрационная флотационная машина
Минерале Сепарэйшн:
1—зона пенообрязования; 2— разгрузка пены в желоб; 3 — пере-
движной порог; 4 — желоб для промежуточных продуктов; 5 —
желоб для концентрата; 6 — зона перемешивания; 7 — поступле-
ние пульпы из предшествующей камеры; 8 — воздушная насадка,
подающая воздух в камеру (200 л/лсин); 9 —конечная разгрузка
из последней камеры машины; 10— разгрузка пульпы в следую-
щую камеру; 11 карман для разгрузки грубых песков; 12 — ре-
шетка; 13 — разгрузка пульпы через окно камеры; 14 — окно, ре-
гулирующее уровень пульпы в камере; 15 —окно для перелива
пульпы
Во многих субаэрационных 'машинах для уменьшения водо-
ворота пульпы выше импеллера расположены горизонтальные
решетки, которые успокаивают бурление пульпы.
Различия в промышленных субаэрационных машинах в ос-
новном касаются их механических деталей. Тем не менее некото-
рые особенности имеют важное значение. Так, раньше некоторые
машины были оборудованы импеллерами с поперечными лопат-
ками, т. е. перемешивание осуществлялось лопатками, располо-
ФЛОТАЦИОННЫЕ МАШИНЫ
499
женными под прямым углом к направлению движения. Затем
было установлено преимущество лопаток, угол наклона которых
к направлению движения составляет 15°. Такое положение лопа-
ток снижает расход энергии. В последнее время получает раз-
витие статор в соединении с импеллером, причем статор распола-
гается либо выше, либо ниже вращающейся части. Одной из
важнейших особенностей соединения ротор — статор является
величина зазора между этими частями. В хорошо сконструиро-
ванной камере должна обеспечиваться возможность регулиро-
вания этого зазора вследствие наблюдающегося износа частей,
особенно при обработке твердых силикатных руд. Регулирование
зазора становится необходимым при эксплуатации фабрики
[28 а]. Предложены две новые конструкции флотационных ма-
шин, одна из которых предусматривает монтаж на одном валу
двух импеллеров, расположенных один выше другого, с различ-
ными по форме лопатками, а другая — монтаж двух импеллеров
на концентрических валах, вращающихся с различными скоро-
стями или в противоположных направлениях.
Разгрузка неотфлотированного материала в субаэрационных
машинах производится через отверстие в межкамерный карман
и оттуда через перегородку. Однако при флотации угля имеются
специальные устройства, при помощи которых достигается боль-
шая производительность и может флотироваться грубый мате-
риал. Скорость прохождения пульпы при флотации изменяется
в широких пределах, и, несмотря на небольшую разницу в напо-
ре питания и разгрузки, может быть создан большой поток.
Уровень пульпы в большинстве машин регулируется вертикаль-
ным перемещением сливных шиберов. При регулировании надо
иметь в виду большую неустойчивость в соотношении количеств
флотирующейся и нефлотирующейся частей пульпы.
Пена свободно переливается через борт машины в том слу-
чае, когда флотирующегося материала много, однако в большин-
стве случаев пена сгребается через борт камеры в желоб, откуда
смывается струей воды. Это сгребание осуществляется с помо-
щью медленно вращающегося вала с продольными гребками, рас-
положенного около борта камеры. Применяются также гребки,
расположенные спиралью.
Аппараты для флотогравитации. Некоторые аппараты не яв-
ляются флотационными [2], тем не менее из них производится от-
деление гидрофобных частиц от гидрофильных. Существенное
отличие этого процесса заключается в том, что пузырьки, слип-
шиеся с частицами, не настолько облегчают эти агрегаты, чтобы
они могли подняться вверх в водной суспензии. Для процесса
флотогравитации наиболее широко применяются рифленые сотря-
сательные столы с быстрым возвратно-поступательным движе-
нием. Плотная пульпа сначала перемешивается с реагентами и
32*
ьОО
ГЛАВА 14
воздухом в барабанном смесителе, затем поступает на стол, где
на материал действует поперечно направленное движение стола
и смывающая струя воды. Гидрофобные частицы прикрепляются
к свободной воздушной поверхности, и благодаря этому происхо-
дит пленочная флотация частиц, которые быстро выносятся к
краю стола. Ввиду того что аэрированные агломераты частиц
легче и крупнее, чем неагломерированные, они транспортируются
потоком воды над неагломерированными частицами и смываются
в приемник, расположенный вдоль сотрясательного стола; в то
же самое время неагломерированный материал, двигаясь по деке
к концу стола, попадает в другой приемник.
Для этого процесса могут быть также использованы ваннеры.
Материал, обработанный реагентами и проаэрированный, пода-
ется на ленту ваннера, по которой текут очень сильные струи во-
ды, заставляющие гидрофобный агломерированный материал
плыть по поверхности струй и отрываться с краев ленты, тогда
как смачиваемый материал проходит по ленте и разгружается у
ее конца.
При применении грохочения под слоем воды пульпа, пред-
варительно обработанная реагентами и насыщенная воздухом,
поступает на погруженный в воду наклонный грохот. Гидрофоб-
ный материал образует слой, движущийся над гидрофильным ми-
нералом; этот последний постепенно проходит через отверстия
грохота, тогда как аэрированный минерал выносится.
Оценка флотационных машин. Относительные преимущества
различных флотационных машин являются предметом обсужде-
ния многих исследователей [7, 8, 22, 26, 30]. Оценить ту или иную
флотационную машину можно, исходя в основном из трех поло-
жений: 1) пригодность машины с точки зрения технологии;
2) экономические показатели работы; 3) безаварийность в ра-
боте.
В основном технологические показатели в большей степени
зависят от химизма процесса, нежели от выбора той или иной
флотационной машины. Однако иногда правильное решение тех-
нологической проблемы требует отказа от определенных типов
машин; в других же случаях выбор типа машин существенного
значения не имеет. В последнее время значительно возрос инте-
рес, особенно в России, к выявлению факторов, которые могут
привести к технически правильному выбору наиболее подходя-
щей машины и к усовершенствованию ее конструкции [18, 19, 25].
Флотация крупного материала требует более интенсивного
перемешивания пульпы (во избежание заиливания) в то же вре-
мя для отделения минерализованных пузырьков от гидрофиль-
ных минералов благоприятны более спокойные условия в камере.
Эти противоречивые требования создают трудности при конструи-
ровании хороших аппаратов. При крупности питания, большей
ФЛОТАЦИОННЫЕ МАШИНЫ
501
чем —35 меш (за исключением угля), целесообразно использо-
вать только аппараты флотогравитации.
Применение субаэрационных машин затруднительно для фло-
тации крупнопескового материала; их целесообразнее применять
для тонкоизмельченной руды.
При эксплуатации машин учитываются непосредственными
замерами потребление мощности [9, 32, 33], данные по износу ча-
стей машины, а также расходы по обслуживанию машин и на-
блюдению за технологическим процессом. В последнее время
имеются значительные достижения не только в усовершенство-
вании конструкции машин, но также в выборе многих материа-
лов для их изготовления.
Для сравнительной оценки различных типов машин (пневма-
тических или субаэрационных) еще не имеется достаточно пол-
ных и проверенных данных. Стоимость первоначального монтажа
пневматических машин ниже, но они не так легко регулируются
при изменении питания и характера руды, как субаэрационные
машины, в которых перемешивание и насыщение пульпы возду-
хом можно изменять в каждой камере отдельно.
Темой многих дискуссий была глубина камер, а именно: ка-
кую глубину камеры (90 или 120 см) считать наилучшей. В ре-
зультате пришли к выводу, что выбор глубины камеры в значи-
тельной мере определяется свойствами руды.
Одно несомненно: при прочих равных условиях камера боль-
ших размеров любого типа машины будет работать лучше [11
Надо также отметить, что оптимальное соотношение между по-
перечным сечением слоя пены и объемом пульпы может быть
разным для разного типа руд.
ЛИТЕРАТУРА
1. Banks Н. R. Experience with flotation machines at the Sullivan Concen-
trator, AIMME, Tech. Publ., 1693, in Mining Technol., 8(2), 5 pp., 1944.
2. Chapman George Albert, and John W. L i 111 e f о r d. U. S. Pa-
tent 1968008/1934.
3. Clemens R. F. U. S. Patent 2375282/1945.
4. Clemmer J. Bruce, and В. H. Clemmons. An improved flotation
test cell, Eng. & Mining J., 144(3), 72—73, 1943.
5. E v a n s L. F. Interaction between surfaces in close proximity, Nature, 172,
776—777, 1953.
6. E v a n s L. F. The flotation of topaz using sodium hexadecyl sulfate as
collector, Australian J. Appl. Sci., 4, 165—173, 1953.
7. Fahrenwald A. W. The aerating capacity of flotation machines, Idaho
Bur. Mines Geol., Pamphl. 64, July. 1943.
8. Fahrenwald A. W. How efficient is a flotation machine? Eng. & Mining
J., 145, 82, 136, January, 1944.
9. Fahrenwald A. W. The submergence factor in the impeller type of flo-
tation machine, AIMME Tech. Publ. 2080, in Mining Technol., 10(6), 8 pp.
1946.
502
ГЛАВА 14
10. Gates J. F., and L. K. Jacobsen. Development and operation of a
50-gram flotation machine, Eng. Mining J., 119, 771—772, 1925.
11. Gaudin A. M. «Principles of Mineral Dressing», McGraw-Hill Book
Company, Inc., New York, 1939.
12. Grund'er, Werner, and Ernst Kadur. Relation between foam sur-
face and volume in flotation cells, Metall u. Erz, 37, 367—372, 1940.
13. H a 11 i m о n d, A. F. Laboratory apparatus for flotation tests, Mining Mag..
70, 87—90, 1944.
14. Hallimond A. F. The role of air in flotation at great dilution, Mining
Mag., 72, 210. 206, 1945.
15. Hansen M. S. Perfection and use of the 50-gram flotation cell, Eng.
Mining J., 133, 28—29, 1932.
’16. Ju ell F. U. S. Patent 2330589/1943.
17. Keiser H. D. New laboratory flotation cell developed at University of
Utah, Eng. Mining J., 126, 504—505, 1928.
18. Классен В. И. Метод оценки распределения воздуха во флотационных
машинах. Цветные металлы 7, 76—79, 1939.
19. Классен В. И. Влияние плотности пульпы на скорость флотации.
Цветные металлы (10, 11), 66—72, 1939.
20. Knapp Е. A. Conditioning for flotation, Mining Mag., 69, 201—207, 1943.
21. Knapp E. A. Reagent handling, Mining Mag., 70, 73—83. 1944.
22. К n a p p E. A. Flotation machines, Mining Mag., 72, 137—151, 206—210,
1945.
23. Leaf, Clyde W., and Alexander Knoll. Laboratory flotation cell.
Small pneumatic cell of all-glass construction, Ind. Eng. Chem., Anal. Ed.,
11, 510—511, 1939.
24. M a j u m d a г К. K. Simple flotation cell, J. Sci. Ind. Research (India)
9B(3), 76—77, 1950.
25. Мит p о ф а н O b С. И. Скорость протекания пульпы через флотацион-
ную машину и скорость флотации. Цветные металлы (8), 30—33, 1939.
26. Myers J. F., and F. М. Lewis. Flotation machines at Tennessee Copper
Company, AIMME Tech. Publ., 1680, in Mining Techno!., 8, 1944.
27. Oberbilling Ernest, and A. W. F a h r e n w a 1 d. Laboratory-size
pneumatic flotation cell, Mining J. (Phoenix, Ariz.), 22(1), 7, 1938.
28. P г у о r E. J. «An Introduction to Mineral Dressing», (a) pp. 500—504,
Mining Publications, Lts., London, 1955.
29. Pryor E. J., and К о u n g-B i L i о u. A simple flotation cell, Bull. Inst.
Mining Met., No. 502, pp. 11—18, 1948.
30. R a b о n e, Philip. Modern methods of flotation. I. Flotation machines,
Mining Mag., 56, 145—153, 208—218, 1937.
31. Richards, Robert H., and Charles E. Locke. «А Textbook of
Ore Dressing», 3d ed., McGraw-Hill Book Company Inc., New York, 1940.
32. Rose E. H. Flotation as a power process, AIMME Tech. Publ. 1702, in
Mining Techno!., 8 (2), 8 pp., 1944.
33. Sutherland K. L. Flotation machine efficiency—a differing viewpoint,
Eng. Mining J., 147(5) 84, 1946.
34. S u t h e r 1 a n d K. L. and I. W. Wark. «Principles of Flotation»,
pp. 58—60, Australasian Institute of Mining and Metallurgy, Melbourne,.
Australia, 1955.
35. Taggart Arthur F. «Handbook of Mineral Dressing», John Wiley &
Sons, Inc., New York. 1945.
36. Taggart Arthur F. «Elements of Ore Dressing», John Wiley & Sons,
Inc., New York, 1951.
Глава 15
СУЛЬФИДНЫЕ РУДЫ
Флотационный метод обогащения с момента своего промыш-
ленного внедрения применялся преимущественно при обогаще-
нии сульфидных руд цветных металлов. К этой же категории
следует отнести руды, в которых металлы находятся в самород-
ном виде, а также сульфидные руды, содержащие благородные
металлы, и руды, характеризующиеся наличием в них сульфидов
мышьяка, сурьмы, висмута и молибдена, встречающихся в от-
дельности, совместно друг с другом или с иными сульфидами.
Сульфидные медные руды
Медные сульфидные руды наиболее пригодны для флотаци-
онного обогащения. Обычно они содержат один или несколько
сульфидных медных минералов, а также сульфиды железа и не-
значительные количества золота и серебра.
Порода представлена кварцем, карбонатами и силикатами,
частично глинами.
Медные минералы могут содержать только медь и серу
(халькозин Cu2S, дигенит CugSs, ковеллин CuS) или медь, желе-
зо и серу (борнит CusFeS.}, халькопирит CuFeS2, кубанит
CuFe2Ss) или же, наконец, медь и серу совместно с мышьяком,
сурьмой, висмутом и оловом (энаргит CU3ASS4, тетраэдрит
Cu12Sb4Si3, станнин Cu2FeSnS4). Первичным медным минералом
является халькопирит.
Халькозин и борнит — главные минералы вторичного обога-
щения.
Пирит FeS2 — наиболее распространенный сульфид железа,
хотя часто встречается и пирротин FenSi2. Золото обычно при-
сутствует в виде включений или примазок в сульфидах, особенно
в пирите и халькопирите. В рудах, подвергавшихся процессам
окисления, золотосодержащие сульфиды могут перейти в тонкие
лимониты, золото в которых останется в виде включений. Сереб-
ро наиболее часто встречается как твердый раствор, замещаю-
щий медь в тетраэдрите. При окислении руд серебро может
осаждаться вновь в виде сульфида совместно с медью, а иногда
в виде твердого раствора во вторичных медных минералах. Се-
ребро также может быть осаждено в виде галлоидного соедине-
ния или как сложный сульфат, например аргентоярозит.
!
504
ГЛАВА 15
Основные минералы породы могут быть различными в зави-
симости от типа месторождения.
В трещиноватых жильных месторождениях порода может
состоять из карбонатов, например кальцита, из кварца, силика-
тов с более или менее значительным количеством глин. Эти гли-
ны могут быть представлены каолином или монтмориллонитом,
который особенно жадно поглощает воду и, по-видимому, усили-
вает явления ионного обмена. Во многих месторождениях Запад-
ной Америки основная масса породы представлена кварцем,
полевыми шпатами и каолином, а в некоторых месторождениях
замещения в сланцах главнейшими породными составляющими
являются ферромагнезиальные силикаты.
Методы обработки руд. Флотация сульфидных медных руд
в большинстве случаев протекает весьма эффективно' и яв-
ляется недорогим процессом. В качестве собирателя применя-
ются сульфгидрильные соединения (например, этиловый ксан-
тогенат натрия), а в качестве пенообразователя — сосновое
масло.
Для контроля pH служит известь. Расход собирателя и пено-
образователя колеблется от нескольких сотых до нескольких
десятых килограмма на тонну руды, а расход извести — от 0,5
до нескольких килограммов на тонну руды.
Известь применяется не только для нейтрализации природ-
ной кислотности руды (обычно образующейся за счет окисления
пирита), но и для создания щелочности пульпы, которая необхо-
дима, чтобы депрессировать пирит при флотации сульфидов ме-
ди (pH = 10—12).
Целесообразно проводить предварительное перемешивание
пульпы с одновременной ее аэрацией. При этом создаются наи-
более благоприятные условия для депрессии пирита в цикле мед-
ной флотации, хотя общий расход извести может несколько уве-
личиться, так как аэрация способствует окислению пирита.
Операция эта часто применяется на фабриках в качестве допол-
нительного мероприятия, но обычно вполне достаточно энергич-
ного перемешивания, происходящего в мельницах, открытых
доступу воздуха.
Иногда для усиления депрессии пирита добавляют небольшие
количества щелочного цианида (несколько сотых килограмма на
тонну руды), но, так как цианид во много раз дороже извести,
обычно стремятся депрессировать основную массу пирита только
одной известью.
Тщательно очищенная поверхность пирита трудно окисляется.
Некоторые чистые образцы пирита подвергались длительному
(в течение нескольких десятков дней) воздействию влаги и воз-
духа, и не претерпевали при этом никаких изменений. С другой
стороны, некоторые руды окисляются очень быстро, даже когда
СУЛЬФИДНЫЕ РУДЫ
505-
приняты все меры, чтобы предотвратить возможность их окисле-
ния. Причины этих явлений в точности не установлены. Можно
предполагать, что окисление отсутствует в тех случаях, когда
зерна пирита прочно защищены какой-либо адсорбированной
на них жировой пленкой, а активное окисление вызывается на-
личием некоторых катализаторов, способных к взаимодействию
даже при минимальных количествах влаги.
Совокупность химических условий, при которых сульфиды
меди способны прилипать к пузырьку воздуха, в то время как
пирит остается смачиваемым водой, являлась объектом много-
численных исследований, особенно австралийской школы Уорка
и его сотрудников, применивших метод контакта минерала с пу-
зырьком. На рис. 4 и 8 гл. 10 представлены результаты этих
исследований (на рис. 4 показано, как сульфгидрильный соби-
ратель и щелочь конкурируют за овладение поверхностью зерен
пирита и арсенопирита; на рис. 8 показано, как при введении
цианида при том же собирателе изменяются условия раз-
деления) .
Однако эти данные являются условными по следующим
причинам: 1) краевые углы меньше 10—15° практически не мо-
гут быть замерены с должной точностью, в то время как углы
даже в 1° теоретически достаточны для извлечения частиц круп-
ностью —400 меш; 2) взаимное воздействие солей из различных
минералов в проводившихся опытах с краевыми углами отсутст-
вовало; 3) во всех этих опытах концентрация ионов собирателя
и депрессора значительно превышала обычную [54].
Основные проблемы флотации медных сульфидных руд. Глав-
ной причиной, снижающей эффективность селективной флотации
этих руд, обычно является значительное преобладание пирита
над сульфидами меди. Например, содержание меди в составе
руды 1% соответствует 2—3% медных сульфидных минералов.
Естественно, что при этом гораздо труднее будет обогащаться
та руда, в которой содержание пирита в 20 раз превышает со-
держание медных минералов, чем та, где эти величины экви-
валентны.
Вторая проблема, с которой часто приходится встречаться на-
практике, заключается в весьма тесном взаимопрорастании суль.
фидов между собой, которое можно наблюдать при цементирова-
нии разрушенного пирита халькопиритом или при активизации
пирита халькозином. Во всех подобных случаях необходимо мик-
роскопическое исследование, что облегчит задачу разработки
схемы и режима разделения минералов на практике.
Третья проблема связана с извлечением благородных метал-
лов. Золото и серебро желательно скон|центрировать в медном
концентрате, однако это может привести к разубоживанию мед-
ного концентрата пиритом, так как в пирите золото присутствует
'506
ГЛАВА 15
в виде тонких включений гораздо чаще, чем в медных минера-
лах.
В подобных случаях необходимо провести особо тщательные
экспериментальные исследования, для того чтобы установить тог
предел, до которого следует перечищать концентрат, а также вы-
явить целесообразность применения комбинированных схем обо-
гащения с целью доизвлечения золота из хвостов флотации гра-
витационными методами.'
Такие же трудности извлечения благородных металлов могут
возникнуть при обогащении смешанных руд. Иногда бывает по-
лезно ради повышения общего извлечения золота дополнить
флотацию операцией выщелачивания или гравитационным (цик-
лом.
В частично окисленных рудах встречается и несульфидная
медь, например карбонатная или силикатная (последняя даже
чаще, нежели карбонатная). Извлечение силикатной меди мето-
дом флотации представляет еще практически не разрешенную
проблему.
Практика обогащения халькозиновых рг/д.Фабрика Моренси
в Аризоне [11] обрабатывается в сутки около 45000 груды, добы-
ваемой открытыми горными работами. После дробления руда из-
мельчается в щелочной среде, создаваемой добавкой известково-
го молока, и подвергается флотации дитиофосфатом в присут-
ствии пенообразователя. Концентрат перечищают несколько раз.
Сточные воды обезвреживают и используют как оборотные. Ха-
рактерной особенностью фабрики является весьма удобное
устройство трубопровода для подачи известкового молока, позво-
ляющее легко регулировать щелочность в любой точке техноло-
гического процесса.
Перерабатываемая на фабрике руда относится к типу прос-
тых. Основным рудным минералом является халькозин, часто
присутствующий в виде оболочек, окружающих пирит. Некото-
рые из этих оболочек настолько толсты, что пирит представляет-
ся в них лишь в виде отдельных включений, другие же столь тон-
ки, что едва могут быть различимы под микроскопом при увели-
чении в 500 раз.
Главной трудностью является достижение достаточно полно-
го разделения халькозина и пирита без снижения извлечения
меди. Принципиальная схема фабрики показана на рис. 1.
Руда измельчается в 27 шаровых мельницах (с разгрузкой
через решетку), работающих в замкнутом цикле с 54 спиральны-
ми классификаторами. Слив классификаторов поступает через
пульподелители во флотацию, которая производится в 36 маши-
нах механического типа по 12 камер в каждой.Черновой концен-
трат доизмельчается в 6 шаровых мельницах со свободной раз-
грузкой, работающих в замкнутом цикле с 6 спиральными клас-
СУЛЬФИДНЫЕ РУДЫ
507
сификаторами, после чего перечищается в 25 машинах по 4 ка-
меры каждая.
При флотации применяются в качестве реагентов известь
(1,75 кг/т руды), сульфгидрильный собиратель — аэрофлот
Медный концентрат
в обезвоживание
Рис. 1. Схема фабрики Моренси
№ 208, представляющий собой смесь диэтилового и вторичного
бутилового дитиофосфата, нейтрализованную содой (15 г/т ру-
ды), пенообразователь — смесь крезиловой кислоты с нефтью
(55 г/т руды). В перечистки концентрата задается дополнитель-
но экстракт квебрахо или сульфированный лигнин (Америкен
Цианамид Ко, реагент 610), представляющий собой органичес-
кий коллоид (50 г/т чернового концентрата).
508
ГЛАВА 15
Фабрика Моренси интересна как предприятие огромного мас-
штаба, где было найдено относительно простое решение пробле-
мы обогащения руды, имеющей сложную ассоциацию пирита и
халькозина.
На фабрике Роан Антилоп в Северной Родезии [27] возникли
трудности иного порядка. Руда здесь почти не содержит пирита,
медные минералы представлены 'халькозином и борнитом, но
связь их с кварцевой породой настолько тесная, что проблема
тонкого измельчения руды приобрела особое значение. Микро-
скопическое изучение хвостов флотации показало, что около
9О°/о содержащихся в них сульфидов представлено сростками,
даже при измельчении руды до —150 меш. И хотя хвосты перет
чистки чернового концентрата доизмельчаются до 98%—
325 меш, в них все еще присутствует значительное количество '
минералов в виде сростков.
За последнее время значительный интерес вызвала фабрика
Уайт Пайн в Мичигане [48]. Руда содержит халькозин, тонко
вкрапленный в кварцевую породу, что вызывает при обработке
руды те же трудности, что и на фабрике Роан Антилоп.
Практика обогащения халькопиритовых руд. Фабрика Мета-
хамбра (Куба) обрабатывает богатую (более 5% Си) руду, со-
держащую в качестве единственного сульфида халькопирит,
крупновкрапленный в кварцевой породе. Извлечение меди в кон-
центрат свыше 95% считается на фабрике обычным. Получае-
мый концентрат представлен почти чистым халькопиритом.
Насколько легко обогащается руда фабрики Метахамбра,
настолько трудно поддается обогащению руда месторождения
Коппер-Хилл (Теннесси). Медь здесь находится в подчиненном
количестве, так как ее содержание в 10 раз меньше, а пирита,
наоборот, в 20 раз больше, чем в руде Метахамбра. Таким обра-
зом, здесь количественное отношение пирита к меди в 200 раз
больше, чем в руде Метахамбра.
После тонкого измельчения [41,42] руда подвергается коллек-
тивной флотации для извлечения сульфидов в слегка кислой сре-
де с применением серной кислоты в качестве регулятора pH.
Этот коллективный концентрат поступает в операцию медной се-
лективной флотации, где создаются условия, вызывающие де-
прессию железных и цинковых минерало-в. Полученный медный
концентрат затем еще дважды перечищается.
Содержание меди в конечном концентрате невысокое, извле-
чение тоже довольно низкое, но, учитывая специфические труд-
ности обогащения данной руды, эти показатели можно признать
удовлетворительными.
В руде, перерабатываемой на фабрике Норанда [37], присут-
ствуют халькопирит, пирит и пирротин в тонком прорастании с
кварцем. Руда тоже содержит золото, которое весьма тонко
СУЛЬФИДНЫЕ РУДЫ
509
вкраплено во все сульфидные минералы. Присутствие пирротина
осложняет технологию извлечения золота, так как он поглощает
цианид в большом количестве.
Принципиальная схема обработки руды на фабрике Норанда
.показана на рис. 2.
Измельченная руда
Основная Си флотация
~К-т ХГ~
Пиритная флотация
Доизмельченае Д оизмельчение
Медно-золотая флотация
(из пирита)
К-т Кв.
Медиа-золотая флотация
(из пирротина)
хв.
Диритный
продукт
Цианирование
Остаток
В отвал
педно- золотой Металлическое
концентрат золото
в плавку
Рис. 2. Схема фабрики Норанда
В руде, перерабатываемой на фабрике Вермонт [35], основ-
ные сульфиды — халькопирит и пирротин. Применяется после-
довательная флотация каждого минерала. Медь извлекают в
первом приеме флотации, а пирротин — в последующем. Всего
имеется три стадии основной медной флотации и три перечистки,
проходящие при рН=9 — 9,8. Пирротин извлекается одностади-
альной основной флотацией с одной перечисткой концентрата.
Частично окисленные медные руды. Значительный интерес
представляет промышленное освоение схем комбинированного
510
ГЛАВА 15
процесса с применением флотации и выщелачивания для извле-
чения меди из частично окисленных сульфидных руд. В таких ру-
дах часть меди может присутствовать в виде солей, раствори-
мых в кислотах, или в виде легко растворимых минералов,
(таких, как основные сульфаты или карбонаты).
В Быотт новые методы горных работ включают в себя систе-
мы с. массовым обрушением больших мощных блоков руды. Не-
которые из этих блоков расположены вблизи поверхности, дру-
гие заключают в себе старые выработки и забои, заполненные
закладкой, которая прежде рассматривалась, как пустая поро-
да. Фактически же эта закладка представляет собой дробленую
руду, видоизмененную рудничным пожаром, ныне уже ликвиди-
рованным. Руда эта значительно беднее по содержанию в ней
меди, чем описанные выше руды, частично окислена и непостоян-
на по составу [20, 32].
Схема переработки такой руды на фабрике Анаконда вклю-
чает следующие операции: 1) кислотное выщелачивание в про-
цессе тонкого измельчения руды в шаровых мельницах, снаб-
женных керамической футеровкой и работающих в замкнутом
цикле с классификаторами; 2) осаждение металлической меди
неочищенным губчатым железом; 3) медную флотацию; 4) пири-
тную флотацию.
Из пиритного концентрата после обжига производят необхо-
димую для процесса серную кислоту и губчатое железо.
Схема фабрики Анаконда показана на рис. 3.
Подобные же методы комбинированной обработки положены
в основу схем фабрик Чуквикамата (Чили) и Нчанга (Родезия),
на которых перерабатываются смешанные сульфидно-окислен-
ные руды [55]. На фабрике Нчанга руда сначала подвергается
флотации с ксантогенатом для извлечения сульфидной меди. Со-
держание меди в получаемом концентрате достигает 50%, при-
чем в нем содержатся не только сульфиды, но и окислы в виде
куприта. Затем следует второй прием флотации с карбоксиль-
ным собирателем, при котором получается низкокачественный
смешанный концентрат, содержащий много малахита. Этот кон-
центрат подвергается кислотному выщелачиванию, после кото-
рого раствор поступает на электролиз, где высаживается катод-
ная медь. Остаток от выщелачивания вновь подвергается суль-
фидной флотации для доизвлечения тех сульфидов, поверхность
которых очищена в процессе кислотного выщелачивания. Полу-
ченный при этом концентрат присоединяется к первому сульфид-
ному концентрату и направляется в плавку.
Схема фабрики показана на рис. 4.
Самородная медь. В течение долгого времени после широко-
го внедрения флотации в практику обогащения сульфидных руд
она не применялась при обработке руд Мичигана, содержащих
СУЛЬФИДНЫЕ РУДЫ
511
самородную медь. Вопрос этот был достаточно изучен Фарен-
волдом [18, 19]. Он вполне обоснованно отверг представлявшее'
ся заманчивым решение проблемы обогащения этих руд с помо-
щью только одной флотации, поскольку медь является ковким.
металлом, встречающимся изредка в виде больших кусков и
очень часто — в виде зерен среднего размера. Фаренволд при-
менил флотацию лишь как дополнение к гравитационным про-
цессам обогащения, которые особо пригодны в качестве основ-
ных для столь тяжелых металлов, как самородная медь. Назна-
чением же флотации, проводившейся с загрузками ксантогената,
являлось лишь доизвлечение из хвостов гравитационного цикла
Ы2
ГЛАВА 15
той части меди, которая присутствовала в руде в виде сульфи-
дов.
Схема этого (ныне уже широко распространенного ) процес-
са [8] показана на рис. 5.
Измельченная руда
Флотация сулыридной меди
К-т ТВ. ~~
Флотация сульриднчв t
окисленной меди
Киа 'оп(ное
выщелачивание
Утделение раствора
"Раствор Твёрдое
Хвосты I
Электричес
кии ток
Электролиз
Металлическая
медь
Серная
кислота
Г ^к^^И^^ль^иВной меди
Медный
/концентрат
в плавку
Хвосты Ж
Рис. 4. Схема фабрики Нчанга для обработки смешан-
ных сульфидно-окисленных медных руд
Сульфидные свинцово-цинковые руды
Описание руд. Эти руды обычно содержат свинец и цинк в
количествах, достаточных для промышленного извлечения каж-
дого из них в отдельный концентрат, хотя иногда встречаются
свинцовые руды с ничтожным содержанием в них цинка, не оп-
равдывающим получения отдельного цинкового концентрата,
СУЛЬФИДНЫЕ РУДЫ
513
и наоборот. Кроме свинца и цинка, эти руды обычно содержат
один или несколько сульфидов железа и незначительные коли-1
> чества меди, серебра и золота в кварцевой или карбонатной по-
роде.
Свинец чаще всего представлен галенитом. В рудах, подверг-
шихся окислению, встречается англезит, церуссит и реже другие
ДробленаярЦ^!^^
ГоаВитация
К-т
Шпаны
Крупный медный
концентрат
Измельчение
Классификация
Пески Слив
Медно-сулыридная флотация
К-т "™"
Тонкий медный
концентрат
Хвосты
Рис. 5. Схема фабрики Лэйк-Супириор для
обработки руд, содержащих самородную
медь
нерастворимые соли. В рудах, содержащих мышьяк, сурьму и
висмут, некоторые из этих окисленных свинцовых минералов
[47] могут,встретиться в значительном количестве.
Цинк обычно присутствует в виде сфалерита или в виде его
железистой разновидности — марматита. Содержание железа в
марматите может достигать 20% от содержания цинка, хотя
обычно оно не превышает 10%. Окисленные цинковые минералы
представлены гемиморфитом (водный силикат) и смитсонитом
(карбонат).
33 А. М. Годэн
514
ГЛАВА 15
Медь большей частью присутствует в подчиненных количес-
* твах в виде халькопирита. Для халькопирита характерно обра-
зование осаждением из раствора, хотя он встречается и в виде
зерен, вкрапленных в марматит. Этот минерал часто находится
в тесной связи с пирротином.
При высоких температурах медь растворяется в марматите,
замещая атомы цинка или железа. В результате этого в течение
медленного охлаждения рудных кристаллов после их образова-
ния медь высаживается из растворов в виде включений халько-
пирита размером 1 — 2р, располагающихся вдоль кристаллогра-
фических осей внутри отдельных кристаллов марматита. Иногда
на гранях кристалов марматита встречаются и несколько более
крупные зерна халькопирита (рис. 14 гл. 13). Флотационное от-
деление частиц халькопирита размером 1—2 ц от вмещающего
их марматита неосуществимо. Но медь, представленная более
крупными частицами халькопирита, а также в виде тетраэдрита
или других сульфосолей, может быть отделена от цинка при фло-
тации.
В этих рудах серебро всегда является минимальной рудной
составляющей по сравнению с количеством других минералов.
Однако с экономической стороны содержание в руде серебра ча-
сто представляет собой весьма существенный фактор. Форма,
в виде которой серебро присутствует в ряде, не всегда бывает
известна.
Наиболее часто оно встречается как продукт замещения ме-
ди в тетраэдрите или в виде одной из многочисленных сульфосо-
лей: серебра, серебра и меди, серебра и олова или серебра и
свинца. Аргентит встречается редко.
Золото главным образом присутствует в самородном виде,
преимущественно как включение в галените и пирите, но факти-
ческих данных об этом недостаточно.
Сульфиды железа представлены пиритом и пирротином. Хотя
эти минералы иногда являются преобладающими, их обычно со-
держится меньше, чем в медных рудах. В свинцово-цинковых
рудах абсолютное количество свинца и цинка больше, чем коли-
чество меди в медных рудах. Вследствие этого отношение сви-
нец : железо и цинк: железо в свинцово-цинковых рудах обычно
много выше, чем отношение медь : железо в медных рудах. Од-
нако весьма желательно наиболее полно удалять сульфиды же-
леза из цинкового концентрата, учитывая трудности металлурги-
ческой переработки цинковых концентратов в присутствии зна-
чительного количества железа.
Порода в свинцово-цинковых рудах может быть представлена
карбонатами (кальцит, доломит, сидерит, анкерит, родохрозит),
сульфатами (барит), кварцем или силикатами с небольшим ко-
личеством глинистых минералов.
СУЛЬФИДНЫЕ РУДЫ
515
Из изложенного следует, что в рудах этого типа минералы
находятся в самых различных сочетаниях, в связи с чем могут
возникнуть многие интересные проблемы.
Наличие растворимых солей, в частности сульфатов железа,
цинка, марганца, магния, кальция и меди, весьма важно
вследствие буферного действия этих солей, а также благодаря
их способности участвовать в реакциях ионного обмена с мине-
ралами.
Методы обработки. Основной задачей селективной флота-
ции свинцово-цинковых руд является выделение свинца, меди и
драгоценных металлов в свинцовый концентрат и выделение
цинка в цинковый концентрат, а иногда и выделение сульфи-
дов железа в отдельный концентрат (пиритный). Пиритный кон-
центрат применяется как сырье для производства серной кисло-
ты и железного огарка, кроме того, его могут выделять, чтобы
более полно извлечь золото.
В последнем случае полученный пиритный продукт может
быть присоединен к свинцовому концентрату при его плавке-
Обычно сначала флотируют свинец, затем цинк и, наконец,
пирит, хотя возможна и другая последовательность. Для флота-
ции свинца применяют сульфгидрильные собиратели типа ксан-
тогенатов, в качестве пенообразователей употребляют сосновое
масло совместно с высшими спиртами и крезиловой кислотой.
Количество этих реагентов составляет несколько десятков грам-
мов на тонну руды. При селективной флотации свинца для деп-
рессирования пирита применяется щелочная среда (pH от 8 до
10). В этих условиях цинк также флотируется слабо, если он не
активирован растворенными солями, главным образом солями
меди. Однако обычно имеется достаточно природных активато-
ров, требующих особых мер для предотвращения минерального
загрязнения свинцового концентрата цинком. Для этой цели луч-
ше всего служит растворимый цианид. Расход его составляет не-
сколько десятков граммо'в на тонну перерабатываемой руды.
Часто бывают полезны добавки щелочного сульфита или дру-
гой подобной соли, обладающей восстанавливающими свойства-
ми, совместно с цианидом или взамен цианида. Сернистый газ
в слабокислой или нейтральной среде также полезен как де-
прессор цинковых минералов. Вероятно, действие сернистого га-
за аналогично действию щелочных сульфитов. Часто вместе с
цианидом'или сульфитом добавляют сульфат цинка или некото-
рые другие соли цинка.
В настоящее время установлено (см. гл. 10), что цианид не
оказывает депрессирующего действия на чистую цинковую об-
манку, но депрессирует марматит, образуя цианогенное покрытие
этого минерала. Цианид дезактивирует все разновидности цин-
ковой обманки активированной медью, образуя комплексное со-
зз*
516
ГЛАВА 15
единение с медью и таким образом удаляя ее с поверхности ми-
нерала. Пока не установлено, действует ли сульфитный ион сам
по себе или только после преобразования его в другой серо-кис-
лородный ион посредством реакций, происходящих между раст-
воренным кислородом и поверхностями минералов- Явление это
представляется достаточно сложным. Ионы цинка при больших
концентрациях препятствуют активации сфалерита ионами свин-
ца [39]. При малых концентрациях, однако, поведение их неясно.
Необходимую щелочность пульпы наиболее удобно поддержи-
вать, добавляя соответствующее количество кальцинированной
соды. Однако вместо соды можно применять значительно более
дешевую известь.
Кальцинированная сода, жидкое стекло, гексаметафосфат
натрия или другие водорастворимые щелочные полифосфаты да-
ют довольно дисперсные суспензии, в то время как известь ока-
зывает флокулирующее действие. В каждом отдельном случае
должно быть установлено эмпирически, какой из регуляторов
щелочности или какая их комбинация могут обеспечить лучшие
результаты.
Для флотации цинка обычно активируют цинковые минералы
растворимыми солями меди, главным образом — сульфатом ме-
ди. Флотацию ведут с сульфгидрильным собирателем (ксантоге-
натом). Эта операция производится в щелочной среде при зна-
чениях рИ= 10—12.
В этих условиях большинство медных солей осаждается вна-
чале в виде гидрата окиси меди или в виде основных медных со-
лей. Осадок этот получается избыточным запасом меди, который
и обеспечивает нужное насыщение раствора. Таким образом,
концентрация ионов меди в растворе поддерживается относи-
тельно низкой, и сульфид цинка активируется весьма селективно.
Как следствие этого, процесс активации протекает довольно
медленно; поэтому необходимо предварительное перемешивание
пульпы с активатором в течение 5—30 мин. Перемешивание це-
лесообразно совмещать с аэрацией, поскольку кислород одновре-
менно способствует депрессии пирита и пирротина.
Собиратель обычно добавляют после этой операции, но его
можно загружать и совместно с активатором.
Расход сульфата меди составляет в среднем от 250 до 500 г/т
руды. Расход, обеспечивающий лучшие результаты, зависит от
величины свободной поверхности минералов и во всех случаях
должен определяться опытным путем.
Количество ксантогената в цинковой флотации обычно гораз-
до меньше количества сульфата меди и составляет в среднем
около 50 г/т. Расход извести или кальцинированной соды за-
висит прежде всего от количества присутствующих растворимых
солей. Он также должен быть установлен опытным путем так,
СУЛЬФИДНЫЕ РУДЫ 517
чтобы обеспечить значение pH пульпы, равное 12 или ниже. На
практике обычный расход этих реагентов составляет от 0,5 до
нескольких килограммов на тонну руды. Количество пенообразо-
вателя незначительно, обычно не свыше 25 г/т руды. Если в пуль-
пе после проведения свинцовой флотации пенообразователь
остается еще в достаточном избытке, то в цинковую флотацию
его можно вообще не добавлять.
Хотя цианид, применяемый в свинцовой флотации, и сульфат
меди, применяемый в цинковой флотации, являются сами по се-
бе противоположными реагентами, это не играет особой роли,
поскольку одной из флотаций цианида является депрессирование
(и возможное покрытие) пирита, в то время как ионы меди пред-
назначаются для воздействия на сфалерит. В связи с этим сле-
дует иметь в виду, что для флотации наиболее полезны реакции,
протекающие только в одном направлении, которые достигают
состояния равновесия при очень продолжительном времени, зна-
чительно превосходящем обычно принятую на практике продол-
жительность операций. Поэтому применение таких «противопо-
ложных» реагентов является вполне допустимым.
Флотация пирита и пирротина после цинковой флотации до-
вольно затруднительна, во всяком случае по сравнению с флота-
цией недепрессированных предварительно железных минералов.
Обычно наиболее простым методом является резкое снижение
значения pH до величины 4 и ниже посредством добавок серной
или сернистой кислоты. Кислота расходуется очень быстро, и
значение pH повышается до 6 или 7; при этих pH и производит-
ся флотация. Добавляя кислоту, очищают поверхность железных
минералов от покрытий окислами и цианистыми комплексами, а
это дает загружаемому сульфгидрильному собирателю возмож-
ность образовать на минералах гидрофобное покрытие. Если же
подавление пирита было не слишком сильным, можно вести пи-
ритную флотацию и в содовой среде при рН~9, применяя при
этом несколько повышенные загрузки собирателя.
Оценка результатов селективной свинцово-цинковой флота-
ции затруднительна не только потому, что получается два или
более конечных ценных продукта, но и вследствие сложного ха-
рактера требований, предъявляемых металлургами к свинцо-
вым и цинковым концентратам. Большую роль играет также со-
держание в концентратах сопутствующих компонентов — золота,
серебра, меди, сульфидов железа.
Особенностью частично окисленных свинцово-цинковых руд
является образование сульфата свинца в виде покрытия на по-
верхности галенита и заполнения в нем трещин. Сфалерит или
марматит, по-видимому, более энергично активируется солями
меди, но возможна также и активация их солями свинца и се-
ребра. Главнейшими окисленными цинковыми минералами яв-
518
ГЛАВА 15
ляются гемиморфит и смитсонит. Они обычно образовывались в
тех случаях, когда процесс окисления руд протекал в нейтраль-
ной или слегка щелочной среде. При кислой среде цинк перехо-
дит в раствор и выносится из месторождения грунтовыми
водами.
Практика обогащения свинцово-цинковых руд. Фабрика
Сулливан [За, 16]. На фабрике перерабатываются руды рас-
положенного поблизости крупного месторождения Кимберлей
(Канада). Основными сульфидами являются галенит, марматит и
пирротин совместно с небольшим количеством пирита. Порода
представлена кварцем. Весьма ценно серебро, которое тесно свя-
зано с галенитом. Значительная его часть присутствует в виде
сульфосолей.
Сложный характер руды усугубляется еще наличием неболь-
ших количеств самородного золота, а также олова, представлен-
ного касситеритом. Все минералы характеризуются весьма тес-
ным взаимопрорагтанием, что весьма осложняет получение цин-
ковых концентрат >в нужного качества.
В настоящее время руда в крупных кусках подвергается
предварительному обогащению в тяжелых суспензиях. Измельче-
ние руды осуществляется стадиально в большой стержневой
мельнице и в нескольких шаровых.
После выделения свинцовой, цинковой и пиритной флотацией
трех концентратов получаются отвальные хвосты, содержащие
незначительное количество олова в виде касситерита. Это олово
извлекается методами гравитационного обогащения.
Разделение в суспензиях I2U осуществляется на материале
крупностью —32 + 5 мм. Удаляемая легкая фракция содержит
свинца и цинка в сумме меньше 0,5%. Выход ее равен 25°/0 и по-
тери в ней обоих металлов меньше 2%. Фактически удаляемый
продукт имеет более низкое содержание металлов, чем флота-
ционные хвосты. При смешивании его с некоторым количеством
пирротина он образует самоцементирующийся материал, при-
годный для рудничной закладки. В качестве разделяющей среды
применяется водная суспензия флотационного свинцового кон-
центрата, добавляемого с расчетом поддержания удельного ве-
са суспензии равным 2,94. Поскольку суспензия дорога, а рас-
ход ее значителен, необходима специальная система полной от-
мывки и извлечения утяжелителя (регенерация). Регенерация
утяжелителя производится флотацией и сгущением при точной
регулировке плотности продукта разгрузки сгустителя [5, 7L
После обогащения в тяжелых суспензиях тяжелая фракция и
отсев подвергаются тонкому измельчению и флотации.
Еще несколько лет назад на фабрике применялись большие
валковые дробилки, работавшие в замкнутом цикле с грохота-
ми, а затем трехстадиальное измельчение в шаровых мельницах.
СУЛЬФИДНЫЕ РУДЫ
519
Сейчас эти валковые дробилки заменены одной очень боль-
шой стержневой мельницей, работающей в открытом цикле. Раз-
грузка стержневой мельницы представляет собой материал, со-
держащий много свободного галенита флотационной крупности-
Поэтому сразу после измельчения в стержневой мельнице произ-
водится первая свинцовая флотация, при которой выделяется до-
вольно крупнозернистый концентрат. Хвосты этой операции,
представляющие собой рудную пульпу лишь с немного меньшим
содержанием свинца, чем в руде, измельчаются окончательно в
шаровых мельницах. Затем последовательно проводится вторая
свинцовая, цинковая и железная флотации. Последнюю перво-
начально предназначали для удаления основной массы пирита и
пирротина с таким расчетом, чтобы обеспечить извлечение кас-
ситерита методами гравитации из несульфидной части хвостов [4].
В настоящее время часть железного концентрата используется
в качестве цементирующего материала крупной легкой фракции,
употребляемой для рудничной закладки, другая часть предназ-
начается для производства серной кислоты. Пиритные огарки
могут найти применение как высокосортная железная руда [30].
Рис. 6 характеризует схему операций фабрики, а на рис. 7 по-
казана схема операций обогащения руды в тяжелых суспензиях.
Черновые концентраты — свинцовый и цинковый — подвер-
гаются двойной перечистке. Цинковый промежуточный продукт
(хвосты первой перечистки цинкового концентрата и концентрат
контрольной флотации) доизмельчаются в отдельной мельнице и
подвергаются повторной свинцовой и цинковой флотации для до-
извлечения свинца и цинка после раскрытия сростков, не извле-
ченных в основных циклах флотации.
Железный концентрат не перечищают.
На рис. 8 показана схема флотации.
На фабрике применяются обычные реагенты. Флотация про-
изводится сульфгидрильными собирателями вместе с известью,
цианидом, кальцинированной содой и сульфатом меди.
Качественные показатели обогащения приведены в табл. 1.
Таблица 1
Средние показатели обогащения руды на фабрике Сулливан
Продукты Содержание, %
Ag г/т РЬ Zn Fe нераствори- мый остаток
Питание Концентраты: 124,8 [8,5 7,5 36,0 12,0
свинцовый .... 748,8 69,5 5,0 6,0 1,2
цинковый .... 62,4 3,3 50,0 11,6 1,4
Грохочение 5мм
~32 *5мм
5-О мм
Разделение' в тяжелой
суспензии
легкая
Фракция
тя/келая\
Фракция
Для рудничной
закладки
I измельчение
Пиритный концентрат Извлечение
вля рудничной закладки олова грави-
или производства серной тацией
кислоты
Рис. 6. Общая схема фабрики Сулливан
Дробленая руда
32-\дмм.
Флотационный РЪ
концентрат
Грохот вибрационный 5мн
'~5-0мм -32+5ММ
Разделение б суспензии
>2. Зв
^2.3V\
' Грохот дренажный
Вода
Г^о^тд^енанхный
I 1 вода
Грохот промывочный
Грохот промывочный
Разбавленная
суспензия
I грохот 0,5мм
Для рудничной
закладки
Д грохот 0,5мм
Сгущение
РЪ срлотация
ПК
Слив
'Г-т
В доизмельчение
Рис. 7. Цикл разделения в тяжелых суспензиях на фабрике
Сулливан
Частично обессвинцованная пульпа
____________ I Вода_____________
И измельчение
Классификация
Основная Ро флота
ция
^т
1~перечистка Контрольная РЪ флотация
~К-т
УВ
К-т
Хв.
Л перечистка
Перемешивание
Свинцовый
концентрат
Основная 2п флотация
Уб^
I перечистка Контрольная 2п флотация
-т
УУ
~Шт
Л перечистка
^пт^^~ХВ |
Пиритная српетация
УВ
Измельчение
Классификация
С гу оценив
На извлечь
ние
олова
Цинковый .
концентрат
Перемешивание
Свинцовая флотация
Слив
Извлечение се
ры и 6 отвал
Цинковая флотация
7?^
Рис. 8. Цикл флотации на фабрике Сулливан
СУЛЬФИДНЫЕ РУДЫ
523
Фабрика И гл. Фабрика Игл в Колорадо [12] является
необычайной в двух отношениях: во-первых, это единственная
фабрика в США, полностью расположенная под землей, во-вто-
рых, она перерабатывает руду, содержащую очень много раство-
римых солей. Главные рудные минералы—галенит, марматит
и пирит, заключенные в смешанной карбонатно-кварцевой поро-
де, содержащей много анкерита. Анкерит представляет собой
двойной карбонат, относящийся к доломитам, но содержащий
железо и некоторое количество ‘Марганца, замещающего магний.
Сопутствующими минералами являются халькопирит и тетраэд-
рит. Руда также содержит немного золота.
Измельченная руда разделяется на пески и шламы. Шламы
подвергаются коллективной флотации для извлечения свинца и
цинка при естественном значении pH пульпы около 6. Это позво-
ляет избавиться от необходимости осаждения основной массы
растворимых солей, так как они выводятся вместе с хвостами
шламовой флотации. Измельченная песковая фракция, к которой
присоединяется шламовый концентрат, подвергается селективной
флотации для извлечения свинца и цинка.
На рис. 9 и 10 указаны основные операции фабрики и пред-
ставлена развернутая схема свинцово-цинковой флотации.
Применяемая схема обеспечивает экономию в расходе изве-
сти по сравнению со схемой без выделения шламов. С другой
стороны, схема более сложна, и благоприятное воздействие боль-
ших количеств имеющегося в руде растворимого цинка здесь не
используется. Кроме извести и ксантогената, применяется ще-
лочной цианид и сульфит натрия для депрессирования мармати-
та, а также сульфат меди для его активации. Для депрессирова-
ния железных минералов прибегают к аэрации.
Извлечение в свинцовый концентрат основной массы присут-
ствующих в руде меди и серебра крайне желательно, однако это
трудно осуществимо из-за текстурных и химических свойств об-
рабатываемой руды.
Другие примеры флотации свинцово-цинковых руд. Приме-
нение щелочных сульфитов при селективной флотации свинцово-
цинковых руд описывалось подробно на примере фабрики Мид-
вэйл 144, 45]. Указывалось также [2], что на фабрике Маунт-
Айза в Австралии для повышения извлечения различных метал-
лов успешно применяли предварительное перемешивание пульпы
с сернистым газом, Описана одна свинцово-цинковая фабрика,
на которой главным собирателем являлся тиокарбоналид [56].
В описании одной из фабрик в Юго-Восточном Миссури от-
мечается эффективность применения комбинированной обработ-
ки руды флотацией и гравитацией 153].
Обработка цианидом цинкового концентрата с последующей
перечисткой может быть использована для повышения качества
Дробленая руда
Вода
Измельчение
Классификация
Промывка б классификаторе
ШламьГ
, (с солями)
Коллективная РЪ = Zn
флотация
ИКЯ8И
Хв.
К-т
Коллективный
шламовый
концентрат
Пески
Хвосты
шламовые
Основная РЪ
Рис. 9. Общая схема фабрики Игл
Измельченная руда и
коллективный шламо-
вый концентрат
I основная РЪ флотация
Хв. |
основная РЪ флотация
Хв.
I перечистка
Хв
л ~пЪречистка
Ш перечистка
^К-т ~~ Хв. |
Перемешивание
Сгущение
Слив
фильтрация
Кек
iam
"1
I основная 2п флотация
| К-т Я
I перечисгт
Хв} ,
Л основная сп
флотация
|л"-/7? Хв.
Свинцовый
концентрат
в плавку
В прудок
Л перечистка
,К-т Хв~^
С гущение
j Слив t
Фильтрация
^дерелив^Кек ^Фильтрат Сгущение
Сушка 8пр^0К
Измельчение
UL основная 2п
флотация
Хв.
Хвосты
в прудок
Цинковый
концентрат
в плавку
Классификация
ГПески СлиТ^
Перемешивание
Г~
I перечистка 2п промпро-
\дукта
-----------JK-m Хв.
Л перечистка 2п промпродук-
та в прудок
Щ перечистка 2п лронлродук------------- 1
Хв.
та
/Гперечистка Еп
промпродукта
1лГ/77
Для опрыскивания
пены в цинковой
флотации
Рис. 10. Цикл свинцовой и цинковой флотации на фабрике Игл
Измельченная руда
РЪ српотацая
К-т
М.
Свинцовый
концентрат
Zn флотация
К-т
М.
Zn концентрат
Хвосты
в отвал
Щелочной цианид
Выщелачивание
Фильтрация
^КеК*****-----
Цианид
Флотация
~К-т
Фильтрат
Свинцовый
продукт
Цинковый
концентрат
Рис. 11. Обессвинцевание цинкового концентрата выщела-
чиванием цианидом
СУЛЬФИДНЫЕ РУДЫ
527
цинкового концентрата, содержание цинка в котором после этой
операции может быть близким к теоретическому содержанию
цинка в минерале [34].
Схема такого процесса показана на рис. 11.
Прочие сульфидные руды
Медно-цинковые сульфидные руды. Большинство медно-цин-
ковых руд состоит из халькопирита, марматита, пирита и сили-
катов, за исключением одной или двух разновидностей, обнару-
женных в Бьютт (Монтана) и в Бельгийском Конго [49, 51], где
основными медными минералами являются халькозин, энаргит
и борнит.
Обычные медно-цинковые руды, количество которых в Во-
сточной и Центральной Канаде весьма значительно, детально ис-
следовались Канадским горным департаментом. Руды эти под-
вергают сначала медной флотации с выделением в концентрат
халькопирита, затем цинковой флотации с получением марма-
титового концентрата. Пирит остается в хвостах цинковой фло-
тации. В первом цикле применяется сульфгидрильный собира-
тель в слегка щелочной пульпе с добавлением щелочного циани-
да. Расход цианида должен регулироваться гораздо более тща-
тельно, чем при флотации свинцово-цинковых руд, поскольку из-
быток его может вызвать депрессию медных минералов.
Диапазон возможных изменений щелочности пульпы также-
значительно уже, чем в свинцовом цикле при флотации свинцо-
во-цинковых руд. Флотация цинка ведется в тех же условиях, что-
и при обогащении свинцово-цинковых руд.
На фабрике Квимонт [38] принята типичная схема селектив-
ной медно-цинковой флотации. Руда содержит 4,6% халькопи-
рита, 3,7% сфалерита, 22% пирита, 32% пирротина и силикаты.
В руде содержится также золото и немного серебра.
Драгоценные металлы частично в виде электрума связаны
главным образом с халькопиритом, пиритом и пирротином, в
меньшей степени со сфалеритом. Руда подвергается последова-
тельно операциям дробления, измельчения и флотации для из-
влечения меди и цинка.
После этого производится флотация золотосодержащего пи-
рита и цианирование пиритного концентрата для извлечения
золота. Так как пиритный концентрат содержит включения халь-
копирита и сфалерита, то он повергается доизмельчению и пере-
чистке для доизвлечения этих ценных компонентов.
Поскольку пирротин, находящийся в хвостах основной пирит-
ной флотации, также содержит золото, производится длительная
контрольная флотация, концентрат которой возвращается в на-
чало пиритной флотации по принципу противотока.
528
ГЛАВА 15
8
сч
<33
=f
s:
ч
о
сч
22
«О
Ci
§
§
'S
5i
Q>
I
Расход г/т о о оо Ю Ю 00 S о 8 о о о о со ’t со «— —- со 100 88 8 888 — со
Точки загрузки Стержневая мельница Стержневая и шаровая мельницы Стержневая мельница и аэратор Аэратор Стержневая мельница, аэратор, флотационные машины По процессу Аэраторы Перечистки Аэраторы Аэраторы Аэраторы, флотационные камеры Аэраторы, флотационные камеры По процессу Флотационные камеры, аэратор, шаровые мельницы Цинковые флотационные камеры Шаровая мельница Флотационные камеры
Назначение Контроль щелочности Депрессор цинка Депрессор цинка и железа' Собиратель Собиратель Пенообразователь Активатор Депрессор железа Активатор цинка Депре:сор железа Собиратель Собиратель Пенообразователь 1 Собиратель Активатор Депрессор железа Собиратель Собиратель Стабилизатор Пенообразователь
Реагенты Кальцинированная сода Сульфит натрия Цианистый натрий Реагент 2081 Калиевый амиловый ксантогенат Сосновое масло Сульфат меди Известь Сернокислый аммоний Цианистый натрий Реагент 2081 Калиевый амиловый ксантогенат Вторичный бутиловый спирт Этиловый ксантогенат натрия Медный купорос Известь Амиловый ксантогенат калия Изопропиловый ксан- тогенат натрия Реагент 2422 Вторичный бутиловый спирт
Пикл Медная флотация Цинковая флотация Пиритная основная и контрольная флотация Доработки пиритного концентрата (с целью доизвлечения сфалерита и халькопирита)
1 Реагент 208—смесь диэтилового и вторичного бутилового дитиофосфата, нейтрализованных содой.
* Реагенат 242—аэрофлот 31, нейтрализованный аммиаком.
СУЛЬФИДНЫЕ РУДЫ
529
На рис. 12 показана схема фабрики Квимо'нт. Флотация меди
производится здесь между операциями вторичного тонкого из-
мельчения и вторичной классификации, что преследует цель из-
влекать возможно более крупный халькопирит. Расход реагентов
приводится в табл. 2, а показатели фабрики — в табл. 3.
Таблица 3
Показатели обогащения руды на фабрике Квимонт
Продукты Содержание, %
Au, г/tn Ag, г/tn Си Zn пирит пирротин
Руда 4,84 29,95 1,60 2,20 22,0 32,0
Концентраты:
медный 47,74 210,29 19,10 3,65 14,0 15,0
цинковый 3,43 50,23 1,04 51,00 1,5 11,5
пиритный 2,34 20,90 0,09 0,65 81,0 15,0
Остаток цианирования . . 1,12 16,54 0,08 0,65 81,0 15,0
Хвосты флотации .... 0,78 10,61 0,08 0,17 5,0 40,0
Продукты Распределение, %
Au Ag Си Zn пирит
Руда Концентрат: 100,0 100,0 100,0 100,0 100,0
медный 77,0 57,0 93,25 13,5 6,7
цинковый 2,2 5,3 2,0 75,0 0,2
Золотой осадок 4,9 2,0 Следы Следы 0,0
Осадок цианирования . . . 5,1 11,3 1,2 6,25 75,0
Хвосты флотации 10,8 14,4 3,85 5,25 18,1
Медно-свинцовые сульфидные руды. Отделение свинца от
меди методом селективной флотации по своему значению не мо-
жет быть приравнено к отделению свинца от цинка и даже меди
от цинка.
Частично это объясняется сравнительно малой распростра-
ненностью руд, в которых и свинец и медь содержатся вместе в
промышленных количествах, а также и тем, что в процессе ме-
таллургического передела разделение свинца и меди происходит
легко и достаточно полно. Поэтому применение селективной фло-
тации для разделения этих металлов не всегда экономически
выгодно. Однако, несмотря на это, иногда считают целесообраз-
ным осуществлять частичное разделение с получением богатого
медного продукта, практически не содержащего свинца — для
•34 А. М. Годэн
Рида
1 дробление
U дробление
I Армельчение
Классификация
Пески I
Слив\
* Z
Яэрацйя
А измельчение
j А Си флотация
К-т Хб.
Медный
концентрат
Л Си флотация
I К-т Хв.
Аэрация Ш Си флотация
1 Хв. \
Шлюз
Перечистка
Шлюз
—С
Хвосты
Аэрация
Классификация
1 Лески Слив |
ЛИ
I Zn флотация
^юз
Аэрация
Цинковый
концентрат
Перечистка
Л-Л2. !SBSSS
Си срмтация
К-т
Аэрация
ПиритНа/ Флотация
---J\ K-m Хв. \
Аэрация Шлюз
Zn флотация
К-т Ttr]
Классификация
..,—,.1 Леска Слид\
гчельчение
Измельчение
Контрольная flu
Флотация
Цианирование
:ка.
Золото
(осадок)
Классификация
ЗПески Слид\
Хвосты
Пиритный
концентрат
Рис. 12. Схема фабрики Квимонт
СУЛЬФИДНЫЕ РУДЫ
531
медной плавки и свинцово-медного продукта — для свинцовой
плавки, в процессе которой медь может быть легко выделена.
В некоторых рудах медь, свинец и цинк присутствуют сов-
местно. При выделении из руды трех различных концентратов и
хвостов трудно получить такое же извлечение металлов, как в
случае обработки свинцово-цинковых руд. Однако за последние
годы интерес к обогащению таких руд сильно возрос, так как при
этом возможно получить значительные экономические выгоды.
Один из методов разделения свинца и меди заключается в
депрессировании медных и цинковых минералов с помощью циа-
нида и флотации галенита сульфгидрильным собирателем. Ре-
гулирование концентрации иона циана в двух резко различных
пределах в две последовательные стадии позволяет осуществлять
разделение всех трех минералов.
Другой метод состоит в обработке общего коллективного
концентрата бихроматом в кислой среде. Это позволяет осуще-
ствить окислительную депрессию галенита и медьсодержащих
сульфидов, в то время как цинковые минералы активируются
растворенной медью (из других присутствующих в Руде мине-
ралов) и флотируются сульфгидрильным собирателем. Таким
методом пользуются, например, на фабрике Тсумеб в Африке [431.
Иногда может быть применен комбинированный метод с исполь-
зованием и цианида и бихромата для разделения трех минера-
лов [28].
Для разделения свинца и меди производится длительное пе-
ремешивание с аэрацией коллективного концентрата в присутст-
вии бихромата. Так работают, например, на фабрике Церро ди
Паско [50, 58L На других фабриках стремятся осуществить раз-
деление медных сульфидных минералов и галенита при помощи
сильных восстановителей—сернистого газа и смеси сернистого
газа с гидросульфитом цинка. Этот метод применяется преиму-
щественно в Мексике [9, 15].
Механизм процесса разделения свинца и меди пока еще мало
изучен. Существует целый ряд вопросов, ответ на которые не
найден. Являются ли бихроматы, широко применяемые на прак-
тике, только окисляющими реагентами, или же хроматная груп-
па действует и как депрессор? Являются ли полезными для этой
же цели высокие концентрации в пульпе сульфатного иона? По-
лезны ли реагенты, удаляющие ионы свинца? Если это так,
можно ли успешно вести флотацию при высокой щелочности
пульпы с образованием плюмбатов? И каким образом можно
примирить окислительные свойства бихромата с восстановитель-
ными свойствами гидросульфита?
Медно-никелевые сульфидные руды. Медно-никелевые суль-
фидные руды, являющиеся в настоящее время основным источ-
ником мировой добычи никеля, представлены месторождения-
34*
532
ГЛАВА 15
ми бассейна Содбери (Онтарио). Главными минералами являют-
ся халькопирит, пентландит NiFeS2 и пирротин совместно с же-
лезо-магниевыми силикатами. Пирротин является частично ни-
кельсодержащим минералом, незначительные количества нике-
ля присутствуют в нем в виде твердого раствора.
Кроме того, некоторая часть пентландита встречается в виде
тонких прорастаний в пирротине, образуя текстуру, схожую с эв-
тектикой a-железа и карбида железа, обнаруженной в углероди-
стых сталях (перлит).
Халькопирит, напротив, не встречается в тесном срастании с
другими сульфидами. Вследствие этих особенностей при обра-
ботке руд Содбери возможно получать раздельно медный кон-
центрат, железо-никелевый концентрат и кварцевые хвосты [1, 3L
Так как при последующей металлургической переработке
продуктов обогащения медь из никелевого концентрата извле-
кается более легко и полно, чем никель из медного концентрата,
процесс ведут так, чтобы получить высококачественный медный
концентрат с минимально возможным содержанием в нем ни-
келя, хотя бы за счет некоторого снижения извлечения меди, и
обеспечить высокое извлечение никеля в никелевый концентрат.
Руды имеют относительно высокое содержание платины, а
также палладия, иридия и осмия. Минералогическая форма этих
металлов изучена недостаточно. Обычно при флотации они из-
влекаются совместно с никелем, а затем отделяются от него.
При флотации руды сначала выделяют коллективный медно-
никелевый концентрат, а после этого никелево-железный. Кол-
лективный концентрат разделяется на никелевый и медный трой-
ной перечисткой с противоточным возвратом промежуточных
продуктов. Полученный при этом никелевый концентрат присо-
единяется к основному никелево-железному концентрату, обра-
зуя общий объединенный продукт, направляемый в никелевую
плавку.
Флотация халькопирита производится обычным методом в
слегка щелочной среде (рН~9) при регулируемых загрузках
сульфгидрильного собирателя и цианида с предварительной аэ-
рацией пульпы.
Получение никелевого продукта интересно в том отношении,
что здесь применяется флотация после плавки [52]. Никелевый
концентрат, выделяемый при флотации пирротина, подвергается
плавке. При этом удаляются железо и сера из сплава меди, ни-
келя, железа и серы. Полученный медно-никелевый штейн сна-
чала должен быть доведен до желаемого состава. Затем его осто-
рожно охлаждают так, чтобы при этом происходило нарастание
кристаллов достаточной величины. Кристаллы эти состоят из
сульфида одновалентной меди, металла и никелевых сульфидов.
Металлическая фракция содержит в себе основную массу дра-
СУЛЬФИДНЫЕ РУДЫ
533
гоценных металлов. Охлажденный штейн поступает в тонкое из-
мельчение, после чего его просеивают на грохоте для удаления
металлических частиц. Просев грохота подвергается селективной
медно-никелевой флотации с получением медного и никелевого
концентратов. На рис. 13 показана принципиальная технологи-
ческая схема фабрики Инко.
В ближайшее время схема, вероятно, будет частично измене-
на, так как на фабрике намерены ввести в действие новую уста-
новку, предназначенную для получения железистых окатышей
из содержащегося в руде пирротина, а это потребует вывода кон-
центрата контрольной флотации или некоторой его части из об-
щего потока, направляемого сейчас в первичную плавку. Этот
пирротиновый концентрат должен быть подвергнут обжигу с по-
следующим выщелачиванием для извлечения из него меди и ни-
келя, а затем окомкованию. Железный продукт предполагается
получать очень высокого качества с содержанием около 70% Fe
при суммарном содержании Си и Ni менее 0,2%. Значительное
количество сернистого газа в течение некоторого времени будет
теряться. Предполагается по возможности организовать его
улавливание и дальнейшее использование.
Медно-кобалытовые сульфидные руды. Интересное сообщение
было опубликовано о процессе обогащения медно-кобальтовой
руды, испытанном на опытной фабрике 1591. Руда состоит из
халькопирита, кобальтина, пирита и пирротина в кварцевой по-
роде. Процесс обогащения руды включает операцию флотации
с промежуточным обжигом. Вначале проводится флотация меди,
а затем кобальта (подобно селективной флотации свинцово-
цинковых руд с последовательным выделением свинцового и
цинкового концентратов). Так как получаемый кобальтовый про-
дукт является низкокачественным и содержит всего около 5%
Со при высоком содержании пирита и пирротина, необходима
его дальнейшая переработка. Кобальтовый продукт подвер-
гается тщательно контролируемому обжигу (425—450°) с после-
дующим охлаждением для перевода железных минералов в окис-
ленные формы (процесс Хэрвуда), после чего производится пе-
речистная флотация кобальта. Из этого цикла вторичной обра-
ботки получается конечный кобальтовый флотационный концент-
рат, содержащий около 20% Со при содержании его в хвостах
0,5%.
Пиритная флотация. В тех случаях, когда пирит извлекается
только ради содержащейся в нем серы, обычно не стремятся к
его полному извлечению, желательно лишь получить наиболее
богатый по содержанию серы концентрат для обеспечения легко-
го обжига. Поэтому при пиритной флотации стараются извлечь
в концентрат лишь свободные чистые зерна пирита, пренебрегая
потерями его в сростках и шламах.
Измельченная руда ,
Коллективная Cu-NL
флотация
| К-т
Разделение Cu-NL концент-
рата
Ж
К-т
ХВ.
NL - пирротиновая
флотация
ТГот Хв.
Медный
концентрат
в плавку
NL концентрат
Хвосты
в отвал
Первичная плавка
Газы
Охлаждение
| Шлаки
NL-Co роштейн
Мокрое измельчение
Производство
серной кислоты ,
Грохочение
Металлические
частицы
Извлечение
благородных
металлов
при плавке
Cu-NL
Разделение
концентрата
Медный
концентрат
в плавку
Никелевый
концентрат
в плавку
Рис. 13. Схема фабрики Инко
СУЛЬФИДНЫЕ РУДЫ
535
При проведении пиритной флотации задается небольшое ко-
личество собирателя и пенообразователя (несколько десятков
граммов на тонну), а иногда и серной кислоты, загружаемой при
кратковременном предварительном перемешивании пульпы пе-
ред флотацией.
Известно много примеров обработки таких руд, в которых пи-
рит не является основным ценным минералом, однако дополни-
тельная пиритная флотация при определенных экономических
условиях вполне рентабельная операция.
Представляет интерес включение пиритной флотации в схему
производства уранового продукта, применяемую в настоящее
время на Ранде при обработке руд, содержащих золото и уран
[26]. Эти руды до 1950 г. перерабатывались лишь ради извлече-
ния из них золота. Однако сейчас они уже рассматриваются как
один из наиболее мощных источников мировой добычи урана.
После того как золото извлечено цианированием, уран рас-
творяется серной кислотой. Остаток от выщелачивания подвер-
гается флотации для извлечения пирита.
Дополнительное извлечение некоторого количества золота и
урана в общем оправдывает расходы на проведение пиритной
флотации и обжиг пиритного концентрата. При этом выделяемый
при обжиге сернистый газ используется для получения необходи-
мой серной кислоты.
Удаление сульфидов из руд, в которых они не являются цен-
ными компонентами. Типичными представителями таких руд яв-
ляются оловянные и вольфрамовые руды. Ценные минералы,
встречающиеся в них, — касситерит, шеелит, вольфрамит —
обычно связаны с гематитом, турмалином, топазом, сидеритом,
баритом, рутилом и другими окисленными минералами, а также
с сульфидами, преимущественно пиритом, пирротином и марма-
титом. Иногда в рудах присутствует также станнин, с которым
может быть связано значительное количество олова.
Гравитационное обогащение подобных руд само по себе не
может обеспечить удовлетворительных показателей вследствие
незначительной разницы в удельных весах сульфидов и ценных
компонентов. Обычно считают целесообразным выделять магне-
тит, лимонит и пирротин магнитной сепарацией (например, ис-
пользуя мокрые магнитные сепараторы), а сульфиды отделять
коллективной флотацией. При этом могут быть применены сле-
дующие три метода [23, 24, 25]: 1) флотация ксантогенатом в
нейтральной или слегка кислой среде (рН~5); 2) флотация ами-
нами, имеющими длинную углеводородную цепь, в щелочной
среде (pH ~ 9); 3) флотация жирными кислотами в сильнокис-
лой среде (рН~2—3).
Последний метод обеспечивает наиболее полное удаление
всех сульфидов, включая и их шламы, но при этом пульпа ока-
536
ГЛАВА 15
зывает корродирующее действие на оборудование. Кроме того,
этот метод неприменим к рудам, содержащим значительные ко-
личества карбонатов, вследствие быстрой реакции их с кислотой.
Выбор между флотацией аминами и ксантогенатом обуслов-
ливается главным образом соображениями экономического по-
рядка и условиями последующей обработки сульфидного кон-
центрата. Оба эти метода обеспечивают хорошее выделение суль-
фидов.
Другим типом руд, относящихся к той же категории, являют-
ся родохрозитовые МпСОз руды, содержащие силикаты и суль-
фиды в качестве сопутствующих минералов. Такие руды могут
обрабатываться, например, флотацией сульфидов с последующей
флотацией родохрозита [31, 57]. Этот метод широко распростра-
нен при обработке марганцевых руд Бьютта (рис. 14).
Возможно также осуществлять предварительную депрессию
сульфидных минералов загрузками щелочных сульфидов (Na2S
и пр.) совместно со щелочными цианидами и карбонатами или
без них, а затем проводить мыльную флотацию родохрозита 122L
Выбор того или иного (метода обусловливается экономическими
соображениями и установившимися традициями, так как всегда
удобнее сначала отфлотировать из пульпы сульфиды.
Золотые руды. Для извлечения из руд золота методом фло-
тации вполне применимы положения, высказанные ранее в от-
ношении проведения флотации пирита.
Очень богатыми рудами считаются те, в которых содержание
золота составляет около 20 г/т. Это означает, что отношение зо-
лота к горной массе составляет по весу всего 2О/юооооо, а по объе-
му не свыше 4/юооооо- Кроме того, большая часть золота при из-
мельчении руды до необходимой для флотации крупности нахо-
дится еще в сростках. Поэтому процесс приходится вести так,
чтобы в концентрат извлекать большую часть пирита, поскольку
пирит является наиболее распространенным основным минера-
лом в золотосодержащих рудах.
Иногда в таких рудах содержатся и другие ценные металлы,
например медь в виде сульфидов или уран в виде окислов. Если
желательно извлечь эти минералы вместе с золотом в один кол-
лективный концентрат, выбор необходимого собирателя опре-
деляется характером этих минералов. Для золотой руды, содер-
жащей халькопирит, применяются сульфгидрильные собиратели.
Для урансодержащей золотой руды целесообразно использовать
при флотации жирные кислоты, например олеиновую, иногда
совместно с сульфгидрильным собирателем. Процесс ведется
при pH около 7 с добавлением небольших количеств диспергато-
ров для глинистых шламов.
Молибденовые руды. Молибденит часто содержится в неболь-
ших количествах во вкрапленных медных рудах. Во многих ме-
СУЛЬФИДНЫЕ РУДЫ
537-
сторождениях Клаймакс в Колорадо молибденит является основ-
ным ценным компонентом при относительно высоком содержа-
нии и весьма малом количестве медных минералов.
^Намельченная руда
Коллективная флотация сульфидов
К-т
Хв.
Свинцовая Флотация
Мп флотация
К-т Хв.
Воздух
I К-т
мродохрозцт)
Хв.
Свинцовый
концентрат
О джиг
Хвосты
кварцевые
Марганцевый
спек
Углекислый
газ в атнюскреру
Zn флотация
Цинковый Хвосты
концентрат '(в основном пирит)
Рис. 14. Схема обработки марганцевых руд месторождения-
Бьютта, Монтана
Когда молибденит присутствует в качестве сопутствующего»
минерала в медных рудах, целесообразно извлечь его во флота-
ционный концентрат совместно с медными минералами. При-
этом в концентрате может содержаться до 25—30% Си и 0,5—
2,0% M0S2. Полученный концентрат затем разделяют на медный,
и молибденовый продукты. Такой метод применяется на фабрике-
Магна АрЧур (Юта) и на многих других.
538
ГЛАВА 15
Для разделения медных и молибденовых минералов разра-
ботано несколько способов. Один из них предусматривает де-
прессию 1молибденита растворимым крахмалом. При этом полу-
чается пенный медистый продукт, почти не содержащий молиб-
дена, и обогащенный молибденом камерный продукт. Последний
подвергается дальнейшей обработке различными методами, при-
чем здесь депрессируется уже медь, а молибден флотируется.
При депрессии медных минералов и извлечении молибдена в
пенный продукт в качестве собирателя для молибденита
применяют жидкие углеводороды. При флотации медных мине-
ралов ксантогенатами (в первом цикле флотации) удаление их
пленки с поверхности минералов может быть осуществлено на-
греванием концентрата.
Если же в качестве собирателя для медных минералов при-
менялись дитиофосфаты, которые, по-видимому, являются более
устойчивыми, чем ксантогенаты, приходилось пользоваться спе-
циальными реагентами для депрессии медных минералов, напри-
мер щелочными сульфидами, цианидами, цианистыми соедине-
ниями меди и солями железосинеродистоводородной НзЕе(СМ)б
и железистосинеродистоводородной H4Fe(CN)6 кислот [46].
Так как техническими кондициями на молибденитовый кон-
центрат жестко лимитируется содержание в нем меди, приходится
поддерживать высокие концентрации цианида в последних пере-
чистных операциях, а также включать в схему обогащения руды
большое число перечисток концентрата.
Один из методов разрушения пленки собирателя на поверх-
ности сфлотированных сульфидов состоит в том, что первичный
концентрат подвергается легкому окислению или обжигу. Если
в качестве собирателя применялся ксантогенат, достаточно бы-
вает обработать первичный медный концентрат кислотой или
прокипятить его при одновременной аэрации пульпы. Могут при-
меняться также и комбинированные методы. На рис. 15 показа-
на типовая схема селективной медно-молибденовой флотации,
применяемая на фабрике Кананеа (Мексика).
Фабрика имеет большую производительность, перерабатывая
в сутки многие тысячи тонн руды, однако для перечистки молиб-
денового концентрата на ней установлены очень небольшие фло-
тационные камеры (почти такие же, как лабораторные). Молиб-
деновый концентрат подвергается многочисленным перечисткам,
что вызывается не только стремлением получить конечный про-
дукт высокого качества, но и необходимостью обеспечить сохра-
нение нужного баланса пульпы при осуществлении перечистных
операций очень малого масштаба.
На фабрике Майами при обработке руды получают медный
концентрат, содержащий также и молибденит 113]. Этот концент-
рат сгущают, перемешивают с добавлением извести, затем обра-
СУЛЬФИДНЫЕ РУДЫ
539
батывают паром и аэрируют при высокой плотности пульпы.
После этого пульпа разбавляется и поступает на флотацию.
Руда, перерабатываемая на фабрике Клаймакс [14, 17, 29],
является чисто молибденовой и содержит другие металлы в ма-
лых количествах, не оправдывающих расходов на извлечение их
в отдельные концентраты. Технологическая схема построена по
Измельченная руда
Си флотация
к-т ~хв.
Удаление
реагента-собирателя Хвосты
6 отвал
Углеводородный
коллектор
Ио флотация
Молибденовый 1 _
концентрат Медный
г концентрат
Рис. 15. Схема селективной медно-молибденовой флотации
принципу всемерного снижения стоимости обработки. Одним из
наиболее существенных факторов является использование прин-
ципа флотации зерен, содержащих чешуйки молибденита
в сростках с пустой породой. Полученный концентрат доизмель-
чается и подвергается перечистной флотации. В результате мно-
гократного применения этого принципа (рис. 16) отвальные хво-
сты являются относительно крупными, а конечный концентрат
получается очень тонким.
При флотации задаются следующие реагенты: предельные уг-
леводороды нефти (0,5 кг!т), сосновое масло (25 г/т) и ионоген-
Дробленая руда.
I измельчение
Кпассисрикация
Пески. Слив
Грубый к-т
I Мо срл стация
ПронпроВукт,
Сгущение
К-т
бый концентрат
Рис. 16. Схема фабрики Клаймакс
СУЛЬФИДНЫЕ РУДЫ
541
ный поверхностно активный реагент, особенно пригодный для
эмульгации углеводородов (Арктик Синтекс М), представляю-
щий собой сульфированный моноглицерин. В очистные операции
в небольших количествах добавляют цианид и кристаллическую
соду для депрессии мелких частиц пирита и халькопирита и для
поддержания pH пульпы в пределах 8,2—8,4.
В последние годы было обнаружено, что в хвостах флотации
фабрики Клаймакс содержится небольшое количество вольфра-
мита. Этот минерал извлекается в отдельной гравитационной
секции, где в качестве основных аппаратов используются спи-
ральные сепараторы Гэмфри. Для перечистки и выделения ко-
нечного концентрата применяются концентрационные столы и
магнитные сепараторы. При этом попутно в небольших количест-
вах извлекается также касситерит и монацит.
При проведении молибденовой флотации весьма важным яв-
ляется использование природной чешуйчатости молибденита —
свойства, которое позволяет извлекать в концентрат даже зерна,
содержащие очень мало молибденита. Другой особенностью
является предпочтительное перед ионогенным собирателем при-
менение смазочного масла. В этом случае, видимо, происходит
адсорбция молекул углеводородов на поверхности частиц молиб-
денита [33].
Краевой угол, измеряемый на поверхности раздела молиб-
денит— масло — вода, значительно больше краевого угла, из-
меряемого на поверхности раздела молибденит — воздух — вода.
Соответственно этому операция перемешивания имеет назна-
чением обеспечить омасливание минералов, а операция флота-
ции— отделить омасленные минералы от минералов неомас-
ленных.
ЛИТЕРАТУРА
1. Anon.: Milling (at International Nickel Co.), Can. Mining J., 67, 423—430,
1946.
2. Anon.: Mt. Isa conditions flotation feed with sulfur dioxide to improve
lead-zinc-silver recovery, Mining World, 14, 31—33, February, 1952.
3. Anon. (Mine Staff). Operation of the Creighton Mill of the International
Nickel Co. of Canada, Ltd,, Trans. Cah. Inst. Mining Met., 55, 153—159,
1952.
3a. Anon. (Management and Staff): Ore dressing. The Sullivan concentrator,
Can. Mining J., 75 (5), 211—223, 1954.
4. Banks H. R. Tin at the Sullivan concentrator, Trans. Can. Inst. Mining
Met., 44, 611—622, 1941,
5. В a n k s H. R. Sink-float at the Sullivan concentrator, Min. Congr. J.,
36, 28—31, July, 1950.
6. Banks H. R. The rod mill in the Sullivan flow, Trans. Can, Inst. Mining
Met.. 55, 245—251, 1952.
7. Banks H. R. Recent developments in practice at the Sullivan concentra-
tor, in «Recent Developments in Mineral Dressing», pp. 633—646, Institu-
tion of Mining and Metallurgy, London, 1953.
542
ГЛАВА 15
8. Benedict С. Harry. «Lake Superior Milling Practice», Michigan Col-
lege of Mining and Technology Press, Houghton, Mich., 1955.
9. Bo eke C. L., and G. G. Gunther. Deleading zinc concentrate at the
Parral and Santa Barnara mills, Mining Eng., 4, 495—498, 1952.
10. Chapman Thomas G. Concentration of copper ores in North Ame-
rica, U. S. Bur. Mines, Bull. 392, 1936.
11. Cody В. H. Milling practice at Morenci Reduction Works, AIMME Tech.
Publ. 2194, 26 pp., in Mining Technol., 11, July, 1947.
12. Craig J. G. Modernizing New Jersey Zinc’s Eagle Mill. Eng. Mining J.,
151 (10), 81—85, 1950, 151 (11), 101—105, 1950.
13. Curtis С. H. Conditioning and treatment of sulfide flotation concentra-
tes preparatory for the separation of molybdenite at the Miami Copper Co.,
Trans. AIMME, 187, 1950; in Mining Eng., 187, 506, 1950.
14. Cuthberston R. E. Climax ore testing program, Mining and Met., 27,
346—349, 1946.
15. Deshler G. O. and T. R. Herndon. New flotation technique in cop-
per-lead separation, Eng. Mining J., 147 (5), 67c69, 1946.
16. Diamond R. W. Ore concentration practice of the Consolidated Mining
and Smelting Co. of Canada Ltd., Trans. AIMME, 87, 95—106, 1928.
17. Duggan E. J. Fine grinding and concentration at Climax, Mining and
Met., 27, 323—329, 1946.
18. F a h r e n w a 1 d A. W. Copper milling research in Michigan, U. S. Bur.
Mines, Rept. Invest. 2878, 1928.
19. Fahrenwald A. W. Xanthate and pine oil float native copper in amyg-
daloid ores, Eng. Mining J., 126, 58—59, 1928.
20. Frick F. F. Plans for treating Greater Butte project ores Min. Congr J.,
37, 42—43, 75, August, 1951.
21. Gaudin A. M. «Principles of Mineral Dressing», chap. XI, pp. 239'—249,
McGraw-Hill Book Company, Inc., New York, 1939
22. Gaudin A. M. U. S. Patent 2,231,265/1941.
23. Gaudin A. M., and Falih Ergunalp. Making tin flotation work. IL
Oploca ore, Eng. Mining J., 147 (3), 70—72, 1947.
24. Gaudin A. M, and Schuhmann, Jr. Making tin flotation work. III.
Colquiri ore, Eng. Mining J., 147 (12), 69—72, 1946, 148 (1), 84— 87, 1947.
25. Gaudin A. M., and R. I. Hukki. Making tin flotation work. IV. San
Jose ore, Eng. Mining J., 148 (3), 70—72, 1947.
26. Gaudin A. M., R. Schuhmann, Jr., and John Dasher. Extraction
Process for Gold-Uranium Ores, J. Metals, 8, 1965—1069, 1956.
27. Goldick M. R. Description of concentrating operations, Roan Antelope
Copper Mines Ltd., Northern Rhodesia, AIMME Tech Publ. 2251, 16 pp.,
in Mining Technology 12(1), January 1948.
28. Gotte A. Selektive Flotation der Blei-Zink-Kupfererze von Tsumeb
durch Kombinierte Cyan-Bichromat Trennung, Arch. Metallkunde 2.
185—187, 1948.
29. Haley D. F., and C. J. Abrams. History of cruching and milling at
Cimax, Mining and Met., 27, 317—319', 1946.
30. Hunt B. G., and A. Turner. Iron and steel produced from pyrrhotite
tailings opens up potential market, J. Metals, 7 (9), 944—947, 1955.
31. Hutt 1 John B. Domestic manganese from Butte helps in emergency,
Eng. Mining J., 143, 56—58, (January, 1942).
32. H u 111 John B. How new leach-float plant handles Greater Butte’s
ore, Eng. Mining J., 154 (6), 90—93, June, 1953.
33. Kainuma Yoshiro, and Ryozi Uyeda. The structure of absorbed
organic longchain molecules on the cleavage surface of molybdenite J.
Phys. Soc. Japan, 5, 199—200, 1950.
34. L a m b 1 у C. A. R., and Frank R. Milliken. De-leading zinc con-
centrates at the Pend Oreille mill, Eng. Mining J„ 141 (3), 29—31, March,
1940.
СУЛЬФИДНЫЕ РУДЫ
54»
35. L u t j е n George P., and John H. Kearney. New life for Vermont's
160-year old copper mine. Eng. Mining J., 154 (10), 72—75, October, 1953.
36. MacDonald William T. Selective flotation mill at Copper Cliff,
Ontario, Eng. Mining J., 130, 465—472, 1930.
37. McLachlan C. G. The development of concentrating operations at
Noranda, Can. Mining J., 55, 175—182, 1934.
38. McLachlan C. G., M. J. S. Bennett, and R. J. Coleman. The
Quemont milling operation, Trans. Can. Inst. Mining Met. 57, 230—245,
1954; also in «Recent Developments in Mineral Dressing», pp. 671—701,
Institution of Mining and Metallurgy, London, 1953.
39. Metzger P. H. Unpublished data obtained at M. I. T., 1955.
40. M о г г о w Bayard S., and George G. Griswold. Production of
highgrade concentrate from Butte copper ores—results of laboratory inves-
tigations, Trans AIMME, 112, 443—423, 1934.
41. Myers J. F„ and F. M. Lewis. Crushing and grinding practice, Tennes-
see Copper Company, Trans. AIMME, 153, 345-—361, 1943.
42. M у e r s J. F. and F. M. Lewis. Fine crushing with a rod mill at the
Tennessee Copper Company, Trans. AIMME, 169, 106—118, 1946.
43. Ong J. N. Tsumeb mill uses five flotation circuits for copper, lead, zinc,
oxide and sulfide ores, Mining World, 14, 34—39, June, 1952.
44. Pal lan ch R. A. Factors governing the separation of lead and zinc in
ore by flotation, Mining and Met., 17, 386—389, 392, 1936.
45. Pallanch R. A Concentration at the Midvale, mill, Mining and Met.,
29, 544—547, 1948.
46. P a p i n J. E. Flotation of molybdenite at the Morenci concentrator, Mi-
ning Eng., 7, 145—147, 1955.
47. Ramdohr P. «Die Erzmineralien und ihre Verwachsungen», Akademische
Verlagsgesellschaft, m. b. H., Leipzig, 1950.
48. Ramsey Robert H. White pine copper, Eng. Mining J., 154 (1), 72—87,
January, 1953.
49. Roger E. Evolution of copper metallurgy at Katanga, Compt. rend, congr.
sci. Elisabethville, 3, 129—168, 1950; cf. Chem. abstr., 46, 7952f, 1952.
50. Saunders D. S. Further notes on milling practice and flowsheet details
(at the Cerro de Pasco enterprise), Mining and Met., 26, 540—544, 1945.
51. Sengier Edgar B. Katanga is well equipped for treating its diverse
ores, Eng. Mining J., 152 (12), 92—96, December, 1951.
52. S p г о u 1 e William K., and George Alan Harcourt. U S. Pa-
tent 2,419,973/1947.
53. Stahl, H. R. Milling practices in southeast Missouri, Mining and Met.,
28, 374—376, 1947.
54. Sutherland K. L., and I. W. Wark. «Principles of Flotation», Austra-
lasian Institute of Mining and Metallurgy, Melbourne, Australia, 1955.
55. Talbot H. L. The development of metallurgical practice for the treatment
of N’Changa mixed oxide-sulfide ores, Bull. Inst. Mining Met., No. 482,
29 pp.. January, 1947.
56. T h о m p s о n R. C. Milling practice at the new lead-zinc concentrator
of Phelps-Dodge Corp., AIMME, Tech. Publ. 2192, 10 pp., 1947, in Mining
TechnoIl., 11, July, 1947.
57. Trauerman Carl J. Manganese in Montana, Min. Cong. J., 27 (6),
23, 1941.
58. Wright T. R. Ore concentration (of Cerro de Pasco Ore), Mining and
Met., 26, 534—539, 1945.
59. Zimmer ley S. R., and S. F. Ravitz. Pilot-plant investigation of
concentration of Blackbird cobalt ore by roast floattion process, Trans.
AIMME, 187, 1044—1046, 1950.
Г л а в a 16
РУДЫ ОКИСЛЕННЫХ МИНЕРАЛОВ
Большое число разнообразных минералов содержит кисло-
род. Поскольку кислород является очень сильным акцептором
электронов, он встречается или в виде иона кислорода, или как
составная часть полиатомного аниона. Относительный размер
иона кислорода или кислородсодержащего полиатомного аниона
определяет структуру минералов и в конечном счете их хими-
ческие свойства и флотационное поведение.
К числу окисленных минералов можно отнести окислы,
гидроокислы, карбонаты, сульфаты, силикаты, вольфраматы,
хроматы, молибдаты и манганаты. Классификация этих окис-
ленных минералов по входящему в их состав аниону или кати-
ону для практических целей менее удовлетворительна, чем клас-
сификация по практически важным элементам, содержащимся
в различных рудах.
В этой главе рассматривается обогащение основных окис-
ленных минералов, состоящих из указанных в табл. 1 составных
частей.
Задачи, требующие разрешения при флотации руд окислен-
ных минералов, сходны с задачами селективной флотации суль-
фидных минералов, поскольку в обоих случаях имеется в виду
разделение минералов различного характера, но одного и того же
широкого класса. При коллективной сульфидной флотации за-
дача значительно проще, так как здесь требуется отделить один
от другого минералы, принадлежащие к разным классам, а
именно, сульфидные от окисленных.
Все приведенные в табл. 1 минералы могут флотироваться
карбоновыми кислотами при условии правильного подбора угле-
водородной группы в молекуле собирателя, а также величины
рИ, концентрации реагентов и характера активатора, если та-
ковой требуется. Кроме того, многие из указанных минералов
можно флотировать при помощи окисленных собирателей, от-
личных от карбоновых кислот, или с применением катионных
собирателей; однако имеющиеся на этот счет сведения отрывоч-
ны и недостаточны. Некоторые минералы медно-свинцово-цин-
ковой группы флотируются сульфгидрильными собирателями;
по имеющимся в настоящее время сведениям из остальных при-
веденных в табл. 1 соединений ни одно не извлекается с по-
мощью указанных собирателей.
РУДЫ ОКИСЛЕННЫХ МИНЕРАЛОВ
545
Таблица 1
Типичные компоненты, входящие в состав обогащаемых
при помощи флотации окисленных минералов
яосится к олову и титану.
** Или полимер Sl^Oy.
Для разделения окисленных минералов необходим точный
подбор расхода собирателя и pH; свобода действий, которой
располагает исследователь в этом случае, много меньше, чем
при коллективной флотации сульфидов, и ближе к той, которая
имеется при селективной флотации сульфидных минералов. Для
дальнейшего усовершенствования процесса разделения реко-
мендуется применение реагентов, селективно осаждающих, свя-
зывающих или образующих внутренние комплексы.
Время от времени появлялись сообщения об эмпирических
открытиях в области флотации окисленных минералов, но они
не вполне понятны даже тем, кто хорошо знаком с этой проб-
лемой.
Окисленные руды меди, свинца и цинка
Наиболее часто в окисленных рудах медные минералы пред-
ставлены двумя основными карбонатами — азуритом и мала-
хитом, но встречаются также и окислы меди и силикат меди —
хризоколла; другой силикат меди — диоптаз, является сравни-
35 А. М. Годэн
546
ГЛАВА 16
тельно редким. В чрезвычайно засушливых областях найдены в
виде минералов различные сульфаты и другие водорастворимые
соли меди. Пустая порода обычно состоит из кварца, глин и дру-
гих силикатов, часто окрашенных гидроокислами железа. Ко-
личество лимонита или гематита может достигать значитель-
ной величины. В некоторых районах встречаются окисленные
медные руды с кальцитовой или доломитовой пустой породой.
Окисленные медно-урановые руды имеются в районе плато Ко-
лорадо: они представляют собой исключение, так как обычно
в окисленных медных рудах отсутствуют побочные ценные ме-
таллы.
Основными окисленными минералами свинца являются кар-
бонат свинца— церуссит, сульфат свинца — англезит, но иногда
существенная часть металла падает на долю хроматов, фосфа-
тов, арсенатов и ванадатов свинца. Окисленные свинцовые руды
часто отличаются высоким содержанием серебра, встречающим-
ся иногда в виде остатков сульфидных или вторичных минералов.
Вообще в этих рудах можно ожидать большого количества гид-
роокислов железа, а также глин совместно с другими силикат-
ными или карбонатными минералами.
В случае цинковых руд термин «каламин» в настоящее вре-
мя относится не к минералу, а к окисленной руде с высоким со-
держанием цинка. В каламинах встречаются два цинковых ми-
нерала, а именно, карбонат цинка — смитсонит и гидратиро-
ванный силикат — гемиморфит. Обычно в окисленных цинковых
рудах эти минералы находятся в виде смеси, проросшей гидра-
тированным кремнеземом, гидроокислами железа и глинами.
Окись цинка (цинкит), цинксодержащая шпинель (франклинит)
и ортосиликат цинка (виллемит) встречаются во Фрэнклин Фэ-
нис Нью-Джерси совместно с магнетитом, кальцитом, гранатом
и множеством других минералов. Эта руда не обогащается при
помощи флотации.
Среди минералов, вызывающих особые затруднения при обо-
гащении, следует отметить цинксодержащую глину, соконит и
сходную с ней медьсодержащую монтмориллонитовую глину,
которая больше известна под названием хризоколлы [38].
Проблемы, связанные с обогащением окисленных руд (не-
благородных металлов), можно классифицировать в соответст-
вии с характером имеющейся в руде пустой породы: силикатной,
карбонатной, глинистой или железистой. Ясно также, что зада-
чи обогащения наиболее просты, когда полезные минералы на-
ходятся в виде окислов или карбонатов, и сильно усложняются
при наличии гидратированных силикатов полезного металла.
Для обогащения различных окисленных руд было предло-
жено пять основных методов, а именно: 1) сульфидизация по-
лезных окисленных минералов, после чего флотация произво-
РУДЫ ОКИСЛЕННЫХ МИНЕРАЛОВ
547
дится так же, как при сульфидных рудах; 2) флотация с приме-
нением в качестве собирателей карбоновых кислот совместно с
депрессорами пустой породы; 3) флотация при помощи сульф-
гидрильных собирателей; 4) флотация с применением в качест-
ве собирателей длинноцепочечных первичных аминов и щелоч-
ных сульфидов для активации; 5) растворение металлов с по-
следующим осаждением их в виде свободных металлов или не-
растворимых соединений, которые затем извлекаются при по-
мощи флотации.
Метод, основанный на применении сульфидизации. Этот ме-
тод является наиболее ранним из указанных выше. Он пригоден
для обогащения в первую очередь свинцовых карбонатных руд
и в меньшей степени для руд, содержащих другие свинцовые
минералы; для медных и цинковых руд этот метод пригоден
лишь в ограниченной степени.
При сульфидизации карбоната свинца реакция обмена меж-
ду твердым карбонатом свинца и сульфидными ионами из ра-
створа протекает быстро. Образовавшийся сульфид свинца при-
липает к минералу так, что реакция не приводит к образова-
нию отслаивающейся пленки. Взаимодействие происходит бы-
стрее, чем это желательно, и поэтому к началу операции суль-
фидизации необходимо обеспечить избыток сульфидных ионов
в растворе.
Флотация сульфидизированного минерала в настоящее вре-
мя лучше всего осуществляется при помощи сульфгидрильного
собирателя (такого, как амиловый ксантогенат), но она может
быть проведена и с карбоксильными собирателями. На практи-
ке подбор условий сульфидизации с ее многочисленными вари-
антами в отношении способа, количества, места и техники до-
бавки реагентов представляется значительно более сложным,
чем подбор условий применения сульфгидрильных собирателей,
в связи с чем метод сульфидизации часто рассматривается от-
дельно от метода применения собирателей, по крайней мере в
патентной литературе1. Из приведенных в патентной литературе
предложений особенно важны те, которые касаются вопросов
подбора разных комбинаций сульфидизатора и собирателя, ре-
гулирования количества и концентрации сероводорода, регули-
рования pH, обесшламливания, подогрева, последовательности
и места загрузки реагентов, измельчения в неметаллической
мельнице и добавок вспомогательных реагентов (таких, как ще-
лочные цианиды или соли).
1 Патенты США: Айер и др., 1916196/1933; Берд, 1686064/1928; Борхердт,
1446375/1923; Кэллоу и др., 1334733/1920, 1334734/1920; Кросдейл, 1573226/1926;
Хендриксон, 1620761/1927; Литлфорд, 1595796/1926; Мартин, 1236856/1917;
Ноукс, 1444552/1923; Рид, 2348360/1944; Терри, 1551588/1925; Спайдер и Грин,
1762364/1930; Терри, 2196233/1940; 2278027/1942; Томпсон и Герри, 1334720/
1920;. Томпсон, 1334721/1920; ван Арсдейл, 1236504/1917; Уилсон, 1733570/1929.
35*
548
ГЛАВА 16
Флотация сульфидизированного минерала при помощи
сульфгидрильного собирателя легко осуществима, если только
концентрация сульфидного иона ниже той, при которой ион со-
бирателя вытесняется с поверхности минерала [43, 71]. Процесс
сульфидизации требует тщательного регулирования концентра-
ции сульфидных ионов, которая должна быть достаточной, но
не чрезмерной; время перемешивания и время флотации долж-
ны быть подобраны так, чтобы соблюдалось правильное соотно-
шение между концентрациями одновременно присутствующих
ионов собирателя и сульфида. Необходимо учитывать, что при
аэрации сульфидный ион, конечно, частично расходуется вслед-
ствие окисления, по всей вероятности, ускоряемого каталити-
ческим действием минеральной поверхности. При правильном
подборе соотношений реагентов и продолжительности операций
флотация протекает удовлетворительно при значительно мень-
ших расходах собирателя, чем без сульфидизации.
Для других окисленных свинцовых минералов (некарбонат-
ных) сульфидизация дает менее удовлетворительные результа-
ты, но все же возможна [43, 71].
Рей и его сотрудники обратили внимание на вредное дей-
ствие солей кальция и бария при сульфидизации церуссита [154].
Они приписывают этот эффект осаждению карбонатов кальция
или бария на границе раздела церуссит — раствор. Для пред-
отвращения осаждения карбонатов они предлагают умягчать
воду или проводить операции при низком значении pH, в связи
с чем кальций удерживается в растворе в виде бикарбоната.
Превосходный краткий обзор промышленного применения суль-
фидизации церусситовых руд сделал Ситиа при описании обо-
гатительной фабрики в Сардинии [174].
Для медных минералов процесс сульфидизации, по-видимо-
му, не изучался, несмотря на то, что имеется патентная литера-
тура. Отрицательные результаты, полученные на медных карбо-
натных рудах, возможно, объясняются несоответствием пара-
метров решетки минерала и поверхностной пленки, характером
микрокристаллов минерала, повышением величины pH в резуль-
тате реакции замещения или несколькими из этих факторов,
вместе взятыми. Для окислов сульфидизация, вероятно, более
перспективна, но для хризоколлы она менее перспективна, чем
для малахита и азурита. Вся проблема в целом требует иссле-
дования, аналогичного тому, которое было проведено Флемин-
гом и его сотрудниками для свинцовых минералов.
Русские исследователи [2, 36] предложили сульфидизировать
карбонатные руды меди и цинка, нагревая сухую руду с серой
или сероводородом при повышенной температуре (250—400°).
Они также нашли возможным [3] проводить сульфидизацию
смитсонита в пульпе при умеренно повышенной температуре
РУДЫ ОКИСЛЕННЫХ МИНЕРАЛОВ
549
(50—60°). В обоих случаях сульфидизация сопровождалась
флотацией с соответствующими собирателями.
Метод, основанный на применении карбоксилсодержащих
кислот. Этот метод весьма успешно используется только при
кремнистой или глинистой пустой породе, но дает неудовлетво-
рительные результаты при карбонатной; высокое содержание
железа в породе также лимитирует возможность его примене-
ния. Метод пригоден для карбонатов или окислов свинца, меди
и цинка и в несколько меньшей степени для других свинцовых
минералов и гемиморфита; для хризоколлы он не пригоден.
Вообще говоря, задача состоит не столько во флотации по-
лезного минерала [37, 179, 192], сколько в предотвращении фло-
тации пустой породы. Поскольку чистый кварц карбоксильным
собирателем не флотируется, а силикаты флотируются только
при наличии на поверхности достаточного количества металли-
ческих ионов, образующих нерастворимые соединения с анио-
ном собирателя (см. раздел этой главы о флотации силикатов),
для депрессии силикатной породы достаточно подобрать усло-
вия, обеспечивающие понижение концентрации активирующих
катионов. Наиболее часто эти ионы представлены кальцием и
магнием, но имеют значение также ионы двухвалентного желе-
за, алюминия, цинка и меди. Для удаления активирующих ка-
тионов целесообразно умягчать оборотную воду и вводить ре-
агенты, образующие с катионами нерастворимые осадки или
комплексные соединения. К числу реагентов, образующих не-
растворимые осадки, относятся щелочные карбонаты, силикаты,
метафосфаты и пирофосфаты, а к числу комплексообразующих—
щелочные цианиды, некоторые фосфаты, этилендиамин, этилен-
диаминтетрауксусная кислота, соли лимонной и винной кислот.
В качестве примера можно, по-видимому, привести крупные
фабрики горного объединения дю О’Катанж [159, 169, 187]. Окис-
ленные медные руды этого объединения различны по составу,
но состоят преимущественно из малахита при силикатной пу-
стой породе, содержащей небольшое количество магния. Вто-
ростепенными медными минералами являются псевдомалахит
(водосодержащий основной фосфат меди) и хризоколла. Некото-
рые руды содержат также водные окислы кобальта.
Вначале медные минералы флотировались олеиновой кисло-
той, но так как этот реагент нужно ввозить из-за границы по до-
рогой цене, его заменили реконструированным пальмовым мас-
лом, представляющим собой смесь из обыкновенного пальмово-
го масла с гидролизованным.'Гидролизованное пальмовое мас-
ло в основном состоит из пальмитиновой кислоты, но содержит
также другие насыщенные кислоты, олеиновую кислоту и очень
немного линолевой. Таким образом, этот реагент является
смесью глицеридов с кислотами, которая менее текуча, чем оле-
550
ГЛАВА 16
иновая кислота, в связи с чем флотация должна проводиться
в слегка подогретой пульпе (до 33—35°). Требуются водоумяг-
чающие реагенты и диспергаторы пустой породы, главным обра-
зом кальцинированная сода и жидкое стекло, количество кото-
рых меняется в зависимости от жесткости воды и обрабатыва-
емой руды.
Как правило, хорошее извлечение получается для малахита
и посредственное — для куприта и кобальтовых минералов; в
отношении хризоколлы результаты неудовлетворительны. На
обогатительной фабрике Панда в 1948 г. обогащались лежалые
хвосты гравитации. Содержание меди в питании 5,36% и в кон-
центрате 24,5% при извлечении 72% (Роджер). В более ранние
годы, когда вместо хвостов гравитации обогащалась исходная
руда, извлечение и качество концентрата были выше.
Метод, основанный на применении сульфгидрильного соби-
рателя. Сульфгидрильные соединения являются собирателями
для окисленных минералов свинца, меди и цинка. Углеводород-
ная группа собирателя должна иметь четыре (и более) атома
углерода. Наиболее употребительны следующие сульфгидриль-
ные собиратели1: ксантогенаты, дитиофосфаты, продукты реак-
ции пентасульфида фосфора с аминами, тиокарбонаты, мерка-
птобензотиазолы, дитиокарбаматы, меркаптаны и тиофенолы.
Алкилксантогенаты, в особенности имеющие от четырех до
шести атомов углерода в радикале, и тиокрезолы являются пре-
восходными собирателями для церуссита и англезита. Как опи-
сывалось выше, эти реагенты служат собирателями для суль-
фидизированных свинцовых минералов, но могут служить соби-
рателями и для несульфидизированных минералов. Они эффек-
тивны также в различной степени для редких окисленных свин-
цовых минералов (хроматы, молибдаты, ванадаты и др.) [43].
Те же самые ксантогенаты вместе с бензилксантогенатами
и алкилмеркаптанами могут быть собирателями медных карбо-
натных минералов даже при карбонатной пустой породе [86, 173].
К сожалению, расход сульфгидрильного собирателя при
обычных условиях флотации велик, в связи с чем этот способ
невыгоден. Данные Мартина показывают [86], что между разны-
ми ксантогенатами имеются существенные различия, но и в луч-
шем случае процесс все же окажется дорогостоящим. Андерсон
установил [78], что значительное сокращение расхода собирате-
ля может быть достигнуто за счет активации карбонатов меди
свинцовой солью, но и при этом условии данный способ не удов-
летворяет практическим требованиям.
1 Патенты США: Христман и Фальконе, 1823316/1931, 1894344/1933,
1997280/1935, 2029156/1936; Фальконе и Христман, 1858007/1932, Ферриз,
178280/1930, 1831808/1931; Льюис, 1668917/1928; Сэйр, 1972247/1934; Вигтон,
1783206/1930; 1783207/1930, 1974885/1934.
РУДЫ ОКИСЛЕННЫХ МИНЕРАЛОВ
551
Имеются некоторые указания на то, что с помощью амил- и
гексилксантогенатов можно осуществить флотацию карбоната
цинка, но на практике этот процесс оказывается недостаточно
селективным. Меркаптаны являются более сильными собирате-
лями, чем ксантогенаты: расход их меньше, они эффективно
флотируют не только смитсонит, но и водный силикат цинка —
гемиморфит [73]. Наиболее пригодными следует, по-видимому,
считать гомологи с шестью и семью атомами углерода: замена
их гомологами с двенадцатью или шестнадцатью-атомами угле-
рода не дает преимуществ. Расход меркаптанов для флотации
окисленных цинковых минералов велик, что, по всей вероят-
ности, вызывается отчасти высокой упругостью пара реагента
и его легкой окисляемостью с переходом в неактивное соедине-
ние, а отчасти трудной диспергируемостью в жидкой фазе фло-
тационной пульпы. Дробная загрузка реагентов, по-видимому,
лучше единовременной.
Предварительную обработку меркаптанами можно произво-
дить и на сухой руде с последующей флотацией в обычных ус-
ловиях.
Метод, основанный на применении катионных и различных
других собирателей. Извлечение окисленных минералов меди,
свинца и цинка при помощи катионных реагентов типа длинно-
цепочечных аминов обстоятельно не изучалось, тем не менее
Кенна с сотрудниками [130] предложили этот способ для обога-
щения цинка при условии предварительной активации минера-
ла щелочным сульфидом.
Алкилзамещенные трифенилметановые красители применя-
лись для флотации силикатов меди [128] и 8-гидроксихинолин
для карбонатов свинца и цинка [65]. Из числа других ре-
агентов, без сомнения полезных при извлечении окисленных ми-
нералов, следует упомянуть о светильном газе [108], ацетилене
139, НИ, щелочных хроматах [186] и фторсиликатах [99]. По-ви-
димому, для подбора собирателей, действующих селективно при
флотации указанных окисленных минералов, требуются даль-
нейшие исследования. Одна из плодотворных идей, которая мо-
жет оказаться полезной вообще, заключается в том, что ион со-
бирателя должен соответствовать по размерам атомам поверх-
ности минерала, для того чтобы прочно и легко закрепляться
на ней.
Метод, основанный на растворении металла с последующим
осаждением и флотацией. Так как различия в свойствах окис-
ленных минералов не обеспечивают условий для достаточно чет-
кого разделения и поскольку окисленные минералы (по крайней
мере меди и свинца) являются сравнительно реакционноспособ-
ными, перспективным следует считать направление, имеющее
целью превращение этих минералов в металл или некоторые
552
ГЛАВА 16
другие флотационно активные соединения с последующей фло-
тацией нового твердого вещества. Этот способ испытывался в
применении к некоторым окисленным рудам, в результате чего
было получено высокое извлечение металла, но при затратах,
ставящих под сомнение выгодность процесса.
Восстановление окисленных минералов можно осуществить
при помощи предварительной обработки в печи или же в горя-
чей пульпе. Восстановление может быть также произведено
электролитическим способом [94] и электрохимическим, как для
железного скрапа [72]. Вместо восстановления до металла при
образовании минералов может оказаться более выгодным осаж-
дение сульфидов или других нерастворимых соединений (таких,
как сульфат окись-закиси меди или ее окисел)1.
Медные руды вначале подвергают восстановительному об-
жигу в печи, а потом флотируют, но препятствием при этом яв-
ляется распыление металлических частиц. Роджер [159] сообща-
ет, что добавка небольшого количества соли во время восстано-
вительного обжига приводит к коалесценции металлических
частиц и к сравнительно легко осуществляемой последующей
флотации. При этом добавляется только часть того количества
соли, которое необходимо для преобразования всей меди в хло-
рид меди. Очевидно, соль и медь переходят в газовую фазу в
виде летучего хлорида меди, который восстанавливается при
контакте с углеродом (например, коксом), что позволяет, если
это необходимо, использовать хлор для многократного перено-
са меди.
Свинцовые руды можно восстанавливать до металла в горя-
чей водной пульпе при помощи железного скрапа, причем неко-
торое количество хлорида в растворе облегчает и ускоряет ре-
акцию [194, 195]. Осажденный металлический свинец можно фло-
тировать так же, как и металлическую медь.
По всей вероятности, эти процессы могут быть распростра-
нены и далее; например, технику восстановительного обжига
можно использовать и для свинца, а восстановление в водной
среде — для меди. Возможно, этим процессам уделялось мень-
ше внимания, чем они заслуживают.
Методы, характеризующиеся осаждением новых нераствори-
мых веществ, отличных от металла, по-видимому, используются
промышленностью в незначительной степени. Это можно объяс-
нить чрезмерным расходом реагентов, необходимых для данно-
го способа, при котором, очевидно, затруднения, связанные с опе-
рацией выщелачивания, усугубляются трудностями флотацион-
ного процесса.
1 Американские патенты: Бэйкон, 1235954/1917; Кристенсен, 1286531/1918»
1316352/1919; Феррис, 1784280/1930; Спайдер н Грии, 1691310/1928.
РУДЫ ОКИСЛЕННЫХ МИНЕРАЛОВ
553
Окисленные минералы щелочноземельных металлов
Несмотря на многообразие окисленных минералов щелочно-
земельных металлов, только некоторые из них имеют экономи-
ческое значение. К ним относятся карбонат магния — магнезит,
двойной карбонат кальция и магния—доломит, карбонат каль-
ция— кальцит, фосфаты кальция, представленные фосфоритом и
апатитом, и сульфат бария — барит. Магнезит является источни-
ком магнезии, кальцит входит в качестве основной составной ча-
сти в портланд-цемент; из фосфорита и апатита добывается фос-
фор, а из барита — барий; кроме того, барит сам по себе исполь-
зуется как дешевый утяжелитель глинистых растворов для буре-
ния нефтяных скважин.
Другие карбонаты, кварц, силикаты и гидроокислы железа
являются примесями, от которых должны быть отделены ука-
занные выше минералы. Иногда встречаются также небольшие
количества сульфидов, например пирита.
Успешно протекает флотация карбоновыми кислотами или
их солями. К числу наиболее употребительных реагентов отно-
сятся олеиновая кислота или другие ненасыщенные карбоновые
кислоты из растительных масел, рыбьих жиров и отходов бу-
мажного производства (талловые масла). Из реагентов этой
группы наиболее дешевы талловые масла, и хотя они несколь-
ко менее эффективны, чем красное масло (олеиновая кислота),
тем не менее им следует отдать предпочтение с экономической
точки зрения.
Флотация производится в слегка щелочной среде. Щелоч-
ность обычно создается с помощью каустической соды или каус-
тической соды совместно с метасиликатом натрия, который при-
меняется из-за своей способности диспергировать пустую поро-
ду. Для той же цели используют и другие реагенты — карбонат
натрия, сульфид натрия и щелочные пирофосфаты, метафосфа-
ты и полифосфаты. Обычно эти реагенты в водном растворе гид-
ролизуются так, что они до некоторой степени способствуют со-
хранению желаемой щелочности, а также диспергированию и
депрессии силикатных, глиноземистых и железистых шламов.
Для предотвращения флотации пустой породы используются
также танины (например, квебрахо) и соли щелочных или ще-
лочноземельных металлов с лигносульфоновыми кислотами'
(например, даксад). Подобно жидкому стеклу эти реагенты счи-
таются депрессорами пустой породы, особенно железосодержа-
щей. Они могут быть полезными при умеренных дозировках
(обычно значительно меньше 0,5 кг/т). При избыточных коли-
чествах депрессируются также и щелочноземельные минералы.
Можно предполагать, что эти реагенты действуют благодаря
образованию комплексов с железными, алюминиевыми и, воз-
'554
ГЛАВА 16
можно, щелочноземельными ионами. Количественные данные
по поводу химизма действия указанных реагентов отсутствуют,
и поэтому в настоящее время можно лишь делать предположе-
ния о качественной стороне вопроса.
Обычно собиратели содержат в качестве примесей доста-
точное количество соединений, которые не обладают собира-
тельными свойствами, но, будучи поверхностно активными, дей-
ствуют как пенообразователи, в связи с чем отпадает необходи-
мость добавки последних. На практике иногда нужно подавить
чрезмерное пенообразование. Это может потребовать примене-
ния более чистого собирателя; в других случаях может оказать-
ся полезной добавка смазочного масла *, не обладающего ни
пенообразующими, ни собирательными свойствами. Масла это-
го типа обволакивают покрытые собирателем поверхности, по-
вышая гидрофобность флотируемого минерала; при этом пу-
зырьки становятся более крупными и хрупкими, а число их
уменьшается. В том же направлении могут хорошо влиять го-
рючие масла (в основном углеводороды), неомыленные глице-
риды и разнообразные неионизированные составные части тал-
лового масла.
В отличие от описанных выше собирателей длинноцепочечные
первичные амины можно использовать для флотации минералов
пустой породы; при этом щелочноземельные минералы остают-
ся в хвостах. Этот метод пригоден для повышения качества маг-
незита, цементного мергеля и фосфоритов, загрязненных сили-
катами, особенно слоистыми.
Удивительно то, что при помощи длинноцепочечных первич-
ных аминов легче отфлотировать барит от силикатов с каркас-
ной структурой, чем эти силикаты от барита, как этого можно
•было бы ожидать по аналогии с кальциевыми минералами.
Магнезит и доломит. Основными примесями в магнезитовой
руде являются тальк, слюды, хлорит, серпентин и изредка
кварц. Флотация карбоновой кислотой вполне возможна1 2, но
при этом возникают затруднения из-за того, что большая часть
руды должна быть отфильтрована, поскольку даже очень за-
грязненные руды состоят главным образом из магнезита.
Кроме того, процесс осложняется из-за наличия талька —
одного из немногих минералов, обладающих естественной фло-
тируемостью и не требующих применения собирателя. В соот-
ветствии с этим наиболее распространенным является метод
флотации талька с одним пенообразователем, как это принято
на фабрике Вермонт, или флотации талька [58, 59] вместе с дру-
1 В некоторых случаях, как будет показано ниже, углеводородные мас-
ла, по-видимому, имеют еще другое назначение.
2 Американские патенты: Вейниг, 2363029/1944, 2363030/4944, 2363031/
1944, 2363104/1944.
РУДЫ ОКИСЛЕННЫХ МИНЕРАЛОВ
555
гими силикатами с помощью катионного собирателя. Лучшими
реагентами, согласно Клеммеру, Дэрнеру и де Ванею [44], яв-
ляются технический лауриламин (ДП 243), продукт конденса-
ции этаноламина и жирной кислоты (ДЛТ 699) и соль лаурил-
пиридиния (эмульсол 903 Л). Последние работы показывают,
что технический октадециламин также хорошо подходит для
указанной цели.
Применяя карбоксильные реагенты, можно отфлотировать
некоторую часть магнезита и доломита от кальцита, но вообще
это разделение неустойчиво и сопряжено с трудностями. Хейл-
ман [104] для осуществления указанного разделения рекоменду-
ет сравнительно высокую щелочность (рН~Ю), создаваемую
за счет извести и одновременно добавляемого силиката натрия
или же при помощи силиката кальция, который мало растворим
в воде; поэтому при одновременном добавлении извести и жид-
кого стекла образующийся силикат кальция может выпадать
в осадок.
Роберт Шенлауб [164, 165] изобрел остроумный способ для
получения карбоната магния и карбоната кальция из доломита.
Исходный материал сначала обжигают до полного обезуглеро-
живания магния; при этом кальцит обезуглероживается только
частично. Обожженный материал сначала гасят, а затем карбо-
натизируют в пульпе, насыщенной газами от операции обжига, и
дополнительно дымовыми газами, если это требуется. Карбонати-
зация производится в две стадии: в первой стадии при рН=10
полностью карбонатизируется кальций, после чего во второй
стадии при pH = 7,2 образуется гидратированный карбонат маг-
ния, а именно, несквегонит MgCO3 ЗН2О. Этот синтетический
минерал образует сравнительно крупные кристаллы величиной
от 10 до 20 ц, в то время как кальцит является микрокристалли-
ческим. Затем пульпа поступает на флотацию карбоксильными
собирателями при pH ~ 8; пенный продукт состоит главным об-
разом из магниевых минералов, а камерный — из кальцита. Оба
концентрата подвергают сушке, и, если это необходимо, магние-
вый продукт дегидратируется. Каждый из этих продуктов можно
также обжигать для получения окислов.
Известняк и цементный мергель. Известняк применяется в
промышленности главным образом для производства портланд-
цемента, который получается спеканием смеси извести, глинозе-
ма и кремнезема в определенных пропорциях. Клинкер состоит
в основном из трикальцийсиликата, дикальцийсиликата, три-
кальцийалюмината и тетракальцийалюминферрита, который яв-
ляется носителем железа в шихте. Лишь очень немногие естест-
венные породы имеют постоянный состав; требуется тщательный
контроль состава шихты. Поэтому на практике обычно смешива-
556
ГЛАВА 16
ются исходные материалы, полученные из различных источников-
при селективной добыче соответствующих пород.
Флотация кальция, осуществленная в 1920 г. [84, 125], позво-
лила удешевить производство цемента, так как новый метод, ос-
нованный на удалении вредных для процесса примесей, эконо-
мичнее старого, основанного на добавлении необходимых ком-
понентов, что часто требовало доставки материалов издалека
при существенных затратах и в постоянно меняющихся коли-
чествах [23, 24L На первых порах обогащение цементного мер-
геля состояло из операции измельчения, обесшламливания с
сохранением шламов из-за повышенного содержания в них каль-
цита, подготовки пескового материала к флотации и фло-
тационного отделения кальцита от кварца и слюды с примене-
нием карбоновых кислот в качестве собирателя [133, 158].
В обогащенном указанным способом материале содержание
кальцита увеличивалось, а содержание окиси магния, щелочей,
глинозема и кремнезема понижалось по сравнению с исходным
материалом. Дальнейшее усовершенствование процесса заклю-
чалось 1 главным образом в использовании более дешевых кар-
боксильных реагентов, в изобретении лучших способов их приме-
нения и в подборе модификаторов, способных повышать селек-
тивность процесса.
Сырые цементные материалы чаще содержат чрезмерно боль-
шие количества глинозема, чем большие количества кремнезема.
Для обогащения этих материалов целесообразна флотация с по-
мощью катионного реагента [64, 193]. Эта операция проводится
в промышленном масштабе на хвостах анионной флотации, т. е.
на смеси белой магниевой слюды, кварца и сравнительно грубо-
го кальцита. Пенный продукт катионной флотации по существу
состоит из слюды, которая идет в отвал. Схема процесса изобра-
жена на рис. 1.
При анионной флотации в качестве реагентов используют
главным образом талловое масло, каустическую соду и жидкое
стекло в количестве от 0,25 до 0,5 кг/т. Для катионной флота-
ции достаточны небольшие количества длинноцепочечных ами-
нов. Можно также загружать незначительные количества вспени-
вателя. Если имеется магнезит или избыточный доломит, коли-
чество их может быть снижено при помощи процесса Хейлмана
[103], описанного выше.
Фосфаты. Обогащение апатитовых руд и фосфоритов [180]’
по своим масштабам является одним из самых ярких примеров
применения флотации к окисленным минералам. Фосфор широко
1 Американские патенты: Брнрвуд, 2162525/1939; Кристмэн и Эриксон,
2335485/1943; Годэн, 2231265/1941; Хейлман, 2349094/1944, 2507012/1950; Рид,.
2163701/1939. 2163702/1939, 2202601/1940; Джейн и др., 2336014/1943; Фогель-
Иоргенсен, 2165268/1937, 2255199/1944.
РУДЫ ОКИСЛЕННЫХ МИНЕРАЛОВ
557
известен как существенная составная часть удобрений. Спрос на
него постоянно увеличивается в связи с ростом населения земно-
го шара, истощением запасов природного фосфора в почве в ре-
зультате ее интенсивного использования, а также благодаря все
возрастающему значению,
которое придается наличию
доступного фосфора в почве.
Имеется очень много
фосфатных минералов.
«Система минералогии» Да-
на [148] насчитывает свыше
100 видов, часть которых
важна благодаря наличию в
них свинца, меди и урана.
Эти минералы здесь рассма-
триваться не будут. Из мно-
гих других фосфатных мине-
ралов только члены группы
апатита имеют экономичное
значение. Фторапатит
Ca5(PO4)3F, хлорапатит
Са5(РО4)3С1 и гидроксила-
патит Саз(РО4)зОН счита-
ются членами этой группы.
Реальные минералы по сво-
ему составу обыкновенно яв-
ляются промежуточными
между крайними членами;в
фосфатном минерале могут
также встречаться некото-
рые карбонаты.
Апатит может быть крупнокристаллическим, как в некоторых
метаморфических горных породах, или микрокристаллическим,
как в фосфоритах Флориды или Марокко. Эта микрокристалли-
ческая разность известна под названием коллофана. Он имеет
пористую структуру с громадной внутренней поверхностью и
окрашен в разные цвета-—от белого до черного, включая жел-
тый и коричневый.
Взвешенная в врве,
измельченная исходная
цементная 'порода
Сдесш.ламл^анив-^Шламы-^1
j
Пески.
Анионной
срлотация------в*. Пена
-------ГТ
Камерный про-
дукт
Пена —- Катионная
срлотация
Кам\рныйч^—г— —»
. . продукт
Слюдистые ' I
кварцевые хвое- |
ты (богатые А1 Концентрат
Му и щелочами) цементного
мергеля
Рис. 1. Упрощенная схема флотации
цементного мергеля с помощью анион-
ных и катионных собирателей
Флотационная активность апатита известна давно [32,179].
Процесс флотации, однако, не применялся, пока не была найде-
на возможность использовать его для руд коллофановых ме-
сторождений. В этих месторождениях фосфатные минералы
содержатся в виде сравнительно крупных зерен, смешанных с
глиной, кварцевым песком и частицами фосфатов алюминия и
железа шламового размера. Для успешного осуществления фло-
тации на данном этапе [188] требуется тщательное обесшдамли-
558
ГЛАВА 16
ванне исходного материала, предпочтительно с очисткой фосфат-
ных частиц при помощи обдирки. Эти операции сопровождают-
ся перемешиванием с реагентами в плотной пульпе, разбавлени-
ем и флотацией.
Широко используются два способа флотации. Согласно пер-
вому из них частицы фосфата флотируются анионным собира-
телем, а именно, жирной или смоляной кислотой; флотация и
предварительная обработка ведутся в слегка щелочной пульпе.
Согласно второму способу в среде, близкой к нейтральной, фло-
тируют кварц катионным собирателем типа длинноцепочечных
аминов.
При обогащении руд, содержащих крупнокристаллические
апатиты, пригодна комбинация реагентов и мыла с небольшими
количествами метасиликата, карбоната, метафосфата или поли-
фосфата натрия. Но если апатит имеет микрокристаллическую
или землисто-пористую структуру, как в фосфорите, добавка не-
обходимого для флотации количества мыла ведет к чрезмер-
ному ледообразованию и снижению показателей. Это затрудне-
ние можно преодолеть, добавив горючее или другое углеродное
масло [51]. Точное назначение добавок масла полностью не вы-
яснено, но можно предполагать, что они уменьшают пенообразо-
вание, вытесняя ионы и молекулы -мыла с поверхности раздела
газ — жидкость, а также увеличивают краевой угол поверхности
минерала благодаря замещению твердого маслом вдоль трех-
фазной границы. При этом масло может заполнить поры на по-
верхности минерала, что также способствует флотации.
Дальнейшее усовершенствование процесса имело целью сни-
зить стоимость реагентов благодаря использованию более деше-
вых источников длинноцепочечных карбоновых кислот или за
счет дополнительных реагентов, функция которых заключается
в усугублении действия собственно собирателя. Рекомендуемые
реагенты включают талловые масла, жирные кислоты рыбьего
жира и растительного масла, нефтяные сульфокислоты.
Процесс флотации, при котором флотируется кварц, а фос-
фат тонет, в промышленности не используется. Отчасти это объ-
ясняется высокой стоимостью катионных собирателей, отчасти
тем, что кварца в руде обычно в несколько раз больше, чем фос-
фата; вследствие этого по данному способу флотируется прео-
бладающая составная часть смеси минералов, что вообще неже-
лательно. Флотация кварца из смеси минералов применяется, од-
нако, для повышения качества фосфатного концентрата.
Во многих месторождениях фосфорита (например, во Флори-
де) полезный минерал встречается в форме зерен, размер кото-
рых колеблется в широких пределах (от одного дюйма до не*
скольких микрон). Частицы размером в 50 и и мельче теряются в
I
РУДЫ ОКИСЛЕННЫХ МИНЕРАЛОВ 559
шламах, а частицы крупнее 0,5 мм выделяются в крупнопесковую>
фракцию; питание флотации в основном состоит из материала’,
крупностью от 50 до 500 Н. Так как различия во флотируемости
между крайними членами этого класса вполне заметны, выгодно»
производить флотацию в два приема, флотируя раньше фосфаты,
а затем кварц, что и применяется в настоящее время на практи-
ке [50]. Этот двухстадиальный метод имеет еще и то преимущест-
во, что некоторые нефосфатные минералы флотируются и с фос-
фатом и с кварцем, что позволяет отделить их от фосфата во.
второй стадии флотации. При осуществлении этого процесса вво-
дится еще одна важная операция, заключающаяся в кислотной
обработке концентрата первой стадии с целью удаления пленки
карбоновой кислоты и связанного с ней масла с поверхности фос-
фатных частиц1. На рис. 2 дана принципиальная схема флота-
ционной секции фосфатной обогатительной фабрики, применяю-
щей описанный выше метод.
Для флотации кварца было предложено много реагентов,
главным образом в результате исследований, имевших целью
снизить затраты. Согласно одному из предложений, очевидно,
представляющему в настоящее время исторический интерес, ани-
онный собиратель применялся вместе с активатором [122, 1231.
Позднее были предложены катионные собиратели, включающие
длинноцепочечные алкиламины, продукты реакции первичных
аминов с цианамидами и окисями алкиленов, алкилированные пи-
перазины, амиды фосфорной кислоты, фосфористые имиды и т. д^
Шламы, выбрасываемые в ходе процесса, содержат значи-
тельное количество фосфата (обычно такое же, как и в питании).
Но только небольшая часть этого фосфата представлена апати-
том, основная же часть содержится, как это принято считать, в
виде фосфатов железа и алюминия. Флотационное извлечение-
фосфата из шламов будет поэтому отличаться от обогащения пе-
сков из-за различия в крупности и минералогическом составе.
Вопрос обогащения шламов от промывки не получил до настоя-
щего времени экономического разрешения из-за наличия боль-
ших запасов руд, в которых принято обогащать лишь песковую
часть.
1 При кислотной обработке карбоновая кислота вытесняется из поверх-
ностных соединений более сильней минеральной кислотой. При этом потеряв-
шая связь с поверхностью минерала карбоновая кислота может в той или
иной мере отслаиваться от поверхности, что, по всей вероятности, связано с
интенсивностью и продолжительностью перемешивания и плотностью пуль-
пы. По вопросу о там, действительно ли имеется отслаивание и в какой мере,
данные отсутствуют. Известны случаи, когда после кислотной обработки с
последующей отмывкой флотация восстанавливается полностью или в значи-
тельной мере, что может служить доказательством неполного удаления кар-
боновой кислоты (см. Гросман Л. И. Депрессия силикатов при разделе-
нии коллективных концентратов, полученных с жирной кислотой, Бюллетень.
ЦИИН, №16, 1956, стр. 21—22). Прим. ред.
.660
ГЛАВА 16
Обогащение крупных фосфатных песков. Крупные пески
(крупностью от 6 до 35 меш) обычно содержат больше фосфа-
та, чем исходная руда в целом, но все же они недостаточно чи-
хты для того, чтобы непосредственно поступать в продажу. Фрак-
Исходный сросфатный
материал
Вода
_ Промывка
'(8 логуошерах)
Крупные ФОс-ч_Мок^ое zpoxove.
(ратныезернаг -
Крупные пески -*—Классификаторы-^~й1памы в отвал
на агломерацию Т - (в отстойник)
[флотограоцтацию) ?
Тонкие пески
(от 50 до 500ja]
6 плотной пульпе)
Кардоновые кие- Кондиционирование
лоты, горючие рР д-р)
масла, щелочи =.. .. с.^_.
Вода----» | кварцевый песок
Флотация фоссрата—* g отвал
Пенный продукт
Серная кис- Удаление реа-
лота --------Агентов рН-У-5
вода~^ Классификация и промывка-^®™™
Вода —г ।
Алинноцепочеч--^^ртация_5^ _ ^Лн^попдукт
нь>е амины Камерный продукт о отдал
I
Обезвоживание
Вода
Фосфатный концентрат
Рис. 2. Схема обогащения фосфоритной гальки месторождения
Флориды
ция указанной крупности трудно обогащается и с помощью
гравитации и с помощью флотации, так как, с одной стороны, от-
сутствует достаточная разница в удельных весах апатита и
кварца, а с другой, частицы слишком крупны для того, чтобы
быть поднятыми на поверхность при энергичном перемешивании
пульпы во флотационной камере.
Чапмэн и Литлфорд [35] разработали процесс, получивший на-
звание агломерации1. По этому способу пульпа сначала переме-
1 В американской литературе термином «агломерация» принято обозна-
чать флотогравитацию. Прим. ред.
РУДЫ ОКИСЛЕННЫХ МИНЕРАЛОВ
561
шивается с реагентами так же, как для флотации, после чего
разделение производится на столах. Осуществление этого про-
цесса становится возможным благодаря тому, что во время пе-
ремешивания с реагентами поверхность частиц полезного мине-
рала становится гидрофобной, и поэтому одновременно поступа-
ющие в пульпу многочисленные пузырьки действуют как связу-
ющее звено между частицами. Рыхло связанные агломераты по-
лезного минерала крупнее и легче, чем неагломерированные ми-
нералы пустой породы, и, попадая на сотрясательный стол, они
отделяются от пустой породы, переходят в верхней слой и смы-
ваются потоком воды. Разделение может быть проведено и на
спиралях Гемпфри или на ленточных аппаратах, а также при
помощи грохочения под водой, во время которого агломерат
скользит вниз по круто наклонному грохоту над неагломериро-
ванной пустой породой [113].
При этих агломерационных процессах разделение происходит
в тонком слое на столе или ленте, в связи с чем создаются бла-
гоприятные условия для того, чтобы агломерированный матери-
ал расположился на поверхности раздела газ — жидкость в ви-
де пленки. Это обычно и наблюдается: значительная или даже
преобладающая часть агломерированного материала образует
на поверхности пленку, которая с большой скоростью удаляется
при помощи разделительных приспособлений. Благодаря этому
эффективность агломерации существенно повышается.
При правильном подборе реагентного режима и крупности
частиц рыхлые агломераты быстро образуются в плотной пуль-
пе при умеренной аэрации. Это справедливо при значительном
содержании минерала, подлежащего агломерации. Если же аг-
ломерируемый материал содержится в пульпе в подчиненном
количестве, желаемые результаты достигаются с трудом и только
после продолжительного и энергичного перемешивания. По-ви-
димому, это связано с увеличением числа столкновений частиц
с пузырьками. Если содержание минерала в пульпе ниже опре-
деленного предела, число столкновений, требуемых для образо-
вания рыхлых агломератов, является функцией второй или бо-
лее высокой степени от концентрации минерала; кроме того, чи-
сло столкновений обратно пропорционально размеру пузырька
и меняется симбатно со скоростью движения составных частей
пульпы одной относительно другой («обдирка»).
Барит. Из-за своего большого удельного веса барит употреб-
ляется как утяжелитель растворов для бурения. Кроме того, он
служит источником получения бария для изготовления чистого
сульфата бария, потребляемого в качестве пигмента в бумаж-
ной промышленности, или смешанного пигмента, литопона
ZnS — BaSCU, используемого для получения некоторых красок.
Для химического производства требуется барит высокой чи-
стоты.
36 А. М. Годэн
562
ГЛАВА 16
Естественный сульфат бария (барит) встречается главным
образом в ассоциации с силикатами и окислами железа, а также
в сочетании с флюоритом и кальцитом. Собирателями для бари-
та служат карбоновые кислоты [18, 19, 52], алкилсульфокислоты
и соли тех и других. Это можно было заранее предвидеть, так
как сульфат и карбонат бария отличаются низкой растворимо-
стью, что наводит на мысль об особенно прочной связи между
Ва и группами С(О)О и SO4 независимо от наличия углеводо-
родных цепочек при этих кислородсодержащих группах. Парал-
лелизм между адсорбцией олеиновой кислоты баритом и его
флотируемостью был установлен Петерсоном [149], значение ал-
килсульфокислот как селективных собирателей для барита по-
казано Дунканом КО], выделившим барит из смеси с флюори-
том при помощи олеилсульфата натрия, и Хоугом [110], кото-
рый использовал комплексный алкилсульфат.
Поскольку силикатная пустая порода легко активируется и
поэтому флотируется карбоксильными реагентами и поскольку
окислы железа, кальцит и флюорит флотируются теми же реа-
гентами, задачи обогащения баритовых руд связаны по сущест-
ву с подбором депрессоров, при помощи которых достигается
селективность.
Отделению барита от окислов железа способствуют следую-
щие условия: умеренно повышенная щелочность пульпы (напри-
мер, pH =Н), добавка метасиликата в количестве 0,5—1 кг/т и
повторные перечистки [140, 153]. Если качество конечного проду-
кта с точки зрения цвета ниже кондиционного, целесообразно
обработать продукт разбавленной серной кислотой. Хоуг счита-
ет, что качество баритового концентрата, полученного в щелоч-
ной среде, может быть также повышено при помощи перечистки
в кислой среде (например, при рН~3), где барит тонет, в то
время как железные минералы и глины флотируются 1109, 1101.
При отделении флюорита от барита хорошие результаты иног-
да получаются при применении олеиновой кислоты совместно с
танином (квебрахо) и солью закисного железа. Считается, что
эта смесь способствует депрессии барита. С другой стороны, мож-
но флотировать барит с алкилсульфатом в качестве собирателя.
Эмульсол XI («натриевая соль комплексного алкилсульфата»)
полезен при отделении барита от флюорита [145]. Перед флота-
цией целесообразно проводить обесшламливание [521, а для по-
вышения селекции обычно полезно применение квебрахо [127].
Окисленные минералы многовалентных металлов
В этой главе рассматриваются окисленные минералы желе-
за, хрома, марганца, титана, вольфрама, алюминия, олова, ура-
РУДЫ ОКИСЛЕННЫХ МИНЕРАЛОВ
563
на, ванадия и тория, которые характеризуются тем, что облада-
ют валентностью, равной трем или выше. Многие из них могут
иметь переменную валентность.
Железные минералы. Из окисленных минералов железа име-
ют значение гематит Fe2O3, магнетит Fe3O4, гетит FeO(OH) и си-
дерит FeCO3. В Америке промышленное значение имеют лишь
первые три. Хотя имеются руды, в которых содержится только
один из этих минералов, но чаще они встречаются вместе в тес-
ной смеси [47]. Пустая порода состоит главным образом из квар-
ца, ферромагниевых и железистых силикатов, глин. В некоторых
рудах заметное распространение имеют кальцит, апатит и гипс.
Кальцит обычно нужен для флюсования, и поэтому наличие
кальцита в руде выгодно, если место добычи руды находится
поблизости от металлургических заводов, в противном случае
кальцит является помехой. Примеси апатита и гипса нежела-
тельны из-за содержащихся в них фосфора и серы.
Поскольку железные руды являются дешевым сырьем, при
обогащении в первую очередь следует стремиться не столько к
увеличению извлечения, сколько к удешевлению процесса. Да-
лее, из-за сильного дутья в доменной печи наличие мелочи в кон-
центрате недопустимо, поэтому флотационный концентрат тре-
бует предварительного спекания. В связи с этим целесообраз-
ность применения флотации для обогащения железных руд опре-
деляется эффективностью и экономичностью не только процесса
флотации, но и процесса спекания.
Для селективной флотации окисленных кремнийсодержащих
железных руд предлагалось по крайней мере три способа [40].
По первому способу минералы железа флотируются анионными
собирателями, а силикаты при этом остаются в хвостах. Имеет-
ся несколько вариантов этого метода. В одном из них собирате-
лем является олеиновая кислота или какая-нибудь другая жир-
ная кислота, предпочтительно ненасыщенная. Этот вариант
впервые исследовался в применении к гематиту [76, 84], затем
для гематита и магнетита северной центральной части США
1116, 117, 168]. Широко изучался этот вариант также в Швеции
Брингом, который написал прекрасную серию статей о своих ис-
следованиях и работе по развитию процесса в этой области
(25—ЗП. Флотация активированных силикатов предотвращается
при помощи депрессора. Вообще лучшие условия для селекции
создаются при значении pH, близком к нейтральному, в особен-
ности при наличии апатита, для депрессии которого необходимо
вести процесс в слегка кислой среде, причем добавка фторидов
или фторсиликатов способствует депрессии. Среди разнообраз-
ных депрессоров, которые предлагались в патентной техничес-
кой литературе, имеются жидкое стекло и коллоидная кремне-
кислота [4]. Предлагалось, кроме того, сначала промывать руду
36*
564
ГЛАВА 16
разбавленной кислотой, а затем флотировать в слабощелочной
среде при pH ~ 8 [56].
По другому варианту того же (первого) метода минералы
железа флотируются сульфонированной жирной кислотой, суль-
фонированным растительным или минеральным маслом. Таким
образом, собирателем может быть вещество, имеющее сильную
полярную группу, а также смесь веществ, содержащих различ-
ные полярные группы. Различные по качеству собиратели типа
сульфонированных кислот и масел представлены так называемы-
ми «зелеными сульфонатами нефти», «махогониевыми сульфона-
тами», «сульфонированным касторовым маслом», сульфониро-
ванной олеиновой кислотой («турецкое красное масло»), алкил-
сульфокислотами. Эти собиратели пригодны для флотации в за-
метно кислой среде (pH — 2—5), в которой кварц дезактивирует-
ся и одновременно создаются неблагоприятные условия для фло-
тации апатита, железистых и железо-магниевых силикатов. Эти-,
ми собирателями, по всей вероятности, нельзя пользоваться при
наличии в руде кальцита и других карбонатов. Превосходные ре-
зультаты по данному вопросу приведены в патентной литерату-
ре
Надлежащее качество пены можно получить при добавке
вспенивателя или смазочного масла или того и другого вместе
взятых, при условии предварительного обесшламливания или
без него. Смазочное масло, по-видимому, более эффективно при
повышенном pH (примерно, так же, как при флотации мылами
жирных кислот).
По второму методу кварц и силикаты активируются известью
или другим растворимым активатором и флотируются карбо-
ксильным собирателем при высоком pH (~ 11). Снижение флоти-
руемости железных минералов достигается при помощи каусти-
фицированных или модифицированных крахмалов, танинов (на-
пример, квебрахо), целлюлозы или ксантогенатов крахмала, ме-
тафосфатов или лигнинсульфоната1 2. Весьма возможно, что эти
реагенты адсорбируются на поверхности железных минералов, но
по этому поводу достоверных данных мало. Некоторые исследо-
ватели рекомендуют высокую плотность пульпы, другие — насы-
щенную известковой водой среду.
По третьему методу кварц и силикаты флотируются катион-
ным собирателем3 без активации известью с применением при-
1 Американские патенты: Бейтс, 2547148/1951; Бус и Херненгоф, 2410376/
1946, 2410377/1946, 2414714/1947, 2439200/1948, 2461875/1949, 2475581/1949.
2 Американские патенты: Браун, 2629493/1953; Браун и Тартарон, 2364618/
1944, 2364777/1944 , 2364778/1944; Клеммер и Клеммонс, 2383467/1945; Клем-
мер и Рампасек, 2403481/1946; Клеммер и Вильямс, 2415416/1947, 2419945'
1947; Херкенгоф, 2423022/1947; Мэртон, 2551893/1951; Сулливан, 2555825/1951'.
3 Американские патенты: Браун, 2629494/1953- де Ваней, 2388471/1945,
2410021/1946, 2450720/1948, 2483890/1949.
РУДЫ ОКИСЛЕННЫХ МИНЕРАЛОВ
565
мерно тех же депрессоров для железных минералов (крахмалов,
белков, смол и танинов).
Комбинированные процессы, включающие различные осо-
бенности описанных выше трех основных методов, были предло-
жены [106, 1071 главным образом с целью снижения стоимости
реагентов, а также и для того, чтобы осуществлять обогащение
при максимально возможной крупности питания.
Сидерит может быть отфлотирован от других минералов (на-
пример, касситерита) с помощью длинноцепочечных первичных
аминов в сильнощелочной среде (при pH ~ 11).
Вообще говоря, классификация или по крайней мере обес-
шламливание дают определенные преимущества. По второму и
третьему методу в пену поднимают пустую породу. Это может
оказаться выгодным при обогащении богатых руд и невыгодным
при бедных рудах. В связи с тем, что амины дороже других соби-
рателей, при обогащении железных руд преимущество отдается
первым двум методам. По второму методу, основанному на акти-
вации известью, можно использовать дешевые реагенты, но при
осуществлении процесса могут возникнуть некоторые неожидан-
ные затруднения.
Если руда содержит небольшие количества апатита, ангидри-
та или гипса, по-видимому, следует предпочесть метод депресси-
рования железа методу его флотации. Так как оптимальные ус-
ловия для флотации указанных примесей не совсем подходят
для флотации основной массы пустой породы, может оказаться
необходимой сначала флотация апатита или гипса, а затем уже
силикатов.
При помощи флотации в основном обогащаются следующие
разновидности железных руд:
1) оолитовые руды, состоящие из гетитовых или гематитовых
оолитов и породы с большим количеством смешанного с квар-
цем кальцита (как в рудах Бирмингама и Алабамы); в этом
случае желательно' отделить кремнезем и глинозем от железа и
кальцита;
2) гипосодержащие руды гетита и гематита (подобные рудам
Мичигана), при обогащении которых основной задачей являет-
ся отделение серы от железа;
3) первичные немагнитные такониты (например, из Минне-
соты), в которых гематит и гетит должны быть отделены от квар-
ца и железистых силикатов;
4) магнитные такониты (например, из Миннесоты), в кото-,
рых гематит и магнетит надлежит отделить от кварца и желези-
стых силикатов;
5) восточные и скандинавские железные руды, в которых
магнетит и гематит («мартит») следует отделить от апатита,
кварца и силикатов.
566
ГЛАВА 16
Выбор метода обогащения зависит от многих технических и
экономических факторов; поэтому в обобщениях следует огра-
ничиваться лишь общими положениями о методах обогащения
и подлежащих разрешению задачах. В каждом отдельном слу-
чае необходимо производить микроскопическое изучение руды,
испытания ее обогатимости, определение стоимости процесса и
уже на основе полученных данных разрабатывать схемы обога-
щения.
Минералы хрома. Из минералов хрома имеет значение толь-
ко хромат железа — хромит, состав которого выражается фор-
мулой FeCr2O4. Благодаря замещению закисного железа маг-
нием и хрома алюминием или окисным железом могут наблю-
даться отклонения от указанного состава. Подобно магнетиту и
франклиниту, хромит относится к минералам группы шпинели
Me Мег О4. По своим флотационным свойствам хромит дол-
жен иметь сходство с магнетитом, но соответствующее критиче-
ское сравнение обоих минералов, по-видимому, еще никем не
производилось.
В патентах, касающихся флотации хромита, рекомендуется
применять в качестве собирателя жирную кислоту в кислой сре-
де после добавки фторида или фторсиликата [101].
Более ранние исследовательские работы [9, 68, 129, 172], про-
веденные в России и США, основывались на использовании ще-
лочных силикатов и метафосфатов для повышения селективно-
сти процесса. Применение алкилсульфатов или сульфонатов в
кислой среде, по-видимому, откроет интересные возможности.
Минералы марганца. К числу окисленных марганцевых ми-
нералов относятся: карбонат марганца — родохрозит, силикат —
родонит и большое число окислов, гидратированных окислов и
сложных окислов марганца и других металлов. В карбонате и
силикате марганец двухвалентен, но в окислах он обычно имеет
более высокую степень окисления и преимущественно четырех-
валентен. Силикат марганца в настоящее время не имеет прак-
тической ценности, в отличие от карбоната, который легко обо-
гащается флотацией и может после обжига давать высокосорт-
ный продукт.
Основными источниками получения марганца служат окислы
и гидратированные окислы марганца с более высокой валентно-
стью. Они обычно встречаются в сложной ассоциации друг с
другом, в связи с чем минералогия марганцевых руд в настоящее
время далеко не ясна. Минералы, состоящие из окислов и гидра-
тированных окислов, содержат только марганец, кислород и во-
дород, как например двуокись МпО2 (пиролюзит или полианит),
промежуточный окисел Мп3О4 (гаусманнит) и окись-гидроокись
МпО(ОН) (манганит); кроме того, имеются сложные окислы,
как например двуокись марганца и кремния — браунит
РУДЫ ОКИСЛЕННЫХ МИНЕРАЛОВ
5S7
(Mn,Si)2O3 и окись-гидроокись бария, Мп11 и МпIv —псиломелан
Ва, Мп11 , Мп8УО[б(ОН)4- Большей частью эти минералы окра-
шены в черный цвет и часто имеют чрезвычайно тонкозернистую
структуру; они удерживают адсорбированную воду и находятся
в прорастании друг с другом и с тонкими гидратированными си-
ликатными и железными минералами и алюмосиликатами [911
(см. рис. 7 и 8 гл. 13).
Отделение родохрозита от ассоциированных с ним минера-
лов пустой породы и сульфидов является относительно простой,
чисто флотационной задачей. В Бьютте (Монтана), где жилы
крупнокристаллического родохрозита вместе с содержащимися в
них сульфидами и силикатами прорезают кварцево-монацитовую
породу, задача заключается в том, чтобы отделить друг от дру-
га минералы трех классов, а именно: сульфиды, карбонаты и си-
ликаты. Предлагались три решения этой задачи:
1) сульфиды флотируют обычным способом, после чего в ще-
лочной среде при помощи мыла флотируют родохрозит, а сили-
каты остаются в хвостах;
2) родохрозит флотируют также при помощи мыла, но в при-
сутствии сульфидов щелочных металлов и других депрессоров
сульфидных минералов, в результате чего в хвосты помимо си-
ликатов переходят и сульфидные минералы 177];
3) сульфиды и силикаты флотируют катионными собирате-
лями, а минералы марганца остаются в камерном продукте.
Первый метод применялся в промышленности [115] и допол-
нялся операцией обжига концентрата для получения высокока-
чественного крупнозернистого продукта, пригодного для далы-
нейшей переработки (см. гл. 15). Второй метод интересен тем,
что в нем (быть может впервые) предлагается депрессировать
легкофлотирующиеся в нормальных условиях сульфиды и в то
же время поднимать в пену труднофлотирующиеся минералы
пустой породы.
Флотационное отделение черных окислов1 марганца от ас-
социированной пустой породы, главным образом силикатной,
осуществить труднее, чем флотацию родохрозита, отчасти из-за
недостаточности наших познаний в области минералогии этих
•соединений и их флотационных свойств, отчасти из-за сложного
минералогического состава руды, но особенно вследствие чрезвы-
чайно тонкой структуры и взаимного прорастания минералов.
В течение многих лет руды Кубинского месторождения обога-
щались при помощи флотации. В качестве реагентов применя-
лись: жирные кислоты рыбьего жира или таллового масла, со-
держащие значительное количество ненасыщенных соединений,
1 Американские патенты: Кристенсен, 1467354/1923; Хиллс, 2259420/1941;
Вид, 2014407/1935; Вейниг, 1911865/1933; Вейниг и др., 1955039/1934.
568
ГЛАВА 16
горючее масло, содержащее много углеводородов и таниновый
продукт (например, квебрахо); pH при флотации поддерживался
на уровне 7—8. Расход горючего масла был значителен и дости-
гал 2,5 кг и более на тонну руды; в связи с этим поры минерала
марганца, покрытого, по-видимому, сначала пленкой собира-
теля, заполнялись мылом, благодаря чему создавались благо-
приятные условия для коалесценции одних частиц с другими,,
аналогичными частицами. Это позволило произвести обесшлам-
ливание руды и сбросить глинистые шламы без больших потерь
марганца даже тогда, когда содержание его в шламах до об-
масливания было выше, чем в питании. Такое явление можно
рассматривать как интересный пример селективной флокуляции,
вызванной действием масла при флотации. Большое количество
масла в концентрате (около 1,5%) полезно при операции обжи-
га, служащей для получения грубого агломерата, который далее
используется в металлургии.
Минералы титана. Титан встречается в виде двуокиси ТЮ2,
известной в трех кристаллографических формах: анатаза, бру-
кита и рутила. Из них наибольшее распространение и значение
имеет рутил. Титан встречается также и как двойной окисел с
железом в ильмените FeTiO3 и с кальцием — в перовските
СаТЮз- Сфен представляет собой титаносиликат кальция
CaTiSiOs; лейкоксен является трудно определяемым окислом ти-
тана, который получается в результате выветривания (естествен-
ного выщелачивания) ильменита.
Значительные запасы руд имеются в россыпях «тяжелых пе-
сков», содержащих рутил и более или менее выветренный иль-
менит, и в жильных месторождениях, в которых титановые ми-
нералы представлены в основном невыветренным ильменитом.
В рутиловых песках основная масса пустой породы состоит
из кварца; обычно его легко отделяют при помощи гравитации,,
но это может быть осуществлено и при помощи флотации. Такое
разделение, по-видимому, не вызывает технических затруднений
1681. Тяжелые пески содержат много отличных от кварца, ильме-
нита и рутила минералов, в том числе ставролит, гранат, циркон,
монацит. Вообще говоря, в этих песках содержатся такие мине-
ралы, которые благодаря своей крупности, твердости, химиче-
ским свойствам и высокому содержанию в исходном материале-
имеют существенное значение для рассматриваемых россыпей.
Состав песков сильно варьирует от месторождения к месторож-
дению, хотя в пределах одной и той же области он может быть
более или менее одинаковым. Отделить тяжелые минералы один
от другого гораздо сложнее, чем отделить их от кварца. Однако
имеются указания об успешном осуществлении разделения,
этих минералов 1971.
Было проведено краткое лабораторное исследование флота-
РУДЫ ОКИСЛЕННЫХ МИНЕРАЛОВ
569’
ционного обогащения брукитовой руды [69]. Полученные резуль-
таты можно считать удовлетворительными, принимая во внима-
ние сложный состав этой руды.
Состав ильменита отвечает теоретической формуле FeTiO3,
но в действительности он несколько отклоняется от нее благо-
даря замещению железа магнием и марганцем и образованию
твердого раствора Fe2O3 в FeTiO3 [до 6% (вес.) при комнатной
температуре]. Кроме того, ильменит выветривается с образова-
нием растворимых соединений железа, которые выщелачивают-
ся, и вторичной окиси титана (лейкоксена). Химический состав
товарных «ильменитов» [148] может поэтому меняться в широ-
ких пределах.
В большинстве случаев ильменит встречается в богатых по-
левым шпатом изверженных породах, называемых анортозито-
выми из-за преобладания в них кальциевого полевого шпата —
анортита. Поэтому основная задача обогащения заключается в
отделении FeTiO3 от CaAl2Si2O8. На практике разделение часто
осложняется из-за наличия железосодержащих силикатов (гра-
натов и пироксено’в) и из-за тесного прорастания ильменита с
гематитом и магнетитом. Это прорастание отчасти является
следствием осаждения из раствора при медленном охлаждении
твердых изверженных пород, в результате чего гематит часто-
прорастает очень тесно с ильменитом, так что каждый из этих
минералов содержит некоторое количество другого минерала в
виде твердого раствора.
Еще одна трудность возникает из-за наличия в руде апатита
(как в нельсонитах Виргинии). Апатит является вредной при-
месью в ильменитовом концентрате, так как он вызывает нару-
шение процесса последующей переработки титанового- концент-
рата для получения пигмента — окиси титана.
В качестве примера -промышленного обогащения ильменит-
содержащей породы с получением титановых концентратов мож-
но привести [134] предприятия Национальной свинцовой ком-
пании (Тэхавус).
Руда состоит главным образом из магнетита, ильменита,
лабрадорита (плагиоклазового полевого шпата, промежуточно-
го между альбитом и анортитом) вместе с небольшими коли-
чествами граната, биотита, роговой обманки, пироксена и апа-
тита. Обогащение включает магнитную сепарацию для выделе-
ния магнетитового продукта (к сожалению, удерживающего
еще много растворенного титана так же, как механически увле-
ченного), гравитационное выделение крупных силикатов и фло-
тацию для удаления тонких силикатов и апатита. Флотация
осуществляется при помощи олеиновой или другой карбоновой
кислоты при pH ~ 6,2 в основной флотации и pH ~ 5,4 в пере-
чистках; для депрессии апатита употребляется фтористый нат-
570
ГЛАВА 16
рий. Большое значение при флотации имеет подбор условий пе-
ремешивания и точное регулирование pH.
В Тэхавусе было установлено, что, если не поддерживать pH
на возможно более низком уровне и не добавлять фтористых
соединений (фтористый натрий или фторсиликат), апатит долж-
ным образом не депрессируется. Для этого же разделения бы-
ли предложены и другие способы
Минералы вольфрама. Промышленное значение имеют два
вольфрамовых минерала—вольфрамит и шеелит. Вольфрамит
является железомарганцевым вольфраматом; при замещении
железа марганцем состав его постепенно изменяется от фербе-
рита FeWO4 до гюбнерита MnWO4. Шеелит представляет собой
вольфрамат кальция CaWO4.
Заслуживает внимания то, что во всех этих минералах по-
ложение атома вольфрама в анионе не отличается от положения
серы в сульфате и фосфора в фосфате. Металлы, содержащиеся
в рассмотренных выше минералах, а именно, железо, марганец,
титан и хром, встречаются в некоторых случаях в виде комп-
лексного аниона. Для вольфрама процесс анионизации метал-
ла прошел до конца, и не приходится ожидать, чтобы вольфра-
мовые минералы проявили свойства, характерные для катиона
вольфрама. С точки зрения флотации вольфрамит и шеелит бо-
лее подвержены действию анионных собирателей, чем минералы
железа, марганца и кальция. Взаимодействие с катионными со-
бирателями соответствующего состава, возможно, специфически
связано с наличием вольфрама в минералах, но данных по это-
му вопросу не имеется.
Шеелит легко флотируется с помощью карбоксильных соби-
рателей; основная задача заключается в предотвращении фло-
тации минералов пустой породы, особенно кварца. Поскольку
вольфрам дорогостоящий металл, приходится обрабатывать ру-
ды с низким содержанием шеелита, что вызывает затруднения
при удалении из шеелитового концентрата минералов, имеющих
второстепенное значение, как например кальцита, доломита,
флюорита, апатита. Кальцит и доломит можно удалить при
помощи кислотной обработки концентрата1 2. В оставшихся пос-
1 Американские патенты: Мойер, 2471341/1949, 2494139/1950, 2557455/1951;
Мак Мэррей и Мойер, 2525146/1951; Мак Мэррей, 2466995/1949.
2 Следует указать, что в СССР Н. С. Петров разработал весьма эффек-
тивный способ, позволяющий удалять кальцит из шеелитового концентрата,
не прибегая к кислотной обработке. Способ заключается в том, что перед пе-
речисткой грубый концентрат подвергается обработке 2—4%-ным раствором
жидкого стекла при т : ж = 1 : 1 и температуре ~80—85° в течение 30—
40 мин. Благодаря этой обработке при последующей перечистке получается
.шеелитовый концентрат кондиционного качества; при этом в хвосты перечи-
стки переходит основная часть кальцита, флюорита и силикатов (см. Пет-
ров Н. С. Флотация шеелита из скарновых руд. Научно-технический инфор-
мационный бюллетень института Механобр, № 1, стр. 16, 1956; Новый метод
РУДЫ ОКИСЛЕННЫХ МИНЕРАЛОВ
571
ле обработки минералах количество сростков уменьшается, что
позволяет повысить качество концентрата при помощи после-
дующей его перечистки [55, 75]. Для повышения селективности
процесса флотации * 1 было предложено большое количество реа-
гентов, в том числе жидкое стекло, основные красители, тани-
ны, сульфонированные продукты, соли металлов, кремнефтори-
стая кислота.
Было установлено, что лучшие условия для разделения соз-
даются в щелочной среде (pH ~ 10,5) при употреблении умяг-
ченной воды и применении в качестве депрессора жидкого стек-
ла, возможно, вместе с небольшим количеством танина. Этот
вопрос неоднократно привлекал к себе внимание. Он был объ-
ектом нескольких исследований Горного бюро США [45, 124],
в СССР [10, 12, 13, 160] и в других местах [68, 135]. Периодиче-
ски появлялись обзоры по флотации шеелита [46, 100, 131]. Бы-
ло также дано описание работы фабрики Иеллоу Пайн Майн в
Неваде 120].
Вольфрамит флотируется хуже шеелита, в связи с чем ассо-
циированные минералы труднее отделить от вольфрамита, чем
от шеелита. Вольфрамиту часто сопутствуют различные сульфи-
ды, касситерит и другие окисленные минералы. Сульфиды мож-
но удалить в первую очередь в основной сульфидной флотации,
после чего вольфрамит флотируют мылами. Если вольфрамит
находится в смеси с монацитом, разделение можно произвести
при помощи аминов в присутствии крахмала [53]; при этих усло-
виях в пенный продукт переходит монацит.
Если сульфиды крупнозернисты, они могут быть удалены
посредством пленочной флотации на столе [1, 111, 132]. Вольфра-
мит и касситерит флотируются вместе и отделяются один от
другого при помощи магнитной сепарации.
Для вольфрамитовой флотации [11, 57, 150, 170], по-видимо-
му, только мыла можно считать в настоящее время эффектив-
ными собирателями. Условия, благоприятствующие флотации
вольфрамита, более ограничены, чем при флотации шеелита;
обычно pH должен быть ниже и концентрация силикатных и
(или) карбонатных ионов требует более строгого контроля.
Минералы алюминия. Из минералов алюминия практическое
значение имеют диаспор и бемит [аллотропические разновидно-
сти А1(О)ОН], а также гиббсит А1(ОН)з. Эти три разновидно-
сти гидратированных окислов алюминия в сочетании с гидрати-
рованными окислами железа, глинами и небольшими количест-
флотации бедных шеелитовых руд, ОБТИ института Механобр, 1940). Прим,
ред.
1 Американские патенты: Бейтс, 2607479/1952; Гисек, 2373305/1945; фран-
цузский патент: Гумбольдт-Дейчмоторен, 811787/1937; германский патент:
Мельцер и Поперле, 736520/1943; советские патенты: Кузнецов, 57959/1940, Ро
зов и др., 67082/1946.
572
ГЛАВА 16
вами ильменита и других сопутствующих минералов образуют
бокситы, которые служат в настоящее время основным источ-
ником получения алюминия.
Бокситы принадлежат к осадочным породам, образовавшим-
ся при выветривании кремнистых и глинистых пород в условиях
тропического или субтропического климата. Сначала из поро-
ды выщелачиваются окись кальция и щелочи, затем—железо и
кремнезем; в остатке содержится глинозем. Вообще бокситы
имеют чрезвычайно большую удельную поверхность и при из-
мельчении дают много шламов; минералы в бокситах плохо оп-
ределяются, за исключением кварца, ильменита или магнетита.
Флотация бокситов связана с некоторыми трудностями. На
данном этапе [41, 162, 163, 1771 основные усилия были направ-
лены на флотацию гидратированных описей алюминия с по-
мошью мыл для отделения от каолина. При этом полезна до-
бавка шелочных метафосфатов и пирофосфатов, расход кото-
рых должен быть равен некоторому промежуточному значению
между предельным, необходимым для предотвращения осажде-
ния солей жесткости из пульпы, и полным стехиометрическим
эквивалентом общей жесткости. Было обнаружено, что катион-
ные собиратели по эффективности действия уступают карбок-
сильным реагентам. Если в руде имеется сидерит, его можно
предварительно удалить флотацией при незначительном расхо-
де собирателей.
Флотации корунда, дегидратированной окиси алюминия, бы-
ло уделено мало внимания главным образом потому, что корунд
представляет ценность только в виде крупных зерен.
Касситерит. Касситерит был объектом многих флотационных
исследований. Этот минерал (SnO2) представляет собой почти
единственный источник олова у нас, хотя в некоторых районах
(например, в Боливии) встречаются незначительные количест-
ва олова в виде сложных сульфидов. Россыпной касситерит
выгодно извлекать с помощью гравитации, в связи с чем на его
флотационные свойства обращалось мало внимания; основные
усилия сосредоточивались на разработке жильных месторожде-
ний олова.
Теоретически SnO2 должен содержать почти 8О°/о олова. Од-
нако в действительности минерал часто содержит небольшие ко-
личества примесей в виде твердого раствора. Кроме того, по-
верхность касситерита часто загрязняется пленками, например
гетита [1891; встречаются также включения других минералов.
Поэтому многие из работ, которые в настоящее время проводят-
ся на оловянных рудах, возможно, не имеют значения с точки
зрения познания флотационных свойств касситерита, как тако-
вого, несмотря на то, что для практических целей они могут
оказаться нужными. Помимо пленочных покрытий, которые бук-
РУДЫ ОКИСЛЕННЫХ МИНЕРАЛОВ
573
вально являются бедствием при флотации касситерита из руд,
большие трудности возникают еще из-за их необычайно слож-
ного минералогического состава. Многие оловянные руды со-
держат от 10 до 15 отличных от касситерита минералов (на-
пример, из сульфидов— пирит, станнин, марматит, халькопи-
рит; из силикатов — кварц, хлориты, слюды, турмалин и топаз;
из рудных окисленных и галоидных минералов — кальцит, до-
ломит, сидерит, барит, рутил и флюорит). Сульфидные минера-
лы затрудняют флотацию касситерита тем, что образуют соли
тяжелых металлов, в особенности сульфат железа; некоторые
окисленные минералы вызывают затруднения другого характе-
ра вследствие близости флотационных свойств этих минералов
к свойствам касситерита.
Кристаллы касситерита состоят из ионов кислорода и ионов
с IV
Sn в октаэдрической координации; каждый атом олова на-
ходится примерно на одинаковом расстоянии от шести атомов
кислорода. Казалось бы, что SnO2 и SiO2 должны быть сходны,
но на самом Деле сходство между ними скорее кажущееся, ибо
SiO2 в отличие -от SnO2 имеет в основном тетраэдрическую
структуру: четыре атома кислорода по существу одинаково уда-
лены от одного атома кремния. Неудивительно поэтому, что
между флотируемостью чистых SnO2 и SiO2 наблюдаются боль-
шие различия, а именно, минералы олова легко флотируются
без активации карбоксильными собирателями [167] в широком
диапазоне значений pH. Между касситеритом и рутилом, имею-
щими сходную структуру, нельзя ожидать таких же больших
различий, несмотря на то что химические свойства олова и ти-
тана могут послужить причиной различий во флотационных
свойствах. С этой точкой зрения согласуются наблюдения Шу-
мана и Пракоша, установивших, что касситерит может флоти-
роваться карбоксильными собирателями в кислой среде, и на-
блюдения [105] австралийской группы исследователей, показав-
ших, что касситерит лучше всего отделяется от ассоциирован-
ных минералов пустой породы алкилсульфатами в сильно кис-
лой среде (pH = 2—3) и что этот реагент адсорбируется мине-
ралом [62]. Изучались также электрокинетические свойства кас-
ситерита [143].
Большое число практических исследований по флотации кас-
ситеритовых руд содержит полезные руководящие указания.
Некоторые исследования касаются германских руд 1701, дру-
гие— руд Боливии [82, 85, 88, 89, 142]; проводились также ис-
следования на рудах Англии [151]. В одной статье затронут
вопрос синтеза -специального собирателя для касситерита [137].
На основании проведенных -работ по флотации касситерита
могут быть сделаны следующие замечания:
1) если в руде присутствуют -сульфидные минералы, их еле-
574
ГЛАВА 16
дует удалить в коллективный концентрат в первую очередь с
помощью ксантогенатов, аминов или жирных кислот в кислой
среде;
2) большая часть растворимых солей, в особенности сульфа-
тов двухвалентного железа, должна быть удалена осаждением
и декантацией, промывкой или с помощью ионообменных смол;
3) влияние оставшихся растворимых солей должно быть
нейтрализовано добавкой реагентов, например ализариновых
красителей, щелочных метафосфатов, цианидов, сульфидов, си-
ликатов; возможно, что действие этих реагентов заключается
в связывании активирующих металлических ионов;
4) в результате мыльной флотации касситерит поднимается
в пену, но подобным же образом ведет себя большое число дру-
гих минералов, особенно рутил, турмалин, топаз, сидерит, барит;
5) если касситерит покрыт пленкой гетита, он флотируется
подобно минералам железа; полученный концентрат может
быть выщелочен минеральной кислотой; остаток от выщелачи-
вания отделяется от железосодержащего раствора и перечища-
ется с оставшимся собирателем для получения высококачест-
венного концентрата;
6) если руды не содержат сульфидных минералов, значи-
тельное улучшение качества концентратов при умеренном из-
влечении достигается с помощью мыльной флотации в слегка
щелочной среде при использовании для депрессии пустой поро-
ды кальцинированной соды, метафосфатов, жидкого стекла или
комбинаций из этих реагентов.
Касситерит легко отделяется от сидерита в сильно щелочной
среде (pH >11) при использовании аминов в качестве собирате-
ля и соснового масла в качестве вспенивателя. Сидерит при этом
флотируется. Механизм разделения не выяснен.
Для извлечения олова многообещающим можно считать про-
цесс, в котором пирометаллургическое восстановление сочетает-
ся с флотацией восстановленной руды; металлические шарики
при этом поднимаются в пену 1102]. Другое интересное предло-
жение включает восстановление поверхности касситерита и ани-
онную флотацию руды при активации ионами свинца [1711.
Урановые и ториевые руды. Имеется много минералов урана
[1381. Все они содержат кислород. Среди наиболее важных мо-
гут быть названы уранинит, смоляная обманка, карнотит, ура-
нофан, отэнит, торбернит. Первые два — окислы, состав кото-
рых колеблется от UO2 до ИзО8; они более или менее гидрати-
рованы и загрязнены продуктами естественного радиоактивно-
го распада и другими веществами. Эти окислы являются основ-
ными первичными минералами урана и часто ассоциируют с
сульфидами и углеводородами. Смесь с углеводородами иногда
носит название тухолита. Карнотит (гидратированный уранова-
РУДЫ ОКИСЛЕННЫХ МИНЕРАЛОВ
575
надат калия) и тюямунит (гидратированный урановадат каль-
ция) — это основные компоненты супергенных месторождений
урана. Уранофан (гидратированный кремнеуранат кальция)
и отэнит (гидратированный фосфатоуранат кальция) обычно от-
носятся к числу вторичных минералов. Торбернит (гидратиро-
ванный фосфатоуранат меди) характерен для сложных урано-
вых минералов, включающих также и другие металлы.
В настоящее время уран считается драгоценным металлом,
хотя и не благородным; в связи с этим даже очень небольшие
количества урановых минералов в руде уже имеют значение.
Поэтому флотационное обогащение урановых руд связано с
чрезвычайно большими трудностями, усугубляемыми неустой-
чивостью урановых соединений и их многообразием. Пустая по-
рода представлена кварцем, слоистыми силикатами, доломи-
том и кальцитом.
Жирные кислоты и мыла до некоторой степени способствуют
извлечению урановых минералов, преимущественно в нейтраль-
ной среде. В настоящее время — это основные собиратели для
урановых минералов [5]. Для улучшения разделения в качест-
ве собирателя предлагалась смесь жирных кислот с жирными
аминами [98] или сочетание их со смесью продуктов взаимодей-
ствия фенолов с окисью этилена [189].
Ввиду склонности карнотитов к легкому ошламованию луч-
ше всего при обогащении карнотитовых руд отделять ценные
урановые минералы от кремнистых при помощи истирания ру-
ды и последующего обесщламливания; при этом шламы сохра-
няются, а пески сбрасываются. Для улучшения этого процесса
в случае известковых руд, при которых возникают затруднения
с точки зрения последующих операций, было предложено обо-
гащать пески с помощью мыльной флотации. Полученный каль-
цитовый концентрат выбрасывался, в то время как кварц и ура-
нат закисного железа, оставшиеся в камерном продукте, выще-
лачивались вместе со шламами для извлечения урана. Этот спо-
соб аналогичен процессу концентрации ванадия, предложенно-
му Вейнигом [190].
В случае кремнисто-сульфидных руд, содержащих уран,,
предлагалась флотация жирной кислотой для получения кол-
лективного урано-сульфидного концентрата и кварцевых хво-
стов. Концентрат выщелачивали для извлечения из него урана,,
а содержавший сульфиды остаток от выщелачивания обжигали,
чтобы получить сернистый ангидрид, из которого приготовляли
необходимую для выщелачивания урана серную кислоту [90].
Наконец, было предложено делить урановую руду на две
фракции — известковый концентрат мыльной флотации и сили-
катный камерный продукт, с последующим выщелачиванием
урана из обеих фракций содой и серной кислотой соответствен-
576
ГЛАВА 16
но; благодаря этому получалась экономия в реагентах, стоимо-
сти топлива и капитальных затратах (содовый процесс требует
повышенной температуры).
Рассматривая все эти схемы, можно сделать вывод о том,
что подлинно селективный собиратель для урана еще не найден.
Редкоземельный фосфат (монацит) является основным про-
мышленным источником получения тория, хотя торит, торианит,
ксенотим и некоторые другие минералы могут в конечном счете
тоже иметь значение [138]. Большая часть нашего монацита до-
бывается из детритовых месторождений, главным образом из
континентальных отложений гравия и песков, хотя богатое
крупное месторождение имеется и в Южной Африке. Из-за вы-
сокого удельного веса монацита и соответствующего размера
частиц минерала детритовых месторождений на первой стадии
обогащения наиболее подходящим является гравитационный
способ. Доводка грубого концентрата обычно производится при
помощи электростатической и электромагнитной сепарации, в
результате чего монацит отделяется от тяжелых минералов,
главным образом от магнетита, ильменита, гематита, граната,
циркона и рутила. Возможность использования флотационного
способа, либо сочетания флотации с обычными методами не
вполне изучена. Однако некоторые обнадеживающие результа-
ты были получены Битрейтом 197] и Кутбертсоном 153], приме-
нявшими соответственно сульфонированные растительные масла
в кислой среде и длинноцепочечные амины вместе с крахмалом
в щелочной среде.
Кремнезем и силикаты
Структуры. Кремнезем и силикаты являются наиболее рас-
пространенными минералами. Физические и химические свойст-
ва силикатов чрезвычайно разнообразны. Правильное понима-
ние структуры силикатов стало возможным благодаря приме-
нению рентгеноструктурного анализа. Читателя, который заин-
тересуется этой темой более детально, полезно направить к ос-
новным работам Брэгга [21, 22], Бермана 115], Струнца [178] или
Уэллса [19П. Много детальных сведений дает последняя моно-
графия Эйтеля 163]. Изучающих глины можно направить к кни-
ге Грима [95]. Интересный научный вклад сделан Грюнером [96].
В настоящее время силикаты рассматриваются как совокуп-
ность ЗЮ4-тетраэдров. Эти тетраэдры могут быть полностью
изолированы один от другого, когда каждый атом кислорода
связан лишь с одним атомом кремния, или могут сочленяться,
если часть атомов кислорода (или даже все они одновременно)
принадлежат к двум смежным атомам кремния. Внутри каждой
.5Ю4-группы расстояние Si — О равняется 1,62 А, а расстояние
РУДЫ ОКИСЛЕННЫХ МИНЕРАЛОВ
577
О — О равно примерно 2,65 А, что вдвое больше радиуса иона
кислорода. Таким образом, силикатную группу можно рассмат-
ривать как состоящую из четырех атомов кислорода в плотной
тетраэдрической упаковке с небольшим атомом кремния в цент-
ре. Силикатные группы несут отрицательный заряд, который
меняется от —4 на один атом кремния при изолированных тет-
раэдрах и до нуля при структурах, в которых каждый атом кис-
лорода является общим для двух соседних тетраэдров.
Силикатная группа может иметь конечные размеры, как в
мономере SiO4—, димере Si2O? , тримере Si3Oo , гексамере
Si6O If~. Это приводит к структурам силикатов, состоящих из
множества катионов, обычно Fe2+ , Mg2 + , Са2+ с различными
конечными силикатными анионами. Примерами, характерными
для мономеров, димеров, тримеров и гексамеров, являются оли-
вин, гемиморфит, волластонит и берилл. Эти минералы, по-ви-
димому, очень близки к не содержащим кремний окисленным
минералам, например к карбонатам, сульфатам, окислам. Это
замечание главным образом относится к мономерным силика-
там, которые обычно называют ортосиликатами.
Силикатная группа может также иметь структуру, не огра-
ниченную в одном, двух или трех измерениях. Бесконечные це-
почки получаются в том случае, когда каждый БЮ4-тетраэдр
соединяется с соседним при помощи двух общих атомов кисло-
рода. Это обнаружено в пироксенах (5Юз)2,г~ . Бесконечные
двойные цепочки имеются в амфиболах (Si4On)®"~ , в которых
две пироксеновые цепочки соединяются между собой так, что
каждый второй атом кремния одной цепочки связан с анало-
гичным атомом второй цепочки через атом кислорода.
Бесконечные слои получаются, если ЗЮ4-тетраэдры соеди-
няются между собой при помощи трех общих атомов кислорода.
Это обнаружено в слюдах, тальке, хлоритах, глинах и приводит
к формуле (Si2O5)2,!_ для бесконечного слоя.
Наконец, когда связь между БЮ4-тетраэдрами осуществляет-
ся всеми четырьмя атомами кислорода, получается каркасная
структура, обозначаемая формулой (SiO2)° . Эта структура
встречается во всех аллотропических формах кремнезема, а
именно, в кварце, тридимите и кристобалите.
Минералогия силикатов сильно усложняется тем, что неко-
торые элементы могут замещать кремний в различных силикат-
ных структурах, причем наибольшее значение имеет замещение
алюминием. Так как алюминий трехвалентен, а кремний четы-
рехвалентен, при замене кремния алюминием для сохранения
электронейтральности требуется соответствующее изменение
среди катионов. Так, при каждом атоме алюминия, замещаю-
37 А. М. Годэн
578
ГЛАВА 16
щем атом кремния, в силикатную структуру должен быть вклю-
чен одновалентный катион, такой как К+ . Na + , Н3О+ . Место
одновалентного катиона может занять двухвалентный катион,
как например Fe2+ или Mg2+, но при этом получается избыток
положительного заряда, который может быть уравновешен до-
полнительным включением в структуру некоторых одновалент-
ных анионов, обычно гидроксила или F-, реже CF, SO42- и еще
реже S2+ . Бор до некоторой степени также может замещать
кремний при тех же самых требованиях в отношении электро-
нейтральности.
Способность алюминия замещать кремний в SiO4, а также
действовать как катион (при этом он находится в октаэдриче-
ской координации с кислородом) наводит на мысль о двойствен-
ном поведении алюминия в силикатах, в которых только некото-
рая часть алюминия проявляет металлические свойства.
Кварц, а-кварц обычно называют просто кварцем. Он обра-
зует кристаллы тригональной сингонии при а = 4,903 А, с =
= 5,393 А, так что при расколе параллельно плоскостям эле-
ментарной ячейки на каждые 20,82 и (или) 26,42 А получается
одна пара одновалентных ионных связей. В среднем при раско-
ле кварца приблизительно на каждые 24 А2, приходится од-
на пара одновалентных ионных связей.
Флотация кварца изучалась больше, чем флотация всех
остальных силикатов, вместе взятых. Это произошло потому, что-
кварц широко распространен в природе, часто встречается в чи-
стом виде и нерастворим в воде.
Ранние исследования показали, что чистый кварц не флоти-
руется мылами или жирными кислотами [74, 84]. Можно также
принять, что чистый кварц не флотируется всеми известными
анионными собирателями, поскольку отсутствуют доказательст-
ва противоположного характера. С другой стороны, многие ме-
таллические ионы (например, кальций, барий, медь, свинец,,
алюминий, железо) активируют кварц при мыльной флотации.
Можно предполагать, что при активации металлами, обладаю-
щими сродством к сере (такими, как медь или свинец) кварц,
может флотироваться гидросульфидными собирателями. Одна-
ко о такого рода флотации сведения отсутствуют.
Механизм активации кварца при мыльной флотации подвер-
гался исследованию. Было показано [871, что количество акти-
ватора и количество собирателя не связаны стехиометрическим
соотношением. Количество активатора превышает количество,
необходимое для образования нерастворимого металлического-
мыла с собирателем. Затем было показано 148, 49], что для ак-
тивации кальций-ионом должны существовать определенные со-
отношения между катионами кальция, водорода и натрия. Мак-
РУДЫ ОКИСЛЕННЫХ МИНЕРАЛОВ 57g
симальная активация достигается, когда 1Са2+] :1Н+] превы-
шает 106 и [Са2 + ]: [Na + ] превышает 10-3, а полное отсутствие
активации наблюдается, если 1Са2+] : [Н+] меньше 105 или
[Са2+ : [Na+J меньше, чем 10-4. Эти результаты приводятся на
рис. 3, где показано, что кварц не флотируется в системе из-
весть— олеат, когда точки лежат ниже заштрихованной обла-
сти, и флотируется, если точки лежат выше этой переходной об-
ласти. Кривая В показывает приблизительные значения раство-
римости гидроокиси кальция. В области pH ~ 11 прилипание
пузырька к частице становится возможным уже при концентра-
ции извести, в 100 000 раз меньшей, чем в насыщенном раство-
ре. Наблюдения Кука и Дигри находятся в соответствии с поло-
жениями о том, что водородный ион играет особую роль в отно-
шении кварца (роль потенциал определяющего иона) и что
ионы кальция и натрия поглощаются кварцем в порядке обмен-
ной адсорбции во внешней части двойного слоя; однако ион
кальция из-за более высокой валентности ближе подходит к по-
верхности минерала и поэтому находится главным образом в
слое Штерна.
Было также проведено физико-химическое исследование более
общего характера по вопросу об активации кварца ионами, обра-
зующими нерастворимые мыла [166], а именно, ионами бария,
алюминия. Это исследование привело к заключению, что актива-
ция может рассматриваться как процесс, при котором растворен-
ные и адсорбированные ионы находятся в состоянии кинетическо-
го равновесия, и что условия равновесия могут быть выражены
с помощью закона действующих масс. На рис. 4 и 5 даны как экс-
периментально полученные, так и вычисленные линии раздела
между областями флотации и отсутствия ее при изменении усло-
вий в широком интервале. Совпадение кривых не полное, но впол-
не достаточное для того, чтобы принять кинетические выводы
этой работы в качестве основы для дальнейшего продвижения
вперед.
Широкий диапазон концентраций, соответствующих, согласно
данным Шумана и Пракоша, критическим условиям флотации,
свидетельствует об отсутствии стехиометрического соотношения
между адсорбированными количествами активатора и собирате-
ля. Последнее было подтверждено экспериментально Чангом [801,
который измерил количество адсорбированных минералом анио-
нов собирателя и катионов активатора. Измерения были произ-
ведены при применении двух радиоактивных изотопов с исполь-
зованием ионов лаурата, меченых С14 и бариевых солей мече-
ных Ва140. Чанг нашел, что обычно несколько адсорбированных
катионов активатора приходятся на каждый адсорбированный
анион собирателя. Работы Чанга приводят к следующим выво-
дам: 1) собиратель не влияет на адсорбцию активатора, но ад-
37*
Рис. 3. Зависимость
флотации кварца от
pH и концентрации
кальция [49]:
ВВ — величина раство-
римости гидроокиси каль-
ция, вычисленная на ос-
новании закона дей-
ствующих масс
5. Теоретиче-
кривые кон-
Рис. 4. Влияние
хлористого бария
и олеиновой кис-
лоты на критиче-
ское значение pH
при флотации
кварца (флотация
наблюдается в об-
ласти, располо-
женной ниже и
правее кривой, со-
ответствующей
данному количе-
ству активатора
[166]):
1 — 3 мг/л BaClt •
• 2Н2О; 2 — 30 мг/л;
3 — 50 мг/л; 4 —
90 мг/л; 5 —
160 мг/л; 6 — 300 мг/л
Рис.
ские
такта для флота-
ции кварца с хло-
ристым барием и
олеиновой
той
должна
области,
женной
правее кривой, со-
ответствующей
данному количест-
ву активатора
[166]):
1 — 3 мг/л BaClt •
2Н2О; 2—30 мг/л;
3—50 мг/л; 4 —
90 мг/л; 5 —
160 мг/л; 6 — 300 мг/л
кисло-
(флотация
быть в
располо-
ниже и
РУДЫ ОКИСЛЕННЫХ .МИНЕРАЛОВ
581
сорбция собирателя обеспечивается наличием активатора;
2) когда кварц становится флотируемым, поверхность его по-
крывается неполными монослоями активатора и собирателя;
3) плотность критического слоя собирателя гораздо меньше, чем
активатора.
Из этих заключений следует, что для предотвращения флота-
ции кварца мылами и другими анионными собирателями доста-
точно уменьшить концентрацию активирующих катионов ниже
соответствующего критического уровня.
Флотация кварца аминами легко осуществляется, особенно в
слегка щелочной среде при pH = 8—10,5. Для полной флотации
минерала требуется образование на поверхности лишь части мо-
нослоя собирателя [79, 81]. Адсорбция додециламина кварцем из-
мерялась [54, 79] в широком диапазоне концентраций собирателя
и значений pH. Обычно адсорбция возрастает с увеличением кон-
центрации собирателя, но не пропорционально ей, а медленнее,
по крайней мере до концентраций, при которых начинается обра-
зование мицелл в непосредственной близости от поверхности раз-
дела минерал — раствор или в двойном слое [83]. Адсорбция воз-
растет также с увеличением щелочности и становится очень
большой при pH > 8.
Так как кварц отрицательно заряжен в воде и даже в слегка
кислой среде (в изоэлектрической точке pH = 3,7), можно легко
себе представить, что катион амина втягивается в двойной слой.
Однако для осуществления роли собирателя ионы амина, по-ви-
димому, должны подойти к поверхности кварца ближе, чем это
соответствует внешней части двойного слоя. Другими словами,
полярная часть иона амина должна прикрепляться к дегидрати-
рованному кислородному участку на поверхности кварца и таким
образом проникать в слой Штерна или во внутреннюю часть
двойного слоя. Очень может быть, что для этого ион амина дол-
жен потерять свою гидратную оболочку. Изучение этого теорети-
ческого вопроса продолжается.
Полевые шпаты. Полевые шпаты подобно кварцу имеют кар-
касную структуру из БЮгтетраэдров; в альбите, ортоклазе и
микроклине одна четверть атомов кремния замещается алюми-
нием, а в анортите замещается половина атомов кремния. Элек-
тронейтральность достигается благодаря внедрению калия или
касную структуру из ЗЮ4-тетраэдров; в альбите, ортоклазе и
микроклина и внедрению Са в случае анортита. Преобладают
твердые растворы, промежуточные между альбитом и анортитом.
Полевые шпаты представляют собой ценное сырье для кера-
мической промышленности; изучалось их отделение от кварца и
слюды. Полевые шпаты флотируются анионным собирателем
примерно так же, как кварц; удовлетворительного метода разде-
ления полевого шпата и кварца с помощью анионных собирате-
582
ГЛАВА 16
лей не найдено. С другой стороны, разделение может быть осу-
ществлено [6, 8, 92, 144, 147] при помощи катионных собирате-
лей 1 в кислой среде с фтористоводородной кислотой, щелочны-
ми фторидами или комплексными фторидами -в качестве модифи-
каторов. Этот процесс лучше всего протекает при pH, близком к
2. Он применим для разделения альбита и кварца или микрокли-
на и кварца. Эти полевые шпаты являются соответственно на-
триевыми и калиевыми шпатами. Относительно кальциевых шпа-
тов данные отсутствуют. Предполагается, что одна из функций
фторида состоит в образовании комплексных соединений с алю-
минием и железом, но механизм разделения еще далеко не ясен.
Другие каркасные силикаты. Среди факторов, которыми опре-
деляется флотируемость силикатов с анионными собирателями,
следующие четыре представляются особенно важными: спай-
ность, относительная плотность металлических узлов на плоско-
сти раскола, характер металла, занимающего эти узлы, и нали-
чие активаторов. В каркасных силикатах спайность несовершен-
на. Активаторы можно пока исключить из рассмотрения, так как
они привносятся извне минерала.
Поскольку плотность упаковки определяется атомами кисло-
рода, относительная плотность атомов металлов характеризуется
отношением атомов металла (представленных в виде атомов од-
новалентных металлов) к общему числу двухвалентных атомов
кислорода. Это отношение для оливина (который не является
каркасным силикатом) должно быть равно 1 : 1; в случае альби-
та (считая, что алюминий фигурирует не в качестве металла, а
как составная часть каркаса) отношение равно 1 : 8, что следует
из формулы [NaJISigAlOsl. Вообще оно колеблется от 1 : в квар-
це до 1 : 2 в гранате, как это можно видеть из табл. 2.
Некоторые силикаты (такие, как кордиерит), с отношениями
колеблющимися между 1 : 2 и 1:1, иногда рассматриваются как
каркасные минералы даже при большей, чем в гранате, степени
замещения атомов кремния алюминием. Но эти минералы могут
также рассматриваться, как алюмосиликаты, состоящие из изо-
лированных силикатных структурных элементов, связанных при
помощи алюминия и других катионов. Это указывает на искус-
ственность принятой здесь классификации и в связи с этим на ее
ограниченность. В действительности гранаты могут также рас-
сматриваться как ортосиликаты.
1 По флотации силикатов (полевого шпата, кварца, слюды и берилла)
катионным собирателем большое и ценное исследование проведено Е. В. Да-
ниловой [см. Данилова Е. В.: а) О некоторых особенностях флотации
кварца катионными реагентами. Флотация полевого шпата лауриламином,
«Обогащение полезных ископаемых», АН СССР, Сб. научных трудов, вып. 1,
1952, стр. 70—94; б) Флотация силикатов лауриламином, диссертация, Ленин-
гсад. Механобр, 1948]. Прим. ред.
РУДЫ ОКИСЛЕННЫХ МИНЕРАЛОВ
583
Отношение между участками, занятыми металлом
(в пересчете на одновалентный), и кислородом
Таблица 2
на плоскости раскола каркасных силикатов
Отношение Минералы Формула
1 : оо Кварц, тридимит, кристобалит sio2
1 :8 Полевые шпаты (альбит) Полевые шпаты (ортоклаз) [Na][Si3A108] !K][Si3A10s]
1:6 Скаполит (мариалит) Фельдшпатоид (лейцит) iNa4||(Si3A10g)3 • Cl] lK][Si2A106j
1:5 Цеолит (натролит) [Na2][(Si3Al2O10)(2H2O)]
1 :4 Полевой шпат (анортит) Полевой шпат (цельзиан) Фельдшпатоид (нефелин) Данбурит [Ca] pi2Al2O8j [Ba]|Si2Al20g] [Na][SiA104] [Ca]|Si2B2O8]
1 :3 Фельдшпатоид (содалит) Скаполит (мейонит) ]Na4][(SiA104)3 (Cl] [Ca4] [(Si 2 Al 2O8)3 SO4)j
1 :2 Гранат (альмандин) Гранат (гроссуляр) [Fe3][Si3Al2C<i2j [Ca3] [Si3 Al2O12j
Во всех каркасных силикатах порядок величины суммарной
плотности кислородных участков на поверхности раскола такой
же, как в кварце, где в среднем одна пара одновалентных ионов
встречается на каждые 24 А2 поверхности раскола. В альбите,
следовательно, надо ожидать один одновалентный ион металла
на каждые 196 А2, а в анортите на каждые 196 А2 приходится
один двухвалентный кальциевый ион или два одновалентных
иона.
Следует полагать, что с повышением указанного отношения
увеличивается реакционная способность каркасных силикатов в
отношении анионных собирателей, в связи с чем гранат наиболее
флотационно активен, а щелочные полевые шпаты наименее фло-
тационноактивны (после кварца). Когда отношение увеличивает-
ся, число участков, на которых ионы собирателя могут закреп-
ляться, возрастает в соответствии с увеличением пригодной для
закрепления поверхности. Вполне вероятно, что флотация стано-
вится невозможной, если отношение падает ниже некоторой пре-
дельной величины, так как даже полное покрытие собирателем
пригодных участков может оказаться в этом случае недостаточ-
ным для того, чтобы обеспечить требуемую гидрофобность по-
верхности.
Кроме того, у этих минералов можно ожидать повышенной
способности фиксировать ионы мыл и других анионных собира-
телей, когда понижается растворимость соединений, образуемых
584
ГЛАВА It
катионами минерала с анионами собирателя. Так, кальций и ба-
рий могут быть более эффективными, чем железо и алюминий,
а ионы щелочных металлов несомненно должны быть менее эф-
фективными, чем поливалентные ионы.
В связи с этим также необходимо напомнить, что большое
значение должен иметь вопрос о возможности замещения одних
ионов другими, что зависит от ряда важных факторов, в том чис-
ле от относительного заряда иона и его размера. Таким образом,
хотя и можно предполагать, что активация каркасных силика-
тов >в общих чертах подобна активации кварца, тем не менее
должны также наблюдаться различия и, возможно, значитель-
ные. Эти явления еще не изучены.
Заманчиво было бы предположить, что каркасные силикаты
по своей флотируемости катионнными собирателями распола-
гаются в ряд, обратный по сравнению с рядом, имеющимся при
анионных собирателях; но это не доказано. Кроме того, имеются
усложняющие факторы, например незначительная селекция при
флотации кварца от полевого шпата в слегка щелочной среде, в
то время как те же минералы четко разделяются в обратном по-
рядке в кислой среде, содержащей ион фтора. Этот факт интере-
сен и с практической и с теоретической точек зрения.
Гранат. Некоторое внимание привлекла флотация граната,
применяемая для отделения его от полевых шпатов, пироксенов
и кварца. В связи с этим было установлено, что весьма полезна
очистка руды в разбавленном растворе кислоты с последующей
отмывкой. Гранат отделяется от других минералов с помощью
флотации жирными кислотами, смесью жирной кислоты с угле-
водородным маслом или длинноцепочечным сульфонатом. Этот
эксперимент был проделан автором тридцать лет назад и с не-
которыми изменениями стал предметом двух новых патентов
(16, 171. Механизм разделения еще не изучался.
Слоистые силикаты. В слоистых силикатах спайность ста-
новится важным флотационным фактором не только потому, что
обеспечивает гладкую поверхность, по которой может расте-
каться соприкасающийся с ней пузырек, но также в связи с тем,
что на поверхности спайности имеется сравнительно небольшое
число нарушенных связей, на которых может проявляться сма-
чиваемость или несмачиваемость.
Слоистые силикаты состоят главным образом из расположен-
ных в виде слоев БЮ4-тетраэдров, в которых каждый атом кис-
лорода основания тетраэдра является общим для двух приле-
гающих атомов кремния, в то время как атом кислорода при
вершине связан лишь с одним атомом кремния и принадлежит,
следовательно, лишь к одному БЮ4-тетраэдру. Как показано ни-
же, структура -получает свое завершение при помощи различ-
ных способов сочленения указанных слоев с параллельными
РУДЫ ОКИСЛЕННЫХ МИНЕРАЛОВ
585
слоями атомов алюминия, связанных с атомами кислорода и
гидроокислами в октаэдрической координации.
В пирофиллите один слой атомов алюминия соединяется с
помощью сильных связей с двумя слоями БЮгтетраэдров, ори-
ентированных своими вершинами по направлению к атомам
алюминия. Для электростатического равновесия на каждый
атом алюминия требуется один гидроксил, который распола-
гается в алюминиевом слое, т. е. внутри сэндвичеподобного па-
кета, который в целом является электростатически нейтраль-
ным и деионизированным. На поверхности спайности пирофил-
лита отсутствуют участки, смачиваемые водой, а также участки,
на которых может закрепляться собиратель. В соответствии с
этим пирофиллит обладает естественной флотируемостью. На
рис. 6 схематически показано расположение пакетов в кристал-
лах четырех характерных классов слоистых силикатов, а имен-
но, в каолине, пирофиллите, мусковите и хлорите, а на рис. 7
суммируются все современные представления о строении паке-
тов и способах их ассоциации для десяти слоистых силикатов,
включая также и те четыре, которые приведены на рис. 6.
В тальке магний замещает алюминий в центральном слое.
Так как в пирофиллите алюминий занимает только две трети из
геометрически возможных участков [191а], требуемая на основа-
нии стехиометрических соотношений замена двух атомов алю-
миния тремя атомами магния является осуществимой с точки
зрения пространственного размещения при переходе от пиро-
филлита к тальку. Поэтому тальк тоже отличается естественной
флотируемостью.
Слюды образуются из тальковых пакетов, видоизмененных
благодаря замещению кремния алюминием в кварцевых солях.
В мусковите замещается одна четверть атомов кремния; заме-
щающие алюминиевые атомы добавляются сверх тех, которые
находятся в октаэдрической координации в центральном слое
пакетов. Для стехиометрического равновесия между пакетами
включаются катионы калия, благодаря чему пакеты превра-
щаются в гигантские анионы. На каждые шесть атомов кисло-
рода, расположенных у оснований силикатных тетраэдров, при-
ходится один одновалентный анионный заряд и один обезво-
женный катион калия, так что на каждых 46,8 А2 (5,2 X 9,0 А)
имеется пара ионов.
Во флогопите, биотите и хрупких слюдах наблюдаются даль-
нейшие изоморфные изменения благодаря замещению в октаэд-
рическом алюминиевом слое части ионов ОН" ионами фтора
(флогопит); далее Mg5+ и Fe2+ занимают место А13+в том же
алюминиевом слое в отношении 3 : 2 (флогопит, биотит), а Са2+
заменяет К+ в ионном слое в отношении 1 : 2 (маргарит).
@-1 0-2 »-3 ®-« O'5 V'5 &~8
Рис. 6. Структура некоторых слоистых силикатов (показано схе-
матически) :
а — каолин; б — пирофиллит; в — мусковит; г — хлорит; / — ОН; 2 — 0;
3 — Si; 4 — Al; 5 — К; 6 — Si илн Al; 7 — Mg нли Al; 8 — Mg или Fe
Si?05
А’г(ОН)г
SizOs
Пирофиллит
SizOj
Мдг(ОН)г
5'205___
Тальк
Флогопит Биотит Вермикулит
Мусковит
[(Мд.АЫ(0М)1г]2 +
6 (SijAVOw (Мд/а)б(ОН)л (Sts Al J 0 io 2—
хлорит А
(нт)
[(МдгАЦ(ОН)|г]М-
(512А1г)О(о
(Мд/е)6(0Н)4 Ц-
($12ДУ0ю
Хлорит В
н*
г АУО H)<i Si2o5(OH) 1-
каолин
SizO*
AkOzCOH)* 1-
SlgO3(OH)
Монтмориллонит
а б
б
Рис. 7. Пакеты в слоистых силикатах:
а _семейство талька; б — семейство слюды; в — семейство хлорита;
г — глины
РУДЫ ОКИСЛЕННЫХ МИНЕРАЛОВ
587
В глинистом минерале иллите кремний замещается алюми-
нием в меньшей степени, чем в мусковите, возможно на ’/е вме-
сто */4, так что плотность ионных участков относительно умень-
шается. Однако в маргарите и других «хрупких слюдах» заме-
щение идет дальше, чем в мусковите, так что ионная плотность
увеличивается.
В вермикулите катионы между пакетами находятся в гидра-
тированном состоянии. Эти катионы, особенно Mg2+ и Са2+, а
обезвоженном состоянии невелики, но становятся большими,
Силикатный
*"? * слой тСиликатный.
; I слой У Силикатный
0 3
о-з ! сдо® ® ® । ------ ।
! Силикатныйл.Силикатный^' Силикатный
А слои слой с/,ои
Рис. 8. Конфигурация промежуточного слоя вермикули-
та (показано схематически согласно представлениям
Уокера [184а]):
а — полностью гидратированного; б — гидратированного пример-
но наполовину после удаления несвязанной воды; в — полно-
стью дегидратированного с атомом магния в полостях сили-
катных слоев; / — нон магния; 2 — «несвязанная» вода; 3 —
«связанная» вода
когда координируются с шестью молекулами воды. В соответст-
вии с этим параметр с в вермикулите много больше, чем в гене-
тически близком ему биотите (около 14 вместо 10 А). Дополни-
тельные молекулы воды занимают промежутки между гидрати-
рованными катионами. Когда вермикулит нагревают, гидратиро-
ванные комплексные катионы диссоциируют, пар раскрывает
пакеты и минерал необратимо переходит в новую форму
(рис. 8).
Можно отметить, что гидроксилы, обычно являющиеся ис-
точниками водородных связей, будучи расположенными внутри
пакетов (как в тальке и пирофиллите), не могут в связи с этим
увеличивать смачиваемость слюд.
Поверхность слюды имеет явно выраженный ионный харак-
тер при относительно небольшом количестве ионных участков на
очень гладких плоских гранях; поэтому можно ожидать, что
слюды будут лучше флотироваться по сравнению с каркасными
силикатами.
В минералах группы хлорита пакеты, имеющие такой же со-
став, как в тальке или пирофиллите (с заменой 1—2 атомов
588
ГЛАВА 16
кремния алюминием), чередуются с пакетами, имеющими состав
брусита Mge(OH)i2 (с частичной заменой магния алюминием).
В тех и других пакетах замена алюминием может наблюдаться
в широких пределах при соблюдении стехиометрических соотно-
шений. Вообще в силикатном слое хлоритов атомы кремния в
большем количестве замещаются атомами алюминия, чем в
слюдах, а именно, степень замещения колеблется от '/4 до '/г, в
то время как в слюдах она меньше или равна V4. Различные ви-
ды хлоритов отличаются один от другого по замещающим ато-
мам, как например Mg2+na Мп2+и Fe2 + , А13+ на Сг3+.
Поверхность раскола хлорита, соприкасающаяся с флотаци-
онной пульпой, состоит или из положительно заряженного бру-
ситового слоя или из отрицательно заряженного алюмосиликат-
ного слоя, причем плотность ионных участков поверхности хло-
рита выше плотности этих участков в слюдах. Соотношение меж-
ду этими плотностями может достигать значения 2 : 1. Брусито-
вые слои гидроксильными группами обращены к жидкости. Сле-
дует ожидать, что между этими группами и атомами кислорода
воды образуются водородные связи, в результате чего поверх-
ность раскола смачивается. Таким образом, хлориты смачи-
ваются заметно лучше, чем слюды; в то же время наличие ато-
мов магния и алюминия в доступных для взаимодействия бру-
ситовых слоях обусловливает способность к сравнительно силь-
ному взаимодействию с карбоксильными анионами, а следова-
тельно, и флотируемость.
Каолин и родственные ему глинистые минералы (диккит и на-
крит) представляют собой накладывающиеся один на другой
пакеты, состоящие из слоя силикатных тетраэдров и слоя алю-
минатных октаэдров; все кислородные атомы у оснований крем-
некислородных тетраэдров, как обычно, полностью связаны вну-
три силикатного слоя, в то время как атомы кислорода у вер-
шин связываются также с алюминием из алюминатного слоя.
Остальные атомы кислорода алюминатного слоя входят в со-
став гидроксильных групп. Согласно этим представлениям, кри-
сталл каолина, состоящий из множества различных пакетов, не
имеет ионов на своей поверхности. Однако поверхностные гид-
роксилы должны проявлять способность к образованию водо-
родных связей, что обычно и наблюдается (см. гл. 2 н 4). То,
что в кристалле каолина имеется сильная водородная связь, под-
тверждается сравнительно коротким расстоянием между паке-
тами каолина; то же самое можно считать правильным в отно-
шении поверхностей кристаллов, находящихся в контакте с во-
дой. Это положение отражено в диаграмме рис. 7, показываю-
щей, что на каждый элементарный пакет приходится одна мо-
лекула ионизированной воды, т. е. один ион водорода и один ион
гидроксила.
РУДЫ ОКИСЛЕННЫХ МИНЕРАЛОВ
589
Монтмориллонит (бентонит) особенно интересен благодаря
тому, что он широко распространен в природе и обладает чрезвы-
чайно высокой емкостью катионного обмена [95, 118], О структу-
ре монтмориллонита было сделано несколько взаимно исклю-
чающих друг друга предположений; требуется провести много
исследований прежде, чем принять предложенную Эдельманом
и Февье структуру, удовлетворительную с точки зрения химии, но
неудовлетворительную с точки зрения минералогии [61].
Следует ожидать, что способные к катионному обмену гли-
ны, в особенности монтмориллонит, должны проявить также ис-
ключительную способность к поглощению катионных собирате-
лей. Это несомненно является одной из наиболее важных при-
чин широкого применения обесшламливания перед флотацией
при помощи аминов. В соответствии со своей структурой глины
легко смачиваются водой и водными растворами, но в то же
время они легко вступают во взаимодействие с катионными со-
бирателями и не только флотируются некоторыми из них, но
также играют роль своеобразного резервуара этих реагентов, ко-
торые, как указано выше, поглощаются глинами в большом ко-
личестве.
Тальк и пирофиллит. В соответствии с рассмотренными выше
структурными особенностями эти минералы отличаются естест-
венной флотируемостью, и для их флотации обычно достаточно
одного вспенивателя [42, 121]. Керосин помогает регулировать
процесс пенообразования (возможно, он растекается на поверх-
ности минерала, повышая ее гидрофобность); жидкое стекло,
кальцинированная сода и (или) калгон способствуют повыше-
нию качества концентрата. В связи с этим следует помнить о
том, что тальк так же, как другие минералы, может быть загряз-
нен прилипшими или вкрапленными примесями [14].
Слюды. При соответствующих условиях слюды могут фло-
тироваться мылами или другими анионными собирателями. По
всей вероятности, это обусловливается активацией (по крайней
мере, частично) [93, 181, 185].
При флотации слюд наибольший интерес представляет ка-
лиевая слюда — мусковит и литиевая слюда — лепидолит. Во-
обще [141] они лучше всего извлекаются с помощью аминов в
кислой среде, предпочтительно в присутствии специально добав-
ляемого алюминия, чем, по-видимому, достигается уменьшение
флотируемости полевых шпатов. Необходимо напомнить, что по-
левые шпаты флотируются из смеси с кварцем аминами в кислой
среде при добавке достаточного количества ионов фтора для
поддержания концентрации АЕ+ на низком уровне. Если же тре-
буется селективно отфлотировать слюду от кварца, наоборот,
может оказаться полезной добавка ионов алюминия. Это спра-
ведливо только для неразрушенных горных пород; для вывет-
590
ГЛАВА 16
ренных горных пород добавка А13+, очевидно, не нужна. Излиш-
не говорить о том, что химизм этого процесса полностью не вы-
яснен.
Глины. Келлог {119] изучал флотационное отделение каолина
от кварца. Он нашел, что оптимум разделения с помощью ами-
нов находится при pH ~3; при этом в ряду каолинит — слюда—
полевой шпат — кварц флотируемость последовательно убыва-
ет; это в значительной степени согласуется с работой Горного
бюро США. Большая склонность глин к катионному обмену так
же, как их способность обменивать свои катионы на углеводо-
родсодержащие катионы (в частности, на ионы амина) была из-
вестна давно. Это свойство было использовано при обесцвечива-
нии с помощью глин, поглощающих сильно окрашенные органи- '
ческие катионы, как например катионы красителей; по своему
химизму этот процесс в значительной степени подобен поглоще-
нию собирателя при флотации.
Цепочечные силикаты и ортосиликаты. Цепочечные силикаты
проявляют свойства ионов в двух направлениях, перпендику-
лярных оси цепочки, концы которой также являются ионными
участками (поскольку в этом месте структура нарушается). Сле-
довательно, если исключить влияние формы кристаллов на гид-
родинамику флотации, между цепочечными силикатами и орто-
силикатами нет большой разницы в их флотационных свойствах.
Поскольку цепочечные силикаты с одиночными и двойными
цепочками близки по своей структуре, ионная плотность не
должна сильно изменяться при переходе от одной разновидно-
сти к другой. Следует полагать, что минералы класса ортоси-
ликатов и класса цепочечных силикатов также не должны рез-
ко отличаться по ионной плотности.
Таким образом, различия между минералами этих классов
обусловливаются в основном характером катионов и их способ-
ностью образовывать нерастворимые соединения с анионами со- S
бирателя. При катионных собирателях флотируемость, по всей
вероятности, определяется главным образом способностью ка-
тионов собирателя конкурировать с имеющимися в растворе ка-
тионами металлов за место на поверхности минерала.
Основными цепочечными силикатами являются амфиболы и
пироксены; типичным представителем первых считается роговая
обманка, вторых — сподумен и диопсид. За исключением споду-
мена, который имеет экономическое значение в качестве источ-
ника лития, эти минералы почти не исследованы.
Очень мало серьезных работ было проведно по изучению
ортосиликатов или других силикатов, имеющих конечную «ос-
тровную» структуру кремнекислородных полимеров. Однако
многие из них уже привлекли к себе некоторое внимание или мо-
гут представить научный интерес в будущем. В табл. 3 приво-
РУДЫ ОКИСЛЕННЫХ МИНЕРАЛОВ
591
дится перечень ионов, которые встречаются в различных минера-
лах, характерных для этих классов силикатов. Приведенные фор-
мулы надо считать лишь приблизительными. Во многих случаях
(например, для кианита) истинная структура является проме-
жуточной между указанной в таблице ионной структурой и кар-
касной или между ионной, цепочечной (благодаря полимери-
зации силикатных ионов, которая не отражена в формуле) и
каркасной структурами (что наблюдается, например, у берилла
и турмалина).
Таблица 3-
Характерные минералы из класса силикатов, имеющих «островную»
структуру, ортосиликатов и родственных им минералов
Минерал Катионы Анионы
Циркон ]Zr]4 + ]SiO4]4-
Торит [ТЬ[4 + [SiO4]4-
Виллемит 2[Zn|2 + [SiO4]4-
Фенакит 2[Ве]2+ ]SiO4]4-
Оливин 2[(Mg, Fe)]2 + |S1O4]4-
Монтичеллит Кианит 1 |Ca]2+, [Mg]2 + |SiO4]4-
Андалузит ) Силлиманит ) 2 ]SiO4]4-[O]2-
Титанит [Ca]2+, ]Ti]4 + ISiO4] 4-> [O]2~
Хондродит 5[Mg]2 + 2[(OH, F)]~, 2[SiOJ4-
Волластонит 3[Ca|2+ [Si3O9]6-
Берилл 3]Be]2+, 2[A1]3+ [Si6O18]12-
Турмалин [ Na)+, 3[Mg]2+ 4[ОНГ, ! Si6B3O27] 3-
Берилл. Много внимания было уделено флотации алюмосили-
ката бериллия — берилла, имеющего сложную структуру. Хотя,
строго говоря, берилл не является каркасным силикатом или си-
ликатом островного строения, его все же удобнее рассматривать-
в этом разделе так же, каки топаз.
Для обогащения берилловых руд западных месторождений
рекомендуется [120, 175] сначала производить обесшламливание
руды, удаление железа, попавшего в руду при ее измельчении,
удаление железных и алюминиевых ионов (например, с помощью
иона фтора), а затем флотацию в среде фтор-ионов при рН~6,5.
Кварц, полевой шпат и мусковит не флотируются при этих уело-
592
ГЛАВА 16
виях. Основные примеси, загрязняющие берилловый концен-
трат,— это гранаты, биотит и турмалин. Слюды целесообразнее
флотировать раньше берилла катионным собирателем с добавкой
плавиковой кислоты до pH = 2,5.
Топаз. Небольшое исследование было проведено по флотации
топаза [66]. Рекомендуемые условия включают применение в
качестве собирателя алкилсульфата в кислой среде.
Сподумен. Имеется значительное число работ эмпирического
характера, в которых изучался вопрос отделения литиевого ми-
нерала — сподумена от ассоциированных с ним пегматитовых
минералов [136, 139]. Так как сподумен не является каркасным
силикатом, а состоит из силикатных частиц, связанных воедино
при помощи катионов, можно предположить, что по флотацион-
ным свойствам он должен быть ближе к ортосиликатам (напри-
мер, к оливину), чем к кварцу и полевому шпату. Отделение его
от кварца и полевого шпата легко достигается при помощи жир-
ной кислоты или мыла, если концентрация активирующих ионов
поддерживается на достаточно низком уровне. Норман и Гесике
рекомендовали обдирку истиранием для очистки поверхности
выветренных минералов и фториды для удаления ионов железа
и алюминия. Согласно Мансону и Эриксону, флотацию следует
проводить в умягченной воде, свободной от избытка щелочи и
сульфатных ионов, при рН~8. Бэнкс предлагал в качестве со-
бирателей реагенты типа сульфонированного масла.
Использование катионных собирателей приводит к новому
способу [7] отделения сподумена от кварца, полевого шпата и
слюд, при котором в пену поднимается вся пустая порода, а спо-
думен вместе с имеющимися ;в руде минералами железа остается
в камерном продукте. Большие трудности возникают при нали-
чии в руде железомагниевого силиката — роговой обманки,
имеющей подобно сподумену силикатно-цепочечную структуру.
Силлиманит и кианит. Эти минералы являются силикатами
алюминия, в которых одна часть алюминия, по-видимому, нахо-
дится в металлических узлах между кремнекислородными тет-
раэдрами, в то время как другая замещает атомы кремния внутри
каркаса (SiO2)n. Различие между этими двоякого рода атомами
алюминия выражено нерезко: в значительной степени оно свя-
зано с различием в величине межатомных расстояний. Силли-
манит и кианит имеют одинаковую формулу AlfeO3-SiO2, так что
их можно считать структурными изомерами алюмосиликатного
полимера.
Значительные успехи имеются в разделении кианита и квар-
ца с помощью мыла в растворе соды или жидкого стекла [36,
46, 144, 146]. Результаты улучшаются, если руда предваритель-
но подвергалась обдирке с кислотой или щелочью [182—184]. Это
улучшение аналогично тому, которое достигается благодаря по-
РУДЫ ОКИСЛЕННЫХ МИНЕРАЛОВ
593
верхностной очистке силикатов при обогащении гранатовых, цир-
коновых, полевошпатовых и других руд. Предполагаемый для
этого процесса механизм «очищающего» действия недостаточно
ясен.
Те же самые схемы в основном пригодны для обогащения
силлиманитовых руд [1521; в качестве собирателя преимуще-
ственно используется опять-таки олеиновая кислота в слегка ще-
лочной среде с добавкой полимеров щелочных фосфатов и жид-
кого стекла для регулирования катионного состава пульпы. Це-
лесообразна предварительная кислотная обдирка.
Турмалин. Для многих обогатителей, работающих в области
флотации несульфидных минералов, наличие турмалина в руде
равносильно настоящему бедствию. Очень часто он встречается
в том же месте и в одно и то же время с различными полезными
минералами — касситеритом, рутилом, бериллом или уранини-
том. Возможно, что это обусловлено сложной и весьма изменчи-
вой структурой турмалина.
ЛИТЕРАТУРА
1. Allan J. С. Table flotation for the removal of sulfides from tin-wolfram
concentrates in Portugal, Bull. Inst. Mining Met., 553, 81—90, 1952.
2. Андреева А. И. Советский патент, 579611, 30, 1940.
3. Андреева А. И. Флотация смитсонита Цветные металлы (9), 46—50,
1940.
4. А г с h i b а 1 d Е. R. U. S. Patent 2,669,355/1954.
5. Armstrong Н. Н., and А. В. Menefee. U. S. Patent 2,000,656/1935.
6. В a nks М. К. U. S. Patent 2,633,241/1953.
7. В a n k s, М. К., William Т. McDaniel, and Philip N. Sales.
A method for the concentration of North Carolina spodumene ores, Trans.
AIMME, 196, in Mining Eng. 5(2), 181—186, 1953.
8. Barr, J. A. U. S. Patent 2,551,197/1951.
9. Batty J. V., T. F. Michell, R. Havens, and R. R. Wells. Benefi-
ciation of chromite ores from the western Unites States, U. S. Bur. Mines,
Rept. Invest., 4079, 26 pp., 1947.
10. Белаш Ф. H. Флотация вольфрамовых руд Букуки, «Цветные металлы»,
16 (13), 16—20, 1943.
11. Белаш Ф. Н. и Пугина О. В. Опыты флотации сульфидно-хобнери-
товой руды. Цветные металлы (7), 72—75, 1939.
1Е. Белаш Ф. Н. и Пугина О. В. Усиление депрессирующего действия
раствора силиката натрия на кальциевые минералы при флотации шеели-
та мылом. Цветные металлы, 19 (6), 22—27, 1946.
13. Белаш Ф. Н. и Вейке ль. Флотация шеелитовых руд гумбейского
месторождения. Цветные металлы (3), 45—50, 1940.
14. Berkelhamer Lewis Н. Petrographic study of domestic talcs, Bull.
Am. Ceram. Soc., 22, 227—230, 1943.
15. Berman H. Constitution and classification of the natural silicates, Am.
Mineralogist. 22, 342—408, 1937
16. Booth R. B„ and R. A. Pickens. U. S. Patent 2,428,763/1947.
17. Booth R. B„ and R. A. Pickens U S. Patent 2,477,402/1949.
18. Borcherdt W. O. U. S. Patent 1,662,633/1928.
19. Borcherdt W. O. U. S. Patent 1,662,634/1928.
38 A. M. Годэн
594
ГЛАВА 16
20. Bradley John D., Joseph A. Me ci a, and Robert E. Baker.
Yellow Pine Mine. Eng. Mining J., 144 (4), 60—66, 1943.
21. Br agg W. L. The structure of silicates, Z. Krist., 74, 237—305, 1930.
22. В r a g g W. L. «The Atomic Structure of Minerals», Cornell University
Press, Ithaca, N. Y., 1937.
23. Breerwood Charles H. U. S. Patent 1,931,921/1933.
24. Breerwood Charles H. U. S. Patent 2,028,313/1936.
25. В r i n g Gust G. Flotation of hematite ore, Jernkontorets Ann., 122,
139—169, 1938.
26. Bring Gust G. Adsorption, flocculation and flotation of oxide minerals,
Jernkontorets Ann., 123, 229—265, 1939.
27. Bring Gust G. Separation of apatite by flotation, Jernkontorets Ann.,
124, 277—312, 1940.
28. В r i n g Gust G. Continuous flotation of hematite, Jernkontorets Ann.,
124, 562—573, 1940.
29. Bring Gust G. Pulp concentration and mixing time in hematite flota-
tion, Jernkontorets Ann., 128, 63—76, 1944.
30. В r i n g Gust G. Recovery of mineral products from tailings from iron
ore concentration plants by flotation, Jernkontorets Ann., 130, 127—160;
605—648, 1946.
31. Bring Gust, G., and I. J a n e 1 i d. Industrial scale test of hematite flo-
tation, Jernkontorets Ann., 126, 143—160, 1942.
32. Broadbridge Walter, and Edwin E d s e r. U. S. Patent
1,547,732/1925.
33. Brown E. H. U. S. Patent 2,629,493/1953.
34. С a г п о с о c h a n R. K-, and R. A. Rogers. Concentration of cyanite from
Death Rapids, В. C., Can. Dept. Mines, Mines Branch, Bull. Rept., 736,
238—240, 1934.
35. Chapman G. A., and J. W. L i 111 e f о r d. U. S. Patent 1,968,008/1934.
36. Ч и ж и к о в Д. М., Запрудский Н. Н. иТурыгин С. М. Концентра-
ция окисленных руд цветных металлов. Цветные металлы, 16 (8), 27—28,
1941.
37. Christensen Niels С. U. S. Patent 1,467,354/1923.
38. Ч у х р о в Ф. В. и Аносов Ф. Я. Природа хризоколлы. Записки Все-
союзного минералогического общества, 79, 127—136, 1950.
39. Clark L. F. U. S. Patent 1,670,021/1928.
40. Clemmer J. Bruce, В. H. Clemmons, Carl Rampace k,
M. F. Williams, and R. H. Stacy. Beneficiation of iron ores by flota-
tion, I. U. S. Bur. Mines, Rept. Invest. 3799, 1945.
41. Clemmer, J. Bruce, В. H. Clemmons, and R. H. Stacy. Prelimi-
nary report on the flotation of bauxite, U, S. Bur. Mines, Rept. Invest.
3586, 1941.
42. С 1 e m m e r J. Bruce, and S. R. B. Cooke. Flotation of Vermont talc —
magnesite ores, U. S. Bur. Mines, Rept. Invest, 2214, 1936.
43. Clemmer J. Bruce, and S. R. B. Cooke. Flotation of complex mo-
lybdenum-vanadium ores from Mammouth Arizona, U. S Bur. Mines
Rept. Invest. 3333, 21—38, 1937.
44. C lemmer J. Bruce, H. A. D о e r n e r, and F. D. De V a n e y. Experi-
mental flotation of Washington magnesite ores, AIMME Tech. Publ. 1448,
10 pp., 1940.
45. Clemmer J. Bruce, and R. G. O’Meara. Flotation and depression
of non sulfides; calcite, silica and silicates, fluorspar, barite, apatite and
tungsten minerals, U. S. Bur. Mines, Rept. Invest. 3239, 1941.
46. С о g h i 11 Will H., and J. Bruce Clemmer. Soap flotation of the
поп-sulfides, Trans. AIMME, 112, 449—465, 1934.
47. Cooke S. R. B. Microscopic structure, and concentratability of the impor-
tant iron ores of the United States, U. S. Bur. Mines Bull., 391, 121 pp..
1936.
РУДЫ ОКИСЛЕННЫХ МИНЕРАЛОВ
595
48. Cooke S. R. В. The flotation of quartz with calcium ion as activator,
Trans. AIMME, 184, Tech. Publ. 2607-B, pp. 306—309, 1949.
49. Cooke S. R. B., and Marcus Digre. Studies on the activation of
quartz with calcium ion, Mining Eng., 1 (8), AIMME 184, 299—305 1949.
50. Crago Arthur. U. S. Patent 2,293,640/1942.
51. Crago Arthur, and Harold S. Martin. U. S. Patent 1,912,433/1933.
52. Crago Arthur, and H. C. Mot singer. U. S. Patent 2,016,105/1935.
53. C u t hb e r t s о n, Robert E. U. S. Patent 2,610,738/1952.
54. De Bruyn, P. L. Flotation of quartz by cationic collectors, Mining Eng.,
7. Tech. Publ. 3983B, March, 1955.
55. Д e м и н о в а О. А. Использование убогих вольфрамовых руд. Цветные
металлы (7), 48—40, 1939.
56. De Vane у. F. D. U. S. Patent 2,470,150/1949.
57. D i с е С. М. Methods and costs of concentrating huebnerite ores at the
Ima tungsten mine, Lemhi County, Idaho, U. S. Bur. Mines, Circ. 7230,
14 pp., 1943.
58. Doerner H. A., and Dwight Harris. Concentration of low grade
magnesite ores by flotation, Wash. State Coll., Min. Expt. Sta, Bull. P. 1,
pp. 1—30, 1938.
59. D о e r n e r H. A., and Dwight Harris. U. S. Patent 2,205,923/1940.
60. Duncan Walter E. U. S. Patent 2,381,120/1945.
61. Edelman С. H„ and J. C. L. Favejes. The crystal structure of
montmorillonite and halloysite, Z. Krist., 102, 417—431, 1940.
62. Edwards G. R., and W. E. Ewers. Adsorption of sodium cetyl sulfate
on cassiterite, Australian J. Sci. Research A4, 627—643, 1951.
63. E i t e 1, Wilhelm. «The Physical Chemistry of the Silicates», University
of Chicago Press, Chicago, 1954,
64. Engel hart G. K. Flotation as applied to modern cement manufacture,
Ind. Eng. Chem., 32, 645—651, 1940.
65. E r 1 e n m e у e r H., J. v. Steiger, and W. Theilheimer. Flotation
experiments with 8-hydroxyquinoline as collector, Helv. Chim Acta, 25,
241—245, 1942.
66. E v a n s L. E. The flotation of topaz using sodium hexadecylsulfate as
collector, Australian J. Appl. Sci, 4, 165—173, 1953.
67. F a 1 с о n e r S. A., and L. J. Christmann. U. S. Patent 1,858,007/1932.
68. Falconer S. A., and Bruce D. Cranford Froth flotation of some
nonsulfide minerals of strategic importance, AIMME, Tech. Publ. 1754,
November, 1944.
69. Fine M. M., and D. W. F г о m m e r. Mineral dressing investigation of
titanium ore from the Christy Property, Hot Spring County, Ark.,
U. S. Bur. Mines, Rept. Invest. 4851, 1952.
70. F i n n, Willy. Tin-tungsten ore flotation at the Schwarzwasser-
Altenberg Mines, Z. Erzbergbau u. Metallhiittenw., 5, 266—270, 1952.
71. Fleming M. G. Effects of alkalinity on the flatation of lead minerals,
Min. Eng., 4, 1231—1236, 1952.
72. G ahi, Rudolf. U. S. Patent 1.217,437/1917.
73. Gaty F. H. deRycker, and H. Thyssen. Study of the flotation of
calamine minerals in the laboratories of the Metallurgical Institute (Uni-
versity of Liege, Belgium), Rev. universelle mines, 89, 155—166, 1946.
74. G a u d i n A. M. The influence of hydrogen-ion concentration on reco-
very in simple flotation systems, Mining and Met., 10, 19—20, 1929.
75. Gaudin A. M. «Flotation» 1st ed., pp. 372—373, McGraw-Hill Book
Company, Inc., New York, 1932.
76. Gaudin A. M. U. S. Patent 1,979,324/1934.
77. Gaudin A. M. U. S. Patent 2,231,265/1941.
78. Gaudin A. M., and Arvid E. Anderson. Flotation fundamentals, V,
Utah Eng. Expt. Sta., Tech. Publ. 9, 1930.
38
596
ГЛАВА 16
79. Gaudin А. М., and F. W. Bloecher, Jr. Concerning the adsorption
of dodecylamine on quartz, Trans. AIMME, 187, 499—505, 1950.
80. G a u d i n A. M., and C. S. Chang. Adsorption on quartz from an aqueous
solution of barium and laurate ions, Min. Eng., 4, 193—201, Trans. AIMME,
Tech. Publ. 3244B, February, 1952.
81. Gaudin A. M., and P. L. de Bruyn. Radioactive tracers in mineral
engineering problems and particulary in flotation, Can. Mining Met. Bull.
447, pp. 331—337, 1949.
82. Gaudin A. M„ and Falih Ergunalp. Making in flotation work. II.
Oploca ore, Eng. Mining, J., 147 (11), 72—74, 1946.
83. Gaudin A. M., and D. W. F u e r s t e n a u. Quartz flotation with anionic
collectors, Min. Eng. Trans. AIMME, 202, 66—72, January, 1955.
84. Gaudin A. M., Harvey Glover, M. S. Hansen, and C. W. Or r.
Flotation fundamentals, I, Univ. Utah Tech. Publ. 3, 1928.
85. Gaudin A. M., and R. T. H u к к i. Making tin flotation work. IV. San
Jose ore, Eng. Mining J., 148 (3), 70—72, 1947.
86. G a u d i n A. M„ and J. Sheldon Martin. Flotation fundamentals, III,
Utah Eng. Expt. Sta., Tech. Publ. 9, 1930.
87. G a u d i n A. M., and Alfonso R i z o-P a t г о n. The mechanism of
activation in flotation, AIMME Tech. Publ. 1453, 1942.
88. Gaudin A. M., and R. Schuhmann, Jr. Making in flotation work.
III. Colquiri ore, Eng. Mining J., 147 (12), 68—72, 1946, 148 (1), 84—87,
1947.
89. Gaudin A. M., R. Schuhmann Jr., and E. G. Brown. Making tin
flotation work. I. Canutillos ores, Eng. Mining J., 147 (10), 54—59, 1946.
90. Gaudin A. M., R. Schuhmann, Jr., and John Dasher. Extraction
process for gold-uranium ores, J. Metals, 8, 1065—1969, 1956.
91. Gaudin A. M„ and H. Rush Spedden. Flotation microscopy of some
Cuban manganese ores, Trans. AIMME, Techn. Publ. 1451, 1942.
92. Gieseke E. W. U. S. Patent 2,483,192/1949.
93. Gieseke E, W. U. S. Patent 2,643,770/1953.
94. Greenawalt William E. U. S. Patent 1,344,127/1920.
95. G r i П Rolph E. «Clay Mineralogy», McGraw-Hill Book Company, Inc.,
New York, 1953.
96. Gruner, J. W. Progress in silicate structures, Am. Mineralogist 33,
679—691, 1948.
97. Gutzeit Gregoire, and P. Koval iv. Selective flotation tests of
minerals constituting black sands, Arch. sci. phys. et nat., 21, 260—269, 1939.
98. H a n d 1 e у R. W., and C. W. Sawyer. U. S. Patents 2,570,119/1951,
2.570,120/1951.
99. Hansen M. S. U. S. Patent 1,972,588/1934.
100. H a r t J. G Flotation of scheelite, Chem. Eng. Mining Rev., 30, 379—381,
1938.
101. Havens Richard. U. S. Patent 2,412,217/1946.
102. Hayward Carle R., and Livingston Wright. Extraction of tin
from ares and concentrates, U. S. Patent 2,547,939/1951.
103. Heilmann, Thorbjorn. U. S. Patent 2.507,012/1950.
104. Heilmann Thorbjorn. U. S. Patent 2,507,013/1950.
105. He r g t H. F. A., J. R о g e r s, and K- L. Sutherland. Principles of flo-
tation— flotation of cassiterite and associated minerals, AIMME Tech.
Publ. 2081, Mining Technol., 11(1), 1947.
106. Herkenhoff E. C. U. S. Patent 2389,727/1945.
107. Herkenhoff E. C. U. S. Patent 2,466,987/1949.
108. Herman John, A. W. Allen, and H. R. N e w i 11. U S. Patent
1,628,046/1927.
109. Hoag E. H. U. S. Patent 2,371,292/1945.
110. Hoag E. H. U. S. Patent 2.378,552/1945.
111. Holladay J. A. U. S. Patent 1,706,293/1929.
РУДЫ ОКИСЛЕННЫХ минералов
597
112. Holman Bros, Ltd., and Francis E. Michell. British Patent
567,000/1945.
113. Hubbell A. H. Phosphate washer reject successfully concentrated on
moving conveyor belts, Eng. Mining J„ 143 (12), 51—54, December, 1942.
114. H u m b e 1 d t-D e u t z m о t о r e n. French Patent 811,787/1937.
115. Hutt 1 John B. Domestic manganese from Butte helps in emergency,
Eng. Mining J., 143 (I), 56—58, January, 1942.
116. Keck W. E., G. C. Eggleston, and W. W. Lowry. A study of the
flotative properties of hematite, AIMME, Tech. Publ. 763, Mining Technol.,
1, January, 1937.
117. Keck W. E., and Paul Jasberg. A study of the flotative properties
of magnetite, AIMME Tech. Publ. 801, Mining Technol, 1, May, 1937.
118. Kelley Walter P. «Cation Exchange in Soils», Reinhold Publishing
Corporation, New York, 1948.
119. Kellogg Herbert H. Flotation of kaolinite for removal of quartz,
Trans. AIMME, 169, 548—555, 1946.
120. Lamb Frank D. Beneficiation of New England beryllium ores, U. S.
Bur. Mines, Rept. Invest. 4040, 1947.
121. Lamb Frank D., and John Ruppert. Flotation of a North Carolina
pyrophyllite ore, U. S. Bureau Mines, Rept. Invest. 4674, 1950.
122. Lange L. H. U. S. Patent 1,914 694/1933.
123. Lan ge L. H. U. S. Patent 1,914,695/1933.
124. Leaver E. S., and M. B. Royer. Flotation for recovery of scheelite
from slimed material, U. S. Bureau Mines, Tech. Paper 585, 24 pp., 1938-
125. Lee Oscar. Flotation of limestone from siliceous gangue, U. S. Bur.
Mines, Rept. Invest. 2744, 1926.
126. Littleford J. W. U. S. Patent 1,780,022/1930.
127. Lowe Richard H. British Patent 554,614/1943.
128. Ludt Randall W. The flotation of copper silicate by alkylsubstituted
triphenylmethane dyes. Univ, Microfilm Publ. 918, Microfilm Abstr. 8(1),
27—29. 1948.
129. MacDonald Wm. T. A useful new selectivity modifier in nonsulfide
flotation, Mining and Met., 18. 285—286, 1937.
130. McKenna William J. Virgil Lessels, and Ernest C Pe-
terson. U. S. Patent 2,482,859/1949.
131. McLaren D. C. Concentration of tungsten ores, Chem. Eng. Mining
Rev., 35, 181—186. 1945.
132. Michell F. B. Table flotation, Mining Mag., 73, 329—337, 1945.
133. M i 11 e r Benjamin L., and Charles H. Breerwood. Flotation
processing of limetsone, AIMME Tech. Publ. 606, 1935.
134. Mi liken Frank R. Metallurgy at National Lead Co., MacIntyre De-
velopment, Tech. Publ. 2355. Mining Technol., 12, May 1948.
135. M i t c h e 11 Will, Jr. C. L. S о 11 e n b e r g e r, and T. G. Kirkland
Flotation tests on Korean scheelite ore, Trans, AIMME, Tech. Publ. 2990-B,
in Mining Eng., 190, 60—64, 1951.
136. Munson Gerald A., and K. L. Erickson. The flotation of spo-
dumene from the Edison Mine, Keystone, South Dakota, U. S. Bur. Mines,
Rept. Invest. 3892, 1946.
137. Neunhoeffer Otto. Synthesis of a flotation agent for cassiterite —
verification of chemical principles of flotation, Metall u. Erz, 40 174—
176, 1943.
138. Nininger Robert D. «Minerals for Atomic Energy», D. Van Nost-
rand Company, Inc., New York, 1954.
139. Norman James, and E. W. Gieseke. Beneficiation of spodumene
rock by froth flotation, AIMME Tech. Publ. 1161, in Mining Technol., 4,
1940.
140. Norman James E., and Benjamin S. Lindsey. Flotation of
598
ГЛАВА 16
barite from Magnet Cove, Ark., Mining Technol. 5, AIMME Tech. Publ.
1326, 1941.
141. Norman James E., and R. G. O'Meara. Froth flotation and agglo-
merate tabling of micas, U. S. Bur. Mines, Rept. Invest, 3558, 1941.
142. О b e r b i 11 i g Ernest, and E. P. Frink. Recovering slime cassite-
rite by soap flotation, Eng. Mining J., 142, 54—57, December, 1941.
143. O’Connor D. J., and A. S. Buchanan. E'.ectrokinetic properties of
cassiterite, Australian J. Chem. 6, 278—293, 1953.
144. O’Meara R. G. U. S. Patent 2,297,689/1942.
145. O’Meara R. G., and G. D. Coe. Concentration of southern barite ores,
U. S. Bur. Mines, Rept. Invest. 3376, 12 pp., 1938.
146. O’Meara R. G., and B. W. G a n d r u d. Concentration of Georgia kya-
nite ore, AIMME Contrib. 98, 1936.
147. О ’M e a r a R. G., J. E. Norman, and Walter E. Hammond. Froth
flotation and agglomerate tabling of feldspars Bull. Am. Ceram. Soq.,
18, 286—292, 1939.
148. Palache Charles, Harry Berman, and Clifford Fronde 1.
«Dana’s system of Mineralogy», 7th ed., vol. II, John Wiley & Sons, Inc.,
New York, 1951.
149. Petersen W. Adsorption of oleic acid and sodium oleate during the
flotation of barium sulfate, Kolloid-Z., 81, 212—222, 1937.
150. Полькин С. И. и Олюнина H. И. Флотация окисленных минералов
(скородита и вольфрамита), Цветные металлы (12), 36—44, 1940.
151. Pryor Е. J., and S. A. Wrobel. Studies in cassiterite flotation, Bull.
Inst. Mining Met., 532, pp. 201—237, 1951.
152. Rampacek Carl, В. H. Clemmons, and J. Bruce Clemmer.
Beneficiation of South Carolina silimanite schists, J. Am. Ceram.. Soc., 28,
197—205, 1945.
153. Rankin H. S., Robert A. Laurence, F. A. W. Davis, Edward
C. Houston, and Lynn I. McMurray. Concentration tests on
Tennessee Valley barite, Mining TehnoL, 2, AIMME, Tech. Publ. 880,
13 pp., 1938.
154. Rey Maurice, Paul Chataignon, and Victor Formanek.
The influence of certain inorganic salts on the flotation of lead carbonate,
Mining Eng., 187, 1126—1130, 1950.
155. Ried Robert C. U. S. Patent 2,163,701/1939.
156. Ried Robert C. U. S. Patent 2,163,702/1939.
157. Ried Robert C. U. S. Patent 2,202,601/1940.
158. Rockwood N. C. Chemistry applied to cement manufacture, Rock Pro-
ducts, 37 (8), 32—37, 1934.
159. Roger E. L’Evolution de la metallurgie de cuivre au Katanga, Compt.
rend, congr. sci. Elizabethville, 3, 129—168, 1950.
160. Розов Б. И. и Мясникова Г. А. Флотация шеелита из руд Тыриы-
Аузского месторождения. Цветные металлы (4), 33—41, 1940.
161 Розов Б. И., Мясникова Г. А. и Б аз лов а Н. И. Советский па-
тент 67,082/1946.
162. Runke S. М., Е. G. Howe, J. S. Kennedy, and Н. Kenworthy.
Pilot plant concentration of Arkansas aluminium ores. U. S. Bur. Mines,
Rept. Invest. 4440, 37 pp., 1949.
163. Runke S. M., and R. G. O’Meara. Concentration of bauxite for mil-
ling in the 50-ton Bureau of Mines pilot plant, Bauxite, Arkansas, U. S.
Bur. Mines, Rept. Invest. 3909, 1946.
164. Schoenlaub Robert A. U. S. Patent 2,414,980/1947.
165. Schoenlaub Robert A. U. S. Patent 2,433,297/1947.
166. Schuhmann R., Jr., and Brahm Prakash. Effect of barium
chloride and other activators on the soap flotation of quartz, Trans.
AIMME, 187, 591—600, 1950.
РУДЫ ОКИСЛЕННЫХ МИНЕРАЛОВ
599
167. Schuhmann R„ J r„ and Brahm Prakash. Effect of activators
and alizarin dyes on soap flotation of cassiterite and fluorite Trans
AIMME Tech. Publ. 2827B, in Mining Eng., 187, 601—618, 1950.
168. Searles John N. Some tests with flotation on Mesabi wash-ore
tailings, Eng. Mining J., 139, 42—43, June, 1938.
169. Sengier Edgar B. Katanga’s mineral empire based on many metals,
Eng. Mining J„ 152 (11), 86—89, 93—96, 1951.
170. Seyer Pierre. Concentration of tungsten ores in France and in the
world, Ann. mines, 141 (a/4), 3—92, 1952.
171. Шор шер И. H. Флотация касситертита. Цветные металлы. 19 (6),
13—19, 1946.
172. Шрадер А. А. Флотация шламов из хромитовых руд, Горнообогати-
тельный журнал, (5), 35—40, 1936.
173. Шведов Д. А. и Андреева А. И. Флотация карбонатных медных
руд бензиловым меркаптаном и причины его высокого расхода. Цвет-
ные металлы (8), 67—75, 1935.
174. Si ti a Giovanni. La flotation des minerals oxydes de plomb dans
les laveries de la Societa mineraria e metallurgica di Pertusola en Sar-
daigne, Congres des laveries des mines metalliques frangaises, F-6, Pa-
ris, October, 1953.
175. Snedden H. D., and H. L. Gibbs. Beneficiation of western beryl
ores, U. S. Bur. Mines, Rept. Invest. 4071, 1947.
176. Snyder E. H„ and W. D. Green. U. S. Patent 1,691,310/1928.
177. Spence Peter & Sons, Ltd., and Arthur P. Laurie. British Pa-
tent 672,216/May, 1952.
178. Strunz H. «Mineralogische Tabellen», pp. 1—56, Akademische Verlags-
gesellschaft, m. b. H., Leipzig, 1941.
179. Sulm ап H. L., and Eawin E d s e r. U. S. Patent 1,492,904/1924.
180. Swainson S. J. Washing and concentrating Florida pebble phosphate
Mining and Met. 25, 469—474, 1944.
181. Tartaron Francis X. U. S. Patent 2,303,962/1942.
182. Tartaron Francis X. U. S. Patent 2,289.741/1942.
183. Tartaron Francis X. U. S. Patent 2,305,502/1942.
184. Tartaron Francis X. U. S. Patent 2,326,807/1943.
185. Tartaron Francis X., and L. R. Harrison. U, S.
Patent 2,226,103/1940.
186. Terry J. T. U. S. Patent 1,709,329/1929.
187. Theys L. L’Evolution de la technique de la flotation des minerais oxy-
des du Katanga, Association ing. Liege, Section coloniale, Congr. 1947,
pp. 439—442.
188. Trotter W., and E. W. Wilkinson. U. S. Patent 1,795,100/1931.
189. Veltman P. L. U. S. Patent 2,647,629/1953.
189a. Walker G. F. Vermiculite and some related mixed-layer minerals,
chap. VII, pp. 192—223, in «Х-ray Identification and Structure of the
Clay Minerals», Mineralogical Society of Great Britain Monograph, 1951.
190. Weinig Arthur J. U. S. Patent 2,464,313/1949 .
191. Wells A. F. «Structural Inorganic Chemistry», (a) pp. 476ff., Oxford
University Press, New York, 1945.
192. Whitaker W i 11 i a m A. U. S. Patent 1,457,680/1923.
193. W i 11 i a m s J. C. Cationic flotation of cement rock, AIMME Tech. Publ.
1901, in Mining Technol., 10, 5 pp., 1946.
194. Zimmerley S. R, Flotation of oxidized silver-lead ores as influenced by
modified grinding, U. S Bur. Mines, Rept. Invest. 3364, pp. 7—30, 1937.
195. Zimmerley S. R. Oxidized lead-ore flotation, Conf. Met. Research,
Met. Div., U. S. Bur. Mines (Salt Lake City), pp. 143—156, May, 1940.
Глава 17
СОЛИ
В этой главе рассматривается обогащение руд или смеси
твердых веществ, содержащих в качестве основных компонентов
галоидные минералы или другие легко растворимые в воде соли.
Приводятся данные по флюоритовым рудам, сильвинитам, ка-
менной соли, нитратам, сульфатам, боратам и борной кислоте.
Все минералы, за исключением флюорита, хорошо раствори-
мы в воде. Так, например, растворимость каменной соли состав-
ляет около 30% по весу при комнатной температуре.
В табл. 1 приводятся данные о растворимости минералов и
солей.
Таблица 1
Растворимость некоторых минералов и солей при 25°
Минералы или соли Формула Растворимость
г; 100 мл воды е/л раствора моль 1л раствора
Флюорит CaF2 0,0017 0,017 0,0002
Криолит Na3AlFe 0,4 4,0 0,02
Галит NaCl 36,0 317,0 5,4
Сильвин CaCl 36,0 312,0 4,2
Борная кислота H3BO3 5,7 55,0 0,89
Арканит '. K2SO4 12,0 116,0 0,67
Кизерит1.2 MgSO4.H2O 48,0 444,0 3,7
Азотнокислый натрий .... NaNO3 91,0 663,0 7,8
Хлористый аммоний NH4Cl 39,0 300,0 5,6
Бишофит1 MgCl2-6H2O 55,0 475,0 5,0
Барит3 BaSO4 0,00023 0,0023 0,00001
Диаспор8 . HA1O2 0,00001 0,0001 0,000001
1 Выражено в граммах безводной соли.
2 При 100°.
• Приведено для сравнения.
Значительная растворимость солей приводит к другим важ-
ным следствиям: 1) высокая ионная сила флотационной среды
способствует сжатию двойного электрического слоя до такого
соли
60 г
размера, что он практически перестает существовать, например
в молярном солевом растворе одно-одновалентного электролита
толщина двойного слоя [уравнение (1а) из гл. 5] составляет око-
ло 3 А; в насыщенном растворе сильвинита толщина двойного
слоя — около 1 А; 2) если ценный минерал легко растворим, то
насыщенный раствор нельзя отбрасывать; наоборот, он должен
возвращаться в процесс. Полнота возврата определяется цен-
ностью продукта; например, если сильвинитовая руда, содержа-
щая 25% КС1, обогащается в пульпе при весовом отношении
Ж: Т = 5 : 1 и если раствор содержит 20% КС1, то в системе на
каждую тонну руды приходится 1 т растворенного КС1. Очевид-
но, раствор содержит калия в четыре раза больше, чем его
имеется в руде. Если извлекается 99% раствора, то при этом в
растворенном виде теряется 4% калия (считая на калий в ис-
ходной руде), а для уменьшения потерь калия до 0,2% тре-
буется извлечь 99,95% раствора.
Именно из-за этих особенностей процесс флотации легко-
растворимых минералов может до некоторой степени отличать-
ся от процесса флотации сульфидных или окисленных минера-
лов, протекающего в среде с невысокой ионной силой; кроме
того, флотационные проблемы, возникающие при флотации рас-
творимых минералов, тесно связаны с гидрометаллургическими.
Вначале предполагали, что высокая растворимость этих
солей и минералов исключает возможность их обогащения мето-
дом флотации. Практически, развитие этой отрасли флотации
началось лишь после того, как Джейнпрост [42] показал, что
операция разделения может проводиться в растворе, насыщен-
ном минеральными солями. В таком растворе поведение мине-
ралов, конечно, отличается от поведения их в воде: они стано-
вятся нерастворимыми.
Скорость насыщения пульпы растворимыми солями может
быть различна (см. закон Фика в гл. 4). При малой раствори-
мости веществ насыщение достигается медленно; если же веще-
ства хорошо растворимы, насыщение наступает быстро. Во всех
случаях весьма вероятно, что соль реагирует с окружающим
раствором, образуя новые твердые вещества; при этом возмож-
но также образование комплексных соединений.
Малорастворимые соли
Флюорит. Поведение флюорита благодаря его относительно’
малой растворимости до некоторой степени приближается к по-
ведению нерастворимых солей, например барита. Для барита
потенциал определяющими ионами являются ионы Ва и
SC>4~ , для флюорита такими являются ионы Са2+ и F-.
602
ГЛАВА 17
Роль pH для этих минералов должна отличаться от той ро-
ли, которую играет pH для кислотных или щелочных окислов
и гидроокислов, где ионы Н+ и ОН~ непосредственно являются
потенциал определяющими ионами.
Флюорит обычно встречается в виде хорошо ограненных ку-
бов с совершенной спайностью по октаэдру. Каждый ион каль-
ция находится в окружении восьми ионов фтора. Ионы кальция
образуют на плоскости спайности гексагональную решетку, где
на каждый ион Са2+ приходится площадь 12,8 А2. Площадь, при-
ходящаяся на один ион фтора, в два раза меньше и составляет
'6,4 А2.
С флюоритом обычно ассоциируются следующие минералы:
кварц, кальцит, барит и некоторые сульфиды, например первич-
ный пирит, сфалерит и галенит.
Флюорит, идущий в химическую промышленность для произ-
водства фтористоводородной кислоты, по техническим условиям
должен быть очень высокой чистоты. Меньшая чистота требует-
ся для металлургического производства, где особое значение
имеет крупность материала.
Давно известно, что флюорит хорошо флотируется карбок-
сильными собирателями [21, 78, 82]. Наиболее распространен-
ным собирателем является олеиновая кислота. Повышение тем-
пературы пульпы (даже до точки кипения) благоприятно отра-
жается на флотации флюорита [10, 66], но физико-химические
факторы, вызывающие улучшение процесса флотации при подо-
греве, не ясны.
Среди новейших реагентов, применяемых для флотации пла-
викового шпата [13, 73], находятся катионные собиратели и раз-
личные алкилсульфаты.
Некоторые исследователи полагают, что целесообразно про-
водить флотацию в воде с пониженной жесткостью, пользуясь
цеолитовыми умягчителями или применяя известковосодовое
умягчение воды [1, 18].
Несмотря на легкую флотируемость флюорита, получение
конечного концентрата необходимой чистоты может быть связа-
но с большими трудностями. Возможно, что подогрев, улучшаю-
щий флотацию флюорита, способствует удалению примесей.
Если флюорит адсорбирует собиратель активнее, чем другие
присутствующие минералы, то продолжительное перемешивание
пульпы при повышенной температуре позволит более полно сор-
бировать собиратель флюоритом.
Сульфидные минералы обычно удаляют предварительной
флотацией, применяя сульфгидрильные собиратели; после этого
проводят флотацию флюорита карбоксильными собирателями.
Можно, однако, поступать так же, как и в случае окисленных
соли
603
минералов, т. е. флотировать щелочноземельные минералы кар-
боксильными собирателями, депрессируя сульфидные минералы
сульфидными и гидросульфидными ионами или ионами циана
[32]. На практике часто пытаются комбинировать эти два мето-
да: сначала флотируют легкофлотирующиеся сульфиды, а затем
флюорит в присутствии депрессоров для сульфидных минералов
[28]. Неотфлотировавшиеся сульфиды практически находятся в
хвостах флюоритовой флотации в виде шламов и сростков.
Трудно удалять барит и кальцит. Иногда решение этого
вопроса ищут, используя окисляющие вещества или добавляя
в пульпу кремнекислые соли тяжелых металлов [19, 34, 67].
Предпочитают, по-видимому, применять танин или родственные
ему соединения (15, 18, 26). Известно, что эти вещества являют-
ся эффективными депрессорами для кальцита и доломита, . но
механизм их действия неизвестен. Обычно предпочитается танин
типа квебрахо, производимый в Южной Америке. Были проведе-
ны опыты для оценки действия различных танинов, получаемых
из красного дерева [65] и установлено влияние зависимости эф-
фективности действия танинов от их молекулярной структуры.
Однако на существующем уровне наших знаний понять меха-
низм депрессии кальцита танином не представляется возмож-
ным; в этом направлении надо еще много работать.
Перечистка флюоритового продукта производится при до-
бавлении мыла и пониженном значении pH (практически значи-
тельно меньшем, чем pH равновесного раствора кальцита), в
результате чего кальцит депрессируется [84]. Некоторые исследо-
ватели утверждают, что при перечистке флюорита целесообраз-
но применять смесь растворимого фторида и жидкого сульфона-
та, а также смесь растворимого фторида и желатинированного
крахмала или декстрина [16, 17]. Опубликовано исследование
воздействия на флотацию флюорита активаторов и красок 181].
Криолит. Двадцать пять лет назад в Европе начали флоти-
ровать криолит. В качестве собирателей применяли жирные
кислоты и мыла, а для депрессии пустой породы — клей. При
этом стремились к осуществлению селективной флотации крио-
лита (натриевого алюмофторида), для отделения его от хагема-
нита (железного алюмофторида), томсенолита (натрийкальци-
евого алюмофторида) и пахнолита (другого натрийкальциевого
алюмофторида), так как эти минералы, благодаря своей окрас-
ке и прочим свойствам уменьшают ценность криолита. Селек-
тивной флотации криолита способствует применение морской
воды и добавка ионов фтора
1 Германские патенты: Фр. Крупп Грузоиверке, 557804/1930; 558965/1932;
французский патент: Гумбольдт Машиненбау-Анштальт 702343/1931; амери-
канский патент Хуберт Шранц и Джозеф Клоуз, 1966649/1934.
604
ГЛАВА )7
Сильвиниты
Калиевые руды. В морской воде и соленых озерах калий
встречается в меньших концентрациях по сравнению с натрием.
Усыхание больших бассейнов соленых вод приводит к насыще-
нию раствора и кристаллизации из него в первую очередь пова-
ренной соли, а затем сульфата кальция. Оставшийся маточный
раствор обогащен калием и магнием; помимо этого он все еще
содержит ионы хлора и натрия, а также некоторое количество
кальциевых и сульфатных ионов. Мировые месторождения ка-
лиевых солей образовались при дальнейшем усыхании таких
маточных растворов. Геологический процесс усыхания не являл-
ся непрерывным из-за периодического поступления новых при-
токов воды. Поэтому отложение калиевых солей могло чередо-
ваться с отложениями слоев галита, гипса и ангидрита. Этот
геологический процесс детально рассмотрен в немецкой литера-
туре [41, 50, 69]; некоторые данные по этому вопросу имеются
в американских и английских книгах [9, 33, 43].
Усыхание маточных растворов ведет к образованию смесей
многих минеральных разновидностей, среди которых в большом
числе присутствуют двойные и тройные соли. Во всех месторож-
дениях содержится много каменной соли, которую можно рас-
сматривать как основную пустую породу. В месторождениях
встречается также гипс CaSO4 • 2Н2О или ангидрит CaSO4, би-
шофит MgCl2 -.6Н2О, кизерит MgSO4 Н2О и элеонит
MgSO-i 7Н2О. Кроме этих солей, являющихся пустой породой,
обычно имеется различное количество глины (часто в виде монт-
мориллонита) и чрезвычайно тонко распределенной окиси желе-
за в виде гетита или гематита.
К калиевым минералам относятся сильвин КС1, арканит
K2SO4, карналлит KMgCl3 6Н2О, глазерит K3Na(SO4)2, шенит
K2Mg(SO4)2 6Н2О, леонит K2Mg(SO4)2 • 4Н2О, лангбейнит
K2Mg(SO4)2, каинит KMg(SO4)Cl • ЗН2О, сингенит
K2Ca(SO4)2 Н2О и полигалит K2MgCa2(SO4)4 2Н2О. Из этих
минералов наибольшее распространение и промышленное зна-
чение имеет сильвин. В Германии имеются большие месторожде-
ния лангбейнита и полигалита. Карналлит, присутствующий во
всех месторождениях, также является промышленно важным
минералом в Европе. Кроме того, надо обратить внимание на
глазерит как на минерал, затрудняющий флотацию вследствие
своей способности кристаллизоваться в пульпе в виде отдельных
скоплений. Другие калиевые минералы встречаются в относи-
тельно малом количестве.
Калиевые руды можно расклассифицировать на сильвинито-
вые, в которых сильвин является основным ценным минералом,
соли
605
карналлитовые, характеризующиеся присутствием кизерита в ка-
честве пустой породы, лангбейнитовые и полигалитовые.
Правила растворимости. Имеется много сведений о раство-
римости различных солей, встречающихся вместе с калиевыми
рудами. Явления растворимости солей детально изучались, осо-
бенно в Германии и Нидерландах, где были выполнены класси-
ческие работы Дж. X. Вант-Гоффа [88, 89], Е. Иенеке [41] и
Дж. д’Анса. Необходимо особенно отметить монументальную
работу д’Анса [241.
Обычно (но не всегда) растворимость солей увеличивается с
повышением температуры. Например, растворимость хлористого
калия в чистой воде увеличивается с 312 до 430 г/л при повы-
шении температуры с 10 до 50°. Растворимость хлористого нат-
рия в воде несколько необычна: при отсутствии примесей она
практически не зависит от изменений температуры. Так, напри-
мер, при повышении температуры с 10 до 50° растворимость
хлористого натрия меняется от 357 до 367 а/д.
Если одновременно присутствует несколько солей, раствори-
мость каждой из них уменьшается, и в этом случае температура
сильно влияет на изменение растворимости. Например, несмотря
на то что растворимость хлористого калия в растворе КС1 —
NaCl ниже, чем его растворимость в чистом растворе КС1,
наблюдается гораздо большее относительное увеличение раство-
римости хлористого калия в смешанном растворе по мере повы-
-шения температуры. Растворимость же NaCl в смешанном рас-
творе при повышении температуры, наоборот, уменьшается; та-
ким образом, поведение NaCl и КО прямо противоположно при
изменении температуры. На рис. 1 приведены данные д’Анса.
Если соли способны кристаллизоваться и в виде безводных
форм, и в виде одной (или более) гидратной формы, то имеется
определенная температура, при которой все три фазы находятся
в равновесии. Например, тенардит NazSCU мирабилит
NazSCXj • ЮН2О и водный раствор NazSC^ находятся в равнове-
сии при 32,38°. Эта температурная точка является характерной
физической константой подобно точке замерзания воды. Добав-
ка других растворов изменяет температуру образования солей.
Например, мирабилит, который при отсутствии в воде других
солей является стабильной фазой сульфата натрия до 32,38°, в
смешанном насыщенном растворе NaCl — NazSOi при любых
соотношениях NaCl: NazSOi является стабильным уже только
до 17,9°.
Область существования двойных и тройных солей, о которых
уже было сказано, лишь до некоторой степени ограничена тем-
пературой. Они могут быть настолько нестабильными при ком-
натной температуре, что получить насыщенный соляный раствор
606
ГЛАВА 17
этих сложных солей при обычном температурном режиме не
представляется возможным.
Система, содержащая более предельного числа твердых
солей в растворе, неизбежно является неустойчивой по отно-
шению к некоторым твердым фазам. Эти твердые фазы при на-
личии достаточного времени должны обязательно исчезнуть.
Вероятно, в общем случае это происходит с рудами, имеющими
Рис. I. Растворимость КС1 и
NaCl в воде и в насыщенных
растворах других хлоридов в
зависимости от температуры
[241:
1 — КС! в воде; 2 — NaCl в воде;
3 — КС! в насыщенном растворе
NaCl; 4 — NaCl в насыщенном рас-
творе КС1; 5 — КС! в насыщенном
растворе NaCl и карналлита; 6 —
NaCl в насыщенном растворе КС)
н карналлита
смешанный соляной состав, хотя время, необходимое для дости-
жения равновесного состояния, может превышать время пребы-
вания руды в процессе.
При обогащении сильвинитовых руд широкое применение
находит флотация.
Основной задачей процесса обогащения является отделение
галита от сильвина, но в некоторых случаях присутствие глин и
сульфата кальция усложняет разделение.
Другие галоидные минералы обычно присутствуют в малых
количествах, поэтому и удаление их не является основной целью.
Однако эти минералы могут играть важную роль, накапливаясь
во флотационной пульпе. Это происходит вследствие перехода
их в раствор, циркуляция которого замкнута. Например, содер-
жание магния и сульфата (относительно которых раствор не на-
соли
607
сыщен) определяется, с одной стороны, количеством второсте-
пенных солевых минералов, солей и степенью их растворимости,,
а с другой, — влиянием убыли раствора, с которым работает си-
стема. Предположим, что убыль раствора в цикле флотации со-
ставляет 10%, и для восполнения этой убыли на каждую тонну
руды поступает 0,1 т свежей воды. Предположим также, что
магниевые и сульфатные минералы растворимы полностью; из
этого следует, что содержание в пульпе ионов Mg2+ и SO42- в
устойчивом состоянии будет в десять раз больше, чем содержа-
ние в обогащаемой руде. Для руды, содержащей только 1 %
карналлита и 1,5% кизерита, раствор системы содержит 27 г/л
магния, т. е. весьма значительное количество магния. Если по-
тери раствора из системы уменьшить до 5%, то содержание маг-
ния в растворе удвоится; при еще меньших потерях, раствор
может стать насыщенным в отношении магния. Увеличение со-
держания в руде карналлита и кизерита дает такой же эффект,
как и уменьшение убыли раствора из системы.
Флотация галита. Для разделения сильвина и галита было-
предложено два основных метода — флотация галита и флота-
ция сильвина. При галитовой флотации в качестве собирателей
применяют карбоксильные кислоты или их щелочные соли при
добавке в качестве активаторов солеи свинца или висмута. Этот
метод, предложенный в СССР С. А. Кузиным и разработанный
затем в США Вейнигом применяется в течение ряда лет на
-предприятиях Поташ Компани оф Америка в Нью Мексике [72].
Этот процесс подробно изучен также в СССР [54]. Так как боль-
шинство сильвинитовых руд содержит больше галита, чем силь-
вина, то в процессе галитовой флотации подлежит флотации
большая часть руды. Это практически невыгодно, и по мере
снижения содержания сильвина в обрабатываемых рудах усили-
ваются возражения против применения этого процесса. С другой
стороны, в этом случае глина, ангидрит и железосодержащие
частицы также флотируются, что упрощает проведение опера-
ции обогащения, тогда как при процессе сильвиновой флотации
необходимо вначале удалить как можно больше глины, а остав-
шуюся часть ее депрессировать, для того чтобы получить высоко-
качественный концентрат.
Механизм флотации соли выяснен недостаточно, в особенно-
сти не ясны роль свинца и висмута. Андерсон [2] утверждает,
что сильвин возможно отфлотировать от галита теми же собира-
телями, которые применял Вейниг, вызвав обратную селектив-
ную флотацию. Это позволяет до некоторой степени объяснить
действие свинцовой или висмутовой соли. Следует, однако, отме-
1 Советский патент: С. А. Кузин, 47678/1936; американские патенты:
А. Дж. Вейниг, 2105295/1938; 2188933/1940; 2211397/1940.
"608
ГЛАВА 17
тить, что получение таких результатов, каких достиг Андерсон,
возможно лишь при очень высокой концентрации в растворе
ионов магния и сульфата [79].
Вейниг указывает, что присутствие свинца или висмута в со-
ляном растворе, насыщенном NaCl и КО, предотвращает кри-
сталлизацию КС1 на холоду, т. е. эти добавки приводят к пере-
насыщению, к состоянию, которое не развивается в отсутствие
этих солей (американский патент 2211396/1940). Можно пред-
положить, что действие свинцовой и висмутовой соли связано с
образованием комплексных анионов [таких, как (РЬС14)2- и
(BiCl5)2~] и с избирательной адсорбцией этих комплексов на по-
верхности хлористого натрия. Казаков [44] предложил для раз-
деления галита и сильвина применять ионы алюминия вместо
ионов свинца.
Совсем недавно Вейниг запатентовал [93] другой класс соби-
рателей для флотации галита, состоящий из веществ, дающих
в растворе ион 2,4-дихлорфенолоксиуксусной кислоты. В каче-
стве собирателей для галита этих веществ, по-видимому, доста-
точно, хотя их можно применять совместно с жирными кислота-
ми, мылами или резинатами. Свинцовые или висмутовые соли
помогают при флотации этими реагентами. Однако добавлять их
не обязательно.
Другое интересное наблюдение было сделано Атвудом и Бур-
ном [8], которые показали, что соли алкилсульфатов с длинной
цепью (например, первичный натрийлаурилсульфат) флотиру-
ют сильвин лучше, чем галит, если в растворе отсутствуют соли
свинца, но это преимущество исчезает при наличии указанных
солей.
Флотация сильвина. Другим основным методом обогащения
сильвинитовых руд является флотация калиевого минерала. Для
этой цели предложены две противоположные группы собирате-
лей. Одна из этих групп представлена щелочными солями алкил-
сульфатов [20, 37] (например, натрийлаурилсульфат), другая —
солями алкиламинов [47]. В первой группе собирателей углево-
дородная цепь входит в состав аниона, имеющего сульфатную
группу, во втором классе собирателей углеводородная цепь
входит в состав катиона, имеющего аминную группу. Оба вида
собирателей лучше действуют при длинных углеводородных це-
почках, обычно содержащих от 8 до 18 атомов углерода.
Удивительно, что два таких изоморфных минерала, как
сильвин и галит, можно разделить флотацией. Следует отметить
еще более поразительное явление, что лучшее разделение этих
минералов, которые отличаются только катионом, достигается
с собирателями, имеющими активный катион. Единственное
объяснение этому явлению заключается в том, что ион амина
замещает ион калия на поверхности сильвина и не замещает
соли
609
иона натрия на поверхности галита. Превосходное доказатель-
ство этого предположения, проверенное на многих других га-
лоидных минералах, было сделано Д. В. Фюрстенау и М. С. Фюр-
стенау [29]. Оно согласуется с тем, что хотя калий и натрий и
являются изоморфными, но КС] и NaCl не образуют смешанных
кристаллов. Обе группы собирателей (в особенности амины)
имеют тот недостаток, что поглощаются глиной и другими шла-
мистыми нерастворимыми примесями, причем эти примеси стано-
вятся флотационно активными. Поэтому при сильвиновой флота-
ции широко применяется обесшламливание [86]. Но это лишь ча-
стично помогает решить вопрос, так как вместе со шламами уда-
ляется нерастворенный сильвин и значительное количество
растворенного КС1. В практике из шламов выщелачивается
твердый сильвин и из соляного раствора выделяются шламы
пустой породы и шламы галита. Подробнее эта операция рас-
сматривается ниже.
Обесшламливание не обеспечивает полного решения вопроса
и по другим причинам, в частности, потому, что некоторое коли-
чество шламов неизбежно остается в песках классификатора.
Даже если больше 90% шламов удалено из пульпы в сливе клас-
сификатора, остаток все же поступает в питание флотации. Это
можно исправить, применив двойную промывку руды раствором,
но на практике обычно не идут по этому пути и вместо промыв-
ки добавляют реагенты, воздействующие на нерастворимые
шламы, уменьшая адсорбцию амина этими шламами К таким
веществам относятся гидролизованный или каустицированный
крахмал, декстрин, производные лигнина, целлюлоза, протеины,
дегидратированный крахмал, манногалактановые смолы, целлю-
лозные ксантогенаты. Большей частью предпочитаются модифи-
цированные крахмалы и манногалактановые смолы, потому что
они не только препятствуют поглощению аминов глинистыми
частицами, но также и флокулируют их. Это облегчает удаление
из раствора глинистых частиц.
Многие депрессоры пустой породы являются веществами,
состоящими из больших молекул, обильно насыщенных гидрок-
силами, которые и придают депрессорам гидрофилизируюшие
свойства. Они представляют класс веществ, рассматриваемых в
коллоидной химии в качестве защитных коллоидов. Можно
лишь предположить, что эти вещества образуют такие же проч-
ные водородные связи с минералами, как и с водой, и если
должным образом контролировать загрузку депрессоров, то они
1 Американские патенты: Дж. Е. Атвуд и Д. Дж. Бурн, 2696912/1954;
Дж. А. Барр и Ф. X. Бунте, 2695106/1954; Е Н. Браун и Т. А. Сесил, 2699255/
1955; А. Т. Коль, 2355365/1944, 2365805/1944; А. Т. Коль, Дж. Б. Дьюк и
К. Ф. Шиллинг, 2322789/1943; А. Т. Коль и В. М. Хаумтон, 2364520/1944;
Ф. X. Тартарон, А. Т. Коль и Дж. Б. Дьюк, 2288497/1942.
39 А. М. Годэн
610
ГЛАВА 17
«защищают» пустую породу, в то время как их концентрации
еще недостаточно, чтобы покрыть в той же степени сильвин, ко-
торый вместо этого покрывается собирателем.
Собирательное действие разных солей аминов в отношении
сильвина проявляется различно, в зависимости от структуры
аминов, значения pH и длины углеводородной цепи. Эта область
почти полностью не исследована. В различных патентах утвер-
ждается, что углеводородная цепь собирателей должна иметь от
7 до 18—20 атомов углерода; также отмечается, что первичные
амины предпочтительнее вторичных или третичных, что четвер-
тичные аммониевые соли непригодны в качестве собирателей и
что алифатические соединения предпочтительнее ароматических
или гетероциклических. Однако систематического изучения ука-
занных явлений не проводится.
Атвуд и его сотрудники нашли, что имеется связь между тем-
пературой раствора и действием первичных алифатических ами-
нов, оптимум которого отвечает определенной температуре.'
Обычно при более низкой температуре среды приходится при-
менять амины с большей степенью ненасыщенности. Поэтому
полагают, что при сезонном изменении температуры раствора
необходимо переходить от одного вида амина к другому или под-
держивать температуру пульпы постоянной [92]. В соответствии
с температурным изменением растворимости КС1 можно предпо-
ложить, что флотируемость сильвина в растворе, температура
которого повышается, и в растворе, температура которого пони-
жается, будет различна. Это предположение является всего
лишь гипотезой.
Процесс выщелачивания. При флотации галита или сильвина
конечными продуктами процесса являются концентрат и хвосты
(или концентрат, хвосты и шламы), увлажненные раствором, со-
держашим хлориды калия и натрия. Предположим, что кек с
фильтров содержит 10% раствора и что раствор имеет темпера-
туру 20°. Тогда можно подсчитать, что вес растворенного КС1 в
фабричных продуктах составляет 10,4 кг/т руды [24а]. Если в
руде содержится 20% КС1 и соотношение между хвостами и кон-
центратом равно 80 : 20 и раствор, остающийся в кеке между
частицами, пропорционально распределен между хвостами и
сильвиновым концентратом (все предположения приемлемы), то
потери КС1 в растворенном виде во влажных хвостах составят
8,3 кг/т руды, или 4% от содержания калия в руде.
В конце процесса влажный сильвиновый концентрат подвер-
гается сушке. Растворенный хлористый натрий, содержащийся
во влаге, понижает качество концентрата. В примере, приведен-
ном в предыдущем абзаце, содержание хлористого натрия в вы-
сушенном калиевом концентрате (за счет NaCl, находящегося
СОЛИ 611
во влаге кека) составляет 20,8 кг/т, что повышает содержание
NaCl в конечном сильвиновом концентрате на 2%.
Чтобы свести к минимуму эти недостатки, а также обеспе-
чить возвращение в процесс растворов, удаляемых с концен-
тратом и хвостами, целесообразно применить промывку продук-
тов. Это особенно важно для снижения потерь растворенного
калия. В зависимости от количества свежей или почти свежей
воды, применяемой для промывки кека фильтров, можно полу-
чить более или менее полную отмывку его от раствора. Обычно
расход промывной воды составляет 30—90% от количества рас-
твора, оставшегося в порах кека. Эффективность отмывки кека,
т. е. отношение фактического удаления к теоретически возможно-
му удалению исходного раствора может составлять 75—90% и
выше и зависит от структуры кека. Сочетание этих двух факто-
ров— количества применяемой воды и качества кека — опреде-
ляет фактическую эффективность отмывки.
Обычно кроме концентрата и хвостов выделяют шламовый
продукт, из которого нерастворенный хлористый калий должен
быть удален выщелачиванием. Легко понять, что для поддержа-
ния постоянства состава соляного раствора рационально приме-
нять для выщелачивания хлористого калия воду после промывки
концентрата и хвостов, даже если промывка была проведена с
эффективностью 100%. Нет необходимости использовать свежую
воду для извлечения хлористого калия.
Для выщелачивания шламов добавляется вода, которая под-
лежит впоследствии выпариванию. Возможно также применять
процесс выщелачивания при нагревании с последующей кри-
сталлизацией при охлаждении. Этот процесс служит для извле-
чения калия из его солей больше столетия. Он подробно описан
в европейской технической литературе, главным образом в не-
мецкой [23, 49, 74, 80, 95], хотя краткие упоминания о нем имеют-
ся и в английской [11, 12]. Этот процесс основан на том, что на-
сыщенный раствор хлористого калия и хлористого натрия содер-
жит повышенное количество калия при возрастании температуры
и повышенное количество натрия при понижении температуры.
Растворимость обеих солей в совместно насыщенном растворе
становится равной при температуре около 75° (см. рис. 1). Про-
цесс протекает следующим образом: руда поступает в холодный
насыщенный раствор и подогревается в нем; при этом происхо-
дит высаживание галита и растворение сильвина. Выщелачива-
ние сильвина при подогреве продолжается до полного его рас-
творения. Выщелоченную руду фильтруют и промывают водой
для удаления раствора; промытый кек является отвальным про-
дуктом. Расход воды при промывке бывает несколько больше,
чем количество удаляемого из системы раствора; в этом случае
избыток воды должен выпариваться в последующей стадии. Го-
39*
R12
ГЛАВА 17
рячий насыщенный раствор ' охлаждается до первоначальной
температуры. При этом осаждается сильвин, который отфиль-
тровывают и высушивают.
Охлаждение выгодно проводить, применяя испарение, так
как это компенсирует избыток воды, добавляемой при промывке
выщелачиваемых остатков и возобновляет насыщение циркули-
рующего раствора галитом, подготовляя его для следующего
цикла.
При применении этого процесса для извлечения относительно
малых количеств сильвиновых шламов, распределенных в очень
большом объеме циркулирующего раствора, достаточно лишь
незначительно повысить температуру раствора. Отсюда ясно,
что всякие посторонние тепловые факторы могут привести к
большим нарушениям в балансе раствора, если не принять спе-
циальных мер предосторожности.
Для растворения многих солей необходимо большое количе-
ство тепла. Хлористый калий и хлористый натрий обладают
‘большой эндотермической теплотой растворения: можно быстро
понизить температуру в сосуде на 10—15° исключительно за
счет простой добавки в воду достаточного количества твердого
хлористого калия с той же температурой, что и вода.
Эти особенности выщелачивания солей, в частности хлористо-
го калия, хорошо известны физико-химикам и производственни-
кам и используются ими при выщелачивании хлористого калия
из сильвинитовых, карналлитовых и кизеритовых руд.
Обширная литература по этому вопросу имеется на немецком
языке. Кейтель [45] изучал скорость растворения сильвина, а
.Сторч и Фрааз [85]— полигалита. Куппер [51—53], Кейтель и
Герлах [46] изучали выщелачивание карналлита, а Фейт [27]
изучал извлечение калия из кизеритовых руд. Виньерон [90] сде-
лал обзор французской калиевой промышленности, а Фульда
[30], Джакоб и Кабидж [40] и Германн [33] — немецкой.
Вейниг предлагает использовать некоторые факторы выще-
лачивания при флотации сильвина Г
Интересно предложение Колин и Бертон [22], направленное
на уменьшение потерь растворенного калия в галитовых продук-
тах. Они предложили применить раствор с предварительно по-
вышенным содержанием магния. Такой раствор должен полу-
чаться естественно, если руда содержит достаточно карналлита;
в этом случае раствор насыщается магнием, так же как натрием
и калием. Такой раствор при той же температуре содержит на-
много меньше КС1, чем раствор, не содержащий магния. Таким
1 Американские патенты: Артур Дж. Вейниг, 2105294/1938; 2188932/1940;
2211396/1940; 2652150/1953; 2672236/1954.
соли
613
образом, при всех прочих равных условиях потери калия в рас-
творенном виде значительно уменьшаются.
Промышленная схема калиевой флотации. На рис. 2 приве-
дена схема калиевой флотационной фабрики. В действительно--
сти это объединенная флотационная фабрика с фабрикой вы-:
щелачивания, где шламы отделяются от песков, а затем выщела-
чиваются, пески же флотируются. Предполагается, что при ра-;
боте по этой схеме промывка кека осуществляется достаточно:
Выщелачивание
Обезвоживание
Раствор 25°С
Дешламация
- Реагенты Пески Шламы
Флотация
Галитовыи про- Сильбинитовый
вода Пр^Кт
Осадок
Кристаллизация
Фильтрация
с проуыокой.
Хвосты
в отвал
Фильтрация
С промывкой
Промытый сильви-
матовый продукт
Осадок
, Фильтрация
с промывкой
Осадок
и ,, Охлажденный растбд ,
на
В отвал
Сильвинитовый
продукт
Рис. 2. Схематическое изображение процесса
сильвинита
флотации и выщелачивания
эффективно и флотационный продукт будет удовлетворительного
качества. Если желательно, можно применять промывку хвостов,
для растворения остатков КС] с целью более полного удаления
калия из галитового продукта. Этим методом пользуются на
фабриках, применяющих галитовую флотацию, при которой
нельзя получить такие высококачественные продукты, как. при
сильвиновой флотации. С другой стороны, при этом возможно,
расходование воды для выщелачивания и промывки кека в коли-,
честве, превышающем 100% замещаемого при промывке раство-
ра. Избыток воды удаляется испарением, которое осуществляет-
ся в отдельной операции. Возможны многие варианты основной
схемы обработки руды.
614
ГЛАВА 17
Прочие соли
Хлориды. Внимание исследователей было привлечено к очист-
ке хлористого натрия от растворимых и нерастворимых загряз-
няющих примесей флотационным методом [54, 56]. В этом слу-
чае флотация не может быть отделена от процесса выщелачива-
ния, так как экономичнее применять комбинированный процесс.
Процесс Пылаева [76] состоит в выщелачивании небогатых ка-
лиевых хвостов, полученных в цикле выщелачивания, и кристал-
лизации сильвинитовой руды ограниченным объемом насыщенно-
го раствора поваренной соли. Так как этот раствор не насыщен
хлористым калием, он является растворителем для остатков
хлористого калия. Затем из пульпы флотацией удаляют в пен-
ный продукт глины и другие нерастворимые примеси, а конеч-
ные хвосты после обезвоживания являются готовым галитовым
продуктом.
Имеются некоторые данные о сравнительной флотационной
активности легко растворимых солей, включающих галоиды
щелочных металлов [29, 35, 56]. По-видимому, эта область пред-
ставляет широкое поле для дальнейших исследований, в особен-
ности в связи с распространением собирателей типа аминов.
В своей статье Гийе и Перрон показали, что несомненно воз-
можно разделение различных пар солей (некоторые из них
имеют взаимно обменивающиеся ионы и способны к двойному
разложению в водной среде). Эксперименты были проведены с
карбоновой кислотой (олеиновой) и алкилсульфатом (утинал В,
лаурилсульфокислота). Действие этих двух собирателей было
различно: например, хлористый аммоний флотировался олеино-
вой кислотой преимущественно перед всеми другими солями,
тогда как с утиналом он флотировался лучше, чем хлористый
калий, но хуже, чем сульфат или нитрат калия.
Представляет интерес раннее применение флотации для раз-
деления солей. Так, Несслер и Андрэ флотировали смесь хлори-
стого аммония и нитратов щелочных металлов-1. При некоторых
температурных условиях в смеси солей со взаимно обмениваю-
щимися ионами происходит двойное разложение с получением
смешанного насыщенного раствора:
NH4NO3 + KC1-»NH4C1 + KNO3. (1)
Разделение смеси двух солей, полученных двойным разло-
жением, достигается флотацией хлористого аммония при при-
менении различных жировых веществ.
1 Американские патенты: Фридрих Несслер, 1967897/1934; немецкие па-
тенты: Фридрих Несслер, 567844/1935; 605746/1935; французские патенты: Луи
Андрэ, 769914/1934; 779317/1935; 780290/1935.
соли
615
Совмещение процесса двойного разложения с флотацией ин-
тересно не только как одно из ранних применений флотации для
разделения легко растворимых солей, но и потому, что исследо-
вания Несслера показали возможность контроля и регулирова-
ния температуры при комбинации процесса флотации с дейст-
вием реакции двойного разложения. На рис. 3 приведена схема
процесса Несслера.
Нитрат Хлористый
аммония калий.
I
Реактор
Реагенты —-----------
L Флотация
Пенный продукт каменный
[ продукт
Выщелачивание
Фильтрация с Фильтрация
Вода промывкой с промывкой вода
Раствор
Суиска
Сущ к а
Раствор
Хлористый Нитрат
аммоний калия
Рис. 3. Процесс Несслера
Нитраты. Одним из первых случаев применения флотации
для очистки растворимых солей было обогащение натровой се-
литры, добываемой в Чили. Вначале было предложено удалять
нерастворимые примеси флотацией [14], позднее Джейнпрост
предложил разделять растворимые соли флотацией в насыщен-
ном растворе, но не дал подробного описания процесса. О при-
менении этого процесса в промышленности нет данных. Возмож-
но, что руды были очень непостоянного или сложного состава,
и оказалось более выгодным применить другие процессы.
Чилийская селитра [70] содержит не только азотнокислый
натрий, но также галит, астраханит (гидратированная двойная
соль сульфата натрия и магния), дарапскит (гидратированная
двойная соль нитрата и сульфата натрия), гипс, ангидрит, поли-
галит, тенардит, безводный сульфат натрия и мирабилит
Na2SO4 10Н2О. Когда содержится так много компонентов, на-
сыщенный раствор имеет, вероятно, очень непостоянный состав.
По-видимому, это полностью объясняет, почему результаты фло-;
616
ГЛАВА 17
тации оставались неудовлетворительными, особенно если учесть,
что влияние температуры не было изучено. В частности, необхо-
димо обратить особое внимание на двойную соль натрия — да-
рапскит, которая в изобилии встречается в месторождении чи-
лийской селитры [91].
Сульфаты. Разделение сульфата натрия и хлористого натрия
исследовал Пай [75]. Он является сторонником применения в ка-
честве активаторов для сульфата натрия щелочноземельных ка-
тионов (таких, как кальций), когда применяется жирная кис-
лота.
Сульфат калия относительно флотационно активен при при-
менении алкилсульфатов или аминов в качестве собирателей.
Сульфат калия обычно не является составной частью природных
солей, но он легко образуется при растворении некоторых солей.
Поэтому в ходе процесса может возникнуть необходимость отде-
ления сульфата калия от других солей.
Флотация сульфата кальция, который значительно менее
растворим, чем другие соли, также легко протекает при приме-
нении жирных кислот. Процесс его обогащения рассматривался
в гл. 16.
Было опубликовано сообщение [25] о быстрой и хорошей
флотации лангбейнита (двойной соли сульфата магния и калия)
при применении собирателей, содержащих авирол 80 (алкилсуль-
фат щелочного металла).
Вначале флотацию лангбейнита не применяли в промышлен-
ном масштабе, потому что был предложен более простой процесс
[36]. Этот процесс основывался на меньшей скорости растворения
лангбейнита в чистой воде по сравнению с галитом.
Процесс флотации алунита, основного алюмокалиевого суль-
фата, также разработан достаточно полно [39].
Отделение глазерита, двойного сульфата калия и натрия
KsNa(SO4)2 и шенита K2Mg(SO4)2 • 6Н2О от хлоридов натрия и
калия было осуществлено Кузиным [57, 60, 61], применившим
для этой цели щелочные соли алкилсульфокислоты и сульфо-
нафтеновую кислоту.
Фосфаты. Двойной фосфат натрия и лития Li2NaPO4 образу-
ется во время процесса усыхания рапы озера Сирлес. Это соеди-
нение кристаллизуется одновременно с буркеитом, двойной
солью карбоната и сульфата натрия Nai0(CO3)2(SO4)3. Разде-
ление твердой литиевой соли и буркеита осуществляется горячим
выщелачиванием суспензии, загрязненной буркеитом, при не-
большом расходе свежей воды; в этих условиях по крайней ме-
ре часть буркеита растворяется. После выщелачивания загряз-
няющих примесей литиевый продукт флотируется при опреде-
ленной температуре. Этот процесс описан сравнительно недав-
но [6, 31]. Из описания следует, что тщательная проверка воз-
соли 617
можности непосредственной флотации соли лития из рапы озера
Сирлес будет весьма полезна.
Карбонаты. Щелочные карбонаты и двойные соли карбона-
тов в большом количестве встречаются в высохших соляных
озерах. Карбонатные соли часто перемешаны с галоидными,
сульфатными, боратными и двойными солями.
Среди карбонатов наиболее важным является гидратный
полуторный карбонат трона Na3H(CO3)2 • 2Н2О, встречается
также часто сода Na2CO3 ЮН2О, термонатрит Na2CO3-H2O,
натрокальциевые карбонаты — гейлюссит Na2Ca(CO3)2-5H2O и
пирссонит Na2Ca(CO3)2-2H2O, а также и буркеит. Системати-
ческого исследования флотационных свойств этих минералов,
по-видимому не проводилось за исключением попутных наблю-
дений, связанных с обработкой рапы из озера Сирлес, обогащен-
ной калием и боратами.
Типл [87] установил в 1919 г. на фабрике Трона, что буркеит
обладает хорошей естественной флотируемостью. Мамфорд [68]
на этой же фабрике разработал способ, при котором не только
не используется, но даже подавляется естественная флотируе-
мость солей.
Андрэ сообщил о разделении бикарбоната натрия и хлорис-
того аммония [5], Кузин [55] привел два примера, показывающих
возможность разделения соды и галита. В обоих случаях в
качестве собирателей применялись карбоновые кислоты.
Бораты. Боратов имеется множество. Отчасти это объясня-
ется тем, что строение многих боркислородных полимеров до не-
которой степени сходно со строением кремнекислородных поли-
меров. Гидратированный борат натрия — бура Na2B4O7- ЮН2О—
очень распространенный минерал, часто ассоциированный с
менее гидратированными ортоборатами натрия, такими как кер-
нит Na2B4O7 • 4Н2О. Другим важным минералом является гидра-
тированный борат кальция — колеманит Са2В60ц • 6Н2О. Маг-
ниевые и магниевокальциевые бораты также многочисленны, на-
пример гидроборацит CaMgB6On 6Н2О, борацит Mg-3B70i3Cl;
известны также двойные бораты натрия и калия и боратохло-
риды натрия.
Эти соли бора встречаются в смеси с галитом, троной, со-
дой, сильвином, буркеитом и многими другими минералами,
упомянутыми в этой главе. Таким образом, обеспечивается ши-
рокое поле деятельности для применения различных вариантов
флотационно-гидрометаллургического метода обогащения этих
РУД.
На озере Сирлес насыщенный раствор, заполняющий проме-
жутки в кристаллической массе высохшего озера, перекачива-
ют на рафинировочную фабрику, где получают хлористый ка-
лий и буру [87]. Исходный раствор, соприкасаясь с массивами
618 ГЛАВА 17
соли, очевидно, близок к равновесному при существующей тем- 'j
тературе; массивы соли состоят из галита, троны, буры, глазе-
рита, ханксита 9Na2SO4 • 2Na2CO3KCl и буркеита.
Примерно третью часть исходного раствора составляют рас-
творенные в нем вещества; из них половина приходится на ука- ’’
занные выше соли, из которых 4,7% представлены хлористым
калием и 1,5% — бурой.
Во время интенсивного испарения, необходимого для полу-
чения сухих солей, происходит обильное вспенивание раствора
и в пену увлекается большое количество хлористого калия и ?
буркеита. Чтобы уменьшить потери, Мамфорд предложил [68]
применять вещества, уничтожающие пену. <
Совсем недавно флотационный передел был включен в про-
цесс выщелачивания [71]. В процессе испарения при повышен-
ной температуре может наступить момент, когда сильвин начнет
кристаллизоваться. При продолжении испарения сильвин и бу-
ра кристаллизуются одновременно. В прежней практике испа- '
рение проводили так быстро, что бура, которая медленно крис-
таллизуется, не выпадала из раствора, в то время как сильвин
из него выкристаллизовывался. В новом процессе вначале уда-
ляют основную массу сильвиновых кристаллов, которые обра-
зовались в первой стадии кристаллизационного процесса, затем ,
совместно кристаллизуются обе соли и смесь солей в маточном
растворе разделяют флотацией при пониженной температуре.
Гаррис отметил в американском патенте 2120277, что бура <
флотируется из смесей с сильвином при применении олеиновой
кислоты, ксилола, терпенов, керосина и нафтеновых кислот, а
сильвин — эмулсолом 480.
Отделение буры от галита и тенардита Na2SO4 или мираби-
лита Na2SO4-10H2O описано в СССР двадцать лет назад s
(4, 55, 62, 63]. Кузин показал, что как олеиновая кислота, так и
асидол А могут применяться в качестве собирателей. Асидол,
который является смесью нафтеновых кислот, особенно эффек-
тивен, в частности при разделении буры и галита. Активация
свинцовыми солями нежелательна, так же как и применение ы
жидкого стекла. Кальциевые и цинковые соли в малых количе-
ствах не дают эффекта. Совсем недавно в США было установ-
лено [64], что активация барием при применении карбоксильных *
собирателей дает хороший эффект при флотации буры.
Борная кислота. Удачное решение вопроса обогащения руд,
содержащих бораты, было найдено благодаря высокой естест-
венной флотационной активности борной кислоты. Необходимо
напомнить, что борная кислота после кристаллизации обладает
слоистой структурой, имеющей состав В(ОН)з, с водородными
связями, скрепляющими отдельные группы в слое одну с дру-
гой и с остаточными вандервальсовыми связями между слоями.
соли
619
Флотация борной кислоты не требует собирателя, он даже мо-
жет быть излишним, хотя обычно его все же применяют. Добав-
ка незначительного количества вспенивателя позволяет полу-
чать высококачественный концентрат.
Процессы, предложенные для обогащения борсодержащих
руд в СССР [3, 4, 55], относятся к борацитовой руде (хлорборат
Коле. чанитовая
руда
302 а
----^Реактор»—
ПГ"
вспениватель ——^-Флотация
Пенный продукт
Вода Фильтрация
—°- с промывкой
Камерный продукт
Ч
вода Фильтрация
“ "«»с промывкой
Раствор
Раствор
Борная
кислота
Хвосты в отвал или в
процесс доя получения
супырата кальция
Рис. 4. Процесс Шелтона—Никкербоккера для по-
лучения борной кислоты
магния), а в США [48, 83]—к колеманитовой руде (гидратиро-
ванный борат кальция). Эти процессы, отличающиеся некоторы-
ми существенными деталями, имеют, однако, одну общую зада-
чу—разложение боратов кислотой, образование борной кислоты
и удаление ее флотацией. В процессе Шелтон — Никкербокке-
ра [48] минеральная суспензия, содержащая бораты в растворе,
почти насыщенном рудными компонентами и борной кислотой,
обрабатывается сернистым газом или другой кислотой
так, чтобы образовалась борная кислота, которая затем удаляет-
ся флотацией. При этом также получается сульфат кальция. Схе-
ма этого процесса показана на рис. 4.
ЛИТЕРАТУРА
1. Anderson Carl О., R. J. Stengl, and J. G. Trewartha. U. S.
Patent 2,263,552/1941.
2. Anderson L. D. U. S. Patent 2,046,312/1936; reissue, 21,566/1940.
3. Андреева А. И. СССР. Патент 53,403/1938.
620
ГЛАВА 17
4. Андреева А. И. и С. А. Кузин, флотация борной кислоты и бора-
тов из продуктов обработки индерских борацитов, ЖПХ (СССР), 10, 845—
852, 1937.
5. Andres Louis. French Patent 838,659/1939.
6. Anon: Lithium salt recovery in American Potash’s soda products plant, Mi-
ning World, 7 (10), 25—28, 1945.
7. Atwood, George E. U. S. Patent 2,689,649/1954.
8. A t w о о d. George E., and Douglas J. Bourne. U S. Pa-
tent 2,696,912/1954.
9. Bateman, Alan M. «Economic Mineral Deposits», pp. 176—198, John
Wiley & Sons, Inc., New York, 1942.
10. Batty J. V., R. Havens, and R. R. Wells. Concentration of Colorado
fluorite ores, U. S. Bur. Mines, Rept. Invest. 4139, 1947.
11. Biasdale Walter C. The separation of the chlorides and sulfates of
sodium and potassium by fractional crystallization. J. Ind. Eng. Chem., 10,
347—353, 1918.
12. Biasdale Walter C. Equilibria in Saturated Salt Solutions, Chemi-
cal Catalog Company, Inc., New York, 1927.
13. Booth R. B„ and J. E. Carpenter. U. S. Patent 2,410,770/1946.
14. Broadbridge Walter, Edwin Edser, and William George
Sellers. U. S. Patent 1,439,061/1922.
15. Clemmer J. B„ and С. O. A n d e r s о n. U. S. Patent 2,168,762/1939.
16. Clemmer J. B., and В. H. Clemmons. U. S. Patent 2,407,651/1946.
17. Clemmer J. B., and В. H. Clemmons. U. S. Patent 2,497,863/1950.
18. Clemmer J. B., W. E. Duncan, F. D. De V a n e y, and M. Gug-
genheim. Flotation of Southern Illinois lead-zinc fluorspar ores,
U. S. Bur. Mines, Rept. Invest. 3437, 1939.
19. Clemmer J. B., and R. G. O’Meara. U. S. Patent 2,120,485/1938.
20. С о g h i 11 Will H., F. D. De V a n e y, J. Bruce Clem--
m e r, and S. R. B. Cooke. Concentration of the potash ores of Carlsbad,
New Mexico, by ore dressing methods, U. S. Bur. Mines, Rept. Invest.
3271, 1935.
21. Coghill Will H., and O. Greeman. Flotation of fluorspar ores for
acid spar, U. S. Bur. Mines, Rept. Invest. 2877, 1928.
22. С о I i n Jules, and Robert Berthon. U. S. Patent 2,702,121/1955.
23. Cornec E., and H. Krombach. Equilibres entre le chlorure de potas-
sium, le chlorure de sodium et I’eau depuis —23° C jusqu’ a +190°C, Ann.
chim. (Paris), 18 (10), 4—31, 1932.
24. D’Ans J. «Die Losungsgleichgewichte der Systeme der Salze ozeanischer
Salzablagerungen», (a) p. 66, Verlagsgesellschaft f. Ackerbau, Berlin, 1933.
25. DeVaney F. D., and S.- R. B. Cooke. Flotation of langbeinite from the
potash field of New Mexico and Texas, U. S. Bur. Mines, Rept. Invest.
3300, 1936.
26. Duncan Walter E. Concentrating operations of the Mahoning Mining
Company, Rosiclare, Illinois, AIMME Tech. Publ. 2040, in Mining Technol.,
10, 1946.
27. Feit W. Uber die Darstallung des Chlorkaliums aus Hartsalz, W. Knapp
Verlag, Halle, Germany, 1910.
28. F i n e M. M., and R. G. O’Meara. Laboratory beneficiation of fluorite
ore from the Minerva Oil Company, AIMME Tech. Publ. 2055, in Mining
Technol., 10, 1946.
29. Fuerstenau D. W., and M. C. Fuerstenau. Ionic size in flotation
collection of alkali halides, Trans, AIMME, 205, 302—307, 1956.
30. F u 1 d a Ernst. «Das Kali», pt- II, Ferd. Enke Verlag, Stuttgart, 1928.
31. Gale W. A. Lithium from Searles Lake, Chem. Ind., 57, 442—446, 1945.
32. Gaudin A. M. U. S. Patent 2,231,265/1941.
33. G r a b a u A. W. «Geology of Non-metallic Mineral Deposits Other than
соли
621
Silicates», Principles of salt deposition, McGraw-Hill Book Company,
Inc., New York, 1920.
34 Greeman Omer W., and Henry A. Lilly. U. S. Pa-
tent 1,926,045/1933.
35. Guyer A., and R. P e r r e n. Trennung von wasserloslichen Salzen durch
Flotation, Helv. Chim. Acta, 25, 1179—1187, 1942.
36. H a r 1 e у G. T., and G. E. Atwood. Langbeinite-mining and proces-
sing, J. Ind. Eng. Chem., 39, 43—47, 1947.
37. Harris Benjamin R. U. S. Patent 2,182,845/1939.
38. H ermann, C. «Fortschritte in der Kali Industrie», T. Steinkopf, Leip-
zig, 1927.
39. Ishikawa Tomekichi. Japanese Patent 100,390/1933.
40. Jacob A., and A. Kabitzsch. «Die Gewinnung der Kalisalze und ihre
Anwendung in der Landwirtschaft», Verlagsgesellschaft f. Ackerbau, Ber-
lin, 1929.
41. J а песке E. «Die Entstehung der deutschen Kalisalzlager», Vieweg-
Verlag, Brunswick, Germany, 1923.
42. Jeanprost Charles. French Patent 669,100/1928.
43. Johnstone Sidney J. «Potash», John Murray, London, 1922.
44. Казаков И. И. Флотация сильвинитовых руд при замене нитрата
свинца солями алюминия. Ж. Химич, пром. (СССР), 16 (2), 19—20,
1939.
45. Keitel Н. The rate of dissolution and displacement of sylvine and rock
salt from natural sylvinite and «hard salt», Mitt. Kali-Forsch.-Anstalt
G. m. b. H., 1922, pp. 95—127.
46. Keitel H., and O. Gerlach. Leaching crude carnallite with final mo-
ther-liquors from previous operations Mitt. Kali-Forsch.-Anstalt
,G. m. b. H., 1922, pp. 189—196.
47. Kirby James Emery. U. S. Patent 2,088,325/1937.
48. К n i с к e r b о с к e r. R. G., and F. K- Shelton. Beneficiation of boron
minerals by flotation as boric acid, U. S. Bur. Mines, Rept. Invest, 3525,
pp. 3—13, 1940,
49. Ko p pen Rudolf. Crystallization processes in potassium chloride solu-
tions, Z. anorg. u. allgem. Chem., 228, 169—174, 1936.
50. К r i s c h e Paul. «Das Kali», pt. I, Ferd. Enke Verlag, Stuttgart, 1923.
51. Kiipper A. Specific heat of carnallite and heat of dissolution of carnal-
lite and potassium chloride in leach liquors, Mitt. Kali-Forsch.-Anstalt
G.m.b.H., 1920, pp. 99—111.
52. Kiipper A. Cold leaching of carnallite Mitt. Kali-Forsch.-Anstalt
G.m.b.H., 1922, pp. 30—41.
53. Kiipper A., and B. Althammer. Leaching carnallite to obtain the
usual mother-liquor, Mitt. Kali-Forsch.-Anstalt G.m.b.H., 1924, pp. 1—15.
54. К У з и н С. А. Применение флотационного метода для разделения солей.
ЖПХ (СССР), 7, 1065—1070, 1934.
55. К у з и н С. А. Флотационный метод разделения боратов и борной кис-
лоты от других солей. Ж. Химич, пром. (СССР), 12, 277—279, 1935.
56. К У з и н С. А. Разделение флотацией натриевых и калиевых хлоритов,
сульфатов и нитратов. ЖПХ (СССР), 9 , 818—833, 1936.
57. Кузин С. А. СССР, патент 47,678/1936.
58. Кузин С. А. Разделение флотацией галита от сильвинита, ЖПХ (СССР),
10, 457—469, 1937.
59. Кузин С. А. Флотация Соликамских сильвинитовых руд, Калий, СССР
(1) 17—27, 1937.
60. Кузин С. А. Разделение смеси шенята, галита и сильвина флотацион-
ным методом, ЖПХ (СССР), 12, 836—843, 1939.
61. Кузин С. А. Выделение глазерита флотацией. ЖПХ (СССР), 12, 381—
387, 1939.
622
ГЛАВА 17
62. Кузин С. А. СССР, патент 56,694/1940.
63. Л и б м а н Е. Выделение естественных баратов методом флотации из
вулканических туфов, Ж- Химич, пром. (СССР), И (6), 58—59, 1934.
64. М а 1 о z е m о f f. Plato, Merrill, W. MacAfee, and Miles W.
Kirk. U. S. Patent 2,184,558/1939.
65. Manuel William A., and Harry F. Lewis. Redwood tannin in the
flotation of fluorspar, J. Ind. Eng. Chem., 36 (12), 1169—1171, 1944.
66. M i t c h e 11 David R., H. E. Gross, and H. E. Oehler. Froth flota-
tion of fluorspar, AIMME Tech. Publ. 999, in Mining Technol., 2, 1938.
67. M о s e r K. Investigations of the flotative separation of fluorite from cal-
cite, Berg- u. hiittenmann. Jahrb. montan. Hochschule Leoben, 83, 109—112,
133—136, 1935.
68. Mumford Russell W. U. S. Patent 1,591,725/1925.
69. Ochsenius Carl. «Die Bildung der Steinsalzlager und ihrer Mutter-
laugensalze», С. E. M. Pfeffer, Halle, Germany, 1877.
70. Palache Charles, Harry Berman and Clifford Fronde 1.
«Dana’s System of Mineralogy», 7th ed., vol. II, John Wiley & Sons, Inc.,
New York, 1951.
71. Pearson E. P. U. S. Patent 2,333,334/1943.
72. Pierce R. A., and L. D. Anderson. Potash Company of America, pro-
ducer of sylvite at Carlsbad, Eng. Mining J., 143 (1), 38—41, 1942.
73. PI ante Enid C. The flotation of fluorite, Trans. AIMME, 183, 126—144,
1949.
74. Precht, H., and B. Wittjen. Loslichkeit von Salzgemischen der Salze
der Alkalien und alkalinischen Erden bei verschiedener Temperatur, Ber.
deut. chem. Ges., 14, 1667—1675, 1881.
75. Py e D a vi d J. U. S. Patent 2,310,315/1943.
76. Пыла ев Б. Ф. Новейшие методы очистки хлористого натрия от остат-
ков сильвина. Обогащение руд (СССР) (1) 22—28, 1932.
77. Ralston О. С., and A. G. Stern. Report of the non-metals division,
fiscal year, 1942. U. S. Bur. Mines, Rept. Invest., 3675, 1942.
78. Richards Robert H., and С. E. Locke. Progress in ore dressing
and coal washing in 1925, Mineral Ind., 34, 752—812, 1925.
79. S ch о eld E. A. Testimony, in Civil Action № 1829; Potash Company of
America, Plaintiff;' International Minerals and Chemical Corp., Defendant;.
District Court, N. M. August, 1952.
80. Scholich Kurt. Ternary systems of potassium chloride, sodium chlo-
ride and the chlorides of the bivalent metals, Neues Jahrb. Mineral. Geol.,
43, 251—294, 1920.
81. Schuhmann R., and Brahm Prakash. Effect of activators and ali-
zarin dyes on soap flotation of cassiterite and fluorite, Trans. AIMME,
Tech. Publ. 2827B, in Mining Eng., 187, 601—608, 1950.
82. Shapley C. U. S. Patent 1,689,693/1928.
83. Shelton F. K- U. S. Patent 2,317,413/1943.
84. Shepard N. L. U. S. Patent 2,238,662/1941.
85. Storch H. H., and F. Fraas. A study of the properties of Texas po-
lyhalite pertaining to the extraction of potash, U. S. Bur. Mines, Rept. In-
vest. 3062, 1931.
86. Tartaron .Francis X. U. S. Patent 2,330,158/1943.
87. Teeple John E. «The industrial Development of Searles Lake Brines
with Equilibrium Data», pp. 36—38, Chemical Catalog Company, Inc., New
York, 1929.
88. V a n ’ t Hoff J. H. «Zur Bildung der ozeanischen Salzablagerungen», Vie-
weg-Verlag, Brunswick, vol. I, 1905, vol. II, 1909.
89. V a n’t Hoff J. Fl. «Untersuchungen tiber die Bildungsverhaltnisse der
ozeanischen Salzablagerungen», Akademische Verlagsgesellschaft m.b.H.,
Leipzig, 1912.
СОЛИ 623-
90. Vi g пег on Henri. The Alsatian potash industry, Chem. & Met Eng
24, 655—661, 1921.
91. Vigneron Henri. New process for the separation of salt mixtures
La Nature (3062), 356—357, 1940.
92. We ini g Arthur J. U. S. Patent 2,105,294/1938.
93. Wein ig Arthur J. U. S. Patent 2,652,150/1953.
94. Williams J. C., and O. W. Gfeeman. U. S. Patent 1,785,992/1930.
95. Wils t J. and E. Lange. Heats of solution and dilution of salts from in-
finite dilution to saturation, Z. physik. Chem., 116, 161—214, 1925.
Глава 18
АПОЛЯРНЫЕ МИНЕРАЛЫ
В этой главе рассматривается группа легко флотируемых
неметаллических минералов, включающая серу, графит и угли.
Элементы, входящие в состав этих минералов, находятся в вос-
становленном состоянии. Термин «аполярный» употребляется в
связи с отсутствием у них сродства к воде (предполагается, что
это результат относительного недостатка ионов на поверхности
раздела минерал — вода).
Сера
Элементарная сера встречается во многих вулканических
породах, а также вместе с нефтью в соляных куполах, особенно
в тесной связи с гипсом и ангидритом. Вероятно, сера соляных
куполов является продуктом реакции восстанавливающих аген-
тов, находящихся в нефти и газе (таких, как сероводородные
воды) с сульфатной серой гипса и ангидрита.
По данным опытов Таггарта и сотрудников [59], сера не име-
ет естественной флотируемости. Согласно их исследованиям,
краевой угол на полированной поверхности серы в воде равен
нулю, если только сера свободна от загрязнений углеводоро-
дами. Очевидно, при экспериментах необходимо принять все
меры к тому, чтобы не допустить загрязнения поверхности, так
как известно, что краевой угол (измеренный через воду) на гра-
нице раздела сера — вода — углеводород весьма велик [58].
Однако имеются веские доказательства наличия естествен-
ной флотируемости серы. Даже Таггарт с сотрудниками при-
знают, что при флотации в цилиндрах с предельно низкой кон-
центрацией одного только чистого пенообразователя всплывает
около 80% серы. Исследователи и практики установили, что для
флотации подавляющей части серы достаточно добавить лишь
один пенообразователь. Возможно, что краевой угол на границе
раздела сера — вода — воздух довольно мал, и измерить его
затруднительно. Следует также напомнить хорошо известный
факт, что серный цвет трудно смачивается водой. Это заставля-
ет предполагать, что при применявшейся технике полировки
шлифов для измерений краевых углов частицы полировочной
пудры вдавливались в мягкую серу, и в действительности испы-
АПОЛЯРНЫЕ МИНЕРАЛЫ
625
туемая поверхность представляла механическую смесь серы и
полировочной пудры, причем частицы пудры выступали над по-
верхностью серы. Наконец, следует указать на факторы, обус-
ловленные кристаллической структурой серы. Как известно,
кристаллическая сера состоит из замкнутых колец молекул Ss,
имеющих только остаточные связи между смежными молекула-
ми. На поверхности излома не должны обнажаться первичные
валентные связи, с которыми может взаимодействовать вода
или ионизированные собиратели.
По данным исследователей, углеводородные масла и серо-
углерод прикрепляются к поверхности серы под водой с обра-
зованием больших краевых углов (измеренных через воду).
Обработка пульпы эмульсией этих масляных собирателей су-
щественно увеличивает флотируемость серы и приводит к повы-
шению качества концентрата за счет снижения механического
выноса частиц пустой породы в пену. Сообщают, что величина
pH при флотации серы особого значения не имеет.
Легкая флотируемость серы мало используется на практике.
Это происходит потому, что сера очень легко плавится и может
быть добыта и одновременно сконцентрирована по способу Фра-
ша. При этом процессе перегретый пар накачивается под давле-
нием непосредственно в рудное тело в количествах, достаточных
для образования массы расплавленной серы. Под давлением
пара поток расплавленной серы поднимается вверх по скважи-
нам и затем застывает на поверхности земли1.
Вследствие этого применение флотации серных руд ограни-
чивается месторождениями, для которых не подходит способ
Фраша. При флотации серных руд трудно получать высокока-
чественные концентраты из-за чрезвычайно тонкой вкраплен-
ности серы [42, 54]. В связи с этим представляет интерес предло-
жение Фэрбата [18]. Он предлагает нагревать серную руду выше
температуры плавления серы с таким расчетом, чтобы заставить
частички серы слиться. Затем руду охлаждают (так, чтобы
только застыла сера), измельчают и флотируют при повышенной
температуре. Конечный концентрат освобождают от загрязне-
ний плавлением и фильтрацией в горячем виде; кек фильтров
возвращается в цикл измельчения.
Графит
Графит в основном встречается как продукт метаморфизма
карбонатных пород, образовавшийся благодаря восстановлению
карбонатных минералов и кристаллизации (температура и дав-
ление неизвестны). Минерал обычно ассоциирован с различными
1 Для подъема расплавленной серы используется также аэролифтный
способ. Прим. ред.
40 А, М. Годэн
626
ГЛАВА 18
силикатами и карбонатами; очень часто он образует хорошо
раскристаллизованные агрегаты.
Как известно, графит — первый минерал, для концентрации
которого применена пенная флотация [2]. Он легко прикрепля-
ется к пузырькам воздуха; так же, как и в случае серы, для его
флотации достаточно добавить один пенообразователь. Однако,
если употребляются нерастворимые углеводородные масла, об-
волакивающие поверхность графита, флотация идет быстрее и
концентраты получаются более чистыми. Таггарт с сотрудника-
ми [59] нашли, что на графите в воде или в растворе ксантоге-
ната невозможно измерить краевой угол. Однако в качествен-
ных опытах они установили слабую флотацию.
По своей кристаллической структуре графит идеально под-
ходит для флотации: он образует плоские слои толщиной в один
атом, которые почти не связаны один с другим, в то время как
внутри каждого слоя атомы связаны очень прочно. Жидкие
углеводороды под водой легко прилипают к графиту, образуя
большой краевой угол (измеряемый через воду). Пары углево-
дородов хорошо адсорбируются на графите. Это согласуется с
большой адсорбционной способностью химически чистого угля
[14].
Поверхность графита трудно очистить, так как он легко ад-
сорбирует различные вещества. Однако автор смог получить
синтетический графит в виде кусков, очищенных продолжитель-
ным нагревом при весьма высокой температуре (2500°). Извест-
но, что при такой температуре все органические соединения не-
устойчивы, большинство окислов металлов также улетучивается
Этот графит был весьма флотационно активен и у него не сма-
чивались водой ни наружная поверхность, ни поверхность из-
лома.
Графит чрезвычайно мягок. Поэтому его частицы легко раз-
мазываются на других частицах, присутствующих в пульпе;
между тем крупные частицы графита ценятся более высоко. Это
и вызывает основные затруднения при его флотации. Между
прочим, на свойстве графита оставлять видимую черту на бума-
ге основано применение его для производства карандашей. Так
как поверхность большинства сортов писчей бумаги состоит из
глины, кальция и протеинового клея, то очевидно, что в процес-
се измельчения графит может размазываться на ассоциирован-
ных с ним минералах. По данным тщательных исследований
обогатимости алабамских графитовых сланцев [9, 20], широко
подтвержденным впоследствии [36, 42], в качестве реагентов
предлагается комбинация растворимых пенообразователей и
углеводородных масел в кислой или слабощелочной среде, со-
здаваемой кальцинированной содой или жидким стеклом. По-
лезно применять повторные перечистки концентрата.
АПОЛЯРНЫЕ МИНЕРАЛЫ
627
Алмаз
Структура алмаза дает основание предполагать, что поверх-
ность измола его должна состоять из положительных и отрица-
тельных ионизированных участков большой реакционной спо-
собности. По-видимому, имеющиеся в окружающем простран-
стве ионы будут легко адсорбироваться на алмазе, будь то гид-
рофильные ионы (такие, как ионы гидроксила и водорода) или
ионы, содержащие углеводороды. Ряд авторов указывает на то
[62], что алмазы можно флотировать жирными кислотами и ами-
нами, эффективными для естественно гидрофильных алмазов,
и одними только пенообразователями, которых вполне доста-
точно для естественно гидрофобных образцов. Максимальный
размер зерен, способных к флотации, составляет около 20 меш.
Флотация алмазов родственна отделению их от сопутствую-
щих минералов на жировых столах. В этом процессе алмазные
пески продвигаются по слегка наклонной деке под действием
воды. Деке сообщают возвратно-поступательные боковые сотря-
сения, поверхность ее покрывают вязким жиром. Состав жира
должен соответствовать температуре воды. Алмазы (а также
пирофиллит и тальк) прилипают к вязкому жиру, поэтому по-
лучаются высококачественные концентраты (с большой сте-
пенью' концентрации).
Угли
Уголь [34] представляет собой основное твердое топливо.
По составу его ни в коем случае нельзя отнести к гомогенным
веществам или к одной из форм углерода. Имеется много разно-
видностей угля, начиная от одеревенелого торфяника и кончая
графитизированным антрацитом. Даже когда отсутствуют по-
сторонние примеси (такие, как сланцы, глина, гипс, марказит и
пирит), состав угля различен не только в разных шахтах, но да-
же в разных кусках.
Все угли содержат, кроме углерода, также в существенных
количествах водород, кислород, азот и серу. Углерод представ-
ляет собой основной компонент угля (и по весу и по числу ато-
мов); водород обязательно входит .в состав угля. Кислород так-
же является весьма существенным компонентом. Сера и азот
могут и не входить в состав угля, но в небольших количествах
всегда содержатся в нем.
Табл. 1 дает представление о содержании в типичных углях
углерода, водорода, кислорода, азота, и серы. Содержание при-
ведено в обычных весовых процентах, а также в виде числа ато-
мов каждого элемента на 100 атомов углерода.
Для сравнения в табл. 2 приводятся атомные отношения раз-
личных органических веществ.
40*
628
Г ЛА В А 18
Таблица 1
Состав типичных углей
Тип Содержание в обеззоленном угле, %
с н о N S
Антрацит 94,5 3 1,25 0,75 0,5
Полубитуминозный .... 91,5 5 1,75 1,25 0,5
Битуминозный А 89 5,5 3,5 1,5 0,5
Битуминозный В 84,5 5,5 8,0 1,5 0,5
Суббитуминознын 79,5 5,5 12,75 1,75 0,5
Лигнит 73,5 5,3 19,0 1,7 0,5
Число атомов каждого элемента на 100 углерода атомов
Антрацит 38 1,0 0,6 0,2
Полубитуминозный .... — 65 1,4 1,2 0,2
Битуминозный А — 74 3,0 1,5 0,2
Битуминозный В __ 78 7,1 1,6 0,2
Суббитуминозный — 83 12 2,0 0,2
Лигнит — 86 19 2,0 0,2
Таблица 2
Атомный состав типичных органических веществ
Число атомов на 100 атомов углерода
Н О N S
Графит 0 0 0 0
Битуминозный уголь 75 6 1,5 0,2
Нафталин С|0Н8 80 0 0 0
Антрацен СцНю 72 0 0 0
Пирен С вН10 63 0 0 0
Нафтол С10Н7ОН 80 10 0 0
Антрахинон СИН8О2 57 14 0 0
Антрон С|4Н10О 72 8 0 0
Бензатрон С17НюО 59 6 0 0
Нгфтиламин CioH7NH2 90 0 10 0
Пипантгитон C9H14O2N 48 7 3 0
Тиодифениламин Cl2HeNS 75 0 8 8
Минералы угля [3, 30, 51]. Даже невооруженным глазом мож-
но установить гетерогенный характер углей. Обычно видны раз-
личного размера включения сланца и пирита совместно со скоп-
лениями блестящего и матового угля. Под микроскопом можно
АПОЛЯРНЫЕ МИНЕРАЛЫ
629
заметить присутствие каолина и прочих глин, гипса, хлорита,
слюды, кальцита, сидерита и различных других минералов.
Обнаружено, что угольному веществу присущи несколько ком-
понентов.
В США [60, 61] различаются три угольных минерала, а имен-
но: ископаемый древесный уголь, или фюзен, углефицированное
дерево, или антраксилон, и обломочная смесь, или аттрит. Вы-
делено два вида аттрита — полупрозрачный и непрозрачный.
В Европе [39, 40, 55] различают четыре угольных компонента,
а именно: фюзен, витрен, кларен и дюрен, которые более или
менее точно соответствуют американским фюзену, антраксило-
ну, полупрозрачному аттриту и непрозрачному аттриту.
Фюзен. Фюзен по существу является ископаемым древесным
углем. Хотя просвечивание лучами Рентгена не дает доказа-
тельства кристаллической структуры фюзена, однако по рассе-
янию лучей он напоминает графит. Так как фюзен весьма по-
рист, то в нем очень велико содержание негорючих примесей,
в основном механически абсорбированных и частично несом-
ненно адсорбированных. Микроскопическое исследование поли-
рованных шлифов фюзена показывает наличие участков с от-
носительно большой отражательной способностью, перемежива-
ющихся с участками с очень низкой отражательной способно-
стью. Это особенно заметно при небольшом увеличении в масля-
ной иммерсии, выявляющей структуру, пористость и гетероген-
ный характер фюзена.
На рис. 1 показаны фотографии двух образцов фюзена, вы-
бранные из числа многих превосходных фотографий книги Ста-
ча [52].
Антраксилон. Антраксилон [53] представляет собой псевдо-
морфоз угольного вещества по древесной ткани. Он состоит из
значительно полимеризованных атомов углерода, водорода и ки-
слорода. Из всех компонентов угля он меньше всего загрязнен
золой, содержание которой в нем может быть ниже О,5°/о. Антра-
ксилон можно рассматривать как пластическое образование,
полученное в результате соответствующего уплотнения атомов,
составляющих лигнин, целлюлозу и протоплазму первичных
растений после значительной перегруппировки, обязанной ча-
стично микробиологической деятельности в анаэробных услови-
ях. При уплотнении происходило выделение воды, затем дву-
окиси углерода и образование других связей (полимеризован-
ных кольцевых структур, а возможно, кетоэнольных) между
карбоксильными радикалами и аминогруппами.
Если выделить и проанализировать антраксилон из различ-
ных углей, то можно установить, что относительный избыток во-
дорода и кислорода по отношению к углероду уменьшается при
переходе от низшего ряда к высшему; наименьшая величина от-
630
ГЛАВА 18
ношения преимущественно у антрацита. Это отражает измене-
ния, которые произошли и, по-видимому, еще происходят в уг-
лях в месторождениях под действием умеренного давления и
умеренно повышенной температуры, постоянно изменяющих
антраксилон за счет удаления воды и двуокиси углерода и об-
разования значительно уплотненных полимеров. Это видно из
табл. 3, в которой приводятся данные о витренах Великобрита-
нии, заимствованные у Хортона [29].
. Рис. 1. Фюзен. Х300 [52]:
а—остаточная структура дерева; б—раздробленная структура дерева
Согласно Хортону, лучшие коксующие свойства имеют витре-
ны промежуточного состава (примерно такие, как витрены ме-
сторождения Танфилд Ли).
Под микроскопом в отраженном свете антраксилон кажется
гладким, с отражательной способностью, низкой по сравнению
с минералами тяжелых металлрв и высокой по сравнению с
большинством силикатов и других компонентов угля (исключая
блестящие части фюзена). На рис. 2 показаны два типичных об-
разца антраксилона в масляной иммерсии. Внешний вид одного
из образцов ясно свидетельствует о его происхождении в резуль-
тате псевдоморфоза по растительной ткани.
АПОЛЯРНЫЕ МИНЕРАЛЫ
631
Таблица 3
Состав типичных витренов из различных углей Великобритании
[29]
Месторождение Зола % Содержание в обезвоженном угле, %
с н О N S
Аллертон Байуотер 0,6 80 5,2 11,8 2,0 1,0
Теаркрофт 0,4 83,5 5,3 8,5 1,7 1,0
Танфилд Ли 0,4 89 5,2 3.0 1,7 0,7
Кумгурач 0,3 91,5 4,0 1,8 1,6 0,8
Нью-Кросс Хэнде 0,5 93 3,2 1,4 1,2 0,7
Число атомов на каждые углерода 00 атомов
Аллертон Байуотер 78 И 2,1 0,5
Теаркрофт — — 76 7,6 1,7 0,4
Танфилд Ли — — 72 2,6 1,6 0,3
Кумгурач — — 52 1,5 1,5 о,з
Нью-Крисс Хэнде — — 41 1,1 1,1 0,3
Соответственно изменениям в составе от весьма накислоро-
женных витренов к низкокислородным и низководородным фор-
мам отражательная способность антраксилона в масле умень-
шается от максимума 4,2% до минимума 0,54% (у цинковой об-
манки 5,1%). Некоторые исследователи [41, 50] указывают, что
отражательная способность витренов изменяется не постепенно
между указанными пределами, а скачками. Это безусловно при-
водит к выводу, что витрены представляют собой хотя родствен-
ные, но вполне определенные химические виды, что не вполне
согласуется с другими представлениями, рассматривающими ме-
тод образования этого основного угольного вещества.
В последние годы было уделено большое внимание выясне-
нию молекулярного состава полимеров антраксилона. В общем
теперь предполагается, что основное значение в последних ста-
диях процесса углефикации имела высокая температура, а не
давление, тогда как в ранних стадиях важную роль играли бак-
терии. Хотя некоторые исследователи считают, что существуют
отдельные молекулы угля [23], большинство рассматривает ан-
траксилон как смесь различных фракций (альфа, гамма и т. д.),
по меньшей мере одна из которых представляет собой накрест
связанный полимер, похожий в общих чертах на некоторые со-
временные синтетические пластмассы, в то время как другая мо-
632
ГЛАВА 18
жет состоять из больших устойчивых слоистых многоатомных
ароматических молекул [47].
Аттриты. По своему составу аттриты довольно различны. В
общем имеется две разновидности аттритов — полупрозрачная
Рис. 2. Антраксилон. X 300 [52]:
а — бесструктурный гель с трещинами сжатия после нагрева до 360°; б — видна струк-
тура дерева
и непрозрачная. В полупрозрачном аттрите, или кларене, бота-
нической основой являются споры, кора и водоросли или внеш-
ние оболочки спор или микроспор. Этот аттрит имеет низкое со-
держание золы (от 1 до 2°/0). Он образует огромные толщи кен-
нельских углей. По составу кларен подобен витрену, только в
первом несколько повышено содержание азота и серы, что
рассматривается как результат относительно высокого содержа-
ния протеина первичных спор и микроспор, остатки которых име-
ются в изобилии в кларене.
Непрозрачный аттрит (дюрен) образовался из веток, кусков
коры, листьев, стеблей, мха, водорослей, семян, перемешанных
с глиной, железистым материалом, зернистыми обломками не-
растворимых минералов и из бактерий, отложившихся в воде.
АПОЛЯРНЫЕ МИНЕРАЛЫ
633
Непрозрачный аттрит содержит много золы (от 10 до 15%) и
образует скопления матового угля.
На рис. 3 представлены фотографии двух погруженных в
масло шлифов типичного матового угля, почти целиком состоя-
щего из аттрита. Видна сложная структура и тонкая вкраплен-
ность компонентов аттрита.
Рис. 3. Аттрит. Х300 [52]:
а — виден разрез листьев с кутикулями; б — склероции, пыльца и обломки
Несмотря на различие в оптических и коксующих свойствах
различных видов аттрита и антраксилона имеется сходство меж-
ду органической частью этих компонентов любых углей. Счита-
ется, что полимер С—Н—О, пропитывающий уголь, представляет
собой одно и то же вещество, независимо от того, является уголь-
ное вещество замещением типа псевдоморфоза по дереву (ан-
траксилон), или оно находится в виде мелких кусочков органи-
ческого материала (аттрит). Отсюда можно сделать вывод, что
различие в свойствах разных кусков одного и того же угля опре-
деляется соотношением и родом остатков, вкрапленных в поли-
мер. В то же время различия в частицах разных углей сходного
петрографического характера определяются составом самого по-
лимера, а именно, степенью обескислороживания и дегидроге-
«34
ГЛАВА 18
низации, происходивших в процессе природного углеобразова-
ния.
Содержание золы в компонентах угля. Различные компонен-
ты угля весьма отличаются по составу содержащейся в них золы.
Это видно из табл. 4, в которой приводятся данные для типич-
ных битуминозных углей 1331.
Таблица 4
Свойства золы из различных составляющих типичного битуминозного
угля [33]
Составляющие Содержание золы % Содержание в золе, %
растворимых в воде не раствори- мых в воде, растворимых в НС1 не раствори- мых в НС1
Блестящий уголь:
витрен 1,1 70 20 10
кларен ^Матовый уголь: 1,2 65 18 17
дюрен 6,2 3 24 73
фюзен 15,6 17 71 13
Фюзен в основном содержит известь и сульфат; витрен и кла-
рен — щелочи, алюминий и сульфат; дюрен — алюминий и крем-
ний. Представляется, что зола в витрене и кларене возникает
преимущественно из материнского растительного материала, то-
гда как в дюрене она представляет механическую примесь, вне-
сенную в процессе отложения, в фюзене же она была осаждена
из раствора благодаря его пористости. Типичные содержания
золы в компонентах очищенного угля следующие: 1 % — витрен,
1,5%—кларен, 7%—дюрен, 15% — фюзен.
В действительности угли содержат золы гораздо больше,
чем чистые компоненты, описанные в предыдущих параграфах.
Так, например, хороший битуминозный уголь может содер-
жать от 10 до 15% золы. Часть загрязнений, увеличивающих
зольность угля, представлена сланцем или другими составляю-
щими горных пород, часть — пиритом или марказитом, а
часть — глиной. Удаление большей части этой золы обычно
является задачей обогащения угля.
Смолы и воски в углях. Угли содержат в подчиненных коли-
чествах смолы и воски, часть из которых встречается в виде
отдельных включений, видимых под микроскопом. Эти вещест-
ва могут быть экстрагированы органическими растворителями,
обычно при умеренной температуре. В качестве таких раство-
рителей наиболее пригодны фенол, бензол, спирт, тетралин (те-
АПОЛЯРНЫЕ МИНЕРАЛЫ
635
трагенизированный нафталин) и пиридин. Горный воск добы-
вается из немецких бурых углей экстракцией тетралином. Он
подобен пчелиному воску и содержит в основном сложные эфи-
ры высокомолекулярных жирных кислот и спиртов.
Коксование. Антраксилон является коксующимся компонен-
том угля. Его коксующие свойства в значительной степени за-
висят от относительного содержания в нем кислорода и водо-
рода. Процесс карбонизации угля для получения кокса заклю-
чается в нагревании угля до температуры, достаточно высокой
для разложения полимера антраксилона и образования новых
связей с выделением таких газов, как пар, окись углерода,
азотсодержащие органические составляющие, фенольные орга-
нические составляющие, аммиак и пр. Для получения кокса с
желаемыми механическими свойствами газовыделяющая часть
коксуемого угля должна иметь некоторую текучесть, достаточ-
ную вязкость при определенной температуре и выделять необ-
ходимый объем газа с соответствующей скоростью.
Так как в антраксилоне изменяются соотношения Н : С и
О : С, качества коксующихся компонентов углей должны раз-
личаться, и действительно они колеблются в широких пре-
делах.
Аттрит типа кларена имеет довольно хорошие коксующие
свойства, но дюрен в общем не подходит для коксования, его
применяют только в виде добавки для углей, антраксилон ко-
торых при коксовании становится слишком текучим. Некоксую-
щий характер дюрена, возможно, обусловлен тем, что слипа-
нию оксигидрокарбоновых частиц препятствуют микроскопи-
ческие и субмикроскопические составляющие, которые заполня-
ют промежутки между этими частицами.
Фюзен не коксуется.
Частицы размером 200 меш, которые в угольной промыш-
ленности рассматриваются как чрезвычайно тонкие, с точки
зрения флотации недостаточно измельчены, так как большей
частью являются сростками. Наиболее чисты частицы антра-
ксилона, которые по существу представляют собой кусочки по-
лимера С — О — Н. Частицы аттрита также являются кусоч-
ками полимера С — О — Н, но с включениями солей натрия и
кальция совместно с каолином и другими силикатами. Части-
цы фюзена представляют собой микрокристаллический амор-
фный углерод (почти графитового типа), ассоциированный с пи-
ритом, гипсом, кальцитом, коллоидальными силикатными и
глинистыми минералами.
Природные флотационные свойства угольных минералов.
Антраксилон имеет естественную флотируемость. Краевые уг-
лы образуются при отсутствии собирателя, особенно на витре-
не, имеющем состав, приблизительно соответствующий опти-
636
ГЛАВА 18
мальным коксующим свойствам. Вообще говоря, краевой угол
зависит от типа угля. Хорсли и Смит 128] сообщают, что крае-
вой угол за короткий период контакта составляет 45° для ант-
рацита, 50° для полубитуминозного угля, 60° для битуминозно-
го, 58° для парабитуминозного, 27° для металигнозного, 10° для
ортолигнозного и 0° для лигнина. Эти данные в основном со-
гласуются с данными Брадо и Гаугера 171.
Краевой угол и флотируемость являются также функцией
содержания золы в угольных минералах и убывают в порядке:
витрен, кларен, дюрен, фюзен. Поэтому представляется воз-
можным отделить угольные минералы не только от породы, но
и один от другого в последовательности: антраксилон, аттрит,
фюзен.
Особенности флотации углей. В связи с применением фло-
тации для обогащения угля полезно вспомнить, что удельный
вес флотируемых компонентов относительно очень низок (ант-
раксилона от 1,25 до 1,30), благодаря чему требуемая подъем-
ная сила пузырьков гораздо меньше, чем обычно. Поэтому
флотации может подвергаться значительно более крупный ма-
териал, чем в случае металлсодержащих руд'. Кроме того,
следует ожидать, что затруднения, вызываемые наличием шла-
мов при флотации углей, должны распространяться на более
крупные частицы, чем при флотации металлсодержащих руд.
На важное значение плотности пульпы, размера частиц и рав-
номерности питания указывал Кроуфорд [12].
Необходимо также иметь в виду, что флотация не исключа-
ет гравитационного обогащения углей. Уголь настолько легко
обогащается гравитацией (по сравнению с другими минерала-
ми) и настолько очевидна промышленная ценность крупного
угля, что на флотацию направляется только мелочь, образо-
вавшаяся при отбойке угля и в некоторых случаях при додраб-
ливании промежуточных продуктов или глинистого угля. Ма-
териал такого типа имеет иное распределение компонентов,
чем выданный из шахты уголь: он содержит больше сланца,
глины, гипса, пирита, фюзена и аттрита, т. е. всех нежелатель-
ных компонентов. Поэтому с помощью коллективной флотации
часто затруднительно получить продукт, сравнимый по качест-
ву с гравитационным концентратом, состоящим из крупного
материала. Селективная флотация дает лучшие результаты, но
ценой снижения извлечения.
Важное значение имеет способность угля, взвешенного в
воде, окисляться. Хорошо известно, что продолжительное сус-
1 Сун и Циммерман [57] обращают внимание на то, что при флотации уг-
лей частицы настолько велики, что несколько пузырьков скорее могут при-
крепиться к одной частице, чем много частиц к одному пузырьку, как эта
обычно наблюдается при флотации металлсодержащих руд.
АПОЛЯРНЫЕ МИНЕРАЛЫ
637
пендирование угля в воде уменьшает его флотируемость, а
складирование сухого угля увеличивает его флотируемость,
возможно, за счет адсорбции способствующих флотации моле-
кул, переходящих от одних частиц к другим через газовую фа-
зу. Поэтому при извлечении угля из пыли и шламов приходит-
ся сталкиваться с различными трудностями [131.
Важно также и то, что объем частиц, закрепляющихся на
пузырьках воздуха, относительно велик. Это объясняется не
только тем, что флотируемый минерал составляет основную
часть исходного продукта (50—80%), но и тем, что он являет-
ся минералом с наименьшим удельным весом, тогда как при
обогащении металлсодержащих руд флотируемый компонент
представляет подчиненную часть питания (часто менее 5%) и
имеет наибольший удельный вес. Кроме того, флотируемые ча-
стицы угля значительно крупнее, чем материал, флотируемый
из металлосодержащих руд. Отсюда следует, что для флотации
углей необходимо изменить конструкцию флотационных
машин.
Масляные собиратели. Хотя антраксилон (и в меньшей сте-
пени аттрит и фюзен) может флотироваться с добавкой одного
воднорастворимого пенообразователя, применение масляных
собирателей, обволакивающих поверхность частиц угля, пред-
ставляет определенные преимущества, по крайней мере для
коллективной флотации [25, 591. При этом не только увеличи-
вается краевой угол, но и уменьшается число флотируемых ча-
стиц за счет образования агрегатов частиц угля. Благодаря
этому значительно возрастает скорость флотации, уменьшается
влажность пены и снижается механический вынос нефлоти-
руемых минералов в концентрат. Все это дает большие пре-
имущества [8, 22, 49].
По мере увеличения расхода углеводородных масел плен-
ка, окружающая частицы, все больше утолщается, и постепен-
но воздух вытесняется из агрегатов уголь — масло. Это явле-
ние представляет собой известное приближение к условиям
процесса Тренти, при котором загрязненный угольный шлам
перемешивается с относительно большим количеством масла.
Короче говоря, желательно употреблять возможно меньшую
загрузку масел-собирателей. Небольшое количество масла мо-
жет дать эффект, если оно перед употреблением эмульгируется
в воде. Это эмульгирование хорошо осуществляется при добав-
ке небольшого количества поверхностно активного вещества
(например, лиссополь N).
Применение ксантогената при флотации углей. Хотя ксан-
тогенат был рекомендован для флотации угля, в настоящее
время установлено, что эффект при его использовании полу-
чается благодаря маслянистым продуктам разложения ксан-
638
ГЛАВА 18
тогената, образовавшимся до (или во время) флотации. Одна-
ко не доказано, что эти маслянистые продукты имеют какое-
либо преимущество перед гораздо более дешевыми керосином
или нейтральными маслами, полученными в качестве побоч-
ного продукта при коксовании [27].
Реагенты для флотации углей. Так как уголь является деше-
вым продуктом, то весьма желательно, чтобы применяемые
реагенты также были дешевыми. В качестве пенообразователей
предпочтительны спирты, терпинеол, сосновое масло и наибо-
лее желательна крезиловая кислота [63] (побочный продукт
производства кокса) и особенно промывные растворы после
промывки побочных продуктов коксования углей [31]. Сообща-
ют, что в штате Незерландс основные реагенты—это растворы
из аммиачных башен коксовых заводов, являющиеся отходами
производства. Эти реагенты, содержащие около 0,04% смоля-
ных кислот и 0,03% нейтральных масел, применяются в коли-
честве 1,3 т на 1 т руды [211.
Там, где нефтяные продукты недороги, подходящим масля-
ным собирателем является керосин, который может применять-
ся совместно с крезиловой кислотой или некоторыми промыв-
ными водами коксовых заводов.
Если пирит загрязняет концентрат, его флотируемость мож-
но понизить известью, которая предпочтительнее цианида
вследствие высокой его стоимости. Также полезно применять
соли закиси и окиси железа вместе с известью [661.
В случае использования крезиловой кислоты и креозота
оптимальная температура пульпы ~25° Ill.
Щелочные алкилсульфаты были рекомендованы для флота-
ции углей как эмульгаторы для углеводородных масел [241.
Морская вода или растворы солей были предложены в качест-
ве среды, в которой уголь флотируется без пенообразователя и
собирателя. Это предложение может представлять интерес для
селективной флотации угольных минералов и заслуживает
дальнейшего изучения [35, 381.
Практические предложения по флотации углей. Для улуч-
шения флотируемости относительно крупного угля Прайс [45]
рекомендует выделять газ из пульпы, содержащей реагенты,
понижением давления над ней после предварительного воздей-
ствия на пульпу высокого давления в присутствии относитель-
но растворимого газа (такого, как СО2). Это интересный ва-
риант вакуумной флотации.
Было предложено несколько вариантов обогащения флота-
цией угольной мелочи, для случаев когда широкий диапазон
крупности продукта препятствовал получению хорошего каче-
ства концентрата и высокого извлечения угля [10, 25]. Большин-
АПОЛЯРНЫЕ МИНЕРАЛЫ
639*
ство решений включало классификацию пульпы с последующей,
раздельной флотацией различных классов крупности.
Эриксон 116] предложил различные комбинации гравита-
ционного и флотационного обогащения для разделения угля,,
пирита и сланца.
Образование вязкой устойчивой пены, которая трудно сгу-
щается и флотируется, представляет одну из трудностей -при.
практическом применении флотации для обогащения углей.
Франц и Бертольд 119] предложили для устранения ее добав-
лять в пену (обработанную кислотой или щелочью) суспен-
зию, в которую входит мука или другой протеин — углеводный
растительный продукт. Предлагалось также устранить это за-
труднение механическим способом, используя звуковые или
ультразвуковые колебания 156].
Поскольку уголь составляет основную массу исходного про-
дукта, подвергаемого флотации, Бишоп 16] предложил обрат-
ную флотацию, при которой уголь депрессируется, а флоти-
руется пустая порода (такая, как силикаты). Следует отме-
тить, что результаты получились посредственные, а расход
реагентов слишком велик, но сама идея довольно заманчива..
Были предложены такие реагенты, как крахмал, или эквива-
лентные ему защитные коллоиды, соль амина и древесной смо-
лы, сосновое масло и керосин при обесшламленном питании
(28—200 меш).
Выделение фюзена 165]. Один из способов отделения антрак-
силона и аттрита от фюзена состоит в том, чтобы создать усло-
вия для относительно медленной флотации, например с одним
пенообразователем, и перечищать пенные продукты. Таким
образом конечный концентрат обогащается желаемыми компо-
нентами и относительно большая часть фюзена переходит в
хвосты.
Согласно Прайсу [44], Эдсеру и Вильямсу 115] и Майеру и
Шранцу [37], крахмал и защитные коллоиды замедляют фло-
тацию фюзена более, чем антраксилона. Их применение по-
этому должно дать лучшие результаты в разделении компо-
нентов угля за счет подавления фюзена. Напротив, Кюловейн
[32] и Шефер и Мертенс Ш] применяли гидролизованный по-
лигексоз для замедления флотации антраксилона. Так как
крахмал тоже представляет собой полигексоз, то этот вопрос
остается невыясненным. Бирбрауэр и Поперле [4, 5] для гидро-
филизации поверхности антраксилона рекомендуют окисляющие
агенты, подавляющие его в первые моменты флотации.
Получение особо чистого угля. Особо чистый уголь необхо-
дим в качестве исходного материала для изготовления элек-
тродов. Эти электроды применяются в больших количествах
для производства алюминия при расходе их около 1 т на 1 т
640
ГЛАВА 18
получаемого металла. Электроды должны быть гораздо чище,
чем обычный кокс. Угольные электроды производятся главным
образом из нефтяных продуктов, но получение их из угля, осо-
бенно в некоторых районах, гораздо экономичнее.
Рядовой уголь
Дробление
Класс ищикация
Крупный класс Тонкий класс
Разделение 8 тяжелых
| с ре Оах |
Антракс слоновая , Сланец в
фракция ____________отвал к
f Фракция короткое
Измельчение пламенных углей
Крупный товар-
ный уголь
И нога ст адсальная
српотация
Ч Флотация
Очищенный
КислотааИтР:Рмипон
~ \ * Раствор
ВыщелачСхЬание
Вода I ~
Фильтрация и
промывка
Брикетирование
‘ Мелкий уголь Мелкая породе
доя топлива
Товарный
уголь
f мывной во-
Антракси- ды
лон для элек-
тродов
Рис. 4. Схема получения сверхчистого угля
Сверхчистый уголь можно получать, применяя флотацию
вместе с другими операциями 110, 641. Например, сначала из
угля подходящего сорта с высоким содержанием антраксилона
выделяется основная часть золы отсадкой, в тяжелых суспен-
зиях и электростатической сепарацией. Затем следует измель-
чение до флотационной крупности, флотация с несколькими
перечистками, чтобы выделить антраксилон, содержащий 0,5—
1,0% золы. Флотационный концентрат окончательно очищает-
ся выщелачиванием кислотой, понижающим содержание золы
до 0,25%. На рис. 4 представлена схема такого процесса.
АПОЛЯРНЫЕ МИНЕРАЛЫ
641
ЛИТЕРАТУРА
1. Bailey R., and Р. F. Whelan. The influence of pulp temperature on the
froth flotation of four British fine coals, J. Inst. Fuel, 25, 304—307, 1953.
2. Bessel Brothers. German Patents 42/1877, 39,369/1886.
3. Bertrand Maurice F. Relation entre les compositions petragra-
phiques et chimiques de quelques charbons purifies, Rev. universelle mines
(Liege), 15, 137—151, 1939.
4. Bierbrauer E., and J. P о p p e r 1 e. Selective flotation of coal from a
chemical standpoint, Gltickauf, 70, 933—940, 1934.
5. В i er b r a u e r, E., and. J. Popper le. German Patents 619,239/1935;
650,031/1937.
6. В i s h о p William T. U. S. Patent 2,633,240/1953.
7. Brady G. A., and A. W. Gauger. Properties of coal surfaces Ind.
Eng. Chem., 32, 1599—1604, 1940.
8. Chapman W. R. Differential froth flotation applied to coal, Fuel, 1, 52—
54, 1922.
9. Clemmer J. Bruce, Richard W. Smith, В. H. Clemmons, and
R. H. Stacy. Flotation of weathered Alabama graphitic schists for cru-
cible flake, Geol. Surv. Alabama, Bull. 49, 101 pp., 1941.
10. Crawford A. Preparation of ultra-clean coal in Germany, Trans.
Inst. Mining Engrs. (London), 111, 204—218, 1952.
11. Crawford Bruce D., and Harry A. Grine. U. S. Pa-
tent 2,136,074/1938.
12. Crawford J. T. Importance of pulp density, particle size and feed regula-
tion in flotation of coal, Trans. AIMME, 119, Contrib. 86, 1936.
13. Davis D. H. Froth flotation of minus 48-mesh bituminous coal slurries,
AIMME Tech. Publ. 2209, Coal Technol., 2 (3), 1947.
14. Deitz Victor R. «Bibliography of Solid Adsorbents, 1900 to 1942», Na-
tional Bureau of Standards, Government Printing Office, Washington,
D. C„ 1944.
15. Edser E., and P. T. Williams. The cleaning of coal by froth flota-
tion, Trans. Fuel Conf., World Power Conf., London, 1928, 1, 374—384,
1929.
16. Erickson, Stephen E. U. S. Patent 2,330,479/1943.
17. Schopf James M. Petrologic methods for application to solid fuels of
the future, Mining Eng., 8, 629—639, 1956.
18. For ba th Thomas P. Sulfur recovery from low-grade surface depo-
sits, Mining Eng., 7 (9), 881—885, 1953.
19. Frantz Philip M., and William E. Bertholf. U. S. Pa-
tent 2,311,527/1943.
20. Gandrud B. W., G. D. Coe, C. S. Benefield, and I. N. Skelton.
The flotation of Alabama graphite ores, U. S, Bur. Mines, Rept. Invest.
3225, 1934.
21. Gandrud B. W., Thomas Fraser, and H. F. Yancey. Froth flo-
tation practice in coal-preparation plants of Western Europe and Great
Britain, U. S. Bur. Mines, Inform. Circ. 7614 17 pp., 1951.
22. Gandrud B. W., and H. L. Riley. A combination cleaning and dewa-
tering process for treating fine sizes of coal, U. S. Bur Mines Rept. In-
vest. 4306, 1948.
23. G i 11 e t Alfred. La molecule de houille, Bull. soc. chim. Beige, 57,
298—306, 1948.
24. Gill son J. L., and С. T. Mentzner, Jr. U. S. Patent 2,112,362/1938.
25. Hedley Norman. U.S. Patent 2,136,341/1938.
26. Horsley R. M. Oily collectors in coal flotation, Trans. Inst. Mining
Engrs (London), 111, 886—894, 1952.
27. Horsley R. M., H. El-Sinbawy, and H. G. Smith. Xanthates in
coal flotation, Fuel, 31, 302—311, 1952.
41 4. M. Годэи
6 42
ГЛАВА 18
28. Horsley R. M., and H. G. Smith. Principles of coal flotation, Fuel,
30, 54—63, 1951.
29. Horton L. Seperation of coals into fractions of different densities, Fuel,
31, 341—354, 1952.
30. Horton L., R. B. Randall, and К. V. Aubrey. The constitution of
coal. Summary of existing knowledge, Fuel, 23, 65—80, 100—109, 1944.
31. Jones F. B., and E. Bury. U. S. Patent 1,388,868/1921.
32. Kuhlwein F. Removal of fusain by selective flotation of coal Gliiskauf,
70, 245—252, 275—277, 1934.
33. L e s s i n g R. The rational preparation of coal, J. Inst. Fuel, 17, 15—24,
33—39, 1943.
34. Lowry H. H., ed. «Chemistry of Coal Utilization», 2 vols., John Wiley &
Sons, Inc., New York, 1945.
35. Л ы с ен к о П. Д. Обогащение лисичанских длиннопламенных углей.
«Кокс и химия», 1935, 5(10), 40—46.
36. Majumdar К. К. Beneficiation of graphite from Mysore State, J. Sci.
Ind. Research (India) 7B, 167—168, 1948.
37. Mayer E. W., and H. Schranz. «Flotation», S. Hirzel Verlag, Leipzig,
1931.
38. Minerals Separation Ltd. German Patent 478,065/1920.
39. M u c k F. «Grundziige und Zeile der Steinkohlen-Chemie», Strauss,
Bonn, 1881.
40. Muck F. «Die Chemie der Steinkohle», W. Engelmann, Leipzig, 1891.
41. Mukherjee В. C. Studies in the reflectance of coal and of some forms
of carbon by vertically incident light, Fuel, 31, 153—158, 1952.
42. Na г а у a n a n P. I. A. The beneficiation of a sulfur ore from Koh-i-
sultan, Baluchistan, Trans. Mining Geol. Met. Inst. India, 41, 129—153,
1946.
43. Narayanan P. I. A., and H. H. Robot tom. The beneficiation of a
flake graphite ore from Kalahandi State, India, Trans. Mining Geol. Met.
Inst. India, 41, 107—127, 1946.
44. P r i c e F. G. U. S. Patent 1,499,872/1924.
45. Price J. D. U. S. Patent 2,142,207/1939.
46. Ralston Oliver C. Comparison of froth with Trent process, Coal Age,
22, 911—914, 1922.
47. R i 1 e у H. L. The macromolecular structure of bitominous coal, Bull. soc.
chim. Beiges, 57, 400—415, 1948 .
48. Schafer W„ and W. Mertens. U. S. Patent 1,944,529/1933.
49. S c h i f f m a n L. E. Kerosene flotatron of bituminous-coal fines. AIMME
187, Tech. Publ. 2918-F, pp. 1047—1056, 1950.
50. Seyler C. A. Characteristic petrological components of coal, Fuel, 31,
159—170, 1952.
51. Sprunk George C., and H. J. O’Donnell. Mineral matter in coal,
U. S. Bur. Mines, Tech. Paper 648, 1942.
52. S t a c h Erich. «Lehrbuch der Kohlenpetrographie», Verlagsbuchhandlung
Gerbruder, Borntrpger, Berlin, 1935.
52a. Stach Erich. «Lehrbuch der Kohlenmikroskopie», vol. 1, Gliickauf.
Kettwig, 1949.
53. S t a c h Erich. Die Vitrit-durit Mischungen in der petrographischen
Kohlenanalysen, Brennstoff-Chem., 33, 361—370, 1952.
54. Staley, W. W. and L. S. Prater. Sulfur in Idaho, Idaho Bur. Mines
Geol., Mining Research Rept. 2, pp. 1—7, 1945.
55. Stopes Marie C. Studies in the composition of coal. I. The four vi-
sible ingredients in banded bituminous coal. Proc. Roy. Soc. (London),
90B, 470—487, 1919.
56. Sun S. C. Destruction of flotation froth with intense high-frequency
sound, Mining Eng., 3, 865—867, 1951.
АПОЛЯРНЫЕ МИНЕРАЛЫ
643
57. Sun S. С., and R. E. Zimmerman. The mechanism of coarse coal and
mineral frath flotations, Trans. AIMME, 187, Tech. Publ 2843-F,
pp. 616—622, 1950.
58. Taggart Arthur F. «Elements of Ore Dressing», (a) p. 249, John Wi-
ley & Sons, Inc., New York, 1951.
59. T a g g a r t Arthur F., G. R. M. del G i u d i с e, A. M. S a d 1 e r, and
M. Hassialis. Oil-air separation of non-sulfide and non-metal
minerals. Trans. AIMME, 134, 180—206, 1939.
60. Thiessen Reinhardt. Complication and composition of bituminous
coal, J. Geol., 28, 185—209, 1920.
61. Thiessen Reinhardt. Structure in paleozoic bituminous coals, U. S.
Bur. Mines, Bull. 117, 292 pp., 1920.
62. We a v in d R. G., I. Wolf, and R. S. Young. Flotation of diamonds,
Mining Eng., 3 (7), 596, 1951.
63. W h e 1 a n P. F. Froth flotation reagents for coal: distillation fractions of
commercial oils and some simple phenolic compounds, J. Appl. Chem. (Lon-
don), 3, 289—301, 1953.
64. Yancey H. F., and J. A. Taylor. Froth flotation of coal; sulfur and
ash reduction, U. S. Bur. Mines, Rept, Invest. 3263, 1935.
65. Yancey H. F., and M. R. Geer. Cleaning of coal, in H. H. Lowry, ed.,
«Chemistry of Coal Utilization», chap. 16, John Wiley & Sons, Inc., New
York, 1945.
66. Y a n с e у H. F., and Thomas Fraser. Coal preparation in Germany
and the Netherlands, AIMME, Tech. Publ. 2112, Coal Technol., 1 (4), 1946.
67. Z i m m e r m a n R. E. Flotation of bituminous coal, AIMME, Tech. Publ.
2397, Coal Technol., 3, 1948.
41»
Глава 19
РАЗЛИЧНЫЕ ОРГАНИЧЕСКИЕ ВЕЩЕСТВА
В последние годы флотационным способом начали пользо-
ваться для обогащения самых разнообразных веществ, глав-
ным образом органического происхождения. В этой области
удалось найти ряд оригинальных решений, которые могут дать
.значительный экономический эффект и заслуживают дальней-
шего изучения.
Нефть, битумы, смолы, каучук
Нефть. Из нефтяных скважин нефть часто поступает вместе
с соляными растворами. Эта эмульгированная смесь обычно
подвергается отстаиванию, после чего производится разделе-
ние при помощи гравитации. Дарси [9] предлагает использо-
вать флотацию для улучшения существующей технологии. Он
полагает, что данный метод можно применить и к сточным во-
дам нефтеочистительных заводов; в этих водах капельки неф-
тепродуктов находятся во взвешенном состоянии в водных рас-
творах разной степени разбавления.
Флотация находящейся в спокойном состоянии жидкости
осуществляется под вакуумом; при этом из раствора выделя-
ются мелкие пузырьки воздуха, объем которых возрастает по
мере увеличения вакуума. Предварительно раствор насыщает-
ся воздухом под давлением, благодаря чему количество рас-
творенного воздуха увеличивается; для образования флокул
добавляют соли окисного железа или квасцы вместе с жидким
стеклом. Вакуумная флотация отличается высокой селектив-
ностью как в данном случае, так и во многих других, описы-
ваемых в настоящей главе. Это объясняется особенностью про-
цесса вакуумной флотации, при которой пузырьки зарождают-
ся на поверхности частиц, в то время как без вакуума встреча
пузырька с частицей затрудняется из-за недостаточной разни-
цы в удельных весах воды и частиц.
Битумы. Бауэр и Меттью [2] предложили способ извлечения
битума из битумсодержащих песков. Процесс состоит: а) в дез-
интеграции материала в водной суспензии при подогреве в
присутствии жидкого стекла и керосина; б) последующей фло-
тации (частично с помощью воздушных пузырьков) для отде-
ления битума от песков и глины.
РАЗЛИЧНЫЕ ОРГАНИЧЕСКИЕ ВЕЩЕСТВА
645
Для выделения битумсодержащих продуктов из горючих
сланцев предложил метод пенной флотации [36]. Основными
реагентами являются такие органические вещества, как масла
древесной смолы и сланцевая смола. Этот процесс может най-
ти применение при не слишком тесном прорастании разделяе-
мых минералов.
Копаловая смола. Разделение копаловой смолы и воска, ко-
торые встречаются в некоторых сортах битуминозного угля,
может быть осуществлено или гравитационным методом с при-
менением соляного раствора надлежащего удельного веса, или
методом пенной флотации. Нагельворт [26] предлагает смачи-
вать уголь раствором дубильной кислоты (1 часть кислоты на
1 000 000 частей воды) и затем выделять флотацией продукт,
представляющий собой смесь смолы, битума и воска. Разделе-
ние этих компонентов производится затем при помощи флота-
ции в тяжелых жидкостях, что осуществляется в водном рас-
творе, имеющем удельный вес выше, чем вода, и содержащем
дубильную кислоту в количестве около 10 частей на
1 000 000 частей раствора. В результате воск флотируется, а
копаловая смола тонет. Окончательная очистка смолы произ-
водится гравитацией.
Каучук. Для выделения каучука из растений, которые не
принадлежат к числу обычных каучуконосов, во время войны
было сделано два предложения, включающие метод флотации.
Тюррол и его сотрудники [35] применяют этот метод к молочаю.
Растения подвергают сушке, выщелачивают кипячением в
1%-ном растворе NaOH для растворения растительных белков
и углеводородов, затем промывают до pH = 9—10 и сгущают
до содержания 10% твердого. После этого пульпу направляют
в шаровую мельницу, в которой каучук собирается в «червеоб-
разные» комочки. Это очень ответственная операция. Измель-
ченную в шаровой мельнице пульпу разбавляют и флотируют
(без добавки реагентов). Извлечение каучука составляет 70—
9О°/о, извлечение сопутствующих смол — около 50—60%.
Эскью [17] выделяет натуральный каучук из кок-сагыза:
Корни этого растения содержат около 10% каучука. Собира-
ют их в определенное время года, -высушивают на воздухе и
выщелачивают кипячением в 0,1%-ном растворе кальциниро-
ванной соды. Остаток от выщелачивания пропускают через
шаровую мельницу для образования «червеобразных» комоч-
ков или капелек каучука, промывают на тонком сите и флоти-
руют для отделения каучука от целлюлозы и других посторон-
них примесей. Флотация должна проводиться в спокойной
среде (пульпе). Пока не установлено, происходит ли при этом
разделение в результате пенной флотации или главным обра-
зом всплывание из-за потери веса в воде.
646
ГЛАВА 19
Семена и продукты растительного происхождения
Бобы. Флотацию применили для очистки бобов перед их
консервированием [27, 28]. Часто в бобах содержатся в качестве
основных примесей некоторые семена и семенные коробочки.
Они имеют примерно тот же размер, что и самые бобы (на-
пример, ягоды паслена и семена сорной травы). Разделение
происходит при загрузке бобов в аэрированную эмульсию очи-
щенного углеводорода в разбавленном растворе лаурилсульфа-
та натрия. При этом бобы смачиваются и тонут, а примеси
флотируются. Эмульсия используется в замкнутом цикле, и
поэтому, кроме добавки моющего средства, никаких реагентов
не требуется. Эмульсия содержит 1°/о масла при концентрации
моющего средства около 0,13 г/л. В начале операции моющее
средство не загружается. В этих условиях весь материал
всплывает. Затем, когда моющее средство начинают добавлять
в возрастающей дозе, полноценные бобы тонут, а поврежден-
ные бобы при нормальных условиях флотируются вместе с
примесями. Особое внимание уделяется тому, чтобы не повре-
дить бобы.
Семена. Томас Эрл [13] запатентовал процесс селективной
флотации, при котором разделяются семена различных расте-
ний или же проросшие и непроросшие семена одинаковых рас-
тений. При этом требуются следующие реагенты: вспениватель
для семян с твердой кожицей или для семян с шелухой (на-
пример, для овса) и вспениватель вместе с карбоксильным со-
бирателем для обмолоченных семян. При разделении непро-
росших и проросших семян наиболее вероятна флотация про-
росших.
Спорынья. Сазерленд с сотрудниками разработал метод от-
деления спорыньи от зерен ржи. с помощью пенной флотации
[30]. Спорынья представляет собой рогообразный гриб, кото-
рый развивается на колосьях ржи, занимая место зерен. Раз-
деление осуществляется в эмульсии углеводородов нефти (ке-
росина) и мыла. При этом зерна ржи флотируются, а спорынья
тонет.
Процесс состоит из следующих операций: 1) отделения
большей части зерен ржи в соляном растворе удельного веса
1,13 (зерна ржи при этом тонут); 2) обезвоживания всплывше-
го продукта, прибавления к нему эмульсии и высушивания до
содержания влаги 15%; 3) погружения обрабатываемого про-
дукта в чан с водой и вспенивателем (при этом спорынья сма-
чивается, а зерна ржи не смачиваются).
Измельченные семена. Богатые маслом фракции семян мож-
но отделить от остальных посредством флотации, используя
природную гидрофобность маслянистых частиц для их закреп-
РАЗЛИЧНЫЕ ОРГАНИЧЕСКИЕ ВЕЩЕСТВА
647
ления на пузырьках воздуха 1211. Перед флотацией семена из-
мельчаются в галечной мельнице, в результате чего освобожда-
ются богатые маслом частицы. Эта операция напоминает опи-
санную выше схему извлечения каучука из корней кок-сагыза
или молочая.
Из Германии сообщалось ПО, 29] про флотационное отделе-
ние крахмала от клейковины.
Очистка растворов сахара. Растворы тростникового сахара
можно очищать при помощи пенной флотации в обычных ма-
шинах, например субаэрационного типа, без добавления ре-
агентов. Обычно даже после добавки извести и осаждения в
сгустителях получаемый раствор сахара не является чистым и
последующая его фильтрация затруднительна вследствие бы-
строго забивания фильтров. После того как раствор подвергал-
ся пенной флотации, вязкость его значительно снижалась, в
связи с чем раствор лучше концентрировался при выпаривании.
Бумажные пульпы
Белая вода. Производство бумаги осуществляется на широ-
ких бесконечных тканых проволочных ситах, имеющих чрезвы-
чайно мелкие отверстия (машины Фордриньера); при этом на
них наливают разбавленную суспензию бумажных волокон.
Эти подвижные сита подвергаются непрерывному поперечно-
му встряхиванию, что способствует сбиванию войлока из во-
локон и удалению воды.
Значительная часть волокон и сопутствующих компонентов
бумажной пульпы — смолистой шихты, клея, казеина, глины
и прочих минеральных пигментов — переходит в «белую воду».
Сбрасывание этой воды в водоемы вызывает не только значи-
тельное их загрязнение, но и дает высокие потери ценных для
бумажного производства материалов.
Во избежание этого обычно «белую воду» сгущают, а слив
возвращают в процесс. Однако такой метод встречает
возражения по ряду причин, одной из которых является то,
что некоторые компоненты бумаги подвергаются при этом бак-
териологическому разрушению.
Применение вакуумной флотации (1, 6] позволяет преодо-
леть эти трудности. Процесс ведется следующим образом:
пульпа, содержащая волокно, при очень небольшом количест-
ве минеральных примесей (не выше 0,05—0,1% от общего ко-
личества твердого) сначала насыщается воздухом; после того
как улетучились большие воздушные пузырьки, суспензию пе-
рекачивают наверх в вакуумную камеру по трубе большого
диаметра. При этом сравнительно медленном переходе (про-
должительностью около 1 мин.) от атмосферного давления к
648
ГЛАВА 19
вакууму выделяется большое количество исключительно мел-
ких пузырьков воздуха, частично на границе раздела твер-
дое — вода. Это способствует образованию плавающей массы,
состоящей из целлюлозы, других твердых веществ и мелких
пузырьков воздуха. Эту массу затем удаляют с поверхности с
помощью особых приспособлений, действие которых также
основано на применении вакуума.
Некоторое внимание было уделено изучению химизма фло-
тации бумажной пульпы [31], однако основные усилия были
направлены на улучшение работы аппаратов и механизмов.
При данном процессе обычно никаких реагентов добавлять
не требуется, так как в «белой воде» имеется значительное ко-
личество поверхностно активных веществ.
Было предложено [4] разделять твердые частицы, содержа-
щиеся в «белой воде», с помощью селективной флотации; од-
нако данных о промышленном применении этого метода не
имеется.
Бумажные отходы. Из пульпы, полученной из бумажных
отходов, флотируют чернила, запачканные чернилами волокна
и пигменты. Бумажные отходы, особенно из лучших сортов
писчей и печатной бумаги, в основном состоят из глины, угле-
кислого кальция in других минеральных пигментов, склеенных
с целлюлозой связующим веществом (наподобие казеина). При
переработке бумажных отходов из них стремятся извлечь толь-
ко целлюлозу. Бумага разваривается при нагревании в до-
вольно крепком растворе щелочи. Перед флотацией чернил
большая часть щелочи и образовавшегося мыла должна быть
предварительно удалена. Данные о полученных при этом ре-
зультатах немногочисленны.
Продукты текстильной промышленности
Сравнительная флотируемость волокон. В связи с примене-
нием флотации в текстильной промышленности было проведе-
но несколько интересных работ. Престон и Саа [32] произвели
сравнение флотируемости различных типов волокон. Волокна,
разрезанные на кусочки длиной до 0,5 мм, испытывались врозь
или в смеси при pH от 2 до 12 и при разбавлении 2000 : 1. Пер-
вые опыты проводились без реагентов; в последующих опытах
добавлялись пенообразователи. При этом было выявлено, что
синтетические волокна и шерсть флотируются хорошо при лю-
бом значении pH без добавки реагентов. Шелк (ацетатный или
вискозный) не флотируется ни при каком значении pH, а на-
туральный шелк, казеиновое волокно и нейлон флотируются в
кислой, но не в щелочной среде. В этой группе лучше всех фло-
тируется казеиновое волокно, хуже всех — нейлон. Очевидно,
флотируемость зависит от структуры волокна.
РАЗЛИЧНЫЕ ОРГАНИЧЕСКИЕ ВЕЩЕСТВА
649
Красители. Флотационное поведение различных красителей,
применяемых в текстильной промышленности, было изучено
Магоффином и его сотрудниками [7, 25, 34]. Красители флоти-
ровались в виде отдельных нерастворившихся частиц. Испыта-
ния проводились главным образом с индиго (цветной индекс
№ 1177), красным конго и сернистым черным (цветной индекс
№ 978). При флотации в качестве реагентов испытывались
натриевые алкилсульфаты и амины. Установлена связь между
флотационными свойствами и электрокинетическими потен-
циалами. Обычно амины обеспечивали лучшие результаты.
Прядильные ванны для вискозного шелка. Основной опера-
цией при производстве вискозного шелка является пропускание
вязкой жидкости через фильеры в кислотную ванну для обра-
зования нитей. При этом происходит реакция, в результате
которой нити затвердевают, а ванна загрязняется примесями.
Для очистки ванны была предложена и использована флотация
как метод непрерывного удаления твердых примесей [3, 8]. За-
дача состоит в удалении серы и ее соединений, выделяющихся
из применяемого для получения ксантогената целлюлозы (вис-
козы) сероуглерода, а также металлических частиц, попадаю-
щих .в ванну в связи с износом аппаратуры. В одном случае
содержание примесей, составлявшее 20 частей на 1 000 000, было
снижено до 1 части, причем в перечищенном пенном продукте
это содержание достигало 500—1000 частей на 1000 000. Хвосты
перечисткой флотации возвращались с новой порцией питания
во флотационную машину. Субаэрационные камеры машины за-
крыли прозрачной крышкой из пластиката, чтобы предотвратить
распыление кислотного раствора в фабричной атмосфере.
Продукты животного происхождения и молочные
Было предложено использовать флотацию для извлечения
протеинов молока [20], а также для обезжиривания животных
тканей, например свиной кожи [37]. Процесс этот также приго-
ден для обработки растворов, получаемых от очистки шерсти [5].
Такие растворы являются щелочными и содержат около 1% жи-
ра— вещества, весьма ценного для производства ланолина. При
коагуляции кислотами, например серной (до концентрации 0,5%
в растворе) и при добавке небольших количеств керосина (до
концентрации 0,2% в растворе) извлечение жира методом пен-
ной флотации достигает 90% и выше.
Весьма интересное сообщение сделано Доньо [11, 12], кото-
рый предлагает применить флотацию для удаления из вод-
ных суспензий различных бактерий. Например, туберкулезные
бациллы Коха флотируются очень легко, в то время как кишеч-
ные палочки флотируются плохо. При соответствующих усло-
виях можно получить очень высокое извлечение бактерий.
650
ГЛАВА 19
Очистка вод
Для очистки воды Хоппер [23] предложил применять фло-
тацию, при которой вредные примеси концентрируются в пен-
ном продукте. В проведенных им опытах из различных проб во-
ды извлекалось до 90—95% содержавшейся в них мути при од-
новременном десятикратном снижении содержания различных
бактерий.
Более поздними исследованиями [24], значительно расширив-
шими область применения флотации при очистке вод, была
установлена возможность удаления из водной среды однокле-
точных организмов, вызывающих дизентерию, а также вирусов
гриппа. В качестве флотационного реагента применялся цетил-
пиридинийхлорид, загружаемый в количестве 10 частей на
1 000 000 частей воды.
Промышленные и хозяйственно-фекальные сточные воды
Метод флотации можно использовать для снижения количе-
ства нежелательных составляющих в промышленных и хозяй-
ственно-фекальных сточных водах при одновременном получе-
нии из части этих отходов ценных удобрений. Обычно эти отхо-
ды чрезвычайно сильно разбавлены и поэтому удаление невод-
ных примесей является одной из трудных проблем при обезвре-
живании сточных вод.
Флотация может быть применена для обработки специфиче-
ских отходов с высоким содержанием жиров (мясохладобоен),
мыл (прачечных), масел и моющих средств (машиностроитель-
ных заводов), а также для обезвреживания хозяйственно-фе-
кальных и прочих сточных вод.
Элиассен и его сотрудники [15,. 16] изучали флотацию приме-
нительно к различным промышленным отходам. Отходы прачеч-
ных, которые имели pH около 10 и содержали всего 1500 частей
твердого на 1 000 000 частей воды, были обработаны методом
вакуумной флотации после добавки специальных реагентов-фло-
кулянтов. Наилучшим из них оказалось хлорное железо, кото-
рое при дозировке 500 частей на 1 000 000 частей воды снижало
pH пульпы до 6,7. Удаление жира и снижение биохимической
потребности в кислороде (ВПК) составляло соответственно око-
ло 95 и 85%. Применение квасцов также давало эффект, но
меньший по сравнению с хлорным железом.
Хансен и Готаас [22] изучали применение флотации к обыч-
ным сточным водам. Они установили исключительную эффек-
тивность действия додециламина, при котором достигалось сни-
жение количества взвешенных твердых частиц на 95—99%, бак-
терий на 99—99,9%, растворенных твердых веществ на 25—
РАЗЛИЧНЫЕ ОРГАНИЧЕСКИЕ ВЕЩЕСТВА
651
35%, БПК примерно на 50%. Однако стоимость этой операции
оказалась очень высокой, так как расход реагента был весьма
значительным.
Возможно, что дальнейшие испытания с применением ами-
нов с длинной углеводородной цепью дадут более экономичные
решения. Интересно, что амины оказывают сильное дезинфици-
рующее действие.
Для флотации сточных вод и промышленных отходов метод
вакуумной флотации представляется наиболее пригодным и
удобным. Прекрасное описание используемой при этом аппара-
туры дано Фишером [18, 19].
На другую возможность применения флотации при очистке
сточных вод указал Рудольф [33], который предложил исполь-
зовать ее для получения шламистого продукта большей плот-
ности, чем это возможно при обычном сгущении. Опыты, кото-
рые он описывает, были проведены с активированным шламом,
полученным, по-видимому, при осаждении, но содержащим все-
го 0,5% твердого. При сгущении этого материала содержание
твердого в нем повысилось только до 2,5—3%. При проведении
же операции вакуумной флотации с добавкой хлорной извести
был получен флотационный пенный продукт, содержащий 6—
10% твердого. Очевидно, при этом выделяется хлор, который
реагируя со шламами, образует пузырьки углекислоты. При
этом значение pH несколько снижается (в одном случае оно
снизилось с 7,2 до 6,4).
Дегидратирующее действие, достигаемое при помощи флота-
ции, вызвало большой интерес к применению процесса Рудольфа.
Растворимые вещества
При некоторых условиях методом пенной флотации можно
разделять вещества, растворенные в воде в небольших количе-
ствах. Если два трудно разделимых вещества одновременно ра-
створены в воде и одно из них является более поверхностно ак-
тивным чем другое, представляется возможным осуществить их
разделение при помощи одного только вспенивателя, получая
отдельно Пенный продукт и остаточный жидкий раствор.
Процесс этот, однако, имеет ограниченное применение, по-
скольку существуют более эффективные методы. Но в отдель-
ных случаях, особенно при научных исследованиях, подобная
схема может представлять несомненный интерес.
ЛИТЕРАТУРА
1. Ansbach Sven. A flotation method for treating white water, Paper
Trade J. 104 (9), 40—44, 1937.
2. Bauer Robert F., and Harold J. Matthews. U. S. Pa-
tent 2,453,060/1948.
652
ГЛАВА 19
3. Booth Robert В. U. S. Patent 2,274,658/1942.
4. Booth Robert B. U. S. Patent 2,347,147/1944.
5. Booth Robert B., and Adolphus Magnum Webb. U. S. Pa-
tent 2,352,365/1944.
6. Brecht W., and K. Scheufelen. Studies with a Sveen Pedersen
flotation save-all, Papier-Fabr., 36, Tech-wiss. Tl., 121—129, 136—
140, 1938.
7. Clanton B. R., F. K. Cameron, and J. E. Magoffin. Removal of
a sulfur black dye from suspension by flotation methods, Textile Research,
10, 201—206, 1940.
8. Costa Joseph L., and William H, Kahler. U . S. Patents
2,336,778/1943; 2,462,948/1949.
9. D’Arcy Nicholas A., Jr. Dissolved-air flotation separates oil from
waste water, Oil Gas J., 50 (27), 319—322, 1951.
10. Deutsche Maizena Ges, German Patent 676,500/1939,
11. Dognon Andre. Concentration et separation des molecules et des par-
ticules par la methode des mousses, Rev. sci., 79, 613—619, 1941.
12. Dognon Andre. Froth concentration and separation. Chemical and ba-
cteriological applications, Bull. soc. chim. biol., 23, 249—262, 1941.
13. Earle Thomas. U. S. Patent 2,155,219/1939; French Patent 846,666/1939.
14. E g u c h i G. M., and Clinton H. Wells. U. S. Patent 2,518,296/1950.
15. Eliassen Rolf, and Henry B. Schulhoff. Laundry-waste treat-
ment by flotation, Water Works & Sewerage, 90, 418—421, 1943.
16. Eliassen Rolf and Henry B. Schulhoff. Grease removal by va-
cuum flotation, Sewage Works J., 16, 287—295, 1944.
17. Eskew Roderick K. Natural rubber from Russian dandelion, India
Rubber World, 113, 517—520, 1946.
18. Fischer A. J. Vacuum flotation of sewage and industrial wastes, Mich.
Eng. Expt. Sta., Bull. 98, pp. 13—28, 1943.
19. Fischer A. J. Vacuum flotation: a new process for sewage and indust-
rial waste treatment, Eng. News-Record, 130, 297—301, 1943.
20. Fritzberg Edward L. U. S. Patent 2,368,919/1945.
21. Grace N. H., and J. B. Palmer. Oil concentration by the froth flota-
tion of pebble-milled seed, Can. J. Research, 24F, 338—347, 1946.
22. Hansen Chris H. and Harold B. Gotaas. Sewage treatment by
flotation, Sewage Works J., 15, 242—252, 1943.
23. Hopper Samuel H. Water purification by flotation, J. Am. Water
Works Assoc., 37, 302, 1945.
24. Hopper Samuel H., and N. Khoobyarian. The flotation method
for the recovery of virus, in press, November, 1955, meeting, American So-
ciety of Tropical Medicine and Hygiene.
25. Magoffin J. E., and B. R. Clanton. Fundamental properties of tex-
tile-processing wates. VIII, Flotation of suspensions. Textile Research,
8, 357—363, 1938.
26. Nagelvoort Adriaan. U. S. Patent 2,378,152/1945.
27. Neubert A. M., Using froth flotation to cleanvined canning peas, Food,
РАЗЛИЧНЫЕ ОРГАНИЧЕСКИЕ ВЕЩЕСТВА
653
Inds., 19, 769—772, 890, 892, 1947.
28. Neubert А. М., and М. К. Veldhuis. Cleaning vined canning peas
by froth flotation, Food Inds., 17, 494—497, 608—616, 1945.
A . 29. Ostwald Wolfgang, Albrecht Siehr, and HansErbring.
German Patent 714,178/1941.
30. P 1 a n t e E n i d C., and K- L. Sutherland. Separation of ergot from
rye corn, J. Council Sci. Ind. Research, 16, 28, 1943.
31. Poor E. N., and H. A. Whiteknight. Flotation treatment of white
water, Paper Trade J„ 114 (9), 21—24, 1942.
32. Preston J. M., and S. Saha. Froth flotation of fibres, J. Textile Inst.
40, T381—388, 1949.
33. Rudolfs Willem. Concentration of activated sludge by compacting and
flotation, Sewage Works J., 15, 642—657, 1943.
34. S u r p r e n a n t L. C., J. E. Magoffin, and F. K- Cameron. Funda-
mental properties of textile wastes. IX. Concentration of sulfur black dyes
' by froth flotation, Textile Research, 11, 126—138, 1941.
35. Tur rail William T., J. Klassen, and H. Smedley. Froth flota-
tion of rubber and resin components of milkweed and other plants that
contain resins, rubber, balata, or related gummy substances. Can. J. Re-
search, 21B, 195—201, 1943.
36. V i s u г a T r e u h a n d-G e s. Swiss Patents 202,474/1939; 205,406/1939.
37. Walter Charles T. U. S. Patent 2,352,154/1944.
Редактор Е. В. Данилова
Редактор издательства М. Л. Ездокова
Технический редактор Е. Б. Вайнштейн
Сдано в производство 11/III 1959 г.
Подписано в печать 7/IX 1959 г.
Бумага 60 X Vie 20,5 + 1 вкл. 0,07.=
= 20,57 бум. л. 41 печ. л.
Уч.-изд. л. 40,36
Т-09923 Заказ 1335
Тираж 2750 Цена 22 р. 15 к.
Типография Металлургиздата,
Москва, Цветной б., 30
ЗАМЕЧЕННЫЕ ОПЕЧАТКИ И ИСПРАВЛЕНИЯ ПЕРЕВОДЧИКОВ
Стра- ница Строка Напечатано Следует чи1аи
24 23 сверху :С1 • :С1-
37 Таблица 3, 1-я графа, 5-я строка сверху РЬ Rb
50 7 сверху Cd (ОН) Cd (ОН)2
54 16 снизу 2п! 4,27 2п! 3,49 4,27 3,49
57 3 сверху °) nd- °и)Р-]
67 16 снизу и ТО
103 1 сверху d/p £/р
3 сверху 1х = То 7о
111 19 сверху диффузия диффузии
126 Рис. 3 Прочно закрепленные;' ионы и вода гидратной оболочки Прочно закрепленные, частично гидратирован- ные ионы
128 10 сверху где р где 8
129 Формула (5) 8 О
130 15 сверху равновесное неравновесное
11 снизу ф = 0 ф0 == 0
132 19 снизу 10"-м., 10~2-м.,
142 6 сверху потенциал раздела - потенциал поверхности раздела
8—7 снизу то же то же
’>4 1 снизу 105" 10" 5
‘>5 18 снизу (е — е у)
58 3 снизу 5 см2 S см2
13 Формула (13) *жг <*жг
; И 5 снизу (13) (13а)
2.52 7 снизу H.,NCNHZ H2NCSN'H,
260 Колонка 1, 9 снизу [i-Нафтиламин а-Нафтиламин
287 14 сверху Очевидно они Очевидно соединения В и С
26 снизу только 97% от этого ко- личества экстрагируется 97% от этого количества не экстрагируется
238 Подпись под рис 4, 3 строка снизу 1 в фильтре 1 в фильтрате
Строка
Напечатано
288
1 строка снизу
ко
*
лонка 4, I строка
13 сверху
1 сверху
Редактор Е. В. Данилова
585
622
Стра
ница
1 снизу
24 снизу
4—3 снизу
11 сверху
Таблица 3, ко-
568
581
305
376
377
517
529
2 сверху
2 и 3 сверху
554
558
невыщелачиваемое
вещество
1 - Na2SO4 = 7- IO”5
радиально
•мплексообразующихся
флотаций
3,85
на Нидерландских госу-
дарственных угольных
шахтах
Редакторы О. С. Богда-
нов и Ц. В. Данилова
исходное количество к
ксантогената. Площадь^
между кривыми 3 и 2 oil
вечает количеству невь<>
щелачиваемого веществ.^
1 — Na2SO4 = 7. !0—4
радикально
комплексообразующих
функций
3,55
Следует читать £
__________________►
--- - >
отфильтрована
и мыла
углеродное
мылом
касную структуру из SiO4
тэтраэдров; в альбите,
ортоклазе и
гидроокислами
Выделение естественных (избл,
боратов методом флота-
ции из вулканических
туфов
в штате Незерланде
снизу
12 снизу
13 сверху
19 сверху
6 сверху
8 снизу
отфлотирована
из мыла
углеводородное
маслом
натрия в структурныр
полости в случае альби-
та, ортоклаза или
гидроксилами
ечение природной
буры из сопочных грязей
методом флотации
638
654
А. М. Г о д эв. Флотация
ПО ОБОГАЩЕНИЮ И АГЛОМЕРАЦИИ РУД
В КНИЖНЫХ МАГАЗИНАХ КНИГОТОРГА
МОЖНО ПРИОБРЕСТИ СЛЕДУЮЩИЕ КНИГИ
Арашкевич В. М. Основы обогащения руд. Допущено
Управлением средних специальных учебных заведений Мини-
стерства высшего образования СССР в качестве учебника для
учащихся цветной металлургии. 1959. Ц. 7 р. 10 к.
Беренов Д. И. Дробильное оборудование обогатитель-
ных и дробильных фабрик. Для горных инженеров-обогатите-
лей, механиков и проектировщиков обогатительных и дробиль-
ных фабрик. 1958. Ц. 11 р. 10 к.
В о з н ы й Р. Ф. Усовершенствование процессов отсадки и
центрифугирования угля на углеобогатительных фабриках. Для
среднего технического персонала и квалифицированных рабо-
чих. 1957. Ц. 95 коп.
Зайцев X. П., МачковскийА. И. Организация и пла-
нирование производства на агломерационных фабриках. Для
инженерно-технических работников агломерационных фабрик.
1959. Ц. 7 р. 70 к.
Кулибин В. А. Подготовка руд к плавке. Издание 2-е пе-
реработанное и дополненное. Для инженеров, техников-обога-
тителей, геологов, горняков, металлургов, экономистов и сту-
дентов вузов. 1959. Ц. 22 р. 35 к.
Никитина. И. иАрбузовВ. А. Агломерация железных
руд. Для мастеров и рабочих агломерационных фабрик и до-
менных цехов. 1957. Ц. 5 р. 60 к.
СвердельИ. С. иКраснянскийЕ. А. Электроисполь-
зование на обогатительных фабриках железорудной промыш-
ленности. Для инженерно-технических работников, мастеров и
высококвалифицированных рабочих. 1959. Ц. 5 р. 85 к.
Циперович М. В. Оборудование углеобогатительных
фабрик. Для инженерно-технических работников производст-
венных предприятий. 1958. Ц. 18 р. 70 к.
ПРИ ОТСУТСТВИИ книг В МЕСТНЫХ книжных
МАГАЗИНАХ ЗАКАЗЫ СЛЕДУЕТ НАПРАВЛЯТЬ ПО
АДРЕСУ: МОСКВА, В-168, 5-я ЧЕРЕМУШКИНСКАЯ
ул., д. 14, „КНИГА ПОЧТОЙ" МАГАЗИН № 93
МОСКНИГОТОРГА.